ПЕРЕКРЕСТНАЯ ССЫЛКА НА СВЯЗАННУЮ ЗАЯВКУ
Настоящая заявка испрашивает приоритет согласно заявкам на патент Китая №201610665625.9 и №201610666564.8, поданными в Национальное управление интеллектуальной собственностью Китая 12 августа 2016 года, обе которые включены в данную заявку посредством ссылки в своей полноте.
ОБЛАСТЬ ТЕХНИКИ
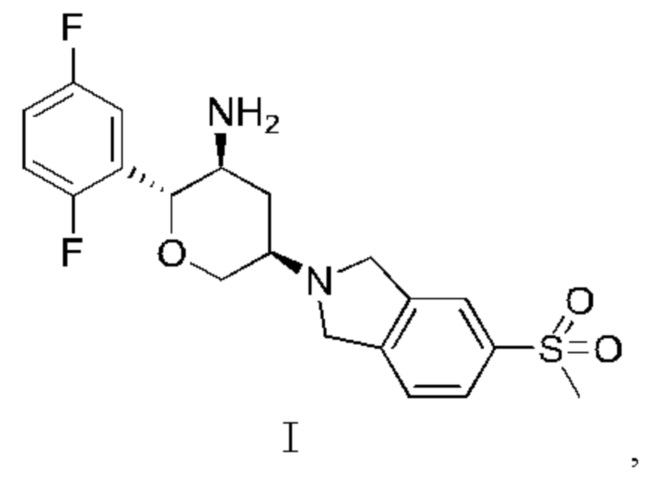
Настоящее изобретение относится к кристаллу (2R,3S,5R)-5-(5-метансульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амина в качестве ингибитора длительного действия, его соли или кристаллу соли, и к фармацевтической композиции, содержащей их, и к их медицинскому применению.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Дипептидилпептидаза-IV (DPP-IV) представляет собой сериновую протеазу, которая может быстро расщеплять белок, в котором аминокислота на N-конце пептидной цепи представляет собой пролин или аланин, и которая является ответственной за метаболической расщепление некоторых эндогенных пептидов (таких как GLP-1 и GIP) in vivo, и, как было показано, обладает протеолитической активностью в отношении множества других пептидов (GHRH, NPY, GLP-2 и VIP) in vitro. Вследствие разрушения ферментом DPP-IV GLP-1 и GIP быстро инактивируются in vivo, таким образом, ингибирование активности DPP-IV могло бы сильно продлить длительность физиологической активности GLP-1 и GIP in vivo, которая опосредованно регулируют секрецию инсулина и в конечном итоге играет роль в контроле уровня глюкозы в крови.
В качестве новых средств для лечения диабета ингибиторы DPP-IV могут глюкозозависимым образом стимулировать секрецию инсулина, не подвержены гипогликемическим побочным эффектам при контроле уровня глюкозы в крови и также имеют некоторые преимущества, такие как сохранение функции островковых β-клеток, обладают малым количеством желудочно-кишечных побочных эффектов, хорошей переносимостью и т.п. Ингибиторы DPP-IV могут быть введены перорально без необходимости инъекций и сравнимы по терапевтической эффективности с существующими гипогликемическими средствами.
Основываясь на вышеуказанных свойствах, ингибиторы DPP-IV являются полезными в лечении и/или профилактике DPP-IV-опосредованных заболеваний и расстройств, таких как диабет, ожирение и т.п., в частности, диабет II типа.
В WO 2016127916 раскрыты замещенные амино шестичленные насыщенные гетероалициклы в качестве ингибиторов DPP-IV длительного действия, включая соединение, представленное Формулой I, и способ его получения, который включен в данную заявку посредством ссылки в своей полноте:
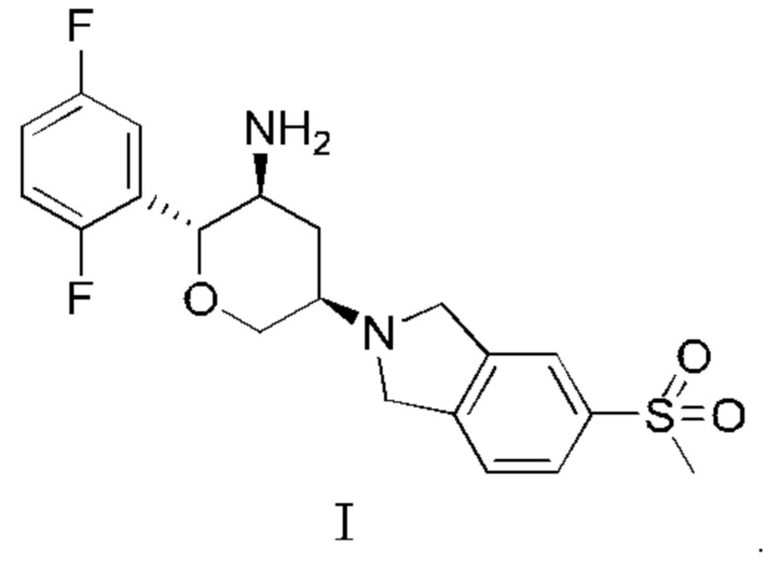
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном из аспектов в настоящем изобретении предложен кристалл соединения, представленного Формулой I, способ получения указанного кристалла, кристаллической композиции, содержащей указанный кристалл, фармацевтической композиции, содержащей указанный кристалл или указанную кристаллическую композицию, и их медицинское применение.
В другом аспекте в настоящем изобретении предложен фосфат соединения, представленного Формулой I, способ получения указанного фосфата, фармацевтическая композиция, содержащая указанный фосфат, и их медицинское применение.
В еще одном аспекте в настоящем изобретении предложен кристалл фосфата соединения, представленного Формулой I, способ получения указанного кристалла фосфата, кристаллическая композиция, включающая указанный кристалл фосфата, фармацевтическая композиция, содержащая указанный кристалл фосфата или указанную кристаллическую композицию, и их медицинское применение.
В еще одном аспекте в изобретении предложен фумарат соединения, представленного Формулой I, способ получения указанного фумарата, фармацевтическая композиция, содержащая указанный фумарат, и их медицинское применение.
В еще одном аспекте в настоящем изобретении предложен кристалл фумарата соединения, представленного Формулой I, способ получения указанного кристалла фумарата, кристаллическая композиция, содержащая указанный кристалл фумарата, фармацевтическая композиция, содержащая указанный кристалл фумарата или указанную кристаллическую композицию, и их медицинское применение.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящее описание включены определенные конкретные детали для обеспечения исчерпывающего понимания многочисленных раскрытых воплощений. Однако специалистам в релевантной области техники будет понятно, что воплощения могут быть осуществлены на практике без одной или более конкретных деталей, или с помощью других способов, компонентов, веществ и т.п.
Если иное явно не следует из контекста, в описании и формуле изобретения термин "содержать" и его вариации, такие как "содержит" и "содержащий", следует понимать в открытом и инклюзивном смысле, то есть, как "содержащий, но не ограничивающийся этим".
В настоящем описании ссылка на "одно из воплощений", или "воплощение", или "другое воплощение", или "некоторые воплощения" означает, что конкретный определяемый элемент, структура или характеристики, описанные в связи с воплощением, включены в по меньшей мере одно воплощение. Соответственно, фразы "в одном из воплощений", или "в воплощении", или "в другом воплощении", или "в некоторых воплощениях" в различных местах в настоящем описании не обязательно относятся к одному и тому же воплощению. Кроме того, конкретные элементы, структуры или характеристики могут быть объединены любым подходящим образом в одном или более воплощениях.
Следует отметить, что, как использовано в настоящем описании и формуле изобретения, формы единственного числа включают множественные определяемые объекты, если из контекста явно не следует обратное. Таким образом, например, ссылка на реакцию, в которой участвует "катализатор", включает единственный катализатор или два или более катализаторов. Если в данной заявке явным образом не указано иное, также следует отметить, что термин "или" обычно используют в смысле "и/или", если из контекста явно не следует обратное.
В одном из аспектов в настоящем изобретении предложен кристалл соединения, представленного Формулой I,

В некоторых воплощениях настоящего изобретения кристалл соединения, представленного Формулой I, имеет дифракционные пики в картине дифракции рентгеновских лучей (XRD) при 2θ, равных 16,4°, 21,8°, 25,3°, и 26,0°±0,2°; типично имеет дифракционные пики при 2θ, равных 16,4°, 19,4°, 21,2°, 21,8°, 25,3°, и 26,0°±0,2°, и более типично имеет дифракционные пики при 2θ, равных 13,0°, 16,4°, 18,5°, 19,4°, 21,2°, 21,8°, 25,3°, и 26,0°±0,2°.
В некоторых воплощениях настоящего изобретения пики дифракции рентгеновских лучей кристалла соединения, представленного Формулой I, имеют следующие характеристики:

