Связанные заявки
По настоящей заявка испрашивается приоритет предварительной патентной заявки США No. 62/221531, поданной 21 сентября 2015; предварительной патентной заявки США No. 62/238511, поданной 7 октября 2015; и предварительной патентной заявки США No. 62/348855, поданной 10 июня 2016. Содержимое вышеуказанных заявок включено в настоящее описание в виде ссылки полностью.
Предшествующий уровень техники изобретения
Множество существующих в настоящее время препаратов имеют ряд недостатков, обусловленных свойствами плохой абсорбции, распределения, метаболизма и/или экскреции (ADME), которые затрудняют их широкое применение или ограничивают их применение при определенных показаниях. Неудовлетворительные ADME свойства также являются главной причиной неудач лекарственных средств кандидатов в клинических исследованиях. Тогда как технологии рецептирования и стратегии пролекарств могут быть использованы в некоторых случаях для улучшения определенных ADME свойств, такие подходы часто не могут решить лежащие в основе ADME проблемы, которые существуют для множества лекарственных средств и лекарственных средств кандидатов. Одной такой проблемой является быстрый метаболизм, который способствует тому, что ряд лекарственных средств, которые иначе были бы высоко эффективными в лечении заболевания, слишком быстро выделяются из организма. Возможным решением быстрого клиренса лекарственного средства является частое введение или введение высокой дозы для достижения достаточно высокого уровня лекарственного средства в плазме. Это, однако, создает ряд потенциальных проблем лечения, таких как плохая приверженность пациента к лечению с использованием схемы лечения, побочные эффекты, которые становятся более острыми при более высоких дозах, и повышенную стоимость лечения. Быстро метаболизирующееся лекарственное средство также может подвергнуть пациентов воздействию нежелательных токсичных или реакционноспособных метаболитов.
Другим ADME ограничением, которое поражает множество препаратов, является образование токсичных или биологически активных метаболитов. В результате, некоторые пациенты, получающие лекарственное средство, могут испытывать токсичность, или безопасное введение таких лекарственных средств может быть ограничено, так что пациенты получают субоптимальное количество активного вещества. В некоторых случаях, модификация интервалов введения или подходов рецептирования могут помочь уменьшить клинические нежелательные эффекты, но часто образование таких нежелательных метаболитов является присущим метаболизму соединения.
В некоторых выбранных случаях метаболический ингибитор вводят совместно с лекарственным средством, которое выводится слишком быстро. Таким является случай с классом лекарственных средств ингибиторов протеаз, которые используют для лечения ВИЧ инфекции. FDA рекомендует, чтобы такие лекарственные средства вводились совместно с ритонавиром, ингибитором фермента цитохром P450 3A4 (CYP3A4), ферментом, обычно отвечающим за их метаболизм (см. Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ритонавир, однако, вызывает нежелательные эффекты и добавляет количество таблеток для пациентов с ВИЧ, которые уже должны принимать комбинацию различных лекарственных средств. Сходным образом ингибитор CYP2D6 квинидин добавляют к декстрометорфану с целью уменьшения быстрого CYP2D6 метаболизма декстрометорфана в лечении аффективной лабильности. Квинидин, однако, имеет нежелательные побочные эффекты, которые существенно ограничивают его применение в потенциальной комбинированной терапии (см. Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at www.accessdata.fda.gov).
В общем, комбинация лекарственных средств с ингибиторами P450 не является удовлетворительной стратегией для уменьшения клиренса лекарственного средства. Ингибирование активности фермента CYP может влиять на метаболизм и выведение других лекарственных средств, метаболизируемых тем же ферментом. Ингибирование CYP может вызывать накопление других лекарственных средств в организме до токсического уровня.
Потенциально привлекательной стратегией улучшения метаболических свойств лекарственного средства является модификация дейтерием. В таком подходе пытаются замедлить CYP-опосредованный метаболизм лекарственного средства или уменьшить образование нежелательных метаболитов путем замены одного или более атомов водорода атомами дейтерия. Дейтерий является безопасным, стабильным, нерадиоактивным изотопом водорода. По сравнению с водородом, дейтерий образует более сильные связи с углеродом. В выбранных случаях, увеличенная сила связи, обусловленная дейтерием, может позитивно влиять на ADME свойства лекарственного средства, создавая потенциал для улучшенной эффективности, безопасности и/и переносимости лекарственного средства. В то же время, так как размер и форма дейтерия являются по существу идентичными таковым водорода, замена водорода дейтерием, вероятно, не будет влиять на биохимическую эффективность и селективность лекарственного средства при сравнении с оригинальным химическим веществом, которое содержит только водород.
В течение последних 35 лет, эффекты замены дейтерия на скорость метаболизма сообщают для очень небольшого процента одобренных лекарственных средств (см., например, Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res, 1985, 14:1-40 (ʺFosterʺ); Kushner, DJ et al, Can J Physiol Pharmacol, 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9:101-09 (ʺFisherʺ)). Результаты были вариабельными и непредсказуемыми. Для некоторых соединений дейтеризация вызывала сниженный метаболический клиренс in vivo. Для других, не было изменений в метаболизме. Другие продемонстрировали увеличенный метаболический клиренс. Вариабельность эффектов дейтерия также привела экспертов к вопросу или отрицанию модификации дейтерием в качестве полезной стратегии дизайна лекарственного средства для ингибирования нежелательного метаболизма (см. Foster на стр. 35 и Fisher на стр. 101).
Эффекты дейтериевой модификации на метаболические свойства лекарственного средства являются непредсказуемыми даже когда атомы дейтерия включают в известные сайты метаболизма. Только посредством точного получения и тестирования дейтерированного лекарственного средства можно определить, будет ли и как скорость метаболизма отличаться от таковой его недейтерированного аналога. См., например, Fukuto et al. (J. Med. Chem., 1991, 34, 2871-76). Множество лекарственных средств имеют множество сайтов, где возможен метаболизм. Сайт(ы), где требуется замещение дейтерием, и степень дейтеризации, необходимая для оказания эффекта на метаболизм, если есть, будут отличаться для каждого лекарственного средства.
Настоящее изобретение относится к новым производным ивакафтора, и его фармацевтически приемлемым солям. Настоящее изобретение также обеспечивает композиции, включающие соединение по настоящему изобретению, и применение указанных композиций в способах лечения заболеваний и состояний, которые предпочтительно лечат путем введения усилителя CFTR (муковосцидозный трансмембранный регулятор проводимости).
Ивакафтор, также известный как VX-770 и по химическому наименованию, N-(2,4-ди-третбутил-5-гидороксифенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамид, действует как усилитель CFTR. Результаты исследований ивакафтора III фазы у пациентов с муковисцидозом, несущих по меньшей мере одну копию мутации G551D-CFTR, продемонстрировали заметное улучшение функции легких и других ключевых индикаторов заболевания, включая уровень хлоридов пота, вероятность раздражения легких и массу тела. Ивакафтор был одобрен FDA в 2012 для лечения муковисцидоза у пациентов, которые имеют G551D-CFTR мутацию. В 2014, ивакафтор был одобрен для лечения муковисцидоза у пациентов, которые имеют одну из восьми дополнительных мутаций (G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P и G1349D) в гене CFTR. В 2015 ивакафтор был одобрен для лечения муковисцидоза у пациентов, которые имеют одну из 10 мутаций в гене CFTR (G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R и R117H). Ивакафтору было разрешено быстрое трековое одобрение и обозначение орфанного лекарственного средства FDA в 2006 и 2007, соответственно, и он продается под торговым наименованием Калидеко®. Ивакафтор также одобрен в комбинации с VX-809 (также известный как люмакафтор, корректор CFTR) для перорального лечения пациентов с муковисцидозом, которые имеют наиболее частую мутацию ΔF508-CFTR; комбинация продается на рынке под торговым наименованием Оркамби®.
Несмотря на полезные активности ивакафтора, существует продолжающаяся необходимость в новых соединениях для лечения вышеупомянутых заболеваний и соединений.
Сущность изобретения
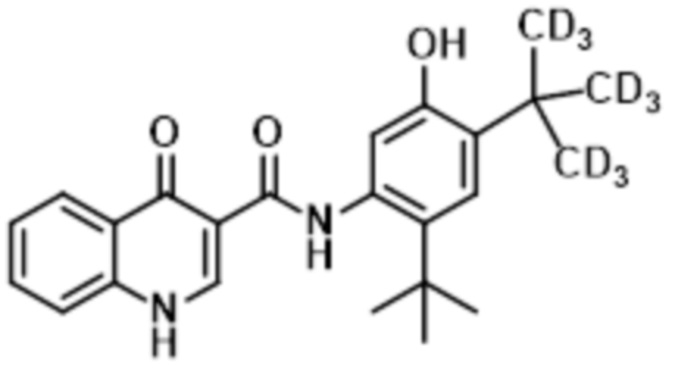
В настоящее время было обнаружено, что дейтерированные аналоги ивакафтора (включая соединение (I), также называемые как CTP-656, D9-ивакафтор или соединение 106, и соединение (II), также называемая как соединение 105 или D18-ивакафтор) имеет улучшенный метаболический профиль, при введении пациенту, при сравнении с ивакафтором. В частности, соотношение исходного к метаболиту соединения (I) больше чем профиль, обнаруживаемый для ивакафтора. Соединение (I) представлено следующей структурной формулой:
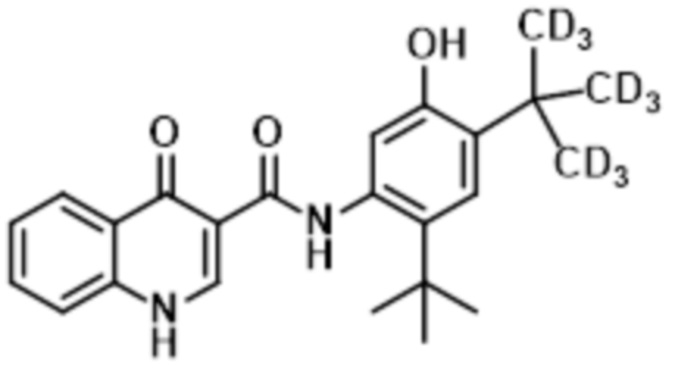
(I),
или его фармацевтически приемлемая соль. Соединение (II) представлено следующей структурной формулой:
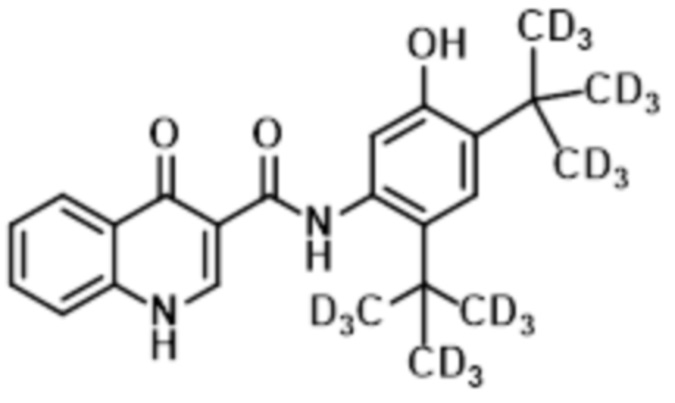
(II)
или его фармацевтически приемлемая соль.
Улучшенный фармакокинетический профиль для соединения (I) относительно ивакафтора предполагает, что соединение (I) может быть эффективным в дозировках в диапазоне от около 50 до около 200 мг один раз в сутки. На основании таких открытий, новые схемы введения с использованием соединения (I) или (II), или его фармацевтически приемлемой соли для лечения состояния, опосредованного CFTR, у пациента, описаны в настоящем описании.
Первый вариант осуществления изобретения представляет собой способ для лечения состояний, которые можно лечить соединениями, которые потенциируют активность CFTR. Способ включает введение пациенту количества соединения (I) или (II), или его фармацевтически приемлемой соли, один раз в сутки, где количество соединения (I) или (II), или его фармацевтически приемлемой соли, находится в диапазоне от около 50 мг до около 200 мг, например, около 50 мг, около 60 мг, около 70 мг, около 80 мг, около 90 мг, около 100 мг, около 110 мг, около 120 мг, около 130 мг, около 140 мг, около 150 мг, около 160 мг, около 170 мг, около 180 мг, около 190 мг, или около 200 мг. В частности, пациентом является человек. В одном аспекте настоящего варианта осуществления изобретения пациентом является человек в возрасте 6 лет или старше. Предпочтительно соединение (I) или (II), или его фармацевтически приемлемую соль вводят перорально в любой из следующих дозировок. В определенных вариантах осуществления изобретения соединение (I) или (II), или его фармацевтически приемлемую соль вводят перорально в любой из вышеуказанных дозировок в фармацевтической композиции, которой является таблетка, включая любую таблетированную композицию, описанную в настоящем описании, или биоэквивалентную таблетированную композицию, или гранулы. В определенных аспектах настоящего варианта осуществления изобретения соединением является соединение (I). В других аспектах настоящего варианта осуществления изобретения соединением является соединение (II).
