ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящей заявкой испрашивается приоритет по предварительной заявке США №62/376703, поданной 18 апреля 2016 г., предварительной заявке США №62/461317, поданной 21 февраля 2017 г. и предварительной заявке США №62/489292, поданной 24 апреля 2017 г., полное содержание каждой из которых тем самым включено посредством ссылки во всей своей полноте для всех назначений.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Воспалительные расстройства пищевода, такие как эозинофильный эзофагит (ЕоЕ), заболевание, характеризующееся высокими уровнями эозинофилов в пищеводе, а также гиперплазией базальной зоны, все чаще диагностируется у детей и взрослых. Многие аспекты этого заболевания остаются неясными, включая его этиологию, естественный ход развития заболевания и оптимальную терапию. ЕоЕ поражает все возрастные группы, но наиболее часто индивидов в возрасте от 20 до 50 лет. Симптомы ЕоЕ часто имитируют симптомы гастроэзофагеальной рефлюксной болезни (GERD) и включают рвоту, дисфагию, боль и затрудненное прохождение пищи. Данное заболевание протекает с болями, вызывает затруднение при глотании и провоцирует у пациентов другие осложнения. Зачастую ЕоЕ ошибочно диагностируется как GERD, что приводит к задержке начала адекватного лечения пациентов с ЕоЕ.
В настоящее время не разработано никаких местно вводимых противовоспалительных препаратов для лечения состояний, ассоциированных с воспалением в верхней части желудочно-кишечного тракта, в частности, воспалительных состояний пищевода, таких как ЕоЕ. Несмотря на то, что методы системного лечения кортикостероидами, такими как преднизолон, эффективны, применение этих терапевтических средств связано со значительными неблагоприятными эффектами, такими как угнетение гипоталамо-гипофизарно-надпочечниковой (НРА) оси, что отражается на уровнях кортизола в слюне, генерализованное угнетение иммунной функции и, особенно у детей, беспокоящие побочные эффекты в результате длительного системного воздействия включают задержку роста.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложены способы введения фармацевтических композиций, содержащих низкодозовый кортикостероид местного действия, для лечения, предупреждения, уменьшения интенсивности или задержки возникновения симптомов и/или воспаления, ассоциированных с воспалительным расстройством желудочно-кишечного тракта. В некоторых воплощениях, воспалительное расстройство желудочно-кишечного тракта локализовано в пищеводе. В некоторых воплощениях, воспалительное расстройство желудочно-кишечного тракта представляет собой эозинофильный эзофагит.
В некоторых воплощениях настоящего изобретения предложены способы лечения эозинофильного эзофагита (ЕоЕ) у пациента, нуждающегося в этом, включающие введение перорального кортикостероида в фазе индукции и поддерживающей фазе. В некоторых воплощениях, в результате осуществления фазы индукции наблюдается улучшение в показателе максимального количества эозинофилов (Eos) по результатам по меньшей мере одной биопсии пищевода, и такое лечение (например, во время фазы индукции) не приводит к какому-либо ухудшению средних еженедельных показателей у пациента в сообщаемой пациентом оценке результатов лечения, которая включает оценку дней без дисфагии. В некоторых воплощениях, пациент демонстрирует значительное улучшение в функции и морфологии пищевода, включая сглаживание борозд пищевода, ослабление процесса очагового сужения пищевода, увеличение диаметра пищевода, увеличение эластичности пищевода, увеличение растяжимости тела пищевода, облегчение при глотании, уменьшение отека, усиление васкуляризации, устранение пищеводных колец, уменьшение или отсутствие экссудата и/или отсутствие стриктуры.
В некоторых воплощениях настоящего изобретения предложены способы лечения эозинофильного эзофагита (ЕоЕ) у пациента, нуждающегося в этом, включающие введение перорального кортикостероида в фазе индукции и поддерживающей фазе, при этом в результате осуществления фазы индукции не наблюдается существенного улучшения в показателе максимального количества эозинофилов по результатам по меньшей мере одной биопсии пищевода. В некоторых воплощениях, пациент испытывает по меньшей мере один эпизод затрудненного прохождения пищи в фазе индукции. В некоторых воплощениях, у пациента проявляются симптомы активного протекания ЕоЕ во время фазы индукции. В некоторых воплощениях, результатом проведения фазы индукции является гистологический ответ, составляющий в показателях максимального количества эозинофилов 15 или более в поле зрения микроскопа под большим увеличением (HPF). В некоторых воплощениях, пациент не демонстрирует значительного улучшения функции и/или морфологии пищевода во время фазы индукции. В некоторых воплощениях, после неэффективной фазы индукции лечение продолжают, в результате чего у пациента наблюдается улучшение в показателе максимального количества эозинофилов по результатам по меньшей мере одной биопсии пищевода, и продолжающееся лечение не приводит к ухудшению средних еженедельных показателей у пациента в сообщаемой пациентом оценке результатов лечения, которая включает оценку дней без дисфагии.
В некоторых воплощениях, в поддерживающей фазе используют дозу, по меньшей мере равную, более высокую или более низкую по сравнению с таковой для фазы индукции. В некоторых воплощениях, индукционная и поддерживающая дозы составляют от примерно 1,5 мг до примерно 3 мг, например, вводимые один или два раза в сутки.
В некоторых воплощениях, фаза индукции включает введение в течение по меньшей мере примерно 6 недель, в течение по меньшей мере примерно 8 недель, в течение по меньшей мере примерно 10 недель или в течение по меньшей мере примерно 12 недель. В некоторых воплощениях, введение осуществляют два раза в сутки. В некоторых воплощениях, результатом фазы индукции является гистологический ответ в показателях максимального количества Eos в HPF менее 6. В некоторых воплощениях, результатом фазы индукции является отсутствие каких-либо эпизодов затрудненного прохождения пищи.
В некоторых воплощениях, у пациента при поддерживающей терапии не наблюдается рецидива с симптомами активного протекания ЕоЕ. В некоторых воплощениях, поддерживающая доза является по существу такой же или ниже индукционной дозы. В некоторых воплощениях, поддерживающая доза выше индукционной дозы. В некоторых воплощениях, у пациента в фазе отмены лечения не наблюдается рецидива с симптомами активного протекания ЕоЕ. В некоторых воплощениях, пациент остается в фазе отмены до возобновления симптомов активного протекания ЕоЕ. В некоторых воплощениях, после повторного проявления симптомов ЕоЕ пациент получает индукционную дозу перорального кортикостероида. В некоторых воплощениях, после повторного проявления симптомов активного протекания ЕоЕ пациент получает поддерживающую дозу перорального кортикостероида. В некоторых воплощениях, после повторного проявления симптомов активного протекания ЕоЕ пациент получает индукционную дозу перорального кортикостероида, затем поддерживающую дозу.
В некоторых воплощениях настоящего изобретения предложено введение композиции на основе перорального кортикостероида пациенту в лежачем положении. В некоторых воплощениях, кортикостероид вводят пациенту в лежачем положении и перед сном (например, примерно за 1 минуту, примерно за 5 минут, примерно за 10 минут, примерно за 15 минут или примерно за 30 минут, включая все значения из указанных выше). В некоторых воплощениях, фармацевтическую композицию вводят пациенту по меньшей мере примерно через 2 часа после вечернего приема пищи. В некоторых воплощениях, кортикостероид вводят пациенту по меньшей мере примерно через 4 часа после вечернего приема пищи.
В некоторых воплощениях, кортикостероид приготовлен в виде перорально растворимой лекарственной формы. В некоторых воплощениях, кортикостероид выбран из группы, состоящей из будесонида, флутиказона, флунизолида, циклесонида, мометазона, беклометазона, тиксокортола и их солей или эфиров и их смесей. В некоторых воплощениях, перорально растворимая лекарственная форма представляет собой таблетку или облатку. В некоторых воплощениях, кортикостероид осаждается местно в верхней части желудочно-кишечного тракта. В некоторых воплощениях, пациент ранее получал терапию ингибиторами протонного насоса (PPI). В некоторых воплощениях, пациент ранее получал PPI терапию в течение восьми недель. В некоторых воплощениях, PPI терапия была неэффективна для существенного улучшения одного или более симптомов ЕоЕ. В некоторых воплощениях, пациент ранее не получал PPI терапии.
В некоторых воплощениях настоящего изобретения предложены способы оценки пригодности субъектов для клинического испытания с целью измерения эффекта перорального кортикостероида на ЕоЕ после введения как в фазе индукции, так и в поддерживающей фазе, при этом набор субъектов для клинического испытания проводят на основании (1) наличия пациентов, имеющих максимальные количества эозинофилов в HPF выше 15, и (2) предшествующего лечения с использованием PPI в течение по меньшей мере примерно 8 недель, которое не было эффективным в плане существенного улучшения одного или более симптомов ЕоЕ.
В некоторых воплощениях настоящего изобретения предложен способ лечения ЕоЕ у пациента, нуждающегося в этом, включающий введение перорального кортикостероида пациенту, при этом пациент находится в лежачем положении, когда вводят пероральный кортикостероид, или пациент ложится сразу же после введения перорального кортикостероида. В других воплощениях настоящего изобретения предложена композиция, содержащая пероральный кортикостероид, для применения в лечении ЕоЕ у пациента, нуждающегося в этом, при этом пациент находится в лежачем положении, когда вводят композицию, или пациент ложится сразу же после введения композиции. В еще одних воплощениях настоящего изобретения предложено применение композиции, содержащей пероральный кортикостероид, для приготовления лекарственного средства для терапевтического применения при ЕоЕ у пациента, нуждающегося в этом, при этом пациент находится в лежачем положении, когда вводят лекарственное средство, или пациент ложится сразу же после введения лекарственного средства.
В различных воплощениях настоящего изобретения предложены способы (или композиции для применения в этих способах) для местного лечения ЕоЕ у пациента, нуждающегося в этом, пероральным кортикостероидом, при этом указанные способы включают: (а) введение перорального кортикостероида, когда пациент находится в лежачем положении, или непосредственно перед тем, как пациент примет лежачее положение. Согласно воплощениям, терапевтически эффективное количество перорального кортикостероида контактирует с пищеводом, тем самым осуществляя местное лечение ЕоЕ.
Согласно воплощениям, лежачее положение представляет собой положение лежа на спине, на животе или на боку. Согласно воплощениям, пероральный кортикостероид вводят примерно за 30 минут или меньше до запланированного времени отхода ко сну. Согласно воплощениям, пероральный кортикостероид вводят по меньшей мере примерно через 30 минут после приема пищи. Согласно воплощениям, пациент не принимает пищу или питье в течение по меньшей мере примерно 30 минут после введения перорального кортикостероида.
Согласно воплощениям, пероральный кортикостероид вводят: (1) один раз в сутки; или (2) два раза в сутки, при этом первую суточную дозу вводят, когда субъект сохраняет вертикальное положение. Согласно воплощениям, кортикостероид обладает системной биодоступностью, меньшей или равной примерно 20% от своей дозы. Согласно воплощениям, введение перорального кортикостероида обеспечивает достижение средней максимальной концентрации в плазме крови (Cmax), меньшей или равной примерно 500 пг/мл, после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида. Согласно воплощениям, введение перорального кортикостероида обеспечивает достижение средней площади под кривой в интервале 0-24 ч (AUC0-24), меньшей или равной примерно 3000 пг*ч/мл, после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида. Согласно воплощениям, пероральный кортикостероид представляет собой будесонид, флутиказон, флунизолид, циклесонид, мометазон или беклометазон или их фармацевтически приемлемые соль, сольват, сложный эфир, полиморф или пролекарство. Согласно воплощениям, пероральный кортикостероид готовят: (1) в виде жидкой композиции; (2) в виде твердой композиции; (3) для образования раствора или суспензии перед пероральным введением; или (4) для образования раствора, суспензии или геля после перорального введения, при этом с использованием форм (1)-(4) доставляют терапевтически эффективное количество перорального кортикостероида в пищевод. Согласно воплощениям, (1) жидкая композиция находится в форме раствора, суспензии или взвеси; и (2) твердая композиция находится в форме геля, пастилки, леденца, шипучей таблетки, порошка, гранул или перорально распадающейся композиции. Согласно воплощениям, перорально распадающаяся композиция представляет собой таблетку, облатку, пленку или лиофилизированную матрицу. Согласно воплощениям, перорально распадающаяся композиция представляет собой таблетку, содержащую: (а) пероральный кортикостероид в количестве от примерно 1,5 мг до примерно 7,5 мг; (б) фармацевтически приемлемый носитель, объединенный с кортикостероидом; и (в) быстро диспергируемые микрогранулы, при этом перорально распадающаяся таблетка распадается в течение 60 секунд при тестировании времени распадаемости с использованием метода, описанного в USP (Фармакопея США) <701>.
Согласно воплощениям, Cmax перорального кортикостероида у пациента составляет примерно 200 пг/мл или менее после перорального введения от 1,5 мг до примерно 7,5 мг перорального кортикостероида. Согласно воплощениям, пероральный кортикостероид представляет собой флутиказона пропионат, и у находящегося в лежачем положении пациента наблюдается Cmax в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 15 пг/мл до примерно 45 пг/мл после перорального введения 6 мг флутиказона пропионата или 3 мг флутиказона пропионата пациенту, находящемуся в лежачем положении. Согласно воплощениям, Cmax кортикостероида в случае пациента, находящегося в лежачем положении, ниже Cmax перорального кортикостероида в случае пациента после приема пищи, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида. Согласно воплощениям, перорального кортикостероида в случае пациента, находящегося в лежачем положении, снижается на величину от примерно 10% до примерно 30% по сравнению с Cmax перорального кортикостероида в случае пациента после приема пищи, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида.
Согласно воплощениям, среднее время достижения максимальной концентрации в плазме крови (Tmax) находится в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 12 ч до примерно 15 ч. Согласно воплощениям, Tmax для кортикостероида в случае пациента, находящегося в лежачем положении, увеличивается по сравнению с Tmax для перорального кортикостероида у пациента, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида. Согласно воплощениям, Tmax для кортикостероида в случае пациента, находящегося в лежачем положении, увеличивается по меньшей мере примерно на 1 час по сравнению со средним значением Tmax для перорального кортикостероида в случае пациента, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида. Согласно воплощениям, Tmax для кортикостероида у пациента, находящегося в лежачем положении, увеличивается на промежуток времени в интервале от примерно 4 ч до примерно 9 ч по сравнению с Tmax для перорального кортикостероида в случае пациента, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида.
Согласно воплощениям, через 12 недель введения перорального кортикостероида в режиме одного раза в сутки воспаление пищевода уменьшается в соответствии с результатами, показывающими снижение количества эозинофилов, увеличение числа дней без дисфагии, уменьшение числа эпизодов дисфагии, улучшение показателя EREFS (эндоскопический референсный показатель), улучшение эластичности пищевода согласно протоколу EndoFLIP, оценке уровня биомаркеров, уменьшению числа эпизодов затрудненного прохождения пищи, улучшению показателей EEsAI (индекс активности эозинофильного эзофагита) (по показателям пациента, врача, эндоскопии, патологического изменения), согласно EoE-QoL-A (опросник по оценке качества жизни взрослых пациентов с эозинофильным эзофагитом), опроснику по визуальной оценке дисфагии (VDQ), показателям избегания, корректировки и медленного приема пищи (Avoidance Modification and Slow eating, AMS) или результатам гистологического исследования. Согласно воплощениям, количество эозинофилов у пациента снижается по меньшей мере примерно на 50%.
Согласно воплощениям, у пациента имеется аллергия на лактозу или аллергия на крахмал.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На Фиг. 1 показано типичное схематичное представление части 1 и 2 плана исследования, фазы 2b, и отмечены фаза скрининга и вводная фаза в части 1 (стадия индукции) и фаза рандомизированной отмены лечения (randomized withdrawal) в части 2.
На Фиг. 2 показано типичное схематичное представление части 3 (поддерживающая фаза) и периода последующего наблюдения в данном исследовании, фазе 2b.
На Фиг. 3 показано другое типичное схематичное представление частей 1 и 2 плана исследования, фазы 2b, и отмечены фаза скрининга и вводная фаза в части 1 (стадия индукции) и фаза рандомизированной отмены лечения в части 2.
На Фиг. 4 показано другое типичное схематичное представление части 3 (поддерживающей фазы) и периода последующего наблюдения в данном исследовании, фазе 2b.
На Фиг. 5 показана схема исследования, представляющая общее описание лечения, которое каждый субъект будет получать на протяжении всего исследования флутиказона при эозинофильном эзофагите (FLUTE).
На Фиг. 6 показан профиль зависимости средней концентрации АРТ-1011 (флутиказона пропионата) в плазме крови от времени в линейных координатах в случае введения после приема пищи, натощак и в режиме HS (перед сном).
На Фиг. 7 показан профиль зависимости средней концентрации АРТ-1011 (флутиказона пропионата) в плазме крови от времени в логарифмических координатах в случае введения после приема пищи, натощак и в режиме HS.
На Фиг. 8 показаны диаграммы «спагетти» (spaghetti plots) для АРТ-1011 (флутиказона пропионата) (условия введения натощак).
На Фиг. 9 показаны диаграммы «спагетти» для АРТ-1011 (флутиказона пропионата) (условия введения после приема пищи).
На Фиг. 10 показаны диаграммы «спагетти» для АРТ-1011 (флутиказона пропионата) (условия введения в режиме HS введения).
ПОДРОБНОЕ ОПИСАНИЕ
Воспалительные расстройства желудочно-кишечного тракта, такие как эозинофильный эзофагит (ЕоЕ), аллергическое/иммунное состояние, при котором субъект страдает от воспаления и/или отека слизистой пищевода, затрагивают способность пациента проглатывать пищу и могут, в связи с этим, быть причиной недоедания и отставания в физическом развитии. Обычно эозинофилы в пищеводе не обнаруживаются, но при ЕоЕ эти клетки накапливаются и вызывают отек, в результате которого уменьшается внутренний диаметр пищевода, что очень затрудняет процесс глотания и приема пищи. Зачастую пациенты испытывают эпизоды затрудненного прохождения пищи, когда пища застревает в пищеводе пациента, что может потребовать неотложной терапии. Вследствие затруднения при глотании и страха перед затрудненным прохождением пищи многие пациенты с ЕоЕ ограничивают себя приемом мягких видов пищи, таких как йогурт, супы и смузи. В тяжелых случаях ЕоЕ пациенты получают парентеральное питание (например, внутривенное кормление), которое может обеспечивать необходимую поддержку, но ограничивает формы деятельности пациента и может приводить к повышенной вероятности заражения в месте расположения катетера.
ЕоЕ чаще всего встречается у мужчин белой европеоидной расы и может встречаться в любом возрасте, при этом симптомы с возрастом изменяются. Младенцы и дети младшего возраста, страдающие от ЕоЕ, могут отказываться от пищи, отставать в физическом развитии или испытывать «рефлюкс» и/или рвоту. У маленьких детей обычно отмечают изжогу/рефлюкс, боль в животе, рвоту, отказ от пищи и/или плохой рост. Для взрослых характерным симптомом является дисфагия (затруднение при глотании), и ЕоЕ имеет отношение более чем к 50% случаев затрудненного прохождения пищи. Обычно взрослые пациенты реже испытывают изжогу или боль в области грудной клетки. Взрослые с ЕоЕ также демонстрируют измененное пищевое поведение, как например, изменение рациона питания, медленный прием пищи, тщательное пережевывание и повышенное потребление жидкостей во время еды.
Несмотря на то, что причины ЕоЕ не известны, многие пациенты с ЕоЕ имеют семейную историю аллергических реакций, астмы и/или симптомов аллергических расстройств (например, астмы, аллергического ринита, атопического дерматита и пищевой аллергии). Кроме того, в развитии ЕоЕ могут играть роль аллергены из окружающей среды, такие как пылевые клещи, алергены животных, пыльца и плесень. Вследствие связи между ЕоЕ и аллергиями, в особенности пищевыми аллергиями, устранение аллергена может оказать помощь в облегчении симптомов. Однако, эти исключения могут быть труднодостижимыми.
Несмотря на то, что лекарственных средств, одобренных в настоящее время к применению для лечения ЕоЕ, не существует, некоторые лекарственные средства, такие как глюкокортикостероиды, антагонисты лейкотриенов, стабилизаторы тучных клеток, иммуномодуляторы, биологические препараты и малые молекулы, могут оказать помощь в облегчение симптомов. Для лечения симптомов у пациентов также были использованы ингибиторы протонного насоса (PPI), которые регулируют количество образующейся кислоты, но они не могут снизить воспаление у пациента. Кроме того, согласно недавним исследованиям обнаружена связь между длительным использованием PPI и деменцией, что делает их применение пациентами с ЕоЕ менее желательным. Для облегчения симптомов также можно использовать эндоскопическую терапию (дилатацию), но это также не влияет на основное воспаление, вызывающее отек пищевода.
Несмотря на то, что эти терапии на некоторое время могут помочь в облегчении симптомов некоторым пациентам, они часто не достигают цели лечения ЕоЕ. Например, современные стероидные лекарственные средства для местного применения не являются оптимальными, при этом 5-50% пациентов классифицируют отсутствие лечебного эффекта. Аналогичным образом, применение элиминационной диеты, которое требует значительного эндоскопического наблюдения за пациентом, показывает отсутствие ответа примерно в 30% случаев.
Необходимы новые способы, не только облегчающие симптомы воспалительных расстройств пищевода, таких как ЕоЕ, но также устраняющие воспаление, являющееся причиной данных симптомов. Согласно настоящему изобретению предложены способы введения фармацевтических композиций, содержащих кортикостероид местного действия, для лечения симптомов и/или воспаления, ассоциированных с воспалительным расстройством желудочно-кишечного тракта. Фармацевтические композиции, содержащие кортикостероид местного действия, для применения в таких способах, также описаны в данной заявке.
Способы, описанные в данной заявке в различных воплощениях, включают по меньшей мере две фазы: фазу индукции и поддерживающую фазу. Во время фазы индукции пациентам вводят дозу фармацевтической композиции по изобретению. На основании ответа пациента по окончании фазы индукции (например, гистологического ответа и согласно отсутствию ухудшения симптомов или отсутствию эпизодов затрудненного прохождения пищи) пациента могут перевести в поддерживающую фазу. Доза фармацевтической композиции может быть одинаковой или разной во время поддерживающей фазы и фазы индукции. После уменьшения интенсивности или ослабления симптомов пациента также могут перевести в фазу отмены, и он не получает никаких доз фармацевтической композиции до момента повторного проявления симптомов.
Согласно настоящему изобретению предложены способы лечения эозинофильного эзофагита (ЕоЕ) у пациента, нуждающегося в этом, включающие введение перорального кортикостероида в фазе индукции и поддерживающей фазе, при этом фаза индукции приводит к улучшению показателя максимального количества эозинофилов по результатам по меньшей мере одной биопсии пищевода, и лечение не приводит к ухудшению средних еженедельных показателей у пациента в сообщаемой пациентом оценке результатов лечения, которая включает оценку дней без дисфагии.
Если не указано иное, все технические и научные термины, использованные в данном описании, имеют значение, обычно понимаемое обычным специалистом в области техники, к которой данное изобретение относится. Хотя при практическом применении или тестировании настоящего изобретения можно использовать способы и материалы, аналогичные или эквивалентные изложенным в данном описании, в данной заявке описаны предпочтительные способы и материалы ниже.
Следует понимать, что формы единственного числа применяются во всей этой заявке для удобства, с тем исключением, однако, когда контекст или подробное изложение указывает иное, при этом подразумевается, что формы единственного числа включают объекты в форме множественного числа. Следует понимать, что все численные диапазоны включают практически каждую выраженную числом точку в пределах численного диапазона, и это следует интерпретировать как упоминание практически каждой выраженной числом точки по отдельности. Включены конечные точки всех диапазонов, относящихся к одному и тому же компоненту или свойству, подразумевается, что их можно независимо комбинировать.
Подразумевается, что использованное в данном описании слово «включать» и его варианты, является неограничивающим, поэтому упоминание в списке объектов не исключает других подобных объектов, которые также могут быть использованы в материалах, композициях, устройствах и способах этой технологии. Аналогичным образом подразумевается, что термины «может» и «возможно» и их варианты являются неограничивающими, поэтому упоминание, что «воплощение может содержать или возможно содержит определенные элементы или признаки», не исключает другие воплощения технологии по настоящему изобретению, которые не содержат этих элементов или признаков. Хотя неограничивающий термин «содержащий», будучи синонимом таким терминам, как «включающий», «включающий в себя» или «имеющий», используется в данном описании для описания и заявления на притязания по настоящему раскрытию изобретения, технология по настоящему изобретению или ее воплощения альтернативно могут быть описаны с использованием более ограничивающих терминов, таких как «состоящий из» или «состоящий по существу из» упомянутых ингредиентов.
Термин «лекарственное средство», «активный» или «активный фармацевтический ингредиент», использованный в данном описании, включает фармацевтически приемлемый и оказывающий местное действие кортикостероид, его фармацевтически приемлемые соли, сложные эфиры, сольваты (в том числе гидраты), полиморфы, стереоизомеры и/или пролекарства, и их смеси. Термин «соли» относится к продукту, образующемуся при взаимодействии подходящей неорганической или органической кислоты с лекарственным средством в форме «свободного основания». Подходящие кислоты включают таковые, имеющие достаточную кислотность для образования стабильной соли, например, кислоты с низкой токсичностью, например, соли, одобренные к применению на людях или животных. Неограничивающие примеры кислот, которые могут быть использованы с образованием соли перорально активного лекарственного средства, включают неорганические кислоты, например, HCl, Н3РО4 и H2SO4. Неограничивающие примеры органических кислот включают алкилсульфоновые кислоты и пропионовую кислоту.
Термины «фармацевтическая композиция» и «фармацевтическая лекарственная форма» используются в данном описании взаимозаменяемо для обозначения пероральной лекарственной формы (суспензии, раствора, порошка, твердого вещества и т.д.), которую можно использовать для введения кортикостероида. Неограничивающие примеры лекарственных форм включают перорально распадающуюся композицию, такую как таблетка, лиофилизированная форма, пленка и облатка, жидкую композицию, гель, суспензию, пастилку, леденец, саше, шипучую таблетку и тому подобное.
Термин «пероральный кортикостероид» и «кортикостероид» используются взаимозаменяемо для обозначения кортикостероида, который вводят перорально, например, в фармацевтической композиции, описанной в данной заявке.
Термины «перорально распадающаяся лекарственная форма», «перорально распадающаяся таблетка», «перорально диспергируемая таблетка» или «ODT» относятся к твердой лекарственной форме/таблетке по настоящему изобретению, которая после введения быстро распадается в ротовой полости пациента без разжевывания с образованием суспензии, содержащей кортикостероид. Скорость распадаемости при пероральном приеме может варьировать, однако существенно превышает скорость распадаемости традиционных твердых лекарственных форм или жевательных твердых лекарственных форм (т.е. таблеток или капсул) при пероральном приеме, которые предназначены для проглатывания сразу же после введения.
Использованные в данном описании термины «подвергание лечению», «лечение» и «лечить» включают в себя (1) предупреждение возникновения конкретного заболевания или расстройства у субъекта, который может быть предрасположен к данному заболеванию или расстройству, но еще без постановки такого диагноза; (2) излечивание, лечение или подавление заболевания, т.е. прекращение его развития; или (3) уменьшение интенсивности симптомов заболевания путем ослабления или устранения симптомов, состояний и/или путем вызывания регрессии заболевания. В некоторых воплощениях, термины «подвергание лечению», «лечение» и «лечить» могут включать в себя применение терапевтически эффективного режима, как определено в данном описании.
Термин «примерно», использованный в данном описании для обозначения численной величины, включает «точное значение» плюс или минус включительно до 10% от значения упоминаемого числового показания. Когда термин «примерно» используют с упоминанием диапазона значений, термин «примерно» относится как к минимальному, так и к максимальному значению этого диапазона (например, «примерно 1-50 мкм» означает «от примерно 1 мкм до примерно 50 мкм»). Термин «равномерно ассоциированные», использованный в данной заявке для описания пространственного расположения двух или более компонентов композиции, относится к компонентам, которые равномерно перемешаны, как например, в смесях, веществах покрытия и матрицах.
Если не указано иное, все процентные доли и соотношения рассчитаны по массе. Если не указано иное, все проценты и соотношения рассчитаны исходя из общего состава композиции.
Термин «не обладающий значительной системной глюкокортикоидной или минералокортикоидной активностью», использованный в данном описании, относится к композициям на основе кортикостероидов, которые не оказывают общего действия на организм вследствие всасывания в кровоток, но оказывают местное действие в результате контакта в месте пораженной заболеванием ткани. Примеры включают флутиказон, флунизолид, будесонид, циклесонид, мометазон, тиксокортол и беклометазон. Кортикостероиды, обладающие высокой системной глюкокортикоидной активностью при пероральном введении, включают, например, гидрокортизон, преднизон, преднизолон, метилпреднизолон, дексаметазон, бетаметазон и т.д., или кортикостероиды с минералокортикоидной активностью (например, альдостерон). Кортикостероиды, обычно обладающие системной глюкокортикоидной или минералокортикоидной активностью при пероральном введении, также могут быть использованы в разбавленных композициях по настоящему изобретению, при использовании которых системное поглощение кортикостероида снижено или подавлено.
«Гистологически обусловленный респондер» может быть определен как субъект, у которого достигается гистологический ответ в показателе максимального количества эозинофилов/HPF 6 или менее (в качестве первичной детерминанты). HPF может быть определено как стандартная площадь, составляющая 0,237 квадратных миллиметров под микроскопом с 40х объективом и окуляром 22 мм.
«Гистологически обусловленный нереспондер»» может быть определен как субъект, у которого не достигается гистологического ответа (т.е. не достигается гистологического ответа в показателе максимального количества эозинофилов/HPF 6 или менее).
Субъекты, у которых в любое время по ходу исследования развивается затрудненное прохождение пищи с дилатацией пищевода или без нее, могут считаться «пациентами с неудачным результатом лечения».
Фармацевтические композиции и введение
Фармацевтические композиции, используемые в (или для применения в) способах, описанных в данной заявке, могут представлять собой любую лекарственную форму, которую можно использовать для местного введения терапевтического агента (например, кортикостероида) в пищевод. Подходящие лекарственные формы включают жидкие композиции (например, растворы, суспензии и взвеси), гели и твердые композиции, которые образуют жидкость или гель после перорального введения. Например, перорально распадающиеся композиции (например, ODT, пленка, лиофилизированная форма или облатка), пастилки и леденцы могут образовывать в ротовой полости пациента раствор, суспензию или гель, содержащие терапевтический агент, и после проглатывания раствора или суспензии растворенный или суспендированный в них кортикостероид контактирует с пищеводом по мере того, как жидкость проходит через пищеводный тракт. В предпочтительном воплощении фармацевтическая композиция находится в форме ODT.
В некоторых воплощениях настоящего изобретения предложена пероральная твердая фармацевтическая композиция, содержащая кортикостероид (например, примерно 10 мг или меньше, включая 7,5 мг, 6,0 мг, 4,5 мг, 3,0 мг, 1,5 мг или 0,75 мг) и по меньшей мере один фармацевтически приемлемый носитель, при этом кортикостероид объединен с фармацевтически приемлемым носителем (например, адсорбирован на или суспендирован в нем). В некоторых воплощениях, лекарственное средство присутствует в количестве меньше примерно 5% (масса лекарственного средства/масса композиции), в частности, меньше 3% по массе. Фармацевтические композиции, описанные в данной заявке, могут быть приготовлены в виде перорально распадающейся таблетки (далее именуемой как ODT), которая распадается в течение 60 секунд (например, в течение 30 секунд) при тестировании с использованием теста на распадаемость USP 701 и/или распадается в течение 60 секунд при попадании в ротовую полость человека.
В некоторых воплощениях, кортикостероид, используемый в композициях и способах, описанных в данной заявке, представляет собой кортикостероид местного действия. В некоторых воплощениях, кортикостероид обладает низким системным действием или по существу не оказывает никакого системного действия. В некоторых воплощениях, кортикостероиды, обладающие низким системным действием или по существу не оказывающие никакого системного действия, представляют собой кортикостероиды, которые не обладают какой-либо значительной системной глюкокортикоидной или минералокортикоидной активностью после перорального введения людям. Выражение «кортикостероид, у которого «отсутствует какая-либо значительная системная глюкокортикоидная или минералокортикоидная активность после перорального введения людям» относится к кортикостероидам или фармацевтическим композициям, содержащим кортикостероиды, которые проявляют меньше, чем примерно 20% системной глюкокортикоидной или минералокортикоидной активности после перорального введения, например, меньше примерно 15%, меньше примерно 10%, меньше примерно 5%, меньше примерно 5%, меньше примерно 4%, меньше примерно 3%, меньше примерно 2% или меньше примерно 1%. Системную глюкокортикоидную или минералокортикоидную активность можно определить, используя способы, известные в данной области техники, такие как измерение утренних уровней кортизола.
В некоторых воплощениях, кортикостероиды для применения в способах и композициях, описанных в данной заявке, имеют системную биодоступность, меньшую или равную примерно 20% от введенной дозы. Неограничивающие примеры пероральных кортикостероидов, которые имеют биодоступность, меньшую или равную примерно 20%, включают флутиказон, флунизолид, будесонид, циклесонид, мометазон, тиксокортол и беклометазон и их фармацевтически приемлемые соли, сольваты, сложные эфиры, полиморфы или пролекарства. В предпочтительных воплощениях пероральный кортикостероид, используемый в способах и композициях, описанных в данной заявке, представляет собой флутиказона пропионат.
В некоторых воплощениях, пероральные кортикостероиды для применения в способах и композициях, описанных в данной заявке, готовят в виде препаратов для получения фармакокинетического профиля, который уменьшает вероятность того, что пациент будет испытывать побочные эффекты, связанные с системным введением кортикостероидов, включая, но не ограничиваясь этим, остеопороз, прирост массы, угнетение иммунной системы (т.е. увеличение числа случаев инфекционных заболеваний), высокое кровяное давление, гипергликемию, мышечную слабость, проблемы с кожей (например, плохое заживление ран, истончение кожи, легкость образования кровоподтеков, растяжки и т.д.), изменения настроения и поведения, повышенный риск развития катаракты и повышенный риск язвы двенадцатиперстной кишки. Иными словами, фармакокинетический профиль кортикостероида может быть модифицирован для получения средней максимальной концентрации в плазме крови (Cmax), среднего времени достижения максимальной концентрации в плазме крови (Tmax) и/или AUC, при которых уменьшаются системные побочные эффекты. Фармакокинетический профиль может быть определен способами, известными в данной области техники, например, способами, описанными в примере 5.
В некоторых воплощениях, пероральные кортикостероиды для применения в способах и композициях, описанных в данной заявке, готовят в виде препаратов для получения средней максимальной концентрации в плазме крови (Cmax), меньшей или равной примерно 10000 пг/мл после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида, например, примерно 9000 пг/мл, примерно 8000 пг/мл, примерно 7000 пг/мл, примерно 6000 пг/мл, примерно 5000 пг/мл, примерно 4000 пг/мл, примерно 3000 пг/мл, примерно 2000 пг/мл, примерно 1000 пг/мл, примерно 900 пг/мл, примерно 800 пг/мл, примерно 700 пг/мл, примерно 600 пг/мл или примерно 500 пг/мл, включая все значения и поддиапазоны между ними. В предпочтительных воплощениях, пероральный кортикостероид готовят в виде препарата для получения Cmax, меньшей или равной примерно 500 пг/мл после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида.
В некоторых воплощениях, пероральные кортикостероиды для применения в способах и композициях, описанных в данной заявке, готовят в виде препаратов для получения средней AUC0-24, меньшей или равной примерно 15000 пг*ч/мл после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида, например, примерно 14000 пг*ч/мл, примерно 13000 пг*ч/мл, примерно 12000 пг*ч/мл, примерно 11000 пг*ч/мл, примерно 10000 пг*ч/мл, примерно 9000 пг*ч/мл, примерно 8000 пг*ч/мл, примерно 7000 пг*ч/мл, примерно 6000 пг*ч/мл, примерно 5000 пг*ч/мл, примерно 4000 пг*ч/мл, примерно 3000 пг*ч/мл, примерно 2000 пг*ч/мл, примерно 1000 пг*ч/мл, включая все значения и поддиапазоны между ними. В предпочтительных воплощениях, пероральные кортикостероиды для применения в способах и композициях, описанных в данной заявке, готовят в виде препаратов для получения средней AUC0-24, меньшей или равной примерно 3000 пг*ч/мл после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида.
В некоторых воплощениях, фармацевтические композиции, описанные в данной заявке, могут быть приготовлены для снижения системной биодоступности, глюкокортикоидной активности и/или минералокортикоидной активности (или их комбинаций) перорального кортикостероида после перорального введения. Снижение системной биодоступности скорее может обеспечить, чтобы кортикостероид проявлял местные терапевтические эффекты, а не абсорбировался системно. Например, кортикостероид, который в других случаях имеет высокую системную биодоступность (например, системную биодоступность выше 20%), может быть приготовлен в ионообменной смоле для снижения системной биодоступности при одновременном усилении местных терапевтических эффектов.
Соли, сольваты, полиморфы и пролекарства можно использовать для модификации кортикостероидов, которые в других случаях имеют системную биодоступность более примерно 20%, для получения «кортикостероида местного действия», имеющего системную активность менее примерно 20% и оказывающего местный эффект более примерно 80% за счет снижения системной биодоступности кортикостероида.
Неограничивающие примеры кортикостероидов, которые могут быть модифицированы (например, в результате образования соли или пролекарства или приготовления перорального кортикостероида в ионообменной смоле) с целью снижения системной биодоступности и усиления местных эффектов, включают гидрокортизон, преднизон, преднизолон. метилпреднизолон, дексаметазон, бетаметазон, альдостерон и тому подобное.
Композиции по настоящему изобретению могут включать водорастворимый или набухающий в воде фармацевтически приемлемый эксципиент, такой как полимер, образующий биогель, или биоадгезивный полимер, который будет усиливать биоадгезию кортикостероида к воспаленной слизистой оболочке пищевода.
Подходящие кортикостероиды местного действия, которые могут быть включены в фармацевтическую композицию по настоящему изобретению, включают будесонид, флутиказон, флунизолид, циклесонид, мометазон, беклометазон, тиксокортол и их соли, сложные эфиры, сольваты, полиморфы или пролекарства и их смеси.
В предпочтительных воплощениях композиция по настоящему изобретению содержит флутиказон (например, флутиказона пропионат). В предпочтительных воплощениях фармацевтическая композиция представляет собой ODT, содержащую флутиказон или его фармацевтически приемлемую соль (например, флутиказона пропионат). В других воплощениях композиция по настоящему изобретению содержит будесонид. В некоторых других воплощениях композиция по настоящему изобретению содержит циклесонид.
В некоторых воплощениях, кортикостероид может быть в форме кристаллов, имеющих средний размер частиц примерно 100 мкм или меньше, примерно 75 мкм или менее, примерно 50 мкм или менее, более конкретно, примерно 25 мкм или менее, либо примерно 15 мкм или менее. В конкретных воплощениях данного изобретения предусмотрено, что кортикостероид подвергают микронизации, чтобы достичь среднего размера частиц менее примерно 10 мкм, менее примерно 8 или менее, менее примерно 6 мкм или, в частности, менее примерно 4 мкм.
Альтернативно, такие кристаллы могут иметь средний размер в субмикронном диапазоне (например, средний размер частиц менее примерно 1 мкм), т.е. могут существовать в виде наночастиц (например, со средним размером частиц в диапазоне примерно 1-100 нм). В некоторых воплощениях, кортикостероид может быть представлен в аморфной форме, например вместе со стабилизирующим агентом, который ограничивает возможность перекристаллизации лекарственного средства, например, поливинилпирролидоном (PVP), гидроксипропилметилцеллюлозой (НРМС), гидроксипропилцеллюлозой, гидроксиэтилцеллюлозой; Soluplus®, Kollidon® VA64, лаурилсульфатом натрия, поверхностно-активными веществами типа твинов, полимером Eudragit® ЕРО и их смесями.
Количество кортикостероида, присутствующего в фармацевтических композициях по настоящему изобретению, выбрано таким образом, чтобы максимально повысить терапевтическую пользу в случае местного введения при одновременном сведении к минимуму побочных эффектов, обусловленных системным всасыванием. В случае твердых фармацевтических композиций по настоящему изобретению количество кортикостероида в композиции составляет менее примерно 5% масс./масс. (масса лекарственного средства/масса композиции). В некоторых воплощениях, количество кортикостероида в фармацевтической композиции составляет менее примерно 4%. В другом воплощении оно составляет менее примерно 3%. В некоторых воплощениях, оно составляет менее примерно 2%, менее примерно 1,5%, менее примерно 1%, менее примерно 0,5% по массе или ниже. В некоторых воплощениях, количество кортикостероида в фармацевтической композиции составляет от примерно 0,50 мг до примерно 18 мг. В некоторых воплощениях, количество кортикостероида в фармацевтической композиции составляет от примерно 0,75 мг до примерно 12 мг. В некоторых воплощениях, количество кортикостероида в фармацевтической композиции составляет от примерно 1,5 мг до примерно 9 мг. В еще одних воплощениях количество кортикостероида составляет примерно 0,01 мг, примерно 0,05 мг, примерно 0,1 мг, примерно 0,15 мг, примерно 0,1 мг, примерно 0,2 мг, примерно 0,25 мг, примерно 0,3 мг, примерно 0,35 мг, примерно 0,4 мг, примерно 0,45 мг, примерно 0,5 мг, примерно 0,6 мг, примерно 0,7 мг, примерно 0,75 мг, примерно 0,8 мг, примерно 1 мг, примерно 1,5 мг, примерно 2 мг, примерно 3 мг, примерно 4 мг, примерно 4,5 мг, примерно 5 мг, примерно 6 мг, примерно 7 мг, примерно 8 мг, примерно 9 мг, примерно 10 мг, примерно 12 мг, примерно 18 мг, включая все диапазоны и поддиапазоны между ними.
В предпочтительных воплощениях количество кортикостероида в фармацевтической композиции, описанной в данной заявке (например, ODT), находится в диапазоне от примерно 1,5 мг до примерно 7,5 мг, включая примерно 3,0 мг, примерно 4,5 мг и примерно 6,0 мг. В предпочтительных воплощениях фармацевтическая композиция представляет собой ODT, и кортикостероид присутствует в количестве примерно 1,5 мг. В других предпочтительных воплощениях фармацевтическая композиция представляет собой ODT, и кортикостероид присутствует в количестве примерно 3,0 мг. В еще одних предпочтительных воплощениях фармацевтическая композиция представляет собой ODT, и кортикостероид присутствует в количестве примерно 4,5 мг. В еще одних предпочтительных воплощениях фармацевтическая композиция представляет собой ODT, и кортикостероид присутствует в количестве примерно 6 мг. В некоторых других предпочтительных воплощениях фармацевтическая композиция представляет собой ODT, и кортикостероид присутствует в количестве примерно 7,5 мг.
В некоторых воплощениях, быстро распадающаяся композиция по изобретению может содержать фармацевтически приемлемые эксципиенты, которые набухают, растворяются или иным образом облегчают распадаемость перорально распадающейся лекарственной формы (например, ODT, пленки, лиофилизированной матрицы или облатки), обеспечивая получение однородной вязкой суспензии, содержащей частицы микронизированного кортикостероида для покрытия воспаленной слизистой оболочки пищевода для лечения эозинофильного эзофагита. Примеры таких фармацевтически приемлемых эксципиентов включают разрыхлители или быстро диспергируемые микрогранулы, которые описаны в данной заявке. Использованный в данном описании термин «быстро диспергируемые микрогранулы» относится к гранулам, содержащим частицы по меньшей мере одного сахарного спирта и/или сахарида в комбинации с частицами по меньшей мере одного разрыхлителя, которые образуются в грануляторе. Согласно воплощениям, кортикостероид (например, частицы микронизированного кортикостероида) может быть подвергнут гранулированию совместно с частицами сахарного спирта и/или сахарида. Альтернативно, в некоторых воплощениях, кортикостероид (например, частицы микронизированного кортикостероида) может быть объединен с фармацевтически приемлемым носителем и затем смешан с быстро диспергируемыми микрогранулами, содержащими частицы по меньшей мере одного сахарного спирта и/или сахарида в комбинации с частицами по меньшей мере одного разрыхлителя.
В некоторых воплощениях настоящего изобретения общую массу лекарственной формы поддерживают в диапазоне от 300 до 900 мг для включения настолько большого количества быстро диспергируемых микрогранул, содержащих по меньшей мере один сахарный спирт и/или сахар ид в комбинации по меньшей мере с одним разрыхлителем, насколько это возможно для максимального покрытия микронизированным кортикостероидом затронутой эозинофильным эзофагитом поверхности. В некоторых воплощениях, быстро диспергируемые микрогранулы содержат по меньшей мере один разрыхлитель в комбинации с сахарным спиртом и/или сахаридом. Количество сахарного спирта и/или сахарида в быстро диспергируемых гранулах находится в диапазоне примерно 99%-90% или примерно 95%-90% от общей массы содержащих разрыхлитель гранул, включая все диапазоны и поддиапазоны между ними. В некоторых воплощениях, средний размер частиц сахарного спирта и/или сахарида составляет примерно 30 мкм или меньше, например, примерно 1-30 мкм, примерно 5-30 мкм, примерно 5-25 мкм, примерно 5-20 мкм, примерно 5-15 мкм, примерно 5-10 мкм, примерно 10-30 мкм, примерно 10-25 мкм, примерно 10-20 мкм, примерно 10-15 мкм, примерно 15-30 мкм, примерно 15-25 мкм, примерно 15-20 мкм, примерно 20-30 мкм, примерно 20-25 мкм или примерно 25-30 мкм.
В некоторых воплощениях, лекарственная форма имеет общую массу 300 мг и содержит примерно 0,05 мг (0,16%), примерно 0,75 мг (0,25% масс./масс.), примерно 1,5 мг (0,5%) масс./масс.), примерно 3 мг (1% масс./масс.), примерно 4,5 мг (1,5%), примерно 6 мг (2% масс./масс.), примерно 7,5 мг (2,5% масс./масс.), примерно 9 мг (3% масс./масс.), примерно 12 мг (4% масс./масс.), примерно 16 мг (5%) кортикостероида.
В некоторых воплощениях, лекарственная форма имеет общую массу 600 мг и содержит примерно 0,75 мг (0,125%) масс./масс.), примерно 1,5 мг (0,25% масс./масс.), примерно 3 мг (0,5%) масс./масс.), примерно 4,5 мг (0,75%), примерно 6 мг (0,1% масс./масс.), примерно 7,5 мг (1,25% масс./масс.), примерно 9 мг (1,5% масс./масс.), примерно 12 мг (2% масс./масс.), примерно 18 мг (3% масс./масс.) кортикостероида. В некоторых воплощениях, кортикостероид местного действия представляет собой флутиказона пропионат, и его содержание в фармацевтической композиции находится в диапазоне от примерно 0,05 до примерно 15 мг при содержании лекарственного средства примерно от 0,16% до 5% от массы композиции.
В некоторых воплощениях, содержание флутиказона пропионата в композиции находится в диапазоне от примерно 0,75 до примерно 7,5 мг при содержании лекарственного средства примерно от 0,25% до 2,5% от массы композиции.
В некоторых воплощениях, содержание флутиказона пропионата в композиции находится в диапазоне от 0,05 до примерно 18 мг при содержании лекарственного средства примерно от 0,125% до 5% от массы композиции.
Фармацевтически приемлемый носитель, используемый в смеси по настоящему изобретению, подходит для объединения с лекарственным средством (например, для адсорбции лекарственного средства или растворения суспензии лекарственного средства в фармацевтически приемлемом носителе), он должен обладать свойствами превосходного носителя в отношении сухих смесей с точки зрения обеспечения сыпучести и пригодности для обработки смесей и предотвращения сегрегации. Он может способствовать обеспечению однородности состава кортикостероида. Подходящие фармацевтически приемлемые носители включают, но не ограничиваются этим, микрокристаллическую целлюлозу, силикатизированную микрокристаллическую целлюлозу, прежелатинизированный крахмал, кукурузный крахмал, коллоидный диоксид кремния или аморфный алюмосиликат магния (имеющийся в продаже под товарным знаком VEEGUM™ или NEUSILIN™). В предпочтительных воплощениях фармацевтически приемлемый носитель для адсорбции кортикостероида представляет собой силикатизированную микрокристаллическую целлюлозу. Предпочтительно, это силикатизированная микрокристаллическая целлюлоза, которая состоит из равномерно ассоциированных частиц микрокристаллической целлюлозы и коллоидного диоксида кремния (PROSOLV®SMCC: 98% МСС и 2% CSD). Использование этого ингредиента в композиции по изобретению улучшает связанные с сыпучестью и смешиванием свойства смеси на основе кортикостероида; улучшенные однородность/гомогенность смеси и физическая стабильность композиций в процессе хранения до момента ее конечной обработки в окончательные лекарственные формы, такие как, например, таблетки или капсулы, также позволяют избежать или свести к минимуму возможное расслоение и сегрегацию кортикостероидных микрочастиц. Наличие этого носителя в смеси с активным ингредиентом также обеспечивает воспроизводимость процессов приготовления композиции по изобретению (в частности, с использованием технологии прямого таблетирования). В некоторых воплощениях, описана смесь, содержащая кортикостероид в низкой дозе и носитель, демонстрирующая высокую однородность смеси, низкую склонность к расслоению и превосходную сыпучесть. Эта смесь особенно подходит для приготовления быстро распадающейся разбавленной композиции кортикостероида. В некоторых воплощениях изобретения данная смесь содержит флутиказона пропионат, адсорбированный на силикатизированной микрокристаллической целлюлозе, и быстро диспергируемые микрогранулы.
Согласно воплощениям, фармацевтически приемлемый носитель для абсорбции кортикостероида микронизируют. В некоторых воплощениях, микронизированный фармацевтически приемлемый носитель имеет диаметр меньше примерно 20 микрон, например, меньше примерно 15 микрон, меньше примерно 10 микрон, меньше примерно 9 микрон, меньше примерно 8 микрон, меньше примерно 7 микрон, меньше примерно 6 микрон, меньше примерно 5 микрон, меньше примерно 4 микрон, меньше примерно 3 микрон, меньше примерно 2 микрон или меньше примерно 1 микрона. В конкретных воплощениях микронизированный фармацевтически приемлемый носитель имеет диаметр меньше примерно 5 микрон.
В некоторых воплощениях, фармацевтически приемлемый носитель присутствует в фармацевтической композиции (например, ODT) в количестве в диапазоне от примерно 1% масс./масс., до примерно 20% масс./масс., например, примерно 2%, примерно 3%, примерно 4%, примерно 5%, примерно 6%, примерно 7%, примерно 8%, примерно 9%, примерно 10%, примерно 11%, примерно 12%, примерно 13%, примерно 14%, примерно 15%, примерно 16%, примерно 17%, примерно 18% или примерно 19%, включая все значения и поддиапазоны этих значений. В предпочтительных воплощениях фармацевтически приемлемый носитель присутствует в фармацевтической композиции (например, ODT) в количестве примерно 10% (масс./масс.).
В некоторых воплощениях, в результате объединения кортикостероида с фармацевтически приемлемым носителем получают партию перорально распадающихся композиций, имеющих однородность содержания кортикостероида в диапазоне от примерно 85% до примерно 115% от дозировки кортикостероида, указанной на этикетке (например, 1,5-7,5 мг, включая 1,5 мг, 3,0 мг, 4,5 мг, 6,0 мг и 7,0 мг). В некоторых воплощениях, партия имеет однородность состава по кортикостероиду в диапазоне от примерно 90% до примерно 110% от дозировки кортикостероида, указанной на этикетке, или в диапазоне от примерно 95% до примерно 105% или от примерно 96% до примерно 104% или от примерно 97% до примерно 103% или от примерно 98% до примерно 102% или от примерно 99% до примерно 101%.
В некоторых воплощениях, скорость распадения распадающихся композиций по настоящему изобретению (например, ODT, облатки, лиофилизированной матрицы, пленки и т.д.) в ротовой полости индивида может составлять порядка примерно 60 секунд или меньше, примерно 50 секунд или меньше, примерно 40 секунд или меньше, примерно 30 секунд или меньше, примерно 20 секунд или меньше либо примерно 10 секунд или меньше.
В некоторых воплощениях, скорость распадения распадающихся композиций по настоящему изобретению (например, ODT, облатки, лиофилизированной матрицы, пленки и т.д.), измеренная с использованием теста на распадаемость из USP 701, составляет примерно 60 секунд или менее, примерно 45 секунд или менее, примерно 30 секунд или менее, примерно 20 секунд или менее либо примерно 10 секунд или менее.
Помимо кортикостероида и носителя, смесь, составляющая композиции или пероральные лекарственные формы по настоящему изобретению, может дополнительно содержать фармацевтически приемлемые ингредиенты, которые набухают, растворяются или иным образом способствуют распадаемости. Такие ингредиенты могут включать, но не ограничиваются этим, разрыхлитель, сахарный спирт, сахарид или их смесь, водорастворимое полимерное связующее вещество, полимер, образующий биогель, или биоадгезивный полимер, который может удерживать частицы кортикостероида в прикрепленном к воспаленным тканям пищевода состоянии дольше, чем в его отсутствие.
В некоторых воплощениях настоящего изобретения предложена твердая фармацевтическая композиция, содержащая кортикостероид и фармацевтически приемлемый образующий биогель полимер, который обеспечивает более длительное удерживание кортикостероида в воспаленных тканях пищевода. Ингредиент, называемый в данном описании как «полимер, образующий биогель» или «биоадгезивный агент», представляет собой агент, который способствует адгезии кортикостероида к биологическим поверхностям, в особенности к воспаленной слизистой оболочке, посредством образования геля в физиологических условиях желудочно-кишечного (ЖК) тракта, например, при контакте с физиологическими жидкостями и/или при физиологической температуре, и включает, но не ограничивается этим, образующие биогель полимеры, перечисленные ниже.
Полимер, образующий биогель, может быть термочувствительным полимером. Подходящие термочувствительные полимеры включают полиакриламиды, такие как поли(N-изопропилакриламид), а также сополимеры на основе полимеров простых и сложных эфиров, такие как поли(этиленгликоль-(DL-молочная кислота-когликолевая кислота)-этиленгликоль). Такие термочувствительные полимеры могут частично или полностью покрывать воспаленные ткани пищевода, одновременно удерживая частицу(ы) кортикостероида вплотную к воспаленным тканям или в тесном контакте с ними, тем самым усиливая контакт с кортикостероидом в месте нахождения воспаленных тканей.
В некоторых воплощениях, композиции по настоящему изобретению включают в себя биоадгезивный агент, такой как липид или полимер. Примерами таких липидов являются глицерофосфолипиды, такие как фосфатидилхолин, и диацилглицерины, такие как глицерин-диолеат. Примеры биоадгезивных полимеров включают хитозан, полиортоэфиры и сополимеры, терполимеры и их смеси.
В некоторых воплощениях, твердые фармацевтические композиции по настоящему изобретению включают адгезивный агент. Подходящие адгезивные агенты включают, но не ограничиваются этим, комплекс сахарозы и сульфата алюминия, хитозан и производные, например триметилхитозан, поливинилпирролидон, метилцеллюлозу, гидроксипропилцеллюлозу, сшитые сополимеры полиакриловой кислоты, сополимер винилпирролидона и винилацетата (например, Kollidon® VA 64 от BASF), Soluplus®, сополимер полиэтиленгликоля 6000, винилкапролактама и винилацетата (13:57:30) от BASF, поливиниловый спирт, полиэтиленоксид, полиамид, альгиновую кислоту и ее соли, каррагинан, ксантановую камедь, аммониометакрилатные сополимеры, полимеры CARBOPOL, мальтодекстрины, пектины, сукралозу и их комбинации. В некоторых воплощениях твердых фармацевтических композиций по настоящему изобретению кортикостероид и адгезивный агент равномерно ассоциированы. В некоторых воплощениях, твердая фармацевтическая композиция содержит кортикостероид, окруженный адгезивным агентом или инкапсулированный в него. В некоторых воплощениях, твердая фармацевтическая композиция содержит кортикостероид, размещенный на поверхности адгезивного агента. В еще одних воплощениях твердая фармацевтическая композиция содержит кортикостероид, смешанный или гранулированный с адгезивным агентом.
В некоторых воплощениях, твердая фармацевтическая композиция включает любую твердую лекарственную форму, которая быстро распадается в полости рта с образованием суспензии порошкообразного кортикостероида, которая, как предполагается, после проглатывания покрывает воспаленную слизистую оболочку пищевода или прилипает к ней.
В некоторых воплощениях, композиция по настоящему изобретению представлена в форме ODT. ODT содержит лекарственное средство в количестве менее примерно 5% (масса лекарственного средства/масса композиции) и фармацевтически приемлемый носитель, при этом композиция не обладает никакой существенной системной глюкокортикоидной или минералокортикоидной активностью после перорального введения людям. Частицы лекарственного средства (например, частицы кортикостероида, описанного в данной заявке, возможно покрытые адгезивным агентом, описанным в данной заявке, или возможно объединенные с ним) объединяют с быстро диспергируемыми микрогранулами. Быстро диспергируемые микрогранулы содержат сахарный спирт, сахарид или их смесь и разрыхлитель как таковой или разрыхлитель в комбинации с фармацевтически приемлемым вспомогательным веществом, обладающим многофункциональной активностью (например, с прежелатинизированным крахмалом, гидроксипропилцеллюлозой или тому подобным).
