ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к 6-карбоксипроизводным бензимидазолов и 4-аза-, 5-аза- и 7-азабензимидазолов в качестве агонистов GLP-1R, способам получения указанных соединений и способам, включающим введение указанных соединений нуждающемуся в них млекопитающему.
УРОВЕНЬ ТЕХНИКИ
Диабет является главной проблемой общественного здравоохранения вследствие его растущего распространения и связанными с ним опасностями для здоровья. Заболевание характеризуется высокими содержаниями глюкозы в крови, обусловленными нарушениями выработки инсулина, воздействия инсулина или обоими этими факторами. Выявлены две основные форма диабета, тип 1 и тип 2. Диабет типа 1 (T1D) развивается, когда иммунная система организма разрушает бета-клетки поджелудочной железы, единственные клетки организма, которые вырабатывают гормон инсулин, который регулирует содержание глюкозы в крови. Для выживания людям с диабетом типа 1 необходим инсулин, вводимый с помощью инъекции или насоса. Сахарный диабет типа 2 (обычно обозначаемый, как T2DM) обычно начинается с резистентности к инсулину или тогда, когда вырабатывается недостаточно инсулина для поддержания приемлемого содержание глюкозы.
В настоящее время доступны фармакологические методики лечения гипергликемии и затем T2DM (Hampp, C. et al. Use of Antidiabetic Drugs in the U.S., 2003-2012, Diabetes Care 2014, 37, 1367-1374). Их можно разделить на шесть основных классов, каждый из которых действует по своему первичному механизму: (A) средства, усиливающее выработку инсулина, включая сульфонилмочевины (например, глипизид, глимепирид, глибурид), меглитиниды (например, натеглидин, репаглинид), ингибиторы дипептидилпептидазы IV (DPP-IV) (например, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин, саксоглиптин) и агонисты рецептора глюкагоноподобного пептида 1 (GLP-1R) (например, лираглутид, албиглутид, эксенатид, ликсисенатид, дилаглутид, семаглутид), которые усиливают выработку инсулина путем воздействия на бета-клетки поджелудочной железы. Сульфонилмочевины и меглитиниды обладают ограниченной эффективностью и переносимостью, вызывают увеличение массы тела и часто вызывают гипогликемию. Ингибиторы DPP-IV обладают ограниченной эффективностью. Имеющиеся в продаже GLP-1R являются пептидами, вводимыми путем подкожной инъекции. Лираглутид дополнительно утвержден для лечения ожирения. (B) Бигуаниды (например, метформин) предположительно в основном действуют путем снижения выработки глюкозы в печени. Бигуаниды часто вызывают желудочно-кишечные нарушения и лактоцидоз, что дополнительно ограничивает их применение. (C) Ингибиторы альфа-глюкозидазы (например, акарбоза) снижают взасывание глюкозы в кишечнике. Эти средства часто вызывают желудочно-кишечные нарушения. (D) Тиазолидиндионы (например, пиоглитазон, росиглитазон) действуют на конкретный рецептор (рецептор гамма, активируемый пероксисомными пролифераторами) в печени, мышцах и жировых тканях. Они регулируют метаболизм липидов и затем усиливают ответ этих тканей на воздействия инсулина. Частое применение этих лекарственных средств может привести к увеличению массы тела и может вызвать отек и анемию. (E) Инсулин используется в более тяжелых случаях по отдельности или в комбинации с указанными выше средствами и частое применение также может привести к увеличению массы тела и вызывает опасность гипогликемии. (F) Ингибиторы натрийзависимого переносчика глюкозы 2 (SGLT2) (например, дапаглифлозин, эмпаглифлозин, канаглифлозин, эртуглифлозин) подавляют обратное всасывание глюкозы в почках и тем самым снижают содержание глюкозы в крови. Этот расширяющийся класс лекарственных средств может быть связан с кетоацидозом и инфекциями мочевых путей.
Однако за исключением агонистов GLP-1R и ингибиторов SGLT2 лекарственные средства обладают ограниченной эффективностью не решают наиболее важные проблемы - ухудшение функции β-клеток и связанное с этим ожирение.
Ожирение является хроническим заболеванием, которое сильно распространено в современном обществе и связано с многочисленными медицинскими проблемами, включая гипертензию, гиперхолестеринемию и ишемическую болезнь сердца. Оно также сильно коррелирует с T2DM и резистентностью к инсулину, последнее из которых обычно сопровождается гиперинсулинемией или гипергликемией, или обоими. Кроме того, T2DM связан с увеличенной в 2-4 раза опасностью заболевания коронарной артерии. В настоящее время единственным лечением, которое устраняет ожирение с высокой эффективностью, является операция при ожирении, но это лечение является дорогостоящим и опасным. Фармакологическое вмешательство обычно менее эффективно и сопряжено с побочными эффектами. Поэтому существует очевидная необходимость в более эффективном фармакологическом вмешательстве с меньшим количеством побочным эффектов и удобным введением.
Хотя T2DM чаще всего связан с гипергликемией и резистентностью к инсулину, другие заболевания, связанные с T2DM, включают резистентность печени к инсулину, нарушенную переносимость глюкозы, диабетическую невропатию, диабетическую нефропатию, диабетическую ретинопатию, ожирение, дислипидемию, гипертензию, гиперинсулинемию и неалкогольную жировую инфильтрацию печени (NAFLD).
NAFLD является проявлением метаболического синдрома в печени и спектр патологических состояний печени включает стеатоз, неалкогольный стеатогепатит (NASH), фиброз, цирроз и в конечном счете печеночно-клеточную карциному. NAFLD и NASH считают первичными жировыми заболеваниями печени, поскольку они наблюдаются в наибольшей части индивидуумов с увеличенным содержанием липидов в печени. Тяжесть NAFLD/NASH основана на наличии липидов, инфильтрата воспалительных клеток, баллонировании гепатоцитов и степени фиброза. Хотя не у всех индивидуумов стеатоз прогрессирует в NASH, это наблюдается у значительной их части.
GLP-1 является содержащим 30 аминокислот гормоном инкретином, вырабатывающимся L-клетками в кишечнике в ответ на потребление пищи. Показано, что GLP-1 стимулирует выработку инсулина физиологическим и зависимым от глюкозы образом, снижает выработку глюкагона, подавляет опорожнение желудка, снижает аппетит и стимулирует пролиферацию бета-клеток. В неклинических экспериментах GLP-1 активирует длительную компетентность бета-клеток путем стимулирования транскрипции генов, важных для глюкоза-зависимой выработки инсулина, и путем активирования неогенеза бета-клеток (Meier, et al. Biodrugs. 2003; 17 (2): 93-102).
У здоровых индивидуумов GLP-1 играет важную роль в регуляции содержания глюкозы в крови после приема пищи путем стимулирования глюкоза-зависимой выаботки инсулина в поджелудочной железе, что приводит к повышенному поглощению глюкозы на периферии. GLP-1 также подавляет выработку глюкагона, что приводит к сниженному выделению глюкозы из печени. Кроме того, GLP-1 задерживает опорожнение желудка и замедляет перистальтику тонкой кишки, задерживая поглощение пищи. У людей с T2DM, нормальное повышение содержания GLP-1 после приема пищи отсутствует или уменьшено (Vilsboll T, et al. Diabetes. 2001. 50; 609-613).
Holst (Physiol. Rev. 2007, 87, 1409) и Meier (Nat. Rev. Endocrinol. 2012, 8, 728) установили, что агонисты рецептора GLP-1, такие как лираглутид и эксендин-4, проявляют 3 основные фармакологические активности с улучшением гликемического контроля у пациентов с T2DM путем уменьшения содержания глюкозы натощак и после приема пищи (FPG и PPG): (i) увеличения глюкоза-зависимой выработки инсулина (улучшение первой и второй фазы), (ii) подавления активности глюкагона при гипергликемических патологических состояниях, (iii) задержки опорожнения желудка, что приводит к замедленному поглощению образовавшейся из пищи глюкозы.
Остается необходимость в легко вводимых средствах предупреждения и/или лечения кардиометаболических и сопутствующих заболеваний.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг. 1 приведена экспериментальная порошковая рентгенограмма для безводной (ангидрат) кристаллической формы (форма 1) соли с Tris соединения примера 7.
На фиг.2 приведена экспериментальная порошковая рентгенограмма для безводной (ангидрат) кристаллической формы (форма A) соли с Tris соединения примера 10.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям формулы I
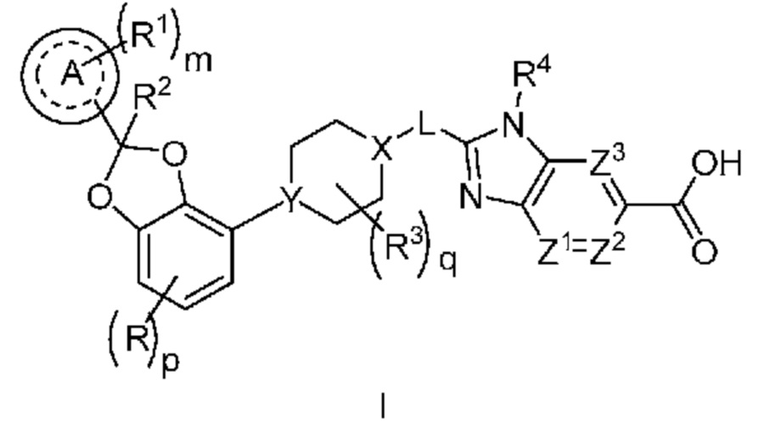
или их фармацевтически приемлемой соли, где
R означает F, Cl или -CN;
p равно 0 или 1;
кольцо A представляет собой фенил или 6-членный гетероарил;
m равно 0, 1, 2 или 3;
каждый R1 независимо выбран из группы, включающей галоген, -CN, -C1-3алкил и -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила замещен 0-3 атомами F;
R2 означает H или -C1-3алкил, где алкил замещен с помощью от 0 до 1 OH;
каждый R3 независимо означает F, -OH, -CN, -C1-3алкил, -OC1-3алкил, и -C3-4циклоалкил, или 2 R3 могут вместе циклизоваться с образованием -C3-4спироциклоалкила, где алкил C1-3алкила и OC1-3алкила, циклоалкила или спироциклоалкила может быть замещен, насколько допускает валентность, 0-3 атомами F и с помощью от 1 до 1 -OH;
q равно 0, 1 или 2;
X-L означает N-CH2, CHCH2 или циклопропил;
Y означает CH или N;
R4 означает -C1-3алкил, -C0-3алкилен-C3-6циклоалкил, -C0-3алкилен-R5 или -C1-3алкилен-R6, где указанный алкил может быть замещен, насколько допускает валентность, 0-3 заместителями, независимо выбранными из группы, включающей 0-3 атома F и 0-1 заместителем, выбранным из группы, включающей -C0-1алкилен-CN, -C0-1алкилен-ORO, -SO2-N(RN)2, -C(O)-N(RN)2, -N(C=O)(RN) и -N(RN)2 и
где указанный алкилен и циклоалкил могут быть независимо замещены, насколько допускает валентность, 0-2 заместителями, независимо выбранными из группы, включающей 0-2 атома F, и 0-1 заместителем, выбранным из группы, включающей -C0-1алкилен-CN, -C0-1алкилен-ORO и -N(R)2;
R5 означает 4- - 6-членный гетероциклоалкил, где указанный гетероциклоалкил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-1 оксогруппу (=O),
0-1 -CN,
0-2 атома F и
0-2 заместителя, независимо выбранные из группы, включающей -C1-3алкил и -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила может быть замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-3 атома F,
0-1 -CN и
0-1 -ORO;
R6 означает 5- - 6-членный гетероарил, где указанный гетероарил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-2 галогены,
0-1 заместителем, выбранным из группы, включающей -ORO и -N(RN)2 и
0-2 -C1-3алкил, где алкил может быть замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-3 атома F и
0-1 -ORO;
каждый RO независимо означает H или -C1-3алкил, где C1-3алкил может быть замещен 0-3 атомами F;
каждый RN независимо означает H или -C1-3алкил;
Z1, Z2 и Z3 все означают -CRZ, или
один из Z1, Z2 и Z3 означает N и два других означают -CRZ; и
каждый RZ независимо означает H, F, Cl или -CH3.
Другой вариант осуществления относится к соединениям формулы II
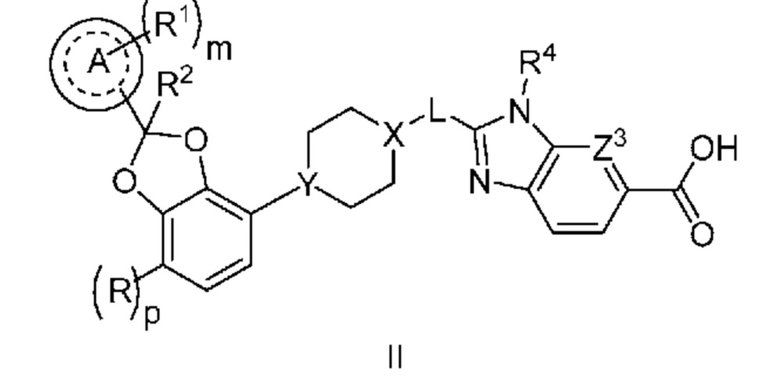
или их фармацевтически приемлемой соли, где
R означает F;
p равно 0 или 1;
кольцо A представляет собой фенил или пиридинил;
m равно 0, 1 или 2;
каждый R1 независимо выбран из группы, включающей галоген, -CN, -C1-3алкил и -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила замещен 0-3 атомами F;
R2 означает H или CH3;
X-L означает N-CH2 или циклопропил;
Y означает CH или N;
Z3 означает -CRZ или N; и
RZ означает H, F, Cl или -CH3.
Другой вариант осуществления относится к соединениям формулы III
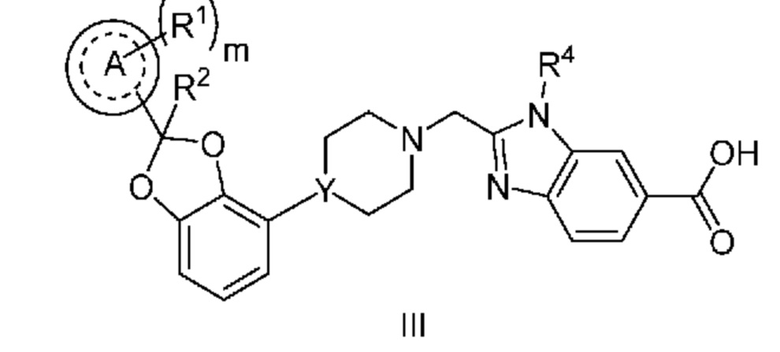
или их фармацевтически приемлемой соли, где
кольцо A представляет собой фенил или пиридинил;
m равно 0, 1 или 2;
каждый R1 независимо выбран из группы, включающей F, Cl и -CN;
R2 означает H или CH3; и
Y означает CH или N.
Другой вариант осуществления относится к соединениям формулы IV
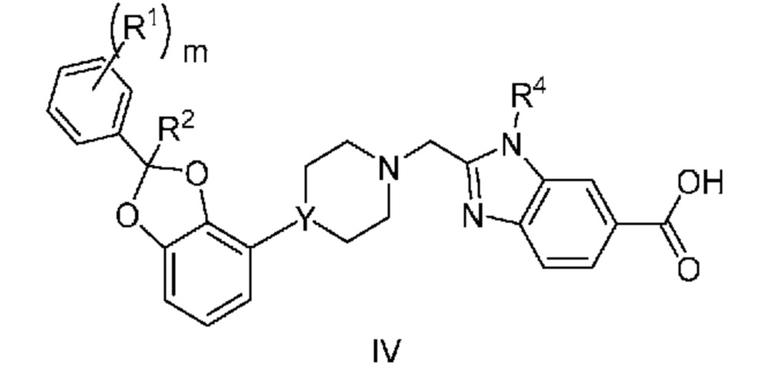
или их фармацевтически приемлемой соли, где
m равно 0, 1 или 2;
каждый R1 независимо выбран из группы, включающей F, Cl и -CN;
R2 означает H или CH3; и
Y означает CH или N.
Другой вариант осуществления относится к соединениям формулы V

или их фармацевтически приемлемой соли, где
m равно 0 или 1;
R1 означает F, Cl или -CN;
R2 означает H или CH3; и
Y означает CH или N.
Другой вариант осуществления относится к соединениям формулы IV или формулы V, где фенил или пиридинил кольца A содержит один заместитель R1 в пара-положении к атому углерода указанного фенила или пиридинила, присоединенному к диоксолану, и образуют:
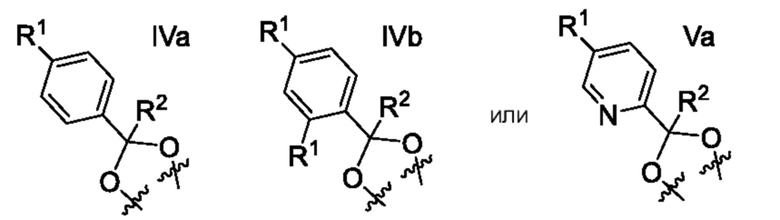
или их фармацевтически приемлемой соли, где
каждый R1 независимо выбран из группы, включающей F, Cl и -CN;
R2 означает H или CH3; и
Y означает CH или N.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I или II, или их фармацевтически приемлемой соли, где X-L означает N-CH2; и Y означает CH или N. В вариантах осуществления, описанных в настоящем изобретении, в таком случае X означает N и L означает CH2.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I или II, или их фармацевтически приемлемой соли, где X-L означает CHCH2; и Y означает N. В вариантах осуществления, описанных в настоящем изобретении, в таком случае X означает CH и L означает CH2.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I или II, или их фармацевтически приемлемой соли, где X-L означает CHCH2; и Y означает CH. В вариантах осуществления, описанных в настоящем изобретении, в таком случае X означает CH и L означает CH2.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I или II, или их фармацевтически приемлемой соли, где X-L означает циклопропил; и Y означает N.
В вариантах осуществления, в которых X-L означает циклопропил, соединения формул I или II представляют собой:
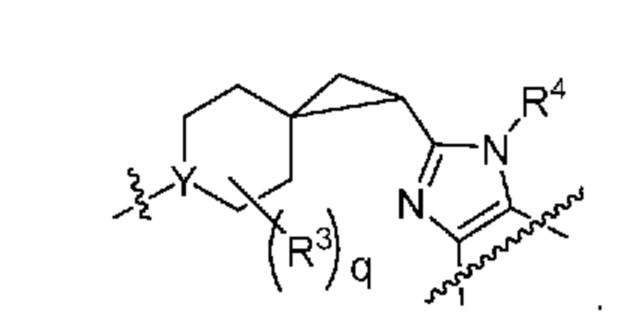 .
.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где R4 означает -CH2CH2OCH3, C1-3алкилен-R5, или C1-3алкилен-R6, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул II, III, IV или V, где R4 является таким, как определено для соединений формулы I, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где R4 означает -C1-3алкил, где указанный алкил может быть замещен, насколько допускает валентность, 0-1 заместителем, выбранным из группы, включающей -C0-1алкилен-ORO и -N(RN)2 или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где R4 означает -(CH2)2OCH3, или -(CH2)2N(CH3)2 или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где
R4 означает -CH2-R5, где R5 означает 4- - 5-членный гетероциклоалкил, где указанный гетероциклоалкил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-2 атома F и
0-1 заместителем, выбранным из группы, включающей -OCH3 и -CH2OCH3;
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероциклоалкил представляет собой
где гетероциклоалкил может быть замещен 0-2 заместителями, насколько допускает валентность, например, замещающими водород, независимо выбранными из группы, включающей:
0-1 оксогруппу (O=),
0-1 -CN,
0-2 атома F и
0-2 заместителя, независимо выбранные из группы, включающей -C1-3алкил и -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила может быть независимо замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-3 атома F,
0-1 -CN и
0-1 -ORO,
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероциклоалкил представляет собой
где гетероциклоалкил может быть замещен 0-2 заместителями, насколько допускает валентность, например, замещающими водород, независимо выбранными из группы, включающей:
0-1 -CN,
0-2 атома F и
0-2 заместителя, независимо выбранные из группы, включающей -C1-3алкил и -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила может быть независимо замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-3 атома F,
0-1 -CN и
0-1 -ORO, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероциклоалкил представляет собой

где гетероциклоалкил может быть замещен 0-1 заместителем, насколько допускает валентность, например, замещающими водород, выбранным из группы, включающей:
-CN,
атом F и
0-1 заместителем, независимо выбранным из группы, включающей -C1-3алкил и -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила может быть замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-3 атома F,
0-1 -CN и
0-1 -ORO,
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероциклоалкил представляет собой
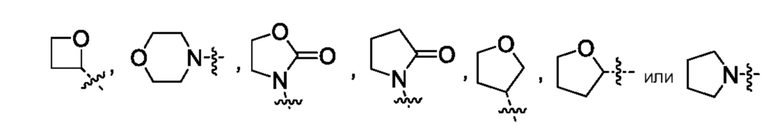
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероциклоалкил представляет собой
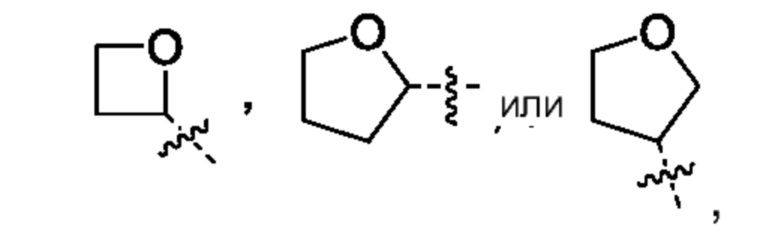 ,
,
где гетероциклоалкил может быть замещен, насколько допускает валентность, 0-1 метилом, где указанный метил может быть замещен 0-3 атомами F, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероциклоалкил представляет собой
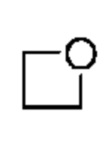
где гетероциклоалкил является незамещенным.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где -CH2-R5 и атом азота, к которому присоединен R4, образуют:
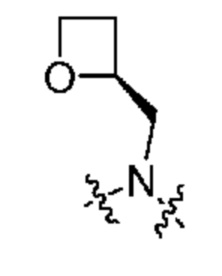
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где
R4 означает -CH2-R6, где R6 означает 5-членный гетероарил, где указанный гетероарил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-2 галогена, где галоген независимо выбран из группы, включающей F и Cl,
0-1 -OCH3 и
0-1 -CH3, -CH2CH3, -CF3 или -CH2CH2OCH3;
или их фармацевтически приемлемой соли.
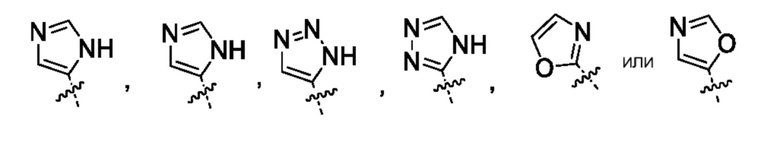
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероарил представляет собой
где указанный гетероарил может быть замещен 0-2 заместителями, насколько допускает валентность, например, замещающими водород, независимо выбранными из группы, включающей:
0-2 галогена, где галоген независимо выбран из группы, включающей F и Cl,
0-1 заместителем, выбранным из группы, включающей -ORO и -N(RN)2 или
0-2 -C1-3алкила, где алкил может быть замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-3 атома F и
0-1 -ORO;
или их фармацевтически приемлемой соли.
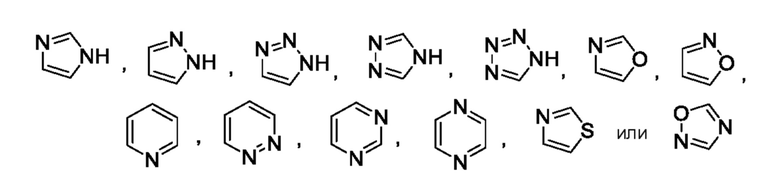
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероарил представляет собой
где указанный гетероарил может быть замещен 0-2 заместителями, насколько допускает валентность, например, замещающими водород, независимо выбранными из группы, включающей:
0-2 галогена, где галоген независимо выбран из группы, включающей F и Cl,
0-1 заместителем, выбранным из группы, включающей -ORO и -N(RN)2 или
0-2 -C1-3алкила, где алкил может быть замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-3 атома F и
0-1 -ORO;
или их фармацевтически приемлемой соли.
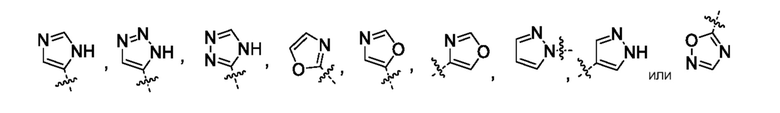
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероарил представляет собой
где указанный гетероарил может быть замещен 0-1 заместителем, насколько допускает валентность,-C1-2алкилом, где алкил может быть замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-3 атома F и
0-1 -ORO; и
каждый RO независимо означает H или -C1-3алкил;
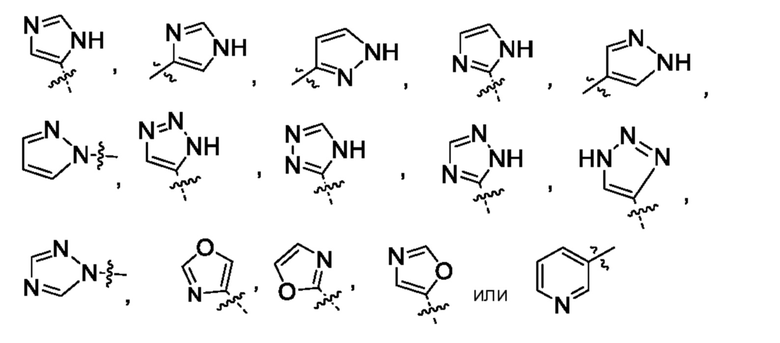
или их фармацевтически приемлемой соли. Следует понимать, что любой заместитель должен замещать H у замещенного атома углерода или азота. Неограниченным примером замещенных гетероарилов являются:
Следует понимать, что H заменен заместителем, например, R6s (заместитель может быть у любого гетероарила, содержащего R6) с образованием:
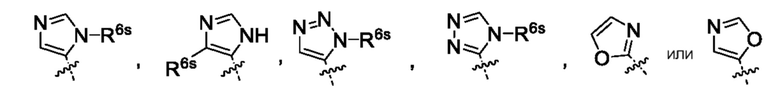
где R6s означает -C1-2алкил, где алкил может быть замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-3 атома F и
0-1 -ORO; и
каждый RO независимо означает H или -C1-3алкил;
или его фармацевтически приемлемую соль.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где гетероарил представляет собой

