ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящая заявка в общем связана с фармацевтическими композициями, содержащими ДГЛК, и способами их применения.
УРОВЕНЬ ТЕХНИКИ
[0002] Дигомогамма-линоленовая кислота (ДГЛК) является незаменимой жирной кислотой, найденной в организме в качестве естественного продукта элонгации гамма-линоленовой кислоты (ГЛК). ГЛК, в свою очередь, является продуктом десатурации линолевой кислоты. Помещение ДГЛК в мягкие желатиновые капсулы проблематично, поскольку она склонна к окислению до альдегидов, которые могут взаимодействовать с аминогруппами желатинового полимера в оболочке капсулы. Это может вызывать замедление высвобождения лекарственного средства из-за поперечного сшивания желатиновых полимеров.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0003] Многие аспекты настоящего документа могут быть лучше поняты со ссылкой на следующие фигуры. Компоненты на фигурах не обязательно масштабированы. Вместо этого, акцент сделан на четкой иллюстрации принципов настоящего документа.
[0004] Фиг. 1 иллюстрирует среднее изменение артериального давления (мм рт. ст) при внутривенном введении доз фенилэфрина после введения на протяжении семи дней подряд Аспирина через зонд в дозе 10 мг/кг/сутки.
[0005] Фиг. 2 иллюстрирует среднее изменение артериального давления (мм рт. ст) при внутривенном введении доз фенилэфрина после введения через зонд на протяжении семи дней подряд ДГЛК в дозе 50 мг/кг+Аспирина в дозе 10 мг/кг.
[0006] Фиг. 3 иллюстрирует среднее изменение артериального давления (мм рт. ст) при внутривенном введении доз фенилэфрина после сопутствующего введения через зонд на протяжении семи дней подряд ДГЛК в дозе 500 мг/кг и Аспирина в дозе 10 мг/кг.
[0007] Фиг. 4 иллюстрирует среднее артериальное давление в исходной точке для шести различных групп после введения веществ на протяжении семи дней подряд через зонд.
[0008] Фиг. 5 иллюстрирует среднее артериальное давление для шести различных групп при внутривенном введении дозы фенилэфрина 20 мкг/кг после введения веществ на протяжении семи дней подряд через зонд.
[0009] Фиг. 6 иллюстрирует среднюю концентрацию в плазме свободной ДГЛК (нг/мл, линейный график) по дозовым когортам (одна доза, популяция ФК).
[0010] Фиг. 7 иллюстрирует среднюю концентрацию свободной ДГЛК в плазме (нг/мл, логарифмически-линейный график) по дозовым когортам (одна доза, популяция ФК).
[0011] Фиг. 8 иллюстрирует среднюю общую концентрацию ДГЛК в плазме (нг/мл, линейный график) по дозовым когортам (одна доза, популяция ФК).
[0012] Фиг. 9 иллюстрирует среднюю общую концентрацию ДГЛК в плазме (нг/мл, логарифмически-линейный график) по дозовым когортам (одна доза, популяция ФК).
[0013] Фиг. 10 иллюстрирует среднюю концентрацию свободной ДГЛК в плазме (нг/мл, линейный график) по дозовым когортам (несколько доз, популяция ФК).
[0014] Фиг. 11 иллюстрирует среднюю концентрацию свободной ДГЛК в плазме (нг/мл, логарифмически-линейный график) по дозовым когортам (несколько доз, популяция ФК).
[0015] Фиг. 12 иллюстрирует среднюю общую концентрацию ДГЛК в плазме (нг/мл, линейный график) по дозовым когортам (несколько доз, популяция ФК).
[0016] Фиг. 13 иллюстрирует среднюю общую концентрацию ДГЛК в плазме (нг/мл, логарифмически-линейный график) по дозовым когортам (несколько доз, популяция ФК).
[0017] Фиг. 14 иллюстрирует среднюю концентрацию свободной ДГЛК в жидкости кожного волдыря (нг/мл, линейный график) по дозовым когортам (несколько доз, популяция ФК).
[0018] Фиг. 15 иллюстрирует среднюю концентрацию свободной ДГЛК в жидкости кожного волдыря (нг/мл, логарифмически-линейный график) по дозовым когортам (несколько доз, популяция ФК).
[0019] Фиг. 16 иллюстрирует среднюю концентрацию общей ДГЛК в жидкости кожного волдыря (нг/мл, линейный график) по дозовым когортам (несколько доз, популяция ФК).
[0020] Фиг. 17 иллюстрирует среднюю концентрацию общей ДГЛК в жидкости кожного волдыря (нг/мл, логарифмически-линейный график) по дозовым когортам (несколько доз, популяция ФК)
[0021] Фиг. 18 иллюстрирует среднюю концентрацию свободной ДГЛК (нг/мл, линейный график) в плазме и жидкости кожного волдыря по дозовым когортам (несколько доз, популяция ФК).
[0022] Фиг. 19 иллюстрирует среднюю концентрацию свободной ДГЛК (нг/мл, логарифмически-линейный график) в плазме и жидкости кожного волдыря по дозовым когортам (несколько доз, популяция ФК).
[0023] Фиг. 20 иллюстрирует среднюю общую концентрацию ДГЛК (нг/мл, линейный график) в плазме и жидкости кожного волдыря по дозовым когортам (несколько доз, популяция ФК).
[0024] Фиг. 21 иллюстрирует среднюю общую концентрацию ДГЛК (нг/мл, логарифмически-линейный график) в плазме и жидкости кожного волдыря по дозовым когортам (несколько доз, популяция ФК).
[0025] Фиг. 22 иллюстрирует среднюю концентрацию дигидротестостерона в плазме (нг/мл, линейный график) по дозовым когортам (несколько доз, популяция ФК).
[0026] Фиг. 23 иллюстрирует среднюю концентрацию дигидротестостерона в плазме (нг/мл, логарифмически-линейный график) по дозовым когортам (несколько доз, популяция ФК).
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0027] В настоящем документе предлагаются фармацевтические композиции, содержащие ДГЛК, подходящие для перорального введения, и способы их применения при лечении различных состояний и расстройств.
[0028] В одном варианте реализации изобретения в настоящем документе предлагается фармацевтическая композиция, содержащая ДГЛК. В одном варианте реализации изобретения композиция заключена в капсульную оболочку. В одном варианте реализации изобретения в настоящем документе предлагается фармацевтическая композиция, содержащая ДГЛК, которая заключена в капсульную оболочку, содержащую желатин, d-сорбит и 1,4-сорбитановые сахарные спирты. В одном варианте реализации изобретения от около 500 мг до около 1 г ДГЛК или ее производного заключены в капсульную оболочку.
[0029] В одном варианте реализации изобретения в настоящем документе предлагается способ лечения кожного заболевания или расстройства у субъекта, нуждающегося в этом, причем способ включает пероральное введение субъекту фармацевтической композиции, содержащей ДГЛК. Необязательно, фармацевтическая композиция содержит ДГЛК, заключенную в капсульную оболочку, содержащую желатин, d-сорбит и 1,4-сорбитановые сахарные спирты. Необязательно, композицию вводят субъекту в количестве, достаточном для обеспечения от около 1 г до около 4 г ДГЛК в сутки. В одном варианте реализации изобретения желатин имеет вязкость гелевой массы от около 9500 мПа до около 11000 мПа, например, около 9775 мПа или около 10500 мПа. В другом варианте реализации изобретения желатин имеет цветность от около 165 г до около 190 г, например, от около 170 г до около 185 г. В другом варианте реализации изобретения желатин имеет зольность > чем около 0,33.
[0030] В одном варианте реализации изобретения в настоящем документе предлагается способ лечения гиперактивного мочевого пузыря у субъекта, нуждающегося в этом, причем способ включает пероральное введение субъекту фармацевтической композиции, содержащей ДГЛК. Необязательно, фармацевтическая композиция содержит ДГЛК, заключенную в капсульную оболочку, содержащую желатин, d-сорбит и 1,4-сорбитановые сахарные спирты. Необязательно, композицию вводят субъекту в количестве, достаточном для обеспечения от около 1 г до около 4 г ДГЛК в сутки. В одном варианте реализации изобретения желатин имеет вязкость гелевой массы от около 9500 мПа до около 11000 мПа, например, около 9775 мПа или около 10500 мПа. В другом варианте реализации изобретения желатин имеет цветность от около 165 г до около 190 г, например, от около 170 г до около 185 г. В другом варианте реализации изобретения желатин имеет зольность > около 0,33. Эти и другие варианты изобретения описаны более подробно ниже.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0031] Несмотря на то, что настоящее изобретение может быть реализовано в различных формах, ниже приведено описание нескольких вариантов реализации изобретения с учетом того, что настоящий документ следует рассматривать как иллюстрацию изобретения, однако он не ограничивает изобретения конкретно проиллюстрированными вариантами реализации изобретения. Заголовки приведены только для удобства чтения и не должны интерпретироваться как ограничивающие изобретение каким-либо образом. Варианты реализации изобретения, проиллюстрированные под каким-либо заголовком, могут быть объединены с вариантами реализации изобретения, проиллюстрированными под любым другим заголовком.
[0032] Использование численных значений в различных количественных значениях, указанных в данной заявке, если четко не указано иное, означает приближение, как если бы минимальным и максимальным значениям в пределах заявленных диапазонов предшествовало слово «около». Таким образом, небольшие отклонения от заявленного значения могут применяться для достижения в существенной мере таких же результатов, что и заявленные значения. Кроме того, раскрытые диапазоны являются непрерывными диапазонами, включающими каждое значение между заявленными минимальными и максимальными значениями, а также любые диапазоны, которые могут быть образованы такими значениями. Кроме того, в настоящем документе раскрыты любые и все соотношения (и диапазоны любых таких соотношений), которые могут быть образованы в результате деления заявленного численного значения на любое другое заявленное численное значение. Соответственно, квалифицированному специалисту будет понятно, что многие из таких соотношений, диапазонов и диапазонов соотношений могут быть получены однозначным образом из численных значений, представленных в настоящем документе, и во всех случаях такие соотношения, диапазоны и диапазоны соотношений представляют различные варианты реализации настоящего изобретения.
Композиции
[0033] В различных вариантах реализации изобретения в настоящем документе предлагаются фармацевтические композиции, содержащие ДГЛК или ее производное, подходящие для перорального введения. Термин ДГЛК в настоящем документе обозначает ДГЛК в форме свободной кислоты. Кроме того, композиции по изобретению могут содержать производное ДГЛК в добавление к или вместо ДГЛК. Такие производные включают алкильные эфиры, эфиры низших алкилов, такие как метиловый или этиловый эфир ДГЛК, или ДГЛК в форме триглицерида. В одном варианте реализации изобретения в настоящем документе предлагается фармацевтическая композиция, содержащая ДГЛК или ее производное, заключенная в капсульную оболочку. В одном варианте реализации изобретения от около 500 мг до около 1 г ДГЛК или ее производного заключено в капсульную оболочку.
[0034] В одном варианте реализации изобретения оболочка капсулы содержит желатин, например, желатин с уменьшенным количеством поперечных связей или обработанный известью костный желатин с более низкой молекулярной массой. В другом варианте реализации изобретения оболочка капсулы содержит желатин с уменьшенным количеством поперечных связей, обработанный протеолитическим ферментом для разрезания желатиновых фрагментов и эффективного уменьшения его молекулярной массы. В другом варианте реализации изобретения фармацевтическая композиция содержит ДГЛК эфиры D-сорбита и 1,4-сорбитана. В одном варианте реализации изобретения оболочка капсулы содержит (a) желатин и (b) пластификаторы, выбранные как один или более из d-сорбита и 1,4-сорбитана. В одном варианте реализации изобретения желатин является таким, как описано в U.S. 7485323, который, таким образом, включен в настоящий документ посредством ссылки в полном объеме.
[0035] В одном варианте реализации изобретения пластификатор содержит 1-4-сорбитаны в количестве от 20% до 30%, например, около 24% и 28% (в пересчете на сухое вещество), и содержит D-сорбит в количестве около 30-50%, например, около 35-45% (в пересчете на сухое вещество).
[0036] В некоторых вариантах реализации изобретения оболочка капсулы дополнительно содержит глицерин, очищенную воду, диоксид титана, среднецепочечные триглицериды и лецитин.
[0037] В различных вариантах реализации изобретения ДГЛК или ее производное присутствует в композиции по изобретению в количестве от около 50 мг до около 5000 мг, от около 75 мг до около 2500 мг или от около 100 мг до около 1000 мг, например, около 75 мг, около 100 мг, около 125 мг, около 150 мг, около 175 мг, около 200 мг, около 225 мг, около 250 мг, около 275 мг, около 300 мг, около 325 мг, около 350 мг, около 375 мг, около 400 мг, около 425 мг, около 450 мг, около 475 мг, около 500 мг, около 525 мг, около 550 мг, около 575 мг, около 600 мг, около 625 мг, около 650 мг, около 675 мг, около 700 мг, около 725 мг, около 750 мг, около 775 мг, около 800 мг, около 825 мг, около 850 мг, около 875 мг, около 900 мг, около 925 мг, около 950 мг, около 975 мг, около 1000 мг, около 1025 мг, около 1050 мг, около 1075 мг, около 1100 мг, около 1025 мг, около 1050 мг, около 1075 мг, около 1200 мг, около 1225 мг, около 1250 мг, около 1275 мг, около 1300 мг, около 1325 мг, около 1350 мг, около 1375 мг, около 1400 мг, около 1425 мг, около 1450 мг, около 1475 мг, около 1500 мг, около 1525 мг, около 1550 мг, около 1575 мг, около 1600 мг, около 1625 мг, около 1650 мг, около 1675 мг, около 1700 мг, около 1725 мг, около 1750 мг, около 1775 мг, около 1800 мг, около 1825 мг, около 1850 мг, около 1875 мг, около 1900 мг, около 1925 мг, около 1950 мг, около 1975 мг, около 2000 мг, около 2025 мг, около 2050 мг, около 2075 мг, около 2100 мг, около 2125 мг, около 2150 мг, около 2175 мг, около 2200 мг, около 2225 мг, около 2250 мг, около 2275 мг, около 2300 мг, около 2325 мг, около 2350 мг, около 2375 мг, около 2400 мг, около 2425 мг, около 2450 мг, около 2475 мг или около 2500 мг. В любом таком варианте реализации изобретения композиция может дополнительно содержать ДГЛК эфиры D-сорбита и 1,4-сорбитана.
[0038] В одном варианте реализации изобретения композиция по изобретению содержит не более чем около 10%, не более чем около 9%, не более чем около 8%, не более чем около 7%, не более чем около 6%, не более чем около 5%, не более чем около 4%, не более чем около 3%, не более чем около 2%, не более чем около 1%, или не более чем около 0,5% масс. общих жирных кислот, жирных кислот, кроме ДГЛК.
[0039] В другом варианте реализации изобретения ДГЛК или ее производное составляет по меньшей мере около 30%, около 40%, около 50%, по меньшей мере около 60%, по меньшей мере около 70%, по меньшей мере около 80%, по меньшей мере около 90%, по меньшей мере около 95%, по меньшей мере около 97%, по меньшей мере около 98%, по меньшей мере около 99% или 100% масс., в пересчете на массу всех жирных кислот, присутствующих в композиции по изобретению.
[0040] В одном варианте реализации изобретения композиция по изобретению, при помещении в условия стандартного испытания на распад, например, как описано в Фарм. США 2040 (Распад и растворение биологически активных пищевых добавок), с водой в качестве среды, демонстрирует скорость высвобождения ДГЛК менее чем около 60 минут, менее чем около 50 минут, менее чем около 40 минут, менее чем около 30 минут или менее чем 20 минут после хранения в течение около 1 месяца, около 2 месяцев или около 3 месяцев при 40°C/75% ОВ.
[0041] В одном варианте реализации изобретения, после хранения в течение около 1 месяца, около 2 месяцев, около 3 месяцев или около 6 месяцев при 40°C/75% ОВ, композиция по изобретению содержит менее чем около 5% эфиров ДГЛК, в пересчете на массу всех жирных кислот, менее чем около 4% эфиров ДГЛК, в пересчете на массу всех жирных кислот, менее чем около 3% эфиров ДГЛК, в пересчете на массу всех жирных кислот, менее чем около 2% эфиров ДГЛК, в пересчете на массу всех жирных кислот, или менее чем около 1% эфиров ДГЛК, в пересчете на массу всех жирных кислот.
Способы
[0042] Любая композиция по изобретению, включая композиции, описанные выше в настоящем документе, или композиции, которые могут быть составлены путем объединения различных вариантов реализации изобретения согласно настоящему документу, может применяться при лечении или предупреждении: кожных расстройств и заболеваний, включая обыкновенные угри, розовые угри, атопический дерматит, псориаз, зуд/зудение, для защиты от воздействия радиации, сухости кожи, для гладкости кожи, для здоровья кожи, против возрастных изменения и для фотозащиты; расстройств и заболеваний мочевых путей, включая рак мочевого пузыря, цистоцеле, гематурию, интерстициальный цистит, нейрогенный мочевой пузырь, болезнь Пейрони, заболевание предстательной железы, недержание, инфекцию мочевых путей и везикоуретеральный рефлюкс; заболевания и расстройств почек, включая почечную недостаточность, острое поражение почек, хроническое заболевание почек и поликистозное заболевание почек; ревматического заболевания, включая анкилозирующий спондилит, фибромиалгию, подагру, инфекционный артрит, волчанку, остеоартрит, ревматическую полимиалгию, псориатический артрит, реактивный артрит, ревматоидный артрит, склеродому; расстройств дыхательных путей, включая воспалительное заболевание легкого, инфекции дыхательных путей, заболевание плевральной полости, заболевание легочных сосудов, пневмонию, легочную эмболию и рак легкого; и сердечно-сосудистых расстройств, включая острые ишемические состояния сердца, острый инфаркт миокарда, стенокардию, аритмию, фибрилляцию предсердий, атеросклероз, артериальную фибрилляцию, сердечную недостаточность, сердечно-сосудистое заболевание, хроническую сердечную недостаточность, хроническую стабильную стенокардию, застойную сердечную недостаточность, заболевание коронарных артерий, заболевание коронарных сосудов сердца, тромбоз глубоких вен, диабет, сахарный диабет, диабетическую невропатию, диастолическую дисфункцию у субъектов с сахарным диабетом, отек, эссенциальную гипертензию, подозрение на легочную эмболию, стеатоз печени, заболевание сердца, сердечную недостаточность, гомозиготную семейную гиперхолестеринемию (ГоСГ), гомозиготную семейную ситостеролемию, гиперхолестеринемию, гиперлипидемию, гипертензию, гипертриглицеридемию, метаболический синдром, смешанную дислипидемию, легкую или умеренную сердечную недостаточность, инфаркт миокарда, контроль ожирения, пароксизмальную предсердную/артериальную фибрилляцию/мерцание/трепетание, пароксизмальную суправентрикулярную тахикардию (ПСВТ), особенно тяжелый или внезапный отек, агрегацию тромбоцитов, первичную гиперхолестеринемию, первичную гиперлипидемию, легочную артериальную гипертензию, легочную гипертензию, возвратную гемодинамически нестабильную вентрикулярную тахикардию (ВТ), возвратные вентрикулярные аритмии, возвратную фибрилляцию желудочков (ФЖ), разрыв аневризмы, ситистеролемию, инсульт, суправентрикулярную тахикардию, симптоматическую фибрилляцию/трепетание предсердий, тахикардию, диабет II типа, заболевание сосудов, венозную тромбоэмболию, вентрикулярные аритмии и другие состояния сердечно-сосудистой системы.
[0043] Термин «лечение» в отношении конкретного заболевания или расстройства, включает, но не ограничиваясь этим, предупреждение заболевания или расстройства, например, остановку развития заболевания или расстройства; облегчение заболевания или расстройства, например, регресс заболевания или расстройства; или облегчение состояния, вызванного или являющегося результатом заболевания или расстройства, например, облегчение, предупреждение или лечение симптомов заболевания или расстройства. Термин «предупреждение» в отношении конкретного заболевания или расстройства обозначает: предупреждение начала развития заболевания, если оно еще не началось, предупреждение возникновения заболевания или расстройства у субъекта, который может быть предрасположен к расстройству или заболеванию, но которому еще не поставлен диагноз расстройства или заболевания, и/или предупреждение дальнейшего развития заболевания/расстройства, если оно уже присутствует.
[0044] В различных вариантах реализации изобретения композиции по изобретению вводят в количестве, достаточном, чтобы обеспечить ежедневную дозу ДГЛК от около 50 мг до около 10000 мг, от около 100 мг до около 7500 мг, или от около 100 мг до около 5000 мг, например, около 200 мг, около 300 мг, около 400 мг, около 500 мг, около 600 мг, около 700 мг, около 800 мг, около 900 мг, около 1000 мг, около 1100 мг, около 1200 мг, около 1300 мг, около 1400 мг, около 1500 мг, около 1600 мг, около 1700 мг, около 1800 мг, около 1900 мг, около 2000 мг, около 2100 мг, около 2200 мг, около 2300 мг, около 2400 мг, около 2500 мга около 2600 мг, около 2700 мг, около 2800 мг, около 2900 мг, около 3000 мг, около 3100 мг, около 3200 мг, около 3300 мг, около 3400 мг, около 3500 мг, 3600 мг, около 3700 мг, около 3800 мг, около 3900 мг, около 4000 мг, около 4100 мг, около 4200 мг, около 4300 мг, около 4400 мг, около 4500 мг, 4600 мг, около 4700 мг, около 4800 мг, около 4900 мг, около 5000 мг, около 5100 мг, около 5200 мг, около 5300 мг, около 5400 мг, около 5500 мг ДГЛК в сутки.
[0045] В одном варианте реализации изобретения предлагается способ лечения атопического дерматита, например, легкого или умеренного атопического дерматита. В одном варианте реализации изобретения способ включает введение субъекту, нуждающемуся в таком лечении, ДГЛК в количестве от около 500 мг до около 3 г в сутки, от около 1 г до около 2,5 г в сутки, около 1 г в сутки или около 2 г в сутки. В одном варианте реализации изобретения ДГЛК вводят субъекту ежедневно в течение периода по меньшей мере около 2 недель, по меньшей мере около 4 недель или по меньшей мере около 8 недель. В родственном варианте реализации изобретения, при лечении в соответствии с настоящим изобретением, например, на протяжении от около 1 до около 12 недель, от около 1 до около 8 недель или от около 1 до около 4 недель, субъект или группа субъектов демонстрирует один или более из следующих результатов:
[0046] (a) уменьшение площади и индекса тяжести экземы (ПЭИТ) относительно исходного значения или контроля плацебо;
[0047] (b) уменьшение процента площади анатомического участка, пораженного атопическим дерматитом относительно исходного или контрольного значения;
[0048] (c) уменьшение баллов общей оценки исследователем относительно исходного значения или контроля плацебо;
[0049] (d) уменьшение интенсивности эритемы, отека/заполненности, выпотевания/струпьев, экскориации, лихенификации и/или сухости относительно исходного значения или контроля плацебо;
[0050] (e) уменьшение эритемы, отека/заполненности, выпотевания/струпьев, экскориации, лихенификации и/или сухости относительно исходного значения или контроля плацебо;
[0051] (f) уменьшение площади поверхности тела (ППТ), пораженной атопическим дерматитом, относительно исходного значения или контроля плацебо;
[0052] (g) уменьшение инсомнии относительно исходного значения или контроля плацебо;
[0053] (h) уменьшение частоты эпизодов зуда (зудения) относительно исходного значения или контроля плацебо;
[0054] (i) уменьшение тяжести зуда как среднее значение для предшествующих трех дней и/или ночей на визуальной аналоговой шкале;
[0055] (j) уменьшение баллов Шкалы атопического дерматита (ШАД) относительно исходного значения или контроля плацебо;
[0056] (k) улучшение показателей пациент-ориентированной экземы меры (ПОЭМ), по сравнению с исходным значением или контролем плацебо;
[0057] (l) уменьшение количества дней на предшествующей неделе, в которые субъект сообщал о кожном зуде в результате экземы;
[0058] (m) уменьшение количества дней на предшествующей неделе, в которые субъект сообщал о нарушении сна из-за экземы;
[0059] (n) уменьшение количества дней на предшествующей неделе, в которые у субъекта возникало кожное кровотечение;
[0060] (o) уменьшение количества дней на предшествующей неделе, в которые у субъекта возникало намокание кожи или выпот прозрачной жидкости;
[0061] (p) уменьшение количества дней на предшествующей неделе, в которые у субъекта трескалась кожа;
[0062] (q) уменьшение количества дней на предшествующей неделе, в которые у субъекта шелушилась кожа;
[0063] (r) уменьшение количества дней на предшествующей неделе, в которые субъект испытывал сухость кожи;
[0064] (s) увеличение трансэпидермальной потери воды, по сравнению с исходным значением или контролем плацебо;
[0065] (t) повышение уровня общей и свободной ДГЛК в плазме, по сравнению с исходным значением;
[0066] (u) увеличение соотношения ДГЛК:АК, по сравнению с исходным значением или контролем плацебо; и/или
[0067] (v) снижение артериального давления крови, по сравнению с исходным значением или контролем плацебо.
[0068] В одном варианте реализации изобретения способы по настоящему изобретению включают измерение исходных уровней одного или более маркеров или показателей, приведенных в (a)-(v) выше до введения дозы субъекту или группе субъектов. В другом варианте реализации изобретения способы включают введение композиции, раскрытой в настоящем документе, субъекту после определения базовых уровней одного или более маркеров или показателей, приведенных в (a)-(v), с последующим дополнительным измерением указанного одного или более маркеров.
[0069] В другом варианте реализации изобретения, при лечении с помощью композиции по настоящему изобретению, например, на протяжении от около 1 до около 12 недель, от около 1 до около 8 недель или от около 1 до около 4 недель, субъект или группа субъектов демонстрирует любые 2 или более, любые 3 или более, любые 4 или более, любые 5 или более, любые 6 или более, любые 7 или более, любые 8 или более, любые 9 или более, любые 10 или более, любые 11 или более, любые 12 или более, любые 13 или более, любые 14 или более, любые 15 или более, любые 16 или более, любые 17 или более, любые 18 или более, любые 19 или более, любые 20 или более, любые 21 или более или все 22 результата (a)-(v), описанные непосредственно выше.
[0070] В другом варианте реализации изобретения, при лечении с помощью композиции по настоящему изобретению, субъект или группа субъектов демонстрирует один или более из следующих результатов:
[0071] (a) уменьшение площади и индекса тяжести экземы (ПЭИТ) относительно исходного значения или контроля плацебо по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0072] (b) уменьшение процента площади анатомического участка, пораженного атопическим дерматитом, относительно исходного или контрольного значения, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0073] (c) уменьшение баллов общей оценки исследователем относительно исходного значения или контроля плацебо по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0074] (d) уменьшение интенсивности эритемы, отека/заполненности, выпотевания/струпьев, экскориации, лихенификации и/или сухости относительно исходного значения или контроля плацебо по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0075] (e) уменьшение эритемы, отека/заполненности, выпотевания/струпьев, экскориации, лихенификации и/или сухости относительно исходного значения или контроля плацебо по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0076] (f) уменьшение площади поверхности тела (ППТ), пораженной атопическим дерматитом, относительно исходного значения или контроля плацебо по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0077] (g) уменьшение инсомнии относительно исходного значения или контроля плацебо по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0078] (h) уменьшение частоты эпизодов зуда (зудения) относительно исходного значения или контроля плацебо по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0079] (i) уменьшение тяжести зуда как среднее значение для предшествующих трех дней и/или ночей на визуальной аналоговой шкале по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0080] (j) уменьшение баллов Шкалы атопического дерматита (ШАД) относительно исходного значения или контроля плацебо по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0081] (k) улучшение показателей пациент-ориентированной экземы меры (ПОЭМ), по сравнению с исходным значением или контролем плацебо, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0082] (l) уменьшение количества дней на предшествующей неделе, в которые субъект сообщал о кожном зуде из-за экземы, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0083] (m) уменьшение количества дней на предшествующей неделе, в которые субъект сообщал о нарушении сна из-за экземы, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0084] (n) уменьшение количества дней на предшествующей неделе, в которые у субъекта возникало кожное кровотечение, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0085] (o) уменьшение количества дней на предшествующей неделе, в которые у субъекта возникало намокание кожи или выпот прозрачной жидкости, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0086] (p) уменьшение количества дней на предшествующей неделе, в которые у субъекта трескалась кожа, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0087] (q) уменьшение количества дней на предшествующей неделе, в которые у субъекта шелушилась кожа, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0088] (r) уменьшение количества дней на предшествующей неделе, в которые субъект испытывал сухость кожи, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0089] (s) увеличение трансэпидермальной потери воды, по сравнению с исходным значением или контролем плацебо, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%;
[0090] (t) повышение уровня общей и свободной ДГЛК в плазме, по сравнению с исходным значением, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%; и/или
[0091] (u) увеличение соотношения ДГЛК:АК, по сравнению с исходным значением или контролем плацебо, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%; и/или
[0092] (v) снижение среднего артериального давления крови по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 65%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90% или по меньшей мере на около 95%.