Неограничивающий список подходящих разрыхлителей для быстро диспергируемых микрогранул включает кросповидон (сшитый PVP), натрия крахмала гликолят, сшитую натрий карбоксиметилцеллюлозу, силикат кальция и гидроксипропилцеллюлозу с низкой степенью замещения.
Количество разрыхлителя в ODT обычно находится в диапазоне от примерно 1% до примерно 10% по массе.
Сахарные спирты представляют собой гидрогенизированные формы углеводов, в которых карбонильная группа (т.е. альдегидная или кетонная) восстановлена до первичной или вторичной гидроксильной группы. Неограничивающие примеры подходящих сахарных спиртов для быстро диспергируемых гранул фармацевтических композиций по настоящему изобретению включают, например, арабит, изомальтит, эритрит, глицерин, лактит, маннит, сорбит, ксилит, мальтит и их смеси.
Термин «сахарид» является синонимом термина «сахар» и включает, но не ограничивается этим, моносахариды, такие как глюкоза, фруктоза и рибоза, и дисахариды, такие как сахароза, лактоза, мальтоза, трегалоза и целлобиоза. Неограничивающие примеры подходящих сахаридов для применения в композициях по настоящему изобретению включают, например, лактозу, сахарозу, мальтозу и их смеси. В предпочтительных воплощениях композиция не содержит лактозу. В некоторых воплощениях, быстро диспергируемые гранулы содержат по меньшей мере один разрыхлитель в комбинации с сахарным спиртом. В некоторых воплощениях, быстро диспергируемые гранулы содержат по меньшей мере один разрыхлитель в комбинации с сахаридом. В некоторых воплощениях, содержащие разрыхлитель гранулы содержат по меньшей мере один разрыхлитель в комбинации с сахарным спиртом и сахаридом.
Количество сахарного спирта и/или сахарида в быстро диспергируемых гранулах изменяется в диапазоне примерно 99%-90% или примерно 95%-90% от общей массы содержащих разрыхлитель гранул, включая все диапазоны и поддиапазоны между ними.
Количество сахарного спирта и/или сахарида в ODT изменяется в диапазоне от примерно 30% до примерно 70% по массе.
В некоторых воплощениях, средний размер частиц сахарного спирта и/или сахарида составляет 30 мкм или менее, например, примерно 1-30 мкм, примерно 5-30 мкм, примерно 5-25 мкм, примерно 5-20 мкм, примерно 5-15 мкм, примерно 5-10 мкм, примерно 10-30 мкм, примерно 10-25 мкм, примерно 10-20 мкм, примерно 10-15 мкм, 15-30 мкм, примерно 15-25 мкм, примерно 15-20 мкм, примерно 20-30 мкм, примерно 20-25 мкм или примерно 25-30 мкм.
Соотношение разрыхлителя и сахарного спирта, сахарида или их смеси в быстро диспергируемых микрогранулах изменяется в диапазоне от примерно 90/10 до примерно 99/01, например, составляет примерно 90/10, примерно 91/9, примерно 92/8, примерно 93/7, примерно 94/6, примерно 95/5, примерно 96/4, примерно 97/3, примерно 98/2, примерно 99/1, включая все значения, диапазоны и поддиапазоны между ними.
Быстро диспергируемые микрогранулы, присутствующие в ODT, способствуют быстрой распадаемости таблетки при ее попадании в ротовую полость, создавая однородную суспензию, содержащую частицы кортикостероидного лекарственного средства. Чтобы обильно покрыть слизистую оболочку пищевода, желательно вводить достаточное количество быстро диспергируемых микрогранул. Это создает проблему однородности состава для таких содержащих низкую дозу ODT (например, в случае ODT по 300 мг, содержащей 12 мг кортикостероида или меньше). Обычно эту проблему преодолевают, используя гранулирование, которое включает распыление разбавленного раствора кортикостероида на слой порошка эксципиента. Частицы лекарственного средства внедряются в гранулы и поэтому могут не экспонироваться к воспаленной слизистой оболочке, что приводит к низкой эффективности. Неожиданно было обнаружено, что существует возможность не только достичь желаемой однородности состава, но также увеличить вероятность экспонирования в значительной степени частиц кортикостероидного лекарственного средства к воспаленной слизистой оболочке в результате адсорбции частиц микронизированного кортикостероидного лекарственного средства местного действия на фармацевтически приемлемом носителе (таком как силикатизированная микрокристаллическая целлюлоза) перед смешиванием с быстро диспергируемыми микрогранулами и другими эксципиентами и прессованием в ODT.
Лекарственная форма, описанная в данной заявке, также может включать в себя фармацевтически приемлемые эксципиенты, обычно используемые в композициях в форме распадающихся таблеток, такие как наполнители, разбавители, глиданты, разрыхлители, связующие вещества и смазывающие вещества.
Примеры подходящих наполнителей, разбавителей и/или связующих веществ включают, но не ограничиваются этим, лактозу (например, высушенную распылением лактозу, такую как FAST-FLO®), микрокристаллическую целлюлозу (различные марки Avicel®, CEOLUS®), гидроксипропилцеллюлозу, L-гидроксипропилцеллюлозу (с низкой степенью замещения), низкомолекулярную гидроксипропилметилцеллюлозу (НРМС) (например, Methocel™ марок Е, F и K от Dow Chemical, MetholoseE SH от Shin-Etsu, Ltd), гидроксиэтилцеллюлозу, натрий карбоксиметилцеллюлозу, карбоксиметилгидроксиэтилцеллюлозу и другие производные целлюлозы, сахарозу, агарозу, сорбит, маннит, декстрины, мальтодекстрины, крахмалы или модифицированные крахмалы (включая картофельный крахмал, кукурузный крахмал и рисовый крахмал), фосфат кальция (например, основный фосфат кальция, гидрофосфат кальция, гидрат фосфорнокислого двузамещенного кальция), сульфат кальция, карбонат кальция, альгинат натрия и коллаген. Предпочтительным наполнителем для композиции по изобретению является маннит, такой как высушенный распылением маннит.
Примеры подходящих разрыхлителей включают, но не ограничиваются этим, кросповидон (сшитый PVP), натрия крахмала гликолят, сшитую карбоксиметилцеллюлозу натрия, силикат кальция и гидроксипропилцеллюлозу с низкой степенью замещения. Предпочтительным разрыхлителем для композиции по изобретению является кросповидон.
Конкретные примеры глидантов и смазывающих веществ включают стеариновую кислоту, стеарат магния, стеарат кальция или стеараты других металлов, тальк, глицерилбегенат, коллоидный диоксид кремния, кукурузный крахмал и возможно стеарат магния или стеарилфумарат натрия (смазывающее вещество, смешиваемое внутригранулярно или используемое для смазывания наружных поверхностей штампа и пуансона). Предпочтительным глидантом для композиции по изобретению является коллоидный диоксид кремния, а предпочтительным смазывающим веществом является стеарилфумарат натрия.
В некоторых воплощениях, способы и композиции, описанные в данной заявке, можно использовать для лечения пациента, страдающего от ЕоЕ, который также испытывает аллергию на лактозу и/или аллергию на крахмал. Фармацевтические композиции, содержащие лактозу, могут усиливать аллергию на лактозу у пациента, страдающего от нее, и это может привести к усилению дискомфорта у пациентов, страдающих также и от ЕоЕ. В некоторых воплощениях, в состав фармацевтических композиций, описанных в данной заявке, не входит лактоза. Аналогично, фармацевтические композиции, содержащие крахмал, могут усиливать аллергию на крахмал у пациента, страдающего от нее, и это может привести к усилению дискомфорта у пациентов, страдающих также и от ЕоЕ. В некоторых воплощениях, в состав фармацевтических композиций, описанных в данной заявке, не входит крахмал. В других предпочтительных воплощениях. В некоторых воплощениях, в состав фармацевтических композиций, описанных в данной заявке, не входит лактоза и сахароза.
Твердые фармацевтические композиции по настоящему изобретению помимо ODT могут включать другие лекарственные формы, такие как облатка, пленка или другая твердая лекарственная форма, которые быстро распадаются в полости рта с образованием суспензии или дисперсии кортикостероида, которая может быть легко проглочена с покрыванием затем поверхности пораженной эозинофильным эзофагитом слизистой оболочки.
Например, облатки могут включать высушенные или лиофилизированные композиции, такие как перорально распадающиеся или растворимые лекарственные формы, приготовленные с использованием технологии лиофилизации Zydis® (например так, как описано в патенте США №6316027), содержащие кортикостероид в качестве активного фармацевтического ингредиента. Лекарственные формы в виде пленок могут включать могут включать съедобные пленки, как например, описанные в патенте США №6596298 или патенте США №6740332, содержащие кортикостероид в качестве активного фармацевтического ингредиента. В некоторых воплощениях, твердая композиция содержит лиофилизированную матрицу, при этом лиофилизированная матрица содержит кортикостероид, носитель и эксципиент. Подходящие эксципиенты включают, но не ограничиваются этим, маннит, ксилит, сорбит, мальтол, мальтит, лактозу, сахарозу, мальтозу и их комбинации.
Местное введение кортикостероида в ротовую полость индивидов ассоциировано с кандидозной инфекцией и кандидозным стоматитом полости рта. Хотя данное изобретение разработано с возможностью уменьшения предрасположенности к таким инфекциям, в некоторых воплощениях данного изобретения фармацевтическая композиция может включать в себя противогрибковый агент. Подходящие противогрибковые агенты включают, но не ограничиваются этим, ингибирующие митоз противогрибковые средства, противогрибковые средства на основе аналогов пиримидина, противогрибковые средства группы полиенов, противогрибковые средства группы бензимидазолов, противогрибковые средства группы имидазолов, противогрибковые средства группы триазолов, противогрибковые средства группы тиазолов, противогрибковые средства группы аллиламинов, противогрибковые средства группы эхинокандинов и другие «некатегоризированные» противогрибковые средства, признанные в данной области техники, которые не попадают ни в одну из упомянутых выше категорий (например, толнафтат и циклопирокс). Например, подходящие противогрибковые агенты, которые могут быть введены в твердые фармацевтические композиции по настоящему изобретению, включают, но не ограничиваются этим, абафунгин, аморолфин, анидулафунгин, бифоназол, бутенафин, бутоконазол, кандицин, каспофунгин, циклопирокс, клотримазол, эконазол, фентиконазол, филипин, флуконазол, флуцитозин, гризеофульвин, изавуконазол, изоконазол, итраконазол, кетоконазол, микафунгин, миконазол, миконазола нитрат, нафтифин, натамицин, нистатин, оксиконазол, позаконазол, прамиконазол, равуконазол, римоцидин, сетаконазол, сулконазол, тербафин, терконазол, тиоконазол, толнафтат, ундециленовую кислоту и вориконазол.
В некоторых воплощениях, фармацевтические композиции по настоящему изобретению включают в себя противовирусный агент. Противовирусные агенты, которые могут быть в введены в твердые фармацевтические композиции по настоящему изобретению, включают интерфероны, нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы, ненуклеозидные ингибиторы обратной транскриптазы, ингибиторы протеаз, ингибиторы интегразы, ингибиторы слияния, ингибиторы созревания, аналоги гуанозина, аналоги пуринов, аналоги пиримидинов и другие «некатегоризированные» противовирусные лекарственные средства, признанные в данной области техники, которые не попадают ни в одну из упомянутых выше категорий (например, фоскарнет и милтефозин). Например, подходящие противовирусные агенты, которые могут быть введены в твердые фармацевтические композиции по настоящему изобретению, включают, но не ограничиваются этим, абакавир, ацикловир, адефовир, амантадин, амдоксовир, ампренавир, аплавирок, априцитабин, арбидол, атазанавир, бевиримат, BMS-488043, боцепревир, бривудин, цидофовир, DCM205, докозанол, делавирдин, диданозин, дарунавир, эфавиренз, элвитегравир, эльвуцитабин, эмтрицитабин, энфувиртид, эпигаллокатехина галлат, этравирин, фамцикловир, фосампренавир, ганцикловир, глобоиднан А, гриффитсин, ибализумаб, идоксуридин, индинавир, ламивудин, лопинавир, ловирид, маравирок, нелфинавир, невирапин, осельтамивир, ПЭГилированный интерферон-альфа-2а, ПЭГилированный интерферон-альфа-2b, пенцикловир, перамивир, плериксафор, PRO 140, рацивир, ралтегравир, ритонавир, рибавирин, римантадин, рилпивирин, саквинавир, стампидин, ставудин, тенофовир, типранавир, TNX-355, трифлуридин, тромантадин, валацикловир, валганцикловир, викривирок, видарабин, вирамидин, вивекон, зальцитабин, занамивир и зидовудин.
Лекарственные формы в виде таблеток, в том числе лекарственные формы в виде ODT, содержащие кортикостероид местного действия в низкой дозе (например, кортикостероиды, имеющие системную биодоступность, меньшую или равную примерно 20% от такой дозы, или кортикостероиды, которые готовят с целью снижения системной биодоступности, каждый из которых описан выше) и фармацевтически приемлемый носитель, при этом лекарственное средство присутствует в количестве меньше примерно 5% (масса лекарственного средства/масса композиции), распадаются быстрее, чем примерно через 30 с (метод из USP), и характеризуются низкой прочностью на истирание, чтобы иметь достаточную долговременную устойчивость к обработке, транспортировке и/или упаковыванию в блистерный упаковочный материал с выдавливаемым защитным слоем. В некоторых воплощениях, прочность на истирание лекарственной формы в виде ODT, описанной в данной заявке, составляет менее примерно 1%, например, менее примерно 0,9%, менее примерно 0,8%, менее примерно 0,7%, менее примерно 0,6%, менее примерно 0,5%, менее примерно 0,4%, менее примерно 0,3% и т.д., включая все диапазоны и поддиапазоны между ними).
В предпочтительных воплощениях ODT, используемая в (или для применения в) способах, описанных в данной заявке, содержит: кортикостероид в количестве от примерно 0,25% до примерно 2,5% масс./масс. (или от примерно 0,75 мг до примерно 7,5 мг); фармацевтически приемлемый носитель в количестве от примерно 5% до примерно 15% масс./масс. (или от примерно 20 мг до примерно 40 мг); и быстро диспергируемые микрогранулы в количестве от примерно 40% до примерно 60% масс./масс. (или от примерно 125 мг до примерно 175 мг), при этом быстро диспергируемые микрогранулы характеризуются соотношением сахарный спирт/сахарид и разрыхлитель от примерно 90:10 до примерно 99:1. Согласно воплощениям, ODT возможно дополнительно содержит: разрыхлитель в количестве от примерно 5% до примерно 10% масс./масс. (или от примерно 5 мг -20 мг до примерно 30 мг); сахарный спирт или сахарид в количестве от примерно 20% до примерно 40% масс./масс. (или от примерно 80 мг до примерно 100 мг); и возможно эксципиенты.
Предпочтительные воплощения фармацевтической композиции, описанной в данной заявке, приведены в Таблице 1 или Таблице 2.
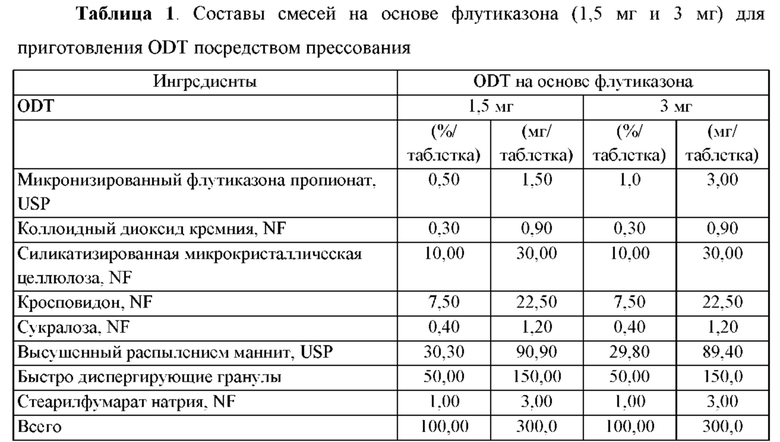
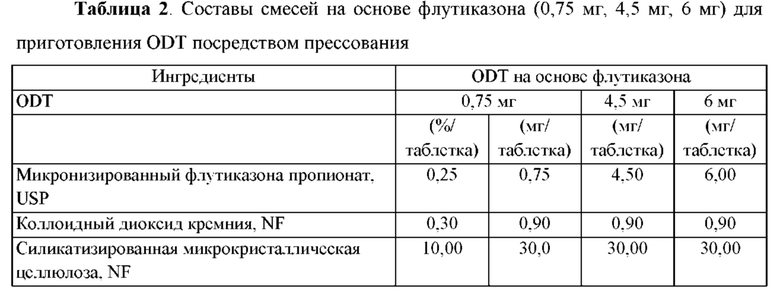
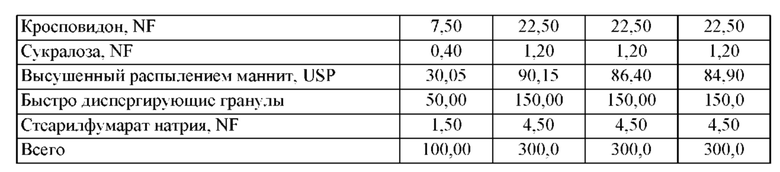
Воспаление желудочно-кишечного тракта и способы лечения
Лечение кортикостероидом местного действия (например, флутиказона пропионатом) приводит к меньшему количеству побочных эффектов, чем при других видах лечения, например, кортикостероидом с сильным системным действием. После введения фармацевтической композиции по настоящему изобретению пациенту композиция распадается в ротовой полости пациента и контактирует с пищеводом без системного всасывания, ограничивая таким образом появление системных эффектов.
В некоторых воплощениях настоящего изобретения предложены способы лечения симптомов, ассоциированных с воспалительным расстройством кишечника. В некоторых воплощениях настоящего изобретения предложены способы лечения воспаления, ассоциированного с воспалительным расстройством желудочно-кишечного тракта. В некоторых воплощениях настоящего изобретения предложены способы лечения как симптомов, так и воспаления, ассоциированного с воспалительным расстройством желудочно-кишечного тракта. В некоторых воплощениях, воспалительное расстройство желудочно-кишечного тракта поражает верхнюю часть желудочно-кишечного тракта. В некоторых воплощениях, верхней частью желудочно-кишечного тракта является пищевод.
В некоторых воплощениях, пероральный кортикостероид контактирует с верхней частью желудочно-кишечного тракта и/или осаждается в ней. В некоторых воплощениях, пероральный кортикостероид контактирует с пищеводом и/или осаждается в нем. В некоторых воплощениях, пероральный кортикостероид контактирует с дистальной частью пищевода и/или осаждается в ней. В некоторых воплощениях, фармацевтическая композиция контактирует с проксимальной частью пищевода и/или осаждается в ней. В некоторых воплощениях, пероральный кортикостероид контактирует с дистальной и проксимальной частью пищевода и/или осаждается в них по существу в одинаковых количествах.
Воспалительные расстройства желудочно-кишечного тракта, которые можно лечить в соответствии с настоящим изобретением, включают, но не ограничиваются этим, воспаление пищевода, воспаление голосовой щели, воспаление надгортанника, воспаление миндалин, воспаление ротоглотки, эозинофильный эзофагит (ЕоЕ), гастроэзофагеальную рефлюксную болезнь (GERD), неэрозивную рефлюксную болезнь (NERD), эрозивный эзофагит, пищевод Баррета, эозинофильный гастроэнтерит, гиперэозинофильный синдром, коррозивный (разъедающий) химический эзофагит, индуцированный облучением эзофагит, индуцированный химиотерапией эзофагит, преходящий индуцированный приемом лекарственных средств эзофагит (также известный как медицинский эзофагит), стойкий индуцированный приемом лекарственных средств эзофагит, поражение пищевода при болезни Крона и псевдомембранозный эзофагит. В некоторых воплощениях, настоящее изобретение включает способ лечения пищевой аллергии, вызываемой идентифицированным аллергеном, например, «атопического IBS (синдром раздраженного кишечника)» и «атопического кишечника». В некоторых воплощениях, настоящее изобретение включает способ лечения пациента, имеющего одно или несколько из указанных выше расстройств желудочно-кишечного тракта, при этом пациент также имеет аллергию на лактозу и/или аллергию на крахмал. В некоторых воплощениях, воспалительное расстройство желудочно-кишечного тракта представляет собой эозинофильный эзофагит (ЕоЕ). В некоторых воплощениях, настоящее изобретение включает способ лечения пациента с ЕоЕ, при этом пациент также страдает аллергией на лактозу и/или аллергией на крахмал.
В некоторых воплощениях, фармацевтические композиции, описанные в данной заявке, вводят до излечения от симптомов и/или воспаления, ассоциированных с воспалением желудочно-кишечного тракта. В некоторых воплощениях, фармацевтические композиции, описанные в данной заявке, продолжают вводить после излечения от симптомов и/или воспаления, ассоциированных с воспалением желудочно-кишечного тракта. В некоторых воплощениях, симптомом является дисфагия, эпизоды затрудненного прохождения пищи, ощущения комка в горле пациента и/или повышенное количество эозинофилов в пищеводе.
Лечение воспаления желудочно-кишечного тракта может быть оценено любым способом, известным в данной области техники. Например, тесты, используемые для оценки пациентов с воспалением пищевода, таким как ЕоЕ, включают, но не ограничиваются этим, биопсии, оценку симптомов (например, по анкете с результатами лечения по оценке пациента (PRO) или по опроснику врача), оценку качества жизни, определение дней без дисфагии у пациента, эндоскопию (например, EREFS), оценку эластичности пищевода и/или улучшения в ремоделировании пищевода (например, с использованием подходящего диагностического теста, такого как EndoFLIP (предоставляемого Crospon Inc.)), оценку уровня биомаркеров, оценку снижения максимального количества эозинофилов, оценку ослабления затрудненного прохождения пищи, EEsAI, определение индекса сильной дисфагии (по опроснику симптомов дисфагии (DSQ)), MDQ-30 (опросник по оценке тяжести дисфагии по шкале Мейо за 30 дней), EoE-QoL-A, VDQ (опросник по визуальной оценке дисфагии), определение показателей избегания, корректировки и медленного приема пищи (AMS) и/или гистологическое исследование.
В некоторых воплощениях, ответ пациента на лечение определяют, измеряя изменения в одном или нескольких показателях из опросника, связанных с биологическим ответом, таких как гистологический показатель (например, количество эозинофилов). Использование сообщаемых пациентами симптомов может дать ложноположительные результаты, поскольку пациенты могут менять стиль своего поведения, чтобы уменьшить число случаев дисфагии (например, избегать проблемных продуктов питания или использовать другие изменения стиля поведения, такие как тщательное пережевывание пищи или употребление жидкости), что может изменять значение показателя из опросника независимо от того, произошло ли улучшение биологических симптомов.
В некоторых воплощениях, ответ пациента оценивают посредством определения гистологических показателей у пациента. В некоторых воплощениях, гистологический показатель оценивают по одному или более разным гистологическим признакам, включая, но не ограничиваясь этим, эозинофильное воспаление, гиперплазию базальной зоны, расширение межклеточного пространства, фиброз собственной пластинки слизистой оболочки, эозинофильный абсцесс, поверхностное расслоение, изменение поверхностного эпителия и наличие некератинизированных эпителиальных клеток.
В некоторых воплощениях, введение перорального кортикостероида согласно способам, описанным в данной заявке, приводит к снижению гистологического показателя у подвергаемого лечению пациента по сравнению с не подвергаемым лечению пациентом или одним и тем же пациентом до лечения. В некоторых воплощениях, гистологический показатель измеряют у подвергаемого лечению пациента в интервале от 1 недели до 10 лет. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению гистологического показателя примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году по сравнению с гистологическим показателем у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению гистологического показателя примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок по сравнению с гистологическим показателем у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, гистологический показатель снижается примерно на 1%, примерно на 5%, примерно на 10%, примерно на 20%, примерно на 30%, примерно на 40%, примерно на 50%, примерно на 60%, примерно на 70%, примерно на 80%, примерно на 90% или примерно на 100% по сравнению с гистологическим показателем у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению гистологического показателя примерно на 1%, примерно на 5%, примерно на 10%, примерно на 20%, примерно на 30%, примерно на 40%, примерно на 50%, примерно на 60%, примерно на 70%, примерно на 80%, примерно на 90% или примерно на 100% примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году по сравнению с гистологическим показателем у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению гистологического показателя примерно на 1%, примерно на 5%, примерно на 10%, примерно на 20%, примерно на 30%, примерно на 40%, примерно на 50%, примерно на 60%, примерно на 70%, примерно на 80%, примерно на 90% или примерно на 100% примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок по сравнению с гистологическим показателем у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, введение перорального кортикостероида согласно способам, описанным в данной заявке, приводит к снижению максимального количества эозинофилов (в поле зрения микроскопа под большим увеличением (HPF), например, как описано в примере 2) по результатам по меньшей мере одной биопсии у подвергаемого лечению пациента по сравнению с максимальным содержанием эозинофилов в HPF у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению максимального количества эозинофилов по результатам по меньшей мере одной биопсии до величины меньше примерно 15/HPF. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению максимального количества эозинофилов по результатам по меньшей мере одной биопсии у подвергаемого лечению пациента до величины меньше примерно 14/HPF, меньше примерно 13/HPF, меньше примерно 12/HPF, меньше примерно 11/HPF, меньше примерно 10/HPF, меньше примерно 9/HPF, меньше примерно 8/HPF, меньше примерно 7/HPF, меньше примерно 6/HPF, меньше примерно 5/HPF, меньше примерно 4/HPF, меньше примерно 3/HPF, меньше примерно 2/HPF, меньше примерно 1/HPF или ниже (например, 0) у пациента. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению максимального количества эозинофилов у пациента по результатам по меньшей мере одной биопсии до величины меньше 1/HPF. В некоторых воплощениях, снижение максимального количества эозинофилов по результатам по меньшей мере одной биопсии у подвергаемого лечению пациента измеряют в интервале от примерно 1 недели до примерно 10 лет. В некоторых воплощениях, снижение максимального количества эозинофилов измеряют примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению максимального количества эозинофилов по результатам по меньшей мере одной биопсии до величины меньше примерно 14/HPF, меньше примерно 13/HPF, меньше примерно 12/HPF, меньше примерно 11/HPF, меньше примерно 10/HPF, меньше примерно 9/HPF, меньше примерно 8/HPF, меньше примерно 7/HPF, меньше примерно 6/HPF, меньше примерно 5/HPF, меньше примерно 4/HPF, меньше примерно 3/HPF, меньше примерно 2/HPF, меньше примерно 1/HPF или ниже (например, 0) у пациента примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению максимального количества эозинофилов по результатам по меньшей мере одной биопсии до величины меньше примерно 14/HPF, меньше примерно 13/HPF, меньше примерно 12/HPF, меньше примерно 11/HPF, меньше примерно 10/HPF, меньше примерно 9/HPF, меньше примерно 8/HPF, меньше примерно 7/HPF, меньше примерно 6/HPF, меньше примерно 5/HPF, меньше примерно 4/HPF, меньше примерно 3/HPF, меньше примерно 2/HPF, меньше примерно 1/HPF или ниже (например, 0) у пациента примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок.
В некоторых воплощениях, введение перорального кортикостероида согласно способам, описанным в данной заявке, приводит к снижению максимального количества эозинофилов (в поле зрения микроскопа под большим увеличением (HPF), например, как описано в примере 2), по результатам по меньшей мере одной биопсии у подвергаемого лечению пациента по сравнению с максимальным количеством эозинофилов в HPF у не подвергаемого лечению пациента или одного и того же пациента до лечения по меньшей мере примерно на 10%, например, примерно на 15%, примерно на 20%, примерно на 25%, примерно на 30%, примерно на 35%, примерно на 40%, примерно на 45%, примерно на 50%, примерно на 55%, примерно на 60%, примерно на 65%, примерно на 70%, примерно на 75%, примерно на 80%, примерно на 85%, примерно на 90%, примерно на 91%, примерно на 92%, примерно на 93%, примерно на 94%, примерно на 95%, примерно на 96%, примерно на 97%, примерно на 98% или примерно на 99%, включая все значения и поддиапазоны между ними. В конкретных воплощениях максимальное количество эозинофилов снижается на величину диапазоне от примерно 50% до примерно 99%, например, примерно на 55%, примерно на 60%, примерно на 65%, примерно на 70%, примерно на 75%, примерно на 80%, примерно на 85%, примерно на 90%, примерно на 91%, примерно на 92%, примерно на 93%, примерно на 94%, примерно на 95%, примерно на 96%, примерно на 97%, примерно на 98%, включая все значения и поддиапазоны между ними.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению максимального количества эозинофилов по результатам по меньшей мере одной биопсии у подвергаемого лечению пациента на 12-й неделе, 26-й неделе или 52-й неделе. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению максимального количества по результатам по меньшей мере одной биопсии у подвергаемого лечению пациента до величины меньше примерно 6/HPF на 12-й неделе, 26-й неделе или 52-й неделе. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению максимального количества эозинофилов по результатам всех протестированных биоптатов у подвергаемого лечению пациента на 12-й неделе, 26-й неделе или 52-й неделе. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению максимального количества эозинофилов по результатам всех протестированных биоптатов у подвергаемого лечению пациента до величины меньше примерно 6/HPF на 12-й неделе, 26-й неделе или 52-й неделе.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя из опросника пациента, заполненного пациентом или врачом. В некоторых воплощениях, в опроснике указывается индекс активности эозинофильного эзофагита (EEsAI), который может отмечаться с периодичностью либо один раз в 7 дней, либо один раз в день для мониторинга тяжести и частоты дисфагии и любых изменений рациона питания. В некоторых воплощениях, данный опросник представляет собой опросник симптомов дисфагии (DSQ), который является дневником для ежедневных записей симптомов, используемым для мониторинга частоты эпизодов дисфагии. В некоторых воплощениях, данный опросник представляет собой индивидуальную сообщаемую пациентом оценку результатов лечения, которая включает оценку числа дней без дисфагии. В некоторых воплощениях, общий показатель согласно опроснику, такому как EEsAI, или анкете с результатами лечения симптомов ЕоЕ по оценке пациента (PROSE), используют в сочетании с другими оценками пациента для определения ответа пациента на лечение. В некоторых воплощениях, корреляция показателя из опросника с улучшением результатов гистологических измерений (например, в количестве эозинофилов) указывает на ответ пациента на лечение.
В некоторых воплощениях, средний показатель из опросника измеряют у подвергаемого лечению пациента в интервале от 1 недели до 10 лет. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя из опросника примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году по сравнению со средним показателем из опросника у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя из опросника примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный по сравнению со средним показателем из опросника у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, средний показатель из опросника снижается примерно на 1%, примерно на 5%, примерно на 10%, примерно на 20%, примерно на 30%, примерно на 40%, примерно на 50%, примерно на 60%, примерно на 70%, примерно на 80%, примерно на 90% или примерно на 100% по сравнению со средним показателем из опросника у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя из опросника примерно на 1%, примерно на 5%, примерно на 10%, примерно на 20%, примерно на 30%, примерно на 40%, примерно на 50%, примерно на 60%, примерно на 70%, примерно на 80%, примерно на 90% или примерно на 100% примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году по сравнению со средним показателем из опросника у подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя из опросника примерно на 1%, примерно на 5%, примерно на 10%, примерно на 20%, примерно на 30%, примерно на 40%, примерно на 50%, примерно на 60%, примерно на 70%, примерно на 80%, примерно на 90% или примерно на 100% примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок по сравнению со средним показателем из опросника у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, в опроснике пациента указывается EEsAI. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EEsAI до значения менее 20. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EEsAI у подвергаемого лечению пациента до значения менее примерно 19, менее примерно 18, менее примерно 17, менее примерно 16, менее примерно 15, менее примерно 14, менее примерно 13, менее примерно 12, менее примерно 11, менее примерно 10, менее примерно 9, менее примерно 8, менее примерно 7, менее примерно 6, менее примерно 5, менее примерно 4, менее примерно 3, менее примерно 2, менее примерно 1 или или примерно до 0. В некоторых воплощениях, снижение среднего показателя EEsAI у подвергаемого лечению пациента измеряют в интервале от примерно 1 недели до примерно 10 лет. В некоторых воплощениях, снижение среднего показателя EEsAI измеряют на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EEsAI до значения менее примерно 20, менее примерно 19, менее примерно 18, менее примерно 17, менее примерно 16, менее примерно 15, менее примерно 14, менее примерно 13, менее примерно 2, менее примерно 11, менее примерно 10, менее примерно 9, менее примерно 8, менее примерно 7, менее примерно 6, менее примерно 5, менее примерно 4, менее примерно 3, менее примерно 2, менее примерно 1 или ниже (например, 0) примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EEsAI до значения менее примерно 20, менее примерно 19, менее примерно 18, менее примерно 17, менее примерно 16, менее примерно 15, менее примерно 14, менее примерно 13, менее примерно 2, менее примерно 11, менее примерно 10, менее примерно 9, менее примерно 8, менее примерно 7, менее примерно 6, менее примерно 5, менее примерно 4, менее примерно 3, менее примерно 2, менее примерно 1 или ниже (например, 0) примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EEsAI у подвергаемого лечению пациента на 12-й неделе, 26-й неделе или 52-й неделе. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EEsAI у подвергаемого лечению пациента до значения менее примерно до 20 на 12-й неделе, 26-й неделе или 52-й неделе.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего эндоскопического референсного показателя (EREFS) для ЕоЕ у подвергаемого лечению пациента, с использованием которого оценивают отек (например, ослабление васкуляризации или бледность), наличие пищеводных колец, экссудатов (например, белых бляшек), борозд и/или стриктуры в пищеводе в соответствии с Таблицей 3. В некоторых воплощениях, показатель EREFS у пациента коррелирует с гистологическим ответом (например, количеством эозинофилов), и показатель EREFS можно использовать вместе с гистологическим ответом для оценки ответа пациента на лечение.
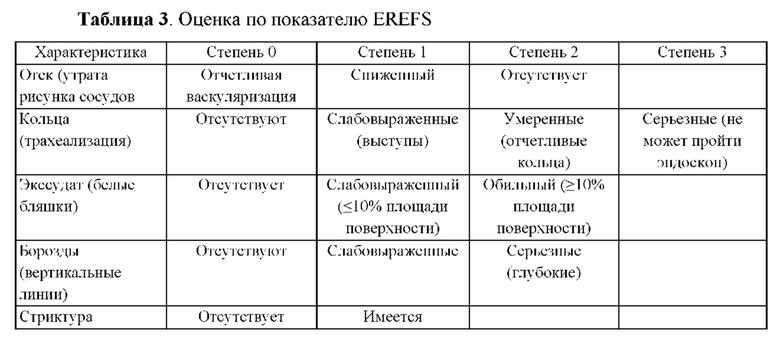
В некоторых воплощениях, композиции и способы, описанные в данной заявке, приводят к снижению показателя EREFS для отека до 1 или 0. В некоторых воплощениях, композиции и способы, описанные в данной заявке, приводят к снижению показателя EREFS для колец до 2, 1 или 0. В некоторых воплощениях, композиции и способы, описанные в данной заявке, приводят к снижению показателя EREFS для экссудатов до 1 или 0. В некоторых воплощениях, композиции и способы, описанные в данной заявке, приводят к снижению показателя EREFS для борозд до 1 или 0. В некоторых воплощениях, композиции и способы, описанные в данной заявке, по существу устраняют стриктуры.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EREFS до значения менее примерно 10, примерно 9, примерно 8, примерно 7, примерно 6, примерно 5, примерно 4, примерно 3, примерно 2, примерно 1 или примерно до нуля у подвергаемого лечению пациента. В некоторых воплощениях, средний показатель EREFS измеряют у подвергаемого лечению пациента в интервале от 1 недели до 10 лет. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EREFS примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EREFS примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок у подвергаемого лечению пациента. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EREFS до значения менее примерно 10, примерно 9, примерно 8, примерно 7, примерно 6, примерно 5, примерно 4, примерно 3, примерно 2, примерно 1 или примерно до нуля примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EREFS до значения менее примерно 10, примерно 9, примерно 8, примерно 7, примерно 6, примерно 5, примерно 4, примерно 3, примерно 2, примерно 1 или примерно до нуля примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок у подвергаемого лечению пациента.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению среднего показателя EREFS у подвергаемого лечению пациента примерно на 12-й неделе, примерно на 26-й неделе или примерно на 52-й неделе.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов дисфагии у подвергаемого лечению пациента. В некоторых воплощениях, снижение числа эпизодов дисфагии у подвергаемого лечению пациента измеряют путем определения количества дней без дисфагии у пациента. В некоторых воплощениях, улучшение в количестве дней без дисфагии у пациента измеряют вместе с другими показателями у пациентов, такими как улучшенные гистологические показатели (например, количества эозинофилов) для оценки ответа пациента на лечение. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к уменьшению числа эпизодов дисфагии у подвергаемого лечению пациента по сравнению с числом эпизодов дисфагии у не подвергаемого лечению субъекта или одного и того же пациента до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к уменьшению числа эпизодов дисфагии до менее примерно 6 в неделю. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к уменьшению числа эпизодов дисфагии до менее примерно 6 в неделю в течение периода времени двух недель. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к уменьшению числа эпизодов дисфагии до менее примерно 6 в неделю, примерно 5 в неделю, примерно 4 в неделю, примерно 3 в неделю, примерно 2 в неделю, примерно одного в неделю или к отсутствию эпизодов дисфагии за неделю. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к уменьшению числа эпизодов дисфагии до менее примерно 6 в неделю, примерно 5 в неделю, примерно 4 в неделю, примерно 3 в неделю, примерно 2 в неделю, примерно одного в неделю или к отсутствию эпизодов дисфагии за неделю в течение периода времени двух недель.
В некоторых воплощениях, число эпизодов дисфагии снижается на величину до примерно 100%. В некоторых воплощениях, число эпизодов дисфагии снижается на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с числом эпизодов дисфагии у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, дисфагия устраняется. В некоторых воплощениях, дисфагию оценивают у подвергаемого лечению пациента в интервале от 1 недели до 10 лет. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов дисфагии примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к уменьшению числа эпизодов дисфагии примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок по сравнению с числом эпизодов дисфагии у не подвергаемого лечению пациента или пациента до лечения. В некоторых воплощениях, число эпизодов дисфагии снижается на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с числом эпизодов дисфагии у не подвергаемого лечению пациента или одного и того же пациента до лечения примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, число эпизодов дисфагии снижается на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с числом эпизодов у не подвергаемого лечению пациента или одного и того же пациента до лечения примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок по сравнению с числом эпизодов у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов дисфагии у подвергаемого лечению пациента на 12-й неделе, 26-й неделе или 52-й неделе по сравнению с числом эпизодов дисфагии у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к улучшению в затрудненном прохождении пищи у подвергаемого лечению пациента по сравнению с числом эпизодов затрудненного прохождения пищи у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов затрудненного прохождения пищи до менее 4 в неделю. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов затрудненного прохождения пищи до менее 4 в неделю в течение периода времени двух недель. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов затрудненного прохождения пищи до менее примерно 4 в неделю, примерно 3 в неделю, примерно 2 в неделю, примерно одного в неделю или к отсутствию эпизодов затрудненного прохождения пищи за неделю. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов затрудненного прохождения пищи до менее примерно 4 в неделю, примерно 3 в неделю, примерно 2 в неделю, примерно одного в неделю или к отсутствию эпизодов затрудненного прохождения пищи за неделю в течение периода времени двух недель.
В некоторых воплощениях, число эпизодов затрудненного прохождения пищи снижается на величину до примерно 100%. В некоторых воплощениях, число эпизодов затрудненного прохождения пищи снижается на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с числом эпизодов затрудненного прохождения пищи у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, затрудненное прохождение пищи устраняется. В некоторых воплощениях, число эпизодов затрудненного прохождения пищи оценивают у подвергаемого лечению пациента в интервале от 1 недели до 10 лет. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов затрудненного прохождения пищи примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов затрудненного прохождения пищи примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок по сравнению с числом эпизодов затрудненного прохождение пищи у не подвергаемого лечению пациента или одного и того же пациента до лечения. В некоторых воплощениях, число эпизодов затрудненного прохождения пищи снижается на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%о, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с числом эпизодов затрудненного прохождение пищи у не подвергаемого лечению пациента или одного и того же пациента до лечения примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, число эпизодов затрудненного прохождения пищи снижается на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с числом эпизодов затрудненного прохождение пищи у не подвергаемого лечению пациента или одного и того же пациента до лечения примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок по сравнению с числом эпизодов затрудненного прохождения пищи у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению числа эпизодов затрудненного прохождения пищи у подвергаемого лечению пациента на 12-й неделе, 26-й неделе или 52-й неделе по сравнению с числом эпизодов затрудненного прохождения пищи у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ (Global ЕоЕ) у подвергаемого лечению пациента по сравнению с общим показателем ЕоЕ у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, снижение общего показателя ЕоЕ у подвергаемого лечению пациента измеряют в интервале от примерно 1 недели до примерно 10 лет. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году по сравнению с не подвергаемым лечению пациентом или одним и тем же пациентом до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет по сравнению с общим показателем ЕоЕ у не подвергаемого лечению пациента или пациента до лечения. В некоторых воплощениях, общий показатель ЕоЕ снижается на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с общим показателем ЕоЕ у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с общим показателем ЕоЕ у не подвергаемого лечению пациента или одного и того же пациента до лечения примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ на величину вплоть до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с общим показателем ЕоЕ у не подвергаемого лечению пациента или одного и того же пациента до лечения примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок.
Общий показатель ЕоЕ оценивают по шкале от 0 до 10, при этом 0 обозначает отсутствие каких-либо симптомов ЕоЕ, а 10 обозначает наличие самых тяжелых симптомов ЕоЕ. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ у подвергаемого лечению субъекта до значения меньше примерно 5. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ до значения меньше примерно 10, меньше примерно 9, меньше примерно 8, меньше примерно 7, меньше примерно 6, меньше примерно 5, меньше примерно 4, меньше примерно 3, меньше примерно 2, меньше примерно 1 или примерно 0. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ у подвергаемого лечению субъекта до значения меньше примерно 10, меньше примерно 9, меньше примерно 8, меньше примерно 7, меньше примерно 6, меньше примерно 5, меньше примерно 4, меньше примерно 3, меньше примерно 2, меньше примерно 1 или примерно 0 примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ у подвергаемого лечению субъекта до значения меньше примерно 10, меньше примерно 9, меньше примерно 8, меньше примерно 7, меньше примерно 6, меньше примерно 5, меньше примерно 4, меньше примерно 3, меньше примерно 2, меньше примерно 1 или примерно 0 примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к снижению общего показателя ЕоЕ у подвергаемого лечению пациента примерно на 12-й неделе, примерно на 26-й неделе или примерно на 52-й по сравнению с общим показателем ЕоЕ у не подвергаемого лечению пациента или одного и того же пациента до лечения.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, улучшает характеристики по результатам измерений с использованием эндоскопии (например, EndoFlip) у подвергаемого лечению пациента по сравнению с не подвергаемым лечению пациентом или одним и тем же пациентом до лечения. Такие характеристики включают, но не ограничиваются этим, диаметр пищевода, эластичность, очаговое сужение пищевода, растяжимость тела пищевода, площадь поперечного сечения (CSA) тела пищевода и диаметр просвета пищевода.
В некоторых воплощениях, характеристики по результатам измерений с использованием эндоскопии оценивают у подвергаемого лечению пациента в интервале от 1 недели до 10 лет. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к улучшению характеристик по результатам измерений с использованием эндоскопии примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году по сравнению с не подвергаемым лечению пациентом или одним и тем же пациентом до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к улучшению характеристик по результатам измерений с использованием эндоскопии примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок по сравнению с не подвергаемым лечению пациентом или одним и тем же пациентом до лечения. В некоторых воплощениях, характеристики по результатам измерений с использованием эндоскопии улучшаются на величину до примерно 100%. В некоторых воплощениях, характеристики по результатам измерений с использованием эндоскопии улучшаются на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с не подвергаемым лечению пациентом или одним и тем же пациентом до лечения. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к улучшению характеристик по результатам измерений с использованием эндоскопии на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с не подвергаемым лечению пациентом или одним и тем же пациентом до лечения примерно на 1-й неделе, примерно 2-й неделе, примерно 3-й неделе, примерно 4-й неделе, примерно 5-й неделе, примерно 6-й неделе, примерно 7-й неделе, примерно 8-й неделе, примерно 9-й неделе, примерно 10-й неделе, примерно 20-й неделе, примерно 30-й неделе, примерно 40-й неделе, примерно 50-й неделе, примерно 60-й неделе, примерно 70-й неделе, примерно 80-й неделе, примерно 90-й неделе, примерно 100-й неделе, примерно к 1-му году, примерно ко 2-му году или примерно к 3-му году. В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к улучшению характеристик по результатам измерений с использованием эндоскопии на величину до примерно 1%, примерно 5%, примерно 10%, примерно 20%, примерно 30%, примерно 40%, примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90% или примерно 100% по сравнению с показателем EndoFlip у не подвергаемого лечению пациента или одного и того же пациента до лечения примерно за 1 неделю, примерно за 1 месяц, примерно за 2 месяца, примерно за 3 месяца, примерно за 4 месяца, примерно за 5 месяцев, примерно за 6 месяцев, примерно за 1 год, примерно за 2 года, примерно за 5 лет или примерно за 10 лет или за более длительный срок.
В некоторых воплощениях, введение фармацевтических композиций, описанных в данной заявке, приводит к улучшению характеристик по результатам измерений с использованием эндоскопии у подвергаемого лечению пациента на 12-й неделе, 26-й неделе или 52-й неделе по сравнению с не подвергаемым лечению пациентом или одним и тем же пациентом до лечения.
В некоторых воплощениях, введение описанных фармацевтических композиций приводит к снижению числа эпизодов, связанных с ЕоЕ, испытываемых пациентом в течение некоторого периода времени. Неограничивающие примеры таких эпизодов включают трудность при глотании пилюли или пищи. Пациенты могут сообщать о возникновении таких эпизодов как об ощущении дискомфорта после глотания пилюли или пищи, и их оценку можно проводить после каждого случая глотания пилюли или пищи либо в течение 24-часового периода или более долгого срока. Для оценки сообщения пациента о дискомфорте можно использовать любой способ, в том числе PROSE.
В некоторых воплощениях, число эпизодов, происходящих в течение указанного периода времени, снижается по меньшей мере на 1 эпизод, по меньшей мере на 2 эпизода, по меньшей мере на 3 эпизода, по меньшей мере на 4 эпизода, по меньшей мере на 5 эпизодов, по меньшей мере на 6 эпизодов, по меньшей мере на 7 эпизодов, по меньшей мере на 8 эпизодов, по меньшей мере на 9 эпизодов, по меньшей мере на 10 эпизодов, по меньшей мере на 11 эпизодов, по меньшей мере на 12 эпизодов, по меньшей мере на 13 эпизодов, по меньшей мере на 14 эпизодов, по меньшей мере на 15 эпизодов, по меньшей мере на 16 эпизодов, по меньшей мере на 17 эпизодов, по меньшей мере на 18 эпизодов, по меньшей мере на 19 эпизодов или по меньшей мере на 20 эпизодов, по меньшей мере на 21 эпизод, по меньшей мере на 22 эпизода, по меньшей мере на 23 эпизода, по меньшей мере на 24 эпизода, по меньшей мере на 25 эпизодов, по меньшей мере на 26 эпизодов, по меньшей мере на 27 эпизодов, по меньшей мере на 28 эпизодов, по меньшей мере на 29 эпизодов или по меньшей мере на 30 эпизодов, по меньшей мере на 31 эпизод, по меньшей мере на 32 эпизода, по меньшей мере на 33 эпизода, по меньшей мере на 34 эпизода, по меньшей мере на 35 эпизодов, по меньшей мере на 36 эпизодов, по меньшей мере на 37 эпизодов, по меньшей мере на 38 эпизодов, по меньшей мере на 39 эпизодов или по меньшей мере на 40 эпизодов, по меньшей мере на 41 эпизод, по меньшей мере на 42 эпизода, по меньшей мере на 43 эпизода, по меньшей мере на 44 эпизода, по меньшей мере на 45 эпизодов, по меньшей мере на 46 эпизодов, по меньшей мере на 47 эпизодов, по меньшей мере на 48 эпизодов, по меньшей мере на 49 эпизодов или по меньшей мере на 50 эпизодов. В некоторых воплощениях, период времени составляет примерно 1 неделю, примерно 2 недели, примерно 3 недели, примерно 4 недели, примерно 5 недель, примерно 6 недель, примерно 7 недель, примерно 8 недель, примерно 9 недель, примерно 10 недель, примерно 20 недель, примерно 30 недель, примерно 40 недель, примерно 50 недель, примерно 60 недель, примерно 70 недель, примерно 80 недель, примерно 90 недель, примерно 100 недель, примерно 1 год, примерно 2 года или примерно 3 года.
Дозирование и введение
Пероральные кортикостероиды, описанные в данной заявке, можно вводить в любой соответствующей дозе и с использованием любой подходящей фармацевтической композиции. Хотя специалист в данной области техники в каждом случае может определить желаемую дозу, подходящая доза терапевтического агента для достижения терапевтической пользы, может находиться, например, в диапазоне от примерно 1 микрограмма (мкг) до примерно 100 миллиграммов (мг) на килограмм массы тела реципиента в сутки, предпочтительно в диапазоне от примерно 10 мкг до примерно 50 мг на килограмм массы тела в сутки и наиболее предпочтительно в диапазоне от примерно 10 мкг до примерно 50 мг на килограмм массы тела в сутки. В некоторых воплощениях, фармацевтическую композицию вводят в низкой дозе, например, примерно 20 мг или менее. В некоторых воплощениях, пероральный кортикостероид вводят в дозе примерно 1 мг на килограмм массы тела в сутки, примерно 3 мг на килограмм массы тела в сутки и/или примерно 9 мг на килограмм массы тела в сутки. Желаемая доза может быть представлена в виде одной дозы либо двух или более субдоз, вводимых с соответствующими интервалами в течение суток. Эти субдозы можно вводить в стандартных лекарственных формах, например, содержащих от примерно 10 мкг до примерно 1000 мг, предпочтительно от примерно 0,05 мкг до примерно 20 мг и наиболее предпочтительно от примерно 0,5 мг до примерно 7,5 мг. В некоторых воплощениях, фармацевтическую композицию вводят в стандартной лекарственной форме, составляющей 0,75 мг, 1,5 мг, 3,0 мг, 4,5 мг, 6,0 мг или 7,5 мг.
В некоторых воплощениях, фармацевтическую композицию, описанную в данной заявке (например, жидкую или твердую композицию, содержащую пероральный кортикостероид) вводят пациенту один раз в сутки перед сном (HS). В некоторых воплощениях, фармацевтическую композицию вводят пациенту два раза в сутки (BID). В некоторых воплощениях, фармацевтическую композицию вводят пациенту один раз утром и один раз вечером. В некоторых воплощениях, фармацевтическую композицию вводят на голодный желудок (например, по меньшей мере через 2 часа после приема пищи или по меньшей мере за 1 час до приема пищи; или по меньшей мере за 30 минут до приема пищи или через 30 минут после приема пищи). В некоторых воплощениях, фармацевтическую композицию вводят пациенту за 30 минут до завтрака и за 30 минут перед сном. В некоторых воплощениях, введение фармацевтической композиции перед сном ослабляет системное всасывание перорального кортикостероида по сравнению с системным всасыванием, наблюдаемым после введения в дневное время.
Таким образом, в некоторых воплощениях, фармацевтическую композицию вводят в вечернее время в интервале от примерно 7 РМ (после полудня) до примерно 10 РМ, например, примерно в 7 РМ, в 7:30 РМ, примерно в 8 РМ, примерно в 8:30 РМ, примерно в 9 РМ или примерно в 9:30 РМ, включая все значения и поддиапазоны между ними. В некоторых воплощениях, фармацевтическую композицию вводят примерно за 30 минут до запланированного времени отхода ко сну. Термин «запланированное время отхода ко сну» может означать время суток, когда пациент намеревается ложиться спать.
Предварительные исследования терапевтической эффективности в лечении ЕоЕ после традиционного перорального введения перорального кортикостероида (т.е. когда пациент находится в вертикальном положении) выявили более сильный клиренс эозинофилов из проксимальной части пищевода (например, ближе к полости рта) и более слабое выведение эозинофилов из дистальной части пищевода (например, ближе к желудку). Без связи с теорией, авторы отмечают, что эти результаты свидетельствовали об усиленном контакте кортикостероида с проксимальной тканью пищевода и ослабленном контакте кортикостероида с дистальной тканью пищевода. В попытке улучшить контакт кортикостероида с дистальной тканью пищевода были проведены различные эксперименты. Неожиданным образом было установлено, что введение перорального кортикостероида, когда пациент находится в лежачем положении, усиливает выведение эозинофилов из дистальной части пищевода при сохранении сильного выведения эозинофилов из проксимальной части пищевода. Без связи с теорией, авторы отмечают, что контакт кортикостероида с дистальной частью пищевода усиливается, когда кортикостероид вводят при нахождении пациента в лежачем положении, поскольку прохождение лекарственного средства через пищевод обусловлено главным образом перистальтикой, тогда как при принятии вертикального положения контакт перорального кортикостероида с дистальной частью пищевода ослабляется, поскольку прохождение лекарственного средства обусловлено, главным образом, силой тяжести и непрерывным потоком жидкостей вниз по пищеводу.
Контакт кортикостероида с пищеводом можно наблюдать непосредственно, проводя сцинтиграфическое исследование, как описано в примере 6. Альтернативно, контакт кортикостероида с пищеводом можно определить опосредованно путем измерения выведения эозинофилов из проксимальной и дистальной частей пищевода. Например, выведение эозинофилов можно определить путем измерения количества эозинофилов в проксимальной и дистальной частях пищевода до лечения (число клеток/мм2 в поле зрения микроскопа под большим увеличением) для установления исходного уровня и сравнения указанного исходного уровня с количеством эозинофилов в проксимальной и дистальной частях пищевода, измеренным через 8 недель лечения, как описано в примере 2.
Соответственно, в конкретных воплощениях пероральный кортикостероид вводят один раз в сутки в ночное время, когда пациент находится в лежачем положении (или когда пациент ложится сразу же после перорального введения). В других конкретных воплощениях пероральный кортикостероид вводят два раза в сутки, при этом пациент может сохранять вертикальное положение при приеме первой суточной дозы, и пациент находится в лежачем положении при приеме второй суточной дозы (или пациент ложится сразу же после перорального введения). В других воплощениях пациент находится в лежачем положении при приеме обеих суточных доз.
В некоторых воплощениях, фармацевтическую композицию вводят пациенту, когда пациент находится в лежачем положении, или непосредственно перед тем, как пациент примет лежачее положение, например, в пределах примерно 1 секунды, примерно 2 секунд, примерно 3 секунд, примерно 4 секунд, примерно 5 секунд, примерно 6 секунд, примерно 7 секунд, примерно 8 секунд, примерно 9 секунд, примерно 10 секунд, примерно 15 секунд, примерно 20 секунд, примерно 25 секунд, примерно 30 секунд, примерно 35 секунд, примерно 40 секунд, примерно 45 секунд, примерно 50 секунд, примерно 60 секунд, примерно 1,1 минуты, примерно 1,2 минуты, примерно 1,3 минуты, примерно 1,4 минуты, примерно 1,5 минуты, примерно 1,6 минуты, примерно 1,7 минуты, примерно 1,8 минуты, примерно 1,9 минуты, примерно 2,0 минут, примерно 2,5 минуты, примерно 3 минут, примерно 4 минут или примерно 5 минут перед тем, как пациент примет лежачее положение, включая все значения и диапазоны между ними.
