Область техники
[0001]
Настоящее изобретение относится к фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль и алкилсульфат натрия, особенно к фармацевтической композиции для перорального введения.
Уровень техники
[0002]
Биодоступность является индикатором, показывающим уровень введенного лекарства для достижения в крови, циркулирующей по телу, и для действия на него, и клинически важным параметром, который тесно связан с лечебными эффектами и токсичностью. В общем случае лекарство, имеющее низкую биодоступность, может не обеспечивать ожидаемые лечебные эффекты или из-за больших колебаний у одного отдельного человека или между отдельными людьми может быть трудно предсказать и/или проконтролировать лечебные эффекты и токсичность. Следовательно, при разработке лекарственных препаратов важно получить соответствующую биодоступность лекарства. В случае лекарства для перорального введения на лекарственное средство влияет коэффициент абсорбции из кишечного тракта и метаболизм в печени и/или кишечнике. В частности, в случае плохо растворимого в воде лекарственного средства становится важным улучшение растворения лекарства из рецептуры или растворимости лекарства в воде, чтобы получить соответствующую биодоступность.
[0003]
В качестве пути улучшения растворения или абсорбции лекарственного средства в целом известно уменьшение размера частиц или солюбилизация лекарственного вещества и способ смешения солюбилизатора, такого как поверхностно-активное вещество, с лекарственным веществом. Однако предпочтительные поверхностно-активные вещества различаются в зависимости от структуры и свойств активного ингредиента и типа препарата, и нелегко найти оптимальную композицию для плохо растворимого в воде лекарственного средства.
[0004]
Известно, что лаурилсульфат натрия, одно из анионных поверхностно-активных веществ, может быть включен в фармацевтический препарат, в качестве стабилизатора, поверхностно-активного вещества, смазывающего вещества, солюбилизатора, основы, связующего вещества, осветлителя, наполнителя, разрыхлителя, эмульгатора, пенообразователя, дисперсанта или т.п. Например, сообщалось, что лаурилсульфат натрия добавляют к гранулам, содержащим определенное соединение, в качестве солюбилизирующего агента для получения фармацевтического препарата (патентный документ 1).
[0005]
С другой стороны, в качестве соединения, имеющего прекрасное FGFR-ингибирующее действие и проявляющего противоопухолевую активность, предложен (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он (далее также называемый как «соединение A») (патентный документ 2-6).
Что касается соединения A, то не сообщалось ни об улучшении его растворимости, ни о комбинированном применении соединения A с алкилсульфатом натрия для других целей.
Список цитирования
Патентные документы
[0006]
Патентный документ 1: Патентная публикация Японии (Kokai) № 2016-104762 A;
Патентный документ 2: Международная публикация № WO2013/108809;
Патентный документ 3: Международная публикация № WO2015/008844;
Патентный документ 4: Международная публикация № WO2015/008839;
Патентный документ 5: Международная публикация № WO2016/159327;
Патентный документ 6: Международная публикация № WO2017/150725.
Сущность изобретения
Техническая задача
[0007]
Хотя соединение A имеет прекрасное FGFR-ингибирующее действие и противоопухолевую активность, при рецептурировании существует пространство для улучшения при обеспечении соответствующей биодоступности. Например, требуется улучшение растворения в интервале нейтральных значений pH и абсорбции соединения A. Таким образом, цель настоящего изобретения состоит в создании фармацевтической композиции, содержащей соединение A или его фармацевтически приемлемую соль, имеющей более прекрасные растворение, стабильность и абсорбцию, и при этом легко производимую.
Решение задачи
[0008]
Заявители настоящего изобретения добавляли различные соединения в композицию, содержащую соединение A или его фармацевтически приемлемую соль, и провели разнообразные исследования, касающиеся наличия или отсутствия эффекта улучшения растворения, стабильности и абсорбции соединения A. В результате установлено, что путем добавления алкилсульфата натрия к соединению A или к его фармацевтически приемлемой соли может быть получена фармацевтическая композиция, проявляющая превосходное растворение, стабильность и абсорбцию, и также имеющую прекрасную технологичность производства. Заявители также провели исследования с целью установления более эффективных наполнителей для использования в фармацевтической композиции, содержащей соединение А или его фармацевтически приемлемую соль и алкилсульфат натрия, тем самым достигнув реализации настоящего изобретения.
[0009]
Говоря конкретно, настоящее изобретение относится к следующим положениям [1]-[15].
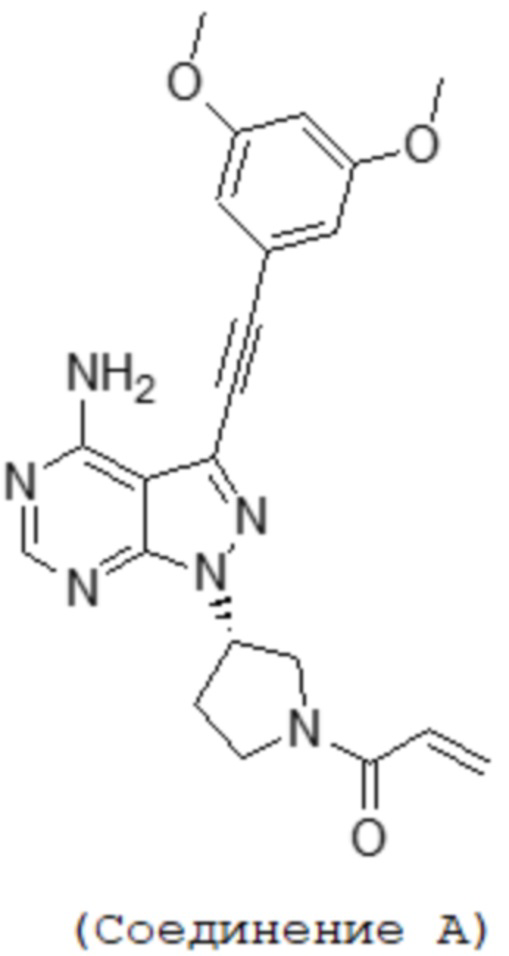
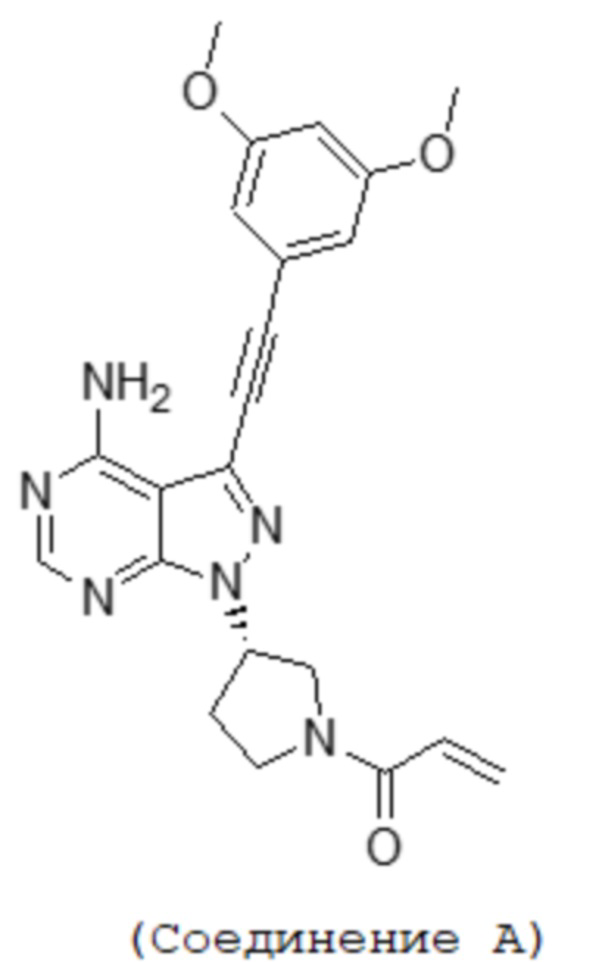
[1] Фармацевтическая композиция, содержащая (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он, имеющий приведенную ниже структуру, или его фармацевтически приемлемую соль и лаурилсульфат натрия:
[Формула 1]
[2] Композиция в соответствии с пунктом [1], содержащая лаурилсульфат натрия в интервале от 0,05 до 15 масс.ч. относительно 1 масс.ч. (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она.
[3] Композиция в соответствии с пунктом [1] или [2], содержащая лаурилсульфат натрия в интервале от 0,2 до 5 масс.ч. относительно 1 масс.ч. (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она.
[4] Композиция в соответствии с любым из пунктов [1]-[3], дополнительно содержащая, по меньшей мере, одно соединение, выбираемое из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция.
[5] Композиция в соответствии с пунктом [4], содержащая кросповидон.
[6] Композиция в соответствии с любым из пунктов [1]-[5], содержащая лаурилсульфат натрия в интервале от 0,2 до 5 масс.ч. и дополнительно содержащая кросповидон в интервале от 0,2 до 5 масс.ч. относительно 1 масс.ч. (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он.
[7] Композиция в соответствии с любым из пунктов [1]-[6], дополнительно содержащая, по меньшей мере, одно соединение, выбираемое из группы, включающей D-маннит и лактозу.
[8] Композиция в соответствии с любым из пунктов [1]-[7] в форме сиропа, порошка, гранулы, таблетки или капсулы.
[9] Способ улучшения растворения (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она из фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль, причем способ включает добавление лаурилсульфата натрия к фармацевтической композиции.
[10] Способ улучшения абсорбции (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она, причем способ включает добавление лаурилсульфата натрия к фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль.
[11] Способ улучшения технологичности производства фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль, причем способ включает добавление лаурилсульфата натрия к фармацевтической композиции.
[12] Применение лаурилсульфата натрия для улучшения растворения (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она или его фармацевтически приемлемой соли.
[13] Применение лаурилсульфата натрия для улучшения абсорбции (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она или его фармацевтически приемлемой соли.
[14] Применение лаурилсульфата натрия для улучшения технологичности производства фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль.
[15] Применение лаурилсульфата натрия для производства фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль.
Настоящее описание включат содержание, раскрытое в патентной публикации Японии № 2018-051620, от которой настоящая заявка испрашивает приоритет.
Положительные эффекты изобретения
[0010]
Настоящее изобретение может предложить фармацевтическую композицию, содержащую соединение A или его фармацевтически приемлемую соль и алкилсульфат натрия, которая имеет прекрасные растворение, стабильность и абсорбцию и также является превосходной с точки зрения технологичности производства, например, с точки зрения смазывающего свойства и текучести.
Краткое описание чертежей
[0011]
ФИГ. 1 показывает результаты оценки усилия выталкивания, требуемого, когда таблетки примера рецептуры 37 и сравнительного примера 20 выгружают из таблеточной машины.
Описание вариантов осуществления
[0012]
Фармацевтическая композиция по настоящему изобретению содержит в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль. Фармацевтическая композиция по настоящему изобретению также может содержать другие активные ингредиенты, пока она проявляет эффекты настоящего изобретения. Структура соединения A ((S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она) показана ниже.
[0013]
[Формула 2]
 .
.
[0014]
Соединение A или его фармацевтически приемлемая соль могут быть сольватированы (например, гидратированы) или не сольватированы. В настоящем изобретении как сольватированная, так и несольватированная формы включены в определение «соединение A или его фармацевтически приемлемая соль». Фармацевтически приемлемая соль соединения A не имеет особенных ограничений, и ее примеры могут включать: соли присоединения с органическими кислотами, такими как соляная кислота и серная кислота; соли присоединения с органическими кислотами, такими как уксусная кислота, лимонная кислота, винная кислота и малеиновая кислота; соли с щелочными металлами, такими как калий и натрий; соли с щелочноземельными металлами, такими как кальций и магний; и соли с органическими основаниями, такие как аммониевые соли, этиламинные соли и аргининовые соли. Соединение A или его фармацевтически приемлемая соль могут быть произведены, например, способом, описанным в патентном документе 2 или 5. В настоящем описании термин «соединение A», как подразумевается, включает фармацевтически приемлемую «соль» соединения A и описанный выше «сольват».