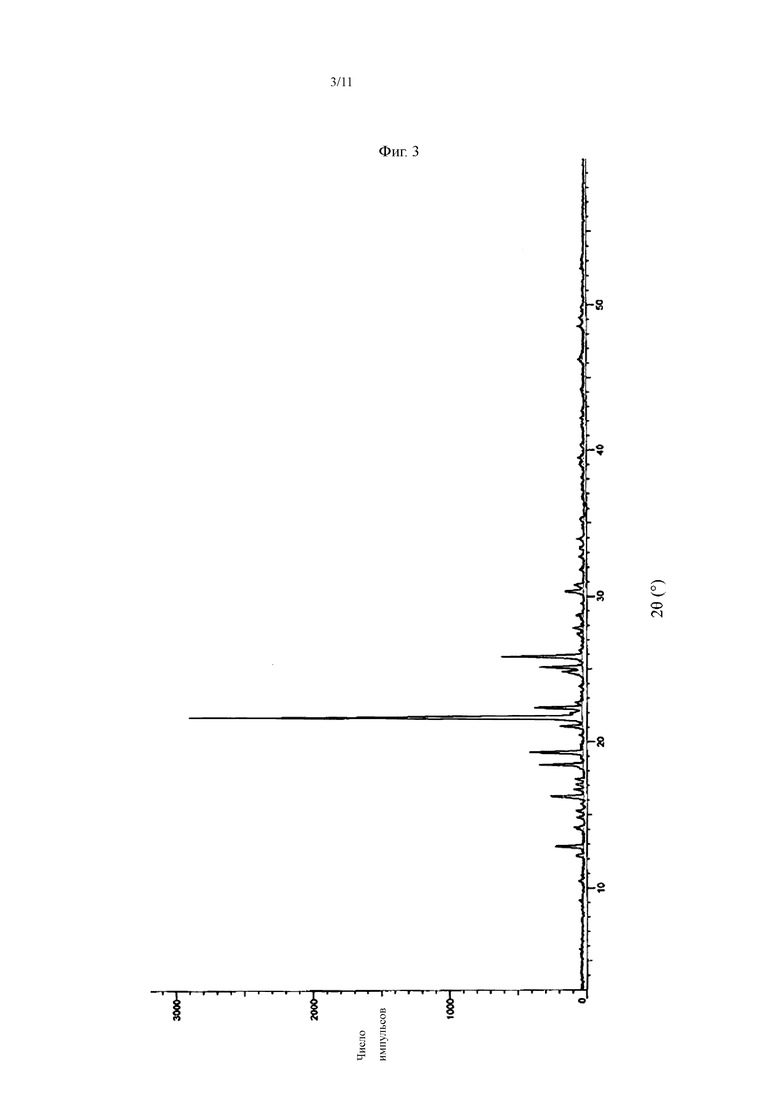
В некоторых воплощениях настоящего изобретения кристалл соединения, представленного Формулой I, имеет картину дифракции рентгеновских лучей, показанную на Фиг. 1, 3, 5 или 6. Из Фиг. 1, 3, 5 и 6 можно увидеть, что кристаллы соединения, представленные Формулой I, полученные в различных кристаллизационных растворителях, имеют по существу одинаковую картину дифракции рентгеновских лучей, и потому они имеют ту же кристаллическую форму.
В некоторых воплощениях настоящего изобретения кристалл соединения, представленного Формулой I согласно настоящей заявке, также может быть охарактеризован с помощью DSC (дифференциальная сканирующая калориметрия): температура начала фазового перехода составляет 193,3±5°С, и пиковая температура составляет 195,2±5°С.
В некоторых воплощениях настоящего изобретения кристалл соединения, представленного Формулой I, имеет картину DSC, как показано на Фиг. 2 или Фиг. 4.
В изобретении предложен способ получения кристалла соединения, представленного Формулой I, включающий:
1) растворение соединения, представленного Формулой I, в кристаллизационном растворителе;
2) охлаждение для кристаллизации и последующую фильтрацию.
В некоторых воплощениях настоящего изобретения кристаллизационный растворитель выбран из группы, состоящей из метанола, этанола, пропанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, ацетона, бутанона, этилацетата, ацетонитрила, дихлорметана, толуола, диоксана, н-гептана, н-гексана, метил-трет-бутило во го эфира, изо про пило во го эфира, изопропилацетата и их смеси.
В некоторых воплощениях настоящего изобретения кристаллизационный растворитель выбран из группы, состоящей из метанола, этанола, пропанола, изопропанола, этилацетата, ацетонитрила, дихлорметана и их смеси; предпочтительно представляет собой метанол.
В некоторых воплощениях настоящего изобретения количество добавленного кристаллизационного растворителя составляет 2 мл-100 мл, предпочтительно 20 мл, 30 мл, 40 мл, 50 мл, 60 мл, 70 мл, 80 мл, 90 мл или 100 мл, и более предпочтительно 20 мл-60 мл, 20 мл-40 мл, или 30 мл-50 мл, в расчете на 1 г соединения, представленного Формулой I.
В настоящем изобретении также предложен другой способ получения кристалла соединения, представленного Формулой I, включающий осаждение кристалла соединения, представленного Формулой I, из растворителя, содержащего метанол.
В настоящем изобретении также предложена кристаллическая композиция, содержащая кристаллическое соединение, представленное Формулой I. В некоторых воплощениях настоящего изобретения кристалл соединения, представленного Формулой I, составляет 50 масс. % или более, предпочтительно 80 масс. % или более, более предпочтительно 90 масс. % или более, и наиболее предпочтительно 95 масс. % или более, от массы кристаллической композиции.
В изобретении также предложена фармацевтическая композиция, содержащая кристаллическое соединение, представленное Формулой I, или кристаллическую композицию, содержащую кристаллическое соединение, представленное Формулой I. Кроме того, фармацевтическая композиция может содержать или может не содержать фармацевтически приемлемый носитель, эксципиент и/или среду.
В настоящем изобретении также предложено применение кристалла соединения, представленного Формулой I, или его кристаллической композиции, или его фармацевтической композиции, в изготовлении лекарственного средства для лечения или предотвращения заболевания, при котором приносит пользу ингибирование DPP-IV. В настоящем изобретении также предложен способ лечения или предотвращения заболевания, при котором приносит пользу ингибирование DPP-IV, включающий введение субъекту, нуждающемуся в этом, кристалла соединения, представленного Формулой I, или его кристаллической композиции, или его фармацевтической композиции. В настоящем изобретении также предложен кристалл соединения, представленного Формулой I, или его кристаллическая композиция, или его фармацевтическая композиция для применения в лечении или предотвращении заболевания, при котором приносит пользу ингибирование DPP-IV. В настоящем изобретении также предложено применение кристалла соединения, представленного Формулой I, или его кристаллической композиции, или его фармацевтической композиции, в лечении или предотвращении заболевания, при котором приносит пользу ингибирование DPP-IV.
В другом аспекте в изобретении предложен фосфат соединения, представленного Формулой I:
В некоторых воплощениях настоящего изобретения молярное соотношение соединения, представленного Формулой I, к ортофосфорной кислоте в фосфате соединения, представленного Формулой I, составляет 1:0,5-2, предпочтительно 1:0,5-1, и более предпочтительно 1:1.
В некоторых воплощениях настоящего изобретения фосфат соединения, представленного Формулой I, находится в кристаллической форме.
В настоящем изобретении также предложен способ получения фосфата соединения, представленного Формулой I, включающий приведение во взаимодействие соединения, представленного Формулой I, с ортофосфорной кислотой, и затем отделение от растворителя. В некоторых воплощениях настоящего изобретения растворитель выбран из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, дихлорметана, ацетонитрила, ацетона, этилацетата, изо пропил ацетата, 1,4-диоксана, н-гептана, н-гексана, метил-трет-бутилового эфира, изо про пило во го эфира, толуола и смеси двух или более из них, предпочтительно представляет собой этанол.
В другом аспекте в изобретении предложен кристалл фосфата соединения, представленного Формулой I:
В некоторых воплощениях настоящего изобретения кристалл фосфата соединения, представленного Формулой I, имеет дифракционные пики в картине дифракции рентгеновских лучей (XRD) при 2θ, равных 6,4°, 11,9°, 18,2°, 21,7°, 22,1°, 22,9°, и 23,2°±0,2°; типично имеет дифракционные пики при 2θ, равных 6,4°, 11,9°, 16,5°, 17,5°, 18,2°, 18,6°, 21,7°, 22,1°, 22,9°, и 23,2°±0,2°, и более типично имеет дифракционные пики при 2θ, равных 6,4°, 10,1°, 11,9°, 16,5°, 17,5°, 18,2°, 18,6°, 19,8°, 21,7°, 22,1°, 22,9°, 23,2°, и 23,8°±0,2°.
В некоторых воплощениях настоящего изобретения пики дифракции рентгеновских лучей кристалла фосфата соединения, представленного Формулой I, имеют следующие характеристики:

