Область изобретения
Настоящее изобретение относится к фармацевтическим композициям, включающим (4-{4-[5-(6-трифторметил-пиридин-3-иламино)-пиридин-2-ил]-фенил}-циклогексил)-уксусную кислоту или ее фармацевтически приемлемую соль, например, ее натриевую соль в качестве активного ингредиента в подходящем носителе. Настоящее изобретение также относится к способам их получения и к их применению в качестве лекарственных средств.
Как раскрыто в WO 2007/126957, род соединений в качестве ингибиторов DGAT1, в том числе в Примере 5-1, соединение (4-{4-[5-(6-трифторметил-пиридин-3-иламино)-пиридин-2-ил]-фенил}-циклогексил)-уксусной кислоты, имеющее структурную формулу (I):
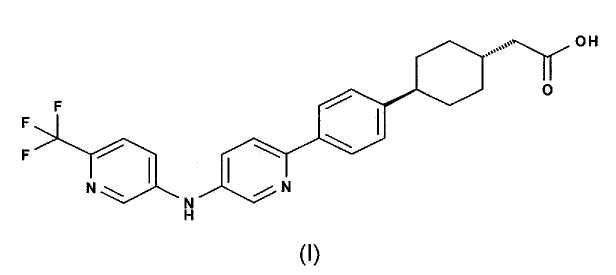
и его натриевую соль
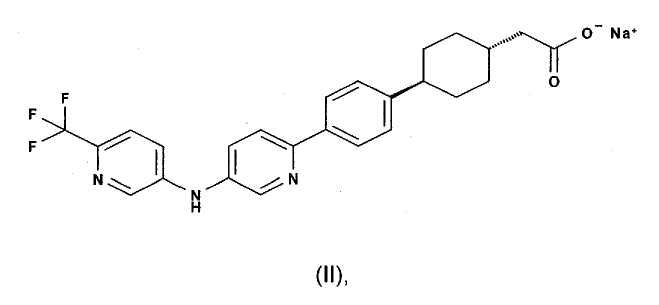
можно использовать в лечении состояния или расстройства, такого как воспалительные состояния, ожирение, диабет и соответствующие метаболические расстройства.
Введение таких фармацевтических средств пероральным путем является более предпочтительным, чем парентеральное введение, поскольку дает возможность пациентам самостоятельно вводить такое средство, тогда как парентеральные композиции в большинстве случаев должен вводить лечащий врач или вспомогательный медицинский персонал. Также важно, что стандартные лекарственные формы, которые изготавливают и которые получает пациент, имеют высокую степень однородности, что касается количества лекарственного вещества, среди отдельных стандартных лекарственных форм. Кроме того, композиция должна иметь хороший профиль растворения и оптимальный профиль высвобождения лекарственного средства in vivo, с минимальной изменчивостью между такими стандартными лекарственными формами.
Однако соединение формулы (I) или его фармацевтически приемлемая соль, в частности его натриевая соль, представляет собой лекарственное вещество, которое трудно составлять в композицию из-за его физико-химических свойств. Более конкретно, натриевая соль соединения формулы (I), описанная выше как соединение формулы (II), является гигроскопичной, плохо растворимой и чрезвычайно проницаемой, с высоким влагопоглощением при 95% относительной влажности. Она также является пластинчатой, очень рыхлой и липкой по своей природе. Она также демонстрирует плохие характеристики текучести.
Эти характеристики лекарственного вещества делают особенно проблематичной разработку композиций, включающих соединение формулы (II), которое должно выдерживать сжимающие силы, необходимые для получения формы таблетки из этой фармацевтической композиции с адекватным окном твердости.
Кроме того, не является обычным получение пероральных композиций соединения формулы (II) в форме таблеток с желаемыми заданными свойствами, такими как хорошая текучесть, характеристики прессуемости (например, отсутствие прилипания в процессе прессования таблеток), хрупкость и/или скорость растворения, надежным и стабильным способом.
Соответственно, существует необходимость в подходящем и твердом галеновом препарате, преодолевающем указанные выше проблемы, связанные со свойствами соединения формулы (II).
В процессе разработки было обнаружено, что достижение такой композиции является трудным. Композиции Примера 1, например, были очень чувствительными к параметрам обработки. Было обнаружено, что прессование при разных твердостях приводило к очень разным профилям растворения. Таким образом, было необходимо снижение изменчивости от таблетки к таблетке, что касается высвобождения лекарственного средства из композиции, поскольку это может иметь большое значение для in vivo доступности лекарственного средства. Также было необходимо разработать композицию, которая была бы более стойкой к параметрам обработки, и которая могла бы избежать одну из основных проблем, связанных с соединением формулы (II), т.е. его липкость.
Сущность изобретения
Неожиданно было обнаружено, что применение комбинации определенных эксципиентов делает возможным получение фармацевтических композиций, в частности, в форме прессованных таблеток, преодолевая, таким образом, указанные выше недостатки.
Изобретение, таким образом, обеспечивает фармацевтическую композицию соединения формулы (I) или его фармацевтически приемлемой соли, которая демонстрирует одну или несколько, например, 1, 2 или 3, из следующих желаемых характеристик:
- профиль растворения, который является подходящим для введения терапевтического средства,
- профиль прессования с широким окном твердости, который, кроме того, обеспечивает приемлемую хрупкость, твердость, время разложения и растворение;
- достаточную стабильность для достижения разумного срока хранения;
- легко достигаемую, если желательно, относительно высокую лекарственную нагрузку.
Композиции по настоящему изобретению также можно получить с использованием стабильного способа получения; который обеспечивает хорошую текучесть, прессуемость и который сводит к минимуму проблемы прилипания и закупоривания при использовании смесей для таблетирования на ротационном прессе. Способ и композиции являются подходящими для крупномасштабного производства, с воспроизводимыми эксплуатационными характеристиками.
Соответственно, настоящее изобретение обеспечивает фармацевтическую композицию, включающую
а) терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли,
b) одно или несколько, например, 1, 2 или 3, поверхностно-активных веществ со свойствами смазывающего вещества;
c) одно или несколько, например, 1, 2 или 3, сухих связующих со свойствами разрыхлителя;
d) один или несколько, например, 1, 2 или 3, наполнителей и
e) один или несколько, например, 1, 2 или 3, разрыхлителей.
Предпочтительные варианты воплощения представляют собой такие, которые определены в настоящей заявке и в зависимых пунктах формулы изобретения.
Подробное описание изобретения
Фармацевтическая композиция в соответствии с настоящим изобретением представляет собой композицию, определенную в пункте 1 формулы изобретения. Применение указанных эксципиентов, к удивлению, преодолевает проблемы формулирования соединения формулы (I) или фармацевтически приемлемой соли, в частности, натриевой соли формулы (II), в твердую пероральную лекарственную форму.
В частности, настоящее изобретение обеспечивает композицию, которая демонстрирует хорошую физическую и химическую стабильность при хранении, которая имеет хороший профиль растворения, которая не является чувствительной к производственным параметрам, и в которой расхождение в высвобождении лекарственного средства между стандартными единицами является минимальным.
В частности, настоящее изобретение также обеспечивает способ, который делает возможным достижение максимальной лекарственной нагрузки, и который не является чувствительным к производственным параметрам.
Авторами настоящего изобретения было обнаружено, что присутствие поверхностно-активного вещества, которое также обладает свойствами смазывающего вещества, существенно снижает тенденцию композиции к прилипанию, а также улучшает свойства растворения и технологические свойства композиций по настоящему изобретению. Таким образом, фармацевтические композиции по настоящему изобретению содержат одно или несколько, например, 1, 2 или 3, поверхностно-активных веществ, которые обладают свойствами смазывающего вещества.
Поверхностно-активные вещества, которые можно использовать в соответствии с настоящим изобретением, включают, без ограничения, лаурилсульфат натрия (SLS), стеариновую кислоту, пальмитиновую кислоту, миристиновую кислоту, полоксамеры и полиэтиленгликоли, такие как PEG 4000-8000, поверхностно-активные вещества серии Tween, поверхностно-активные вещества серии Brij (т.е. Brij 80), Triton X-100 и комбинации таких веществ, предпочтительно лаурилсульфат натрия (SLS).
