Область техники
Изобретение относится к химико-фармацевтической промышленности и касается способа получения аморфных ко-эвапоратов кладрибина и способа их применения.
Уровень техники
Кладрибин (2-хлордезоксиаденозин, 2-CdA) был разработан в конце 1970-х гг. научно-исследовательским институтом Скрипса (Ла-Хойя, штат Калифорния) и использовался, в частности, для лечения гемато-онкологических заболеваний. Кладрибин представляет собой синтетический хлорированный аналог дезоксиаденозина со следующей структурой:

Помимо химиотерапевтического действия, кладрибин также оказывает иммуносупрессивное действие, особенно на адаптивный компонент иммунной системы, характеризующийся выраженной лимфопенией периферических В-клеток (CD19+) и CD4+, а также частично CD8+ Т клеток.
Кладрибин является пролекарством, поглощаемым клетками со специфическими транспортными белками. В клетках он подвергается фосфорилированию дезоксицитидинкиназой (DCK) с образованием мононуклеотидного 2-хлордезоксиаденозин-5'-монофосфата (2-CdAMP), который впоследствии фосфорилируется с образованием 2-хлордезоксиаденозин-5'-трифосфата (2-CdATP), который сам по себе является активным соединением.
В покоящихся клетках 2-CdATP вызывает разрывы одиночных спиралей ДНК, быстрое потребление никотинамидадениндинуклеотида, истощение АТФ и гибель клеток. В делящихся клетках 2-CdATP вмешивается в синтез ДНК посредством ингибирования рибонуклеотидредуктазы и конкурирует с дезоксиаденозинтрифосфатом за включение в ДНК ДНК-полимеразами.
В большинстве типов клеток активная трифосфорилированная форма инактивируется дефосфорилированием 5β-нуклеотидазы, однако в лимфоцитах, особенно в B-клетках, соотношение киназа:фосфатаза высокое, так что препарат остается активным и преимущественно ингибирует данные клетки, в то время как другие типы клеток остаются нетронутыми.
Изменения в уровнях экспрессии деоксицитидинкиназы (DCK) и 5′- нуклеотидаза (5'-NT) между подтипами иммунных клеток могут объяснить различия в чувствительности иммунных клеток к кладрибину. Ввиду таких уровней экспрессии клетки врожденной иммунной системы страдают меньше, чем клетки адаптивной иммунной системы.
Одним из характерных свойств кладрибина является его нестабильность в кислой среде. Кладрибин относится к так называемым «кислотолабильным лекарствам», то есть веществам, которые быстро разлагаются в кислой среде. Даже при физиологической температуре 37°C значение t1/2 кладрибина определяется как равное 1,6 ч для растворов с pH 2,0 и всего 0,37 ч для растворов с pH 1,0 (Tarasiuk A., Skierski J., Kazimierczuk Z. Stability of 2-chloro-2'-deoxyadenosine at various pH and temperature. Archivum Immunologiae et Therapiae Experimentalis, 01 Jan 1994, 42(1):13-15).
Поскольку основным местом всасывания пероральных лекарств является желудок с низким pH, всасывание чувствительных к кислоте лекарств в желудке затруднено. Кислый pH желудка можно регулировать, например, с помощью энтеросолюбильных составов или с помощью буфера, или веществ, снижающих pH желудка. Эти стандартные процедуры оказались неэффективными для кладрибина (F. Albertioni, G. Juliusson & J. Liliemark. On the bioavailability of 2-chloro-2′-deoxyadenosine (CdA). The influence of food and omeprazole. European Journal of Clinical Pharmacology v. 44, p. 579-582, 1993).
Для защиты кладрибина при низких рН Шульц и др. предложили использовать комплекс циклодекстрина с клабрибином (US patent № 6,194,395). Циклодекстрины в значительной мере содействуют стабильности кладрибина в растворе с pH 1,4 и поддерживают более 80% действующего вещества в течение почти 5 часов и примерно 70% действующего вещества в течение 8 часов, т.е. на протяжении всего эксперимента, в то время как потери кладрибина, растворенного в растворе, в отсутствие циклодекстринов составляли около 50% примерно через 3 часа, а через 8 часов в растворе оставалось около 15% действующего вещества. На основании полученных результатов было предложено использовать комплексы кладрибина с циклодекстринами для получения стабильных жидких и твердых лекарственных форм.
Однако Шульц и др. описали только способ получения твердой лекарственной формы путем экструзии из расплава, в котором кладрибин и циклодекстрин смешивают с другими вспомогательными ингредиентами, а затем нагревают до расплавления. Широкий диапазон доз от 1 до 15 мг кладрибина и от 100 до 500 мг циклодекстрина, представленных в патенте, включает в себя множество комбинаций, которые могут быть пригодными в качестве смесей, но не для образования комплексов. Например, в соотношении 1 мг кладрибина к 500 мг циклодекстрина содержится слишком много циклодекстрина, так что лекарство не может легко покинуть комплекс и выполнить свою терапевтическую функцию. С другой стороны, 15 мг кладрибина и только 100 мг циклодекстрина будет недостаточно для образования комплекса данного количества кладрибина.
Кроме того, соотношение кладрибина к циклодекстрину 1:500 фактически не позволяет получить пероральную лекарственную форму с более высоким содержанием действующего вещества. Даже заявленная защита кладрибина от разложения при низком pH, вероятно, зависит от соотношения АФИ (активный фармацевтический ингредиент) : циклодекстрин и не является общей характеристикой циклодекстрина.
Другой способ получения комплекса кладрибин-циклодекстрин заключается в следующем: (i) растворение циклодекстрина в воде, (ii) добавление кладрибина и перемешивание в течение 15-18 часов с последующим (iii) удалением нерастворенного кладрибина фильтрацией и (iv) лиофилизацией фильтрата в течение 97-118 часов (US № 8785415).