[0015]
С точки зрения растворения, стабильности, абсорбции, технологичности производства и др., количество соединения A или его фармацевтически приемлемой соли, используемое в настоящем изобретении, составляет предпочтительно от 1 до 50% масс., более предпочтительно от 2 до 30% масс. и также предпочтительно от 3 до 18% масс. из расчета на общее количество фармацевтической композиции.
[0016]
Для улучшения биодоступности соединения A после введения требуется улучшить, в частности, растворение из препарата in vivo и абсорбцию в организм соединения A.
Как описано выше, солюбилизатор, как правило, может быть использован в случае фармацевтической композиции, содержащей плохо растворимый в воде активный ингредиент. Примеры солюбилизаторов могут включать поверхностно-активное вещество, простое полиэфирное соединение и полоксамер. Примеры поверхностно-активных веществ могут включать алкилсульфат, эфир сахарозы и жирной кислоты (DK эфир и др.), полисольват (Tween 20, Tween 60, Tween 80 и др.) и полиоксиэтиленовое касторовое масло (Cremophor, Cremophor EL и др.). Примеры простых полиэфирных соединений включают полиэтиленгликоль (PEG400, PEG4000, PEG6000, Macrogol и др.). Полоксамером может быть, например, Lutrol (Lutrol F68 и др.).
[0017]
Когда алкилсульфат натрия используют в качестве солюбилизатора, растворимость соединения A может быть существенно повышена по сравнению с использованием других солюбилизаторов. Кроме того, использование алкилсульфата натрия может не только поддержать химическую стабильность соединения A и физическую стабильность дозированной лекарственной формы фармацевтической композиции, но также может повысить абсорбцию соединения A при пероральном введении.
[0018]
Алкилсульфат натрия может представлять собой, например, алкилсульфаты натрия, имеющие алкильную группу, содержащую от 10 до 18 атомов углерода. Его конкретные примеры могут включать децилсульфат натрия, лаурилсульфат натрия (также обозначаемый как «SLS» или «додецилсульфат натрия (SLS)»), тетрадецилсульфат натрия, цетилсульфат натрия (гексадецилсульфат натрия) и стеарилсульфат натрия (октадецилсульфат натрия). С точки зрения растворения, стабильности, адсорбции, технологичности производства и др. алкилсульфат натрия, используемый в настоящем изобретении, предпочтительно представляет собой лаурилсульфат натрия. В качестве лаурилсульфата натрия может быть приобретен и соответствующим образом использован NIKKOL SLS (производства компании Nikko Chemicals Co., Ltd.), Emal OS (Kao Corporation), или Kolliphor SLS (BASF Corporation).
[0019]
С упомянутой выше точки зрения алкилсульфат натрия может быть использован в количестве в интервале от 0,01 до 25 масс.ч. относительно 1 масс.ч. соединения A, предпочтительно от 0,05 до 15 масс.ч., более предпочтительно от 0,1 до 10 масс.ч., даже более предпочтительно от 0,2 до 5 масс.ч., также предпочтительно от 0,25 до 3 масс.ч., еще более предпочтительно от 0,75 до 1,5 масс.ч. и особенно предпочтительно 1 масс.ч. относительно 1 масс.ч. соединения A. Более того, алкилсульфат натрия предпочтительно используют в количестве от 1 до 50% масс. из расчета на общее количество фармацевтической композиции и более предпочтительно от 2 до 30% масс., даже более предпочтительно от 3 до 18% масс., также предпочтительно от 4 до 12% масс. и особенно предпочтительно от 4 до 5% масс., от 6 до 7% масс. или от 9 до 10% масс. из расчета на общее количество фармацевтической композиции.
[0020]
Термин «растворение», используемый в данном документе, означает растворение соединения A из композиции, содержащей соединение A (из фармацевтического препарата). Растворение может быть исследовано в соответствии с методом испытания на растворение (метод с использованием лопастной мешалки) (Japanese Pharmacopoeia, 16th Edition). Улучшение растворения может быть оценено по снижению времени распадаемости или по коэффициенту растворения при достижении равновесного состояния. За счет улучшения растворения соединения A из фармацевтической композиции лечебные эффекты соединения A в качестве активного ингредиента могут быть проявлены более подобающим образом.
Термин «стабильность», используемый в данном документе, включает как стабильность препарата, содержащего фармацевтическую композицию, так и химическую стабильность соединения A. Улучшение стабильности может быть оценено при сравнении состояния фармацевтического препарата до и после хранения фармацевтического препарата при одинаковых условиях, и также путем сравнения химической чистоты соединения A с использованием высокоэффективной жидкостной хроматографии или т.п. С учетом хранения и распространения фармацевтических продуктов улучшение стабильности всегда является чрезвычайно важной задачей для фармацевтических композиций.
[0021]
Термин «абсорбция», используемый в данном документе, означает абсорбцию соединения A в организм субъекта, которому было введено соединение A. Абсорбция может быть подтверждена с использованием площади под кривой (концентрация в крови)-время (AUC), максимальной концентрации в крови (Cмакс) и др. после того, как растворенное соединение A было абсорбировано в организм субъекта, как описано выше. Улучшение абсорбции может быть подтверждено на основании увеличения значения AUC или Cмакс. Более того, скорость абсорбции соединения A после введения фармацевтического препарата может быть оценена на основании времени достижения максимальной концентрации в крови (Tмакс). В результате улучшения абсорбции, оцениваемой с помощью этих параметров, предполагаемые эффекты соединения A могут быть провялены более выгодно, что в результате приводит к оптимизации схемы назначения.
[0022]
Кроме того, термин «технологичность производства», используемый в данном документе, означает свойство, допускающее простое производство фармацевтической композиции, содержащей активный ингредиент и алкилсульфат натрия, и он включает свойство, допускающее простое изготовление фармацевтической композиции, имеющей прекрасные смазывающее свойство или текучесть. В настоящем изобретении обнаружено, что ингредиентом, удовлетворяющим всем требованиям по растворению, абсорбции и технологичности производства, является алкилсульфат натрия, как описано выше.
[0023]
Термин «смазывающее свойство», используемый в данном документе, означает свойство, что порошки, такие как гранулированные продукты или гранулы, используемые при производстве таблетки, не прилипают к таблеточной машине или т.п. Смазывающее свойство может быть подтверждено отсутствием «прилипания», за счет которого фармацевтический препарат прикрепляется к поршню таблеточной машины, или отсутствием «связывания», за счет которого фармацевтический препарат прикрепляется к ее матрице. Смазывающее свойство также может быть подтверждено тем фактом, что усилие выталкивания, создаваемое при выталкивании таблетки, не растет. За счет улучшения смазывающего свойства при производстве фармацевтического препарата таблетки могут быть произведены без повреждения полученных таблеток или производственных машин, таких как таблеточная машина.
[0024]
Термин «текучесть», используемый в данном случае, означает легкое течение фармацевтической композиции перед гранулированием. Текучесть может быть оценена на основании угла естественного откоса или коэффициента прессуемости. На стадии гранулирования в псевдоожиженном слое трудно переводить в псевдоожиженное состояние порошки, имеющие очень низкую текучесть, и, следовательно, такие порошки может быть невозможно гранулировать. За счет улучшения текучести порошков может быть стимулировано гранулирование порошков, так что могут быть получены однородные гранулированные продукты.
[0025]
Наполнители, используемые в фармацевтической композиции по настоящему изобретению, особенно не ограничены описанными выше наполнителями, при условии, что их обычно используют для препаратов в фармацевтической области. Например, могут быть использованы глидант, наполнитель, связующее вещество, лубрикант, красящее вещество, разрыхлитель и т.п.
[0026]
Примеры глидантов могут включать диоксид кремния, силикат натрия, тальк и стеарат магния.
Примеры наполнителей могут включать лактозу (в том числе гидрат лактозы), кукурузный крахмал, микрокристаллическую целлюлозу и D-маннит.
[0027]
Примеры связующих веществ могут включать гидроксипропилцеллюлозу, гипромеллозу и поливиниловый спирт.
Примеры лубрикантов могут включать гидрогенизированное масло, эфир сахарозы и жирной кислоты, лаурилсульфат натрия, стеарат магния и стеариновую кислоту.
Примеры красящих веществ могут включать пищевой желтый пигмент № 5, пищевой голубой пигмент № 2, пищевой красочный пигмент, полуторный оксид железа, желтый полуторный оксид железа и оксид титана.
[0028]
Примеры покрывающих агентов могут включать гидроксипропил-метилцеллюлозу (гипромеллозу, TC-5, METOLOSE и др.) и полиэтиленгликоль (PEG400, PEG1500, PEG4000, PEG6000, Macrogol 400, Macrogol 1500, Macrogol 4000, Macrogol 6000 и др.).
[0029]
Примеры разрыхлителей могут включать низкозамещенную гидроксипропилцеллюлозу, кукурузный крахмал, частично прежелатинизированный крахмал, микрокристаллическую целлюлозу, кармеллозу натрия, кармеллозу кальция, D-маннит и кросповидон. Из них предпочтительны микрокристаллическая целлюлоза, D-маннит или кросповидон.
[0030]
В настоящем изобретении фармацевтическая композиция, содержащая A или его фармацевтически приемлемую соль и алкилсульфат натрия? может также содержать разрыхлитель.
Кросповидон (поперечно сшитый поливинилпирролидон (PVP)), используемый в качестве разрыхлителя, представляет собой коммерчески доступный наполнитель фармацевтического продукта. В настоящем изобретении количество кросповидона составляет от 1 до 20% масс. и предпочтительно от 2 до 15% масс. из расчета на количество всей фармацевтической композиции.
[0031]
Кроме того, кросповидон используют в количестве от 0,1 до 20 масс.ч., предпочтительно от 0,2 до 5 масс.ч., более предпочтительно от 0,2 до 3 масс.ч. и особенно предпочтительно от 0,9 до 1,1 масс.ч., от 1,4 до 1,6 масс.ч. или от 1,9 до 2,1 масс.ч. относительно 1 масс.ч. соединения A.
Более того, кросповидон используют в количестве от 0,1 до 20 масс.ч., предпочтительно от 0,2 до 5 масс.ч., более предпочтительно от 0,2 до 3 масс.ч. и особенно предпочтительно от 0,9 до 1,1 масс.ч., от 1,4 до 1,6 масс.ч. или от 1,9 до 2,1 масс.ч. относительно 1 масс.ч. алкилсульфата натрия.
[0032]
В настоящем изобретении фармацевтическая композиция, содержащая соединение A или его фармацевтически приемлемую соль и алкилсульфат натрия, может также содержать кармеллозу натрия в качестве разрыхлителя.
Кармеллоза натрия представляет собой наполнитель фармацевтического продукта. В настоящем изобретении количество кармеллозы натрия составляет 1 до 10% масс. из расчета на количество всей фармацевтической композиции.
[0033]
Кроме того, кармеллозу натрия используют в количестве от 0,1 до 5 масс.ч., предпочтительно от 0,2 до 2 масс.ч. и особенно предпочтительно от 0,2 до 0,4 масс.ч. или от 0,9 до 1,2 масс.ч. относительно 1 масс.ч. соединения A.