Поверхностно-активное вещество или комбинацию поверхностно-активных веществ можно использовать в количестве в пределах от около 0,1% до около 5%, предпочтительно от около 0,5% до около 3%, например 2%, в расчете на массу таблетки (до процедуры нанесения какого-либо пленочного покрытия, которое является необязательным). Эти проценты указаны в расчете на соединение формулы (I), и, если используют соль, проценты должны быть соответственно отрегулированы.
Проблемы прилипания в процессе прессования порошка прокаткой и последующего таблетирования (при силах 5 кN или меньше) также преодолеваются в результате присутствия сухого связующего со свойствами смазывающего вещества. Кроме того, присутствие такого связующего дает более стабильные показатели вращательного усилия в процессе прессования порошка прокаткой по сравнению с теми, которые получали для композиций без такого связующего. Таким образом, фармацевтические композиции по настоящему изобретению, кроме того, содержат одно или несколько, например, 1, 2 или 3, связующих со свойствами смазывающего вещества.
Сухие связующие, которые можно использовать в соответствии с настоящим изобретением, включают, без ограничения, полиэтиленгликоли (PEG), например, PEG 4000; предварительно желатинизированный крахмал; крахмал; хитозан; гуаровую камедь, микрокристаллическую целлюлозу; метилцеллюлозу; кальциевую соль карбоксиметилцеллюлозы; натриевую соль карбоксиметилцеллюлозы, альгиновую кислоту и/или ее натриевую соль; гидроксипропилметилцеллюлозу или гидроксипропилцеллюлозу, обе предпочтительно от средней до высокой вязкости, например, степени вязкости 3 или 6 сП, например, низкозамещенную гидроксипропилцеллюлозу (L-HPC LH-21); и комбинации таких веществ. Наиболее предпочтительным связующим является низкозамещенная гидроксипропилцеллюлоза (L-HPC LH-21).
Сухое связующее или комбинацию сухих связующих можно использовать в количестве в пределах от около 2% до около 20%, предпочтительно от около 5% до около 15%, например, около 10% в расчете на массу таблетки (до процедуры нанесения какого-либо пленочного покрытия, которое является необязательным). Эти проценты указаны в расчете на соединение формулы (I), и, если используют соль, проценты должны быть соответственно отрегулированы.
Наполнители, которые можно использовать в соответствии с настоящим изобретением, включают, без ограничения, микрокристаллическую целлюлозу (например, целлюлозу MK GR и продукты, доступные под зарегистрированными торговыми марками AVICEL, FILTRAK, HEWETEN или PHARMACEL, Vivapur, emcocel, tabulose), низкозамещенную гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилметилцеллюлозу, безводный дикальций фосфат, дикальцийфосфат, лактозу, безводную лактозу и комбинации таких веществ.
Предпочтительно, наполнитель представляет собой микрокристаллическую целлюлозу, безводный дикальцийфосфат и безводную лактозу или смесь таких веществ. Можно использовать комбинацию наполнителей, такую как комбинации микрокристаллической целлюлозы и безводного дикальцийфосфата, и комбинации микрокристаллической целлюлозы и лактозы.
Наполнитель или комбинацию наполнителей можно использовать в количестве в пределах от около 4% до около 85%, предпочтительно от около 20% до около 85%, наиболее предпочтительно от около 50-80%, например, 50-65% или 70-80% в расчете на массу таблетки (до процедуры нанесения какого-либо пленочного покрытия, которое является необязательным). Эти проценты указаны в расчете на соединение формулы (I), и, если используют соль, проценты должны быть соответственно отрегулированы.
Когда используют комбинации наполнителей, их можно использовать в соотношении от 1:1 до 1:5, предпочтительно 1:2.
В одном варианте воплощения, наполнитель представляет собой комбинацию микрокристаллической целлюлозы и другого наполнителя, например, безводного дикальцийфосфата или лактозы, где отношение микрокристаллической целлюлозы к лактозе или микрокристаллической целлюлозы к безводному дикальцийфосфату составляет 1:2.
Разрыхлители, которые можно использовать в фармацевтических композициях по настоящему изобретению, могут быть экстрагранулярными или интрагранулярными, или и теми и другими. Примеры разрыхлителей, которые можно использовать в соответствии с настоящим изобретением, включают, без ограничения, кальциевую соль карбоксиметилцеллюлозы (CMC-Ca), натриевую соль карбоксиметилцеллюлозы (CMC-Na) или натрий кроскармеллозу, например, AC-DI-SOL, натрий крахмалгликолят (SSG); альгиновую кислоту, альгинат натрия и гуаровую камедь; предпочтительно натрий кроскармеллозу, например, AC-DI-SOL, поперечно сшитый поливинилпирролидон (например, CROSPOVIDONE, POLYPLASDONE или KOLLIDON XL), натрий крахмалгликолят (SSG).
Наиболее предпочтительным разрыхлителем является натрий крахмалгликолят (SSG).
Разрыхлитель или комбинацию разрыхлителей можно использовать в количестве в пределах от около 0,5% до около 25%, предпочтительно от около 1% до около 10%, наиболее предпочтительно от около 1% до около 6%, в расчете на массу таблетки (до процедуры нанесения какого-либо пленочного покрытия, которое является необязательным). В одном варианте воплощения, разрыхлитель присутствует в количестве, которое составляет 2, 6 или 9% в расчете на массу таблетки. Эти проценты указаны в расчете на соединение формулы (I), и, если используют соль, проценты должны быть соответственно отрегулированы.
Смазывающие вещества могут обеспечить преимущества при формулировании фармацевтической композиции, когда лекарственное вещество плохо растворяется в воде, и используют способ уплотнения, такой как способ уплотнения прокаткой, с лекарственными нагрузками вплоть до 25% масс/масс. Таким образом, фармацевтические композиции по настоящему изобретению, кроме того, могут содержать одно или несколько, например, 1, 2 или 3, смазывающих веществ.
Смазывающие вещества, которые можно использовать в соответствии с настоящим изобретением, включают, без ограничения, стеарат магния, силикат алюминия или кальция, стеариновую кислоту, cutina, PEG 4000-8000, тальк и комбинации таких веществ, предпочтительно стеарилфумарат натрия или стеарат магния, более предпочтительно стеарилфумарат натрия.
Смазывающее вещество или смазывающие вещества можно использовать в количестве в пределах от около 0,1% до около 10%, предпочтительно от около 0,5% до около 5%, например, 2-3%, в расчете на массу таблетки (до процедуры нанесения какого-либо пленочного покрытия, которое является необязательным). Эти проценты указаны в расчете на соединение формулы (I), и, если используют соль, проценты должны быть соответственно отрегулированы.
Соединение формулы (I) или фармацевтически приемлемую соль можно использовать в количестве в пределах от около 0,1% до около 50%, предпочтительно от около 0,5% до около 30%, наиболее предпочтительно от около 1-30%, в расчете на массу фармацевтических композиций (до процедуры нанесения какого-либо пленочного покрытия, которое является необязательным). Соединение формулы (I) может присутствовать в фармацевтических композициях в количестве 2, 10, 15, 20, 25 и 30% масс. Эти проценты указаны в расчете на соединение формулы (I), и, если используют соль, проценты должны быть соответственно отрегулированы.
Фармацевтически приемлемые добавки, подходящие для применения в фармацевтических композициях, в частности, в форме таблеток в соответствии с настоящим изобретением, включают, без ограничения, агенты скольжения, красители и комбинации таких веществ. Количество каждой добавки в фармацевтической комбинации, включающей пероральную фиксированную дозу, может варьировать в пределах, которые являются общепринятыми в данной области.
Подходящие агенты скольжения включают, без ограничения, коллоидный диоксид кремния (например, Aerosil 200), трисиликат магния, порошкообразную целлюлозу, крахмал, тальк и комбинации таких веществ. В случае присутствия, агент скольжения или агенты скольжения в слое, содержащем компонент a), можно использовать в количестве в пределах от около 0,05% до около 5%, предпочтительно от около 0,1% до около 1%, более предпочтительно от около 0,25% до около 1%, например 0,25 или 0,5%, в расчете на массу таблетки (до процедуры нанесения какого-либо пленочного покрытия, которое является необязательным). Эти проценты указаны в расчете на соединение формулы (I), и, если используют соль, проценты должны быть соответственно отрегулированы.
