Родственная заявка
Настоящая заявка претендует на приоритет согласно 35 U.S.C. §119 от предварительной заявки на патент США No. 62/579,416, поданной 31 октября 2017 г., все содержание которой включено сюда путем ссылки.
Уровень техники
IgE играет центральную роль в опосредовании реакций гиперчувствительности I-го типа, которые вызывают аллергические заболевания, включая аллергическую астму, аллергический ринит, атопический дерматит и др. Аллергические реакции – это ответ иммунной системы на безвредные вещества окружающей среды типа пылевых клещей, пыльцы деревьев и трав, определенных продуктов и лекарств, а также на укусы пчел и огненных муравьев. При таких реакциях связывание аллергена с IgE на поверхности базофилов и тучных клеток вызывает сшивание IgE и агрегацию соответствующих рецепторов IgE.Fc: рецепторов IgE.Fc I-го типа или FcεRI. После агрегации рецепторов активируется сигнальный путь, ведущий к экзоцитозу гранул и высвобождению таких фармакологических медиаторов, как гистамин, лейкотриены, триптаза, цитокины и хемокины. Высвобождение этих медиаторов из тучных клеток и базофилов вызывает различные патологические проявления аллергии.
Существует два типа молекул IgE: свободный (или растворимый) IgE и мембраносвязанный IgE (mIgE). Свободные молекулы IgE циркулируют в крови и интерстициальной жидкости. mIgE экспрессируется на поверхности B-лимфобластов и B-клеток памяти. Считается, что воздействие на mIgE эффективно ингибирует выработку антиген-специфичного IgE и тем самым подавляет опосредованные IgE иммунные ответы.
Сущность изобретения
Настоящее изобретение основывается на неожиданном открытии того, что однократная доза антитела FB825, которое нацелено на домен CεmX в mIgE на клетках B-лимфоцитов человека, успешно снижает уровень общего IgE у человека на протяжении по меньшей мере трех месяцев.
Соответственно, в одном аспекте настоящего изобретения предусмотрен способ лечения связанных с IgE заболеваний, причем способ включает введение нуждающемуся в этом субъекту первой дозы антитела, связывающегося с доменом CεmX мембраносвязанного IgE; и введение субъекту второй дозы антитела. Вторая доза вводится по меньшей мере через 8 недель (например, по меньшей мере через 10 недель, 12 недель или 3 месяца) после первой дозы.
В любом из описанных здесь способов первая доза, вторая доза или обе могут быть в диапазоне от 0,5 мг/кг до 15 мг/кг (например, от 1 мг/кг до 15 мг/кг). Например, первая доза, вторая доза или обе дозы находятся в диапазоне от 1 мг/кг до 8 мг/кг (например, от 1,5 мг/кг до 10 мг/кг). Первая доза, вторая доза или обе могут вводиться посредством внутривенной инъекции.
Субъектами, подлежащими лечению описанным здесь способом, могут быть люди, страдающие или с подозрением на наличие заболевания, связанного с IgE, например, аллергической астмы, аллергического ринита, синдрома гипер-IgE или атопического дерматита. В некоторых воплощениях заболевание представляет собой вызванную простудой крапивницу, хроническую крапивницу, холинергическую крапивницу, хронический риносинусит, системный мастоцитоз, кожный мастоцитоз, аллергический бронхолегочный аспергиллёз, рецидивирующий идиопатический ангионевротический отек и интерстициальный цистит, связанные с эозинофилами желудочно-кишечные заболевания, пищевую аллергию или лекарственную аллергию.
В другом аспекте настоящего изобретения предусмотрен способ лечения атопического дерматита, включающий введение нуждающемуся в этом субъекту первой дозы антитела, связывающегося с доменом CεmX мембраносвязанного IgE; причем первая доза составляет примерное от 1 мг/кг до 10 мг/кг (например, от 3 мг/кг до 8 мг/кг, например, 5 мг/кг). Способ может дополнительно включать введение субъекту второй дозы антитела примерно через 3 месяца после первой дозы, если ко времени второй дозы изменение уровня общего IgE у субъекта составляет менее 50% от уровня общего IgE перед первой дозой. В некоторых случаях вторая доза может быть идентична первой дозе, например, 5 мг/кг. Первая доза антитела, вторая доза антитела или обе могут вводиться посредством внутривенной инфузии.
В любом из описанных выше способов субъекту наносят увлажняющий крем по меньшей мере два раза в день на протяжении по меньшей мере семи последовательных дней перед первой дозой. Или же способ может дополнительно включать введение субъекту местного кортикостероида. В некоторых случаях местный кортикостероид наносят на активный очаг ежедневно. Такой местный кортикостероид может представлять собой крем с 0,05% флутиказона пропионата, крем с 0,1% монетазона фуроата, мазь с 0,06% бетаметазона валерата или 1% гидрокортизона. В других случаях местный кортикостероид может представлять собой крем с 0,05% флуоцинонида, мазь с 0,25% дезоксиметазона или мазь с 0,05% клобетазола пропионата.
В некоторых воплощениях субъект не подвергается местному применению такролимуса, местному применению пимекролимуса, системному применению кортикостероидов, применению ингибиторов лейкотриенов, аллергеновой иммунотерапии, лечению, включающему иммунодепрессанты или иммуномодуляторы, применению вакцин, лечению при помощи традиционной китайской медицины, хирургическим процедурам, ультрафиолетовым процедурам или загару.
В любом из описанных здесь способов лечения антитело против CεmX, описанное здесь, может связываться с фрагментом mIgE GLAGGSAQSQRAPDRVL (SEQ ID NO: 1) или фрагментом mIgE GLAGGSAQSQRA (SEQ ID NO: 7). В некоторых случаях антитело связывается с тем же эпитопом, что и антитело 4B12 (FB825), или конкурирует с антителом FB825 за связывание с доменом CεmX мембраносвязанного IgE. В некоторых примерах антитело содержит те же определяющие комплементарность участки тяжелой цепи, что и антитело FB825, и/или те же определяющие комплементарность участки легкой цепи, что и антитело FB825. Таким антителом может быть гуманизованное антитело 4B12, к примеру, FB825. Антитело может содержать вариабельную область тяжелой цепи, имеющую аминокислотную последовательность SEQ ID NO: 2, и/или вариабельную область легкой цепи, имеющую аминокислотную последовательность SEQ ID NO: 3. Любое из антител, используемых в любом из описанных здесь способов, может представлять собой полноразмерное антитело или его антиген-связывающий фрагмент. Антитело может представлять собой человеческое антитело, гуманизованное антитело, химерное антитело или одноцепочечное антитело.
В любом из описанных здесь способов антитело против CεmX может входить в состав фармацевтической композиции, содержащей антитело, буфер (например, буфер, содержащий аминокислоту типа гистидина), соль (например, хлорид натрия) и неионогенное ПАВ (например, полисорбат 80). В некоторых воплощениях фармацевтическая композиция представляет собой водный раствор со значением рН от 5 до 8. В некоторых примерах антитело в фармацевтической композиции составляет от 10 мг/мл до 30 мг/мл (например, около 20 мг/мл), гистидиновый буфер имеет концентрацию 10-30 мМ (например, около 20 мМ), хлорид натрия имеет концентрацию около 120-160 мМ (например, около 140 мМ), а полисорбат 80 имеет концентрацию 0,01-0,03% (например, около 0,02%). Любые из описанных здесь фармацевтических композиций также входят в объем настоящего изобретения.
В другом аспекте изобретения предусмотрены водные составы, содержащие любое из описанных здесь антител против CεmX (например, FB825 или его функциональный вариант) в концентрации от 10 мг/мл до 30 мг/мл, буфер, содержащий аминокислоту (например, гистидин) в концентрации 10-30 мМ, соль (например, хлорид натрия) в концентрации 120-160 мМ, и неионогенное ПАВ (например, полисорбат 80) в концентрации 0,01-0,03%, причем водный состав имеет рН примерно 5-8. В одном примере водный состав содержит антитело в концентрации около 20 мг/мл, гистидиновый буфер в концентрации около 20 мМ, хлорид натрия в концентрации около 140 мМ, и полисорбат 80 в концентрации около 0,02%, причем водный состав имеет рН около 6,5.
Также в объем настоящего изобретения входят: (i) фармацевтические композиции для применения при лечении связанных с IgE заболеваний, как описано здесь, причем фармацевтические композиции содержат антитело против CεmX и фармацевтически приемлемый носитель, при этом фармацевтические композиции вводятся нуждающимся в лечении субъектам по меньшей мере в двух дозах, которые вводятся с интервалом по меньшей мере 8 недель (например, по меньшей мере 10 недель, 12 недель или трех месяцев либо с интервалом от 12 недель до 6 месяцев); и (ii) применение антител против CεmX для изготовления медикаментов для применения при лечении связанных с IgE заболеваний, причем медикамент может вводиться нуждающимся в лечении субъектам по меньшей мере в двух дозах, с интервалом по меньшей мере в три месяца.
Подробности одного или нескольких воплощений изобретения изложены в приведенном ниже описании. Другие признаки или преимущества настоящего изобретения станут очевидными из следующих чертежей и подробного описания некоторых воплощений, а также из прилагаемой формулы изобретения.
Краткое описание фигур
На фиг. 1 представлена диаграмма среднего (± SD) изменения от исходного уровня общего IgE по времени у субъектов, получавших однократную дозу FB825. Общий IgE определяли в образцах крови. Исходный уровень устанавливали по последнему непропущенному анализу (включая повторные и внеплановые анализы) перед введением исследуемого препарата.
На фиг. 2 представлена диаграмма среднего (± SD) изменения в процентах от исходного уровня общего IgE по времени у субъектов, получавших однократную дозу FB825. Общий уровень IgE определяли в образцах крови, взятых у субъектов. Исходный уровень устанавливали по последнему непропущенному анализу (включая повторные и внеплановые анализы) перед введением исследуемого препарата.
На фиг. 3 представлена диаграмма среднего (± SD) изменения от исходного уровня антител против лекарственного средства (ADA) по времени у субъектов, получавших однократную дозу FB825. ADA определяли в образцах крови, взятых у субъектов. Исходный уровень устанавливали по последнему непропущенному анализу (включая повторные и внеплановые анализы) перед введением исследуемого препарата.
На фиг. 4 представлено выравнивание аминокислотных последовательностей VH и VL моноклонального антитела 4B12 и моноклонального антитела FB825, которое является гуманизованным антителом 4B12. Выделены отличия между двумя антителами. Определяющие комплементарность участки VH и VL (CDR) выделены жирным шрифтом и подчеркнуты. VH 4B12: SEQ ID NO: 4; VL 4B12: SEQ ID NO: 5; VH FB825: SEQ ID NO: 2; VL FB825: SEQ ID NO: 3.
На фиг. 5 представлена диаграмма изменения в процентах от исходного уровня индекса площади и тяжести экземы (EASI) в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169 в открытом поисковом исследовании для оценки безопасности и эффективности FB825 у взрослых с атопическим дерматитом.
На фиг. 6 представлена диаграмма изменения в процентах от исходного уровня глобальной оценки исследователя (IGA) в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169 в открытом поисковом исследовании для оценки безопасности и эффективности FB825 у взрослых с атопическим дерматитом.
На фиг. 7 представлена диаграмма изменения в процентах от исходного уровня показателя степени тяжести атопического дерматита (SCORAD) в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169 в открытом поисковом исследовании для оценки безопасности и эффективности FB825 у взрослых с атопическим дерматитом.
На фиг. 8 представлена диаграмма изменения в процентах от исходного уровня зуда по визуально-аналоговой шкале (VAS) в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169 в открытом поисковом исследовании для оценки безопасности и эффективности FB825 у взрослых с атопическим дерматитом.
Раскрытие сущности изобретения
У индивидуумов с атопическими проявлениями имеющих повышенный риск возникновения аллергии, концентрация IgE в системе кровообращения может превышать нормальный уровень более чем в 10 раз. Концентрация аллерген-специфичных антител типа IgE тесно связана с клиническими симптомами и может быть в 1000 раз выше у больных аллергическими заболеваниями, чем у здоровых людей. Иммуноглобулин E сенсибилизирует эффекторные клетки, как-то базофилы, тучные клетки и активированные эозинофилы, занимая высокоаффинный рецептор IgE, FcεRI, который на них экспрессируется. При гиперчувствительности I-го типа аллергены поперечно сшивают молекулы IgE, связавшиеся с FcεRI, после чего запускают дегрануляцию эффекторных клеток, высвобождающих такие провоспалительные медиаторы, как гистамин и лейкотриены. Опосредованный IgE аллергический путь, который вызывает связанные с медиатором аллергические симптомы, запускает иммунную активность локально или системно. Базофилы и тучные клетки также выделяют целый ряд воспалительных цитокинов и хемокинов, которые не только прямо вызывают клинические симптомы, но также активируют и рекрутируют различные типы клеток для усиления воспалительного состояния. Следовательно, терапия против IgE может ослабить как опосредованный IgE путь, так и воспалительные состояния.
Здесь описаны антитела против CεmX, предназначенные для снижения общего уровня IgE и тем самым лечения опосредованных IgE заболеваний. Такие антитела можно вводить нуждающимся в лечении субъектам по меньшей мере двумя дозами, которые могут быть с интервалом по меньшей мере 8 недель (например, 10 недель, 12 недель или 3 месяца).
Антитела, способные связываться с доменом CεmX мембраносвязанного IgE
CεmX представляет собой сегмент из 52 аминокислот, расположенный между доменом CH4 и C-концевым заякоренным в мембране сегментом мембраносвязанной ε-цепи (mε) человека. Аминокислотная последовательность типичного фрагмента CεmX mIgE человека представлена ниже (SEQ ID NO: 6):
GLAGGSAQSQ RAPDRVLCHS GQQQGLPRAA GGSVPHPRCH CGAGRADWPG PP
Описанные здесь антитела могут связываться с доменом CεmX mIgE, к примеру, mIgE, экспрессированного на поверхности B-клеток. Такие антитела могут индуцировать клеточную гибель B-клеток, экспрессирующих mIgE, к примеру, посредством антителозависимой клеточной цитотоксичности и/или апоптоза клетки, тем самым устраняя B-клетки, что приведет к снижению продукции свободного IgE. Соответственно, антитела против CεmX, описанные здесь, могут снижать общий уровень IgE у субъекта (например, пациента), получающего антитело.
Антитело (взаимозаменяемо применяется и во множественном числе) представляет собой молекулу иммуноглобулина, способную специфически связываться с мишенью типа углевода, полинуклеотида, липида, полипептида и т.д., через по меньшей мере один сайт распознавания антигена, расположенный в вариабельной области молекулы иммуноглобулина. В настоящем изобретении термин “антитело” охватывает не только интактные (т.е. полноразмерные) поликлональные или моноклональные антитела, но также их антигенсвязывающие фрагменты (типа Fab, Fab′, F(ab′)2, Fv), одноцепочечные (scFv), их мутанты, слитые белки, содержащие часть антитела, гуманизованные антитела, химерные антитела, диатела, линейные антитела, одноцепочечные антитела, мультиспецифичные антитела (например, биспецифичные антитела) и любые другие модифицированные конфигурации молекул иммуноглобулина, содержащие сайт распознавания антигена с требуемой специфичностью, включая гликозилированные варианты антител, варианты аминокислотных последовательностей антител и ковалентно модифицированные антитела. Антитела включают антитела любого класса, как-то IgD, IgE, IgG, IgA или IgM (либо их подкласса), но антитело не обязательно должно относиться к какому-либо определенному классу. В зависимости от аминокислотной последовательности константного домена тяжелых цепей антител иммуноглобулины могут относиться к различным классам. Существует пять основных классов иммуноглобулинов: IgA, IgD, IgE, IgG и IgM, а некоторые из них могут еще подразделяться на подклассы (изотипы), например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2. Константные домены тяжелой цепи, которые соответствуют различным классам иммуноглобулинов, называются альфа, дельта, эпсилон, гамма и мю, соответственно. Субъединичные структуры и трехмерные конфигурации различных классов иммуноглобулинов хорошо известны.
Антитела для применения в описанных здесь способах могут быть мышиными, крысиными, человеческими или любого другого происхождения (включая химерные или гуманизованные антитела).
Любые из описанных здесь антител могут быть либо моноклональными, либо поликлональными. “Моноклональное антитело” относится к однородной популяции антител, а “поликлональное антитело” относится к гетерогенной популяции антител. Эти два термина не ограничивают источник антитела или способ его получения.
В одном примере антитела, используемые в описанных здесь способах, представляют собой гуманизованные антитела. Гуманизованные антитела означают такие формы антител не человека (например, мышиных), которые представляют собой специфические химерные иммуноглобулины, цепи иммуноглобулинов либо их антигенсвязывающие фрагменты, содержащие минимальную последовательность, происходящую из иммуноглобулина не человека. По большей части гуманизованные антитела представляют собой иммуноглобулины человека (антитела-реципиенты), в которых остатки из определяющих комплементарность участков (CDR) реципиента заменены остатками из CDR не человека (донорского антитела) типа мыши, крысы или кролика, обладающих требуемой специфичностью, аффинностью и ёмкостью. В некоторых случаях остатки каркасных участков (FR) Fv иммуноглобулина человека заменены соответствующими остатками не от человека. Кроме того, гуманизованные антитела могут содержать остатки, которые не встречаются ни в антителе-реципиенте, ни в импортированных CDR или каркасных последовательностях, но включены для дальнейшего совершенствования и оптимизации характеристик антител. В общем, гуманизованные антитела должны содержать практически все из по меньшей мере одного или обычно двух вариабельных доменов, в которых все или практически все участки CDR соответствуют таковым иммуноглобулина не человека, а все или практически все участки FR представлены консенсусными последовательностями иммуноглобулина человека. Оптимально гуманизованные антитела также должны содержать по меньшей мере часть константной области или домена (Fc) иммуноглобулина, как правило, иммуноглобулина человека. Антитела могут содержать участки Fc, модифицированные, как описано в WO 99/58572. Другие формы гуманизованных антител содержат один или несколько CDR (один, два, три, четыре, пять, шесть), измененных относительно исходного антитела, которые также именуются как один или несколько CDR, “происходящих из” одного или нескольких CDR исходного антитела. Гуманизованные антитела также могут включать созревание аффинности.
В другом примере описанные здесь антитела представляют собой химерные антитела, которые могут включать константную область тяжелой цепи и константную область легкой цепи антител человека. Химерные антитела означают такие антитела, которые содержат вариабельную область или часть вариабельной области от первого вида и константную область от второго вида. Как правило, в этих химерных антителах вариабельные области и легкой, и тяжелой цепи воспроизводят вариабельные области антител, происходящие из одного вида млекопитающих (например, других млекопитающих, чем человек, типа мыши, кролика и крысы), тогда как константные области гомологичны последовательностям антител, происходящим от других млекопитающих типа человека. В некоторых воплощениях могут проводиться аминокислотные модификации в вариабельной области и/или константной области.
В некоторых примерах описанные здесь антитела специфически связываются с доменом CεmX мембраносвязанного IgE, который может экспрессироваться на поверхности B-клеток. Термин антитело, которое “специфически связывается” с мишенью или эпитопом (применяются здесь взаимозаменяемо), хорошо известен в данной области, а способы определения такого специфического связывания также хорошо известны. Говорят, что молекула проявляет “специфическое связывание”, если она реагирует или связывается чаще, быстрее, с большей продолжительностью и/или с большим аффинностью с определенным целевым антигеном, чем с альтернативными мишенями. Антитело “специфически связывается” с целевым антигеном, если оно связывается с большим аффинностью, авидностью, более легко и/или с большей продолжительностью, чем оно связывается с другими веществами. Например, антитело, которое специфически (или предпочтительно) связывается с эпитопом домена CεmX, представляет собой антитело, которое связывает этот эпитоп домена CεmX с большим аффинностью, авидностью, более легко и/или с большей продолжительностью, чем оно связывается с другими эпитопами домена CεmX или эпитопами не домена CεmX. Из этого определения также понятно, к примеру, что антитело, которое специфически связывается с первым антигеном-мишенью, может не связываться специфически или предпочтительно со вторым антигеном-мишенью. При этом “специфическое связывание” или “предпочтительное связывание” не обязательно означает (хотя и может включать) исключительное связывание. Обычно, хотя и не обязательно, ссылка на связывание означает преимущественное связывание.
Аффинность связывания описанных здесь антител против CεmX может составлять менее 100 нМ, например, менее 50 нМ, менее 10 нМ, менее 1 нМ, менее 500 пМ, менее 100 пМ или менее 50 пМ вплоть до 2 пМ. Аффинность связывания может выражаться в виде KD или константы диссоциации, а повышение аффинности связывания соответствует снижению KD. Одним из способов определения аффинности связывания антител к CεmX является измерение аффинности связывания монофункциональных Fab-фрагментов антител. Для получения монофункциональных Fab-фрагментов можно расщепить антитело (например, IgG) папаином или экспрессировать рекомбинантным путем. Аффинность Fab-фрагмента антитела против CεmX можно определить методом поверхностного плазмонного резонанса (система поверхностного плазмонного резонанса (SPR) BIAcore3000, BIAcore, Inc., Piscaway, N.J.). Получают скорости кинетической ассоциации (kon) и скорости диссоциации (koff) (обычно измеряют при 25°C); и рассчитывают значения равновесной константы диссоциации (KD) в виде koff/kon.
В некоторых воплощениях антитела связываются с доменом CεmX IgE человека и практически не связываются с IgE из другого вида млекопитающих. В некоторых воплощениях антитела связываются с IgE человека, а также с одним или несколькими IgE из других видов млекопитающих. Эпитопы, с которыми связываются антитела, могут быть непрерывными или прерывистыми.
В некоторых воплощениях описанные здесь антитела против CεmX связываются с N-концевой часть домена CεmX, например, GLAGGSAQSQRAPDRVL (SEQ ID NO: 1) или GLAGGSAQSQRA (SEQ ID NO: 7). Такие антитела могут иметь такие же CDR тяжелой цепи и/или легкой цепи, что и антитело 4B12/FB825, приведенное на фиг.4. Также см. U.S. Patent No. 8,460,664, соответствующее содержание которого включено сюда посредством ссылки. Антитело против CεmX может представлять собой гуманизованное антитело 4B12 (например, FB825). В некоторых примерах антителом против CεmX для применения в описанных здесь способах является FB825, которое представляет собой гуманизованное антитело 4B12 (фиг. 4) или его функциональный вариант. Также см. U.S. Patent No. 8,460,664, соответствующее содержание которого включено сюда посредством ссылки.
Функциональные варианты (эквиваленты) FB825 обладают практически такой же специфичностью связывания эпитопа, что и FB825, и проявляют практически такую же биоактивность, что и FB825, включая активность устранения B-клеток, экспрессирующих mIgE, и снижение уровня общего IgE у субъекта. В некоторых воплощениях функциональные варианты FB825 содержат такие же участки/остатки, ответственные за связывание антигена, что и FB825, как-то такие же определяющие специфичность остатки в CDR или же целые CDR. В других воплощениях функциональные варианты FB825 содержат цепь VH, которая включает CDR1 VH, CDR2 VH и CDR3 VH, которые по меньшей мере на 75% (например, 80%, 85%, 90%, 95% или 98%) идентичны соответствующим CDR VH у FB825, и цепь VL, которая включает CDR1 VL, CDR2 VL и CDR3 VL, которые по меньшей мере на 75% (например, 80%, 85%, 90%, 95% или 98%) идентичны соответствующим CDR VL у FB825. Например, функциональный вариант FB825 может содержать цепь VH, которая включает до 5 (например, 1, 2, 3, 4 или 5)вариаций аминокислотных остатков в участках CDR VH (CDR1, CDR2 и/или CDR3 VH в целом) по сравнению с CDR VH mAb7E, и/или цепь VL, которая включает до 5 (например, 1, 2, 3, 4 или 5) вариаций аминокислотных остатков в участках CDR VL (CDR1, CDR2 и/или CDR3 VL в целом) по сравнению с CDR VL mAb7E.
