ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому пиррольному производному шестичленного гетероарильного кольца, способу его получения, лекарственной композиции, содержащей его производное, и терапевтическому агенту с его использованием и, в частности, к фармацевтическому применению в качестве ингибитора JAK и при получении иммуносупрессора.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Множество протеинкиназ составляют большое семейство киназ, которые контролируют трансдукцию различных сигналов в клетки посредством катализа. Многие заболевания связаны с нарушенными внутриклеточными ответами, вызванными регуляцией протеинкиназы, включая аутоиммунные заболевания, воспалительные заболевания, заболевания костей, метаболические заболевания, неврологические и нейродегенеративные заболевания, виды рака и сердечно-сосудистые заболевания.
Янус-киназа (JAK) представляет собой тип тирозинкиназ, включающий четыре члена JAK1, JAK2, JAK3 и TYK2. JAK играют важную роль в трансдукции сигнала множества цитокинов. JAK1, JAK2 и TYK2 широко встречаются в различных тканях и клетках, тогда как JAK3 в основном обнаруживают в лимфоцитах. JAK3 может быть специфически не ковалентно связана с обычной γ цепью (Fcγ) цитокинового рецептора, тогда как JAK1 связана с бета цепью, причем обе они активируются цитокинами IL-2 (интерлейкин-2), IL-4, IL-7, IL-9 и IL-15. JAK2 играет важную роль в сигнальном эритропоэтиновом пути (ЕРО), включающем стимуляцию дифференциации эритроцитов и активацию STAT5 (сигнальный трансдуктор и активатор транскрипции).
Сигнальный трансдуктор и активатор транскрипции (STAT) представляет собой группу белков цитоплазмы, которые могут быть связаны с ДНК в регуляторной области гена-мишени. В качестве последующих субстратов JAK STAT могут быть активированы с помощью самофосфорилирования по тирозину при стимуляции внешним сигналом, затем могут переноситься в ядро и регулировать транскрипцию генов.
Цитокин связывается с ассоциированным рецептором, что приводит к димеризации рецептора, JAK, связанные с рецептором, находятся близко друг к другу и активируются путем фосфорилирования взаимодействующих остатков тирозина. Активированные JAK катализируют фосфорилирование остатков тирозина самого рецептора, тем самым образуя соответствующие "стыковочные сайты" для связывания STAT с рецепторным комплексом. SH2 домены STAT связываются с фосфотирозиновыми остатками рецептора, и под действием JAK достигается фосфорилирование С-концевых тирозиновых остатков. Две фосфорилированные молекулы STAT взаимодействуют друг с другом с образованием гомологичных/гетерологичных димеров, которые передают молекулы рецепторов в клеточное ядро, связываются с промоторными областями гена-мишени и регулируют транскрипцию и экспрессию генов.
Многие нарушенные иммунные ответы, такие как аутоиммунные заболевания, включающие аллергии, астму, (аллогенное) отторжение трансплантата, ревматоидный артрит, боковой амиотрофический склероз и рассеянный склероз, миелопролиферативные расстройства и гематологические злокачественные новообразования, включающие лейкоз и лимфому, их модуляции связаны с сигнальным путем JAK/STAT.
Дефицит JAK3 связан с фенотипом тяжелого комбинированного иммунодефицита (SCID) как у грызунов, так и людей. Фенотип SCID млекопитающих JAK3-/- и специфическая экспрессия JAK3 в лимфоидных клетках являются двумя благоприятными свойствами, приводящими к тому, что JAK3 является мишенью для иммуносупрессанта. Т-клетки мышей с дефицитом JAK3 не могут отвечать на IL-2, а Т-клетки мышей с дефицитом JAK1 проявляют слабый ответ на IL-2. IL-2 играет решающую роль в модуляции Т-клеток, например, в том случае, когда антитело связывается с внеклеточной частью рецептора IL-2, они могут эффективно предотвращать отторжение трансплантата.
Дальнейшие исследования на животных указали на то, что JAK3 не только играют решающую роль в созревании В- и Т-лимфоцитов, но также существенны для поддержания функции Т-клеток. Модуляция иммунной активности посредством данного нового механизма может быть полезна для лечения Т-клеточного пролиферативного расстройства, такого как отторжение трансплантата и аутоиммунные заболевания.
Ингибиторы JAK-киназ, в частности, ингибиторы JAK3-киназ, могли бы препятствовать активации Т-клеток и предотвращать отторжение трансплантата после трансплантации и также могли бы предоставить благоприятный терапевтический эффект при других аутоиммунных расстройствах. JAK3 также вовлечена во многие биологические процессы. Например, было показано, что пролиферация и выживание тучных клеток мышей, индуцируемые IL-4 и IL-9, зависят от JAK3- и гамма-цепь-сигналинга (Suzuki et al., 2000, Blood 96:2172-2180). JAK3 также играет важную роль в реакциях деградации тучных клеток, опосредованной рецептором IgE (иммуноглобулин Е), ингибирование JAK3 также приводит к иммуносупрессии при отторжении трансплантата. JAK3 играет ключевую роль в IgE-рецептор-опосредованных ответах деградации тучных клеток (Malaviya et al., 1999, Biochem. Biophys. Res. Commun. 257:807-813), и, было показано, что ингибирование активности JAK3-киназ предотвращает гиперчувствительность I типа, включая анафилаксию (Malaviya et al., 1999, J. Biol. Chem. 274:27028-27038). Также было показано, что ингибирование JAK3 приводит к иммунной супрессии при отторжении аллотрансплантата (Kirken, 2001, Transpl. Proc. 33:3268-3270). JAK3-киназы также вовлечены в механизм данных заболеваний, таких как ранние и поздние стадии ревматоидного артрита; наследственный боковой амиотрофический склероз; лейкоз; фунгоидный микоз и нарушенный клеточный рост. Будучи важной протеинкиназой, JAK3 может регулировать функцию лимфоцитов, макрофагов и тучных клеток. Предполагается, что ингибиторы JAK3 включены в лечение или предупреждение заболеваний, связанных с функциями лимфоцитов, макрофагов или тучных клеток.
Для подтипов JAK2, JAK2-киназа - JAK2 V617F (мутация вызывает аномальную активность JAK2-киназы), мутация соматических клеток, при которой белковый продукт экспрессии мутантного гена приобретает новые и патологические функции, была обнаружена в классических миелопролиферативных новообразованиях, негативных по отношению к филадельфийской хромосоме (Ph), которые включают первичный тромбоцитоз, истинную полицитемию и первичный миелофиброз, так что имеется большой интерес к разработке терапий данных заболеваний, направленных на JAK2. В некоторых исследованиях было обнаружено, что у пациентов, страдающих от фиброза костного мозга, мутация JAK2-киназы происходила у более чем 50% пациентов in vivo, а симптомы, связанные с заболеванием, такие как анемия, спленомегалия и риск трансформации в острый миелоидный лейкоз (AML), были связаны с повышенной активностью и гиперактивным сигнальным путем JAK-STAT, являющимся результатом мутации в гене JAK2. В то же время, активность JAK2 была аномально повышена в целом ряде солидных опухолей и в гематологической опухоли (глиобластома, рак молочной железы, множественная миелома, рак предстательной железы, AML и т.д.). Таким образом, разработка селективного ингибитора JAK2 при терапии миелопролиферативных новообразований и лейкоза имеет огромное медицинское значение и рыночный потенциал (по имеющимся оценкам, миллиарды долларов). Недавно, селективный ингибитор JAK2, названный Ruxolitinib (INCB-018424), разработанный INCYTE при сотрудничестве с NOVARTIS, был одобрен FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов) и успешно появился на рынке. (Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, в Myelofibrosis. Srdan Verstovsek, M.D., Ph. D., Hagop Kantarjian, M.D., RubenA. Mesa, M.D, et al. N EngI J Med 2010; 363:1117-1127).
Серия ингибиторов JAK была описана в некоторых патентных заявках, включая WO 2001042246, WO 2002000661, WO 2009054941 и WO 2011013785 и т.д.
Несмотря на то, что описана серия ингибиторов JAK-киназ, обладающих действием в отношении иммунных заболеваний, остается необходимость в разработке новых соединений, обладающих лучшей эффективностью. После продолжительных попыток согласно настоящему изобретению предложены соединения формулы (I), и обнаружено, что соединения, обладающие такой структурой, проявляют превосходные эффекты и действия.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на предложение соединения формулы (I) или его фармацевтически приемлемой соли, и его таутомера, рацемата, мезомера, рацема, энантиомера, диастереомера и их смеси, и их фармацевтически приемлемой соли, а также его метаболитов, метаболических предшественников или пролекарств. Согласно настоящему изобретению предложено соединение формулы (I), имеющее следующую структуру:
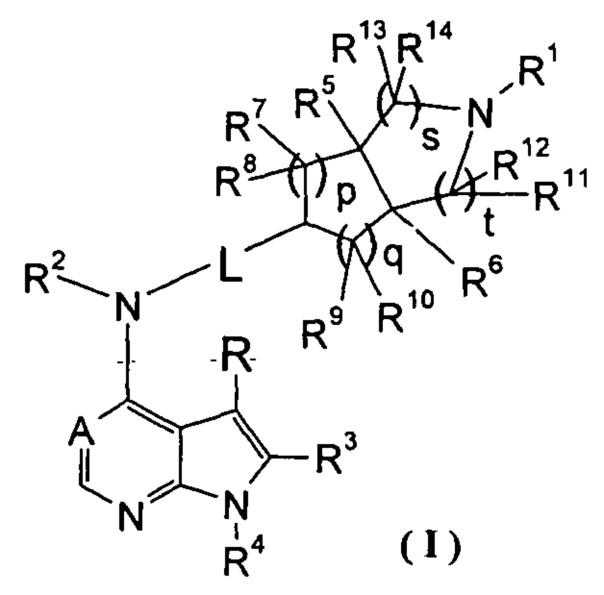 ,
,
где:
А представляет собой СН или N;
L представляет собой связь или алкил;
R1 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила, гетероарила, -(СН2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17 и -S(O)mR15, где каждый из алкила, циклоалкила, гетероциклила, арила или гетероарила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)(O)R15 и -NHS(O)mR15;
Каждый из R2 или R4 независимо выбран из группы, состоящей из водорода и алкила;
Каждый из R или R3 независимо выбран из группы, состоящей из водорода, галогена и алкила;
Каждый из R5 или R6 независимо выбран из группы, состоящей из водорода, алкила и арила, где каждый из алкила или арила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила и галогена;
Каждый из R7, R8, R9 или R10 независимо выбран из группы, состоящей из водорода, алкила, гидроксиалкила и галогена, или, R7 и R8 или R9 и R10, взятые вместе, образуют оксогруппу;
Каждый из или R11, R12, R13 или R14 независимо выбран из группы, состоящей из водорода, алкила и галогена, или, R11 и R12 или R13 и R14, взятые вместе, образуют оксогруппу;
R15 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, алкенила, алкинила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила или гетероарила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидрокси, циано, амино, нитро, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)nC(O)OR18, -OC(O)R18, -C(O)R18, -C(O)NR19R20, -NHC(O)R18, -NR19R20, -OC(O)NR19R20, -NHC(O)NR19R20, -S(O)mR18, -NHC(O)OR18 и -NHS(O)mR18;
Каждый из R16 или R17 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила или гетероарила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидрокси, циано, амино, алкокси, циклоалкила, гетероциклила, гидроксиалкила, алкинила, арила, гетероарила, карбоксила, алкоксикарбонила и -OR18;
R18 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, гидроксиалкила, арила и гетероарила;
Каждый из R19 или R20 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила;
m равен 0, 1 или 2;
n равен 0, 1 или 2;
р равен 0, 1 или 2;
q равен 0, 1 или 2;
s равен 0, 1 или 2; и
t равен 0, 1 или 2.
В предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль выбраны из соединения формулы (II) или его фармацевтически приемлемой соли:
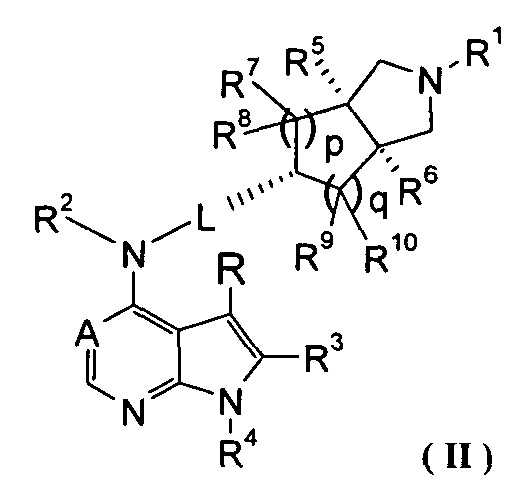 ,
,
где A, L, R, R1-R10, р и q представляют собой такие, как описано в формуле (I).
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастеремер и их смесь, и его фармацевтически приемлемая соль выбраны из соединения формулы (III) или его фармацевтически приемлемой соли:
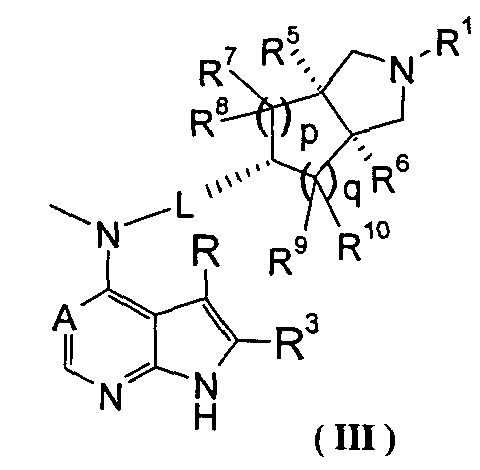 ,
,
где А, L, R, R1, R3, R5-R10, р и q представляют собой такие, как описано в формуле (I).
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль выбраны из соединения формулы (IV-a) или (IV-b), или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли:
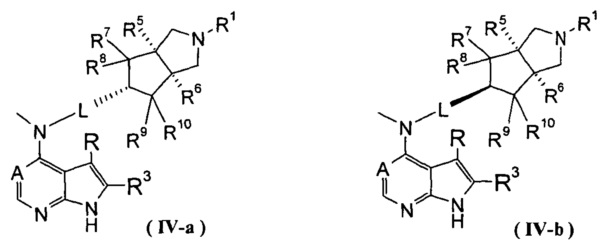 ,
,
где А, L, R, R1, R3 и R5-R10 представляют собой такие, как описано в формуле (I).
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль выбраны из соединения формулы (V-a) или (V-b) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли:
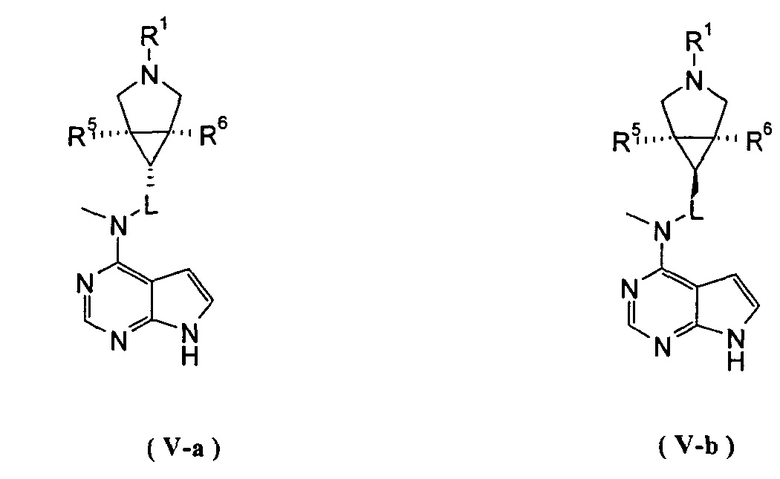 ,
,
где R1, R5-R10 и L представляют собой такие, как описано в формуле (I).
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль выбраны из соединения формулы (VI-a) или (VI-b) или его фармацевтически приемлемой соли:
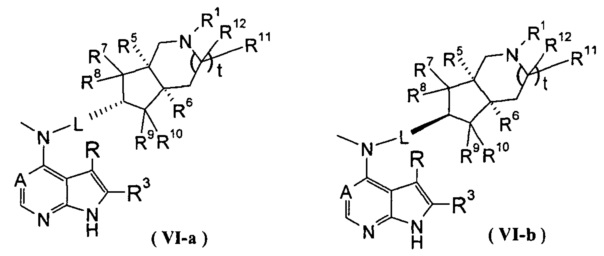 ,
,
где А, L, R, R1 и R5-R12 представляют собой такие, как описано в формуле (I).
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль выбраны из соединения формулы (VII-a) или (VII-b) или его фармацевтически приемлемой соли:
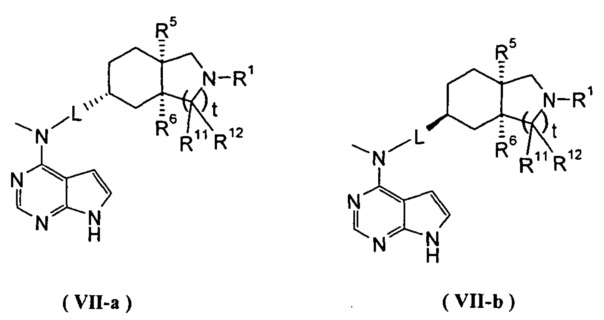 ,
,
где R1, R5, R6, R11, R12, L и t представляют собой такие, как описано в формуле (I).
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где L представляет собой связь или алкил, предпочтительно связь; указанный алкил предпочтительно представляет собой -CH2- или -СН(СН3)-.
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из алкила, гетероарила, -(CH2)nC(O)OR15, -C(O)R15, -C(O)NR16R17 и -S(O)2R15, где каждый из указанного алкила или гетероарила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидроксила, циано и -(CH2)nC(O)OR15;
R15 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, алкенила, алкинила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила или гетероарила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидрокси, циано, амино, нитро, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)nC(O)OR18, -OC(O)R18, -C(O)R18, -S(O)2R18, -NHC(O)(O)R18, -NHS(O)2R18 и -NR19R20; предпочтительно R11 выбран из группы, состоящей из алкила и циклоалкила, где каждый из указанного алкила, циклоалкила или гетероциклила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, гидроксила, циано, амино, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)nC(O)OR18, -OC(O)R18, -C(O)R18, -S(O)2R18, -NHC(O)OR18, -NHS(O)2R18 и -NR19R20.
Каждый из R16 или R17 независимо выбран из группы, состоящей из водорода, алкила и гетероарила; где указанный гетероарил возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкокси, циклоалкила, гидроксиалкила, алкинила и -OR18;
R18 выбран из группы, состоящей из водорода, алкила и гидроксиалкила;
Каждый из R19 или R20 независимо выбран из группы, состоящей из водорода и алкила;
и n равен 0, 1 или 2.
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где каждый из R5 или R6 независимо выбран из группы, состоящей из водорода и алкила, предпочтительно водорода или метила.
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где каждый из R7, R8, R9 или R10 независимо выбран из группы, состоящей из водорода, алкила и гидроксиалкила, предпочтительно водорода, метила или гидроксиметила, более предпочтительно водорода.
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер, и их смесь, и его фармацевтически приемлемая соль, где каждый из R11, R12, R13 или R14 независимо представляет собой водород.
В другом предпочтительном воплощении изобретения соединение формулы (I), или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где R11 и R12 или R13 и R14, взятые вместе, образуют оксогруппу.
В другом предпочтительном воплощении изобретения соединение формулы (I) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где А представляет собой N.
Соединения по настоящему изобретению включают все конформационные изомеры, например цис-изомеры и транс-изомеры; и все их оптические изомеры и стереоизомеры и их смеси. Соединения по настоящему изобретению имеют асимметрические центры, и, таким образом, существуют различные энантиомерные и диастереомерные изомеры. Настоящее изобретение относится к применению соединений по изобретению и фармацевтических композиций, содержащих соединения по изобретению, и его терапевтическому способу. С этой точки зрения соединения по настоящему изобретению включают Z-изомер и Е-изомер. Соединения формулы (I) могут существовать в виде таутомеров. Настоящее изобретение относится к применению всех таких таутомеров и их смесей.
Типичное соединение по настоящему изобретению или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь включают, но не ограничиваются следующим: см. таблицу А в конце описания, или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемую соль.
В другом аспекте данного изобретения предложено соединение формулы (1В) или таутомер, мезомер, рацем, энантиомер, диастереомер и их смесь, используемые в качестве промежуточного соединения для получения соединения формулы (I), где:
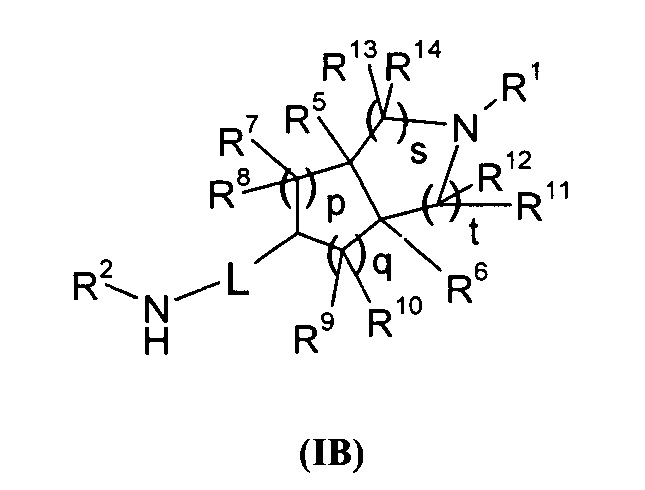
L представляет собой связь или алкил;
R1 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17 и -S(O)mR15, где каждый из алкила, циклоалкила, гетероциклила, арила или гетероарила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -(СН2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)(O)R15 и -NHS(O)mR15;
R2 выбран из группы, состоящей из водорода и алкила;
Каждый из R5 или R6 независимо выбран из группы, состоящей из водорода, алкила и арила, где каждый из алкила или арила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила и галогена;
Каждый из R7, R8, R9 или R10 независимо выбран из группы, состоящей из водорода, алкила, гидроксиалкила и галогена; или, R7 и R8 или R9 и R10, взятые вместе, образуют оксогруппу;
Каждый из R11, R12, R13 или R14 независимо выбран из группы, состоящей из водорода, алкила и галогена, или, R11 и R12 или R13 и R14, взятые вместе, образуют оксогруппу;
R15 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, алкенила, алкинила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила или гетероарила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидрокси, циано, амино, нитро, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)nC(O)OR18, -OC(O)R18, -C(O)R18, -C(O)NR19R20, -NHC(O)R18, -NR19R20, -OC(O)NR19R20, -NHC(O)NR19R20, -S(O)mR18, -NHC(O)OR18 и -NHS(O)mR18;
Каждый из R16 или R17 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила или гетероарила возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидрокси, циано, амино, алкокси, циклоалкила, гетероциклила, гидроксиалкила, алкинила, арила, гетероарила, карбоксила, алкоксикарбонила и -OR18;
R18 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, гидроксиалкила, арила и гетероарила;
Каждый из R19 или R20 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила;
m равен 0, 1 или 2;
n равен 0, 1 или 2;
р равен 0, 1 или 2;
q равен 0, 1 или 2;
s равен 0, 1 или 2; и
t равен 0, 1 или 2.
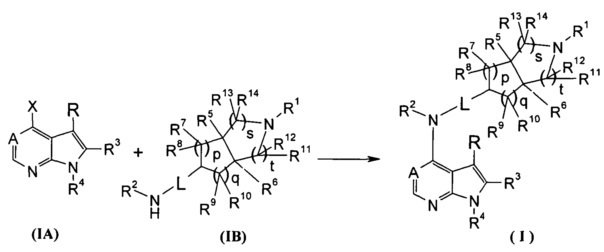
Согласно другому аспекту данного изобретения предложен способ получения соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, включающий стадии:
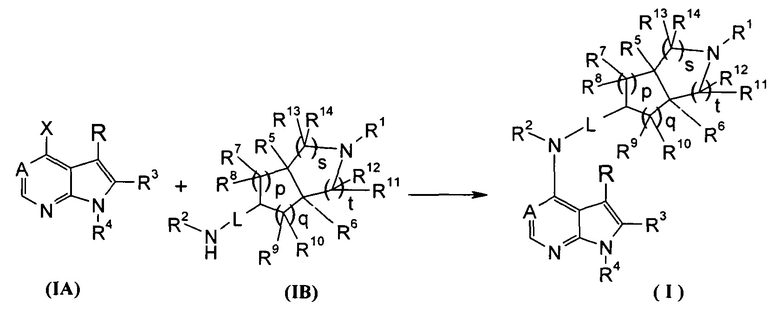
в щелочных условиях, при этом соединения формулы (IA) реагируют с соединениями формулы (IB) с получением соединений формулы (I);
щелочные условия обеспечивают при помощи органического основания и неорганического основания, где указанное органическое основание включает триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, трет-бутил калия алкоксид, тетрабутиламмония бромид, но не ограничивается ими, и указанное неорганическое основание включает гидрид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, но не ограничивается ими;
где Х представляет собой галоген, A, L, R, R1-R14, p, q, s и t представляют собой такие, как определено в формуле (I); предпочтительно, R1 представляет собой трет-бутоксикарбонил.
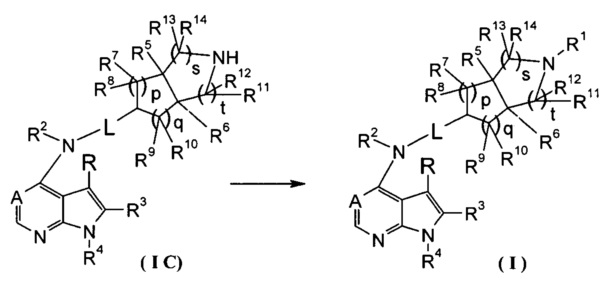
Согласно другому аспекту данного изобретения предложен способ получения соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, включающий стадии:
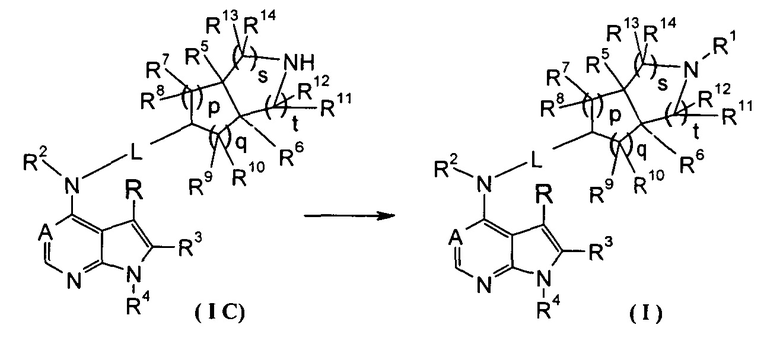
в щелочных условиях, при этом соединение формулы (IC) или его фармацевтически приемлемая соль реагирует с карбоновой кислотой, хлорангидридом карбоновой кислоты, сульфонилхлоридом, сложными эфирами карбоновой кислоты, производным этиленоксида или галогенидом с получением соединений формулы (I);
где: в том случае, когда R1 представляет собой трет-бутоксикарбонил, тогда трет-бутоксикарбонил дополнительно возможно удаляют из соединения формулы (I) с получением соединения формулы (IC) или его фармацевтически приемлемой соли;
реакционный растворитель включает: тетрагидрофуран, этанол, метанол, н-бутанол, дихлорметан, 1,4-диоксан или N,N-диметилформамид, но не ограничивается ими;
щелочные условия обеспечивают при помощи органического основания и неорганического основания, где указанное органическое основание включает триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, трет-бутил калия алкоксид, но не ограничивается ими, и указанное неорганическое основание включает гидрид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, но не ограничивается ими;
где A, L, R, R1-R14, p, q, s и t представляют собой такие, как определено в формуле (I).
Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли вместе с фармацевтически приемлемым носителем или эксципиентом.
Настоящее изобретение также относится к применению соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или содержащей их фармацевтической композиции при получении лекарственного средства для ингибирования JAK-киназы; предпочтительно ингибирования JAK1, JAK2 или JAK3.
Настоящее изобретение также относится к применению соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или содержащей их фармацевтической композиции при получении лекарственного средства для ингибирования JAK-киназы; где лекарственное средство возможно содержит дополнительно один или более чем один реагент для регуляции иммунной системы млекопитающего, противораковые агенты или противовоспалительные агенты.
Настоящее изобретение также относится к применению соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или содержащей их фармацевтической композиции при получении лекарственного средства для ингибирования JAK-киназы; где лекарственное средство полезно для лечения или предупреждения следующих расстройств или заболеваний: заболевания иммунной системы, включающие такие, как отторжение трансплантированных органов (например, отторжение аллотрансплантата и реакция «хозяин против трансплантата»); аутоиммунные заболевания, включающие такие, как волчанка, рассеянный склероз, ревматоидный артрит, болезнь Стилла, псориаз, неспецифический язвенный колит, болезнь Крона, аутоиммунное заболевание щитовидной железы и т.д.; заболевания кожи, включающие такие, как болезнь, сопровождающуюся зудом, высыпание, атопический дерматит и т.д.; аллергические расстройства, включающие такие, как астма, ринит и т.д.; вирусные заболевания, включающие такие, как гепатит В, гепатит С, вирус ветряной оспы и т.д.; сахарный диабет I типа и осложнения, связанные с диабетом; болезнь Альцгеймера; сухость глаз; фиброз костного мозга; тромбоцитоз; полицитемия или лейкоз; виды рака, включая, например, солидные опухоли (такие как рак предстательной железы, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак молочной железы, рак легкого, рак головы и шеи, рак щитовидной железы, глиобластома, меланома и т.д.), рак крови (такой как лимфома, лейкоз и т.д.), рак кожи (такой как Т-клеточная лимфома кожи, В-клеточная лимфома кожи) и т.д.
Настоящее изобретение также относится к применению соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или содержащей их фармацевтической композиции при получении лекарственного средства для ингибирования JAK-киназы; где лекарственное средство возможно содержит дополнительно один или более чем один реагент для регуляции иммунной системы млекопитающего, противораковые агенты или противовоспалительные агенты, где указанное лекарственное средство представляет собой средство для лечения или предупреждения следующих расстройств или заболевания: заболевания иммунной системы, включая такие, как отторжение трансплантированных органов (например, отторжение аллотрансплантата и реакция «хозяин против трансплантата»); аутоиммунные заболевания, включая такие, как волчанка, рассеянный склероз, ревматоидный артрит, болезнь Стилла, псориаз, неспецифический язвенный колит, болезнь Крона, аутоиммунное заболевание щитовидной железы и т.д.; заболевания кожи, включая такие, как болезнь, сопровождающуюся зудом, высыпание, атопический дерматит, и т.д.; аллергические расстройства, включая такие, как астма, ринит, и т.д.; вирусные заболевания, включая такие, как гепатит В, гепатит С, вирус ветряной оспы и т.д.; сахарный диабет I типа и осложнения, связанные с диабетом; болезнь Альцгеймера; сухость глаз; фиброз костного мозга; тромбоцитоз; полицитемия или лейкоз; виды рака, включая, например, солидные опухоли (такие как рак предстательной железы, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак молочной железы, рак легкого, рак головы и шеи, рак щитовидной железы, глиобластома, меланома и т.д.), рак крови (такой как лимфома, лейкоз и т.д.), рак кожи (такой как Т-клеточная лимфома кожи, В-клеточная лимфома кожи), и т.д. Указанное млекопитающее представляет собой человека.
Настоящее изобретение также относится к соединению формулы (I) или таутомеру, мезомеру, рацему, энантиомеру, диастереомеру и их смеси, и его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в качестве лекарственного средства для ингибирования JAK-киназы. Указанная JAK-киназа предпочтительно выбрана из группы, состоящей из JAK1, JAK2 и JAK3.
Настоящее изобретение также относится к применению соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или содержащей их фармацевтической композиции для применения в качестве лекарственного средства, комбинированного с дополнительным одним или более чем одним реагентом для регуляции иммунной системы млекопитающего, противораковыми агентами или противовоспалительными агентами.
Настоящее изобретение также относится к соединению формулы (I) или таутомеру, мезомеру, рацему, энантиомеру, диастереомеру и их смеси, и его фармацевтически приемлемой соли или содержащей их фармацевтической композиции для применения в качестве лекарственного средства для лечения или предупреждения следующих расстройств или заболеваний: заболевания иммунной системы, включая такие, как отторжение трансплантированных органов (например, отторжение аллотрансплантата и реакция «хозяин против трансплантата»); аутоиммунные заболевания, включая такие, как волчанка, рассеянный склероз, ревматоидный артрит, болезнь Стилла, псориаз, неспецифический язвенный колит, болезнь Крона, аутоиммунное заболевание щитовидной железы и т.д.; заболевания кожи, включая такие, как болезнь, сопровождающуюся зудом, высыпание, атопический дерматит и т.д.;
аллергические расстройства, включая такие, как астма, ринит и т.д.; вирусные заболевания, включая такие, как гепатит В, гепатит С, вирус ветряной оспы и т.д.; сахарный диабет I типа и осложнения, связанные с диабетом; болезнь Альцгеймера; сухость глаз; фиброз костного мозга; тромбоцитоз; полицитемия или лейкоз; виды рака, включая, например, солидные опухоли (такие как рак предстательной железы, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак молочной железы, рак легкого, рак головы и шеи, рак щитовидной железы, глиобластома, меланома и т.д.), рак крови (такой как лимфома, лейкоз и т.д.), рак кожи (такой как Т-клеточная лимфома кожи, В-клеточная лимфома кожи) и т.д.
Настоящее изобретение также относится к соединению формулы (I) или таутомеру, мезомеру, рацему, энантиомеру, диастереомеру и их смеси, и его фармацевтически приемлемой соли или содержащей их фармацевтической композиции, дополнительно комбинированным с одним или более чем одним реагентом для регуляции иммунной системы млекопитающего, противораковыми агентами или противовоспалительными агентами, для применения в качестве лекарственного средства для лечения или предупреждения следующих расстройств или заболеваний: заболевания иммунной системы, включая такие, как отторжение трансплантированных органов (например, отторжение аллотрансплантата и реакция «хозяин против трансплантата»); аутоиммунные заболевания, включая такие, как волчанка, рассеянный склероз, ревматоидный артрит, болезнь Стилла, псориаз, неспецифический язвенный колит, болезнь Крона, аутоиммунное заболевание щитовидной железы и т.д.; заболевания кожи, включая такие, как болезнь, сопровождающуюся зудом, высыпание, атопический дерматит и т.д.; аллергические расстройства, включая такие, как астма, ринит и т.д.; вирусные заболевания, включая такие, как гепатит В, гепатит С, вирус ветряной оспы и т.д.; сахарный диабет 1 типа и осложнения, связанные с диабетом; болезнь Альцгеймера; сухость глаз; фиброз костного мозга; тромбоцитоз; полицитемия или лейкоз; виды рака, включая, например, солидные опухоли (такие как рак предстательной железы, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак молочной железы, рак легкого, рак головы и шеи, рак щитовидной железы, глиобластома, меланома и т.д.), рак крови (такой как лимфома, лейкоз и т.д.), рак кожи (такой как Т-клеточная лимфома кожи, В-клеточная лимфома кожи) и т.д.
Другими словами, настоящее изобретение относится к способу ингибирования JAK-киназы, который включает стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединений формулы (I) или таутомеров, рацематов, энантиомеров, диастереомеров и их смесей, и их фармацевтически приемлемых солей или содержащей их фармацевтической композиции. Кроме того, соединения формулы (I) или таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, и их фармацевтически приемлемые соли или содержащая их фармацевтическая композиция могут быть дополнительно комбинированы с одним или более чем одним реагентом для регуляции иммунной системы млекопитающего, противораковыми агентами или противовоспалительными агентами.
Настоящее изобретение относится к способу лечения или предупреждение расстройств или заболеваний иммунной системы, включающему стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или содержащей их фармацевтической композиции; где указанные расстройства или заболевания иммунной системы включают такие, как отторжение трансплантированных органов (например, отторжение аллотрансплантата и реакция «хозяин против трансплантата»); аутоиммунные заболевания, включающие такие, как волчанка, рассеянный склероз, ревматоидный артрит, болезнь Стилла, псориаз, неспецифический язвенный колит, болезнь Крона, аутоиммунное заболевание щитовидной железы и т.д.; заболевания кожи, включающие такие, как болезнь, сопровождающуюся зудом, высыпание, атопический дерматит и т.д.; аллергические расстройства, включающие такие, как астма, ринит и т.д.; вирусные заболевания, включающие такие, как гепатит В, гепатит С, вирус ветряной оспы и т.д.; сахарный диабет I типа и осложнения, связанные с диабетом; болезнь Альцгеймера; сухость глаз; фиброз костного мозга; тромбоцитоз; полицитемия или лейкоз; виды рака, включающие, например, солидные опухоли (такие как рак предстательной железы, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак молочной железы, рак легкого, рак головы и шеи, рак щитовидной железы, глиобластома, меланома и т.д.), рак крови (такой как лимфома, лейкоз и т.д.), рак кожи (такой как Т-клеточная лимфома кожи, В-клеточная лимфома кожи) и т.д.
Настоящее изобретение также относится к способу лечения или предупреждения расстройств или заболеваний иммунной системы, включающему стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или таутомера, мезомера, рацема, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или содержащей их фармацевтической композиции, и дополнительно одного или более чем одного реагента для регуляции иммунной системы млекопитающего, противораковых агентов или противовоспалительных агентов, где указанные расстройства или заболевания иммунной системы включают такие, как отторжение трансплантированных органов (например, отторжение аллотрансплантата и реакция «хозяин против трансплантата»); аутоиммунные заболевания, включающие такие, как волчанка, рассеянный склероз, ревматоидный артрит, болезнь Стилла, псориаз, неспецифический язвенный колит, болезнь Крона, аутоиммунное заболевание щитовидной железы и т.д.; заболевания кожи, включающие такие, как псориаз, высыпание, атопический дерматит и т.д.; аллергические расстройства, включающие такие, как астма, ринит и т.д.; вирусные заболевания, включающие такие, как гепатит В, гепатит С, вирус ветряной оспы и т.д.; сахарный диабет I типа и осложнения, связанные с диабетом; болезнь Альцгеймера; сухость глаз; фиброз костного мозга; тромбоцитоз; полицитемия или лейкоз; виды рака, включающие например, солидные опухоли (такие как рак предстательной железы, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак молочной железы, рак легкого, рак головы и шеи, рак щитовидной железы, глиобластома, меланома и т.д.), рак крови (такой как лимфома, лейкоз и т.д.), рак кожи (такой как Т-клеточная лимфома кожи, В-клеточная лимфома кожи) и т.д.
Композиции по данному изобретению могут быть приготовлены обычным образом с использованием одного или более чем одного фармацевтически приемлемого носителя. Таким образом, активные соединения по данному изобретению могут быть приготовлены в виде различных лекарственных форм для перорального, буккального введения, интраназального, парентерального (например, внутривенного, внутримышечного или подкожного) или ректального введения, или введения путем ингаляции или вдувания. Соединения по данному изобретению могут быть также приготовлены в виде лекарственных форм с длительным высвобождением.
Для перорального введения фармацевтические композиции, например, могут быть приготовлены в виде таблеток или капсул с фармацевтически приемлемыми эксципиентами с помощью традиционных способов, где эксципиенты включают такие, как связывающие агенты (например, крахмал, желатин, поливинилпирролидон или аравийская камедь) и наполнители (например, лактоза, микрокристаллическая целлюлоза или фосфат кальция), смазывающие вещества (например, стеарат магния, тальк или диоксид кремния), разрыхлители (например, картофельный крахмал или натрия крахмал гликолят) или увлажнители (такие как лаурилсульфат натрия). Таблетки могут быть покрыты способами, хорошо известными в данной области техники. Жидкие препараты для перорального введения могут представлять собой растворы, сиропы или суспензии, или могут представлять собой сухой продукт для разведения водой или другим подходящим носителем перед применением. Такие жидкие препараты могут быть приготовлены традиционными средствами с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сорбитный сироп, метилцеллюлоза или гидрированные пищевые жиры), эмульгаторы (например, лецитин или аравийская камедь), неводные носители (например, миндальное масло, масляные сложные эфиры или этанол) и консерванты (например, метил или пропил пара-гидроксибензоат).
Для буккального введения композиции могут быть приготовлены в виде таблеток или пастилок обычным образом.
Активные соединения по данному изобретению могут быть приготовлены для парентерального введения путем инъекции, включая использование обычных способов катетеризации или инфузии. Инъекция может быть представлена в стандартной лекарственной форме, например, в ампулах или в многоемкостных контейнерах с добавлением консерванта. Композиции могут представлять собой суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать препаратообразующие агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. В качестве альтернативы, активный ингредиент может быть представлен в форме порошка для разведения перед применением подходящим носителем, например, стерильной апирогенной водой.
Активные соединения по данному изобретению также могут быть приготовлены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основания суппозиториев, такие как масло какао или другие глицериды.
Для интраназального введения или введения путем ингаляции активные соединения по настоящему изобретению обычно доставляют в форме раствора или суспензии, высвобождаемой из контейнера с пульверизатором, который сжимается или нагнетается пациентом, или в виде аэрозольного спрея, высвобождаемого из контейнера под давлением или распылителя, с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением стандартная доза может быть определена с помощью клапана, высвобождающего отмеренное количество. Контейнер под давлением или распылитель может содержать раствор или суспензию активного соединения. Капсулы или картриджи (например, изготовленные из желатина) для применения в ингаляторе или инсуффляторе могут быть приготовлены содержащими порошкообразную смесь по настоящему изобретению и подходящее порошковое основание, такое как лактоза или крахмал.
Соединения по данному изобретению могут быть введены в фармацевтически приемлемой лекарственной форме или отдельно, или в комбинации с одним или более чем одним агентом для регуляции иммунной системы млекопитающего или противовоспалительными агентами. Данные агенты могут включать циклоспорин А (такой как Sandimmune® или Neroal®), рапамицин, FK-506 (такролимус), лефлуномид, дезоксиспергуалин, соли микофенолата (например, Cellcept®), азатиоприн (например, Imuran®), даклизумаб (например, Zenapax®), OKT3 (например, Orthoclone®), AcGam, аспирин, ацетаминофен, ибупрофен, напроксен, мелоксикам пиррол и противовоспалительные стероиды (такие как преднизон или дексаметазон), но не ограничиваются ими. Данные агенты могут быть введены как часть одной и той же или отдельных лекарственных форм одним и тем же или разными путями введения согласно стандартной фармацевтической практике, основанной на одинаковых или различных схемах введения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
"Алкил" относится к насыщенной алифатической углеводородной группе, включающей группы C1-C20 с прямой цепью и разветвленной цепью. Предпочтительно алкильная группа представляет собой алкил, имеющий от 1 до 12 атомов углерода. Типичные примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втop-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-ди метил гексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этил гексил, 2-метил-3-этил гексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил, и их изомеры с разветвленной цепью, но не ограничиваются ими. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода. Типичные примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, emop-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.д., но не ограничиваются ими. Алкильная группа может быть замещенной или незамещенной. Будучи замещенной, группа(пы) заместителя(ей) может(гут) быть замещена(ны) в любой свободной точке присоединения, предпочтительно, группа(пы) заместителя(ей) представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкиксоила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкиоксила, гетероциклического алкиоксила, циклоалкилтио, гетероциклического алкилтио, оксогруппы, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)OR15 и -NHS(O)mR15.
"Алкенил" относится к алкилу, определенному выше, имеющему по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную двойную связь, например, винилу, 1-пропенилу, 2-пропенилу, 1-, 2-или 3-бутенилу и т.д. Алкенильная группа может быть замещенной или незамещенной. Будучи замещенной, группа(пы) заместителя(ей) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкиксоила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкиоксила, гетероциклического алкиоксила, циклоалкилтио, гетероциклического алкилтио, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)OR15 и -NHS(O)mR15.
"Алкинил" относится к алкилу, определенному выше, имеющему по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную тройную связь, например, этинилу, 1-пропинилу, 2-пропинилу, 1-, 2-или 3-бутинилу и т.д., предпочтительно алкинилу С2-10, более предпочтительно алкинилу С2-6 и наиболее предпочтительно алкинилу С2-4. Алкинильная группа может быть замещенной или незамещенной. Будучи замещенной, группа(пы) заместителя(ей) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкиксоила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкиоксила, гетероциклического алкиоксила, циклоалкилтио, гетероциклического алкилтио, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)OR15 и -NHS(O)mR15.
"Циклоалкил" относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе и имеет от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода, и наиболее предпочтительно от 3 до 8 атомов углерода или от 3 до 6 атомов углерода. Типичные примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.д., но не ограничиваются ими. Полициклический циклоалкил включает циклоалкил, имеющий спирокольцо, конденсированное кольцо или связанное мостиковыми связями кольцо.
"Спироциклоалкил" относится к 5-20-членной полициклической группе с кольцами, связанными посредством одного общего атома углерода (названного спироатомом), где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не обладает полностью конъюгированной системой пи-электронов. Предпочтительно, спироциклоалкил является 6-14-членным, более предпочтительно, 7-10-членным. Согласно числу общих спироатомов спироциклоалкил делится на моно-спироциклоалкил, ди-спироциклоалкил или поли-спироциклоалкил, предпочтительно относится к моно-спироциклоалкилу или ди-спироциклоалкилу, более предпочтительно 4-членному/4-членному, 4-членному/6-членному, 4-членному/6-членному, 5-членному/5-членному или 5-членному/6-членному моно-спироциклоалкилу. Типичные примеры спироциклоалкила включают, но не ограничиваются следующими группами:
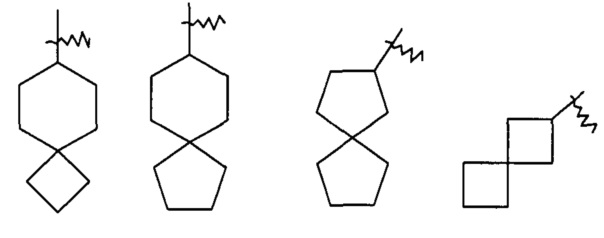 и
и  .
.
"Конденсированный циклоалкил" относится к 5-20-членной полициклической углеводородной группе, где каждое кольцо в системе имеет с другим кольцом общую соседнюю пару атомов углерода, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не обладает полностью конъюгированной системой пи-электронов. Предпочтительно, конденсированная циклоалкильная группа является 6-14-членной, более предпочтительно 7-10-членной. Согласно числу членных колец конденсированный циклоалкил делится на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, предпочтительно относится к бициклическому или трициклическому конденсированному циклоалкилу, более предпочтительно, 5-членному/5-членному или 5-членному/6-членному бициклическому конденсированному циклоалкилу. Типичные примеры конденсированного циклоалкила включают, но не ограничиваются следующими группами:
 и
и  .
.
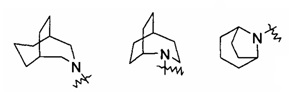
"Связанный мостиковыми связями циклоалкил" относится к 5-20-членной полициклической углеводородной группе, где каждые два кольца в системе имеют общими два несвязанных атома углерода. Указанные кольца могли бы иметь одну или более чем одну двойную связь, но не обладают полностью конъюгированной системой пи-электронов. Предпочтительно связанный мостиковыми связями циклоалкил является 6-14-членным, более предпочтительно 7-10-членным. Согласно числу членных колец связанный мостиковыми связями циклоалкил делится на бициклический, трициклический, тетрациклический или полициклический связанный мостиковыми связями циклоалкил, предпочтительно относится к бициклическому, трициклическому или тетрациклическому связанному мостиковыми связями циклоалкилу, более предпочтительно, бициклическому или трициклическому связанному мостиковыми связями циклоалкилу. Типичные примеры связанного мостиковыми связями циклоалкила включают, но не ограничиваются следующими группами:
 и
и  .
.
Указанный циклоалкил может быть конденсирован с арилом, гетероарилом или кольцом гетероциклического алкила, где кольцо, связанное с первичной структурой, представляет собой циклоалкил. Типичные примеры включают инданилуксусную группу, тетрагидронафталин, бензоцидогептил и т.п., но не ограничиваются ими. Указанный циклоалкил может быть возможно замещен или незамещен. Будучи замещенной, группа(пы) заместителя(ей) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкиксоила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкиоксила, гетероциклического алкиоксила, циклоалкилтио, гетероциклического алкилтио, оксогруппы, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)OR15 и -NHS(O)mR15.
Тетероциклил" относится к 3-20-членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более чем один гетероатом, выбранный из группы, состоящей из N, О, и S(O)m (где m равен 0, 1 или 2) в качестве кольцевых атомов, но за исключением -O-O-, -O-S- или -S-S- в кольце, причем оставшиеся кольцевые атомы представляют собой С. Предпочтительно, гетероциклил является 3-12-членным, имея от 1 до 4 указанных гетероатомов; более предпочтительно, 3-10-членным, имея от 1 до 3 указанных гетероатомов; наиболее предпочтительно, 5-6-членным, имея от 1 до 2 указанных гетероатомов. Типичные примеры моноциклического гетероциклила включают пирролидил, пиперидил, пиперазинил, морфолинил, сульфоморфолинил, гомопиперазинил и т.п., но не ограничиваются ими. Полициклический гетероциклил включает гетероциклил, имеющий спирокольцо, конденсированное кольцо или связанное мостиковыми связями кольцо.
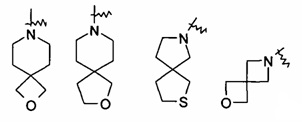
"Спиро-гетероциклил" относится к 5-20-членному полициклическому гетероциклилу с кольцами, связанными посредством одного общего атома углерода (названного как спироатом), где указанные кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О, и S(O)m (где m равен 0, 1 или 2) в качестве кольцевых атомов, причем оставшиеся кольцевые атомы представляют собой С, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не обладает полностью конъюгированной системой пи-электронов. Предпочтительно, спиро-гетероциклил является 6-14-членным, более предпочтительно 7-10-членным. Согласно числу общих спироатомов спиро-гетероциклил делится на моно-спиро-гетероциклил, ди-спиро-гетероциклил или поли-спиро-гетероциклил, предпочтительно относится к моно-спиро-гетероциклилу или ди-спиро-гетероциклилу, более предпочтительно 4-членному/4-членному, 4-членному/5-членному, 4-членному/6-членному, 5-членному/5-членному или 5-членному/6-членному моно-спиро-гетероциклилу. Типичные примеры спиро-гетероциклила включают, но не ограничиваются следующими группами:
 и
и  .
.
"Конденсированный гетероциклил" относится к 5-20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую соседнюю пару атомов углерода с другим кольцом, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не обладает полностью конъюгированной системой пи-электронов, и где указанные кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О, и S(O)p (где р равен 0, 1 или 2) в качестве кольцевых атомов, причем оставшиеся кольцевые атомы представляют собой С. Предпочтительно, конденсированный гетероциклил является 6-14-членным, более предпочтительно 7-10-дленным. Согласно числу членных колец конденсированный гетероциклил делится на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, предпочтительно относится к бициклическому или трициклическому конденсированному гетероциклилу, более предпочтительно 5-членному/5-членному или 5-членному/6-членному бициклическому конденсированному гетероциклилу. Типичные примеры конденсированного гетероциклила включают, но не ограничиваются следующими группами:
 и
и 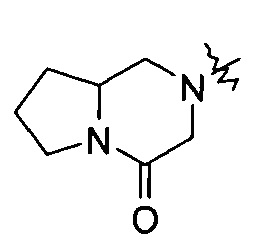 .
.
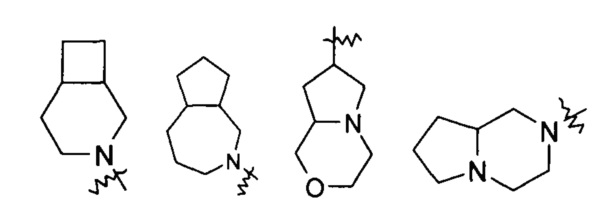
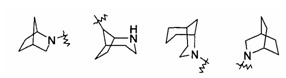
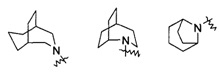
"Связанный мостиковыми связями гетероциклил" относится к 5-14-членной полициклической гетероциклической алкильной группе, где каждые два кольца в системе имеют общими два несвязанных атома, где указанные кольца могли бы иметь одну или более чем одну двойную связь, но не обладают полностью конъюгированной системой пи-электронов, и указанные кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О, и S (O)m (где m равен 0, 1 или 2) в качестве кольцевых атомов, причем оставшиеся кольцевые атомы представляют собой С. Предпочтительно связанный мостиковыми связями гетероциклил является 6-14-членным, более предпочтительно 7-10-членным. Согласно числу членных колец связанный мостиковыми связями гетероциклил делится на бициклический, трициклический, тетрациклический или полициклический связанный мостиковыми связями гетероциклил, предпочтительно относится к бициклическому, трициклическому или тетрациклическому связанному мостиковыми связями гетероциклилу, более предпочтительно бициклическому или трициклическому связанному мостиковыми связями гетероциклилу. Типичные примеры связанного мостиковыми связями гетероциклила включают, но не ограничиваются следующими группами:
 и
и  .
.
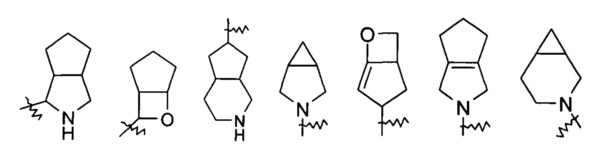
Указанное кольцо гетероциклила может быть конденсировано с кольцом арила, гетероарила или циклоалкила, где кольцо, связанное с первичной структурой, представляет собой гетероциклил. Типичные примеры включают, но не ограничиваются следующими группами:
 и
и  и т.д.
и т.д.
Гетероциклил может быть возможно замещенным или незамещенным. Будучи замещенной, группа(пы) заместителя(ей) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкиксоила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкиоксила, гетероциклического алкиоксила, циклоалкилтио, гетероциклического алкилтио, оксогруппы, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)OR15 и -NHS(O)mR15.
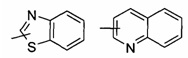
"Арил" относится к 6-14-членной полностью состоящей из атомов углерода моноциклической кольцевой группе или полициклической конденсированной кольцевой группе ("конденсированная" кольцевая система означает, что каждое кольцо в системе имеет общими соседнюю пару атомов углерода с другим кольцом в системе), и обладает полностью конъюгированной системой пи-электронов. Предпочтительно арил является 6-10-членным, например, фенилом и нафтилом, наиболее предпочтительно фенилом. Указанный арил может быть конденсирован с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с первичной структурой, представляет собой арил. Типичные примеры включают, но не ограничиваются следующими группами:
 и
и 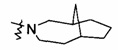 .
.
Арильная группа может быть замещенной или незамещенной. Будучи замещенной, группа(пы) заместителя (ей) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкиксоила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкиоксила, гетероциклического алкиоксила, циклоалкилтио, гетероциклического алкилтио, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)OR15 и -NHS(O)mR15.
"Гетероарил" относится к гетероарильной системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N, в качестве кольцевых атомов, и имеющей от 5 до 14 кольцевых атомов. Предпочтительно гетероарил является 5-10-членным, более предпочтительно 5- или 6-членным, например, тиадиазолилом, пиразолилом, оксазолилом, оксадиазолилом, имидазолилом, триазолилом, тиазолилом, фурилом, тиенилом, пиридилом, пирролилом, N-алкилпирролилом, пиримидинилом, пиразинилом, имидазолилом, тетразолилом и т.п. Указанный гетероарил может быть конденсирован с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с первичной структурой, представляет собой гетероарил. Типичные примеры включают, но не ограничиваются следующими группами:
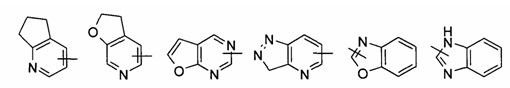
 и
и  .
.
Гетероарильная группа может быть замещенной или незамещенной. Будучи замещенной, группа(пы) заместителя(ей) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкиксоила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкиоксила, гетероциклического алкиоксила, циклоалкилтио, гетероциклического алкилтио, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)OR15 и -NHS(O)mR15.
"Алкоксил" относится как к группе -О-(алкил), так и группе -O-(незамещенный циклоалкил), где алкил определен выше. Типичные примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и т.п., но не ограничиваются ими. Алкоксил может быть возможно замещенным или незамещенным. Будучи замещенным, заместитель предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкиксоила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкиоксила, гетероциклического алкиоксила, циклоалкилтио, гетероциклического алкилтио, -(CH2)nC(O)OR15, -OC(O)R15, -C(O)R15, -C(O)NR16R17, -NHC(O)R15, -NR16R17, -OC(O)NR16R17, -NHC(O)NR16R17, -S(O)mR15, -NHC(O)OR15 и -NHS(O)mR15.
"Связь" относится к ковалентной связи с использованием обозначения "-".
"Гидроксилалкил" относится к алкильной группе, замещенной гидроксильной группой, где алкил представляет собой такой, как определено выше.
"Гидрокси" относится к группе -ОН.
"Галоген" относится к атомам фтора, хлора, брома или йода.
"Амино" относится к группе -NH2.
"Циано" относится к группе -CN.
"Нитро" относится к группе -NO2.
"Оксогруппа" относится к группе =O.
"Карбоксил" относится к группе -С(O)ОН.
"Алкоксикарбонил" относится к группе -С(O)O(алкил) или (циклоалкил), где алкил и циклоалкил определены выше.
Энантиомер:
"Возможный" или "возможно" означает, что событие или обстоятельство, описанное далее, может произойти, но не обязательно, и описание включает примеры события или обстоятельства, которое может произойти или не произойти. Например, фраза "гетероциклическая группа, возможно замещенная алкилом" означает, что алкильная группа может присутствовать, но не обязательно, и описание включает случай, когда гетероциклическая группа замещена алкилом, и когда гетероциклическая группа не замещена алкилом.
"Замещенный" относится к одному или более чем одному атому водорода в группе, предпочтительно вплоть до 5, более предпочтительно от 1 до 3 атомов водорода, независимо замещенных соответствующим числом заместителей. Совершенно очевидно, что заместители существуют только в своем возможном химическом положении. Специалист в данной области техники способен определить, возможно ли или не возможно замещение без того, чтобы прилагать чрезмерные усилия посредством эксперимента или теории. Например, комбинация амино- или гидроксильной группы, имеющей свободный водород, и атомов углерода, имеющих ненасыщенные связи (такие как олефиновые), может быть нестабильной.
"Фармацевтическая композиция" относится к смеси одного или более чем одного соединения, описанного в настоящем изобретении, или его физиологически/фармацевтически приемлемых солей или его пролекарств, и других химических компонентов, таких как физиологически/фармацевтически приемлемые носители и эксципиенты. Задача фармацевтической композиции заключается в том, чтобы облегчить введение соединения в организм, что благоприятствует абсорбции активного ингредиента, и, таким образом, демонстрируя биологическую активность.
"Фармацевтически приемлемые соли" относятся к солям соединений по изобретению, причем такие соли являются безопасными и эффективными при использовании в отношении млекопитающего и обладают соответствующей биологической активностью.
N, m и R15-R17 представляют собой такие, как определено для соединения формулы (I).
СПОСОБ СИНТЕЗА СОЕДИНЕНИЯ ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Для достижения цели изобретения в настоящем изобретении применяется без ограничения следующее техническое решение:
способ получения соединения формулы (I) по изобретению или его фармацевтически приемлемой соли, включающий следующие стадии:
 ,
,
причем соединение формулы (IA) реагирует с соединением формулы (IB) в щелочных условиях с получением соединения формулы (I);
щелочные условия обеспечивают посредством органического основания и неорганического основания, где указанное органическое основание включает триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, трет-бутил калия алкоксид, но не ограничивается ими, и указанное неорганическое основание включает гидрид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, но не ограничивается ими;
где Х представляет собой галоген, A, R, L, R1-R14, p, q, s и t представляют собой такие, как определено в формуле (I); предпочтительно, R1 представляет собой трет-бутоксикарбонил; предпочтительно L представляет собой связь.
Способ получения соединения или соли формулы (I) по изобретению, включающий следующие стадии:
 ,
,
когда R1 представляет собой трет-бутоксикарбонил, трет-бутоксикарбонил возможно дополнительно удаляют из соединения формулы (I) с получением соединения формулы (IC) или его фармацевтически приемлемой соли; соединение формулы (IC) или его фармацевтически приемлемая соль реагирует с карбоновой кислотой, хлорангидридом карбоновой кислоты, сульфонилхлоридом, сложными эфирами карбоновой кислоты, производным этиленоксида или галогенидом в щелочных условиях с получением соединений формулы (I);
реакционный растворитель включает: тетрагидрофуран, этанол, метанол, н-бутанол, дихлорметан, 1,4-диоксан или N,N-диметилформамид, но не ограничивается ими;
щелочные условия обеспечивают посредством органического основания и неорганического основания, где указанное органическое основание включает триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, трет-бутилкалия алкоксид, тетрабутиламмония бромид, но не ограничивается ими, и указанное неорганическое основание включает гидрид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, но не ограничивается ими;
где A, R, L, R1-R14, p, q, s и t представляют собой такие, как определено в формуле (I); предпочтительно L представляет собой связь.
ПРЕДПОЧТИТЕЛЬНЫЕ ВОПЛОЩЕНИЯ
Следующие примеры служат для иллюстрации изобретения, но примеры не следует рассматривать как ограничивающие объем изобретения. Если конкретные условия экспериментального способа не указаны в примерах настоящего изобретения, они в общем соответствуют обычным условиям или рекомендованным условиям производителя сырья и продукта. И реагенты без указания конкретного источника имеются в продаже, обычные реагенты.
Структуру соединений идентифицировали с помощью NMR (ядерного магнитного резонанса) и/или MS (масс-спектрометрии). Химические сдвиги NMR (δ) приведены в 10-6 (млн-1). NMR определяли с помощью аппарата Bruker AVANCE-400. Растворители представляли собой дейтерированный диметилсульфоксид (DMSO-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD) с тетраметилсиланом (TMS) в качестве внутреннего стандарта.
MS определяли с помощью масс-спектрометра FINNIGAN LCQAd (ESI (электрораспылительная ионизация)) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
HPLC (высокоэффективная жидкостная хроматография) осуществляли на жидкостном хроматографе-спектрометре высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire C18 150×4,6 мм) и жидкостном хроматографе-спектрометре высокого давления Waters 2695-2996 (хроматографическая колонка Gimini C18 150×4,6 мм).
Среднюю скорость ингибирования киназы и величину IC50 (концентрация полумаксимального ингибирования) определяли с помощью микропланшета-ридера (BMG company, Германия).
Для тонкослойной хроматографии на силикагеле использовали пластину со слоем силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размеры пластин, использованных в TLC (тонкослойная хроматография), составили от 0,15 мм до 0,2 мм, и размеры пластин, использованных в тонкослойной хроматографии для очистки продукта, составили от 0,4 мм до 0,5 мм.
Для колоночной хроматографии в качестве носителя как правило использовали силикагель Yantai Huanghai от 200 до 300 меш.
Известное исходное вещество по изобретению может быть получено с помощью традиционного способа синтеза предшествующего уровня техники или приобретено у ABCR GmbH & Со. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc или Dari chemical Company и т.д.
Если в примерах не указано иное, следующие реакции осуществляли в атмосфере аргона или атмосфере азота.
Термин "атмосфера аргона" или "атмосфера азота" относится к тому, что реакционная колба оборудована баллоном, содержащим 1 л аргона или азота.
Термин "атмосфера водорода" относится к тому, что реакционная колба оборудована баллоном, содержащим 1 л водорода.
Реакции гидрирования при высоком давлении проводили с помощью аппарата для гидрирования Parr 3916EKX и генератора водорода clear blue QL-500 или аппарата для гидрирования HC2-SS.
В реакциях гидрирования реакционную систему, как правило, вакуумировали и заполняли водородом, и вышеприведенное действие повторяли три раза.
Микроволновые реакции проводили с помощью микроволнового реактора СЕМ Discover-S 908860.
Если в примерах не указано иное, раствор, используемый в следующих реакциях, относится к водному раствору.
Если в примерах не указано иное, температура реакции в следующей реакции представляла собой комнатную температуру.
Комнатная температура представляла собой наиболее правильную температуру реакций, которая составляла от 20°С до 30°С.
За процессом реакции наблюдали с помощью тонкослойной хроматографии (TLC), система проявляющих растворителей включала: (А) систему дихлорметана и метанола, (Б) систему н-гексана и этилацетата, (В) систему петролейного эфира и этилацетата, (Г) ацетон. Отношение объемов растворителей корректировали в соответствии с полярностью соединений.
Система элюции при очистке соединений с помощью колоночной хроматографии и тонкослойной хроматографии включала: (А) систему дихлорметана и метанола, (Б) систему н-гексана и этилацетата, (В) систему дихлорметана и ацетона, отношение объемов растворителей корректировали в соответствии с полярностью соединений, и иногда также может быть добавлено небольшое количество щелочного реагента, такого кактриэтиламин, или кислотного реагента, такого как уксусная кислота.
Пример 1
(3aR,5R,6aS/3aS,5S,6aR)-трет-бутил-3а-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
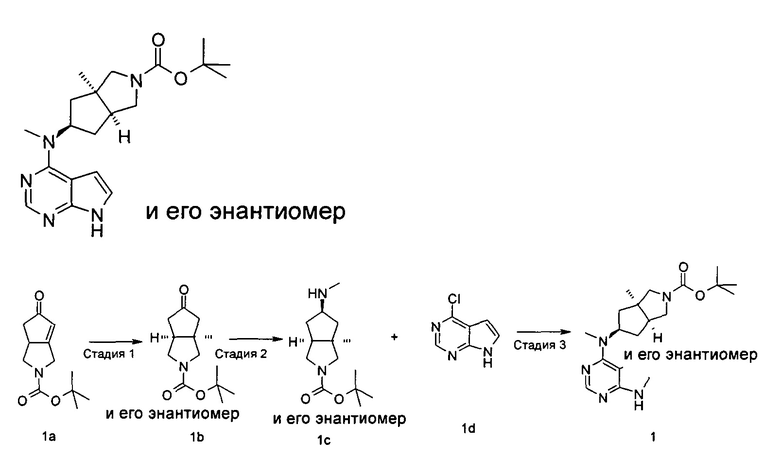
Стадия 1
(3aR,6aS/3aS,6aR)-трет-Бутил 3а-метил-5-оксогексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
Йодид меди (770 мг, 4 ммоль) растворяли в 10 мл тетрагидрофурана. После охлаждения до -78°С в реакционную смесь добавляли 3 М раствор бромистого метилмагния в диэтиловом эфире (30 мл, 6,7 ммоль). После взаимодействия в течение 30 минут реакционную смесь нагревали до -35°С, а затем по каплям добавляли 10 мл раствора трет-бутил 5-оксо-3,3а,4,5-тетрагидроциклопента[с]пиррол-2(1H)-карбоксилата 1а (500 мг, 2,24 ммоль) в тетрагидрофуране. После взаимодействия в течение 30 минут реакционную смесь нагревали до комнатной температуры, а затем добавляли 10 мл насыщенного раствора хлорида аммония для остановки реакции. Реакционную смесь экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (3aR,6aS/3aS,6aR)-трет-бутил 3а-метил-5-оксогексагидроциклопента[с]пиррол-2(1Н)-карбоксилата 1b (500 мг, коричневый жир), который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
(3aR,5R,6aS/3aS,5S,6aR)-трет-Бутил 3а-метил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
Неочищенный продукт (3aR,6aS/3aS,6aR)-трет-бутил 3а-метил-5-оксогексагидроциклопента[с]пиррол-2(1Н)-карбоксилат 1b (200 мг, 0,84 ммоль) растворяли в 5 мл метанола, а затем добавляли 2 мл 37% раствора метилэтаноламина и триацетоксиборгидрид натрия (532 мг, 2,5 ммоль). После взаимодействия в течение 16 часов 10 мл насыщенного раствора хлорида аммония добавляли в реакционную смесь для остановки реакции. Реакционную смесь экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке продукта (3aR,5R,6aS/3aS,5S,6aR)-трет-бутил 3а-метил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 1 с (130 мг, выход 61,0%) в виде коричневого жира.
MS m/z(ESI): 255,2 [М+1]
Стадия 3
(3aR,5R,6aS/3aS,5S,6aR)-трет-Бутил-3а-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
4-Хлор-7H-пирроло[2,3-d]пиримидин 1d (60 мг, 0,39 ммоль) растворяли в 5 мл H2O, а затем добавляли (3aR,5R,6aS/3aS,5S,6aR)-трет-бутил 3а-метил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат 1с (100 мг, 0,39 ммоль) и карбонат калия (322 мг, 2,34 ммоль). После взаимодействия в течение 16 часов при 100°С реакционную смесь экстрагировали этилацетатом (10 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта (3aR,5R,6aS/3aS,5S,6aR)-трет-бутил-3а-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 1 (40 мг, выход 28,8%) в виде белого твердого вещества.
MS m/z (ESI): 372,5 [M+1]
1H NMR (400 МГц, CDCl3): δ 10,38 (s, 1H), 8,30 (s, 1H), 7,05-7,04 (m, 1H), 6,57-6,56 (m, 1H), 5,57-5,52 (m, 1H), 3,56-3,49 (m, 3H), 3,37 (s, 3H), 3,27-3,25 (m, 1H), 2,25-2,21 (m, 2H), 1,89-1,87 (m, 2H), 1,67-1,65 (m, 2H), 1,49 (s, 9H), 0,88-0,86 (m, 2H)
Пример 2
N-((3aR,5R,6aS/3aS,5S,6aR)-2-(Этилсульфонил)-3а-метилоктагидроциклопента[с]пиррол-5-ил)-N-метил-7H-пирроло[2,3-о]пиримидин-4-амин
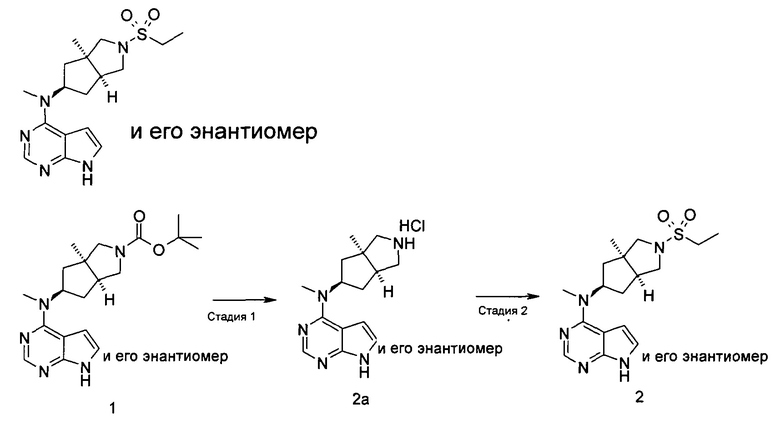
Стадия 1 N-Метил-N-((3aR,5R,6aS)-3а-метилоктагидроциклопента[с]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорид
(3aR,5R,6aS/3aS,5S,6aR)-трет-Бутил-3а-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат 1 (1 г, 2,7 ммоль) растворяли в 15 мл раствора 6 М хлорида водорода в метаноле. После взаимодействия в течение 12 часов реакционную смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта N-метил-N-((3aR,5R,6aS)-3а-метилоктагидроцикпопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорида 2а (1,2 г, белое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 272,2 [М+1]
Стадия 2
N-((3aR,5R,6aS/3aS,5S,6aR)-2-(Этилсульфонил)-3а-метилоктагидроциклопента[с]пиррол-5-ил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амин
N-Метил-N-((3aR,5R,6aS/3aS,5S,6aR)-3a-метилоктагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 2а (100 мг, 0,37 ммоль) и триэтиламин (112 мг, 1,11 ммоль) растворяли в 5 мл тетрагидрофурана, а затем по каплям добавляли этилсульфонилхлорид (95 мг, 0,74 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли 20 мл H2O и экстрагировали этилацетатом (20 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта N-((3aR,5R,6aS/3aS,5S,6aR)-2-(этилсульфонил)-3а-метилоктагидроциклопента[с]пиррол-5-ил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амина 2 (28 мг, выход 21,5%) в виде белого твердого вещества.
MS m/z (ESI): 364,2 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 12,56 (s, 1H), 8,25-8,23 (m, 1H), 7,95-7,93 (m, 1H), 7,21-7,20 (m, 1H), 5,51-5,49 (m, 1H), 4,02 (m, 2H), 3,75-3,58 (m, 6H), 3,38 (s, 3H), 3,22-3,16 (m, 2H), 2,06 (s, 3H), 1,97-1,95 (m, 1H), 1,43-1,41 (m, 3H)
Пример 3
3-((3aR,5R,6aS/3aS,5S,6aR)-3а-Метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
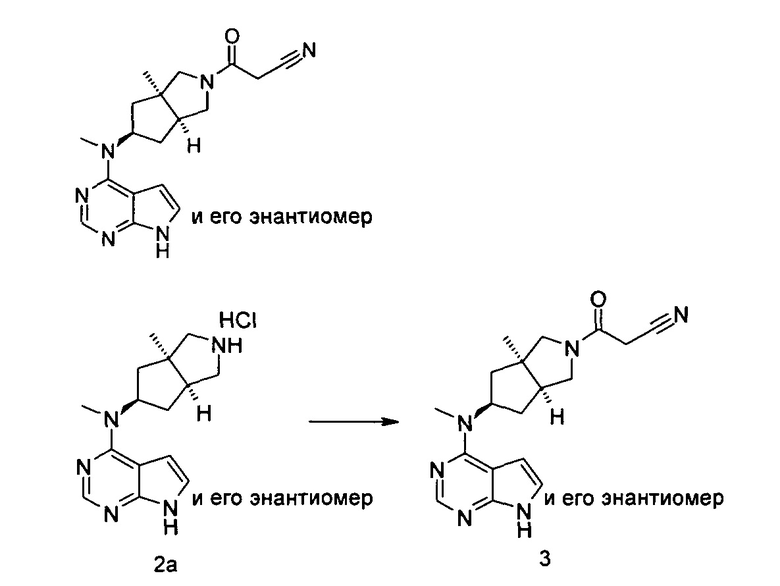
N-Метил-N-((3aR,5R,6aS/3aS,5S,6aR)-3а-метилоктагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 2а (50 мг, 0,18 ммоль) и этил 2-цианоацетат (49 мг, 0,36 ммоль) растворяли в 3 мл этанола, а затем по каплям добавляли 1,4-диазабициклооктан (56 мг, 0,36 ммоль). После взаимодействия в течение 16 часов при 40°С в реакционную смесь добавляли 20 мл Н2О и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 3-((3aR,5R,6aS/3aS,5S,6aR)-3а-метил-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)-3-оксопропаннитрила 3 (46 мг, выход 56,8%) в виде белого твердого вещества.
MS m/z (ESI): 339,1 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 12,58 (s, 1H), 8,27-8,25 (m, 1H), 7,97-7,95 (m, 1H), 7,26-7,24 (m, 1H), 5,58-5,53 (m, 1H), 4,33 (s, 2H), 3,75-3,58 (m, 6H), 3,39 (s, 3H), 3,22-3,16 (m, 2H), 2,08 (s, 3H), 1,99-1,97 (m, 1H)
Пример 4
2-((3aR,5R,6aS/3aS,5S,6aR)-3а-Метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)ацетонитрил
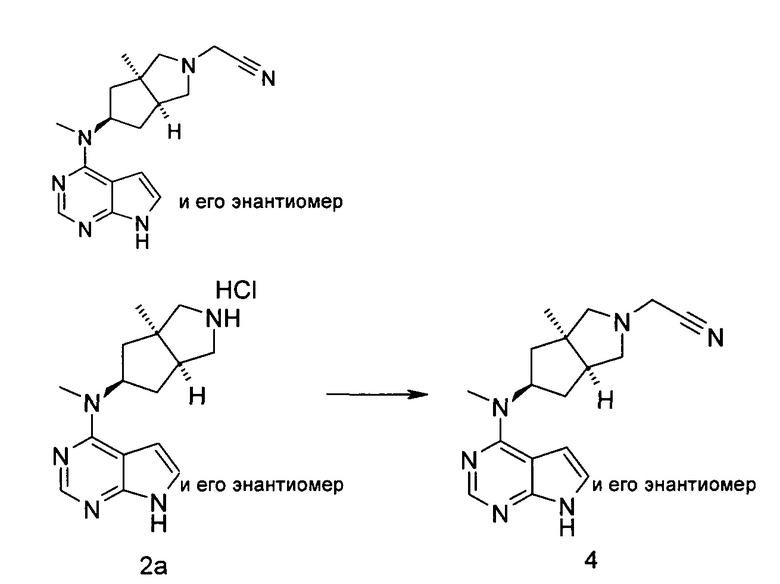
N-Метил-N-({3aR,5R,6aS/3aS,5S,6aR)-3a-метилоктагидроциклопента[с]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 2а (50 мг, 0,18 ммоль) растворяли в 5 мл ацетонитрила, а затем добавляли карбонат калия (75 мг, 0,54 ммоль), бромацетонитрил (24 мг, 0,2 ммоль) и 5 мл дихлорметана. После взаимодействия в течение 16 часов в реакционную смесь добавляли небольшое количество H2O для остановки реакции. Водную фазу и органическую фазу разделяли. Водную фазу экстрагировали дихлорметаном (10 мл × 3). Органические фазы объединяли, последовательно промывали водой (5 мл), насыщенным раствором хлорида натрия (5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 2-((3aR,5R,6aS/3aS,5S,6aR)-3a-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)ацетонитрила 4 (20 мг, выход 35,1%) в виде белого твердого вещества.
MS m/z (ESI): 311,5 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 11,64 (s, 1H), 8,12-8,10 (m, 1H), 7,14 (s, 1H), 6,54 (s, 1H), 3,86-3,85 (m, 2H), 3,14-3,13 (m, 3H), 2,77-2,75 (m, 1H), 2,62-2,60 (m, 1H), 2,08-2,06 (m, 3H), 1,86-1,80 (m, 1H), 1,70-1,69 (m, 1H), 1,58-1,55 (m, 1H), 1,30-1,18 (m,2H), 0,86 (s, 3H)
Пример 5
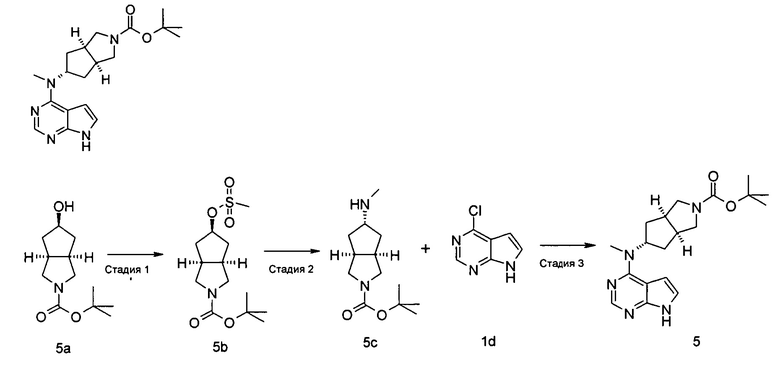
(3aR,5s,6aS)-трет-Бутил-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
Стадия 1
(3aR,5R,6aS)-трет-Бутил 5-((метилсульфонил)окси)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
(3aR,5R,6aS)-трет-Бутил 5-гидроксигексагидроциклопента[с]пиррол-2(1H)-карбоксилат 5а (9 г, 40 ммоль) растворяли в 150 мл дихлорметана, а затем при 0°С добавляли метилсульфонилхлорид (4,70 мл, 60 ммоль) и триэтиламин (11,20 мл, 80 ммоль). После взаимодействия в течение 2 часов при комнатной температуре в реакционную смесь добавляли 200 мл насыщенного раствора бикарбоната натрия. Водную фазу и органическую фазу разделяли. Органическую фазу промывали насыщенным раствором хлорида натрия (200 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке продукта (3aR,5R,6aS)-трет-бутил 5-((метилсульфонил)окси)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилата 5b (12,00 г, выход 98,4%) в виде желтой жидкости.
Стадия 2
(3aR,5s,6aS)-трет-Бутил 5-(метиламино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
(3aR,5R,6aS)-трет-Бутил 5-((метилсульфонил)окси)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат 5b (60 мг, 0,2 ммоль) растворяли в 10 мл метанола, а затем добавляли 5 мл метиламина. После взаимодействия в течение 16 часов при 40°С реакционную смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (3aR,5s,6aS)-трет-бутил 5-(метиламино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилата 5 с (60 мг, коричневый жир), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 241,5 [M+1]
Стадия 3
(3aR,5s,6aS)-трет-Бутил 5-(метил(7Н-пирроло[2,3-с(]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
(3aR,5s,6aS)-трет-Бутил 5-(метиламино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат 5 с (200 мг, 0,8 ммоль) и 4-хлор-7Н-пирроло[2,3-d]пиримидин 1d (127 мг, 0,8 ммоль) растворяли в 5 мл н-бутанола, а затем добавляли триэтиламин (168 мг, 1,6 ммоль). После взаимодействия в течение 48 часов при 100°С реакционную смесь концентрировали при пониженном давлении, а затем добавляли 10 мл H2O и 10 мл этилацетата. Водную фазу и органическую фазу разделяли. Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта (3aR,5s,6aS)-трет-бутил 5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 5 (5 мг, выход 5,0%) в виде белого твердого вещества.
MS m/z (ESI): 358,5 [M+1]
1H NMR (400 МГц, CDCl3): δ 10,07 (s, 1Н), 8,31 (s, 1H), 7,50 (s, 1H), 6,55(s, 1Н), 5,58-5,54 (m, 1H), 3,65-3,62 (m, 2H), 3,27-3,23 (m,5H), 2,86-2,81 (m, 2H), 2,06-2,02 (m, 2H), 1,93-1,91 (m, 2H), 1,49 (s, 6H)
Пример 6
2-Гидрокси-1-((3aR,5s,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)этанон
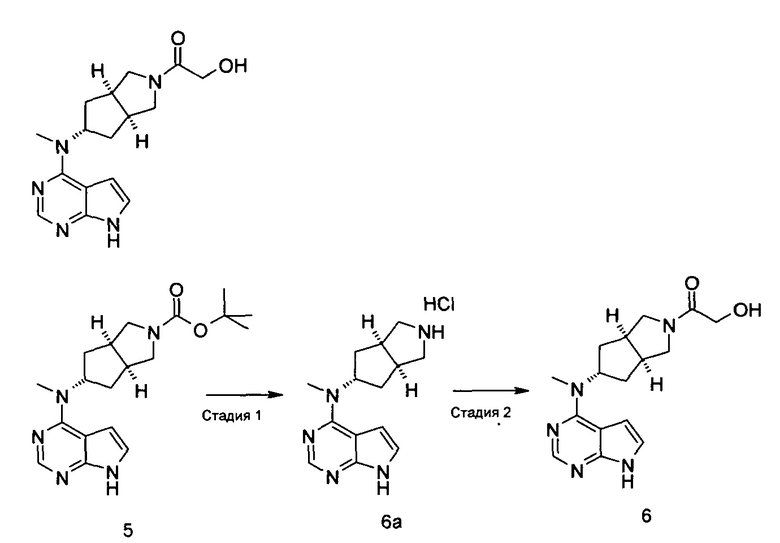
Стадия 1
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид
(3aR,5s,6aS)-трет-Бутил 5-(метил(7H-пирроло[2,3-о]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат 5 (1,5 г, 4,2 ммоль) растворяли в 20 мл 1 М раствора хлорида водорода в метаноле. После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта N-метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорида 6а (1,5 г, коричневое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 258,1 [М+1]
Стадия 2
2-Гидрокси-1-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанон
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,3 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли 2-гликолевую кислоту (26 мг, 0,3 ммоль), триэтиламин (103 мг, 1,02 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (194 мг, 0,45 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли 10 мл H2O. Водную фазу и органическую фазу разделяли. Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта 2-гидрокси-1-((3aR,5s,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)этанона 6 (10 мг, выход 9,7%) в виде белого твердого вещества.
MS m/z (ESI): 316,2 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,60 (s, 1H), 8,09 (s, 1H), 7,11 (s, 1H), 6,53 (s, 1H), 5,47-5,44 (m, 1H), 4,52-4,49 (m, 1H), 4,03-3,99 (m, 2H), 3,60-3,57 (m, 2H), 3,24-3,22 (m, 2H), 3,15 (s, 3H), 2,80-2,75 (m, 2H), 2,02-1,94 (m, 2H), 1,78-1,75 (m, 2H)
Пример 7
2-Метил-1-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)пропан-2-ол
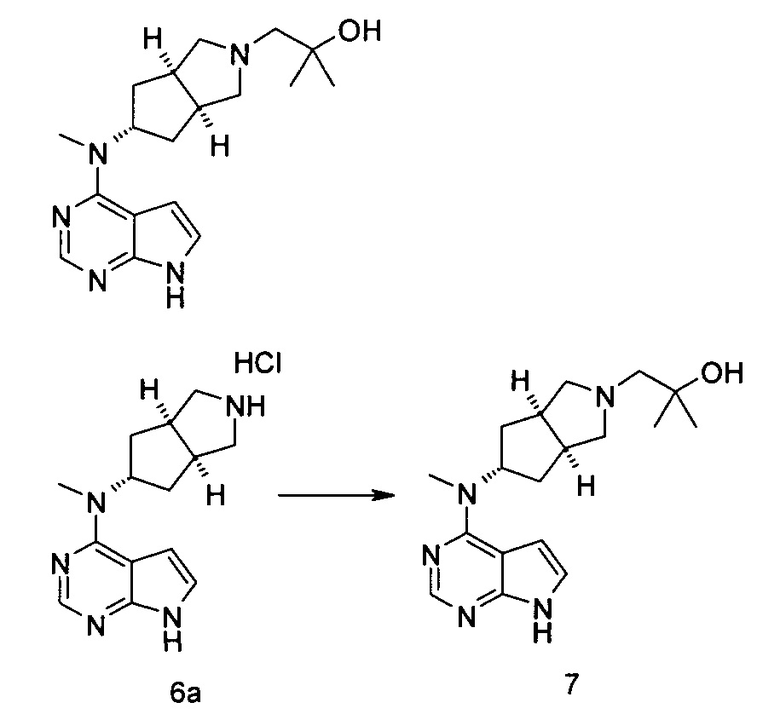
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл метанола, а затем добавляли 2,2-диметилэпоксиэтан (42 мг, 0,58 ммоль). После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 2-метил-1-((3aR,5s,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)пропан-2-ола 7 (50 мг, выход 39,1%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 330,2 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 11,68 (s, 1H), 8,11 (s, 1H), 7,14-713 (m, 1H), 6,60 (s, 1H), 5,46 (s, 1H), 5,12 (s, 1H), 3,86 (m, 2H), 3,18 (s, 4H), 3,07-3,01 (m, 3H), 2,84 (d, 2H), 1,95-1,91 (m, 2H), 1,68 (s, 2H), 1,33-1,18 (m, 6H)
Пример 8
3-((3aR,5s,6aS)-5-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)-3-оксопропаннитрил
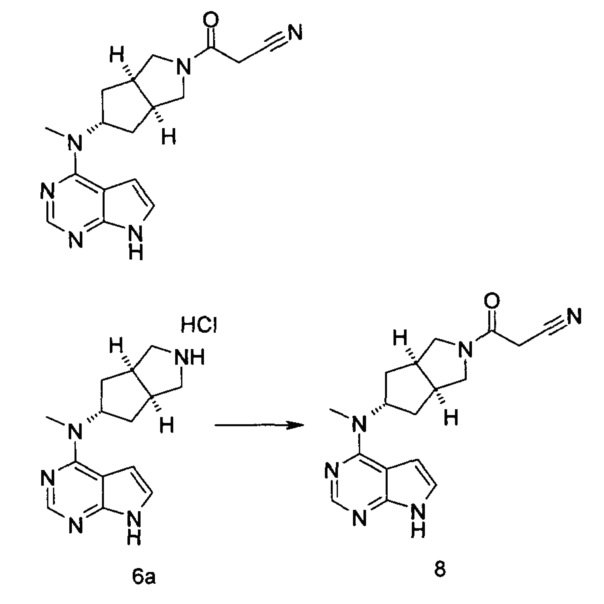
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл н-бутанола, а затем добавляли 2-этилцианоацетат (77 мг, 0,68 ммоль) и DBU (1,8-диазабицикло[5.4.0]ундец-7-ен) (103 мг, 0,68 ммоль). После взаимодействия в течение 15 часов при 40°С реакционную смесь концентрировали при пониженном давлении. В остаток добавляли 50 мл этилацетата и промывали насыщенным раствором хлорида натрия (15 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта 3-((3aR,5s,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)-3-оксопропаннитрила 8 (15 мг, выход 13,6%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 325,1 [М+1]
1H NMR (400 МГц, CDCl3): δ 9,79 (s, 1H), 8,28 (s, 1H), 7,06-7,05 (m, 1H), 6,55-6,54 (m, 1H), 5,68-5,64 (m, 1H), 3,85-3,79 (m, 2H), 3,49 (s, 2H), 3,47-3,37 (m, 2H), 3,27 (s, 3H), 3,08-2,91 (m, 2H), 2,09-2,05 (m, 2H), 1,96-1,94 (m, 2H)
Пример 9
(3aR,5s,6aS)-N-Изопропил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамид
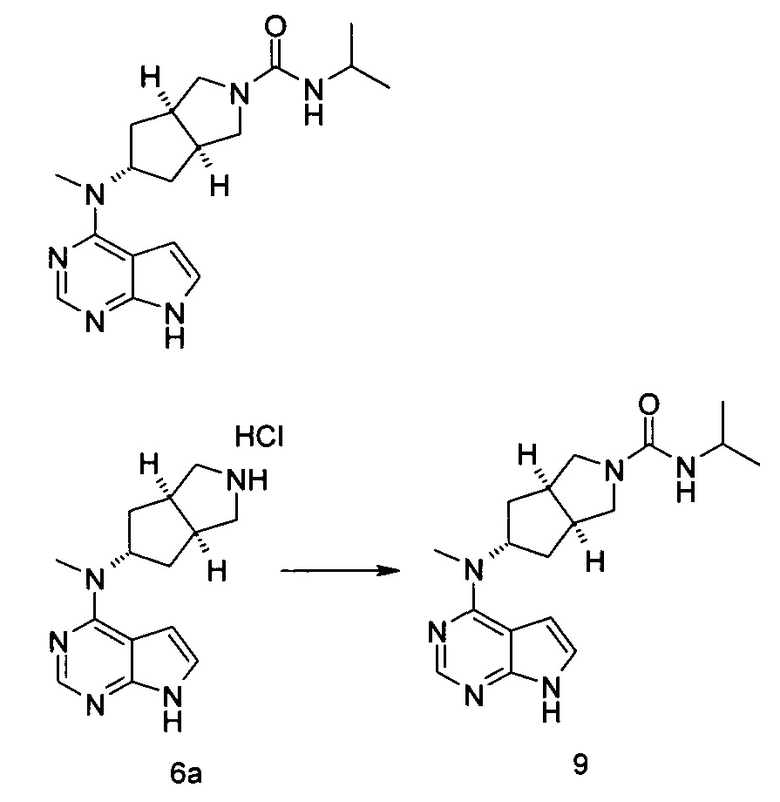
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,39 ммоль) растворяли в 5 мл N,N-диметилформамида, а затем по каплям добавляли триэтиламин (39 мг, 0,39 ммоль). После взаимодействия в течение 30 минут в реакционную смесь добавляли изоцианатопропан (33 мг, 0,39 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли 10 мл H2O и 10 мл этилацетата. Водную фазу и органическую фазу разделяли. Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта (3aR,5s,6aS)-N-изопропил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида 9 (15 мг, выход 11,3%) в виде белого твердого вещества.
MS m/z(ESI): 343,3 [М+1]
1H NMR (400 МГц, CDCl3): δ 11,18 (s, 1H), 8,31 (s, 1H), 7,06 (s, 1H), 6,54 (s, 1H), 5,63-5,58 (m, 1H), 4,06-4,01 (m, 2H), 3,63-3,58 (m, 2H), 3,26-3,21 (m, 5H), 2,92-2,88 (m, 2H), 2,04-2,01 (m, 2H), 1,94-1,89 (m, 2H), 1,89-1,74 (m, 6H)
Пример 10
(3aR,5S,6aS/3aS,5R,6aR)-трет-Бутил 3а-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
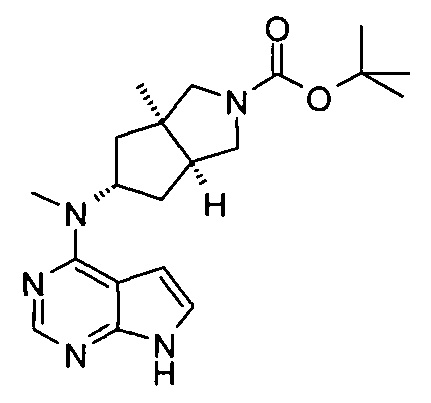 и его энантиомер
и его энантиомер
Стадия 1
(3aR,5S,6aS/3aS,5R,6aR)-трет-Бутил 5-гидрокси-3а-метилгексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
Неочищенный продукт (3aR,6aS/3aS,6aR)-трет-бутил 3а-метил-5-оксогексагидроциклопента[с]пиррол-2(1H)-карбоксилат 1b (1 г, 4,2 ммоль) растворяли в 10 мл метанола, а затем добавляли боргидрид натрия (320 мг, 8,4 ммоль). После взаимодействия в течение 1 часа реакционную смесь выливали в 50 мл насыщенного раствора хлорида аммония и экстрагировали этилацетатом (20 мл × 3). Органическую фазу объединяли, последовательно промывали H2O (5 мл × 3) и насыщенным раствором хлорида натрия (5 мл × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (3aR,5S,6aS/3aS,5R,6aR)-трет-бутил 5-гидрокси-3а-метилгексагидроциклопента[с]пиррол-2(1H)-карбоксилата 10а (900 мг, бесцветный жир), который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
(3aR,5R,6aS/3aS,5S,6aR)-трет-Бутил 3а-метил-5-((метилсульфонил)окси)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
Неочищенный продукт (3aR,5R,6aS/3aS,5S,6aR)-трет-бутил 5-гидрокси-3а-метилгексагидроциклопента[с]пиррол-2(1H)-карбоксилат 10а (1 г, 4,2 ммоль) растворяли в 10 мл дихлорметана, а затем добавляли триэтиламин (1,27 г, 12,6 ммоль). В реакционную смесь при 0°С по каплям добавляли 5 мл раствора метилсульфонилхлорида в дихлорметане (700 мг, 6,2 ммоль). После взаимодействия в течение 16 часов реакционную смесь выливали в 10 мл H2O. Водную фазу и органическую фазу разделяли. Органическую фазу объединяли и последовательно промывали насыщенным бикарбонатом натрия (10 мл × 3) и насыщенным раствором хлорида натрия (10 мл × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке продукта (3aR,5R,6aS/3aS,5S,6aR)-трет-бутил 3а-метил-5-((метилсульфонил)окси)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 10b (1,2 г, выход 92,3%) в виде светло-желтого жира.
Стадия 3
(3aR,5S,6aS/3aS,5R,6aR)-трет-Бутил 3а-метил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
(3aR,5R,6aS/3aS,5S,6aR)-трет-Бутил 3а-метил-5-((метилсульфонил)окси)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат 10b (500 мг, 1,6 ммоль) растворяли в 10 мл 1 М раствора метиламина в метаноле. После взаимодействия в течение 16 часов при 40°С реакционную смесь концентрировали при пониженном давлении при 40°С с получением неочищенного указанного в заголовке продукта (3aR,5S,6aS/3aS,5R,6aR)-трет-бутил 3а-метил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 10с (350 мг, светло-желтый жир), который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 4
(3aR,5S,6aS/3aS,5R,6aR)-трет-Бутил 3а-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
Неочищенный продукт (3aR,5S,6aS/3aS,5R,6aR)-трет-бутил 3а-метил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат 10с (350 мг, 1,37 ммоль) растворяли в 10 мл н-бутанола, а затем добавляли 4-хлор-7Н-пирроло[2,3-d]пиримидин 1d (320 мг, 2,06 ммоль) и триэтиламин (410 мг, 4,13 ммоль). После взаимодействия в течение 16 часов при 100°С реакционную смесь охлаждали до комнатной температуры и выливали в 50 мл Н2О, а затем добавляли 20 мл этилацетата. Водную фазу и органическую фазу разделяли. Водную фазу экстрагировали этилацетатом (10 мл). Органическую фазу объединяли, последовательно промывали H2O (5 мл × 3) и насыщенным раствором хлорида натрия (5 мл × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта (3aR,5S,6aS/3aS,5R,6aR)-трет-бутил 3а-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 10 (20 мг, выход 3,9%) в виде светло-желтого жира.
MS m/z (ESI): 372,2 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,61 (s, 1H), 8,09 (s, 1H), 7,10 (s, 1H), 6,53 (s, 1H), 5,47-5,44 (m, 1H), 3,52 (t, 1H), 3,44 (m, 1H), 3,28 (d, 1H), 3,20 (d, 1H), 3,17 (s, 3H), 2,33-2,30 (m, 1H), 2,13-2,07 (m, 1H), 1,96-1,91 (m, 1H), 1,73-1,67 (m, 2H), 1,42 (s, 9H), 1,22 (s, 3H)
Пример 11
1-((3aR,5s,6aS)-5-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)этанон
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (200 мг, 0,68 ммоль) растворяли в 20 мл дихлорметана, а затем добавляли триэтиламин (206 мг, 2,04 ммоль). После взаимодействия в течение 0,5 часа в реакционную смесь по каплям добавляли ацетилхлорид (53 мг, 0,68 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли 10 мл H2O. Водную фазу и органическую фазу разделяли. Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта 1-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанона 11 (20 мг, выход 9,8%) в виде белого твердого вещества.
MS m/z (ESI): 300,3 [М+1]
1H NMR (400 МГц, CDCl3): δ 11,32 (s, 1H), 8,30 (s, 1H), 7,08 (s, 1H), 6,52 (s, 1H), 5,66-5,61 (m, 1H), 3,77-3,71 (m, 2H), 3,43-3,40 (m, 2H), 3,33 (s, 3H), 3,87-2,95(m, 2H), 2,02-2,00 (m, 2H), 1,94-1,89 (m, 2H)
Пример 12
Этил 2-((3aR,5s,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)ацетат
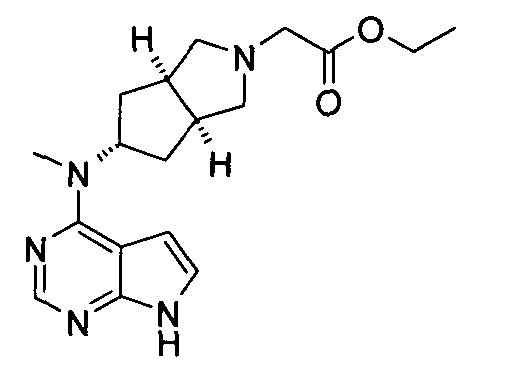
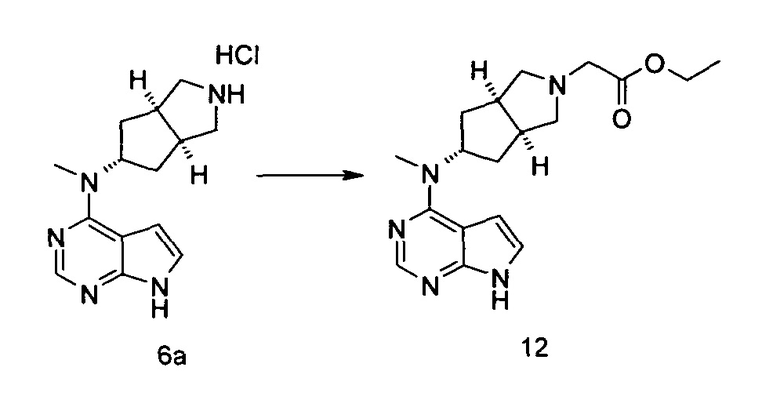
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) и карбонат калия (94 мг, 0,68 ммоль) растворяли в 5 мл 1,4-диоксана. После взаимодействия в течение 0,5 часа в реакционную смесь по каплям добавляли 2-этилбромацетат (56 мг, 0,34 ммоль). После взаимодействия в течение 4 часов в реакционную смесь для растворения остатка добавляли 50 мл дихлорметана. Реакционную смесь промывали насыщенным раствором хлорида натрия (15 мл). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта этил 2-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)ацетата 12 (24 мг, выход 21,4%) в виде белого твердого вещества.
MS m/z (ESI): 344,4 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 11,54 (s, 1H), 8,09 (s, 1H), 7,07-7,06 (m, 1H), 6,76-6,75 (m, 1H), 5,36-5,34 (m, 1H), 4,11 (q, 2H), 3,28 (s, 2H), 3,08 (s, 3H), 2,67-2,65 (m, 4H), 1,95-1,93 (m, 2H), 1,63-1,58 (m, 2H), 1,25-1,18 (m, 5H)
Пример 13
Циклопропил((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)метанон
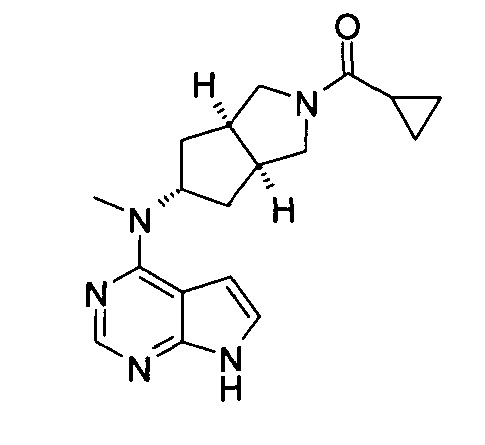
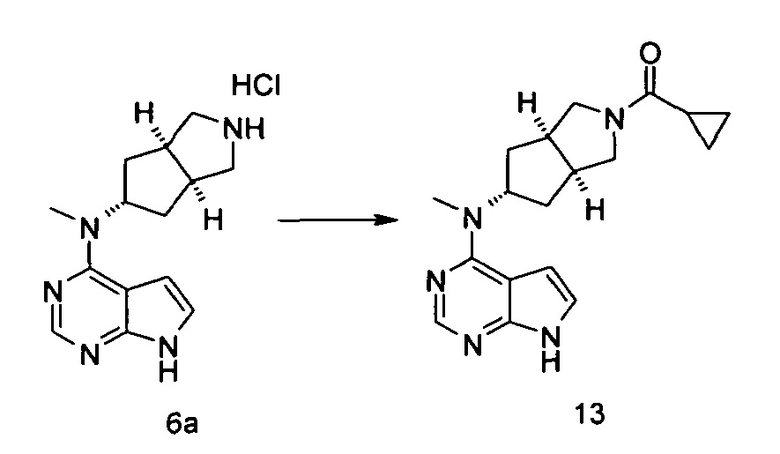
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем по каплям добавляли триэтиламин (69 мг, 0,68 ммоль). После взаимодействия в течение 0,5 часа в реакционную смесь добавляли хлористый циклопропилформил (39 мг, 0,37 ммоль). После взаимодействия в течение 3 часов в реакционную смесь для растворения остатка добавляли 50 мл дихлорметана. Реакционную смесь промывали насыщенным раствором хлорида натрия (15 мл). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта циклопропил ((3aR,5s,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)метанона 13 (26 мг, выход 23,6%) в виде белого твердого вещества.
MS m/z (ESI): 326,1 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 11,60 (s, 1H), 8,09 (s, 1H), 7,11-7,10 (m, 1H), 6,52-6,51 (m, 1H), 5,48-5,44 (m, 1H), 3,85-3,83 (m, 1H), 3,55-3,49 (m, 2H), 3,24-3,21 (m, 1H), 3,15 (s, 3H), 2,93-2,91 (m, 1H), 2,81-2,79 (m, 1H), 2,03-1,98 (m, 2H), 1,83-1,78 (m,3H), 0,78-0,71 (m, 4H)
Пример 14
2-Гидрокси-1-((3aS,5R,6aR/3aS,5R,6aR)-3а-метил-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)этанон
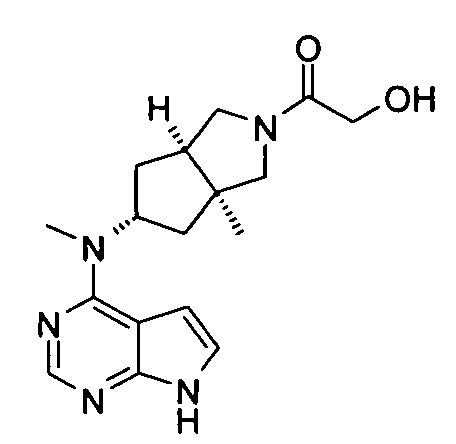 и его энантиомер
и его энантиомер
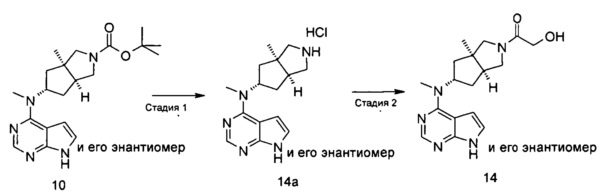
Стадия 1
N-Метил-N-((3aR,5S,6aS/3aS,5R,6aR)-3a-метилоктагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид
(3aR,5S,6aS/3aS,5R,6aR)-трет-Бутил 3а-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат 10 (1,56 г, 4,2 ммоль) растворяли в 20 мл 1 М раствора хлорида водорода в метаноле. После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта N-метил-N-((3aR,5S,6aS/3aS,5R,6aR)-3a-метилоктагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорида 14а (1,5 г, коричневое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
2-Гидрокси-1-((3aS,5R,6aR/3aS,5R,6aR)-3а-метил-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанон
N-Метил-N-((3aR,5s,6aS/3aS,5R,6aR)-3a-метилоктагидроциклопента[с]пиррол-5-ил)-7H-пиррол[2,3-d]пиримидин-4-амина гидрохлорид 14а (70 мг, 0,22 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли триэтиламин (67 мг, 0,66 ммоль). После взаимодействия в течение 0,5 часа в реакционную смесь добавляли 2-гликолевую кислоту (25 мг, 0,33 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния гексафторфосфат (210 мг, 0,33 ммоль). После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта 2-гидрокси-1-((3aS,5R,6aR/3aS,5R,6aR)-3а-метил-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанона 14 (11 мг, выход 13,9%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 330,4 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 11,62 (s, 1H), 8,09 (s, 1H), 7,12 (s, 1H), 6,54(s, 1H), 5,51 (s, 1H), 4,56 (s, 1H), 4,01-3,66 (m, 2H), 3,64-3,58 (m, 1H), 3,23 (s, 3H), 2,11 (d, 1H), 2,10-2,08 (m, 1H), 1,99-1,97 (m, 1H), 1,76-1,70 (m, 2H), 1,30-1,19 (m, 5H)
Пример 15
N-((3aR,5s,6aS)-2-(Циклопропилсульфонил)октагидроциклопента[с]пиррол-5-ил)-N-метил-7Н-пирроло[2,3-d]пиримидин-4-амин
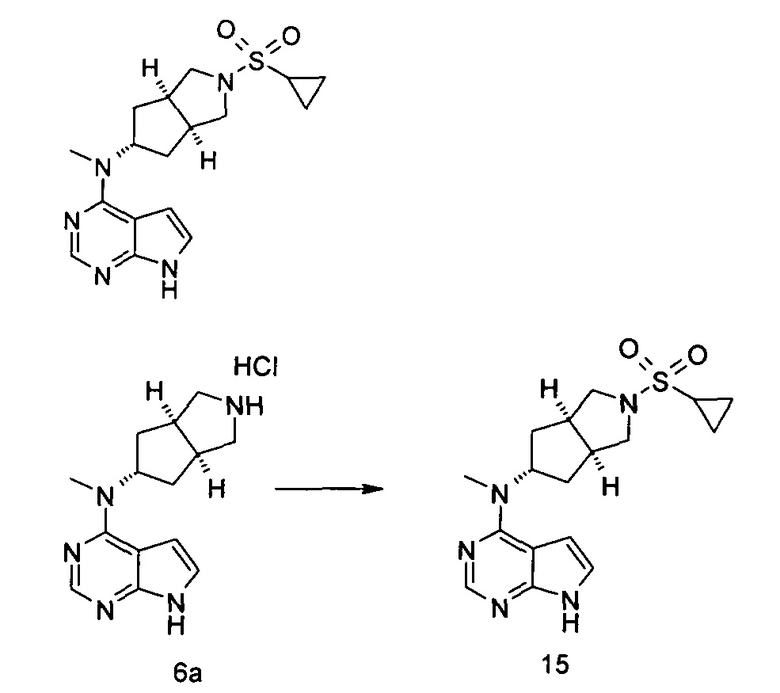
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли триэтиламин (78 мг, 0,7 ммоль). После взаимодействия в течение 0,5 часа в реакционную смесь по каплям добавляли циклопропилсульфонилхлорид (54 мг, 0,4 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли 10 мл H2O. Водную фазу и органическую фазу разделяли. Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта N-((3aR,5s,6aS)-2-(циклопропилсульфонил)октагидроциклопента[с]пиррол-5-ил)-N-метил-7Н-пирроло[2,3-d]пиримидин-4-амина 15 (20 мг, выход 14,3%) в виде белого твердого вещества.
MS m/z (ESI): 362,3 [M+1]
1H NMR (400 МГц, CDCl3): δ 10,11 (s, 1H), 8,31 (s, 1H), 7,08 (s, 1H), 6,70 (s, 1H), 5,52 (s, 1H), 3,57(s, 2H), 3,31-3,25 (m, 4H), 2,95-2,93 (m, 2H), 2,39-2,27 (m, 2H), 1,25-1,20 (m, 4H), 1,04-1,03 (m, 2H), 0,87-0,86 (m, 2H)
Пример 16
трет-Бутил (2-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)-2-оксоэтил)карбамат
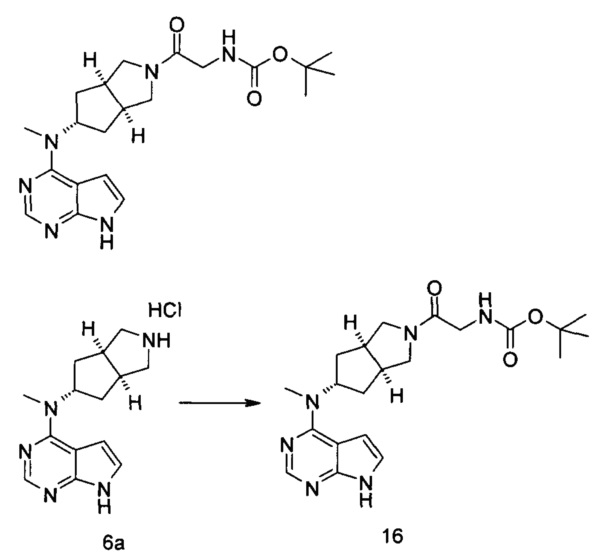
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (500 мг, 1,7 ммоль) растворяли в 20 мл дихлорметана, а затем добавляли триэтиламин (0,47 мл, 3,4 ммоль). После взаимодействия в течение 1 часа в реакционную смесь добавляли 2-(трет-бутоксиформамид)уксусную кислоту (0,36 г, 2,04 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния гексафторфосфат (0,76 г, 2,04 ммоль). После взаимодействия в течение 11 часов в реакционную смесь добавляли 100 мл дихлорметана. Реакционную смесь промывали насыщенным раствором хлорида натрия (20 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта трет-бутил (2-((3aR,5s,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)-2-оксоэтил)карбамата 16 (320 мг, выход 45,5%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 415,4 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,60 (s, 1H), 8,10 (s, 1H), 7,13-7,12 (m, 1H), 6,76-6,75 (m, 1H), 6,54 (s, 1H), 5,49-5,47 (m, 1H), 3,73-3,59 (m, 4H), 3,28-3,20 (m, 2H), 3,16 (s, 3H), 2,91-2,90 (m, 1H), 2,79-2,78 (m, 1H), 2,02-1,95 (m, 2H), 1,81-1,76 (m, 2H), 1,39 (s, 9H)
Пример 17
(S)-2-Гидрокси-1-((3aR,5R,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)пропан-1-он
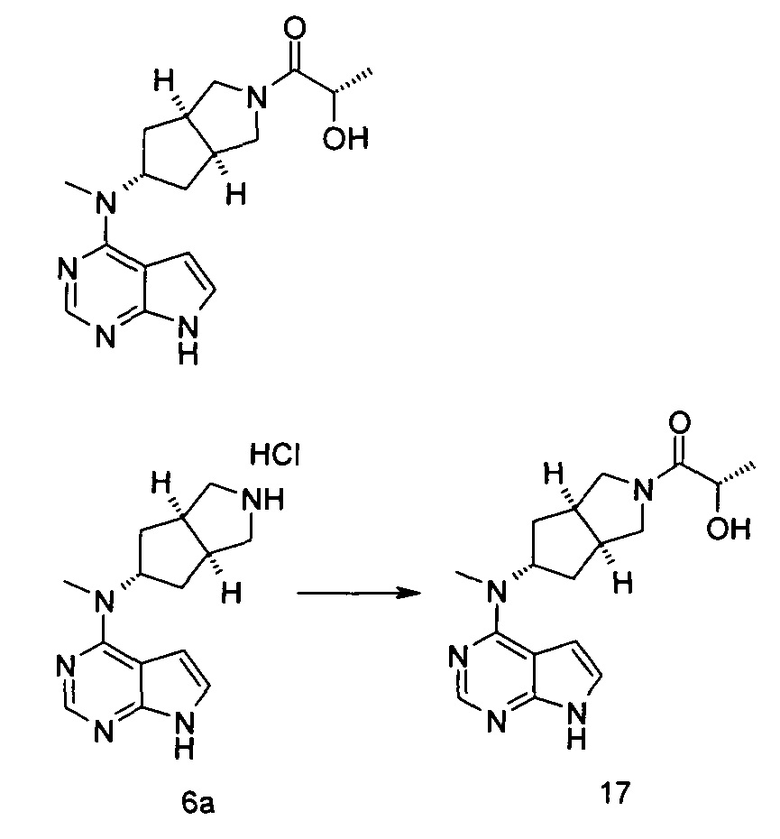
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид ба (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли триэтиламин (101 мг, 1 ммоль), (3)-2-гидракриловую кислоту (46 мг, 0,5 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния гексафторфосфат(190 мг, 0,5 ммоль). После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта (S)-2-гидрокси-1-((3aR,5R,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)пропан-1-она 17 (13 мг, выход 11,6%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 330,3 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,61 (s, 1H), 8,09-8,07 (m, 1H), 7,12-7,10 (m, 1H), 6,53 (s, 1H), 5,46-5,44 (m, 1H), 4,88-4,83 (m, 1H), 4,32-4,28 (m, 1H), 3,65-3,60 (m, 3H), 3,32-3,23 (m, 2H), 3,22-3,15 (m, 2H), 2,88-2,79 (m, 2H), 2,01-1,95 (m, 2H), 1,77-1,74 (m, 2H), 1,23-1,17 (m, 3H)
Пример 18
2-Метокси-1-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанон
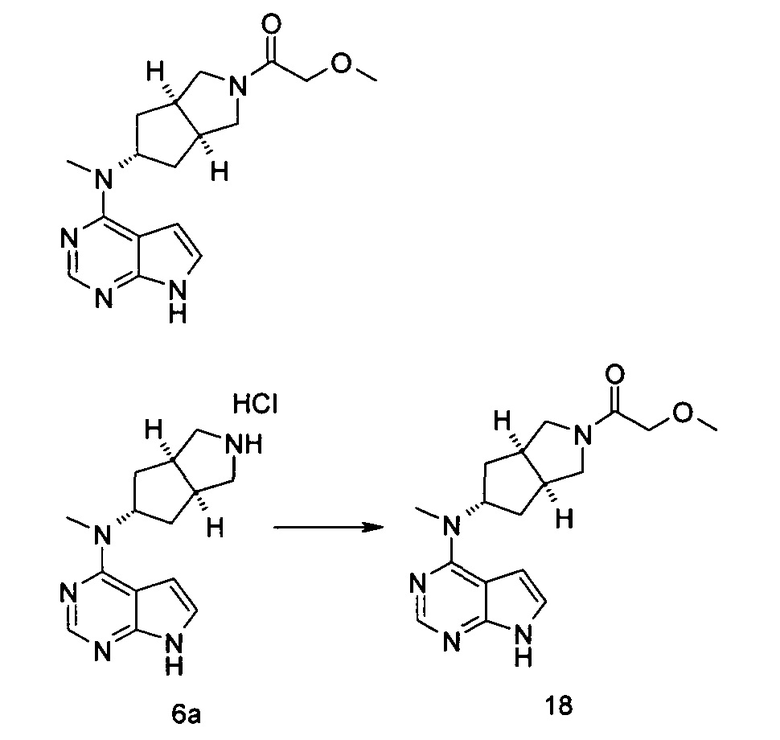
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли триэтиламин (101 мг, 1,02 ммоль). После взаимодействия в течение 0,5 часа в реакционную смесь по каплям добавляли 2-метоксиуксусную кислоту (30 мг, 0,33 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли 10 мл Н2О. Водную фазу и органическую фазу разделяли. Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта 2-метокси-1-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанона 18 (20 мг, выход 17,9%) в виде белого твердого вещества.
MS m/z (ESI): 330,3 [М+1]
1H NMR (400 МГц, CDCl3): δ 10,51 (s, 1H), 8,30 (s, 1H), 7,06 (s, 1H), 6,54 (s, 1H), 5,66-5,58 (m, 1H), 4,07 (s, 2H), 3,84-3,72 (m, 2H), 3,50-3,46 (m, 4H), 3,45-3,36 (m, 1H), 3,26 (s, 3H), 2,97-2,86 (m, 2H), 2,08-2,04 (m, 2H), 1,95-1,93 (m, 2H)
Пример 19
(R)-2-Гидрокси-1-((3aR,5S,6aS)-5-(метил(7Н-пирроло(2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)пропан-1-он