В различных воплощениях фармацевтическую композицию вводят в режиме HS, когда пациент находится в лежачем положении (или пациент ложится сразу же после введения). В других воплощениях фармацевтическую композицию вводят (например, в режиме введения BID или QD (один раз в день)), когда пациент находится в лежачем положении (или пациент ложится сразу же после введения). В различных воплощениях пациент сохраняет лежачее положение после введения в течение промежутка времени, достаточного для локального осаждения и/или контакта кортикостероида в пищеводе для лечения воспаления пищевода (например, времени, достаточного для улучшения ЕоЕ по результатам измерений, изложенных в данном описании, как например, для уменьшения числа эпизодов дисфагии). В некоторых таких воплощениях пациент сохраняет лежачее положение после введения в течение промежутка времени в интервале от примерно 1 минуты до примерно 8 часов, включая 5 минут, примерно 10 минут, примерно 15 минут, примерно 20 минут, примерно 25 минут, примерно 30 минут, примерно 35 минут, примерно 40 минут, примерно 45 минут, примерно 50 минут, примерно 55 минут, примерно 1 ч, примерно 1,1 ч, примерно 1,2 ч, примерно 1,3 ч, примерно 1,4 ч, примерно 1,5 ч, примерно 1,6 ч, примерно 1,7 ч, примерно 1,8 ч или примерно 1,9 ч, примерно 2 ч, примерно 2,1 ч, примерно 2,2 ч, примерно 2,3 ч, примерно 2,4 ч, примерно 2,5 ч, примерно 2,6 ч, примерно 2,7 ч, примерно 2,8 ч или примерно 2,9 ч, примерно 3 ч, примерно 3,1 ч, примерно 3,2 ч, примерно 3,3 ч, примерно 3,4 ч, примерно 3,5 ч, примерно 3,6 ч, примерно 3,7 ч, примерно 3,8 ч или примерно 3,9 ч, примерно 4 ч, примерно 4,1 ч, примерно 4,2 ч, примерно 4,3 ч, примерно 4,4 ч, примерно 4,5 ч, примерно 4,6 ч, примерно 4,7 ч, примерно 4,8 ч, примерно 4,9 ч, примерно 5 ч, примерно 5,1 ч, примерно 5,2 ч, примерно 5,3 ч, примерно 5,4 ч, примерно 5,5 ч, примерно 5,6 ч, примерно 5,7 ч, примерно 5,8 ч, примерно 5,9 ч, примерно 6 ч, примерно 6,1 ч, примерно 6,2 ч, примерно 6,3 ч, примерно 6,4 ч, примерно 6,5 ч, примерно 6,6 ч, примерно 6,7 ч, примерно 6,8 ч или примерно 6,9 ч, примерно 7 ч, примерно 7,1 ч, примерно 7,2 ч, примерно 7,3 ч, примерно 7,4 ч, примерно 7,5 ч, примерно 7,6 ч, примерно 7,7 ч, примерно 7,8 ч или примерно 7,9 ч, включая все значения и поддиапазоны между ними. Согласно воплощениям, когда фармацевтическую композицию вводят в дневное время, пациент сохраняет лежачее положение в течение промежутка времени в интервале от примерно 1 минуты до примерно 60 минут, включая примерно 5 минут, примерно 10 минут, примерно 15 минут, примерно 20 минут, примерно 25 минут, примерно 30 минут, примерно 35 минут, примерно 40 минут, примерно 45 минут, примерно 50 минут, примерно 55 минут, включая все значения и поддиапазоны между ними. В некоторых воплощениях, пациент сохраняет лежачее положение в течение от примерно 5 до примерно 10 минут.
Использованные в данном описании термины «лежачее положение», «ложится» и их производные и варианты, относятся к пациенту, принимающему положение лежа на спине, на животе или на боку на кровати или на полу, или относятся по существу к горизонтальному положению тела, в результате чего кортикостероид (после проглатывания) контактирует с пищеводом и локально осаждается в пищеводе, например, в месте воспаления. Как использовано в данном описании, «по существу горизонтальное» относится положению тела, которое отклоняется от вертикального по меньшей мере на 10°, например, меньше чем примерно на 15°, меньше чем примерно на 20°, меньше чем примерно на 25°, меньше чем примерно на 30°, меньше чем примерно на 35°, меньше чем примерно на 40°, меньше чем примерно на 45°, меньше чем примерно на 50°, меньше чем примерно на 55°, меньше чем примерно на 65°, меньше чем примерно на 70°, меньше чем примерно на 75°, меньше чем примерно на 80°, меньше чем примерно на 85° или примерно на 90° от вертикального, включая все значения и диапазоны между ними. Например, если композицию готовят в виде ODT, то ODT быстро распадается в полости рта лежащего на спине пациента с образованием суспензии, содержащей кортикостероид, которую пациент проглатывает. Затем суспензия проходит через пищевод пациента, посредством чего обеспечивается местный контакт кортикостероида с пищеводом для местного лечения воспаления пищевода. Использованный в данном описании термин «в вертикальном положении» относится к пациенту, занимающему по существу любое другое положение, включая, но не ограничиваясь этим, положение стоя или сидя.
В некоторых воплощениях, фармацевтическую композицию вводят пациенту перед сном. В некоторых воплощениях, фармацевтическую композицию вводят пациенту перед сном, когда пациент находится в лежачем положении. В некоторых воплощениях, фармацевтическую композицию вводят пациенту, находящемуся в лежачем положении и перед сном (например, в пределах от примерно 1 минуты до примерно 1 часа до сна, например, примерно за 1 минуту, примерно за 5 минут, примерно за 10 минут, примерно за 15 минут, примерно за 20 минут, примерно за 25 минут, примерно за 30 минут, примерно за 35 минут, примерно за 40 минут, примерно за 45 минут, примерно за 50 минут, примерно за 55 минут, включая все входящие в них значения). В предпочтительных воплощениях фармацевтическую композицию вводят пациенту в лежачем положении примерно за 30 минут перед сном. В некоторых воплощениях, фармацевтическую композицию вводят пациенту после вечернего приема пищи, например, в пределах от примерно 1 минуты до примерно 5 часов после вечернего приема пищи (например, в пределах примерно 5 минут, примерно 10 минут, примерно 15 минут, примерно 20 минут, примерно 25 минут, примерно 30 минут, примерно 35 минут, примерно 40 минут, примерно 45 минут, примерно 50 минут, примерно 55 минут, примерно 1 часа, примерно 1,5 часа, примерно 2 часов, примерно 2,5 часа, примерно 3 часов, примерно 3,5 часа, примерно 4 часов, примерно 4,5 часа, включая все значения и поддиапазоны между ними. В предпочтительных воплощениях фармацевтическую композицию вводят по меньшей мере примерно через 30 минут после вечернего приема пищи.
В некоторых воплощениях, введение фармацевтической композиции, содержащей кортикостероид, когда пациент находится в лежачем положении, приводит к усилению контакта кортикостероида с верхней частью желудочно-кишечного тракта (например, пищеводом), при этом также ослабляется системное всасывание кортикостероида. Таким образом, согласно воплощениям, введение фармацевтической композиции, когда пациент лежит, улучшает эффективность и (в других воплощениях) приводит к снижению побочных эффектов, ассоциированных с системным введением кортикостероидов.
Время прохождения жидкой фармацевтической композиции по пищеводу обычно находится в диапазоне от примерно 1 до примерно 5 секунд. Без ограничения теорией, следует отметить, что введение фармацевтической композиции, описанной в данной заявке, когда пациент находится в лежачем положении, увеличивает время пребывания (также называемое временем задержки) кортикостероида в пищеводе, что увеличивает время, необходимое для достижения кортикостероидом желудка, и уменьшает количество кортикостероида, которое достигает желудка и всасывается системно. В некоторых воплощениях, увеличение времени пребывания кортикостероида в пищеводе усиливает контакт кортикостероида с пищеводом. В некоторых воплощениях, время пребывания кортикостероида в пищеводе увеличивается и/или контакт кортикостероида с пищеводом усиливается у пациента, находящегося в лежачем положении, по сравнению со временем пребывания кортикостероида в пищеводе при введении кортикостероида в составе аналогичной композиции, когда пациент находится в вертикальном положении.
В некоторых воплощениях, время пребывания кортикостероида в пищеводе после введения пациенту, находящемуся в лежачем положении, находится в интервале от примерно 1 секунды до примерно 5 минут, например, составляет примерно 2 секунды, примерно 3 секунды, примерно 5 секунд, примерно 6 секунд, примерно 7 секунд, примерно 8 секунд, примерно 9 секунд, примерно 10 секунд, примерно 15 секунд, примерно 20 секунд, примерно 25 секунд, примерно 30 секунд, примерно 35 секунд, примерно 40 секунд, примерно 45 секунд, примерно 50 секунд, примерно 55 секунд, примерно 60 секунд, примерно 65 секунд, примерно 70 секунд, примерно 75 секунд, примерно 80 секунд, примерно 85 секунд, примерно 90 секунд, примерно 95 секунд, примерно 100 секунд, примерно 105 секунд, примерно 110 секунд, примерно 115 секунд, примерно 120 секунд, примерно 125 секунд, примерно 130 секунд, примерно 135 секунд, примерно 140 секунд, примерно 145 секунд, примерно 150 секунд, примерно 155 секунд, примерно 160 секунд, примерно 165 секунд, примерно 170 секунд, примерно 175 секунд, примерно 180 секунд, примерно 185 секунд, примерно 190 секунд, примерно 195 секунд, примерно 200 секунд, 205 секунд, примерно 210 секунд, примерно 215 секунд, примерно 220 секунд, примерно 225 секунд, примерно 230 секунд, примерно 235 секунд, примерно 240 секунд, примерно 245 секунд, примерно 250 секунд, примерно 255 секунд, примерно 260 секунд, примерно 265 секунд, примерно 270 секунд, примерно 275 секунд, примерно 280 секунд, примерно 285 секунд, примерно 290 секунд или примерно 295 секунд, включая все значения и поддиапазоны между ними. В конкретных воплощениях время перемещения кортикостероида по пищеводу после введения пациенту в лежачем положении находится в интервале от примерно 5 секунд до примерно 60 секунд.
В некоторых воплощениях, введение фармацевтической композиции, когда пациент находится в лежачем положении, приводит к увеличению времени пребывания кортикостероида в пищеводе (по сравнению со временем пребывания кортикостероида в пищеводе в случае нахождения пациента в вертикальном положении) примерно на 5 секунд, примерно на 10 секунд, примерно на 15 секунд, примерно на 20 секунд, примерно на 25 секунд, примерно на 30 секунд, примерно на 35 секунд, примерно на 40 секунд, примерно на 45 секунд, примерно на 50 секунд, примерно на 55 секунд, примерно на 60 секунд, примерно на 65 секунд, примерно на 70 секунд, примерно на 75 секунд, примерно на 80 секунд, примерно на 85 секунд, примерно на 90 секунд, примерно на 95 секунд, примерно на 100 секунд, примерно на 105 секунд, примерно на 110 секунд, примерно на 115 секунд, примерно на 120 секунд, примерно на 125 секунд, примерно на 130 секунд, примерно на 135 секунд, примерно на 140 секунд, примерно на 145 секунд, примерно на 150 секунд, примерно на 155 секунд, примерно на 160 секунд, примерно на 165 секунд, примерно на 170 секунд, примерно на 175 секунд, примерно на 180 секунд, примерно на 185 секунд, примерно на 190 секунд, примерно на 195 секунд, примерно на 200 секунд, 205 секунд, примерно на 210 секунд, примерно на 215 секунд, примерно на 220 секунд, примерно на 225 секунд, примерно на 230 секунд, примерно на 235 секунд, примерно на 240 секунд, примерно на 245 секунд, примерно на 250 секунд, примерно на 255 секунд, примерно на 260 секунд, примерно на 265 секунд, примерно на 270 секунд, примерно на 275 секунд, примерно на 280 секунд, примерно на 285 секунд, примерно на 290 секунд, примерно на 295 секунд или примерно на 300 секунд. В конкретных воплощениях время пребывания кортикостероида в пищеводе после введения пациенту, находящемуся в лежачем положении, увеличивается на промежуток времени в интервале от примерно 5 секунд до примерно 60 секунд.
В некоторых воплощениях, среднее время достижения максимальной концентрации кортикостероида в плазме крови (Tmax) для пациента в лежачем положении находится в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 8 ч до примерно 20 ч, например, примерно 4,0 ч, примерно 4,5 ч, примерно 5 ч, примерно 5,5 ч, примерно 6 ч, примерно 6,6 ч, примерно 7 ч, примерно 7,5 ч, примерно 8 ч, примерно 8,5 ч, примерно 9,0 ч, примерно 9,5 ч, примерно 10,0 ч, примерно 10,5 ч, примерно 11 ч, примерно 11,5 ч, примерно 12 ч, примерно 12,5 ч, примерно 13 ч, примерно 13,5 ч, примерно 14 ч, примерно 14,5 ч, примерно 15 ч, примерно 15,5 ч, примерно 16 ч, примерно 16,5 ч, примерно 17 ч, примерно 17,5 ч, примерно 18 ч, примерно 18,5 ч, примерно 19 ч, примерно 19,5 ч, примерно 20 ч, примерно 20,5 ч, примерно 21 ч, примерно 21,5 ч или примерно 22 ч, примерно 22,5 ч, примерно 23 ч или примерно 23,5 ч, примерно 24 ч, примерно 24,5 ч, примерно 25 ч, примерно 25,5 ч или примерно 26 ч, включая все значения и поддиапазоны между ними. В предпочтительных воплощениях Tmax для перорального кортикостероида для пациента в лежачем положении находится в диапазоне от примерно 80% до примерно 125% от значения примерно 14 ч ± 6 ч, включая все значения и поддиапазоны между ними.
В некоторых воплощениях, Tmax для перорального кортикостероида в случае пациента в лежачем положении увеличивается на промежуток времени в интервале от примерно 1 ч до примерно 15 ч по сравнению с Tmax для пациента в вертикальном положении (например, в условиях введения до полудня (AM) после приема пищи или натощак), например, примерно на 1,5 ч, примерно на 2,5 ч, примерно на 3 ч, примерно на 3,5 ч, примерно на 4 ч, примерно на 4,5 ч, примерно на 5 ч, примерно на 5,5 ч, примерно на 6 ч, примерно на 6,5 ч, примерно на 7 ч, примерно на 7,5 ч, примерно на 8 ч, примерно на 8,5 ч, примерно на 9 ч, примерно на 9,5 ч, примерно на 10 ч, примерно на 10,5 ч, примерно на 11 ч, примерно на 11,5 ч, примерно на 12 ч, примерно на 12,5 ч, примерно на 13 ч, примерно на 13,5 ч, примерно на 14 ч, примерно на 14,5 ч, включая все значения и поддиапазоны между ними.
В некоторых воплощениях, Tmax для перорального кортикостероида при введении пациенту в вертикальном положении в после приема пищи (и пациент не ложится сразу же после этого) находится в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 1 ч до примерно 10 ч, например, составляет примерно 1,5 ч, примерно 2,5 ч, примерно 3 ч, примерно 3,5 ч, примерно 4 ч, примерно 4,5 ч, примерно 5 ч, примерно 5,5 ч, примерно 6 ч, примерно 6,5 ч, примерно 7 ч, примерно 7,5 ч, примерно 8 ч, примерно 8,5 ч, примерно 9 ч, примерно 9,5 ч, примерно 10 ч, примерно 10,5 ч, примерно 11 ч, примерно 11,5 ч, примерно 12 ч, примерно 12,5 ч, примерно 13 ч, включая все значения и поддиапазоны между ними.
В некоторых воплощениях, Tmax для перорального кортикостероида при введении пациенту в вертикальном положении в состоянии натощак (и пациент не ложится сразу же после этого) находится в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 2 ч до примерно 30 ч, например, составляет примерно 1 ч, примерно 1,5 ч, примерно 2,5 ч, примерно 3 ч, примерно 3,5 ч, примерно 4 ч, примерно 4,5 ч, примерно 5 ч, примерно 5,5 ч, примерно 6 ч, примерно 6,5 ч, примерно 7 ч, примерно 7,5 ч, примерно 8 ч, примерно 8,5 ч, примерно 9 ч, примерно 9,5 ч, примерно 10 ч, примерно 10,5 ч, примерно 11 ч, примерно 11,5 ч, примерно 12 ч, примерно 12,5 ч, примерно 13 ч, примерно 13,5 ч, примерно 14 ч, примерно 14,5 ч, примерно 15 ч, примерно 15,5 ч, примерно 16 ч, примерно 16,5 ч, примерно 17 ч, примерно 17,5 ч, примерно 18 ч, примерно 18,5 ч, примерно 19 ч, примерно 19,5 ч, примерно 20 ч, примерно 20,5 ч, примерно 21 ч, примерно 21,5 ч, примерно 22 ч, примерно 22,5 ч, примерно 23 ч, примерно 23,5 ч, примерно 24 ч, примерно 24,5 ч, примерно 25 ч, примерно 25,5 ч, примерно 26 ч, примерно 26,5 ч, примерно 27 ч, примерно 27,5 ч, примерно 28 ч, примерно 28,5 ч, примерно 29 ч, примерно 29,5 ч, примерно 30 ч, примерно 30,5 ч, примерно 31 ч, примерно 31,5 ч, примерно 32 ч, примерно 32,5 ч, примерно 33 ч, включая все значения и поддиапазоны между ними.
Традиционно, прием пищи обычно приводит к увеличению Tmax, и поэтому значения Tmax после приема пищи обычно превышают Tmax в состоянии натощак. Однако фармакокинетические исследования показали, что значения Tmax в состоянии натощак превышают Tmax после приема пищи. Так, в некоторых воплощениях, значения Tmax в состоянии натощак превышают Tmax после приема пищи примерно на 1 ч, примерно на 1,5 ч, примерно на 2 ч, примерно на 2,5 ч, примерно на 3 ч, примерно на 3,5 ч, примерно на 4 ч, примерно на 4,5 ч, примерно на 5 ч, примерно на 5,5 ч, примерно на 6 ч, примерно на 6,5 ч, примерно на 7 ч, примерно на 7,5 ч, примерно на 8 ч, примерно на 8,5 ч, примерно на 9 ч, примерно на 9,5 ч, примерно на 10 ч, примерно на 10,5 ч, примерно на 11 ч, примерно на 11,5 ч, примерно на 12 ч, примерно на 12,5 ч, примерно на 13 ч, примерно на 13,5 ч, примерно на 14 ч, примерно на 14,5 ч, примерно на 15 ч, примерно на 15,5 ч, примерно на 16 ч, примерно на 16,5 ч, примерно на 17 ч, примерно на 17,5 ч, примерно на 18 ч, примерно на 18,5 ч, примерно на 19 ч, примерно на 19,5 ч, примерно на 20 ч или больше.
Вследствие местного контакта перорального кортикостероида с пищеводом и применения кортикостероидов, обладающих низкой (или модифицированных с возможностью обладания низкой) системной биодоступностью, описанные в данной заявке способы и композиции пригодны для лечения ЕоЕ, при этом уровни кортикостероида в плазме крови неожиданным образом оказываются низкими. Преимущество низких уровней кортикостероида в плазме крови заключается в уменьшении или избегании побочных эффектов, ассоциированных с системным введением кортикостероидов. Таким образом, в некоторых воплощениях способов и композиций, описанных в данной заявке, осуществляется местное введение кортикостероида в верхнюю часть желудочно-кишечного тракта (например, в пищевод) в количестве кортикостероида, эффективном для местного лечения воспаления пищевода (например, ЕоЕ). В некоторых воплощениях, пероральное введение кортикостероида обеспечивает достижение средней максимальной концентрации в плазме крови (Cmax), меньшей или равной примерно 1000 пг/мл после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида, например, меньшей или равной примерно 950 пг/мл, примерно 900 пг/мл, примерно 850 пг/мл, примерно 800 пг/мл, примерно 750 пг/мл, примерно 700 пг/мл, примерно 650 пг/мл, примерно 600 пг/мл, примерно 550 пг/мл, примерно 500 пг/мл, примерно 450 пг/мл, примерно 400 пг/мл, примерно 350 пг/мл, примерно 300 пг/мл, примерно 250 пг/мл, примерно 200 пг/мл, примерно 150 пг/мл, примерно 100 пг/мл или примерно 50 пг/мл. В предпочтительных воплощениях введение перорального кортикостероида обеспечивает достижение Cmax, меньшей или равной примерно 500 пг/мл, после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида.
В предпочтительных воплощениях у пациента в лежачем положении Cmax находится в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 15 пг/мл до примерно 40 пг/мл после введения 6 мг флутиказона пропионата пациенту, находящемуся в лежачем положении, например, составляет примерно 10 пг/мл, примерно 11 пг/мл, примерно 12 пг/мл, примерно 13 пг/мл, примерно 14 пг/мл, примерно 15 мг/мл, примерно 16 пг/мл, примерно 17 пг/мл, примерно 18 пг/мл, примерно 19 пг/мл, примерно 20 пг/мл, примерно 21 пг/мл, примерно 22 пг/мл, примерно 23 пг/мл, примерно 24 пг/мл, примерно 25 пг/мл, примерно 26 пг/мл, примерно 27 пг/мл, примерно 28 пг/мл, примерно 29 пг/мл, примерно 30 пг/мл, примерно 31 пг/мл, примерно 32 пг/мл, примерно 33 пг/мл, примерно 34 пг/мл, примерно 35 пг/мл, примерно 36 пг/мл, примерно 37 пг/мл, примерно 38 пг/мл, примерно 39 пг/мл, примерно 40 пг/мл, примерно 41 пг/мл, примерно 42 пг/мл, примерно 43 пг/мл, примерно 44 пг/мл, примерно 45 пг/мл, примерно 46 пг/мл, примерно 47 пг/мл, примерно 48 пг/мл, примерно 49 пг/мл, примерно 50 пг/мл, примерно 51 пг/мл, примерно 52 пг/мл, примерно 53 пг/мл, примерно 54 пг/мл или примерно 55 пг/мл, включая все значения и поддиапазоны между ними. В некоторых воплощениях, низкие уровни в плазме крови, достигаемые для лечения ЕоЕ, также приводят к ослаблению побочных эффектов, ассоциированных с системным введением кортикостероидов. Например, согласно воплощениям, включающим HS введение, при местной доставке кортикостероида ослабляется системное всасывание и ассоциированные с ним побочные эффекты, даже несмотря на то, что общеизвестно, что при HS введении усиливается системное всасывание.
В некоторых воплощениях, Cmax перорального кортикостероида для пациента в лежачем положении по меньшей мере примерно на 1% ниже по сравнению с Cmax перорального кортикостероида для пациента после приема пищи, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида, например, по меньшей мере примерно на 2%, по меньшей мере примерно на 3%, по меньшей мере примерно на 4%, по меньшей мере примерно на 5%, по меньшей мере примерно на 6%, по меньшей мере примерно на 7%, по меньшей мере примерно на 8%, по меньшей мере примерно на 9%, по меньшей мере примерно на 10%, по меньшей мере примерно на 11%, по меньшей мере примерно на 12%, по меньшей мере примерно на 13%, по меньшей мере примерно на 14%, по меньшей мере примерно на 15%, по меньшей мере примерно на 16%, по меньшей мере примерно на 17%, по меньшей мере примерно на 18%, по меньшей мере примерно на 19%, по меньшей мере примерно на 20%, по меньшей мере примерно на 25%, по меньшей мере примерно на 30%, по меньшей мере примерно на 35%, по меньшей мере примерно на 40%, по меньшей мере примерно на 45% или по меньшей мере примерно на 50%, включая все значения и поддиапазоны между ними.
В некоторых воплощениях, введение перорального кортикостероида обеспечивает достижение средней AUC0-24, меньшей или равной примерно 10000 пг*ч/мл, после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида, например, примерно 9000 пг*ч/мл, примерно 8000 пг*ч/мл, примерно 7000 пг*ч/мл, примерно 6000 пг*ч/мл, примерно 5000 пг*ч/мл, примерно 4000 пг*ч/мл, примерно 3000 пг*ч/мл, примерно 2000 пг*ч/мл, примерно 1000 пг*ч/мл, примерно 900 пг*ч/мл, примерно 800 пг*ч/мл, примерно 700 пг*ч/мл, примерно 600 пг*ч/мл, примерно 500 пг*ч/мл, примерно 400 пг*ч/мл, примерно 300 пг*ч/мл, примерно 200 пг*ч/мл или примерно 100 пг*ч/мл. В конкретных воплощениях введение перорального кортикостероида обеспечивает достижение средней AUC0-24, меньшей или равной примерно 3000 пг*ч/мл, после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида.
В некоторых воплощениях, значение AUC0-24 (пг*ч/мл) для пациента находится в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 50 пг⋅ч/мл до примерно 1000 пг⋅ч/мл после введения 1,5, 3,0, 4,5, 6,0 или 7,5 мг флутиказона пропионата пациенту, находящемуся в лежачем положении, например, составляет примерно 10 пг⋅ч/мл, примерно 20 пг⋅ч/мл, примерно 30 пг⋅ч/мл, примерно 40 пг⋅ч/мл, примерно 50 пг⋅ч/мл, примерно 60 пг⋅ч/мл, примерно 70 пг⋅ч/мл, примерно 80 пг⋅ч/мл, примерно 90 пг⋅ч/мл, примерно 100 пг⋅ч/мл, примерно 110 пг⋅ч/мл, примерно 120 пг⋅ч/мл, примерно 130 пг⋅ч/мл, примерно 140 пг⋅ч/мл, примерно 150 пг⋅ч/мл, примерно 160 пг⋅ч/мл, примерно 170 пг⋅ч/мл, примерно 180 пг⋅ч/мл, примерно 190 пг⋅ч/мл, примерно 200 пг⋅ч/мл, примерно 210 пг⋅ч/мл, примерно 220 пг⋅ч/мл, примерно 230 пг⋅ч/мл, примерно 240 пг⋅ч/мл, примерно 250 пг⋅ч/мл, примерно 260 пг⋅ч/мл, примерно 270 пг⋅ч/мл, примерно 280 пг⋅ч/мл, примерно 290 пг⋅ч/мл, примерно 300 пг⋅ч/мл, примерно 310 пг⋅ч/мл, примерно 320 пг⋅ч/мл, примерно 330 пг⋅ч/мл, примерно 340 пг⋅ч/мл, примерно 350 пг⋅ч/мл, примерно 360 пг⋅ч/мл, примерно 370 пг⋅ч/мл, примерно 380 пг⋅ч/мл, примерно 390 пг⋅ч/мл, примерно 400 пг⋅ч/мл, примерно 410 пг⋅ч/мл, примерно 420 пг⋅ч/мл, примерно 430 пг⋅ч/мл, примерно 440 пг⋅ч/мл, примерно 450 пг⋅ч/мл, примерно 460 пг⋅ч/мл, примерно 470 пг⋅ч/мл, примерно 480 пг⋅ч/мл, примерно 490 пг⋅ч/мл, примерно 500 пг⋅ч/мл, примерно 510 пг⋅ч/мл, примерно 520 пг⋅ч/мл, примерно 530 пг⋅ч/мл, примерно 540 пг⋅ч/мл, примерно 550 пг⋅ч/мл, примерно 560 пг⋅ч/мл, примерно 570 пг⋅ч/мл, примерно 580 пг⋅ч/мл, примерно 590 пг⋅ч/мл, примерно 600 пг⋅ч/мл, примерно 610 пг⋅ч/мл, примерно 620 пг⋅ч/мл, примерно 630 пг⋅ч/мл, примерно 640 пг⋅ч/мл, примерно 650 пг⋅ч/мл, примерно 660 пг⋅ч/мл, примерно 670 пг⋅ч/мл, примерно 680 пг⋅ч/мл, примерно 690 пг⋅ч/мл, примерно 700 пг⋅ч/мл, примерно 710 пг⋅ч/мл, примерно 720 пг⋅ч/мл, примерно 730 пг⋅ч/мл, примерно 740 пг⋅ч/мл, примерно 750 пг⋅ч/мл, примерно 760 пг⋅ч/мл, примерно 770 пг⋅ч/мл, примерно 780 пг⋅ч/мл, примерно 790 пг⋅ч/мл, примерно 800 пг⋅ч/мл, примерно 810 пг⋅ч/мл, примерно 820 пг⋅ч/мл, примерно 830 пг⋅ч/мл, примерно 840 пг⋅ч/мл, примерно 850 пг⋅ч/мл, примерно 860 пг⋅ч/мл, примерно 870 пг⋅ч/мл, примерно 880 пг⋅ч/мл, примерно 890 пг⋅ч/мл, примерно 900 пг⋅ч/мл, примерно 910 пг⋅ч/мл, примерно 920 пг⋅ч/мл, примерно 930 пг⋅ч/мл, примерно 940 пг⋅ч/мл, примерно 950 пг⋅ч/мл, примерно 960 пг⋅ч/мл, примерно 970 пг⋅ч/мл, примерно 980 пг⋅ч/мл, примерно 990 пг⋅ч/мл, примерно 1000 пг⋅ч/мл, примерно 1010 пг⋅ч/мл, примерно 1020 пг⋅ч/мл, примерно 1030 пг⋅ч/мл, примерно 1040 пг⋅ч/мл, примерно 1050 пг⋅ч/мл, примерно 1060 пг⋅ч/мл, примерно 1070 пг⋅ч/мл, примерно 1080 пг⋅ч/мл, примерно 1090 пг⋅ч/мл, примерно 1100 пг⋅ч/мл, примерно 1110 пг⋅ч/мл, примерно 1120 пг⋅ч/мл, примерно 1130 пг⋅ч/мл, примерно 1140 пг⋅ч/мл, примерно 1150 пг⋅ч/мл, примерно 1160 пг⋅ч/мл, примерно 1170 пг⋅ч/мл, примерно 1180 пг⋅ч/мл, примерно 1190 пг⋅ч/мл, примерно 1200 пг⋅ч/мл, примерно 1210 пг⋅ч/мл, примерно 1220 пг⋅ч/мл, примерно 1230 пг⋅ч/мл, примерно 1240 пг⋅ч/мл, примерно 1250 пг⋅ч/мл, примерно 1260 пг⋅ч/мл, примерно 1270 пг⋅ч/мл, примерно 1280 пг⋅ч/мл, примерно 1290 пг⋅ч/мл, примерно 1300 пг⋅ч/мл, включая все значения и поддиапазоны между ними.
В некоторых воплощениях, значение AUC0-24 (пг*ч/мл) для пациента находится в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 50 пг⋅ч/мл до примерно 1000 пг⋅ч/мл после введения 1,5, 3,0, 4,5, 6,0 или 7,5 мг флутиказона пропионата, когда пациент находится в вертикальном положении (в условиях введения до полудня (AM) после приема пищи или натощак), например, составляет примерно 10 пг⋅ч/мл, примерно 20 пг⋅ч/мл, примерно 30 пг⋅ч/мл, примерно 40 пг⋅ч/мл, примерно 50 пг⋅ч/мл, примерно 60 пг⋅ч/мл, примерно 70 пг⋅ч/мл, примерно 80 пг⋅ч/мл, примерно 90 пг⋅ч/мл, примерно 100 пг⋅ч/мл, примерно 110 пг⋅ч/мл, примерно 120 пг⋅ч/мл, примерно 130 пг⋅ч/мл, примерно 140 пг⋅ч/мл, примерно 150 пг⋅ч/мл, примерно 160 пг⋅ч/мл, примерно 170 пг⋅ч/мл, примерно 180 пг⋅ч/мл, примерно 190 пг⋅ч/мл, примерно 200 пг⋅ч/мл, примерно 210 пг⋅ч/мл, примерно 220 пг⋅ч/мл, примерно 230 пг⋅ч/мл, примерно 240 пг⋅ч/мл, примерно 250 пг⋅ч/мл, примерно 260 пг⋅ч/мл, примерно 270 пг⋅ч/мл, примерно 280 пг⋅ч/мл, примерно 290 пг⋅ч/мл, примерно 300 пг⋅ч/мл, примерно 310 пг⋅ч/мл, примерно 320 пг⋅ч/мл, примерно 330 пг⋅ч/мл, примерно 340 пг⋅ч/мл, примерно 350 пг⋅ч/мл, примерно 360 пг⋅ч/мл, примерно 370 пг⋅ч/мл, примерно 380 пг⋅ч/мл, примерно 390 пг⋅ч/мл, примерно 400 пг⋅ч/мл, примерно 410 пг⋅ч/мл, примерно 420 пг⋅ч/мл, примерно 430 пг⋅ч/мл, примерно 440 пг⋅ч/мл, примерно 450 пг⋅ч/мл, примерно 460 пг⋅ч/мл, примерно 470 пг⋅ч/мл, примерно 480 пг⋅ч/мл, примерно 490 пг⋅ч/мл, примерно 500 пг⋅ч/мл, примерно 510 пг⋅ч/мл, примерно 520 пг⋅ч/мл, примерно 530 пг⋅ч/мл, примерно 540 пг⋅ч/мл, примерно 550 пг⋅ч/мл, примерно 560 пг⋅ч/мл, примерно 570 пг⋅ч/мл, примерно 580 пг⋅ч/мл, примерно 590 пг⋅ч/мл, примерно 600 пг⋅ч/мл, примерно 610 пг⋅ч/мл, примерно 620 пг⋅ч/мл, примерно 630 пг⋅ч/мл, примерно 640 пг⋅ч/мл, примерно 650 пг⋅ч/мл, примерно 660 пг⋅ч/мл, примерно 670 пг⋅ч/мл, примерно 680 пг⋅ч/мл, примерно 690 пг⋅ч/мл, примерно 700 пг⋅ч/мл, примерно 710 пг⋅ч/мл, примерно 720 пг⋅ч/мл, примерно 730 пг⋅ч/мл, примерно 740 пг⋅ч/мл, примерно 750 пг⋅ч/мл, примерно 760 пг⋅ч/мл, примерно 770 пг⋅ч/мл, примерно 780 пг⋅ч/мл, примерно 790 пг⋅ч/мл, примерно 800 пг⋅ч/мл, примерно 810 пг⋅ч/мл, примерно 820 пг⋅ч/мл, примерно 830 пг⋅ч/мл, примерно 840 пг⋅ч/мл, примерно 850 пг⋅ч/мл, примерно 860 пг⋅ч/мл, примерно 870 пг⋅ч/мл, примерно 880 пг⋅ч/мл, примерно 890 пг⋅ч/мл, примерно 900 пг⋅ч/мл, примерно 910 пг⋅ч/мл, примерно 920 пг⋅ч/мл, примерно 930 пг⋅ч/мл, примерно 940 пг⋅ч/мл, примерно 950 пг⋅ч/мл, примерно 960 пг⋅ч/мл, примерно 970 пг⋅ч/мл, примерно 980 пг⋅ч/мл, примерно 990 пг⋅ч/мл, примерно 1000 пг⋅ч/мл, примерно 1010 пг⋅ч/мл, примерно 1020 пг⋅ч/мл, примерно 1030 пг⋅ч/мл, примерно 1040 пг⋅ч/мл, примерно 1050 пг⋅ч/мл, примерно 1060 пг⋅ч/мл, примерно 1070 пг⋅ч/мл, примерно 1080 пг⋅ч/мл, примерно 1090 пг⋅ч/мл, примерно 1100 пг⋅ч/мл, примерно 1110 пг⋅ч/мл, примерно 1120 пг⋅ч/мл, примерно 1130 пг⋅ч/мл, примерно 1140 пг⋅ч/мл, примерно 1150 пг⋅ч/мл, примерно 1160 пг⋅ч/мл, примерно 1170 пг⋅ч/мл, примерно 1180 пг⋅ч/мл, примерно 1190 пг⋅ч/мл, примерно 1200 пг⋅ч/мл, примерно 1210 пг⋅ч/мл, примерно 1220 пг⋅ч/мл, примерно 1230 пг⋅ч/мл, примерно 1240 пг⋅ч/мл, примерно 1250 пг⋅ч/мл, примерно 1260 пг⋅ч/мл, примерно 1270 пг⋅ч/мл, примерно 1280 пг⋅ч/мл, примерно 1290 пг⋅ч/мл, примерно 1300 пг⋅ч/мл, включая все значения и поддиапазоны между ними.
В некоторых воплощениях, значение AUC0-24 (пг*ч/мл) для пациента находится в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 250 пг⋅ч/мл до примерно 475 пг⋅ч/мл после введения 6 мг флутиказона пропионата пациенту, находящемуся в лежачем положении, например, составляет примерно 150 пг⋅ч/мл, примерно 160 пг⋅ч/мл, примерно 170 пг⋅ч/мл, примерно 180 пг⋅ч/мл, примерно 190 пг⋅ч/мл, примерно 200 пг⋅ч/мл, примерно 210 пг⋅ч/мл, примерно 220 пг⋅ч/мл, примерно 230 пг⋅ч/мл, примерно 240 пг⋅ч/мл, примерно 250 пг⋅ч/мл, примерно 260 пг⋅ч/мл, примерно 270 пг⋅ч/мл, примерно 280 пг⋅ч/мл, примерно 290 пг⋅ч/мл, примерно 300 пг⋅ч/мл, примерно 310 пг⋅ч/мл, примерно 320 пг⋅ч/мл, примерно 330 пг⋅ч/мл, примерно 340 пг⋅ч/мл, примерно 350 пг⋅ч/мл, примерно 360 пг⋅ч/мл, примерно 370 пг⋅ч/мл, примерно 380 пг⋅ч/мл, примерно 390 пг⋅ч/мл, примерно 400 пг⋅ч/мл, примерно 410 пг⋅ч/мл, примерно 420 пг⋅ч/мл, примерно 430 пг⋅ч/мл, примерно 440 пг⋅ч/мл, примерно 450 пг⋅ч/мл, примерно 460 пг⋅ч/мл, примерно 470 пг⋅ч/мл, примерно 480 пг⋅ч/мл, примерно 490 пг⋅ч/мл, примерно 500 пг⋅ч/мл, примерно 510 пг⋅ч/мл, примерно 520 пг⋅ч/мл, примерно 530 пг⋅ч/мл, примерно 540 пг⋅ч/мл, примерно 550 пг⋅ч/мл, примерно 560 пг⋅ч/мл, примерно 570 пг⋅ч/мл, примерно 580 пг⋅ч/мл, примерно 590 пг⋅ч/мл, примерно 600 пг⋅ч/мл, включая все значения и поддиапазоны между ними.
В некоторых воплощениях, значение AUC0-24 (пг*ч/мл) для пациента находится в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 250 пг⋅ч/мл до примерно 475 пг⋅ч/мл после введения 6 мг флутиказона пропионата пациенту, находящемуся в вертикальном положении, например, составляет примерно 150 пг⋅ч/мл, примерно 160 пг⋅ч/мл, примерно 170 пг⋅ч/мл, примерно 180 пг⋅ч/мл, примерно 190 пг⋅ч/мл, примерно 200 пг⋅ч/мл, примерно 210 пг⋅ч/мл, примерно 220 пг⋅ч/мл, примерно 230 пг⋅ч/мл, примерно 240 пг⋅ч/мл, примерно 250 пг⋅ч/мл, примерно 260 пг⋅ч/мл, примерно 270 пг⋅ч/мл, примерно 280 пг⋅ч/мл, примерно 290 пг⋅ч/мл, примерно 300 пг⋅ч/мл, примерно 310 пг⋅ч/мл, примерно 320 пг⋅ч/мл, примерно 330 пг⋅ч/мл, примерно 340 пг⋅ч/мл, примерно 350 пг⋅ч/мл, примерно 360 пг⋅ч/мл, примерно 370 пг⋅ч/мл, примерно 380 пг⋅ч/мл, примерно 390 пг⋅ч/мл, примерно 400 пг⋅ч/мл, примерно 410 пг⋅ч/мл, примерно 420 пг⋅ч/мл, примерно 430 пг⋅ч/мл, примерно 440 пг⋅ч/мл, примерно 450 пг⋅ч/мл, примерно 460 пг⋅ч/мл, примерно 470 пг⋅ч/мл, примерно 480 пг⋅ч/мл, примерно 490 пг⋅ч/мл, примерно 500 пг⋅ч/мл, примерно 510 пг⋅ч/мл, примерно 520 пг⋅ч/мл, примерно 530 пг⋅ч/мл, примерно 540 пг⋅ч/мл, примерно 550 пг⋅ч/мл, примерно 560 пг⋅ч/мл, примерно 570 пг⋅ч/мл, примерно 580 пг⋅ч/мл, примерно 590 пг⋅ч/мл, примерно 600 пг⋅ч/мл, включая все значения и поддиапазоны между ними.
В некоторых воплощениях, фармацевтическую композицию вводят пациенту по меньшей мере примерно через 2 часа после вечернего приема пищи (без каких-либо перекусов) пациенту, находящемуся в лежачем положении. В некоторых воплощениях, фармацевтическую композицию вводят пациенту по меньшей мере примерно через 4 часа после вечернего приема пищи (без каких-либо перекусов) пациенту, находящемуся в лежачем положении. В некоторых воплощениях, фармацевтическую композицию вводят пациенту в пределах примерно 2-х часов после вечернего приема пищи (без каких-либо перекусов) пациенту, находящемуся в лежачем положении. В некоторых воплощениях, фармацевтическую композицию вводят пациенту в пределах примерно 4-х часов после вечернего приема пищи (без каких-либо перекусов) пациенту, находящемуся в лежачем положении. В некоторых воплощениях, после введения фармацевтической композиции пациенту, находящемуся в лежачем положении, пациент засыпает. В некоторых воплощениях, после введения фармацевтической композиции пациенту, находящемуся в лежачем положении, пациент не поднимается по меньшей мере в течение одного часа.
В некоторых воплощениях, пациент удерживает фармацевтическую композицию в ротовой полости в течение периода времени, достаточного для проглатывания композиции (например, в течение примерно 1 секунды, примерно 2 секунд, примерно 3 секунд, примерно 4 секунд, примерно 5 секунд, примерно 6 секунд, примерно 7 секунд, примерно 8 секунд, примерно 9 секунд или примерно 10 секунд). В других воплощениях пациент удерживает фармацевтическую композицию в ротовой полости в течение периода времени, достаточного для растворения фармацевтической композиции в слюне. Специалисту в данной области техники будет очевидно, что такой период времени, необходимый для фармацевтической композиции, частично зависит от лекарственной формы. Например, перорально распадающиеся композиции распадаются в слюне в пределах примерно 60 секунд (например, примерно 50 секунд, примерно 40 секунд, примерно 30 секунд, примерно 20 секунд или примерно 10 секунд) с образованием суспензии, которую затем проглатывают. В воплощениях, где фармацевтическая композиция представляет собой пастилку или леденец, пастилка или леденец могут оставаться в ротовой полости пациента в течение примерно 1 минуты, примерно 2 минут, примерно 3 минут, примерно 4 минут, примерно 5 минут, примерно 6 минут, примерно 7 минут, примерно 8 минут, примерно 9 минут или примерно 10 минут.
В некоторых воплощениях, фармацевтическую композицию вводят пациенту от одного до пяти раз в сутки. В некоторых воплощениях, фармацевтическую композицию вводят пациенту по меньшей мере один раз в сутки, по меньшей мере два раза в сутки, по меньшей мере три раза в сутки, по меньшей мере 4 раза в сутки или по меньшей мере пять раз в сутки. В некоторых воплощениях, фармацевтическую композицию вводят пациенту по меньшей мере от одного до пяти раз в сутки в течение от одной недели до 10 лет или более. В некоторых воплощениях, фармацевтическую композицию вводят пациенту по меньшей мере один раз в сутки, по меньшей мере два раза в сутки, по меньшей мере три раза в сутки, по меньшей мере 4 раза в сутки или по меньшей мере пять раз в сутки в течение по меньшей мере одной недели, по меньшей мере двух недель, по меньшей мере трех недель, по меньшей мере четырех недель, по меньшей мере пяти недель, по меньшей мере шести недель, по меньшей мере семи недель, по меньшей мере восьми недель, по меньшей мере девяти недель, по меньшей мере десяти недель, по меньшей мере пятнадцати недель, по меньшей мере двадцати недель, по меньшей мере тридцати недель, по меньшей мере сорока недель, по меньшей мере пятидесяти недель, по меньшей мере пятидесяти двух недель, по меньшей мере шестидесяти недель, по меньшей мере семидесяти недель, по меньшей мере восьмидесяти недель, по меньшей мере девяноста недель или по меньшей мере ста недель или более. В некоторых воплощениях, фармацевтическую композицию вводят пациенту в течение неограниченного периода времени. В некоторых воплощениях, фармацевтическую композицию вводят два раза в сутки в течение по меньшей мере примерно 6 недель, по меньшей мере примерно 8 недель, по меньшей мере примерно 10 недель или по меньшей мере примерно 12 недель. В некоторых воплощениях, фармацевтическую композицию вводят два раза в сутки во время фазы индукции и/или поддерживающей фазы. В некоторых воплощениях, фармацевтическую композицию вводят два раза в сутки в течение по меньшей мере примерно 6 недель, по меньшей мере примерно 8 недель, по меньшей мере примерно 10 недель или по меньшей мере примерно 12 недель во время фазы индукции.
В некоторых воплощениях, фармацевтическую композицию вводят пациенту в дозе 0,75 мг, 1,5 мг, 3,0 мг, 4,5 мг или 6,0 мг по меньшей мере один раз в сутки, по меньшей мере два раза в сутки, по меньшей мере три раза в сутки, по меньшей мере четыре раза в сутки или по меньшей мере пять раз в сутки. В некоторых воплощениях, фармацевтическую композицию вводят пациенту в одной и той же дозе несколько раз в сутки. В некоторых воплощениях, фармацевтическую композицию вводят пациенту в одной и той же дозе по меньшей мере два раза в сутки, по меньшей мере три раза в сутки, по меньшей мере 4 раза в сутки или по меньшей мере пять раз в сутки. В некоторых воплощениях, фармацевтическую композицию вводят пациенту в одной и той же дозе от двух до пяти раз в сутки в течение от одной недели до 10 лет или более. В некоторых воплощениях, фармацевтическую композицию вводят пациенту по меньшей мере в одной и той же дозе по меньшей мере два раза в сутки, по меньшей мере три раза в сутки, по меньшей мере 4 раза в сутки или по меньшей мере пять раз в сутки в течение по меньшей мере одной недели, в течение по меньшей мере двух недель, в течение по меньшей мере трех недель, по меньшей мере четырех недель, по меньшей мере пяти недель, по меньшей мере шести недель, по меньшей мере семи недель, по меньшей мере восьми недель, по меньшей мере девяти недель, по меньшей мере десяти недель, по меньшей мере пятнадцати недель, по меньшей мере двадцати недель, по меньшей мере тридцати недель, по меньшей мере сорока недель, по меньшей мере пятидесяти недель, по меньшей мере пятидесяти двух недель, по меньшей мере шестидесяти недель, по меньшей мере семидесяти недель, по меньшей мере восьмидесяти недель, по меньшей мере девяноста недель или по меньшей мере ста недель либо дольше или в течение неограниченного периода времени.
В некоторых воплощениях, фармацевтическую композицию вводят два раза в сутки в разных дозах. В некоторых воплощениях, фармацевтическую композицию вводят два раза в сутки, при этом утренняя доза выше вечерней дозы. В некоторых воплощениях, фармацевтическую композицию вводят дважды в сутки, при этом утренняя доза ниже вечерней дозы.
В некоторых воплощениях, пациенту вводят разные дозы фармацевтической композиции в зависимости от фазы схемы лечения. Например, схема лечения может быть разделена по меньшей мере на фазы индукции, лечения, отмены или поддерживающую фазу. В некоторых воплощениях, схема лечения включает по меньшей мере одну из этих фаз. В некоторых воплощениях, схема лечения включает комбинацию одной или нескольких из этих фаз. В некоторых воплощениях, схема лечения включает все эти фазы.
В некоторых воплощениях, схема лечения включает фазы индукции и отмены. В некоторых воплощениях, схема лечения по мере необходимости включает несколько циклов индукции и отмены. В некоторых воплощениях, схема лечения включает несколько циклов индукции и отмены, повторяющихся в течение неограниченного периода времени. В некоторых воплощениях, период индукции не приводит к повторному проявлению симптомов.
Соответствующая продолжительность фаз схемы лечения может быть любой. В некоторых воплощениях, фаза индукции длится от примерно 1 недели и до примерно 10 недель, примерно 12 недель, примерно 15 недель, примерно 20 недель, примерно 30 недель, примерно 40 недель или примерно 50 недель. В некоторых воплощениях, фаза индукции длится примерно 14 недель. В некоторых воплощениях, фаза отмены длится от примерно 1 недели и до примерно 10 недель, примерно 15 недель, примерно 20 недель, примерно 30 недель, примерно 40 недель, примерно 50 недель, примерно 1 года, примерно 2 лет, примерно 5 лет, примерно 10 лет или в течение неограниченного периода времени. В некоторых воплощениях, фаза отмены длится до момента повторного проявления симптомов. В некоторых воплощениях, фаза отмены длится примерно 14 недель. В некоторых воплощениях, поддерживающая фаза длится от примерно 1 недели и до примерно 15 недель, примерно 20 недель, примерно 30 недель, примерно 40 недель, примерно 50 недель, примерно 1 года, примерно 2 лет, примерно 5 лет, примерно 10 лет или более. В некоторых воплощениях, поддерживающая фаза длится примерно 28 недель. В некоторых воплощениях, поддерживающая фаза имеет неограниченную продолжительность.
В некоторых воплощениях, пациенту вводят более высокую дозу в одной фазе или нескольких фазах схемы лечения по сравнению с другими фазами. В некоторых воплощениях, пациенту вводят одну и ту же дозу в одной фазе или нескольких фазах схемы лечения. В некоторых воплощениях, пациенту вводят одну и ту же дозу в каждой фазе схемы лечения. В некоторых воплощениях, пациенту не вводят никакой дозы во время одной фазы или нескольких фаз.
В некоторых воплощениях, пациенту вводят более высокую дозу во время стадии индукции по сравнению с поддерживающей стадией. В некоторых воплощениях, пациенту вводят более низкую дозу во время стадии индукции по сравнению с поддерживающей стадией. В некоторых воплощениях, пациенту не вводят никакой дозы или во время стадии индукции, или во время поддерживающей стадии. В некоторых воплощениях, пациенту не вводят никакой дозы как во время стадии индукции, так и во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят одну и ту же дозу во время стадий индукции и поддерживающей стадии. В некоторых воплощениях, пациенту вводят по существу одну и ту же дозу во время стадий индукции и поддерживающей стадии. В некоторых воплощениях, пациенту вводят 3,0 мг BID во время стадии индукции и 1,5 мг BID во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 3,0 мг BID во время стадии индукции и 1,5 мг HS во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 1,5 мг BID во время стадии индукции и 3,0 мг BID во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 1,5 мг HS во время стадии индукции и 3,0 мг BID во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 1,5 мг BID как во время стадии индукции, так и во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 1,5 мг HS во время стадии индукции и поддерживающей стадии. В некоторых воплощениях, пациенту вводят 3,0 мг BID во время стадии индукции и поддерживающей стадии. В некоторых воплощениях, пациенту вводят 6,0 мг BID во время стадии индукции и 3,0 или 1,5 мг BID во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 6,0 мг BID во время стадии индукции и 3,0 или 1,5 мг HS во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 1,5 или 3,0 мг BID во время стадии индукции и 6,0 мг BID во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 1,5 или 3,0 мг HS во время стадии индукции и 6,0 мг BID во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 6,0 или 3,0 мг BID как во время стадии индукции, так и во время поддерживающей стадии. В некоторых воплощениях, пациенту вводят 6,0 или 3,0 мг HS во время стадии индукции и поддерживающей стадии. В некоторых воплощениях, пациенту вводят 6,0 мг BID во время стадии индукции и поддерживающей стадии.
В некоторых воплощениях, пациенту не вводят совместно сильный ингибитор цитохрома Р4503А4. В некоторых воплощениях, пациенту не вводят совместно ритонавир или кетоконазол.
В некоторых воплощениях, пациентом является человек, но в других воплощениях может быть не являющееся человеком млекопитающее, такое как домашнее животное (например, собака или кошка) или домашний скот или сельскохозяйственное животное (например, лошадь, корова, овца или свинья).
Популяции пациентов
Фармацевтические композиции по настоящему изобретению можно вводить любому пациенту, которому поставлен диагноз воспалительное расстройство желудочно-кишечного тракта или который предположительно страдает от него. В некоторых воплощениях, пациентом является взрослый человек. В некоторых воплощениях, пациентом является подросток. В некоторых воплощениях, пациентом является ребенок. В некоторых воплощениях, пациентом является младенец.
В некоторых воплощениях, воспалительное расстройство желудочно-кишечного тракта представляет собой ЕоЕ. Пациенту может быть поставлен диагноз с использованием любого из соответствующих средств в данной области техники. В некоторых воплощениях, пациенту поставлен диагноз ЕоЕ на основании симптомов, результатов гистологического исследования и/или документально подтвержденных данных о неудовлетворительных результатах применения ингибиторов протонного насоса. В некоторых воплощениях, пациент получал PPI терапию до введения фармацевтической композиции по настоящему изобретению. В некоторых воплощениях, пациент не получал PPI терапии до введения фармацевтической композиции по настоящему изобретению. В некоторых воплощениях, пациент не чувствовал улучшения через 8 недель применения PPI в высокой дозе (например, 40 мг). Отсутствие ответа на PPI терапию может быть определено как максимальное количество эозинофилов/HPF, равное 15 или больше, по меньшей мере в одном подвергнутом биопсии месте, через 8 недель лечения с использованием PPI в высокой дозе. В некоторых воплощениях, неудачу PPI терапии документально фиксируют до введения фармацевтической композиции по настоящему изобретению. В некоторых воплощениях, неудачу PPI терапии документально фиксируют после введения фармацевтической композиции по настоящему изобретению. В некоторых воплощениях, пациентам, которые оказались невосприимчивыми к предыдущей PPI терапии, вводят высокую дозу перорального кортикостероида в соответствии со способами (или для применения в способах), описанными(ых) в данной заявке, такую как 6,0 мг; 7,5 мг или больше (например, от примерно 9,0 мг до примерно 20 мг; включая примерно 9 мг; примерно 10 мг; примерно 11 мг; примерно 12 мг; примерно 13 мг; примерно 14 мг; примерно 15 мг; примерно 16 мг; примерно 17 мг; примерно 18 мг и примерно 19 мг, включая все значения и поддиапазоны между ними).
В некоторых воплощениях, пациент с диагнозом ЕоЕ имеет стриктуру пищевода. В некоторых воплощениях, указанному пациенту вводят высокую дозу перорального кортикостероида в соответствии со способами (или для применения в способах), описанными(ых) в данной заявке, такую как 6,0 мг; 7,5 мг или больше (например, от примерно 9,0 мг до примерно 20 мг; включая примерно 9 мг; примерно 10 мг; примерно 11 мг; примерно 12 мг; примерно 13 мг; примерно 14 мг; примерно 15 мг; примерно 16 мг; примерно 17 мг; примерно 18 мг и примерно 19 мг, включая все значения и поддиапазоны между ними).
В некоторых воплощениях, пациент с диагнозом ЕоЕ имеет тяжелую пищевую аллергию (например, аллергию на лактозу или крахмал). В некоторых воплощениях, указанному пациенту вводят высокую дозу перорального кортикостероида в соответствии со способами (или для применения в способах), описанными(ых) в данной заявке, такую как 6,0 мг; 7,5 мг или больше (например, от примерно 9,0 мг до примерно 20 мг; включая примерно 9 мг; примерно 10 мг; примерно 11 мг; примерно 12 мг; примерно 13 мг; примерно 14 мг; примерно 15 мг; примерно 16 мг; примерно 17 мг; примерно 18 мг и примерно 19 мг, включая все значения и поддиапазоны между ними).
В некоторых воплощениях, у пациента диагностирован ЕоЕ на основании гистологического анализа. В некоторых воплощениях, пациента диагностируют как имеющего максимальное количество эозинофилов в HPF (увеличение 400×), равное 15 или больше, по результатам по меньшей мере одной биопсии. В некоторых воплощениях, у пациента взято как минимум 6 биоптатов. В некоторых воплощениях, взято по меньшей мере по 3 биоптата из каждой проксимальной и дистальной части пищевода.
В некоторых воплощениях, у пациента диагностируют наличие ЕоЕ по EEsAI, общему показателю ЕоЕ, EREFS, PROSE и/или по результатам измерения характеристик пищевода с использованием эндоскопии (например, EndoFlip). В некоторых воплощениях, у пациента диагностируют наличие ЕоЕ на основании 7-дневного показателя EEsAI больше 30. В некоторых воплощениях, у пациента диагностируют наличие ЕоЕ на основании 7-дневного общего показателя ЕоЕ, равного 5 или больше. В некоторых воплощениях, у пациента диагностируют наличие ЕоЕ на основании показателя EREFS, с помощью которого оцениваются выявленные при эндоскопическом обследовании характеристики (см. Таблицу 3). В некоторых воплощениях, диагноз ЕоЕ ставится пациенту с показателем, превышающим степень 0 для любой из пяти характеристик, приведенных в Таблице 3.
В некоторых воплощениях, у пациента диагностируют наличие ЕоЕ на основании симптомов, включая, но не ограничиваясь этим, эпизоды затрудненного прохождения пищи, эпизоды затрудненного прохождения пищи, требующие проведения эндоскопии, отказ от пищи, рвоту, рефлюкс и/или дисфагию. В некоторых воплощениях, у пациента диагностируют наличие ЕоЕ на основании дисфагии (затрудненного глотания). В некоторых воплощениях, у пациента диагностируют наличие ЕоЕ на основании проявления дисфагии по меньшей мере 3 раза в неделю в течение 2-х недель.
Результат лечения пациента и ответ пациента на введение фармацевтической композиции по настоящему изобретению можно контролировать или оценивать с использованием любых соответствующих средств в данной области техники (например, эндоскопии, гистологии, опросников).
Пациенты, у которых наблюдается улучшение симптомов и/или гистологического ответа после начала лечения, классифицируются как респондеры. В некоторых воплощениях, пациенты, у которых наблюдается максимальное количество эозинофилов/HPF меньше 15, классифицируются как респондеры. В некоторых воплощениях, пациенты, у которых наблюдается максимальное количество эозинофилов/HPF меньше 6, классифицируются как респондеры. В некоторых воплощениях, пациенты, у которых наблюдается максимальное количество эозинофилов/HPF меньше 6 и отсутствие ухудшения симптомов (например, нет увеличения еженедельного показателя EEsAI по сравнению с исходным значением; отсутствует стриктура, требующая дилатации), классифицируются как респондеры. В некоторых воплощениях, пациенты, у которых наблюдается максимальное количество эозинофилов/HPF меньше 6 и отсутствие эпизодов затрудненного прохождения пищи, классифицируются как респондеры. В некоторых воплощениях, респондеры демонстрируют признаки ремиссии воспаления по результатам эндоскопии, как например, отсутствие белого экссудата и/или борозд. В некоторых воплощениях, респондеры демонстрируют признаки ремиссии фиброза, включая отсутствие стриктур и колец или наличие колец в степени от умеренной до тяжелой. В некоторых воплощениях, респондеры демонстрируют усиление васкуляризации. В некоторых воплощениях, респондеры демонстрируют улучшенные уровни биомаркеров (например, уровни интерлейкина 5 (IL-5), иммуноглобулина Е (IgE)).