В качестве альтернативы функциональный вариант FB825 содержит цепь VH, которая по меньшей мере на 75% (например, 80%, 85%, 90%, 95% или 98%) идентична цепи VH у FB825, и цепь VL, которая по меньшей мере на 75% ( например, 80%, 85%, 90%, 95% или 98%) идентична цепи VL у FB825. Вариации аминокислотной последовательности могут встречаться только в одном или нескольких каркасных участках VH и/или VL.
“Процент идентичности” двух аминокислотных последовательностей определяют с помощью алгоритма Karlin and Altschul, Proc. Natl. Acad. Sci. USA 87: 2264-68, 1990, модифицированного как в Karlin and Altschul, Proc. Natl. Acad. Sci. USA 90: 5873-77, 1993. Такой алгоритм встроен в программы NBLAST и XBLAST (версия 2.0) по Altschul et al., J. Mol. Biol. 215: 403-10, 1990. Поиск белков по BLAST может проводиться с помощью программы XBLAST, счет = 50, длина слов = 3, получая аминокислотные последовательности, гомологичные данным белковым молекулам. Если существуют разрывы между двумя последовательностями, можно использовать Gapped BLAST, как описано в Altschul et al., Nucleic Acids Res. 25(17): 3389-3402, 1997. При использовании программ BLAST и Gapped BLAST можно использовать параметры по умолчанию соответствующих программ (например, XBLAST или NBLAST).
Получение антител
Антитела, способные связываться с доменом CεmX мембраносвязанного IgE, как описано здесь, могут быть получены любым способом, известным в данной области. К примеру, см. Harlow and Lane (1988) Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, New York.
В некоторых воплощениях антитела, специфичные к целевому антигену (например, к CεmX mIgE типа mIgE человека), могут быть получены по стандартной гибридомной технологии. Для получения антител, связывающихся с таким антигеном, можно использовать полноразмерный целевой антиген или его фрагмент, необязательно связанный с белком-носителем типа KLH, для иммунизации животного-хозяина. Способы и графики иммунизации животного-хозяина обычно соответствуют общепринятым и стандартным методам стимуляции и получения антител, как дополнительно описано здесь. Общие методики получения мышиных, гуманизованных и человеческих антител известны в данной области и описаны здесь. Предполагается, что можно работать с любым субъектом-млекопитающим, включая человека, или с клетками, продуцирующими антитела, чтобы они служили основой для продуцирования линий клеток гибридомы млекопитающего, включая человека. Как правило, животным-хозяевам вводят некоторое количество иммуногена внутрибрюшинно, внутримышечно, перорально, подкожно, интраплантарно и/или внутрикожно, в том числе, как описано здесь.
Гибридомы могут быть получены из лимфоцитов или иммортализованных клеток миеломы по общей методике гибридизации соматических клеток Kohler B. and Milstein C. (1975) Nature 256: 495-497 или в модификации Buck DW et al., In Vitro, 18: 377-381 (1982). Для гибридизации можно использовать доступные линии миеломы, в том числе, без ограничения, X63-Ag8.653 и из Salk Institute, Cell Distribution Center, San Diego, Calif., США. Обычно методика включает слияние клеток миеломы и лимфоидных клеток с помощью такого фузогена, как полиэтиленгликоль, или с помощью электрических средств, хорошо известных специалистам. После слияния клетки отделяют от среды для слияния и культивируют в селективной среде роста типа среды гипоксантин-аминоптерин-тимидин (HAT), чтобы устранить негибридизованные исходные клетки. Для культивирования гибридом, секретирующих моноклональные антитела, можно использовать любые из описанных здесь сред с добавлением сыворотки или без нее. В качестве другой альтернативы методике слияния клеток для получения моноклональных антител против CεmX по настоящему изобретению можно использовать иммортализованные B-клетки EBV. Гибридомы размножают и субклонируют, если нужно, а супернатанты анализируют на активность против иммуногена стандартными методами иммуноанализа (например, радиоиммуноанализа, ферментного иммуноанализа или флуоресцентного иммуноанализа).
Гибридомы, которые можно использовать в качестве источника антител, охватывают все производные и потомство клеток исходных гибридом, которые вырабатывают моноклональные антитела, способные связываться с доменом CεmX. Гибридомы, которые вырабатывают такие антитела, можно культивировать in vitro или in vivo по известным методикам. Моноклональные антитела можно выделить из культуральной среды или биологических жидкостей по стандартным методикам очистки иммуноглобулинов, таким как осаждение сульфатом аммония, гель-электрофорез, диализ, хроматография и ультрафильтрация, если нужно. Нежелательную активность, если она есть, можно удалить, к примеру, пропусканием препарата через адсорбенты, составленные из иммуногена, прикрепленного к твердой фазе, с элюированием или высвобождением требуемых антител из иммуногена. При иммунизации животных-хозяев целевым антигеном или фрагментом, содержащим целевую аминокислотную последовательность, конъюгированную с белком, который является иммуногенным для иммунизируемого вида, например, гемоцианином морского блюдечка, сывороточным альбумином, бычьим тироглобулином или соевым ингибитором трипсина с помощью бифункционального или дериватизирующего реагента, к примеру, малеимидобензоилсульфосукцинимидного эфира (конъюгирование через остатки цистеина), N-гидроксисукцинимида (через остатки лизина), глутаральдегида, янтарного ангидрида, SOCl или R1N=C=NR, где R и R1 означают различные алкильные группы, можно получить популяцию антител (например, моноклональных антител).
Если нужно, представляющее интерес антитело (моноклональное или поликлональное) (например, вырабатываемое гибридомой) можно секвенировать, а затем клонировать полинуклеотидную последовательность в вектор для экспрессии или размножения. Последовательность, кодирующая данное антитело, может находиться в векторе в клетке-хозяине, а затем клетки -хозяева можно размножить и заморозить для использования в будущем. С другой стороны, полинуклеотидную последовательность можно использовать для генетической манипуляции с целью “гуманизации” антитела либо для улучшения аффинности (созревания аффинности) или других характеристик антитела. Например, можно подвергнуть инженерии константную область так, чтобы она больше походила на константную область человека, чтобы избежать иммунного ответа при использовании антитела в клинических испытаниях и при лечении людей. Может понадобиться генетически изменить последовательность антитела для получения большей аффинности к целевому антигену и более эффективного снижения общего IgE. Специалистам должно быть ясно, что в антитело можно вносить изменения одного или нескольких полинуклеотидов и при этом сохранить его специфичность связывания с целевым антигеном.
В других воплощениях можно получить полностью человеческие антитела с использованием коммерчески доступных мышей, подвергнутых инженерии для экспрессии специфических белков иммуноглобулина человека. Также для получения гуманизованных антител или человеческих антител можно использовать трансгенных животных, разработанных для получения более желательных (например, полностью человеческих) антител или более сильного иммунного ответа. Примеры таких технологий: Xenomouse™ от Amgen, Inc. (Fremont, CA), HuMAb-Mouse™ и TC Mouse™ фирмы Medarex, Inc. (Princeton, N.J.). С другой стороны, антитела можно получать рекомбинантным путем по технологии фагового дисплея. Например, см. U.S. Pat. Nos. 5,565,332; 5,580,717; 5,733,743; 6,265,150; а также Winter et al. (1994) Annu. Rev. Immunol. 12:433-455. К тому же технологию фагового дисплея можно использовать (McCafferty et al. (1990) Nature 348: 552-553) для получения человеческих антител и фрагментов антител in vitro из репертуара генов вариабельного (V) домена иммуноглобулина от неиммунизированных доноров.
Антигенсвязывающие фрагменты интактных антител (полноразмерных антител) можно получить обычными методами. Например, F(ab′)2-фрагменты можно получить путем расщепления пепсином молекул антитела, а Fab-фрагменты можно получить путем восстановления дисульфидных мостиков у F(ab′)2-фрагментов.
Генно-инженерные антитела типа гуманизованных антител, химерных антител, одноцепочечных антител и биспецифичных антител можно получить, например, по стандартной рекомбинантной технологии. В одном примере можно легко выделить и секвенировать ДНК, кодирующую моноклональные антитела, специфичные к целевому антигену, по стандартной методике (например, с помощью олигонуклеотидных зондов, способных специфически связываться с генами, кодирующими тяжелые и легкие цепи моноклональных антител). Предпочтительным источником такой ДНК служат клетки гибридомы. После выделения можно вставить ДНК в один или несколько экспрессирующих векторов, а затем трансфецировать ими клетки-хозяева типа клеток E. coli, обезьяньих клеток COS, клеток яичников китайского хомячка (CHO) или клеток миеломы, которые иначе не вырабатывают белок иммуноглобулина, чтобы провести синтез моноклональных антител в рекомбинантных клетках-хозяевах. Например, см. PCT Publication WO 87/04462. Затем можно модифицировать ДНК, к примеру, подставляя кодирующую последовательность для константных доменов тяжелой и легкой цепи человека вместо гомологичных последовательностей мыши, см. Morrison et al. (1984) Proc. Nat. Acad. Sci. 81:6851, или путем ковалентного присоединения к кодирующей последовательности иммуноглобулина всей или части кодирующей последовательности для неиммуноглобулинового полипептида. При этом можно получить генно-инженерные антитела типа “химерных” или “гибридных” антител, обладающих специфичностью связывания с целевым антигеном.
Методы, разработанные для получения “химерных антител”, хорошо известны в данной области. Например, см. Morrison et al. (1984) Proc. Natl. Acad. Sci. USA 81, 6851; Neuberger et al. (1984) Nature 312, 604; и Takeda et al. (1984) Nature 314:452.
Методы получения гуманизованных антител также хорошо известны в данной области. Например, см. Queen et al., Proc. Natl. Acad. Sci. USA, 86: 10029-10033 (1989). В одном примере проводят анализ вариабельных областей VH и VL исходного антитела не человека методом трехмерного молекулярного моделирования известными способами. Затем тем же методом трехмерного молекулярного моделирования идентифицируют те каркасные аминокислотные остатки, которые должны быть важными для формирования правильных структур CDR. Параллельно этому идентифицируют цепи VH и VL человека, имеющие аминокислотные последовательности, гомологичные последовательностям исходного антитела не от человека, из любой базы данных по генам антител, используя исходные последовательности VH и VL в качестве запросов для поиска. Затем отбирают акцепторные гены VH и VL человека.
Участки CDR в выбранных акцепторных генах человека можно заменить на участки CDR из исходного антитела не человека или его функциональных вариантов. При необходимости для замены соответствующих остатков в акцепторных генах человека можно использовать те остатки в каркасных участках исходной цепи, которые признаны важными для взаимодействия с участками CDR (см. описание выше).
Одноцепочечные антитела можно получать по рекомбинантной технологии путем соединения последовательности нуклеотидов, кодирующей вариабельную область тяжелой цепи, и последовательности нуклеотидов, кодирующей вариабельную область легкой цепи. Предпочтительно между двумя вариабельными областями вставляется гибкий линкер. С другой стороны, можно адаптировать методы, описанные для получения одноцепочечных антител (U.S. Patent Nos. 4,946,778 и 4,704,692), для получения фаговой библиотеки scFv и идентифицировать из неё обычными методами клоны scFv, специфичные к IgE.
Антитела, полученные известными и описанными здесь способами, можно охарактеризовать хорошо известными в данной области методами. Например, одним из методов является определение эпитопа, с которым связывается антиген, или “картирование эпитопа”. Существует много известных способов картирования и характеристики расположения эпитопов на белках, включая определение кристаллической структуры комплекса антитело-антиген, анализ конкуренции, анализ экспрессии генных фрагментов и методы на основе синтетических пептидов, как описано, к примеру, в главе 11, Harlow and Lane, Using Antibodies, a Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1999. В другом примере картирование эпитопа может использоваться для определения последовательности, с которой связывается антитело. Эпитоп может быть линейным эпитопом, т.е. содержаться в одной цепочке аминокислот, или конформационным эпитопом, образованным при трехмерном взаимодействии аминокислот, которые необязательно могут содержаться в одной цепочке (линейной последовательности первичной структуры). Можно выделить или синтезировать (например, рекомбинантно) пептиды различной длины (например, длиной по меньшей мере 4-6 аминокислот) и использовать для анализа связывания с антителом. В другом примере эпитоп, с которым связывается антитело, можно определить при систематическом скрининге с использованием перекрывающихся пептидов, полученных из последовательности антигена-мишени, и определения их связывания с антителом. При анализе экспрессии генных фрагментов открытую рамку считывания, кодирующую целевой антиген, фрагментируют случайным образом либо при помощи специфических генетических конструкций и определяют способность экспрессированных фрагментов антигена реагировать с исследуемым антителом. Генные фрагменты можно получить, к примеру, методом ПЦР, а затем транскрибировать и транслировать в белок in vitro в присутствии радиоактивных аминокислот. Затем определяют связывание антитела с мечеными радиоактивно фрагментами антигена методами иммунопреципитации и гель-электрофореза. Некоторые эпитопы также можно идентифицировать с использованием больших библиотек случайных последовательностей пептидов, экспонированных на поверхности фаговых частиц (фаговые библиотеки). С другой стороны, можно протестировать определенную библиотеку перекрывающихся пептидных фрагментов на связывание с исследуемым антителом простыми методами связывания. В другом примере можно провести мутагенез антигенсвязывающего домена, эксперименты по обмену доменов и сканирующий мутагенез по аланину для идентификации остатков, требуемых, достаточных и/или необходимых для связывания эпитопа. Например, можно провести эксперименты по обмену доменов с использованием мутантов целевого антигена, у которых различные фрагменты полипептида IgE были заменены (обменены) на последовательности из близкородственного, но отличающегося по антигенам белка (типа другого представителя семейства белков иммуноглобулинов). При определении связывания антитела с мутантным иммуноглобулином можно оценить важность определенного фрагмента антигена для связывания с антителом.
С другой стороны, можно провести анализ конкуренции с использованием других антител, которые связываются с тем же антигеном, чтобы определить, связывается ли антитело с тем же эпитопом, что и другие антитела. Конкурентные методы хорошо известны специалистам в данной области.
Фармацевтические композиции
Одно или несколько из описанных выше антител против CεmX можно смешать с фармацевтически приемлемым носителем (наполнителем), включающим буфер, получая фармацевтические композиции для применения при лечении заболеваний, связанных с IgE. “Приемлемый” означает то, что носитель должен быть совместимым с активным ингредиентом композиции (и предпочтительно способным стабилизировать активный ингредиент) и не причинять вреда подлежащему лечению субъекту. Фармацевтически приемлемые наполнители (носители), включающие буферы, хорошо известны в данной области. Например, см. Remington: The Science and Practice of Pharmacy, 20th Ed. (2000) Lippincott, Williams and Wilkins, Ed. K.E. Hoover. В одном примере фармацевтическая композиция содержит более одного антитела против CεmX, которые распознают различные эпитопы антигена-мишени.
Фармацевтические композиции для применения в настоящих способах могут содержать фармацевтически приемлемые носители, наполнители или стабилизаторы в виде лиофилизованных составов или водных растворов, см. Remington: The Science and Practice of Pharmacy, 20th Ed. (2000) Lippincott, Williams and Wilkins, Ed. K.E. Hoover. Приемлемые носители, наполнители или стабилизаторы являются нетоксичными для получателей при используемых дозировках и концентрациях и могут содержать буферы типа фосфатных, цитратных и других органических кислот; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (как-то октадецилдиметилбензиламмонийхлорид; гексаметонийхлорид; бензалконийхлорид, бензетонийхлорид; фенол, бутиловый или бензиловый спирт; алкилпарабены типа метил- или пропилпарабена; катехол; резорцинол; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее 10 остатков) полипептиды; такие белки, как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры типа поливинилпирролидона; такие аминокислоты, как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстраны; хелатообразующие агенты типа ЭДТА; такие сахара, как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы типа натрия; комплексы металлов (например, комплексы Zn-белок); и/или неионные поверхностно-активные вещества типа Tween™, Pluronics™ или полиэтиленгликоля (PEG). Фармацевтически приемлемые наполнители еще дополнительно описаны здесь.
В некоторых примерах описанные здесь фармацевтические композиции содержат липосомы, содержащие антитело против CεmX, которые могут быть получены способами, известными в данной области, типа описанных в Epstein et al., Proc. Natl. Acad. Sci. USA 82:3688 (1985); Hwang et al., Proc. Natl. Acad. Sci. USA 77:4030 (1980); и U.S. Pat. Nos. 4,485,045 и 4,544,545. Липосомы с повышенным временем циркуляции в кровотоке раскрыты в U.S. Pat. No. 5,013,556. Особенно полезные липосомы могут быть получены методом обратнофазового испарения с липидной композицией, содержащей фосфатидилхолин, холестерин и дериватизованный PEG фосфатидилэтаноламин (PEG-PE). Липосомы пропускают через фильтры с заданным размером пор, получая липосомы требуемого диаметра.
Антитела против CεmX также могут быть заключены в микрокапсулы, полученные, к примеру, методами коацервации или межфазной полимеризации, например, в микрокапсулы из гидроксиметилцеллюлозы или желатина и микрокапсулы из поли(метилметакрилата), соответственно, в коллоидные системы доставки лекарств (например, в липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие методы известны в данной области, например, см. Remington, The Science and Practice of Pharmacy, 20th Ed. Mack Publishing (2000).
В других примерах описанные здесь фармацевтические композиции могут быть составлены в формате замедленного высвобождения. Подходящие примеры препаратов с замедленным высвобождением включают полупроницаемые матриксы из твердых гидрофобных полимеров, содержащие антитело, причем эти матриксы имеют вид фасонных изделий, например, пленок или микрокапсул. Примеры матриксов с замедленным высвобождением включают полиэфиры, гидрогели (к примеру, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт)), полилактиды (U.S. Pat. No. 3,773,919), сополимеры L-глутаминовой кислоты и 7-этил-L-глутамата, неразлагаемый этиленвинилацетат, разлагаемые сополимеры молочной кислоты и гликолевой кислоты типа Lupron Depot™ (микросферы для инъекций, состоящие из сополимера молочной кислоты и гликолевой кислоты и лейпролидацетата), ацетат-изобутират сахарозы и поли-D-(-)-3-гидроксимасляную кислоту.
В некоторых воплощениях фармацевтические композиции, содержащие описанные здесь антитела против CεmX, например, FB825 или его функциональные варианты, которые тоже описаны здесь, могут представлять собой водные составы, которые также могут содержать буфер (который может содержать аминокислоту типа гистидина или аргинина), соль (например, хлорид натрия) и/или поверхностно-активное вещество типа неионогенного ПАВ. Например, водный состав может содержать антитело в концентрации 10-30 мг/мл, буфер, содержащий аминокислоту (например, гистидин или аргинин) в концентрации 10-30 мМ, ПАВ типа полисорбата 80 в концентрации 0,01-0,03% и/или хлорид натрия в концентрации 120-160 мМ. Такой водный состав может иметь рН от 5 до 8. В одном конкретном примере водный состав может содержать антитело FB825 в концентрации около 20 мг/мл, L-гистидин в концентрации около 20 мМ, хлорид натрия в концентрации около 140 мМ, полисорбат 80 в концентрации около 0,02% и рН около 6,5.
Термин “около” или “примерно” означает в пределах допустимого диапазона ошибок для определенного значения при определении рядовым специалистом в данной области, который будет частично зависеть от того, как измеряется или определяется это значение, то есть ограничения измерительной системы. Например, “примерно” может означать в пределах допустимого стандартного отклонения, принятого на практике в данной области. С другой стороны, “примерно” может означать диапазон вплоть до ± 20%, предпочтительно до ± 10%, более предпочтительно до ± 5% и еще более предпочтительно до ± 1% от заданного значения. С другой стороны, особенно в отношении биологических систем или процессов, термин может означать и в пределах порядка данной величины, предпочтительно в 2 раза от значения. Если в заявке и формуле изобретения описаны конкретные значения, то, если не указано иначе, термин “примерно” подразумевается и в данном контексте означает в пределах допустимого диапазона ошибок для данной величины.
Фармацевтические композиции для применения in vivo должны быть стерильными. Это легко осуществляется, к примеру, фильтрованием через стерильные фильтрующие мембраны. Композиции терапевтических антител обычно помещают в контейнер со стерильным отверстием для доступа, к примеру, в мешок для внутривенного раствора или флакон с пробкой, прокалываемой иглой для подкожных инъекций.
Описанные здесь фармацевтические композиции могут быть в виде стандартных дозовых форм типа таблеток, пилюль, капсул, порошков, гранул, растворов или суспензий либо свечей для перорального, парентерального или ректального введения либо введения путем ингаляции или инсуффляции.
Для приготовления твердых композиций типа таблеток основной активный ингредиент смешивают с фармацевтическим носителем, например, с обычными ингредиентами для таблетирования, такими как кукурузный крахмал, лактоза, сахароза, сорбит, тальк, стеариновая кислота, стеарат магния, дикальцийфосфат или камеди, и другими фармацевтическими разбавителями, например, с водой, получая твердую предварительную композицию, содержащую однородную смесь соединения по настоящему изобретению или его нетоксичной фармацевтически приемлемой соли. Когда эти предварительные композиции называют однородными, это значит, что активный ингредиент равномерно распределен по всей композиции, так что композицию можно легко разделить на одинаково эффективные стандартные дозовые формы типа таблеток, пилюль и капсул. Такую твердую предварительную композицию затем разделяют на стандартные дозовые формы описанного выше типа, содержащие от 0,1 до 500 мг активного ингредиента по настоящему изобретению. Таблетки или пилюли из новой композиции могут быть покрыты оболочкой или уплотнены иным образом, получая дозовую форму, дающую преимущество пролонгированного действия. Например, таблетки или пилюли могут содержать внутренний дозирующий и наружный дозирующий компонент, причем последний имеет вид оболочки над первым. Два компонента могут быть разделены энтеросолюбильным слоем, который служит для противодействия распаду в желудке и позволяет внутреннему компоненту проходить без изменений в двенадцатиперстную кишку или задерживаться при высвобождении. Для таких энтеросолюбильных слоев или покрытий могут использоваться различные материалы, включая ряд полимерных кислот и смеси полимерных кислот с такими материалами, как шеллак, цетиловый спирт и ацетат целлюлозы.
Подходящие поверхностно-активные вещества включают, в частности, неионные средства типа полиоксиэтиленсорбитанов (например, Tween™ 20, 40, 60, 80 или 85) и других сорбитанов (например, Span™ 20, 40, 60, 80 или 85). Композиции с поверхностно-активным веществом обычно содержат от 0,05 до 5% ПАВ или же от 0,1 до 2,5%. Понятно, что при необходимости можно добавлять и другие ингредиенты, к примеру, маннит или другие фармацевтически приемлемые носители.
