Область техники, к которой относится изобретение
[0001]
Настоящее изобретение относится к способу производства катализатора для аммоксидирования, предназначенного для использования при производстве акрилонитрила в результате проведения реакции между пропиленом, молекулярным кислородом и аммиаком, и способу производства акрилонитрила при использовании катализатора для аммоксидирования, произведенного при использовании данного способа.
Уровень техники
[0002]
Способ производства акрилонитрила в результате проведения реакции между пропиленом, молекулярным кислородом и аммиаком известен под наименованием «реакции аммоксидирования». Данную реакцию используют по всему миру в качестве промышленного технологического процесса производства акрилонитрила.
[0003]
В данной реакции для достижения благоприятного выхода акрилонитрила используют катализатор на основе сложного оксида. Например, в промышленных масштабах в качестве существенного компонента используют катализатор, содержащий Mo-Bi-Fe или Fe-Sb. В целях достижения более благоприятного выхода акрилонитрила были продолжены исследования в отношении состава металлов (смотрите, например, патентные документы 1 и 2).
[0004]
Между тем, были предприняты попытки улучшения выхода акрилонитрила не только в результате улучшения состава металлов, но и в результате улучшения стадии получения катализатора. Например, в патентном документе 3 раскрывается способ производства катализатора для производства акрилонитрила, включающий получение суспензии, содержащей молибден, висмут, железо, вольфрам и тому подобное, при температуре в диапазоне от 30 до 70°С. В патентном документе 4 раскрывается способ производства катализатора для производства акрилонитрила, включающий выдерживание суспензии на протяжении заданного времени в конкретных условиях во время одной стадии.
[0005]
В патентном документе 5 раскрывается способ производства катализатора для производства акрилонитрила, включающий подстраивание размера частиц агрегата, содержащегося в суспензии предшественника катализатора, в результате проведения обработки при использовании гомогенизатора и обработки при использовании ультразвука в отношении суспензии предшественника.
Перечень документов предшествующего уровня техники
Патентный документ
[0006]
Патентный документ 1: японский патент № 5919870
Патентный документ 2: японский патент № 4954750
Патентный документ 3: японский патент № 4823950
Патентный документ 4: японский патент № 4425743
Патентный документ 5: японский патент № 5378041
Сущность изобретения
Проблемы, разрешаемые в изобретении
[0007]
Однако, данные способы производства катализатора все еще являются недостаточными для улучшения выхода акрилонитрила несмотря на некоторую степень эффективности данных способов в отношении улучшения. Таким образом, требуется еще большее улучшение.
[0008]
В патентном документе 5 раскрывается способ контроля размера частиц агрегата в суспензии в результате проведения измельчения в порошок при использовании гомогенизатора. В общем случае измельчение в порошок при использовании гомогенизатора называется раскалыванием, и был предложен механизм, при использовании которого агрегат дезинтегрируется на более мелкие агрегаты. Однако, первичные частицы, составляющие агрегат, измельчают в порошок при использовании физического соударения и поверхность частиц активируется в результате формирования новой поверхности или дефекта кристаллической решетки и тому подобного таким образом, что взаимодействие между частицами увеличивается. В качестве результата суспензия утрачивает стабильность в результате повторного агрегирования первичных частиц, измельченных в порошок. Это может ухудшить эксплуатационные характеристики катализатора.
[0009]
Настоящее изобретение было сделано в свете проблем, описанных выше. Одна цель настоящего изобретения заключается в предложении способа производства катализатора для аммоксидирования, который демонстрирует высокий выход акрилонитрила, и способа производства акрилонитрила.
Средства разрешения проблем
[0010]
Изобретатели настоящего изобретения провели кропотливые исследования, направленные на достижение данной цели, и, следовательно, совершили настоящее изобретение в результате установления возможности достижения данной цели при использовании способа производства катализатора, включающего подстраивание размеров частиц для первичных частиц металла, составляющих агрегат, содержащих металл и носитель, в суспензии предшественника с доведением их до конкретного диапазона в результате оптимизирования условий получения суспензии предшественника.
[0011]
Говоря конкретно, настоящее изобретение представляет собой нижеследующее.
[1]
Способ производства катализатора для аммоксидирования, включающий стадии:
получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу;
высушивания суспензии предшественника катализатора в целях получения сухих частиц; и
прокаливания сухих частиц в целях получения катализатора для аммоксидирования,
где твердая фаза суспензии предшественника катализатора содержит агрегат, содержащий металл и носитель, первичные частицы металла, составляющие агрегат, характеризуются размером частиц, составляющим 1 мкм и менее, и средний размер частиц для первичных частиц металла находится в диапазоне от 40 нм и более до 200 нм и менее.
[2]
Способ производства катализатора для аммоксидирования, соответствующий позиции [1], где катализатор для аммоксидирования содержит сложный оксид металлов, характеризующийся составом, описывающимся следующей далее общей формулой (1):
Mo12BiaFebXcYdZeOf, (1)
где Х представляет собой один или несколько элементов, выбираемых из группы, состоящей из никеля, кобальта, магния, кальция, цинка, стронция и бария; Y представляет собой один или несколько элементов, выбираемых из группы, состоящей из церия, хрома, лантана, неодима, иттрия, празеодима, самария, алюминия, галлия и индия; Z представляет собой один или несколько элементов, выбираемых из группы, состоящей из калия, рубидия и цезия; а представляет собой атомное соотношение между висмутом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤а≤2,0; b представляет собой атомное соотношение между железом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤b≤3,0; c представляет собой атомное соотношение между Х и 12 атомами молибдена и удовлетворяет неравенству 0,1≤c≤10,0; d представляет собой атомное соотношение между Y и 12 атомами молибдена и удовлетворяет неравенству 0,1≤d≤3,0; e представляет собой атомное соотношение между Z и 12 атомами молибдена и удовлетворяет неравенству 0,01≤e≤2,0; и f представляет собой атомное соотношение между кислородом и 12 атомами молибдена и представляет собой количество атомов кислорода, необходимое для удовлетворения требований по валентности у других присутствующих элементов.
[3]
Способ производства катализатора для аммоксидирования, соответствующий позициям [1] или [2], где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, концентрация свободной кислоты в суспензии находится в диапазоне от 0,1% и более до 1,2% и менее.
[4]
Способ производства катализатора для аммоксидирования, соответствующий любой позиции от [1] до [3], где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, концентрация свободной кислоты в суспензии находится в диапазоне от 0,8% и более до 1,2% и менее.
[5]
Способ производства катализатора для аммоксидирования, соответствующий любой позиции от [1] до [4], где катализатор для аммоксидирования содержит носитель, и уровень содержания носителя в катализаторе для аммоксидирования находится в диапазоне от 35 до 45% (масс.).
[6]
Способ производства катализатора для аммоксидирования, соответствующий любой позиции от [1] до [5], где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, доля совокупной массы металлического компонента и носителя в качестве исходных материалов по отношению к массе полной суспензии предшественника катализатора находится в диапазоне от 10% (масс.) и более до 40% (масс.) и менее.
[7]
Способ производства катализатора для аммоксидирования, соответствующий любой позиции от [1] до [6], где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, доля совокупной массы металлического компонента и носителя в качестве исходных материалов по отношению к массе полной суспензии предшественника катализатора находится в диапазоне от 20% (масс.) и более до 35% (масс.) и менее.
[8]
Способ производства катализатора для аммоксидирования, соответствующий любой позиции от [1] до [7], где на стадии высушивания суспензии предшественника катализатора в целях получения сухих частиц сушилку выдерживают при температуре воздуха на впуске в диапазоне от 180 до 250°С и при температуре на выпуске в диапазоне от 100 до 150°С.
[9]
Способ производства катализатора для аммоксидирования, соответствующий любой позиции от [1] до [8], где стадия прокаливания сухих частиц в целях получения катализатора для аммоксидирования включает денитрационную обработку до прокаливания, и денитрационная обработка включает проведение нагревания при температуре в диапазоне от 150 до 450°С на протяжении от 1,5 до 3 часов.
[10]
Способ производства катализатора для аммоксидирования, соответствующий любой позиции от [1] до [9], где на стадии прокаливания сухих частиц в целях получения катализатора для аммоксидирования температура прокаливания находится в диапазоне от 550 до 650°С.
[11]
Способ производства акрилонитрила, включающий стадии:
получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу;
высушивания суспензии предшественника катализатора в целях получения сухих частиц;
прокаливания сухих частиц в целях получения катализатора для аммоксидирования; и
предварительной подачи катализатора для аммоксидирования в реакционную емкость с псевдоожиженным слоем и при одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проведения реакции между пропиленом, молекулярным кислородом и аммиаком в целях получения акрилонитрила,
где твердая фаза суспензии предшественника катализатора содержит агрегат, содержащий металл и носитель, первичные частицы металла, составляющие агрегат, характеризуются размером частиц, составляющим 1 мкм и менее, и средний размер частиц для первичных частиц металла находится в диапазоне от 40 нм и более до 200 нм и менее.
[12]
Способ производства акрилонитрила, соответствующий позиции [11], где
источник молекулярного кислорода представляет собой воздух, и
молярное соотношение между аммиаком и воздухом и пропиленом находится в диапазоне 1/(от 0,8 до 1,4)/(от 7 до 12) применительно к соотношению пропилен/аммиак/воздух.
