ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к кристаллической форме аналога пиридо[1,2-a]пиримидона, способу его получения и его промежуточному соединению.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Сигнальный путь PI3K в раковых клетках человека является центром наиболее частого происхождения мутаций, которые могут приводить к клеточной пролиферации, активации и усилению передачи сигнала.
PI3K-киназа (фосфатидилинозитол-3-киназа, PI3K) принадлежит к семейству липидкиназ и может фосфорилировать 3’-OH-конец инозитольного кольца в составе фосфатидилинозитола. PI3K представляет собой липидкиназу, состоящую из регуляторной субъединицы p85 или p101 и каталитической субъединицы p110, и играет ключевую роль в клеточной пролиферации, выживании, метаболизме и т. д. за счет катализирования фосфорилирования фосфатидилинозитол-4,5-бифосфата (PIP2) с образованием фосфатидилинозитол-3,4,5-трифосфата (PIP3), активируя тем самым Akt далее по сигнальному пути и т. п. Таким образом, ингибирование фосфатидилинозитол-3-киназы может влиять на сигнальный путь PI3K, и, следовательно, ингибировать пролиферацию и активацию раковых клеток.
Ген-супрессор опухолей PTEN (гомолог фосфатазы и тензина, ассоциированный с делецией в десятой хромосоме) дефосфорилирует PIP3 с образованием PIP2, таким образом осуществляя негативную регуляцию сигнального пути PI3K/Akt, ингибируя клеточную пролиферацию и стимулируя апоптоз. Частые случаи мутации гена PI3K и увеличения числа его копий, а также утрата PTEN при раке и т. д. указывают на то, что путь PI3K тесно связан с онкогенезом.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предусмотрен способ получения соединения 1:
 ,
,
который включает следующие стадии:
 ,
,
где
X выбран из Cl или Br;
щелочь С выбрана из пиридина, 2,6-лутидина, Et3N, 4-DMAP, LiOH, Cs2CO3 или K2CO3;
растворитель с выбран из пиридина, дихлорметана, толуола, ацетонитрила, ацетона, DMF или THF;
молярное соотношение соединения 7 и соединения 8 составляет 1:1-3;
молярное соотношение соединения 7 и щелочи С составляет 1:1-3.
В некоторых вариантах осуществления настоящего изобретения молярное соотношение соединения 7 и соединения 8 составляет 1:1,2-1,6.
В некоторых вариантах осуществления настоящего изобретения получение вышеупомянутого соединения 1 включает следующие стадии:
 ,
,
где
щелочь A выбрана из карбоната калия, карбоната натрия, карбоната цезия, гидроксида калия или гидроксида натрия;
растворитель a выбран из DMF, DMSO или NMP.
В данном документе 2-диметиламиноэтилхлорид или 2-диметиламиноэтилбромид можно применять в виде их солей, таких как гидрохлорид 2-диметиламиноэтилхлорида или гидрохлорид 2-диметиламиноэтилбромида.
В некоторых вариантах осуществления настоящего изобретения молярное соотношение соединения 5 и 2-диметиламиноэтилхлорида (или его хлористоводородной соли) или 2-диметиламиноэтилбромида (или его хлористоводородной соли) составляет 1:1-2.
В некоторых вариантах осуществления настоящего изобретения молярное соотношение соединения 5 и 2-диметиламиноэтилхлорида (или его хлористоводородной соли) или 2-диметиламиноэтилбромида (или его хлористоводородной соли) составляет 1:1,1-1,3.
В некоторых вариантах осуществления настоящего изобретения получение вышеупомянутого соединения 1 включает следующие стадии:
 ,
,
где
щелочь B выбрана из карбоната калия, карбоната натрия, гидроксида бария, фосфата калия, карбоната цезия, фторида калия, фторида цезия, гидроксида натрия, трет-бутоксида калия, трет-бутоксида натрия, ацетата калия или ацетата натрия;
растворитель b выбран из 1,4-диоксана, DMSO, THF, 1,4-диоксана/воды или THF/воды;
объемное соотношение 1,4-диоксана или THF и воды в растворителе b составляет 3-6:1, предпочтительно 5:1;
катализатор выбран из Pd(dppf)Cl2 или Pd(PPh3)4.
В некоторых вариантах осуществления настоящего изобретения объемное соотношение 1,4-диоксана или THF и воды в вышеупомянутом растворителе b составляет 5:1.
В некоторых вариантах осуществления настоящего изобретения получение вышеупомянутого соединения 1 включает следующие стадии:
 .
.
В вышеуказанной реакционной схеме реакцию соединения 5 с гидрохлоридом 2-диметиламиноэтилхлорида для получения соединения 6 предпочтительно осуществляют в присутствии щелочи A и растворителя a, где щелочь A выбрана из карбоната калия, карбоната натрия, карбоната цезия, гидроксида калия или гидроксида натрия; и растворитель a выбран из DMF, DMSO или NMP. В некоторых вариантах осуществления настоящего изобретения молярное соотношение соединения 5 и гидрохлорида 2-диметиламиноэтилхлорида составляет 1:1-2. В некоторых вариантах осуществления настоящего изобретения молярное соотношение соединения 5 и гидрохлорида 2-диметиламиноэтилхлорида составляет 1:1,1-1,3.
В вышеуказанной реакционной схеме реакцию соединения 6 с 2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-амином для получения соединения 7 предпочтительно осуществляют в присутствии щелочи B, растворителя b и катализатора, где щелочь B выбрана из карбоната калия, карбоната натрия, гидроксида бария, фосфата калия, карбоната цезия, фторида калия, фторида цезия, гидроксида натрия, трет-бутоксида калия, трет-бутоксида натрия, ацетата калия или ацетата натрия; растворитель b выбран из 1,4-диоксана, DMSO, THF, 1,4-диоксана/воды или THF/воды, где объемное соотношение 1,4-диоксана или THF и воды в растворителе b составляет 3-6:1, предпочтительно 5:1; и катализатор выбран из Pd(dppf)Cl2 или Pd(PPh3)4.
В вышеуказанной реакционной схеме реакцию соединения 7 с 2-хлор-4-фторбензолсульфонилхлоридом для получения соединения 1 предпочтительно осуществляют в присутствии щелочи С и растворителя с, где щелочь С выбрана из пиридина, 2,6-лутидина, Et3N, 4-DMAP, LiOH, Cs2CO3 или K2CO3; растворитель с выбран из пиридина, дихлорметана, толуола, ацетонитрила, ацетона, DMF или THF; и молярное соотношение соединения 7 и 2-хлор-4-фторбензолсульфонилхлорида составляет 1:1-3; и молярное соотношение соединения 7 и щелочи С составляет 1:1~3. В некоторых вариантах осуществления настоящего изобретения молярное соотношение соединения 7 и 2-хлор-4-фторбензолсульфонилхлорида составляет 1:1,2~1,6.
В настоящем изобретении также предусмотрены соединения, выступающие в качестве промежуточных соединений для получения соединения 1, которые представлены следующими формулами:
 ,
,  .
.
В настоящем изобретении предусмотрена кристаллическая форма IX соединения 1, характеризующаяся наличием дифракционных пиков на дифракционной рентгенограмме (XRD) при 2θ = 7,947°, 10,073°, 14,531°, 19,187°, 21,237°, 24,055°, 25,497°; преимущественно при 2θ = 7,947°, 10,073°, 11,970°, 13,468°, 14,531°, 15,911°, 19,187°, 21,237°, 24,055°, 25,497°; более преимущественно при 2θ = 7,947°, 10,073°, 11,970°, 13,468°, 14,531°, 15,911°, 19,187°, 19,561°, 21,237°, 23,446°, 24,055°, 25,497°, 27,074°.
В настоящем изобретении предусмотрена кристаллическая форма IX соединения 1, которая характеризуется XRPD-рентгенограммой, показанной на фиг. 25.
В некоторых вариантах осуществления настоящего изобретения данные анализа XRPD-рентгенограммы вышеупомянутой кристаллической формы IX соединения 1 показаны в таблице 1.
Таблица 1. Данные анализа XRPD-рентгенограммы кристаллической формы IX соединения 1

В некоторых вариантах осуществления настоящего изобретения профиль DSC вышеупомянутой кристаллической формы IX соединения 1 является таким, как показано на фиг. 26.
В некоторых вариантах осуществления настоящего изобретения профиль TGA вышеупомянутой кристаллической формы IX соединения 1 является таким, как показано на фиг. 27.
Кристаллическая форма IX соединения 1 может быть представлена в виде кристалла несольватной формы или кристалла сольвата. Сольват в данном документе относится к сольвату, образованному органическим растворителем и/или водой с соответствующим соединением.
В настоящем изобретении предусмотрена кристаллическая композиция на основе кристаллической формы IX. В некоторых вариантах осуществления настоящего изобретения кристаллическая форма IX составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более, наиболее предпочтительно 95% или более по весу кристаллической композиции.
В настоящем изобретении предусмотрена фармацевтическая композиция на основе кристаллической формы IX, содержащая терапевтически эффективное количество кристаллической формы IX или кристаллической композиции на основе кристаллической формы IX. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носители, наполнители и/или среды или не содержать их.
В настоящем изобретении предусмотрено соединение 2, представленное следующей формулой:
 .
.
В настоящем изобретении предусмотрена кристаллическая форма I соединения 2, характеризующаяся наличием дифракционных пиков на дифракционной рентгенограмме (XRD) при 2θ = 10,154°, 12,285°, 14,511°, 16,328°, 24,311°, 26,188°; преимущественно при 2θ = 7,270°, 10,154°, 12,285°, 13,206°, 14,511°, 16,328°, 24,311°, 26,188°, 27,724°; более преимущественно при 2θ = 7,270°, 10,154°, 12,285°, 13,206°, 14,511°, 16,328°, 19,008°, 20,702°, 21,259°, 24,311°, 26,188°, 27,724°.
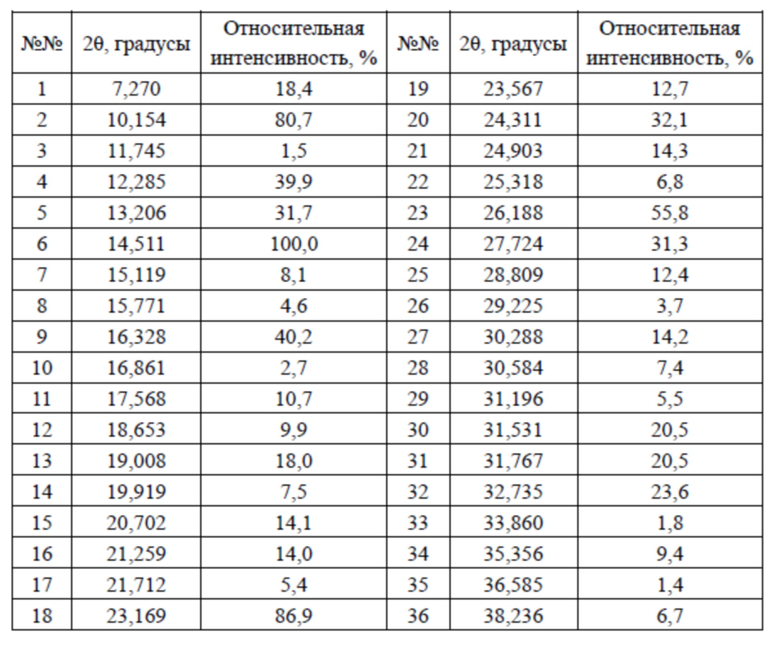
В настоящем изобретении предусмотрена кристаллическая форма I соединения 2, которая характеризуется XRPD-рентгенограммой, показанной на фиг. 1.
В некоторых вариантах осуществления настоящего изобретения данные анализа XRPD-рентгенограммы вышеупомянутой кристаллической формы I соединения 2 показаны в таблице 2.
Таблица 2. Данные анализа XRPD-рентгенограммы кристаллической формы I соединения 2
В некоторых вариантах осуществления настоящего изобретения профиль DSC кристаллической формы I соединения 2 является таким, как показано на фиг. 2.
В некоторых вариантах осуществления настоящего изобретения профиль TGA кристаллической формы I соединения 2 показан на фиг. 3.
Кристаллическая форма I соединения 2 может быть представлена в виде кристалла несольватной формы или кристалла сольвата. Сольват в данном документе относится к сольвату, образованному органическим растворителем и/или водой с соответствующим соединением.
В настоящем изобретении предусмотрена кристаллическая композиция на основе кристаллической формы I. В некоторых вариантах осуществления настоящего изобретения кристаллическая форма I составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более, наиболее предпочтительно 95% или более по весу кристаллической композиции.
В настоящем изобретении предусмотрена фармацевтическая композиция на основе кристаллической формы I, содержащая терапевтически эффективное количество кристаллической формы I или кристаллической композиции на основе кристаллической формы I. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носители, наполнители и/или среды или не содержать их.
В настоящем изобретении предусмотрена кристаллическая форма II соединения 2, характеризующаяся наличием дифракционных пиков на дифракционной рентгенограмме (XRD) при 2θ = 6,524°, 7,782°, 13,895°, 15,495°, 17,487°, 19,322°; преимущественно при 2θ = 6,524°, 7,782°, 11,628°, 13,895°, 15,495°, 17,487°, 19,322°, 20,962°, 23,269°; более преимущественно при 2θ = 6,524°, 7,782°, 11,628°, 13,895°, 15,495°, 17,487°, 19,322°, 20,962°, 23,269°, 24,257°, 26,009°, 31,533°.
В настоящем изобретении предусмотрена кристаллическая форма II соединения 2, которая характеризуется XRPD-рентгенограммой, показанной на фиг. 4.
В некоторых вариантах осуществления настоящего изобретения данные анализа XRPD-рентгенограммы кристаллической формы II соединения 2 показаны в таблице 3.
Таблица 3. Данные анализа XRPD-рентгенограммы кристаллической формы II соединения 2

В некоторых вариантах осуществления настоящего изобретения профиль DSC кристаллической формы II соединения 2 является таким, как показано на фиг. 5.
В некоторых вариантах осуществления настоящего изобретения профиль TGA кристаллической формы II соединения 2 является таким, как показано на фиг. 6.
Кристаллическая форма II соединения 2 может быть представлена в виде кристалла несольватной формы или кристалла сольвата. Сольват в данном документе относится к сольвату, образованному органическим растворителем и/или водой с соответствующим соединением.
В настоящем изобретении предусмотрена кристаллическая композиция на основе кристаллической формы II. В некоторых вариантах осуществления настоящего изобретения кристаллическая форма II составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более, наиболее предпочтительно 95% или более по весу кристаллической композиции.
В настоящем изобретении предусмотрена фармацевтическая композиция на основе кристаллической формы II, содержащая терапевтически эффективное количество кристаллической формы II или кристаллической композиции на основе кристаллической формы II. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носители, наполнители и/или среды или не содержать их.
В настоящем изобретении предусмотрена кристаллическая форма III соединения 2, характеризующаяся наличием дифракционных пиков на дифракционной рентгенограмме (XRD) при 2θ = 6,979°, 9,939°, 14,392°, 16,107°, 20,982°, 25,990°; преимущественно при 2θ = 6,187°, 6,979°, 9,939°, 11,910°, 14,392°, 16,107°, 20,982°, 22,755°, 25,990°; более преимущественно при 2θ = 6,187°, 6,979°, 9,939°, 11,910°, 13,148°, 14,392°, 16,107°, 20,982°, 22,755°, 23,975°, 25,990°, 29,006°.
В настоящем изобретении предусмотрена кристаллическая форма III соединения 2, которая характеризуется XRPD-рентгенограммой, показанной на фиг. 7.
В некоторых вариантах осуществления настоящего изобретения данные анализа XRPD-рентгенограммы вышеупомянутой кристаллической формы III соединения 2 показаны в таблице 4.
Таблица 4. Данные анализа XRPD-рентгенограммы кристаллической формы III соединения 2