Более того, кармеллозу натрия используют в количестве от 0,1 до 5 масс.ч., более предпочтительно от 0,2 до 2 масс.ч. и особенно предпочтительно от 0,2 до 0,4 масс.ч. или от 0,9 до 1,2 масс.ч. относительно 1 масс.ч. алкилсульфата натрия.
[0034]
В настоящем изобретении фармацевтическая композиция, содержащая соединение A или его фармацевтически приемлемую соль и алкилсульфат натрия, может также содержать кармеллозу кальция в качестве разрыхлителя. В настоящем изобретении количество кармеллозы кальция составляет от 1 до 10% масс. из расчета на количество всей фармацевтической композиции.
[0035]
Кроме того, кармеллозу кальция используют в количестве от 0,1 до 5 масс.ч., предпочтительно от 0,2 до 2 масс.ч. и особенно предпочтительно от 0,2 до 0,4 масс.ч. или от 0,9 до 1,2 масс.ч. относительно 1 масс.ч. соединения A.
Более того, кармеллозу кальция используют в количестве от 0,1 до 5 масс.ч., более предпочтительно от 0,2 до 2 масс.ч. и особенно предпочтительно от 0,2 до 0,4 масс.ч. или от 0,9 до 1,2 масс.ч. относительно 1 масс.ч. алкилсульфата натрия.
[0036]
В настоящем изобретении фармацевтическая композиция, содержащая соединение A или его фармацевтически приемлемую соль и алкилсульфат натрия, может также содержать D-маннит в качестве разрыхлителя.
D-маннит, используемый в качестве разрыхлителя, известен как разрыхлитель, используемый в пероральном быстродействующем разрыхлителе. Количество D-маннита, которое может быть использовано в настоящем изобретении, составляет от 10 до 80% масс., предпочтительно от 15 до 70% масс. и более предпочтительно от 20 до 60% масс. из расчета на всю фармацевтическую композицию.
[0037]
Более того, количество D-маннита, которое может быть использовано в настоящем изобретении, составляет от 1 до 20 масс.ч., предпочтительно от 2 до 15 масс.ч., более предпочтительно от 2 до 12 масс.ч. и особенно предпочтительно от 2 до 4 масс.ч., от 6 до 8 масс.ч. или от 9 до 11 масс.ч., относительно 1 масс.ч. соединения A.
[0038]
В настоящем изобретении фармацевтическая композиция, содержащая соединение A или его фармацевтически приемлемую соль и алкилсульфат натрия, может также содержать лактозу в качестве наполнителя.
Количество лактозы, которое может быть использовано в настоящем изобретении, составляет от 1 до 80% масс., предпочтительно от 2 до 70% масс. и более предпочтительно от 3 до 60% масс. из расчета на всю фармацевтическую композицию.
[0039]
Более того, количество лактозы, которое может быть использовано в настоящем изобретении, составляет от 1 до 30 масс.ч., предпочтительно от 1 до 10 масс.ч., более предпочтительно от 1 до 5 масс.ч. и особенно предпочтительно от 1 до 2 масс.ч. или от 4 до 5 масс.ч. относительно 1 масс.ч. соединения A.
[0040]
Фармацевтическая композиция по настоящему изобретению может представлять собой, например, фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую алкилсульфат натрия. Фармацевтическая композиция по настоящему изобретению представляет собой предпочтительно фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую лаурилсульфат натрия.
[0041]
Фармацевтическая композиция по настоящему изобретению более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и содержащую от 0,05 до 15 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A.
Фармацевтическая композиция по настоящему изобретению даже более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и содержащую от 0,1 до 10 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A.
[0042]
Фармацевтическая композиция по настоящему изобретению также предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и содержащую от 0,2 до 5 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A.
Фармацевтическая композиция по настоящему изобретению также предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и содержащую от 0,25 до 3 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A
[0043]
Фармацевтическая композиция по настоящему изобретению еще более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,75 до 1,5 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A.
Фармацевтическая композиция по настоящему изобретению еще более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую 1 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A.
[0044]
Фармацевтическая композиция по настоящему изобретению особенно предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую 1 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A и дополнительно содержащую один наполнитель, выбираемый из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция.
[0045]
В другом варианте осуществления фармацевтическая композиция по настоящему изобретению предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция.
Фармацевтическая композиция по настоящему изобретению более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую от 0,1 до 20 масс.ч. одного наполнителя, выбираемого из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция, относительно 1 масс.ч. соединения A.
[0046]
Фармацевтическая композиция по настоящему изобретению даже более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую от 0,2 до 5 масс.ч. кросповидона относительно 1 масс.ч. соединения A.
Фармацевтическая композиция по настоящему изобретению также предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую от 0,2 до 3 масс.ч. кросповидона относительно 1 масс.ч. соединения A.
[0047]
Фармацевтическая композиция по настоящему изобретению особенно предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую от 0,9 до 1,1 масс.ч., от 1,4 до 1,6 масс.ч. или от 1,9 до 2,1 масс.ч. кросповидона, относительно 1 масс.ч. соединения A.
[0048]
В другом варианте осуществления фармацевтическая композиция по настоящему изобретению предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую кросповидон.
[0049]
Фармацевтическая композиция по настоящему изобретению более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую от 0,1 до 10 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A и дополнительно содержащую от 0,1 до 20 масс.ч. кросповидона относительно 1 масс.ч. соединения A.
[0050]
Фармацевтическая композиция по настоящему изобретению даже более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую от 0,25 до 3 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A и дополнительно содержащую от 0,2 до 5 масс.ч. кросповидона относительно 1 масс.ч. соединения A.
[0051]
Фармацевтическая композиция по настоящему изобретению также предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую от 0,75 до 1,5 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A и дополнительно содержащую от 0,2 до 3 масс.ч. кросповидона относительно 1 масс.ч. соединения A.
[0052]
Фармацевтическая композиция по настоящему изобретению особенно предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую 1 масс.ч. лаурилсульфата натрия относительно 1 масс.ч. соединения A и дополнительно содержащую от 0,9 до 1,1 масс.ч., от 1,4 до 1,6 масс.ч. или 1,9 до 2,1 масс.ч. кросповидона относительно 1 масс.ч. соединения A.
[0053]
В другом варианте осуществления фармацевтическая композиция по настоящему изобретению предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция.
Фармацевтическая композиция по настоящему изобретению более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция, и дополнительно содержащую D-маннит.
[0054]
Фармацевтическая композиция по настоящему изобретению даже более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция, и дополнительно содержащую от 1 до 20 масс.ч. D-маннита относительно 1 масс.ч. соединения A.
Фармацевтическая композиция по настоящему изобретению также предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция, и дополнительно содержащую от 2 до 12 масс.ч. D-маннита относительно 1 масс.ч. соединения A.
[0055]
Фармацевтическая композиция по настоящему изобретению особенно предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция, и дополнительно содержащую от 2 до 4 масс.ч., от 6 до 8 масс.ч. или от 9 до 11 масс.ч. D-маннита относительно 1 масс.ч. соединения A.
[0056]
В другом варианте осуществления фармацевтическая композиция по настоящему изобретению предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция, и дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей D-маннит и лактозу.
[0057]
Фармацевтическая композиция по настоящему изобретению более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция, дополнительно содержащую D-маннит и дополнительно содержащую от 1 до 30 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0058]
Фармацевтическая композиция по настоящему изобретению также предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция, дополнительно содержащую D-маннит и дополнительно содержащую от 1 до 10 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0059]
Фармацевтическая композиция по настоящему изобретению особенно предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция, дополнительно содержащую от 2 до 12 масс.ч. D-маннита относительно 1 масс.ч. соединения A и дополнительно содержащую от 1 до 5 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0060]
В другом варианте осуществления фармацевтическая композиция по настоящему изобретению предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую D-маннит.
[0061]
Фармацевтическая композиция по настоящему изобретению более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую от 1 до 20 масс.ч. D-маннита относительно 1 масс.ч. соединения A.
Фармацевтическая композиция по настоящему изобретению также предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую от 2 до 15 масс.ч. D-маннита относительно 1 масс.ч. соединения A.
[0062]
Фармацевтическая композиция по настоящему изобретению особенно предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую от 2 до 12 масс.ч. D-маннита относительно 1 масс.ч. соединения A и дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция.
[0063]
В другом варианте осуществления фармацевтическая композиция по настоящему изобретению предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую лактозу.
[0064]
Фармацевтическая композиция по настоящему изобретению более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую от 1 до 30 масс.ч. лактозы относительно 1 масс.ч. соединения A.
Фармацевтическая композиция по настоящему изобретению также предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия и дополнительно содержащую от 1 до 10 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0065]
Фармацевтическая композиция по настоящему изобретению особенно предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль, также содержащую лаурилсульфат натрия, дополнительно содержащую от 1 до 5 масс.ч. лактозы относительно 1 масс.ч. соединения A и дополнительно содержащую один или несколько наполнителей, выбираемых из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция.
[0066]
В другом варианте осуществления фармацевтическая композиция по настоящему изобретению предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую лаурилсульфат натрия, кросповидон, лактозу и D-маннит.
[0067]
Фармацевтическая композиция по настоящему изобретению более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,01 до 25 масс.ч. лаурилсульфата натрия, от 0,1 до 20 масс.ч. кросповидона, от 0,1 до 20 масс.ч. D-маннита и от 0,1 до 30 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0068]
Фармацевтическая композиция по настоящему изобретению даже более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,1 до 10 масс.ч. лаурилсульфата натрия, от 0,1 до 20 масс.ч. кросповидона, от 0,1 до 20 масс.ч. D-маннита и от 0,1 до 30 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0069]
Фармацевтическая композиция по настоящему изобретению также предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,2 до 5 масс.ч. лаурилсульфата натрия, от 0,1 до 20 масс.ч. кросповидона, от 0,1 до 20 масс.ч. D-маннита и от 0,1 до 30 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0070]
Фармацевтическая композиция по настоящему изобретению еще более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,25 до 3 масс.ч. лаурилсульфата натрия, от 0,1 до 20 масс.ч. кросповидона, от 0,1 до 20 масс.ч. D-маннита и от 0,1 до 30 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0071]
Фармацевтическая композиция по настоящему изобретению еще более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,75 до 1,5 масс.ч. лаурилсульфата натрия, от 0,1 до 20 масс.ч. кросповидона, от 0,1 до 20 масс.ч. D-маннита и от 0,1 до 30 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0072]
Фармацевтическая композиция по настоящему изобретению еще более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,75 до 1,5 масс.ч. лаурилсульфата натрия, от 0,2 до 5 масс.ч. кросповидона, от 0,1 до 20 масс.ч. D-маннита и от 0,1 до 30 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0073]
Фармацевтическая композиция по настоящему изобретению также более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,75 до 1,5 масс.ч. лаурилсульфата натрия, от 0,2 до 3 масс.ч. кросповидона, от 0,1 до 20 масс.ч. D-маннита и от 0,1 до 30 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0074]
Фармацевтическая композиция по настоящему изобретению также более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,75 до 1,5 масс.ч. лаурилсульфата натрия, от 0,2 до 3 масс.ч. кросповидона, от 2 до 15 масс.ч. D-маннита и от 1 до 10 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0075]
Фармацевтическая композиция по настоящему изобретению намного более предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 0,75 до 1,5 масс.ч. лаурилсульфата натрия, от 0,2 до 3 масс.ч. кросповидона, от 2 до 12 масс.ч. D-маннита и от 1 до 5 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0076]
Фармацевтическая композиция по настоящему изобретению особенно предпочтительно представляет собой фармацевтическую композицию, содержащую в качестве активного ингредиента соединение A или его фармацевтически приемлемую соль и также содержащую от 1 масс.ч. лаурилсульфата натрия, от 0,9 до 1,1 масс.ч., от 1,4 до 1,6 масс.ч. или от 1,9 до 2,1 масс.ч. кросповидона, от 2 до 4 масс.ч., от 6 до 8 масс.ч. или от 9 до 11 масс.ч. D-маннита и от 1 до 2 масс.ч. или от 4 до 5 масс.ч. лактозы относительно 1 масс.ч. соединения A.