В первом альтернативном варианте осуществления изобретения способ включает введение пациенту количества соединения (I) или (II), или его фармацевтически приемлемой соли, один раз в сутки, где количество соединения (I) или (II) или его фармацевтически приемлемой соли находится в диапазоне от около 25 мг до около 75 мг, например, около 25 мг, около 37,5 мг, около 50 мг, около 62,5 мг, или около 75 мг, где пациентом является человек в возрасте от 2 до менее чем 6 лет и менее чем 14 кг; или альтернативно, является человек в возрасте от 2 до менее чем 6 лет и 14 кг или более. В одном аспекте дозировка для человека 2 или менее чем 6 лет и менее чем 14 кг составляет 25 мг. В одном аспекте дозировка для человека от 2 до менее чем 6 лет и более чем 14 кг составляет 37,5 мг. Предпочтительно соединение (I) или (II) или его фармацевтически приемлемую соль вводят перорально в любой из вышеуказанных дозировок. Предпочтительно, Соединение (I) или (II) или его фармацевтически приемлемую соль вводят перорально в любой из вышеуказанных дозировок в фармацевтической композиции, которой является гранула. В определенных аспектах настоящего варианта осуществления изобретения соединением является Соединение (I). В других аспектах настоящего варианта осуществления изобретения соединением является Соединение (II).
Вторым вариантом осуществления изобретения является Соединение (I) или (II) или его фармацевтически приемлемая соль, для лечения состояний, которые можно лечить соединениями, которые потенциируют активность CFTR. Соединение можно вводить по схеме введения, описанной в настоящем описании. В определенных аспектах настоящего варианта осуществления изобретения соединением является Соединение (I). В других аспектах настоящего варианта осуществления изобретения соединением является Соединение (II).
Третьим вариантом осуществления настоящего изобретения является применение Соединения (I) или (II) или его фармацевтически приемлемой соли для получения лекарственного препарата для лечения состояний, которые можно лечить соединениями, которые потенциируют активность CFTR. Соединение можно вводить по схеме введения, описанной в настоящем описании, например, в количестве в диапазоне от 50 мг до 200 мг, один раз в сутки. В определенных аспектах настоящего варианта осуществления изобретения соединением является Соединение (I). В других аспектах настоящего варианта осуществления изобретения соединением является Соединение (II).
Четвертым вариантом осуществления изобретения является фармацевтическая композиция, включающая фармацевтически приемлемый носитель или разбавитель и от около 50 мг до около 200 мг Соединения (I) или (II), или его фармацевтически приемлемой соли. Специфически фармацевтическая композиция включает около 50 мг, около 60 мг, около 70 мг, около 80 мг, около 90 мг, около 100 мг, около 110 мг, около 120 мг, около 130 мг, около 140 мг, около 150 мг, около 160 мг, около 170 мг, около 180 мг, около 190 мг, или около 200 мг Соединения (I) или (II) или его фармацевтически приемлемой соли. Более специфически, например, фармацевтическая композиция включает 75, 100, или 150 мг Соединения I для введения один раз в сутки. В определенном варианте осуществления изобретения фармацевтическая композиция включает 100-150 мг Соединения I для введения один раз в сутки. В определенном варианте осуществления изобретения фармацевтическая композиция включает 100 мг Соединения I для введения один раз в сутки. В определенном аспекте фармацевтической композицией является таблетка. Альтернативным четвертым вариантом осуществления изобретения является фармацевтическая композиция, включающая фармацевтически приемлемый носитель или разбавитель и от около 25 мг до около 75 мг Соединения (I) или (II), или его фармацевтически приемлемой соли. Специфически, фармацевтическая композиция включает около 25 мг, около 37,5 мг, около 50 мг, около 62,5 мг, или около 75 мг Соединения (I) или (II) или его фармацевтически приемлемой соли. В определенном аспекте фармацевтической композицией являются гранулы. В определенных аспектах настоящего варианта осуществления изобретения соединением является Соединение (I). В других аспектах настоящего варианта осуществления изобретения соединением является Соединение (II).
Дополнительные варианты осуществления изобретения описаны ниже.
Краткое описание чертежей
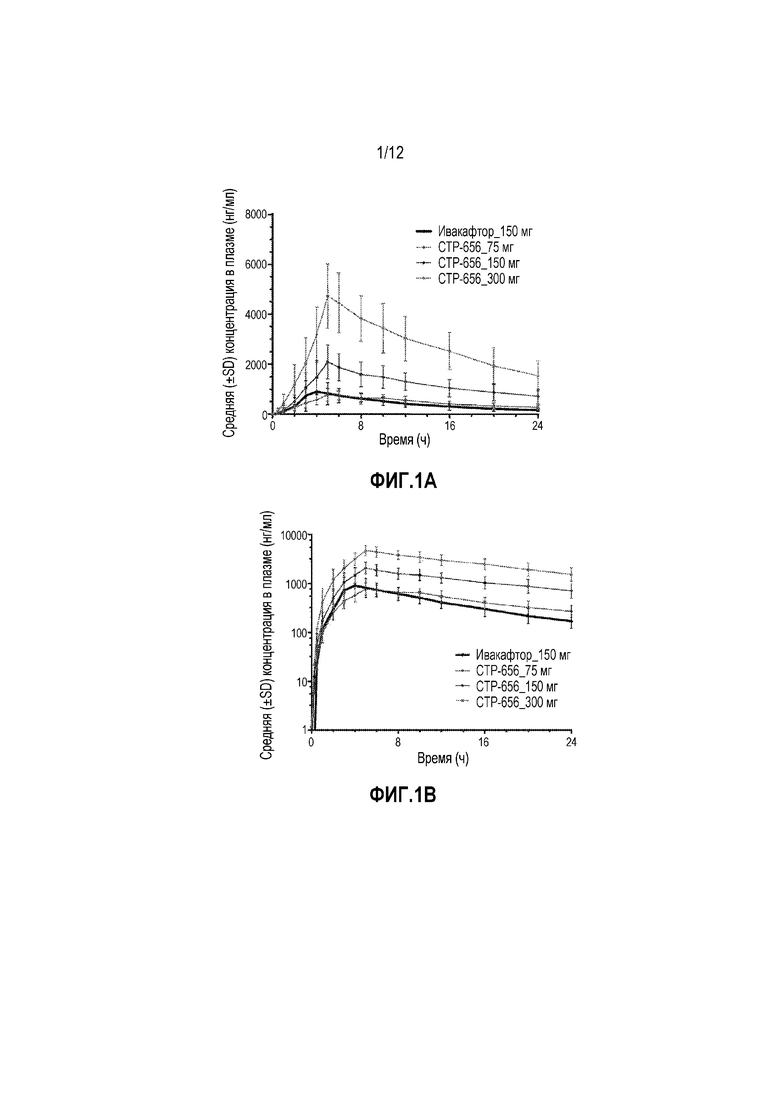
На фиг. 1A изображена средняя концентрация в плазме (нг/мл) для CTP-656 и ивакафтора в исследовании разовой нарастающей дозы.
На фиг. 1B изображена средняя концентрация в плазме (нг/мл) для CTP-656 и ивакафтора в исследовании разовой нарастающей дозы.
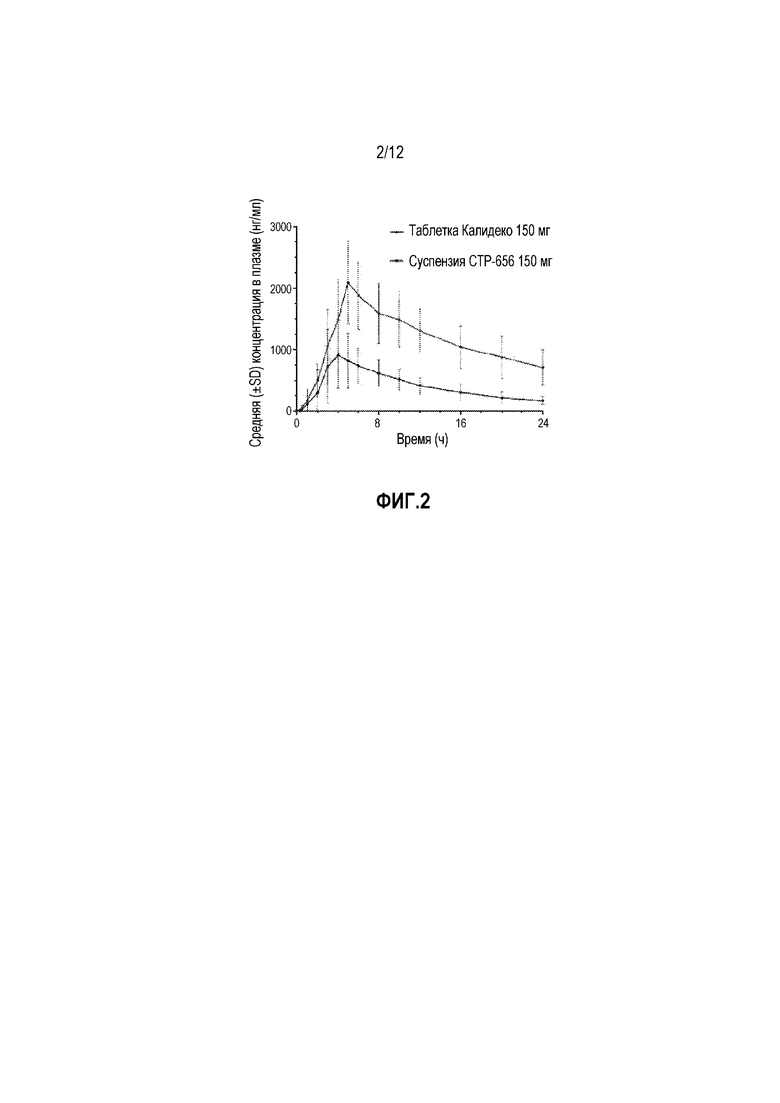
На фиг. 2 изображена средняя концентрация в плазме (нг/мл) для CTP-656 и ивакафтора после пероральной дозы 150 мг.
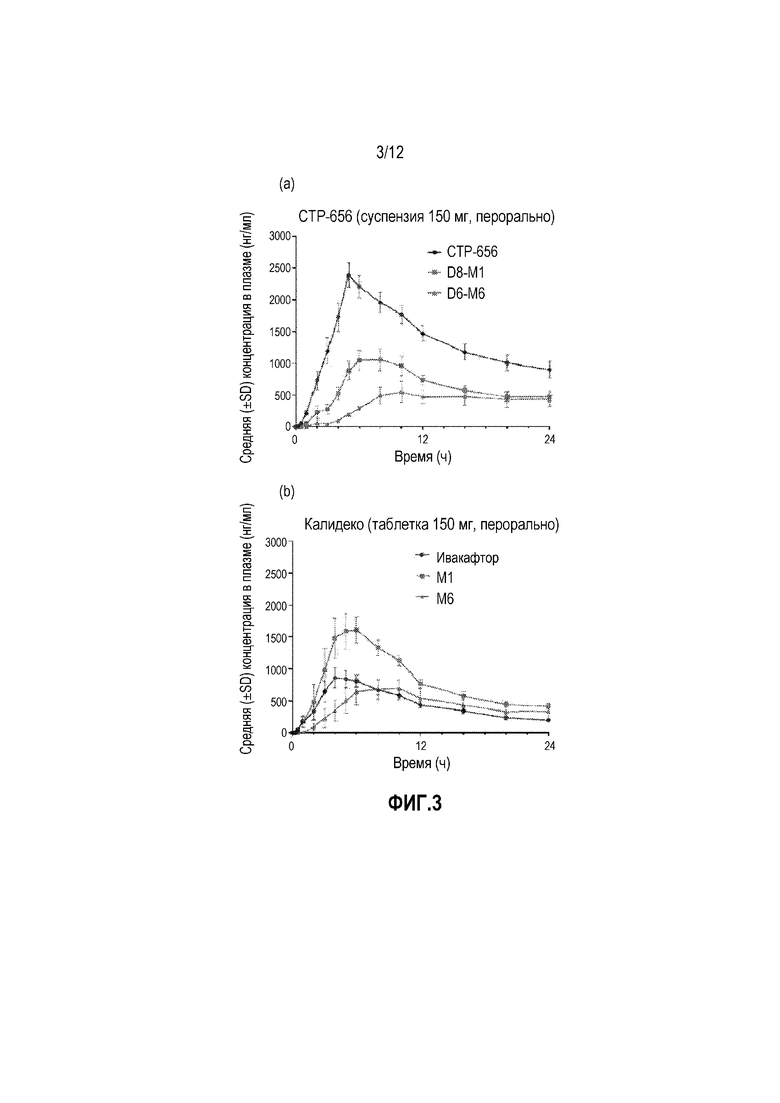
На фиг. 3 изображен фармакокинетический профиль исходного соединения по сравнению с метаболитом для (a) CTP-656 и (b) Ивакафтора (Калидеко) после пероральной дозы 150 мг.
