Настоящее изобретение относится к медицине, в частности к медицине катастроф, трансфузиологии, представлено в качестве биопрепарата и предназначено для получения биоплазмозаменителя гемодинамического действия для лечения состояний организма, связанных с массивной острой кровопотерей, а также различными видами шока.
Замена внутрисосудистой жидкости относится к важным мерам при профилактике и терапии гиповолемии, возникающей в большинстве случаях в результате непосредственной потери крови при острых кровотечениях, травмах, операциях, ожогах и других сходных патологических состояниях. Применимые для этих показаний инфузионные растворы должны восстанавливать нормальный объем крови и поддерживать перфузию жизненно важных органов, а также периферический кровоток. Одновременно эти растворы не должны чрезмерно загружать кровоток, и они не должны иметь побочных действий. В этом отношении все существующие в настоящее время растворы кровезаменителей обнаруживают преимущества и недостатки. Так называемые кристаллоидные растворы в значительной степени лишены непосредственных побочных эффектов, однако обеспечивают только краткосрочную или неадекватную стабилизацию внутрисосудистого объема и гемодинамики, прежде всего за счет наполнения объема, а не восстановления и поддержания онкотического давления. При выраженной или длительной гиповолемии необходима их инфузия в избыточных количествах, поскольку они не остаются во внутрисосудистом пространстве и при дефиците гидрофильных полимеров быстро выходят во внесосудистое пространство. Прогрессивный отток из внутрисосудистого пространства не только снижает наполняющее действие кристаллоидных растворов, но имеет риск возникновения периферических и легочных отеков. В противоположность этому коллоидные растворы растительного, биологического или синтетического происхождения удерживают воду в кровотоке и предотвращают ее отток в межтканевое пространство. Одним из существенных недостатков коллоидных растворов синтетического или растительного происхождения является высокий риск, до 10%, нежелательных реакций, вплоть до анафилактического шока с летальным исходом. К этому можно добавить и то, что коллоидные препараты биологического происхождения, например альбумин, несут риск заражения вирусными заболеваниями. При этом доступность альбумина является ограниченной и его применение в качестве плазмозаменителя является чрезвычайно дорогим. В случае синтетических коллоидов, тяжелые анафилактические реакции и массивное нарушение свертывания крови привели к тому, что препараты декстрана практически полностью исключили из терапии. Растворы гидроксиэтилкрахмала (ГЭК) обладают меньшим потенциалом в запуске анафилактических реакций и влиянии на свертывание крови. В сравнении с растворами желатина, которые также применяются в качестве плазмозаменителей и позволяют в значительной степени уменьшить влияние на систему свертывания крови, высокомолекулярные и среднемолекулярные разновидности растворов ГЭК имеют преимущество за счет более длительного периода выведения из организма и большую эффективность по стабилизации онкотического давления.
Полисахариды на основе декстрана широко используют в качестве высокомолекулярных растворов плазмозаменителей с преимущественно гемодинамическим действием. Известны основные распространенные препараты декстранов, такие как полиглюкин, реополиглюкин, макродекс и др. Водные растворы нативного декстрана представляют собой высоковязкие жидкости, в связи с чем он не применяется в качестве плазмозаменителя без предварительной модификации. Для этого нативный декстран подвергают какому-либо виду химической или физической обработки: кислотному или щелочному гидролизу, ультразвуковому, термическому или радиционному воздействию. Низкомолекулярный декстран, используемый при производстве плазмозаменителей, после модификации нуждается в сложной и дорогостоящей очистке.
Известен способ получения низкомолекулярного декстрана, не требующий последующей очистки, с возможностью непосредственного использования как основы плазмозаменителя (Способ получения низкомолекулярного декстрана. Патент РФ 2039754 от 31.07.1992. Авторы: П.Т. Петров, В.Н. Гапанович, В.М. Царенков, В.У. Заборонок, М.П. Лапковский, В.И. Тюрин, Т.Н. Забелло (17). Согласно описанию патента, модификацию декстрана производят гамма облучением. Выход препарата составляет 100%.
Существенным недостатком этого способа является то, что в результате облучения происходит радиолиз воды с выходом частиц, наиболее реакционно способными из которых являются ОН-радикал, гидратированный электрон 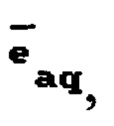 Н-радикал. Гидратированный электрон практически не взаимодействует с декстраном, но с большой вероятностью вступает в реакцию с H2CO2, приводя к резкому увеличению выхода ОН-радикалов. Таким образом, конечный раствор декстранов содержит повышенную концентрацию свободных радикалов - факторов, вызывающих оксидативный стресс и наряду с гемодинамическим действием его введение при массивных кровопотерях может привести резкому понижению антиоксидантной защиты. Побочный эффект полученного таким способом декстрана является прямым противопоказанием к его применению в качестве плазмозаменителя.
Н-радикал. Гидратированный электрон практически не взаимодействует с декстраном, но с большой вероятностью вступает в реакцию с H2CO2, приводя к резкому увеличению выхода ОН-радикалов. Таким образом, конечный раствор декстранов содержит повышенную концентрацию свободных радикалов - факторов, вызывающих оксидативный стресс и наряду с гемодинамическим действием его введение при массивных кровопотерях может привести резкому понижению антиоксидантной защиты. Побочный эффект полученного таким способом декстрана является прямым противопоказанием к его применению в качестве плазмозаменителя.
Известен «Препарат гемодинамического действия с функцией нормализации кислотно-основного равновесия и электролитного баланса», патент РФ 2185173 (19), на основе декстрана с молекулярным весом 40000 Да в водном растворе натрия хлорида, отличающийся тем, что препарат дополнительно содержит калия хлорид, кальция хлорид, магния хлорид и натрия ацетат при определенном соотношении компонентов. Согласно описанию авторов, заявляемый препарат по всем физико-химическим препаратам оказался совместимым и стабильным, а значение рН неизменно удерживалось в пределах 6,0-7,0. Более длительное удержание солевого раствора вокруг молекул декстрана (до 3-х суток) в сосудистом русле обеспечивает продолжительное время предотвращения ацидоза и нормализует окислительно-восстановительный потенциал.
Недостатком препарата является, прежде всего, его относительная совместимость, поскольку проведенные испытания на кошках вряд ли можно считать доказательством совместимости. При том что испытания гемодинамических свойств препарата подтвердили его эффективность, необходимо отметить, что использование препаратов на основе декстранов сопряжено с доказанным высоким риском развития острых трансфузионных осложнений и применение линейки этих препаратов в настоящее время практически прекращено.
Известен гидроксиэтилкрахмал - (ГЭК) - высокополимерное соединение, на основе полимеризованных остатков декстрозы, который получают из природного амилопектина в процессе гидролиза и гидрооксиэтилирования. Технология получения ГЭК дает возможность получения молекул с заданным молекулярным весом и степенью полимерности, что определяет чрезвычайно важные перспективные направления использования в медицине, в частности для применения в качестве плазмозаменителя. Известны многочисленные препараты на основе ГЭК, которые применяются в качестве плазмозаменителей, в частности, такие как: «Инфукол», «Волювен», «Плазмастабил» и др. Все эти и подобные им препараты на основе ГЭК объединяют общие механизмы их биологического действия. Экспериментальными и клиническими наблюдениями установлено, что ГЭК длительное время циркулирует в кровяном русле, не менее 6 часов, стабильно увеличивает объем циркулирующей крови, причем пропорционально практически всему объему вводимого препарата, восстанавливает гемодинамику при массивной кровопотере (9). Молекулярный вес различных производных ГЭК колеблется в сравнительно больших пределах, от 120 кДальтон до 500 кДальтон и выше. Молекулы с молекулярной массой ниже 50000 быстро элиминируются путем почечной экскреции. После однократного в/в введения дозы около 500 мл (примерно 50 гр.) с мочой выводится 70-80% дозы в течение первых 24 часов.
Эти параметры определяют основные возможные биологические эффекты, связанные с такими процессами, как скорость выведения из организма, степень накопления в органах и тканях, волемический индекс и пр. Растворы ГЭК стабильны как при обычной, так и при пониженной температуре, что позволило применять их при шоках различной этиологии, массивных кровопотерях.
Общими для всей группы представителей ГЭК являются крайне серьезные недостатки, связанные с опасностью их применения, включая даже риск смертельного исхода (18, 20). Препараты этой группы вызывают трудно купирующиеся анафилактоидные реакции, гиперчувствительность, тахикардия и бронхоспазм и отек легких (21).
Более того, Комитет по оценке рисков, связанных с безопасностью лекарственных средств, Европейского Агентства по лекарственным средствам (PRAC ЕМА) завершил анализ данных клинических исследований применения инфузионных препаратов гидроксиэтилкрахмала у пациентов в критическом состоянии (22, 26, 27) и принял решение рекомендовать Европейской Комиссии приостановить на территории Европейского Союза обращение данных лекарственных средств (23, 24, 25).
Касаясь способов получения ГЭК можно выделить патент DE №2373222 (7), в котором описывается способ получения высокополимерного ГЭК с молекулярным весом 500 и 900 кДальтон, соответственно, из крахмала. Заявленный авторами технологический процесс последовательно реализуется путем проведения следующих операций: 30 кг жидкокипящего крахмала восковидной кукурузы суспендировали в 52,2 кг воды для инъекционных целей при комнатной температуре при сильном перемешивании. Для оптимального гидролиза крахмала эту суспензию сразу после этого клейстеризовали нагреванием до по меньшей мере 85°С. После многократных созданий атмосферы инертного газа в суспензии азотом посредством 10-минутного пропускания азота и последующей откачки воздуха вакуумированием крахмал активировали добавлением 5,1 кг NaOH. После этого в реактор вводили 4,159 кг охлажденного этиленоксида в жидкой форме и температуру медленно повышали до 40°С и оставляли эту реакционную смесь на 2 часа при продолжающемся перемешивании при этой температуре. Непревращенный этиленоксид обрабатывали инертным газом, как описано выше, и удаляли из реакционной смеси. Затем из этого исходного крахмала получали при помощи ступенчатого кислотного гидролиза ГЭК-препарата. Для уменьшения молекулярной массы этот раствор доводили 20%-ной НСl до рН 2,0, нагревали до 75±1°С и оставляли при этой температуре, пока средняя молекулярная масса ГЭК-коллоида, не снижалась до 865 кД. Треть этой смеси для гидролиза вынимали из реактора и тотчас же охлаждали до температур ниже 50°С. После обесцвечивания раствора обработкой активированным углем раствор фильтровали с использованием предварительного фильтра и стерильного фильтра и после разбавления до 12% очищали ультрафильтрацией. Для этого использовали полиэфирсульфоновые мембраны фирмы Millipore с пределом молекулярной массы 10 кД. Оставшуюся после извлечения первой трети смесь для гидролиза продолжали обрабатывать далее, пока средняя молекулярная масса Mw не снизилась до 460 кД. После этого вторую треть обрабатывали таким же образом, как и первую треть. Оставшуюся треть параллельно с этим гидролизовали далее до 95 кД и подвергали тому же самому способу обработки, какой использовали для двух других частичных смесей. Из частичной смеси 1 был получен ГЭК 900/0,42 (С2/С6-отношение = 4,83), из частичной смеси 2 был получен ГЭК500/0,42 (С2/С6-отношение = 4,83) и из частичной смеси 3 был получен ГЭК130/0,42 (С2/С6-отношение = 4,83).