[0077]
Для фармацевтической композиции по настоящему изобретению предпочтительным является обычный способ введения, такой как пероральное введение, трансдермальное введение, внутрибрюшинное введение или внутривенное введение. Среди них предпочтителен пероральный способ введения. Соответственно, в предпочтительном варианте осуществления фармацевтическая композиция по настоящему изобретению представляет собой фармацевтическую композицию для перорального введения, содержащую соединение A и алкилсульфат натрия.
[0078]
Примеры фармацевтических композиций для перорального введения могут включать, но без ограничения ими, сироп, порошок, гранулу, таблетку и капсулу.
Фармацевтическая композиция по настоящему изобретению может быть произведена известным способом производства фармацевтического препарата. Например, гранулированный материал может быть произведен способом гранулирования, способом гранулирования в псевдоожиженном слое, способом гранулирования с перемешиванием, способом ротационного гранулирования в псевдоожиженном слое, способом экструзионного гранулирования, способом распылительного гранулирования, способом гранулирования размолом и т.п.
[0079]
Когда фармацевтическую композицию по настоящему изобретению рецептурируют в таблетку, поверхность таблетки может быть покрыта, чтобы производить фармацевтическую композицию для перорального введения, которая стабильна и проста для приема. Покрытие включает пленочную оболочку и покрытие с сахарной оболочкой. Примеры покрывающего агента могут включать гипромеллозу, этилцеллюлозу, гидроксипропилцеллюлозу, поливиниловый спирт и белый сахар.
Кроме того, для получения фармацевтической композиции для перорального введения, которую легко принимать, различные типы вкусоароматических добавок, таких как апельсиновая и лимонная вкусоароматические добавки, могут быть использованы в качестве вкусовых агентов, и также l-ментол, камфора, мята и т.п. могут быть использованы в качестве корригирующих веществ для фармацевтической композиции по настоящему изобретению.
[0080]
Так как соединение A имеет прекрасную EGFR-ингибирующую активность, фармацевтическая композиция по настоящему изобретению может быть полезна в качестве противоопухолевого средства. Рак как мишень особо не ограничен, и примеры рака могут включать рак головы и шеи, рак желудочно-кишечного тракта [например, рак пищевода, рак желудка, стромальную опухоль желудочно-кишечного тракта, рак двенадцатиперстной кишки, рак печени, рак желчных путей (например, рак желчного пузыря и/или рак желчного протока и др.), рак поджелудочной железы, рак тонкого кишечника, рак толстой кишки (например, колоректальный рак, рак толстой кишки, рак прямой кишки и др.), и др.], рак легких, рак груди, рак яичников, рак матки (например, рак шейки матки, рак эндометрия и др.), рак почки, рак мочевого пузыря, рак простаты, уротелиальную карциному, саркому костей и мягких тканей, рак крови (например, В-клеточную лимфому, хронический лимфолейкоз, периферическую Т-клеточную лимфому, миелодиспластический синдром, острый миелолейкоз, острый лимфолейкоз и др.), множественную миелому, рак кожи и мезотелиому.
[0081]
Соответственно, настоящее изобретение предлагает фармацевтическую композицию для использования при лечении или предупреждении опухоли, выбираемой из рака головы и шеи, рака желудочно-кишечного тракта [например, рака пищевода, рака желудка, стромальной опухоли желудочно-кишечного тракта, рака двенадцатиперстной кишки, рака печени, рака желчных путей (например, рака желчного пузыря и/или рака желчных протоков и др.), рака поджелудочной железы, рака тонкого кишечника, рака толстой кишки (например, колоректального рака, рака толстой кишки, рака прямой кишки и др.) и др.], рака легких, рака груди, рака яичников, рака матки (например, рака шейки матки, рака эндометрия и др.), рака почки, рака мочевого пузыря, рака простаты, уротелиальной карциномы, саркомы костей и мягких тканей, рака крови (например, В-клеточной лимфомы, хронического лимфоцитарного лейкоза, периферической Т-клеточной лимфомы, миелодиспластического синдрома, острого миелогенного лейкоза, острого лимфолейкоза и др.), множественной миеломы, рака кожи и мезотелиомы.
[0082]
В другом аспекте настоящее изобретение предлагает способ улучшения растворения соединения A из фармацевтической композиции, содержащей соединение A или его фармацевтически приемлемую соль, который отличается тем, что он включает добавление лаурилсульфата натрия к фармацевтической композиции.
[0083]
В другом аспекте настоящее изобретение предлагает способ улучшения абсорбции соединения A, который отличается тем, что включает добавление лаурилсульфата натрия к фармацевтической композиции, содержащей соединение A или его фармацевтически приемлемую соль.
[0084]
В другом аспекте настоящее изобретение предлагает способ улучшения технологичности производства, который отличается тем, что включает добавление лаурилсульфата натрия к фармацевтической композиции, содержащей соединение A или его фармацевтически приемлемую соль.
[0085]
В другом аспекте настоящее изобретение предлагает применение лаурилсульфата натрия для улучшения растворения соединения A или его фармацевтически приемлемой соли.
[0086]
В другом аспекте настоящее изобретение предлагает применение лаурилсульфата натрия для улучшения абсорбции соединения A или его фармацевтически приемлемой соли.
[0087]
В другом аспекте настоящее изобретение предлагает применение лаурилсульфата натрия для улучшения технологичности производства фармацевтической композиции, содержащей соединение A или его фармацевтически приемлемую соль.
[0088]
В другом аспекте настоящее изобретение предлагает применение лаурилсульфата натрия для производства фармацевтической композиции, содержащей соединение A или его фармацевтически приемлемую соль.
Примеры
[0089]
Далее настоящее изобретение описано более конкретно с помощью примеров. Однако эти примеры не предназначены для ограничения объема настоящего изобретения. Настоящее изобретение в достаточной степени описано в примерах, но понятно, что различные измерения или модификации могут быть выполнены специалистом в данной области техники. Соответственно, такие изменения или модификации включены в настоящее изобретение, если они не отклоняются от объема настоящего изобретения.
Различные типы реагентов, используемых в примерах, являются коммерчески доступными продуктами, если не указано иное.
[0090]
Пример испытания 1: Испытание на растворимость
Как описано в примерах рецептуры 1 и 2 и в сравнительных примерах 1-15, испытуемые растворы, в которых (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он (соединение A) объединен с различными типами поверхностно-активных веществ, готовят следующим образом, и полученные испытуемые растворы используют в испытаниях на растворимость, как описано ниже.
[0091]
Пример рецептуры 1
Растворяют 0,05 г лаурилсульфата натрия (производства компании SERVA, высокой чистоты) в 50 мМ фосфатном буфере (50 мл) с pH 6,8 и затем суспендируют в растворе 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0092]
Пример рецептуры 2
Растворяют 0,5 г лаурилсульфата натрия в 50 мМ фосфатном буфере (50 мл) с pH 6,8 и затем суспендируют в растворе 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0093]
Сравнительный пример 1
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) суспендируют 25 мг соединения A и полученную суспензию нагревают при 37°C в течение 60 мин с получением испытуемого образца.
[0094]
Сравнительный пример 2
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,05 г моноэфира жирной кислоты и сахарозы (DK Ester SS, производства компании DKS Co. Ltd.) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0095]
Сравнительный пример 3
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,5 г моноэфира жирной кислоты и сахарозы (DK Ester SS) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0096]
Сравнительный пример 4
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,05 г PEG6000 (Macrogol 6000, производства компании NOF CORPORATION) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0097]
Сравнительный пример 5
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,5 г PEG6000 и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0098]
Сравнительный пример 6
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,05 г Poloxamer (Lutrol F68, производства компании BASF Corporation) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0099]
Сравнительный пример 7
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,5 г Poloxamer (Lutrol F68) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0100]
Сравнительный пример 8
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,05 г полиоксиэтилен-сорбитанмонолаурата (Tween 20, производства компании Tokyo Chemical Industry Co., Ltd.) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0101]
Сравнительный пример 9
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,5 г полиоксиэтилен-сорбитанмонолаурата (Tween 20) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0102]
Сравнительный пример 10
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,05 г полиоксиэтилен-сорбитанмоностеарата (Tween 60, производства компании Tokyo Chemical Industry Co., Ltd.) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0103]
Сравнительный пример 11
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,5 г полиоксиэтилен-сорбитанмоностеарата (Tween 60) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0104]
Сравнительный пример 12
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,05 г полиоксиэтилен-сорбитанмоноолеата (Tween 80, производства компании Tokyo Chemical Industry Co., Ltd.) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0105]
Сравнительный пример 13
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,5 г полиоксиэтилен-сорбитанмоноолеата (Tween 80) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0106]
Сравнительный пример 14
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,05 г полиоксиэтиленового касторового масла (Cremophor EL, производства компании Sigma-Aldrich) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0107]
Сравнительный пример 15
В растворе 2 (50 мл) с pH 6,8 для испытания на растворение (Japanese Pharmacopeia) растворяют 0,5 г полиоксиэтиленового касторового масла (Cremophor EL) и затем в растворе суспендируют 25 мг соединения A, после чего следует нагревание суспензии при 37°C в течение 60 мин с получением испытуемого образца.
[0108]
Описанные выше примеры рецептуры 1 и 2 и сравнительные примеры 1-15 оценивают с точки зрения растворимости с использованием высокоэффективной жидкостной хроматографии.
Прибор: LC-2010C (Shimadzu Corporation)
Длина волны измерения: 300 нм
Работу с устройствами, включая обработку данных, проводят в соответствии с методом и методиками, указанными для каждого устройства. Композиции примеров рецептуры 1 и 2 и сравнительных примеров 1-15 и результаты данного испытания представлены в таблицах 1 и 2.
[0109]
Таблица 1
Испытание на растворение
[0110]
Таблица 2
(Japanese Pharmacopeia)
Испытание на растворение
[0111]
Как показано в таблицах 1 и 2, при сравнении со сравнительным примером 1, в котором не используют поверхностно-активное вещество, хотя растворимость соединения A едва ли меняется при добавлении некоторых поверхностно-активных веществ, эффект улучшения растворимости выявлен при добавлении некоторых поверхностно-активных веществ, включая лаурилсульфат натрия. Среди других лаурилсульфат натрия и моноэфир жирной кислоты и сахарозы обеспечивают высокий эффект улучшения растворимости. В частности, лаурилсульфат натрия показывает высокую растворимость в 0,1% растворе (пример рецептуры 1), и установлено, что растворимость соединения A в 1,0% растворе (пример рецептуры 2) становится приблизительно в 500 раз выше, чем без добавления поверхностно-активного вещества (сравнительный пример 1).
[0112]
Пример испытания 2: Испытание на абсорбцию
С использованием лаурилсульфата натрия и моноэфира жирной кислоты и сахарозы, которые обеспечивают благоприятный эффект улучшения растворимости соединения A в примере испытания 1, испытание на абсорбцию проводят следующим образом.