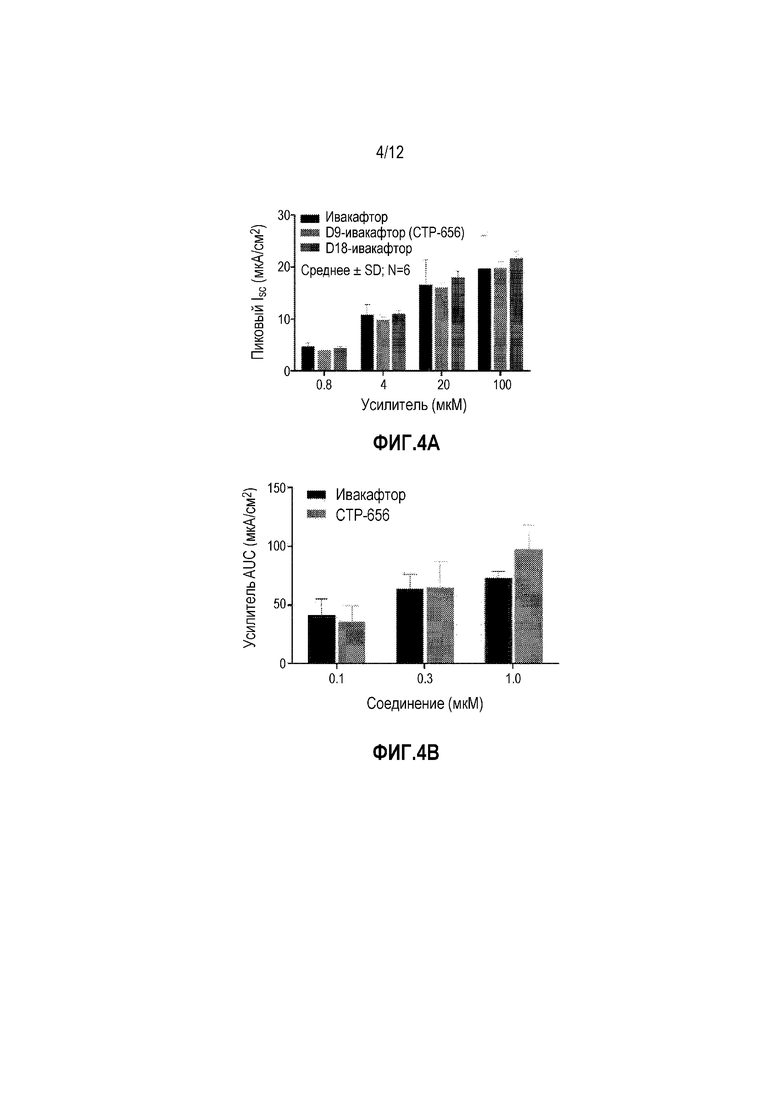
На фиг. 4A изображен пиковый ток, потенциированный последовательными добавлениями тестируемых изделий.
На фиг. 4B изображена AUC ответа усилителя.
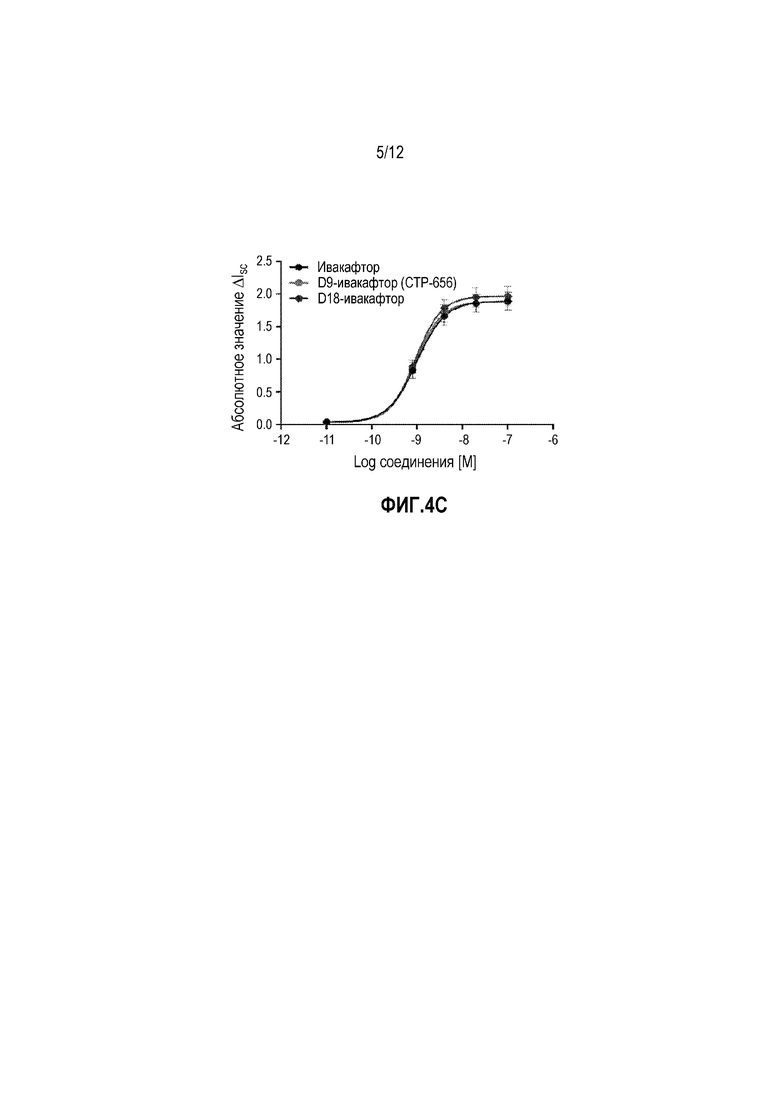
На фиг. 4C изображена ΔISC ответа усилителя для ивакафтора, CTP-656, и D18-ивакафтора.
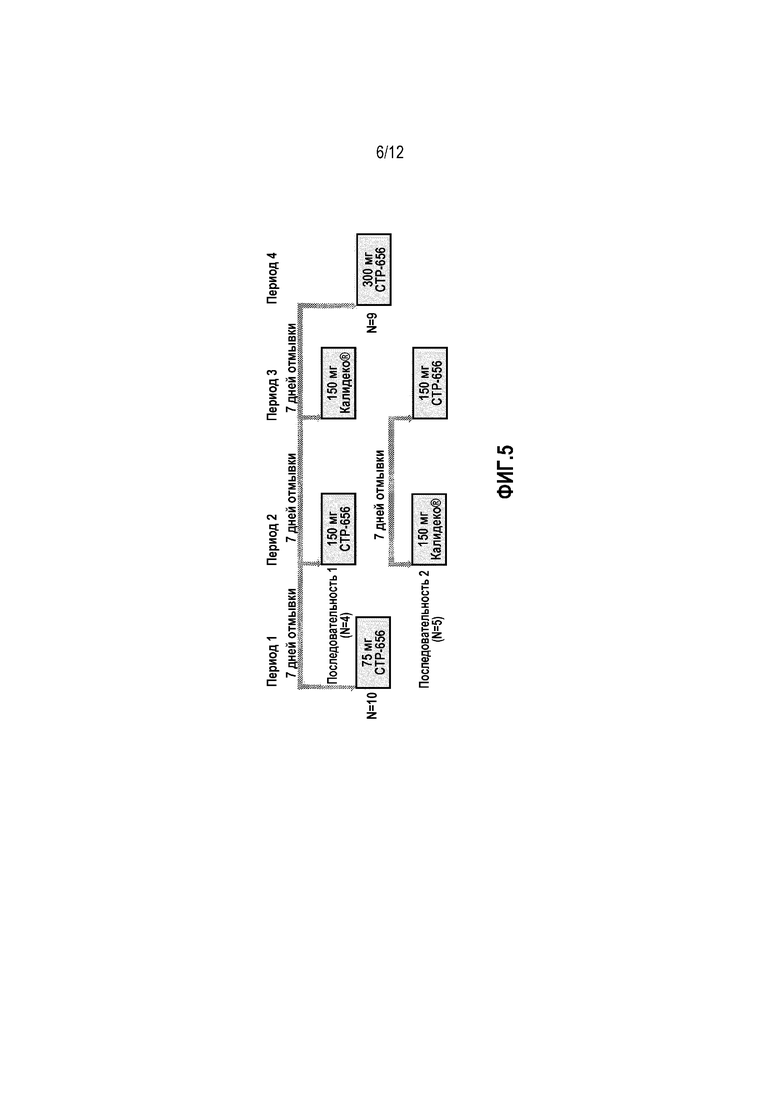
Фиг. 5 представляет собой схему исследования разовой нарастающей дозы.
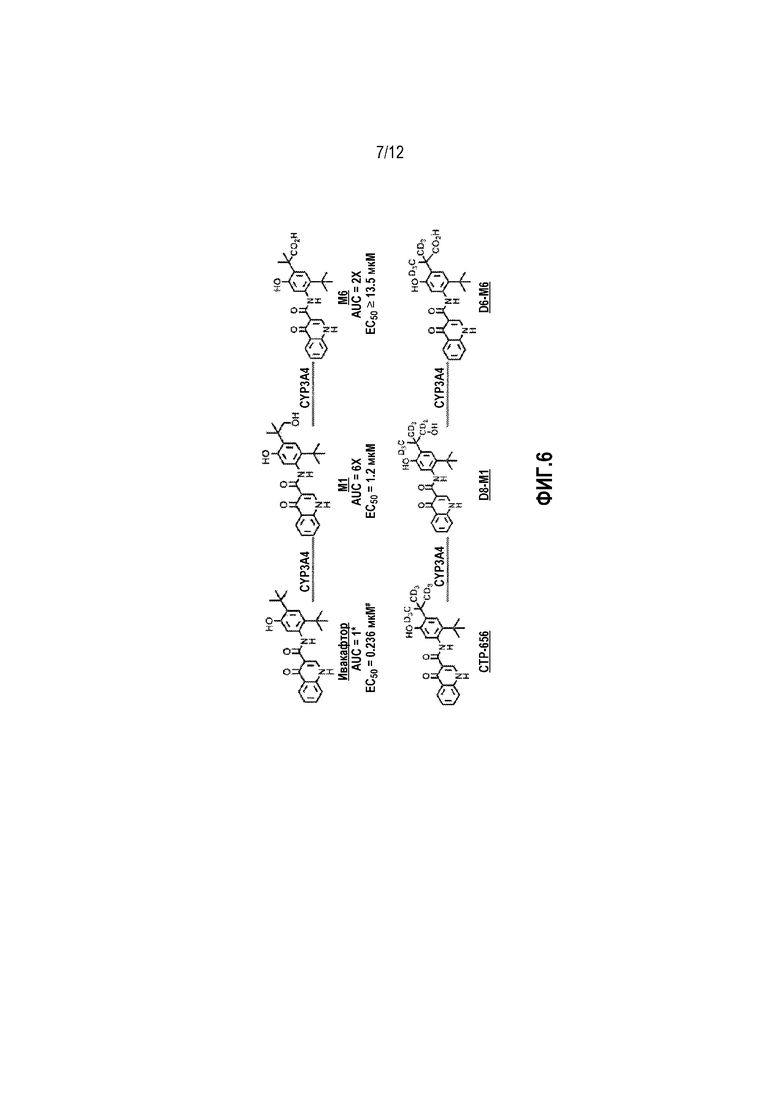
Фиг. 6 представляет собой схему метаболитов ивакафтора и CTP-656.