[0093] В другом варианте реализации изобретения, при лечении с помощью композиции по настоящему изобретению, после введения однократной дозы или введения нескольких доз, например, на протяжении от около 1 до около 12 недель, от около 1 до около 8 недель или от около 1 до около 4 недель, субъект или группа субъектов демонстрирует любые 2 или более, любые 3 или более, любые 4 или более, любые 5 или более, любые 6 или более, любые 7 или более, любые 8 или более, любые 9 или более, любые 10 или более, любые 11 или более, любые 12 или более, любые 13 или более, любые 14 или более, любые 15 или более, любые 16 или более, любые 17 или более, любые 18 или более, любые 19 или более, любые 20 или более, любые 21 или более или все 22 результата (a)-(v) описанные непосредственно выше.
[0094] В другом варианте реализации изобретения при лечении субъекта или группы субъектов (после приема пищи или натощак) композицией, содержащей от около 200 мг ДГЛК до около 8000 мг ДГЛК (в виде одной или более дозированных единиц, например, в виде дозированных единиц по 500 мг или 1 г, которые в сумме составляют суточные дозы ДГЛК около 500 мг, около 1000 мг, около 2000 мг, около 3000 мг, около 4000 мг, около 5000 мг, около 6000 мг, около 7000 мг или около 8000 мг), после введения одной дозы или после введения нескольких доз субъект или группа субъектов демонстрируют один или более из следующих результатов:
[0095] (a) Cmax свободной ДГЛК (или среднее значение или медиана Cmax) от около 400 нг/мл до около 4500 нг/мл, от около 500 нг/мл до около 3400 нг/мл, от около 600 нг/мл до около 3300 нг/мл, от около 700 нг/мл до около 3200 нг/мл, например, около 900 нг/мл, около 1000 нг/мл, около 1100 нг/мл, около 1200 нг/мл, около 1300 нг/мл, около 1400 нг/мл, около 1500 нг/мл, около 1600 нг/мл, около 1700 нг/мл, около 1800 нг/мл, около 1900 нг/мл, около 2000 нг/мл, около 2100 нг/мл, около 2200 нг/мл, около 2300 нг/мл, около 2400 нг/мл, около 2500 нг/мл, около 2600 нг/мл, около 2700 нг/мл, около 2800 нг/мл, около 2900 нг/мл, около 3000 нг/мл, около 3100 нг/мл, около 3200 нг/мл, около 3300 нг/мл, около 3400 нг/мл, около 3500 нг/мл, около 3600 нг/мл, около 3700 нг/мл, около 3800 нг/мл, около 3900 нг/мл, около 4000 нг/мл, около 4100 нг/мл, около 4200 нг/мл, около 4300 нг/мл, около 4400 нг/мл или около 4500 нг/мл;
[0096] (b) Cmax свободной ДГЛК/доза (или среднее значение или медиана Cmax/доза) от около 0,5 (1/кл) до около 3 (1/кл), от около 0.6 (1/кл) до около 2,5 (1/кл) или от около 0,7 (1/кл) до около 2 (1/кл), например, около 0,7 (1/кл), около 0,8 (1/кл), около 0,9 (1/кл), около 1 (1/кл), около 1,5 (1/кл), около 1,6 (1/кл), около 1,7 (1/кл) или около 1,8 (1/кл);
[0097] (c) AUC0-24 свободной ДГЛК (или среднее значение или медиана AUC0-24) от около 1500 нг⋅час/мл до около 12000 нг⋅час/мл, от около 2000 нг⋅час/мл до около 11000 нг⋅час/мл или от около 2500 нг⋅час/мл до около 10000 нг⋅час/мл, например, около 1000 нг⋅час/мл, около 1500 нг⋅час/мл, около 2000 нг⋅час/мл, около 2500 нг⋅час/мл, около 3000 нг⋅час/мл, около 3500 нг⋅час/мл, около 4000 нг⋅час/мл, около 4500 нг⋅час/мл, около 5000 нг⋅час/мл, около 5500 нг⋅час/мл, около 6000 нг⋅час/мл, около 6500 нг⋅час/мл, около 7000 нг⋅час/мл, около 7500 нг⋅час/мл, около 8000 нг⋅час/мл, около 8500 нг⋅час/мл, около 9000 нг⋅час/мл, около 9500 нг⋅час/мл, около 10000 нг⋅час/мл, около 10500 нг⋅час/мл, около 11000 нг⋅час/мл, около 11500 нг⋅час/мл или около 12000 нг⋅час/мл.
[0098] (d) AUC0-24 свободной ДГЛК/доза (или среднее значение или медиана AUC0-24 /доза) от около 1,5 до около 10 час/кл, от около 1,7 до около 8 час/кл или от около 2 до около 6 час/кл, например, около 2 час/кл, около 2,5 час/кл, около 3 час/кл, около 3,5 час/кл, около 4 час/кл, около 4,5 час/кл, около 5 час/кл или около 5,5 час/кл;
[0099] (e) tmax(час)свободной ДГЛК от около 2 до около 10 часов, от около 3 до около 8 часов, например, около 3 часов, около 4 часов, около 5 часов, около 6 часов, около 7 часов или около 8 часов;
[00100] (f) Cmax общей ДГЛК (или среднее значение или медиана Cmax общей ДГЛК) от около 4000 нг/мл до около 45000 нг/мл, от около 5000 нг/мл до около 34000 нг/мл, от около 6000 нг/мл до около 33000 нг/мл или от около 7000 нг/мл до около 32000 нг/мл, например, около 7000 нг/мл, около 7200 нг/мл, около 7500 нг/мл, около 8000 нг/мл, около 8500 нг/мл, около 9000 нг/мл, около 9500 нг/мл, около 10000 нг/мл, около 11000 нг/мл, около 12000 нг/мл, около 13000 нг/мл, около 14000 нг/мл, около 15000 нг/мл, около 16000 нг/мл, около 17000 нг/мл, около 18000 нг/мл, около 19000 нг/мл, около 20000 нг/мл, около 21000 нг/мл, около 22000 нг/мл, около 23000 нг/мл, около 24000 нг/мл, около 25000 нг/мл, около 26000 нг/мл, около 27000 нг/мл, около 28000 нг/мл, около 29000 нг/мл, около 30000 нг/мл, около 31000 нг/мл, около 32000 нг/мл, около 33000 нг/мл, около 34000 нг/мл или около 35000 нг/мл;
[00101] (g) Cmax общей ДГЛК/доза (или среднее значение или медиана Cmax общей ДГЛК/доза) от около 2 (1/кл) до около 25 (1/кл), от около 4 (1/кл) до около 20 (1/кл) или от около 5 (1/кл) до около 17 (1/кл), например, около 6 (1/кл), около 9 (1/кл), около 14 (1/кл) или около 16 (1/кл);
[00102] (h) AUC0-24 общей ДГЛК (или среднее значение или медиана AUC0-24 общей ДГЛК) от около 15000 нг⋅час/мл до около 900000 нг⋅час/мл, от около 20000 нг⋅час/мл до около 250000 нг⋅час/мл или от около 25000 нг⋅час/мл до около 225000 нг⋅час/мл, например, около 40000 нг⋅час/мл, около 210000 нг⋅час/мл, около 215000 нг⋅час/мл или около 435000 нг⋅час/мл;
[00103] (i) AUC0-24 общей ДГЛК/доза (или среднее значение или медиана AUC0-24/доза общей ДГЛК) от около 50 до около 400 час/кл, от около 60 до около 250 час/кл или от около 70 до около 225 час/кл, например, около 80 час/кл, около 100 час/кл, около 110 час/кл или около 215 час/кл;
[00104] (j) tmax(час) общей ДГЛК от около 2 до около 25 часов или от около 3 до около 20 часов, например, около 8 часов, около 10 часов или около 18 часов;
[00105] (k) соотношение Cmax общей ДГЛК к Cmax свободной ДГЛК от около 5:1 до около 12:1, от около 6:1 до около 10:1 или от около 7:1 до около 9:1, например, около 7,7:1, около 8,6:1, около 8,8:1 или около 9,8:1;
[00106] (l) уровень свободной ДГЛК в плазме в стационарной фазе (Cavg) или среднее значение или медиана уровня свободной ДГЛК в стационарной фазе (Cavg), через от 1 до около 30, от 1 до около 28, от 1 до около 14 или от 1 до около 10 последовательных дней ежедневного введения, до около 2000 нг/мл, до около 750 нг/мл или до около 700 нг/мл, например, около 385 нг/мл или около 675 нг/мл;
[00107] (m) уровень общей ДГЛК в плазме в стационарной фазе (Cavg) или среднее значение или медиана уровня общей ДГЛК в плазме (Cavg), через от 1 до около 30, от 1 до около 28, от 1 до около 14 или от 1 до около 10 последовательных дней ежедневного введения, до 250000 нг/мл, до 180000 нг/мл, до 150000 нг/мл, до 125000 нг/мл или до 100000 нг/мл; и/или
[00108] (n) соотношение содержания свободной ДГЛК в плазме к ДГЛК в коже (например, по данным определения в жидкости кожного волдыря) от около 0,2:1 до около 5:1, от около 0,5:1 до около 2,5:1 или от около 0,6:1 до около 1,5:1. В другом варианте реализации изобретения, при лечении с помощью композиции по настоящему изобретению, например, на протяжении периода от около 1 до около 12 недель, от около 1 до около 8 недель или от около 1 до около 4 недель, субъект или группа субъектов демонстрирует любые 2 или более, любые 3 или более, любые 4 или более, любые 5 или более, любые 6 или более, любые 7 или более, любые 8 или более, любые 9 или более, любые 10 или более, любые 11 или более, любые 12 или более, любые 13 или более, любые 14 или более, любые 15 или более, любые 16 или более, любые 17 или более, любые 18 или более, любые 19 или более, любые 20 или более, любые 21 или более или все 22 результата (a)-(n), описанные непосредственно выше.
[00109] В другом варианте реализации изобретения, при лечении субъектов натощак или после приема пищи или групп субъектов натощак или после приема пищи композицией, содержащей от около 200 мг ДГЛК до около 8000 мг ДГЛК (в виде одной или более дозированных единиц, например, в виде дозированных единиц по 500 мг или 1 г, которые в сумме составляют общие суточные дозы ДГЛК около 500 мг, около 1000 мг, около 2000 мг, около 3000 мг, около 4000 мг, около 5000 мг, около 6000 мг, около 7000 мг или около 8000 мг), после введения одной дозы или после введения нескольких доз, субъект или группа субъектов демонстрирует один или более из следующих результатов:
[00110] (a) соотношение Cmax свободной ДГЛК натощак: после приема пищи от около 1:1 до около 5:1, например, около 2,5:1, около 3:1 или около 3,5:1;
[00111] (b) соотношение AUC0-24 свободной ДГЛК натощак: после приема пищи от около 1:1 до около 5:1, например, около 1,5:1, около 2:1 или около 2,5:1;
[00112] (c) соотношение Cmax общей ДГЛК натощак: после приема пищи от около 1:1 до около 5:1, например, около 1:1, около 1,5:1 или около 2:1; и/или
[00113] (d) соотношение AUC0-24 общей ДГЛК натощак: после приема пищи от около 1:1 до около 5:1, например, около 1,5:1, около 2:1 или около 2,5:1.
[00114] В одном варианте реализации изобретения ДГЛК-содержащая композиция по изобретению содержит следующий характерный профиль жирных кислот:
[00115] В одном варианте реализации изобретения ДГЛК-содержащая композиция по изобретению содержит следующий характерный профиль жирных кислот:
20:3ω6 - ДГЛК
Родственные примеси
20:2ω6
20:3ω3
20:4ω6
20:4ω3
20:5ω3
Общее содержание неидентифицированных родственных примесей
<НПО - <5%
<НПО - <5%
<НПО - <5%
<НПО - <5%
<НПО - <5%
Не более 2
[00116] Иллюстративная ДГЛК-содержащая композиция по изобретению содержит следующий характерный профиль жирных кислот:
[00117] В одном варианте реализации изобретения ДГЛК-содержащая композиция по изобретению содержит следующий характерный профиль жирных кислот:
20:3ω6 - ДГЛК
[00118] В одном варианте реализации изобретения ДГЛК-содержащая композиция по изобретению содержит следующий характерный профиль жирных кислот:
C20:3 n-6 (ДГЛК-триглицериды) Мин. 30%
C16:0 Макс. 26%
C18:0 Макс. 12%
C18:1 n-9 Макс. 15%
C18:2 n-6 Макс. 15%
C18:3 n-6 Макс. 5%
C20;4 n·6 Макс. 1%
C22:0 Макс. 5%
C24:0 Макс. 15%
ПРИМЕРЫ
Пример 1
[00119] Три серии фармацевтических композиций, содержащих ДГЛК (с 2000 промилле dl-альфа токоферола) и расфасованных в желатиновые капсулы, получали, как проиллюстрировано в Табл. 1.
Таблица 1.
[00120] Оболочки капсул содержали следующие вспомогательные вещества: желатин, вода очищенная, глицерин, титана диоксид, а также технологические добавки лецитин и среднецепочечный триглицерид.
[00121] Кроме того, были получены дополнительные серии капсул ДГЛК, в том числе СЖК ДГЛК (стабилизированная номинально 2000 промилле dl-альфа токоферола) в капсулах, содержащих желатин, полисорб или смесь глицерина/полисорба, воду очищенную, титана диоксид, а также технологические добавки лецитин и среднецепочечный триглицерид, как проиллюстрировано в Табл. 2.
Таблица 2.
[00122] Состав капсульной оболочки для каждой из серий проиллюстрирован в Табл. 3 и 4 ниже.
Таблица 3.
1 Желатин УПС имеет более низкое содержание высокомолекулярных полимеров массы (> 200000 Да, ~5% ).
Таблица 4.
[00123] Проводили тестирование стабильности вышеупомянутых капсул. Капсулы из каждой серии хранили до 6 месяцев и оценивали с применением качественного или количественного протокола испытания Фарм. США 2040 Распад и растворение. Результаты приведены в Табл. 5-7.
Таблица 5. Данные стабильности для капсул ДГЛК Softgel: результаты качественного испытания на разрыв
60% ОВ
60% ОВ
65% ОВ
01
02
Таблица 6. Проценты ДГЛК глицерида
Таблица 7.
30 мин
45 мин
60 мин
94
102
102
102
30 мин
45 мин
60 мин
н/о
92
96
97
30 мин
45 мин
60 мин
81
91
96
102
30 мин
45 мин
60 мин
н/о
97
99
98
30 мин
45 мин
60 мин
95
101
101
102
30 мин
45 мин
60 мин
92
97
97
98
30 мин
45 мин
60 мин
88
97
99
99
30 мин
45 мин
60 мин
87
94
95
95
[00124] Как проиллюстрировано выше, с течением времени наблюдалось замедление скорости растворения в воде для капсул, составленных с глицерином и стандартным кислым говяжьим желатином (E09726/01 и E09727). Зарегистрирована скорость высвобождения ДГЛК более чем 30 минут через 6 месяцев при 40°C/75% ОВ в симулированном желудочном соке (pH 1,2, пепсин).
[00125] Скорость высвобождения ДГЛК менее чем 30 минут через 6 месяцев при 40 °C/75% ОВ была достигнута только в симулированном желудочном соке (pH 1,2, пепсин) для капсул, содержащих обработанный известью костный желатин с более низкой молекулярной массой (М. м.) (E09777/02).
[00126] Зарегистрировано значительное увеличение образования ДГЛК глицерида через некоторое время в оболочках капсул ДГЛК, содержащих глицерин (Табл. 4). Этот феномен был зависимым от температуры, с образованием самых высоких концентраций ДГЛК при 40°C и 75% ОВ.
[00127] Полисорб обычно применяется в качестве гидрофильного пластификатора, с целью ограничения обмена между средой, заполняющей капсулу, и оболочкой. D-сорбит и 1,4-сорбитан имеют более высокую М. м., чем глицерин, что ограничивает его проникновение сквозь желатиновую оболочку. Несмотря на это, по-прежнему происходило взаимодействие D-сорбита и 1,4-сорбитана с образованием эфиров СЖК ДГЛК в сериях E09777 1/2 и 3.
[00128] Не зарегистрировано снижения кислотности ДГЛК для серий, составленных с D-сорбитом и 1,4-сорбитаном (E09777 1/02/3), тогда как для E09778, составленной с глицерином, наблюдалось снижение кислотности.
[00129] Не зарегистрировано снижения скорости растворения в воде с течением времени для капсул, составленных с D-сорбитом и 1,4-сорбитаном (E09777/03). Скорость высвобождения ДГЛК в воде составила менее чем 30 минут через 3 месяца при 40°C/75% ОВ.
Пример 2
[00130] Будет проведено рандомизированное, двойное слепое, плацебо-контролируемое исследование фазы II для оценки эффективности и безопасности перорально введенной ДГЛК у пациентов с умеренным или тяжелым атопическим дерматитом. Приблизительно 100 субъектов мужского или женского пола, в возрасте 18 лет или старше, с умеренным или тяжелым атопическим дерматитом (АД) будут включены в данное исследование.
[00131] Атопический дерматит представляет собой хроническое воспалительное расстройство кожи, характеризующееся зудом, папулами, лихенификацией, экскориацией, ксерозом и намоканием. Распространенность АД увеличилась в последние несколько десятилетий, затрагивая до 20 процентов маленьких детей, причем большинство случаев начинается у детей в возрасте до 5 лет. В большинстве случаев при взрослении наступает улучшение.
[00132] АД представляет собой многофакторное заболевание, причем доминирующими факторами являются наследственность, окружающая среда и нарушенная иммунная реакция. Дендритные клетки, T-лимфоциты, макрофаги, кератиноциты, тучные клетки и эозинофилы - все играют роль в АД, высвобождая провоспалительные цитокины и хемокины, которые индуцируют воспалительный ответ, характерный для очагов атопического дерматита.
[00133] Большинство одобренных на данное время терапевтических средств для лечения АД являются местными и включают кортикостероиды, пимекролимус и такролимус. До сих пор кортикостероиды были базовым компонентом лечения АД, причем они доступны в различных дозировках и лекарственных формах. Местные кортикостероиды эффективны при лечении АД, но их применение ограничено потенциалом местных побочных реакций, таких как атрофия кожи и стрии. Кроме того, системная абсорбция кортикостероидов может индуцировать диабет, катаракту, остеопороз и подавление гипоталамически-гипофизарной оси. Дополнительно, за временным улучшением часто следует внезапное обострение болезни при прекращении лечения. Другие доступные терапевтические средства включают местные ингибиторы кальциневрина (например, пимекролимус, такролимус) и препараты дегтя. Сообщалось о случаях лимфомы у пациентов, получавших лечение ингибиторами кальциневрина. Пациентов с более тяжелой формой лечат фототерапией в ультрафиолетовом диапазоне B и A или агентами для перорального введения, такими как кортикостероиды, циклоспорин, микофенолят мофетил, метотрексат и азатиоприн.
[00134] Дигомо-гамма-линоленовая кислота (ДГЛК) представляет собой незаменимую жирную кислоту, найденную в организме в качестве естественного продукта 2-углеродной элонгации гамма-линоленовой кислоты (ГЛК). ДГЛК метаболизируется ферментами циклооксигеназой (ЦОГ) и липоксигеназой (ЛПО) с образованием эйкозаноидов, простагландинов (ПГ) серии 1, а также гидрокси-жирной кислоты, такой как 15-гидроксиэйкозатриеновая кислота ДГЛК (15-ГЭТрЕ).
ГИПОТЕЗА И ЗАДАЧИ ИССЛЕДОВАНИЯ
[00135] DS107G превосходит плацебо с точки зрения улучшения оценки ПЭИТ у пациентов с умеренным или тяжелым атопическим дерматитом.
[00136] Основная цель: сравнить эффективность двух доз капсул DS107G для перорального введения против плацебо при лечении взрослых пациентов с умеренным или тяжелым атопическим дерматитом.
[00137] Вторая цель: оценить безопасность двух доз капсул DS107G для перорального введения против плацебо у взрослых пациентов с умеренным или тяжелым атопическим дерматитом.
КОНЕЧНЫЕ ТОЧКИ ИССЛЕДОВАНИЯ
Первичная конечная точка
[00138] Доля больных, достигающих ООИ (Общей оценки исследователем) 0 (чистая) или 1 (практически чистая) и уменьшение ООИ по меньшей мере на 2 пункта ООИ на 8 неделе.
Вторичные конечные точки
[00139] Изменение по сравнению с исходным значением ООИ на 2, 4 и 8 неделе.
[00140] Изменение по сравнению с исходным значением ПЭИТ (Площадь поверхности и индекс тяжести экземы) на 2, 4 и 8 неделе.
[00141] Доля больных, достигающих уменьшения ООИ по меньшей мере на 1 пункт на 8 неделе.
[00142] Изменение по сравнению с исходным значением Пациент-ориентированной экземы меры (ПОЭМ) на 2, 4 и 8 неделе.
[00143] Изменение по сравнению с исходным значением баллов Дерматологического индекса качества жизни (ДИКЖ) на 2, 4 и 8 неделе.
[00144] Изменение по сравнению с исходным значением баллов ШАД на 2, 4 и 8 неделе.
[00145] Изменение по сравнению с исходным значением баллов Визуальной аналоговой шкалы зуда (ВАШ) пациента на 2, 4 и 8 неделе.
[00146] Изменение по сравнению с исходным значением площади поверхности тела (ППТ) на 2, 4 и 8 неделе.
[00147] Количество вызванных лечением побочных реакций (ВЛПР) в каждой группе лечения.
Исследовательские конечные точки
[00148] Изменение по сравнению с исходным значением трансэпидермальной потери воды (ТЭПВ) на 2, 4 и 8 неделе (только выбранные центры).
[00149] Концентрация общей и свободной ДГЛК в плазме в исходной точке, на 4 и 8 неделе.
[00150] Профиль общих жирных кислот в плазме в исходной точке, на 4 неделе и 8 неделе (образец хранят и анализируются позже).
ДИЗАЙН ИССЛЕДОВАНИЯ
[00151] Приблизительно 100 пациентов c умеренным или тяжелым атопическим дерматитом будут включены в данное многоцентровое, двойное слепое, плацебо-контролируемое исследование фазы IIa. Все субъекты будут подписывать информированное согласие и проходить скрининг для определения пригодности. Субъекты будут рандомизированы (1:1) в ходе визита исходного уровня для получения перорально 2 г DS107G (капсулы ДГЛК, предоставляемые в виде непрозрачных, овальных мягких желатиновых капсул, содержащих 500 мг ДГЛК), 1 г DS107G или плацебо один раз в сутки в течение 8 недель натощак. Включение в исследование будет стратифицировано по тяжести (на основании ООИ) заболевания, причем будет набрано не более 30% пациентов с легким атопическим дерматитом (что определяется ООИ 2 балла).
[00152] Субъекты будут посещать клинику 6 раз: для скрининга, визит исходного уровня, 2 неделя, 4 неделя, 8 неделя (окончание лечения/досрочное прекращение участия) и 10 неделя (последующее наблюдение). Участие всех субъектов в исследовании завершится визитом на 10 неделе. Первичной переменной эффективности будет доля пациентов, достигших ООИ 0 (чистая) или 1 (практически чистая) и уменьшения ООИ по меньшей мере на 2 балла на 8 неделе. Вторичные переменные эффективности будут включать ООИ в ходе других визитов, зуд (по данным визуальной аналоговой шкалы ШАД), ПЭИТ, ППТ, ПОЭМ, ДИКЖ, ШАД и ТЭПВ (только выбранные центры). Безопасность будет оценена на основе побочных реакций, медицинского осмотра, основных показателей жизнедеятельности и лабораторных анализов безопасности (включая тесты на беременность для женщин, способных к деторождению). Образцы для анализа фармакокинетики будут получены в ходе визита исходного уровня (0 день), визитов на 4 неделе и 8 неделе для измерения минимальных уровней общей и свободной ДГЛК в плазме. Отдельные образцы плазмы будут храниться для дальнейшего анализа профиля общих жирных кислот и профиля интерлейкинов.
ПОПУЛЯЦИЯ ИССЛЕДОВАНИЯ
[00153] Приблизительно 100 субъектов с умеренным или тяжелым атопическим дерматитом по данным баллов ООИ и ППТ по меньшей мере 10% могут быть включены в настоящее исследование. Субъекты будут мужчинами или женщинами, в возрасте 18 лет или старше.
Критерии включения:
[00154] Субъекты мужского или женского пола, в возрасте 18 лет и старше на день подписания формы информированного согласия (ФИС).
[00155] Клинически подтвержденный диагноз активного атопического дерматита согласно критериям Hanifin и Rajka (Приложение G).
[00156] Умеренный или тяжелый атопический дерматит на момент начала исследования по данным ООИ по меньшей мере 3 в ходе визита исходного уровня.
[00157] Атопический дерматит, покрывающий минимум 10% площади поверхности тела на момент начала исследования.
[00158] Индекс массы тела (ИМТ) от 18 до 35 кг/м2 включительно.
[00159] Пациенты женского пола, способные к деторождению, должны применять соответствующие меры контрацепции или иметь стерилизованного партнера на протяжении исследования: системные гормональные контрацептивы, внутриматочное устройство или барьерный метод контрацепции в сочетании со спермицидом, или согласиться на сексуальное воздержание. Гормональные контрацептивы должны применяться в стабильной дозе по меньшей мере на протяжении одного месяца перед началом исследования. Примечание: Женщинами, не обладающими способностью к деторождению, являются:
женщины после хирургической стерилизации (гистерэктомия или двусторонняя овариэктомия или перевязка труб);
женщины в возрасте старше 60 лет;
женщины в возрасте от 40 до 60 лет, у которых менструации отсутствовали по меньшей мере в течение 12 месяцев, и тест на фолликулостимулирующий гормон (ФСГ) подтвердил отсутствие способности к деторождению (ФСГ ≥ 40 мМЕ/мл), или менструации отсутствовали по меньшей мере в течение 24 месяцев без подтвержденных уровней ФСГ.
[00160] Пациенты, которые могут и готовы приостановить лечение атопического дерматита на период исследования (за исключением разрешенных смягчающих средств).
[00161] Способность и готовность предоставить письменное информированное согласие, причем согласие должно быть получено до проведения любых процедур, связанных с исследованием.
Критерии исключения
[00162] Пациенты женского пола с положительным тестом на беременность на момент скрининга или визита исходного уровня (начало исследования) в 0 день или лактирующие женщины.
[00163] Любое клинически значимое медицинское состояние, контролируемое или неконтролируемое, или отклонение лабораторных показателей, которое, по мнению исследователя, будет подвергать пациента ненужному риску или мешать интерпретации результатов исследования.
[00164] Клинически значимое нарушение почечной или печеночной функции.
[00165] Другие состояния кожи, которые могут воспрепятствовать постановке диагноза атопического дерматита и/или оценке (например, псориаз или присутствующие вирусные, бактериальные и грибковые инфекции кожи).
[00166] Гиперчувствительность к любому веществу в капсулах DS107G или плацебо.
[00167] Применение биологических средств за 3 месяца (начало исследования) или 5 периодов полувыведения (в зависимости от того, что длится дольше) до начала лечения/визита в 0 день.
[00168] Применение системного лечения (кроме биологических средств), которое может повлиять на атопический дерматит менее чем за 4 недели до визита исходного уровня (0 день), например, ретиноидов, ингибиторов кальциневрина, метотрексата, циклоспорина, гидроксикарбамида (гидроксимочевины), азатиоприна и пероральных/инъекционных кортикостероидов; разрешено применение интраназальных кортикостероидов и ингаляционных кортикостероидов при стабильных медицинских состояниях.
[00169] Лечение любым экспериментальным лекарственным средством в пределах 30 дней или 5 периодов полувыведения (в зависимости от того, что длится дольше) до визита в 0 день (начало исследования).
[00170] Чрезмерное воздействие солнечных лучей, использование вертикальных соляриев или других источников ультрафиолетового (УФ) света за 4 недели до визита в 0 день (начало исследования) и/или запланированное путешествие в регион с солнечным климатом или использование вертикальных соляриев или других источников УФ в период между скринингом и последующими визитами.
[00171] Применение любого местного медикаментозного лечения атопического дерматита на протяжении 2 недель до начала лечения/визита в 0 день (начало исследования), в том числе, но не ограничиваясь этим, местных кортикостероидов, ингибиторов кальциневрина, смол, отбеливающего вещества, противомикробных средств и отбеливающих ванн.
[00172] Местное применение продуктов, содержащих мочевину, церамиды или гиалуроновую кислоту, за 2 недели до 0 дня.
[00173] Применение антигистаминных средств для лечения атопического дерматита в пределах 2 недель до начала исследования.
[00174] Значимое, неконтролируемое сердечно-сосудистое (анамнез отклонений на ЭКГ, которые, по мнению исследователя, являются клинически значимыми), неврологическое, злокачественное, психиатрическое, респираторное или гипертоническое заболевание, а также диабет и артрит.
[00175] Медицинский анамнез хронического инфекционного заболевания (например, гепатит B, гепатит C или инфицирование вирусом иммунодефицита человека).