После завершения ультрафильтрации концентрацию коллоида доводили до 6% и рН доводили до 5,5, этот раствор делали изотоническим добавлением NaCl, помещали в стеклянные склянки на 500 мл и стерилизовали 20 минут при 121°С.
Недостатками указанного выше способа являются технические особенности технологии, связанные с множеством операций, усложняющих процесс получения ГЭК, определяющих высокую себестоимость конечного продукта, экономически неоправданный количественный выход конечного продукта.
Основным недостатком полученного по этому способу продукта является высокий риск осложнений, на который обращено внимание в вышеописанных документах.
Известны препараты на основе природных пектинов, предложенных для получения плазмозаменителей. Технология их выделения из доступного растительного сырья и применения в медицинских целях достаточно проста и не требует применения сложного и дорогостоящего оборудования (8). В частности, имеется описание «Способ получения плазмозаменителя». Патент SU 539040 (1). Цель изобретения - получение препарата, не обладающего кумулятивными свойствами, улучшения его растворимости, расширения ассортимента и увеличения выхода целевого продукта. Растительное сырье - свекловичный жом, сначала подвергают кислотному гидролизу при рН1 с последующим пропусканием гидролизата через анионит в муравьинокислой форме и целевой продукт осаждают ацетоном в соотношении 1:1-1:2. Гидролиз пектиновых веществ проводят при рН1 путем добавления концентрированной серной кислоты. Преимуществом предлагаемого способа является высокий выход пектиновых веществ (80%), хорошая растворимость препарата в воде, более низкий молекулярный вес и следовательно отсутствие кумулятивного действия, а также почти повсеместное наличие большого количества сырья. Недостатком данного способа является практически полное отсутствие процедуры очистки, достаточной для получения очищенного препарата, пригодного для парентерального применения в качестве плазмозаменителя.
Известен способ, сходный по технологии с вышеописанным патентом, а именно «Способ получения пектина» (патент SU 1666457)(11). Целью изобретения является получение пектина с повышенной растворимостью в воде и пониженной вязкостью его раствора для применения в качестве плазмозаменителя. Для этого приводится пример, описывающий технологический процесс его получения, который заключается в следующем. К 10 г отходов лепестков розы (Rosa canina) после извлечения эфирных масел прибавляют 500 мл 0,25% оксалатного буфера с рН 2,86 и экстракцию проводят при 60С в течение 6 часов. Экстракт отделяют от осадка (целлюлозно-лигниновая фракция) фильтрованием через плотную ткань. В дальнейшем экстракт очищают от низкомолекулярных примесей на Сефадексе. В колонку, заполненную Сефадекс-25, помещают 50 мл экстракта (содержит смесь моно, олиго и полисахаридов) и элюируют буфером рН 4,5, который состоит из 10 мл уксусной кислоты, 4 мл пиридина и 1000 мл воды. Фракции анализируют орцин-сернокислым методом и собирают элюенты, содержащие полисахариды. Полисахаридный элюент гомогенен по молекулярной массе. В мембранную трубку помещают элюенты и диализуют против дистиллированной воды в течение 2 часов при трехкратной смене воды. Температура комнатная. Очищенный диализом экстракт, содержащий гомогенную полисахаридную фракцию в объеме 150 мл, заливают в круглодонную форму на 500 мл и ее содержимое замораживают путем вращения в сосуде, заполненном охлаждающим раствором, и концентрируют, удаляя затем лед без экстракта.
Недостатком указанного способа является применение сорбентов, в частности сефадекс-25, которые обладают низкой производительностью и пригодны для получения пектинов только в лабораторных целях Дополнительно к этому следует отметить, что исходя из описания патента, применяемые реактивы, в частности пиридин, после процедуры диализа сохраняется в конечном продукте, поскольку другой процедуры его целенаправленного и полного удаления из препарата не предусмотрено.
Известен способ получения пектина для медицинских целей в качестве плазмозаменителя («Способ получения пектата глицерина». Патент SU 584012 (2), который заключается в следующем. В примере способа, описанного в этом патенте, суспензируют 15 г пектовой кислоты в 50 мл метанола, содержащего 1,5 мл воды и 12 мл глицерина. Реакцию проводят при 50-55С в течение 24 часов при постоянном перемешивании. Полученный сырой эфир отфильтровывают, тщательно промывают метанолом, ацетоном и эфиром и высушивают на воздухе. Готовят 4% водный раствор пектата глицерина. Раствор центрифугируют, эфир осаждают ацетоном. Осадок растворяют в дистиллированной воде, переосаждают ацетоном. Чистый эфир промывают смесью ацетона и эфира. Выход сырого эфира составил 14,5 г, чистого 10 г. Недостатком этого способа является применение опасных веществ в процессе выделения пектата глицерина, отсутствие в описании патента данных, подтверждающих получение препарата, который имеет степень очистки и свойства, пригодные для применения в качестве плазмозаменителя.
Общими основными недостатками предлагаемых способов получения плазмозаменителя на основе пектина или его производных является полное отсутствие биосовместимости с ферментными системами организма человека и млекопитающих. Галауроновая кислота, составляющая основу пектинов растительного происхождения, не утилизируется в организме, что создает риск ее депонирования и блокады детоксикационных систем. Следует добавить, что в настоящее время в доступной литературе нет сведений о клиническом применении препаратов на основе пектинов.
Одним из перспективных направлений является получение биоплазмозаменителей из доступных природных биологических продуктов, конкретно из мочи человека.
Наиболее пригодными для этих целей являются протеогликаны (ПГ) и гликозаминогликаны (ГАГ), которые содержатся в моче человека в значительных количествах. Эти высокополимерные вещества обладают уникальными свойствами, прежде всего по способности удерживать воду в своей структуре в соотношении 1:100 и более, создавая тем самым возможность использования гемодинамического эффекта. Эти вещества не обладают иммуногенностью, токсичностью, не имеют групповых различий и могут применяться без ограничений при трансфузионном лечении пострадавших, имеющих разные группы крови или другие изоантигенные системы. Важной особенностью ПГ-ГАГ является их полная биосовместимость, а также отсутствие кумулятивного эффекта, который представляет серьезную опасность при использовании практически всех известных синтетических полимерных плазмозаменителей.
Использование мочи дает возможность неограниченного получения исходного сырья, исключить зависимость от поставок импортных и дорогостоящих препаратов, обеспечить экстренное получение необходимого объема плазмозаменителей как для индивидуальных целей, так и для промышленного производства. Известны биопрепараты на основе ПГ-ГАГ, выделенных из тканей животных, которые нашли широкое применение в медицине и косметологии.
Протеогликан (ПГ) - это общее название, относящееся к гликопротеину с разнообразными типами структуры и обычно состоящему из одного корового белка с по меньшей мере с двумя и более ковалентно присоединенных линейных полисахаридных цепей (6). ПГ является комплексным соединением, однако поскольку соединение полисахаридной цепи и корового белка имеет слабую силу связывания, они имеют тенденцию к легкому отделению друг от друга. По этой причине выделение или очистка ПГ являются чрезвычайно тонким процессом, требующим максимально щадящего проведения отделения на всех этапах. Это требование подразумевает максимальное исключение процедур, повреждающих структуру ПГ, в частности высокоскоростного центрифугирования, а также фильтрации через мелкопористый фильтр, не говоря уже об ультрафильтрации и ультрацентрифугировании. Таким образом, обычные способы не подходят для выделения ПГ из-за сложных или ручных действий.
Полисахаридные цепи ГАГ чрезвычайно гидрофильны, вследствие чего они удерживают большое количество воды и даже в очень низких концентрациях образуют гидратированный гель. Это свойство еще более усиливает высокая плотность отрицательных зарядов, которые притягивают осмотически активные катионы. Такой гидрофильный эффект растворов ГАГ способствует поддержанию онкотического давления в сосудистом русле и давления во внутриклеточном матриксе.
Известно, что гиалуроновая кислота, которая является самым крупным полисахаридом из всех ГАГ, содержит 20-30 тысяч дисахаридных звеньев и высокую молекулярную массу - до 15 млн. дальтон, представляет гидрофильное соединение, одна молекула которого может удерживать 200-500 молекул воды (3). Особенностью хондроитина среди ПГ его способность сохранять воду в толще хряща в виде водных полостей (12).
В настоящее время разработано множество способов получения и приготовления ПГ из природных источников. Однако практически все из них предполагают использование биологического сырья, полученного из тканей животных или рыб и моллюсков, имеющих низкую биосовместимость для человека, и требуют высокую степень очистки.
Известен способ получения ПГ «Способ получения агрегатов протеогликанов из соединительной ткани животного», RU 2401839 (13), Способ предусматривает использование в качестве исходного материала хрящевую ткань рыб, моллюсков и млекопитающих. Измельченный продукт подвергают иммерсии в 0,00025-0,05 н. растворе соли щелочного металла, центрифугированию с последующим отделением твердой и масляной фазы. Жидкую фазу вновь центрифугируют и фильтруют, промывают водой с последующим отделением и концентрированием целевого продукта. Изобретение позволяет получить ПГ в неизмененной, нераспавшейся форме, который предлагается использовать как материал для получения лекарственных средств, материалов медицинского назначения, косметики.
Для выделения используют 0,00025-0,1 н. раствор гидроксида натрия при 0-10°С. Экстракция контролировалась определением уроновой кислоты. Дополнительная очистка проводилась фильтрацией или ультрафильтрацией. ПГ получали в гелеобразном виде после добавления этанола и в дальнейшем лиофилизирован или получен после распылительной сушки в качестве порошка.
Недостатками способа являются:
1. Многоэтапность процесса получения ПГ - не менее 12 этапов.
2. Длительность всего процесса получения ПГ - более 50 часов.
3. Множественность различных сложных процедур способа: эстракция, диализ, дополнительные процедуры применения этанола и эфира, обработки катионообменной смолой.
4. Длительность этапа диализа для удаления солей, длительность второго этапа диализа для удаления лимонной кислоты.
5. Применение исходного бионесовместимого сырья из тканей животных.