В некоторых воплощениях, пациентов, которые классифицируются как респондеры, зачисляют в поддерживающую стадию схемы лечения. В некоторых воплощениях, пациентам, которые классифицируются как респондеры, вводят разную дозу фармацевтической композиции по настоящему изобретению после распределения по классам. В некоторых воплощениях, респондеры получают более высокую дозу после распределения по классам. В некоторых воплощениях, респондеры получают более низкую дозу после распределения по классам. В некоторых воплощениях, респондеры получают одну и ту же дозу после распределения по классам. В некоторых воплощениях, респондеры получают по существу одну и ту же дозу после распределения по классам.
В некоторых воплощениях, пациента классифицируют как респондер, если фармацевтические композиции, описанные в данной заявке, вводят в фазе индукции и поддерживающей фазе, и во время фазы индукции наблюдают улучшение в показателе максимального количества эозинофилов по результатам по меньшей мере одной биопсии пищевода и/или у пациента не наблюдают по меньшей мере никакого ухудшения средних еженедельных показателей EEsAI, и при этом в поддерживающей фазе используют дозу, по меньшей мере равную, более высокую или более низкую по сравнению с таковой для фазы индукции.
Пациенты, которые не удовлетворяют определению респондера, изложенному выше, классифицируются как нереспондеры. Пациенты с ухудшением гистологического показателя и/или симптомов классифицируются как пациенты с рецидивом. В некоторых воплощениях, пациенты с ухудшением гистологического показателя и/или симптомов в любой момент во время лечения классифицируются как пациенты с рецидивом. В некоторых воплощениях, пациенты, которые испытывают затрудненное прохождение пищи, требующее проведения эндоскопии, и/или клинически значимое ухудшение симптомов, классифицируются как пациенты с рецидивом. В некоторых воплощениях, пациентам, которые классифицируются как нереспондеры или пациенты с рецидивом заболевания, вводят разные дозы фармацевтической композиции по настоящему изобретению после распределения по классам. В некоторых воплощениях, нереспондеры и/или пациенты с рецидивом получают более высокую дозу после распределения по классам. В некоторых воплощениях, нереспондеры и/или пациенты с рецидивом получают более низкую дозу после распределения по классам. В некоторых воплощениях, нереспондеры и/или пациенты с рецидивом получают одну и ту же дозу после распределения по классам. В некоторых воплощениях, нереспондеры и/или пациенты с рецидивом получают по существу одну и ту же дозу после распределения по классам.
В некоторых воплощениях, пациента классифицируют как респондер, если фармацевтические композиции, описанные в данной заявке, вводят в фазе индукции и поддерживающей фазе, и во время фазы индукции не наблюдают никакого улучшения в показателе максимального количества эозинофилов по результатам по меньшей мере одной биопсии пищевода и/или у пациента не наблюдают по меньшей мере никакого ухудшения средних еженедельных показателей EEsAI, и при этом в поддерживающей фазе используют дозу, по меньшей мере равную, более высокую или более низкую по сравнению с таковой для фазы индукции.
В некоторых воплощениях настоящего изобретения предложены способы оценки пригодности субъектов для клинического испытания с целью определения влияния перорального кортикостероида на ЕоЕ после введения как в фазе индукции, так и в поддерживающей фазе. В некоторых воплощениях, набор субъектов для клинического испытания проводят на основании оценки количества эозинофилов у пациента и предшествующего лечения. В некоторых воплощениях, пациенты, отобранные для клинического испытания, имеют максимальные количества эозинофилов в HPF более чем примерно 6, более чем примерно 10, более чем примерно 15 или более чем примерно 20. В некоторых воплощениях, пациенты, отобранные для клинического испытания, имеют максимальные количества эозинофилов в HPF более чем примерно 15. В некоторых воплощениях, у пациентов, отобранных для клинического испытания, предшествующее лечение не увенчалось успехом. В некоторых воплощениях, предшествующее лечение представляло собой введение PPI в течение по меньшей мере примерно 8 недель, которое не было эффективным в плане существенного улучшения одного или более симптомов ЕоЕ.
Целевые продукты
В соответствии с изложенным выше, согласно настоящему изобретению предложен пероральный кортикостероид для применения в способе лечения ЕоЕ у пациента, при этом пероральный кортикостероид вводят непосредственно перед тем, как пациент примет лежачее положение, или когда пациент находится в лежачем положении. Согласно воплощениям, с пищеводом пациента контактирует терапевтически эффективное количество перорального кортикостероида. Термин «терапевтически эффективное количество» может означать количество, способное вызывать полезную биологическую активность in vivo и, в одном из воплощений, способное излечивать ЕоЕ по результатам измерений с использованием любого из показателей, изложенных в данном описании. Дозы перорального кортикостероида, изложенные в данном описании, соответственно доставляют терапевтически эффективное количество перорального кортикостероида пациенту.
Пероральный кортикостероид, способ лечения и пациент могут быть такими, как описано в данной заявке. Действительно, каждый признак и все признаки, описанные в данной заявке для раскрытых композиций, способов лечения, дозирования, введения и популяций пациентов, могут быть использованы применительно к раскрытым целевым продуктам. Каждая комбинация и все комбинации таких признаков в явной форме охватываются раскрытым изобретением, за исключением комбинаций, в которых по меньшей мере некоторые из таких признаков являются взаимоисключающими.
Таким образом, согласно воплощениям, лежачим положением может быть положение лежа на спине, на животе или на боку.
Пероральный кортикостероид можно вводить примерно за 30 минут или меньше до запланированного времени отхода ко сну. Пероральный кортикостероид можно вводить по меньшей мере примерно через 30 минут после приема пищи. В одном из воплощений пациент может не принимать пищу или питье в течение по меньшей мере примерно 30 минут после введения перорального кортикостероида.
Пероральный кортикостероид можно вводить один раз в сутки. Его можно вводить два раза в сутки, при этом первую суточную дозу вводят, когда пациент сохраняет вертикальное положение.
Пероральный кортикостероид можно вводить в дозе от примерно 0,01 мг до примерно 20 мг. Пероральный кортикостероид может представлять собой флутиказона пропионат, вводимый в дозе от примерно 1,5 мг до примерно 7,5 мг и предпочтительно примерно 1,5, 3,0, 4,5, 6,0 или 7,5 мг.
В одном из воплощений пациент может иметь аллергию на лактозу или аллергию на крахмал.
Пероральный кортикостероид может обладать системной биодоступностью, меньшей или равной примерно 20%, меньше примерно 15%, меньше примерно 10%, меньше примерно 5% или меньше примерно 1% от своей дозы.
Введение перорального кортикостероида может обеспечить достижение средней максимальной концентрации в плазме крови (Cmax), меньшей или равной примерно 500 пг/мл после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида.
Введение перорального кортикостероида может обеспечить достижение средней AUC0-24, меньшей или равной примерно 3000 пг*ч/мл после перорального введения от примерно 0,01 мг до примерно 20 мг перорального кортикостероида.
Согласно воплощениям, пероральный кортикостероид может представлять собой будесонид, флутиказон, флунизолид, циклесонид, мометазон, тиксокортол или беклометазон или их фармацевтически приемлемые соль, сольват, сложный эфир, полиморф или пролекарство.
В одном из воплощений пероральный кортикостероид приготовлен в виде жидкой композиции. В другом воплощении, он может быть приготовлен в виде твердой композиции. Он может быть приготовлен в виде композиции для образования раствора или суспензии перед пероральным введением. Он может быть приготовлен в виде композиции для образования раствора, суспензии или геля после перорального введения. В случае приготовления в виде жидкой композиции пероральный кортикостероид может быть в форме раствора, суспензии или взвеси. В случае приготовления в виде твердой композиции пероральный кортикостероид может быть в форме геля, пастилки, леденца, шипучей таблетки, порошка, гранул или перорально распадающейся композиции. Перорально распадающаяся композиция может быть в форме таблетки, облатки, пленки или лиофилизированной формы.
В одном из воплощений пероральный кортикостероид готовят в виде перорально распадающейся таблетки, содержащей пероральный кортикостероид в количестве от примерно 1,5 до примерно 7,5 мг, фармацевтически приемлемый носитель, объединенный с кортикостероидом, и быстро диспергируемые микрогранулы, при этом перорально распадающаяся таблетка распадается в течение 60 секунд при тестировании с использованием теста на распадаемость USP <701>.
В одном из воплощений пациент может иметь Cmax перорального кортикостероида, меньшую или равную примерно 200 пг/мл после перорального введения от примерно 1,5 до примерно 7,5 мг перорального кортикостероида.
Среднее время достижения максимальной концентрации в плазме крови (Tmax) для перорального кортикостероида может находиться в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 12 ч до примерно 15 ч.
В одном из воплощений пероральный кортикостероид может представлять собой флутиказона пропионат, и значение Cmax может находиться в диапазоне от примерно 80% до примерно 125% от значений в диапазоне от примерно 15 пг/мл до примерно 40 пг/мл после введения 6 мг флутиказона пропионата или 3 мг флутиказона пропионата.
Согласно воплощениям, через 12 недель ежесуточного введения перорального кортикостероида воспаление пищевода может уменьшаться согласно результатам, показывающим снижение количества эозинофилов, повышение числа дней без дисфагии, уменьшение числа эпизодов дисфагии, улучшение показателя EREFS, улучшение эластичности пищевода согласно протоколу EndoFLIP, оценке уровня биомаркеров, уменьшению числа эпизодов затрудненного прохождения пищи, улучшению показателей EEsAI (по показателям пациента, врача, эндоскопии, патологического изменения), согласно EoE-QoL-A, опроснику по визуальной оценке дисфагии (VDQ), показателям избегания, корректировки и медленного приема пищи (AMS) или результатам гистологического исследования.
В одном из воплощений количество эозинофилов у пациента может быть снижено по меньшей мере примерно на 50%.
Комбинированные терапии
Один или более терапевтических агентов можно «вводить совместно», т.е. вводить субъекту скоординированным образом либо в виде отдельных фармацевтических композиций, либо в виде смеси в единой фармацевтической композиции. Посредством «совместного введения» один или более терапевтических агентов также можно вводить вместе с фармацевтическими композициями по настоящему изобретению или вводить по отдельности, в том числе в разные моменты времени и с разной частотой. Один или более терапевтических агентов можно вводить любым известным путем, как например, перорально, внутривенно, внутримышечно, назально, подкожно, интравагинально, интраректально и тому подобное; и терапевтический агент также можно вводить любым удобным путем.
Когда в комбинации используют два или более лекарственных средств, дозировка каждого лекарственного средства обычно идентична дозировке лекарственного средства при его независимом использовании, но когда лекарственное средство мешает метаболизму других лекарственных средств, дозировка каждого лекарственного средства должным образом подбирается. Каждое из лекарственных средств можно вводить совместно или по отдельности через соответствующий интервал времени.
Фармацевтические композиции, описанные в данной заявке, могут быть введены совместно с различными терапевтическими средствами, используемыми для лечения, предупреждения, задержки и/или уменьшения интенсивности симптомов воспалительных состояний желудочно-кишечного тракта, включая, но не ограничиваясь этим, воспаление пищевода, воспаление голосовой щели, воспаление надгортанника, воспаление миндалин, воспаление ротоглотки, эозинофильный эзофагит (ЕоЕ), гастроэзофагеальную рефлюксную болезнь (GERD), неэрозивную рефлюксную болезнь (NERD), эрозивный эзофагит, пищевод Баррета, эозинофильный гастроэнтерит, гиперэозинофильный синдром, коррозивный (разъедающий) химический эзофагит, индуцированный облучением эзофагит, индуцированный химиотерапией эзофагит, преходящий индуцированный приемом лекарственных средств эзофагит (также известный как медицинский эзофагит), стойкий индуцированный приемом лекарственных средств эзофагит, пищеводная болезнь Крона и псевдомембранозный эзофагит.
Одним или более чем одним терапевтическим агентом может быть любое соединение, молекула или вещество, которое оказывает терапевтический эффект субъекту, нуждающемуся в этом.
В некоторых воплощениях, фармацевтические композиции, описанные в данной заявке, вводят совместно с одним или несколькими кортикостероидами. Подходящие кортикостероиды включают, но не ограничиваются этим, гидрокортизон, преднизон, преднизолон, метилпреднизолон, дексаметазон, бетаметазон и т.д., или кортикостероиды с минералокортикоидной активностью (например, альдостерон), будесонид, флутиказон, флунизолид, циклесонид, мометазон, беклометазон, тиксокортол и их соли или сложные эфиры и их смеси.
В некоторых воплощениях, фармацевтические композиции, описанные в данной заявке, вводят вместе с одним или несколькими ингибиторами протонного насоса (PPI). Подходящие PPI включают, но не ограничиваются этим, омепразол, лансопразол, декслансопразол, рабепразол, пантопразол и эзомепразол. В некоторых воплощениях, PPI вводят в высоких дозах.
В некоторых воплощениях, фармацевтические композиции, описанные в данной заявке, вводят вместе с противогрибковым агентом. Подходящие противогрибковые агенты включают, но не ограничиваются этим, ингибирующие митоз противогрибковые средства, противогрибковые средства на основе аналогов пиримидина, противогрибковые средства группы полиенов, противогрибковые средства группы бензимидазолов, противогрибковые средства группы имидазолов, противогрибковые средства группы триазолов, противогрибковые средства группы тиазолов, противогрибковые средства группы аллиламинов, противогрибковые средства группы эхинокандинов и другие «некатегоризированные» противогрибковые средства, признанные в данной области техники, которые не попадают ни в одну из упомянутых выше категорий (например, толнафтат и циклопирокс). Например, подходящие противогрибковые агенты, которые могут быть введены в твердые фармацевтические композиции по настоящему изобретению, включают, но не ограничиваются этим, абафунгин, аморолфин, анидулафунгин, бифоназол, бутенафин, бутоконазол, кандицин, каспофунгин, циклопирокс, клотримазол, эконазол, фентиконазол, филипин, флуконазол, флуцитозин, гризеофульвин, изавуконазол, изоконазол, итраконазол, кетоконазол, микафунгин, миконазол, миконазола нитрат, нафтифин, натамицин, нистатин, оксиконазол, позаконазол, прамиконазол, равуконазол, римоцидин, сетаконазол, сулконазол, тербафин, терконазол, тиоконазол, толнафтат, ундециленовую кислоту и вориконазол.
В других воплощениях фармацевтические композиции, описанные в данной заявке, вводят вместе с противовирусным агентом. Противовирусные агенты, которые могут быть использованы в настоящем изобретении, включают, но не ограничиваются этим, интерфероны, нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы, ненуклеозидные ингибиторы обратной транскриптазы, ингибиторы протеаз, ингибиторы интегразы, ингибиторы слияния, ингибиторы созревания, аналоги гуанозина, аналоги пуринов, аналоги пиримидинов и другие «некатегоризированные» противовирусные лекарственные средства, признанные в данной области техники, которые не попадают ни в одну из упомянутых выше категорий (например, фоскарнет и милтефозин). Например, подходящие противовирусные агенты, которые могут быть введены в твердые фармацевтические композиции по настоящему изобретению, включают, но не ограничиваются этим, абакавир, ацикловир, адефовир, амантадин, амдоксовир, ампренавир, аплавирок, априцитабин, арбидол, атазанавир, бевиримат, BMS-488043, боцепревир, бривудин, цидофовир, DCM205, докозанол, делавирдин, диданозин, дарунавир, эфавиренз, элвитегравир, эльвуцитабин, эмтрицитабин, энфувиртид, эпигаллокатехина галлат, этравирин, фамцикловир, фосампренавир, ганцикловир, глобоиднан А, гриффитсин, ибализумаб, идоксуридин, индинавир, ламивудин, лопинавир, ловирид, маравирок, нелфинавир, невирапин, осельтамивир, ПЭГилированный интерферон-альфа-2а, ПЭГилированный интерферон-альфа-2b, пенцикловир, перамивир, плериксафор, PRO 140, рацивир, ралтегравир, ритонавир, рибавирин, римантадин, рилпивирин, саквинавир, стампидин, ставудин, тенофовир, типранавир, TNX-355, трифлуридин, тромантадин, валацикловир, валганцикловир, викривирок, видарабин, вирамидин, вивекон, зальцитабин, занамивир и зидовудин.
В некоторых воплощениях, фармацевтические композиции, описанные в данной заявке, вводят вместе с одним или несколькими иммуносупрессорами. Подходящие иммуносупрессоры включают, но не ограничиваются этим, циклоспорин, такролимус, преднизолон, гидрокортизон, сиролимус, эверолимус, азатиоприн, микофеноловую кислоту, метотрексат, базиликсимаб, даклизумаб, ритуксимаб, антитимоцитарный глобулин и антилимфоцитарный глобулин.
Наборы
Согласно изобретению также предложены наборы для лечения воспалительных расстройств желудочно-кишечного тракта. В некоторых воплощениях, наборы включают в себя фармацевтическую композицию по настоящему изобретению (флутиказона пропионат, ODT) в стандартной лекарственной форме. В некоторых воплощениях, наборы включают в себя фармацевтическую композицию по настоящему изобретению в дозе 1,5 мг или 3,0 мг. В некоторых воплощениях, наборы включают в себя поддерживающую дозировку фармацевтической композиции по настоящему изобретению. В некоторых воплощениях, наборы включают в себя индукционную дозировку фармацевтической композиция по настоящему изобретению. В некоторых воплощениях, наборы включают в себя как индукционную дозировку, так и поддерживающую дозировку фармацевтической композиция по настоящему изобретению. Такой набор может дополнительно содержать этикетку или распечатанные инструкции с информацией относительно применения описанных реагентов. Набор может дополнительно содержать информацию о необходимых методах тестирования.
ВКЛЮЧЕНИЕ ССЫЛОК
Все процитированные публикации, патенты и патентные публикации включены в данное описание посредством ссылки во всей своей полноте для всех назначений.
Данная заявка включает посредством ссылки следующие публикации и заявки во всей своей полноте для всех назначений: заявку US 8771729, поданную 1 октября 2010 г.; заявку US 2016/0206627, поданную 5 сентября 2014 г., заявку US 61/874450, поданную 6 сентября 2013 г., заявку WO 2015/034678, поданную 21 августа 2014 г и заявку WO 2015/035114, поданную 5 сентября 2014 года.
Данное описание дополнительно проиллюстрировано приведенными далее неограничивающими примерами.
ПРИМЕРЫ
Пример 1. Рандомизированное двойное слепое исследование по подбору дозы таблетки флутиказона пропионата, растворяющейся в полости рта
В это исследование, фазу 2b, будут включены 320 пациентов с эозинофильным эзофагитом (EoE) в возрасте 14-75 лет. Чтобы принять участие в этом исследовании, пациенты должны иметь диагноз ЕоЕ (максимальное содержание эозинофилов в HPF более 15) после подтверждения неудачного лечения в течение 8 недель или более ингибитором протонного насоса (PPI) в высоких дозах (т.е. 40 мг эзомепразола, BID). Во время этого исследования PPI терапия может быть продолжена, только если субъект получал PPI терапию в момент включения в исследование. В ходе исследования не допускается ни начала новой PPI терапии, ни изменения имеющейся PPI терапии. Кроме того, субъекты должны проявлять симптомы и иметь 7-дневный общий показатель симптомов ЕоЕ, составляющий 5 или более, и 7-дневный показатель EEsAI более 20, для зачисления во вводную фазу исследования с применением плацебо, и повышенный показатель EEsAI согласно ежедневнику.
Первичные задачи: оценить эффективность перорально распадающейся таблетки флутиказона пропионата (АРТ-1011) у подростков и взрослых с эозинофильным эзофагитом (ЕоЕ).
Вторичные задачи: оценить безопасность АРТ-1011 (флутиказона пропионата, ODT) у подростков и взрослых с ЕоЕ. Определить зависимость ответа от дозы АРТ-1011. Оценить фармакокинетику (ФК) АРТ-1011 для пациентов с ЕоЕ. Оценить эффект отмены лечения, включая период времени до рецидива и эффект повторного лечения. Оценить поддержание эффективности лечения и долгосрочную безопасность. Оценить влияние на выявленные при эндоскопическом обследовании визуальные характеристики, используя эндоскопический референсный показатель (ERES) для ЕоЕ.
Обоснование и план исследования: чтобы определить ответ на воздействие АРТ-1011 и минимальную эффективную дозу, будут исследованы дозы от 1,5 мг HS до 3,0 BID. Это рандомизированное двойное слепое, плацебо-контролируемое исследование по подбору дозы для трех суточных доз (1,5 мг; 3 мг и 6 мг) АРТ-1011, вводимых в режиме 1,5 мг HS, 1,5 мг BID или 3,0 мг BID, в сравнении с соответствующим плацебо. Также будет проведена оценка эффекта отмены лечения, повторного лечения и долгосрочного поддержания лечения.
Составы АРТ-1011 (флутиказона пропионата, ODT), использованного в клинических испытаниях, приведены в следующих далее таблицах.
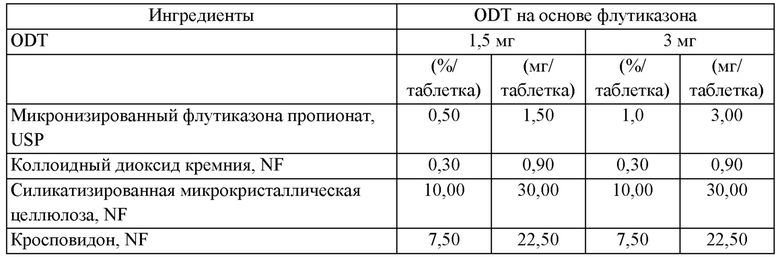
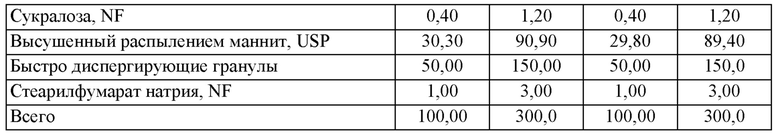
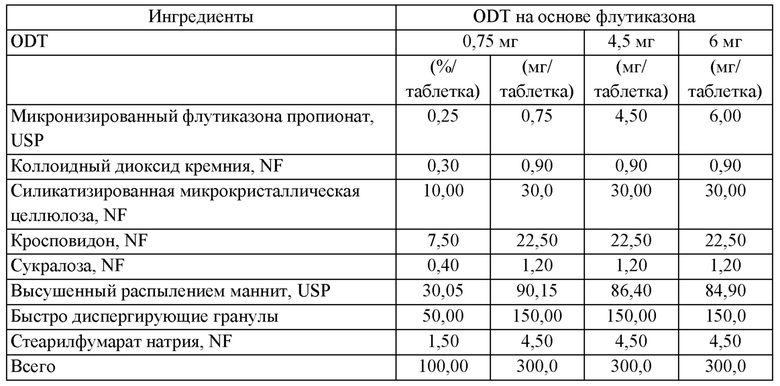
Введение доз: для 3-х получающих дозы групп АРТ-1011 будут представлены в виде замаскированных таблеток в дозировках 1,5 мг и 3,0 мг. Для четвертой получающей дозу группы это будет соответствующее плацебо. Таблетки будут введены в режимах BID - за 30 минут до завтрака и HS (перед сном). Обратите внимание, что в группе, получающей дозу 1,5 мг, субъекты будут получать таблетки с плацебо за 30 минут до завтрака и 1,5 мг АРТ-1011 HS (перед сном). Для поддержания условий слепого исследования все таблетки будут маркированы для приема «до завтрака» и «перед сном».
Скрининг: субъекты будут подвергнуты скринингу с использованием эндоскопии верхних отделов пищевода (EGD (эзофагогастродуоденоскопия)) в течение 2-4 недель. Субъекты, которые удовлетворяют другим критериям включения и имеют максимальное содержание эозинофилов в HPF, составляющее 15 или более, и имеют общий показатель симптомов больше 5, и 7-дневный показатель EEsAI больше 20, могут быть зачислены во вводную фазу исследования с применением плацебо. Субъекты, которые имеют повышенный показатель EEsAI согласно ежедневнику, комплаентность ежедневнику более 80% и комплаентность исследуемому лекарственному средству более 90%, и удовлетворяют всем другим критериям включения, будут рандомизированы. Субъекты будут рандомизированы в соотношении 1:1:1:1 с попаданием в группы, получающие 3 дозы активного средства и плацебо.
Проводимое исследование будет состоять из трех частей, как показано на Фиг. 1 и 2. Часть 1 и Часть 2 представляют связанную с отменой Часть плана лечения. В Части 1 оценивается эффективность лечения. В Части 2 оценивается эффект отмены лечения, включая период времени до рецидива и разницу в частоте рецидивов между лечением активным средством и плацебо.
Часть 1: лечение (с 1-х суток по 14-ю неделю). Субъекты будут разделены согласно возрасту (младше 18 лет и 18 лет и старше) и параллельно в соответствии с применением PPI во время этого исследования («да» или «нет»); затем случайным образом распределены с попаданием в 1 из 4-х получающих дозы групп (в соотношении 1:1:1:1).
Все субъекты будут ежемесячно осуществлять регулярные визиты, а для внеплановых визитов должно иметь место ухудшение симптомов между визитами. Субъекты будут получать лечение в течение 12 недель и проходить EGD для оценки улучшения по результатам гистологического и эндоскопического анализов. Улучшение симптомов будет оцениваться постоянно. ЕоЕ-респондеры, определяемые как субъекты, которые достигают гистологического ответа в показателях максимального количества Eos менее 6 в HPF, и не имеющие ухудшения симптомов (например, увеличения еженедельного показателя EEsAI по сравнению с исходными данными) или эпизодов затрудненного прохождения пищи, будут рандомизированы для проведения Части 2 на 14-й неделе. Нереспондеры, определяемые как субъекты, которые не удовлетворяют определению респондера, будут получать лечение с открытой маркировкой (OL) в режиме по 3 мг BID до 28-й недели с EGD на 28-й неделе. ЕоЕ-респондеры к окончанию периода OL лечения будут удовлетворять критериям зачисления в Часть 3. Нереспондеры к OL лечению будут зачислены для прохождения 2-недельного периода последующего наблюдения.
Часть 2: отмена лечения (с 14-й недели по 28-ю неделю). ЕоЕ-респондеры из Части 1 будут рандомизированы либо в группу, продолжающую принимать свою дозу, либо в группу, получающую плацебо, в соотношении 3:1. Респондеры к «лечению» с использованием плацебо, которых, как ожидается, будет немного, будут по-прежнему получать плацебо. Субъекты, у которых наблюдается ухудшение симптомов (например, с возвратом к исходному еженедельному показателю EEsAI или жалобами на ухудшение симптомов) или затрудненное прохождение пищи, будут проходить эндоскопию при внеплановом визите. Субъекты с затрудненным прохождением пищи, которым требуется проведение эндоскопии или у которых имеется требующая дилатации стриктура, будут проходить 2-недельный период последующего наблюдения и покидать исследование. Субъекты, считающиеся имеющими рецидив, будут снова получать свою первоначальную дозу, кроме тех, кто ранее принимал плацебо, которые будут получать 3 мг, BID. Субъекты с рецидивом на исследуемое лекарственное средство также будут получать 3 мг, BID, чтобы избежать процедуры демаскирования при проведении исследования. Пациенты с рецидивом заболевания будут зачислены в Часть 3. Все принимающие участие в исследовании субъекты на 12-й, 26-й и 52-й неделях будут проходить эндоскопию. ЕоЕ-респондеры на 28-й неделе будут переходить к Части 3. Нереспондеры по окончании Части 2, когда они будут зачислены в Часть 3, будут получать 3 мг, BID.
Часть 3: поддерживающая фаза (с 28-й недели по 54-ю неделю). Нереспондеры из Части 1, которые являются восприимчивыми к лечению с открытой маркировкой (3 мг, BID), будут продолжать принимать эту дозу в Части 3. Обратите внимание, что эта группа не будет принимать участия в Части 2. ЕоЕ-респондеры из Части 2 продолжат прием той же дозы. Пациенты с рецидивом заболевания и нереспондеры из Части 2 будут получать 3 мг, BID, в Части 3. Субъекты с рецидивом заболевания (например, испытывающие затрудненное прохождение пищи, требующее проведения эндоскопии, или испытывающие клинически значимое ухудшение симптомов) в Части 3 будут проходить внеплановую эндоскопию, проходить 2-недельное последующее наблюдение и покидать исследование. Все продолжающие лечение субъекты будут проходить эндоскопию на 52-й неделе и совершат свой заключительный визит лечения на 54-й неделе.
Последующее наблюдение (через 2 недели после приема последней дозы). Все субъекты должны пройти заключительную эндоскопию в пределах 3 недель до начала последующего наблюдения. Исключения включают отзыв согласия или противопоказание к проведению эндоскопии. Причины для начала последующего наблюдения включают завершение 54-й недели, нежелательное явление, требующее преждевременной отмены лечения, включающее затрудненное прохождение пищи, требующее проведения эндоскопии, невосприимчивость к лечению с открытой маркировкой (3 мг, BID) или рецидив в Части 3.
Фармакокинетика: прием всех доз АРТ-1011 будет проводиться два раза в сутки за 30 минут до завтрака и перед сном, за исключением дозы 1,5 мг, прием которой будет осуществлен в режиме HS (перед сном) или за 30 минут до завтрака. Популяционный ФК анализ будет проведен на основе комбинации данных о концентрации в плазме крови из серийного и выборочного вариантов анализа.
Популяционная ФК: отбор образцов для выборочного ФК контроля будет проведен для всех субъектов (за исключением субъекта +/- 3 суток), кому показан повторный прием дозы. На 4-й неделе, в 1-е сутки (+/- 3 суток) периода повторного приема дозы отбор ФК образцов будет проведен не ранее, чем за 15 минут до приема утренней дозы и через 0,5, 1,5 и 3 часа после введения утренней дозы. Время приема дозы в ночное время (например, перед сном) наряду с фактическим временем отбора ФК образцов будет задокументировано.
Подгруппа многократного ФК контроля для Части 1: в определенных исследовательских центрах для 24 оцениваемых субъектов (по 6 субъектов из расчета на одну получающую дозу группу) будет проведен многократный серийный ФК контроль после приема разовой дозы на 1-й неделе, в 1-е сутки, и после повторного приема дозы на 4-й неделе, в 1-е сутки (+/- 3 суток). Утром на 1-й неделе в 1-е сутки субъекты получат разовую дозу APT-1011, и ФК образцы будут отобраны не позднее, чем за 15 минут до AM дозы и через 0,5, 1, 2, 4, 8, 24 и 48 часов после введения дозы. ПРИМЕЧАНИЕ: 48-часовой образец следует отбирать после введения разовой дозы и до начала периода повторного приема дозы. Этот образец будет отобран во время визита, когда субъекты вернутся для получения лекарственного средства в амбулаторных условиях в период повторного приема дозы.
Период повторного приема дозы начнется после отбора 48-часового ФК образца на 1-й неделе, в 3-й сутки. На 4-й неделе, в 1-е сутки (+/- 3 суток) периода повторного приема дозы ФК образцы будут отобраны не ранее, чем за 15 минут до AM дозы и через 0,5, 1, 2, 4, 8 и 12 часов после AM дозы, непосредственно перед РМ дозой. ПРИМЕЧАНИЕ: 12-часовой ФК образец после AM дозы является «необязательным». Этот 12-часовой ФК образец следует отбирать непосредственно перед РМ дозой. Время приема дозы (перед сном в предыдущую ночь и следующим утром) наряду с фактическими временами отбора ФК образцов будет задокументировано).
Продолжительность исследования: в этом исследовании ожидаемый период набора в группы составляет 12 месяцев. Субъекты, которые будут зачислены в исследование и выполнят его (Часть 1, Часть 2 и Часть 3), будут участвовать в исследовании до 15 месяцев включительно.
Исследуемая популяция: в данное исследование будет включено приблизительно 320 подростков и взрослых субъектов с диагнозом ЕоЕ, которые удовлетворяют критериям зачисления, и которые будут рандомизированы с попаданием в 1 из 4-х получающих дозы групп (включая плацебо) по 80 субъектов в одной получающей дозу группе.
Критерии включения: субъекты мужского и женского пола в возрасте от 14 до 75 лет включительно. Пациенты должны иметь поставленный или предположительный диагноз ЕоЕ, включая таких пациентов с рецидивом, должны иметь подтверждение в виде наличия симптомов и данных гистологического исследования; наличия документа, подтверждающего предыдущее неудачное лечение в течение 8 недель или более ингибитором протонного насоса (PPI) в высоких дозах (например, 40 мг, BID) либо до постановки первоначального диагноза, либо задокументированного впоследствии. Отсутствие ответа на PPI терапию определяется как максимальное количество эозинофилов в HPF, составляющее 15 или более, по меньшей мере в одном подвергнутом биопсии месте, через 8 недель лечения с использованием высокой дозы. Во время этого исследования PPI терапия может быть продолжена, только если субъект получал PPI терапию в момент отбора биоптатов при проведении эндоскопического скринингового обследования. В ходе исследования не допускается ни начала новой PPI терапии, ни изменения имеющейся PPI терапии.
Субъекты должны иметь доказательства эозинофильного эзофагита, определяемые по максимальному количеству эозинофилов в слизистой оболочке пищевода в поле зрения микроскопа с большим увеличением (HPF, увеличение 400x), составляющему 15 или более, по меньшей мере в одном (1) из биоптатов, взятых из разных мест пищевода (из числа 3 и более биопсий из каждой проксимальной и дистальной частей пищевода; в целом как минимум из 6 биопсий). Процедуры биопсии должны быть проведены не ранее, чем за 30 суток до скринингового визита, и предметные стекла должны быть получены главным специалистом по лабораторной диагностике в пределах 30 суток после скринингового визита или за 2 недели до рандомизации. Пригодность будет основываться исключительно на заключении главного специалиста по лабораторной диагностике.
Субъекты должны иметь 7-дневный общий показатель ЕоЕ, равный 5 или более, по результатам измерений по шкале от 0 до 10 (0 означает отсутствие симптомов, а 10 означает наиболее тяжелые симптомы).
Субъекты должны иметь 7-дневный показатель EEsAI более 20, чтобы быть зачисленными во вводную фазу исследования с применением плацебо. Для рандомизации необходимо наличие более высокого значения показателя EEsAI согласно ежедневнику и наличие клинических симптомов дисфагии (затруднения при глотании) по меньшей мере 3 раза в неделю в течение 2 недель.
Критерии исключения: известное противопоказание, гиперчувствительность к кортикостероидам или их непереносимость; любое физическое, психическое или социальное состояние, история болезни или отклонения лабораторных показателей от нормы, которые по мнению исследователя могут помешать проведению процедур исследования или способности субъекта присоединиться к исследованию и участвовать в нем; наличие любого типа инфекции полости рта или слизистой оболочки пищевода; любое состояние, затрагивающее слизистую оболочку пищевода или изменяющее моторику пищевода, кроме ЕоЕ, включая эрозивный эзофагит, грыжу пищеводного отверстия длиной более 3 см и пищевод Барретта; применение системных, пероральных или парентеральных кортикостероидов в течение 30 суток или ингаляционное или продолжительное применение высокоэффективных вводимых интрадермальной инъекцией кортикостероидов для местного применения в течение 30 суток перед: биопсией пищевода, необходимой для зачисления в это исследование, или EGD, если проводится в период до скрининга; утренний (с 07:00 до 08:00 часов или по возможности ближе к этому интервалу) уровень кортизола в сыворотке крови 5 мкг/дл или менее (138 нмоль/л); уровень кортизола в плазме крови менее 18 мкг/мл (497 нмоль/л) через 60 минут после теста стимуляции адренокортикотропным гормоном (АСТН) с использованием 250 мкг косинтропина, потребление грейпфрутового сока во время лечения запрещено; применение биологических иммуномодуляторов в течение последних 6 месяцев; применение ингибиторов кальциневрина, аналогов пуринов (азатиоприна, 6-меркаптопурина) в течение последних 3 месяцев; противопоказание к EGD или биопсии пищевода либо сужение пищевода, препятствующее проведению EGD со стандартным 9-мм эндоскопом; желудочно-кишечное кровотечение в пределах 1 месяца до скринингового визита или между скрининговым визитом и визитом для рандомизации; текущая хроническая инфекция, иммуносупрессия или иммунодефицит; анамнез или наличие болезни Крона, глютеновой болезни или другого воспалительного заболевания желудочно-кишечного тракта, включая эозинофильный гастроэнтерит; текущее злоупотребление алкоголем или наркотиками; беременные или кормящие грудью субъекты женского пола; живущие половой жизнью женщины детородного возраста, которые не соглашаются использовать высокоэффективные методы контрацепции во время исследования, включая период последующего наблюдения (для подростков допускается воздержание); для субъектов женского пола с хирургической менопаузой или менопаузой, подтвержденной содержанием фолликулостимулирующего гормона (FSH), нет необходимости в контрацепции или тестировании определения беременности во время данного исследования; участие в клиническом исследовании с вовлечением какого-либо экспериментального лекарственного средства в течение 30 суток после скринингового визита.
Методология/методики исследования показана(ы) в Таблицах 4-7.
Первичные конечные точки эффективности: имеются 2 сопервичные конечные точки эффективности для оценки ЕоЕ-ответа:
процентная доля субъектов с максимальным количеством эозинофилов/HPF <6 во всех подвергнутых биопсии местах пищевода на 12-й неделе и отклонение от исходного значения среднего еженедельного показателя EEsAI. Полученный с применением эндоскопии ЕоЕ-ответ и отсутствие ухудшения среднего еженедельного показателя EEsAI будут служить основанием для зачисления в Часть 2.
Вторичные конечные точки эффективности: ЕоЕ-ответ, также будет оцениваться на 26-й неделе по сравнению с плацебо на предмет оценки эффекта отмены лечения. ЕоЕ-ответ, также будет оцениваться на 52-й неделе по дозе и подгруппе. Ремиссия ЕоЕ и ЕоЕ-ответ, также будут оцениваться через 12 недель OL лечения у нереспондеров Части 1.
Устойчивый ЕоЕ-ответ, будет оцениваться на 24-й и 52-й неделях для субъектов, продолжающих принимать ту же дозу: оценка рецидива и неудачного результата лечения; отсутствие ответа в каждой конечной точке по данным эндоскопии; рецидив при приеме плацебо в Части 2; процентная доля субъектов, нуждающихся в неотложном эндоскопическом освобождении от пищи в зависимости от дозы и Части исследования; процентная доля субъектов, нуждающихся в дилатации пищевода.
Эффект повторного лечения будет определяться путем оценки ЕоЕ-ответа у пациентов с рецидивом заболевания в Части 2 на 52-й неделе в зависимости от подвергаемой лечению группы.
Процентная доля субъектов с максимальным количеством Eos в HPF менее 1 и менее 15 для всех основных моментов времени, когда оценивается ЕоЕ-ответ.
Процентная доля субъектов со средним еженедельным показателем EEsAI менее 20 для всех основных моментов времени, когда оценивается ЕоЕ-ответ.
Изменения по данным эндоскопии будут основаны на показателе EREFS, учитывающем 5 эндоскопических признаков: отек, образование борозд, экссудаты, пищеводные кольца, стриктуры, и оценке врачом общей активности заболевания (заболевание отсутствует, степень слабая, умеренная, тяжелая): процентной доли субъектов с общей оценкой «улучшение», «отсутствие изменений» и «ухудшение» по сравнению с исходным состоянием (при скрининговом визите) 12-й, 26-й и 52-й неделях, а также по окончании OL лечения нереспондеров из Части 1; на изменении, по сравнению с исходным состоянием, 7-дневного общего показателя ЕоЕ, который будет оцениваться перед вводной фазой, по сравнению с исходным состоянием, на 12-й неделе, 26-й неделе и 52-й неделе.
Фармакокинетические конечные точки: популяционные ФК (выборочный ФК и многократный ФК) анализы для стационарного состояния; ФК анализы разовой дозы.
Эксплоративные конечные точки: гистологический показатель Коллина для ЕоЕ для всех проведенных биопсий, будет оцениваться изменение по сравнению с исходными данными; оценка качества жизни на основании EoE-QoL-A на 12-й, 26-й, 52-й неделе и 12-й неделе OL лечения по дозе и подгруппе; оценка пациентом симптомов по сравнению с предыдущим визитом. Этот вопрос будет оцениваться на 8-й, 12-й, 18-й, 26-й, 30-й и 52-й неделях.
Безопасность: безопасность будет оцениваться посредством мониторинга и регистрации всех возникающих при лечении нежелательных явлений (ТЕАЕ), ТЕАЕ, приводящих к отмене лечения, и серьезных нежелательных явлений (SAE). Все ТЕАЕ получат код на основании классификации MedDRA (Словарь по нормативно-правовой деятельности в области медицины), версии 14.0, для нежелательных явлений (АЕ) и будут классифицированы исследователем по тяжести (слабое, умеренное, тяжелое) и связи с исследуемым лекарственным средством (по типу связь есть или связи нет). ТЕАЕ, возникающие в течение 2 суток после изменения дозы, будут отнесены к предыдущей дозе. Это время отнесения может быть изменено на основании периода полувыведения дозы. Для документирования исходного состояния субъекта и выявления связанных с АЕ изменений будут проведены физические обследования. При всех визитах также будут оценены жизненно важные признаки и будет сообщено о клинически значимых отклонениях.
На протяжении всего исследования будут проводиться стандартные лабораторные анализы, включая общий анализ крови, биохимический анализ крови, анализ мочи, снятие электрокардиограмм (ЭКГ), как указано в графиках обследований. Клинически значимые изменения в результатах лабораторных анализов или ЭКГ будут обобщены.
Проблемы с кортизолом: после получения субъектом разрешения неизбежно могут обнаруживаться аномальные AM уровни кортизола, глюкозы в моче или глюкозы в сыворотке крови. В регистрационных картах пациентов (CRF) должно быть зафиксировано наличие или отсутствие известных обусловленных глюкокортикоидами АЕ, таких как лунообразное лицо, акне, гирсутизм, перепады настроения, бессонница или депрессия. В Форме информированного согласия (ICF) следует обратить особое внимание на то, что во время серьезных медицинских заболеваний может потребоваться применение стероидных гормонов стресса. Тест стимуляции АСТН (250 мкг косинтропина) должен быть выполнен для всех субъектов в начальный момент времени и на 12-й неделе, в момент преждевременной отмены лечения и на 52-й неделе. АСТН тест должен быть выполнен для всех субъектов с концентрацией кортизола в плазме крови более 5 мкг/дл (138 ммоль/л). Все субъекты с положительными результатами теста должны быть исключены. Любые положительные результаты к моменту окончания лечения должны сопровождаться восстановлением функции надпочечников. Число субъектов, прерывающих лечение по причине угнетения НРА оси или положительных результатов теста стимуляции АСТН, будет обобщено. Заказчик клинического исследования (спонсор) предоставит каждому исследовательскому центру и исследователю руководящие указания по обеспечению безопасности последующего наблюдения и задокументирует восстановление функции надпочечников у всех субъектов, демонстрирующих признаки гиперадренокортицизма или угнетения НРА оси в ходе исследования. Субъектов в возрасте до 18 лет будут оценивать по таким ростовым параметрам, как рост, вес, индекс массы тела (BMI) и соответствующие z-показатели. Представляющими интерес конечными точками безопасности являются: частота возникающих при лечении нежелательных явлений (ТЕАЕ), ТЕАЕ, приводящие к отмене лечения, и возникающие при лечении серьезные нежелательные явления (SAE), а также процентная доля субъектов с уровнем кортизола в сыворотке крови 5 мкг/дл (138 нмоль/л) или менее или положительным результатом теста стимуляции АСТН (содержанием кортизола в сыворотке крови менее 18 мкг/мл (497 нмоль/л) через 60 минут). Число субъектов, прерывающих лечение по причине угнетения НРА оси, будет зарегистрировано.
Статистические методы: определение объема выборки: для Части 1 определение объема выборки основано на запланированных сравнениях в отношении ЕоЕ-ответа между каждой из получающих дозу групп: 1) сравнения процентной доли субъектов с максимальным количеством эозинофилов/HPF менее 6 во всех подвергнутых биопсии местах пищевода на 12-й неделе; и 2) изменения по сравнению с исходными данными в показателе EEsAI. В случае настоящего исследования через 12 недель лечения процентная доля субъектов, подвергаемых лечению плацебо, с максимальным количеством эозинофилов/HPF менее 6 во всех подвергнутых биопсии местах пищевода, предположительно составляет 15% для плацебо и 60% для групп с лечением активным средством с ожидаемой разницей 45. В случае среднего еженедельного показателя EEsAI, ЕоЕ-ответа для симптомов на 12-й неделе, авторы изобретения предполагают уровень ответа на плацебо 35% с ожидаемой эффективностью 65% в группах с лечением активным средством с ожидаемой разницей 30%. На основании равномерной рандомизации приблизительно 60% субъектов будут зачислены в Часть 2 данного исследования. Чтобы гарантировать достаточность объема выборки для обнаружения разницы 30% в Части 1, и чтобы гарантировать зачисление в Часть 2 достаточного для анализа количества субъектов, в предположении, что у 20% пациентов в группах с лечением активным средством и у 80% пациентов в группах с лечением плацебо разовьется рецидив симптомов, в исследование будут включены минимум 320 субъектов (по 80 на одну группу). С учетом 320 на одну группу и общего индекса выбывших из исследования, равного 20%, авторы изобретения предполагают, что приблизительно у 35 субъектов из расчета на одну группу с лечением активным средством будет оцениваться устойчивая ремиссия на 52-й неделе.
Первичной анализируемой в отношении эффективности популяцией является предназначенная для лечения (ITT) популяция, определяемая как «все рандомизированные субъекты». Анализируемой в отношении безопасности популяцией является популяция для оценки безопасности, определяемая как «все субъекты, которые получают по меньшей мере одну дозу исследуемого лекарственного средства». Популяция для изучения ФК разовой дозы, будет определена как «все субъекты в ITT популяции из Части 1, которые рандомизированы с попаданием в одну трех получающих АРТ-1011 групп, принимают свою первую дозу АРТ-1011 и имеют по меньшей мере один ФК образец, включенный в завершающий популяционный ФК анализ разовой дозы». Популяция для изучения ФК в стационарном состоянии, будет определена как «все субъекты в ITT популяции из Части 1, которые рандомизированы с попаданием в одну из трех получающих АРТ-1011 групп, принимают свою дозу АРТ-1011, соответствующую периоду ФК контроля в стационарном состоянии, и имеют по меньшей мере один ФК образец, включенный в завершающий популяционный ФК анализ в стационарном состоянии». В дополнение к анализу по протоколу популяции, которые завершают Части 1, 2 и 3 и OL лечение, могут быть оценены согласно Плану статистического анализа (SAP). Исходные и демографические данные будут обобщены с использованием описательной статистики для непрерывных и порядковых переменных (например, возраста, веса, роста) и количеств и процентов для категориальных переменных (например, пола, расовой принадлежности).
Первичная эффективность для ЧАСТИ 1
Имеются две сопервичные конечные точки эффективности для ЧАСТИ 1: процентная доля субъектов с максимальным количеством эозинофилов/HPF менее 6 во всех подвергнутых биопсии местах пищевода на 12-й неделе; и изменение по сравнению с исходными данными в среднем еженедельном показателе EEsAI на 12-й неделе. Чтобы сохранить общий уровень значимости для данного исследования при 0,05, каждая из этих двух сопервичных конечных точек эффективности будет оцениваться на уровне 0,05.
Для каждой сопервичной конечной точки представляющими интерес сравнениями, связанными с первичным лечением, являются сравнения каждой получающей дозу АРТ-1011 группой с группой, получающей плацебо. Гипотезы эффективности АРТ-1011 по сравнению с плацебо будут протестированы в следующем порядке:
H0: μ для АРТ-1011 (3,0 мг, BID) = μ для плацебо,
H0: μ для АРТ-1011 (1,5 мг, BID) = μ для плацебо,
H0: μ для АРТ-1011 (1,5 мг, HS) = μ для плацебо,
где для одной сопервичной конечной точки эффективности μ обозначает долю субъектов с максимальным количеством эозинофилов/HPF менее 15 во всех подвергнутых биопсии местах пищевода на 12-й неделе, а для другой сопервичной конечной точки эффективности μ означает изменение по сравнению с исходными данными среднего еженедельного показателя EEsAI на 12-й неделе.
Статистическое тестирование начнется со сравнения АРТ-1011 в дозе 3,0 мг, BID, с плацебо в отношении каждой из двух сопервичных конечных точек эффективности. Если результаты обоих сравнений являются статистически значимыми с уровнем 0,05, то следующая более низкая доза будет протестирована по сравнению с плацебо для обеих сопервичных конечных точек эффективности. Процедура останавливается после того, как первое попарное сравнение перестает давать статистически значимый результат для одной или обеих сопервичных конечных точек эффективности. Поскольку статистическое тестирование различий в лечении между АРТ-1011 и плацебо должно выполняться с использованием априори упорядоченных гипотез, никакой корректировки уровня значимости для множественности сравнений не требуется.
Попарные сравнения между каждой группой, получающей АРТ-1011, и группой, получающей плацебо, в отношении доли субъектов с максимальным количеством эозинофилов/HPF менее 6 во всех подвергнутых биопсии местах пищевода на 12-й неделе будут проводиться с использованием критерия Кохрана-Мантеля-Гензеля (СМН) с поправкой на два фактора рандомизационной стратификации: возрастную группу и применение PPI; а также с поправкой на географический регион (Северная Америка; не Северная Америка). Попарные сравнения между каждой группой, получавшей АРТ-1011, и группой, получавшей плацебо, в отношении изменения по сравнению с исходными данными в среднем еженедельном показателе EEsAI на 12-й неделе, будут выполнены путем сравнения соответствующих средних значений, полученных методом наименьших квадратов, из ковариационного анализа (ANCOVA) с лечением, двух факторов рандомизационной стратификации и географического региона в модели в качестве фиксированных эффектов и исходного показателя согласно симптоматическому опроснику (SQ) в качестве независимой переменной. Данный метод будет использоваться для корректировки множественных сравнений. Объем выборки увеличен соответствующим образом.
Если продемонстрировано превосходство по меньшей мере одной из групп, получающих АРТ-1011, над группой, получающей плацебо, по двум сопервичным конечным точкам, то зависимость ответа от дозы будет оцениваться для данных двух сопервичных конечных точек с общесуточной дозой среди независимых переменных в соответствующих статистических моделях.
Эффективность OL лечения
Будет использован подход, аналогичный подходу в Части 1 для нереспондеров из Части 1, которые получают OL лечение в дозе 3,0 мг, BID.
Эффективность для Части 2
Существует одна первичная конечная точка эффективности для Части 2, процентная доля субъектов с рецидивом заболевания во время или перед окончанием двойного слепого лечения в Части 2, определяемая как возврат к исходному показателю EEsAI или устная жалоба, что «Мои симптомы ухудшились и являются примерно такими же, как в момент, когда я был зачислен в исследование», и максимальное количество эозинофилов/HPF составляет 15 или более по меньшей мере в одном подвергнутом биопсии месте пищевода во время или перед окончанием обследования на 26-й неделе Части 2. Чтобы сохранить общий уровень значимости для данного исследования при 0,05, статистическое тестирование для конечной точки эффективности в Части 2 будет проводиться только в том случае, если продемонстрирована статистическая значимость в пользу по меньшей мере одной получающей дозу АРТ-1011 группы по сравнению с плацебо для двух сопервичных конечных точек эффективности в Части 1. Гипотеза эффективности АРТ-1011 (3,0 мг, QD) по сравнению с плацебо выглядит так, как приведено ниже:
H0: μ для АРТ-1011 (3,0 мг, QD) = μ для плацебо,
где μ означает долю субъектов с рецидивом заболевания во время или перед окончанием 12-недельного двойного слепого лечения в Части 2. Тест будет проводиться с уровнем значимости 0,05 с использованием критерия СМН с поправкой на два фактора рандомизационной стратификации (из Части 1), географический регион и предшествующее лечение, перед которым субъект проходил рандомизацию в двойное слепое лечение в Части 2. Предшествующее лечение состояло в приеме плацебо, АРТ-1011 в дозах 1,5 мг, QD; 1,5 мг, BID; и 3,0 мг, BID, для рандомизированных субъектов в Части 1.
Эффективность для Части 3
Устойчивый ЕоЕ-ответ будет оцениваться в подгруппе субъектов, у которых был получен ЕоЕ-ответ в Части 1 и Части 2, и для которых были проведены оценки на 52-й неделе.
Вторичная и эксплоративная эффективность
Статистические тесты для сравнения каждой получающей дозу АРТ-1011 группы с группой, получающей плацебо, будут проводиться для вторичных конечных точек эффективности, но соответствующие р-значения будут рассматриваться как описательные, а не логически обоснованные. Вторичные конечные точки будут проанализированы аналогичным образом, как описано выше для первичных конечных точек эффективности, а именно, с использованием критерия СМН для категориальных конечных точек и ANCOVA для изменения или выраженного в процентах изменения по сравнению с исходными конечными точками, за исключением конечной точки периода времени до рецидива после начала двойного слепого лечения в Части 2, которая будет проанализирована с использованием метода Каплана-Мейера. Никакого статистического тестирования эксплоративных конечных точек эффективности проводиться не будет.
Безопасность
Число случаев ТЕАЕ будет обобщено согласно классу по поражению органов и систем органов и предпочтительной терминологии. Будут предоставлены отдельные сводные данные для случаев максимальной тяжести и связи с исследуемым лекарственным средством. Число случаев ТЕАЕ, приводящих к прекращению исследования, и возникающих при лечении SAE также будет обобщено. У субъектов с изменением получающих дозу групп ТЕАЕ будут отнесены к предыдущей дозе, если они происходят в течение 2 суток после изменения. Период отнесения может быть изменен в зависимости от периода полувыведения дозы. Клинически значимые изменения, представляющие возможный клинический интерес в клинических тестах, будут обобщены, включая общий анализ крови, биохимический анализ крови, анализ мочи, ЭКГ, содержание кортизола, показатели жизненно важных функций, оценки роста для подростков и минеральную плотность костной ткани. Никакого статистического тестирования конечных точек безопасности проведено не будет. При необходимости могут быть приведены таблицы изменений.
Фармакокинетический и фармакодинамический анализ
В этом исследовании прием всех доз АРТ-1011 будет проводиться два раза в сутки (до завтрака и перед сном) за исключением дозы 1,5 мг один раз в сутки, прием которой будет осуществлен в режиме QD перед сном, с плацебо, вводимым после завтрака. Популяционный ФК анализ будет проведен на основе комбинации данных о концентрации в плазме крови для серийного и выборочного вариантов анализа. Предыдущие серийные ФК данные для APT-1011 могут быть включены в этот анализ для облегчения разработки основной ФК модели.
Популяционный ФК анализ будет проведен с использованием программного обеспечения для расчета нелинейных моделей со смешанными эффектами, NONMEM, версии 7.2.0, или более поздней версии (ICON Development Solutions, Ellicott City, MD), или другого соответствующего программного обеспечения для расчета нелинейных моделей со смешанными эффектами. Структурная ФК модель будет включать CL/F (системный клиренс) и V/F (объем распределения) в качестве параметров с фиксированным эффектом. В дополнение к этому межсубъектная вариабельность (между субъектами) в оценках параметров и случайная остаточная ошибка в данных будут оцениваться с применением соответствующей модели ошибок. Оптимальная базовая модель будет выбрана в соответствии со стандартными критериями, такими как минимальное значение целевой функции и диагностические графики. Учитывая небольшое количество субъектов в этом исследовании, проведение анализа независимых переменных не планируется. Релевантная информация из биоаналитических и клинических баз данных (например, времена введения доз и времена отбора проб) будет извлечена и объединена для создания файл(ов) с вводными данными для популяционной ФК. Будут приложены все возможные усилия для сбора недостающей информации. Любая дополнительная информация, полученная относительно неполных данных и методик, применяемых для обработки любых неполных данных, будет задокументирована и обсуждена в отчете о клиническом исследовании. После построения окончательной модели будут рассчитаны вторичные параметры, такие как AUC и Cmax, чтобы охарактеризовать степень системного воздействия FP (флутиказона пропионат). При необходимости может быть проведено дополнительное численное моделирование с целью информирования о принятии решений для будущих исследований. В общем случае стадия численного моделирования будет включать создание файлов данных с использованием фиктивных субъектов с желаемыми временами отбора проб и режимами введения, осуществление численного моделирования с желаемый числом повторений с использованием выходных параметров окончательной модели в контрольном файле. Будут сформированы выходные данные окончательных популяционных моделей, включая соответствующие диагностические графики, списки и сводные данные по ФК параметрам. В дополнение к этому будут приведены графические и табличные представления всех данных, связанных с численным ФК моделированием.
Если позволяют данные, также могут быть выполнены эксплоративные анализы, в которых оценивается взаимосвязь между системным воздействием FP и изменениями в уровнях кортизола, как описано выше. Возможные эксплоративные ФК/ФД (фармакодинамический) анализы будут использованы для облегчения выбора безопасных и эффективных доз для будущих исследований.