Подходящие эмульсии могут быть получены с использованием коммерчески доступных жировых эмульсий, таких как Intralipid™, Liposyn™, Infonutrol™, Lipofundin™ и Lipiphysan™. Активный ингредиент может быть растворен в заранее смешанной эмульсионной композиции или же он может быть растворен в масле (например, в соевом масле, сафлоровом масле, хлопковом масле, кунжутном масле, кукурузном масле или миндальном масле), а эмульсия образуется при смешивании с фосфолипидом (например, с яичными фосфолипидами, соевыми фосфолипидами или соевым лецитином) и водой. Понятно, что можно добавлять и другие ингредиенты, к примеру, глицерин или глюкозу для регулирования тоничности эмульсии. Подходящие эмульсии обычно содержат до 20% масла, к примеру, от 5 до 20%.
Эмульсионные композиции могут быть получены смешиванием антитела против CεmX с Intralipid™ или его компонентами (соевым маслом, яичными фосфолипидами, глицерином и водой).
Фармацевтические композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях либо их смесях и порошках. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые наполнители, приведенные выше. В некоторых воплощениях композиции вводят через ротовые или носовые дыхательные пути для местного или системного действия.
Композиции предпочтительно в стерильных фармацевтически приемлемых растворителях могут распыляться при помощи газов. Распыленные растворы можно вдыхать прямо из распыляющего устройства или же прикрепить распыляющее устройство к лицевой маске, палатке или к аппарату перемежающейся вентиляции легких с положительным давлением. Композиции в виде растворов, суспензий или порошков могут вводиться предпочтительно через рот или нос из устройств, доставляющих композицию соответствующим образом.
Применение антител против CεmX для лечения заболеваний, связанных с IgE
Для практического применения изложенного здесь способа нуждающимся в лечении субъектам (например, людям) можно вводить эффективное количество описанной выше фармацевтической композиции подходящим способом, как-то внутривенно, например, болюсом или непрерывным вливанием в течение периода времени, внутримышечно, внутрибрюшинно, интрацереброспинально, подкожно, внутрисуставно, интрасиновиально, интратекально, перорально, посредством ингаляции или топически. Для введения пригодны коммерчески доступные распылители для жидких составов, в том числе струйные распылители и ультразвуковые распылители. Жидкие составы можно распылять непосредственно, а лиофилизованные порошки можно распылять после восстановления. С другой стороны, антитела против CεmX можно вводить в аэрозоле с помощью фторуглеродного состава и ингалятора с отмеренной дозой либо вдыхать в виде лиофилизованного и размолотого порошка.
Субъектами для лечения описанными здесь способами могут быть млекопитающие, более предпочтительно люди. Млекопитающие включают, без ограничения, сельскохозяйственных животных, спортивных животных, комнатных животных, приматов, лошадей, собак, кошек, мышей и крыс. Субъектами, нуждающимися в лечении, могут быть пациенты, болеющие, подверженные риску или с подозрением на заболевания, связанные с IgE (например, аллергическую астму, а также другие заболевания, известные в данной области и/или приведенные здесь). Субъектов с заболеваниями, связанными с IgE типа аллергической астмы, можно идентифицировать при обычном медицинском обследовании, например, по лабораторным анализам. Субъекты с подозрением на заболевания, связанные с IgE, могут проявлять один или несколько симптомов заболевания, например, повышение уровня IgE и/или гиперреактивность на аллерген и/или антиген. Субъектами, подверженными риску заболевания, могут быть субъекты с одним или несколькими факторами риска для этого заболевания.
Типичными заболеваниями, связанными с IgE, являются, без ограничения, астма, аллергический ринит, синдром гипер-IgE, атопический дерматит, крапивница, вызванная простудой, хроническая крапивница, холинергическая крапивница, хронический риносинусит, системный мастоцитоз, кожный мастоцитоз, аллергический бронхолегочный аспергиллёз, рецидивирующий идиопатический ангионевротический отек и интерстициальный цистит, связанные с эозинофилами желудочно-кишечные заболевания, пищевая аллергия или лекарственная аллергия.
“Эффективное количество” в настоящем изобретении означает количество каждого активного средства, необходимое для оказания терапевтического действия на субъекта, по отдельности или в сочетании с одним или несколькими другими активными средствами. Эффективные количества варьируются, как это понятно специалистам в данной области, в зависимости от конкретного заболевания, подлежащего лечению, тяжести заболевания, индивидуальных параметров пациента, включая возраст, физическое состояние, размер, пол и вес, продолжительности лечения, характера параллельной терапии (если таковая имеется), конкретного способа введения и тому подобных факторов в пределах знаний и опыта практикующего врача. Эти факторы хорошо известны рядовым специалистам и могут быть установлены просто обычным экспериментированием. Обычно предпочтительно используются максимальные дозы отдельных компонентов либо их комбинаций, то есть самые высокие безопасные дозы в соответствии со здравым медицинским суждением. Однако рядовым специалистам следует иметь в виду, что пациент может настаивать на более низкой дозе или переносимой дозе по медицинским причинам, психологическим причинам или практически по любым другим причинам.
Определению дозировки обычно способствуют эмпирические соображения типа периода полураспада. Например, для продления периода полужизни антител и предотвращения атаки их иммунной системой хозяина можно использовать антитела, которые совместимы с иммунной системой человека, как-то гуманизованные антитела или полностью человеческие антитела. Частоту введения можно определять и корректировать в течение курса терапии, причем она обычно, хотя и не обязательно, исходит из лечения и/или подавления и/или ослабления и/или замедления заболевания, связанного с IgE. С другой стороны, могут подойти составы с замедленным непрерывным высвобождением антител против CεmX. В данной области известны различные составы и устройства для замедленного высвобождения.
В одном примере дозировки для антител против CεmX, как описано здесь, можно определять эмпирически у лиц, которым один или несколько раз вводили антитела против CεmX. Людям вводят постепенно возрастающие дозы антител против CεmX. Для оценки эффективности антител против CεmX можно отслеживать показатель заболевания, связанного с IgE (типа уровня IgE).
В целях настоящего изобретения надлежащая дозировка антител против CεmX будет зависеть от конкретного используемого антитела против CεmX (или его композиции), типа и тяжести заболевания, связанного с IgE, вводится ли антитело в профилактических или терапевтических целях, предшествующего лечения, клинической истории и реакции пациента на антитело, а также усмотрения лечащего врача. Как правило, врач будет вводить антитело против CεmX типа FB825 до тех пор, пока не будет достигнута доза, дающая требуемый результат. Введение антитела против CεmX может быть непрерывным или прерывистым, в зависимости, к примеру, от физиологического состояния получателя, терапевтической или профилактической цели введения и других факторов, известных специалистам.
В некоторых воплощениях описанные здесь антитела против CεmX (например, FB825) вводятся нуждающимся в лечении субъектам в количестве, достаточном для снижения уровня общего IgE по меньшей мере на 20% (например, 30%, 40%, 50%, 60%, 70%, 80%, 90% или больше).
В настоящем изобретении термин “лечение” означает применение или введение композиции, включающей одно или несколько активных средств, субъектам с заболеванием, связанным с IgE, симптомом заболевания, связанного с IgE, или с предрасположенностью к заболеванию с целью лечения, исцеления, ослабления, облегчения, изменения, исправления, смягчения, улучшения или воздействия на заболевание, симптом заболевания или предрасположенность к заболеванию.
Ослабление заболевания, связанного с IgE, включает замедление развития или прогрессирования заболевания либо снижение тяжести заболевания. Ослабление заболевания не обязательно требует лечебных результатов. В настоящем изобретении “замедление” развития заболевания (типа заболевания, связанного с IgE) означает задерживание, препятствование, замедление, задержку, стабилизацию и/или отсрочку прогрессирования заболевания. Это замедление может быть различной продолжительности, в зависимости от истории болезни и/или проходящих лечение лиц. Способ, который “замедляет” или ослабляет развитие заболевания либо задерживает начало заболевания, – это способ, который снижает вероятность развития одного или нескольких симптомов заболевания в данный период времени и/или уменьшает степень симптомов в данный период времени по сравнению с неиспользованием способа. Такие сравнения обычно основываются на клинических исследованиях с количеством субъектов, достаточным для получения статистически значимого результата.
“Развитие” или “прогрессирование” заболевания означает начальные проявления и/или следующее за ними прогрессирование заболевания. Развитие заболевания может выявляться и оцениваться по стандартным клиническим методикам, хорошо известным в данной области. Однако развитие означает и такое прогрессирование, которое может не выявляться. В целях настоящего изобретения развитие или прогрессирование относится к биологическому течению симптомов. “Развитие” включает в себя возникновение, рецидив и начало. В настоящем изобретении термин “начало” или “возникновение” заболевания, связанного с IgE, включает в себя исходное начало и/или повторное появление.
Для осуществления способов, как описано здесь, нуждающимся в лечении субъектам (например, больным людям) можно вводить любые из антител против CεmX типа FB825 в виде однократной дозы или множественных доз подходящим способом, к примеру, путем внутривенного вливания или подкожной инъекции. Дозировка антител против CεmX для каждого введения может составлять от 0,5 до 25 мг/кг (например, от 1 мг/кг до 20 мг/кг, от 5 мг/кг до 15 мг/кг или от 10 мг/кг до 20 мг/кг), в зависимости от различных факторов, в том числе описанных здесь. Для повторного введения в течение нескольких дней или дольше, в зависимости от заболевания, лечение продолжается до тех пор, пока не произойдет желательное подавление симптомов или не будут достигнуты достаточные терапевтические уровни для ослабления связанного с IgE заболевания или его симптома.
Введение антител против CεmX (например, FB825) может быть практически непрерывным в течение заданного периода времени или может происходить в виде серии разделенных доз, например, до, во время или после развития заболевания, связанного с IgE. Типичная схема введения включает введение нуждающемуся в лечении субъекту первой дозы антитела против CεmX (например, в дозе 3 мг/кг, 5 мг/кг, 10 мг/кг, 15 мг/кг, 20 мг/кг или 25 мг/кг) с последующим введением второй дозы антитела по меньшей мере через 3 месяца после первой дозы (например, через 4, 5 или 6 месяцев). Дозировка при втором введении может быть выше, такой же или ниже, чем при первом введении. Могут применяться и другие схемы дозировки в зависимости от того профиля фармакокинетического распада, которого желает достичь лечащий врач.
В некоторых воплощениях нуждающемуся в лечении субъекту можно вводить первую дозу антитела в подходящем количестве (например, в дозе 3 мг/кг, 5 мг/кг, 10 мг/кг, 15 мг/кг, 20 мг/кг или 25 мг/кг). Затем субъекта периодически наблюдают на появление симптомов, указывающих на связанное с IgE заболевание, например, аллергических реакций и/или повышение уровня общего IgE. Когда наблюдается такой симптом, субъекту можно вводить вторую дозу антитела.
Также в объем настоящего изобретения входит профилактическое лечение связанных с IgE заболеваний с помощью любых антител против CεmX, чтобы уменьшить риск возникновения таких заболеваний. Подходящими субъектами для такого профилактического лечения могут быть пациенты со связанными с IgE заболеваниями в анамнезе и/или с семейной историей связанных с IgE заболеваний.
Для введения фармацевтических композиций субъектам можно использовать стандартные методы, известные рядовым специалистам в области медицины, в зависимости от типа подлежащего лечению заболевания или локализации заболевания. Такие композиции можно вводить и другими стандартными способами, например, перорально, парентерально, посредством ингаляции, топически, ректально, назально, буккально, вагинально или через имплантированный резервуар. Термин “парентерально” в настоящем изобретении включает методы подкожного, внутрикожного, внутривенного, внутримышечного, внутрисуставного, внутриартериального, интрасиновиального, интрастернального, интратекального, внутриочагового и внутричерепного введения или вливания. Кроме того, их можно вводить субъектам в виде инъекционного депо типа 1-, 3- или 6-месячных депо из инъекционных или биоразлагаемых материалов.
Композиции для инъекций могут содержать различные носители, как-то растительные масла, диметиллактамид, диметилформамид, этиллактат, этилкарбонат, изопропилмиристат, этанол и полиолы (глицерин, пропиленгликоль, жидкий полиэтиленгликоль и др.). Для внутривенного введения водорастворимые антитела можно вводить капельным способом, при этом вливают фармацевтическую композицию, содержащую антитело и физиологически приемлемые наполнители. Физиологически приемлемые наполнители могут включать, к примеру, 5% декстрозу, 0,9% физраствор, раствор Рингера или другие подходящие наполнители. Внутримышечные препараты, например, стерильные композиции подходящей растворимой солевой формы антитела, можно растворять и вводить в таком фармацевтическом наполнителе, как вода для инъекций, 0,9% физраствор или 5% раствор глюкозы.
В одном воплощении антитела против CεmX вводят методами сайт-специфической или адресной локальной доставки. Примеры методов сайт-специфической или адресной локальной доставки включают различные имплантируемые источники типа депо антител против CεmX или такие катетеры для локальной доставки, как инфузионные катетеры, постоянные катетеры или катетеры с иглой, синтетические имплантаты, адвентициальные обмотки, шунты и стенты или другие имплантируемые устройства, сайт-специфические носители, прямые инъекции или непосредственное нанесение. Например, см. PCT Publication WO 00/53211 и U.S. Pat. No. 5,981,568. Может применяться и адресная доставка терапевтических композиций, содержащих антисмысловые полинуклеотиды, экспрессирующие векторы или субгеномные полинуклеотиды. Описаны методы опосредованной рецепторами доставки ДНК, к примеру, в Findeis et al., Trends Biotechnol. (1993) 11:202; Chiou et al., Gene Therapeutics: Methods and Applications of Direct Gene Transfer (J.A. Wolff, ed.) (1994); Wu et al., J. Biol. Chem. (1988) 263:621; Wu et al., J. Biol. Chem. (1994) 269:542; Zenke et al., Proc. Natl. Acad. Sci. USA (1990) 87:3655; Wu et al., J. Biol. Chem. (1991) 266:338. Для местного введения при проведении генной терапии применяются терапевтические композиции, содержащие полинуклеотиды в диапазоне от 100 нг до 200 мг ДНК. В некоторых воплощениях при проведении генной терапии концентрации также могут составлять от 500 нг до 50 мг, от 1 мкг до 2 мг, от 5 мкг до 500 мкг и от 20 мкг до 100 мкг ДНК или больше.
Терапевтические полинуклеотиды и полипептиды, описанные здесь, могут быть доставлены с помощью носителей для доставки генов. Носители для доставки генов могут иметь вирусное или невирусное происхождение (в общем см. Jolly, Cancer Gene Therapy (1994) 1:51; Kimura, Human Gene Therapy (1994) 5:845; Connelly, Human Gene Therapy (1995) 1:185; и Kaplitt, Nature Genetics (1994) 6:148. Экспрессия таких кодирующих последовательностей может индуцироваться с помощью эндогенных или гетерологичных промоторов и/или энхансеров млекопитающих. Экспрессия кодирующей последовательности может быть конститутивной или регулируемой.
Вирусные векторы для доставки нужных полинуклеотидов и экспрессии в нужных клетках хорошо известны в данной области. Типичными вирусными векторами являются, без ограничения, рекомбинантные ретровирусы (например, см. PCT Publications WO 90/07936; WO 94/03622; WO 93/25698; WO 93/25234; WO 93/11230; WO 93/10218; WO 91/02805; U.S. Pat. Nos. 5,219,740 and 4,777,127; GB Patent No. 2,200,651; и EP Patent No. 0 345 242), векторы на основе альфа-вируса (например, векторы из вируса Sindbis, вируса Semliki forest (ATCC VR-67; ATCC VR-1247), вируса Ross River (ATCC VR-373; ATCC VR-1246) и вируса венесуэльского лошадиного энцефалита (ATCC VR-923; ATCC VR-1250; ATCC VR-1249; ATCC VR-532)) и векторы из аденоассоциированных вирусов (AAV) (например, см. PCT Publications WO 94/12649, WO 93/03769; WO 93/19191; WO 94/28938; WO 95/11984 и WO 95/00655). Также может применяться введение ДНК, связанной с убитым аденовирусом, как описано в Curiel, Human Gene Ther. (1992) 3:147. Могут применяться и невирусные носители и методы доставки, включая, без ограничения, конденсированную поликатионами ДНК, связанную или несвязанную с одним только убитым аденовирусом (например, см. Curiel, Human Gene Ther. (1992) 3:147); связанную с лигандом ДНК (например, см. Wu, J. Biol. Chem. (1989) 264: 16985); клетки-носители для доставки в эукариотические клетки (например, см. U.S. Pat. No. 5,814,482; PCT Publications WO 95/07994; WO 96/17072; WO 95/30763; и WO 97/42338) и нейтрализацию заряда нуклеиновой кислоты или слияние с клеточной мембраной. Также можно использовать голую ДНК. Типичные методы введения голой ДНК описаны в PCT Publication WO 90/11092 и U.S. Pat. No. 5,580,859. Липосомы, которые могут действовать в качестве носителей для доставки генов, описаны в U.S. Pat. No. 5,422,120; PCT Publications WO 95/13796; WO 94/23697; WO 91/14445; и EP Patent No. 0524968. Дополнительные подходы описаны в Philip, Mol. Cell. Biol. (1994) 14: 2411; и Woffendin, Proc. Natl. Acad. Sci. (1994) 91:1581.
Также понятно, что экспрессирующие векторы можно использовать и для управления экспрессией любых белков на основе описанных здесь антител против CεmX (например, FB825). Например, известны и другие фрагменты антител против CεmX, которые способны связывать CεmX и/или биологическую активность IgE.
Конкретная схема дозировки, т.е. доза, время и повторность в описанном здесь способе, будет зависеть от конкретного субъекта и истории болезни этого субъекта. Любые из описанных здесь антител против CεmX можно использовать в сочетании с другими средствами (например, другими средствами для лечения заболеваний, связанных с IgE), которые служат для усиления и/или дополнения их эффективности.
В некоторых воплощениях описанные здесь антитела против CεmX, к примеру, FB825, применяются для лечения атопического дерматита следующим образом. Атопический дерматит, также известный как экзема, является хроническим заболеванием кожи, характеризующимся покраснением и/или зудом. Он часто встречается у детей, но может возникнуть в любом возрасте. Нуждающихся в лечении пациентов можно выявить при плановом медосмотре по наличию одного или нескольких симптомов атопического дерматита, включая сухость кожи, зуд, пятна от красных до коричневато-серых, небольшие выпуклые шишки, из которых может вытекать жидкость, и струпья на поцарапанной, утолщенной, потрескавшейся, шелушащейся коже и/или сырая, чувствительная, опухшая от царапин кожа. В некоторых случаях при медосмотре субъекта-кандидата можно исследовать общий уровень IgE и уровень аллерген-специфичного IgE. Если уровень IgE у субъекта-кандидата (например, общего IgE, аллерген-специфичного IgE или обоих) выше, чем в норме (представляющей средний уровень IgE у субъектов того же вида, например, человека, у которых нет атопического дерматита или других аллергических заболеваний, связанных с IgE).
Нуждающимся в лечении пациентам можно вводить первую дозу антител, которая может составлять от 3 мг/кг до 8 мг/кг, стандартным способом, как описано здесь. В некоторых случаях первая доза составляет 5 мг/кг. После первой дозы можно отслеживать общий уровень IgE у пациента. Если снижение уровня IgE через 3-4 недели после первой дозы составляет менее 50%, то пациенту можно вводить вторую дозу антител через 3-4 недели после первой дозы. Вторая доза может быть идентична первой дозе или меньше первой дозы. В некоторых случаях как первая доза, так и вторая доза составляет 5 мг/кг и вводится путем внутривенного вливания в течение 1-2 часов. В ходе лечения можно отслеживать и другие биомаркеры, указывающие на эффективность и/или безопасность. Такие биомаркеры включают, без ограничения, тимусный и регулируемый активацией хемокин (TARC), эотаксин-3, тимусный стромальный лимфопоэтин (TSLP), периостин, IL-1a, IL-4, IL-5, IL-13, IL-16, IL-31, M-CSF либо их комбинации.
Вышеуказанному лечению подвергаются пациенты, у которых отмечается хронический атопический дерматит на протяжении по меньшей мере 3 лет, который диагностирован при плановом медосмотре, к примеру, установлен по пересмотренным Эйнифилдом критериям Ханнифина и Райки и подтвержден положительным результатом на аллерген-специфичный IgE. У пациента может быть один или несколько из следующих признаков: (i) индекс площади и тяжести экземы (EASI) более 14 баллов, (ii) глобальная оценка исследователя (IGA) более 3 баллов (по 5-балльной шкале), (iii) поражено более 10% площади поверхности тела (BSA), (iv) история неадекватной реакции на постоянный режим топических кортикостероидов или ингибиторов кальциневрина в течение по меньшей мере одного месяца или по меньшей мере трех месяцев до лечения. Кроме того, пациент может получать постоянные дозы смягчающего средства по два раза в день в течение по меньшей мере 7 дней до лечения.
В некоторых случаях описанные здесь антитела против CεmX, например, FB825, могут применяться вместе с увлажнителями (например, при постоянных дозах типа по меньшей мере два раза в день) и/или местными кортикостероидами (TCS). TCS со средней активностью можно наносить на участки с активными поражениями, а после того, как поражения будут под контролем, можно переключаться на TCS с низкой активностью. Если же поражения повторяются, то можно возобновить лечение TCS со средней активностью с понижающим подходом. Если поражения сохраняются или ухудшаются после ежедневного лечения TCS со средней активностью, то можно использовать TCS с высокой или сверхвысокой активностью, если только это не будет небезопасным. TCS с низкой активностью можно использовать на участках с тонкой кожей (например, на лице, шее, местах опрелостей, в области гениталий или на участках с атрофией кожи) или на таких участках, где дальнейшее применение TCS со средней активностью считается небезопасным.
TCS с низкой, средней и высокой или сверхвысокой активностью хорошо известны в данной области. Типичным TCS средней активности является крем с 0,05% флутиказона пропионата, крем с 0,1% мометазона фуроата или крем с 0,06% бетаметазона валерата. Типичным TCS низкой активности является мазь с 1% гидрокортизона. Типичным TCS высокой активности является крем с 0,05% флуоцинонида или мазь с 0,25% дезоксиметазона. Типичным TCS сверхвысокой активности является мазь с 0,05% клобетазола пропионата.
В некоторых случаях описанному здесь лечению подвергаются пациенты, которым не проводится одна или несколько из следующих терапий: (i) местного применения такролимуса и пимекролимуса, (ii) системного применения кортикостероидов, (iii) ингибиторов лейкотриенов, (iv) аллергеновой иммунотерапии, (v) системного применения иммунодепрессантов или иммуномодуляторов (например, циклоспорина, микофенолата-мофетила, IFN-γ, азатиоприна, метотрексата или биологических препаратов), (vi) живых (например, ослабленных) вакцин и/или (vii) традиционной китайской медицины. Пациенты также могут не получать каких-либо хирургических процедур и/или УФ-процедур.
Любые из описанных здесь способов могут дополнительно включать определение наличия снижения гемоглобина, инфекций верхних дыхательных путей, инфекций мочевыводящих путей либо их комбинаций у субъектов после первой дозы. Если наблюдается одно или несколько из них, то количество антител против CεmX (например, FB825) во второй дозе можно уменьшить. С другой стороны, можно остановить лечение.
