Область техники, к которой относится изобретение
Изобретение вообще относится к области перорального введения лекарственных средств, таких как человеческий инсулин, которые являются неустойчивыми в желудочно-кишечном тракте (GI) или плохо всасываются в нем.
Многие лекарственные средства являются неэффективными, когда вводятся перорально, из-за неустойчивости лекарственного средства в GI и слабой способности проникать через поверхности GI. Переваривающие белки ферменты, такие как пепсин, и сильная кислота могут дополнительно вносить вклад в неустойчивость пептидных и белковых лекарственных средств в желудке, что может также подавлять устойчивость и эффективность таких лекарственных средств. Кроме того, лекарственные средства, имеющие низкую липофильность и/или высокую молекулярную массу, нелегко всасываются через эпителиальные слои в GI тракте.
Сообщается о нескольких стратегиях для улучшения устойчивости и биодоступности вводимых перорально активных средств. Конкретно, предполагается, что в захваченных лекарственных средствах в носителях, таких как липосомы, мицеллы, наночастицы, эмульсии вода-в-масле (w/o) или вода-в-масле-в-воде (w/o/w) или микроэмульсии, или капсулах с энтеросолюбильным покрытием, активные соединения защищены от воздействия нежелательного химического окружения (например, низкого рН или пищеварительных ферментов). Такие подходы могут иметь такие недостатки, как низкая устойчивость лекарственного средства, низкая нагрузка лекарственного средства, неэффективность, сложные требования к переработке и высокая стоимость.
В патенте Соединенных Штатов 6191105 раскрывается получение w/o эмульсионных препаратов инсулина. Эмульсия w/o может быть неустойчивой из-за фазового перехода, который происходит после пероральной доставки, с последующим воздействием непосредственно на лекарственное средство раздражающей окружающей среды GI.
В патенте Соединенных Штатов 6277413 раскрываются эмульсии w/o/w, в которых водорастворимые лекарственные средства включены во внутреннюю водную фазу. Такие эмульсии показывают низкую нагрузку лекарственных средств.
В патенте Соединенных Штатов 5552156 раскрывается применение липосом и мицелл в качестве носителей лекарственных средств. Получение таких препаратов является сложным и дорогостоящим.
В патенте Австралии 2004305395 раскрываются композиции и получение наночастиц водорастворимых лекарственных средств для перорального введения. Способ получения композиций включает сушку наночастиц вымораживанием, которая может повысить стоимость получения.
В заявке на патент Соединенных Штатов, регистрационный № 13/561195, раскрываются содержащие катионные наночастицы капсулы с энтеросолюбильным покрытием для предотвращения кислотного разложения активных веществ, таких как инсулин. Раскрытый способ получения капсул является сложным, включающим сушку вымораживанием и получение капсул с энтеросолюбильным покрытием.
В заявке на патент Соединенных Штатов, регистрационный № 13/521377, раскрываются композиции для перорального введения инсулиновых пептидов с использованием самомикроэмульгирующихся систем доставки (SMEDDS) в капсуле с энтеросолюбильным покрытием. Инсулиновый пептид в композиции SMEDDS является еще неустойчивым (разлагающимся или инактивируемым) в кислой среде желудка. Для того, чтобы охватить устойчивость, инсулиновый пептид в SMEDDS помещают в носитель с энтеросолюбильным покрытием для защиты активных соединений от расщепления или другого разрушения в желудке. Однако носитель с энтеросолюбильным покрытием показывает нежелательное замедление начала действия при пероральном введении. Кроме того, время опорожнения желудка различается у людей, и это будет влиять на время высвобождения инсулина из препарата и соответствующее всасывание в кишечнике. Такие вариации вызывают широкие вариации во всасывании инсулина, потенциально ведущие к неконтролируемым уровням сахара в крови.
SMEDDS в жидких лекарственных формах имеет ограничение, такое как несовместимость эксципиента и капсулы (см., например, Mu et al., 2013, Int. J. Pharm., 453(1): 215-224, и Kallakunta et al., 2012, Powd. Technol., 221: 375-382).
В публикации заявки на патент Соединенных Штатов № 2011/0293714 раскрываются композиции, которые включают полярный органический растворитель и липофильный компонент, и которые применимы для перорального введения дериватизированных инсулиновых пептидов. Необходимо использовать высокую пероральную дозу (840 МЕ/кг) таких композиций для снижения глюкозы в крови.
В публикации заявки на патент Соединенных Штатов № 2009/0176691 раскрываются однофазные композиции, которые включают буферирующее вещество и белковое активное средство в свободной форме. Такие однофазные композиции предназначены для введения перорально, причем после перорального введения буферирующее вещество вызывает рН в желудке и/или кишечнике, забуференный в интервале рН 4-8.
По меньшей мере некоторые технологии, описанные другими авторами, дают композиции, из которых осуществляется всасывание лекарственных средств (например, инсулина) после перорального введения (например, Wong, 2010, J. Drug Target., 18(2): 79-92; Arbit et al., 2009, Diabetes Sci. Technol., 3(3): 562-567). Однако заявители полагают, что не созданы пероральные композиции, которые показывают быстрое начало, высокую биодоступность и, необязательно, краткое время активности, которые были бы особенно полезны для лекарственных средств, таких как инсулины. Сообщается, что обычные инсулиновые препараты имеют длительное время начала (до 1,5 часов) и длительное время действия (до 5 часов). Для врачей и пациентов было бы благоприятным иметь возможность быстрой активации (начало в пределах 15 минут) и кратковременное действие (менее 5 часов) лекарственных средств, таких как инсулин, для того, чтобы обеспечить эффективный метаболический контроль с использованием обычной вводимой перорально лекарственной формы.
В настоящем изобретении описываются композиции, которые преодолевают по меньшей мере некоторые недостатки известных композиций, и описываются быстродействующие с небольшой длительностью действия лекарственные композиции даже для лекарственных средств, которые неустойчивы или имеют плохую биодоступность, когда вводятся перорально с использованием обычных препаратов.
Сущность изобретения
Изобретение относится к лекарственной форме для перорального введения гидрофильного лекарственного средства в кровоток млекопитающего. Лекарственная форма включает болюс антацида, достаточный для повышения рН в желудке млекопитающего до по меньшей мере примерно 3 (предпочтительно, по меньшей мере до примерно 3-4) после проглатывания лекарственной формы (например, болюс может быть способен нейтрализовать 1-7 миллиэквивалентов желудочных кислот). Лекарственная форма также включает по существу гомогенную комбинацию терапевтически эффективного количества лекарственного средства и поверхностно-активной системы. Поверхностно-активная система включает неионогенное поверхностно-активное вещество.
Идентичность и количество поверхностно-активной системы выбирают как достаточные для индукции спонтанного эмульгирования после контакта между комбинацией и водной средой в условиях слабого механического перемешивания, таких как условия, которые встречаются в желудке млекопитающего или в контейнере (например, небольшой чашке) до введения, в которых комбинация вращается с небольшим количеством водной среды. Как пример, идентичность и количество поверхностно-активной системы можно выбрать как достаточные для индукции спонтанного эмульгирования после контакта между комбинацией и девятикратным избытком дистиллированной воды в условиях механического перемешивания, характерных для желудка млекопитающего. (Точный выбранный объективный стандарт не является критичным; можно выбрать поверхностно-активную систему, чтобы она была достаточной для индукции спонтанного эмульгирования после контакта с четырех- или двукратным избытком дистиллированной воды или имитированной желудочной жидкости по USP, например.) Предпочтительно идентичность и количество поверхностно-активной системы выбирают такими, чтобы средний размер капель эмульсии, образовавшейся после контакта между комбинацией и водной средой, не превышал примерно 2000 нанометров (или меньше, например, не больше примерно 800, 500 или 300 нанометров).
В лекарственной форме болюс может быть включен в по существу гомогенную комбинацию. С другой стороны, болюс и комбинация могут присутствовать в различных частях лекарственной формы, например, в форме разных твердых веществ, порошков или жидкостей.
Лекарственная форма применима для введения ряда гидрофильных лекарственных средств, включая лекарственные средства, которые обычно являются недостаточно биологически доступными, когда вводятся перорально. Примеры таких лекарственных средств включают инсулиновые пептиды (например, антротерапевтические инсулины, такие как изолированные или синтезированные человеческие инсулиновые пептиды), гормоны роста, гентамицин, гемцитабин, пенициллины и ванкомицин.
Лекарственная форма может быть поставлена в форме набора, который включает лекарственную форму и количество водной среды, достаточное для растворения или суспендирования болюса антацида и для эмульгирования комбинации. С другой стороны, она может быть поставлена в форме набора, который включает первую лекарственную форму, которая включает болюс антацида, и вторую лекарственную форму, включающую по существу гомогенную комбинацию лекарственного средства и поверхностно-активной системы.
Изобретение также относится к способу перорального введения гидрофильного лекарственного средства в кровоток млекопитающего. Способ выполняют путем комбинирования терапевтически эффективного количества лекарственного средства и поверхностно-активной системы, описанных в настоящем описании, смешивания комбинации, водной среды и болюса антацида, достаточного для повышения рН в желудке млекопитающего до по меньшей мере примерно 3, для получения эмульгированной смеси; и после этого перорального введения эмульгированной смеси млекопитающему.
В альтернативном способе лекарственное средство и поверхностно-активную систему соединяют и получают комбинацию, болюс антацида вводят перорально млекопитающему, и комбинацию вводят перорально млекопитающему достаточно близко по времени к введению болюса, который поддерживает рН в желудке по меньшей мере примерно 3, пока вводят комбинацию.
Краткое описание фигур
Фигура 1 представляет собой график зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для стрептозотоцин(STZ)-индуцированных диабетических мышей, которым перорально вводили 200 МЕ/кг инсулина в 3,78% (мас./об.) водном растворе бикарбоната натрия (NaHCO3) (заштрихованные кружочки), в забуференном фосфатом физиологическом растворе (PBS) (незаштрихованные кружочки) или в композиции 1, суспендированной в растворе NaHCO3 (конечная концентрация 3,78%; треугольники). Приведенные результаты являются средними со стандартными отклонениями для групп по 8 мышей в каждой.
Фигура 2 представляет собой график зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для STZ-индуцированных диабетических мышей, которым перорально вводили 200 МЕ/кг инсулина в композиции 1, суспендированной в растворах NaHCO3 различных конечных концентраций. Конечные концентрации NaHCO3: 0,90% (заштрихованные кружочки), 1,80% (незаштрихованные кружочки), 2,70% (заштрихованные треугольники) и 3,78% (незаштрихованные треугольники). Приведенные результаты являются средними со стандартными отклонениями для групп по 8 мышей в каждой.
Фигура 3 представляет собой график зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для собак, которым перорально вводили 150 МЕ/кг инсулина в композиции 1, суспендированной в растворе NaHCO3 (конечная концентрация 3,78%; незаштрихованные кружочки), или для необработанных контрольных собак (заштрихованные кружочки), Приведенные результаты являются средними со стандартными отклонениями для групп из двух биглей каждая.
Фигура 4 представляет собой график зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для STZ-индуцированных диабетических мышей, которым перорально вводили 200 МЕ/кг инсулина в композиции 2, суспендированной в растворе NaHCO3 (конечная концентрация 3,78%). Приведенные результаты являются средними со стандартными отклонениями для групп по 8 мышей в каждой.
Фигура 5 представляет собой график зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для STZ-индуцированных диабетических мышей, которым перорально вводили 200 МЕ/кг инсулина в композиции 3, суспендированной в растворе NaHCO3 (конечная концентрация 3,78%). Приведенные результаты являются средними со стандартными отклонениями для групп по 8 мышей в каждой.
Фигура 6, состоящая из фигур 6А и 6В, представляет собой пару графиков зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови (фиг.6А), и концентрации инсулина в плазме со временем (фиг. 6В) для здоровых крыс Wistar, которым перорально вводили 200 МЕ/кг инсулина в композиции 4, суспендированной в 3,00% растворе NaHCO3. Приведенные результаты являются средними со стандартными отклонениями для групп по 3 крысы в каждой. Для сравнения также приводятся результаты для крыс, которым вводили свободный инсулин, суспендированный в 3% растворе NaHCO3.
Фигуры 7 и 8 представляют собой графики зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для STZ-индуцированных диабетических мышей, которым перорально вводили 50 МЕ/кг инсулина в композиции 5, которая включает 8% NaHCO3 (фиг. 7), или 50 МЕ/кг инсулина в композиции 6, которая включает 2,1% NaHCO3 и 0,9% Mg(OH)2 (фиг. 8),
Фигура 9, состоящая из фигур 9А и 9В, представляет собой пару графиков зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови (фиг.9А), и концентрации инсулина в плазме со временем (фиг. 9В) для STZ-индуцированных диабетических крыс Wistar, которым перорально вводили 200 МЕ/кг инсулина в композиции 4. Приведенные результаты являются средними со стандартными отклонениями для групп по 3 крысы в каждой.
Фигура 10 представляет собой график зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для STZ-индуцированных диабетических крыс Wistar, которым перорально вводили 200 МЕ/кг инсулина в суспензии после диспергирования композиции 7 в воде.
Фигура 11 представляет собой график зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для STZ-индуцированных диабетических мышей, которым перорально вводили 200 МЕ/кг инсулина в суспензии после диспергирования композиции 8 в воде.
Фигура 12 представляет собой график концентрации инсулина в плазме со временем для STZ-индуцированных диабетических крыс Wistar, которым перорально вводили 116 МЕ/кг в композиции 9. Приведенные результаты являются средними со стандартными отклонениями для групп по 5 крыс в каждой.
Фигура 13 представляет собой график зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для стрептозотоцин(STZ)-индуцированных диабетических мышей, которым перорально вводили 200 МЕ/кг инсулина в 3,78% (мас./об.) водном растворе бикарбоната натрия (NaHCO3), который был свежеприготовленным (заштрихованные квадраты), или который до введения хранился при 5 градусах по Цельсию в течение трех месяцев (заштрихованные кружочки).
Фигура 14 представляет собой график зависимости уровней глюкозы в крови от времени, вычисленных в виде процентов от начальных значений уровней глюкозы в крови, для стрептозотоцин(STZ)-индуцированных диабетических мышей, которым перорально вводили 200 МЕ/кг или свободного инсулина в растворе (заштрихованные квадраты) или быстродействующей пероральной композиции инсулина, описанной в примере в настоящем описании (заштрихованные кружочки).
Фигура 15 представляет собой график зависимости концентрации инсулина в плазме от времени для собак биглей, которым перорально вводили 25 МЕ/кг инсулина в композиции 8, и для других, которым подкожной (SC) инъекцией вводили 0,5 МЕ/кг свободного инсулина. Приведенные результаты являются средними со стандартными отклонениями для групп из трех биглей в каждой.
Подробное описание
Настоящее изобретение относится к пероральным композициям и лекарственным формам лекарственных средств (особенно гидрофильных лекарственных средств), которые являются неустойчивыми в кислых растворах, которые чувствительны к перевариванию пепсином или другими пищеварительными протеазами/пептидазами, которые показывают слабую гастроинтестинальную проницаемость, которые показывают нежелательно замедленное начало действия, когда вводятся перорально, которые показывают нежелательную длительность действия, когда вводятся перорально, или любую комбинацию таких свойств (все вместе «гастрально непрактичные лекарственные средства»). Пероральные композиции включают как i) само(микро)эмульгирующуюся систему доставки лекарственного средства (SMEDDS), которая включает лекарственное средство, так и ii) болюс антацида, которого достаточно для повышения рН в желудке до по меньшей мере примерно 3, и предпочтительно до 3-4 или выше после перорального введения композиции субъекту, такому как человек или другое млекопитающее.