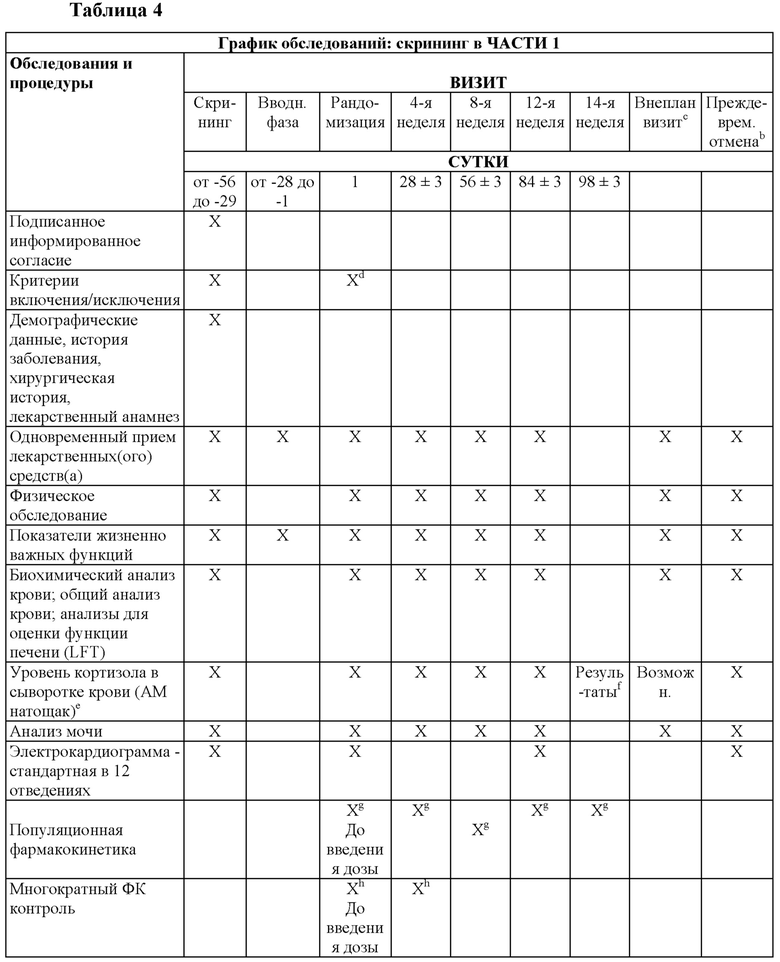
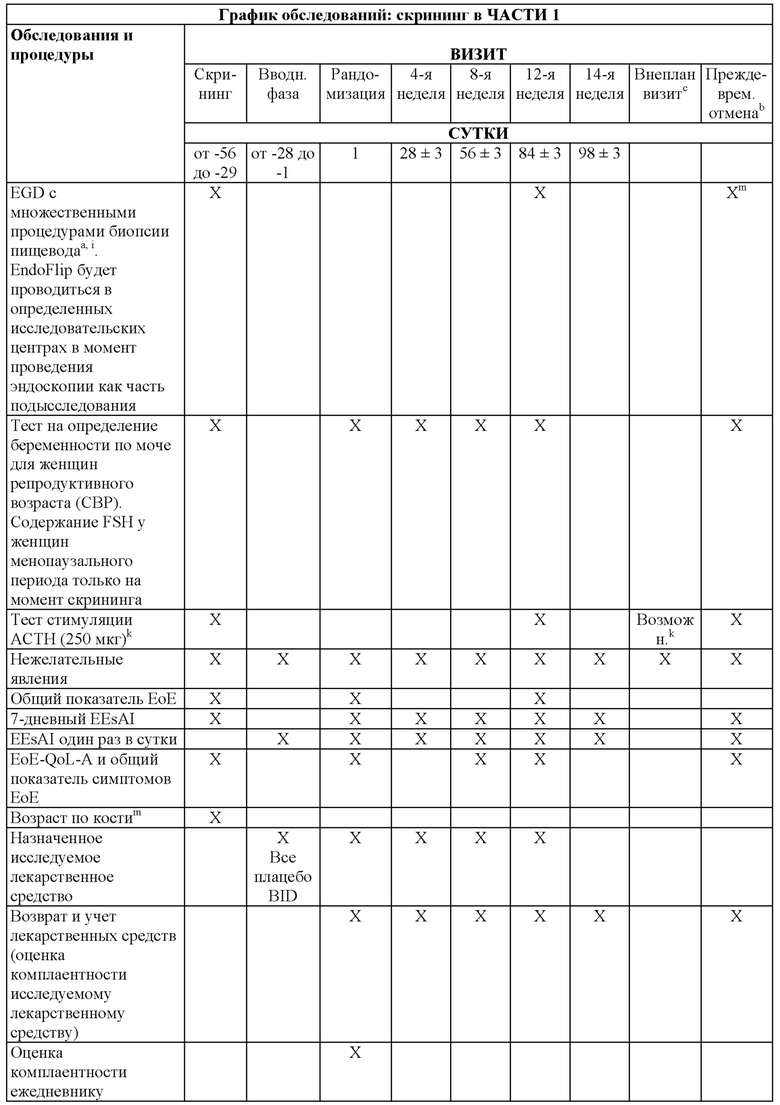
а. Эндоскопия может быть проведена во время отдельного визита. Результаты гистологического анализа потребуются при рандомизации и на 14-й неделе.
b. Пациент должен появиться в кабинете не позднее 7 дней с момента определения необходимости отмены лечения для данного пациента. Если это невозможно сделать ввиду SAE или другого непредвиденного обстоятельства, визит может быть совершен пациентом или членом его семьи по телефону. Документальное подтверждение того, почему пациент не смог прийти в исследовательский центр, должно быть внесено в историю болезни пациента.
c. Причиной для внепланового визита будет необходимость формирования процедур по усмотрению руководителя клинических испытаний (PI).
d. Подтверждение того, что пациент все еще удовлетворяет критериям включения/исключения на основании ежедневного EEsAI в дополнение к другим критериям.
e. Отборы образцов необходимо проводить как можно ближе по времени к 08:00. Пациенты не должны принимать пищу в течение восьми(8)-часового периода времени до проведения оценок уровня кортизола в сыворотке крови. Кровь для анализа AM уровня кортизола в сыворотке крови может быть взята в пределах +/- 2 дней от планового визита, чтобы соответствовать точному временному графику. Другие отборы крови, запланированные на время данного визита, могут быть выполнены в то же самое время.
f. Если во время заключительного визита лечения отмечается аномальный уровень кортизола в сыворотке крови, то может потребоваться проведение дополнительного мониторинга и АСТН теста.
g. Образец будет получен на 4-й неделе для всех субъектов за исключением субъектов из подгруппы многократного ФК контроля не ранее, чем за 15 минут до AM дозы и через 0,5, 1,5 и 3 часа после AM дозы. Время введения РМ дозы (перед сном) и AM дозы наряду со временем фактического отбора ФК образцов будет задокументировано.
h. Приблизительно по 6 субъектов из расчета на одну получающую дозу группу будут проходить многократный ФК контроль в определенных исследовательских центрах в интервале между 2-й и 4-й неделями: ФК образцы следует отбирать не ранее, чем за 15 минут до AM дозы и через 0,5, 1, 2, 4, 8 и 12 часов после AM дозы, непосредственно перед РМ дозой. ПРИМЕЧАНИЕ: 12-часовой ФК образец после AM дозы является «необязательным». 12-часовой ФК образец следует отбирать непосредственно перед РМ дозой. Время приема РМ дозы (перед сном) и AM дозы наряду со временем фактического отбора ФК образцов будет задокументировано.
i. Может быть проведена за 30 суток до скринингового визита (при отсутствии приема системных или ингалируемых, интраназальных или обладающих высокой эффективностью вводимых интрадермальной инъекцией кортикостероидов для местного применения в течение этих 30 суток до скринингового визита). Если результаты предыдущего гистологического исследования считаются неприемлемыми, должен быть запланирован дополнительный скрининговый визит для проведения EGD и биопсии во время периода скрининга. Результаты от главного специалиста по лабораторной диагностике должны быть получены до рандомизации. Должен быть получен показатель EREFS, или в равной степени он может быть получен из отчета по эндоскопии, соответствующего любым стандартам качества выполнения эндоскопии.
j. Если пациент покидает исследование ввиду отсутствия эффективности или по другим причинам, исследователь может провести EGD, если к этому есть клинические показания.
k. Все субъекты проходят тест стимуляции АСТН (250 мкг) в начальный момент времени и по окончании лечения (EOT) или в момент преждевременной отмены лечения. Во время данного исследования также будет проведен тест стимуляции АСТН при возможном визите для субъектов, уровень кортизола в сыворотке крови которых подтверждается двумя отборами крови, как составляющий 5 мкг/дл (138 нмоль/л) или менее, или если у них имеются признаки и симптомы гиперкортицизма, для оценки угнетения гипоталамо-гипофизарно-надпочечниковой (НРА) оси, вызывающего клиническую озабоченность.
l. Для всех подростков (в возрасте до 18 лет на момент рандомизации) должно быть выполнено определение возраста по кости; исключение составляют субъекты женского пола, которые перестали прибавлять в росте, или, по усмотрению PI, субъекты мужского пола, которые не перестали прибавлять в росте к 18 годам.
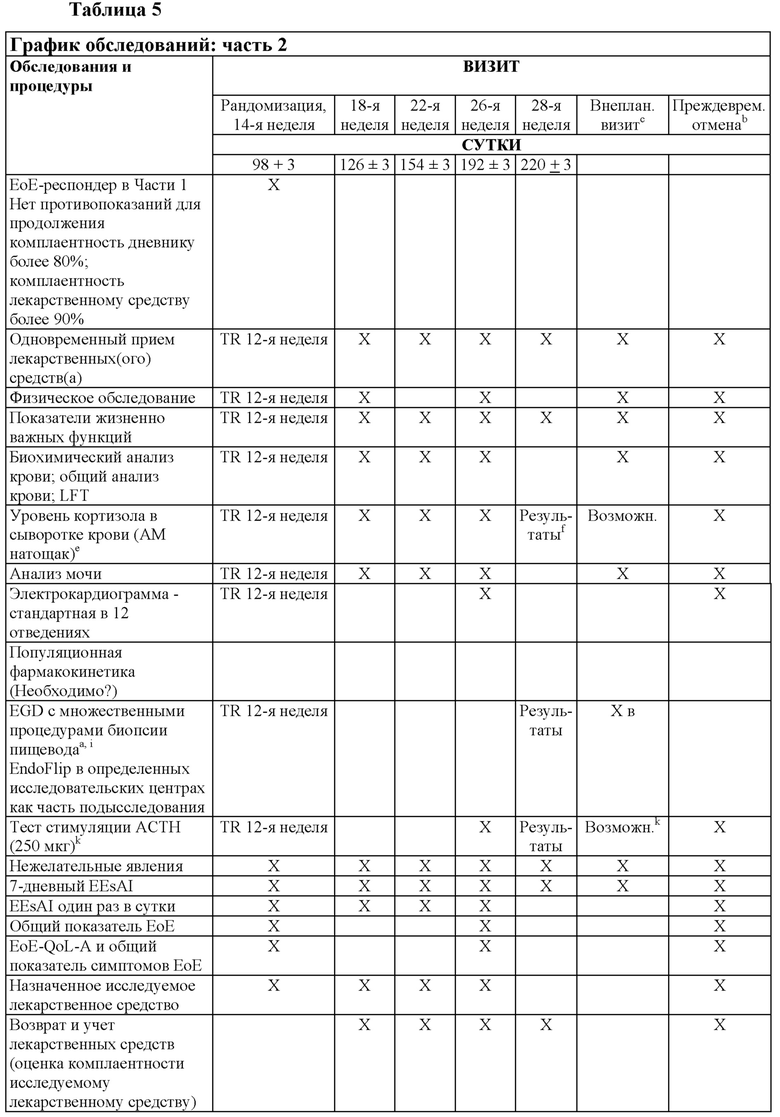
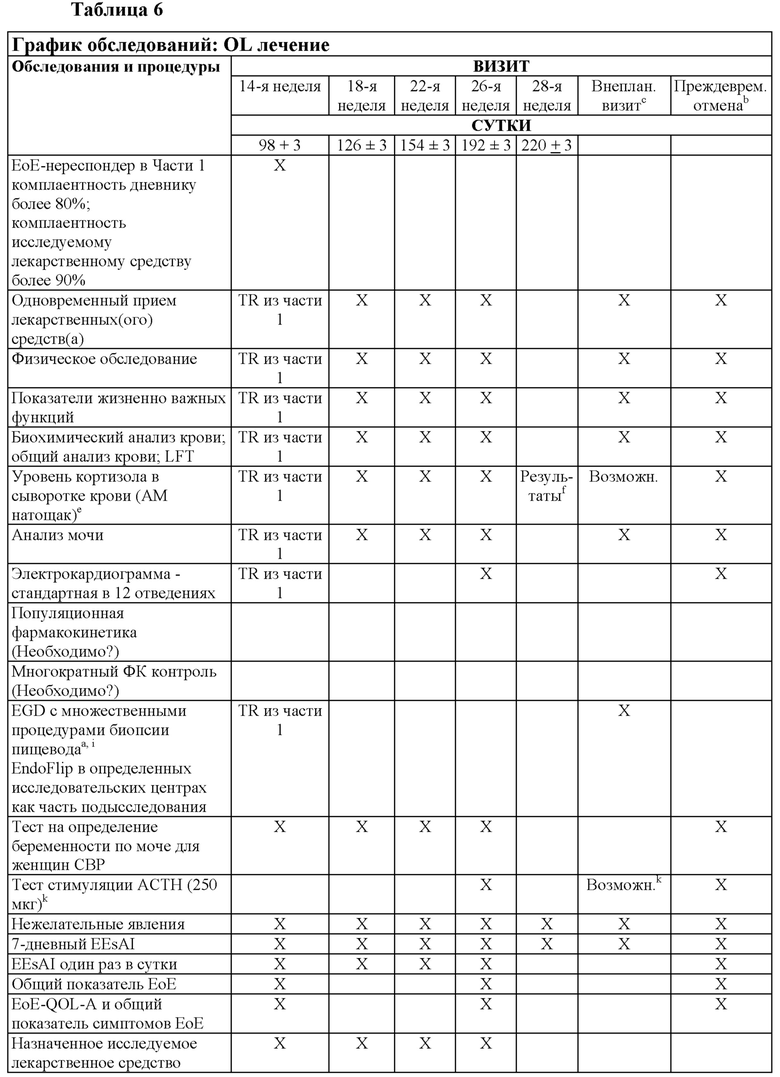
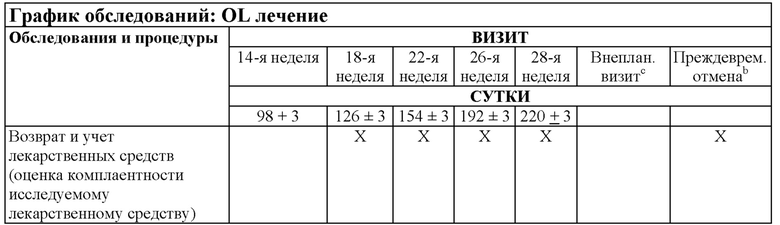
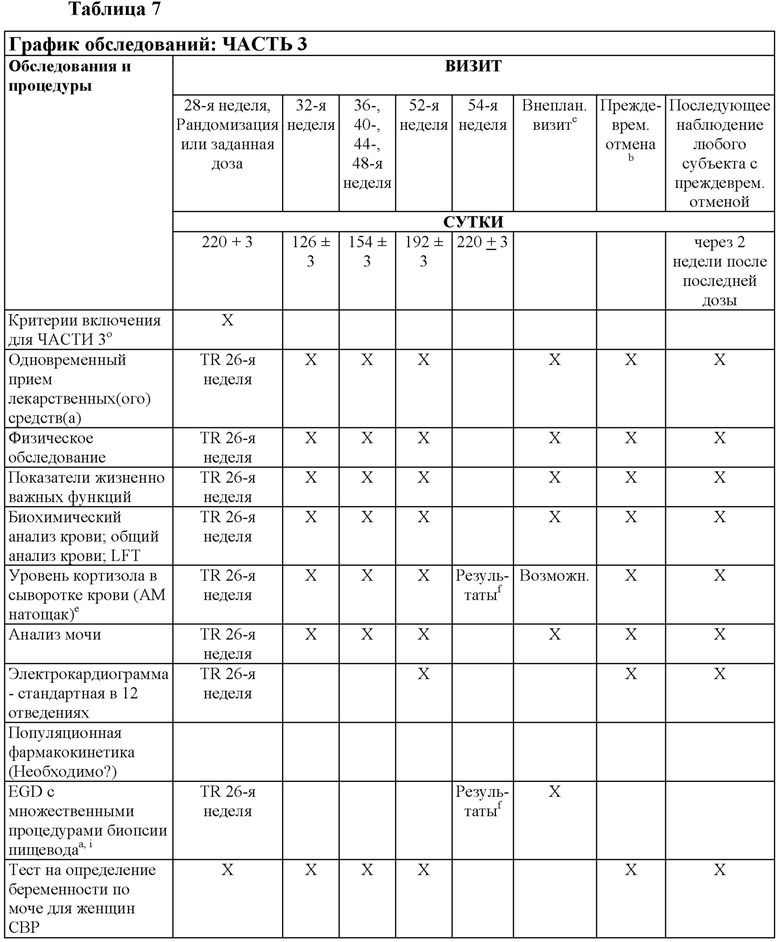
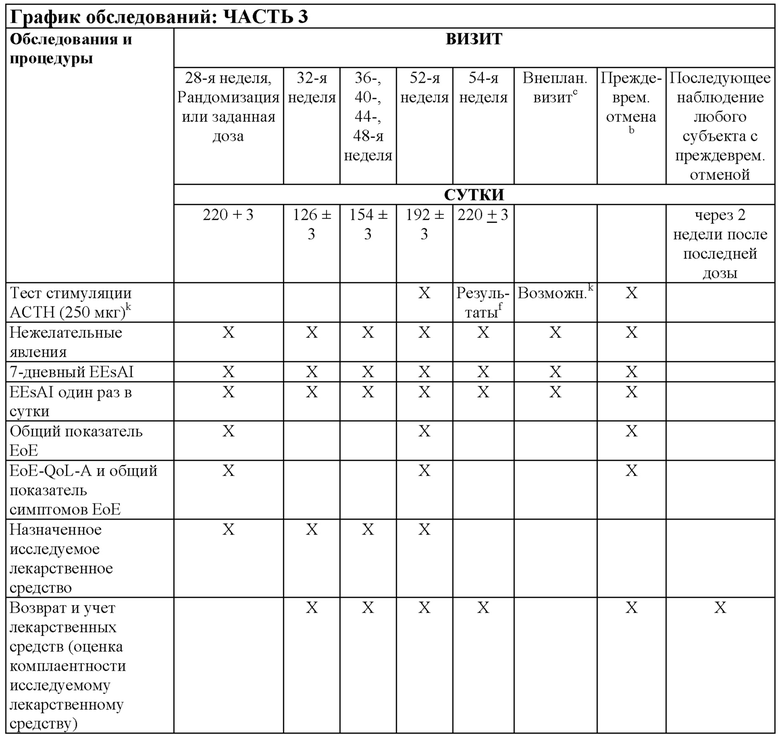
a-m - см. предыдущие сноски.
o. Субъекты удовлетворяют критериям включения для ЧАСТИ 3.
Завершение ЧАСТИ 2 в качестве респондера - продолжение с использованием той же дозы.
Завершение ЧАСТИ 2 в качестве нереспондера - назначение дозы 3 мг, BID.
Рецидив в случае приема плацебо - возврат к предшествующей дозе в ЧАСТИ 3.
Рецидив в части 2 в случае приема плацебо - назначение дозы 3 мг, BID в ЧАСТИ 3.
Завершение OL лечения в качестве респондера - продолжение, доза 3 мг, BID.
Завершение OL лечения в качестве нереспондера - выход из исследования и зачисление в период последующего наблюдения.
Пример 2. Рандомизированное, двойное слепое, плацебо-контролируемое испытание перорально распадающейся таблетки флутиказона пропионата на пациентах взрослого и подросткового возраста с эозинофильным эзофагитом: исследование безопасности и переносимости, фаза 1/2а
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ И ЦЕЛИ: эозинофильный эзофагит (ЕоЕ) представляет собой хроническое иммунно-опосредуемое заболевание, характеризующееся наличием тканевой эозинофилии и симптомов дисфункции пищевода. Данное исследование было первым клиническим исследованием АРТ-1011, новой перорально распадающейся таблетки (ODT) флутиказона пропионата (FP), в котором оценивали переносимость и безопасность 2-х режимов введения АРТ-1011 по сравнению с плацебо (РВО) на подростках и взрослых пациентах с ЕоЕ.
СПОСОБЫ: субъектов рандомизировали в соотношении 1:1:1 для получения либо 1,5 мг, BID (группа BID) АРТ-1011 (n=8), либо 3,0 мг, QD (группа QD) АРТ-1011 (n=8), либо РВО (n=8). Пациентам была проведена эзофагогастродуоденоскопия (EGD) с биопсией пищевода в исходный момент времени (BL) и по окончании лечения (EOT) (на 8-й неделе) для оценки изменения среднего количества эозинофилов. Вторичные конечные точки включали в себя улучшение выявленных в результате эндоскопии признаков по результатам измерений эндоскопического референсного показателя (EREFS) для ЕоЕ и общей оценки тяжести заболевания у пациента (PatGA). Также проводили оценку безопасности.
РЕЗУЛЬТАТЫ: двадцать четыре субъекта (включая 9 пациентов подросткового возраста, по 3 в каждой подвергаемой лечению группе) выполнили 8-недельный период двойного слепого исследования. Показаны исходные количества и средние на момент EOT количества эозинофилов в пищеводе (число клеток/мм2 поля зрения микроскопа под большим увеличением), среднее значение показателя EREFS, а также средняя PatGA для групп РВО, BID и QD (Таблица 8). На момент EOT средние значения количества эозинофилов в пищеводе были значительно снижены по сравнению с BL в биоптатах пациентов, получавших АРТ-1011 (для обоих режимов), но не у тех пациентов, которые получали плацебо. Наблюдалось значительное уменьшение одного (l) среднего показателя EREFS (при сравнении с BL) в обеих группах пациентов, получавших АРТ-1011, но не плацебо. Наблюдалось значительное улучшение в плане общей оценки тяжести заболевания у пациентов, получавших АРТ-1011 BID (тенденция в группе с режимом QD), но не плацебо. В Таблице 8 показаны средние значения (и итерквартильная широта (IQR)) количества эозинофилов в пищеводе, а также среднее значение показателя EREFS. р-значение менее 0,05 считается значимым.
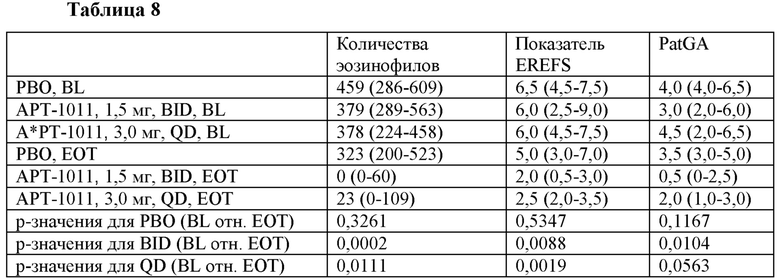
Частота возникающих при лечении нежелательных явлений (ТЕАЕ) составляла 75% (РВО), 75% (АРТ-1011) (QD). Все ТЕАЕ были небольшой степени, за исключением 1 случая утомляемости средней тяжести и случая депрессии средней тяжести у 1 субъекта в группе, получающей плацебо.
ЗАКЛЮЧЕНИЕ: лечение с использованием АРТ-1011 в течение восьми недель хорошо переносится. Применение АРТ-1011 у пациентов взрослого и подросткового возраста с ЕоЕ приводило к значительному ослаблению эозинофилии пищевода и улучшениям в степени тяжести заболевания по результатам ЕоЕ-ассоциированных данных эндоскопии.
Пример 3. Рандомизированное двойное слепое, плацебо-контролируемое исследование по подбору и поддержанию дозы АРТ-1011 у подростков и взрослых с эозинофильным эзофагитом (ЕоЕ)
Первичная задача: оценить эффективность АРТ-1011 у подростков и взрослых с эозинофильным эзофагитом (ЕоЕ).
Вторичные задачи: оценить безопасность АРТ-1011 у подростков и взрослых с ЕоЕ; определить зависимость ответа от дозы АРТ-1011; выбрать дозу(ы) для Фазы 3; оценить индивидуальную фармакокинетику АРТ-1011 в подгруппе взрослых и подростков с ЕоЕ; оценить популяционную фармакокинетику (PopPK) АРТ-1011 для пациентов с ЕоЕ; оценить поддержание эффективности лечения и долгосрочную безопасность; оценить влияние на выявленные при эндоскопическом обследовании визуальные характеристики, используя эндоскопический референсный показатель (EREFS) для ЕоЕ (Hirano, 2015; van Rhijn, 2014); оценить изменение по сравнению с исходными данными 7-дневного показателя EEsAI и общего показателя симптомов ЕоЕ.
Задачи по оценке безопасности: определить профиль безопасности при неотложном и длительном введении FP и его влияние на HP ось.
Эксплоративные задачи: оценить изменение по сравнению с исходными данными показателя ЕоЕ по гистологической балльной системе (EoEHSS) (Collins, 2016); оценить качество жизни в соответствии с опросником по оценке качества жизни взрослых пациентов с эозинофильным эзофагитом (EoE-QoL-A) (Taft, 2011); оценить симптомы по сравнению с предыдущим(ими) визитом(ами) (по 7-балльной шкале); оценить ФК/ФД (кортизол) и взаимосвязи ответа (эффективности) и воздействия.
Обоснование исследования: будут исследованы дозы от 1,5 мг HS до 3,0 мг BID для определения ответа на воздействие АРТ-1011 и минимальная эффективная доза, оказывающая влияние ниже любого клинически значимого на гипоталамо-гипофизарно-надпочечниковую (HPA) ось.
План исследования
Это рандомизированное двойное слепое, плацебо-контролируемое исследование по подбору дозы для трех общесуточных доз (1,5; 3 и 6 мг) АРТ-1011, вводимых в режиме 1,5 мг HS, 1,5 мг BID и 3,0 мг BID, в сравнении с соответствующим плацебо на 320 субъектах взрослого и подросткового возраста с ЕоЕ. Также будет проведена оценка поддержания эффективности и долгосрочной безопасности.
Подходящими будут субъекты в возрасте 14-75 лет (включительно), которые имеют диагноз ЕоЕ (максимальное количество Eos в HPF составляет 15 или более) после подтверждения неудачного лечения в течение 8 недель или более ингибитором протонного насоса (PPI) в высоких дозах (например, с введением два раза в сутки). Субъекты, у которых не было надлежащей проверки в отношении PPI терапии, в случае этого исследования должны проходить ее перед скринингом. Во время этого исследования PPI терапия может быть продолжена, только если субъект получал PPI терапию во время подписания информированного согласия. В ходе исследования не допускается ни начала новой PPI терапии, ни изменения имеющейся PPI терапии. Кроме того, субъекты должны иметь клинические проявления дисфагии, иметь 7-дневный общий показатель симптомов ЕоЕ более 3, сообщать по меньшей мере о 3 эпизодах дисфагии за последние 7 дней и не иметь никаких исключений, связанных с проведением данного исследования, чтобы быть зачисленными для проведения 2-недельной оценки исходных симптомов. Субъекты должны сообщать в ежедневнике по меньшей мере о 3 эпизодах дисфагии в неделю в течение каждой из двух недель проведения оценки исходных симптомов, чтобы быть пригодными к рандомизации.
В Части 1 субъекты будут разделены согласно возрасту (младше 18 и 18 лет и старше) и одновременно в соответствии с применением PPI во время этого исследования («да» или «нет»); затем случайным образом распределены с попаданием в 1 из 4-х получающих дозы групп (в соотношении (1,7):1:1:1).
Все субъекты будут ежемесячно осуществлять регулярные визиты, а для внеплановых визитов должно иметь место ухудшение симптомов между визитами.
Данное исследование будет состоять из нескольких частей:
Скрининг:
продолжительностью 1-4 недели, включая эндоскопию верхних отделов пищевода (EGD).
Оценка исходных симптомов: в течение 2 недель.
Необходимо иметь общий показатель симптомов ЕоЕ более 3, сообщать по меньшей мере о 3 эпизодах дисфагии за последние 7 дней и не иметь никаких исключений, связанных с проведением данного исследования, чтобы быть зачислеными для проведения оценки исходных симптомов.
Рандомизация: субъекты должны сообщать в ежедневнике по меньшей мере о 3 эпизодах дисфагии в неделю в течение каждой из двух недель проведения оценки исходных симптомов, иметь боее 75% комплаентности ежедневнику, иметь максимальное количество Eos в HPF, составляющее 15 или более, и удовлетворять всем другим критерия включения и исключения, чтобы быть рандомизированными. Субъекты будут рандомизированы в соотношении 1,7:3 в группы, получающие плацебо и дозы активного соединения. Регулируемая в отношении ответов рандомизация будет использована среди получающих дозы активного соединения, которая будет обновляться при каждом промежуточном анализе.
Субъекты будут разделены согласно возрасту и текущему применению PPI.
Общий протокол показан на Фиг. 3 и 4. Часть 1 представляет собой фазу индукции ЕоЕ-ответа, а в Частях 2 и 3 будет оцениваться поддержание ответа.
Нереспондерам в Части 1 будет предложено лечение с открытой маркировкой.
ЧАСТЬ 1. Лечение (с 1-й по 14-ю неделю)
Субъекты будут получать лечение в течение 12 недель и проходить EGD для оценки улучшения по результатам гистологического и эндоскопического анализов. Оценка улучшения симптомов будет проводиться постоянно. ЕоЕ-респондеры, определяемые как субъекты, которые достигают гистологического ответа в показателях максимального количества Eos в HPF 6 или менее, и не имеющие ухудшения симптомов (т.е., никакого усиления дисфагии согласно ежедневнику по сравнению с исходными данными) или эпизодов затрудненного прохождения пищи, будут зачислены в Часть 2 (поддерживающую фазу) на 14-й неделе. Эта Часть будет включать регулируемый объем выборки и регулируемую в отношении ответов рандомизацию в группах с лечением активным средством. Промежуточные анализы часто будут проведены после рандомизации 160 субъектов, для которых изменены вероятности рандомизации и оценены критерии преждевременной остановки исследования.
Лечение с открытой маркировкой (OL) (с 14-й по 28 неделю)
Нереспондеры (субъекты, которые не имеют гистологического ответа, и/или у которых имеется ухудшение дисфагии) на 14-й неделе будут получать лечение с открытой маркировкой (OL), 3 мг BID до 28-й недели включительно с EGD на 28-й неделе и затем, если они являются респондерами к окончанию OL периода (определяется так же, как и выше), они будут зачислены в Часть 3. Нереспондеры к OL лечению будут зачислены для прохождения 2-недельного периода последующего наблюдения и затем покинут исследование.
ЧАСТЬ 2. Поддерживающая фаза (с 14-й по 28 неделю)
ЕоЕ-респондеры (наличие гистологического ответа и отсутствие ухудшения дисфагии) из Части 1 будут продолжать принимать ту же самую дозу, что и в Части 1. Плацебо-респондеры, которые, как ожидается, окажутся малочисленны, будут продолжать принимать плацебо. К необходимости проведения эндоскопии при внеплановом визите будет приводить либо ухудшение дисфагии, либо затрудненное прохождение пищи. После эндоскопии все «пациенты с рецидивом заболевания» незамедлительно будут зачислены в Часть 3, за исключением пациентов с затрудненным прохождением пищи, требующих проведения эндоскопии и дилатации стриктуры. Субъекты с затрудненным прохождением пищи, которым требуется проведение эндоскопии или у которых имеется требующая дилатации стриктура, будут зачислены в 2-недельный период последующего наблюдения и покинут исследование. Все субъекты в этом исследовании на 26-й неделе будут проходить эндоскопию. ЕоЕ-респондеры на 28-й неделе будут переходить к Части 3. Нереспондеры по окончании Части 2 или пациенты с преждевременным рецидивом заболевания будут получать 3 мг, BID, когда они будут зачислены в Часть 3.
ЧАСТЬ 3. Поддерживающая фаза (с 28-й по 54 неделю)
Нереспондеры из Части 1, которые восприимчивы к лечению с открытой маркировкой, 3 мг, BID, будут продолжать принимать эту дозу в Части 3. Обратите внимание, что эта группа не будет принимать участия в Части 2.
ЕоЕ-респондеры из Части 2 продолжат прием той же дозы.
Нереспондеры и пациенты с преждевременным рецидивом заболевания из Части 2 будут получать лечение с открытой маркировкой, 3 мг BID, в Части 3.
Субъекты с рецидивом заболевания (затрудненным прохождением пищи, требующим проведения эндоскопии, или с клинически значимыми рецидивирующими симптомами) в Части 3 будут проходить внеплановую эндоскопию и будут зачислены в 2-недельное последующее наблюдение и покинут исследование.
Все продолжающие лечение субъекты будут проходить эндоскопию на 52-й неделе и совершат свой заключительный визит лечения на 54-й неделе.
Последующее наблюдение (через 2 недели после приема последней дозы)
Все субъекты должны пройти заключительную эндоскопию в пределах 3 недель до начала последующего наблюдения. Исключения включают: отзыв согласия или противопоказание к проведению эндоскопии. Причины для начала фазы последующего наблюдения включают: завершение 54-й недели, нежелательное явление, требующее преждевременной отмены лечения, включая затрудненное прохождение пищи, требующее проведения эндоскопии, невосприимчивость к лечению с открытой маркировкой (3 мг, BID) или рецидив в Части 3.
Фармакокинетика: в этом исследовании дозы АРТ-1011 1,5 мг BID и 3,0 мг BID будут введены по меньшей мере через 30 минут ПОСЛЕ завтрака и перед сном (по меньшей мере через 2 часа после вечернего приема пищи). Доза 1,5 мг будет введена перед сном (по меньшей мере через 2 часа после вечернего приема пищи). Субъекты, получающие дозу 1,5 мг, будут получать плацебо утром по меньшей мере через 30 минут ПОСЛЕ завтрака. Будет запрещен прием любой еды или питья по меньшей мере в течение 1 часа после приема дозы.
Чтобы охарактеризовать воздействие FP на популяцию пациентов, будет собрана комбинация образцов для многократного ФК контроля у подгруппы субъектов и образцов для выборочного ФК контроля у остальных субъектов. Для обработки данных многократного ФК контроля будет проведен некомпартментный анализ (NCA), а на основе комбинации данных о концентрации в плазме крови из образцов для многократного серийного и выборочного контроля будет проведен популяционный ФК анализ.
Многократный ФК контроль: многократный серийный ФК контроль образцов будет проведен для начального момента исследования и в стационарном состоянии в подгруппе взрослых субъектов (у 8 субъектов из расчета на одну получающую дозу группу) и в подгруппе субъектов подросткового возраста (приблизительно у 5 субъектов из расчета на одну получающую дозу группу) в предварительно определенных центрах проведения ФК исследований. Эти субъекты будут принимать суточную дозу (обе таблетки) в 1-е сутки. Образцы крови для определения концентраций флутиказона в плазме крови следует собирать до приема дозы и через 0,5, 1, 2, 4, 8 и 12 часов после утренней дозы в 1-е сутки и на 4-й неделе. Образцы для стационарного состояния могут быть собраны во время визита на 8-й или 12-й неделе, если субъект не может оставаться в течение 12 часов на 4-й неделе. Сбор образцов для многократного ФК контроля в течение 12-часового периода связан с режимом введения BID и необходимостью избегать пребывания в стационаре этих пациентов с ЕоЕ.
Выборочный ФК контроль: отбор образцов для выборочного ФК контроля будет проведен среди всех взрослых субъектов и субъектов подросткового возраста (за исключением субъектов из подгруппы многократного ФК контроля). Исходный образец «до приема дозы» будет отобран перед приемом дозы в 1-е сутки. На 4-й, 8-й и 12-й неделях повторного приема доз субъекты будут принимать свою AM дозу дома и будут иметь два образца, взятых во время своего планового визита. Один образец будет взят по прибытии в исследовательский центр, а другой образец будет взят на 1-1,5 часа позже, непосредственно перед тем, как покинуть исследовательский центр. Время приема дозы предыдущим вечером (перед сном предыдущей ночью) и время утреннего приема дозы (в день запланированного визита) будет регистрироваться. Времена фактического отбора ФК образцов также будут задокументированы. Исследовательскими центрами будет приветствоваться варьирование визитов в исследовательские центры в течение дня. Благодаря вариабельности времени визитов в исследовательские центры процесс взятия образцов для выборочного ФК контроля обычно будет занимать большую часть 12-часового периода после приема дозы.
Продолжительность исследования: в этом исследовании ожидаемый период набора в группы составляет приблизительно 12-18 месяцев. Субъекты, которые зачислены в исследование и выполнят его (Часть 1, Часть 2 и Часть 3), будут участвовать в исследовании до 14 месяцев включительно.
Исследуемая популяция: в данном исследовании используют регулируемый объем выборки, в диапазоне 160-320 взрослых субъектов с диагнозом ЕоЕ, которые удовлетворяют критериям отбора. Ожидается, что в данном исследовании примут участие приблизительно 25% женщин и 5% пожилых субъектов.
Критерии включения
Субъекты мужского и женского пола в возрасте 14 лет и старше до 75 лет на момент информированного согласия.
Наличие подписанной формы информированного согласия (ICF) (там, где это приемлемо, должен подписать родитель или опекун) и формы согласия несовершеннолетнего пациента (требуемой для подростков в возрасте младше 18 лет или возрасте, установленном в соответствии с местным законодательством) и желание и возможность соблюдать все процедуры исследования.
Поставленный или предположительный диагноз ЕоЕ, который должен быть подтвержден наличием симптомов и данными гистологического исследования; и документом, подтверждающим предыдущее неудачное лечение в течение 8 недель или более ингибитором протонного насоса (PPI) в высоких дозах (т.е. 20 мг омепразола, BID, или 20-40 мг, BID, одобренного к применению предписанного PPI. Поддерживающие дозы или дозы продаваемого без рецепта (ОТС) лекарственного средства являются слишком малыми). Подтвержденное документами неудачное лечение с применением PPI до первоначальной диагностики или подтвержденное документами неудачное лечение с применением PPI на момент скрининга. Отсутствие ответа на PPI терапию определяется как максимальное количество эозинофилов/HPF, составляющее 15 и более по меньшей мере в одном подвергнутом биопсии месте, через 8 недель лечения с использованием высокой дозы. Во время этого исследования PPI терапия может быть продолжена, только если субъект получал PPI терапию в момент отбора биоптатов при проведении эндоскопического скринингового обследования. Субъекты, принимающие PPI, могут либо прекратить прием после проведения эндоскопии, либо уменьшить дозу до той, которая была перед начальным периодом. После такого изменения дозы субъекты должны оставаться принимать эту постоянную дозу. После прекращения приема PPI он не должен быть возобновлен. В ходе исследования не допускается начала новой PPI терапии.
Наличие признаков эозинофильного эзофагита, определяемого по максимальному количеству эозинофилов в слизистой оболочке пищевода, составляющему 15 и более в поле зрения микроскопа с большим увеличением (HPF, увеличение 400х (0,3 мм2), по меньшей мере в 1 из подвергнутых биопсии мест пищевода (по результатам 3 и более биопсий из каждой проксимальной и дистальной частей пищевода; в целом как минимум 6 биопсий). Дополнительные биоптаты из средней части пищевода могут быть отобраны по усмотрению исследователя. Необходимо провести процедуры биопсии, и биоптаты должны быть получены главным специалистом по лабораторной диагностике в течение 30 дней после скринингового визита или за 2 недели до рандомизации и перед начальным периодом. Пригодность будет основываться исключительно на заключении главного специалиста по лабораторной диагностике. Могут быть отобраны «необязательные» биоптаты и обработаны на месте для местного использования, если это оговорено в принятой в данном месте ICF. Субъекты должны иметь 7-дневный общий показатель ЕоЕ более 3; субъекты должны сообщать по меньшей мере о 3 эпизодах дисфагии за неделю для каждой недели при оценке исходных симптомов; должны иметь желание и возможность соблюдать связанные с исследованием схемы лечения, процедуры и график визитов.
Критерии исключения: известное противопоказание, гиперчувствительность к кортикостероидам или их непереносимость. Любое физическое, психическое или социальное состояние, история болезни или отклонения лабораторных показателей от нормы, которые по мнению исследователя могут помешать проведению процедур исследования или способности субъекта присоединиться к исследованию и участвовать в нем. Наличие любого типа инфекции полости рта или слизистой оболочки пищевода. Любое состояние полости рта или зубов, мешающее нормальному приему пищи. Любое состояние, затрагивающее слизистую оболочку пищевода или изменяющее моторику пищевода, кроме ЕоЕ, включая эрозивный эзофагит (степени В согласно LA (Лос-Анджелесская классификация) или выше), грыжу пищеводного отверстия длиной более 3 см, пищевод Барретта и ахалазию. Применение системных, пероральных или парентеральных кортикостероидов в течение 30 суток либо ингаляционное или продолжительное применение высокоэффективных вводимых интрадермальной инъекцией кортикостероидов для местного применения в течение 30 суток перед: биопсией пищевода, необходимой для зачисления в это исследование; EGD, если проводится в период до скрининга. Применение стероидов для проглатывания/местного применения в течение 30 дней. Начало элиминационной диеты или элементной диеты в течение 30 дней скрининга. Во время этого исследования нельзя начинать никакой диетотерапии. Утренний (с 07:00 до 08:00 часов или по возможности ближе к этому интервалу) уровень кортизола в сыворотке крови 5 мкг/дл (138 нмоль/л) или менее. Уровень кортизола в сыворотке крови менее 18 мкг/мл (497 нмоль/л) через 60 минут после теста стимуляции адренокортикотропным гормоном (АСТН) с использованием 250 мкг косинтропина. Потребление грейпфрутового сока во время лечения запрещено. Применение биологических иммуномодуляторов в течение последних 6 месяцев. Применение ингибиторов кальциневрина, аналогов пуринов (азатиоприна, 6-меркаптопурина) в течение последних 3 месяцев. Противопоказание к эзофагогастродуоденоскопии (EGD) или биопсии пищевода либо сужение пищевода, препятствующее проведению EGD со стандартным 9-мм эндоскопом. Открытое желудочно-кишечное кровотечение в пределах 1 месяца до скринингового визита или между скрининговым визитом и визитом для рандомизации. Текущая хроническая инфекция, иммуносупрессия или иммунодефицит. Анамнез или наличие болезни Крона, глютеновой болезни или другого воспалительного заболевания желудочно-кишечного тракта, включая эозинофильный гастроэнтерит. Имеющееся злоупотребление алкоголем или наркотиками. Беременные или кормящие грудью субъекты женского пола. Живущие половой жизнью женщины репродуктивного возраста, которые не соглашаются использовать высокоэффективные методы контрацепции во время исследования, включая период последующего наблюдения. Для подростков допускается воздержание. Для субъектов женского пола с хирургической менопаузой или менопаузой, подтвержденной содержанием FSH, нет необходимости в контрацепции или тестировании определения беременности во время данного исследования. Участие в клиническом исследовании с вовлечением какого-либо другого экспериментального лекарственного средства в течение 30 суток скринингового визита.
Введение исследуемого лекарственного средства: для 3-х получающих дозы групп АРТ-1011 (флутиказона пропионат, ODT) будут представлены в виде замаскированных таблеток в дозировках 1,5 мг и 3,0 мг. Для четвертой получающей дозу группы это будет соответствующее плацебо. Таблетки будут введены в режимах BID - через 30 минут после завтрака и HS (перед сном) по меньшей мере через 2 часа после вечернего визита пищи. Обратите внимание, что в группе, получающей дозу 1,5 мг, субъекты будут получать таблетки с плацебо через 30 минут ПОСЛЕ завтрака и 1,5 мг АРТ-1011 HS. Для поддержания условий слепого исследования все таблетки будут маркированы для введения «после завтрака» и «перед сном». Субъекты не должны принимать пищу или питье в течение 1 часа после введения исследуемого лекарственного средства. Дозу для приема перед сном следует вводить после употребления всех напитков, перекусов, чистки зубов и непосредственно перед сном.
ПЕРВИЧНЫЕ КОНЕЧНЫЕ ТОЧКИ ЭФФЕКТИВНОСТИ на 12-й неделе
Имеются две сопервичные конечные точки эффективности для оценки ЕоЕ-ответа: максимальное количество эозинофилов/HPF не более 6 во всех подвергнутых биопсии местах пищевода и изменение по сравнению с исходными данными числа эпизоды дисфагии в течение предществующих 14 суток.
ВТОРИЧНЫЕ КОНЕЧНЫЕ ТОЧКИ ЭФФЕКТИВНОСТИ
Изменение по сравнению с исходными данными в числе дней без дисфагии в течение предшествующих 14 суток; устойчивый ЕоЕ-ответ, который будет оцениваться на 26-й и 52-й неделях на основании сопервичной конечной точки; изменение по сравнению с исходными данными в 7-дневном общем показателе ЕоЕ, который будет определен до оценки исходных симптомов, в исходном состоянии, на 12-й неделе, 26-й неделе и 52-й неделе. 7-Дневный общий показатель симптомов ЕоЕ будет оцениваться при каждом визите. Изменение по сравнению с исходными данными показателя EREFS на 12-й, 26-й и 52-й неделях. Изменения по данным эндоскопии будут основаны на показателе EREFS, учитывающем 5 эндоскопических признаков: отек, образование борозд, экссудаты, пищеводные кольца, стриктуры. Процентная доля субъектов с максимальным количеством Eos в HPF менее 1 и менее 15 для всех основных моментов времени, когда оценивается ЕоЕ-ответ. Изменение по сравнению с исходными данными в 7-дневном показателе EEsAI, и процентная доля субъектов со средним еженедельным показателем EEsAI менее 20, и изменение по сравнению с исходившими субпоказателями будут оцениваться на 12-й, 26-й и 52-й неделях. 7-Дневный EEsAI будет оцениваться при каждом визите.
Оценка рецидива и неудачного результата лечения
(а) Отсутствие ответа по данным эндоскопии в любой момент времени.
(б) Процентная доля субъектов, нуждающихся в неотложном эндоскопическом освобождении от пищи в зависимости от дозы и Части исследования.
(в) Процентная доля субъектов, нуждающихся в дилатации пищевода.
OL лечение: ЕоЕ-ответ (сопервичный, см. выше) также будет оцениваться через 12 недель OL лечения у нереспондеров из Части 1.
ФАРМАКОКИНЕТИЧЕСКИЕ КОНЕЧНЫЕ ТОЧКИ
Приведенные далее ФК параметры будут рассчитаны для субъектов, проходящих многократный ФК контроль, с использованием некомпартментных методов, если позволяют данные: Cmax, Tmax, AUC0-12, CLss/F (кажущийся клиренс в стационарном состоянии) и коэффициент накопления.
Популяционные фармакокинетические параметры, включая клиренс (CL/F) и объем распределения (V/F) при пероральном приеме, будут оценены, если позволяют данные. Дополнительные параметры при необходимости будут оценены на основании окончательной структурной фармакокинетической модели.
ЭКСПЛОРАТИВНЫЕ КОНЕЧНЫЕ ТОЧКИ
Показатель ЕоЕ по гистологической бальной системе для всех проведенных биопсий. Будет оценено изменение по сравнению с исходными данными. Оценка качества жизни на основании EoE-QoL-A на 12-й, 26-й, 52-й неделе и 12-й неделе OL лечения в зависимости от дозы и подгруппы. Оценка пациентом симптомов по сравнению с предыдущим визитом. Этот вопрос будет оцениваться на 8-й, 12-й, 18-й, 26-й, 36-й и 52-й неделях. Оценка ФК/ФД (для кортизола) и взаимосвязей воздействие-ответ (эффективности).
Конечные точки безопасности: оценка безопасности будет проведена посредством мониторинга и регистрации всех возникающих при лечении нежелательных явлений (ТЕАЕ), ТЕАЕ, приводящих к отмене лечения, и серьезных нежелательных явлений (SAE). Все ТЕАЕ получат код на основании классификации MedDRA, версии 14.0, для нежелательных явлений (АЕ) и будут классифицированы исследователем по тяжести (слабое, умеренное, тяжелое) и связи с исследуемым лекарственным средством (по типу связь есть или связи нет). ТЕАЕ, возникающие в течение 3 суток после изменения дозы, будут отнесены к предыдущей дозе. Для документирования исходного состояния субъекта и выявления связанных с АЕ изменений будут проведены физические обследования. При всех визитах также будут оценены жизненно важные признаки и будет сообщено о клинически значимых отклонениях.
На протяжении всего исследования будут проводиться стандартные лабораторные анализы, включая общий анализ крови, биохимический анализ крови, анализ мочи, снятие электрокардиограмм (ЭКГ), как указано в графиках обследований. Клинически значимые изменения в результатах лабораторных анализов или ЭКГ будут обобщены.
Проблемы с кортизолом: после получения субъектом разрешения неизбежно могут обнаруживаться аномальные AM уровни кортизола, глюкозы в моче или глюкозы в сыворотке крови. В CRF должно фиксироваться наличие или отсутствие известных связанных с глюкокортикоидами АЕ, таких как лунообразное лицо, акне, гирсутизм, перепады настроения, бессонница или депрессия. В ICF должно обращаться особое внимание на то, что во время серьезных медицинских заболеваний может потребоваться применение стероидных гормонов стресса. Тест стимуляции АСТН (250 мкг косинтропина) должен быть выполнен для всех субъектов в начальный момент времени и на 12-й неделе, в момент преждевременной отмены и на 52-й неделе. АСТН тест должен быть выполнен для всех субъектов с AM концентрацией кортизола в сыворотке крови выше 5 мкг/дл (138 ммоль/л). Все субъекты с положительными результатами теста должны быть исключены. Любые положительные результаты к моменту окончания лечения должны сопровождаться восстановлением функции надпочечников. Число субъектов, прерывающих лечение по причине угнетения НРА оси или положительных результатов тестов стимуляции АСТН, будет обобщено. Субъекты в возрасте до 18 лет будут оцениваться по таким ростовым параметрам, как рост, вес, индекс массы тела (BMI) и соответствующие z-показатели. Также будут определены стадия Таннера и возраст по кости у подростков в возрасте младше 18 лет, которые не перестали прибавлять в росте.
Представляющими интерес конечными точками безопасности являются: частота возникающих при лечении нежелательных явлений (ТЕАЕ), ТЕАЕ, приводящие к отмене лечения и возникающие при лечении серьезные нежелательные явления (SAE), а также процентная доля субъектов с уровнем кортизола в сыворотке крови 5 мкг/дл или менее (138 нмоль/л или менее) или положительным результатом теста стимуляции АСТН (концентрация кортизола в сыворотке крови менее 18 мкг/мл (497 нмоль/л) через 60 минут). Число субъектов, прерывающих лечение по причине угнетения НРА оси, будет зарегистрировано.
Статистические методы
Определение объема выборки
Часть 1 включает регулируемый объем выборки в диапазоне от 160 до 320 взрослых пациентов. После проведения рандомизации по меньшей мере для 160 субъектов будут проведены многократные промежуточные анализы, в которых оцениваются критерии успеха и бесполезности. Если доказательства эффективности лечения по сравнению с сопервичными результатами недостаточны, данное исследование может быть остановлено преждевременно по причине бесполезности. Если прогностическая вероятность получения достаточных доказательств для демонстрации эффективности лечения очень высока, в данном исследовании можно остановить набор испытуемых в ожидании успеха, при этом решающий анализ выполняется, когда все включенные в исследование пациенты завершат 12 недель периода последующего наблюдения (Broglio, 2014). Для расчета ожидаемого объема выборки, ошибки 1-го типа и статистической мощности при различных предполагаемых профилях лечения (которые будут представлены в SAP) используется численное моделирование.
Ожидается, что приблизительно 60% субъектов будут зачислены в Часть 2 данного исследования. С учетом минимум 102 пациентов, принимающих участие в лечении активным средством в Части 1, ожидается, что по меньшей мере 61 пациент будет зачислен в Часть 2. Ожидается, что приблизительно 17 плацебо-респондеров будут зачислены в Часть 2. Ожидается, что 70% плацебо-респондеров и приблизительно 20% пациентов, принимающих участие в лечении активным средством, могут давать рецидив до 26-й недели. Ожидается, что приблизительно 82 субъекта, принимающих участие в лечении активным средством, будут зачислены в Часть 3.
В дополнение к этому, при условии коэффициента отсева, составляющего 20% в Части 3, ожидается, что по меньшей мере 66 субъектов (22 из расчета на одну дозу) будут оцениваться в группах с лечением активным средством на устойчивую ремиссию на 52-й неделе, и по меньшей мере 4 пациента ожидается в контрольной группе. Это обеспечивает минимальную ожидаемую эффективность 73% в случае сравнения степеней устойчивой ремиссии, составляющих 80%, против с 20% для каждой дозы активного средства относительно контроля, и минимальную ожидаемую эффективность 80% в случае сравнения степеней устойчивой ремиссии для объединенного лечения активным средством относительно контроля. Однако, основная направленность Части 3 будет носить описательный характер.
Статистическая методология
Сводные данные по статистике будут представлены в табличной форме для получающей дозу группы и подгрупп, соответственно, или для каждой Части данного исследования, включая OL лечение.
Первичной анализируемой в отношении эффективности популяцией является предназначенная для лечения (ITT) популяция, определяемая как «все рандомизированные субъекты». Анализируемой в отношении безопасности популяцией является популяция для оценки безопасности, определяемая как «все субъекты, которые получают по меньшей мере одну дозу исследуемого лекарственного средства». Популяция для изучения ФК разовой дозы будет определена как «все субъекты в ITT популяции из Части 1, которые рандомизированы с попаданием в одну трех получающих АРТ-1011 групп, принимают свою первую дозу АРТ-1011 и имеют по меньшей мере один ФК образец, включенный в завершающий популяционный ФК анализ разовой дозы. Популяция для изучения стационарной ФК, будет определена как «все субъекты в ITT популяции из Части 1, которые рандомизированы с попаданием в одну трех получающих АРТ-1011 групп, принимают дозу АРТ-1011, соответствующую периоду ФК контроля в стационарном состоянии, и имеют по меньшей мере один ФК образец, включенный в завершающий популяционный ФК анализ в стационарном состоянии». В дополнение к анализу по протоколу популяции, которые завершают Части 1, 2 и 3 и OL лечение, могут быть оценены согласно Плану статистического анализа (SAP).
Исходные и демографические данные будут обобщены с использованием описательной статистики для непрерывных и порядковых переменных (например, возраста, веса, роста) и количеств и процента для категориальных переменных (например, пола, расовой принадлежности).
Анализ фармакокинетики и фармакодинамики
В этом исследовании дозы 1,5 мг BID и 3,0 мг BID будут введены по меньшей мере через 30 минут после завтрака и перед сном (по меньшей мере через 2 часа после вечернего визита пищи). Доза HS 1,5 мг будет введена перед сном (по меньшей мере через 2 часа после вечернего визита пищи). Субъекты, получающие 1,5 мг HS один раз в сутки, также будут получать плацебо по меньшей мере через 30 минут после завтрака.
С использованием ФК данных от субъектов с многократным отбором ФК образцов будут рассчитаны параметры для FP с применением, где это возможно, некомпартментных методов, как приведено ниже.
Cmax: максимальная наблюдаемая концентрация, определяемая посредством анализа графиков зависимости концентрации в плазме крови от времени у отдельных участников исследования.
Tmax: время достижения максимальной наблюдаемой концентрации, определяемое непосредственно из данных зависимости наблюдаемой концентрации от времени.
AUC0-12: площадь под кривой зависимости концентрации в плазме крови от времени в интервале от 0 до 12 часов после приема дозы, рассчитанная с использованием комбинации линейного и логарифмического методов трапеций (до максимальной концентрации метод трапеций применяется к самой функции, после максимальной - к ее логарифму).
CLss/F: кажущийся клиренс в стационарном состоянии.
Коэффициент накопления: коэффициент накопления, рассчитанный из AUC0-12 в стационарном состоянии и AUC0-12 после однократного приема дозы.
Будут получены индивидуальные и усредненные кривые зависимости концентрации в плазме крови от времени (как в линейном, так и полулогарифмическом масштабе). Подробная методология, касающаяся сводной статистики для данных по концентрации и ФК параметров, будет задокументирована в SAP.
Популяционный ФК анализ будет проведен на основе комбинации данных о концентрации в плазме крови для серийного и выборочного вариантов анализа. Предыдущие серийные ФК данные для APT-1011 могут быть включены в этот анализ для облегчения разработки основной ФК модели.
Популяционные ФК параметры, включая клиренс (CL/F) и объем распределения (V/F) при пероральном приеме, будут оценены, если позволяют данные. Дополнительные параметры при необходимости будут оценены на основании окончательной структурной ФК модели.
На основе окончательных популяционных моделей будут получены выходные данные, включая соответствующие диагностические графики, списки и сводные данные по ФК параметрам. В дополнение к этому будут подготовлены презентации в виде графиков и таблиц для всех результатов численного моделирования ФК. Будут создан отдельный отчет по PopPK и объединен с отчетом о клиническом исследовании.
Если позволяют данные, также могут быть выполнены эксплоративные анализы, в которых оценивается взаимосвязь между системным воздействием FP и изменениями в уровнях кортизола, как описано выше. Для облегчения выбора безопасных и эффективных доз для будущих исследований и клинического применения могут быть проведены дополнительные эксплоративные анализы взаимосвязи воздействие-ответ на основании сопервичной конечной точки или ее компонентов.