[13]
Способ производства акрилонитрила, соответствующий позиции [11], где
источник молекулярного кислорода представляет собой воздух, и
молярное соотношение между аммиаком и воздухом и пропиленом находится в диапазоне 1/(от 0,9 до 1,3)/(от 8 до 11) применительно к соотношению пропилен/аммиак/воздух.
[14]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [13], где температура, при которой пропилен, молекулярный кислород и аммиак вступают в реакцию в присутствии катализатора для аммоксидирования, находится в диапазоне от 350 до 550°С.
[15]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [13], где температура, при которой пропилен, молекулярный кислород и аммиак вступают в реакцию в присутствии катализатора для аммоксидирования, находится в диапазоне от 400 до 500°С.
[16]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [15], где катализатор для аммоксидирования содержит сложный оксид металлов, характеризующийся составом, описывающимся следующей далее общей формулой (1):
Mo12BiaFebXcYdZeOf, (1)
где Х представляет собой один или несколько элементов, выбираемых из группы, состоящей из никеля, кобальта, магния, кальция, цинка, стронция и бария; Y представляет собой один или несколько элементов, выбираемых из группы, состоящей из церия, хрома, лантана, неодима, иттрия, празеодима, самария, алюминия, галлия и индия; Z представляет собой один или несколько элементов, выбираемых из группы, состоящей из калия, рубидия и цезия; а представляет собой атомное соотношение между висмутом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤а≤2,0; b представляет собой атомное соотношение между железом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤b≤3,0; c представляет собой атомное соотношение между Х и 12 атомами молибдена и удовлетворяет неравенству 0,1≤c≤10,0; d представляет собой атомное соотношение между Y и 12 атомами молибдена и удовлетворяет неравенству 0,1≤d≤3,0; e представляет собой атомное соотношение между Z и 12 атомами молибдена и удовлетворяет неравенству 0,01≤e≤2,0; и f представляет собой атомное соотношение между кислородом и 12 атомами молибдена и представляет собой количество атомов кислорода, необходимое для удовлетворения требований по валентности у других присутствующих элементов.
[17]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [16], где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, концентрация свободной кислоты в суспензии находится в диапазоне от 0,1% и более до 1,2% и менее.
[18]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [17], где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, концентрация свободной кислоты в суспензии находится в диапазоне от 0,8% и более до 1,2% и менее.
[19]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [18], где катализатор для аммоксидирования содержит носитель, и уровень содержания носителя в катализаторе для аммоксидирования находится в диапазоне от 35 до 45% (масс.).
[20]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [19], где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, доля совокупной массы металлического компонента и носителя в качестве исходных материалов по отношению к массе полной суспензии предшественника катализатора находится в диапазоне от 10% (масс.) и более до 40% (масс.) и менее.
[21]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [20], где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, доля совокупной массы металлического компонента и носителя в качестве исходных материалов по отношению к массе полной суспензии предшественника катализатора находится в диапазоне от 20% (масс.) и более до 35% (масс.) и менее.
[22]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [21], где на стадии высушивания суспензии предшественника катализатора в целях получения сухих частиц сушилку выдерживают при температуре воздуха на впуске в диапазоне от 180 до 250°С и при температуре на выпуске в диапазоне от 100 до 150°С.
[23]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [22], где стадия прокаливания сухих частиц в целях получения катализатора для аммоксидирования включает денитрационную обработку до прокаливания, и денитрационная обработка включает проведение нагревания при температуре в диапазоне от 150 до 450°С на протяжении от 1,5 до 3 часов.
[24]
Способ производства акрилонитрила, соответствующий любой из позиций от [11] до [23], где на стадии прокаливания сухих частиц в целях получения катализатора для аммоксидирования температура прокаливания находится в диапазоне от 550 до 650°С.
Преимущества от изобретения
[0012]
Настоящее изобретение может предложить способ производства катализатора для аммоксидирования, который демонстрирует высокий выход акрилонитрила в реакции аммоксидирования для пропилена, и способ производства акрилонитрила при высоком выходе.
Краткое описание чертежа
[0013]
[Фигура] Фигура представляет собой концептуальную диаграмму для одного примера результатов измерения при проведении измерения в отношении агрегата в суспензии предшественника катализатора при использовании метода затухания ультразвука.
Режим осуществления изобретения
[0014]
Ниже в настоящем документе будет описываться режим осуществления настоящего изобретения (ниже в настоящем документе просто обозначаемый термином «настоящий вариант осуществления»). Однако, на настоящее изобретение представленным ниже вариантом осуществления ограничений не накладывают. В настоящем документе могут быть сделаны различные изменения или модификации без отклонения от сущности настоящего изобретения.
[0015]
Способ производства катализатора для аммоксидирования, соответствующий изобретению, включает стадии:
получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу;
высушивания суспензии предшественника катализатора в целях получения сухих частиц; и
прокаливания сухих частиц в целях получения катализатора для аммоксидирования,
где твердая фаза суспензии предшественника катализатора содержит агрегат, содержащий металл и носитель, первичные частицы металла, составляющие агрегат, характеризуются размером частиц, составляющим 1 мкм и менее, и средний размер частиц для первичных частиц металла находится в диапазоне от 40 нм и более до 200 нм и менее.
[0016]
В способе производства катализатора для аммоксидирования, соответствующем настоящему варианту осуществления, предпочтительным является сначала получение суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, в результате смешивания исходных материалов для компонентов, составляющих катализатор, таких как металл и носитель. Для случая использования, например, диоксида кремния в качестве носителя металлические частицы и диоксид кремния, которые являются нерастворимыми в растворителе в суспензии, накапливаются в суспензии предшественника катализатора, составляя агрегат. Состояние данного агрегата можно наблюдать при использовании метода, известного на современном уровне техники. Агрегирование первичных частиц металла и частиц диоксида кремния можно наблюдать, например, в результате извлечения агрегата на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании сканирующего электронного микроскопа (СЭМ). При данной операции в комбинации с этим может быть использована и энергодисперсионная рентгеновская спектроскопия (ЭДРС) в целях подтверждения формирования частиц, составляющих агрегат, из металла и диоксида кремния.
[0017]
В настоящем варианте осуществления размеры частиц для первичных частиц металла, составляющих агрегат, могут быть рассчитаны в результате проведения измерения для суспензии предшественника катализатора в неразбавленной форме при использовании устройства для измерения распределения частиц по размерам на основе метода затухания ультразвука, где данный метод является методом, известным на современном уровне техники. Метод затухания ультразвука представляет собой подход, который включает генерирование ультразвуковой волны от ультразвукового генератора, наблюдение ультразвуковой волны, которая дошла до детектора через суспензию в кювете для образца, и определение распределения частиц по размерам исходя из результатов относительно степени затухания ультразвуковой волны (спектра затухания ультразвука) в суспензии. Метод затухания ультразвука характеризуется возможностью проведения измерения в отношении высококонцентрированной суспензии в неразбавленной форме без проведения разбавления, и, таким образом, ему свойственно преимущество, заключающееся в возможности исключения воздействия изменения состояния суспензии, обусловленного разбавлением. Другие методы измерения, такие как метод лазерной дифракции или метод динамического светорассеяния, которые требуют разбавления суспензии во время получения образца для измерения, могут привести к агрегированию частиц металла и/или диоксида кремния в результате разбавления и не являются подходящими для использования при измерении размера первичных частиц металла. В настоящем варианте осуществления измерение размера первичных частиц металла проводят при использовании устройства AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. На фигуре продемонстрирована концептуальная диаграмма для одного примера результатов измерения при проведении измерения в отношении агрегата в суспензии предшественника катализатора, использованной в настоящем варианте осуществления, при использовании метода затухания ультразвука. Как это продемонстрировано на фигуре, на графике с нанесением по оси абсцисс размера частиц (при логарифмическом представлении) и по оси ординат частоты при расчете на объем наблюдаются два пика. В свете доминирования левого пика нанометрового порядка на фигуре с точки зрения количества частиц, результатов наблюдения агрегата в суспензии при использовании вышеупомянутого метода и маленького значения % (масс.) носителя (например, диоксида кремния) в агрегате, как это упоминается ниже, левый пик нанометрового порядка определяется как пик, произведенный от первичных частиц металла, а правый пик микрометрового порядка определяется как пик, произведенный от самого агрегата, содержащего металл и носитель (например, диоксид кремния). В настоящем варианте осуществления модальный диаметр для левого пика нанометрового порядка рассматривается в качестве «среднего размера частиц для первичных частиц металла». Как это считается, первичные частицы металла, составляющие агрегат, осциллируют под воздействием ультразвукового облучения во время измерения таким образом, что могут быть измерены размеры частиц для первичных частиц металла. Детальные условия измерения и анализа будут упомянуты ниже. В способе производства катализатора для аммоксидирования в соответствии с настоящим вариантом осуществления диапазон среднего размера частиц для первичных частиц металла, составляющих агрегат, содержащий металл и носитель (например, диоксид кремния), в суспензии предшественника катализатора демонстрирует 40 нм в качестве нижнего предельного значения и 200 нм в качестве верхнего предельного значения. Средний размер частиц для первичных частиц металла предпочтительно находится в диапазоне от 45 нм и более до 180 нм и менее, более предпочтительно от 45 нм и более до 150 нм и менее, еще более предпочтительно от 50 нм и более до 130 нм и менее. Размеры частиц для первичных частиц металла подстраивают, доводя до 1 мкм и менее. На нижнее предельное значение у размеров частиц для первичных частиц металла конкретных ограничений не накладывают, и оно, например, составляет 10 нм и более. При нахождении размеров частиц для первичных частиц металла в пределах описанного выше диапазона распределение частиц по размерам для первичных частиц металла не является чрезмерно широким и может предотвратить искажение структуры агрегата. Поэтому выход акрилонитрила улучшается.
