ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] В общем, настоящее изобретение относится к улучшенному способу получения амфифильных соединений имидазолиния, таких как хлорид 1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния (DOTIM). DOTIM и подобные соединения могут быть приготовлены в виде катионных липосом, которые пригодны в качестве химических векторов для доставки нуклеиновых кислот в генной терапии.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
[0002] Генная терапия использует нуклеиновые кислоты в качестве лечения генетических нарушений и большого разнообразия приобретенных заболеваний и включает в себя большие молекулы ДНК (плазмидная ДНК; пДНК), а также малые молекулы ДНК (олигонуклеотиды; ОДН) и РНК (рибозимы, миРНК и мРНК). Успех генной терапии во многом зависит от разработки вектора доставки генов, который может быть вирусным вектором или невирусным вектором, таким как химический носитель или доставка оголенной ДНК физическими способами. Невирусные векторы имеют много преимуществ по сравнению с вирусными, включая простое крупномасштабное производство, отсутствие иммуногенности и низкую токсичность.
[0003] Катионные липиды, способные образовывать положительно заряженные липосомы, являются одним из наиболее широко используемых невирусных векторов для доставки генов (Zhi et al., Bioconjugate Chemistry, 2013, 24: 478-519). Катионные липиды представляют собой амфифильные молекулы и, как правило, состоят из гидрофобного домена (например, алифатических цепей, стероидных колец), гидрофильной головной группы (например, аминов, четвертичных аммониевых солей, гуанидиниев, гетероциклов) и линкерной группы (например, простого эфира, сложного эфира, карбамата или амидной связи), соединяющей эти два домена. Гидрофильная головная группа обеспечивает конденсацию нуклеиновых кислот посредством электростатических взаимодействий с отрицательно заряженными фосфатными группами генов и дополнительно регулирует эффективность трансфекции. Обычно катионные липиды составляют в виде катионных липосом с нейтральным со-липидом, таким как диолеоилфосфатидилэтаноламин (DOPE) или холестерином для улучшения трансфекции. При смешивании с отрицательно заряженной ДНК положительно заряженные липосомы спонтанно образуют однозначно уплотненные структуры, называемые липоплексами.
[0004] У Солодина и соавторов сообщается об использовании катионных липидов имидазолиния в качестве синтетических носителей для доставки генов в клетки (Solodin et al., Biochemistry, 1995, 34(41): 13537-13544). Эти липиды включали хлорид 1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния (DOTIM) и его аналоги хлорид 1-[2-(гексадеканоилокси)этил]-2-пентадецил-3-(2-гидроксиэтил)имидазолиния (DPTIM) и хлорид 1-[2-(тетрадеканоилокси)этил]-2-тридецил-3-(2-гидроксиэтил)-имидазолиния (DMTIM). Было установлено, что DOTIM является наиболее эффективным среди трех соединений как для трансфекции in vitro, так и для доставки генов in vivo. DMTIM, DPTIM и DOTIM имеют следующие структуры:

[0005] Способы получения алифатических соединений имидазолиния исходя из многофункциональных соединений N,N'-бис(2-гидроксиэтил)этилендиамина были описаны в Патентах США №5,705,655 (Heath), 5,830,878 (Gorman) и 8,044,215 (Yu). Тем не менее, эти способы из известного уровня техники обладают различными недостатками. Например, чтобы ацилировать первичные гидроксильные группы без сопутствующего ацилирования более нуклеофильных вторичных аминов, последние защищают трет-бутилоксикарбонильными группами. Обычно и в настоящей заявке эти защитные группы называют группами "ВОС". Для этой стадии требуется реагент ди-трет-бутилдикарбонат, который является дорогостоящим и токсичным соединением. К тому же, защитные группы ВОС должны быть удалены путем кислотного гидролиза в последующей стадии, что приводит к дополнительному количеству органических и водных отходов.
[0006] Кроме того, процедуры ацилирования этого, защищенного ВОС промежуточного продукта, требуют использования галогенангидридов в присутствии основания (например, триэтиламина), или реакции с карбоновой кислотой в присутствии N,N'-дициклогексилкарбодиимида (DCC) и 4-диметиламинопиридина (DMAP). Потребность в триэтиламине или DMAP в реакциях ацилирования приводит к дополнительным расходам и отходам. Помимо этого, процедура DCC/DMAP приводит к образованию дициклогексилмочевины (DCU) в виде побочного продукта, и в большинстве случаев требует очищение образующегося сложного эфира, которое может быть трудоемким.
[0007] Поэтому существует потребность в улучшенных способах получения и очищения DOTIM и других амфифильных соединений имидазолиния. В частности, существует потребность в таких способах, которые могут быть легко масштабируемыми, экономически эффективными, экологически чистыми и способными постоянно обеспечивать соединения высокой чистоты.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0008] Поэтому вкратце, настоящее изобретение относится к улучшенным способам получения амфифильных соединений имидазолиния, включая хлорид 1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния (DOTIM).
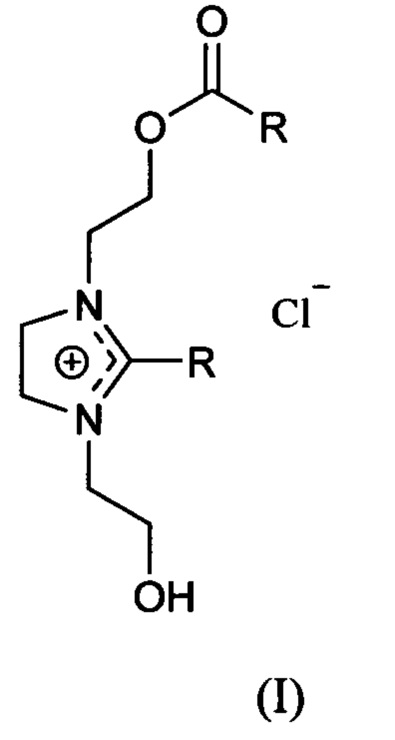
[0009] В различных вариантах осуществления, настоящее изобретение относится к способу получения амфифильного соединения имидазолиния формулы (I)

в которой R представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода. Способ включает в себя реакцию соединения формулы (II) с галогенидом водорода (НХ) с получением соединения формулы (III), в которой X представляет собой Cl, Br или I.

Соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты [RC(O)Y], где Y выбирают из группы, которая включает Cl, Br, F, и I, или ангидридом карбоновой кислоты [RC(O)OC(O)R2], где R имеет значение, определенное выше для формулы (I) и R2 представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода, с получением соединения формулы (IV).

Соединение формулы (IV) нагревают с получением соединения формулы (I).

[0010] В других разных вариантах осуществления, настоящее изобретение относится к способу получения соединения формулы (I)

в которой R представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода. Способ включает в себя нагревание соединения формулы (IV) в реакционной смеси, содержащей органический растворитель и основание с получением соединения формулы (I), в которой R имеет определенное выше значение и X представляет собой Cl, Br или I.

