ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение относится к С-арилглюкозидам, которые являются ингибиторами натрий-зависимых переносчиков глюкозы, обнаруживаемых в кишечнике и почке (SGLT2), и к способу лечения диабета, главным образом, диабета II типа, а также гипергликемии, гиперинсулинемии, ожирения, гипертриглицеридемии, синдрома X, осложенения диабета, атеросклероза и родственных заболеваний, использующему такие С-арильные глюкозиды, одни или в комбинации с одним, двумя или более антидиабетических веществ другого типа и/или с двумя или более терапевтических агентов, таких как гиполипидемические агенты.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Примерно, 100 миллионов человек во всем мире страдают от диабета II типа (NIDDM), который характеризуется гипергликемией вследствие выработки печенью избыточной глюкозы и периферической устойчивости к инсулину, истинные причины которых до сих пор неизвестны. Считают, что гипергликемия является основным фактором риска для развития осложнений диабета и, по-видимому, вносит вклад непосредственно в ослабление секреции инсулина, наблюдаемое при запущенном NIDDM. Можно предположить, что нормализация содержания глюкозы в плазме у больных усилит действие инсулина и отодвинет развитие осложнений диабета. Ожидается, что ингибитор натрий-зависимого переносчика глюкозы способствует нормализации уровня глюкозы в плазме и, вероятно, веса тела за счет усиления секреции глюкозы.
Создание новых безопасных и активных при пероральном приеме антидиабетических агентов также требуется для дополнения существующих лекарственных средств, включающих сульфонилмочевины, тиазолидиндионы, метформин и инсулин, и для того, чтобы избежать возможные побочные эффекты, связанные с применением этих других агентов.
Гипергликемия является признаком диабета II типа (NIDDM); постоянный контроль уровня глюкозы в плазме при даибете может предотвратить развитие осложнений диабета и восполнить недостаток бета-клеток, наблюдаемый при запущенном заболевании. Глюкоза плазмы обычно фильтруется в клубочках почки и активно снова всасывается в проксимальном канальце. По-видимому, SGLT2 является основным переносчиком, ответственным за повторное поглощение (усвоение) глюкозы на этом участке. SGLT-специфический ингибитор флоризин или близкородственные аналоги ингибируют этот процесс повторного поглощения у больных диабетом грызунов и собак, что приводит к нормализации уровня глюкозы в плазме за счет стимулирования выделения глюкозы без побочного эффекта - гипогликемии. Продолжительное (6 месяцев) лечение больных диабетом крыс Zucker с помощью ингибитора SGLT2, как сообщалось, усиливает реакцию инсулина на гликемию, повышает восприимчивость инсулина и приостанавливает наступление (начало) нефропатии и невропатии у этих животных при отсутствии заметной патологии в почках и электролитного дисбаланса в плазме. Селективное ингибирование SGLT2 у больных диабетом, как ожидают, нормализует уровень глюкозы в плазме за счет увеличения выделения глюкозы с мочой, тем самым повышается восприимчивость к инсулину, и предупреждается развитие осложнений диабета.
Девяносто процентов повторного поглощения глюкозы в почках происходит в эпителиальных клетках раннего S1 сегмента почечного кортикального проксимального канальца и, по-видимому, SGLT2 является основным переносчиком, ответственным за это повторное поглощение. SGLT2 представляет собой белок из 672 аминокислот, содержащий 14 заполняющих мембрану сегментов, который преимущественно экспрессирует в раннем S1 сегменте почечных проксимальных канальцев. Специфичность в отношении субстрата, зависимость от натрия и локализация SGLT2 согласуются со свойствами высокой емкости, низкого сродства натрий-зависимого переносчика глюкозы, ранее охарактеризованного как в кортикальных проксимальных почечных канальцах. Кроме того, исследования гибридного истощения вовлекают SGLT2 как предпочтительного переносчика Na+/глюкозы в сегменте S1 проксимального канальца, так как фактически вся активность Na-зависимого транспорта глюкозы, кодируемая в мРНК коркового вещества почки крыс, ингибируется антисмысловым олигонуклеотидом, специфичным к SGLT2 крыс. SGLT2 представляет собой возможный ген некоторых форм глюкозурии, генетического отклонения, при которой реабсорбция (повторное всасывание) глюкозы в почках ослаблено в различной степени. Ни один из этих синдромов, исследованных до настоящего времени, не относят к локусу SGLT2 на хромосоме 16. Однако исследования в высокой степени гомологичных SGLT грызунов в значительной степени указывают на SGLT2 как на основного почечного натрий-зависимого переносчика глюкозы и наводят на мысль, что локус глюкозурии, который картирован, кодирует регулятор SGLT2. Ингибирование SGLT2, как было предсказано, снижает уровень глюкозы в плазме за счет выведения глюкозы у больных диабетом.
SGLT1, другой Na-зависимый сопереносчик глюкозы, который на 60% идентичен SGLT2 на аминокислотном уровне, экспрессирует в тонком кишечнике и в более дистальном сегменте S3 почечного проксимального канальца. Несмотря на сходство их последовательностей, SGLT1 и SGLT2 человека отличаются биохимически. Для SGLT1 молярное соотношение Na+ и транспортируемой глюкозы составляет 2:1, тогда как для SGLT2 это соотношение составляет 1:1. Кm для Na+ равны 32 и 250-300 мМ для SGLT1 и SGLT2 соответственно. Значения Кm для поглощения глюкозы и неметаболизируемого аналога глюкозы α-метил-D-глюкопиранозида (AMG) похожи для SGLT1 и SGLT2, т.е. 0,8 и 1,6 мМ (глюкоза) и 0,4 и 1,6 мМ (AMG) для переносчиков SGLT1 и SGLT2 соответственно. Однако два переносчика отличаются по их субстратной специфичности в отношении сахаров, таких как галактоза, которая является субстратом только для SGLT1.
Введение флоризина, специфичного ингибитора SGLT-активности, доказывает это in vivo, промотируя выведение глюкозы, снижая содержание глюкозы в плазме натощак и после еды и стимулируя использование глюкозы без гипогликемии - побочного эффекта - у нескольких моделей грызунов и у одной модели собак, больных диабетом. Не наблюдалось никакого вредного побочного действия на ионный баланс плазмы, почечную функцию или почечную морфологию в результате двухнедельного применения флоризина. Кроме того, никаких гипогликемических или других вредных эффектов не наблюдалось, когда флоризин вводят нормальным животным, несмотря на наличие глюкозурии. Введение ингибитора SGLT в течение 6 месяцев (Tanabe Seiyaku), как сообщалось, улучшает уровень глюкозы в плазме натощак и после еды, улучшает секрецию и использование инсулина при ожирении у NIDDM-моделей крыс, противостоит развитию нефропатии и невропатии при отсутствии гипогликемии или побочного действия на почки.
Сам флоризин непривлекателен в качестве перорального лекарственного средства, так как он является неспецифичным ингибитором SGLT1/SGLT2, который гидролизуется в кишечнике до его агликона флоритина, который является мощным ингибитором легкого (с меньшими ограничениями) транспорта глюкозы. Конкурентное ингибирование веществ, способствующих переносу глюкозы (GLUT), нежелательно,так как, как можно предсказать, такие ингибиторы обостряют периферическую устойчивость сосудов к инсулину, а также стимулируют гипогликемию в CNS. Ингибирование SGLT1 может также иметь серьезные последствия, как это проиллюстрировано на примере наследственного синдрома недостаточного всасывания глюкозы/галактозы (GGM), при котором мутации в SGLT1-сопереносчике приводят к ослабленному всасыванию глюкозы в кишечнике и к опасной для жизни диарее и обезвоживанию. Биохимические различия между SGLT2 и SGLT1, а также степень дивергенции последовательностей позволяют идентифицировать избирательные ингибиторы SGLT2.
Синдромы семейной глюкозурии представляют собой состояния, при которых кишечный транспорт глюкозы и почечный транспорт других ионов и аминокислот является нормальным. Больные с синдромом глюкозурии, по-видимому, развиваются нормально, имеют нормальные уровни глюкозы в плазме крови и, по-видимому, не страдают от недостаточно хорошего общего состояния здоровья вследствие своего нарушения, несмотря на достаточно высокие, время от времени (110-114 г/день) уровни выведения глюкозы. Основные симптомы, очевидные у этих больных, включают полифагию, полиурию и полидипсию, а почки, по-видимому, являются нормальными по структуре и функции. То есть из доказательств, полученных до настоящего времени, недостатки повторного всасывания глюкозы почками, по-видимому, оказывают минимальное продолжительное отрицательное воздействие на нормальных в других отношениях индивидуумов.
Следующие ссылки описывают O-арилгликозидные ингибиторы SGLT2 для лечения диабета.
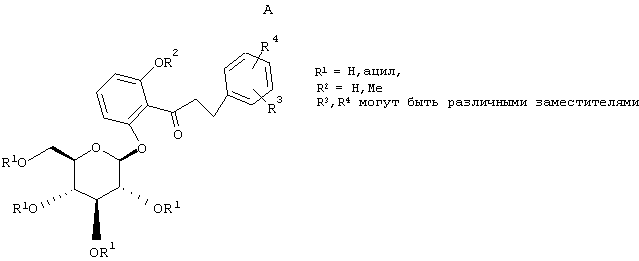
Европейская заявка ЕР 598359 А1 (а также японский патент JP 035988) (Tanabe Seiyaku) описывает соединения следующей структуры А
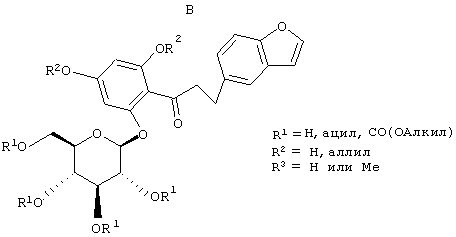
Европейская заявка ЕР 0850948 А1 описывает структуры вида В
Японская патентная заявка JP 09188625 A относится к структуре В, включая те примеры В, где R3 обозначает Н и где пятичленный цикл является насыщенным, а также является частью бензотиофенов (О=S) и инденов (О=СН2).
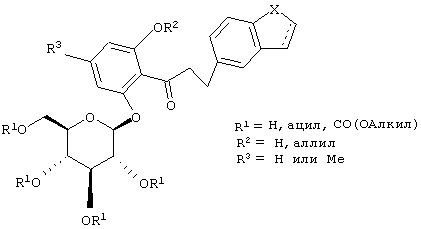
Японская патентная заявка JP 09124685 A распространяется на структуру В с R3=Н, включая производные моноацилированных С6 гидроксилов, где ацильная группа замещена на бензойную или пиридилкарбоновую кислоту или уретан, образованных из соответствующего фенола.
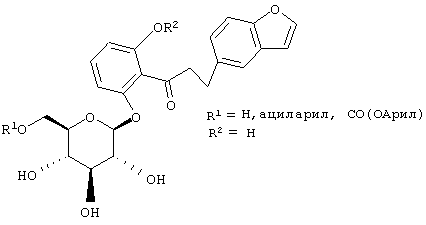
В японской заявке JP 09124684 описаны производные структуры В
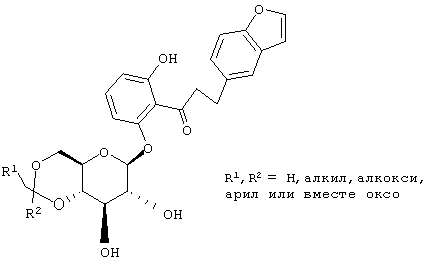
В европейской заявке ЕР 773226-А1 описаны производные структуры В
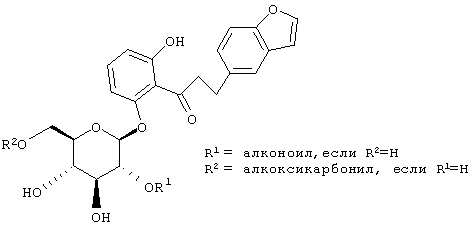
В японской заявке JP 08027006-А описаны производные структуры А, в которых ацилированы различные комбинации гидроксилов глюкозы и, по-видимому, она аналогична ЕР 598359 А1.
Европейская заявка ЕР 684254-А1, по-видимому, охватывает производные структуры В, описанные в JP 09188625 A.
Другие патенты и публикации, которые описывают ингибиторы SGLT2, включают следующие:
К. Tsujihara et al., Chem. Pharm. Bull. 44, 1174-1180 (1996)
M. Hongu et al., Chem. Pharm. Bull. 46, 22-33 (1998)
M. Hongu et al., Chem. Pharm. Bull. 46, 1545-1555 (1998)
A. Oku et al., Diabetes, 48, 1794-1800 (1999)
В японском патенте JP 10245391 (Dainippon) описано 500 структур в качестве гипогликемических агентов для лечения диабета. Эти соединения являются O-глюкозидами гидроксилированных кумаринов.
В Международной заявке WO 98/31697 описаны соединения строения

где Ar включает среди прочего, фенил, бифенил, дифенилметан, дифенилэтан и дифениловый эфир, и R1 обозначает глюкозид, R2 обозначает Н, ОН, амино, галоген, карбокси, алкил, циклоалкил или карбоксамидо, а R3 обозначает водород, алкил или ацил, и k, m и n обозначают независимо 1-4. Группа соединений, описанных в WO 98/31697, содержит соединения следующего строения

которые, как описано, предназначены для лечения или предупреждения воспалительных заболеваний, аутоиммунных заболеваний, инфекций, рака и метастаз при раке, нарушений реперфузии, тромбоза, язвы, ран, остеопороза, сахарного диабета и атеросклероза, среди прочих.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
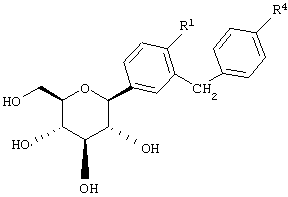
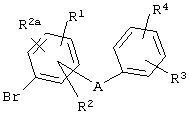
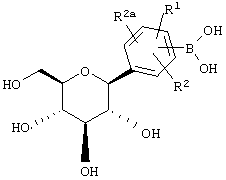
Согласно данному изобретению созданы С-арилглюкозидные соединения формулы I
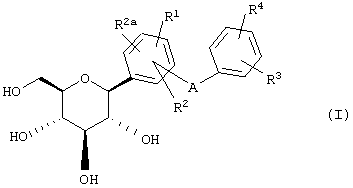
где R1, R2 и R2a независимо обозначают водород, ОН, OR5, алкил, CF3, OCHF2, OCF3, SR5i или галоген и два из R1, R2 и R2a вместе с прилегающими атомами углерода образуют аннелированный пятичленный гетероцикл, который может содержать 2 атома кислорода в цикле,
R3 и R4 независимо обозначают водород, ОН, OR5, ОАрил, ОСН2Арил, алкил, циклоалкил, CF3, -OCHF2, -OCF3, галоген, -CN, -СО2R5b, -CO2H, -COR6b, -CH(OH)R6c, -CH(OR5h)R6d, -CONR6R6a, -NHCOR5c, -NHSO2R5d, -NHSO2Арил, арил, -SR5e, -SOR5f, -SO2R5g, -SO2Арил или пяти-, шести- или семичленный гетероцикл, который может содержать 1-4 гетероатома в цикле, представляющие собой N, О, S, SO и/или SO2, или R3 и R4 вместе с прилегающими к ним атомами углерода образуют аннелированный пятичленный гетероцикл, который может содержать 2 атома кислорода в цикле,
R5, R5a, R5b, R5c, R5d, R5e, R5f, R5g, R5h и R5i независимо обозначают алкил;
R6, R6a, R6b, R6c и R6d независимо обозначают водород, алкил, арил, алкиларил или циклоалкил, или R6 и R6a вместе с прилегающими к ним атомами азота образуют аннелированный пяти-, шести- или семичленный гетероцикл, который может содержать 1-4 гетероатома в цикле, которыми являются N, О, S, SO и/или SO2;
А обозначает О, S, NH или (СН2)n, где n обозначает 0-3, и их фармацевтически приемлемые соли.
Соединения формулы I по изобретению, как определено выше, также включают условие, что если А обозначает (СН2)n, где n обозначает 0, 1, 2 или 3 или А обозначает О, и, по меньшей мере, один из R1, R2 и R2a обозначает ОН или OR5, тогда, по меньшей мере, один из R1, R2 и R2a обозначает CF3, OCF3 или OCHF2 и/или, по меньшей мере, один из R3 и R4 обозначает CF3, -OCHF2, -OCF3, -СН(OR5h)R6d, -CH(OH)R6c, -COR6b, -CN, -СО2R5b, -NHCOR5c, -NHSO2R5d, -NHSO2Арил, арил, -SR5e, -SOR5f, -SO2R5g или -SO2Арил.
Предпочтительные соединения формулы I, как определено выше, включают условие, что если А обозначает (СН2)n, где n обозначает 0, 1, 2 или 3 или А обозначает О, и, по меньшей мере, один из R1, R2, R2a, R3 и R4 обозначат ОН или OR5, то, по меньшей мере, один из R1, R2 и R2a обозначают CF3, OCF3 или OCHF2 и/или, по меньшей мере, один из R 3 и R4 обозначает CF3, -OCHF2, -OCF3, -CN, -CO2R5b, -CH(OR5h)R6d, -NHCOR5c, -NHSO2R5d, -NHSO2Арил, арил, -SR5e, -SOR5f, -SO2R5g, -SO2Арил или галоген.
Соединения формулы I обладают активностью как ингибиторы натрий-зависимых переносчиков глюкозы, обнаруживаемых в кишечнике и почках млекопитающих, и применимы при лечении диабета и микро- и макроваскулярных осложнений диабета, таких как ретинопатия, невропатия, нефропатия.
Данное изобретение охватывает соединения формулы I и способы применения таких соединений.
Кроме того, согласно данному изобретению предлагается способ лечения или замедления прогрессирования или начала диабета, в особенности диабета типа I или типа II, включая осложнения диабета, в том числе ретинопатию, невропатию, нефропатию, при котором больному человеку, нуждающемуся в лечении, вводят терапевтически эффективное количество соединения формулы I.
Кроме того, по данному изобретению, предлагается способ лечения диабета, причем терапевтически эффективное количество комбинации соединения структуры I и антидиабетического агента другого типа и/или терапевтического агента другого типа, такого как гиполипидемический агент, вводят больному человеку, нуждающемуся в лечении.
Термин "другой тип терапевтических агентов", применяемый в данном описании, относится к одному или более антидиабетических агентов (иным, нежели ингибиторы SGLT2 формулы I), одному или более агентов против ожирения, антигипертензивных агентов, антитромбоцитных агентов, противоатеросклеротических агентов и/или одному или более понижающих содержание липидов агентов (включая, противоатеросклеротические агенты).
В вышеуказанном способе по изобретению соединение формулы I по изобретению применяется в весовом соотношении с одним, двумя или более антидиабетическим агентом и/или одним, двумя или более терапевтическим агентом (в зависимости от их способа действия) в интервале, примерно, от 0,01:1 до, примерно, 300:1, предпочтительно, примерно, от 0,1:1 до, примерно, 1:1.
Предпочтительными являются соединения формулы IA
IA
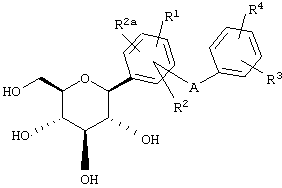
где А обозначает СН2 или О или S и в м-положении относительно глюкозида;
R1, R2 и R2a независимо выбирают из Н, низшего алкила, галогена, OR5 или OCHF2 или два из R1, R2 и R2a обозначают Н, а другой обозначает низший алкил, галоген, OR5 или OCHF2;
R3 и R4 независимо выбирают из низшего алкила, OR5 или -OCHF2, -SR5e, ОН, -CO2R5b, -3,4-(OCH2O)-, -COR6b, -CH(OH)R6c, -CH(OR5b)R6d, CF3, 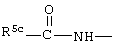 , -SOR5f, -SO2R5g, арил, -NHSO2Арил, -NHSO2R5d, COOH, тиадиазол, тетразол, -ОСН2Арил, -OCF3, ОАрил, или Н.
, -SOR5f, -SO2R5g, арил, -NHSO2Арил, -NHSO2R5d, COOH, тиадиазол, тетразол, -ОСН2Арил, -OCF3, ОАрил, или Н.
Более предпочтительны соединения формулы I, где А обозначает СН2;
R1 обозначает водород, галоген или низший алкил;
R2 и R2a каждый обозначает Н;
R3 обозначает Н;
R4 обозначает низший алкил, -COR6b, -СН(OH)R6c, -CH(OR5b)R6d, R5aO, -OCHF2, -OCF3 или -SR5e.
Наиболее предпочтительны соединения формулы I структуры IB
IB
где R1 обозначает водород, галоген или низший алкил и R4 обозначает низший алкил, R5aO, -OCHF2 или-SR5e. Предпочтительно, когда R1 находится в пара-положении к глюкозидной связи и заместитель R4 находится в пара-положении.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
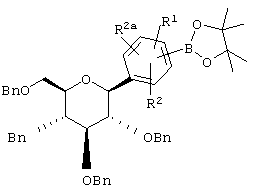
Соединение формулы I по изобретению можно получать, как показано на нижеприведенных реакционных схемах и дано их описание, где температура выражена в градусах Цельсия. Соединения формулы I можно получать, как показано на Схеме 1, обработкой соединений формулы II
II
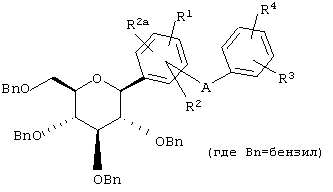
водородом в присутствии катализатора, такого как 1) Pd/C с применением растворителя, такого как МеОН или EtOH или 2) предпочтительно, Pd(OH)2, используя растворитель, такой как EtOAc. Или же соединения формулы I можно получать обработкой соединений формулы II кислотой Льюиса, такой как BBr3, BCl3 или BCl3·Me2S, в растворителе, таком как CH2Cl2, при -78°С. Соединения формулы I также можно получать обработкой соединений формулы II в растворителе, таком как EtSH, BF3·Et2O при 20°С.
Соединения формулы II (которые являются новыми интермедиатами) можно получать обработкой соединений формулы III силанами, такими как Et3SiH или, предпочтительно, (iPr)3SiH, в растворителе, таком как MeCN или смесь MeCN/CH2Cl2, содержащем кислоту Льюиса, такую как BF3·Et2O при -30°С.
III
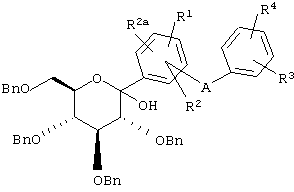
Соединения формулы III (которые являются новыми интермедиатами) можно получать реакцией соединения формулы IV
IV
с соединением V
V
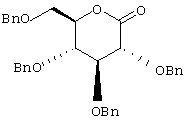
Соединения формулы IV активируются с целью присоединения обработкой н-BuLi или трет.-BuLi при -78°С в растворителе, таком как ТГФ, перед присоединением лактона V. Получение лактона V описано в R. Benhaddou, S. Czernecki, et al., Carbohydr. Res., 260 (1994), 243-250.
Соединения формулы IV, где А обозначает (СН2)n, где n=1-3, можно получать, как показано на Схеме 2, обработкой соединений формулы VI
VI
с силанами, такими как Et3SiH, в растворителе, таком как MeCN или CH2Cl2, содержащем кислоту Льюиса, такую как BF3·Et2О или TFA (ТФК) при -30°С е +60°С.
Соединения формулы VI можно получать присоединением получаемых промышленно бромбензальдегидов формулы VII
VII
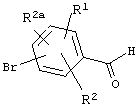
либо к металлоорганическим производным лития или магния соединений формулы VIII
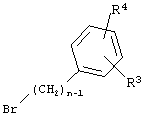
в растворителе, таком как Et2O или ТГФ, в условиях, известным специалистам в данной области техники.
Соединения формулы I, где R4 обозначает CH(OR5b)R6d, можно получать обработкой соединений формулы I, где R4 обозначает COR6b, последовательно, действием 1) ацетилирующего агента, такого как Ас2О, в растворителе, таком как пиридин один или СН2Cl2, содержащий 1,5 эквивалента основания, такого как Et3N, 2) восстановителя, такого как NaBH4, в растворителе, таком как EtOH, 3) алкилирующего агента, такого как R5hBr R5hI, в присутствии основания, такого как NAH, в растворителе, таком как ДМФА, и 4) в условиях щелочного гидролиза сложного эфира, например в присутствии LiOH в смеси 2:3:1 ТГФ/МеОН/Н2О.
Соединения формулы I, где R4 обозначает CH(OH)R6e, можно получать обработкой соединений формулы I, где R4 обозначает COR6b, восстановителем, таким как NaBH4, в растворителе, таком как EtOH.
Соединения формулы I, где R4 обозначает COR6b, можно получать обработкой соединений формулы II, где R4 обозначает COR6b, кислотой Льюиса, такой как BCl3 или BBr3, при -78°С в растворителе, таком как СН2Cl2.
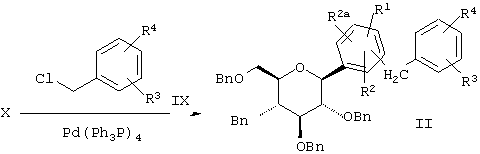
Соединения формулы II, где А обозначает СН2 и R4 обозначает COR6b, можно получать, как показано на Схеме 3, при взаимодействии выпускаемых промышленно или легкодоступных соединений формулы IX
IX

где Z обозначает Br или Cl, с соединениями формулы X,
Х
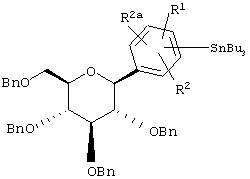
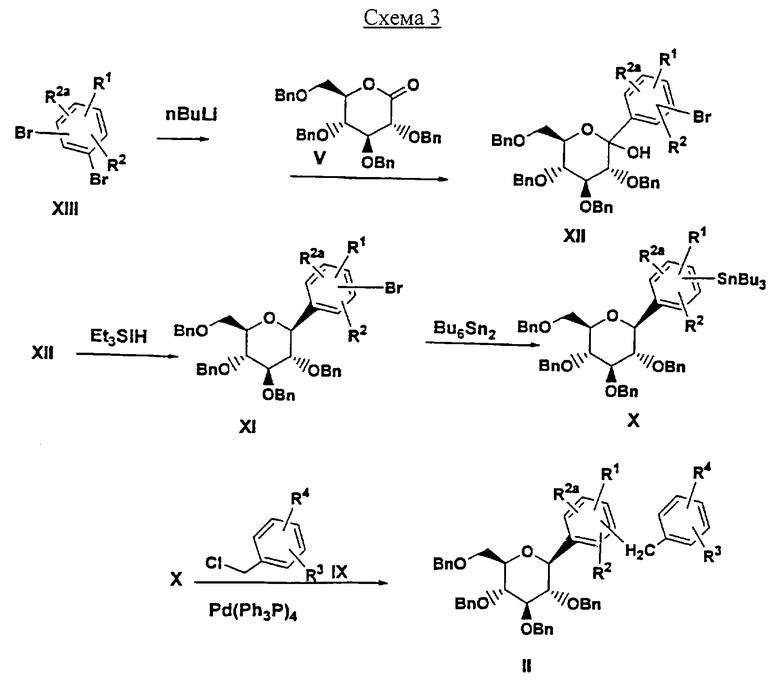
нагревая оба компонента в растворителе, таком как PhMe, в присутствии катализатора, такого как Pd(PPh3)4. Соединения формулы Х (которые являются новыми интермедиатами) можно получать из соединений формулы XI
XI
обработкой (Bu3Sn)2 и катализатором, таким как Pd(PPh3)4, в растворителе, таком как толуол.