[00176] Анамнез клинически значимого злоупотребления наркотиками или спиртным на протяжении года до 0 дня (начало исследования).
Ограничения исследования
[00177] Каждый субъект будет опрошен относительно конкретных пунктов, перечисленных в списке ниже, перед введением лекарственного средства. Если субъект не соблюдает указанные ограничения, руководитель клинических испытаний (или уполномоченное лицо) и/или спонсор будет принимать решение, позволить ли субъекту продолжать участие в исследовании. Несоблюдение указанных ограничений будет регистрироваться.
[00178] Субъекты будут проинструктированы о необходимости воздержаться от планирования путешествий в регионы с солнечным климатом или использования оборудования для загара в период между скринингом и последующими визитами.
[00179] Субъекты будут проинструктированы о необходимости воздержаться от применения любых лекарственных средств/терапевтических средств, которые могут повлиять на атопический дерматит (ссылка на критерии исключения и раздел запрещенных терапевтических средств или процедур), на протяжении исследования.
[00180] Субъекты должны будут прекратить прием пищи по меньшей мере за 8 часов до введения лекарственного средства при пробуждении. Голодание будет длиться по меньшей мере 60 минут после введения лекарственного средства, после чего субъект может позавтракать. Будет разрешено пить воду в любое время в ходе периода воздержания от приема пищи, но прием других жидкостей не разрешается. Лечение других состояний, которое разрешено в ходе исследования, можно проводить как обычно.
[00181] В ходе визитов исходного уровня (0 день), на 4 неделе и 8 неделе, будет взят образец крови для анализа ФК. Образцы для изучения ФК следует получать до введения дозы; поэтому введение исследуемого лекарственного средства будет происходить в ходе визита в 0 день и визита на 4 неделе. Поскольку введение дозы будет происходить в клинике на 0 день и 4 неделе, субъекты должны будут воздержаться от приема пищи по меньшей мере в течение 8 часов до введения исследуемого лекарственного средства, и им будет разрешено поесть через 60 минут после введения исследуемого лекарственного средства.
Прекращение участия в исследовании
[00182] Субъекты имеют право прекратить участие в исследовании в любое время, по любой причине, без штрафа. Кроме того, исследователь имеет право прекратить участие субъектов в исследовании, если он считает, что это в интересах субъекта, или если субъект отказывается сотрудничать или не соблюдает предписания. Все заинтересованные лица должны понимать, что большое количество случаев прекращения участия в исследовании может сделать исследование недействительным; таким образом, ненужного прекращения участия субъектов следует избегать. Если субъект решит прекратить участие, будут приложены все усилия для завершения исследования и регистрации результатов наблюдений, особенно последующего обследования, настолько тщательно, насколько это возможно.
[00183] Исследователь или один из его или ее сотрудников должен связаться с субъектом по телефону или посредством личного визита, чтобы определить настолько полно, насколько это возможно причину прекращения участия и зарегистрировать причину в первичном документе субъекта и индивидуальной регистрационной карте (ИРК). Полная заключительная оценка досрочного прекращения участия (неделя 8) должна быть проведена на момент прекращения участия субъекта, с объяснением того, почему субъект прекращает участие в исследовании. Если причиной прекращения участия субъекта является побочная реакция или отклонение в лабораторных показателях, будет зарегистрирована конкретная основная побочная реакция или показатель. Субъектов, которые прекращают участие до визита на 8 неделе, попросят, если они согласятся, прийти в клинику для окончательной оценки (визит досрочного прекращения участия).
Причины прекращения участия в исследовании включают:
[00184] Принятие решения исследователем о прекращении участия субъекта. Если такое решение принято в результате тяжелого или устойчивого побочной реакции, отклонения лабораторных показателей или интеркуррентного заболевания, введение исследуемого лекарственного средства следует прекратить и предпринять соответствующие меры. Исследователь немедленно сообщит Спонсору или уполномоченному лицу.
[00185] Запрос субъекта или лечащего врача о прекращении участия субъекта в исследовании.
[00186] Субъект по какой-либо причине нуждается в лечении другим терапевтическим агентом, который продемонстрировал свою эффективность при лечении исследуемого показания. В данном случае, прекращение участия в исследовании произойдет немедленно после введения нового агента.
[00187] Связь с субъектом утрачена, в данном случае должны быть предприняты разумные усилия наладить связь с субъектом и выяснить его/ее состояние, причем такие попытки должны быть задокументированы.
[00188] Серьезное нарушение протокола, включая постоянное несоблюдение предписаний.
[00189] Спонсор или регуляторные органы по какой-либо причине прекращают исследование. Участие всех субъектов в исследовании будет прекращено, и субъектам будет сообщена причина прекращения.
[00190] Беременность в любой момент в ходе исследования.
[00191] Другое: субъект может прекратить участие в исследовании по любой другой причине, включая отзыв своего согласия.
ЛЕЧЕНИЕ
Проведение лечения
[00192] Субъекты, которые соответствуют всем критериям включения и не соответствуют ни одному критерию исключения, могут быть включены в исследование. Каждый субъект должен прочитать и подписать форму информированного согласия до начала проведения любых процедур скрининга. Данное исследование включает сравнение DS107G (2 г) с плацебо, которые вводят перорально один раз в сутки при пробуждении общей продолжительностью 8 недель. Последнее введение исследуемого лекарственного средства должно происходить в день, предшествующий визиту на 8 неделе/визит досрочного прекращения участия (ДПУ). Субъекты будут рандомизированы в одну из двух групп лечения в соотношении 1:1:
[00193] Группа лечения A: 2 г DS107G (4 капсулы).
[00194] Группа лечения B: 2 г капсул плацебо (4 капсулы).
[00195] Субъекты должны будут прекратить прием пищи по меньшей мере за 8 часов до введения лекарственного средства при пробуждении. Голодание будет длиться по меньшей мере 60 минут после введения лекарственного средства, после чего субъект может позавтракать. Будет разрешено пить воду в любое время в ходе периода воздержания от приема пищи, но прием других жидкостей не разрешается. Лечение других состояний, которое разрешено в ходе исследования, можно проводить как обычно.
[00196] Блистерные упаковки будут состоять из 7 рядов по 4 капсулы. Каждый ряд содержит одну суточную дозу. Субъекты будут проинструктированы принимать 4 капсулы слева направо, сверху вниз.
Исследуемое лечение
[00197] Капсулы DS107G будут предоставлены в виде непрозрачных, овальных мягких желатиновых капсул, содержащих 500 мг свободной жирной кислоты (СЖК) ДГЛК.
[00198] Капсулы плацебо также будут предоставлены в виде непрозрачных, овальных мягких желатиновых капсул, содержащих 500 мг жидкого парафина.
[00199] Капсулы DS107G будут предоставляться в заводской форме (с маскировкой), упакованные блистеры из алюминиевой фольги по 28 штук. Плацебо будет предоставляться в идентичных блистерах и упаковках и храниться/упаковываться так же, как капсулы DS107G. Исследуемое терапевтическое средство будет промаркировано согласно законоположениям США и Канады.
[00200] Исследуемое лекарственное средство будет предоставлено спонсором исследователю и будет храниться в исследовательском центре, в закрытой на замок комнате или кабинете с ограниченным доступом. Капсулы DS107G и плацебо следует хранить при контролируемой комнатной температуре, 15-30°C, и выдавать только субъектам, принимающим участие в исследовании, под наблюдением исследователя.
[00201] Исследуемое лекарственное средство будет выдаваться субъекту в исследовательском центре, в ходе каждого визита исследования. Субъекты должны вернуть все блистерные упаковки исследуемого лекарственного средства (использованные и неиспользованные) в исследовательский центр. Капсулы в блистерных упаковках будут пересчитаны перед отпуском и при возврате, причем результаты подсчета будут зарегистрированы в первичных документах и электронной индивидуальной регистрационной карточке (эИРК). Каждый субъект должен быть проинструктирован относительно важности возврата исследуемого лекарственного средства в ходе следующего визита исследования. Если субъект не возвращает исследуемое лекарственное средство, он/она будет инструктирован сделать это как можно быстрее.
[00202] Исследователь несет ответственность за аккуратную регистрацию исследуемого лекарственного средства, полученного вначале, выданного/использованного исследуемого лекарственного средства, возвращенного субъектами лекарственного средства и уничтоженного или возвращенного Спонсору или уполномоченному лицу лекарственного средства. Все формы учета исследуемого лекарственного средства и журналы лечения должны храниться у исследователя в документации исследования. Эти записи в любое время должны быть доступны для инспекции Спонсором, его уполномоченными лицами или контролирующими органами.
[00203] Использованные коробки/блистерные упаковки лекарственного средства будут храниться в безопасных условиях до уничтожения, причем исследователь должен вести их учет. Лицо, контролирующее проведение исследования будет вести учет лекарственного средства для всего исследуемого лекарственного средства в исследовательском центре и будет помогать в возврате исследуемого лекарственного средства, в том числе использованного и неиспользованного исследуемого лекарственного средства Спонсору или уполномоченному лицу. После верификации учета лекарственного средства спонсором, исследователь гарантирует надлежащее уничтожение или возврат остатков исследуемого продукта.
[00204] Будет учитываться любое случайное или намеренное уничтожение исследуемого лекарственного средства. Будет объяснено любое отличие между выданным и возвращенным количеством.
[00205] Инвентаризационные и учетные записи лекарственного средства будут вестись в каждом исследовательском центре в соответствии с директивами Надлежащей клинической практики/Международной конференции по гармонизации (НКП/МКГ). Приблизительно 100 пациентов будут рандомизированы в двойные слепые группы лечения в соотношении 1:1 Интерактивной системой с доступом через Интернет (ИСДИ) или Системой интерактивного голосового ответа (СИГО), как указано ниже:
[00206] Группа лечения A: 2 г DS107G (4 капсулы).
[00207] Группа лечения B: 2 г капсул плацебо (4 капсулы).
[00208] Переставленные местами блоки списка рандомизации, стратифицированные по центру, будут генерироваться Dignity Sciences или уполномоченным ею лицом. Расписание рандомизации с назначениями исследуемого лекарственного средства будет генерироваться до начала исследования и будет известно только лицам, ответственным за маркировку исследуемого лекарственного средства. ИСДИ или СИГО будет присваивать номер набора исследуемого лекарственного средства каждому субъекту, и его содержимое будет базироваться на коде рандомизации.
[00209] В исследовательском центре каждому субъекту в ходе скрининга будет присвоен идентификационный номер пациента, который будет использоваться для всей документации пациента. Идентификационный номер пациента будет включать номер центра и номер пациента, присвоенный по порядку в ходе скринингового визита (например: 02-010 для десятого пациента, который проходит скрининг в центре № 02). Номера будут присвоены в порядке увеличения, начиная с 001.
Обоснование выбора и расчета времени введения доз в исследовании
[00210] Дозы до 4 г хорошо переносятся здоровыми субъектами. Доза 2 г выбрана как исследуемая доза в данном исследовании, в первую очередь на основании фармакокинетических результатов исследования Фазы I, которые наводят на мысль о насыщаемых уровнях общей ДГЛК в коже при повторном пероральном введении доз более чем 2 г в сутки. Кроме того, рассматривались следующие факторы:
[00211] меньшая частота транзиторных нарушений со стороны желудочно-кишечного тракта при дозе 2 г, по сравнению с 4 г в сутки.
[00212] количество капсул (4) для ежедневного введения. Более высокая доза возможна, но считается менее желательной, поскольку слишком большое количество капсул может отрицательно повлиять на соблюдение пациентом схемы лечения.
Раскрытие данных клинического исследования
[00213] В течение всего времени, информация о лечении и рандомизации будет оставаться конфиденциальной, и не будет раскрываться исследователю, персоналу исследования, контрактной исследовательской организации (КИО) или исследовательской команде спонсора до завершения исследования.
[00214] Замаскированные коды могут быть раскрыты только в экстремальных ситуациях из соображений безопасности субъекта. Способ будет осуществляться вручную или электронными средствами. В случае раскрытия маскированных данных субъекта, причина должна быть полностью документирована. При любой возможности, исследователь должен связаться со Спонсором или его уполномоченным лицом перед раскрытием данных. Если данные раскрыты, исследователь должен немедленно проинформировать медицинского наблюдателя. Документирование раскрытия маскированных данных должно осуществляться с датой и временем, когда данные были раскрыты, а также именами всех задействованных сотрудников.
[00215] Участие в исследовании субъекта, данные которого были раскрыты, будет прекращено, и будет проведена процедура досрочного прекращения участия (ДПУ) (визит на 8 неделе). В случаях, в которых существуют этические причины для продолжения участия субъекта в исследовании, исследователь должен получить конкретное одобрение Спонсора или его уполномоченного лица для продолжения участия субъекта в исследовании.
Сопутствующая терапия
[00216] Все лекарственные средства (в том числе, безрецептурные лекарственные средства, витамины и антацидные средства), принятые менее чем за 4 недели (включая 4 неделю) до скрининга и в ходе исследования должны быть зарегистрированы. Все лекарственные средства, принятые для лечения атопического дерматита в течение 2 месяцев до скрининга, должны быть зарегистрированы.
[00217] Записи о лекарственных средствах должны содержать специфичное генерическое название. Торговое название товара может применяться в случае комбинированных лекарственных средств. Записи будут включать дозу, лекарственную форму и частоту введения, способ введения, дату начала, дату окончания и показание. Если лечение прекращено или доза изменена, эти детали должны быть зарегистрированы.
[00218] Исследователь должен оценивать любые сопутствующие процедуры, лекарственные средства и биологически активные добавки, которые явно не запрещены, на предмет приемлемости.
Разрешенные терапевтические средства
[00219] Смягчающие средства
[00220] Субъекты могут наносить на кожу, в том числе, пораженную АД, мягкое смягчающее средство по своему выбору, при условии, что применение смягчающего средства начато по меньшей мере за 2 недели до 0 дня и продолжается с такой же частотой и на такой же площади поверхности кожи на протяжении исследования. Субъектов попросят избегать применения смягчающих средств, содержащих любой из следующих ингредиентов:
[00221] (1) Мочевина
[00222] (2) Церамид
[00223] (3) Гиалуроновая кислота
[00224] Следует приложить все усилия для применения одного и того же смягчающего средства на протяжении исследования. Коммерческое название выбранных(ого) смягчающих(его) средств(а) будет занесено в первичный документ и эИРК. Никакие другие продукты нельзя наносить на поврежденные участки в ходе исследования.
Другие разрешенные терапевтические средства
[00225] Применение неседативных антигистаминных средств (например, лоратадин, фексофенадин) в ходе исследования разрешается только в случае применения для лечения других медицинских состояний, кроме атопического дерматита. Применение таких лекарственных средств разрешается в ходе исследования только в том случае, если субъект применял стабильную дозу по меньшей мере в течение 2 недель до 0 дня и продолжает применять такой же агент ежедневно в ходе исследования.
[00226] Разрешено применение ингаляционных и интраназальных кортикостероидов при стабильных медицинских состояниях.
Запрещенные терапевтические средства или процедуры
[00227] Следующие местные терапевтические средства или процедуры запрещены в ходе исследования для всех субъектов:
[00228] Лекарственные средства для местного применения, которые могли бы повлиять на атопический дерматит, в том числе, но не ограничиваясь этим:
[00229] местные кортикостероиды
[00230] ингибиторы кальциневрина
[00231] смолы
[00232] отбеливающее вещество
[00233] противомикробные агенты
[00234] ванны с отбеливающим веществом
[00235] любые продукты для местного применения содержащие мочевину, церамиды или гиалуроновую кислоту
[00236] системная терапия, которая могла бы повлиять на атопический дерматит, например, ретиноиды, ингибиторы кальциневрина, метотрексат, циклоспорин, гидроксикарбамид (гидроксимочевина), азатиоприн и пероральные/инъекционные кортикостероиды
[00237] антигистаминные средства (кроме неседативного антигистаминного средства)
[00238] любой биологический агент
[00239] УФА или УФB фототерапия
[00240] лечение псораленом+ультрафиолетом A (ПУВА)
[00241] чрезмерное воздействие солнечных лучей или использование солярия
[00242] любой исследуемый агент
Оценка соблюдения предписаний
[00243] Соблюдение предписаний относительно лечения будет оцениваться в ходе каждого визита путем прямого опроса, обзора журнала соблюдения предписаний субъектом и подсчета капсул, и будет основываться на последнем. При каждом визите субъекты будут получать, наряду с исследуемым лекарственным средством, бумажный дневник. Субъекты будут указывать любые пропущенные дозы в дневнике, а также время последнего приема пищи перед введением исследуемого лекарственного средства и приема пищи после введения исследуемого лекарственного средства. Субъекты будут инструктированы приносить все капсулы и блистерные упаковки (использованные и неиспользованные) и журнал соблюдения предписаний на следующий визит исследования. Любое отклонение от назначенной схемы введения будет регистрироваться в первичном документе и в эИКР. Субъектов, которые допускают серьезные отклонения от схемы лечения, будут консультировать.
ПРОЦЕДУРЫ ИССЛЕДОВАНИЯ
[00244] См. Приложение A относительно технологической карты процедур, выполняемых в ходе каждого визита.
Скрининг, Визит 1 (от дня -30 до дня -1)
[00245] Скрининговая оценка будет выполняться только после того, как субъект согласился принимать участие, подписал и датировал форму информированного согласия. Никакое лечение или связанные с исследованием процедуры не будет начинаться до подписания информированного согласия. Визиты 0 дня должны выполняться не позднее 30 дней после скринингового визита.
[00246] Скрининговая оценка будет выполняться в соответствии с критериями включения и исключения. Если субъект соответствует всем критериям включения и не соответствует ни одному из критериев исключения, субъект может быть включен в исследование.
[00247] Следующие процедуры будут проводиться в ходе скринингового визита:
[00248] информированное согласие;
[00249] обзор критериев включения-исключения, включая обзор критериев Hanifin и Rajka (Приложение G);
[00250] присвоение субъекту идентификационного номера (номер центра - номер субъекта);
[00251] демографические данные;
[00252] сопутствующая терапия;
[00253] анамнез медикаментозного лечения/хирургических вмешательств;
[00254] медицинский осмотр;
[00255] основные показатели жизнедеятельности;
[00256] ИМТ;
[00257] лабораторные показатели безопасности (химические анализы, коагуляция, анализ крови и анализ мочи);
[00258] сывороточный тест на беременность (только для женщин, способных к деторождению) и тест на уровень ФСГ для женщин в возрасте от 40 до 60 лет, у которых менструации отсутствуют по меньшей мере в течение 12 месяцев, но менее чем 24 месяца;
[00259] оценка ППТ;
[00260] ООИ.
Исходный уровень, Визит 2 (0 день)
[00261] Субъекты должны будут воздержаться от приема пищи по меньшей мере в течение 8 часов до введения исследуемого лекарственного средства. Им будет разрешено поесть через 60 минут после введения исследуемого лекарственного средства.
[00262] Следующие процедуры будут проводиться в ходе данного визита:
[00263] (1) подтверждение права на участие на основании критериев включения и исключения
[00264] (2) обновление или подтверждение анамнеза медикаментозного лечения/хирургических вмешательств
[00265] (3) сопутствующая терапия
[00266] (4) Основные показатели жизнедеятельности
[00267] (5) ИМТ
[00268] (6) лабораторные показатели безопасности и биомаркеры воспаления (химические анализы, коагуляция, анализ крови, анализ мочи и профиль интерлейкинов)
[00269] (7) анализ мочи на беременность (только женщины, способные к деторождению)
[00270] (8) отбор образца крови для определения фармакокинетических показателей до начала лечения
[00271] (9) отбор образца крови для определения общего профиля жирных кислот
[00272] (10) оценка ППТ
[00273] (11 ) ООИ
[00274] (12) оценка ПЭИТ
[00275] (13) оценка ШАД (в том числе, оценка зуда ВАШ)
[00276] (14) опросник ПОЭМ
[00277] (15) опросник ДИКЖ
[00278] (16) оценка ТЭПВ (только выбранные центры)
[00279] (17) рандомизация субъекта с помощью ИСДИ/СИГО
[00280] (18) введение исследуемого лекарственного средства
[00281] (19) выдача исследуемого лекарственного средства
[00282] (20) выдача субъектам журнала соблюдения предписаний
[00283] (21) оценка побочных реакций (после первого введения исследуемого лекарственного средства)
Неделя 2, Визит 3 (14 день ± 2 дня)
[00284] Следующие процедуры будут выполняться в ходе данного визита:
[00285] (1) Основные показатели жизнедеятельности
[00286] (2) Тест мочи на беременность (только для женщин, способных к деторождению)
[00287] (3) оценка ППТ
[00288] (4) ООИ
[00289] (5) оценка ПЭИТ
[00290] (6) оценка ШАД (включая оценку зуда ВАШ)
[00291] (7) опросник ПОЭМ
[00292] (8) опросник ДИКЖ
[00293] (9) оценка ТЭПВ (только выбранные центры)
[00294] (10) сбор и выдача исследуемого лекарственного средства; обзор, сбор и выдача субъектам журнала соблюдения предписаний
[00295] (11) подсчет капсул
[00296] (12) сопутствующая терапия
[00297] (13) оценка побочных реакций
Неделя 4, Визит 4 (28 день ± 2 дня).
[00298] Субъекты должны будут воздержаться от приема пищи по меньшей мере в течение 8 часов до введения исследуемого лекарственного средства. Им будет разрешено поесть через 60 минут после введения исследуемого лекарственного средства.
[00299] Следующие процедуры будут проводиться в ходе данного визита:
[00300] (1) основные показатели жизнедеятельности
[00301] (2) тест мочи на беременность (только женщины, способные к деторождению)
[00302] (3) медицинский осмотр
[00303] (4) лабораторные показатели безопасности (химические анализы, коагуляция, анализ крови, анализ мочи и профиль интерлейкинов)
[00304] (5) отбор образца крови до введения дозы для анализа фармакокинетики (если исследуемое лекарственное средство было принято до визита, субъект должен вернуться в центр на следующий день)
[00305] (6) отбор образца крови для определения общего профиля жирных кислот
[00306] (7) оценка ППТ
[00307] (8) ООИ
[00308] (9) оценка ПЭИТ
[00309] (10) оценка ШАД (включая оценку зуда ВАШ)
[00310] (11) опросник ПОЭМ
[00311] (12) опросник ДИКЖ
[00312] (13) оценка ТЭПВ (только выбранные центры)
[00313] (14) введение исследуемого лекарственного средства (инструктирование субъекта относительно того, что последняя доза должна быть введена в день, предшествующий визиту на 8 неделе)
[00314] (15) сбор и выдача исследуемого лекарственного средства
[00315] (16) обзор, сбор и выдача субъектам журнала соблюдения предписаний
[00316] (17) подсчет капсул
[00317] (18) сопутствующая терапия
[00318] (19) оценка побочных реакций.
Неделя 8, Визит 5 (56 день ± 2 дня) (Завершение лечения/Визит досрочного прекращения участия).
[00319] Следующие процедуры будут проводиться в ходе данного визита:
[00320] (1) текущий обзор медицинского анамнеза
[00321] (2) основные показатели жизнедеятельности
[00322] (3) медицинский осмотр
[00323] (4) ИМТ
[00324] (5) лабораторные показатели безопасности (химические анализы, коагуляция, анализ крови и анализ мочи)
[00325] (6) сывороточный тест на беременность (только женщины, способные к деторождению)
[00326] (7) отбор образца крови для изучения фармакокинетики (если исследуемое лекарственное средство было введено до визита, субъект должен вернуться в центр на следующий день)
[00327] (8) отбор образца крови для определения общего профиля жирных кислот
[00328] (9) оценка ППТ
[00329] (10) ООИ
[00330] (11) оценка ПЭИТ
[00331] (12) оценка ШАД (включая оценку зуда ВАШ)
[00332] (13) опросник ПОЭМ
[00333] (14) опросник ДИКЖ
[00334] (15) оценка ТЭПВ (только выбранные центры)
[00335] (16) сбор исследуемого лекарственного средства
[00336] (17) обзор и сбор журналов соблюдения предписаний субъектом
[00337] (18) подсчет капсул
[00338] (19) сопутствующая терапия
[00339] (20) оценка побочных реакций
Наблюдение/Неделя 10, Визит 6 (70 день ± 3 дня).
[00340] Следующие процедуры будут проводиться в ходе данного визита:
[00341] (1) текущий обзор медицинского анамнеза
[00342] (2) основные показатели жизнедеятельности
[00343] (3) медицинский осмотр
[00344] (4) лабораторные показатели безопасности (химические анализы, коагуляция, анализ крови и анализ мочи): только в случае, если клинически значимое изменение лабораторных показателей безопасности по сравнению с исходным уровнем зарегистрировано на 8 неделе
[00345] (5) тест мочи на беременность (только женщины, способные к деторождению)
[00346] (6) оценка ППТ
[00347] (7) ООИ
[00348] (8) оценка ПЭИТ
[00349] (9) оценка ШАД (включая оценку зуда ВАШ)
[00350] (10) опросник ПОЭМ
[00351] (11) опросник ДИКЖ
[00352] (12) оценка ТЭПВ (только выбранные центры)
[00353] (13) сопутствующая терапия
[00354] (14) оценка побочных реакций
[00355] (15) визит досрочного прекращения участия.
[00356] В случае, если субъект прекращает участие в исследовании до его завершения, следует провести процедуры по списку для визита на 8 неделе.
ОЦЕНКА В ХОДЕ ИССЛЕДОВАНИЯ
Оценка эффективности
[00357] Клиническая оценка атопического дерматита будет выполняться опытным и квалифицированным дерматологом (сертифицированным советом или эквивалентным). Для обеспечения систематичности и уменьшения вариабельности, при любой возможности один и тот же эксперт должен выполнять все оценки для конкретного субъекта.
Общая оценка исследователем
[00358] Общая оценка исследователем (ООИ) тяжести заболевания (Приложение B) будет проводиться в ходе каждого визита. ООИ представляет собой глобальную оценку текущего состояния заболевания. Это морфологическая оценка общей тяжести заболевания из 6 пунктов, которая будет осуществляться в соответствии со следующими определениями: 0 (чистая), 1 (практически чистая), 2 (мягкий), 3 (умеренный), 4 (тяжелый) и 5 (очень тяжелый). Для включения в исследование субъекты должны получить ООИ ≥ 3 баллов в ходе визита исходного уровня (0 день).
Площадь экземы и индекс тяжести (ПЭИТ)
[00359] Площадь экземы и индекс тяжести (ПЭИТ) будет оцениваться в ходе каждого визита, кроме скринингового визита. Этот показатель количественно определяет тяжесть атопического дерматита у субъекта, на основании как тяжести поражения, так и процента пораженной ППТ. ПЭИТ представляет собой комплексный балл, варьирующий в диапазоне 0-72, который учитывает степень эритемы, уплотнения/образования папул, экскориации и лихенификации (каждому в отдельности присваиваются баллы от 0 до 3) для каждого из четырех участков тела, с корректировкой на процент пораженной ППТ для каждого участка тела и доли участка тела по отношению ко всему телу. Методика вычисления балла ПЭИТ подробно описана в Приложении C.
Площадь поверхности тела (ППТ)
[00360] Общая ППТ, пораженная АД, будет оцениваться (от 0 до 100%) в ходе каждого визита. Одна ладонь пациента представляет 1% от его/ее общей ППТ. В ходе всех визитов исследования, кроме скринингового, ППТ для затронутой кожи будет измеряться с помощью измерения ШАД (см. описание ниже) и оцениваться, как отдельная конечная точка. Для того, чтобы быть пригодными к участию в исследовании, показатель ППТ у субъекта должен составлять по меньшей мере 10% в ходе визита исходного уровня (0 день).
[00361] Шкала атопического дерматита (ШАД). ШАД будет измеряться в ходе каждого визита, кроме скринингового визита. Система ранжирования ШАД была разработана Европейской рабочей группой по атопическому дерматиту (1993) и является стандартным инструментом для оценки тяжести АД в клинических исследованиях в Европе. Шесть показателей (эритема, отек/образование папул, намокание/образование корой, экскориация, лихенификация и сухость) будут выбраны для оценки тяжести АД. Общая ППТ, пораженная АД, будет оценена (от 0 до 100%) и включена в балл ШАД. Нарушение сна и зуд будет оценено пациентами по визуальной аналоговой шкале (0-10). Сумма этих показателей составляет ШАД, которая может варьировать от 0 до 103. Методика вычисления балла ШАД подробно описана в Приложении D.
Визуальная аналоговая шкала зуда
[00362] В ходе всех визитов исследования, кроме скринингового, балл тяжести зуда будет зарегистрирован при измерении ШАД и будет оцениваться, как отдельная конечная точка. Для его оценки субъектов попросят указать на 10 см шкале оценки (0-10) точку, соответствующую среднему значению на протяжении последних трех дней/ночей.