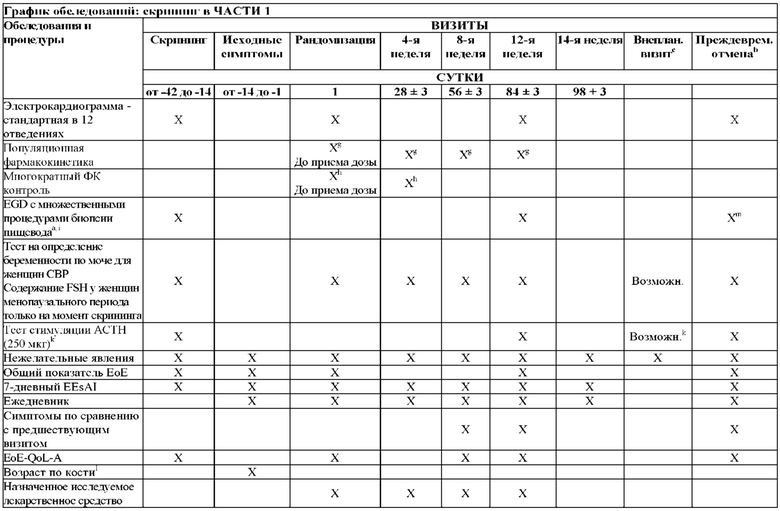
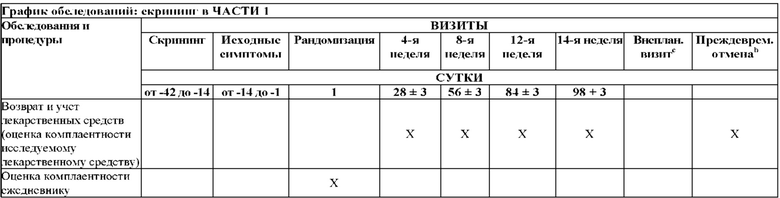
а Эндоскопия может быть проведена во время отдельного визита. Результаты гистологического анализа потребуются при рандомизации и на 14-й неделе.
b Пациент должен появиться в кабинете не позднее 7 дней с момента определения необходимости отмены лечения для данного пациента. Если это невозможно сделать ввиду SAE или другого непредвиденного обстоятельства, визит может быть совершен пациентом или членом его семьи по телефону. Документальное подтверждение того, почему пациент не смог прийти в исследовательский центр, должно быть внесено в историю болезни пациента.
с Причина для внепланового визита будет формировать процедуры по усмотрению PI.
d Подтверждение того, что пациент все еще удовлетворяет критериям включения/исключения на основании ежедневного показателя EEsAI в дополнение к другим критериям.
е Отборы образцов необходимо проводить как можно ближе по времени к 08:00. Пациенты не должны принимать пищу в течение восьми(8)-часового периода времени до проведения оценок уровня кортизола в сыворотке крови. Кровь для анализа AM уровня кортизола в сыворотке крови может быть взята в пределах +/- 2 дней от планового визита, чтобы соответствовать точному временному графику. Отбор крови для проведения других анализов, запланированных на время данного визита, может быть выполнен одновременно.
f Если во время заключительного визита лечения отмечается аномальный уровень кортизола в сыворотке крови, то может потребоваться проведение дополнительного мониторинга и АСТН теста. См. раздел «Безопасность».
g Отбор образцов для выборочного ФК контроля будет проведен среди субъектов за исключением субъектов из подгруппы многократного ФК контроля. Исходный образец «до приема дозы» будет отобран перед приемом первой дозы в 1-е сутки. На 4-й, 8-й и 12-й неделях повторного введения доз субъекты будут принимать свою утреннюю дозу дома и будут иметь два образца, взятых во время своего планового визита. Один образец будет взят по прибытии в исследовательский центр, а другой образец будет взят на 1-1,5 часа позже, непосредственно перед тем, как покинуть исследовательский центр. Время введения дозы предыдущим вечером (перед сном предыдущей ночью) и время утреннего введения дозы (в день запланированного визита) будет регистрироваться. Времена фактического отбора ФК образцов также будут задокументированы.
h Приблизительно по 8 взрослых субъектов и 5 подростков из расчета на одну получающую дозу группу будут подвергаться многократному ФК контролю в определенных исследовательских центрах. После получения обеих доз (таблетки AM и HS), многократные ФК образцы будут отобраны в 1-е сутки и на 4-й неделе. Во время каждого из этих визитов ФК образцы будут отобраны до введения дозы (не ранее, чем за 15 минут до введения AM дозы) и через 0, 5, 1, 2, 4, 8 и 12 часов после введения AM дозы. ФК образцы, запланированные на 4-ю неделю, могут быть отобраны во время визита на 8-й неделе или 12-й неделе, если субъект не может оставаться в течение 12 часов на 4-й неделе.
i Может быть проведена за 30 суток до скринингового визита (при отсутствии приема системных или ингалируемых, интраназальных или обладающих высокой эффективностью вводимых интрадермальной инъекцией кортикостероидов для местного применения в течение этих 30 суток до скринингового визита). Должен быть определен EREFS и взяты 3 биоптата по меньшей мере на 2-х уровнях пищевода.
j Если пациент покидает исследование ввиду отсутствия эффективности или по другим причинам, исследователь может провести EGD, если к этому есть клинические показания.
k Все субъекты проходят тест стимуляции АСТН (250 мкг) в начальный момент времени и при EOT или в момент преждевременной отмены лечения. Тест стимуляции АСТН также будет проведен во время исследования при «необязательном» визите для субъектов, уровень кортизола в сыворотке крови которых подтверждается двумя отборами крови, как составляющий 5 мкг/дл (138 нмоль/л) или менее, или если у них имеются признаки и симптомы гиперкортицизма, для оценки угнетения гипоталамо-гипофизарно-надпочечниковой (HPA) оси, вызывающего клиническую озабоченность.
l Для всех подростков (в возрасте<18 лет на момент рандомизации) должно быть выполнено определение возраста по кости; исключение составляют субъекты женского пола, которые перестали прибавлять в росте, или, по усмотрению PI, субъекты мужского пола, которые не перестали прибавлять в росте к 18 годам.
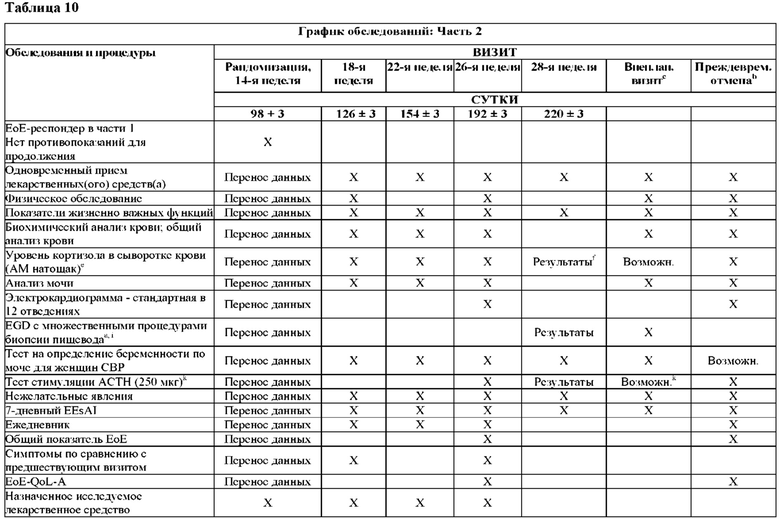

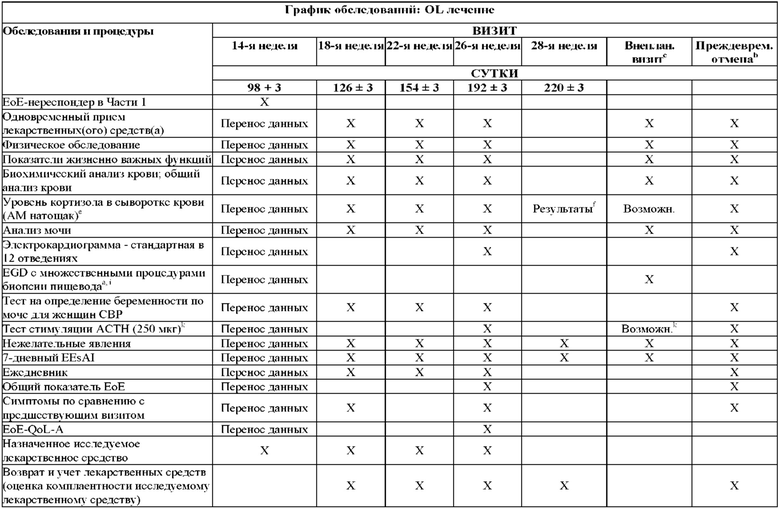
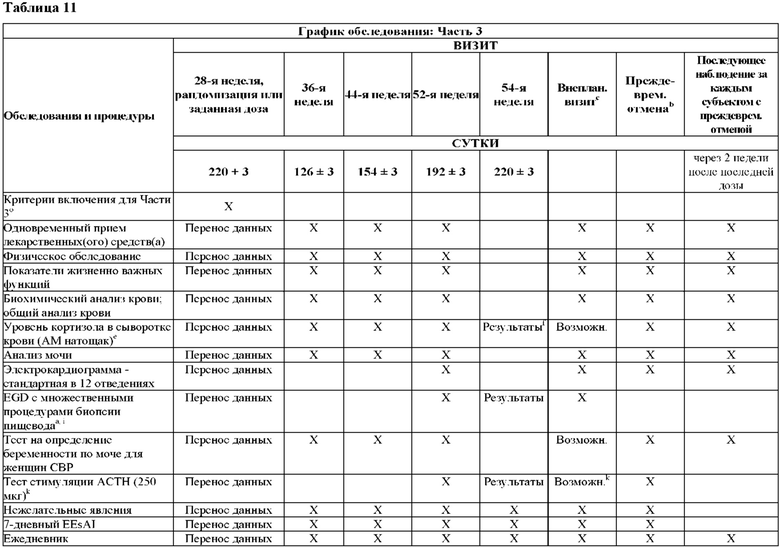
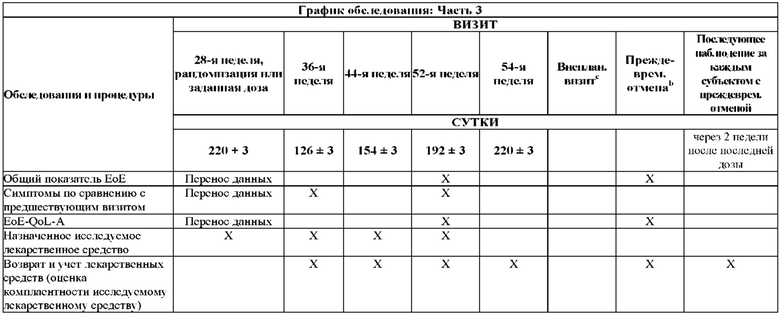
Сноски a-m - см. предыдущие сноски.
°. Субъекты удовлетворяют критериям включения для Части 3:
Завершение Части 2 в качестве респондера - продолжение с применением той же дозы; завершение Части 2 в качестве нереспондера - назначение дозы 3 мг, BID.
Рецидив в случае приема плацебо - возврат к предшествующей дозе из Части 1 при зачислении в Часть 3; рецидив в Части 2 в случае приема плацебо - назначение дозы 3 мг, BID, в Части 3.
Завершение OL лечения в качестве респондера - продолжение с применением дозы 3 мг, BID.
Пример 4. Флутиказон при эозинофилъном эзофагите (FLUTE): Рандомизированное двойное слепое, плацебо-контролируемое исследование по подбору и поддержанию дозы АРТ-1011 на субъектах с эозинофильным эзофагитом
Первичная задача: первичная задача данного исследования заключается в оценке эффективности (гистологического ответа) АРТ-1011 у взрослых (в возрасте 18 лет и старше) с эозинофильным эзофагитом (ЕоЕ).
Вторичные задачи: вторичные задачи данного исследования приведены ниже: определить зависимость ответа от дозы АРТ-1011; выбрать дозу(ы) АРТ-1011 для фазы 3; оценить влияние АРТ-1011 на гистологические и выявленные при эндоскопическом обследовании визуальные характеристики; оценить поддержание эффективности и долгосрочную безопасность АРТ-1011; оценить популяционную фармакокинетику (PopPK) АРТ-1011; оценить влияние АРТ-1011 на эпизоды дисфагии.
Эксплоративные задачи: эксплоративные задачи данного исследования приведены ниже;
оценить влияние АРТ-1011 на дисфагию и другие симптомы ЕоЕ; оценить качество жизни; оценить симптоматику во времени; оценить фармакокинетические (ФК)/фармакодинамические (ФД) взаимосвязи (с использованием кортизола, в качестве первичной конечной точки); оценить зависимость ответа от дозы для гистологического ответа и ответа в виде проявления симптомов; разработать схему выставления балльных оценок и различные конечные точки, исходя из анкеты с результатами лечения симптомов ЕоЕ по оценке пациента (PROSE); оценить возможность количественного представления данных PROSE, включая надежность, конструктивную валидность, чувствительность к изменению; разработать руководящие указания для интерпретации клинически значимого изменения и определить порог ответа на лечение исходя из PROSE или определения респондера.
ПЛАН ИССЛЕДОВАНИЯ
Это рандомизированное двойное слепое, плацебо-контролируемое исследование по подбору дозы для 4-х общесуточных доз APT-1011 в сравнении с приемом плацебо на 100 взрослых субъектах (в возрасте 18 лет и старше) с диагнозом ЕоЕ.
Во время простой слепой вводной фазы/оценки исходных симптомов субъекты будут получать плацебо через 30 минут после завтрака и hora somni (HS; перед сном). Будут введены четыре дозы исследуемого лекарственного средства: плацебо через 30 минут после завтрака и 1,5 мг АРТ-1011 hora somni (HS; перед сном); 1,5 мг АРТ-1011 два раза в сутки (BID) (через 30 минут после завтрака и перед сном; общесуточная доза 3 мг); плацебо через 30 минут после завтрака и АРТ-1011 в дозе 3 мг HS (перед сном) и 3 мг BID (через 30 минут после завтрака и перед сном; общесуточная доза 6 мг), и соответствующая доза плацебо через 30 минут после завтрака и HS (перед сном).
100 субъектов будут рандомизированы в соотношении 1:1:1:1:1 для получения плацебо или одной из доз активного соединения в Части 1 данного исследования. Как описано ниже, лечение, которое субъект получает в Части 2, зависит от статуса его гистологического ответа на 12-й неделе.
Рандомизация будет проведена двойным слепым способом с использованием единой интерактивной системы веб-ответа (IWRS), и пациенты будут разделены согласно наличию или отсутствию ранее стриктур пищевода либо имеющимся на момент скрининга стриктурам пищевода и согласно имеющемуся ранее положительному стероидному ответу на любое лечение кортикостероидами, ранее полученному при лечении ЕоЕ у субъекта с зарегистрированными демографическими данными.
Будут исследованы эффективность (включая устойчивый ЕоЕ-ответ и сообщаемые пациентом результаты лечения), безопасность и ФК параметры для АРТ-1011.
Анализ FLUTE будет проводиться в несколько этапов (скрининг (в течение 4 недель), затем 4-недельная простая слепая вводная фаза исследования с применением плацебо и оценка исходных симптомов и состоящее из 2-х Частей лечение (Части 1 и Части 2)) с визитом в рамках последующего наблюдения, который должен состояться через 2 недели после приема заключительной дозы исследуемого лекарственного средства. Скрининг займет до 28 дней включительно. Эзофагогастродуоденоскопия (EGD) для определения пригодности (критерий включения №7: признаки ЕоЕ, определяемого по максимальному количеству эозинофилов/поле зрения микроскопа под большим увеличением (HPF) 15 или более по результатам по меньшей мере 5-6 биопсий, включая образцы как из проксимальной, так и из дистальной части (по 3 каждого)) будет проведена во время периода скрининга, и биоптаты должны быть получены главным специалистом по лабораторной диагностике в сроки, указанные в этом критерии включения. Чтобы быть зачисленными в 4-недельный период оценки исходных симптомов, субъект должен удовлетворять всем критериям пригодности, включая общий показатель ЕоЕ боле 3 (критерий включения №5), кроме тех, которые должны получить подтверждение во время этой фазы (критерий включения №7: признаки ЕоЕ, определяемого по максимальному количеству эозинофилов/HPF 15 или более по результатам по меньшей мере 5-6 биопсий, включая биоптаты как из проксимальной, так и из дистальной части пищевода (по 3 каждого); критерий включения №8: наличие в ежедневнике сообщения об эпизодах дисфагии на протяжении 3 иоли более дней в неделю в течение последних 2-х недель 4-недельного периода оценки исходных симптомов; критерий включения №9: оформление записей относительно случаев эпизодов в течение по меньшей мере 5 дней из каждых 7 дней во время 4-недельного периода оценки исходных симптомов; и критерий исключения №23: уровень кортизола в сыворотке крови менее 18 мкг/дл (497 нмоль/л) через 60 минут после теста стимуляции адренокортикотропным гормоном (АСТН) с использованием 250 мкг косинтропина (т.е. в случае положительного результата теста стимуляции АСТН)). Субъекты будут получать плацебо в соответствии с их электронным дневником в начале 4-недельной вводной фазы исследования с применением плацебо/оценки исходных симптомов.
Во время 4-недельного периода оценки исходных симптомов будет проведено определение тяжести исходных симптомов и будет оценена способность субъекта следовать записям в дневнике. Субъекты должны иметь максимальное количество эозинофилов/HPF 15 или более по их результатам биопсии пищевода, чтобы быть рандомизированы. Чтобы гарантировать постановку диагноза, необходимо провести по меньшей мере 5-6 биопсий как проксимальной, так и дистальной частей пищевода (по 3 каждой). Наличие или отсутствие анамнеза стриктуры пищевода либо имеющаяся в настоящий момент стриктура пищевода по данным EGD, наряду с наличием в анамнезе положительного ответа на любое полученное лечение кортикостероидами для лечения ЕоЕ у субъекта с зарегистрированными демографическими данными, будут являться стратификационными переменными при рандомизации.
После подтверждения этих критериев пригодности подходящие субъекты могут быть рандомизированы так, как описано выше.
Во время лечения все субъекты будут повторно посещать исследовательский центр приблизительно один раз в месяц для плановых визитов и для внеплановых визитов при наличии значительных нежелательных явлений или при ухудшении симптомов, включая затрудненное прохождение пищи.
Определения гистологического ответа, отсутствия гистологического ответа и неудачного результата лечения
Статус респондера или нереспондера будет оцениваться за 2 недели до планируемого окончания лечения для Части 1 (на 12-й неделе) и Части 2 (на 26-й и 52-й неделях).
Гистологически обусловленный респондер будет определен как субъект, который достигает гистологического ответа в показателях максимального количества эозинофилов/HPF 6 или менее (в качестве первичной детерминанты). HPF будет определена как стандартная площадь, составляющая 0,235 квадратных миллиметров в микроскопе с объективом 40х и окуляром 22 мм.
Гистологически обусловленный нереспондер будет определен как субъект, который не дает гистологического ответа (т.е. который не достигает гистологического ответа в показателях максимального количества эозинофилов/HPF 6 или менее).
Субъекты, у которых развивается затрудненное прохождение пищи, требующее или не требующее дилатации пищевода в любой момент во время данного исследования, будут считаться пациентами с неудачным результатом лечения, для них будут проведены оценки на предмет преждевременного завершения, и они покинут исследование по окончании 2-недельного проводимого после курса лечения периода последующего наблюдения. Субъекты, которые добровольно покидают исследование ввиду ухудшения симптомов перед проведением оценки на 12-й неделе или позже в этом исследовании, также будут считаться пациентами с неудачным результатом лечения. Необходимо приложить все усилия для проведения EGD у субъектов, желающих покинуть исследование ввиду ухудшения симптомов. Кроме того, для них должны быть проведены оценки на предмет преждевременного завершения, и они должны покинуть исследование по окончании 2-недельного проводимого после курса лечения периода последующего наблюдения.
Часть 1. Фаза индукции (с 1-х суток по 14-ю неделю)
Во время Части 1 субъекты будут проходить лечение в течение 14 недель с применением исследуемого лекарственного средства (Фиг. 5). На 12-й неделе у субъектов будет выполняться оценка ответа на лечение, включая EGD для оценки эндоскопического и гистологического статуса.
Гистологически обусловленные респондеры и гистологически обусловленные нереспондеры (на 12-й неделе) будут зачислены в Часть 2.
Часть 2. Поддерживающая фаза (с 14-й недели по 52-ю неделю)
В Части 2 все субъекты, классифицированные как гистологически обусловленные респондеры на 12-й неделе, будут продолжать лечение согласно получающей дозу группе, в которую они были рандомизированы для Части 1, в течение еще 14 недель с началом на 14-й неделе (Фиг. 5). Субъекты могут продолжать принимать эту дозу в течение до 9 месяцев включительно после завершения Части 1.
Субъекты, являющиеся гистологически обусловленными нереспондерами на 12-й неделе, будут получать лечение в рамках простого слепого исследования 3 мг, BID, в Части 2.
На 26-й неделе у субъектов будет проведена оценка ответа на лечение, включая EGD для оценки гистологического ответа. Также будут оценены симптомы. Будет проведено сравнение состояния в течение 14 суток до 26-й недели и 14 суток до рандомизации. Субъекты, которые являются гистологически обусловленными нереспондерами, прекратят лечение на 28-й неделе, будут зачислены в 2-недельный период последующего наблюдения и покинут исследование. Гистологически обусловленные респондеры продолжат прием той же дозы.
Для субъектов, которые завершают лечение на 52-й неделе, будет выполнена оценка ответа на лечение, включая EGD для оценки эндоскопического и гистологического статуса. Также будут оценены симптомы. Будет проведено сравнение состояния в течение 14 суток до 52-й недели и 14 суток до рандомизации.
Субъекты будут совершать визит в рамках последующего наблюдения через 2 недели после получения заключительной дозы исследуемого лекарственного средства.
Визит в рамках последующего наблюдения
Субъекты будут совершать визит в рамках последующего наблюдения по одной или нескольким из указанных ниже причин: субъект завершил лечение на 52-й неделе (после проведения EGD); субъект испытал нежелательное явление (АЕ), требующее преждевременного прерывания лечения, включая затрудненное прохождение пищи, требующее проведения EGD; у субъекта отсутствует гистологический ответ на 26-й неделе, включая субъектов, получающих лечение в режиме простого слепого исследования 3 мг, BID; у субъекта имеется ухудшение симптомов, в результате чего он добровольно покидает исследование. Визит в рамках последующего наблюдения будет совершен через 2 недели после приема субъектом заключительной дозы исследуемого лекарственного средства. Все субъекты должны пройти заключительную EGD в течение 3 недель до выполнения визита в рамках последующего наблюдения, за исключением тех случаев, когда субъект аннулировал соглашение или имеет противопоказание к проведению EGD.
Фармакокинетика
Выборочный ФК контроль будет проведен для того, чтобы охарактеризовать воздействие флутиказона пропионата (FP) на исследуемую категорию пациентов. PopPK анализ будет проведен на основании данных по концентрации в плазме крови для выборочного варианта анализа.
Отбор фармакокинетических образцов будет осуществлен у субъектов во всех 5 получающих дозу группах с сохранением условий слепого исследования. Образцы, отобранные у субъектов получающей плацебо группы, анализироваться не будут. Образцы, отобранные у субъектов, получающих дозы активного соединения, будут проанализированы в отношении результатов PopPK.
Для проведения такого выборочного ФК контроля образец «до приема дозы» будет отобран в 1-е сутки. На 4-й неделе, 8-й неделе и 12-й неделе субъекты свою дозу «после завтрака» будут принимать в соответствии с графиком в день визита (скорее всего, дома), а 2 образца будут отобраны во время их планового визита: по прибытии в исследовательский центр и приблизительно через 1-1,5 часа после отбора первого образца (непосредственно перед тем, как покинуть исследовательский центр). Исследовательскими центрами будет приветствоваться регистрация визитов субъектов в течение суток. Благодаря такой вариабельности выборочные ФК образцы, как ожидается, будут отражать большую часть 12-часового интервала после введения дозы.
Планируемое количество субъектов
Приблизительно 100 субъектов будут рандомизированы в Части 1 в соотношении 1:1:1:1:1 со разделением согласно наличию или отсутствию ранее стриктур пищевода либо имеющимся в настоящий момент стриктурам пищевода и согласно имеющемуся ранее положительному стероидному ответу на любое лечение кортикостероидами, ранее полученному при лечении ЕоЕ у субъекта.
Несмотря на то, что к участию в исследовании будут рекомендованы субъекты обоих полов, ожидается, что приблизительно 25% субъектов, принявших участие в исследовании, будут составлять женщины. Хотя разрешенный возраст субъектов должен составлять до 75 лет включительно, ожидается, что 5% принявших участие в исследовании субъектов будут лицами пожилого и старческого возраста (65 лет и старше).
Никаких промежуточных анализов не планируется.
Критерии включения
Перед зачислением в 4-недельный период оценки исходных симптомов субъекты должны удовлетворять всем приведенным далее критериям.
Это должны быть мужчина или женщина в возрасте от 18 до 75 лет включительно в момент подписания информированного согласия; иметь подписанную форму информированного согласия (ICF) и желание и возможность соблюдать все процедуры исследования; иметь поставленный или предположительный диагноз ЕоЕ; диагноз ЕоЕ должен быть подтвержден симптомами, результатами гистологического исследования и документом, подтверждающим предыдущее неудачное лечение в течение 8 недель или более ингибиторами протонного насоса (PPI) в высоких дозах. Для целей FLUTE, в качестве PPI в высокой дозе используют 20 мг, BID, омепразола или от 20 до 40 мг, BID, любого поставляемого на рынок PPI; поддерживающие дозы PPI не приемлемы. Отсутствие ответа на PPI терапию определяют как максимальное количество эозинофилов/HPF 15 или более по результатам по меньшей мере 5 биопсий, в том числе в образцах, взятых как из проксимальной, так и дистальной частей пищевода, через 8 недель лечения с использованием PPI в высокой дозе.
До проведения первоначальной диагностики необходимо иметь подтвержденную документально информацию относительно неудачного применения PPI или подтвержденную документально информацию относительно неудачного применения PPI во время скрининга. Субъекты могут пройти предварительное скрининговое обследование, но не должны давать согласие, подписывать ICF или быть приглашенными для участия в FLUTE, если они не удовлетворяют диагностическим критериям в отношении ЕоЕ, для подтверждения чего необходимо, чтобы они потерпели неудачу в 8-недельном испытании с использованием PPI в высоких дозах, за исключением тех, которые принимали PPI в течение 8 недель с применением EGD в этом исследовании для этого документального подтверждения. Исследователь и возможный субъект должны принять решение о завершении испытания с использованием PPI независимо от любых связанных с данным исследованием соображений. В рамках настоящего исследования времени для проведения такого 8-недельного испытания недостаточно. Если субъект соглашается с ошибкой, и из-за этого имеет место неудача при скрининге, он может пройти повторный скрининг, как описано в протоколе. Субъекту необходимо иметь зафиксированную в сообщении историю 3 или более эпизодов дисфагии (затруднения при прохождении пищи) в течение 7 суток перед скринингом; иметь 7-дневный общий показатель симптомов ЕоЕ >3, взятый в качестве исходного уровня (показатель ЕоЕ должен оставаться >3 в каждый из визитов 1, 2 и 3 до рандомизации). Это будет зафиксировано «на бумаге» при прохождении скринингового визита; иметь желание и возможность соблюдать связанные с исследованием схемы лечения, процедуры и график визитов.
До рандомизации: до рандомизации следует определить: наличие признаков ЕоЕ, что определяется по максимальному количеству эозинофилов/HPF 15 или более по результатам по меньшей мере 5-6 биопсий, включая как проксимальные, так и дистальные образцы (по 3 каждого); никакие данные EGD и биопсий, выполненных вне FLUTE, не приемлемы в плане удовлетворения критериям пригодности. Могут быть отобраны «необязательные» биоптаты и обработаны на месте для местного использования, если это оговорено в принятой в данном месте ICF. Получение биоптатов должно предшествовать 4-недельному периоду оценки исходных симптомов. Пригодность с гистологической точки зрения будет основываться исключительно на заключении главного специалиста по лабораторной диагностике.
До рандомизации следует определить: наличие в ежедневнике сообщения по меньшей мере о 3 эпизодах дисфагии (затруднении с прохождением пищи) для каждого из последних 7 дней в течение последних 2 недель 4-недельного периода оценки исходных симптомов.
До рандомизации следует определить: наличие оформленных записей относительно случаев эпизодов в течение по меньшей мере 5 дней из каждых 7 дней в течение последних 14 дней 4-недельного периода оценки исходных симптомов.
Критерии исключения
Субъекты не будут зачислены в FLUTE по любой из приведенных далее причин.
Перед зачислением в 4-недельный период оценки исходных симптомов: имеют известное противопоказание, гиперчувствительность к кортикостероидам или их непереносимость; любое физическое, психическое или социальное состояние либо история болезни или отклонения лабораторных показателей от нормы, которые по мнению исследователя могут помешать проведению процедур исследования или способности субъекта присоединиться к исследованию и участвовать в нем; любого типа инфекции полости рта или слизистой оболочки пищевода; имеют любое состояние полости рта или зубов, мешающее нормальному приему пищи; имеют любое состояние, затрагивающее слизистую оболочку пищевода или изменяющее моторику пищевода, кроме ЕоЕ, включая эрозивный эзофагит (степени В или выше согласно Лос-Анджелесской классификации гастроэзофагеальной рефлюксной болезни), грыжу пищеводного отверстия длиной более 3 см, пищевод Барретта и ахалазию; применение системных (пероральных или парентеральных) кортикостероидов в течение 60 суток перед скринингом; применение ингалируемых/проглатываемых кортикостероидов в течение 30 суток перед скринингом или продолжительное применение высокоэффективных вводимых интрадермальной инъекцией кортикостероидов для местного применения в течение 30 суток перед скринингом; начало элиминационной диеты или элементной диеты в течение 30 дней перед скринингом; утренний (с 07:00 до 08:00 часов или по возможности ближе к этому интервалу) уровень кортизола в сыворотке крови 5 мкг/дл (138 нмоль/л) или менее; применение биологических иммуномодуляторов в течение 24 недель перед скринингом; применение ингибиторов кальциневрина или аналогов пуринов (азатиоприна, 6-меркаптопурина) в течение 12 недель перед скринингом; применение сильнодействующих ингибиторов цитохрома Р450 (CYP), изоформы 3А4 (например, ритановира и кетоконазола) в течение 12 недель перед скринингом; имеют противопоказание к EGD или факторы, существенно повышающие риск EGD или биопсии пищевода, или имеют сужение пищевода, препятствующее проведению EGD со стандартным 9-мм эндоскопом; имеют в анамнезе стриктуру пищевода, требующую проведения дилатации, за предыдущие 12 недель перед скринингом; субъекты, которые начали, прервали или изменили режим приема PPI, Н2 антагонистов (блокаторов Н2-гистаминовых рецепторов), антацидных средств или антигистаминных препаратов при любых состояниях, таких как GERD при аллергическом рините, в течение 4 недель перед отборочной эндоскопией. Эти лекарственные средства должны следует применять постоянно на протяжении всего исследования. Наличие инфекции вирусом гепатита В, гепатита С или иммунодефицита человека (должно быть протестировано во время скрининга).
Для определения инфекции, вызываемой вирусами гепатита В и гепатита С, будут использованы следующие параметры: положительный результат анализа на поверхностный антиген вируса гепатита В (HBsAg), суммарные антитела к капсидному антигену вируса гепатита В (анти-НВс) или антитела к вирусу гепатита С. Однако, субъекты, которые являются положительными в отношении антитела к поверхностному антигену вируса гепатита В, но отрицательными в отношении HBsAg и анти-НВс, примут участие в исследовании.
Имеют желудочно-кишечное кровотечение в пределах 4 недель до скринингового визита или между скрининговым визитом и визитом для рандомизации; наличие текущей хронической инфекции, иммуносупрессии или иммунодефицита; имеют анамнез или болезнь Крона, глютеновую болезнь или другое воспалительное заболевание ЖК тракта, включая эозинофильный гастроэнтерит; имеют текущее злоупотребление алкоголем или наркотиками по мнению исследователя; субъекты женского пола, являющиеся беременными, кормящими грудью или планирующие беременность во время исследования; результаты теста на определение беременности по крови на момент скрининга и теста на определение беременности по моче в течение 4-недельного периода оценки исходных симптомов у женщин детородного возраста должны быть отрицательными. Живущие половой жизнью женщины детородного возраста, которые не соглашаются использовать высокоэффективные методы контрацепции во время визита в период последующего наблюдения; в случае приема системного контрацептивного средства применение должно быть постоянным в течение 28 суток или более перед скринингом. Для субъектов женского пола с хирургической менопаузой или менопаузой, подтвержденной содержанием фолликулостимулирующего гормона/лютеинизирующего гормона, нет необходимости в контрацепции или тестировании определения беременности во время данного исследования. Участие в клиническом исследовании с вовлечением какого-либо другого экспериментального продукта в течение 30 суток скрининга (или интервала времени, соответствующего 5 периодам полувыведения, в зависимости от того, что является более продолжительным).
До рандомизации
Уровень кортизола в сыворотке крови менее 18 мкг/мл (497 нмоль/дл) через 60 минут после теста стимуляции адренокортикотропным гормоном (АСТН) с использованием 250 мкг косинтропина (т.е. положительный результат в тесте стимуляции АСТН).
Тестируемый продукт, доза и способ введения
АРТ-1011 представляет собой перорально распадающуюся таблетку, которая в себя включает FP в качестве своего активного ингредиента.
Для задач этого протокола использованный термин «исследуемое лекарственное средство» относится к любому вводимому без маркировки лекарственному средству (т.е. любой дозировке АРТ-1011 или плацебо).
Субъекты будут проинструктированы о необходимости перорального приема исследуемого лекарственного средства без использования воды или других жидкостей. Таблетку следует поместить в полость рта, выполнять манипуляции в ротовой полости между языком и небом и позволить ей полностью распасться на языке субъекта. Проглатывать следует после полного распадения, не кусая или разжевывая. После введения запивание водой или жидкостями не допускается.
Прием дозы будет осуществляться утром («после завтрака»; через 30 минут или более после завтрака) и перед сном («перед сном»; через 2 или более часов после вечернего приема пищи). Доза исследуемого лекарственного средства «перед сном» будет введена непосредственно перед сном, лежа в постели. Все действия, а именно прием пищи, напитков и чистка зубов, должны быть завершены перед приемом дозы.
Исследуемое лекарственное средство будет введено BID (через 30 минут после завтрака и перед сном) во всех Частях исследования. Во время вводной фазы исследования с применением плацебо субъекты будут получать плацебо, BID. Субъекты в HS группах будут получать плацебо утром через 30 минут после завтрака и свои дозы средства перед сном. Субъекты в группах, принимающих плацебо, после рандомизации будут получать плацебо, BID.
Субъекты в группах, получающих АРТ-1011 в дозе 1,5 мг, BID, АРТ-1011 в дозе 3 мг, BID, и плацебо, будут принимать это же исследуемое лекарственное средства в виде доз «после завтрака» и «перед сном». Субъекты в группах, получающих АРТ-1011 в дозах 1,5 мг, HS, и 3 мг, HS, будут принимать плацебо «после завтрака» и АРТ-1011 в дозе 1,5 мг или 3 мг «перед сном».
Субъекты должны воздерживаться от перорального приема твердых продуктов или жидкостей в течение 1 часа или более после приема дозы.
Критерии для оценки
Эффективность будет оцениваться гистологическими методами (по количеству эозинофилов в HPF), эндоскопическими методами (по эндоскопическому референсному показателю (EREFs) для эозинофильного эзофагита), клиническими методами в виде эксплоративной конечной точки (с использованием анкеты PROSE, заполняемой для каждого эпизода дисфагии и по окончании каждого дня) и с использованием приведенных далее дополнительных сообщаемых пациентом результатов лечения: общего показателя симптомов ЕоЕ, общего впечатления пациента относительно тяжести заболевания (PGIS) и общего впечатления пациента об изменении состояния (PGIC), 7-дневного индекса активности эозинофильного эзофагита (EEsAI), общего показателя и субпоказателей, и оценки симптомов субъектом. Связанное с состоянием здоровья качество жизни (HRQoL) будет оцениваться как эксплоративная конечная точка согласно опроснику по оценке качества жизни взрослых пациентов с эозинофильным эзофагитом (EoE-QoL-A).
Первичная конечная точка эффективности: приведенная ниже первичная конечная точка эффективности будет оцениваться на 12-й неделе для оценки ЕоЕ-ответа.
Гистология: процентная доля субъектов с максимальным количеством эозинофилов/HPF 6 или менее после оценки по меньшей мере 5-6 биоптатов из проксимальной и дистальной частей пищевода (приблизительно по 3 каждого), при этом HPF площадь составляет 23 5 квадратных микрон (увеличение объектива 40 с окуляром 22 мм).
Вторичные конечные точки эффективности: будут оценены следующие вторичные конечные точки эффективности:
устойчивый ЕоЕ-ответ: процентная доля субъектов, которые соответствуют первичной конечной точке на 12-й неделе и сохраняют показатели первичной конечной точки на 26-й неделе и 52-й неделе;
изменение по сравнению с исходным EREFs на 12-й неделе, 26-й неделе и 52-й неделе;
изменения по данным эндоскопии, которые будут основаны на оценке показателя EREFS, учитывающего следующие эндоскопические признаки: отек, пищеводные кольца, экссудаты, борозды, стриктуру и несколько вспомогательных признаков («гофрированный» пищевод, узкий диаметр пищевода и эрозии пищевода);
процентная доля субъектов с максимальным количеством эозинофилов/HPF менее 1 и менее 15 на 12-й неделе, 26-й неделе и 52-й неделе;
изменение по сравнению с исходным общим показателем симптомов ЕоЕ, определенным до рандомизации, который будет оцениваться в течение 7-дневного периода перед следующими визитами по ходу исследования: на 4-й неделе, 8-й неделе, 12-й неделе, 18-й неделе, 22-й неделе, 26-й неделе, 28-й неделе, 36-й неделе, 44-й неделе и 52-й неделе;
в отношении дисфагии: изменение числа эпизодов дисфагии, взятого в качестве исходного уровня (в течение 14-дневного периода до рандомизации), по сравнению с таковым для 14-дневного периода перед представляющим интерес моментом времени (на 12-й неделе, 26-й неделе и 52-й неделе);
изменение по сравнению с исходным 7-дневным общим показателем EEsAI на 12-й неделе, 26-й неделе и 52-й неделе;
изменение по сравнению с исходными 7-дневными субпоказателями EEsAI на 12-й неделе, 26-й неделе и 52-й неделе;
процентная доля субъектов со средним 7-дневным общим показателем EEsAI менее 20 на 12-й неделе, 26-й неделе и 52-й неделе;
изменение впечатлений, оцененных на 4-й, 8-й, 12-й, 14-й, 18-й, 22-й, 26-й, 28-й, 36-й, 44-й и 52-й неделях, по сравнению с исходным оцененным до рандомизации PGIS;
PGIC на 4-й, 8-й, 12-й, 14-й, 18-й, 22-й, 26-й, 28-й, 36-й, 44-й и 52-й неделях.
Оценка неудачного результата лечения и рецидива, включающая:
процентную долю гистологически обусловленных нереспондеров в зависимости от дозы на 12-й неделе, 26-й неделе и 52-й неделе;
процентную долю субъектов, нуждающихся в неотложном эндоскопическом освобождении от пищи, в зависимости от дозы до 14-й недели, в интервале с 14-й недели по 28-ую неделю и в интервале с 28-й недели по 52-ю неделю;
процентную долю субъектов, нуждающихся в дилатации пищевода, в зависимости от получающей дозу группы и Части исследования.
Эксплоративные конечные точки эффективности, будет проведена оценка следующих эксплоративных конечных точек эффективности:
в отношении дисфагии: изменение числа эпизодов дисфагии, взятого в качестве исходного уровня (в течение 14-дневного периода до рандомизации), по сравнению с таковым для 14-дневного периода перед представляющим интерес моментом времени (на 12-й, 26-й и 52-й неделях);
изменение по сравнению с исходным числом дней без дисфагии в течение 14-дневного периода перед следующими визитами по ходу исследования: на 12-й неделе, 26-й неделе и 52-й неделе;
устойчивый ЕоЕ-ответ (в отношении дисфагии): процентная доля всех субъектов, которые соответствовали вторичной конечной точке по дисфагии на 12-й неделе и сохраняли связанный с дисфагией ответ на 26-й неделе и 52-й неделе;
оценка ФК/ФД (кортизола) и взаимосвязей ответа и воздействия (эффективности);
оценка субъектом симптомов по сравнению с предыдущим визитом на 4-й неделе, 8-й неделе, 12-й неделе, 14-й неделе, 18-й неделе, 22-й неделе, 26-й неделе, 28-й неделе, 36-й неделе, 44-й неделе и 52-й неделе и визитом по поводу преждевременного завершения исследования (если таковой предусмотрен);
оценка HRQoL на основании EoE-QoL-A при рандомизации, на 12-й неделе, 26-й неделе, 52-й неделе для всех субъектов в зависимости от дозы и подгруппы;
данные о субъектах, получающих простое слепое (для субъекта) лечение (в Части 2) будут сведены отдельно;
процентная доля субъектов, которые были классифицированы как гистологически обусловленные нереспондеры на 12-й неделе и имеют максимальное количество эозинофилов/HPF 6 или менее во всех подвергнутых биопсии местах пищевода на 26-й неделе и 52-й неделе;
изменение по сравнению с исходным числом эпизодов дисфагии в течение 14-дневного периода до 26-й недели и 52-й недели для субъектов, которые были классифицированы как нереспондеры на 12-й неделе;
процентная доля субъектов, которые были классифицированы как гистологически обусловленные нереспондеры на 12-й неделе и соответствуют первичной конечной точке на 26-й неделе и 52-й неделе;
схема выставления балльных оценок и различные конечные точки будут получены исходя из DEDI;
будут оценены возможности PROSE в отношении определения психометрических характеристик;
анализы с использованием якорных точек и анализы распределения для оценки значимых изменений на основании PROSE.
Нежелательные явления
Любое АЕ или любая сопутствующая болезнь, испытываемые субъектом во время любой части FLUTE, должны быть подробно описаны и в полной мере оценены исследователем. Исследователь несет ответственность за регистрацию всех АЕ, наблюдаемых или упоминаемых во время исследования, независимо от связи с исследуемым препаратом и/или клинической значимости.
Оценка безопасности будет проведена посредством мониторинга и регистрации всех возникающих при лечении нежелательных явлений (ТЕАЕ), ТЕАЕ, приводящих к прерыванию лечения, и серьезных нежелательных явлений (SAE). Все ТЕАЕ получат код на основании классификации с использованием Словаря по нормативно-правовой деятельности в области медицины, версии 14.0, и будут классифицированы по тяжести (слабое, умеренное, тяжелое). Сообщение относительно связи SAE с исследуемым лекарственным средством (по типу связь есть или связи нет) будет сделано только исследователем. Нежелательные явления при лечении, возникающие в пределах 3 суток после изменения дозы, будут отнесены к предыдущей дозе.
Ожидается, что АРТ-1011 (флутиказона пропионат, ODT), кортикостероид с минимальной всасываемостью, будет действовать локально в пищеводе. Ввиду этого существует возможность снижения эффективности, если субъект запивает водой сразу после приема дозы. Поскольку применение флутиказона только в очень редких случаях ассоциируется с кандидозом полости рта, необходимости в полоскании и сплевывании, так как это может быть непреднамеренно связано с проглатыванием воды, не будет.
Тем не менее, кандидоз полости рта и пищевода будут считаться АЕ, представляющими особый интерес. Субъекты могут продолжать принимать участие в этом исследовании во время лечения этих АЕ. Этим субъектам исследователь может разрешить осуществлять полоскание и сплевывание через 30 минут после приема доз. Субъекты должны быть проинструктированы о том, чтобы не глотать воду для полоскания.
Симптомы гиперадренокортицизма (см. ниже) также относятся к АЕ, представляющим особый интерес.
Лабораторные анализы
На протяжении всего исследования будут проводиться стандартные лабораторные анализы и обследования, включая общий анализ крови, биохимический анализ крови, анализ мочи и снятие электрокардиограмм (ЭКГ). Клинически значимые изменения в результатах лабораторных анализов или ЭКГ будут обобщены.
Физическое обследование и жизненно важные функции
Для документирования исходного состояния субъекта и выявления связанных с АЕ изменений будут проведены физические обследования. Также будут оценены жизненно важные признаки и будет сообщено о клинически значимых отклонениях.
Связанные с кортизолом данные
Все субъекты будут проходить тест стимуляции АСТН (250 мкг) во время 4-недельного периода оценки исходных симптомов после достижения утреннего уровня кортизола в сыворотке крови во время периода скрининга. Этот тест также будет проведен на 12-й неделе (в 6-й визит), 26-й неделе (в 10-й визит) и 52-й неделе (в 14-й визит).
Во время всех визитов (плановых или внеплановых) особое внимание будет уделено возможным изменениям, связанным с кортикостероидами, а также симптомам гиперадренокортицизма. Если субъект подвергается хирургическому вмешательству или получает травму во время исследования, то следует соблюдать особую осторожность при наблюдении за субъектами в отношении признаков неадекватного ответа надпочечников.
Если есть подозрения на гиперадренокортицизм или угнетение надпочечников, то для подтверждения или исключения этих данных должно быть проведено адекватное обследование. Конкретно, для мониторинга угнетения гипоталамо-гипофизарно-надпочечниковой (HPA) оси, вызывающего клиническую озабоченность, после скрининга также будет проведен тест стимуляции АСТН (250 мкг), если имеет место любое из приведенного ниже:
во время проведения стандартных лабораторных анализов, проводимых в рамках данного исследования, у субъекта наблюдается утренний уровень кортизола в сыворотке крови 5 мкг/дл (138 нмоль/л) или менее (подтвержденный 2-мя отборами крови), в том числе при последнем визите в ходе лечения субъекта и при визите, если таковой предусмотрен, по поводу преждевременного завершения;
субъект сообщает о наличии симптомов гиперадренокортицизма;
субъект прерывает лечение по причине угнетения НРА оси.
Положительный результат теста стимуляции АСТН определяется как уровень кортизола в сыворотке крови менее 18 мкг/дл (497 нмоль/л) через 60 минут после обработки 250 мкг косинтропина. Этот результат является критерием исключения, если он имеет место в период скрининга/4-недельный период оценки исходных симптомов, и требует последующего наблюдения за восстановлением функции надпочечников, если после этого произошло ее нарушение. Обсуждение лечения угнетения НРА оси приводится в полном протоколе. Заказчик клинического исследования предоставит руководящие указания по обеспечению безопасности последующего наблюдения и задокументирует восстановление функции надпочечников у всех субъектов, демонстрирующих признаки гиперадренокортицизма или угнетения НРА оси в ходе исследования.
Электрокардиограмма
Будут оценены показания электрокардиограмм и будет сообщаться о клинически значимых отклонениях.
Конечные точки безопасности, представляющими интерес конечными точками безопасности являются:
частота случаев ТЕАЕ;
ТЕАЕ, приводящие к прерыванию лечения;
число возникающих при лечении SAE;
процентная доля субъектов с уровнем кортизола в сыворотке крови 5 мкг/дл или менее (138 нмоль/л или менее) или с положительными результатами теста стимуляции АСТН (уровень кортизола в сыворотке крови менее 18 мкг/мл (497 нмоль/л или менее) через 60 минут);
число субъектов, прервавших лечение по причине угнетения НРА оси, которое будет зарегистрировано;
частота случаев кандидоза полости рта и пищевода.
Фармакокинетические переменные: приведенные далее параметры PopPK будут оцениваться с использованием выборочного контроля образцов, если позволяют данные: клиренс при пероральном приеме препарата; объем распределения. Дополнительные параметры PopPK будут оцениваться, при необходимости, на основании окончательной структурной ФК модели.
Статистические методы
Определение объема выборки: Часть 1 будет включать в себя объем выборки с диапазоном из 100 субъектов, в котором 20 пациентов рандомизированы в группы, получающие 1,5 мг HS, 1,5 мг BID, 3 мг HS, 3 мг BID и плацебо (1:1:1:1:1). В соответствии с этими соотношениями при рандомизации приблизительно 80% всех субъектов в Части 1 будут проходить лечение в режиме введения АРТ-1011, а приблизительно 20% всех субъектов в Части 1 будут проходить лечение с использованием плацебо.
Анализируемые популяции
Вся участвующая в исследовании популяция включает в себя всех субъектов, которые подписывают ICF и участвуют в исследовании. Популяция для оценки безопасности включает в себя всех субъектов, которые получают 1 дозу или более исследуемого лекарственного средства. Предназначенная для лечения (ITT) популяция включает в себя всех субъектов, которые получают 1 дозу или более исследуемого лекарственного средства и имеют 1 оценку или более эффективности после приема дозы. Субъект, который участвует в исследовании и получает исследуемое лекарственное средство, но терпит неудачу при проведении лечения, будет считаться выбывшим. Подгруппа для выборочного ФК контроля включает в себя всех субъектов, которые имеют 1 или более поддающегося количественному определению ФК образца, отобранного для выборочных оценок ФК.
Дополнительные анализируемые популяции (например, упомянутые в протоколе популяции, включающие субъектов, которые завершают Часть 1, 26-ю и 52-ю недели Части 2) могут быть определены в Плане статистического анализа.
Статистическая методология
Характеристики субъектов: исходные и демографические данные будут обобщены с использованием описательной статистики для непрерывных и порядковых переменных (например, возраста и веса) и количеств и процента для категориальных переменных (наличия или отсутствия стриктур, предварительного ответа на стероиды, пола, расовой принадлежности).
Первичный анализ эффективности для Части 1
Пусть р0 представляет собой долю пациентов, которые удовлетворяют гистологическому ответу на плацебо, и пусть pj представляет собой долю пациентов, которые удовлетворяют гистологическому ответу для дозы j, при этом j равно 1, 2, 3, 4, согласуясь с дозами 1,5 мг HS, 1,5 мг BID, 3 мг HS и 3 мг BID, соответственно. Имеются 4 гипотезы, соответствующие 4 дозам активного соединения, которые будут протестированы с использованием стратегии входного контроля, чтобы сохранить ошибку I типа для каждого анализа.
1) Первичная гипотеза №1
Н0: р4≤р0,
Н1: р4=р0.
Для проверки первичной гипотезы №1, т.е. 3 мг, BID, по сравнению с плацебо, будет использован критерий хи-квадрат пропорций. Если соответствующее р-значение меньше или равно 0,05, нулевая гипотеза будет отклонена и после этого будет проверена следующая гипотеза:
2) первичная гипотеза №2
Н0: р3≤р0,
Н1: р3=р0.
Для проверки первичной гипотезы №2, т.е. 3 мг, HS, по сравнению с плацебо, будет использован критерий хи-квадрат пропорций. Если соответствующее р-значение меньше или равно 0,05, нулевая гипотеза будет отклонена и после этого будет проверена следующая гипотеза:
3) первичная гипотеза №3
Н0: р2≤р0,
Н1: р2=р0.
Для проверки первичной гипотезы №3, т.е. 1,5 мг, BID, по сравнению с плацебо, будет использован критерий хи-квадрат пропорций. Если соответствующее р-значение меньше или равно 0,05, нулевая гипотеза будет отклонена и после этого будет проверена следующая гипотеза:
4) первичная гипотеза №4
Н0: p1≤р0,
Н1: p1=р0.
Для проверки первичной гипотезы №4, т.е. 1,5 мг, HS, по сравнению с плацебо, будет использован критерий хи-квадрат пропорций. Если соответствующее р-значение меньше или равно 0,05, нулевая гипотеза будет отклонена.
Обратите внимание, что стратегия входного контроля позволяет провести только формальную проверку гипотез 1,5 мг, HS, или 1,5 мг, BID, если более высокие дозы соответствуют статистической значимости.
Анализ эффективности для Части 2
Устойчивый ЕоЕ-ответ будет оцениваться у субъектов, которые завершили и Часть 1, и Часть 2, и для которых были проведены оценки на 26-й и 52-й неделе. Оценка этого будет проведена по первичной конечной точке. Другие показатели эффективности будут оценены на 26-й неделе. Для гистологически обусловленных нереспондеров из Части 1, которые проходят лечение в Части 2, будет обобщена эффективность. Другие показатели эффективности будут оценены на 52-й неделе.
Анализ вторичных и эксплоративных конечных точек эффективности
Статистические тесты для сравнения каждой получающей дозу АРТ-1011 группы с группой, получающей плацебо, будут проводиться для вторичных конечных точек эффективности, но соответствующие р-значения будут рассматриваться как описательные, а не логически обоснованные.
Вторичные конечные точки будут проанализированы с использованием критерия Кохрана-Мантеля-Гензеля (СМН) для категориальных конечных точек и ковариационного анализа для изменения по сравнению с исходными конечными точками, за исключением конечной точки периода времени до рецидива после начала двойного слепого лечения в Части 2, которая будет проанализирована с использованием метода Каплана-Мейера.
Никакого статистического тестирования эксплоративных конечных точек эффективности проводиться не будет.
Анализ безопасности
Число случаев ТЕАЕ будет обобщено согласно классу по поражению органов и систем органов и предпочтительной терминологии. Будут предоставлены отдельные сводные данные для случаев максимальной тяжести (всех АЕ) и связи с исследуемым лекарственным средством (только для SAE). Число случаев ТЕАЕ, приводящих к прерыванию исследования, и число возникающих при лечении SAE также будут обобщены. У субъектов с изменением получающих дозу групп ТЕАЕ будут отнесены к предыдущей дозе, если они происходят в течение 3 суток после изменения.
Клинически значимые изменения, представляющие возможный клинический интерес в клинических тестах, будут обобщены, включая общий анализ крови, биохимический анализ крови, анализ мочи, ЭКГ, содержание кортизола, показатели жизненно важных функций и минеральную плотность костной ткани. Никакого статистического тестирования конечных точек безопасности проведено не будет. При необходимости могут быть приведены таблицы изменений. Число субъектов, прерывающих лечение по причине угнетения НРА оси или положительных результатов теста стимуляции АСТН, будет обобщено.
Популяционная фармакокинетика
PopPK анализ будет проведен на основании выборочных данных для концентрации в плазме крови. Он будет проведен с использованием программного обеспечения для расчета нелинейных моделей со смешанными эффектами, NONMEM, версия 7.2.0, или более поздней версии (ICON Development Solutions, Ellicott City, MD), или другого соответствующего программного обеспечения для расчета нелинейных моделей со смешанными эффектами. Структурная ФК модель будет включать клиренс при пероральном приеме препарата и объем распределения в качестве параметров с фиксированным эффектом. В дополнение к этому вариабельность между субъектами в оценках параметров и случайная остаточная ошибка в данных будут оцениваться с применением соответствующей модели ошибок. Оптимальная базовая модель будет выбрана в соответствии со стандартными критериями, такими как минимальное значение целевой функции и диагностические графики. В качестве приложения к отчету о клиническом исследовании будет создан отдельный отчет по PopPK.
Эксплоративный ФК/ФД анализ
Если позволяют данные, также могут быть проведены эксплоративные ФК/ФД анализы, в которых оценивается взаимосвязь между системным воздействием FP и изменениями в уровнях кортизола, как описано выше. Для облегчения выбора безопасных и эффективных доз для будущих исследований и клинического применения могут быть проведены дополнительные эксплоративные ФК/ФД анализы.
ОБОСНОВАНИЕ ИССЛЕДОВАНИЯ И АНАЛИЗ СООТНОШЕНИЯ РИСК-ПОЛЬЗА
Цель исследования действия флутиказона (FLUT) при эозинофильном эзофагите (FLUTE) состоит в изучении 3 общесуточных доз АРТ-1011 для определения ответа на воздействие АРТ-1011 и минимальной эффективной дозы при одновременном сведении к минимуму любых клинически значимых влияний на НРА ось. Ожидается, что АРТ-1011 обеспечит следующие преимущества для пациентов с ЕоЕ: пероральное введение, как правило, является более приемлемым и более надежным с точки зрения точного введения доз. В настоящее время единственной доступной формой композиции на основе FP является дозированный аэрозоль для ингаляций, который распыляется в полость рта и проглатывается пациентом. Пероральное введение АРТ-1011 характеризуется очень низкой биодоступностью даже по сравнению с похожими соединениями, такими как будесонид, что дополнительно снижает его потенциал в плане системной кортикостероидной токсичности несмотря на то, что он может быть более эффективным в расчете на один мг.
Настоящее исследование представляет собой исследование по подбору первой дозы АРТ-1011. С учетом низкой биодоступности маловероятно, что АРТ-1011 будет оказывать какие-либо значительные системные кортикостероидные эффекты.