или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I, II, III, IV или V, где Z1, Z2 и Z3 все означают CRZ, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I, II, III, IV или V, где RZ означает H, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I, II, III, IV или V, где Z1, Z2 и Z3 все означают CH, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I, II, III, IV или V, где R3 означает -CH3 или -CF3; и q равно 1, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I, II, III, IV или V, где каждый R1 независимо означает F, Cl или -CN, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I, II, III, IV или V, где R4 означает -CH2-R5, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I, II, III, IV или V, где R4 означает -CH2-R6, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления настоящего изобретения, например, соединениям формул I, II, III, IV или V, где соединением является свободная кислота.
Другой вариант осуществления относится к любому варианту осуществления соединений формул I, II, III, IV или V, где кольцо A и R2 образуют:
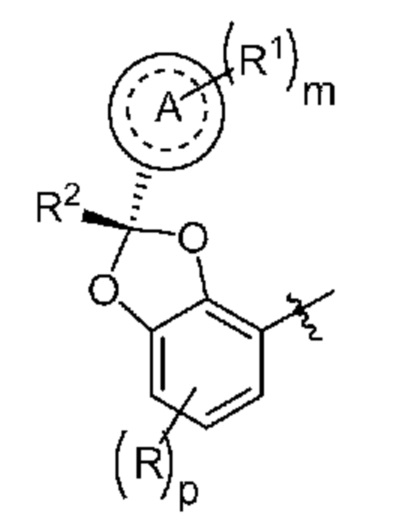
или их фармацевтически приемлемой соли, где
R означает F, Cl или -CN;
p равно 0 или 1;
m равно 0, 1 или 2; и
каждый R1 независимо выбран из группы, включающей галоген, -CN, -C1-3алкил и -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила замещен 0-3 атомами F.
Другой вариант осуществления относится к соединениям формул I, II, III, IV или V, где R2 означает H, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, предлагаемым в настоящем изобретении, в которых R2 означает H, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, предлагаемым в настоящем изобретении, в которых соединением является
2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, предлагаемым в настоящем изобретении, в которых соединением является
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, предлагаемым в настоящем изобретении, в которых R2 означает CH3, или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, предлагаемым в настоящем изобретении, в которых соединением является
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-Циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-3-(1,3-оксазол-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-4-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(пиридин-3-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-1,2,3-триазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-7-фтор-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, предлагаемым в настоящем изобретении, в которых соединением является
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, предлагаемым в настоящем изобретении, в которых соединением является
2-({4-[(2S)-2-(4-Циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, предлагаемым в настоящем изобретении, в которых соединением является
2-({4-[(2R)-2-(4-Циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2R)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2R)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, предлагаемым в настоящем изобретении, в которых соединением является
2-({4-[2-(4-Циано-2-фторфенил)-2*-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, где хиральность 2* обусловлена C56;
2-({4-[2-(5-хлорпиридин-2-ил)-2*-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, где хиральность 2* обусловлена P9;
2-({4-[2-(4-хлор-2-фторфенил)-2*-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота, где хиральность 2* обусловлена 17;
2-({4-[2-(4-хлор-2-фторфенил)-7-фтор-2*-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, где хиральность 2* обусловлена 96; или
2-({4-[2-(4-циано-2-фторфенил)-2*-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота, где хиральность 2* обусловлена C82;
или их фармацевтически приемлемой соли.
Другой вариант осуществления включает соединение, которым является 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, или его фармацевтически приемлемую соль, где солью является соль с Tris.
Другой вариант осуществления включает соединение, которым является 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота в виде свободной кислоты.
Другой вариант осуществления включает соединение, которым является
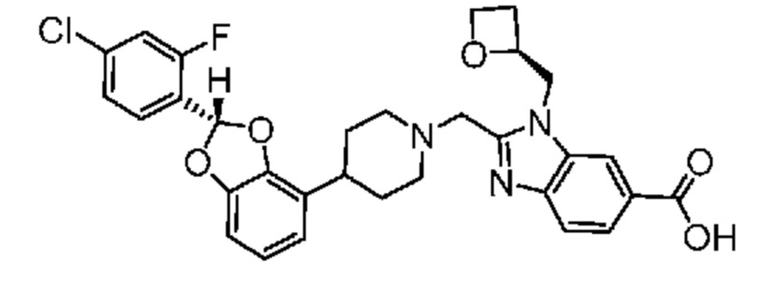
или его фармацевтически приемлемая соль.
Другой вариант осуществления включает соединение, которым является 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, или фармацевтически приемлемая соль, где солью является соль с Tris {соль с Tris этого соединения также известна, как: 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминий 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат}.
В некоторых вариантах осуществления настоящее изобретение относится к кристаллической форме безводной соли с Tris 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты. В некоторых дополнительных вариантах осуществления кристаллическая форма безводной (ангидрат) соли с Tris 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты обозначена, как "форма I", которая охарактеризована в соответствии с ее уникальными характеристиками твердого состояния, например, по данным порошковой рентгенографии (PXRD), описанными в настоящем изобретении (в основном такими, как приведенные на фиг. 1). В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей по меньшей мере два характеристических пика с углами 2θ, выбранными из числа расположенных при 3,7±0,2°; 7,3±0,2°; 8,5±0,2°; 10,1± 0,2°; 14,7± 0,2°; и 16,9± 0,2°. В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей по меньшей мере три характеристических пика с углами 2θ, выбранными из числа расположенных при 3,7±0,2°; 7,3±0,2°; 8,5±0,2°; 10,1± 0,2°; 14,7± 0,2°; и 16,9± 0,2°. В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей по меньшей мере четыре характеристических пика с углами 2θ, выбранными из числа расположенных при 3,7±0,2°; 7,3±0,2°; 8,5±0,2°; 10,1± 0,2°; 14,7± 0,2°; и 16,9± 0,2°. В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей по меньшей мере пять характеристических пиков с углами 2θ, выбранными из числа расположенных при 3,7±0,2°; 7,3±0,2°; 8,5±0,2°; 10,1± 0,2°; 14,7± 0,2°; и 16,9± 0,2°.
В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей характеристические пики с углами 2θ при 3,7±0,2° и 7,3±0,2°.
В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей пики с углами 2θ при 3,7±0,2°; 7,3±0,2°; и 14,7±0,2°. В некоторых других вариантах осуществления форма I обладает порошковой рентгенограммой, дополнительно содержащей по меньшей мере один пик с углом 2θ, выбранным из числа расположенных при 8,5± 0,2°; 10,1± 0,2°; и 16,9± 0,2°.
В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей пики с углами 2θ при 3,7±0,2°; 7,3±0,2°; 14,7±0,2°; и 16,9± 0,2°.
В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей пики с углами 2θ при 3,7±0,2°; 7,3±0,2°; 8,5±0,2°; 10,1± 0,2°; 14,7± 0,2°; и 16,9± 0,2°.
В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, в основном такой, как приведенная на фиг. 1. Перечень дифракционных пиков, описанных углами 2θ, и относительных интенсивностей с относительной интенсивностью ≥ 3,0% приведен выше в таблице X1.
Как хорошо известно в области порошковой рентгенографии, относительные интенсивности пиков (отражений) могут меняться в зависимости от методики приготовления образца, методики закрепления образца и конкретного использующегося прибора. Кроме того, различия приборов и другие факторы могут влиять на значения 2-тета. Поэтому положения пиков в XRPD могут меняться примерно на±0,2°.
Другой вариант осуществления включает соединение, которым является 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота в виде свободной кислоты.
Другой вариант осуществления включает соединение, которым является
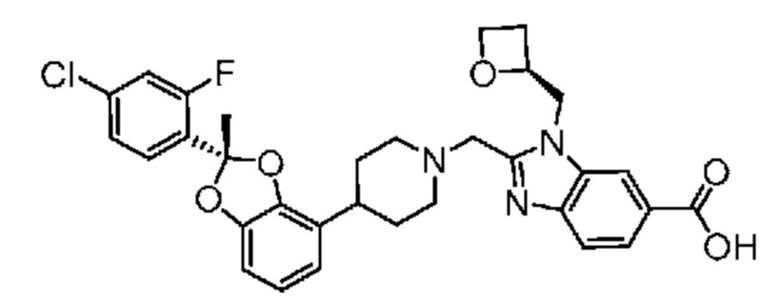
или его фармацевтически приемлемую соль.
Другой вариант осуществления включает соединение, которым является
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2R)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота в виде свободной кислоты.
Другой вариант осуществления включает соединение, которым является
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2R)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или его фармацевтически приемлемую соль, где солью является соль с Tris.
Другой вариант осуществления включает соединение, которым является 2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, DIAST-X2:
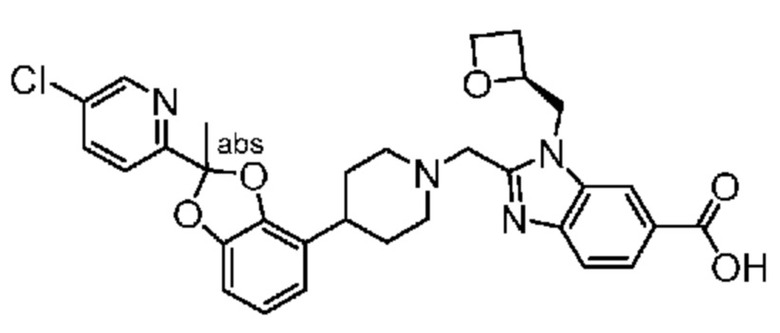 DIAST-X2,
DIAST-X2,
или его фармацевтически приемлемую соль. В некоторых дополнительных вариантах осуществления настоящее изобретение относится к соединению, которым является 2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, DIAST-X2 или его соль с Tris [т. е. соль с 1,3-дигидрокси-2-(гидроксиметил)пропан-2-амином]. Хиральный центр в левой части структуры соединения отмечен, как "abs" для указания на то, что хиральный центр обладает только одной стереохимической конфигурацией (т. е. не является рацематом по этому хиральному центру).
В некоторых вариантах осуществления настоящее изобретение относится к кристаллической форме безводной соли с Tris 2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, DIAST-X2. В некоторых дополнительных вариантах осуществления кристаллическая форма безводной (ангидрат) соли с Tris 2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, DIAST-X2, обозначена, как "форма A", которая охарактеризована в соответствии с ее уникальными характеристиками твердого состояния, например, по данным порошковой рентгенографии (PXRD), описанными в настоящем изобретении (в основном такими, как приведенные на фиг. 2). В некоторых вариантах осуществления форма A обладает порошковой рентгенограммой, содержащей по меньшей мере два характеристических пика с углами 2θ, выбранными из числа расположенных при 7,7±0,2°; 15,2±0,2°; 15,7± 0,2°; и 17,6± 0,2°. В некоторых вариантах осуществления форма A обладает порошковой рентгенограммой, содержащей по меньшей мере три характеристических пика с углами 2θ, выбранными из числа расположенных при 7,7±0,2°; 15,2±0,2°; 15,7± 0,2°; и 17,6± 0,2°. В некоторых вариантах осуществления форма A обладает порошковой рентгенограммой, содержащей характеристические пики с углами 2θ, выбранными из числа расположенных при 7,7±0,2°; 15,2±0,2°; 15,7± 0,2°; и 17,6± 0,2°.
В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей характеристические пики с углами 2θ при 7,7±0,2° и 17,6±0,2°.
В некоторых вариантах осуществления форма A обладает порошковой рентгенограммой, содержащей пики с углами 2θ при 7,7±0,2°; 15,2±0,2°; и 17,6±0,2°.
В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей пики с углами 2θ при 7,7±0,2°; 15,2±0,2°; и 15,7±0,2°.
В некоторых вариантах осуществления форма I обладает порошковой рентгенограммой, содержащей пики с углами 2θ при 7,7±0,2°; 15,2±0,2°; 15,7± 0,2°; и 17,6± 0,2°.
В некоторых вариантах осуществления форма A обладает порошковой рентгенограммой, в основном такой, как приведенная на фиг. 2. Перечень дифракционных пиков, описанных углами 2θ, и относительных интенсивностей с относительной интенсивностью ≥ 3,0% приведен выше в таблице X2.
Как хорошо известно в области порошковой рентгенографии, относительные интенсивности пиков (отражений) могут меняться в зависимости от методики приготовления образца, методики закрепления образца и конкретного использующегося прибора. Кроме того, различия приборов и другие факторы могут влиять на значения 2-тета. Поэтому положения пиков в XRPD могут меняться примерно на±0,2°.
В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение формул I, II, III, IV или V, или его фармацевтически приемлемую соль, определенное в любом из вариантов осуществления, описанных в настоящем изобретении, в смеси по меньшей мере с одним фармацевтически приемлемым инертным наполнителем. Она включает фармацевтическую композицию, содержащую соединение формул I, II, III, IV или V, или его фармацевтически приемлемую соль, определенное в любом из вариантов осуществления, описанных в настоящем изобретении, в смеси по меньшей мере с одним фармацевтически приемлемым инертным наполнителем и один или большее количество других терапевтических средств, рассмотренных в настоящем изобретении.
Настоящее изобретение также включает следующие варианты осуществления:
соединение формул I, II, III, IV или V, или его фармацевтически приемлемую соль, определенное в любом из вариантов осуществления, описанных в настоящем изобретении, для применения в качестве лекарственного средства;
соединение формул I, II, III, IV или V, или его фармацевтически приемлемую соль, определенное в любом из вариантов осуществления, описанных в настоящем изобретении, для применения для предупреждения и/или лечения кардиометаболических и сопутствующих заболеваний, рассмотренных в настоящем изобретении, включая T2DM, преддиабет, NASH и сердечно-сосудистое заболевание;
способ лечения заболевания, для которого показан агонист GLP-1R, у субъекта, нуждающегося в таком предупреждении и/или лечении, включающий введение субъекту терапевтически эффективного количества соединения формул I, II, III, IV или V, или его фармацевтически приемлемой соли, определенного в любом из вариантов осуществления, описанных в настоящем изобретении;
применение соединения формул I, II, III, IV или V, или его фармацевтически приемлемой соли, определенного в любом из вариантов осуществления, описанных в настоящем изобретении для приготовления лекарственного средства для лечения заболевания или патологического состояния, для которого показан агонист GLP-1R;
соединение формул I, II, III, IV или V, или его фармацевтически приемлемая соль, определенное в любом из вариантов осуществления, описанных в настоящем изобретении, для применения для лечения заболевания или патологического состояния, для которого показан агонист GLP-1R; или
фармацевтическая композиция для лечения заболевания или патологического состояния, для которого показан агонист GLP-1R, включающая соединение формул I, II, III, IV или V, или его фармацевтически приемлемую соль, определенное в любом из вариантов осуществления, описанных в настоящем изобретении.
Соединение каждого примера или его фармацевтически приемлемую соль может быть заявлено по отдельности или в сгруппированном виде в любой комбинации с любым количеством каждых и всяких вариантов осуществления, описанных в настоящем изобретении.
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формул I, II, III, IV или V, или ее фармацевтически приемлемую соль, определенное в любом из вариантов осуществления, описанных в настоящем изобретении, для применения для лечения и/или предупреждения кардиометаболических и сопутствующих заболеваний, рассмотренных в настоящем изобретении, включая T2DM, преддиабет, NASH и сердечно-сосудистое заболевание.
Другой вариант осуществления настоящего изобретения относится к соединению формул I, II, III, IV или V, или его фармацевтически приемлемой соли, определенному в любом из вариантов осуществления, описанных в настоящем изобретении, для применения для лечения и/или предупреждения кардиометаболических и сопутствующих заболеваний, включая диабет (T1D и/или T2DM, включая преддиабет), идиопатический T1D (тип 1b), латентный аутоиммунный диабет взрослых (LADA), юношеский T2DM (EOD), юношеский атипичный диабет (YOAD), диабет зрелого возраста у юношей (MODY), диабет, связанный с недостаточным питанием, гестациозный диабет, гипергликемию, резистентность к инсулину, резистентность печени к инсулину, нарушенную переносимость глюкозы, диабетическую невропатию, диабетическую нефропатию, заболевание почек (например, острое почечное нарушение, дисфункцию канальцев, провоспалительные изменения в проксимальных канальцах), диабетическую ретинопатию, дисфункцию адипоцитов, висцеральное отложение жира, апноэ во сне, ожирение (включая гипоталамическое ожирение и моногенное ожирение) и связанные с ними сопутствующие заболевания (например, остеоартрит и недержание мочи), пищевые расстройства (включая синдром компульсивного переедания, нервную булимию и синдромное ожирение, такое как синдром Прадера - Вилли и синдром Барде - Бидля), увеличение массы тела вследствие применения других средств (например, вследствие применения стероидов и антипсихотических средств), чрезмерное пристрастие к сладкому, дислипидемию (включая гиперлипидемию, гипертриглицеридемию, повышенное содержание общего холестерина, высокое содержание LDL холестерина и низкое содержание HDL холестерина), гиперинсулинемию, NAFLD (включая сопутствующие заболевания, такие как стеатоз, NASH, фиброз, цирроз, и печеночно-клеточная карцинома), сердечно-сосудистое заболевание, атеросклероз (включая заболевание коронарной артерии), заболевание периферических сосудов, гипертензию, эндотелиальную дисфункцию, нарушенную эластичность сосудов, застойную сердечную недостаточность, инфаркт миокарда (например, некроз и апоптоз), удар, геморрагический удар, ишемический удар, травматическое повреждение головного мозга, легочную гипертензию, рестеноз после ангиопластики, перемежающуюся хромоту, липемию после еды, метаболический ацидоз, кетоз, артрит, остеопороз, болезнь Паркинсона, гипертрофию левого желудочка, заболевание периферической артерии, дегенерацию желтого пятна, катаракту, гломерулосклероз, хроническую почечную недостаточность, метаболический синдром, синдром X, предменструальный синдром, стенокардию, тромбоз, атеросклероз, преходящие ишемические нарушения, рестеноз сосудов, нарушенный метаболизм глюкозы, патологические состояния с нарушенным содержанием глюкозы в плазме натощак, гиперурикемию, подагру, эректильную дисфункцию, кожные нарушения и нарушения соединительной ткани, псориаз, изъязвления ног, язвенный колит, гипер-апо-B-липопротеинемию, болезнь Альцгеймера, шизофрению, нарушенную познавательную способность, воспалительную болезнь кишечника, синдром короткой кишки, болезнь Крона, колит, синдром раздраженной толстой кишки, предупреждения или лечения синдрома поликистоза яичников и лечения привыкания (например, к алкоголю и/или злоупотребления лекарственным средством или наркотиком).
Аббревиатуры, использующиеся в настоящем изобретении, являются следующими:
Термин "алкил" при использовании в настоящем изобретении означает обладающую линейной или разветвленной цепью одновалентную углеводородную группу формулы -CnH(2n+1). Неограничивающие примеры включают метил, этил, пропил, бутил, 2-метилпропил, 1,1-диметилэтил, пентил и гексил.
Термин "алкилен" при использовании в настоящем изобретении означает обладающую линейной или разветвленной цепью двухвалентную углеводородную группу формулы -CnH2n-. Неограничивающие примеры включают этилен и пропилен.
Термин "циклоалкил" при использовании в настоящем изобретении означает циклическую одновалентную углеводородную группу формулы -CnH(2n-1), содержащую по меньшей мере три атома углерода. Неограничивающие примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
Термин "галоген" при использовании в настоящем изобретении означает фторид, хлорид, бромид или йодид.
Термин "гетероциклоалкил" при использовании в настоящем изобретении означает циклоалкильную группу, в которой одна или большее количество кольцевых метиленовых групп (-CH2-) замещены группой, выбраной из группы, включающей -O-, -S- или азот, где азот может быть положением присоединения или может быть замещен, как указано в каждом варианте осуществления. Если азот является положением присоединения, изображение структуры гетероциклоалкила должно включать водород у указанного азота. Обычно гетероциклоалкил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей оксогруппу, -CN, галоген, алкил и -Oалкил и алкил может быть дополнительно замещенным. Следует отметить, что, если имеется 0 замещений, то гетероциклоалкил является незамещенным.
Термин "гетероарил" при использовании в настоящем изобретении означает моноциклический ароматический углеводород, содержащий от 5 до 6 атомов углерода, в котором по меньшей мере один из кольцевых атомов углерода заменен гетероатомом, выбранным из группы, включающей кислород, азот и серу. Такая гетероарильная группа может быть присоединена через кольцевой атом углерода или, если допускает валентность, через кольцевой атом азота. Обычно гетероарил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей галоген, OH, алкил, O-алкил и аминогруппу (например, NH2, NHалкил, N(алкил)2), и алкил может быть дополнительно замещен. Следует отметить, что, если имеется 0 замещений, то гетероарил является незамещенным.
Комнатная температура: RT (от 15 до 25°C).
Метанол: MeOH.
Этанол: EtOH.
Изопропанол: iPrOH.
Этилацетат: EtOAc.
Тетрагидрофуран: THF.
Толуол: PhCH3.
Карбонат цезия: Cs2CO3.
Бис(триметилсилил)амид лития: LiHMDS.
Трет-бутоксид натрия: NaOtBu.
Трет-бутоксид калия: KOtBu.
Диизопропиламид лития: LDA.
Триэтиламин: Et3N.
N, N-диизопропилэтиламин: DIPEA.
Карбонат калия: K2CO3.
Диметилформамид: DMF.
Диметилацетамид: DMAc.
Диметилсульфоксид: DMSO.
N-Метил-2-пирролидинон: NMP.
Гидрид натрия: NaH.
Трифторуксусная кислота: TFA.
Трифторуксусный ангидрид: TFAA.
Уксусный ангидрид: Ac2O.
Дихлорметан: DCM.
1,2-Дихлорэтан: DCE.
Хлористоводородная кислота: HCl.
1,8-Диазабицикло[5.4.0]ундец-7-ен: DBU.
Комплекс боран-диметилсульфид: BH3-DMS.
Комплекс боран-тетрагидрофуран: BH3-THF.
Алюмогидрид лития: LAH.
Уксусная кислота: AcOH.
Ацетонитрил: MeCN.
п-Толуолсульфоновая кислота: pTSA.
Дибензилидинацетон: DBA.
2,2′-Бис(дифенилфосфино)-1,1′-бинафталин: BINAP.
1,1′-Ферроцендиил-бис(дифенилфосфин): dppf.
1,3-Бис(дифенилфосфино)пропан: DPPP.
3-Хлорпербензойная кислота: m-CPBA.
Трет-бутилметиловый эфир: MTBE.
Метансульфонил: Ms.
N-Метилпирролидинон: NMP.
Тонкослойная хроматография: TLC.
Надкритическая жидкостная хроматография: SFC.
4-(Диметиламино)пиридин: DMAP.
Трет-бутилоксикарбонил: Boc.
1-[Бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфат: HATU.
Петролейный эфир: PE.
2-(1H-Бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат: HBTU.
2-Амино-2-(гидроксиметил)пропан-1,3-диол: Tris.
Трис(дибензилиденацетон)дипалладий: Pd2(dba)3.
Спектры 1H ядерного магнитного резонанса (NMR) во всех случаях согласовались с предложенными структурами. Характеристики химических сдвигов (δ) приведены в частях на миллион относительно остаточного сигнала протона дейтерированного растворителя (CHCl3 при 7,27 част./млн; CD2HOD при 3,31 част./млн; MeCN при 1,94 част./млн; DMSO при 2,50 част./млн) и представлены с использованием обычных аббревиатур для обозначения основных пиков: например, s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; br, широкий. Символ ^ указывает, что площадь пика 1H NMR является предположительной, поскольку пик частично закрыт пиком воды. Символ ^^ указывает, что площадь пика 1H NMR является предположительной, поскольку пик частично закрыт пиком растворителя.
Специалист с общей подготовкой
При использовании в настоящем изобретении волнистая линия "  " обозначает положение присоединения заместителя к другой группе.
" обозначает положение присоединения заместителя к другой группе.
Соединения и промежуточные продукты, описанные ниже, названы с использованием соглашения о названиях, представленного в ACD/ChemSketch 2012, File Version C10H41, Build 69045 (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada). Соглашение о названиях, представленное ACD/ChemSketch 2012, хорошо известно специалистам в данной области техники и предполагается, что соглашение о названиях, представленное ACD/ChemSketch 2012, обычно согласуется с рекомендациями IUPAC (International Union for Pure and Applied Chemistry) по номенклатуре органической химии и с правилами CAS Index. Следует отметить, что химические названия могут включать только круглые скобки или могут включать круглые скобки и квадратные скобки. Стереохимические обозначения также могут находиться на разных участках самого названия в зависимости от соглашения о названиях. Специалист с общей подготовкой в данной области техники должен знать эти варианты формата и понимать, что они описывают одну и ту же химическую структуру.
Фармацевтически приемлемые соли соединений формул I, II, III, IV или V включают соли присоединения с кислотами и основаниями.
Подходящие соли присоединения с кислотами образуются из кислот, которые образуют нетоксичные соли. Примеры включают ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, цикламат, эдизилат, эзилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напзилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат, 1,5-нафталиндисульфонат и ксинафоат.
Подходящие соли присоединения с основаниями образуются из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, бис(2-гидроксиэтил)амина (диоламина), глицина, лизина, магния, меглумина, 2-аминоэтанола (оламина), калия, натрия, 2-амино-2-(гидроксиметил)пропан-1,3-диол (Tris или трометамина) и цинка.
Также могут образоваться гемисоли кислот и оснований, например, гемисульфат и гемисоли кальция. Обзор подходящих солей, см. Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002).
Фармацевтически приемлемые соли соединений формулы I можно получить по одной или большему количеству из трех методик:
(i) по реакции соединения формулы I с желательной кислотой или основанием;
(ii) путем удаления лабильной в кислоте или основании защитной группы из подходящего предшественника соединения формулы I или путем раскрытия цикла подходящего циклического предшественника, например, лактона или лактама, с использованием желательной кислоты или основания; или
(iii) путем превращения одной соли соединения формулы I в другую по реакции с подходящей кислотой или основанием или с помощью подходящей ионообменной колонки.
Все три реакции обычно проводят в растворе. Полученную соль можно осадить и собрать фильтрованием или можно выделить путем выпаривания растворителя. Степень ионизации полученной соли моет меняться от полностью ионизированной до почти неионизированной.
Соединения формулы I и их фармацевтически приемлемые соли могут существовать в несольватированной и сольватированной формат. Термин "сольват" используется в настоящем изобретении для описания молекулярного комплекса, включающего соединение формулы I или его фармацевтически приемлемую соль и одну или большее количество молекул фармацевтически приемлемого растворителя, например, этанола. Термин "гидрат" используется, когда указанным растворителем является вода.
Принятая в настоящее время система классификации органических гидратов определяет понятия гидратов в виде изолированного центра, канального или координированного с ионом металла - см. Polymorphism in Pharmaceutical Solids by K. R. Morris (Ed. H. G. Brittain, Marcel Dekker, 1995). Гидраты в виде изолированного центра представляют собой такие, в которых молекулы воды изолированы от непосредственного контакта друг с другом расположенными между ними органическими молекулами. В канальных гидратах молекулы воды находятся в каналах решетки, где они расположены после других молекул воды. В гидратах, координированных с ионом металла, молекулы воды связаны с ионом металла.
Если растворитель или вода связаны прочно, комплекс может обладать хорошо определенной стехиометрией независимо от влажности. Однако, если растворитель или вода связаны слабо, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя может зависеть от влажности и условий сушки. В таких случаях нормой является отсутствие стехиометрии.
В объем настоящего изобретения также входят многокомпонентные комплексы (кроме солей и сольватов), в которых лекарственное средство и по меньшей мере один другой компонент содержатся в стехиометрических или нестехиометрических количествах. Комплексы этого типа включают клатраты (комплексы включения лекарственное средство-хозяин) и совместные кристаллы. Последние обычно определяются, как кристаллические комплексы нейтральных молекулярных компонентов, которые связаны друг с другом нековалентными взаимодействиями, но также могут представлять собой комплекс нейтральной молекулы с солью. Совместные кристаллы можно получить путем кристаллизации расплава, путем перекристаллизации из растворителей или путем совместного механического размола компонентов - см. Chem Commun, 17, 1889-1896, O. Almarsson and M. J. Zaworotko (2004). Общий обзор многокомпонентных комплексов см. в J Pharm Sci, 64 (8), 1269-1288, by Haleblian (August 1975).
Соединения, предлагаемые в настоящем изобретении, могут существовать в непрерывном наборе твердофазных состояний в диапазоне от полностью аморфного до полностью кристаллического. Термин "аморфное" означает состояние, в котором у вещества отсутствует дальний порядок на молекулярном уровне, и, в зависимости от температуры, оно может обладать физическими характеристиками твердого вещества или жидкости. Обычно такие вещества не дают четкие картины дифракции рентгеновского излучения и, если они обладают физическими характеристиками твердого вещества, их более формально описывают, как жидкости. При нагревании происходит изменение характеристик от твердого вещества к жидкости, что характеризуется фазовым переходом, обычно второго порядка ("стеклование"). Термин "кристаллическая" означает твердую фазу, в которой вещество обладает регулярно упорядоченной внутренней структурой на молекулярном уровне и дает четкую рентгенограмму с выраженными пиками. Такие вещества при достаточном нагревании также обладают характеристиками жидкости, но переход от твердого состояния в жидкое характеризуется фазовым переходом, обычно первого порядка ("плавление").
Соединения формулы I при определенных условиях также могут существовать в мезоморфном состоянии (мезофаза или жидкий кристалл). Мезоморфное состояние является промежуточным между истинно кристаллическим состоянием и истинно жидким состоянием (расплав или раствор). Мезоморфизм, возникающий вследствие изменения температуры, описывается, как "термотропный" и возникающий вследствие добавления второго компонента, такого как вода или другой растворитель, описывается, как "лиотропный". Соединения, которые обладают способностью образовывать лиотропные мезофазы, описываются, как "амфифильные" и состоят из молекул, которые обладают ионной (такой как -COO-Na+, -COO-K+ или -SO3-Na+) или неионной (такой как -N-N+(CH3)3) полярной головной группой. Дополнительная информация приведена в Crystals and the Polarizing Microscope by N. H. Hartshorne and A. Stuart, 4th Edition (Edward Arnold, 1970).
Соединения формулы I могут обладать полиморфизмом и/или одним или большим количеством типов изомерии (например, оптической, геометрической или таутомерной изомерией). Соединения формулы I также могут быть изотопно-мечеными. Такое изменение применительно к соединениям формулы I определено, как относящееся к их структурным особенностям, и поэтому входит в объем настоящего изобретения.
Соединения формулы I, содержащее один или большее количество асимметрических атомов углерода, может существовать в виде двух или большего количества стереоизомеров. Если соединение формулы I содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс (или Z/E) изомеры. Если структурные изомеры превращаются друг в друга с низким энергетическим барьером, может происходить таутомерная изомерия ("таутомерия"). Она может проявляться в виде протонной таутомерии в соединениях формулы I, содержащих, например, иминогруппу, кето- или оксимную группу, или так называемой валентной таутомерии в соединениях, которые содержат ароматический фрагмент. Следовательно, одно соединение может обладать более, чем одним типом изомерии.
Фармацевтически приемлемые соли соединений формулы I также могут содержать противоион, который является активным (например, d-лактат или l-лизин) или рацемическим (например, dl-тартрат или dl-аргинин).
Цис/транс изомеры можно разделить по обычным методикам, хорошо известным специалистам в данной области техники, например, с помощью хроматографии и фракционной кристаллизации.
Обычные методики получения/выделения отдельных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с помощью, например, хиральной высокоэффективной жидкостной хроматографии (HPLC). Альтернативно, рацемический предшественник, содержащий хиральную сложноэфирную группу, можно разделить путем ферментативного разделения (см., например, Int J Mol Sci 29682-29716 by A. C. L. M. Carvaho et. al. (2015)). В случае, если соединение формулы I содержит кислотный или основной фрагмент, соль можно образовать с оптически чистым основанием или кислотой, таким как 1-фенилэтиламин или винная кислота. Полученную диастереоизомерную смесь можно разделить с помощью фракционной кристаллизации и одну или обе диастереоизомерные соли превратить в соответствующий чистый энантиомер(ы) по методикам, хорошо известным специалисту в данной области техники. Альтернативно, рацемат (или рацемический предшественник) можно ввести в реакцию с образованием ковалентной связи с подходящим оптически активным соединением, например, спиртом, амином или бензилхлоридом. Полученную диастереоизомерную смесь можно разделить с помощью хроматографии и/или фракционной кристаллизации по методикам, хорошо известным специалисту в данной области техники, и получить разделенные диастереоизомеры в виде отдельных энантиомеров с 2 или большим количеством хиральных центров. Хиральные соединения формулы I (и их хиральные предшественники) можно получить в энантиомерно-обогащенной форме с помощью хроматографии, обычно HPLC, на асимметрической смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего от 0 до 50 об.% изопропанола, обычно от 2% до 20%, и от 0 до 5 об.% алкиламина, обычно 0,1% диэтиламина. Концентрирование элюата дает обогащенную смесь. Можно использовать хиральную хроматографию с применением суб- и надкритических жидкостей. Методики хиральной хроматографии, применимые в некоторых вариантах осуществления настоящего изобретения, известны в данной области техники (см., например, Smith, Roger M., Loughborough University, Loughborough, UK; Chromatographic Science Series (1998), 75 (SFC with Packed Columns), pp. 223-249 и и цитированную там литературу). В некоторых соответствующих примерах в настоящем изобретении, колонки получали у фирмы Chiral Technologies, Inc, West Chester, Pennsylvania, USA, subsidiary of Daicel® Chemical Industries, Ltd., Tokyo, Japan.
Если какой-либо рацемат кристаллизуется, возможны кристаллы двух разных типов. Первым типом является рацемическое соединение (истинный рацемат), указанное выше, где образуется одна однородная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Вторым типом является рацемическая смесь или конгломерат, где две формы кристалла образуются в эквимолярных количествах, каждая из которых содержит один энантиомер. Хотя обе кристаллические формы, содержащиеся в рацемической смеси, обладают одинаковыми физическими характеристиками, они могут обладать физическими характеристиками, отличающимися от характеристик истинного рацемата. Рацемические смеси можно разделить по обычным методикам, хорошо известным специалистам в данной области техники - см., например, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, 1994).
Следует подчеркнуть, что соединения формулы I изображены в настоящем изобретении в одной таутомерной форме, все возможные таутомерные формы входят в объем настоящего изобретения.
Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченые соединения формулы I, в которых один или большее количество атомов заменено атомами, обладающими таким не атомным номером, но атомной массой ил массовым числом, отличающимися от атомной массы или атомного числа, преобладающего в природе.
Примеры изотопов, подходящих для включения в соединения, предлагаемые в настоящем изобретении, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, и серы, такие как 35S.
Некоторые изотопно-меченые соединения формулы I, например, включающие радиоактивный изотоп, применимы для исследования распределения лекарственного средства и/или субстрата в ткани. Радиоактивные изотопы трития, т. е. 3H, и углерода-14, т. е. 14C, являются особенно подходящими для этой цели вследствие легкости включения и имеющихся средств детектирования.
Замещение более тяжелыми изотопами, такими как дейтерий, т. е. 2H, может обеспечить некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличением периода полувыведения или возможностью использования меньших доз.
Замещение излучающими позитроны изотопами, такими как 11C, 18F, 15O и 13N, можно использовать в исследованиях с помощью позитронной эмиссионной томографии (PET) для изучения занятости рецепторов субстрата.
Изотопно-меченые соединения формулы I обычно можно получить по обычным методикам, хорошо известным специалистам в данной области техники или по методикам, аналогичным описанным в прилагаемых примерах и синтезах, с использованием подходящего изотопно-меченого реагента вместо применявшегося ранее немеченого реагента.
Фармацевтически приемлемые сольваты в контексте настоящего изобретения включают такие, в которых растворитель для кристаллизации может быть изотопно замещенным, например, D2O, d6-ацетон, d6-DMSO.
Одним путем осуществления настоящего изобретения является введение соединение формулы I в форме пролекарства. Так, некоторые производные соединения формулы I, которые сами могут обладать низкой фармакологической активностью или не обладать такой активностью, при введении в организм или нанесении на него могут превратиться в соединение формулы I, обладающее желательной активностью, например, вследствие гидролитического расщепления, в частности, гидролитического, стимулированного ферментом эстеразой или пептидазой. Такие производные называются "пролекарствами". Дополнительная информация о применении пролекарств приведена в публикациях "Pro-drugs as Novel Delivery Systems", Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) и "Bioreversible Carriers in Drug Design", Pergamon Press, 1987 (Ed. E. B. Roche, American Pharmaceutical Association). Также дается ссылка на Nature Reviews/Drug Discovery, 2008, 7, 355 и Current Opinion in Drug Discovery and Development, 2007, 10, 550.
Пролекарства в контексте настоящего изобретения, например, можно получить путем замены соответствующих функциональных групп, содержащихся в соединениях формулы I, некоторыми фрагментами, известными специалистам в данной области техники, как "профрагменты", описанные, например, в "Design of Prodrugs" by H. Bundgaard (Elsevier, 1985) и Y. M. Choi-Sledeski and C. G. Wermuth, "Designing Prodrugs and Bioprecursors" in Practice of Medicinal Chemistry, (Fourth Edition), Chapter 28, 657-696 (Elsevier, 2015).
Таким образом, пролекарством в контексте настоящего изобретения является (a) сложноэфирное или амидное производное карбоновой кислоты в соединении формулы I; (b) сложноэфирное, карбонатное, карбаматное, фосфатное или простое эфирное производное гидроксигруппы в соединении формулы I; (c) амидное, иминное, карбаматное или аминное производное аминогруппы в соединении формулы I; (d) оксимное или иминное производное карбонильной группы в соединении формулы I; или (e) метильная, первичная гидроксильная или альдегидная группа, которая может быть метаболически окислена в карбоксигруппу в соединении формулы I.
Некоторые конкретные примеры пролекарств в контексте настоящего изобретения включают такие случаи:
(i) когда соединение формулы I содержит карбоксигруппу (-COOH), ее сложноэфирное производное, такое как соединение, в котором водород карбоксигруппы соединения формулы I заменен на C1-C8 алкил (например, этил) или (C1-C8 алкил)C(=O)OCH2- (например, tBuC(=O)OCH2-);
(ii) когда соединение формулы I содержит гидроксигруппу (-OH), ее сложноэфирное производное, такое как соединение, в котором водород гидроксигруппы соединения формулы I заменен на -CO(C1-C8 алкил) (например, метилкарбонил) или гидроксигруппа этерифицирована аминокислотой;
(iii) когда соединение формулы I содержит гидроксигруппу (-OH), ее простой эфир, такое как соединение, в котором водород гидроксигруппы соединения формулы I заменен на (C1-C8 алкил)C(=O)OCH2- или -CH2OP(=O)(OH)2;
(iv) когда соединение формулы I содержит гидроксигруппу (-OH), ее фосфат, такое как соединение, в котором водород гидроксигруппы соединения формулы I заменен на -P(=O)(OH)2 или -P(=O)(ONa)2 или -P(=O)(O-)2Ca2+;
(v) когда соединение формулы I содержит (-NH2 или -NH, где R ≠ H), ее амид, например, соединение, в котором, как это может быть, один или оба водорода аминогруппы соединения формулы I заменен/заменены на (C1-C10)алканоил, -COCH2NH2 или аминогруппа дериватизирована аминокислотой;
(vi) когда соединение формулы I содержит первичную или вторичную аминогруппу (-NH2 или -NHR, где R ≠ H), ее амин, например, соединение, в котором, как это может быть, один или оба водорода аминогруппы соединения формулы I заменен/заменены на -CH2OP(=O)(OH)2;
(vii) когда карбоксигруппа в соединении формулы I заменена на метильную группу, группу -CH2OH или альдегидную группу.
Некоторые соединения формулы I сами могут действовать, как пролекарства других соединений формулы I. Также может быть, что два соединения формулы I объединяются в форме пролекарства. В некоторых случаях пролекарство соединения формулы I может быть образовано путем внутреннего связывания двух функциональных групп в соединении формулы I, например, с образованием лактона.
Указания на соединения формулы I включают сами соединения и их пролекарства. Настоящее изобретение включает такие соединения формулы I, а также фармацевтически приемлемые соли таких соединений и фармацевтически приемлемые сольваты и соли указанных соединений.
Введение и дозирование
Обычно соединение, предлагаемое в настоящем изобретении, вводят в количестве, эффективном для лечения патологического состояния, описанного в настоящем изобретении. Соединения, предлагаемые в настоящем изобретении, можно вводить, как сами соединения, или, альтернативно, в виде фармацевтически приемлемой соли. Для введения и дозирования само соединение или его фармацевтически приемлемую соль ниже просто называют, как соединения, предлагаемые в настоящем изобретении.
Соединения, предлагаемые в настоящем изобретении, вводят любым подходящим путем в виде фармацевтической композиции, адаптированной для такого пути, в дозе, эффективной для назначенного лечения. Соединения, предлагаемые в настоящем изобретении, можно вводить перорально, ректально, вагинально, парентерально или местно.
Соединения, предлагаемые в настоящем изобретении, можно вводить перорально. Пероральное введение может включать проглатывание, так что соединение попадает в желудочно-кишечный тракт, или можно использовать буккальное или сублингвальное введение, с помощью которого соединение попадает в кровоток непосредственно изо рта.
В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, также можно вводить непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие пути для парентерального введения включают внутривенный, внутриартериальный, внутрибрюшинный, интратекальный, внутрижелудочковый, внутриуретральный, надчревный, внутричерепной, внутримышечный и подкожный. Подходящие устройства для парентерального введения включают игольчатые (включая микроигольчатые) шприцы, безыгольные шприцы и устройства для вливания.
В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, также можно вводить местно на кожу или слизистую оболочку, т. е. накожно или чрескожно. В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, также можно вводить интраназально или путем ингаляции. В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, можно вводить ректально или вагинально. В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, также можно вводить непосредственно в глаз или ухо.
Режим дозирования соединений, предлагаемых в настоящем изобретении, и/или композиций, содержащих указанные соединения основан на множестве факторов, включая тип, возраст, массу, пол и состояние здоровья пациента; тяжесть патологического состояния; путь введения; и активность конкретного использующегося соединения. Таким образом, режим дозирования может значительно меняться. В одном варианте осуществления полная суточная доза соединения, предлагаемого в настоящем изобретении, обычно равна от примерно 0,001 до примерно 100 мг/кг (т. е. количество миллиграммов соединения, предлагаемого в настоящем изобретении, на 1 кг массы тела) для лечения указанных патологических состояний, рассмотренных в настоящем изобретении. В другом варианте осуществления полная суточная доза соединения, предлагаемого в настоящем изобретении, равна от примерно 0,01 до примерно 30 мг/кг, и в другом варианте осуществления от примерно 0,03 до примерно 10 мг/кг, и в еще одном варианте осуществления от примерно 0,1 до примерно 3. Не является необычным, если введение соединений, предлагаемых в настоящем изобретении, повторяют несколько раз в сутки (обычно не более 4 раз). Несколько доз в сутки при желании обычно можно использовать для увеличения полной суточной дозы.
Для перорального введения композиции можно использовать в форме таблеток, содержащих 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 30,0 50,0, 75,0, 100, 125, 150, 175, 200, 250 и 500 мг активного ингредиента для симптоматического подбора дозы для пациента. Лекарственное средство обычно содержит от примерно 0,01 мг до примерно 500 мг активного ингредиента, или в другом варианте осуществления от примерно 1 мг до примерно 100 мг активного ингредиента. Внутривенные дозы могут находиться в диапазоне от примерно 0,01 до примерно 10 мг/кг/мин при постоянной скорости вливания.
Подходящие субъекты в контексте настоящего изобретения включают млекопитающих. В одном варианте осуществления подходящими субъектами являются люди. Люди могут быть любого пола и на любой стадии развития.
Фармацевтические композиции
В другом варианте осуществления настоящее изобретение относится к фармацевтическим композициям. Такие фармацевтические композиции содержат соединение, предлагаемое в настоящем изобретении, вместе с фармацевтически приемлемым носителем. Также могут содержаться другие фармакологически активные вещества. При использовании в настоящем изобретении "фармацевтически приемлемый носитель" включает любые и все растворители, диспергирующие среды, покрытия, антибактериальные и фунгицидные агенты, изотонические агенты и агенты, задерживающие всасывание и т. п., которые являются физиологически совместимыми. Примеры фармацевтически приемлемых носителей включают один или большее количество следующих: вода, физиологический раствор, забуференный фосфатом физиологический раствор, декстроза, глицерин, этанол и т. п., а также их комбинации, и могут содержать в композиции изотонические агенты, например, сахара, хлорид натрия или многоатомные спирты, такие как маннит, или сорбит. Примеры также включают фармацевтически приемлемые вещества, такие как смачивающие агенты или небольшие количества вспомогательных веществ, таких как смачивающие или эмульгирующие агенты, консерванты или буферы, которые увеличивают длительность действия или эффективность антител или фрагментов антител.
Композиции, предлагаемые в настоящем изобретении, могут находиться в разных формах. Они включают, например, жидкие, полужидкие и твердые дозированные формы, такие как жидкие растворы (например, растворы для инъекции и вливания), дисперсии или суспензии, таблетки, пилюли, порошки, липосомы и суппозитории. Форма зависит от назначенного пути введения и терапевтического применения.
Типичные композиции находятся в форме растворов для инъекции или вливания, такие как композиции, аналогичные используемым для пассивной общей иммунизации людей антителами. Одним путем введения является парентеральный (например, внутривенный, подкожный, внутрибрюшинный, внутримышечный). В другом варианте осуществления антитела вводят путем внутривенного вливания или инъекции. В еще одном варианте осуществления антитела вводят путем внутримышечной или подкожной инъекции.
Дозированная форма для перорального введения твердого вещества может, например, быть представлена в отдельных порциях, таких как твердые или мягкие капсулы, пилюли, облатки, пастилки или таблетки, каждая из которых содержат заданное количество по меньшей мере одного соединения, предлагаемого в настоящем изобретении. В другом варианте осуществления пероральное введение может проводиться в форме порошка или гранулы. В другом варианте осуществления пероральная дозированная форма является сублингвальной, такой как, например, пастилка. В таких твердых дозированных формах, соединения формулы I обычно объединяют с одним или большим количеством вспомогательных веществ. Такие капсулы или таблетки могут представлять собой препарат регулируемого высвобождения. В случае капсул, таблеток и пилюль дозированные формы также могут содержать буферные реагенты или их можно приготовить с энтеросолюбильными покрытиями.
В другом варианте осуществления пероральное введение может проводиться с помощью жидкой дозированной формы. Жидкие дозированные формы для перорального введения включают, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно использующиеся в данной области техники (например, воду). Такие композиции также могут содержать вспомогательные вещества, такие как смачивающие, эмульгирующие, суспендирующие, придающие вкус (например, подсластители) и/или отдушки.
В другом варианте осуществления настоящее изобретение относится к парентеральной дозированной форме. "Парентеральное введение" включает, например, подкожные инъекции, внутривенные инъекции, внутрибрюшинные, внутримышечные инъекции, надчревные инъекции и вливание. Препараты для инъекции (т. е. стерильные водные или масляные суспензии для инъекции) можно приготовить в соответствии с известным уровнем техники с использованием подходящих диспергирующих, смачивающих агентов и/или суспендирующих агентов.
В другом варианте осуществления настоящее изобретение относится к местной дозированной форме. "Местное введение" включает, например, чрескожное введение, такое как с помощью чрескожных пластырей или устройств для ионтофореза, внутриглазное введение, или интраназальное или ингаляционное введение. Композиции для местного введения также включают, например, местные гели, спреи, мази и кремы. Местный препарат может включать соединение, которое усиливает поглощение или проникновение активного ингредиента через кожу или другие пораженные участки. Если соединения, предлагаемые в настоящем изобретении, вводят с помощью чрескожного устройства, введение проводят с помощью пластыря резервуарного и пористого мембранного типа, или разных твердых матриц. Типичные препараты для этой цели включают гели, гидрогели, примочки, растворы, кремы, мази, присыпки, повязки, пенки, пленки, кожные пластыри, пластинки, имплантаты, губки, волокна, перевязочные материалы и микроэмульсии. Также можно использовать липосомы. Типичные носители включают спирт, воду, минеральное масло, жидкое вазелиновое масло, белое вазелиновое масло, глицерин, полиэтиленгликоль и пропиленгликоль. Можно включать средства, улучшающие проницаемость - см., например, B. C. Finnin and T. M. Morgan, J. Pharm. Sci., vol. 88, pp. 955-958, 1999.
Препараты, подходящие для местного введения в глаза, включают, например, глазные капли, в которых соединение, предлагаемое в настоящем изобретении, растворено или суспендировано в подходящем носителе. Типичный препарат, подходящий для введения в глаза или уши, может находиться в форме капель микронизированной суспензии или раствора в изотоническом с установленным pH стерильном физиологическом растворе. Другие препараты, подходящие для введения в глаза или уши, включают мази, биологически разрушающиеся (т. е. впитывающие гель губки, коллаген) и биологически неразрушающиеся (т. е. силиконовые) имплантаты, пластинки, линзы и измельченные или везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновую кислоту, полимер целлюлозы, например, гидроксипропилметилцеллюлозу, гидроксиэтилцеллюлозу или метилцеллюлозу, или полимерный гетерополисахарид, например, геллановую камедь, можно включить вместе с консервантом, таким как бензалконийхлорид. Такие препараты также можно доставить с помощью ионтофореза.
Для интраназального введения или введения путем ингаляции, соединения, предлагаемые в настоящем изобретении, обычно доставляют в форме раствора или суспензии из контейнера с насосом для распыления, который сжимает или использует для накачивания пациент или системы распыления аэрозоля из контейнера под давлением или распылителя с использованием подходящего пропеллента. Препараты, подходящие для интраназального введения обычно вводят в форме сухого порошка (чистого, в виде смеси, например, сухой смеси с лактозой, или смешанных частиц компонента, например, в смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора для сухого порошка или с распылением аэрозоля из контейнера под давлением, с помощью насоса, распылителя, атомизатора (предпочтительно атомизатора с использованием электродинамического устройства для получения тонкого тумана), или распылителя с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Для интраназального применения, порошок может содержать биоадгезивный агент, например, хитозан или циклодекстрин.
В другом варианте осуществления настоящее изобретение относится к ректальной дозированной форме. Такая ректальная дозированная форма может находиться в форме, например, суппозитория. Масло какао является традиционной основой для суппозитория, но можно использовать разные альтернативы, если они являются подходящими.
Также можно использовать другие материалы носителей и пути введения, известные в фармацевтике. Фармацевтические композиции, предлагаемые в настоящем изобретении, можно получить по любой из хорошо известных методик фармацевтик, таким как эффективные процедуры получения и введения. Приведенные выше соображения в отношении эффективных препаратов и методик введения хорошо известны в данной области техники и описаны в стандартных учебниках. Составление лекарственных средств рассмотрено, например, в Hoover, John E., Remington"s Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999.
Совместное введение
Соединения, предлагаемые в настоящем изобретении, можно использовать по отдельности или в комбинации с другими терапевтическими средствами. Настоящее изобретение относится к любым из применений, способов или композиций, определенных в настоящем изобретении, где соединение любого варианта осуществления формулы I в настоящем изобретении или его фармацевтически приемлемую соль, или фармацевтически приемлемый сольват указанного соединения или соли используют в комбинации с одним или большим количеством других терапевтических средств, рассмотренных в настоящем изобретении. Это включает фармацевтическую композицию для лечения заболевания или патологического состояния, для которого показан агонист GLP-1R, содержащую соединение формул I, II, III, IV или V, или его фармацевтически приемлемую соль, определенное в любом из вариантов осуществления, описанных в настоящем изобретении, и один или большее количество других терапевтических средств, рассмотренных в настоящем изобретении.
Введение двух или большего количества соединений "в комбинации" означает, что все соединения вводят в близкие моменты времени, так что каждое может оказать биологическое воздействие в один и тот же заданный интервал времени. Наличие одного средства может изменить биологические воздействия другого соединения (соединений). Два или большее количество соединений можно вводить одновременно, совместно или последовательно. Кроме того, одновременное введение можно провести путем смешивания соединений до введения или путем введения соединений в один и тот же момент времени, но в виде отдельных дозированных форм на один или разные участки введения.
Выражения "одновременное введение", "совместное введение", "синхронное введение" и "введение одновременно" означает, что соединения вводят в комбинации.
В другом варианте осуществления настоящее изобретение относится к способам лечения, которые включают введение соединений, предлагаемых в настоящем изобретении, в комбинации с одним или большим количеством других фармацевтических средств, где одно или большее количество других фармацевтических средств можно выбрать из числа средств, рассмотренных в настоящем изобретении.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, вводят с противодиабетическим средством, включая, но не ограничиваясь только ими, бигуанид (например, метформин), сульфонилмочевина (например, толбутамид, глибенкламид, гликлазид, хлорпропамид, толазамид, ацетогексамид, гликлопирамид, глимепирид или глипизид), тиазолидиндион (например, пиоглитазон, росиглитазон или лобеглитазон), глитазар (например, сароглитазар, алеглитазар, мураглитазар или тезаглитазар), меглитинид (например, натеглинид, репаглинид), ингибитор дипептидилпептидазы 4 (DPP-4) (например, ситаглиптин, вилдаглиптин, саксаглиптин, линаглиптин, гемиглиптин, анаглиптин, тенелиглиптин, алоглиптин, трелаглиптин, дутоглиптин, или омариглиптин), глитазон (например, пиоглитазон, росиглитазон, балаглитазон, ривоглитазон или лобеглитазон), ингибитор натрийзависимого переносчика глюкозы 2 (SGLT2) (например, эмпаглифлозин, канаглифлозин, дапаглифлозин, ипраглифлозин, ипраглифлозин, тофоглифлозин, серглифлозинэтабонат, ремоглифлозинэтабонат или эртуглифлозин), ингибитор SGLTL1, агонист GPR40 (агонист FFAR1/FFA1, например, фасиглифам), глюкозозависимый инсулинотропный полипептид (GIP) и его аналоги, ингибитор альфа-глюкозидазы (например, воглибоза, акарбоза или миглитол) или инсулин или аналог инсулина, включая фармацевтически приемлемые соли конкретных названных средств и фармацевтически приемлемые сольваты и соли указанных средств.
В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, вводят со средством против ожирения, включая, но не ограничиваясь только ими, пептид YY или его аналог, агонист рецептора нейропептида Y типа 2 (NPYR2), антагонист NPYR1 или NPYR5, антагонист каннабиноидного рецептора типа 1 (CB1R), ингибитор липазы (например, орлистат), человеческий про-островковый пептид (HIP), агонист меланокортинового рецептора 4 (например, сетмеланотид), антагонист рецептора 1 меланин-концентрирующего гормона, агонист фарнезоидного рецептора X (FXR) (например, обетихолевая кислота), зонисамид, фентермин (по отдельности или в комбинации с топираматом), ингибитор обратного захвата норэпинефрина/допамина (например, бупроприон), антагонист опиоидного рецептора (например, налтрексон), комбинация ингибитора обратного захвата норэпинефрина/допамина и антагониста опиоидного рецептора (например, комбинация бупропиона и налтрексона), аналог GDF-15, сибутрамин, агонист олецистокинина, амилин и его аналоги (например, прамлинтид), лептин и его аналоги (например, метролептин), серотонинергическое средство (например, лоркасерин), метионин ингибитор аминопертидазы 2 (MetAP2) (например, белораниб или ZGN-1061), фендиметразин, диэтилпропион, бензфетамин, ингибитор SGLT2 (например, эмпаглифлозин, канаглифлозин, дапаглифлозин, ипраглифлозин, ипраглифлозин, тофоглифлозин, серглифлозинэтабонат, ремоглифлозин этабонат или эртуглифлозин), ингибитор SGLTL1, двойной ингибитор SGLT2/SGLT1, модулятор рецептора фактора роста фибробластов (FGFR), активатор AMP-активированной протеинкиназы (AMPK), биотин, модулятор рецептора MAS, или агонист глюкагонового рецептора (по отдельности или в комбинации с другим агонистом GLP-1R, например, лираглутидом, эксенатидом, дилаглутидом, албиглутидом, ликсисенатидом или семаглутидом), включая фармацевтически приемлемые соли конкретных названных средств и фармацевтически приемлемые сольваты и соли указанных средств.
В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, вводят в комбинации с одним или большим количеством следующих: средство для лечения NASH включая, но не ограничиваясь только ими, PF-05221304, агонист FXR (например, обетихолевая кислота), агонист PPAR α/δ (например, элафибранор), коньюгат синтетической жирной кислоты−желчной кислоты (например, арамхол), ингибитор каспазы (например, эмриказан), моноклональные антитела к гомологу 2 антилизилоксидазы (LOXL2) (например, симтузумаб), ингибитор галектина 3 (например, GR-MD-02), ингибитор MAPK5 (например, GS-4997), двойной антагонист хемокинового рецептора 2 (CCR2) и CCR5 (например, ценикривирок), агонист фактора роста фибробластов 21 (FGF21) (например, BMS-986036), антагонист лейкотриенового рецептора D4 (LTD4) (например, типелукаст), аналог ниацина (например, ARI 3037MO), ингибитор ASBT (например, воликсибат), ингибитор ацетил-CoA карбоксилазы (ACC) (например, NDI 010976 или PF-05221304), ингибитор кетогексокинызы (KHK), ингибитор диацилглицерилацилтрансферазы 2 (DGAT2), антагонист рецептора CB1, антитела к CB1R или ингибитор регулирующей апоптоз киназы 1 (ASK1), включая фармацевтически приемлемые соли конкретных названных средств и фармацевтически приемлемые сольваты и соли указанных средств.
Некоторые конкретные соединения, которые можно использовать в комбинации с соединениями, предлагаемыми в настоящем изобретении, для лечения заболеваний или нарушений, описанных в настоящем изобретении (например, NASH) включают:
4-(4-(1-Изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)безойную кислоту, которая является примером селективного ингибитора ACC и получена в виде свободной кислоты в примере 9 патента U.S. № 8859577, который является национальной фазой в США Международной заявки № PCT/IB2011/054119, раскрытия которых во всей их полноте включены в настоящее изобретение в качестве ссылки для всех объектов. Кристаллические формы 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты, включая безводную моно-Tris форму (форма 1) и тригидрат моно-соли с Tris (форма 2), описаны в международной заявке PCT №. PCT/IB2018/058966, раскрытие которой во всей его полноте включено в настоящее изобретение в качестве ссылки для всех объектов;
(S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль и его твердые кристаллические формы (форма 1 и форма 2) является примером ингибитора DGAT2, описанного в примере 1 патента U.S. № 10071992, раскрытие которого во всей его полноте включено в настоящее изобретение в качестве ссылки для всех объектов;
[(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусную кислоту или ее фармацевтически приемлемая соль (включая ее кристаллическую форму свободной кислоты) является примером ингибитора кетогексокиназы и описана в примере 4 патента U.S. № 9809579, раскрытие которого во всей его полноте включено в настоящее изобретение в качестве ссылки для всех объектов; и
агонист FXR тропифексор или его фармацевтически приемлемую соль, описан в примере 1-1B патента U.S. № 9150568, раскрытие которого во всей его полноте включено в настоящее изобретение в качестве ссылки для всех объектов.
Эти средства и соединения, предлагаемые в настоящем изобретении, можно объединять с фармацевтически приемлемыми разбавителями, такими как физиологический раствор, раствор Рингера, раствор декстрозы и т. п. Конкретный режим введения, т. е. доза, время и количество повторов, зависит от конкретного индивидуума и истории болезни индивидуума.
Приемлемые носители, инертные наполнители или стабилизаторы нетоксичны для реципиента в использующихся дозах и концентрациях и могут содержать буферы, такие как фосфатный, цитратный и включающий другие органические кислоты; соли, такие как хлорид натрия; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензилхлорид аммония; гексаметонийхлорид; бензалконийхлорид, бензэтонийхлорид; фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метил- или пропилпарабен; пирокатехин; резорцин; циклогексанол; 3-пентанол; и м-крезол); обладающие низкой молекулярной массой (менее примерно 10 остатков) полипептиды; балки, такие как сывороточный альбумин, желатин или Igs; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатные агенты, такие как EDTA; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как ионы натрия; комплексы металлов (например, комплексы Zn-белок); и/или неионогенные поверхностно-активные вещества, такие как TWEENTM, PLURONICSTM или полиэтиленгликоль (PEG).
Липосомы, содержащие эти средства и/или соединения, предлагаемые в настоящем изобретении, получают по методикам, известным в данной области техники, таким как описанные в патентах U.S. №№ 4485045 и 4544545. Липосомы с увеличенным временем обращения раскрыты в патенте U.S. № 5013556. Особенно полезные липосомы можно получить по методике обращенно-фазового выпаривания с использованием липидной композиции, содержащей фосфатидилхолин, холестерин и дериватизированный с помощью PEG фосфатидилэтаноламин (PEG-PE). Липосомы экструдируют через фильтры с определенным размером отверстий и получают липосомы определенного диаметра.
Эти средства и/или соединения, предлагаемые в настоящем изобретении, также можно поместить в микрокапсулы, получаемые, например, по методикам коацервации или с помощью межфазной полимеризации, например, гидроксиметилцеллюлозы, или желатиновые микрокапсулы и поли(метилметакрилатные) микрокапсулы соответственно для коллоидных систем доставки лекарственного средства (например, липосомы, микросферы альбумина, микроэмульсии, наночастицы и нанокапсулы) или включить в макроэмульсии. Такие методики раскрыты в Remington, The Science and Practice of Pharmacy, 20th Ed., Mack Publishing (2000).
Можно использовать препараты пролонгированного высвобождения. Подходящие примеры препаратов пролонгированного высвобождения включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие соединение формул I, II, III, IV или V, и эти матрицы находятся в виде формованных изделий, например, пленок или микрокапсул. Примеры матриц пролонгированного высвобождения включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт)), полилактиды (патент U.S. № 3773919), сополимеры L-глутаминовой кислоты и 7-этил-L-глутамата, неразлагающиеся сополимеры этилен-винилацетат, разлагающиеся сополимеры молочная кислота-гликолевая кислота, такие как использующиеся в LUPRON DEPOTTM (микросферы для инъекции, состоящие из сополимера молочная кислота-гликолевая кислота и лейпролидацетата), ацетат-изобутират сахарозы и поли-D-(-)-3-гидроксимасляную кислоту.
Препараты для использования для внутривенного введения должны быть стерильными. Это легко обеспечить, например, путем стерильного фильтрования через мембраны. Соединения, предлагаемые в настоящем изобретении, обычно помещают в контейнер, содержащий стерильное впускное отверстие, например, мешок с раствором для внутривенного введения или флакон с пробкой, прокалываемой иглой шприца для инъекции.
Подходящие эмульсии можно получить с использованием имеющихся в продаже эмульсии жиров, таких как IntralipidTM, LiposynTM, InfonutrolTM, LipofundinTM и LipiphysanTM. Активный ингредиент можно растворить в предварительно смешанной композиции эмульсии или, альтернативно, его можно растворить в масле (таком как, соевое масло, сафлоровое масло, хлопковое масло, кунжутное масло, кукурузное масло или миндальное масло) и эмульсии, образовавшейся после перемешивания с фосфолипидом (таким как яичные фосфолипиды, соевые фосфолипиды или соевый лецитин) и водой. Следует понимать, что можно добавить другие ингредиенты, например, глицерин или глюкозу, для регулирования тоничности эмульсии. Подходящие эмульсии обычно содержат до 20% масла, например, от 5 до 20%. Эмульсия жира может содержать капельки жира размером от 0,1 до 1,0 мкм, предпочтительно от 0,1 до 0,5 мкм, и обладают pH в диапазоне от 5,5 до 8,0.
Композиции эмульсии могут быть такими, которые готовят путем смешивания соединения, предлагаемого в настоящем изобретении, с IntralipidTM или его компонентами (соевое масло, яичные фосфолипиды, глицерин и вода).
Композиции для ингаляции или вдувания включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях или их смеси, и порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые инертные наполнители, указанные выше. В некоторых вариантах осуществления композиции вводят пероральным или назальным респираторным путем для обеспечения локального или системного эффекта. Композиции, предпочтительно в стерильных фармацевтически приемлемых растворителях, можно распылять путем использования газов. распыляемые растворы можно вдыхать непосредственно из распыляющего устройства или распыляющее устройство можно присоединить к лицевой маске, тенту или дыхательному аппарату, периодически создающему избыточное давление. Раствор, суспензию или порошкообразные композиции можно вводить, предпочтительно перорально или назально, из устройств, которые доставляют препарат подходящим образом.
Наборы
Другим объектом настоящего изобретения являются наборы, включающие соединение формул I, II, III, IV или V, или фармацевтические композиции, содержащие соединение формул I, II, III, IV или V, предлагаемое в настоящем изобретении. Набор может включать, в дополнение к соединению формул I, II, или III, предлагаемому в настоящем изобретении, или содержащей его фармацевтической композиции, диагностические или терапевтические средства. Набор также может включать инструкции для применения в диагностической или терапевтической методике. В некоторых вариантах осуществления набор включает соединение формул I, II, III, IV или V, или содержащую его фармацевтическую композицию и диагностическое средство. В других вариантах осуществления набор включает соединение формул I, II, III, IV или V, или содержащую его фармацевтическую композицию.
В еще одном варианте осуществления настоящее изобретение относится к набору, который являются подходящими для применения при осуществлении способов лечения, описанных в настоящем изобретении. В одном варианте осуществления набор включает первую дозированную форму, содержащую одно или большее количество соединений, предлагаемых в настоящем изобретении, в количествах, достаточных для осуществления способов, предлагаемых в настоящем изобретении. В другом варианте осуществления набор включает одно или большее количество соединений, предлагаемых в настоящем изобретении, в количествах, достаточных для осуществления способов, предлагаемых в настоящем изобретении, и контейнер для дозы.
ПОЛУЧЕНИЕ
Соединения формул I, II, III, IV или V можно получить по общим и конкретным методикам, описанным ниже, с использованием обычного общего уровня знаний специалиста в области синтетической органической химии. Такой обычный общий уровень знаний приведен в стандартных учебниках, таких как Comprehensive Organic Chemistry, Ed. Barton and Ollis, Elsevier; Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Larock, John Wiley and Sons; и Compendium of Organic Synthetic Methods, Vol. I-XII (published by Wiley-Interscience). Исходные вещества, использующиеся в настоящем изобретении, имеются в продаже или их можно получить по стандартным методикам, известным в данной области техники.
Следует отметить, что при получении соединений формул I, II, III, IV или V в некоторых из методик получения, описанных в настоящем изобретении может потребоваться защита удаленных функциональных групп (например, первичной аминогруппы, вторичной аминогруппы, карбоксигруппы в предшественниках формулы I). Необходимость такой защиты меняется в зависимость от природы удаленной функциональной группы и условий в методиках получения. Необходимость такой защиты легко определит специалист в данной области техники. Применение таких методик введения/удаления защитных групп также известно специалисту в данной области техники. Общее описание защитных групп и их применение, см. T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Например, некоторые соединения содержат первичные аминогруппы или карбоксигруппы, которые могут мешать реакциям на других центрах молекулы, если останутся незащищенными. Соответственно, такие функциональные группы можно защитить подходящей защитной группой, которую можно удалить на следующей стадии. Подходящие защитные группы для защиты аминогруппы или карбоксигруппы включают такие защитные группы, которые обычно используют в синтезе пептидов (такие как N-трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz) и 9-флуоренилметиленоксикарбонил (Fmoc) для аминогрупп и низшие алкильные или бензильные сложноэфирные группы для карбоксигрупп), которые обычно не являются химически реакционноспособными при описанных условиях проведения реакций и обычно можно удалить без химического изменения другой функциональной группы в соединениях формулы I.
Схемы, приведенные ниже, предназначены для предоставления общего описания методологии, использующейся для получения соединений, предлагаемых в настоящем изобретении. Некоторые из соединений, предлагаемых в настоящем изобретении, могут содержать один или несколько хиральных центров со стереохимическим обозначением (R) или (S). Для специалиста в данной области техники должно быть очевидно, что все синтетические превращения можно провести родственным образом, если вещества являются энентиомерно обогащенными или рацемическими. Кроме того разделение желательного оптически активного вещества можно провести на любой стадии последовательности по хорошо известным методикам, таким как описанные в настоящем изобретении и в химической литературе. Например, промежуточные продукты (например, S4, S7, S8, S24, S40 и S41) и конечные соединения (например, S25 и S42) можно разделить с помощью хиральных хроматографических методик. Альтернативно, хиральные соли можно использовать для выделения энантиомерно обогащенных промежуточных продуктов и конечных соединений.
На приведенных ниже схемах переменные X, Y, Z1, Z2, Z3, R, R1, R2, R3, R4, m, p и q являются такими, как описано в настоящем изобретении для соединений формул I, II, III, IV или V, если не указано иное. Для простоты переменную A используют для обозначения кольца A и его необязательного заместителя R1. На приведенных ниже схемах все X1, X2, X3 и X4 могут независимо означать отщепляющуюся группу, такую как любой алкил или арилсульфонат (например, мезилат, тозилат или трифлат), или галоген, или любую другую группу, которую можно заменить аминогруппой или использовать в катализируемой металлом реакции сочетания. X4 также может означать защищенную карбоксигруппу (т. е. сложноэфирную группу). Если защитная группа обозначена, как Pg1, она может представлять собой защитную группу алкиламина, такую как бензил, бензгидрил, аллил и т. п.; защитную группу карбамата, такую как Boc, Cbz и т. п.; или защитную группу амида, такую как трифторацетамид. Если защитная группа обозначена, как Pg2, она может представлять собой защитную группу карбоксигруппы, такую как метил, этил, бензил, трет-бутил и т. п. Если защитная группа обозначена, как Pg3, она может представлять собой защитную группу гидроксигруппы, такую как триметилсилилэтоксиэтил; или ацильную группу, такую как ацетил, бензоил и т. п.; или триалкилсилильную группу, такую как триметилсилил, трет-бутилдиметилсилил, триизопропилсилил и т. п. R2a означает H или -C1-2алкил, где алкил может содержать 0-1 OH. R4a означает C1-2алкил, C0-2алкилен-C3-6циклоалкил, C0-2алкилен-R5, или C1-22алкилен-R6, где указанный алкил, алкилен или циклоалкил может быть независимо замещен, насколько допускает валентность, 0-3 атомами F и 0-1 заместителем, независимо выбранным из группы, включающей C0-1алкилен-ORO и -N(RN)2.
Замещенный пиперидин S8, в котором R2=H и Y=CH, можно получить, как показано на схеме 1. Арил- или гетероарилбромид S1 можно обработать алкиллитием, например, бутиллитием или трет-бутиллитием, и получить арил- или гетероариллитиевые соединения, которые можно ввести в реакцию с альдегидом S2 с получением диола S3. Другие арильные или гетероарильные металлоорганические реагенты, такие как, но не ограничиваясь только ими, реагенты Гриньяра, также можно использовать для получения S3. Реакцию обычно проводят при температуре около −70°C. Затем диол S3 можно окислить с помощью NaIO4 и получить ацеталь S4 (R2=H). Соединение S4 затем можно ввести в реакцию с замещенной бороновой кислотой или боронатом (S5) в присутствии палладиевого катализатора и сложного лиганда по схеме реакции Судзуки (Maluenda and Navarro, Molecules, 2015, 20, 7528-7557) и получить соединения общей формулы S6. Восстановление олефина с получением соединений общей структуры S7 можно провести в атмосфере водорода (15-100 фунт-сила/дюйм2 H2) в спиртовом растворителе, таком как MeOH или EtOH, или, альтернативно, апротонном органическом растворителе, таком как EtOAc или THF, в присутствии подходящего катализатора, такого как палладий на угле, Pd(OH)2 на угле (катализатор Перлмана), PtO2 (катализатор Адамса) или хлорид трис(трифенилфосфин)родия(I) (катализатор Уилкинсона). Реагенты для гидрирования с переносом, например, формиат аммония или дигидробензол или аналогичные, можно применять с использованием подходящего катализатора. Альтернативно, восстановление можно провести по альтернативным методикам, известным специалистам в данной области техники с использованием реагентов, таких как триэтилсилан или другие силаны, при кислотном катализе или катализе металлами, или металлическими восстановителями, таких как магний или аналогичные. Альтернативно, олефин можно функционализировать по методикам, известным специалисту в данной области техники для введения групп R3. Например, олефин можно гидроборировать и получить спирт, который можно алкилировать или дополнительно превратить в нитрил, F или алкильную группу. Удаление Pg1 можно провести по многим методикам, описанным в литературе, и получить амины S8.
Схема 1
На схеме 2 представлено альтернативное получение соединений общей структуры S4. Реакция подходящим образом замещенных диолов общей структуры S9 с альдегидами или кетонами общей структуры S10a в присутствии кислоты, такой как п-толуолсульфоновая кислота или пиридиний-п-толуолсульфонат, в апротонном органическом растворителе, таком как толуол или бензол, может дать соединения общей структуры S4. Реакционную смесь обычно кипятят с обратным холодильником с использованием ловушки Дина-Штарка для азеотропного удаления воды. Диолы S9 также можно ввести в реакцию с циклическими (имеется пунктирная линия) или ациклическими (отсутствует пунктирная линия) ацеталями или кеталями общей структуры S10b при кислотном катализе. Это применимо и к циклическим или ациклическим тиоацеталям или тиокеталям общей структуры S10c при воздействии солей ртути, мягких окислителей или алкилирующих реагентов с получением соединений S4. Альтернативно, диолы общей формулы S9 можно ввести в реакцию с подходящим образом замещенным алкином S11 в апротонном растворителе, таком как толуол, в присутствии додекакарбонила трирутения при температуре около 100°C и получить соединения общей структуры S4, в котором R2=CH2R2a. В случаях, когда R2 содержит гидроксигруппу, такую как CH2OH, защитную группу гидроксигруппы (Pg3), такую как ацетат, можно ввести в соединения общей структуры S10. Затем защитную группу можно удалить на последующей стадии. Затем промежуточный продукт S4 можно дополнительно модифицировать по методикам, описанным на схеме 1, и получить амины общей структуры S8.
Схема 2
Как показано на схеме 3, превращение S4 в соединения общей структуры S7, в которой Y=N, можно провести таким образом, как сочетание C-N по Бухвальду-Хартвигу соединений общей структуры S4 и подходящим образом замещенного и защищенного пиперазина S12 в присутствии палладиевого или медного катализатора и сложного лиганда. Эти реакции обычно проводят при температуре от 0 до 110°C в апротонных органических растворителях, таких как, но не ограничиваясь только ими, 1,4-диоксан и PhCH3, с добавлением основания, такого как Cs2CO3, LiHMDS или NaOtBu. Удаление Pg1 можно провести по многим методикам, описанным в литературе, и получить амины S8, в которых Y=N.
Схема 3
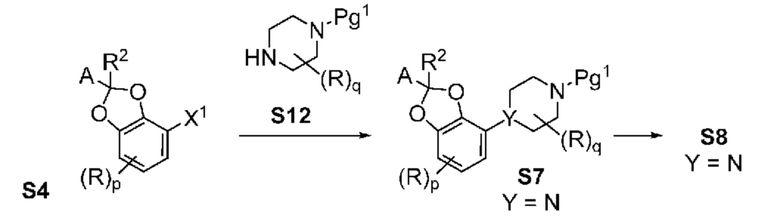
Амины S8, полученные по методикам, описанным на схемах 1-3, можно алкилировать защищенным 2-бромацетатом в присутствии подходящего основания, такого как K2CO3, Et3N, NaH или LiHMDS, в полярном апротонном растворителе, таком как, но не ограничиваясь только ими, DMF, DMAc, DMSO или NMP, и получить соединения общей структуры S13 (X=N, L=CH2). Можно провести стандартный гидролиз сложных эфиров и получить кислоты S14. Если Pg2 означает трет-бутил, стандартные кислотные методики удаления защитных групп, такие как использующие TFA/DCM, HCl/1,4-диоксан, HCl/EtOAc или другие подходящие условия, можно использовать для получения кислот S14. Если Pg2 означает метил или этил, стандартные щелочные методики удаления защитных групп, такие как использующие водный раствор NaOH в метаноле или этаноле, или другие подходящие условия, можно использовать для получения кислот S14.
Схема 4
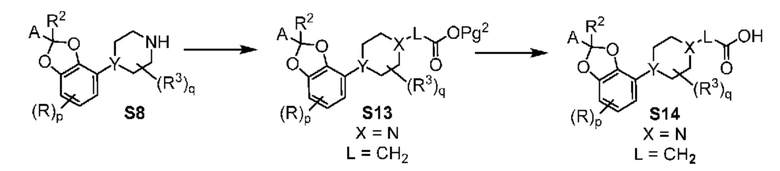
Соединения общей структуры S15 (схема 5) можно ввести в реакцию с аминами R4NH2 в присутствии оснований, таких как карбонат, бикарбонат, гидроксид, ацетат натрия, калия или цезия, или основания - органического амина, такого как Et3N, DIPEA, DBU и т. п., в полярном апротонном растворителе, таком как, но не ограничиваясь только ими, THF, DMF, DMAc, DMSO или NMP, или протонном растворителе, таком как вода, MeOH, EtOH или iPrOH или их смесь, и получить соединения общей структуры S16. Следует отметить, что, если пример дает R4 с разделенным энантиомерным центром, другой энантиомер или их рацемическую смесь можно получить путем выбора подходящего исходного вещества. Предпочтительные заместители X3 включают F, Cl и Br, предпочтительные группы X4 включают Cl, Br, и -CO2-Pg2. Восстановление нитрогруппы можно провести путем гидрирования при 1-6 атм. H2 с помощью металлического катализатора, такого как палладий на угле или никель Ренея, в протонном растворителе, таком как MeOH или EtOH, или в апротонном растворителе, таком как DMF, THF или EtOAc. Альтернативно, нитрогруппу можно восстановить с помощью железа, цинка, SnCl2 или другого подходящего металла в кислых средах, таких как 1 н. HCl, AcOH, или водным раствором NH4Cl в THF или метаноле и получить соединения общей структуры S17 (схема 5a). Соединения, такие как S18, можно ацилировать ацилгалогенидами стандартным образом или карбоксилатами по стандартным методикам сочетания с образованием амида и получить соединения S19. Восстановление в соединения S20 можно провести при стандартных условиях восстановительными реагентами, такими как LAH или BH3-THF, или BH3-DMS (схема 5b).
Схема 5
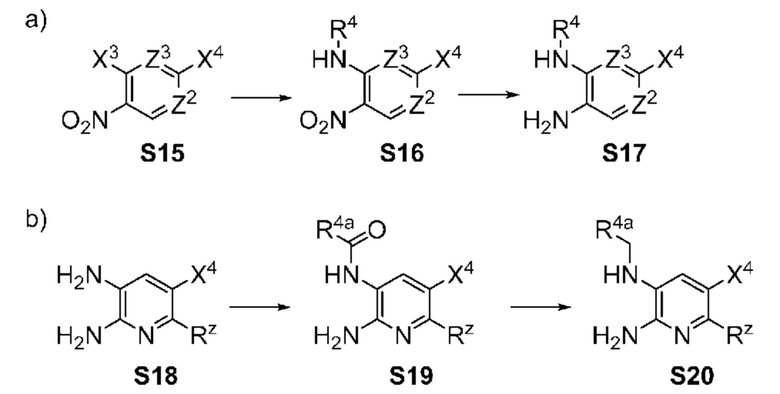
Диамины S17 и S20, полученные по методикам, описанным на схемах 5a и 5b, совместно обозначенные, как диамин S21 (схема 6), можно ацилировать кислотами общей структуры S14 по стандартным методикам сочетания с образованием амида и получить амиды S22, которые существуют в виде смеси от 100% S22a до 100% S22b. Эту смесь аминов S22 можно циклизовать и получить соединения общей структуры S23 по разным методикам. Амины S22 можно нагреть с дегидратирующим реагентом, таким как T3P®, или алкиловым спиртом, таким как н-бутанол, микроволновым излучением (10-60 мин при 120-180°C) и получить соединения S23. Альтернативно, смесь соединений S22 можно нагреть в кислой среде, такой как AcOH, при 60-100°C или в щелочной среде, такой как водный раствор NaOH или KOH в 1,4-диоксане при 60-100°C и получить S23. Соединения общей структуры S23 (X4=Cl, Br или I) можно превратить в сложные эфиры структуры S24 посредством катализируемого палладием карбонилирования в атмосфере монооксид углерода при давлении 15-100 фунт-сила/дюйм2, при температуре 20-100 подходящим спиртом, таким как MeOH или EtOH или другим алкиловым спиртом. Гидролиз сложного эфира S24 можно провести, как описано на схеме 4 и получить кислоты S25. Для соединений S22, в которых X4=CO2-Pg2 превращение в сложный эфир S24 протекает при условиях, аналогичных описанным выше, но с использованием щелочной методики циклизации, в которой соединение S25 можно выделить прямо из реакционной смеси. Для соединений S24, в которых X4 означает CO2tBu, удаление защитной группы с получением кислоты S25 можно провести в кислой среде, как показано на схеме 4. Альтернативно, для соединений S24, в которых Pg2 означает C1-C8 алкил, такой как метил, этил, гексил или октил, удаление сложноэфирной защитной группы можно провести с помощью различных ферментов, включая эстеразы, протеазы, пептидазы, липазы и гликолидазы, которые хорошо известны специалистам в данной области техники. Гидролиз также можно провести путем обработки сложного эфира водным раствором 1,5,7-триазабицикло[4.4.0]дец-5-ена при КТ.
Схема 6
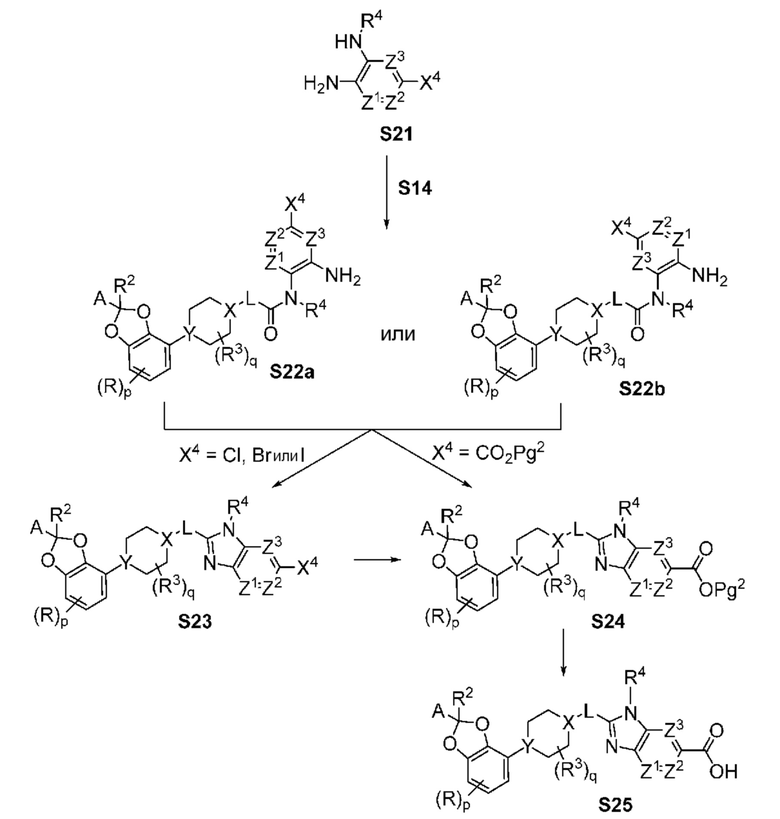
Кроме того, диамин S21 можно превратить в 2-хлорметилбензимидазол S26 (схема 7) по различным методикам. Обработка 2-хлорацетилхлоридом или хлоруксусным ангидридом в апротонном растворителе, таком как 1,4-диоксан, с последующим нагреванием при 40-100°C в течение 2-18 ч может дать искомый бензимидазол S26, в котором Z1, Z2 и Z3 означают CH. В случаях, когда Z1, Z2 и Z3 не все означают CRz, после обработки 2-хлорацетилхлоридом в апротонном растворителе, таком как 1,4-диоксан, в течение от 30 мин до 4 ч, растворитель заменяют на кислые среды, такие как AcOH или TFA, с последующим нагреванием при 40-100°C в течение 2-18 ч и получают искомое соединение S26. Диамин S21 также можно обработать хлоруксусным ангидридом при температуре от 0 и 80°C в апротонном растворителе таком как, но не ограничиваясь только ими, 1,4-диоксан, THF или MeCN, с последующим нагреванием в течение от 2 до 18 ч при 60-100°C и получить искомое соединение S26. Кроме того, диамин S21 можно обработать 2-хлор-1,1,1-триметоксиэтаном в апротонном растворителе таком как, но не ограничиваясь только ими, 1,4-диоксан, THF или MeCN, или протонном растворителе, например, MeOH или EtOH, в присутствии кислотного катализатора, например, pTSA, при 20-100°C. Альтернативно, диамины S21 можно нагреть 100-180°C м 2-гидроксиуксусной кислотой в апротонном растворителе, таком как, но не ограничиваясь только ими, мезитилен, и получить промежуточный гидроксиметил. Превращение гидроксиметильной группы в хлорметил S26 можно провести по стандартным методикам, включая обработку с помощью SOCl2 в апротонном растворителе. Соединения общей структуры S26 можно ввести в реакцию с соединениями S8 в присутствии оснований, таких как карбонат, бикарбонат натрия, калия или цезия, или основания - органического амина, такого как Et3N, DIPEA, DBU и т. п. в полярном апротонном растворителе, таком как, но не ограничиваясь только ими, THF, MeCN, DMF, DMAc, DMSO или NMP, и получить соединения S23 (X4=Cl, Br, I) или соединения S24 (X4=CO2-Pg2), которые затем используют для получения соединений S25 по методикам, описанным на схеме 6.
Схема 7
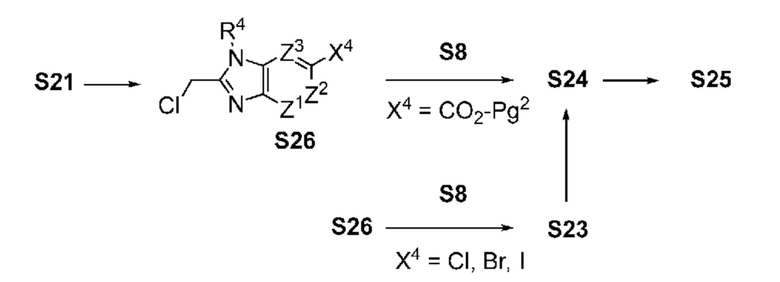
Альтернативно (схема 8), соединения общей структуры S26 можно ввести в реакцию с подходящим образом замещенными и защищенными пиперазинами S12, в присутствии оснований, таких как карбонат, бикарбонат натрия, калия или цезия, или основания - органического амина, такого как Et3N, DIPEA, DBU и т. п. в полярном апротонном растворителе, таком как, но не ограничиваясь только ими, THF, MeCN, DMF, DMAc, DMSO или NMP, и получить соединения S27 (схема 8). Удаление Pg1 можно провести по многим методикам, описанным в литературе, и получить амины S28. Превращение в соединения общей структуры S23 (X4=Cl, Br или I) или S24 (X4=CO2-Pg2) можно провести таким образом, как сочетание C-N по Бухвальду-Хартвигу соединений общей структуры S4 и как показано выше на схеме 3. Соединения общей структуры S23 или S24 затем можно использовать и получить соединения структуры S25 по методикам, описанным на схеме 6.
Схема 8
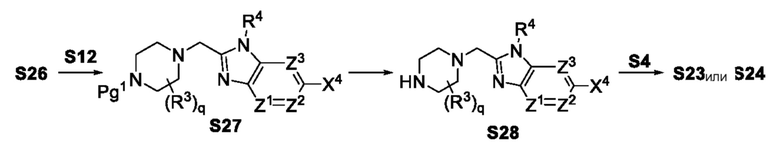
Соединения общей структуры S25 также можно получить, как показано на схеме 9. Диол S9 можно защитить и получить S29. Триметилсилилэтоксиметильная группа является предпочтительной защитной группой. Защита диола в виде соответствующего ацеталя, например, ацеталя формальдегида, также является предпочтительной. Затем соединение S29 можно ввести в реакцию с замещенной бороновой кислотой или боронатом (S5) и затем олефин восстановить по методикам, описанным на схеме 1, и получить соединения общей формулы S31, в которых Y=CH. Альтернативно, соединение S29 можно ввести в реакцию сочетания с пиперазинами общей структуры S12 по методикам, описанным на схеме 3, и получить S31, в котором Y=N. Из соединений общей структуры S31 можно удалить защитные группы и затем ввести в реакцию сочетания с S26 и получить соединения общей структуры S33 по методикам, описанным на схеме 7. Альтернативно, соединения общей структуры S33 можно получить из S32 путем превращения S32 в соответствующее производное N-уксусной кислоты и с последующей конденсацией с диаминами S21, описанными на схемах 4 и 6. Удаление защитной группы из S33 по методикам, известным специалистам в данной области техники, может дать диолы общей структуры S34, которые затем можно ввести в реакцию с алкинами общей структуры S11 по методикам, описанным на схеме 2, и получить S23 или S24. Альтернативно, S34 можно превратить в S23 или S24 с использованием альдегидов, кетонов или их производных, как показано на схеме 2. Затем можно использовать соединения общей структуры S23 или S24 и получить соединения структуры S25 по методикам, описанным на схеме 6.
Схема 9