Пациент-ориентированная экземы мера (ПОЭМ)
[00363] Пациент-ориентированную экземы меру (ПОЭМ) будут оценивать в ходе каждого визита, кроме скринингового визита. ПОЭМ, разработанная Charman и др., представляет собой самостоятельную оценку тяжести заболевания пациентом. ПОЭМ имеет максимальное значение двадцать восемь на основании ответа пациента на семь вопросов, который оценивается в баллах согласно следующей шкале:
[00364] Ни одного дня=0
[00365] 1-2 дня=1
[00366] 3-4 дня=2
[00367] 5-6 дней=3
[00368] Ежедневно=4
[00369] Подробное описание оценки ПОЭМ приведено в Приложении E.
Опросник Дерматологиеского индекса качества жизни (ДИКЖ)
[00370] ДИКЖ представляет собой простой валидированный опросник из 10 вопросов, который будет заполняться при каждом визите, кроме скринингового. Опросник приведен в Приложении F.
Трансэпидермальная потеря воды (ТЭПВ) (только выбранные центры)
[00371] Клиническая тяжесть АД и сопутствующее влияние на барьерную функцию кожи будет оцениваться в ходе каждого визита, кроме скринингового визита. Эта оценка будет проводиться в выбранных центрах, которые продемонстрировали предыдущий опыт работы с данным устройством.
[00372] В начале исследования (0 день) исследователь выберет три характерных участка активного АД для каждого субъекта; расположение этих участков будет зарегистрировано. В ходе последующих визитов показатели ТЭПВ для каждого участка с АД будут измеряться в стандартных комнатных условиях (22-25°C, 40-60% относительной влажности); для анализов будет использоваться среднее значение ТЭПВ.
Оценки безопасности
Основные показатели жизнедеятельности
[00373] Следующие основные показатели жизнедеятельности будут регистрироваться в ходе каждого визита в положении сидя, после того, как сидящий спокойно просидит по меньшей мере 5 минут: систолическое и диастолическое давление крови (мм рт. ст), пульс (уд/мин), температура тела (°C) и частота дыхательных движений (дыхательные движения/мин).
[00374] Масса тела (кг) и рост (см) будут измерены для вычисления ИМТ, и будут зарегистрированы в ходе визитов: скринингового, исходного уровня и на 8 неделе. Рост будет регистрироваться только один раз в ходе скринингового визита, и это значение будет использоваться для вычисления ИМТ в ходе визитов исходного уровня и на 8 неделе.
[00375] Любой аномальный показатель, связанный с основными показателями жизнедеятельности, который исследователь считает клинически значимым, должен быть зарегистрирован как ПР.
Медицинский осмотр
[00376] Следующие участки/системы будут включены в медицинский осмотр. Каждая система будет оценена как норма/аномалия (клинически незначимая или клинически значимая). Соответствующие детали должны быть зарегистрированы для любых клинически значимых данных.
[00377] (1) внешний вид
[00378] (2) дерматологический осмотр (кроме атопического дерматита)
[00379] (3) голова, глаза, уши, нос, горло (ГГУНГ)
[00380] (4) дыхательные пути
[00381] (5) сердечно-сосудистая система
[00382] (6) органы брюшной полости
[00383] (7) неврологический осмотр
[00384] (8) опорно-двигательная система
[00385] (9) лимфатическая система
Клинические лабораторные анализы
[00386] Лабораторные анализы будут выполняться в ходе скрининга, в 0 день, на 4 неделе и 8 неделе. Если результаты на 8 неделе будут показывать клинически значимое отклонение от исходного уровня, лабораторные анализы дополнительно будут выполняться на 10 неделе. Анализы будут включать анализ мочи, анализ крови с дифференциальным подсчетом и тестом коагуляции, стандартную химическую панель (химические анализы, включая печеночные пробы и холестерин), коагуляцию, сывороточный тест на беременность (скрининг и 8 неделя/Визиты досрочного прекращения участия) для женщин, способных к деторождению (ЖСДР). В ходе визитов исходного уровня (0 день), на 2 неделе, 4 неделе и 10 неделе, тест мочи на беременность будет выполняться для женщин, способных к деторождению (проводимый в исследовательском центре). В ходе скринингового визита, уровни ФСГ будут определяться у женщин в возрасте от 40 и до 60 лет, у которых менструации отсутствуют по меньшей мере в течение 12 месяцев, но менее чем 24 месяца. Конкретные анализы в этих панелях приведены в Табл. 8 ниже.
Таблица 8. Клинические лабораторные анализы
Уровни общей и свободной ДГЛК в плазме
[00387] В ходе визитов исходного уровня, на 4 неделе и на 8 неделе будут отбираться образцы крови до введения исследуемого лекарственного средства (исследуемое лекарственное средство не вводится в ходе визита на 8 неделе). Если субъект прибывает в центр после принятия ежедневной дозы исследуемого лекарственного средства, от данного субъекта потребуется вернуться в центр на следующий день для отбора образца крови для изучения ФК. Будут определяться минимальные уровни общей и свободной ДГЛК в плазме. Второй образец крови будет отбираться для более поздней оценки общего профиля жирных кислот в плазме. Образец крови для химического анализа сыворотки будет разделен на две аликвоты для химического анализа и более поздней оценки интерлейкинов.
[00388] Дата и время приема субъектом последней дозы дома перед визитом будет точно регистрироваться. В исследовательском центре субъектов проинструктируют не принимать ежедневную дозу исследуемого лекарственного средства дома перед визитом на 4 неделе. Доза будет вводиться в центре в ходе визита исследования. Должно быть зарегистрировано точное время отбора образцов.
[00389] Образцы крови будут обрабатываться как можно быстрее, не позже 1 часа после отбора образца крови. Полученная плазма будет перенесена в полипропиленовые пробирки. Каждая пробирка будет маркирована для идентификации анализируемого вещества, которое будут определять. Все образцы будут заморожены в вертикальном положении. Подробная инструкция относительно сбора, обработки, хранения и отгрузки образцов для изучения ФК, общих жирных кислот и интерлейкинов будет приведена в лабораторном руководстве для центра. Маркировка на каждой пробирке будет включать по меньшей мере следующую информацию:
[00390] (1) протокол исследования
[00391] (2) номер центра
[00392] (3) идентификационный номер субъекта
[00393] (4) название визита
[00394] (5) название анализируемого вещества
[00395] (6) основной (A) или дубликат (B)
[00396] Отгрузка опытных образцов будет осуществляться Центральными лабораториями ICON.
[00397] Подробные инструкции по отгрузке будут приведены в лабораторном руководстве для центра. Далее образцы будут отгружены аналитическому учреждению и будут проанализированы с применением валидированного аналитического метода в соответствии со стандартными операционными процедурами учреждения.
[00398] Образцы крови для изучения ФК будут храниться маскированными.
ПОБОЧНЫЕ ЭФФЕКТЫ
[00399] Побочной реакцией является любое неблагоприятное медицинское событие у пациента, которому вводят фармацевтический продукт, независимо от возможности причинно-следственной связи с данным лечением.
[00400] Исследователи несут ответственность за мониторинг безопасности субъектов, которые принимают участие в данном исследовании, а также за оповещение Спонсора о любом событии, которое выглядит необычным, даже если такое событие может рассматриваться как непредвиденная польза для субъекта. Исследователь несет ответственность за надлежащий медицинский уход за субъектами в ходе исследования.
[00401] Исследователь остается ответственным за наблюдение посредством подходящей возможности здравоохранения за побочными реакциями, которые являются серьезными или которые вынудили субъекта досрочно прекратить участие в исследовании. За состоянием субъекта необходимо наблюдать до тех пор, пока побочная реакция исчезнет или стабилизируется. Частота последующего наблюдения определяется решением исследователя.
[00402] Безопасность будет оцениваться посредством сбора информации о побочных реакциях, основных показателях жизнедеятельности, проведения медицинских осмотров и оценки лабораторных результатов. Зарегистрированные побочные реакции будут кодироваться согласно терминологии Медицинского словаря нормативно-правовой деятельности (МСНПД).
[00403] До включения в исследование, персонал исследовательского центра будет отмечать случаи и природу медицинского(их) состояния(й) каждого субъекта в соответствующем разделе первичного документа и ИРК. В ходе исследования, персонал центра будет и далее отмечать любое изменение в состоянии(ях) и случаи и природу любых побочных реакций.
[00404] Если у субъекта возникает побочная реакция после приема первой дозы исследуемого лекарственного средства, событие будет зарегистрировано как побочная реакция в первичном документе и ИРК. Все ПР будут описаны в первичных документах и ИРК.
Причинно-следственная связь для побочных реакций
[00405] Исследователь будет устанавливать степень причинно-следственной связи ПР с экспериментальным лечением. Исследователь должен принимать во внимание анамнез субъекта, последние результаты медицинского осмотра и сопутствующую терапию.
[00406] Следующие определения будут применяться для определения причинно-следственной связи для ПР:
[00407] не связанный: временное соотношение возникновения ПР относительно экспериментального лечения неправдоподобно, или другая причина может объяснять случай ПР;
связанный: временное соотношение возникновения ПР относительно экспериментального лечения правдоподобно, наблюдается известный паттерн ответа на лечение, и альтернативная причина является невероятной.
Тяжесть побочных реакций
[00408] Интенсивность ПР представляет собой оценку относительной тяжести события, сделанную исследователем на основании его или ее клинического опыта и знакомства с литературой. Следующие определения следует применять для оценки тяжести ПР:
[00409] мягкий: симптом просто привлекает внимание субъекта, но не влияет на ежедневную деятельность. Лечение обычно не показано.
[00410] умеренный: симптом достаточно выраженный, чтобы причинять субъекту неудобства и влиять на ежедневную деятельность. Может понадобиться лечение.
[00411] тяжелый: симптом вызывает серьезный дискомфорт, причем ежедневная деятельность существенно нарушается или не может быть осуществлена. Может понадобиться лечение.
Серьезные побочные реакции
[00412] Если у субъекта возникает серьезная побочная реакция после приема первой дозы исследуемого лекарственного средства, событие будет зарегистрировано как серьезная побочная реакция. Все ПР будут описаны в первичных документах и ИРК.
[00413] Серьезная побочная реакция (опыт) или эффект представляет собой любое неблагоприятное медицинское событие, которое в любой дозе:
• приводит к смерти,
• угрожает жизни,
• требует госпитализации или продления госпитализации субъекта,
• приводит к постоянной или значительной инвалидности/нетрудоспособности, или
• представляет собой врожденную аномалию/порок развития.
[00414] Термин «угрожающий жизни» в определении «серьезного» обозначает событие, при котором субъект подвергается риску смерти во время события; он не обозначает события, которое гипотетически могло бы вызвать смерть, если бы оно было более тяжелым.
[00415] Медицинское и научное заключение должно быть принято для решения о том, будет ли подходящей экспресс-отчетность в других ситуациях, таких как значимые медицинские события, которые могут не быть непосредственно угрожающими жизни или не приводить к смерти или госпитализации, но могут подвергать субъекта опасности или могут потребовать вмешательства для предупреждения одного из других исходов, перечисленных в определении выше. Обычно их также считают серьезными.
[00416] Примерами таких событий является интенсивная терапия в отделении неотложной помощи или дома аллергического бронхоспазма; дискразий крови или судорог, которые не приводят к госпитализации; или развитие зависимости от лекарственного средства или злоупотребление лекарственным средством.
Сообщение о беременности
[00417] Если у субъекта возникает беременность в ходе исследования, субъект должен как можно быстрее сообщить в исследовательский центр. В случае подтверждения беременности субъект должен прекратить прием исследуемого лекарственного средства, но может продолжать участие в исследовании. При подтверждении беременности исследователь должен заполнить связанную с исследованием Форму беременности и отправить ее в Innovaderm Research в пределах 24 часов после подтверждения беременности. Innovaderm Research будет сообщать обо всех случаях беременности Спонсору в установленные сроки (контакты для связи такие же, как для сообщений о СПР). После завершения лечения следует осуществлять наблюдение, чтобы гарантировать безопасность субъекта. Беременность сама по себе не является ПР или СПР; однако, осложнения или аномалии у матери/эмбриона будут зарегистрированы как ПР или СПР, в соответствующих обстоятельствах. Исследователь будет наблюдать за беременностью до завершения (или прерывания беременности) и сообщит исход Innovaderm Research в качестве продолжения к исходной Форме беременности.
ОБЕСПЕЧЕНИЕ КАЧЕСТВА ДАННЫХ/МОНИТОРИНГ ЦЕНТРОВ
[00418] В ходе исследования визиты мониторинга будут проводиться с регулярными промежутками. Визиты мониторинга будут проводиться с целью обеспечения соблюдения протокола, качества данных, точности информации в эИРК, учета лекарственного средства, соответствия регулярным требованиям и постоянного соответствия исследовательского центра и его оборудования требованиям.
[00419] Аудит и/или мониторинг центров может проводиться сотрудником отдела обеспечения качества, который выбран Спонсором. и/или регуляторными органами, которые могут изъявить желание к проведению аудита в исследовательском центре. Исследователь будет извещен до начала проверки, и от него ожидается сотрудничество с любой проверкой, обеспечение помощи и документации (включая первичные данные), по запросу.
СБОР И ХРАНЕНИЕ ДАННЫХ
[00420] Данные субъекта будут вводиться персоналом сайта с применением Medrio eClinical Overnight, веб-ориентированной системы сбора и регистрации данных в электронной форме (СРЭ). Данное приложение будет установлено для удаленного входа. Medrio Inc. является разработчиком и владельцем Medrio eClinical Overnight. Программное обеспечение СРЭ полностью валидировано и соответствует требованиям 21 Свода федеральных законоположений (СФЗ), Часть 11. Персонал исследовательского центра не получит доступа к системе СРЭ, пока он не будет полностью обучен Спонсором или уполномоченным лицом. Уполномоченный персонал исследователя будет вводить данные, требуемые протоколом, в эИРК с применением данного веб-ориентированного приложения. Автоматические программы валидации проверяют расхождение данных в эИРК и, генерируя соответствующие сообщения об ошибке, позволяют внести изменения или осуществить проверку введенных данных персоналом исследователя до подтверждения данных. Исследователь должен удостоверить полноту и точность данных с помощью электронной подписи к эИРК.
[00421] Исследователь должен вести первичные документы для каждого субъекта, включенного в исследование, состоящие из индивидуальной карточки и отметок о визитах (клинические медицинские записи), которые содержат демографическую и медицинскую информацию и результаты любых анализов или оценок. Вся информация в эИРК должна быть доступной для прослеживания этих первичных документов в файле субъекта. Данные, не требующие письменной или электронной регистрации, будут определены до начала исследования и будут занесены непосредственно в эИРК, которая будет документироваться как первичные данные.
[00422] Собранные данные будут кодироваться и храниться электронным способом в системе баз данных. Валидированные данные впоследствии могут быть переданы спонсору.
КОНФИДЕНЦИАЛЬНОСТЬ ДОКУМЕНТАЦИИ ИССЛЕДОВАНИЯ И ЗАПИСЕЙ СУБЪЕКТОВ
[00423] Исследователь должен обеспечить сохранение анонимности субъектов, а также защиту их идентификационной информации от несанкционированных сторон. В ИРК или другом документе, предоставляемом Спонсору, субъекты не должны быть идентифицированы по их именам, а только по идентификационному коду. Исследователь должен вести журнал включения субъектов в исследование, в котором коды будут связаны с именами субъектов. Исследователь должен хранить документы, которые не предоставляются спонсору, например, формы письменного согласия субъектов, в строжайшей конфиденциальности.
ФАЙЛЫ ИССЛЕДОВАТЕЛЯ/ХРАНЕНИЕ ДОКУМЕНТОВ
[00424] Исследователь должен вести надлежащие и точные записи, чтобы обеспечить полное документирование исследования и данные исследования, которые впоследствии будут верифицированы. Такие записи включают, но не ограничиваясь этим, идентификационную информацию всех субъектов, принимающих участие, все оригиналы подписанных документов информированного согласия, копии всех ИРК, формы отчетов о безопасности, первичную документацию и подробные записи об утилизации лекарственного средства, а также надлежащим образом документированную переписку по теме исследования. Указанные документы следует классифицировать в две отдельные категории: Файл исследования Исследователя и Первичная клиническая документация субъектов.
[00425] Исследователь должен хранить записи в соответствии с локальными законоположениями Международной конференции по гармонизации (МКГ) или как определено в Соглашении о проведении клинических исследований (СКИ), в зависимости от того, где указан более поздний срок.
ОБЪЕМ ВЫБОРКИ И СТАТИСТИЧЕСКИЕ МЕТОДЫ
Определение объема выборки
[00426] Первичная конечная точка может быть интерпретирована как анализ респондеров (пациентов, ответивших на лечение), в котором субъект будет классифицирован как Респондер, если он/она достигает балла ООИ 0 (чистая) или 1 (практически чистая) на 8 неделе, учитывая уменьшение на 2 балла от исходного значения. Объему выборки 45 субъектов будет соответствовать мощность 80% для обнаружения статистически значимой разницы 25% между респондерами из группы лекарственного средства и из группы плацебо, на основании теста хи-квадрат и альфа 0,05. На основании обзора литературы ожидается, что эффективность плацебо может составлять до 7%, таким образом, минимальная доля, ожидаемая в группе лекарственного средства, должна составить по меньшей мере 32%. Допуская выбывание 10%, всего необходимо включить в исследование 100 субъектов.
Статистические и аналитические планы
[00427] Непрерывные переменные будут суммироваться в таблицах и будут включать количество субъектов, среднее значение, стандартное отклонение, медиану, минимум, максимум и межквартильный диапазон. Категориальные переменные будут представлены в таблицах как значения частоты и проценты.
[00428] Все статистические анализы будут двухсторонними и будут выполняться с уровнем значимости 0,05, если не определено иное.
Распределение субъектов
[00429] Эффективность будет оцениваться на основании совокупности всех рандомизированных субъектов согласно назначенному лечению, которым была введена хотя бы 1 доза исследуемого лекарственного средства (популяция РНЛ), и анализы будут проводиться на основании назначенного лечении, а не на основании полученного лечения.
[00430] Популяция анализа по протоколу (ПП) будет включать всех субъектов, которые были рандомизированы без существенных отклонений от протокола. Специфические критерии для популяции ПП и популяции РНЛ будут подробно описаны в отдельном плане статистического анализа.
[00431] Популяция безопасности будет определена как совокупность всех субъектов, получивших по меньшей мере одну дозу лекарственного средства. Анализ будет проведен в соответствии с фактическим лечением, которое они получали.
Анализ эффективности
[00432] Первичная конечная точка может быть интерпретирована как анализ респондеров, где субъект будет классифицирован как Респондер, если он/она достигает отметки ООИ 0 (чистая) или 1 (практически чистая) на 8 неделе, с учетом уменьшения на 2 балла от исходного значения. Сравнение между группами для первичной конечной точки будет проводиться с применением теста хи-квадрат. Анализ чувствительности для проверки вклада центра в сравнения будет проводиться с применением теста Кохрана-Мантеля-Гензеля, где центр будет использоваться в качестве фактора стратификации. Первичный анализ эффективности будет проводиться с применением наблюдаемых значений, и поддерживающий анализ будет проводиться с применением анализа точки перегиба, стратегии, которая применяется для оценки влияния пропущенных значений. Пропущенные значения будут установлены для респондера/нереспондера последовательно, и решения, основанные на значениях p, будут нанесены на график. Подробное описание данной стратегии будет представлено в плане статистического анализа. Не будет осуществляться подстановки данных для отсутствующих данных в других визитах. Анализы будут проводиться с применением популяции РНЛ и будут служить первичным анализом, в то время как анализ первичной конечной точки с применением популяции ПП, будет использоваться в качестве анализа чувствительности.
[00433] Вторичные конечные точки, включающие изменения от исходного уровня, будут проанализированы с применением анализа ковариации (АНКОВА), включая изменение от исходного уровня в качестве зависимого фактора, центр и группу лечения или центр в качестве фиксированных факторов и исходное значение в качестве ковариаты. Будут представлены пределы среднего значения и 95% ДИ, наряду с соответствующим значением p, полученным в результате сравнения схем лечения. Вторичные конечные точки, включающие долю, будут проанализированы с применением теста Кохрана-Мантеля-Гензеля, со стратификацией по центру, и будет представлено значение p. Анализы для вторичных конечных точек будут проведены с применением наблюдаемых данных, причем не будет проводиться подстановка данных в случае пропущенных наблюдений.
[00434] Все подробности статистических анализов будут включены в отдельный план статистического анализа.
Анализ безопасности
[00435] Все побочные реакции (ПР), которые возникнут после введения первой дозы исследуемого лекарственного средства в ходе исследования, будут классифицированы на основании Медицинского словаря нормативно-правовой деятельности (МСНПД). Описание ПР будет включать дату появления, дату исчезновения ПР (если он исчезнет), тяжесть и степень серьезности ПР, связь ПР с исследуемым лекарственным средством и исход. Фокус в данном протоколе будет ограничен побочными реакциями, возникающими в ходе лечения.
[00436] Зарегистрированные ПР будут суммироваться по количеству субъектов, сообщающих об эпизодах, а также по Классу органов и систем, предпочтительному термину, тяжести, серьезности и связи с исследуемым лекарственным средством. Для резюме ПР по тяжести, каждый пациент будет учитываться только один раз в пределах Класса органов и систем или предпочтительного термина, с использованием ПР с наибольшей интенсивностью в пределах каждой категории для каждого анализа. Для резюме связи ПР с исследуемым лекарственным средством, каждый пациент будет учитываться только один раз в пределах Класса органов и систем или предпочтительного термина, с использованием ПР с наиболее выраженной зарегистрированной связью в пределах каждой категории. Для резюме ПР и тяжести по связи с исследуемым лекарственным средством, каждый пациент будет учитываться только один раз в пределах Класса органов и систем или предпочтительного термина, с использованием (1) наиболее выраженной зарегистрированной связи, и затем (2) наибольшей зарегистрированной интенсивности.
[00437] Вся информация, имеющая отношение к ПР, отмеченным в ходе исследования, будет приведена по пациенту с указанием подробной дословной информации, класса органов и систем, предпочтительного термина, даты появления, даты исчезновения, интенсивности, исхода и связи с исследуемым лекарственным средством. Кроме того, возникновение ПР будет показано относительно (в количестве дней) дня введения исследуемого продукта. Серьезные побочные реакции (СПР) будут приведены в таблице по группам лечения, связи с исследуемым продуктом, со ссылкой на встречаемость СПР до относительного дня введения.
[00438] Сопутствующая терапия будет кодироваться с помощью Глоссария ВОЗ по лекарственным средствам и будет приведена по субъектам.
[00439] Кроме того, будет приведен список субъектов, которые прекратили участие в исследовании, и список субъектов, у которых возникали СПР.
[00440] Результаты лабораторных анализов и основные показатели жизнедеятельности будут сведены в таблицы с применением описательной статистики. Значение на момент визита, а также отклонение от исходного уровня будет представлено описательно.
[00441] Статистика вывода не будет применяться для переменных безопасности (ВЛПР, сопутствующая терапия, лабораторные показатели и основные показатели жизнедеятельности).
ЭТИКА
Локальные законоположения/Хельсинская декларация
[00442] Данное исследование будет проводиться в соответствии с этическими принципами, описанными в Хельсинской декларации (2008), которые также согласуются с трехсторонней Директивой МКГ «Надлежащая клиническая практика» (июль 2002) и соответствующими законами и законоположениями страны, в которой проводится исследование, в зависимости от того, какой документ предоставляет большую степень защиты индивидууму.
Обзор этических норм
[00443] Спонсор понимает, что данный протокол (и любая поправка), а также соответствующие процедуры предоставления согласия будут рассмотрены и одобрены Этическим советом по исследованиям/Экспертным советом организации (ЭСИ/ЭСО). Данный Совет должен действовать в соответствии с действующими Федеральными законоположениями. Письмо или аттестационный сертификат будет отправлено Исследователем Спонсору до начала исследования, а также каждый раз при последующем внесении изменений в протокол.
Информированное согласие
[00444] Обязанностью Исследователя или лица, уполномоченного Исследователем (если это приемлемо согласно локальным законоположениям) является получение письменного информированного согласия от каждого субъекта данного исследования в отдельности, после соответствующего объяснения целей, методов, задач и потенциального риска исследования. Кроме того, субъектам следует объяснить, что они полностью свободны отказаться от участия в исследовании или отозвать свое согласие в любое время по любой причине.
[00445] Если новая информация относительно безопасности приведет к существенным изменениям в оценке риска/пользы, или будет получена любая новая информации, которая может повлиять на готовность продолжать участие, форма согласия должна, при необходимости, быть пересмотрена и обновлена Этическим советом по исследованиям/Экспертным советом организации. До сведения всех субъектов (в том числе, уже получающих лечение) должна быть доведена новая информация, предоставлена копия исправленной формы согласия, и всех субъектов следует попросить дать согласие на продолжение участия в исследовании.
ПРИЛОЖЕНИЕ A Расписание исследования
¥: Для женщин в возрасте от 40 до 60 лет, у которых менструации отсутствуют по меньшей мере в течение 12 месяцев, но менее чем 24 месяца
**: Только если на 8 неделе возникает клинически значимое отклонение лабораторных показателей безопасности от исходного уровня
*: Сывороточный тест на беременность в ходе скрининга и на 8 неделе/Визиты ДПУ, тест мочи на беременность для всех других визитов
††: Если субъект принял исследуемое лекарственное средство до визита, от него/нее потребуют вернуться на следующий день для отбора образца крови для изучения ФК.
€: Субъекты должны воздерживаться от приема пищи по меньшей мере в течение 8 часов перед и 60 минут после введения лекарственного средства
: Субъекты будут проинструктированы о приеме последней дозы исследуемого лекарственного средства за дань до визита на 8 неделе.
†: Сбор информации о ПР начнется после первого введения исследуемого лекарственного средства
Ɣ: Оценка, если какое-либо из существующих состояний улучшилось по сравнению с исходным уровнем.
Φ: Профиль интерлейкинов не будет оцениваться в ходе скрининга и на 10 неделе.
ПРИЛОЖЕНИЕ B - Общая оценка Исследователем
ПРИЛОЖЕНИЕ C - Площадь экземы и индекс тяжести (ПЭИТ)
[00446] Четыре анатомических отдела - голова, верхние конечности, туловище и нижние конечности - оцениваются на предмет эритемы, отвердения (папулы), экскориаций и лихенификации, наблюдаемых в день обследования. Тяжесть каждого признака оценивают с применением 4-бальной шкалы:
[00447] 0=Симптомы отсутствуют
[00448] 1=Легкая
[00449] 2=Умеренная
[00450] 3=Значительная
[00451] Площадь поверхности, пораженной атопическим дерматитом в пределах конкретного анатомического отдела, оценивают как процент от общей площади данного анатомического отдела и присваивают цифровое значение согласно степени поражения атопическим дерматитом, как указано ниже:
[00452] 0=Поражение отсутствует
[00453] 1=< 10%
[00454] 2=от 10 до < 30%
[00455] 3=от 30 до < 50%
[00456] 4=от 50 до < 70%
[00457] 5=от 70 до < 90%
[00458] 6=90-100%
[00459] Баллы ПЭИТ получают с применением формулы:
[00460] ПЭИТ=0,1 (Eh+Ih+Exh+Lh) Ah+0,2 (Eu+Iu+Exu+Exu) Au+0,3 (Et+It+Ext+Ext) At+0,4 (El+Il+Exl+Exl) Al
где E, I, Ex, L и A обозначают эритему, затвердение, экскориации, лихенификацию и площадь, соответственно, а h, u, t и l обозначают голову, верхние конечности, туловище и нижние конечности, соответственно.
ПРИЛОЖЕНИЕ D - Шкала атопического дерматита (ШАД)
[00461] Шесть показателей (эритема, отек/образование папул, намокание/образование корок, экскориации, лихенификация и сухость) выбраны для оценки тяжести АД. Интенсивность каждого показателя ранжируют с применением 4-бальной шкалы:
[00462] 0=Симптомы отсутствуют
[00463] 1=Мягкий
[00464] 2=Умеренный
[00465] 3=Тяжелый
[00466] Участок, выбранный для ранжирования, должен быть характерным (средняя интенсивность) для каждого показателя. Индивидуальные баллы интенсивности для каждого показателя далее будут суммированы (диапазон 0-18) и умножены на 3,5, что дает максимум 63 балла.
[00467] Общую ППТ, пораженную АД (от 0 до 100%) оценивают и делят на 5. Одна ладонь пациента представляет 1% от его/ее общей ППТ. Максимум составляет 20.
[00468] Субъективные показатели включают нарушение сна и эпизоды зуда. Для их оценки пациентов просят указать на 10 см шкале (0-10) оценки точку, соответствующую среднему значению на протяжении последних трех дней/ночей. Объединенный максимальный балл для этих двух показателей составляет 20.
[00469] Сумма упомянутых выше данных представляет значение ШАД, которое может варьировать от 0 до 103. Если субъективные оценки зуда и нарушения сна исключают, ШАД становится объективным ШАД (диапазон баллов 0-83).
ПРИЛОЖЕНИЕ E - Пациент-ориентированная экземы мера (ПОЭМ)
Идентификационный № пациента ___-___ Инициалы пациента: ______
День визита: ______ Дата Визита (дд-мм-гг): __________
Пожалуйста, обведите кружком один ответ для каждого из семи вопросов ниже относительно вашей экземы. Пожалуйста, оставьте пустыми любые вопросы, на которые, как вы чувствуете, вы не в состоянии ответить.