[0011] Другие объекты и признаки отчасти будут очевидны и частично указаны в дальнейшем.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0012] В целом настоящее изобретение относится к способам получения амфифильных соединений имидазолиния, которые обеспечивают преимущества по сравнению с предшествующим уровнем техники. Например, в способах отказываются от применения защитных групп ВОС, и отсутствует потребность в основании во время процедуры ацилирования, тем самым уменьшая стоимость способа, проблемы токсичности и количество образующихся отходов.
[0013] В частности, настоящее изобретение обеспечивает способы получения амфифильных соединений имидазолиния формулы (I):

в которой R представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода, или с 12-25 атомами углерода. Если ненасыщенная, то группа R может иметь одну или большее количество этиленненасыщенных связей.
[0014] Иллюстративные группы R вместе с карбонильной группой, к которой он присоединен (т.е. RC(O)-) включают олеоил, лауроил, миристоил, пальмитоил, стеароил, линолеоил, эйкозаноил, трикозаноил и нонакозаноил (происходят от кислот жирного ряда соответствующего названия: олеиновой, лауриновой, миристиновой и т.д.). При наличии системных названий только для групп R соответствующие названия гидрокарбильной группы, полученной из олеиновой кислоты представляют собой цис-8-гептадеценил; из лауриновой кислоты - ундецил; из миристиновой - тридецил; из пальмитиновой кислоты - пентадецил; из стеариновой кислоты - гептадецил; из ленолевой кислоты - это цис,цис-8,11-гептадецидиенил; из эйкозановой кислоты - это нонадецил; из трикозановой кислоты - это дикозанил; и из триаконтановой кислоты - это нонакозанил. Особенно предпочтительным соединением формулы (I) является хлорид 1-[2-(9(Z)-октадеценоилоксиэтил]-2-[8(Z)-гептадеценил]-3-гидроксиэтилимидазолиния (DOTIM):

[0015] Соединения имидазолиния в соответствии с настоящим изобретением представляют собой соли, которые имеют фармацевтически приемлемый анион. Обычно, соль имидазолиния, образованная согласно данным процессам представляет собой хлоридную соль. Тем не менее, анион может быть обменен с получением соли с другим анионом. Например, хлоридную соль имидазолиния можно растворить в пригодном растворителе и промыть раствором, содержащим целевой анион. Несмотря на то, что хлорид является предпочтительным анионном, также приемлемыми являются бромид и другие физиологически приемлемые анионы, включая ацетат, сукцинат и цитрат.
[0016] В соответствии с настоящим изобретением, общий способ получения амфифильного соединения имидазолиния формулы (I) является следующим:

[0017] Указанные выше формулы (IVa) и формула (IVb) в качестве продукта стадии 2, в данной заявке, как правило, относятся к (включая приложенную формулу изобретения) формуле (IV).
Стадия 1: Защита вторичных амино-групп
[0018] Первой стадией в способе является получение гидрогалогенидной соли N,N'-бис(2-гидроксиэтил)этилендиамина (Формула (III)). Этого достигают посредством реакции N,N'-бис(2-гидроксиэтил)этилендиамина (Формула (II)) с галогенидом водорода (НХ) в реакционной смеси, обычно включая пригодный растворитель, где X означает Cl, Br, или I. В различных предпочтительных вариантах осуществления, галогенид водорода представляет собой хлорид водорода (где X означает Cl) или бромид водорода (где X означает Br). В различных особенно предпочтительных вариантах осуществления, галогенид водорода представляет собой бромид водорода. Как правило, галогенид водорода вводят в реакционную смесь, содержащую N,N'-бис(2-гидроксиэтил)этилендиамин. Это можно осуществить, например, посредством введения галогенида водорода в виде газа в реакционную смесь, которая обычно находится в виде раствора N,N'-бис(2-гидроксиэтил)этилендиамина, растворенного в органическом растворителе. В качестве альтернативы, к реакционной смеси может быть добавлен раствор галогенида водорода в органическом растворителе (например, неводном HBr в уксусной кислоте, или растворе HCl в метаноле, этаноле, диоксане или простом диэтиловом эфире). Как правило, предпочтительным является введение галогенида водорода в реакционную смесь в жидкой фазе.
[0019] Пригодные органические растворители для получения гидрогалогенидной соли формулы (III) включают, но не ограничены ними, С2-С6 карбоновые кислоты; С2-С6 нитрилы; C1-С6 спирты; С2-С10 простые эфиры; С3-С6 алкилацетаты; С3-С10 кетоны; C5-C8 алифатические углеводороды; C1-С6 хлорированные углеводороды; С3-С8 алкилкарбонаты; сульфолан; диметилсульфоксид; толуол; хлорбензол; а также их моно- или полифазные смеси.
[0020] Конкретные примеры таких растворителей включают, но не ограничены ними, уксусную кислоту, пропионовую кислоту, ацетонитрил, пропионитрил, метанол, этанол, изопропанол, трет-бутанол, простой диэтиловый эфир, тетрагидрофуран, диоксан, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, метилацетат, этилацетат, трет-бутилацетат, ацетон, метилэтилкетон, гексан, гептан, циклогексан, дихлорметан, хлороформ, 1,2-дихлорэтан, пропиленкарбонат, сульфолан, диметилсульфоксид, толуол, хлорбензол и их комбинации. Предпочтительно растворитель выбирают из группы, которая включает уксусную кислоту, метанол, этанол, изопропанол, этилацетат и их комбинации.
[0021] Реакцию для образования гидрогалогенидной соли формулы (III) обычно проводят при температуре приблизительно от 0°С до приблизительно 60°С, более типично при температуре приблизительно от 10°С до приблизительно 30°С, например, посредством контролируемого добавления к реакционной смеси галогенида водорода.
[0022] Обычно, исходное вещество превращают в гидрогалогенидную соль формулы (III) в течение реакционного времени приблизительно от 10 до приблизительно 120 минут, и более обычно приблизительно от 30 до приблизительно 60 минут после добавления реагента.
[0023] Соединение формулы (III), в виде гидрогалогенидной соли, легко образует осадок в реакционной смеси и может быть выделено при помощи фильтрации. После чего восстановленный продукт обычно промывают и сушат под действием вакуума.
Стадия 2: Ацилирование первичных гидроксильных групп
[0024] Первичные гидроксильные группы соединения формулы (III) ацилируют с вторичными аминогруппами N,N'-бис(2-гидроксиэтил)этилендиамина, защищенного как галогенидная соль, для получения сложного диэфира.
[0025] Ацилирующий агент может представлять собой активированное производное карбоновой кислоты, такое как галогенангидрид карбоновой кислоты (RC(O)Y) или ангидрид карбоновой кислоты (RC(O)OC(O)R2), где R имеет значение, определенное выше для формулы (I) и R2 представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода, или от 12 до 25 атомов углерода. В ангидриде карбоновой кислоты, R и R2 могут быть одинаковыми (т.е., симметричный ангидрид карбоновой кислоты). Тем не менее, обычно R и R2 не являются одинаковыми. Обычно, R2 означает C1-С10 неразветвленный или разветвленный алифатический углеводород, и более предпочтительно R2 означает С3-С10 разветвленный алифатический углеводород. В настоящее время считается, что стерически затрудненные или разветвленные группы R2 (например, трет-бутил или изопропил) приводят к получению целевого продукта, при этом минимизируя образование нежелательных смешанных с продуктом сложных эфиров. В галогенангидриде карбоновой кислоты, Y обычно выбирают из группы, включающей Cl, Br, F и I. Более предпочтительно, Y означает Cl или Br. Y может быть идентичным противоиону, X, соединения формулы (III), но это не требуется. Как правило, хлориды карбоновых кислот являются предпочтительными по сравнению с бромидами, фторидами и йодидами, а также ангидриды карбоновых кислот в качестве ацилирующего агента из-за их более низкой стоимости и доступности. Для получения DOTIM, галогенангидрид карбоновой кислоты предпочтительно представляет собой хлорид олеиновой кислоты.
[0026] Ацилирующие агенты галогенангидриды карбоновой кислоты являются коммерчески доступными. Тем не менее, было обнаружено, что более высокие выходы соединения формулы (IV) могут быть достигнуты при одновременном получении галогенангидрида карбоновой кислоты или незадолго до его применения в способе в соответствии с настоящим изобретением. Один из пригодных способов получения ацилирующего агента галогенангидрида карбоновой кислоты (хлорид олеиновой кислоты) приведен в Примере 2.
[0027] Для гидробромидных солей формулы (III), реакция с бромидом карбоновой кислоты или ангидридом карбоновой кислоты дает соединение формулы (IVa), представленное выше. Подобным образом, для гидрохлоридных солей формулы (III), реакция с хлорангидридом кислоты или ангидридом карбоновой кислоты дает соединение формулы (IVb).
[0028] Для стадии ацилирования, гидрогалогенидную соль N,N'-бис(2-гидроксиэтил)этилендиамина (Формула (III)) разбавляют в органическом растворителе с последующим добавлением ацилирующего агента активированной карбоновой кислоты. Пригодные органические растворители для стадии ацилирования включают, но не ограничены ними, С2-С6 нитрилы; С2-С10 простые эфиры; С3-С6 алкилацетаты; С3-С10 кетоны; C5-C8 алифатические углеводороды; C1-С6 хлорированные углеводороды; С3-C8 алкилкарбонаты; сульфолан; диметилсульфоксид; толуол; хлорбензол; а также их моно- или полифазные смеси.
[0029] Конкретные примеры таких растворителей включают, но не ограничены ними, ацетонитрил, пропионитрил, простой диэтиловый эфир, тетрагидрофуран, диоксан, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, метилацетат, этилацетат, трет-бутилацетат, ацетон, метилэтилкетон, гексан, гептан, циклогексан, дихлорметан, хлороформ, 1,2-дихлорэтан, пропиленкарбонат, сульфолан, диметилсульфоксид, толуол и хлорбензол. Предпочтительно, растворитель выбирают из ацетонитрила, пропионитрила, дихлорметана, хлороформа, тетрагидрофурана и их комбинации.
[0030] Ацилирование обычно осуществляют при температуре приблизительно от 0°С до приблизительно 120°С, более предпочтительно приблизительно от 20°С до приблизительно 100°С и еще более предпочтительно, приблизительно от 40°С до приблизительно 80°С.
[0031] Реакцию ацилирования обычно осуществляют в течение периода времени приблизительно от 1 до приблизительно 12 часов, более предпочтительно приблизительно от 2 до приблизительно 6 часов.
[0032] Если ацилирующий агент представляет собой галогенангидрид карбоновой кислоты, то в ходе реакции выделяется газ галогенид водорода и может быть поглощен с помощью газового скруббера. Если ацилирующий агент является ангидридом карбоновой кислоты, то получается побочный продукт карбоновая кислота и удерживается в органическом растворителе.
[0033] Ацилирование гидрогалогенидной соли N,N'-бис(2-гидроксиэтил)этилендиамина (Формула (III)) происходит равномерно, и обеспечивает необходимый сложный диэфир формулы (IV) с высоким выходом. По завершении реакции, реакционную смесь обычно охлаждают до температуры приблизительно от 20°С до приблизительно 40°С, и она может быть разбавлена ацетоном для улучшения фильтрации образовавшегося осадка. Осажденный продукт формулы (IV) затем легко восстанавливают, фильтрат промывают, и восстановленное твердое вещество сушат под действием вакуума.