Данный способ получения комплекса кладрибин : циклодекстрин занимает от 112 до 136 часов.
Похожий способ получения комплекса кладрибин-циклодекстрин описан в US № 7,888,328. Полученный продукт характеризуется авторами как смесь, состоящая на 30-40% из аморфного комклекса циклодекстрина и кладрибина, где кладрибин расположен внутри “поры” циклодекстрина, и на 60-70%, где кладрибин расположен вне “поры” циклодекстрина (inclusion and non-inclusion complexes). К сожалению, соотношение компонентов в известных комплексах не подтверждено каким-либо аналитическим методом.
Также не доказано, что аморфные формы циклодекстрина влияют на растворимость кладрибина. Для четкого вывода, например, отсутствует сравнение растворимости самой аморфной формы или других полиморфных форм с аморфной формой в виде комплекса циклодекстрина.
Оба упомянутых выше способа получения комплекса кладрибина с циклодекстрином, независимо от природы образованного комплекса, практически идентичны, и в обоих для получения комплекса циклодекстрина с кладрибином используется лиофилизация.
Лиофилизация - это трудоемкий процесс, характеризующийся высокими капитальными затратами на техническое оборудование и эксплуатацию. Ввиду своей стоимости лиофилизация используется скорее при получении, например, парентеральных лекарств, вакцин, белков или веществ, подверженных повреждению от перегрева в целом с использованием стандартных методов. Она редко используется в производстве пероральных препаратов ввиду стоимости и, по существу, ограничивается производством составов, диспергируемых в полости рта.
Цель настоящего изобретения заключалась в том, чтобы устранить недостатки вышеупомянутых способов получения комплекса кладрибина и предоставить надежную, рентабельную, безопасную и простую в применении промышленную технологию производства комплекса кладрибин-циклодекстрин, которая обеспечивает получение продукта с такими же физико-химическими свойствами, как лиофилизированный кладрибиновый комплекс.
Техническим результатом заявляемого изобретения является разработка простого в инструментальном исполнении способа получения аморфного комплекса кладрибин-циклодекстрин (ко-эвапорат), используемого для приготовления пероральной лекарственной формы.
В ходе экспериментов, направленных на изучение возможности замены лиофилизации более дешевым и надежным методом, было обнаружено, что (i) использование водно-спиртового раствора снижает температуру, необходимую для растворения кладрибина, и значительно сокращает время, необходимое для растворения кладрибина в растворе циклодекстрина, (ii) ко-эвапорат кладрибина с циклодекстринами позволяют получить высокостабильный аморфный комплекс с удовлетворительными солюбилизирующими свойствами, что (iii) полученный аморфный ко-эвапорат проявляет физико-химические свойства, идентичные образцу, полученному в соответствии с примером, приведенным в патенте США 8785415, а в 7 888 328 представлен такой же профиль растворения, как и у исходного состава в буферах с pH 1,0, 2,0, 4,5 и 6,8, при этом общее время получения аморфного ко-эвапората, а также затраты на его получение на порядок ниже, чем при получении комплекса кладрибина в соответствии с вышеупомянутыми патентами.
Краткое изложение сущности изобретения
Настоящее изобретение относится к способу получения аморфного комплекса кладрибина с циклодекстрином (ко-эвапората), включающего в себя:
а) кладрибин,
b) циклодекстрин и
c) дополнительный остаточный органический растворитель.
При этом способ получения аморфного ко-эвапората включает в себя стадии:
(i) растворения циклодекстрина в водно-спиртовом растворе,
(ii) растворения кладрибина в водно-спиртовом растворе циклодекстрина, и
(iii) полное удаление растворителя в вакуумном испарителе.
Еще одним предметом настоящего изобретения является пероральная лекарственная форма, содержащая:
(а) аморфный ко-эвапорат по настоящему изобретению; а также
(b) фармацевтически приемлемые вспомогательные вещества.
Предпочтительно пероральная лекарственная форма представлена в виде таблеток, более предпочтительно - в виде таблеток без оболочки. Таблетки предпочтительно готовят прямым прессованием.
Ко-эвапорат представляет собой комплекс АФИ (активного фармацевтического ингредиента) и полимерного наполнителя(ов) (эксципиента(ов)), полученный путем удаления летучего органического растворителя, в котором и лекарственное средство (АФИ), и наполнитель были предварительно растворены.
Термин «кладрибин» в контексте настоящего документа относится к кладрибину в форме его фармацевтически приемлемых сольватов, гидратов, энантиомеров, полиморфов или их смесей. Предпочтительно кладрибин используется в кристаллической форме.
Помимо кладрибина, ко-эвапорат содержит циклодекстрин и, в некоторых случаях, остатки органических растворителей.
Как правило, термин «циклодекстрин» относится к циклическим олигосахаридам с молекулярной массой около 1000 Да, которые состоят из шести-восьми единиц глюкозы, связанных α-1,4-гликозидной связью. Молекулярная структура циклодекстрина напоминает усеченный конус с гидрофильной внешней поверхностью и неполярной внутренней полостью.
В настоящем изобретении предпочтительно используется гидроксипропил-β-циклодекстрин (гидроксипропилированный бета-циклодекстрин, HPβCD). HP-β-циклодекстрин представляет собой кольцевую молекулу, состоящую из семи единиц глюкозы, связанных α-1,4-гликозидными связями, со средней степенью замещения от 3 до 6, более предпочтительно от 4 до 5, особенно 4,6. Средняя степень замещения означает количество заместителей на циклодекстриновое кольцо. Средняя степень замещения от 4 до 5, в частности 4,6, приводит к отличным свойствам растворения.