Наиболее близким по техническому решению, принятый как прототип, является способ получения ПГ, описанный в патенте РФ №2621311 «Способ получения протеогликана» (4). Способ реализуется следующим образом. Моча, полученная ex tempore, проходит предварительную обработку, путем охлаждения в течение 60 мин, при -1°С, до образования агрегатов и начала выпадения осадка солей, липопротеидов, фосфолипидов и белков, макрокомпонентов мочи - клеток крови, гиалиновых цилиндров, бактерий, слизи с последующим центрифугированием при 3000 об/мин в течение 10 минут, при -1°С. Не прекращая режим охлаждения надосадочную жидкость отделяют и используют для дальнейшего выделения ПГ, для чего коррегируют ее кислотно-щелочной показатель добавлением 1 N раствора едкого натра и 1 N раствора соляной кислоты до значений рН 6,0-6,2, при котором происходит максимальное осаждение ПГ. Затем 1 объем надосадочной жидкости смешивают с 1,6 объемами 96° этанола и оставляют не изменяя температурный режим на срок не менее 2 часов и не более 4 часов, предпочтительно 3 часов, для образования преципитата, который отделяют центрифугированием при 1500 об/мин. Осадок размешивают в 40° этаноле, насыщенным 2-3 М водном раствором хлористого натрия, в объемном соотношении 1:5, не выходя из установленного температурного режима, выдерживают в течение 10 мин до образования преципитата и центрифугируют при 1500 об/мин в 3 мин, для повышения степени очистки за счет удаления остатков надосадочной жидкости. Повторная процедура переосаждения ПГ не требуется, поскольку она не приводит к существенному изменению степени очистки этим осадителем.
Недостатком способа является то, что он не предназначен для применения в качестве биоплазмозаменителя (БПЗ), поскольку это требует от конечного продукта дополнительную очистку от сопутствующих примесей до состояния, пригодного для парентерального введения. При этом повторная процедура переосаждения применяемым составом осадителя не приводит к существенному изменению степени очистки конечного продукта и является основной причиной, исключающей возможность повышение степени очистки только за счет технологических решений, которые используются в этом способе.
Предлагаемый способ позволяет устранить указанные недостатки. В известной нам научно-технической и патентной литературе не обнаружены способы с подобной совокупностью признаков. Полученный результат, обусловленный совокупностью этих признаков, не достигался в известных решениях.
Целью предлагаемого нами решения является создание простого в реализации и эффективного способа получения БПЗ с гемодинамическим действием, на основе ПГ-ГАГ.
Согласно предлагаемому изобретению исходный продукт, содержащий высокую концентрацию ПГ-ГАГ, получают из биосовместимого и практически неограниченного, постоянно доступного сырья - мочи, после ее обработки, основанной на применении физического принципа охлаждения, с целью предварительного удаления подавляющей массы сопутствующих основных нежелательных компонентов мочи по способу, описанному в патенте РФ №2621311(4), в нашей модификации, не затрагивающий состав этого продукта.
В проведенных ранее исследованиях был подробно описан состав и качественные характеристики целевого продукта, состоящего из ПГ и ГАГ, полученных способом, описанным в патенте РФ №2621311(4). Поскольку в заявляемом способе этот же продукт использован нами в качестве исходного, целью наших исследований было изучение степени качественной и количественной очистки получаемого целевого продукта, прежде всего в контексте его применения в качестве высокоочищенного продукта, пригодного для получения БПЗ.
Для этих целей были использованы следующие методы исследования.
Электрофорез на ацетат целлюлозных мембранах по Gold, 1981, (в модификации Пауль, Русова, 1995), определение общего белка по Лоури, определение липопротеинов низкой плотности (ЛПНП) (14), определение липопротеинов высокой плотности (ЛПВП) (15), определение фосфолипидов - прямой ферментативный колориметрический тест. Набор производства "Sentinel", определение растворимых фибрин мономерных комплексов (РФМК) (16). Общее содержание ГАГ, определяли по уровню уроновых кислот карбазоловым методом.
Исследования основных, количественно превалирующих гормонов, проводили на ИФА анализаторе Multiskan FC, "Thermo Scientific". Концентрацию эстрадиола (Е2) определяли с помощью набора «Эстрадиол - ИФА», "Хема-Хема-Medica". Результат выражали в пг/мл. Концентрацию прогестерона, тестостерона и кортизола определяли с помощью наборов «Диагностические системы», Н. Новгород, РФ. Результаты исследования прогестерона и тестостерона выражали в нмоль/л, кортизола в мкг/сутки.
Полученные показатели обработаны методом вариационной статистики с определением среднего арифметического значения (М) и стандартной ошибки среднего арифметического (m). Средние величины сравнивали с использованием U-критерия Манна-Уитни и W-критерия Вилкоксона. Различия двух сравниваемых величин считали достоверным, если вероятность их тождества была менее 5% (p<0,05).
Ранее в проведенных нами исследованиях был установлен состав исходного продукта. Эти результаты представлены в таблице №1, где приводится содержание общих ГАГ и их фракций в исследуемых образцах продукта, полученного после выделения по способу патента РФ №2621311(4).

Примечание - различия достоверны, p<0,05.
Состав фракций в исследуемых образцах исходного продукта показывает присутствие ГАГ в виде хондроитинсульфата С, хондроитинсульфатов А+В (I - фракция), кератансульфата (II-фракция), гепарансульфата, гепарина (III-фракция).
Результаты исследований состава ПГ-ГАГ после выделения по способу патента РФ №2621311(4) показывают наличие в образцах исходного продукта уроновых кислот, аминосахаров, сульфатов, белковых компонентов, что согласуется с данными таблицы №1, отражающими качественный и количественный состав ГАГ.
Дополнительное изучение состава осадка ПГ-ГАГ показало наличие в образцах широкого спектра нежелательных компонентов в концентрации, неприемлемой для применения в качестве плазмозаменителя. Основными представителями этих веществ являются фосфолипиды, липопротеиды высокой и низкой плотности, денатурированные белки, стероидные гормоны, а также пигменты. Эти сведения представлены в таблице №2.
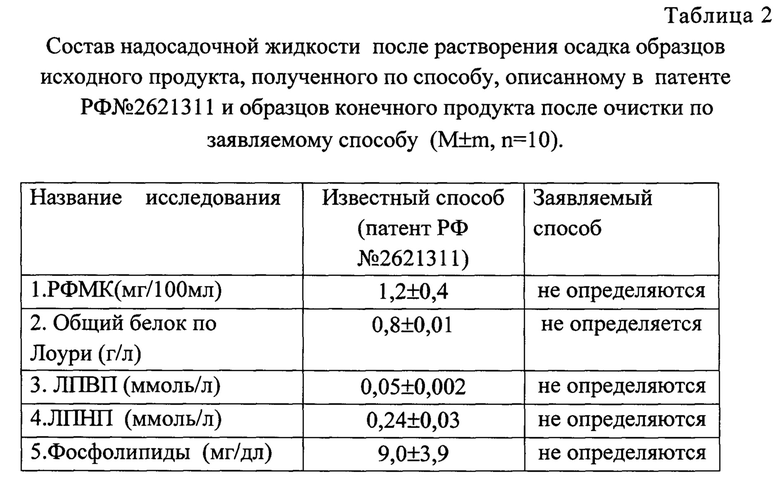
Примечание - различия достоверны, p<0,05. Проведенные нами исследования показали, что объемное содержание ПГ и ГАГ в общем объеме осадка исходного продукта доминирует и составляет, в среднем, 68%, при колебаниях в зависимости от источника сырья от 56% до 72%. Остальную часть составляют примеси, основные из которых описаны выше.
Перед дальнейшим обсуждением предлагаемого изобретения представляется необходимым конкретизировать и более подробно раскрыть понятия и терминологию, используемые нами в описательной части, затрагивающие характеристики различных форм биогеля БПЗ.
1. Понятие нерастворимая щелочная гелевая форма применяется для характеристики как исходного, так и промежуточного состояния биогеля в процессе его обработки, рН раствора, в котором находится суспензия биогеля, составляет 9,8-10.
2. Понятие пребиоформа применяется на промежуточном этапе подготовки биогеля и предусматривает название щелочной нерастворимой формы концентрата биогеля после всех этапов его обработки, готовой к заключительному розливу. Эта форма биогеля устойчива к длительному хранению (экспериментально доказано не менее 3 лет) и транспортировке при условии предотвращения ее смешиванием с нейтрализатором.
3. Понятие растворимая активная (кисло-солевая) форма концентрата биогеля применяется для его характеристики после смешивания суспензии концентрата пребиоформы с нейтрализатором во флаконе перед непосредственным применением, рН растворимой активной формы концентрата биогеля составляет 5,6-6.0. Эта форма представляет полностью растворенный концентрат биогеля, сохраняющий биологическую активность с оптимальным сроком применения до 6 часов.
4. Понятие активная форма раствора биогеля биоплазмозаменителя применяется после смешивания активной формы растворимого концентрата биогеля с 0,9% (физиологическим) раствором хлористого натрия или раствором других кристаллоидов с доведением концентрации раствора биогеля биоплазмозаменителя предпочтительно, до 7% в емкости, предназначенной для инфузии.
В приведенной ниже описательной части предлагаемого изобретения рассмотрены в отдельности каждый из перечисленных свойств и отличительных признаков предлагаемого БПЗ, а также их совокупность, позволяющие выделить их принципиальные и существенные отличия от аналогов и выбранного прототипа.
В нижеперечисленной части описания представлены и обобщены основные этапы технологического процесса способа получения основного компонента биоплазмозаменителя, конкретно ПГ-ГАГ. Они следующие:
1. Этап дополнительной очистки исходного продукта от сопутствующих примесей нежелательных компонентов.
2. Этап выделения и обработки ПГ-ГАГ для получения биогеля.
3. Этап технологических операций, выполняемых в двух альтернативных вариантах, отличающихся расфасовкой и хранением БПЗ в виде преформы или в виде лиофильно высушенного препарата.
При этом этап дополнительной очистки разделен на два последовательно выполняемые подэтапа:
1.1. Обработка мочи охлаждением до -2°С с целью криоагрегации, применяемой для удаления из исходного продукта основной массы примесей нежелательных компонентов, таких как: крупномолекулярные белковые агрегаты, липидные вещества, бактерии и их фрагменты, слизь, форменные элементы и другие надмолекулярные образования.
1.2. Охлаждение мочи до -10°С с целью вымораживания криоконцентрата, применяемой для отделения и удаления из исходного продукта оставшихся примесей нежелательных компонентов, таких как: стероидные гормоны, пигменты, пептиды и другие низкомолекулярные вещества, представляющие естественные метаболиты.