Соединения формулы XI (которые являются новыми интермедиатами) можно получать из соединений формулы XII
XII
обработкой силанами, такими как iPr3SiH или Et3SiH, в растворителе, таком как MeCN, содержащем кислоту Льюиса, такую как BF3·Et2O, при -30°С.
Соединения формулы XII (которые являются новыми интермедиатами) можно получать при взаимодействии соединения V с литийорганическими соединениями, полученными обработкой соединений формулы XIII
XIII
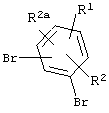
и н-BuLi или трет.-BuLi при -78°С в ТГФ.
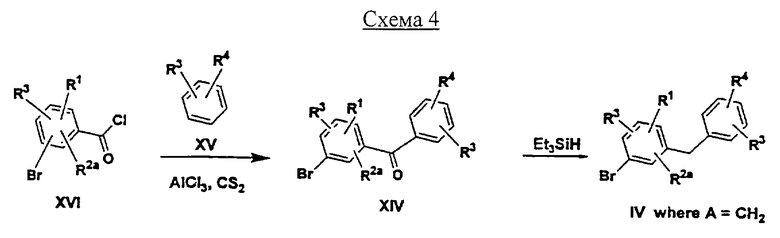
Альтернативный синтез (Схема 4) соединений формулы IV, где А обозначает СН2, включает восстановление соединений формулы XIV
XIV
восстановителем, таким как Et3SiH, в растворителе, таком как MeCN или СН2Cl2 или их смеси, содержащим катализатор, такой как BF3·Et2O.
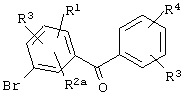
Соединения формулы XIV можно получать ацилированием по Фриделю-Крафтсу выпускаемых промышленностью углеводородов формулы XV
XV
легкодоступными хлорангидридами кислот формулы XVI
XVI
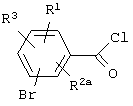
в растворителе, таком как CS2, содержащем два эквивалента кислоты Льюиса, такой как AlCl3 или AlBr3.
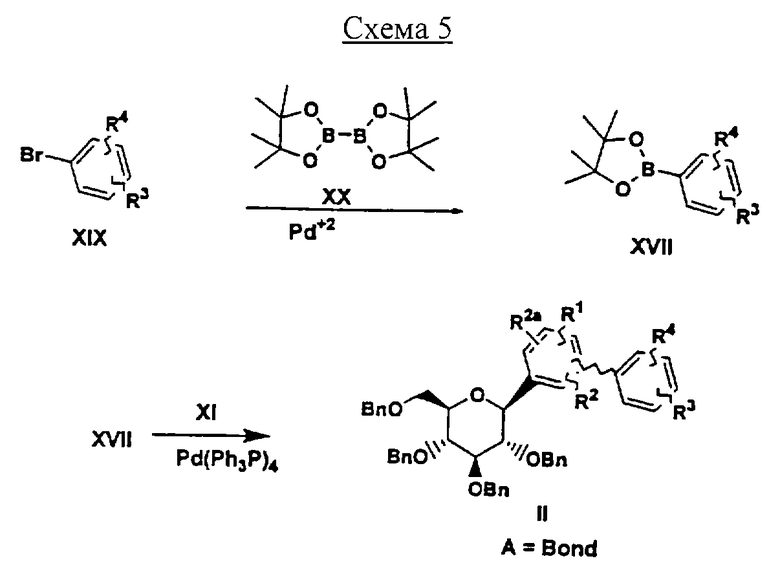
Соединения формулы II, где А обозначает связь, можно получать, как показано на Схеме 5, взаимодействием соединений формулы XI с соединениями формулы XVII
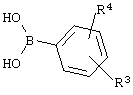
XVII
или соответствующей (арил)борной кислотой XVIII.
XVIII
Взаимодействие влечет за собой нагревание в присутствии катализатора, такого как Pd(PPh3)4, в растворителе, таком как PhMe/EtOH (3:1), содержащем Na2СО3. Соединения формулы XVII либо выпускаются промышленностью, либо их можно получать обработкой соединений формулы XVII BCl3 в растворителе, таком как CH2Cl2. Соединения формулы XVII можно получать нагреванием соединений формулы XIX
XIX
в растворителе, таком как ДМСО, содержащем катализатор, такой как PdCl2·dppf, и основание, такое как КОАс, с соединением XX.
XX
Соединения формулы II, где А=СН2 и R2=ОН, можно получать, как показано на Схеме 6, последовательно обрабатывая соединения формулы XXI
XXI
основанием, таким как NaH с последующим нагреванием с соединениями формулы IX в растворителе, таком как PhMe. Соединения формулы XXI можно получать из соединений формулы XXII
XXII
обработкой силанами, такими как Et3SiH или iPr3SiH, в растворителе, таком как MeCN, содержащем кислоту Льюиса, такую как BF3·Et2O, при -30°С.
Соединения формулы XXII можно получать взаимодействием соединения формулы V с активированными металлопроизводными соединения формулы XXIII
которые получают последовательной обработкой XXIII основанием, таким как NaH, КН или КОтрет.-Bu, а затем алкиллитием, таким как н-BuLi, или в растворителе, таком как сухой ТГФ.

Соединения формулы I, где А обозначает О или NH, можно получать, как показано на Схеме 7, взаимодействием соединений формулы XXIV
XXIV
с выпускаемыми промышленностью соединениями формулы XXV, где Х обозначает О или NH,
нагреванием в растворителе, таком как пиридин, содержащий основание, такое как Et3N, катализатор, такой как Cu(ОАс)2, и молекулярные сита.
Соединения формулы XXIV (которые являются новыми интермедиатами) можно получать, обрабатывая соединения формулы XXVI BCl3 в растворителе, таком как CH2Cl2, при -78°С.
XXVI
Соединения формулы XXVI (которые являются новыми интермедиатами) можно получать, нагревая соединения формулы XI с соединениями формулы XX в растворителе, таком как ДМСО, содержащем катализатор, такой как PdCl2·dppf, и основание, такое как КОАс.
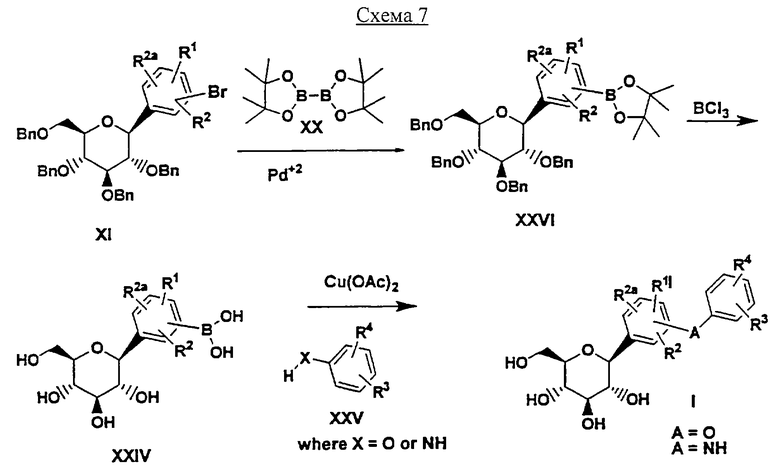
Соединения формулы IV, где А обозначает О или NH, можно получать, как показано на Схеме 8, взаимодействием соединений формулы XVIII
XVIII
с соединениями формулы XXVII, где Х обозначает О или NH,
XXVII
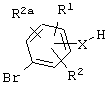
нагреванием в растворителе, таком как пиридин, содержащий основание, такое как Et3N, катализатор, такой как Cu(ОАс)2, и молекулярные сита.
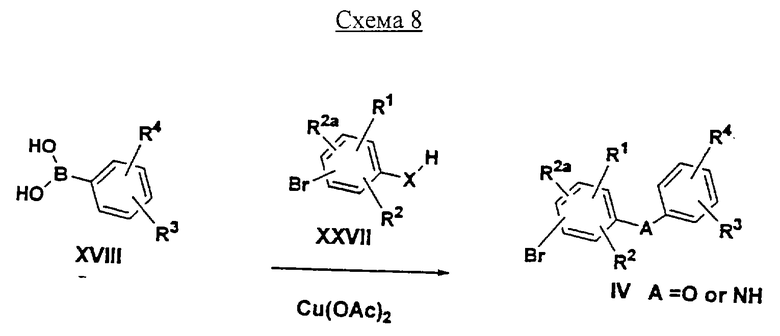
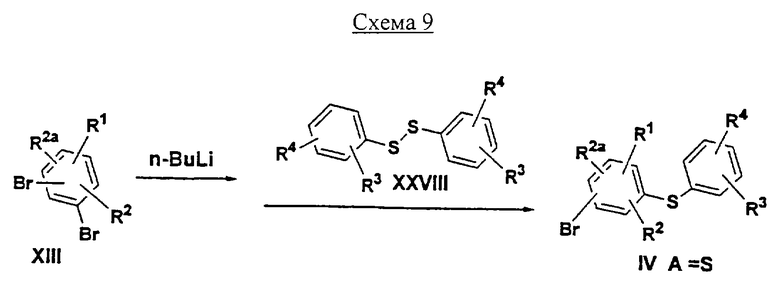
Соединения формулы IV, где А обозначает S, можно получать, как показано на Схеме 9, взаимодействием арилдисульфидов формулы XXVIII
XXVIII
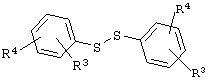
с литийорганическими соединениями, полученными металлированием соединений формулы XIII н-BuLi или трет.-BuLi при -78°С в ТГФ.
Ниже представлен перечень определений различных терминов, применяемых в описании данного изобретения. Эти определения применяются в отношении этих терминов по всему описанию (если они как-либо иначе не ограничены в конкретных примерах) или индивидуально, или как часть большой группы.
Следующие сокращения используются в данном описании:
Ph = фенил
Bn = бензил
t-Bu = трет.-бутил
Me = метил
Et = этил
TMS = триметилсилил
TMSN3 = триметилсилил азид
TBS = трет.-бутилдиметилсилил
ТГФ = тетрагидрофуран
Et2O = диэтиловый эфир
EtOAc = этилацетат
ДМФА = диметилформамид
МеОН = метанол
EtOH = этанол
i-PrOH = изопропанол
НОАс или АсОН = уксусная кислота
TFA = трифторуксусная кислота
i-Pr2NEt = диизопропилэтиламин
Et3N = триэтиламин
DMAP = 4-диметиламинопиридин
NaBH4 = боргидрид натрия
LiAlH4 = литийалюминий гидрид
n-BuLi = н-бутиллитий
Pd/C = палладий на угле
КОН = гидроксид калия
NaOH = гидроксид натрия
LiOH = гидроксид лития
К2СО3 = углекислый калий
NaHCO3 = бикарбонат натрия
EDC (или EDC·HCl) или EDCl (или EDCl·HCl) или EDAC = гидрохлорид 3-этил-3'(диметиламинопропил)-3-этилкарбодиимида
НОВТ или НОВТ·Н2О = гидрат 1-гидроксибензотриазола
НОАТ = 1-гидрокси-7-азабензотриазол
Ph3Р = трифенилфосфин
Pd(ОАс)2 = ацетат палладия
(Ph3Р)4Pd° = тетракис трифенилфосфин палладий
Ar = аргон
N2 = азот
мин = минута(ы)
час = час(ы)
л = литр
мл = миллилитр
мкл = микролитр
г = грамм(ы)
мг = миллиграмм(ы)
мол = моль
ммол = миллимоль(и)
мэкв = миллиэквивалент
RT = комнатная температура
sat или sat'd = насыщенный
aq. = водный
TLC = тонкослойная хроматография
HPLC = высокоэффективная жидкостная хроматография
LC/MS = высокоэффективная жидкостная хроматомасс-спектрометрия
MS или Mass Spec = масс-спектрометрия
NMR = ядерный магнитный резонанс
mp = точка плавления
dppf = дифенилфосфинферроцен
Если не указано иначе, термин "низший алкил", употребляемый в данном описании один или как часть другой группы, включает как линейные, так и с разветвленной цепью углеводороды, содержащие 1-8 атомов углерода, а термины "алкил" и "alk" ("алк"), употребляемые в данном описании одни или как часть другой группы, включают углеводороды с линейной или разветвленной цепью, содержащие 1-20 атомов углерода, предпочтительно, 1-10 атомов углерода, более предпочтительно, 1-8 атомов углерода, в нормальной цепи, такие как метил, этил, пропил, изопропил, бутил, трет.-бутил, изобутил, пентил, гексил, изогексил, гептил, 4,4-диметилпентил, октил, 2,2,4-триметилпентил, нонил, децил, ундецил, додецил, различные их изомеры с разветвленной цепью и т.п., а также такие группы, включающие 1-4 заместетеля, таких как галоид, например F, Br, Cl или I или CF3, алкил, алкокси, арил, арилокси, арил(алкил) или диарил, арилалкил, арилалкокси, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкилалкил, циклоалкилалкилокси, при необходимости содержащие заместители амино, гидрокси, гидроксиалкил, ацил, алканоил, гетероарил, гетероарилокси, циклогетероалкил, арилгетероарил, арилалкоксикарбонил, гетероарилалкил, гетероарилалкокси, арилоксиалкил, арилоксиарил, алкиламидо, алканоиламино, арилкарбониламино, нитро, циано, тиол, галоалкил, тригалоалкил и/или алкилтио.
Если не указано иначе, термин "циклоалкил", употребляемый в данном описании один или как часть другой группы, включает насыщенные или частично ненасыщенные (содержащие 1 или 2 двойных связи) циклические углеводородные группы, содержащие до 3 циклов, включая моноциклоалкил, бициклоалкил и трициклоалкил, содержащие в целом 3-20 атомов углерода, образующих циклы, предпочтительно, 3-10 атомов углерода, образующих цикл и которые могут быть конденсированы с 1 или 2 ароматическими циклами, описанными при пояснении для арила, которые включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и циклододецил, циклогексенил,
 ,
,  ,
,  ,
,  ,
,  ,
,
любая из этих групп может иметь 1-4 заместителя, таких как галоген, алкил, алкокси, гидрокси, арил, арилокси, арилалкил, циклоалкил, алкиламидо, алканоиламино, оксо, ацил, арилкарбониламино, амино, нитро, циано, тиол и/или алкилтио и/или любой из алкильных заместителей.
Термин "циклоалкенил", употребляемый в данном описании один или как часть другой группы, относится к циклическим углеводородам, содержащим 3-12 атомов углерода, предпочтительно, 5-10 атомов углерода, и 1 или 2 двойные связи. Примеры циклоалкенильных групп включают циклопентенил, циклогексенил, циклогептенил, циклооктенил, циклогексадиенил и циклогептадиенил, которые при необходимости имеют заместители, приведенные при определении циклоалкила.
Термин "алканоил", употребляемый в данном описании один или как часть другой группы, относится к алкилу, соединенному с карбонильной группой.
Если не указано иначе, термин "низший алкенил", употребляемый в данном описании один или как часть другой группы, относится к радикалам с линейной или разветвленной цепью, содержащим 2-8 атомов углерода, а термин "алкенил", употребляемый в данном описании сам по себе или как часть другой группы, относится к радикалам с линейной или разветвленной цепью, содержащим 2-20 атомов углерода, предпочтительно, 2-12 атомов углерода, и более предпочтительно, 2-8 атомов углерода в нормальной цепи, которые имеют одну-шесть двойных связей в нормальной цепи, таким как винил, 2-пропенил, 3-бутенил, 2-бутенил, 4-пентенил, 3-пентенил, 2-гексенил, 3-гексенил, 2-гептенил, 3-гептенил, 4-гептенил, 3-октенил, 3-ноненил, 4-деценил, 3-ундеценил, 4-додеценил, 4,8,12-тетрадекатриенил и т.п. и которые, при необходимости, могут иметь 1-4 заместетеля, а именно галоид, галоалкил, алкил, алкокси, алкенил, алкинил, арил, арилалкил, циклоалкил, амино, гидрокси, гетероарил, циклогетероалкил, алканоиламино, ариламидо, арилкарбониламино, нитро, циано, тиол, алкилтио и/или любой из алкильных заместителей, представленных в данном описании.
Если не указано иначе, термин "низший алкинил", употребляемый в данном описании один или как часть другой группы, относится к линейным или разветвленным радикалам, содержащим 2-8 атомов углерода, а термин "алкинил", употребляемый в данном описании сам по себе или как часть другой группы, относится к радикалам с линейной или разветвленной цепью, содержащим 2-20 атомов углерода, предпочтительно, 2-12 атомов углерода, и более предпочтительно, 2-8 атомов углерода в нормальной цепи, которые включают одну тройную связь в нормальной цепи, таким как 2-пропинил, 3-бутинил, 2-бутинил, 4-пентинил, 3-пентинил, 2-гексинил, 3-гексинил, 2-гептинил, 3-гептинил, 4-гептинил, 3-октинил, 3-нонинил, 4-децинил, 3-ундецинил, 4-додецинил, и т.п. и которые, при необходимости, могут иметь 1-4 заместетеля, а именно галоид, галоалкил, алкил, алкокси, алкенил, алкинил, арил, арилалкил, циклоалкил, амино, гетероарил, циклогетероалкил, гидрокси, алканоиламино, ариламидо, арилкарбониламино, нитро, циано, тиол, алкилтио и/или любой из алкильных заместителей, представленных в данном описании.
Термины "арилалкил", "арилалкенил" и "арилалкинил", употребляемые в данном описании одни или как часть другой группы, относятся к алкильной, алкенильной и алкинильной группам по определению, приведенному выше, имеющим арильный заместитель.
Если алкильные группы по вышеприведенному определению связываются с другими группами ординарными связями при двух различных атомах углерода, они называются "алкилен", "алкиленовая группа" и могут при необходимости иметь заместители, приведенные выше при определении понятия "алкил".
Если алкенильные группы по вышеприведенному определению и алкинильные группы по вышеприведенному определению, соответственно, связаны с другими группами ординарными связями при двух различных атомах углерода, они называются "алкениленовая группа" и "алкиниленовая группа", соответственно, и могут при необходимости иметь заместители, приведенные выше при определении понятия "алкенил" и "алкинил".
Соответствующие алкиленовые, алкениленовые или алкиниленовые группы (СН2)m или (СН2)р (где р равно 1-8, предпочтительно 1-5, и m равно 1-5, предпочтительно 1-3, которая включает алкиленовые, алкениленовые или алкиниленовые группы) по определению см. выше в данном описании могут, при необходимости иметь 1, 2 или 3 заместителя, которые включают алкил, алкенил, галоген, циано, гидрокси, алкокси, амино, тиоалкил, кето, С3-С6 циклоакил, алкилкарбониламино или алкилкарбонилокси.
Примеры (СН2)m, или (СН2)р, алкилена, алкенилена и алкинилена включают -СН2-, -CH2CH2-,
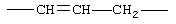 ,
, 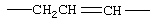 ,
, 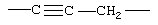 ,
, 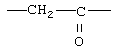 ,
,
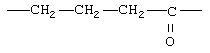 ,
, 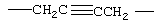 ,
, 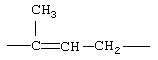 ,
, 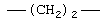 ,
, 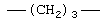 ,
, 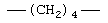 ,
, 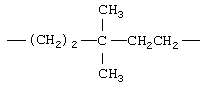 ,
, 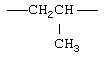 ,
, 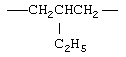 ,
, 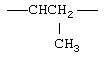 ,
, 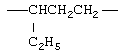 ,
, 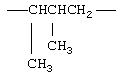 ,
, 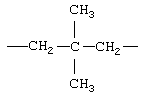 ,
, 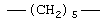 ,
, 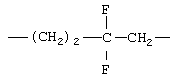 ,
,  ,
,  ,
, 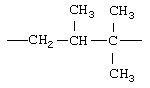 ,
, 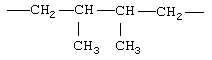 ,
, 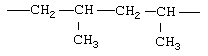 ,
, 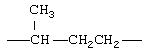 ,
, 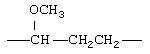 ,
, 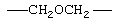 ,
, 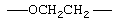 ,
, 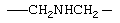 ,
, 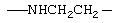 ,
, 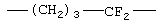 ,
, 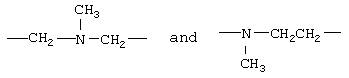 .
.
Термин "галоген" или "галоид" ("гало"), употребляемый в данном описании один или как часть другой группы, относится к хлору, брому, фтору и иоду, причем предпочтительными являются хлор или фтор.
Термин "ион металла" относится к ионам щелочных металлов, таких как натрий, калий или литий, и ионам щелочно-земельных металлов, таких как магний и кальций, а также к ионам цинка и алюминия.
Если не указано иначе, термин "арил" или "Арил", употребляемый в данном описании один или как часть другой группы, относится к моноциклическим или бициклическим ароматическим группам, содержащим 6-10 атомов углерода в циклическом фрагменте (таком как фенил или нафтил, включая 1-нафтил и 2-нафтил), и может, при необходимости, включать один-три дополнительных цикла, конденсированных с карбоциклическим кольцом или гетероциклическим кольцом (таким как арильный, циклоалкильный, гетероарильный или циклогетероалкильный циклы, например,
 ,
,  ,
,  ,
,  ,
, 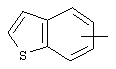 ,
, 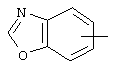 ,
, 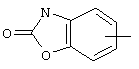 ,
, 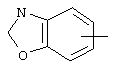 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,
и может при необходимости замещаться при соответствующих атомах углерода 1, 2 или 3 группами, выбранными из водорода, гало, галоалкила, алкила, галоалкила, алкокси, галоалкокси, алкенила, трифторметила, трифторметокси, алкинила, циклоалкилалкила, циклогетероалкила, циклогетероалкилалкила, арила, гетероарила, арилалкила, арилокси, арилоксиалкила, арилалкокси, алкоксикарбонила, арилкарбонила, арилалкенила, аминокарбониларила, арилтио, арилсульфинила, арилазо, гетероарилалкила, гетероарилалкенила, гетероарилгетероарила, гетероарилокси, гидрокси, нитро, циано, амино, замещенного амина, где аминогруппа содержит 1 или 2 заместителя (которые представляют собой алкил, арил или любые из других арильных соединений, указанных в определениях), тиола, алкилтио, арилтио, гетероарилтио, арилтиоалкил, алкоксиарилтио, алкилкарбонила, арилкарбонила, алкиламинокарбонила, ариламинокарбонила, алкоксикарбонила, аминокарбонила, алкилкарбонилокси, арилкарбонилокси, алкилкарбониламино, арилкарбониламино, арилсульфинила, арилсульфинилалкила, арилсульфониламино и арилсульфонаминокарбонила и/или любого из алкильных заместителей, представленных в данном описании.
Если не указано иначе, термин "низший алкокси", "алкокси", "арилокси" или "аралкокси", употребляемый в данном описании один или как часть другой группы, включает любую из вышеприведенных алкильных, аралкильных или арильных групп, связанную с атомом кислорода.
Если не указано иначе, термин "замещенный амино" ("замещенная аминогруппа"), употребляемый в данном описании один или как часть другой группы, относится к аминогруппе, имеющей один или два заместителя, которые могут быть одинаковыми или различными, такие как алкил, арил, арилалкил, гетероарил, гетероарилалкил, циклогетероалкил, циклогетероалкилалкил, циклоалкил, циклоалкилалкил, галоалкил, гидроксиалкил, алкоксиалкил и тиоалкил. Эти заместители могут, в свою очередь, иметь в качестве заместителей карбоновую кислоту и/или любой из вышеприведенных алкильных заместителей. Кроме того, заместители в аминогруппе могут вместе с атомом азота, с которым они соединены, образовывать 1-пирролидинил, 1-пиперидинил, 1-азепинил, 4-морфолинил, 4-тиаморфолинил, 1-пиперазинил, 4-алкил-1-пиперазинил, 4-арилалкил-1-пиперазинил, 4-диарилалкил- 1-пиперазинил, 1-пирролидинил, 1-пиперидинил или 1-азепинил, при необходимости имеющие в качестве заместителя алкил, алкокси, алкилтио, гало, трифторметил или гидрокси.
Если не указано иначе, термин "низший алкилтио", "алкилтио", "арилтио" или "аралкилтио", употребляемый в данном описании один или как часть другой группы, включает любую из вышеприведенных алкильных, аралкильных или арильных групп, связанную с атомом серы.
Если не указано иначе, термин "низший алкиламино", "алкиламино", "ариламино" или "аралкиламино", употребляемый в данном описании один или как часть другой группы, включает любую из вышеприведенных алкильных, аралкильных или арильных групп, связанную с атомом азота.
Если не указано иначе, термин "ацил", употребляемый в данном описании сам по себе или как часть другой группы по определению в данном описании, относится к органическому радикалу, связанному с карбонильной (С=O) группой; примеры ацильных групп включают любой из алкильных заместителей, связанный с карбонилом, такой как алканоил, алкеноил, ароил, аралканоил, гетероароил, циклоалканоил, циклогетероалканоил и т.п.