Кроме того, предпочтительно используется циклодекстрин с объемной плотностью 150-700 миллиграммов/см2, более предпочтительно 200-350 миллиграммов/см2.
Кроме того, особенно предпочтительно, чтобы циклодекстрины в форме аморфного гидрата циклодекстрина использовали во всех вариантах настоящего изобретения.
В дополнение к кладрибину и декстрину, ко-эвапорат содержит остаточный органический растворитель. В общем, любой органический растворитель, способный растворять кладрибин и циклодекстрин (по крайней мере, частично, но предпочтительно полностью), подходит для получения соиспаряемого вещества.
Вода не считается органическим растворителем.
В предпочтительном варианте используется органический растворитель с относительной диэлектрической проницаемостью (ε) 3-40, предпочтительно 4-38, более предпочтительно 5-35, еще более предпочтительно 8-32, измеренной при 20°C.
В общем, подходящие органические растворители могут быть выбраны из кетонов C3-C6, алифатических или ароматических углеводородов C5-C9, необязательно замещенных, например, галогеном, сложным эфиром C3-C6, спиртом C2-C6, простым эфиром C2-C6, диметилацетомида, диметилформамида, диметилсульфоксида, N-метил-2-пирролидон (NMP) и их смесями. В предпочтительном варианте органический растворитель представляет собой спирт, предпочтительно спирт C2-C6, более предпочтительно этанол и изопропиловый спирт. Этанол особенно предпочтителен.
Наконец, еще одним аспектом настоящего изобретения является использование органического растворителя с относительной диэлектрической проницаемостью 3-40, измеренной при 20°C.
Как правило, органический растворитель может содержать смеси двух или более из указанных выше растворителей.
Способ приготовления ко-эвапората по настоящему изобретению включает в себя стадии:
i) растворения циклодекстрина в водно-спиртовом растворе;
ii) растворения кладрибина в водно-спиртовом циклодекстрине;
iii) удаления растворителя (полностью или частично).
На стадии (i) циклодекстрин полностью растворяется в водно-спиртовом растворе.
На стадии ii) кладрибин постепенно добавляют и полностью растворяют в растворе органического растворителя и воды при перемешивании. Растворение можно проводить при повышенной температуре. Предпочтительно используются температуры 25-80°C, более предпочтительно 25-60°C, оптимально 25-40°C.
Количество органического растворителя, используемого на стадиях i) и ii) для растворения кладрибина и декстрина, обычно влияет на процесс в отношении времени и стоимости процесса.
В предпочтительном варианте кладрибин растворяют в количестве 0,1-38 мг/мл водно-спиртового раствора, более предпочтительно 0,5-30 мг/мл, еще более предпочтительно 0,5-25 мг/мл, наиболее предпочтительно 1-20 мг/мл. При этом содержание спирта в водно-спиртовом растворе составляет от не менее 5 об.% до не более 95 об.%.
В предпочтительном варианте декстрин растворяют в количестве 10-1000 мг/мл водно-спиртового раствора, более предпочтительно 20-500 мг/мл, еще более предпочтительно 35-400 мг/мл, наиболее предпочтительно 40-350 мг/мл.
Органический растворитель полностью или частично удаляют, так, что оставшееся количество растворителя содержится в получившемся ко-эвапорате. В предпочтительном варианте в ко-эвапорате содержится остаточный органический растворитель в количестве 0,1-10% масса/масса, более предпочтительно 0,5 - 5% масса/масса, еще более предпочтительно 0,6-4% масса/масса, оптимально 0,7-2,0% масса/масса в расчете на общую массу испарившихся твердых веществ.
В другом предпочтительном варианте исходные соединения и условия процесса выбирают так, чтобы в полученном аморфном ко-эвапорате содержалось:
(a) 0,1-30 мас.%, предпочтительно 1-20 мас.%, более предпочтительно 5-15 мас.% кладрибина,
(b) 70-99,9 мас.%, предпочтительно 80-99 мас.%, более предпочтительно 85-95 мас.% циклодекстрина, и
(c) 0-20 мас.%, предпочтительно 0,1-10 мас.%, более предпочтительно 0,5-5 мас.% органического растворителя от общей массы ко-эвапората.
На стадии iii) органический растворитель можно удалить при повышенной температуре и/или при пониженном давлении. Растворитель можно удалить под давлением 0,1-1000 мбар, предпочтительно 1-200 мбар, более предпочтительно 5-100 мбар, еще более предпочтительно 10-50 мбар, в частности 30-40 мбар. В лабораторном масштабе время стадии iii) может составлять примерно 0,5-2,0 ч, предпочтительно приблизительно 90 минут. Удаление органического растворителя можно проводить, например, в вакуумном ротационном испарителе, например, Rotavapor Büchi®.
В предпочтительном варианте растворитель удаляют при температуре 30-70°C, предпочтительно 30-65°C, более предпочтительно 30-45°C. Время испарения длится от десятков минут до часов. Остаточный растворитель в аморфном ко-эвапорате определяют методом газовой хроматографии.
Способ по настоящему изобретению, как показано выше, приводит к образованию аморфного ко-эвапората, содержащего кладрибин, циклодекстрин и остаточное количество органического растворителя.
Термин «аморфный ко-эвапорат» используется в целях заявки на патент для обозначения твердого вещества, которое не проявляет свойств кристаллической структуры в условиях РПД (рентгеновской порошковой дифрактометрии). По сравнению с кристаллическими формами имеет неоспоримое преимущество ввиду своей более высокой растворимости.