2. Этап выделения и обработки ПГ-ГАГ для получения биогеля биоплазмозаменителя разделен на три основных подэтапа:
2.1. Переосаждение комплекса концентрата нерастворимой щелочной гелевой формы биогеля 0,5N раствором гидроксида натрия с целью дополнительной очистки, стерилизации и повышения концентрации в исходном продукте.
2.2. Центрифугирование и выделение из осадка фракции, содержащей преимущественно ПГ-ГАГ.
2.3. Формирование пребиоформы в виде концентрата нерастворимой щелочной гелевой формы биогеля на основе ПГ-ГАГ.
3. Технологические операции, выполняемы в двух альтернативных вариантах:
3.1. Вариант 1. Связан с розливом и фасовкой пребиоформы во флаконы, закрывающимися специальным устройством - «Устройство для хранения, транспортировки, дезактивации консерванта и применения биологической жидкости», патент РФ №2709511(5), в дальнейшем (КУ) с последующей нейтрализацией консерванта и образованием активной растворимой формы концентрата биогеля биоплазмозаменителя при непосредственной необходимости его применения.
3.2. Вариант 2. Связан с розливом и фасовкой пребиоформы в емкость для нейтрализации консерванта и образования активной растворимой формы концентрата биогеля биоплазмозаменителя, дегидратации концентрата биогеля ацетоном с последующей сублимацией или лиофилизацией, расфасовкой и укупоркой целевого продукта, готового для применения.
Для получения БПЗ, согласно заявляемому способу, исходный продукт очищают в ходе нескольких последовательных этапов.
На этапе 1, подэтап 1.1 полученную ex tempore мочу охлаждают в проточном режиме до -1°С - -3°С, предпочтительно -2°С со скоростью 15-25 мл /мин, предпочтительно 20 мл/мин. Выбор предпочтительных, взаимосвязанных между собой параметров скорости и температуры охлаждения обусловлен тем, что при температуре -1°С не во всех образцах исследуемой мочи происходит криоагрегация, что требует большего охлаждения или уменьшения скорости пропускания мочи до 15 мл/мин. Напротив, при охлаждении мочи до -4°С и ниже, в некоторых образцах начинается образование льда, что требует снижения температуры до -2°С и увеличения скорости пропускания мочи 25 мл/мин. На этапе 1, подэтап 1.2 полученные на выходе криоагрегаты, не содержащие ПГ-ГАГ, отделяют от мочи фильтрацией или центрифугированием в проточном режиме, а мочу дополнительно охлаждают до -10°С с образованием в процессе охлаждения двух фракций, одна из них жидкая, представляет концентрат мочи, содержащий преимущественно фосфолипиды, стероидные гормоны, пигменты и другие нежелательные компоненты, которую удаляют, а другая в твердом состоянии, представляет лед, состоящий из замерзшей мочи, которую размораживают до жидкого состояния при комнатной температуре. На Фиг. 1 видны две фракции мочи после охлаждения до -10°С, одна из них представляет концентрат мочи, темно-коричневого цвета, а вторая светлая, в виде льда. Соотношение объема жидкой фракции, содержащей концентрат, к объему твердой фракции после размораживания находится в интервале 1:8-1:14, составляет, в среднем, 1:11.
На этапе 2, подэтап 2.1 для осаждения ПГ-ГАГ к размороженной фракции мочи добавляют осадитель в виде 1N раствора гидроксида натрия, в соотношении 10:1, смесь оставляют на 10 минут при комнатной температуре для образования преципитата.
На этапе 2, подэтап 2.2 преципитат осаждают центрифугированием при 1000 об/мин в течение 20-40 секунд, предпочтительно 30 секунд, затем отделяют надосадочную жидкость, а полученный осадок, содержащий ПГ-ГАГ, и используют как исходный продукт для получения БПЗ.
Выбор предпочтительного времени центрифугирования обусловлен тем, что за 20 секунд центрифугирования не происходит полного отделения осадка, а при центрифугировании свыше 40 секунд осадок становится плотным, что затрудняет работу с ним.
Нами неожиданно установлено, что после центрифугирования в осадке определяются две основные фракции, из которых верхняя, светло-белового оттенка, полупрозрачная, слегка опалесцирующая состоит преимущественно из ПГ-ГАГ (Ф1), а нижняя (Ф2) - белого цвета, плотной консистенции за счет грубодисперсных частиц, состоит преимущественно из солей жирных кислот, фосфолипидов, денатурированных белков. На Фиг. 2 в осадке видны две фракции с четкой границей раздела между ними. Отделение фракции Ф1 от Ф2, содержащей нежелательные компоненты, позволило существенно снизить их содержание и также существенно увеличить объемное содержание основных компонентов, а именно: ПГ-ГАГ. Изучение различных параметров эффективности очистки показало следующее. На таблице №3 показаны результаты определения объемного соотношения фракций Ф1 и Ф2 до и после проведения очистки продукта заявляемым способом.

Как видно из представленных на таблице №3 данных, объемное соотношение фракций Ф1 и Ф2 после очистки существенно изменилось за счет максимального увеличения доли Ф1, содержащей ПГ и ГАГ, при 225-кратном уменьшении объема Ф2, содержащей нежелательные компоненты.
Для выделения верхней фракции Ф1 к осадку после центрифугирования добавляют раствор 0,5N гидроксида натрия в соотношении 1:1 и осторожно размешивают так, чтобы размешивание затронуло только верхнюю фракцию Ф1.
На этапе 2, подэтап 2.3 суспензию фракции Ф1 переносят в другую емкость, снабженную на дне выпускным штуцером и краном, в которую добавляют насыщенный раствор хлористого натрия в 0,5N растворе гидроксида натрия в соотношении 1:10, перемешивают в течение 5-10 секунд и экспонируют в течение 5-15 минут, предпочтительно 10 минут. Экспозиция в течение 5 минут недостаточна для окончательного формирования преципитата, а экспозиция свыше 15 минут нецелесообразна, поскольку окончательное формирование преципитата заканчивается к 10 минутам. По истечению 10 минут преципитат поднимается вверх и располагается над жидкостью в виде кольца белого цвета (Фиг. 3). Жидкость, находящуюся ниже преципитата сливают через штуцер открытием крана. Затем к преципитату добавляют раствор 0,5N гидроксида натрия в соотношении 1:1 и осторожно размешивают до образования однородной суспензии, которая представляет пребиоформу нерастворимой щелочной гелевой формы ПГ-ГАГ.
На этапе 3, подэтап 3.1, согласно варианту 1, пребиоформу нерастворимой щелочной гелевой формы ПГ-ГАГ в асептических условиях переносят в стерильные флаконы, которые герметично закрывают крышкой, представляющей устройство, описанное в патенте РФ №2709511(5), (КУ) (Фиг. 4). Описание устройства, чертеж (Фиг. 5) и способ его применения размещены в Приложении 1.
Перед непосредственным применением БПЗ получают растворимую активную (кисло-солевую) форму концентрата БПЗ, путем смешивания суспензии концентрата пребиоформы с нейтрализатором во флаконе с крышкой «КУ» способом, описанным в этом же патенте РФ №2709511(5). Для этого с соблюдением асептики снимают установленный сверху крышки защитный колпачок, надавливают на заглушку на штоке, который выталкивает пробку-дозатор с реагентом (нейтрализатором) во флакон с раствором компонентов. После химической реакции нейтрализации реагента с консервантом (0,5N гидроксидом натрия) в емкости, шток возвращают в исходное положение. Далее снимают заглушку, установленную на штоке «ПУ», вместо которой присоединяют внешние шприц или магистраль с аналогичным соединением типа Luer-lock. Отобранный из флакона шприцом или через внешнюю магистраль активную форму концентрата БПЗ переносят в емкость для инфузии с 0,9% (физиологическим) раствором хлористого натрия или раствором других кристаллоидов с доведением концентрации БПЗ до 7%. В этом состоянии растворенный 7% БПЗ соответствует понятию активная форма раствора биогеля биоплазмозаменителя и готов к непосредственному применению.
На этапе 3, подэтап 3.2, согласно варианту 2 заявляемый способ предусматривает альтернативный вариант получения растворимой активной формы БПЗ, пригодной для длительного хранения в сублимированном или лиофилизированном состоянии.
Для этого проводят дегидратацию нерастворимой щелочной формы геля ПГ-ГА в виде концентрата пребиоформы с исходной рН10, путем добавления охлажденного до -2°С ацетона в соотношении 1:10, смесь экспонируют в течение 5-15 минут, предпочтительно 10 минут, при температуре в интервале от -1°С до -3°С, предпочтительно -2°С, до образования грубодисперсного преципитата, после чего центрифугируют при 1000 об/мин в течение 30 сек. Надосадочную жидкость удаляют, осадок разбавляют повторно охлажденным до -2°С ацетоном в соотношении 1:10, экспонируют не меняя температурный режим в течение 10 минут и повторно центрифугируют при 1000 об/мин в течение 30 сек. Надосадочную жидкость удаляют, к осадку добавляют 96° этиловый спирт в соотношении 1:1, размешивают и помещают в эксикатор под вакуумом на 1-3 часа, предпочтительно 2 часа при температуре 0°С для максимального удаления спирта. После вакуум экстракции на дне эксикатора образуется белый мелкодисперсный гигроскопичный порошок, объем которого в 250-300 раз меньше объема первоначального концентрата. Длительность вакуум экстракции ограничивают 2 часами, поскольку последующее увеличение времени ее применения приводит к существенному изменению свойств полученного продукта, что, прежде всего, проявляется в ухудшении его растворимости, а меньшее время было недостаточно для необходимого уменьшения объема конечного продукта. Полученный сублимированный или лиофильно высушенный порошок БПЗ расфасовывают в стеклянные пенициллиновые флаконы объемом предпочтительно 40 мл, герметично укупоривают и хранят до применения. Проведенные испытания растворимости сублимированного высушенного порошка БПЗ показали его полную растворимость и стабильность, без образования осадка в динамике наблюдения в течение 24 часов.
На таблице №2 приведены результаты изучения состава надосадочной жидкости после растворения осадка образцов исходного продукта, полученного по способу патента РФ №2621311(4) и образцов конечного продукта после очистки по заявляемому способу. Как видно, все исследованные нами нежелательные компоненты, такие как РФМК, ЛПВП, ЛПНП, фосфолипиды, после очистки не определяются. Не менее важно, что после очистки не установлено наличие общего белка, что, в определенной степени, свидетельствует об удалении примеси нежелательных нативных белков в процессе очистки.
Электрофорез на ацетатцеллюлозных мембранах по Gold, 1981 (в модификации Пауль, Русова, 1995) показал высокую степень очистки ПГ-ГАГ, полученных заявляемым способом, от сопутствующих нежелательных белковых компонентов. Белковые фракции не выявлены.