Соединения общей структуры S24 и S33, в которых Y=N и X-L=циклопропил, можно получить, как показано на схеме 10. Защищенный пиперидинон S35 можно превратить а ненасыщенный сложный эфир S36 по методикам, хорошо известным специалистам в данной области техники. Например, олефинирование S42 по Хорнеру-Уадсворту-Эммонсу фосфонатом, таким как этил(диэтоксифосфорил)ацетат, который депротонирован сильным основанием, таким как трет-бутоксид лития, натрия или калия, может дать S36. Реакцию обычно проводят в апротонном растворителе, таком как THF или DME, при температуре примерно от 0 до −50°C. Превращение S36 в производное циклопропана S37 можно провести путем обработки сульфоксонийилидом, полученным из триметилсульфоксониййодида и основания, такого как трет-бутоксид калия или гидрид натрия. Удаление защитной группы из S37 и последующее сочетание полученной карбоновой кислоты S38 с S21, где X4=CO2Pg2, по методикам, описанным на схеме 6, может дать соединения общей формулы S39. Удаление защитной группы из S39 и сочетание с S4 по методикам, описанным на схеме 3, может дать соединения общей структуры S24, в которых Y=N и X-L означает циклопропил. Затем можно использовать соединения общей структуры S24 и получить соединения структуры S25 по методикам, описанным на схеме 6. Альтернативно, S40 можно ввести в реакцию с S29 по методикам, описанным на схеме 3, и получить S33, в котором Y=N и X-L=циклопропил. Затем можно использовать соединения общей структуры S33 и получить соединения структуры S25 по методикам, описанным на схеме 6 и 9.
Схема 10
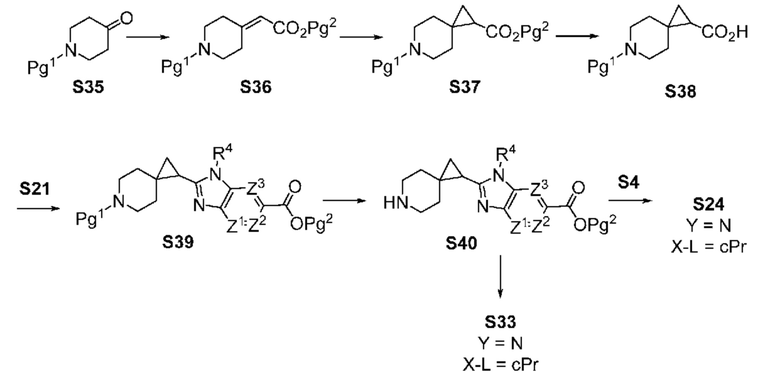
Альтернативно, соединения общей структуры S25, в которых Y=N и X-L означает циклопропил, можно получить, как описано на схеме 11. Удаление Pg1 из S37 дает производное пиперидина S43. Сочетание S43 с S4 по методике, аналогичной описанной на схеме 3, дает S13, в котором Y=N и X-L означает циклопропил. Удаление защитной группы может дать соединения общей структуры S14, которые затем можно использовать для получения S25, как описано на схеме 6.
Схема 11
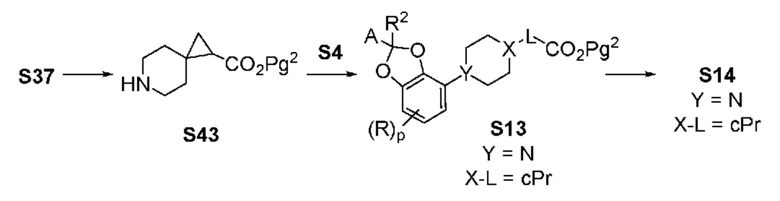
ПРИМЕРЫ
Ниже проиллюстрирован синтез неограничивающих соединений, предлагаемых в настоящем изобретении. Дополнительные соединения, предлагаемые в настоящем изобретении, можно получить по методикам, приведенным в этих примерах, по отдельности или в комбинации с методиками, общеизвестными в данной области техники.
Эксперименты обычно поводили в инертной атмосфере (азот или аргон), в особенности в случаях, когда используются чувствительные к действию кислорода или влаги реагенты или промежуточные продукты. Имеющиеся в продаже растворители и реагенты обычно использовали без дополнительной очистки. Безводные растворители использовали, если это было подходящим, обычно продукты AcroSeal® фирмы Acros Organics, Aldrich® Sure/Seal™ фирмы Sigma-Aldrich или DriSolv® продукты фирмы EMD Chemicals. В других случаях имеющиеся в продаже растворители пропускали через колонки, заполненные 4Å молекулярными ситами, пока обеспечивались следующие стандарты QC (контроль качества) для содержания воды: a) <100 част./млн для дихлорметана, толуола, N, N-диметилформамида и тетрагидрофурана; b) <180 част./млн для метанола, этанола, 1,4-диоксана и диизопропиламина. Для очень чувствительных реакций растворители дополнительно обрабатывали металлическим натрием, гидридом кальция или молекулярными ситами и перед использованием их перегоняли. Продукты обычно сушили в вакууме до использования в последующей реакции или для биологических исследований. Приведены данные масс-спектрометрии, полученные на приборах для жидкостной хроматографии-масс-спектрометрии (LCMS), химической ионизации при атмосферном давлении (APCI) или газовой хроматографии-масс-спектрометрии (GCMS). Значок ♦ показывает, что в масс-спектре наблюдали сигнал изотопа хлора.
Хиральные разделения использовали для разделения энантиомеров или диастереоизомеров некоторых промежуточных продуктов при получении соединений, предлагаемых в настоящем изобретении. Если проводили хиральное разделение, разделенные энантиомеры обозначали, как ENT-1 или ENT-2 (или DIAST-1 или DIAST-2) в соответствии с порядком их элюирования. В некоторых вариантах осуществления энантиомеры, обозначенные, как ENT-1 или ENT-2, можно использовать в качестве исходных веществ для получения других энантиомеров или диастереоизомеров. В таких случаях полученные энантиомеры обозначали, как ENT-X1 и ENT-X2 соответственно в соответствии с их исходными веществами; аналогичным образом, полученные диастереоизомеры обозначали, как DIAST-X1 и DIAST-X2 соответственно (или DIAST-) в соответствии с их исходными веществами. Обозначения DIAST-Y и DIAST-Z использовали аналогичным образом в синтезах с использованием нескольких промежуточных продуктов.
Для соединений с двумя хиральными центрами стереоизомеры по каждому стереоцентру выделяли в разные времена. Обозначение ENT-1 или ENT-2 (или DIAST-1 или DIAST-2) промежуточного продукта или соединения примера соответствует порядку элюирования для разделения, проводимого на каждой стадии. Следует понимать, что, если стереоизомеры по хиральному центру разделены в соединении с двумя или большим количеством центров, разделенные энантиомеры являются диастереоизомерами друг друга. Например, без наложения ограничений, соединения примеров 15 и 16 содержат два хиральных центра. Хиральный центр циклопропильного фрагмента разделяли, когда промежуточный продукт C36 разделяли на ENT-1 и получали промежуточный продукт P17, и на ENT-2 и получали промежуточный продукт P18. Затем P18 использовали для получения C70, который содержал один стереоизомер, обогащенный по циклопропильному хиральному атому углерода, и смесь стереоизомеров по диоксолановому атому углерода. Затем C70 разделяли на DIAST-Y1 по диоксолановому атому углерода и получали промежуточный продукт C71, и DIAST-Y2 по диоксолановому атому углерода и получали промежуточный продукт C72, где эти промежуточные продукты обогащены одним стереоизомером. Затем C71 использовали для получения соединения примера 15, которое обладает названием 2-{6-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-6-азаспиро[2.5]окт-1-ил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота, DIAST-X1, трифторацетат [из P18 через C71]. В этих процедурах после разделения хиральный центр обозначен, как "abs" вблизи центра, причем следует понимать, что разделенные энантиомеры могут не быть энантиомерно чистыми. Обычно обогащенный энантиомер по каждому хиральному центру составляет >90% от выделенного вещества. Предпочтительно, если обогащенный энантиомер по каждому центру составляет >98% от смеси.
В некоторых примерах оптическое вращение энантиомера измеряли с помощью поляриметра. В соответствии с наблюдающимися данными по его вращению (или данными по его удельному вращению), энантиомер с вращением по часовой стрелке обозначали, как (+)-энантиомер и энантиомер с вращением против часовой стрелки обозначали, как (-)-энантиомер. Рацемические соединения указаны посредством отсутствия изображения или описания стереохимии, или посредством (+/-) рядом со структурой; в этом последнем случае указанная стереохимическая конфигурация характеризует относительную (а не абсолютную) стереохимическую конфигурацию заместителей соединения.
Реакции, протекающие через регистрируемые промежуточные продукты, обычно отслеживали с помощью LCMS и им давали протекать до полного превращения до добавления последующих реагентов. Для указанных процедур синтеза в других примерах или методиках условия проведения реакций (длительность проведения реакции и температура) могут меняться. Обычно за реакциями следили с помощью тонкослойной хроматографии или масс-спектрометрии, и проводили обработку, если это было подходящим. Очистки могут быть разными для разных экспериментов: обычно выбирали растворители и отношения растворителей, использующихся для элюирования/градиентных режимов и получали подходящие Rfs или времена удерживания. Все исходные вещества, использующиеся в этих процедурах и примерах, имеются в продаже или их можно получить по методикам, известным в данной области техники или как описано в настоящем изобретении.
Процедура P1
трет-Бутил-4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилат (P1)

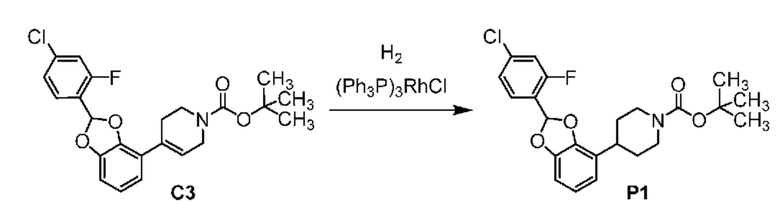
Стадия 1. Синтез 2-бром-6-[(4-хлор-2-фторфенил)(гидрокси)метил]фенола (C1).
Этот эксперимент проводили с двумя партиями одинаковой величины. н-Бутиллитий (2,5 M раствор в гексанах; 32,8 мл, 82,0 ммоля) медленно добавляли при −70°C к раствору 1-бром-4-хлор-2-фторбензола (17,2 г, 82,1 ммоля) в диэтиловом эфире (100 мл) при поддержании температуры реакционной смеси ниже −60°C. После перемешивания реакционной смеси при −70°C в течение 20 мин медленно добавляли раствор 3-бром-2-гидроксибензальдегида (5,5 г, 27 ммоля) в диэтиловом эфире (100 мл) при поддержании температуры реакционной смеси ниже −60°C. После перемешивания в течение еще при −70°C реакцию останавливали путем добавления водного раствора хлорида аммония (50 мл) при −70°C и полученную смесь разбавляли водой (100 мл). Затем эти 2 партии объединяли и экстрагировали этилацетатом (400 мл); органический слой промывали насыщенным водным раствором хлорида натрия (200 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиентный режим: от 0% до 7% этилацетата в петролейном эфире) давало C1 в виде белого твердого вещества. Суммарный выход: 15,7 г, 47,4 ммоля, 88%. 1H NMR (400 MHz, хлороформ-d) δ 7,44 (dd, J=8,0, 1,5 Hz, 1H), 7,37 (dd, J=8,1, 8,1 Hz, 1H), 7,15 (br dd, J=8,5, 2,1 Hz, 1H), 7,12-7,05 (m, 2H), 6,80 (dd, J=7,8, 7,8 Hz, 1H), 6,78 (s, 1H), 6,31 (d, J=4,8 Hz, 1H), 3,02 (br d, J=4,9 Hz, 1H).
Стадия 2. Синтез 4-бром-2-(4-хлор-2-фторфенил)-1,3-бензодиоксола (C2).
К раствору C1 (15,7 г, 47,4 ммоля) в метаноле (450 мл) добавляли раствор перйодата натрия (25,4 г, 119 ммоля) в воде (105 мл) и реакционную смесь перемешивали при 30°C в течение 16 ч, затем ее концентрировали в вакууме. После разбавления остатка дихлорметаном (500 мл), его промывали водой (500 мл). Затем раствор в дихлорметане сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка с помощью хроматографии на силикагеле (элюент: петролейный эфир) давала C2 в виде белого твердого вещества. Выход: 10,0 г, 30,3 ммоля, 64%. Приведенные ниже данные 1H NMR получали из эксперимента, проводимого таким же образом, но в меньшем масштабе. 1H NMR (400 MHz, DMSO-d6) δ 7,67-7,61 (m, 2H), 7,50 (s, 1H), 7,43 (br dd, J=8, 2 Hz, 1H), 7,09 (dd, J=8,3, 1,1 Hz, 1H), 7,01 (dd, J=7,9, 1,1 Hz, 1H), 6,86 (dd, J=8,1, 8,1 Hz, 1H).
Стадия 3. Синтез трет-бутил-4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]-3,6-дигидропиридин-1(2H)-карбоксилата (C3).
Реакционный сосуд, содержащий суспензию C2 (8,00 г, 24,3 ммоля), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилата (9,01 г, 29,1 ммоля), карбоната натрия (5,15 г, 48,6 ммоля), и [1,1"-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) [Pd(dppf)Cl2; 888 мг, 1,21 ммоля] в 1,4-диоксане (80 мл) и воде (32 мл), откачивали и заполняли азотом. Этот цикл откачки повторяли дважды и затем реакционную смесь перемешивали при 90°C в течение 16 ч. После удаления растворителя в вакууме остаток подвергали распределению между этилацетатом (200 мл) и водой (200 мл). Органический слой промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Хроматография на силикагеле (градиентный режим: от 0% до 4,3% этилацетата в петролейном эфире) давала продукт, который объединяли с веществом, полученным в аналогичной реакции, проведенной с использованием C2 (2,00 г, 6,07 ммоля) и получали C3 в виде светло-желтого смолообразного вещества. Суммарный выход: 10,3 г, 23,8 ммоля, 78%. 1H NMR (400 MHz, хлороформ-d) δ 7,53 (dd, J=8,3, 7,8 Hz, 1H), 7,23-7,16 (m, 3H), 6,88-6,83 (m, 2H), 6,81-6,76 (m, 1H), 6,34-6,28 (br m, 1H), 4,10-4,05 (m, 2H), 3,61 (br dd, J=6, 5 Hz, 2H), 2,59-2,50 (br m, 2H), 1,48 (s, 9H).
Стадия 4. Синтез трет-бутил-4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилата (P1).
Раствор C3 (10,3 г, 23,8 ммоля) и хлорида трис(трифенилфосфин)родия(I) (катализатор Уилкинсона; 1,54 г, 1,66 ммоля) в метаноле (100 мл) перемешивали при 50°C в атмосфере водорода (45 фунт-сила/дюйм2) в течение 18 ч. Затем реакционную смесь фильтровали через слой диатомовой земли и фильтрат концентрировали при пониженном давлении и обрабатывали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 9% этилацетата в петролейном эфире). Полученное вещество объединяли с полученным в аналогичной реакции, проведенной с использованием C3 (1,67 г, 3,87 ммоля) и получали P1 в виде бесцветного смолообразного вещества. Суммарный выход: 10,3 г, 23,7 ммоля, 86%. LCMS m/z 456,1♦ [M+Na+]. 1H NMR (400 MHz, хлороформ-d) δ 7,52 (dd, J=8,5, 7,6 Hz, 1H), 7,23-7,17 (m, 2H), 7,16 (s, 1H), 6,83 (dd, J=7,8, 7,8 Hz, 1H), 6,78-6,69 (m, 2H), 4,35-4,10 (br m, 2H), 2,89-2,71 (m, 3H), 1,89-1,77 (m, 2H), 1,77-1,63 (m, 2H), 1,47 (s, 9H).
Процедура P2
трет-Бутил-4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилат (P2)
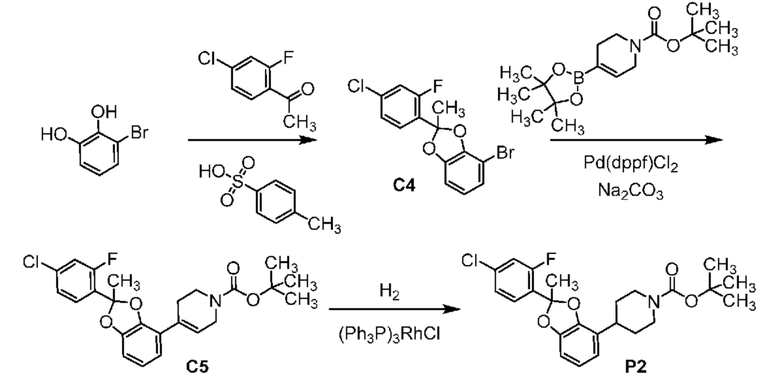
Стадия 1. Синтез 4-бром-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксола (C4).
К раствору 3-бромбензол-1,2-диола (330 г, 1,75 моля) в толуоле (1,5 л) добавляли 1-(4-хлор-2-фторфенил)этанон (316 г, 1,83 моля) и п-толуолсульфоновую кислоту (6,02 г, 35,0 ммоля). Реактор снабжали ловушкой Дина-Штарка и реакционную смесь нагревали при 140°C в течение 60 ч, затем раствор концентрировали в вакууме и очищали с помощью хроматографии на силикагеле (элюент: петролейный эфир); C4 получали в виде смеси желтого масла и твердого вещества. Выход: 158 г, 460 ммоля, 26%. 1H NMR (400 MHz, хлороформ-d): δ 7,54 (dd, J=8,4, 8,4 Hz, 1H), 7,17-7,10 (m, 2H), 6,95 (dd, J=7,9, 1,4 Hz, 1H), 6,75 (dd, компонент системы ABX, J=7,8, 1,4 Hz, 1H), 6,70 (dd, компонент системы ABX, J=7,9, 7,9 Hz, 1H), 2,11 (d, J=1,1 Hz, 3H).
Стадия 2. Синтез трет-бутил-4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-3,6-дигидропиридин-1(2H)-карбоксилата (C5).
трет-Бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (62 г, 200 ммоля) и карбонат натрия (100 г, 940 ммоля) добавляли к раствору C4 (58,0 г, 169 ммоля) в 1,4-диоксане (600 мл). После добавления [1,1"-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (6,0 г, 8,2 ммоля), реакционную смесь нагревали при 90°C и перемешивали в течение 16 ч. Затем добавляли воду (500 мл) и полученную смесь экстрагировали этилацетатом (2×500 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (2×500 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиентный режим: от 0% до 9% этилацетата в петролейном эфире) давала C5 в виде желтого масла. Выход: 56,0 г, 126 ммоля, 75%. 1H NMR (400 MHz, хлороформ-d) δ 7,50 (dd, J=8,2, 8,2 Hz, 1H), 7,17-7,09 (m, 2H), 6,83-6,77 (m, 2H), 6,74 (dd, компонент системы ABX, J=5,4, 3,6 Hz, 1H), 6,39-6,33 (br m, 1H), 4,14-4,08 (m, 2H), 3,70-3,56 (m, 2H), 2,66-2,45 (m, 2H), 2,07 (d, J=1,1 Hz, 3H), 1,50 (s, 9H).
Стадия 3. Синтез трет-бутил-4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилата (P2).
К раствору C5 (56,0 г, 126 ммоля) в метаноле (200 мл) добавляли хлорид трис(трифенилфосфин)родия(I) (катализатор Уилкинсона; 8,10 г, 8,75 ммоля) и реакционную смесь нагревали при 50°C в течение 18 ч в атмосфере водорода (45 фунт-сила/дюйм2). Затем ее охлаждали до 25°C и фильтровали через диатомовую землю. Фильтрат концентрировали в вакууме и дважды очищали с помощью хроматографии на силикагеле (первая колонка - градиентный режим: от 0% до 9% этилацетата в петролейном эфире; вторая колонка - градиентный режим: от 0% до 2% этилацетата в петролейном эфире) и получали P2 в виде желтого твердого вещества. Выход: 37,0 г, 82,6 ммоля, 66%. LCMS m/z 392,1♦ [(M − 2-метилпроп-1-ен)+H]+. 1H NMR (400 MHz, хлороформ-d) δ 7,51 (dd, J=8,3, 8,0 Hz, 1H), 7,17-7,09 (m, 2H), 6,77 (dd, компонент системы ABC, J=7,8, 7,8 Hz, 1H), 6,70 (dd, компонент системы ABC, J=7,7, 1,3 Hz, 1H), 6,66 (dd, компонент системы ABC, J=7,8, 1,3 Hz, 1H), 4,37-4,13 (br m, 2H), 2,92-2,73 (m, 3H), 2,05 (d, J=1,1 Hz, 3H), 1,90-1,63 (m, 4H), 1,49 (s, 9H).
Процедура P3
4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин, п-толуолсульфонат (P3)