Пожалуйста, обведите кружком один ответ для каждого из семи вопросов ниже относительно вашей экземы. Пожалуйста, оставьте пустыми любые вопросы, на которые, как вы чувствуете, вы не в состоянии ответить.
1. Как часто за прошлую неделю вы ощущали кожный зуд из-за экземы?
Ни одного дня 1-2 дня 3-4 дня 5-6 дней Ежедневно
2. Как часто за прошлую неделю ваш сон был нарушен из-за экземы?
Ни одного дня 1-2 дня 3-4 дня 5-6 дней Ежедневно
3. Как часто за прошлую неделю у вас возникали кожные кровотечения из-за экземы?
Ни одного дня 1-2 дня 3-4 дня 5-6 дней Ежедневно
4. Как часто за прошлую неделю ваша кожа намокала, или выделялась прозрачная жидкость из-за экземы?
Ни одного дня 1-2 дня 3-4 дня 5-6 дней Ежедневно
5. Как часто за прошлую неделю ваша кожа трескалась из-за экземы?
Ни одного дня 1-2 дня 3-4 дня 5-6 дней Ежедневно
6. Как часто за прошлую неделю ваша кожа шелушилась из-за экземы?
Ни одного дня 1-2 дня 3-4 дня 5-6 дней Ежедневно
7. Как часто за прошлую неделю вы ощущали сухость или огрубелость кожи из-за экземы?
Ни одного дня 1-2 дня 3-4 дня 5-6 дней Ежедневно
[00470] © CR Charman, AJ Venn, HC Williams, декабрь 2004 года.
ПРИЛОЖЕНИЕ F - Дерматологический индекс качества жизни (ДИКЖ)
[00471] Цель данного опросника - оценить насколько ваша проблемная кожа влияла на вашу жизнь НА ПРОТЯЖЕНИИ ПОСЛЕДНЕЙ НЕДЕЛИ. Пожалуйста, отметьте один вариант ответа для каждого вопроса.

[00472] Цель данного опросника - оценить насколько ваша проблемная кожа влияла на вашу жизнь НА ПРОТЯЖЕНИИ ПОСЛЕДНЕЙ НЕДЕЛИ. Пожалуйста, отметьте один вариант ответа для каждого вопроса.
ПРИЛОЖЕНИЕ G - Диагностические критерии атопического дерматита
[00473] Согласно Критерию включения 2, субъекту должен быть поставлен клинический диагноз атопического дерматита в соответствии с критериями Hanifin и Rajka. Критериями являются следующие:
Основные критерии (должны присутствовать по меньшей мере три)
[00474] Зуд
[00475] Типичная морфология и распространение:
[00476] Взрослые: флексионная лихенификация или линейность
[00477] Дети и младенцы: поражение лица и разгибательных поверхностей
[00478] Хронический или рецидивирующий дерматит
[00479] Личный или семейный анамнез атопии
Минорные критерии (должно присутствовать по меньшей мере три)
[00480] Ксероз
[00481] Ихтиоз/фолликулярный кератоз/ладонная гиперлинейность
[00482] Реакция в тесте на немедленную кожную гиперчувствительность (тип 1)
[00483] Повышенный уровень IgE в сыворотке
[00484] Ранний возраст возникновения
[00485] Тенденция к кожным инфекциям (золотистый стафилококк, простой герпес)/нарушенный клеточный
[00486] Иммунитет
[00487] Дерматит кистей/стоп
[00488] Экзема сосков
[00489] Конъюнктивит
[00490] Складка Дени-Моргана
[00491] Кератоконус
[00492] Передняя субкапсулярная катаракта
[00493] Орбитальное помутнение
[00494] Бледность/покраснение лица
[00495] Себорейная экзема
[00496] Складки на передней поверхности шеи
[00497] Зуд при потоотделении
[00498] Непереносимость шерсти и растворителей липидов
[00499] Околофолликулярная акцентуация
[00500] Непереносимость пищевых продуктов
[00501] На протекания влияют факторы окружающей среды/эмоциональные факторы
[00502] Принадлежность к белой расе/реакция бланш-задержки
Пример 3
[00503] Проводилось исследование антигипертензивной эффективности in vivo после сопутствующего введения ДГЛК и аспирина у спонтанно гипертонических крыс. В данном эксперименте изучали эффективность ДГЛК при хроническом сопутствующем введении с аспирином с целью уменьшения выраженности гипертонической реакции, вызванной острым действием фенилэфрина у спонтанно гипертонических крыс. Для данного испытания использовали спонтанно гипертонических, взрослых крыс-самцов (250-300 г), разведенных Charles River Laboratories. Животных маркировали по прибытии в соответствии с директивами Канадской ассоциации производителей химреагентов (КАПХ). Животных поселяли группами по два (низкая доза+Аспирин, высокая доза+Аспирин или только Аспирин) на период перорального введения через зонд и до хирургического вмешательства. Все процедуры ухода и содержания животных в виварии документировали, и документацию хранили в исследовательском центре.
[00504] Модель на крысах представляет собой хорошо описанную, высокочувствительную модель для оценки эффектов сосудистого напряжения и оценки эффективности последующего хронического воздействия исследуемого продукта. Данный дизайн исследования основан на действующих гармонизированных трехсторонних директивах Международной конференции по гармонизацию (МКГ) [МКГ S7A] и общепринятых методиках тестирования фармацевтических соединений. Данное неклиническое лабораторное исследование было разработано как исследование за рамками Надлежащей лабораторной практики (GLP)и не требовало гарантий качества (ГК).
[00505] Исследуемые концентрации ДГЛК (50 и 500 мг/кг), а также концентрация аспирина (10 мг/кг) были выбраны Спонсором на основании информации, доступной на момент дизайна данного исследования. Они были выбраны таким образом, чтобы охватить интервал концентраций, пригодных для идентификации механизма действия исследуемого продукта.
[00506] Концентрации положительного контроля (карведилол 0,1 мг/кг) и прессорного агента (фенилэфрин 3, 10 и 20 мкг/кг) были выбраны на основании литературных источников. Приготовление всех растворов документировалось в Форме содержания раствора, определяющей информацию для маркировки и всю уместную информацию относительно реактивов: номер серии, условия хранения, содержание и даты приготовления/окончания срока годности.
[00507] Исследуемый продукт составляли с применением оливкового масла в качестве основы. В дальнейшем указанный базовый раствор перорально вводили через зонд один раз в сутки на протяжении семи дней подряд. Тестировали низкую и высокую дозу (50 мг/кг и 500 мг/кг).
[00508] Базовый раствор Аспирина 5 мг/мл готовили путем растворения подходящего количества аспирина в 100% ДМСО, и далее разбавляли водой. Концентрация ДМСО составляла менее чем 1%. Подходящий объем перорально вводили через зонд каждому животному один раз в сутки на протяжении семи дней подряд. Базовый раствор хранили при комнатной температуре, причем он считался стабильным на протяжении периода введения через зонд.
[00509] Базовый раствор карведилола 0,4 мг/мл готовили в фосфатно-солевом буфере (ФСБ, pH=4,00 ± 0,05). Подходящий объем вводили каждому животному внутривенной инъекцией в конце эксперимента, в день хирургического вмешательства. Базовый раствор хранили при 4°C и считали стабильным на протяжении эксперимента.
[00510] Базовый раствор фенилэфрина 1 мг/мл готовили в ФСБ (pH=7,4 ± 0,05). Базовый раствор хранили при 4°C. Дата окончания срока годности была установлена как 14 дней после приготовления.
[00511] Любые остатки исследуемого продукта после эксперимента будут уничтожены после завершения исследования.
[00512] Самцы спонтанно гипертонических крыс массой около 300 г прибывали в центр по меньшей мере за два дня до начала перорального введения через зонд. Животных рандомизировали в одну из трех групп (низкая доза+аспирин, высокая доза+аспирин и аспирин) и поселяли парами на период акклиматизации. Каждая группа (n=4) получала соответствующее соединение перорально через зонд с применением игл для кормления 16G. Базовый раствор вводили один раз в сутки на протяжении семи дней подряд. На седьмой день введения животным проводили хирургическое вмешательство и мониторинг гипертензии.
[00513] Крысам проводили анестезию с помощью 2,5-3,0% смеси изофлурана-O2 в индукционном боксе. После извлечения животных из индукционной коробки им устанавливали назальный конус для поддержания анестезии в ходе трахеотомии. Животным проводили трахеотомию с помощью эндотрахеальной трубки (длина 7 см от соединителя до наконечника, изготовлена из трубки PE205 производства BD и иглы 16G), чтобы облегчить самопроизвольное дыхание, стабилизировать гемодинамику и поддерживать крысу под анестезией смесью изофлурана-O2. Были установлены два катетера: один в правой бедренной артерии для измерения системного артериального давления (САД), и один в правой бедренной вене для введения прессорного агента и положительного контроля. Сигналы ЭКГ получали с помощью контактов ЭКГ, размещенных в конфигурации Отведение-1 на анестезированном животном, и пульсовый оксиметр был присоединен к задней лапе животного для осуществления непрерывного мониторинга общего состояния крысы в ходе хирургического вмешательства. После измерения исходных показателей, три дозы фенилэфрина (3, 10 и 20 мкг/кг) вводили с помощью инъекции в/в болюса, с промежутком 5 минут между дозами. Вводили одну дозу положительного контроля, карведилола (0,1 мг/кг), за которой следовала самая высокая концентрация фенилэфрина. После положительного контроля снова вводили 10 мкг/кг фенилэфрина. Артериальное давление, системное артериальное давление (САД) и частоту сердцебиения непрерывно контролировали на протяжении получаса. Повышенное артериальное давление обнаруживали как повышение диастолического, систолического или среднего артериального давления. В конце эксперимента, животных подвергали эвтаназии путем обескровливания.
[00514] Для анализа применяли программное обеспечение Clampfit 10.2.0.14 Axon Instruments, установленное на подключенных к сети персональных компьютерах с Microsoft Windows. Clampfit 10.2.0.14 полностью валидировано в контексте соединения, в котором оно использовалось. Графическим программным обеспечением для иллюстраций было Microsoft Office Excel 2007, установленное на подключенных к сети персональных компьютерах с Microsoft Windows. Применяли непрерывную регистрацию системного артериального давления (САД) для вычисления среднего системного артериального давления (сСАД) для каждого состояния. Частоту сердцебиения непрерывно контролировали с момента проведения анестезии. Проведен анализ методом одностороннего АНОВА для сравнения показателей до и после воздействия в экспериментальных группах. Статистическая значимость была подтверждена на уровне p≤0,05.
[00515] В Табл. 9 представлено артериальное давление и средние значения системного артериального давления (мм рт. ст) после семи дней введения через зонд, перед внутривенным введением фенилэфрина.
Таблица 9.
Аспирин 10 мг/кг
* По сравнению с исходным значением
** По сравнению с ФЭ 20 мкг/кг
† По сравнению со вторым исходным значением
По сравнению с карведилолом
[00516] Табл. 10 иллюстрирует изменения среднего системного артериального давления при внутривенном введении фенилэфрина после перорального введения через зонд Аспирина в дозе 10 мг/кг/сутки на протяжении семи дней подряд.
Таблица 10.
(мм рт. ст)
[00517] Фиг. 1 иллюстрирует изменение среднего системного артериального давления в мм рт. ст после введения фенилэфрина. Пять крыс получили внутривенно дозы фенилэфрина после введения через зонд Аспирина в дозе 10 мг/кг/сутки на протяжении семи дней подряд. Среднее системное артериальное давление для каждой из трех доз фенилэфрина статистически сравнивали с первым исходным значением, в то время как состояние после введения карведилола статистически сравнивали с исходным значением непосредственно перед введением, и состояние после завершающей дозы фенилэфрина (10 мкг/кг) сравнивали с состоянием положительного контроля непосредственно перед ее ведением.
[00518] Полученные результаты демонстрируют повышение среднего системного артериального давления с повышением доз фенилэфрина. Все полученные данные были статистически отличными от исходного значения, подтверждая влияние прессорного агента фенилэфрина на модели спонтанно гипертонических крыс, получавших аспирин. Влияние фенилэфрина было полностью обратимым; среднее артериальное давление в точке второго исходного значения (после дозы 20 мкг/кг) фактически было подобным первому исходному значению.
[00519] Карведилол вызывал значительное снижение при добавлении непосредственно после фенилэфрина, подтверждая его известную способность уменьшать выраженность гипертензии (карведилол является неспецифическим бета-блокатором/альфа-1 блокатором и блокирует связывание фенилэфрина с альфа-1 адренергическими рецепторами). Последнее добавление фенилэфрина, после карведилола, вызывало ограниченное увеличение среднего системного артериального давления.
[00520] Фиг. 2. Изменение среднего артериального давления (мм рт. ст) при внутривенном введении фенилэфрина после введения через зонд ДГЛК в дозе 50 мг/кг+Аспирина в дозе 10 мг/кг на протяжении семи дней подряд.
[00521] Таблица 11. Изменение среднего артериального давления по сравнению с исходным уровнем при внутривенном введении фенилэфрина после введения через зонд ДГЛК в дозе 50 мг/кг+Аспирина в дозе 10 мг/кг на протяжении семи дней подряд.
Таблица 11.
[00522] Результаты, представленные на Фиг. 2, демонстрируют изменение среднего артериального давления в мм рт. ст после внутривенного введения фенилэфрина у пяти самопроизвольно гипертонических крыс после семи дней подряд перорального введения ДГЛК в дозе 50 мг/кг+Аспирина в дозе 10 мг/кг. Результаты для исследуемого продукта сравнивали с данными, полученными в группе введения только аспирина (10 мг/кг). Карведилол сравнивали со вторым исходным значением, и последнюю дозу фенилэфрина статистически сравнивали с карведилолом.
[00523] Животные продемонстрировали дозозависимое повышение артериального давления с увеличением концентрации фенилэфрина. Крысы, которым вводили ДГЛК в дозе 50 мг/кг и аспирин, продемонстрировали для данной дозы фенилэфрина повышение артериального давления, которое было по существу таким, как измеренное у крыс, получавших Аспирин на протяжении 10 дней. Для крыс, которые получали ДГЛК в дозе 50 мг/кг в дополнение к 10 мг/кг аспирина, не зарегистрировано существенной разницы в среднем артериальном давлении, по сравнению с группой аспирина, в любой из концентраций фенилэфрина. Возможно, это следствие того, что ежедневный прием Аспирина нивелирует благоприятное влияние ежедневного введения ДГЛК в дозе 50 мг/кг.
[00524] Карведилол вызывал значительное уменьшение антериального давления у животных, получавших аспирин+ДГЛК. Из-за проблемы с анестезией, крыса №3 не получила последнюю дозу фенилэфрина (n=4 для данного условия).
[00525] Таблица 12. Изменение среднего артериального давления при внутривенном введении фенилэфрина после введения через зонд ДГЛК в дозе 500 мг/кг параллельно с Аспирином в дозе 10 мг/кг на протяжении семи дней подряд.
Таблица 12.
[00526] Фиг. 3 иллюстрирует изменение среднего артериального давления после внутривенного введения фенилэфрина четырем самопроизвольно гипертоническим крысам после введения через зонд на протяжении семи дней подряд ДГЛК в дозе 500 мг/кг параллельно с аспирином в дозе 10 мг/кг. Как и в случае более низкой дозы ДГЛК+аспирина, состояние после введения фенилэфрина сравнивали с состоянием в группе введения только аспирина. Карведилол статистически сравнивали с исходным уровнем непосредственно перед введением, тогда как последнюю дозу фенилэфрина (10 мкг/кг) сравнивали с положительным контролем.
[00527] Несмотря на наблюдаемые более низкие значения среднего артериального давления, не зарегистрировано статистически значимой разницы между средним системным артериальным давлением у крыс, получавших ДГЛК и аспирин, и крыс, получавших только аспирин. Таким образом, возможно, Аспирин снова нивелировал благоприятное влияние ежедневного введения 500 мг/кг ДГЛК. Положительный контроль успешно демонстрировал статистически значимое снижение артериального давления у животных, как наблюдалось и в других группах. Повышение, вызванное последней дозой фенилэфрина, не считалось значительным.
[00528] Табл. 13 иллюстрирует среднее артериальное давление в исходной точке после введения через зонд на протяжении семи дней подряд для шести различных групп.
Таблица 13.
[00529] На Фиг. 4 представлено среднее артериальное давление в исходной точке для шести групп, которые получали лечение в ходе двух частей данного исследования. Трахеотомия стабилизировала гемодинамику, но также вызывала изменения артериального давления у крыс. Значения артериального давления у крыс, которые не получали аспирина (Основа, ДГЛК 50 мг/кг и ДГЛК 500 мг/кг), были получены в ходе первой фазы данного исследования (исследование № 20131022-1), и значения для других групп были зарегистрированы в ходе настоящего исследования (различные партии животных от Charles River, но крысы такой же породы и размера). Статистические t тесты были проведены для групп, которым вводили ДГЛК в дозе 50 или 500 мг/кг и эквивалентные дозы с добавлением аспирина в дозе 10 мг/кг. Наблюдалось статистически значимое повышение среднего артериального давления в исходной точке между только ДГЛК в дозе 50 мг/кг и ДГЛК 50 мг/кг параллельно с аспирином в дозе 10 мг/кг, а также между группой ДГЛК 500 мг/кг по сравнению с группой ДГЛК 500 мг/кг параллельно с аспирином 10 мг/кг (значительное повышение, обозначенное *).
[00530] Таким образом, ДГЛК, по-видимому, успешно снижает артериальное давление у спонтанно гипертонических крыс, причем данный эффект нивелируется Аспирином.
[00531] Табл. 14 иллюстрирует среднее артериальное давление при внутривенном введении фенилэфрина в дозе 20 мкг/кг для шести различных групп после введения через зонд на протяжении семи дней подряд.
Таблица 14.
[00532] Фиг. 5 представляет среднее артериальное давление во всех группах, проанализированных на протяжении обоих исследований для изучения ДГЛК. Указанные значения артериального давления были получены после внутривенного введения фенилэфрина в дозе 20 мкг/кг. Как и в случае результатов, полученных в исходной точке, каждую дозу ДГЛК статистически сравнивали с эквивалентным условием в группе только аспирина. Значительный прирост среднего артериального давления отмечали между обеими соответствующими группами (обозначены *).
[00533] Данное исследование было разработано с целью изучения эффективности ДГЛК, в отдельности и при хроническом параллельном введении с аспирином, с точки зрения снижения артериального давления, измеренного у гипертонических крыс, а также уменьшения выраженности гипертонической реакции, вызванной острым действием фенилэфрина у спонтанно гипертонических крыс такой же породы.
[00534] Спонтанно гипертоническая крыса (СГК) представляет собой широко применяемую модель в исследовании гипертензии, поскольку она демонстрирует, в пределах каждой колонии, однородные изменения в ответ на прямые и косвенные эффекторы сердечно-сосудистой системы. Крысы были отобраны и разведены на протяжении поколений, с определением гипертензии как систолического артериального давления выше 150 мм рт. ст, сохраняющегося более чем на протяжении одного месяца.
[00535] В ходе исследования было обнаружено, что хроническое параллельное введение ДГЛК и аспирина мешало ДГЛК снижать среднее системное артериальное давление (снижение до 25%), что наблюдалось ранее у спонтанно гипертонических крыс, леченных ДГЛК.
[00536] Изученные дозы ДГЛК (50 и 500 мг/кг) в сочетании с аспирином в дозе 10 мг/кг не вызывали значительного уменьшения дозозависимого повышения среднего артериального давления последующей инъекцией фенилэфрина, по сравнению с животными, которые получали перорально только аспирин. Две дозы ДГЛК вызывали сходные эффекты, наводя на мысль о том, что благоприятное влияние более высоких доз ДГЛК может быть минимальным в случае параллельного введения аспирина. Если обе дозы ДГЛК сравнивали с эквивалентными условиями «только аспирин», то Аспирин вызывал очевидное повышение среднего артериального давления в исходном состоянии, а также при внутривенном введении крысам фенилэфрина в дозе 20 мкг/кг.
[00537] В данном исследовании карведилол применяли в качестве положительного контроля. Он представляет собой бета-блокатор, применяемый для лечения сердечной недостаточности и гипертонии. Его действие осуществляется путем релаксации кровеносных сосудов и замедления частоты сердцебиения, с улучшением таким образом заполнения желудочков, кровотока и уменьшением артериального давления. Влияние карведилола при внутривенной инъекции спонтанно гипертоническим крысам подтвердило его способность снижать артериальное давление после каждой из трех доз фенилэфрина и продемонстрировало чувствительность тестовой системы.
ПРИМЕР 4
[00538] Одноцентровое, двойное слепое, рандомизированное, плацебо-контролируемое исследование Фазы I, состоящее из двух частей, проводили с участием здоровых субъектов мужского и женского пола в возрасте от 18 до 45 лет включительно. Основной целью данного исследования была оценка безопасности и фармакокинетики (ФК) в плазме при пероральном введении однократных доз и пероральном введении множественных доз капсул DS107G (один раз в сутки на протяжении 28 дней) у здоровых субъектов.
[00539] Вторичные задачи данного исследования состояли в оценке следующего:
[00540] Влияние приема пищи на ФК перорально введенной однократной дозы капсул DS107G у здоровых субъектов.
[00541] ФК ДГЛК в коже человека после введения множественных доз капсул DS107G при введении один раз в сутки на протяжении 28 дней здоровым субъектам.
[00542] Всего были включены 48 субъектов (в том числе 32 для однодозового исследования и 16 для многодозового исследования). Однодозовое и многодозовое исследование состояло из 8 субъектов на когорту (идеально 4 мужчины и 4 женщины, но не менее, чем 3 на пол).
[00543] Субъекты были рандомизированы в соотношении 3:1 в группу DG107G или плацебо методом блочной рандомизации. Однодозовая часть исследования включала три когорты по восемь субъектов в каждой, с дополнением Когорты четыре на основании оценки данных безопасности Когорты три Комитетом по мониторингу безопасности (КМБ). Однократную пероральную дозу DS107G или соответствующую дозу плацебо параллельно вводили субъектам натощак в Когортах с первой по третью (500, 1000 и 2000 мг, соответственно); субъектам в Когорте четыре вводили дозу 4000 мг после завершения исследования в Когорте три. Субъекты в Когорте два получали 1000 мг или соответствующую дозу плацебо натощак через ≥ 14 дней после введения первой дозы.
[00544] Многодозовая вторая часть исследования включала две когорты (Когорты пять и шесть) по восемь субъектов в каждой, которым натощак вводили множественные дозы исследуемого лекарственного средства на протяжении 28 дней. Субъекты в Когортах пять и шесть получали DS107G в дозах 2000 или 4000 мг, соответственно, или соответствующую дозу плацебо один раз в сутки ежедневно на протяжении 28 дней. Исследование в Когорте шесть было начато после того, как данные безопасности Когорты пять были оценены КМБ. Проведение временного анализа не планировалось.
[00545] В первой Части исследования, анализ ФК дигомо-гамма-линоленовой кислоты (ДГЛК) и 15-гидроксиэйкозатриеновой кислоты (15-ГЭТрЕ) по отдельности в плазме субъектов натощак проводили для Когорт с первой по четвертую на протяжении 312 часов после введения дозы в 1-5, 8, 14 дни исследования и в ходе визита наблюдения. Во второй Части проводили анализ ФК ДГЛК в плазме для Когорт пять и шесть на протяжении 168 часов после введения дозы в 1, 2, 3, 5 и 8 Дни исследования. Показатели безопасности контролировали на протяжении исследования.
[00546] Применяли рандомизированный, плацебо-контролируемый, двойной слепой дизайн для минимизации предвзятости при оценке показателей безопасности и переносимости.
[00547] Первую часть исследования проводили в соответствии с дизайном однократных нарастающих доз с повышением дозы до 4000 мг. За исключением дозы 4000 мг, все дозы в первой Части исследования были предварительно протестированы у человека. Вызванных лечением побочных реакций (ВЛПР) не наблюдали в ранее тестированных многодозовых схемах, таких как 150 мг/сутки на протяжении 28 дней, 450 мг/сутки на протяжении 28 дней, 1000 мг/сутки на протяжении 14 дней и 2000 мг/сутки на протяжении 10 дней. Выбор дозы был основан на пероральных дозах ДГЛК, протестированных в предыдущих клинических исследованиях и характеристике ФК и безопасности ранее протестированных доз, причем более высокие дозы рассматривались как основная задача.
[00548] Дополнительно оценивали влияние пищи на биодоступность ДГЛК при пероральном введении.
[00549] Здоровые субъекты женского пола были включены в данное исследование с целью оценки потенциального гендерного компонента на биомаркер дигидротестостерон, поскольку ДГЛК принимает участие в метаболизме стероидов.
[00550] Общая продолжительность участия для каждого субъекта в первой Части исследования составила около 14 дней, исключая период скрининга. В Части 2 исследования, продолжительность составила около 42 дней.
[00551] Скрининговые процедуры проводили для обеих частей, первой и второй, до начала исследования в -1 день (исходный уровень), оценивая показатели безопасности. Показатели безопасности включали побочные реакции (ПР), клинические лабораторные анализы (анализ крови, биохимические анализы, вирусология [поверхностный антиген гепатита B, антитела к вирусу иммунодефицита человека (ВИЧ), антитела к вирусу гепатита C], а также анализ мочи), результаты теста на употребление наркотиков (УН) и тест на беременность для субъектов женского пола (β-хорионический гонадотропин человека [βХГЧ] в моче), основные показатели жизнедеятельности (артериальное давление [АД], пульс, температура), электрокардиограммы (ЭКГ) с 12 отведениями, медицинские осмотры и оценку сопутствующей терапии (только вторая часть исследования).
[00552] От всех субъектов требовалось соответствие критериям включения и исключения, описанным ниже. Однако, незначительные отклонения, которые были клинически незначимыми и которые не создавали каких-либо проблем с безопасностью, считались приемлемыми Исследователем и Спонсором, в соответствии с протоколом.
[00553] От всех кандидатов на включение в исследование требовалось соответствие следующим критериям включения:
1. Субъект был мужского или женского пола.
2. Субъект женского пола или партнер женского пола субъекта мужского пола:
• Не должен быть беременным (для включения в исследование результаты теста мочи на беременность у субъектов женского пола должны быть отрицательными).
• Не должен кормить грудью.
• Не должен планировать беременность в ходе периода исследования или в пределах 3 месяцев после исследования.
• Должен применять надлежащую форму контрацепции, начиная до включения в исследование и еще в ходе 3 месяцев после визита наблюдения.
3. Возраст субъекта должен составлять от 18 до 45 лет включительно.
4. Субъект предоставил подписанную форму информированного согласия.
5. Индекс массы тела (ИМТ) субъекта должен составлять от 18,0 до 30,0 кг/м2 включительно.
6. С точки зрения Исследователя, субъект является здоровым на основании оценки показателей безопасности.
7. Субъекты должны быть способны поддерживать надлежащую связь с Исследователем, понимать и выполнять требования исследования, а также понять и подписать письменную ФИС. Субъектов исключали из исследования при наличии признаков любого из следующих критериев: у субъекта присутствовало клинически значимое заболевание на протяжении 4 недель перед скринингом.
8. Женщины, способные к деторождению, партнеры женского пола субъектов мужского пола, которые не применяли безопасный метод контрацепции на протяжении 3 месяцев до начала исследования и не были готовы применять безопасный метод контрацепции на протяжении исследования; примеры безопасного метода контрацепции включали внутриматочные или пероральные контрацептивы, диафрагму или презерватив, при условии применения в комбинации со спермицидом.
9. Субъект применял назначенные препараты на протяжении 2 недель перед введением или применял безрецептурные препараты (в том числе витамины и биологически активные добавки) на протяжении 1 недели перед введением (за исключением парацетамола, который разрешалось применять за 48 часов до введения) и гормональные контрацептивы.
10. Субъект применял биологически активные добавки, богатые омега-3 или омега-6 жирными кислотами.
11. У субъекта присутствовал значимый анамнез злоупотребления наркотиками/растворителями, или результаты скринингового теста на УН были положительными.
12. По мнению Исследователя, у субъекта присутствовал анамнез злоупотребления спиртным, или он употреблял более 28 порций в неделю (мужчины) или 21 порции в неделю (женщины) на момент скрининга.
13. По мнению Исследователя, субъект был непригоден для участия в исследовании.
14. Субъект принимал участие в другом клиническом исследовании с применением исследуемого лекарственного средства/медицинского устройства в пределах 3 месяцев до первого дня введения исследуемого лекарственного средства.
15. Результаты теста на антитела против ВИЧ, поверхностный антиген гепатита B или антитела против вируса гепатита C у субъекта являются положительными на момент скрининга.
16. У субъекта возникала серьезная побочная реакция или значимая гиперчувствительность к какому-нибудь лекарственному средству.
17. Субъект сдавал кровь или продукты крови в пределах 3 месяцев перед скринингом.
18. У субъекта присутствует известная гиперчувствительность к каким-нибудь ингредиентам исследуемого лекарственного средства.