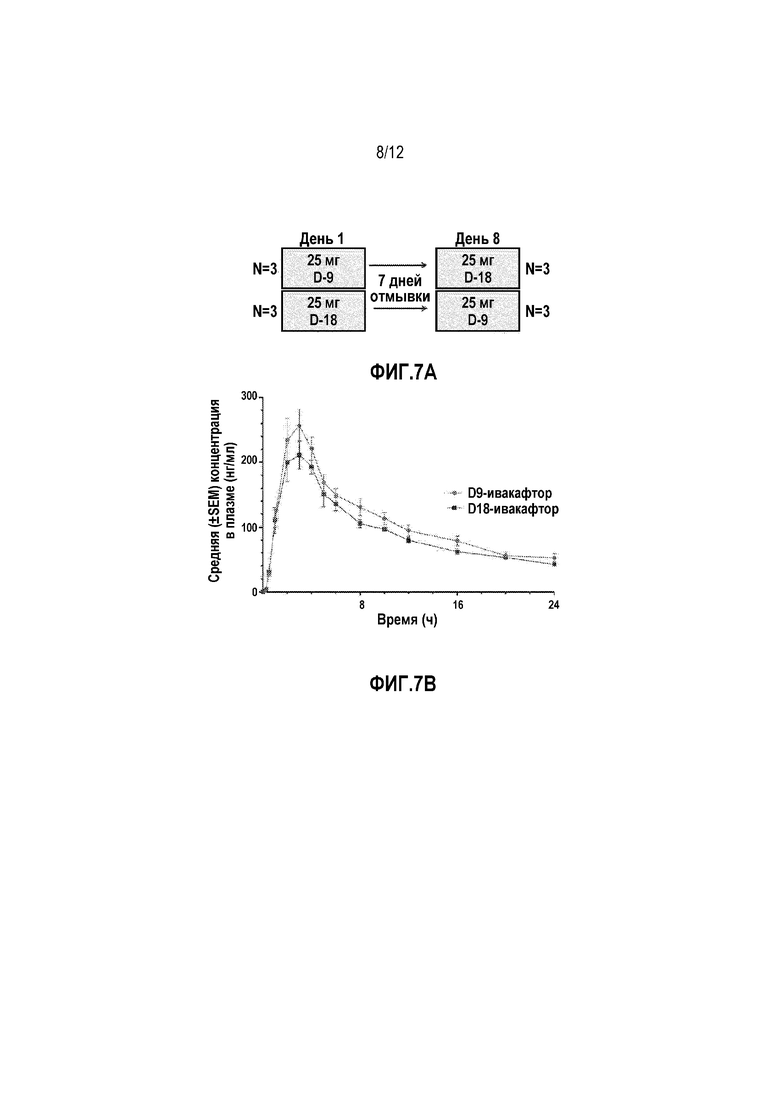
Фиг. 7A представляет собой схему перекрестного исследования для D9-ивакафтора и D18-ивакафтора.
На фиг. 7B изображена средняя концентрация в плазме (нг/мл) для D9-ивакафтора и D18-ивакафтора после пероральной дозы 25 мг.
На фиг. 8 показана схема дизайна исследования множественной нарастающей дозы для CTP-656 (D9-ивакафтор). Часть A: фармакокинетическое сравнение разовой дозы (с перекрестом) 150 мг CTP-656 (2x 75 мг таблетки) по сравнению с 150 мг ивакафтора. Часть B: оценка трех доз CTP-656 (75 мг, 150 мг, и 225 мг плацебо, вводимого один раз в сутки в течение семи дней.
Фиг. 9 представляет собой график, демонстрирующий концентрацию в плазме CTP-656 и ивакафтора после разовой дозы CTP-656 или ивакафтора.
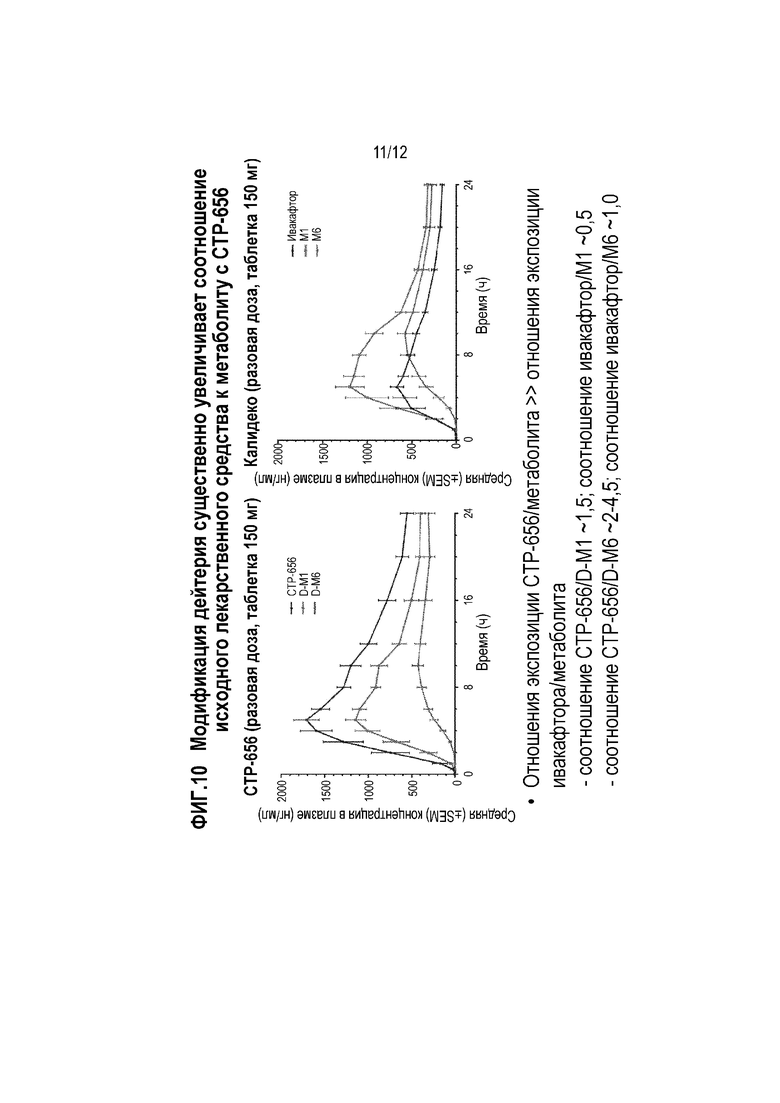
Фиг. 10 представляет собой график, показывающий концентрацию в плазме CTP-656 и метаболитов (левая панель) и график, показывающий концентрацию в плазме ивакафтора и метаболитов (правая панель), после разовой дозы CTP-656 или ивакафтора.
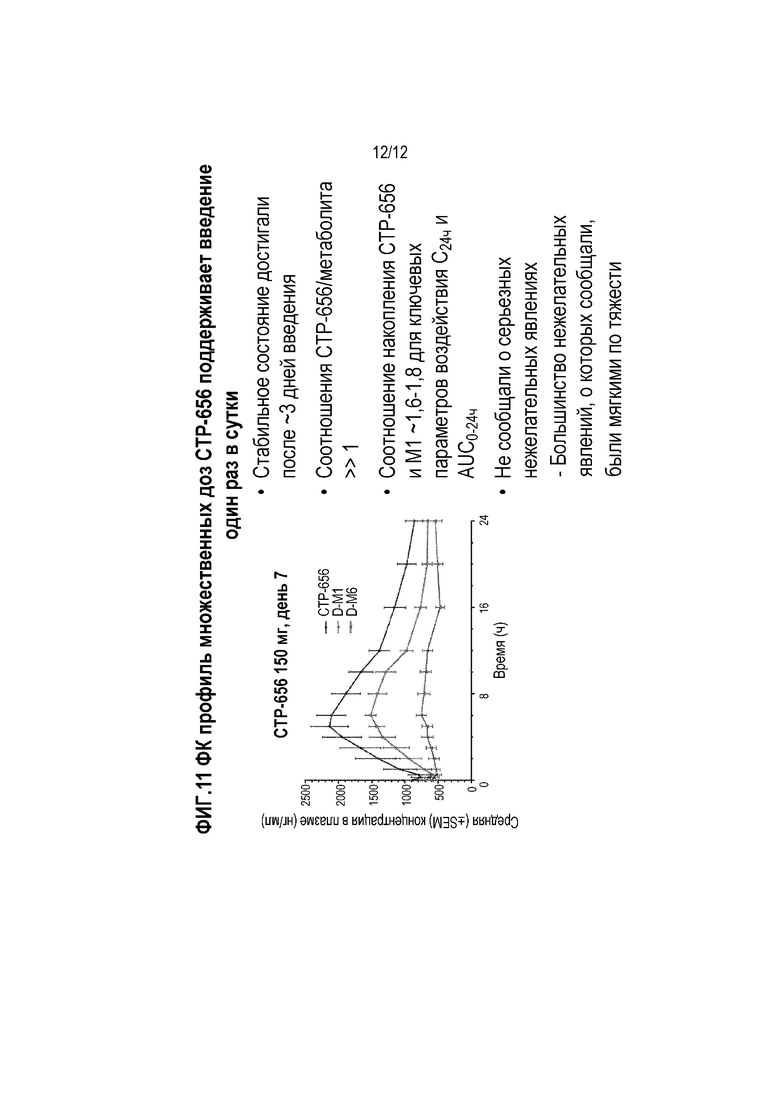
Фиг. 11 представляет собой график, показывающий концентрацию в плазме CTP-656 и метаболитов после многократного введения (один раз в сутки в течение семи дней) CTP-656.
Подробное описание изобретения
Настоящее изобретение в одном варианте осуществления изобретения относится к способам применения Соединения (I) или (II) или его фармацевтически приемлемой соли, включающим определенные схемы введения и определенные фармацевтические композиции, включающие Соединение (I) или (II) или его фармацевтически приемлемую соль. Фармацевтические композиции и схемы введения являются полезными для лечения состояний, опосредованных CFTR (муковисцидозным регулятором трансмембранной проводимости). В частности, Соединение (I) или (II) или его фармацевтически приемлемая соль и фармацевтические композиции и способы являются полезными для лечения состояний, которые могут быть лечены соединениями, которые усиливают активность CFTR.
В одном варианте осуществления изобретение обеспечивает способ для лечения состояний, которые могут быть лечены соединениями, которые усиливают активность CFTR. Способ включает введение пациенту количества Соединения (I) или (II), или его фармацевтически приемлемой соли в диапазоне от 50 мг до 200 мг один раз в сутки. В другом варианте осуществления изобретение обеспечивает способ лечения состояния, которое опосредовано CFTR, у пациента, способ включает введение пациенту композиции, включающей Соединение I:
(I),
или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, где вводимое количество соединения находится в диапазоне от 50 мг/сутки до 200 мг/сутки и композицию вводят один раз в сутки. Специфически в вышеуказанных вариантах осуществления изобретения, например, 50 мг, 60 мг, 70 мг, 80 мг, 90 мг, 100 мг, 110 мг, 120 мг, 130 мг, 140 мг, 150 мг, 160 мг, 170 мг, 180 мг, 190 мг, или 200 мг Соединения I можно вводить один раз в сутки. Более специфически, например, 75, 100, или 150 мг Соединения I можно вводить один раз в сутки. В определенном варианте осуществления изобретения 100-150 мг Соединения I можно вводить один раз в сутки. В определенном варианте осуществления изобретения 100 мг Соединения I можно вводить один раз в сутки. В определенных вариантах осуществления изобретения пациентом является человек. Альтернативно, способ включает введение пациенту количества Соединения (I) или (II), или его фармацевтически приемлемой соли, один раз в сутки, где количество Соединения (I) или (II) или его фармацевтически приемлемой соли находится в диапазоне от 25 мг до 75 мг, например, 25 мг, 37,5 мг, 50 мг, 62,5 мг, или 75 мг, где пациентом является человек от 2 до менее чем 6 лет и менее чем 14 кг; или альтернативно, является человек от 2 до менее чем 6 лет и 14 кг или более. В одном аспекте доза для человека от 2 до менее чем 6 лет и менее чем 14 кг составляет 25 мг. В одном аспекте дозировка для человека от 2 до менее чем 6 лет и более чем 14 кг составляет 37,5 мг.