[0113]
Пример рецептуры 3
В воде (40 мл) растворяют 2,4 г лаурилсульфата натрия (Wako Corporation, для биохимического применения) и затем в растворе суспендируют 0,8 г соединения A, получают суспензию соединения A.
[0114]
Сравнительный пример 16
Суспендируют 0,8 г соединения A в 0,5%-ном водном растворе гипромеллозы (40 мл), обычно используемом в испытании на абсорбцию для фармацевтических продуктов, получают суспензию соединения A.
[0115]
Сравнительный пример 17
В воде (40 мл) растворяют 2,4 г моноэфира жирной кислоты и сахарозы (DK Ester SS) и затем в растворе суспендируют 0,8 г соединения A, получают суспензию соединения A.
[0116]
Пример рецептуры 3 и сравнительные примеры 16 и 17 подвергают следующему испытанию на абсорбцию.
Условия абсорбционного опыта
Животные: Собаки породы бигль (KITAYAMA LABES CO., LTD., 3 самца)
Условия питания: Воздержание от приема пищи накануне в течение 20 час
Доза: 100 мг/тело
Введение образца: 0,82 г каждого из примера рецептуры 3 и сравнительных примеров 16 и 17
Способ введения: Пероральное введение с 50 мл воды с использование зонда.
[0117]
Предварительная подготовка: За тридцать минут до приема вводимого образца раствор сульфата атропина для внутривенной инъекции (10 мкг/0,1 мл/кг) и раствор пентагастрина для внутримышечной инъекции (10 мкг/0.1 мл/кг) вводят собакам внутримышечно или внутривенно, а затем раствор пентагастрина для внутримышечной инъекции (10 мкг/0,1 мл/кг) вводят собакам дважды внутримышечно с интервалом 45 мин.
Через тридцать минут, 1 час, 1,5 часа, 2 часа, 4 часа и 8 часов после перорального введения примера рецептуры и сравнительных примеров от каждого животного отбирают образцы крови, измеряют концентрацию в крови соединения A (с помощью жидкостной хроматографии/масс-спектрометрии) и рассчитывают значения AUC и Cмакс. Результаты представлены в таблице 3.
[0118]
Таблица 3
[0119]
Как показано в таблице 3, сравнительный пример 17, содержащий моноэфир жирной кислоты и сахарозы вместе с соединением A, проявляет сравнимую абсорбцию, как и сравнительный пример 16, который представляет собой суспензию одного соединения А. С другой стороны, пример рецептуры 3, содержащий лаурилсульфат натрия вместе с соединением A, показывает абсорбцию соединения A, которая намного выше, чем в сравнительном примере 17, содержащем такое же количество моноэфира жирной кислоты и сахарозы. Таким образом, становится ясно, что лаурилсульфат натрия может быть полезен для улучшения абсорбции соединения A.
[0120]
Пример испытания 3: Испытание на абсорбцию
Гранулы, содержащие соединение A и лаурилсульфат натрия, готовят следующим образом, и испытание на абсорбцию проводят так же, как в примере испытания 2.
[0121]
Пример рецептуры 4
В стеклянной колбе в течение 1 мин смешивают друг с другом 2 г соединения A, 0,5 г лаурилсульфата натрия, 9,5 г лактозы и 4 г кукурузного крахмала. Все количество полученной смеси просеивают через сито с отверстием 500 мкм и затем снова смешивают в стеклянной колбе в течение 1 мин. При перемешивании смеси с использованием пестика и ступки к смеси добавляют 3400 мкл 10%-ной низковязкой гидроксипропилцеллюлозы (HPC-SL). Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт сушат с использованием влагомера (AND, MX-50) при 70°C. Затем все количество полученного продукта дополнительно просеивают через сито с отверстием 1000 мкм, получают гранулы соединения A.
[0122]
Пример рецептуры 5
В стеклянной колбе в течение 1 мин смешивают друг с другом 2 г соединения A, 2 г лаурилсульфата натрия, 8,4 г лактозы и 3,6 г кукурузного крахмала. Все количество полученной смеси просеивают через сито с отверстием 500 мкм и затем смешивают в стеклянной колбе в течение 1 мин. При смешении смеси использованием пестика и ступки к смеси добавляют 3200 мкл 10%-ной низковязкой гидроксипропилцеллюлозы (HPC-SL). Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт сушат с использованием влагомера (AND, MX-50) при 70°C. Затем все количество полученного продукта дополнительно просеивают через сито с отверстием 1000 мкм, получают гранулы соединения A.
[0123]
Пример рецептуры 6
В стеклянной колбе в течение 1 мин смешивают друг с другом 1,4 г соединения A, 4,2 г лаурилсульфата натрия, 3,9 г лактозы и 1,9 г кукурузного крахмала. Все количество полученной смеси просеивают через сито с отверстием 500 мкм и затем смешивают в стеклянной колбе в течение 1 мин. Когда 6,4 г полученной смеси смешивают с использованием пестика и ступки, к смеси добавляют 1330 мкл 10%-ной низковязкой гидроксипропилцеллюлозы (HPC-SL). Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт сушат с использованием влагомера (AND, MX-50) при 70°C. Затем все количество полученного продукта дополнительно просеивают через сито с отверстием 1000 мкм, получают гранулы соединения A.
[0124]
Примеры рецептур 4-6 и сравнительного примера 16, используемые в примере испытания 2, подвергают следующему испытанию на абсорбцию.
Условия абсорбционного опыта
Животные: Собаки породы бигль (KITAYAMA LABES CO., LTD., 3 самца)
Условия питания: Воздержание от приема пищи накануне в течение 20 час
Доза: 100 мг/тело
Введение образца: 0,82 г каждого из примеров рецептуры 3-6 и сравнительного примера 16
Способ введения: Пероральное введение с 50 мл воды.
[0125]
Предварительная подготовка: За тридцать минут до приема вводимого образца раствор сульфата атропина для внутривенной инъекции (10 мкг/0,1 мл/кг) и раствор пентагастрина для внутримышечной инъекции (10 мкг/0.1 мл/кг) вводят собакам внутримышечно или внутривенно, а затем раствор пентагастрина для внутримышечной инъекции (10 мкг/0,1 мл/кг) вводят собакам дважды внутримышечно с интервалом 45 мин.
Таким же образом, как и в примере испытания 2, через 30 минут, 1 час, 1,5 часа, 2 часа, 4 часа и 8 часов после перорального введения примеров рецептуры и сравнительного примера от каждого животного отбирают образцы крови, измеряют концентрацию в крови соединения A (с помощью жидкостной хроматографии/масс-спектрометрии) и рассчитывают значения AUC и Cмакс. Результаты представлены в таблице 4.
[0126]
Таблица 4
[0127]
Как показано в таблице 4, установлено, что все типы гранул, содержащих лаурилсульфат натрия, добавленный в 0,25-кратном, эквивалентном и 3-кратном количестве от количества соединения A (примеры рецептуры 4, 5 и 6, соответственно), показывают более высокую абсорбцию, чем сравнительный пример 16, в котором суспендировано только соединение A. Хотя пример рецептуры 6 проявляет самое высокое значение Cмакс, его значение AUC находится на том же уровне, что и значение примера рецептуры 5. Следовательно, считают, что более высокая абсорбция могла быть получена путем добавления лаурилсульфата натрия в количестве, равном или большем, чем количество соединения A.
[0128]
Пример испытания 4: Оценка формуемости и распадаемости таблеток
Проведено исследование с целью улучшения распадаемости таблетки, содержащей соединение A, чтобы выбрать разрыхлитель. Используют пять типов кандидатов в наполнители: низкозамещенная гидроксипропилцеллюлоза (LH-21, производства компании Shin-Etsu Chemical Co., Ltd.), кросповидон (Kollidon CL-SF, BASF Corporation), кармеллоза натрия (KICCOLATE, Asahi Kasei Corporation), кармеллоза кальция (E.C.G-505, GOTOKU CHEMICAL COMPANY LTD.) и карбоксиметилкрахмал натрия (Glycolith, производства компании ROQUETTE), каждый добавляемый в количестве 3% или 10% относительно общей массы таблетки, содержащей соединение A. Затем с использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) готовят таблетки с композициями, показанными ниже в таблице 5. В этом время оценивают давление сжатия, необходимое для получения целевой твердости (65 Н), и распадаемость таблеток, с тем чтобы проверить разрыхлители. Распадаемость таблеток оценивают в соответствии с испытанием на распадаемость (Japanese Pharmacopoeia, 16th Edition) с использованием воды в качестве тестового растворителя.
[0129]
Пример рецептуры 7
В полиэтиленовом пакете в течение 1 мин смешивают друг с другом 120 г соединения A, 120 г лаурилсульфата натрия, 516 г лактозы и 276 г кукурузного крахмала. Все количество полученной смеси просеивают через сито с отверстием 500 мкм и полученный продукт снова смешивают в полиэтиленовом пакете в течение 5 мин. Помещают 340 г полученных смешанных порошков в гранулятор с псевдоожиженным слоем (Freund Corporation) и затем гранулируют, при этом на порошок распыляют 161 г 7,5%-ной низковязкой гидроксипропилцеллюлозы, получают гранулы.
Затем к полученному гранулированному материалу добавляют микрокристаллическую целлюлозу, кросповидон и стеарат магния, после чего смешивают в стеклянной колбе. Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт снова смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) получают таблетки соединения A при давлении сжатия, подходящем для получения целевой твердости (65 Н).
[0130]
Пример рецептуры 8
Микрокристаллическую целлюлозу, кросповидон и стеарат магния добавляют к гранулированному материалу, полученному таким же образом, как и в примере рецептуры 7, после чего смешивают в стеклянной колбе. Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт снова смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), получают таблетки соединения A при давлении сжатия, подходящем для получения целевой твердости (65 Н).
[0131]
Пример рецептуры 9
Микрокристаллическую целлюлозу, кармеллозу натрия и стеарат магния добавляют к гранулированному материалу, полученному таким же образом, как и в примере рецептуры 7, после чего смешивают в стеклянной колбе. Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт снова смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), получают таблетки соединения A при давлении сжатия, подходящем для получения целевой твердости (65 Н).
[0132]
Пример рецептуры 10
Микрокристаллическую целлюлозу, кармеллозу натрия и стеарат магния добавляют к гранулированному материалу, полученному таким же образом, как и в примере рецептуры 7, после чего смешивают в стеклянной колбе. Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт снова смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), получают таблетки соединения A при давлении сжатия, подходящем для получения целевой твердости (65 Н).
[0133]
Пример рецептуры 11
Микрокристаллическую целлюлозу, кармеллозу кальция и стеарат магния добавляют к гранулированному материалу, полученному таким же образом, как и в примере рецептуры 7, после чего смешивают в стеклянной колбе. Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт снова смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), получают таблетки соединения A при давлении сжатия, подходящем для получения целевой твердости (65 Н).
[0134]
Пример рецептуры 12
Микрокристаллическую целлюлозу, кармеллозу кальция и стеарат магния добавляют к гранулированному материалу, полученному таким же образом, как и в примере рецептуры 7, после чего смешивают в стеклянной колбе. Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт снова смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), получают таблетки соединения A при давлении сжатия, подходящем для получения целевой твердости (65 Н).
[0135]
Пример рецептуры 13
Микрокристаллическую целлюлозу, карбоксиметилкрахмал натрия и стеарат магния добавляют к гранулированному материалу, полученному таким же образом, как и в примере рецептуры 7, после чего смешивают в стеклянной колбе. Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт снова смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), получают таблетки соединения A при давлении сжатия, подходящем для получения целевой твердости (65 Н).