[00554] Субъекты могли свободно отозвать согласие принимать участие в исследовании в любое время, по любой причине. Кроме того, Исследователь мог прекратить участие субъекта в исследовании, если, по мнению Исследователя, этого требовали интересы субъекта. Участие в исследовании могло быть прекращено по любой из следующих причин:
• Отзыв согласия в любое время
• Отклонения от протокола
• Интеркуррентное заболевание
• ПР (побочная реакция).
[00555] Хотя субъект не был обязан сообщать причину досрочного прекращения участия, Исследователь должен был приложить все усилия для выяснения причины, при полном уважении к правам субъекта. Если причина отзыва согласия была медицинской, субъект должен был оставаться под наблюдением Исследователя до тех пор, пока состояние здоровья субъекта не станет удовлетворительным; Исследователь должен был провести дополнительную оценку.
[00556] Если Исследователь считал, что это в интересах благополучия субъекта, Исследователь должен был уведомить лечащего врача общей практики субъекта о медицинской причине прекращения участия в исследовании. Необходимо было приложить все усилия для связи с субъектом, который не в состоянии вернуться в центр для последующих назначений, чтобы гарантировать, что состояние здоровья субъекта является удовлетворительным.
[00557] В первой Части исследования, субъектам в Когортах с первой по третью параллельно вводили однократную дозу исследуемого лекарственного средства (500, 1000 или 2000 мг в виде капсул DS107G по 500 мг или соответствующее количество капсул плацебо) в 1 день исследования по меньшей мере после 8-часового голодания, в соответствии с Табл. 15. Исследование в Когорте 4 было начато после рассмотрения КМБ данных безопасности Когорты 3.
[00558] Влияние пищи оценивали во второй Когорте по меньшей мере через 14 дней после введения первой дозы, во время введения второй однократной дозы. Субъектам вводили дозу 1000 мг исследуемого лекарственного средства с 240 мл воды после 10-часового голодания и через 30 минут после приема пищи. Затем субъекты воздерживались от приема пищи по меньшей мере на протяжении 4 часов после введения дозы. Прием пищи был стандартизированным по меньшей мере на протяжении 12 часов после введения дозы с применением стандартизированной пищи с высоким содержанием жира (800-1000 кКал, в том числе 500-600 кКал в виде жиров и 250 кКал в виде углеводов). Типичная стандартная тестовая пища состояла из двух яиц, поджаренных в масле, двух полосок бекона, двух ломтиков тоста с маслом, 120 мл смеси коричневого картофеля и 240 мл цельного молока.
[00559] Во второй Части исследования (Когорты пять и шесть), субъектам вводили исследуемое лекарственное средство (капсулы DS107G по 500 мг или соответствующее количество капсул плацебо) один раз в сутки натощак на протяжении 28 дней. Вначале исследуемое лекарственное средство получали субъекты в пятой Когорте и, если они переносили ежедневную дозу 2000 мг на протяжении первых 14 дней, субъекты в шестой Когорте начинали с ежедневной дозы 4000 мг на протяжении 28 дней.
Таблица 15. Когорты лечения
Примечание: Каждая капсула DS107G содержала 500 мг ДГЛК.
[00560] Решение о любом повышении уровня дозы или начале исследования в следующей когорте принималось Главным Исследователем. По меньшей мере 5 субъектов с доступными для оценки данными безопасности из третьей Когорты (доза 2000 мг) требовалось до начала исследования в четвертой Когорте (доза 4000 мг). В Части 2, по меньшей мере 5 субъектов из Когорты 5 с доступными для оценки данными безопасности за 14 дней (2000 мг/сутки) требовалось до начала исследований в шестой Когорте (4000 мг/сутки).
[00561] Были разработаны капсулы ДГЛК DS107G в одной дозировке, содержащие 500 мг свободной жирной кислоты ДГЛК (СЖК). Капсулы содержали следующие вспомогательные вещества: СЖК ДГЛК (стабилизированная номинальным содержанием 2000 промилле dl-альфа токоферола). Все вспомогательные вещества в капсульной оболочке обычно применялись в мягких желатиновых продуктах и включают сертифицированную на отсутствие трансмиссивной губчатой энцефалопатии (ТГЭ) желатиновую оболочку, содержащую следующие ингредиенты: вода очищенная, пластификатор глицерин, краситель титана диоксид и технологические добавки лецитин и среднецепочечный триглицерид. Капсула плацебо (Капсула плацебо DS107G) для клинических исследований состояла из жидкого парафина, инкапсулированного в мягкую желатиновую оболочку, и по внешнему виду была идентичной капсуле ДГЛК.
[00562] Субъекты, отвечающие критериям пригодности, были рандомизированы для получения ДГЛК (дозы 500, 1000, 2000, 4000 мг) или соответствующего количества капсул плацебо, с применением списка рандомизации. Рандомизация представляла собой блочную рандомизацию с соотношением активного лечения и плацебо 3:1. Список рандомизации генерировался Planimeter с применением SAS® 9.1.3 SP4.
[00563] Субъектам не разрешалось применять назначенное лечение на протяжении 2 недель до введения дозы или применять безрецептурные препараты (в том числе витамины и биологически активные добавки) и гормональные контрацептивы на протяжении 1 недели до введения дозы, за исключением парацетамола, который разрешалось принимать за 48 часов до введения дозы. Кроме того, субъектам не разрешалось применять биологически активные добавки, богатые омега-3 или омега-6 жирными кислотами. Субъектам не разрешалось употреблять более чем 28 порций спиртного в неделю (субъекты мужского пола) или 21 порции в неделю (субъекты женского пола).
[00564] Субъектам сообщали о необходимости избегать употребления биологически активных добавок, богатых омега-3 или омега-6 жирными кислотами (например, капсулы с рыбьим жиром) в течение 4 недель до скрининга и на протяжении исследования.
[00565] Субъектам сообщали о необходимости избегать употребления маковых зерен и продуктов, содержащих маковые зерна, по меньшей мере на протяжении 24 часов перед отбором образца мочи для теста на УН, поскольку маковые зерна иногда могут давать ложно положительный результат теста.
[00566] Потребление пищи субъектами во второй Когорте, которые проходили оценку влияния пищи на перорально вводимые капсулы ДГЛК 1000 мг, ограничивали по меньшей мере на протяжении 4 часов после введения дозы. Потребление пищи было стандартизированным по меньшей мере на протяжении 12 часов после введения дозы, с применением стандартизированной пищи с высоким содержанием жиров.
[00567] Субъекты должны были воздерживаться от приема любых системных или безрецептурных лекарственных средств (в том числе витаминов и биологически активных добавок) на протяжении исследования, за исключением гормональных контрацептивов. Прием парацетамола (в дозе до 4 г/сутки) разрешался за 48 часов до введения дозы. Кроме того, субъекты должны были воздерживаться от употребления спиртного, начиная с 48 часов до введения первой дозы исследуемого лекарственного средства (1 день) и до визита наблюдения.
[00568] Не налагалось ограничений на употребление кофеина или использование табака до начала или на протяжении исследования.
[00569] От субъектов требовалось избегать физических нагрузок и энергичной физической активности по меньшей мере на протяжении 3-4 часов до отбора образцов крови для клинического лабораторного анализа.
[00570] Анализ концентраций ДГЛК (свободной и общей) и свободной 15-ГЭТрЕ в плазме проводили на образцах крови, полученных от субъектов. Кроме того, образцы жидкости кожного волдыря получали в 1, 7, 14 и 28 день для анализа свободной и общей ДГЛК.
[00571] Концентрации дигидрокситестостерона (ДГТ) в плазме оценивали в Части 2 в качестве биомаркера или исследовательской конечной точки эффективности.
[00572] Анализ всех образцов плазмы и жидкости кожного волдыря проводили с применением валидированных методов. Концентрации свободной и общей ДГЛК определяли в плазме и жидкости кожного волдыря жидкостной хроматографией с тандемной масс-спектрометрией (ЖХ/МС/МС); диапазон количественной оценки составил 100-10000 нг/мл для свободной ДГЛК и 5000-500000 нг/мл для общей ДГЛК. Концентрации в плазме свободной 15-ГЭТрЕ определяли методом ЖХ/МС/МС с диапазоном количественной оценки 100-10000 нг/мл. Концентрации ДГТ в плазме определяли методом ЖХ/МС с диапазоном количественной оценки 0,02-1,5 нг/мл.
[00573] Состояние субъектов контролировали на протяжении действия ограничений в части Фазы 1 на предмет побочных реакций на исследуемое лекарственное средство и/или процедуры.
[00574] Фармакокинетические показатели, применяемые в данном исследовании, были стандартными для оценки потенциальных терапевтических агентов. Показатели безопасности включали методы, которые считаются стандартными для клинического исследования Фазы 1.
[00575] Исследование было исследовательским, и формальных вычислений мощности не проводилось. Количество субъектов, запланированное для каждой когорты (8 субъектов), считалось достаточным для оценки безопасности и системного контакта с капсулами DS107G.
[00576] Анализ популяций включал следующие:
• Популяция субъектов, рандомизированных согласно назначенному лечению (РНЛ), которую составляют все рандомизированные субъекты, которым была введена по меньшей мере 1 доза исследуемого лекарственного средства.
• Популяция анализа по протоколу (ПП), которую составляют все субъекты популяции РНЛ, для которых не зарегистрировано существенных отклонений от протокола, как определено в ПСА.
• популяцию ФК составляют все субъекты, входящие в популяцию ПП, для которых получены пригодные для оценки данные ФК для плазмы. Наблюдение концентрации в плазме считалось валидным, пригодным для оценки показателей, если были доступны следующие данные: идентификационный номер исследования, номер рандомизации, дата и время отбора образца, доза и концентрация. Серии таких измерений для одного образца считались завершенными, если концентрация в каждой точке согласно протоколу была пригодной для оценки. Данные ФК в плазме были пригодными для оценки по определению, если они содержали данные полной серии показателей. Отсутствие любого показателя ФК в плазме приводило бы к неполноте данных ФК, поэтому субъект с любым пропущенным показателем ФК в плазме исключался из популяции ФК. Кроме того, субъекты, рандомизированные для получения плацебо, были исключены из популяции ФК.
[00577] Популяции, определенные выше, генерировались отдельно для данных Части 1 и Части 2 исследования. Анализ безопасности выполняли на популяции РНЛ.
[00578] Анализы безопасности выполняли на популяции безопасности, представленной в таблицах по группе лечения, когорте и визиту. Все данные безопасности характеризовали с помощью инструментов описательной статистики. В протоколе исследования не было установлено никакой гипотезы для изучения. Оценку показателей безопасности выполняли в описательной форме. Непрерывные переменные характеризовали средним значением, стандартным отклонением (СО), медианой, минимальными и максимальными значениями; дискретные переменные характеризовали абсолютным (частота) и относительным (процент [%]) распространением. Группы лечения были также.
[00579] Первичные конечные точки (ФК характеристики, полученные в результате перорального введения однократной и множественных доз капсул DS107G) были получены с помощью некомпартментального моделирования ФК. Вторичные конечные точки состояли из характеристики влияния пищи на ФК однократной пероральной дозы капсул DS107G и характеристики ФК ДГЛК в коже человека после перорального введения множественных доз капсул DS107G).
[00580] Вторичные конечные точки регистрировали с применением следующих описательных инструментов анализа: количество валидных показателей, среднее значение, стандартное отклонение (СО), медиана, минимальные и максимальные значения, полученные для непрерывных показателей, сгруппированных по группе лечения, визиту и когорте.
[00581] В ходе оценки данных исследования не выполнялось никакой формальной проверки гипотезы. В Части 1 (однократная нарастающая доза), системный контакт составил 1 день по определению (в случае успешного введения исследуемого лекарственного средства). В Части 2 (множественные нарастающие дозы) системный контакт вычисляли как день последнего введения исследуемого лекарственного средства минус день первого введения исследуемого лекарственного средства+1 (если не было задокументировано перерывов во введении исследуемого лекарственного средства). В случае перерыва(ов), результат упомянутой выше формулы уменьшали на количество перерывов.
[00582] Статистический анализ данных ФК выполняли с применением программного обеспечения SAS (версия 9.1.3). Анализ фармакодинамики не проводили.
[00583] Показатели ФК ДГЛК и 15-ГЭТрЕ в плазме оценивали с применением независимых от модели методик (некомпартментальный анализ), причем указанные показатели включали: данные Cmax, tmax, Clast, Tlast, AUC0-24, AUC0-inf, AUClast, λz, CL, V, а также t½ для Части 1 и 2, и tmin, cmin, CLss, Vss, Cavg, а также %PTF только для Части 2 (стационарная фаза).
[00584] Все показатели ФК суммировали описательно, в связи с исследовательской природой исследования формального статистического тестирования не проводилось.
[00585] В условиях введения однократной дозы (Часть 1), прекращение приема исследуемого лекарственного средства не было потенциальным результатом в первой, третьей и четвертой Когортах; таким образом, предоставления информации в этих когортах не требовалось. В Когорте 2 зарегистрированные субъекты получали исследуемое лекарственное средство дважды: введение первой однократной дозы натощак и второй однократной дозы после приема пищи.
[00586] Кроме того, следующие исправления были внесены в протокол.
• Добавлена Таблица клинических исследований, с целью освещения предыдущих клинических исследований с пероральным введением ДГЛК и их выводов относительно безопасности.
• Добавлена оценка риска/пользы, с целью освещения оцененных рисков и пользы от предлагаемого исследования в интересах Исследователя и Комитета по этике.
• Критерии включения были изменены таким образом, что субъекты, которые были включены в Часть 1 исследования (когорта однократной дозы), могли вернуться и быть повторно включены в Часть 2 (когорта множественных доз), при условии, что у них не возникало ПР, связанных с исследуемым лекарственным средством и период вымывания длился по меньшей мере 14 дней до начала схемы введения множественных доз. Обоснование для данного изменения состояло в содействии рекрутингу без подвергания добровольцев риску.
• Клинические лабораторные анализы для коагуляции (протромбиновое время и активированное парциальное протромбиновое время [АПТТ]) были добавлены в качестве показателей во все временные точки клинической лабораторной оценки в Части 2 исследования (Когорты множественных доз 5 и 6). Указанные показатели были добавлены с целью контроля любых потенциальных изменений в факторах свертываемости в качестве исследовательского биомаркера для будущих исследований.
• Неоднозначное выражение было заменено, с целью уточнения того, что будут получены единичные ЭКГ.
• 15-ГЭТрЕ в качестве анализируемого вещества была исключена из тестирования в Части 2 (когорты множественных доз), поскольку предварительные данные ФК для когорт Части 1 выявили, что все образцы для анализа 15-ГЭТрЕ содержали это вещество в количестве ниже предела количественного определения (ПКО).
• В дополнение к свободной ДГЛК (неэтерифицированой), количественное определение «общей» ДГЛК было добавлено ко всем анализам, поскольку ФК профиль «общей» в плазме может отличаться от «свободной».
• Кроме того, модификации были введены в запланированные анализы, с целью обеспечения дополнительных анализов и статистических результатов.
РЕЗУЛЬТАТЫ
Однодозовое исследование.
[00587] Был проведен скрининг сорока субъектов; из этих 40, 4 субъекта были исключены за невыполнение требований включения/исключения, и 4 субъекта отозвали согласие. Распределение 32 субъектов, рандомизированных для получения исследуемого лекарственного средства, приведено в Табл. 16.
Таблица 16 Сводная таблица распределения субъектов - популяция РНЛ (однократная доза).
РНЛ=рандомизация согласно назначенному лечению
Из 8 субъектов, получавших плацебо, по 2 субъекта были рандомизированы в каждую из когорт введения DS107G.
[00588] Концентрации свободной и общей ДГЛК в плазме в начале исследования, до введения DS107G, сведены в Табл. 17. В общем, указанные концентрации были высоко вариабельными.
Таблица 17 Концентрации ДГЛК в плазме в начале исследования (однократная доза, популяция фармакокинетики)
Мин, Макс
150, 357
268, 347
133, 327
134, 204
241, 538
Мин, Макс
22000, 57100
26700, 63900
28400, 66600
22700, 43200
19100, 45500
[00589] Средние концентрации в плазме после однократной дозы DS107G графически проиллюстрированы по когорте дозы для свободной ДГЛК на Фиг. 6 (линейный график) и Фиг. 7 (логарифмически-линейный график), а для общей ДГЛК на Фиг. 8 (линейный график) и Фиг. 9 (логарифмически-линейный график).
[00590] После однократной дозы капсул DS107G (500, 1000, 2000 и 4000 мг), межсубъектная вариабельность (измеренная по СО) свободной и общей ДГЛК была высокой как для концентраций в плазме, так и для скорректированных с учетом исходного уровня показателей ФК. Натощак, скорректированные с учетом исходного уровня средние значения Cmax и AUC0-24 как свободной, так и общей ДГЛК линейно возрастали (Табл. 18, Табл. 19). Медиана времени до максимальной концентрации (Tmax) составила 4 часа для свободной ДГЛК (Табл. 18) и не коррелировала с дозой, со значениями 8 часов для 2 более низких доз и 18 часов для 2 более высоких доз (Табл. 19). Хотя скорректированные с учетом исходного уровня показатели ФК элиминации в некоторых когортах могли быть определены менее чем для половины субъектов, не наблюдалось доказательств нелинейной фармакокинетики с точки зрения элиминационного периода полувыведения или клиренса для свободной или общей ДГЛК (Табл. 19 и Табл. 20). Хотя этот показатель не оценивали статистически, введение однократной дозы DS107G 1000 мг натощак приводило к около 50% повышению скорости и степени абсорбции общей ДГЛК, по данным скорректированных с учетом исходного уровня средних значений Cmax и AUC0-24 (Табл. 19).
Таблица 18 Скорректированные с учетом исходного уровня показатели фармакокинетики свободной ДГЛК в плазме после введения натощак (однократная доза, популяция фармакокинетики)
(n=5)
(n=6)
(n=6)
(n=6)
Медиана
Мин, Макс
4,00
4,0, 4,0
4,00
4,0, 4,0
4,00
4,0, 8,0
4,00
4,0, 6,0
Среднее
СО
934,6
776,87
1602,8
1202,43
2074,0
1276,03
3021,8
1581,97
Среднее
СО
1,869
1,5537
1,603
1,2024
1,037
0,6380
0,755
0,3955
Среднее
СО
2728,1
2253,49
3998,5
1885,27
6429,0
2119,39
9436,8
2227,25
Среднее
СО
5,46
4,507
4,00
1,885
3,21
1,060
2,36
0,557
Среднее
СО
0,0035
0,00101
0,0487
0,05133
0,0200
0,02445
0,0603
0,10642
Медиана
Мин, Макс
206,33
164,3, 248,4
28,30
6,2, 3226,3
57,33
11,0, 181,1
67,53
2,8, 80,9
Среднее
СО
21483,3
14902,37
218955,6
474291,75
26877,0
19394,67
17113,6
3545,72
Среднее
СО
42,97
29,805
218,96
474,292
13,44
9,697
4,28
0,886
Среднее
СО
0,0306
0,02126
0,1866
0,18154
0,1339
0,11952
0,2420
0,05066
Среднее
СО
10,035
8,9574
7,356
9,1148
8,977
4,0610
17,460
11,5516
Примечание: Концентрация ДГЛК до начала введения была вычтена из последующих концентраций до расчета показателей; отрицательные значения были заменены нулевыми.
Таблица 19 Скорректированные с учетом исходного уровня показатели фармакокинетики общей ДГЛК в плазме после введения натощак (однократная доза, популяция фармакокинетики)
(n=5)
(n=6)
(n=6)
(n=6)
Медиана
Мин, Макс
8,00
6,0, 312,0
8,00
0,0, 312,0
18,00
6,0, 24,0
18,00
8,0, 24,0
Среднее
СО
7200,0
2801,79
15750,0
16326,02
17733,3
9867,66
25883,3
13658,32
Среднее
СО
14,400
5,6036
15,750
16,3260
8,867
4,9338
6,471
3,4146
Среднее
СО
39556,6
37969,05
215369,7
346249,37
210508,9
167660,31
437754,0
278838,30
Среднее
СО
79,11
75,938
215,37
346,249
105,25
83,830
109,44
69,710
Среднее
СО
0,0013
0,00042
0,0136
0,01381
0,0084
0,00301
0,0102
0,00993
Медиана
Мин, Макс
552,07
428,3, 675,9
105,42
29,7, 181,2
88,04
65,8, 110,3
71,03
26,7, 464,7
Среднее
СО
4415966,6
723917,02
4031410,9
5304874,41
2276495,1
1948487,62
5973518,8
7261476,90
Среднее
СО
8831,93
1447,834
4031,41
5304,874
1138,25
974,244
1493,38
1815,369
Среднее
СО
0,0001
0,00002
0,0018
0,00243
0,0014
0,00119
0,0036
0,00519
Среднее
СО
0,089
0,0140
0,093
0,0843
0,203
0,2136
0,275
0,1569
Примечание: Концентрация ДГЛК до начала введения была вычтена из последующих концентраций до расчета показателей; отрицательные значения были заменены нулевыми.
[00591] Влияние пищи на скорректированную с учетом исходного уровня однодозовую ФК свободной и общей ДГЛК с применением дозы 1000 мг (вторая Когорта) было оценено и приведено в Табл. 21. Если коротко, среднее значение скорректированной с учетом исходного уровня Cmax ДГЛК было приблизительно в 3 раза выше и возникало на 1 час (медиана) быстрее натощак, чем после приема пищи (Табл. 21). Среднее значение скорректированной с учетом исходного уровня AUC0-24 свободной ДГЛК натощак было приблизительно в 2 раза выше, чем значение после приема пищи. Таким образом, наблюдалось повышение скорости и степени абсорбции ДГЛК натощак. С точки зрения элиминации свободной ДГЛК (Табл. 20), не наблюдалось очевидных отличий между состоянием натощак и после приема пищи.
[00592] В случае общей ДГЛК, среднее значение скорректированной с учетом исходных значений Cmax натощак было приблизительно в 1,5 раза выше, и время tmax наступало приблизительно на 50% быстрее (медиана, 8 против 15 часов), чем после приема пищи (Табл. 20). Среднее значение скорректированной с учетом исходных значений AUC0-24 натощак было приблизительно в 1,8 раза выше, чем после приема пищи. Данные немного менее чем половины субъектов (2/6 натощак, 3/6 после приема пищи) были достаточными, чтобы оценить λz, t1/2, клиренс и объем распределения общей ДГЛК. Указанные данные наводят на мысль о повышении скорости и степени абсорбции общей ДГЛК натощак. Не существует достоверного заключения относительно элиминации или объема распределения общей ДГЛК из-за маленькой популяции данных.
[00593] На основании показателей ФК, приведенных в Табл. 20, среднее значение скорректированной с учетом исходного значения Cmax было выше ~ в 10 раз (натощак) и ~ в 20 раз (после приема пищи) для общей ДГЛК, чем для свободной ДГЛК. Среднее значение скорректированной с учетом исходного значения AUC0-24 было ~ в 54 раза (натощак) и ~ в 56 раз (после приема пищи) выше для общей ДГЛК, чем для свободной ДГЛК.
Таблица 20 Скорректированные с учетом исходного уровня фармакокинетические показатели ДГЛК в плазме при введении натощак и после приема пищи (однократная доза, популяция фармакокинетики)
Медиана
Мин, Макс
4,00
4,0, 4,0
5,00
4,0, 8,0
8,00
0,0, 312,0
15,00
6,0, 24,0
Среднее
СО
1602,8
1202,43
520,2
235,54
15750,0
16326,02
10316,7
2043,93
Среднее
СО
1,603
1,2024
0,520
0,2355
15,750
16,3260
10,317
2,0439
Среднее
СО
3998,5
1885,27
2102,7
1174,23
215369,7
346249,37
117389,7
70263,85
Среднее
СО
4,00
1,885
2,10
1,174
215,37
346,249
117,39
70,264
Среднее
СО
0,0487
0,05133
0,0403
0,06154
0,0136
0,01381
0,0805
0,06155
Медиана
Мин, Макс
28,30
6,2, 3226,3
140,86
6,2, 149,4
105,42
29,7,181,2
6,62
5,5,66,0
Среднее
СО
218955,6
474291,75
29150,8
23323,42
4031410,9
5304874,41
267053,2
335666,87
Среднее
СО
218,96
474,292
29,15
23,323
4031,41
5304,874
267,05
335,667
Среднее
СО
0,1866
0,18154
0,1658
0,24651
0,0018
0,00243
0,0096
0,00702
Среднее
СО
7,356
9,1148
4,623
0,5166
0,093
0,0843
0,128
0,0209
Примечание: Концентрация ДГЛК до начала введения была вычтена из последующих концентраций до расчета показателей; отрицательные значения были заменены нулевыми.
[00594] DSI07G хорошо переносилась здоровыми добровольцами в виде однократной дозы в количествах 500, 1000, 2000 или 4000 мг. Наиболее частой из зарегистрированных ВЛПР была легкая или умеренная диарея (сообщалось о послаблении стула), причем процент субъектов был подобным для субъектов в группах активного лечения и контроля плацебо (частота: 5/24 [20,8%] субъектов в группе активного лечения против 2/8 [25,0%] субъектов в контрольной группе плацебо). Эпизоды диареи были относительно короткими, причем все из них (включая возникавшие у субъектов контрольной группы плацебо) рассматривались Исследователем, как возможно связанные с исследуемым лекарственным средством. Примечательно, что у субъектов, которые получали вторую однократную дозу DS107G, и у которых возникали ВЛПР в виде диареи при приеме натощак, диарея не возникала повторно. Все другие ВЛПР возникали только у 1 субъекта каждый и только в группах активного лечения, в том числе: легкая инфекция, боль в орофарингеальной области и фарингит; а также умеренная пирексия и инфекция мочевых путей при введении доз после приема пищи.
МНОГОДОЗОВЫЕ РЕЗУЛЬТАТЫ - ЧАСТЬ 2 ИССЛЕДОВАНИЯ
[00595] Средние концентрации в плазме и средняя концентрация в стационарной фазе для Части 2 графически проиллюстрированы для свободной ДГЛК по когортам дозы на Фиг. 10 (линейный график) и Фиг. 11 (логарифмически-линейный график) и для общей ДГЛК на Фиг. 12 (линейный график) и Фиг. 13 (логарифмически-линейный график). В 1 и 28 дни, средние концентрации свободной ДГЛК достигали пика через около 4 часов после введения, тогда как для общей ДГЛК не наблюдалось выраженной пиковой концентрации. Средние концентрации как свободной, так и общей ДГЛК возрастали во времени с введением повторных доз, однако увеличение для общей ДГЛК было более выраженным. По-видимому, концентрации в плазме достигают стационарной фазы приблизительно на 14 день для обеих доз (2000 и 4000 мг ежедневно) и анализируемых веществ (свободная и общая ДГЛК), на основании визуальной инспекции графиков средней концентрации. Если доза удваивалась с 2000 до 4000 мг в сутки, то средняя концентрация в стационарной фазе повышалась в 1,6 раза для свободной ДГЛК, но только в 1,2 для общей ДГЛК, наводя на мысль об одном или более насыщаемых процессах при более высокой дозе.
[00596] Показатель ФК вычисляли после коррекции дозовых концентраций ДГЛК с учетом исходного уровня концентраций ДГЛК.
[00597] Фармакокинетическое показатели, скорректированные с учетом исходного уровня, для свободной ДГЛК в плазме приведены в Табл. 21. Если коротко, скорректированное с учетом исходного уровня среднее значение Cmax и AUC для свободной ДГЛК было выше в когорте более высокой дозы DS107G в оба дня проведения оценки. Скорректированное с учетом исходного уровня среднее значение Cmax для дозы 4000 мг было ~ в 3 раза выше, чем для дозы 2000 мг в 1 день, но только ~ в 1,4 раза выше в 28 день. Скорректированное с учетом исходного уровня среднее значение AUC0-24 для дозы 4000 мг было ~ в 2,5 раза выше, чем для дозы 2000 мг в 1 день, и только ~ в 1,7 раза выше в 28 день. Изменения с дозой были линейными для скорректированных с учетом исходного уровня Cmax и AUC0-24 в 1 день, но только для скорректированной с учетом базового уровня AUC0-24 в 28 день. Причиной такой несистематичности могла стать высокая межсубъектная вариабельность. Медиана tmax была подобной для обеих дозовых когорт как в 1 день, так и в 28 день, со значениями 4 или 5 часов. Медиана t1/2 элиминации была длиннее в 28 день, чем в 1 день, причем значение в 28 день зависело от оцениваемого временного интервала. Средний клиренс уменьшался, и средний объем распределения увеличивался при введении множественных доз.