Технология SMEDDS известна и принята (см., например, Khan et al. 2012, J. Pharmacy Alt. Med., 1: 13-19; публикация заявки на патент США № 2003/0022944; публикация заявки на патент США № 2010/0273730). SMEDDS представляют собой изотропные смеси одного или нескольких относительно гидрофобных растворителей, одного или нескольких поверхностно-активных веществ и лекарственного средства, которые показывают способность образовывать тонкие микроэмульсии (например, мицеллы или липосомы) при слабом перемешивании после разбавления в (например, контакте с) водной фазой. Усиление биодоступности гидрофобных и гидрофильных лекарственных средств путем включения таких лекарственных средств в SMEDDS описано ранее. Однако просто включение в SMEDDS лекарственного средства, которое показывает нежелательную биодоступность или фармакокинетику, когда вводится перорально, обычно не придает надежно биодоступность или фармакокинетику, адекватные фармацевтическим целям. Это особенно так в случае лекарственных средств, которые чувствительны (т.е. разрушаются, расщепляются или инактивируются) к кислой среде желудка или к действию одной или нескольких протеаз или пептидаз (например, пепсина), которое обычно происходит в желудке.
В нестоящем описании раскрывается композиция, которая включает как содержащую лекарственное средство композицию SMEDDS, так и болюс антацида. Эти два компонента можно перорально совместно вводить пациенту млекопитающему (или вводить перорально пациенту достаточно близко по времени, когда антацидное действие болюса перекрывает период, когда содержащая лекарственное средство композиция SMEDDS остается в желудке) как для более эффективной доставки лекарственного средства через желудочно-кишечный барьер, так и для уменьшения или устранения разложения лекарственного средства желудочной кислотой и/или ферментами.
Предмет изобретения, раскрытый в настоящем описании, включает лекарственные формы для перорального введения млекопитающему (например, человеку) лекарственного средства (предпочтительно гидрофильного лекарственного средства), которое плохо доступно для желудка, когда вводится млекопитающему перорально в простой форме с немедленным высвобождением (например, таблетке, капсуле, наборе гранул или растворе). Лекарственная форма включает болюс антацида, достаточный для повышения рН в желудке животного до по меньшей мере примерно 3 после проглатывания лекарственной формы. Лекарственная форма также включает (или в виде дополнительной части единой лекарственной формы или в виде составной части многочастной лекарственной формы) комбинацию (предпочтительно, по существу гомогенную комбинацию) i) терапевтически эффективного количества лекарственного средства, ii) необязательно, полиольного растворителя и iii) поверхностно-активной системы, которая включает неионогенное поверхностно-активное вещество. Композиция является «по существу гомогенной», когда она тщательно перемешана или соединена, так что, как представляется фармацевту, он имеет визуально однородную композицию (т.е. даже если композиция включает визуально различимые компоненты, эти компоненты оказываются равномерно распределенными в композиции). Идентичность и количества лекарственного средства, полиольного растворителя и поверхностно-активной системы выбирают так, что когда комбинацию вводят в контакт с водной фазой в условиях слабого механического перемешивания, комбинация эмульгируется спонтанно. Так, например, когда комбинацию перемешивают в чашке с водой (или другой водной жидкостью, такой как напиток) перед пероральным введением, или когда комбинация контактирует с водным содержимым желудка, комбинация эмульгируется, образуя содержащую лекарство эмульсию в GI тракте млекопитающего. Поскольку болюс антацида снижает кислотность в желудке и, следовательно, снижает активность желудочных протеолитических ферментов, устойчивость лекарственного средства в эмульсии усиливается, и всасывание лекарственного средства млекопитающим (например, в кровоток млекопитающего) также усиливается.
Композиции, описанные в настоящем описании, могут предотвращать разложение лекарственного средства в присутствии сильной кислоты и пищеварительных фрагментов, естественно находящихся в желудочной среде млекопитающих. Композиции также могут улучшить всасывание лекарственного средства в GI тракте. Такие композиции могут увеличить скорость поглощения и могут, необязательно, ограничить по времени действие лекарственного средства (например, путем включения ограниченного количества антацида). В контексте некоторых лекарственных средств желательны быстрое начало действия (для инсулина, например, в пределах 15-30 минут после введения) и относительно короткая длительность действия (для инсулина, например, на уровне менее 25% максимальной активности менее, чем через 5 часов, и предпочтительно менее, чем через примерно 4 часа после введения). Таким образом, например, вводимые перорально инсулиносодержащие композиции, описанные в настоящем описании, могут имитировать характеристики относительной незамедлительности и кратковременности действия инсулина, инъецируемого подкожно. В других воплощениях лекарственное средство может быть включено в композицию, из которой лекарственное средство будет высвобождаться в течение длительного периода времени (например, 0-24 часа, например, за счет выбора компонентов композиции, из которой лекарственное средство переносится в водные желудочно-кишечные жидкости только постепенно).
Ниже подробнее раскрываются компоненты композиций и способы, описанные в настоящем описании.
Композиции, описанные в настоящем описании для перорального введения лекарственных средств, которые обычно плохо абсорбируются из GI тракта, имеют два основных компонента, которые можно комбинировать в единую лекарственную форму, упакованную в виде набора, включающего два или более компонентов, или предоставить отдельно врачу или пациенту для комбинированного применения. Двумя основными компонентами композиций являются «композиция SMEDDS» (т.е. комбинация концентрата микроэмульсии и лекарственного средства, которая может быть, необязательно, гомогенной) и болюс антацида. Болюс антацида вводят больному млекопитающему, такому как человек, для повышения рН в желудке (и, необязательно, в других частях GI тракта). Композиция SMEDDS включает лекарственное средство и спонтанно эмульгируется после контакта с водной средой с образованием капель (например, мицелл), которые включают или контактируют с лекарственным средством и которые облегчают доставку сквозь слои клеток (например, эпителии желудка или тонкой кишки) вдоль GI тракта. Лекарственное средство, доставленное через такие клеточные слои, может поступить в общее кровообращение и распространиться по организму.
Компоненты композиция SMEDDS и антацидный болюс можно вводить субъекту в лекарственной форме, в которой два компонента объединены. Как пример, их можно вводить в лекарственной форме, которая включает жидкость, в которой оба компонента суспендированы или растворены, в лекарственной форме, которая включает отдельно порошкообразные формы двух компонентов, которые смешаны, или в лекарственной форме, которая включает адсорбент (например, нерастворимый минеральный порошок, такой как частицы диоксида кремния), в котором адсорбированы один или оба компонента. С другой стороны, два компонента можно вводить в лекарственной форме, в которой два компонента находятся в разных местах (например, в двухслойной таблетке или многокамерной капсуле, в которой два компонента находятся в отдельных камерах). Как другая альтернатива, два компонента можно вводить в отдельных лекарственных формах до тех пор, пока композицию SMEDDS вводят в период времени, в который антацидный болюс вызывает рН в желудке выше примерно 3. Композиции в форме жидкостей, которые получают перед пероральным введением, или в форме порошков, можно преимущественно вводить пациентам, имеющим затруднения при глотании таблеток или капсул.
Композиция SMEDDS
Важная часть композиций и способов, описанных в настоящем описании, связана с содержащей лекарственное средство композицией, которая спонтанно эмульгируется после контакта с водой или водной средой с образованием капель, которые включают лекарственное средство. Так как настоящее изобретение относится в первую очередь к улучшению доставки лекарственных средств, практически нецелесообразных для доставки через желудок (например, относительно гидрофильных лекарственных средств), таких как полипептиды (например, антротерапевтические инсулины), капли, которые образуются, предпочтительно представляют собой мицеллы, которые включают лекарственное средство или фракцию лекарственного средства. Такие мицеллы суспендируют в водной среде, и суспензию вводят субъекту. С другой стороны, мицеллы могут образовываться с GI тракте за счет введения композиции SMEDDS, необязательно (т.е. в случае, если желудок содержит относительно мало жидкости) вместе с достаточным количеством водной жидкости (например, антацидный болюс растворяют в воде) для облегчения эмульгирования в желудке. В GI субъекта мицеллы облегчают продвижение лекарственного средства через клеточные слои GI, предпочтительно в кровоток субъекта, откуда они могут достигнуть нужного места действия (например, в кровь или оттуда в место в организме, удаленное от GI тракта).
Композиция SMEDDS представляет собой комбинацию терапевтически эффективного количества вводимого лекарственного средства и поверхностно-активной системы. Лекарственное средство может быть растворено или суспендировано в водном растворе, полиольном растворителе или том и другом перед объединением его с поверхностно-активной системой. Лекарственное средство также можно, например, комбинировать с поверхностно-активной системой в порошкообразной (например, безводного или гидратированного порошка) форме. Поверхностно-активная система включает неионогенное поверхностно-активное вещество. Идентичность и количество поверхностно-активной системы выбирают так, что комбинация SMEDDS спонтанно эмульгируется после контакта с водной средой в условиях слабого механического перемешивания, когда композицию SMEDDS осторожно перемешивают в емкости (например, стакане для напитков или формочке для стандартной дозы) после ее объединения со средой. В одном воплощении композицию SMEDDS поддерживают дискретной (например, в форме порошкообразной или гранулированной композиции, содержащейся в капсуле, которую проглатывают целиком, и которая затем растворяется в GI тракте) непосредственно до или после ее проглатывания субъектом, и композиция SMEDDS эмульгируется в желудочно-кишечном (GI) тракте субъекта, когда она контактирует с водными жидкостями в нем (например, в форме порошкообразной или гранулированной композиции, содержащейся в капсуле, которую проглатывают целиком, и которая затем растворяется в желудке или тонкой кишке). Время комбинирования лекарственного средства и поверхностно-активной системы не является критичным. Когда вызывает беспокойство устойчивость, лекарственное средство можно смешать (например, равномерно) с поверхностно-активной системой; с другой стороны, лекарственное средство и поверхностно-активную систему можно объединить (например, перемешиванием порошкообразного лекарственного средства с жидкой поверхностно-активной системой) непосредственно перед или одновременно с объединением того и другого с антацидным болюсом.
Композиция SMEDDS также может включать полиольный растворитель, такой как глицерин, пропиленгликоль или полиэтиленгликоль (или другой простой полиэфир), который является жидким в своем чистом состоянии при 20 градусах по Цельсию и атмосферном давлении. Полиольный растворитель может функционировать, способствуя соединению лекарственного средства и поверхностно-активной системы, образованию покрытия лекарство/полиол/поверхностно-активная система на адсорбенте или растворению лекарственного средства в водной среде, например. Количество полиола, которое включают в композицию SMEDDS, не является критичным и может быть легко определено эмпирически специалистом в данной области в зависимости от цели(ей) назначения полиола. Как пример, композиция SMEDDS может включать примерно 0-80 мас.% полиола перед соединением с водной средой. В композициях, которые включают человеческий инсулин в качестве лекарственного средства, желательны композиции SMEDDS, которые включают по меньшей мере примерно 40 мас.% полиола.
Важной характеристикой композиции SMEDDS является то, что по меньшей мере некоторые капли, образовавшиеся после контакта между ней и водной средой, имеют размер, который соответствует переходу через или сквозь клеточные слои GI тракта. Капли должны иметь размер (т.е. диаметр) не больше примерно 500 нанометров, предпочтительно не больше примерно 300 нанометров, и предпочтительно иметь размер по меньшей мере примерно 10 нанометров. Размер капель, которые образуются, определяется составом композиции SMEDDS, главным образом, поверхностно-активной системы.
Получение самоэмульгирующихся композиций известно в технике (см., например, Khan et al., 2012, J. Pharmacy Alt. Med., 1: 13-19; публикация заявки на патент США № 2003/0022944; публикация заявки на патент США № 2010/0273730; и другие работы). Неотъемлемыми при составлении самоэмульгирующихся композиций, имеющих выбранное распределение капель по размерам, являются обычные испытания множества комбинаций и пропорций ингредиентов, например, идентичности и концентраций используемых поверхностно-активных веществ. Обычно до некоторой степени выполняют эмпирические проверки при выборе идентичности и концентраций используемых компонентов.
Композицию SMEDDS можно получить, упаковать и/или ввести субъекту в виде по существу гомогенной композиции, представляющей собой или по существу гомогенную однофазную жидкость, или по существу однородный порошок или гранулы (например, сухие спрессованные в таблетку или содержащиеся в капсуле) или по существу гомогенную эмульсию (например, эмульсию w/o или эмульсию w/o/w). В форме эмульсии композиция SMEDDS предпочтительно содержит лекарственное средство в дисперсной водной фазе эмульсии (т.е. в водной фазе эмульсии w/o). В одном воплощении композицию SMEDDS получают, упаковывают и/или вводят в форме порошкообразной или гранулированной смеси (необязательно включающей антацидный болюс), которая предназначена для смешивания с водой или другой водной жидкостью (для облегчения эмульгирования композиции SMEDDS) незадолго (в пределах 24 часов, предпочтительно в пределах 2 часов) или непосредственно перед пероральным введением.
Композиция SMEDDS применима для улучшения доставки лекарственных средств сквозь оболочки желудка и кишечника, например, через плотные соединения, которые, как известно, существуют между эпителиальными клетками кишечника. Композиции можно использовать для улучшения доставки лекарственных средств по существу любой гидрофильности/гидрофобности, но настоящее изобретение фокусируется особенно на относительно гидрофильных лекарственных средствах, которые обычно подвергаются действию кислот и ферментов, которые содержатся в водной среде желудочных жидкостей млекопитающего. Примеры таких лекарственных средств включают инсулиновые пептиды, гормоны роста, эритропоэтин, антитела (например, моноклональные антитела) и фрагменты антител, гентамицин, гемицитабин, пенициллин и ванкомицин.
Инсулиновые пептиды представляют собой особенно важный класс лекарственных средств, которые, как известно, чувствительны к разложению и/или инактивации в желудочных жидкостях. Известно, что семейство протеаз желудка, отнесенных по общему характеру к пепсину, расщепляет инсулиновые пептиды в определенных местах в кислых средах (рН < 3,4), которые обычны в желудках млекопитающих. Пепсин имеет максимальную активность при примерно рН 2,0 и по существу неактивен при рН 6,5 или выше. Таким образом, можно выбрать антацидный болюс для получения рН в желудке выше примерно 3, предпочтительно выше примерно 3,4 и предпочтительнее даже выше (может быть незначительное дополнительное преимущество при индукции рН в желудке > 6,5). Также известно, что инсулиновые пептиды подвергаются деамидированию в кислых средах. Расщепленные и/или деамидированные инсулиновые пептиды проявляют меньшую благоприятную фармацевтическую активность, чем интактный инсулин, что вероятно отвечает за неэффективность инсулина для лечения восприимчивых к инсулину расстройств (например, диабета), когда инсулин вводят пероральным путем. Композиции, описанные в настоящем описании, защищают инсулин (и другие лекарства) от инактивирующего действия желудочной кислоты и протеаз и также облегчают переход инсулина через оболочки GI. Поэтому композиции и способы, описанные в настоящем описании, особенно применимы при улучшении биодоступности инсулиновых пептидов, когда их вводят пероральным путем.