В некоторых вариантах осуществления настоящего изобретения профиль DSC кристаллической формы III соединения 2 является таким, как показано на фиг. 8.
В некоторых вариантах осуществления настоящего изобретения профиль TGA кристаллической формы III соединения 2 является таким, как показано на фиг. 9.
Кристаллическая форма III соединения 2 может быть представлена в виде кристалла несольватной формы или кристалла сольвата. Сольват в данном документе относится к сольвату, образованному органическим растворителем и/или водой с соответствующим соединением.
В настоящем изобретении предусмотрена кристаллическая композиция на основе кристаллической формы III. В некоторых вариантах осуществления настоящего изобретения кристаллическая форма III составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более, наиболее предпочтительно 95% или более по весу кристаллической композиции.
В настоящем изобретении предусмотрена фармацевтическая композиция на основе кристаллической формы III, содержащая терапевтически эффективное количество кристаллической формы III или кристаллической композиции на основе кристаллической формы III. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носители, наполнители и/или среды или не содержать их.
В настоящем изобретении предусмотрена кристаллическая форма IV соединения 2, характеризующаяся наличием дифракционных пиков на дифракционной рентгенограмме (XRD) при 2θ = 6,388°, 7,278°, 11,076°, 15,454°, 21,256°; преимущественно при 2θ = 6,388°, 7,278°, 11,076°, 12,102°, 15,454°, 16,091°, 18,912°, 21,256°; более преимущественно при 2θ = 6,388°, 7,278°, 11,076°, 12,102°, 15,103°, 15,454°, 16,091°, 18,912°, 21,256°, 21,846°.
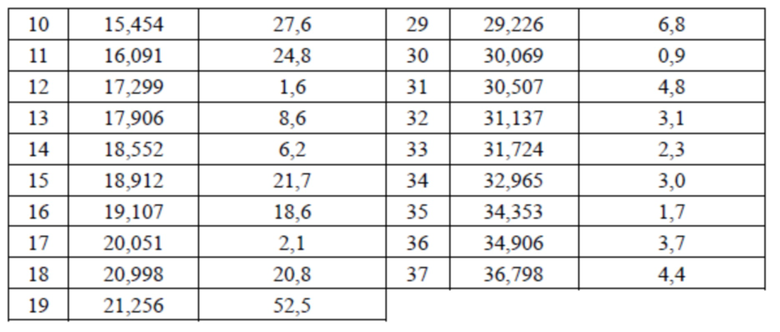
В настоящем изобретении предусмотрена кристаллическая форма IV соединения 2, которая характеризуется XRPD-рентгенограммой, показанной на фиг. 10.
В некоторых вариантах осуществления настоящего изобретения данные анализа XRPD-рентгенограммы вышеупомянутой кристаллической формы IV соединения 2 показаны в таблице 5.
Таблица 5. Данные анализа XRPD-рентгенограммы кристаллической формы IV соединения 2

В некоторых вариантах осуществления настоящего изобретения профиль DSC кристаллической формы IV соединения 2 является таким, как показано на фиг. 11.
В некоторых вариантах осуществления настоящего изобретения профиль TGA кристаллической формы IV соединения 2 является таким, как показано на фиг. 12.
Кристаллическая форма IV соединения 2 может быть представлена в виде кристалла несольватной формы или кристалла сольвата. Сольват в данном документе относится к сольвату, образованному органическим растворителем и/или водой с соответствующим соединением.
В настоящем изобретении предусмотрена кристаллическая композиция на основе кристаллической формы IV. В некоторых вариантах осуществления настоящего изобретения кристаллическая форма IV составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более, наиболее предпочтительно 95% или более по весу кристаллической композиции.
В настоящем изобретении предусмотрена фармацевтическая композиция на основе кристаллической формы IV, содержащая терапевтически эффективное количество кристаллической формы IV или кристаллической композиции на основе кристаллической формы IV. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носители, наполнители и/или среды или не содержать их.
В настоящем изобретении предусмотрена кристаллическая форма V соединения 2, характеризующаяся наличием дифракционных пиков на дифракционной рентгенограмме (XRD) при 2θ = 7,116°, 14,137°, 15,911°, 22,223°, 24,610°; преимущественно при 2θ = 7,116°, 14,137°, 15,911°, 21,691°, 22,223°, 24,213°, 24,610°, 28,987°.
В настоящем изобретении предусмотрена кристаллическая форма V соединения 2, которая характеризуется XRPD-рентгенограммой, показанной на фиг. 13.
В некоторых вариантах осуществления настоящего изобретения данные анализа XRPD-рентгенограммы вышеупомянутой кристаллической формы V соединения 2 показаны в таблице 6.
Таблица 6. Данные анализа XRPD-рентгенограммы кристаллической формы V соединения 2
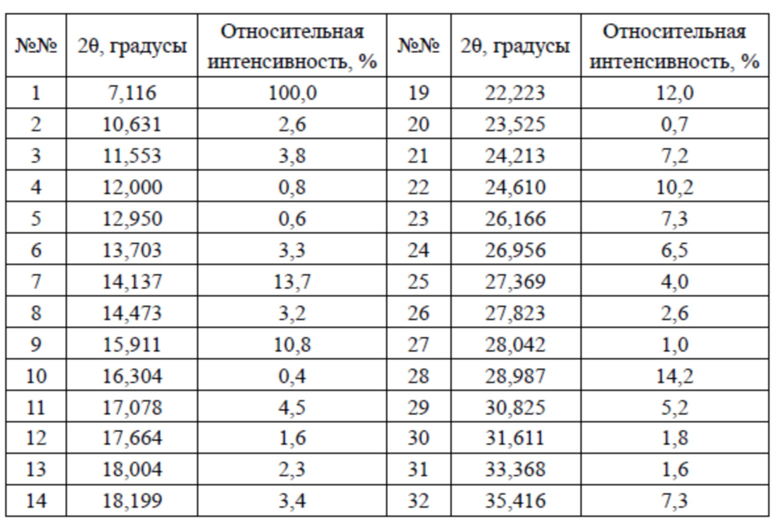

В некоторых вариантах осуществления настоящего изобретения профиль DSC кристаллической формы V соединения 2 является таким, как показано на фиг. 14.
В некоторых вариантах осуществления настоящего изобретения профиль TGA кристаллической формы V соединения 2 является таким, как показано на фиг. 15.
Кристаллическая форма V соединения 2 может быть представлена в виде кристалла несольватной формы или кристалла сольвата. Сольват в данном документе относится к сольвату, образованному органическим растворителем и/или водой с соответствующим соединением.
В настоящем изобретении предусмотрена кристаллическая композиция на основе кристаллической формы V. В некоторых вариантах осуществления настоящего изобретения кристаллическая форма V составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более, наиболее предпочтительно 95% или более по весу кристаллической композиции.
В настоящем изобретении предусмотрена фармацевтическая композиция на основе кристаллической формы V, содержащая терапевтически эффективное количество кристаллической формы V или кристаллической композиции на основе кристаллической формы V. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носители, наполнители и/или среды или не содержать их.
В настоящем изобретении предусмотрена кристаллическая форма VI соединения 2, характеризующаяся наличием дифракционных пиков на дифракционной рентгенограмме (XRD) при 2θ = 5,775°, 11,770°, 14,415°, 15,753°, 22,518°, 26,623°; преимущественно при 2θ = 5,775°, 11,770°, 14,415°, 15,753°, 17,132°, 20,939°, 22,518°, 26,623°; более преимущественно при 2θ = 5,775°, 11,770°, 14,415°, 15,753°, 17,132°, 20,939°, 22,518°, 23,745°, 26,623°, 31.295°.
В настоящем изобретении предусмотрена кристаллическая форма VI соединения 2, которая характеризуется XRPD-рентгенограммой, показанной на фиг. 16.
В некоторых вариантах осуществления настоящего изобретения данные анализа XRPD-рентгенограммы вышеупомянутой кристаллической формы VI соединения 2 показаны в таблице 7.
Таблица 7. Данные анализа XRPD-рентгенограммы кристаллической формы VI соединения 2

В некоторых вариантах осуществления настоящего изобретения профиль DSC кристаллической формы VI соединения 2 является таким, как показано на фиг. 17.
В некоторых вариантах осуществления настоящего изобретения профиль TGA кристаллической формы VI соединения 2 является таким, как показано на фиг. 18.
Кристаллическая форма VI соединения 2 может быть представлена в виде кристалла несольватной формы или кристалла сольвата. Сольват в данном документе относится к сольвату, образованному органическим растворителем и/или водой с соответствующим соединением.
В настоящем изобретении предусмотрена кристаллическая композиция на основе кристаллической формы VI. В некоторых вариантах осуществления настоящего изобретения кристаллическая форма VI составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более, наиболее предпочтительно 95% или более по весу кристаллической композиции.
В настоящем изобретении предусмотрена фармацевтическая композиция на основе кристаллической формы VI, содержащая терапевтически эффективное количество кристаллической формы VI или кристаллической композиции на основе кристаллической формы VI. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носители, наполнители и/или среды или не содержать их.
В настоящем изобретении предусмотрено соединение 3, представленное следующей формулой:
 .
.
В настоящем изобретении предусмотрена кристаллическая форма VII соединения 3, характеризующаяся наличием дифракционных пиков на дифракционной рентгенограмме (XRD) при 2θ = 6,325°, 12,677°, 15,813°, 21,395°, 22,519°, 27,133°; преимущественно при 2θ = 6,325°, 12,677°, 13,251°, 15,813°, 18,954°, 21,395°, 22,519°, 25,161°, 27,133°; более преимущественно при 2θ = 6,325°, 12,677°, 13,251°, 15,813°, 16,565°, 18,954°, 21,395°, 22,519°, 24,117°, 25,161°, 26,405°, 27,133°.
В настоящем изобретении предусмотрена кристаллическая форма VII соединения 3, которая характеризуется XRPD-рентгенограммой, показанной на фиг. 19.
В некоторых вариантах осуществления настоящего изобретения данные анализа XRPD-рентгенограммы вышеупомянутой кристаллической формы VII соединения 3 показаны в таблице 8.
Таблица 8. Данные анализа XRPD-рентгенограммы кристаллической формы VII соединения 3
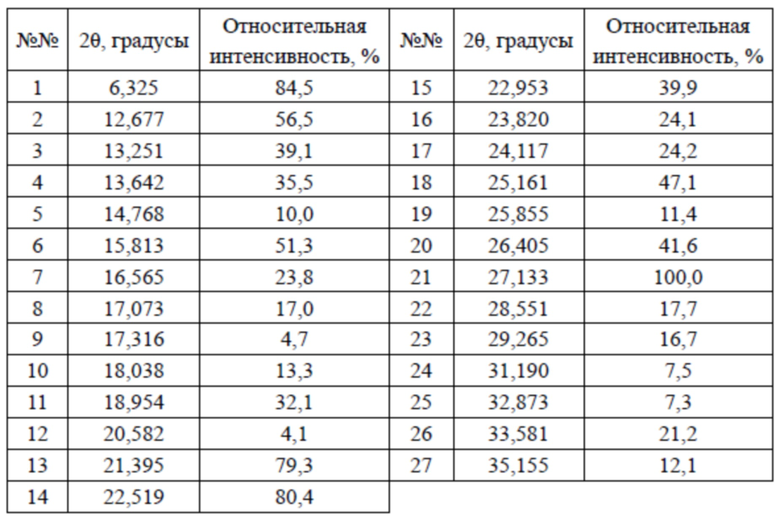
В некоторых вариантах осуществления настоящего изобретения профиль DSC кристаллической формы VII соединения 3 является таким, как показано на фиг. 20.
В некоторых вариантах осуществления настоящего изобретения профиль TGA кристаллической формы VII соединения 3 является таким, как показано на фиг. 21.
Кристаллическая форма VII соединения 3 может быть представлена в виде кристалла несольватной формы или кристалла сольвата. Сольват в данном документе относится к сольвату, образованному органическим растворителем и/или водой с соответствующим соединением.
В настоящем изобретении предусмотрена кристаллическая композиция на основе кристаллической формы VII. В некоторых вариантах осуществления настоящего изобретения кристаллическая форма VII составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более, наиболее предпочтительно 95% или более по весу кристаллической композиции.
В настоящем изобретении предусмотрена фармацевтическая композиция на основе кристаллической формы VII, содержащая терапевтически эффективное количество кристаллической формы VII или кристаллической композиции на основе кристаллической формы VII. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носители, наполнители и/или среды или не содержать их.
В настоящем изобретении предусмотрено соединение 4, представленное следующей формулой:

В настоящем изобретении предусмотрена кристаллическая форма VIII соединения 4, характеризующаяся наличием дифракционных пиков на дифракционной рентгенограмме (XRD) при 2θ = 5,889°, 11,002°, 12,518°, 14,906°, 17,825°, 22,814°, 25,555°; преимущественно при 2θ = 5,889°, 7,173°, 11,002°, 11,396°, 12,518°, 12,895°, 14,906°, 17,825°, 22,814°, 25,555°; более преимущественно при 2θ = 5,889°, 7,173°, 11,002°, 11,396°, 12,518°, 12,895°, 14,906°, 16,169°, 17,825°, 19,875°, 21,574°, 22,814°, 25,555°, 27,254°.
В некоторых вариантах осуществления настоящего изобретения XRPD-рентгенограмма кристаллической формы VIII соединения 4 является такой, как показано на фиг. 22.
В некоторых вариантах осуществления настоящего изобретения данные анализа XRPD-рентгенограммы вышеупомянутой кристаллической формы VIII соединения 4 показаны в таблице 9.
Таблица 9. Данные анализа XRPD-рентгенограммы кристаллической формы VIII соединения 4
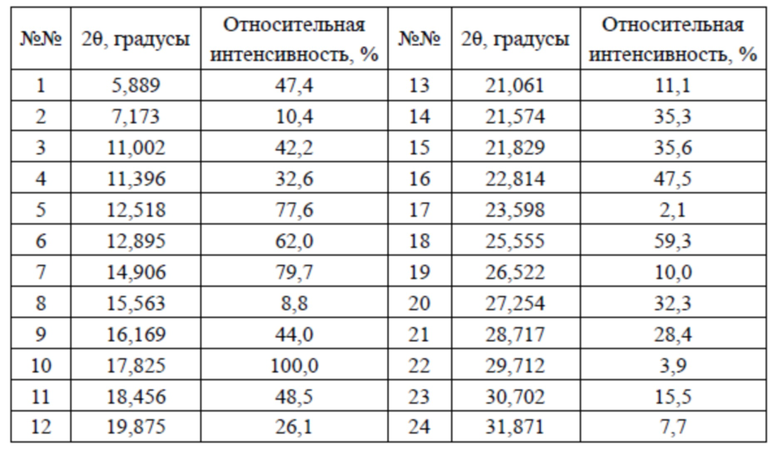
В некоторых вариантах осуществления настоящего изобретения профиль DSC кристаллической формы VIII соединения 4 является таким, как показано на фиг. 23.
В некоторых вариантах осуществления настоящего изобретения профиль TGA кристаллической формы VIII соединения 4 является таким, как показано на фиг. 24.
Кристаллическая форма VIII соединения 4 может быть представлена в виде кристалла несольватной формы или кристалла сольвата. Сольват в данном документе относится к сольвату, образованному органическим растворителем и/или водой с соответствующим соединением.
В настоящем изобретении предусмотрена кристаллическая композиция на основе кристаллической формы VIII. В некоторых вариантах осуществления настоящего изобретения кристаллическая форма VIII составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более, наиболее предпочтительно 95% или более по весу кристаллической композиции.
В настоящем изобретении предусмотрена фармацевтическая композиция на основе кристаллической формы VIII, содержащая терапевтически эффективное количество кристаллической формы VIII или кристаллической композиции на основе кристаллической формы VIII. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носители, наполнители и/или среды или не содержать их.
Целью настоящего изобретения также является обеспечение применения соединений 1, 2, 3 и 4 или фармацевтических композиций на их основе в изготовлении лекарственного препарата для лечения заболеваний, связанных с рецептором PI3K.
Другой целью настоящего изобретения является обеспечение применения кристаллических форм I, II, III, IV, V, VI, VII, VIII и IX, кристаллических композиций на их основе и фармацевтической композиции на их основе в изготовлении лекарственного препарата для лечения заболеваний, связанных с PI3K-киназой.
В некоторых вариантах осуществления настоящего изобретения заболевания, связанные с PI3K-киназой, выбраны из рака, такого как рак толстой кишки, рак желудка и т. п.
Целью настоящего изобретения является обеспечение способа лечения заболеваний, связанных с PI3K-киназой, предусматривающего введение терапевтически эффективного количества соединений 1, 2, 3 и 4 или фармацевтической композиции на их основе пациенту, нуждающемуся в этом.
Другой целью настоящего изобретения является обеспечение способа лечения заболеваний, связанных с PI3K-киназой, предусматривающего введение терапевтически эффективного количества кристаллических форм I, II, III, IV, V, VI, VII, VIII и IX по настоящему изобретению, кристаллической композиции на их основе и фармацевтической композиции на их основе пациенту, нуждающемуся в этом.
В некоторых вариантах осуществления настоящего изобретения заболевания, связанные с PI3K-киназой, выбраны из рака, такого как рак толстой кишки, рак желудка и т. п.
Определение и описание
Если не указано иное, предполагается, что используемые в данном документе термины и выражения имеют следующие значения. Конкретное выражение или термин не следует считать неопределенным или неясным ввиду отсутствия конкретного определения, а следует понимать в соответствии с его общепринятым значением. Предполагается, что при упоминании в данном документе торгового названия, оно относится к соответствующему продукту или его активному ингредиенту.
Промежуточные соединения по настоящему изобретению можно получать с помощью ряда способов синтеза, хорошо известных специалистам в данной области техники, в том числе конкретных вариантов осуществления, проиллюстрированных ниже, вариантов осуществления в сочетании с другими способами химического синтеза и эквивалентов, хорошо известных специалистам в данной области техники. Предпочтительные варианты осуществления включают без ограничения примеры настоящего изобретения.
Химические реакции в конкретных вариантах осуществления настоящего изобретения осуществляют в подходящих растворителях, которые должны быть подходящими для химических превращений, а также с реагентами и материалами, необходимыми в настоящем изобретении. Для того, чтобы получить соединения по настоящему изобретению, иногда специалисту в данной области техники необходимо модифицировать или выбрать стадии синтеза или схемы реакций на основании данных вариантов осуществления, представленных в данном документе.
Важным фактором, который необходимо учитывать при любом планировании пути синтеза в данной области техники, является выбор подходящей защитной группы для реакционноспособной функциональной группы (такой как аминогруппа согласно настоящему изобретению). (Protective Groups In Organic Synthesis, Wiley and Sons, 1991) под редакцией Greene и Wuts является авторитетным источником для квалифицированных специалистов-практиков в данной области техники. Все ссылки, приведенные в настоящем изобретении, включены в данный документ посредством ссылки во всей своей полноте.
Настоящее изобретение будет конкретно описано ниже с помощью примеров, однако предполагается, что настоящее изобретение не ограничивается данными примерами.
Все растворители, применяемые согласно настоящему изобретению, являются коммерчески доступными и их применяют без дополнительной очистки. Реакции, как правило, проводят в атмосфере инертного газообразного азота в безводном растворителе. Данные протонного ядерного магнитного резонанса регистрировали на спектрометре Bruker Avance III 400 (400 МГц), где значения химических сдвигов обозначены как (ppm) тетраметилсилана при слабом поле. Масс-спектры измеряли на Agilent 1200 Series plus 6110 (&1956A). LC/MS или Shimadzu MS содержит детектор DAD: SPD-M20A (LC) и детектор Shimadzu Micromass 2020. Масс-спектрометр оснащен источником ионизации путем электрораспыления (ESI), работающим в режиме образования положительных или отрицательных ионов.
Следует отметить, что если речь идет о спектре рентгеновской дифракции, дифракционная рентгенограмма, полученная для кристаллического соединения, часто является характеристической для конкретной кристаллической формы, при этом относительная интенсивность полос (особенно под малым углом) может варьировать вследствие эффекта доминирующей ориентации, вызванного различиями в условиях кристаллизации, размере частиц и других условиях измерения. Таким образом, относительная интенсивность дифракционных пиков не является характеристической для целевой кристаллической формы. При оценке того, является ли кристаллическая форма идентичной уже известной кристаллической форме, следует обращать внимание на относительные расположения пиков, а не на их относительные интенсивности. Кроме того, для каждой конкретной кристаллической формы может присутствовать небольшая погрешность в расположениях пиков, что также хорошо известно в области кристаллографии. Например, положение пика может сдвигаться вследствие изменений температуры, изменений положения образца или калибровки измерительных приборов в ходе анализа образца, и погрешность измерения значения 2θ в некоторых случаях составляет приблизительно ±0,5°, предпочтительно приблизительно ±0,3°, более предпочтительно приблизительно ±0,2°. Таким образом, при определении структуры каждой кристаллической формы следует учитывать такую погрешность, и значения 2θ в пределах погрешности также попадают в пределы объема настоящего изобретения. На XRD-рентгенограмме положение пика обычно представляется величиной угла 2θ или межплоскостного расстояния d, и существует простая формула преобразования между ними: d=λ/2sinθ, где d представляет собой межплоскостное расстояние, λ представляет собой длину волны падающих рентгеновских лучей, и θ представляет собой дифракционный угол. В случае одинаковой кристаллической формы соединения одного и того же типа расположения пиков на их спектрах XRD в целом сходны, а относительная интенсивность может иметь большую погрешность. Также следует отметить, что при идентификации смеси части дифракционных линий могут быть утрачены вследствие таких факторов, как низкое содержание и т. п. В данном случае даже одна полоса также может быть характеристической для конкретного кристалла, и при этом не обязательно учитывать все полосы, как наблюдается в случае образца с высокой степенью чистоты.
Следует иметь в виду, что в ходе получения кристаллической формы лекарственного средства при приведении молекулы лекарственного средства и молекулы растворителя в контакт друг с другом, трудно избежать того, что молекула растворителя будет образовывать эвтектическую смесь с молекулой соединения и оставаться в виде твердого вещества вследствие внешних условий и внутренних факторов, образуя тем самым сольват, в том числе конкретно стехиометрический и нестехиометрический сольваты. Такие сольваты охватываются объемом настоящего изобретения.
Стехиометрический показатель для хлорид-иона в соединении 2 (гидрохлорид), полученном согласно настоящему изобретению, можно определять с помощью ионообменной хроматографии. Применяемым измерительным прибором был 883 Basic IC plus 1; выбранной колонкой была Metrosep A Supp 5 - 150/4,0; расход составлял 0,700 мл/мин; общее время анализа составляло 10 мин.
В настоящем изобретении используются следующие сокращения: DCM представляет собой дихлорметан; PE представляет собой петролейный эфир; EA представляет собой этилацетат; DMF представляет собой N,N-диметилформамид; DMSO представляет собой диметилсульфоксид; THF представляет собой тетрагидрофуран; MeOH представляет собой метанол; NMP представляет собой N-метилпирролидон; Et3N представляет собой триэтиламин; 4-DMAP представляет собой 4-диметиламинопиридин; LiOH представляет собой гидроксид лития; Cs2CO3 представляет собой карбонат цезия; K2CO3 представляет собой карбонат калия; PPh3 представляет собой трифенилфосфин; Pd(PPh3)4 представляет собой тетра(трифенилфосфин)палладий; Pd(dppf)Cl2 представляет собой хлорид 1,1'-бис(дифенилфосфино)ферроценпалладия.
Способ с применением порошкового рентгеновского дифрактометра (XRPD) согласно настоящему изобретению:
модель измерительного прибора: рентгеновский дифрактометр Bruker D8 advance;
условия испытаний: подробные параметры XRPD являются следующими:
генератор рентгеновского излучения: Cu, kα, (λ=1,54056 Ǻ);
напряжение на рентгеновской трубке: 40 кВ, сила тока на рентгеновской трубке: 40 мА;
рассеивающая щель: 0,60 мм;
щель детектора: 10,50 мм;
антирассеивающая щель: 7,10 мм;
диапазон сканирования: 4-40 градуса;
длина шага: 0,02 градуса;
скорость: 0,1 с;
скорость вращения планшета с образцом: 15 об/мин.
Способ с применением дифференциального сканирующего калориметра (DSC) согласно настоящему изобретению:
модель измерительного прибора: дифференциальный сканирующий калориметр TA Q2000;
условия испытаний: образец (0,5~1 мг) берут и помещают в алюминиевый тигель для DSC для проведения анализа, способ представляет собой: нагревание от комнатной температуры до 300°C при скорости нагревания 10°C/мин.
Способ с применением термогравиметрического анализатора (TGA) согласно настоящему изобретению:
модель измерительного прибора: термогравиметрический анализатор TA Q5000IR;
условия испытаний: образец (2~5 мг) берут и помещают в платиновый тигель для TGA для проведения анализа, способ представляет собой: нагревание от комнатной температуры до 300°C при скорости нагревания 10°C/мин.
Технический эффект
Предусмотренные в настоящем изобретении соединения 2, 3 и 4, кристаллическая форма IX соединения 1, кристаллические формы I, II, III, IV, V и VI соединения 2, кристаллическая форма VII соединения 3 и кристаллическая форма VIII соединения 4 являются стабильными по своей природе и обладают хорошей растворимостью и превосходной гигроскопичностью, за счет чего имеют многообещающие перспективы в качестве лекарственного средства.
Предусмотренные в настоящем изобретении способы синтеза соединения 1 и его промежуточных соединений, для которых исходные материалы являются дешевыми и доступными, преодолевают недостатки, связанные с высокой токсичностью применяемых реагентов, жесткими условиями реакций, трудностями выделения и очистки и непростым осуществлением производства в промышленных масштабах и т. п.
Описание прилагаемых графических материалов
На фиг. 1 показана XRPD-рентгенограмма кристаллической формы I соединения 2 с применением Cu-Kα излучения.
На фиг. 2 показан профиль DSC кристаллической формы I соединения 2.
На фиг. 3 показан профиль TGA кристаллической формы I соединения 2.
На фиг. 4 показана XRPD-рентгенограмма кристаллической формы II соединения 2 с применением Cu-Kα излучения.
На фиг. 5 показан профиль DSC кристаллической формы II соединения 2.
На фиг. 6 показан профиль TGA кристаллической формы II соединения 2.
На фиг. 7 показана XRPD-рентгенограмма кристаллической формы III соединения 2 с применением Cu-Kα излучения.
На фиг. 8 показан профиль DSC кристаллической формы III соединения 2.
На фиг. 9 показан профиль TGA кристаллической формы III соединения 2.
На фиг. 10 показана XRPD-рентгенограмма кристаллической формы IV соединения 2 с применением Cu-Kα излучения.
На фиг. 11 показан профиль DSC кристаллической формы IV соединения 2.
На фиг. 12 показан профиль TGA кристаллической формы IV соединения 2.
На фиг. 13 показана XRPD-рентгенограмма кристаллической формы V соединения 2 с применением Cu-Kα излучения.
На фиг. 14 показан профиль DSC кристаллической формы V соединения 2.
На фиг. 15 показан профиль TGA кристаллической формы V соединения 2.
На фиг. 16 показана XRPD-рентгенограмма кристаллической формы VI соединения 2 с применением Cu-Kα излучения.
На фиг. 17 показан профиль DSC кристаллической формы VI соединения 2.
На фиг. 18 показан профиль TGA кристаллической формы VI соединения 2.
На фиг. 19 показана XRPD-рентгенограмма кристаллической формы VII соединения 3 с применением Cu-Kα излучения.
На фиг. 20 показан профиль DSC кристаллической формы VII соединения 3.
На фиг. 21 показан профиль TGA кристаллической формы VII соединения 3.
На фиг. 22 показана XRPD-рентгенограмма кристаллической формы VIII соединения 4 с применением Cu-Kα излучения.
На фиг. 23 показан профиль DSC кристаллической формы VIII соединения 4.
На фиг. 24 показан профиль TGA кристаллической формы VIII соединения 4.
На фиг. 25 показана XRPD-рентгенограмма кристаллической формы IX соединения 1 с применением Cu-Kα излучения.
На фиг. 26 показан профиль DSC кристаллической формы IX соединения 1.
На фиг. 27 показан профиль TGA кристаллической формы IX соединения 1.
ПОДРОБНОЕ ОПИСАНИЕ
С целью лучшего понимания содержания настоящего изобретения оно будет дополнительно описываться путем комбинирования следующих конкретных примеров, которые никоим образом не ограничивают содержание настоящего изобретения.
Справочный пример 1. Получение соединения 5
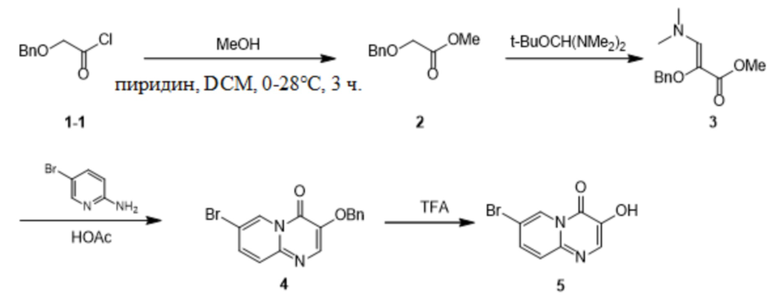
Получение метил-2-(бензилокси)ацетата (2)

Дихлорметан (960 мл) добавляли в трехгорлую круглодонную колбу объемом 3,0 л, добавляли метанол (197,6 г, 247 мл) и пиридин (304,78 мл, 311 моль) и смесь охлаждали до 0°C на водяной бане со льдом. В защитной атмосфере газообразного азота в круглодонную колбу по каплям добавляли 2-(бензилокси)ацетилхлорид (300 г, 1,54 моль) и в ходе добавления температуру поддерживали на уровне 0-10°C. После добавления убирали водяную баню со льдом и реакционный раствор перемешивали при 20°C в течение 1,5 часа. После отбора образцов и выявления TLC (петролейный эфир/этилацетат = 5/1) показала, что реакция завершилась. В круглодонную колбу добавляли воду (1,5 л) и перемешивали в течение 10 минут, обеспечивали разделение слоев и собирали органический слой; затем органический слой промывали с помощью 1,0 моль/л разведенной хлористоводородной кислоты (900 мл × 2), обеспечивали разделение слоев и собирали органический слой; органический слой дополнительно промывали 20% раствором карбоната натрия (600 мл), обеспечивали разделение слоев, органический слой собирали, высушивали с помощью безводного сульфата натрия (150 г) и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта в виде бесцветного масла (284 г, 1,53 моль, выход 97%, чистота 99%). 1H ЯМР (400 МГц, хлороформ-d) ppm 7,37-7,32 (m, 5H), 4,63 (s, 2H), 4,11 (s, 2H), 3,76 (s, 3H); LCMS (ESI) масса/заряд: 202,8 (M+23).
Получение метил-2-(бензилокси)-3-(диметиламино)акрилата (3)

Метил-2-(бензилокси)ацетат (506 г, 2,72 моль) добавляли в круглодонную колбу объемом 3 л, добавляли трет-бутокси-бис-(диметиламино)метан (569 г, 3,26 моль) и поддерживали температуру реакции на уровне 90°C - 100°C с обеспечением ее протекания в течение 14 часов. После отбора образцов и выявления TLC (PE/EA = 5/1) показала, что реакция завершилась. Реакционный раствор охлаждали до 60°C и концентрировали с применением насоса для масла с получением продукта в виде желтого масла (699 г, неочищенный продукт), который непосредственно применяли на следующей стадии.
1H ЯМР (400 МГц, хлороформ-d) ppm 7,44-7,2 (m, 2H), 7,37-7,28 (m, 3H), 6,87 (s, 1H), 4,72 (s, 2H), 3,73 (s, 3H), 2,98 (s, 6H).
Получение 3-(бензилокси)-7-бром-4H-пиридо[1,2-a]пиримидин-4-она (4)