N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли (Р)-2-гидракриловую кислоту (46 мг, 0,5 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния гексафторфосфат (190 мг, 0,5 ммоль). После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта (R)-2-гидрокси-1-((3aR,5S,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)пропан-1-она 19 (6 мг, выход 5,4%) в виде желтого твердого вещества.
MS m/z (ESI): 330,4 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 11,61 (s, 1H), 8,09-8,08 (m, 1H), 7,12-7,11 (m, 1H), 6,53 (s, 1H), 5,47-5,45 (m, 1H), 4,87-4,82 (m, 1H), 4,31-4,27 (m, 1H), 3,67-3,60 (m, 3H), 3,32-3,24 (m, 1H), 3,22-3,15 (m, 3H), 2,89-2,79 (m, 2H), 2,03-1,95 (m, 2H), 1,79-1,74 (m, 2H), 1,23-1,17 (m, 3H)
Пример 20
2-Амино-1-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)этанон
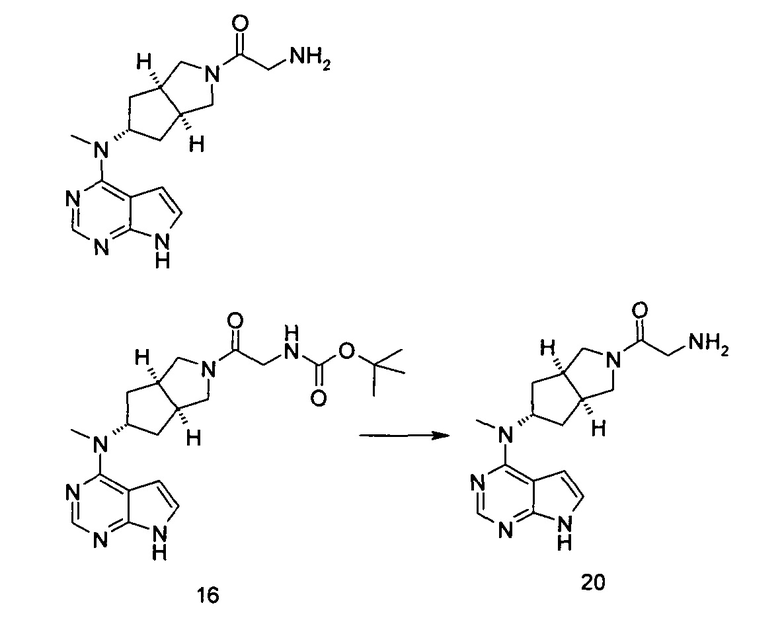
трет-Бутил (2-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)-2-оксоэтил)карбамат 16 (261 мг, 0,63 ммоль) растворяли в 8 мл дихлорметана, а затем по каплям добавляли 2 мл трифторуксусной кислоты. После взаимодействия в течение 1 часа в реакционную смесь добавляли 10 мл H2O и экстрагировали дихлорметаном (20 мл × 2). 1 М раствор гидроксида натрия добавляли в водную фазу для регуляции величины рН до 9, и экстрагировали дихлорметаном (30 мл × 5). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (15 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 2-амино-1-((3aR,5s,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанона 20 (145 мг, выход 73,2%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 315,5 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,93 (s, 1H), 8,30 (s, 2H), 8,13 (s, 1H), 7,21-7,20 (m, 1H), 6,63-6,62 (m, 1H), 5,49-5,45 (m, 1H), 3,83-3,65 (m, 4H), 3,40-3,35 (m, 2H), 3,20 (s, 3H), 2,93-2,83 (m, 2H), 2,03-1,97 (m, 2H), 1,83-1,81 (m, 2H)
Пример 21
N-(2-((3aR,5s,6aS)-5-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)-2-оксоэтил)метансульфонамид
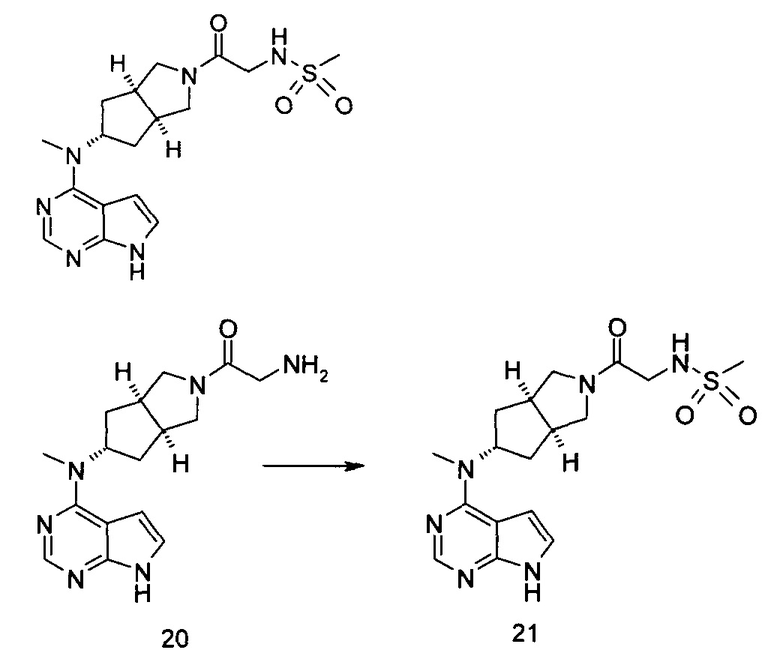
2-Амино-1-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанон 20 (60 мг, 0,19 ммоль) растворяли в 10 мл дихлорметана, а затем по каплям в ледяной бане добавляли триэтиламин (38 мг, 0,38 ммоль) и метилсульфонилхлорид (24 мг, 0,29 ммоль). После взаимодействия в течение 2 часов реакционную смесь концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта N-(2-((3aR,5s,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)-2-оксоэтил)метансульфонамида 21 (8 мг, выход 11,3%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 393,3 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 11,60 (s, 1H), 8,09 (s, 1H), 7,13-7,10 (m, 2H), 6,55-6,53 (m, 1H), 5,50-5,46 (m, 1H), 3,88-3,82 (m, 2H), 3,65-3,60 (m, 2H), 3,32-3,25 (m, 2H), 3,15 (s, 3H), 2,96 (s, 3H), 2,93-2,73 (m, 2H), 1,99-1,94 (m, 2H), 1,78-1,75 (m, 2H)
Пример 22
4-((3aR,5s,6aS)-5-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)-4-оксобутаннитрил