В настоящей заявке описаны различные пронумерованные варианты воплощения настоящего изобретения. Должно быть понятно, что характерные признаки, определенные в каждом варианте воплощения, можно объединять с другими определенными характерными признаками, где определенные характерные признаки представляют собой такие, которые описаны в каждом варианте воплощения, а также в настоящем описании, для обеспечения дополнительных вариантов воплощения настоящего изобретения.
В первом варианте воплощения 1, изобретение обеспечивает фармацевтическую композицию, описанную в пункте 1.
Вариант воплощения 2: фармацевтическая композиция в соответствии с вариантом воплощения 1, где поверхностно-активное вещество со свойствами смазывающего вещества выбрано из лаурилсульфата натрия (SLS), стеариновой кислоты, пальмитиновой кислоты, миристиновой кислоты, полоксамеров и полиэтиленгликолей, таких как PEG 4000-8000, поверхностно-активных веществ серии Tween, поверхностно-активных веществ серии Brij (т.е. Brij 80), Triton X-100 и комбинации таких веществ, предпочтительно лаурилсульфата натрия (SLS).
Вариант воплощения 3: фармацевтическая композиция в соответствии с вариантом воплощения 1 или 2, где сухое связующее выбрано из полиэтиленгликоля (PEG), например, PEG 4000; предварительно желатинизированного крахмала; крахмала; хитозана; гуаровой камеди, микрокристаллической целлюлозы; метилцеллюлозы; кальциевой соли карбоксиметилцеллюлозы; натриевой соли карбоксиметилцеллюлозы, альгиновой кислоты и/или ее натриевой соли; гидроксипропилметилцеллюлозы или гидроксипропилцеллюлозы, обе предпочтительно от средней до высокой вязкости, например, степени вязкости 3 или 6 сП, например, низкозамещенной гидроксипропилцеллюлозы (L-HPC LH-21); и комбинаций таких веществ, наиболее предпочтительно, низкозамещенной гидроксипропилцеллюлозы (L-HPC LH-21).
Вариант воплощения 4: фармацевтическая композиция в соответствии с любым из вариантов воплощения 1-3, где наполнитель выбран из микрокристаллической целлюлозы (например, целлюлозы MK GR и продуктов, доступных под зарегистрированными торговыми марками AVICEL, FILTRAK, HEWETEN или PHARMACEL, Vivapur, emcocel, tabulose), низкозамещенной гидроксипропилцеллюлозы, гидроксиэтилцеллюлозы, гидроксипропилметилцеллюлозы, безводного дикальцийфосфата, дикальцийфосфата, лактозы, безводной лактозы и комбинаций таких веществ.
Вариант воплощения 5: фармацевтическая композиция в соответствии с любым из вариантов воплощения 1-4, где разрыхлитель выбран из кальциевой соли карбоксиметилцеллюлозы (CMC-Ca), натриевой соли карбоксиметилцеллюлозы (CMC-Na) или натрий кроскармеллозы, например, AC-DI-SOL, натрий крахмалгликолята (SSG); альгиновой кислоты, альгината натрия и гуаровой камеди; предпочтительно натрий кроскармеллозы, например, AC-DI-SOL, поперечно сшитого поливинилпирролидона (например, CROSPOVIDONE, POLYPLASDONE или KOLLIDON XL), натрий крахмалгликолята (SSG).
В других вариантах воплощения настоящего изобретения, фармацевтические композиции, представленные в настоящей заявке, могут в дополнение к этому содержать смазывающие вещества, агенты скольжения, красители и комбинации таких веществ, как подробно описано выше.
В предпочтительном аспекте, количества каждого из эксципиентов и количество соединения формулы (I) или его фармацевтически приемлемой соли представляют собой такие, которые указаны в настоящем описании и указаны в Примерах.
Различные термины, используемые в тексте настоящей заявки, определены ниже:
Твердость: Термин “твердость”, который также обычно имеет значение “разрушающая сила” или “стойкость к раздавливанию”, как он используется в настоящей заявке, относится к усилию, необходимому для повреждения таблетки (т.е. разрушения) в определенной плоскости.
Твердость измеряют стандартными способами, которые известны специалистам в данной области, см. согласованную процедуру, описанную в фармакопеях USP <1217> и EP 2.9.8 и JP. Если таблетка слишком мягкая, она не сможет выдерживать манипуляции в процессе последующей обработки, такие как процедуры нанесения покрытия или упаковки и транспортировку. Подобным образом, если таблетка слишком твердая, она не может разлагаться в течение необходимого периода времени или соответствовать требованиям растворимости. Основным принципом испытания твердости является принцип: чем больше таблетка, тем выше твердость. Таким образом, целью специалиста в области формулирования является улучшение профиля сжатия/твердости, чтобы свести к минимуму влияние твердости на время разложения и растворения и максимально увеличить лекарственную нагрузку.
Профиль высвобождения: Термин “высвобождение”, как он используется в настоящей заявке, относится к способу, которым фармацевтическая комбинация, включающая пероральную фиксированную дозу, приводится в контакт с жидкостью, и жидкость транспортирует лекарственное средство(средства) из лекарственной формы в жидкость, которая окружает лекарственную форму. Сочетание скорости доставки и продолжительности доставки, демонстрируемых данной лекарственной формой в организме пациента, может быть определено как профиль высвобождения in vivo. Профили высвобождения/растворения лекарственной формы могут демонстрировать различные скорости и продолжительности высвобождения, и могут быть непрерывными. Непрерывные профили высвобождения включают профили высвобождения, когда один или несколько, например, 1, 2 или 3 активных ингредиента высвобождаются постоянно, либо с постоянной, либо переменной скоростью. Приемлемым профилем высвобождения лекарственного средства для фармацевтической композиции может быть, например, 80% в течение 45 минут.
Разложение: Термин “разложение”, как он используется в настоящей заявке, относится к способу, где фармацевтическая комбинация, включающая пероральную фиксированную дозу, типично посредством жидкости, распадается на отдельные частицы и диспергируется. Разложение достигается, когда твердая пероральная лекарственная форма находится в таком состоянии, при котором любой остаток твердой пероральной лекарственной формы, за исключением фрагментов нерастворимого покрытия или оболочки капсулы, в случае их присутствия, остающийся на сите испытательного оборудования, представляет собой мягкую массу, не имеющую никакого ощутимого твердого ядра, в соответствии с USP<701>. Жидкость для определения свойства разложения представляет собой воду, такую как водопроводная вода или деионизированная вода. Время разложения измеряют стандартными способами, которые известны специалистам в данной области, см. согласованную процедуру, описанную в фармакопеях USP <701> и EP 2.9.1 и JP.
Скорость растворения: Термин “растворение”, как он используется в настоящей заявке, относится к тому, каким образом твердое вещество, в данном случае активные ингредиенты, диспергируется в молекулярной форме в среде. Скорость растворения активных ингредиентов фармацевтической комбинации, включающей пероральную фиксированную дозу, по настоящему изобретению определяют по количеству лекарственного вещества, которое переходит в раствор, на единицу времени в стандартизированных условиях границы раздела жидкость/твердое вещество, температуры и композиции растворителя. Скорость растворения измеряют стандартными способами, которые известны специалистам в данной области, см. согласованную процедуру, описанную в фармакопеях USP <711> и EP 2.9.3 и JP. Для целей настоящего изобретения, испытание для измерения скорости растворения индивидуальных активных ингредиентов осуществляли в соответствии с фармакопеей USP <711> при pH 4,5 с использованием лопастного перемешивающего элемента при 75 об/мин (оборотов в минуту). Растворяющая среда предпочтительно представляет собой буфер, типично фосфатный буфер, в частности, описанный в примере “Испытание на растворимость”. Молярность буфера предпочтительно 0,1 M.