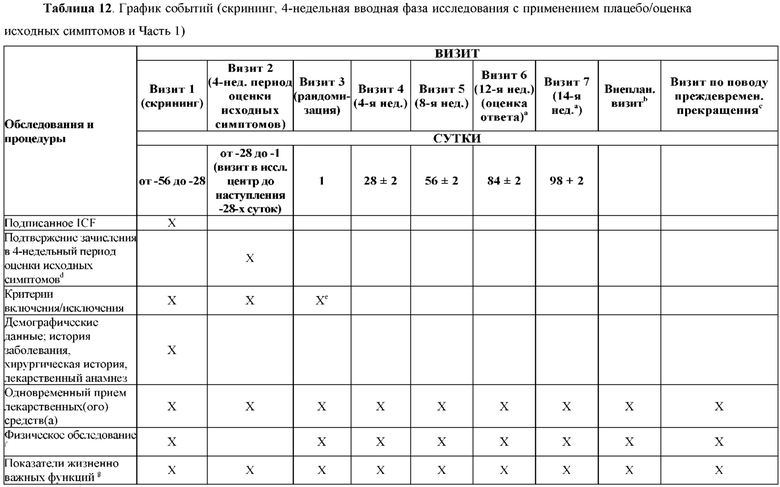

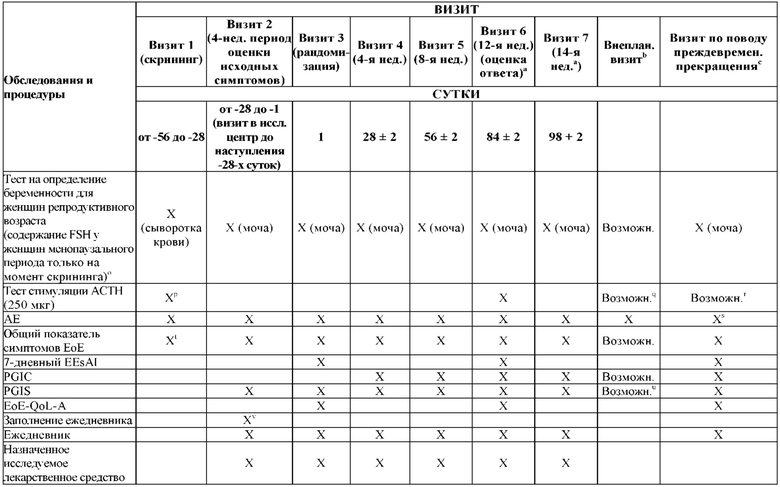
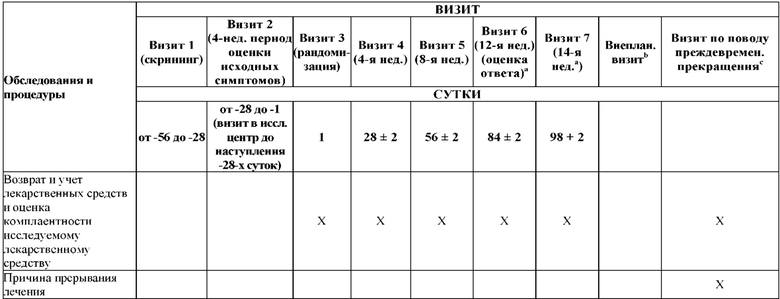
Сокращения: АСТН = адренокортикотропный гормон; АЕ = нежелательное явление; ЭКГ = электрокардиограмма; eCRF = электронная форма карты информированного согласия; EEsAI = индекс активности эозинофильного эзофагита; EGD = эзофагогастродуоденоскопия; ЕоЕ = эозинофильный эзофагит; EoE-QoL-A = опросник по оценке качества жизни взрослых пациентов с эозинофильным эзофагитом; EREFs = эндоскопический референсный показатель для эозинофильного эзофагита; FSH = фолликулостимулирующий гормон; НРА = гипоталамо-гипофизарно-надпочечниковая ось; ICF = форма информированного согласия; PGIC = общее впечатление пациента об изменении состояния; PGIS = общее впечатление пациента относительно тяжести заболевания; ФК = фармакокинетика; popPK = популяционная фармакокинетика; SAE = серьезное нежелательное явление.
а Данные и образцы, собранные в течение 14-й недели согласно графику событий Части 1, также будут применимы для визита согласно графику событий в Части 2 (сообразно обстоятельствам), и процедуры не будут повторяться. Гистологически обусловленные нереспондеры на 12-й неделе будут получать простое слепое лечение (3 мг, BID) в Части 2.
b Причиной для внепланового визита будет необходимость формирования процедур по усмотрению исследователя.
с Субъект должен появиться в исследовательском центре не позднее 7 дней с момента определения необходимости прерывания лечения. Если это невозможно сделать ввиду SAE или другого непредвиденного обстоятельства, визит может быть совершен субъектом или членом его семьи по телефону. В eCRF должно быть зафиксировано, почему субъект не смог посетить исследовательский центр.
d Как описано в разделе 4.1.4, чтобы быть зачисленными 4-недельный период оценки исходных симптомов, субъекты должны удовлетворять всем критериям включения, в том числе значению общего показателя ЕоЕ более 3 (критерию включения №5), за исключением тех, которые должны оцениваться во время 4-недельного периода оценки исходных симптомов (критерию включения №7: наличие признаков ЕоЕ, что определяется по максимальному количеству эозинофилов/HPF 15 или более, критерию включения №8: наличие в ежедневнике сообщений о 3 или более днях с эпизодами дисфагии из 7 дней в течение последних 14 суток 4-недельного периода оценки исходных симптомов, критерию включения №9: ведение ежедневника по меньшей мере в течение 5 дней из каждых 7 дней в течение последних 14 суток оценки исходных симптомов, и критерию исключения №23: уровень кортизола в сыворотке крови менее 18 мкг/дл (497 нмоль/л) через 60 минут после теста стимуляции адренокортикотропным гормоном (АСТН) с применением 250 мкг косинтропина (т.е. случаю положительного результата теста стимуляции АСТН).
е Подтверждение того, что субъект удовлетворяет критериям пригодности, включает следующие критерии, которые должны быть подтверждены в течение 4-недельного периода оценки исходных симптомов: критерий включения №7: наличие признаков ЕоЕ, что определяется по максимальному количеству эозинофилов/HPF 15 или более, критерий включения №8: наличие в ежедневнике сообщений о 3 или более днях с эпизодами дисфагии из 7 дней в течение последних 14 суток 4-недельного периода оценки исходных симптомов, критерий включения №9: ведение ежедневника по меньшей мере в течение 5 дней из каждых 7 дней в течение последних 14 суток периода оценки исходных симптомов, и критерий исключения №23: уровень кортизола в сыворотке крови менее 18 мкг/дл (497 нмоль/л) через 60 минут после теста стимуляции АСТН с применением 250 мкг косинтропина (т.е. случай положительного результата теста стимуляции АСТН).
f Физическое обследование будет включать обследования роста на момент скрининга (только 1-е сутки), веса, внешнего вида пациента, головы и шеи, глаз и ушей, носа и горла, грудной клетки, легких, сердца, области живота, конечностей и суставов, лимфатических узлов, кожи и неврологический осмотр. Расчет индекса массы тела (BMI) будет выполнен на 1-е сутки.
g Показатели жизненно важных функций, которые должны быть отмечены, включают частоту сердечных сокращений, частоту дыхательных движений, температуру (°С; после 5-минутного отдыха в сидячем положении) и кровяное давление (измеряемое на одной и той же руке на протяжении всего исследования).
h Отборы образцов по времени необходимо проводить в 07:00-8:00 или как можно ближе к это интервалу, насколько это возможно. Субъекты не должны принимать пищу в течение в течение 8-часового периода времени до проведения оценок уровня кортизола в сыворотке крови. Кровь для анализа уровня кортизола в сыворотке крови (утром натощак) может быть взята в пределах ±3 дней от планового визита, чтобы соответствовать точному временному графику. При желании отбор крови для проведения других анализов, запланированных на время данного визита, может быть выполнен одновременно.
i Если при последнем визите в ходе лечения субъекта сообщается об аномальном уровне кортизола в сыворотке крови, то может потребоваться проведение дополнительного мониторинга и АСТН теста (см. раздел 6.3.4).
j Отбор образцов для выборочного ФК контроля будет проведен для всех субъектов. Образец следует собирать до приема дозы на 1-е сутки. На 4-й неделе, 8-й неделе и 12-й неделе субъекты будут принимать свою дозу «после завтрака» дома, а 2 образца будут отобраны во время их планового визита: по прибытии в исследовательский центр и через 1-1,5 часа после отбора первого образца (непосредственно перед тем, как покинуть исследовательский центр). Субъекты должны записать время приема своей утренней дозы и информировать сотрудников исследовательского центра. Время приема дозы «после завтрака» (в день планируемого визита) и время приема дозы непосредственно «перед сном» (вечером перед планируемым визитом) будет зафиксировано. Реальные времена отбора ФК образцов будут задокументированы.
k Ожидается, что обычно EGD будет проведена во время, не совпадающее со временен выполнения других процедур для заданной временной точки. Как EGD, так и любые другие процедуры, показанные для визита по ходу исследования, должны быть выполнены в пределах временного интервала для этого визита по ходу исследования. В случае скрининга EGD должна быть проведена после подписания ICF и в течение 2 недель или более до даты ожидаемой рандомизации.
l При проведении EGD эндоскопист определит EREFs и получит приблизительно 3 биоптата как из проксимальной, так и дистальной частей пищевода (в общей сложности 5-6 биоптатов). Гистологические образцы будет оценивать специалист по лабораторной диагностике.
m EGD для определения пригодности к лечению должна быть проведена до зачисления в 4-недельную (в течение 28-х суток) простую слепую вводную фазу/оценку исходных симптомов. Оценку патологии в EGD-биоптатах также можно выполнить во время 4-недельного периода оценки исходных симптомов (или раньше, если это возможно).
n Если субъект прерывает исследование ввиду отсутствия эффективности или по другим причинам, исследователь может провести EGD, если к этому есть клинические показания.
o Тест на определение беременности по крови будет проведен на момент скрининга, а после этого тесты на определение беременности по моче. Результаты тестирования на беременность женщин репродуктивного возраста на беременность должны быть отрицательными (см. раздел 6.3.2.1).
р Все субъекты проходят тест стимуляции АСТН (250 мкг) как часть обследований по пригодности к лечению. Такая оценка будет проведена после достижения утреннего уровня кортизола в сыворотке крови во время периода скрининга. Она должна быть проведена до зачисления в 4-недельную (в течение 28-х суток) простую слепую вводную фазу/оценку исходных симптомов. Положительный результат теста стимуляции АСТН (уровень кортизола в сыворотке крови менее 18 мкг/дл (497 нмоль/л) через 60 минут после обработки 250 мкг косинтропина) будет представлять собой критерий исключения (см. критерий исключения №9: применение биологических иммуномодуляторов в течение 24 недель до скрининга, в разделе 4.2.2).
q Тест стимуляции АСТН (250 мкг) будет проведен (во время внепланового визита) для оценки угнетения НРА оси, вызывающего клиническую озабоченность, как приведено ниже:
1) для любого момента времени, когда субъект имеет утренний уровень кортизола в сыворотке крови 5 мкг/дл (138 нмоль/л) или менее (подтвержденный 2-мя отборами крови);
2) для любого момента времени, когда субъект отмечает симптомы гиперадренокортицизма (приложение 6).
r Субъекты, которые прерывают исследование ввиду подтверждения угнетения НРА оси, будут проходить тест стимуляции АСТН (250 мкг) при визите по поводу преждевременного завершения.
s Для визита по поводу преждевременного завершения исследователь должен зафиксировать наличие любого АЕ (через 14±5 суток после прерывания исследования). Субъекты, прервавшие исследование по причине проблем с НРА, должны отслеживаться до получения разрешения.
t Общий показатель симптомов ЕоЕ в момент скрининга будет зафиксирован «на бумаге». Показатель ЕоЕ должен оставаться на уровне более 3 во время каждого из визитов 1, 2 и 3 до рандомизации, или будет считаться, что результаты скрининга субъекта являются неудачными.
u Только если визит связан с изменением симптомов ЕоЕ.
v Регистрация записей в ежедневнике начнется не ранее 28-х суток до планируемой даты рандомизации.
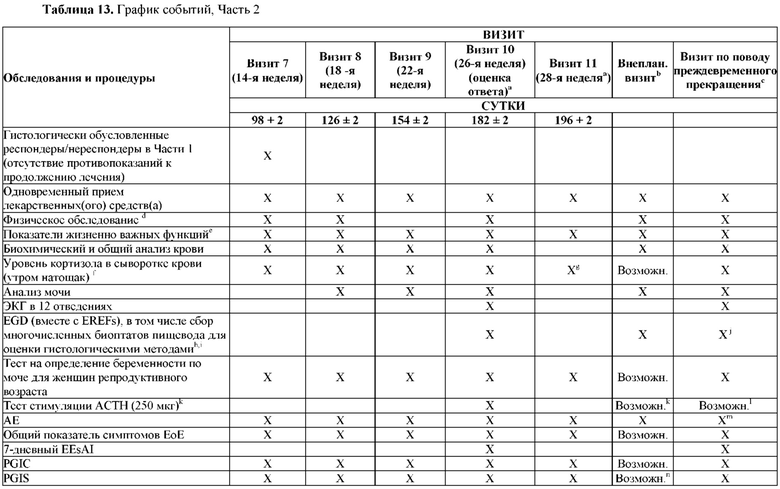
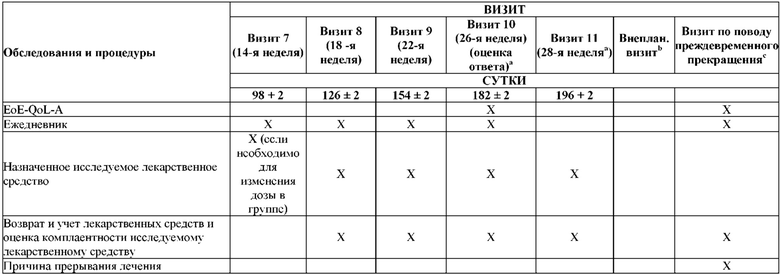
Сокращения: АСТН = адренокортикотропный гормон; АЕ = нежелательное явление; ЭКГ = электрокардиограмма; eCRF = электронная форма карты информированного согласия; EEsAI = индекс активности эозинофильного эзофагита; EGD = эзофагогастродуоденоскопия; ЕоЕ = эозинофильный эзофагит; EoE-QoL-A = опросник по оценке качества жизни взрослых пациентов с эозинофильным эзофагитом; EREFs = эндоскопический референсный показатель для эозинофильного эзофагита; НРА = гипоталамо-гипофизарно-надпочечниковая ось; PGIC = общее впечатление пациента об изменении состояния; PGIS = общее впечатление пациента относительно тяжести заболевания; SAE = серьезное нежелательное явление.
а Данные и образцы, собранные в течение 28-й недели согласно графику событий Части 1, также будут применимы для визита согласно графику событий в Таблице 4-3 в Части 2 (сообразно обстоятельствам), и процедуры не будут повторяться. На 26-й неделе субъекты, представляющие собой гистологически обусловленные нереспондеры, прекратят лечение на 28-й неделе и будут переведены к 2-недельному периоду последующего наблюдения. Гистологически обусловленные респондеры продолжат исследование с использованием той же дозы.
b Причиной для внепланового визита будет необходимость формирования процедур по усмотрению исследователя.
с Субъект должен появиться в исследовательском центре не позднее 7 дней с момента определения необходимости прерывания лечения. Если это невозможно сделать ввиду SAE или другого непредвиденного обстоятельства, визит может быть совершен субъектом или членом его семьи по телефону. В eCRF должно быть зафиксировано, почему субъект не смог посетить исследовательский центр.
d Физическое обследование будет включать обследования веса, внешнего вида пациента, головы и шеи, глаз и ушей, носа и горла, грудной клетки, легких, сердца, брюшной полости, конечностей и суставов, лимфатических узов, кожи и неврологический осмотр.
е Показатели жизненно важных функций, которые должны быть отмечены, включают частоту сердечных сокращений, частоту дыхательных движений, температуру (°С; после 5-минутного отдыха в сидячем положении) и кровяное давление (измеряемое на одной и той же руке на протяжении всего исследования).
f Отборы образцов по времени необходимо проводить в интервале 07:00-8:00 или как можно ближе к это интервалу, насколько это возможно. Субъекты не должны принимать пищу в течение в течение 8-часового периода времени до проведения оценок уровня кортизола в сыворотке крови. Кровь для анализа уровня кортизола в сыворотке крови (утром натощак) может быть взята в пределах ±3 дней от планового визита, чтобы соответствовать точному временному графику. При желании отбор крови для проведения других анализов, запланированных на время данного визита, может быть выполнен одновременно.
g Если при последнем визите в ходе лечения субъекта сообщается об аномальном уровне кортизола в сыворотке крови, то может потребоваться проведение дополнительного мониторинга и АСТН теста (см. раздел 6.3.4).
h Ожидается, что обычно EGD будет проведена во время, не совпадающее со временен выполнения других процедур для заданной временной точки. Как EGD, так и любые другие процедуры, показанные для визита по ходу исследования, должны быть выполнены в пределах временного интервала для этого визита по ходу исследования. В случае скрининга EGD должна быть проведена после подписания ICF и в течение 2 или более недель до даты ожидаемой рандомизации.
i При проведении EGD эндоскопист определит EREFs и получит приблизительно 3 биоптата как из проксимальной, так и дистальной частей пищевода (в общей сложности 5-6 биоптатов). Гистологические образцы будет оценивать специалист по лабораторной диагностике.
j Если субъект прерывает исследование ввиду отсутствия эффективности или по другим причинам, исследователь может провести EGD, если к этому есть клинические показания.
k Тест стимуляции АСТН (250 мкг) будет проведен (во время внепланового визита) для оценки угнетения НРА оси, вызывающего клиническую озабоченность, как приведено ниже:
1) для любого момента времени, когда субъект имеет утренний уровень кортизола в сыворотке крови 5 мкг/дл (138 нмоль/л) или менее (подтвержденный 2-мя отборами крови);
2) для любого момента времени, когда субъект отмечает симптомы гиперадренокортицизма (приложение 6).
l Субъекты, которые прерывают исследование ввиду подтверждения угнетения НРА оси, будут проходить тест стимуляции АСТН (250 мкг) при визите по поводу преждевременного завершения.
m Для визита по поводу преждевременного завершения исследователь должен зафиксировать любое наличие АЕ (14±5 суток после прерывания лечения). Субъекты, прервавшие лечение по причине проблем с НРА, должны отслеживаться до получения разрешения.
n Только если визит связан с изменением симптомов ЕоЕ.
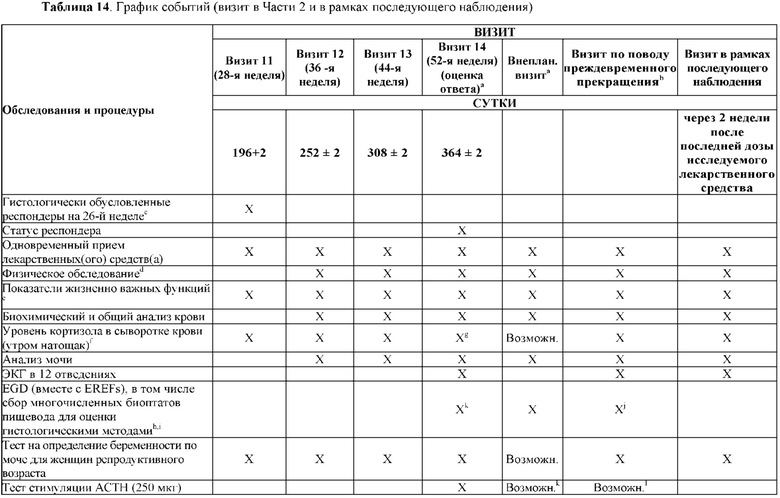
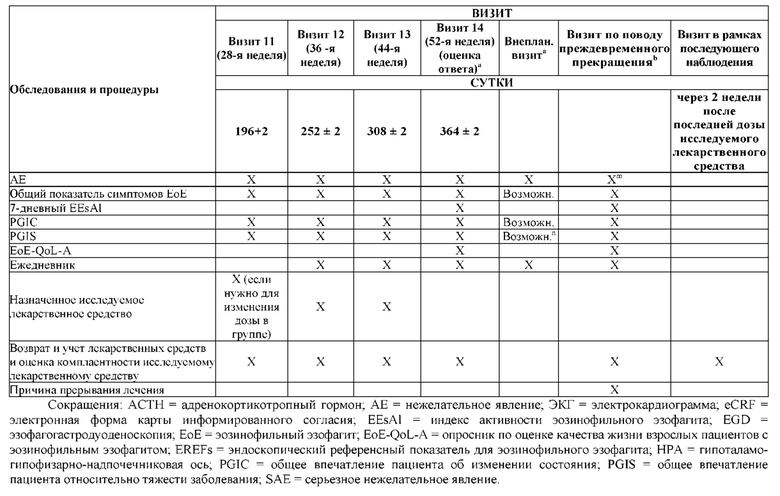


Скрининг
Период скрининга составляет 4 недели (28 суток). Наряду с отчетами, подтверждающими первичный диагноз ЕоЕ у субъекта, исследователь будет оценивать критерии пригодности (см. раздел 4.2.1 и раздел 4.2.2) субъекта на основе результатов скрининга. Общий показатель ЕоЕ должен быть более 3, чтобы субъект мог продолжить участие в этом исследовании.
Процедуры EGD для определения пригодности (см. критерий включения №7: наличие признаков ЕоЕ, что определяется по максимальному количеству эозинофилов/HPF 15 или более. Чтобы гарантировать проведение диагностики, необходимо будет выполнить по меньшей мере 5-6 процедур биопсии, включая получение образцов как из проксимальной, так и дистальной части пищевода (приблизительно по 3 из каждой)), которые будут проведены во время периода скрининга, и эти биоптаты должны быть получены главным специалистом по лабораторной диагностике в сроки, указанные в этом критерии включения. EGD должна быть проведена перед визитом 2 (до 4-недельного периода оценки исходных симптомов).
Тест стимуляции АСТН также должен быть проведен перед визитом 2 (до 4-недельного периода оценки исходных симптомов).
С одобрения медицинского наблюдателя субъект может быть подвергнут повторному скринингу еще один раз, если предыдущей причины неудачного скрининга больше нет, или субъект был исключен по личным или семейным причинам, которые с тех пор были устранены. Если субъект проходит повторный скрининг, то будет подписана новая форма информированного согласия (ICF), и ей будет присвоен новый номер. Все тесты и обследования должны быть проведены повторно.
4-Недельный период оценки исходных симптомов (вводная фаза исследования с применением плацебо)
Чтобы быть зачисленными в эту фазу, субъекты должны:
удовлетворять всем критериям включения, которые возможны для оценки перед 4-недельным периодом оценки исходных симптомов, в том числе значению общего показателя ЕоЕ более 3 (критерию включения №5; показатель ЕоЕ должен оставаться более 3 в каждый из визитов 1, 2 и 3 до рандомизации, или будет считаться, что результаты скрининга субъекта являются неудачными), за исключением тех, которые должны оцениваться во время 4-недельного периода оценки исходных симптомов: критерию включения №7: наличие признаков ЕоЕ, что определяется по максимальному количеству эозинофилов/HPF 15 или более; критерию включения №8: наличие в ежедневнике сообщений о 3 или более днях с эпизодами дисфагии из 7 дней в течение последних 14 суток 4-недельного периода оценки исходных симптомов; критерию включения №9: ведение ежедневника по меньшей мере в течение 5 дней из каждых 7 дней в течение последних 14 суток оценки исходных симптомов,
удовлетворять отсутствию критериев исключения, что будет оцениваться перед 4-недельным периодом оценки исходных симптомов (т.е. всех, за исключением критерия исключения №23: уровень кортизола в сыворотке крови менее 18 мкг/дл (497 нмоль/л) через 60 минут после теста стимуляции адренокортикотропным гормоном (АСТН) с применением 250 мкг косинтропина (т.е. случай положительного результата теста стимуляции АСТН)).
Субъект совершит визит в исследовательский центр в начале 4-недельного периода оценки исходных симптомов для выполнения процедур и обследований, указанных в Таблице 4-1.
Во время 4-недельного периода оценки исходных симптомов будет проведено определение тяжести исходных симптомов и будет оценена способность субъекта соответствовать записям в дневнике. Также можно оценить патологию по EGD-биоптатам (или раньше, если это возможно).
Рандомизация
Подтверждение пригодности
Чтобы быть пригодными к рандомизации, субъекты должны удовлетворять всем критериям включения/исключения, в том числе приведенным далее критериям включения, которые, как ожидается, должны быть подтверждены во время 4-недельного периода оценки исходных симптомов, а именно: критерию включения №7: наличие признаков ЕоЕ, что определяется по максимальному количеству эозинофилов/HPF 15 или более; критерию включения №8: наличие в ежедневнике сообщений о 3 или более днях с эпизодами дисфагии за 7 дней в течение последних 14 суток 4-недельного периода оценки исходных симптомов; критерию включения №9: ведение ежедневника по меньшей мере в течение 5 дней из каждых 7 дней в течение последних 14 суток периода оценки исходных симптомов; и критерию исключения №23: уровень кортизола в сыворотке крови менее 18 мкг/дл (497 нмоль/л) через 60 минут после теста стимуляции адренокортикотропным гормоном (АСТН) с применением 250 мкг косинтропина (т.е. случаю положительного результата теста стимуляции АСТН).
Если исследователь подтверждает удовлетворение критериям пригодности, то он или она будет проводить рандомизацию субъекта с использованием интерактивной системы веб-ответа (IWRS). IWRS подтвердит пригодность субъекта в плане результатов гистологического исследования (т.е. подтвердит гистологические доказательства диагноза ЕоЕ, соответствующие критерию включения № 7: доказательства наличия ЕоЕ, что определяется по максимальному количеству эозинофилов/HPF 15 или более) и предоставит рандомизационый номер исследователю и спонсору.
Схема рандомизации
В общей сложности 100 взрослых субъектов будут рандомизированы в соответствии с получением 1-й из 4-х доз АРТ-1011 (1,5 мг HS, 1,5 мг BID, 3 мг HS и 3 мг BID) или плацебо. Субъекты в группах с лечением в режиме HS будут получать плацебо через 30 минут после завтрака с целью сохранения режима слепого введения.
Рандомизация будет проведена двойным слепым способом с использованием единой системы IWRS, и пациенты будут разделены согласно наличию или отсутствию ранее стриктур пищевода либо имеющимся на момент скрининга стриктурам пищевода и согласно имеющемуся ранее положительному стероидному ответу на любое полученное ранее лечение кортикостероидами для лечения ЕоЕ у субъекта с зарегистрированными демографическими данными. Рандомизация для субъектов в Части 1 будет проведена с разделением согласно применению, в результате чего во время исследования на каждую из 5-ти получающих дозу групп будет приходиться сопоставимая процентная доля субъектов.
Лечение
На Фиг. 5 приведено общее представление лечения, которое будет получать каждый субъект, определяемое для всего FLUTE (на основании статуса респондера). Оценка улучшения или ухудшения симптомов будет проводиться постоянно.
Фаза индукции (с 1-х суток по 14-ю неделю)
Во время Части 1 субъекты будут получать лечение согласно своей рандомизации в течение 14 недель.
На 12-й неделе субъекты будут проходить процедуру оценки ответа, включая EGD для оценки эндоскопического и гистологического статуса. После представления способа в разделе 6.2 необходимо будет определить статус респондера (как определено в разделе 4.1.1) и информировать свой исследовательский центр. Также будут оценены симптомы. Результаты, полученные за период 14 суток до 12-й недели, будут сравниваться с таковыми для 14 суток до рандомизации.
Гистологически обусловленные респондеры и нереспондеры (на 12-й неделе) будут зачислены в Часть 2 (см. раздел 4.1.6.2).
Поддерживающая фаза (с 14-й недели по 52-ю неделю)
В Части 2 все субъекты, классифицированные как гистологически обусловленные респондеры на 12-й неделе, будут продолжать лечение согласно получающей дозу группе, в которую они были рандомизированы для Части 1. Субъекты могут продолжать принимать эту дозу в течение до 9 месяцев включительно по завершении Части 1.
Субъекты, являющиеся гистологически обусловленными нереспондерами (см. раздел 4.1.1) на 12-й неделе, будут получать лечение в режиме простого слепого исследования 3 мг, BID, в Части 2.
На 26-й неделе субъекты будут проходить EGD с биопсией для оценки гистологического ответа. После представления способа в разделе 6.2 необходимо будет определить статус респондера (как определено в разделе 4.1.1) и информировать свой исследовательский центр. Также будут оценены симптомы. Все субъекты, классифицированные как гистологически обусловленные респондеры, будут продолжать лечение согласно получающей дозу группе, в которую они были рандомизированы для части 1 до 52-й недели включительно. Результаты, полученные за период 14 суток до 26-й недели, будут сравниваться с таковыми для 14 суток до рандомизации.
Субъекты, являющиеся гистологически обусловленными нереспондерами (см. раздел 4.1.1) на 26-й неделе, прекратят лечение на 28-й неделе, будут зачислены в 2-недельный период последующего наблюдения и покинут исследование.
Для субъектов, которые завершают исследование на 52-й неделе, будет выполнена оценка ответа на лечение, включая EGD для оценки эндоскопического и гистологического статуса. После представления способа в разделе 6.2 необходимо будет определить статус респондера (как определено в разделе 4.1.1) и информировать свой исследовательский центр. Также будут оценены симптомы. Субъект будет проходить лечение исследуемым лекарственным средством исследуемым лекарственным средством до проведения EGD, ассоциированной с визитом на 52-й неделе. Результаты, полученные за период 14 суток до 52-й недели, будут сравниваться с таковыми для 14 суток до рандомизации.
Субъекты будут совершать визит в рамках последующего наблюдения через 2 недели после получения заключительной дозы исследуемого лекарственного средства (см. раздел 4.1.8).
ИССЛЕДОВАНИЯ ЭФФЕКТИВНОСТИ, БЕЗОПАСНОСТИ И ФАРМАКОКИНЕТИКИ
Исходные и демографические данные, рекомендованные для обобщения, включают возраст, пол, вес, расовую принадлежность (белый европеоид, негроид, монголоид, коренной американец или другие), этническую принадлежность (испаноязычный, не испаноязычный или другие) и отношение к курению.
Оценка эффективности и сообщаемых пациентом результатов лечения
Эзофагогастродуоденоскопия: для задач настоящего исследования ожидается, что основное внимание при проведении процедур EGD будет уделено пищеводу.
По возможности все процедуры EGD в данном исследовании должен проводить один и тот же эндоскопист.
Множественные процедуры биопсии пищевода
Как минимум 3 биоптата будут получены как из проксимальной, так и дистальной частей пищевода (в общей сложности 6 или более биоптатов) во время проведения EGD. Следует соблюдать осторожность при получении биоптатов, которые должны быть достаточного размера и быть непрозрачными. При отборе неоптимальных биоптатов следует предпринимать дополнительные попытки. Предполагается, что для достижения оптимальных результатов биоптаты должны быть получены по отдельности.
Все биоптаты следует хранить при комнатной температуре.
Гистология эозинофилъного эзофагита
Главный специалист по лабораторной диагностике проведет оценку всех биоптатов пищевода и подсчет максимального количества эозинофилов/HPF. Требования к пригодности с гистологической точки зрения описаны в абзаце Критерии включения (например, наличие признаков ЕоЕ, что определяется по максимальному количеству эозинофилов/HPF 15 или более по меньшей мере с использованием 5-6 биоптатов, включая образцы как из проксимальной, так и дистальной частей пищевода (по 3 из каждой)).
Эндоскопический референсный показатель для эозинофильного эзофагита
Эндоскопист зарегистрирует значение наблюдаемого эндоскопического референсного показателя (EREFs) для эозинофильного эзофагита 27, учитывающего отек, образование борозд, экссудаты, кольца, стриктуры, несколько вспомогательных признаков, и оценку врачом общей активности заболевания (заболевание отсутствует, степень слабая, умеренная, тяжелая) для каждого случая EGD. Показано, что EREFs является надежным диагностическим инструментом как для диагностирования ЕоЕ, так и для оценки ответа на лечение.
Заполнение рабочего журнала для EREFs будет проведено эндоскопистом, а перенесение данных в соответствующую eCRF будет выполнено медицинским персоналом исследовательского центра/координатором исследования. Рабочий журнал должен быть сохранен в исследовательском центре в качестве исходного документа.
Ежедневник
Субъект будет делать ежедневные записи в дневнике для оценки наличия дисфагии и отметки вопросов, связанных с ее тяжестью и ассоциированной с ней болью. Субъект будет заполнять дневник ежедневно на протяжении всего исследования.
Эти данные будут самостоятельно сообщены субъектом в электронном виде, автоматически перенесены в провайдер для хранения сообщаемых в электронном виде пациентом результатов лечения (ePRO) и после этого переданы в клиническую базу данных.
Общий показатель симптомов эозинофильного эзофагита
Для определения общего показателя симптомов ЕоЕ субъект будет отвечать на следующий вопрос:
насколько тяжелыми по шкале от 0 до 10 (0 соответствует отсутствию каких-либо симптомов, а 10 соответствует наиболее тяжелым симптомам), были у него симптомы эозинофильного эзофагита за последние 7 суток?
Эти данные будут самостоятельно сообщены субъектом в электронном виде, автоматически перенесены в ePRO провайдер и после этого переданы в клиническую базу данных.
7-Дневный индекс оценки симптомов эозинофильного эзофагита
Оценка симптомов будет проводиться периодически с использованием опросника для 7-дневного EEsAI и будут рассчитаны как общий, так и субпоказатели. Субпоказатели будут включать такие симптомы, как дисфагия, отказ от пищи и изменение рациона питания и болезненное глотание (одинофагия).
Также будут оценены дополнительные вопросы, используемые в качестве «якорных точек» для 7-дневного показателя EEsAI. Эти данные будут самостоятельно сообщены субъектом «на бумаге» и после этого переданы в клиническую базу данных исследовательским центром.
Оценка симптомов субъектом
Для изучения оценки субъектом симптомов ему будут заданы приведенные далее вопросы согласно анкете «Общее впечатление пациента об изменении состояния» (PGIC):
по сравнению с началом исследования, до начала Вами лечения, имеющиеся у Вас на сегодня симптомы ЕоЕ: намного ухудшились; ухудшились в средней степени; немного ухудшились; остались прежними; немного улучшились; улучшились в средней степени; намного улучшились. Пожалуйста, подумайте обо всех испытываемых вами симптомах, обусловленных ЕоЕ, и сделайте общее заключение, выбрав 1 из приведенных выше вариантов;
по сравнению с началом исследования, до начала Вами лечения, имеющееся у Вас на сегодня затруднение, связанное с прохождением пищи или таблеток: намного ухудшилось; ухудшилось в средней степени; немного ухудшилось; осталось прежним; немного улучшилось; улучшилось в средней степени; намного улучшилось. Пожалуйста, подумайте об имеющемся у вас затруднении, связанном с прохождением пищи или таблеток, и сделайте общее заключение, выбрав 1 из приведенных выше вариантов.
Эти данные будут самостоятельно сообщены субъектом в электронном виде, автоматически перенесены в ePRO провайдер и после этого переданы в клиническую базу данных.
Оценка субъектом тяжести симптомов
Далее приведены вопросы согласно анкете «Общее впечатление пациента относительно тяжести заболевания» (PGIS), которые будут заданы для изучения оценки субъектом тяжести симптомов.
Пожалуйста, выберите ответ, который лучше всего описывает тяжесть испытываемых Вами симптомов ЕоЕ за последнюю неделю (отметьте один ответ): симптомы отсутствуют; слабые; умеренные; тяжелые; очень тяжелые. Пожалуйста, подумайте обо всех испытываемых Вами симптомах, обусловленных ЕоЕ, и сделайте общее заключение об их тяжести, выбрав 1 из приведенных выше вариантов.
Пожалуйста, выберите ответ, который лучше всего описывает имеющееся у Вас на сегодня затруднение, связанное с прохождением пищи или таблеток: за последнюю неделю (отметьте один ответ): затруднение отсутствует; степень слабая; умеренная; тяжелая; очень тяжелая. Пожалуйста, подумайте об имеющемся у Вас затруднении, связанном с прохождением пищи или таблеток, и сделайте общее заключение о его тяжести, выбрав 1 из приведенных выше вариантов.
Эти данные будут самостоятельно сообщены субъектом в электронном виде, автоматически перенесены в ePRO провайдер и после этого переданы в клиническую базу данных.
Опросник по оценке качества жизни взрослых пациентов с эозинофилъным эзофагитом
Опросник по оценке качества жизни взрослых пациентов с эозинофильным эзофагитом (EoE-QoL-A) 29 позволяет узнать оценку, которая может быть дана непосредственно субъектами для определения того, как ЕоЕ влияет на качество их жизни. Он включает 30 вопросов по 5-бальной шкале Лайкерта; вопросы касаются 5 факторов: влияния на прием пищи/диету, социального воздействия, эмоционального воздействия, болезненного тревожного состояния и связанного с удушьем тревожного состояния, и было подтверждено, что они коррелируют с установленными связанными с состоянием здоровья показателями качества жизни. Более высокие показатели указывают на лучшее качество жизни.
Эти данные будут самостоятельно сообщены субъектом в электронном виде, автоматически перенесены в ePRO провайдер и после этого переданы в клиническую базу данных.
Конечные точки эффективности
Первичная конечная точка эффективности, для оценки ЕоЕ-ответа на 12-й неделе будет рассмотрена следующая первичная конечная точка эффективности.
Гистология: процентная доля субъектов с максимальным количеством эозинофилов/HPF 6 или менее после оценки по меньшей мере 5-6 биоптатов из проксимальной и дистальной частей пищевода, при этом HPF площадь составляет 0,235 квадратных миллиметров (увеличение объектива 40 с окуляром 22 мм).
Вторичные конечные точки эффективности
Будут оценены следующие вторичные конечные точки эффективности: устойчивый ЕоЕ-ответ: процентная доля субъектов, которые соответствовали первичной конечной точке (гистология) на 12-й неделе и сохраняли показатели первичной конечной точки на 26-й неделе и 52-й неделе; изменение по сравнению с исходными EREFs на 12-й неделе, 26-й неделе и 52-й неделе; изменения по данным эндоскопии, которые будут основаны на оценке EREFS, учитывающем следующие эндоскопические признаки: отек, пищеводные кольца, экссудаты, борозды, стриктуру и несколько вспомогательных признаков («гофрированный» пищевод, узкий диаметр пищевода и эрозии пищевода); процентная доля субъектов с максимальным количеством эозинофилов/FIPF менее 1 и менее 15 на 12-й неделе, 26-й неделе и 52-й неделе; изменение по сравнению с исходным общим показателем симптомов ЕоЕ, который будет оцениваться в течение 7-дневного периода перед следующими визитами по ходу исследования: на 12-й неделе, 26-й неделе и 52-й неделе; в отношении дисфагии: изменение числа эпизодов дисфагии, взятого в качестве исходного уровня (в течение 14-дневного периода до рандомизации), по сравнению с таковым для 14-дневного периода перед представляющим интерес моментом времени (на 12-й неделе, 26-й неделе и 52-й неделе); изменение по сравнению с исходным 7-дневным общим показателем EEsAI на 12-й неделе, 26-й неделе и 52-й неделе; изменение по сравнению с исходными 7-дневными субпоказателями EEsAI на 12-й неделе, 26-й неделе и 52-й неделе; процентная доля субъектов со средним 7-дневным общим показателем EEsAI менее 20 на 12-й неделе, 26-й неделе и 52-й неделе; изменение по сравнению с исходным PGIS, оцениваемое до рандомизации на 4-й, 8-й, 12-й, 14-й, 18-й, 22-й, 26-й, 28-й, 36-й, 44-й и 52-й неделях; pGIC на 4-й, 8-й, 12-й, 14-й, 18-й, 22-й, 26-й, 28-й, 36-й, 44-й и 52-й неделях. Оценка неудачного результата лечения и рецидива, включая: процентную долю нереспондеров в соответствии с дозой на 14-й недели, в интервале от 14-й до 28-й недели и в интервале от 28-й до 52-й недели; процентную долю субъектов, нуждающихся в неотложном эндоскопическом освобождении от пищи, в соответствии с дозой на 12-й неделе, 26-й неделе и 52-й неделе; процентную долю субъектов, нуждающихся в дилатации пищевода, в соответствии с получающей дозу группой и Частью исследования.
Эксплоративные конечные точки эффективности
Будет проведена оценка следующих эксплоративных конечных точек эффективности:
в отношении дисфагии: изменение числа эпизодов дисфагии, взятого в качестве исходного уровня (в течение 14-дневного периода до рандомизации), по сравнению с таковым для 14-дневного периода перед представляющим интерес моментом времени (на 12-й, 26-й и 52-й неделях); изменение по сравнению с исходным числом дней без дисфагии для 14-дневного периода перед следующими визитами по ходу исследования: на 4-й неделе, 8-й неделе, 12-й неделе, 14-й неделе, 18-й неделе, 22-й неделе, 26-й неделе, 28-й неделе, 36-й неделе, 44-й неделе и 52-й неделе; устойчивый ЕоЕ-ответ (в отношении дисфагии): процентная доля всех субъектов, которые соответствовали вторичной конечной точке по дисфагии на 12-й неделе и сохраняли связанный с дисфагией ответ на 26-й неделе и 52-й неделе; оценка ФК/ФД (кортизола) и взаимосвязей ответа и воздействия (эффективности); оценка субъектом симптомов по сравнению с предыдущим визитом на 4-й неделе, 8-й неделе, 12-й неделе, 14-й неделе, 18-й неделе, 22-й неделе, 26-й неделе, 28-й неделе, 36-й неделе, 44-й неделе и 52-й неделе и визит по поводу преждевременного завершения (если такой предусмотрен); оценка HRQoL на основании показателя EoE-QoL-A, взятого в качестве исходного уровня, на 12-й неделе, 26-й неделе, 52-й неделе для всех субъектов в зависимости от дозы и подгруппы; данные о субъектах, получающих простое слепое (для субъекта) лечение (в Части 2) будут сведены отдельно;
процентная доля субъектов, которые были классифицированы как гистологически обусловленные нереспондеры на 12-й неделе и имеют максимальное количество эозинофилов/HPF 6 или менее во всех подвергнутых биопсии местах пищевода на 26-й неделе и 52-й неделе;
изменение по сравнению с исходным числом эпизодов дисфагии в течение 14-дневного периода до 26-й недели и 52-й недели для субъектов, которые были классифицированы как нереспондеры на 12-й неделе;
процентная доля субъектов, которые были классифицированы как гистологически обусловленные нереспондеры на 12-й неделе и соответствуют первичной конечной точке на 26-й неделе и 52-й неделе;
схема выставления балльных оценок и различные конечные точки будут получены исходя из DEDI. Подлежащие анализу популяции будут подробно описаны в Плане статистического анализа (SAP). Анализ подгрупп может быть проведен с учетом возраста, пола, PPI статуса и других исследуемых субпопуляций.
Пример 5. Фаза 1 исследования для оценки фармакокинетики, безопасности и переносимости разовой дозы флутиказона пропионата, вводимой в условиях после приема пищи или натощак либо перед сном, в рандомизированном трехстороннем перекрестном формате
Первичная задача, оценивали фармакокинетику (ФК) флутиказона пропионата (FP) в плазме крови после введения в условиях после приема пищи или натощак утром.
Вторичные задачи, проводили оценку содержание FP в плазме крови (ФК) после введения перед сном (HS). Проводили оценку безопасности/переносимости после введения разовой дозы FP здоровым взрослым добровольцам мужского и женского пола, для случая введения в условиях натощак, после приема пищи или HS.
Первичная конечная точка, определяли Cmax, AUClast и Тmax для FP после введения FP в условиях после приема пищи и натощак в случае утреннего приема дозы.
Вторичные конечные точки, определяли AUC0-24, AUCinf, AUCext, λz, t1/2, CL/F и Vz/F для FP после введения FP в условиях после приема пищи и натощак. Определяли AUC0-24, AUCinf, AUCext, λz, t1/2, CL/F и Vz/F для FP после введения FP в условиях введения в режиме HS. Рассчитывали дополнительные ФК параметры, которые необходимы для адекватного исследования фармакокинетики FP после введения утром (натощак и после приема пищи) или введения в режиме HS. При необходимости рассчитывали число случаев возникающих при лечении нежелательных явлений (АЕ), АЕ, приводящих к отмене лечения, и серьезных нежелательных явлений (SAE), сгруппированных согласно классу по поражению органов и систем органов (SOC). При необходимости определяли клинически значимые аномальные значения для клинической лаборатории, AM уровень кортизола (138 нмоль/л или ниже), проводили анализ мочи, снимали показатели жизненно важных функций и электрокардиограммы (ЭКГ).
План и продолжительность исследования: это исследование представляло собой рандомизированное трехстороннее перекрестное исследование разовой дозы по оценке эффектов введения 6 мг FP утром (AM) в условиях после приема пищи или натощак либо перед сном (HS), через 4 часа после вечернего приема пищи. В общей сложности были включены и рандомизированы 24 субъекта с попаданием в одну из шести групп с различной последовательностью применения каждой из этих трех схем лечения в течение трех периодов времени с 7-дневным периодом «вымывания» между приемом доз. Эти три схемы лечения включали введение 6 мг FP (2 перорально распадающиеся таблетки по 3 мг) в следующих условиях:
A) натощак: 6,0 мг АРТ-1011 (например, FP) (две перорально растворимые таблетки по 3 мг) после воздержания от приема пищи в течение ночи (по меньшей мере в течение 10 часов), с воздержанием от приема пищи, продолжающимся в течение 4 часов после приема дозы;
B) после приема пищи: 6,0 мг АРТ-1011 (две перорально растворимые таблетки по 3 мг) через 30 минут после начала завтрака с высоким содержанием жиров;
C) HS: 6,0 мг АРТ-1011 (две перорально растворимые таблетки по 3 мг) перед сном приблизительно через 4 часа после обычного вечернего приема пищи.
• Субъектам, получающим лечение в условиях «натощак» (по схеме лечения А), вводили по 6,0 мг АРТ-1011 после воздержания от приема пищи в течение ночи (по меньшей мере в течение 10 часов). Эти субъекты продолжали воздерживаться от приема пищи в течение по меньшей мере 4 часов после приема дозы.
• Субъекты, получающие лечение в условиях «после приема пищи» (по схеме лечения В), принимали завтрак с высоким содержанием жира через 30 минут после воздержания от приема пищи в течение ночи по меньшей мере в течение 10 часов. Субъектам вводили по 6,0 мг АРТ-1011 через 30 минут после начала приема завтрака с высоким содержанием жира.
• Субъекты, получающие лечение «в режиме HS» (по схеме лечения С), получали стандартное питание приблизительно за 4 часа перед сном. Субъектам вводили по 6,0 мг АТР-1011 непосредственно перед перед сном (через 4 часа после обычного приема пищи). Субъектам вводили дозу FP в лежачем положении.
Рандомизация (1-е сутки): субъектов рандомизировали с попаданием в одну из шести групп с различной последовательностью (см. Таблицу 15) получения разовой дозы согласно каждой из этих схем лечения в течение трех периодов времени с 7-дневным периодом «вымывания» между приемом доз.
Периоды лечения: для каждого периода времени субъекты находились в стационаре в течение 5 суток (сутки 0-4, сутки 7-11, сутки 14-18), при этом ФК образцы собирали в течение 72 часов после каждой дозы. Регистрацию субъектов проводили в CRU (отделение клинических исследований) вечером перед каждым приемом дозы (в 0-е, 7-е и 14-е сутки), и они получали лечение в соответствии с последовательностью для группы, в которую они были рандомизированы в 1-е сутки. АРТ-1011 вводили в разовых дозах в 1-е, 8-е и 15-е сутки. Времена всех приемов пищи (AM и за 4 часа перед сном) были стандартизированы для всех субъектов и всех периодов времени. Субъекты завершали приемы пищи через 30 минут. Серийные образцы плазмы крови для ФК исследований FP отбирали до приема дозы и в моменты времени 0,5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, 20, 24, 30, 36, 42, 48, 60 и 72 часа после каждой дозы. Субъекты, получающие дозу HS, следовали тому же ФК графику для ФК образцов, запланированных относительно дозы, принимаемой перед сном.
Субъектов выписывали из CRU после проведения ФК контроля и обследований в отношении безопасности для каждого периода лечения (на 4-е сутки, 11-е сутки, 18-е сутки).
Последующее наблюдение (25-е сутки ±2 суток): после 3-го периода субъекты возвращались в клинику в пределах 7 суток (±2 суток) после выписки из отделения клинических исследований для определения критериев последующего наблюдения.
Критерии включения: субъекты женского и мужского пола были пригодны для включения в это исследование, только если удовлетворяли всем из приведенных ниже критериев.
1. Субъект считался здоровым на основании медицинской карты, физического обследования, результатов клинических лабораторных анализов и ЭКГ в 12 отведениях. Субъектов с историей легкого острого или хронического медицинского заболевания или с лабораторными показателями, выходящими за пределы нормального референсного диапазона, включали только в случае получения согласия исследователя, что эти данные вряд ли внесут дополнительный риск и не повлияют на процедуры исследования.
2. Утвержденное Экспертным советом медицинского учреждения (IRB) информированное согласие было подписано и датировано до начала любых относящихся к исследованию действий.
3. Возраст субъекта составлял от 18 до 55 лет включительно.
4. До рандомизации у субъекта женского пола репродуктивного возраста был отрицательный результат теста на определение беременности по моче или была проведена хирургическая стерилизация, было диагностировано бесплодие или постменопаузальное состояние. Женщины репродуктивного возраста, чтобы удовлетворять критериям включения, должны быть готовы применять адекватный контроль за рождаемостью в течение по меньшей мере 28 суток до первого приема исследуемого лекарственного средства, во время исследования и в течение по меньшей мере 30 суток после приема последней дозы исследуемого лекарственного средства.
5. Субъекты мужского пола, которые вели половую жизнь, должны дать согласие на применение презерватива и спермицида во время исследования.
6. Индекс массы тела от 19 до 30 кг/м2 (вес/(рост)2) включительно.
7. В момент скрининга определяются нормальные (или аномальные, но клинически несущественные) значения лабораторных показателей.
8. Субъект обладал способностью понимать требования исследования и готовностью соблюдать все процедуры исследования.
9. Субъект не употреблял и дает согласие воздержаться от приема любых пищевых добавок, фитотерапевтических средств или отпускаемых без рецепта лекарственных средств (за исключением случаев, разрешенных исследователем) в течение 7 суток до 1-го поступления в CRU (для 1-го периода) и на протяжении последующего наблюдения.
10. Субъект не употреблял и давал согласие воздержаться от приема любых отпускаемых по рецепту лекарственных средств (за исключением случаев, разрешенных исследователем) в течение 28 суток до 1-го поступления в CRU (для 1-го периода) и на протяжении последующего наблюдения.
11. Субъект не употреблял спиртосодержащие напитки в течение 3 суток до 1 -го поступления в CRU (для 1-го периода) и на протяжении последующего наблюдения.
12. Субъект давал согласие воздержаться от употребления продуктов, содержащих кофеин или шоколад, с момента поступления в CRU и до выписки в каждый период исследования.
13. Субъект давал согласие воздержаться от чрезмерных физических нагрузок с момента подписания формы информированного согласия и до заключительного визита в период последующего наблюдения.
Ключевые критерии исключения: субъект был не пригоден для включения в это исследование, если удовлетворял любому из приведенных ниже критериев.
1. Имел в анамнезе астму, требующую периодического применения ингаляционных кортикостероидов.
2. Имел недавнюю (в течение 30 суток) историю применения кортикостероидов, в том числе, но не ограничиваясь этим, кортикостероидов для интраназального, ингаляционного, интрадермального, офтальмологического или внутрисуставного путей введения.
3. Имел в анамнезе злоупотребление запрещенными лекарствами в прошлом году или текущие признаки такого злоупотребления по мнению исследователя на основе критериев, установленных в DSM-IV (4-е издание Руководства по диагностике и статистике психических расстройств).
4. Использовал табачные изделия в пределах 3 месяцев до начала 1-х суток этого исследования.
5. Имел положительные результаты анализа мочи на наличие наркотических средств или положительный результат теста на котинин.
6. Имел в анамнезе регулярное употребление алкоголя в течение 6 месяцев исследования, определяемое как среднее еженедельное потребление более 14 порций в неделю для мужчин или более 10 порций в неделю для женщин. Одна порция эквивалентна 12 г алкоголя: 5 унциям (150 мл) вина, или 12 унциям (360 мл) пива, или 1,5 унции (45 мл) 80-градусного дистиллированного спирта. Тест на содержание этанола в выдыхаемом воздухе проводили дважды и использовали для документального подтверждения отсутствия применения этанола при проведении скрининга и в 0-е сутки, а в случае положительного результата тест был основанием к исключению из исследования.
7. При проведении скрининговых обследований давал положительный результат (т.е. имел признаки текущей инфекции) на наличие вируса иммунодефицита человека (ВИЧ), гепатита В и/или гепатита С.
8. Субъект представлял собой женщину, которая была беременной или вскармливающей грудью.
9. Представлял собой субъект мужского пола, партнерша которого, как известно, была беременной.
10. Имел заболевание в острой форме в пределах 28 суток до первого поступления в CRU.
11. Имел какой-либо анамнез туберкулеза и/или профилактики туберкулеза.
12. Был донором плазмы крови в предшествующие 7 суток перед 1-ми сутками этого исследования.
13. Сдавал 500 мл крови или более (Canadian Blood Services, Hema-Quebec, клинические исследования и т.д.) в предшествующие 56 суток перед 1-ми сутками этого исследования.
14. Принимал индуктор или ингибитор ферментов семейства цитохрома Р450 3А (CYP3А) в течение 28 суток до приема первой дозы.
15. Употреблял грейпфрутовый сок в течение 7 суток до приема первой дозы.
16. Принимал участие в исследовании какого-либо другого экспериментального лекарственного средства в течение 30 суток до первого поступления в CRU, или у субъекта не прошел интервал времени, соответствующий 5 периодам полувыведения любого предыдущего экспериментального лекарственного средства, перед первым поступлением в CRU (в зависимости от того, что является более продолжительным).
17. Имел любое физическое, психическое или социальное состояние, историю болезни или отклонения лабораторных показателей от нормы, которые по мнению исследователя могут помешать процедурам исследования или возможности субъекта присоединиться к исследованию и завершить его.
18. Результаты клинических лабораторных анализов указывали на наличие релевантной почечной или печеночной недостаточности по мнению исследователя.
19. Имел в анамнезе любые состояния желудочно-кишечного тракта, почек или печени, которые могли бы повлиять на процесс всасывания или метаболизма/выделения экспериментального соединения.
20. Не мог или не хотел бы употреблять продукты из рациона обычной пищи с высоким содержанием жира из-за диетических предпочтений или ограничений.
21. Имел в анамнезе чувствительность к исследуемому лекарственному средству (флутиказона пропионату или другим кортикостероидам) или его компонентам либо имел в анамнезе лекарственную или другую аллергию (например, на яйца), которая по мнению исследователя служит противопоказанием для участия.
Анализ
Фармакокинетика: результаты описательной статистики ФК параметров для образцов плазмы крови обобщены после обработки. Для изучения ФК FP в состояниях натощак (как контрольного) и после приема пищи (как тестируемого) проводили дисперсный анализ (ANOVA) с точки зрения фиксированного эффекта в отношении лечения (в состоянии после приема пищи или натощак), с точки зрения фиксированного эффекта в отношении Периода (1, 2, 3), с точки зрения фиксированного эффекта в отношении последовательности (ABC, ВСА, CAB, СВА, АСВ, ВАС) и субъекта в последовательности в качестве случайного эффекта на преобразованные в логарифмической шкале параметры AUClast, AUC0-24, AUCinf и Cmax. Относительную биодоступность оценивали путем возведения в степень разницы в полученных методом наименьших квадратов средних значениях (тест - контроль) и соответствующего 90%-ного доверительного интервала (ДИ). Проводили дополнительные сравнения введения дозы в режиме HS и введения после приема пищи и натощак.


Значения концентрации FP обобщали с использованием описательной статистики (включающей n, среднее значение, стандартное отклонение (SD), коэффициент вариации (CV, %), медиану, минимальное и максимальное значения) для каждого варианта лечения.
Приведенные далее ФК параметры оценивали с использованием некомпартментных методов для образцов плазмы крови. Концентрации ниже предела количественного определения принимали равными нулю для всего ФК анализа. Для оценки ФК параметров учитывали все отклонения от запланированного по графику времени отбора образцов, составляющие 2 минуты или больше. В том случае, когда концентрации FP невозможно было определить по клиническим или биоаналитическим причинам, эти значения отмечали как пропущенные для фармакокинетического анализа.
Для определения площади под кривой использовали правило трапеций (линейную интерполяцию применительно к линейному правилу трапеций) и заключительную фазу оценивали, определяя максимальное значение коэффициента корреляции оцениваемого по модели логарифмически-линейной регрессии. Тем не менее, оценку параметров распределения для индивидуальных профилей зависимости концентрации от времени не проводили в том случае, когда конечную логарифмически-линейную фазу было невозможно надежно охарактеризовать.
Если концентрация FP до введения дозы детектировалась, то данные для такого субъекта включали в фармакокинетический анализ без корректировки, если концентрация до приема дозы была равна или была меньше 5% от значения Сmax соответствующего периода. Если концентрация до приема дозы составляла более 5% от значения Сmax, то данные для такого субъекта исключали из всех фармакокинетических оценок.
С учетом низкой ожидаемой системной биодоступности FP (примерно 1%) имеется вероятность, что расчет некоторых ФК параметров был проведен недостоверно ввиду низких концентраций FP в плазме крови (например, концентраций ниже предела количественного определения (BLQ)). Там, где было возможно, рассчитывали ФК параметры.