Проведенные исследования содержания 4 различных стероидных гормонов в исходной моче, а также в криоконцентрате показали существенные различия. Результаты представлены в таблице 4.
Как видно из представленных в таблице 4 данных, уровень эстрадола в криоконцентрате превысил 90000 пг/мл и был практически в 15 раз выше показателей в исходной моче. Аналогично этому, из представленных в таблице 4 данных видно, что уровень кортизола при исходных значениях 430,9±26,4 мкг/сутки, в криоконцентрате превысил 8000 мкг/сутки.
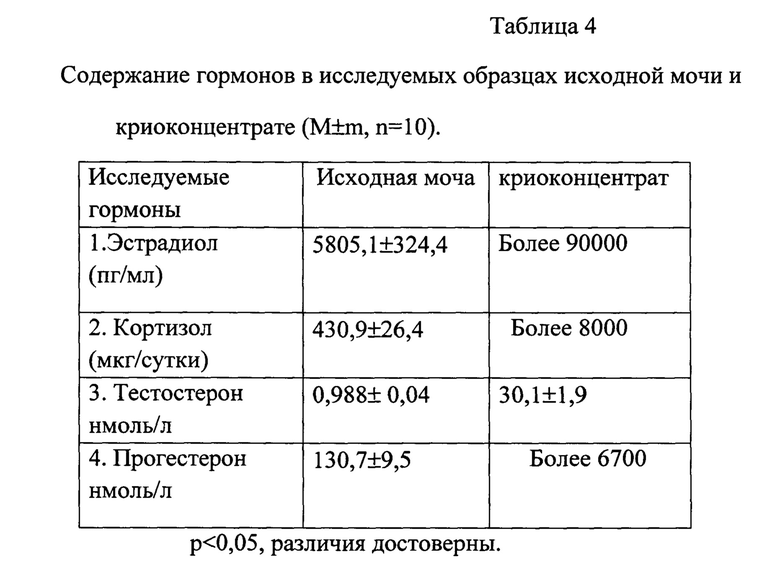
Результаты исследования содержания других гормонов, таких как: тестостерон и прогестерон показали, что их концентрация в гормональном концентрате также существенно выше таковой, определяемой в исходной моче.
Проведенные исследования показали, что после очистки исходной мочи путем применения в заявляемом способе вымораживания криоконцентрата, содержание исследуемых стероидных гормонов в конечном продукте снижается до нерегистрируемых значений.
Приведенные в таблицах №2, №3 и №4 результаты исследований свидетельствуют о высокой эффективности технического решения комплексной очистки, которая применена в заявляемом способе. Степень очистки конечного продукта достаточна для применения в качестве БПЗ и безопасна по составу и содержанию остаточных примесей. Для определения одного из важных свойств полученного целевого продукта, а именно, степени его гидрофильности (способности удерживать воду), был выбран принцип предварительной дегидратации для получения продукта, путем удаления из него воды. После определения объема и веса сублимированного продукта, его потом насыщали водой, и по разнице объемов и веса продукта до и после насыщения водой определяли степень гидрофильности, выражая ее значения коэффициентом гидрофильности (Кг).
Для реализации этой цели нами была использована методика дегидратации, ранее разработанная для дегидратации древесины (10), которая в нашей модификации оказалась применимой для дегидратации ПГ-ГАГ. В доступной нам литературе мы не нашли описания подобных или других способов дегидратации этих веществ. Способ дегидратации заключается в следующем.
К нерастворимой щелочной форме геля ПГ-ГА в виде концентрата пребиоформы (после очистки и отделения Ф1) с исходной рН10, добавляют охлажденный до -2°С ацетон в соотношении 1:10, экспонируют в течение 5-15 минут, предпочтительно, 10 минут, при температуре в интервале от -1°С до -3°С, предпочтительно -2°С, до образования грубодисперсного преципитата, после чего центрифугируют при 1000 об/мин в течение 30 сек. Надосадочную жидкость удаляют, осадок разбавляют повторно охлажденным до -2°С ацетоном в соотношении 1:10, экспонируют не меняя температурный режим в течение 10 минут и повторно центрифугируют при 1000 об/мин в течение 30 сек. Надосадочную жидкость удаляют, осадок разбавляют 96° этиловым спиртом в соотношении 1:1, размешивают и помещают в эксикатор под вакуумом на 1-3 часа, предпочтительно, 2 часа при температуре 0°С для максимального удаления спирта. После вакуум экстракции на дне эксикатора образуется белый мелкодисперсный гигроскопичный порошок, объем которого в 250-300 раз меньше объема первоначального концентрата. Длительность вакуум экстракции ограничивают 2 часами, поскольку последующее увеличение времени ее применения приводит к существенному изменению свойств полученного продукта, что, прежде всего, проявляется в ухудшении его растворимости, а меньшее время было недостаточно для необходимого уменьшения объема конечного продукта.
Полученный продукт взвешивают, после чего к нему в соотношении 1:1 доливают раствор 0,9% хлористого натрия с 0,1N раствором гидроксида натрия с конечным рН раствора 10,0 и размешивают до образования суспензии на дне эксикатора, затем вновь добавляют в соотношении 1:10 раствор 0,9% хлористого натрия с 0,1N раствором гидроксида натрия с конечным рН раствора 10,0 и оставляют на 2 часа в термостате при 37°С при постоянном медленном перемешивании. Затем суспензию переносят в мерный цилиндр, добавляют в соотношении 1:50 раствор 0,9% хлористого натрия с 0,1N раствором гидроксида натрия с конечным рН раствора 10,0 и оставляют на 24 часа при температуре 37°С.
После завершения экспозиции отмечают появление разделительной линии между слоем геля (снизу) и слоем раствора хлористого натрия (сверху), которая стабильно сохраняется на одном и том же уровне. Согласно уровню разделительной линии определяют конечный объем и вес набухшей нерастворимой формы. Эти показатели превышают исходный объем и вес, в среднем, в 250 раз. Рассчитанный таким образом коэффициент гидрофильности (Кг) составляет: Кг = 250,4.
Проведенные исследования подтвердили высокую степень гидрофильности полученного заявляемым способом конечного продукта (нерастворимая щелочная форма ПГ-ГАГ после очистки и отделения Ф1 (в виде концентрата пребиоформы). Таким образом, подтверждено одно из основных свойств полученного продукта, определяющее его назначение к применению, а именно, возможность его использования для стабилизации онкотического давления за счет удержания воды и, в конечном итоге, для создания гемодинамического эффекта.
Исследования целевого продукта находящегося во флаконах в виде пребиоформы во временной динамике, конкретно ежемесячно в первый год, а также во 2 и 3 годы - каждые три месяца, не выявили изменений стабильности и свойств препарата.
Бактериологические исследования целевого продукта находящегося во флаконах в виде пребиоформы во временной динамике, конкретно ежемесячно в первый год, а также во 2 и 3 годы - каждые три месяца, не выявили появления микробной флоры во флаконах. Этот факт объясняется тем, что консервант - 0,5N раствор гидроксида натрия, находящийся во флаконах оказывает мощное антибактериальное действие и способствует устранению причин возникновения микробной контаминации.
Способ реализуется следующим образом. Для получения БПЗ, согласно заявляемому способу, полученную ех tempore мочу охлаждают в проточном режиме до -1°С - -3°С, предпочтительно -2°С со скоростью 15-25 мл /мин, предпочтительно 20 мл/мин. Полученные на выходе криоагрегаты, не содержащие ПГ-ГАГ, отделяют от мочи фильтрацией, или центрифугированием в проточном режиме, а моча дополнительно охлаждается до -10°С с образованием в процессе охлаждения двух фракций, одна из них жидкая, представляет концентрат мочи, содержащий нежелательные компоненты, которую удаляют, а другая в твердом состоянии, представляет лед, состоящий из замерзшей мочи, которую размораживают до жидкого состояния при комнатной температуре. Для осаждения ПГ-ГАГ к размороженной фракции мочи добавляют осадитель в виде 1N раствора гидроксида натрия, в соотношении 10:1, смесь оставляют на 10 минут при комнатной температуре для образования преципитата. Затем преципитат осаждают центрифугированием при 1000 об/мин, в течение предпочтительно 30 секунд, затем отделяют надосадочную жидкость, а полученный осадок, содержащий ПГ-ГАГ, и используют как исходный продукт для получения БПЗ.
После центрифугирования в осадке определяются две основные фракции, из которых верхняя (Ф1), состоит преимущественно из ПГ-ГАГ, а нижняя (Ф2) содержит нежелательные компоненты.
Для выделения верхней фракции Ф1 к осадку после центрифугирования добавляют раствор 0,5N гидроксида натрия в соотношении 1:1 и осторожно размешивают так, чтобы размешивание затронуло только верхнюю фракцию Ф1. После этого суспензию фракции Ф1 переносят в другую емкость, снабженную на дне выпускным штуцером и краном, в которую добавляют насыщенный раствор хлористого натрия в 0,5N растворе гидроксида натрия в соотношении 1:10, перемешивают в течение 5-10 секунд и экспонируют в течение, предпочтительно 10 минут. По истечении 10 минут преципитат поднимается вверх и располагается над жидкостью, которую сливают через штуцер открытием крана. Затем к преципитату добавляют раствор 0,5N гидроксида натрия в соотношении 1:1 и осторожно размешивают до образования однородной суспензии, которая представляет пребиоформу нерастворимой щелочной гелевой формы ПГ-ГАГ. Эту пребиоформу в асептических условиях переносят в стерильные флаконы, герметично закрывают крышкой, представляющей устройство, описанное в патенте РФ №2709511(5)(КУ). Описание устройства и способ его применения размещены в Приложении 1.
Перед непосредственным применением БПЗ готовят растворимую активную (кисло-солевую) форму концентрата БПЗ, путем смешивания суспензии концентрата пребиоформы с нейтрализатором во флаконе с крышкой «КУ» способом, описанным в этом же патенте РФ №2709511(5). Для этого с соблюдением асептики снимают установленный сверху крышки защитный колпачок, надавливают на заглушку на штоке, который выталкивает пробку-дозатор с реагентом (нейтрализатором в виде порока лимонной кислоты) во флакон с концентратом пребиоформы. После химической реакции нейтрализации реагента с консервантом (0,5N гидроксидом натрия) в емкости, шток возвращают в исходное положение. Далее снимают установленную на штоке «КУ» заглушку, вместо которой присоединяют внешние шприц или магистраль с аналогичным соединением типа Luer-lock. Отобранную из флакона шприцом или через внешнюю магистраль растворимую активную форму концентрата БПЗ переносят в емкость для инфузии с 0,9% (физиологическим) раствором хлористого натрия или раствором других кристаллоидов с доведением концентрации БПЗ до 7%. В этом состоянии растворенный 7% БПЗ соответствует понятию активная форма раствора биогеля БПЗ, который готов к непосредственному применению.