Заявляемый способ приводит к получению стабильного, быстро растворимого аморфного получаемого путем со-испарения комплекса кладрибина и циклодекстрина (ко-эвапората) в форме материала белого или полупрозрачного (молочно-белого) цвета. Ко-эвапорат проявляет физико-химические свойства (спектры FTIR (инфракрасная спектроскопия на основе преобразования Фурье), РПД, 13 C ЯМР, ТГ/ДТА), эквивалентные образцу, полученному в соответствии с примером, приведенным в патентах США 8785415 и 7888328.
Настоящее изобретение также относится к другим ко-эвапоратам, которые могут быть получены заявляемым способом. Что касается указанных ко-эвапоратов, все вышеупомянутые замечания, касающиеся предпочтительных вариантов, применяются (например, относительно типа и количества органического растворителя, типа и количества кладрибина, а также типа и количества циклодекстрина, условий удаления растворителя и т.д.).
Полученный ко-эвапорат по настоящему изобретению можно вводить в форме фармацевтических препаратов.
Предпочтительно пероральная лекарственная форма представлена в виде таблеток, более предпочтительно - в виде таблеток без оболочки. Таблетки предпочтительно готовят прямым прессованием.
В фармацевтическом составе по настоящему изобретению могут использоваться одно или несколько фармацевтически приемлемых вспомогательных веществ, таких как наполнители, связующие, смазывающие, скользящие вещества, антиадгезивные вещества и разрыхлители. Что касается вышеупомянутых фармацевтически приемлемых вспомогательных веществ, в заявке приводятся ссылки на книгу «Lexikon der Hilfsstoffe fur Pharmazie, Kosmetik und angrenzende Gebiete», опубликованную Г.П. Фидлером, 4-е издание, изд-во «Edito Cantor», Аулендорф и более ранние издания, а также на «Руководстве по фармацевтическим вспомогательным веществам», третье издание, под редакцией Артура Х. Киббе, Американская фармацевтическая ассоциация, Вашингтон, США, и сервис «Pharmaceutical Press», Лондон.
Краткое описание чертежей
Изобретение поясняется следующими чертежами.
На фиг. 1 представлены структуры гидрата кладрибина и его расположение в базальной клетке.
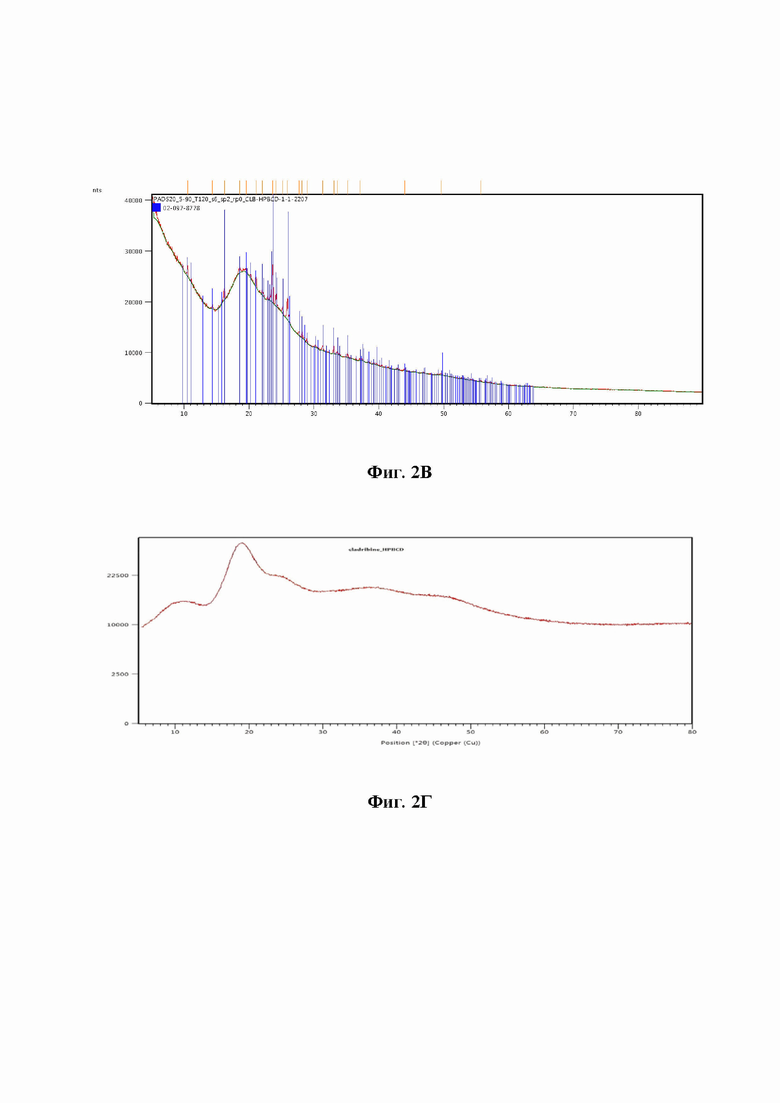
На фиг. 2 представлено сравнение РПД используемых форм кладрибина, механической смеси, HPbCD и дифрактограммы полученного аморфного ко-эвапората. На основе РПД невозможно отличить ко-эвапорат, полученный в соответствии с настоящим патентом, от образца в соответствии с патентом США 7888328, поэтому приводится только одна типичная дифрактограмма конечного продукта независимо от способа получения.
На фиг. 3 представлены результаты анализа ко-эвапората и образца методом ТГ/ДТА в соответствии с патентом США 7,888,328.
На фиг. 4 представлены спектры FTIR кладрибина, HPbCD, механической смеси, ко-эвапората и образца в соответствии с патентом США 7,888,328.