Известно много инсулиновых пептидов, и этот термин используется в настоящем описании для обозначения как встречающихся в природе форм инсулина (например, обычного немодифицированного человеческого инсулина), так и синтетических инсулинов и инсулиноподобных пептидов (например, полученных модификацией встречающегося в природе инсулина или небиотическими синтетическими способами). Точная идентичность инсулинового пептида не является критичной. Предпочтительно инсулиновый пептид представляет собой антротерапевтический инсулин, так как он вызывает один или больше физиологических эффектов у человека, которому его вводят, которые схожи или идентичны эффектам, вызываемым при инъекции (например, внутривенной, внутримышечной или подкожной) встречающегося в природе человеческого инсулина.
Количества лекарственного средства и любых полиольных или водных растворителей, которые включаются в композицию SMEDDS, не являются критичными до тех пор, пока композиция SMEDDS сохраняет способность к спонтанному эмульгированию после контакта с избытком водной жидкости. Композиция SMEDDS должна включать по меньшей мере достаточно лекарственного средства для того, чтобы обладать нужным фармацевтическим действием на субъекта, когда субъекту вводят стандартную дозу композиции SMEDDS. Водный раствор следует выбирать так, чтобы он был совместим с лекарственным средством (т.е. не вызывал существенного разложения или инактивации) в период и в условиях ожидаемого хранения от изготовления до введения композиции SMEDDS. Соответствующие количества водного раствора могут улучшить обработку композиции SMEDDS во время изготовления и типично не будут превышать примерно 30% (мас./мас.) композиции SMEDDS, и предпочтительно составляют до 20%, 10% или меньше от композиции SMEDDS. Композицию SMEDDS можно получить в объеме и распределить по аликвотам в стандартных лекарственных формах, подходящих для введения отдельным субъектам; в таких случаях объемная композиция SMEDDS будет включать множество эффективных доз лекарственного средства, в то время как стандартная доза будет включать одну эффективную дозу. Как пример, объемную композицию SMEDDS можно получить и упаковать в одно отделение многих лекарственных форм, включающих два отделения (например, капсул).
Композиция SMEDDS может включать полиольный растворитель, такой как растворитель из одного или нескольких компонентов: глицерина, пропиленгликоля и полиэтиленгликоля (ПЭГ). Подобным образом можно использовать другие подобные соединения (например, другие простые полиэфиры). Полиольные растворители могут облегчать растворение или суспендирование лекарственных средств (например, инсулина) в других компонентах композиции SMEDDS, причем посредством этого улучшается вызывающее всасывание лекарства действие композиции SMEDDS. Когда включают полиольный растворитель, композиция SMEDDS предпочтительно включает по меньшей мере примерно 5% (мас./мас.) полиольного(ых) растворителя(лей) и предпочтительно не более примерно 50% (мас./мас.). В некоторых композициях SMEDDS содержание соответствующего(их) полиола(ов) в композиции находится в интервале примерно 20-30% (мас./мас.) от композиции.
Композиция SMEDDS включает поверхностно-активную систему, которая, в комбинации с лекарственным средством и любым включенным водным или полиольным растворителем, делает композицию SMEDDS спонтанно эмульгирующейся после контакта с водной средой. Не требуется определенная степень или скорость эмульгирования, но предпочтительно, чтобы по существу вся композиция SMEDDS становилась эмульсией в пределах одного часа, когда ее соединяют с девятикратным избытком дистиллированной воды при 20 градусах по Цельсию при слабом перемешивании (т.е. девять частей воды и одну часть композиции SMEDDS перемешивают в стакане с регулируемой температурой при вращении стержня для перемешивания со скоростью 10 оборотов в минуту). Поверхностно-активная система включает по меньшей мере одно неионогенное поверхностно-активное вещество и предпочтительно включает по меньшей мере одно поверхностно-активное вещество, выбранное из группы, включающей полигликозилированные глицериды, имеющие по меньшей мере одну ацильную группу, и эфиры полиэтиленгликоля и жирных кислот. Полигликозилированным глицеридом при использовании в настоящем описании называют смесь моноглицеридов, диглицеридов и триглицеридов с моноэфирами жирных кислот и/или диэфирами жирных кислот полиэтиленгликоля (ПЭГ), имеющими величины гидрофильно-липофильного баланса (ГЛБ) между и включая 4 и 19 (предпочтительно от 6 до 14). Ацильная(ые) группа(ы) представляет(ют) собой линейные или разветвленные алканы или алкены (предпочтительно имеющие не более двух алкенильных связей), включающие от 8 до 18 атомов углерода. Предпочтительные ацильные группы включают –CO–(CH2)7CH3, –CO–(CH2)9CH3, –CO–(CH2)11CH3, –CO–(CH2)13CH3, –CO–(CH2)7–CH=CH–(CH2)7CH3 и –CO–(CH2)7–CH=CH–CH2–CH=CH–(CH2)4CH3. Примеры полигликозилированных глицеридов включают олеоилполиоксилглицериды (олеоилполиоксил-6-глицериды, такие как лаброфил® М-1944CS), линолеоилполиоксилглицериды (линолеоилполиоксил-6-глицериды, такие как лаброфил® М-2125CS), каприлокапроилполиоксилглицериды (ПЭГ-6-каприловые/каприновые глицериды, такие как софтиген® 767), каприлокапроилполиоксил-8-глицериды (например, лабрасол®), лауроилполиоксилглицериды (гелуцир® 44/14) и их комбинации.
Эфиры пропиленгликоля и жирных кислот при использовании в настоящем описании обозначают смесь моно- и диэфиров пропиленгликоля и насыщенных и ненасыщенных жирных кислот, предпочтительно полученных из съедобных масел и жиров, которые можно получить или прямой этерификацией пропиленгликоля жирными кислотами или переэтерификацией эфиров пропиленгликоля жирами или маслами. При получении переэтерификацией продукт может содержать остаточные моно- и диглицериды и глицерин, и за этим процессом может следовать молекулярная перегонка на отдельные моноэфиры. Примеры эфиров пропиленгликоля и жирных кислот включают монокаприлат пропиленгликоля, дилаурат пропиленгликоля, монолаурат пропиленгликоля, дикаприлокапрат пропиленгликоля, лаурат пропиленгликоля, каприлат пропиленгликоля.
Поверхностно-активная система может также включать дополнительное поверхностно-активное вещество, такое как выбранное из группы, включающей полисорбат, полоксамеры, полиоксиэтиленпроизводные касторового масла, простые полиоксиэтиленалкилэфиры, сорбитановые эфиры жирных кислот, глицерилмоноолеат, глицерилмонолинолеат, среднецепные триглицериды, полиглицерилолеат, ланоилполиоксилглицерид, стеароилполиоксилглицериды и их комбинации.
Получение самоэмульгирующихся композиций известно в технике, включая выбор идентичности(ей) и концентрации поверхностно-активного(ых) вещества(веществ), включенного(ых) в такие композиции. Идентичность и концентрация поверхностно-активного(ых) вещества(веществ), включенного(ых) в такие поверхностно-активные системы, не являются критичными, выбирают и другие, которые придают композиции SMEDDS способность к спонтанному эмульгированию после контакта с водной средой (необязательно при слабом перемешивании). Предпочтительно поверхностно-активная система составляет до примерно 5-90% (мас./мас.) композиции SMEDDS, предпочтительнее, по меньшей мере примерно 20%, по меньшей мере примерно 30%, по меньшей мере примерно 40%, по меньшей мере примерно 50% и еще предпочтительнее, по меньшей мере примерно 60% (например, 20-70%, 40-70% или 50-70%, мас./мас.). Как известно в технике, комбинации поверхностно-активных веществ подходят для придания способности к спонтанному эмульгированию композициям, содержащим лекарственные средства, и такие комбинации подходят для поверхностно-активной системы, описанной в настоящем описании. Комбинации поверхностно-активных веществ можно выбрать, например, обратившись к величине их гидрофильно-липофильного баланса (ГЛБ) (см., например, публикации заявок на патент США №№ 2003/0022944 и 2010/0273730). Когда для выбора поверхностно-активной системы используют величины ГЛБ, величины ГЛБ в интервале от 8 до 19 рассматривают как подходящие для композиций, описанных в настоящем описании.
Композицию SMEDDS можно получить путем объединения ее компонентов в любом порядке, который эффективен для получения композиции, которая спонтанно эмульгируется после контакта с водной средой. В соответствующем способе лекарственное средство растворяют или суспендируют в водном растворе, и затем этот раствор/суспензию соединяют с полиольным(и) растворителем(ями) с образованием по существу гомогенной однофазной смеси. К смеси добавляют поверхностно-активное(ые) вещество(а) поверхностно-активной системы (по отдельности или после предварительного объединения поверхностно-активных веществ). В зависимости от выбранной поверхностно-активной системы, слабое перемешивание, встряхивание или иное возбуждение может дать или вторую по существу гомогенную однофазную смесь (например, если включают мало или не включают водный или полиольный растворитель(и)) или многофазную смесь, такую как эмульсия w/o или эмульсия w/o/w (особенно, если включают большие количества водного(ых) растворителя(ей)).
Композиция SMEDDS может быть ассоциирована с адсорбентом. Адсорбенты представляют собой твердые композиции, которые наиболее часто используют в форме тонкоизмельченных порошков, которые действуют преимущественно как эксципиенты для облегчения обращения с композициями с лекарственными средствами, описанными в настоящем описании. Поскольку имеется тенденция представлять адсорбенты в виде свободно текущих порошков или других веществ, с которыми легко обращаться, связывание, адсорбция, сушка или прилипание компонента к, на или в адсорбенте облегчает обращение с таким компонентом. Использование адсорбентов хорошо известно в фармацевтических областях. Примеры таких подходящих адсорбентов включают диоксид кремния (например, порошки диоксида кремния, такие как высокодисперсный диоксид кремния) и другие минеральные порошки, которые по существу не растворяются в воде, целлюлозы (например, порошки монокристаллической целлюлозы) и крахмалы.
Как пример, композицию SMEDDS (включающую лекарственное средство) можно отвердить на гранулах микрокристаллической целлюлозы (например, путем контактирования гранул с композицией SMEDDS, включающей летучий растворитель, и затем выпаривая часть или весь растворитель), и гранулы с покрытием из композиции SMEDDS можно объединить с порошкообразным антацидом и получить лекарственную форму. С другой стороны, антацид можно отвердить на гранулах (например, в виде наружного слоя). Как еще одна альтернатива, композицию SMEDDS (пока не включающую лекарственное средство) можно нанести как покрытие на гранулы и затем порошкообразное лекарственное средство и порошкообразный антацид вместе можно смешать с гранулами. Специалист в этой области примет к сведению, что многие известные конформации адсорбентов и другие компоненты композиций с лекарственными средствами, описанные в настоящем описании, можно использовать без отхода от сущности изобретения, описанного в настоящем описании.
Композицию SMEDDS можно хранить (предпочтительно при регулируемой температуре, такой как менее 20 градусов по Цельсию, и предпочтительно выше температуры замерзания любой водной фазы, присутствующей в ней, например, при 4-5 градусах по Цельсию) в виде по существу гомогенной смеси (эмульгированной или нет), или ее можно объединить с водной средой (например, водой или водным раствором/суспензией антацида) для образования эмульсии перед хранением. Известно, что сухие или с низким содержанием влаги композиции показывают превосходные свойства хранения в широком интервале условий. Лекарственные формы, описанные в настоящем описании, в которых один или несколько компонентов присутствуют в форме сухого порошка (например, покрытия на или прилипшие к адсорбенту), могут таким образом выдержать более суровые условия хранения, такие как хранение при регулируемой температуре (например, ниже 30 градусов по Цельсию и выше 0 градусов по Цельсию) в течение продолжительных периодов.
Композицию SMEDDS можно вводить непосредственно субъекту (т.е. так, что она будет спонтанно эмульгироваться после контактирования с водной жидкостью желудка субъекта), или ее можно ввести в контакт с водной средой (например, стакане с водой, стакане с водой, в которой растворен антацидный болюс, описанный в настоящем описании, или ароматизированном напитке) перед введением среды субъекту (т.е. так, что композиция SMEDDS будет полностью или частично эмульгирована в среде перед ее введением субъекту.
Антацидный болюс
Соединения и способы, описанные в настоящем описании, включают болюс одного или нескольких антацидов, который достаточен для повышения рН в желудке животного, которому вводят болюс, до по меньшей мере примерно 3 после (т.е. предпочтительно не более чем через 3-5 минут после) перорального введения болюса. Идентичность антацида не является критичной, и подходящие примеры включают бикарбонат натрия, гидроксид магния, карбонат кальция и гидроксид алюминия. Другие подходящие примеры описаны, например, в публикации заявки на патент США № 2014/0127296. В альтернативном воплощении антацид добавляют в форме, которая не высвобождается как один болюс после перорального введения, но вместо этого высвобождает антацид в течение протяженного периода времени (например, 2-24 часов) после перорального введения. Препараты с протяженным высвобождением антацида известны.
Выбор соответствующего антацида (или комбинации антацидов) и соответствующих количеств антацидов для достижения рН в желудке не менее примерно 3 (предпочтительно не менее примерно 3,4) входит в круг познаний специалиста в данной области техники, и в расчет принимают предполагаемое количество кислоты, присутствующей в желудке субъекта. Как пример, предполагается, что у здорового проголодавшегося человека в ее или его желудке имеется примерно 1-7 миллиэквивалентов (мэкв) желудочных кислот. Специалист в данной области техники способен вычислить количество антацидов, необходимое для достижения желательного рН в желудке субъекта в течение желательного периода времени.
Форма, в которой антацидный болюс упаковывается и/или вводится субъекту, не является критичной. Жидкости являются относительно объемными, и имеются трудности в отношении упаковки и хранения, но являются относительно простыми для введения. Твердые формы более компактны и обычно стойкие в хранении, но требуют гидратации и растворения или перед введением или после введения пациенту.
Количество антацида в болюсе будет влиять на длительность периода, в течение которого достигается рН в желудке выше 3. Вообще говоря, большие количества антацида будут приводить к более длительному периоду до по меньшей мере цели. Когда для лекарства желательно относительно быстрое действие, включение в лекарственную форму только достаточного количества антацида для повышения рН в желудке в течение желательного периода времени может служить для ограничения длительности действия лекарства, за счет возможности желудочных секреций, которые преодолевают эффект повышения рН антацидом, разрушая или инактивируя лекарственное средство.
Фармацевтическое средство, эффективное для снижения секреции желудочной кислоты, можно вводить с (или перекрывая по времени с) композициями, описанными в настоящем описании, если желательна более длительная защита лекарственного средства от действия желудочной кислоты.
Лекарственная форма
Точная форма или характер лекарственной формы, в которой композиции, описанные в настоящем описании, вводят субъекту, не являются критичными. Можно использовать любую из самых разных известных лекарственных форм, включая таблетки, капсулы, жидкие носители и многослойные или многокамерные лекарственные формы. Другие рассматриваемые лекарственные формы включают порошки, гранулы и капсулы (dosage cups), имеющие твердый материал, содержащийся или присоединенный к ним, каждая из которых может быть соединена с водной жидкостью перед введением для того, чтобы образовалась эмульсия композиции SMEDDS, суспензия или раствор антацида или то и другое. Важно то, что лекарственное средство высвобождается из лекарственной формы, которая содержит композицию SMEDDS, в период времени, который перекрывает период времени, в течение которого антацидный болюс повышает рН в желудке выше примерно 3.