[0018]
При среднем размере частиц для первичных частиц металла, равном или большем в сопоставлении с нижним предельным значением, точка разложения акрилонитрила уменьшается по величине при уменьшении поверхности частиц металла для катализатора таким образом, что подавляется вторичное разложение целевого акрилонитрила. Таким образом, выход акрилонитрила улучшается.
[0019]
При среднем размере частиц для первичных частиц металла, равном или меньшем в сопоставлении с верхним предельным значением, точка реакции для синтезирования акрилонитрила увеличивается по величине при увеличении поверхности частиц металла. Таким образом, выход акрилонитрила улучшается. При среднем размере частиц для первичных частиц металла, равном или меньшем в сопоставлении с верхним предельным значением, агрегат, содержащий металл и носитель (например, диоксид кремния), не является чрезмерно большим, и может быть предотвращено его осаждение в емкости для получения суспензии или технологической линии для подачи жидкости во время производства катализатора.
[0020]
С другой стороны, на размеры частиц для частиц неметаллов конкретных ограничений не накладывают. Для случая использования, например, золя кремниевой кислоты в качестве исходного материала для носителя средний размер частиц диоксида кремния может быть подстроен, принимая во внимание активность катализатора и тому подобное.
[0021]
Примеры способа контроля размеров частиц для первичных частиц металла, составляющих агрегат в суспензии предшественника катализатора, включают следующий далее способ, но конкретно не ограничиваются только этим: например, для случая получения суспензии предшественника катализатора, содержащей твердую фазу и жидкую фазу, в результате диспергирования или растворения исходных материалов в целях получения растворов и смешивания множества растворов в целях получения агрегата его примеры включают способ задания концентрации свободной кислоты в суспензии предшественника катализатора, массового соотношения между металлом и носителем, значений % (масс.) металла и носителя при расчете на полную суспензию во время получения суспензии предшественника катализатора, перемешивающей способности во время смешивания или степени смешивания растворов исходных материалов находящимися в пределах конкретного диапазона и выбора добавки для получения суспензии предшественника катализатора.
В частности, размеры частиц для первичных частиц металла могут быть заданы находящимися в конкретном диапазоне в результате надлежащего комбинирования перемешивающей способности для золя кремниевой кислоты, раствора молибдена и водного раствора нитрата металла, упомянутых ниже, со временем добавления водного раствора нитрата металла. Время добавления водного раствора нитрата металла предпочтительно находится в диапазоне, например, от 10 до 80 секунд.
[0022]
Для случая использования исходных материалов в твердой форме его примеры включают способ контроля размеров частиц твердых исходных материалов в результате измельчения в порошок.
[0023]
На стадию получения суспензии предшественника катализатора подходом к получению конкретных ограничений не накладывают до тех пор, пока удовлетворяются требования, относящиеся к размерам частиц для первичных частиц металла, составляющих агрегат, в суспензии предшественника катализатора, описанной выше. Может быть надлежащим образом выбран и использован способ получения, известный на современном уровне техники.
[0024]
На композицию катализатора для аммоксидирования, использованную в настоящем варианте осуществления, конкретных ограничений не накладывают, и в рамках одного примера предпочтительно ею является композиция, содержащая молибден, висмут и железо и описывающаяся общей формулой (1), представленной ниже. Молибден играет роль центра адсорбирования для пропилена и активного центра для аммиака. Висмут играет роль при активировании пропилена, извлечении атома водорода в положении α и формировании π-аллильных частиц. Железо играет роль при подаче кислорода, присутствующего в паровой фазе, к каталитической активной точке в результате прохождения окисления-восстановления между трехвалентным/двухвалентным состояниями. Такая композиция имеет тенденцию к дополнительному улучшению степени селективности по акрилонитрилу.
Mo12BiaFebXcYdZeOf, (1)
В формуле (1) Х представляет собой один или несколько элементов, выбираемых из группы, состоящей из никеля, кобальта, магния, кальция, цинка, стронция и бария, Y представляет собой один или несколько элементов, выбираемых из группы, состоящей из церия, хрома, лантана, неодима, иттрия, празеодима, самария, алюминия, галлия и индия, и Z представляет собой один или несколько элементов, выбираемых из группы, состоящей из калия, рубидия и цезия. а представляет собой атомное соотношение между висмутом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤а≤2,0, предпочтительно 0,15≤а≤1,0, более предпочтительно 0,2≤а≤0,7. b представляет собой атомное соотношение между железом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤b≤3,0, предпочтительно 0,5≤b≤2,5, более предпочтительно 1,0≤b≤2,0. c представляет собой атомное соотношение между Х и 12 атомами молибдена и удовлетворяет неравенству 0,1≤c≤10,0, предпочтительно 3,0≤c≤9,0, более предпочтительно 5,0≤c≤8,5. d представляет собой атомное соотношение между Y и 12 атомами молибдена и удовлетворяет неравенству 0,1≤d≤3,0, предпочтительно 0,2≤d≤2,0, более предпочтительно 0,3≤d≤1,5. e представляет собой атомное соотношение между Z и 12 атомами молибдена и удовлетворяет неравенству 0,01≤e≤2,0, предпочтительно 0,05≤e≤1,0. f представляет собой атомное соотношение между кислородом и 12 атомами молибдена и представляет собой количество атомов кислорода, необходимое для удовлетворения требований по валентности у других присутствующих элементов.
[0025]
Способ производства катализатора для аммоксидирования, соответствующий настоящему варианту осуществления, включает стадии: (1) получение суспензии предшественника (суспензии предшественника катализатора), содержащей жидкую фазу и твердую фазу и исполняющую функцию предшественника катализатора; (2) высушивание суспензии предшественника катализатора в целях получения сухих частиц; и (3) прокаливания сухих частиц в целях получения катализатора для аммоксидирования.
[0026]
Стадия (1) является стадией получения суспензии предшественника, содержащей жидкую фазу и твердую фазу и исполняющей функцию предшественника катализатора, например, в результате смешивания каждого элемента с носителем. Примеры источника элемента для каждого элемента включают аммониевые соли, нитрат и соли органических кислот, растворимые в воде или кислом водном растворе. Данные источники являются предпочтительными, поскольку данные источники не приводят к появлению ни остаточного хлорида из использованного гидрохлорида, ни остаточной серы из использованного сульфата.
[0027]
На исходный материал для носителя конкретных ограничений не накладывают до тех пор, пока исходный материал является материалом, используемым обычно. Его примеры включают оксиды, такие как диоксид кремния, оксид алюминия, диоксид титана и диоксид циркония. В их числе предпочтительным является диоксид кремния. Диоксид кремния в сопоставлении с другими оксидами является неактивным сам по себе и характеризуется благоприятным связывающим действием в отношении активных компонентов катализатора.
[0028]
На порядок смешивания компонентов при получении суспензии предшественников катализатора конкретных ограничений не накладывают. Например, один показательный вариант осуществления композиции, описывающейся общей формулой (1), соответствует представленному ниже описанию изобретения. Сначала к золю кремниевой кислоты (ниже в настоящем документе обозначаемому термином «раствор диоксида кремния») добавляют аммониевую соль молибдена, растворенную в теплой воде, (ниже в настоящем документе обозначаемую термином «раствор молибдена»). После этого сюда же добавляют раствор, содержащий нитрат в качестве источника элемента для каждого элемента, такого как висмут, церий, железо, хром, никель, магний, цинк, марганец, кобальт, рубидий, цезий или калий, растворенного в водном растворе азотной кислоты, (ниже в настоящем документе обозначаемый термином «водный раствор нитрата металла») в целях получения суспензии предшественника катализатора. В альтернативном варианте, от суспензии предшественника катализатора не требуется обязательное содержание всех элементов, составляющих катализатор, и исходный материал для элемента, который не содержится в суспензии предшественника катализатора, может быть добавлен на каждой стадии до стадии высушивания или может быть добавлен к катализатору после высушивания при использовании способа, такого как импрегнирование.
[0029]
В способе получения вышеупомянутой суспензии исходного материала значение рН суспензии может быть изменено в результате подстраивания концентрации использованной азотной кислоты или добавления аммиачной воды к золю кремниевой кислоты, раствору молибдена или водному раствору нитрата металла. Также суспензия предшественника может быть получена в результате надлежащего добавления растворимого в воде полимера, такого как полиэтиленгликоль, метилцеллюлоза, поливиниловый спирт, полиакриловая кислота или полиакриламид, аминов, карбоновых кислот, аминокарбоновых кислот или других органических кислот к золю кремниевой кислоты, раствору молибдена или водному раствору нитрата металла. В числе данных добавок предпочтительными являются имидазол или карбоновая кислота, более предпочтительными являются нитрилотриуксусная кислота или щавелевая кислота, а еще более предпочтительной является щавелевая кислота. Первичные или вторичные амины, содержащие NH, могут генерировать гель раствора молибдена. Для случая использования данных аминов в качестве добавок размеры первичных частиц металла в суспензии предшественника катализатора имеют тенденцию к увеличению. Также предпочтительным является предварительное смешивание исходного материала для диоксида кремния и исходного материала для щавелевой кислоты.