Наборы для применения при лечении связанных с IgE заболеваний
Настоящим изобретением также предусмотрены наборы для применения при лечении связанных с IgE заболеваний. Такие наборы могут включать один или несколько контейнеров, содержащих описанные здесь антитела против CεmX (например, FB825).
В некоторых воплощениях набор может содержать инструкции по применению в соответствии с любым из описанных здесь способов. Включенные инструкции могут содержать описание введения антител против CεmX для лечения, замедления начала или ослабления связанного с IgE заболевания в соответствии с любым из описанных здесь способов. Набор также может содержать описание выбора лиц, подходящих для лечения, на основе определения наличия, подозрения на наличие или риска заболевания. В других воплощениях инструкции содержат описание введения антител против CεmX субъектам, нуждающимся в лечении, для снижения риска развития связанного с IgE заболевания.
Инструкции по применению антител против CεmX обычно включают информацию о дозировке, схеме дозирования и пути введения для предполагаемого лечения. Контейнеры могут представлять собой стандартные дозы, объемные упаковки (например, многодозовые упаковки) или дробные дозы. Инструкции, вложенные в наборы по изобретению, обычно представляют собой письменные инструкции на этикетке или вкладыше (например, на листе бумаги, включенном в комплект), но также приемлемы машиночитаемые инструкции (например, инструкции, хранящиеся на магнитном или оптическом диске).
Этикетка или вкладыш в упаковку указывает, что композиция применяется для лечения, замедления начала и/или ослабления связанного с IgE заболевания. Инструкции могут предоставляться для реализации любого из описанных здесь способов.
Наборы по изобретению находятся в подходящей упаковке. Подходящие упаковки включают, без ограничения, флаконы, бутылки, банки, гибкие упаковки (например, герметичные пакеты из майлара или пластика) и т.п. Также предусмотрены упаковки для применения вместе со специальным устройством типа ингалятора, устройства для назального введения (например, распылителя) или инфузионного устройства типа мининасоса. Набор может иметь стерильный порт для доступа (например, контейнер может представлять собой мешок для внутривенного раствора или флакон с пробкой, прокалываемой иглой для шприца). По меньшей мере одно активное средство в композиции представляет собой антитело против CεmX типа FB825.
Наборы могут необязательно содержать дополнительные компоненты типа буферов и пояснительной информации. Обычно набор содержит контейнер и этикетку либо вкладыш на нем или связанный с ним. В некоторых воплощениях настоящего изобретения предусмотрены изделия, содержащие содержимое наборов, описанных выше.
Общие методы
В практике настоящего изобретения будут применяться, если не указано иначе, стандартные методы молекулярной биологии (включая рекомбинантные методы), микробиологии, клеточной биологии, биохимии и иммунологии, которые известны специалистам в данной области. Такие методы полностью изложены в литературе, как-то: Molecular Cloning: A Laboratory Manual, second edition (Sambrook et al., 1989) Cold Spring Harbor Press; Oligonucleotide Synthesis (M.J. Gait, ed., 1984); Methods in Molecular Biology, Humana Press; Cell Biology: A Laboratory Notebook (J.E. Cellis, ed., 1998) Academic Press; Animal Cell Culture (R.I. Freshney, ed., 1987); Introduction to Cell and Tissue Culture (J.P. Mather and P.E. Roberts, 1998) Plenum Press; Cell and Tissue Culture: Laboratory Procedures (A. Doyle, J.B. Griffiths, and D.G. Newell, eds., 1993-8) J. Wiley and Sons; Methods in Enzymology (Academic Press, Inc.); Handbook of Experimental Immunology (D.M. Weir and C.C. Blackwell, eds.); Gene Transfer Vectors for Mammalian Cells (J.M. Miller and M.P. Calos, eds., 1987); Current Protocols in Molecular Biology (F.M. Ausubel et al., eds., 1987); PCR: The Polymerase Chain Reaction (Mullis et al., eds., 1994); Current Protocols in Immunology (J.E. Coligan et al., eds., 1991); Short Protocols in Molecular Biology (Wiley and Sons, 1999); Immunobiology (C.A. Janeway and P. Travers, 1997); Antibodies (P. Finch, 1997); Antibodies: a practical approach (D. Catty., ed., IRL Press, 1988-1989); Monoclonal antibodies: a practical approach (P. Shepherd and C. Dean, eds., Oxford University Press, 2000); Using antibodies: a laboratory manual (E. Harlow and D. Lane (Cold Spring Harbor Laboratory Press, 1999); The Antibodies (M. Zanetti and J. D. Capra, eds., Harwood Academic Publishers, 1995).
Без дальнейшего уточнения считается, что специалисты в данной области на основании приведенного выше описания смогут применить настоящее изобретение в самой полной степени. Поэтому следующие конкретные воплощения следует истолковывать как чисто иллюстративные и никоим образом не ограничивающие остальное изложение. Все процитированные здесь публикации включены путем ссылки для приведенных здесь целей или предметов обсуждения.
Примеры
Пример 1. Исследования токсичности FB825 на макаках-крабоедах
Материалы и методы
Лабораторные анализы
Брали образцы крови и мочи для гематологического анализа, исследования показателей коагулограммы, биохимического состава сыворотки (включая функциональные пробы печени), функциональных проб щитовидной железы и анализа мочи собирали и анализировали в ходе проведения рутинных клинических лабораторных исследований
Аномальные клинико-лабораторные значения отмечали как высокие или низкие (либо нормальные или аномальные) на основании контрольных диапазонов для каждого лабораторного параметра. Клиническую значимость определяли как любые вариации результатов, которые имели медицинское значение и могли привести к изменению медицинской тактики (например, активное наблюдение, диагностические меры или терапевтические меры). Если отмечалось клинически значимое изменение от результатов скрининга, то регистрировали клинически значимое значение и причины клинической значимости. Проходивших обработку макак-крабоедов продолжали отслеживать по дополнительным оценкам до тех пор, пока значения не достигали контрольного диапазона или значений при скрининге или же пока в последующем наблюдении больше не было медицинской необходимости.
Фармакодинамические измерения
Отбирали пробы крови для определения общего IgE и антител против лекарственного средства (ADA) в пробирки для забора крови на 3,5 мл (пробирки для отделения сыворотки BD Vacutainer® SST™) фирмы Vince and Associates Clinical Research. Пробы брали так, чтобы был минимальный объем крови на один размер пробирки для забора крови. В каждой временной точке получали как минимум 1,0 мл сыворотки. После получения проб крови пробирки переворачивали 5 раз и оставляли для образования сгустка крови на 30 мин при окружающей среды температуре (19°C-24°C). Пробы центрифугировали при 2200 об/мин в течение 10 мин при комнатной температуре в центрифуге с подвесными стаканами. Двойные аликвоты сыворотки примерно равного объема (как минимум 500 мкл на пробу) переносили по стандартной лабораторной методике в 2 маркированные соответствующим образом пробирки для хранения (полипропиленовые криопробирки на 2 мл) фирмы Vince and Associates Clinical Research.
Пробы на иммуногенность для измерения ADA анализировали валидированным методом ELISA.
Аспартатаминотрансфераза и аланинаминотрансфераза в сыворотке
Функцию печени у обработанных FB825 макак-крабоедов отслеживали по измерению уровней аспартатаминотрансферазы (AST) и аланинаминотрансферазы (ALT). Активности этих двух ферментов выражали в единицах на литр (ед./л).
Результаты
Исследования однократных доз FB825 на макаках-крабоедах
Изучали токсичность однократных доз FB825 в не связанных с GLP исследованиях по токсичности однократных доз, проводившихся на макаках-крабоедах. В первом исследовании определяли связанные с препаратом эффекты на макаках-крабоедах, получавших однократное 10-минутное внутривенное вливание FB825. Во втором исследовании определяли связанные с препаратом эффекты на макаках-крабоедах, получавших одну подкожную инъекцию FB825.
В исследовании по определению диапазона доз при внутривенном введении самцы и самки макак-крабоедов хорошо переносили однократное 10-минутное вливание FB825 по 30, 100 и 300 мг/кг. Не отмечалось никаких связанных с препаратом эффектов на клинические параметры, потребление пищи, массу тела или смертность. Связанные с FB825 эффекты ограничивались минимальным повышением ALT и AST, которое восстанавливалось на 57-й день, и минимальным повышением интерлейкина-6 и интерлейкина-10 через 6 часов и/или 1 час после введения дозы у животных, получавших по меньшей мере 100 мг/кг FB825. Исходя из этих результатов, уровень отсутствия наблюдаемых неблагоприятных эффектов (NOAEL) посчитали равным 300 мг/кг.
В другом токсикологическом исследовании однократных доз макаки-крабоеды хорошо переносили введение FB825 посредством однократной подкожной инъекции при 300 мг/кг. Не наблюдалось никаких связанных с FB825 клинических признаков или эффектов на массу тела или параметры клинической патологии (гематология, коагулограмма и клиническая биохимия).
Исследования повторных доз FB825 на макаках-крабоедах
Изучали токсичность повторных доз FB825 на макаках-крабоедах. Определяли связанные с препаратом эффекты на макаках-крабоедах, получавших однократное 10-минутное внутривенное вливание FB825 раз в неделю, в общей сложности 4 дозы.
Макаки-крабоеды хорошо переносили введение FB825 при 10-минутном внутривенном вливании раз в неделю в общей сложности 4 дозы по 30, 100 и 300 мг/кг. В этом исследовании оценивались следующие параметры и конечные точки: клинические признаки, масса тела, потребление пищи, офтальмология, электрокардиология, параметры клинической патологии (гематология, коагулограмма и клиническая биохимия), биоанализ и токсикокинетические измерения, анализ антител против препарата, проточная цитометрия, анализ IgE, уровень гормонов щитовидной железы, общие результаты вскрытия, вес органов и гистопатологические исследования.
Не отмечалось никаких связанных с FB825 клинических признаков или эффектов на массу тела, потребление пищи, офтальмологические и электрокардиографические исследования, параметры свертывания крови и уровни гормонов щитовидной железы у обоих полов в дозах вплоть до 300 мг/кг. Кроме того, не отмечалось никаких заметных макроскопических изменений, связанных с FB825, при дозах до 300 мг/кг включительно.
Связанные с FB825 эффекты ограничивались обратимым заметным повышением ALT при дозах FB825, превышающих или равных 100 мг/кг, и обратимым умеренным повышением AST, частично обратимым минимальным снижением уровня альбумина и соответствующего соотношения альбумин:глобулин и обратимым минимальным повышением числа моноцитов при дозе FB825 в 300 мг/кг. Степень повышения уровней ALT и AST при 300 мг/кг посчитали неблагоприятной. Эффекты на органы-мишени наблюдались при уровнях ≥30 мг/кг и состояли из неадекватного истощения коллоида в фолликулах щитовидной железы и снижения массы щитовидной железы. Однако снижение массы щитовидной железы было в пределах диапазона, наблюдавшегося исторически у контрольных макак. Поскольку изменения в характере окрашивания коллоида, изменения размера фолликулов щитовидной железы и дегенерация вакуолей отмечались как спонтанные у макак-крабоедов (Ishida 2000; Hatakeyama 2011), и так как не было никакого связанного с препаратом воздействия на уровни тироксина и тиреотропного гормона, то результаты по щитовидной железе посчитали неблагоприятными в условиях этого исследования.
Исходя из повышения уровней ALT и AST при 300 мг/кг, посчитали, что NOAEL составляет 100 мг/кг (Cmax = 5330 мкг/мл и AUC(0−168ч) = 520 мг⋅час/мл для самцов и Cmax = 5220 мкг/мл и AUC(0−168ч) = 487 мг⋅час/мл для самок).
Пример 2. Клинические исследования на людях
Главной задачей этого исследования была оценка безопасности и переносимости однократных возрастающих внутривенных доз FB825 у нормальных, здоровых лиц. Вторичные задачи этого исследования включают определение ФК-профиля при однократных возрастающих внутривенных дозах FB825, изучение влияния на общий IgE после однократных возрастающих внутривенных доз FB825 и исследование наличия антител против FB825 после однократных возрастающих внутривенных доз FB825.
План исследования
Это было рандомизованное, двойное слепое, контролируемое плацебо исследование 1-й фазы, впервые проведенное на людях (FIH), для оценки безопасности, переносимости, фармакокинетики и иммуногенности однократных возрастающих внутривенных доз FB825. Расписание мероприятий, реализуемых во время клинического исследования FB825 на людях, приведено в таблице 1. Субъектов, которые соответствовали критериям для включения в исследование, определяли в текущую дозовую когорту и назначали случайным образом на получение FB825 или плацебо (носителя). Все дозы FB825 вводились в виде 1-часового внутривенного вливания.
Перед переходом к когорте со следующей возрастающей дозой рассматривали данные по безопасности для каждой дозовой когорты. Дозирование в когорте со следующей возрастающей дозой разрешали только после подтверждения его безопасности. Уровень дозы можно было корректировать или повторять, если нужно. Полученные вслепую данные по ФК рассматривали одновременно с данными по безопасности.
Исследование включало 6 когорт и в общей сложности 54 нормальных, здоровых субъекта (по 7 субъектов [препарат: 4, плацебо: 3] в когортах A и B и по 10 субъектов [препарат: 7, плацебо: 3] в группах C, D, E, и F). Начальная доза FB825 составляла 0,003 мг/кг в/в с запланированным возрастанием в последующих когортах до 0,03, 0,3, 1,5, 5 и 10 мг/кг в/в. Исследование состояло из периода скрининга (дни от −28 до −2), регистрации (день −1), периода лечения/последующего наблюдения (дни от 1 до 140) и посещения в конце исследования (день 140).
Субъекты регистрировались в клинике в день −1 и проходили процедуры проверки. Когда процедуры перекрывались и приходились на один и тот же момент времени, то порядок проведения процедур заключался в измерении основных показателей состояния организма, электрокардиограммы, а затем фармакокинетических пробах крови.
Субъекты получали исследуемый препарат либо плацебо в день 1. Первые 2 субъекта из 2 когорт с наименьшей дозой (когорты A и B) получали либо плацебо, либо FB825 (т.е. должны были быть представлены как плацебо, так и исследуемый препарат). Остальным субъектам в этих когортах вводили дозу через 48 часов после введения первым двум субъектам. Всем субъектам в 4 когортах с самыми высокими дозами вводили дозы одновременно. Проходило как минимум 14 дней между дозированием последнего субъекта в группе и принятием решения о переходе к дозированию следующей группы. Перед переходом к когорте со следующей возрастающей дозой рассматривали слепым методом данные по безопасности для каждой дозовой когорты. Дозирование в когорте со следующей возрастающей дозой разрешали только после подтверждения его безопасности. Полученные вслепую данные по ФК рассматривали одновременно с данными по безопасности.
Пробы крови для оценки ФК брали в день 1 за 30 мин (± 5 мин) до начала вливания; через 30 мин (± 2 мин) после начала вливания; через 1, 1,25 и 2 часа (± 2 мин) после начала вливания; через 4 и 8 часов (± 5 мин) после начала вливания; и через 24 и 48 часов (± 10 мин) после начала вливания. Кроме того, брали отдельные пробы крови на 5, 14, 29, 85 и 140-й день после вливания. Пробы крови для оценки иммуногенности брали в день 1 (за 30 мин [± 5 мин] до начала вливания) и в дни 5, 14, 29, 85 и 140.
Субъекты содержались в клинике со дня −1 до выписки в день 3 (через 48 часов после введения дозы) и возвращались в клинику в дни 5, 14, 29, 85 и 140 для амбулаторного посещения. Продолжительность исследования, исключая скрининг, составляла около 140 дней.
Популяция пациентов
Здоровые мужчины и женщины в возрасте от 18 до 55 лет включительно, с массой тела больше или равной 50 кг и индексом массы тела от 18,0 до 30,0 кг/м2 включительно, и давали письменное информированное согласие. Всего записалось 54 субъекта, причем 41 субъект (75,9%) прошел исследование полностью, а 13 субъектов (24,1%) прервали. 7 субъектов (13,0%) прекратили по выбору субъекта, в том числе по 1 субъекту в группах, получавших 0,3, 1,5, 5 и 10 мг/кг FB825, и 3 субъекта в группе, получавшей плацебо. 6 субъектов (11,1%) не пришли для последующего наблюдения, в том числе по 1 субъекту в группах, получавших 0,003, 0,3 и 1,5 мг/кг FB825 и плацебо, и 2 субъекта в группе, получавшей 10 мг/кг FB825.
Введение FB825
В день 1 испытуемые получали однократное внутривенное вливание FB825 (0,003, 0,03, 0,3, 1,5, 5 или 10 мг/кг) либо плацебо в течение примерно 1 часа.
Все дозы вводили утром запланированного дня дозирования (день 1) в виде однократного вливания в течение примерно 1 часа после голодания примерно 2 часа. Легкий завтрак разрешали за 2 часа и более до дозировки. Вода была разрешена в любое время, кроме 1 часа до и 1 часа после дозировки. Растворы FB825 разбавляли перед введением.
В день 1 в 0 часов проводили однократное внутривенное вливание FB825 или плацебо в течение примерно 1 часа. Переход к следующему уровню дозы зависел от одобрения группы проверки безопасности. Первые 2 субъекта из 2 когорт с наименьшей дозой (когорты A и B) получали либо плацебо, либо FB825 (т.е. должны были быть представлены как плацебо, так и исследуемый препарат). Остальным субъектам в этих когортах вводили дозу через 48 часов после введения первым двум субъектам. Всем субъектам в 4 когортах с самыми высокими дозами вводили дозы одновременно.
Фармакокинетические (ФК) измерения
Пробы крови для анализа ФК FB825 брали у всех субъектов в следующие временные точки: в день 1 за 30 мин (± 5 мин) до начала вливания; через 30 мин (± 2 мин) после начала вливания; через 1, 1,25 и 2 часа (± 2 мин) после начала вливания; через 4 и 8 часов (± 5 мин) после начала вливания; и через 24 и 48 часов (± 10 мин) после начала вливания. Кроме того, брали отдельные пробы крови на 5, 14 (± 1 день), 29 (± 2 дня), 85 (± 3 дня) и 140-й (± 5 дней) день после вливания.
По данным о концентрации в сыворотке для каждого субъекта рассчитывали следующие параметры ФК при однократной дозе FB825 с применением некомпартментных методов:
AUC0-t: площадь под кривой концентрации в сыворотке от времени (AUC) со времени 0 до последней измеряемой концентрации, рассчитанная по линейной формуле трапеций;
AUC0-inf: AUC со времени 0 с экстраполяцией до бесконечности, рассчитанная по следующей формуле:
AUC0-inf = AUC0-t + Ct/Kel, где Ct – последняя измеряемая концентрация в сыворотке, а Kel – терминальная константа скорости выведения. Если экстраполированная площадь (Ct/Kel) была больше, чем 20% от AUC0-inf, то AUC0-inf и связанные с ней параметры (CL и Vd) считались отсутствующими;
%AUCex: процент площади, экстраполированной для расчета AUC0-inf;
Cmax: максимальная наблюдаемая концентрация в сыворотке;
Tmax: время максимальной наблюдаемой концентрации в сыворотке;
Kel: терминальная константа скорости выведения, где Kel – величина наклона графика линейной регрессии логарифма концентрации от времени в течение терминальной фазы; Kel сохраняли только, если R2≥0,80 и 3 точки в терминальной фазе не включали Cmax;
t1/2: терминальный период полужизни (когда это возможно), рассчитанный как (ln)2/Kel;
MRT: среднее время удержания, рассчитанное как AUMC/AUC;
CL: кажущийся клиренс, рассчитанный как доза/AUC0-inf;
Vd: кажущийся объем распределения, рассчитанный следующим образом: доза/(AUC0-inf × Kel).
Пробы крови для фармакокинетики
Пробы крови для анализа FB825 отбирали в пробирки для забора крови на 3,5 мл (пробирки для отделения сыворотки BD Vacutainer® SST™) фирмы Vince and Associates Clinical Research. Пробы брали так, чтобы был минимальный объем крови на один размер пробирки для забора крови. По каждой точке времени получали как минимум 1,0 мл сыворотки. После получения проб крови пробирки переворачивали 5 раз и оставляли для образования сгустка крови на 30 мин при температуре окружающей среды (19°C-24°C). Пробы центрифугировали при 2200 об/мин в течение 10 мин при комнатной температуре в центрифуге с подвесными стаканами. Двойные аликвоты сыворотки примерно равного объема (как минимум 500 мкл на пробу) переносили по стандартной лабораторной методике в 2 маркированные соответствующим образом пробирки для хранения (полипропиленовые криопробирки на 2 мл) фирмы Vince and Associates Clinical Research. К каждой пробирке прикрепляли этикетку, которая содержала следующую информацию:
Тип образца: сыворотка человека
Тип анализа: ФК
Протокол: FB825CLCT01
Номер субъекта: номер при рандомизации
Точка времени: см. протокол
Аликвота сыворотки: 1 или 2.
В пределах 90 минут после сбора обе аликвоты проб хранили в вертикальном положении при -70°C ± 10°C. Фармакокинетическое определение FB825 в сыворотке проводили проверенным методом ELISA.
Фармакодинамические измерения
Пробы крови для определения общего IgE и антител против препарата (ADA) отбирали в день 1 за 30 мин (± 5 мин) до начала вливания и на 5-й, 14 (± 1 день), 29 (± 2 дня), 85 (± 3 дня) и 140 (± 5 дней) день после вливания или при раннем прекращении.
Пробы крови для анализа уровня ADA отбирали в пробирки для забора крови на 3,5 мл (пробирки для отделения сыворотки BD Vacutainer® SST™) фирмы Vince and Associates Clinical Research. Пробы брали так, чтобы был минимальный объем крови на один размер пробирки для забора крови. По каждой точке времени получали как минимум 1,0 мл сыворотки. После получения проб крови пробирки переворачивали 5 раз и оставляли для образования сгустка крови на 30 мин при обычной температуре (19°C-24°C). Пробы центрифугировали при 2200 об/мин в течение 10 мин при комнатной температуре в центрифуге с подвесными стаканами. Двойные аликвоты сыворотки примерно равного объема (как минимум 500 мкл на пробу) переносили по стандартной лабораторной методике в 2 маркированные соответствующим образом пробирки для хранения (полипропиленовые криопробирки на 2 мл). Пробы на иммуногенность для измерения ADA анализировали валидированным методом ELISA.
Оценка безопасности
Безопасность и переносимость оценивали путем мониторинга и регистрации AE, результатов клинических лабораторных анализов (гематологии, коагулограмма, биохимический состав сыворотки, включая тесты на функцию печени, тесты на функцию щитовидной железы и исследование мочи), основных показателей состояния организма, результатов ЭКГ в 12 отведениях, данных по кардиотелеметрии и результатов медицинского осмотра.
Клинические лабораторные анализы
Брали образцы крови и мочи для гематологического анализа, коагуолограммы, биохимического анализа сыворотки (включая тесты на функцию печени), тестов на функцию щитовидной железы, исследования мочи и проверки на наркотики натощак (голодание в течение 2 или больше часов) в временные точки, указанные в графике мероприятий (таблица 1). Клинические лабораторные анализы (гематология, коагулограмма, биохимический анализ сыворотки [включая тесты на функцию печени] и исследование мочи) проводили при скрининге; регистрации; в день 1 до начала вливания и через 1 час (конец вливания), 8 и 24 часа (± 15 мин) после начала вливания; и в отдельные временные точки (± 15 мин до 48 часов после дозы) в дни 3, 5, 14, 29, 85 и 140.