На фиг. 5 представлены спектры 13 C ЯМР механической смеси кладрибин/HPbCD, стандарта HPbCD, ко-эвапората и образца в соответствии с патентом США 7,888,328.
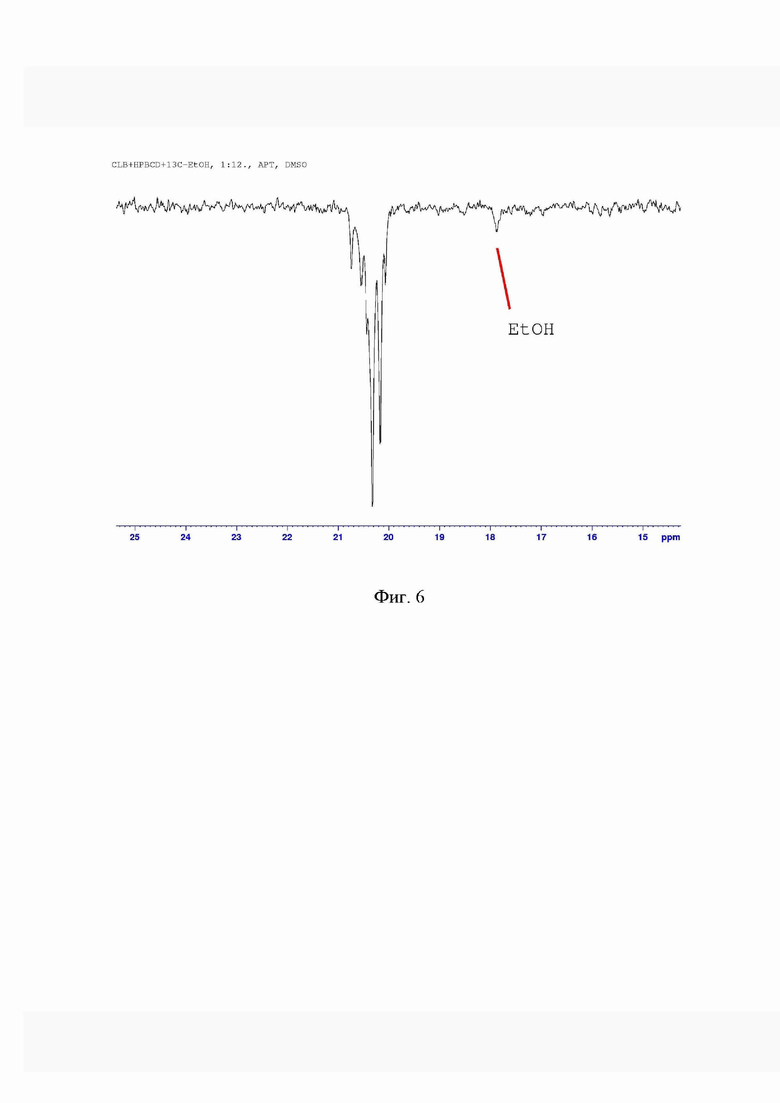
На фиг. 6 представлено обнаружение остаточного 13C этанола в ко-эвапорате жидкофазным ЯМР.
На фиг. 7 представлены результаты сравнительного растворения ко-эвапората и образца в соответствии с патентом США 7,888,328 в растворах для растворения с pH 1,2 и 6,8.
Осуществление изобретения
Ниже представлено более подробное описание заявляемого изобретения. Настоящее изобретение может подвергаться различным изменениям и модификациям, понятным специалисту на основе прочтения данного описания. Такие изменения не ограничивают объем притязаний.
Все используемые реагенты являются коммерчески доступными, все процедуры, если не оговорено особо, осуществляли при комнатной температуре или температуре окружающей среды, то есть в диапазоне от 18 до 25°C.
Пример 1. Получение ко-эвапората кладрибина с циклодекстрином с использованием известной кристаллической формы кладрибина I (TW 201117816 A): 6000 мг гидроксипропил-β-циклодекстрина (HPBCD) растворяли в 15 мл дистиллированной воды. После растворения к раствору добавляли 96% этанол в объемном соотношении вода : этанол 3:2. В заключение добавляли 500 мг кристаллической формы кладрибина I, полученную суспензию нагревали и перемешивали при температуре 25-35°C с помощью магнитной мешалки до получения прозрачного раствора. Время, необходимое для растворения кладрибина, составляло приблизительно 15 минут. Весовое соотношение HPBCD : АФИ составляло 12:1. Затем образец помещали на роторный испаритель с температурой водяной бани 40°C, где его сушили в вакууме в течение 1 часа. После чего образец помещали в сухожаровой шкаф и сушили в течение ночи при температуре 45°C.
Пример 2. Получение ко-эвапората кладрибина с циклодекстрином с использованием аморфной формы кладрибина: 6000 мг гидроксипропил-β-циклодекстрина (HPBCD) растворяли в 15 мл дистиллированной воды. После растворения к раствору добавляли 96% этанол в объемном соотношении вода:этанол 3:2. В заключение добавляли 500 мг аморфной формы кладрибина, полученную суспензию нагревали и перемешивали при температуре примерно 25-35°C с помощью магнитной мешалки до получения прозрачного раствора. Время, необходимое для растворения кладрибина, составляло приблизительно 7 минут. Весовое соотношение HPBCD : АФИ составляло 12:1. Затем образец помещали на роторный испаритель с температурой водяной бани 40°C, где его сушили в вакууме в течение 1 часа. После чего образец помещали в сухожаровой шкаф и сушили в течение ночи при температуре 45°C.
Аморфную форму кладрибина получали в соответствии с Примером 3 в патентной заявке WO 2011/054179 A1. Наличие аморфной формы кладрибина подтверждено РПД.