[0030]
Уровень содержания карбоновой кислоты в суспензии предшественника катализатора предпочтительно находится в диапазоне от 0,01 до 0,10 молярного эквивалента при расчете на сумму металлических элементов, составляющих катализатор для аммоксидирования. Уровень содержания более предпочтительно находится в диапазоне от 0,02 до 0,07 молярного эквивалента. При уровне содержания карбоновой кислоты, составляющем 0,01 молярного эквивалента и более, получающийся в результате катализатор имеет тенденцию к дополнительному улучшению выхода акрилонитрила. При уровне содержания карбоновой кислоты, составляющем 0,10 молярного эквивалента и менее, на ступени производства катализатора подавляются выработка тепла, приписываемая разложению карбоновой кислоты, и возникновение трещин в частицах катализатора. Таким образом, прочность получающегося в результате катализатора имеет тенденцию к дополнительному улучшению.
[0031]
Ниже в настоящем документе будет подробно описываться способ контроля размеров частиц для первичных частиц металла, составляющих агрегат, в вышеупомянутой суспензии предшественника катализатора.
[0032]
Концентрация свободной кислоты представляет собой следующий далее параметр.
Концентрация свободной кислоты (%) = молекулярная масса кислоты × (количество молей использованной кислоты - количество молей использованного основания) / масса полной суспензии предшественника катализатора × 100
В данном контексте термин «кислота» относится к сильной кислоте и не соответствует слабой кислоте, такой как карбоновая кислота. При высокой концентрации свободной кислоты увеличивается количество металла, растворенного в жидкой фазе суспензии предшественника катализатора. Поэтому уменьшаются размеры первичных частиц металла в агрегате в суспензии. С другой стороны, при низкой концентрации свободной кислоты увеличивается количество осажденного металла. Поэтому увеличиваются размеры первичных частиц металла в агрегате в суспензии предшественника катализатора. Концентрация свободной кислоты предпочтительно находится в диапазоне от 0,1% и более до 1,2% и менее, более предпочтительно от 0,8% и более до 1,2% и менее.
[0033]
При использовании летучих кислоты и основания концентрация свободной кислоты может быть изменена вследствие улетучивания данных компонентов во время нагревания и перемешивания в ходе получения суспензии предшественника катализатора. Таким образом, предпочтительными являются проведение получения раствора исходного материала или перемешивание суспензии предшественника катализатора в закрытом контейнере.
[0034]
Массовое соотношение между оксидом металла и носителем в катализаторе для аммоксидирования предпочтительно находится в диапазоне оксид металла : носитель=от 55 : 45 до 65 : 35. Для случая увеличения уровня содержания носителя размер первичных частиц металла имеет тенденцию к уменьшению несмотря на отсутствие ясности в отношении причин для этого. С точки зрения прочности, такой как сопротивление разрушению или сопротивление истиранию, в практических условиях уровень содержания носителя предпочтительно составляет 35% (масс.) и более.
[0035]
Доля совокупной массы металлического компонента и носителя в качестве исходных материалов по отношению к массе полной суспензии предшественника катализатора предпочтительно находится в диапазоне от 10% (масс.) и более до 40% (масс.) и менее, более предпочтительно от 20% (масс.) и более до 35% (масс.) и менее. При значении % (масс.) металла и носителя при расчете на полную суспензию предшественника катализатора, равном или большем в сопоставлении с нижним предельным значением, увеличивается абсолютное количество металла в суспензии предшественника катализатора при одновременном уменьшении количества металла, растворенного в жидкой фазе. Поэтому увеличиваются размеры частиц для первичных частиц металла, составляющих агрегат. При значении % (масс.) металла и носителя при расчете на полную суспензию предшественника катализатора, равном или меньшем в сопоставлении с верхним предельным значением, уменьшается вязкость суспензии предшественника катализатора таким образом, что суспензия предшественника катализатора характеризуется благоприятной текучестью. Таким образом, во время распылительного высушивания может быть подавлено неудовлетворительное профилирование порошкообразного катализатора.
[0036]
Перемешивающая способность во время смешивания раствора предпочтительно находится в диапазоне от 50 об/мин и более до 400 об/мин и менее. При перемешивающей способности, равной или большей в сопоставлении с нижним предельным значением, растворы смешиваются в достаточной степени таким образом, что размер первичных частиц металла уменьшается. При перемешивающей способности, равной или меньшей в сопоставлении с верхним предельным значением, во время перемешивания суспензии предотвращается захватывание воздушных пузырьков. Таким образом, во время распылительного высушивания может быть подавлено неудовлетворительное профилирование порошкообразного катализатора.
[0037]
Предпочтительным является постепенное увеличение перемешивающей способности во время смешивания растворов. Предпочтительным является постепенное увеличение перемешивающей способности таким образом, чтобы она находилась бы в диапазоне от 120 об/мин и более до 300 об/мин и менее на ступени (2) добавления водного раствора молибдена к водному раствору диоксида кремния и в диапазоне от 150 об/мин и более до 400 об/мин и менее на ступени (3) добавления водного раствора нитрата металла к водному раствору диоксида кремния-молибдена. Перемешивающая способность более предпочтительно находится в диапазоне от 150 об/мин и более до 250 об/мин и менее на ступени (2) и в диапазоне от 150 об/мин и более до 300 об/мин и менее на ступени (3), еще более предпочтительно в диапазоне от 180 об/мин и более до 200 об/мин и менее на ступени (2) и в диапазоне от 200 об/мин и более до 300 об/мин и менее на ступени (3).
[0038]
При смешивании растворов исходных материалов (например, в вышеупомянутом показательном варианте осуществления при смешивании раствора диоксида кремния и раствора молибдена или при смешивании смешанного раствора диоксида кремния и молибдена и водного раствора нитрата) предпочтительным является завершение смешивания вновь добавленного раствора в течение от 15 секунд и более до 3 минут и менее. При времени смешивания, равном или большем в сопоставлении с нижним предельным значением, растворы смешиваются однородно таким образом, что улучшаются эксплуатационные характеристики для получающегося в результате катализатора. При времени смешивания, равном или меньшем в сопоставлении с верхним предельным значением, уменьшаются размеры частиц для первичных частиц металла, составляющих агрегат, несмотря на отсутствие ясности в отношении причин для этого.
[0039]
В настоящем варианте осуществления размеры частиц для первичных частиц металла, составляющих агрегат, измеряют при использовании устройства AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. Измерение проводят при использовании суспензии предшественника катализатора в неразбавленной форме и жидкой фазы суспензии. В качестве жидкой фазы определяется фильтрат, полученный в результате фильтрования суспензии предшественника катализатора при использовании фильтра, имеющего отверстие 1 мкм, а в качестве твердой фазы определяется остаток в фильтрате, который не прошел через фильтр. При фактическом проведении измерения при использовании сканирующего электронного микроскопа (СЭМ) в отношении пленки реплики жидкой фазы, полученной при использовании вышеупомянутого способа получения реплик с замороженных срезов, в жидкой фазе не наблюдали ни агрегата, содержащегося в суспензии предшественника катализатора в неразбавленной форме, ни первичных частиц металла, составляющих агрегат. Агрегат суспензии становится похожим на лепешку и остается в качестве остатка на фильтровальной бумаге во время фильтрования. Говоря вкратце, твердая фаза может быть определена в качестве кластера агрегатов в суспензии предшественника катализатора. В данном контексте жидкое вещество, содержащееся в остатке в фильтрате непосредственно после фильтрования, рассматривается в качестве жидкой фазы (прикрепившейся жидкой фазы) и определяется исходя из уменьшения массы после вакуумного высушивания остатка в фильтрате при 60°С на протяжении 18 часов. В данном отношении состав прикрепившейся жидкой фазы является тем же самым, что и состав фильтрата. Масса твердой фазы является массой после высушивания вышеупомянутого остатка в фильтрате, а масса жидкой фазы является совокупной массой фильтрата и прикрепившейся жидкой фазы. В целях получения спектра затухания ультразвука, произведенного из агрегата в суспензии предшественника катализатора, спектр затухания ультразвука для одной только жидкой фазы вычитают из спектра затухания ультразвука для суспензии предшественника катализатора. Полученный спектр затухания ультразвука для твердой фазы может быть проанализирован при использовании сопутствующего программного обеспечения, используя значение % (масс.) агрегата в суспензии предшественника катализатора, истинную плотность агрегата и проницаемость агрегата в целях вычисления размеров частиц. Значение % (масс.) агрегата в суспензии предшественника катализатора рассчитывают в результате деления массы после высушивания твердой фазы на массу полной суспензии предшественника катализатора. Истинную плотность и проницаемость агрегата измеряют при использовании порошка, полученного в результате размалывания высушенной твердой фазы при использовании способа, известного на современном уровне техники.