Согласно варианту 2 заявляемый способ предусматривает альтернативный вариант получения растворимой активной формы БПЗ, пригодной для длительного хранения в лиофилизированном состоянии.
Для этого проводят дегидратацию нерастворимой щелочной формы геля ПГ-ГА в виде концентрата пребиоформы с исходной рН10, путем добавления охлажденного до -2°С ацетона в соотношении 1:10, смесь экспонируют в течение 5-15 минут, предпочтительно, 10 минут, при температуре в интервале от -1°С до -3°С, предпочтительно -2°С, до образования грубодисперсного преципитата, после чего центрифугируют при 1000 об/мин в течение 30 сек. Надосадочную жидкость удаляют, осадок разбавляют повторно охлажденным до -2°С ацетоном в соотношении 1:10, экспонируют не меняя температурный режим, в течение 10 минут и повторно центрифугируют при 1000 об/мин в течение 30 сек. Надосадочную жидкость удаляют, к осадку добавляют 96° этиловый спирт в соотношении 1:1, осторожно размешивают до однородной массы и помещают в эксикатор под вакуумом, предпочтительно, на 2 часа при температуре, предпочтительно, 0°С для максимального удаления спирта. После вакуум экстракции на дне эксикатора образуется белый мелкодисперсный порошок БПЗ, который расфасовывают в стеклянные пенициллиновые флаконы объемом предпочтительно 20 мл, герметично укупоривают и хранят до применения.
Необходимо дополнить, что варианты комбинаций композиции компонентов БПЗ, и их объемные соотношения, которые могут варьировать между собой, изложены ниже, они следующие.
Так, вместо 0.9% раствора хлористого натрия, для разведения концентрата растворимого геля ПГ-ГАГ могут использоваться другие изотонические растворы, какими являются растворы кристаллоидов, или другие, разрешенные к применению растворы, или комбинации этих растворов, не содержащие органических компонентов.
Также то, что объем флакона для хранения и транспортировки концентрата БПЗ не ограничен, и может варьировать в сторону уменьшения или увеличения, в зависимости от предпочтения исполнителя.
Также то, что предлагаемая к применению концентрация растворимого геля БПЗ в емкости для инфузии до 7% не ограничена обозначенными выше значениями, и может варьировать в сторону уменьшения или увеличения в зависимости от предпочтения исполнителя.
Отдельно следует отметить, что применение мочи в качестве исходного продукта требует обязательного поведения мероприятий, связанных с обеззараживанием исходного продукта от бактерий и вирусов и предотвращения в последующем микробной контаминации.
Из имеющейся доступной нам технической литературы известно применение гидроксида натрия в качестве высокоэффективного бактерицидного средства в медицине. В частности известно его использование для стерилизации крови и ее компонентов в аппаратах системы "Allegro", США, которыми оснащены многие станции переливания крови. С этих позиций, использование гидроксида натрия в описанной в предлагаемом способе концентрации, обеспечивает в полной мере требуемый бактерицидный эффект. Согласно документу, известному как «Правила проведения исследований биологических лекарственных средств Евразийского экономического союза. Утверждены решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. №89» рекомендуется проводить испытание способности производственных процессов элиминировать или инактивировать вирусы, а также проводить; испытание препарата на соответствующих этапах производства на предмет отсутствия обнаруживаемых вирусов. В документе также указывается, что ни один из подходов сам по себе не дает достаточной гарантии, поэтому в целях ее достижения необходимо использовать их комбинацию. В этом контексте, предлагаемый способ на каждом этапе его технологического процесса предполагает присутствие вещества - гидроксида натрия, обладающего доказанным антимикробным и антивирусным действием. Помимо этого, на одном из этапов технологического процесса заявляемого способа присутствует применение физического метода элиминации патогенных источников, конкретно криоагрегация, способствующая удалению бактерий из исходного продукта. На заключительных этапах технологического процесса применяются ацетон и этиловый спирт в качестве дегидратирующих агентов, бактерицидное действие которых также известно. Таким образом, в заявленном способе и технологическом процессе его реализации представлена комбинация известных и доказанных средств, а также способов элиминации и инактивации бактерий и вирусов, которые обязательны для обеспечения безопасности биологических препаратов, каким является предлагаемый БПЗ.
Представляется целесообразным обратить внимание на сравнительную характеристику основных параметров предлагаемого БПЗ с аналогами. Сравнительная оценка совокупных признаков известных плазмозаменителей гемодинамического действия и предлагаемого БПЗ показала следующее. Эти данные представлены в таблице №5.
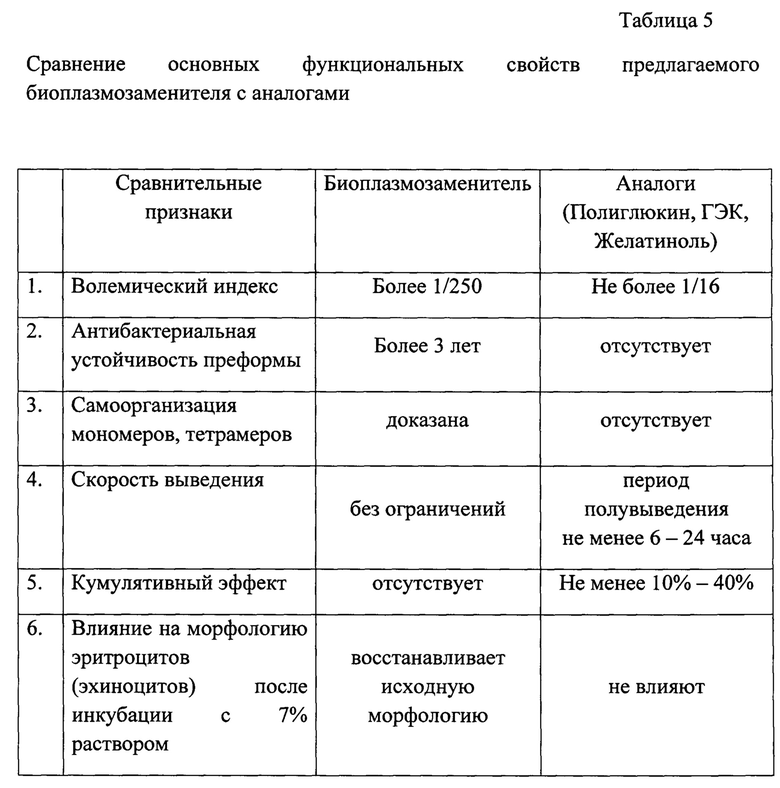

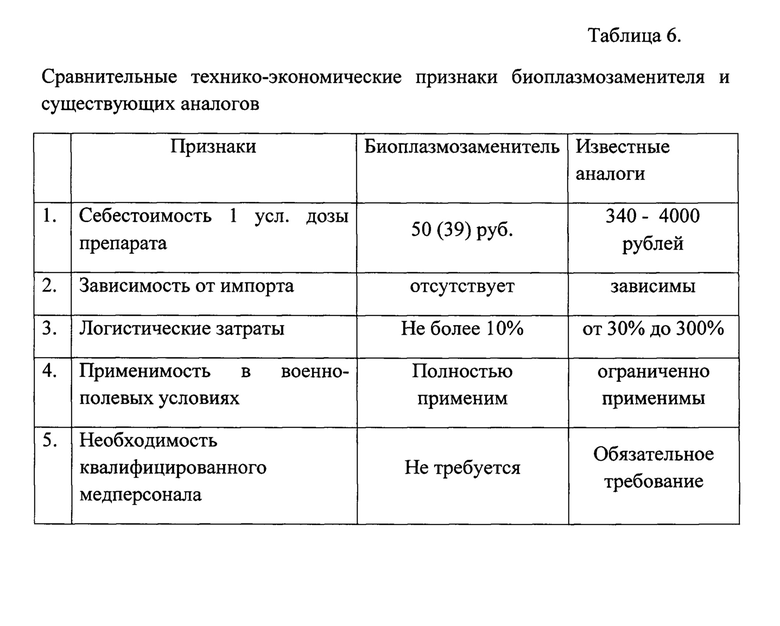
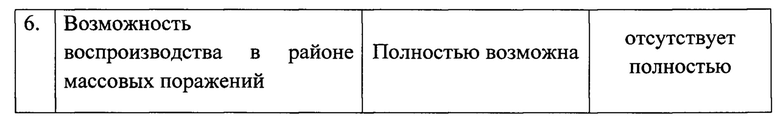
Представленные данные на таблицах №5 и №6 позволяют сделать заключение о том, что предлагаемый и прошедший предварительные испытания на крупных лабораторных животных БПЗ гемодинамического действия по основным сопоставимым признакам и их совокупности существенно превосходит известные зарубежные или отечественные плазмозаменители аналогичного действия.
Техническим результатом, достигаемым при использовании предложенного способа, является получение БПЗ на основе высокоочищенного ПГ-ГАГ, который может широко использоваться для получения фармацевтических субстанций и биопрепаратов, пригодных для парентерального применения.
Разработанный и заявляемый способ позволяет использовать описанный выше исходный продукт для приготовления БПЗ путем его очистки, достаточной для применения конечного продукта для парентерального введения при геморрагическом шоке, связанном с массивной кровопотерей и других видах шока. Получение БПЗ на основе ПГ-ГАГ представляет новый перспективный подход для патогенетического лечения этих состояний. Следует добавить, что в заявленных целях патента РФ №2621311(4) не было ссылок на возможность применения конечного продукта в качестве плазмозаменителя.
Технический результат предложенного способа, имеющий существенные преимущества перед уже известными аналогам и прототипом, заключается в следующем:
Технический результат предложенного способа.
1. Предложен способ получения БПЗ из биосырья, содержащего ПГ-ГАГ в растворимой гелевой форме, создающие гемодинамический эффект.
2. Полученный целевой продукт имеет высокую гидрофильность, позволяющую его использование для стабилизации онкотического давления за счет удержания воды и создания гемодинамического эффекта.
3. Способ позволяет применить физические способы очистки для удаления подавляющего количество сопутствующих примесей различных по составу нежелательных компонентов: липопротеидов, фосфолипидов, денатурированных белков и их дериватов, стероидных гормонов и пигментов до минимальных значений, которые уже не влияют на применение выделенных ПГ и ГАГ в качестве биоплазмозаменителя.
4. Способ позволяет исключить дополнительные процедуры, такие как ультрафильтрация, гельфильтрация, диализ и др. для очистки конечного продукта.
5. Способ позволяет осуществить выделение очищенных ПГ и ГАГ для биотехнологического промышленного производства.