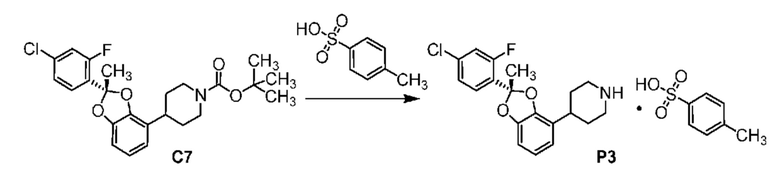
Стадия 1. Выделение трет-бутил-4-[(2R)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилата (C6) и трет-бутил-4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилата (C7).
Разделение P2 (75,2 г, 168 ммоля) на его компоненты-энантиомеры проводили с помощью SFC (надкритическая жидкостная хроматография) [колонка: Chiral Technologies Chiralpak AD-H, 5 мкм; подвижная фаза: 4:1 диоксид углерода/(2-пропанол, содержащий 0,2% 1-аминопропан-2-ола)]. Первое элюирующееся соединение обозначали, как C6, и второй элюирующийся энантиомер, как C7. Показанные абсолютные конфигурации устанавливали на основании рентгенографического определения структуры монокристалла, проводимого для C8, который получили из C6 (см. ниже).
C6 - Выход: 38,0 г, 84,8 ммоля, 50%. Время удерживания 3,64 мин [колонка: Chiral Technologies Chiralpak AD-H, 4,6×250 мм, 5 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: 2-пропанол, содержащий 0,2% 1-аминопропан-2-ола; градиентный режим: 5% B в течение 1,00 мин, затем от 5% до 60% B за 8,00 мин; скорость потока: 3,0 мл/мин; противодавление: 120 бар].
C7 - Выход: 36,8 г, 82,2 ммоля, 49%. Время удерживания 4,19 мин (условия проведения анализа с помощью SFC такие же, как использованные для C6).
Стадия 2. Синтез 4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин, п-толуолсульфоната (P3).
Раствор C7 (1,62 г, 3,62 ммоля) в этилацетате (36 мл) обрабатывали моногидратом п-толуолсульфоновой кислоты (791 мг, 4,16 ммоля) и нагревали при 45°C. Через 23 ч реакционной смеси давали охладиться до комнатной температуры и твердое вещество собирали фильтрованием. Его промывали смесью этилацетата и гептана (1:1, 2×15 мл) и получали P3 в виде белого твердого вещества. Выход: 1,37 г, 2,63 ммоля, 73%. LCMS m/z 348,1♦ [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8,53 (v br s, 1H), 8,29 (v br s, 1H), 7,65-7,55 (m, 2H), 7,47 (d, J=8,1 Hz, 2H), 7,35 (dd, J=8,4, 2,0 Hz, 1H), 7,11 (d, J=7,8 Hz, 2H), 6,88-6,81 (m, 2H), 6,75-6,68 (m, 1H), 3,42-3,33 (m, 2H), 3,11-2,93 (m, 3H), 2,29 (s, 3H), 2,03 (s, 3H), 1,98-1,82 (m, 4H).
Превращение C6 в метансульфонат 4-[(2R)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидина (C8) для определения абсолютной стереохимической конфигурации

п-Толуолсульфоновую кислоту (377 мг, 2,19 ммоля) добавляли к раствору C6 (490 мг, 1,09 ммоля) в этилацетате (5,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. После дополнительного разбавления этилацетатом реакционную смесь последовательно промывали водным раствором бикарбоната натрия, водой и насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Выход: 375 мг, 1,08 ммоля, 99%. 1H NMR (400 MHz, метанол-d4) δ 7,59 (dd, J=8,3. 8,3 Hz, 1H), 7,27 (dd, J=10,9, 2,0 Hz, 1H), 7,20 (br dd, J=8,4, 2,1 Hz, 1H), 6,81-6,75 (m, 1H), 6,74-6,67 (m, 2H), 3,18-3,09 (m, 2H), 2,88-2,77 (m, 1H), 2,77-2,67 (m, 2H), 2,02 (d, J=0,7 Hz, 3H), 1,85-1,73 (m, 4H).
Получали 0,1 M раствор этого свободного основания (4-[(2R)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидина) в этилацетате и проводили скрининг солей. В настоящем изобретении описано только образование метансульфоната. Смесь метансульфоновой кислоты (25 мкл, 39 мкмоля) и раствора субстрата (0,1 M; 0,25 мл, 25 мкмоля) перемешивали в течение ночи. Затем добавляли количество метанола, достаточное для растворения содержащегося твердого вещества, и этилацетат (3 мл) добавляли. Полученному раствору давали медленно испаряться без перемешивания и получали кристаллы C8; один из них использовали для рентгенографического определения структуры монокристалла, описанного ниже.
Рентгенографическое определение структуры монокристалла C8
Рентгенографический анализ монокристалла
Сбор данных проводили на дифрактометре Bruker D8 Quest при комнатной температуре. Сбор данных включал сканирование по углам омега и фи.
Структуру определяли с помощью внутреннего фазирования с использованием программного обеспечения SHELX в орторомбическом классе пространственной группы P212121. Затем структуру уточняли по методике наименьших квадратов в полноматричном приближении. Обнаружены все атомы, не являющиеся атомами водорода, и их положения уточнены с использованием параметров анизотропного смещения.
Образование метансульфоната было подтверждено с помощью N1_H1X_O4 протонного переноса.
Атомы водорода, расположенные на азоте и кислороде, определены по карте разностей Фурье и их положение уточнено с ограничениями на расстояния. Остальные атомы водорода помещали в рассчитанных положениях и им было разрешено смещаться на их атомах-носителях. Заключительное уточнение включало параметры изотропного смещения для всех атомов водорода.
Анализ абсолютной структуры с помощью методик правдоподобия (Hooft, 2008) проводили с использованием PLATON (Spek). Результаты показывают, что абсолютная структура определена правильно; расчетом по методике показано, что вероятность правильности структуры равна 100%. Параметр Хоофта найден равным 0,02 с оцененным стандартным отклонением, равным 0,0012 и параметр Парсона найден равным 0,07 с оцененным стандартным отклонением, равным 0,009. Абсолютная конфигурация по C7 была подтверждена, как (R).
Асимметричная ячейка состоит из одной молекулы протонированного свободного основания C8 и одной молекулы депротонированной метансульфоновой кислоты. Конечный индекс R равнялся 4,6%. Конечная разность Фурье подтвердила, что нет отсутствующей или неправильно расположенной электронной плотности.
Данные для соответствующего кристалла, собранные данные и информация по уточнению представлены в таблице A. Координаты атомов, длины связей, валентные углы и параметры смещения представлены в таблицах B - D.
Программное обеспечение и литература
SHELXTL, Version 5.1, Bruker AXS, 1997.
PLATON, A. L. Spek, J. Appl. Cryst. 2003, 36, 7-13.
MERCURY, C. F. Macrae, P. R. Edington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler, and J. van de Streek, J. Appl. Cryst. 2006, 39, 453-457.
OLEX2, O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, and H. Puschmann, J. Appl. Cryst. 2009, 42, 339-341.
R. W. W. Hooft, L. H. Straver, and A. L. Spek, J. Appl. Cryst. 2008, 41, 96-103.
H. D. Flack, Acta Cryst. 1983, A39, 867-881.
Таблица A. Данные для кристалла и уточнения структуры C8.
Таблица B. Координаты атомов (×104) и эквивалентные параметры изотропного смещения (Å2×103) для C8. U(eq) определяется, как одна треть следа ортогонализованного тензора Uij.
Таблица C. Длины связей [Å] и валентные углы [°] для C8.
Преобразования симметрии, использованные для генерации эквивалентных атомов.
Таблица D. Параметры анизотропного смещения (Å2×103) для C8. Экспонента коэффициента анизотропного смещения имеет вид: −2π2[h2 a*2U11 +... + 2 h k a* b* U12 ].
Получение ди-п-толуоил-L-тартрата P3
ди-п-толуоил-L-тартрат 4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидина (ди-п-толуоил-L-тартрат P3).
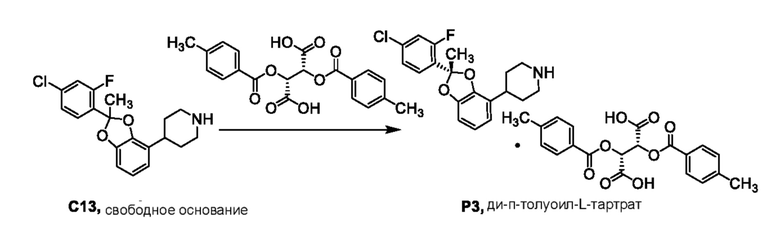
Раствор свободного основания C13 (519 мг, 1,49 ммоля) и ди-п-толуоил-L-винной кислоты (278 мг, 0,719 ммоля) в ацетонитриле (7,5 мл) перемешивали при 50°C в течение 1,5 ч. Смеси давали охладиться до комнатной температуры со скоростью 0,2°C/мин. Через 15 ч при комнатной температуре, смесь нагревали при 65°C и добавляли ацетонитрил (15 мл). Смеси давали охладиться до комнатной температуры при 0,2°C/мин. Через 15 ч при комнатной температуре смесь нагревали при 54°C. Через 3 ч твердое вещество собирали фильтрованием и сушили в вакуумном сушильном шкафу при 35°C в атмосфере азота и получали ди-п-толуоил-L-тартрат P3 в виде белого твердого вещества (217 мг, 0,296 ммоля, 20%, 82% ee).
Раствору ди-п-толуоил-L-тартрата P3 (217 мг, 0,296 ммоля, 82% ee) в ацетонитриле (8,0 мл) при 50°C давали охладиться до комнатной температуры со скоростью 0,2°C/мин. Через 15 ч твердое вещество собирали фильтрованием и сушили в вакуумном сушильном шкафу при 35°C в атмосфере азота и получали ди-п-толуоил-L-тартрат P3 в виде белого твердого вещества (190 мг, 0,259 ммоля, 88%, 88% ee). LCMS m/z 348,1♦ [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8,9-8,5 (br s, 2H), 7,79 (d, J=8,1 Hz, 4H), 7,64-7,54 (m, 2H), 7,34 (dd, J=8,4, 2,1 Hz, 1H), 7,26 (d, J=8,0 Hz, 4H), 6,87-6,78 (m, 2H), 6,69 (dd, J=6,7, 2,5 Hz, 1H), 5,58 (s, 2H), 3,37-3,28 (m, 2H, предположительно; частично закрыт пиком воды), 3,05-2,89 (m, 3H), 2,33 (s, 6H), 2,02 (s, 3H), 1,92-1,80 (m, 4H). Время удерживания: пик 1 (4,97 мин, второстепенный) и пик 2 (5,31 мин, основной) {колонка: Chiralpak IC-U 3,0×50 мм, 1,6 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: 0,1% изопропиламина в метаноле; градиентный режим: от 10% B в течение 5,00 мин, затем 45% B в течение 0,6 мин; скорость потока: 1,7 мл/мин; противодавление: 130 бар}.
Процедура P4
трет-Бутил-4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилат (P4)
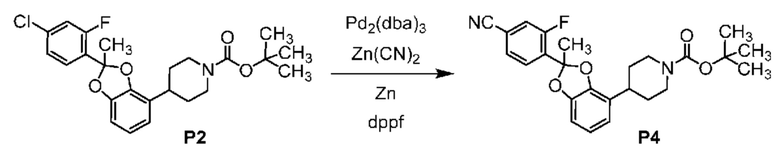
Суспензию P2 (2,00 г, 4,46 ммоля), цианида цинка (734 мг, 6,25 моля), цинка (70,1 мг, 1,07 ммоля), 1,1"-бис(дифенилфосфино)ферроцена (dppf; 198 мг, 0,357 ммоля) и трис(дибензилиденацетон)дипалладия(0) (164 мг, 0,179 ммоля) в N, N-диметилацетамиде (20 мл) перемешивали при 120°C в течение 16 ч, затем ее фильтровали. Фильтрат смешивали с водой (50 мл) и экстрагировали этилацетатом (3×50 мл); затем объединенные органические слои последовательно промывали водой (30 мл) и насыщенным водным раствором хлорида натрия (20 мл) и концентрировали в вакууме. Хроматография на силикагеле (градиентный режим: от 0% до 30% этилацетата в петролейном эфире) давала твердое вещество, которое обрабатывали ацетонитрилом (15 мл) и водой (15 мл) и лиофилизировали. Это давало P4 в виде светло-желтого твердого вещества. Выход: 1,17 г, 2,67 ммоля, 60%. LCMS m/z 461,3 [M+Na+]. 1H NMR (400 MHz, хлороформ-d) 7,71 (dd, J=7,7, 7,6 Hz, 1H), 7,45 (dd, J=8,0, 1,6 Hz, 1H), 7,42 (dd, J=10,0, 1,5 Hz, 1H), 6,79 (dd, компонент системы ABC, J=7,7, 7,6 Hz, 1H), 6,72 (dd, компонент системы ABC, J=7,8, 1,3 Hz, 1H), 6,68 (dd, компонент системы ABC, J=7,8, 1,3 Hz, 1H), 4,37-4,14 (br m, 2H), 2,91-2,73 (m, 3H), 2,07 (d, J=1,1 Hz, 3H), 1,89-1,62 (m, 4H), 1,49 (s, 9H).
Процедуры P5 и P6
4-Бром-2-фенил-1,3-бензодиоксол, ENT-1 (P5), и 4-бром-2-фенил-1,3-бензодиоксол, ENT-2 (P6)

Стадия 1. Синтез 2-бром-6-[гидрокси(фенил)метил]фенола (C9).
Фениллитий (1,9 M раствор в 1-бутоксибутане; 78,5 мл, 149 ммоля) при температуре −70°C медленно добавляли к раствору 3-бром-2-гидроксибензальдегида (10,0 г, 49,7 ммоля) в тетрагидрофуране (70 мл) со скоростью, обеспечивающей поддержание температуры реакционной смеси ниже −60°C. Полученную суспензию перемешивали при −70°C в течение 1 ч и затем давали нагреваться до комнатной температуры в течение ночи, затем ее при температуре 0°C выливали в водный раствор хлорида аммония (30 мл). Эту смесь экстрагировали этилацетатом (3×30 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиентный режим: от 0% до 5% этилацетата в петролейном эфире) давала C9 в виде желтого твердого вещества. Выход: 6,11 г, 21,9 ммоля, 44%. 1H NMR (400 MHz, хлороформ-d) δ 7,45-7,28 (m, 6H), 7,22-7,18 (m, 1H), 7,06 (br d, J=7,7 Hz, 1H), 6,77 (dd, J=7,9, 7,8 Hz, 1H), 6,06 (br s, 1H), 2,89 (br s, 1H).
Стадия 2. Синтез 4-бром-2-фенил-1,3-бензодиоксола (C10).
К раствору C9 (6,11 г, 21,9 ммоля) в метаноле (370 мл) добавляли раствор перйодата натрия (11,7 г, 54,7 ммоля) в воде (175 мл). Реакционную смесь перемешивали при 30°C в течение 40 ч, затем большую часть метанола удаляли концентрированием в вакууме. Полученную смесь экстрагировали дихлорметаном (5×100 мл) и объединенные органические слои последовательно промывали водным раствором сульфита натрия (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Хроматография на силикагеле (элюент: петролейный эфир) давала C10 в виде бесцветного масла. Выход: 4,50 г, 16,2 ммоля, 74%. LCMS m/z 278,5 (наблюдали сигналы изотопа брома) [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 7,62-7,57 (m, 2H), 7,49-7,43 (m, 3H), 7,04 (s, 1H), 7,00 (dd, J=8,0, 1,4 Hz, 1H), 6,79 (dd, компонент системы ABX, J=7,8, 1,4 Hz, 1H), 6,75 (dd, компонент системы ABX, J=7,9, 7,8 Hz, 1H).
Стадия 3. Выделение 4-бром-2-фенил-1,3-бензодиоксола, ENT-1 (P5), и 4-бром-2-фенил-1,3-бензодиоксола, ENT-2 (P6).
Энантиомеры содержащие C10 (5,00 г, 18,0 ммоля), разделяли с помощью SFC [колонка: Chiral Technologies ChiralCel OD, 10 мкм; подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся энантиомер обозначали, как ENT-1 (P5), и второй элюирующийся энантиомер, как ENT-2 (P6); оба получали в виде желтого масла.
P5 Выход: 2,20 г, 7,94 ммоля, 44%. LCMS m/z 277,0 (наблюдали сигналы изотопа брома) [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 7,63-7,55 (m, 2H), 7,51-7,42 (m, 3H), 7,04 (s, 1H), 7,00 (dd, J=8,0, 1,3 Hz, 1H), 6,80 (dd, компонент системы ABX, J=7,8, 1,4 Hz, 1H), 6,75 (dd, компонент системы ABX, J=7,9, 7,8 Hz, 1H). Время удерживания 3,28 мин (колонка: Chiral Technologies ChiralCel OD-H, 4,6×150 мм, 5 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: метанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 5,5 мин; скорость потока: 2,5 мл/мин).
P6 Выход: 2,00 г, 7,22 ммоля, 40%. LCMS m/z 276,9 (наблюдали сигналы изотопа брома) [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 7,63-7,55 (m, 2H), 7,50-7,42 (m, 3H), 7,04 (s, 1H), 7,00 (dd, J=8,0, 1,4 Hz, 1H), 6,80 (dd, компонент системы ABX, J=7,8, 1,4 Hz, 1H), 6,75 (dd, компонент системы ABX, J=7,9, 7,9 Hz, 1H). Время удерживания 3,73 мин (условия проведения анализа такие же, как использованные для P5).
Процедура P7
трет-Бутил-4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилат (P7)
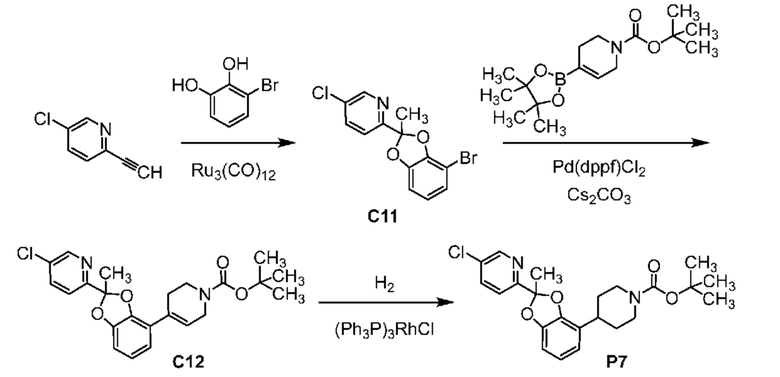
Стадия 1. Синтез 2-(4-бром-2-метил-1,3-бензодиоксол-2-ил)-5-хлорпиридина (C11).
Смесь 5-хлор-2-этинилпиридина (1,80 г, 13,1 ммоля), 3-бромбензол-1,2-диола (2,47 г, 13,1 ммоля) и додекакарбонила трирутения (167 мг, 0,261 ммоля) в толуоле (25 мл) дегазировали в течение 1 мин и затем нагревали при 100°C в течение 16 ч. Реакционную смесь разбавляли этилацетатом (30 мл) и фильтровали через слой диатомовой земли; фильтрат концентрировали в вакууме и очищали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 1% этилацетата в петролейном эфире) и получали C11 в виде желтого масла. Выход: 1,73 г, 5,30 ммоля, 40%. LCMS m/z 325,6 (наблюдали сигналы изотопа брома-хлора) [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 8,63 (dd, J=2,4, 0,7 Hz, 1H), 7,71 (dd, компонент системы ABX, J=8,4, 2,4 Hz, 1H), 7,60 (dd, компонент системы ABX, J=8,4, 0,7 Hz, 1H), 6,97 (dd, J=8,0, 1,4 Hz, 1H), 6,76 (dd, компонент системы ABX, J=7,8, 1,4 Hz, 1H), 6,72 (dd, компонент системы ABX, J=8,0, 7,8 Hz, 1H), 2,10 (s, 3H).
Стадия 2. Синтез трет-бутил-4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]-3,6-дигидропиридин-1(2H)-карбоксилата (C12).
[1,1"-Бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (388 мг, 0,530 ммоля) добавляли к суспензии C11 (1,73 г, 5,30 ммоля), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилата (1,64 г, 5,30 ммоля) и карбоната цезия (5,18 г, 15,9 ммоля) в 1,4-диоксане (35 мл) и воде (6 мл). Реакционную смесь перемешивали при 90°C в течение 4 ч, затем ее разбавляли этилацетатом (30 мл) и водой (5 мл). Органический слой концентрировали в вакууме и остаток обрабатывали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 5% этилацетата в петролейном эфире) и получали C12 в виде желтого смолообразного вещества. Выход: 1,85 г, 4,31 ммоля, 81%. LCMS m/z 451,0♦ [M+Na+]. 1H NMR (400 MHz, хлороформ-d) δ 8,62 (dd, J=2,5, 0,8 Hz, 1H), 7,69 (dd, компонент системы ABX, J=8,4, 2,4 Hz, 1H), 7,57 (dd, компонент системы ABX, J=8,4, 0,8 Hz, 1H), 6,84-6,79 (m, 2H), 6,78-6,73 (m, 1H), 6,39-6,33 (br m, 1H), 4,13-4,07 (m, 2H), 3,68-3,58 (m, 2H), 2,60-2,51 (br m, 2H), 2,07 (s, 3H), 1,49 (s, 9H).
Стадия 3. Синтез трет-бутил-4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилата (P7).
Раствор C12 (2,61 г, 6,08 ммоля) и хлорида трис(трифенилфосфин)родия(I) (катализатор Уилкинсона; 563 мг, 0,608 ммоля) в метаноле (100 мл) дегазировали в вакууме и затем продували водородом; этот цикл откачка-продувка проводили всего трижды. Затем реакционную смесь перемешивали при 60°C в атмосфере водорода (50 фунт-сила/дюйм2) в течение 16 ч, затем ее фильтровали. Фильтрат концентрировали в вакууме и остаток очищали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 10% этилацетата в петролейном эфире); полученное вещество объединяли с веществом после аналогичного гидрирования, проводимого для C12 (110 мг, 0,256 ммоля) и получали P7 в виде светло-желтого смолообразного вещества. Суммарный выход: 2,05 г, 4,76 ммоля, 75%. LCMS m/z 431,3♦ [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 8,62 (d, J=2,3 Hz, 1H), 7,69 (dd, компонент системы ABX, J=8,4, 2,4 Hz, 1H), 7,57 (d, половина квартета AB, J=8,4 Hz, 1H), 6,79 (dd, компонент системы ABC, J=7,8, 7,7 Hz, 1H), 6,72 (dd, компонент системы ABC, J=7,8, 1,3 Hz, 1H), 6,68 (br d, компонент системы ABC, J=7,9 Hz, 1H), 4,32-4,12 (br m, 2H), 2,91-2,73 (m, 3H), 2,05 (s, 3H), 1,90-1,62 (m, 4H), 1,48 (s, 9H).
Процедуры P8 и P9
трет-Бутил-4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилат, ENT-1 (P8), и трет-бутил-4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилат, ENT-2 (P9)

Разделение P7 (500 мг, 1,16 ммоля) на его компоненты-энантиомеры проводили с помощью SFC {колонка: Phenomenex Lux Amylose-1, 5 мкм; подвижная фаза: 9:1 диоксид углерода/[2-пропанол, содержащий 0,2% (7 M аммиака в метаноле)]}. Первый элюирующийся энантиомер обозначали, как ENT-1 (P8), и второй элюирующийся энантиомер, как ENT-2 (P9).
P8 Выход: 228 мг, 0,529 ммоля, 46%. Время удерживания 4,00 мин {колонка: Phenomenex Lux Amylose-1, 4,6×250 мм, 5 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: [2-пропанол, содержащий 0,2% (7 M аммиака в метаноле)]; градиентный режим: 5% B в течение 1,00 мин, затем от 5% до 60% B за 8,00 мин; скорость потока: 3,0 мл/мин; противодавление: 120 бар}.
P9 Выход: 229 мг, 0,531 ммоля, 46%. Время удерживания 4,50 мин (условия проведения анализа такие же, как использованные для P8).
Процедура P10
{4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}уксусная кислота (P10)
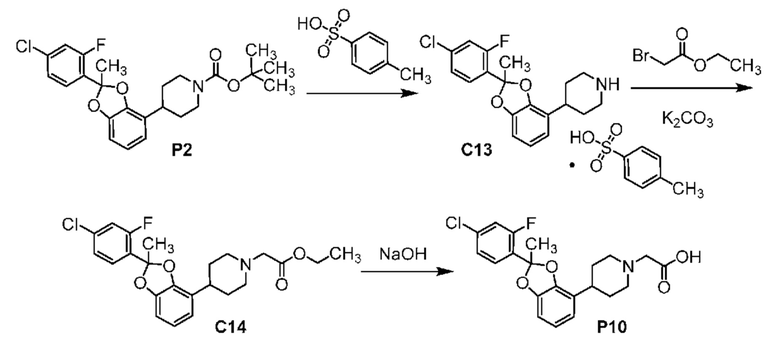
Стадия 1. Синтез 4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин, п-толуолсульфоната (C13).
Раствор P2 (5,0 г, 11 ммоля) и п-толуолсульфоновой кислоты (4,81 г, 27,9 ммоля) в этилацетате (100 мл) перемешивали при 60°C в течение 2 ч, затем его концентрировали в вакууме и получали C13 в виде желтого смолообразного вещества. Это вещество сразу направляли на следующую стадию. LCMS m/z 347,9♦ [M+H]+.
Стадия 2. Синтез этил {4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}ацетата (C14).
Карбонат калия (7,71 г, 55,8 ммоля) и этилбромацетат (1,86 г, 11,2 ммоля) добавляли к раствору C13 (из предыдущей стадии; ≤11 ммоля) в ацетонитриле (150 мл) и реакционную смесь перемешивали при 55°C в течение 16 ч. Затем ее фильтровали и фильтрат концентрировали в вакууме и очищали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 30% этилацетата в петролейном эфире) и получали C14 в виде желтого смолообразного вещества. По данным анализа с помощью1H NMR это вещество не было совершенно чистым. Выход: 3,57 г, 8,23 ммоля, 75% за 2 стадии. 1H NMR (400 MHz, хлороформ-d), только пики C14: δ 7,52 (dd, J=8,4, 8,0 Hz, 1H), 7,17-7,07 (m, 2H), 6,77 (dd, компонент системы ABC, J=7,8, 7,8 Hz, 1H), 6,72-6,67 (m, 2H), 4,21 (q, J=7,1 Hz, 2H), 3,27 (s, 2H), 3,07 (m, 2H), 2,70 (tt, J=12,1, 3,8 Hz, 1H), 2,35 (ddd, J=11,5, 11,5, 2,7 Hz, 2H), 2,04 (d, J=1,1 Hz, 3H), 2,02-1,76 (m, 4H), 1,29 (t, J=7,1 Hz, 3H).
Стадия 3. Синтез {4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}уксусной кислоты (P10).
Раствор C14 (3,57 г, 8,23 ммоля) и водного раствора гидроксида натрия (3 M; 13,7 мл, 41,1 ммоля) в смеси метанола (80 мл) и тетрагидрофурана (40 мл) перемешивали при 25°C в течение 16 ч. После удаления растворителей в вакууме водный остаток подкисляли до pH 7 путем добавления 1 M хлористоводородной кислоты и затем экстрагировали смесью дихлорметана и метанола (10:1, 2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении и получали P10 в виде желтого твердого вещества. Выход: 2,95 г, 7,27 ммоля, 88%. LCMS m/z 406,2♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 7,61 (dd, J=8,3, 8,3 Hz, 1H), 7,29 (dd, J=10,9, 2,0 Hz, 1H), 7,22 (ddd, J=8,4, 2,0, 0,8 Hz, 1H), 6,82 (dd, компонент системы ABC, J=8,3, 7,1 Hz, 1H), 6,78-6,72 (m, 2H), 3,65-3,54 (br m, 2H), 3,51 (s, 2H), 3,04-2,88 (m, 3H), 2,23-2,07 (m, 2H), 2,07-1,93 (m, 2H), 2,04 (d, J=1,1 Hz, 3H).
Процедура P11
Метил-2-(хлорметил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат (P11)
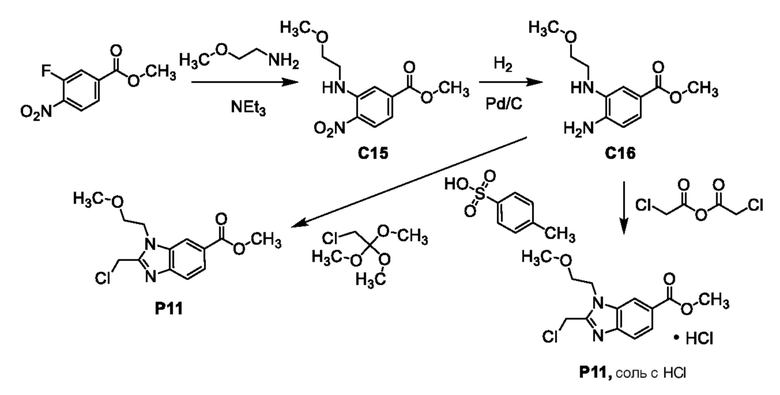
Стадия 1. Синтез метил-3-[(2-метоксиэтил)амино]-4-нитробензоата (C15).
К бесцветному раствору метил-3-фтор-4-нитробензоата (50 г, 250 ммоля) в тетрагидрофуране (400 мл) добавляли триэтиламин (40,7 г, 402 ммоля, 55,8 мл), затем по каплям, при комнатной температуре добавляли 2-метоксиэтанамин (30,2 г, 402 ммоля) в тетрагидрофуране (100 мл). Полученный желтый раствор перемешивали при 55°C в течение 18 ч. Раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении для удаления тетрагидрофуран. Полученное желтое твердое вещество растворяли в этилацетате (800 мл) и промывали насыщенным водным раствором хлорида аммония (250 мл). Водную фазу отделяли и экстрагировали этилацетатом (200 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (3×250 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении и получали C15 (60,2 г, 94%) в виде желтого твердого вещества. 1H NMR (600 MHz, хлороформ-d) δ 8,23 (d, 1H), 8,17 (br s, 1H), 7,58 (d, 1H), 7,25 (dd, 1H), 3,95 (s, 3H), 3,69-3,73 (m, 2H), 3,56 (m, 2H), 3,45 (s, 3H); LCMS m/z 255,4 [M+H]+.
Стадия 2. Синтез метил-4-амино-3-[(2-метоксиэтил)амино]бензоата (C16).
К раствору C15 (30 г, 118 ммоля) в метаноле (500 мл) добавляли Pd/C (10 г, 94 ммоля). Эту реакционную смесь перемешивали при комнатной температуре в атмосфере водорода под давлением 15 фунт-сила/дюйм2 в течение 18 ч. Черную суспензию фильтровали через диатомовую землю и осадок на фильтре промывали метанолом (500 мл). Объединенные фильтраты концентрировали в вакууме и получали C16 (26,5 г, количественный выход) в виде коричневого масла, которое затвердевало при выдерживании. 1H NMR (400 MHz, хлороформ-d) δ 7,48 (dd, 1H), 7,36 (d, 1H), 6,69 (d, 1H), 3,87 (s, 3H), 3,77 (br s, 2H), 3,68 (t, 2H), 3,41 (s, 3H), 3,32 (t, 2H); LCMS m/z 224,7 [M+H]+.
Стадия 3. Синтез метил-2-(хлорметил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (P11).
К раствору C16 (5,00 г, 22,3 ммоля) в тетрагидрофуране (100 мл) добавляли 2-хлор-1,1,1-триметоксиэтан (3,31 мл, 24,6 ммоля), затем моногидрат п-толуолсульфоновой кислоты (84,8 мг, 0,446 ммоля). Реакционную смесь нагревали при 45°C в течение 5 ч, затем ее концентрировали в вакууме; оставшееся масло растворяли в этилацетате (10 мл) и нагревали до образования раствора. Его медленно перемешивали с охлаждением до комнатной температуры в течение ночи. Осадок собирали фильтрованием и промывали гептаном и получали P11 в виде серого твердого вещества. Выход: 5,73 г, 20,3 ммоля, 91%. 1H NMR (600 MHz, хлороформ-d) δ 8,12 (br s, 1H), 8,01 (br d, J=8,6 Hz, 1H), 7,79 (d, J=8,4 Hz, 1H), 4,96 (s, 2H), 4,52 (t, J=5,1 Hz, 2H), 3,96 (s, 3H), 3,74 (t, J=5,1 Hz, 2H), 3,28 (s, 3H).
Стадия 4. Синтез метил-2-(хлорметил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилатгидрохлорида (P11, соль с HCl).
Раствор C16 (5,0 г, 24 ммоля) в 1,4-диоксане (100 мл) нагревали при 100°C, раствор хлоруксусного ангидрида (4,1 г, 24,5 ммоля) в 1,4-диоксане (60 мл) добавляли через капельную воронку в течение 10 ч и реакционную смесь перемешивали в течение еще 12 ч при 100°C. На следующий день реакционную смесь охлаждали до комнатной температуры и 1,4-диоксан удаляли при пониженном давлении. Неочищенную реакционную смесь растворяли в этилацетате и промывали насыщенным водным раствором бикарбоната натрия. Этилацетатный слой отделяли, сушили над сульфатом натрия и фильтровали. 4 M Раствор хлорида водорода в 1,4-диоксане (1,1 экв.) добавляли к раствору в этилацетате при постоянном перемешивании. Гидрохлорид P11 осаждался в виде бледно-желтого твердого вещества. Суспензию перемешивали в течение 1 ч и затем гидрохлорид P11 собирали фильтрованием и получали светло-желтое твердое вещество (6,1 г, 86%). 1H NMR (600 MHz, CD3OD) δ 8,64 (s, 1H), 8,30 (d, 1H), 7,92 (d, 1H), 5,32 (s, 2H), 4,84 (m, 2H), 3,99 (s, 3H), 3,83 (t, 2H), 3,31 (s, 3H). LCMS m/z 283,2 [M+H]+.
Процедура P12
Метил-1-(2-метоксиэтил)-2-(пиперазин-1-илметил)-1H-бензимидазол-6-карбоксилат (P12)
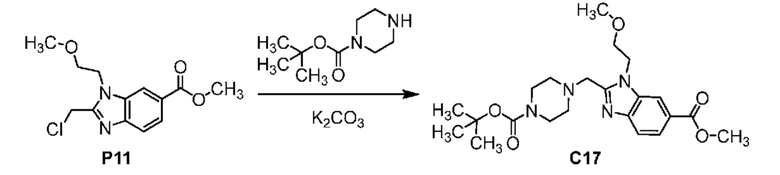
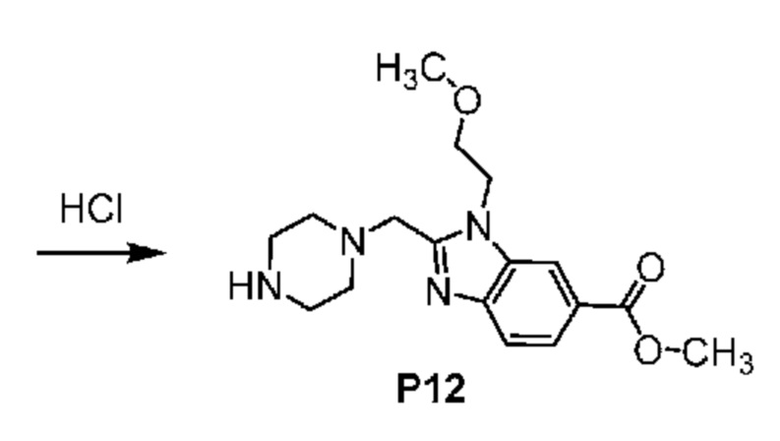
Стадия 1. Синтез метил-2-{[4-(трет-бутоксикарбонил)пиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (C17).
Соединение P11 (1,59 г, 5,62 ммоля) добавляли при 15°C к смеси трет-бутилпиперазин-1-карбоксилата (1,00 г, 5,37 ммоля) и карбоната калия (2,97 г, 21,5 ммоля) в ацетонитриле (15 мл) и реакционную смесь перемешивали при 55°C в течение 12 ч. Затем ее объединяли с продуктом аналогичной реакции, проведенной с помощью P11 и трет-бутилпиперазин-1-карбоксилата (200 мг, 1,07 ммоля), и смесь фильтровали. Затем фильтрат концентрировали в вакууме, остаток очищали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 60% этилацетата в петролейном эфире) и получали C17 в виде бледно-желтого твердого вещества. Суммарный выход: 2,30 г, 5,32 ммоля, 83%. LCMS m/z 433,0 [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 8,12 (d, J=1,5 Hz, 1H), 7,96 (dd, J=8,4, 1,5 Hz, 1H), 7,73 (d, J=8,5 Hz, 1H), 4,58 (t, J=5,4 Hz, 2H), 3,95 (s, 3H), 3,89 (s, 2H), 3,73 (t, J=5,4 Hz, 2H), 3,46-3,37 (br m, 4H), 3,28 (s, 3H), 2,54-2,44 (br m, 4H), 1,45 (s, 9H).
Стадия 2. Синтез метил-1-(2-метоксиэтил)-2-(пиперазин-1-илметил)-1H-бензимидазол-6-карбоксилата (P12).
К раствору C17 (2,30 г, 5,32 ммоля) в дихлорметане (80 мл) добавляли раствор хлорида водорода в этилацетате (20 мл). Реакционную смесь перемешивали при 20°C в течение 2 ч, затем ее концентрировали в вакууме. Остаток разбавляли водой (20 мл), pH устанавливали равным от 9 до 10 путем добавления насыщенного водного раствора бикарбоната натрия и экстрагировали смесью этилацетата и метанола (10:1, 15×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме и получали P12 в виде бледно-желтого твердого вещества. Выход: 1,68 г, 5,05 ммоля, 95%. LCMS m/z 332,8 [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 8,13 (br s, 1H), 7,96 (br d, J=8,5 Hz, 1H), 7,72 (d, J=8,5 Hz, 1H), 4,59 (t, J=5,5 Hz, 2H), 3,95 (s, 3H), 3,86 (s, 2H), 3,75 (t, J=5,5 Hz, 2H), 3,29 (s, 3H), 2,87 (t, J=4,8 Hz, 4H), 2,50 (br m, 4H).
Процедура P13
6-Бром-2-(хлорметил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин (P13)
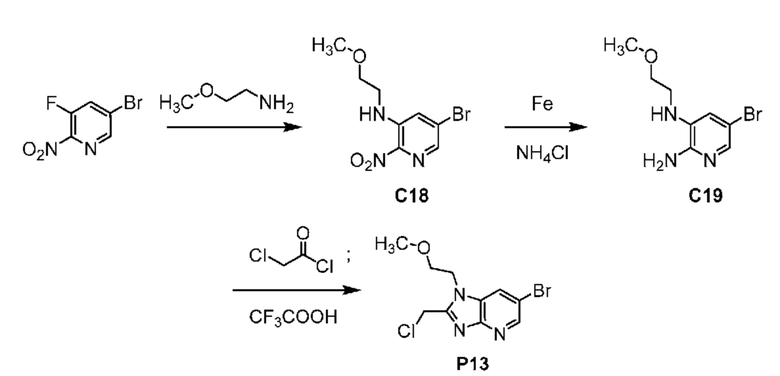
Стадия 1. Синтез 5-бром-N-(2-метоксиэтил)-2-нитропиридин-3-амина (C18).
Раствор 5-бром-3-фтор-2-нитропиридина (400 мг, 1,81 ммоля) и 2-метоксиэтанамина (408 мг, 5,43 ммоля) в тетрагидрофуране (10 мл) перемешивали при 25°C в течение 2 ч, затем его разбавляли этилацетатом (100 мл) и промывали водой (50 мл). Органический слой промывали насыщенным водным раствором хлорида натрия (50 мл), сушили над сульфатом магния, фильтровали и концентрировали и получали C18 в виде желтого твердого вещества. Выход: 430 мг, 1,56 ммоля, 86%.
Стадия 2. Синтез 5-бром-N3-(2-метоксиэтил)пиридин-2,3-диамина (C19).
Раствор C18 (430 мг, 1,56 ммоля), хлорида аммония (833 мг, 15,6 ммоля), и порошкообразное железо (870 мг, 15,6 ммоля) в смеси метанола (10 мл) и воды (2 мл) перемешивали при 80°C в течение 30 мин. Полученную суспензию выливали в воду (50 мл) и экстрагировали этилацетатом (2×50 мл); объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали и получали C19 в виде коричневого твердого вещества. Выход: 350 мг, 1,42 ммоля, 91%. 1H NMR (400 MHz, хлороформ-d) δ 7,63 (d, J=2,1 Hz, 1H), 6,88 (d, J=2,0 Hz, 1H), 4,33-4,19 (br s, 2H), 3,65 (dd, J=5,6, 4,6 Hz, 2H), 3,40 (s, 3H), 3,22 (br t, J=5 Hz, 2H).
Стадия 3. Синтез 6-бром-2-(хлорметил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридина (P13).
Раствор C19 (400 мг, 1,63 ммоля) в 1,4-диоксане (8 мл) обрабатывали хлорацетилхлоридом (0,284 мл, 3,57 ммоля) и перемешивали при комнатной температуре, пока анализ с помощью LCMS не указывал на полное превращение C19 в промежуточный амид. После удаления 1,4-диоксана в вакууме остаток растворяли в трифторуксусной кислоте (8 мл) и нагревали при 80°C в течение 18 ч, затем реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученное масло растворяли в этилацетате (50 мл) и нейтрализовывали путем добавления насыщенного водного раствора бикарбоната натрия. Водный слой экстрагировали этилацетатом (20 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиентный режим: от 0% до 80% этилацетата в гептане) давала P13 в виде твердого вещества. Выход: 176 мг, 0,578 ммоля, 35%. LCMS m/z 306,1 (наблюдали сигналы изотопа брома-хлора) [M+H]+. 1H NMR (600 MHz, хлороформ-d) δ 8,58 (br s, 1H), 7,89 (br s, 1H), 4,92 (s, 2H), 4,44 (t, J=5,0 Hz, 2H), 3,71 (t, J=5,0 Hz, 2H), 3,28 (s, 3H).
Процедура P14
Метил-2-{[4-(2,3-дигидроксифенил)пиперидин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат (P14)