В круглодонную колбу объемом 5 л добавляли метил-2-(бензилокси)-3-(диметиламино)акрилат (318 г, 1,35 моль) и к нему добавляли уксусную кислоту (3 л) и 2-амино-5-бромпиридин (246 г, 1,35 моль). Обеспечивали протекание реакции в реакционном растворе в течение 14 часов при перемешивании с поддержанием температуры на уровне 120°C - 130°C. После отбора образцов и выявления LCMS показала, что реакция практически завершилась. Реакционный раствор охлаждали до 60°C, концентрировали и выпаривали для удаления растворителя, добавляли этилацетат (750 мл), перемешивали в течение 10 мин и фильтровали. К полученному фильтрационному осадку добавляли этилацетат (500 мл), перемешивали в течение 10 мин и фильтровали. Дополнительный полученный фильтрационный осадок промывали этилацетатом (150 мл) и высушивали на центрифуге с получением соединения в виде желтого твердого вещества (319 г, чистота 95%, выход 67,79%).
1H ЯМР (400 МГц, хлороформ-d) d=9,13 (d, J=2,0 Гц, 1H), 8,05 (s, 1H), 7,56 (dd, J=2,0, 9,6 Гц, 1H), 7,46-7,42 (m, 3H), 7,37-7,33 (m, 3H), 5,30 (s, 2H); LCMS (ESI) масса/заряд: 332,6 (изотоп M+1).
Получение 7-бром-3-гидрокси-4H-пиридо[1,2-a]пиримидин-4-она (5)
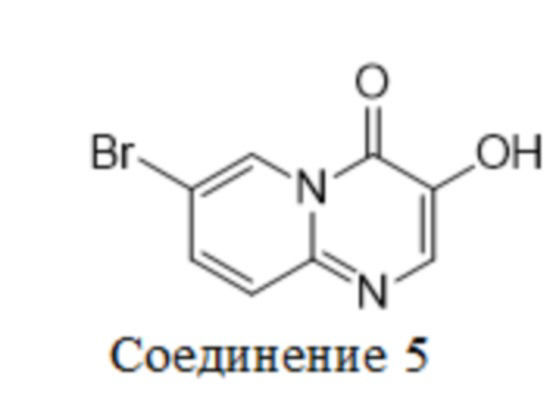
В круглодонную колбу объемом 3 л добавляли трифторуксусную кислоту (1,2 л), к ней добавляли 3-(бензилокси)-7-бром-4H-пиридо[1,2-a]пиримидин-4-он (313 г, 897,9 ммоль) и обеспечивали протекание реакции в реакционном растворе в течение 2 часов при перемешивании с поддержанием температуры на уровне 80°C - 90°C. После отбора образцов и выявления LCMS показала, что реакция практически завершилась. Реакционный раствор охлаждали до 60°C, концентрировали и выпаривали для удаления растворителя, добавляли этилацетат (1,2 л), перемешивали в течение 60 мин и фильтровали. К полученному фильтрационному осадку добавляли этилацетат (400 мл), перемешивали в течение 60 мин и фильтровали. Дополнительный полученный фильтрационный осадок высушивали при пониженном давлении при 40°C в течение 70 часов с получением соединения в виде желтого твердого вещества (191 г, содержание 95,6%, чистота 100%, выход 84,59%).
1H ЯМР (400 МГц, DMSO-d6) d=9,92 (br, 1H), 8,90 (s, 1H), 8,07 (s, 1H), 7,73 (dd, J=2,0, 9,6 Гц, 1H), 7,53 (d, J=9,6 Гц, 1H); MS масса/заряд: 240,9 (M+1), 242,8 (изотоп M+1).
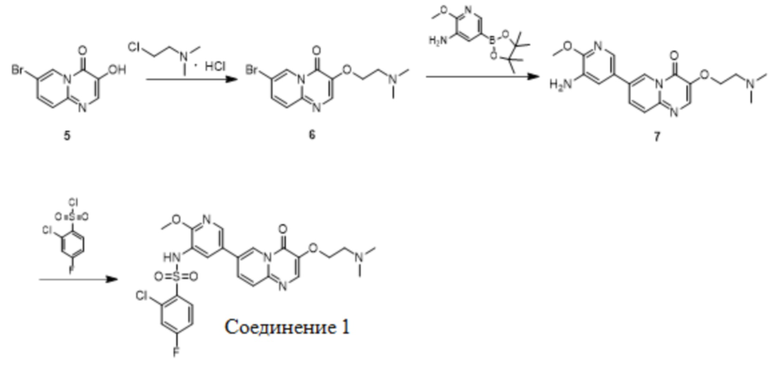
Пример 1. Получение соединения 1
Получение 7-бром-3-(2-(диметиламино)этокси)-4H-пиридо[1,2-a]пиримидин-4-она (6)
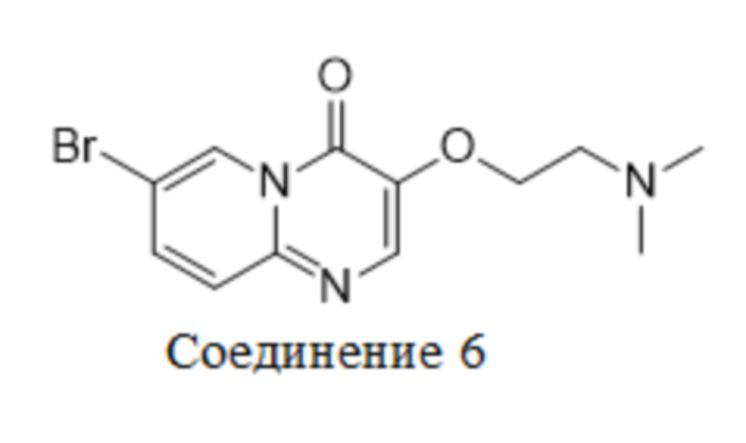
В круглодонную колбу добавляли 7-бром-3-гидрокси-4H-пиридо[1,2-a]пиримидин-4-он (300 г, 1,2 моль) и N,N-диметилформамид (3 л) и температуру в реакторе доводили до 95°C - 100°C. В реакционную колбу добавляли карбонат калия (497,4 г, 3,6 моль) и перемешивали в течение 30 мин. Затем гидрохлорид 2-диметиламиноэтилхлорида разделяли на три порции и вносили их следующим образом: добавление гидрохлорида 2-диметиламиноэтилхлорида (70,6 г, 0,49 моль) в реакционную колбу и перемешивание в течение 30 мин; добавление гидрохлорида 2-диметиламиноэтилхлорида (70,6 г, 0,49 моль) в реакционную колбу и перемешивание в течение 30 мин; и добавление гидрохлорида 2-диметиламиноэтилхлорида (70,6 г, 0,49 моль) в реакционную колбу и перемешивание в течение 2-2,5 часов.
После завершения реакции (что контролировали с помощью HPLC) температуру в реакторе доводили до 15±5°C. Реакционный раствор добавляли в воду (15 л) с последующим экстрагированием с помощью дихлорметана (4,5 л × 4). Органическую фазу объединяли и концентрировали при пониженном давлении при 35±5°C до достижения постоянной массы. К полученному после концентрирования продукту добавляли н-гептан (1,8 л) и перемешивали при 15±5°C в течение 15-16 часов. После фильтрации полученный фильтрационный осадок выпаривали на центрифуге при пониженном давлении при 35±5°C с получением продукта в виде зеленого твердого вещества (280 г, выход 74,09%, чистота 98,22%).
1H ЯМР (400 МГц, CDCl3) d=2,35 (s, 6 H), 2,78 (t, J=5,6 Гц, 2H), 4,25 (t, J=6,0 Гц, 2H), 7,45 (d, J=9,6 Гц, 1H), 7,55 (dd, J=9,6 Гц, 2 Гц, 1H), 8,13 (s, 1H), 9,09 (d, J=2,0 Гц, 1H); LCMS (ESI) масса/заряд: 312 (изотоп M+1).
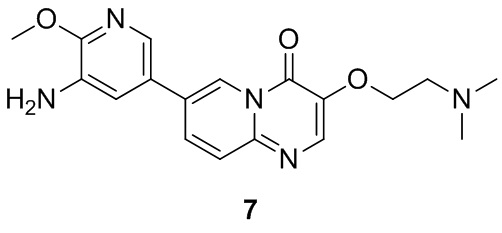
Получение 7-(5-амино-6-метоксипиримидин-3-ил)-3-(2-(диметиламино)этокси)-пиридо[1,2-a]пиримидин-4-она (7)

В реакционную колбу последовательно добавляли 7-бром-3-(2-(диметиламино)этокси)-4H-пиридо[1,2-a]пиримидин-4-он (275 г, 0,87 моль), 2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-амин (249 г, 0,96 моль), 1,4-диоксан (2,75 л), воду (550 мл) и карбонат калия (362 г, 2,62 моль); после барботирования в течение 30-60 мин в реакционную колбу добавляли Pd(dppf)Cl2 (19,2 г, 26 ммоль) и атмосферу газообразного азота в реакционной колбе заменяли 5 раз; температуру в реакционной колбе доводили до 95±5°C и смесь перемешивали в течение 2-2,5 часов при данной температуре. После завершения реакции (что контролировали с помощью HPLC) температуру в реакторе доводили до 15±5°C. В реакционный раствор добавляли н-гептан (6,6 л) и после доведения температуры до 15±5°C смесь перемешивали в течение 2-2,5 часов при данной температуре. После фильтрации полученный фильтрационный осадок высушивали на центрифуге при пониженном давлении при 45±5°C. К полученному остатку добавляли дихлорметан/метанол (V/V = 8/1, 2,75 л), перемешивали при 15±5°C в течение 30-60 мин и фильтровали с получением фильтрационного осадка; и к фильтрационному осадку добавляли дихлорметан/метанол (V/V = 8/1, 1,375 л), перемешивали в течение 30-60 мин при 15±5°C и фильтровали снова с получением другого фильтрационного осадка, который затем промывали с помощью дихлорметана/метанола (V/V = 8/1, 1,375 л). Два полученных фильтрата объединяли и концентрировали при пониженном давлении при 45±5°C. К концентрированному остатку добавляли дихлорметан/метанол (V/V = 2/1, 4,125 л) и растворяли путем перемешивания. К полученному раствору добавляли тиоциануровую кислоту (13,93 г) и активированный уголь (27,5 г), перемешивали при 15±5°C в течение 15-16 часов и фильтровали с помощью диатомовой земли (137,5 г), и полученный фильтрационный осадок промывали с помощью дихлорметана/метанола (V/V = 2/1, 1,375 л x 2). Фильтрат концентрировали при пониженном давлении при 45±5°C. К концентрированному остатку добавляли метанол (1,1 л), перемешивали при 15±5°C в течение 2-3 часов и фильтровали, и полученный фильтрационный осадок промывали с помощью метанола (137,5 мл) и выпаривали на центрифуге при пониженном давлении при 45±5°C с получением продукта в виде желтого твердого вещества (270 г, чистота 97,98%, выход 84,24%).
1H ЯМР (400 МГц, DMSO-d6) d=8,92 (d, J=1,6 Гц, 1H), 8,24 (s, 1H), 8,04 (dd, J=9,6 Гц, 2 Гц, 1H), 7,80 (d, J=2 Гц, 1H), 7,67 (d, J=9,6 Гц, 1H), 7,27 (d, J=2,0 Гц, 1H), 5,24 (s, 2H), 4,19 (t, J=6,0 Гц, 2H), 3,93 (s, 3H), 2,66 (t, J=6,0 Гц, 2H), 2,25 (s, 6H); LCMS (ESI) масса/заряд: 356 (M+1).
Получение соединения 1