Приемлемый профиль растворения для медленно растворяющегося или слаборастворимого в воде лекарственного средства (BCS класс 2) может означать, например, более чем 80%, например, 85% растворение в течение 30, 45 или 60 минут, см. например, Guidance for Industry: Dissolution Testing of Immediate Immediate Release Solid Oral Dosage Forms, Aug 1997, p. 5.
Термин "в форме частиц", как он используется в настоящей заявке, относится к состоянию вещества, которое характеризуется присутствием отдельных частиц, небольших сфер, шариков или гранул, независимо от их размера, формы или морфологии. Когда присутствует множество твердых частиц, такие вещества называют мультикорпускулярными. Типично, твердые частицы имеют средний размер меньше чем около 3 мм, предпочтительно в пределах от около 1 мкм до 3 мм. Фраза "средний размер частиц" означает, что по меньшей мере 50% твердых частиц имеют размер частиц меньше чем около указанного значения, в расчете на массу. Размер частиц можно определить на основании среднемассового размера частиц, измеренного традиционным методом измерения размера частиц, который хорошо известен специалистам в данной области. Такие методы включают, например, седиментационное фракционирование потока, фотонно-корреляционную спектроскопию, светорассеяние и дисковое центрифугирование.
Термины “эффективное количество” или “терапевтически эффективное количество” относятся к количеству активного ингредиента или средства, которое задерживает или снижает прогрессирование состояния, подлежащего лечению, или которое иным образом полностью или частично вылечивает или паллиативно действует на такое состояние.
Термин "профилактически эффективное количество" относится к количеству активного ингредиента или средства, которое предотвращает проявление заболевания, состояния или расстройства.
Термины “теплокровное животное” или “пациент” используются взаимозаменяемо в настоящей заявке и включают, но не ограничиваются этим, людей, собак, кошек, лошадей, свиней, коров, обезьян, кроликов, мышей и лабораторных животных. В одном варианте воплощения млекопитающим является человек.
Термин "лечение" означает оказание помощи и уход за пациентом в целях профилактики, борьбы с заболеванием, состоянием или расстройством или замедления его развития, предпочтительно в целях борьбы с заболеванием, состоянием или расстройством, и, в частности, также включает профилактическое лечение.
Термины “профилактика”/”профилактический” следует рассматривать как означающие профилактическое введение лекарственного средства, такого как комбинированный препарат или фармацевтическая композиция, здоровым пациентам для предотвращения вспышки заболевания, состояния или расстройства.
Термины “замедление развития”/“замедляющий развитие” следует рассматривать как означающие введение лекарственного средства, такого как комбинированный препарат или фармацевтическая композиция, пациентам, находящимся на стадии, непосредственно предшествующей проявлению заболевания, состояния или расстройства.
Термины “лекарственное средство”, “активное вещество”, “активный ингредиент”, “активное средство” следует рассматривать как означающие соединение в свободной форме или в форме фармацевтически приемлемой соли, в частности, как определено в настоящей заявке.
Когда используют форму множественного числа для соединений, солей, эксципиентов, фармацевтических композиций, заболеваний, расстройств и т.п., предполагается, что это означает одно или несколько, например, 1, 2 или 3, отдельных соединений, солей, эксципиентов, фармацевтических композиций, заболеваний, расстройств или т.п., когда используют единственное число или неопределенный артикль (“a”, “an”), это означает, что включена форма множественного или единственного числа (“один”).
В следующем варианте воплощения настоящее изобретение преодолевает недостатки, связанные с формулированием композиции лекарственного вещества, и обеспечивает специальный способ получения фармацевтической композиции, включающей соединение формулы (I) или его фармацевтически приемлемую соль.
Изобретение обеспечивает, еще в одном из его аспектов, способ получения твердой пероральной лекарственной формы, описанной выше. Такую твердую пероральную лекарственную форму можно получить путем обработки конечной композиции, определенной выше, в соответствующих количествах с получением стандартных единиц лекарственной формы.
В одном варианте воплощения, обеспечивается способ получения фармацевтической композиции в соответствии с любым из предшествующих пунктов патентной формулы, включающий стадии смешивания соединения формулы (I) или его фармацевтически приемлемой соли с, по меньшей мере, одним фармацевтически приемлемым эксципиентом с получением смеси; уплотнение, такое как уплотнение прокаткой, указанной смеси; необязательно, смешивание с дополнительными фармацевтически приемлемыми эксципиентами, и, необязательно, прессование конечной смеси в твердую пероральную лекарственную форму.
Также обеспечивается способ получения твердых пероральных лекарственных форм, описанных выше, включающий стадии
(a) смешивания соединения формулы (I) или его фармацевтически приемлемой соли с, по меньшей мере, одним фармацевтически приемлемым эксципиентом, с получением смеси;
(b) уплотнения прокаткой, затем перемалывания указанной смеси;
(c) смазку полученной смеси, и
(d) прессование полученной смеси в твердую пероральную лекарственную форму.
Количества ингредиентов, представленные как процент в расчете на массу фармацевтической композиции, используемые в каждом примере, указаны в соответствующих таблицах, представленных после соответствующих описаний. Еще один вариант воплощения настоящего изобретения представляет способ получения таблетки в соответствии с настоящим изобретением.
Фармацевтические композиции, включающие пероральные фиксированные дозы, по настоящему изобретению представляют собой таблетки с низкой хрупкостью. Предпочтительно хрупкость не превышает 0,8%. Хрупкость измеряют стандартными способами, которые известны специалистам в данной области, см. согласованную процедуру, описанную в фармакопеях USP <1216> и EP 2,9,7 и JP.
Фармацевтические композиции, включающие пероральные фиксированные дозы, по настоящему изобретению представляют собой таблетки подходящей твердости (например, средняя твердость в пределах от около 30 N до около 110 N). Такую среднюю твердость определяют до нанесения какого-либо пленочного покрытия на фармацевтические комбинации, включающие пероральные фиксированные дозы. Соответственно, предпочтительный вариант воплощения настоящего изобретения направлен на фармацевтические пероральные композиции, которые имеют пленочное покрытие. Подходящие пленочные покрытия являются известными и коммерчески доступными, или их можно получить в соответствии с известными способами. Типично, материал пленочного покрытия представляет собой полимерный пленочный материал для покрытия, включающий вещества, такие как гидроксипропилметилцеллюлоза или поливиниловый спирт, полиэтиленгликоль, лецитин, тальк и краситель. Типично, материал пленочного покрытия наносят в таком количестве, чтобы обеспечить пленочное покрытие, составляющее от около 1% до около 6% в расчете на массу имеющей пленочное покрытие таблетки. Покрытие, включающее поливиниловый спирт и вещества, такие как полиэтиленгликоль, тальк и красители (такие как Opadry AMB или Opadry II 85F) можно наносить в виде гидроизолирующего слоя для обеспечения дополнительной влагозащиты для предотвращения конверсии активного ингредиента в другие полиморфные формы. Достаточную влагозащиту также можно получить при помощи различных упаковок, в том числе, но не ограничиваясь этим: HDPE бутыли с тепло-индукционным уплотнением с или без осушающего вещества и блистерные упаковочные материалы, известные в промышленности как имеющие низкие скорости проникновения паров влаги (т.е. алюминий/алюминий, PVC/PCTFE (поливинилхлорид/полихлортрифторэтилен), ACLAR).
Изобретение обеспечивает способ получения фармацевтических пероральных композиций, описанных в настоящей заявке выше. Такую фармацевтическую комбинацию, включающую пероральную фиксированную дозу, можно получить путем обработки компонентов, определенных выше, в соответствующих количествах с получением стандартных единиц фармацевтической комбинации, включающей пероральную фиксированную дозу.