Образцы использовались для клинических лабораторных анализов, включающих гематологический анализ, коагулограмму, биохимию сыворотки, функцию щитовидной железы и исследование мочи.
Для всех субъектов женского пола проводили анализ сыворотки на беременность (хорионический β-гонадотропин человека) при скрининге, регистрации, на 3-й день (через 48 часов после дозы) и при посещении под конец исследования (день 140). Субъектам женского пола, которые находились в постменопаузе, делали анализ FSH в сыворотке при скрининге.
Проводили анализ на поверхностный антиген гепатита B, антитела против вируса гепатита C и антитела против вируса иммунодефицита человека (1-го или 2-го типа) при скрининге.
При скрининге и в день −1 проводили проверку мочи на наркотики на предмет алкоголя, амфетаминов, барбитуратов, бензодиазепинов, метаболитов кокаина, никотина, метилендиоксиметамфетамина, опиатов, фенциклидина, пропоксифена и тетрагидроканнабинола.
Аномальные клинико-лабораторные значения отмечали как высокие или низкие (либо нормальные или аномальные) на основании контрольных диапазонов для каждого лабораторного параметра. Клиническую значимость определяли как любые вариации результатов, которые имели медицинское значение и могли привести к изменению медицинской тактики (например, активное наблюдение, диагностические меры или терапевтические меры). Если отмечалось клинически значимое изменение от результатов скрининга, то регистрировали клинически значимое значение и причины клинической значимости на странице AE в eCRF. Испытуемых продолжали отслеживать по дополнительным оценкам до тех пор, пока значения не достигали контрольного диапазона или значений при скрининге или же пока в последующем наблюдении больше не было медицинской необходимости.
Основные показатели состояния организма
Основные показатели состояния организма включали систолическое и диастолическое артериальное давление, частоту сердечных сокращений, частоту дыхания и температуру тела в полости рта. Перед проведением всех измерений испытуемые находились в сидячем положении не менее 5 минут, за исключением ортостатических измерений. В временные точки для ортостатических измерений, после выполнения всех измерений в сидячем положении, испытуемых переводили в положение лежа на спине на 5 минут, после чего у них измеряли артериальное давление и частоту сердечных сокращений; затем испытуемые стояли в течение 1 минуты, после чего у них опять измеряли артериальное давление и частоту сердечных сокращений.
Основные показатели состояния организма (систолическое и диастолическое артериальное давление, частоту сердечных сокращений, частоту дыхания и температуру тела в полости рта) получали при скрининге; регистрации; в день 1 до начала вливания и через 1 час (конец вливания), 2 и 4 часа (± 15 мин) и 8 и 24 часа (± 30 мин) после начала вливания; и в дни 3 (± 15 мин до 48 часов после дозы), 5, 14, 29, 85 и 140. Во время вливания основные показатели состояния организма получали через каждые 15 минут (± 5 мин). Ортостатические измерения проводили при регистрации; в день 1 до начала вливания и через 2, 4, 8 и 24 часа (± 15 мин) после начала вливания; и на 5-й день.
Клиническую значимость определяли как любые вариации результатов, которые имели медицинское значение и могли привести к изменению медицинской помощи (например, активного наблюдения, диагностических мер или терапевтических мер). Если отмечалось клинически значимое изменение от результатов скрининга, то регистрировали клинически значимое значение и причины клинической значимости на странице AE в eCRF субъекта. Испытуемых могли продолжать отслеживать по дополнительным оценкам до тех пор, пока значения не достигнут контрольного диапазона или значений при скрининге или же пока последующее наблюдение больше не будет необходимым с медицинской точки зрения.
Электрокардиограмма в 12 отведениях
Одиночные ЭКГ в 12 отведениях получали после того, как субъект находился в положении лежа на спине в течение не менее 5 минут в временные точки, указанные в графике мероприятий (таблица 1). Электрокардиограммы в 12 отведениях снимали при скрининге; регистрации; в день 1 до начала вливания и через 1 час (конец вливания), 8 и 24 часа (± 15 мин) после начала вливания; и в дни 3 (± 15 мин до 48 часов после дозы), 5, 14 и 140.
Оценка электрокардиограмм включала комментарии о том, были ли записи нормальными или аномальными, а также о ритме, наличии аритмии или дефектов проводимости, морфологии, признаках инфаркта миокарда и аномалиях сегмента ST, зубца T и зубца U. Кроме того, проводили и регистрировали измерения следующих интервалов: интервала RR, интервала PR, ширины QRS, интервала QT и QTcF.
Клиническую значимость определяли как любые вариации результатов, которые имели медицинское значение и могли привести к изменению медицинской тактики (например, активное наблюдение, диагностические меры или терапевтические меры). Если отмечалось клинически значимое изменение от результатов скрининга, то регистрировали клинически значимое значение и причины клинической значимости на странице AE в eCRF субъекта. Испытуемых продолжали отслеживать по дополнительным оценкам до тех пор, пока значения не достигали контрольного диапазона или значений при скрининге или же пока в последующем наблюдении больше не было медицинской необходимости.
Кардиотелеметрия
Кардиотелеметрия проводилась в временные точки, указанные в графике мероприятий (таблица 1). Мониторинг по кардиотелеметрии начинали в день 1 (начиная примерно за 30 мин до начала вливания) и продолжали в течение 4 часов после окончания вливания.
Клиническую значимость определяли как любые вариации результатов, которые имели медицинское значение и могли привести к изменению медицинской помощи (например, активного наблюдения, диагностических мер или терапевтических мер). Если отмечалось клинически значимое изменение от результатов исходной телеметрии в день 1 (примерно за 30 мин до начала вливания), то регистрировали клинически значимое значение и причины клинической значимости на странице AE в eCRF субъекта. Испытуемых продолжали отслеживать по дополнительным оценкам до тех пор, пока последующее наблюдение больше не было необходимым с медицинской точки зрения.
Медицинский осмотр
Проводили полный медосмотр и краткий медосмотр в временные точки, указанные в графике мероприятий (таблица 1). Полный медосмотр (включая оценку кожной эритемы) проводили при скрининге и в дни 3, 14 и 140. Краткий медосмотр (включая оценку кожной эритемы) проводили при регистрации (день −1) и в дни 5, 29 и 85. Медосмотр включал рост и вес при скрининге и только вес в другие дни.
Полный медосмотр включал оценку кожи (включая любые признаки кожной эритемы), головы, ушей, глаз, носа, горла, шеи, щитовидной железы, легких, сердца, сердечно-сосудистой системы, брюшной полости, лимфатических узлов, а также опорно-двигательного аппарата/конечностей. Промежуточный медосмотр проводился для оценки AEs или клинико-лабораторных отклонений.
Краткий медосмотр включал оценку состояния кожи (включая любые признаки кожной эритемы), легких, сердечно-сосудистой системы и брюшной полости (печень, селезенка).
Измеряли рост и вес и рассчитывали индекс массы тела только при скрининге. Вес измеряли во все другие времена медосмотра, указанные в графике мероприятий.
Подбор дозы для исследований на людях
Начальная доза в этом исследовании FIH 1-й фазы составляла 0,003 мг/кг, которая была установлена по NOAEL из 4-недельного токсикологического исследования с повторными дозами на нечеловекообразных приматах, самой близкой и наиболее подходящей модели для людей, с применением соответствующего коэффициента безопасности, а также из расчета минимального ожидаемого уровня биологического действия (MABEL) для FB825 на основе фармакологических данных in vitro и in vivo и токсикокинетических данных.
Значение NOAEL для FB825 в 4-недельном токсикологическом исследовании с повторными дозами у макак-крабоедов было принято равным 100 мг/кг. Эквивалентная доза для человека, рассчитанная в соответствии с Руководством для промышленности от Управления по контролю за продуктами и лекарствами (FDA) «Оценка максимальной безопасной начальной дозы при начальных клинических испытаниях терапевтических средств на взрослых здоровых добровольцах (июль 2005 г.)», составляет 38,8 мг/кг; а с применением соответствующего коэффициента безопасности (т.е. в 100 раз) максимальная рекомендуемая начальная доза для людей будет примерно равна 0,39 мг/кг.
Значение MABEL для FB825 было принято равным 0,1 мкг/мл. Было подсчитано, что однократная внутривенная доза в 0,003 мг/кг у людей дает системный уровень FB825 в пределах от 0,06 мкг/мл до 0,09 мкг/мл; таким образом, MABEL не должен превышаться во время исследования. Принимая более высокое значение из этого диапазона, ожидаемая максимальная концентрация FB825 в кровотоке при этой начальной дозе будет примерно в 4,4×104 раз меньше, чем Cmax для FB825 при уровне дозы NOAEL (100 мг/кг/день) у макак-крабоедов, получавших однократную внутривенную дозу FB825 (исследование 20031008). Это дает запас прочности в 3,7×104 раз в пересчете на мг/кг, что намного выше эквивалентной дозы в 111,6 мг/кг для человека при NOAEL (300 мг/кг).
Результаты
В этом исследовании оценивали безопасность, переносимость, фармакокинетику и иммуногенность однократных возрастающих доз FB825 при внутривенной инъекции в рандомизированном контролируемом плацебо двойном слепом исследовании на здоровых взрослых людях. Планировка и выбор исследуемой популяции для запланированного первого на людях (FIH) клинического исследования 1-й фазы исходили из необходимости получения начальных данных по безопасности, переносимости, ФК и иммуногенности FB825 для будущих клинических исследований. Данные получали в соответствии с графиком мероприятий, приведенным в таблице 1.
Введение FB825 безопасно для пациентов
Однократное 1-часовое внутривенное вливание FB825 в дозах 0,003, 0,03, 0,3, 1,5, 5 и 10 мг/кг было безопасным и хорошо переносилось здоровыми субъектами в клиническом испытании. Не было случаев смерти и испытуемые не бросали из-за возникающих при лечении неблагоприятных событий (TEAE). За исключением снижения гемоглобина, инфекции верхних дыхательных путей, инфекции мочевыводящих путей и огнестрельного ранения, все TEAE проходили к концу исследования.
Не отмечалось связанных с лечением или дозой тенденций по результатам клинико-лабораторных анализов, основным показателям состояния организма, результатам ЭКГ в 12 отведениях или по данным медосмотра.
Общий IgE снижается при введении FB825
Общий IgE снижался во всех временных точках после дозы в группах, получавших 1,5 и 10 м/кг FB825. В группах, получавших другие дозы (0,003, 0,03, 0,3 и 5 мг/кг FB825) и плацебо, общий IgE снижался в некоторые моменты времени после дозы, но не было общих тенденций (фиг. 1 и фиг. 2).
Только у 4 субъектов (по 1 в группах, получавших 0,03, 0,3 и 5 мг/кг FB825 и плацебо) были детектируемые ADA, причем только у 1 субъекта в группе, получавшей 0,03 мг/кг FB825, были активные ADA (фиг. 3).
Пример 3. Открытое поисковое исследование для оценки безопасности и эффективности FB825 у взрослых с атопическим дерматитом
Настоящее исследование спланировано для оценки изменения от исходного уровня общего IgE и аллерген-специфичного IgE у пациентов с атопическим дерматитом после внутривенного введения FB825 и для оценки клинической эффективности у этих пациентов. Это исследование также направлено на оценку безопасности FB825 у пациентов, получающих такое лечение, мониторинг изменений в клинической гематологии после внутривенного введения FB825 и изучение изменений от исходного уровня биомаркеров, включая регулируемый тимусом и активацией хемокин (TARC), эотаксин-3, тимусный стромальный лимфопоэтин (TSLP), периостин, IL-1a, IL-4, IL-5, IL-13, IL-16, IL-31 и M-CSF после внутривенного введения FB825.
FB825 представляет собой гуманизованный моноклональный иммуноглобулин G1 (IgG1), нацеленный на домен CεmX в B-лимфоцитах человека, экспрессирующих мембраносвязанный IgE (mIgE). FB825 может блокировать биологический путь синтеза IgE, тем самым способствуя лечению опосредованных IgE аллергических заболеваний. FB825 готовится в виде водного раствора и вводится пациентам посредством 1-часового внутривенного вливания по 5 мг/кг.
Процедуры исследования
Это было открытое поисковое исследование для оценки безопасности и эффективности FB825 у взрослых с атопическим дерматитом. В исследование были записаны 12 пациентов с атопическим дерматитом (AD), которые соответствовали критериям для включения в исследование. Все подходящие пациенты получали FB825 по 5 мг/кг посредством 1-часового внутривенного вливания в 1-й и 85-й день (таблица 2). После получения FB825 испытуемых госпитализировали в день 1 и день 85 и выписывали из больницы на следующий день для наблюдения за безопасностью (не менее 12 часов). Пациенты возвращались на место исследования в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169 для оценки безопасности и эффективности, а также для определения биомаркеров (таблица 2).
В некоторых случаях пациентов могли попросить посетить участок в день 78 для взятия проб крови. Целью взятия проб крови было измерение общего IgE, чтобы определить, нуждаются ли субъекты во второй дозе FB825. Испытуемые с изменением общего IgE от исходного уровня менее чем на 50% в день 78 могли получать вторую дозу FB825 (5 мг/кг). Испытуемым с изменением общего IgE от исходного уровня на 50% в день 78 могли назначить последнее посещение в день 85 и на этом завершить исследование. Для пациентов, получавших вторую дозу FB825, данные, собранные по второй дозе, анализировали и обобщали так же, как и полученные для первой дозы. В некоторых случаях пациенты получали вторую дозу FB825 в 5 мг/кг путем 1-часового внутривенного вливания в день 85. Перед получением FB825 в день 85 пациентов госпитализировали и выписывали из больницы на следующий день для наблюдения за безопасностью (не менее 12 часов). Пациенты возвращались на место исследования в дни 92, 99, 113, 141 и 169 для оценки безопасности и эффективности.
При запланированных посещениях измеряли общий IgE в сыворотке и определяли антиген-специфичный IgE для изучения изменений от исходного уровня после внутривенного введения 5 мг/кг FB825.
Пациентов обследовали для проведения оценки клинической эффективности при запланированных посещениях, включая визуально-аналоговую шкалу (VAS) зуда, индекс площади и тяжести экземы (EASI), показатель степени тяжести атопического дерматита (SCORAD), глобальную оценку исследователя (IGA) для AD и площадь поверхности тела (BSA) с симптомами AD.
Данные по безопасности, включая AEs и лабораторные анализы, рассматривались главным исследователем (PI). Продолжительность участия субъектов в исследовании составляла примерно 24 недели.
Отбор популяции для исследования
Испытуемых мужчин и женщин с атопическим дерматитом записывали в одном и том же месте исследования. В общей сложности было зарегистрировано 15 субъектов для получения по меньшей мере 12 оцениваемых субъектов.
Критерии отбора
(i) Основные критерии включения:
• испытуемые мужчины или женщины в возрасте от 20 до 65 лет включительно;
• подтвержденный врачом диагноз хронического атопического дерматита на основании 3-летней истории симптомов по пересмотренным Эйнифилдом критериям Ханнифина и Райки и подтвержденный положительным результатом на аллерген-специфичный IgE при скрининге;
• индекс площади и тяжести экземы (EASI)  14 баллов при скрининге и основном посещении;
14 баллов при скрининге и основном посещении;
• глобальная оценка исследователя (IGA) 3 баллов (по 5-балльной шкале) при скрининге и основном посещении;
• поражено AD 10% площади поверхности тела (BSA) при скрининге и основном посещении;
• история неадекватной реакции на постоянный (1 месяц) режим топических кортикостероидов или ингибиторов кальциневрина при лечении AD в пределах 3 месяцев до скрининга (схема местного применения кортикостероидов означает от средней до высокой активности при нанесении по меньшей мере 28 дней или при максимальной продолжительности, рекомендованной при назначении препарата);
• пациенты должны применять постоянные дозы смягчающего средства, предусмотренного для атопического дерматита, по два раза в день в течение по меньшей мере 7 дней до основного посещения;
• испытуемые женщины с детородным потенциалом должны применять как минимум две формы контроля над рождаемостью: одна – барьерная защита (т.е. презерватив или женский презерватив), а другая – один из приемлемых методов контроля над рождаемостью (т.е. диафрагма, внутриматочное устройство, гормональные контрацептивы или воздержание) на протяжении всего исследования; хирургически стерильные субъекты (т.е. гистерэктомия, двусторонняя перевязка маточных труб или двусторонняя овариэктомия) или в постменопаузе (определяется как аменорея в течение 12 месяцев подряд и документированный уровень фолликулостимулирующего гормона в сыворотке >40 мЕ/мл) должны рассматриваться как не обладающие детородным потенциалом; все испытуемые женщины должны иметь отрицательный анализ сыворотки на беременность перед дозированием; они должны применять указанный выше метод контрацепции на протяжении всего исследования и в течение 16 недель или 5 периодов полураспада после последнего введения FB825;
• вес тела ≥ 40 кг при скрининге и индекс массы тела от 18,0 до 30,0 кг/м2 включительно;
• нормальная, как определено исследователем, электрокардиограмма (ЭКГ) в 12 отведениях с нормальными параметрами сердечной проводимости:
– частота сердечных сокращений от 45 до 100 ударов в минуту,
– интервал QT с коррекцией по Fridericia (QTcF) ≤ 450 миллисекунд (мужчины) или ≤ 470 миллисекунд (женщины) и
– интервал QRS менее 120 миллисекунд;
• субъект здоров, за исключением атопических заболеваний, установленных исследователем на основании результатов клинико-лабораторных анализов, проведенных при скрининге, а если результаты выходят за пределы нормальных контрольных диапазонов, то субъект может быть включен только, если исследователь посчитает эти аномалии или отклонения от нормы не значимыми клинически, причем это определение должно быть записано в исходном документе субъекта и подписано исследователем; это не относится к лабораторным отклонениям, перечисленным в критериях исключения (по критериям Отдела микробиологии и инфекционных заболеваний);
• субъект способен представить письменное информированное согласие;
• субъект согласен соблюдать все требования протокола.
(ii) Основные критерии исключения:
• испытуемые женщины, которые беременны или кормят грудью;
• субъект находится на диете или плохо питается;
• в анамнезе у субъекта была сердечная аритмия (любая клинически значимая);
• у субъекта положительный результат анализа на поверхностный антиген гепатита B, антитела к вирусу гепатита C или антитела к вирусу иммунодефицита человека при скрининге;
• в анамнезе у субъекта было злоупотребление алкоголем или наркотиками, которое, по мнению исследователя, могло бы помешать или поставить под угрозу полное участие пациентов в исследовании;
• субъект находится под судебным надзором или кураторством;
• у субъекта имеется клинически значимое, активное в настоящее время или сопутствующее желудочно-кишечное, сердечно-сосудистое, неврологическое, психиатрическое, метаболическое, почечное, печеночное, респираторное (за исключением неосложненного аллергического ринита), воспалительное, иммунологическое, эндокринное, инфекционное заболевание или диабет и он, по мнению исследователя, не подходит для участия в исследовании;
• в анамнезе у субъекта была предыдущая анафилактическая реакция;
• у субъекта есть такое заболевание, которое, по мнению исследователя, может поставить под угрозу исследование или благополучие субъекта либо помешать ему выполнить или выполнять требования исследования;
• субъект получал какие-либо препараты иммуноглобулина или препараты крови в пределах 3 месяцев до введения дозы;
• субъект получал биологические препараты:
– он получал какие-либо истощающие клетки средства, не ограничиваясь лишь ритуксимабом, в течение 6 месяцев до введения дозы или до нормализации числа лимфоцитов, смотря что было дольше,
– он получал другие биологические препараты в течение 5 периодов полураспада (если он известен) или 16 недель, смотря что было дольше, до введения дозы;
• у субъекта была одна или несколько следующих лабораторных аномалий при скрининге, как определено Отделом микробиологии и инфекционных заболеваний в Таблице токсичности для взрослых, 2007 [лабораторные значения могут быть преобразованы в эквивалентные стандартные единицы, а повторное тестирование аномальных лабораторных показателей, которые могут привести к исключению, разрешается один раз (без предварительного согласия спонсора), причем повторное тестирование должно проводиться во время незапланированного посещения на этапе скрининга (до основного посещения)]:
– аспартатаминотрансфераза либо аланинаминотрансфераза более чем в 2 раза больше верхнего предела нормы [ULN]) или выше,
– общий билирубин ≥ 1,5×ULN,
– креатинин в сыворотке ≥ 1,6×ULN,
– любые другие лабораторные аномалии, превышающие или равные 2-кратным, за исключением уровня IgE, числа эозинофилов, катионного белка эозинофилов (ECP) и лабораторных показателей, приведенных выше, причем лабораторные значения могут быть преобразованы в эквивалентные стандартные единицы, а повторное тестирование аномальных лабораторных показателей, которые могут привести к исключению, разрешается один раз (без предварительного согласия спонсора), причем повторное тестирование должно проводиться во время незапланированного посещения на этапе скрининга (до основного посещения);
• субъект получал какие-либо одобренные или неутвержденные (т.е. исследовательские) иммунотерапевтические препараты в пределах 3 последних месяцев;
• субъект применял любые из следующих классов лекарств (по рецепту или без рецепта):
– интраназальные кортикостероиды (например, флутиказон пропионат) в пределах 30 дней до введения дозы,
– системные кортикостероиды (например, преднизон) в пределах 30 дней до введения дозы,
– модификаторы лейкотриенов (например, монтелукаст) в пределах 30 дней до введения дозы,
– иммунодепрессанты (например, соли золота, метотрексат, азатиоприн, циклоспорин) в пределах последних 30 дней до введения дозы,
– иммуномодулирующие препараты (например, IFN-γ) в пределах последних 30 дней до введения дозы,
– антитела против IgE (например, омализумаб) в пределах последнего 1 года до введения дозы,
– аллергеновую иммунотерапию в пределах последнего 1 года до введения дозы,
– пероральную ингаляцию кортикостероидов (например, будесонида) в пределах последних 30 дней до введения дозы;
• субъект проходил фототерапию в пределах 4 недель до введения дозы;
• субъект получал живую вакцину в пределах 12 недель до введения дозы;
• у субъекта в анамнезе, по мнению исследователя, была известная или предполагаемая иммуносупрессия, в том числе оппортунистические инфекции (например, туберкулез) в анамнезе;
• у субъекта в анамнезе была злокачественная опухоль в пределах 5 лет до периода скрининга;
• высокий риск заражения паразитами: факторы риска паразитарных заболеваний (проживание в эндемической области, хронические желудочно-кишечные симптомы, поездки в течение последних 6 месяцев в регионы, где эндемичны геогельминтозы и/или хроническая иммуносупрессия) И свидетельства колонизации или заражения паразитами при анализе кала на яйца и паразиты, причем анализ кала на яйца и паразиты должен проводиться только у пациентов с факторами риска и числом эозинофилов, более чем в два раза превышающим верхний предел у нормальных субъектов.