В реакционную колбу добавляли 7-(5-амино-6-метоксипиримидин-3-ил)-3-(2-(диметиламино)этокси)-пиридо[1,2-a]пиримидин-4-он (265 г, 0,72 моль) и пиридин (2,65 л); после охлаждения до 5±5°C в реакционную колбу по каплям добавляли раствор 2-хлор-4-фторбензолсульфонилхлорида (252 г, 1,08 моль) в пиридине (504 мл); после завершения добавления реакционный раствор доводили до температуры, составляющей 30°C - 35°C, и перемешивали в течение 2-3 часов при данной температуре. После завершения реакции (что контролировали с помощью HPLC) реакционный раствор концентрировали при пониженном давлении при 45±5°C до достижения веса, превышающего в 2,5-3 раза вес соединения 7. К концентрированному продукту добавляли дихлорметан (3,7 л), перемешивали при 25±5°C в течение 30 мин и затем концентрировали при пониженном давлении при 45±5°C до достижения веса, превышающего в 2,5-3 раза вес соединения 7. К концентрированному продукту добавляли дихлорметан (3,7 л) и суспендировали при 25±5°C в течение 2-3 часов. После фильтрации фильтрационный осадок собирали и выпаривали на центрифуге при 45±5°C до достижения веса, превышающего в 1,3-1,7 раза вес соединения 7. К образованному в результате выпаривания на центрифуге продукту добавляли дихлорметан (1,85 л) и суспендировали при 25±5°C в течение 2-3 часов. После фильтрации фильтрационный осадок собирали, выпаривали на центрифуге при 45±5°C до достижения веса, превышающего в 1,2-1,4 раза вес соединения 7, и затем высушивали в вакууме при 45°C в течение 3-4 часов до достижения веса, превышающего в 1,2-1,3 раза вес соединения 7. К полученному неочищенному продукту добавляли ацетонитрил (2,12 л) и суспендировали при 55±5°C в течение 15-16 часов. После суспендирования раствор охлаждали до 25±5°C и фильтровали, и полученный фильтрационный осадок собирали и выпаривали на центрифуге при 45±5°C до достижения веса, превышающего в 1,1-1,2 раза вес соединения 7. К образованному в результате выпаривания на центрифуге продукту добавляли ацетонитрил (1,9 л) и суспендировали при 55±5°C в течение 15-16 часов. После суспендирования раствор охлаждали до 25±5°C и фильтровали, и полученный фильтрационный осадок выпаривали на центрифуге при 45±5°C до достижения веса, превышающего в 1,0-1,1 раза вес соединения 7. К образованному в результате выпаривания на центрифуге продукту добавляли метанол (5,3 л) и активированный уголь (53 г) и перемешивали при 75±5°C в течение 2-3 часов. Фильтрацию осуществляли с помощью диатомовой земли (132 г) и собирали полученный фильтрационный осадок, добавляли смешанный растворитель, содержащий дихлорметан и метанол (V/V = 4/1, 7,95 л), и перемешивали при 25±5°C в течение 30-60 мин. Снова осуществляли фильтрацию и два полученных фильтрата объединяли и концентрировали при 45±5°C до достижения веса, превышающего в 1,01-1,03 раза вес соединения 7. К концентрированному продукту добавляли воду (4,24 л) и этанол (1,06 л), перемешивали при 25±5°C в течение 5-10 мин, по каплям добавляли насыщенный водный раствор гидрокарбоната натрия (1,3 л) и дополнительно перемешивали в течение 2-3 часов. После фильтрации фильтрационный осадок выпаривали на центрифуге при 45±5°C до достижения веса, превышающего в 1,1-1,3 раза вес соединения 7. К образованному в результате выпаривания на центрифуге продукту добавляли этанол (1,59 л) и суспендировали при 75±5°C в течение 15-16 часов. После суспендирования раствор охлаждали до 25±5°C и фильтровали, и фильтрационный осадок собирали и выпаривали на центрифуге при 45±5°C до достижения веса, превышающего в 0,89-0,92 раза вес соединения 7. К образованному в результате выпаривания на центрифуге продукту добавляли этанол (1,59 л) и суспендировали при 75±5°C в течение 15-16 часов. После суспендирования раствор охлаждали до 25±5°C и фильтровали, и полученный фильтрационный осадок собирали и выпаривали на центрифуге при 45±5°C до достижения веса, превышающего в 0,87-0,9 раза вес соединения 7. К образованному в результате выпаривания на центрифуге продукту добавляли воду (2,35 л) и перемешивали при 45±5°C в течение 61±1 часа. Смешанный раствор охлаждали до 25±5°C и фильтровали. Полученный фильтрационный осадок собирали, добавляли воду (2,35 л), перемешивали при 25±5°C в течение 2-3 часов и фильтровали. Полученный фильтрационный осадок собирали, высушивали в вакууме при 60°C в течение 15-16 часов и затем просеивали через сито 60 меш с получением продукта в виде светло-желтого твердого вещества (190 г, чистота 98,33%, выход 47,36%).
1H ЯМР (400 МГц, DMSO-d6-d) d=2,96 (t, J=6,0 Гц, 2H), 3,72 (s, 3H), 4,27 (t, J=5,2 Гц, 2H), 7,32 (td, J=8,8, 2,8 Гц, 1H), 7,60 (dd, J=8,4, 2,4 Гц, 1H), 7,70 (d, J=9,2 Гц, 1H), 7,75 (d, J=2,0 Гц, 1H), 7,95-8,05 (m, 2H), 8,15 (d, J=1,6 Гц, 1H), 8,27 (s, 1H), 8,85 (s, 1H); LCMS (ESI) масса/заряд: 548 (M+1).
Пример 2. Получение кристаллической формы IX соединения 1
7-(5-Амино-6-метоксипиримидин-3-ил)-3-(2-(диметиламино)этокси)-пиридо[1,2-a]пиримидин-4-он (2,5 г, 6,75 ммоль, 1,0 экв.) растворяли в пиридине (25 мл), по каплям добавляли 2-хлор-4-фторбензолсульфонилхлорид (2,01 г, 8,78 ммоль, 1,3 экв.) при 0°C и перемешивали при 10°C - 20°C в течение 16 часов. После завершения реакции растворитель выпаривали на центрифуге с получением неочищенного продукта. Неочищенный продукт очищали с помощью колонки (DCM/MeOH: 10/1-4/1). Получали продукт в виде желтого твердого вещества (2,4 г, чистота 98,31%, выход 63,79%). Вышеуказанное желтое твердое вещество (1,3 г, 2,37 ммоль) отделяли с помощью препаративной HPLC (в нейтральной среде). Жидкость, полученную при разделении с помощью препаративной HPLC (в нейтральной среде), экстрагировали с помощью DCM (500 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия (100 г), а затем фильтровали и полученный фильтрат высушивали на центрифуге с получением продукта в виде белого твердого вещества, являющегося кристаллической формой IX соединения 1 (970 мг, 1,75 ммоль, чистота 99%, выход 73,94%).
Пример 3. Получение кристаллической формы I соединения 2
В трехгорлую круглодонную колбу R1 объемом 1,0 л, оснащенную мешалкой, добавляли 7-(5-амино-6-метоксипиримидин-3-ил)-3-(2-(диметиламино)этокси)-пиридо[1,2-a]пиримидин-4-он (29,0 г, 81,60 ммоль, 1,0 экв.) и пиридин (290 мл). R1 помещали на ледяную баню и охлаждали до 0-5°C. Раствор 2-хлор-4-фторбензолсульфонилхлорида (24,30 г, 106,08 ммоль, 1,3 экв.) в пиридине (60 мл) по каплям добавляли в R1 на протяжении приблизительно 30 мин, и обеспечивали нагревание реакционного раствора естественным путем до 20°C, и обеспечивали реакцию при перемешивании в течение 16 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении для удаления пиридина с получением 80 г неочищенного продукта в виде красного твердого вещества. Отбирали 64 г вышеуказанного неочищенного продукта и помещали в круглодонную колбу R2 объемом 1,0 л, и в R2 добавляли дихлорметан (350 мл). Затем содержимое R2 перемешивали при 15°C в течение 2 часов и фильтровали, и полученный фильтрационный осадок собирали и высушивали с получением светло-красного твердого вещества (33,4 г, выход 77%, чистота 99,4%). Отбирали 30 г вышеуказанного твердого вещества и помещали в круглодонную колбу R3 объемом 1 л, и в R3 добавляли метанол (600 мл) и активированный уголь (6 г, 20%). Смесь помещали на масляную баню при 70°C и перемешивали в течение 12 часов. Смесь фильтровали с помощью диатомовой земли (15 г) пока она была горячей. Фильтрат собирали и высушивали на центрифуге с получением продукта в виде желтого твердого вещества (22,6 г, чистота 97,47%). К вышеуказанному твердому веществу добавляли ацетонитрил (150 мл); полученную смесь перемешивали на масляной бане при 85°C в течение 12 часов, охлаждали до 20°C и фильтровали; и фильтрационный осадок собирали и высушивали с получением указанного в заголовке продукта в виде белого твердого вещества, являющегося кристаллической формой I соединения 2 (21 г, выход 44,3%, чистота 100%). Молярное соотношение хлорид-иона соединения 2 и хлорид-иона соединения 1 составляло 1:1, что определяли с помощью ионообменной хроматографии.
1H ЯМР (400 МГц, DMSO-d6-d) d=2,91 (s, 6H), 3,53 (t, 2H), 3,71 (s, 3H), 4,52 (t, 2H), 7,38 (m, 1H), 7,77 (m, 2H), 7,97 (m, 2H), 8,16 (m, 1H), 8,45 (m, 2H), 8,98 (s, 1H).
Пример 4. Получение кристаллической формы II соединения 2
Отбирали приблизительно 50 мг кристаллической формы I соединения 2 и добавляли 0,4 мл ацетона с образованием суспензии. Образец суспензии помещали в смешивающий прибор при постоянной температуре (40°C) и встряхивали в течение 2 дней (с защитой от света). Остаточное твердое вещество центрифугировали и высушивали в вакуумном сушильном шкафу при 40°C в течение ночи с получением кристаллической формы II соединения 2.
Пример 5. Получение кристаллической формы III соединения 2
Процедура получения кристаллической формы III является идентичной процедуре получения кристаллической формы II, за исключением того, что растворитель ацетон меняли на изопропанол. Получали кристаллическую форму III соединения 2.
Пример 6. Получение кристаллической формы IV соединения 2
Процедура получения кристаллической формы IV является идентичной процедуре получения кристаллической формы II, за исключением только того, что растворитель ацетон меняли на этилацетат. Получали кристаллическую форму IV соединения 2.
Пример 7. Получение кристаллической формы V соединения 2
Кристаллическую форму I соединения 2 (2,0 г, 3,42 ммоль) помещали в одногорлую колбу R1 объемом 500 мл и твердое вещество полностью растворяли путем добавления DCM/MeOH (2/1, 200 мл) при перемешивании. Раствор подвергали воздействию пониженного давления при 40°C для удаления растворителя с получением 2,0 г желтого твердого вещества; отбирали 1 г твердого вещества и помещали в одногорлую колбу объемом 50 мл с последующим добавлением этанола (6 мл); и полученную смесь помещали на масляную баню при 80°C при перемешивании в течение 12 часов и затем нагревание прекращали. Смесь охлаждали до 20°C при перемешивании и затем фильтровали, и фильтрационный осадок высушивали с получением кристаллической формы V соединения 2.
Пример 7. Получение кристаллической формы VI соединения 2
В трехгорлую круглодонную колбу R1 объемом 2,0 л, оснащенную механической мешалкой, добавляли 7-(5-амино-6-метоксипиримидин-3-ил)-3-(2-(диметиламино)этокси)-пиридо[1,2-a]пиримидин-4-он (70,0 г, 222,90 ммоль, 1,0 экв., чистота 99,4%) и пиридин (700 мл) и R1 помещали на ледяную баню и охлаждали до 0-5°C. Раствор 2-хлор-4-фторбензолсульфонилхлорида (70,81 г, 293,67 ммоль, 1,5 экв., чистота 95%) в пиридине (140 мл) по каплям добавляли в R1 в течение приблизительно 30 мин. R1 помещали на масляную баню при 30°C и обеспечивали реакцию при перемешивании в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении для удаления растворителя пиридина с получением неочищенного продукта в виде красного твердого вещества (200 г). К остатку добавляли дихлорметан (1,0 л) и перемешивали при 20°C в течение 3 часов. После фильтрации собирали фильтрационный осадок. К фильтрационному осадку добавляли ацетонитрил (1,2 л); полученный реакционный раствор нагревали с обратным холодильником при 85°C в течение 12 часов, охлаждали до 20°C и фильтровали; и другой фильтрационный осадок собирали и высушивали с получением твердого вещества (92 г). К твердому веществу фильтрационного осадка добавляли метанол (2 л) и активированный уголь (14 г), нагревали с обратным холодильником при перемешивании в течение 3 часов, фильтровали с помощью диатомовой земли (40 г) пока он все еще был горячим и промывали 500 мл. Фильтрат высушивали на центрифуге при пониженном давлении при 40°C с получением твердого вещества (83 г). К полученному твердому веществу добавляли ацетонитрил (800 мл) и смесь нагревали с обратным холодильником при 85°C в течение ночи, охлаждали до 20°C и фильтровали, и полученный фильтрационный осадок высушивали с получением 77 г белого твердого вещества. Отбирали 72 г белого твердого вещества и полностью растворяли в метаноле и высушивали на центрифуге с получением кристаллической формы VI соединения 2.
Пример 8. Получение кристаллической формы VII соединения 3
Соединение 1 (997,34 мг, 1,82 ммоль, 1,00 экв.) помещали в стеклянный флакон объемом 5 мл, добавляли этанол/воду (7,5 мл/2,5 мл) и перемешивали при комнатной температуре (15°C) в течение 0,1 часа, при этом значительное количество твердого вещества не растворялось. К смеси добавляли малеиновую кислоту (211,25 мг, 1,82 ммоль, 1,00 экв.), и перемешивали при комнатной температуре (15°C) в течение 18 часов, и обеспечивали полное растворение твердого вещества и образование желтого раствора. Полученный раствор высушивали на центрифуге при пониженном давлении при 40°C до достижения объема, составляющего 2 мл, добавляли EA (20 мл), перемешивали в течение 0,5 часа и фильтровали; образованный фильтрационный осадок высушивали на центрифуге при пониженном давлении при 40°C с получением кристаллической формы VII соединения 3.
1H ЯМР (400 МГц, DMSO-d6) d ppm 2,94 (s, 6H), 3,51 - 3,56 (m, 2H), 3,71 (s, 3H), 4,36 - 4,59 (m, 2H), 6,03 (s, 2H), 7,18 - 7,48 (m, 1H), 7,65 - 7,90 (m, 2H), 7,92 - 8,09 (m, 2H), 8,17 (dd, J=9,29, 1,76 Гц, 1H), 8,35 - 8,55 (m, 2H), 8,99 (s, 1H).
Пример 9. Получение кристаллической формы VIII соединения 4
Соединение 1 (997,34 мг, 1,82 ммоль, 1,00 экв.) помещали в стеклянный флакон объемом 5 мл, добавляли этанол/воду (7,5 мл/2,5 мл) и перемешивали при комнатной температуре (15°C) в течение 0,5 часа, при этом значительное количество твердого вещества не растворялось. К смеси добавляли лимонную кислоту (382,45 мг, 1,82 ммоль, 1,00 экв.) и перемешивали при комнатной температуре (15°C) в течение 18 часов с получением молочно-белой суспензии в реакционном флаконе. Полученный смешанный раствор высушивали на центрифуге при пониженном давлении при 40°C до достижения объема, составляющего 2 мл, добавляли EA (20 мл), перемешивали в течение 0,5 часа и фильтровали; образованный фильтрационный осадок высушивали на центрифуге при пониженном давлении при 40°C с получением кристаллической формы VIII соединения 4.
1H ЯМР (400 МГц, DMSO-d6) d ppm 2,56-2,68 (m, 4H), 2,76 (s, 6 H), 3,31 (m, 2H), 3,72 (s, 3H), 4,37 - 4,40 (m, 2H), 7,35-7,37 (m, 1H), 7,71 - 7,78 (m, 2H), 7,91 - 7,95 (m, 1H), 7,95 - 8,09 (m, 1H), 8,11 - 8,13 (m, 1H), 8,36 - 8,37 (m, 2H), 8,96 (d, J=1,6, 1H).
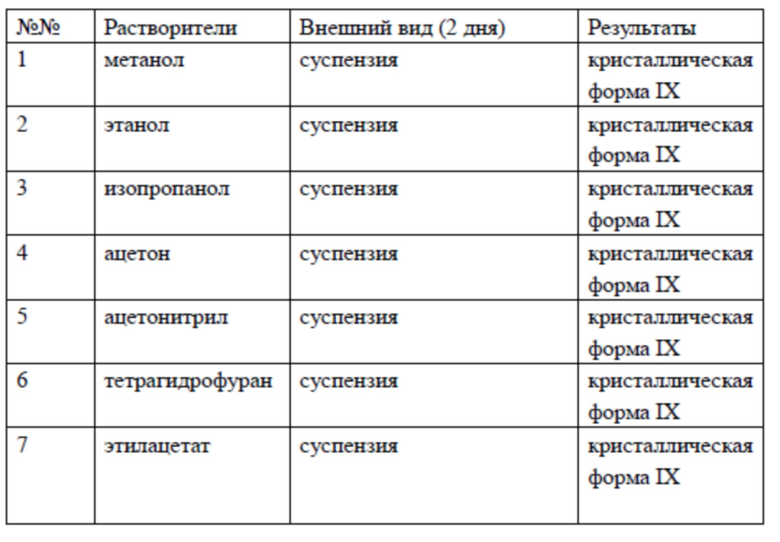
Пример тестирования 1. Тестирование стабильности кристаллической формы IX соединения 1 в разных растворителях
Отбирали несколько образцов кристаллической формы IX соединения 1 в подходящем количестве, соответственно добавляли к 0,3-0,4 мл простого растворителя или смешанного растворителя, как показано в следующей таблице, и перемешивали при 40°C. После перемешивания в течение 2 дней образцы центрифугировали. Собирали твердое вещество в образцах и его кристаллическое состояние выявляли с помощью XRPD. Результаты показаны в таблице 10.
Таблица 10. Эксперименты в отношении стабильности кристаллической формы IX в виде свободного основания в разных растворителях

Пример тестирования 2. Тестирование стабильности кристаллической формы IX соединения 1 в твердом состоянии в условиях высокой температуры, высокой влажности и яркого света
Взвешивали приблизительно 10 мг кристаллической формы IX соединения 1 и помещали на дно стеклянного сосуда для образцов с образованием тонкого слоя. В случае образцов, которые помещали в условия с температурой 60°C и комнатной температурой/92,5% RH, горловины сосудов накрывали алюминиевой фольгой и в алюминиевой фольге проделывали несколько отверстий, что обеспечивало свободный доступ воздуха из внешней среды к образцам; в случае образцов, которые подвергали воздействию яркого света (5 клк), сосуды герметично закупоривали с помощью завинчивающихся крышек. Отбирали другой образец, содержащий 15 мг кристаллической формы IX, и помещали его согласно вышеописанному способу для выявления кристаллической формы образца. Образцы, помещенные в разные условия, отбирали и выявляли в день 5 и день 10, и результаты выявления сравнивали с результатами первоначального выявления в день 0. Результаты тестирования показаны в таблице 11 ниже.
Таблица 11. Тестирование стабильности кристаллической формы IX соединения 1 в твердом состоянии

Результаты экспериментов: кристаллические формы по настоящему изобретению обладают хорошей стабильностью и с ними можно легко изготовлять лекарственный препарат.
Пример тестирования 3. Тестирования ферментативной активности in vitro
Анализ I с помощью ADP-Glo
Разбавление соединений
Тестируемые соединения разбавляли с получением трехкратного градиента концентрации и в общей сложности получали 10 значений концентрации (от 10000 нМ до 0,5 нМ).
Экспериментальный способ
В реакционный планшет (PerkinElmer № 6007299) переносили 50 нл тестируемых соединений по настоящему изобретению и добавляли3 мкл фермент/субстратной смеси (0,33 нМ PI3K-альфа, Millipore № 14-602-K/166,5 мкМ PIP2); после инкубации в течение 20 мин добавляли 2 мкл раствора АТФ (100 мкМ) для инициирования реакции; после 2 часов проведения реакции при комнатной температуре реакцию с киназой останавливали путем добавления 5 мкл реагента ADP-Glo с последующей инкубацией при комнатной температуре в течение 60 мин для обеспечения возможности полного расщепления оставшихся непрореагировавших молекул АТФ; и к полученному раствору добавляли 10 мкл реагента для выявления киназы и инкубировали при комнатной температуре в течение 40 мин и затем измеряли уровень флуоресценции с помощью Envision. Все из PIP2, АТФ, реагента ADP-Glo и реагентов для анализа киназы были взяты из набора для анализа киназы ADP-Glo (Promega № V1792).
Анализ данных
IC50 рассчитывали с применением стандартного 4-параметрического способа подгонки (Model 205, XL-fit, iDBS).
Активность тестируемых соединений по настоящему изобретению в отношении mTOR-киназы тестировали с помощью нижеприведенного способа тестирования.
Реакционный буфер: 20 мМ Hepes (pH 7,5), 10 мМ MgCl2, 2 мМ MnCl2, 1 мМ EGTA, 0,02% Brij35, 0,02 мг/мл BSA, 0,1 мМ Na3VO4, 2 мМ DTT, 2% DMSO.
Фермент для реакции: экспрессируемый в клетках насекомых гуманизированный рекомбинантный фрагмент mTOR с GST-меткой на N-конце (аминокислоты 1360-2549, молекулярная масса = 163,9 кДа).
Реакционный субстрат: экспрессируемый в бактериях гуманизированный рекомбинантный полноразмерный 4EBP1 с His-меткой на N-конце (молекулярная масса = 13,6 кДа).
Условия реакции: 3 мкМ 4EBP1 и 10 мкМ АТФ.
Процедура проведения реакции
1. Реакционный субстрат и другие факторы, обеспечивающие реакцию, добавляли к свежеполученному реакционному буферу.
2. К реакционному субстрату добавляли киназу и осторожно перемешивали.
3. Соединение, растворенное в 100% DMSO, переносили в реакционный раствор, содержащий киназу, с применением технологии акустического переноса жидкостей (Echo 550; нанолитровый интервал) и затем инкубировали при комнатной температуре в течение 20 мин.
4. К реакционной системе добавляли 32P-АТФ в соответствующей концентрации.
5. Инкубировали при комнатной температуре в течение 2 часов.
6. Активность киназы выявляли с помощью способа связывания на фильтре P81.
Результаты экспериментов показаны в таблице 12.
Таблица 12. Результаты тестирования ферментативной активности in vitro