Состояния, которые лечат способами, описанными в настоящем описании, включают муковисцидоз, врожденную эмфизему, врожденный гемохроматоз, дефициты коагуляции-фибринолиза, такие как дефицит протеина С, врожденный ангиоотек 1 типа, дефициты процессинга липидов, такие как семейная гиперхолестеринемия, хиломикронемия 1 типа, абеталипопротеинемия, заболевания липосомального хранения, такая как I-клеточная болезнь/Псевдо-Гурлер, мукополисахаридоз, Сандхоф/Тай-Сакс, Криглер-Найар типа II, полиэндокринопатия/гиперинсулемия, сахарный диабет, карликовость Ларона, дефицит миелопероксидазы, первичный гипопаратиреоз, меланома, гликаноз CDG типа 1, врожденная эмфизема, врожденный гипотиреоз, несовершенный остеогенез, врожденная гиперфибриногенемия, дефицит ACT, несахарный диабет (DI), нейрофизеальный DI, нефрогенный DI, синдром Шарко-Мари-Тота, болезнь Перлизауса-Мерцбахера, нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз, прогрессирующий надъядерный паралич, болезнь Пика, некоторые полиглютаминовые неврологические расстройства, такие как Хантингтон, спиномозжечковая атаксия типа I, спинальная и бульбарная мышечная атрофия, дентаторубральный паллидопаралич, и миотоническая дистрофия, а также губчатые энцефалопатии, такие как врожденная болезнь Крейтцфельда-Якоба, болезнь Фабри, синдром Штраусслера-Шейнкера, хроническая обструктивная болезнь легких (ХОБЛ), синдром сухого глаза, болезнь Шегрена, и расстройства желчевоводящих путей или расстройства ионных каналов почки, включая, без ограничения, синдром Барттера и болезнь Дента.
В специфическом варианте осуществления изобретения состоянием является муковисцидоз у пациента, такого как человек, нуждающийся в этом. В другом специфическом варианте осуществления изобретения состоянием является хроническая обструктивная болезнь легких у пациента, такого как человек, нуждающийся в этом. В определенных вариантах осуществления изобретения пациентом является человек, имеющий мутацию G551D-CFTR. В определенных вариантах осуществления изобретения пациентом является человек, имеющий одну из следующих мутаций в гене CFTR: G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P или G1349D. В определенных вариантах осуществления изобретения пациентом является человек, имеющий одну из следующих мутаций в гене CFTR: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R и R117H. В другом примере вышеуказанных вариантов осуществления изобретения соединение вводят перорально один раз в сутки.
В вышеуказанных вариантах осуществления изобретения соединение вводят необязательно в комбинации со вторым агентом. В определенных вариантах осуществления изобретения пациентом является человек, имеющий мутацию ΔF508-CFTR. Примеры вторых агентов включают корректоры CFTR, такие как лумакафтор (VX-809) или тезакафтор (VX-661). В некоторых вариантах осуществления, где Соединение (I) или (II) или его фармацевтически приемлемую соль вводят необязательно в комбинации со вторым агентом, количество Соединения (I) или (II) или его фармацевтически приемлемой соли, вводят один раз в сутки от 50 мг до 200 мг каждый раз, например, 50 мг, по 60 мг, 70 мг, 80 мг, 90 мг, 100 мг, 110 мг, 120 мг, 130 мг, 140 мг, 150 мг, 160 мг, 170 мг, 180 мг, 190 мг, или 200 мг. Альтернативно, когда Соединение (I) или (II), или его фармацевтически приемлемую соль вводят необязательно в комбинации со вторым агентом, количество Соединения (I) или (II) или его фармацевтически приемлемой соли вводят один раз в сутки от 25 мг до 75 мг каждый раз, например, 25 мг, 37,5 мг, 50 мг, 62,5 мг, или 75 мг.
Эффективные дозы также варьируются, как понимает специалист в области техники, в зависимости от заболеваний, которые лечат, тяжести заболевания, пути введения, пола, возраста и общего состояния здоровья пациента, использования вспомогательных веществ, возможности совместного применения с другими терапевтическими вариантами лечения, такими как применение других агентов, и мнением лечащего врача.
Для фармацевтических композиций, которые включают второе терапевтическое средство, эффективное количество второго терапевтического средства составляет от около 20% до 100% дозировки, обычно используемой в режиме монотерапии с использованием только такого агента. Предпочтительно, эффективное количество составляет от около 70% до 100% нормальной монотерапевтической дозы. Нормальные монотерапевтические дозы таких вторых терапевтических средств хорошо известны в области техники. См., например, Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), каждая из указанных ссылок включена в настоящее описание в виде ссылки полностью.
Ожидают, что некоторые из вторых терапевтических средств, указанных выше, будут действовать синергически с соединениями по настоящему изобретению. Когда это развивается, это позволит снизить эффективную дозировку второго терапевтического средства и/или Соединения (I) или (II) или его фармацевтически приемлемой соли, от таковой, требуемой в монотерапии. Это имеет преимущества минимизации токсических побочных эффектов или второго терапевтического средства или Соединения (I) или (II) или его фармацевтически приемлемой соли, синергические улучшения эффективности, улучшенной простоты введения или применения и/или сниженной общей стоимости получения или рецептирования соединения.
В другом варианте осуществления изобретения любой из вышеуказанных способов лечения включает дополнительную стадию совместного введения пациенту, нуждающемуся в этом, одного или более вторых терапевтических средств. Выбор второго терапевтического средства может быть сделан из любого второго терапевтического средства, известного как применимый для совместного введения с ивакафтором. Выбор второго терапевтического средства также зависит от определенного заболевания или состояния, которое лечат. Примерами вторых терапевтических средств, которые могут быть использованы в способах по настоящему изобретению, являются таковые, указанные выше для применения в комбинированных композициях, включающих Соединение (I) или (II) или его фармацевтически приемлемую соль, и второе терапевтическое средство.
В частности, комбинированная терапия по настоящему изобретению включает совместное введение Соединения (I) или (II), или его фармацевтически приемлемой соли, и второго терапевтического средства, такого как VX-809 (лумакафтор) или VX-661 (тезакафтор), пациенту, нуждающемуся в этом для лечения. В определенных вариантах осуществления изобретения пациентом является человек, имеющий ΔF508-CFTR мутацию (в частности, человек, гомозиготный по мутации F508del).
Термин ʺсовместное введениеʺ, как используется в настоящем описании, обозначает, что второе терапевтическое средство можно вводить вместе с Соединением (I) или (II), или его фармацевтически приемлемой солью, в качестве части стандартной лекарственной формы (такой как композиция по настоящему изобретению, включающая соединение по изобретению и второе терапевтическое средство, как описано выше) или в виде отдельных, множественных лекарственных форм. Альтернативно, дополнительное средство можно вводить до, последовательно с или после введения Соединения (I) или (II), или его фармацевтически приемлемой соли. В таком лечении комбинированной терапией любое Соединение (I) или (II) или его фармацевтически приемлемую соль и второе терапевтическое средство(а) вводят обычными способами. Введение пациенту композиции по настоящему изобретению, включающей и Соединение (I) или (II) или его фармацевтически приемлемую соль, и второе терапевтическое средство, не мешает отдельному введению такого же терапевтического средства или любого другого второго терапевтического средства или Соединения (I) или (II) или его фармацевтически приемлемой соли указанному пациенту в другое время в течение курса лечения.
Эффективные количества таких вторых терапевтических средств хорошо известны специалистам в области техники и руководства для введения могут быть обнаружены в патентах и опубликованных патентных заявках, на которые ссылаются в настоящем описании, а также в Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), и других медицинских текстах. Однако в рамках знаний специалиста в области техники определить оптимальный диапазон эффективного количества второго терапевтического средства.
В одном варианте осуществления изобретения эффективное количество второго терапевтического средства будет меньше, чем его эффективное количество, когда Соединение (I) или (II) или его фармацевтически приемлемую соль не вводят. Таким образом, нежелательные побочные эффекты, ассоциированные с высокими дозами каждого агента, могут быть минимизированы. Другие потенциальные преимущества (включая без ограничения улучшенные схема введения и/или сниженную стоимость лекарственного средства) будут очевидны специалисту в области техники.
В еще одном аспекте изобретение обеспечивает применение Соединения (I) или (II) или его фармацевтически приемлемой соли отдельно или вместе с одним или более из вышеописанных вторых терапевтических средств в получении лекарственного препарата, или в виде разовой композиции или в виде отдельных лекарственных форм, для лечения или профилактики у пациента заболевания, расстройства или симптома, указанного выше. Другим аспектом изобретения является Соединение (I) или (II) или его фармацевтически приемлемая соль для применения в лечении или профилактике у пациента заболевания, расстройства или его симптома, обозначенного в настоящем описании.
В одном варианте осуществления изобретения любой атом, не обозначенный как дейтерий, присутствует в его натуральной изотопной обогащенности в Соединении (I) или (II) или его фармацевтически приемлемой соли.
Синтез Соединения (I) или (II) или его фармацевтически приемлемой соли может быть легко осуществлен методами, описанными в патенте США No. 8865902, содержание которого включено в настоящее описание в виде ссылки. В частности, в примере 3 патента США No. 8865902 описан синтез Соединения (I) и в примере 4 описан синтез Соединения (II).
Такие способы могут быть осуществлены с использованием соответствующих дейтерированных и необязательно содержащих другие изотопы реагентов и/или промежуточных продуктов для синтеза соединений, обозначенных в настоящем описании, или используя стандартные синтетические протоколы, известные в области техники для внесения изотопных атомов в химическую структуру.
Изобретение также обеспечивает фармацевтические композиции, включающие эффективное количество Соединения (I) или (II) или его фармацевтически приемлемой соли; и фармацевтически приемлемый носитель. Носитель(и) является ʺприемлемым(и)ʺ в смысле совместимости с другими ингредиентами композиции и в случае фармацевтически приемлемого носителя, не вредным для реципиента в количестве, используемом в лекарственном препарате.
Фармацевтически приемлемые носители, добавки и основы, которые могут быть использованы в фармацевтических композициях по настоящему изобретению, включают, без ограничения, ион обменники, квасцы, стеарат алюминия, лецитин, белки сыворотки, такие как человеческий альбумин сыворотки, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота, сорбат калия, смеси частичных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, гидрофосфат динатрия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, карбоксиметилцеллюлоза натрия, полиакрилаты, воска, блокполимеры поли этилена-полиоксипропилена, полиэтиленгликоль и ланолин.
Если требуется, растворимость и биодоступность соединений по настоящему изобретению в фармацевтических композициях может быть усилена способами, хорошо известными в области техники. Один способ включает применение жидких вспомогательных веществ в композиции. См. ʺOral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs (Drugs and the Pharmaceutical Sciences),ʺ David J. Hauss, ed. Informa Healthcare, 2007; и ʺRole of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery: Basic Principles and Biological Examples,ʺ Kishor M. Wasan, ed. Wiley-Interscience, 2006.
Другим известным способом увеличения биодоступности является применение аморфной формы соединения по настоящему изобретению, необязательно рецептированного с полоксамером, таким как LUTROLTM и PLURONICTM (BASF Corporation), или блоксополимеров этиленоксида и пропиленоксида. См. Патент Соединенных Штатов 7014866; и патентные публикации Соединенных Штатов 20060094744 и 20060079502.
Фармацевтические композиции по изобретению включают таковые, подходящие для перорального введения. Другие композиции могут в общем быть представлены в стандартной лекарственной форме, например, таблетках, капсулах замедленного высвобождения, гранулах и в липосомах, и могут быть получены любыми способами, хорошо известными в области фармации. См., например, Remington: The Science and Practice of Pharmacy, Lippincott Williams & Wilkins, Baltimore, MD (20th ed. 2000).
Такие способы получения включают стадию ассоциации с молекулой, которую вводят, ингредиентов, таких как носитель, который содержит один или более дополнительных ингредиентов. В общем, композиции получают путем однородного и тесного взаимодействия активных ингредиентов с жидкими носителями или мелко измельченными твердыми носителями, или обоими, и затем, если необходимо, формования продукта.
В определенных вариантах осуществления изобретения соединение вводят перорально. Композиции по настоящему изобретению, подходящие для перорального введения, могут быть представлены в виде отдельных единиц, таких как капсулы, саше или таблетки, каждая содержащая заранее определенное количество активного ингредиента; порошка или гранул; раствора или суспензии в водной жидкости или неводной жидкости; эмульсии типа масло-в-воде; жидкой эмульсии типа вода-в-масле; упакованной в липосомы; или в виде болюса и др. Мягкие желатиновые капсулы могут быть полезными для содержания таких суспензий, которые могут преимущественным образом увеличивать скорость абсорбции соединения. В специфическом варианте осуществления изобретения соединение вводят перорально в виде таблетки. Альтернативно соединение вводят перорально в виде гранул.
В случае таблеток для перорального применения носители, которые обычно используют, включают лактозу и кукурузный крахмал. Смазывающие агенты, такие как стеарат магния, также обычно добавляют. Для перорального введения в форме капсул, полезные разбавители включают лактозу и высушенный кукурузный крахмал. Когда водные суспензии вводят перорально, активный ингредиент комбинируют с эмульгирующим и суспендирующими агентами. При желании, могут быть добавлены определенные подсластители и/или ароматизаторы и/или красители. В другом варианте осуществления изобретения композиция находится в форме таблеток. В определенных вариантах осуществления изобретения примерные композиции для таблеток описаны в патенте США No. 8754224, содержание которого таким образом включено в виде ссылки.