N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли триэтиламин (101 мг, 1 ммоль), 3-цианопропионовую кислоту (37 мг, 0,37 ммоль) и 0-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния гексафторфосфат (194 мг, 0,5 ммоль). После взаимодействия в течение 24 часов в реакционную смесь для остановки реакции добавляли 10 мл насыщенного хлорида аммония. Водную фазу и органическую фазу разделяли. Водную фазу экстрагировали дихлорметаном (15 мл × 2). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (15 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 4-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)-4-оксобутаннитрила 22 (6 мг, выход 5,2%) в виде белого твердого вещества.
MS m/z (ESI): 339,4 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 11,61 (s, 1H), 8,10-8,09 (m, 1H), 7,12-7,11 (m, 1H), 6,54-6,53 (m, 1H), 3,69-3,57 (m, 2H), 3,36-3,35 (m, 6H), 3,24-3,21 (m, 1H), 3,16-3,15 (m, 3H), 2,65-2,64 (m, 3H), 1,25-1,24 (m, 3H)
Пример 23
2-Гидрокси-1-((3aR,5R,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанон
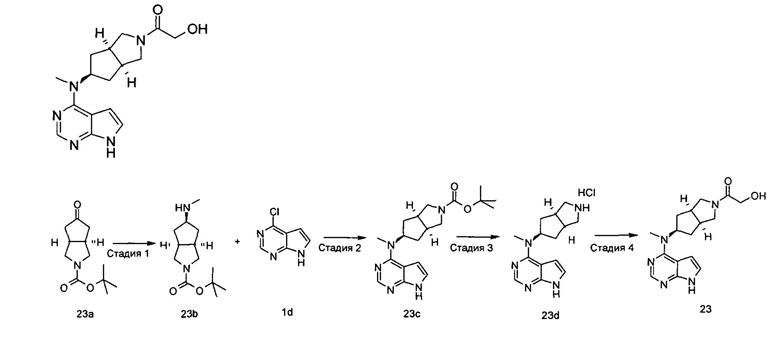
Стадия 1 (3aR,5R,6aS)-трет-Бутил 5-(метиламино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
(3aR,6aS)-трет-Бутил 5-оксогексагидроциклопента[с]пиррол-2(1H)-карбоксилат 23а (1,0 г, 4,44 ммоль) растворяли в 20 мл 2 М метиламина. После взаимодействия в течение 1 часа в реакционную смесь партиями добавляли цианоборгидрид натрия (420 мг, 6,66 ммоль). После взаимодействия в течение 1 часа реакционную смесь концентрировали при пониженном давлении, а затем добавляли 50 мл дихлорметана для растворения остатка, затем промывали насыщенным раствором хлорида натрия (20 мл). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (3aR,5R,6aS)-трет-бутил 5-(метиламино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 23b (1,23 г, желтый жир), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 241,3 [M+1]
Стадия 2
(3aR,5R,6aS)-трет-Бутил 5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
(3aR,5R,6aS)-трет-Бутил 5-(метиламино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат 23b (1,06 г, 4,41 ммоль) растворяли в 20 мл н-бутанола, а затем добавляли 4-хлор-7Н-пирроло[2,3-d]пиримидин (670 мг, 4,41 ммоль) и карбонат калия (1,22 г, 8,82 ммоль). После взаимодействия в течение 24 часов при 110°С реакционную смесь концентрировали при пониженном давлении, и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (3aR,5R,6aS)-трет-бутил 5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 23 с (830 мг, выход 52,9%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 358,2 [M+1]
Стадия 3
N-Метил-N-((3aR,5R,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид
(3aR,5R,6aS)-трет-Бутил 5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат 23 с (830 мг, 2,32 ммоль) растворяли в 10 мл 6 М раствора хлорида водорода в метаноле. После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении, промывали безводным диэтиловым эфиром (20 мл) и сушили в вакууме с получением указанного в заголовке продукта N-метил-N-((3aR,5R,6aS)-октагидроцикпопента[с]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорида 23d (630 мг, выход 92,6%) в виде серого твердого вещества.
Стадия 4
2-Гидрокси-1-((3aR,5R,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)этанон
N-Метил-N-((3aR,5R,6aS)-октагидроциклопента[с]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 23d (100 мг, 0,34 ммоль) растворяли в 5 мл н-бутанола, а затем добавляли 2-гидроксиметилацетат (61 мг, 0,68 ммоль) и 1,8-диазабицикло-бицикло[5,4,0]-7-гендецен (103 мг, 0,68 ммоль). После взаимодействия в течение 10 часов при 60°С реакционную смесь нагревали до 60°С в течение 10 часов. Реакционную смесь концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 2-гидрокси-1-((3aR,5R,6aS)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)этанона 23 (8 мг, выход 7,5%) в виде серого твердого вещества.
MS m/z (ESI): 316,2 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,61 (s, 1Н), 8,09 (s, 1H), 7,12 (s, 1H), 6,58 (s, 1H), 5,32-5,30 (m, 1H), 4,52-4,49 (m, 1H), 4,08-3,99 (m, 2H), 3,54-3,51 (m, 2H), 3,42-3,37 (m, 2H), 3,16 (s, 3H), 2,75-2,63 (m, 2H), 2,02-1,99 (m, 2H), 1,57-1,52 (m, 2H)
Пример 24
Метил 2-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)ацетат
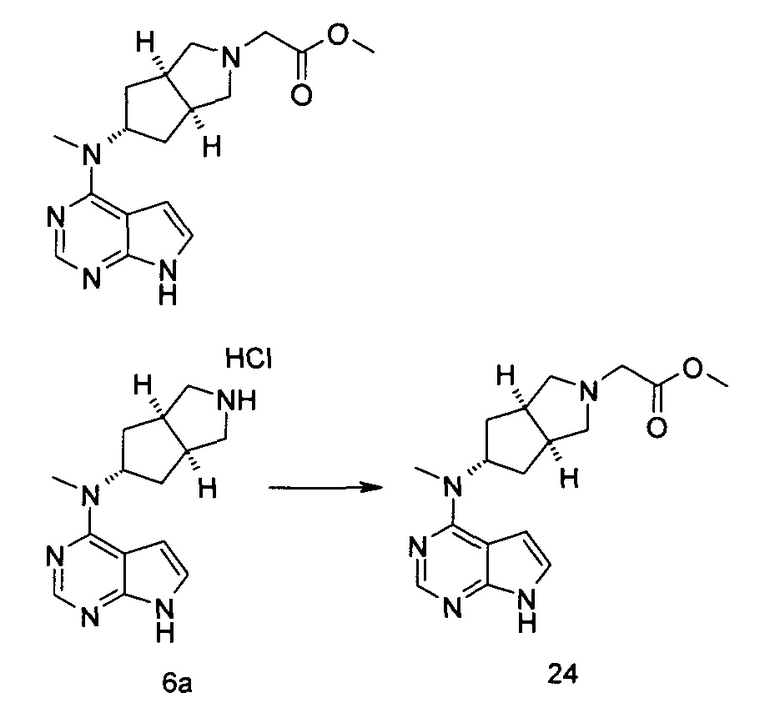
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли триэтиламин (101 мг, 1 ммоль) и 2-метилбромацетат (54 мг, 0,35 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли 15 мл насыщенного хлорида аммония для остановки реакции. Водную фазу и органическую фазу разделяли. Водную фазу экстрагировали дихлорметаном (15 мл × 2). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (25 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта метил 2-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)ацетата 24 (6 мг, выход 5,4%) в виде белого твердого вещества.
MS m/z (ESI): 330,4 [М+1]
1H NMR (400 МГц, CDCl3): δ 10,61 (s, 1H), 8,27-8,26 (m, 1H), 7,10-7,09 (m, 1H), 6,91 (s, 1H), 5,49-5,46 (m, 1H), 3,78 (s, 3H), 3,40-3,33 (m, 2H), 3,23 (s, 3H), 2,90-2,83 (m, 4H), 2,57-2,55 (m, 1H), 2,02-1,94 (m, 2H), 1,83-1,78 (m, 2H), 1,47-1,46 (m, 1H)
Пример 25
(3aR,5s,6aS)-Изопропил 5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
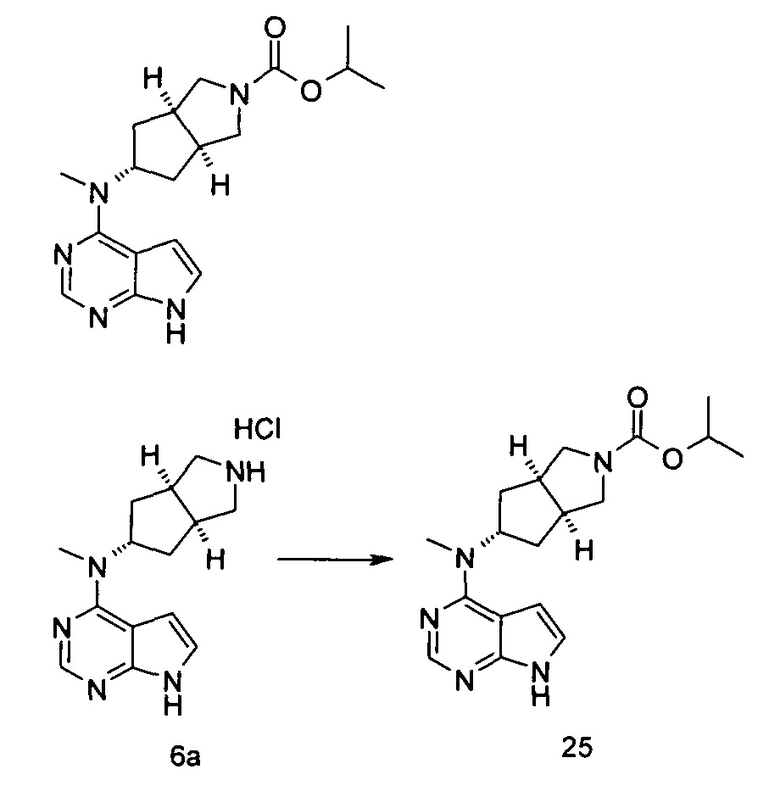
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли триэтиламин (101 мг, 1 ммоль) и изопропилхлорформиат (46 мг, 0,37 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли 15 мл насыщенного хлорида аммония для остановки реакции. Водную фазу и органическую фазу разделяли. Водную фазу экстрагировали дихлорметаном (15 мл × 2). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (15 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта (3aR,5s,6aS)-изопропил 5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 24 (6 мг, выход 5,1%) в виде белого твердого вещества.
MS m/z (ESI): 344,4 [M+1]
1H NMR (400 МГц, CDCl3): δ 10,61 (s, 1H), 8,34 (s, 1H), 7,10-7,09 (m, 1H), 6,60-6,59 (m, 1H), 5,64-5,60 (m, 1H), 5,01-4,95 (m, 1H), 3,32-3,30 (m, 4H), 2,91-2,90 (m, 1H), 2,11-2,03 (m, 2H), 1,98-1,93 (m, 2H), 1,31-1,30 (m, 10Н)
Пример 26
2-((3aR,5s,6aS)-5-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-ил)ацетонитрил
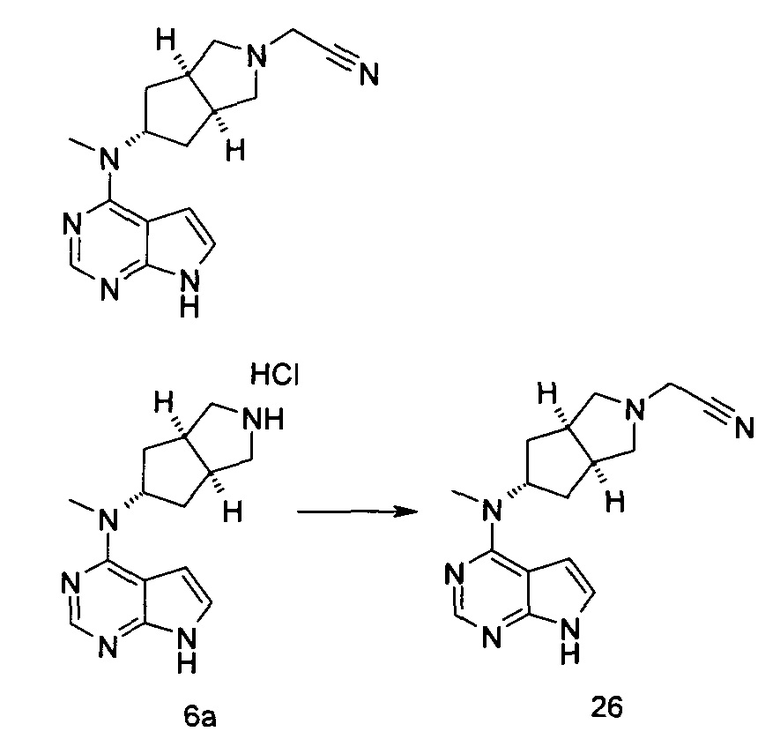
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли триэтиламин (101 мг, 1 ммоль) и бромацетонитрил (41 мг, 0,34 ммоль). После взаимодействия в течение 24 часов в реакционную смесь добавляли небольшое количество насыщенного раствора хлорида аммония для остановки реакции. Водную фазу и органическую фазу разделяли. Водную фазу экстрагировали дихлорметаном (10 мл × 3). Органическую фазу объединяли, последовательно промывали насыщенным раствором хлорида аммония (10 мл) и насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 2-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)ацетонитрила 26 (10 мг, выход 9,9%) в виде белого твердого вещества.
MS m/z (ESI): 297,2 [М+1]
1H NMR (400 МГц, CDCl3): δ 10,43 (s, 1H), 8,33-8,32 (m, 1H), 7,10-7,09 (m, 1H), 6,72-6,71 (m, 1H), 5,50-5,43 (m, 1H), 3,22 (s, 3H), 2,87-2,86 (m, 4H), 2,73-2,66 (m, 2H), 2,09-2,04 (m, 2H), 1,85-1,80 (m, 4H)
Пример 27
3-((3aR,5s,6aS)-5-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)пропаннитрил
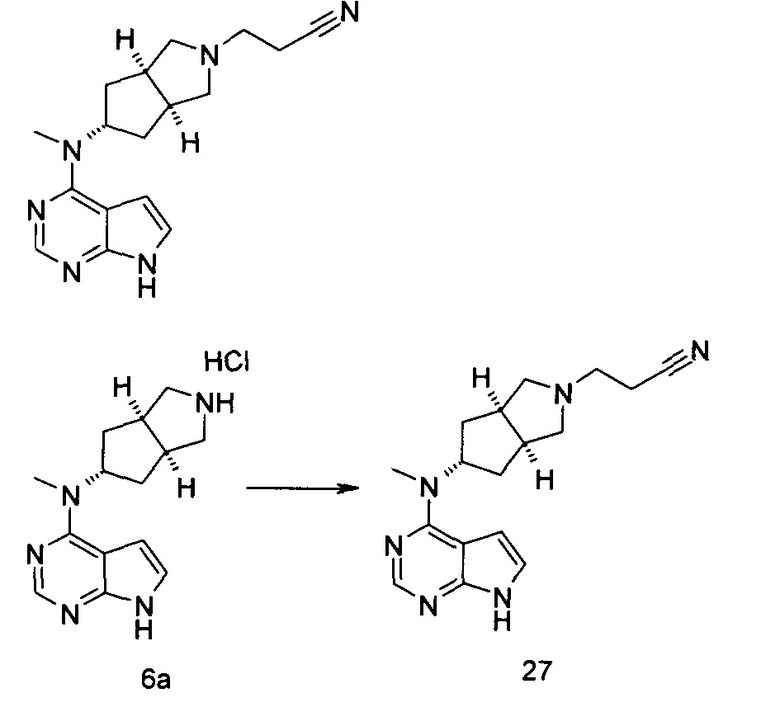
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (100 мг, 0,34 ммоль) растворяли в 5 мл дихлорметана, а затем добавляли триэтиламин (101 мг, 1 ммоль) и 3-бромпропионитрил (46 мг, 0,34 ммоль). После взаимодействия в течение 24 часов в реакционную смесь добавляли небольшое количество насыщенного раствора хлорида аммония для остановки реакции. Водную фазу и органическую фазу разделяли. Водную фазу экстрагировали дихлорметаном (10 мл × 3). Органическую фазу объединяли, последовательно промывали насыщенным раствором хлорида аммония (10 мл) и насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 3-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-ил)пропаннитрила 27 (10 мг, выход 9,4%) в виде белого твердого вещества.
MS m/z (ESI): 311,3 [M+1]
1H NMR (400 МГц, CDCl3): δ 10,77 (s, 1H), 8,35-8,34 (m, 1H), 7,12-7,11 (m, 1H), 6,75-6,74 (m, 1H), 5,50-5,41 (m, 1H), 3,22 (s, 3H), 2,83-2,80 (m, 4H), 2,63-2,59 (m, 4H), 1,98-1,81 (m, 4H), 1,81-1,78 (m, 2H)
Примеры 28, 29
N-((3aR,5s,6aS)-2-(4,6-Дихлорпиримидин-2-ил)октагидроциклопента[с]пиррол-5-ил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амин
N-((3aR,5s,6aS)-2-(2,6-Дихлорпиримидин-4-ил)октагидроциклопента[с]пиррол-5-ил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амин
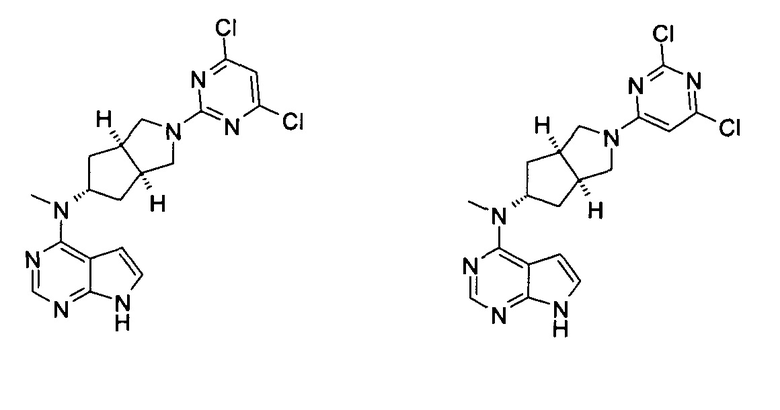
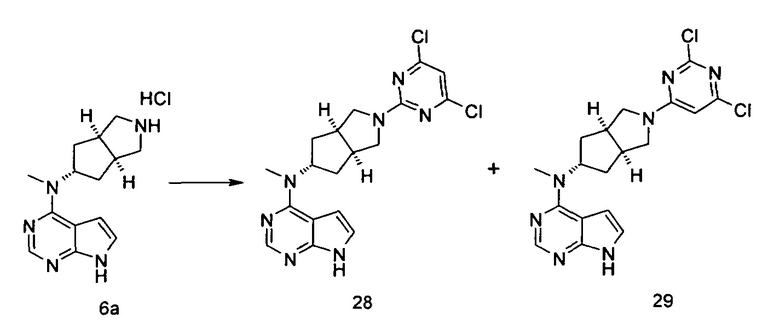
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (55 мг, 0,2 ммоль) растворяли в 5 мл этанола, а затем добавляли триэтиламин (52 мг, 0,5 ммоль). После взаимодействия в течение 0,5 часа в реакционную смесь добавляли 2,4,6-трихлорпиримидин (32 мг, 0,2 ммоль). После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанных в заголовке продуктов N-((3aR,5s,6aS)-2-(4,6-дихлорпиримидин-2-ил)октагидроциклопента[с]пиррол-5-ил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амина 28 (10 мг, выход 20,0%) в виде белого твердого вещества) и N-((3aR,5s,6aS)-2-(2,6-дихлорпиримидин-4-ил)октагидроциклопента[с]пиррол-5-ил)-N-метил-7Н-пирроло[2,3-d]пиримидин-4-амина 29 (20 мг, выход 40,0%) в виде белого твердого вещества.
MS m/z (ESI): 404,3 [M+1]
1H NMR (400 МГц, CDCl3): δ 9,81 (s, 1H), 8,28 (s, 1H), 7,01 (s, 1H), 6,57 (s, 1H), 6,50 (d, 1H), 5,64-5,60 (m, 1H), 3,90-3,85 (m, 2H), 3,59-3,56 (m, 2H), 3,25 (s, 3H), 3,04-2,96 (m, 2H), 2,13-2,05 (m, 2H), 1,99-1,95 (m, 2H)
1H NMR (400 МГц, CDCl3): δ 9,97 (s, 1H), 8,28 (s, 1H), 7,04 (s, 1H), 6,52 (d, 1H), 6,26 (d, 1H), 5,69-5,65 (m, 1H), 3,96-3,92 (m, 1H), 3,68-3,64 (m, 2H), 3,28-3,24 (m, 4H), 3,06-3,01 (m, 2H), 2,09-2,00 (m, 2H), 1,98-1,95 (m, 2H).
Пример 30
(3aR,5s,6aS)-Метил 5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
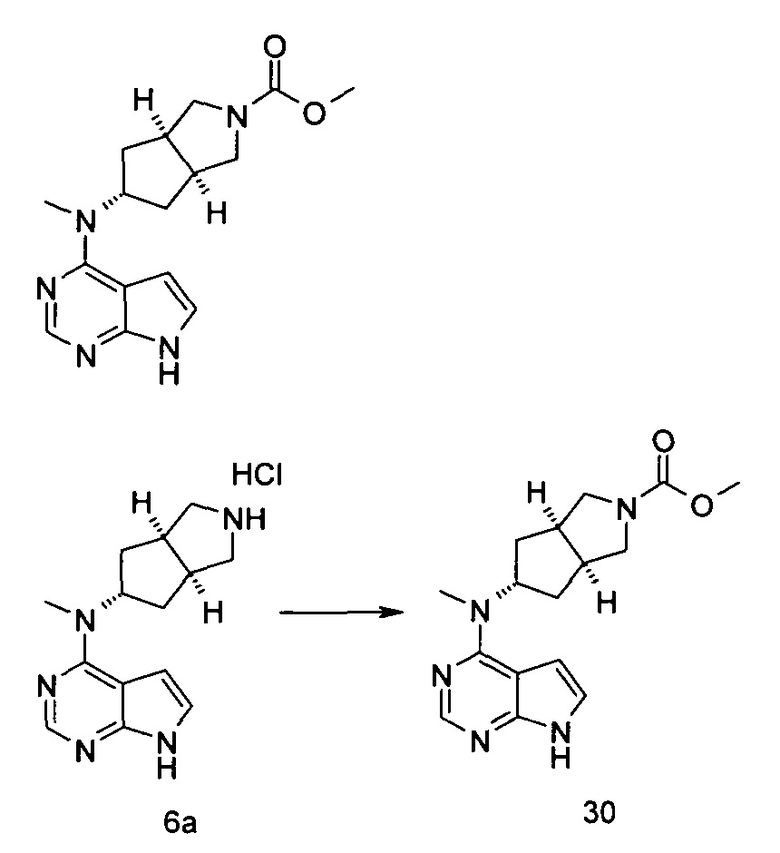
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид ба (100 мг, 0,34 ммоль) растворяли в 15 мл дихлорметана, а затем в ледяной бане добавляли метилхлорформиат (44 мг, 0,47 ммоль) и 3-триэтиламин (59 мг, 0,58 ммоль). После взаимодействия в течение 16 ч в реакционную смесь добавляли небольшое количество насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (20 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта (3aR,5s,6aS)-метил 5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 30 (50 мг, выход 41,0%) в виде белого твердого вещества.
MS m/z (ESI): 316,3 [М+1]
1H NMR (400 МГц, CDCl3): δ 10,60 (s, 1Н), 8,26 (s, 1H), 7,06-7,05 (m, 1H), 6,54-6,53 (m, 1H), 5,60-5,56 (m, 1H), 3,72 (s, 3H), 3,70-3,65 (m, 2H), 3,32-3,25 (m, 2H), 3,24 (s, 3H), 2,91-2,85 (m, 2H), 2,06-2,00 (m, 2H), 1,92-1,82 (m, 2H)
Пример 31
2-Гидрокси-1-((1R,5S,6s)-6-((метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)метил)-3-азабицикло[3.1.0]гексан-3-ил)этанон
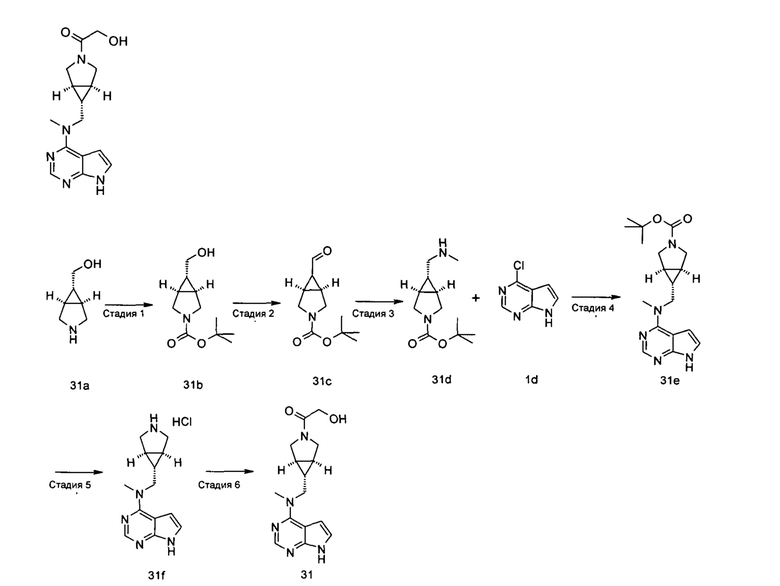
Стадия 1
(1R,5S,6r)-трет-Бутил 6-(гидроксиметил)-3-азабицикло[3.1.0]гексан-3-карбоксилат
(1R,5S,6r)-3-Азабицикло[3.1.0]гексан-6-илметанол 31а (10 г, 88,4 ммоль) растворяли в смеси растворителей диоксана и H2O (V/V=3/2), а затем добавляли гидроксид натрия (4,2 г, 106 ммоль) и ди-трет-бутилдикарбонат (28,9 г, 132,6 ммоль). После взаимодействия в течение 10 часов в реакционную смесь добавляли 50 мл H2O. Реакционную смесь экстрагировали этилацетатом (150 мл × 3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (50 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции С с получением указанного в заголовке продукта (1R,5S,6r)-трет-бутил 6-(гидроксиметил)-3-азабицикло[3.1.0]гексан-3-карбоксилата 31b (16,9 г, выход 89,9%) в виде светло-желтой жидкости.
Стадия 2
(1R,5S)-трет-Бутил 6-формил-3-азабицикло[3,1,0]гексан-3-карбоксилат
Оксалилхлорид (2,41 мл, 28,1 ммоль) растворяли в 100 мл дихлорметана, а затем при -78°С по каплям добавляли диметилсульфоксид (4,32 мл, 60,8 ммоль). После взаимодействия в течение 30 минут в реакционную смесь добавляли (1R,5S,6r)-трет-бутил 6-(гидроксиметил)-3-азабицикло[3.1.0]гексан-3-карбоксилат 31b (5,0 г, 23,4 ммоль). После взаимодействия в течение 1 часа в реакционную смесь добавляли триэтиламин (16,23 мл, 117 ммоль). После взаимодействия в течение 30 минут при комнатной температуре в реакционную смесь добавляли 200 мл дихлорметана, 50 мл Н2О, 50 мл 1 М соляной кислоты и 50 мл насыщенного раствора бикарбоната натрия. Водную фазу и органическую фазу разделяли, органическую фазу промывали насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции С с получением указанного в заголовке продукта (1R,5S)-трет-бутил 6-формил-3-азабицикло[3.1.0]гексан-3-карбоксилата 31с (4,12 г, выход 82,4%) в виде светло-желтой жидкости.
Стадия 3
(1R,5S,6s)-трет-Бутил 6-((метиламино)метил)-3-азабицикло[3.1.0]гексан-3-карбоксилат
(1R,5S)-трет-Бутил 6-формил-3-азабицикло[3.1.0]гексан-3-карбоксилат 31с (3,05 г, 14,4 ммоль) растворяли в 20 мл раствора метиламина в метаноле. После взаимодействия в течение 24 часов в реакционную смесь в ледяной бане партиями добавляли цианоборгидрид натрия (1,09 г, 17,3 ммоль). После взаимодействия в течение 2 часов при комнатной температуре в реакционную смесь добавляли 15 мл насыщенного раствора хлорида натрия и 15 мл H2O. Реакционную смесь экстрагировали этилацетатом (30 мл × 5). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (15 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке продукта (1R,5S,6s)-трет-бутил 6-((метиламино)метил)-3-азабицикло[3.1.0]гексан-3-карбоксилата 31d (3,95 г) в виде бесцветного жира, который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 227,46 [M+1]
Стадия 4
(1R,5S,6s)-трет-Бутил 6-((метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)метил)-3-азабицикло[3.1.0]гексан-3-карбоксилат
4-Хлор-7H-пирроло[2,3-d]пиримидин 1d (2,21 г, 14,4 ммоль) растворяли в 320 мл н-бутанола, а затем добавляли (1R,5S,6s)-трет-бутил 6-((метиламино)метил)-3-азабицикло[3,1,0]гексан-3-карбоксилат 31d (3,25 г, 14,4 ммоль) и карбонат калия (3,97 г, 28,8 ммоль). После взаимодействия в течение 24 часов при 120°С в реакционную смесь добавляли 150 мл этилацетата и 20 мл H2O. Водную фазу и органическую фазу разделяли, органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (1R,5S,6s)-трет-бутил 6-((метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)метил)-3-азабицикло[3.1.0]гексан-3-карбоксилата 31е (1,82 г, выход 36,8%) в виде белого твердого вещества.
MS m/z (ESI): 344,2 [M+1]
Стадия 5
N-((1R,5S,6r)-3-Азабицикло[3.1.0]гексан-6-илметил)-N-метил-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорид
(1R,5S,6s)-трет-Бутил 6-((метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)метил)-3-азабицикло[3.1.0]гексан-3-карбоксилат 31е (1,72 г, 5,01 ммоль) растворяли в 20 мл 1 М раствора хлорида водорода в метаноле. После взаимодействия в течение 14 часов реакционную смесь концентрировали при пониженном давлении, и образующийся остаток промывали смесью растворителей дихлорметана и диэтилового эфира (V/V=1/1), сушили в вакууме с получением указанного в заголовке продукта N-((1R,5S,6r)-3-азабицикло[3.1.0]гексан-6-илметил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорида 31f (1,17 г, выход 95,9%) в виде серого твердого вещества.
MS m/z (ESI): 244,46 [M+1]
Стадия 6
2-Гидрокси-1-((1R,5S,6s)-6-((метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)метил)-3-азабицикпо[3.1.0]гексан-3-ил)этанон
N-((1R,5S,6r)-3-Азабицикло[3.1.0]гексан-6-илметил)-N-метил-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 31f (100 мг, 0,36 ммоль) растворяли в 5 мл тетрагидрофурана, а затем добавляли триэтиламин (111 мг, 1,1 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния гексафторфосфат (204 мг, 0,54 ммоль). После взаимодействия в течение 30 минут в реакционную смесь добавляли 2-гликолевую кислоту (30 мг, 0,39 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли небольшое количество насыщенного раствора хлорида аммония для остановки реакции. Водную фазу и органическую фазу разделяли. Водную фазу экстрагировали дихлорметаном (10 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 2-гидрокси-1-((1R,5S,6s)-6-((метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)метил)-3-азабицикло[3.1.0]гексан-3-ил)этанона 31 (5 мг, выход 4,7%) в виде желтого твердого вещества.
MS m/z (ESI): 300,45 [М-1]
1H NMR (400 МГц, CDCl3): δ 10,64(s, 1Н), 8,27 (s, 1Н), 7,12-7,11 (m, 1H), 6,61-6,60 (m, 1Н), 5,34-5,33 (m, 1Н), 4,09-4,04 (m, 2H), 3,83-3,81 (m, 2H), 3,49 (s, 3H), 1,79-1,72 (m, 2H), 1,53-1,45 (m, 2H), 1,31-1,28 (m, 1Н), 1,00-0,89 (m, 2H)
Пример 32
2-((1R,5S,6r)-6-((Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)метил)-3-азабицикло[3.1.0]гексан-3-ил)ацетонитрил
N-((1R,5S,6r)-3-Азабицикпо[3.1.0]гексан-6-илметил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 31f (100 мг, 0,36 ммоль) растворяли в 5 мл ацетонитрила, а затем добавляли триэтиламин (109 мг, 1,1 ммоль) и бромацетонитрил (47,5 мг, 0,4 ммоль). После взаимодействия в течение 16 часов в реакционную смесь добавляли небольшое количество насыщенного раствора хлорида аммония для остановки реакции. Водную фазу и органическую фазу разделяли. Водную фазу экстрагировали дихлорметаном (10 мл × 3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 2-((1R,5S,6r)-6-((метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)метил)-3-азабицикло[3.1.0]гексан-3-ил)ацетонитрила 32 (15 мг, выход 14,9%) в виде белого твердого вещества.
MS m/z (ESI): 283,2 [М+1]
1H NMR (400 МГц, CDCl3): δ 10,64 (s, 1H), 8,31 (s, 1H), 7,09-7,08 (m, 1H), 6,64-6,63 (m, 1H), 3,76-3,74 (m, 2H), 3,63 (s, 2H), 3,47 (s, 3H), 3,00-2,98 (m, 2H), 2,75-2,73 (m, 2H), 1,57-1,53 (m, 2H), 1,33-1,30 (m, 1H)
Пример 33
N-(((1R,5S,6s)-3-(2-Хлорпиримидин-4-ил)-3-азабицикло[3.1.0]гексан-6-ил)метил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амин
N-((1R,5S,6r)-3-Азабицикло[3.1.0]гексан-6-илметил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 31f (100 мг, 0,36 ммоль) растворяли в 5 мл этанола, а затем добавляли триэтиламин (108 мг, 1,07 ммоль) и 4,6-дихлорпиримидин (26,6 мг, 0,17 ммоль). После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта N-(((1R,5S,6s)-3-(2-хлорпиримидин-4-ил)-3-азабицикло[3.1.0]гексан-6-ил)метил)-N-метил-7H-пирроло[2,3-d]пиримидин-4-амина 33 (5 мг, выход 7,9%) в виде белого твердого вещества.
MS m/z (ESI): 356,1 [М+1]
1H NMR (400 МГц, CDCl3): δ 10,46 (s, 1H), 8,25 (s, 1H), 7,97 (s, 1H), 7,07 (s, 1H), 6,59 (s, 1H), 6,13 (s, 1H), 4,03-4,01 (m, 1H), 3,84-3,81 (m, 2H), 3,49-3,47 (m, 3H), 3,42 (s, 3H), 1,96-1,94 (m, 2H), 1,07-1,05 (m, 1H)
Пример 34
(3aR,55,6aS)-N-(3-Метокси-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамид

Стадия 1
Фенил (3-метокси-1,2,4-тиадиазол-5-ил)карбамат
3-Метокси-1,2,4-тиадиазол-5-амин 34а (500 мг, 3,82 ммоль) и фенилкарбонохлоридат 34b (600 мг, 3,82 ммоль) растворяли в 20 мл дихлорметана, а затем добавляли триэтиламин (0,8 мл, 5,73 ммоль). После взаимодействия в течение 16 часов в реакционную смесь для разбавления раствора добавляли 30 мл H2O. Водную фазу и органическую фазу разделяли, водную фазу экстрагировали дихлорметаном (20 мл × 2), и органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта фенил (3-метокси-1,2,4-тиадиазол-5-ил)карбамата 34 с (200 мг, выход 20,8%) в виде белого твердого вещества.
MS m/z (ESI): 252,0 [М+1]
Стадия 2
(3aR,5s,6aS)-N-(3-Метокси-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамид
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (120 мг, 0,47 ммоль) растворяли в 15 мл тетрагидрофурана, а затем добавляли фенил (3-метокси-1,2,4-тиадиазол-5-ил)карбамат 34 с (117 мг, 0,47 ммоль) и триэтиламин (0,13 мл, 0,94 ммоль). После взаимодействия в течение 5 часов при 60°С в реакционную смесь добавляли 30 мл H2O и экстрагировали дихлорметаном (50 мл × 3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (50×2 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (3aR,5s,6aS)-N-(3-метокси-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамида 34 (50 мг, выход 25,9%) в виде белого твердого вещества.
MS m/z (ESI): 412,9 [М-1]
1H NMR (400 МГц, DMSO-d6): δ 11,60 (m, 2Н), 8,08 (s, 1Н), 7,06-7,05 (m, 1Н), 6,53-6,51 (m, 1Н), 5,48-5,44 (m, 1Н), 3,90 (s, 3H), 3,69-3,65 (m, 2Н), 3,37-3,32 (m, 2Н), 3,16 (s, 3H), 2,90-2,88 (m, 2Н), 2,02-1,99 (m, 2Н), 1,80-1,77 (m, 2Н)
Пример 35
1-((4aS,7aR)-6-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-2(3H,4Н,4aH,5H,6H,7H,7aH)-ил)этанон
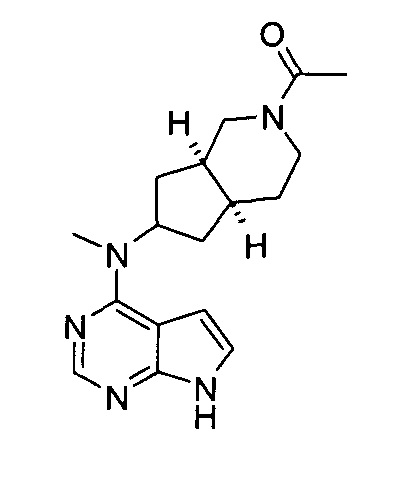
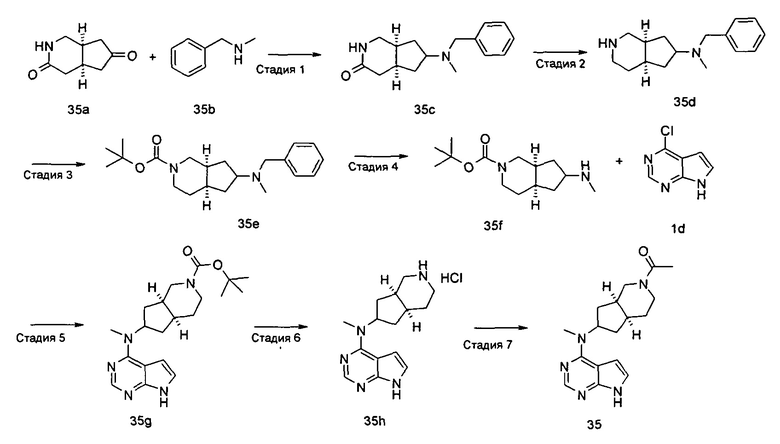
Стадия 1
(4aS,7aS)-6-(Бензил(метил)амино)гексагидро-1Н-циклопента[с]пиридин-3(2Н)-он
(4aS,7aS)-Тетрагидро-1Н-циклопента[с]пиридин-3,6(2Н,4Н)-дион 35а (3 г, 19,60 ммоль, полученный хорошо известным способом "Tetrahedron: Asymmetry, 1997, 8 (17), 2893-2904") и N-метил-1-фенилметанамин 35b (2,37 г, 19,60 ммоль) растворяли в 5 мл метанола, а затем добавляли две капли уксусной кислоты. После перемешивания и взаимодействия в течение 3 часов в реакционную смесь добавляли цианоборгидрид натрия (1,80 г, 29,40 ммоль). После взаимодействия в течение 12 часов реакционную смесь фильтровали и концентрировали при пониженном давлении, и образующийся остаток очищали при помощи колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (4aS,7aS)-6-(бензил(метил)амино)гексагидро-1Н-циклопента[с]пиридин-3(2Н)-она 35 с (1,99 г, выход 39,8%) в виде желтого твердого вещества. MS m/z (ESI): 259,2 [М+1]
Стадия 2
(4aR,7aS)-N-Бензил-N-метилоктагидро-1H-циклопента[с]пиридин-6-амин
(4aS,7aS)-6-(Бензил(метил)амино)гексагидро-1Н-циклопента[с]пиридин-3(2H)-он 35 с (3,90 г, 15,11 ммоль) растворяли в 150 мл тетрагидрофурана, а затем партиями в ледяной бане добавляли алюмогидрид лития (3 г, 78,60 ммоль). После перемешивания в течение 48 часов в реакционную смесь добавляли 10 мл ледяной воды. Реакционную смесь фильтровали, промывали дихлорметаном (50 мл) и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (4aR,7aS)-N-бензил-N-метилоктагидро-1H-циклопента[с]пиридин-6-амина 35d (3,69 г, белое твердое вещество), которое использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 245,2 [М+1]
Стадия 3
(4aR,7aS)-трет-Бутил 6-(бензил(метил)амино)гексагидро-1Н-циклопента[с]пиридин-2(3H)-карбоксилат
(4aR,7aS)-N-Бензил-N-метилоктагидро-1H-циклопента[с]пиридин-6-амин 35d (3,69 г, 15,11 ммоль) растворяли в 50 мл дихлорметана, а затем добавляли ди-трет-бутил дикарбонат (4,94 г, 22,60 ммоль) и этиламин (3,81 г, 37,75 ммоль). После перемешивания в течение 12 часов в реакционную смесь добавляли 20 мл H2O и экстрагировали дихлорметаном (30 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (4aR,7aS)-трет-бутил 6-(бензил(метил)амино)гексагидро-1Н-циклопента[с]пиридин-2(3H)-карбоксилата 35е (3,28 г, выход 63%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 345,3 [М+1]
Стадия 4
(4aR,7aS)-трет-Бутил 6-(метиламино)гексагидро-1Н-циклопента[с]пиридин-2(3H)-карбоксилат
(4aR,7aS)-трет-Бутил 6-(бензил(метил)амино)гексагидро-1Н-циклопента[с]пиридин-2(3H)-карбоксилат 35е (2 г, 5,81 ммоль) растворяли в 150 мл метанола, а затем добавляли гидроксид палладия на углероде (500 мг, 25%). После продувки реактора водородом, осуществляемой три раза, реакционную смесь перемешивали в течение 72 часов, и затем фильтровали, промывали метанолом (20 мл) и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (4aR,7aS)-трет-бутил 6-(метиламино)гексагидро-1Н-циклопента[с]пиридин-2(3H)-карбоксилата 35f (1,72 г) в виде бесцветной вязкой жидкости, который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 255,2 [М+1]
Стадия 5
(4aS,7aR)-трет-Бутил 6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-циклопента[с]пиридин-2(3H)-карбоксилат
4-Хлор-7H-пирроло[2,3-d]пиримидин 1d (1,78 г, 11,65 ммоль) растворяли в 50 мл 1,4-диоксана, а затем добавляли (4aR,7aS)-трет-бутил 6-(метиламино)гексагидро-1H-циклопента[с]пиридин-2(3H)-карбоксилат 35f (2,96 г, 11,65 ммоль) и карбонат калия (3,22 г, 23,30 ммоль). После взаимодействия в течение 48 часов при 110°С реакционную смесь концентрировали при пониженном давлении, а затем добавляли 50 мл H2O, и экстрагировали этилацетатом (30 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке (4aS,7aR)-трет-бутил 6-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-циклопента[с]пиридин-2(3H)-карбоксилата 35g (1,34 г, выход 31%) в виде белого твердого вещества.
MS m/z (ESI): 372,2 [М+1]
Стадия 6
N-Метил-N-((4aS,7aR)-октагидро-1H-циклопента[c]пиридин-6-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид
(4aS,7aR)-трет-Бутил 6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-циклопента[с]пиридин-2(3H)-карбоксилат 35g (1,34 г, 3,61 ммоль) растворяли в 15 мл раствора 6 М раствора хлорида водорода в растворе метанола. После взаимодействия в течение 12 часов реакционную смесь концентрировали при пониженном давлении, а затем добавляли 20 мл Н2О и 10% раствор гидроксида натрия до тех пор, пока рН реакционной смеси не составил от 9 до 10. Реакционную смесь экстрагировали дихлорметаном (20 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке N-метил-N-((4aS,7aR)-октагидро-1H-циклопента[с] пиридин-6-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорида 35h (1 г, белое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 272,2 [М+1]
Стадия 7
1-((4aS,7aR)-6-(Метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-2(3H,4Н,4aH,5H,6H,7H,7аН)-ил)этанон
N-Метил-N-((4aS,7aR)-октагидро-1H-циклопента[с]пиридин-6-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 35h (100 мг, 0,37 ммоль) растворяли в 5 мл N,N-диметилформамида, а затем добавляли безводную уксусную кислоту (22 мг, 0,37 ммоль), триэтиламин (112 мг, 1,10 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (106 мг, 0,55 ммоль) и 1-гидроксибензотриазол (74 мг, 0,55 ммоль). После взаимодействия в течение 12 часов в реакционную смесь добавляли 10 мл H2O. Реакционную смесь экстрагировали дихлорметаном (20 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта 1-((4aS,7aR)-6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-2(3H,4H,4aH,5Н,6Н,7H,7aH)-ил)этанона 35 (7 мг, выход 5%) в виде белого твердого вещества.
MS m/z (ESI): 314,2 [М+1]
1H NMR (400 МГц, CDCl3): δ 11,34 (s, 1H), 8,32 (s, 1H), 7,08 (s, 1H), 6,56 (s, 1H), 5,43-5,45 (m, 1H), 3,88-3,91 (m, 1H), 3,27-3,55 (m, 6H),2,28-2,30 (m, 2H), 1,91-1,96 (m, 6H), 1,58-1,60 (m, 3H)
Пример 36
(4aS,7aR)-Метил 6-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-2(3H)-карбоксилат
N-Метил-N-((4aS,7aR)-октагидро-1H-циклопента[с]пиридин-6-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 35h (100 мг, 0,37 ммоль) растворяли в 10 мл дихлорметана, а затем в ледяной бане добавляли метилхлорформиат (35 мг, 0,37 ммоль) и триэтиламин (56 мг, 0,55 ммоль). После взаимодействия в течение 12 часов в реакционную смесь добавляли 10 мл H2O. Реакционную смесь экстрагировали дихлорметаном (10 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта (4aS,7aR)-метил 6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-циклопента[с]пиридин-2(3H)-карбоксилата 36 (20 мг, выход 16,5%) в виде белого твердого вещества.
MS m/z (ESI): 330,3 [М+1]
1H NMR (400 МГц, CDCl3): δ 10,57 (s, 1H), 8,24 (s, 1H), 7,05 (s, 1H), 6,56 (s, 1H), 5,38-5,39 (m, 1H), 3,70 (s, 3H), 3,61-3,62 (m, 2H), 3,39-3,40 (m, 1H), 3,23-3,27 (m, 4H), 2,20-2,23 (m, 2H), 2,05-2,09 (m, 1H), 1,83-1,84 (m, 2H), 1,52-1,57 (m, 3H)
Пример 37
2-Гидрокси-1-((4aS,7aR)-6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-2(3H,4H,4aH,5H,6H,7H,7aH)-ил)этанон
N-Метил-N-((4aS,7aR)-октагидро-1H-циклопента[с]пиридин-6-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 35h (80 мг, 0,30 ммоль) растворяли в 5 мл N,N-диметилформамида, а затем добавляли 2-гликолевую кислоту (22 мг, 0,30 ммоль), триэтиламин (89 мг, 0,89 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (84 мг, 0,44 ммоль) и 1-гидроксибензотриазол (60 мг, 0,44 ммоль). После взаимодействия в течение 12 часов реакционную смесь концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта 2-гидрокси-1-((4aS,7aR)-6-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-2(3H,4H,4аН,5H,6H,7Н,7aH)-ил)этанона 37 (8 мг, выход 8,2%) в виде белого твердого вещества.
MS m/z (ESI): 330,2 [М+1]
1H NMR (400 МГц, CDCl3): δ 11,57 (s, 1H), 8,30 (s, 1H), 7,07 (s, 1H), 6,56 (s, 1H), 5,40-5,44 (m, 1H), 4,14-4,18 (m, 1H), 3,34-3,39 (m, 2H), 3,27(s, 3H), 3,18-3,22(m, 1H), 2,30-2,32 (m, 2H), 1,95-2,13 (m, 3H), 1,66-1,68 (m, 1H), 1,52-1,57 (m, 2H)
Пример 38
(4aS,7aR)-N-(3-Метокси-1,2,4-тиадиазол-5-ил)-6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-2(3H)-карбоксамид
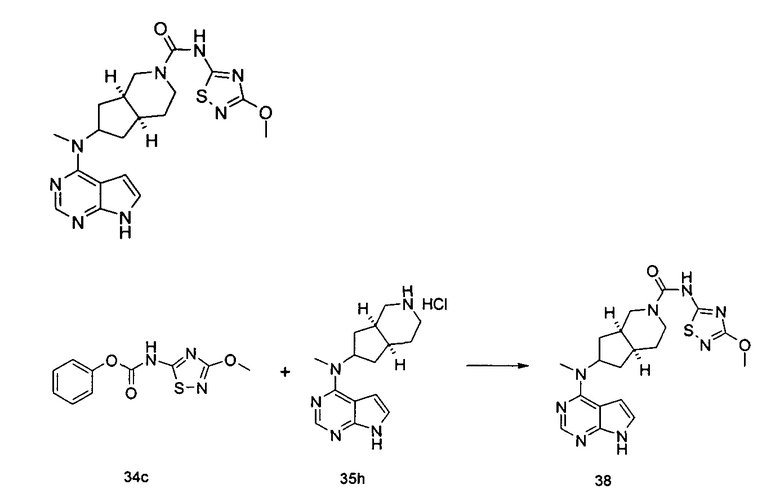
N-Метил-N-((4aS,7aR)-октагидро-1Н-циклопента[с]пиридин-6-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 35h (80 мг, 0,30 ммоль) растворяли в 20 мл тетрагидрофурана, а затем добавляли фенил(3-метокси-1,2,4-тиадиазол-5-ил)карбамат 34с (75 мг, 0,30 ммоль) и триэтиламин (0,06 мл, 0,44 ммоль). После взаимодействия в течение 3 часов при 60°С в реакционную смесь добавляли 30 мл Н2О. Реакционную смесь экстрагировали дихлорметаном (30 мл × 2). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (20 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (4aS,7aR)-N-(3-метокси-1,2,4-тиадиазол-5-ил)-6-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-2(3H)-карбоксамида 38 (30 мг, выход 23,8%) в виде белого твердого вещества.
MS m/z (ESI): 429,3 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,63 (s, 1H), 11,61 (s, 1H), 8,08 (s, 1H), 7,12 (d, 1H), 6,58 (d, 1H), 5,26-4,30 (m, 1H), 3,84 (s, 3H), 3,69-3,73 (m, 1H), 3,59-3,64 (m, 1H), 3,38-3,40 (m, 2H), 3,17 (s, 3H), 2,20-2,23 (m, 2H), 1,82-1,92 (m, 3H), 1,60-1,65 (m, 2H), 1,40-1,48 (m, 1H)
Пример 39
3-((4aS,7aR)-6-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-циклопента[с]пиридин-2(3H,4Н,4aH,5H,6Н,7H,7аН)-ил)-3-оксопропаннитрил
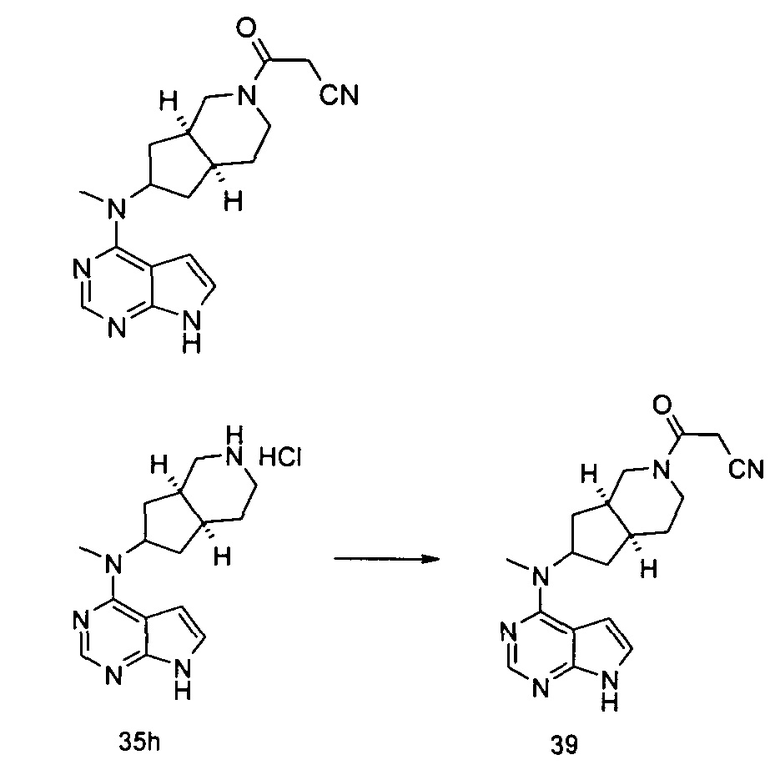
N-Метил-N-((4aS,7aR)-октагидро-1H-циклопента[с]пиридин-6-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 35h (100 мг, 0,37 ммоль) растворяли в 5 мл н-бутанола, а затем добавляли 2-этилцианоацетат (83 мг, 0,74 ммоль) и DBU (113 мг, 0,74 ммоль). После взаимодействия в течение 15 часов при 50°С реакционную смесь концентрировали при пониженном давлении, экстрагировали этилацетатом (20 мл × 2) и промывали насыщенным раствором хлорида натрия (15 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта 3-((4aS,7aR)-6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-2(3H,4Н,4аН,5Н,6H,7Н,7аН)-ил)-3-оксопропаннитрила 39 (35 мг, выход 28,0%) в виде светло-розового твердого вещества.
MS m/z (ESI): 339,3 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,6 (s, 1Н), 8,09 (s, 1H), 7,13 (s, 1H), 6,58 (s, 1H), 5,27-5,29 (m, 1H), 4,01-4,09 (m, 2H), 3,60-3,63 (m, 1H), 3,43-3,48 (m, 2H), 3,18 (s, 3H), 2,08-2,10 (m, 2H), 1,81-1,88 (m, 3H), 1,23-1,43 (m, 4H)
Примеры 40, 41
(4aR,6R,7aR)-6-(Метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-3(2H)-он
(4aR,6S,7aR)-6-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-3(2Н)-он
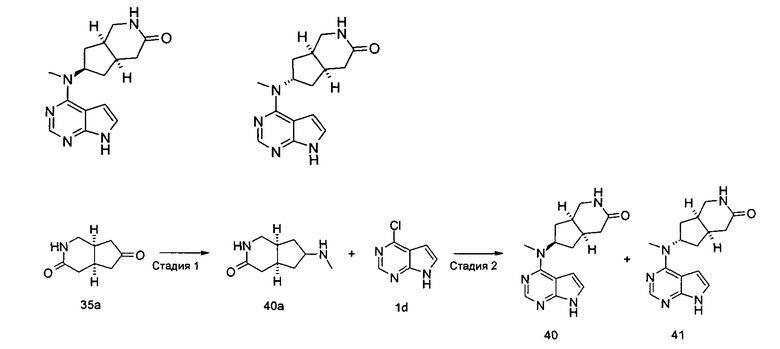
Стадия 1
(4aS,7aS)-6-(Метиламино)гексагидро-1H-циклопента[с]пиридин-3(2H)-он
(4aS,7aS)-Тетрагидро-1H-циклопента[с]пиридин-3,6(2Н,4Н)-дион 35а (1 г, 6,54 ммоль, полученный хорошо известными способами "Tetrahedron: Asymmetry, 1997, 8 (17), 2893-2904") и метиламина гидрохлорид (200 мг, 6,54 ммоль) растворяли в 20 мл метанола, а затем добавляли две капли уксусной кислоты. После перемешивания и взаимодействия в течение 1 часа в реакционную смесь добавляли цианоборгидрид натрия (600 мг, 9,68 ммоль). После взаимодействия в течение 12 часов реакционную смесь фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке продукта (4aS,7aS)-6-(метиламино)гексагидро-1Н-циклопента[с]пиридин-3(2H)-она 40а (1 г) в виде коричневого жира, который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 169,2 [М+1]
Стадия 2
(4aR,6R,7aR)-6-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-3(2H)-он
(4aR,6S,7aR)-6-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-3(2Н)-он
4-Хлор-7Н-пирроло[2,3-d]пиримидин 1d (910 г, 5,95 ммоль) растворяли в 30 мл H2O, а затем добавляли (4aS,7aS)-6-(метиламино)гексагидро-1H-циклопента[с]пиридин-3(2Н)-он 40а (1 г, 5,95 ммоль) и карбонат калия (1,6 г, 11,59 ммоль). После взаимодействия в течение 12 часов при 100°С реакционную смесь концентрировали при пониженном давлении, добавляли 50 мл H2O, и экстрагировали этилацетатом (30 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали при помощи хиральной препаративной HPLC с получением указанных в заголовке продуктов (4aR,6S,7aR)-6-(метил (7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-циклопента[с]пиридин-3(2H)-она 40 (15 мг, выход 0,9%) в виде белого твердого вещества и (4aR,63,7aR)-6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-3(2Н)-она 41 (15 мг, выход 0,9%) в виде белого твердого вещества.
MS m/z (ESI): 286,4 [М+1]
1H NMR (400 МГц, CDCl3): δ 11,37 (s, 1H), 8,28 (s, 1H), 7,06 (s, 1H), 6,98 (s, 1H), 6,53 (s, 1H), 5,24-5,27 (m, 1H), 3,41-3,44 (m, 1H), 3,21 (s, 3H), 3,10-3,13 (m, 1H), 2,56-2,58 (m, 1H), 2,50-2,52 (m, 2H), 2,30-2,31 (m, 1H), 2,07-2,09 (m, 2H), 1,50-1,64 (m, 2H)
1H NMR (400 МГц, CDCl3): δ 11,37 (s, 1H), 8,28 (s, 1H), 7,06 (s, 1H), 6,98 (s, 1H), 6,53 (s, 1H), 5,24-5,27 (m, 1H), 3,41-3,44 (m, 1H), 3,21 (s, 3H), 3,10-3,13 (m, 1H), 2,56-2,58 (m, 1H), 2,50-2,52 (m, 2H), 2,30-2,31 (m, 1H), 2,07-2,09 (m, 2H), 1,50-1,64 (m,2H)
Пример 42
(4aR,6R,7aR)-2-Метил-6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-3(2H)-он
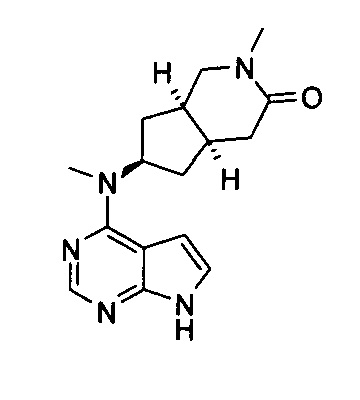
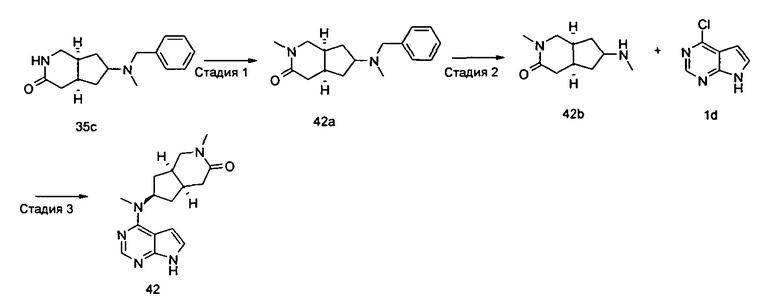
Стадия 1
(4aS,7aS)-6-(Бензил(метил)амино)-2-метилгексагидро-1H-циклопента[с]пиридин-3(2H)-он
В ледяной бане (4aS,7aS)-6-(бензил(метил)амино)гексагидро-1Н-циклопента[с]пиридин-3(2Н)-он 35с (400 мг, 1,55 ммоль) растворяли в 10 мл тетрагидрофурана, а затем добавляли гидрид натрия (56 мг, 2,33 ммоль). После перемешивания в течение 30 минут в реакционную смесь добавляли метилйодид (241 мг, 1,71 ммоль). После взаимодействия в течение 12 часов в реакционную смесь добавляли 50 мл H2O и экстрагировали этилацетатом (20 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (4aS,7aS)-6-(бензил(метил)амино)-2-метилгексагидро-1Н-циклопента[с]пиридин-3(2H)-она 42а (500 мг) в виде бесцветной жидкости, который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 273,2 [М+1]
Стадия 2
(4aS,7aS)-2-Метил-6-(метиламино)гексагидро-1H-циклопента[с]пиридин-3(2Н)-он
(4aS,7aS)-6-(Бензил(метил)амино)-2-метилгексагидро-1Н-циклопента[с]пиридин-3(2H)-он 42а (500 г, 1,84 ммоль) растворяли в 10 мл метанола, а затем добавляли гидроксид палладия на углероде (125 мг, 25%). После продувки реактора водородом, осуществляемой три раза, реакционную смесь перемешивали в течение 6 часов и затем фильтровали, промывали метанолом (10 мл) и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (4aS,7aS)-2-метил-6-(метиламино)гексагидро-1H-циклопента[с]пиридин-3(2H)-она 42b (300 мг) в виде бесцветного жира, который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 183,2 [М+1]
Стадия 3
(4aR,6R,7aR)-2-Метил-6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-3(2Н)-он
4-Хлор-7H-пирроло[2,3-d]пиримидин 1d (168 мг, 1,10 ммоль) растворяли в 20 мл H2O, а затем добавляли (4aS,7aS)-2-метил-6-(метиламино)гексагидро-1Н-циклопента[с]пиридин-3(2Н)-он 42b (200 мг, 1,10 ммоль) и карбонат калия (303 мг, 2,20 ммоль). После взаимодействия в течение 48 часов при 100°С в реакционную смесь добавляли 30 мл H2O и экстрагировали этилацетатом (20 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной HPLC с получением указанного в заголовке продукта (4aR,6R,7aR)-2-метил-6-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-циклопента[с]пиридин-3(2Н)-она 42 (8 мг, выход 5%) в виде белого твердого вещества.
MS m/z (ESI): 300,3 [М+1]
1H NMR (400 МГц, CDCl3): δ 11,61 (s, 1H), 8,09 (s, 1H), 7,12 (s, 1H), 6,56 (s, 1H), 5,10-5,15 (m, 1H), 3,40-3,44 (m, 1H), 3,15-3,16 (m, 1H), 3,11 (s, 3H), 2,89 (s, 3H), 2,40-2,44 (m, 3H), 2,14-2,16 (m, 1H), 1,90-1,92 (m, 2H), 1,35-1,47 (m, 2H)
Примеры 43, 45
2-Гидрокси-1-((3aS,5R,7aR)-5-(метил(7Н-пирроло[2,3-сУ]пиримидин-4-ил)амино)гексагидро-1H-изоиндол-2(3H,3aR,4H,5H,6Н,7H,7aH)-ил)этанон
2-Гидрокси-1-((3aS,5S,7aR)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-изоиндол-2(3H,3aR,4H,5H,6H,7H,7аН)-ил)этанон
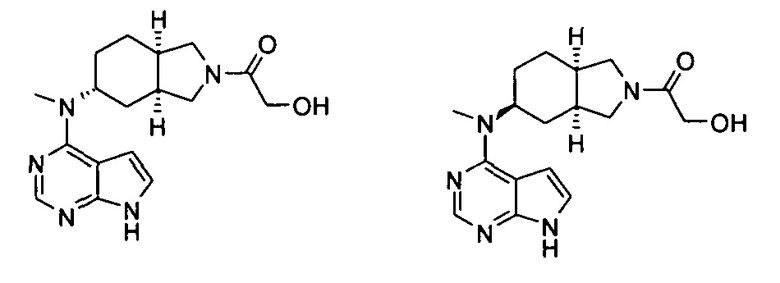
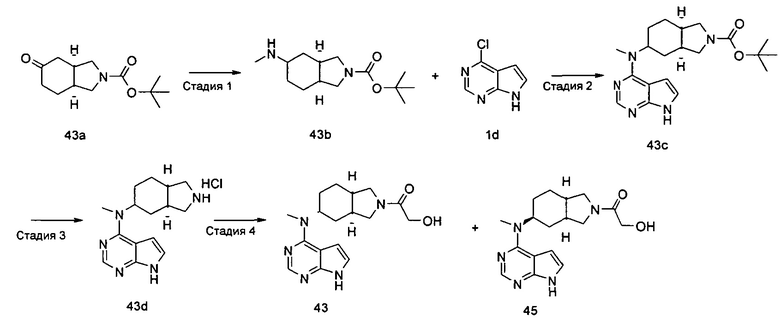
Стадия 1
(3aR,7aS)-трет-Бутил 5-(метиламино)гексагидро-1Н-изоиндол-2(3H)-карбоксилат
(3aR,7aS)-трет-Бутил 5-оксогексагидро-1H-изоиндол-2(3H)-карбоксилат 43а (3,77 г, 15,75 ммоль, полученный хорошо известным способом "Journal of the American Chemical Society, 2000, 122(44), 10743-10753") и метиламина спиртовой раствор (0,73 г, 23,63 ммоль) растворяли в 50 мл метанола. После перемешивания в течение 2 часов при 70°С в реакционную смесь добавляли цианоборгидрид натрия (2 г, 31,51 ммоль). После взаимодействия в течение 1 часа при 70°С в реакционную смесь добавляли 100 мл H2O и экстрагировали дихлорметаном (30 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке продукта (3aR,7aS)-трет-бутил 5-(метиламино)гексагидро-1H-изоиндол-2(3H)-карбоксилата 43b (3,2 г, коричневый жир), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 255,2 [М+1]
Стадия 2
(3aS,7aR)-трет-Бутил 5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-изоиндол-2(3H)-карбоксилат
4-Хлор-7H-пирроло[2,3-d]пиримидин 1d (1,93 г, 12,58 ммоль) растворяли в 50 мл н-бутанола, а затем добавляли (3aR,7aS)-трет-бутил 5-(метиламино)гексагидро-1H-изоиндол-2(3H)-карбоксилат 43b (3,20 г, 12,58 ммоль) и карбонат калия (3,47 г, 25,16 ммоль). После взаимодействия в течение 12 часов при 110°С в реакционную смесь добавляли 100 мл H2O и экстрагировали этилацетатом (100 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (3aR,7aR)-трет-бутил 5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-изоиндол-2(3H)-карбоксилата 43с (2,5 г, выход 53,2%) в виде белого твердого вещества.
MS m/z (ESI): 372,2 [М+1]
Стадия 3
N-Метил-N-((3aS,7aR)-октагидро-1H-изоиндол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид
(3aR,7aR)-трет-Бутил 5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-изоиндол-2(3H)-карбоксилат 43с (2 г, 5,38 ммоль) растворяли в 15 мл 6 М раствора хлорида водорода в метаноле. После взаимодействия в течение 12 часов реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке продукта N-метил-N-((3aS,7aR)-октагидро-1H-изоиндол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорида 43d (1,1 г, белое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 272,3 [М+1]
Стадия 4
2-Гидрокси-1-((3aS,5R,7aR)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-изоиндол-2(3H,3aR,4H,5H,6H,7H,7аН)-ил)этанон
2-Гидрокси-1-((3aS,5S,7aR)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-изоиндол-2(3H,3aR,4H,5H,6H,7H,7аН)-ил)этанон
N-Метил-N-((3aS,7aR)-октагидро-1H-изоиндол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 43d (100 мг, 0,37 ммоль) растворяли в 10 мл N,N-диметилформамида, а затем добавляли 2-гликолевую кислоту (33 мг, 0,44 ммоль), триэтиламин (120 мг, 1,11 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния гексафторфосфат (0,21 г, 0,55 ммоль). После взаимодействия в течение 12 часов в реакционную смесь добавляли 30 мл H2O и экстрагировали этилацетатом (20 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанных в заголовке продуктов 2-гидрокси-1-((3aS,5R,7aR)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-изоиндол-2(3H,3aH,4Н,5H,6H,7Н,7аН)-ил)этанона 43 (10 мг, выход 8,3%) в виде белого твердого вещества и 2-гидрокси-1-((3aS,5S,7aR)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-изоиндол-2-(3H,3aH,4H,5H,6Н,7H,7aH)-ил)этанона 45 (5 мг, выход 4,2%) в виде белого твердого вещества.
MS m/z (ESI): 330,3 [М+1]
1H NMR (400 МГц, CD3OD-d4): δ 8,12 (s, 1Н), 7,06 (d, 1H), 6,58 (d, 1H), 4,96-4,99 (m, 1H), 4,60-4,64 (m, 1H), 4,18-4,24 (m, 2H), 3,52-3,57 (m, 1H), 3,48 (s, 3H), 3,26 (d, 2H), 2,72-2,74 (m, 1H), 2,16-2,26 (m, 1H), 1,96-1,98 (m, 1H), 1,83-1,87 (m, 2H), 1,69-1,72 (m, 1H), 1,44-1,52 (m, 1H), 1,23-1,33 (m, 3H)
1H NMR (400 МГц, CD3OD-d4): δ 8,10 (s, 1H), 7,11 (d, 1H), 6,66 (d, 1H), 4,71-4,75 (m, 1H), 4,58-4,62 (m, 1H), 4,10-4,22 (m, 2H), 3,43-3,57 (m, 1H), 3,48 (s, 3H), 3,26 (d, 2H), 2,50-2,54 (m, 1H), 2,44-2,48 (m, 1H), 1,96-1,98 (m, 2H), 1,58-1,68 (m, 2H), 1,28-1,33 (m, 4H)
Пример 44
3-((3aS,7aR)-5-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1Н-изоиндол-2(3H,3aH,4H,5Н,6H,7H,7аН)-ил)-3-оксопропаннитрил
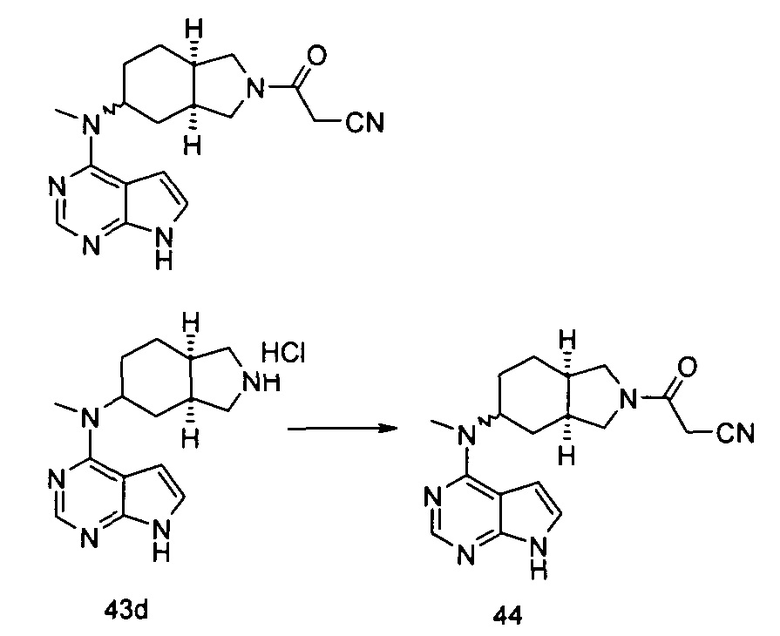
N-Метил-N-((3aS,7aR)-октагидро-1H-изоиндол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 43d (110 мг, 0,41 ммоль) растворяли в 15 мл N,N-диметилформамида, а затем добавляли 2-цианоуксусную кислоту (42 мг, 0,49 ммоль), триэтиламин (82 мг, 0,82 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния гексафторфосфат (231 мг, 0,61 ммоль). После перемешивания в течение 12 часов в реакционную смесь добавляли 15 мл H2O и экстрагировали этилацетатом (20 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта 3-((3aS,7aR)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидро-1H-изоиндол-2(3H,3aH,4H,5Н,6Н,7H,7aH)-ил)-3-оксопропаннитрила 44 (13 мг, выход 9,5%) в виде белого твердого вещества.
MS m/z (ESI): 339,3 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 10,68 (s, 1H), 9,01 (s, 1H), 7,79 (d, 1H), 7,30 (d, 1H), 5,65-4,69 (m, 1H), 4,17-4,43 (m, 6H), 4,02 (s, 3H), 3,31-3,34 (m, 1H), 3,25-3,28 (m, 1H), 2,61-2,71 (m, 2H), 2,24-2,32 (m, 2H), 1,98-2,05 (m, 2H)
Пример 46
2-Гидрокси-1-((4aS,8aR)-6-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)октагидроизохинолин-2(1H)-ил)этанон
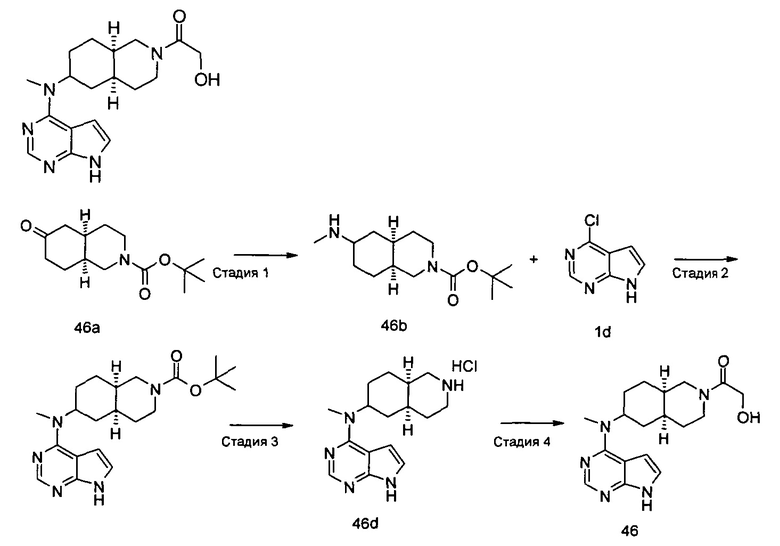
Стадия 1
(4aR,8aS)-трет-Бутил 6-(метиламино)октагидроизохинолин-2(1Н)-карбоксилат
(4aR,8aS)-трет-Бутил 6-оксооктагидроизохинолин-2(1Н)-карбоксилат 46а (2,50 г, 9,87 ммоль, полученный хорошо известным способом "Journal of the American Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1995, 20, 2535-2542") и метиламина спиртовой раствор (0,92 г, 29,61 ммоль) растворяли в 50 мл метанола. После перемешивания в течение 2 часов при 70°С в реакционную смесь добавляли цианоборгидрид натрия (1,24 г, 19,74 ммоль). После взаимодействия в течение 2 часов при 70°С в реакционную смесь добавляли 50 мл H2O и экстрагировали дихлорметаном (50 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (4aR,8aS)-трет-бутил 6-(метиламино)октагидроизохинолин-2(1H)-карбоксилата 46b (2 г, коричневый жир), который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
(4aS,8aR)-трет-Бутил 6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)октагидроизохинолин-2(1H)-карбоксилат
4-Хлор-7Н-пирроло[2,3-d]пиримидин 1d (1,14 г, 7,45 ммоль) растворяли в 50 мл 1,4-диоксана, а затем добавляли (4aR,8aS)-трет-бутил 6-(метиламино)октагидроизохинолин-2(1H)-карбоксилат 46b (2 г, 7,45 ммоль) и карбонат калия (2 г, 14,90 ммоль). После взаимодействия в течение 24 часов при 100°С в реакционную смесь добавляли 100 мл H2O и экстрагировали этилацетатом (100 мл × 3). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (4aS,8aR)-трет-бутил 6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)октагидроизохинолин-2(1H)-карбоксилата 46с (1,4 г, выход 48,3%) в виде не совсем белого твердого вещества.
MS m/z (ESI): 386,0 [М+1]
Стадия 3
(4aS,8aR)-N-Метил-N-(7Н-пирроло[2,3-d]пиримидин-4-ил)декагидроизохинолин-6-амина гидрохлорид
(4aS,8aR)-трет-Бутил 6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)октагидроизохинолин-2(1H)-карбоксилат 46с (300 мг, 0,78 ммоль) растворяли в 15 мл 6 М раствора хлорида водорода в растворе метанола. После взаимодействия в течение 12 часов реакционную смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (4aS,8aR)-N-метил-N-(7Н-пирроло[2,3-d]пиримидин-4-ил)декагидроизохинолин-6-амина гидроглорида 46d (220 мг, не совсем белое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 286,2 [М+1]
Стадия 4
2-Гидрокси-1-((4aS,8aR)-6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)октагидроизохинолин-2(1H)-ил)этанон
((4aS,8aR)-N-Метил-N-(7H-пирроло[2,3-d]пиримидин-4-ил)декагидроизохинолин-6-амина гидрохлорид 46d (100 мг, 0,35 ммоль) растворяли в 10 мл дихлорметана, а затем добавляли гликолевую кислоту (32 мг, 0,42 ммоль), триэтиламин (106 мг, 1,05 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (100 мг, 0,53 ммоль) и 1-гидроксибензотриазол (69 мг, 0,53 ммоль). После взаимодействия в течение 12 часов в реакционную смесь добавляли 30 мл H2O и экстрагировали дихлорметаном (30 мл × 2). Органическую фазу объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта 2-гидрокси-1-((4aS,8aR)-6-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)октагидроизохинолин-2(1H)-ил)этанона 46 (15 мг, выход 12,5%) в виде белого твердого вещества.
MS m/z (E8I): 344,2 [М+1]
1H NMR (400 МГц, CDCl3): δ 10,87 (s, 1H), 8,28 (s, 1H), 7,06 (d, 1H), 6,55 (d, 1H), 4,90-4,95 (m, 1H), 4,18 (s, 2H), 3,48-3,51 (m, 1H), 3,30 (s, 3H), 3,12-3,32 (m, 4H), 1,82-1,85 (m, 2H), 1,70-1,80 (m, 2H), 1,58-1,61 (m, 2H), 1,21-1,29 (m, 4H)
Пример 47
1-((3aS,4R,5S,6aS)-4-Метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)-3,3a,R4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-2-ил)-2-гидрокси-этанон
Стадия 1
(3aR,6aS)-Бензил 5-оксогексагидроциклопента[с]пиррол-2(1H)-карбоксилат
(3aR,6aS)-Гексагидроциклопента[с]пиррол-5(1H)-он 47а (16,80 г, 0,13 моль, полученный хорошо известным способом "Patent EP2246347") растворяли в 200 мл дихлорметана в ледяной бане, а затем добавляли триэтиламин (16,2 мл, 0,16 моль) и по каплям добавляли бензил хлорформиат (25,22 г, 0,15 моль). Реакционную смесь нагревали до комнатной температуры. После взаимодействия в течение 3 часов в реакционную смесь добавляли 200 мл H2O и экстрагировали дихлорметаном (100 мл × 2). Органическую фазу объединяли, промывали насыщенным бикарбонатом натрия (50 мл), насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле с системой элюции В с получением указанного в заголовке продукта (3aR,6aS)-бензил 5-оксогексагидроциклопента[с]пиррол-2(1H)-карбоксилата 47b (18,5 г, выход 54,9%) в виде белого твердого вещества.
MS m/z (ESI): 260,1 [М+1]
Стадия 2
(3aS,4R,6aS)-Бензил 4-метил-5-оксогексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
(3aR,6aS)-Бензил 5-оксогексагидроциклопента[с]пиррол-2(1H)-карбоксилат 47b (5 г, 19,28 ммоль) при -78°С растворяли в 70 мл тетра гидрофура на, а затем по каплям добавляли литий би(триметилсилил)амид (19,7 мл, 19,67 ммоль). После перемешивания в течение 1 часа при -78°С в реакционную смесь добавляли метилйодид (3,01 г, 21,21 ммоль). Реакционную смесь нагревали до комнатной температуры. После взаимодействия в течение 2 часов в реакционную смесь добавляли 20 мл насыщенного хлорида аммония и экстрагировали этилацетатом (50 мл × 2). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (20 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле с системой элюции В с получением указанного в заголовке продукта (3aS,4R,6aS)-бензил 4-метил-5-оксогексагидроциклопента[с]пиррол-2(1H)-карбоксилата 47с (1,08 г, выход 20,5%) в виде чисто желтой суспензии.
MS m/z (ESI): 274,1 [М+1]
Стадия 3
(3aS,4R,5R,6aS)-Бензил 4-метил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
(3aS,4R,6aS)-Бензил 4-метил-5-оксогексагидроциклопента[с]пиррол-2(1H)-карбоксилат 47с (1,08 г, 3,95 ммоль) растворяли в 30 мл спиртового раствора метиламина. После перемешивания в течение 24 часов при 50°С в реакционную смесь партиями добавляли цианоборгидрид натрия (500 мг, 7,90 ммоль). После взаимодействия в течение 24 часов при 50°С реакционную смесь концентрировали при пониженном давлении, а затем добавляли 50 мл H2O и экстрагировали дихлорметаном (50 мл × 2). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (20 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта (3aS,4R,5R,6aS)-бензил 4-метил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 47d (0,95 г, бледно-желтое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 289,2 [М+1]
Стадия 4
(3aS,4R,5S,6aS)-Бензил 4-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат
4-Хлор-7H-пирроло[2,3-d]пиримидин 1d (176 мг, 1,14 ммоль) растворяли в 15 мл Н20, а затем добавляли (3aS,4R,5R,6aS)-бензил 4-метил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат 47d (300 мг, 1,04 ммоль) и карбонат калия (287 мг, 2,08 ммоль). После взаимодействия в течение 24 часов при 100°С в реакционную смесь добавляли 10 мл H2O и экстрагировали дихлорметаном (20 мл × 2). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (10 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (3aS,4R,5S,6aS)-бензил 4-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилата 47е (0,21 г, выход 49,9%) в виде чисто желтого твердого вещества.
М8 m/z (ESI): 406,3 [М+1]
Стадия 5
N-Метил-N-((3aS,4R,5S,6aS)-4-метилоктагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид
(3aS,4R,5S,6aS)-Бензил 4-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксилат 47е (280 мг, 0,69 ммоль) растворяли в 20 мл метанола, а затем добавляли гидроксид палладия на углероде (10 мг, 4%). После продувки реактора водородом, осуществляемой три раза, реакционную смесь перемешивали в течение 2 часов, и затем фильтровали, промывали метанолом (10 мл) и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта N-метил-N-((3aS,4R,5S,6aS)-4-метилоктагидроциклопента[с]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорида 47f (160 мг, чисто желтое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 272,3 [М+1]
Стадия 6
1-((3aS,4R,5S,6aS)-4-Метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-ил)-2-гидрокси-этанон
N-метил-N-((3aS,4R,5S,6aS)-4-метилоктагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 47f (160 мг, 0,59 ммоль) растворяли в 10 мл N,N-диметилформамида, а затем добавляли 2-гликолевую кислоту (54 мг, 0,71 ммоль), триэтиламин (119 мг, 1,18 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (169 мг, 0,88 ммоль) и 1-гидроксибензотризол (119 мг, 0,88 ммоль). После взаимодействия в течение 15 часов реакционную смесь концентрировали при пониженном давлении, добавляли (20 мл) и затем экстрагировали дихлорметаном (30 мл × 2). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (10 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью хиральной препаративной HPLC с получением указанного в заголовке продукта 21-((3aS,4R,5S,6aS)-4-метил-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-ил)-2-гидрокси-этанона 47 (32 мг, выход 16,5%) в виде белого твердого вещества.
MS m/z (ESI): 330,2 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,6 (s, 1H), 8,08 (s, 1H), 7,13 (s, 1H), 6,61 (s, 1H), 4,94-4,96 (m, 1H), 4,47-4,50 (m, 1H), 4,00-4,07 (m, 2H), 3,61-3,63 (m, 1H), 3,41-3,51 (m, 2H), 3,37-3,39 (m, 1H), 3,13 (s, 3H), 2,60-2,70 (m, 1H), 2,08-2,18 (m, 3H), 1,50-1,51 (m, 1H), 1,47-1,49 (m, 1H), 0,86-0,89 (m, 3H)
Пример 48
(3aR,5s,6aS)-N-(3-Этил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-карбоксамид
Стадия 1
Фенил (3-этил-1,2,4-тиадиазол-5-ил)карбамат
3-Этил-1,2,4-тиадиазол-5-амин 48а (1,29 г, 9,98 ммоль, полученный хорошо известным способом "Collection of Czechoslovak Chemical Communications, 1971, 36, 4091-4098") растворяли в 50 мл тетрагидрофурана в ледяной бане, а затем добавляли безводный карбонат калия (1,79 г, 12,80 ммоль) и по каплям добавляли фенилкарбонохлоридат 34b (1,72 г, 10,98 ммоль). Реакционную смесь нагревали до комнатной температуры. После взаимодействия в течение 12 часов реакционную смесь фильтровали и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта фенил (3-этил-1,2,4-тиадиазол-5-ил)карбамата 48b (2,14 г, желтое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 250,2 [М+1]
Стадия 2
(3aR,5s,6aS)-N-(3-Этил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамид
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (120 мг, 0,47 ммоль) растворяли в 50 мл тетрагидрофурана, а затем добавляли фенил (3-этил-1,2,4-тиадиазол-5-ил)карбамат 48b (2,14 г, 8,58 ммоль) и по каплям добавляли триэтиламин (2,4 мл, 17,17 ммоль). После взаимодействия в течение 12 часов при 60°С в реакционную смесь добавляли 30 мл H2O и экстрагировали дихлорметаном (50 мл × 3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (50×2 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта (3aR,5s,6aS)-N-(3-этил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамида 48 (1,25 г, выход 81,7%) в виде белого твердого вещества.
MS m/z (ESI): 413,4 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 11,60 (s, 1H), 11,52 (s, 1H), 8,08 (s, 1H), 7,07-7,02 (m, 1H), 6,55-6,5 (m, 1H), 5,56-5,38 (m, 1H), 3,78-3,60 (m, 2H), 3,48-3,36 (m, 2H), 3,16 (s, 3H), 2,90 (m, 2H), 2,74 (q, 2H), 2,10-1,94 (m, 2H), 1,8-1,7 (m, 2H), 1,25 (t, 3H)
Пример 49
(3aR,5s,6aS)-N-(3-Циклопропил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамид
Стадия 1
3-Циклопропил-1,2,4-тиадиазол-5-амин
Тиоцианат натрия (527 мг, 6,50 ммоль) растворяли в 20 мл метанола и помещали на -20°С, а затем добавляли циклопропилкарбамидин гидрохлорид (603 мг, 5 ммоль) и триэтиламин (0,8 мл, 5,74 ммоль). После перемешивания в течение 45 минут в реакционную смесь по каплям добавляли триэтиламин (0,7 мл, 5,02 ммоль) и 8% раствор гипохлорита натрия (4,2 мл, 5 ммоль). После взаимодействия в течение 2 часов при -20°С реакционную смесь нагревали до комнатной температуры. После взаимодействия в течение 12 часов реакционную смесь концентрировали при пониженном давлении, а затем добавляли 35 мл H2O и экстрагировали этилацетатом (30 мл × 3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта 3-циклопропил-1,2,4-тиадиазол-5-амина 49а (243 мг, белое твердое вещество), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 142,2 [М+1]
Стадия 2
Фенил (3-циклопропил-1,2,4-тиадиазол-5-ил)карбамат
3-Циклопропил-1,2,4-тиадиазол-5-амин 49а (212 мг, 1,50 ммоль) растворяли в 5 мл тетрагидрофурана в ледяной бане, а затем добавляли безводный карбонат калия (270 мг, 1,95 ммоль) и по каплям добавляли фенил карбонохлоридат 34b (246 мг, 1,58 ммоль), затем реакционную смесь нагревали до комнатной температуры. После взаимодействия в течение 12 часов реакционную смесь фильтровали и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке продукта фенил (3-циклопропил-1,2,4-тиадиазол-5-ил)карбамата 49b (350 мг, бесцветный жир), который использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 262,3 [М+1]
Стадия 3
(3aR,5s,6aS)-N-(3-Циклопропил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамид
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d]пиримидин-4-амина гидрохлорид 6а (344 мг, 1,34 ммоль) растворяли в 6 мл тетрагидрофурана, а затем добавляли фенил (3-циклопропил-1,2,4-тиадиазол-5-ил)карбамат 49b (350 мг, 1,34 ммоль) и по каплям добавляли триэтиламин (0,6 мл, 4,02 ммоль). После взаимодействия в течение 12 часов при 50°С в реакционную смесь добавляли 20 мл H2O и экстрагировали дихлорметаном (20 мл × 3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (20×2 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта (3aR,5s,6aS)-N-(3-циклопропил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамида 49 (120 мг, выход 21,1%) в виде чисто желтого твердого вещества.
MS m/z (ESI): 425,4 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,53 (d, 2H), 8,07 (s, 1H), 7,05-7,03 (m, 1H), 6,53-6,52 (m, 1H), 5,47-5,43 (m, 1H), 3,70-3,65 (m, 2H), 3,37-3,32 (m, 2H), 3,15 (s, 3H), 2,90-2,89 (m, 2H), 2,12-2,07 (m, 1H), 2,05-1,98 (m, 2H), 1,80-1,74 (m, 2H), 0,97-0,93 (m, 4H)
Пример 50
(3aR,5s,6aS)-N-(3-(Гидроксиметил)-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамид

Стадия 1
Метил 5-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)октагидроциклопента[с]пиррол-2-карбоксамидо)-1,2,4-тиадиазол-3-карбоксилат
N-Метил-N-((3aR,5s,6aS)-октагидроциклопента[с]пиррол-5-ил)-7H-пирроло[2,3-d|пиримидин-4-амина гидрохлорид 6а (90 мг, 0,35 ммоль) растворяли в 2 мл N,N-диметилформамида, а затем добавляли метил 5-((феноксикарбонил)амино)-1,2,4-тиадиазол-3-карбоксилат 50а (97 мг, 0,35 ммоль, полученный хорошо известным способом "патентная заявка WO 2004103980") и по каплям добавляли триэтиламин (105 мг, 1,04 ммоль). После взаимодействия в течение 12 часов при 100°С в реакционную смесь добавляли 10 мл Н2О, и реакционную смесь экстрагировали дихлорметаном (10 мл × 3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (10×2 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюции А с получением указанного в заголовке продукта метил 5-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)октагидроциклопента[с]пиррол-2-карбоксамидо)-1,2,4-тиадиазол-3-карбоксилата 50b (99,5 мг, выход 65,0%) в виде белого твердого вещества.
MS m/z (ESI): 443,4 [M+1]
Стадия 2
(3aR,5s,6aS)-N-(3-(Гидроксиметил)-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамид
Метил 5-((3aR,5s,6aS)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)октагидроциклопента[с]пиррол-2-карбоксамидо)-1,2,4-тиадиазол-3-карбоксилат 50b (95 мг, 0,21 ммоль) растворяли в 3 мл этанола, а затем партиями добавляли боргидрид натрия (49 мг, 1,29 ммоль). После взаимодействия в течение 2 часов реакционную смесь концентрировали при пониженном давлении, а затем добавляли 10 мл H2O и экстрагировали дихлорметаном (10 мл × 2). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (10×2 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и образующийся остаток очищали с помощью тонкослойной хроматографии с системой элюции А с получением указанного в заголовке продукта (3aR,5s,6aS)-N-(3-(гидроксиметил)-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамида 50 (10 мг, выход 11,2%) в виде белого твердого вещества.
MS m/z (ESI): 415,4 [М+1]
1H NMR (400 МГц, DMSO-d6): δ 11,56 (s, 1H), 8,08 (s, 1H), 6,99-7,04 (m, 1H), 6,50-6,54 (m, 1H), 5,37-5,50 (m, 1H), 4,70 (br. s, 1H), 4,26-4,34 (m, 2H), 3,55-3,66 (m, 2H), 3,21-3,29 (m, 2H), 3,14 (s, 3H), 2,74-2,85 (m, 2H), 1,92-2,05 (m, 2H), 1,67-1,76 (m, 2H)
Соединения по примерам 51-156 синтезировали со ссылкой на способы в примере 1 и примере 2 с использованием соответствующих реагентов согласно способу синтеза соединений по настоящему изобретению.
Номера их примеров, их структуры и характеризующие данные приведены в таблице В в конце описания.
ПРИМЕРЫ ТЕСТОВ
БИОЛОГИЧЕСКИЕ АНАЛИЗЫ
Пример теста 1. Анализ с целью определения активности соединений по настоящему изобретению в отношении ингибирования JAK1-киназы.
Активность соединений по настоящему изобретению in vitro в отношении ингибирования JAK1-киназы определяли следующим способом.
Анализы киназы in vitro, описанные ниже, можно использовать для определения активности тестируемого соединения в отношении ингибирования активности JAK1-киназы. Тестируемые соединения растворяли в диметилсульфоксиде и разбавляли водой для получения серийного концентрационного градиента, как требовалось в эксперименте. Субстраты JAK1 (cell signaling technology, номер по каталогу: 1305s) и раствор АТР (аденозин-5'-трифосфат) (2 мМ) разбавляли водой с получением раствора с конечной концентрацией 20 мкМ АТР и 1,2 мкМ субстрата. Достаточное количество JAK1-киназы (Invitrogen, номер по каталогу: pv4774) смешивали с 4 × буфером (приготовленным пользователем, и содержащим 50 мМ HEPES, рН 7,3, 125 мМ NaCl, 24 мМ MgCl2, 1,25 мМ DTT (дитиотреитол)) до конечной концентрации 8 нг/мкл. В каждую лунку микропланшета [DELFIA®, покрытый стрептавидином прозрачный планшет (Perkin Elmer, номер по каталогу: AAAND-0005)] добавляли 17,5 мкл смеси АТР/субстрата, 5 мкл водного раствора тестируемого соединения (в контрольную и холостую пробы добавляли только 5 мкл чистой воды) и 7,5 мкл раствора киназы, приготовленного как описано выше (в контроль добавляли только 4 × буфер). Содержимое каждой лунки перемешивали в достаточной степени, затем инкубировали при комнатной температуре (27°С) в течение 50 минут, промывали промывочным буфером и сушили три раза, затем добавляли антитело, конъюгированное с HRP (пероксидаза хрена) [фосфотирозиновое мышиное mAb (Р-Tyr-100) (конъюгат HRP, Cell signaling Technology, номер по каталогу: 5465)], и инкубировали в течение 1 часа. Микропланшет промывали промывочным буфером и сушили три раза, и затем добавляли ТМВ (тетраметилбензидин) (Sigma, номер по каталогу: Т4444) и окрашивали в течение 5-15 минут. Для остановки реакции добавляли блокирующий раствор (раствор 1 н. серной кислоты). Поглощение измеряли на микропланшете-ридере novostar при длине волны 450 нм. Значения IC50 тестируемых соединений могут быть рассчитаны с помощью данных для тестируемых соединений в отношении ингибирования активности JAK1-киназы при различных концентрациях.
Активность соединений по настоящему изобретению
Биохимическую активность соединений по настоящему изобретению определяли с помощью вышеописанного анализа, и значения IC50 показаны в следующей таблице 1.
Вывод: соединения по настоящему изобретению обладали значительной активностью в отношении ингибирования пролиферации JAK1-киназы.
Пример теста 2. Анализ с целью определения активности соединений по настоящему изобретению в отношении ингибирования иАК2-киназы
Активность соединений по настоящему изобретению in vitro в отношении ингибирования JAK2-киназы определяли следующим способом.
Анализы киназы in vitro, описанные ниже, можно использовать для определения активности тестируемого соединения в отношении ингибирования активности JAK2-киназы. Тестируемые соединения растворяли в диметилсульфоксиде и разбавляли водой для получения серийного концентрационного градиента, как требовалось в эксперименте. Субстраты JAK2 (cell signaling technology, номер по каталогу: 1305s) и раствор АТР (2 мМ) разбавляли водой с получением раствора с конечной концентрацией 20 мкМ АТР и 1,2 мкМ субстрата. Достаточное количество иАК2-киназы (Invitrogen, номер по каталогу: pv4210) смешивали с 4 × буфером (приготовленным пользователем и содержащим 50 мМ HEPES, рН 7,3, 125 мМ NaCl, 24 мМ MgCl2, 1,25 мМ DTT) до конечной концентрации 8 нг/мкл. В каждую лунку микропланшета [DELFIA®, покрытый стрептавидином прозрачный планшет (Perkin Elmer, номер по каталогу: AAAND-0005)] добавляли 17,5 мкл смеси АТР/субстрата, 5 мкл водного раствора тестируемого соединения (в контрольную и холостую пробы добавляли только 5 мкл чистой воды) и 7,5 мкл раствора киназы, приготовленного как описано выше (в контроль добавляли только 4 × буфер). Содержимое каждой лунки перемешивали в достаточной степени, затем инкубировали при комнатной температуре (27°С) в течение 50 минут, промывали промывочным буфером и сушили три раза, затем добавляли антитело, конъюгированное с HRP [фосфотирозиновое мышиное mAb (Р-Туг-100) (конъюгат HRP, Cell signaling Technology, номер по каталогу: 5465)], и инкубировали в течение 1 часа. Микропланшет промывали промывочным буфером и сушили три раза, и затем добавляли ТМВ (Sigma, номер по каталогу: Т4444) и окрашивали в течении 5-15 минут. Для остановки реакции добавляли блокирующий раствор (раствор 1 н. серной кислоты). Поглощение измеряли на микропланшете-ридере novostar при длине волны 450 нм. Значения IC50 тестируемых соединений могут быть рассчитаны с помощью данных для тестируемых соединений в отношении ингибирования активности JAK2-киназы при различных концентрациях.
Активность соединений по настоящему изобретению
Биохимическую активность соединений по настоящему изобретению определяли с помощью вышеописанного анализа, и значения IC50 показаны в следующей таблице 2.
Вывод: соединения по настоящему изобретению обладали значительной активностью в отношении ингибирования пролиферации JAK2-киназы.
Пример теста 3. Анализ с целью определения активности соединений по настоящему изобретению в отношении ингибирования JAK3-киназы
Активность соединений по настоящему изобретению in vitro в отношении ингибирования JAK3-киназы определяли следующим способом.
Анализы киназы in vitro, описанные ниже, можно использовать для определения активности тестируемого соединения в отношении ингибирования активности JAK3-киназы. Тестируемые соединения растворяли в диметилсульфоксиде и разбавляли водой для получения серийного концентрационного градиента, как требовалось в эксперименте. Субстраты JAK3 (cell signaling technology, номер по каталогу: 1305s) и раствор АТР (2 мМ) разбавляли водой с получением раствора с конечной концентрацией 20 мкМ АТР и 1,2 мкМ субстрата. Достаточное количество JAK3-киназы (Invitrogen, номер по каталогу: pv3855) смешивали с 4 × буфером (полученным пользователем и содержащим 50 мМ HEPES, рН 7,3, 125 мМ NaCl, 24 мМ MgCl2, 1,25 мМ DTT) до конечной концентрации 8 нг/мкл. В каждую лунку микропланшета [DELFIA®, покрытый стрептавидином прозрачный планшет (Perkin Elmer, номер по каталогу: AAAND-0005)] добавляли 17,5 мкл смеси АТР/субстрата, 5 мкл водного раствора тестируемого соединения (в контрольную и холостую пробы добавляли только 5 мкл чистой воды) и 7,5 мкл раствора киназы, приготовленного как описано выше (в контроль добавляли только 4 × буфер). Содержимое каждой лунки перемешивали в достаточной степени, затем инкубировали при комнатной температуре (27°С) в течение 50 минут, промывали промывочным буфером и сушили три раза, затем добавляли антитело, конъюгированное с HRP [фосфотирозиновое мышиное mAb (P-Tyr-100) (конъюгат HRP, Cell signaling Technology, номер по каталогу; 5465)], и инкубировали в течение 1 часа. Микропланшет промывали промывочным буфером и сушили три раза, и затем добавляли ТМВ (Sigma, номер по каталогу: Т4444) и окрашивали в течение 5-15 минут. Для остановки реакции добавляли блокирующий раствор (раствор 1 н. серной кислоты). Поглощение измеряли на микропланшет-ридере novostar при длине волны 450 нм. Значения IC50 тестируемых соединений могут быть рассчитаны с помощью данных для тестируемых соединений в отношении ингибирования активности JAK3-киназы при различных концентрациях.
Активность соединений по настоящему изобретению
Биохимическую активность соединений по настоящему изобретению определяли с помощью вышеописанного анализа, и значения IC50 показаны в следующей таблице 3.
Вывод: соединения по настоящему изобретению обладали значительной активностью в отношении ингибирования пролиферации JAK3-киназы.
Пример теста 4: Анализ соединений по настоящему изобретению в отношении ингибирования пролиферации на клеточной линии человеческого эритроидного лейкоза TF-1
Следующий анализ in vitro предназначен для определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации клеточной линии человеческого эритроидного лейкоза TF-1.
Следующий клеточный анализ in vitro может быть использован для определения активности тестируемого соединения в отношении ингибирования пролиферации, опосредованной IL-4 (указанный IL-4 может опосредовать путь JAK3). Активность представлена значением IC50, которое может отражать не только активность тестируемого соединения в отношении ингибирования киназы JAK2 и JAK3, но также отражать селективность тестируемого соединения в отношении киназы JAK2 и JAK3.
Общий протокол анализа приведен ниже: во-первых, клетки TF-1 (приобретены у АТСС (Американская коллекция типовых культур), номер по каталогу: CRL 2003) высевали в 96-луночный планшет для культуры клеток в подходящей концентрации клеток (8000 клеток/мл среды), и в каждую лунку добавляли 10 нг/мл IL-4 (Invitrogen, номер по каталогу: РНС0044). Затем готовили 10 × серийный концентрационный градиент растворов тестируемых соединений (10000, 1000, 100, 10, 1 и 0,1 нМ), и затем 10 × растворы соединений, приготовленные ранее, добавляли в 96-луночный планшет для культуры клеток, содержащий IL-4. После непрерывной культивации планшетов с клетками в течение 72 часов активность тестируемых соединений в отношении ингибирования пролиферации клеток определяли с использованием набора 8 для подсчета клеток (приобретен у Dojindo, номер по каталогу: СК04). Значения IC50 рассчитывали с помощью данных по скорости ингибирования при разных концентрациях тестируемых соединений.
Биологическую активность соединений по настоящему изобретению тестировали с помощью использования вышеописанного анализа. Измеряли значения IC50, и они показаны ниже в таблице 4:
Вывод: соединения по настоящему изобретению обладали значительной активностью в отношении ингибирования пролиферации клеток TF-1, опосредованной путем JAK3 /JAK2, опосредованным IL-4.
Пример теста 5: Анализ соединений по настоящему изобретению в отношении ингибирования пролиферации на Т-клетках
Следующий анализ in vitro предназначен для определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации Т-клеток.
Следующий клеточный анализ in vitro может быть использован для определения активности тестируемого соединения в отношении ингибирования пролиферации Т-клеток. Активность представлена значением IC50.
Общий протокол анализа приведен ниже: во-первых, линию клеток РВМС (приобретенную у Shanghai Blood Center) центрифугировали, удаляли супернатант и проводили их подсчет. Затем клетки инкубировали в среде [RPMI-1640 (Hyclone, номер по каталогу: SH30809,01B)+10% фетальная телячья сыворотка (GIBCO, номер по каталогу: 10099)+1% Pen Strep (пенициллин/стрептомицин) (GIBCO, номер по каталогу: 15140)], а также с 200 мкг функциональных очищенных антител против человеческого CD3 (eBioscience, номер по каталогу: 16-0037-81) и в инкубаторе с 5% диоксида углерода (CO2) при 37°С в течение 3 суток до тех пор, пока концентрация клеток не достигала 2×106/мл. Затем клетки промывали 3 раза, ресуспендировали, разбавляли до 2×106/мл, и затем добавляли рекомбинантный человеческий IL-2 (приобретенный у Peprotech, номер по каталогу: 200-02) до концентрации 10 нг/мл, культивировали в течение еще 3 суток. Клетки снова промывали 3 раза и разводили до 5×105/мл. 80 мкл клеток высевали в каждую лунку 96-луночного планшета для клеточной культуры. Исходная концентрация лекарственного средства составляла 1 мМ. Затем лекарственное средство разбавляли культуральной средой до 10000, 2500, 625, 156, 39, 9,8, 2,4, 0,6 мкМ, и 10 мкл разбавленного лекарственного средства добавляли в соответствующую лунку. В контрольные лунки добавляли 10 мкл культуральной среды. Планшет помещали в инкубатор. После инкубирования в течение 1 часа в лунки с отрицательным контролем добавляли 10 мкл культуральной среды, а в остальные лунки добавляли IL-2 до концентрации 10 нг/мл, инкубировали непрерывно в течение 72 часов. Через 3 суток активность тестируемых соединений в отношении ингибирования пролиферации клеток определяли с использованием набора ATPlite™ Luminescence Assay System (PerkinElmer, номер по каталогу: 6016947). Значения IC50 рассчитывали с помощью данных по скорости ингибирования при различных концентрациях тестируемых соединений.
Биологическую активность соединений по настоящему изобретению тестировали с помощью использования вышеописанного анализа. Измеряли значения IC50, и они показаны ниже в таблице 5.
Вывод: соединения по настоящему изобретению обладали значительной активностью в отношении ингибирования пролиферации Т-клеток.
ФАРМАКОКИНЕТИЧЕСКИЙ АНАЛИЗ
Пример теста 6: Фармакокинетический анализ соединений по настоящему изобретению.
1. Аннотация
В качестве подопытных животных использовали крыс. Соединения по примеру 6, примеру 17, примеру 22, примеру 34, примеру 35, примеру 40, примеру 48, примеру 49, примеру 99, примеру 114, примеру 121, примеру 125, примеру 128 и примеру 148 вводили крысам внутрижелудочно для определения концентрации лекарственного средства в плазме в разные моменты времени с помощью способа LC/MS/MS (жидкостная хроматография/тандемная масс-спектрометрия). Фармакокинетическое поведение соединений по настоящему изобретению изучали и оценивали на крысах.
2. Протокол
2.1 Образцы
Соединения по примеру 6, примеру 17, примеру 22, примеру 34, примеру 35, примеру 40, примеру 48, примеру 49, примеру 99, примеру 114, примеру 121, примеру 125, примеру 128 и примеру 148.
2.2 Подопытные животные
56 здоровых взрослых крыс SD, самцов и самок поровну, приобретенных у SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., CO, № сертификата: SCXK (Shanghai) 2003-0002, делили на 14 групп, по 4 крысы в каждой группе.
2.3 Получение тестируемых соединений
Взвешивали достаточные количества тестируемых соединений, и к ним добавляли 1,0 мл диметилсульфоксида с получением суспензии 1,0 мг/мл.
2.4 Введение
После ночного голодания крысам SD внутрижелудочно вводили дозу 10,0 мг/кг, и вводимый объем составлял 10 мл/кг.
3. Способ
Соединения по примеру 6, примеру 17, примеру 22, примеру 34, примеру 35, примеру 40, примеру 48, примеру 49, примеру 99, примеру 114, примеру 121, примеру 125, примеру 128 и примеру 148 вводили крысам внутрижелудочно. Образцы крови (0,2 мл) отбирали из орбитального синуса перед введением и через 0,5 ч, 1,0 ч, 2,0 ч, 3,0 ч, 4,0 ч, 6,0 ч, 8,0 ч, 12,0 ч, 24,0 ч и 36,0 ч после введения, хранили в пробирках с гепарином и центрифугировали в течение 10 минут при 10000 об./мин при 4°С для отделения плазмы крови. Образцы плазмы крови хранили при -20°С. Крыс кормили через 2 часа после введения.
Определение содержания тестируемых соединений в плазме крыс после внутрижелудочного введения в различных концентрациях: к 50 мкл крысиной плазмы, отобранной в разное время после введения, добавляли 50 мкл раствора внутренних стандартов и 100 мкл метанола, и смешивали в течение 3 минут с помощью вортекса. Смесь центрифугировали в течение 10 минут при 13500 об./мин. 10 мкл супернатанта отбирали из образца плазмы и анализировали с помощью LC-MS/MS.
4. Результаты фармакокинетических параметров Фармакокинетические параметры соединений по настоящему изобретению представлены ниже:
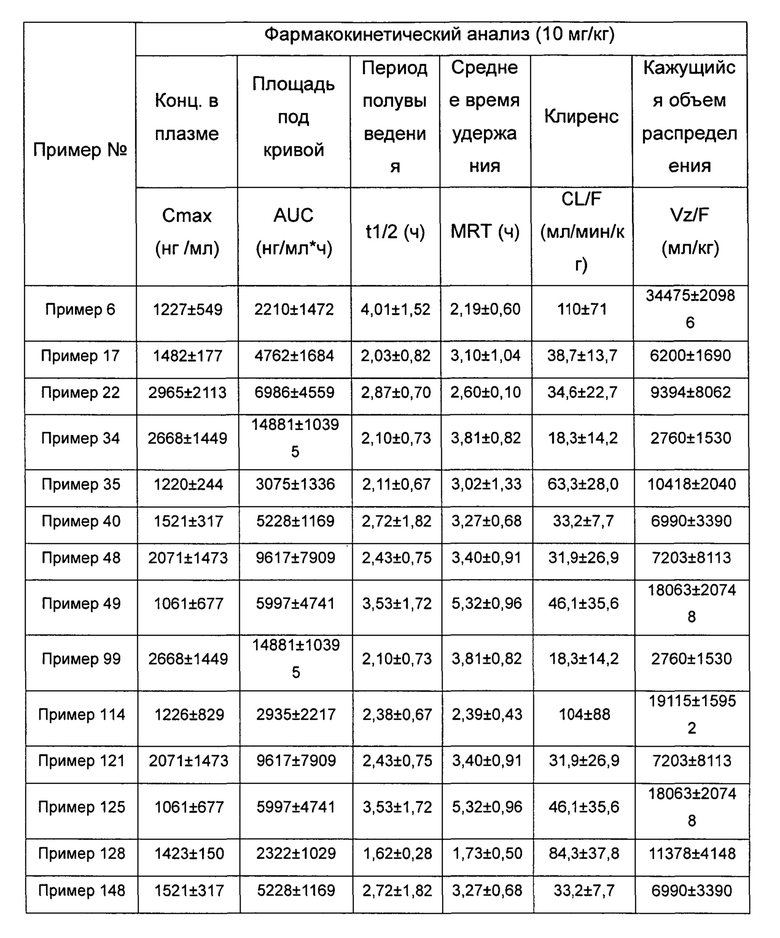
Вывод: Соединения по настоящему изобретению обладали улучшенными фармакокинетическими данными и значительным преимуществом в фармакокинетических свойствах.
Следующие примеры демонстрируют сравнительные данные активности соединений по настоящему изобретению и соединений, раскрытых в документах предшествующего уровня техники: WO 2006069080, WO 2002000661 и WO 1999065908.
Сравнительный пример 1
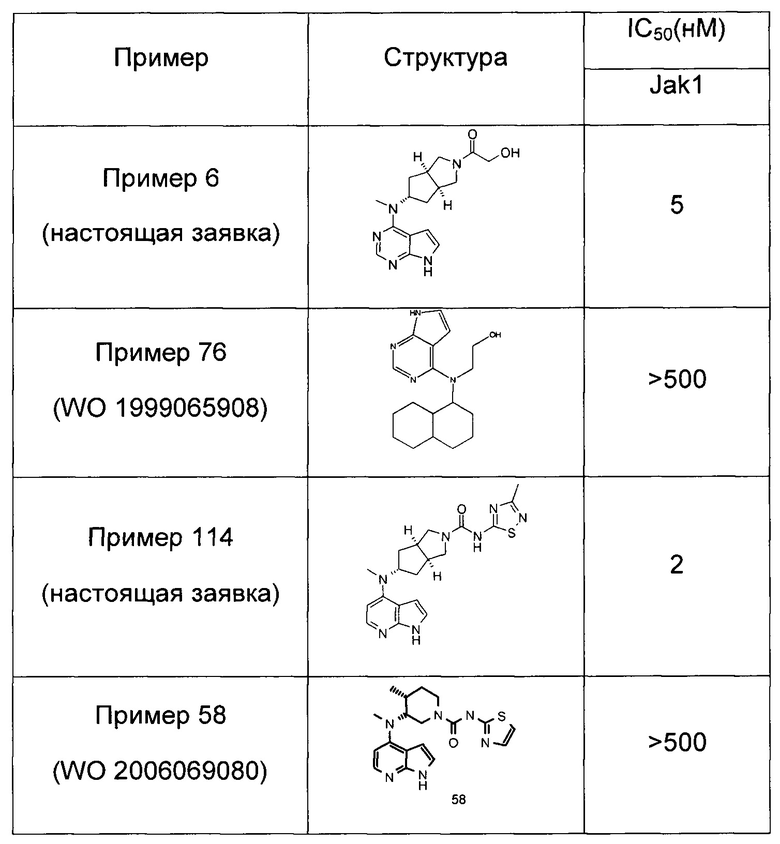
Сравнительный пример 2
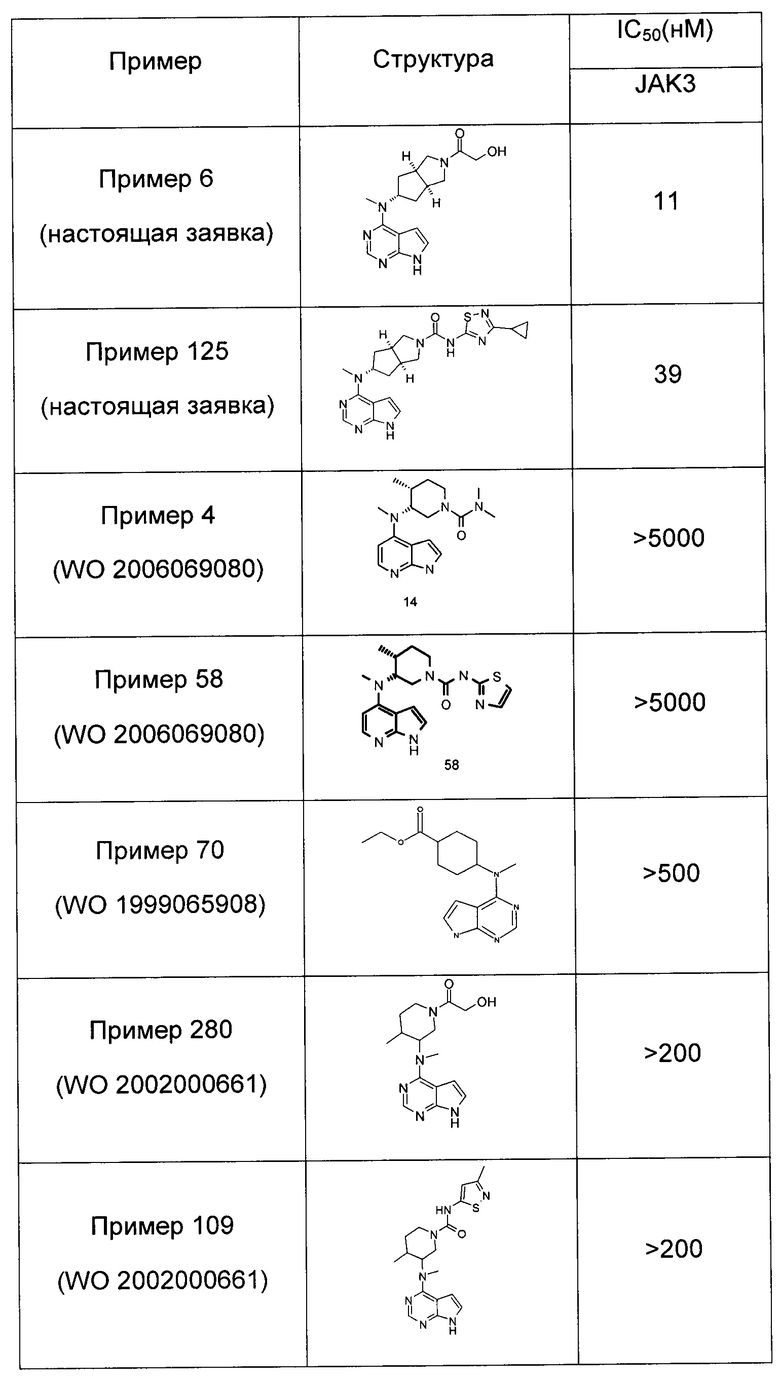
| название | год | авторы | номер документа |
|---|---|---|---|
| БИСУЛЬФАТ ИНГИБИТОРА ЯНУС-КИНАЗЫ (JAK) И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2014 |
|
RU2665680C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТНОГО ИНГИБИТОРА JAK-КИНАЗЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2716260C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТА ИНГИБИТОРА JAK И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2704795C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| УСИЛИТЕЛЬ ПРОТИВООПУХОЛЕВОГО ВОЗДЕЙСТВИЯ, СОДЕРЖАЩИЙ СОЕДИНЕНИЕ ПИРРОЛОПИРИМИДИНА | 2016 |
|
RU2710380C1 |
| НОВОЕ ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕЕ, В ЧАСТНОСТИ, АГЕНТ ДЛЯ ПРЕДОТВРАЩЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ОПУХОЛЕЙ, И ТОМУ ПОДОБНОЕ, НА ОСНОВЕ ИНГИБИТОРНОГО ВОЗДЕЙСТВИЯ НА NAE | 2015 |
|
RU2658008C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДОНА ИЛИ ПИРРОЛОТРИАЗОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2014 |
|
RU2674977C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ИНГИБИТОР ЯНУС-КИНАЗЫ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМУЮ СОЛЬ | 2017 |
|
RU2744432C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ОПОСРЕДОВАНИЯ АКТИВНОСТИ ТИРОЗИНКИНАЗЫ 2 | 2020 |
|
RU2826012C2 |