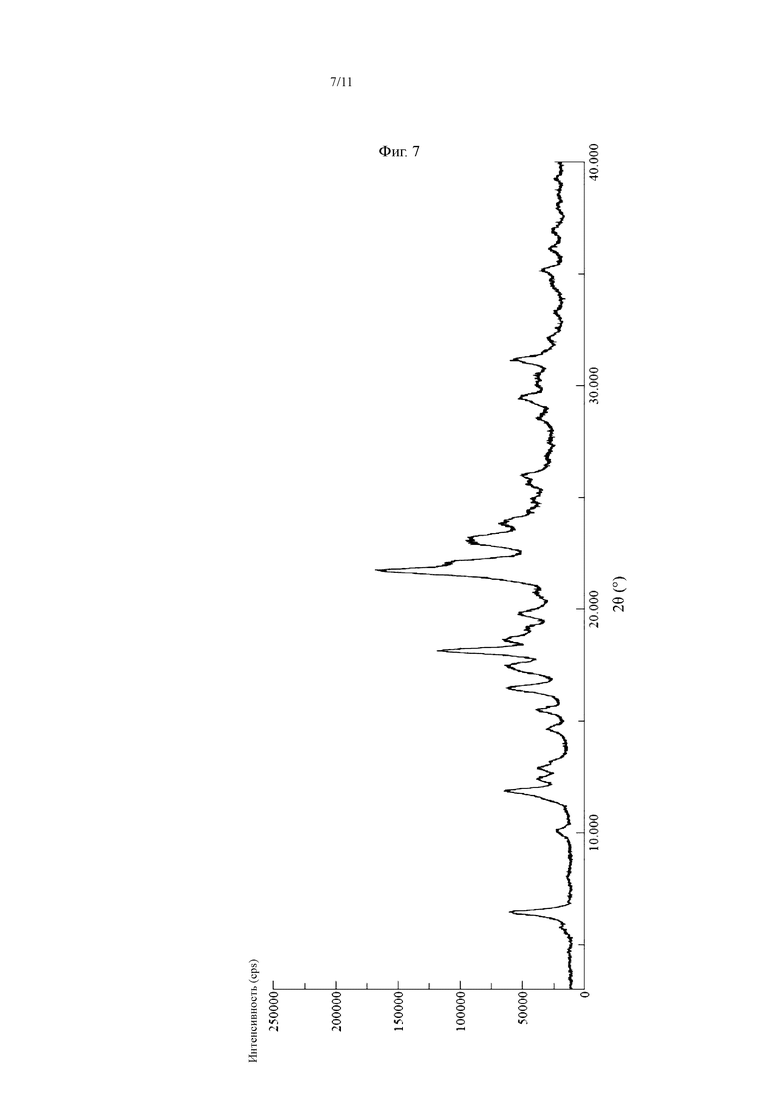
В некоторых воплощениях настоящего изобретения кристалл фосфата соединения, представленного Формулой I, имеет картину дифракции рентгеновских лучей, как показано на Фиг. 7.
В некоторых воплощениях настоящего изобретения кристалл фосфата соединения, представленного Формулой I, имеет картину DSC, как показано на Фиг. 8.
В настоящем изобретении также предложен способ получения кристалла фосфата соединения, представленного Формулой I, включающий приведение во взаимодействие соединения, представленного Формулой I, с ортофосфорной кислотой и затем кристаллизацию из растворителя. В некоторых воплощениях настоящего изобретения растворитель выбран из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, дихлорметана, ацетонитрила, ацетона, этилацетата, изопр опил ацетата, 1,4-диоксана, н-гептана, н-гексана, метил-трет-бутилового эфира, изо про пило во го эфира, толуола и смеси двух или более из них, предпочтительно представляет собой этанол.
В настоящем изобретении также предложена кристаллическая композиция, содержащая кристалл фосфата соединения, представленного Формулой I. В некоторых воплощениях настоящего изобретения кристалл фосфата соединения, представленного Формулой I, составляет 50 масс. % или более, предпочтительно 80 масс. % или более, более предпочтительно 90 масс. % или более, и наиболее предпочтительно 95 масс. % или более от массы кристаллической композиции.
В изобретении также предложена фармацевтическая композиция, содержащая фосфат соединения, представленного Формулой I, или кристалл фосфата соединения, представленного Формулой I, или кристаллическую композицию, содержащую кристалл фосфата соединения, представленного Формулой I. В некоторых воплощениях настоящего изобретения фармацевтическая композиция содержит терапевтически эффективное количество фосфата соединения, представленного Формулой I, или кристалла фосфата соединения, представленного Формулой I. Кроме того, фармацевтическая композиция может содержать или не содержать фармацевтически приемлемый носитель, эксципиент и/или среду.
В настоящем изобретении также предложено применение фосфата соединения, представленного Формулой I, или кристалла фосфата соединения, представленного Формулой I, или его кристаллической композиции, или его фармацевтической композиции в изготовлении лекарственного средства для лечения или предотвращения заболевания, при котором приносит пользу ингибирование DPP-IV. В настоящем изобретении также предложен способ лечения или предотвращения заболевания, при котором приносит пользу ингибирование DPP-IV, включающий введение субъекту, нуждающемуся в этом, фосфата соединения, представленного Формулой I, или кристалла фосфата соединения, представленного Формулой I, или его кристаллической композиции, или его фармацевтической композиции. В настоящем изобретении также предложен фосфат соединения, представленного Формулой I, или кристалл фосфата соединения, представленного Формулой I, или его кристаллическая композиция, или его фармацевтическая композиция для применения в лечении или предотвращении заболевания, при котором приносит пользу ингибирование DPP-IV. В настоящем изобретении также предложено применение фосфата соединения, представленного Формулой I, или кристалла фосфата соединения, представленного Формулой I, или его кристаллической композиции, или его фармацевтической композиции, в лечении или предотвращении заболевания, при котором приносит пользу ингибирование DPP-IV.
В другом аспекте в изобретении предложен фумарат соединения, представленного Формулой I:

В некоторых воплощениях настоящего изобретения молярное соотношение соединения, представленного Формулой I, к фумаровой кислоте в фумарате соединения, представленного Формулой I, составляет 1:0,5-2, предпочтительно 1:0,5-1, и более предпочтительно 1: 0,5.
В некоторых воплощениях настоящего изобретения фумарат соединения, представленного Формулой I, может быть в кристаллической форме.
В настоящем изобретении также предложен способ получения фумарата соединения, представленного Формулой I, включающий приведение во взаимодействие соединения, представленного Формулой I, с фумаровой кислотой, и затем выделение из растворителя. В некоторых воплощениях настоящего изобретения растворитель выбран из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, дихлорметана, ацетонитрила, ацетона, этилацетата, изо пропил ацетата, 1,4-диоксана, н-гептана, н-гексана, метил-трет-бутилового эфира, изо про пило во го эфира, толуола и смеси двух или более из них, предпочтительно, представляет собой этанол.
В другом аспекте в изобретении предложен кристалл фумарата соединения, представленного Формулой I:

В некоторых воплощениях настоящего изобретения кристалл фумарата соединения, представленного Формулой I, имеет дифракционный пик в картине дифракции рентгеновских лучей (XRD) при 2θ, равном 20,67°±0,2°. В некоторых воплощениях настоящего изобретения кристалл фумарата соединения, представленного Формулой I, имеет картину дифракции рентгеновских лучей, как показано на Фиг. 9.
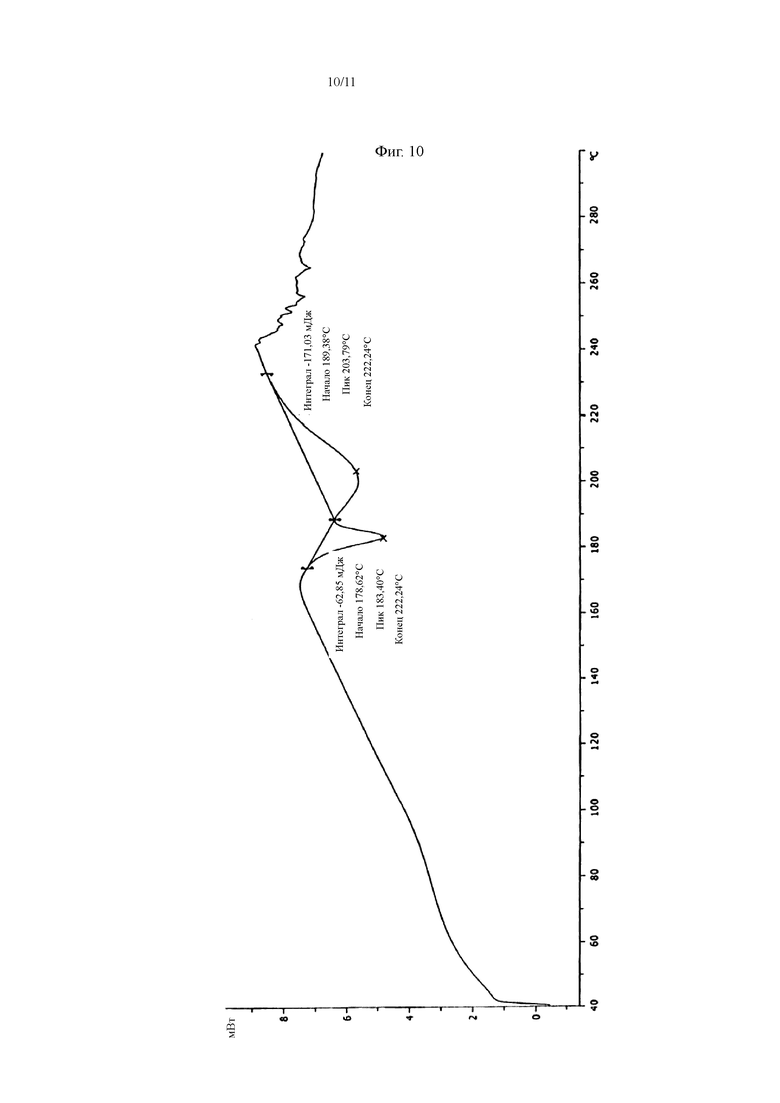
В некоторых воплощениях настоящего изобретения кристалл фумарата соединения, представленного Формулой I, имеет картину DSC, как показано на Фиг. 10.
В настоящем изобретении также предложен способ получения кристалла фумарата соединения, представленного Формулой I, включающий приведение во взаимодействие соединения, представленного Формулой I, с фумаровой кислотой и затем кристаллизацию из растворителя. В некоторых воплощениях настоящего изобретения растворитель выбран из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, дихлорметана, ацетонитрила, ацетона, этилацетата, изо пропил ацетата, 1,4-диоксана, н-гептана, н-гексана, метил-трет-бутилового эфира, изопропилового эфира, толуола и смеси двух или более из них, предпочтительно, представляет собой этанол.
В настоящем изобретении также предложена кристаллическая композиция, содержащая кристалл фумарата соединения, представленного Формулой I. В некоторых воплощениях настоящего изобретения кристалл фумарата соединения, представленного Формулой I, составляет 50 масс. % или более, предпочтительно 80 масс. % или более, более предпочтительно 90 масс. % или более, и наиболее предпочтительно 95 масс. % или более от массы кристаллической композиции.
В изобретении также предложена фармацевтическая композиция, содержащая фумарат соединения, представленного Формулой I, или кристалл фумарата соединения, представленного Формулой I, или кристаллическую композицию, содержащую кристалл фумарата соединения, представленного Формулой I. В некоторых воплощениях настоящего изобретения фармацевтическая композиция содержит терапевтически эффективное количество фумарата соединения, представленного Формулой I, или кристалла фумарата соединения, представленного Формулой I. Кроме того, фармацевтическая композиция может содержать или не содержать фармацевтически приемлемый носитель, эксципиент и/или среду.
В настоящем изобретении также предложено применение фумарата соединения, представленного Формулой I, или кристалла фумарата соединения, представленного Формулой I, или его кристаллической композиции, или его фармацевтической композиции, в изготовлении лекарственного средства для лечения или предотвращения заболевания, при котором приносит пользу ингибирование DPP-IV. В настоящем изобретении также предложен способ лечения или предотвращения заболевания, при котором приносит пользу ингибирование DPP-IV, включающий введение субъекту, нуждающемуся в этом, фумарата соединения, представленного Формулой I, или кристалла фумарата соединения, представленного Формулой I, или его кристаллической композиции, или его фармацевтической композиции. В настоящем изобретении также предложен фумарат соединения, представленного Формулой I, или кристалл фумарата соединения, представленного Формулой I, или его кристаллическая композиция, или его фармацевтическая композиция для применения в лечении или предотвращении заболевания, при котором приносит пользу ингибирование DPP-IV. В настоящем изобретении также предложено применение фумарата соединения, представленного Формулой I, или кристалла фумарата соединения, представленного Формулой I, или его кристаллической композиции, или его фармацевтической композиции, в лечении или предотвращении заболевания, при котором приносит пользу ингибирование DPP-IV.
В некоторых воплощениях настоящего изобретения заболевание, при котором приносит пользу ингибирование DPP-IV, выбрано из группы, состоящей из инсулинорезистентности, гипергликемии, диабета II типа, диабетической дислипидемии, нарушения переносимости глюкозы (IGT), нарушения гликемии натощак (IFG), метаболического ацидоза, кетоза, регуляции аппетита, ожирения, различных раковых заболеваний, неврологических расстройств, расстройств иммунной системы и тому подобного, предпочтительно представляет собой диабет II типа или ожирение.
В настоящем изобретении спектры дифракции рентгеновских лучей измеряют следующим способом: прибор: рентгеновский дифрактометр Bruker D8 ADVANCE; способ: мишень: Cu: K-альфа; длина волны 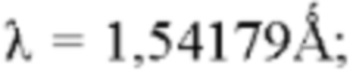 напряжение трубки: 40 кВ; ток трубки: 40 мА; диапазон сканирования: 4-40°; скорость вращения образцца: 15 об/мин; скорость сканирования: 10°/мин. Альтернативно, они также могут быть измерены следующим способом: прибор: рентгеновский дифрактометр Bruker D8 ADVANCE; способ: мишень: Cu; длина волны
напряжение трубки: 40 кВ; ток трубки: 40 мА; диапазон сканирования: 4-40°; скорость вращения образцца: 15 об/мин; скорость сканирования: 10°/мин. Альтернативно, они также могут быть измерены следующим способом: прибор: рентгеновский дифрактометр Bruker D8 ADVANCE; способ: мишень: Cu; длина волны 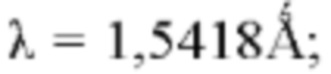 ; напряжение трубки: 40 кВ; ток трубки: 40 мА; диапазон сканирования: 3-40°; скорость сканирования: 0,1 сек/шаг, и 0,02°/шаг.
; напряжение трубки: 40 кВ; ток трубки: 40 мА; диапазон сканирования: 3-40°; скорость сканирования: 0,1 сек/шаг, и 0,02°/шаг.
В настоящем изобретении использован следующий способ дифференциальной сканирующей калориметрии (DSC): прибор: дифференциальный сканирующий калориметр ТА Q2000; способ: образец (~1 мг) помещают в алюминиевую емкость для DSC и измеряют при температуре от 25°С до 300°С при скорости нагрева 10°С/мин.
Соотношение соединения, представленного Формулой I, к соответствующей кислоте в фосфате или фумарате соединения, представленного Формулой I, согласно настоящей заявке, может быть измерено титрационным способом. Титратор: METTLER Т50; титрующий раствор: титрующий раствор 0,1 моль/л гидроксида натрия; титрующий растворитель: вода.
Следует отметить, что в спектре дифракции рентгеновских лучей дифракционная картина кристаллического соединения обычно является характеристической для конкретной кристаллической формы. Относительные интенсивности полос (особенно при малых углах) могут варьироваться в зависимости от предпочтительных ориентационных эффектов, происходящих из условий кристаллизации, размера частиц и других условий измерения. Таким образом, относительные интенсивности дифракционных пиков не являются характеристичными для конкретной кристаллической формы. Именно на относительные положения пиков, а не на их относительные интенсивности, следует обращать внимание при оценке того, является ли данная кристаллическая форма такой же, как известная кристаллическая форма. Кроме того, для любой заданной кристаллической формы может существовать небольшая ошибка в положении пиков, что также хорошо известно в области кристаллографии. Например, при измерении положение пика может сдвигаться вследствие изменения температуры, движения образца или калибровки прибора и тому подобного, и ошибка измерения значения 2θ иногда составляет примерно ± 0,2°. Соответственно, эту ошибку следует учитывать при идентификации структуры кристалла. Обычно положение пика выражается в терминах угла 2θ или периода решетки d в спектре XRD, и простое соотношение превращения между ними представляет собой
d=λ/2sinθ, где d представляет собой период решетки, λ представляет собой длину волны падающего рентгеновского луча, и θ представляет собой дифракционный угол. Для одной и той же кристаллической формы одного и того же соединения положение пиков в его спектре XRD в основном является сходным, и ошибка относительных интенсивностей может быть более большой. Кроме того, необходимо отметить, что вследствие некоторых факторов, таких как сниженное содержание, части дифракционных линий могут отсутствовать при идентификации смеси. При этом даже одна линия может быть характеристичной для заданной кристаллической формы вне зависимости от всех линий образца с высокой чистотой.
Следует отметить, что DSC используют для измерения температуры теплового перехода при поглощении или высвобождении тепла вследствие изменения кристаллической структуры или плавления кристалла. При непрерывном анализе одной и той же кристаллической формы одного и того же соединения ошибка температуры теплового перехода и температуры плавления обычно находится в диапазоне примерно ±5°С. Когда говорят, что данное соединение имеет данный пик DSC или температуру плавления, это значит, что пик DSC или температура плавления может варьироваться в пределах диапазона ±5°С. DSC дает дополнительный способ различения разных кристаллических форм. Различные кристаллические формы могут быть идентифицированы по их характеристически различным температурам тепловых переходов.
В настоящей заявке термин "фармацевтическая композиция" относится к композиции, которая содержит активное соединение согласно настоящему изобретению и носитель, эксципиент и/или среду, обычно применяемую в данной области для переноса биоактивного соединения в организм (например, человека). Цель фармацевтической композиции заключается в облегчении введения в организм соединения согласно настоящему изобретению.
В настоящей заявке термин "фармацевтически приемлемый носитель" относится к носителю и разбавителю, которые не оказывают значительного влияния на организм (например, человека), и не будут ухудшать биоактивность и свойства активного соединения. "Фармацевтически приемлемый эксципиент и/или носитель" относится к инертному веществу, которое вводят вместе с активным ингредиентом, и которое является полезным для введения активного ингредиента. "Фармацевтически приемлемый носитель, эксципиент, и/или среда" включает, но не ограничивается этим, любые носители, эксципиенты, среды, глиданты, подсластители, разбавители, консерванты, пигменты/красители, корригенты, поверхностно-активные вещества, увлажняющие вещества, диспергирующие агенты, разрыхлители, суспендирующие агенты, стабилизаторы, изотонические агенты, растворители и эмульгаторы и т.п., которые являются приемлемыми для применения у людей или животных (таких как сельскохозяйственные животные). Неограничивающие примеры эксципиента включают карбонат кальция, фосфат кальция, различных сахара и крахмалы, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли, и т.п.
Соединение согласно настоящему изобретению или его соли, или его кристаллы, или его кристаллические композиции, могут быть введены в их чистых формах или в форме подходящих фармацевтических композиций любыми приемлемыми путями введения лекарственного средства, которое имеет сходное применение. Фармацевтические композиции согласно настоящему изобретению могут быть получены путем объединения соединения согласно настоящему изобретению или его солей, или его кристаллов, или его кристаллических композиций, с подходящим фармацевтически приемлемым носителем, разбавителем, средой или эксципиентом. Фармацевтические композиции согласно настоящему изобретению могут быть изготовлены в виде твердых, полутвердых, жидких и газообразных композиций, таких как таблетки, пилюли, капсулы, порошки, гранулы, мази, эмульсии, суспензии, растворы, суппозитории, инъекционные препараты, ингаляционные препараты, гели, микросферы, аэрозоли и т.п.
Типичные пути введения соединения согласно настоящему изобретению или его солей, или его кристаллов, или его кристаллических композиций, или его фармацевтических композиций, включают, но не ограничиваются этим, пероральное, ректальное, чресслизистое, кишечное введение, или местное, чрескожное, ингаляционное, парентеральное, подъязычное, интравагинальное, интраназальное, интраокулярное, внутрибрюшинное, внутримышечное, подкожное, внутривенное введение и т.п. Предпочтительным путем введения является пероральное введение.
Фармацевтические композиции согласно настоящему изобретению могут быть получены с использованием способов, хорошо известных специалистам в данной области техники, таких как традиционный способ смешивания, способ растворения, способ гранулирования, способ получения драже, способ размалывания, способ эмульгирования, способ сублимационной сушки и т.п.
В предпочтительных воплощениях фармацевтические композиции находятся в пероральной форме. Для перорального введения фармацевтические композиции могут быть приготовлены путем смешивания активного соединения с фармацевтически приемлемым носителем, эксципиентом и/или средой, хорошо известными в данной области. Такой носитель, эксципиент и среда позволяют приготавливать соединение согласно настоящему изобретению или его соли, или его кристаллы, или его кристаллические композиции, в виде таблеток, пилюль, пастилок, драже, капсул, жидкостей, гелей, сиропов, суспензий и т.п., для перорального введения пациентам.
Твердая пероральная фармацевтическая композиция может быть получена путем традиционного способа смешивания, заполнения или таблетирования. Например, она может быть получена путем смешивания активного соединения с твердым эксципиентом, необязательно размалывания полученной смеси, добавления других подходящих эксципиентов, если это необходимо, и затем переработки смеси в гранулы для получения таблеток или ядер драже. Подходящие эксципиенты включают, но не ограничиваются этим, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; целлюлозы, такие как микрокристаллическая целлюлоза, кукурузный крахмал, пшеничный крахмал, рисовый крахмал и картофельный крахмал; и также могут быть использованы другие вещества, такие как пектин, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза натрия и/или поливинилпирролидон; разрыхлители, такие как карбоксиметилкрахмал натрия, кросс-сшитая карбоксиметилцеллюлоза натрия, кросс-сшитый поливинилпирролидон, агар или альгиновая кислота, и соль, такая как альгинат натрия. Ядра драже могут быть необязательно покрыты покрытием согласно хорошо известным способам в фармацевтической практике, в частности, с использованием кишечно-растворимого покрытия.
Все растворители, использованные в настоящей заявке, доступны в продаже и могут быть использованы без дальнейшей очистки. Реакцию обычно осуществляют в инертной атмосфере, такой как атмосфера азота, и в безводном растворителе.
Кристалл соединения, представленного Формулой I, предложенный в настоящей заявке, имеет одно или более преимуществ, таких как высокая чистота, высокая степень кристалличности, хорошая стабильность и т.д. Кроме того, способ получения кристалла соединения, представленного Формулой I, предложенный в настоящей заявке, имеет одно или более преимуществ, таких как упрощенный процесс, недорогой и легко доступный растворитель, мягкие условия кристаллизации и т.д., а также является подходящим для промышленного производства. Способ получения солей соединения, представленного Формулой I, предложенный в настоящей заявке, легок в осуществлении, и полученные коли соединения, представленного Формулой I, имеют высокую чистоту и хорошие фармакокинетические свойства, а также являются подходящими для получения в виде требуемой фармацевтической композиции.
ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 показана картина XRD кристалла соединения, представленного Формулой I.
На Фиг. 2 показана картина DSC кристалла соединения, представленного Формулой I.
На Фиг. 3 показана картина XRD кристалла соединения, представленного Формулой I.
На Фиг. 4 показана картина DSC кристалла соединения, представленного Формулой I.
На Фиг. 5 показана картина XRD кристалла соединения, представленного Формулой I.
На Фиг. 6 показана картина XRD кристалла соединения, представленного Формулой I.
На Фиг. 7 показана картина XRD кристалла фосфата соединения, представленного Формулой I.
На Фиг. 8 показана картина DSC кристалла фосфата соединения, представленного Формулой I.
На Фиг. 9 показана картина XRD кристалла фумарата соединения, представленного Формулой I.
На Фиг. 10 показана картина DSC кристалла фумарата соединения, представленного Формулой I.
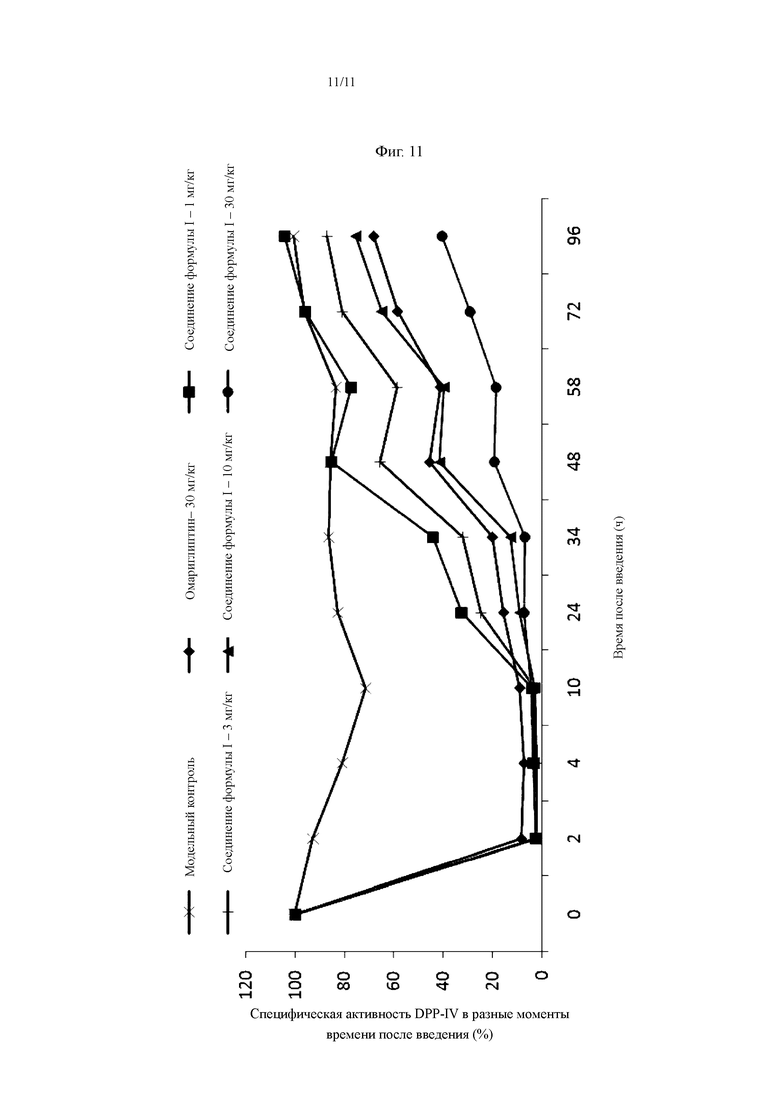
На Фиг. 11 показан ингибирующий эффект соединения, представленного Формулой I, на активность DPP-IV в сыворотке у мышей ob/ob.
ПРИМЕРЫ
Настоящее изобретение проиллюстрировано ниже со ссылкой на конкретные примеры, однако эти конкретные примеры не ограничивают объем настоящей заявки.
Пример 1: 5-Метансульфонилизоиндолина гидрохлорид (2)