В определенном варианте осуществления изобретения таблетка, содержащая 150 мг Соединения (I) или (II), или его фармацевтически приемлемой соли, и следующие неактивные ингредиенты: коллоидный диоксид кремния, кроскармеллоза натрия, ацетатсукцинат гипромеллозы, моногидрат лактозы, стеарат магния, микрокристаллическая целлюлоза, и лаурилсульфат натрия. В определенных вариантах осуществления изобретения пленочная оболочка таблетки содержит карнаубский воск, FD&C синий #2, PEG 3350, поливиниловый спирт, тальк, и диоксид титана. В определенных вариантах осуществления изобретения таблетку печатают с помощью печатных чернил; печатные чернила могут содержать гидроксид аммония, черный оксид железа, пропиленгликоль и шеллак. В другом определенном варианте осуществления изобретения таблетка содержит 75 мг Соединения I (CTP-656, D9-ивакафтор), вместе со следующими неактивными ингредиентами: микрокристаллическая целлюлоза, моногидрат лактозы, коллоидный диоксид кремния, кроскармеллоза, стеарат магния и лаурилсульфат натрия. Множество таблеток можно вводить для обеспечения подходящей суточной дозы (например, две таблетки по 75 мг, вводимые вместе для 150 мг суточной дозы). В другом определенном варианте осуществления изобретения, таблетка включает гранулы, прессованные с экстрагранулярным материалом; гранулы включают около 17,1 процентов (по массе таблетки) аморфной дисперсии Соединения I (где аморфная дисперсия включает около 80% по существу аморфного Соединения I по массе дисперсии, ацетат сукцинат гипромеллозы (HPMCAS) (около 19,5 процентов по массе дисперсии) и лаурелсульфат натрия (около 0,5 процентов по массе дисперсии)), компрессионную добавку, такую как микрокристаллическая целлюлоза (например, Avicel PH101) (около 39,0 процентов по массе таблетки), разбавитель/наполнитель, такой как моногидрат лактозы 316 (около 38,9 процентов по массе таблетки) поверхностно-активное вещество, такое как лаурилсульфат натрия (SLS) (около 0,50 процентов по массе таблетки), дезинтегрирующее вещество, такое как кроскармеллоза натрия (например, Ac-di-sol) (около 1,50 процентов по массе таблетки) и смазывающее вещество, такое как стеарат магния Hyqual (около 0,20 процентов по массе таблетки), и внеклеточная матрица включает дезинтегрирующее вещество, такое как кроскармеллоза натрия (например, Ac-di-sol) (около 1,50 процентов по массе таблетки), глидант, такой как коллоидный диоксид кремния (около 0,50 процентов по массе таблетки) и дополнительное смазывающее вещество, такое как стеарат магния (например, Hyqual (около 0,80 процентов по массе таблетки). Следовательно, в одном варианте осуществления изобретение обеспечивают фармацевтическую композицию, включающую около 17,1 масс% твердой дисперсии по массе композиции, где дисперсия включает около 80 масс% по существу аморфного Соединения I (CTP-656) по массе дисперсии, около 19,5 масс% ацетатсукцината гипромеллозы (HPMCAS) по массе дисперсии, и около 0,5 масс% SLS по массе дисперсии; около 39,0 масс% микрокристаллической целлюлозы по массе композиции; около 38,9 масс% моногидрата лактозы по массе композиции; около 3 масс% кроскармеллозы натрия по массе композиции; около 0,5 масс% SLS по массе композиции; около 0,5 масс% коллоидного диоксида кремния по массе композиции; и около 0,8 масс% стеарата магния по массе композиции. В определенных вариантах осуществления изобретения таблетка включает 75 мг Соединения I (CTP-656). В других вариантах осуществления изобретения таблетка включает 100 мг Соединения I (CTP-656). В еще других вариантах осуществления изобретения таблетка включает 150 мг Соединения I (CTP-656).
В другом определенном варианте осуществления изобретения гранулу заключают в стандартный пакет, содержащий 25 мг, 50 мг или 75 мг Соединения (I) или (II), или его фармацевтически приемлемой соли. Каждый стандартный однодозовый пакет пероральных гранул Соединения (I) или (II) содержит 25 мг Соединения (I) или (II), 50 мг Соединения (I) или (II) или 75 мг Соединения (I) или (II) и следующие неактивные ингредиенты: коллоидный диоксид кремния, кроскармеллозу натрия, ацетатсукцинат гипромеллозы, моногидрат лактозы, стеарат магния, маннит, сукралозу, и лаурилсульфат натрия. В другом варианте осуществления изобретения композиция находится в форме гранул. В определенных вариантах осуществления изобретения примерные композиции для гранул описаны в патенте США No. 8883206, содержание которого включено в настоящее описание в виде ссылки.
В другом варианте осуществления изобретения композиция по настоящему изобретению дополнительно включает второе терапевтическое средство. Второе терапевтическое средство может быть выбрано из любого соединения или терапевтического средства, известного как имеющее или которое демонстрирует преимущественные свойства при введении с соединением, имеющим такой же механизм действия, как ивакафтор.
Предпочтительно, вторым терапевтическим средством является средство, применимое в лечении множества состояний, включая муковисцидоз, врожденную эмфизему, врожденный гемохроматоз, дефициты коагуляции-фибринолиза, такие как дефицит протеина С, врожденный ангиоотек типа 1, дефициты процессинга липидов, такие как семейная гиперхолестеринемия, хиломикронемия типа 1, абеталипопротеинемия, заболевания хранения лизосом, такие как I-клеточное заболевание/Псевдо-Гурлер, мукополисахаридозы, Сандхоф/Тай-Сакс, Криглер-Найяр типа II, полиэндокринопатия/гиперинсулинемия, сахарный диабет, карликовость Ларона, дефицит миелопероксидазы, первичный гипопаратиреоз, меланома, гликаноз CDG типа 1, врожденная эмфизема, врожденный гипертиреоз, несовершенный остеогенез, врожденная гиперфибриногенемия, ACT дефицит, несахарный диабет (DI), нейрофизеальный DI, нефрогенный DI, синдром Шарко-Мари-Тута, болезнь Перлиза-Мерцбахера, нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз, прогрессирующий надъядерный паралич, болезнь Пика, некоторые полиглютаминовые неврологические расстройства, такие как Хантингтон, спиноцеребеллярная атаксия типа I, спинальная и бульбарная мышечная атрофия, дентаторубральная паллидо-льюисова, и миотоническая дистрофия, а также губчатые энцефалопатии, такие как врожденная болезнь Крейтцфельда-Якоба, болезнь Фабри, синдром Штраусслера-Шейнкера, ХОБЛ, болезнь сухого глаза, болезнь Шегрена, и заболевание желчных путей или заболевания ионных каналов почки, включая, без ограничения, синдром Барттера и болезнь Дента.
В специфическом варианте осуществления изобретения вторым терапевтическим средством является средство, полезное в лечении муковисцидоза. В определенных вариантах осуществления изобретения вторым терапевтическим средством является средство применимое в лечении муковисцидоза у человека, имеющего G551D-CFTR мутацию. В определенных вариантах осуществления изобретения вторым терапевтическим средством является средство, применимое в лечении муковисцидоза у человека, имеющего любую из следующих мутаций в гене CFTR: G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P или G1349D. В определенных вариантах осуществления изобретения вторым терапевтическим средством является средство, применимое в лечении муковисцидоза у человека, имеющего любую из следующих мутаций в гене CFTR: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R и R117H.
В одном варианте осуществления изобретения вторым терапевтическим средством является VX-809 (лумакафтор) или VX-661 (тезакафтор). В определенных вариантах осуществления изобретения пациентом является человек, имеющий мутацию ΔF508-CFTR (в частности, человек, гомозиготный по мутации F508del).
В другом варианте осуществления изобретение обеспечивает отдельные лекарственные формы Соединения (I) или (II), или его фармацевтически приемлемой соли, и одно или более из любых из вышеописанных вторых терапевтических средств, где Соединение (I) или (II), или его фармацевтически приемлемая соль, и второе терапевтическое средство ассоциированы друг с другом. Термин ʺассоциированный друг с другомʺ, как используется в настоящем описании, обозначает, что различные лекарственные формы упаковывают вместе или они иным образом прикреплены друг к другу, так что очевидно, что отдельные лекарственные формы предназначены для продажи и введения вместе (в течение менее чем 24 часов один от другого, последовательно или одновременно).
В фармацевтических композициях по изобретению Соединение (I) или (II), или его фармацевтически приемлемая соль, присутствует в эффективном количестве. Как используется в настоящем описании, ʺэффективное количествоʺ относится к количеству, которое при введении по соответствующей схеме введения, достаточно для лечения целевого расстройства.
Изобретение также обеспечивает продукт, который включает a) фармацевтическую композицию, включающую Соединение I или II, или его соль, в количестве в диапазоне от 50 мг до 200 мг; и b) предписывающую информацию для введения Соединения I или II, или его соли. Предписывающая информация включает инструкции для введения от 50 мг до 200 мг Соединения I или II или его соли, один раз в сутки пациенту, нуждающемуся в таком лечении, например, пациенту, страдающему от или предрасположенному к состоянию, которое опосредовано CFTR, такому как муковисцидоз.
Взаимоотношения дозировок для животных и людей (на основании миллиграмм на метр квадратный поверхности тела) описаны в Freireich et al., Cancer Chemother. Rep, 1966, 50: 219. Площадь поверхности тела может быть приблизительно определена из роста и массы тела пациента. См. например, Scientific Tables, Geigy Pharmaceuticals, Ardsley, N.Y., 1970, 537.
Определения
Термин ʺлечитьʺ обозначает снижение, подавление, облегчение, улучшение, остановку или стабилизацию развития или прогрессирования заболевания (например, заболевания или расстройства, указанного в настоящем описании), уменьшение тяжести заболевания или улучшение симптомов, ассоциированных с заболеванием.
ʺЗаболеваниеʺ обозначает любое состояние или расстройство, которое повреждает или вмешивается в нормальную функцию клетки, ткани или органа.
Понимают, что некоторые вариации натуральной изотопной обогащенности появляются в синтезированном соединении в зависимости от происхождения химических материалов, используемых в синтезе. Следовательно, получение ивакафтора по природе содержит небольшие количества дейтерированных изотопологов. Концентрация натурально обогащенных стабильных изотопов водорода и углерода, несмотря на такие вариации, является небольшой и нематериальной по сравнению со степенью стабильного изотопного замещения соединений по настоящему изобретению. См., например, Wada, E et al., Seikagaku, 1994, 66:15; Gannes, LZ et al., Comp Biochem Physiol Mol Integr Physiol, 1998, 119:725.
В Соединении (I) или (II) любой атом, специфически не обозначенный как определенный изотоп, представляет собой любой стабильный изотоп такого атома. Если не указано иначе, когда положение определяется специфически как ʺHʺ или ʺводородʺ, положение понимают, как имеющее водород в его натуральной изотопной обогащенной композиции. Также если не указано иначе, когда положение определяют, как ʺDʺ или ʺдейтерийʺ, положение понимают, как имеющее дейтерий в обогащении, которое составляет по меньшей мере в 3000 раз более чем натуральное обогащение дейтерием, которое составляет 0,015% (т.е. по меньшей мере 45% включения дейтерия).
Термин ʺфактор изотопного обогащенияʺ, как используется в настоящем описании, обозначает соотношение между изотопным обогащением и натуральным обогащением специфическим изотопом.
В других вариантах осуществления изобретения соединение по настоящему изобретению (т.е., Соединение I или Соединение II) имеет фактор изотопного обогащения для каждого обозначенного атома дейтерия по меньшей мере 3500 (52,5% включения дейтерия в каждом обозначенном атоме дейтерия), по меньшей мере 4000 (60% включения дейтерия), по меньшей мере 4500 (67,5% включения дейтерия), по меньшей мере 5000 (75% дейтерия), по меньшей мере 5500 (82,5% включения дейтерия), по меньшей мере 6000 (90% включения дейтерия), по меньшей мере 6333,3 (95% включения дейтерия), по меньшей мере 6466,7 (97% включения дейтерия), по меньшей мере 6600 (99% включения дейтерия), или по меньшей мере 6633,3 (99,5% включения дейтерия).
Термин ʺизотопологʺ относится к видам, в которых химическая структура отличается от Соединения (I) или (II) только по изотопному составу.