[0136]
Пример рецептуры 14
Микрокристаллическую целлюлозу, низкозамещенную гидрокси-пропилцеллюлозу и стеарат магния добавляют к гранулированному материалу, полученному таким же образом, как и в примере рецептуры 7, после чего смешивают в стеклянной колбе. Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт снова смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), получают таблетки соединения A при давлении сжатия, подходящем для получения целевой твердости (65 Н).
[0137]
Пример рецептуры 15
Микрокристаллическую целлюлозу и стеарат магния добавляют к гранулированному материалу, полученному таким же образом, как и в примере рецептуры 7, после чего смешивают в стеклянной колбе. Все количество полученной смеси просеивают через сито с отверстием 850 мкм и полученный продукт снова смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), получают таблетки соединения A при давлении сжатия, подходящем для получения целевой твердости (65 Н).
Результаты для примеров рецептур 7-15, относящихся к испытанию на распадаемость, представлены в таблице 5.
[0138]
Таблица 5
[0139]
Как показано в таблице 5, установлено, что с любым типом разрыхлителя таблетки, имеющие постоянную твердость, могут быть получены при давлении сжатия 13 кН или ниже, которое находится в интервале давления сопротивления поршня, и что время распадаемости находится в пределах 10 мин. В частности, когда используют кросповидон, кармеллозу натрия или кармеллозу кальция, таблетки с постоянной твердостью могут быть получены при давлении сжатия 10 кН или ниже, и время распадаемости находится в пределах 9 мин. Кроме того, кросповидон обеспечивает снижение времени распадаемости в сравнении с примером рецептуры 15, которое не сравнимо с любым из кандидатов в наполнители, и требуемое давление сжатия является низким, показывая эффект улучшения формуемости. С другой стороны, низкозамещенная гидроксипропилцеллюлоза и карбоксиметилкрахмал натрия не обеспечивают снижение времени распадаемости, и они требуют высокого давления сжатия, показывая тенденцию снижения формуемости. По результатам все типы разрыхлителей являются полезными для таблеток, но особенно подтверждено, что кросповидон, кармеллоза натрия и кармеллоза кальция являются исключительно полезными.
[0140]
Пример испытания 5: Испытание на формуемость и распадаемость таблеток
С использованием кросповидона и кармеллозы натрия, которые, как установлено в примере испытания 4, обеспечивают высокую формуемость и эффект улучшения распадаемости таблеток, содержащих соединение A и лаурилсульфат натрия, и с использованием других компонентов, каждый из которых включен при одинаковой молекулярной массе во все из примеров рецептуры, готовят таблетки следующим образом при давлении сжатия, приемлемом для получения целевой твердости таблетки (60 Н) с помощью ротационной таблеточной машины, а затем сравнивают. Результаты представлены в таблице 6.
[0141]
Пример рецептуры 16
В полиэтиленовом пакете в течение 1 мин смешивают друг с другом 200 г соединения A, 200 г лаурилсульфата натрия (NIKKOL SLS, производства компании Nikko Chemicals Co., Ltd.), 860 г лактозы и 460 г кукурузного крахмала. Все количество полученной смеси просеивают через сито с отверстием 500 мкм и полученный продукт затем снова смешивают в полиэтиленовом пакете в течение 1 мин. Полученные смешанные порошки помещают в гранулятор с псевдоожиженным слоем (Freund Corporation) и гранулируют, причем на порошок распыляют 800 г 7,5%-ной низковязкой гидроксипропилцеллюлозы, получают гранулированный материал. Все количество полученного гранулированного материала просеивают через сито с отверстием 850 мкм.
Затем 25 г микрокристаллической целлюлозы, 7,5 г кросповидона и 2,5 г стеарата магния добавляют к 222,5 г просеянного продукта гранулированного материала и смешивают друг с другом в полиэтиленовом пакете. После этого полученную смесь таблетируют с помощью таблеточной машины (KIKUSUI SEISAKUSHO LTD.) с получением таблеток.
[0142]
Пример рецептуры 17
К 222,5 г просеянного продукта гранулированного материала, полученного таким же образом, как и в примере рецептуры 16, добавляют 25 г микрокристаллической целлюлозы, 25 г кросповидона и 2,5 г стеарата магния и смешивают друг с другом в полиэтиленовом пакете. После этого полученную смесь таблетируют с помощью таблеточной машины (KIKUSUI SEISAKUSHO LTD.) с получением таблеток.
[0143]
Пример рецептуры 18
К 222,5 г просеянного продукта гранулированного материала, полученного таким же образом, как и в примере рецептуры 16, добавляют 25 г микрокристаллической целлюлозы, 7,5 г кармеллозы натрия и 2,5 г стеарата магния и смешивают друг с другом в полиэтиленовом пакете. После этого полученную смесь таблетируют в таблеточной машине (KIKUSUI SEISAKUSHO LTD.) с получением таблеток.
[0144]
Пример рецептуры 19
К 445 г просеянного продукта гранулированного материала, полученного таким же образом, как и в примере рецептуры 16, добавляют 50 г микрокристаллической целлюлозы и 5 г стеарата магния и смешивают друг с другом в полиэтиленовом пакете. После этого полученную смесь таблетируют в таблеточной машине (KIKUSUI SEISAKUSHO LTD.) с получением таблеток.
[0145]
Таблица 6
CEOLUS KG-802
[0146]
Как показано в таблице 6, установлено: что получена такая же тенденция, как и в примере испытания 4; что пример рецептуры 19, не содержащий ни кросповидон, ни кармеллозу натрия, имеет самое длительное время распадаемости, а также порядок времени распадаемости следующий - пример рецептуры 18 (6 масс.ч. кармеллозы натрия) > пример рецептуры 16 (6 масс.ч. кросповидона) > пример рецептуры 17 (20 масс.ч. кросповидона); и что порядок давления сжатия для получения таблеток следующий - пример рецептуры 18 > пример рецептуры 19 > пример рецептуры 16 > пример рецептуры 17. В соответствии с настоящим исследованием показано, что кросповидон является наиболее полезным для улучшения распадаемости и формуемости при сравнении с добавлением такого же количества кармеллозы натрия.
[0147]
Пример испытания 6: Испытание на абсорбцию
Готовят таблетки с пленочной оболочкой примера рецептуры 20 (содержащие 20 мг соединения A) и примера рецептуры 21 (содержащие 4 мг соединения A), в которых и те, и другие таблетки содержат соединение A и лаурилсульфат натрия, а содержание соединения A отличается друг от друга, и затем подвергают такому же испытанию на абсорбцию, как и в примере испытания 2.
[0148]
Пример рецептуры 20
Раствор для пленочной оболочки, содержащий 6,8 г покрывающего агента и 81,2 г очищенной воды, распыляют на 180 г таблеток, полученных в примере рецептуры 19, с использованием аппарата для нанесения оболочек (FREUND CORPORATION), так что получают таблетку с пленочной оболочкой примера рецептуры 20. В качестве покрывающего агента используют обычный покрывающий агент, содержащий гипромеллозу, полиэтиленгликоль, оксид титана и красящее вещество.
[0149]
Пример рецептуры 21
В полиэтиленовом пакете в течение 1 мин смешивают друг с другом 12 г соединения A, 12 г лаурилсульфата натрия (NIKKOL SLS, производства компании Nikko Chemicals Co., Ltd.), 354 г лактозы и 138 г кукурузного крахмала. Все количество полученной смеси просеивают через сито с отверстием 500 мкм и полученный продукт затем снова смешивают в полиэтиленовом пакете в течение 1 мин. Полученные смешанные порошки помещают в гранулятор с псевдоожиженным слоем (Powrex Corporation) и гранулируют, при этом на порошок распыляют 239 г 7,5%-ной низковязкой гидроксипропилцеллюлозы, получают гранулированный материал. Все количество полученных гранул просеивают через сито с отверстием 850 мкм.
После этого 20 г микрокристаллической целлюлозы и 2 г стеарата магния добавляют к 178 г просеянных гранул, затем смешивают в полиэтиленовом пакете. Полученную смесь таблетируют в таблеточной машине (KIKUSUI SEISAKUSHO LTD.) с получением таблеток. Раствор для пленочной оболочки, содержащий 6,8 г покрывающего агента и 81,2 г очищенной воды, распыляют на 180 г полученных таблеток с использованием аппарата для нанесения оболочек (FREUND CORPORATION), получают таблетку с пленочной оболочкой примера рецептуры 21.
[0150]
Эти примеры рецептуры подвергают испытанию на абсорбцию следующим образом.
Условия абсорбционного опыта
Животные: Собаки породы бигль (KITAYAMA LABES CO., LTD., 6 самцов)
Условия питания: Воздержание от приема пищи накануне в течение 20 час
Доза: 20 мг/тело
Введение образца: Примеры рецептуры 20 и 21
Способ введения: Пероральное введение с 50 мл воды.
[0151]
Предварительная подготовка: За тридцать минут до приема вводимого образца раствор сульфата атропина для внутривенной инъекции (10 мкг/0,1 мл/кг) и раствор пентагастрина для внутримышечной инъекции (10 мкг/0,1 мл/кг) вводят собакам внутримышечно или внутривенно, а затем раствор пентагастрина для внутримышечной инъекции (10 мкг/0,1 мл/кг) вводят собакам дважды внутримышечно с интервалом 45 мин.
Через тридцать минут, 1 час, 1,5 часа, 2 часа, 4 часа и 8 часов после перорального введения примеров рецептуры 20 и 21 от каждого животного отбирают образцы крови, измеряют концентрацию в крови соединения A (с помощью жидкостной хроматографии/масс-спектрометрии) и рассчитывают значения AUC, Cмакс и Тмакс. Результаты представлены в таблице 7.
[0152]
Таблица 7
[0153]
По результатам таблицы 7 установлено, что как 20 мг таблетка (пример рецептуры 20), так и 4 мг таблетка (пример рецептуры 21) в собаках показывают абсорбцию при отсутствии проблем. Сравнение свойств двух типов таблеток указывает на то, что 20 мг таблетка представляет собой рецептуру с медленным Tмакс, тогда как 4 мг таблетка является рецептурой, которая относительно мгновенно абсорбируется, хотя ее значения Tмакс меняются.
[0154]
Пример испытания 7: Испытание на распадаемость
Чтобы найти фармацевтический препарат, имеющий небольшое колебание значений Tмакс и мгновенное поглощение, таблетки примеров рецептуры 22-32, композиции которых представлены в таблице 8, готовят следующим образом и оценивают типы и количества дополнительных добавок, которые должны быть добавлены к гранулированным продуктам, содержащим соединение A и лаурилсульфат натрия. Распадаемость оценивают в соответствии с испытанием на распадаемость с использованием воды в качестве тестового растворителя. Результаты представлены в таблице 8.
[0155]
Пример рецептуры 22
В полиэтиленовом пакете в течение 1 мин смешивают друг с другом 60 г соединения A, 60 г лаурилсульфата натрия (NIKKOL SLS, производства компании Nikko Chemicals Co., Ltd.), 258 г лактозы и 138 г кукурузного крахмала. Все количество полученной смеси просеивают через сито с отверстием 500 мкм и полученный продукт смешивают снова в полиэтиленовом пакете в течение 1 мин. Полученные смешанные порошки помещают в гранулятор с псевдоожиженным слоем (Powrex Corporation) и затем гранулируют, при этом на порошки распыляют 241 г 7,5%-ной низковязкой гидроксипропилцеллюлозы, получают гранулы. Все количество полученных гранул просеивают через сито с отверстием 850 мкм.