Процедуры исследования
(i) Дозировка и внутривенное введение исследуемого препарата
Субъектам с атопическим дерматитом вводили две дозы FB825 по 5 мг/кг. Субъекты получали FB825 путем 1-часового внутривенного вливания утром в день 1 и день 85. В некоторых случаях требовалось голодание не менее 2 часов. Прием воды не допускается в течение 1 часа перед дозировкой и 1 часа после дозировки. Вводимое количество FB825 можно регулировать в зависимости от массы тела субъекта. Соответствующее количество препарата разбавляли 250 мл 0,9% раствора хлорида натрия. FB825 вводили внутривенно на протяжении 1 часа с помощью программируемого мерного инфузионного устройства. Конечный разбавленный продукт в 0,9% хлориде натрия после восстановления нужно использовать как можно скорее, в пределах 8 часов. Восстановленный FB825 может храниться при температуре от 2°C до 25°C максимум 8 часов перед использованием.
(ii) Предыдущие, сопутствующие и запрещенные лекарства
• Предыдущие лекарства и терапии
Информация о приеме предшествующих лекарств субъектом в пределах 30 дней до получения информированного согласия регистрируется в CRF субъекта.
• Предварительные/сопутствующие лекарства и процедуры
Любое лечение (включая пищевые добавки) или процедуры, проводимые с момента подписания ICF до конца исследования, считаются сопутствующими и регистрируются в CRF. Это включает разрешенные лекарства, действующие на момент получения согласия.
Базальная терапия AD во время исследования описана ниже:
– все пациенты должны наносить увлажняющие средства по меньшей мере два раза в день в течение по меньшей мере 7 дней подряд перед дозированием и продолжать лечение на всем протяжении исследования, причем разрешаются все типы увлажнителей, но пациенты не могут начинать лечение с рецептурными увлажнителями или увлажнителями, содержащими добавки, в период скрининга или во время исследования;
– пациенты могут продолжать применение стабильных доз рецептурных увлажняющих средств или увлажняющих средств, содержащих добавки, если они начаты до скрининга, а начиная с 1-го дня/основного посещения все пациенты должны начинать лечение местным кортикостероидом (TCS) по стандартизированной схеме в соответствии со следующими инструкциями:
– ежедневно наносить TCS со средней активностью на участки с активными поражениями, а на участки с тонкой кожей (лицо, шея, зоны опрелостей и область гениталий, участки с атрофией кожи и т.п.) или на участки, где продолжение применения TCS со средней активностью считается небезопасным, следует наносить TCS с низкой активностью;
– после того, как поражения будут под контролем (чистые или почти чистые), перейти с TCS со средней активностью на TCS с низкой активностью и обрабатывать ежедневно в течение 7 дней, а затем остановиться;
– если поражения вернутся, то возобновить лечение с помощью TCS со средней активностью, применяя описанный выше понижающий подход после прекращения поражения;
– при поражениях, сохраняющихся или ухудшающихся при ежедневном применении TCS со средней активностью, пациентов можно лечить (спасать) с помощью TCS с высокой или сверхвысокой активностью, если только TCS с высокой активностью не будут считаться небезопасными;
– наблюдать пациентов на признаки локальной или системной токсичности TCS и при необходимости понижать или прекращать лечение;
– регистрировать тип и количество топических препаратов, используемых во время исследования, причем количество использованного TCS определяется путем взвешивания тюбика при каждом посещении (см. подробное руководство по исследованию);
– рекомендуется, чтобы в качестве TCS со средней активностью пациенты использовали крем с 0,05% флутиказона пропионата, крем с 0,1% мометазона фуроата или крем с 0,06% бетаметазона, а в качестве TCS с низкой активностью – мазь с 1% гидрокортизона;
– если требуется спасение с помощью TCS, то пациентам в качестве TCS с высокой активностью рекомендуется использовать крем с 0,05% флуоцинонида, мазь с 0,25% дезоксиметазона, а в качестве TCS со сверхвысокой активностью – мазь с 0,05% клобетазола пропионата;
– не использовать увлажнители и TCS на одних и тех же участках в одно и то же время в течение дня, а на участки, не обработанные TCS, следует наносить увлажнители два раза в день – утром и вечером.
Предварительные лекарства/процедуры: лекарства, принимаемые или процедуры, выполняемые перед дозированием.
Сопутствующие лекарства/процедуры: лекарства, принимаемые или процедуры, выполняемые после внутривенного введения исследуемого препарата вплоть до последнего посещения (EOS).
• Запрещенные сопутствующие лекарства:
– топический такролимус и пимекролимус;
– системные кортикостероиды, если только субъекты не получают неотложное лечение;
– ингибиторы лейкотриенов;
– аллергеновая иммунотерапия;
– системное лечение AD с помощью иммунодепрессантов/иммуномодуляторов (включая, без ограничения, циклоспорин, микофенолат-мофетил, IFN-γ, азатиоприн, метотрексат или биологические препараты);
– лечение с помощью живых (аттенюированных) вакцин;
– традиционная китайская медицина.
• Запрещенные сопутствующие процедуры:
– хирургические процедуры;
– сопутствующие ультрафиолетовые (УФ) процедуры (фототерапия [NBUVB, UVB, UVA1 или PUVA]);
– не разрешается загорать в кровати/кабинке во время исследования;
– пациентам разрешается не более двух отбеливающих ванн в неделю во время участия в исследовании.
(iii) Обработка при инфузионной или аллергической реакции
Внутривенное введение исследуемого препарата должно проводиться под наблюдением обученного медицинского персонала и там, где имеются средства для лечения аллергических реакций. Если субъект испытывает связанную с вливанием реакцию, то он должен проходить симптоматическое лечение с поддерживающей терапией, дополнительным мониторингом и соответствующей медицинской терапией, которая может включать антигистаминные препараты и/или кортикостероиды, если нужно. Вливание при исследовании можно остановить, а субъекта отслеживать до окончания исследования. Отметить введенное количество. Если субъект испытывает симптомы, типичные для аллергической реакции (например, одышка, анафилаксия, крапивница, ангионевротический отек), то внутривенное введение исследуемого препарата следует немедленно и окончательно прекратить.
Подозрение на анафилаксию следует оценивать по клиническим диагностическим критериям, установленным Национальным институтом аллергии и инфекционных заболеваний, которые приведены в Приложении 12-2.
При этих и других обстоятельствах субъекты могут получать надлежащую медицинскую помощь по усмотрению исследователя.
В случае аллергических реакций пациентов можно спасать с помощью запрещенных лекарств или процедур для лечения недопустимых симптомов AD:
– при медицинской необходимости (т.е. для контроля недопустимых симптомов AD) участвующим в исследовании пациентам после 2-й недели может предоставляться неотложное лечение AD системными кортикостероидами при дозах преднизолона менее 1 мг/кг/день и не более 3 дней;
– непосредственно перед проведением любого неотложного лечения пациентов подвергают оценке эффективности и безопасности (например, оценке тяжести заболевания, безопасности в лаборатории).
Процедуры исследования и методы оценки
В следующих разделах описаны процедуры исследования и получаемые данные. Субъектов оценивал один и тот исследователь или персонал, когда это было возможно. График и оценки представлены в таблице 2. Исходные характеристики приведены в таблице 3.
(i) Конечные показатели
• Первичные показатели:
– изменение от исходного уровня общего IgE в день 85 и в день 169/под конец исследования (EOS);
– изменение от исходного уровня аллерген-специфичного IgE в день 85 и в день 169/EOS.
• Показатели по биомаркерам:
– изменение от исходного уровня общего IgE в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169;
– изменение от исходного уровня аллерген-специфичного IgE в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169;
– изменение от исходного уровня биомаркеров, включая регулируемый тимусом и активацией хемокин (TARC), эотаксин-3, тимусный стромальный лимфопоэтин (TSLP), периостин, IL-1a, IL-4, IL-5, IL-13, IL-16, IL-31 и M-CSF после внутривенного введения FB825 в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169.
• Показатели эффективности:
– изменение от исходного уровня зуда по визуально-аналоговой шкале (VAS) в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169;
– изменение от исходного уровня индекса площади и тяжести экземы (EASI) в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169;
– изменение от исходного уровня показателя степени тяжести атопического дерматита (SCORAD) в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169;
– изменение от исходного уровня глобальной оценки исследователя (IGA) для атопического дерматита в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169;
– изменение от исходного уровня площади поверхности тела (BSA), пораженной атопическим дерматитом, в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169;
– изменение от исходного уровня биомаркеров, включая регулируемый тимусом и активацией хемокин (TARC), эотаксин-3, тимусный стромальный лимфопоэтин (TSLP), периостин, IL-1a, IL-4, IL-5, IL-13, IL-16, IL-31 и M-CSF после внутривенного введения FB825 в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169.
Безопасность оценивали путем мониторинга и регистрации неблагоприятных событий (AE) и серьезных неблагоприятных событий (SAE); результатов медосмотра и основных показателей состояния организма (систолическое и диастолическое артериальное давление, частота сердечных сокращений, частота дыхания и температура тела), результатов клинических лабораторных анализов (гематология, коагулограмма, биохимический анализ сыворотки [включая тесты на функцию печени, уровень глюкозы в крови] и исследование мочи); ЭКГ в 12 отведениях.
(ii) Определение биомаркеров
Пробы крови для измерения общего и аллерген-специфичного IgE, регулируемого тимусом и активацией хемокина (TARC), эотаксина-3, тимусного стромального лимфопоэтина (TSLP), периостина, IL-1a, IL-4, IL-5, IL-13, IL-16, IL-31 и M-CSF анализировали по методологии, описанной в отдельном отчете.
Брали пробы крови. Фактическое время взятия пробы и проблемы с отбором проб, если они были, отмечали в CRF. На 1 пробу отбирали примерно 5 мл венозной крови в следующие временные точки.
• Общий иммуноглобулин/аллерген-специфичный IgE:
– период скрининга: в любое время;
– день 1 и день 85: за 2 часа до начала вливания;
– в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169 после внутривенного введения FB825: в любое время дня.
• TARC, эотаксин-3, TSLP, периостин, IL-1a, IL-4, IL-5, IL-13, IL-16, IL-31 и M-CSF:
– в день 1 и день 85 за 2 часа до начала вливания и в любое время в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169.
(iii) Оценка клинической эффективности
• Визуально-аналоговая шкала (VAS) зуда
Для измерения зуда применяли визуально-аналоговую шкалу (VAS) зуда. Пациентам предлагали дать числовую оценку, отражающую интенсивность их симптомов, по шкале от 0 до 10, где 0 – нет симптомов, а 10 – самые худшие симптомы. Субъектам предлагали выполнить измерение при скрининге, в день 1 и день 85 перед дозированием, в день 2 и день 86 перед выпиской и в любое время в дни 8, 15, 29, 57, 92, 99, 113, 141 и 169.
• Показатель степени тяжести атопического дерматита (SCORAD)
Показатель SCORAD – проверенная система оценки при атопическом дерматите (AD). Для измерения степени AD применяется правило девяток на переднем/заднем чертеже воспалительных поражений пациента. Степень оценивается от 0 до 100. SCORAD по интенсивности состоит из 6 пунктов: эритема, отек/появление папул, исцарапанность, лихенизация, выделения/корки и сухость. Каждый пункт может быть оценен по шкале от 0 (нет) до 3 (тяжелый). Субъективные пункты включают ежедневный зуд и бессонницу. Формула показателя SCORAD: A/5 + 7B/2 + C. В этой формуле A означает степень (0–100), B означает интенсивность (0–18), а C – субъективные симптомы (0–20). Максимальная оценка индекса SCORAD равна 103. Субъектам предлагали выполнить измерение при скрининге, в день 1 и день 85 перед дозированием, в день 2 и день 86 перед выпиской и в любое время в дни 8, 15, 29, 57, 92, 99, 113, 141 и 169.
• Индекс площади и тяжести экземы (EASI)
В системе оценки EASI используется определенный процесс для оценки тяжести признаков экземы и степени поражения. Степень и тяжесть признаков экземы оценивали в 4 частях тела, а общий балл представляет собой сумму баллов по 4 участкам с коррекцией по множителям. Оценка EASI составляет от 0 до 72. Субъектам предлагали выполнить измерение при скрининге, в день 1 и день 85 перед дозированием, в день 2 и день 86 перед выпиской и в любое время в дни 8, 15, 29, 57, 92, 99, 113, 141 и 169.
• Глобальная оценка исследователя (IGA) для AD
IGA позволяет исследователям оценивать общую тяжесть заболевания в каждый данный временной точке и состоит из 5-балльной шкалы тяжести от чистого тела до очень тяжелого заболевания (0 = чисто, 1 = почти чисто, 2 = легкое заболевание, 3 = умеренное заболевание и 4 = тяжелое заболевание). Субъектам предлагали выполнить измерение при скрининге, в день 1 и день 85 перед дозированием, в день 2 и день 86 перед выпиской и в любое время в дни 8, 15, 29, 57, 92, 99, 113, 141 и 169.
• Площадь поверхности тела (BSA) с симптомами AD
Её измеряли как часть A (степень) SCORAD. Субъектам предлагали выполнить измерение при скрининге, в день 1 и день 85 перед дозированием, в день 2 и день 86 перед выпиской и в любое время в дни 8, 15, 29, 57, 92, 99, 113, 141 и 169.
(iii) Оценка безопасности
Безопасность оценивали путем мониторинга и регистрации неблагоприятных событий (AE) и серьезных неблагоприятных событий (SAE); результатов медосмотра и основных показателей состояния организма (систолическое и диастолическое артериальное давление, частота сердечных сокращений, частота дыхания и температура тела), результатов клинических лабораторных анализов (гематология, коагулограмма, биохимический анализ сыворотки [включая тесты на функцию печени] и исследование мочи) и ЭКГ в 12 отведениях. Общая сводка по числу пациентов с неблагоприятными событиями за дни 15-168 периода лечения-SAF представлена в таблице 4.
• Неблагоприятные события
В разделе «Неблагоприятные события» (раздел 7) описаны неблагоприятные события (AE), серьезные неблагоприятные события (SAE) и неблагоприятные события, представляющие особый интерес, которые были собраны во время исследования.
• Медицинский осмотр
Полный медосмотр проводили в временные точки, указанные в графике мероприятий.
Полный медосмотр включал оценку кожи (включая любые признаки кожной эритемы), головы, ушей, глаз, носа, горла, шеи, щитовидной железы, легких, сердца, сердечно-сосудистой системы, брюшной полости, лимфатических узлов, а также опорно-двигательного аппарата/конечностей. Промежуточный медосмотр проводился по усмотрению исследователя, при необходимости, для оценки AEs или клинико-лабораторных отклонений. Измеряли рост и вес и рассчитывали индекс массы тела только при скрининге. Вес также измеряли при медосмотре во все другие временные точки, как указано в графике мероприятий для исследования. Вес тела записывали в килограммах (кг) с точностью до 1 десятичного знака в домашней одежде (без пальто и обуви), а рост (без обуви) измеряли в сантиметрах (см) без десятичных знаков.
• Основные показатели состояния организма
Основные показатели состояния организма включают систолическое и диастолическое артериальное давление, частоту сердечных сокращений, частоту дыхания и температуру тела. Перед проведением всех измерений субъекты находились в сидячем положении не менее 5 минут. Основные показатели состояния организма измеряли в временные точки, указанные в графике мероприятий.
Когда процедуры перекрываются и приходятся на один и тот же момент времени, то порядок проведения процедур заключается в измерении основных показателей состояния организма, а затем ЭКГ.
Исследователь может определить, является ли какой-либо из основных показателей состояния организма клинически значимым или нет. Клиническая значимость определяется как любые вариации результатов, которые имеют медицинское значение и могут привести к изменению медицинской помощи (например, активного наблюдения, диагностических мер или терапевтических мер). Если отмечается клинически значимое изменение от результатов скрининга, то клинически значимое значение и причины клинической значимости заносятся на страницу AE в CRF субъекта. Исследователь может продолжить отслеживать испытуемых по дополнительным оценкам до тех пор, пока значения не достигнут контрольного диапазона или значений при скрининге или же пока исследователь не установит, что в последующем наблюдении больше нет медицинской необходимости.
• Клинические лабораторные анализы
Клинические лабораторные анализы проводили на месте. Брали кровь и мочу натощак (голодание в течение 2 или больше часов) в временные точки, указанные в графике мероприятий.
Проводили следующие анализы по гематологии, коагуляции крови, биохимии сыворотки (включая тесты на функцию печени и функцию щитовидной железы) и исследованию мочи:
– гематология: гематокрит (Hct), гемоглобин (Hb), среднее содержание гемоглобина в эритроцитах (MCH), средняя концентрация гемоглобина в эритроцитах (MCHC), средний объем эритроцитов (MCV), число тромбоцитов, число эритроцитов (RBC) и лейкоцитов (WBC) и дифференциальная формула (абсолютно и в процентах);
– коагулограмма: международное нормализованное соотношение (INR), частичное тромбопластиновое время (PTT) и протромбиновое время (PT);
– биохимический анализ сыворотки: ALT, альбумин, щелочная фосфатаза, амилаза, анионная разница, AST, бикарбонат, билирубин (общий и прямой), азот мочевины крови (BUN), кальций, диоксид углерода, хлорид, холестерин (общий, липопротеинов высокой плотности и расчетный липопротеинов низкой плотности), креатинфосфокиназа, креатинин, гамма-глутамилтрансфераза (γ-GT), глобулин, глюкоза, лактатдегидрогеназа (LDH), липаза, магний, фосфор, калий, натрий, общий белок, триглицериды, тропонин I или T и мочевая кислота;
– функция щитовидной железы: свободный T4 и тиреотропный гормон;
– исследование мочи: внешний вид, билирубин, цвет, глюкоза, кетоны, лейкоцитарная эстераза, микроскопия (проводится, если индикаторная полоска положительна; включает бактерии, цилиндры, кристаллы, эпителиальные клетки, эритроциты и лейкоциты), нитриты, скрытая кровь, pH, белок, удельный вес, мутность и уробилиноген;
Анализ сыворотки на беременность (хорионический β-гонадотропин человека) должен проводиться у всех субъектов женского пола с детородным потенциалом при скрининге с подтверждением результата перед дозированием. Анализы мочи на беременность проводились в дни 8, 15, 29, 57, 85, 92, 99, 113, 141 и 169. У женщин, которые находились в постменопаузе, при скрининге проводился анализ на фолликулостимулирующий гормон в сыворотке.
При скрининге проводится анализ на поверхностный антиген гепатита B, антитела против вируса гепатита C и антитела против вируса иммунодефицита человека.
Аномальные клинико-лабораторные значения отмечали как высокие или низкие (либо нормальные или аномальные) на основании контрольных диапазонов для каждого лабораторного параметра. Исследователь может определить, являются ли какие-либо аномально высокие или низкие результаты клинически значимыми или нет. Клиническая значимость определяется как любые вариации результатов, которые имеют медицинское значение и могут привести к изменению медицинской помощи (например, активного наблюдения, диагностических мер или терапевтических мер). Если отмечается клинически значимое изменение от результатов скрининга, то клинически значимое значение и причины клинической значимости заносятся на страницу AE в CRF. Исследователь может продолжить отслеживать испытуемых по дополнительным оценкам до тех пор, пока значения не достигнут контрольного диапазона или значений при скрининге или же пока исследователь не установит, что в последующем наблюдении больше нет медицинской необходимости.
Составляли список клинически значимых лабораторных значений для отдельных субъектов. Представляли сводку о количестве и проценте субъектов с клинически значимыми лабораторными показателями в каждой временной точке.
• Электрокардиограммы
Одиночные ЭКГ в 12 отведениях получали после того, как субъект находился в положении лежа на спине не менее 5 минут в временные точки, указанные в графике мероприятий. Анализ электрокардиограмм включает комментарии о том, были ли записи нормальными или аномальными, ритме, наличии аритмии или дефектов проводимости, морфологии, любых признаках инфаркта миокарда или аномалии сегмента ST, зубца T и зубца U. Кроме того, проводили измерения и отмечали следующие интервалы: интервал RR, интервал PR, ширина QRS, интервал QT и QTcF.
Неблагоприятные события
Общая сводка по числу пациентов с неблагоприятными событиями за дни 15-168 периода лечения-SAF представлена в таблице 4.
(i) Определения
• Неблагоприятные события (AE)
AE определяется как любое нежелательное медицинское явление, связанное с применением лекарства у людей, независимо от того, считается оно связанным с препаратом или нет. Субъектам будет предложено связаться с исследователем в любое время в период исследования, если возникнут какие-либо симптомы.
TEAE определяется как любое явление, которого не было до воздействия исследуемого препарата, или любое уже существующее явление, которое ухудшается по интенсивности или частоте после воздействия.
Неблагоприятной реакцией является любое AE, вызванное препаратом. Неблагоприятные реакции представляют собой подмножество всех предполагаемых нежелательных реакций, для которых есть основания сделать вывод, что они вызваны препаратом.
Предполагаемой неблагоприятной реакцией является такое AE, для которого существует разумная вероятность того, что оно вызвано исследуемым препаратом. В целях отчетности о безопасности новых исследовательских препаратов “разумная вероятность” означает то, что имеются данные, свидетельствующие о причинно-следственной связи между исследуемым препаратом и AE. Предполагаемая неблагоприятная реакция подразумевает меньшую степень уверенности в отношении причинности, чем неблагоприятная реакция, которая означает любое AE, вызванное исследуемым препаратом.
AE или предполагаемая неблагоприятная реакция считается “неожиданной”, если она не приведена в IB или с такой специфичностью или серьезностью, которая наблюдалась при тестировании исследуемого препарата; или же, если IB не требуется или не доступен, она не согласуется с информацией о рисках, описанной в общем плане исследования или где-либо еще в текущей заявке. Например, по этому определению некроз печени будет неожиданным (в силу большей тяжести), если в IB указано только повышение печеночных ферментов или гепатит. Точно так же церебральная тромбоэмболия и церебральный васкулит будет неожиданным (в силу большей специфичности), если в IB указано только расстройство церебральных сосудов. “Неожиданными” по этому определению будут и такие AE или предполагаемые неблагоприятные реакции, которые приведены в IB как встречающиеся у целого класса лекарств или как ожидаемые из фармакологических свойств препарата, но конкретно не упоминаются как встречающиеся с данным препаратом при исследовании.
• Серьезные неблагоприятные события (SAE)
AE или предполагаемая неблагоприятная реакция считается SAE, если, по мнению исследователя или спонсора, она приводит к любому из следующих результатов:
– смерти;
– опасному для жизни AE;
– госпитализации в стационаре или продлению существующей госпитализации;
– постоянной или значительной нетрудоспособности или существенному нарушению способности выполнять нормальные жизненные функции;
– врожденной аномалии или врожденному дефекту.
Важные медицинские события, которые могут не приводить к смерти, угрожать жизни или требовать госпитализации, могут считаться серьезными, если на основании соответствующего медицинского суждения они могут поставить субъекта под угрозу и потребовать медицинского или хирургического вмешательства для предотвращения одного из результатов, перечисленных в этом определении. Примеры таких медицинских событий включают аллергический бронхоспазм, требующий интенсивного лечения в отделении неотложной помощи или на дому, дискразию крови или судороги, которые не приводят к госпитализации в стационаре, или появление зависимости от наркотиков или злоупотребления наркотиками.
AE или предполагаемая неблагоприятная реакция считается “опасной для жизни”, если, по мнению исследователя или спонсора, её возникновение ставит субъекта под непосредственный риск смерти. Это не включает такие AE или предполагаемые неблагоприятные реакции, которые, если бы они произошли в более тяжелой форме, могли бы привести к смерти.