[0034] В отличие от процессов ацилирования, описанных в известном уровне техники, для этого преобразования не требуется добавления основания. К тому же, стадию ацилирования согласно данному способу обычно проводят при отсутствии любого кислотного катализатора. В частности, в этой стадии не используют кислотные катализаторы, такие как n-толуол сульфоновой кислоты, бензолсульфоновая кислота, сульфоуксусная кислота, фосфорная кислота и трихлорид фосфора.
[0035] В различных предпочтительных вариантах осуществления, X в Формуле III означает Br и гидробромидная соль N,N'-бис(2-гидроксиэтил)этилендиамина (Формула (III)) вступает в реакцию с галогенангидридом карбоновой кислоты (например, хлорид олеиновой кислоты). Было обнаружено, что эта реакция легко образует соединение формулы (IVb). Поскольку X в Формуле III означает Cl, то гидрохлоридная соль N,N'-бис(2-гидроксиэтил)этилендиамина (Формула (III)) также может вступать в реакцию с галогенангидридом карбоновой кислоты (например, олеиновой кислоты хлорид). Исходя из его растворимости, тем не менее, может возникнуть необходимость растворения хлоридной соли N,N'-бис(2-гидроксиэтил)этилендиамина в сильнополярном растворителе, таком как, например, сульфолан, диметилформамид (DMF) и диметилацетамид (DMAC). В целом, эта процедура менее желательна из-за необходимости дополнительной обработки и дополнительных затрат.
Превращение гидрогалогенидной соли IVa в гидрохлоридную соль IVb
[0036] Так как хлорид является противоионом в конечном продукте формулы (I), то необходима дополнительная стадия промывания, когда соединение формулы (IV) представляет собой гидрогалогенидную соль, отличную от гидрохлоридной соли (например, когда соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты RC(O)Y, когда Y не означает Cl). Тем не менее, если гидрохлоридную соль (IVb) получают из соединения формулы (III), то промывание является излишним.
[0037] Для любой необходимой стадии промывания, соединение формулы (IVa) растворяют в пригодном растворителе и промывают концентрированным водным раствором хлорида натрия. Часто используют несколько промываний, например, можно применять 2 или большее количество промываний. Обычно это промывание для осуществления анионного обмена осуществляют при температуре приблизительно от 0°С до приблизительно 60°С, более предпочтительно приблизительно от 10°С до приблизительно 30°С и еще более предпочтительно, приблизительно от 15°С до приблизительно 25°С. После отделения конечного водного слоя хлорида натрия (соляного раствора), органическую фазу концентрируют с получением целевой гидрохлоридной соли формулы (IVb).
[0038] Пригодные растворители для стадии анионного обмена включают, но не ограничены ними, С2-С6 нитрилы; С2-С10 простые эфиры; С3-С6 алкилацетаты; С3-С10 кетоны; C5-C8 алифатические углеводороды; C1-С6 хлорированные углеводороды; толуол; хлорбензол; а также их моно- или полифазные смеси.
[0039] Конкретные примеры таких растворителей включают, но не ограничены ними, ацетонитрил, пропионитрил, простой диэтиловый эфир, тетрагидрофуран, диоксан, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, метилацетат, этилацетат, трет-бутилацетат, ацетон, метилэтилкетон, гексан, гептан, циклогексан, дихлорметан, хлороформ, 1,2-дихлорэтан, толуол, хлорбензол и их смеси. Предпочтительно, растворитель выбирают из дихлорметан, хлороформ, 1,2-дихлорэтан, метилацетат, этилацетат, толуол и их комбинации.
Стадия 3: Реакция перегруппировки
[0040] Для осуществления реакции перегруппировки, соединение формулы (IVb) растворяют в реакционной смеси, содержащей пригодный растворитель, и эту реакционную смесь нагревают и перемешивают, после чего происходит миграция ацильной группы с последующей конденсацией, что приводит к образованию соединения имидазолиния формулы (I).
[0041] Пригодные растворители для реакции перегруппировки включают, но не ограничены ними, С2-С6 нитрилы; C1-С6 спирты; С2-С10 простые эфиры; С3-С6 алкилацетаты; С3-С10 кетоны; C5-C8 алифатические углеводороды; C1-С6 хлорированные углеводороды; С3-C8 алкилкарбонаты; сульфолан; диметилсульфоксид; толуол; хлорбензол; а также их моно- или полифазные смеси.
[0042] Конкретные примеры таких растворителей включают, но не ограничены ними, ацетонитрил, пропионитрил, метанол, этанол, изопропанол, трет-бутанол, простой диэтиловый эфир, тетрагидрофуран, диоксан, метил трет-бутиловый эфир, 1,2-диметоксиэтан, метилацетат, этилацетат, трет-бутилацетат, ацетон, метилэтилкетон, гексан, гептан, циклогексан, дихлорметан, хлороформ, 1,2-дихлорэтан, пропиленкарбонат, сульфолан, диметилсульфоксид, толуол, хлорбензол и их смеси.
[0043] В некоторых вариантах осуществления, растворитель представляет собой смесь хлорированного углеводорода и спирта, такого как, например, смесь хлороформа и метанола. В таких случаях, весовое соотношение хлороформа к метанолу обычно составляет приблизительно от 4:1 до приблизительно 10:1, или приблизительно от 6:1 до приблизительно 8:1. Дополнительно или в качестве альтернативы, объемное соотношение хлороформа к метанолу обычно составляет приблизительно от 2:1 до приблизительно 6:1, или приблизительно от 3:1 до приблизительно 5:1.
[0044] В некоторых вариантах осуществления, вместе с растворителем или смесью растворителей реакционная смесь дополнительно содержит основание. В некоторых вариантах осуществления используют слабое основание. Пригодные слабые основания включают, но не ограничены ними, бикарбонат натрия (гидрокарбонат натрия), бикарбонат калия (гидрокарбонат калия), дигидрофосфат калия (монофосфат калия), гидрофосфат дикалия (дикалий фосфат), третичные амины и их смеси.
[0045] Как правило, основание вводят в молярном соотношении к соединению формулы (IVb) приблизительно от 0.25:1 до приблизительно 1.75:1 или приблизительно от 0.8:1 до приблизительно 1.5:1. В некоторых вариантах осуществления основание вводят в молярном соотношении к соединению формулы (IVb) приблизительно от 1:1 до приблизительно 1:5:1, или приблизительно от 1:1 до приблизительно 1.2:1 (например, приблизительно 1.1:1).
[0046] Вдобавок к органическому растворителю или смеси растворителей, и необязательно основания, к реакционной смеси перед нагреванием может быть добавлен осушитель. Пригодные осушители включают, например, молекулярные сита (например, сита, имеющие размер пор от 2 до 5), хлорид кальция, сульфат магния и сульфат натрия. Особенно пригодные осушители включают в себя молекулярные сита (например, сита, имеющие размер пор от 2 до 5 или приблизительно 3) и сульфат магния. Также пригодным осушителем является активированный уголь. Количество осушителя не является исключительно критичным, но должно быть достаточным для поглощения воды, образующейся во время реакции перегруппировки/конденсации.
до 5), хлорид кальция, сульфат магния и сульфат натрия. Особенно пригодные осушители включают в себя молекулярные сита (например, сита, имеющие размер пор от 2 до 5 или приблизительно 3) и сульфат магния. Также пригодным осушителем является активированный уголь. Количество осушителя не является исключительно критичным, но должно быть достаточным для поглощения воды, образующейся во время реакции перегруппировки/конденсации.
[0047] Реакцию, как правило, проводят при температуре приблизительно от 20°С до приблизительно 100°С, более предпочтительно приблизительно от 40°С до приблизительно 80°С и еще более предпочтительно, приблизительно от 50°С до приблизительно 70°С.
[0048] Стадию перегруппировки обычно осуществляют в течение периода времени приблизительно от 2 до приблизительно 48 часов, более предпочтительно приблизительно от 12 до приблизительно 36 часов. Хотя такие периоды времени является приемлемыми, в некоторых вариантах осуществления, когда в реакционную смесь вводят основание, реакция перегруппировки протекает с большей скоростью. В таких вариантах осуществления стадию перегруппировки проводят в течение периода времени приблизительно от 2 до приблизительно 12 часов, или приблизительно от 4 до приблизительно 10 часов, или даже приблизительно 5 часов или приблизительно 6 часов.
[0049] После этого реакционную смесь охлаждают или оставляют охладиться до комнатной температуры, смесь отфильтровывают, чтобы удалить фракцию твердых частиц и обеспечить фильтрат, содержащий соединение имидазолиния формулы (I). Фильтрат концентрируют с получением сырого продукта обычно в виде воскообразного твердого вещества. Сырой продукт может быть очищен с помощью пригодных способов, известных в данной области, включая, например, промывание растворителем, посредством перекристаллизации или даже с помощью приемлемого хроматографического метода.