Если не указано иначе, термин "циклогетероарил", употребляемый в данном описании один или как часть другой группы, относится к 5-, 6- или 7-членному насыщенному или частично ненасыщенному циклу, который содержит 1-2 гетероатома, таких как азот, кислород и/или сера, соединенному через атом углерода или гетероатом, где возможно, при необходимости через линкер (СН2)р (где р означает 1, 2 или 3), такому как
 ,
,  ,
,  ,
, 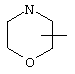 ,
,
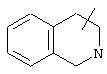 ,
, 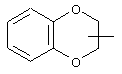 ,
, 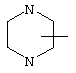 ,
,  ,
,
 ,
,  ,
,  ,
,
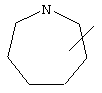 ,
, 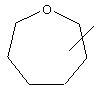 ,
, 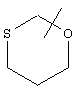 ,
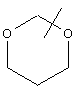
, 
и т.п. Вышеприведенные группы могут включать 1-4 заместителя, таких как алкил, галоид, оксо и/или любой из алкильных заместителей, представленных в данном описании. Кроме того, любой из циклогетероалкильных колец может быть конденсирован с циклоалкильным, арильным, гетероарильным или циклогетероалкильным циклом.
Если не указано иначе, термин "гетероарил", употребляемый в данном описании один или как часть другой группы, относится к 5- или 6-членному ароматическому циклу, который содержит 1, 2, 3 или 4 гетероатома, таких как азот, кислород или сера, и к таким циклам, конденсированным с арильным, циклоалкильным, гетероарильным или циклогетероалкильным циклом (например, бензотиофенильным или индолильным), и включает возможные N-оксиды. Гетероарильная группа, может включать, при необходимости, 1-4 заместителя, таких как любой из вышеприведенных алкильных заместителей. Примеры гетероарильных групп включают следующие:
 ,
,  ,
,  ,
,  ,
,
 ,
,  ,
, 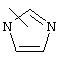 ,
, 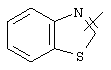 ,
, 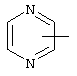 ,
, 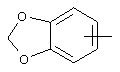 ,
,
 ,
,  ,
,  ,
,  ,
, 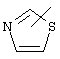 ,
, 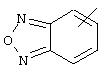 ,
,
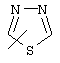 ,
, 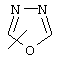 ,
, 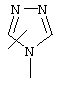 ,
, 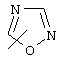 ,
, 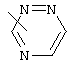 , и т.п.
, и т.п.
Термин "циклогетероалкилалкил", употребляемый в данном описании один или как часть другой группы, относится к циклогетероалкильным группам по определению см. выше, связанных через С-атом или гетероатом с (СН2)р цепью.
Термин "гетероарилалкил" или "гетероарилалкенил", употребляемый в данном описании один или как часть другой группы, относится к гетероарильной группе по определению см. выше, связанной через С-атом или гетероатом с -(СН2)р- цепью, алкиленом или алкениленом по определению см. выше.
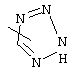
Термин "пяти-, шести- или семичленный карбоцикл или гетероцикл", употребляемый в данном описании, относится к циклоалкильным или циклоалкенильным группам по определению см. выше, или гетероарильным группам или циклогетероарильным группам по определению см. выше, таким как тиадиазол, тетразол, имидазол или оксазол.
Термин "полигалоалкилокси", употребляемый в данном описании, относится к "алкильной" группе по определению см. выше, которая включает 2-9, предпочтительно, 2-5 галоидных заместителей, таких как F и Cl, предпочтительно, F, такой как CF3СН2О, CF3О или CF3CF2CH2О.
Термин "пролекарственный сложные эфиры", употребляемый в данном описании, включает сложные эфиры и карбонаты, образующиеся реакцией одной или более гидроксильных групп в соединении формулы I с алкил-, алкокси- или арилзамещенным ацилирующим агентом по известным специалистам в данной области техники методикам для получения ацетатов, пивалатов, метилкарбонатов, бензоатов и т.п., кроме того, пролекарственных сложных эфиров, которые известны в технике для сложных эфиров карбоновых и фосфорных кислот, таких как метиловые, этиловые, бензиловые и т.п. эфиры.
Примеры таких пролекарственных сложных эфиров включают
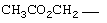 ,
,  ,
,  , или
, или
 .
.
Если соединения структуры I находятся в форме кислоты, они могут образовывать фармацевтически приемлемую соль, такую как соли щелочных металлов, лития, натрия или калия, соли щелочно-земельных металлов, кальция или магния, а также цинка или алюминия и других катионов, таких как аммоний, холин, диэтаноламин, лизин (D или L), этилендиамин, трет.-бутиламин, трет.-октиламин, трис-(гидроксиметил)аминометан (TRIS), N-метилгликозамин (NMG), триэтаноламин и дегидроабиетиламин.
Рассматриваются все стереоизомеры соединений по данному изобретению либо в смеси, либо в чистом, либо в практически чистом виде. Соединения по данному изобретению могут иметь асимметрические центры при любом из атомов углерода, имеющем любой из заместителей R. Следовательно, соединения формулы I могут существовать в энантиомерной и диастереомерной формах или в виде их смесей. При получении могут применяться в качестве исходных веществ рацематы, энантиомеры или диастереомеры. Когда получают диастереомерные или энантиомерные продукты, их можно делить обычными методами, например хроматографией или фракционной кристаллизацией.
Когда требуется, соединения структуры I можно использовать в сочетании с одним или более антидиабетических агентов других типов и/или с одним или более терапевтических агентов других типов, которые можно вводить перорально в виде той же самой лекарственной формы или с помощью инъекции.
Другой тип антидиабетического агента, который можно при необходимости применять в сочетании с ингибитором SGLT2 формулы I, может представлять собой 1, 2, 3 или более антидиабетических агентов или антигипергликемических агентов, включая средства, усиливающие секрецию инсулина, или сенсибилизаторы инсулина, или другие антидиабетические агенты, предпочтительно, имеющие механизм действия, отличный от SGLТ2-ингибирования, и могут включать бигуанидины, сульфонилмочевины, ингибиторы глюкозидазы, PRAP γ-агонисты, такие как тиазолидиндионы, ингибиторы аР2, PRAP α/γ-двойные агонисты, ингибиторы дипептидилпетидазы IV (DP4) и/или меглитиниды, а также инсулин, глюкагонподобный пептид-1 (GLP-1), ингибиторы РТР1В, ингибиторы глюкогенфосфорилазы и/или ингибиторы глюкозо-6-фосфатазы.
Другие типы терапевтических агентов, которые можно при необходимости применять в комбинации с ингибиторами SGLT2 формулы I, включают агенты против ожирения, антигипертензивные агенты, антитромбоцитные агенты, противоатеросклерозные агенты и/или агенты, понижающие содержание липидов.
Ингибиторы SGLT2 формулы I можно также при необходимости применять в сочетании с агентами для лечения осложнений при диабете. Такие агенты включают ингибиторы РКС и/или ингибиторы AGE.
Полагают, что применение соединений структуры I в сочетании с 1, 2, 3 или более других антидиабетических агентов даст лучший антигипергликемический результат, чем достигался бы при приеме каждого из этих препаратов, и лучший, чем объединенные аддитивные гипергликемические эффекты, даваемые этими препаратами.
Другой антидиабетический агент может быть пероральным антигипергликемическим агентом, предпочтительно, бигуанидом, таким как метформин НСТ.
Если другой антидиабетический агент представляет собой бигуанид, соединение структуры I можно применять в весовом отношении к бигуаниду в интервале, примерно, от 0,01:1 до, примерно, 100:1, предпочтительно, примерно, от 0,1:1 до, примерно, 5:1.
Другой антидиабетический агент также может, предпочтительно, представлять собой сульфонилмочевину, такую как глибурид (также известный каглибенкламид), глимепирид (описанный в Патенте США 4379785), глипизид, дликлазид или хлорпропамид, другие известные сульфонилмочевины или другие антигипергликемические агенты, которые действуют на АТР-зависимый канал β клеток, причем предпочтительными являются глибурид и глипизид, которые можно вводить в тех же самых или отдельных пероральных лекарственных формах.
Соединения структуры I применяются в весовом отношении с сульфонилмочевиной в интервале, примерно, от 0,01:1 до, примерно, 100:1, предпочтительно, примерно, от 0,2:1 до, примерно, 10:1.
Пероральнай антидиабетический агент может быть также ингибитором глюкозидазы, таким как акарбоза (acarbose) (описываемая в Патенте США 4904769) или маглитол (описываемый в Патенте США 4639436), которые можно вводить в виде одной или раздельных лекарственных форм.
Соединения структуры I применяются в весовом отношении к ингибитору глюкозидазы в интервале, примерно, от 0,01:1 до, примерно, 100:1, предпочтительно, примерно, от 0,5:1 до, примерно, 50:1.
Соединения структуры I можно применять в сочетании с агонистом PRAP γ, таким как тиазолидиндионовый пероральный антидиабетический агент, или с другими сенсибилизаторами инсулина (которые вызывают у больных NIDDM эффект чувствительности к инсулину), такими как троглитазон (RezulinТ, фирмы Warner-Lambert, описываемый в Патенте США 4572912), розиглитазон (SKB), пиоглитазон (Takeda), МСС-555 фирмы Mitsubishi (описываемый в Патенте США 5594016), GL-262570 фирмы Glaxo-Welcome, энглитазон (СР-68722, Pfizer) или дарглитазон (СР-86325, Pfizer), изаглитазон (izaglitazone, MIT/J&J), JTT-501 (JPNT/P&U), L-895645 (Merck), R-119702 (Sankyo/WL), NN-2344 (Dr. Reddy/NN) или YM-440 (Yamanouchi), предпочтительно, розиглитазон и пиоглитазон.
Соединения структуры I применяются в весовом отношении с тиазолидиндионом в интервале, примерно, от 0,01:1 до, примерно, 100:1, предпочтительно, примерно, от 0,2:1 до, примерно, 10:1.
Сульфонилмочевина и тиазолидиндион в количествах менее 150 мг перорального антидиабетического агента можно вводить в виде единой таблетки с соединениями формулы I.
Соединения структуры I можно также применять в комбинации с антигипергликемическим агентом, таким как инсулин, или с глюкагонподобным пептидом-1 (GLP-1), таким как GLP-1 (1-36) амид, GLP-1 (7-36) амид, GLP-1 (7-37) амид (как показано в Патенте США 5614492, выданном Habener, содержание которого вводится в данное описание в качестве ссылки), а также АС2993 (Amylen) и LY-315902 (Lilly), которые можно вводить с помощью инъекций, интраназально или с помощью трансдермальных или трансбуккальных (защечных) устройств.
Если присутствуют метформин, сульфонилмочевины, такие как глибурин, глимепирид, глипирид, глипизид, хлорпропамид и гликлазид, и ингибиторы глюкозидазы акарбоза, или миглитол, или инсулин (в инъецируемой, легочной, защечной или пероральной форме), они могут применяться в рецептурах, как описано выше, и в количествах и дозировках, указанных в справочнике врача (PDR).
Если присутствуют метформин или его соль, их можно применять в количествах, примерно, от 500 до, примерно, 2000 мг в день, которые можно вводить в виде однократной или раздельных доз от одного до четырех раз в день.
Если присутствует тиазолидиндионовый антидиабетический агент, его можно применять в количествах, примерно, от 0,01 до, примерно, 2000 мг в день, которые можно вводить в виде однократной или раздельных доз от одного до четырех раз в день.
Если присутствует инсулин, то его можно применять в рецептурах, количествах и дозах, указанных в справочнике врача.
Если присутствуют GLP-1 пептиды, то их можно вводить в виде пероральных защечных препаратов, назально или парентерально, как описано в Патентах США 5346701 (TheraTech), 5614492 и 5631224, вводимых в данное описание в качестве ссылки.
Другим антидиабетическим агентом может быть также двойной агонист PPAR α/γ, такой как AR-HО39242 (Astra/Zeneca), GW-409544 (Glaxo-Wellcome), KRP297 (Kyorin Merck), a также (препараты), описываемые Murakami et al., "A Novel Insulin Sensitizer Acts As a Coligand for Peroxisome Proliferation - Activated Receptor Alpha (PPAR alpha) and PPAR gamma. Effect on PPAR alpha Actovation on Abnormal Lipid Metabolism in Liver of Zucker Fatty Rats" (Новый сенсибилизатор инсулина действует как ко(совместимый)лиганд для Активируемого Пролиферацией Пероксисомы Рецептора Альфа (PPAR alpha) и PPAR гамма. Влияние активации PPAR альфа на аномальный метаболизм липидов в печени страдающих ожирением крыс Zucker), Diabetes 47, 1841-1847 (1988) и в предварительной заявке на патент США N 60/155400, поданной 22 сентября 1999 г. (файл у поверенного LA29), которая вводится в данное описание в качестве ссылки, с применением указанных там дозировок, причем соединения, признанные как предпочтительные, являются предпочтительными по данному изобретению.
Другим антидиабетическим агентом может быть ингибитор аР2, таким как описан в патентной заявке США 09/391053, поданной 7 сентября 1999 г., и в предварительной заявке на Патент США 60/127745, поданной 5 апреля 1999 г. (файл у поверенного LA27*), с применением указанных в данном описании дозировок. Предпочтительны соединения, признанные предпочтительными в вышеуказанной заявке.
Другим антидиабетическим агентом может быть ингибитор DP4, такой как описан в Международных заявках WО99/38501, WО99/46292, WО99/67279 (PROBIODRUG), WО99/67278 (PROBIODRUG), WО99/61431 (PROBIODRUG), NVP-DPP728A (1-[[[2-[(5-цианопиридин-2-ил)амино]этил]амино]ацетил]-2-циано-(S)-пирролидин) (Novartis) (предпочтительный) как описано Hughes et al., Biochemistry, 38(36), 11597-11603, 1999; TSL-225 (триптофил-1,2,3,4-тетрагидроизохинолин-3-карбокси кислота (описанная Yamada et al., Bioorg. and Med. Chem. Lett., 8 (1998) 1537-1540; 2-цианопирролидиды и 4-цианопирролидиды, как описано Ashworth et al., Bioorg. and Med. Chem. Lett., Vol.6, No.22, pp.1163-1166 и 2745-2748 (1996) с применением дозировок, указанных в вышеприведенных ссылках.
Меглитинид, который при необходимости можно применять в сочетании с соединением формулы I по изобретению, может быть репаглинидом, натеглинидом (Novartis) или KAD1229 (PF/Kissei), при этом репаглинид является предпочтительным.
Ингибитор SGLT2 формулы I применяется в весовом отношении с меглитинидом, агонистом PPAR γ, двойньм агонистом PPAR α/γ, ингибитором аР2 или ингибитором DP4 в интервале, примерно, от 0,01:1 до, примерно, 100:1, предпочтительно, примерно, от 0,2:1 до, примерно, 10:1.
Гиполипидемический агент, или агент, понижающий содержание липидов, который при необходимости можно применять в сочетании с соединениями формулы I по изобретению, могут включать 1, 2, 3 или более ингибиторов МТР, ингибиторов HMG СоА редуктазы, ингибиторов синтетазы сквалена, производные фибриновых кислот, ингибиторы АСАТ, ингибиторы липоксигеназ, ингибиторы всасывания холестерина, идеальные ингибиторы (совместного) сопереносчика N+/желчных кислот, активаторы (позитивные регуляторы) LDL-рецепторной активности, вещества, усиливающие экскрецию желчных кислот, и/или никотиновую кислоту и ее производные.
Ингибиторы МТР, применяемые в данном описании, включают ингибиторы МТР, описанные в Патентах США 5595872, 5739135, 5712279, 5760246, 5827875, 5885983 и в заявке на Патент США 09/175180, поданной 20 октября 1998 г., в настоящее время Патент США 5962440. Предпочтительными являются каждый из МТР-ингибиторов, описанных в каждом вышеуказанном патенте и заявке, как предпочтительный. Все вышеуказанные Патенты и патентная заявка США вводятся в данное описание в качестве ссылок.
Гиполипидемический агент может быть ингибитором редуктазы HMG СоА, включающим, без ограничения, мевастатин и родственные соединения, такие как описанные в Патенте США 3983140, ловастатин (мевинолин) и родственные соединения, такие как описанные в Патенте США 4231938, правастатин и родственные соединения, такие как описанные в Патенте США 4346227, симвастатин и родственные соединения, такие как описанные в Патентах США 4448784 и 4450171. Гиполипидемический агент может также быть соединением, описанным в предварительных заявках на Патент США 60/211594 и 60/211595. Другие ингибиторы редуктазы HMG СоА, которые можно применять по данному изобретению включают, но без ограничения, флувастатин, описанный в Патенте США 5354772; церивастатин, описанный в Патентах США 5006530 и 5177080; аторвастатин, описанный в Патентах США 4681893, 5273995, 5385929 и 5686104, атавастатин (нисвастатин фирмы Nissan/Sankyo (NK-104)), описанный в Патенте США 5011930; висастатин (visastatin) фирмы Shionogi-Astra/Zeneca (Zd-4522), описываемый Патенте США 5260440, и родственные статиновые соединения, описанные в Патенте США 5753675; пиразольные аналоги производных мевалонолактона, как описано в Патенте США 4613610; инденовые аналоги мевалонолактона, как описываемые в Международной заявке РСТ WO 86/03488; 6-[2-(замещенные-пиррол-1-ил)-алкил-пиран-2-оны и их производные, описываемые в Патенте США 4647576; SC-45355 фирмы Searle, дихлорацетат производного 3-замещенной пентандионовой кислоты; имидазольные аналоги мевалонолактона, описываемые в Международной заявке РСТ WO 86/07054; производные 3-карбокси-2-гидрокси-пропан-фосфониевой кислоты, описываемые во Французском Патенте 2596393; производные 2,3-дизамещенного пиррола, фурана и тиофена, как описано в Европейской патентной заявке 0221025; нафтильные аналоги мевалонолактона, как описано в Патенте США 4686237; октагидронафталины, такие как описанные в Патенте США 4499289; кетоаналоги мевинолина (ловастатина), как описано в Европейской патентной заявке 0142146А2; и производные хинолина и пиридина, описанные в Патентах США 5506219 и 5691322.
Кроме того, соединения фосфиновых кислот, применимые для ингибирования HMG СоА редуктазы, пригодные для применения по данному изобретению, описаны в английском Патенте GB 2205837.
Ингибиторы синтетазы сквалена, которые можно применять по данному изобретению, включают, но без ограничения, α-фосфоносульфокислоты, описанные в Патенте США 5712396; α-фосфоносульфонаты, описанные Biller et al., J. Med. Chem., 1988, том 31, №10, с.1869-1871, включая изопреноидные (фосфинилметил)-фосфонаты, а также другие известные ингибиторы синтетазы сквалена, например, описанные в Патентах США 4871 и 4924024 и в Biller S.A., Neuenschwander К., Ponpipom M.M., и Poulter C.D., Current Pharmaceutical Design, 2, 1-40 (1996).
Кроме того, другие ингибиторы синтетазы сквалена, пригодные для применения по данному изобретению, включают терпеноидные пирофосфатазы, описанные Ortiz de Montellano et al., J. Med. Chem., 1977, 20, 243-249; аналог А фарнезилдифосфата и аналоги прескваленпирофосфата (PSQ-PP), описанные Corey and Volante, J. Am. Chem. Soc., 1976, 98, 1291-1293; фосфинилфосфонаты, описанные McClard R.W. et al., J.A.C.S., 1987, 109, 5544 и циклопропаны, описанные Capson T.L., Ph.D. dissertation, June, 1987, Dept. Med. Chem. U of Utah, Abstract (реферат), Tables of Contents, pp.16, 17, 40-43, 48-51, Summary.
Другие гиполипидемические агенты, пригодные для применения, но без ограничения, производные фибриновых кислот, такие как фенофибрат, гемфиброзил, клофибрат и т.п., пробукол и родственные соединения, описанные в Патенте США 3674836, при этом предпочтительными являются пробукол и гемфиброзил; вещества, усиливающие действие желчных кислот, экскрецию, такие как холестирамин, холестипол и DEAE-Sephadex (SecholexT, PolicexideT), а также липостабил (Rhone-Poulenc), Eisai E-5050 (производное N-замещенного этаноламина), иманиксил (НОЕ-402), тетрагидролипстатин (THL), истигмастанилфосфорилхолин (SPC, Roche), аминоциклодекстрин (Tanabe Seiyoku), Ajinomoto AJ-814 (производное азулена), мелинамид (Sumitomo), Sandoz 58-035, CL-277082 и CL-283546 фирмы American Cyanamid (производные дизамещенной мочевины), никотиновая кислота, аципимокс, ацифран, неомицин, п-аминосалициловая кислота, аспирин, производные поли(диаллилметиламина), как описано в Патенте США 4027009, и другие известные понижающие уровень холестирина в сыворотке агенты.
Другим гиполипидемическим агентом может быть ингибитор АСАТ, такой как описанный в "Drugs of the Future" 24, 9-15 (1999), (Avasimible); "Ингибитор АСАТ, С1-1011 эффективен для предупреждения и регрессии жировых полос на поверхности аорты у хомяка", Nicolosi et al., Atherosclerosis (Shannon, Irel). (1988), 137(1), 77-85; "Фармакологический профиль FCE 27677: новый ингибитор АСАТ с высокой гиполипидемической активностью, опосредуемой избирательным торможением секреции АроВ100-содержащего липопротеина", Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16(1) 16-30; "RP 73163: биодоступный алкилсульфинилдифенил-имидазольный ингибитор АСАТ", Smith С., et al., Bioorg. Med. Chem. Lett. (1996), 6(1), 47-50; "АСАТ inhibitors: phisiological mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals" ("Ингибиторы АСАТ: физиологические механизмы гиполипидемической и антиатеросклеротической активности у подопытных животных"); Krause et al., Editor(s): Ruffolo, Robert R., Ir.; Hollinger, Mannfred A., Inflammation: Mediators Paphways ("Воспаление: метаболические пути медиаторов", (1995), 173-98, Publisher: CRC, Boca Raton, Fla.; "Ингибиторы АСАТ: потенциальные антиатеросклеротические агенты", Sliskovic et al., Curr. Med. Chem. (1994) 1(3), 204-25; "Ингибиторы ацил-СоА: холестирин O-ацилтрансферазы (АСАТ) как гипохолистеринемические агенты. 6. Первый водорастворимый ингибитор АСАТ с липид-регуляторной активностью. Ингибиторы ацил-СоА: холестирин O-ацилтрансферазы (АСАТ) как гипохолистеринемические агенты. 7. Получение серии замещенных N-фенил-N'-[(1-фенилциклопентил)метил] мочевин с повышенной гипохолистеринемической активностью", Stout et al., Химические издания: Org. Chem. (1995), 8(6), 359-62 или TS-962 (Taisho Pharmaceutical Co. Ltd).
Гиполипидемическим агентом может быть позитивный регулятор LD2-рецепторной активности, такой как MD-700 (Taisho Pharmaceutical Co. Ltd) и LY295427 (Eli Lilly).
Гиполипидемическим агентом может быть ингибитор всасывания (поглощения, адсорбции) холистерина, предпочтительно, SCH48461 фирмы Schering-Plough, а также ингибиторы адсорбции холистерина, описанные в Atherosclerosis 115, 45-63 (1995) и J. Med. Chem., 41, 973 (1998).
Гиполипидемическим агентом может быть идеальный ингибитор сопереносчика Na+/ желчных кислот, такой как описываемый в Drugs of the Future, 24, 425-430 (1999).
Предпочтительными гиполипидемическими агентами являются правастатин, ловастатин, симвастатин, аторвастатин, флувастатин, церивастатин, атавастатин и розувастатин.
Вышеуказанные патенты США вводятся в данное описание в качестве ссылки. Применяемые количества и дозировки указаны во "Врачебном справочнике" ("Справочник врача") и/или в вышеуказанных патентах.
Соединения формулы I по изобретению применяются в весовом соотношении с гиполипидемическим агентом (если он присутствует) в интервале, примерно, от 500:1, примерно, до 1:500, предпочтительно, примерно от 100:1 до, примерно 1:100.
Вводимую дозу следует тщательно скорректировать в соответствии с возрастом, весом и состоянием больного, а также в соответствии со способом введения, лекарственной формой и схемой и желаемым результатом.
Дозировки и рецептуры гиполипидемического агента такие, которые описываются в различных обсуждаемых выше патентах и заявках.
Дозировки и рецептуры другого применяемого гиполипидемического агента, когда он применяется, таковы, какими они представлены в самом последнем издании "Справочника врача".
При пероральном применении удовлетворительный результат можно получить при использовании ингибитора МТР в количестве, примерно, 0,01 мг/кг - 500 мг и, предпочтительно, примерно, 0,1 мг/кг - 100 мг, один-четыре раза в день.
Предпочтительная пероральная лекарственная форма, например таблетки или капсулы, содержит МТР-ингибитор в количестве, примерно, 1-500 мг, предпочтительно, примерно 2-400 мг, и более предпочтительно, около 5-250 мг, один-четыре раза в день.
При пероральном введении удовлетворительный результат можно получить, используя ингибитор HMG СоА-редуктазы, например правастатин, ловастатин, симвастатин, аторвастатин, флувастатин или церивастатин, в дозировках, указанных в "Справочнике врача", так что количество составляет около 1-2000 мг, и предпочтительно, около 4-200 мг.
Ингибитор сквален-синтетазы можно применять в дозировках, примерно, 10-2000 мг, и предпочтительно, около 25-200 мг.
Предпочтительная пероральная лекарственная форма, например таблетки или капсулы, содержит ингибитор HMG СоА-редуктазы в количестве, примерно, 0,1-100 мг, предпочтительно, примерно 5-80 мг, и более предпочтительно, около 10-40 мг.
Предпочтительная пероральная лекарственная форма, такая как таблетки или капсулы, содержит ингибитор сквален-синтетазы в количестве, около, 10-500 мг, предпочтительно, примерно 25-200 мг.
Другой гиполипидемический агент может также являться ингибитором липоксигеназы, включая ингибитор 15-липоксигеназы (15-LO), такой как производные бензимидазола, как описано в Международной заявке WO 97/12615, ингибиторы 15-LO, описанные в WO 97/12613, изотиазолоны, описанные в WO 96/38144 и ингибиторы 15-LO, описанные Sendobry et al., "Ослабление вызванного диетой атеросклероза у кроликов под действием высокоселективного ингибитора 15-липоксигеназы с недостаточно заметными противоокислительными свойствами", Brit. J. Pharmacology (1998) 120, 1199-1206, и Cornicelli et al., "15-липоксигеназа и ее ингибирование: Новая терапевтическая цель для сосудистого заболевания". Current Pharmaceutical Design, 1999, 5, 11-20.