[0040]
В примерах и сравнительных примерах, упомянутых ниже, модальный диаметр левого пика нанометрового порядка, продемонстрированного на фигуре в результатах анализа, полученных при использовании метода, описанного выше, определяли в качестве среднего размера частиц для первичных частиц металла. Это имеет в своей основе доминирование левого пика нанометрового порядка на фигуре с точки зрения количества частиц, результаты наблюдения агрегата в суспензии при использовании вышеупомянутого метода и маленькое значение % (масс.) носителя (например, диоксида кремния) при расчете на совокупную массу агрегатов, полученных при использовании способа получения суспензии предшественника катализатора в примерах или сравнительных примерах, (3% (масс.) и менее). Значение % (масс.) носителя в агрегате рассчитывали в результате полного растворения высушенного продукта твердой фазы в смешанном водном растворе, содержащем 5 частей хлористо-водородной кислоты при 36% (масс.), 10 частей иодисто-водородной кислоты при 57% (масс.) и 2,5 части фтористо-водородной кислоты при 47% (масс.), и впоследствии перевода в цифровое выражение количества кремния при использовании эмиссионного спектрофотометра с индуктивно-связанной плазмой.
[0041]
Стадия (2) является стадией высушивания суспензии предшественника катализатора в целях получения сухих частиц. Данная стадия предпочтительно является стадией распылительного высушивания суспензии предшественника катализатора в целях получения сухих частиц. В результате распылительного высушивания суспензии предшественника катализатора могут быть получены сферические мелкие частицы, подходящие для использования при реакции в псевдожиженном слое. В качестве аппаратуры для распылительного высушивания может быть использована общераспространенная аппаратура, такая как аппаратура, относящаяся к типу с вращающимся диском или типу с соплом. Размер частиц получающегося в результате катализатора для аммоксидирования может быть подстроен в результате подстраивания условий распылительного высушивания. Для использования в качестве катализатора в псевдоожиженном слое размер частиц катализатора для аммоксидирования предпочтительно находится в диапазоне от 25 до 180 мкм. Один пример условий получения частиц катализатора для аммоксидирования, характеризующихся предпочтительным размером частиц, включает распылительное высушивание, которое проводят при использовании аппаратуры для центробежного распыления, снабженной тарелкообразным ротатором, установленным в центре верхней части сушилки, и выдерживание сушилки при температуре воздуха на впуске в диапазоне от 180 до 250°С и при температуре на выпуске в диапазоне от 100 до 150°С.
[0042]
Данная стадия (3) является стадией прокаливания сухих частиц, полученных в результате высушивания, в целях получения катализатора для аммоксидирования. Вследствие возможности содержания сухими частицами азотной кислоты предпочтительным является проведение денитрационной обработки до прокаливания. Денитрационная обработка предпочтительно включает проведение нагревания при температуре в диапазоне от 150 до 450°С на протяжении от 1,5 до 3 часов. Прокаливание может быть проведено в воздушной атмосфере. Температура прокаливания предпочтительно находится в диапазоне от 550 до 650°С. При температуре прокаливания, составляющей 550°С и более, рост кристаллов протекает в достаточной степени таким образом, что селективность по акрилонитрилу для получающегося в результате катализатора имеет тенденцию к дополнительному улучшению. При температуре прокаливания, составляющей 650°С и менее, увеличивается площадь поверхности у получающегося в результате катализатора для аммоксидирования таким образом, что активность в реакции для пропилена имеет тенденцию к дополнительному улучшению. Атмосфера газа для использования при денитрации и прокаливании может быть атмосферой окисленного газа, содержащей кислород, или может быть атмосферой инертного газа, например, азота. Обыкновенно используют воздух.
[0043]
Способ производства акрилонитрила, соответствующий настоящему варианту осуществления, включает стадию реакции, заключающуюся в проведении реакции между пропиленом, молекулярным кислородом и аммиаком, (реакции аммоксидирования) в присутствии катализатора для аммоксидирования, полученного при использовании вышеупомянутого способа, в целях производства акрилонитрила. Производство акрилонитрила в результате прохождения реакции аммоксидирования может быть проведено при использовании реактора с неподвижным слоем или реактора с псевдоожиженным слоем (реакционной емкости с псевдоожиженным слоем). В их числе с точки зрения эффективного отвода тепла, выработанного во время реакции, и улучшения выхода акрилонитрила предпочтительным является реактор с псевдоожиженным слоем (реакционная емкость с псевдоожиженным слоем). Для случая проведения стадии реакции в реакционной емкости с псевдоожиженным слоем предпочтительными являются предварительная подача катализатора для аммоксидирования в реакционную емкость с псевдоожиженным слоем и при одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проведение реакции аммоксидирования. От исходных материалов для пропилена и аммиака при реакции аммоксидирования не требуется обязательная демонстрация высокой степени чистоты, и может быть использован технический сорт. При наличии источника молекулярного кислорода в виде воздуха молярное соотношение между пропиленом, аммиаком и воздухом (пропилен/аммиак/воздух) в исходном материале для газа предпочтительно находится в диапазоне 1/(от 0,8 до 1,4)/(от 7 до 12), более предпочтительно в диапазоне 1/(от 0,9 до 1,3)/(от 8 до 11). Температура реакции находится в диапазоне предпочтительно от 350 до 550°С, более предпочтительно от 400 до 500°С. Давление реакции предпочтительно находится в диапазоне от нормального атмосферного давления до 0,3 МПа. Время контактирования между исходным материалом для газа и катализатором для аммоксидирования предпочтительно находится в диапазоне от 2 до 7 секунд, более предпочтительно от 3 до 6 секунд.
[0044]
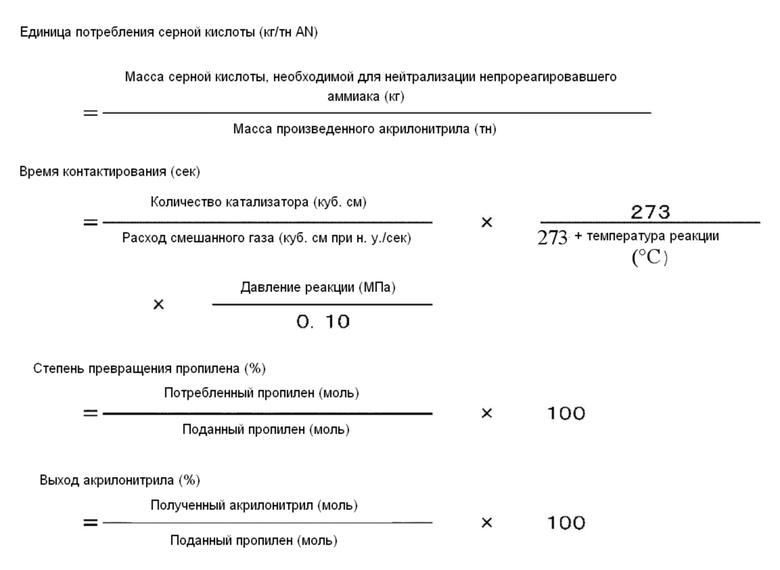
На реакционную трубку для использования при реакции аммоксидирования для пропилена конкретных ограничений не накладывают. Например, может быть использована стеклянная трубка Pyrex(R), имеющая внутренний диаметр 25 мм и включающая шестнадцать листов проволочной сетки с 10 отверстиями на один линейный дюйм (0,393 отверстия на один миллиметр) с интервалами между листами 1 см. На один конкретный пример реакции аммоксидирования конкретных ограничений не накладывают. Например, сначала количество катализатора для аммоксидирования задают равным 50 куб. см, температуру реакции задают равной 430°С и давление реакции задают равным 0,17 МПа. Через стеклянную трубку обеспечивают прохождение смешанного газа (пропилена, аммиака, кислорода и гелия), содержащего 9% (об.) пропилена. После этого объемное соотношение между аммиаком и пропиленом задают таким образом, чтобы единица потребления серной кислоты, определенная в соответствии с выражением, представленным ниже, составляла бы 20 кг/тн AN. В данном отношении молярное соотношение аммиак/пропилен определяется в виде N/C. Объемное соотношение между кислородом и пропиленом задают таким образом, чтобы концентрация кислорода в газе на выпуске реактора составляла бы 0,2±0,02% (об.). В данном отношении молярное количество кислорода преобразуется в молярное количество воздуха при том условии, что воздух содержит 21% кислорода. В данном отношении молярное соотношение воздух/пропилен определяется в виде А/С. Время контактирования, определенное в соответствии с выражением, представленным ниже, может быть изменено в результате изменения расхода смешанного газа. Тем самым, степень превращения пропилена, определенная в соответствии с выражением, представленным ниже, может быть задана равной 99,3±0,2%. Единица потребления серной кислоты, время контактирования, степень превращения пропилена и выход акрилонитрила определяются в соответствии со следующими далее выражениями.
Примеры
[0045]
Ниже в настоящем документе настоящий вариант осуществления будет описываться более подробно при обращении к примерам. Однако, на настоящий вариант осуществления примерами, представленными ниже, ограничений не накладывают. Состав катализатора, описанный в примерах и сравнительных примерах, имеет то же самое значение, что и состав каждого добавленного элемента.