[0050] Предпочтительные протоколы очистки/восстановления продукта включают обработку фильтрата сырого продукта ацетоном с последующей фильтрацией и концентрированием для выделения целевого твердого продукта. Другой предпочтительный протокол очистки/восстановления продукта включает фильтрацию с использованием колонки с диоксидом кремния и дихлорметана или хлороформа. Примечательно, что эти предпочтительные протоколы очистки/восстановления продукта не являются хроматографическими методами и поэтому намного проще и более подходят для использования в промышленном масштабе, чем методы восстановления на основе хроматографии, обычно используемые в данной области техники. Выгодным является то, что протоколы очистки/восстановления продукта, используемые в настоящих способах, которые не являются хроматографическими, обеспечивают чистоту продукта более 95% или выше (например, превышающую 97%).
[0051] Как правило, стадия перегруппировки обеспечивает соединение формулы (I) с выходом (в пересчете на соединение формулы (IVb)) по меньшей мере приблизительно в 70%, по меньшей мере приблизительно 75%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 85%, по меньшей мере приблизительно 90%, или по меньшей мере приблизительно 95% (например, приблизительно 97% или выше).
[0052] Подробно описав изобретение, будет очевидно, что возможны модификации и варианты, не выходящие за пределы объема изобретения, определенного в прилагаемой формуле изобретения.
ПРИМЕРЫ
[0053] Следующие неограничивающие примеры приведены для дополнительной иллюстрации настоящего изобретения.
Пример 1: Синтез дибромида N,N'-бис(2-гидроксиэтил)этан-1,2-диаминия
[0054] N,N'-Бис(2-гидроксиэтил)этилендиамин (20 г, 0.135 моль) растворяли в 100 мл уксусной кислоты (экзотермическая). После охлаждения этого раствора приблизительно до 20°С, бромисто-водородную кислоту (0.283 моль; 50.3 мл, 33% раствор в уксусной кислоте) медленно добавляли к реакционной смеси в течение 45 минут, так что температура реакционной смеси не превышала 30°С. Во время добавления к реакционной смеси бромисто-водородной кислоты образовался белый осадок. После полного добавления, реакционную смесь перемешивали в течение 30 минут, с последующей фильтрацией осадка. Затем осадок промывали ацетонитрилом (2х 25 мл) и сушили при 30°С под действием вакуума (5 мбар) с получением 40.58 г (0.131 моль, 97% выход) дибромида N,N'-бис(2-гидроксиэтил)этан-1,2-диаминия в виде белого твердого вещества. Структуру продукта подтверждали с помощью ЯМР-спектроскопии: 1Н-ЯМР (ДМСО-d6, 400 МГц) δ=8.62 (4Н, bs), 5.34 (2Н, bs), 3.67 (4Н, bs), 3.29 (4Н, bs), и 3.07 (4Н, bs) част. на млн.
Пример 2: Синтез дибромида N,N'-бис{2-[(9Z)-октадец-9-еноилокси]этил}этан-1,2-диаминия
[0055] Дибромид N,N'-бис{2-[(9Z)-октадец-9-еноилокси]этил}этан-1,2-диаминия получали путем ацилирования дибромида N,N'-бис(2-гидроксиэтил)этан-1,2-диаминия со свежеполученным хлоридом олеиновой кислоты.
[0056] Хлорид олеиновой кислоты получали путем растворения олеиновой кислоты (9.0 г, 0.0319 моль, 99% чистота) в 20 мл дихлорметана, с последующим добавлением оксалилхлорида (8.08 г, 0.064 моль) при 20°С. После добавления, реакционную смесь перемешивали в течение 60 минут. Дихлорметан и избыток оксалилхлорида выпаривали при 30°С под действием вакуума (750 мбар, постепенно до 50 мбар) с получением 9.5 г (0.0315 моль, 99% выход) хлорида олеиновой кислоты в виде бесцветного масла. Структуру продукта подтверждали с помощью ЯМР-спектроскопии: 1Н-ЯМР (CDCl3, 400 МГц) δ=5.34 (2Н, m), 2.88 (2Н, t), 2.01 (4Н, m), 1.71 (2Н, m), 1.31 (20Н, m), и 0.88 (3Н, t) част, на млн.
[0057] Дибромид N,N'-бис(2-гидроксиэтил)этан-1,2-диаминия (25 г, 0.0806 моль), полученный, как описано в Примере 1 добавляли при 20°С в 200 мл ацетонитрила для получения белой суспензии. После нагревания до приблизительно 82°С, свежеполученный хлорид олеиновой кислоты (72.8 г, 0.242 моль) добавляли к реакционной смеси в течение 1 часа, с последующим дополнительным перемешиванием приблизительно при 82°С в течение 3 часов. В течение этого времени из реакционной смеси выделялся газообразный гидрохлорид, который был поглощен газовым скруббером. После охлаждения до 40°С, реакционную смесь разбавляли с 150 мл ацетона, чтобы улучшить фильтрационные свойства образовавшегося осадка. При 20°С, суспензию фильтровали и последовательно промывали ацетоном (1×20 мл), водой (1×40 мл) и ацетоном (2×25 мл). Оставшееся твердое вещество сушили при 40°С под действием вакуума (5 мбар) с получением 54.1 г (0.0645 моль, 80% выход) дибромида N,N'-бис{2-[(9Z)-октадец-9-еноилокси]этил}этан-1,2-диаминия в виде не совсем белого твердого вещества. Соотношение бромид/хлорид в продукте было обнаружено как составляющее 18:1, как определено ионной хроматографией. Структуру продукта подтверждали с помощью ЯМР-спектроскопии: 1Н-ЯМР (CDCl3, 400 МГц) δ=9.50 (4Н, bs), 5.34 (4Н, m), 4.52 (4Н, m), 3.81 (4Н, bs), 3.45 (4Н, bs), 2.47 (4Н, m), 2.00 (8Н, m), 1.62 (4Н, m), 1.29 (40Н, m), и 0.88 (6Н, m) част. на млн.
Пример 3: Синтез дихлорида N,N'-бис{2-[(9Z)-октадец-9-еноилокси]этил}этан-1,2-диаминия
[0058] Дибромид N,N'-бис{2-[(9Z)-октадец-9-еноилокси]этил}этан-1,2-диаминия (42 г, 0.050 моль) растворяли в хлороформе (500 мл). К этому раствору при 20°С добавляли 250 мл воды и 500 мл концентрированного водного раствора хлорида натрия. Реакционную смесь перемешивали в течение одного часа, а затем органическую фазу отделяли от водного слоя. Предварительное центрифугирование (при от 3000 до 5000 об. в мин.) реакционной смеси обычно улучшало разделение фаз. Органический слой снова обрабатывали (2х) водным раствором хлорида натрия тем же способом. В конце концов, органический слой промывали водой, сушили над 2 г хлорида кальция, и концентрировали при 40°С под действием вакуума (500 мбар, постепенно до 20 мбар) с получением 31.9 г (0.0425 моль, 85%) дихлорида N,N'-бис{2-[(9Z)-октадец-9-еноилокси]этил}этан-1,2-диаминия в виде не совсем белого твердого вещества. Структуру продукта подтверждали с помощью ЯМР-спектроскопии: 1Н-ЯМР (CDCl3, 400 МГц) δ=10.10 (4Н, bs), 5.35 (4Н, m), 4.49 (4Н, m), 3.69 (4Н, bs), 3.38 (4Н, m), 2.45 (4Н, m), 1.99 (8Н, m), 1.61 (4Н, m), 1.29 (40Н, m), и 0.88 (6Н, m) част. на млн.
[0059] Соотношение бромид/хлорид продукта, полученного после 3-го промывания концентрированным водным раствором хлорида натрия, обнаруживали как составляющее 1:180.
Пример 4: Синтез хлорида 1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния
[0060] Дихлорид N,N'-бис{2-[(9Z)-октадец-9-еноилокси]этил}этан-1,2-диаминия (20 г, 0.0266 моль) растворяли в смеси из хлороформа (160 мл) и метанола (40 мл), затем к этой смеси добавляли 3 молекулярные сита (5 г). Реакционную смесь нагревали приблизительно до 54°С и перемешивали в течение 24 часов. Затем реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления твердых веществ. Фильтрат концентрировали в вакууме (500 мбар, постепенно до 5 мбар) при 40°С с получением желтоватого воскового твердого вещества. Сырой продукт обрабатывали при помощи 30 мл ацетона и перемешивали в течение 30 минут при 40°С. Полученную суспензию фильтровали и фильтрат снова концентрировали под действием вакуума с получением 14.4 г (0.0207 моль, 78% выход) хлорид 1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния в виде бесцветного воскообразного твердого вещества. Структуру продукта подтверждали с помощью ЯМР-спектроскопии: 1Н-ЯМР (CDCl3, 400 МГц) δ=6.12 (1H, bs), 5.34 (4Н, m), 4.34 (2Н, m), 4.09 (4Н, s), 3.87 (2Н, m), 3.77 (2Н, m), 3.52 (2Н, m), 2.78 (2Н, m), 2.32 (2Н, m), 2.00 (8Н, m), 1.59 (4Н, m), 1.30 (40Н, m), и 0.88 (6Н, m) част, на млн.
Пример 5: Синтез хлорида (1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния) с добавлением основания
[0061] Дихлорид N,N'-бис{2-[(9Z)-октадец-5-еноилокси]этил}этан-1,2-диаминия (140 г, 0.186 моль) и бикарбонат натрия (гидрокарбонат натрия) (15.68 г, 0.205 моль) суспендировали в смеси из хлороформа (1120 мл) и метанола (280 мл) при 20°С. Реакционную смесь нагревали до температуры приблизительно в 50-52°С и перемешивали в течение 5 часов. Затем смесь концентрировали в вакууме (40°С; 150 мбар), чтобы удалить растворители и получить остаток (196 г). Сырой продукт остатка растворяли в ацетоне (1120 мл) и затем добавляли активированный уголь (28 г, высушенный при 120°С), и суспензию перемешивали в течение 30 минут при комнатной температуре. Полученную суспензию фильтровали и фильтровальную лепешку промывали ацетоном (150 мл). Объединенный прозрачный желтый фильтрат концентрировали под действием вакуума (40°С; 4 мбар), чтобы получить 120.3 г (0.173 моль, 92.7% выход) хлорида (1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния).
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ
[0062] Для дополнительной иллюстрации, дополнительные неограничивающие варианты осуществления настоящего раскрытия приведены ниже.
[0063] Вариант осуществления А1 представляет собой способ получения соединения формулы (I)