Пример 3. Получение ко-эвапората кладрибина с циклодекстрином с использованием нового гидратированного полиморфа кладрибина: 1200 мг гидроксипропил-β-циклодекстрина (HPBCD) растворяли в 15 мл дистиллированной воды. После растворения к раствору добавляли 96% этанол в объемном соотношении вода : этанол 3:2. В заключение добавляли 100 мг новой кристаллической формы гидрата кладрибина, полученную суспензию нагревали и перемешивали при температуре приблизительно 25-35°C с помощью магнитной мешалки до получения прозрачного раствора. Весовое соотношение HPBCD : АФИ составляло 12:1. Время, необходимое для растворения кладрибина, составляло около 8 минут. Затем образец помещали на роторный испаритель с температурой водяной бани 45°C, где его сушат в вакууме в течение 1 часа. После чего образец помещали в сухожаровой шкаф и сушат в течение ночи при температуре 40°C.
Кристаллическая структура нового полиморфа кладрибина и его пространственная структура подтверждены РСА и РПД (фиг. 1).
Использование кладрибина разной кристалличности не влияет на получение ко-эвапората, а влияет только на время, необходимое для растворения кристаллической формы в растворе циклодекстрина в водно-спиртовом растворе. Для получения новой кристаллической формы 100 мг формы I кладрибина, растворяли при перемешивании в избытке растворителя - 10 мл 96% EtOH, 15 мл дистиллированной H2O при повышенной температуре (60°C). Раствор концентрировали выпариванием в вакууме до объема 10 мл. Концентрированный раствор выливали непрерывным тонким слоем на чашку Петри и дают ему свободно испариться при комнатной температуре.
Структура моногидрата кладрибина охарактеризована с помощью структурного анализа монокристалла с источником излучения Cu-Kα и при температуре измерения 200 K. Моногидрат кладрибина кристаллизуется в орторомбической системе в пространственной группе P212121. Конфигурация хиральных центров - C11-R; C13-S; C14-R; C31-R; C33-S; C44-R. Параметры решетки моногидрата кладрибина:
a = 7,1448 (2) Å
b = 13,4926 (4) Å
c = 26,5439 (6) Å
α = 90,00°
β = 90,00°
γ = 90,00°
V = 2558,88 (12) Å3
Z = 8
R = 0,0395
Плотность (г/см3) = 1,577
Пример 4. Получение образца в соответствии с примером 2 патента США 7888328 (сравнительный пример)
17,25 г гидроксипропил-β-циклодекстрина (с получением приблизительно 17% раствора) растворяли в 82,5 мл дистиллированной воды, затем добавляли 1875 мг кладрибина, смесь перемешивали при температуре 48°C в течение девяти часов, после чего перемешивали в течение еще 9 часов при комнатной температуре. Весь нерастворенный кладрибин удаляли фильтрацией. После фильтрации растворы разливали во флаконы для лиофилизации, частично закрывали пробкой и лиофилизировали в соответствии с условиями, описанными в патенте.
Пример 5. Pентгеновская порошковая дифрактометрия
Pентгеновскую порошковую дифрактометрию кладрибина, ко-эвапоратов кладрибина с циклодекстрином, полученных в соответствии с Примерами 1и 3, и образца, полученного в соответствии с Примером 2 патента США № 7,888,328 выполняли с использованием стандартного нормального излучения Cu-Kα. Пики отражения регистрировали в диапазоне углов 2-тета 5-40 градусов и 5-80 градусов соответственно. Результаты XRD обеих испытанных кристаллических форм кладрибина приведены на фиг. 2a (рентгенограмма формы I показана в верхней части рисунка, рентгенограмма моногидрата кладрибина - в нижней части). Фиг. 2b иллюстрирует РПД стандарта HPbCD (Гидроксипропил-бета-циклодекстрин). Фиг. 2c иллюстрирует рентгенограмму механической смеси, полученную простым смешиванием кристаллической формы I кладрибина и HPbCD. Из-за высокой концентрации HPbCD, которая частично маскирует отклик кладрибина, пики, соответствующие кладрибину, обозначены вертикальными линиями для лучшей ориентации. Фиг. 2d иллюстрирует XRD ко-эвапората. Независимо от кристаллической структуры исходного кладрибина, все образцы ко-эвапоратов, и образец, приготовленный в соответствии с примером патента США № 7,888,328 демонстрируют XRD аморфного продукта.
На основании полученных данных можно однозначно утверждать, что ни использование вещества с другой кристаллической структурой, ни способ получения образца не влияют на аморфную природу конечного продукта.
Пример 6. Термогравиметрический анализ/Дифференциальный термический анализ (ТГА/ДТА)
ТГА-профили кладрибина, HPbCD, механической смеси, ко-эвапората и образца, приготовленного в соответствии с патентом США 7888328 при 25-300°C приведены на фиг. 3а. Кривая ТГА кладрибина свидетельствует о том, что процесс разложения начинается при температуре приблизительно 200°C (фиг. 3а). В случае HPbCD ввиду дегидратации потеря веса приблизительно составила 6% в диапазоне температур от 30 до 140°C. ТГ анализ образца в соответствии с патентом США 7888328 продемонстрировал потерю воды в диапазоне 20-100°C и процесс разложения, начинающийся примерно при 250-200°C). Кривая ТГ механической смеси показывает многоступенчатый путь разложения. Первая фаза в диапазоне комнатная температура - 100°C связана с потерей воды из HPbCD, а вторая фаза, наблюдаемая при температурах выше 200°C, связана с разложением кладрибина. На основании сравнения фазы разложения ТГ для соиспаряемого вещества и образца в соответствии с патентом США 7888328 получается, что оба образца разлагаются медленнее, чем смесь. Продемонстрированная более высокая термостойкость комплекса является признаком взаимодействия HPbCD и кладрибина. ТГА ко-эвапората показывает результат, аналогичный результату ТГА образца в соответствии с патентом США 7888328. Чтобы лучше понять термическое поведение образцов, был проведен ДТА (фиг. 3b). Кладрибин-стандарт при дифференциальной сканирующей калориметрии (ДСК) демонстрирует два эндотермических явления. Первое эндотермическое явление при температуре приблизительно 206°C характеризует начало термического разложения кладрибина и соответствует переходу в расплавленное состояние. При втором эндотермическом явлении, приблизительно при 212°C, вероятно образование продукта разложения кладрибина.