Соединения формулы I и гиполипидемический агент можно применять вместе в той же самой пероральной лекарственной форме или в раздельных пероральных лекарственных формах, даваемых одновременно.
Вышеописанные композиции можно вводить в лекарственных формах, как описано выше, в виде однократной дозы или раздельных доз один-четыре раза в день. Может быть целесообразно начинать больному с комбинации низких доз и постепенно доводить до комбинации высоких доз.
Предпочтительными гиполипидемическими агентами являются правастатин, ловастатин, симвастатин, аторвастатин, флувастатин, церивастатин, атавастатин и розувастатин.
Если другой тип терапевтического агента, который при необходимости применяется с ингибитором SGLT2 формулы I, представляет собой 1, 2, 3 или более агентов против ожирения, он может включать агонист бета 3 адренергического агента, ингибитор липазы, ингибитор повторного усвоения серотонина (допанина), препарат тироидного рецептора бета, лекарственное средство, снижающее аппетит, антагонист NPY, аналог лептина и/или агонист МС4.
Агонист бета 3 адренергического агента, который, при необходимости, можно применять в комбинации с соединением формулы I, может быть AJ9677 (Takeda/Dainippon), L750355 (Merck) или СР331648 (Pfizer) или другими известными бета 3 агонистами, как описано в Патентах США 5541204, 5770615, 5491134, 5776983 и 5488064, при этом предпочтительными являются AJ9677, L750355 и СР331648.
Ингибитор липазы, который, при необходимости, можно применять в комбинации с соединением формулы I, может представлять собой орлистат или ATL-962 (Alizyme), при этом орлистат является предпочтительным.
Ингибитор повторного поглощения серотонина (и допамина), который, при необходимости, можно применять в комбинации с соединением формулы I, может представлять собой сибутрамин, топирамат (Johnson & Johnson) или аксокин (Regeneron), при этом сибутрамин и топирамат являются предпочтительными.
Соединением тироидного рецептора бета, который, при необходимости, можно применять в комбинации с соединением формулы I, может быть лигандом тироидного рецептора, описанным в Международной заявке WO 97/21993 (U. Cal SF), WO 99/00353 (Karossio) и в английской Заявке GB 98/284425 (Karossio), при этом соединения заявок фирмы Karossio являются предпочтительными.
Агент, снижающий аппетит, который можно применять в комбинации с соединением формулы I, может представлять собой дексамфетамин, фентермин, фенилпропаноламин или мазиндол, при этом дексамфетамин является предпочтительным.
Различные агенты против ожирения, описанные выше, могут применяться в той же самой лекарственной форме с соединением формулы I или в разных лекарственных формах, и в дозировках и по схеме, обычно известных в технике или приведенных в PDR.
Примеры антитромбоцитного(ых) агента(ов), которые можно применять в сочетаниях по данному изобретению, включают абциксимат, тиклопидин, эптифибатид, дипиридамоль, аспирин, анагремид, тирофибан и/или клопидогрел.
Примеры антигипертензивного(ых) агента(ов), которые, при необходимости, можно применять в сочетаниях по данному изобретению, включают ингибиторы АСЕ, антагонисты кальция, альфа-блокаторы, диуретики, "центрально" действующие агенты, антагонисты ангиотензина-II, бета-блокаторы и ингибиторы вазопептидазы.
Примеры ингибиторов АСЕ включают лизиноприл, эналоприл, квинаприл, беназеприл, фозиноприл, рамиприл, каптоприл, эналоприл, моэксиприл, трандолаприл и периндоприл: примеры антагонистов кальция включают амлодипин, дилтиазем (diltiazem), нифедипин, верапамил, фелодипин, низолдипин (nisoldipine), исрадипин (isradipine) и никарадипин; примеры альфа-блокаторов включают теразозин (terazosin), доксазозин и празозин; примеры диуретиков включают гидрохлортиазид, торасемид, фуросемид, спиронолактон и индапамид; примеры "центрально" действующих агентов включают клонидин и гуанфацин; примеры антагонистов ангиотензина-II включают лосартан, валсартан, ирбесартан, кандесартан и телмисартан; примеры бета-блокаторов включают мотопролол, пропранолол, антенолол, карведилол и соталол; и примеры ингибиторов вазопептидазы включают омапатрилат и гемопатрилат.
При осуществлении способа по изобретению применяемая фармацевтическая композиция содержит соединения структуры I с другим антидиабетическим агентом или без него, и/или с антигиперлипидемическим агентом или без него, или с терапевтическим агентом другого типа или без него, вместе (в "ассоциации") с фармацевтическим носителем или разбавителем. Фармацевтическую композицию можно готовить, применяя обычные твердые или жидкие носители или разбавители и фармацевтические добавки, соответствующие требуемому способу введения. Соединения можно вводить млекопитающим, включая человека, обезьян, собак и т.д., пероральным способом, например, в форме таблеток, капсул, гранул или порошков, или их можно вводить парентерально в форме инъецируемых препаратов, или их можно вводить интраназально или в виде трансдермальных "накладок" (пластырей). Доза для взрослых составляет, предпочтительно, 10-2000 мг в день, их можно вводить в виде разовой (однократной) дозы или в виде отдельных доз 1-4 раза в день.
Типичный препарат для инъекций готовят, помещая 250 мг соединения структуры I в асептических условиях в пузырек, в асептических условиях его лиофилизируют и плотно закрывают. Для целей использования содержимое пузырька смешивают с 2 мл физиологического раствора, получая препарат для инъекции.
SGLТ2-ингибиторную активность соединений по изобретению можно определить с помощью аналитической системы, как представлено ниже.
Анализ на SGLT2-активность.
мРНК последовательность SGLT2 человека (GenBank N M95549) клонируют с помощью обратной транскрипции и амплификации мРНК почки человека, используя стандартные методики молекулярной биологии. Последовательность кДНК устойчиво трансфицируют в клетки СНО, и клоны анализируют на SGLT2-активность, в основном, как описано у Ryan et al. (1994). Оценку ингибирования SGLT2-активности в клонально отобранной клеточной линии осуществляют, в основном, как описано у Ryan et al. со следующими модификациями. Клетки выращивают в 96-луночных планшетах 2-4 дня до 75000 или 30000 клеток на лунку в питательной смеси F-12 (F-12 Ham), 10% фетальной бычьей сыворотке, 300 мкг/мл генетицина и пенициллина-стрептомицина. При достижении монослоя клетки отмывают дважды с помощью 10 мМ Hepes/Tris, pH 7,4; 137 мМ N-метил-D-глюкамина, 5,4 мМ KCl, 2,8 мМ CaCl2, 1,2 мМ MgSO4. Затем клетки инкубируют с 10 мкМ [14C]AMG и 10 мкМ ингибитора (конечный ДМСО = 0,5%) в 10 мМ Hepes/Tris, pH 7,4; 137 мМ NaCl, 5,4 мМ KCl, 2,8 мМ CaCl2, 1,2 мМ MgSO4 при 37°С в течение 1,5 часов. Анализ поглощения прекращают (разбавляют) ледяным 1X PBS, содержащим 0,5 мМ флоризина, и затем клетки лизируют 0,1% NaOH. После добавления сцинтилляционной жидкости MicroScint клетки встряхивают 1 ч, а затем [14C]AMG количественно определяют с помощью сцинтилляционного счетчика TopCount. Контроль осуществляют с и без NaCl. Для определения значений ЕС50 используют 10 концентраций ингибитора через 2 log-интервала (с разницей в 102) в соответствующем интервале ответа, и значения для трех планшетов усредняют.
Ryan M.J., Johnson G., Kirk J., Fuerstenberg S.M., Zager R.A. and Torok-Storb B. 1994. HK-2: иммортализованная эпителиальная клеточная линия проксимальных канальцев нормальной почки взрослого человека Kidney International 45; 48-57.
Следующие рабочие примеры дают представление о предпочтительных вариантах данного изобретения. Все значения температур даны в градусах Цельсия, если иначе не указано.
Пример 1
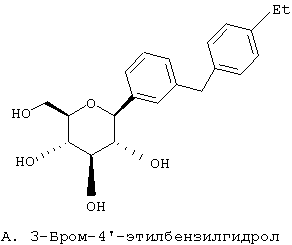
Сухую магниевую стружку (4,4 г, 0,178 мол) перемешивают в атмосфере аргона в течение ночи, после чего добавляют 100 мл сухого Et2O, затем добавляют в течение 1 ч п-бромэтилбензола (22 г, 0,119 мол) в 20 мл Et2O. (Если реакция не начинается, добавляют 0,5 мл 1,2-дибромэтана). После перемешивания в течение ночи медленно добавляют м-бромбензальдегид (11 г, 0,06 мол) в 20 мл Et2O. Полученный светлый раствор анализируют с помощью ВЭЖХ в течение 4-6 ч, чтобы определить окончание реакции. Реакцию гасят насыщенным водным NH4Cl, экстрагируют трижды EtOAc. Объединенные органические слои промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя. Полученное желтое масло хроматографируют на силикагеле с использованием 5% EtOAc/гексан, чтобы элюировать неполярные примеси и 7-9% EtOAc/гексан, чтобы элюировать 12,4 г (71%) 3-бром-4'-этилбензгидрола в виде светло-желтого масла.
В. 3-Бром-4'-этилдифенилметан
К перемешиваемому при -30°С раствору 3-бром-4'-этилбензгидрола (12,4 г, 0,0426 мол) из Раздела А в 120 мл MeCN добавляют BF3·Et2O (6,04 г, 0,426 мол) и затем Et3SiH (9,9 г, 0,852 мол). Темную реакционную смесь после перемешивания в течение 1 ч при -30°С медленно разогревают до -5°С. По окончании (ТСХ-контроль) реакцию прекращают добавлением насыщенного водного К2СО3. После добавления 100 мл Н2О, смесь трижды экстрагируют Et2O. Объединенные органические слои промывают рассолом, сушат над Na2SO4. После концентрирования с использованием роторного испарителя получают 3-бром-4'-этилдифенилметан (11,17 г, 95%) в виде светло-желтого масла, которое используют без дополнительной очистки.
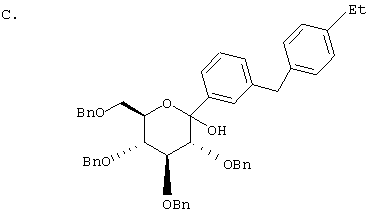
К перемешиваемому при -78°С раствору 3-бром-4'-этилдифенилметана (10,9 г, 0,04 мол) из Раздела В в 100 мл сухого ТГФ в атмосфере аргона добавляют в течение 20 мин 25,7 мл 1,7 М трет.-BuLi в гексане. Через 1 ч добавляют 2,3,4,6-тетра-O-бензил-β-D-глюколактон (23,5 г, 0,0437 мол) в 30 мл ТГФ в течение 15 мин. Раствор перемешивают в течение 1 ч при -78°С, после чего гасят насыщенным водным NH4Cl. Температуру доводят до 20°С, реакционную массу разбавляют в два раза EtOAc, затем промывают водой и рассолом. После высушивания над Na2SO4 и концентрирования в роторном испарителе получают 29,2 г вышеприведенного лактола в виде бесцветного сиропа, который используют далее без дополнительной очистки.
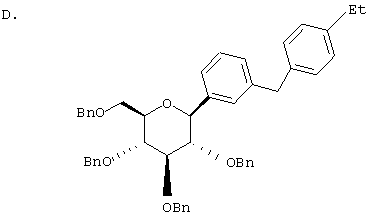
К перемешиваемому при -30°С раствору лактола (29,1 г, 0,04 мол) из Раздела В в 100 мл MeCN добавляют BF3·Et2O (5,62 г, 0,4 мол) и затем Et3SiH (9,21 г, 0,08 мол). Через 2 ч, когда по данным ТСХ реакция завершена, добавляют насыщенный водный К2СО3 и суспензию перемешивают в течение 1 ч при 20°С, после чего разбавляют Н2О и Et2O. Объединенные органические слои из 3 эфирных экстракций промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя, получают 28,3 г светло-желтого сиропа. Хроматографируют на силикагеле с использованием 5% EtOAc/гексан, чтобы элюировать неполярные примеси и затем медленно требуемый бета аномер и альфа аномер. Фракции, обогащенные бета аномером, необходимо дополнительно очищать растиранием гексаном или перекристаллизацией из EtOH, чтобы получить 6 г требуемого бета тетра-O-бензил С-глюкозида. (Примечание: когда восстановителем является Et3SiH, получают смесь бета/альфа аномеров 5:1, в то время как при использовании iPr3SiH получают смесь 30:1).
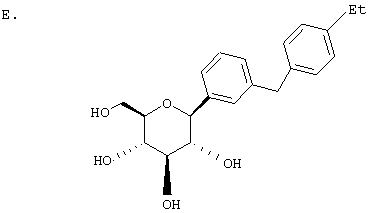
Раствор тетра-O-бензил С-глюкозида (2,4 г, 3,35 ммол) из Раздела D в EtOAc (100 мл), содержащий 10% Pd(OH)2/C (0,35 г) перемешивают в течение ночи при 1 атм Н2. После того как реакция завершена (ВЭЖХ), катализатор отфильтровывают и растворитель удаляют с использованием роторного испарителя, получают 1,1 г нужного бета С-глюкозида (92%) в виде белого кристаллического вещества.
Время удерживания ВЭЖХ: 7,04 мин, 100% чистота, колонка 4,6×50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 8 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (500 МГц, CD3OD) δ 7,27 (с, 1Н), 7,23 (д, 2Н, J=4,95 Гц), 7,1-7,0 (м, 5Н), 4,08 (д, 1H, J=9,3 Гц), 3,91 (с, 2Н), 3,9 (дд, 1H, J=2,2, 11 Гц), 3,68 (дд, 1Н, J=5,5, 11 Гц), 3,5-3,35 (м, 4Н), 2,57 (к, 2Н, J=7,2 Гц), 1,18 (т, 3Н, J=7,2 Гц).
13С NMR (125 МГц, CD3OD) δ 143, 142,8, 141, 140, 129,9, 129,6, 129,5, 129,1, 128,8, 126,7, 83,8, 82,3, 79,9, 76,4, 72,0, 63,2, 42,5, 29,4, 16,2.
Анализ: Вычислено для C21H26O5 LC-MS [M+NH4] 376; найдено 376.
Пример 2
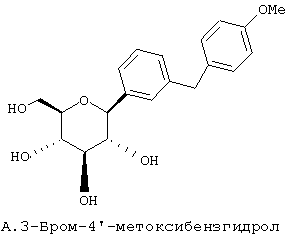
К перемешиваемому при -78°С раствору м-дибромбензола (70,9 г, 0,3 мол) в 200 мл сухого ТГФ в атмосфере аргона добавляют 117 мл 2,56 М н-BuLi (0,3 мол) в гексане в течение 10 мин. Через 30 мин добавляют п-метоксибензальдегид (27,2 г, 0,02 мол) в 50 мл ТГФ в течение 20 мин. Раствор перемешивают в течение 1 ч при -78°С (контроль по ТСХ), после чего гасят насыщенным водным NH4Cl. Температуру доводят до 20°С, реакционную массу разбавляют в два раза EtOAc, затем промывают водой и рассолом. После высушивания над Na2SO4 и концентрирования в роторном испарителе получают 103 г 3-бром-4'-метоксибензгидрола в виде желтого масла, которое используют далее без дополнительной очистки.
В. 3-Бром-4'-метоксидифенилметан
К перемешиваемому при -40°С раствору 3-бром-4'-метоксибензгидрола (103 г, 0,2 мол) из Раздела А в 300 мл MeCN добавляют Et3SiH (64 мл, 0,4 мол) и затем BF3·Et2O (27,7 г, 0,2 мол). По окончании (ТСХ-контроль) реакцию прекращают добавлением насыщенного водного К2СО3 (25 мл). После добавления 100 мл Н2О, смесь трижды экстрагируют EtOAc. Объединенные органические слои промывают рассолом, сушат над Na2SO4. После концентрирования с использованием роторного испарителя полученный вышеназванный сырой 3-бром-4'-метоксидифенилметан (92 г) хроматографируют на силикагеле с использованием в качестве элюента EtOAc/гексан с получением 17 г прозрачного продукта, который используют без дополнительной очистки.
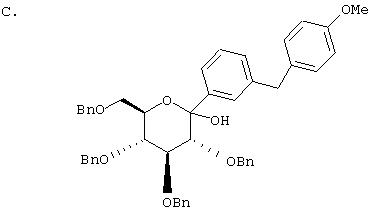
К перемешиваемому при -78°С раствору 3-бром-4'-метоксидифенилметана (9,6 г, 0,035 мол) из Раздела В в 50 мл сухого ТГФ в атмосфере аргона добавляют в течение 5 мин 14 мл 2,5 М трет.-BuLi в гексане. После перемешивания в течение 30 мин добавляют 2,3,4,6-тетра-О-бензил-β-D-глюколактон (12,5 г, 0,023 мол) в 20 мл ТГФ в течение 10 мин. Раствор перемешивают в течение 1 ч при -78°С, после чего ТСХ-анализ показывает, что реакция завершена. После гашения насыщенным водным NH4Cl (25 мл) температуру доводят до 20°С, реакционную массу разбавляют EtOAc (200 мл). Органический слой промывают водой и рассолом. После высушивания над Na2SO4 и концентрирования в роторном испарителе, вышеназванный требуемый лактол хроматографируют на силикагеле с использованием 12,5% EtOAc/гексан, чтобы элюировать 8,1 г >90% лактола и дополнительно >80% чистоты.
К перемешиваемому при -40°С раствору лактола (7,8 г, 0,019 мол) из Раздела В в 100 мл MeCN добавляют Et3SiH (3,42 мл, 0,04 мол) и затем BF3·Et2O (1,37 мл, 0,02 мол). Через 1 ч, когда по данным ТСХ реакция завершена, добавляют насыщенный водный К2СО3 (10 мл) и суспензию перемешивают в течение 1 ч при 20°С, после чего трижды экстрагируют EtOAc. Объединенные органические слои промывают Н2О, рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя, получают 8 г сырого продукта. Хроматографией на силикагеле с использованием 5% EtOAc/гексан элюируют неполярные примеси и затем медленно 0,92 г чистого β-тетра-O-бензил С-глюкозида и затем 6,5 г, содержащих оба аномера.
Вышеполученные две фракции соединения из Раздела D гидрируют по отдельности над 10% Pd(OH)2/C (2% по весу) в течение ночи при 1 атмосфере H2 в EtOAc (12,5 мл/г соединения из Раздела D). Затем катализатор отфильтровывают и растворитель - продукт гидрогенолиза смешанных фракций - очищают препаративной ВЭЖХ с использованием колонки с YMC S5 для обращенно-фазовой хроматографии. Объединенное вещество дает 1,85 г чистого β аномера в виде белого твердого вещества.
Время удерживания ВЭЖХ: 6,04 мин, колонка 6×75 мм с Zorbax С-18, 2,5 мл/мин, детектирование при 220 нм; 8 мин градиент 0-100% В, 3 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3РО4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3РО4.
1Н NMR (400 МГц, СО3OD) δ 7,28 (с, 1Н), 7,24 (д, 2Н, J=3 Гц), 7,09 (м, 3Н), 6,69 (д, 2Н, J=7 Гц), 4,08 (д, 1Н, J=8,8 Гц), 3,88 (с, 2Н), 3,75 (д, 1Н, J=12 Гц), 3,73 (с, 3Н), 3,65 (дд, 1Н, J=12,3 Гц), 3,4 (м, 4Н).
13С NMR (105 МГц, CD3OD) δ 158,6, 142,1, 140,2, 133,8, 130,0, 128,7, 128,6, 128,3, 125,8, 82,9, 81,3, 79,0, 75,5, 71,1, 63,5, 55,1, 41,1.
Анализ: Вычислено для С20Н24О6 LC-MS [M-Н] 359; найдено 359.
Пример 3
К перемешиваемому при -78°С раствору м-дибромбензола (12,6 г, 53 ммол) в 50 мл сухого ТГФ в атмосфере аргона добавляют 20 мл 2,56 М н-BuLi (51 ммол) в гексане в течение 10 мин. Через 40 мин добавляют 2,3,4,6-тетра-O-бензил-β-D-глюколактона (12 г, 22 ммол) в 30 мл ТГФ в течение 15 мин. Раствор перемешивают в течение 1 ч при -78°С (контроль по ТСХ), после чего гасят насыщенным водным NH4Cl (40 мл). Температуру доводят до 20°С, реакционную массу разбавляют в два раза EtOAc, затем промывают водой и рассолом. После высушивания над Na2SO4 и концентрирования в роторном испарителе получают 20 г требуемого сырого лактола в виде масла, которое используют далее без дополнительной очистки.
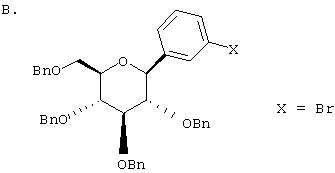
К перемешиваемому при -45°С раствору лактола (20 г, 0,2 мол) из Раздела А в 60 мл MeCN добавляют Et3SiH (7,8 мл, 45 ммол) и затем медленно добавляют в течение 20 мин BF3·Et2O (4,2 мл, 22 ммол). По окончании (ТСХ-контроль) реакцию прекращают добавлением насыщенного водного К2СО3 (25 мл) и смесь трижды экстрагируют EtOAc. Объединенные органические слои промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя. Полученное масло растирают с 50 мл гексана, в результате чего после стояния в течение 1 часа получают твердый остаток. Это вещество отделяют фильтрованием, промывают дважды холодным гексаном и сушат на воздухе с получением 8,9 г нужного представленного выше β-м-бромфенил-С-глюкозида.
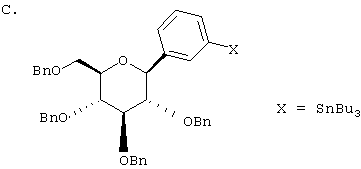
Раствор полученного выше β-м-бромфенил-С-глюкозида (1,36 г, 2 ммол) из Раздела В, Pd(PPh3)4 (70 мг, 0,06 ммол) и гексабутилдиолово (2,724 г, 6 ммол) в сухом толуоле (10 мл) нагревают при перемешивании в атмосфере аргона при 80°С в течение 15 ч. После удаления толуола с использованием роторного испарителя остаток хроматографируют на силикагеле, используя смесь 12:1 EtOAc/гексан, чтобы элюировать нужное представленное выше арилолово (761 мг), плюс смесь фракций, которые после повторной хроматографии на колонке дают дополнительно 92 мг чистого вышеназванного соединения олова с общим выходом 48%, и затем 230 мг регенерированного исходного β-м-бромфенил-С-глюкозида из Раздела В.
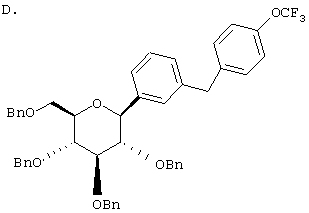
Смесь соединения олова (2,66 г, 3 ммол) из Раздела Е, п-трифторметоксибензилхлорида (1,04 г, 6 ммол) и Pd(PPh3)4 (100 мг, 0,09 ммол) кипятят с обратным холодильником в атмосфере аргона в ТГФ (1 мл) в течение 15 ч. После удаления ТГФ с использованием роторного испарителя, остаток хроматографируют на силикагеле с использованием смеси 10:1 EtOAc/гексан, чтобы элюировать 1,3 г требуемого представленного выше тетрабензилового эфира.
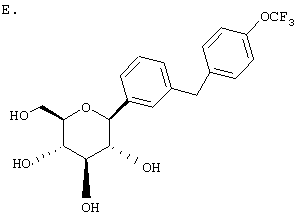
Превращение в свободный конечный глюкозид достигается перемешиванием 295 мг тетрабензилового эфира из Раздела D с Pd(OH)2 (15 мг) в EtOAc (3 мл) в течение 15 ч при 1 атмосфере Н2. Конечный продукт выделяют после фильтрования, препаративной ВЭЖХ и удаления растворителя.
Время удерживания ВЭЖХ: 7,21 мин, колонка 6×75 мм с Zorbax С-18, 2,5 мл/мин, детектирование при 220 нм; 8 мин градиент 0-100% В, 3 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3РО4.
1H NMR (400 МГц, CD3OD) δ 7,3 (м, 5Н), 7,15 (м, 3Н), 4,10 (д, 1Н, J=8,8 Гц), 3,99 (с, 2Н), 3,9 (д, 1Н, J=12 Гц), 3,7 (дд, 1Н, J=12, 3 Гц), 3,4 (м, 4Н).
Анализ: Вычислено для С20H21F3O6 LC-MS [М-Н] 413; найдено 413.
Пример 4
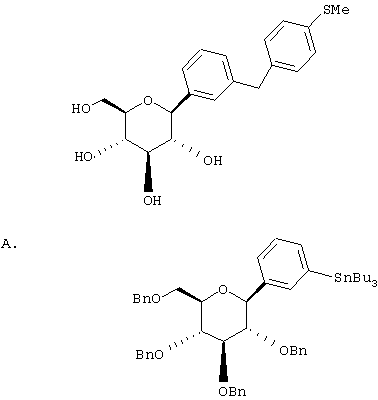
Раствор β-м-бромфенил-С-глюкозида (3,0 г, 4,41 ммол) из Примера 3, Pd(PPh3)4 (153 мг, 0,13 ммол) и гексабутилдиолова (6,0 г, 13,2 ммол) в сухом толуоле (5 мл) нагревают при перемешивании в атмосфере аргона при 88°С в течение 3 ч, после чего ТСХ-анализ показывает, что реакция прошла на 90%. Реакцию завершают через 5 часов общего времени. После удаления толуола с использованием роторного испарителя остаток хроматографируют на силикагеле, используя смесь 1:8 EtOAc/гексан, чтобы элюировать 2,95 г нужного арилолова.