6. Способ позволяет исключить использование опасных химических реактивов, а также их полное отсутствие на выходе в конечном продукте.
7. Способ приводит к снижению антигенности конечного продукта, за счет повышения степени очистки от примеси нативных и денатурированных белков, а также ЛПВП, ЛПНП и фосфолипидов, которые практически полностью удаляются, а примеси других нежелательных компонентов, таких как стероидные гормоны уменьшаются до следовых количеств, которые приемлемы для применения БПЗ.
8. Полученный целевой продукт имеет высокую биосовместимость, что позволяет снизить число осложнений при его использовании в виде БПЗ.
9. Полученный продукт в виде биопреформы, находящийся во флаконах стерилен и сохраняет биологическую активность в течение не менее 3 лет.
10. Технологический процесс заявляемого способа предусматривает применение веществ, способствующих стерилизации исходного и целевого продуктов и предотвращению в них микробной контаминации.
11. Способ простой в исполнении, безопасен, количество этапов минимально, а для его реализации не требуется дорогостоящего оборудования.
12. Исходный продукт и все реактивы для его применения полностью доступны и максимально дешевые.
13. Способ позволяет исключить процедуры синтеза или модификации уже существующих синтетических веществ, применяемых для производства плазмозаменителей.
14. Предлагаемый БПЗ не содержит растительных или синтетических веществ, используемых в качестве плазмозаменителей гемодинамического действия.
Настоящее изобретение промышленно применимо, освоено в лабораторных и полупромышленных условиях. Результаты предклинических испытаний показали высокую эффективность полученного БПЗ, превосходящую по большинству показателей существующие аналоги, основу которых представляют синтетические вещества или продукты растительного происхождения. Полученные положительные результаты предклинических испытаний препарата БПЗ на крупных лабораторных животных не включены в описание заявляемого способа получения БПЗ, поскольку касаются способа его применения.
Примеры реализации способа.
Пример 1. Для проведения испытаний на крупных лабораторных животных, полученную ex tempore мочу охлаждают в проточном режиме до -2°С со скоростью 20 мл/мин. Полученные на выходе криоагрегаты, не содержащие ПГ-ГАГ, отделяют от мочи фильтрацией в проточном режиме, а моча дополнительно охлаждается до -10°С с образованием в процессе охлаждения двух фракций, одна из них жидкая, представляет концентрат мочи, которую удаляют, а другая в твердом состоянии представляет лед, который размораживают до жидкого состояния при комнатной температуре. Для осаждения ПГ-ГАГ к размороженной фракции мочи добавляют осадитель в виде 1N раствора гидроксида натрия, в соотношении 10:1, смесь оставляют на 10 минут при комнатной температуре для образования преципитата. Затем преципитат осаждают центрифугированием при 1000 об/мин, в течение 30 секунд, отделяют надосадочную жидкость, а полученный осадок, содержащий ПГ-ГАГ, используют как исходный продукт для получения БПЗ.
После центрифугирования в осадке определяются две основные фракции, из которых верхняя (Ф1) и нижняя (Ф2). Для выделения верхней фракции Ф1 к осадку после центрифугирования добавляют раствор 0,5N гидроксида натрия в соотношении 1:1 и осторожно размешивают так, чтобы размешивание затронуло только верхнюю фракцию Ф1. После этого суспензию фракции Ф1 переносят в другую емкость, снабженную на дне выпускным штуцером и краном, в которую в соотношении 1:10 добавляют насыщенный раствор хлористого натрия в 0,5N растворе гидроксида натрия, перемешивают в течение 10 секунд и экспонируют в течение 10 минут. По истечении 10 минут преципитат поднимается вверх и располагается над жидкостью, которую сливают через штуцер открытием крана. Затем к преципитату добавляют раствор 0,5N гидроксида натрия в соотношении 1:1 и осторожно размешивают до образования однородной суспензии, которая представляет пребиоформу нерастворимой щелочной гелевой формы ПГ-ГАГ. Эту пребиоформу в количестве 70 мл в асептических условиях переносят в стерильные флаконы объемом 100 мл, герметично закрывают крышкой, представляющей устройство КУ. Перед непосредственным применением БПЗ готовят растворимую активную форму концентрата БПЗ, путем смешивания суспензии концентрата пребиоформы с нейтрализатором во флаконе с крышкой «КУ» способом, описанным в этом же патенте РФ №2709511(5). Для этого с соблюдением асептики снимают установленный сверху крышки защитный колпачок, надавливают на заглушку на штоке, который выталкивает пробку-дозатор с реагентом (нейтрализатором в виде порошка лимонной кислоты) во флакон с концентратом пребиоформы. После химической реакции нейтрализации реагента с консервантом (0,5N гидроксидом натрия) в емкости, шток возвращают в исходное положение. Далее снимают установленную на штоке «КУ» заглушку, вместо которой присоединяют внешние шприц или магистраль с аналогичным соединением типа Luer-lock. Отобранную из флакона шприцом или через внешнюю магистраль растворимую активную форму концентрата БПЗ переносят в емкость для инфузии с 0,9% раствором хлористого натрия с доведением концентрации БПЗ до 7%. С учетом веса животного и объема экссангвинированной крови (при моделировании острой кровопотери 50% ОЦК), в емкость для инфузии вводили раствор 0,9% хлористого натрия до 350 мл. В этом состоянии растворенный БПЗ соответствует понятию активная форма раствора биогеля БПЗ, и используется в эксперименте путем парентерального внутривенного введения.
Пример 2. Для проведения испытаний на крупных лабораторных животных, полученную ex tempore мочу охлаждают в проточном режиме до -2°С со скоростью 20 мл/мин. Полученные на выходе криоагрегаты, не содержащие ПГ-ГАГ, отделяют от мочи фильтрацией в проточном режиме, а моча дополнительно охлаждается до -10°С с образованием в процессе охлаждения двух фракций, одна из них жидкая, представляет концентрат мочи, которую удаляют, а другая, в твердом состоянии, представляет лед, который размораживают до жидкого состояния при комнатной температуре. Для осаждения ПГ-ГАГ к размороженной фракции мочи добавляют осадитель в виде 1N раствора гидроксида натрия, в соотношении 10:1, смесь оставляют на 10 минут при комнатной температуре для образования преципитата. Затем преципитат осаждают центрифугированием при 1000 об/мин, в течение 30 секунд, отделяют надосадочную жидкость, а полученный осадок, содержащий ПГ-ГАГ и используют как исходный продукт для получения БПЗ. После центрифугирования в осадке определяются две основные фракции, из которых верхняя (Ф1) и нижняя (Ф2). Для выделения верхней фракции Ф1 к осадку после центрифугирования добавляют раствор 0,5N гидроксида натрия в соотношении 1:1 и осторожно размешивают так, чтобы размешивание затронуло только верхнюю фракцию Ф1. После этого суспензию фракции Ф1 переносят в другую емкость, снабженную на дне выпускным штуцером и краном, в которую добавляют насыщенный раствор хлористого натрия в 0,5N растворе гидроксида натрия в соотношении 1:10, перемешивают в течение 10 секунд и экспонируют в течение 10 минут. За это время преципитат поднимается вверх и располагается над жидкостью, которую сливают через штуцер открытием крана. Затем к преципитату добавляют раствор 0,5N гидроксида натрия в соотношении 1:1 и осторожно размешивают до образования однородной суспензии, которая представляет пребиоформу нерастворимой щелочной гелевой формы ПГ-ГАГ. Эту пребиоформу переносят отдельную емкость, где проводят дегидратацию нерастворимой щелочной формы геля ПГ-ГА в виде концентрата пребиоформы с исходной рН10, путем добавления охлажденного до -2°С ацетона в соотношении 1:10, смесь экспонируют в течение 10 минут, при температуре -2°С до образования грубодисперсного преципитата, после чего центрифугируют при 1000 об/мин в течение 30 сек. Надосадочную жидкость удаляют, осадок разбавляют повторно охлажденным до -2°С ацетоном в соотношении 1:10, экспонируют не меняя температурный режим в течение 10 минут и повторно центрифугируют при 1000 об/мин в течение 30 сек. Надосадочную жидкость удаляют, к осадку добавляют 96° этиловый спирт в соотношении 1:1 и помещают в эксикатор под вакуумом на 2 часа при температуре 0°С. После вакуум экстракции образованный на дне эксикатора белый мелкодисперсный гигроскопичный сублимированный порошок БПЗ в количестве 1000 мг расфасовывают в стеклянный флакон объемом 40 мл и герметично укупоривают. Перед непосредственным применением во флакон с сублимированным порошком БПЗ вводят шприцом 20 мл 0,9% раствора хлористого натрия, осторожно перемешивают в течение 2 минут, после чего отбирают весь растворимый концентрат активного биогеля БПЗ и переносят его в емкость для инфузии.
С учетом веса животного и объема экссангвинированной крови (при моделировании острой кровопотери 50% ОЦК), в емкость для инфузии вводили 350 мл 0,9% раствора хлористого натрия для разведения в ней растворимого концентрата активного биогеля БПЗ, полученного из 1000 мг сублимированного БПЗ. В этом состоянии активная форма раствора биогеля БПЗ используется в эксперименте путем парентерального внутривенного введения.
Литература и другие источники информации
1. Г.Б. Аймухамедова, Н.П. Шелухина, Р.Ш. Шабаева, А.А. Алтымашев. Способ получения плазмозаменителя. Патент SU 539040 от 13.10.1975 г.
2. Г.Б. Аймухамедова, Н.П. Шелухина, Д.Э. Алиева. Способ получения пектата глицерина. Патент SU 584012 от 05.06.1977 г.
3. Б. Альберте, Д. Брей, Д. Льюис, М. Рэфф, К. Робертс, Д. Уотсон. Молекулярная Биология клетки. Т. 3, изд. Мир, 1978, 297 с.
4. Б.И. Асатуров. Способ получения протеогликана. Патент РФ №2621311 от 09 декабря 2015 г.
5. Б.И. Асатуров. Устройство для хранения, транспортировки, дезактивации и применения биологической жидкости. Патент РФ №2709511 от 25 марта 2019 г.
6. Т.Т. Березов, Б.Ф. Коровкин. Биологическая химия. 3 издание. Изд. Медицина, 2008, 704 с.
7. Б. Браун, А.Г. Мельзунген. Гидроксиэтилкрахмал. Патент DE №2373222, от 11.03.2019 г.
8. Голубев В.Н. Пектин: химия, технология, применение / В.Н. Голубев, Н.П. Шелухина. - Москва, 1995. - 317 с.
9. А.В. Крупин. «Медико-биологическое обоснование эффективности и безопасности восполнения объема циркулирующей крови холодными инфузионными растворами при острой кровопотере в чрезвычайных ситуациях (экспериментальное исследование)». Автореф. к.м.н., Санкт-Петербург, 2018.