Профиль ДТА HPbCD соответствует потере воды.
Кривые ДТА ко-эвапората и образца в соответствии с патентом США 7888328 демонстрируют эндотермическое явление ввиду потери воды в HPbCD (40-140°C). Кроме того, оба образца демонстрируют эндотермические явления при приблизительно 200°C, которые могут быть вызваны остатком несвязанного кладрибина. Отсутствие термических явлений, типичных для чистого кладрибина, означает потерю кристаллической природы кладрибина при его включении в комплекс. Это является косвенным свидетельством взаимодействия кладрибина с HPbCD. Оба испытуемых образца, т.е. как ко-эвапорат, так и образец в соответствии с патентом США 7888328, могут считаться сходными на основании результатов анализов ТГ/ДТА.
Пример 7. Фурье-спектроскопия (спектры FTIR)
Для получения образца отвешивали 0,58 г бромида калия и измельчали в ступке с 0,005 г образца лекарственной формы, полученной в соответствии с Примером 1 и Примером 4. Смесь снова взвешивали и количественно переносили в пресс, превращали в таблетку, помещали в чашку Петри и хранили в осушителе. Таблетку, содержащую 0,58 г бромида калия, использовали для измерения эталонного спектра. Эталонный спектр таблетки бромида калия, служивший фоном, измеряли с использованием спектрометра Bruker Vertex 80. Затем измеряли спектры таблеток обоих приготовленных образцов с фоновым считыванием. Результаты представлены на фиг. 4a-4e. На фиг. 4а представлен спектр FTIR кристаллической Формы I кладрибина. На фиг. 4b представлен спектр FTIR стандарта HPbCD. На фиг. 4с представлена механическая смесь, полученная простым смешиванием кладрибина с HPbCD. ИК-спектр механической смеси можно интерпретировать как «сумму» спектров чистого кладрибина и чистого HPbCD с резкими колебательными полосами для чистого кладрибина, что указывает на то, что молекулы структурно определены и регулярны (в кристаллическом поле). Это наглядная демонстрация наличия двух отдельных фаз - кристаллического кладрибина и HPbCD. Напротив, спектры FTIR ко-эвапората (фиг. 4d) и образца в соответствии с патентом США 7,888,328 (фиг. 4e) демонстрируют четкие различия, особенно в отношении маркеров кристаллической фазы кладрибина, наблюдаемых в механической смеси. Происхождение аморфной фазы можно объяснить взаимодействием кладрибина с HPbCD, которое защищает молекулы кладрибина от кристаллизации. Как видно из фиг. 4d и 4e, спектры FTIR сравниваемых образцов не показывали значительных изменений, и образцы можно считать очень похожими на основе спектров FTIR.
Пример 8. Поведение твердой фазы 13 C ЯМР исследуют с использованием твердотельного ЯМР-спектрометра Bruker Avance III HD 500 WB/ЯМР. Твердотельные спектры ЯМР механической смеси кладрибина с HPbCD, ко-эвапората кладрибина и образца, полученного в соответствии с патентом США № 7 888 328 представлены на фиг. 5a и 5b и указывают на то, что межмолекулярного взаимодействия между кладрибином и HPbCD нет и что смесь состоит из по-разному расположенных молекулярных доменов каждого отдельного компонента. Сравнение спектров на фиг. 5b (сверху вниз - спектр образца в соответствии с патентом США 7888328, стандарт HPbCD, ко) свидетельствует о том, что для сигналов HPbCD отсутствует сдвиг, а резонансы кладрибина практически не обнаруживаются (в спектральной области приблизительно 40 ppm, 120 ppm и 160 ppm). Это указывает на отсутствие кристаллического домена кладрибина и на то, что образец состоит из кладрибина, диспергированного на молекулярном уровне в HPbCD. Как видно из фиг. 5b, различий между спектрами ко-эвапората и образца в соответствии с патентом США 7888328 обнаружено не было, и поэтому образцы можно считать схожими.
Пример 9. Обнаружение остаточного растворителя
Остаточный растворитель определяли методом газовой хроматографии, и полученные данные подтверждали жидкостным ЯМР. Для определения методом газовой хроматографии использовали газовый хроматограф с пламенно-ионизационным детектором и гелием (99,999%) в качестве газа-носителя. С помощью газового хроматографа продемонстрировано присутствие следового количества этанола в количестве, близком к пределу обнаружения прибора. Даже метод ЯМР в жидкой фазе не показал присутствия остаточного этанола. Только использование 13С-Этанола при получении соиспаряемого вещества позволило обнаружить следовые количества растворителя в образце (фиг. 6). Основываясь на данных измерений, можно утверждать, что ко-эвапорат, полученный заявляемым способом, содержит только следовые количества остаточного растворителя и, следовательно, приближается к составу образца приготовленным в соответствии со способом по патенту США 7888328, который получали только из водного раствора циклодекстрина.