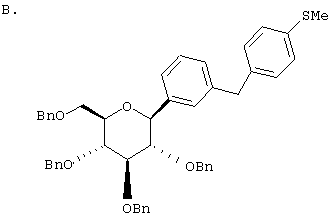
Смесь соединения олова (2,66 г, 3 ммол) из Раздела А, п-метилтиобензилхлорида (1,04 г, 6 ммол) и Pd(PPh3)4 (100 мг, 0,09 ммол) кипятят с обратным холодильником в атмосфере аргона в ТГФ (1 мл) в течение 15 ч. После удаления ТГФ с использованием роторного испарителя остаток хроматографируют на силикагеле с использованием смеси 6:1 EtOAc/гексан, чтобы элюировать 1,2 г представленного выше тетра-O-бензилового эфира и затем еще 600 мг тетра-О-бензилового эфира, содержащего Ph3P.
1 М BCl3/СН2Cl2 (6 мл, 8 ммол) добавляют в течение 5 мин к перемешиваемому при -78°С раствору тетрабензилового эфира (295 мг, 0,4 ммол) из Раздела В в CH2Cl2 (0,25 мл) в атмосфере аргона. Через 30 мин, когда ТСХ-анализ показывает, что реакция завершена, добавляют 30 мл смеси 2:1 CH2Cl2/PhMe и затем 2 мл МеОН. Объем уменьшают наполовину с помощью роторного испарителя и добавляют 10 мл МеОН. После повторения этой процедуры 3 раза все летучие продукты удаляют в вакууме. Остаток хроматографируют на силикагеле с использованием 5% MeOH/CH2Cl2, чтобы элюировать 143 мг нужного глюкозида с 90%-ной чистотой. Это вещество далее очищают с помощью обращенной фазовой препаративной хроматографии с получением 104 мг конечного требуемого глюкозида.
Время удерживания ВЭЖХ: 6,69 мин, колонка 6×75 мм с Zorbax С-18, 2,5 мл/мин, детектирование при 220 нм; 8 мин градиент 0-100% В, 3 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (400 МГц, CD3OD) δ 7,27 (с, 1Н), 7,25 (д, 2Н, J=2 Гц), 7,15 (м, 5Н), 4,09 (д, 1Н, J=8,8 Гц), 3,92 (с, 2Н), 3,86 (д, 1Н, J=12 Гц), 3,68 (дд, 1Н, J=12, 3 Гц), 3,4 (м, 4Н), 2,43 (с, 3Н).
Анализ: Вычислено для C20H24O6S LC-MS [М-Н] 375; найдено 375.
Пример 5
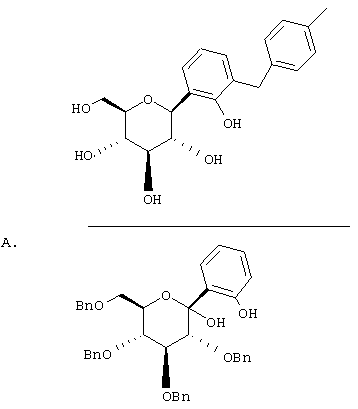
К перемешиваемой суспензии 60% NaH (180 мг, 4,5 ммол) в ТГФ (7 мл) в атмосфере аргона добавляют 2-бромфенол (350 мкл, 3 ммол). После перемешивания в течение 15 мин реакцию охлаждают до -78°С и добавляют по каплям 1,4 М трет.-BuLi/гексан (2,36 мл, 3,3 ммол). Через 10 мин раствор переносят с помощью шприца в перемешиваемый при -78°С раствор 2,3,4,6-тетра-O-бензил-β-D-глюколактона (1,62 г, 3,0 ммол) в ТГФ (5 мл). Реакцию гасят через 15 мин медленным добавлением насыщенного NH4Cl/H2O и затем доводят температуру до 20°С, после чего добавляют 200 мл EtOAc. Органический слой промывают последовательно Н2О и рассолом, сушат над MgSO4 и концентрируют. Хроматография на силикагеле с использованием смеси 3:1 гексан/EtOAc дает 390 мг нужного представленного выше лактола.
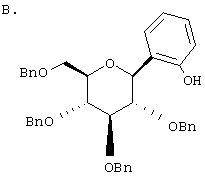
К перемешиваемой смеси 3:1 MeCN/CH2Cl2 (4 мл), содержащей лактол (390 мг, 0,62 ммол) из Раздела А, добавляют при -30°С Et3SiH (197 мкл, 1,23 ммол) и BF3·Et2O (78 мкл, 0,62 ммол). Через 1 ч реакцию гасят насыщенным водным К2СО3, доводят температуру до 20°С и разбавляют 100 мл EtOAc. Органический слой промывают последовательно Н2О и рассолом, сушат над MgSO4 и концентрируют. Хроматография на силикагеле с использованием смеси 3:1 гексан/EtOAc дает 269 мг нужного представленного выше фенольного глюкозида.
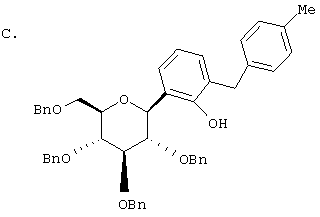
К толуольному раствору (1,1 мл) фенола из Раздела В (139 мг, 0,22 ммол) в атмосфере аргона добавляют 60% NaH (11 мг, 0,27 ммол). Через 10 мин добавляют 4-метилбензилбромид (46 мг, 0,25 ммол), после чего раствор, который становится голубым, нагревают при 80°С в течение 3,5 ч, пока реакция по данным ТСХ-анализа не завершится. После охлаждения добавляют водный NH4Cl, реакцию разбавляют EtOAc. Органический слой промывают последовательно Н2О и рассолом, сушат над MgSO4 и концентрируют. Хроматография на силикагеле с использованием смеси 5:1 гексан/EtOAc дает 71 мг нужного представленного выше тетра-O-бензилглюкозида.
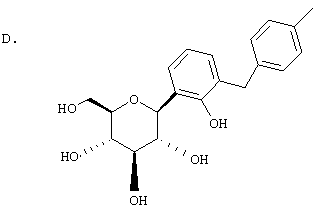
Последующий гидрогенолиз тетра-O-бензилового эфира из Раздела С над Pd/C в МеОН при 1 атмосфере Н2 дает конечный приведенный выше продукт, который очищают препаративной ВЭЖХ с использованием колонки С 18 для обращенной фазовой хроматографии, градиент 45-90% МеОН/Н2О в течение 10 мин, чтобы получить нужный β-С-глюкозид (2 мг).
Время удерживания ВЭЖХ: 6,754 мин, 100% чистота, колонка 4,6×50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 8 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (500 МГц, CD3OD) δ 7,15 (дд, 1Н, J=1,1, 7,7 Гц), 7,07 (д, 2Н, J=8,3 Гц), 7,02 (д, 2Н, J=8,3 Гц), 6,96 (дд, 1H, J=1,2, 7,7 Гц), 6,77 (т, 1Н, J=7,7 Гц), 4,44 (д, 1Н, J=8,8 Гц), 3,89 (с, 2Н), 3,87 (д, 1Н, J=2,2 Гц), 3,75 (дд, 1Н, J=4,9, 12,1 Гц), 3,49-3,41 (м, 4Н), 2,26 (с, 3Н).
Анализ: Вычислено для С20H26O6 LC-MS [М-Н] 361; найдено 361.
Пример 6
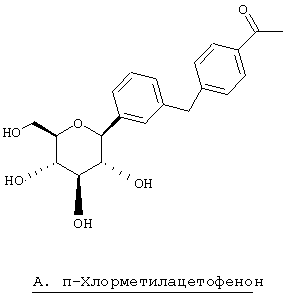
К перемешиваемому раствору п-хлорметилбензоил хлорида (390 мг, 2,06 ммол) в 8 мл ТГФ при -20°С в атмосфере аргона добавляют трибутилфосфин (406 мг, 2,29 ммол). После перемешивания полученного желтого раствора в течение 20 мин при -20° ÷ -15°С добавляют одной порцией 0,3 мл 3 М раствора метилмагнийбромида в эфире (2,1 ммол), при этом получают красный раствор, который через 10 мин становится оранжевым. Реакцию гасят добавлением 1 N водной HCl. После разбавления водой смесь экстрагируют трижды EtOAc, промывают водой, сушат над Na2SO4. Остаток, полученный после удаления летучих примесей, хроматографируют на силикагеле с использованием 5% EtOAc/гексан, чтобы элюировать 171 мг (50%) п-хлорметилацетофенона.
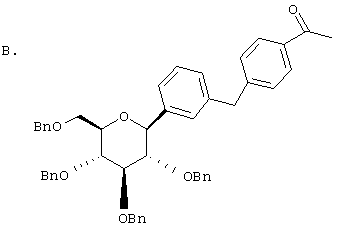
Смесь соединения олова, описанного в Примере 3 Раздел С (300 мг, 0,33 ммол), п-хлорметилацетофенона (114 мг, 0,66 ммол), Pd(PPh3)4 (20 мг, 0,09 ммол), окиси трифенилфосфина (180 мг, 0,65 ммол), K2CO3 (75 мг, 0,55 ммол) нагревают при 70°С в атмосфере аргона в ТГФ (0,3 мл) в течение 16 часов. После удаления ТГФ в роторном испарителе, остаток хроматографируют на силикагеле с использованием смеси от 20:1 до 10:1 гексан/EtOAc, чтобы элюировать нужный тетрабензиловый эфир (170 мг, 70%).
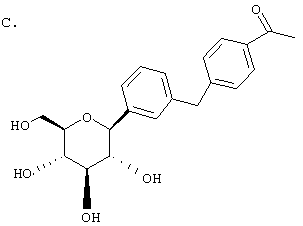
Раствор тетрабензилового эфира из Раздела В (60 мг, 0,08 ммол) в CH2Cl2 (0,25 мл) в атмосфере аргона охлаждают до -78°С и затем добавляют 0,8 мл 1 М раствора BCl3 в CH2Cl2. После перемешивания в течение 1 ч при -78°С добавляют вторые 0,8 мл 1 М раствора BCl3 к перемешиваемой реакционной смеси. Через два часа добавляют 0,5 мл PhMe и затем по каплям 0,5 мл МеОН. Летучие примеси удаляют на роторном испарителе; повторяют процедуру после добавления смеси 2:1 CH2Cl2/MeOH. Хроматография на силикагеле с использованием 5% MeOH/CH2Cl2 дает 20 мг тетраола - конечного продукта с выходом 67%.
Время удерживания ВЭЖХ: 2,35 мин, 100% чистота, колонка 4,6×50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 4 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (500 МГц, CD3OD) δ 7,88 (д, 2Н), 7,27-7,34 (м, 5Н), 7,13 (д, 1Н), 4,09 (д, 1Н), 3,91 (с, 2Н), 4,03 (с, 2Н), 3,85 (д, 1Н), 3,68 (дд, 1Н), 3,35-3,48 (м, 4Н), 2,55 (с, 3Н).
13C NMR (500 МГц,CD3OD) δ 200,3, 148,8, 141,4, 141,2, 136,3, 130,2, 129,7, 129,6, 129,3, 127,0, 83,6, 82,2, 79,8, 76,4, 71,9, 63,1, 42,7, 26,6.
Анализ: Вычислено для С21Н24О6 LC-MS [M+NH4 +] 390,2; найдено 390,2.
Пример 7
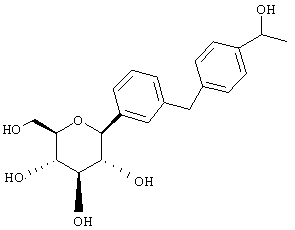
Перемешиваемый раствор конечного продукта из Примера 6 (15 мг, 0,04 ммол) в 5 мл EtOH охлаждают до -20°С, затем добавляют NaBH4 (5 мг, 0,13 ммол). Через 20 мин, когда по данным ТСХ-анализа реакция завершена, ее гасят несколькими каплями насыщенного водного NH4Cl. После удаления летучих примесей хроматографируют на силикагеле. Элюирование 5% MeOH/EtOAc приводит к получению 10 мг (67%) нужного продукта.
Время удерживания ВЭЖХ: 5,2 мин, 100% чистота, колонка 4,6×50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 8 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (500 МГц, CD3OD) δ 7,21-7,32 (м, 5Н), 7,16 (д, 2Н), 7,10-7,11 (м, 1Н), 4,77 (к, 1Н), 4,08 (д, 1Н), 3,94 (с, 2Н), 3,86 (дд, 1Н), 3,68 (дд, 1Н), 3,34-3,48 (м, 4Н), 1,40 (д, 3Н).
13С NMR (500 МГц, CD3OD) δ 145,2, 142,5, 141,5, 140,9, 129,8, 129,6, 129,5, 129,2, 126,7, 126,6, 83,7, 82,2, 79,8, 76,4, 72,0, 63,2, 42,5, 25,5.
Анализ: Вычислено для С21Н26О6 LC-MS [M+NH4 +] 392,2; найдено 392,2.
Пример 8
Смесь о-толуиловой кислоты (28 г, 206 ммол), порошок железа (0,74 г, 13 ммол) и Br2 (42 г, 260 ммол) перемешивают при 0°С в течение 2 ч. В этот момент, когда реакция проходит на ˜40%, ее разбавляют 25 мл CH2Cl2, чтобы облегчить перемешивание. Затем реакцию нагревают при 45°С в течение 16 ч, чтобы довести ее до конца. После охлаждения реакционную смесь разбавляют CH2Cl2, промывают дважды 10%-ным NaHSO3, один раз рассолом, затем сушат над Na2SO4. После удаления летучих примесей, остаток представляет собой смесь 2:1 5-бром- и 3-бромтолуиловой кислоты, которую перекристаллизовывают из 95% EtOH с получением 14,4 г 5-бром-2-метилбензойной кислоты.
В. 5-Бром-2-метил-4'-метоксибензофенон
К перемешиваемой суспензии 5-бром-2-метилбензойной кислоты (1,29 г, 6 ммол) в 12 мл CH2Cl2, содержащей оксалил хлорид (8 ммол) добавляют 2 капли ДМФА. После прекращения энергичного выделения газа реакционную смесь перемешивают в течение 6 ч и затем удаляют в роторном испарителе летучие примеси. После растворения сырого 5-бром-2-метилбензоилхлорида в 15 мл CS2 перемешиваемую смесь охлаждают до 4°С, затем добавляют анизол (0,7 г, 6,6 ммол) и следом AlCl3 (1,7 г, 12 ммол). Реакционную смесь доводят до 20°С в течение 1 ч и затем перемешивают в течение 15 ч, после чего гасят 1 N HCl. Далее, суспензию разбавляют 50 мл Н2О и перемешивают, пока весь осадок не перейдет в раствор. Смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают по одному разу 1 N HCl, Н2О, водным NaHCO3 и рассолом, а затем сушат над Na2SO4. После удаления летучих примесей полученное рыжевато-коричневое твердое вещество перекристаллизовывают из 95% EtOH с получением 1,6 г 5-бром-2-метил-4'-метоксибензофенона.
С. 5-Бром-2-метил-4'-метоксидифенилметан
Раствор Et3SiH (2,5 мл, 15,5 ммол), BF3·Et2O (1,3 мл, 10 ммол) и 5-бром-2-метил-4'-метоксибензофенона (1,6 г, 5,25 ммол) в 11 мл смеси 1:4 СН2Cl2/MeCN перемешивают в течение ночи при 20°С. Когда по ВЭЖХ-анализу остается 5% исходного кетона, раствор нагревают при 40°С в течение 1 часа, после чего гасят 10%-ным NaOH. После разбавления Н2O, реакционную смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают два раза Н2О и один раз рассолом, а затем сушат над Na2SO4. После удаления летучих примесей, остаток хроматографируют на силикагеле, используя гексан, чтобы элюировать 5-бром-2-метил-4'-метоксидифенилметан в виде бесцветного масла (1,4 г, 95%).
К перемешиваемому при -78°С раствору 5-бром-2-метил-4'-метоксидифенилметана из Раздела С (0,43 г, 1,5 ммол) в 7 мл сухого ТГФ в атмосфере аргона добавляют по каплям 0,9 мл 1,8 М раствора н-BuLi в гексане. Через 2 часа добавляют 2,3,4,6-тетра-О-бензил-β-D-глюколактона (0,88 г, 1,6 ммол) в 3 мл ТГФ в течение 1 мин. Раствор перемешивают 2 часа при -78°С и затем гасят насыщенным водным NH4Cl. Затем температуру доводят до 20°С, реакционную смесь разбавляют в два раза Н2О, после чего трижды экстрагируют EtOAc. Объединенные органические экстракты промывают рассолом и сушат над Na2SO4. После концентрирования с использованием роторного испарителя получают 1,1 г нужного приведенного выше лактола в виде бесцветного сиропа, который используют далее без дополнительной очистки.
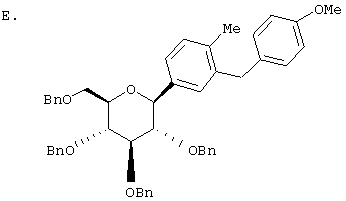
К перемешиваемому при -30°С раствору лактола (1,1 г, 1,47 ммол) из Раздела D в 10 мл MeCN добавляют iPr3SiH (0,7 мл, 4,5 ммол) и затем BF3·Et2O (0,38 мл, 2,6 мол). Через 3 ч при -40° ÷ -30°С реакция, как показывает ТСХ-контроль, заканчивается. Добавляют насыщенный водный К2СО3 и суспензию перемешивают 1 ч при 20°С, после чего разбавляют Н2О и EtOAc. Объединенные органические слои из трех экстракций EtOAc промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя с получением 1,2 г светло-желтого сиропа. Хроматография на силикагеле с использованием 10% EtOAc/гексан после элюирования неполярных примесей дает нужный бета С-арилглюкозид (0,54 г).

Раствор тетра-О-бензил С-глюкозида (515 мг, 0,7 ммол) из Раздела Е в EtOAc (10 мл), содержащий 10% Pd(OH)2/C (80 мг) перемешивают в течение ночи при 1 атм Н2. После того как реакция завершена (ВЭЖХ), катализатор отфильтровывают и растворитель удаляют с использованием роторного испарителя, получают белое стекловидное вещество, которое далее очищают с помощью препаративной ВЭЖХ с использованием колонки С 18 для обращенной фазовой хроматографии, чтобы получить 220 мг нужного бета С-глюкозида в виде бесцветного сиропа.
Время удерживания ВЭЖХ: 6,43 мин, 100% чистота, колонка 4,6×50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 8 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3РО4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3РО4.
1H NMR (500 МГц, СО3OD) δ 7,20 (с, 1Н), 7,18 (д, 2Н, J=7 Гц), 7,11 (д, 2Н, J=7 Гц), 6,89 (АВк, 4Н), 4,07 (д, 1Н, J=9 Гц), 3,90 (с, 2Н), 3,87 (м, 1Н), 3,70 (с, 3Н), 3,68 (дд, 1Н), 3,48-3,30 (м, 4Н), 2,16 (с, 3Н).
13C NMR (125 МГц, CD3OD) δ 159,3, 140,3, 138,3, 137,4, 133,7, 131,0, 130,8, 130,6, 126,9, 114,7, 83,5, 82,1, 79,8, 76,3, 71,9, 63,1, 55,6, 59,6, 19,5.
Анализ: Вычислено для С21Н26О6 LC-MS [M-H] 373; найдено 373.
Пример 9
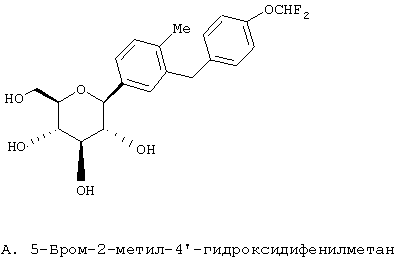
К перемешиваемому при -78°С раствору 5-бром-2-метил-4'-метоксидифенилметана (1,0 г, 3,4 ммол) (получение см. Пример 8, Раздел С) в 10 мл CH2Cl2 добавляют 4,12 мл 1 М раствора BBr3/CH2Cl2. Через 2 ч реакцию выдерживают при -40°С в течение 20 ч, после чего ВЭЖХ-анализ не обнаруживает следов исходного эфира. Реакцию гасят водной NaOH, экстрагируют трижды CH2Cl2, промывают рассолом и затем сушат над Na2SO4. После удаления летучих примесей получают 0,84 г 5-бром-2-метил-4'-гидроксидифенилметана в виде сиропа, который используют далее без дополнительной очистки.
В. 5-Бром-2-метил-4'-бензилоксидифенилметан
Раствор 10 мл ДМФА, содержащий 5-бром-2-метил-4'-гидроксидифенилметан из Раздела А (735 мг, 2,65 ммол), бензилбромид (548 мг, 3,2 ммол) и K2CO3 (732 мг, 5,3 ммол), перемешивают в течение ночи. Реакцию затем нагревают при 60°С в течение 6 ч, чтобы достичь конверсии от 80 до 100%. После разбавления Н2O реакционную смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают Н2О и рассолом, а затем сушат над Na2SO4. Остаток после удаления летучих примесей в вакууме хроматографируют на силикагеле, используя 3% EtOAc/гексан, чтобы элюировать 785 мг 5-бром-2-метил-4'-бензилоксидифенилметана в виде бесцветного сиропа.
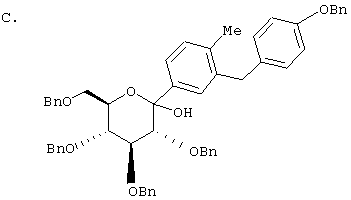
К перемешиваемому при -78°С раствору 5-бром-2-метил-4'-бензилоксидифенилметана из Раздела В (0,43 г, 1,2 ммол) в 7 мл сухого ТГФ в атмосфере аргона добавляют по каплям 0,68 мл 1,9 М раствора н-BuLi в гексане. Через 30 мин добавляют 2,3,4,6-тетра-О-бензил-β-D-глюколактона (0,7 г, 1,3 ммол) в 3 мл ТГФ в течение 1 мин. Раствор перемешивают 0,75 ч при -78°С и затем гасят насыщенным водным NH4Cl. Затем температуру доводят до 20°С, реакционную смесь разбавляют в два раза Н2О, после чего трижды экстрагируют EtOAc. Объединенные органические экстракты промывают рассолом и сушат над Na2SO4. После концентрирования с использованием роторного испарителя получают 0,96 г нужного приведенного выше лактола в виде бесцветного сиропа, который используют далее без дополнительной очистки.
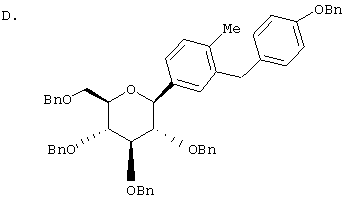
К перемешиваемому при -30°С раствору лактола (0,96 г, 1,16 ммол) из Раздела D в 10 мл MeCN добавляют iPr3SiH (0,37 мл, 2,3 ммол) и затем BF3·Et2O (0,2 мл, 1,4 мол). Через 3 ч при -40° ÷ -30°С реакция, как показывает ТСХ-контроль, заканчивается. Добавляют насыщенный водный К2СО3 и суспензию перемешивают 1 ч при 20°С, после чего разбавляют Н2O и EtOAc. Объединенные органические слои из трех экстракций EtOAc промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя с получением 1,2 г светло-желтого сиропа. Хроматография на силикагеле с использованием 10% ЕЮАс/гексан после элюирования неполярных примесей дает нужный бета С-арилглюкозид (0,26 г).
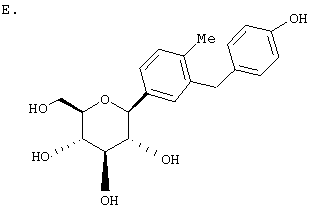
Раствор пента-O-бензил С-глюкозида (255 мг, 0,31 ммол) из Раздела D в EtOAc (10 мл), содержащий 10% Pd(OH)2/C (65 мг), перемешивают в течение 24 ч при 1 атм Н2. После того как реакция завершена (ВЭЖХ), катализатор отфильтровывают и растворитель удаляют с использованием роторного испарителя, получают 115 мг белого стекловидного вещества, которое используют далее без дополнительной очистки.
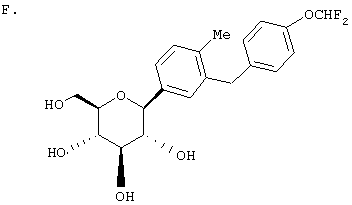
Трехгорлую колбу, содержащую магнитную мешалку, 4 мл iPrOH и фенольный глюкозид из Раздела Е (80 мг, 0,16 ммол) охлаждают до -78°С, после чего добавляют CHClF2 в виде конденсированного газа. После добавления 3 мл 25% водного NaOH, колбу закрывают тефлоновым затвором и нагревают при 70°С в течение 2 часов. ВЭЖХ-анализ показывает, что реакционная смесь содержит смесь 2:3 исходного фенола и нужного эфира. (Попытки повысить конверсию за счет более продолжительного времени реакции оказались безуспешными). После охлаждения добавляют 1 N НС в количестве, достаточном для достижения рН 2, после чего большую часть летучих продуктов удаляют в роторном испарителе. Остаток, после растворения в смеси 2:1 МеОН/Н2О, очищают с помощью препаративной ВЭЖХ с использованием колонки YMC S5 С-18 для обращенной фазовой хроматографии (20х100 мм) при 10 мин линейном градиенте 45%-90% водного МеОН при 20 мл/мин, чтобы получить 400 мг нужного фенольного эфира.
Время удерживания ВЭЖХ: 6,6 мин, 95% чистота, колонка 4,6 х 50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 8 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% H3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (400 МГц, CD3OD) δ 7,22 (с, 1Н), 7,20 (м, 1Н), 7,12(м, 1Н), 7,06 (АВк, 4Н), 6,73 (т, 1Н, J=27 Гц), 4,09 (д, 1Н, J=9 Гц), 3,98 (д, 1Н), 3,68 (дд, 1Н), 3,47-3,30 (м, 4Н), 2,17 (с, 3Н).
13C NMR (100 МГц, CD3OD) δ 138,7, 138,2, 137,7, 136,6, 130,3, 130,2, 130,1, 126,4, 119,3, 117,0, 82,7, 81,4, 79,0, 75,6, 71,1, 62,3, 49,0, 38,8, 18,6.
Анализ: Вычислено для С21Н24F2O6 LC-MS [M+NH4] 428; найдено 428.