Таблица 21 Скорректированные с учетом исходного уровня фармакокинетические показатели свободной ДГЛК в плазме (множественные дозы, популяция фармакокинетики)
0-24 часа
0-24 часа
0-168 часов
(n=6)
(n=6)
(n=6)
(n=6)
(n=6)
(n=6)
Медиана
Мин, Макс
4,00
4,0, 6,0
5,00
2,0, 8,0
4,00
2,0, 4,0
4,00
4,0, 4,0
4,00
2,0, 4,0
4,00
4,0, 4,0
Среднее
СО
850,5
458,84
2641,5
1734,46
1999,3
1003,19
2873,2
1246,97
1999,3
1003,19
2873,2
1246,97
Среднее
СО
0,425
0,2294
0,660
0,4336
1,000
0,5016
0,718
0,3117
1,000
0,5016
0,718
0,3117
Среднее
СО
4548,7
3330,51
11441,4
5313,29
9248,1
2559,31
16155,2
6367,97
9248,1
2559,31
16155,2
6367,97
Среднее
СО
2,27
1,665
2,86
1,328
4,62
1,280
4,04
1,592
4,62
1,280
4,04
1,592
Среднее
СО
0,2133
0,16812
0,1504
0,10843
0,1282
0,15619
0,0596
0,02019
0,0250
0,02983
0,0404
0,05960
Медиана
Мин, Макс
4,71
2,1, 7,3
5,92
2,0, 18,3
14,04
2,2, 25,6
12,72
7,5, 16,5
63,59
9,0, 132,1
48,18
4,3, 65,7
Среднее
СО
5955,6
5216,07
13900,3
4993,97
10389,0
2677,43
22926,3
6485,37
43024,8
35641,74
43184,3
21443,83
Среднее
СО
2,98
2,608
3,48
1,248
5,19
1,339
5,73
1,621
21,51
17,821
10,80
5,361
Среднее
СО
0,5448
0,47711
0,3325
0,16310
0,2002
0,04493
0,1865
0,05432
0,0949
0,08636
0,1266
0,08671
Среднее
СО
2,426
0,3245
3,063
1,9716
3,592
2,5139
3,493
1,5255
4,470
1,5179
5,273
2,1568
Примечание: Концентрация ДГЛК до начала введения была вычтена из последующих концентраций до расчета показателей; отрицательные значения были заменены нулевыми.
[00598] Скорректированные с учетом исходного уровня фармакокинетические показатели стационарной фазы для свободной ДГЛК в плазме приведены в Табл. 22. Если коротко, концентрации в плазме свободной и общей ДГЛК возрастают с введением повторных доз и достигают стационарной фазы приблизительно на 14 день. При достижении стационарной фазы, по оценкам, на 28 день (0-24 часа), колебания от пика до минимума на графике (КПМ) были достаточно высокими для обоих дозовых когорт (среднее значение, ~ 430% и ~ 490%; Табл. 9). Среднее соотношение аккумуляции (СА) было выше для дозовой когорты 2000 мг, чем для 4000 мг, как в показателях Cmax, так и AUC (СА ~ 2,8 и ~ 3,3 для 2000 мг против ~ 1,4 и ~ 1,6 для 4000 мг. Данные наводят на мысль о насыщаемой кинетике и/или присутствии изменений в объеме распределения при введении повторных доз свободный ДГЛК.
Таблица 22 Скорректированные с учетом исходного уровня фармакокинетические показатели в стационарной фазе свободной ДГЛК в плазме (множественные дозы, популяция фармакокинетики)
Уровень дозы DS107G
(n=6)
(n=6)
Медиана
Мин, Макс
8,00
6,0,24,0
8,00
0,0,12,0
Среднее
СО
100,8
103,34
147,0
230,37
Среднее
СО
0,2295
0,05874
0,2858
0,12493
Среднее
СО
5,3096
4,02532
5,7563
3,12495
Среднее
СО
385,3
106,64
673,1
265,33
Среднее
СО
492,54
212,957
431,97
207,312
Среднее
СО
2,796
1,7385
1,385
0,7726
Среднее
СО
3,340
2,4958
1,561
0,6377
Примечание: Концентрация ДГЛК до начала введения была вычтена из последующих концентраций до расчета показателей; отрицательные значения были заменены нулевыми.
[00599] Скорректированные с учетом исходных значений фармакокинетические показатели для общей ДГЛК в плазме приведены Табл. 23. Если коротко, средние значения скорректированных с учетом исходных значений Cmax и AUC0-24 общей ДГЛК были выше в когорте более высокой дозы DS107G в оба дня проведения оценки, как и ожидалось. Скорректированное с учетом исходных уровней среднее значение Cmax и AUC0-24 для дозы 4000 мг было ~ в 1,5 и ~ в 1,5 раза выше, соответственно, чем для дозы 2000 мг в 1 день, но только ~ в 1,.2 и ~ в 1,4 раза выше, чем для дозы 2000 мг в 28 день.
[00600] Изменения скорректированных с учетом исходного уровня Cmax и AUC4 с дозой не были линейными для общей ДГЛК в любой из дней проведения оценки. Причиной такой несистематичности могла стать высокая межсубъектная вариабельность. Медиана Tmax возникала быстрее в случае множественных доз (8-10 часов), чем в случае однократной дозы (10-18 часов) в обеих дозовых когортах. Медиана t1/2 элиминации общей ДГЛК для когорты дозы 2000 мг составила 34,4-44,0 часа при оценке периода 24 часа (1 и 28 дни, соответственно) и 62,6 часа в 28 день при оценке периода свыше 0-168 часов. Средний клиренс и объем распределения уменьшались при введении множественных доз.
Таблица 23. Скорректированные с учетом исходного уровня фармакокинетические показатели общей ДГЛК в плазме (множественные дозы, популяция фармакокинетики)
0-24 часа
0-24 часа
0-168 часов
(n=6)
(n=6)
(n=6)
(n=6)
(n=6)
(n=6)
Медиана
Мин, Макс
10,00
8,0, 12,0
18,00
12,0, 24,0
8,00
6,0, 24,0
10,00
8,0, 24,0
8,00
6,0, 24,0
10,0
8,0, 24,0
Среднее
СО
18100,0
15113,44
27866,7
10391,66
75583,3
30385,55
90866,7
33000,16
75583,3
30385,55
90866,7
33000,16
Среднее
СО
9,050
7,5567
6,967
2,5979
37,792
15,1928
22,717
8,2500
37,792
15,1928
22,717
8,2500
Среднее
СО
298184,2
336969,41
456199,5
151586,31
1351372,5
379053,12
1860574,3
760386,93
1351372,5
379053,12
1860574,3
760386,93
Среднее
СО
149,09
168,485
114,05
37,897
675,69
189,527
465,14
190,097
675,69
189,527
465,14
190,097
Среднее
СО
0,221
0,02180
0,0199
0,00966
0,0471
0,05409
0,0169
0,01064
0,0194
0,00927
Медиана
Мин, Макс
44,04
14,9, 162,5
34,40
22,2, 89,2
36,57
6,3, 53,8
62,64
20,0, 74,9
39,21
19,7, 70,9
Среднее
СО
4161212,0
6203131,92
5159152,1
4269186,29
4779645,9
2974257,92
5027822,8
2286762,41
6303903,1
4913744,09
Среднее
СО
2080,61
3101,566
2579,58
2134,593
1194,91
743,564
2513,91
1143,381
1575,98
1228,436
Среднее
СО
0,0027
0,00266
0,0006
0,00041
0,0012
0,00099
0,0005
0,00027
0,0009
0,00056
Среднее
СО
0,106
0,0597
0,029
0,0095
0,035
0,0114
0,032
0,0133
0,046
0,0133
Примечание: Концентрация ДГЛК до начала введения была вычтена из последующих концентраций до расчета показателей; отрицательные значения были заменены нулевыми.
[00601] Скорректированные с учетом исходного уровня фармакокинетические показатели в стационарной фазе для свободной ДГЛК в плазме приведены в Табл. 24. Если коротко, в стационарной фазе, согласно оценкам на 28 день (0-24 часа), колебания от пика до минимума на графике (КПМ) были достаточно высокими для обеих дозовых когорт (среднее значение, 62,5% и 44,9%). Средняя СА была выше для дозовой когорты 2000 мг, чем для 4000 мг, как в показателях Cmax, так и AUC. Данные наводят на мысль о насыщаемой кинетике и/или изменениях в объеме распространения при повторном введении доз общей ДГЛК.
Таблица 24 Скорректированные с учетом исходных значений фармакокинетические показатели в стационарной фазе общей ДГЛК в плазме (множественные дозы, популяция фармакокинетики)
Уровень дозы DS107G
(n=6)
(n=6)
Медиана
Мин, Макс
1,50
0,0, 6,0
2,00
2,0, 24,0
Среднее
СО
37933,3
10890,12
59216,7
37002,29
Среднее
СО
0,0016
0,00051
0,0025
0,00107
Среднее
СО
0,0980
0,04161
0,1047
0,07339
Среднее
СО
56307,2
15793,88
77523,9
31682,79
Среднее
СО
62,50
31,403
44,90
38,967
Среднее
СО
5,156
2,2542
3,440
1,2090
Среднее
СО
7,991
5,9537
4,163
1,2331
[00602] Средние концентрации свободной ДГЛК в жидкости кожного волдыря приведены по когортам дозы на в Фиг. 14 (линейный график) и Фиг. 15 (логарифмически-линейный график). Средние концентрации возрастали практически вдвое при удвоении дозы (на основании концентраций в 1, 8, 14 и 28 дни) и аккумулировались при введении повторных доз в обоих схемах. Средние концентрации свободной ДГЛК в 28 день были приблизительно в 3 раза выше, чем показатели в 1 день для обеих групп 2000 и 4000 мг в сутки.
[00603] Средние концентрации общей ДГЛК в жидкости кожного волдыря приведены по дозовым когортам на Фиг. 16 (линейный график) и Фиг. 17 (логарифмически-линейный график). Средние концентрации общей ДГЛК возрастали приблизительно в 1,4 раза с удвоением дозы (на основании концентрации в 1, 8, 14 и 28 дни). Средние концентрации общей ДГЛК в 28 день были приблизительно в 2,5 и 3 раза выше, чем показатели 1 дня для обеих групп 2000 и 4000 мг в сутки, соответственно.
[00604] Профили концентрации в плазме и жидкости кожного волдыря накладывались, причем средние концентрации свободной ДГЛК в плазме и жидкости кожного волдыря были в некоторой степени подобными для одной и той же дозы DS107G в 8 день и 14 день (но не 28 день). Фиг. 18 [линейный график] и Фиг. 19 [логарифмически-линейный график]) наводят на мысль о том, что свободная ДГЛК распределяется в плазме и коже подобным образом.
[00605] В случае общей ДГЛК, средние концентрации в плазме были намного выше, чем в жидкости кожного волдыря для одной и той же дозы DS107G после 1 дня (Фиг. 20 [линейный график] и Фиг. 21 [логарифмически-линейный график]), указывая на то, что общую ДГЛК проще обнаружить в плазме, чем в коже. Механизм ограниченного распространения общей ДГЛК в коже наиболее вероятно связан с более низким количеством липидов в коже, по сравнению с плазмой.