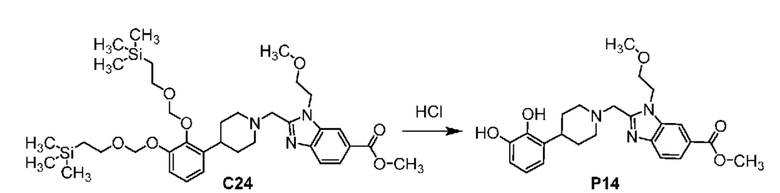
Стадия 1. Синтез [(3-бромбензол-1,2-диил)бис(оксиметандиилоксиэтан-2,1-диил)]бис(триметилсилана) (C20).
Эту реакцию проводили с двумя партиями одинаковой величины. N, N-Диизопропилэтиламин (37,8 мл, 217 ммоля) по каплям добавляли к раствору 3-бромбензол-1,2-диола (10,0 г, 52,9 ммоля) в тетрагидрофуране (300 мл). Затем смеси перемешивали в течение 10 мин при 20°C, в течение 5 мин по каплям добавляли [2-(хлорметокси)этил](триметил)силан (19,2 мл, 108 ммоля) и перемешивание продолжали в течение 16 ч при комнатной температуре (18°C). Повторно добавляли N, N-диизопропилэтиламин (27,6 мл, 158 ммоля), затем по каплям добавляли [2-(хлорметокси)этил](триметил)силан (14,0 мл, 79,1 ммоля) при комнатной температуре (18°C). Еще через 2,5 ч при комнатной температуре реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Затем неочищенные продукты из двух партий объединяли и очищали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 7% этилацетата в петролейном эфире) и получали C20 в виде бесцветного масла. По данным анализа с помощью1H NMR это вещество не было совершенно чистым. Суммарный выход: 22,9 г, 50,9 ммоля, 48%. 1H NMR (400 MHz, хлороформ-d), только пики C20: δ 7,19 (dd, J=8,1, 1,5 Hz, 1H), 7,12 (dd, J=8,3, 1,4 Hz, 1H), 6,90 (dd, J=8,2. 8,2 Hz, 1H), 5,26-5,19 (m, 4H), 4,00-3,92 (m, 2H), 3,80-3,73 (m, 2H), 1,00-0,91 (m, 4H), 0,03 (s, 9H), 0,00 (s, 9H).
Стадия 2. Синтез трет-бутил-4-(2,3-бис{[2-(триметилсилил)этокси]метокси}фенил)-3,6-дигидропиридин-1(2H)-карбоксилата (C21).
Реакционный сосуд, содержащий суспензию C20 (6,11 г, 13,6 ммоля), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилата (5,04 г, 16,3 ммоля), водного раствора карбоната натрия (1 M; 40,8 мл, 40,8 ммоля) и [1,1"-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (497 мг, 0,679 ммоля) в 1,4-диоксане (100 мл), откачивали и заполняли азотом. Этот цикл откачки повторяли дважды и затем реакционную смесь перемешивали при 85°C в течение 16 ч, затем реакционную смесь разбавляли водой (40 мл) и экстрагировали этилацетатом (3×150 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка с помощью хроматографии на силикагеле (градиентный режим: от 0% до 8% метанола в дихлорметане) давала C21 в виде желтого масла. Выход: 5,47 г, 9,91 ммоля, 73%. 1H NMR (600 MHz, хлороформ-d) δ 7,10 (br d, J=8,2 Hz, 1H), 6,98 (dd, J=7,9, 7,9 Hz, 1H), 6,81 (br d, J=7,7 Hz, 1H), 5,79 (br s, 1H), 5,23 (s, 2H), 5,07 (s, 2H), 4,03 (br s, 2H), 3,83-3,74 (m, 4H), 3,59 (br s, 2H), 2,52 (br s, 2H), 1,49 (s, 9H), 1,01-0,89 (m, 4H), 0,01 (s, 9H), 0,01 (s, 9H).
Стадия 3. Синтез трет-бутил-4-(2,3-бис{[2-(триметилсилил)этокси]метокси}фенил)пиперидин-1-карбоксилата (C22).
Раствор C21 (12,5 г, 22,6 ммоля) в метаноле (300 мл) обрабатывали с помощью 10% палладия на угле (2,94 г, 2,76 ммоля) и гидрировали в течение 16 ч при 40 фунт-сила/дюйм2 и 25°C. В этот момент анализ с помощью LCMS указывал на превращение в продукт: LCMS m/z 576,0 [M+Na+]. Затем реакционную смесь фильтровали и осадок на фильтре промывали метанолом (2×100 мл), объединенные фильтраты концентрировали в вакууме и получали C22 в виде бесцветного масла. Выход: 11,2 г, 20,1 ммоля, 89%. 1H NMR (400 MHz, хлороформ-d) δ 7,05-6,97 (m, 2H), 6,83 (dd, J=6,9, 2,5 Hz, 1H), 5,22 (s, 2H), 5,13 (s, 2H), 4,38-4,10 (br m, 2H), 3,90-3,82 (m, 2H), 3,81-3,73 (m, 2H), 3,22 (tt, J=12,2, 3,5 Hz, 1H), 2,79 (br dd, J=12,8, 12,8 Hz, 2H), 1,78 (br d, J=13 Hz, 2H), 1,65-1,52 (m, 2H), 1,48 (s, 9H), 1,04-0,91 (m, 4H), 0,03 (s, 9H), 0,00 (s, 9H).
Стадия 4. Синтез 4-(2,3-бис{[2-(триметилсилил)этокси]метокси}фенил)пиперидина (C23).
При комнатной температуре (15°C) к раствору C22 (7,23 г, 13,0 ммоля) в дихлорметане (90 мл) добавляли 2,6-диметилпиридин (2,39 г, 22,3 ммоля), затем по каплям добавляли триметилсилилтрифторметансульфонат (3,80 г, 17,1 ммоля). Реакционную смесь перемешивали при 15°C в течение 16 ч, затем дополнительно добавляли 2,6-диметилпиридин (909 мг, 8,48 ммоля) и триметилсилилтрифторметансульфонат (1,45 г, 6,52 ммоля). После перемешивания при комнатной температуре (15°C) в течение еще 5 ч анализ реакционной смеси с помощью LCMS указывал на наличие продукта: LCMS m/z 454,1 [M+H]+. Реакционную смесь концентрировали в вакууме и остаток последовательно промывали водным раствором хлорида аммония (3×100 мл) и насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении и получали C23 в виде коричневого масла (6,6 г). Это вещество сразу направляли на следующую стадию.
Стадия 5. Синтез метил-2-{[4-(2,3-бис{[2-(триметилсилил)этокси]метокси}фенил)пиперидин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (C24).
К раствору C23 (из предыдущей стадии; 6,6 г, ≤13 ммоля) в ацетонитриле (150 мл) добавляли P11 (3,08 г, 10,9 ммоля), затем карбонат калия (10,1 г, 73,1 ммоля) и реакционную смесь перемешивали при комнатной температуре (15°C) в течение 16 ч. В этот момент анализ с помощью LCMS указывал на наличие продукта: LCMS m/z 700,2 [M+H]+. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме; очистка с помощью хроматографии на силикагеле (градиентный режим: от 34% до 56% этилацетата в петролейном эфире) давало C24 в виде желтого масла. Выход: 5,4 г, 7,7 ммоля, 59% за 2 стадии. 1H NMR (400 MHz, хлороформ-d) δ 8,16-8,12 (m, 1H), 7,96 (dd, J=8,5, 1,5 Hz, 1H), 7,73 (d, J=8,5 Hz, 1H), 7,04-6,96 (m, 2H), 6,86 (dd, J=6,7, 2,6 Hz, 1H), 5,21 (s, 2H), 5,12 (s, 2H), 4,63 (t, J=5,5 Hz, 2H), 3,95 (s, 3H), 3,93-3,83 (m, 4H), 3,80-3,72 (m, 4H), 3,31 (s, 3H), 3,17-3,06 (m, 1H), 2,99 (br d, J=11,2 Hz, 2H), 2,35-2,22 (m, 2H), 1,81 (br d, половина квартета AB, J=12,6 Hz, 2H), 1,75-1,61 (m, 2H), 1,04-0,91 (m, 4H), 0,05 (s, 9H), −0,01 (s, 9H).
Стадия 6. Синтез метил-2-{[4-(2,3-дигидроксифенил)пиперидин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (P14).
Раствор хлорида водорода в 1,4-диоксане (4 M; 96 мл, 384 ммоля) при комнатной температуре(18°C) добавляли к раствору C24 (6,40 г, 9,14 ммоля) в 1,4-диоксане (120 мл). После завершения добавления реакционную смесь перемешивали при комнатной температуре (18°C) в течение 16 ч, объединяли с продуктом аналогичной реакции, проведенной с использованием C24 (1,00 г, 1,43 ммоля), и концентрировали в вакууме. Остаток обрабатывали смесью дихлорметана и метанола (20:1, 150 мл) и перемешивали при комнатной температуре (18°C) в течение 1 ч, затем твердое вещество (4,85 г) собирали фильтрованием. Это вещество обрабатывали водой (100 мл) и в смеси устанавливали pH от 7 до 8 путем добавления водного раствора бикарбоната натрия, перемешивали при комнатной температуре (18°C) в течение 30 мин и фильтровали. Осадок на фильтре промывали водой (2×20 мл), затем смешивали с метанолом (100 мл) и концентрировали в вакууме. Полученное вещество обрабатывали петролейным эфиром (100 мл) и перемешивали при комнатной температуре (18°C) в течение 30 мин. После фильтрования осадок на фильтре смешивали с толуолом (30 мл) и концентрировали в вакууме и получали P14 в виде серого твердого вещества. Суммарный выход: 2,92 г, 6,64 ммоля, 63%. LCMS m/z 440,1 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8,21 (d, J=1,6 Hz, 1H), 7,81 (dd, J=8,5, 1,6 Hz, 1H), 7,66 (d, J=8,5 Hz, 1H), 6,64-6,51 (m, 3H), 4,63 (t, J=5,3 Hz, 2H), 3,88 (s, 3H), 3,84 (s, 2H), 3,75 (t, J=5,3 Hz, 2H), 3,22 (s, 3H), 2,97-2,78 (m, 3H), 2,18 (br dd, J=11, 11 Hz, 2H), 1,75-1,64 (m, 2H), 1,64-1,49 (m, 2H).
Процедура P15
Метил-2-(хлорметил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат (P15)

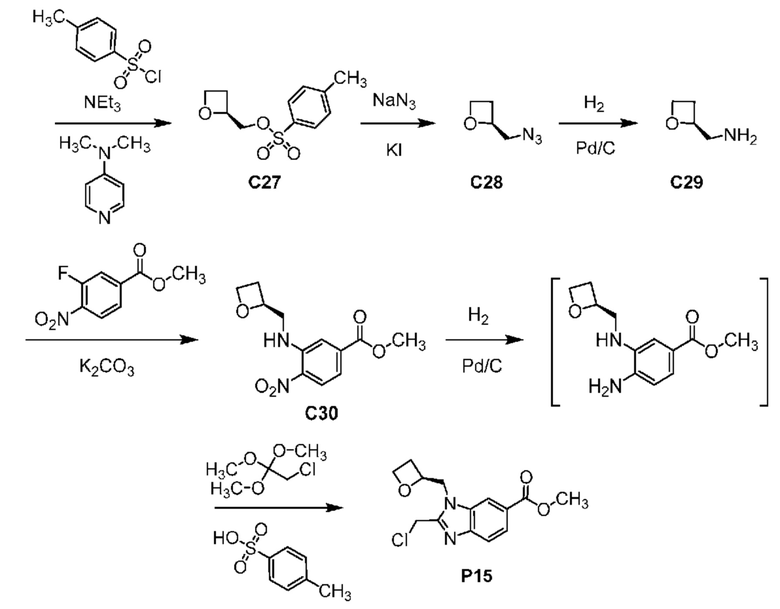 Всю эту последовательность проводили в крупном масштабе. Обычно до реакций, а также после добавления реагентов, реакторы откачивали до давления, равного от −0,08 до −0,05 МПа и затем заполняли азотом до нормального давления. Эту процедуру обычно повторяли 3 раза и затем определяли содержание кислорода, чтобы гарантировать, что оно составляло ≤1,0%. Для экстракции и промывки органических слоев смеси обычно перемешивали в течение от 15 до 60 мин и затем им давали отстояться в течение от 15 до 60 мин до разделения слоев.
Всю эту последовательность проводили в крупном масштабе. Обычно до реакций, а также после добавления реагентов, реакторы откачивали до давления, равного от −0,08 до −0,05 МПа и затем заполняли азотом до нормального давления. Эту процедуру обычно повторяли 3 раза и затем определяли содержание кислорода, чтобы гарантировать, что оно составляло ≤1,0%. Для экстракции и промывки органических слоев смеси обычно перемешивали в течение от 15 до 60 мин и затем им давали отстояться в течение от 15 до 60 мин до разделения слоев.
Стадия 1. Синтез (2S)-2-[(бензилокси)метил]оксетана (C25).
Эту реакцию проводили с тремя партиями примерно одинаковой величины. В облицованный стеклом реактор объемом 2000 л помещали 2-метилпропан-2-ол (774,7 кг). Трет-бутоксид калия (157,3 кг, 1402 моля) добавляли через встроенную воронку для добавления и смесь перемешивали в течение 30 мин. Затем таким же образом добавляли триметилсульфоксониййодид (308,2 кг, 1400 моля) и реакционную смесь нагревали при температуре от 55°C до 65°C в течение от 2 до 3 ч, затем добавляли (2S)-2-[(бензилокси)метил]оксиран (92,1 кг, 561 моля) со скоростью от 5 до 20 кг/ч. После поддержания температуры реакционной смеси равной от 55°C до 65°C в течение 25 ч ее охлаждали до температуры от 25°C до 35°C и фильтровали через диатомовую землю (18,4 кг). Осадок на фильтре промывали трет-бутилметиловым эфиром (3×340 кг) и объединенные фильтраты переносили в реактор объемом 5000 л, обрабатывали очищенной водой (921 кг) и перемешивали в течение от 15 до 30 мин при температуре от 15°C до 30°C. Затем органический слой дважды промывали раствором хлорида натрия (230,4 кг) в очищенной воде (920,5 кг) и концентрировали при пониженном давлении (≤ −0,08 МПа) при температуре ≤45°C. Добавляли н-гептан (187 кг) и полученную смесь концентрировали при пониженном давлении (≤ −0,08 МПа) при температуре ≤45°C; органическую фазу очищали с помощью хроматографии на силикагеле (280 кг) с хлоридом натрия (18,5 кг) в верхней части колонки. Неочищенное вещество загружали в колонку с использованием н-гептана (513 кг) и затем элюировали смесью н-гептана (688,7 кг) и этилацетата (64,4 кг). Эти 3 партии объединяли и получали C25 в виде светло-желтого масла чистоты 85% (189,7 кг, 906 ммоля, 54%). 1H NMR (400 MHz, хлороформ-d), только пики C25: δ 7,40-7,32 (m, 4H), 7,32-7,27 (m, 1H), 4,98 (dddd, J=8,1, 6,7, 4,9, 3,7 Hz, 1H), 4,72-4,55 (m, 4H), 3,67 (dd, компонент системы ABX, J=11,0, 4,9 Hz, 1H), 3,62 (dd, компонент системы ABX, J=11,0, 3,7 Hz, 1H), 2,72-2,53 (m, 2H).
Стадия 2. Синтез (2S)-оксетан-2-илметанола (C26).
10% Палладий на угле (30,7 кг) добавляли через воронку для добавления при температуре от 10°C до 30°C к раствору C25 чистоты 85% (из предыдущей стадии; 185,3 кг, 884,8 моля) в тетрагидрофуране (1270 кг) в реактор-автоклав из нержавеющей стали объемом 3000 л. Воронку для добавления промывали очищенной водой и тетрагидрофураном (143 кг) и промывочные растворы добавляли к реакционной смеси. Затем содержимое реактора продували азотом, аналогичным образом продували водородом с повышением давления до равного от 0,3 до 0,5 МПа и затем давление сбрасывали до равного 0,05 МПа. Эту продувку водородом повторяли 5 раз, затем давление водорода повышали до равного от 0,3 до 0,4 МПа. Затем реакционную смесь нагревали при температуре от 35°C до 45°C. Через 13 ч, причем в течение этого периода давление водорода поддерживали равным от 0,3 до 0,5 МПа, давление в смеси сбрасывали до 0,05 МПа и пять раз продували азотом с повышением давления до равного от 0,15 до 0,2 МПа и затем давление сбрасывали до равного 0,05 МПа. После охлаждения смеси до температуры, равной от 10°C до 25°C, ее фильтровали и реактор промывали тетрагидрофураном (2×321 кг). Осадок на фильтре дважды замачивали в этой промывочной жидкости и затем отфильтровывали; концентрирование при пониженном давлении (≤ −0,06 МПа) проводили при температуре ≤40°C и получали C26 (62,2 кг, 706 моля, 80%) в тетрагидрофуране (251 кг)
Стадия 3. Синтез (2S)-оксетан-2-илметил-4-метилбензолсульфоната (C27).
4-(Диметиламино)пиридин (17,5 кг, 143 моля) добавляли при температуре от 10°C до 25°C к раствору C26 (из предыдущей стадии; 62,2 кг, 706 моля) в тетрагидрофуране (251 кг) и триэтиламина (92,7 кг, 916 моля) в дихлорметане (1240 кг). Через 30 мин порциями добавляли п-толуолсульфонилхлорид (174,8 кг, 916,9 моля) с интервалами от 20 до 40 мин и реакционную смесь перемешивали при от 15°C до 25°C в течение 16 ч и 20 мин. Добавляли очищенную воду (190 кг); после перемешивания, органический слой промывали водным раствором бикарбоната натрия (получен с использованием 53,8 кг бикарбоната натрия и 622 кг очищенной воды) и затем промывали водным раствором хлорида аммония (получен с использованием 230 кг хлорида аммония и 624 кг очищенной воды). После заключительной промывки очищенной водой (311 кг) органический слой фильтровали через нуч-фильтр из нержавеющей стали, в который предварительно помещали силикагель (60,2 кг). Осадок на фильтре замачивали в дихлорметане (311 кг) в течение 20 мин и затем фильтровали; объединенные фильтраты концентрировали при пониженном давлении (≤ −0,05 МПа) и температуре ≤40°C, пока не оставалось от 330 до 400 л. Затем добавляли тетрагидрофуран (311 кг) при температуре от 15°C до 30°C и смесь концентрировали таким же образом до конечного объема, равного от 330 до 400 л. Добавление тетрагидрофуран и концентрирование повторяли, опять до объема, равного от 330 до 400 л и получали светло-желтый раствор C27 (167,6 кг, 692 ммоля, 98%) в тетрагидрофуране (251,8 кг). 1H NMR (400 MHz, хлороформ-d), только пики C27: δ 7,81 (d, J=8,4 Hz, 2H), 7,34 (d, J=8,1 Hz, 2H), 4,91 (ddt, J=8,0, 6,7, 3,9 Hz, 1H), 4,62-4,55 (m, 1H), 4,53-4,45 (m, 1H), 4,14 (d, J=3,9 Hz, 2H), 2,75-2,63 (m, 1H), 2,60-2,49 (m, 1H), 2,44 (s, 3H).
Стадия 4. Синтез (2S)-2-(азидометил)оксетана (C28).
N, N-Диметилформамид (473 кг), азид натрия (34,7 кг, 534 моля) и йодид калия (5,2 кг, 31 моля) объединяли в облицованном стеклом реакторе объемом 3000 л при температуре от 10°C до 25°C. После добавления C27 (83,5 кг, 344,6 моля) в тетрагидрофуране (125,4 кг) реакционную смесь нагревали при температуре от 55°C до 65°C в течение 17 ч и 40 мин, затем ее охлаждали до температуры от 25°C до 35°C, через клапан на дне пропускали азот в течение 15 мин. Затем добавляли трет-бутилметиловый эфир (623 кг) и очищенную воду (840 кг) и полученный водный слой дважды экстрагировали трет-бутилметиловым эфиром (312 кг и 294 кг). Объединенные органические слои промывали очищенной водой (2×419 кг), поддерживая температуру равной от 10°C до 25°C и получали C28 (31,2 кг, 276 моля, 80%) в растворе указанного выше органического слоя (1236,8 кг).
Стадия 5. Синтез 1-[(2S)-оксетан-2-ил]метанамина (C29).
10% Палладий на угле (3,7 кг) добавляли через воронку для добавления при температуре от 10°C до 30°C к раствору C28 [из предыдущей стадии; 1264 кг (31,1 кг C28, 275 моля)] в тетрагидрофуране (328 кг) в реакторе-автоклаве из нержавеющей стали объемом 3000 л . Воронку для добавления промывали тетрагидрофураном (32 кг) и промывочный раствор добавляли к реакционной смеси. Затем содержимое реактора продували азотом, аналогичным образом продували водородом с повышением давления до равного от 0,05 до 0,15 МПа и затем давление сбрасывали до равного от 0,03 до 0,04 МПа. Эту продувку водородом повторяли 5 раз, затем давление водорода повышали до равного от 0,05 до 0,07 МПа. Температуру реакционной смеси повышали до равной от 25°C до 33°C и давление водорода поддерживали равным от 0,05 до 0,15 МПа в течение 22 ч, заменяя водород каждые 3-5 ч. Затем смесь пять раз продували азотом с повышением давления до равного от 0,15 до 0,2 МПа и затем давление сбрасывали до равного 0,05 МПа. После фильтрования тетрагидрофуран (92 кг и 93 кг) использовали для промывки реактора и затем замачивали осадок на фильтре. Объединенные фильтраты концентрировали при пониженном давлении (≤ −0,07 МПа) и температуре ≤45°C и получали C29 (18,0 кг, 207 моля, 75%) в тетрагидрофуране (57,8 кг). 1H NMR (400 MHz, DMSO-d6), только пики C29: δ 4,62 (ddt, J=7,6, 6,6, 5,1 Hz, 1H), 4,49 (ddd, J=8,6, 7,3, 5,6 Hz, 1H), 4,37 (dt, J=9,1, 5,9 Hz, 1H), 2,69 (d, J=5,1 Hz, 2H), 2,55-2,49 (m, 1H), 2,39 (m, 1H).
Стадия 6. Синтез метил-4-нитро-3-{[(2S)-оксетан-2-илметил]амино}бензоата (C30).
Карбонат калия (58,1 кг, 420 моля) добавляли к раствору метил-3-фтор-4-нитробензоата (54,8 кг, 275 моля) в тетрагидрофуране (148 кг) в облицованном стеклом реакторе объемом 100 л и смесь перемешивали в течение 10 мин. Раствор C29 (29,3 кг, 336 моля) в тетрагидрофуране (212,9 кг) добавляли и реакционную смесь перемешивали при от 20°C до 30°C в течение 12 ч, затем этилацетат (151 кг) добавляли, и смесь фильтровали через силикагель (29 кг). Осадок на фильтре промывали этилацетатом (от 150 кг до 151 кг) и объединенные фильтраты концентрировали при пониженном давлении (≤ −0,08 МПа) и температуре ≤45°C до объема, равного от 222 до 281 л. Затем смеси охлаждали до температуры от 10°C до 30°C, добавляли н-гептан (189 кг), перемешивание проводили в течение 20 мин и смесь концентрировали при пониженном давлении (≤ −0,08 МПа) и температуре ≤45°C до объема, равного 222 л. К смеси повторно добавляли н-гептан (181 кг) со стандартной скоростью, равной от 100 до 300 кг/ч и перемешивание продолжали в течение 20 мин. Смесь анализировали, пока содержание оставшегося тетрагидрофуран не становилось ≤5% и оставшегося этилацетата не становилось равным от 10% до 13%. Смесь нагревали при температуре от 40°C до 45°C и перемешивали в течение 1 ч, затем ее охлаждали до температуры от 15°C до 25°C со скоростью от 5°C до 10°C в час и затем перемешивали при температуре от 15°C до 25°C в течение 1 ч. Фильтрование с помощью центрифуги из нержавеющей стали давало осадок на фильтре, который промывали смесью этилацетата (5,0 кг) и н-гептана (34 кг) и затем перемешивали с тетрагидрофураном (724 кг) при температуре от 10°C до 30°C в течение 15 мин; фильтрование давало желтое твердое вещество, в основном состоящее из C30 (57,3 кг, 210 моля, 76%). 1H NMR (400 MHz, DMSO-d6) 8,34 (t, J=5,8 Hz, 1H), 8,14 (d, J=8,9 Hz, 1H), 7,63 (d, J=1,7 Hz, 1H), 7,13 (dd, J=8,9, 1,8 Hz, 1H), 4,99 (dddd, J=7,7, 6,7, 5,3, 4,1 Hz, 1H), 4,55 (ddd, J=8,6, 7,3, 5,8 Hz, 1H), 4,43 (dt, J=9,1, 6,0 Hz, 1H), 3,87 (s, 3H), 3,67-3,61 (m, 2H), 2,67 (dddd, J=11,1, 8,6, 7,7, 6,2 Hz, 1H), 2,57-2,47 (m, 1H).
Стадия 7. Синтез метил-2-(хлорметил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (P15).
Раствор C30 (из предыдущей стадии; 51,8 кг, 190 моля) в тетрагидрофуране (678 кг) в реакторе-автоклаве объемом 3000 л обрабатывали 10% палладием на угле (5,2 кг) при температуре от 10°C до 30°C. Трубу для прибавления промывали тетрагидрофураном (46 кг) и промывочную жидкость добавляли к реакционной смеси. Затем содержимое реактора продували азотом, аналогичным образом продували водородом с повышением давления до равного от 0,1 до 0,2 МПа и затем давление сбрасывали до равного от 0,02 до 0,05 МПа. Эту продувку водородом повторяли 5 раз, затем давление водорода повышали до равного от 0,1 до 0,25 МПа. Реакционную смесь перемешивали при температуре от 20°C до 30°C и каждые 2-3 ч, смесь трижды продували азотом и затем пять раз продували водородом; после каждой заключительной замены водородом давление водорода повышали до равного от 0,1 до 0,25 МПа. После проведения реакции в течение всего 11,25 ч давление в реакционной смеси сбрасывали до нормального и пять раз продували азотом с повышением давления до равного от 0,15 до 0,2 МПа и затем давление сбрасывали до равного 0,05 МПа. Затем ее фильтровали и осадок на фильтре дважды промывали тетрагидрофураном (от 64 кг до 63 кг); объединенную промывочную жидкость и фильтрат концентрировали при пониженном давлении (≤ −0,08 МПа) и при температуре ≤40°C до объема, равного от 128 до 160 л. Добавляли тетрагидрофуран (169 кг) и смесь повторно концентрировали до объема, равного от 128 до 160 л; эту процедуру повторяли всего 4 раза и получали раствор промежуточного продукта метил-4-амино-3-{[(2S)-оксетан-2-илметил]амино}бензоата.
К этому раствору добавляли тетрагидрофуран (150 кг), затем 2-хлор-1,1,1-триметоксиэтан (35,1 кг, 227 моля) и моногидрат п-толуолсульфоновой кислоты (1,8 кг, 9,5 моля). Затем реакционную смесь перемешивали в течение 25 мин, ее нагревали при температуре от 40°C до 45°C в течение 5 ч, затем ее концентрировали при пониженном давлении до объема, равного от 135 до 181 л. Добавляли 2-пропанол (142 кг) и смесь повторно концентрировали до объема, равного от 135 до 181 л, затем добавляли 2-пропанол (36,5 кг) и очищенную воду (90 кг) и перемешивание продолжали до образования раствора. Его фильтровали через встроенный жидкостный фильтр и затем обрабатывали очищенной водой (447 кг) со стандартной скоростью, равной от 150 до 400 кг/ч, при температуре от 20°C до 40°C. Затем смесь охлаждали до температуры от 20°C до 30°C, ее перемешивали в течение 2 ч и твердое вещество собирали фильтрованием с помощью центрифуги. Осадок на фильтре промывали раствором 2-пропанол (20,5 кг) и очищенной водой (154 кг); после сушки получали P15 в виде белого твердого вещества (32,1 кг, 109 моля, 57%). 1H NMR (400 MHz, хлороформ-d) δ 8,14-8,11 (m, 1H), 8,01 (dd, J=8,5, 1,1 Hz, 1H), 7,79 (br d, J=8,6 Hz, 1H), 5,26-5,18 (m, 1H), 5,04 (s, 2H), 4,66-4,58 (m, 2H), 4,53 (dd, компонент системы ABX, J=15,7, 2,7 Hz, 1H), 4,34 (dt, J=9,1, 6,0 Hz, 1H), 3,96 (s, 3H), 2,82-2,71 (m, 1H), 2,48-2,37 (m, 1H).
Процедура P16
Метил-2-(хлорметил)-1-метил-1H-бензимидазол-6-карбоксилат (P16)
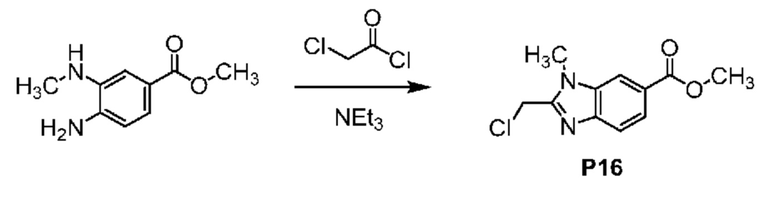
Метил-4-амино-3-(метиламино)бензоат (206 мг, 1,14 ммоля) растворяли в 1,4-диоксане (11,5 мл) и обрабатывали хлорацетилхлоридом (109 мкл, 1,37 ммоля). Смесь перемешивали при 100°C в течение 3 ч и охлаждали до комнатной температуры. Добавляли триэтиламин (0,8 мл, 7 ммоля) и гептан (10 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали с помощью хроматографии на силикагеле (элюент: 40% этилацетата в гептане) и получали 120 мг P16 (44%). 1H NMR (400 MHz, хлороформ-d) δ 8,14 (s, 1H), 8,01 (d, 1H), 7,78 (d, 1H), 4,87 (s, 2H), 3,97 (s, 3H), 3,94 (s, 3H); LCMS m/z 239,1 [M+H]+.
Процедуры P17 и P18
Метил-2-(6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат, ENT-1 (P17) и метил-2-(6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат, ENT-2 (P18)
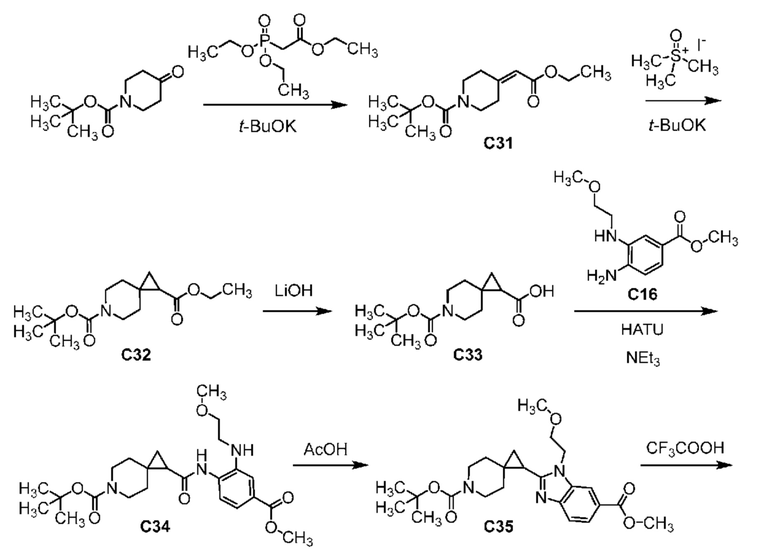
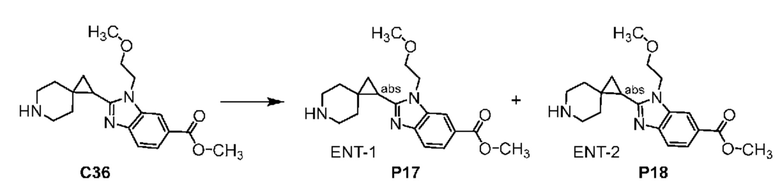
Стадия 1. Синтез трет-бутил-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилата (C31).
Раствор трет-бутоксида калия (65,9 г, 587 ммоля) в тетрагидрофуране (500 мл) при 0°C добавляли к раствору этил(диэтоксифосфорил)ацетата (132 г, 589 ммоля) в тетрагидрофуране (500 мл) и полученную суспензию перемешивали при 0°C в течение 1 ч, затем ее охлаждали до −50°C. По каплям добавляли раствор трет-бутил-4-оксопиперидин-1-карбоксилата (90,0 г, 452 ммоля) в тетрагидрофуране (1,5 л) при −50°C и затем реакционной смеси давали медленно нагреваться до 20°C и затем перемешивали в течение 16 ч при 20°C. После добавления воды (1 л) смесь концентрировали в вакууме для удаления тетрагидрофурана. Водный остаток экстрагировали этилацетатом (2×800 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (500 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученное вещество несколько раз промывали петролейным эфиром (200 мл) и получали C31 в виде белого твердого вещества. Выход: 95,0 г, 353 ммоля, 78%. 1H NMR (400 MHz, хлороформ-d) δ 5,71 (s, 1H), 4,16 (q, J=7,2 Hz, 2H), 3,55-3,43 (m, 4H), 2,94 (br t, J=5,5 Hz, 2H), 2,28 (br t, J=5,5 Hz, 2H), 1,47 (s, 9H), 1,28 (t, J=7,0 Hz, 3H).
Стадия 2. Синтез 6-трет-бутил-1-этил-6-азаспиро[2.5]октан-1,6-дикарбоксилата (C32).
К раствору триметилсульфоксониййодида (140 г, 636 ммоля) в диметилсульфоксиде (800 мл) добавляли трет-бутоксид калия (71,2 г, 634 ммоля) одной порцией при 20°C. Затем реакционную смесь перемешивали при 20°C в течение 1,5 ч, по каплям добавляли раствор C31 (95,0 г, 353 ммоля) в диметилсульфоксиде (800 мл) и перемешивание продолжали при 20°C в течение 16 ч. Затем добавляли насыщенный водный раствор хлорида натрия (2,0 л); полученную смесь нейтрализовывали путем добавления хлорида аммония и экстрагировали этилацетатом (3,0 л). Объединенные органические слои последовательно промывали водой (2×1,0 л) и насыщенным водным раствором хлорида натрия (2,0 л), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка с помощью хроматографии на силикагеле (элюент: 10:1 петролейный эфир/этилацетат) давало C32 в виде желтого масла. Анализ с помощью 1H NMR показал, что содержится загрязняющее алифатическое вещество. Выход: 80 г, 280 ммоля, 79%. 1H NMR (400 MHz, хлороформ-d), только пики C32: δ 4,19-4,09 (m, 2H), 3,55-3,39 (m, 3H), 3,27 (ddd, J=13,0, 7,0, 4,5 Hz, 1H), 1,76-1,64 (m, 2H), 1,56 (dd, J=8,0, 5,5 Hz, 1H, предположительно; частично закрыт пиком воды), 1,47 (s, 9H), 1,47-1,37 (m, 2H), 1,27 (t, J=7,0 Hz, 3H), 1,17 (dd, J=5,0, 5,0 Hz, 1H), 0,93 (dd, J=8,0, 4,5 Hz, 1H).
Стадия 3. Синтез 6-(трет-бутоксикарбонил)-6-азаспиро[2.5]октан-1-карбоновой кислоты (C33).
К смеси C32 (80 г, 280 ммоля) в тетрагидрофуране (500 мл) и воде (500 мл) одной порцией добавляли моногидрат гидроксида лития (37,4 г, 891 ммоля). Реакционную смесь перемешивали при 25°C в течение 16 ч, затем ее разбавляли водой (600 мл) и промывали этилацетатом (3×300 мл). Органические слои отбрасывали и водный слой подкисляли до pH 3-4 путем добавления 6 M хлористоводородной кислоты. Полученную смесь экстрагировали этилацетатом (3×600 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Растирание остатка с петролейным эфиром (300 мл) давало C33 в виде белого твердого вещества. Выход: 42,0 г, 164 ммоля, 59%. LCMS m/z 278,2 [M+Na+]. 1H NMR (400 MHz, DMSO-d6) δ 12,15-12,03 (br s, 1H), 3,43-3,25 (m, 3H, предположительно; частично закрыт пиком воды), 3,23-3,12 (m, 1H), 1,64-1,50 (m, 2H), 1,52 (dd, J=7,5, 5,5 Hz, 1H), 1,39 (s, 9H), 1,39-1,28 (m, 2H), 0,96-0,88 (m, 2H).
Стадия 4. Синтез трет-бутил-1-({4-(метоксикарбонил)-2-[(2-метоксиэтил)амино]фенил}карбамоил)-6-азаспиро[2.5]октан-6-карбоксилата (C34).
Раствор C33 (570 мг, 2,23 ммоля), C16 (500 мг, 2,23 ммоля) и O-(7-азабензотриазол-1-ил)-N, N,N",N"-тетраметилуронийгексафторфосфата (HATU; 1,27 г, 3,34 ммоля) в N, N-диметилформамиде (10 мл) перемешивали при 30°C в течение 30 мин, затем добавляли триэтиламин (902 мг, 8,91 ммоля) и перемешивание продолжали при 30°C в течение 16 ч. Затем реакционную смесь выливали в воду (60 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (3×50 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (элюент: 1:1 петролейный эфир/этилацетат) давала C34 в виде коричневого масла, которое сразу направляли на следующую стадию.
Стадия 5. Синтез метил-2-[6-(трет-бутоксикарбонил)-6-азаспиро[2.5]окт-1-ил]-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (C35).
Раствор C34 (из предыдущей стадии, ≤2,23 ммоля) в уксусной кислоте (15 мл) перемешивали при 50°C в течение 16 ч, затем его концентрировали в вакууме и получали C35 в виде коричневого масла. Это вещество сразу использовали на следующей стадии. LCMS m/z 444,1 [M+H]+.
Стадия 6. Синтез метил-2-(6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (C36).
Трифторуксусную кислоту (5 мл) добавляли к раствору C35 (из предыдущей стадии; ≤2,23 ммоля) в дихлорметане (10 мл) и реакционную смесь перемешивали при 25°C в течение 2 ч. После удаления растворителей в вакууме остаток подщелачивали путем добавления насыщенного водного раствора карбоната калия (40 мл) и экстрагировали смесью дихлорметана и метанола (10:1, 3×40 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали, концентрировали в вакууме и обрабатывали с помощью хроматографии на силикагеле (элюент: 10:1:0,1 дихлорметан/метанол/концентрировали гидроксид аммония) и получали C36 в виде желтого твердого вещества. Выход: 640 мг, 1,86 ммоля, 83% за 3 стадии. LCMS m/z 344,1 [M+H]+.
Стадия 7. Выделение метил-2-(6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата, ENT-1 (P17), и метил-2-(6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата, ENT-2 (P18).
Разделение C36 (630 мг, 1,83 ммоля) на его компоненты-энантиомеры проводили с помощью SFC [колонка: Chiral Technologies Chiralpak AD, 10 мкм; подвижная фаза: 55:45 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся пик обозначали, как ENT-1 (P17), и второй элюирующийся энантиомер, как ENT-2 (P18); оба выделяли в виде бледно-желтого твердого вещества.
P17 Выход: 300 мг, 0,874 ммоля, 48%. LCMS m/z 344,1 [M+H]+. Время удерживания: 5,10 мин (колонка: Chiral Technologies Chiralpak AD-3, 4,6×150 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: этанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 5,5 мин, затем непрерывно при 40% B в течение 3,0 мин; скорость потока: 2,5 мл/мин).
P18 Выход: 240 мг, 0,699 ммоля, 38%. LCMS m/z 344,1 [M+H]+. Время удерживания: 7,35 мин (условия проведения анализа такие же, как использованные для P17).
Процедура P19
Метил-4-амино-3-{[(1-этил-1H-имидазол-5-ил)метил]амино}бензоат (P19)
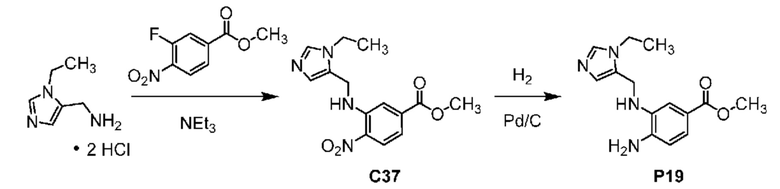
Стадия 1. Синтез метил-3-{[(1-этил-1H-имидазол-5-ил)метил]амино}-4-нитробензоата (C37).
Триэтиламин (3,65 мл, 26,2 ммоля) добавляли к раствору метил-3-фтор-4-нитробензоата (1,00 г, 5,02 ммоля) и 1-(1-этил-1H-имидазол-5-ил)метанаминдигидрохлорида (1,00 г, 5,05 ммоля) в смеси тетрагидрофурана (12 мл) и метанола (8 мл). Реакционную смесь перемешивали при 60°C в течение 40 ч, затем ее концентрировали в вакууме и очищали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 2% метанола в дихлорметане) и получали C37 в виде оранжевого твердого вещества. Выход: 1,27 г, 4,17 ммоля, 83%. 1H NMR (400 MHz, хлороформ-d) δ 8,24 (d, J=8,8 Hz, 1H), 7,98-7,91 (m, 1H), 7,68 (d, J=1,7 Hz, 1H), 7,57 (br s, 1H), 7,33 (dd, J=8,8, 1,7 Hz, 1H), 7,11 (br s, 1H), 4,53 (d, J=4,9 Hz, 2H), 3,99 (q, J=7,3 Hz, 2H), 3,95 (s, 3H), 1,47 (t, J=7,3 Hz, 3H).
Стадия 2. Синтез метил-4-амино-3-{[(1-этил-1H-имидазол-5-ил)метил]амино}бензоата (P19).
Смесь влажного палладия на угле (144 мг) и C37 (412 мг, 1,35 ммоля) в метаноле (13 мл) перемешивали при подаче водорода из баллона в течение 16 ч при 25°C. Затем реакционную смесь фильтровали через слой диатомовой земли и фильтрат концентрировали в вакууме и получали P19 в виде серого твердого вещества. Выход: 340 мг, 1,24 ммоля, 92%. 1H NMR (400 MHz, метанол-d4) δ 7,66 (br s, 1H), 7,38-7,29 (m, 2H), 6,97 (br s, 1H), 6,67 (d, J=7,9 Hz, 1H), 4,35 (s, 2H), 4,11 (q, J=7,3 Hz, 2H), 3,81 (s, 3H), 1,44 (t, J=7,3 Hz, 3H).
Процедура P20
Метил-4-амино-3-(метиламино)бензоат (P20)
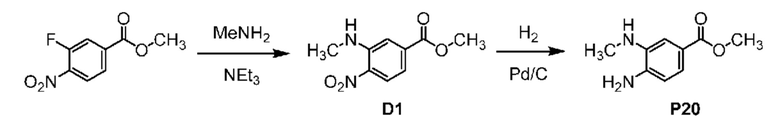
Стадия 1. Синтез метил-3-(метиламино)-4-нитробензоата (D1).
К раствору метил-3-фтор-4-нитробензоата (5,10 г, 25,6 ммоля) в тетрагидрофуране (60 мл) по каплям в течение 10 мин добавляли метиламин (38,4 мл, 76,8 ммоля, 2 M в тетрагидрофуране). Бледно-желтый раствор становился темно-оранжевым сразу после добавления и его перемешивали в течение 2 ч при комнатной температуре. Затем реакционную смесь разбавляли диэтиловым эфиром (100 мл) и органический слой последовательно промывали водой (50 мл) и насыщенным водным раствором хлорида натрия (50 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении и получали 5,26 г метил-3-(метиламино)-4-нитробензоата (98%) в виде темно-оранжевого твердого вещества. LCMS m/z 211,1 [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 8,22 (d, J=8,9 Hz, 1H), 8,00 (br s, 1H), 7,56 (d, J=1,7 Hz, 1H), 7,25 (dd, J=8,9, 1,7 Hz, 1H, предположительно; частично закрыт пиком растворителя), 3,95 (s, 3H), 3,09 (d, J=5,1 Hz, 3H).
Стадия 2. Синтез метил-4-амино-3-(метиламино)бензоата (P20).
Раствор D1 (5,26 г, 25,0 ммоля) в этаноле (150 мл) добавляли во флакон Парра® объемом 500 мл, в который предварительно помещали 10% палладий на угле (50% воды; 1 г). Смесь встряхивали при давлении 50 фунт-сила/дюйм2 в атмосфере водорода в течение 1 ч при комнатной температуре, затем ее фильтровали и осадок на фильтре промывали этанолом (100 мл). Фильтрат концентрировали при пониженном давлении и получали 4,38 г P20 (97%) в виде почти белого твердого вещества. LCMS m/z 181,1 [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 7,46 (dd, J=8,0, 1,9 Hz, 1H), 7,34 (d, J=1,8 Hz, 1H), 6,68 (d, J=8,0 Hz, 1H), 3,87 (s, 3H), 3,72 (br s, 2H), 3,21 (br s, 1H), 2,91 (s, 3H).
Процедуры P21 и P22
5-Бром-N3-метилпиридин-2,3-диамин (P21) и
5-Бром-N3,6-диметилпиридин-2,3-диамин (P22)
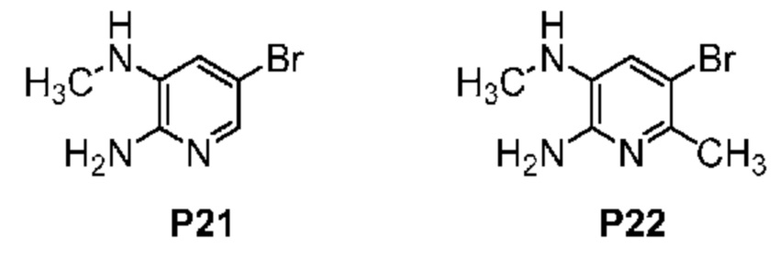
Промежуточный продукт P21 синтезировали по описанной в литературе методике (Choi, J. Y. et al. J. Med. Chem. 2012, 55, 852−870). Промежуточный продукт P22 синтезировали по такой же методике.
Процедура P23
Метил-2-(хлорметил)-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилат
(P23)