[0046]
[Размер первичных частиц металла]
В суспензиях предшественников катализаторов, полученных в примерах и сравнительных примерах, размеры частиц для первичных частиц металла, составляющих агрегат, измеряли следующим далее образом при использовании устройства AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. Измерение проводили при использовании суспензии предшественника катализатора в неразбавленной форме и жидкой фазы суспензии. В качестве жидкой фазы определялся фильтрат, полученный в результате фильтрования суспензии предшественника катализатора при использовании фильтра, имеющего отверстие 1 мкм, а в качестве твердой фазы определялся остаток в фильтрате, который не прошел через фильтр. Говоря вкратце, твердая фаза определялась в качестве кластера агрегатов в суспензии предшественника катализатора. В данном контексте жидкое вещество, содержащееся в остатке в фильтрате непосредственно после фильтрования, рассматривалось в качестве жидкой фазы (прикрепившейся жидкой фазы) и определялось исходя из уменьшения массы после вакуумного высушивания остатка в фильтрате при 60°С на протяжении 18 часов. В данном отношении состав прикрепившейся жидкой фазы являлся тем же самым, что и состав фильтрата. Масса твердой фазы являлась массой после высушивания вышеупомянутого остатка в фильтрате, а масса жидкой фазы являлась совокупной массой фильтрата и прикрепившейся жидкой фазы. В целях получения спектра затухания ультразвука, произведенного из агрегата в суспензии предшественника катализатора, спектр затухания ультразвука для одной только жидкой фазы вычитали из спектра затухания ультразвука для суспензии предшественника катализатора. Полученный спектр затухания ультразвука для твердой фазы анализировали при использовании программного обеспечения, используя значение % (масс.) агрегата в суспензии предшественника катализатора, истинную плотность агрегата и проницаемость агрегата в целях вычисления размеров частиц. Значение % (масс.) агрегата в суспензии предшественника катализатора рассчитывали в результате деления массы после высушивания твердой фазы на массу полной суспензии предшественника катализатора. Истинную плотность агрегата измеряли при использовании пикнометра, относящегося к типу Wadon, и порошка, полученного в результате размалывания высушенной твердой фазы. Проницаемость агрегата определяли в результате добавления порошка, полученного в результате размалывания высушенной твердой фазы, в различные растворители со следующим далее измерением при использовании аппаратуры для измерения проницаемости. В данном отношении измеряли проницаемости растворителя, содержащего порошок, полученный в результате размалывания высушенной твердой фазы, и одного только растворителя, и растворитель выбирался при условии отсутствия разницы между ними. Проницаемость растворителя гипотетически предполагалась проницаемостью высушенного продукта твердой фазы.
[0047]
Фигура демонстрирует концептуальную диаграмму для одного примера результатов измерения при проведении измерения для агрегата в суспензии предшественника катализатора при использовании метода затухания ультразвука. Модальный диаметр левого пика нанометрового порядка, продемонстрированного на фигуре в результатах анализа, полученных при использовании метода, описанного выше, определяли в качестве «среднего размера частиц для первичных частиц металла». Это имеет в своей основе доминирование левого пика нанометрового порядка на фигуре с точки зрения количества частиц, результаты наблюдения агрегата в суспензии при использовании вышеупомянутого метода и маленькое значение % (масс.) носителя (например, диоксида кремния) при расчете на совокупную массу агрегатов, полученных при использовании способа получения суспензии предшественника катализатора в примерах или сравнительных примерах, (3% (масс.) и менее). Значение % (масс.) носителя в агрегате рассчитывали в результате полного растворения высушенного продукта твердой фазы в смешанном водном растворе, содержащем 5 частей хлористо-водородной кислоты при 36% (масс.), 10 частей иодисто-водородной кислоты при 57% (масс.) и 2,5 части фтористо-водородной кислоты при 47% (масс.), и впоследствии перевода в цифровое выражение количества кремния при использовании эмиссионного спектрофотометра с индуктивно-связанной плазмой.
[0048]
[Единица потребления серной кислоты, время контактирования, степень превращения пропилена и выход акрилонитрила]
Реакционная трубка для использования при реакции аммоксидирования для пропилена являлась стеклянной трубкой Pyrex(R), имеющей внутренний диаметр 25 мм и включающей шестнадцать листов проволочной сетки с 10 отверстиями на один линейный дюйм (0,393 отверстия на один миллиметр) с интервалами между листами в 1 см. При реакции аммоксидирования количество катализатора для аммоксидирования задавали равным 50 куб. см, температуру реакции задавали равной 430°С и давление реакции задавали равным 0,17 МПа. Через стеклянную трубку обеспечивали прохождение смешанного газа (пропилена, аммиака, кислорода и гелия), содержащего 9% (об.) пропилена. После этого объемное соотношение между аммиаком и пропиленом задавали таким образом, чтобы единица потребления серной кислоты, определенная в соответствии с выражением, представленным ниже, составляла бы 20 кг/тн AN. В данном отношении молярное соотношение аммиак/пропилен определяли в виде N/C. Объемное соотношение между кислородом и пропиленом задавали таким образом, чтобы концентрация кислорода в газе на выпуске реактора составляла бы 0,2±0,02% (об.). В данном отношении молярное количество кислорода преобразовывалось в молярное количество воздуха при том условии, что воздух содержал 21% кислорода. В данном отношении молярное соотношение воздух/пропилен определяли в виде А/С. Время контактирования, определенное в соответствии с выражением, представленным ниже, изменяли в результате изменения расхода смешанного газа. Тем самым, степень превращения пропилена, определенную в соответствии с выражением, представленным ниже, задавали равной 99,3±0,2%. Единица потребления серной кислоты, время контактирования, степень превращения пропилена и выход акрилонитрила определялись в соответствии со следующими далее выражениями.
[0049]
[Пример 1]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г дигидрата щавелевой кислоты, растворенного в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 200 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 250 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,1% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 61 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее.
Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0050]
[Пример 2]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г дигидрата щавелевой кислоты, растворенного в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 200 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 275 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,1% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 57 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее.
Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0051]
[Пример 3]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 200 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 250 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,1% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 64 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее.
Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0052]
[Пример 4]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г дигидрата щавелевой кислоты, растворенного в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 200 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 15 секунд при перемешивании со скоростью вращения при перемешивании 250 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,6% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 52 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее. Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0053]
[Пример 5]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г дигидрата щавелевой кислоты, растворенного в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 200 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 75 секунд при перемешивании со скоростью вращения при перемешивании 250 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,6% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 67 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее. Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0054]
[Пример 6]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 667 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 12,5 г дигидрата щавелевой кислоты, растворенного в 143,8 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 1310,8 г теплой воды при 60°С растворяли 238,3 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 17,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 297,65 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 25,8 г нитрата висмута [Bi(NO3)3⋅5H2O], 48,0 г нитрата церия [Ce(NO3)3⋅6H2O], 85,6 г нитрата железа [Fe(NO3)3⋅9H2O], 101,6 г нитрата никеля [Ni(NO3)2⋅6H2O], 129,4 г нитрата кобальта [Co(NO3)2⋅6H2O] и 2,5 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 200 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 250 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 10,6% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 55 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее. Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 605°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0055]
[Пример 7]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г нитрилотриуксусной кислоты, растворенной в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 200 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 250 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 21,7% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 55 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее. Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0056]
[Пример 8]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г щавелевой кислоты, растворенной в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 200 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 250 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой и, кроме того, сюда же добавляли 20 г водного аммиачного раствора при 28% (масс.) в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 19,0% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 97 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее. Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 590°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0057]
[Пример 9]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г щавелевой кислоты, растворенной в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 200 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 250 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой и, кроме того, сюда же добавляли 28 г водного аммиачного раствора при 28% (масс.) в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 22,5% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 123 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее. Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 590°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0058]
[Пример 10]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г дигидрата щавелевой кислоты, растворенного в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 150 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 160 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,1% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 183 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее.
Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0059]
[Пример 11]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 120 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 150 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,1% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 188 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее.
Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0060]
[Пример 12]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г дигидрата щавелевой кислоты, растворенного в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 180 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 200 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,6% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 176 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее. Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0061]
[Сравнительный пример 1]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г дигидрата щавелевой кислоты, растворенного в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 120 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,1% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 217 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее.
Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0062]
[Сравнительный пример 2]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 120 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 1 минуты при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,1% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 220 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее.
Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0063]
[Сравнительный пример 3]
При использовании следующих далее методик производили катализатор, у которого оксид металла, характеризующийся составом металлического компонента, описывающимся формулой Mo12,00Bi0,47Ce0,99Fe1,88Ni3,08Co3,90Rb0,15, наносили на носитель в виде диоксида кремния (оксид металла: 60% (масс.), диоксид кремния: 40% (масс.)).