• Неблагоприятные события, представляющие особый интерес
Представляющие особый интерес неблагоприятные события включают следующие события:
– субъект испытывает возникающее при лечении AE (TEAE) – анафилаксию;
– субъект испытывает SAE (раздел 7.1.2);
– субъект испытывает устойчивое удлинение интервала QT (> 500 миллисекунд или изменение на ≥60 миллисекунд от исходного уровня) в течение не менее 30 минут либо ишемические изменения при повторных ЭКГ или постоянную симптоматическую аритмию;
– реакции гиперчувствительности, включая анафилаксию, нарушения щитовидной железы, кожная эритема, тромбоцитопения, анемия, гемолиз, нейтропения, гепатотоксичность и нефротоксичность;
– у отдельных субъектов встречаются следующие лабораторные параметры:
ALT или AST ≥ 5×ULN,
билирубин ≥ 2×ULN,
число тромбоцитов, 1 степень ≤ 99,999×109/л,
гемоглобин ≤ 105 г/л,
абсолютное число нейтрофилов, 1 степень ≤ 1,5×109/л,
азот мочевины или креатинин в крови повышен до > 2×ULN.
(ii) Выявление неблагоприятных событий
Исследователь несет ответственность за то, чтобы все AE и SAE были отмечены в CRF и переданы в Fountain. Неблагоприятные события оценивали от времени проведения скрининга до завершения всех процедур исследования и выписки из исследования.
При каждом посещении для исследования или оценки субъектам задавали стандартные вопросы, чтобы выявить любые связанные с лечением изменения в их благополучии. Их также спрашивали, были ли они госпитализированы, были ли у них несчастные случаи, использовали какие-либо новые лекарства или изменяли схемы приема сопутствующих лекарств (как отпускаемых по рецепту, так и без рецепта).
Наряду с наблюдением испытуемых, случаи AE устанавливали по данным, собранным на странице AE в CRF (например, лабораторные значения, результаты медосмотра и изменения ЭКГ) или в других документах, имеющих отношение к безопасности субъектов.
Оценка тяжести. Для оценки тяжести (или интенсивности) AE использовали Общую терминологию критериев для нежелательных явлений (CTCAE) при классификации степени тяжести инфузионных реакций.
– 1-я степень, легкая: преходящий или легкий дискомфорт (<48 часов); не требуется медицинское вмешательство/терапия;
– 2-я степень, средняя: ограничение активности от легкой до умеренной – может потребоваться некоторая помощь; не требуется или требуется минимальное медицинское вмешательство/терапия;
– 3-я степень, сильная: заметное ограничение активности, обычно требуется некоторая помощь; требуется медицинское вмешательство/терапия, возможна госпитализация;
– 4-я степень, угрожающая жизни: сильное ограничение активности, требуется значительная помощь; требуется существенное медицинское вмешательство/терапия, возможна госпитализация или уход в приюте.
Изменения степени тяжести AE должны быть документированы, чтобы можно было провести оценку продолжительности события на каждом уровне интенсивности. Если AE характеризуется как периодическое, то требуется документирование начала и продолжительности каждого эпизода.
• 7.3.2 Оценка причинно-следственной связи
Оценка исследователем отношения AE к исследуемому препарату является частью процесса документирования, но не является фактором, определяющим, что отмечается или не отмечается в исследовании.
Исследователь оценивает причинно-следственную связь (т.е. имеется ли разумная вероятность того, что исследуемый препарат вызвал событие) для всех AE и SAE. Связи характеризуются по следующей классификации:
– нет связи: такое отношение предполагает отсутствие связи между исследуемым препаратом и зарегистрированным событием;
– возможная: такая связь основывается на данных, свидетельствующих о причинно-следственной связи между исследуемым препаратом и AE, т.е. имеется разумная вероятность того, что препарат вызвал событие, причем событие следует разумной временной последовательности со времени внутривенного введения препарата или же следует известной схеме реакции на исследуемый препарат, но также могло быть вызвано и другими факторами;
– вероятная: такая связь предполагает, что существует разумная временная зависимость события от внутривенного введения препарата, и на основании известного фармакологического действия препарата, известных или ранее отмеченных нежелательных реакций на препарат или класс препаратов либо суждения, основанного на клиническом опыте исследователя, связь события с исследуемым препаратом представляется вероятной;
– определенная: такая связь предполагает, что существует определенная причинно-следственная связь между внутривенным введением препарата и AE, а другие условия (сопутствующее заболевание, прогрессирование/проявление заболевания или реакция на сопутствующее лекарство) не могут объяснить данное событие.
Статистический анализ
(i) Общая статистика
Все статистические анализы подробно описаны в плане статистического анализа. Все собранные данные были представлены в списках данных. Данные от субъектов, исключенных из популяции для анализа, были представлены в списках данных, но не включались в расчеты по сводной статистике.
Для категориальных переменных были представлены частоты и проценты. Непрерывные переменные обобщали по описательной статистике (количество субъектов, среднее, медиана, SD, минимум и максимум).
Исходные демографические и фоновые переменные обобщали по дозе и в целом по всем субъектам. Было представлено количество субъектов, включенных в исследование, а также количество и процент субъектов, завершивших исследование. Также были представлены частота и процент субъектов, прервавших или вышедших из исследования, и причины выхода или прекращения. Анализ по этому исследованию был представлен по описательной статистике для каждого периода и группы. Критерии значимости не применялись.
Был составлен план статистического анализа (SAP) для подробного учета работы по статистическому анализу. Клиническая база данных блокировалась после того, как все данные были выверены (т.е. “подчищены”) после того, как последний пациент завершил исследование.
(ii) Расчет размера выборки
Всего для этого исследования было запланировано около 12 оцениваемых субъектов. Размер выборки для этого исследования исходил из клинических и практических соображений, а не из формального расчета статистической мощности.
(iii) Комплекты для анализа
Полный комплект анализа (FAS) определяется как субъекты, получавшие как минимум 1 дозу исследуемого препарата. Все оценки эффективности должны проводиться на популяции FAS.
Популяция по безопасности определяется как субъекты, получавшие как минимум 1 дозу исследуемого препарата. Все оценки безопасности должны проводиться на популяции по безопасности.
(iv) Статистический анализ
• Анализ биомаркеров
Концентрации и изменения от исходного уровня общего и аллерген-специфичного IgE и биомаркеров, перечисленных в качестве конечных точек, обобщали по посещениям и представляли графически. Изменения от исходного уровня общего и аллерген-специфичного IgE обобщали по исходной концентрации общего IgE (сывороточный IgE > 1500 МЕ/мл либо сывороточный IgE ≤ 1500 МЕ/мл) и вводимым дозам FB825 (1 доза или 2 дозы).
У субъектов, получавших вторую дозу FB825, данные, полученные для второй дозы, анализировали и обобщали так же, как и данные, полученные для первой дозы.
• Анализ клинической эффективности
Индекс оценки определялся PI или помощником во время каждого посещения для каждого субъекта. Клинические конечные показатели анализировали по описательной статистике (средние значения EASI, SCORAD, IGA, VAS и BSA; SD, CV, количество субъектов) по времени посещения и вводимым дозам FB825 (1 доза или 2 дозы).
Профили изменения среднего значения индекса в зависимости от времени планового посещения представляли графически.
У субъектов, получавших вторую дозу FB825, данные, полученные для второй дозы, анализировали и обобщали так же, как и данные, полученные для первой дозы.
• Анализ безопасности
Неблагоприятные события кодировали по предпочтительным классам терминов и систем органов по последней версии Медицинского словаря для регулирующих действий и обобщали по лечению, уровню дозы и в целом. Неблагоприятные события также обобщали по тяжести, связи с исследуемым препаратом, SAEs и AEs, приводящим к отмене исследуемого препарата.
Фактические значения и изменения от исходного уровня для результатов клинических лабораторных анализов, основных показателей состояния организма и результатов ЭКГ в 12 отведениях обобщали по лечению и дозам в каждой временной точке по описательной статистике (количество субъектов, среднее значение, SD, медиана, минимум и максимум). Для результатов клинических лабораторных анализов составляли таблицы смещения. Клинико-лабораторные данные, основные показатели состояния организма, результаты ЭКГ в 12 отведениях и данные медосмотров представляли в виде таблицы данных.
(v) Обработка недостающих данных
Концентрации, которые ниже предела количественного определения (BLQ), принимались за ноль для описательной статистики. В качестве BLQ представляли средние концентрации BLQ, а SD и CV указывали как неприменимые. Недостающие концентрации исключали из расчетов.
Для обработки отсутствующих данных применяли перенос последнего наблюдения (LOCF). Для данных по безопасности не использовали никаких расчетных данных.
(vi) Обеспечение качества данных
Все аспекты исследования проверяли на соответствие применимым нормативным актам в отношении действующего согласованной трехсторонней директивы E6 (R1) Международной конференции по гармонизации (ICH): надлежащая клиническая практика и действующие стандартные рабочие процедуры. Возможна внутренняя проверка качества данных и дополнительные проверки по клиническим мониторам.
Таблица 3. Исходные характеристики
значений
(1534,95 – 4029,40)
Таблица 4. Общая сводка по числу пациентов с неблагоприятными событиями за дни 15-168 периода лечения-SAF
+ TCS (n=12)
Результаты
Это было открытое поисковое исследование для оценки безопасности и эффективности FB825 у взрослых с атопическим дерматитом (AD). В исследование записались 12 подходящих пациентов с AD, которые получали FB825 по 5 мг/кг путем 1-часового внутривенного вливания в день 1 и день 85. Пациенты должны были возвращаться на место исследования в соответствии с графиком мероприятий, представленным в таблице 2, для оценки безопасности и эффективности.
Введение FB825 безопасно для пациентов с AD
Пациентам вводили FB825 путем внутривенного вливания на протяжении 1 часа по 5 мг/кг в дни 1 и 85. Не было случаев смерти и испытуемые не бросали из-за возникающих при лечении неблагоприятных событий (TEAE) или возникающих при лечении тяжелых неблагоприятных событий (TE-SAE). У 6 испытуемых возникали TEAE, три из которых были возможно связаны с препаратом. У 5 испытуемых возникали инфекции верхних дыхательных путей, а 2 субъекта подхватили герпесвирусные инфекции. Только у одного субъекта отмечалось появление одного из следующего: конъюнктивит, ринорея, приступ астмы, лихорадка, кашель или удлинение QTC (таблица 4).
Не отмечалось связанных с лечением или дозой тенденций по результатам клинико-лабораторных анализов, основным показателям состояния организма, результатам ЭКГ в 12 отведениях или по данным медосмотра.
Эффективность
Эффективность FB825 определяли путем регистрации изменений от исходного уровня индекса площади и тяжести экземы (EASI), глобальной оценки исследователя (IGA), показателя степени тяжести атопического дерматита (SCORAD) и зуда по визуально-аналоговой шкале (VAS) в день 1 и день 85 перед дозированием, в день 2 и день 86 перед выпиской и в любое время в дни 8, 15, 29, 57, 92, 99, 113, 141 и 169. Исходные демографические и фоновые переменные обобщены по дозе и в целом по всем субъектам в таблице 3.
(i) FB825 снижает EASI по сравнению с исходным уровнем
Субъекты проявляли прогрессирующее снижение EASI после введения FB825 в день 1 примерно до дня 55 (снижение примерно на 65%). Показатель EASI начинал возрастать после дня 55 до второго введения FB825 в день 85, но все еще был примерно на 40% меньше исходного уровня. Субъекты проявляли дальнейшее снижение EASI до уровня примерно на 70% в день 113 и сохраняли этот уровень до конца исследования (EOS) (фиг. 5).
(ii) FB825 снижает IGA по сравнению с исходным уровнем
Субъекты проявляли прогрессирующее снижение EASI после введения FB825 в день 1 до дня 55 (снижение примерно на 30%). Показатель IGA начинал возрастать после дня 55 до второго введения FB825 в день 85, но все еще был примерно на 15% меньше исходного уровня. Субъекты проявляли дальнейшее снижение IGA до уровня примерно на 45% как минимум в день 113 и сохраняли снижение на уровне 35% от исходного до конца исследования (EOS) (фиг. 6).
(iii) FB825 снижает SCORAD по сравнению с исходным уровнем
Субъекты проявляли прогрессирующее снижение SCORAD после введения FB825 в день 1 примерно до дня 55 (снижение примерно на 45%). Показатель SCORAD начинал возрастать после дня 55 до второго введения FB825 в день 85, но все еще был примерно на 30% меньше исходного уровня. Субъекты проявляли дальнейшее снижение SCORAD до уровня примерно на 55% в день 113 и сохраняли этот уровень до конца исследования (EOS) (фиг. 7).
(iv) FB825 снижает VAS по сравнению с исходным уровнем
Субъекты проявляли прогрессирующее снижение VAS после введения FB825 в день 1 примерно до дня 15 (снижение примерно на 45%). Показатель VAS начинал возрастать после дня 15 и оставался на 25% меньше исходного уровня до второго введения FB825 в день 85. Субъекты проявляли дальнейшее снижение VAS до уровня примерно на 55% в день 113 и сохраняли этот уровень до конца исследования (EOS) (фиг. 8).
Другие воплощения