Результаты описательной статистики ФК параметров для образцов плазмы крови сведены в таблицы после обработки. Описательная статистика для ФК параметров включала арифметическое и геометрическое среднее значение, CV (%), SD среднего арифметического, медиану, минимальное, максимальное значения и п, при необходимости.
Для изучения ФК FP проводили дисперсный анализ (ANOVA) с точки зрения фиксированного эффекта в отношении лечения, с точки зрения фиксированного эффекта в отношении Периода (1, 2, 3), с точки зрения фиксированного эффекта в отношении последовательности (ABC, ВСА, CAB, СВА, АСВ, ВАС) и субъекта в последовательности в качестве случайного эффекта на преобразованные в логарифмической шкале параметры AUClast, AUC0-24, AUCinf и Cmax. Относительную биодоступность оценивали путем возведения в степень разницы в полученных методом наименьших квадратов средних значениях (тест - контроль) и соответствующего 90%-ного доверительного интервала (ДИ).
Анализ влияния пищи: 90%-ный доверительный интервал для экспоненты, показателем которой является разница в LSmeans (средние значения, рассчитанные методом наименьших квадратов) между В (после приема пищи) и А (натощак) рассчитывали для параметров, преобразованных по шкале натуральных логарифмов (геометрических LSmeans для отношения теста к контролю).
Анализ режима HS по сравнению с режимом натощак: 90%-ный доверительный интервал для экспоненты, показателем которой является разница в LSmeans между С (HS) и А (натощак) рассчитывали для параметров, преобразованных по шкале натуральных логарифмов (геометрических LSmeans для отношения теста к контролю).
Анализ режима HS по сравнению с режимом после приема пищи: 90%-ный доверительный интервал для экспоненты, показателем которой является разница в LSmeans между С (HS) и В (после приема пищи) рассчитывали для параметров, преобразованных по шкале натуральных логарифмов (геометрических LSmeans для отношения теста к контролю).
Статистические анализы осуществляли, используя SAS® (версию 9).
Объем выборки: с использованием оценки 54%, определенной с применением общей оценки вариабельности, составляющей 60%, и в предположении, что интра-субъектная вариабельность по оценкам составляет примерно на 10% меньшее значение. Тогда, в случае двустороннего 90%-ного доверительного интервала для нормального среднего объема выборки, составляющего 24, получается, что значение ширины на полувысоте составляет не более 0,25 с условной вероятностью 0,98.
Обоснование для проведения настоящего исследования: подход к лечению ЕоЕ посредством перорального введения APT-1011 заключался в обеспечении локального воздействия FP на пищевод при сведении к минимуму системного воздействия и ассоциированных с ним системных фармакологических эффектов. Предыдущие исследования продемонстрировали значительное ослабление воздействия при введении лекарственного средства после завтрака; например, введение АРТ-1011 после приема пищи с высоким содержанием жира значительно ослабляло системное воздействие по сравнению с введением в состоянии натощак. Кроме того, введение АРТ-1011 в дозе 6 мг, QD, приводило приблизительно к такому же доза-нормированному системному воздействию в стационарной состоянии (AUCtau/доза), что и в режиме 3 мг, BID. Однако данные сильно варьировали, причем межсубъектная вариабельность (геометрическое среднее CV %) в отношении AUC(0-24 ч) для FP после введения разовой дозы составляла более 55%.
Предыдущее ФК исследование продемонстрировало существенно более низкие уровни лекарственного средства при QD введении по сравнению с той же дозой, разделенной и введенной BID. Причина этого не выяснена.
В настоящем исследовании оценивали влияние пищи на фармакокинетику АРТ-1011. Воздействие АРТ-1011 сравнивали для случаев, когда утреннюю дозу вводят либо в состоянии натощак, либо через 30 минут после начала приема пищи с высоким содержанием жира. Кроме того, в настоящем исследовании охарактеризована фармакокинетика АРТ-1011 при введении в виде разовой дозы перед сном (HS).
Это исследование предоставляет руководство по приему лекарственного средства в зависимости от приема пищи и времени суток, выбранных для введения.
СХЕМЫ ЛЕЧЕНИЯ
Применяемые схемы лечения: введение исследуемого лекарственного средства выполняли участвующие в проведении исследования квалифицированные сотрудники только в соответствии с процедурами, описанными в этом протоколе. Все дозы АРТ-1011 составляли 6 мг, которые вводили в виде двух перорально распадающихся таблеток по 3 мг. Все субъекты получали инструкции по приему таблетки исследуемого лекарственного средства перорально без использования воды или других жидкостей. Субъекты помещали обе таблетки исследуемого лекарственного средства на верхнюю поверхность языка и осторожно растирали таблетку исследуемого лекарственного средства в полости рта между языком и нёбом, пока она не распадалась в степени, достаточной для осуществления обычного проглатывания со слюной в течение от одной до пяти минут. Субъекты получали инструкции не разжевывать или не разламывать таблетку исследуемого лекарственного средства и не глотать какую-либо часть неповрежденной таблетки исследуемого лекарственного средства до ее полного растворения. Субъекты не пили в течение по меньшей мере 1 часа после приема дозы и не принимали пищи в течение по меньшей мере 4 часов после приема дозы.
Ввиду низкой биодоступности АРТ-1011, для этого исследования выбирали дозу 6 мг. С учетом предыдущих данных концентрации FP после приема дозы 6 мг обеспечивают детектируемые уровни FP в плазме крови для большинства временных точек при использовании предложенного метода биоаналитического анализа.
Выбор дозы и временной график приема дозы для каждого субъекта
Все дозы АРТ-1011 составляли 6 мг, которые принимали одновременно в виде двух перорально распадающихся таблеток по 3 мг. В случае лечения «натощак» (по схеме лечения А) субъектам вводили дозы после в условиях отсутствия приема пищи в течение ночи, по меньшей мере в течение 10 часов. Стандартный прием пищи предлагали через 4 часа после введения доз. В случае лечения в условиях после приема пищи (по схеме лечения В), субъекты начинали принимать высококалорийную пищу с высоким содержанием жира в условиях отсутствия приема пищи в течение ночи, по меньшей мере в течение 10 часов, и за 30 минут до введения доз. Пищу потребляли в течение 30 минут или меньше.
В случае лечения в режиме HS субъектам вводили АРТ-1011 приблизительно через 4 часа после обычного приема пищи без каких-либо закусок после обеда. После приема лекарственного средства субъект сразу же ложился и не вставал в течение по меньшей мере 1 часа.
Фармакокинетика
Отбор, обработка и доставка фармакокинетических образцов: отбирали образцы крови (по 6 мл на один образец) и проводили их обработку для получения плазмы. Полученную плазму крови (приблизительно 3 мл) подразделяли на 2 образца (приблизительно по 1,5 мл каждый). Один из этих образцов использовали, а другой хранили при температуре -20°С или ниже в CRU до проведения исследования.
Биоаналитический метод для количественного определения флутиказона пропионата: концентрации флутиказона пропионата измеряли, используя метод с применением жидкостной хроматографии в сочетании с тандемной масс-спектометрией (LC/MS/MS), утвержденный в соответствии с руководящими нормативными указаниями США. Этот метод был разработан для 1 мл плазмы крови, собранной из крови, содержащей оксалат калия/фторид натрия в качестве антикоагулянта/консерванта. Данный метод является линейным в заданном диапазоне концентраций 1,00-500 пг/мл. Предел количественного определения составляет 1 пг/мл 20. Проводили анализ образцов от всех субъектов.
Оценка безопасности
Оценки безопасности включают оценку следующего: нежелательных явлений, как описанных в литературе, так и наблюдаемых, независимо от степени тяжести или серьезности; массы тела; показателей жизненно важных функций (кровяного давления, частоты сердечных сокращений, температуры, частоты дыхания); данных ЭКГ; результатов клинических лабораторных анализов и анализа мочи; данных физического обследования.
Клинические лабораторные анализы: использовали приведенные ниже клинические лабораторные анализы. Общий анализ крови, биохимический анализ крови, анализ свертывания крови и анализ мочи проводили при скрининге, в 0-е сутки (при поступлении в CRU), через 12 часов после приема дозы в каждый Период, перед выпиской из CRU в каждый Период и во время визита в рамках последующего наблюдения.
Общий анализ крови: количество эритроцитов (число красных кровяных телец (RBC)), гемоглобин, гематокрит, средний объем эритроцитов (MCV), среднее содержание гемоглобина в эритроците (МСН), средняя концентрация гемоглобина в эритроците (МСНС), количество лейкоцитов в крови (WBC) и лейкоцитарная форма, абсолютное содержание нейтрофилов (ANC) и содержание тромбоцитов.
Биохимический анализ крови/анализ свертывания крови: содержание аланинаминотрансферазы (ALT; SGPT (глутамат-пируват-трансаминаза из сыворотки крови)) и аспартатаминотрансферазы (AST; SGOT (глутамат-оксалоацетат-трансаминаза из сыворотки крови), общего билирубина, связанного билирубина, азота мочевины в крови (BUN), креатинина, щелочной фосфатазы, ионов натрия, калия, кальция, хлорида, глюкозы и альбумина.
Тест на определение беременности по моче: анализ мочи на содержание β-hCG (бета-субъединица хорионического гонадотропина человека) у женщин в фазах скрининга, проверки для каждого периода времени (т.е. в 0-е сутки, на 7-е сутки, на 14-е сутки) и последующего наблюдения.
Тест на содержание фолликуло-стимулирующего гормона (FSH)/лютеинизирующего гормона (LH): это тест выполняли для всех женщин менопаузального периода, включая женщин с хирургической менопаузой.
Анализ мочи: анализ мочи включал макроскопический анализ и микроскопический анализ только при указании на это посредством тест-полоски. Анализы включали: определение цвета, мутности, относительной плотности, рН, содержания глюкозы, белка, кетонов, уробилиногена, билирубина, наличия крови, нитритов и лейкоцитарной эстеразы.
Серология: комбинированное определение антигена и антитела (Ag/Ab Combo) к ВИЧ, определение гепатита В (HBsAg (В) (гепатит В)) и гепатита С (HCV (С)) (проводится только во время скрининга).
Проверка мочи на содержание наркотических средств: мочу для тестирования на содержание наркотических средств собирали у всех субъектов в течение периода скрининга и в 0-е сутки, на 7-е сутки и 14-е сутки. Образец тестировали на предмет присутствия котинина, кокаина, тетрагидроканнабинола, барбитуратов, амфетаминов, бензодиазепинов и опиатов. Субъектов с положительными результатами скринингового тестирования наличия наркотических средств в моче в отношении любого из указанных выше веществ, исключали из этого исследования.
Тест на содержание этанола в выдыхаемом воздухе: употребление алкоголя запрещалось в течение 3 дней до первого поступления в CRU и на протяжении последующего наблюдения. Тест на содержание этанола в выдыхаемом воздухе проводили во время скрининга и при поступлении в CRU в каждый период времени (т.е. в 0-е сутки, на 7-е сутки и 14-е сутки). Субъектов с положительными результатами теста на содержание этанола в выдыхаемом воздухе во время скрининга не включали в это исследование, чтобы избежать неумышленного зачисления субъекта с хроническим алкоголизмом. Субъектов с положительными результатами теста на содержание этанола в выдыхаемом воздухе в 0-е сутки (при поступлении в CRU) также отстраняли от дальнейшего участия в этом исследовании.
НЕЖЕЛАТЕЛЬНЫЕ ЯВЛЕНИЯ
Серьезное нежелательное явление (SAE) определяли, как любое нежелательное медицинское явление, которое при любой дозе; приводит к смерти; является опасным для жизни; требует госпитализации в стационар или продления существующей госпитализации, за исключением госпитализации по причинам административного характера (например, субъект не имеет возможности передвигаться по дому) или госпитализации для планового лечения имеющегося ранее состояния, которое не ухудшалось во время исследования, если только не наблюдается возникновение осложнения во время госпитализации; приводит к постоянному или существенному снижению трудоспособности/дееспособности; или приводит к врожденной(ому) аномалии/пороку развития.
РЕЗУЛЬТАТЫ
На Фиг. 6 и 7 показаны средние концентрации АРТ-1011 (флутиказона пропионата) в плазме крови для схем лечения в случае введения натощак, после приема пищи и в режиме HS. На Фиг. 8-10 показаны «спагетти»-кривые концентрации АРТ-1011 (флутиказона пропионата) в плазме крови в случае введения в состояниях натощак (Фиг. 8), после приема пищи (Фиг. 9) и в режиме HS (Фиг. 10). Неожиданным образом, введение в режиме HS находящемуся в лежащем положении пациенту перед сном демонстрирует устойчивый уровень лекарственного средства. Вместо резкого, быстрого повышения, которое наблюдалось при введении после приема пищи и натощак, HS введение в положении лежа на животе приводило к устойчивой концентрации АРТ-1011 в крови пациента. Это может быть связано с длительным воздействием на дистальную часть пищевода. Такое длительное воздействие на дистальную часть пищевода имеет дополнительное преимущество, заключающееся также в ослаблении системного воздействия АРТ-1011, что, как ожидается, уменьшит число неблагоприятных побочных эффектов.
В данное исследование были зачислены в общей сложности 24 участника, и 22 (92%) завершили исследование. Сводные данные по ФК параметрам в зависимости от тестируемой группы представлены в Таблице 18. AM введение доз ассоциировалось с более высокой скоростью всасывания в условиях введения после приема пищи по сравнению с введением натощак. После приема пищи с высоким содержанием жира обнаруживали более высокую максимальную концентрацию (для Сmax: соотношение [90% ДИ (доверительный интервал)] = 120,65% [99,84%-145,79%]) и меньшее время достижения максимальной концентрации по сравнению с введением в состоянии натощак (Тmax: после приема пищи = 5,00 ч; натощак = 10,00 ч). Тем не менее, наблюдали более слабое общее воздействие при введении в состоянии после приема пищи по сравнению с введением натощак (для AUClast: соотношение [90% ДИ] = 76,97% [67,64%-87,59%]). При приеме доз в режиме HS была обнаружена более низкая скорость всасывания по сравнению с условиями AM приема доз. В частности время достижения максимальной концентрации в случае приема в режиме HS (Тmax = 14 ч) было больше, чем в случае AM приема доз (Тmax: натощак = 10 ч, после приема пищи = 5 ч). Прием доз в режиме HS ассоциировался с более сильным общим воздействием (для AUCinf: соотношение [90% ДИ] = 122,36% [ 107,02%-139,88%] и более низким значением Сmax (для Сmax: соотношение [90% ДИ] = 67,79% [56,29%-81,64%]) по сравнению со схемой введения после приема пищи. По сравнению со схемой введения натощак для приема доз в режиме HS получали более слабое общее воздействие (для AUCinf: соотношение [90% ДИ] = 87,00% [75,24%-100,59%] и более низкое значение Сmax (для Сmax: соотношение [90% ДИ] = 81,78% [67,93%-98,47%]). Для всех режимов приема доз Сmax флутиказона при использовании АРТ-1011 изменялась в диапазоне 5,97-200 пг/мл.
Не сообщали о каких-либо представляющих специальный интерес АЕ (AESI), серьезных нежелательных явлениях (SAE) и случаях смерти ни для одного из субъектов, включенных в это исследование. Ни один субъект не был отстранен исследователем по причинам безопасности.
В общей сложности было зарегистрировано 12 возникших при лечении нежелательных явлений (ТЕАЕ) у 7 (29%) из 24 субъектов, которые приняли участие в этом исследовании. Из этих событий 4 происходили после введения по схеме лечения А, 1 после введения по схеме лечения В, а другие 7 после введения по схеме лечения С. Тяжесть всех ТЕАЕ была признана легкой. Считалось, что большинство ТЕАЕ (92%) возможно были связаны с лечением.
ТЕАЕ, зарегистрированные в этом исследовании, проявлялись в небольшом числе случаев, по 1 субъекту на подвергаемую лечению группу (5% для схемы лечения А, 5% для схемы лечения В и 4% для схемы лечения С). Они включали: тошноту, дискомфорт в области грудной клетки, инфекцию верхних дыхательных путей и головную боль для схемы лечения А, дискомфорт в области грудной клетки для схемы лечения В и запор, герпес полости рта, головокружение, кровоподтек в месте разрыва сосуда, повышенный уровень аспартат-аминотрансферазы, заложенность носа и приливы для схемы лечения С.
Число случаев ТЕАЕ составляло 9% для схемы лечения А, 5% для схемы лечения В и 22% для схемы лечения С. При регистрации связанных с приемом лекарственного средства ТЕАЕ наблюдали сходную тенденцию, поскольку общее число случаев ТЕАЕ: 9% для схемы лечения А, 5% для схемы лечения В и 17% для схемы лечения С.
Как правило, отклонение аномальных значений клинических лабораторных показателей в большую или меньшую сторону от своих референсных диапазонов было незначительным, и в большинстве случаев они рассматривались исследователем как не имеющие клинической значимости. Тем не менее, для одного субъекта был показан аномальный результат по аспартат-аминотрансферазе во время визита после окончания исследования, который был признан исследователем клинически значимым и зарегистрирован как ТЕАЕ легкой степени (повышенный уровень аспартат-аминотрансферазы). Последней терапией, назначаемой этому субъекту, была схема лечения С. Конечный результат остался неизвестным к окончанию исследования; данный субъект был недоступен для последующего наблюдения.
В целом, средние значения лабораторных показателей, показателей жизненно важных функций и ЭКГ для разных видов лечения были сопоставимы. Помимо этого, сопоставимы были средние значения оцениваемых параметров для всех субъектов во время скрининга и после проведения исследования.
Кроме того, в этом исследовании у субъектов не было никаких клинически значимых отклонений в показателях жизненно важных функций и ЭКГ.
В целом, наиболее распространенными ответами в плане оценок вкусовой привлекательности были ответы «не очень нравится» и «ни нравится, ни не нравится».
ЗАКЛЮЧЕНИЕ
Пероральный АРТ-1011 был безопасен и хорошо переносился при введении в условиях AM натощак, AM после приема пищи или в режиме HS здоровым субъектам.
В фазе 1 этого исследования было продемонстрировано, что после перорального введения АРТ-1011 (в общей дозе 6 мг) в условиях введения дозы натощак, после приема пищи и перед сном системное воздействие FP является очень слабым. Осуществление ФК контроля образцов позволило произвести точную оценку профиля выведения. Благодаря этому было осуществлено сравнение степени системного воздействия в течение 24 часов, 72 часов и с аппроксимацией к бесконечному промежутку времени.
Максимальные значения для случаев приема доз утром натощак и в режиме HS были сопоставимы, однако максимальные концентрации достигались быстрее при приеме АРТ-1011 на фоне пищи с высоким содержанием жира.
Межиндивидуальная вариабельность (CV, %) в случае Сmax была наивысшей для схемы введения в режиме HS, затем следовали схемы введения доз утром натощак и после приема пищи. По причине ожидаемой низкой биодоступности этой композиции (ожидаемого минимального системного воздействия) такой высокий наблюдаемый уровень вариабельности является ожидаемым.
Влияние пищи и влияние времени суток обратно пропорциональны максимальному воздействию FP по сравнению с приемом доз утром натощак. По сравнению со схемой приема утром натощак, максимальное воздействие на 20% превышало таковое для случая схемы введения утром после приема пищи и на 19% ниже в случае введения в режиме HS. Более низкая скорость всасывания подразумевается, когда APT-1011 принимают перед сном, в то время как более высокая скорость всасывания наблюдается тогда, когда прием осуществляется утром на фоне пищи с высоким содержанием жиров.
Несмотря на то, что наблюдалась более быстрое всасывание АРТ-1011 в условиях введения после приема пищи и более медленное всасывание при приеме перед сном, в целом всасывание флутиказона в составе этой композиции является низким (ниже 200 пг/мл). Более медленное всасывание в случае введения доз в режиме HS предполагает возможность более длительного времени пребывания в пищеводе; взаимосвязь приема доз перед сном с гистологической эффективностью как в проксимальной, так и в дистальной частях пищевода будет выяснена в будущих исследованиях АРТ-1011 в отношении ЕоЕ.
В целом, наблюдали влияние приема пищи, поскольку соотношения для Сmax и AUC и/или 90% ДИ не все находились в пределах предварительно определенного стандартного диапазона 80,00-125,00%).
При этом полное отсутствие влияния времени суток на биодоступность FP не могло быть установлено, поскольку нижние границы 90% ДИ для Сmax и AUCinf были ниже предварительно определенного стандартного диапазона, составляющего 80,00%.
Общая степень системного воздействия в течение 72 часов (AUClast) была примерно на 24% ниже для случая введения доз после приема пищи по сравнению со статистически незначимым уменьшением, составляющим 8%, при приеме в режиме HS по сравнению с приемом утром натощак.
Полученное экстраполированием к бесконечности значение степени системного воздействия (AUCinf) было ниже для обеих схем введения (после приема пищи: уменьшение на 29%; и для HS: уменьшение на 13%) по сравнению с приемом доз утром натощак.
И наконец, для введения в режиме HS было показано более существенное ослабление воздействия (уменьшения Сmax на 33%) по сравнению со схемой введения утром после приема пищи, чем со схемой введения утром натощак. Кроме того, степень воздействия в течение 72 часов и полученное экстраполированием к бесконечности значение было примерно на 20% выше после введения перед сном по сравнению с введением утром после приема пищи, и это указывает на то, что введение перед сном оказывает более устойчивое воздействие с более низкими максимальными значениями во времени.
Сводные данные по фармакокинетическим параметрам для исследований в условиях натощак, после приема пищи и в режиме HS приведены в Таблицах 18-21.


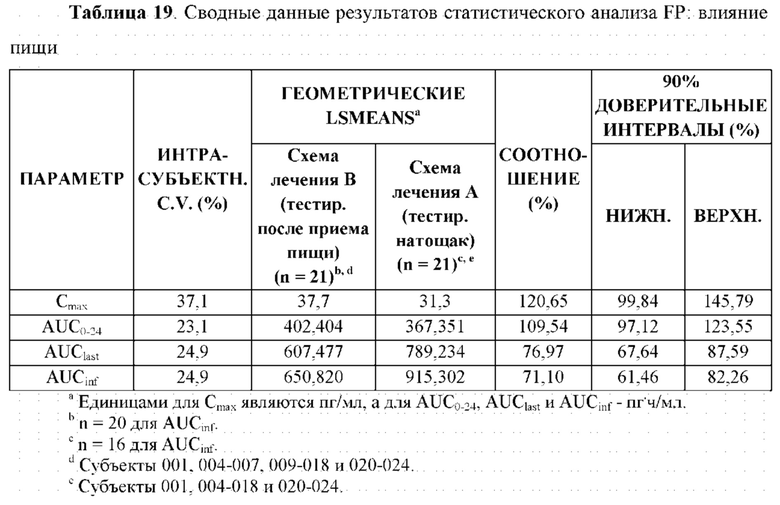
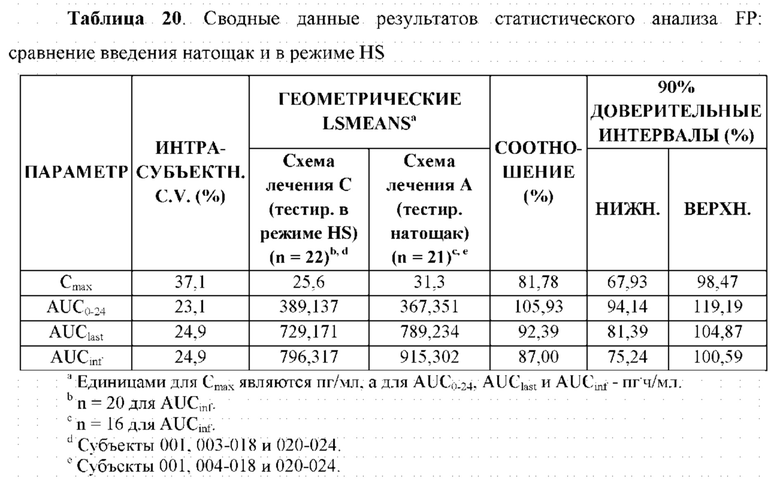
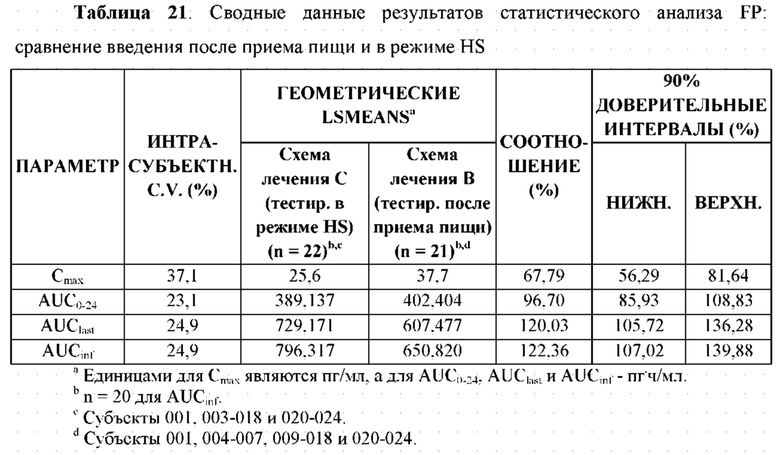
Пример 6. Сцинтиграфическое исследование анатомического контакта перорального кортикостероида после перорального введения пациенту, находящемуся в лежачем положении, по сравнению с находящимся в вертикальном положении пациентом
Пероральный кортикостероид (например, флутиказона пропионат) будет помечен радиоактивной меткой путем присоединения радиоактивных изотопов, и на основе такого кортикостероида будет приготовлена фармацевтическая композиция, такая как перорально распадающаяся таблетка. Меченный радиоактивной меткой пероральный кортикостероид будет введен пациенту, при этом пациент находится в лежачем положении. Независимо, меченный радиоактивной меткой пероральный кортикостероид будет введен пациенту, который находится в вертикальном положении.
Радиоактивные изотопы испускают гамма-излучение. Внешние детекторы (гамма-камеры) фиксируют гамма-излучение, исходящее от меченного радиоактивной меткой кортикостероида, когда он проходит через пищевод, которое преобразуется в изображения. Локализацию и относительное количество кортикостероида в дистальной и проксимальной частях пищевода для пациента, находящегося в лежачем положении, по сравнению с пациентом в вертикальном положении можно определить на основании указанных изображений.
Результаты сцинтиграфического исследования будут указывать на то, что при введении перорального кортикостероида пациенту, находящемуся в лежачем положении, с проксимальной и дистальной частями пищевода контактируют одинаковые количества кортикостероида, в то время как находящийся в вертикальном положении пациент имеет большее количество кортикостероида в проксимальной части пищевода по сравнению с дистальной частью пищевода.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ АКТИВНОГО ЭОЗИНОФИЛЬНОГО ЭЗОФАГИТА | 2018 |
|
RU2776651C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЭОЗИНОФИЛЬНОГО ЭЗОФАГИТА С ПОМОЩЬЮ ИНГИБИТОРА ИЛ-4Р | 2014 |
|
RU2679141C2 |
| ПРИМЕНЕНИЕ СОЛИФЕНАЦИНА И СТИМУЛЯТОРОВ СЛЮНООТДЕЛЕНИЯ В ЛЕЧЕНИИ ГИПЕРАКТИВНОГО МОЧЕВОГО ПУЗЫРЯ | 2012 |
|
RU2671575C2 |
| ЛЕЧЕНИЕ АЛЛЕРГИЧЕСКИХ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ IgE | 2018 |
|
RU2800765C2 |
| КОРТИКОСТЕРОИДНЫЕ КОМПОЗИЦИИ | 2008 |
|
RU2491074C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ДГЛК, И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2714323C2 |
| СПОСОБЫ УМЕНЬШЕНИЯ ПСИХОГЕННОГО ИЛИ КОМПУЛЬСИВНОГО ПЕРЕЕДАНИЯ | 2011 |
|
RU2620913C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ АГОНИСТОВ FXR | 2017 |
|
RU2743075C2 |
| СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЖЕЛУДОЧНО-КИШЕЧНОГО ТРАКТА ТРАДИПИТАНТОМ | 2018 |
|
RU2797248C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2015 |
|
RU2713406C2 |
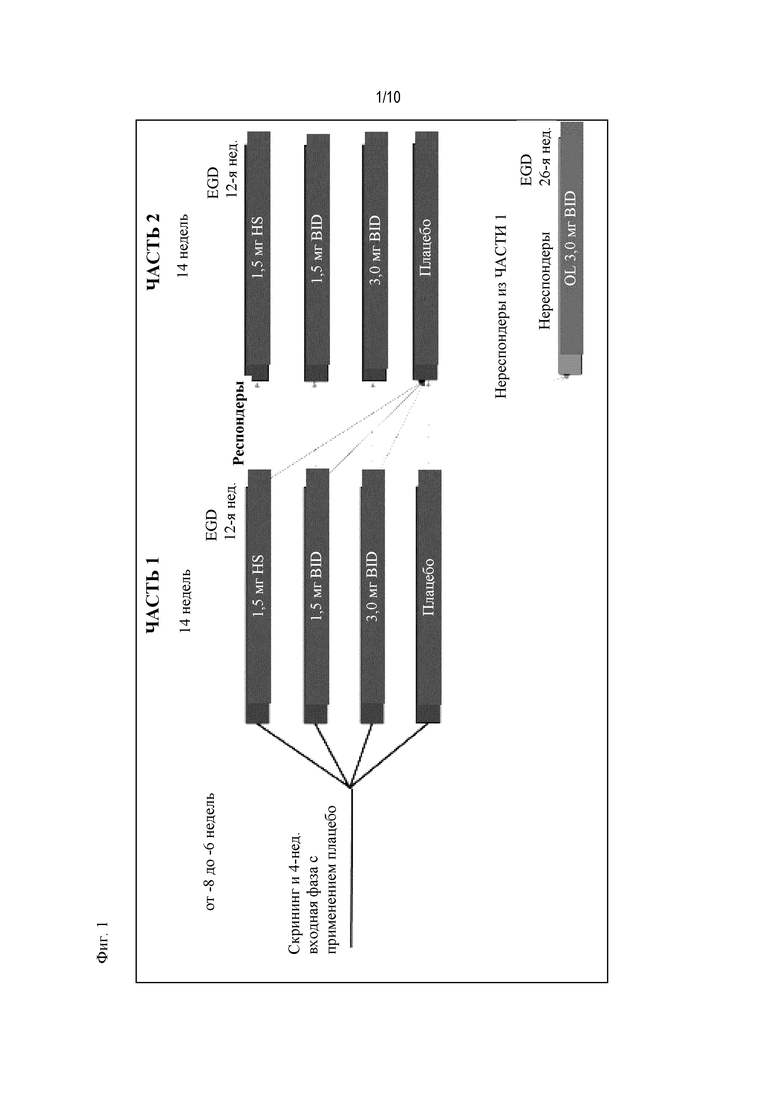
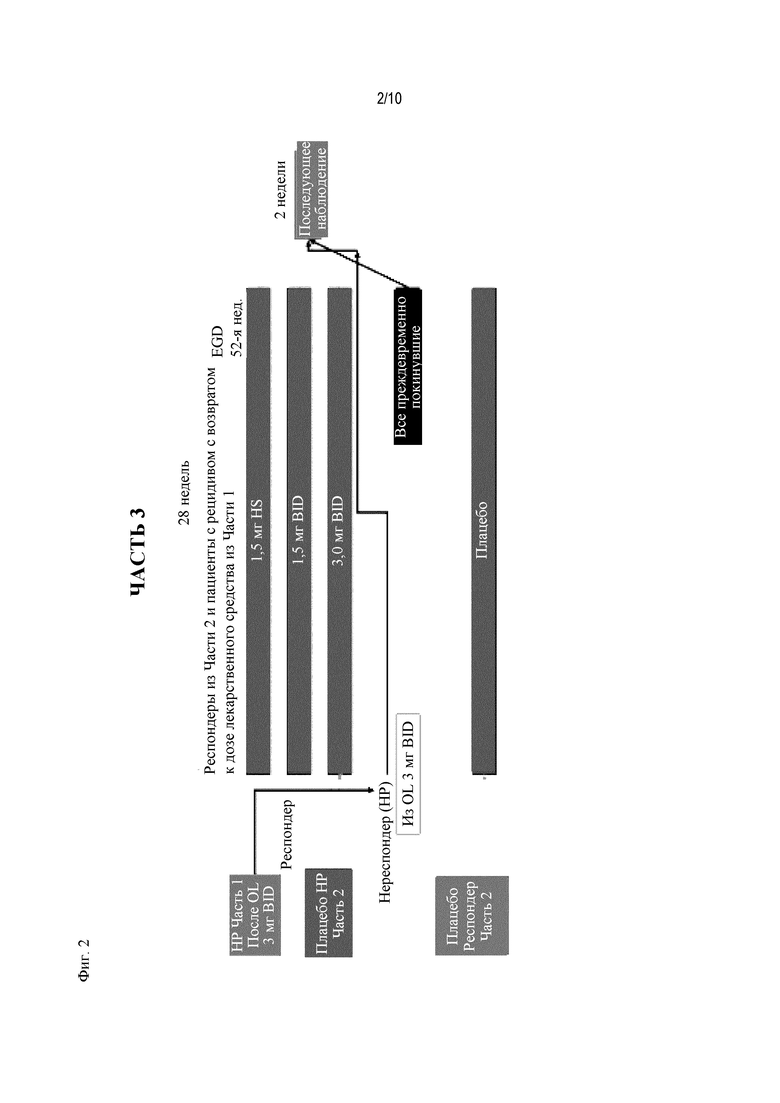
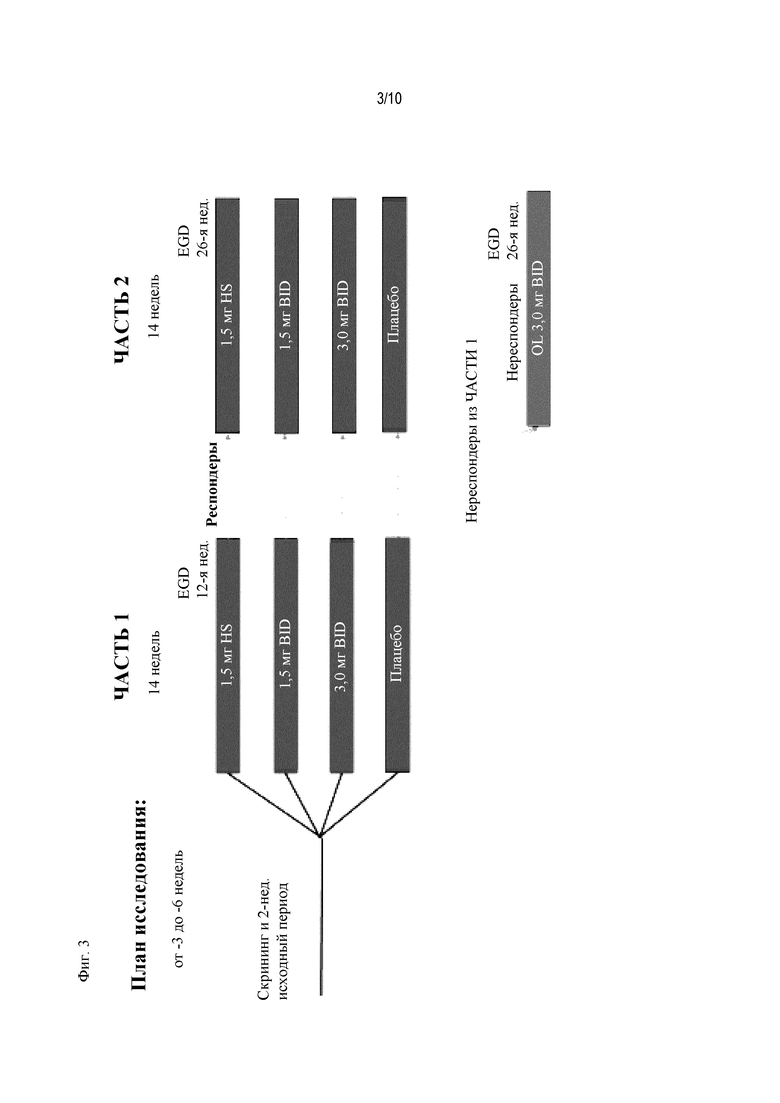


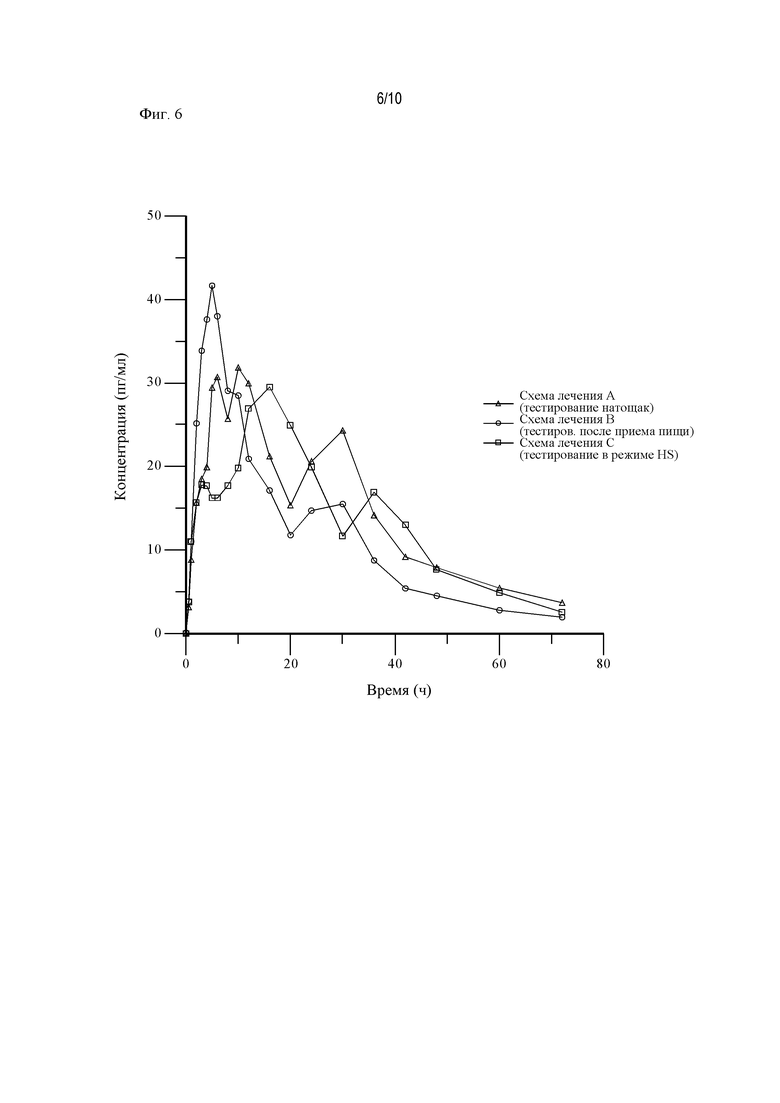
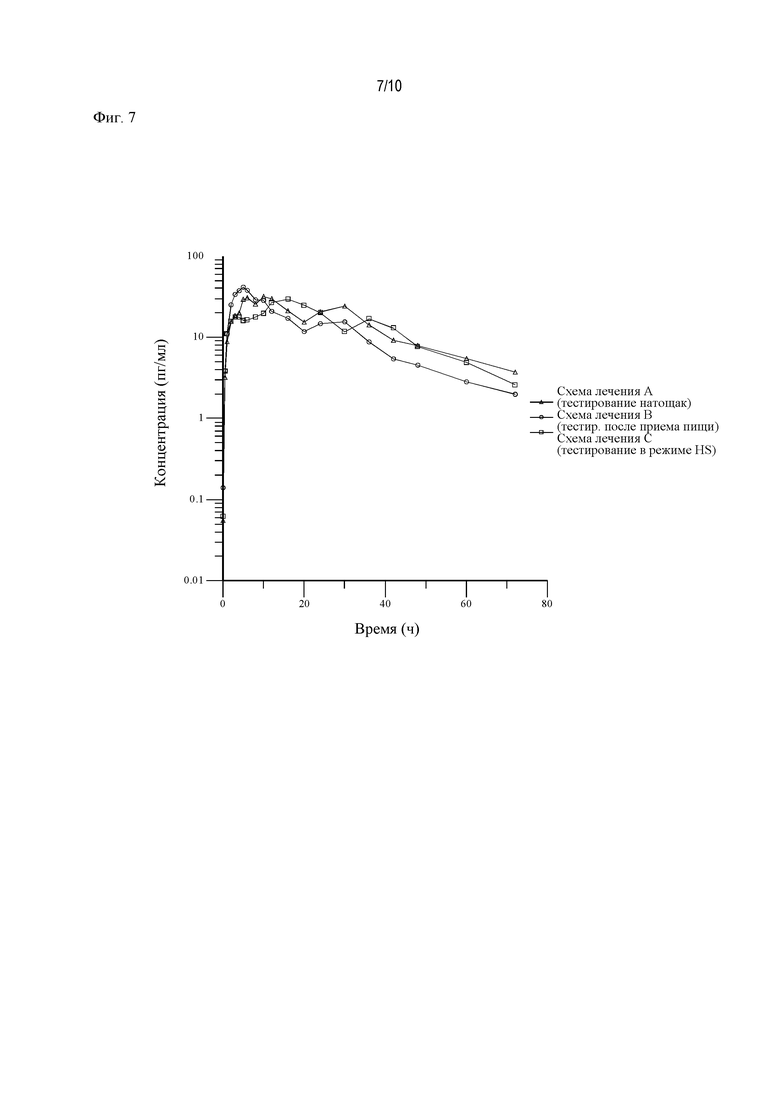
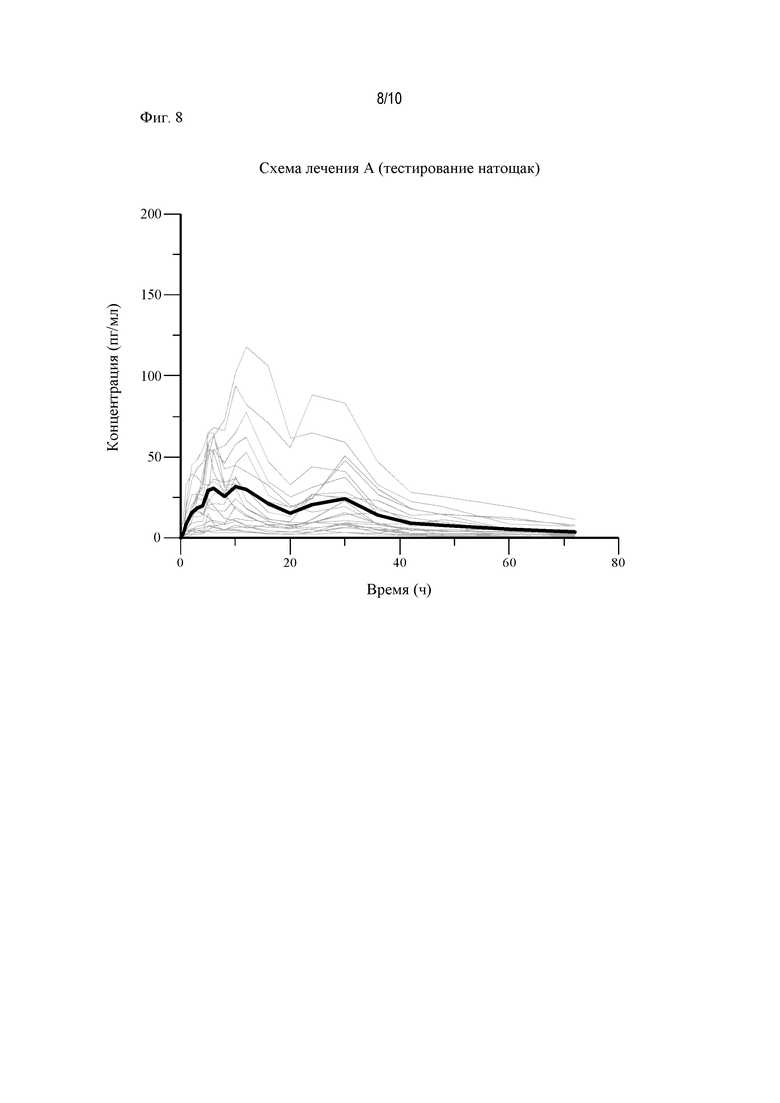
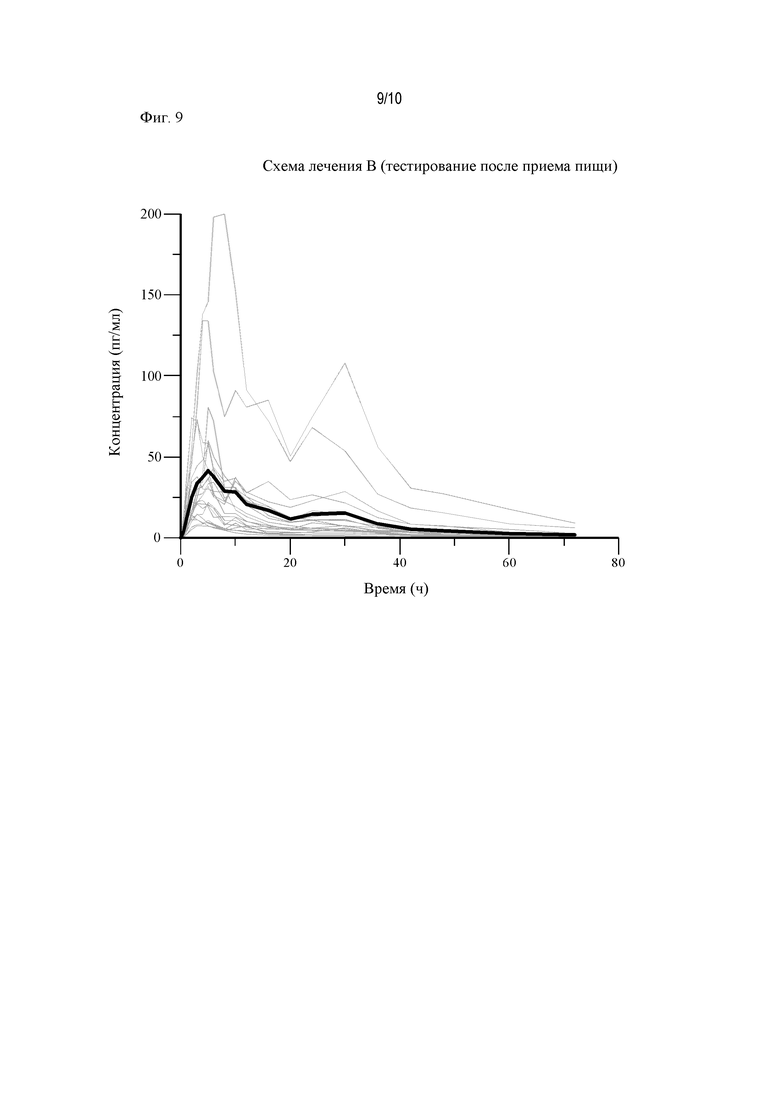
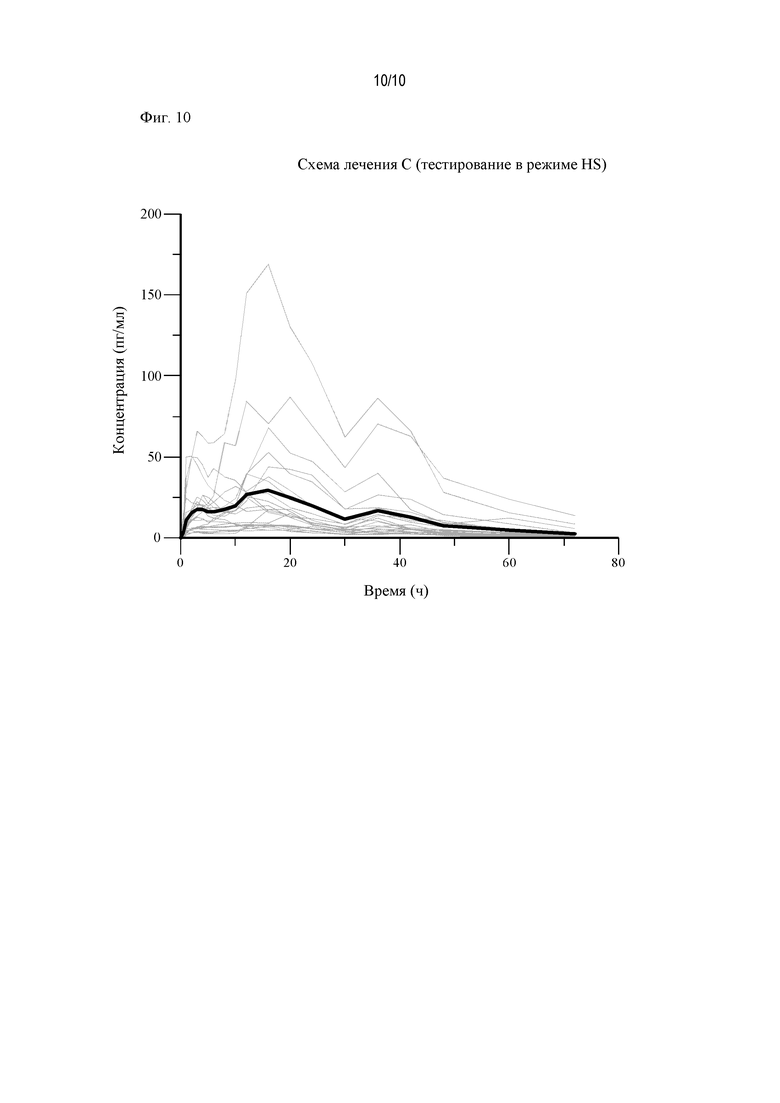
Изобретение относится к области медицины. Способ местного лечения эозинофильного эзофагита (EoE) у пациента, нуждающегося в этом, перорально распадающейся фармацевтической композицией, содержащей пероральный кортикостероид, включает введение фармацевтической композиции, когда пациент находится в лежачем положении или в пределах 5 минут перед тем, как пациент примет лежачее положение, при этом терапевтически эффективное количество перорального кортикостероида контактирует с пищеводом, тем самым осуществляя местное лечение EoE. Изобретение обеспечивает эффект увеличения клиренса эозинофилов в дистальном отделе пищевода при сохранении высокого клиренса эозинофилов в проксимальном отделе пищевода. 27 з.п. ф-лы, 10 ил., 21 табл., 6 пр.
1. Способ местного лечения эозинофильного эзофагита (EoE) у пациента, нуждающегося в этом, перорально распадающейся фармацевтической композицией, содержащей пероральный кортикостероид, включающий:
а) введение фармацевтической композиции, когда пациент находится в лежачем положении или в пределах 5 минут перед тем, как пациент примет лежачее положение,
при этом терапевтически эффективное количество перорального кортикостероида контактирует с пищеводом, тем самым осуществляя местное лечение EoE.
2. Способ по п. 1, где лежачее положение представляет собой положение лежа на спине, на животе или на боку.
3. Способ по п. 1, где перорально распадающуюся фармацевтическую композицию вводят за 30 минут или менее до запланированного времени отхода ко сну.
4. Способ по п. 1, где перорально распадающуюся фармацевтическую композицию вводят по меньшей мере через 30 минут после приема пищи.
5. Способ по п. 1, где пациент не принимает пищу или питье в течение по меньшей мере 30 минут после введения перорально распадающейся фармацевтической композиции.
6. Способ по п. 1, где перорально распадающуюся фармацевтическую композицию вводят:
(1) один раз в сутки; или
(2) два раза в сутки, при этом первую суточную дозу вводят, когда субъект сохраняет вертикальное положение.
7. Способ по п. 1, где перорально распадающаяся фармацевтическая композиция обладает системной биодоступностью, менее или равной 20% от своей дозы.
8. Способ по п. 1, где пероральный кортикостероид обеспечивает среднюю максимальную концентрацию в плазме крови (Cmax), менее или равную 500 пг/мл, после перорального введения от 0,01 до 20 мг перорального кортикостероида.
9. Способ по п. 1, где пероральный кортикостероид обеспечивает достижение средней площади под кривой в интервале 0-24 ч (AUC0-24), менее или равной 3000 пг*ч/мл, после перорального введения от 0,01 до 20 мг перорального кортикостероида.
10. Способ по п. 1, где пероральный кортикостероид представляет собой будесонид, флутиказон, флунизолид, циклесонид, мометазон или беклометазон либо их фармацевтически приемлемые соль, сольват или сложный эфир.
11. Способ по п. 1, где перорально распадающаяся фармацевтическая композиция приготовлена:
(1) в виде твердой композиции;
(2) для образования раствора или суспензии перед пероральным введением; или
(3) для образования раствора, суспензии или геля после перорального введения,
при этом формы (1)-(3) доставляют терапевтически эффективное количество перорального кортикостероида в пищевод.
12. Способ по п. 1, где перорально распадающаяся композиция представляет собой таблетку, облатку, пленку или лиофилизированную матрицу.
13. Способ по п. 12, где перорально распадающаяся композиция представляет собой таблетку, содержащую:
а) пероральный кортикостероид в количестве от 1,5 до 7,5 мг;
б) фармацевтически приемлемый носитель, объединенный с кортикостероидом; и
в) быстро диспергируемые микрогранулы,
при этом перорально распадающаяся таблетка распадается в течение 60 секунд при тестировании с использованием метода на время распадения, описанного в USP (Фармакопея США) <701>.
14. Способ по п. 1, где пациент имеет Cmax перорального кортикостероида, менее или равную 200 пг/мл, после перорального введения от 1,5 до 7,5 мг перорального кортикостероида.
15. Способ по п. 1, где пероральный кортикостероид представляет собой флутиказона пропионат и находящийся в лежачем положении пациент имеет Cmax в диапазоне от 80% до 125% от значений в диапазоне от 15 до 45 пг/мл после перорального введения 6 мг флутиказона пропионата или 3 мг флутиказона пропионата пациенту, находящемуся в лежачем положении.
16. Способ по п. 1, где Cmax кортикостероида у пациента, находящегося в лежачем положении, ниже Cmax перорального кортикостероида у пациента после приема пищи, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида.
17. Способ по п. 15, где Cmax перорального кортикостероида у пациента, находящегося в лежачем положении, снижается на величину от 10 до 30% по сравнению с Cmax перорального кортикостероида у пациента после приема пищи, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида.
18. Способ по п. 1, где среднее время достижения максимальной концентрации в плазме крови (Tmax) находится в диапазоне от 80 до 125% от значений в диапазоне от 12 до 15 ч.
19. Способ по п. 1, где Tmax для кортикостероида в случае пациента, находящегося в лежачем положении, увеличивается по сравнению с Tmax для перорального кортикостероида в случае пациента, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида.
20. Способ по п. 19, где Tmax для кортикостероида у пациента, находящегося в лежачем положении, увеличивается по меньшей мере на 1 час по сравнению со средним значением Tmax для перорального кортикостероида у пациента, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида.
21. Способ по п. 20, где Tmax для кортикостероида у пациента, находящегося в лежачем положении, увеличивается на промежуток времени в интервале от 4 до 9 ч по сравнению с Tmax для перорального кортикостероида у пациента, который находится в вертикальном положении и не ложится сразу же после введения перорального кортикостероида.
22. Способ по п. 1, где через 12 недель ежесуточного введения перорально распадающейся фармацевтической композиции воспаление пищевода ослабляется согласно результатам, показывающим снижение количества эозинофилов, увеличение числа дней без дисфагии, уменьшение числа эпизодов дисфагии, улучшение показателя EREFS (эндоскопический референсный показатель), улучшение эластичности пищевода согласно протоколу EndoFLIP, оценке уровня биомаркеров, уменьшению числа эпизодов затрудненного прохождения пищи, улучшению показателей EEsAI (по показателям пациента, врача, эндоскопии, патологического изменения), согласно EoE-QoL-A (опросник по оценке качества жизни взрослых пациентов с эозинофильным эзофагитом), опроснику по визуальной оценке дисфагии (VDQ), показателям избегания, корректировки и медленного приема пищи (AMS) или гистологического исследования.
23. Способ по п. 22, где количество эозинофилов у пациента снижается по меньшей мере на 50%.
24. Способ по п. 1, где пациент имеет аллергию на лактозу или аллергию на крахмал.
25. Способ по п. 1, где перорально распадающуюся фармацевтическую композицию вводят в пределах 4 минут перед тем, как пациент примет лежачее положение.
26. Способ по п. 1, где перорально распадающуюся фармацевтическую композицию вводят в пределах 3 минут перед тем, как пациент примет лежачее положение.
27. Способ по п. 1, где перорально распадающуюся фармацевтическую композицию вводят в пределах 2 минут перед тем, как пациент примет лежачее положение.
28. Способ по п. 1, где перорально распадающуюся фармацевтическую композицию вводят в пределах 1 минуты перед тем, как пациент примет лежачее положение.
| US 2015231156 А1, 20.08.2015 | |||
| US 2010216754 А1, 26.08.2010 | |||
| US 2016213681 А1, 28.07.2016 | |||
| US 20120164080 А1, 28.06.2012 | |||
| KOUKOURAKIS M.I | |||
| ET AL | |||
| Oral Administration of Recombinant Human Granulocyte Macrophage Colony-stimulating Factor in the Management of Radiotherapy-induced Esophagitis | |||
| Clinical Cancer Research, December 1999, vol.5, |