в которой R представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода, способ включает в себя: реакцию соединения формулы (II) с галогенидом водорода (НХ) с получением соединения формулы (III), в которой X означает Cl, Br, или I;

реакцию соединения формулы (III) с галогенангидридом карбоновой кислоты [RC(O)Y], в которой Y выбирают из группы, которая включает Cl, Br, F и I, или ангидридом карбоновой кислоты [RC(O)OC(O)R2], в которой R имеет значение, определенное выше для формулы (I) и R2 представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода, с получением соединения формулы (IV); и

нагревание соединения формулы (IV) с получением соединения формулы (I)

[0064] Вариант осуществления А2 представляет собой способ варианта осуществления А1, в котором когда соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты [RC(O)Y], и X не означает Cl и обеспечивают соединение формулы (IVa), то способ дополнительно содержит промывание соединения формулы (IVa) водным раствором хлорида натрия с получением соединения формулы (IVb)

[0065] Вариант осуществления A3 представляет собой способ варианта осуществления А1, в котором если соединение формулы (III) вступает в реакцию с ангидридом карбоновой кислоты [RC(O)OC(O)R2], или галогенангидридом карбоновой кислоты [RC(O)Y] и X=Cl и получают соединение формулы (IVb).
[0066] Вариант осуществления А4 представляет собой способ варианта осуществления А2 или A3, в котором соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты или ангидридом карбоновой кислоты в отсутствие кислотного катализатора.
[0067] Вариант осуществления А5 представляет собой способ варианта осуществления А2 или A3, в котором соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты или ангидридом карбоновой кислоты в отсутствие кислотного катализатора, выбранного из группы, которая включает n-толуолсульфоновую кислоту, бензолсульфоновую кислоту, сульфоуксусную кислоту, фосфорную кислоту и трихлорид фосфора.
[0068] Вариант осуществления А6 представляет собой способ любого из вариантов осуществления от А1 до А5, в котором соединение формулы (II) вступает в реакцию с галогенидом водорода (НХ) в реакционной смеси, дополнительно содержащей органический растворитель.
[0069] Вариант осуществления А7 представляет собой способ варианта осуществления А6, в котором органический растворитель выбирают из С2-С6 карбоновых кислот, С2-С6 нитрилов, C1-С6 спиртов, С2-С10 простых эфиров, С3-С6 алкилацетатов, С3-С10 кетонов, C5-C8 алифатических углеводородов, C1-С6 хлорированных углеводородов, С3-C8 алкилкарбонатов, сульфоланов, диметилсульфоксида, толуола, хлорбензола и их комбинаций.
[0070] Вариант осуществления А8 представляет собой способ варианта осуществления А7, в которой органический растворитель выбирают из группы, которая включает уксусную кислоту, пропионовую кислоту, ацетонитрил, пропионитрил, метанол, этанол, изопропанол, трет-бутанол, простой диэтиловый эфир, тетрагидрофуран, диоксан, метил-трет-бутиловый эфир, диметоксиэтан, метилацетат, этилацетат, трет-бутилацетат, ацетон, метилэтилкетон, гексан, гептан, циклогексан, дихлорметан, хлороформ, дихлорэтан, пропиленкарбонат, сульфолан, диметилсульфоксид, толуол, хлорбензол и их комбинации.
[0071] Вариант осуществления А9 представляет собой способ варианта осуществления А8, в которой органический растворитель выбирают из группы, которая включает уксусную кислоту, метанол, этанол, изопропанол, этилацетат, и их комбинации.
[0072] Вариант осуществления А10 представляет собой способ любого из вариантов осуществления от А1 до А9, в котором соединение формулы (II) вступает в реакцию с галогенидом водорода при температуре приблизительно от 0°С до приблизительно 60°С.
[0073] Вариант осуществления А11 представляет собой способ варианта осуществления А10, в которой температура составляет приблизительно от 10°С до приблизительно 30°С.
[0074] Вариант осуществления А12 представляет собой способ любого из вариантов осуществления от А1 до А11, в котором R2 представляет собой C1-С10 неразветвленный или разветвленный алифатический углеводород.
[0075] Вариант осуществления А13 представляет собой способ любого из вариантов осуществления от А1 до А11, в которой R2 представляет собой С3-С10 разветвленный алифатический углеводород.
[0076] Вариант осуществления А14 представляет собой способ любого из вариантов осуществления от А1 до А13, в котором соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты [RC(O)Y], где R означает С11-С29 гидрокарбильную группу с прямой цепью, и Y означает Cl или Br.
[0077] Вариант осуществления А15 представляет собой способ варианта осуществления А14, где R означает С12-С25 неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу.
[0078] Вариант осуществления А16 представляет собой способ варианта осуществления А14, в котором галогенангидрид карбоновой кислоты представляет собой хлорид олеиновой кислоты.
[0079] Вариант осуществления А17 представляет собой способ любого из вариантов осуществления от А1 до А13, в котором соединение формулы (III) вступает в реакцию с ангидридом карбоновой кислоты [RC(O)C(O)R2], где R означает а С11-С29 неразветвленную насыщенную или ненасыщенную алифатическую гидрокарбильную группу и R2 представляет собой C1-С10 неразветвленный или разветвленный алифатический углеводород.
[0080] Вариант осуществления А18 представляет собой способ варианта осуществления А17, где R означает С12-С25 неразветвленную насыщенную или ненасыщенную гидрокарбильную группу.
[0081] Вариант осуществления А19 представляет собой способ любого из вариантов осуществления от А1 до А18, в котором соединение формулы (III) вступает в реакцию с ангидридом карбоновой кислоты [RC(O)OC(O)R2], или галогенангидридом карбоновой кислоты [RC(O)Y] в реакционной смеси, дополнительно содержащей органический растворитель.
[0082] Вариант осуществления А20 представляет собой способ варианта осуществления А19, в котором органический растворитель выбирают из группы, которая включает С2-С6 нитрилы, С2-С10 простые эфиры, С3-С6 алкилацетаты, С3-С10 кетоны, C5-C8 алифатические углеводороды, C1-С6 хлорированные углеводороды, С3-С8 алкилкарбонаты, сульфолан, диметилсульфоксид, толуол, хлорбензол и их комбинации.
[0083] Вариант осуществления А21 представляет собой способ варианта осуществления А20, в котором органический растворитель выбирают из группы, которая включает ацетонитрил, пропионитрил, простой диэтиловый эфир, тетрагидрофуран, диоксан, метил-трет-бутиловый эфир, диметоксиэтан, метилацетат, этилацетат, трет-бутилацетат, ацетон, метилэтилкетон, гексан, гептан, циклогексан, дихлорметан, хлороформ, дихлорэтан, пропиленкарбонат, сульфолан, диметилсульфоксид, толуол, дихлорбензол и их комбинации.
[0084] Вариант осуществления А22 представляет собой способ варианта осуществления А21, в котором органический растворитель содержит ацетонитрил, пропионитрил, дихлорметан, хлороформ, тетрагидрофуран, и их комбинации.
[0085] Вариант осуществления А23 представляет собой способ любого из вариантов осуществления от А1 до А22, в котором соединение формулы (III) вступает в реакцию с ангидридом карбоновой кислоты или галогенангидридом карбоновой кислоты при температуре приблизительно от 0°С до приблизительно 120°С.
[0086] Вариант осуществления А24 представляет собой способ варианта осуществления А23, в котором температура составляет приблизительно от 20°С до приблизительно 120°С.
[0087] Вариант осуществления А25 представляет собой способ варианта осуществления А24, в котором температура составляет приблизительно от 40°С до приблизительно 85°С.
[0088] Вариант осуществления А26 представляет собой способ по любому из вариантов осуществления от А2 до А25, в котором соединение формулы (IVa) вводят в контакт с водным раствором хлорида натрия, чтобы получить соединение формулы (IVb).
[0089] Вариант осуществления А27 представляет собой способ варианта осуществления А26, в котором соединение формулы (IVa) вводят в контакт с водным раствором хлорида натрия в реакционной смеси, дополнительно содержащей органический растворитель.
[0090] Вариант осуществления А28 представляет собой способ варианта осуществления А27, в котором органический растворитель выбирают из группы, которая включает С2-С6 нитрилы, С2-С10 простые эфиры, С3-С6 алкилацетаты, С3-С10 кетоны, C5-C8 алифатические углеводороды, C1-С6 хлорированные углеводороды, толуол, хлорбензол и их комбинации.
[0091] Вариант осуществления А29 представляет собой способ варианта осуществления А28, в котором органический растворитель выбирают из группы, которая включает ацетонитрил, пропионитрил, простой диэтиловый эфир, тетрагидрофуран, диоксан, метил-трет-бутиловый эфир, ацетон, метилэтилкетон, гексан, гептан, циклогексан, дихлорметан, хлороформ, дихлорэтан, толуол, хлорбензол и их комбинации.
[0092] Вариант осуществления А30 представляет собой способ варианта осуществления А29, в котором органический растворитель содержит дихлорметан, хлороформ, 1,2-дихлорэтан, метилацетат, этилацетат, толуол, и их комбинации.
[0093] Вариант осуществления A31 представляет собой способ по любому из вариантов осуществления от А27 до А30, в котором соединение формулы (IVa) вводят в контакт с водным раствором хлорида натрия при температуре приблизительно от 0°С до приблизительно 60°С.
[0094] Вариант осуществления А32 представляет собой способ варианта осуществления A31, в котором температура составляет приблизительно от 10°С до приблизительно 30°С.
[0095] Вариант осуществления А33 представляет собой способ по любому из вариантов осуществления от А2 до A32, в котором соединение формулы (IVb) нагревают в реакционной смеси, содержащей соединение формулы (IVb) и органический растворитель.
[0096] Вариант осуществления А34 представляет собой способ варианта осуществления А33, в котором органический растворитель выбирают из группы, которая включает С2-С6 нитрилы, C1-С6 спирты, С2-С10 простые эфиры, С3-С6 алкилацетаты, С3-С10 кетоны, C5-C8 алифатические углеводороды, C1-С6 хлорированные углеводороды, С3-C8 алкилкарбонаты, сульфолан, диметилсульфоксид, толуол, хлорбензол и их комбинации.
[0097] Вариант осуществления A35 представляет собой способ варианта осуществления А34, в котором органический растворитель выбирают из группы, которая включает ацетонитрил, пропионитрил, метанол, этанол, изопропанол, трет-бутанол, простой диэтиловый эфир, тетрагидрофуран, диоксан, метил-трет-бутиловый эфир, диметоксиэтан, метилацетат, этилацетат, трет-бутилацетат, ацетон, метилэтилкетон, гексан, гептан, циклогексан, дихлорметан, хлороформ, дихлорэтан, пропиленкарбонат, сульфолан, диметилсульфоксид, толуол, хлорбензол и их комбинации.
[0098] Вариант осуществления A36 представляет собой способ варианта осуществления А35, в котором органический растворитель содержит хлороформ и метанол.
[0099] Вариант осуществления A37 представляет собой способ по любому из вариантов осуществления от А33 до А36 в которой реакционную смесь, содержащую соединение формулы (IVb) дополнительно содержит основание.
[00100] Вариант осуществления A38 представляет собой способ варианта осуществления A37, в котором основание является слабым основанием.
[00101] Вариант осуществления A39 представляет собой способ варианта осуществления A38, в котором основание представляет собой бикарбонат натрия, бикарбонат калия, дигидрофосфат калия, гидрофосфат дикалия, третичный амин, или их смесь.
[00102] Вариант осуществления А40 представляет собой способ по любому из вариантов осуществления от А33 до A39, в котором реакционная смесь, содержащая соединение формулы (IVb) и органический растворитель, дополнительно содержит осушитель, выбранный из группы, которая включает молекулярные сита, хлорид кальция, сульфат магния, сульфат натрия, активированный уголь, или их комбинацию.
[00103] Вариант осуществления А41 представляет собой способ по любому из вариантов осуществления от А33 до А40, в котором реакционную смесь, содержащую соединение формулы (IVb) и органический растворитель нагревают до температуры приблизительно от 20°С до приблизительно 100°С.
[00104] Вариант осуществления А42 представляет собой способ варианта осуществления А41, в котором реакционную смесь, содержащую соединение формулы (IVb) и органический растворитель нагревают до температуры приблизительно от 40°С до приблизительно 80°С.
[00105] Вариант осуществления А43 представляет собой способ варианта осуществления А42, реакционную смесь, содержащую соединение формулы (IVb) и органический растворитель нагревают до температуры приблизительно от 50°С до приблизительно 70°С.
[00106] Вариант осуществления А44 представляет собой способ любого из вариантов осуществления от А1 до А43, в котором нагревание соединение формулы (IV) образует смесь продуктов, содержащую соединение формулы (I) и способ дополнительно включает в себя фильтрацию смеси продуктов с образованием фракции твердых веществ и фильтрата, содержащего соединение формулы (I).
[00107] Вариант осуществления А45 представляет собой способ варианта осуществления А44, в котором соединение формулы (I) восстанавливают из фильтрата смеси продуктов и очищают путем промывания растворителем, посредством перекристаллизации или хроматографическим методом.
[00108] Вариант осуществления А46 представляет собой способ варианта осуществления А44, в котором фильтрат смеси продуктов промывают растворителем с образованием суспензии и продукт соединение формулы (I) восстанавливают из суспензии фильтрацией, продукт имеет чистоту по меньшей мере 95%.
[00109] Вариант осуществления А47 представляет собой способ варианта осуществления А44, в котором фильтрат смеси продуктов пропускают через колонку с диоксидом кремния и продукт соединение формулы (I) восстанавливают, продукт имеет чистоту по меньшей мере 95%.
[00110] Вариант осуществления А48 представляет собой способ любого из вариантов осуществления от А1 до А47, в котором соединение формулы (I) представляет собой хлорид 1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния (DOTIM):