Пример 10. Сравнительное растворение
Сравнительное растворение проводили в трех основных буферах для растворения (0,1 M HCl, pH 1,0; в ацетатном буфере, pH 4,5 и фосфатном буфере, pH 6,8) с использованием прибора для растворения II, при 50 об/мин, при 37°C. Образцы отбирали с интервалом в 5 минут. В качестве эталонного продукта использовали Мавенклад 10 мг, испытуемый продукт представлял собой таблетки, полученные из аморфного ко-эвапората с использованием вспомогательных веществ, перечисленных в листке-вкладыше к оригинальному продукту (Мавенклад). На фиг. 7 приводится сравнение профилей растворения в буферах с рН 1,0 и 6,8, причем профиль растворения в буфере с рН 1,0 считается дискриминативным из-за высокой чувствительности кладрибина к низкому уровню рН. Как видно из фиг. 7, оба испытуемых состава демонстрируют соответствие параметров растворения в обоих используемых буферах для растворения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Фармацевтическая композиция, содержащая твердые дисперсии аморфного кладрибина и фармацевтически приемлемый водорастворимый носитель | 2020 |
|
RU2748311C1 |
| СОЛИ ДАСАТИНИБА В АМОРФНОЙ ФОРМЕ | 2014 |
|
RU2655435C2 |
| ТВЕРДЫЕ ФОРМЫ (1,1-ДИОКСО-4-ТИОМОРФОЛИНИЛ)-[6-[[3-(4-ФТОРФЕНИЛ)-5-МЕТИЛ-4-ИЗОКСАЗОЛИЛ]МЕТОКСИ]-3-ПИРИДИНИЛ]-МЕТАНОНА | 2012 |
|
RU2618524C2 |
| СОЛИ ДАСАТИНИБА В КРИСТАЛЛИЧЕСКОЙ ФОРМЕ | 2014 |
|
RU2662805C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2719450C1 |
| СОСТАВ НА ОСНОВЕ КОМПЛЕКСА АРИПИПРАЗОЛА | 2003 |
|
RU2342931C2 |
| АЭРОЗОЛЬНЫЕ ФТОРХИНОЛОНЫ И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2603638C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУСПЕНЗИИ, СОДЕРЖАЩЕЙ НАНОРАЗМЕРНЫЕ ЧАСТИЦЫ ЛЕКАРСТВА | 2020 |
|
RU2756757C1 |
| КОМПЛЕКСЫ ВКЛЮЧЕНИЯ ПЕРИНДОПРИЛА | 2005 |
|
RU2372353C2 |
| КОМПЛЕКС ВКЛЮЧЕНИЯ ДИСУЛЬФИРАМА С ЦИКЛОДЕКСТРИНОМ | 2015 |
|
RU2592625C1 |







Изобретение относится к способу получения аморфного ко-эвапората, содержащего (а) кладрибин, (b) циклодекстрин и (c) органический растворитель, причем указанный способ включает стадии: (i) растворения циклодекстрина в водно-спиртовом растворе, (ii) растворения кладрибина в водно-спиртовом растворе циклодекстрина, (iii) удаления растворителя с получением конечного аморфного соиспаряемого вещества, содержащего менее 2% остаточного растворителя. Технический результат - разработан новый способ получения аморфного комплекса кладрибина с циклодекстрином, который позволяет получить высокостабильный аморфный комплекс, который может найти применение в медицине для производства твердых лекарственных форм, содержащих кладрибин. 7 з.п. ф-лы, 7 ил., 10 пр.
1. Способ получения аморфного комплекса кладрибина с циклодекстрином, включающий:
(i) растворение циклодекстрина в водно-спиртовом растворе,
(ii) растворение кладрибина в водно-спиртовом растворе циклодекстрина,
(iii) полное или частичное удаление растворителя.
2. Способ по п. 1, отличающийся тем, что полученный аморфный комплекс содержит:
a) кладрибин,
b) циклодекстрин и
c) необязательно остаточный органический растворитель.
3. Способ по пп. 1 и 2, отличающийся тем, что полученный аморфный комплекс содержит:
a) 1-20 мас.%, предпочтительно 5-15 мас.% кладрибина,
b) 80-99 мас.%, предпочтительно 85-95 мас.% циклодекстрина и
c) 0-10 мас.%, предпочтительно 0,5-2 мас.% органических растворителей.
4. Способ по любому из пп. 1-3, отличающийся тем, что для приготовления водно-спиртового раствора используют спирт C2-C6.
5. Способ по п. 4, отличающийся тем, что в качестве спирта используют этаноловый или изопропиловый спирт.
6. Способ по любому из пп. 1-5, отличающийся тем, что на стадии (iii) растворитель удаляют при температуре 30-60°C и давлении 0,1-1000 мбар, предпочтительно 1-200 мбар, более предпочтительно 5-100 мбар, еще более предпочтительно 10-50 мбар, в частности 30-40 мбар.
7. Способ по любому из предшествующих пунктов, отличающийся тем, что циклодекстрин представляет собой гидроксипропилированный бета-циклодекстрин.
8. Способ по любому из пунктов, отличающийся тем, что полученный аморфный комплекс кладрибина с циклодекстрином используют для получения фармацевтических составов для перорального введения.
| US 8785415 B2, 22.07.2014 | |||
| US 7888328 B2, 15.02.2011 | |||
| US 6194395 B1, 27.02.2001 | |||
| Способ приготовления сварочного порошка для чугуна | 1927 |
|
SU9944A1 |
| Предохранительное устройство впереди повозки | 1928 |
|
SU9714A1 |
| RU 2005133196 A, 10.03.2006. | |||