Пример 10
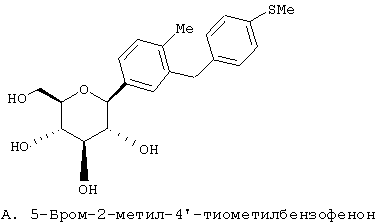
AlCl3 (535 мг, 4 ммол) добавляют при 4°С к перемешиваемому раствору в 5 мл CS2 сырого 5-бром-2-метилбензоил хлорида (466 мг, 2 ммол) (получение см. Пример 8, Раздел В) и тиоанизола (270 мг, 3,3 ммол). Реакционную смесь доводят до 20°С в течение 1 ч и затем перемешивают в течение 2 ч, после чего гасят 1 N HCl. Далее, суспензию разбавляют 50 мл Н2О и перемешивают, пока весь осадок не перейдет в раствор. Смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают по одному разу 1 N HCl, Н2О, водным NaHCO3 и рассолом, а затем сушат над Na2SO4. После удаления летучих примесей остаток хроматографируют на силикагеле с использованием 15% EtOAc/гексан, чтобы элюировать 450 мг 5-бром-2-метил-4'-тиометилбензофенона в виде белого твердого вещества.
В. 5-Бром-2-метил-4'-тиометилдифенилметан
Раствор Et3SiH (0,45 мл, 2,85 ммол), BF3·Et2O (0,3 мл, 2,4 ммол) и 5-бром-2-метил-4'-тиометилбензофенона из Раздела А (450 мг, 1,4 ммол) в 3 мл смеси 1:9 CH2Cl2/MeCN перемешивают в течение ночи при 20°С. После обработки 10%-ным NaOH и разбавления Н2О, реакционную смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают два раза Н2О и один раз рассолом, а затем сушат над Na2SO4. После удаления летучих примесей остаток хроматографируют на силикагеле, используя 5% EtOAc/гексан, чтобы элюировать 416 мг 5-бром-2-метил-4'-тиометилдифенилметана в виде бесцветного масла.
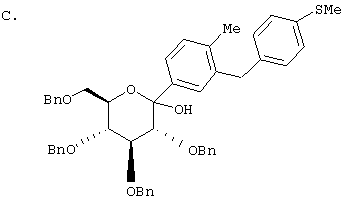
К перемешиваемому при -78°С раствору 5-бром-2-метил-4'-тиометилдифенилметана из Раздела В (200 мг, 0,65 ммол) в 10 мл сухого ТГФ в атмосфере аргона добавляют по каплям 0,42 мл 1,8 М раствора н-BuLi в гексане. Через 2 часа этот раствор вносят с помощью шприца в перемешиваемый при -78°С раствор 2,3,4,6-тетра-O-бензил-β-D-глюколактона (0,88 г, 1,6 ммол) в 5 мл ТГФ. Раствор перемешивают 2 ч при -78°С и затем гасят насыщенным водным NH4Cl. Затем температуру доводят до 20°С, реакционную смесь разбавляют в два раза Н2О, после чего трижды экстрагируют EtOAc. Объединенные органические экстракты промывают рассолом и сушат над Na2SO4. После концентрирования с использованием роторного испарителя получают 550 мг нужного приведенного выше лактола в виде бесцветного сиропа, который используют далее без дополнительной очистки.
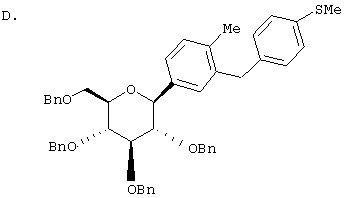
К перемешиваемому при -40°С раствору лактола (550 мг, 0,72 ммол) из Раздела С в 6 мл MeCN добавляют iPr3SiH (0,22 мл, 1,0 ммол) и затем BF3·Et2O (0,11 мл, 0,8 мол). Через 1,5 ч при -40° ÷ -30°С реакцию, когда, как показывает ТСХ-контроль, реакция заканчивается, добавляют насыщенный водный K2CO3 и суспензию перемешивают 1 час при 20°С, после чего разбавляют Н2О и EtOAc. Объединенные органические слои из трех экстракций EtOAc промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя с получением 1,2 г светло-желтого сиропа. Хроматография на силикагеле с использованием 9% EtOAc/гексан в качестве элюента дает 240 мг нужного бета С-арилглюкозида.
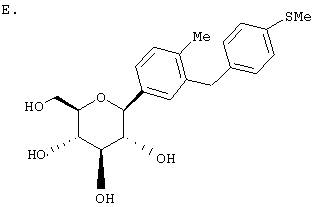
Раствор тетра-О-бензил С-глюкозида из Раздела D (70 мг, 0,1 ммол) в EtSH (1,5 мл), содержащий BF3·Et2O (0,24 мл, 2 ммол) перемешивают при 20°С в течение 2 ч. Спустя еще один час после добавления дополнительных 0,12 мл BF3·Et2O реакция завершается. Реакционную смесь гасят, медленно добавляя 0,4 мл пиридина, после чего разбавляют водным NH4Cl. Объединенные органические экстракты после трех экстракций EtOAc промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя. Остаток очищают с помощью препаративной ВЭЖХ с использованием колонки YMC S5 С-18 для обращенной фазовой хроматографии, чтобы получить 20 мг бета С-глюкозида в виде белого лиофилизата после лиофилизирования.
Время удерживания ВЭЖХ: 3,8 мин, 95% чистота, колонка 4,6×50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 4 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2O+0,2% Н3PO4.
1H NMR (500 МГц, CD3OD) δ 7,21-7,11 (м, 5Н), 7,05 (д, 2Н, J=8,0 Гц), 4,08 (д, 1Н, J=9,1 Гц), 3,98 (с, 2Н), 3,87 (д, 1Н, J=12,6 Гц), 3,68 (дд, 1Н, J=5,2, 12,1 Гц), 3,49-3,30 (м, 4Н), 2,41 (с, 3Н).
13C NMR (125 МГц, CD3OD) δ 139,8, 138,9, 138,4, 137,5, 137,1, 131,1, 130,9, 129,1, 130,3, 127,8, 127,1, 83,6, 82,2, 79,8, 76,4, 72,0, 62,3, 39,9, 19,5, 16,1.
Анализ: Вычислено для C21H26O5S LC-MS [M+NH4] 408; найдено 408.
Пример 11
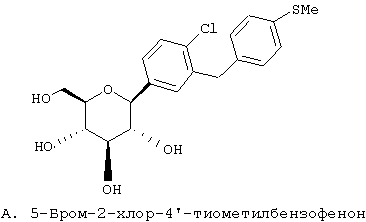
К перемешиваемой суспензии промышленной 5-бром-2-хлорбензойной кислоты (506 мг, 2,12 ммол) в 10 мл CH2Cl2, содержащей оксалил хлорид (2,4 ммол) добавляют 2 капли ДМФА. После прекращения энергичного выделения газа реакционную смесь перемешивают в течение 1,5 ч и затем удаляют в роторном испарителе летучие примеси. После растворения сырого 5-бром-2-хлорбензоилхлорида в 8 мл CS2 перемешиваемую смесь охлаждают до 4°С, затем добавляют тиоанизол (260 мг, 2,12 ммол) и следом AlCl3 (566 мг, 4,25 ммол). Реакционную смесь доводят до 20°С в течение 1 ч и затем перемешивают в течение 20 ч, после чего гасят 1 N HCl. Далее, суспензию разбавляют 50 мл Н2О и перемешивают, пока весь осадок не перейдет в раствор. Смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают по одному разу 1 N HCl, Н2О, водным NaHCO3 и рассолом, а затем сушат над Na2SO4. После удаления летучих примесей получают 710 мг сырого 5-бром-2-хлор-4'-метоксибензофенона, который далее не очищают.
В. 5-Бром-2-хлор-4'-тиометилдифенилметан
Раствор Et3SiH (1,45 мл, 8,8 ммол), BF3·Et2O (0,83 мл, 6,6 ммол) и 5-бром-2-хлор-4'-тиометилбензофенона из Раздела А (710 мг, 2,1 ммол) в 10 мл смеси 1:4 CH2Cl2/MeCN перемешивают в течение 2 ч при 20°С. После обработки 10%-ным NaHCO3 и разбавления Н2О, реакционную смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают два раза Н2О и один раз рассолом, а затем сушат над Na2SO4. После удаления летучих примесей остаток хроматографируют на силикагеле, используя 5% EtOAc/гексан, чтобы элюировать 630 мг 5-бром-2-хлор-4'-тиометилдифенилметана в виде бесцветного масла.
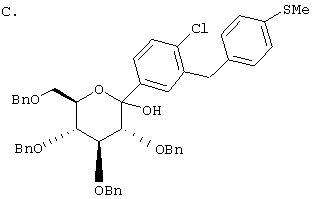
К перемешиваемому при -78°С раствору 5-бром-2-хлор-4'-тиометилдифенилметана из Раздела В (200 мг, 0,61 ммол) в 6 мл сухого ТГФ в атмосфере аргона добавляют по каплям 0,48 мл 1,5 М раствора н-BuLi в гексане. Через 35 мин этот раствор вносят с помощью шприца в перемешиваемый при -78°С раствор 2,3,4,6-тетра-O-бензил-β-D-глюколактона (361 мг, 0,67 ммол) в 5 мл ТГФ. Раствор перемешивают 1,5 ч при -78°С и затем гасят насыщенным водным NH4Cl. Затем температуру доводят до 20°С, реакционную смесь разбавляют в два раза Н2О, после чего трижды экстрагируют EtOAc. Объединенные органические экстракты промывают рассолом и сушат над Na2SO4. После концентрирования с использованием роторного испарителя остаток хроматографируют с использованием 20% EtOAc/гексан, чтобы получить 250 мг нужного приведенного выше лактола.
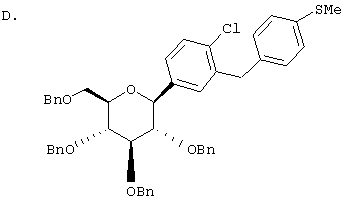
К перемешиваемому при -30°С раствору лактола (250 мг, 0,32 ммол) из Раздела С в 5 мл MeCN добавляют iPr3SiH (0,10 мл, 0,56 ммол) и затем BF3·Et2O (0,048 мл, 0,38 мол). Через 0,5 ч при -30°С реакцию, когда, как показывает ТСХ-контроль, реакция заканчивается, добавляют насыщенный водный NaHCO3 и суспензию перемешивают 1 ч при 20°С, после чего разбавляют Н2О и EtOAc. Объединенные органические слои из трех экстракций EtOAc промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя с получением 1,2 г светло-желтого сиропа. Хроматография на силикагеле с использованием 9% EtOAc/гексан в качестве элюента дает 200 мг нужного бета С-арилглюкозида.
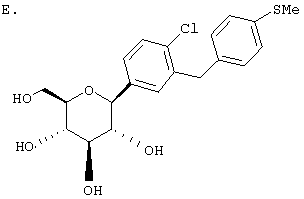
Раствор тетра-O-бензил С-глюкозида из Раздела D (60 мг, 0,1 ммол) в EtSH (2 мл), содержащий BF3·Et2O (0,24 мл, 2 ммол) перемешивают при 20°С в течение 3 ч. Реакционную смесь гасят, медленно добавляя 0,4 мл пиридина, после чего разбавляют водным NH4Cl. Объединенные органические экстракты после трех экстракций EtOAc промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя. Остаток очищают с помощью препаративной ВЭЖХ с использованием колонки YMC S5 С-18 для обращенной фазовой хроматографии, чтобы получить 21,5 мг нужного бета С-глюкозида в виде белого лиофилизата после лиофилизирования.
Время удерживания ВЭЖХ: 3,96 мин, 95% чистота, колонка 4,6×50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 4 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (400 МГц, CD3OD) δ 7,36-7,27 (м, 3Н), 7,15 (д, 2Н, J=8,3 Гц), 7,11 (д, 2Н, J=8,3 Гц), 4,10-4,04 (м, 3Н), 3,98 (с, 2Н), 3,87 (д, 1Н, J=12 Гц), 3,70 (дд, 1Н, J=7,1, 11,8 Гц), 3,47-3,26 (м, 4Н), 2,42 (с, 3Н).
13С NMR (100 МГц, CD3OD) δ 140,1, 139,3, 138,0, 137,5, 134,5, 132,0, 130,4, 130,2, 128,4, 128,0, 82,9, 82,8, 82,2, 79,7, 76,5, 71,8, 63,1, 39,5, 16,1.
Анализ: Вычислено для C20H23ClO5S LC-MS [M-H] 409; найдено 409.
Пример 12
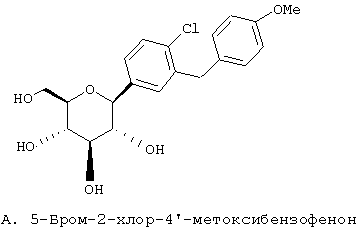
К перемешиваемой суспензии промышленной 5-бром-2-хлорбензойной кислоты (506 мг, 2,12 ммол) в 10 мл СН2Cl2, содержащей оксалил хлорид (2,4 ммол) добавляют 2 капли ДМФА. После прекращения энергичного выделения газа реакционную смесь перемешивают в течение 1,5 ч и затем удаляют в роторном испарителе летучие примеси. После растворения сырого 5-бром-2-хлорбензоилхлорида в 8 мл CS2 перемешиваемую смесь охлаждают до 4°С, затем добавляют анизол (240 мг, 2.12 ммол) и следом AlCl3 (566 мг, 4,25 ммол). Реакционную смесь доводят до 20°С в течение 1 ч и затем перемешивают в течение 20 ч, после чего гасят 1 N HCl. Далее, суспензию разбавляют 50 мл Н2О и перемешивают, пока весь осадок не перейдет в раствор. Смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают по одному разу 1 N HCl, Н2О, водным NaHCO3 и рассолом, а затем сушат над Na2SO4. После удаления летучих примесей остаток хроматографируют на силикагеле с использованием 15% EtOAc/гексан, чтобы элюировать 450 мг 5-бром-2-хлор-4'-метоксибензофенона.
В. 5-Бром-2-хлор-4'-метоксидифенилметан
Раствор Et3SiH (0,45 мл, 2,85 ммол), BF3·Et2O (0,3 мл, 2,4 ммол) и 5-бром-2-хлор-4'-метоксибензофенона из Раздела А (450 мг, 1,4 ммол) в 3 мл смеси 1:9 CH2Cl2/MeCN перемешивают в течение ночи при 20°С. После обработки 10%-ным NaHCO3 и разбавления Н2О реакционную смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают два раза Н2О и один раз рассолом, а затем сушат над Na2SO4. После удаления летучих примесей, остаток хроматографируют на силикагеле, используя 2% EtOAc/гексан, чтобы элюировать 630 мг 5-бром-2-хлор-4'-метоксидифенилметана в виде бесцветного масла.
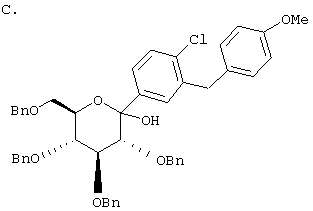
К перемешиваемому при -78°С раствору 5-бром-2-хлор-4'-метоксидифенилметана из Раздела В (212 мг, 0,68 ммол) в 8 мл сухого ТГФ в атмосфере аргона добавляют по каплям 0,36 мл 1,9 М раствора н-BuLi в гексане. Через 30 мин этот раствор вносят с помощью шприца в перемешиваемый при -78°С раствор 2,3,4,6-тетра-O-бензил-β-D-глюколактона (0,39 г, 0,71 ммол) в 5 мл ТГФ. Раствор перемешивают 2 ч при -78°С и затем гасят насыщенным водным NH4Cl. Затем температуру доводят до 20°С, реакционную смесь разбавляют в два раза Н2О, после чего трижды экстрагируют EtOAc. Объединенные органические экстракты промывают рассолом и сушат над Na2SO4. После концентрирования с использованием роторного испарителя остаток хроматографируют с использованием 20% EtOAc/гексан, чтобы получить 142 мг нужного приведенного выше лактола.
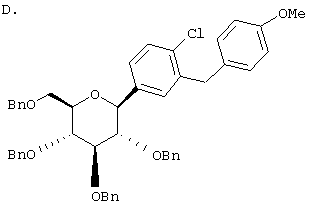
К перемешиваемому при -40°С раствору лактола (142 мг, 0,18 ммол) из Раздела С в 1,5 мл MeCN добавляют iPr3SiH (0,041 мл, 0,2 ммол) и затем BF3·Et2О (0,026 мл, 0,2 мол). Через 2 ч при -40°С реакцию, когда, как показывает ТСХ-контроль, реакция заканчивается, добавляют насыщенный водный NaHCO3 и суспензию перемешивают 1 ч при 20°С, после чего разбавляют Н2О и CH2Cl2. Объединенные органические слои из трех экстракций CH2Cl2 промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя. Хроматография остатка на силикагеле с использованием 25% EtOAc/гексан в качестве элюента дает 139 мг нужного бета С-арилглюкозида.
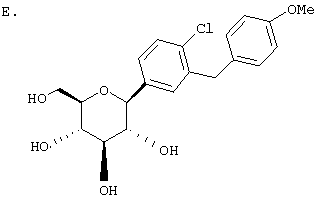
Раствор тетра-O-бензил С-глюкозида из Раздела D (136 мг, 0,18 ммол) в EtSH (1 мл), содержащий BF3·Et2O (0,46 мл, 3,6 ммол), перемешивают при 20°С в течение 4 ч. Реакционную смесь разбавляют CH2Cl2 и затем концентрируют с использованием роторного испарителя. Остаток после растворения в CH2Cl2 промывают водным NH4Cl, Н2О, рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя. Остаток очищают с помощью препаративной ВЭЖХ с использованием колонки YMC S5 С-18 для обращенной фазовой хроматографии, чтобы получить 26 мг нужного бета С-глюкозида в виде белого лиофилизата после лиофилизирования.
Время удерживания ВЭЖХ: 3,07 мин, 95% чистота, колонка 4,6×50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 4 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3РО4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (500 МГц, CD3OD) δ 7,35-7,28 (м, 3Н), 7,1 (д, 2Н, J=8,8 Гц), 6,8 (д, 2Н, J=8,3 Гц), 4,05-3,90 (м, 3Н), 3,80 (д, 1Н, J=12,3 Гц), 3,67 (с, 3Н), 3,61 (дд, 1H, J=4,8, 11,9 Гц), 3,42-3,25 (м, 4Н).
13C NMR (125 МГц,CD3OD) δ 159,6, 140,0, 139,9, 134,5, 133,3, 131,9, 130,8, 130,1, 114,8, 82,9, 82,8, 82,2, 79,8, 76,5, 71,9, 63,1, 55,6, 39,2.
Анализ: Вычислено для С20Н23ClO6 LC-MS [M+NH4] 412; найдено 412.
Пример 13
К перемешиваемому при -78°С раствору п-бромэтилбензола (2,03 г, 11 ммол) в 10 мл сухого ТГФ в атмосфере аргона добавляют в течение 10 мин 5 мл 2,5 М раствора н-BuLi в гексане. Температуре дают подняться до -10°С в течение 2 ч, после чего реакционную смесь охлаждают до -78°С и добавляют твердый 5-бром-2-метоксибензальдегид (2,15 г, 10 ммол). После перемешивания в течение ночи при 20°С реакционную смесь гасят насыщенным водным NH4Cl и разбавляют в 5 раз Н2О, после чего трижды экстрагируют EtOAc. Объединенные органические экстракты промывают рассолом и сушат над Na2SO4. После концентрирования с использованием роторного испарителя остаток хроматографируют с использованием 10% EtOAc/гексан, чтобы получить 1,44 г 5-бром-2-метокси-4'-этилбензгидрола.
В. 5-Бром-2-метокси-4'-этилдифенилметан
9 мл раствора 1:8 CH2Cl2/MeCN, содержащего сырой 5-бром-2-метокси-4'-этилбензгидрол из Раздела А (1,44 г, 4,5 ммол), Et3SiH (0,75 мл, 5 ммол) и BF3·Et2O (0,6 мл, 6,4 ммол) перемешивают в течение ночи при 20°С. После обработки насыщенным водным раствором NaOH реакционную смесь экстрагируют трижды EtOAc. Объединенные органические экстракты промывают рассолом и сушат над Na2SO4. После концентрирования с использованием роторного испарителя, остаток хроматографируют на силикагеле, используя 2% EtOAc/гексан, чтобы элюировать 1,28 г 5-бром-2-метокси-4'-этилдифенилметана.
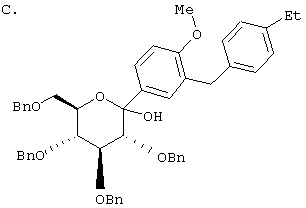
К перемешиваемому при -78°С раствору 5-бром-2-метокси-4'-этилдифенилметана из Раздела В (0,25 г, 0,82 ммол) в 7 мл сухого ТГФ в атмосфере аргона добавляют по каплям 0,5 мл 1,8 М раствора н-BuLi в гексане. Через 2 часа добавляют 2,3,4,6-тетра-О-бензил-β-D-глюколактона (0,48 г, 0,9 ммол) в 3 мл ТГФ в течение 1 мин. Раствор перемешивают 2 ч при -78°С и затем гасят насыщенным водным NH4Cl. Затем температуру доводят до 20°С, реакционную смесь разбавляют в два раза Н2О, после чего трижды экстрагируют EtOAc. Объединенные органические экстракты промывают рассолом и сушат над Na2SO4. После концентрирования с использованием роторного испарителя, получают 0,67 г нужного приведенного выше лактола в виде светло-желтого сиропа, который используют далее без дополнительной очистки.
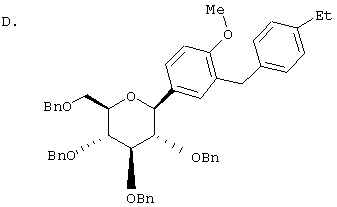
К перемешиваемому при -30°С раствору лактола (450 мг, 0,59 ммол) из Раздела С в 10 мл MeCN добавляют iPr3SiH (0,2 мл, 0,9 ммол) и затем BF3·Et2O (0,1 мл, 0,7 мол). Через 1,5 ч при -40°С реакцию, когда, как показывает ТСХ-контроль, реакция заканчивается, ее гасят добавлением насыщенного водного NaHCO3 и трижды экстрагируют EtOAc. Объединенные органические слои промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя. Хроматография остатка на силикагеле с использованием 10% EtOAc/гексан в качестве элюента дает 320 мг нужного бета С-арилглюкозида.

Раствор тетра-O-бензил С-глюкозида (320 мг, 0,7 ммол) из Раздела D в EtOAc (15 мл), содержащий 10% Pd(OH)2/C (30 мг) перемешивают в течение ночи при 1 атм Н2. После того как реакция завершена (ВЭЖХ), катализатор отфильтровывают и растворитель удаляют с использованием роторного испарителя. Остаток очищают с помощью препаративной ВЭЖХ с использованием колонки YMC S5 С-18 для обращенной фазовой хроматографии, чтобы получить 24 мг нужного бета С-глюкозида в виде белого лиофилизата после лиофилизирования.
Время удерживания ВЭЖХ: 3,84 мин, 95% чистота, колонка 4,6×50 мм с YMC S5 С-18, 2,5 мл/мин, детектирование при 220 нм; 4 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (500 МГц, CD3OD) δ 7,23 (д, 1H, J=7 Гц), 7,17 (с, 1Н), 7,05 (АВк, 4Н), 6,89 (д, 1H, J=87 Гц), 4,02 (д, 2Н, J=9 Гц), 3,92-3,83 (м, 3Н), 3,76 (с, 3Н), 3,66 (дд, 1H), 3,45-3,29 (м, 4Н), 2,55 (к, 2Н), 1,16 (т, 3Н).
Анализ: Вычислено для C22H28O6 LC-MS [M+NH4] 406; найдено 406.
Пример 14
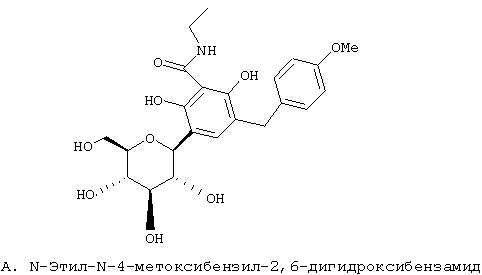
К перемешиваемому раствору N-этил-4-метоксибензиламина (1,07 г, 6,49 ммол) в ДМФА (10 мл) добавляют 2,6-дигидроксибензойную кислоту (1,0 г, 6,49 ммол) и затем HOAt (0,97 г, 7,14 ммол) и EDC (1,31 г, 6,81 ммол). После перемешивания в течение ночи реакционную смесь разбавляют EtOAc, после чего промывают трижды Н2O. Объединенные водные слои экстрагируют один раз EtOAc. Органические фракции объединяют, промывают один раз рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя. Остаток хроматографируют на силикагеле с использованием 75% EtOAc/гексан в качестве элюента. Полученные смешанные фракции с примесями дополнительно очищают хроматографией на силикагеле. Общий выход нужного N-этил-N-4-метоксибензил-2,6-дигидроксибензамида - 631 мг.
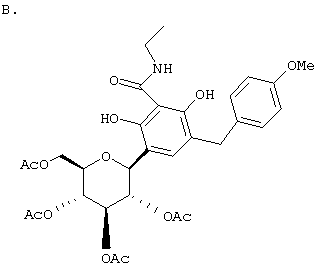
Перемешиваемую суспензию амида из Раздела А (630 мг, 2,09 ммол), CdCO3 (939 мг, 5,44 ммол) в толуоле (30 мл) кипятят с обратным холодильником в течение 1,5 ч, используя насадку Дина-Старка, после чего добавляют 2,3,4,6-тетра-O-ацетил-α-D-глюкозопиранозил бромид (1,12 г, 2,72 ммол). Через 15 ч кипячения ТСХ-анализ не обнаруживает остатков исходного амида. Горячую суспензию фильтруют через целит, который промывают горячим PhMe и затем трижды горячим CHCl3. После удаления летучих примесей с помощью роторного испарителя остаток хроматографируют на силикагеле. С использованием 1: EtOAc/гексан сначала элюируют смесь O-глюкозидов, а затем тетраацетат нужного приведенного выше С-глюкозида; получают 172 мг несколько загрязненного вышеназванного С-глюкозида.