[00111] Вариант осуществления В1 представляет собой способ получения соединения формулы (I)
в которой R представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода, способ включает в себя:
нагревание соединения формулы (IV) в реакционной смеси, содержащей органический растворитель и основание с получением соединения формулы (I)

в которой R имеет значение, определенное выше для формулы (I) и X означает Cl, Br, или I.
[00112] Вариант осуществления В2 представляет собой способ варианта осуществления В1, дополнительно включающий промывание соединения формулы (IVa), в которой X не означает Cl, водным раствором хлорида натрия получением соединения формулы (IVb), и реакционная смесь содержит соединение формулы IV(b)

[00113] Вариант осуществления В3 представляет собой способ варианта осуществления В1 или В2, в котором основание является слабым основанием.
[00114] Вариант осуществления В4 представляет собой способ по любому из вариантов осуществления от В1 до В3, в котором основание представляет собой бикарбонат натрия, бикарбонат калия, дигидрофосфат калия, гидрофосфат дикалия, третичный амин, или их смесь.
[00115] При введении элементов настоящего изобретения или их предпочтительных вариантов осуществления, учитывают то, что существует один или несколько элементов. Понятия «содержащий», «включающий» и «имеющий» предназначены для обозначения того, что могут быть дополнительные элементы, кроме перечисленных элементов.
[00116] Ввиду вышеизложенного будет очевидно, что достигнуты несколько задач изобретения и получены другие выгодные результаты.
[00117] Поскольку в вышеуказанных композициях и способах могут быть внесены различные изменения, не выходящие за рамки настоящего изобретения, то предполагается, что весь материал, содержащийся в приведенном выше описании и представленный в настоящей заявке, должен интерпретироваться как иллюстративный, а не в ограничивающем смысле.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 5-(АЛЬФА-ГАЛОГЕНАЦЕТИЛ)-8-(ЗАМЕЩЕННЫЙ ОКСИ)-(1Н)-ХИНОЛИН-2-ОНОВ | 2004 |
|
RU2339621C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНОПИРИДАЗИНОВ | 2016 |
|
RU2778306C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНОПИРИДАЗИНОВ | 2016 |
|
RU2742663C2 |
| ЭНАНТИОСЕЛЕКТИВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИНА | 2005 |
|
RU2383534C2 |
| ПИРРОЛОПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ | 2001 |
|
RU2254335C2 |
| РАЦЕМОСЕЛЕКТИВНОЕ ПОЛУЧЕНИЕ ПОДДАЮЩИХСЯ ВЫДЕЛЕНИЮ АНСА-МЕТАЛЛОЦЕНОВЫХ БИФЕНОКСИДНЫХ КОМПЛЕКСОВ, ИМЕЮЩИХ ОТНОСИТЕЛЬНО КОРОТКОЕ ВРЕМЯ ИЗОМЕРИЗАЦИИ | 2003 |
|
RU2329272C2 |
| СПОСОБ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2456296C2 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИПЕРИДИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2309147C9 |
| СПОСОБ ВЗАИМОДЕЙСТВИЯ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ПРИГОДНЫЕ ДЛЯ ПОЛУЧЕНИЯ ЦЕФАЛОСПОРИНОВ | 2001 |
|
RU2237670C1 |
| ПОЛУЧЕНИЕ ГЕТЕРОЦИКЛИЧЕСКИХ КЕТОНОВ | 2003 |
|
RU2325383C2 |
Изобретение относится к улучшенным способам получения амфифильных соединений имидазолиния, таких как хлорид 1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния (DOTIM). В частности, изобретение относится к способам синтеза таких соединений, в которых отказываются от использования токсичных реагентов, которые являются более экономичными и приводят к меньшему количеству отходов, чем обычные методы. DOTIM и подобные соединения могут быть приготовлены в виде катионных липосом, которые пригодны в качестве химических векторов для доставки нуклеиновых кислот в генной терапии. 2 н. и 19 з.п. ф-лы, 5 пр.
1. Способ получения соединения формулы (I)