Фармацевтические композиции являются полезными в лечении или профилактике состояния или расстройства, связанного с активностью DGAT1. Состояния, для которых настоящее изобретение является полезным, включают, без ограничения, метаболические расстройства, такие как ожирение, диабет, нервная анорексия, булимия, кахексия, синдром X, резистентность к инсулину, гипогликемия, гипергликемия, гиперурикемия, гиперинсулинемия, гиперхолестеринемия, гиперлипидемия, дислипидемия, дислипидемия смешанного типа, гипертриглицеридемия, хиломикронемия, семейная хиломикронемия и неалкогольное ожирнение печени; сердечно-сосудистые заболевания, такие как атеросклероз, артериосклероз, острая сердечная недостаточность, застойная сердечная недостаточность, заболевание коронарных артерий, кардиомиопатия, инфаркт миокарда, стенокардия, гипертензия, гипотензия, удар, ишемия, ишемическое реперфузионное поражение, аневризма, рестеноз и сосудистый стеноз; опухолевые заболевания, такие как солидные опухоли, рак кожи, меланома, лимфома и эндотелиальные опухоли, например, рак молочной железы, рак легкого, колоректальный рак, рак желудка, другие опухоли желудочно-кишечного тракта (например, рак пищевода и рак поджелудочной железы), рак предстательной железы, рак почки, рак печени, рак мочевого пузыря, цервикальный рак, рак матки, тестикулярный рак и рак яичников; дерматологические состояния, такие как обыкновенные угри. Еще в одном аспекте, фармацевтические композиции являются полезными в качестве лекарственного средства, снижающего аппетит.
Настоящее изобретение, таким образом, обеспечивает способ лечения или профилактики состояния или расстройства, связанного с активностью DGAT1, включающий введение животному, в том числе человеку, который является пациентом, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции в соответствии с настоящим изобретением.
Настоящее изобретение также обеспечивает применение фармацевтической композиции в соответствии с настоящим изобретением для получения лекарственного средства для лечения или профилактики состояния или расстройства, связанного с активностью DGAT1.
Настоящее изобретение также обеспечивает фармацевтическую композицию в соответствии с настоящим изобретением для применения в лечении или профилактике состояния или расстройства, связанного с активностью DGAT1.
В конечном счете, точная доза активного вещества и конкретная композиция для введения зависят от различных факторов, например, состояния, подлежащего лечению, желательной продолжительности лечения и скорости высвобождения активного вещества. Например, необходимое количество активного вещества и скорость его высвобождения можно определить на основании известных методов in vitro или in vivo, определяя, как долго концентрация конкретного активного вещества в плазме крови остается на приемлемом уровне для терапевтического эффекта.
Приведенное выше описание полностью раскрывает изобретение, включая предпочтительные варианты его воплощения. Модификации и улучшения вариантов воплощения, конкретно раскрытых в настоящей заявке, охватываются объемом представленной далее формулы изобретения. Без дополнительного уточнения, считается, что специалист в данной области сможет, с использованием представленного выше описания, использовать настоящее изобретение в самом полном объеме. Поэтому примеры, представленные в настоящей заявке, следует рассматривать просто как иллюстративные, а не ограничивающие каким-либо образом объем настоящего изобретения.
ПРИМЕРЫ:
Методологический Пример: ИСПЫТАНИЕ НА РАСТВОРИМОСТЬ
Таблетки, описанные в Примерах, испытывали на растворение в 900 мл pH 6,8 фосфатного буфера со скоростью вращения лопастей 75 об/мин.
Комплект оборудования состоял из следующего: закрытый сосуд из стекла или другого инертного прозрачного материала; мотор и лопасть, образованная из крыла и стержня, в качестве перемешивающего элемента. Сосуд частично погружали в подходящую водяную баню любого удобного размера или помещали в нагревательный кожух. Водяная баня или нагревательный кожух позволяли поддерживать температуру внутри сосудов при 37±0,5° в процессе испытания и поддерживать жидкость бани в постоянном и ровном движении. Ни одна из частей этого оборудования, включая окружение, в котором находилось это оборудование, не способствовала каким-либо существенным образом усилению движения, взбалтывания или вибрации выше того, что получали в результате работы ровно вращающегося перемешивающегося элемента. Устройство, которое позволяло наблюдать за образцом и перемешивающим элементом в процессе испытания, имело следующие размеры и емкость: высота от 160 мм до 210 мм и внутренний диаметр от 98 мм до 106 мм. Стенки имели фланцы в верхней части. Можно использовать подогнанную крышку для задержки испарения. Положение стержня такое, чтобы его ось находилась на расстоянии не более чем 2 мм в любой точке от вертикальной оси сосуда и вращалась ровно без существенного колебания. Вертикальная центральная линия крыла лопасти проходила через ось стержня таким образом, чтобы нижняя часть крыла лопасти была вровень с нижней частью стержня. Конструкция лопасти представляла собой такую, как показано в USP <711>, Фиг. 2. В процессе испытания поддерживали расстояние 25±2 мм между лопастями и внутренней стороной донной части сосуда. Металлические или достаточно инертные жесткие крыло лопасти и стержень составляли единое целое. Можно использовать подходящую разъемную конструкцию, состоящую из двух частей, при условии, что блок остается прочным в процессе испытания. Крыло и стержень лопасти могут иметь покрытие из подходящего инертного материала. Лекарственной форме давали опуститься на дно сосуда до того, как лопасть начинала вращаться. Небольшой свободный кусок нереакционноспособного вещества, например, не более чем несколько витков проволочной спирали, можно прикрепить к стандартным лекарственным формам, которые иначе могут всплывать. Можно использовать другие общепринятые устройства для погружения.
1 л забуференного водного раствора, доведенного до pH 6,8±0,05 (0,05M фосфатно-буферного раствора, полученного путем растворения 6,805 г дигидрофосфата калия и 0,896 г гидроксида натрия и разбавления до 1000 мл водой и доведения pH до 6,80±0,05 с использованием 0,2M гидроксида натрия или 1M фосфорной кислоты; указан далее в настоящей заявке как “Растворяющая среда”) помещали в сосуд этого устройства, осуществляли сборку устройства, среду для растворения уравновешивали к 37±0,5° и термометр удаляли. 1 лекарственную форму (например, таблетку или капсулу) помещали в устройство, при этом следили, чтобы не было пузырьков воздуха с поверхности лекарственной формы, и сразу включали устройство со скоростью 75+2 об/мин. С определенными интервалами времени (например, 10, 20, 30, 45, 60, 90 и 120 минут) или в каждой определенной точке времени образец(>1 мл) извлекали из зоны в середине между поверхностью среды для растворения и верхней частью вращающейся лопасти, не меньше чем 1 см от стенки сосуда. [Примечание: аликвоты, взятые для анализа, заменяли равными объемами свежей среды для растворения при 37°, или, когда было показано, что замена среды не является необходимой, изменение объема корректировали путем расчета. Сосуд поддерживали закрытым на протяжении всего испытания и температуру испытываемой смеси контролировали через подходящие промежутки времени]. Образец фильтровали через подходящий фильтр, например, 0,45 мкм PVDF фильтр (Millipore) и первые миллилитры (2-3 мл) фильтрата сливали. Анализ осуществляли при помощи ВЭЖХ или УФ детекции. Испытание повторяли, по меньшей мере, 6 раз с дополнительными стандартными единицами лекарственной формы.
Методологический Пример B: ИСПЫТАНИЕ НА ТВЕРДОСТЬ
Устройство для испытания твердости, Schleuniger 8M Hardness tester, использовали для осуществления испытания твердости таблеток. Таблетки помещали на платформу устройства. Каждая таблетка была расположена продольно в одинаковом положении в соответствии с отличительными маркировочными знаками (когда это было подходящим). Испытание осуществляли для 10 таблеток из каждой партии и каждой силы сжатия.