Все признаки, раскрытые в данном описании, можно комбинировать в любом сочетании. Каждый признак, раскрытый в данном описании, может быть заменен альтернативным признаком, служащим такой же, эквивалентной или аналогичной цели. Таким образом, если прямо не указано иначе, каждый раскрытый признак является лишь примером обобщенной серии эквивалентных или близких признаков.
Из приведенного выше описания специалисты в данной области могут легко определить основные характеристики настоящего изобретения и, не отступая от его сущности и не выходя за его объем, могут вносить различные изменения и модификации в изобретение, чтобы адаптировать его к различным применениям и условиям. Таким образом, другие воплощения также входят в формулу изобретения.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Fountain Biopharma Inc.
<120> ЛЕЧЕНИЕ IGE-ОПОСРЕДОВАННЫХ АЛЛЕРГИЧЕСКИХ ЗАБОЛЕВАНИЙ
<130> F0720.70004WO00
<140> Еще не назначено
<141> Одновременно с этим
<150> US 62/579,416
<151> 2017-10-31
<160> 7
<170> PatentIn версия 3.5
<210> 1
<211> 17
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетическая последовательность
<400> 1
Gly Leu Ala Gly Gly Ser Ala Gln Ser Gln Arg Ala Pro Asp Arg Val
1 5 10 15
Leu
<210> 2
<211> 117
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетическая последовательность
<400> 2
Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu
1 5 10 15
Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Tyr Ser Ile Thr Ser Asp
20 25 30
Tyr Ala Trp Asn Trp Ile Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp
35 40 45
Ile Gly Ser Ile Ser Tyr Ser Gly Ile Thr Gly Tyr Asn Pro Ser Leu
50 55 60
Lys Ser Arg Val Thr Ile Ser Val Asp Thr Ser Lys Asn Gln Phe Ser
65 70 75 80
Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Met Gly Tyr Asp Gly Leu Ala Tyr Trp Gly Gln Gly Thr Leu
100 105 110
Val Thr Val Ser Ser
115
<210> 3
<211> 113
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетическая последовательность
<400> 3
Asp Ile Val Met Thr Gln Thr Pro Leu Ser Leu Ser Val Thr Pro Gly
1 5 10 15
Gln Pro Ala Ser Ile Ser Cys Arg Ser Ser Gln Ser Ile Val His Ser
20 25 30
Asn Gly Asn Thr Tyr Leu Glu Trp Tyr Leu Gln Lys Pro Gly Gln Ser
35 40 45
Pro Gln Leu Leu Ile Tyr Lys Val Ser Asn Arg Phe Ser Gly Val Pro
50 55 60
Asp Arg Phe Ser Gly Ser Gly Ser Gly Thr Glu Phe Thr Leu Lys Ile
65 70 75 80
Ser Arg Val Glu Ala Glu Asp Val Gly Val Tyr Tyr Cys Phe Gln Gly
85 90 95
Ser His Val Pro Pro Thr Phe Gly Gly Gly Thr Lys Val Glu Ile Lys
100 105 110
Arg
<210> 4
<211> 117
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетическая последовательность
<400> 4
Asp Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Gln
1 5 10 15
Ser Leu Ser Leu Thr Cys Thr Val Thr Gly Tyr Ser Ile Thr Ser Asp
20 25 30
Tyr Ala Trp Asn Trp Ile Arg Gln Phe Pro Gly Asn Lys Leu Glu Trp
35 40 45
Met Gly Ser Ile Ser Tyr Ser Gly Ile Thr Gly Tyr Asn Pro Ser Leu
50 55 60
Lys Ser Arg Ile Ser Val Thr Arg Asp Thr Ser Lys Asn Gln Phe Phe
65 70 75 80
Leu Gln Leu Asn Ser Val Thr Thr Glu Asp Thr Ala Thr Tyr Tyr Cys
85 90 95
Ala Arg Met Gly Tyr Asp Gly Leu Ala Tyr Trp Gly His Gly Thr Thr
100 105 110
Val Thr Val Ser Ala
115
<210> 5
<211> 113
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетическая последовательность
<400> 5
Asp Val Leu Met Thr Gln Thr Pro Leu Ser Leu Pro Val Ser Leu Gly
1 5 10 15
Asp Gln Ala Ser Ile Ser Cys Arg Ser Ser Gln Ser Ile Val His Ser
20 25 30
Asn Gly Asn Thr Tyr Leu Glu Trp Tyr Leu Gln Lys Pro Gly Gln Ser
35 40 45
Pro Lys Leu Leu Ile Tyr Lys Val Ser Asn Arg Phe Ser Gly Val Pro
50 55 60
Asp Arg Phe Ser Gly Ser Gly Ser Gly Thr Glu Phe Thr Phe Lys Ile
65 70 75 80
Ser Arg Val Glu Ala Glu Asp Leu Gly Val Tyr Tyr Cys Phe Gln Gly
85 90 95
Ser His Val Pro Pro Thr Phe Gly Gly Gly Thr Lys Leu Glu Ile Lys
100 105 110
Arg
<210> 6
<211> 52
<212> БЕЛОК
<213> Homo sapiens
<400> 6
Gly Leu Ala Gly Gly Ser Ala Gln Ser Gln Arg Ala Pro Asp Arg Val
1 5 10 15
Leu Cys His Ser Gly Gln Gln Gln Gly Leu Pro Arg Ala Ala Gly Gly
20 25 30
Ser Val Pro His Pro Arg Cys His Cys Gly Ala Gly Arg Ala Asp Trp
35 40 45
Pro Gly Pro Pro
50
<210> 7
<211> 12
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетическая последовательность
<400> 7
Gly Leu Ala Gly Gly Ser Ala Gln Ser Gln Arg Ala
1 5 10
<---
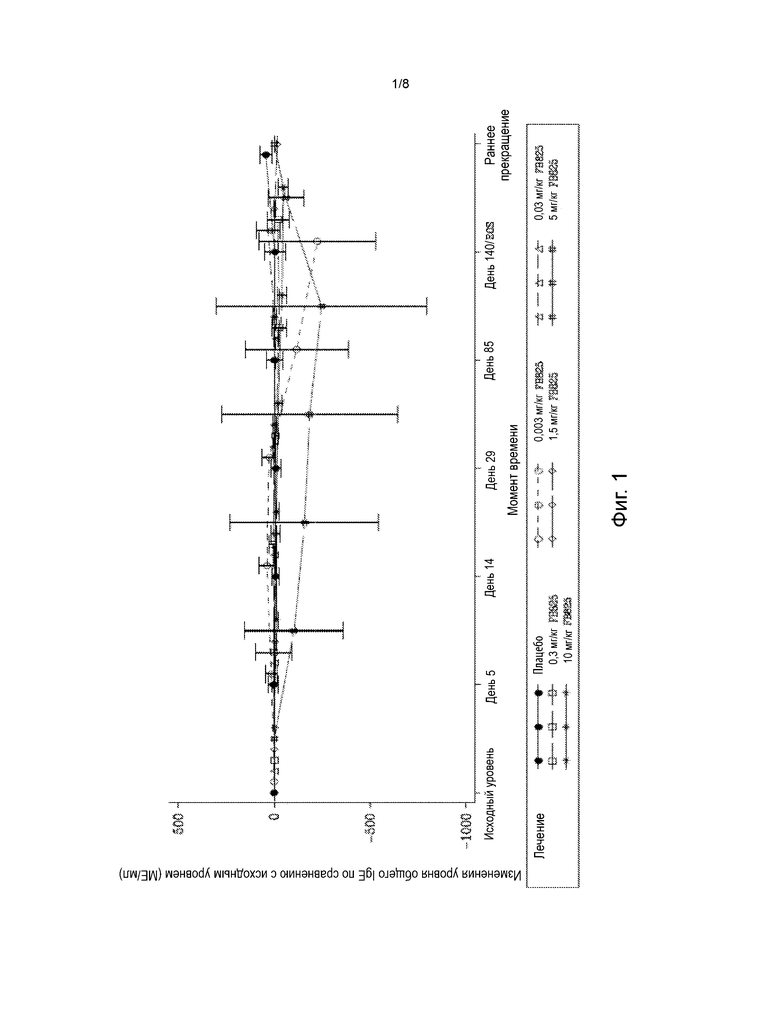
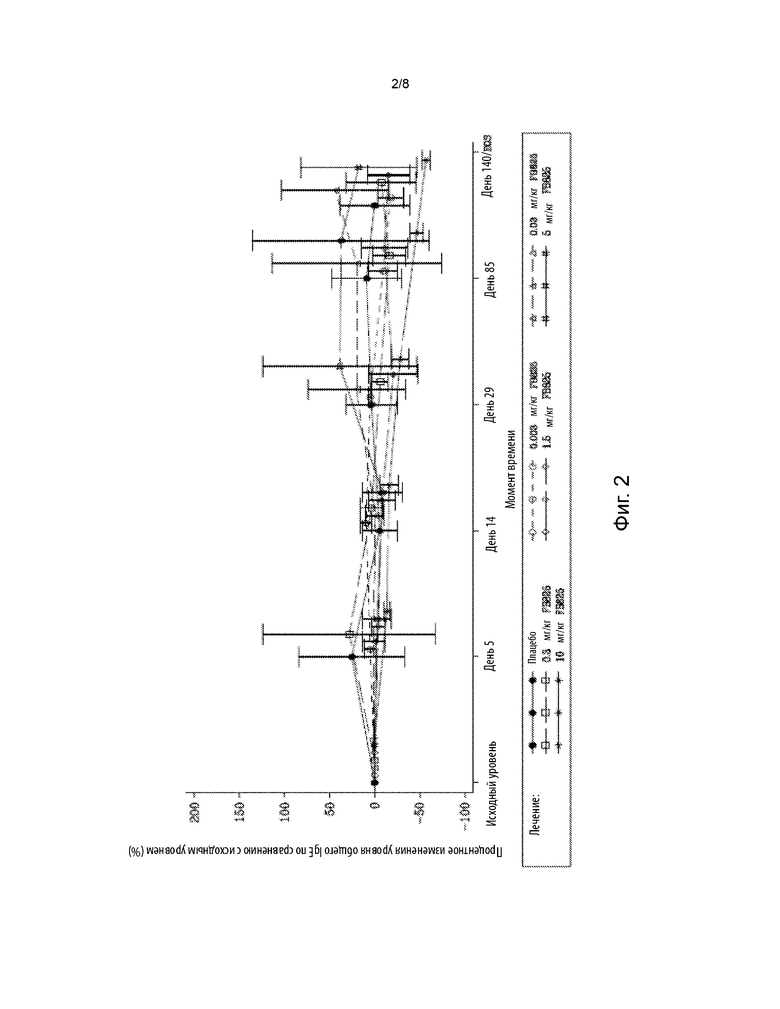

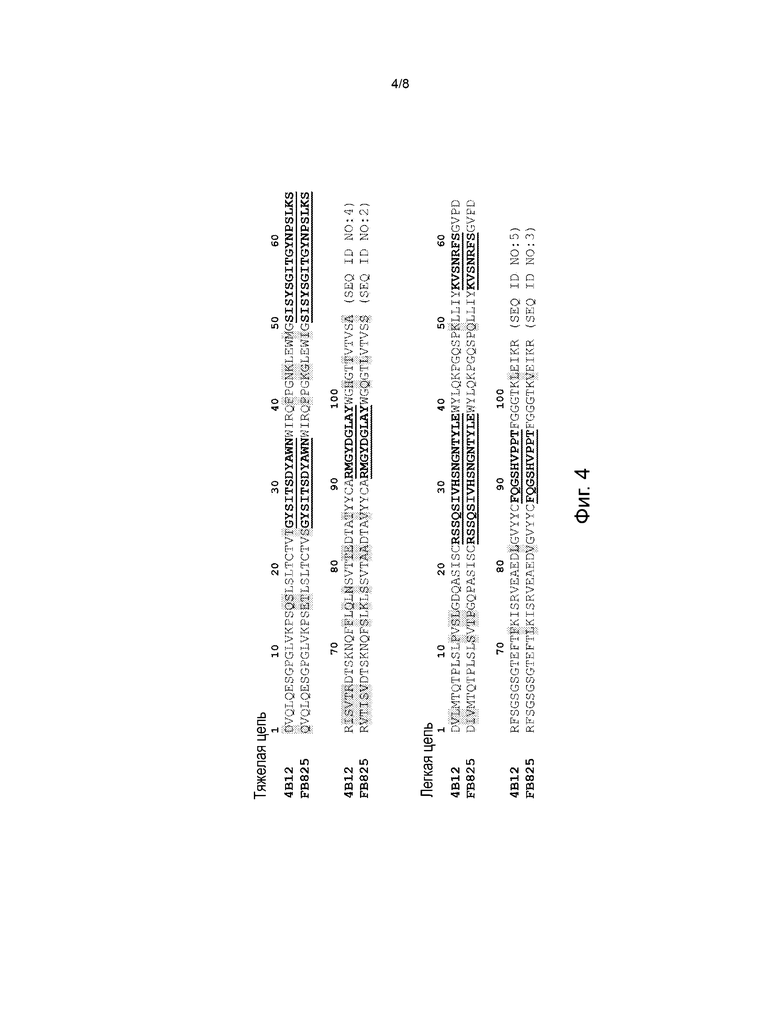
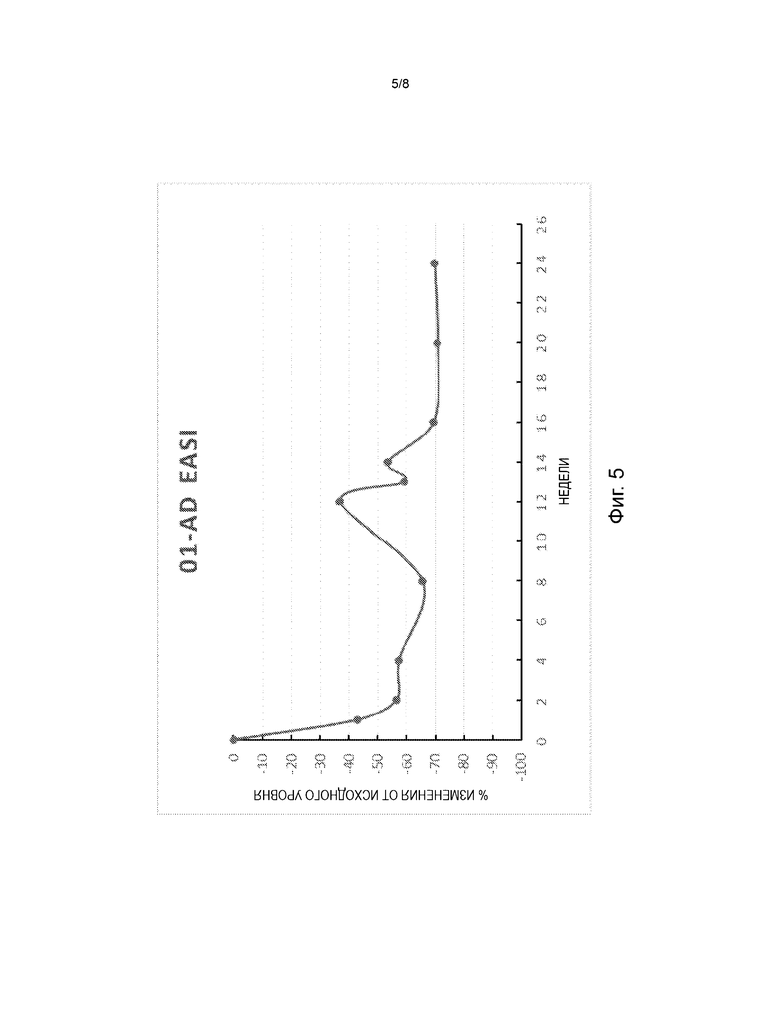
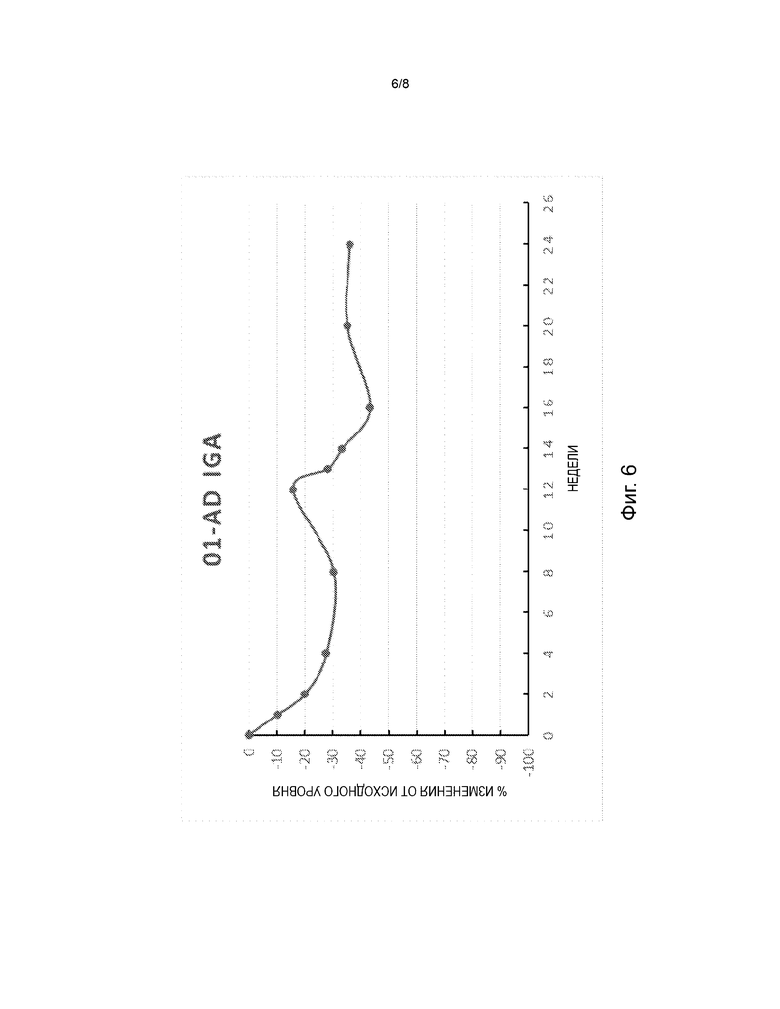
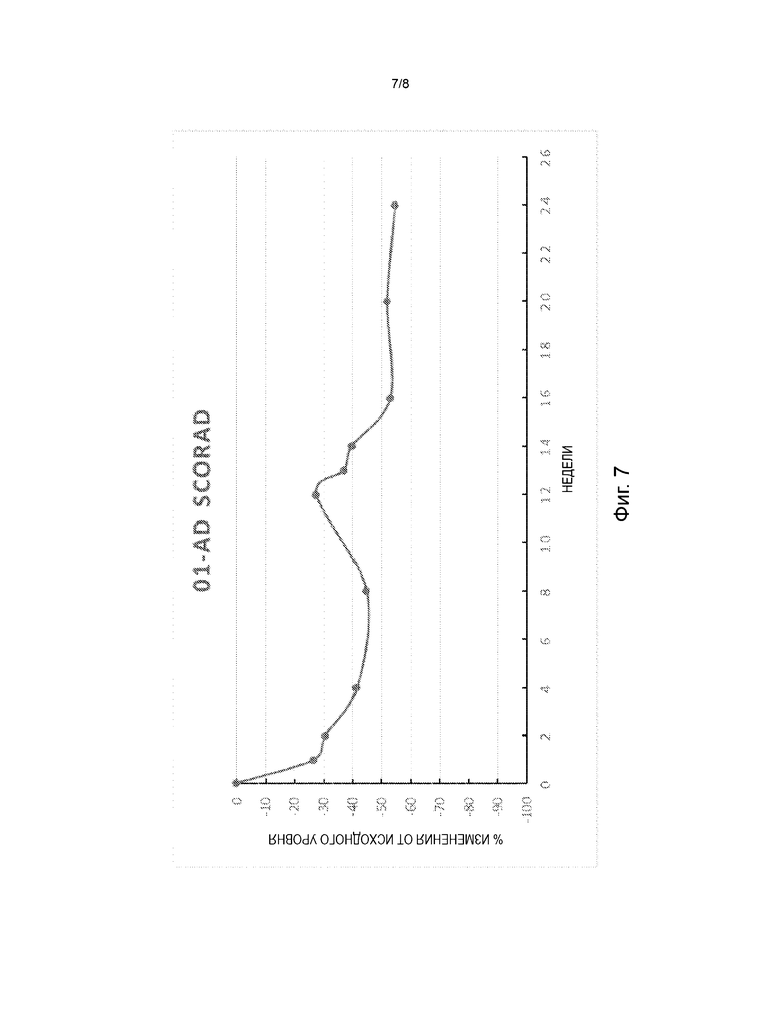
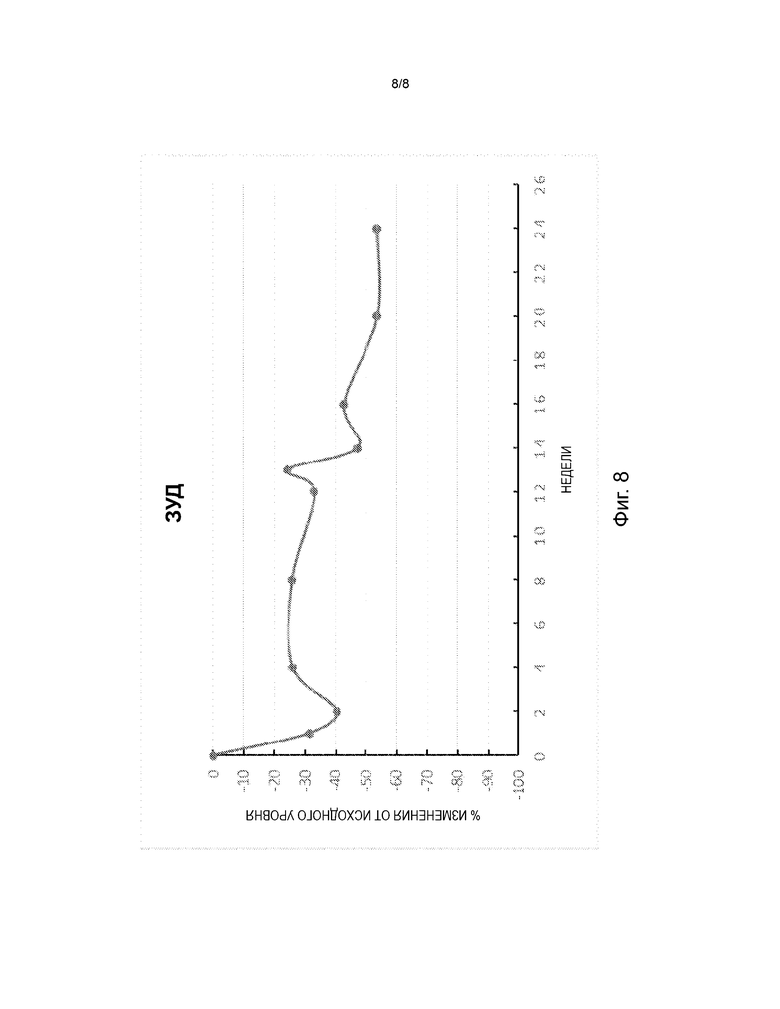
Группа изобретений относится к области фармацевтики и медицины, а именно к способам лечения заболевания, связанного с иммуноглобулином E (IgE) и представляющего собой аллергическую астму, аллергический ринит, атопический дерматит или синдром гипер-IgE, которые включают (i) введение нуждающемуся в этом субъекту первой дозы антитела, связывающегося с доменом CεmX мембраносвязанного IgE, и (ii) введение субъекту второй дозы антитела, причем вторая доза вводится по меньшей мере через 8 недель и вплоть до 6 месяцев после первой дозы, и первая доза, вторая доза или обе находятся в диапазоне от 1 мг/кг до 10 мг/кг. Технический результат заключается в снижении уровня общего IgE у человека на протяжении по меньшей мере трех месяцев при введении однократной дозы антитела. 2 н. и 24 з.п. ф-лы, 8 ил., 4 табл., 3 пр.
1. Способ лечения заболевания, связанного с иммуноглобулином E (IgE), который включает:
(i) введение нуждающемуся в этом субъекту первой дозы антитела, связывающегося с доменом CεmX мембраносвязанного IgE; и
(ii) введение субъекту второй дозы антитела, причем вторая доза вводится по меньшей мере через 8 недель и вплоть до 6 месяцев после первой дозы,
где антитело содержит вариабельную область тяжелой цепи, которая содержит (a) определяющий комплементарность участок (CDR) 1 тяжелой цепи, CDR2 тяжелой цепи и CDR3 тяжелой цепи, и (b) вариабельную область легкой цепи, содержащую CDR1 легкой цепи, CDR2 легкой цепи и CDR3 легкой цепи; где:
CDR1 тяжелой цепи содержит аминокислотную последовательность, соответствующую остаткам 26-36 в SEQ ID NO: 2,
CDR2 тяжелой цепи содержит аминокислотную последовательность, соответствующую остаткам 51-66 в SEQ ID NO: 2,
CDR3 тяжелой цепи содержит аминокислотную последовательность, соответствующую остаткам 98-106 в SEQ ID NO: 2,
CDR1 легкой цепи содержит аминокислотную последовательность, соответствующую остаткам 24-39 в SEQ ID NO: 3,
CDR2 легкой цепи содержит аминокислотную последовательность, соответствующую остаткам 55-61 в SEQ ID NO: 3, и
CDR3 легкой цепи содержит аминокислотную последовательность, соответствующую остаткам 94-102 в SEQ ID NO: 3; и
где тяжелая цепь содержит вариабельную область тяжелой цепи (VH), по меньшей мере на 90% идентичную SEQ ID NO: 2, и где легкая цепь содержит вариабельную область легкой цепи (VL), по меньшей мере на 90% идентичную SEQ ID NO: 3,
где вариации аминокислотной последовательности могут встречаться только в одном или нескольких каркасных участках VH и/или VL;
где первая доза, вторая доза или обе находятся в диапазоне от 1 мг/кг до 10 мг/кг; и
где заболевание, связанное с IgE, представляет собой аллергическую астму, аллергический ринит, атопический дерматит или синдром гипер-IgE.
2. Способ по п. 1, при этом вторая доза вводится по меньшей мере через 3 месяца после первой дозы.
3. Способ по п. 2, при этом вторая доза вводится через 12 недель - 6 месяцев после первой дозы.
4. Способ по любому из пп. 1-3, при этом первая доза, вторая доза или обе составляют 5 мг/кг.
5. Способ по любому из пп. 1-3, при этом первая доза, вторая доза или обе составляют от 1 мг/кг до 8 мг/кг.
6. Способ лечения атопического дерматита, который включает введение нуждающемуся в этом субъекту первой дозы антитела, связывающегося с доменом CεmX мембраносвязанного IgE; причем первая доза составляет от 1 мг/кг до 10 мг/кг,
где антитело содержит вариабельную область тяжелой цепи, которая содержит (a) определяющий комплементарность участок (CDR) 1 тяжелой цепи, CDR2 тяжелой цепи и CDR3 тяжелой цепи, и (b) вариабельную область легкой цепи, содержащую CDR1 легкой цепи, CDR2 легкой цепи и CDR3 легкой цепи; где:
CDR1 тяжелой цепи содержит аминокислотную последовательность, соответствующую остаткам 26-36 в SEQ ID NO: 2,
CDR2 тяжелой цепи содержит аминокислотную последовательность, соответствующую остаткам 51-66 в SEQ ID NO: 2,
CDR3 тяжелой цепи содержит аминокислотную последовательность, соответствующую остаткам 98-106 в SEQ ID NO: 2,
CDR1 легкой цепи содержит аминокислотную последовательность, соответствующую остаткам 24-39 в SEQ ID NO: 3,
CDR2 легкой цепи содержит аминокислотную последовательность, соответствующую остаткам 55-61 в SEQ ID NO: 3, и
CDR3 легкой цепи содержит аминокислотную последовательность, соответствующую остаткам 94-102 в SEQ ID NO: 3,
где тяжелая цепь содержит вариабельную область тяжелой цепи (VH), по меньшей мере на 90% идентичную SEQ ID NO: 2, и где легкая цепь содержит вариабельную область легкой цепи (VL), по меньшей мере на 90% идентичную SEQ ID NO: 3,
где вариации аминокислотной последовательности могут встречаться только в одном или нескольких каркасных участках VH и/или VL;
где субъекту далее вводят вторую дозу антитела, причем вторая доза вводится по меньшей мере через 8 недель и вплоть до 3 месяцев после первой дозы, если ко времени введения второй дозы изменение общего уровня IgE у субъекта от общего уровня IgE перед первой дозой составляет менее 50%.
7. Способ по п. 6, при этом первая доза антитела составляет 5 мг/кг.
8. Способ по п. 6 или 7, при этом способ дополнительно включает введение субъекту второй дозы антитела через 8 недель после первой дозы, если ко времени второй дозы изменение общего уровня IgE у субъекта составляет менее 50% от общего уровня IgE перед первой дозой.
9. Способ по п. 6 или 7, при этом способ дополнительно включает введение субъекту второй дозы антитела через 3 месяца после первой дозы, если ко времени второй дозы изменение общего уровня IgE у субъекта составляет менее 50% от общего уровня IgE перед первой дозой.
10. Способ по п. 9, при этом вторая доза составляет 5 мг/кг.
11. Способ по любому из пп. 1-10, при этом первая доза, вторая доза или обе вводятся посредством внутривенной инъекции.
12. Способ по любому из пп. 1-11, при этом антитело представляет собой человеческое антитело или гуманизованное антитело.
13. Способ по любому из пп. 1-12, при этом антитело представляет собой полноразмерное антитело или его антигенсвязывающий фрагмент.
14. Способ по любому из пп. 1-13, при этом антитело содержит вариабельную область тяжелой цепи, имеющую аминокислотную последовательность, по меньшей мере на 95% идентичную SEQ ID NO: 2, и вариабельную область легкой цепи, имеющую аминокислотную последовательность, по меньшей мере на 95% идентичную SEQ ID NO: 3.
15. Способ по п. 14, при этом антитело содержит вариабельную область тяжелой цепи, имеющую аминокислотную последовательность, по меньшей мере на 98% идентичную SEQ ID NO: 2, и вариабельную область легкой цепи, имеющую аминокислотную последовательность, по меньшей мере на 98% идентичную SEQ ID NO: 3.
16. Способ по любому из пп. 1-15, при этом субъектом является человеческий пациент.
17. Способ по любому из пп.1-5, при этом заболевание представляет собой атопический дерматит или аллергическую астму.
18. Способ по любому из пп. 1-17, при этом субъекту наносится увлажняющий крем по меньшей мере два раза в день на протяжении по меньшей мере семи последовательных дней перед первой дозой.
19. Способ по любому из пп. 1-18, дополнительно включающий нанесение субъекту местного кортикостероида.
20. Способ по п. 19, при этом местный кортикостероид наносится на активные очаги ежедневно, а местный кортикостероид представляет собой крем с 0,05% флутиказона пропионата, крем с 0,1% монетазона фуроата, мазь с 0,06% бетаметазона валерата или 1% гидрокортизона.
21. Способ по п. 20, при этом местный кортикостероид представляет собой крем с 0,05% флуоцинонида, мазь с 0,25% дезоксиметазона или мазь с 0,05% клобетазола пропионата.
22. Способ по любому из пп. 1-21, при этом субъект не получает местного применения такролимуса, местного применения пимекролимуса, системного применения кортикостероидов, применения ингибиторов лейкотриенов, аллергеновой иммунотерапии, лечения, включающего иммунодепрессанты или иммуномодуляторы, применения вакцин, лечения при помощи традиционной китайской медицины, хирургических процедур, ультрафиолетовых процедур или загара.
23. Способ по любому из пп. 1-22, при этом антитело входит в состав фармацевтической композиции, содержащей антитело, буфер, соль и неионное поверхностно-активное вещество (ПАВ).
24. Способ по п. 23, при этом фармацевтическая композиция представляет собой водный раствор со значением pH от 5 до 8.
25. Способ по п. 23 или 24, при этом буфер представляет собой гистидиновый буфер, соль – хлорид натрия и/или неионное ПАВ – полисорбат 80.
26. Способ по п. 25, при этом антитело в фармацевтической композиции составляет от 10 мг/мл до 30 мг/мл, гистидиновый буфер имеет концентрацию 10-30 мМ, хлорид натрия имеет концентрацию 120-160 мМ, а полисорбат 80 имеет концентрацию 0,01-0,03%.
| US 2015086558 A1, 26.03.2015 | |||
| АНТИ-CemX АНТИТЕЛА, СПОСОБНЫЕ СВЯЗЫВАТЬСЯ С migE ЧЕЛОВЕКА НА В ЛИМФОЦИТАХ | 2010 |
|
RU2550270C2 |
| LIOUR S.S | |||
| et al | |||
| Treating IgE‐mediated diseases via targeting IgE‐expressing B cells using an anti‐CεmX antibody // Pediatric Allergy and Immunology | |||
| Токарный резец | 1924 |
|
SU2016A1 |
| - Vol | |||
| Прибор с двумя призмами | 1917 |
|
SU27A1 |
| - No | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| - P | |||
| Способ пропитывания дерева | 1921 |
|
SU446A1 |
| EZZO D | |||
| Treatment and Managed Care Issues of Atopic Dermatitis // The American journal of managed | |||