Стадия 1. Синтез метил-3-{[(1-метил-1H-имидазол-5-ил)метил]амино}-4-нитробензоата (D2).
К бесцветному раствору метил-3-фтор-4-нитробензоата (1,0 г, 5,0 ммоля) в N, N-диметилформамиде (10 мл) медленно добавляли 1-(1-метил-1H-имидазол-5-ил)метанамин (670 мг, 6,0 ммоля) и триэтиламин (762 мг, 7,53 ммоля). Реакционную смесь перемешивали при 60°C в течение 16 ч, затем ее выливали в воду (30 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали с помощью хроматографии на силикагеле (элюент: 20% метанола в дихлорметане). Полученное желтое твердое вещество растирали с 30:1 смесью петролейный эфир/этилацетат и получали D2 (1,2 г, 82%) в виде желтого твердого вещества. LCMS m/z 290,9 [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 8,25 (d, J=8,9 Hz, 1H), 7,98-7,92 (m, 1H), 7,70 (d, J=1,7 Hz, 1H), 7,49 (s, 1H), 7,34 (dd, J=8,9, 1,7 Hz, 1H), 7,12 (s, 1H), 4,54 (d, J=5,0 Hz, 2H), 3,96 (s, 3H), 3,67 (s, 3H).
Стадия 2. Синтез метил-4-амино-3-{[(1-метил-1H-имидазол-5-ил)метил]амино}бензоата (D3).
К суспензии D2 (5,46 г, 18,8 ммоля) в метаноле (160 мл) добавляли влажный 10% палладий на угле (1 г). Смесь перемешивали при равном 1 атм. давлении в атмосфере водорода в течение 36 ч при 20°C. Реакционную смесь фильтровали и осадок на фильтре промывали метанолом (200 мл). Фильтрат концентрировали при пониженном давлении и получали D3 (4,8 г, 98%) в виде коричневого твердого вещества. LCMS m/z 260,9 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 7,56 (s, 1H), 7,18 (br d, J=8,1 Hz, 1H), 7,12 (br s, 1H), 6,87 (s, 1H), 6,55 (d, J=8,2 Hz, 1H), 5,50 (s, 2H), 4,84 (t, J=5,2 Hz, 1H), 4,23 (d, J=5,0 Hz, 2H), 3,73 (s, 3H), 3,63 (s, 3H).
Стадия 3. Синтез метил-2-(гидроксиметил)-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилата (D4).
Смесь D3 (780 мг, 3,00 ммоля) и 2-гидроксиуксусной кислоты (342 мг, 4,49 ммоля) в 1,3,5-триметилбензоле (8 мл) перемешивали при 140°C в течение 14 ч и при 25°C в течение 48 ч. Прозрачный желтый раствор декантировали и получали коричневый остаток, который растворяли в метаноле (50 мл) и концентрировали при пониженном давлении. Неочищенное вещество очищали с помощью хроматографии на силикагеле (элюент: 20% метанола в дихлорметане) и получали D4 (318 мг, 35%) в виде желтого вспененного вещества. LCMS m/z 300,9 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8,13-8,11 (m, 1H), 7,83 (dd, J=8,4, 1,6 Hz, 1H), 7,71 (d, J=8,5 Hz, 1H), 7,59 (s, 1H), 6,58 (s, 1H), 5,69 (s, 2H), 4,75 (s, 2H), 3,84 (s, 3H), 3,53 (s, 3H).
Стадия 4. Синтез метил-2-(хлорметил)-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилата (P23).
К суспензии D4 (500 мг, 1,66 ммоля) в дихлорметане (10 мл) и N, N-диметилформамиде (3 мл) по каплям, при комнатной температуре добавляли тионилхлорид (990 мг, 0,60 мл, 8,32 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали при пониженном давлении. Полученный коричневый остаток растирали с дихлорметаном (10 мл). Твердые вещества собирали фильтрованием и промывали дихлорметаном (5 мл) и получали P23 (431 мг, 73%) в виде почти белого твердого вещества. LCMS m/z 318,9♦ [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 9,17 (s, 1H), 8,31 (s, 1H), 7,93 (br d, J=8,5 Hz, 1H), 7,82 (d, J=8,5 Hz, 1H), 7,11 (s, 1H), 5,92 (s, 2H), 5,13 (s, 2H), 3,87 (s, 3H), 3,87 (s, 3H).
Процедура P24
5-хлор-2-(хлорметил)-3-метил-3H-имидазо[4,5-b]пиридин (P24)
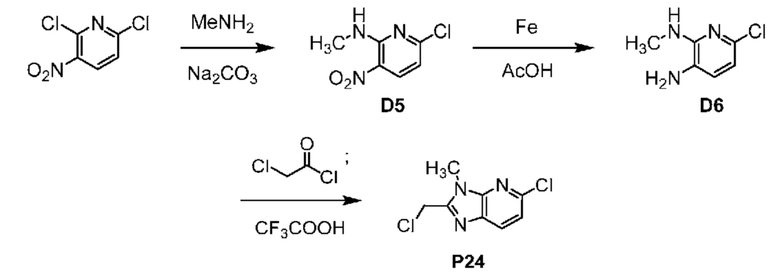
Стадия 1. Синтез 6-хлор-N-метил-3-нитропиридин-2-амина (D5).
К суспензии 2,6-дихлор-3-нитропиридина (200 г, 1,04 моля) и Na2CO3 (132 г, 1,24 моля) в этаноле (1 л) по каплям при 0°C шприцем добавляли раствор метиламина в тетрагидрофуране (2,0 M; 622 мл,1,24 моля). После завершения добавления реакционную смесь перемешивали при 18°C в течение 6 ч. Смесь фильтровали и фильтрат концентрировали при пониженном давлении и получали светло-желтое твердое вещество. Неочищенное вещество очищали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 5% этилацетата в петролейном эфире) и получали D5 (158 г, выход 81%) в виде желтого твердого вещества. 1H NMR (400 MHz, DMSO-d6) δ 8,72 (br s, 1H), 8,41 (d, J=8,6 Hz, 1H), 6,76 (d, J=8,6 Hz, 1H), 3,00 (d, J=4,8 Hz, 3H).
Стадия 2. Синтез 6-хлор-N2-метилпиридин-2,3-диамина (D6).
К смеси D5 (15,8 г, 84,2 ммоля) в уксусной кислоте (100 мл) добавляли порошкообразное железо (15,4 г, 276 ммоля). Реакционную смесь перемешивали при 80°C в течение 3 ч, затем ее охлаждали до комнатной температуры и фильтровали. Осадок на фильтре промывали этилацетатом (2×100). Объединенные органические слои концентрировали при пониженном давлении и неочищенное вещество очищали с помощью хроматографии на силикагеле (элюент: 1:1 этилацетат/петролейный эфир) и получали D6 (8,40 г, 63% выход) в виде коричневого твердого вещества. 1H NMR (400 MHz, хлороформ-d) δ 6,79 (d, J=7,7 Hz, 1H), 6,49 (d, J=7,7 Hz, 1H), 3,00 (s, 3H).
Стадия 3. Синтез 5-хлор-2-(хлорметил)-3-метил-3H-имидазо[4,5-b]пиридина (P24).
К раствору D6 (50,0 г, 317 ммоля) в 1,4-диоксане (1,2 л) добавляли хлорацетилхлорид (55,5 мл, 698 ммоля) и реакционную смесь перемешивали при 15°C в течение 50 мин. Затем ее концентрировали при пониженном давлении и получали коричневое твердое вещество, которое переносили в трифторуксусную кислоту (1,2 л) и перемешивали при 80°C в течение 60 ч. Смесь концентрировали при пониженном давлении и получали коричневое масло, которое разбавляли этилацетатом (1 л) и нейтрализовывали путем добавления насыщенного водного раствора бикарбоната натрия. Когда прекращалось выделение диоксида углерода, слои разделяли и водный слой экстрагировали этилацетатом (200 мл). Органические экстракты объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали с помощью хроматографии на силикагеле (градиентный режим: от 10% до 25% этилацетата в петролейном эфире) и получали P24 (61,0 г, 79% выход) в виде желтого твердого вещества. LCMS m/z 215,7 (наблюдали сигнал изотопов хлора) [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8,13 (d, J=8,3 Hz, 1H), 7,37 (d, J=8,4 Hz, 1H), 5,11 (s, 2H), 3,84 (s, 3H).
Примеры 1 и 2
2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперазин-1-ил}метил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота, ENT-X1, трифторацетат (1) [из C39]; и 2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперазин-1-ил}метил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота, ENT-X2, трифторацетат (2) [из C40]
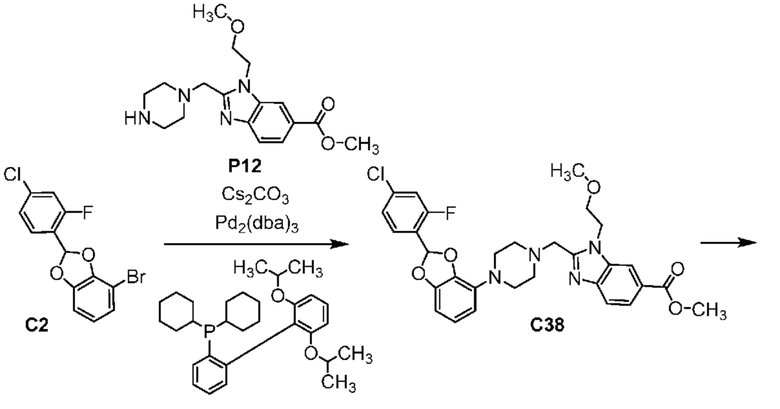
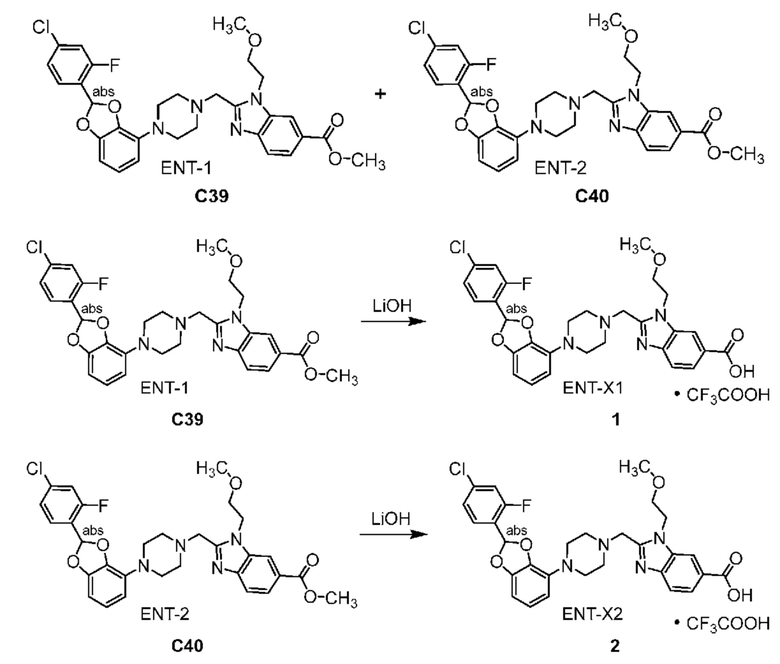
Стадия 1. Синтез метил-2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперазин-1-ил}метил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (C38).
Этот эксперимент проводили с двумя партиями одинаковой величины. Реакционный сосуд, содержащий смесь C2 (500 мг, 1,52 ммоля), P12 (530 мг, 1,59 ммоля), [2',6'-бис(пропан-2-илокси)бифенил-2-ил](дициклогексил)фосфана (Ruphos; 142 мг, 0,304 ммоля), трис(дибензилиденацетон)дипалладия(0) (139 мг, 0,152 ммоля) и карбоната цезия (1,48 г, 4,54 ммоля) в толуоле (15 мл), откачивали и заполняли азотом. Этот цикл откачки повторяли дважды, затем реакционную смесь перемешивали при 100°C в течение 16 ч, объединяли со второй партией и фильтровали. Фильтрат концентрировали и остаток обрабатывали с помощью хроматографии на силикагеле (градиентный режим: от 0% до 60% этилацетата в петролейном эфире), затем препаративной тонкослойной хроматографии (элюент: 1:1 петролейный эфир/этилацетат) и получали C38 в виде бледно-желтого твердого вещества. Суммарный выход: 600 мг, 1,03 ммоля, 34%. LCMS m/z 581,0♦ [M+H]+.
Стадия 2. Выделение метил-2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперазин-1-ил}метил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата, ENT-1 (C39), и метил-2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперазин-1-ил}метил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата, ENT-2 (C40).
Разделение C38 (780 мг, 1,34 ммоля) на его компоненты-энантиомеры проводили с помощью SFC [колонка: Chiral Technologies Chiralpak AD, 10 мкм; подвижная фаза: 3:2 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся энантиомер, обозначенный, как ENT-1 (C39), получали в виде белого твердого вещества. Выход: 282 мг, 0,485 ммоля, 36%. LCMS m/z 581,0♦ [M+H]+. Время удерживания 1,90 мин (колонка: Chiral Technologies Chiralpak AD-3, 4,6×50 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: этанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% B в течение 0,20 мин, затем от 5% до 40% B за 1,4 мин, затем непрерывно при 40% B в течение 1,05 мин; скорость потока: 4,0 мл/мин).
Второй элюирующийся энантиомер, обозначенный, как ENT-2, (C40), подвергали второй очистке с помощью SFC [колонка: Chiral Technologies Chiralpak AD, 10 мкм; подвижная фаза: 3:2 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Это давало C40 в виде бледно-коричневого твердого вещества. Выход: 280 мг, 0,482 ммоля, 36%. LCMS m/z 581,0♦ [M+H]+. Время удерживания 2,18 мин (условия проведения анализа такие же, как использованные для C39).
Стадия 3. Синтез трифторацетата 2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперазин-1-ил}метил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновой кислоты, ENT-X1 (1) [из C39].
Водный раствор гидроксида лития (2 M; 0,30 мл, 0,60 ммоля) добавляли к раствору C39 (70 мг, 0,12 ммоля) в смеси метанола (3 мл) и тетрагидрофурана (3 мл). Затем реакционную смесь перемешивали при 25°C в течение 16 ч, повторно добавляли водный раствор гидроксида лития (2 M; 0,30 мл, 0,60 ммоля) и перемешивание продолжали в течение еще 20 ч. Затем значение pH реакционной смеси устанавливали равным 7 путем добавления 1 M хлористоводородной кислоты и затем концентрировали в вакууме для удаления метанола и тетрагидрофурана. Значение pH остатка устанавливали равным 5-6 путем добавления трифторуксусной кислоты и затем очищали с помощью HPLC с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,1% трифторуксусной кислоты в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 30% до 60% B) и получали 1 в виде белого твердого вещества. Выход: 40,5 мг, 59,5 мкмоля, 50%. LCMS m/z 567,0♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 8,37 (br s, 1H), 8,07 (dd, J=8,5, 1,5 Hz, 1H), 7,79 (d, J=8,6 Hz, 1H), 7,59 (dd, J=8,0, 8,0 Hz, 1H), 7,34 (dd, J=10,2, 2,0 Hz, 1H), 7,30 (br dd, J=8,3, 2,0 Hz, 1H), 7,22 (s, 1H), 6,87 (dd, J=8,1, 8,1 Hz, 1H), 6,63 (br d, J=8 Hz, 1H), 6,60 (br d, J=8 Hz, 1H), 4,70 (s, 2H), 4,65 (t, J=4,8 Hz, 2H), 3,75 (t, J=4,8 Hz, 2H), 3,59-3,42 (m, 8H), 3,29 (s, 3H).
Стадия 4. Синтез трифторацетата 2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперазин-1-ил}метил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновой кислоты, ENT-X2 (2) [из C40].
Водный раствор гидроксида лития (2 M; 0,30 мл, 0,60 ммоля) добавляли к раствору C40 (69 мг, 0,12 ммоля) в смеси метанола (3 мл) и тетрагидрофурана (3 мл). Затем реакционную смесь перемешивали при 25°C в течение 16 ч, повторно добавляли водный раствор гидроксида лития (2 M; 0,30 мл, 0,60 ммоля) и перемешивание продолжали в течение еще 20 ч. Значение pH реакционной смеси устанавливали равным 7 путем добавления 1 M хлористоводородной кислоты и затем концентрировали в вакууме для удаления метанола и тетрагидрофурана. Значение pH остатка устанавливали равным 5-6 путем добавления трифторуксусной кислоты и затем очищали с помощью HPLC с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,1% трифторуксусной кислоты в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 30% до 60% B) и получали 2 в виде белого твердого вещества. Выход: 22,9 мг, 33,6 мкмоля, 28%. LCMS m/z 567,0♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 8,40-8,35 (m, 1H), 8,07 (dd, J=8,6, 1,5 Hz, 1H), 7,79 (d, J=8,6 Hz, 1H), 7,59 (dd, J=8,0, 8,0 Hz, 1H), 7,35 (dd, J=10,2, 2,0 Hz, 1H), 7,31 (br dd, J=8, 2 Hz, 1H), 7,22 (s, 1H), 6,87 (dd, J=8,3, 8,0 Hz, 1H), 6,63 (br d, J=8 Hz, 1H), 6,60 (br d, J=8 Hz, 1H), 4,68 (s, 2H), 4,65 (t, J=4,9 Hz, 2H), 3,76 (t, J=4,8 Hz, 2H), 3,57-3,40 (m, 8H), 3,29 (s, 3H).
Пример 3
Трифторацетат 2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин-6-карбоновой кислоты (3)
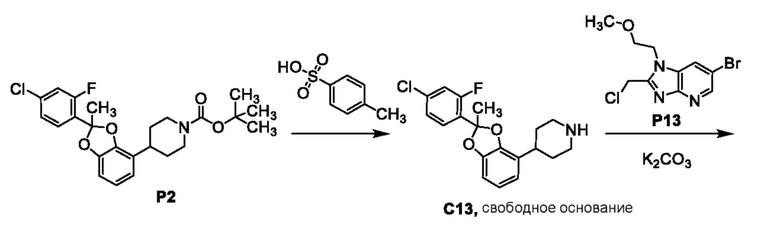
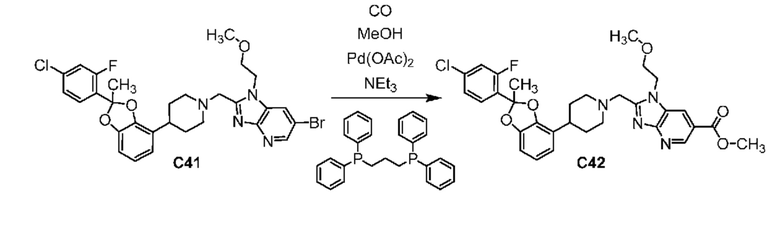
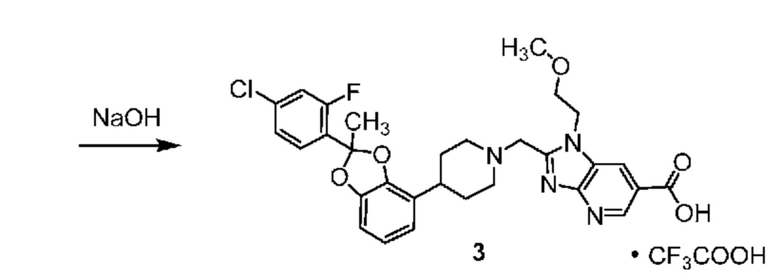
Стадия 1. Синтез 4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидина (C13, свободное основание).
К раствору P2 (300 мг, 0,670 ммоля) в этилацетате (3,5 мл) добавляли моногидрат п-толуолсульфоновой кислоты (318 мг, 1,67 ммоля). Реакционную смесь перемешивали при 60°C в течение 1 ч, затем ее подщелачивали путем добавления насыщенного водного раствора карбоната калия (20 мл) и экстрагировали смесью дихлорметана и метанола (10:1, 3×50 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме и получали свободное основание C13 в виде коричневого твердого вещества. Выход: 230 мг, 0,661 ммоля, 99%.
Стадия 2. Синтез 6-бром-2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридина (C41).
Суспензию свободного основания C13 (130 мг, 0,374 ммоля), P13 (130 мг, 0,427 ммоля) и карбоната калия (172 мг, 1,24 ммоля) в ацетонитриле (2 мл) перемешивали при 50°C в течение 16 ч. Затем реакционную смесь очищали с помощью препаративной тонкослойной хроматографии (элюент: этилацетат) и получали C41 в виде коричневого масла. Выход: 114 мг, 0,185 ммоля, 49%. LCMS m/z 617,1 (наблюдали сигналы изотопа брома-хлора) [M+H]+.
Стадия 3. Синтез метил-2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин-6-карбоксилата (C42).
Раствор C41 (114 мг, 0,185 ммоля), 1,3-бис(дифенилфосфино)пропана (15,3 мг, 37,1 мкмоля), ацетата палладия(II) (8,3 мг, 37 мкмоля) и триэтиламина (187 мг, 1,85 ммоля) в смеси метанола (5 мл) и N, N-диметилформамида (1 мл) перемешивали при 80°C в атмосфере монооксида углерода (50 фунт-сила/дюйм2) в течение 16 ч. Затем реакционную смесь разбавляли этилацетатом (50 мл), ее промывали насыщенным водным раствором хлорида натрия (2×50 мл), сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Очистка с помощью препаративной тонкослойной хроматографии (элюент: этилацетат) давала C42 в виде бесцветного масла. Выход: 60,0 мг, 0,101 ммоля, 55%. LCMS m/z 617,2 (наблюдали сигналы изотопа хлора [M+Na+].
Стадия 4. Синтез трифторацетата 2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин-6-карбоновой кислоты (3).
К раствору C42 (60,0 мг, 0,101 ммоля) в метаноле (2,0 мл) добавляли водный раствор гидроксида натрия (3 M; 1,0 мл, 3,0 ммоля) и реакционную смесь перемешивали при 20°C в течение 2 ч. Затем ее значение pH устанавливали равным 7 путем добавления 1 M хлористоводородной кислоты и экстрагировали смесью дихлорметана и метанола (10:1, 3×30 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали, концентрировали в вакууме и очищали с помощью HPLC с обращенной фазой (колонка: Boston Green ODS, 5 мкм; подвижная фаза A: 0,1% трифторуксусной кислоты в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 10% до 95% B) и получали 3 в виде белого твердого вещества. Выход: 29,6 мг, 42,6 мкмоля, 42%. LCMS m/z 581,0♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 9,13 (d, J=1,9 Hz, 1H), 8,74 (d, J=1,9 Hz, 1H), 7,63 (dd, J=8,3, 8,3 Hz, 1H), 7,30 (dd, J=10,9, 2,0 Hz, 1H), 7,24 (ddd, J=8,4, 2,0, 0,7 Hz, 1H), 6,89-6,84 (m, 1H), 6,82-6,77 (m, 2H), 4,98-4,89 (m, 2H, предположительно; значительно закрыт пиком воды), 4,64 (t, J=4,8 Hz, 2H), 4,04-3,92 (br m, 2H), 3,75 (dd, J=5,4, 4,2 Hz, 2H), 3,51-3,39 (m, 2H), 3,31 (s, 3H), 3,19-3,06 (m, 1H), 2,41-2,24 (m, 2H), 2,24-2,12 (m, 2H), 2,06 (d, J=1,0 Hz, 3H).
Примеры 4 и 5
2-({4-[(2R)-2-(4-Хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (4) и 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (5)
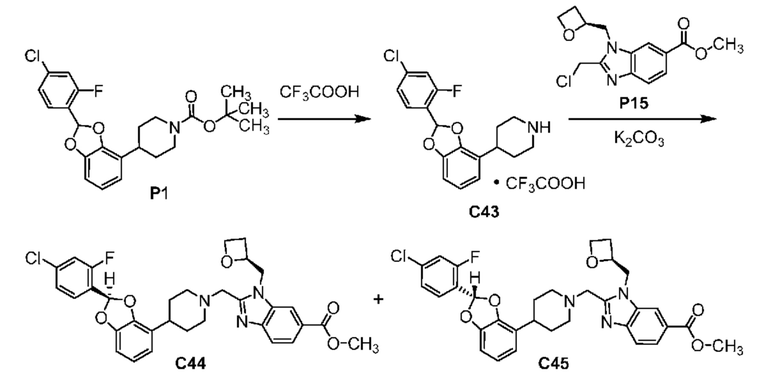

Стадия 1. Синтез трифторацетата 4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидина (C43).
К раствору P1 (300 мг, 0,691 ммоля) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1,3 мл). Реакционную смесь перемешивали при 29°C в течение 2 ч, затем ее концентрировали в вакууме и получали C43 в виде коричневого масла, которое сразу использовали на следующей стадии.
Стадия 2. Синтез метил-2-({4-[(2R)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C44) и метил-2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C45).
К раствору C43 (из предыдущей стадии, ≤0,691 ммоля) в ацетонитриле (10 мл) добавляли P15 (204 мг, 0,692 ммоля), затем карбонат калия (956 мг, 6,92 ммоля). Реакционную смесь перемешивали при 29°C в течение 16 ч, затем ее фильтровали; фильтрат концентрировали в вакууме и получали остаток, который очищали с помощью препаративной тонкослойной хроматографии (элюент: 2:1 петролейный эфир/этилацетат) и получали смесь диастереоизомерных продуктов в виде желтого смолообразного вещества (178 мг). Разделение на два продукта проводили с помощью SFC [колонка: Chiral Technologies ChiralCel OD, 5 мкм; подвижная фаза: 55:45 диоксид углерода/(метанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся диастереоизомер, полученный в виде желтого масла, обозначали, как C44. Выход: 44,3 мг, 74,8 мкмоля, 11% за 2 стадии. LCMS m/z 592,1♦ [M+H]+. Время удерживания 4,26 мин (колонка: Chiral Technologies ChiralCel OD-3, 4,6×100 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: метанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 4,5 мин, затем непрерывно при 40% B в течение 2,5 мин; скорость потока: 2,8 мл/мин).
Второй элюирующийся диастереоизомер подвергали второй очистке с помощью SFC [колонка: Chiral Technologies ChiralCel OD, 5 мкм; подвижная фаза: 3:2 диоксид углерода/(метанол, содержащий 0,1% гидроксида аммония)] и получали второй элюирующийся диастереоизомер в виде бесцветного масла, который обозначали, как C45. Выход: 38 мг, 64 мкмоля, 9% за 2 стадии. LCMS m/z 592,1♦ [M+H]+. Время удерживания 4,41 мин (условия проведения анализа такие же, как использованные для C44).
Показанные абсолютные стереохимические конфигурации по диоксолану определяли по корреляции активности 5 с активностью образца свободной кислоты 5, синтезированной из промежуточного продукта C48; абсолютную стереохимическую конфигурацию этого промежуточного продукта устанавливали на основании рентгенографического определения структуры монокристалла (см. ниже) C49, гемисульфата C48.
Стадия 3. Синтез 2-({4-[(2R)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (4).
Водный раствор гидроксида лития (2 M; 0,80 мл, 1,6 ммоля) добавляли к раствору C44 (44,3 мг, 74,8 мкмоля) в смеси метанола (1 мл) и тетрагидрофурана (1 мл) и реакционную смесь перемешивали при 26°C в течение 3 ч. Затем ее значение pH устанавливали равным 7 путем добавления трифторуксусной кислоты и полученную смесь концентрировали в вакууме и очищали с помощью HPLC с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксида аммония в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 30% до 50% B) и получали 4 в виде белого твердого вещества. Выход: 26,6 мг, 44,7 мкмоля, 60%. LCMS m/z 578,0♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 8,31 (d, J=1,4 Hz, 1H), 7,96 (dd, J=8,5, 1,6 Hz, 1H), 7,66 (d, J=8,5 Hz, 1H), 7,57 (dd, J=8,0, 8,0 Hz, 1H), 7,34 (dd, J=10,1, 2,0 Hz, 1H), 7,29 (br dd, J=8,3, 2,0 Hz, 1H), 7,20 (s, 1H), 6,86-6,79 (m, 1H), 6,77 (br dd, компонент системы ABC, J=7,9, 1,3 Hz, 1H), 6,73 (dd, компонент системы ABC, J=7,5, 1,4 Hz, 1H), 5,29-5,18 (m, 1H), 4,9-4,78 (m, 1H, предположительно; частично закрыт пиком воды), 4,68 (dd, J=15,3, 2,7 Hz, 1H), 4,54 (td, J=8,0, 5,9 Hz, 1H), 4,44 (dt, J=9,2, 5,9 Hz, 1H), 4,02 (AB квартет, JAB=13,9 Hz, ΔνAB=49,0 Hz, 2H), 3,18-3,08 (m, 1H), 3,05-2,96 (m, 1H), 2,81-2,68 (m, 2H), 2,56-2,45 (m, 1H), 2,45-2,30 (m, 2H), 2,03-1,88 (m, 2H), 1,88-1,79 (m, 2H).
Стадия 4. Синтез 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (5).
Водный раствор гидроксида лития (2 M; 0,80 мл, 1,6 ммоля) добавляли к раствору C45 (38 мг, 64 мкмоля) в смеси метанола (1 мл) и тетрагидрофурана (1 мл) и реакционную смесь перемешивали при 24°C в течение 2,5 ч. Затем ее значение pH устанавливали равным 7 путем добавления 1 M хлористоводородной кислоты и полученную смесь концентрировали в вакууме и очищали с помощью HPLC с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксида аммония в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 29% до 49% B) и получали 5 в виде белого твердого вещества. Выход: 27,9 мг, 46,9 мкмоля, 73%. LCMS m/z 577,9♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 8,32 (d, J=1,4 Hz, 1H), 7,96 (dd, J=8,5, 1,5 Hz, 1H), 7,66 (d, J=8,5 Hz, 1H), 7,56 (dd, J=8,0, 8,0 Hz, 1H), 7,34 (dd, J=10,2, 2,0 Hz, 1H), 7,29 (br dd, J=8,3, 2,0 Hz, 1H), 7,20 (s, 1H), 6,85-6,80 (m, 1H), 6,77 (dd, компонент системы ABC, J=8,0, 1,3 Hz, 1H), 6,73 (dd, компонент системы ABC, J=7,5, 1,4 Hz, 1H), 5,30-5,20 (m, 1H), 4,9-4,79 (m, 1H, предположительно; частично закрыт пиком воды), 4,68 (dd, J=15,4, 2,7 Hz, 1H), 4,62-4,54 (m, 1H), 4,44 (dt, J=9,2, 5,9 Hz, 1H), 4,02 (AB квартет, JAB=13,9 Hz, ΔνAB=44,6 Hz, 2H), 3,18-3,09 (m, 1H), 3,06-2,97 (m, 1H), 2,80-2,67 (m, 2H), 2,55-2,30 (m, 3H), 2,02-1,78 (m, 4H).
Альтернативный синтез свободной кислоты соединения примера 5
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота (5, свободная кислота)
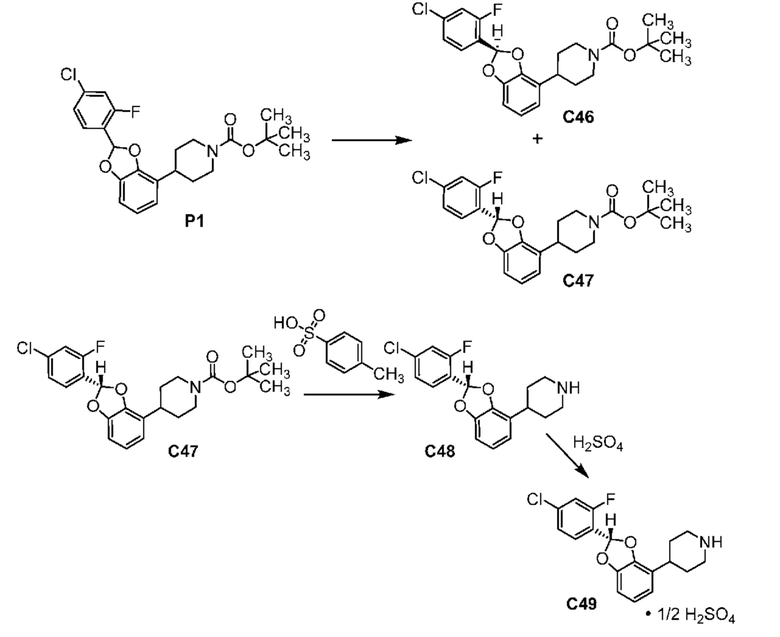
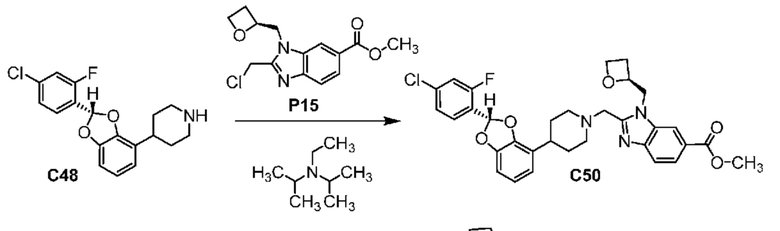
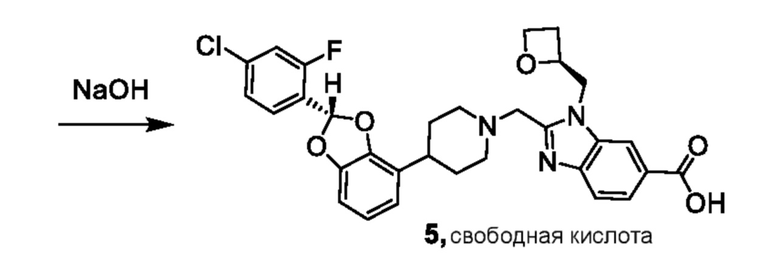
Стадия 1. Выделение трет-бутил-4-[(2R)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилата (C46) и трет-бутил-4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилата (C47).
Разделение P1 (10 г, 23 ммоля) на его компоненты-энантиомеры проводили с помощью HPLC с обращенной фазой [колонка: Phenomenex Lux Amylose-1, 5 мкм; подвижная фаза: 9:1 диоксид углерода/(2-пропанол, содержащий 0,2% 1-аминопропан-2-ола)]. Первый элюирующийся энантиомер обозначали, как C46, и второй элюирующийся энантиомер, как C47; оба получали в виде бесцветного масла. Абсолютные стереохимические конфигурации, указанные для C46 и C47, устанавливали на основании рентгенографического определения структуры монокристалла, проводимого для C49, который синтезировали из C47 (см. ниже).
C46 Выход: 4,47 г, 10,3 ммоля, 45%. Время удерживания: 3,98 мин [колонка: Phenomenex Lux Amylose-1, 4,6×250 мм, 5 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: 2-пропанол, содержащий 0,2% 1-аминопропан-2-ола; градиентный режим: 5% B в течение 1,00 мин, затем от 5% до 60% B за 8,00 мин; скорость потока: 3,0 мл/мин; противодавление: 120 бар].
C47 Выход: 4,49 г, 10,3 ммоля, 45%. Время удерживания: 4,32 мин (условия проведения анализа с помощью SFC такие же, как использованные для C46).
Стадия 2. Синтез 4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидина (C48).
Моногидрат п-толуолсульфоновой кислоты (566 мг, 2,98 ммоля) добавляли к раствору C47 (1,12 г, 2,58 ммоля) в этилацетате (26 мл). Затем реакционную смесь нагревали при 45°C в течение 16 ч, ее концентрировали в вакууме, растворяли в этилацетате и промывали насыщенным водным раствором бикарбоната натрия. Водные слои экстрагировали этилацетатом и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении и получали C48 в виде пенообразного белого твердого вещества (947 мг), LCMS m/z 334,0♦ [M+H]+. Порцию этого вещества, которое еще содержало некоторое количество п-толуолсульфоновой кислоты, использовали ниже для синтеза C50.
Вторую порцию пенообразного белого твердого вещества (440 мг) растворяли в этилацетате (25 мл) и промывали насыщенным водным раствором бикарбоната натрия (2×15 мл); органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме и получали C48 (350 мг) в виде бесцветного масла, которое больше не содержало п-толуолсульфоновую кислоту. Исправленный выход: 350 мг, 1,05 ммоля, 88%. 1H NMR (400 MHz, хлороформ-d) δ 7,53 (dd, J=8,4, 7,8 Hz, 1H), 7,22-7,13 (m, 3H), 6,87-6,80 (m, 1H), 6,79-6,71 (m, 2H), 3,23-3,14 (m, 2H), 2,86-2,69 (m, 3H), 1,90-1,68 (m, 4H).
Стадия 3. Синтез гемисульфата 4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидина (C49).
Готовили 0,1 M раствор C48 (бесцветное масло, полученное выше) в этилацетате и проводили скрининг солей. В настоящем изобретении описано только образование сульфата. Смесь серной кислоты (25 мкмоля) и раствора субстрата (0,1 M, 250 мкл, 25 мкмоля) нагревали при 45°C в течение 1 ч, давали охладиться до комнатной температуры и перемешивали в течение 15 ч. Полученную суспензию обрабатывали метанолом (примерно 150 мкл) до образования раствора; ему давали медленно испаряться в течение ночи, пока не оставалось примерно 50 мкл растворителя. Один из полученных кристаллов анализировали с помощью рентгенографического определения структуры монокристалла и устанавливали приведенную абсолютную стереохимическую конфигурацию.
Рентгенографическое определение структуры монокристалла C49
Рентгенографический анализ монокристалла
Сбор данных проводили на дифрактометре Bruker D8 Venture при комнатной температуре. Сбор данных включал сканирование по углам омега и фи.
Структуру определяли с помощью внутреннего фазирования с использованием программного обеспечения SHELX в триклинном классе пространственной группы P1. Затем структуру уточняли по методике наименьших квадратов в полноматричном приближении. Обнаружены все атомы, не являющиеся атомами водорода, и их положения уточнены с использованием параметров анизотропного смещения.
Атомы водорода, расположенные на азоте, определены по карте разностей Фурье и их положение уточнено с ограничениями на расстояния. Остальные атомы водорода помещали в рассчитанных положениях и им было разрешено смещаться на их атомах-носителях. Заключительное уточнение включало параметры изотропного смещения для всех атомов водорода.
Асимметричная ячейка состоит из двух молекул протонированного C48, одной молекулы дважды депротонированной серной кислоты и одной молекулы полностью занятой воды. Таким образом, структура представляет собой гемисульфат и полугидрат. Хлорфторфенильное кольцо разупорядочено и моделировано с занятостью 60/40, где кольцо находится в двух зеркальных положениях.
Анализ абсолютной структуры с помощью методик правдоподобия (Hooft, 2008) проводили с использованием PLATON (Spek). Результаты показывают, что абсолютная структура определена правильно; расчетом по методике показано, что вероятность правильности структуры равна 100,0. Параметр Хоофта найден равным 0,061 с оцененным стандартным отклонением, равным 0,004 и параметр Парсона найден равным 0,063 с оцененным стандартным отклонением, равным 0,005.
Конечный индекс R равнялся 3,1%. Конечная разность Фурье подтвердила, что нет отсутствующей или неправильно расположенной электронной плотности.
Данные для соответствующего кристалла, собранные данные и информация по уточнению представлены в таблице E. Координаты атомов, длины связей, валентные углы и параметры смещения представлены в таблицах F - H.
Программное обеспечение и литература
SHELXTL, Version 5.1, Bruker AXS, 1997.
PLATON, A. L. Spek, J. Appl. Cryst. 2003, 36, 7-13.
MERCURY, C. F. Macrae, P. R. Edington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler, and J. van de Streek, J. Appl. Cryst. 2006, 39, 453-457.
OLEX2, O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, and H. Puschmann, J. Appl. Cryst. 2009, 42, 339-341.
R. W. W. Hooft, L. H. Straver, and A. L. Spek, J. Appl. Cryst. 2008, 41, 96-103.
H. D. Flack, Acta Cryst. 1983, A39, 867-881.
Таблица E. Данные для кристалла и уточнения структуры C49.
Таблица F. Координаты атомов (×104) и эквивалентные параметры изотропного смещения (Å2×103) для C49. U(eq) определяется, как одна треть следа ортогонализованного тензора Uij.
Таблица G. Длины связей [Å] и валентные углы [°] для C49.
Преобразования симметрии, использованные для генерации эквивалентных атомов.
Таблица H. Параметры анизотропного смещения (Å2×103) для C49. Экспонента коэффициента анизотропного смещения имеет вид: −2π2[h2 a*2U11 +... + 2 h k a* b* U12 ].
Стадия 4. Синтез метил-2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат (C50).
Раствор C48 (500 мг пенообразного белого твердого вещества, полученного выше, с поправкой на содержание п-толуолсульфоновой кислоты: 1,25 ммоля) в ацетонитриле (6 мл) обрабатывали N, N-диизопропилэтиламином (0,68 мл, 3,9 ммоля) и перемешивали в течение 5 мин при 45°C. После добавления P15 (319 мг, 1,08 ммоля) перемешивание при 45°C продолжали в течение 7,25 ч, затем реакционную смесь разбавляли водой (6 мл) и ацетонитрилом (2 мл) при 45°C. Полученной гетерогенной смеси давали охладиться до комнатной температуры и перемешивали в течение 72 ч. Дополнительно добавляли воду (5 мл) и после еще 30 мин перемешивания твердое вещество собирали фильтрованием и промывали смесью ацетонитрила и воды (15:85, 3×5 мл) и получали C50 в виде белого твердого вещества со светло-розовым оттенком. Выход: 605 мг, 1,02 ммоля, 82%. LCMS m/z 592,0♦ [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 8,17 (d, J=1,6 Hz, 1H), 7,96 (dd, J=8,5, 1,5 Hz, 1H), 7,73 (d, J=8,4 Hz, 1H), 7,51 (dd, J=8,0, 8,0 Hz, 1H), 7,19 (br s, 1H), 7,18-7,14 (m, 2H), 6,85-6,79 (m, 1H), 6,76-6,71 (m, 2H), 5,26-5,18 (m, 1H), 4,73 (dd, компонент системы ABX, J=15,3, 5,9 Hz, 1H), 4,67 (dd, компонент системы ABX, J=15,3, 3,5 Hz, 1H), 4,63-4,55 (m, 1H), 4,38 (ddd, J=9,1, 6,0, 5,9 Hz, 1H), 3,94 (s, 5H), 3,03-2,89 (m, 2H), 2,77-2,65 (m, 2H), 2,51-2,39 (m, 1H), 2,34-2,20 (m, 2H), 1,91-1,76 (m, 4H).
Стадия 5. Синтез 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты (5, свободная кислота).
Суспензию C50 (595 мг, 1,00 ммоля) в метаноле (10 мл) нагревали при 45°C и обрабатывали водным раствором гидроксида натрия (1 M; 2,01 мл, 2,01 ммоля). Через 21 ч при 45°C реакционной смеси давали охладиться до комнатной температуры; затем ее обрабатывали водным раствором лимонной кислоты (1 M, 1 мл), устанавливали pH, равный 5-6. Добавляли воду (10 мл) и смесь перемешивали в течение 1 ч, затем твердое вещество собирали фильтрованием. Его промывали смесью метанола и воды (1:10, 3×5 мл) и получали твердое вещество (433 мг). Порцию этого вещества (300 мг) перемешивали смесью гептана и этилацетата (1:3, 5 мл) при 40°C в течение 1 ч; после охлаждения до комнатной температуры при постоянном перемешивании твердое вещество собирали фильтрованием и промывали смесью гептана и этилацетата (3:1, 3×3 мл) и получали 5, свободную кислоту, в виде белого твердого вещества. Выход: 260 мг, 0,450 ммоля, что соответствовало 65% для всей реакции. LCMS m/z 578,0♦ [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 12,75 (v br s, 1H), 8,26 (br s, 1H), 7,79 (dd, J=8,4, 1,6 Hz, 1H), 7,66-7,56 (m, 3H), 7,40 (dd, J=8,3, 2,0 Hz, 1H), 7,35 (s, 1H), 6,87-6,75 (m, 3H), 5,13-5,03 (m, 1H), 4,76 (dd, компонент системы ABX, J=15,3, 7,2 Hz, 1H), 4,62 (dd, компонент системы ABX, J=15,2, 2,8 Hz, 1H), 4,46-4,38 (m, 1H), 4,34 (ddd, J=9,0, 5,9, 5,8 Hz, 1H), 3,84 (AB квартет, JAB=13,5 Hz, ΔνAB=67,7 Hz, 2H), 3,00 (br d, J=11,2 Hz, 1H), 2,84 br (d, J=11,3 Hz, 1H), 2,71-2,56 (m, 2H), 2,45-2,34 (m, 1H), 2,28-2,08 (m, 2H), 1,84-1,65 (m, 4H).
То, что это вещество обладало такой же абсолютной конфигурацией, как в приведенном выше примере 5, устанавливали путем сопоставления его биологической активности с активностью 4 и 5: в анализе 2 этот образец свободной кислоты 5 обладал значением EC50, равным 25 нМ (среднее геометрическое значение 3 повторных анализов). В анализе 2 активность аммониевых солей соединения примера 4 и примера 5 составляла >20000 нМ (2 повторных анализа) и 20 нМ (среднее геометрическое значение 3 повторных анализов) соответственно.
Синтез 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли соединения примера 5
1,3-Дигидрокси-2-(гидроксиметил)пропан-2-аминий 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат (1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевая соль 5).
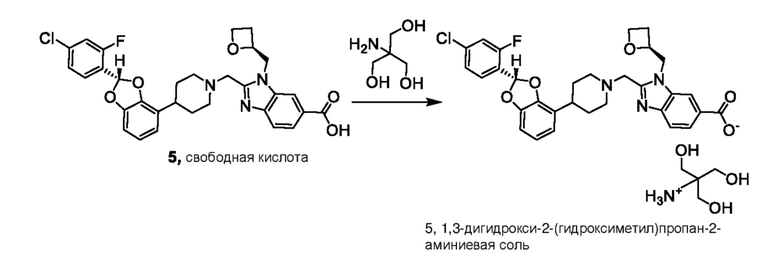
Смесь свободной кислоты 5 (0,50 г, 0,86 ммоля) в тетрагидрофуране (4 мл) обрабатывали водным раствором 2-амино-2-(гидроксиметил)пропан-1,3-диола (Tris, 1,0 M; 0,5 мл, 1,0 ммоля). Через 20 ч смесь концентрировали в вакууме этанолом (2×6 мл). Смесь обрабатывали этанолом (4 мл). После перемешивания в течение 48 ч твердое вещество собирали фильтрованием, промывали этанолом (2×10 мл) и сушили в вакууме и получали 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевую соль 5 в виде белого твердого вещества. Выход: 410 мг, 0,586 ммоля, 68%. 1H NMR (600 MHz, DMSO-d6), характеристические пики: δ 8,19 (s, 1H), 7,78 (br d, J=8,4 Hz, 1H), 7,62-7,58 (m, 2H), 7,55 (d, J=8,3 Hz, 1H), 7,40 (dd, J=8,4, 2,0 Hz, 1H), 7,35 (s, 1H), 6,85-6,80 (m, 2H), 6,79 (dd, J=6,9, 2,4 Hz, 1H), 5,11-5,05 (m, 1H), 4,73 (dd, J=15,2, 7,2 Hz, 1H), 4,60 (dd, J=15,3, 2,9 Hz, 1H), 4,45-4,39 (m, 1H), 4,34 (ddd, J=9,0, 6,0, 5,8 Hz, 1H), 3,91 (d, J=13,5 Hz, 1H), 3,74 (d, J=13,5 Hz, 1H), 2,99 (br d, J=11,1 Hz, 1H), 2,85 (br d, J=11,3 Hz, 1H), 2,68-2,59 (m, 2H), 2,44-2,37 (m, 1H), 2,25-2,18 (m, 1H), 2,17-2,10 (m, 1H), 1,80-1,69 (m, 4H). Температура плавления=от 168°C до 178°C.
Примеры 6 и 7
2-({4-[(2R)-2-(4-Хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (6) и 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (7)