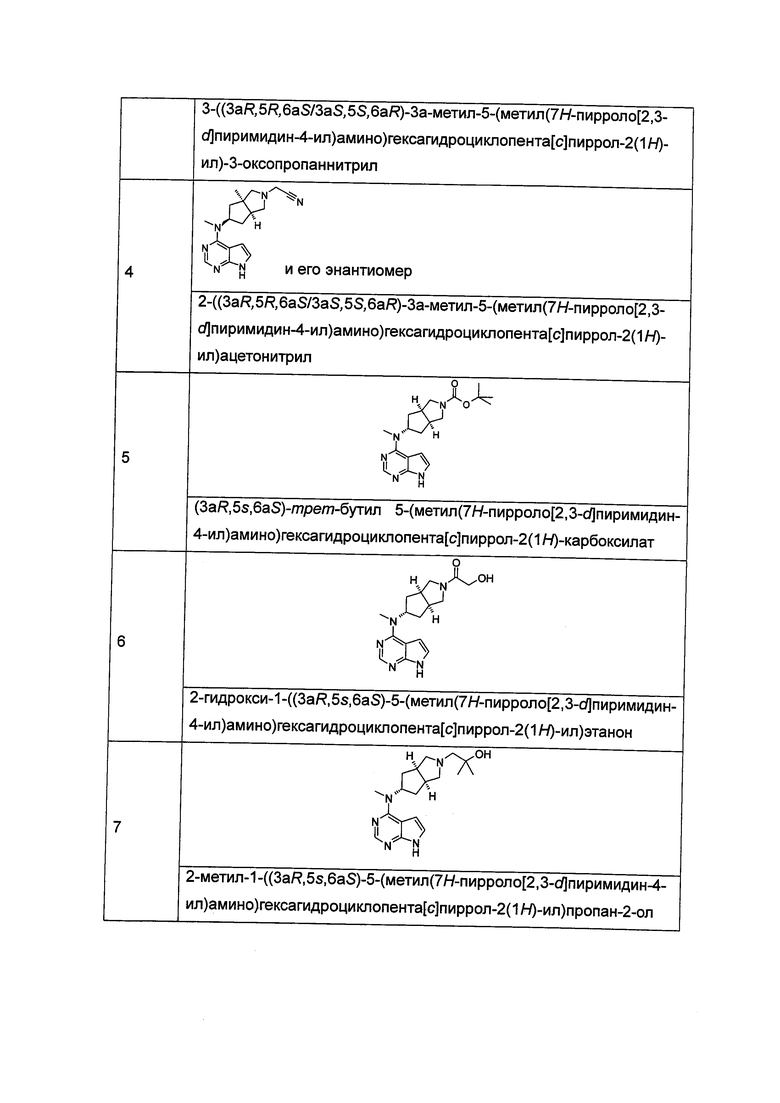
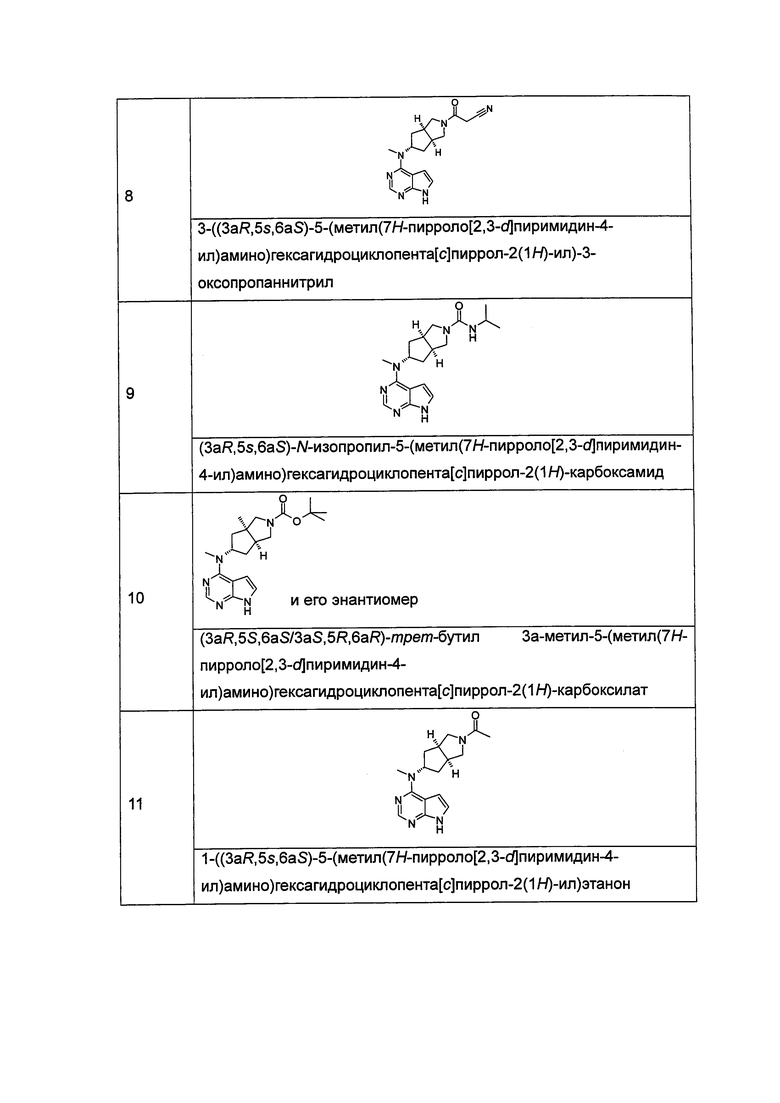
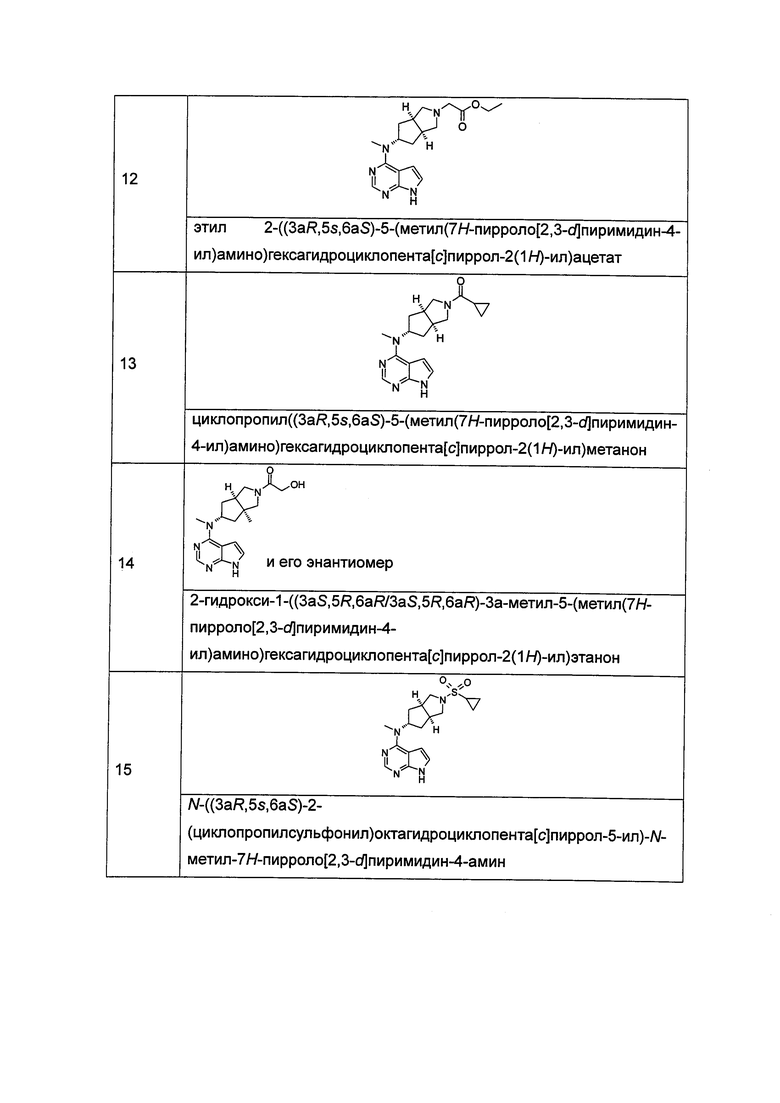
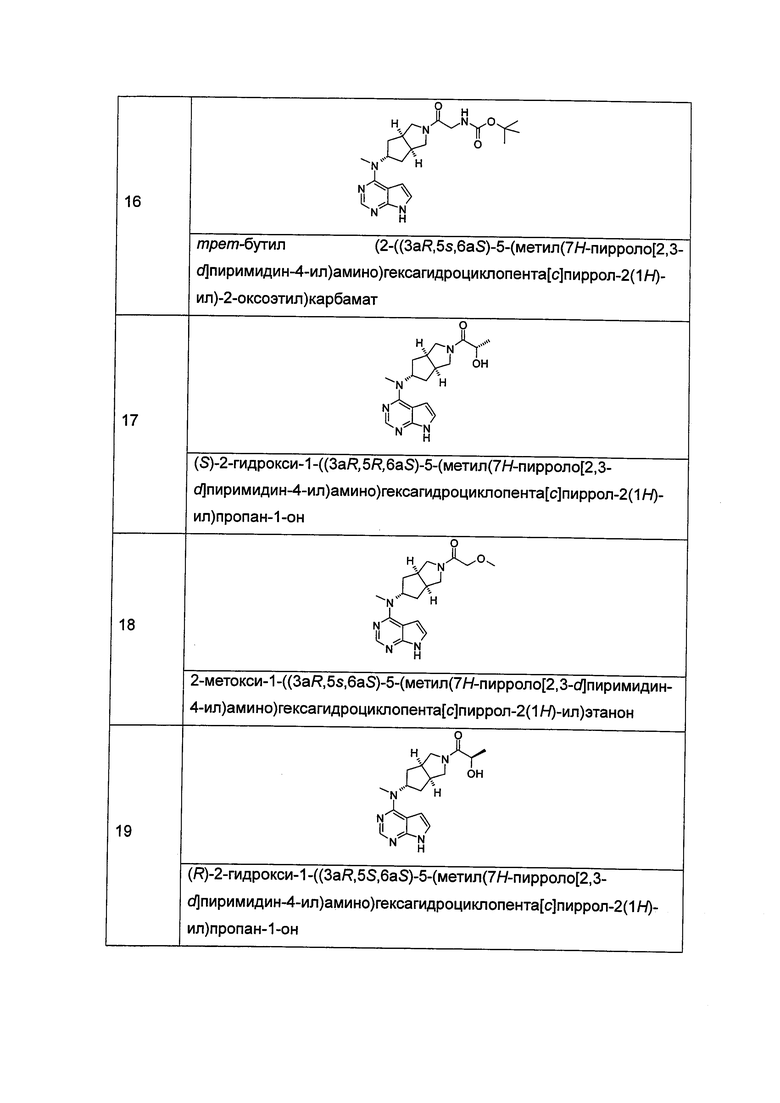
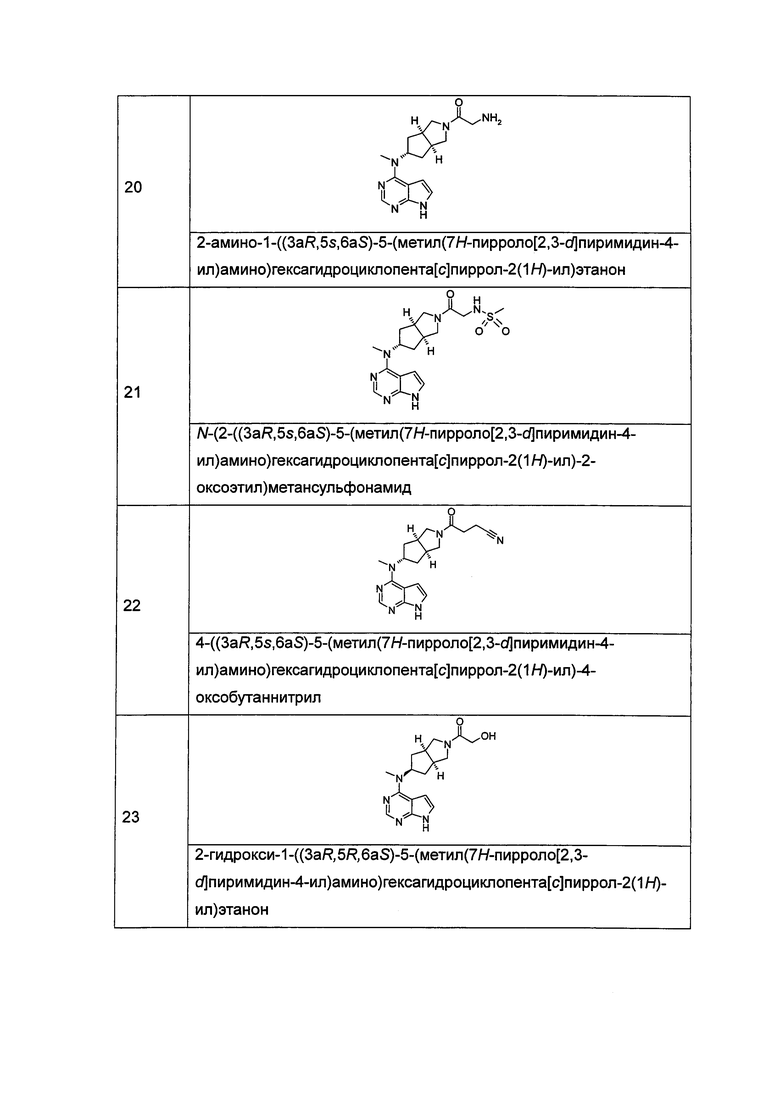
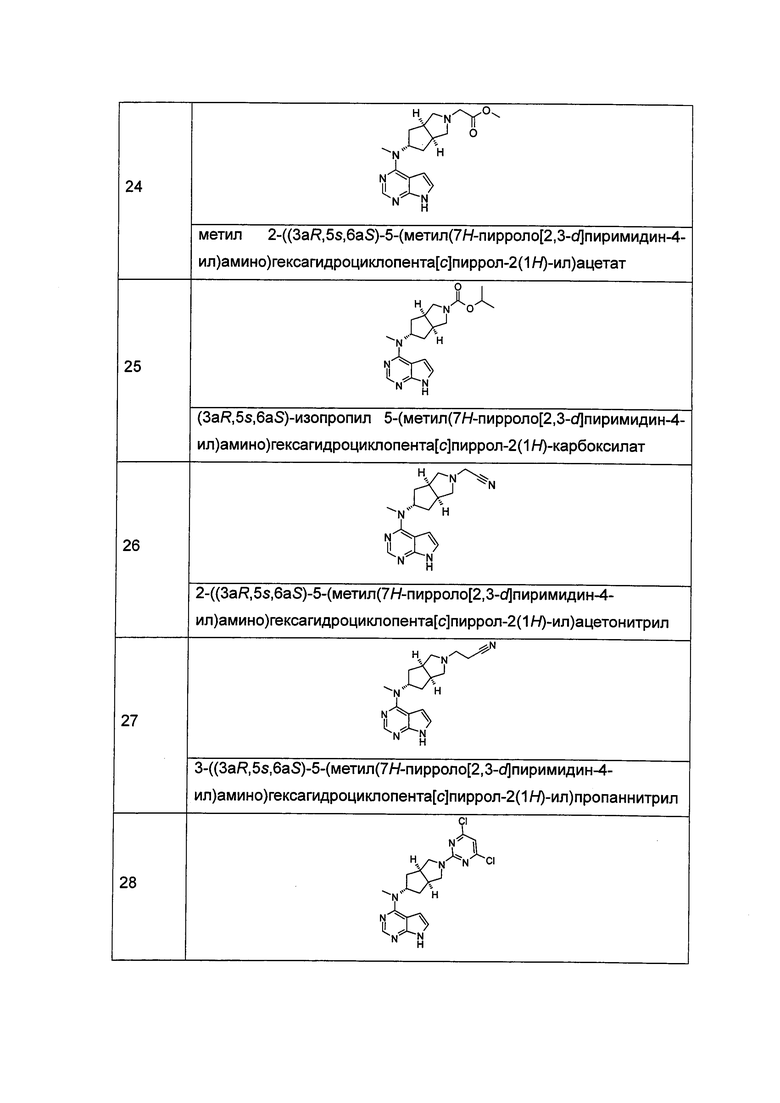
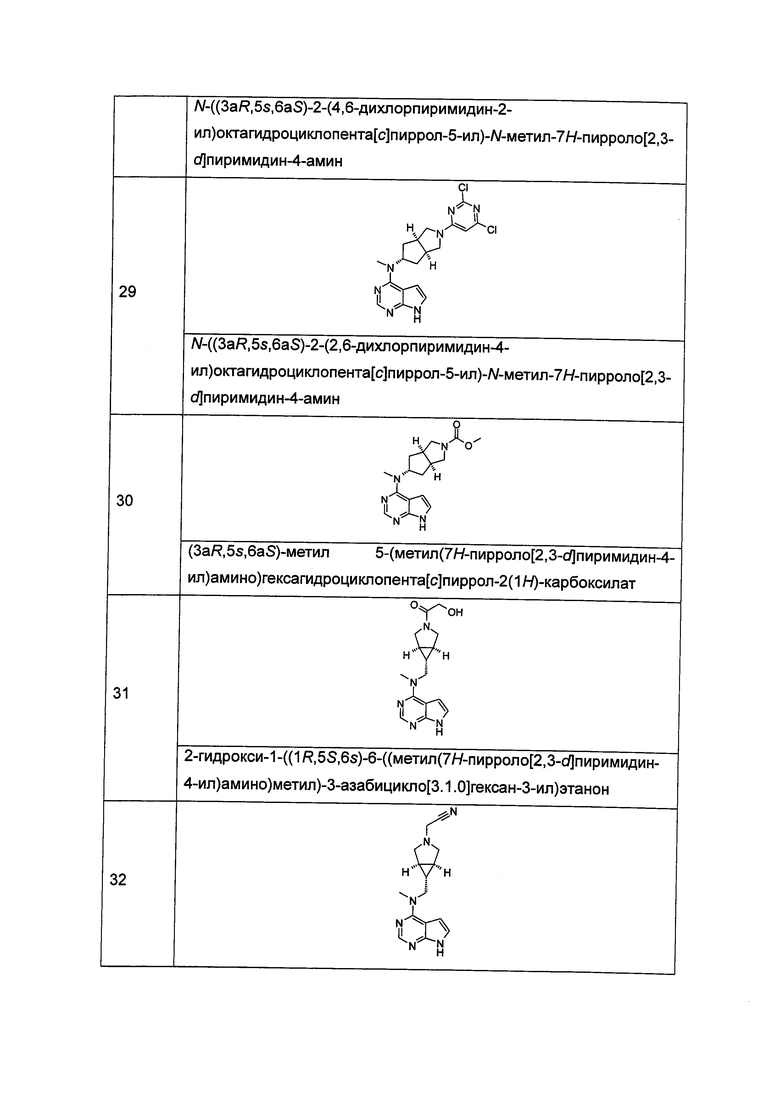
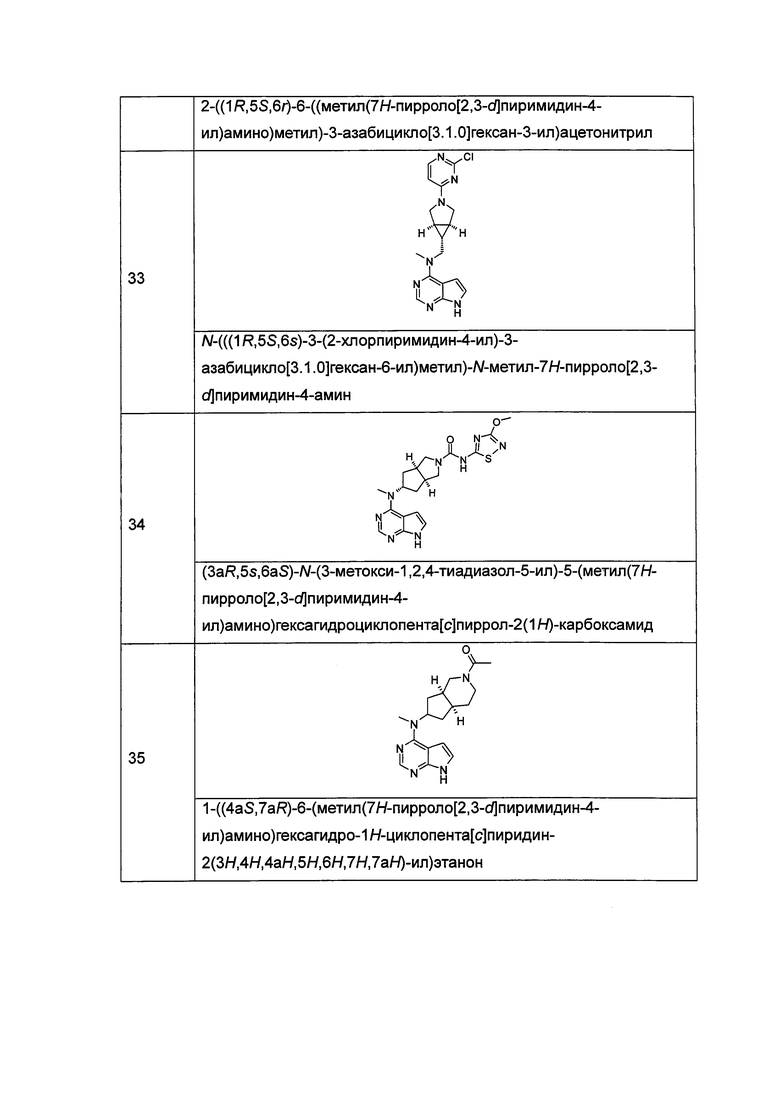
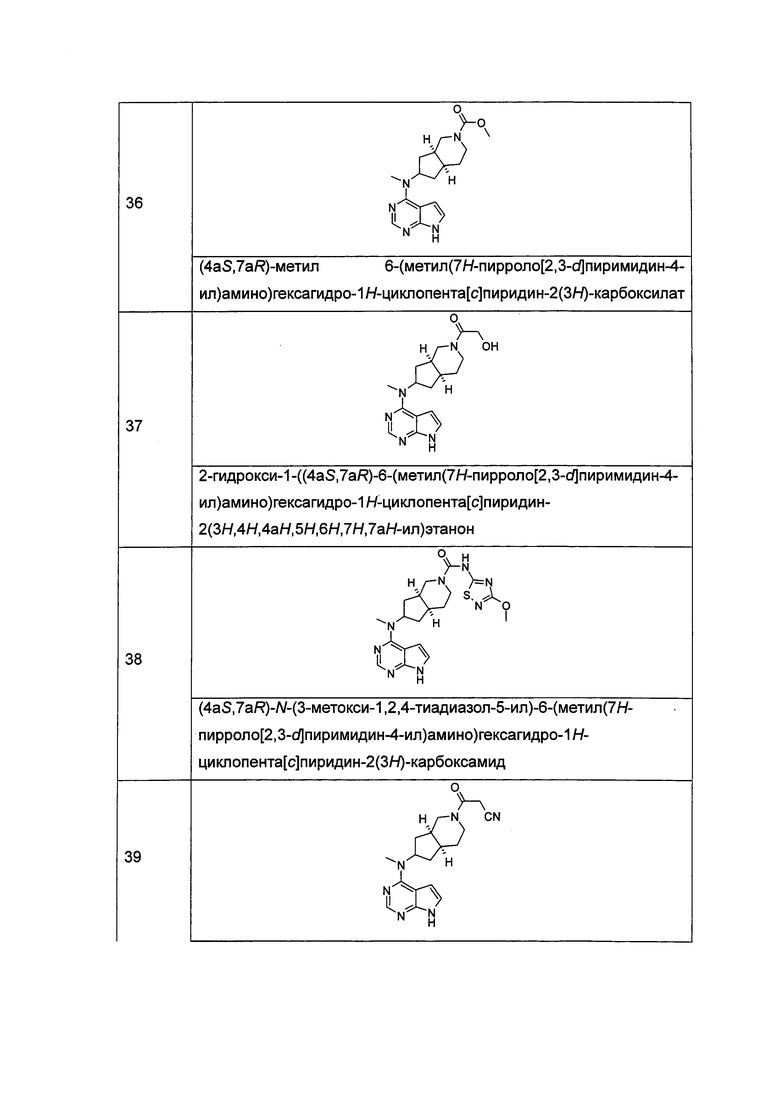
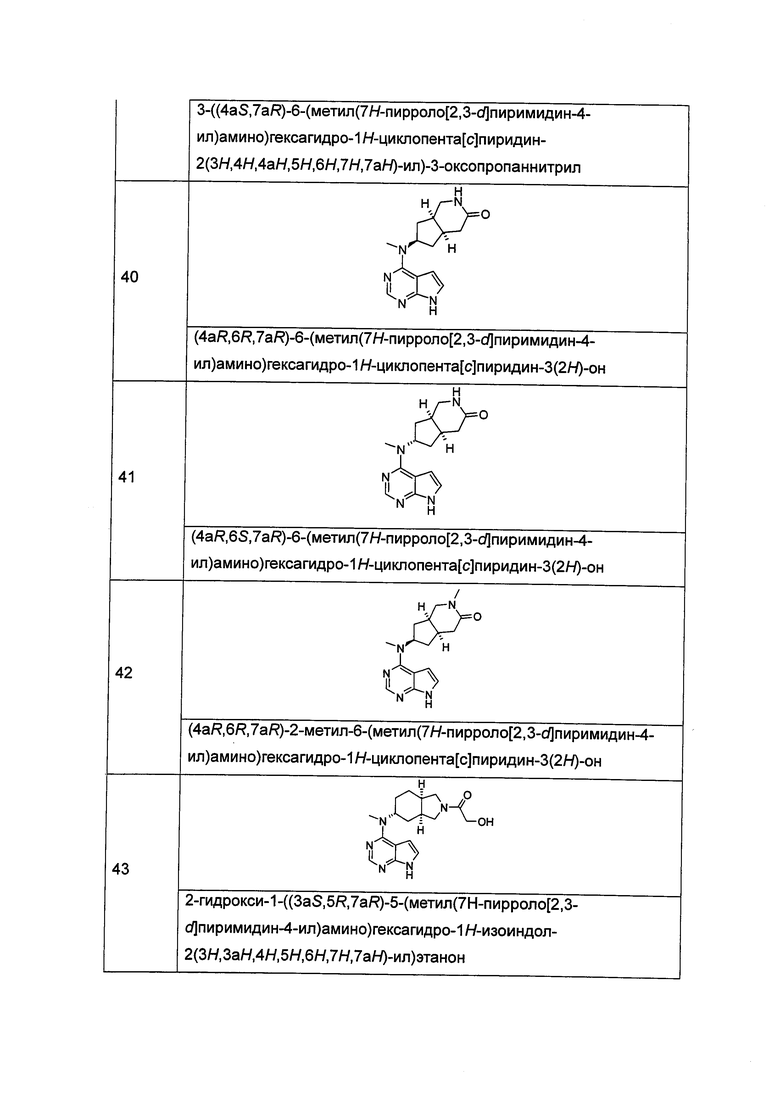
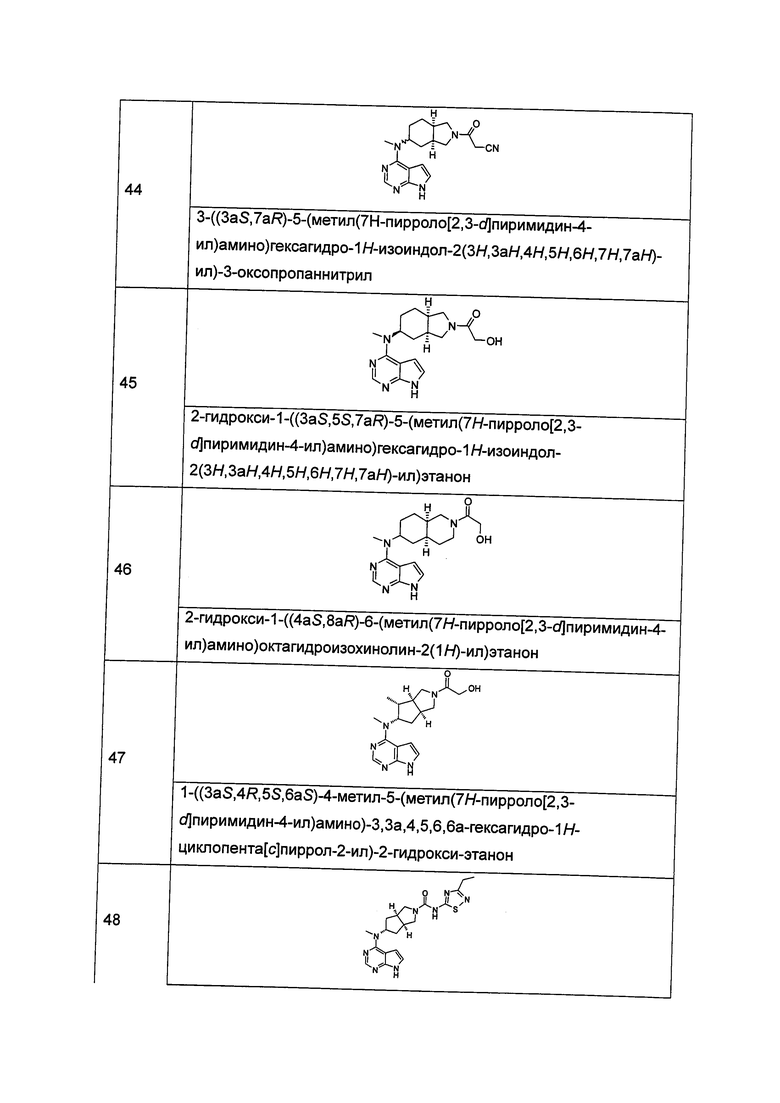
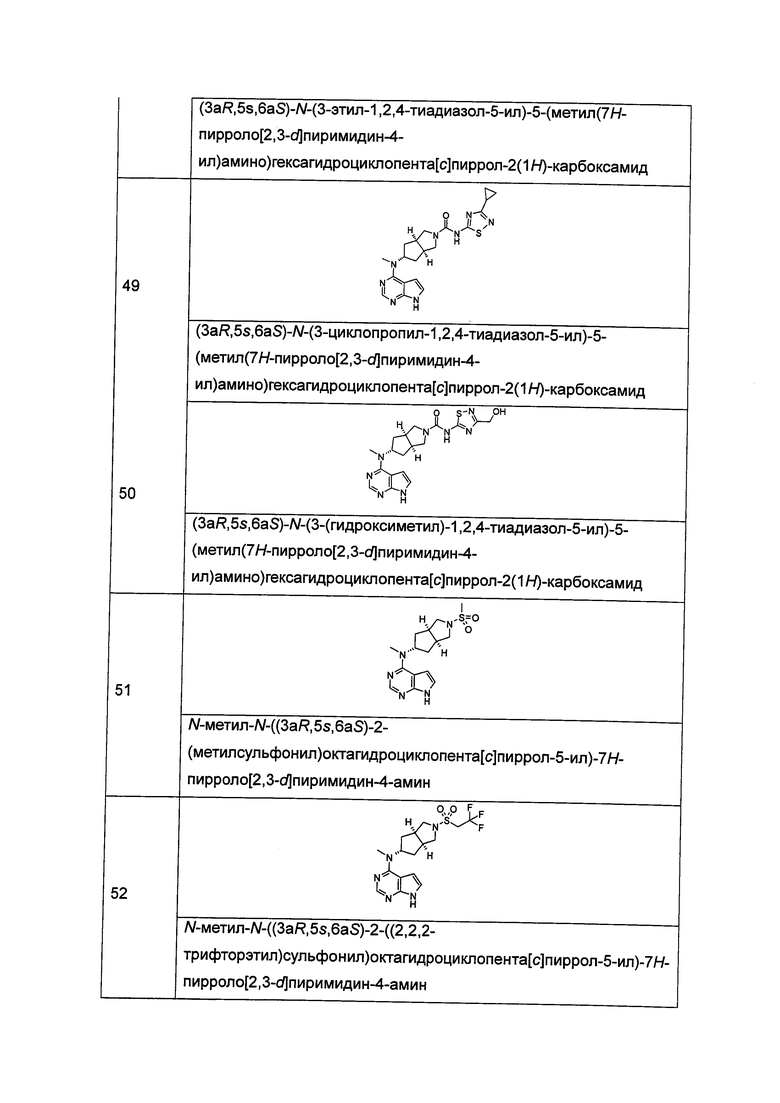
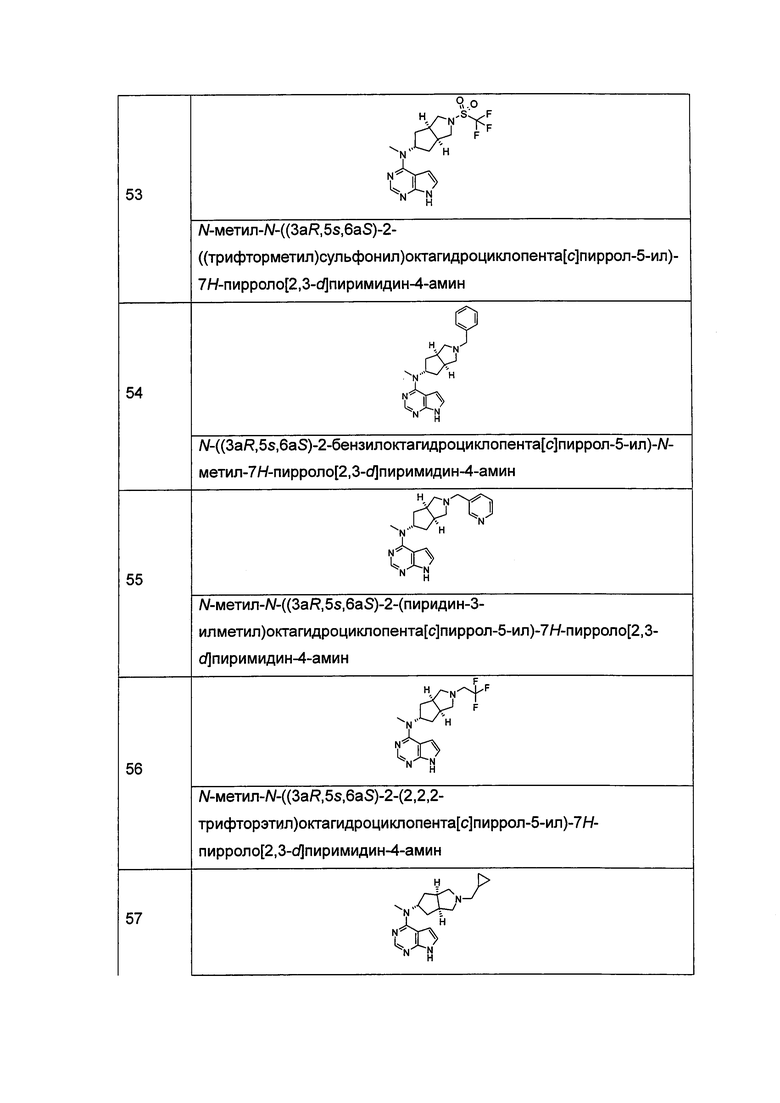


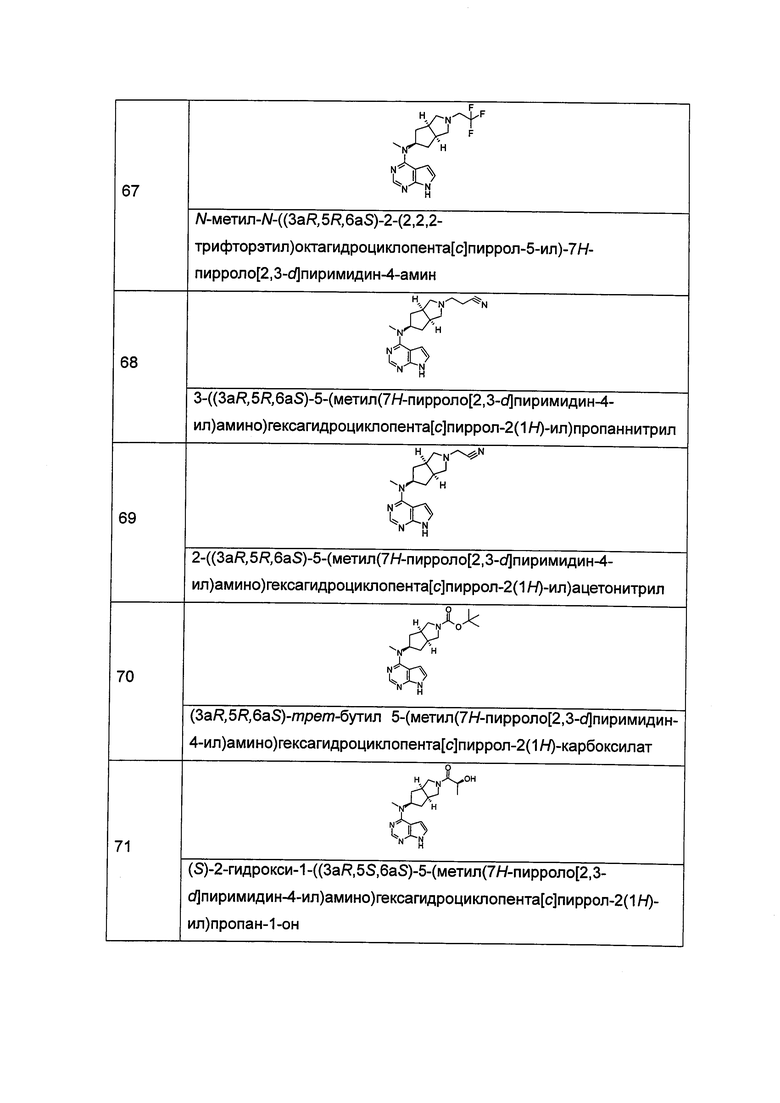

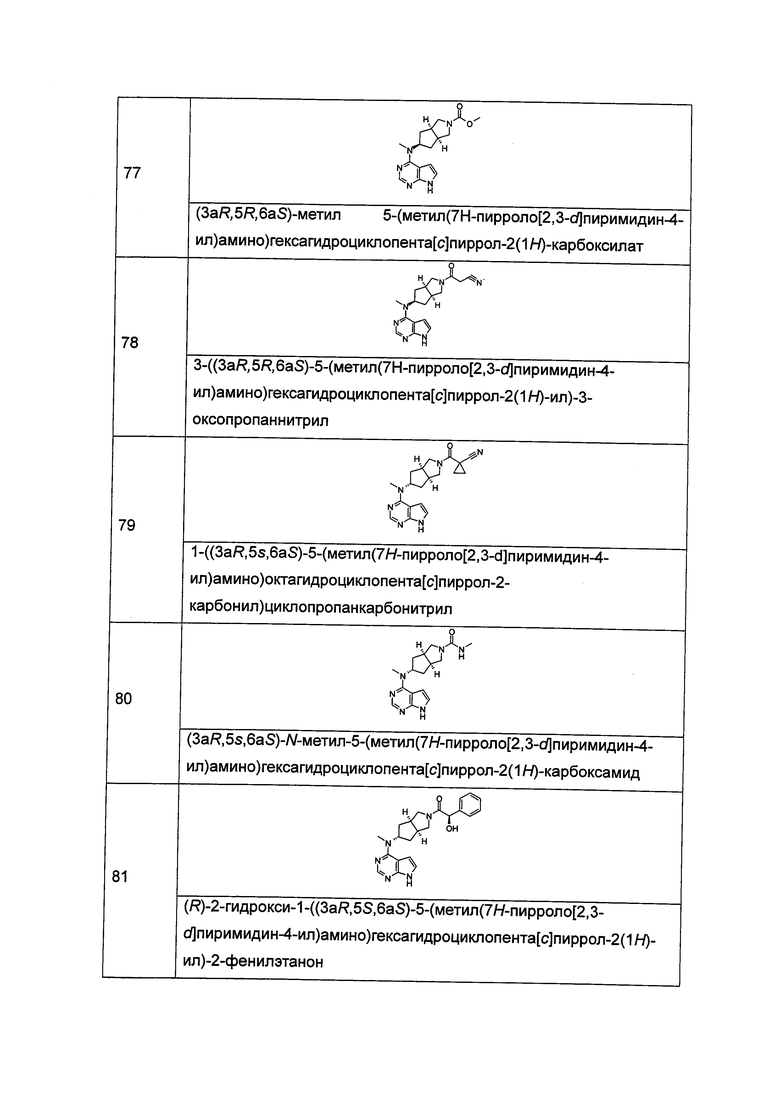

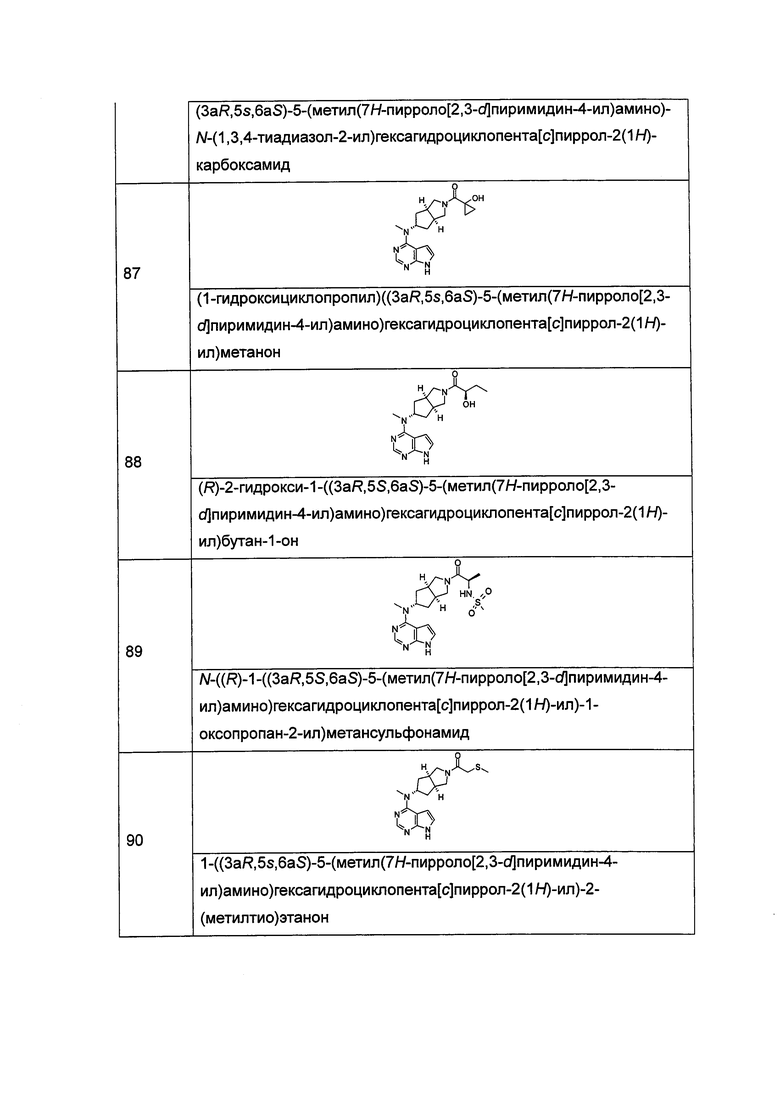

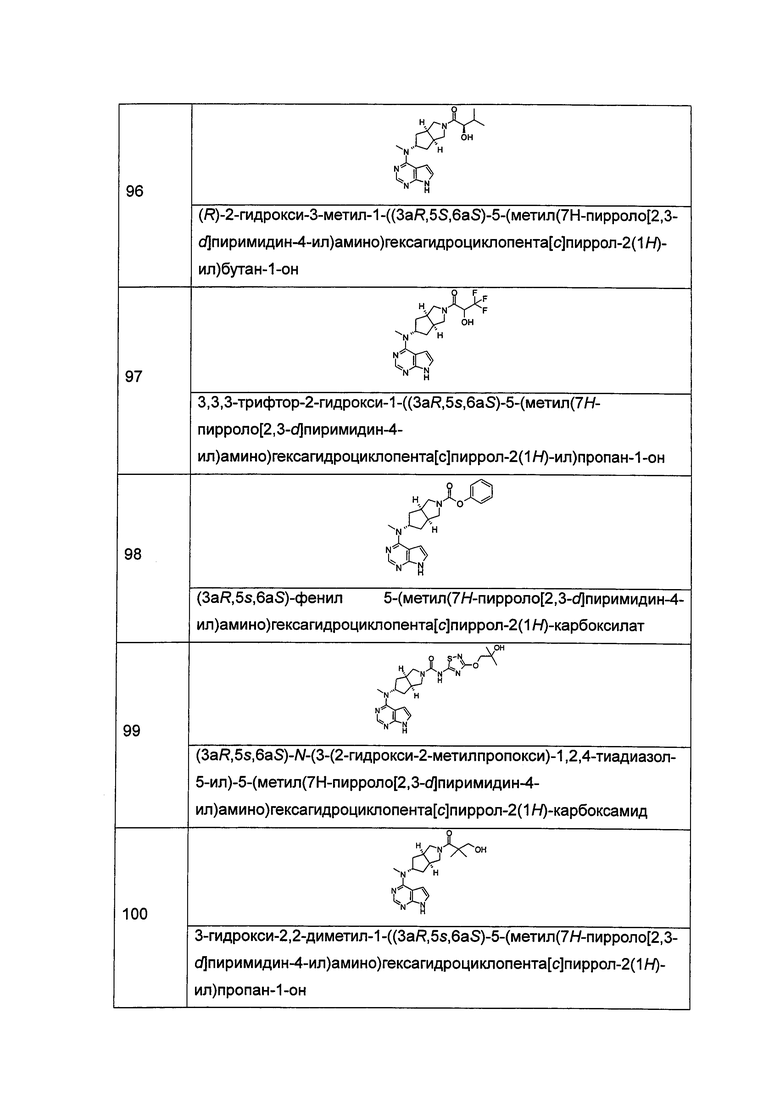
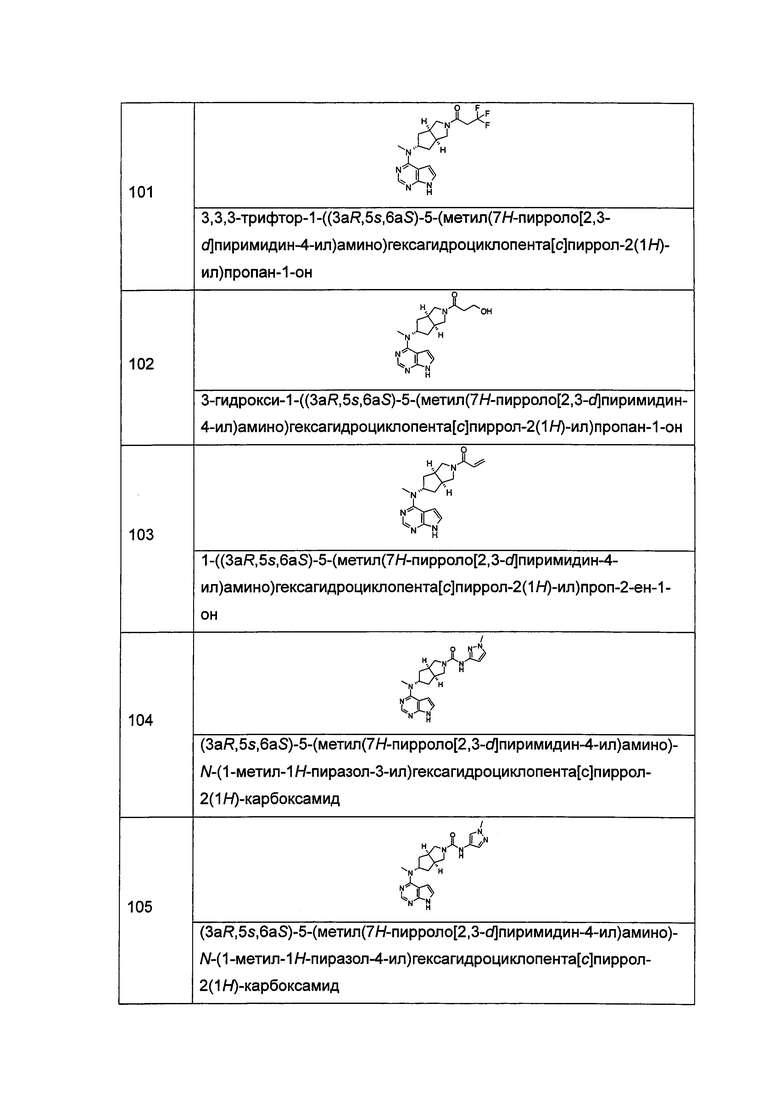


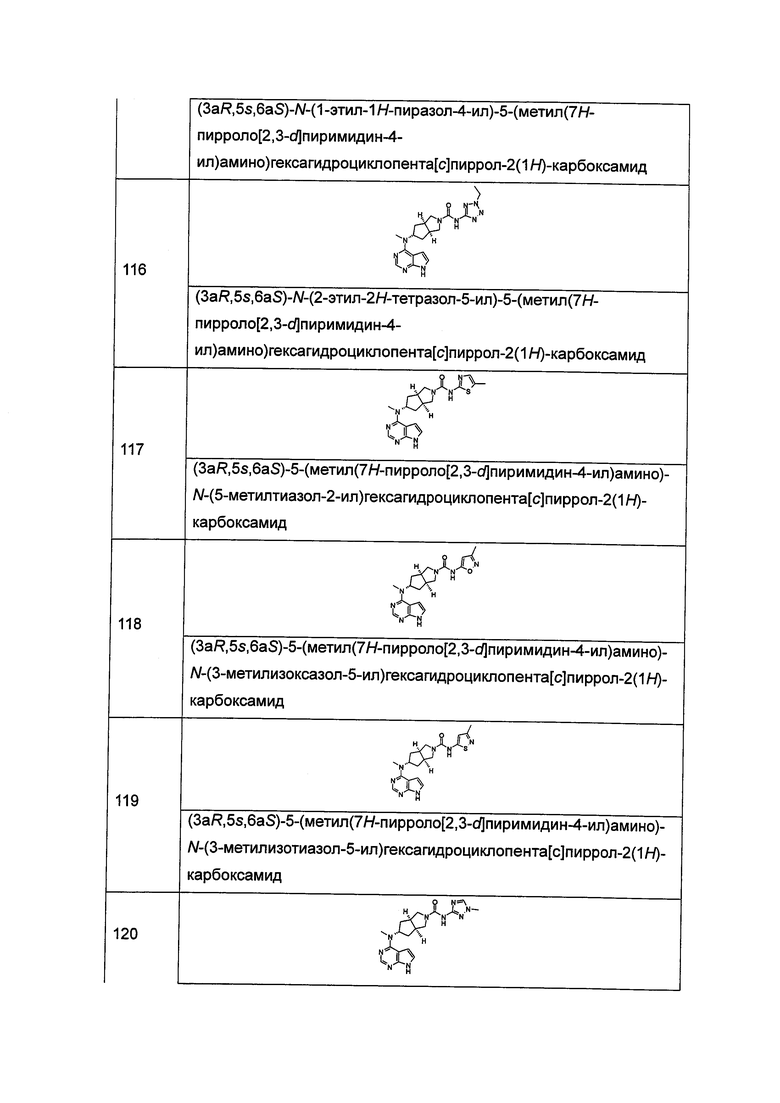

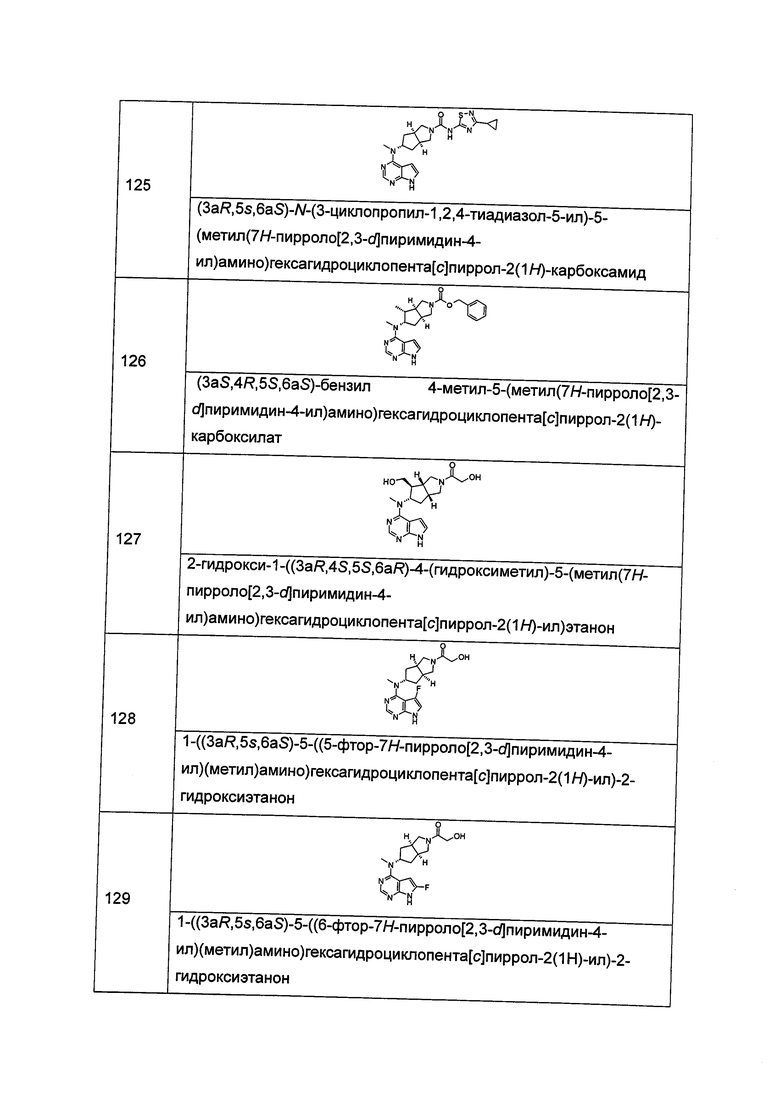
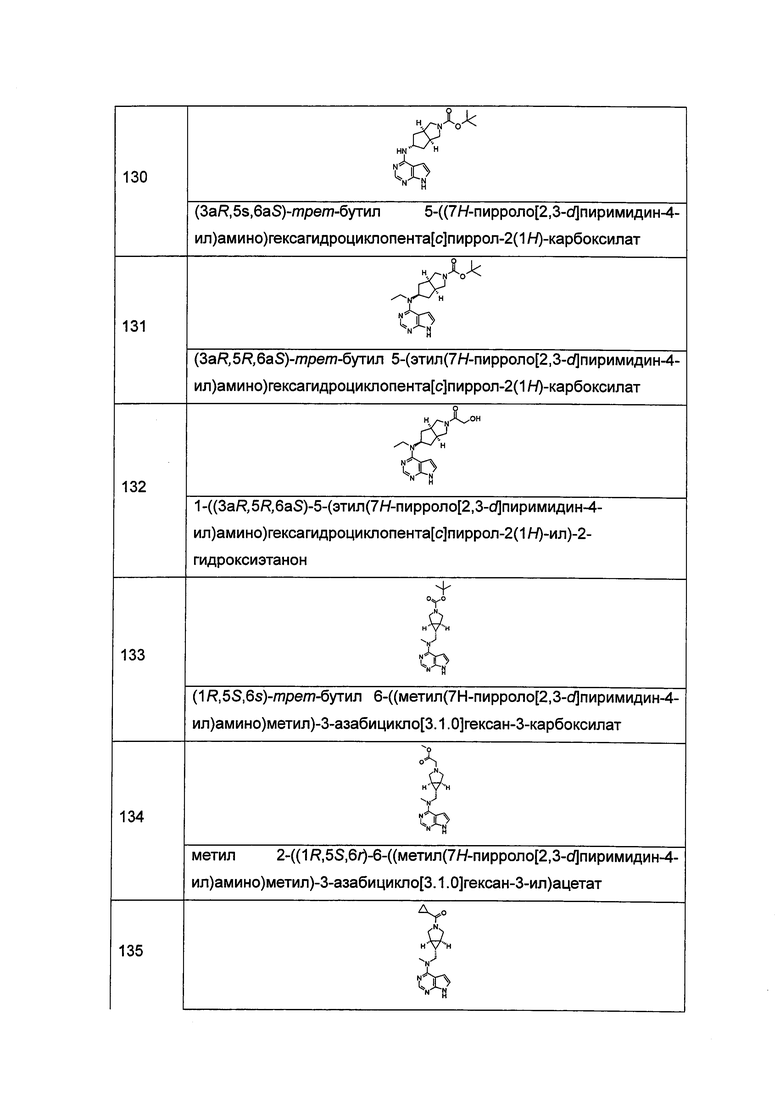