Сначала в контейнере, имеющем крышку, располагали и при 40°С выдерживали 1333 г золя кремниевой кислоты, содержащей 30% (масс.) SiO2, и сюда же при перемешивании со скоростью вращения при перемешивании 120 об/мин добавляли 25,0 г дигидрата щавелевой кислоты, растворенного в 287,5 г воды. После закрытия крышки смесь перемешивали на протяжении 10 минут в целях получения водного раствора диоксида кремния. В еще одном контейнере, имеющем крышку, располагали и в 850,8 г теплой воды при 60°С растворяли 476,7 г парамолибдата аммония [(NH4)6Mo7O24⋅4H2O]. После охлаждения до 45°С сюда же добавляли 35,8 г водного аммиачного раствора при 15% (масс.) в целях получения водного раствора молибдена. В одном дополнительном альтернативном контейнере, имеющем крышку, в 393,3 г раствора азотной кислоты при 16,6% (масс.) растворяли и при 40°С выдерживали 51,6 г нитрата висмута [Bi(NO3)3⋅5H2O], 96,1 г нитрата церия [Ce(NO3)3⋅6H2O], 171,2 г нитрата железа [Fe(NO3)3⋅9H2O], 203,4 г нитрата никеля [Ni(NO3)2⋅6H2O], 258,8 г нитрата кобальта [Co(NO3)2⋅6H2O] и 5,0 г нитрата рубидия [RbNO3] в целях получения водного раствора нитрата. К водному раствору диоксида кремния, выдерживаемому при 40°С, при перемешивании со скоростью вращения при перемешивании 180 об/мин в течение 1 минуты добавляли водный раствор молибдена в целях получения водного раствора диоксида кремния-молибдена. Полученный раствор перемешивали на протяжении 5 минут, а после этого непрерывно выдерживали при 40°С и к водному раствору диоксида кремния-молибдена в течение 2 минут при перемешивании со скоростью вращения при перемешивании 250 об/мин добавляли водный раствор нитрата в целях получения суспензии исходного материала. Суспензию исходного материала перемешивали при 40°С на протяжении 45 минут при одновременном покрытии крышкой в целях получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу. Агрегирование первичных частиц металла и частиц диоксида кремния можно было наблюдать в результате извлечения агрегатов в суспензии предшественника катализатора на пленку реплики при использовании способа получения реплик с замороженных срезов и проведения измерения при использовании микроскопа СЭМ. При данной операции в комбинации с этим могло быть использовано измерение при использовании метода ЭДРС в целях подтверждения того, что частицы, составляющие агрегат, представляют собой металл и диоксид кремния. Полученную суспензию предшественника катализатора использовали при измерении, которое соответствует представленному выше описанию изобретения, в целях выявления значения % (масс.) агрегатов в суспензии предшественника катализатора, равного 16,6% (масс.), истинной плотности агрегатов, равной 2,7 г/мл, и проницаемости агрегата, равной 6. При использовании данных параметров анализировали спектр затухания для суспензии предшественника катализатора, измеренный при использовании метода затухания ультразвука, используя устройство AcoustoSizer IIX от компании Kyowa Interface Science Co., Ltd.. В качестве результата средний размер частиц для первичных частиц металла, составляющих агрегаты, рассчитали равным 212 нм. Диапазон размеров частиц для первичных частиц металла соответствовал 1 мкм и менее. Полученную суспензию предшественника катализатора высушивали при использовании распылительной сушилки, относящейся к типу с вращающимся диском, в целях получения сухих частиц. При данной операции температуру воздуха на впуске в сушилку задавали равной 230°С, а температуру воздуха на выпуске задавали равной 110°С. Скорость вращения диска задавали равной 12500 об/мин. Полученные сухие частицы выдерживали при 200°С на протяжении 5 минут, нагревали при 2,5°С/мин от 200°С до 450°С и выдерживали при 450°С на протяжении 20 минут для денитрации. Подвергнутые денитрации сухие частицы прокаливали при 595°С на протяжении 2 часов в целях получения катализатора для аммоксидирования. Полученный катализатор для аммоксидирования предварительно подавали в реакционную емкость с псевдоожиженным слоем. При одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проводили реакцию между пропиленом, молекулярным кислородом и аммиаком (реакцию аммоксидирования) в целях производства акрилонитрила. Определяли молярное соотношение аммиак/пропилен (N/C), молярное соотношение воздух/пропилен (А/С) и выход акрилонитрила. Результаты продемонстрированы в таблице 1.
[0064]
[Таблица 1]
[0065]
Как это с очевидностью следует исходя из таблицы 1, все катализаторы для аммоксидирования, полученные в примерах от 1 до 12, были способны обеспечить синтезирование акрилонитрила при высоком выходе. С другой стороны, катализаторы для аммоксидирования, полученные в сравнительных примерах от 1 до 3, характеризовались низким выходом акрилонитрила в сопоставлении тем, что имеет место в примерах от 1 до 12, несмотря на наличие того же самого состава металлов, что и в примерах от 1 до 12.
[0066]
Настоящая заявка имеет в своей основе японскую патентную заявку, поданную 23 августа 2018 года, (японскую патентную заявку № 2018-156514), содержание которой во всей своей полноте посредством ссылки на нее включается в настоящий документ.
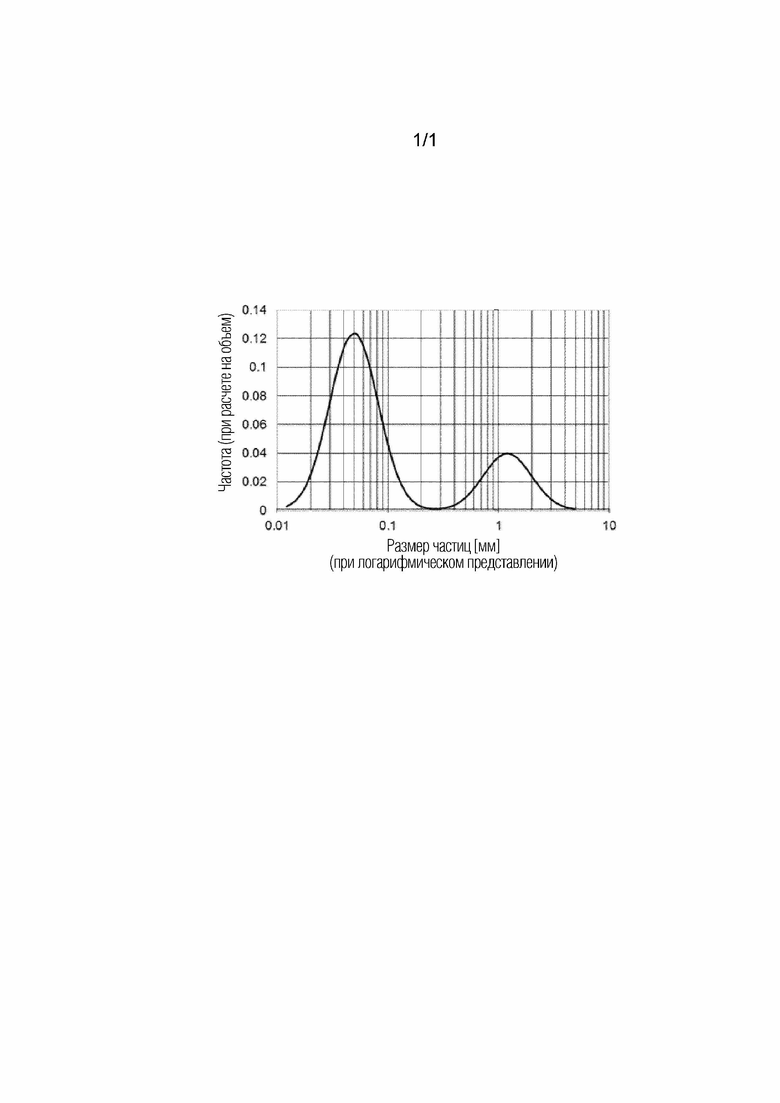
В настоящем изобретении предлагается способ производства катализатора для аммоксидирования. Способ производства катализатора для аммоксидирования включает стадии: получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу; высушивания суспензии предшественника катализатора в целях получения сухих частиц; и прокаливания сухих частиц в целях получения катализатора для аммоксидирования, где твердая фаза суспензии предшественника катализатора содержит агрегат, содержащий металл и носитель, первичные частицы металла, составляющие агрегат, характеризуются размером частиц, составляющим 1 мкм и менее, и средний размер частиц для первичных частиц металла находится в диапазоне от 40 нм и более до 200 нм и менее, носитель содержит диоксид кремния, где катализатор для аммоксидирования содержит сложный оксид металлов, характеризующийся составом, описывающимся следующей далее общей формулой (1): Mo12BiaFebXcYdZeOf (1), где Х представляет собой один или несколько элементов, выбираемых из группы, состоящей из никеля, кобальта, магния, кальция, цинка, стронция и бария; Y представляет собой один или несколько элементов, выбираемых из группы, состоящей из церия, хрома, лантана, неодима, иттрия, празеодима, самария, алюминия, галлия и индия; Z представляет собой один или несколько элементов, выбираемых из группы, состоящей из калия, рубидия и цезия; а представляет собой атомное соотношение между висмутом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤а≤2,0; b представляет собой атомное соотношение между железом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤b≤3,0; c представляет собой атомное соотношение между Х и 12 атомами молибдена и удовлетворяет неравенству 0,1≤c≤10,0; d представляет собой атомное соотношение между Y и 12 атомами молибдена и удовлетворяет неравенству 0,1≤d≤3,0; e представляет собой атомное соотношение между Z и 12 атомами молибдена и удовлетворяет неравенству 0,01≤e≤2,0; и f представляет собой атомное соотношение между кислородом и 12 атомами молибдена и представляет собой количество атомов кислорода, необходимое для удовлетворения требований по валентности у других присутствующих элементов, на стадии высушивания суспензии предшественника катализатора в целях получения сухих частиц сушилку выдерживают при температуре воздуха на впуске в диапазоне от 180 до 250°С и при температуре на выпуске в диапазоне от 100 до 150°С, на стадии прокаливания сухих частиц в целях получения катализатора для аммоксидирования температура прокаливания находится в диапазоне от 550 до 650°С. Технический результат – создание способа производства катализатора для аммоксидирования, который демонстрирует высокий выход акрилонитрила, и способа производства акрилонитрила. 2 н. и 16 з.п. ф-лы, 1 ил., 1 табл., 12 пр.