в которой R представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода, где способ включает в себя:
реакцию соединения формулы (II) с галогенидом водорода (НХ) с получением соединения формулы (III), в которой X означает Cl, Br или I;

реакцию соединения формулы (III) с галогенангидридом карбоновой кислоты [RC(O)Y], где Y представляет собой Cl, Br, F или I и где значение R определено выше для формулы (I), с получением соединения формулы (IVa) или формулы (IVb),

и если X не является Cl и получают соединение формулы (IVa), то способ дополнительно включает в себя промывание соединения формулы (IVa) водным раствором хлорида натрия с получением соединения формулы (IVb);
и нагревание соединения формулы (IVb) с получением соединения формулы (I)

2. Способ по п. 1, в котором, когда соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты [RC(O)Y] и X=Cl, получают соединение формулы (IVb).
3. Способ по п. 2, в котором соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты в отсутствие кислотного катализатора.
4. Способ по п. 2, в котором соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты в отсутствие кислотного катализатора, выбранного из группы, включающей n-толуолсульфоновую кислоту, бензолсульфоновую кислоту, сульфоуксусную кислоту, фосфорную кислоту и трихлорид фосфора.
5. Способ по п. 1, в котором соединение формулы (II) вступает в реакцию с галогенидом водорода (НХ) в реакционной смеси, дополнительно содержащей органический растворитель, выбранный из C2-C6 карбоновых кислот, C2-C6 нитрилов, C1-С6 спиртов, С2-С10 простых эфиров, С3-С6 алкилацетатов, С3-С10 кетонов, С5-С8 алифатических углеводородов, C1-С6 хлорированных углеводородов, С3-C8 алкилкарбонатов, сульфолана, диметилсульфоксида, толуола, хлорбензола и их комбинаций.
6. Способ по любому из предыдущих пунктов, в котором соединение формулы (II) вступает в реакцию с галогенидом водорода при температуре приблизительно от 0°С до приблизительно 60°С.
7. Способ по любому из предыдущих пунктов, в котором R2 представляет собой C1-С10 неразветвленный или разветвленный алифатический углеводород.
8. Способ по любому из предыдущих пунктов, в котором соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты [RC(O)Y], где R означает С11-С29 гидрокарбильную группу с прямой цепью, и Y означает Cl или Br.
9. Способ по любому из предыдущих пунктов, в котором соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты [RC(O)Y] в реакционной смеси, дополнительно содержащей органический растворитель, выбранный из группы, включающей C2-C6 нитрилы, С2-С10 простые эфиры, С3-С6 алкилацетаты, С3-С10 кетоны, С5-С8 алифатические углеводороды, C1-С6 хлорированные углеводороды, С3-C8 алкилкарбонаты, сульфолан, диметилсульфоксид, толуол, хлорбензол и их комбинации.
10. Способ по любому из предыдущих пунктов, в котором соединение формулы (III) вступает в реакцию с галогенангидридом карбоновой кислоты при температуре приблизительно от 0°С до приблизительно 120°С.
11. Способ по любому из предыдущих пунктов, в котором соединение формулы (IVa) вводят в контакт с водным раствором хлорида натрия для получения соединения формулы (IVb).
12. Способ по п. 11, в котором соединение формулы (IVa) вводят в контакт с водным раствором хлорида натрия в реакционной смеси, дополнительно содержащей органический растворитель, выбранный из группы, включающей С2-С6 нитрилы, С2-С10 простые эфиры, С3-С6 алкилацетаты, С3-С10 кетоны, С5-С8 алифатические углеводороды, C1-C6 хлорированные углеводороды, толуол, хлорбензол и их комбинации.
13. Способ по п. 12, в котором соединение формулы (IVa) вводят в контакт с водным раствором хлорида натрия при температуре приблизительно от 0°С до приблизительно 60°С.
14. Способ по любому из пп. 1-13, в котором соединение формулы (IVb) нагревают в реакционной смеси, содержащей соединение формулы (IVb) и органический растворитель, выбранный из группы, включающей С2-С6 нитрилы, C1-С6 спирты, С2-С10 простые эфиры, С3-С6 алкилацетаты, С3-С10 кетоны, C5-C8 алифатические углеводороды, C1-С6 хлорированные углеводороды, С3-C8 алкилкарбонаты, сульфолан, диметилсульфоксид, толуол, хлорбензол и их комбинации.
15. Способ по п. 14, в котором реакционная смесь, содержащая соединение формулы (IVb), дополнительно содержит слабое основание, выбранное из группы, включающей бикарбонат натрия, бикарбонат калия, дигидрофосфат калия, гидрофосфат дикалия, третичный амин или их смесь.
16. Способ по п. 14 или 15, в котором реакционная смесь, содержащая соединение формулы (IVb) и органический растворитель, дополнительно содержит осушитель, выбранный из группы, включающей молекулярные сита, хлорид кальция, сульфат магния, сульфат натрия, активированный уголь или их комбинацию.
17. Способ по любому из пп. 14-16, в котором реакционную смесь, содержащую соединение формулы (IVb) и органический растворитель, нагревают до температуры приблизительно от 20°С до приблизительно 100°С.
18. Способ по любому из предыдущих пунктов, в котором при нагревании соединения формулы (IV) образуется смесь продукта, содержащая соединение формулы (I), и способ дополнительно включает фильтрацию смеси продукта для образования фракции твердых веществ и фильтрата, содержащего соединение формулы (I).
19. Способ по п. 18, в котором соединение формулы (I) восстанавливают из фильтрата смеси продукта и очищают промыванием растворителем, посредством перекристаллизации или хроматографическим методом.
20. Способ по любому из предыдущих пунктов, в котором соединение формулы (I) представляет собой хлорид 1-[2-(9(Z)-октадеценоилокси)этил]-2-(8(Z)-гептадеценил)-3-(2-гидроксиэтил)имидазолиния (DOTIM):

21. Способ получения соединения формулы (I)

в которой R представляет собой неразветвленную, алифатическую, насыщенную или ненасыщенную гидрокарбильную группу с 11-29 атомами углерода, где способ включает в себя:
нагревание соединения формулы (IV), в котором X означает Cl, в реакционной смеси, содержащей органический растворитель, включающий хлороформ и метанол, и основание, которое представляет собой бикарбонат натрия, с получением соединения формулы (I)

в которой значение R определено выше для формулы (I) и X означает Cl, и
если X означает не Cl, а представляет собой Br или I, то способ дополнительно включает промывание водным раствором хлорида натрия соединения формулы (IVa) с получением соединения формулы (IVb), и тогда реакционная смесь содержит соединение формулы IV(b)

| US 2010280258 A1, 04.11.2010 | |||
| US 5705655 A, 06.01.1998 | |||
| US 5830878 A, 03.11.1998 | |||
| U S8044215 B2, 25.10.2011 | |||
| Соловьева В | |||
| В., Кудряшова Н | |||
| В., Ризванов А | |||
| А | |||
| "ПЕРЕНОС РЕКОМБИНАНТНЫХ НУКЛЕИНОВЫХ КИСЛОТ В КЛЕТКИ (ТРАНСФЕКЦИЯ) С ПОМОЩЬЮ ГИСТОНОВ И ДРУГИХ ЯДЕРНЫХ БЕЛКОВ", Гены & Клетки, 2011, Т | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Солесос | 1922 |
|
SU29A1 |