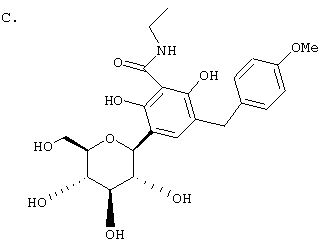
Неочищенный эфир из Раздела В перемешивают в смеси 6:1 EtOH/H2O (1,4 мл), содержащей КОН (140 мг, 2,5 ммол), в течение 16 ч. Полученный раствор охлаждают до 4°С, подкисляют до рН 5 и затем экстрагируют дважды EtOAc. Объединенные EtOAc слои промывают рассолом, сушат над Na2SO4 и концентрируют с использованием роторного испарителя. Остаток очищают препаративной ВЭЖХ с использованием колонки С18 для обращенной фазовой хроматографии, градиент 45-90% МеОН/Н2О в течение 30 мин, чтобы получить нужный приведенный выше С-глюкозид (7,8 мг).
ВЭЖХ: 99,1%; колонка (6,0×150 мм) Shimadzu LC-6A, YMC S3 ODS, 1,5 мл/мин, детектирование при 220 нм; 30 мин градиент элюирования 0-100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4. Время удерживания = 23,4 мин.
1H NMR (400 МГц, CD3OD) δ 1,22 (3Н, т, J=7,2 Гц), 3,4-3,5 (6Н, м), 3,73 (3Н, с), 3,74 (1Н, м), 3,77 (1Н, м), 3,8-3,9 (2Н, м), 4,36 (1Н, д, J=9,3 Гц), 6,77 (1Н, д, J=8,6 Гц), 7,11 (2Н, д, J=8,6 Гц), 7,18 (1Н, с).
13C NMR (125 МГц, CD3OD) δ 14,9, 35,1, 35,1, 55,7, 62,5, 71,2, 75,8, 79,6, 80,3, 82,3, 104,8, 114,7, 117,1, 122,7, 130,7, 134,5, 134,6, 151, 159,3, 161, 171,9.
Анализ: Вычислено для C23H29NO9 LC-MS [M-H] 462; найдено 462.
Пример 15
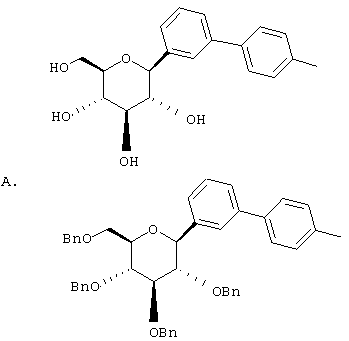
Смесь β-м-бромфенил-С-глюкозида (100 мг, 0,14 ммол) из Примера 3 Раздел В, п-метилфенилборной кислоты (59 мг, 0,43 ммол), Na2CO3 (46 мг, 0,43 ммол) и Pd(PPh3)4 (153 мг, 0,13 ммол) в смеси 3:1 PhMe/EtOH перемешивают в атмосфере аргона при 80°С в течение 15 ч. После удаления летучих примесей с использованием роторного испарителя остаток хроматографируют на силикагеле, используя смесь 10:1 гексан/EtOAc, чтобы элюировать нужный представленный выше бифенил-С-глюкозид (90 мг) в виде бесцветного масла.
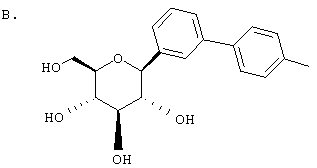
К перемешиваемому при -78°С раствору тетра-O-бензилового эфира (65 мг, 0,09 ммол) из Раздела А в CH2Cl2 (0,4 мл) в атмосфере аргона добавляют 0,37 мл 1 М BCl3 в СН2Cl2. Через 1 ч реакционную смесь гасят 2 мл МеОН и температуру поднимают до 20°С. После того как рН доводят до ˜7 с помощью NaHCO3, суспензию экстрагируют дважды CH2Cl2. Объединенные органические слои сушат над MgSO4 и концентрируют. Полученный остаток после очистки с помощью препаративной ВЭЖХ с использованием колонки С-18 для обращенной фазовой хроматографии дает 6,6 мг конечного требуемого продукта. (Примечание: продукт частично разрушается сильнокислой средой, получающейся после того, как BCl3 гасят МеОН.)
Время удерживания ВЭЖХ: 6,353 мин, 100% чистота, колонка 4,6×50 мм с Zorbax С-18, 2,5 мл/мин, детектирование при 220 нм; 8 мин градиент 0-100% В, 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0,2% Н3РО4. Растворитель В: 90% МеОН/Н2О + 0,2% Н3PO4.
1H NMR (400 МГц, CD3OD) δ 7,65 (с, 1Н), 7,53-7,50 (м, 3Н), 7,39-3,37 (м, 2Н), 7,23 (д, 2Н, J=7,9 Гц), 4,20 (д, 1Н, J=9,3 Гц), 3,89 (дд, 1Н, J=2,2, 11,9 Гц), 3,71 (дд, 1Н, J=5,7, 11,9 Гц), 3,50-3,40 (м, 4Н), 2,36 (с, 3Н).
Анализ: Вычислено для C19H22O5 LC-MS [М-Н] 329; найдено 329.
Примеры 16-80
Соединения Примеров 16-80, представленные в Таблицах 1 и 2, получают по методикам в Примерах 1-15 и реакционным Схемам 1-9. Следует понимать, что эти соединения, в которых А могут быть соединены в орто-, мета- и пара-положении арильного кольца, присоединенного к глюкозиду, и могут быть любым из (СН2)n, О, NH или S, тогда как R1, R2, R2a, R3 и R4 могут быть любым из вышеуказанных заместителей, можно получать по методикам Примеров 1-15 и по реакционным Схемам 1-9.
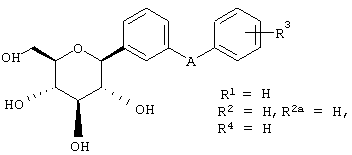
N
Ki (nM)
(M+NH4)
(M+NH4)
(M+NH4)
(M+NH4)
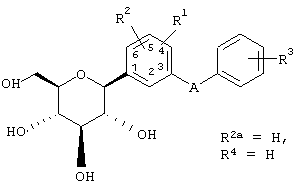
N
Ki (nM)
(M+Na)
(М-Н)
(М-Н)
(M+NH4)
(М-Н)
(М-Н)
(М-Н)
(M+NH4)
(M+NH4)
(М-Н)
(М-Н)
(M+NH4)
(M+NH4)
(M+NH4)
(М-Н)
(М-Н)
(М+Н)
(M+NH4)
(M+NH4)
(M+Na)
Представленные в таблицах данные по ингибирующей SGLT2 активности были получены в результате следующего анализа.
Последовательность мРНК человеческого SGLT2 (GenBank #M95549) клонируют обратной транскрипцией и амплификации при использовании мРНК почки человека стандартными методами молекулярной биологии. Последовательность кДНК устойчиво трансфецируют в клетки СНО и клоны анализируют на SGLT2 активность, в основном как описано в Ryan et al. (Ryan M.J., Johnson G., Kirk J., Fuerstenberq S.M., Zager R.A. and Torok- Storb B. 1994. HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney international 45: 48-57). Оценку ингибирования SGLT2 активности в клонально выбранной клеточной линии осуществляют, главным образом, как описано в Ryan et al., с нижеприведенными модификациями. Клетки помещают по 10000 или 20000 клеток на лунку и культивируют в среде Хэма F-12, содержащей 10% фетальной бычьей сыворотки и 500 мкг/мл генетицина. Клетки, конфлуэнтные, примерно, на 90%. Анализируют через 2 или 3 дня после засевания. Клетки отмывают один раз буфером, не содержащим натрий, который включает 10 мМ Hepes/Tris, 137 мМ N-метил-D-глюкамина, 5.4 мМ KCl, 2.8 мМ CaCl2 и 1.2 мМ MgSO4, pH 7.4. Ингибиторы анализируют в присутствии 10 мкМ [14С] AMG (а-метил-D-глюкопиранозида) при 8 концентрациях после 120-минутной инкубации в буфере, не содержащем белка, включающем 10 мМ Hepes/Tris, 137 мМ NaCl, 5.4 мМ KCl, 2.8 мМ CaCl2 и 1.2 мМ MgSO4 pH 7.4. Кривую ответа строят по четырем параметрам эмпирической модели с целью определения концентрации ингибитора при полумаксимальном ответе, обозначенной IC50. Определение проводят в тройном повторе. Анализ прекращают, отмывая 3 раза ледяным IX фосфатно-солевым буферным раствором (PBS), содержащим 0.5 мМ флоризина и затем клетки лизируют в 50 мкл 0.1% NaOH. Прибавляют 200 мкл сцинтилляционной жидкости MicroSeint- 40, клетки встряхивают 1 час, а затем количественно определяют [14C]AMG с помощью сцинтилляционного счетчика TopCount. Контрольный анализ в отсутствие ингибитора осуществляют с и без NaCl и кривую ответа на дозу для флоризина строят для каждого анализа в качестве позитивного контроля.
Пример 81
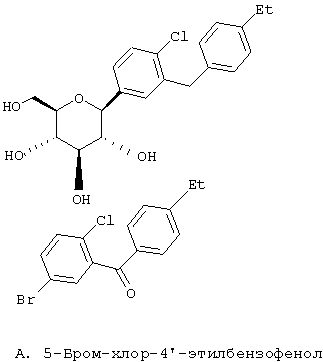
В 2-литровую круглодонную колбу, содержащую перемешиваемую магнитной мешалкой суспензию продажной 5-бром-2-хлорбензойной кислоты (410 г, 1.74 моля) в 700 мл СН2Cl2, прибавляют оксалилхлорид (235 г, 1.85 моля), а затем 1.5 л ДМФА. Для улавливания выделяющейся HCl колба снабжена отводной трубкой, чтобы газ проходил над поверхностью перемешиваемого раствора КОН. Когда бурное выделение газа прекращается через два часа, гомогенную реакционную смесь перемешивают в течение ночи прежде, чем удалить летучие в вакууме на роторном испарителе. Полученное масло при последующей эвакуации.
После растворения сырого 5-бром-2-хлорбензоилхлорида в 530 мл этилбензола желтый раствор охлаждают до -3°С и добавляют AlCl3 (257 г, 1.93 моля) порциями по 30 г в течение 60 мин, следя за тем, чтобы температура не поднималась выше 10°С. Большое количество газообразного HCl, которое начинает выделяться после прибавления 60% AlCl3, улавливают, пропуская газ через перемешиваемый конц. раствор NaOH. Если реакционная смесь становится более концентрированной, магнитная мешалка не промешивает по окончании прибавления AlCl3. После перемешивания в течение 1 ч, когда баня нагревается до -15(?)°С, баню отставляют. После 4 ч при 20°С густой сироп выливают в лед (1.5 кг). Затем, по охлаждении суспензии, прибавляют Н2О (1 л) и экстрагируют 4 × EtOAc. Объединенные органические вытяжки промывают 2 × 1 N HCl, 3× 1 М КОН и 2× рассолом и сушат Na2SO4. Летучие отгоняют сначала на роторном испарителе, а затем нагревают при ˜60°С при 1 Торр (1 мм Hg, 133.322 Па). 1 Н ЯМР спектр полученного темного масла показывает, что остаток представляет собой смесь орто/пара изомеров в соотношении 1:14. После растворения в гексане и последующего фильтрования через слой силикагеля раствор становится почти бесцветным.
Упариванием элюента получают 560 г (99%) 14:1 смеси 5- бром- 2- хлор-4'-этилбензофенона/5- бром- 2- хлор- 2'-этилбензофенона.
ВЭЖХ время удерживания: 4.7 мин, колонка YMC S5 С-18 4.6×50 мм, 2.5 мл/мин, детектирование при 220 нМ; 4 мин градиент 0-100% В, выдержка 2 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0.2% Н3PO4. Растворитель В: 90% МеОН/Н2O+ 0.2% Н3РО4.
5-Бром-2-хлор-4'-этилбензофенон
1Н ЯМР (400 МГц, CDCl3) δ 7.73 (1, 2Н, JAB=8.2 Гц), 7.54 (дд, 1H, J=2.2 Гц, J=8.8 Гц), 7.32 (д, 1H, J=8.8 Гц), 7.295 (д, 2Н, JAB=8.2 Гц), 2.72 (кв, 2Н, J=7.7 Гц), 1.27 (т, 3Н, J=7.7 Гц).
13С ЯМР (100 МГц, CDCl3) δ 193.13, 151.33, 140.49, 133.8, 133.52, 131.6, 131.44, 130.34, 130.16, 128.28, 120.44, 29.04, 15.02.
5-Бром-2-хлор-2'-этилбензофенон (характерные сигналы)
1Н ЯМР (400 МГц, CDCl3) δ 2.64 (кв, 2Н, J=7.7 Гц), 1.23 (т, 3Н, J=7.7 Гц).
13С ЯМР (100 МГц, CDCl3) δ 28.9, 15.5.
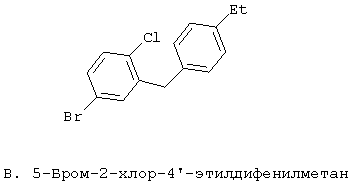
К раствору Et3SiH (400 г, 3.45 моля) и 5-бром-2-хлор-4'-этилбензофенона (534 г, 1.65 моля), содержащего ˜7% изомерного кетона, в 300 мл ТФК ( TFA), при перемешивании при 30°С прибавляют CF3SO3Н (1.5 г, 0.01 моля). За несколько минут температура повышается, раствор начинает бурно кипеть. Эту экзотермическую реакцию следует охлаждать в бане со льдом. Через 1 час, по показаниям ВЭЖХ, реакция проходит на 90%. После добавления дополнительного количества Е t3SiH (20 г) и нагревания в течение ночи при 70°С реакция (ВЭЖХ) проходит более чем на 95%. По охлаждении летучие удаляют перегонкой в вакууме из колбы в колбу. Получают 1 л светло-серого масла, выливают его в 1 л воды. Смесь экстрагируют 3× гексаном, объединенные органические вытяжки промывают 3× Н2О, 2× Na2СО3 и 2× рассолом, сушат Na2SO4. После упаривания на роторном испарителе остается ˜1 л прозрачного светло-желтого масла. Этот продукт дополнительно упаривают; (Et3Si)2О (450 мл) отгоняют перегонкой при 0.6 мм Hg. Когда температура головной фракции при перегонке достигает 75°С, колбу охлаждают. 1 Н ЯМР спектр остатка показывает, что он содержит ˜8:1 смесь диарилметана и (Et3Si)2O. Перекристаллизацию смеси осуществляют, выливая продукт в энергично перемешиваемый при 10°С 85% EtOH/H2O (1.2 л). После перемешивания в течение нескольких часов кристаллы отфильтровывают, промывают холодным 1:1 EtOH/H2O и сушат в вакууме. 5-Бром-2-хлор-4'-этилдифенилметан (500 г), получаемый в виде низкоплавкого твердого вещества, содержащего ˜1% (Et3Si)2O, используют без дополнительной очистки.
ВЭЖХ время удерживания: 5.3 мин, колонка YMC S5 С-18 4.6×50 мм, 2.5 мл/мин, детектирование при 220 нМ; 4 мин градиент 0-100% В, выдержка 2 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0.2% Н3РО4. Растворитель В: 90% МеОН/Н2О + 0.2% Н3РО4.
1Н ЯМР (125 МГц, CDCl3) δ 7.27-7.23 (м, 3Н), 7.14 (д, 2Н, JAB=7.7 Гц), 7.09 (д. 2Н, JAB=7.7 Гц), 2.63 (кв, 2Н, J=7,7 Гц), 1.23 (т, 3Н, J=7.7 Гц).
13C ЯМР (100 МГц, CDCl3) δ 142.46, 141.08, 135.68, 133.64, 133.13, 130.85, 130.55, 128.63, 128.1, 120.0, 38.62, 28.43, 15.51.
С. 2,3,4,6-тетра-О-Триметилсилил-D-глюколактон
К раствору глюколактона (239 г, 1.34 моля) и N-метилморфолина (1180 мл, 10.73 моля) в 2.4 л ТГФ в атмосфере Ar при перемешивании при -5°С прибавляют триметилсилилхлорид (1022 мл, 8.05 моля) из капельной воронки с такой скоростью, чтобы температура в колбе не превышала 5°С. Через 1 час реакционную смесь при перемешивании нагревают при 35°С и греют в течение 5 ч, затем оставляют на ночь охлаждаться до 20°С. Прибавляют 3.6 л толуола, смесь охлаждают до 0-5°С и осторожно прибавляют 7 л H2O с такой скоростью, чтобы температура не превышала 10°С. Внимание! При добавлении первой порции Н2O начинается бурная экзотермическая реакция. После перемешивания происходит разделение, а затем расслоение фаз. Органический слой промывают водн. NaH2PO4 (2 л), Н2О (1 л) и рассолом (1 л). Затем органический слой упаривают в вакууме на роторном испарителе, оставшееся светло-желтое масло дважды растворяют в 250 мл толуола и снова упаривают. Получают 616 г.
К раствору 5-бром-2-хлор-4'-этилдифенилметана (раздел В) (88 г, 0.26 моля) в 450 мл смеси 1:2 сухого ТГФ/толуола при перемешивании под аргоном медленно прибавляют 2.5 М н-BuLi (136 мл, 0.34 моля) в гексане с такой скоростью, чтобы температура в колбе была ниже -55°С. После 10-минутного перемешивания по окончании прибавления раствор переносят с помощью иглы в раствор (раздел С) 2,3,4,6-тетра-О-триметилсилил-D-глюколактона (153 г, 0.33 моля) в толуоле (350 мл) с такой скоростью, чтобы температура реакционной смеси была ниже -55°С. Раствор перемешивают в течение 30 мин при -78°С и добавляют 400 мл МеОН, содержащий метансульфоновую кислоту (28 мл, 0.45 моля). Реакционную смесь перемешивают 18 ч в течение ночи при 20°С. ВЭЖХ анализ показывает новый пик, который, по показаниям LC/MS, соответствует массе ожидаемого О-метилглюкозида. По окончании реакции к реакционной смеси прибавляют Na2СО3 (42 г, 0.5 моля) в 200 мл Н2О. Если значение рН не является слабоосновным, прибавляют дополнительное количество Na2СО3 и разбавляют в 2 раза водой и 3 раза экстрагируют EtOAc. Объединенные EtOAc вытяжки промывают рассолом и сушат Na2SO4. После упаривания на роторном испарителе масло (140 г, 90% чистота по ВЭЖХ анализу) дополнительно не очищают, а вместо этого используют далее неочищенную смесь диастереоизомеров.
1Н ЯМР (400 МГц, CDCl3) δ 7,37 (м, 1Н), 7.23 (м, 2Н), 7.02 (м, 4Н), 5.14 (м, 1H), 5.06 (м, 1Н), 4.07(м. 1Н), 4,03 (д, 1Н, JAB=15.4 Гц), 3,97 (д, 1Н, JAB=15.4 Гц), 3.80-3.70 (м, 4Н), 3.60 (м, 1Н), 3.48 (м, 1Н), 3.31 (м, 1Н), 2.84 (с, 3Н), 2.53 (кв, 2Н, J=7.6 Гц), 1.14 (т, 3Н, J=7.5 Гц).
13С ЯМР (100 МГц, CDCl3) δ 144.4, 140.7, 138.94, 136.9, 132.51, 131.6, 130.96, 130.6, 130.2, 129.16, 103.36, 77.0, 74,86, 72.48, 6(?)4.27, 51.57, 41.33, 30.75, 17,9.
ВЭЖХ время удерживания: 4.23 мин, колонка YMC S5 C-18 4.6×50 мм, 2.5 мл/мин, детектирование при 220 нМ; 4 мин градиент 0-100% В, выдержка 2 мин при 100% В. Растворитель А: 10% MeOH/H2O + 0.2% Н3PO4. Растворитель В: 90% МеОН/Н2O + 0.2% Н3РО4.
LC/MS: [М-ОМе]+ 391, 393; [M+Na]+ 445, 447.
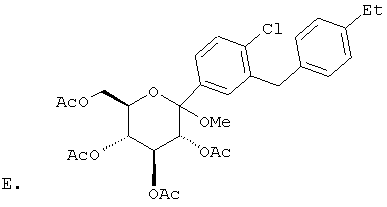
Раствор О-метилглюкозида (раздел D) (206 г, 0.49 моля) в ТГФ (1 л), содержащий диизопропилэтиламин (465 г, 3.6 моля) и DMAP (0.5, 4.1 ммоля), охлаждают до 0°С. Медленно прибавляют уксусную кислоту (326 г, 3.19 моля) с такой скоростью, чтобы температура не поднималась выше 5°С. Температуру раствора медленно доводят до 20°, перемешивают в течение 10 ч, после чего анализ показывает полное превращение в тетраацетат. К реакционной смеси прибавляют EtOAc (1.5 л) и 10%-ный водн. Н3PO4 (1-5 л). После расслаивания водную фазу экстрагируют 2× EtOAc. Объединенные органические вытяжки промывают 1× рассолом, сушат Na2SO4 и упаривают в вакууме. Полученное масло растворяют дважды в 300 мл толуола и снова упаривают, получают вязкое масло (300 г, 95% чистота по ВЭЖХ), которое используют без дополнительной очистки полученной неочищенной смеси диастереоизомеров.
1Н ЯМР (400 МГц, CDCl3) δ 7.38 (д, 1Н, J=8.3 Гц), 7.28 (дд, 1Н, J=8.3 Гц, J=2.2 Гц), 7.24 (д, 1H, J=2.2 Гц), 7.11 (д, 2Н, JAB=8.3 Гц), 7.04 (д, 2Н, JAB=8.3 Гц), 5.56 (т, 1Н, J=9.7 Гц), 5.21 (т, 1H, J=10.1 Гц), 4.93 (т, 1H, J=10.1 Гц), 4.20 (дд, 1H, J=12 Гц, J=2 Гц), 4.12 (д, 1Н, JAB=15.4Гц), 4.02 (м, 1H), 4.018 (д, 1H, JAB = 15.4 Гц), 3.10 (с, 3Н), 2.606 (кв, 2Н, J=7.7 Гц), 2.097 (с, 3Н), 2.05 (с, 3Н), 1.94 (с, 3Н), 1.72 sd (с, 3Н), 1.21 (т, 3Н, J=7.7 Гц).
13С ЯМР (100 МГц, CDCl3) δ 170.7, 170.05, 169.47, 168.9, 142.2, 138.74, 136.4, 135.1, 134.7, 129.8, 129.4, 128.6, 128.0, 126.0, 100.02, 73.83, 71.33, 68.87, 68.77, 62.11, 49.43, 38.75, 28.4, 22.64, 20.68, 20.58, 20.16, 15.5.
ВЭЖХ время удерживания: 4.81 мин, колонка YMC S5 С-18 4.6×50 мм, 2.5 мл/мин, детектирование при 220 нМ; 4 мин градиент 0-100% В, выдержка 2 мин при 100% В. Растворитель А: 10% МеОН/Н2O + 0.2% Н3PO4. Растворитель В: 90% МеОН/Н2О+ 0.2% Н3РО4.
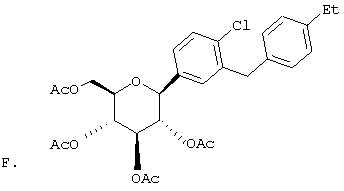
Раствор полученного выше сырого масла (301 г, 0.51 моля) в CH2Cl2 (500 мл), содержащий один эквивалент Н2О (9 г, 0.5 моля) и Et3SiH (188 г, 1.62 моля), при перемешивании охлаждают до 20°С и добавляют BF3·Et2O (145 г, 1.02 моля). Во время прибавления температуру поддерживают <0°С. Затем реакционную смесь перемешивают 2 ч при 10°С и 18 ч при 15-20°С и прибавляют СН2Cl2 (500 мл) и Н2О (500 мл). После разделения фаз вод. слой экстрагируют один раз СН2Cl2. Объединенные органические вытяжки промывают 1 х водн. NaHCO3 и рассолом и сушат Na2SO4. Отфильтровывают Na2SO4, прибавляют Ас2О (6.4 г, 65 ммолей), диизопропилэтиламин (9.5 г, 74 ммоля) и DMAP (100 мг, 0.8 ммоля). Раствор перемешивают при 20°С в течение 18 ч, чтобы быть уверенными, что все глюкозидные гидроксильные группы, которые гидролизовались в процессе восстановления и обработки, снова проацетилировались. Масло, полученное после упаривания в вакууме, кристаллизуется при добавлении EtOH. После фильтрования чистота этого продукта по показаниям ВЭЖХ составляет 98%; перекристаллизацией из EtOH получают тетраацетилированный бета-С-глюкозид в виде белого твердого вещества (180 г, чистота 99.8%). Общая конверсия на стадиях D- F составляет 61%.
ВЭЖХ время удерживания: 4.74 мин, 100% чистота, колонка YMC S5 С-18 4.6×50 мм, 2.5 мл/мин, детектирование при 220 нМ; 4 мин градиент 0- 100% В, выдержка 2 мин при 100% В. Растворитель А: 10%МеОН/Н2О + 0.2% Н3РО4. Растворитель В: 90% МеОН/Н2О + 0.2% Н3РО4.
1Н ЯМР (500 МГц, CDС13) δ 7.35 (д, 1Н, J=8.2 Гц), 7.19 (дд, 1Н, J=8.2 Гц, J=2.2 Гц), 7.11 (д, 2Н, JAB=8.5 Гц), 7.086 (д, 1Н, J=2.2 Гц), 7.06 (д, 2Н, JAB=8.5 Гц), 5.28 (т, 1Н, J=9.7 Гц), 5.20 (т, 1Н, J=9.7 Гц), 5.04 (т, 1Н, J=9.7 Гц), 4.31 (д, 1Н. J=9.9 Гц), 4,26 (дд, 1H, J=12 Гц, J=5 Гц), 4.135 (дд, 1Н, J=12 Гц, J=5 Гц), 4.095 (д, 1Н, JAB=7.7Гц), 3.995 (д, 1H, JAB=7.7Гц), 3.79 (м, 1H), 2.605 (кв, 2Н, J=7.7 Гц), 2.069 (с, 3Н), 2.04 (с, 3Н), 1.96 (с, 3Н), 1.67 (с, 3Н), 1.21 (т, 3Н, J=7.7 Гц).
13С ЯМР (100 МГц, CDCl3) δ 170.64, 170.3, 169.4, 168.7, 142.2, 138.78, 136.4, 135.1, 134.6, 129.9, 129.8, 128.7, 128.0, 125.9, 79.45, 76.1, 74.1, 72.5, 68.45, 62.2, 38.6, 28.4, 20.7, 20.6, 20.59, 20.2, 15.55. LC-MS [M+NH4 +] при m/z 578.3.
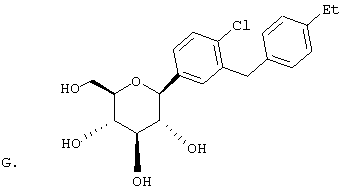
К белой суспензии, образующейся при перемешивании тетраацетилированного бета-С-глюкозида из раздела F (25 г, 44.6 ммоля) в течение 5 мин в смеси 2:3 ТГФ/МеОН (350 мл) под азотом при 20°С, прибавляют LiOH·H2O (2.0 г, 550 ммолей) в Н2О (70 мл). Через 15 мин реакционная смесь превратилась в мутный раствор; через 2.5 часа, по показаниям ВЭЖХ, реакция прошла на 98%. Конверсия повышается до 99% после стояния в течение ночи. Летучие отгоняют на роторном испарителе, так что объем уменьшается до 150 мл. К остатку прибавляют 10% водн. KHSO4 (100 мл), прибавляют еще 100 мл воды и экстрагируют 3 × EtOAc. Сушат Na2SO4, летучие отгоняют на роторном испарителе и полученное масло в минимальном количестве EtOAc превращается в пену в вакууме. Количество EtOAc, впитанное этим материалом, можно понизить сушкой в вакууме. Это стекловидное грязно-белое вещество соскребают и дополнительно сушат при 0.15 Торр (мм Hg) при 25°С в течение 24 ч, получают 17.3 г нужного С-арилглюкозида, содержащего 6.7 мол.% EtOAc.
ВЭЖХ время удерживания: 4.21 мин, 98.8% чистота, колонка YMC S5 С-18 4.6×50 мм, 2.5 мл/мин, детектирование при 220 нМ; 4 мин градиент 0-100% В, выдержка 2 мин при 100% В. Растворитель А: 10%МеОН/Н2O + 0.2% Н3PO4. Растворитель В: 90% МеОН/Н2O + 0.2% Н3PO4.
1Н ЯМР (500 МГц, CD3OD) δ 7.34 (д, 1Н, J=8.2 Гц), 7.33 (д, 1Н, J=1.7 Гц), 7.27 (дд, 1H, J=8.2 Гц, J=1,7 Гц), 7,08 (частично перекрываемый АВ квартет, 4Н), 4.1-4.0 (м, 3Н), 3.86 (д, 1Н, J=11.6 Гц), 3.68 (дд, 1H, J=5.3, 10.6 Гц), 3 46-3.26 (м, 4Н) Гц), 2.57 (кв, 2H, J=7 Гц), 1,19 (т, 3Н, J=7 Гц).
13C ЯМР (125 МГц, CD3OD) δ 143.2, 140.0, 139.7, 138.1, 134.5, 131.98, 130.1, 129.8, 128.8, 128.2, 82.8, 82,14, 79.7, 76.4, 71.9, 63.1, 39.7, 29.4, 16.25.
MS [M+Na+] m/z теоретически 415.1288; найдено 415,1293.
Анализ для C21H26ClO8 • 0.07 EtOAc • 0,19 Н2О. Вычислено С 63.51, Н 6.50, Cl 8.80. Найдено С 63.63, Н 6.63, Cl 8.82.
Пример 82
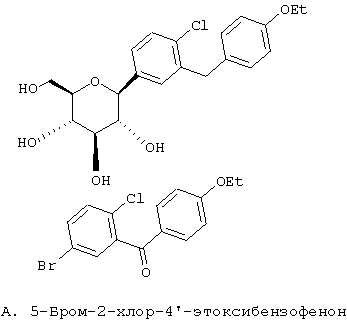
К суспензии продажной 5-бром-2-хлорбензойной кислоты (250 мг, 1.06 моля) в 450 мл CH2Cl2, содержащей оксалилхлорид (1.1. моля) при перемешивании прибавляют 1.5 мл ДМФА. По окончании интенсивного выделения газа реакционную смесь перемешивают в течение ночи и удаляют летучие на роторном испарителе. Растворяют сырой 5-бром-2- хлорбензоилхлорид в 200 мл CH2Cl2, желтый раствор переносят в 3-горлую колбу на 2 л, снабженную высокой мешалкой и внутренним термометром. Смесь при перемешивании охлаждают до -3°С и прибавляют фенетол (130 г, 1.08 моля). Через воронку для сыпучих прибавляют AlCl3 в течение 30 мин, так чтобы температура не превышала 4°С. Газообразный HCl, который начинает интенсивно выделяться после прибавления 60% AlCl3, улавливают, пропуская его через конц. раствор NaOH при перемешивании. Через 10 мин после окончания прибавления ВЭЖХ показывает, что реакция прошла на 95%. Смесь перемешивают 1 ч при 4°С, реакцию прекращают, выливая реакционную смесь на лед. Затем к суспензии прибавляют Н2O (1 л) и экстрагируют 3 × CH2Cl2. Объединенные органические вытяжки промывают 2 × 1 N HCl, 1 × H2O, 2 × 1 M NaOH и 2× рассолом, сушат Na2SO4. Удаляют летучие, по данным ВЭЖХ продукт представляет собой 1:7 смесь орто/пара изомеров. Перекристаллизацией 2× из 400 мл абсолютного EtOH получают 230 г (64%) 5-бром-2-хлор-4'-этоксибензофенона.
В. 5-Бром-2-хлор-4'-этоксидифенилметан
К раствору Et3SiH (400 г, 3.45 моля) и 5-бром-2-хлор-4'-этоксибензофенона (390 г, 1.15 моля) в 900 мл смеси 1:2 1,2-дихлорэтана/MeCN при 10°С прибавляют BF3 Et2O (150 мл, 1.58 моля) с такой скоростью, чтобы температура не превышала 20°С. Осторожно! Реакция экзотермическая. Перемешиваем в течение ночи при 20°С, по данньм ВЭЖХ реакция прошла на 90%. Прибавляем еще 40 мл Et3SiH и 15 мл BF3 Et2O, нагреваем реакционную смесь 3 ч при 50°С (замечание: повышенная температура увеличивает образование продукта реакции Риттера 5-бром-2-хлор-4'-этоксидифенилметиламина). По охлаждению к реакционной смеси прибавляют 120 г КОН в 300 мл Н2О. Перемешивают в течение 2 ч, происходит расслоение. Водный слой экстрагируют 2× СН2Cl2; объединенные органические вытяжки промывают 1× порциями 300 мл 2 М КОН, 2× Н2О, содержащей 10% рассола для лучшего разделения фаз, и 2× рассолом, сушат Na2SO4. Удаляют летучие, остаток перекристаллизовывают из абсолютного EtOH, получают 230 г 5-бром-хлор-4-этоксидифенилметан в виде твердого вещества белого цвета.
С. 2,3,4,6-тетра-О-Триметилсилил-β-D-глюколактон
К раствору глюколактона (239 г, 1.34 моля) и N-метилморфолина (1180 мл, 10.73 моля) в 2.4 л ТГФ в атмосфере Ar при перемешивании при -5°С прибавляют триметилсилилхлорид (1022 мл, 8.05 моля) из капельной воронки с такой скоростью, чтобы температура в колбе не превышала 5°С. Через 1 час реакционную смесь при перемешивании нагревают при 35°С и греют в течение 5 ч, затем оставляют на ночь охлаждаться до 20°С. Прибавляют 3.6 л толуола, смесь охлаждают до 0-5°С и осторожно прибавляют 7 л Н2О с такой скоростью, чтобы температура не превышала 10°С. Внимание! При добавлении первой порции Н2O начинается бурная экзотермическая реакция. После перемешивания происходит разделение, а затем расслоение фаз. Органический слой промывают водн. NaH2РО4 (2 л), Н2О (1 л) и рассолом (1 л). Затем органический слой упаривают в вакууме на роторном испарителе, оставшееся светло-желтое масло дважды растворяют в 250 мл толуола и снова упаривают. Получают 616 г титульного соединения.
К раствору 5-бром-2-хлор-4'-этоксидифенилметана (раздел В) (150 г, 0.46 моля) в 1.2 мл смеси 1:2 сухого ТГФ/толуола при перемешивании под аргоном при -78°С по каплям прибавляют 184 мл 2.5 М н- BuLi в гексане так, чтобы температура в колбе была ниже -70°С. После 30-минутного перемешивания по окончании прибавления раствор переносят с помощью иглы в раствор (раздел С) 2,3,4,6-тетра-О-триметилсилил-β-D-глюколактона (236 г, 0.51 моля) в 1.1 л толуола с такой скоростью, чтобы температура реакционной смеси была ниже -70°С. Раствор перемешивают в течение 30 мин при -78°С и добавляют 1 л МеОН, содержащий метансульфоновую кислоту (41.8 мл, 0.64 моля). Реакционную смесь перемешивают в течение ночи при 20°С. ВЭЖХ анализ показывает два новых пика, которые соответствуют по массе ожидаемому О-метилглюкозиду: соотношение, как правило, меняется от 95:5 до 80:20. Нужный продукт соответствует основному пику с более коротким временем удерживания. Замечание: более продолжительное время реакции или прибавление 50% избытка метансульфоновой кислоты приводит к полному превращению изомерного продукта в нужный О-метилглюкозид. По окончании реакции к реакционной смеси прибавляют NaHCO3 (37 г, 0.37 моля) в 200 мл Н2О. Если значение рН не является слабоосновным, прибавляют дополнительное количество NaHCO3 и разбавляют в 2 раза водой и 3 раза экстрагируют EtOAc. Объединенные EtOAc вытяжки промывают рассолом и сушат Na2SO4. После упаривания на роторном испарителе остаток растворяют в горячем толуоле (150 мл). Полученный раствор при перемешивании выливают в литр гексана. Осадок отфильтровывают в вакууме, промывают на фильтре 2× 500 мл гексана и сушат на воздухе, получают 171 г титульного соединения в виде твердого вещества белого цвета.
К раствору О-метилглюкозида (Раздел D) (123 г, 0.28 моля) в 1.2 л смеси 1:1 CH2Cl2/MeCN при перемешивании при -10°С прибавляют Et3SiH (65.27 г, 0.56 моля), а затем BF3·Et2О (59.75 г, 0.42 моля) с такой скоростью, чтобы температура была -5° ÷ -10°С. Раствор оставляют нагреваться до 0°С при перемешивании в течение 5 ч. Когда ВЭЖХ показывает окончание реакции, к реакционной смеси прибавляют насыщ. водн. NaHCO (310 мл). Органические летучие вещества отгоняют в вакууме на роторном испарителе. Остаток распределяют между 2 л EtOAc и 2 л H2О. После разделения слоев Н2О слой экстрагируют 2× порциями по 2 л EtOAc. Объединенные органические вытяжки промывают Н2О (2 л) и рассолом (2 л) и сушат MgSO4, а затем упаривают на роторном испарителе, получают 104.6 г желтой затвердевающей пены. Растворяют этот остаток в СН2Cl2 (750 мл), прибавляют пиридин (200 г, 2.53 моля), а затем Ас2О (261.1 г, 2.56 моля) одной порцией. Температура поднимается с 28° до 47°С, а затем начинает падать. Прибавляют DMAP (1.56 г, 13 ммоля). Когда через 1.5 час ВЭЖХ анализ показывает окончание реакции, добавляют Н2O (1.8 л). Смесь экстрагируют 2× СН2Cl2 (общий объем 2.7 л); объединенные органические вытяжки промывают 2 × 1 N HCl (1.8 л), 2× рассолом (1.8 л), сушат MgSO4. Упаривают на роторном испарителе, остаток перекристаллизовывают из абсолютного EtOH (750 мл), получают 89.5 г заданного β-С-глюкозида в виде твердого вещества белого цвета. Маточный раствор, содержащий соответствующий α-С-глюкозиду, а также более полярной изомерной фуранозной форме.
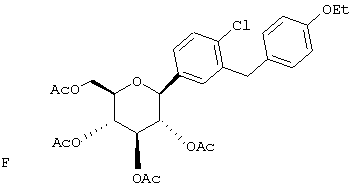
Или же О-метилглюкозид из Раздела D можно сначала проацетилировать, а затем восстановить и получить нужный тетраацетилированный С-арилглюкозид, используя нижеприведенную методику.
Раствор О-метилглюкозида (3.0 г, 6.8 ммоля) из Раздела D в толуоле(45 мл), содержащий диизопропилэтиламин (6.9 мл, 40 ммоля), охлаждают до 0°С и прибавляют уксусный ангидрид (3.35 мл, 35.5 ммоля) и DMAP (84 мг, 0.68 ммоля). Раствор оставляют постепенно нагреваться до 20°С, через шесть часов данные анализа показывают полное превращение в тетраацетат. Реакцию "гасят", добавляя 50 мл 20% Н3PO4. После расслаивания водную фазу экстрагируют толуолом 2×. Объединенные органические вытяжки промывают 1× 50 мл Н2О и упаривают в вакууме. Полученное масло растворяют в 20 мл толуола и снова упаривают, получают вязкое масло (4.15 г), которое используют без дополнительной очистки.
Раствор полученного выше сырого масла (4.15 г, 6.8 ммоля) в MeCN (60 мл), содержащий один эквивалент Н2О (123 мг, 6.8 ммоля), охлаждают до 0°С и добавляют Et3SiH (3.27 мл, 20.5 ммоля), а затем BF3·Et2O (1.73 мл, 13.7 ммоля). Перемешивают 1 ч, раствор оставляют нагреваться до 20°С. Через 4 ч, когда проводимый периодически анализ ВЭЖХ показывает, что реакция не проходит далее выше 60%, прибавляют дополнительно 2 мл Et3SiH и 1 мл BF3·Et2О. Через два часа по данным ВЭЖХ реакционная смесь не содержит исходных. Прибавляют aq. NaHCO3, перемешивают 30 мин, экстрагируют 3 × EtOAc. Объединенные органические вытяжки промывают 1× aq. NaHCO3 и рассолом и сушат Na2SO4. Масло, полученное после упаривания в вакууме, растворяют в 70 мл горячей смеси 25% EtOAc/гексан. По охлаждении 2.45 г нужного тетраацетилированного β-С-арилглюкозида кристаллизуют и отфильтровывают.
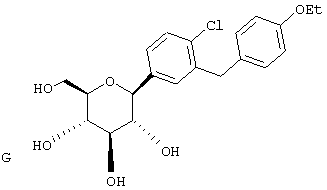
К раствору тетраацетилированного β-С-арилглюкозида (27.2 г, 49 ммоля) (полученного как описано в Разделе Е) при 20°С в 480 мл смеси 2.3:1 ТГФ/МеОН/Н2О прибавляют LiOH H2О (2.3 г, 57 ммоля). Перемешивают в течение ночи, летучие отгоняют на роторном испарителе. Остаток растворяют в EtOAc (300 мл), промывают 1× рассолом (150 мл), 1× рассолом (50 мл), содержащим 10 мл 5% aq. KHSO4 и, наконец, рассолом (50 мл), сушат Na2SO4. Летучие отгоняют на роторном испарителе и полученное масло в минимальном количестве CH2Cl2 в вакууме превращается в пену, получают 20.4 г ожидаемого титульного С-арилгликозида в виде стеклообразного бело-серого твердого вещества, содержащего 0.11 мол.% EtOAc.
ВЭЖХ время удерживания: 7.08 мин, 94% чистота, колонка YMC S5 С-18 4.6×50 мм, 2.5 мл/мин, детектирование при 220 нМ; 8 мин градиент 0-100% В, выдержка 5 мин при 100% В. Растворитель А: 10% МеОН/Н2О + 0.2% Н3PO4. Растворитель В: 90% МеОН/Н2О + 0.2% Н3PO4.
1H ЯМР (500 МГц, CD3OD) δ 7.33 (д. 1Н, J=6 Hz), 7.31 (д, 1Н, J=2.2 Гц), 7.31 (дд, 1Н, J=6 Гц, J=2.2 Гц), 7.07 (д, 2Н, J=8.8 Гц), 6.78 (д, 2Н, J=8.8 Гц), 4.07-3.90 (м, 7Н), 3.85 (д, 1Н, J=10.6 Гц), 3.69 (дд, 1Н, J=5.3, 10.6 Гц), 3.42-3.25 (м, 4Н), 1.34 (т, 3Н, J=7 Гц).
13C ЯМР (125 МГц, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2.
Анализ: Вычислено для С21H25ClO3 LC-MS [M+Na+] 431; найдено 431.
| название | год | авторы | номер документа |
|---|---|---|---|
| О-АРИЛГЛЮКОЗИДНЫЕ ИНГИБИТОРЫ SGLT2 И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2269540C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ И ИСПОЛЬЗОВАНИЯ ИНГИБИТОРА SGLT2 | 2013 |
|
RU2643764C2 |
| С-АРИЛ ГЛЮКОЗИДНЫЕ SGLT2 ИНГИБИТОРЫ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2746132C1 |
| С-АРИЛ ГЛЮКОЗИДНЫЕ SGLT2 ИНГИБИТОРЫ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2800510C1 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV НА ОСНОВЕ КОНДЕНСИРОВАННЫХ ЦИКЛОПРОПИЛПИРРОЛИДИНОВ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2286986C2 |
| ОКСА- И ТИАЗОЛПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТИДИАБЕТИЧЕСКИХ АГЕНТОВ И АГЕНТОВ ПРОТИВ ОЖИРЕНИЯ | 2000 |
|
RU2279427C2 |
| C-АРИЛ ГЛЮКОЗИДНЫЕ SGLT2 ИНГИБИТОРЫ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2003 |
|
RU2337916C2 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНКИНАЗ | 2000 |
|
RU2260592C9 |
| НЕНУКЛЕОЗИДНЫЕ ИНГИБИТОРЫ ОБРАТНОЙ ТРАНСКРИПТАЗЫ | 2007 |
|
RU2451676C2 |
| НОВЫЕ ИНГИБИТОРЫ S-НИТРОЗОГЛУТАТИОНРЕДУКТАЗЫ | 2011 |
|
RU2585763C2 |
Предлагаются соединения, ингибирующие SGLT2, имеющие формулу (I), где R1, R2 и R2a независимо обозначают водород, ОН, OR5, алкил, CF3, OCHF2, OCF3, CR5i или галоген, или два из R1, R2 и R2a вместе с прилегающими к ним атомами углерода могут образовывать аннелированный пятичленный гетероцикл; R3 и R4 независимо обозначают водород, ОН, OR5, ОАрил, ОСН2Арил, алкил, циклоалкил, CF3, -OCHF2, -OCF3, галоген, -CN, -СО2R5b, -СО2Н, -CONR6R6a, -NHCOR5c, -NHSO2R5d, -NHSO2Арил, арил, -SR5e, -SOR5f, -SO2R5g, -SO2Арил или пяти-, шести- или семичленный карбоцикл или гетероцикл; R5, R5a, R5b, R5c, R5d, R5e, R5f, R5g, R5h, R5i независимо обозначают алкил; R6, R6a, R6b, R6c и R6d независимо обозначают водород, алкил, арил, алкиларил или циклоалкил, или R6 и R6a вместе с прилегающими к ним атомами азота образуют аннелированный пяти-, шести- или семичленный гетероцикл; А обозначает О, S, NH или (СН2)n, где n обозначает 0-3. Также предлагается способ лечения диабета и родственных заболеваний с применением SGLT2-ингибирующего количества вышеописанного соединения, одного или в комбинации с другими терапевтическими агентами. 3 н. и 16 з.п. ф-лы, 2 табл.
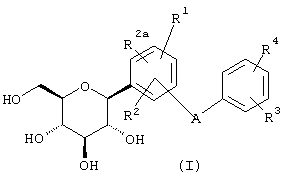
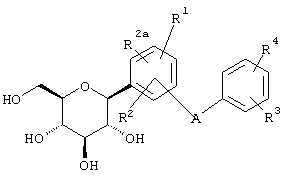
где R1, R2 и R2a независимо обозначают водород, ОН, OR5, алкил, CF3, OCHF2, OCF3, SR5i или галоген или два из R1, R2 и R2a вместе с прилегающими атомами углерода образуют аннелированный пятичленный гетероцикл, который может содержать 2 атома кислорода в цикле;
R3 и R4 независимо обозначают водород, ОН, OR5a, ОАрил, ОСН2Арил, алкил, циклоалкил, CF3, -OCHF2, -OCF3, галоген, -CN, -CO2R5b, -CO2H, -COR6b, -CH(OH)R6c, -CH(OR5h)R6d, -CONR6R6a, -NHCOR5c, -NHSO2R5d, -NHSO2Арил, арил, -SR5e, -SOR5f, -SO2R5g, -SO2Арил или пяти-, шести- или семичленный гетероцикл, который может содержать 1-4 гетероатома в цикле, представляющие собой N, О, S, SO и/или SO2, или R3 и R4 вместе с прилегающими к ним атомами углерода образуют аннелированный пятичленный гетероцикл, который может содержать 2 атома кислорода в цикле;
R5, R5a, R5b, R5c, R5d, R5e, R5f, R5g, R5h, R5i независимо обозначают алкил;
R6, R6a, R6b, R6c и R6d независимо обозначают водород, алкил, арил, алкиларил или циклоалкил или R6 и R6a вместе с прилегающими к ним атомами азота образуют аннелированный пяти-, шести- или семичленный гетероцикл, который может содержать 1-4 гетероатома в цикле, которыми являются N, О, S, SO и/или SO2;
А обозначает О, S, NH или (CH2)n, где n обозначает 0 - 3, или его фармацевтически приемлемые соли;
при условии, что если А обозначает (СН2)n, где n обозначает 0, 1, 2 или 3, или А обозначает О и, по меньшей мере, один из R1, R2 и R2a обозначает ОН или OR5, тогда, по меньшей мере, один из R1, R2 и R2a обозначает CF3, OCF3 или OCHF2 и/или, по меньшей мере, один из R3 и R4 обозначает CF3, -OCHF2, -OCF3, -CN, -CO2R5b, CH(OR5h)R6d, CH(OH)R6c, COR6b, -NHCOR5c, -NHSO2R5d, -NHSO2Арил, арил, -SR5e, -SOR5f, -SO2R5g, -SO2Арил.
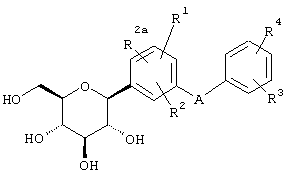
где радикалы имеют значения, представленные в п.1.
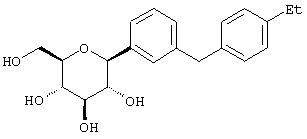
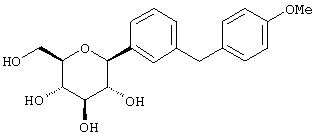
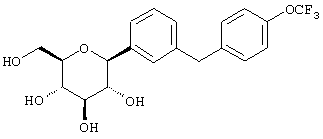
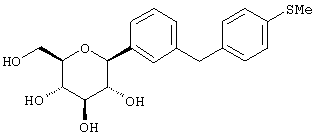
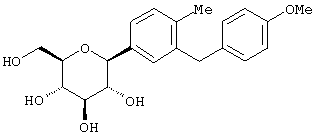
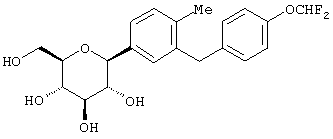
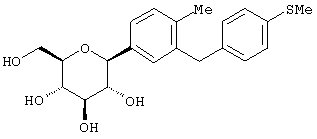
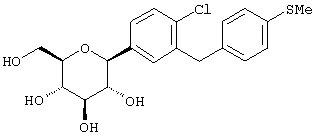
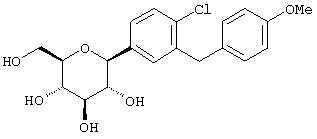
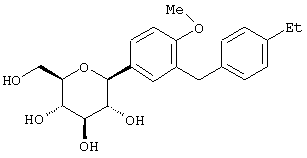
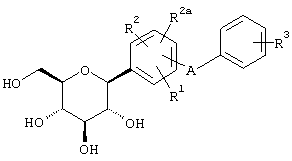
где А обозначает СН2 и находится в положении мета относительно глюкозида, R1, R2 и R2a каждый обозначает Н и R3 обозначает нижеследующее:
4-Ме, 4-ОН, 3-Ме, Н, 3-ОМе, 4-CO2Ме, 3,4-(ОСН2O), 4-CF3, 4-NHAc, 4-SO2Me, 4-Ph, 4-NHSO2Ph-4'-Me, 4-NHSO2Me, 4-CO2H, 4-тиадиазол, 4-тегразол, 4-OCH2Ph-4'-CN, 4-OCHF2, 4-изопропил, 2-изопропил, 4-O-н-пропил, 4-тетразол-2'-Ме, 4-тетразол-1'-Ме, 4-OPh, 4-н-пропил, 4-н-бутил, 4-SO2Et, 4-SO2-н-пропил, 4-SO2Ph или 4-SOMe.
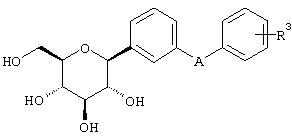
где
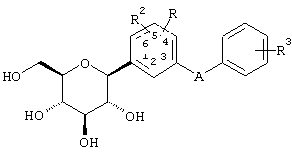
где
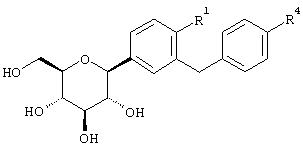
где радикалы имеют значения, представленные в п.1.

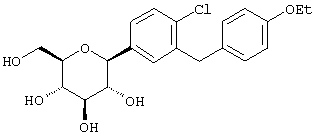
где радикалы имеют значения, представленные в п.1.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| ЕР 0598359, А1, 25.05.1994 | |||
| Способ получения @ - @ -фенилтиоксилозидов | 1989 |
|
SU1780538A3 |