Термин ʺсоединениеʺ, когда относится к соединению по настоящему изобретению, относится к группе молекул, имеющих идентичную химическую структуру, за исключением того, что могут быть изотопные варианты среди составляющих атомов молекулы. Следовательно, очевидно специалисту в области техники, что соединение, представленное определенной химической структурой, содержащей указанные атомы дейтерия, также содержит меньше изотопологов, имеющих атомы водорода в одном или более обозначенных положениях дейтерия в такой структуре. Относительное количество таких изотопологов в соединении по настоящему изобретению будет зависеть от ряда факторов, включающих изотопную чистоту дейтерированных реагентов, используемых для получения соединения и эффективности включения дейтерия в различные стадии синтеза, используемые для получения соединения.
Изобретение также обеспечивает соли соединения (I) или (II). Соль соединения по настоящему изобретению получают между кислой и основной группой соединения, такой как аминофункциональная группа, или основной и кислой группой соединения, такой как карбоксильная функциональная группа. В соответствии с другим вариантом осуществления изобретения соединение является фармацевтически приемлемой аддитивной солью кислоты.
Термин ʺфармацевтически приемлемыйʺ, как используется в настоящем описании, относится к компоненту, который является, в рамках озвученного медицинского суждения, подходящим для применения в контакте с тканями человека и других млекопитающих без нежелательной токсичности, раздражения, аллергического ответа и подобного, и соответствует приемлемому соотношению польза/риск. ʺФармацевтически приемлемая сольʺ обозначает любую нетоксичную соль, которая, при введении реципиенту, способна обеспечивать или прямо или опосредованно, соединение по настоящему изобретению. ʺФармацевтически приемлемый противоионʺ представляет собой ионную часть, которая не является токсичной при высвобождении из соли при введении реципиенту.
Кислоты, обычно используемые для получения фармацевтически приемлемых солей, включают неорганические кислоты, такие как бисульфит водорода, соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота и фосфорная кислота, а также органические кислоты, такие как пара-толуолсульфоновая кислота, салициловая кислота, виннокаменная кислота, бивиннокаменная кислота, аскорбиновая кислота, малеиновая кислота, безиловая кислота, фумаровая кислота, глюконовая кислота, глюкуроновая кислота, муравьиная кислота, глютамовая кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, молочная кислота, щавелевая кислота, пара-бромфенилсульфоновая кислота, карбоновая кислота, янтарная кислота, лимонная кислота, бензойная кислота и уксусная кислота, а также связанные неорганические и органические кислоты. Такие фармацевтически приемлемые соли, следовательно, включают сульфат, бисульфит, фосфат, моногидрофосфат, дигидрофосфат, метанфосфат, пирофосфат, хлорид, бромид, йодид, ацетат, пропионат, деканоат, каприлат, акрилат, формат, изобутират, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, бутин-1,4-диоат, гексин-l,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, терфталат, сульфонат, ксилен сульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, ?-гидроксибутират, гликолат, малеат, тартрат, метансульфонат, пропансульфонат, нафталан-1-сульфонат, нафталан-2-сульфонат, манделат и другие соли. В одном варианте осуществления изобретения фармацевтически приемлемые аддитивные соли кислоты включают таковые, образованные с минеральными кислотами, такими как соляная кислота и бромистоводородная кислота, и особенно таковые, полученные с органическими кислотами, такими как малеиновая кислота.
Термин ʺстабильные соединенияʺ, как используется в настоящем описании, относится к соединениям, которые обладают стабильностью, достаточной для возможности их получения, и которые сохраняют целостность соединения в течение достаточного периода времени, чтобы быть применимыми для целей, перечисленных в настоящем описании (например, рецептирование в терапевтические продукты, промежуточные соединения для применения в получении терапевтических соединений, лечение заболевания или состояния, отвечающего на терапевтические средства).
Термин ʺбиоэквивалентʺ, как используется в настоящем описании, обозначает лекарственный продукт, демонстрирующий отсутствие значительных различий в скорости и степени, с которой активный ингредиент или активный фрагмент в фармацевтическом эквиваленте лекарственного продукта становится доступным в месте действия лекарственного средства при введении в той же молярной дозе в сходных условиях в соответствующим образом спланированном исследовании, где ʺдостоверные различияʺ обозначают, что 90% доверительные интервалы (ДИ) тестируемого лекарственного продукта должны соответствовать 80%-125% контрольного лекарственного продукта (см. онлайн тренировочный семинар: ʺThe FDA Process for Approving Generic Drugsʺ; www.fda.gov/Training/For HealthProfessionals/ucm090320.htm). The Food and Drug Administration (FDA) выпустила руководства, касающиеся биоэквивалентных лекарственных продуктов, включающие специфические рекомендации по переносимой вариации неактивных ингредиентов в лекарственном продукте, который вероятно вводит его в фармацевтически эквивалентную форму. См., например, FDA's Guidance for Industry: Submission of Summary Bioequivalence Data for ANDAs от мая 2011, полное содержание которого включено в настоящее описание.
ʺDʺ и ʺdʺ оба относятся к дейтерию. ʺСтереоизомерʺ относится к обоим энантиомерам и диастереомерам. ʺТретʺ и ʺт-ʺ каждый относится к третичному. ʺСШАʺ относится к Соединенным Штатам Америки.
ʺЗамещенный дейтериемʺ относится к замене одного или более атомов водорода соответствующим количеством атомов дейтерия.
Примеры
Пример 1. Клиническое исследование разовой нарастающей дозы (SAD) 1 фазы
Десять здоровых волонтеров мужчин и женщин включали в исследование разовой нарастающей дозы из 3 доз CTP-656 (75, 150 и 300 мг), с перекрестным сравнением 150 мг CTP-656 и 150 мг Калидеко® (торговое наименование ивакафтора) (фиг. 5). Дозы CTP-656 вводили в водной суспензии и Калидеко вводили в виде таблетки. Все дозы CTP-656 и Калидеко вводили в течение 30 минут после начала завтрака, содержащего большое количество жира. Между дозами был период отмывки 7 дней. Задачей настоящего исследования было сравнить фармакокинетику разовых нарастающих доз (75, 150, и 300 мг) CTP-656, сравнить фармакокинетику разовой дозы 150 мг CTP-656 и 150 мг Калидеко и оценить безопасность и переносимость CTP-656.
В общем, CTP-656 вводили в виде разовой дозы в дозах 75 мг, 150 мг и 300 мг (после приема высокожировой пищи) обычно хорошо переносился у здоровых мужчин и женщин. Таблетки Калидеко®, вводимые в виде разовой пероральной дозы 150 мг (после приема высокожировой пищи) также обычно хорошо переносились здоровыми мужчинами и женщинами.
Не наблюдали клинически достоверных различий в показателях безопасности между CTP-656 (75 мг, 150 мг или 300 мг) и Калидеко® (150 мг) и не было очевидных связанных с дозой тенденций у пациентов после CTP-656.
Tmax для CTP-656 было сходным между каждым из вариантов лечения, с очевидным терминальным периодом полужизни 14-17 часов. Очевидный терминальный период полужизни 150 мг Калидеко® (11,18 часов) был короче, чем для CTP-656.
Для CTP-656, взаимоотношения между дозой и воздействием были приблизительно линейными с использованием log-log регрессионного анализа с увеличением воздействия более, чем доза пропорционально уровню дозы (75 мг, 150 мг и 300 мг) (фиг. 1a и 1b).
Резюме фармакокинетических свойств CTP-656 и Калидеко представлено в таблице ниже:
150 мг
300 мг
a Среднее (диапазон) для 150 мг CTP-656, Cmax была приблизительно в 2 раза, AUC0-inf была приблизительно в 3,5-раза, и C24hr составила в 4,2 раза чем таблетки Калидеко®. CL/F (клиренс относительно биодоступности, оценка клиренса лекарственного средства, вводимого перорально) для CTP-656 составила приблизительно 30% таковой, как Калидеко® (фиг. 2).
Сравнение фармакокинетических свойств CTP-656 и Калидеко для дозы 150 представлено в таблице ниже:
а диапазон
Следовательно, на основании улучшенного ФК профиля для CTP-656 относительно Калидеко®, CTP-656 обладает потенциалом проявлять эффективность в дозировках в диапазоне 50-200 мг QD (один раз в сутки).
Пример 2. Фармакокинетический профиль исходного вещества относительно метаболита
Как показано на фиг. 3, дейтерирование драматически влияет на метаболизм дейтерированного аналога ивакафтора CTP-656 по сравнению с ивакафтором после разовой дозы. Ивакафтор, CTP-656, и их метаболиты показаны на фиг. 6. Существует достоверное уменьшение продукции метаболитов D8-M1 и D6-M6 из CTP-656 относительно продукции метаболитов M1 и M6 из ивакафтора. Следовательно, соотношение исходного вещества к M1 AUC0-24ч для CTP-656/D8-M1 равно 2,0, по сравнению с 0,58 для соотношения ивакафтора к M1. Соотношение исходного вещества к M1 Cmax и C24hr для CTP-656/D8-M1 составляет 2,1 и 2,2, соответственно, по сравнению с 0,54 и 0,55, соответственно, для ивакафтора/M1. Дополнительно соотношение исходного соединения к M6 AUC0-24ч для CTP-656/D6-M6 составляет 4,0, по сравнению с 1,5 для соотношения ивакафтора к M6. Соотношение исходного соединения к M6 Cmax и C24ч для CTP-656/D6-M6 составляет 4,3 и 2,5 соответственно, по сравнению с 1,4 и 0,97, соответственно, для ивакафтора/M6. Как видно на фиг. 3 (a), CTP-656 является наиболее частым видом в плазме во все измеренные времена, в противоположность к ивакафтору (b), где M1 метаболит преобладает через 2 часа и уровень M6 метаболита достигает уровня ивакафтора через около 8 часов. В результате, наиболее фармакологически активные виды (т.е. исходные вещества) являются наиболее частыми видами для CTP-656, но не для ивакафтора.
В общем, CTP-656 демонстрирует превосходный фармакокинетический профиль по сравнению с Калидеко, существующим в настоящее время стандартом для лечения пациентов с муковисцидозом. Результаты исследования 1 фазы также показали, что CTP-656 хорошо переносился и его профиль безопасности был сравнимым с Калидеко. В перекрестном сравнении фазы 1 CTP-656 и Калидеко, CTP-656 продемонстрировал превосходный фармакокинетический профиль по сравнению с Калидеко, включая сниженную скорость клиренса, более длительный период полужизни, по существу усиленное воздействие и большие уровни в плазме через 24 часа. Анализ метаболитов в плазме также показал, что профиль общего воздействия CTP-656 отличался от такового Калидеко тем, что большую часть воздействия в плазме в случае CTP-656 было опосредовано исходным лекарственным средством, тогда как с Калидеко большая часть воздействия в плазме происходит из-за менее активного метаболита, также наблюдали и приблизительно эквивалентное количество неактивного метаболита. Считают, что более длительный период полужизни получали для CTP-656, что дает преимущество дозирования для пациента.
Пример 3- Измерение активности CTP-656 и ивакафтора
Хлоридный транспорт G551D-CFTR, избыточно экспрессируемого в клетках щитовидной железы крыс Fischer измеряли в камере прибора Ussing. Ток короткого замыкания (ISC) отслеживали после базолатеральной пермеабилизации с использованием 100 мкм амфотерицина и активации CFTR посредством 10 мкM форсколина. Тестирование проводили в присутствии градиента хлоридов при 35°C. Тестируемые изделия наносили аддитивным и последовательным образом на эпителий по 0,0008 мкM, 0,004 мкM, 0,02 мкM, и 0,1 мкM вместе с 0,5 мкл, 2,0 мкл, 0,5 мкл, и 2,5 мкл добавлениям DMSO носителя. Значения были средними ответов для каждой тестируемой концентрации, наносимой на шесть эпителиев. (Фиг. 4A)
Хлоридный транспорт гомозиготных F508del-CFTR человеческих бронхиальных эпителиальных клеток монослоев (код пациента CFFT027G) измеряли в камере аппарата Using. Ток короткого замыкания (ISC) отслеживали после блокады натриевого тока через эпителиальный натриевый канал (ENaC) с 30 мкM амилорида и активации CFTR посредством 10 мкM форсколина. Симметричные физиологические солевые растворы при 27°C использовали для температурной коррекции F508del-CFTR. Тестируемые изделия наносили дополнительным и последовательным образом на эпителий по 0,0008 мкM, 0,004 мкM, 0,02 мкM, и 0,1 мкM вместе с 0,5 мкл, 2,0 мкл, 0,5 мкл, и 2,5 мкл добавлениями DMSO носителя. Значения представляют собой средние ответа для каждой тестируемой концентрации, наносимой на шесть эпителиев. (Фиг. 4B и фиг. 4C).