Стадия 1: 5-бромизоиндолин (4)
К соединению, представленному Формулой 3 (22,6 г, 100 ммоль), в безводном тетрагидрофуране (250 мл) по каплям добавляли боран-диметилсульфидный комплекс (51 мл, 500 ммоль), перемешивали в течение 2 часов при комнатной температуре и затем нагревали с обратным холодильником в течение ночи. После охлаждения аккуратно по каплям добавляли метанол для гашения избытка борана. Полученную смесь упаривали и концентрировали, и затем остаток очищали посредством колоночной хроматографии на силикагеле с получением 5-бромизоиндолина (10,36 г). Выход: 52%. MS m/z [ESI]: 198.0 [М+1].
Стадия 2: 5-бром-2-трет-бутоксикарбонилизоиндолин (5)
Соединение, представленное Формулой 4 (10,36 г, 52,3 ммоль), растворяли в 80 мл дихлорметана и охлаждали на ледяной бане. По каплям добавляли Вое ангидрид (22,8 г, 104,6 ммоль), затем добавляли карбонат натрия (16,6 г, 156,9 ммоль) и воду (150 мл) и перемешивали в течение 4 часов на ледяной бане. Органическую фазу отделяли, промывали рассолом и концентрировали, и затем остаток очищали с помощью колоночной хроматографии на силикагеле с получением продукта 5-бром-2-трет-бутоксикарбонилизоиндолина (13,3 г). Выход: 85%. MS m/z [ESI]: 298.0 [М+1]. 1H-ЯМР (400 МГц, CDCl3): δ=7.37 (2Н, m), 7.11 (1H, m), 4.62 (4Н, m), 1.51 (9Н, s).
Стадия 3: 5-метансульфонил-2-трет-бутоксикарбонилизоиндолин (6)
Соединение, представленное Формулой 5 (5,96 г, 20 ммоль), метилсульфинат натрия (90%, 2,94 г, 26 ммоль), йодид меди (762 мг, 4 ммоль) и L-пролин (920 мг, 8 ммоль) добавляли к диметилсульфоксиду (80 мл), продували азотом для удаления воздуха и перемешивали в течение 2 суток при 120°С. После охлаждения полученную смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу сушили, упаривали и концентрировали, и затем остаток очищали с помощью хроматографии на силикагеле с получением 5-метансульфонил-2-трет-бутоксикарбонилизоиндолина (5,46 г). Выход: 92%. MS m/z [ESI]: 298.1 [М+1].
Стадия 4: 5-метансульфонилизоиндолина гидрохлорид (2)
Раствор соединения, представленного Формулой 6 (5,46 г, 18,4 ммоль) в смеси метанол/дихлорметан (1:1, 80 мл) продували газообразным хлористым водорода до насыщения и перемешивали в течение 1 часа при комнатной температуре. После того, как реакционную смесь выливали в 800 мл этилового эфира, остаток собирали путем фильтрации, промывали этиловым эфиром и сушили с получением продукта 5-метансульфонилизоиндолина гидрохлорида (3,44 г). Выход: 80%. MS m/z [ESI]: 198.0 [М+1]. 1H-ЯМР (400 МГц, CDCl3): δ=7.82 (1H, s), 7.81 (1Н, d, J=8,0 Гц), 7.43 (1Н, d, J=8,0 Гц), 4.31 (4Н, s), 3.05 (3Н, s), 2.30 (2Н, brs).
Пример 2: неочищенный продукт (2R,3S,5R)-5-(5-метансульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
Стадия 1: трет-бутил-(2R,3S,5R)-5-(5-метансульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат (8)

К 2,25 л растворителя N,N-диизопропилацетамида добавляли соединение, представленное Формулой 7 (150 г, 458,27 ммоль), и соединение, представленное Формулой 2 (117,82 г, 504,09 ммоль), равномерно перемешивали и охлаждали до -10°С, и затем к реакционной системе медленно по каплям добавляли уксусную кислоту (26,26 мл, 458,27 ммоль). После того, как добавление завершали, добавляли NaBH(AcO)3 (194,25 г, 916,54 ммоль), и затем полученную смесь подвергали реакции в течение 1 ч при перемешивании, поддерживая эту температуру. Температуру контролировали ниже 20°С, и реакционную систему доводили до рН=10 водным раствором аммиака, перемешивали в течение 15 мин и затем фильтровали под вакуумом. Осадок на фильтре суспендировали и промывали очищенной водой и затем фильтровали под вакуумом. Полученный осадок на фильтре сушили сжатым воздухом при 60°C с получением 223,5 г соединения, представленного Формулой 8. Выход: 95%.
Стадия 2: неочищенный продукт (2R,3S,5R)-5-(5-метансульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин (I)