Пример 1: Ссылочный пример
Натриевую соль транс-(4-{4-[5-(6-трифторметил-пиридин-3-иламино)-пиридин-2-ил]-фенил}-циклогексил)-уксусной кислоты вместе с микрокристаллической целлюлозой (частью) и кросповидоном (интрагранулярный) смешивали в смесительном устройстве с низким сдвигом. Смешиваемое содержимое вместе с остальной частью микрокристаллической целлюлозы пропускали через вибрационную мельницу, снабженную подходящим ситом. Просеянное содержимое смешивали в смесительном устройстве с низким сдвигом в течение подходящего времени. Коллоидный диоксид кремния, просеянный через соответствующее сито, смешивали со смесью с предыдущей стадии и все содержимое смешивали в течение подходящего времени. Стеарат магния, просеянный через сито подходящего размера, добавляли к предварительной смеси и смешивали в течение подходящего времени. Смазанную интрагранулярную предварительную смесь пропускали через систему уплотнения прокаткой для уплотнения при оптимизированных параметрах для скорости подачи, скорости вращения роликов и вращательного усилия. Полученные таким способом полоски собирали и пропускали через вибрационную мельницу, снабженную подходящим ситом, с получением желаемого измельченного вещества. Измельченное вещество затем смешивали с экстрагранулярным предварительно просеянным кросповидоном и смешивали в смесительном устройстве с низким сдвигом в течение подходящего времени. К этой смеси добавляли предварительно просеянный стеарат магния и смешивали в течение подходящего времени. Конечную смесь затем прессовали в таблетки с желаемой массой с достижением оптимизированных толщины, твердости и времени разложения.
Пример 1.A Таблетка без покрытия, включающая ингибитор DGAT1, (5 мг активного ингредиента, в расчете на форму свободной кислоты соединения 1)
Пример 1.B Таблетка без покрытия, включающая ингибитор DGAT1, (10 мг активного ингредиента, в расчете на форму свободной кислоты соединения 1)
Таблица ниже представляет растворение таблеток Примера 1.A, которые прессовали при двух разных твердостях, т.е. 6 кN и 12 кN. Растворение для разных партий осуществляли с использованием лопастной мешалки USP-2/0,4% CTAB/pH 6,8 буфер/50 об/мин.
Показатели растворения Примера 1.А (при двух уровнях твердости)
Пример 2: Эффект поверхностно-активного вещества со свойствами смазывающего вещества
Способ получения фармацевтической композиции:
Микрокристаллическую целлюлозу (Avicel), поверхностно-активное вещество (лаурилсульфат натрия), разрыхлитель (внутренний) и агент скольжения (Aerosil 200) (внутренний) добавляли к терапевтическому средству. Смесь просеивали через сито и смешивали до введения смазывающего вещества. Затем добавляли смазывающее вещество (внутреннее) в смесительный бункер и смешивали в течение соответствующего периода времени. Смесь подвергали уплотнению прокаткой с использованием роликового уплотнителя и затем перемалывали. К смеси добавляли разрыхлитель (внешний) и агент скольжения (Aerosil 200) (внешний) и смешивали в смесительном бункере. Затем полученную смесь смешивали со смазывающим веществом (внешний эксципиент), просеивали через сито в смесительном бункере. Полученную конечную смесь затем прессовали в таблетку с массой около 100 мг.
Таблица ниже представляет эффект поверхностно-активного вещества со свойствами смазывающего вещества. Следующая далее Таблица представляет две композиции, одну с добавлением, а другую без лаурилсульфата натрия.
L-HPC LH 21: низкозамещенная гидроксипропилцеллюлоза
Профили растворения определенных выше композиций представлены ниже.
Показатели растворения Примера 2 (с использованием и без поверхностно-активного вещества)
Сравнение показателей растворения ясно показывает более быструю скорость растворения в первые две точки времени для композиции с 2% масс/масс SLS. Кроме того, в процессе обработки партии, содержащей SLS, отмечали, что тенденция прилипания к роликам уплотнителя также существенно снижалась. Следовательно, с точки зрения преимуществ, касающихся растворения и обработки, включение поверхностно-активного вещества со свойствами смазывающего вещества в композицию имеет критическое значение.
Пример 3: Эффект сухого связующего со свойствами разрыхлителя
Эффект сухого связующего со свойствами разрыхлителя можно определить следующим образом. В Примере 3B, невключение L-HPC LH-21 компенсировали таким же количеством Avicel PH-102 во внешней фазе.
L-HPC LH 21: низкозамещенная гидроксипропилцеллюлоза
Таблица ниже представляет профиль растворения определенных выше композиций и образцов с низкой и высокой твердостью каждой из партий.
(твердость 35 N)
(твердость 89 N)
(твердость 38 N)
(твердость 90 N)
Из представленной выше таблицы можно видеть, что нет никакой явной разницы между профилями растворения при изменениях композиции, вызванных присутствием и отсутствием L-HPC LH-21. Однако, с точки зрения обработки, партия Примера 3B имела проблемы прилипания в процессе уплотнения прокаткой и последующего таблетирования (при силах 5 кN или меньше). Более того, отмечали, что для композиции без рассматриваемого сухого связующего показатели вращательного усилия в процессе уплотнения прокаткой были менее стабильными по сравнению с композицией Примера 3A.
Хрупкость композиции, содержащей сухое связующее со свойствами смазывающего вещества, была ниже.
Поэтому сухое связующее со свойствами смазывающего вещества является существенно важным компонентом композиций по настоящему изобретению.
Пример 4: Эффект используемого типа наполнителя
(внутренний)
Avicel PH 102: микрокристаллическая целлюлоза (MCC)
Безводный дикальцийфосфат: DCP
Профили растворения Примеров 4A и 4B представлены в Таблице ниже.
Сравнение показателей растворения, представленных в таблице, ясно показывает, что ядра таблеток из партии, содержащей MCC:DCP в соотношении 1:2, демонстрировали более медленный профиль растворения по сравнению с партией, содержащей MCC:безводную лактозу в соотношении 1:2.
Пример 5. 25-мг варианты без покрытия с различными типами разрыхлителей
Терапевтическое средство в этих примерах представляло собой соединение формулы (II). Таблица ниже представляет композицию для примеров 5A, 5B и 5C, содержащую 25 мг терапевтического средства, где указанные 25 мг относятся к количеству соединения формулы (I). Примеры 5A, 5B и 5C обеспечивают возможные варианты воплощения лекарственной формы в виде таблетки с использованием различных разрыхлителей.
Микрокристаллическую целлюлозу (Avicel), лаурилсульфат натрия, разрыхлитель (внутренний) и Aerosil 200 (внутренний) добавляли к терапевтическому средству. Смесь просеивали через сито и смешивали до введения смазывающего вещества. Затем в смесительный бункер добавляли стеарилфумарат натрия (внутренний) и смешивали в течение соответствующего периода времени. Смесь подвергали уплотнению прокаткой с использованием роликового уплотнителя и затем перемалывали. К смеси добавляли разрыхлитель (внешний) и Aerosil 200 (внешний) и смешивали в смесительном бункере. Затем полученную смесь смешивали со стеарилфумаратом натрия (внешний эксципиент), просеивали через сито в смесительном бункере. Полученную конечную смесь затем прессовали в таблетку с массой около 100 мг. Показатели растворения этих примеров при pH 6,8 представлены в Таблице ниже.
25-мг варианты без покрытия с различными типами разрыхлителей
PVP- XL: Поперечно-сшитый поливинилпирролидон
Ac-di-sol: Натрий кроскармеллоза;
L- HPC 21: низкозамещенная гидроксипропилцеллюлоза
Профили растворения для примеров 5.1-5.3.
Растворяющая среда: 900 мл pH 6,8 Фосфат+0,05% CTAB; скорость вращения лопасти 75 об/мин; ВЭЖХ метод; быстрое перемешивание 45-60 минут.
(SSG)(8,3 кN)
(PVP- XL) (2,8 кN)
(PVP- XL) (8,0 кN)
(Ac-di-sol)
(2,5 кN)
(Ac-di-sol)(8,0 кN)
Из представленной выше таблицы можно видеть, что ядра таблеток, сформулированных с поперечно-сшитым поливинилпирролидоном (PVP-XL), демонстрируют более медленное высвобождение по сравнению с композициями, в которых использовали натрий крахмалгликолят (SSG) и натрий кроскармеллозу (Ac-Di-Sol). Кроме того, было обнаружено, что поперечно-сшитый поливинилпирролидон (PVP-XL) не совместим с лекарственным веществом из-за присутствия остаточного формальдегида. Также было обнаружено, что композиции, содержащие натрий кроскармеллозу (Ac-Di-Sol), желтеют после их получения и воздействия условий 50°C в сухой атмосфере, а также 50°C/75% остаточной влажности в течение 4 недель.