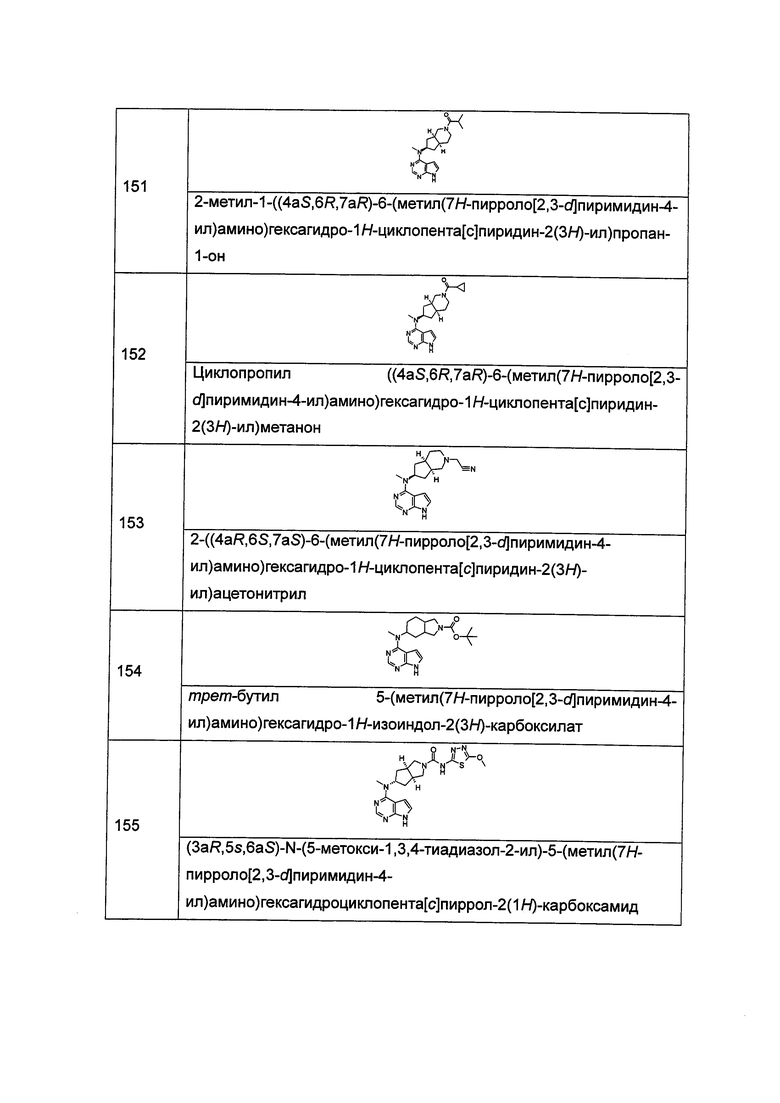

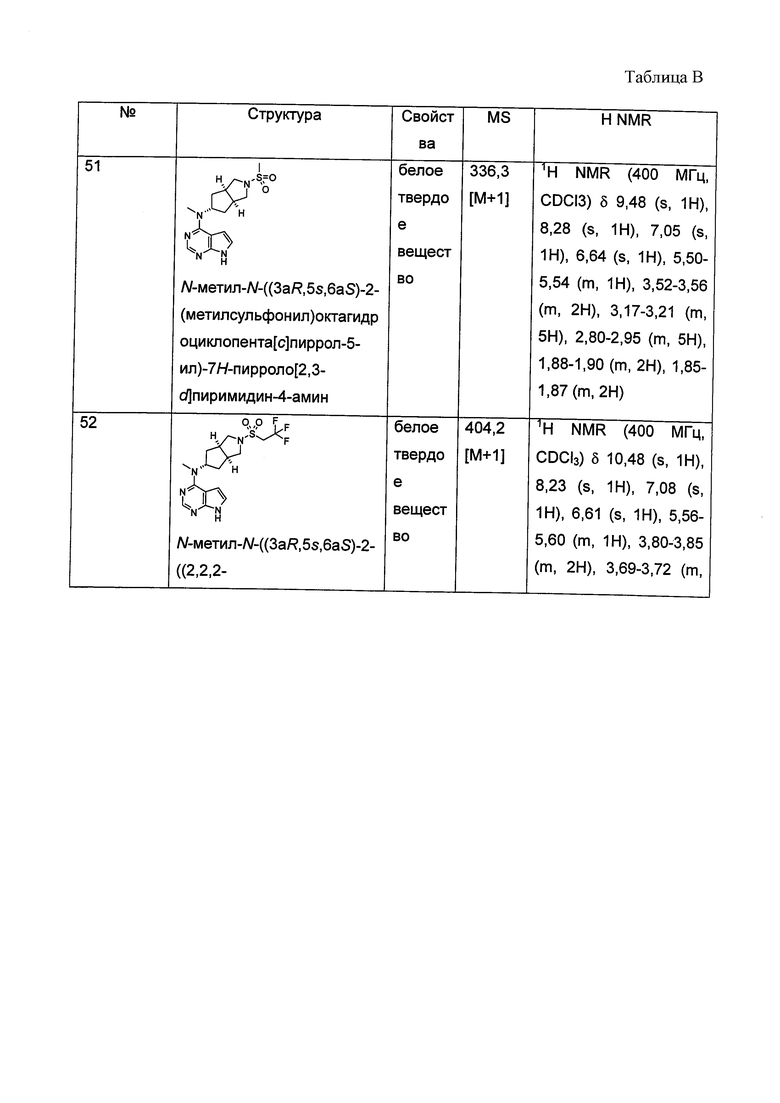
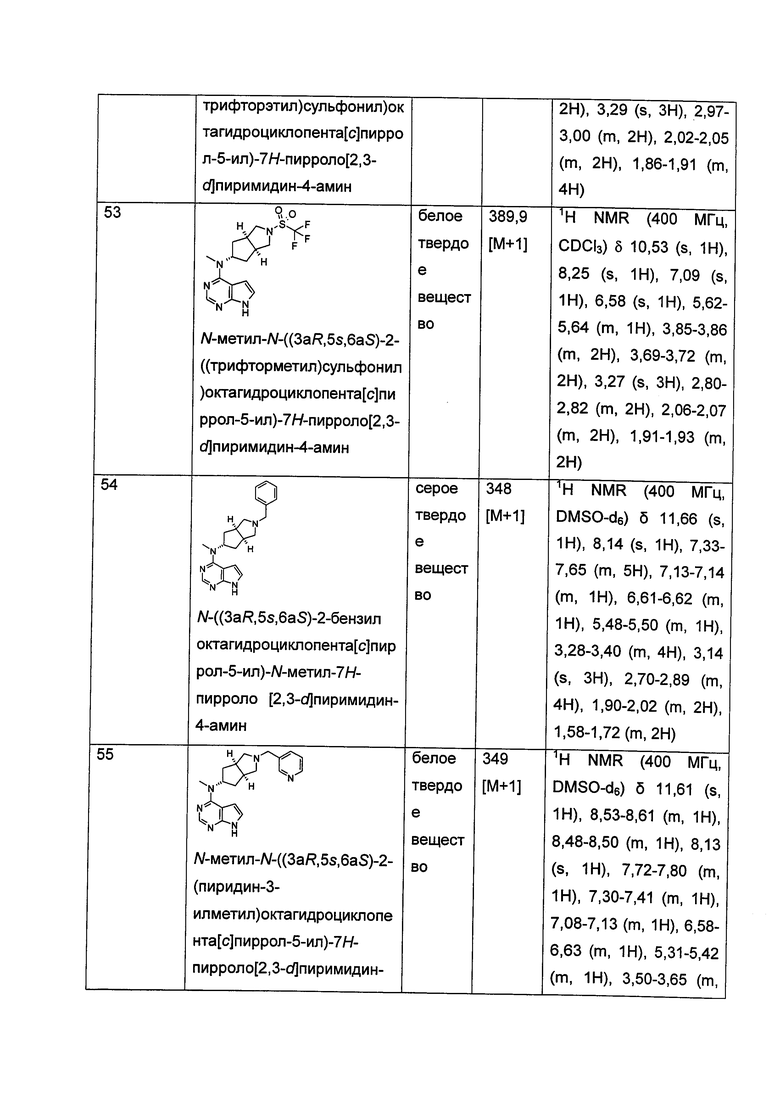
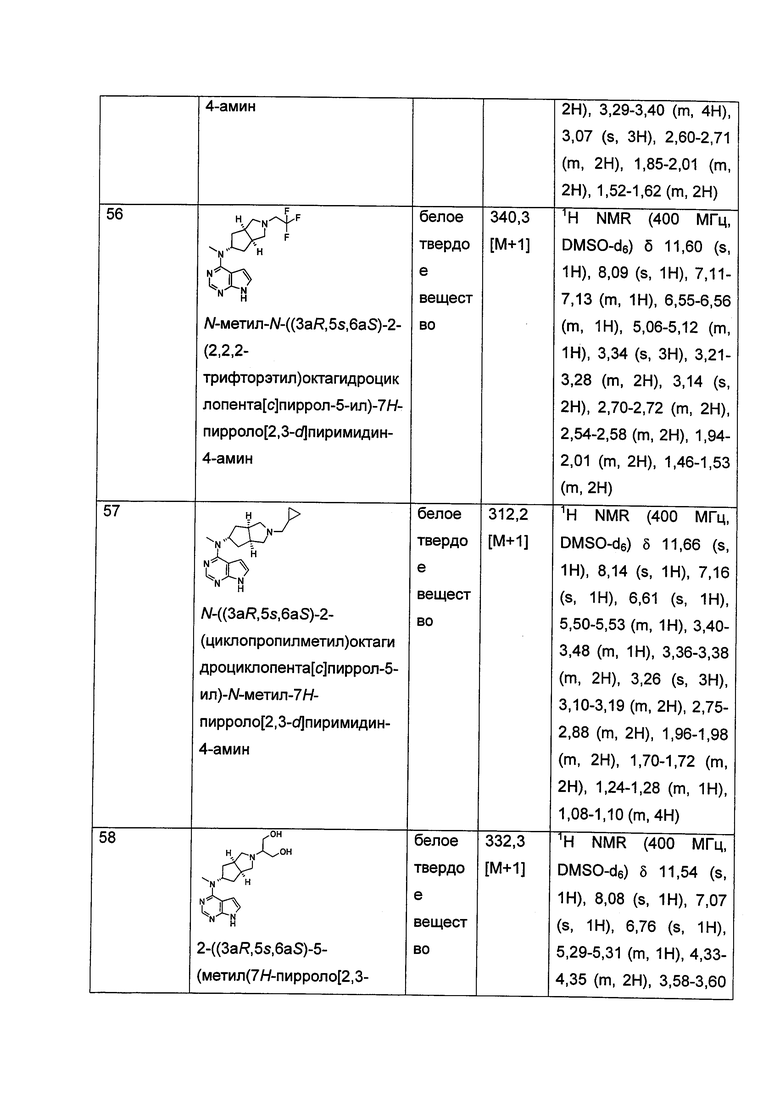
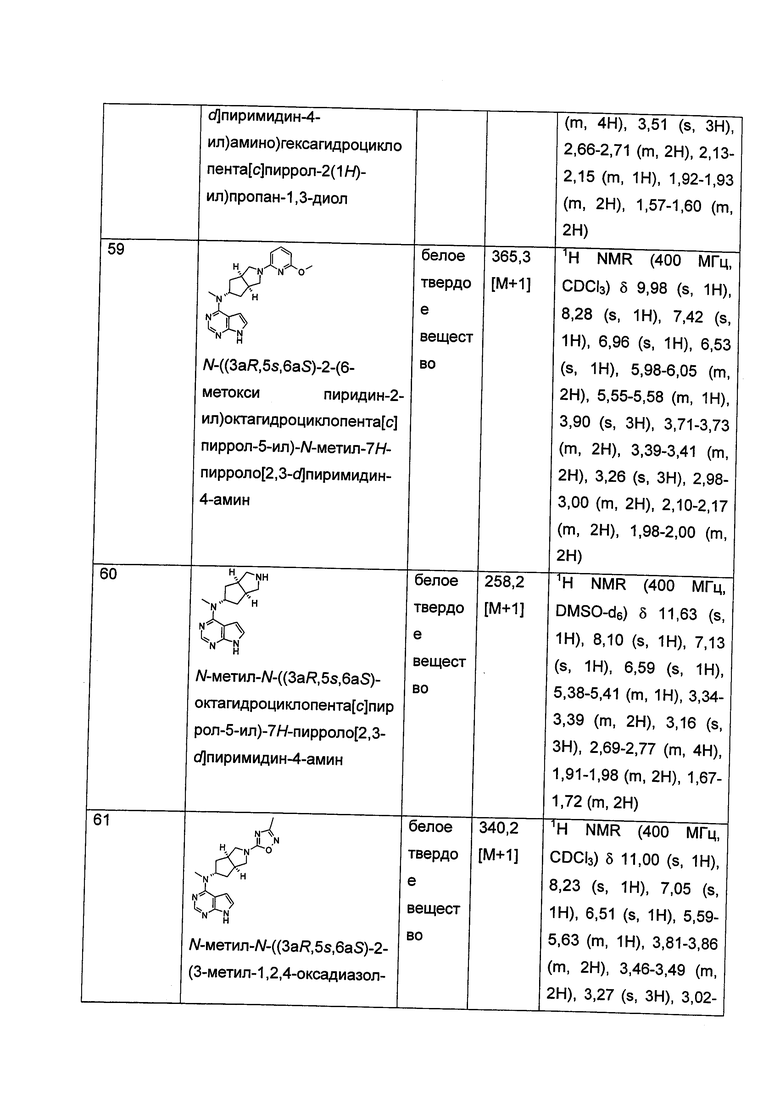
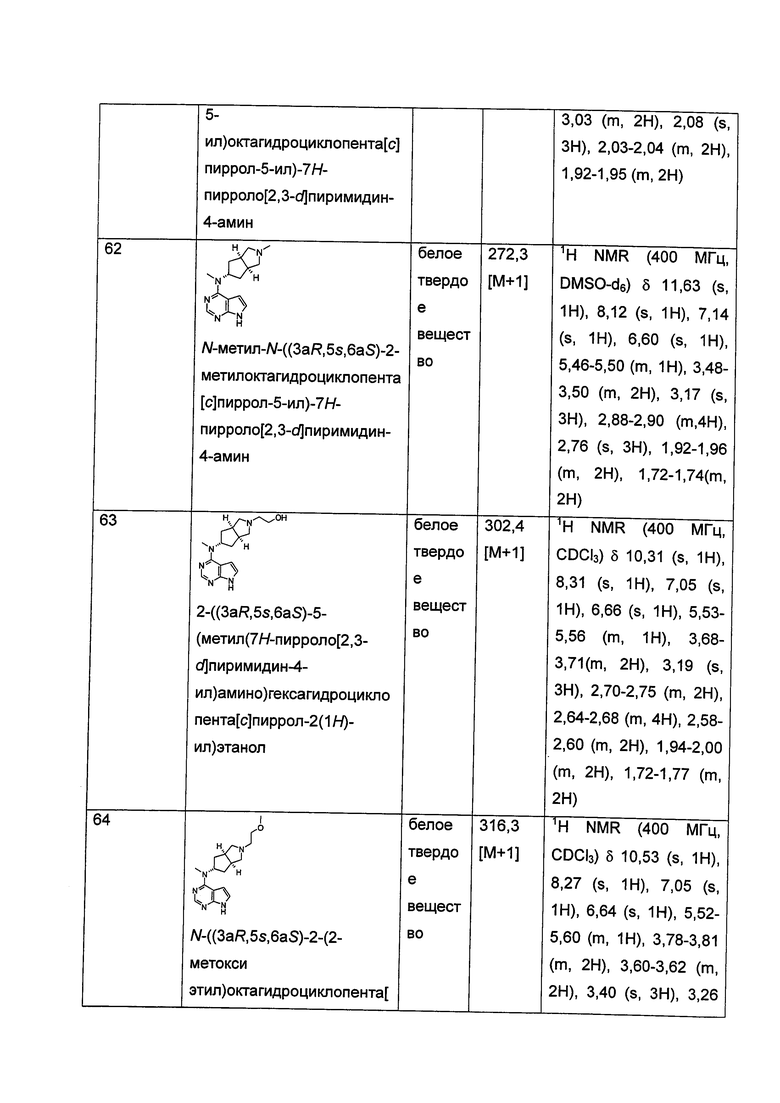
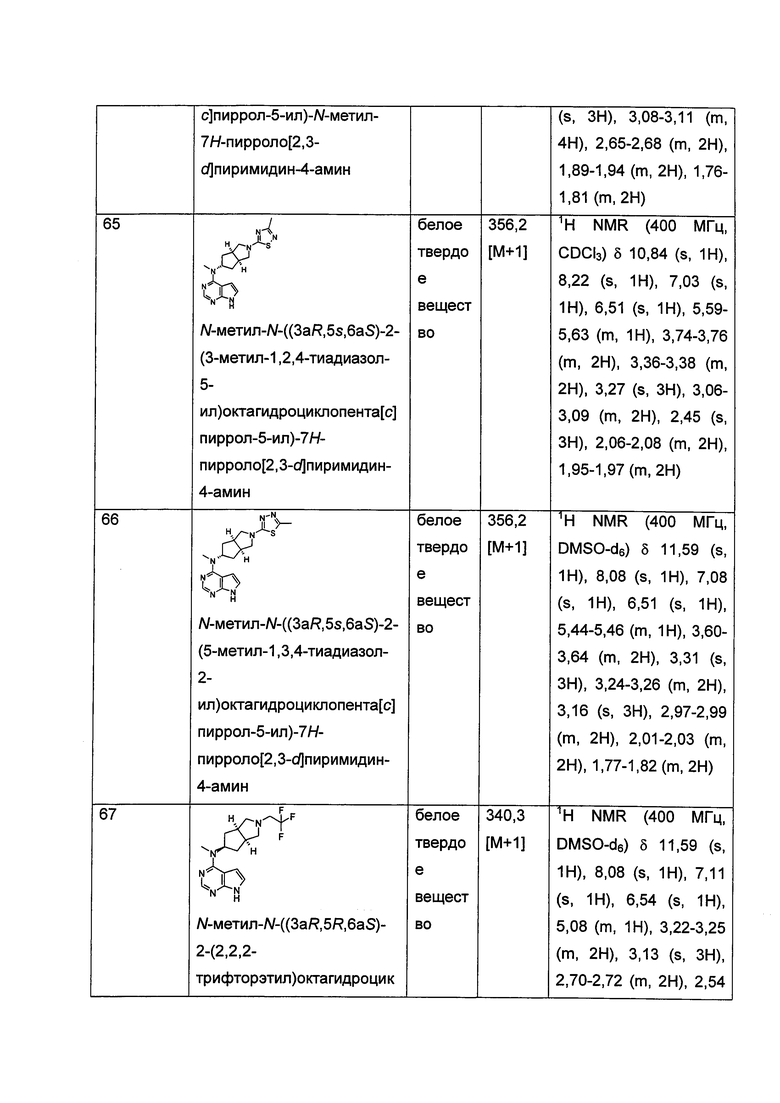
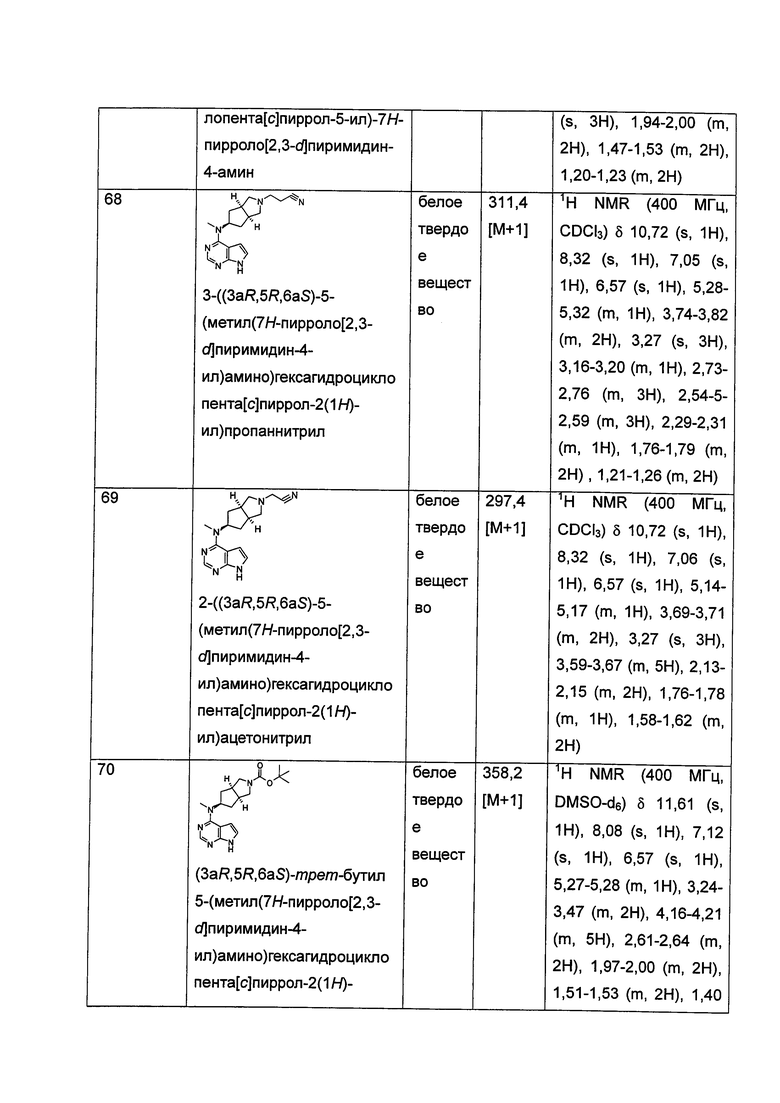
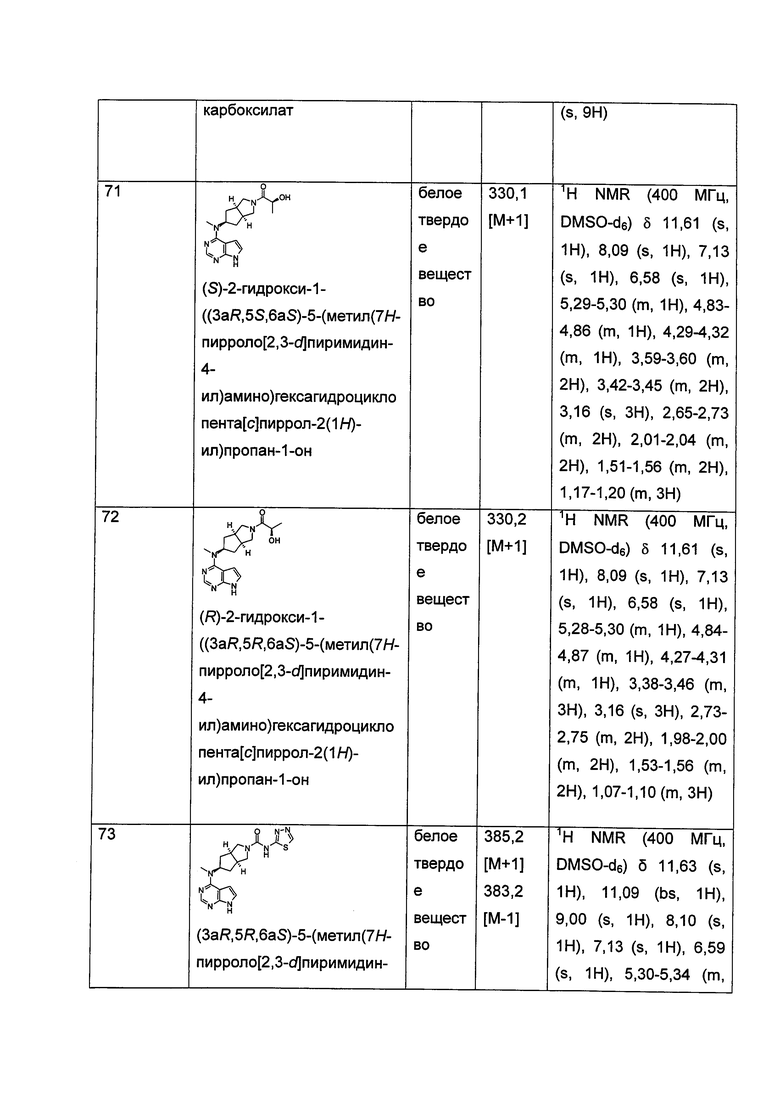

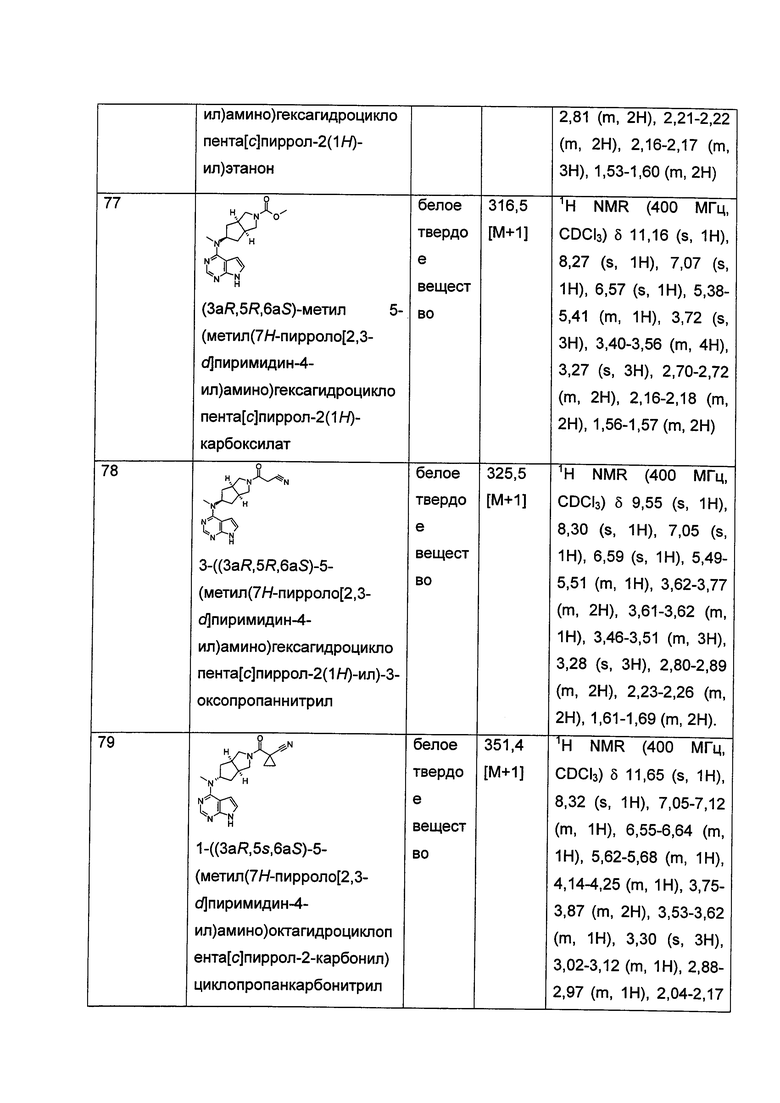
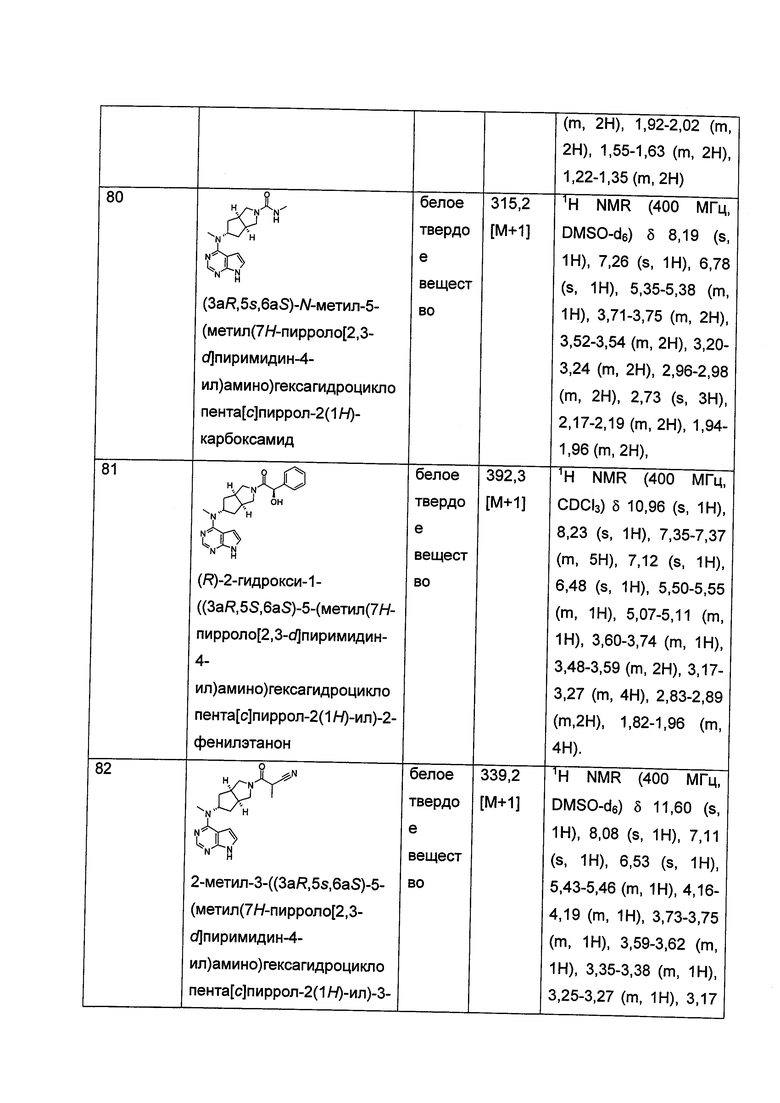
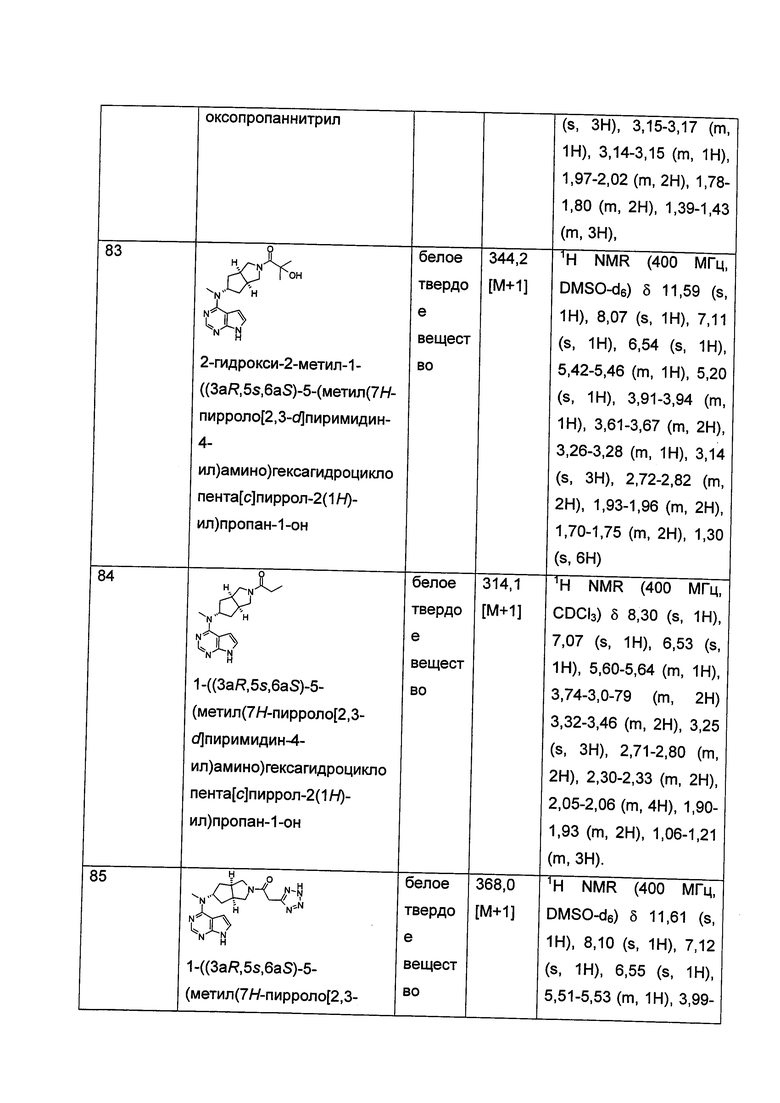
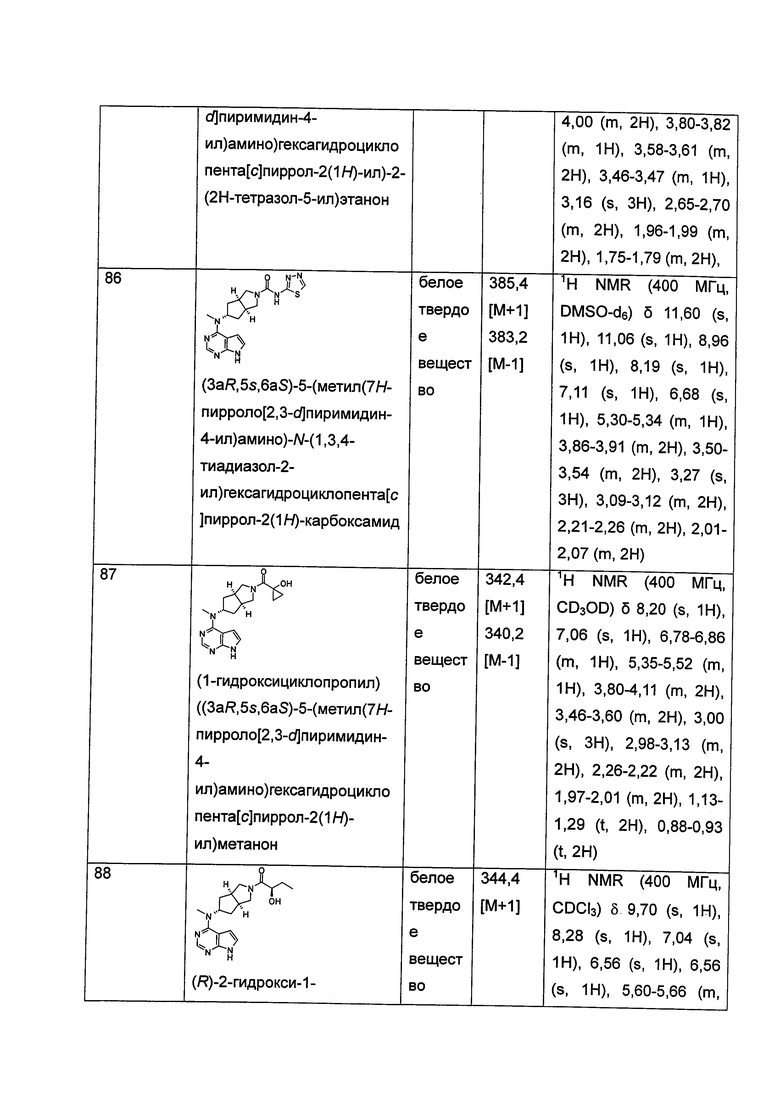
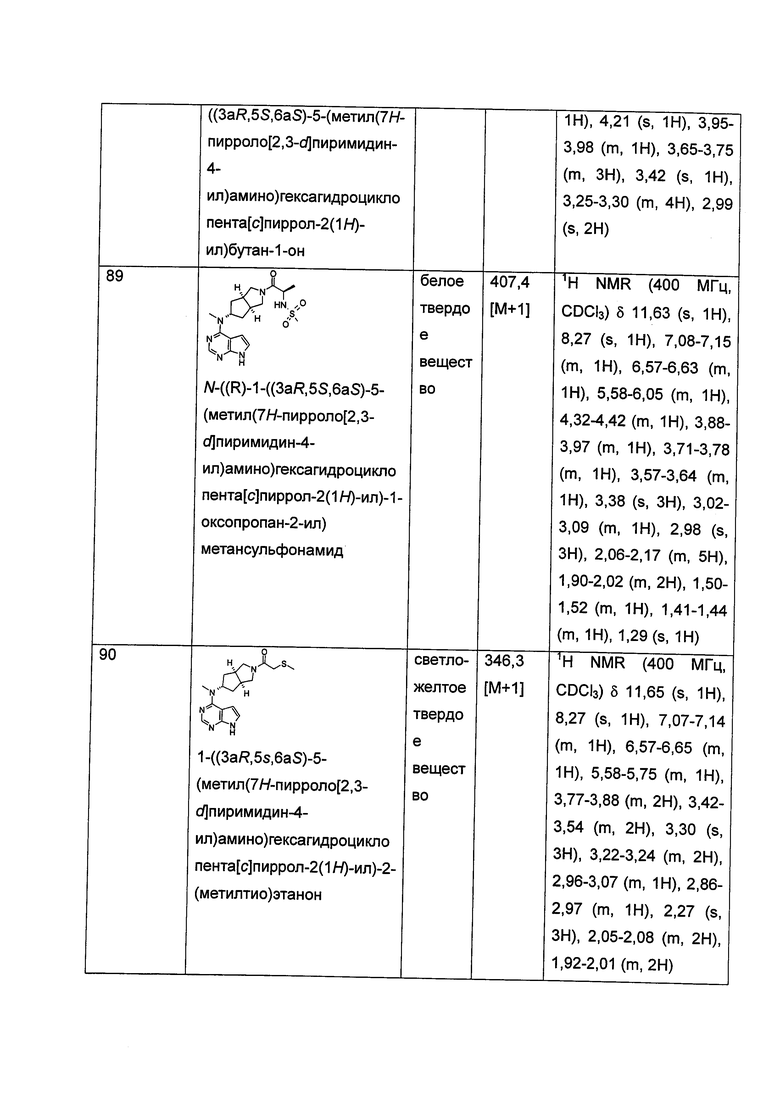
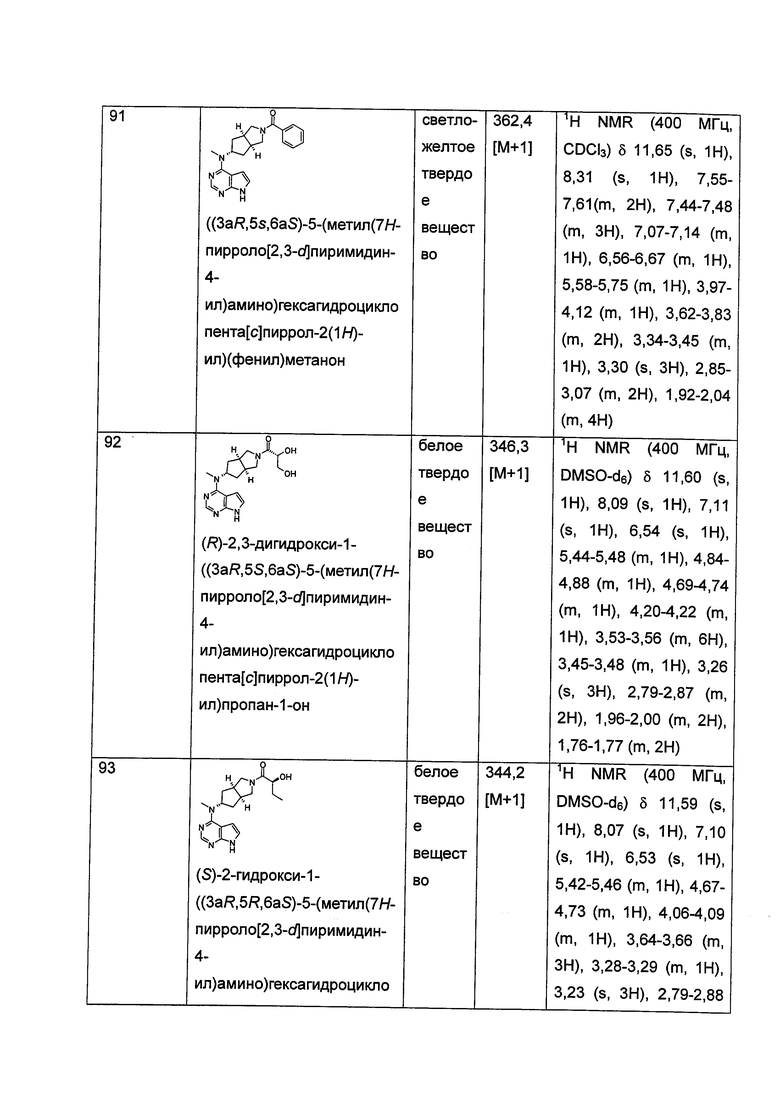
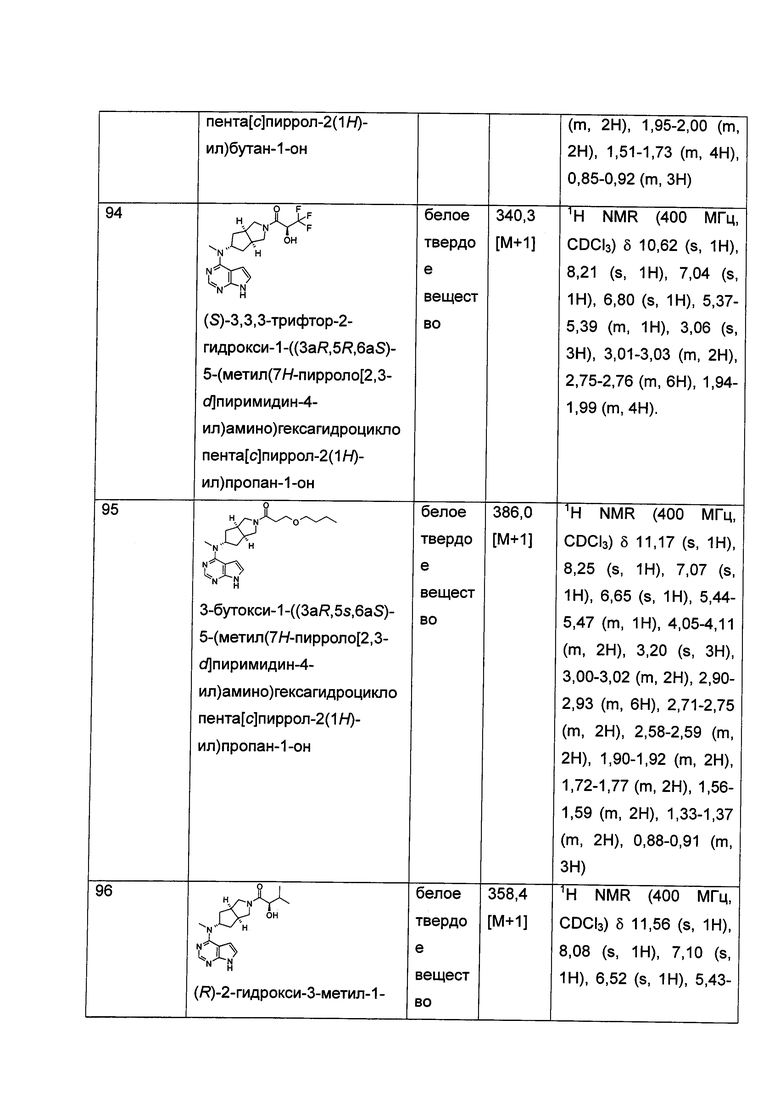
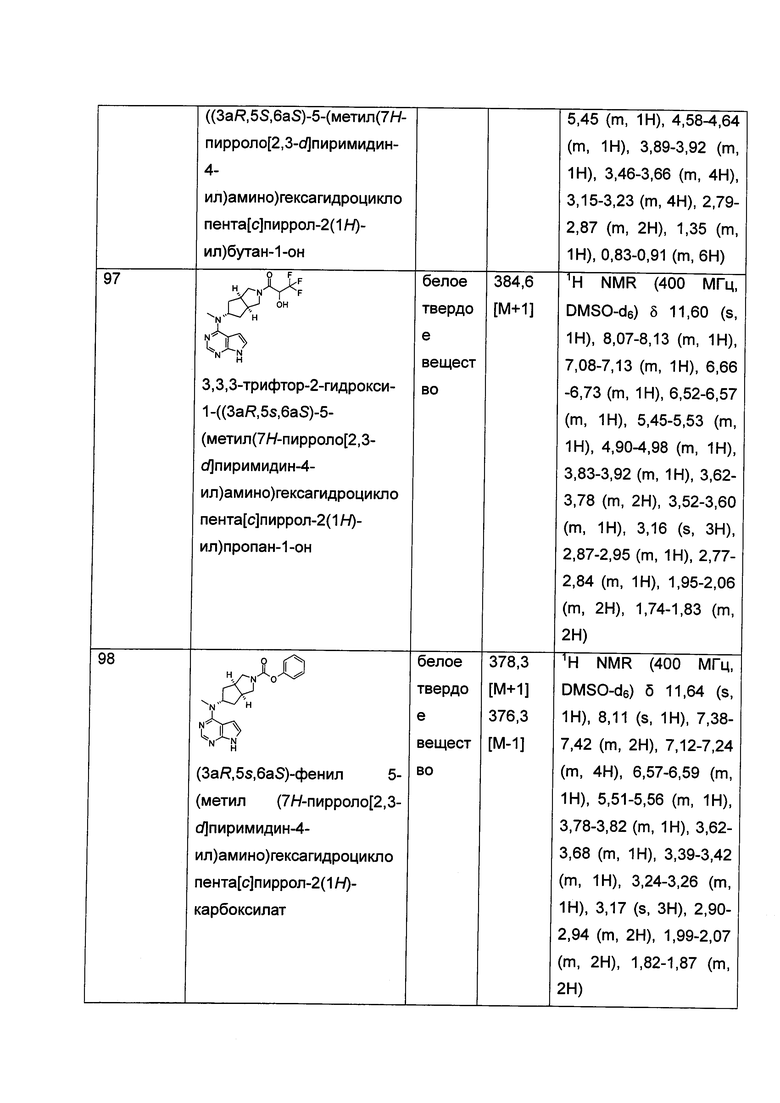
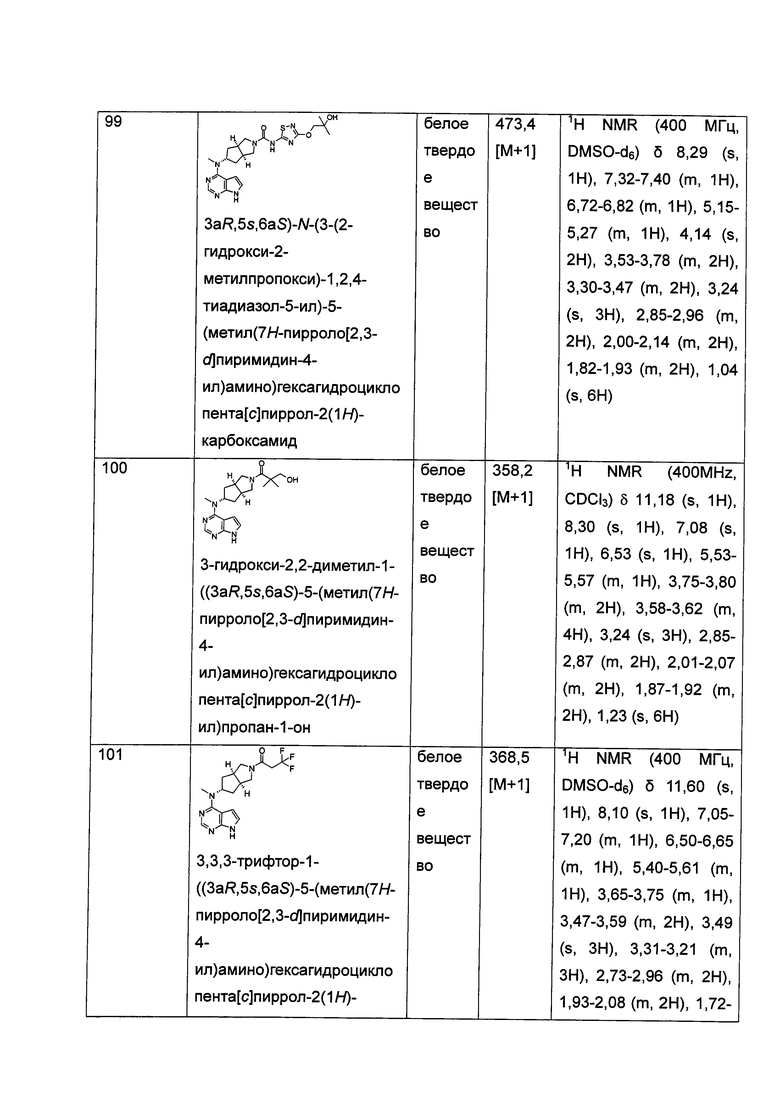
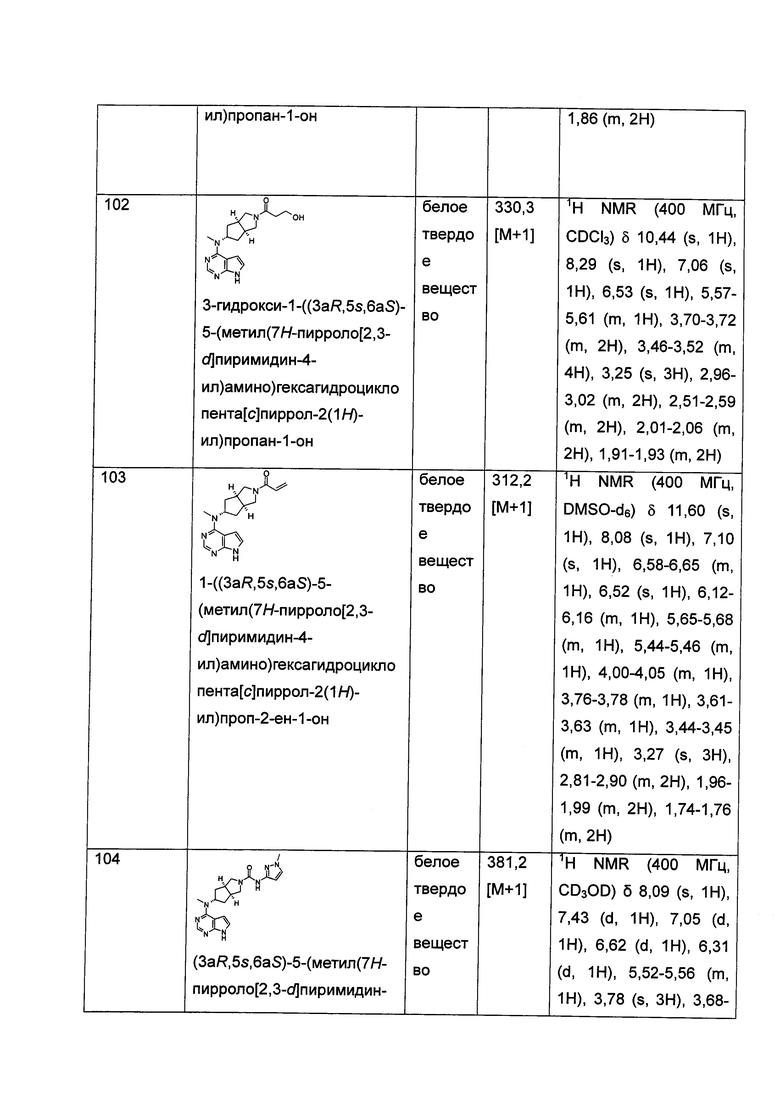
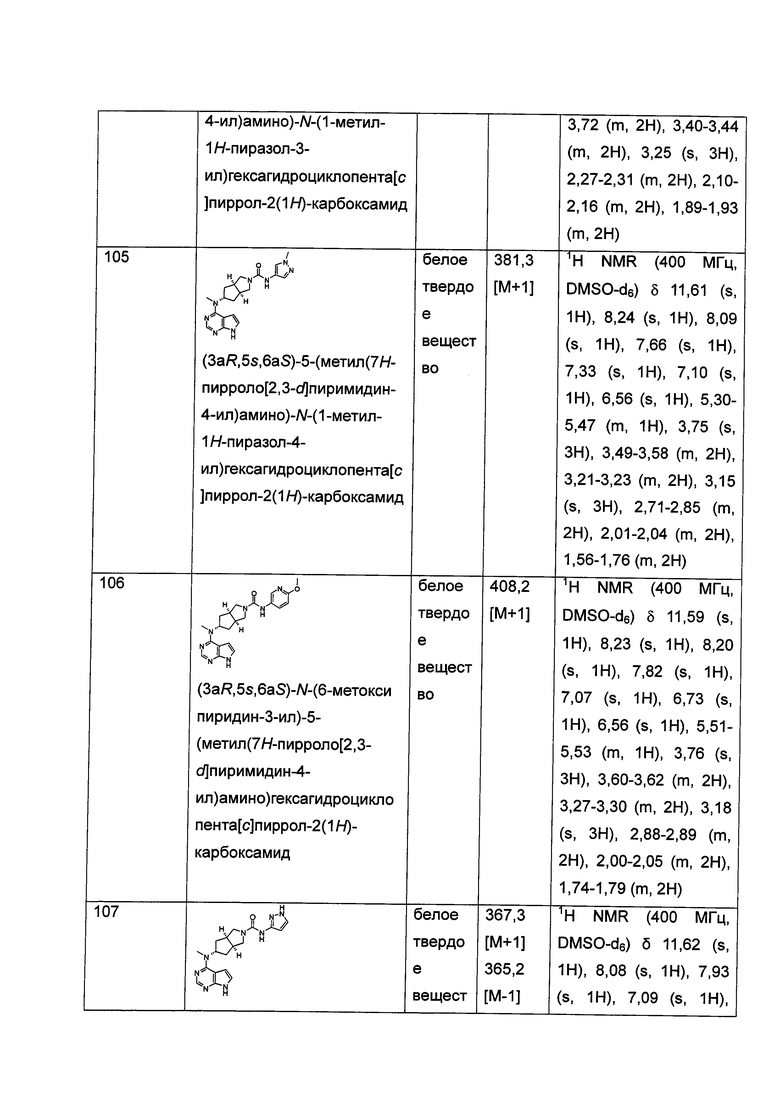