Стадия 1. Синтез п-толуолсульфоната 4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидина (C13).
Раствор P2 (150 мг, 0,335 ммоля) и моногидрата п-толуолсульфоновой кислоты (159 мг, 0,836 ммоля) в этилацетате (2,0 мл) перемешивали при 60°C в течение 3,5 ч. Реакционную смесь концентрировали в вакууме и получали C13 в виде коричневого масла, которое сразу использовали на следующей стадии. LCMS m/z 348,1♦ [M+H]+.
Стадия 2. Синтез метил-2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C51).
К суспензии C13 (из предыдущей стадии; ≤0,335 ммоля) и карбоната калия (232 мг, 1,68 ммоля) в ацетонитриле (5,0 мл) добавляли P15 (99,1 мг, 0,336 ммоля). Реакционную смесь перемешивали при 60°C в течение 10 ч, затем ее фильтровали и фильтрат концентрировали в вакууме. Затем остаток (390 мг) объединяли с продуктом аналогичной реакции, проведенной с использованием C13 (≤0,11 ммоля), его разбавляли водой (20 мл) и экстрагировали смесью дихлорметана и метанола (10:1, 3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и обрабатывали с помощью препаративной тонкослойной хроматографии (элюент: 1:1 дихлорметан/метанол) и получали C51, смесь диастереоизомеров, в виде бесцветного масла. Суммарный выход: 80,6 мг, 0,133 ммоля, 30% за 2 стадии. LCMS m/z 606,2♦ [M+H]+.
Стадия 3. Выделение метил-2-({4-[(2R)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C52) и метил-2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C53).
Разделение C51 (180 мг, 0,297 ммоля) на компоненты-диастереоизомеры проводили с помощью повторяющейся SFC [колонка: Chiral Technologies Chiralpak AD, 10 мкм; подвижная фаза: 65:35 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся диастереоизомер обозначали, как C52. Выход: 61,2 мг, 0,101 ммоля, 34%. LCMS m/z 627,9♦ [M+Na+]. Время удерживания: 5,03 мин (колонка: Chiral Technologies Chiralpak AD-3, 4,6×150 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: этанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 5,5 мин, затем непрерывно при 40% B в течение 3,0 мин; скорость потока: 2,5 мл/мин).
Второй элюирующийся диастереоизомер обозначали, как C53. По данным анализа это вещество было загрязнено соответствующим этиловым эфиром; его направляли на стадию гидролиза (для получения 7) в виде этой смеси. Выход: 40,0 мг, 66,0 мкмоля, 22%. LCMS m/z 606,0♦ [M+H]+. Время удерживания: 5,19 мин (условия проведения анализа такие же, как использованные для C52).
Показанные абсолютные стереохимические конфигурации по диоксолану определяли по корреляции активности 7 с активностью образца свободной кислоты 7, синтезированной из промежуточного продукта P3 (см. ниже, альтернативный синтез соединения примера 7, свободная кислота); абсолютную стереохимическую конфигурацию P3 анализировали с помощью рентгенографического определения структуры монокристалла C8 (см. выше).
Стадия 4. Синтез 2-({4-[(2R)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (6).
Водный раствор гидроксида лития (2 M; 0,990 мл, 1,98 ммоля) добавляли к раствору C52 (60 мг, 99 мкмоля) в смеси метанола (1,0 мл) и тетрагидрофурана (1,0 мл) и реакционную смесь перемешивали при 20°C в течение 16 ч. Трифторуксусную кислоту добавляли, пока значение pH реакционной смеси не становилось равным 7, затем ее концентрировали в вакууме и остаток очищали с помощью HPLC с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксида аммония в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 29% до 49% B) и получали 6 в виде белого твердого вещества. Выход: 14,4 мг. 23,6 мкмоля, 24%. LCMS m/z 592,0♦ [M+H]+. 1H NMR (400 MHz, метанол-d4), характеристические пики: δ 8,35 (d, J=1,3 Hz, 1H), 7,97 (dd, J=8,5, 1,5 Hz, 1H), 7,67 (d, J=8,5 Hz, 1H), 7,58 (dd, J=8,3, 8,3 Hz, 1H), 7,28 (dd, J=10,9, 2,0 Hz, 1H), 7,21 (br dd, J=8,4, 1,9 Hz, 1H), 6,81-6,75 (m, 1H), 6,74-6,68 (m, 2H), 5,33-5,25 (m, 1H), 4,72 (dd, J=15,4, 2,7 Hz, 1H), 4,49 (dt, J=9,1, 6,0 Hz, 1H), 4,03 (AB квартет, JAB=13,9 Hz, ΔνAB=47,8 Hz, 2H), 3,14 (br d, J=11 Hz, 1H), 3,02 (br d, J=11,5 Hz, 1H), 2,88-2,78 (m, 1H), 2,77-2,68 (m, 1H), 2,60-2,50 (m, 1H), 2,47-2,32 (m, 2H), 2,03 (d, J=1,1 Hz, 3H), 2,01-1,87 (m, 2H), 1,87-1,78 (br m, 2H).
Стадия 5. Синтез 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (7).
Водный раствор гидроксида лития (2 M; 0,642 мл, 1,28 ммоля) добавляли к раствору C53 (38,9 мг, 64,2 мкмоля) в смеси метанола (1,0 мл) и тетрагидрофурана (1,0 мл). Затем реакционную смесь перемешивали при 20°C в течение 16 ч, значение pH устанавливали равным 7 путем добавления трифторуксусной кислоты, концентрировали в вакууме и очищали с помощью HPLC с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксида аммония в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 0% до 80% B) и получали 7 в виде белого твердого вещества. Выход: 25,1 мг, 41,2 мкмоля, 64%. LCMS m/z 591,9♦ [M+H]+. 1H NMR (400 MHz, метанол-d4), характеристические пики: δ 8,34 (d, J=1,5 Hz, 1H), 7,98 (dd, J=8,5, 1,6 Hz, 1H), 7,68 (d, J=8,5 Hz, 1H), 7,58 (dd, J=8,3, 8,3 Hz, 1H), 7,28 (dd, J=10,9, 2,0 Hz, 1H), 7,20 (br dd, J=8,4, 1,9 Hz, 1H), 6,81-6,74 (m, 1H), 6,74-6,67 (m, 2H), 5,33-5,23 (m, 1H), 4,73 (dd, J=15,4, 2,7 Hz, 1H), 4,68-4,61 (m, 1H), 4,48 (dt, J=9,1, 5,9 Hz, 1H), 4,05 (AB квартет, JAB=13,9 Hz, ΔνAB=44,1 Hz, 2H), 3,15 (br d, J=11,7 Hz, 1H), 3,03 (br d, J=11,6 Hz, 1H), 2,87-2,69 (m, 2H), 2,60-2,49 (m, 1H), 2,48-2,33 (m, 2H), 2,03 (br s, 3H), 2,01-1,77 (m, 4H).
Альтернативный синтез свободной кислоты соединения примера 7
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота (7, свободная кислота)
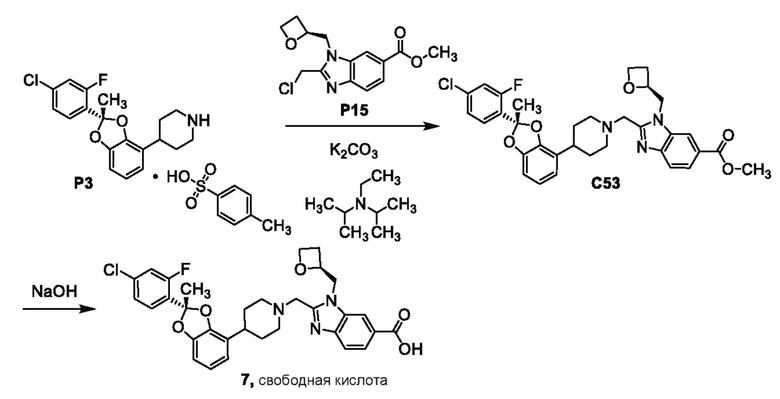 Стадия 1. Синтез метил-2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C53).
Стадия 1. Синтез метил-2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C53).
N, N-Диизопропилэтиламин (15,1 мл, 86,9 ммоля) добавляли к смеси P3 (8,22 г, 15,8 ммоля) в ацетонитриле (185 мл); после перемешивания в течение 5 мин добавляли P15 (4,57 г, 15,5 ммоля) и реакционную смесь нагревали при 45°C. Через 4 ч реакционную смесь концентрировали в вакууме до половины ее исходного объема и полученную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали водой (50 мл), сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиентный режим: от 50% до 100% этилацетата в гептане) давала C53 в виде белого твердого вещества. Выход: 8,4 г, 13,9 ммоля, 88%. LCMS m/z 606,1♦ [M+H]+. 1H NMR (600 MHz, DMSO-d6) δ 8,30 (s, 1H), 7,82 (br d, J=8,4 Hz, 1H), 7,67 (d, J=8,4 Hz, 1H), 7,58-7,53 (m, 2H), 7,33 (dd, J=8,4, 2,1 Hz, 1H), 6,80-6,76 (m, 2H), 6,76-6,72 (m, 1H), 5,14-5,07 (m, 1H), 4,81 (dd, J=15,2, 7,2 Hz, 1H), 4,67 (dd, J=15,3, 2,8 Hz, 1H), 4,51-4,44 (m, 1H), 4,37 (ddd, J=8,9, 5,9, 5,9 Hz, 1H), 3,97 (d, J=13,6 Hz, 1H), 3,87 (s, 3H), 3,78 (d, J=13,5 Hz, 1H), 3,02 (br d, J=11,1 Hz, 1H), 2,86 (br d, J=11,3 Hz, 1H), 2,74-2,60 (m, 2H), 2,48-2,41 (m, 1H), 2,29-2,22 (m, 1H), 2,21-2,14 (m, 1H), 2,02 (s, 3H), 1,83-1,73 (m, 2H), 1,73-1,64 (m, 2H).
Стадия 2. Синтез 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты (7, свободная кислота).
Смесь C53 (8,40 г, 14,0 ммоля) в метаноле (135 мл) нагревали при 45°C, и обрабатывали водным раствором гидроксида натрия (1 M; 27,7 мл, 27,7 ммоля). Через 20 ч реакционную смесь концентрировали в вакууме до половины ее исходного объема. Полученную смесь разбавляли водой (100 мл), и водный раствор лимонной кислоты (1 M, 15 мл) использовали для установления pH равным 5-6. Полученное твердое вещество отфильтровывали, промывали водой (2×15 мл) и переносили в делительную воронку в виде раствора в этилацетате (50 мл); оставшуюся воду удаляли таким образом. Органический слой сушили над сульфатом магния, фильтровали, объединяли с четырьмя партиями, полученными ранее по аналогичной методике (количество C53, использованного в этих реакциях, равнялось 987 мг, 1,63 ммоля; 1,15 г, 1,90 ммоля; 8,57 г, 14,1 ммоля; и 12,6 г, 20,8 ммоля) и концентрировали в вакууме. Полученное липкое твердое вещество обрабатывали с помощью 10% этилацетата в гептане (500 мл). Через 4 ч твердое вещество собирали фильтрованием и промывали с помощью 10% этилацетата в гептане (2×25 мл) и получали 7, свободной кислоты в виде белого твердого вещества. Выход 29,4 г, 0,527 ммоля, 74% для суммарных реакций. LCMS 592,2♦ [M+H]+. 1H NMR (600 MHz, DMSO-d6): δ 12,74 (br s, 1H), 8,28 (s, 1H), 7,80 (br d, J=8,4 Hz, 1H), 7,64 (d, J=8,4 Hz, 1H), 7,59-7,52 (m, 2H), 7,33 (dd, J=8,4, 2,1 Hz, 1H), 6,81-6,76 (m, 2H), 6,76-6,72 (m, 1H), 5,14-5,07 (m, 1H), 4,79 (dd, J=15,3, 7,3 Hz, 1H), 4,65 (dd, J=15,2, 2,8 Hz, 1H), 4,51-4,45 (m, 1H), 4,38 (ddd, J=9,0, 5,9, 5,9 Hz, 1H), 3,96 (br d, J=13,6 Hz, 1H), 3,78 (br d, J=13,5 Hz, 1H), 3,02 (br d, J=11,1 Hz, 1H), 2,86 (br d, J=11,1 Hz, 1H), 2,74-2,60 (m, 2H), 2,48-2,41 (m, 1H), 2,29-2,21 (m, 1H), 2,21-2,14 (m, 1H), 2,02 (s, 3H), 1,83-1,74 (m, 2H), 1,74-1,64 (m, 2H). То, что это вещество обладало такой же абсолютной конфигурацией, как в приведенном выше примере 7, устанавливали путем сопоставления его биологической активности с активностью 6 и 7: в анализе 2 этот образец свободной кислоты 7 обладал значением EC50, равным 4,3 нМ (среднее геометрическое значение 3 повторных анализов). В анализе 2 активность аммониевых соединения примера 6 и примера 7 составляла 2400 нМ (среднее геометрическое значение 5 повторных анализов) и 2,9 нМ (среднее геометрическое значение 8 повторных анализов) соответственно.
Синтез 7S-1. Синтез 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли соединения примера 7
1,3-Дигидрокси-2-(гидроксиметил)пропан-2-аминий 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат (7, 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевая соль).
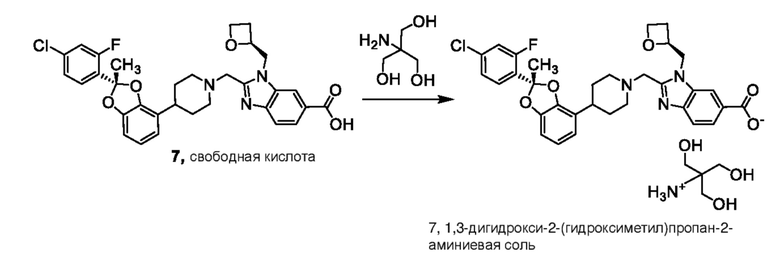
Смесь свободной кислоты 7 (2,00 г, 3,38 ммоля) в тетрагидрофуране (16 мл) обрабатывали водным раствором 2-амино-2-(гидроксиметил)пропан-1,3-диола (Tris,1,0 M; 3,55 мл, 3,55 ммоля). Через 18 ч реакционную смесь концентрировали в вакууме и обрабатывали этанолом (30 мл). Затем эту смесь перемешивали в течение 23 ч, твердое вещество собирали фильтрованием и промывали этилацетатом (2×10 мл) и получали 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевую соль 7 в виде белого твердого вещества. Выход: 1,41 г, 1,98 ммоля, 59%. LCMS m/z 592,3♦ [M+H]+. 1H NMR (600 MHz, DMSO-d6), характеристические пики: δ 8,20 (s, 1H), 7,79 (d, J=8,4 Hz, 1H), 7,59-7,52 (m, 3H), 7,33 (br d, J=8,5 Hz, 1H), 6,81-6,72 (m, 3H), 5,14-5,07 (m, 1H), 4,76 (dd, J=15,2, 7,2 Hz, 1H), 4,63 (br d, J=15,4 Hz, 1H), 4,50-4,44 (m, 1H), 4,37 (ddd, J=8,9, 5,9, 5,9 Hz, 1H), 3,94 (d, J=13,4 Hz, 1H), 3,76 (d, J=13,4 Hz, 1H), 3,01 (br d, J=11,1 Hz, 1H), 2,86 (br d, J=11,2 Hz, 1H), 2,73-2,60 (m, 2H), 2,5-2,41 (m, 1H), 2,27-2,20 (m, 1H), 2,20-2,13 (m, 1H), 2,02 (s, 3H), 1,83-1,64 (m, 4H). Температура плавления=от 175°C до 180°C.
Синтез 7S-2. Альтернативный синтез 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли соединения примера 7
3,3 M Раствор 2-амино-2-(гидроксиметил)-1,3-пропандиола (1,0 экв., 1,93 л) а воде добавляли к раствору 7, свободной кислоты (3,74 кг) в изопропаноле (20 л) при 65°C. Дополнительно добавляли изопропанол (19 л), затем метанол (19 л), поддерживая температуру равной 65°C. Смесь медленно охлаждали до 45°C в течение 2 ч, затем выдерживали при 45°C в течение не менее 12 ч. Затем смесь охлаждали до 5°C в течение 3 ч, затем выдерживали при 5°C в течение не менее 3 ч. Затем смесь фильтровали и твердое вещество собирали, промывали этилацетатом (2×10 мл) и получали 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевую соль 7 в виде белого твердого вещества (выход: 3,64 кг, 80,9%). Получали данные LCMS и 1H NMR, которые были в основном такими же, как для синтеза 7S-1, приведенного выше.
Сбор данных порошковой рентгенографии (PXRD) для формы I 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли соединения примера 7
Белую твердую соль с Tris соединения примера 7 (синтез 7S-1 и синтез 7S-2) исследовали с помощью PXRD и установили, что она представляет собой кристаллическое вещество (которое обозначено, как форма I этой безводной кристаллической формы). Анализ с помощью порошковой рентгенографии проводили с использованием дифрактометра Bruker AXS D8 Endeavor, снабженного источником излучения Cu. Щель расходимости устанавливали на 15 мм с непрерывным освещением. Дифрагированное излучение регистрировали детектором PSD-Lynx Eye с углом раскрытия детектора PSD, установленным равным 2,99 градусов. Напряжение и силу тока на рентгеновской трубке устанавливали равными 40 кВ и 40 мА соответственно. Данные собирали при длине волны Cu (CuKᾱ=1,5418 λ) в тета-тета гониометре от 3,0 до 40,0 градусов 2-тета с использованием размера шага, равного 0,01 градуса, и длительности шага, равной 1,0 с. Антирассеивающий экран устанавливали на фиксированное расстояние, равное 1,5 мм. При сборе данных образцы вращали. Образцы готовили путем их помещения в кремниевый держатель образца с низким уровнем фона и при сборе их вращали. Данные собирали с использованием программного обеспечения Bruker DIFFRAC Plus и анализы проводили с помощью программного обеспечения EVA diffract plus. Файл с данными PXRD не обрабатывали до поиска пиков. С использованием алгоритма поиска пиков в программном обеспечении EVA пики, выбранные с пороговым значением 1, использовали для проведения предварительного отнесения пиков. Для обеспечения правильности регулировки проводили вручную; результаты автоматических отнесений проверяли визуально и положения пиков корректировали по максимумам пиков. Обычно выбирали пики с относительной интенсивностью ≥ 3%. Обычно пики, которые не разрешены или согласовывались с шумом, не выбирали. Типичная погрешность для положения пика по данным PXRD в USP установлена равной до +/- 0,2° 2-тета (USP-941). Одна дифракционная картина наблюдалась постоянно и приведена на фиг. 1. Перечень дифракционных пиков, описанных углами 2θ, и относительных интенсивностей с относительной интенсивностью ≥ 3,0% в PXRD образца, полученного с помощью синтеза 7S-2, приведен ниже в таблице X1.
Таблица X1
Примеры 8 и 9
2-({4-[2-(4-Циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, DIAST-X1 (8) [из C56]; и 2-({4-[2-(4-Циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, DIAST-X2 (9) [из C57]

Стадия 1. Синтез п-толуолсульфоната 3-фтор-4-[2-метил-4-(пиперидин-4-ил)-1,3-бензодиоксол-2-ил]бензонитрила (C54).
К раствору P4 (161 мг, 0,367 ммоля) в этилацетате (8 мл) добавляли п-толуолсульфоновую кислоту (158 мг, 0,919 ммоля) и реакционную смесь перемешивали при 65°C в течение 16 ч. Удаление растворителя в вакууме давало C54 в виде темно-желтого смолообразного вещества; это вещество сразу использовали на следующей стадии.
Стадия 2. Синтез метил-2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C55).
К раствору C54 (из предыдущей стадии; ≤0,367 ммоля) в ацетонитриле (3,7 мл) добавляли карбонат калия (219 мг, 1,58 ммоля), затем P15 (115 мг, 0,390 ммоля). Реакционную смесь перемешивали при 50°C в течение 20 ч, затем ее разбавляли этилацетатом (10 мл) и фильтровали. Осадок на фильтре промывали этилацетатом (3×10 мл) и объединенные фильтраты концентрировали в вакууме. Хроматография на силикагеле (градиентный режим: от 0% до 100% этилацетата в петролейном эфире) давала C55 в виде темно-желтого масла. Выход: 191,0 мг, 0,320 ммоля, 87% за 2 стадии. LCMS m/z 619,1 [M+Na+].
Стадия 3. Выделение метил-2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата, ENT-1 (C56), и метил-2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата, ENT-2 (C57).
Разделение C55 (191 мг, 0,320 ммоля) на его компоненты-стереоизомеры по диоксолану проводили с помощью SFC [колонка: Chiral Technologies ChiralCel OD, 5 мкм; подвижная фаза: 3:2 диоксид углерода/2-пропанол]. Первый элюирующийся изомер, полученный в виде белого смолообразного вещества, обозначали, как ENT-1 (C56). Выход: 114 мг; это вещество содержало остаточный этанол. LCMS m/z 597,1 [M+H]+. Время удерживания 4,40 мин (колонка: Chiral Technologies ChiralCel OD-3, 4,6×100 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: 2-пропанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 4,5 мин, затем непрерывно при 40% B в течение 2,5 мин; скорость потока: 2,8 мл/мин).
Второй элюирующийся изомер повторно очищали с помощью SFC [колонка: Chiral Technologies ChiralCel OD, 5 мкм; подвижная фаза: 55:45 диоксид углерода/(2-пропанол, содержащий 0,1% гидроксида аммония)] и получали бесцветное смолообразное вещество, которое обозначали, как ENT-2 (C57). Выход: 50 мг, 83,8 мкмоля, 26%. LCMS m/z 597,1 [M+H]+. Время удерживания 4,74 мин (условия проведения анализа такие же, как использованные для C56).
Стадия 4. Синтез 2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, DIAST-X1 (8) [из C56].
Раствор C56 (114 мг, 0,191 ммоля) в ацетонитриле (10 мл) обрабатывали водным раствором 1,3,4,6,7,8-гексагидро-2H-пиримидо[1,2-a]пиримидина (0,97 M, 394 мкл, 0,382 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 23 ч. Дополнительно добавляли водный раствор 1,3,4,6,7,8-гексагидро-2H-пиримидо[1,2-a]пиримидина (0,97 M, 394 мкл, 0,382 ммоля) и перемешивание продолжали в течение 6 ч, затем значение pH осторожно устанавливали равным 7-8 путем добавления 1 M хлористоводородной кислоты. После удаления летучих веществ в вакууме, очистку проводили с помощью HPLC с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксида аммония в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 30% до 50% B) и получали 8 в виде белого твердого вещества. Выход: 22,2 мг, 38,1 мкмоля, 20%. LCMS m/z 583,3 [M+H]+. 1H NMR (400 MHz, метанол-d4): δ 8,19 (d, J=1,4 Hz, 1H), 7,94 (dd, J=8,4, 1,5 Hz, 1H), 7,77 (dd, J=7,7, 7,7 Hz, 1H), 7,64 (dd, J=10,6, 1,6 Hz, 1H), 7,58 (d, J=8,4 Hz, 1H), 7,57 (dd, J=8,0, 1,5 Hz, 1H), 6,81-6,75 (m, 1H), 6,75-6,68 (m, 2H), 5,34-5,25 (m, 1H), 4,73 (dd, J=15,3, 3,0 Hz, 1H), 4,67-4,59 (m, 1H), 4,49 (dt, J=9,2, 6,0 Hz, 1H), 3,96 (AB квартет, JAB=13,7 Hz, ΔνAB=41,2 Hz, 2H), 3,06 (br d, J=11 Hz, 1H), 2,95 (br d, J=11 Hz, 1H), 2,87-2,76 (m, 1H), 2,71 (tt, J=12,0, 3,9 Hz, 1H), 2,61-2,50 (m, 1H), 2,36-2,21 (m, 2H), 2,06 (s, 3H), 1,95-1,72 (m, 4H).
Стадия 5. Синтез 2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, DIAST-X2 (9) [из C57].
Раствор C57 (50 мг, 84 мкмоля) в ацетонитриле (10 мл) обрабатывали водным раствором 1,3,4,6,7,8-гексагидро-2H-пиримидо[1,2-a]пиримидина (0,97 M; 173 мкл, 0,168 ммоля). Реакционную смесь перемешивали при комнатной температуре (примерно 25°C) в течение 16 ч, затем добавляли дополнительное количество водного раствора 1,3,4,6,7,8-гексагидро-2H-пиримидо[1,2-a]пиримидина (0,97 M; 173 мкл, 0,168 ммоля) и перемешивание продолжали при 25°C в течение 29 ч. Затем значение pH реакционной смеси осторожно устанавливали равным 7-8 путем добавления 1 M хлористоводородной кислоты; полученную смесь концентрировали в вакууме и обрабатывали с помощью HPLC с обращенной фазой (колонка: Xtimate™ C18, 5 мкм; подвижная фаза A: 0,05% гидроксида аммония в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 27% до 67% B) и получали 9 в виде белого твердого вещества. Выход: 18,0 мг, 30,9 мкмоля, 37%. LCMS m/z 583,3 [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 8,36-8,33 (m, 1H), 7,97 (dd, J=8,5, 1,5 Hz, 1H), 7,78 (dd, J=7,7, 7,7 Hz, 1H), 7,70-7,63 (m, 2H), 7,57 (dd, J=8,0, 1,5 Hz, 1H), 6,83-6,76 (m, 1H), 6,76-6,71 (m, 2H), 5,34-5,25 (m, 1H), 4,95-4,85 (m, 1H, предположительно; частично закрыт пиком воды), 4,73 (dd, компонент системы ABX, J=15,3, 2,7 Hz, 1H), 4,68-4,60 (m, 1H), 4,50 (dt, J=9,2, 6,0 Hz, 1H), 4,02 (AB квартет, JAB=13,8 Hz, ΔνAB=48,2 Hz, 2H), 3,13 (br d, J=11 Hz, 1H), 3,01 (br d, J=11,5 Hz, 1H), 2,89-2,78 (m, 1H), 2,78-2,68 (m, 1H), 2,60-2,50 (m, 1H), 2,45-2,30 (m, 2H), 2,07 (br s, 3H), 2,00-1,86 (m, 2H), 1,83 (m, 2H).
Пример 10
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, DIAST-X2 (10) [из P9]
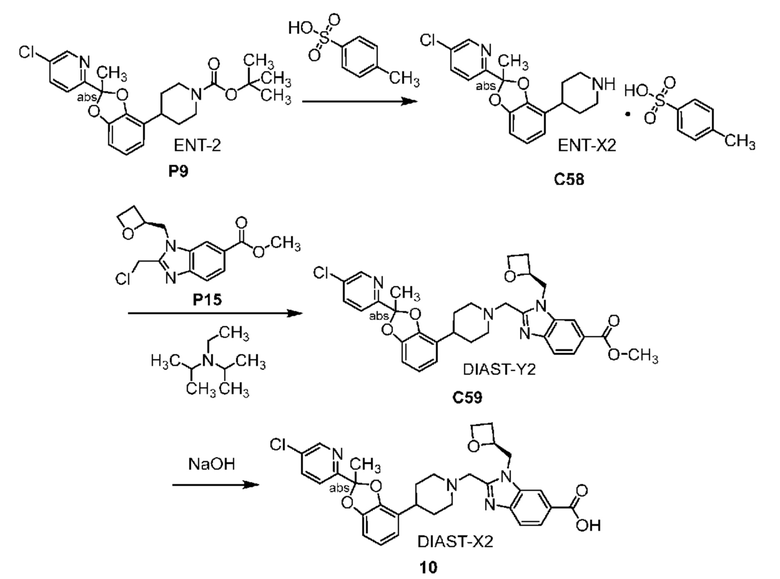
Стадия 1. Синтез п-толуолсульфоната 5-хлор-2-[2-метил-4-(пиперидин-4-ил)-1,3-бензодиоксол-2-ил]пиридина, ENT-X2 (C58) [из P9].
Раствор P9 (228 мг, 0,529 ммоля) в этилацетате (2,7 мл) обрабатывали моногидратом п-толуолсульфоновой кислоты (116 мг, 0,610 ммоля) и реакционную смесь нагревали при 50°C в течение 16 ч. Затем ее перемешивали при комнатной температуре в течение ночи, затем осадок собирали фильтрованием и промывали смесью этилацетата и гептана (1:1, 2×20 мл) и получали C58 в виде белого твердого вещества. Выход: 227 мг, 0,451 ммоля, 85%. LCMS m/z 331,0♦ [M+H]+. 1H NMR (400 MHz, DMSO-d6): δ 8,73 (d, J=2,4 Hz, 1H), 8,61-8,46 (br m, 1H), 8,35-8,18 (br m, 1H), 8,02 (dd, J=8,5, 2,5 Hz, 1H), 7,64 (d, J=8,5 Hz, 1H), 7,47 (d, J=7,8, 2H), 7,11 (d, J=7,8 Hz, 2H), 6,89-6,81 (m, 2H), 6,72 (пентет, J=4,0 Hz, 1H), 3,45-3,27 (m, 2H, предположительно; частично закрыт пиком воды), 3,10-2,91 (m, 3H), 2,28 (s, 3H), 2,02 (s, 3H), 1,97-1,80 (m, 4H).
Стадия 2. Синтез метил-2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата, DIAST-Y2 (C59) [из P9].
N, N-Диизопропилэтиламин (0,234 мл, 1,34 ммоля) добавляли к раствору C58 (225 мг, 0,447 ммоля) в ацетонитриле (2,2 мл). Затем эту смесь перемешивали в течение 5 мин при 45°C, добавляли P15 (120 мг, 0,407 ммоля) и перемешивание продолжали при 45°C в течение 16 ч, затем дополнительно добавляли P15 (11 мг, 37 мкмоля). После еще 3 ч перемешивания, реакционную смесь обрабатывали водой (2,5 мл) и давали охладиться до комнатной температуры. Дополнительно добавляли воду (5 мл) и полученную суспензию перемешивали в течение 2 ч, затем твердое вещество собирали фильтрованием и промывали смесью ацетонитрила и воды (15:85, 3×5 мл) и получали C59 в виде почти белого твердого вещества (252 мг). Это вещество, по данным анализа с помощью1H NMR содержащее некоторое количество N, N-диизопропилэтиламин сразу направляли на следующую стадию. LCMS m/z 589,1♦ [M+H]+. 1H NMR (400 MHz, хлороформ-d) 8,61 (d, J=2,3 Hz, 1H), 8,18 (d, J=1,5 Hz, 1H), 7,96 (dd, J=8,5, 1,5 Hz, 1H), 7,74 (d, J=8,5 Hz, 1H), 7,67 (dd, компонент системы ABX, J=8,4, 2,4 Hz, 1H), 7,59-7,51 (m, 1H), 6,82-6,75 (m, 1H), 6,74-6,66 (m, 2H), 5,28-5,19 (m, 1H), 4,75 (dd, компонент системы ABX, J=15,3, 6,0 Hz, 1H), 4,68 (dd, компонент системы ABX, J=15,3, 3,4 Hz, 1H), 4,67-4,58 (m, 1H), 4,41 (ddd, J=9,1, 5,9, 5,9 Hz, 1H), 3,95 (s, 2H), 3,95 (s, 3H), 3,07-2,89 (m, 2H), 2,81-2,69 (m, 2H), 2,53-2,41 (m, 1H), 2,37-2,22 (m, 2H), 2,05 (s, 3H), 1,93-1,74 (m, 4H).
Стадия 3. Синтез 2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, DIAST-X2 (10) [из P9].
Суспензию C59 (из предыдущей стадии; 250 мг, ≤0,407 ммоля) в метаноле (2 мл) нагревали при 40°C, затем добавляли водный раствор гидроксида натрия (1 M; 0,81 мл, 0,81 ммоля). Через 17 ч реакционной смеси давали охладиться до комнатной температуры и 1 M водным раствором лимонной кислоты значение pH устанавливали равным 5-6. Полученную смесь разбавляли водой (2 мл), перемешивали в течение 2 ч и экстрагировали этилацетатом (3×5 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия (5 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме и получали пенообразное твердое вещество. Это вещество переносили в смесь этилацетата и гептана (1:1, 4 мл), нагревали при 50°C и затем ему давали охладиться и перемешивали в течение ночи. Фильтрование давало 10 в виде белого твердого вещества. Выход: 179 мг, 0,311 ммоля, 76% за 2 стадии. LCMS m/z 575,1♦ [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 12,73 (br s, 1H), 8,71 (d, J=2,5 Hz, 1H), 8,27 (d, J=1,5 Hz, 1H), 8,00 (dd, J=8,5, 2,5 Hz, 1H), 7,80 (dd, J=8,4, 1,6 Hz, 1H), 7,64 (d, J=8,4 Hz, 1H), 7,60 (d, J=8,5 Hz, 1H), 6,83-6,72 (m, 3H), 5,14-5,06 (m, 1H), 4,77 (dd, компонент системы ABX, J=15,2, 7,2 Hz, 1H), 4,63 (dd, компонент системы ABX, J=15,2, 2,8 Hz, 1H), 4,50-4,42 (m, 1H), 4,37 (ddd, J=9,0, 5,9, 5,9 Hz, 1H), 3,85 (AB квартет, JAB=13,6 Hz, ΔνAB=71,5 Hz, 2H), 3,01 (br d, J=11,2 Hz, 1H), 2,85 (br d, J=11,2 Hz, 1H), 2,74-2,57 (m, 2H), 2,47-2,38 (m, 1H), 2,29-2,10 (m, 2H), 2,01 (s, 3H), 1,81-1,64 (m, 4H).
Синтез 10S-1. Синтез 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли соединения примера 10
Синтез 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминий-2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата, DIAST-X2 (1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли 10) [из P9].
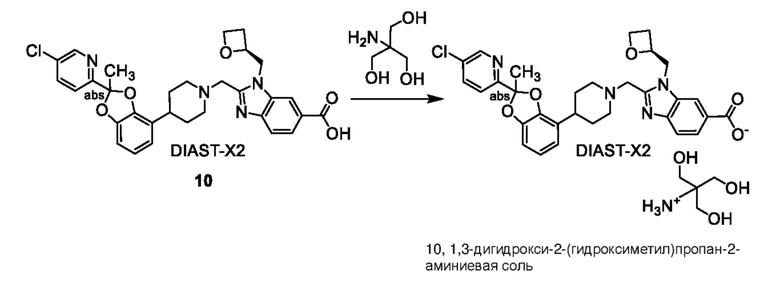
Смесь 10 (1,54 г, 2,68 ммоля) в тетрагидрофуране (10 мл) обрабатывали водным раствором 2-амино-2-(гидроксиметил)пропан-1,3-диола (Tris, 1,0 M; 2,81 мл, 2,81 ммоля). Через 24 ч реакционную смесь концентрировали в вакууме этанолом (2×50 мл). Остаток обрабатывали этанолом (15 мл). После перемешивания в течение 20 ч твердое вещество собирали фильтрованием и промывали холодным этанолом (5 мл) и получали 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевую соль 10 в виде белого твердого вещества. Выход: 1,41 г, 2,03 ммоля, 76%. LCMS m/z 575,3♦ [M+H]+. 1H NMR (600 MHz, DMSO-d6) δ 8,71 (d, J=2,5 Hz, 1H), 8,21 (br s, 1H), 8,00 (dd, J=8,5, 2,5 Hz, 1H), 7,79 (br d, J=8,4 Hz, 1H), 7,60 (d, J=8,5 Hz, 1H), 7,57 (d, J=8,4 Hz, 1H), 6,82-6,73 (m, 3H), 5,13-5,07 (m, 1H), 4,74 (dd, J=15,3, 7,2 Hz, 1H), 4,61 (dd, J=15,3, 2,9 Hz, 1H), 4,49-4,43 (m, 1H), 4,37 (ddd, J=9,0, 5,9, 5,9 Hz, 1H), 3,93 (d, J=13,6 Hz, 1H), 3,75 (d, J=13,5 Hz, 1H), 3,01 (br d, J=11,3 Hz, 1H), 2,86 (br d, J=11,4 Hz, 1H), 2,73-2,59 (m, 2H), 2,48-2,37 (m, 1H), 2,27-2,20 (m, 1H), 2,19-2,12 (m, 1H), 2,01 (s, 3H), 1,82-1,66 (m, 4H). Температура плавления=от 184°C до 190°C.
Синтез 10S-2. Альтернативный синтез 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли соединения примера 10
Смесь 10 (8,80 г, 15,3 ммоля) в 2-метилтетрагидрофуране (90 мл) концентрировали в вакууме в роторном испарителе при 37°C на водяной бане для уменьшения полного объема до ~54 мл. Изопропанол (90 мл) добавляли к смеси и затем повторно концентрировали полученную смесь до объема, равного ~54 мл. К смеси добавляли изопропанол (135 мл), затем добавляли водный раствор трис-амина (3M; 5,0 мл, 0,98 экв.). Полученную смесь/раствор перемешивали при температуре окружающей среды; и твердый осадок начинал образовываться не позже, чем через ~15 мин. Затем смесь перемешивали при температуре окружающей среды в течение еще 5 ч. Полученную смесь/суспензию охлаждали до 0°C и охлажденную суспензию перемешивали в течение еще примерно 2 ч. Суспензию фильтровали и промывали холодным изопропанолом (3×15 мл). Собранному твердому веществу давали сохнуть на воздухе в воронке для сбора в течение примерно 90 мин и затем переносили в вакуумный сушильный шкаф дл сушки в течение ночи. Через ~16 ч при 50°C/в вакууме при давлении 23 мм рт. ст. (слабой подаче азота) 8,66 г 10, 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли получали в виде белого твердого вещества; 99,8% площади по данным UPLC (выход:12,5 ммоля, 81%). Получали данные LCMS и 1H NMR, которые были в основном такими же, как для синтеза 10S-1, приведенного выше.
Сбор данных порошковой рентгенографии (PXRD) для формы A 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли соединения примера 10 (также известной, как форма A безводной соли с Tris соединения примера 10)
Белую твердую соль с Tris соединения примера 10 (синтез 10S-1 и синтез 10S-2) исследовали с помощью PXRD и установили, что она представляет собой кристаллическое вещество (которое обозначено, как форма A). Анализ с помощью порошковой рентгенографии проводили с использованием дифрактометра Bruker AXS D8 Endeavor, снабженного источником излучения Cu. Щель расходимости устанавливали на 15 мм с непрерывным освещением. Дифрагированное излучение регистрировали детектором PSD-Lynx Eye с углом раскрытия детектора PSD, установленным равным 2,99 градусов. Напряжение и силу тока на рентгеновской трубке устанавливали равными 40 кВ и 40 мА соответственно. Данные собирали при длине волны Cu (CuKᾱ=1,5418 λ) в тета-тета гониометре от 3,0 до 40,0 градусов 2-тета с использованием размера шага, равного 0,01 градуса, и длительности шага, равной 1,0 с. Антирассеивающий экран устанавливали на фиксированное расстояние, равное 1,5 мм. При сборе данных образцы вращали. Образцы готовили путем их помещения в кремниевый держатель образца с низким уровнем фона и при сборе их вращали. Данные собирали с использованием программного обеспечения Bruker DIFFRAC Plus и анализы проводили с помощью программного обеспечения EVA diffract plus. Файл с данными PXRD не обрабатывали до поиска пиков. С использованием алгоритма поиска пиков в программном обеспечении EVA пики, выбранные с пороговым значением 1, использовали для проведения предварительного отнесения пиков. Для обеспечения правильности регулировки проводили вручную; результаты автоматических отнесений проверяли визуально и положения пиков корректировали по максимумам пиков. Обычно выбирали пики с относительной интенсивностью ≥ 3%. Обычно пики, которые не разрешены или согласовывались с шумом, не выбирали. Типичная погрешность для положения пика по данным PXRD в USP установлена равной до +/- 0,2° 2-тета (USP-941). Перечень дифракционных пиков, описанных углами 2θ, и относительных интенсивностей с относительной интенсивностью ≥ 3,0% в PXRD образца, полученного с помощью синтеза 10S-2 приведен ниже в таблице X2.
Таблица X2
Пример 11
Формиат 1-(2-метоксиэтил)-2-({4-[2-метил-2-(пиридин-3-ил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1H-бензимидазол-6-карбоновой кислоты (11)
Всю последовательность синтеза проводили в библиотечном формате.
Стадия 1. Синтез метил-1-(2-метоксиэтил)-2-({4-[2-метил-2-(пиридин-3-ил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1H-бензимидазол-6-карбоксилат (C60).
Смесь P14 (44 мг, 100 мкмоля) и 3-этинилпиридина (21 мг, 200 мкмоля) в толуоле (800 мкл) обрабатывали бикарбонатом натрия (100 мкмоля), затем додекакарбонилом трирутения (6 мг, 9 мкмоля). Затем реакционный сосуд закрывали и встряхивали при 120°C в течение 16 ч. Удаление растворителя с помощью концентратора Speedvac® давало C60, который сразу направляли на следующую стадию.
Стадия 2. Синтез формиата 1-(2-метоксиэтил)-2-({4-[2-метил-2-(пиридин-3-ил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1H-бензимидазол-6-карбоновой кислоты (11).
Водный раствор гидроксида натрия (1,0 M; 200 мкл, 200 мкмоля) добавляли к раствору C60 (из предыдущей стадии, ≤100 мкмоля) в смеси метанола (400 мкл) и тетрагидрофурана (400 мкл). Реакционный сосуд закрывали и встряхивали при 80°C в течение 16 ч, затем реакционную смесь выпаривали с помощью концентратора Speedvac® и очищали с помощью HPLC с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,225% муравьиной кислоты в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 12% до 52% B) и получали 11. Выход: 2,2 мг, 4,2 мкмоля, 4% за 2 стадии. LCMS m/z 529 [M+H]+. Время удерживания: 2,47 мин (колонка: Waters XBridge C18, 2,1×50 мм, 5 мкм; подвижная фаза A: 0,0375% трифторуксусной кислоты в воде; подвижная фаза B: 0,01875% трифторуксусной кислоты в ацетонитриле; градиентный режим: от 1% до 5% B за 0,6 мин; от 5% до 100% B за 3,4 мин; скорость потока: 0,8 мл/мин).
Пример 12
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[2-(диметиламино)этил]-1H-бензимидазол-6-карбоновая кислота (12) 
Всю последовательность синтеза проводили в библиотечном формате.
Стадия 1. Синтез метил-3-{[2-(диметиламино)этил]амино}-4-нитробензоата (C61).
Метил-3-фтор-4-нитробензоат (0,2 M раствор в N, N-диметилформамиде; 1 мл, 200 мкмоля) обрабатывали N, N-диметилэтан-1,2-диамином (18 мг, 200 мкмоля) и N, N-диизопропилэтиламином (78 мг, 600 мкмоля). Затем реакционный сосуд закрывали и встряхивали при 50°C в течение 16 ч, затем реакционную смесь выпаривали с помощью концентратора Speedvac® и получали C61. Это вещество сразу направляли на следующую стадию.
Стадия 2. Синтез метил-4-амино-3-{[2-(диметиламино)этил]амино}бензоаат (C62).
Цинковую пыль активировали разбавленной хлористоводородной кислотой. Метанол (2 мл) добавляли к C61 (из предыдущей стадии, ≤200 мкмоля), затем водный раствор хлорида кальция (1,0 M; 200 мкл, 200 мкмоля) и активированную цинковую пыль (130 мг, 2,0 ммоля). Реакционный сосуд закрывали и встряхивали при 70°C в течение 16 ч, затем реакционную смесь фильтровали. Фильтрат концентрировали с помощью концентратора Speedvac® и остаток переносили в воду (2 мл) и затем экстрагировали этилацетатом (2×3 мл). Объединенные органические слои выпаривали с помощью концентратора Speedvac® и получали C62 (по оценкам 150 мкмоля), который сразу использовали на следующей стадии.
Стадия 3. Синтез метил-4-[({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}ацетил)амино]-3-{[2-(диметиламино)этил]амино}бензоата (C63).
Соединение P10 (41 мг, 100 мкмоля) добавляли к C62 (из предыдущей стадии, примерно 150 мкмоля) и смесь обрабатывали раствором в N, N-диметилацетамиде 2-гидроксипиридин 1-оксида и 1-[3-(диметиламино)пропил]-3-этилкарбодиимидгидрохлорида (0,1 M в каждом; 1 мл, 100 мкмоля каждого). Затем добавляли N, N-диизопропилэтиламин (39 мг, 300 мкмоля) и реакционный сосуд закрывали и встряхивали при 50°C в течение 16 ч. Затем реакционную смесь концентрировали с помощью концентратора Speedvac® и очищали с помощью препаративной тонкослойной хроматографии и получали C63, который сразу направляли на следующую стадию.
Стадия 4. Синтез метил-2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[2-(диметиламино)этил]-1H-бензимидазол-6-карбоксилата (C64).
Смесь уксусной кислоты (500 мкл) и C63 (из предыдущей стадии, ≤100 мкмоля) встряхивали в закрытом сосуде при 150°C в течение 2 ч, затем реакционную смесь выпаривали с помощью концентратора Speedvac®. Полученный C64 сразу направляли на следующую стадию.
Стадия 5. Синтез 2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[2-(диметиламино)этил]-1H-бензимидазол-6-карбоновой кислоты (12).
Раствор C64 (из предыдущей стадии, ≤100 мкмоля) в этаноле (500 мкл) обрабатывали водным раствором гидроксида лития (2,0 M; 500 мкл, 1 ммоля) и реакционную смесь встряхивали при 50°C в течение 2 ч в герметизированном сосуде. Затем pH смеси устанавливали равным 7 путем добавления 1,0 M хлористоводородной кислоты, полученную смесь концентрировали с помощью концентратора Speedvac® и затем очищали с помощью HPLC с обращенной фазой [колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: гидроксид аммония в воде (pH 10); подвижная фаза B: ацетонитрил; градиентный режим: от 25% до 65% B] и получали 12. Выход: 7,0 мг, 12 мкмоля, 12% за 3 стадии. LCMS m/z 593 [M+H]+. Время удерживания: 2,45 мин (колонка: Waters XBridge C18, 2,1×50 мм, 5 мкм; подвижная фаза A: 0,0375% трифторуксусной кислоты в воде; подвижная фаза B: 0,01875% трифторуксусной кислоты в ацетонитриле; градиентный режим: от 10% до 100% B за 4,0 мин; скорость потока: 0,8 мл/мин).
Пример 13
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-3-(1,3-оксазол-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота (13)
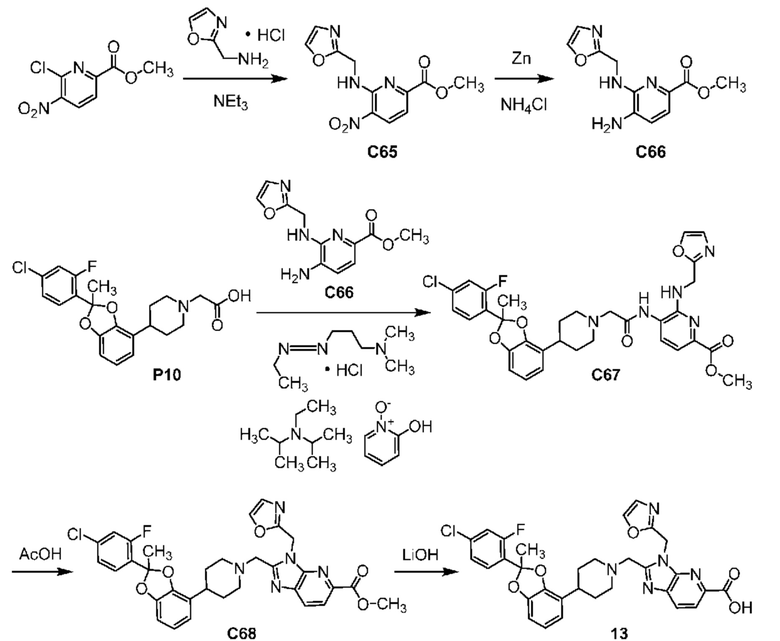 Стадия 1. Синтез метил-6-[(1,3-оксазол-2-илметил)амино]-5-нитропиридин-2-карбоксилата (C65).
Стадия 1. Синтез метил-6-[(1,3-оксазол-2-илметил)амино]-5-нитропиридин-2-карбоксилата (C65).
Триэтиламин (532 мг, 5,26 ммоля) добавляли к суспензии 1-(1,3-оксазол-2-ил)метанамингидрохлорида (236 мг, 1,75 ммоля) и метил-6-хлор-5-нитропиридин-2-карбоксилата (386 мг, 1,78 ммоля) в тетрагидрофуране (5 мл). Затем реакционную смесь перемешивали при 25°C в течение 14 ч, ее выливали в воду (30 мл) и экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме; хроматография на силикагеле (градиентный режим: от 0% до 5% метанола в дихлорметане) давала C65 в виде желтого твердого вещества. Выход: 310 мг, 1,11 ммоля, 63%. LCMS m/z 278,7 [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 8,69-8,61 (br m, 1H), 8,58 (d, J=8,4 Hz, 1H), 7,65 (d, J=0,8 Hz, 1H), 7,46 (d, J=8,4 Hz, 1H), 7,11 (d, J=1,0 Hz, 1H), 5,07 (d, J=5,3 Hz, 2H), 3,97 (s, 3H).
Остальную часть этой последовательности синтеза проводили в библиотечном формате.
Стадия 2. Синтез метил-5-амино-6-[(1,3-оксазол-2-илметил)амино]пиридин-2-карбоксилат (C66).
Водный раствор хлорида аммония (5,0 M; 400 мкл, 2,0 ммоля), затем активированный цинк (131 мг, 2,0 ммоля) добавляли к раствору C65 (56 мг, 200 мкмоля) в метаноле (2,0 мл). Затем реакционный сосуд закрывали и встряхивали при 30°C в течение 16 ч, затем реакционную смесь фильтровали. Фильтрат концентрировали с помощью концентратора Speedvac®, затем смешивали с водой (1,0 мл) и экстрагировали дихлорметаном (3×1,0 мл); объединенные органические слои выпаривали с помощью концентратора Speedvac® и получали C66, который сразу направляли на следующую стадию.
Стадия 3. Синтез метил-5-[({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}ацетил)амино]-6-[(1,3-оксазол-2-илметил)амино]пиридин-2-карбоксилата (C67).
Смесь P10 (81 мг, 200 мкмоля) и C66 (из предыдущей стадии, ≤200 мкмоля) смешивали с N, N-диметилацетамидом и затем обрабатывали N, N-диизопропилэтиламином (100 мкл, 600 мкмоля). Добавляли раствор, содержащий 1-[3-(диметиламино)пропил]-3-этилкарбодиимидгидрохлорид (0,24 M) и 2-гидроксипиридин 1-оксид (0,1 M) в N, N-диметилацетамиде (1,0 мл, содержащем 240 мкмоля 1-[3-(диметиламино)пропил]-3-этилкарбодиимидгидрохлорид и 100 мкмоля 2-гидроксипиридин 1-оксид) и реакционный сосуд закрывали и встряхивали при 50°C в течение 16 ч. Затем летучие вещества удаляли с помощью концентратора Speedvac® и остаток обрабатывали с помощью препаративной тонкослойной хроматографии и получали C67, который сразу направляли на следующую стадию.
Стадия 4. Синтез метил-2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-3-(1,3-оксазол-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоксилата (C68).
Смесь уксусной кислоты (1,0 мл) и C67 (из предыдущей стадии, ≤200 мкмоля) встряхивали при 150°C в течение 2 ч, затем реакционную смесь выпаривали с помощью концентратора Speedvac®. Полученный C68 сразу использовали на следующей стадии.
Стадия 5. Синтез 2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-3-(1,3-оксазол-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоновой кислоты (13).
Водный раствор гидроксида лития (2,0 M; 1,0 мл, 2,0 ммоля) добавляли к смеси C68 (из предыдущей стадии, ≤200 мкмоля) в тетрагидрофуране (1,0 мл). После добавления метанола (500 мкл), реакционный сосуд закрывали и встряхивали при 50°C в течение 16 ч. После удаления летучих веществ с помощью концентратора Speedvac® добавляли диметилсульфоксид (1,0 мл) и значение pH устанавливали равным 7-8 концентрированной хлористоводородной кислотой. Полученную смесь очищали с помощью HPLC с обращенной фазой [колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: гидроксид аммония в воде (pH 10); подвижная фаза B: ацетонитрил; градиентный режим: от 24% до 64% B] и получали 13. Выход: 3,9 мг, 6,5 мкмоля, 3% за 4 стадии. LCMS m/z 604 [M+H]+. Время удерживания: 3,14 мин (колонка: Waters XBridge C18, 2,1×50 мм, 5 мкм; подвижная фаза A: 0,0375% трифторуксусной кислоты в воде; подвижная фаза B: 0,01875% трифторуксусной кислоты в ацетонитриле; градиентный режим: от 1% до 5% B за 0,6 мин; от 5% до 100% B за 3,4 мин; скорость потока: 0,8 мл/мин).
Пример 14
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-метил-1H-бензимидазол-6-карбоновая кислота (14)
 Стадия 1. Синтез метил-2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-метил-1H-бензимидазол-6-карбоксилата (C69).
Стадия 1. Синтез метил-2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-метил-1H-бензимидазол-6-карбоксилата (C69).
N, N-Диизопропилэтиламин (683 мкл, 3,92 ммоля) добавляли к смеси P3 (680 мг, 1,31 ммоля) в ацетонитриле (5,2 мл); смесь перемешивали в течение 5 мин при 45°C, затем добавляли P16 (319 мг, 1,34 ммоля). Перемешивание продолжали при 45°C в течение 2,75 ч и затем добавляли воду (6 мл) и реакционной смеси давали охладиться до комнатной температуры и перемешивали в течение 30 мин. Твердые вещества собирали фильтрованием и промывали смесью ацетонитрила и воды (1:4, 3×5 мл) и получали C69 в виде белого твердого вещества. Выход: 635 мг, 1,15 ммоля, 88%. LCMS m/z 550,1♦ [M+H]+. 1H NMR (400 MHz, хлороформ-d) δ 8,15-8,12 (m, 1H), 7,97 (dd, J=8,5, 1,6 Hz, 1H), 7,74 (d, J=8,5 Hz, 1H), 7,50 (dd, J=8,2, 8,2 Hz, 1H), 7,16-7,07 (m, 2H), 6,79-6,73 (m, 1H), 6,72-6,65 (m, 2H), 3,98 (s, 3H), 3,96 (s, 3H), 3,88 (s, 2H), 3,04-2,93 (m, 2H), 2,76-2,66 (m, 1H), 2,37-2,25 (m, 2H), 2,04 (br s, 3H), 1,89-1,78 (m, 4H).
Стадия 2. Синтез 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-метил-1H-бензимидазол-6-карбоновой кислоты (14).
Смесь C69 (600 мг, 1,09 ммоля) в метаноле (11 мл) нагревали при 45°C и затем обрабатывали водным раствором гидроксида натрия (1 M; 2,2 мл, 2,2 ммоля). Через 24 ч значение pH реакционной смеси устанавливали равным 5-6 путем добавления водного раствора лимонной кислоты (1 M; 1,1 мл) и затем разбавляли водой (10 мл). Полученной смеси давали охладиться до комнатной температуры и перемешивали в течение 1 ч, затем осадившееся твердое вещество собирали фильтрованием и промывали смесью метанола и воды (1:4; 3×5 мл). Это давало 14 в виде белого твердого вещества. Выход: 535 мг, 0,998 ммоля, 92%. LCMS m/z 536,1♦ [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8,16 (d, J=1,5 Hz, 1H), 7,81 (dd, J=8,4, 1,6 Hz, 1H), 7,64 (d, J=8,4 Hz, 1H), 7,59-7,52 (m, 2H), 7,33 (dd, J=8,3, 2,1 Hz, 1H), 6,81-6,70 (m, 3H), 3,94 (s, 3H), 3,84 (s, 2H), 3,01-2,91 (m, 2H), 2,70-2,59 (m, 1H), 2,28-2,16 (m, 2H), 2,02 (s, 3H), 1,73 (m, 4H).
Примеры 15 и 16
Трифторацетат 2-{6-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-6-азаспиро[2.5]окт-1-ил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновой кислоты, DIAST-X1 (15) [из P18 через C71]; и трифторацетат 2-{6-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-6-азаспиро[2.5]окт-1-ил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновой кислоты, DIAST-X2 (16) [из P18 через C72]
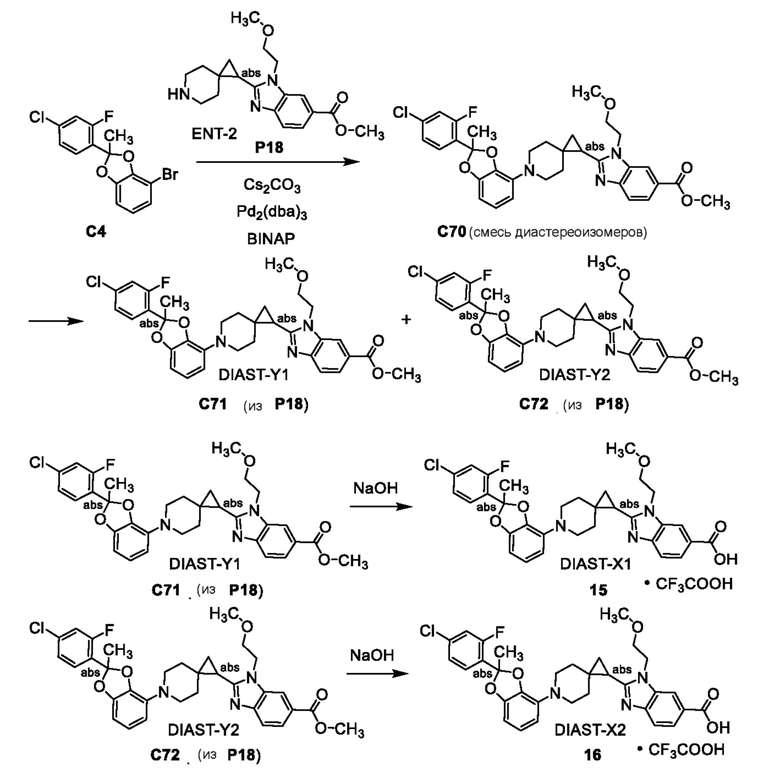
Стадия 1. Синтез метил-2-{6-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-6-азаспиро[2.5]окт-1-ил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (C70) [из P18].
Смесь P18 (240 мг, 0,699 ммоля), C4 (275 мг, 0,800 ммоля), карбоната цезия (455 мг, 1,40 ммоля), трис(дибензилиденацетон)дипалладия(0) (40,0 мг, 43,7 мкмоля) и 1,1'-бинафталин-2,2'-диилбис(дифенилфосфана) (BINAP; 52,2 мг, 83,8 мкмоля) в толуоле (5 мл) дегазировали азотом в течение 5 мин и затем перемешивали при 90°C в течение 16 ч. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме; препаративная тонкослойная хроматография (элюент: 1:1 петролейный эфир/этилацетат) давала C70, смесь диастереоизомеров, в виде желтого масла. Выход: 165 мг, 0,272 ммоля, 39%. LCMS m/z 628,1♦ [M+Na+].
Стадия 2. Выделение метил-2-{6-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-6-азаспиро[2.5]окт-1-ил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата, DIAST-Y1 (C71) [из P18]; и метил-2-{6-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-6-азаспиро[2.5]окт-1-ил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата, DIAST-Y2 (C72) [из P18].
Разделение стереоизомеров C70 (165 мг, 0,272 ммоля) по диоксолану проводили с помощью SFC [колонка: Chiral Technologies Chiralpak AD, 10 мкм; подвижная фаза: 65:35 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся изомер обозначали, как DIAST-Y1 (C71) и второй элюирующийся изомер, как DIAST-Y2 (C72); оба выделяли в виде белого твердого вещества.
C71 Выход: 55,0 мг, 90,7 мкмоля, 33%. LCMS m/z 605,9♦ [M+H]+. Время удерживания 4,47 мин (колонка: Chiral Technologies Chiralpak AD-3, 4,6×100 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: этанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 4,5 мин, затем непрерывно при 40% B в течение 2,5 мин; скорость потока: 2,8 мл/мин).
C72 Выход: 58,0 мг, 95,7 мкмоля, 35%. LCMS m/z 628,0♦ [M+Na+]. Время удерживания 4,88 мин (условия проведения анализа такие же, как использованные для C71).
Стадия 3. Синтез трифторацетата 2-{6-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-6-азаспиро[2.5]окт-1-ил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновой кислоты, DIAST-X1 (15) [из P18 через C71].
К раствору C71 (55,0 мг, 90,7 мкмоля) в смеси метанола (2,0 мл) и тетрагидрофурана (1,0 мл) добавляли водный раствор гидроксида натрия (3 M; 1,0 мл, 3,0 ммоля). Затем реакционную смесь перемешивали при 20°C в течение 2 ч, значение pH устанавливали равным 7 путем добавления 1 M хлористоводородной кислоты и полученную смесь экстрагировали смесью дихлорметана и метанола (10:1, 3×30 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. HPLC с обращенной фазой (колонка: Boston Green ODS, 5 мкм; подвижная фаза A: 0,1% трифторуксусной кислоты в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 10% до 95% B) давала 15 в виде белого твердого вещества. Выход: 35,8 мг, 50,7 мкмоля, 56%. LCMS m/z 592,3♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 8,46 (s, 1H), 8,21 (d, J=8,6 Hz, 1H), 7,78 (d, J=8,6 Hz, 1H), 7,54 (dd, J=8,3, 8,3 Hz, 1H), 7,16-7,08 (m, 2H), 6,76 (dd, J=8,2, 8,1 Hz, 1H), 6,55-6,47 (m, 2H), 4,9-4,70 (m, 2H, предположительно; частично закрыт пиком воды), 3,82 (t, J=4,9 Hz, 2H), 3,66-3,56 (m, 1H), 3,50-3,41 (m, 1H), 3,19-3,09 (m, 1H), 3,15 (s, 3H), 3,08-2,99 (m, 1H), 2,63-2,57 (m, 1H), 2,27-2,17 (m, 1H), 2,01 (s, 3H), 1,76-1,66 (m, 2H), 1,62-1,50 (m, 2H), 1,35-1,26 (m, 1H).
Стадия 4. Синтез трифторацетата 2-{6-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-6-азаспиро[2.5]окт-1-ил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновой кислоты, DIAST-X2 (16) [из P18 через C72].
К раствору C72 (58,0 мг, 95,7 мкмоля) в смеси метанола (2,0 мл) и тетрагидрофурана (1,0 мл) добавляли водный раствор гидроксида натрия (3 M; 1,0 мл, 3,0 ммоля). Затем реакционную смесь перемешивали при 20°C в течение 2 ч, значение pH устанавливали равным 7 путем добавления 1 M хлористоводородной кислоты и полученную смесь экстрагировали смесью дихлорметана и метанола (10:1, 3×30 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. HPLC с обращенной фазой (колонка: Boston Green ODS, 5 мкм; подвижная фаза A: 0,1% трифторуксусной кислоты в воде; подвижная фаза B: ацетонитрил; градиентный режим: от 35% до 95% B) давала 16 в виде белого твердого вещества. Выход: 33,4 мг, 47,3 мкмоля, 49%. LCMS m/z 592,2♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 8,53-8,50 (m, 1H), 8,25 (dd, J=8,6, 1,4 Hz, 1H), 7,80 (br d, J=8,6 Hz, 1H), 7,57 (dd, J=8,4, 8,2 Hz, 1H), 7,25 (dd, J=10,8, 2,0 Hz, 1H), 7,19 (br dd, J=8,4, 2,1 Hz, 1H), 6,77 (dd, J=8,2, 8,1 Hz, 1H), 6,55-6,50 (m, 2H), 4,9-4,72 (m, 2H, предположительно; частично закрыт пиком воды), 3,93-3,80 (m, 2H), 3,68-3,58 (m, 1H), 3,41-3,3 (m, 1H, предположительно; частично закрыт пиком растворителя), 3,25 (s, 3H), 3,22-3,12 (m, 1H), 3,07-2,97 (m, 1H), 2,67 (dd, J=8,3, 5,8 Hz, 1H), 2,28-2,17 (m, 1H), 2,01 (d, J=1,0 Hz, 3H), 1,86-1,71 (m, 2H), 1,69-1,56 (m, 2H), 1,36-1,26 (m, 1H).
Примеры 17 и 18
2-({4-[2-(4-Хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилат аммония, ENT-1 (17) и 2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилат аммония, ENT-2 (18)
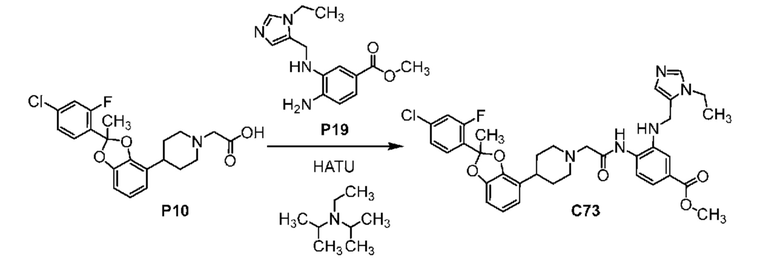