Таким образом, наиболее предпочтительным разрыхлителем является натрий крахмалгликолят, который также дает хороший уровень хрупкости.
Пример 6. Таблетка 25 мг
Терапевтическое средство в этих примерах представляет собой соединение формулы (II). Таблица ниже представляет композицию для примеров 6.1-6.4, содержащую 25 мг терапевтического средства, где указанные 25 мг относятся к количеству соединения формулы (I). Таблетки для примеров 6.1-6.4 были получены таким же способом, как описано в Примере 2.
25-мг варианты без покрытия с различными уровнями разрыхлителя
(внутренний)
круглая
Данные in vitro скорости растворения примеров 6.1-6.4 представлены в Таблице ниже.
In vitro скорость растворения примеров 6.1-6.4
Растворяющая среда: 900 мл pH 6,8 Фосфата+0,05% CTAB (цетилтриметиламмонийбромид); скорость вращения лопасти 75 об/мин; ВЭЖХ метод; быстрое перемешивание 45-60 минут.
Профили растворения демонстрируют явный ранговый порядок в том, что касается уровня содержания разрыхлителя и скорости растворения до уровня 6%. Не было никакой значительной разницы в скоростях высвобождения на уровне от 6% до 9%. Больше чем 90% терапевтического средства высвобождалось из Примеров с разрыхлителем. Разрыхлители обычно снижают прессуемость композиции, и более высокие уровни будут снижать твердость таблетки и/или создавать проблемы хрупкости. Таким образом, необходимо соблюдение точного баланса между количеством разрыхлителя и другими свойствами композиции.
Разрыхлитель распределяли равномерно между внутренними и внешними компонентами. Например, 6% распределяли как 3% внутреннего и 3% внешнего компонентов, соответственно, и 2% распределяли как 1% внутреннего и 1% внешнего компонентов, соответственно.
Пример 7. Таблетка 2 мг
Терапевтическое средство в этом примере представляет собой соединение формулы (II). Таблицы ниже представляют композицию для примера 7, содержащую 2 мг терапевтического средства; где указанные 2 мг относятся к количеству соединения формулы (I).
Пример 8: Таблетки без покрытия, содержащие 20 мг активного вещества, с 6% разрыхлителя и 2% разрыхлителя
Терапевтическое средство в этих примерах представляет собой соединение формулы (II). Таблица ниже представляет композицию для примеров 8.1 и 8.2, содержащую 20 мг терапевтического средства, где указанные 20 мг относятся к количеству соединения формулы (I).
(внутренний)
Пример 9: Техническая стабильность примеров
Данные, характеризующие техническую стабильность, представленные в Таблицах ниже, показывают, что таблетки по настоящему изобретению имеют хорошую стабильность даже без осушителя. Таблетки упаковывали в 90 см3 HPDE бутыль с тепло-индукционным уплотнением.
Химические данные: 25 мг, 2% разрыхлителя,
таблетка - Пример 6.2
анализ
Физические данные: 25 мг, 2% разрыхлитель,
таблетка - Пример 6.2
анализ
Химические данные: 25 мг, 6% разрыхлитель,
таблетка - Пример 6.3
ингредиента
[% Соединения формулы (II)]
анализ
Физические данные: 25 мг, 6% разрыхлитель,
таблетка - Пример 6.3
анализ
Пример 10: Биодоступность композиций таблеток
Осуществляли испытание в едином центре, рандомизированное, с открытой этикеткой, однодозовое в параллельных группах для оценки относительной биодоступности терапевтического средства после перорального введения одной дозы соединения формулы (II), сформулированного в форму таблетки (20 мг), которую вводили здоровым субъектам в условиях голодания или сытости (стандартный FDA завтрак). В испытании участвовало всего 120 человек, и их поровну распределяли на 5 групп обработки (24 субъекта/группа обработки, в соотношении 1:1:1:1:1).
• T1: одна 20 мг таблетка (Пример 8.1) в условиях голодания
• T2: одна 20 мг таблетка (Пример 8.2) в условиях голодания
• T3: две 10 мг таблетки (Ссылочный пример 1.B) в условиях голодания
• T4: одна 20 мг таблетка (Пример 8.1) в условиях сытости
• T5: одна 20 мг таблетка (Пример 8.2) в условиях сытости
Все участники испытания прошли период отбора до 20-дней (День -21 до -2), период определения базовой линии (День -1), период лечения с введением одной дозы, с последующим 36-дневным периодом наблюдения при визитах к врачу находящихся вне стационара пациентов и оценкой по завершении испытания (День 36). Субъекты, которые соответствовали критериям включения/исключения при отборе, допускались для определения базовой линии в день перед введением дозы. Все результаты определения базовой линии безопасности должны быть доступны до введения средства.
Серийные образцы крови для фармакокинетических (PK) исследований собирали в Дни с 1 по 36 после введения лекарственного средства для определения PK профиля различных композиций по настоящему изобретению.
Оценки безопасности включают физическое обследование, ЭКГ, основные показатели состояния организма, стандартные клинические лабораторные анализы гематологии, химического состава крови, мочи, мониторинг неблагоприятных результатов и серьезных неблагоприятных результатов.
Фармакокинетические оценки:
• PK сбор крови (3 мл в EDTA пробирках (плазма)): До введения дозы, 1, 2, 4, 6, 8, 10, 12, 24, 48, 72, 96, 120, 144, 168, 240, 312, 408, 504, 672, 840 часов после введения дозы.
• Точное время сбора крови регистрировали на eCRF.
• Вещества, определяемые в анализе, среда и методы: соединение формулы (II) в плазме, утвержденным методом LC-MS/MS.
• PK параметры соединения формулы (II) (определяемые для каждой композиции таблеток): Cmax, Tmax, AUC0-last, AUC0-inf, t1/2, CL/F и Vd/F
• PK определения: Представлена описательная статистика всех рассчитанных PK параметров.
Фармакокинетические параметры после одного перорального введения 20 мг терапевтического средства, такого как таблетка Примера 8.1, Примера 8.2 или Ссылочного примера 1.B в условиях голодания или Примера 8.1, Примера 8.2 в условиях сытости здоровым субъектам
голодание
голодание
таблетка
сытость
сытость
Среднее
(мин., макс.)
23,5
(9,95, 144)
17,0
(4,00, 120)
36,0
(4,00, 144)
12,0
(2,00, 119)
12,0
(4,00, 48,0)
(нг/мл)а
Среднее значение±
(% CV)
141±85,5
(60,5)
161±132
(81,7)
131±84,0
(64,3)
260±235
(90,4)
219±190
(86,7)
Среднее значение±
(% CV)
136±28,2
(20,8)
153±59,2
(38,6)
140±45,1
(32,3)
164±95,0
(57,8)
139±34,4
(24,7)
Среднее значение±
(% CV)
18400±
11300
(61,0)
22100±
14500
(65,7)
20300±
13000
(64,3)
33100±
23200
(70,1)
29700±
16800
(56,4)
Среднее значение±
(% CV)
19400±
11400
(58,7)
23100±
15500
(67,1)
21800±
13800
(63,3)
34000±
23300
(68,6)
30300±
16900
(55,7)
час)а,b
Среднее значение±
(% CV)
1310±
572
(43,6)
1330±
882
(66,2)
1260±
716
(57,0)
812±
392
(48,3)
880±
536
(61,0)
Среднее значение±
(% CV)
252000±
113000
(45,1)
269000±
169000
(62,8)
254000±
192000
(75,6)
194000±
170000
(87,9)
172000±
93000
(54,2)
bЗначения для субъектов с выверенными значениями Rsq<0,75 и/или экстраполированными значениями AUC %>25 не были включены в эту таблицу
После перорального введения 20 мг таблеток соединения формулы (II), таких как Пример 8.1 и Пример 8.2 или Ссылочный пример 1.B, голодающим здоровым субъектам профили концентрации в плазме-время были приблизительно совпадающими, особенно в последних точках времени, что указывает на одинаковые скорости и степень абсорбции и выведения из организма для этих трех композиций. Были небольшие различия фармакокинетических параметров (Cmax, AUClast и AUCinf) этих трех композиций. Однако эти различия не были статистически значимыми.