1. Способ производства катализатора для аммоксидирования, включающий стадии:
получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу;
высушивания суспензии предшественника катализатора в целях получения сухих частиц; и
прокаливания сухих частиц в целях получения катализатора для аммоксидирования,
где
твердая фаза суспензии предшественника катализатора содержит агрегат, содержащий металл и носитель,
первичные частицы металла, составляющие агрегат, характеризуются размером частиц, составляющим 1 мкм и менее, и
средний размер частиц для первичных частиц металла находится в диапазоне от 40 нм и более до 200 нм и менее,
носитель содержит диоксид кремния, где катализатор для аммоксидирования содержит сложный оксид металлов, характеризующийся составом, описывающимся следующей далее общей формулой (1):
Mo12BiaFebXcYdZeOf, (1)
где Х представляет собой один или несколько элементов, выбираемых из группы, состоящей из никеля, кобальта, магния, кальция, цинка, стронция и бария; Y представляет собой один или несколько элементов, выбираемых из группы, состоящей из церия, хрома, лантана, неодима, иттрия, празеодима, самария, алюминия, галлия и индия; Z представляет собой один или несколько элементов, выбираемых из группы, состоящей из калия, рубидия и цезия; а представляет собой атомное соотношение между висмутом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤а≤2,0; b представляет собой атомное соотношение между железом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤b≤3,0; c представляет собой атомное соотношение между Х и 12 атомами молибдена и удовлетворяет неравенству 0,1≤c≤10,0; d представляет собой атомное соотношение между Y и 12 атомами молибдена и удовлетворяет неравенству 0,1≤d≤3,0; e представляет собой атомное соотношение между Z и 12 атомами молибдена и удовлетворяет неравенству 0,01≤e≤2,0; и f представляет собой атомное соотношение между кислородом и 12 атомами молибдена и представляет собой количество атомов кислорода, необходимое для удовлетворения требований по валентности у других присутствующих элементов,
на стадии высушивания суспензии предшественника катализатора в целях получения сухих частиц сушилку выдерживают при температуре воздуха на впуске в диапазоне от 180 до 250°С и при температуре на выпуске в диапазоне от 100 до 150°С,
на стадии прокаливания сухих частиц в целях получения катализатора для аммоксидирования температура прокаливания находится в диапазоне от 550 до 650°С.
2. Способ производства катализатора для аммоксидирования по п. 1, где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, концентрация свободной кислоты в суспензии находится в диапазоне от 0,1% и более до 1,2% и менее.
3. Способ производства катализатора для аммоксидирования по любому одному из пп. 1, 2, где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, концентрация свободной кислоты в суспензии находится в диапазоне от 0,8% и более до 1,2% и менее.
4. Способ производства катализатора для аммоксидирования по любому одному из пп. 1-3, где катализатор для аммоксидирования содержит носитель, и уровень содержания носителя в катализаторе для аммоксидирования находится в диапазоне от 35 до 45% (масс.).
5. Способ производства катализатора для аммоксидирования по любому одному из пп. 1-4, где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, доля совокупной массы металлического компонента и носителя в качестве исходных материалов по отношению к массе полной суспензии предшественника катализатора находится в диапазоне от 10% (масс.) и более до 40% (масс.) и менее.
6. Способ производства катализатора для аммоксидирования по любому одному из пп. 1-5, где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, доля совокупной массы металлического компонента и носителя в качестве исходных материалов по отношению к массе полной суспензии предшественника катализатора находится в диапазоне от 20% (масс.) и более до 35% (масс.) и менее.
7. Способ производства катализатора для аммоксидирования по любому одному из пп. 1-6, где стадия прокаливания сухих частиц в целях получения катализатора для аммоксидирования включает денитрационную обработку до прокаливания, и денитрационная обработка включает проведение нагревания при температуре в диапазоне от 150 до 450°С на протяжении от 1,5 до 3 ч.
8. Способ производства акрилонитрила, включающий стадии:
получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу;
высушивания суспензии предшественника катализатора в целях получения сухих частиц;
прокаливания сухих частиц в целях получения катализатора для аммоксидирования; и
предварительной подачи катализатора для аммоксидирования в реакционную емкость с псевдоожиженным слоем и при одновременном циркулировании катализатора в реакционной емкости с псевдоожиженным слоем проведения реакции между пропиленом, молекулярным кислородом и аммиаком в целях получения акрилонитрила,
где твердая фаза суспензии предшественника катализатора содержит агрегат, содержащий металл и носитель, первичные частицы металла, составляющие агрегат, характеризуются размером частиц, составляющим 1 мкм и менее, и средний размер частиц для первичных частиц металла находится в диапазоне от 40 нм и более до 200 нм и менее,
носитель содержит диоксид кремния,
где катализатор для аммоксидирования содержит сложный оксид металлов, характеризующийся составом, описывающимся следующей далее общей формулой (1):
Mo12BiaFebXcYdZeOf, (1)
где Х представляет собой один или несколько элементов, выбираемых из группы, состоящей из никеля, кобальта, магния, кальция, цинка, стронция и бария; Y представляет собой один или несколько элементов, выбираемых из группы, состоящей из церия, хрома, лантана, неодима, иттрия, празеодима, самария, алюминия, галлия и индия; Z представляет собой один или несколько элементов, выбираемых из группы, состоящей из калия, рубидия и цезия; а представляет собой атомное соотношение между висмутом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤а≤2,0; b представляет собой атомное соотношение между железом и 12 атомами молибдена и удовлетворяет неравенству 0,1≤b≤3,0; c представляет собой атомное соотношение между Х и 12 атомами молибдена и удовлетворяет неравенству 0,1≤c≤10,0; d представляет собой атомное соотношение между Y и 12 атомами молибдена и удовлетворяет неравенству 0,1≤d≤3,0; e представляет собой атомное соотношение между Z и 12 атомами молибдена и удовлетворяет неравенству 0,01≤e≤2,0; и f представляет собой атомное соотношение между кислородом и 12 атомами молибдена и представляет собой количество атомов кислорода, необходимое для удовлетворения требований по валентности у других присутствующих элементов,
на стадии высушивания суспензии предшественника катализатора в целях получения сухих частиц сушилку выдерживают при температуре воздуха на впуске в диапазоне от 180 до 250°С и при температуре на выпуске в диапазоне от 100 до 150°С,
на стадии прокаливания сухих частиц в целях получения катализатора для аммоксидирования температура прокаливания находится в диапазоне от 550 до 650°С.
9. Способ производства акрилонитрила по п. 8, где
источник молекулярного кислорода представляет собой воздух, и
молярное соотношение между аммиаком и воздухом и пропиленом находится в диапазоне 1/(от 0,8 до 1,4)/(от 7 до 12) применительно к соотношению пропилен/аммиак/воздух.
10. Способ производства акрилонитрила по п. 9, где
источник молекулярного кислорода представляет собой воздух, и
молярное соотношение между аммиаком и воздухом и пропиленом находится в диапазоне 1/(от 0,9 до 1,3)/(от 8 до 11) применительно к соотношению пропилен/аммиак/воздух.
11. Способ производства акрилонитрила по любому одному из пп. 8-10, где температура, при которой пропилен, молекулярный кислород и аммиак вступают в реакцию в присутствии катализатора для аммоксидирования, находится в диапазоне от 350 до 550°С.
12. Способ производства акрилонитрила по любому одному из пп. 8-11, где температура, при которой пропилен, молекулярный кислород и аммиак вступают в реакцию в присутствии катализатора для аммоксидирования, находится в диапазоне от 400 до 500°С.
13. Способ производства акрилонитрила по любому одному из пп. 8-12, где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, концентрация свободной кислоты в суспензии находится в диапазоне от 0,1% и более до 1,2% и менее.
14. Способ производства акрилонитрила по любому одному из пп. 8-13, где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, концентрация свободной кислоты в суспензии находится в диапазоне от 0,8% и более до 1,2% и менее.
15. Способ производства акрилонитрила по любому одному из пп. 8-14, где катализатор для аммоксидирования содержит носитель, и уровень содержания носителя в катализаторе для аммоксидирования находится в диапазоне от 35 до 45% (масс.).
16. Способ производства акрилонитрила по любому одному из пп. 8-15, где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, доля совокупной массы металлического компонента и носителя в качестве исходных материалов по отношению к массе полной суспензии предшественника катализатора находится в диапазоне от 10% (масс.) и более до 40% (масс.) и менее.
17. Способ производства акрилонитрила по любому одному из пп. 8-16, где на стадии получения суспензии предшественника катализатора, содержащей жидкую фазу и твердую фазу, доля совокупной массы металлического компонента и носителя в качестве исходных материалов по отношению к массе полной суспензии предшественника катализатора находится в диапазоне от 20% (масс.) и более до 35% (масс.) и менее.
18. Способ производства акрилонитрила по любому одному из пп. 8-17, где стадия прокаливания сухих частиц в целях получения катализатора для аммоксидирования включает денитрационную обработку до прокаливания, и денитрационная обработка включает проведение нагревания при температуре в диапазоне от 150 до 450°С на протяжении от 1,5 до 3 ч.
| JP 2015188801 A, 02.11.2015 | |||
| JP 2010172851 A, 12.08.2010 | |||
| JP 2010240593 A, 28.10.2010 | |||
| СМЕШАННЫЕ МЕТАЛЛООКСИДНЫЕ КАТАЛИЗАТОРЫ | 2012 |
|
RU2612976C2 |
| Способ получения акрилонитрила | 1979 |
|
SU957763A3 |
| WO 2018043007 A1, 08.03.2018 | |||
| JP 5491037 B1, 14.05.2014. | |||