Следовательно, D9-, D18-ивакафтор и ивакафтор обеспечивали эквивалент в in vitro потенциировании CFTR.
Пример 4- Человеческое перекрестное исследование D9-ивакафтора и D18-ивакафтора
Шесть здоровых волонтеров включали в перекрестное сравнение 25 мг D9-ивакафтора (CTP-656) и 25 мг D18-ивакафтора (Фиг. 7A). Задачей исследования было сравнить фармакокинетику разовой дозы 25 мг D9-ивакафтора и 25 мг D18-ивакафтора. Дозы D9-ивакафтора (CTP-656) и D18-ивакафтора вводили в виде водной суспензии. Все дозы D9-ивакафтора (CTP-656) и D18-ивакафтора вводили пациентам натощак (три пациента на группу). Имелся 7 дневный период отмывки между дозами.
Сравнение фармакокинетических свойств D9-ивакафтора и D18-ивакафтора представлено в таблице ниже:
a среднее (диапазон)
Было обнаружено, что D9-ивакафтор проявлял превосходный ФК профиль по сравнению с D18-ивакафтором (фиг. 7B).
Пример 5- Получение таблетированной формы
CTP-656 (D9-ивакафтор) (17,1 масс% 80% аморфной дисперсии) смешивали с микрокристаллической целлюлозой (Avicel PH101) (39,00 процентов по массе), моногидратом лактозы 316 (38,9 процентов по массе) лаурилсульфатом натрия (0,50 процентов по массе), кроскармеллозой натрия (Ac-di-sol) (1,50 процентов по массе) и стеаратом магния Hyqual (0,20 процента по массе) в шейкере. Смесь прессовали и измельчали для получения гранул, которые просеивали через сита размером #20 и #80. Гранулы и оставшиеся измельченные остатки смешивали с дополнительной кроскармеллозой натрия (Ac-di-sol) (1,50 процента по массе), коллоидным диоксидом кремния (0,50 процента по массе) и дополнительным стеаратом магния Hyqual (0,80 процента по массе), и окончательную смесь прессовали в таблетки с использованием ротационного пресса. Каждая таблетка содержала 75 мг CTP-656 (D9-ивакафтора).
Пример 6 - Перекрестное исследование D9-ивакафтора и ивакафтора у человека
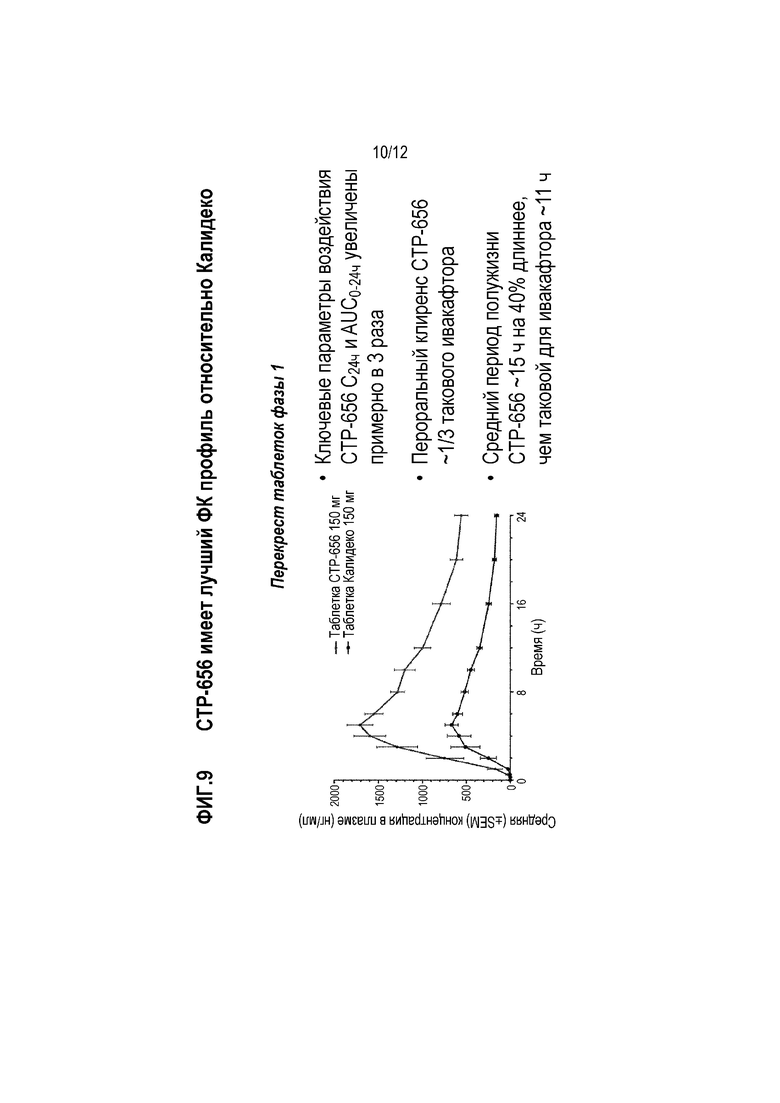
Здоровых волонтеров включали в перекрестное сравнение 150 мг D9-ивакафтора (CTP-656) и 150 мг ивакафтора (Калидеко) (фиг. 8, левая панель). Задачей исследования было сравнить безопасность, переносимость и фармакокинетику разовой дозы 150 мг D9-ивакафтора и 150 мг ивакафтор. Дозы D9-ивакафтора (CTP-656) и ивакафтора вводили в виде таблеток (D9-ивакафтор вводили в виде двух 75 мг таблеток). Все D9-ивакафтор (CTP-656) и D18-ивакафтор вводили пациентам после еды (высокожировой завтрак) (четыре пациента на последовательность). Имелось 7 дней отмывки между дозами. Образцы крови получали с интервалами.
Результаты показаны на фиг. 9 и 10. Было обнаружено, что CTP-656 имел приблизительно 3-кратное увеличение C24ч и AUC0-24ч по сравнению с ивакафтором (Фиг. 9). Пероральный клиренс CTP-656 оставил около одной трети такового ивакафтора. Период полужизни CTP-656 составил около 15 часов, что было около 40% больше чем период полужизни ивакафтора (около 11 часов). Кроме того, соотношение CTP-656 к метаболитам D-M1 и D-M6 (см. Фиг. 6) было выше, чем соотношение ивакафтора к метаболитам M1 и M6 (Фиг. 10).
Пример 7- Исследование множественной нарастающей дозы D9-ивакафтора у человека
Здоровых волонтеров включали в исследование множественной нарастающей дозы D9-ивакафтора (CTP-656) (фиг. 8, правая панель). Задачей исследования было сравнить безопасность, переносимость и фармакокинетику D9-ивакафтора в трех дозах в течение семи дней, по сравнению с плацебо. Дозы D9-ивакафтора (CTP-656) и плацебо вводили в виде таблеток (D9-ивакафтор вводили в виде одной, двух или трех 75 мг таблеток для доз 75 мг, 150 мг, или 225 мг). Все дозы D9-ивакафтор (CTP-656) вводили пациентам после еды (высокожировой завтрак) (8 пациентов на последовательность получали CTP-656, два пациента на последовательность получали плацебо). Имелась 7 дневная отмывка между дозами. Образцы крови получали с интервалами и концентрацией в плазме CTP-656.
Результаты показаны на фиг. 11. Было обнаружено, что уровня в плазме в стабильном состоянии CTP-656 достигали после трех дней введения. CTP-656 проявлял дозозависимое увеличение воздействия при повторном введении для 150 мг дозы относительно дозы 75 мг. Группа дозы 225 мг показала более высокое дозопропорциональное воздействие. Соотношение CTP-656 к метаболитам D-M1 и D-M6 в плазме было больше чем один. Соотношение накопления CTP-656 и D-M1 составило около 1-6 до 1,8 для ключевых параметров воздействия C24ч и AUC0-24ч. Не сообщали о серьезных нежелательных явлениях; большинство указанных нежелательных явлений были мягкими по тяжести.
Без дополнительного описания считают, что обычный специалист в области техники может, с использованием предшествующего описания и иллюстративных примеров, осуществить и использовать соединения по настоящему изобретению и применить заявленные способы. Необходимо понимать, что вышеуказанное обсуждение и примеры представляют только подробное описание определенных предпочтительных вариантов осуществления изобретения. Очевидно специалисту в области техники, что различные модификации и эквиваленты могут быть осуществлены без отклонения от рамок и тенденции изобретения.

Изобретение относится к способу лечения состояния, которое опосредовано муковосцидозным трансмембранным регулятором проводимости (CFTR), у пациента, включающему введение пациенту Соединения (I), представленного следующей структурной формулой:
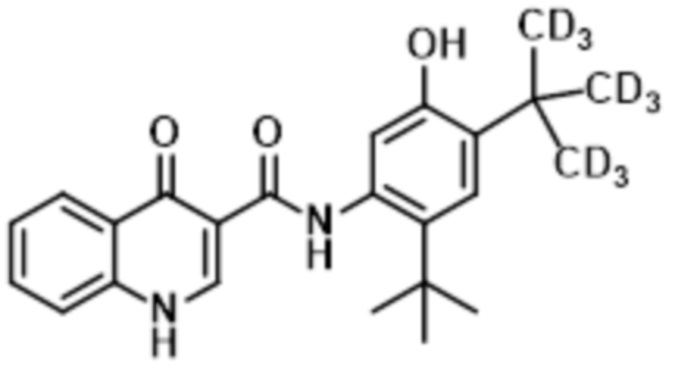 , или его фармацевтически приемлемой соли в количестве от 150 мг до 200 мг один раз в сутки, а также к фармацевтической композиции, предназначенной для лечения состояния, опосредованного CFTR, у пациента, включающей фармацевтически приемлемый носитель или разбавитель и от 150 мг до 200 мг соединения, представленного следующей структурной формулой: , или его фармацевтически приемлемой соли. 2 н. и 13 з.п. ф-лы, 4 табл., 7 пр., 12 ил.
, или его фармацевтически приемлемой соли в количестве от 150 мг до 200 мг один раз в сутки, а также к фармацевтической композиции, предназначенной для лечения состояния, опосредованного CFTR, у пациента, включающей фармацевтически приемлемый носитель или разбавитель и от 150 мг до 200 мг соединения, представленного следующей структурной формулой: , или его фармацевтически приемлемой соли. 2 н. и 13 з.п. ф-лы, 4 табл., 7 пр., 12 ил.
1. Способ лечения состояния, которое опосредовано муковосцидозным трансмембранным регулятором проводимости (CFTR), у пациента, включающий введение пациенту Соединения (I), представленного следующей структурной формулой:
 ,
,
или его фармацевтически приемлемой соли
в количестве от 150 мг до 200 мг один раз в сутки.
2. Способ по п. 1, где состоянием является муковисцидоз.
3. Способ по п. 1, включающий введение пациенту 150 мг Соединения (I) или его фармацевтически приемлемой соли в сутки.
4. Способ по п. 1, включающий введение пациенту 200 мг Соединения (I) или его фармацевтически приемлемой соли в сутки.
5. Способ по п. 1, где соединение или его фармацевтически приемлемую соль вводят перорально.
6. Способ по п. 5, где соединение или его фармацевтически приемлемую соль вводят в составе фармацевтической композиции, которая является таблеткой.
7. Способ по п. 5, где соединение или его фармацевтически приемлемую соль вводят в составе фармацевтической композиции, которая является гранулами.
8. Способ по любому из пп. 1-7, где любой атом, не обозначенный как дейтерий в любом из вариантов осуществления изобретения, указанных выше, присутствует в его натуральной изотопной обогащенности.
9. Фармацевтическая композиция, предназначенная для лечения состояния, опосредованного CFTR, у пациента, включающая фармацевтически приемлемый носитель или разбавитель и от 150 мг до 200 мг соединения, представленного следующей структурной формулой:
,
Соединение I
или его фармацевтически приемлемой соли.
10. Фармацевтическая композиция по п. 9, включающая 150 мг Соединения (I).
11. Фармацевтическая композиция по п. 9, включающая 200 мг Соединения (I).
12. Фармацевтическая композиция по п. 9, где фармацевтическая композиция подходит для перорального введения.
13. Фармацевтическая композиция по п. 12, где композицией является таблетка.
14. Фармацевтическая композиция по п. 12, где композицией является гранула.
15. Фармацевтическая композиция по любому из пп. 9-14, где композицию вводят один раз в сутки.
| WO 2014078842 A1, 22.05.2014 | |||
| EA 201391615 A1, 31.03.2014. |