В одном воплощении композиции, описанные в настоящем описании, упаковываются в виде порошкообразных стандартных лекарственных форм, в которых стандартная доза твердой композиции SMEDDS (например, порошкообразной композиции SMEDDS или адсорбента с композициями SMEDDS, адсорбированными на нем или высушенных на нем) объединена со стандартными лекарственными формами лекарственного средства (в твердой форме) и антацида (также в твердой форме). Твердые компоненты лекарственной формы соединяют с аликвотой водной жидкости (или предоставленной субъектом или включенной отдельно с твердой лекарственной формой), для того, чтобы образовать дисперсию, эмульсию или (предпочтительно) наноэмульсию, суспендированную в водной жидкости перед введением суспензии пациенту.
В другом воплощении композиции, описанные в настоящем описании, упаковывают в виде стандартных лекарственных форм, в которых стандартная доза композиции SMEDDS эмульгирована путем контактирования ее с водной средой, в которой растворена стандартная доза антацида, и эмульсию вводят субъекту перорально (например, путем вливания или выжимания содержимого лекарственной формы в рот субъекта или путем проглатывания субъектом всей лекарственной формы, такой как форма капсулы).
В другом воплощении композиции используются врачами и/или пациентами в форме набора, который содержит стандартную дозу антацида, упакованную отдельно (например, в виде таблетки или жидкости) от стандартной дозы композиции SMEDDS (например, предоставленной в капсуле); полную лекарственную форму вводят пациенту путем введения пациенту как стандартной дозы антацида, так и стандартной дозы композиции SMEDDS.
В еще одном воплощении стандартную дозу антацида и стандартную дозу композиции SMEDDS упаковывают в отдельные камеры или слои единой лекарственной формы (например, многокамерного контейнера, с покрытием или двухслойной таблетки или с покрытием или многокамерной капсулы), так что прием внутрь субъектом полной лекарственной формы приведет к высвобождению антацидного болюса из его камеры (т.е. повышению посредством этого рН в желудке до >3) и высвобождению композиции SMEDDS из ее камеры (ведущему к высвобождению или образованию содержащей лекарство эмульсии в GI тракте).
В еще одном воплощении лекарственное средство и антацид объединяют и получают в форме таблетки или порошка и предоставляют пациенту вместе с композицией SMEDDS, которая уже объединена с водной жидкостью (т.е. так, что она находится в форме суспендированной нано- или микроэмульсии). В таком воплощении лекарственную форму вводят пациенту путем объединения (или объединения пациентом) таблетки или порошка с суспендированной эмульсией (для растворения или суспендирования в ней лекарственного средства и антацида) и введения полученной жидкости пациенту.
В еще некоторых формах лекарственная форма может представлять собой капсулу, которая содержит композицию SMEDDS, и на которую нанесено покрытие с быстрорастворяющимся антацидным болюсом. Лекарственную форму можно погрузить в стакан с водой для образования раствора антацида (необязательно ароматизированного), и этот раствор можно использовать или как среду для облегчения проглатывания капсулы субъектом или как среду, в которой капсула растворяется (с образованием посредством этого содержащей лекарство эмульсии в среде), и затем среда потребляется субъектом.
Этот перечень примеров воплощения не является ограничивающим. По существу можно использовать любую лекарственную форму или способ для перорального введения двух композиций одному и тому же субъекту. Однако введение композиции SMEDDS должно осуществляться в течение периода, когда контакт между композицией SMEDDS и желудочными жидкостями происходит в то время, когда рН в желудке достигает выше примерно 3 за счет введения антацидного болюса.
Применение композиций
Композиции, описанные в настоящем описании, можно использовать для перорального введения лекарственного средства, практически нецелесообразного для введения в кровоток млекопитающего через желудок (т.е. плохо доступного в желудке лекарственного средства, такого как гидрофильное лекарственное средство). Для того, чтобы этого достигнуть, терапевтически эффективное количество лекарственного средства растворяют или суспендируют в воде или водном растворе. Растворенное или суспендированное лекарственное средство соединяют с поверхностно-активной системой, описанной в настоящем описании, и, необязательно, полиольным растворителем. Эти компоненты осторожно перемешивают с образованием по существу гомогенной композиции SMEDDS, которая может представлять собой по существу однофазную жидкость, эмульсию w/o (гидрофильные лекарственные средства склонны локализоваться в водной фазе) или эмульсии w/o/w (гидрофильные лекарственные средства склонны локализоваться в одной или обеих водных фазах). В другом воплощении лекарственное средство соединяют с поверхностно-активной системой и адсорбентом с образованием смеси в твердой форме (например, твердой гранулы или порошка), которую объединяют с водной жидкостью (например, водой или суспензией антацидного болюса) перед введением.
Как описано в настоящем описании, идентичности и количества лекарственного средства, любого водного растворителя, любого полиольного растворителя и поверхностно-активной системы выбирают такими, чтобы композиция SMEDDS эмульгировалась спонтанно после контактирования с водной средой в условиях слабого механического перемешивания. Композицию SMEDDS вводят в контакт с такой водной средой для того, чтобы создать эмульсию (или для разбавления существующей эмульсии в композиции SMEDDS), и такую эмульсию можно, необязательно, хранить при пониженной температуре (предпочтительно выше температуры замерзания водных фаз эмульсии). Эмульсию, полученную таким образом, перорально вводят млекопитающему вместе с (т.е. незадолго до, вскоре после или одновременно с введением) болюсом антацида, достаточным для повышения рН в желудке животного до по меньшей мере примерно 3 (и предпочтительно, выше 3,4).
Необязательно, эмульсию можно формировать в желудке субъекта путем введения субъекту лекарственной формы, которая высвобождает композицию SMEDDS непосредственно в желудок. Если композиция SMEDDS в лекарственной форме еще не в форме эмульсии, она может спонтанно образовать эмульсию в желудочных жидкостях субъекта. Прием внутрь воды вместе с лекарственной формой может увеличить вероятность того, что содержимое желудка субъекта включает достаточно воды для образования эмульсии.
Композицию SMEDDS следует вводить перорально млекопитающему достаточно близко по времени к введению болюса, пока рН в желудке животного остается по меньшей мере примерно 3 в то время как эмульсия, образованная из композиции SMEDDS, остается в желудке млекопитающего.
Диабет и другие расстройства, которые лечат путем введения субъекту инсулиновых пептидов, являются расстройствами, для которых композиции и способы, описанные в настоящем описании, рассматриваются как особенно подходящие. Эффективное неотложное лечение инсулиновосприимчивых расстройств может требовать быстрого начала действия после введения инсулинового пептида, и может быть желательно, чтобы длительность действия инсулинового пептида не выходила за предел в несколько часов. По этим причинам обычно используют инъецируемые композиции инсулиновых пептидов, поскольку инсулиновые пептиды имеют склонность к плохой биодоступности (если вообще биодоступны), когда их вводят другими (например, пероральным) путями. Как указывает обсуждение и примеры в настоящем описании, инсулиновые пептиды, которые вводятся перорально в одной из композиций, описанных в настоящем описании, могут проявлять очень быстрое (<30 минут) начало действия и длительность действия, которая не выходит за 2-4 часа. По этим причинам композиции, описанные в настоящем описании, можно использовать или для доставки инсулиновых пептидов на регулярной основе или, когда требуется, на неотложной основе субъектам, нуждающимся в инсулиновой терапии. Способность субъектов принимать композиции перорально, а не через инъекцию, также может улучшить комфорт и облегчить введение, поддерживая принятие пациентом предписанных схем приема лекарств или положений инструкций.
Примеры
Предмет настоящего изобретения теперь описывается с обращением к следующим далее примерам. Эти примеры предлагаются только с целью пояснения, и предмет изобретения не ограничивается этими примерами, но охватывает все вариации, которые очевидны как результат указаний, предоставленных в настоящем описании.
Пример 1
Композиции
В этом примере описываются композиции, содержащие инсулин.
Каждую из композиций 1, 2 и 3 получают следующим образом.
Немодифицированный человеческий инсулин (28,8 МЕ/мг) взвешивают в 7-миллилитровом флаконе, и во флакон добавляют указанное количество 0,05 нормальной HCl для растворения инсулина. Во флакон добавляют полиольный растворитель (пропиленгликоль, глицерин и/или ПЭГ 400), и содержимое осторожно перемешивают для соединения ингредиентов. Затем во флакон добавляют три поверхностно-активных вещества, и содержимое осторожно перемешивают до тех пор, пока не образуется прозрачная смесь. Прозрачную смесь перед введением суспендируют в примерно 160-230 миллилитрах 3,78% (мас./мас.) раствора NaHCO3 (десятикратное разведение), вызывая эмульгирование.
Композиция, названная в настоящем описании композицией 1, имеет указанный далее состав.
Композиция, названная в настоящем описании композицией 2, имеет указанный далее состав.
Композиция, названная в настоящем описании композицией 3, имеет указанный далее состав.
Лабразол™ является товарным знаком Gattefosse, США, и представляет собой смесь ПЭГ-8-каприловых/каприновых глицеридов, имеющих общую формулу R-(CH2-CH2O)8-R, где каждый -R представляет собой или -CO-(CH2)6-CH3 или -CO-(CH2)8-CH3. Лаурогликоль™ FCC является товарным знаком Gattefosse, США, и представляет собой монолаурат пропиленгликоля (C15H30O3). Твин™ 80 является товарным Sigma-Aldrich Chemical Company и представляет собой смесь моноолеатов полиоксиэтилен(20)сорбитана. Софтиген™ 767 является товарным знаком Sasol Olefins & Surfactants GmbH и представляет собой смесь ПЭГ-6-каприловых/каприновых глицеридов, имеющих общую формулу R-(CH2-CH2O)6-R, где каждый -R представляет собой или -CO-(CH2)6-CH3 или -CO-(CH2)8-CH3. Кремофор™ RH40 является товарным знаком BASF Group и представляет собой поверхностно-активные вещества гидрогенизированного касторового масла. Основным составляющим кремофораTM RH 40 является гидроксистеарат глицеролполиэтиленгликоля, который вместе с глицеролполигликолевыми эфирами жирных кислот образует гидрофобную часть продукта. Гидрофильная часть состоит из полиэтиленгликолей и глицеролэтоксилата, имеющего формулу C57H110O9(CH2CH2O)n. Лабрафил™ 1944CS является товарным знаком Gattefosse, США, и имеет общую структуру HO-(CH2-CH2O)6-CO-(CH2)7-CH=CH-(CH2)7-CH3.
Поверхностно-активные вещества, также рассматриваемые как приемлемые, но не использованные в этих экспериментах, включают лабрафил™ M-2125CS, который является товарным знаком Gattefosse, США, и имеет общую структуру HO-(CH2-CH2O)6-CO-(CH2)7-CH=CH-CH2-CH=CH-(CH2)4-CH3, и гелюцир 44/41, который является товарным знаком Gattefosse, США, и имеет общую формулу R-(CH2-CH2O)32-R, где каждый -R представляет собой или -CO-(CH2)10-CH3 или -CO-(CH2)12-CH3.
Композицию 4 получают, смешивая в 20-миллилитровом стакане 8 миллиграмм немодифицированного человеческого инсулина, 300 миллиграмм бикарбоната натрия, 100 миллиграмм глицерина и 600 миллиграмм лабразола™. Затем в стакан добавляют 8992 миллиграмма воды, и объединенные ингредиенты перемешивают с осторожностью до образования наноэмульсии.
Композиция, названная в настоящем описании композицией 4, имеет указанный далее состав.
Композицию 5 получают следующим образом. В 20-миллилитровом стакане соединяют и смешивают 8 миллиграмм инсулина, 800 миллиграмм бикарбоната натрия, 100 миллиграмм глицерина и 600 миллиграмм лабразола™. В стакан добавляют 8492 миллиграмма воды, и содержимое перемешивают с осторожностью до образования наноэмульсии.
Композиция, названная в настоящем описании композицией 5, имеет указанный далее состав.
Композицию 6 получают следующим образом. В 20-миллилитровом стакане соединяют и смешивают 8 миллиграмм инсулина, 210 миллиграмм бикарбоната натрия, 90 миллиграмм гидроксида магния, 100 миллиграмм глицерина и 600 миллиграмм лабразола™. Затем в стакан добавляют 8992 миллиграмма воды, и содержимое перемешивают с осторожностью до образования наноэмульсии.
Композиция, названная в настоящем описании композицией 6, имеет указанный далее состав.
Композицию 7 (порошок, предназначенный для перорального введения после образования суспензии из порошка) получают, добавляя по частям лабразол™ к аэросилу™ 200, содержащемуся в ступке. После добавления полученную смесь гомогенизируют с использованием соответствующего пестика для уверенности в равномерном распределении. После этого к смеси лабразол™-аэросил™ 200 добавляют инсулин и порошок бикарбоната натрия, и полученную комбинацию перемешивают. Полученный порошок пропускают через сито №16 (номинальное отверстие сита 1,19 миллиметров), сушат при температуре окружающей среды и хранят до дальнейшего использования. Затем 1,158 грамм хранившегося порошка соединяют с 9 миллилитрами воды перед введением, получая дисперсию на водной основе.
Композиция, названная в настоящем описании композицией 7, имеет указанный далее состав.
Композицию 8 (порошок, предназначенный для перорального введения после образования водной суспензии из порошка) получают, добавляя по частям жидкий лабразол™ к Neusilin® US2 (гранулированный алюмометасиликат магния – продукт, продаваемый Fuji Chemical Industry Co., Ltd., имеющий средний размер частиц примерно 60-120 микрон), содержащемуся в ступке. После добавления полученную смесь гомогенизируют с использованием соответствующего пестика для уверенности в равномерном распределении. После этого к смеси лабразол™-Neusilin® US2 добавляют порошки инсулина и бикарбоната натрия, и полученную комбинацию перемешивают. Полученный порошок пропускают через сито №16, сушат при температуре окружающей среды и хранят до дальнейшего использования. Затем 1,108 грамм этого порошка соединяют с 9 миллилитрами воды перед введением, получая дисперсию на водной основе.
Композиция, названная в настоящем описании композицией 8, имеет указанный далее состав.
Композицию 9 (композиция из двух частей) получают следующим образом.
Компоненты части А
Часть А (гранулы) получают методом влажной грануляции, при котором все компоненты части А взвешивают и пропускают через сито №20. Инсулин растворяют в 0,05 N HCl и затем помещают в ступку, содержащую другие компоненты части А. После добавления инсулина смесь гомогенизируют с использованием пестика 100 движениями для уверенности в равномерном распределении композиции, и гранулы сушат с использованием сушилки с псевдоожиженным слоем при 25 градусах по Цельсию в течение 30 минут. Сухие гранулы пропускают через сито 16 и хранят при 4 градусах по Цельсию до дальнейшего использования.
Компоненты части В
Часть В получают, аккуратно взвешивая лабразол® и глицерин, и затем эти компоненты суспендируют в воде для образования дисперсии.
Композицию 9 получают, объединяя часть А и часть В перед пероральным введением (например, в пределах 2 часов перед пероральным введением).
Пример 2
Размер капель
Преимуществом композиций, описанных в настоящем описании, является то, что инсулин содержится в водной сердцевине капель (которые могут включать мицеллы и/или липосомы), которые образуются спонтанно (необязательно при слабом перемешивании) после контакта композиции с водой или водной средой. Поскольку размер таких капель влияет на их способность пересекать гастроинтестинальные поверхности (и таким образом влиять на скорость и степень биодоступности лекарственного средства, содержащегося в липосомах), размер капель, образовавшихся после самоэмульгирования композиций, описанных в настоящем описании, анализируют.
В отдельных образцах одну часть каждой из композиций 1, 2, 3 и 4 соединяют с 500 частями дистиллированной воды при осторожном перемешивании и допускают образование дисперсии или эмульсии. Размер образовавшихся таким образом капель измеряют с использованием анализатора дзета-потенциала Zetasizer Nano ZS (Malvern Instruments, Ltd.). Этот же прибор используют для измерения индекса полидисперсности (PDI) для указанных эмульсий. Вычисления параметра PDI определяются в документе стандарте ISO 13321:1996 E и ISO 22412:2008. Одну часть композиции 1 соединяют с 500 частями 0,1 N HCl или забуференного фосфатом физиологического раствора (PBS), такие образцы показывают, что размеры капель как правило не превышают примерно 2000 нанометров, и другие композиции, описанные в настоящем описании, имеют капли, размер которых как правило не превышает примерно 800 нанометров.
Результаты этих экспериментов приводятся далее.
Композиция 4
115
Средний размер капель композиции 1 в 0,1 N HCl составляет 1822 нанометра. Средний размер капель композиции 1 в стандартном забуференном фосфатом физиологическом растворе составляет 873 нанометра.
Пример 3
Исследования нейтрализации кислоты
Композицию 1 разбавляют в десять раз 4,2% (мас./мас.) раствором бикарбоната натрия (т.е. одну часть композиции 1 соединяют с девятью частями раствора бикарбоната натрия; конечная концентрация 3,78%). Величина рН такой разбавленной композиции 8,2. Величина рН 0,1 N HCl 1,2.
Соединяют 4,0 миллилитра 0,1 N HCl с выбранными количествами разбавленной композиции, и измеряют рН полученного комбинированного раствора. Когда HCl соединяют с 1,0 миллилитром разбавленной композиции, полученный рН равен 5,64. Когда HCl соединяют с 1,5 миллилитрами разбавленной композиции, полученный рН равен 6,27. Когда HCl соединяют с 2 миллилитрами разбавленной композиции, полученный рН равен 6,48. Такие результаты показывают нейтрализующее кислоту действие комбинаций композиция/антацид, описанных в настоящем описании. Специалист в данной области техники может выбрать соответствующее количество антацида для нейтрализации предполагаемых количеств кислоты в желудке у людей и других субъектов (например, для повышения рН в желудке до по меньшей мере 3,0, до по меньшей мере 3,4 или любой другой желательной величины).
Пример 4
Протеолитические исследования
Пепсин представляет собой пищеварительный фермент желудка протеазу, которая проявляет значительную протеолитическую активность (включая инсулининактивирующую активность) при рН между рН 1 и рН 3. Следующие далее эксперименты выполняют для исследования влияния рН на пепсинопосредуемую инактивацию инсулина, содержащегося в композициях с инсулином, описанных в настоящем описании.
Отдельно 1 миллилитр композиции 1 объединяют с 9 миллилитрами стандартного забуференного фосфатом физиологического раствора с образованием дисперсии или эмульсии (точная природа композиции не рассматривается как критичная, и в настоящем описании далее называется «эмульсией»). Величина рН такой эмульсии 6,9. Затем эмульсию соединяют с 0,5 миллилитрами имитированной желудочной жидкости (1 грамм хлорида натрия, 3,5 миллилитра 37% HCl в 500 миллилитрах воды), которая включает 1650 единиц пепсина, при рН или 1,4 или 3,4 (рН регулируют 0,1 N раствором NaOH). Затем смеси инкубируют при 37°С. Через 5, 30 или 90 минут инкубацию в аликвотах смесей прекращают, добавляя 0,1 N раствор NaOH (для изменения рН до интервала примерно 6,-6,5 и остановки таким образом действия пепсина).
Пепсинопосредуемое расщепление инсулина оценивают в каждой аликвоте с использованием ВЭЖХ, обнаруживая ожидаемые продукты расщепления инсулина. Не обнаруживают интактный инсулин в аликвотах, инкубированных при рН 1,4 в течение 5, 30 или 90 минут, что предполагает, что происходит быстрое протеолитическое расщепление инсулина. По существу весь инсулин остается интактным в аликвотах, которые инкубировали в течение 30 и 90 минут при рН 3,4 (инсулин не оценивали в аликвоте, инкубированной при рН 3,4 в течение только 5 минут), что предполагает, что рН 3,4 делает пепсин достаточно инактивным для сохранения инсулина в нерасщепленной форме в течение указанных периодов. Такие результаты также предполагают, что повышение рН в желудке субъекта до рН 3,4 (или по меньшей мере до 3,0) дает возможность инсулину оставаться интактным в пространстве желудка, когда вводится перорально в композициях, описанных в настоящем описании.
Пример 5
Исследования проницаемости кишечника in vitro
Эксперименты, описанные в этом примере, показывают, что инсулин можно транспортировать сквозь монослои клеток Сасо-2, которые, как известно, походят на слои энтероцитов, которые выстилают тонкую кишку. Поэтому такие эксперименты рассматриваются как показывающие способность композиций, описанных в настоящем описании, транспортировать гидрофильные лекарственные средства, такие как инсулин, сквозь выстилку кишечника.
Клетки Сасо-2 культивируют при 37±2 градусах по Цельсию в минимальной эссенциальной среде (МЕМ) с солью Игла и 1-глутамином с добавлением 15% сыворотки плода коровы, с 1% не-незаменимой аминокислоты и 1% антибиотка-антимикотика в инкубаторе в атмосфере с 5% диоксида углерода для имитирования клеток выстилки кишечника. Для этого исследования используют клеточные монослои, которые через 21-28 дней после посева показывают величины трансэпителиального электрического сопротивления больше 300 ом на квадратный сантиметр.
В момент ноль среду с одной стороны клеточного монослоя заменяют на 0,5 миллилитра раствора, который включает одну из композиций 1, 2 или 3 или не включающую инсулин в SMEDDS-содержащей композиции. Раствор разбавляют в десять раз средой (т.е. одна часть инсулиносодержащего раствора и девять частей буфера) перед нанесением на монослой. Через тридцать минут после такой замены анализируют ВЭЖХ содержание инсулина на базолатеральной стороне монослоя. Коэффициент проницаемости (Рарр) вычисляют из следующего уравнения: Papp = (dQ/dt) / ( C0 × площадь), где dQ/dt – линейная скорость появления, полученная из профиля перенесенного количества субстрата со временем (измеренная в микрограммах в секунду); C0 – измеренная начальная концентрация в донорской камере (измеренная в микрограммах на миллилитр), и «площадь» – площадь поверхности мембраны из клеточного монослоя.
Отсутствие переноса свободного инсулина (т.е. инсулина, не объединенного со SMEDDS-содержащей композицией) можно обнаружить после 30-минутной инкубации. Определяют величины Papp для монослоев, к которым применяют композицию 1, 12×10-6 сантиметров в секунду; для монослоев, к которым применяют разбавленную композицию 2, 16×10-6 сантиметров в секунду; и для монослоев, к которым применяют разбавленную композицию 3, 9×10-6 сантиметров в секунду.
Такие результаты показывают, что введение инсулина в монослои культивированных Сасо-2 в разбавленных композициях с SMEDDS, описанных в настоящем описании, существенно усиливает перенос инсулина сквозь монослой. Поскольку монослои клеток Сасо-2 образуют плотные соединения и, как полагают, являются соответствующей моделью слоев энтероцитов, которые выстилают тонкую кишку, результаты этих экспериментов показывают способность композиций, описанных в настоящем описании, транспортировать гидрофильные лекарственные средства, такие как инсулин, сквозь выстилку кишечника.
Пример 6
Гипогликемическое исследование in vivo с SMEDDS, содержащими инсулин, объединенными с антацидами, после перорального введения диабетическим мышам, здоровым крысам и здоровым собакам бигль
Диабет вызывают у самцов мышей С57BL/6JNarl (возраст 8 недель, масса тела примерно 20 грамм) двумя инъекциями в хвостовую вену стрептозотоцина (STZ) (первая 75 мг/кг, вторая 150 мг/кг). Индукцию диабета у мышей подтверждают путем измерения концентрации глюкозы в образце крови, которую берут из хвостовой вены. Уровни глюкозы в крови выше 300 мг/мл считаются подтверждением индукции диабета, и мышей, показывающих такие уровни («STZ-индуцированные диабетические мыши»), используют в экспериментах, описанных в этом примере.
Композицию, содержащую 200 МЕ/кг инсулина, вводят через пероральный зонд каждой из восьми STZ-индуцированных диабетических мышей в трех группах (т.е. всего 24 мышам). В первой группе каждая мышь получает свободный немодифицированный инсулин, суспендированный в примерно 160-230 микролитрах (в зависимости от массы животного) 3,78% (мас./мас.) растворе антацида NaHCO3. Во второй группе каждая мышь получает инсулин в форме композиции 1, суспендированной в примерно 160-230 микролитрах забуференного фосфатом физиологического раствора (PBS). Третья группа получает инсулин в композиции 1 в форме композиции 1, суспендированной в примерно 160-230 микролитрах 3,78% (мас./мас.) растворе антацида NaHCO3 (т.е. десятикратное разбавление композиции 1). Предполагают, что рН в желудке после зондирования быстро (т.е. в пределах 0-3 минут) повысится выше 3,4 в первой и третьей группах, и во второй группе не ожидают существенного изменения рН в желудке после зондирования.
Образцы крови берут у каждой мыши после зондирования, и определяют уровни глюкозы в крови в этих образцах. Результаты таких определений глюкозы в крови показаны на фигуре 1.
STZ-индуцированные диабетические мыши в третьей группе (которой вводили инсулин в композиции 1 вместе с антацидом) показывают существенное падение уровня глюкозы в крови, начинающееся через 15 минут после перорального зондирования и удерживающееся не долее, чем примерно 4 часа. В течение первых 15 минут после введения уровень глюкозы в крови снижается примерно на 10% по сравнению с уровнем в момент ноль. Уровни глюкозы в крови у STZ-индуцированных диабетических мышей в первой (свободный инсулин + антацид) и второй (инсулин в композиции 1 без антацида) существенно не изменяются в течение остального периода исследования.
Приблизительно пропорциональную зависимость от дозы наблюдают в группах STZ-индуцированных диабетических мышей, которым по отдельности вводят перорально 50, 100 и 200 МЕ/кг инсулина в композиции 4, разбавленной 3% раствором NaHCO3, как описано в настоящем описании. Из этих мышей мыши, которые получали 50 МЕ/кг инсулина, показывают приблизительно 11% максимальное снижение уровней глюкозы в крови через 30 минут после введения относительно исходных уровней; мыши, которые получают 100 МЕ/кг инсулина, показывают приблизительно 25% максимальное снижение уровней глюкозы в крови после введения; мыши, которые получают 200 МЕ/кг инсулина, показывают приблизительно 45% максимальное снижение уровней глюкозы в крови после введения.
Подобные результаты получают у STZ-индуцированных диабетических мышей, которым с помощью перорального зонда вводят композицию 2 в 3,78% растворе NaHCO3, как показано на фигуре 4 (200 МЕ/кг инсулина). Подобные результаты также наблюдают, когда STZ-индуцированным диабетическим мышам с помощью перорального зонда вводят композицию 5 вместе с 8% раствором NaHCO3 (см. фигуру 7) или композицию 6, которая включает 2,1% NaHCO3 и 0,9% Mg(OH)2 (см. фигуру 8). У STZ-индуцированных диабетических мышей, которым перорально вводят 200 МЕ/кг инсулина в композиции 3 (десятикратное разбавление в 3,78% растворе NaHCO3), также наблюдают снижение уровня глюкозы в крови, хотя снижение относительно слабое (см. фигуру 5). С другой стороны, у STZ-индуцированных диабетических крыс Wistar, которым вводят перорально 200 МЕ/кг инсулина в композиции 4, наблюдают подобный профиль сниженного уровня глюкозы в крови (см. фиг. 9А), когда повышаются уровни инсулина в крови (см. фиг. 9В).
Композиции 7 и 8 также эффективны для быстрого снижения уровня глюкозы в крови у STZ-индуцированных диабетических крыс Wistar или STZ-индуцированных диабетических мышей после введения, как видно на фиг. 10 и 11. Кроме того, инсулин (116 МЕ/кг) в композиции 9, вводимой STZ-индуцированным диабетическим крысам Wistar, вызывает повышение концентрации инсулина в крови (см. фиг. 12), показывая эффективное всасывание инсулина из этой композиции.
Результаты экспериментов, в которых изменяют содержание NaHCO3 в композиции, вводимой STZ-индуцированным диабетическим мышам с помощью перорального зонда, показаны на фигуре 2. В интервале концентрации NaHCO3 в композиции 1,80-3,78% (мас./мас.) получают небольшое различие в уровнях глюкозы в крови, и некоторое различие в полученных уровнях глюкозы в крови можно наблюдать, когда концентрацию NaHCO3 снижают до 0,90% (мас./мас.). Такие результаты предполагают, что количество антацида, достаточное для быстрого достижения рН в желудке 3,4 (или по меньшей мере примерно 3) после перорального введения, благоприятно для биодоступности инсулина по пероральному пути. Уменьшение болюса антацида, который вводят вместе с инсулином, может ограничить общую биодоступность и сократить длительность действия инсулина. Поэтому использование различного количества антацида в композиции может титровать (или регулировать) длительность действия инсулина.
Как поясняется на фигуре 13, эффективность композиции 1, смешанной с антацидом, перорально введенной STZ-индуцированным диабетическим мышам, является неизменной, если композицию перед введением хранят в течение трех месяцев при 5 градусах по Цельсию (относительно свежеприготовленной композиции). Такие данные поясняют, что композиции, описанные в настоящем описании, подходят для хранения.
Когда немодифицированный инсулин (150 МЕ/кг) вводят здоровым собакам бигль через пероральный зонд в композиции 1, десятикратно разбавленной в 3,78% растворе NaHCO3 (общий объем 50 миллилитров), наблюдают подобные результаты, как показано на фигуре 3. На фигуре 3 быстрое начало действия инсулина иллюстрируется падением уровней глюкозы в крови у обработанных собак (незаштрихованные кружочки) через 15 минут после введения относительно необработанных собак (заштрихованные кружочки). Оказывается, что длительность действия у собак (не долее примерно 1 часа) короче, чем у мышей, возможно по причине большего продуцирования желудочной кислоты у собак относительно мышей.
Когда немодифицированный инсулин (2050 МЕ/кг) вводят здоровым крысам Wistar через пероральный зонд в композиции 4 в 3% растворе NaHCO3 (общий объем 2,5-3,3 миллилитра), получают результаты, схожие с результатами, наблюдаемыми для мышей и собак, как иллюстрирует фигура 6А. Фигура 6В показывает уровни инсулина, измеренные у таких крыс, подтверждающие, что снижающее глюкозу в крови действие может быть отнесено к повышенным уровням инсулина в крови.
Одну из двух композиций с инсулином вводят собакам бигль для измерения концентрации инсулина в крови. Первая композиция это композиция 8, описанная в настоящем описании, и ее вводят в количестве 25 МЕ на килограмм пероральным путем после суспендирования в жидкости. Вторая композиция представляет собой регулярный инсулин, вводимый подкожно (SC) в водной суспензии в количестве 0,5 МЕ на килограмм. Берут образцы крови у собак, получающих композиции, во время 0 (т.е. непосредственно перед введением) и через 15, 30, 60, 90, 120, 150, 180 и 240 минут после введения. Результаты этих исследований показаны на фигуре 15.
Все вместе результаты, показанные в примерах, описанных в настоящем описании, демонстрируют, что композиция инсулина в SMEDDS-содержащей композиции и комбинации с антацидным болюсом, достаточным для повышения рН в желудке выше примерно 3,0, быстро (т.е. в пределах 15-30 минут или меньше) придает инсулину доступность у млекопитающих и может привести к длительности действия инсулина примерно 2-4 часа. Такие данные показывают, что композиции, описанные в настоящем описании, можно эффективно применять для введения гидрофильных лекарственных средств, которые чувствительны к кислой и/или протеолитической среде в желудке, и что такие композиции можно выбрать для получения быстрого начала и ограниченной длительности действия лекарственных средств, доставляемых таким образом.
Пример 7
Быстродействующую пероральную композицию с инсулином получают, используя растворы наноэмульсии, содержащие немодифицированный инсулин (регулярный инсулин). Быстродействующая пероральная композиция с инсулином начинает проявлять свое физиологическое действие в пределах 15 минут после перорального введения, и максимальные уровни инсулина в крови имеют место в 30 минут. Длительность активности составляет менее 3 часов. Кроме того, глюкодинамические реакции пероральной композиции с инсулином пропорциональны вводимой дозе. Исследования устойчивости показывают, что эффективность и фармакологическое действие такой композиции с инсулином стабильны после хранения при 5 градусах по Цельсию в течение трех месяцев.
Инсулин обычно вводят парентеральным путем из-за протеолитического расщепления и низкой проницаемости в желудочно-кишечном тракте. Инъекции обычно болезненны и могут привести к слабой уступчивости пациента. По сравнению с неудобством и дискомфортом введения инъекцией пероральный путь приема лекарственных средств является комфортным и удобным путем введения. Описаны многие стратегии для улучшения пероральной доставки инсулина, такие как липосомы, наночастицы, микроэмульсия или капсула с энтеросолюбильным покрытием для улучшения. По меньшей мере некоторые из таких платформ обеспечивают эффективное всасывание инсулина (Wong, 2010, J. Drug Target., 18(2): 79-92; Arbit et al., 2009, J. Diabetes Sci. Technol., 3(3): 562-567). Однако ни одна из таких пероральных композиций не является быстродействующей и кратковременной по длительности действия.
Существующие экспериментальные пероральные композиции с инсулином имеют медленное начало (свыше 1,5 часов) и долгую длительность действия (свыше 5 часов). Быстродействие (начало в пределах 15 минут) и кратковременная длительность (менее 5 часов) перорально вводимого инсулина с действием, аналогичным инсулину, вводимому IV инъекцией, могли бы предоставить больший контроль метаболизма (Mannucci et al., 2009, Diabetes Obes. Metab., 11(1): 53-59). Это имеет место, поскольку соответствующая координация введения инсулина приводит к соответствию с всасыванием углеводов после приема пищи. Поэтому целью экспериментов, описанных в этом примере, является разработка быстродействующей кратковременной длительности пероральной композиции с инсулином для перорального введения для обеспечения более хорошего контроля метаболизма для больных диабетом.
В исследованиях, описанных в этом примере, используют быстродействующую пероральную композицию, включающую регулярный (т.е. немодифицированный) инсулин («композиция с инсулином»). Выполняют исследование гипогликемии in vivo для изучения действия свободного инсулина и композиции с инсулином, вводимых через пероральный зонд, на стрептозотицин-индуцированных диабетических мышей (200 МЕ/кг) и здоровых собак бигль (150 МЕ/кг). Также исследуют соразмерность дозы (дозы 50, 100 и 200 МЕ/кг) с глюкодинамическими реакциями у стрептозотицин-индуцированных диабетических мышей.
В этом исследовании также изучают устойчивость быстродействующей композиции с инсулином. Образцы композиции с инсулином хранят в закрытых навинчивающейся крышкой стеклянных флаконах при 5 градусах по Цельсию в течение трех месяцев. Лекарственное средство, остающееся в композиции, анализируют, и также оценивают эффективность in vivo путем введения композиции с инсулином стрептозотицин-индуцированным диабетическим мышам и измерения глюкозы в крови глюкометром.
Ниже описываются результаты, полученные в этом исследовании.
Уровни глюкозы в крови у стрептозотицин-индуцированных диабетических мышей после перорального введения 200 МЕ/кг свободного инсулина в растворе и быстродействующей пероральной композиции с инсулином показаны на фигуре 14. Максимальное снижение от начальной величины (примерно 45%) глюкозы в крови наблюдают через 0,5 часа после перорального зондирования композиции с инсулином. Длительность активности составляет менее трех часов. Существенных различий в уровнях глюкозы в крови в любой момент времени, когда через пероральный зонд вводят раствор свободного инсулина, не обнаруживают.
Снижение глюкозы у здоровых собак бигль после обработки быстродействующей композицией с инсулином показано на фигуре 3. Быстродействующая пероральная композиция с инсулином вызывает среднее снижение уровня глюкозы в крови (примерно на 25%) относительно контроля через 0,5 часа после дозирования. Такой результат схож с наблюдениями за гипергликемическими мышами.
Приведенная далее таблица показывает соразмерность дозы (дозы 50, 100 и 200 МЕ/кг) быстродействующей пероральной композиции с инсулином с глюкодинамическими реакциями. Среднее снижение глюкозы через 0,5 часа после дозирования показывает линейное соотношение дозы и реакции.
Оценивают эффективность in vitro композиций с инсулином, описанных в настоящем описании, после хранения при 5 градусах по Цельсию в течение трех месяцев и показывают, что 101,3±0,16% инсулина остается интактным и доступным (данные не приводятся). Эффективность in vivo оценивают путем введения композиции с инсулином диабетическим мышам с помощью перорального зонда (результаты показаны на фигуре 13). Существенного различия между гипогликемическими профилями с использованием свежеприготовленной композиции с инсулином и композиции с инсулином, хранившейся в течение трех месяцев, не имеется. Такой результат подтверждает эффективность композиции с инсулином in vitro после хранения при 5 градусах по Цельсию в течение по меньшей мере 3 месяцев.
Информация в этом примере показывает, что быстродействующая кратковременной длительности пероральная композиция с инсулином («композиция с инсулином»), описанная в этом примере, показывает соразмерность дозы при использовании доз 50, 100 и 200 МЕ/кг. Исследования как in vitro, так и in vivo не показывают существенной потери эффективности и биологической активности инсулина в композиции, хранившейся при 5 градусах по Цельсию в течение трех месяцев.
Изобретение каждого патента, заявки на патент и публикации, цитированных в настоящем описании, полностью входит в настоящее описание в качестве ссылки.
Хотя предмет настоящего изобретения раскрыт с обращением к конкретным воплощениям, очевидно, что другие воплощения и вариации могут быть разработаны другими специалистами в данной области техники без отхода от сущности и объема предмета изобретения, описанного в настоящем описании. Прилагаемая формула изобретения включает все такие воплощения и эквивалентные вариации.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ лечения гипергликемии | 2017 |
|
RU2739255C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА, СОДЕРЖАЩАЯ КОНЪЮГАТ ИНСУЛИНА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ И КОНЪЮГАТ ИНСУЛИНОТРОПНОГО ПЕПТИДА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2012 |
|
RU2606840C2 |
| ПРИМЕНЕНИЕ БЕНЗОАТА ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 2017 |
|
RU2780450C2 |
| ЖИДКАЯ КОМПОЗИЦИЯ ДЛИТЕЛЬНО ДЕЙСТВУЮЩИХ ИНСУЛИНА И ИНСУЛИНОТРОПНОГО ПЕПТИДА | 2013 |
|
RU2643766C2 |
| ЭФФЕКТОРЫ ДИПЕПТИДИЛПЕПТИДАЗУ IV | 1999 |
|
RU2309161C2 |
| ЖИДКАЯ КОМПОЗИЦИЯ ДЛИТЕЛЬНО ДЕЙСТВУЮЩЕГО КОНЪЮГАТА ИНСУЛИНА | 2013 |
|
RU2670270C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ ДИАБЕТА И СВЯЗАННЫХ С НИМ МЕТАБОЛИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2021 |
|
RU2809286C1 |
| КОМПОЗИЦИИ ДЛЯ ВВЕДЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2009 |
|
RU2554814C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2010 |
|
RU2532330C2 |
| Ацилированное производное GLP-1 | 2019 |
|
RU2773242C2 |
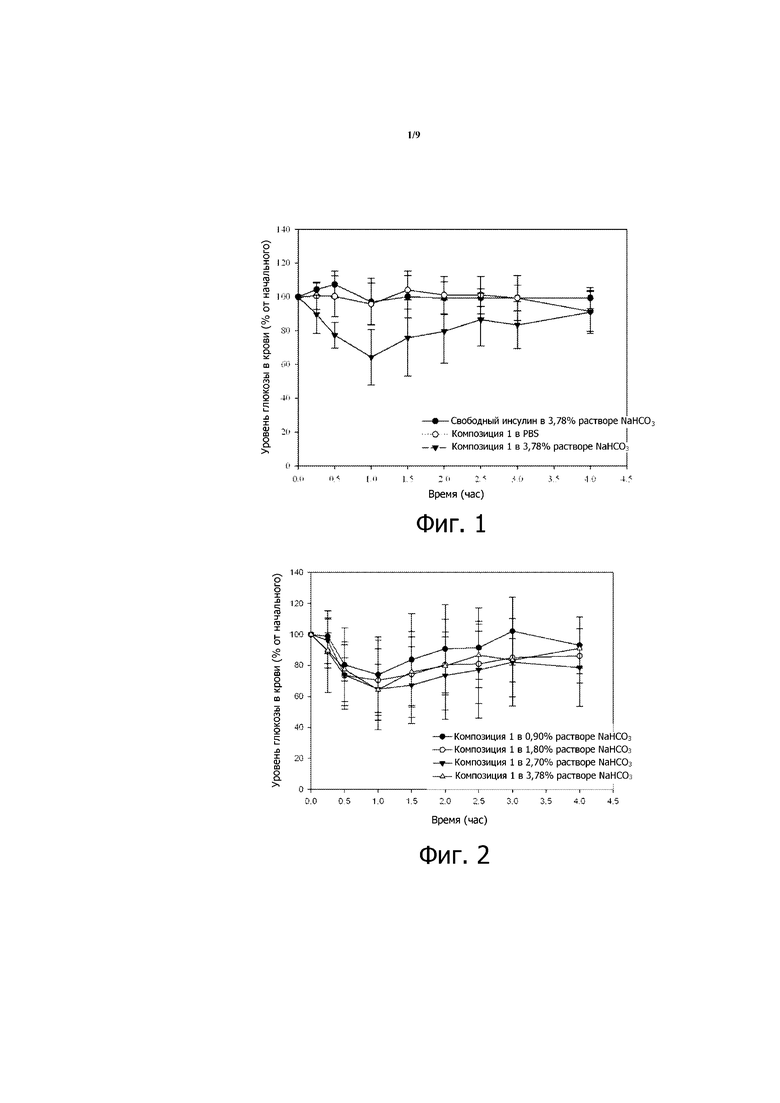

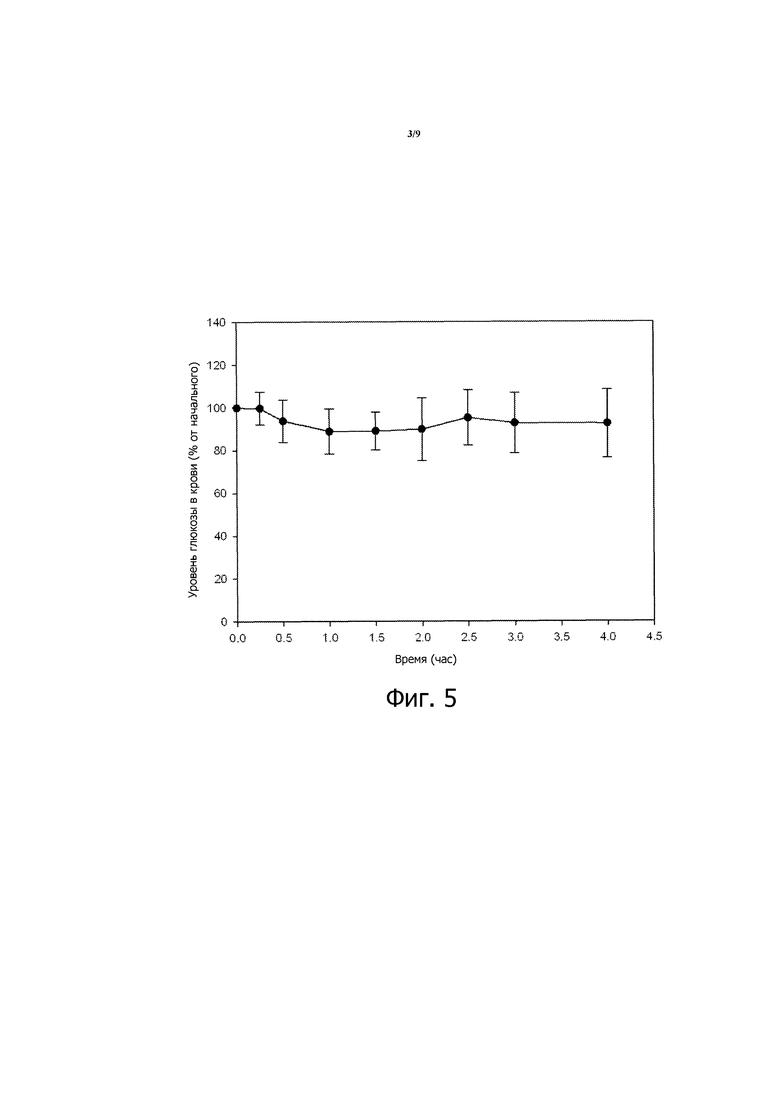
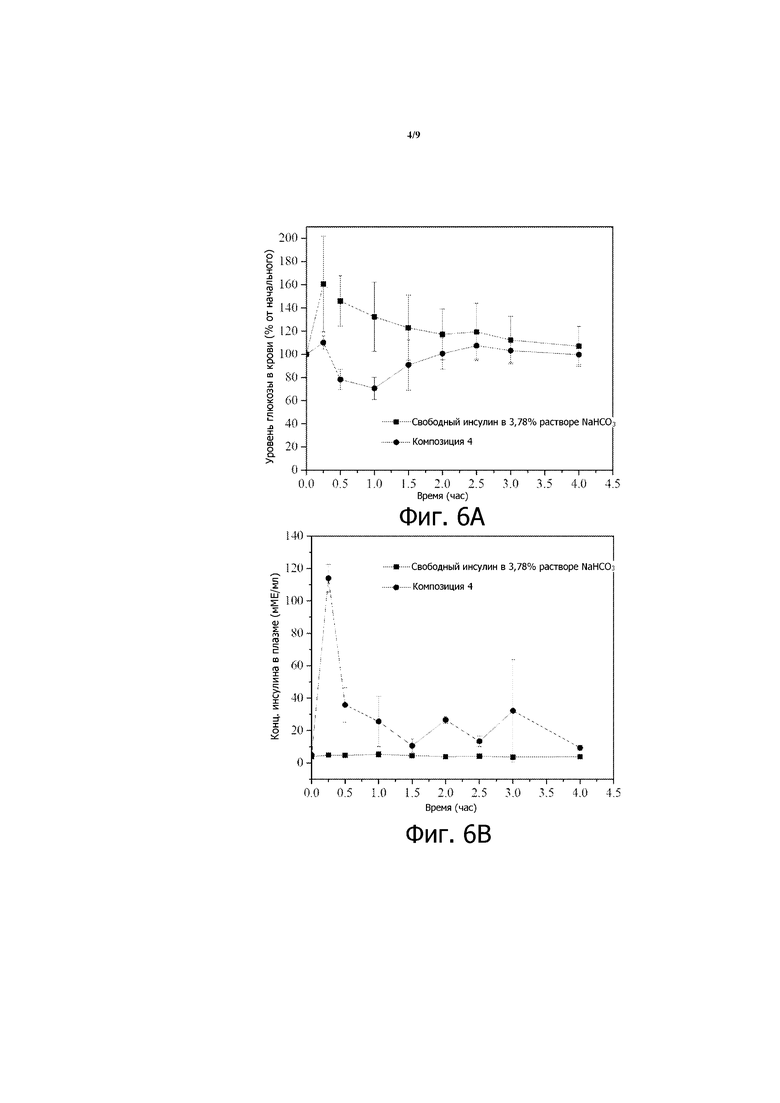
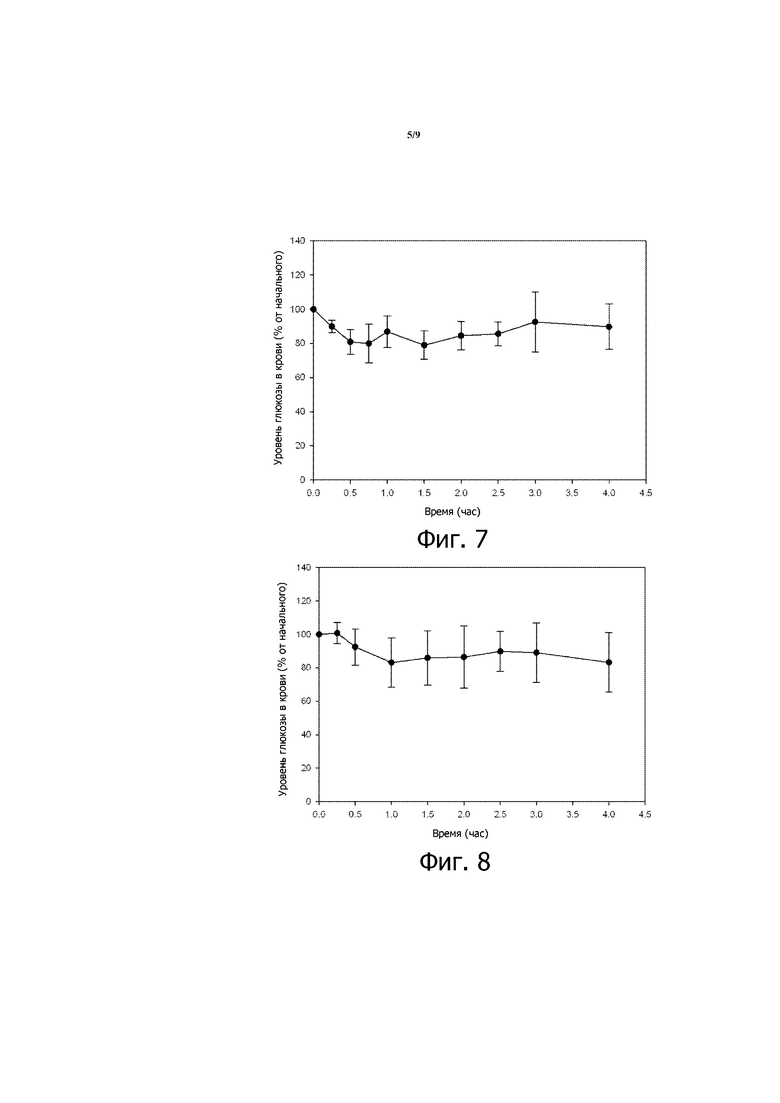
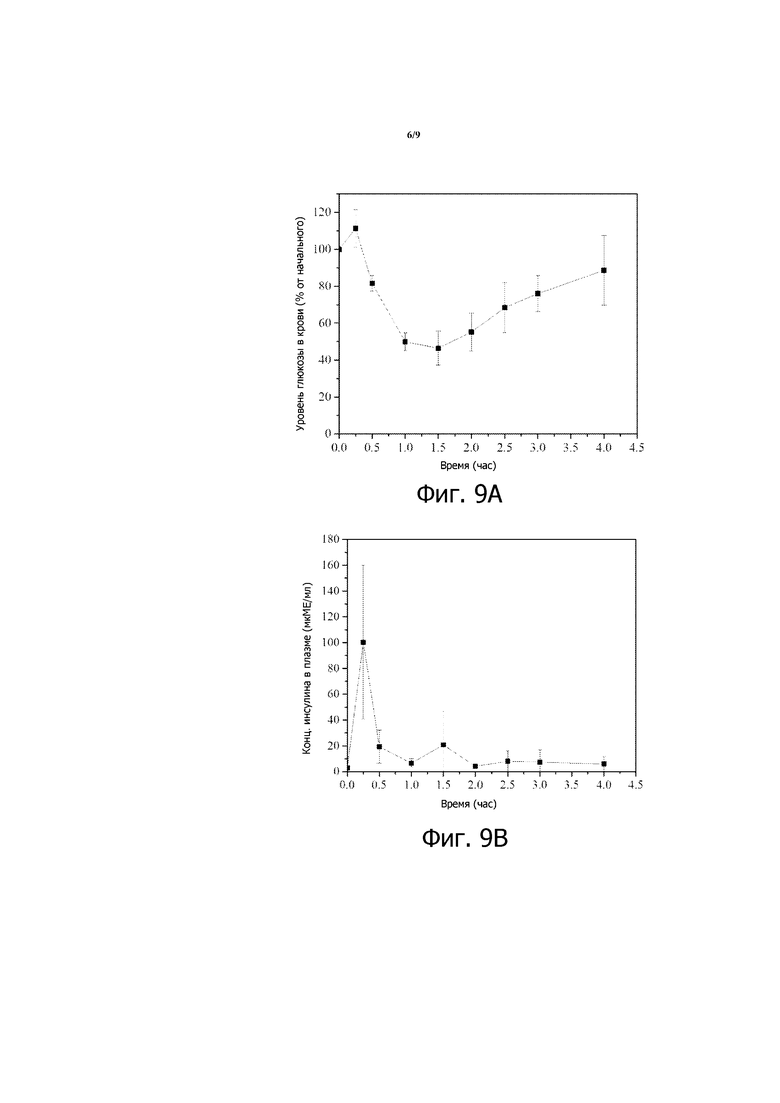
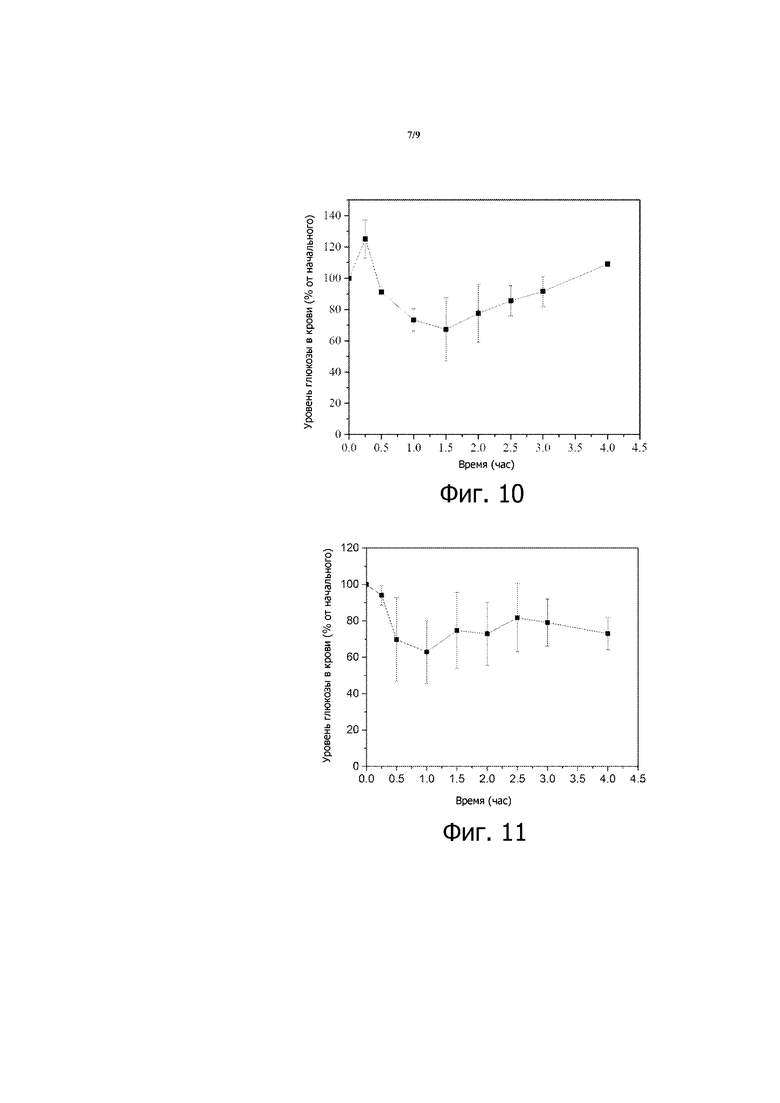
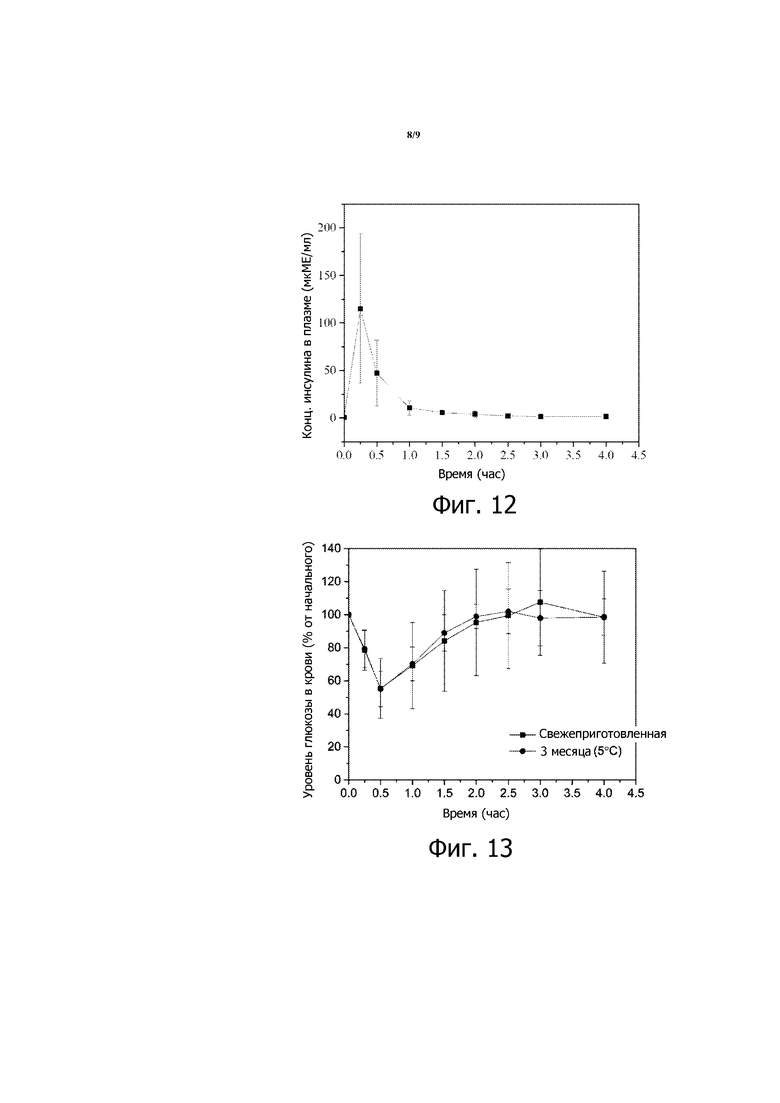
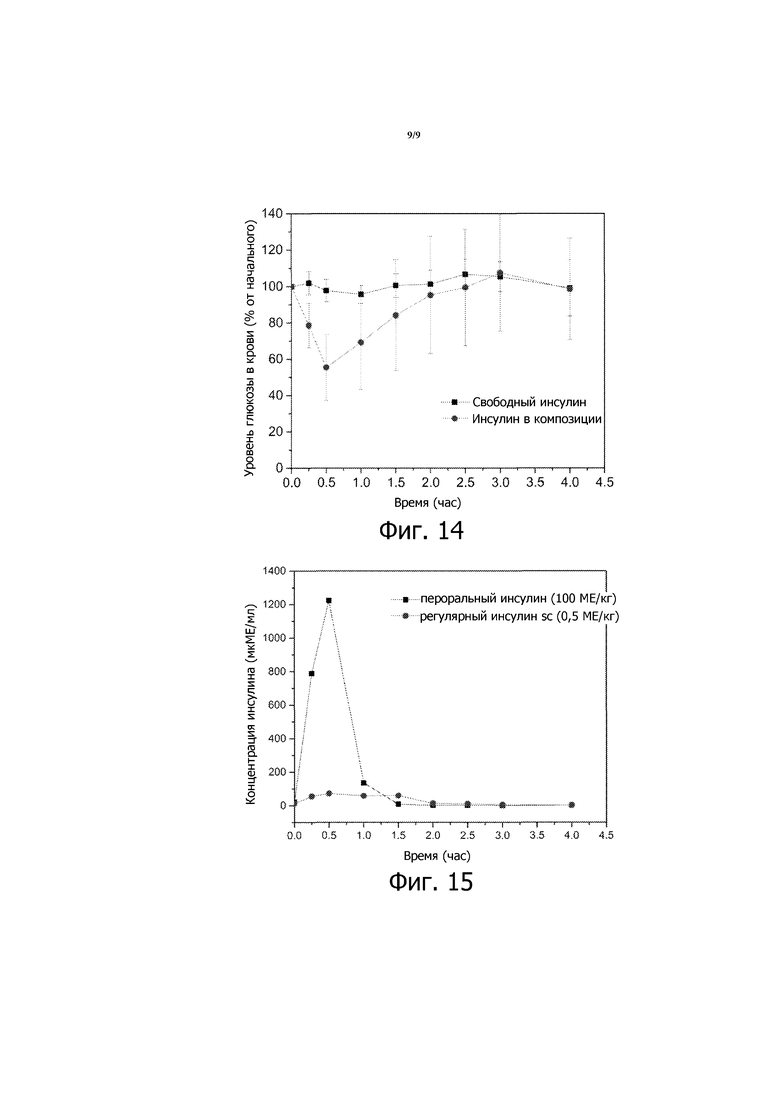
Группа изобретений относится к области медицины, а именно к пероральной лекарственной форме для быстрого снижения глюкозы в крови, которая содержит: антацид, достаточный для повышения желудочного рН млекопитающего, по меньшей мере, приблизительно до 3 после приема лекарственной формы, и комбинацию терапевтически эффективного количества лекарственного средства для быстрого снижения глюкозы в крови и одно или несколько неионогенных поверхностно-активных веществ в качестве самомикроэмульгирующейся системы доставки лекарственного средства (SMEDDS), которая индуцировала спонтанное эмульгирование при контакте между комбинацией и водной средой в условиях мягкого механического перемешивания, также относится к набору для перорального введения лекарственного средства для снижения глюкозы в крови, также относится к способу перорального введения лекарственного средства для снижения глюкозы в крови, который включает объединение терапевтически эффективного количества лекарственного средства и одного или нескольких неионогенных поверхностно-активных веществ в комбинацию в качестве самомикроэмульгирующейся системы доставки лекарственного средства (SMEDDS) так, что комбинация самопроизвольно эмульгируется при контакте с водной средой в условиях легкого механического перемешивания; смешивание комбинации, водной среды и антацида, достаточных для повышения желудочного рН млекопитающего по меньшей мере до 3, чтобы получить эмульгированную смесь; и после этого перорально вводят эмульгированную смесь млекопитающему, также относится к способу перорального введения лекарственного средства для снижения глюкозы в крови, который включает объединение терапевтически эффективного количества лекарственного средства и одного или нескольких неионогенных поверхностно-активных веществ, в комбинацию в качестве самомикроэмульгирующейся системы доставки лекарственного средства (SMEDDS) так, что комбинация самопроизвольно эмульгируется при контакте с водной средой в условиях легкого механического перемешивания; пероральное введение млекопитающему антацида, достаточного для повышения желудочного рН млекопитающего по меньшей мере до 3; и пероральное введение комбинации млекопитающему достаточно близко по времени к введению антацида, чтобы желудочный рН млекопитающего оставался по меньшей мере 3, пока вводят комбинацию. Группа изобретений обеспечивает разработку быстродействующих лекарственных композиций короткого действия. 6 н. и 38 з.п. ф-лы, 15 ил., 7 пр.
1. Пероральная лекарственная форма для быстрого снижения глюкозы в крови, которая содержит:
антацид, достаточный для повышения желудочного рН млекопитающего, по меньшей мере, приблизительно до 3 после приема лекарственной формы, и
комбинацию терапевтически эффективного количества лекарственного средства для быстрого снижения глюкозы в крови и одно или несколько неионогенных поверхностно-активных веществ в качестве самомикроэмульгирующейся системы доставки лекарственного средства (SMEDDS), которая индуцировала спонтанное эмульгирование при контакте между комбинацией и водной средой в условиях мягкого механического перемешивания.
2. Лекарственная форма по п. 1, в которой водная среда является двукратным, четырехкратным или девятикратным избытком дистиллированной воды.
3. Лекарственная форма по п. 1, в которой комбинация включает антацид.
4. Лекарственная форма по п. 1, в которой антацид является достаточным для повышения желудочного рН млекопитающего по меньшей мере приблизительно до 3,4 при приеме лекарственной формы.
5. Лекарственная форма по п. 1, в которой комбинация находится в форме первой композиции, а антацид находится в форме второй композиции.
6. Лекарственная форма по п. 5, в которой первая и вторая композиции присутствуют в отдельных частях лекарственной формы.
7. Лекарственная форма по п. 5 или 6, в которой первая композиция и вторая композиция находятся в жидкой или твердой форме.
8. Лекарственная форма по любому из пп. 1-6, в которой эмульгация образует капли среднего размера капель не больше 2000 нанометров.
9. Лекарственная форма по п. 8, в которой средний размер капель составляет не более 800 нанометров.
10. Лекарственная форма по любому из пп. 1-6,
где лекарственное средство представляет собой пептиды инсулина; или
где лекарственное средство представляет собой природные формы инсулина, синтетических инсулинов или инсулиноподобных пептидов.
11. Лекарственная форма по п. 10, в которой лекарственное средство выбрано из группы, состоящей из антротерапевтических инсулинов.
12. Лекарственная форма по п. 11, где лекарственное средство представляет собой человеческий пептид инсулина.
13. Лекарственная форма по п. 1, в которой лекарственное средство представляет собой гидрофильное лекарственное средство.
14. Лекарственная форма по любому из пп. 1-6, в которой одно или несколько неионогенных поверхностно-активных веществ включает, по меньшей мере, одно поверхностно-активное вещество, выбранное из группы, состоящей из полигликолизированных глицеридов, имеющих, по меньшей мере, один ацильный фрагмент, и сложных эфиров пропиленгликоля и жирных кислот.
15. Лекарственная форма по п. 1, в которой комбинация дополнительно содержит полиольный растворитель.
16. Лекарственная форма по п. 15, в которой полиольный растворитель выбран из группы, состоящей из глицерина, пропиленгликоля и полиэтиленгликолей, которые являются жидкими при 20°С.
17. Лекарственная форма по п. 1, в которой антацид является достаточным для повышения желудочного pH животного до по меньшей мере 3 после приема лекарственной формы.
18. Лекарственная форма по любому из пп. 1-6, в которой антацид выбран из группы, состоящей из бикарбоната натрия, гидроксида магния, карбоната кальция, гидроксида алюминия и их комбинаций.
19. Лекарственная форма по п. 1, в которой антацид является достаточным для нейтрализации, по меньшей мере, приблизительно 1 миллиэквивалента кислот желудка.
20. Лекарственная форма по любому из пп. 1-6,
где лекарственное средство представляет собой пептиды инсулина;
где одно или несколько неионогенных поверхностно-активных веществ включают
по меньшей мере один полигликолизированный глицерид, содержащий, по меньшей мере, один ацильный фрагмент, и сложные эфиры пропиленгликоля и жирных кислот;
где комбинация дополнительно включает полиоловый растворитель; и
где антацид выбран из группы, состоящей из бикарбоната натрия, гидроксида магния, карбоната кальция, гидроксида алюминия и их комбинаций.
21. Лекарственная форма по п. 20, в которой, по меньшей мере, один полигликолизированный глицерид, имеющий, по меньшей мере, одну ацильную группу и сложные эфиры пропиленгликоля и жирных кислот, выбран из группы, состоящей из полигликолизированных глицеридов, включая олеоилполиоксилглицериды (олеоилполиоксил-6 глицериды), линолеоил полиоксилглицериды (линолеоилполиоксил-6 глицериды), каприлокапроил полиоксилглицериды (PEG-6 каприловые/каприновые глицериды), каприлокапроилполиоксил-8 глицериды и лауроилполиоксилглицериды.
22. Лекарственная форма по п. 20 или 21, где лекарственная форма дополнительно включает одно или несколько дополнительных поверхностно-активных веществ.
23. Лекарственная форма по п. 22, где одно или несколько дополнительных поверхностно-активных веществ выбраны из группы, состоящей из полисорбата, полоксамеров, полиоксиэтиленовых производных касторового масла, полиоксиэтиленалкиловых эфиров, сложных эфиров сорбитана и жирных кислот, глицерилмоноолеата, глицерилмонолинолеата, среднецепочечных триглицеридов, полиглицерилолеата, лауроилполиоксилглицерида, стеароилполиоксилглицеридов и их комбинации.
24. Лекарственная форма по любому из пп. 20-23, где растворитель на основе полиола выбран из группы, состоящей из глицерина, пропиленгликоля и полиэтиленгликолей, которые являются жидкими при 20°С.
25. Лекарственная форма по любому из пп. 20-24, где лекарственная форма представляет собой стандартную лекарственную форму или многокамерную лекарственную форму.
26. Лекарственная форма по любому из пп. 20-25, где лекарственная форма включает таблетки, капсулы, порошок, гранулы, раствор и/или суспензию.
27. Лекарственная форма по любому из пп. 20-25, где лекарственная форма представляет собой по существу гомогенную композицию.
28. Набор для перорального введения лекарственного средства для снижения глюкозы в крови, содержащий лекарственную форму по любому из пп. 1-27 и аликвоту водной среды в количестве, достаточном для растворения или суспендирования антацида и для эмульгирования комбинации.
29. Набор для перорального введения лекарственного средства для снижения глюкозы в крови, включающий
первую лекарственную форму, содержащую антацид, достаточный для повышения желудочного рН млекопитающего, по меньшей мере, до 3 после приема первой лекарственной формы, и
вторую лекарственную форму, содержащую комбинацию терапевтически эффективного количества лекарственного средства и одного или нескольких неионогенных поверхностно-активных веществ, которая включает неионогенное поверхностно-активное вещество, в качестве системы самомикроэмульгирующейся доставки лекарственного средства (SMEDDS), которая вызывает спонтанное эмульгирование при контакте второй лекарственной формы и водной среды в условиях легкого механического перемешивания.
30. Способ перорального введения лекарственного средства для снижения глюкозы в крови, причем способ включает:
объединение терапевтически эффективного количества лекарственного средства и одного или нескольких неионогенных поверхностно-активных веществ в комбинацию в качестве самомикроэмульгирующейся системы доставки лекарственного средства (SMEDDS) так, что комбинация самопроизвольно эмульгируется при контакте с водной средой в условиях легкого механического перемешивания;
смешивание комбинации, водной среды и антацида, достаточных для повышения желудочного рН млекопитающего по меньшей мере до 3, чтобы получить эмульгированную смесь; и
после этого перорально вводят эмульгированную смесь млекопитающему.
31. Способ по п. 30, где лекарственное средство представляет собой пептиды инсулина; или
где лекарственное средство представляет собой природные формы инсулина, синтетических инсулинов или инсулиноподобных пептидов.
32. Способ по п. 30, в котором лекарственное средство выбрано из группы, состоящей из антротерапевтических инсулинов.
33. Способ по п. 30, в котором лекарственное средство представляет собой человеческий пептид инсулина.
34. Способ перорального введения лекарственного средства для снижения глюкозы в крови, причем способ включает:
объединение терапевтически эффективного количества лекарственного средства и одного или нескольких неионогенных поверхностно-активных веществ в комбинацию в качестве самомикроэмульгирующейся системы доставки лекарственного средства (SMEDDS) так, что комбинация самопроизвольно эмульгируется при контакте с водной средой в условиях легкого механического перемешивания;
пероральное введение млекопитающему антацида, достаточного для повышения желудочного рН млекопитающего по меньшей мере до 3; и
пероральное введение комбинации млекопитающему достаточно близко по времени к введению антацида, чтобы желудочный рН млекопитающего оставался по меньшей мере 3, пока вводят комбинацию.
35. Способ по п. 34, где лекарственное средство представляет собой пептиды инсулина; или
где лекарственное средство представляет собой природные формы инсулина, синтетических инсулинов или инсулиноподобных пептидов.
36. Способ по п. 34, в котором лекарственное средство выбрано из группы, состоящей из антротерапевтических инсулинов.
37. Способ по п. 34, где лекарственное средство представляет собой человеческий пептид инсулина.
38. Лекарственная форма быстрого снижения глюкозы в крови, включающая:
антацид, достаточный для повышения желудочного рН млекопитающего, по меньшей мере, до 3 после приема лекарственной формы,
одно или несколько неионогенных поверхностно-активных веществ, в качестве самомикроэмульгирующейся системы доставки лекарственного средства (SMEDDS), которая индуцирует самопроизвольное эмульгирование при контакте с водной средой в условиях легкого механического перемешивания, и
терапевтически эффективное количество лекарственного средства для снижения глюкозы в крови.
39. Лекарственная форма по п. 38, где лекарственное средство представляет собой пептиды инсулина; или
где лекарственное средство представляет собой природные формы инсулина, синтетических инсулинов или инсулиноподобных пептидов.
40. Лекарственная форма по п. 38 или 39, в которой лекарственная форма находится в стандартной лекарственной форме или многокамерной лекарственной форме.
41. Лекарственная форма по п. 38 или 39, где лекарственная форма включает таблетки, капсулы, порошок, гранулы, раствор и/или суспензию.
42. Лекарственная форма по п. 38 или 39, где лекарственная форма представляет собой по существу гомогенную композицию.
43. Лекарственная форма по п. 1 или 38, где лекарственная форма выбрана из группы, которую составляют:
(i) лекарственная форма, содержащая пептид инсулина, каприлокапроилполиоксил-8 глицериды, пропиленгликоль, гидрохлорид (HCl), пропиленгликоль монолаурат и полиоксиэтилен (20) сорбитана моноолеаты, в сочетании с бикарбонатом натрия (NaHCO3);
(ii) лекарственная форма, содержащая пептид инсулина, PEG-6 каприловые/каприновые глицериды, пропиленгликоль, гидрохлорид (HCl), пропиленгликоль монолаурат и полиоксиэтилен (20) сорбитана моноолеаты, в сочетании с бикарбонатом натрия (NaHCO3);
(iii) лекарственная форма, содержащая пептид инсулина, олеоилполиоксил-6 глицериды, глицерин, полиэтиленгликоль, гидрохлорид (HCl), масло касторовое гидрогенизированное PEG-40 и полиоксиэтилен (20) сорбитана моноолеаты, в сочетании с бикарбонатом натрия (NaHCO3);
(iv) лекарственная форма, содержащая пептид инсулина, каприлокапроилполиоксил-8 глицериды, глицерин и бикарбонат натрия;
(v) лекарственная форма, содержащая пептид инсулина, каприлокапроилполиоксил-8 глицериды, глицерин, бикарбонат натрия и гидроксид магния;
(vi) лекарственная форма, содержащая пептид инсулина, каприлокапроилполиоксил-8 глицериды, бикарбонат натрия и адсорбент; и
(vii) лекарственная форма, содержащая первую часть, содержащую пептид инсулина, бикарбонат натрия, маннит и повидон К-30, и вторую часть, содержащую каприлокапроилполиоксил-8 глицериды, глицерин и воду.
44. Лекарственная форма по п. 43, где лекарственная форма дополнительно содержит воду.
| US 2010273730 A1, 28.10.2010 | |||
| US 2009175959 A1, 09.07.2009 | |||
| US 3591680 A, 06.07.1971 | |||
| US 2009176691 A1, 09.07.2009 | |||
| Энциклопедический словарь медицинских терминов | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| / Гл.редактор В.И | |||
| Покровский | |||
| // М.: "Медицина" | |||
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Устройство для сцепления и расцепления конических фрикционных муфт автомобилей | 1918 |
|
SU960A1 |
| Способ получения борнеола из пихтового или т.п. масел | 1921 |
|
SU114A1 |
| Способ получения композиции инсулина для орального введения | 1988 |
|
SU1814559A3 |