10. В.В. Кузнецов. Физическая и коллоидная химия. Изд. 2е. Изд. «Высшая школа», Москва, 1968.
11. Е.П. Кухта, В.Я. Чирва, В.В. Шепа, A.M. Сабелко, М.П. Скрыльник. Способ получения пектина. Патент SU 1666457 А1.
12. А. Ленинджер. Основы биохимии: В 3-х т. Т. 2. Пер. с англ. - М.: Мир. 1985. Глава: Гликопротеины и протеогликаны. Р. Марри, стр. 307-308.
13. Наруми Масаки, Кудо Йосиаки. Способ получения протеогликана. Патент JP 2401839 от 14.02.2007 г.
14. Ольвекс диагностикум. Холестерин-ЛПНП-Ольвекс (013.004). Набор реагентов для определения концентрации липопротеинов низкой плотности (ЛПНП). https://www.olvex-d.ru/catalog/biohim-nabori/holesterin/holesterin-lpnp-olveks-013-0-id658/.
15. Ольвексдиагностикум. Холестерин-ЛПВП-Ольвекс (013.004). Набор реагентов для определения концентрации липопротеинов высокой плотности (ЛПВП). https://www.olvex-d.ru/catalog/biohim-nabori/holesterin/holesterin-lpvp-olveks-013-0-id43/.
16. Ольвексдиагностикум. Набор реагентов для определения растворимых фибрин-мономерных комплексов в плазме крови. https://www.olvex-d.ru/catalog/lab-obomdovanie/gemostaz/gemostaz-tehnologija-standart/rfmk-test-flakonnyi-variant-id424/.
17. П.Т. Петров, В.Н. Гапанович, В.М. Царенков, В.У. Заборонок, П. Лапковский, В.И. Тюрин, Т.Н. Забелло. Способ получения низкомолекулярного декстрана. Патент РФ 2039754 от 31.07.1992 г.
18. Письмо Федеральной службы по надзору в сфере здравоохранения от 10 июля 2013 г. N 16И-746/13 «О новых данных лекарственных препаратов гидроксиэтилкрахмала».
19. К.Г. Хлябич, Г.Т. Черненко, В.П. Мерзлов. Препарат гемодинамического действия с функцией нормализации кислотно-основного равновесия и электролитного баланса. Патент РФ 2185173.
20. R. Zarychanski, AM. Abou-Setta, A.F. Turgeon, Houston, BL; McIntyre, L; Marshall, JC; Fergusson, D.A. Association of hydroxy ethyl starch administration with mortality and acute kidney injury in critically ill patients requiring volume resuscitation: a systematic review and metaanalysis. // JAMA: the Journal of the American Medical Association: journal. - 2013. - 20 February, vol. 309, no. 7. - P. 678-688. - DOI:10.1001/jama.2013.430. - PMID 23423413.
21. P. Perel, I. Roberts, K. Ker. Colloids versus crystalloids for fluid resuscitation in critically ill patients. // The Cochrane database of systematic reviews: journal. - 2013. - 28 February (vol. 2). - P. CD000567. - DOI:10.1002/14651858.CD000567. pub6. - PMID 23450531.
22. J. Downar, S.E. Lapinsky. Pro/con debate: Should synthetic colloids be used in patients with septic shock? // Critical Care: journal. - 2009. - 29 January (vol. 13, no. 1). - P. 203. - DOI:10.1186/cc7147. - PMID 19226441.
23. http://www.nejm.org/doi/full/10.1056/NEJMoa1204242.
24. http://www.nejm.org/doi/full/10.1056/NEJMoa070716.
25. http://www.nejm.org/doi/full/10.1056/NEJMoa1209759.
26. http://www.bfarm.de/SharedDocs/1 Downloads/DE/BfArM/publ/bulletin/2013/1-2013.pdf? blob=publicationFile.
27. http://www.ema.europa.eu/ema/index.isp?curl=pages/medicines/human/referral s/Hydroxvethyl starchcontaining solutions/human referral prac 000012.jsp&mid=WC0b01ac05805c516f.



Группа изобретений относится к медицине, а именно к способам получения биоплазмозаменителя из биологического сырья. Способ получения биоплазмозаменителя из биологического сырья, включающий охлаждение полученной ex tempore мочи, её фильтрацию или центрифугирование в проточном режиме, далее охлаждают мочу до образования двух фракций, жидкой и твердой, твердую фракцию отделяют и размораживают до жидкого состояния, к размороженной фракции мочи добавляют раствор гидроксида натрия, выдерживают для образования преципитата, который осаждают центрифугированием, затем отделяют надосадочную жидкость, полученный осадок центрифугируют, верхнюю фракцию выделяют добавлением раствора гидроксида натрия, в суспензию верхней фракции приливают насыщенный раствор хлористого натрия в растворе гидроксида натрия и экспонируют, затем к полученному преципитату добавляют раствора гидроксида натрия до образования однородной суспензии нерастворимой щелочной гелевой формы протеогликанов и гликозаминогликанов, суспензию в асептических условиях переносят в стерильные флаконы, содержащие порошок лимонной кислоты, а перед непосредственным применением биоплазмозаменителя готовят его растворимую кисло-солевую форму, при определенных условиях (варианты). Вышеописанные способы позволяют получить высокоочищенный биоплазмозаменитель гемодинамического действия для парентерального введения. 2 н. и 3 з.п. ф-лы, 6 табл., 2 пр., 4 ил.
1. Способ получения биоплазмозаменителя из биологического сырья путем осаждения гидроксидом натрия, отличающийся тем, что полученную ex tempore мочу охлаждают в проточном режиме до -2°С со скоростью 20 мл/мин, полученные на выходе криоагрегаты отделяют от мочи фильтрацией или центрифугированием в проточном режиме, моча дополнительно охлаждается до -10°С с образованием в процессе охлаждения двух фракций, жидкой, которую удаляют, и твердой, которую размораживают до жидкого состояния при комнатной температуре, далее к размороженной фракции мочи добавляют 1N раствора гидроксида натрия, в соотношении 10:1, смесь оставляют на 10 минут при комнатной температуре для образования преципитата, который осаждают центрифугированием при 1000 об/мин в течение 30 секунд, затем отделяют надосадочную жидкость, а полученный осадок центрифугируют, верхнюю фракцию выделяют добавлением раствора 0,5N гидроксида натрия в соотношении 1:1, размешивают, и суспензию верхней фракции переносят в емкость, приливают насыщенный раствор хлористого натрия в 0,5N растворе гидроксида натрия в соотношении 1:10, перемешивают в течение 5-10 секунд и экспонируют в течение 10 минут, затем к полученному преципитату добавляют раствора 0,5N гидроксида натрия в соотношении 1:1, размешивают до образования однородной суспензии нерастворимой щелочной гелевой формы протеогликанов и гликозаминогликанов, суспензию в асептических условиях переносят в стерильные флаконы, содержащие порошок лимонной кислоты, а перед непосредственным применением биоплазмозаменителя готовят его растворимую кисло-солевую форму, растворимую форму биоплазмозаменителя переносят в емкость для инфузии с 0,9% раствором хлористого натрия с доведением концентрации биоплазмозаменителя до 7%.
2. Способ по п. 1, отличающийся тем, что вместо 0,9% раствора хлористого натрия применяются растворы других кристаллоидов, не содержащие органических веществ, которые могут влиять на свойства биоплазмозаменителя.
3. Способ по п. 1, отличающийся тем, что для охлаждения мочи с целью вымораживания криоконцентрата применяется любое устройство и способ, позволяющее ускорить и оптимизировать процесс вымораживания.
4. Способ получения биоплазмозаменителя из биологического сырья путем осаждения гидроксидом натрия, отличающийся тем, что полученную ex tempore мочу охлаждают в проточном режиме до -2°С со скоростью 20 мл/мин, полученные на выходе криоагрегаты отделяют от мочи фильтрацией или центрифугированием в проточном режиме, моча дополнительно охлаждается до -10°С с образованием в процессе охлаждения двух фракций: жидкой, которую удаляют, и твердой, которую размораживают до жидкого состояния при комнатной температуре, далее к размороженной фракции мочи добавляют 1N раствора гидроксида натрия в соотношении 10:1, смесь оставляют на 10 минут при комнатной температуре для образования преципитата, который осаждают центрифугированием при 1000 об/мин в течение 30 секунд, затем отделяют надосадочную жидкость, а полученный осадок центрифугируют, верхнюю фракцию выделяют добавлением раствора 0,5N гидроксида натрия в соотношении 1:1, размешивают и суспензию верхней фракции переносят в емкость, и приливают насыщенный раствор хлористого натрия в 0,5N растворе гидроксида натрия в соотношении 1:10, перемешивают в течение 5-10 секунд и экспонируют в течение 10 минут, затем к полученному преципитату добавляют раствора 0,5N гидроксида натрия в соотношении 1:1 размешивают до образования однородной суспензии нерастворимой щелочной гелевой формы протеогликанов и гликозаминогликанов, далее проводят дегидратацию нерастворимой щелочной формы суспензии, путем добавления охлажденного до -2°С ацетона в соотношении 1:10, смесь экспонируют в течение 10 минут при температуре -2°С, до образования преципитата, после чего центрифугируют при 1000 об/мин в течение 30 сек, надосадочную жидкость удаляют, осадок разбавляют повторно охлажденным до -2°С ацетоном в соотношении 1:10, экспонируют не меняя температурный режим в течение 10 минут и центрифугируют при 1000 об/мин в течение 30 сек, надосадочную жидкость удаляют, к осадку добавляют 96° этиловый спирт в соотношении 1:1, размешивают до однородной массы и помещают в эксикатор под вакуумом на 2 часа при температуре 0°С, порошок биоплазмозаменителя расфасовывают в стеклянные пенициллиновые флаконы.
5. Способ по п. 4, отличающийся тем, что предусматривается получение биоплазмозаменителя в лиофилизированном состоянии.
| СПОСОБ ПОЛУЧЕНИЯ ПРОТЕОГЛИКАНА | 2015 |
|
RU2621311C1 |
| УСТРОЙСТВО ДЛЯ ХРАНЕНИЯ, ТРАНСПОРТИРОВКИ, ДЕЗАКТИВАЦИИ КОНСЕРВАНТА И ПРИМЕНЕНИЯ БИОЛОГИЧЕСКОЙ ЖИДКОСТИ | 2019 |
|
RU2709511C1 |
| Способ получения плазмозаменителя | 1975 |
|
SU539040A1 |
| Способ получения плазмозаменителяраствора гемоглобина | 1974 |
|
SU629854A3 |
| US 20030153734 А1, 14.08.2003. | |||