Примечание: A≤1 нМ; 200 нМ<D.
Вывод: соединение 1 обладает значительным ингибирующим эффектом в отношении PI3K (p110α), но более слабым ингибирующим эффектом в отношении mTOR.
Анализ II с помощью ADP-Glo
Процедура проведения эксперимента
1) Соединение разбавляли с применением Echo от компании Labcyte, 50 нл соединения переносили в аналитический планшет и центрифугировали при скорости 1000 об/мин в течение 10 сек.
2) Получали смешанный раствор киназы/липидного субстрата и смешанный раствор киназного реакционного буфера/липидного субстрата; смешанный раствор киназы/липидного субстрата добавляли в колонки 3-24 аналитического планшета по 3 мкл на лунку; смешанный раствор киназного реакционного буфера/липидного субстрата добавляли в колонки 1-2 аналитического планшета по 3 мкл на лунку; и планшет центрифугировали при скорости 1000 об/мин в течение 10 сек.
3) Получали раствор АТФ и добавляли в аналитический планшет по 2 мкл на лунку; планшет центрифугировали при скорости 1000 об/мин в течение 10 сек, встряхивали и перемешивали в режиме 2-й передачи на планшетном шейкере в течение 1 мин, дополнительно центрифугировали при скорости 1000 об/мин в течение 10 сек и инкубировали в течение 120 мин при 23°C.
4) Получали реагент ADP-Glo® и добавляли в аналитический планшет по 5 мкл на лунку; планшет центрифугировали при скорости 1000 об/мин в течение 10 сек, встряхивали и перемешивали в режиме 2-й передачи на планшетном шейкере в течение 1 мин, дополнительно центрифугировали при скорости 1000 об/мин в течение 10 сек и инкубировали в течение 60 мин при 23°C.
5) Получали реагент для выявления киназы, добавляли в аналитический планшет по 10 мкл на лунку; планшет центрифугировали при скорости 1000 об/мин в течение 10 сек, встряхивали и перемешивали в режиме 2-й передачи на планшетном шейкере в течение 1 мин, дополнительно центрифугировали при скорости 1000 об/мин в течение 10 сек, инкубировали в течение 30 мин при 23°C и затем считывали на мультимаркерном детекторе Envision.
Анализ данных
Результаты IC50 анализировали с помощью XLfit5 (Formula 205) от компании IDBS.
Соответствующие эксперименты и анализ осуществляли с использованием вышеописанной процедуры проведения эксперимента с соединением BKM-120 в качестве положительного контроля лекарственного средства.
Результаты экспериментов
Значения IC50 в отношении ингибирования соединением 1 активности PI3Kα, PI3Kβ, PI3Kδ и PI3Kγ составляли соответственно 0,6±0,2 нМ, 9,9±2,7 нМ, 0,5±0,1 нМ и 7,0±0,9 нМ (n = 2). В отличие от этого, значения IC50 в отношении ингибирования положительным контролем лекарственного средства BKM120 (ингибитор PI3K, бупарлизиб) активности PI3Kα, PI3Kβ, PI3Kδ и PI3Kγ составляли соответственно 24,7±4,7 нМ, 241,6±50,6 нМ, 68,8±25,0 нМ и 111,9±15,2 нМ соответственно.
Вывод: соединение 1 проявляет крайне высокую ингибирующую активность в отношении всех четырех подтипов PI3K.
Пример тестирования 4. Тестирования клеточной активности in vitro
Экспериментальные стадии и способы
1) Клетки MCF-7 инокулировали при плотности 2,5×104 клеток/лунка в 96-луночные планшеты (применяемая культуральная среда должна быть полной культуральной средой, содержащей 10% FBS).
2) На следующий день среду в каждой лунке удаляли. Определенную концентрацию (предварительный скрининг) или серию концентраций (тестирование IC50) тестируемых соединений растворяли в культуральной среде без сыворотки крови и добавляли в 96-луночные планшеты с культивированием клеток в течение 2 часов.
3) Инсулин растворяли в культуральной среде без сыворотки крови, добавляли к культивируемым клеткам и инкубировали в течение 30 минут, при этом конечная концентрация инсулина составляла 10 мг/мл.
4) В ходе периода ожидания прохождения реакции получали лизирующий раствор согласно следующему способу:
a) раствор усилителя доставали из холодильника для предварительного оттаивания;
b) раствор усилителя разбавляли в 10 раз с помощью 5× лизирующего буфера с получением концентрированного лизирующего раствора;
c) концентрированный лизирующий раствор разбавляли в 5 раз с помощью бидистиллированной воды с получением лизирующего раствора.
5) Полностью удаляли среду из каждой лунки и каждую лунку один раз быстро промывали с помощью PBS.
6) В каждую лунку добавляли 150 мкл свежеполученного лизирующего раствора и встряхивали при комнатной температуре в течение 10 мин.
7) После подтверждения того, что все клетки отделились, лизирующий раствор вместе с клеточным дебрисом переносили в пробирку объемом 1,5 мл.
8) Пробирку несколько раз центрифугировали на вортексе для обеспечения полного смешивания лизирующего раствора и клеток и затем смесь центрифугировали при 4°C при 12000g в течение 10 мин.
9) Рассчитывали число необходимых полосок в микролуночном планшете для ELISA-one. Лишние полоски убирали из корпуса, возвращали в пакет для хранения и герметично закрывали. Перед применением полосок микролуночного планшета каждую лунку промывали с помощью 200 мкл бидистиллированной воды для удаления консерванта.
10) В каждую лунку добавляли 50 мкл смеси антител. (Раствор смеси антител получали путем смешивания в равных объемах реагента на основе антител к среде и реагента на основе антител, меченных ферментом. Для получения смеси антител не требовалось центрифугирование на вортексе.)
11) В каждую лунку микролуночного планшета для ELISA-one добавляли 25 мкл клеточных лизатов. Микролуночный планшет накрывали клейкой герметизирующей пленкой и инкубировали на осцилляторе для микролуночных планшетов при комнатной температуре в течение 1 часа.
12) Каждую лунку 3 раза промывали с помощью 150 мкл 1× промывочного буфера. После последнего промывания промывочный буфер полностью удаляли из лунок. При необходимости можно допускать нахождение 1× промывочного буфера в микролуночном планшете в течение не более 30 мин с тем, чтобы в течение данного периода времени можно было получить смешанный раствор субстрата.
13) Смешанный раствор субстрата необходимо получать непосредственно перед каждым применением, 100 мкл смешанного раствора субстрата добавляли в каждую лунку и микролуночный планшет герметично закрывали с помощью оловянной фольги и инкубировали на осцилляторе для микролуночных планшетов при комнатной температуре в течение 10 мин.
14) В каждую лунку добавляли 10 мкл останавливающего раствора и слегка перемешивали (5-10 сек) на осцилляторе для микролуночных планшетов.
15) Соответствующую отсортированную группу ELISA-one формировали и использовали для считывания интенсивности сигнала флуоресценции.
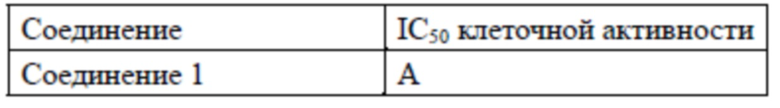
Результаты экспериментов показаны в таблице 13.
Таблица 13. Результаты тестирования клеточной активности in vitro
Примечание: A≤50 нМ.
Пример тестирования 5. Эксперимент по оценке эффективности in vivo
Исследования проводили для определения того, обладают ли тестируемые лекарственные средства эффективностью in vivo на животной модели рака толстой кишки человека CO-04-0032 и животной модели рака желудка ST-02-0013. Описания питания животных, ингредиентов корма, экспериментальных наблюдений, экспериментальных показателей, прекращения эксперимента, а также анализа данных в тестированиях были следующими.
Питание животных. После доставки животных следует кормить в экспериментaльных условиях в течение 3-7 дней перед началом эксперимента. Животных содержали в клетках с IVC (системой индивидуальной вентиляции) (по 5 животных в клетке) в вивариях класса SPF. Все клетки, подстилку и питьевую воду было необходимо стерилизовать перед применением, и записи о стерилизации показаны в приложении. Весь лабораторный персонал в вивариях должен носить защитную спецодежду и латексные перчатки во время работы. В информационной карте каждой клетки должно быть указано число животных в клетке, пол, линия, дата прибытия, режим приема препарата, номер эксперимента, группа и дата начала экспериментов. Клетки, корм и питьевую воду меняли два раза в неделю. Климат при кормлении и условия освещения были следующими:
температура: 20~26°C;
влажность: 40~70%;
светотемновой цикл: 12 ч с освещением, 12 ч без освещения.
Ингредиенты корма. Корм соответствовал стандарту идентификации продуктов питания для лабораторных животных. Максимальное содержание загрязняющих примесей находится в пределах контролируемого диапазона, и производитель ответственен за регулярную проверку. В качестве питьевой использовали питьевую воду, стерилизованную в автоклаве.
Группирование животных. Перед осуществлением введения животных взвешивали и измеряли объем опухоли. Животных группировали случайным образом согласно объему опухоли (схема рандомизированных блоков).
Наблюдение. Экспериментальный протокол и его любые модификации проводили с одобрением Институционального комитета по содержанию и использованию лабораторных животных (IACUC) WuXi AppTec, Шанхай. Использование и условия содержания экспериментальных животных осуществлялись согласно правилам Ассоциации по аттестации и аккредитации содержания лабораторных животных (AAALAC). Ежедневно вели наблюдения за состоянием здоровья и случаями гибели животных, и регулярные обследования включали наблюдение роста опухоли и влияния лечения с применением лекарственного средства на ежедневное поведение животных и показатели, такие как поведение, прием пищи и воды, изменение массы тела (с измерением массы тела два раза в неделю), внешний вид или другие отклоняющиеся от нормы обстоятельства. Случаи гибели животных и побочные эффекты регистрировали с учетом числа животных в каждой группе, и соответствующие записи показаны в приложении.
Экспериментальные показатели. Экспериментальные показатели использовали для установления того, подавлялся, замедлялся или останавливался ли рост опухоли. Диаметр опухоли измеряли два раза в неделю с помощью штангенциркуля. Объем опухоли рассчитывали по формуле: V = 0,5a × b2, где a и b представляют собой соответственно диаметр по длинной оси и диаметр по короткой оси опухоли. Подавление роста опухоли (TGI) соединением оценивали с помощью показателей T-C (дни) и T/C (%). T-C (дни) означает показатель замедления роста опухоли, где T представляет собой среднее число дней, необходимое для достижения предварительно заданного объема опухоли (например, 1,000 мм3) в группе с введением, и C представляет собой среднее число дней, необходимое для достижения того же объема опухоли в контрольной группе. Процентное значение T/C (%) означает скорость подавления роста опухоли, где T и C представляют собой соответственно вес опухоли (объем опухоли) в группе с введением и контрольной группе в определенный день.
Скорость подавления роста опухоли рассчитывали по следующей формуле: TGI (%) = [1-(Ti-T0)/(Vi-V0)] × 100, где Ti представляет собой средний объем опухоли в группе с введением в определенный день, и T0 представляет собой средний объем опухоли в группе с введением в момент начала введения; Vi представляет собой средний объем опухоли в контрольной группе, получающей среду-носитель, в определенный день (такой же день, как и для Ti), и V0 представляет собой средний объем опухоли в контрольной группе, получающей среду-носитель, в момент начала введения. В конце эксперимента измеряли вес опухоли и рассчитывали процентное отношение T/C, где T и C представляют собой соответственно вес опухоли в группе с введением и контрольной группе, получающей среду-носитель.
Прекращение эксперимента. Если состояние здоровья животного продолжает ухудшаться, или объем опухоли у животного составляет более чем 2000 мм3, или у животного имеется тяжелое недомогание или боль, то животное должно быть подвергнуто эвтаназии. Ветеринара уведомляют и животных подвергают эвтаназии при появлении следующих состояний:
явная исхудалость, потеря веса более чем на 20%;
неспособность свободного доступа к пище и воде;
достижение в контрольной группе среднего объема опухоли 2000 мм3, в случае чего, эксперимент следует прекратить;
животное имеет следующие клинические проявления, и они продолжают ухудшаться:
- пилоэрекция,
- выгнутая спина,
- бледные уши, нос, глаза или ступни,
- быстрое дыхание,
- припадки,
- постоянная диарея,
- обезвоживание,
- замедленные движения,
- издавание звуков.
Анализ данных. Для сравнения трех или более групп применяли однофакторный ANOVA. При наличии значимых различий в величине значения F после анализа ANOVA следует осуществлять множественные сравнения. Все данные анализировали с помощью SPSS 17.0. Значение p < 0,05 указывало на наличие статистически значимой разницы.
Исследования фармакодинамики in vivo тестируемых лекарственных средств на моделях с подкожным ксенотрансплантатом опухоли из клеток рака толстой кишки человека CO-04-0032
Схема эксперимента
Создание трансплантационной модели опухоли человека: модель рака толстой кишки человека CO-04-0032 изначально получали из образцов опухоли, удаленных хирургическим путем. Приобретение и использование образцов строго удовлетворяло требованиям этических норм и нормативным актам государства, госпиталей и компаний, включая информированное согласие пациента. Способ создания модели строго соответствовал внутреннему SOP компании. Правила присвоения названия поколению пассажа состояли в следующем: после инокуляции образца опухоли «голой» мыши опухоли присваивали название поколения P0, следующему пассажу присваивали название поколения P1 и т. д.; и размороженному после криоконсервации образцу присваивали название FP. В данном эксперименте использовали опухолевые ткани из поколения FP4.
Животные: «голые» мыши BALB/c, самки, возрастом 6-8 недель, с массой тела 18-20 г, предоставленные Shanghai Sippr-BK Laboratory Animal Co., Ltd.
Инокуляция опухоли: объем, составляющий около 30 мм3 опухолевой массы CO-04-0032, инокулировали подкожно в правую часть спины каждой мыши; и мышей разделяли на группы, и каждой группе вводили лекарственные средства при достижении среднего объема опухоли, составляющего от приблизительно 100 до 200 мм3.
Результаты экспериментов: практически не наблюдалось увеличения объема опухоли в период от дня 15 до дня 30 введения соединения 1 (включая его кристаллическую форму IX) по настоящему изобретению; и в сравнении с лекарственным средством BKM120 в качестве положительного контроля соединение 1 (включая его кристаллическую форму IX) по настоящему изобретению обладает более превосходной противоопухолевой активностью в отношении рака толстой кишки.
Исследования фармакодинамики in vivo тестируемых лекарственных средств на моделях с подкожным ксенотрансплантатом опухоли из клеток рака желудка человека ST-02-0013
Схема эксперимента
Создание трансплантационной модели опухоли человека: PDX-модель ST-02-0013 изначально получали из удаленного хирургическим путем клинического образца, который имплантировали «голым» мышам и присваивали название поколения P0. Следующему поколению, полученному в результате имплантации опухолей P0, присваивали название поколения P1 и т. д. для опухолей с продолжающейся имплантацией из поколения в поколение у мышей. Опухоли FP2 размораживали после криоконсервации с получением опухолей FP3. Опухоли FP3 пассировали с получением опухолей FP4, которые применяли в данном исследовании.
Животные: «голые» мыши BALB/c, самки, возрастом 6-8 недель, с массой тела 18-22 г, предоставленные Shanghai Ling Chang Biotechnology Co., Ltd.
Инокуляция опухоли: объем, составляющий около 30 мм3 опухолевой ткани FP4 ST-02-0013, инокулировали подкожно в правую часть спины каждой мыши; и мышей разделяли на группы, и каждой группе вводили лекарственные средства при достижении среднего объема опухоли от приблизительно 150 до 200 мм3.
Результаты экспериментов: практически не наблюдалось увеличения объема опухоли в период от дня 15 до дня 30 введения соединения 1 (включая его кристаллическую форму IX) по настоящему изобретению; и в сравнении с лекарственным средством BKM120 в качестве положительного контроля соединение 1 (включая его кристаллическую форму IX) по настоящему изобретению обладает более превосходной противоопухолевой активностью в отношении рака желудка.
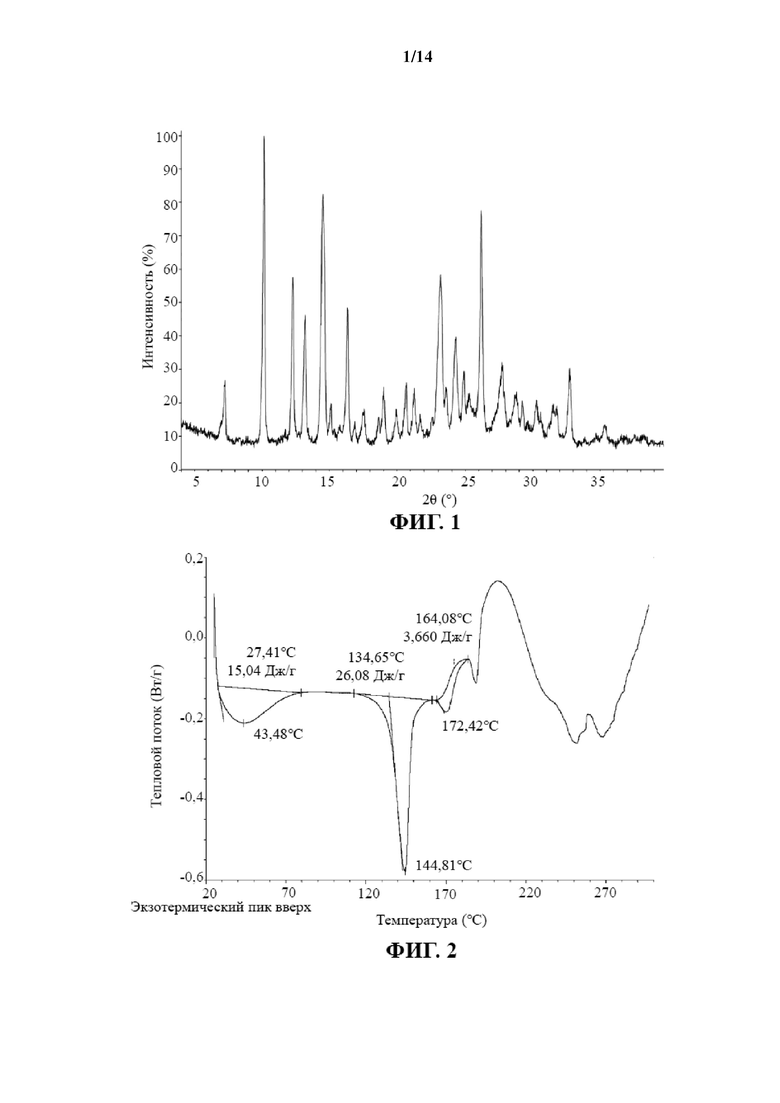
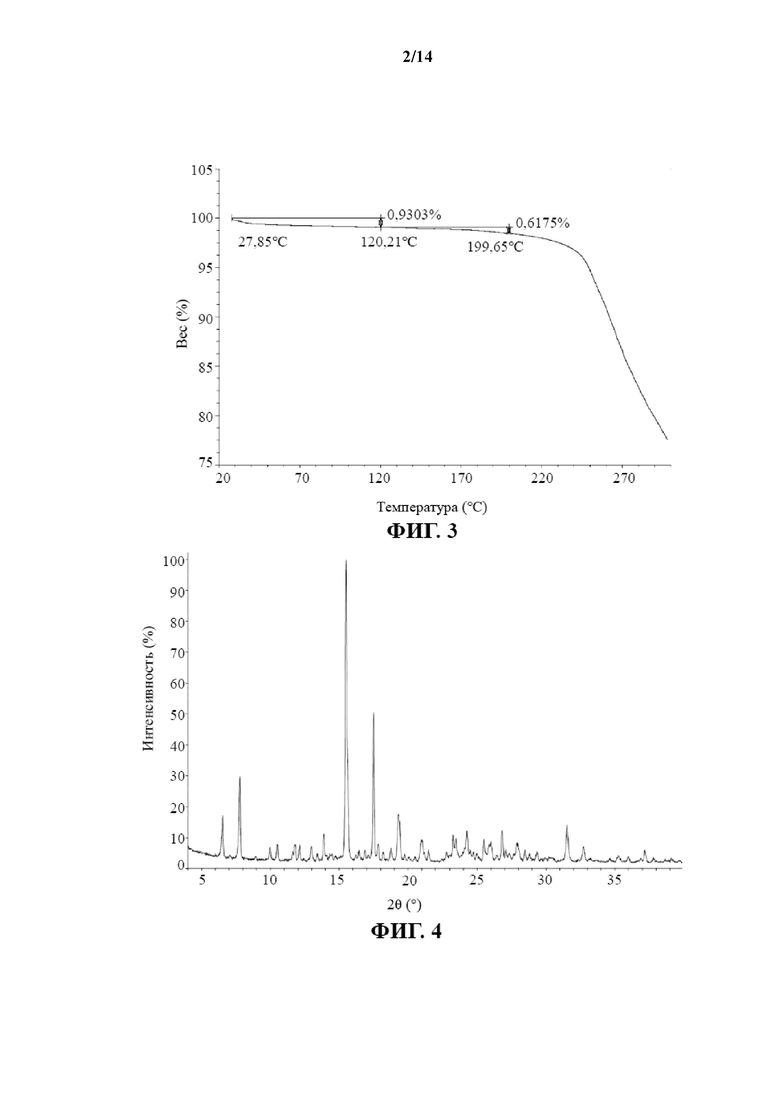