После этого гидрат лактозы (Super Tab 11SD, DFE Pharma), микрокристаллическую целлюлозу (CEOLUS pH-102, Asahi Kasei Corporation), кросповидон (Kollidon CL, BASF Corporation) и стеарат магния добавляют к полученным гранулам и полученную смесь смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) производят 500 г таблеток.
[0156]
Пример рецептуры 23
D-Маннит (Pearlitol 100SD, производства компании Roquette), микрокристаллическую целлюлозу (CEOLUS pH-102), кросповидон (Kollidon CL) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) производят 500 г таблеток.
[0157]
Пример рецептуры 24
Микрокристаллическую целлюлозу (CEOLUS pH-102), кросповидон (Kollidon CL) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) производят 300 г таблеток.
[0158]
Пример рецептуры 25
D-Маннит (Pearlitol 100SD), микрокристаллическую целлюлозу (CEOLUS KG-802, производства компании Asahi Kasei Corporation), кросповидон (Kollidon CL) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) производят 500 г таблеток.
[0159]
Пример рецептуры 26
D-Маннит (Pearlitol 100SD), микрокристаллическую целлюлозу (CEOLUS KG-802), кросповидон (Kollidon CL-SF) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) производят 300 г таблеток.
[0160]
Пример рецептуры 27
D-Маннит (Pearlitol 100SD), микрокристаллическую целлюлозу (CEOLUS KG-802), кросповидон (Kollidon CL-SF) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) производят 300 г таблеток.
[0161]
Пример рецептуры 28
D-Маннит (Pearlitol 100SD), микрокристаллическую целлюлозу (CEOLUS KG-802), кросповидон (Kollidon CL-SF) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) производят 400 г таблеток.
[0162]
Пример рецептуры 29
D-Маннит (Pearlitol 100SD), микрокристаллическую целлюлозу (CEOLUS KG-802), кросповидон (Kollidon CL-SF) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) производят 400 г таблеток.
[0163]
Пример рецептуры 30
D-Маннит (Pearlitol 100SD), микрокристаллическую целлюлозу (CEOLUS KG-802), кросповидон (Kollidon CL-SF) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation) производят 500 г таблеток.
[0164]
Пример рецептуры 31
D-Маннит (Pearlitol 100SD), микрокристаллическую целлюлозу (CEOLUS KG-802) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), производят 500 г таблеток.
[0165]
Пример рецептуры 32
Маннит (Pearlitol 100SD), микрокристаллическую целлюлозу (CEOLUS pH-102) и стеарат магния добавляют к гранулам, полученным таким же образом, как и в примере рецептуры 22, и полученную смесь затем смешивают в стеклянной колбе, получают смесь для прессования. С использованием универсальной машины для испытаний на растяжение/сжатие (Shimadzu Corporation), производят 500 г таблеток.
[0166]
Таблица 8
[0167]
Вначале оценивают влияние добавления моногидрата лактозы и D-маннита. В итоге, как показывают результаты примеров рецептуры 22, 23 и др., распадаемость таблеток в воде значительно улучшается за счет добавления D-маннита.
Затем оценивают сорт микрокристаллической целлюлозы и влияние добавления кросповидона. В итоге, как показывают результаты примеров рецептуры 23 и 25, примеры рецептуры 31 и 32 и др., сорт микрокристаллической целлюлозы едва ли влияет на распадаемость таблеток. С другой стороны, результаты примеров рецептуры 25, 30, 31 и др., показывают, что добавление кросповидона вносит большой вклад в улучшение свойства распадаемости.
[0168]
И, наконец, всесторонне оценивают влияние вспомогательных количеств D-маннита, микрокристаллической целлюлозы и кросповидона. Как результат, время распадаемости всех из примеров рецептуры 26-30 было более длительным, чем для примера рецептуры 25. Распадаемость примера рецептуры 27 (сверх добавленное количество: 122 мг, кросповидон: 10%), примера рецептуры 29 (сверх добавленное количество: 222 мг, кросповидон: 10%) и примера рецептуры 30 (сверх добавленное количество: 322 мг, кросповидон: 5%) хуже, чем распадаемость примера рецептуры 25 (сверх добавленное количество: 322 мг, кросповидон: 10%). Таким образом, это подтверждает, что общее количество дополнительных добавок, необходимое для приготовления таблетки, имеющей быструю распадаемость, составляет приблизительно 1,5-кратное количество гранулированного продукта, а также, что разрыхлитель (кросповидон) необходимо добавлять в количестве приблизительно 10% из расчета на массу таблетки.
[0169]
Пример испытания 8
Таблетки примеров рецептуры 33 и 34, каждая содержащая по 20 мг соединения A, в которых количество дополнительной добавки отличается друг от друга, и таблетку примера рецептуры 35, содержащую 4 мг соединения A, готовят следующим образом, и оценивают на поглощение in vivo аналогично примеру испытания 2. Результаты представлены в таблице 9.
[0170]
Пример рецептуры 33
В полиэтиленовом пакете в течение 1 мин смешивают друг с другом 80 г соединения A, 80 г лаурилсульфата натрия (NIKKOL SLS, производства компании Nikko Chemicals Co., Ltd.), 108 г лактозы и 120 г кукурузного крахмала. Все количество полученной смеси просеивают через сито с отверстием 500 мкм и полученный продукт смешивают снова в полиэтиленовом пакете в течение 1 мин. Полученную смесь помещают в гранулятор с псевдоожиженным слоем (Powrex Corporation) и затем гранулируют, при этом на смесь распыляют 241 г 7,5%-ной гипромеллозы, получают гранулы. Все количество полученных гранул просеивают через сито с отверстием 850 мкм.
После этого 137 г D-маннита (Pealitol 100SD), 30 г микрокристаллической целлюлозы (CEOLUS KG-802), 30 г кросповидона (Kollidon CL) и 3 г стеарата магния добавляют к 100 г просеянных гранул, затем смешивают в полиэтиленовом пакете. Полученную смесь таблетируют в таблеточной машине (KIKUSUI SEISAKUSHO LTD.) с получением таблеток. Раствор для пленочной оболочки, содержащий 7,7 г покрывающего агента и 92,3 г очищенной воды, распыляют на 180 г полученных таблеток с использованием аппарата для нанесения оболочек (FREUND CORPORATION), получают таблетку с пленочной оболочкой примера рецептуры 33. В качестве покрывающего агента используют обычный покрывающий агент, содержащий низковязкую гидроксипропил-целлюлозу, полиэтиленгликоль, оксид титана и красящее вещество.
[0171]
Пример рецептуры 34
К 125 г просеянного продукта гранул, полученных таким же образом, как и в примере рецептуры 33, добавляют 72,5 г D-маннита (Pealitol 100SD), 25 г микрокристаллической целлюлозы (CEOLUS KG-802), 25 г кросповидона (Kollidon CL) и 2,5 г стеарата магния и смешивают друг с другом в полиэтиленовом пакете. После этого полученную смесь таблетируют в таблеточной машине (KIKUSUI SEISAKUSHO LTD.) с получением таблеток. Раствор для пленочной оболочки, содержащий 7,2 г покрывающего агента и 86,4 г очищенной воды, распыляют на 180 г полученных таблеток с использованием аппарата для нанесения оболочек (FREUND CORPORATION), получают таблетку с пленочной оболочкой примера рецептуры 34.
[0172]
Пример рецептуры 35
К 60 г просеянного продукта гранул, полученных таким же образом, как и в примере рецептуры 33, добавляют 129,6 г D-маннита (Pealitol 100SD), 24 г микрокристаллической целлюлозы (CEOLUS KG-802), 24 г кросповидона (Kollidon CL) и 2,4 г стеарата магния и смешивают друг с другом в полиэтиленовом пакете. После этого полученную смесь таблетируют в таблеточной машине (KIKUSUI SEISAKUSHO LTD.) с получением таблеток. Раствор для пленочной оболочки, содержащий 7,5 г покрывающего агента и 90,4 г очищенной воды, распыляют на 180 г полученных таблеток с использованием аппарата для нанесения оболочек (FREUND CORPORATION), получают таблетку с пленочной оболочкой примера рецептуры 35.
[0173]
Эти примеры рецептуры подвергают испытанию на абсорбцию следующим образом.
Условия абсорбционного опыта
Животные: Собаки породы бигль (KITAYAMA LABES CO., LTD., 6 самцов)
Условия питания: Воздержание от приема пищи накануне в течение 20 час
Доза: 20 мг/тело
Введение образца: Примеры рецептуры 33, 34 и 35
Способ введения: Пероральное введение с 50 мл воды.
[0174]
Предварительная подготовка: За тридцать минут до приема образцов раствор сульфата атропина для внутривенной инъекции (10 мкг/0,1 мл/кг) и раствор пентагастрина для внутримышечной инъекции (10 мкг/0,1 мл/кг) вводят собакам внутримышечно или внутривенно, а затем раствор пентагастрина для внутримышечной инъекции (10 мкг/0,1 мл/кг) вводят собакам дважды внутримышечно с интервалом 45 мин.
Через тридцать минут, 1 час, 2 часа, 3 часа, 4 часа, 6 часов и 8 часов после перорального введения примеров рецептуры 33, 34 и 35 от каждого животного отбирают образцы крови, измеряют концентрацию в крови соединения A (с помощью жидкостной хроматографии/масс-спектрометрии) и рассчитывают значения AUC, Cмакс и Тмакс. Результаты представлены в таблице 9.
[0175]
Таблица 9
[0176]
Как показано в таблице 9, значение Tмакс имеет тенденцию сокращаться во всех примерах рецептуры, и какие-либо различия в фармакокинетическом профиле не обнаружены. Следовательно, это показывает, что все примеры рецептуры дают композиции препарата, имеющие небольшое колебание в значениях Tмакс и мгновенную абсорбцию.
[0177]
Пример испытания 9
Гранулы, не содержащие алкилсульфат натрия, готовят следующим образом и оценивают влияние алкилсульфата натрия на процесс производства гранулированного материала. Результаты представлены в таблице 10.
[0178]
Пример рецептуры 36
Через сито с отверстием 1700 мкм просеивают 60 г соединения A, 60 г лаурилсульфата натрия, 81 г лактозы и 90 г кукурузного крахмала и помещают в гранулятор с псевдоожиженным слоем (Freund Corporation). Затем смесь гранулируют, при этом на смесь распыляют 180 г 5%-ной низковязкой гидроксипропилцеллюлозы, получают гранулы.
[0179]
Сравнительный пример 18
Через сито с отверстием 1700 мкм просеивают 60 г соединения A, 141 г лактозы и 90 г кукурузного крахмала и помещают в гранулятор с псевдоожиженным слоем (Freund Corporation). После этого, как и следует, проводят стадию гранулирования при распылении 5%-ной низковязкой гидроксипропилцеллюлозы. Однако порошки не были текучими в контейнере и не могли быть подвергнуты гранулированию.
[0180]
Сравнительный пример 19
Через сито с отверстием 1700 мкм просеивают 30 г соединения A, 70,5 г лактозы и 45 г кукурузного крахмала и помещают в гранулятор с псевдоожиженным слоем (Freund Corporation). Перед распылением связующей жидкости распыляют воду для флюидизации смеси. Затем смесь гранулируют, при этом распыляя 90 г 5%-ной низковязкой гидроксипропилцеллюлозы, получают гранулированный материал.
[0181]
Оценка физических свойств порошков
Смешанные порошки перед гранулированием примера рецептуры 36 и сравнительного примера 19 оценивают с использованием прибора для испытания порошков (HOSOKAWA MICRON CORPORATION) относительно физических свойств порошков (угол естественного откоса, угол обрушения, насыпная плотность, насыпная плотность после уплотнения и коэффициент прессуемости). Результаты представлены в таблице 10.