К 1,2 л растворителя, смешанного из N,N-диизопропилацетамида и очищенной воды (об./об.=1/1) добавляли соединение, представленное Формулой 8 (202 г, 396,44 ммоль), и равномерно перешивали и к полученной смеси медленно по каплям добавляли раствор серной кислоты (1,1 л, 5,95 моль). После того, как добавление завершали, реакционную систему нагревали до 40°С и проводили реакцию в течение 2 ч при перемешивании. Затем полученный раствор доводили приблизительно до рН=10 путем добавления по каплям водного раствора аммиака. После того, как добавление по каплям завершали, полученную смесь перемешивали в течение 1 ч и затем фильтровали под вакуумом. Осадок на фильтре промывали очищенной водой и затем сушили сжатым воздухом при 60°C с получением 133,1 г соединения, представленного Формулой I, в виде неочищенного продукта. Выход: 83%.
Пример 3: Кристалл (2R,3S,5R)-5-(5-метансульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амина (I)
Способ I
130 г неочищенного продукта добавляли к 650 мл безводного метанола, нагревали и растворяли с получением прозрачного раствора. Затем раствор обесцвечивали с помощью активированного угля и фильтровали в горячем виде под вакуумом. Фильтрат охлаждали до комнатной температуры, кристаллизовали в течение 2 ч и затем фильтровали под вакуумом. Осадок на фильтре сушили сжатым воздухом при 60°C с получением 94,2 г кристалла. Выход: 72,4%.
Способ II
Безводный метанол (26,8 л) нагревали до температуры флегмообразования и затем к нему добавляли неочищенный продукт (670 г), растворяли и фильтровали. Фильтрат охлаждали до значений от ~5°С до 5°С, кристаллизовали в течение 1 ч и затем фильтровали под вакуумом. Осадок на фильтре промывали безводным метанолом и сушили сжатым воздухом при 50°С-60°С в течение 10-12 ч с получением 528 г кристалла. Выход: 78%.
Типичная картина XRD кристалла, полученного способом I с использованием метанола в качестве кристаллизационного растворителя, показана на Фиг. 1, а картина DSC показана на Фиг. 2.
Другая типичная картина XRD кристалла, полученного способом II, с использованием метанола в качестве кристаллизационного растворителя показана на Фиг. 3, а картина DSC показана на Фиг. 4.
Со ссылкой на процедуру, сходную с процедурой в Способе I или Способе II в Примере 3, кристаллизационный растворитель заменяли, и полученные кристаллы показаны в таблице ниже.
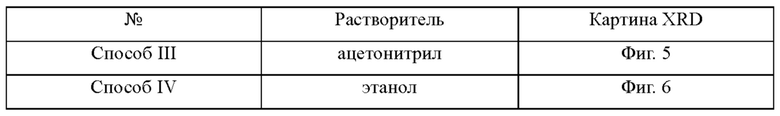
В Примере 3 все кристаллы соединения, представленного Формулой I, полученные с использованием различных кристаллизационных растворителей, принадлежат к одной и той же кристаллической форме.
Пример 4: Кристалл фосфата (2R,3S,5R)-5-(5-метансульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амина

Соединение, представленное Формулой I (7 г, 17,1 ммоль), добавляли к 350 мл растворителя этанола, нагревали до температуры флегмообразования и растворяли с получением прозрачного раствора. Затем полученную смесь обесцвечивали в течение 10 мин с помощью 1,0 г активированного угля и фильтровали в горячем виде под вакуумом. Затем к фильтрату по каплям добавляли раствор ортофосфорной кислоты (1,8 мл, 34,2 ммоль), и осаждалось большое количество белого твердого вещества. После того, как добавление по каплям завершали, реакционную систему охлаждали до комнатной температуры и перемешивали в течение 2 ч, и твердое вещество непрерывно осаждалось. Полученную смесь фильтровали под вакуумом и твердое вещество сушили сжатым воздухом в течение ночи с получением 8,3 г кристалла фосфата соединения, представленного Формулой I, (1:1, измерено способом титрования). Выход: 95,8%, Чистота: 99,12%. Полученный продукт имел типичную картину XRD, как показано на Фиг. 7, и картину DSC, как показано на Фиг. 8.
Пример 5: Фумарат (2R,3S,5R)-5-(5-метансульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амина
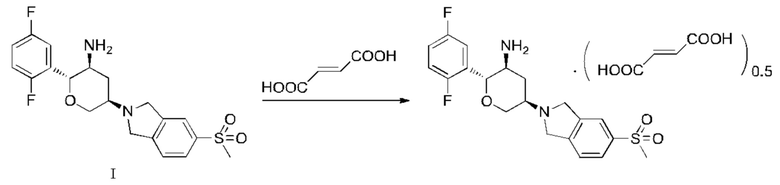
Соединение, представленное Формулой I (7 г, 17,1 ммоль), добавляли к 350 мл растворителя этанола, нагревали до температуры флегмообразования и растворяли с получением прозрачного раствора. Затем полученную смесь обесцвечивали в течение 10 мин с помощью 1,0 г активированного угля и фильтровали в горячем виде под вакуумом. Затем к фильтрату добавляли фумаровую кислоту (3,97 г, 34,2 ммоль). После того, как добавление завершали, реакционную систему охлаждали до комнатной температуры и затем перемешивали в течение 2 ч на водо-ледяной бане, и твердое вещество осаждалось. Полученную смесь фильтровали под вакуумом, и твердое вещество сушили сжатым воздухом в течение 6 ч при 50°C с получением 7,2 г фумарата соединения, представленного Формулой I, (1:0,5, как измерено способом титрования). Выход: 80,3%, Чистота: 97,7%. Полученный продукт имел типичную картину XRD, как показано на Фиг. 9, и картину DSC, как показано на Фиг. 10.
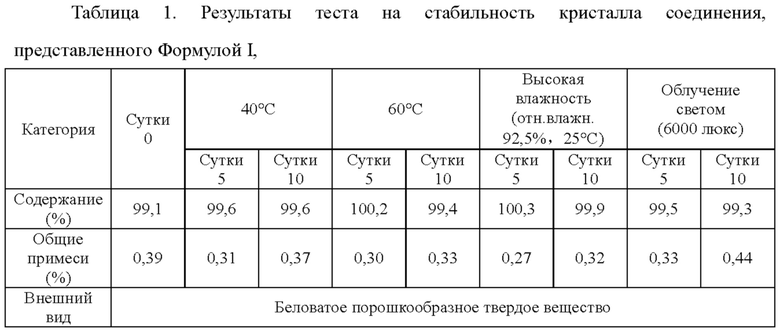
Экспериментальный пример 1: Исследование стабильности кристалла соединения, представленного Формулой I,
Стабильность кристалла соединения, представленного Формулой I, согласно настоящей заявке, при температуре 40°С или 60°С, или при высокой влажности (отн. влажн. 92,5%), или в условиях облучения светом тестировали в соответствии с "Руководством по исследованиям на стабильность активных фармацевтических ингредиентов и фармацевтических препаратов" (Фармакопея Китая, издание 2010 года, Приложение XIXC). Образцы отбирали и тестировали на сутки 5 или сутки 10, соответственно, и результаты сравнивали с исходными результатами. Результаты исследования показаны в Таблице 1 ниже.
Экспериментальный пример 2: Фармакокинетика соединения, представленного Формулой I, и его солей в кристаллической форме
Самцов собак Бигль (масса тела 10±1 кг) случайным образом делали на 3 группы (по 3 собаки в группе) по прошествии 7 суток адаптации, и вводили кристалл соединения, представленного Формулой I, кристалл фумарата соединения, представленного Формулой I, и кристалл фосфата соединения, представленного Формулой I, в дозировке 2 мг/кг массы тела (в свободной форме), соответственно.
Самцов собак Бигль не кормили в течение примерно 12 ч перед введением, и у них был свободный доступ к воде. Собак также не кормили в течение 4 часов после введения. Образцы крови (0,8 мл) отбирали из вены в передней конечности подопытных собак Бигль через 0,25, 0,5, 1, 2, 4, 6, 8, 10, 24, 30, 48 и 72 ч после введения. Затем образцы помещали в пробирки для центрифугирования EDTA-K2, хранили при 4°С и центрифугировали в течение 10 мин со скоростью 4000 об/мин при 4°С в течение 0,5 ч после забора крови для отделения плазмы. Плазму хранили при -20°С в течение 1 ч после сбора всей плазмы.
300 мкл раствора вещества внутреннего стандарта в метаноле добавляли к 50 мкл образца плазмы для тестирования и в образец для калибровочной кривой, соответственно. Полученную смесь равномерно перемешивали путем встряхивания в течение 5 мин и центрифугировали в течение 10 мин со скоростью 13000 об/мин. Затем отбирали 80 мкл надосадочной жидкости и 5 мкл надосадочной жидкости отбирали пипеткой для определения с помощью LC/MS/MS (жидкостная хроматография/масс-спектрометрия/масс-спектрометрия), и регистрировали хроматограмму.
Пероральная биодоступность соединения, представленного Формулой I, согласно настоящему изобретению, и его солей была оценена в ходе фармакокинетического эксперимента in vivo на собаках Бигль. Фармакокинетические параметры соединения, представленного Формулой I, и его солей показаны в таблице ниже.