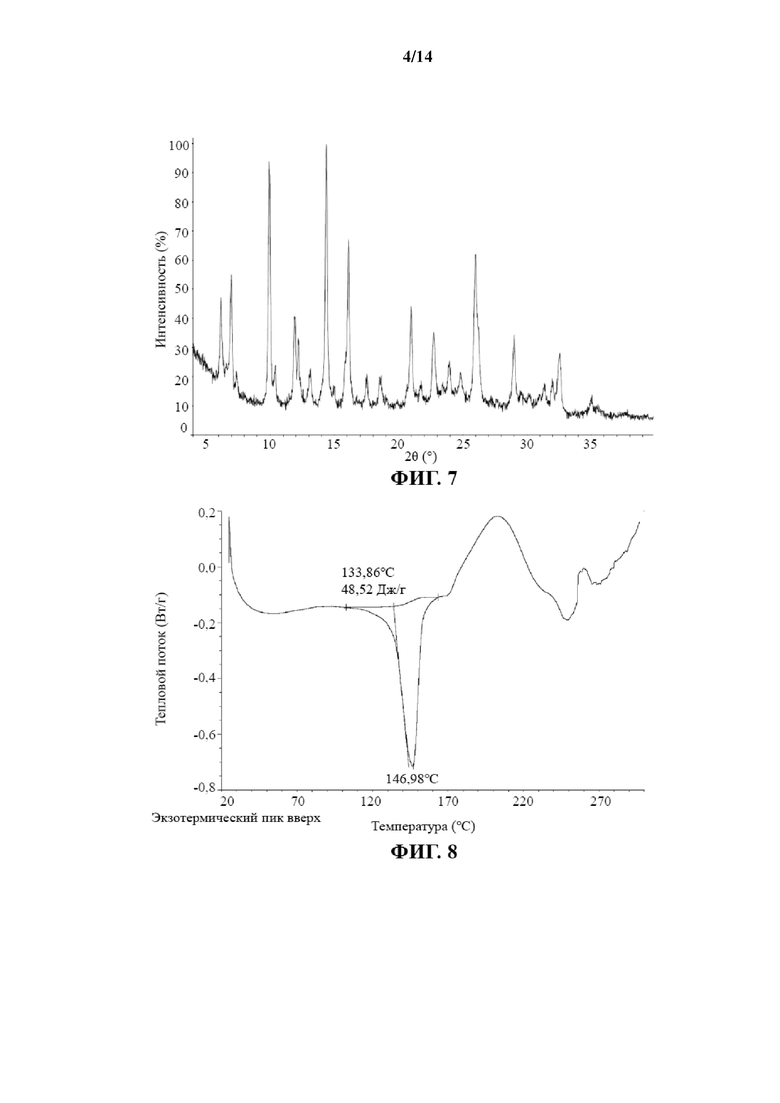

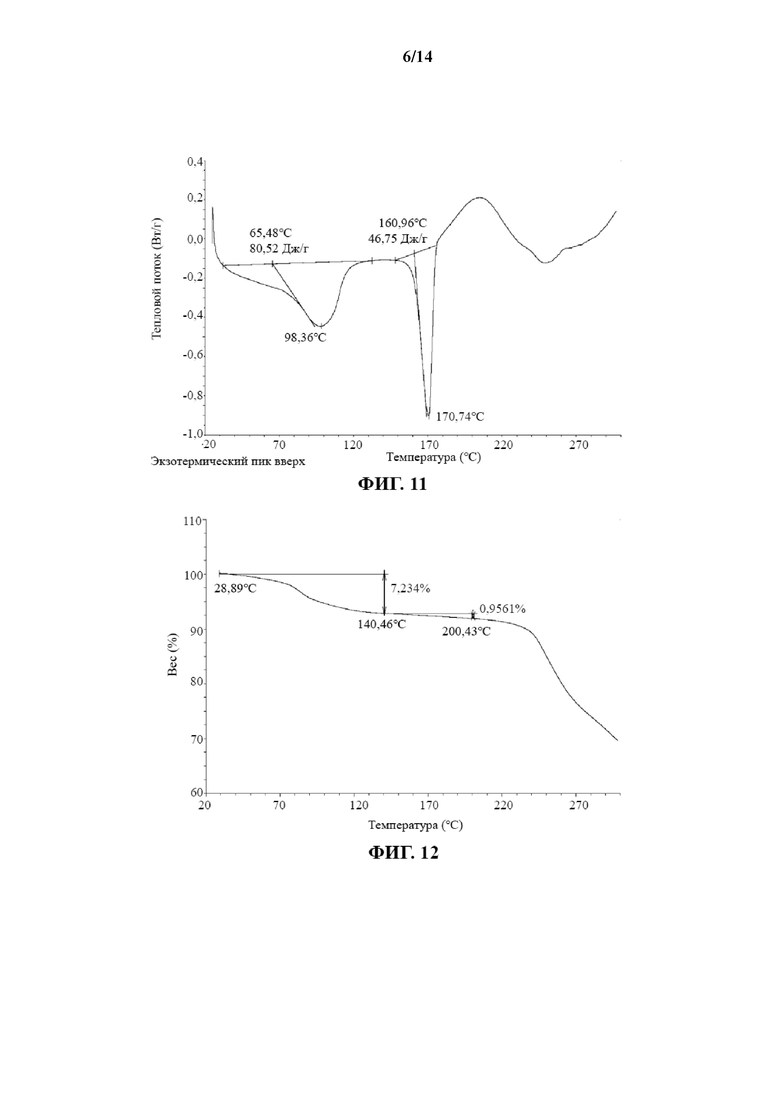




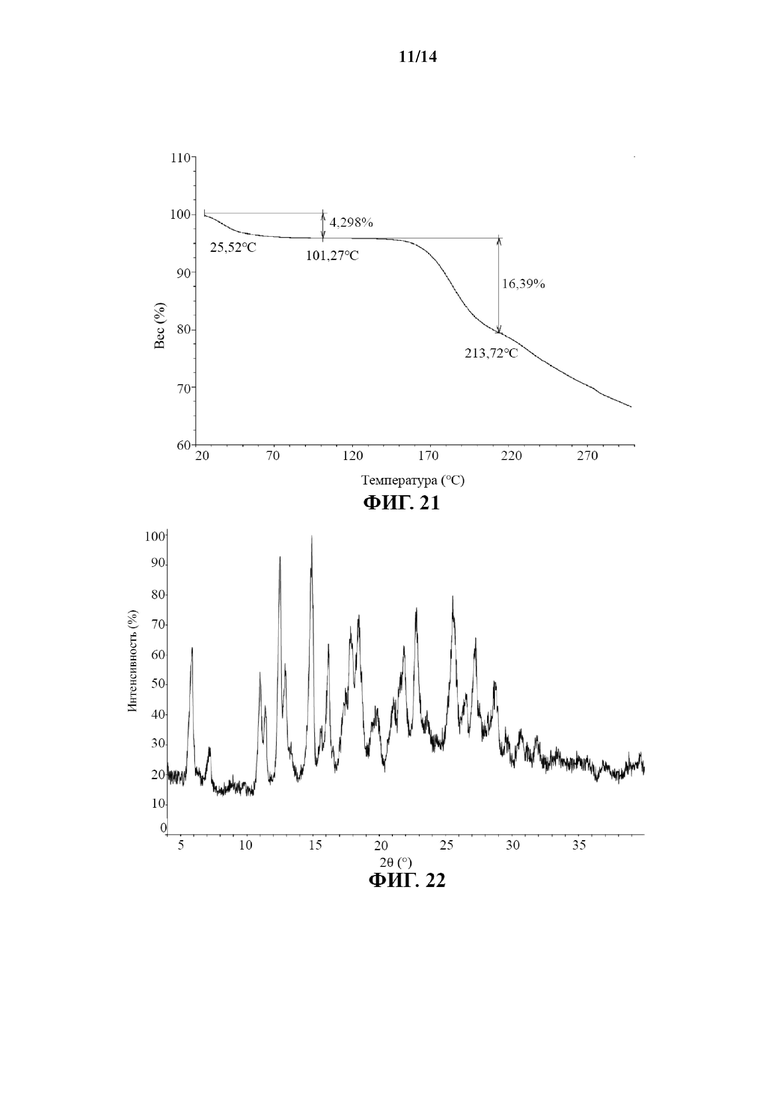

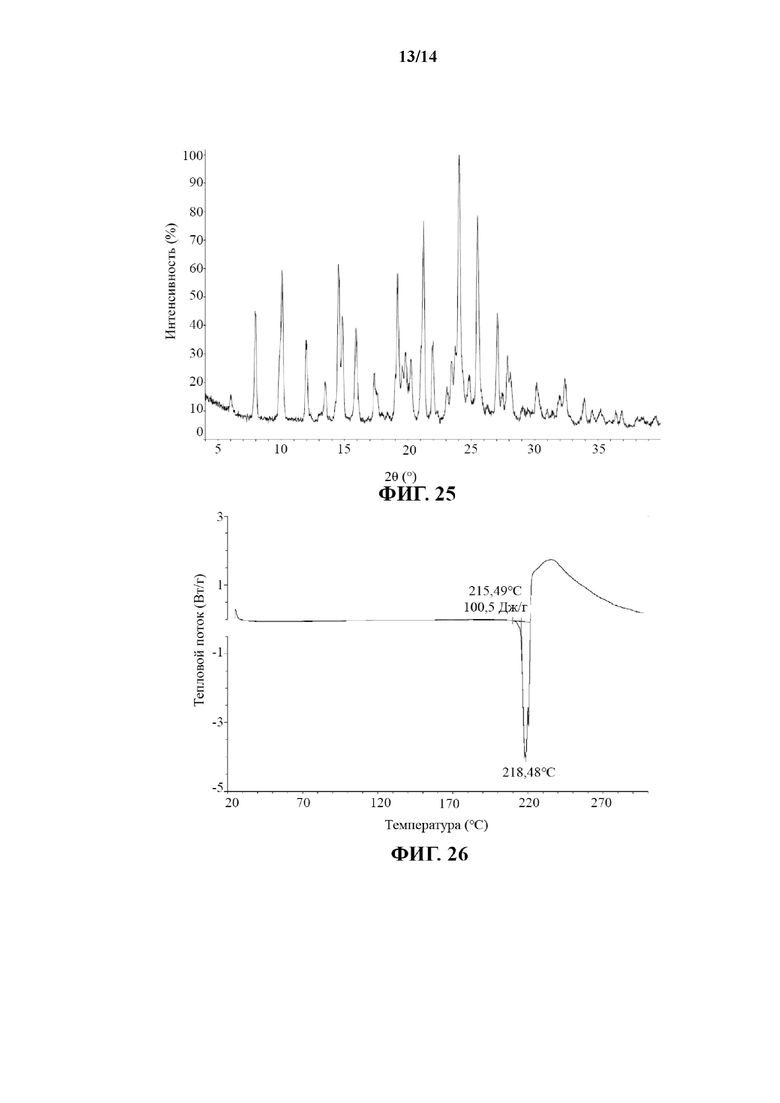

Изобретение относится к формам соединения 1, выбранным из группы, состоящей из кристаллической формы IX соединения 1, гидрохлоридной соли соединения 1 (соединение 2), кристаллических форм I-VI соединения 2, малеиновой соли соединения 1 (соединение 3), кристаллической формы VII соединения 3, лимонной соли соединения 1 (соединение 4), кристаллической формы VIII соединения 4, где указанные кристаллические формы характеризуются картинами дифракции рентгеновских лучей на порошке такими, как указано в формуле изобретения. Также изобретение относится к фармацевтической композиции, содержащей указанные формы, их применению и способу лечения рака. Технический результат – формы соединения 1, ингибирующие PI3K-киназу и пригодные для лечения рака, связанного с PI3K-киназой. 15 н. и 29 з.п. ф-лы, 13 табл., 27 ил., 14 пр.