[0182]
Таблица 10
[0183]
Что касается физических свойств порошков, то испытание, проведенное в соответствии с международными требованиями по гармонизации, описано в публикации «26. Fluidity of Powders» (Japanese Pharmacopoeia, 17th Edition). В этой публикации коэффициент прессуемости, насыпную плотность (ρ насыпная плотность) и насыпную плотность после уплотнения (ρ насыпная плотность после уплотнения) определяют следующим образом:
Коэффициент прессуемости = [(ρ после уплотнения - ρ насыпная)]/[ρ после уплотнения] × 100.
[0184]
Более того, в публикации Japanese Pharmacopoeia, 17th Edition, если ссылаться на «Таблица 1. Текучие свойства и соответствующие углы естественного откоса», то в ней описано, что «когда угол естественного откоса превышает 50°, сыпучесть редко является приемлемой для производственных целей». Исходя из этого описания, когда пример рецептуры 36 сравнивают со сравнительным примером 19, то угол естественного откоса составляет меньше чем 50° и улучшен из-за присутствия лаурилсульфата натрия. Следовательно, установлено, что в настоящем изобретении добавление алкилсульфата натрия, включая лаурилсульфат натрия, обеспечивает эффект глиданта (скользящего вещества).
[0185]
Пример испытания 10
Гранулированный материал, не содержащий алкилсульфат натрия, таблетируют с получением таблеток следующим образом и оценивают влияние алкилсульфата натрия на процесс производства таблеток.
[0186]
Пример рецептуры 37
Все количество гранул, полученных в примере рецептуры 36, просеивают через сито с отверстием 600 мкм. Добавляют 261,61 г D-маннита (Pearlitol 100SD), 48 г микрокристаллической целлюлозы (CEOLUS KG-802), 48 г кросповидона (Kollidon CL) и 2,4 г стеарата магния к 120 г просеянных гранул и смесь смешивают в полиэтиленовом пакете. После этого полученную смесь таблетируют в таблеточной машине (KIKUSUI SEISAKUSHO LTD.) с получением таблеток.
[0187]
Сравнительный пример 20
Используя гранулы, полученные в сравнительном примере 19, получают таблетки таким же образом, как и в примере рецептуры 37.
[0188]
Оценка усилия выталкивания после таблетирования
Таблетки примера рецептуры 37 и сравнительного примера 20 оценивают с точки зрения усилия, требуемого для выталкивания таблеток из таблеточной машины. Результаты представлены на ФИГ. 1.
[0189]
При проведении таблетирования в течение 60 мин усилие выталкивания для примера рецептуры 37 было постоянным, но усилие выталкивания для сравнительного примера 20 выросло через 5 мин после начала таблетирования, дополнительно росло со временем до 30 мин после начала таблетирования, а затем все еще было необходимо высокое усилие выталкивания.
[0190]
Таблетки примера рецептуры 37 и сравнительного примера 20 также сравнивают друг с другом с точки зрения условий для матрицы и пуансона через 30 мин после начала таблетирования. В обеих таблетках порошки не прилипают к пуансону и никакого залипания не подтверждено. Однако в сравнительном примере 20 наблюдается адгезия порошков к матрице. То есть, подтверждено много вертикальных штрихов на боковой поверхности полученных таблеток, и имеет место трение матрицы, как один из недостатков таблетирования, в таблетках сравнительного примера 20, которые не содержат лаурилсульфат натрия. Напротив, такое трение матрицы не наблюдается в примере рецептуры 37.
[0191]
На основании этих результатов установлено, что в настоящем изобретении добавление алкилсульфата натрия, в том числе лаурилсульфата натрия, обеспечивает эффект улучшения трения матрицы, как одного из недостатков при таблетировании.
Все публикации, патенты и патентные заявки, цитируемые в настоящем описании, включены в него посредством ссылки во всей их полноте.
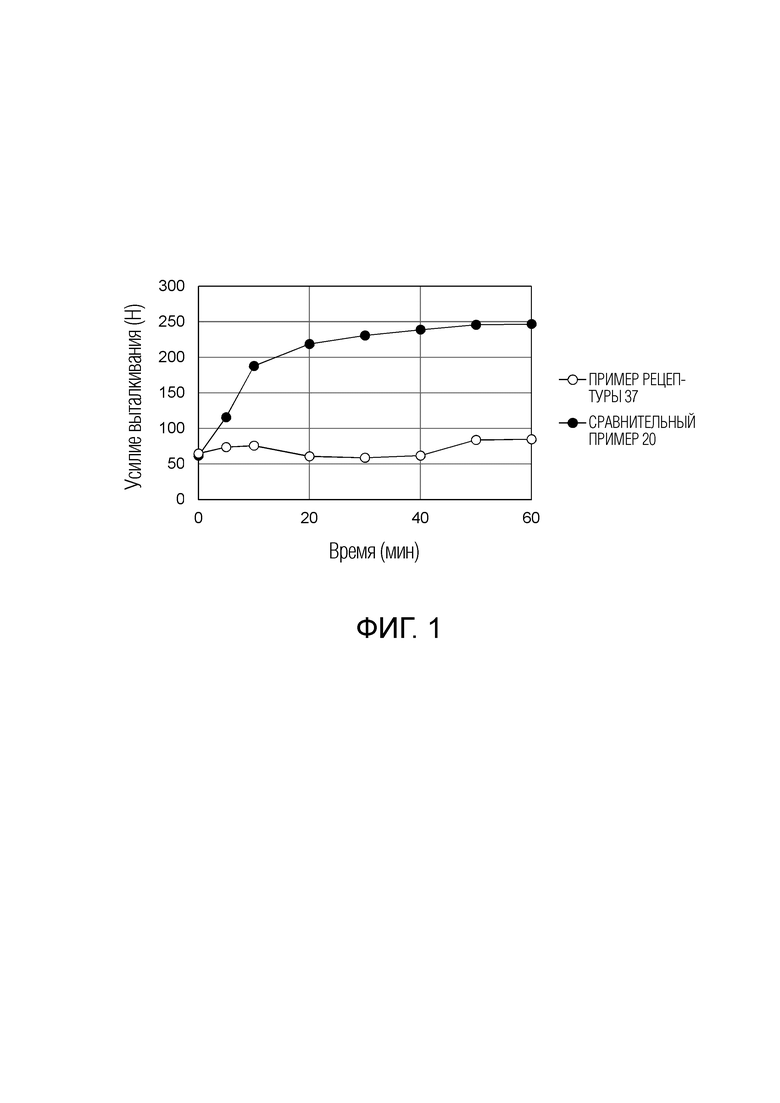
Группа изобретений относится к области химии и фармацевтики. 1 объект представляет собой фармацевтическую композицию, содержащую (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль и лаурилсульфат натрия. 2-4 объекты – способы улучшения растворения и абсорбции (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она и улучшения технологичности производства фармацевтической композиции, включающие добавление лаурилсульфата натрия к фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль. 5-8 объекты – применение лаурилсульфата натрия для улучшения растворения и абсорбции (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она или его фармацевтически приемлемой соли, для производства и улучшения технологичности производства фармацевтической композиции. Технический результат заключается в улучшении растворения, стабильности, абсорбции и производства фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль, за счет добавления лаурилсульфата натрия. 8 н. и 6 з.п. ф-лы, 1 ил., 10 табл., 10 пр.
1. Фармацевтическая композиция, содержащая (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он, имеющий приведенную ниже структуру, или его фармацевтически приемлемую соль, и лаурилсульфат натрия:
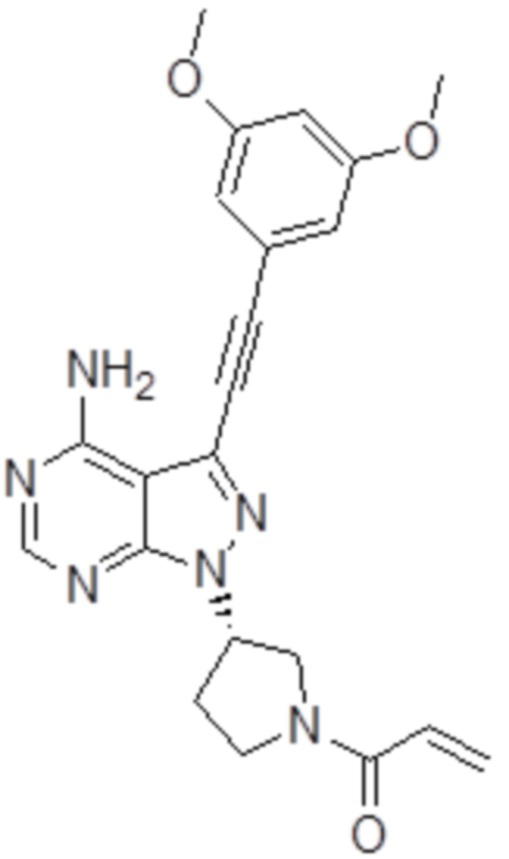
2. Композиция по п. 1, содержащая лаурилсульфат натрия в интервале от 0,05 до 15 масс.ч. относительно 1 масс.ч. (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]-пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она.
3. Композиция по п. 1 или 2, содержащая лаурилсульфат натрия в интервале от 0,2 до 5 масс.ч. относительно 1 масс.ч. (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она.
4. Композиция по п. 1 или 2, дополнительно содержащая, по меньшей мере, одно соединение, выбираемое из группы, включающей кросповидон, кармеллозу натрия и кармеллозу кальция.
5. Композиция по п. 4, содержащая кросповидон.
6. Композиция по п. 1 или 2, дополнительно содержащая, по меньшей мере, одно соединение, выбираемое из группы, включающей D-маннит и лактозу.
7. Композиция по п. 1 или 2 в форме сиропа, порошка, гранулы, таблетки или капсулы.
8. Способ улучшения растворения (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она из фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль, где способ включает добавление лаурилсульфата натрия к фармацевтической композиции.
9. Способ улучшения абсорбции (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она, где способ включает добавление лаурилсульфата натрия к фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль.
10. Способ улучшения технологичности производства фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль, где способ включает добавление лаурилсульфата натрия к фармацевтической композиции.
11. Применение лаурилсульфата натрия для улучшения растворения (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она или его фармацевтически приемлемой соли.
12. Применение лаурилсульфата натрия для улучшения абсорбции (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-она или его фармацевтически приемлемой соли.
13. Применение лаурилсульфата натрия для улучшения технологичности производства фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль.
14. Применение лаурилсульфата натрия для производства фармацевтической композиции, содержащей (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил-1H-пиразоло[3,4-d]пиримидин-1-ил-1-пирролидинил)-2-пропен-1-он или его фармацевтически приемлемую соль.
| US 2016136168 A1, 19.05.2016 | |||
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР DGAT1 | 2011 |
|
RU2595866C2 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ТВЕРДОГО, ОРАЛЬНО ПРИМЕНИМОГО ФАРМАЦЕВТИЧЕСКОГО СОСТАВА | 2009 |
|
RU2493850C2 |
| WO 2017013160 A1, 26.01.2017 | |||
| WO 2007127630 A1, 08.11.2007 | |||
| QIANG D | |||
| et al., Evaluation of the impact of sodium lauryl sulfate source variability on solid oral dosage form development // Drug development and industrial pharmacy | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| - Vol | |||
| Коридорная многокамерная вагонеточная углевыжигательная печь | 1921 |
|
SU36A1 |
| - No | |||
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
| - P | |||