Экспериментальный пример 3: Определение ингибирующей активности по отношению к ферменту DPP-IV
Ингибирующую активность соединения, представленного Формулой I, согласно настоящего изобретения, в отношении фермента DPP-IV в плазме определяли с использованием следующего способа. Ингибирующую активность выражали как значения IC50, т.е., концентрацию соединения, требуемую для достижения 50%-ного ингибирования активности фермента DPP-IV.
Материалы и методы:
Материалы:
a. Белый 384-луночный планшет (Perkin Elmer, каталожный №607290/99)
b. Буфер HEPES: использование 1М буфера HEPES (Invitrogen, каталожный №15630-080) для получения 50 мл 0,5М HEPES буфера в результате стадий взятия 25 мл 1 М буфера HEPES, добавления подходящего количества ddH2O (бидистиллированной воды), доведения рН до 7,8 с помощью NaOH, и наконец добавления ddH2O до 50 мл.
c. Плазма крыс: отбор образцов крови из глазницы крыс, добавление гепарина для антикоагуляции, центрифугирование в течение 10 минут при 4000 об/мин, отбор надосадочной плазмы, являющейся источником фермента DPP-IV.
d. H-Gly-Pro-AMC (глицин-пролин-7-амино-4-метилкумарин) в качестве реакционного субстрата DPP-IV, который синтезировал один из авторов изобретения, растворяли в DMSO с получением маточного раствора 100 мМ.
e. 1М MgCl2
f. 1,5M NaCl
g. 10% BAS
h. DMSO (диметилсульфоксид)
i. ddH2O
j. Тестовые соединения: Омариглиптин в качестве соединения положительного контроля и соединение, представленное Формулой I, согласно настоящему изобретению. Придерживались следующей последовательности действий:
1. Получали реакционный буфер фермента DPP-IV (50 мМ HEPES (рН=7,8), 80 мМ MgCl2, 150 мМ NaCl, 1% BSA) и хранили на льду для использования;
2. Тестовые соединения разбавляли DMSO от 10 мМ до 1 мМ (100-кратная конечная рабочая концентрация) и затем градиентно разбавляли 3-кратно в 96-луночном планшете с получением 11 концентраций; DMSO добавляли к двенадцатой лунке в качестве холостого контроля, и затем разбавляли в 25 раз реакционным буфером фермента вплоть до 4-кратной конечной концентрации для использования;
3. Реакционный субстрат фермента DPP-IV H-Gly-Pro-AMC растапливали и разбавляли до 160 мкМ (4-кратная рабочая концентрация) реакционным буфером фермента, и затем хранили на льду для использования;
4. Плазму крыс растапливали и разбавляли 100-кратно (2-кратная рабочая концентрацияы) реакционным буфером фермента, и затем хранили на льду для использования;
5. 5 мкл тестовых соединений (4-кратная концентрация) добавляли в 384-луночный планшет и затем добавляли 10 мкл крысиной плазмы (2-кратная рабочая концентрация), центрифугировали и тщательно смешивали;
6. добавляли 5 мкл реакционного субстрата фермента H-Gly-Pro-AMC (4-кратная рабочая концентрация), центрифугировали и тщательно смешивали, и затем 384-луночный планшет герметизировали пленкой;
7. Полученную смесь инкубировали в инкубаторе (22-23°С) в течение 1 часа;
8. Сигнал флуоресценции определяли с использованием микропланшетного ридера FlexStationI3 (Molecular devices) (возбуждали при 380 нм, и спектр эмиссии определяли при длине волны 460 нм);
9. Определяли значения IC50 тестовых соединений в ингибировании активности фермента DPP-IV, т.е., вычисляли значения IC50 соединений с использованием программного обеспечения GraFit6.


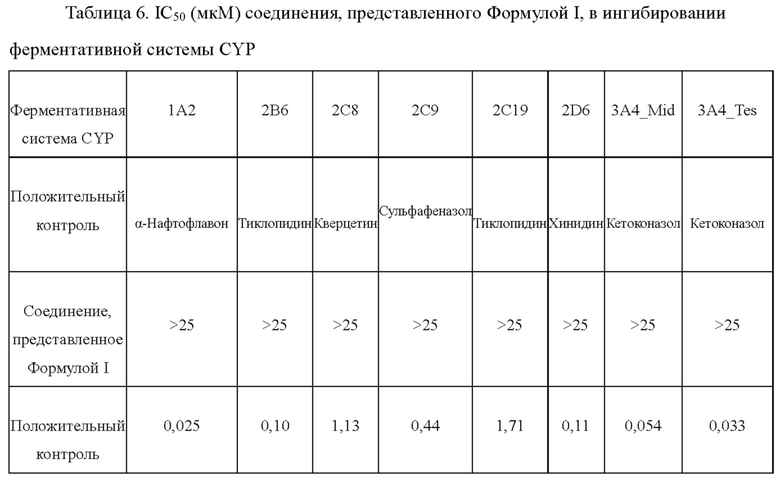
Экспериментальный пример 4: Определение значения IC50 в отношении ингибирования ферментативной системы CYP
Значение IC50 соединения, представленного Формулой I, согласно настоящему изобретению, в ингибировании ферментативной системы CYP, определяли с использованием следующего метода.
Человеческие печеночные микросомы, замороженные при -80°С, помещали на лед для оттаивания, из них 100 мкл помещали в генератор постоянной температуры для инкубированиия (1 час) при 60°С и 100 об/мин, при немедленной оттаивании, и остаток немедленно замораживали при -80°С. По прошествии одного часа, отбирали 100 мкл инактивированных печеночных микросом, и к ним добавляли 400 мкл фосфатного буфера и непрерывно перемешивали с получением раствора 4 мг/мл инактивированных печеночных микросом. В это же время, человеческие печеночные микросомы, замороженные при -80°С, помещали на лед для оттаивания, из них 100 мкл отбирали при немедленном оттаивании, и к ним добавляли 400 мкл фосфатного буфера и непрерывно перемешивали с получением раствора 4 мг/мл печеночных микросом. Смеси для инкубирования для положительного контроля, тестовые соединения и отрицательный контроль получали согласно Таблице 4 ниже:

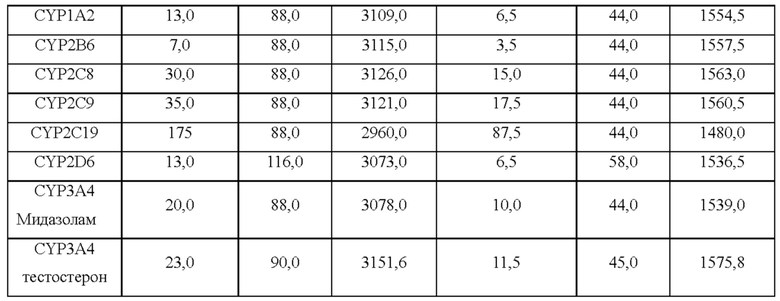
Вышеуказанные смеси для инкубирования инкубировали в течение 5 минут в генераторе постоянной температуры при 37°С и 100 об/мин.
К 2,5 мкл рабочего раствора тестовых соединений или положительного контроля (отрицательный контроль добавляли к рабочему раствору тестовых соединений) добавляли 91,5 мкл смесей для инкубирования и 6 мкл раствора NADPH, и затем реакцию инициировали при перемешивании на вортексе. Полученные растворы инкубировали в осцилляторе постоянной температуры при 37°С и 100 об/мин, и время инкубирования показано в Таблице 5 ниже:

После инкубирования 200 мкл раствора внутреннего стандарта (раствор внутреннего стандарта CYP2C19 представлял собой раствор 100 нг/мл хлорамфеникола в ацетонитриле, и другие растворы внутренниих стандартов представляли собой раствор 250 нг/мл варфарина в ацетонитриле и раствор 500 нг/мл пропранолола в ацетонитриле) добавляли для завершения реакции. Образцы завершенной реакции центрифугировали при 12000 об/мин в течение 10 минут и надосадочные жидкости отбирали для анализа.
Для обработки данных использовали программное обеспечение Analyst 1.4.2 или эквивалентное программное обеспечение. Определяли интегралы для того, чтобы убедиться в том, что все пики были должным образом проинтегрированы, и, если необходимо, корректировали интегральные параметры.
Количественное определение аналита определяли как соотношение пиковой области аналита к пиковой области внутреннего стандарта. Для анализа использовали метод LC-MS/MS. Такие параметры, как IC50, и тому подобные вычисляли с использованием программного обеспечения Graphpad Prism (Version 5.03). Результаты были показаны в Таблице 6 ниже:
Экспериментальный пример 5: Метаболическая стабильность печеночных микросом
Метаболическую стабильность соединения, представленного Формулой I, в печеночных микросомах определяли с использованием следующего способа.
8 мкл человеческих печеночных микросом (20 мг/мл), 20 мкл NADPH и 368 мкл 0,1 М фосфатного буфера смешивали и затем предварительно инкубировали при 37°С в течение 5 минут. Добавляли 4 мкл рабочих растворов (тестовые соединения или положительный контроль), соответственно. Будучи предварительно инкубированными при 37°С, 50 мкл растворов для инкубирования отбирали через 0, 10, 20, 30, 45 и 60 минут и к ним добавляли 150 мкл раствора внутреннего стандарта (0,25М варфарина) в ацетонитриле. 4 мкл микросом печени крыс (20 мг/мл), 10 мкл NADPH и 184 мкл 0,1 М фосфатного буфера смешивали и затем предварительно инкубировали при 37°С в течение 5 минут. Добавляли 2 мкл рабочих растворов (тестовые соедиинения или положительный контроль), соответственно. Будучи предварительно инкубированными при 37°С, 20 мкл растворов для инкубирования отбирали через 0, 10, 20, 30, 45 и 60 минут, и к ним добавляли 180 мл раствора внутреннего стандарта (0,25 М варфарин) в ацетонитриле. Все образцы подвергали вихревому перемешиванию и центрифугировали при 4000 об/мин в течене 15 мин, и 150 мкл надосадочных жидкостей добавляли в 96-луночный планшет, и затем 5 мкл надосадочных жидкостей детектировали в системе LC/MS/MS. Хроматографическая колонка для анализа представляла собой С18 1,7 мкм 2,1×50 мм (Waters). Для детектированиия использовали тройную квадрупольную масс-спектрометрию (API4000, АВ Company). Отношение области пика СТ-1225 к области пика внутреннего стандарта определяли в положительном ионном режиме. Полураспад был представлен как соотношение области пика тестовых соединений/внутреннего стандарта к времени. Результаты показаны в Таблице 7 ниже:

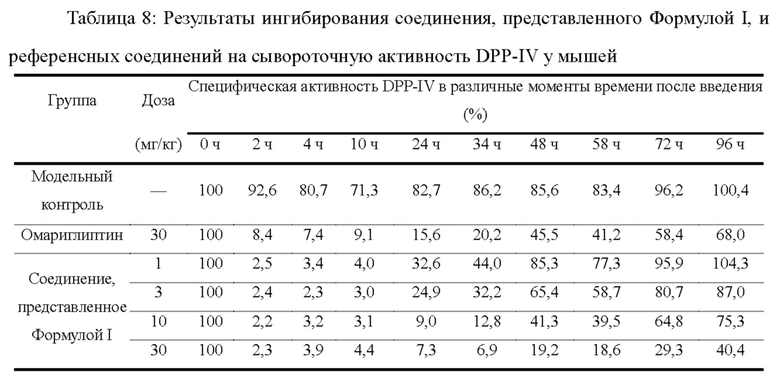
Экспериментальный пример 6: Ингибирующий эффект однократной дозы на активность сыворотки DPP-IV у мышей ob/ob
36 самок мышей ob/ob случайным образом разделяли на 6 групп (6 мышей в каждой группе), которые представляли собой модельную контрольную группу, группу, получающую 1 мг/кг соединения, представленного Формулой I, группу получающую 3 мг/кг соединения, представленного Формулой I, группу получающую 10 мг/кг соединения, представленного Формулой I, группу, получающую 30 мг/кг соединения, представленного Формулой I, и группу, получающую 30 мг/кг омариглиптина (в качестве положительного контроля). Мышам перорально вводили соединение, представленное Формулой I, или омариглиптин в разных дозах, за исключением того, что мышам в контрольной группе перорально вводили 0,25% CMC-Na. Образцы крови отбирали перед введением и через 2, 4, 10, 24, 34, 48, 58, 72 и 96 ч после введения, и сыворотку отделяли для определения активности DPP-IV.
Способ определения активности DPP-IV в сыворотке: к 5 мкл образца сыворотки добавляли 45 мкл 80 мМ буфера MgCl2, тщательно перемешивали и предварительно инкубировали при комнатной температуре в течение 5 минут; к ней добавляли 10 мкл 0,1 мМ реакционного субстрата Gly-Pro-7-AMC и 40 мкл буфера и держали в темноте; после тщательного перемешивания осуществляли определение флуоресценции (волна возбуждения 380 нм/волна испускания 460 нм) каждые 3 минуты в течение 18 минут в общей сложности 6 раз; получали кривую время-флуоресценция на основании результатов определения за вычетом холостого фона, в которой наклон представлял собой значение активности; активность DPP-IV в момент часа 0 перед введенем устанавливали как 100%; и специфическую активность в каждый момент времени после введения вычисляли согласно следующей формуле: специфическая активность (%) = активность после введения/активность перед введенем × 100%.
Результаты эксперимента: после того, как мышам ob/ob однократно перорально вводили соединение, представленное Формулой I, в различных дозах, активность DPP-IV в сыворотке была значительно ингибирована дозозависимым и времязависимым образом. Степень ингибирования сывороточной активности DPP-IV у мышей составляла более 70% на протяжении 10 часов после введения 1 мг/кг соединения, представленного Формулой I. Степень ингибирования сывороточной актвиностии DPP-IV у мышей составляла более 70% на протяжении 24 часов после введения 3 мг/кг соединения, представленного Формулой I. Степень ингибирования сывороточной активности DPP-IV у мышей составляла более 70% на протяжении 34 часов после введения 10 мг/кг соединения, представленного Формулой I. Степень ингибирования сывороточной актвиностии DPP-IV у мышей составляла более 70% на протяжении 72 часов после введения 30 мг/кг соединения, представленного Формулой I. Степень ингибирования сывороточной актвиностии DPP-IV у мышей в группе 30 мг/кг омариглиптина (в качестве положительного контроля) составляла более, чем 70% на протяжении 34 часов после введения.







Изобретение относится к кристаллу соединения (2R,3S,5R)-5-(5-метансульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амина, представленному формулой (I). Также изобретение относится к фосфату соединения формулы (I), при молярном отношении соединения формулы (I) к ортофосфорной кислоте, равном 1:1, к фумарату соединения формулы (I), при молярном отношении соединения формулы (I) к фумаровой кислоте, равном 1:0,5, а также к кристаллам фосфата и фумарата соединения формулы (I). Соединения по изобретению предназначены в качестве ингибитора DPP-IV длительного действия. 6 н. и 9 з.п. ф-лы, 8 табл., 11 ил., 11 пр.
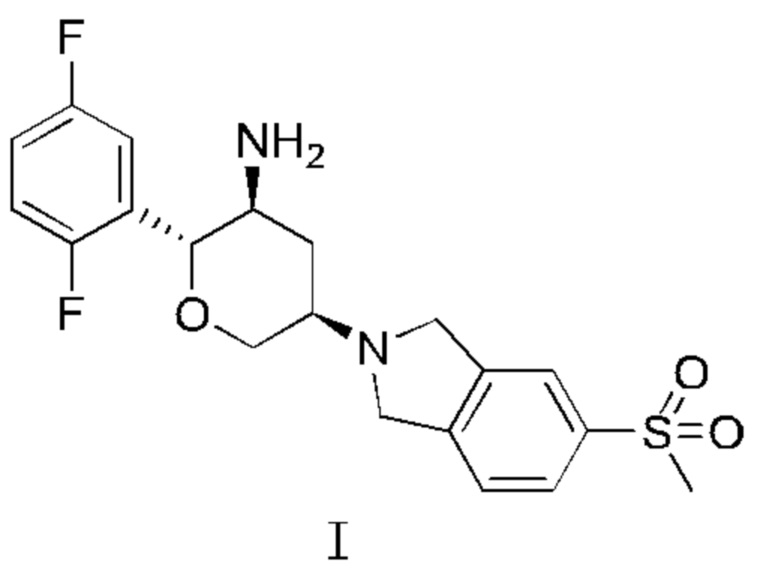
1. Кристалл соединения, представленного Формулой I,
имеющий в картине дифракции рентгеновских лучей дифракционные пики при 2θ, равные 16,4°, 21,8°, 25,3° и 26,0°±0,2°.
2. Кристалл соединения, представленного Формулой I, по п.1, имеющий в картине дифракции рентгеновских лучей дифракционные пики при 2θ, равные 16,4°, 19,4°, 21,2°, 21,8°, 25,3° и 26,0°±0,2°.
3. Кристалл соединения, представленного Формулой I, по п.1, имеющий в картине дифракции рентгеновских лучей дифракционные пики при 2θ, равные 13,0°, 16,4°, 18,5°, 19,4°, 21,2°, 21,8°, 25,3° и 26,0°±0,2°.
4. Кристалл соединения, представленного Формулой I, по п. 1, имеющий температуру начала фазового перехода 193,3±5°С и пиковую температуру 195,2±5°С при характеризации посредством дифференциальной сканирующей калориметрии (DSC).
5. Кристалл соединения, представленного Формулой I, по п. 1, где его пики дифракции рентгеновских лучей имеют следующие характеристики:

6. Фосфат соединения, представленного Формулой I,

где молярное отношение соединения, представленного Формулой I, к ортофосфорной кислоте составляет 1:1.
7. Кристалл фосфата соединения, представленного Формулой I, по п. 6, имеющий дифракционные пики в картине дифракции рентгеновских лучей при 2θ, равные 6,4°, 11,9°, 18,2°, 21,7°, 22,1°, 22,9° и 23,2°±0,2°.
8. Кристалл фосфата соединения, представленного Формулой I, по п. 7, имеющий дифракционные пики в картине дифракции рентгеновских лучей при 2θ, равные 6,4°, 11,9°, 16,5°, 17,5°, 18,2°, 18,6°, 21,7°, 22,1°, 22,9° и 23,2°±0,2°.
9. Кристалл фосфата соединения, представленного Формулой I, по п. 7, имеющий дифракционные пики в картине дифракции рентгеновских лучей при 2θ, равные 6,4°, 10,1°, 11,9°, 16,5°, 17,5°, 18,2°, 18,6°, 19,8°, 21,7°, 22,1°, 22,9°, 23,2° и 23,8°±0,2°.
10. Фумарат соединения, представленного Формулой I,

где молярное отношение соединения, представленного Формулой I, к фумаровой кислоте составляет 1:0,5.
11. Кристалл фумарата соединения, представленного Формулой I, по п. 10, имеющий дифракционный пик в картине дифракции рентгеновских лучей при 2θ, равный 20,67°±0,2°.
12. Фармацевтическая композиция, содержащая терапевтически эффективное количество кристалла соединения, представленного Формулой I, по любому из пп. 1-5, или терапевтически эффективное количество фосфата соединения, представленного Формулой I, по п. 6, либо кристалла фосфата соединения Формулы I по любому из пп.7-9, или терапевтически эффективное количество фумарата соединения, представленного Формулой I, по п. 10, либо кристалла фумарата соединения Формулы I по п.11, для применения в лечении или предотвращении заболевания, при котором полезно ингибирование дипептидилпептидазы-IV (DPP-IV), где заболевание, при котором полезно ингибирование DPP-IV, выбрано из группы, состоящей из инсулинорезистентности, гипергликемии, диабета II типа, диабетической дислипидемии, нарушения переносимости глюкозы, нарушенной гликемии натощак, метаболического ацидоза, кетоза, регуляции аппетита, ожирения, различных раковых заболеваний, неврологических расстройств и расстройств иммунной системы.
13. Фармацевтическая композиция по п.12, где заболевание, при котором полезно ингибирование DPP-IV, представляет собой диабет II типа или ожирение.
14. Кристалл соединения, представленного Формулой I, по любому из пп. 1-5, или фосфат соединения, представленного Формулой I, по п. 6, или кристалл фосфата соединения Формулы I по любому из пп.7-9, или фумарат соединения, представленного Формулой I, по п. 10, или кристалл фумарата соединения Формулы I по п.11, для применения в лечении или предотвращении заболевания, при котором полезно ингибирование DPP-IV, где заболевание, при котором полезно ингибирование DРР-IV, выбрано из группы, состоящей из инсулинорезистентности, гипергликемии, диабета II типа, диабетической дислипидемии, нарушения переносимости глюкозы, нарушенной гликемии натощак, метаболического ацидоза, кетоза, регуляции аппетита, ожирения, различных раковых заболеваний, неврологических расстройств и расстройств иммунной системы.
15. Кристалл соединения, представленного Формулой I, по п. 14, или фосфат соединения, представленного Формулой I, по п. 14, или кристалл фосфата соединения Формулы I по п. 14, или фумарат соединения, представленного Формулой I, по п. 14, или кристалл фумарата соединения Формулы I по п. 14, где заболевание, при котором полезно ингибирование DPP-IV, представляет собой диабет II типа или ожирение.
| WO 2016127916 A1, 18.08.2016 | |||
| WO 2007097931 A2, 30.08.2007 | |||
| Способ устройства водонепроницаемой верхней пароходной палубы | 1929 |
|
SU18613A1 |