Примеры 8.1 и 8.2, таким образом, являются аналогичными Ссылочному примеру 1.B в том, что касается скорости и степени абсорбции терапевтического средства.
Изобретение, таким образом, обеспечивает фармацевтическую композицию соединения формулы (I) или его фармацевтически приемлемой соли, которая демонстрирует одну или несколько, например, 1, 2 или 3, из следующих желательных характеристик:
- профиль растворения, который является подходящим для введения терапевтического средства,
- профиль сжатия с широким окном твердости, которое, однако, обеспечивает приемлемую хрупкость, твердость, время разложения и растворение;
- достаточная стабильность для достижения разумного срока хранения;
- относительно высокая лекарственная нагрузка, если желательно, легко достигаемая.
Композиции по настоящему изобретению также можно получить стабильным способом получения; который дает хорошую текучесть, прессуемость и который сводит к минимуму проблемы прилипания и закупоривания при обработке смесей для таблетирования на ротационном прессе. Способ и композиции являются подходящими для крупномасштабного производства, с воспроизводимыми эксплуатационными характеристиками.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИЯ ТАБЛЕТКИ ЛЕНАЛИДОМИДА ДЛЯ ПЕРОРАЛЬНОГО ПРИЕМА | 2017 |
|
RU2725074C1 |
| ТАБЛЕТКА МЕЛАТОНИНА И СПОСОБЫ ИЗГОТОВЛЕНИЯ И ПРИМЕНЕНИЯ | 2008 |
|
RU2485949C2 |
| НОВАЯ КОМПОЗИЦИЯ ЛАПАТИНИБА В ВИДЕ ТВЕРДОЙ ЛЕКАРСТВЕННОЙ ФОРМЫ ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ И СПОСОБ ЕЕ ИЗГОТОВЛЕНИЯ | 2019 |
|
RU2821950C2 |
| СПОСОБ СУХОГО ГРАНУЛИРОВАНИЯ ДЛЯ ПОЛУЧЕНИЯ КОМПОЗИЦИЙ МЕТФОРМИНА В ВИДЕ ТАБЛЕТОК И ЕГО КОМПОЗИЦИИ | 2013 |
|
RU2647421C2 |
| Твёрдые лекарственные формы палбоциклиба | 2016 |
|
RU2686840C1 |
| СОСТАВЫ В ФИКСИРОВАННЫХ ДОЗАХ | 2018 |
|
RU2810163C2 |
| ТАБЛЕТКИ И ГРАНУЛИРОВАННЫЕ ПОРОШКИ, СОДЕРЖАЩИЕ 6-ФТОР-3-ГИДРОКСИ-2-ПИРАЗИНКАРБОКСАМИД | 2010 |
|
RU2527766C2 |
| ЛЕКАРСТВЕННАЯ ФОРМА С УЛУЧШЕННЫМИ ХАРАКТЕРИСТИКАМИ pH-ЗАВИСИМОГО ВЫСВОБОЖДЕНИЯ ПРЕПАРАТА, СОДЕРЖАЩАЯ ЭЗОМЕПРАЗОЛ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМУЮ СОЛЬ | 2017 |
|
RU2716025C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИРБЕСАРТАН | 2008 |
|
RU2465900C2 |
| Композиции перорально распадающихся таблеток, содержащие кортикостероиды, для лечения эозинофильного эзофагита | 2014 |
|
RU2678695C2 |
Настоящее изобретение относится к фармацевтической композиции в форме таблетки, включающей а) терапевтически эффективное количество натриевой соли (4-{4-[5-(6-трифторметил-пиридин-3-иламино)-пиридин-2-ил]-фенил}-циклогексил)-уксусной кислоты, b) лаурилсульфат натрия в качестве поверхностно-активного вещества со свойствами смазывающего вещества в количестве от 0,1 до 5%, с) низкозамещенную гидроксипропилцеллюлозу в качестве сухого связующего вещества со свойствами разрыхлителя в количестве от 2 до 20%, d) смесь микрокристаллической целлюлозы и безводной лактозы в качестве наполнителя в соотношении от 1:5 до 1:1 и е) натрий крахмалгликолят в качестве разрыхлителя в количестве от 1 до 10% в расчете на массу таблетки до нанесения пленочного покрытия. Также изобретение относится к способу получения фармацевтической композиции и к ее применению в качестве лекарственного средства. Композиции по изобретению демонстрируют профиль сжатия с широким окном твердости, которое обеспечивает приемлемую хрупкость, твердость, время разложения и растворения, а также достаточную стабильность для достижения разумного срока хранения. 3 н. и 7 з.п. ф-лы, 10 пр.
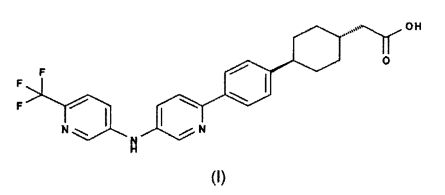
1. Фармацевтическая композиция в форме таблетки, содержащая
a) терапевтически эффективное количество натриевой соли соединения формулы (I)
b) лаурилсульфат натрия в качестве поверхностно-активного вещества со свойствами смазывающего вещества, где поверхностно-активное вещество присутствует в количестве, составляющем от 0,1 до 5% в расчете на массу таблетки до нанесения какого-либо пленочного покрытия;
c) низкозамещенную гидроксипропилцеллюлозу (L-HPC LH-21) в качестве сухого связующего вещества со свойствами разрыхлителя, где сухое связующее вещество присутствует в количестве, составляющем от 2 до 20% в расчете на массу таблетки до нанесения какого-либо пленочного покрытия;
d) смесь безводной лактозы и микрокристаллической целлюлозы в качестве наполнителя, где соотношение микрокристаллической целлюлозы и безводной лактозы составляет от 1:5 до 1:1, и
e) натрий крахмалгликолят в качестве разрыхлителя, где разрыхлитель присутствует в количестве, составляющем от 1 до 10% в расчете на массу таблетки до нанесения какого-либо пленочного покрытия.
2. Фармацевтическая композиция по п. 1, дополнительно содержащая одно или несколько смазывающих веществ, где смазывающее вещество выбирают из стеарата магния, силиката алюминия, силиката кальция, стеариновой кислоты, кутина, PEG 4000-8000, тальк или их комбинации,
где смазывающее вещество присутствует в количестве, составляющем от 0,1 до 10% в расчете на массу таблетки до нанесения какого-либо пленочного покрытия.
3. Фармацевтическая композиция по п. 1, где поверхностно-активное вещество присутствует в количестве от 0,1 до 5% в расчете на массу композиции.
4. Фармацевтическая композиция по п. 1, где сухое связующее вещество присутствует в количестве от 5 до 15% в расчете на массу композиции.
5. Фармацевтическая композиция по п. 1, где разрыхлитель присутствует в количестве 2-10% в расчете на массу таблетки.
6. Фармацевтическая композиция по п. 1, где разрыхлитель присутствует в количестве 2, 6 или 9% в расчете на массу таблетки.
7. Фармацевтическая композиция по п. 1, где соединение формулы (I) присутствует в количестве в пределах от около 0,1 до около 50%, предпочтительно от около 0,5 до около 30%, наиболее предпочтительно от около 1-30% в расчете на массу таблетки до нанесения какого-либо пленочного покрытия.
8. Способ лечения или профилактики состояния или расстройства, связанного с активностью DGAT1, включающий введение животному, в том числе человеку, который является пациентом, нуждающимся в таком лечении, терапевтически эффективного количества фармацевтической композиции по любому из предшествующих пунктов.
9. Фармацевтическая композиция по п. 1 для применения в лечении или профилактике состояния или расстройства, связанного с активностью DGAT1.
10. Способ получения фармацевтической композиции по любому из предшествующих пунктов, включающий стадии:
(a) смешивания соединения формулы (I) или его фармацевтически приемлемой соли с по меньшей мере одним фармацевтически приемлемым эксципиентом с получением смеси;
(b) уплотнения прокаткой, затем перемалывания указанной смеси;
(c) смазки полученной смеси и
(d) прессования полученной смеси в твердую пероральную лекарственную форму.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |