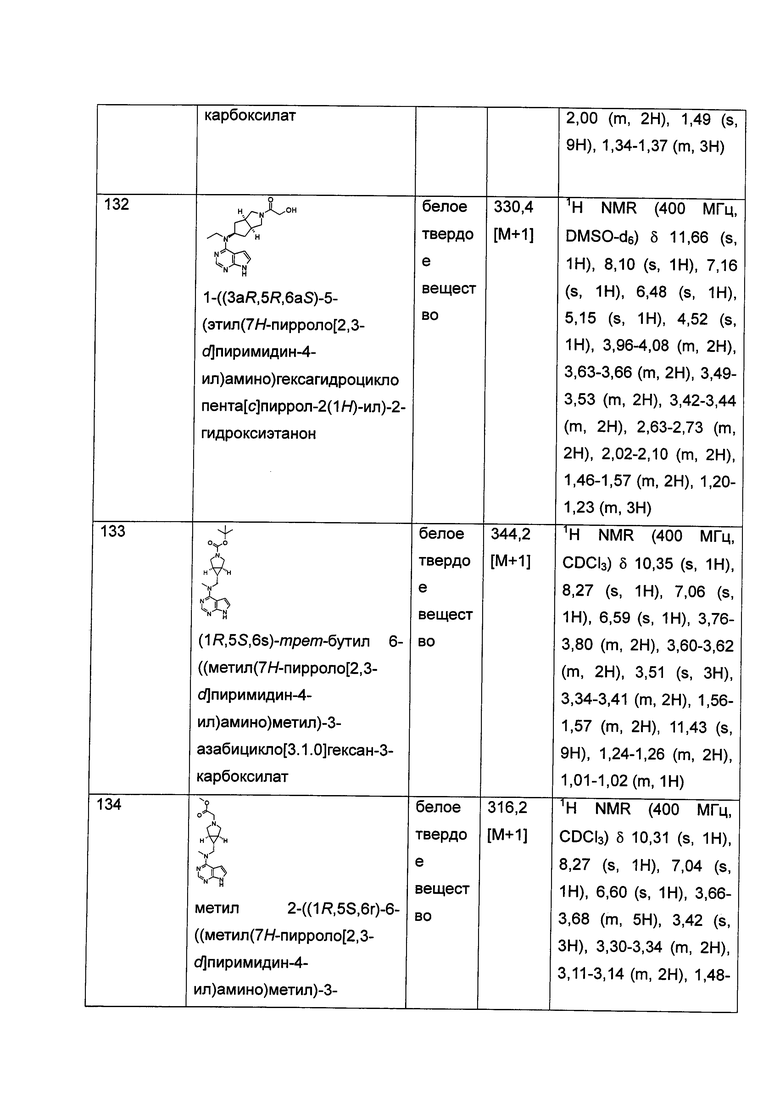
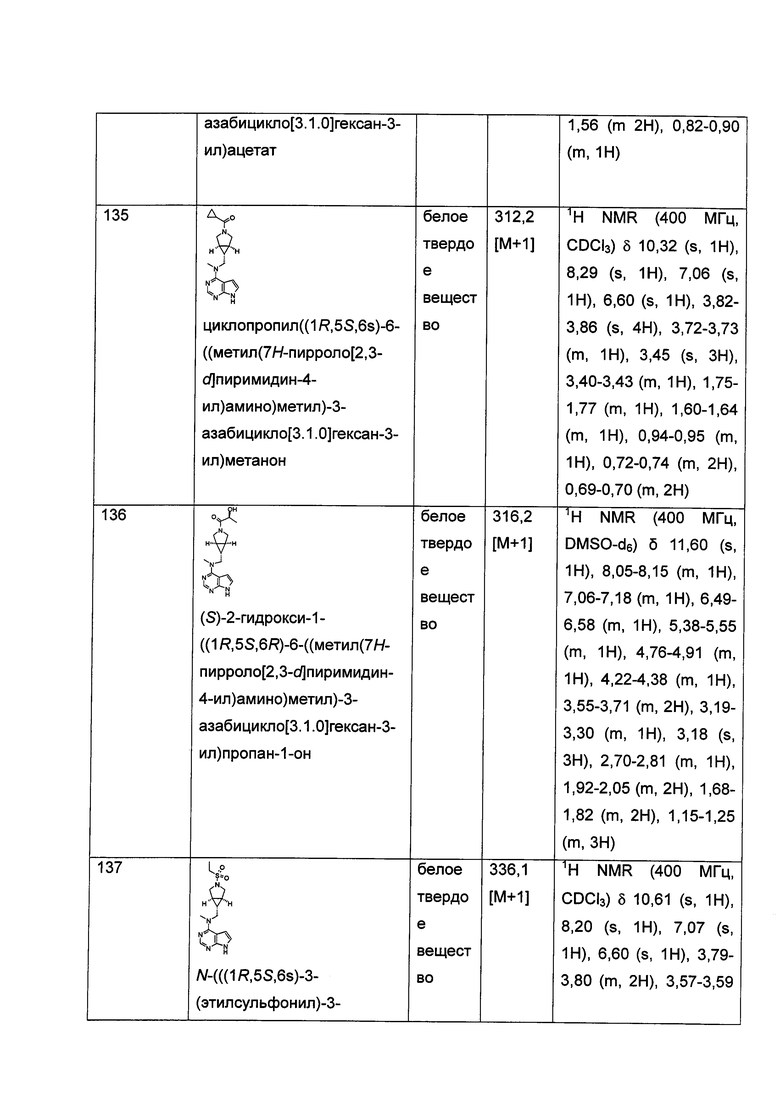
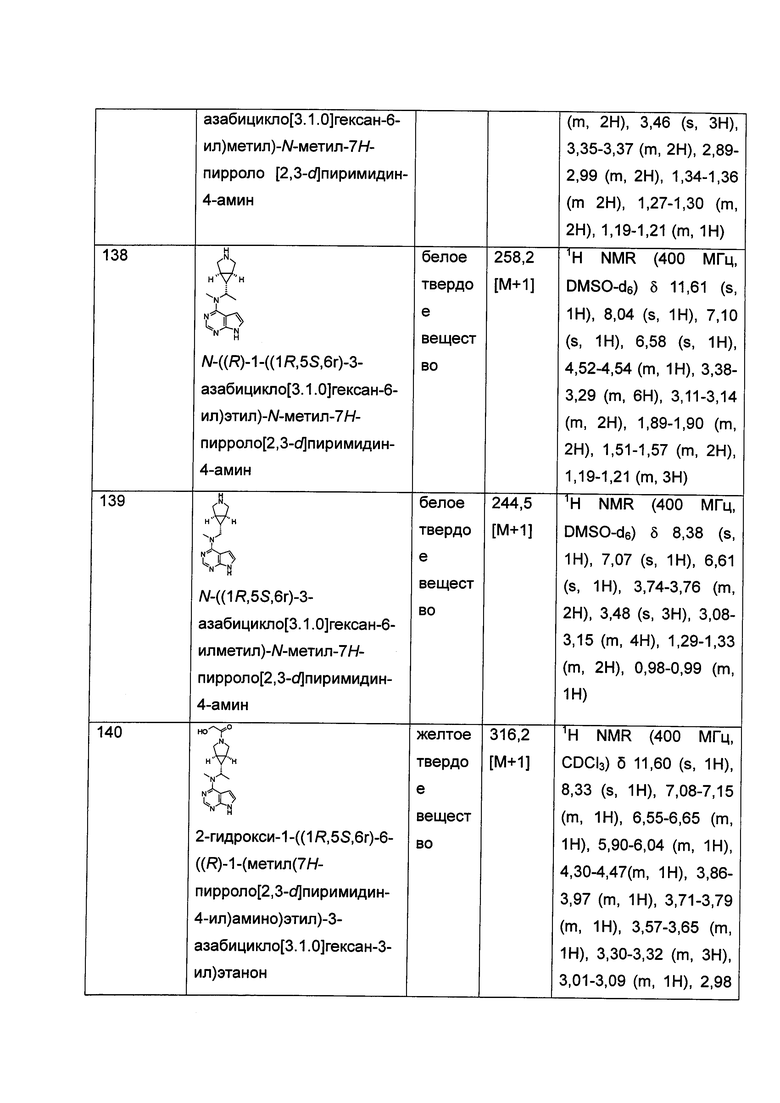
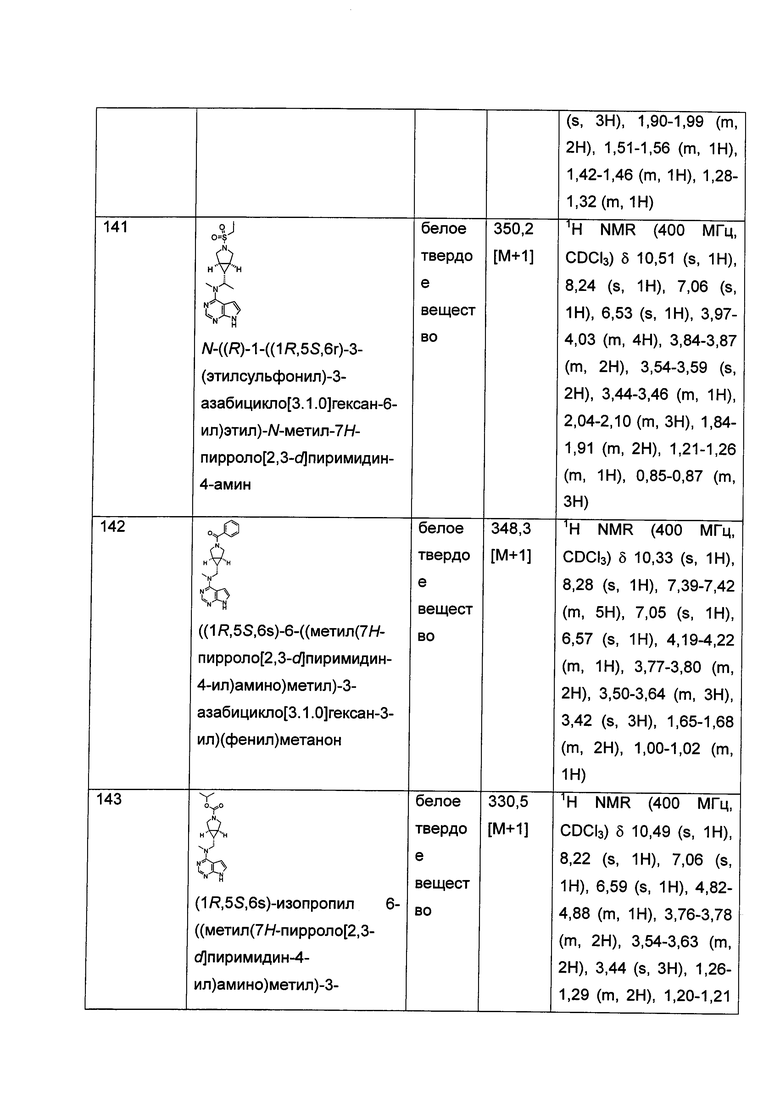
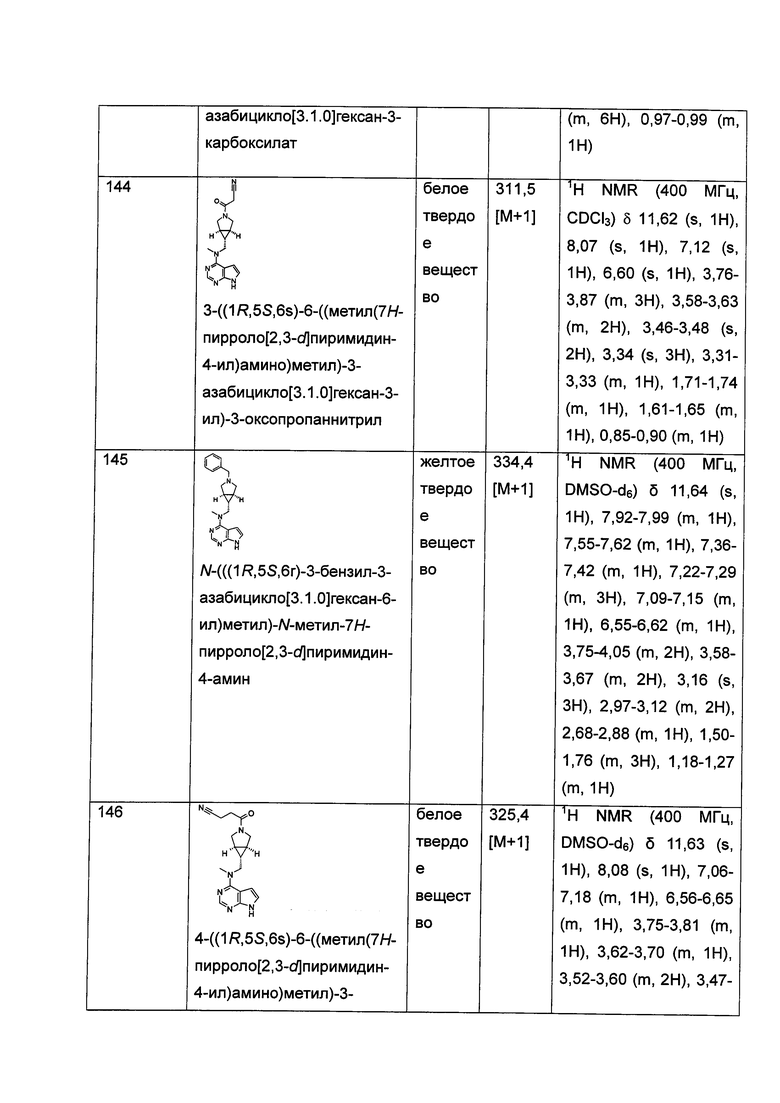
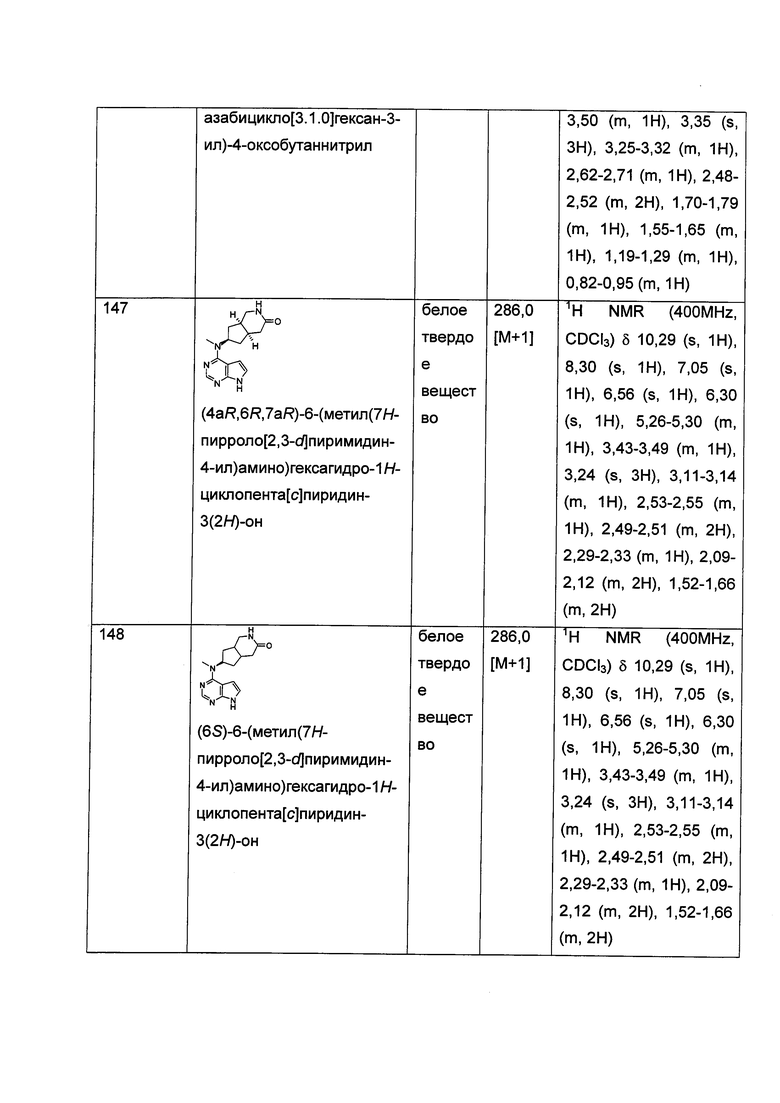
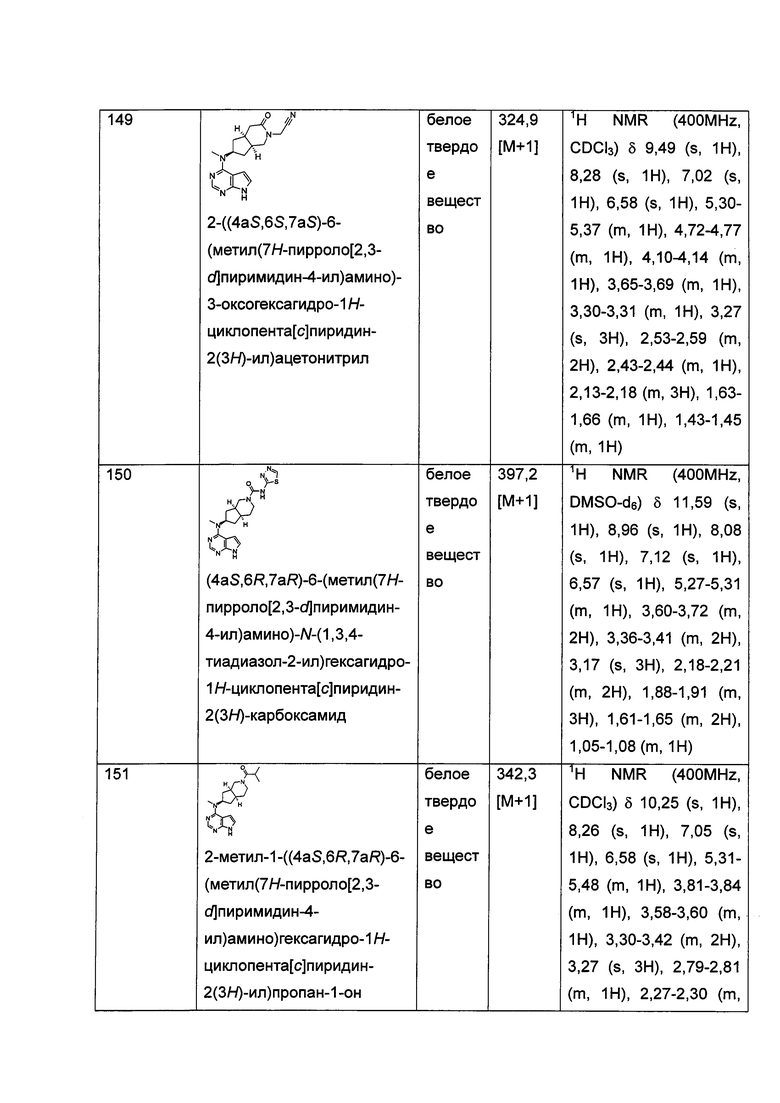
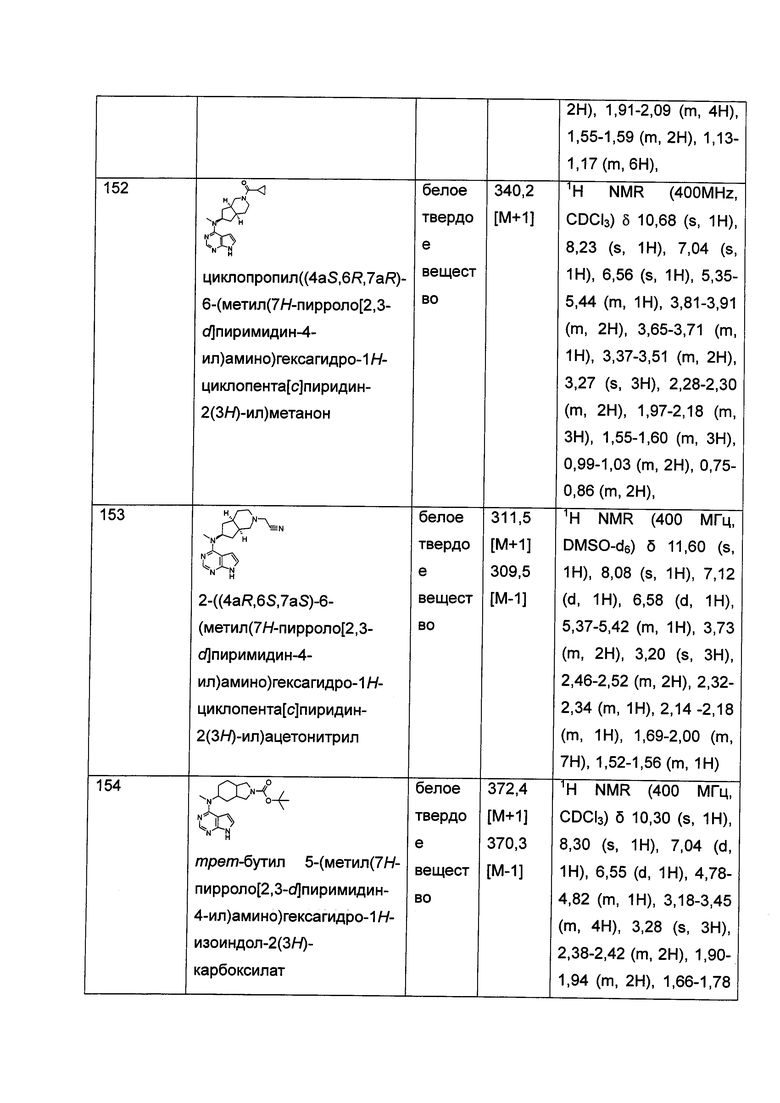
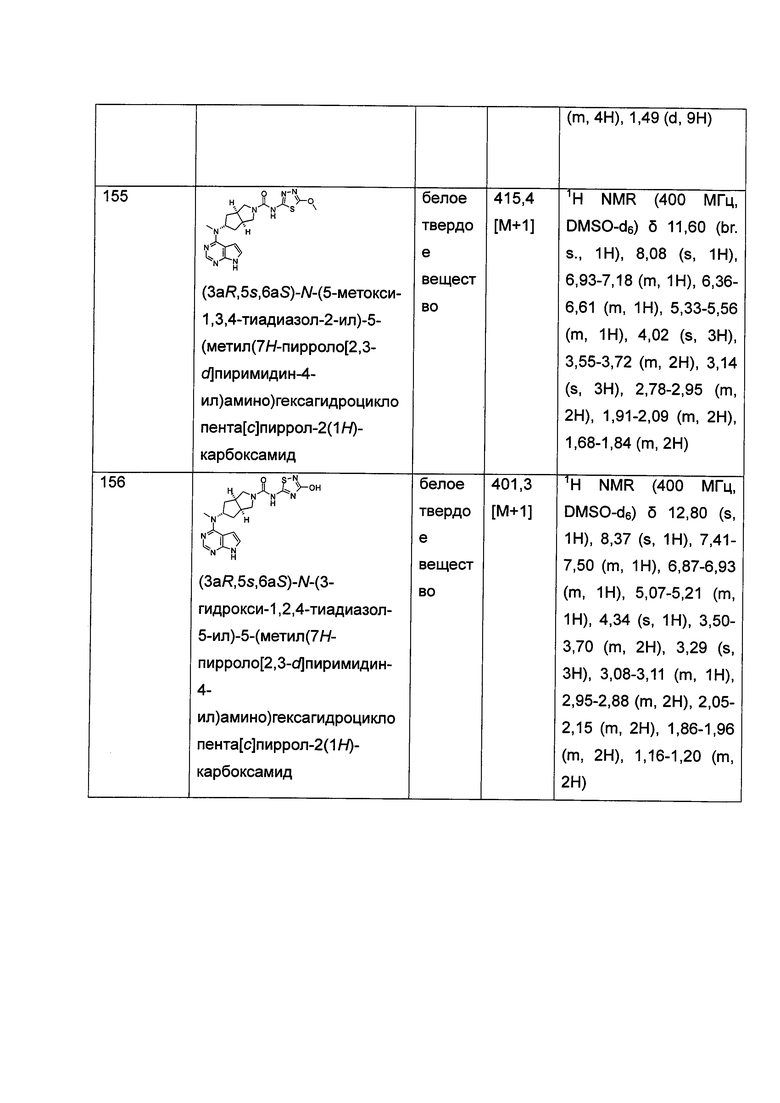
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I) или к его энантиомеру, диастереомеру и их смеси и к его фармацевтически приемлемой соли, где А: СН или N; L: связь или С1-2алкил; R1 выбран из водорода, С1-4алкила, гетероарила, -(СН2)nC(O)OR15, -C(O)R15, -C(O)NR16R17, и -S(O)mR15, где каждый из алкила или гетероарила возможно замещен 1-3 группами, выбранными из галогена, гидрокси, циано, С1-2алкила, С1-2алкокси, С3циклоалкила, С6арила, пиридина и -(CH2)nC(O)OR15; R2 или R4 независимо выбран из водорода и C1-2алкила; R или R3 независимо выбран из водорода и галогена; R5 или R6 независимо выбран из водорода и C1-2алкила; R7, R8, R9 или R10 независимо выбран из водорода, С1-2алкила и гидроксиC1-2алкила; R11, R12, R13 или R14 независимо выбран из водорода или R11 и R12 или R13 и R14, взятые вместе, образуют оксогруппу; R15 выбран из С1-4алкила, С3циклоалкила, С2алкенила, С6арила и пиридина, где каждый из алкила, циклоалкила или гетероарила возможно замещен 1-4 группами, выбранными из галогена, гидрокси, циано, С1-4алкокси, С6арила, тетразола, -NR19R20, -S(O)mR18, -NHC(O)OR18 и -NHS(O)mR18; R16 или R17 независимо выбран из водорода, С1-3алкила и гетероарила, где гетероарил возможно замещен одной группой, выбранной из С1-2алкила, гидрокси, С1-2алкокси, С3циклоалкила, гидроксиC1алкила и -OR18; R18 выбран из С1-4алкила и гидроксиС1-4алкила; R19 или R20 независимо выбран из группы, состоящей из водорода; m равен 0, 1 или 2; n равен 0 или 1; р равен 0, 1 или 2; q равен 0 или 1; s равен 1 или 2; и t равен 1 или 2. Также изобретение относится к конкретным промежуточным соединениям, способу получения соединения формулы (I), фармацевтической композиции на его основе, применению соединения формулы (I), способу ингибирования JAK-киназы и способу лечения некоторых заболеваний иммунной системы. Технический результат: получены новые гетероциклические соединения, обладающие активностью ингибитора JAK-киназы. 10 н. и 12 з.п. ф-лы, 7 табл., 158 пр.
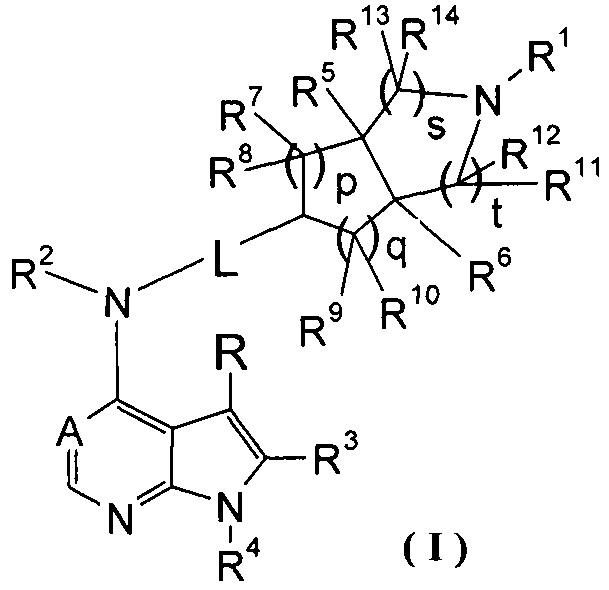
1. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль:
 ,
,
где:
А представляет собой СН или N;
L представляет собой связь или С1-2алкил;
R1 выбран из группы, состоящей из водорода, С1-4алкила, гетероарила (выбранного из пиримидина, пиридина, оксадиазола и тиадиазола), -(СН2)nC(O)OR15, -C(O)R15, -C(O)NR16R17, и -S(O)mR15, где каждый из алкила или гетероарила возможно замещен одной, двумя или тремя группами, выбранными из группы, состоящей из галогена, гидрокси, циано, С1-2алкила, С1-2алкокси, С3циклоалкила, С6арила, гетероарила (выбранного из пиридина) и -(CH2)nC(O)OR15;
каждый из R2 или R4 независимо выбран из группы, состоящей из водорода и C1-2алкила;
каждый из R или R3 независимо выбран из группы, состоящей из водорода и галогена;
каждый из R5 или R6 независимо выбран из группы, состоящей из водорода и C1-2алкила;
каждый из R7, R8, R9 или R10 независимо выбран из группы, состоящей из водорода, С1-2алкила и гидроксиC1-2алкила;
каждый из R11, R12, R13 или R14 независимо выбран из группы, состоящей из водорода, или R11 и R12 или R13 и R14, взятые вместе, образуют оксогруппу;
R15 выбран из группы, состоящей из С1-4алкила, С3циклоалкила, С2алкенила, С6арила и гетероарила (выбранного из пиридина), где каждый из алкила, циклоалкила или гетероарила возможно замещен от одной до четырех групп, выбранных из группы, состоящей из галогена, гидрокси, циано, С1-4алкокси, С6арила, гетероарила (выбранного из тетразола), -NR19R20, -S(O)mR18, -NHC(O)OR18 и -NHS(O)mR18;
каждый из R16 или R17 независимо выбран из группы, состоящей из водорода, С1-3алкила и гетероарила (выбранного из тиадиазола, пиразола, пиридина, тетразола, тиазола, оксазола, оксадиазола, имидазола, изоксазола, изотиазола и триазола), где гетероарил возможно замещен одной группой, выбранной из группы, состоящей из С1-2алкила, гидрокси, С1-2алкокси, С3циклоалкила, гидроксиC1алкила и -OR18;
R18 выбран из группы, состоящей из С1-4алкила и гидроксиС1-4алкила;
каждый из R19 или R20 независимо выбран из группы, состоящей из водорода;
m равен 0, 1 или 2;
n равен 0 или 1;
р равен 0, 1 или 2;
q равен 0 или 1;
s равен 1 или 2; и
t равен 1 или 2.
2. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по п.1, выбранные из соединения формулы (II) или его фармацевтически приемлемой соли:
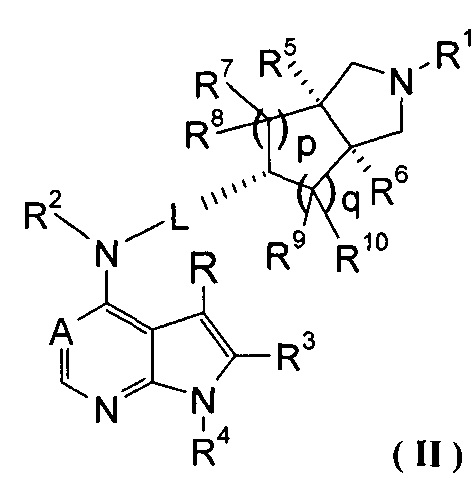 ,
,
где A, L, R, R1 - R10, р и q представляют собой такие, как определено в п.1.
3. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по п.1 или 2, выбранные из соединения формулы (III) или его фармацевтически приемлемой соли:
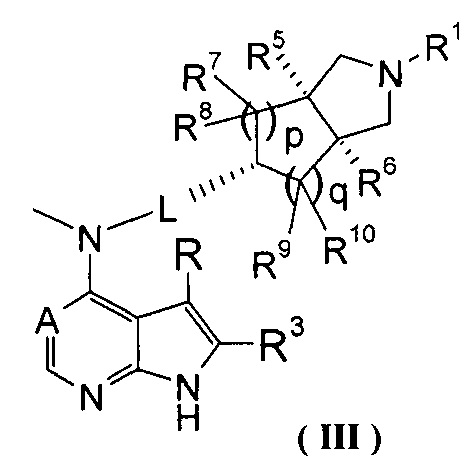 ,
,
где A, L, R, R1, R3, R5-R10, р и q представляют собой такие, как определено в п.1.
4. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по п.1 или 2, выбранные из соединения формулы (IV-a), или формулы (IV-b), или их фармацевтически приемлемой соли:
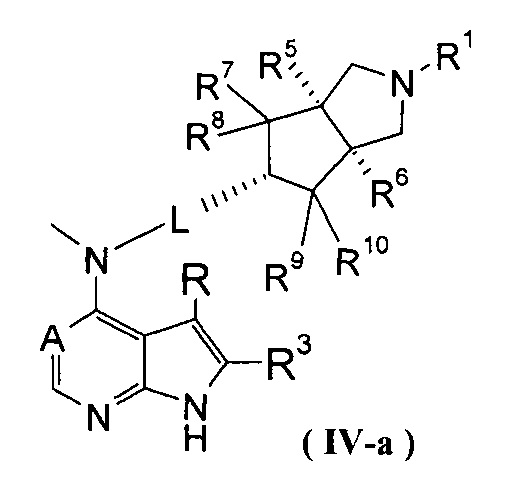
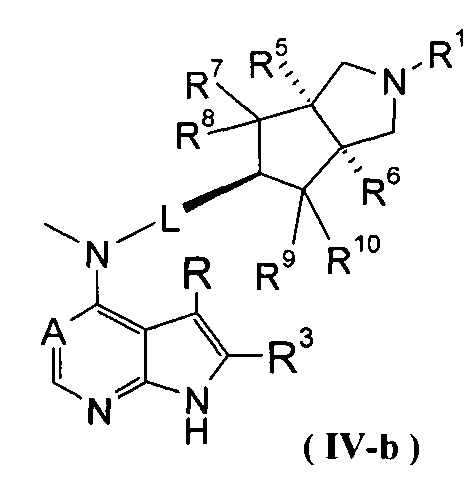 ,
,
где A, L, R, R1, R3 и R5-R10 представляют собой такие, как определено в п.1.
5. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по п.1 или 2, выбранные из соединения формулы (V-a), или формулы (V-b), или их фармацевтически приемлемой соли:
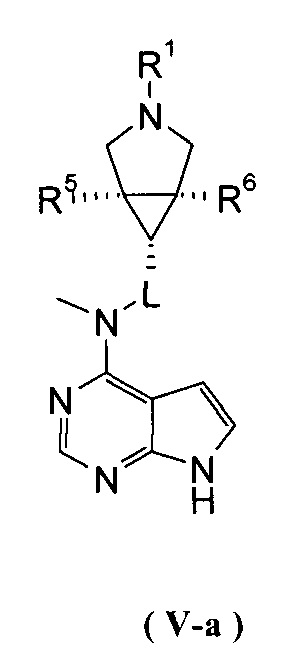
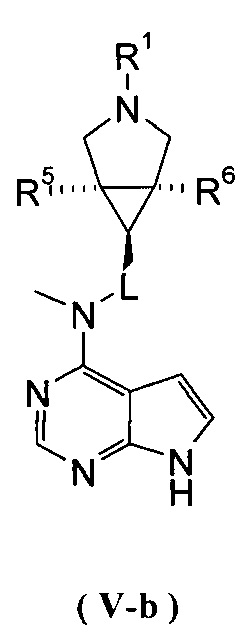 ,
,
где R1, R5, R6 и L представляют собой такие, как определено в п.1.
6. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по п.1 или 2, выбранные из соединения формулы (VI-a), или формулы (VI-b), или их фармацевтически приемлемой соли:
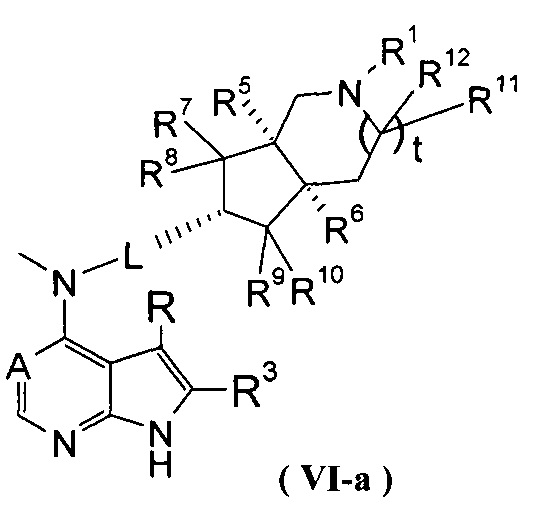
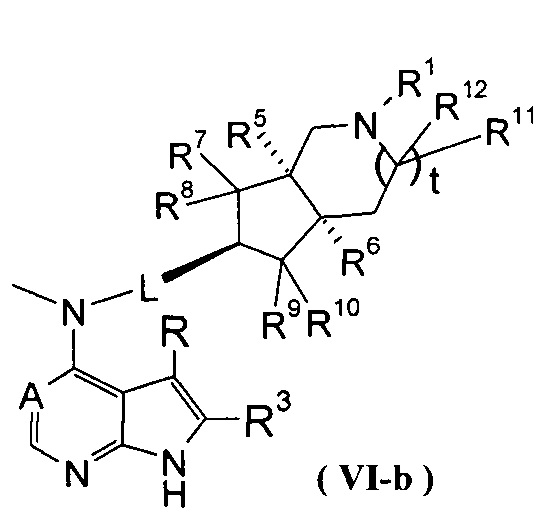 ,
,
где:
t равно 1;
A, L, R, R1, R3 и R5-R12 представляют собой такие, как определено в п.1.
7. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по п.1 или 2, выбранные из соединения формулы (VII-a), или формулы (VII-b), или их фармацевтически приемлемой соли:
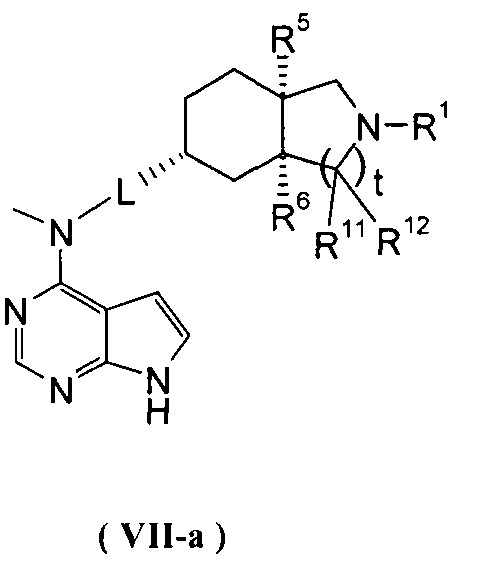
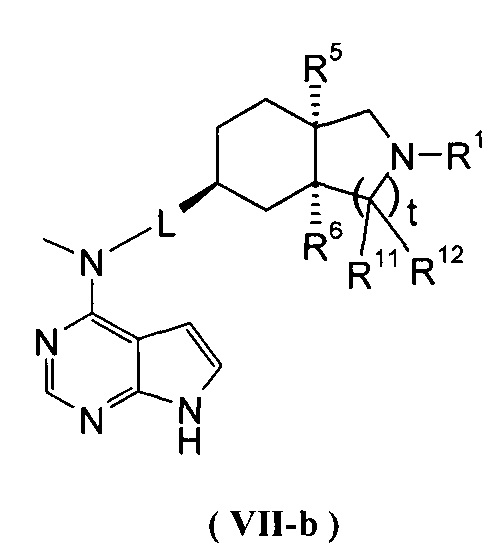 ,
,
где R1, R5, R6, R11, R12, L и t представляют собой такие, как определено в п.1.
8. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по любому из пп.1 или 2, где L представляет собой связь.
9. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по любому из пп.1 или 2, где R1 выбран из группы, состоящей из С1-4алкила, гетероарила (выбранного из пиримидина, пиридина, оксадиазола и тиадиазола), -(CH2)nC(O)OR15, -C(O)R15, -C(O)NR16R17 и -S(O)2R15, где каждый из алкила или гетероарила возможно замещен одной, двумя или тремя группами, выбранными из группы, состоящей из галогена, гидроксила, циано и -(CH2)nC(O)OR15;
R15 выбран из группы, состоящей из С1-4алкила, С3циклоалкила, С2алкенила, С6арила и гетероарила (выбранного из пиридина), где каждый из алкила, циклоалкила или гетероарила возможно замещен от одной до четырех групп, выбранных из группы, состоящей из галогена, гидрокси, циано, С1-4алкокси, С6арила, гетероарила (выбранного из тетразола), -S(O)2R18, -NHC(O)(O)R18, -NHS(O)2R18 и -NR19R20;
каждый из R16 или R17 независимо выбран из группы, состоящей из водорода, С1-3алкила и гетероарила (выбранного из тиадиазола, пиразола, пиридина, тетразола, тиазола, оксазола, оксадиазола, имидазола, изоксазола, изотиазола и триазола); где указанный гетероарил возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из С1-2алкокси, С3циклоалкила, гидроксиC1алкила и -OR18;
R18 выбран из группы, состоящей из С1-4алкила и гидроксиС1-4алкила;
каждый из R19 или R20 независимо выбран из группы, состоящей из водорода;
и n равен 0 или 1.
10. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по п.1, где каждый из R11, R12, R13 или R14 независимо представляет собой водород.
11. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по п.1, где R11 и R12 или R13 и R14, взятые вместе, образуют оксогруппу.
12. Соединение формулы (I) или энантиомер, диастереомер и их смесь и его фармацевтически приемлемая соль по любому из пп.1 или 2, где соединение выбрано из группы, состоящей из:
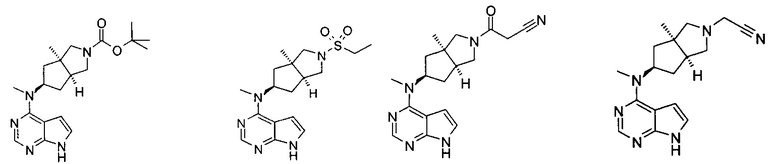
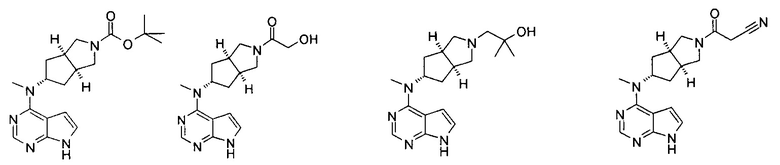
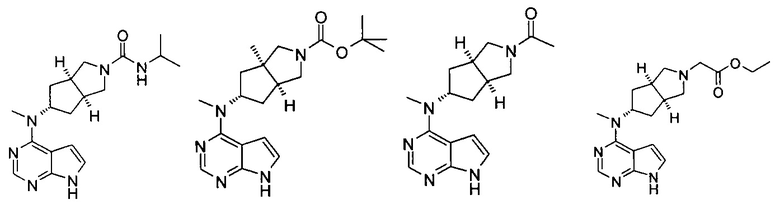
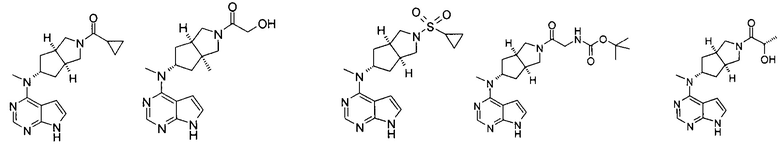

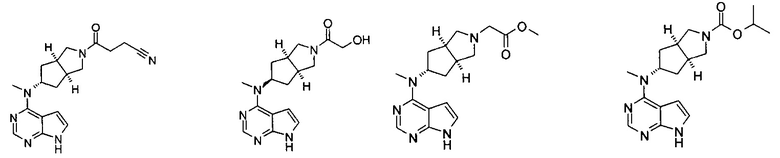
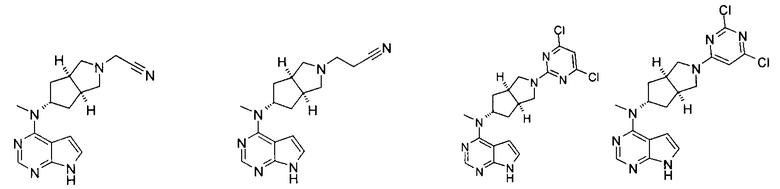
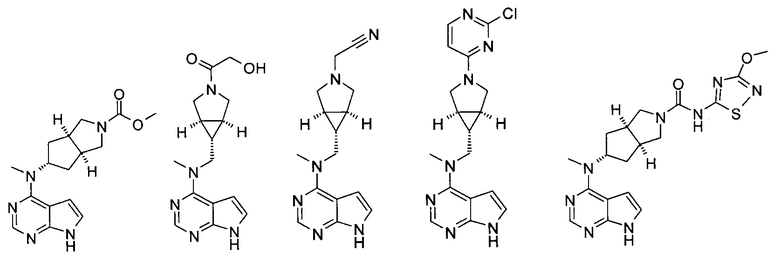
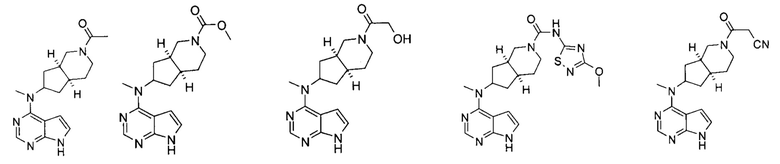
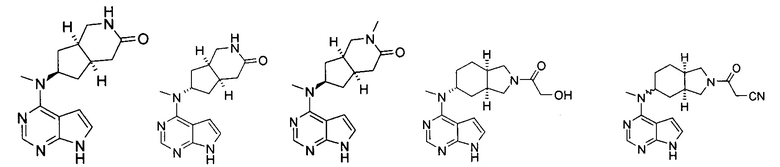
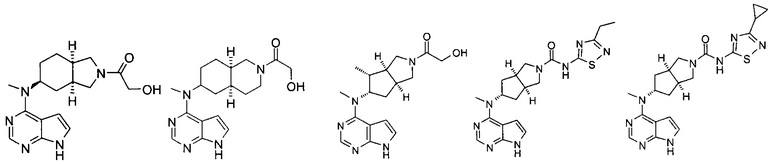
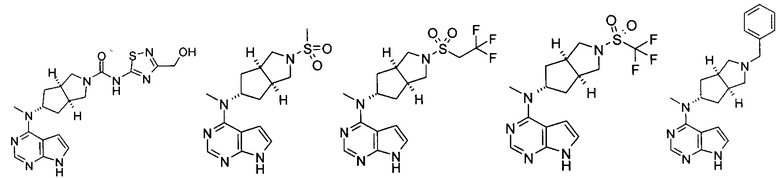
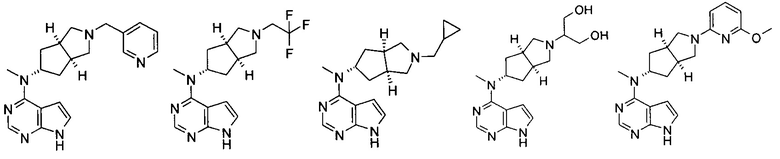
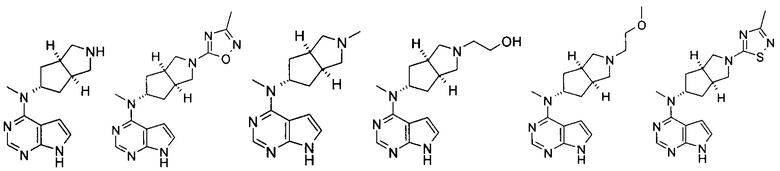
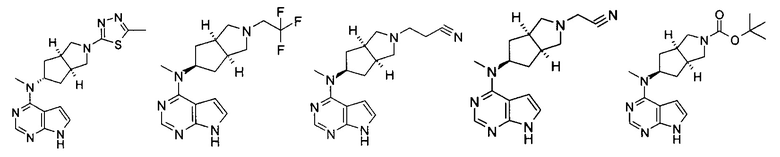
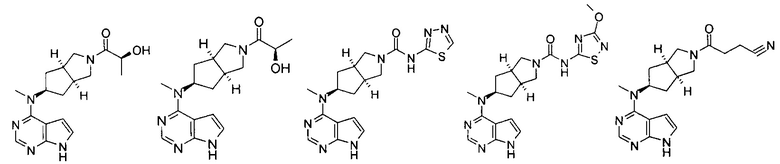
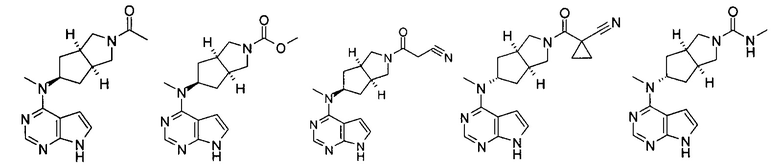
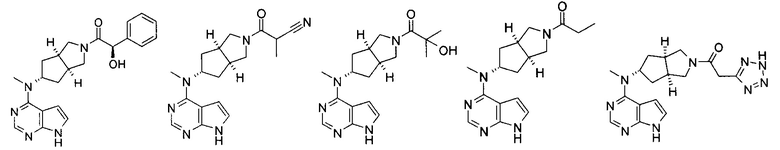
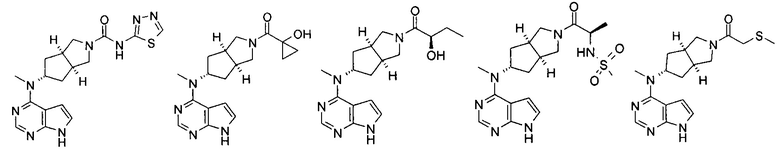
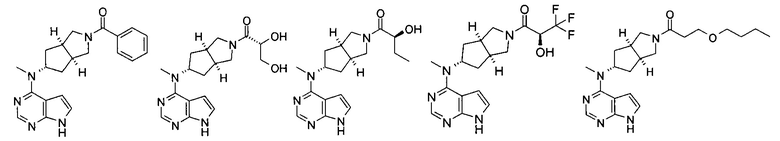
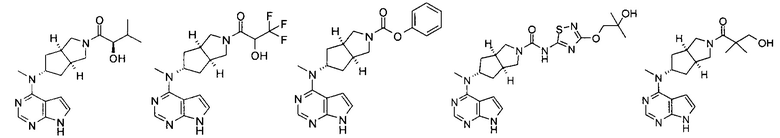
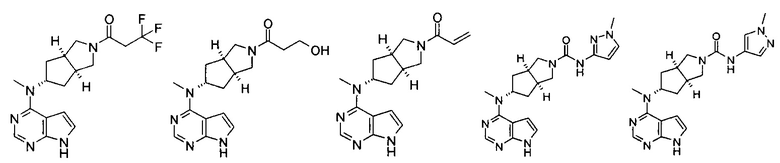

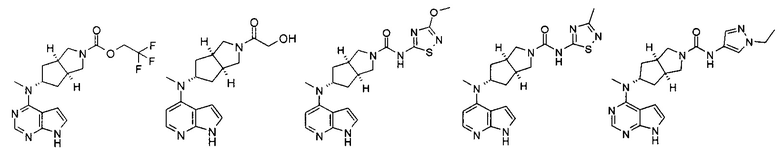
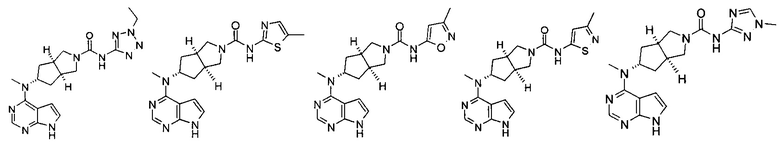
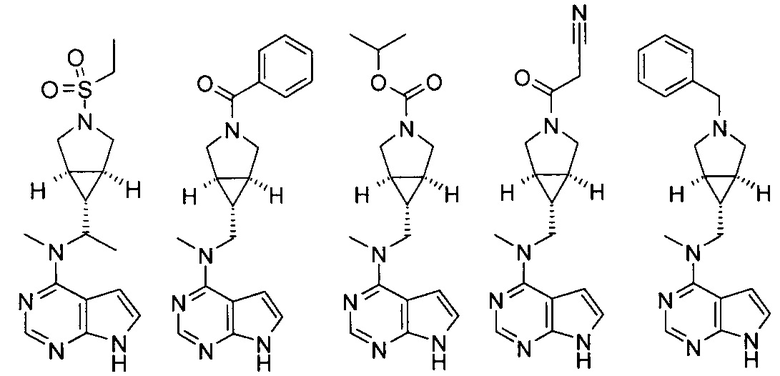
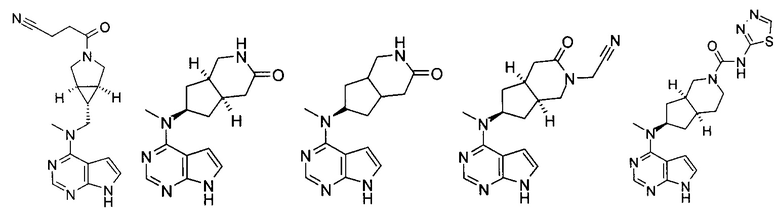
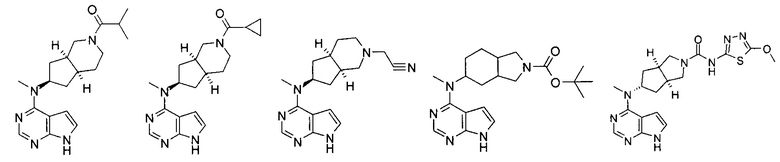 и
и 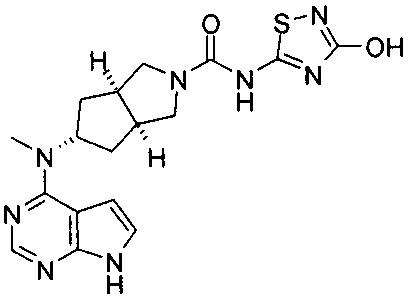 .
.
13. Соединение, выбранное из группы, состоящей из:
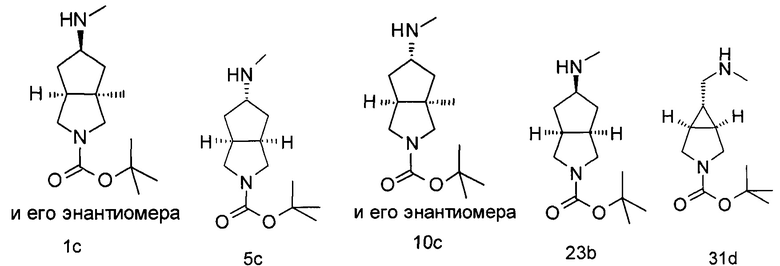
 и
и  .
.
14. Способ получения соединения формулы (I) или энантиомера, диастереомера и их смеси и его фармацевтически приемлемой соли, включающий стадии:
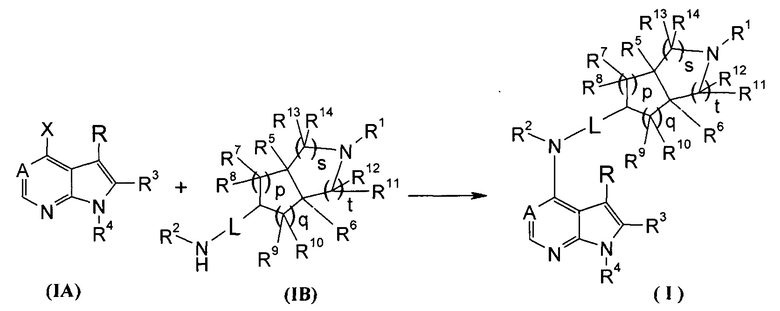
в щелочных условиях, причем соединение формулы (IA) реагирует с соединением формулы (IB) с получением соединения формулы (I);
где X представляет собой галоген, A, L, R, R1-R14, р, q, s и t представляют собой такие, как определено в п.1.
15. Способ получения соединения формулы (I) или энантиомера, диастереомера и их смеси и его фармацевтически приемлемой соли, включающий стадии:
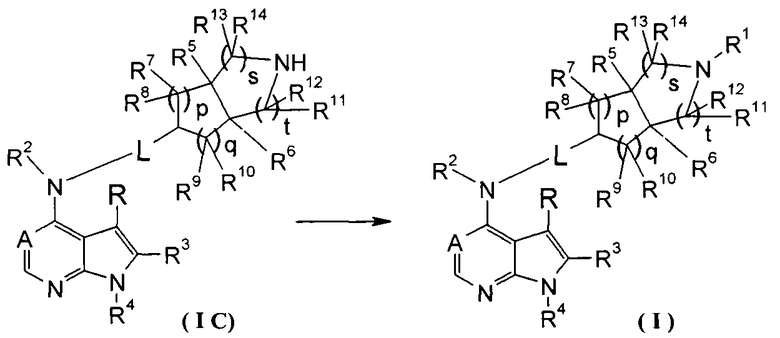
в щелочных условиях, причем соединение формулы (IC) или его фармацевтически приемлемая соль реагирует с карбоновой кислотой, хлорангидридом карбоновой кислоты, сульфонилхлоридом, сложным эфиром карбоновой кислоты, производным этиленоксида или галогенидом с получением соединения формулы (I);
где A, L, R, R1-R14, р, q, s и t представляют собой такие, как определено в п.1.
16. Фармацевтическая композиция, обладающая активностью ингибитора JAK-киназы, содержащая терапевтически эффективное количество соединения формулы (I) или энантиомера, диастереомера и их смеси и его фармацевтически приемлемой соли по любому из пп.1-12, и фармацевтически приемлемый носитель.
17. Применение соединения формулы (I) или энантиомера, диастереомера и их смеси и его фармацевтически приемлемой соли по любому из пп.1-12 или фармацевтической композиции по п.16 при получении лекарственного средства для ингибирования JAK-киназы, предпочтительно ингибирования JAK1, JAK2 или JAK3.
18. Применение по п.17, где лекарственное средство пригодно для лечения или предупреждения следующих расстройств или заболеваний: отторжение аллотрансплантата, реакция «хозяин против трансплантата», ревматоидный артрит, псориаз, атопический дерматит, рак поджелудочной железы, рак молочной железы, лимфома, лейкоз, Т-клеточная лимфома кожи или В-клеточная лимфома кожи.
19. Применение соединения формулы (I) или энантиомера, диастереомера и их смеси и его фармацевтически приемлемой соли по любому из пп.1-12 или фармацевтической композиции по п.16 в качестве лекарственного средства для ингибирования JAK-киназы, предпочтительно ингибирования JAK1, JAK2 или JAK3.
20. Применение соединения формулы (I) или энантиомера, диастереомера и их смеси и его фармацевтически приемлемой соли по любому из пп.1-12 или фармацевтической композиции по п.16 в качестве лекарственного средства для лечения или предупреждения следующих расстройств или заболеваний: отторжение аллотрансплантата, реакция «хозяин против трансплантата», ревматоидный артрит, псориаз, атопический дерматит, рак поджелудочной железы, рак молочной железы, лимфома, лейкоз, Т-клеточная лимфома кожи или В-клеточная лимфома кожи.
21. Способ ингибирования JAK-киназы, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или энантиомера, диастереомера и их смеси и его фармацевтически приемлемой соли по любому из пп.1-12 или фармацевтической композиции по п.16.
22. Способ лечения или предупреждения расстройства или заболевания иммунной системы, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или энантиомера, диастереомера и их смеси и его фармацевтически приемлемой соли по любому из пп.1-12 или фармацевтической композиции по п.16, где расстройство или заболевание иммунной системы выбрано из группы, состоящей из отторжения аллотрансплантата, реакции «хозяин против трансплантата», ревматоидного артрита, псориаза, атопического дерматита, рака поджелудочной железы, рака молочной железы, лимфомы, лейкоза, Т-клеточной лимфомы кожи или В-клеточной лимфомы кожи.
| M | |||
| Ogata и др | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Oleksandr O | |||
| Grygorenko и др | |||
| "Bicyclic Conformationally Restricted Diamines", Chem | |||
| Rev., 2011, 111 (9), стр.5506-5568 | |||
| WO 2009098283 A1, 13.08.2009 | |||
| US 2005227960 A1, 13.10.2005 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO 2006012396 A1, 02.02.2016 | |||
| WO 2006069080 A2, 29.06.2006 | |||
| WO 2002000661 A1,03.01.2002 | |||
| WO 1999065908 A1, 23.12.1999 | |||
| ПИРРОЛОПИРИМИДИНЫ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2006 |
|
RU2434871C2 |