1. Кристаллическая форма IX соединения 1, представленная следующей формулой:
 ,
,
отличающаяся тем, что кристаллическая форма IX характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 7,947°, 10,073°, 14,531°, 19,187°, 21,237°, 24,055°, 25,497°.
2. Кристаллическая форма IX по п. 1, отличающаяся тем, что кристаллическая форма IX характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 7,947°, 10,073°, 11,970°, 13,468°, 14,531°, 15,911°, 19,187°, 21,237°, 24,055°, 25,497°.
3. Кристаллическая форма IX по п. 1, отличающаяся тем, что кристаллическая форма IX характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 7,947°, 10,073°, 11,970°, 13,468°, 14,531°, 15,911°, 19,187°, 19,561°, 21,237°, 23,446°, 24,055°, 25,497°, 27,074°.
4. Кристаллическая форма IX по п. 1, отличающаяся тем, что данные анализа ее XRPD-рентгенограммы представляют собой следующие:


5. Кристаллическая форма IX по п. 1, отличающаяся тем, что ее XRPD-рентгенограмма является такой, как показано на фиг. 25.
6. Соединение 2, представленное следующей формулой:
 .
.
7. Кристаллическая форма I соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма I характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 10,154°, 12,285°, 14,511°, 16,328°, 24,311°, 26,188°.
8. Кристаллическая форма I соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма I характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 7,270°, 10,154°, 12,285°, 13,206°, 14,511°, 16,328°, 24,311°, 26,188°, 27,724°.
9. Кристаллическая форма I соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма I характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 7,270°, 10,154°, 12,285°, 13,206°, 14,511°, 16,328°, 19,008°, 20,702°, 21,259°, 24,311°, 26,188°, 27,724°.
10. Кристаллическая форма I соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма I характеризуется следующими данными анализа XRPD-рентгенограммы:
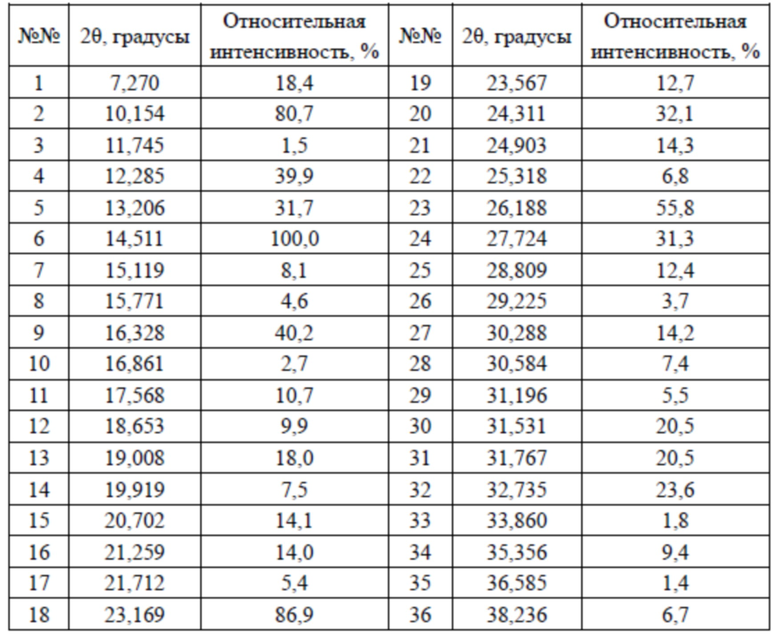
11. Кристаллическая форма II соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма II характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,524°, 7,782°, 13,895°, 15,495°, 17,487°, 19,322°.
12. Кристаллическая форма II соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма II характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,524°, 7,782°, 11,628°, 13,895°, 15,495°, 17,487°, 19,322°, 20,962°, 23,269°.
13. Кристаллическая форма II соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма II характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,524°, 7,782°, 11,628°, 13,895°, 15,495°, 17,487°, 19,322°, 20,962°, 23,269°, 24,257°, 26,009°, 31,533°.
14. Кристаллическая форма II соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма II характеризуется следующими данными анализа XRPD-рентгенограммы:

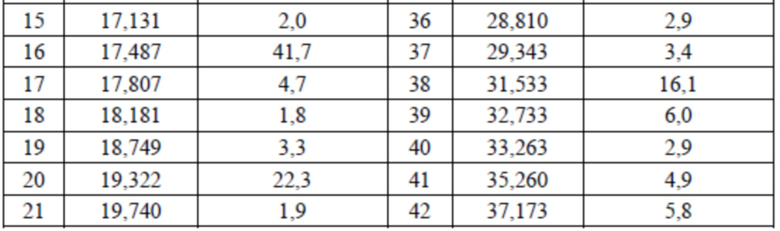
15. Кристаллическая форма III соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма III характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,979°, 9,939°, 14,392°, 16,107°, 20,982°, 25,990°.
16. Кристаллическая форма III соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма III характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,187°, 6,979°, 9,939°, 11,910°, 14,392°, 16,107°, 20,982°, 22,755°, 25,990°.
17. Кристаллическая форма III соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма III характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,187°, 6,979°, 9,939°, 11,910°, 13,148°, 14,392°, 16,107°, 20,982°, 22,755°, 23,975°, 25,990°, 29,006°.
18. Кристаллическая форма III соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма III характеризуется следующими данными анализа XRPD-рентгенограммы:

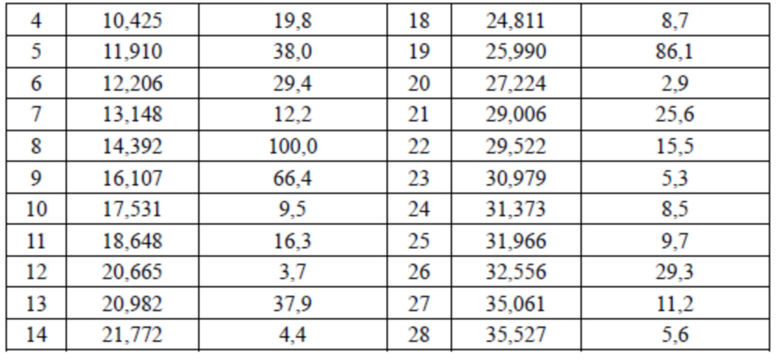
19. Кристаллическая форма IV соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма IV характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,388°, 7,278°, 11,076°, 15,454°, 21,256°.
20. Кристаллическая форма IV соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма IV характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,388°, 7,278°, 11,076°, 12,102°, 15,454°, 16,091°, 18,912°, 21,256°.
21. Кристаллическая форма IV соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма IV характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,388°, 7,278°, 11,076°, 12,102°, 15,103°, 15,454°, 16,091°, 18,912°, 21,256°, 21,846°.
22. Кристаллическая форма IV соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма IV характеризуется следующими данными анализа XRPD-рентгенограммы:
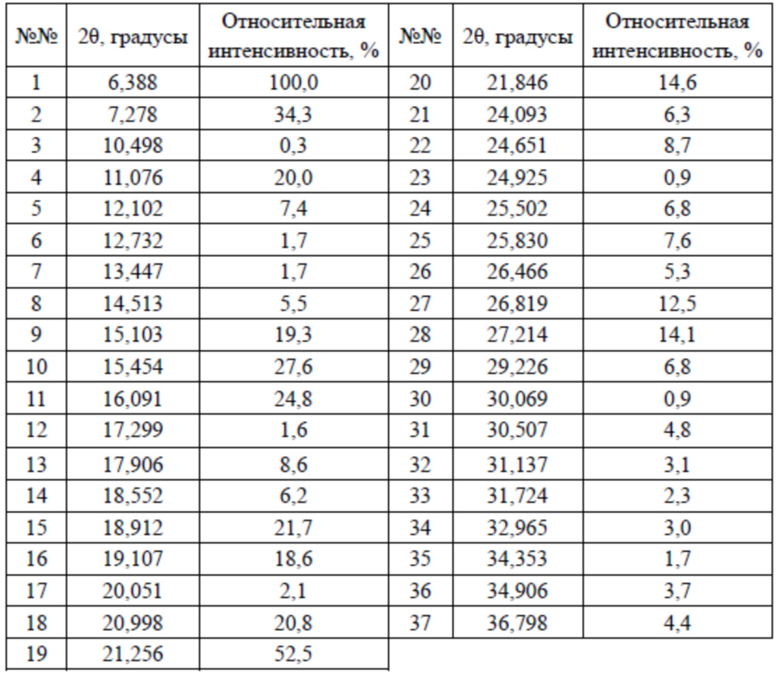
23. Кристаллическая форма V соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма V характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 7,116°, 14,137°, 15,911°, 22,223°, 24,610°.
24. Кристаллическая форма V соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма V характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 7,116°, 14,137°, 15,911°, 21,691°, 22,223°, 24,213°, 24,610°, 28,987°.
25. Кристаллическая форма V соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма V характеризуется следующими данными анализа XRPD-рентгенограммы:

26. Кристаллическая форма VI соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма VI характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 5,775°, 11,770°, 14,415°, 15,753°, 22,518°, 26,623°.
27. Кристаллическая форма VI соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма VI характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 5,775°, 11,770°, 14,415°, 15,753°, 17,132°, 20,939°, 22,518°, 26,623°.
28. Кристаллическая форма VI соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма VI характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 5,775°, 11,770°, 14,415°, 15,753°, 17,132°, 20,939°, 22,518°, 23,745°, 26,623°, 31,295°.
29. Кристаллическая форма VI соединения 2 по п. 6, отличающаяся тем, что кристаллическая форма VI характеризуется следующими данными анализа XRPD-рентгенограммы:
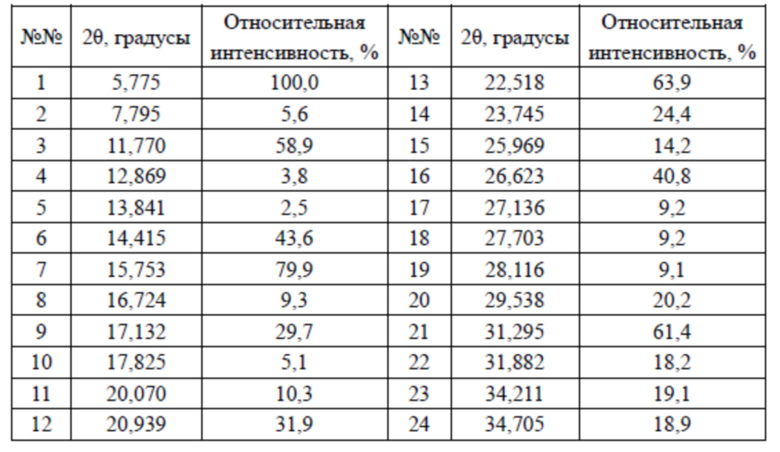
30. Соединение 3, представленное следующей формулой:
 .
.
31. Кристаллическая форма VII соединения 3 по п. 30, отличающаяся тем, что кристаллическая форма VII характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,325°, 12,677°, 15,813°, 21,395°, 22,519°, 27,133°.
32. Кристаллическая форма VII соединения 3 по п. 30, отличающаяся тем, что кристаллическая форма VII характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,325°, 12,677°, 13,251°, 15,813°, 18,954°, 21,395°, 22,519°, 25,161°, 27,133°.
33. Кристаллическая форма VII соединения 3 по п. 30, отличающаяся тем, что кристаллическая форма VII характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 6,325°, 12,677°, 13,251°, 15,813°, 16,565°, 18,954°, 21,395°, 22,519°, 24,117°, 25,161°, 26,405°, 27,133°.
34. Кристаллическая форма VII соединения 3 по п. 30, отличающаяся тем, что кристаллическая форма VII характеризуется следующими данными анализа XRPD-рентгенограммы:

35. Соединение 4, представленное следующей формулой:
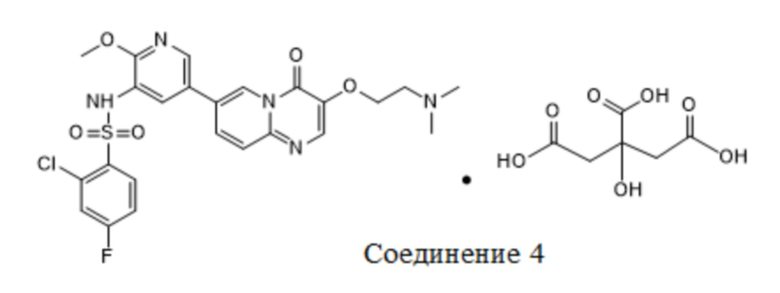 .
.
36. Кристаллическая форма VIII соединения 4 по п. 35, отличающаяся тем, что кристаллическая форма VIII характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 5,889°, 11,002°, 12,518°, 14,906°, 17,825°, 22,814°, 25,555°.
37. Кристаллическая форма VIII соединения 4 по п. 35, отличающаяся тем, что кристаллическая форма VIII характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 5,889°, 7,173°, 11,002°, 11,396°, 12,518°, 12,895°, 14,906°, 17,825°, 22,814°, 25,555°.
38. Кристаллическая форма VIII соединения 4 по п. 35, отличающаяся тем, что кристаллическая форма VIII характеризуется дифракционными пиками на дифракционной рентгенограмме при 2θ = 5,889°, 7,173°, 11,002°, 11,396°, 12,518°, 12,895°, 14,906°, 16,169°, 17,825°, 19,875°, 21,574°, 22,814°, 25,555°, 27,254°.
39. Кристаллическая форма VIII соединения 4 по п. 35, отличающаяся тем, что кристаллическая форма VIII характеризуется следующими данными анализа XRPD-рентгенограммы:

40. Фармацевтическая композиция для лечения рака, связанного с PI3K-киназой, содержащая соединение по любому из пп. 6, 30 и 35 или кристаллическую форму по любому из пп. 1-5, 7-29, 31-34 и 36-39, где фармацевтическая композиция содержит терапевтически эффективное количество соединения или кристаллической формы и необязательно фармацевтически приемлемые носители, наполнители и/или среды.
41. Применение кристаллической формы по любому из пп. 1-5, 7-29, 31-34 и 36-39, или соединения 1, представленного следующей формулой, или соединения по любому из пп. 6, 30 и 35, или фармацевтической композиции на их основе в изготовлении лекарственного препарата для лечения рака, связанного с PI3K-киназой,
 .
.
42. Применение по п. 41, где рак, связанный с PI3K-киназой, выбран из рака толстой кишки и рака желудка.
43. Способ лечения рака, связанного с PI3K-киназой, предусматривающий введение терапевтически эффективного количества кристаллической формы по любому из пп. 1-5, 7-29, 31-34 и 36-39, или соединения по любому из пп. 6, 30 и 35, или фармацевтической композиции на их основе пациентам, нуждающимся в этом.
44. Способ по п. 43, где рак, связанный с PI3K-киназой, выбран из рака толстой кишки и рака желудка.
| WO 2014022128 A1, 06.02.2014 | |||
| СN 103539777 A, 29.01.2014. |