[00606] Концентрации дигидротестостерона (ДГТ) в плазме количественно определяли как исследовательскую конечную точку эффективности или биомаркер. Межсубъектная вариабельность данных концентрации была высокой, по данным СО в большинстве временных точек. Средние концентрации ДГТ в плазме приведены по когорте дозы на Фиг. 22 (линейный) и Фиг. 23 (логарифмически-линейный).
[00607] Ни один из образцов не содержал измеримых концентраций свободной 15-ГЭТрЕ; все концентрации были ниже нижнего предела количественного определения (НПКО, 100 нг/мл).
[00608] В ходе многодозового исследования не было зарегистрировано смертельных случаев. При введении здоровым добровольцам дозы 2000 или 4000 мг один раз в сутки на протяжении 28 дней подряд, DSI07G хорошо переносился, причем наихудшим ВЛПР была легкая или умеренная диарея (сообщалось о послаблении стула), относительно кратковременная. Большинство эпизодов диареи (частота 7/12 [43,8%] среди субъектов в группе активного лечения против 0/4 [0.0%] среди субъектов в контрольной группе плацебо) рассматривалось Исследователем, как возможно связанные с исследуемым лекарственным средством. Более высокая доля субъектов сообщала о диарее в группе 4000 мг (4/6 [66,7%]), чем в группе 2000 мг (3/6 [50%]). Частота ВЛПР среди субъектов в группе активного лечения была намного выше, чем среди субъектов в контрольной группе плацебо (11/12 [91,73%] субъектов, которые сообщили об общем количестве 52 ВЛПР против 1/4 [25,0%] субъектов, сообщивших об общем количестве 1 ВЛПР, соответственно). Не было зарегистрировано тяжелых ВЛПР, и, за исключением ВЛПР в виде диареи, все эпизоды рассматривались, как не связанные или маловероятно связанные с исследуемым лекарственным средством. Тошнота была вторым по распространенности зарегистрированным ВЛПР (10 эпизодов у 4/6 [66,7%] субъектов в группе лечения 4000 мг); 9/10 эпизодов тошноты были легкими, а один был умеренным по своей тяжести. За исключением диареи, все остальные из ВЛПР были зарегистрированы у 2 субъектов каждый (бронхит и назофарингит) или 1 субъекта каждый (боль в животе, астения, пирексия, повышенная КФК крови, повышенный C-реактивный белок (CРБ), сниженный уровень лейкоцитов в крови, головокружение, головная боль, кашель и гематома), причем большинство из них рассматривались Исследователем, как маловероятно связанные или не связанные с исследуемым лекарственным средством. Другие ВЛПР, которые рассматривались, как возможно связанные с исследуемым лекарственным средством, были боль в животе и астения (сообщалось о «слабости»), обе из которых по времени ассоциировались с эпизодами послабления стула.
[00609] В ходе многодозового исследования ни у одного пациента не наблюдалось клинически значимых отклонений основных показателей жизнедеятельности или ЭКГ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ДЕКСТРОМЕТОРФАНОВОЕ СОЕДИНЕНИЕ И ХИНИДИН ДЛЯ ЛЕЧЕНИЯ ВОЗБУЖДЕНИЯ ПРИ ДЕМЕНЦИИ | 2020 |
|
RU2838188C2 |
| БЫСТРОЕ КУПИРОВАНИЕ ДВИГАТЕЛЬНЫХ ФЛУКТУАЦИЙ ПРИ БОЛЕЗНИ ПАРКИНСОНА | 2014 |
|
RU2698330C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, ИХ КОМПОЗИЦИИ И ПРИМЕНЕНИЯ | 2017 |
|
RU2766155C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ 15-HEPE, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2737089C2 |
| СПОСОБЫ ЛЕЧЕНИЯ СИНДРОМА ШЕГРЕНА С ПРИМЕНЕНИЕМ ИНГИБИТОРА ТИРОЗИНКИНАЗЫ БРУТОНА | 2020 |
|
RU2824354C2 |
| ЛИКСИСЕНАТИД И МЕТФОРМИН ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА ТИПА 2 | 2012 |
|
RU2623023C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЭОЗИНОФИЛЬНОГО ЭЗОФАГИТА | 2017 |
|
RU2766577C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ХРОНИЧЕСКОЙ СПОНТАННОЙ КРАПИВНИЦЫ С ПРИМЕНЕНИЕМ ИНГИБИТОРА ТИРОЗИНКИНАЗЫ БРУТОНА | 2020 |
|
RU2818678C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, КОМПОЗИЦИИ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2731000C2 |
| АНТИТЕЛО К РЕЦЕПТОРУ IL-6 ДЛЯ ЛЕЧЕНИЯ ЮВЕНИЛЬНОГО ИДИОПАТИЧЕСКОГО АРТРИТА | 2020 |
|
RU2822089C2 |
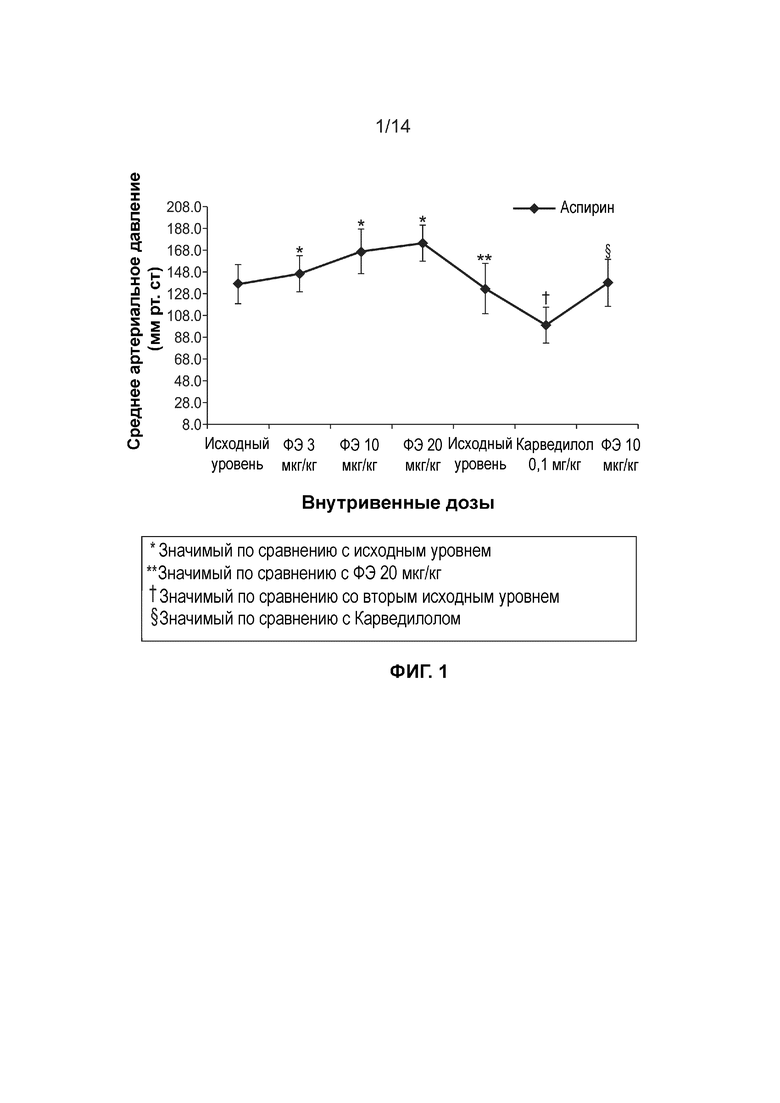
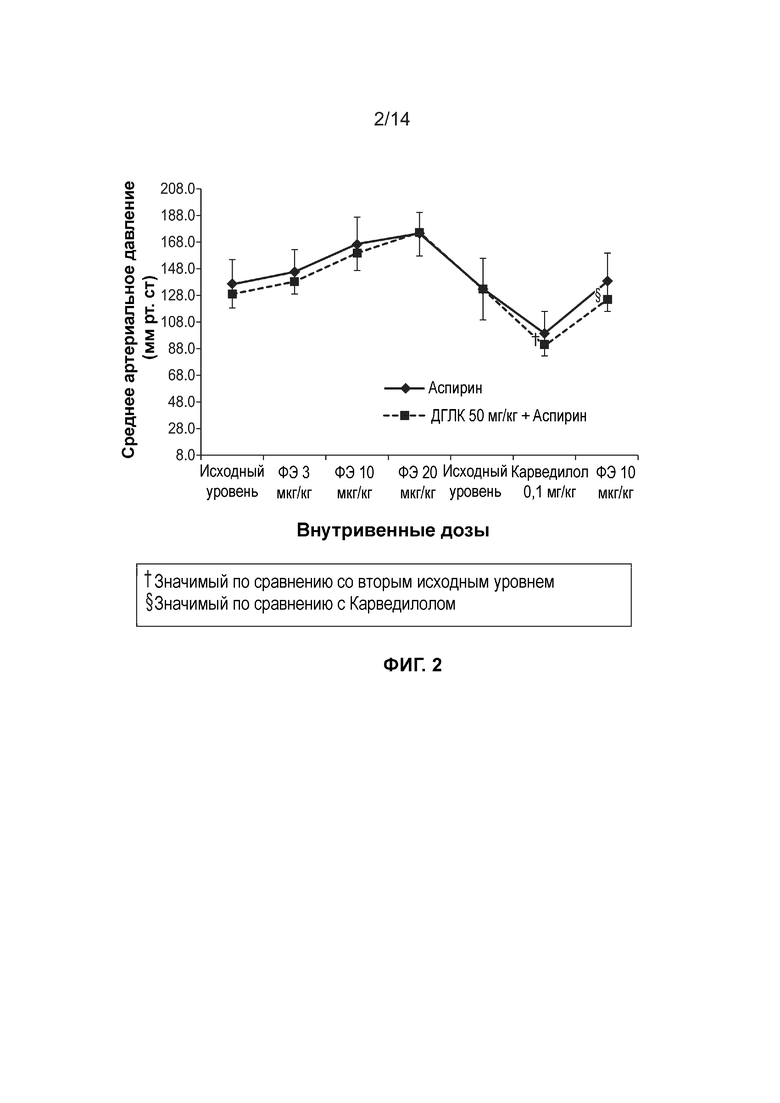
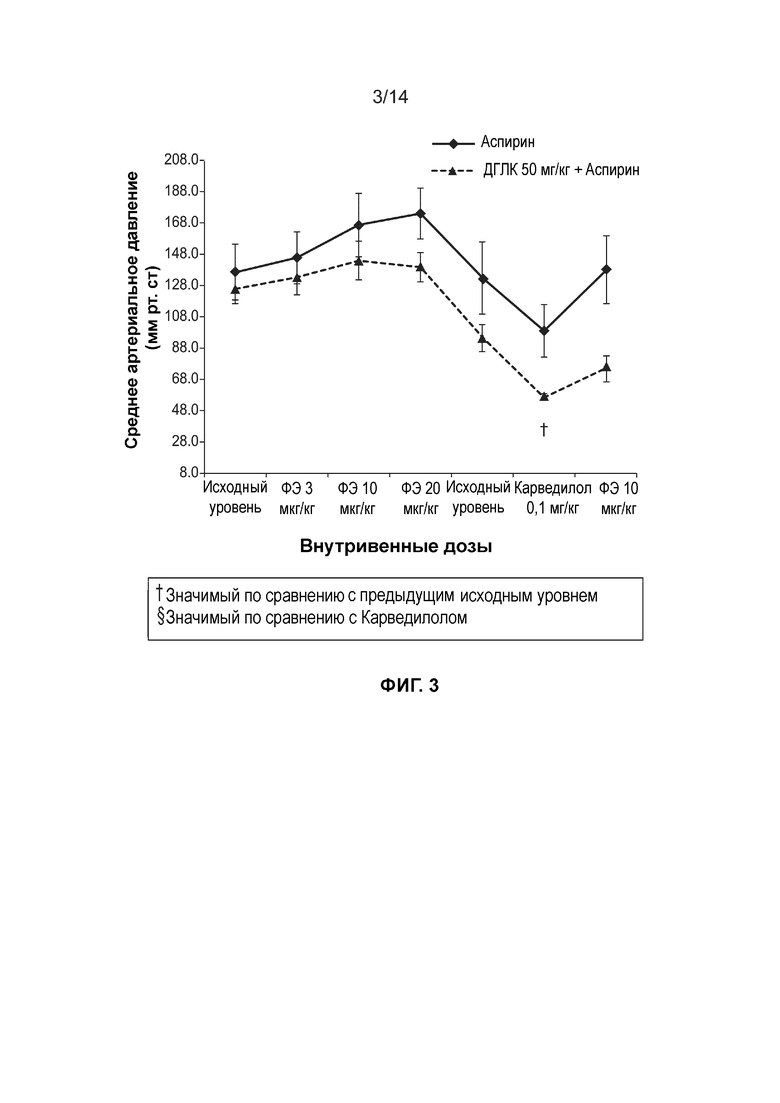
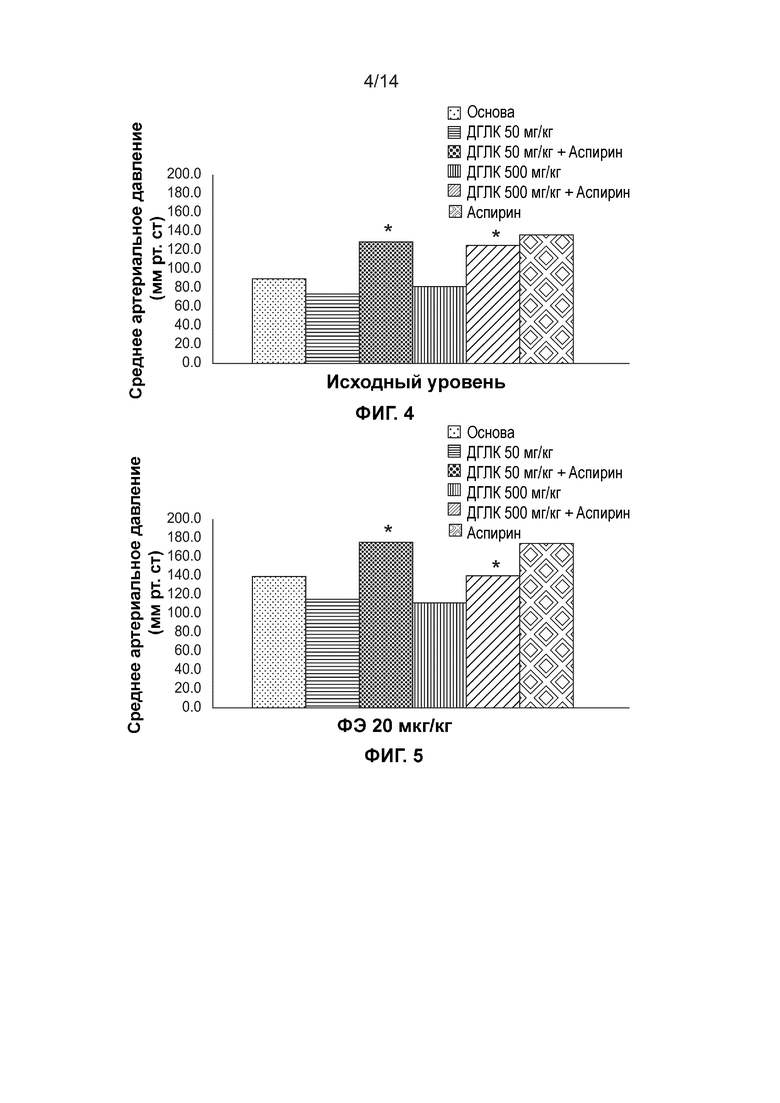
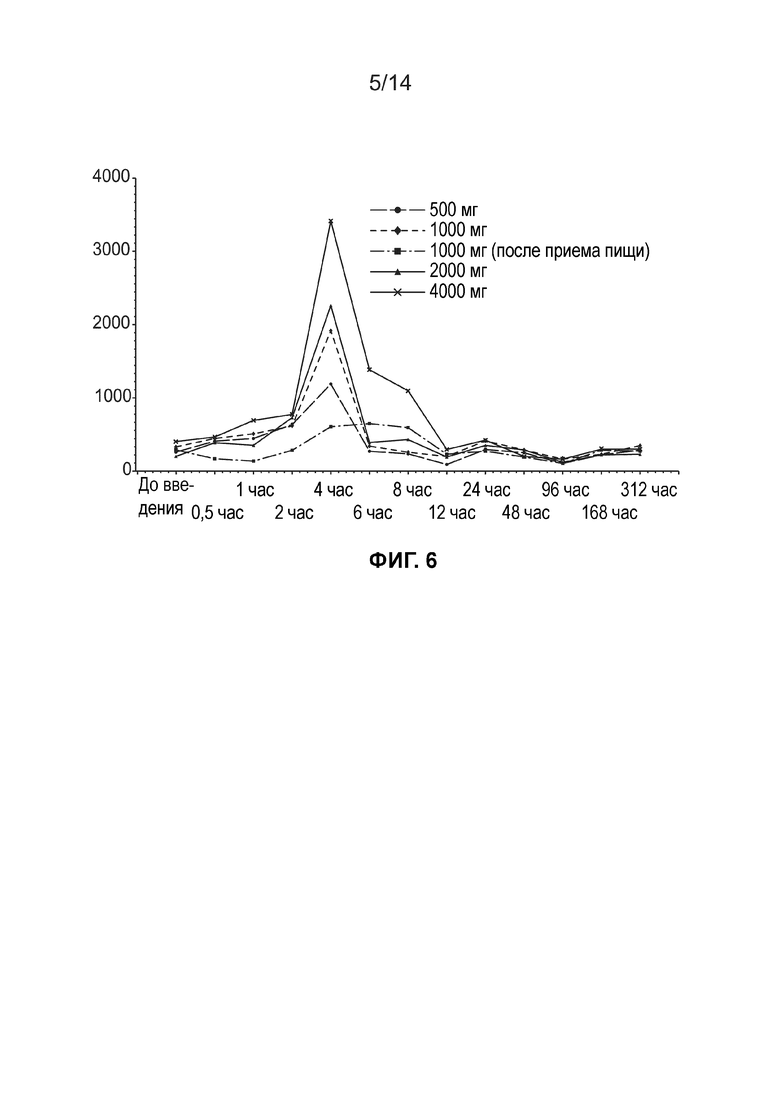
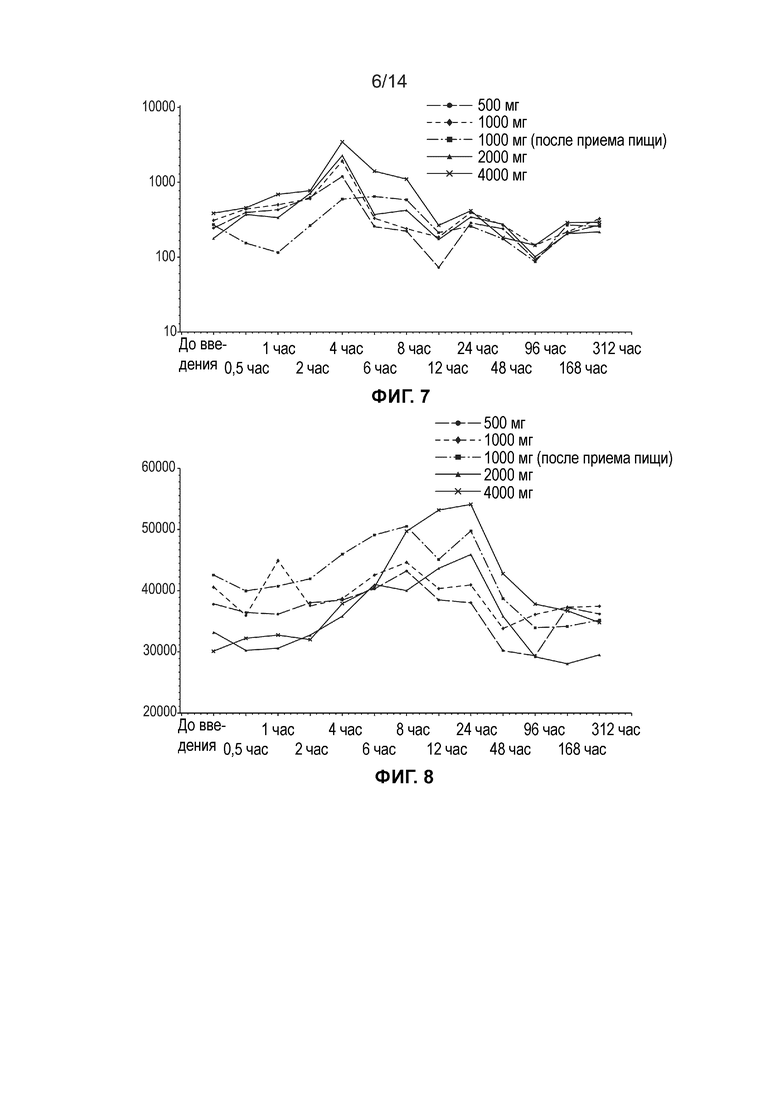
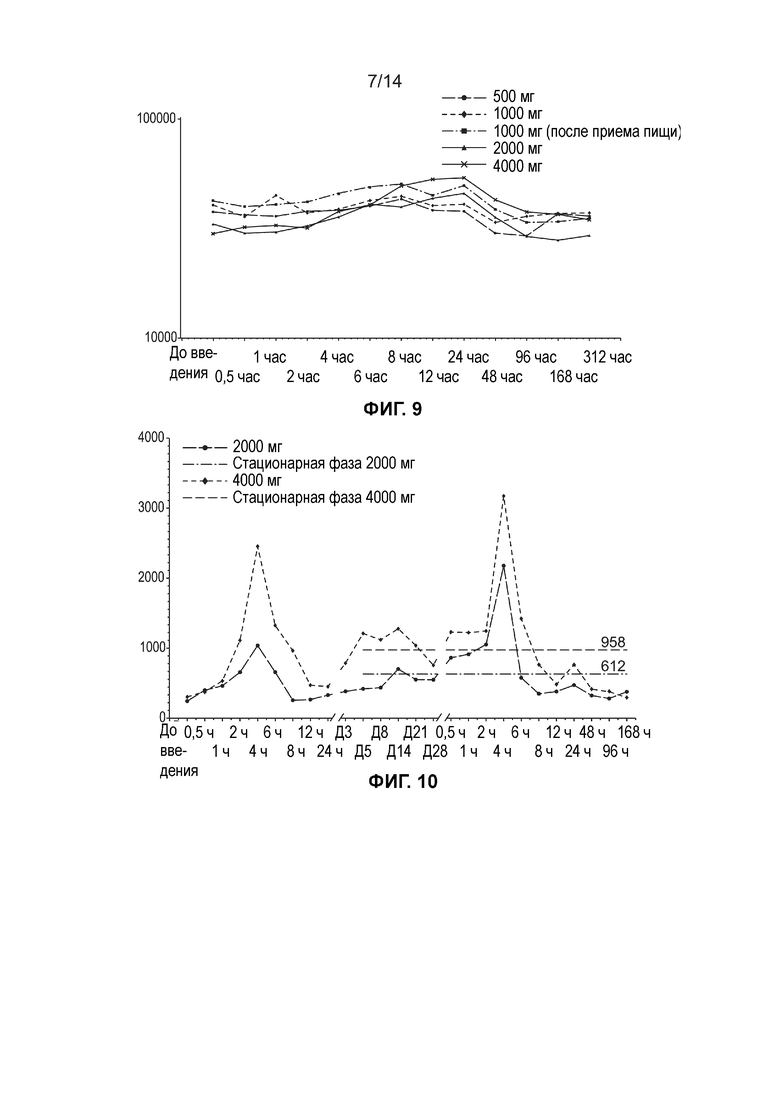
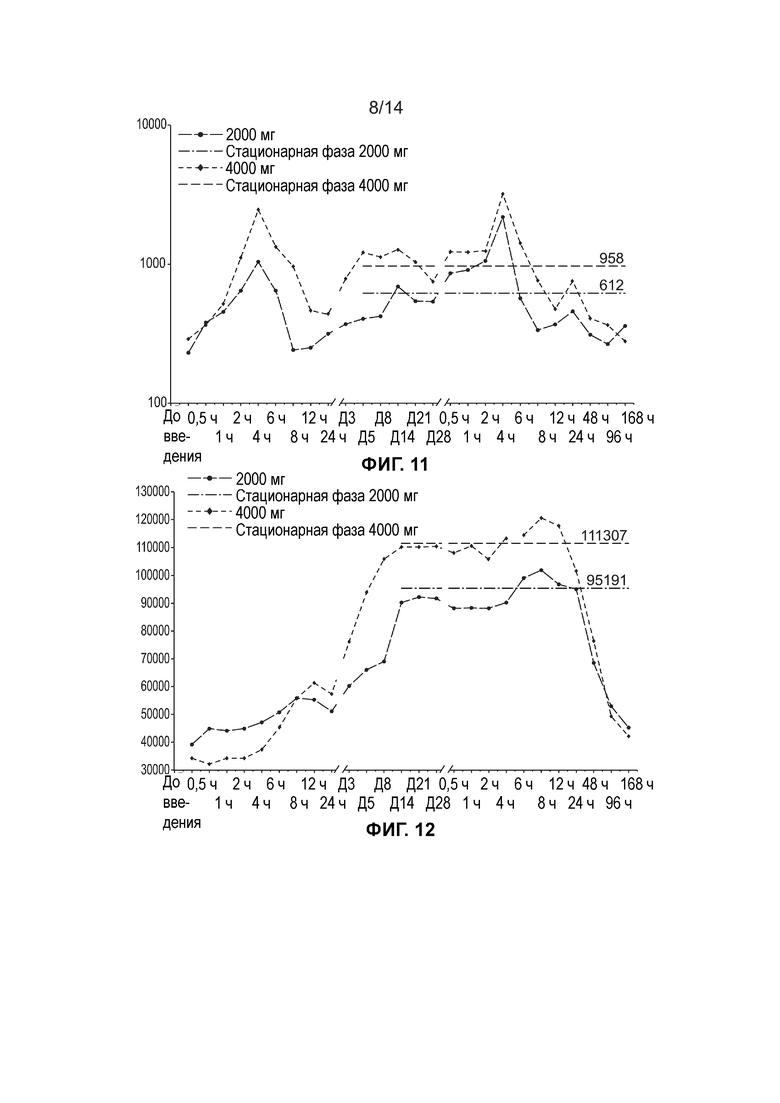
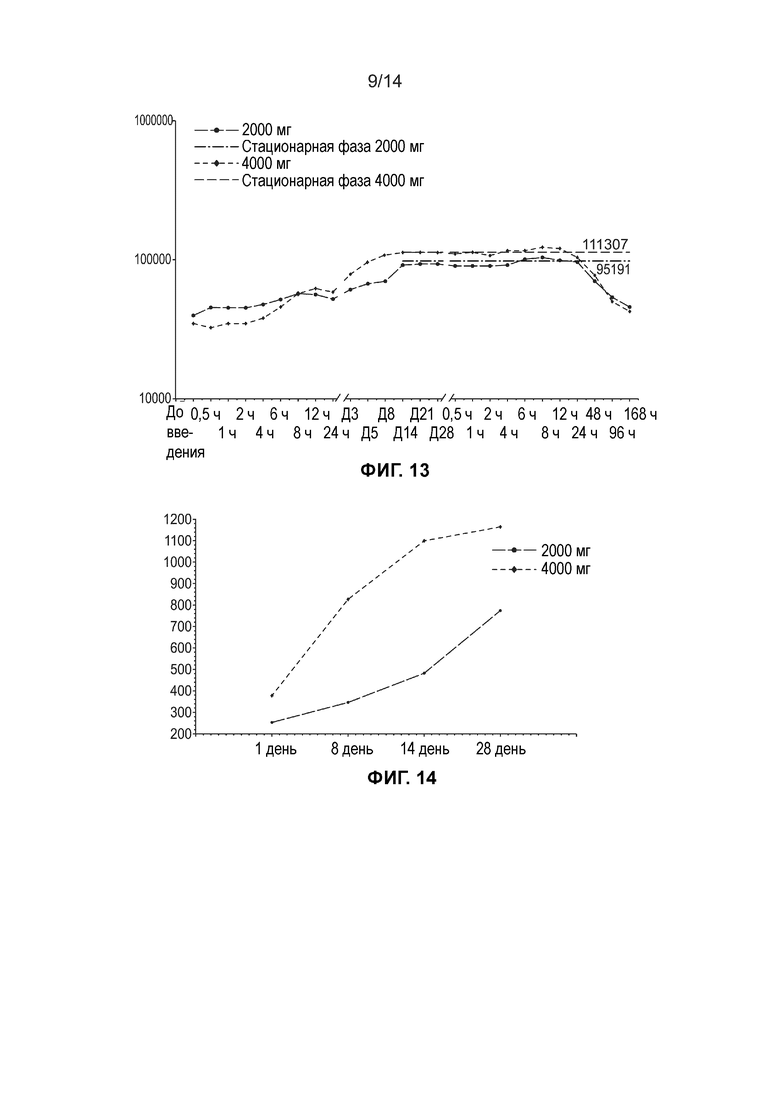
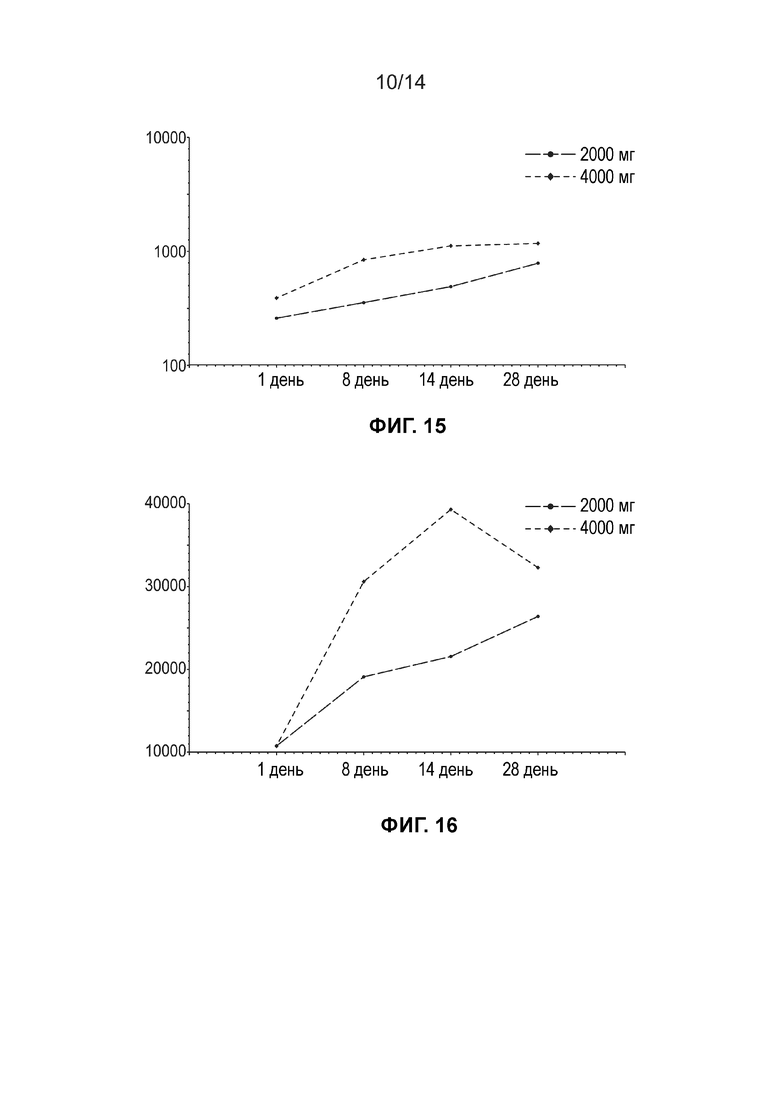
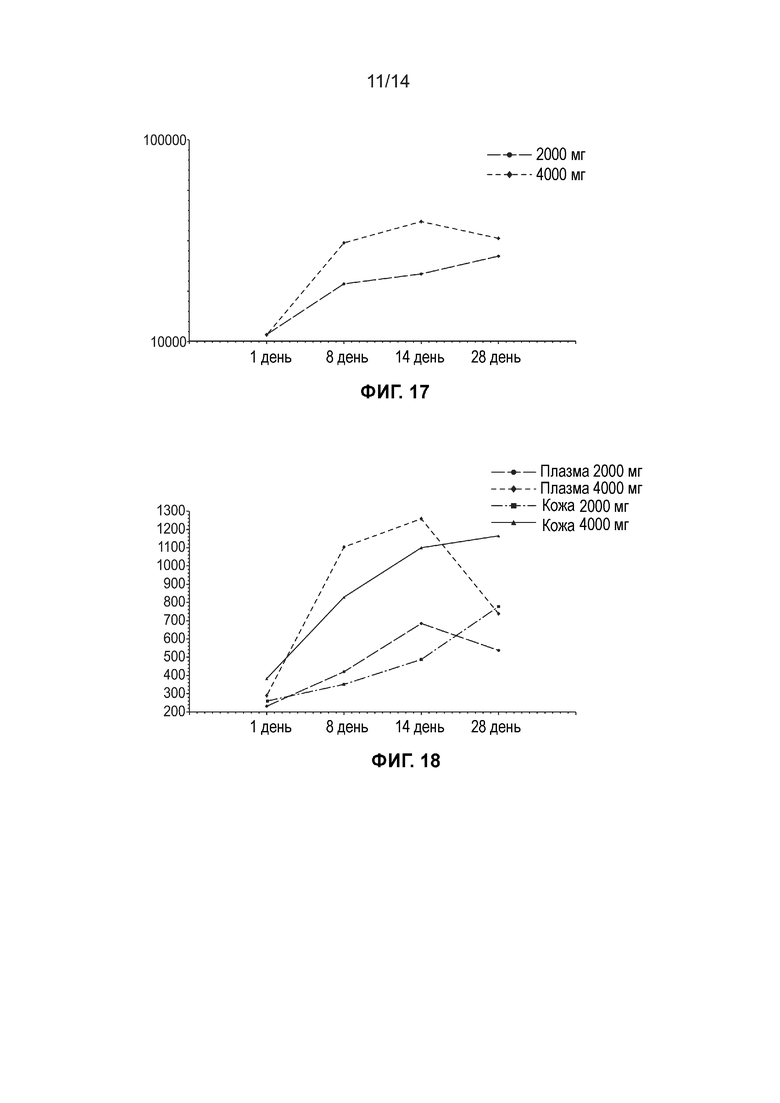
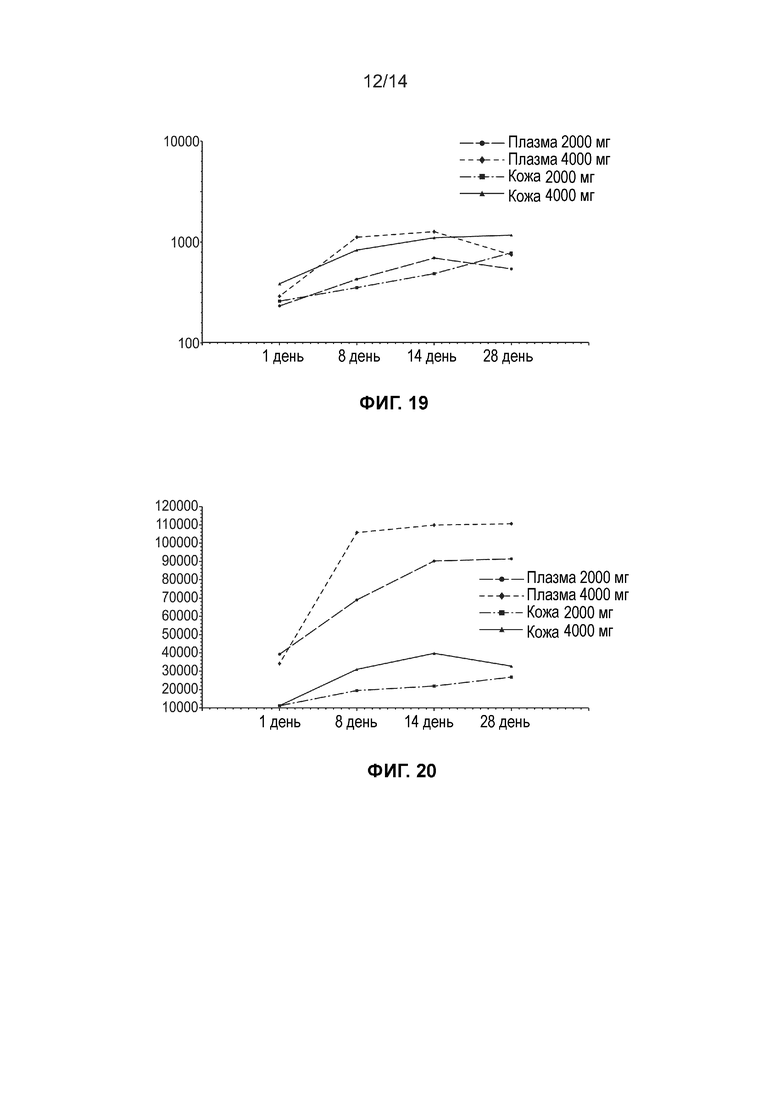
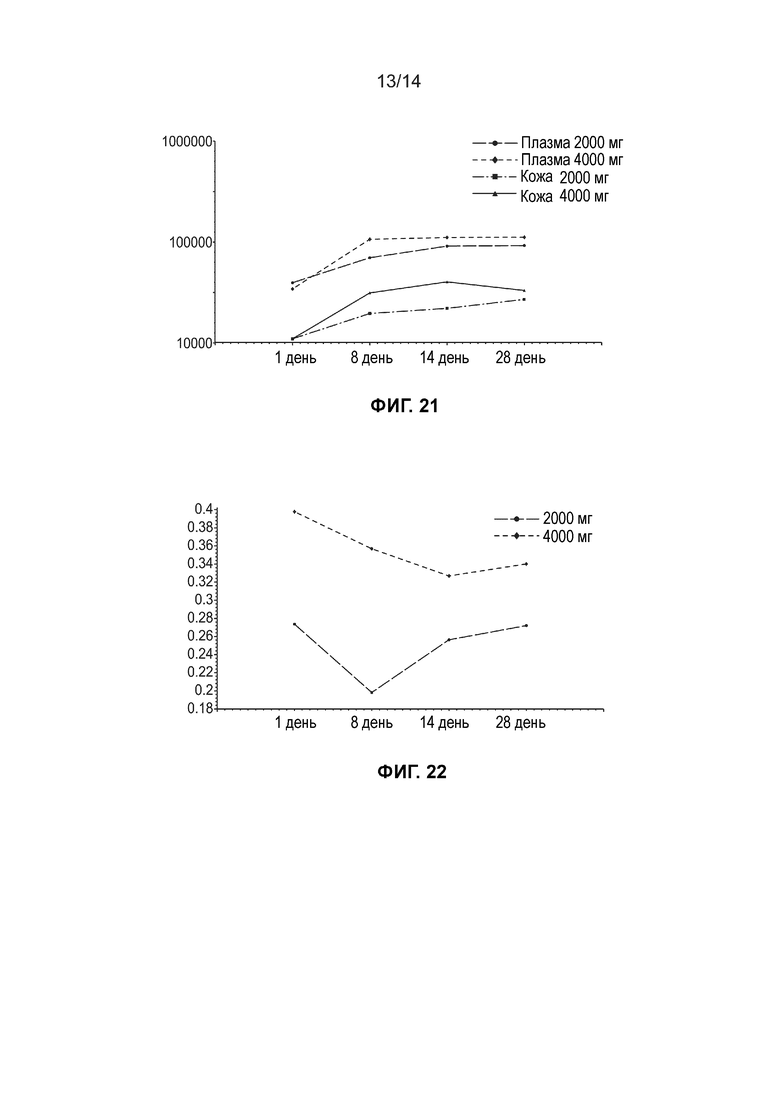
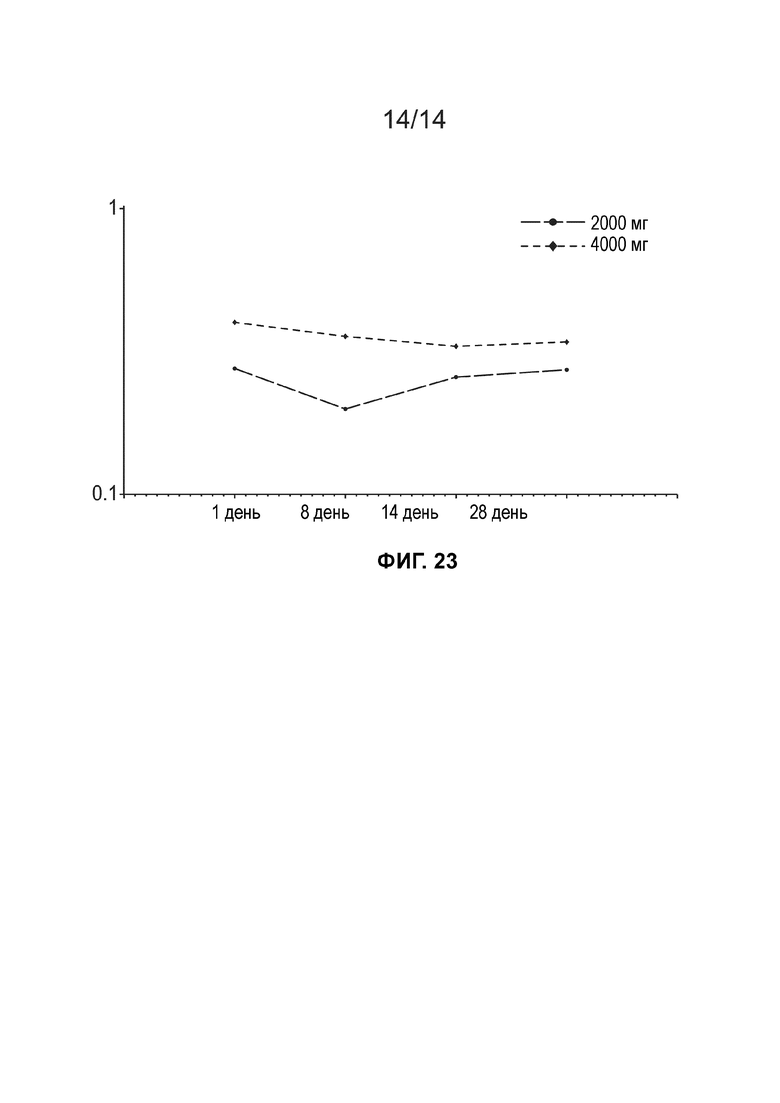
Описана фармацевтическая композиция для применения при лечении атопического дерматита. Фармацевтическая композиция содержит дигомогамма-линоленовую кислоту (DGLA) или алкиловый эфир DGLA, эйкозатетраеновую кислоту, эйкозатриеновую кислоту, олеиновую кислоту, линолевую кислоту и эйкозадиеновую кислоту, инкапсулированные в капсульную оболочку. DGLA или алкиловый эфир DGLA составляет по меньшей мере 95% по массе от общего количества жирных кислот в фармацевтической композиции. Указанная фармацевтическая композиция доставляется перорально. В капсульную оболочку инкапсулировано от 200 мг до 1 г DGLA или алкилового эфира DGLA. Фармацевтическая композиция вводится в количестве от 1 до 8 капсул в день. Капсульная оболочка содержит желатин и глицерин. Атопический дерматит представляет собой тяжелый атопический дерматит. Пероральное введение фармацевтической композиции по изобретению обеспечивает лечение атопического дерматита. 2 н. и 9 з.п. ф-лы, 23 ил., 24 табл., 4 пр.
1. Фармацевтическая композиция для применения при лечении атопического дерматита, где фармацевтическая композиция содержит дигомогамма-линоленовую кислоту (DGLA) или алкиловый эфир DGLA, эйкозатетраеновую кислоту, эйкозатриеновую кислоту, олеиновую кислоту, линолевую кислоту и эйкозадиеновую кислоту, инкапсулированные в капсульную оболочку, где DGLA или алкиловый эфир DGLA составляет по меньшей мере 95% по массе от общего количества жирных кислот в фармацевтической композиции, и где фармацевтическая композиция доставляется перорально.
2. Фармацевтическая композиция для применения при лечении атопического дерматита, где фармацевтическая композиция содержит дигомогамма-линоленовую кислоту (DGLA) или алкиловый эфир DGLA, эйкозатетраеновую кислоту, эйкозатриеновую кислоту, олеиновую кислоту, линолевую кислоту и эйкозадиеновую кислоту, где DGLA или алкиловый эфир DGLA составляет по меньшей мере 95% по массе от общего количества жирных кислот в фармацевтической композиции, и где фармацевтическая композиция доставляется перорально.
3. Фармацевтическая композиция по п. 2, где фармацевтическая композиция содержит капсульную оболочку и DGLA или алкиловый эфир DGLA, инкапсулированый в капсульную оболочку.
4. Фармацевтическая композиция по п. 1, где от 200 мг до 1 г DGLA или алкилового эфира DGLA инкапсулировано в капсульную оболочку.
5. Фармацевтическая композиция по п. 1 или 3, где фармацевтическая композиция вводится в количестве от 1 до 8 капсул в день.
6. Фармацевтическая композиция по п. 5, где каждая капсула содержит до 1 г DGLA или алкилового эфира DGLA.
7. Фармацевтическая композиция по любому из предшествующих пунктов, где фармацевтическая композиция содержит не более чем 5% любой индивидуальной жирной кислоты, выбранной из 20:2ω6 (эйкозадиеновая кислота), 20:3ω3 (эйкозатриеновая кислота), 20:4ω6 (арахидоновая кислота), 20:4ω3 (эйкозатетраеновая кислота) и 20:5ω3 (эйкозапентаеновая кислота), и не более чем 2% масс. неидентифицированных родственных примесей.
8. Фармацевтическая композиция по п. 5, где каждая капсула содержит от 200 мг до 1 г DGLA или алкилового эфира DGLA.
9. Фармацевтическая композиция для применения по любому из пп. 1 или 3-8, где капсульная оболочка содержит желатин и глицерин.
10. Фармацевтическая композиция для применения по п. 9, где желатин содержит обработанный известью костный желатин, а капсульная оболочка дополнительно содержит один или несколько из следующих компонентов: глицерин, очищенная вода, диоксид титана, триглицериды со средней длиной цепи и/или лецитин.
11. Фармацевтическая композиция по любому из предыдущих пунктов, где атопический дерматит представляет собой тяжелый атопический дерматит.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| СПОСОБ КОНТРОЛЯ ОТКЛЮЧЕНИЯ ВВОДНОГО ВЫКЛЮЧАТЕЛЯ ШИН И ЛОЖНОГО ОТКЛЮЧЕНИЯ ВВОДНОГО ВЫКЛЮЧАТЕЛЯ ТРАНСФОРМАТОРА ПОДСТАНЦИИ | 2012 |
|
RU2504061C1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| ТЕРАПЕВТИЧЕСКИЕ И ДИЕТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ НЕОБХОДИМЫЕ ЖИРНЫЕ КИСЛОТЫ И БИОЛОГИЧЕСКИ АКТИВНЫЕ ДИСУЛЬФИДЫ | 1998 |
|
RU2205004C2 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| RU 94002337 A1, 27.06.1996 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