Стадия 1. Синтез метил-4-[({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}ацетил)амино]-3-{[(1-этил-1H-имидазол-5-ил)метил]амино}бензоата (C73).
O-(7-Азабензотриазол-1-ил)-N, N,N",N"-тетраметилуронийгексафторфосфат (566 мг, 1,49 ммоля) добавляли к смеси P19 (340 мг, 1,24 ммоля) в N, N-диметилформамиде (10 мл) и смесь перемешивали при 25°C в течение 10 мин. Затем добавляли раствор P10 (503 мг, 1,24 ммоля) и N, N-диизопропилэтиламин (615 мкл, 3,53 ммоля) в N, N-диметилформамиде (7,7 мл) и реакционную смесь перемешивали при 25°C в течение 16 ч, затем ее выливали в воду (10 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои последовательно промывали водным раствором хлорида аммония (3×20 мл) и насыщенным водным раствором хлорида натрия (2×20 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После очистки с помощью хроматографии на силикагеле (градиентный режим: от 0% до 5% метанола в этилацетате) получали C73 в виде бледно-коричневого смолообразного вещества. Выход: 316 мг, 0,477 ммоля, 38%. LCMS m/z 662,2♦ [M+H]+.
Стадия 2. Синтез метил-2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилата (C74).
Раствор C73 (316 мг, 0,477 ммоля) в уксусной кислоте (14 мл) перемешивали при 55°C в течение 16 ч. Растворитель удаляли в высоком вакууме и остаток очищали с помощью препаративной тонкослойной хроматографии (элюент: 10:1 дихлорметан/метанол) и получали C74 в виде бесцветного масла. Выход: 200 мг, 0,310 ммоля, 65%. LCMS m/z 644,3♦ [M+H]+.
Стадия 3. Синтез 2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилата аммония, ENT-1 (17) и 2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилата аммония, ENT-2 (18).
Смесь C74 (150 мг, 0,233 ммоля) и водного раствора гидроксида натрия (2 M; 233 мкл, 0,466 ммоля) в смеси метанола (3 мл) и тетрагидрофурана (3 мл) перемешивали при 45°C в течение 16 ч. Затем значение pH реакционной смеси устанавливали равным 7 путем добавления 1 M хлористоводородной кислоты, ее концентрировали в вакууме и получали смесь 17 и 18. Эти энантиомеры разделяли с помощью SFC [колонка: Chiral Technologies ChiralCel OD, 10 мкм; подвижная фаза: 1:1 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся энантиомер обозначали, как ENT-1 (17), и второй элюирующийся энантиомер, как ENT-2 (18); оба выделяли в виде белого твердого вещества.
17 Выход: 45,0 мг, 69,5 мкмоля, 30%. LCMS m/z 630,3♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 8,15 (br s, 1H), 8,00 (br d, J=8,4 Hz, 1H), 7,81 (s, 1H), 7,72 (d, J=8,2 Hz, 1H), 7,56 (dd, J=8,3, 8,3 Hz, 1H), 7,28 (dd, J=10,9, 2,0 Hz, 1H), 7,21 (dd, J=8,3, 2,1 Hz, 1H), 6,77 (dd, компонент системы ABC, J=8,0, 7,7 Hz, 1H), 6,69 (dd, компонент системы ABC, J=7,8, 1,2 Hz, 1H), 6,67-6,60 (m, 2H), 5,82 (s, 2H), 4,12 (q, J=7,2 Hz, 2H), 3,89 (AB квартет, JAB=14,3 Hz, ΔνAB=6,9 Hz, 2H), 3,00-2,90 (m, 2H), 2,74-2,64 (m, 1H), 2,32-2,21 (m, 2H), 2,02 (s, 3H), 1,82-1,61 (m, 4H), 1,29 (t, J=7,3 Hz, 3H). Время удерживания 5,66 мин (колонка: Chiral Technologies Chiralpak AD-3, 4,6×150 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: метанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 5,5 мин, затем непрерывно при 40% B в течение 3,0 мин; скорость потока: 2,5 мл/мин).
18 Выход: 32,8 мг, 50,7 мкмоля, 22%. LCMS m/z 630,3♦ [M+H]+. 1H NMR (400 MHz, метанол-d4) δ 8,15 (s, 1H), 8,00 (d, J=8,5 Hz, 1H), 7,81 (s, 1H), 7,72 (d, J=8,5 Hz, 1H), 7,56 (dd, J=8,3, 8,3 Hz, 1H), 7,28 (dd, J=10,9, 2,0 Hz, 1H), 7,21 (dd, J=8,3, 2,0 Hz, 1H), 6,77 (dd, компонент системы ABC, J=7,8, 7,8 Hz, 1H), 6,69 (dd, компонент системы ABC, J=7,9, 1,2 Hz, 1H), 6,67-6,60 (m, 2H), 5,82 (s, 2H), 4,12 (q, J=7,3 Hz, 2H), 3,89 (AB квартет, JAB=14,1 Hz, ΔνAB=7,4 Hz, 2H), 3,01-2,90 (m, 2H), 2,74-2,63 (m, 1H), 2,31-2,21 (m, 2H), 2,02 (s, 3H), 1,82-1,60 (m, 4H), 1,29 (t, J=7,3 Hz, 3H). Время удерживания 5,34 мин (условия проведения анализа с помощью SFC такие же, как использованные для 17).
Соединения, приведенные в таблице 1, получали по методикам, аналогичным использованным в примерах, указанных в таблице 2, с использованием подходящего промежуточного продукта (продуктов), указанного в таблице 2. Соединения очищали по методикам, рассмотренным в настоящем изобретении. Конечные соединения можно было выделить в виде нейтральных или кислых, или основных солей.
Таблица 1. Структура и названия IUPAC соединений примеров 19-102
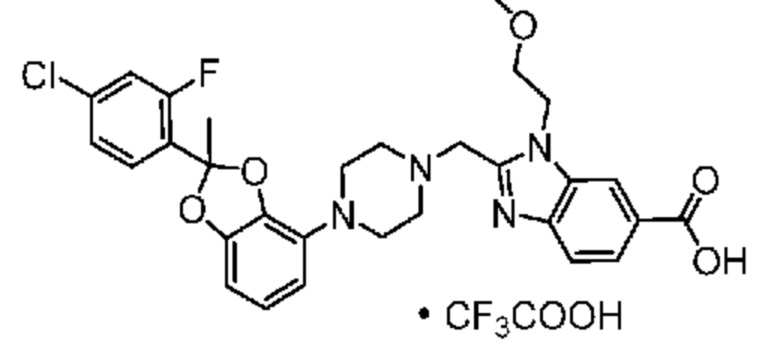
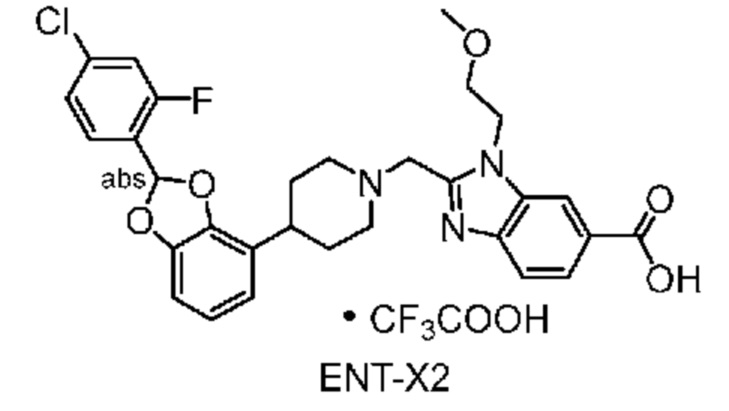

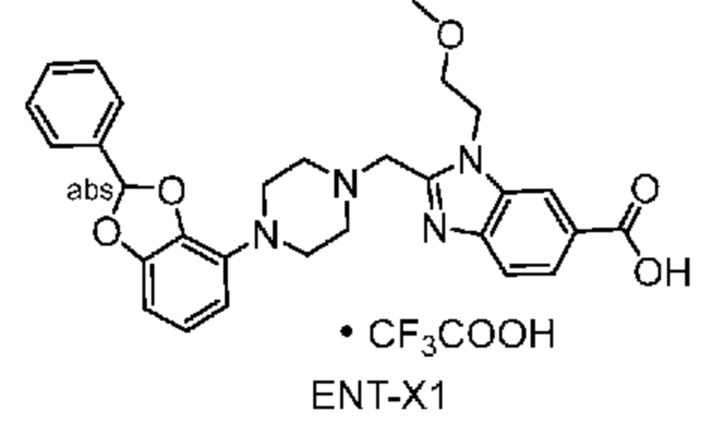


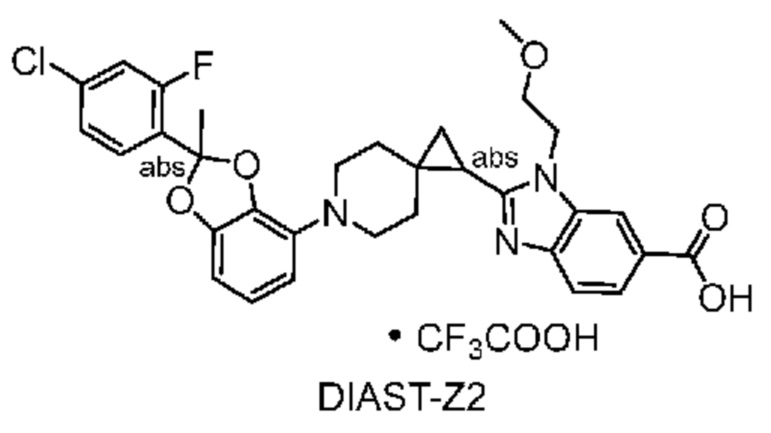
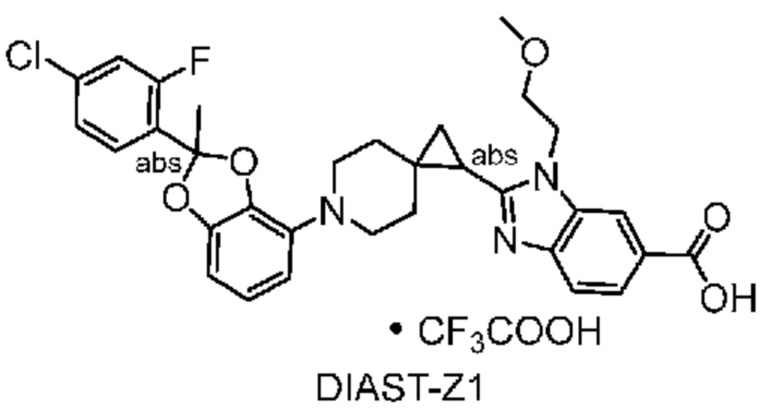
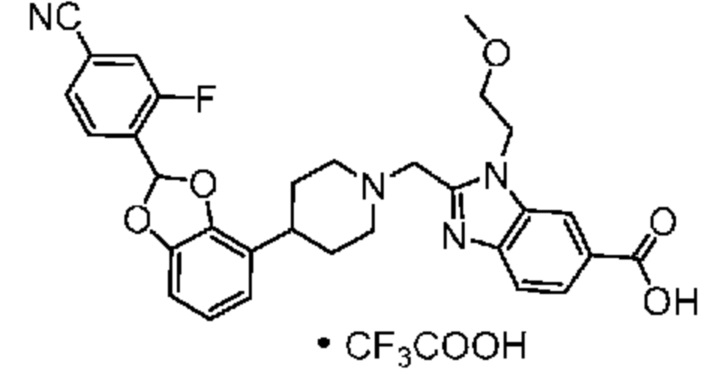
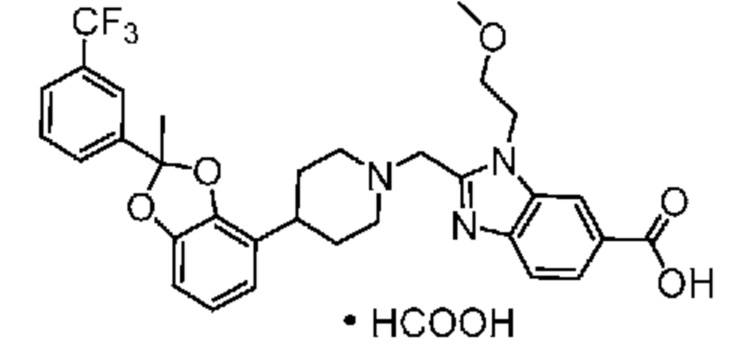

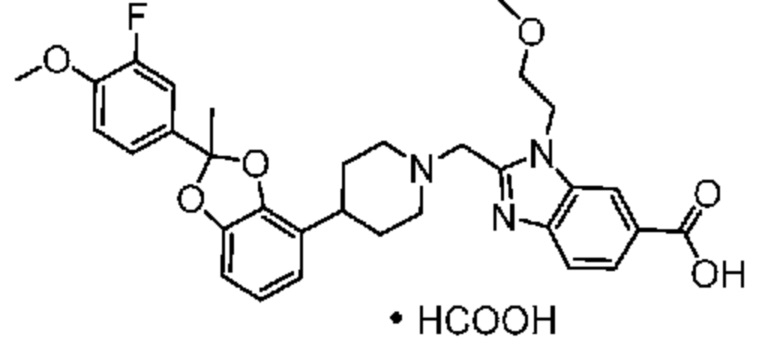



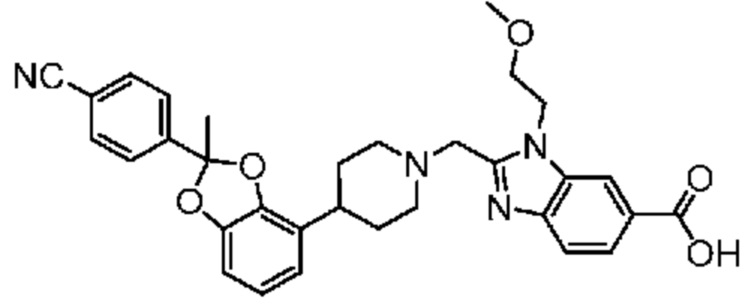

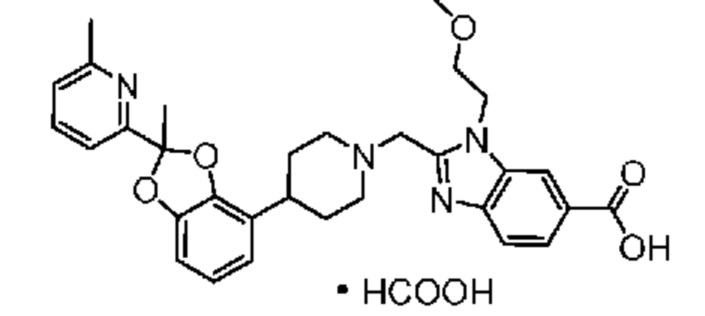

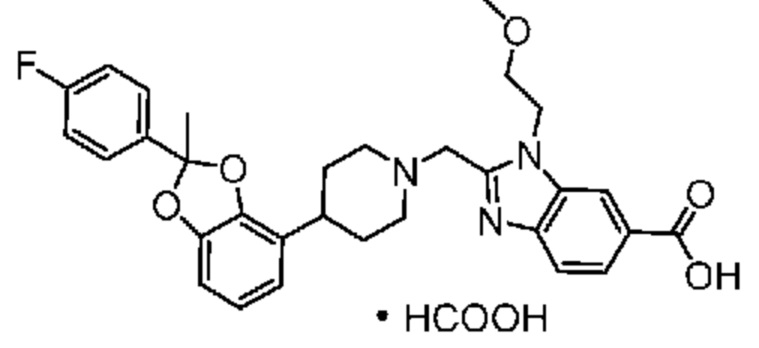
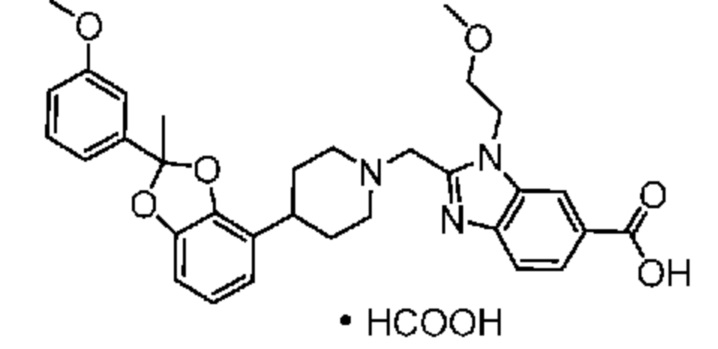
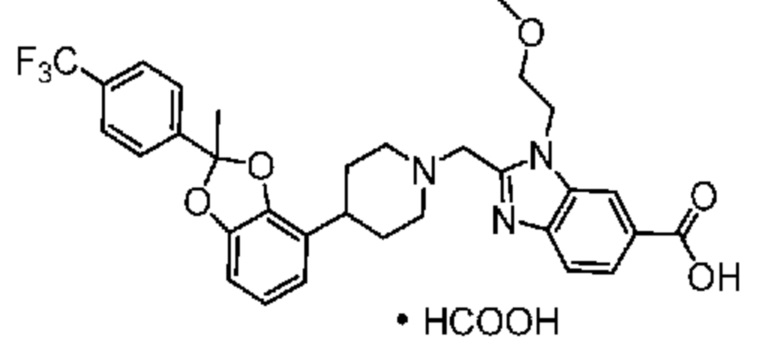

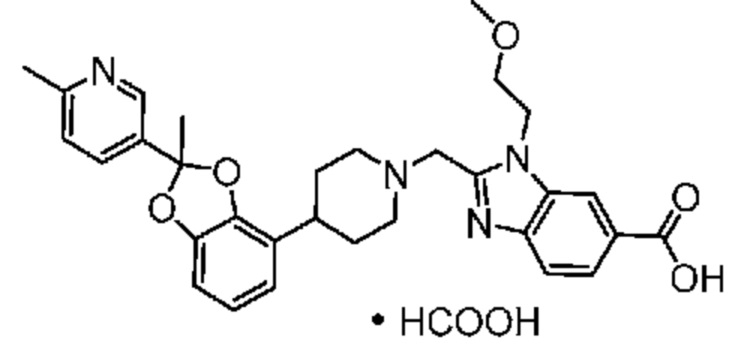


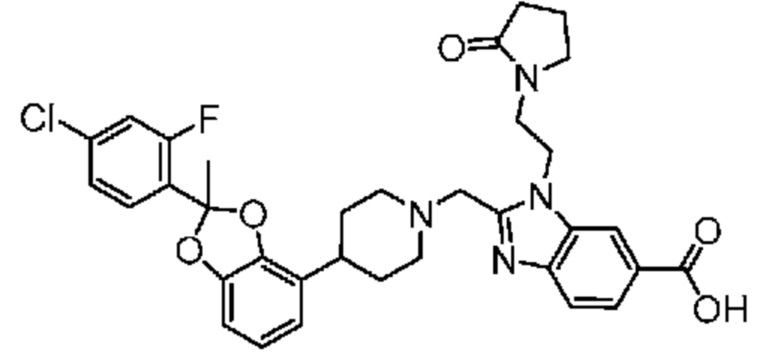

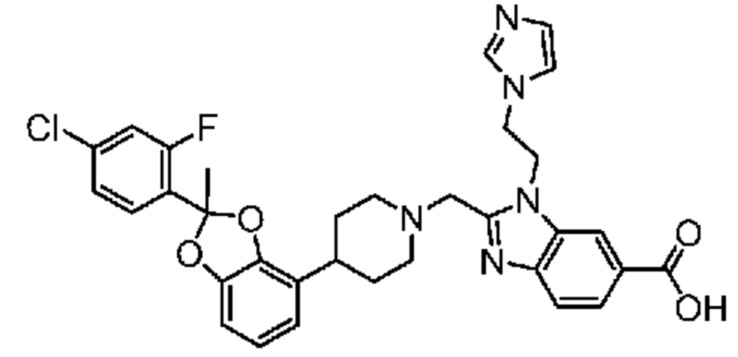

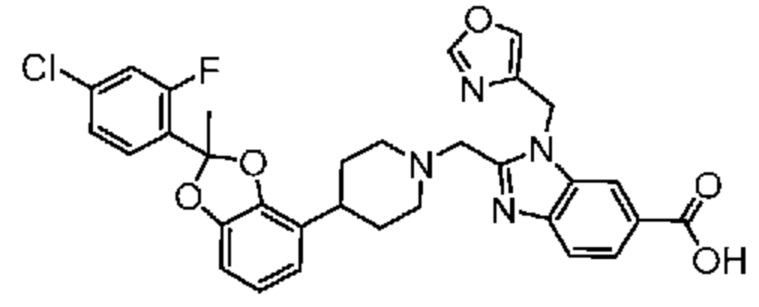

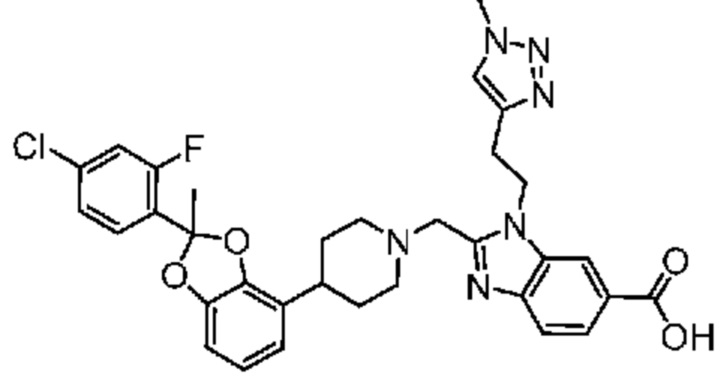

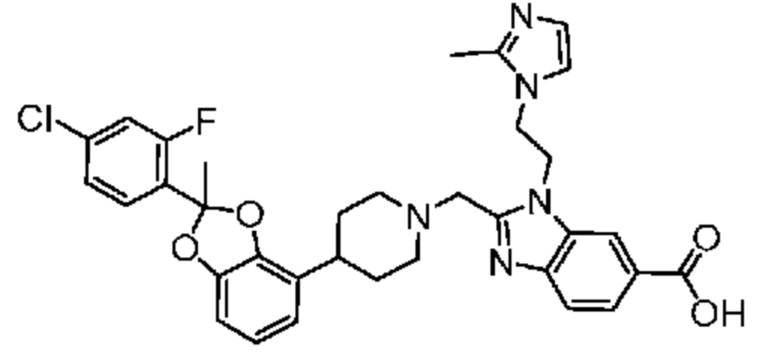
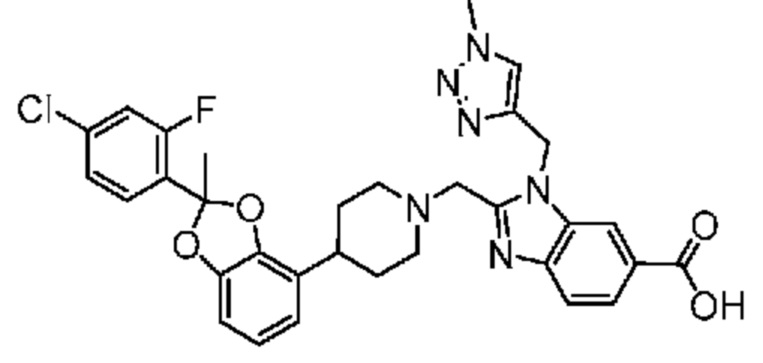
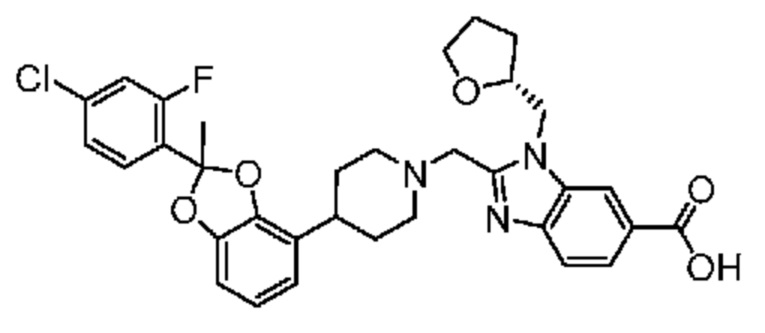



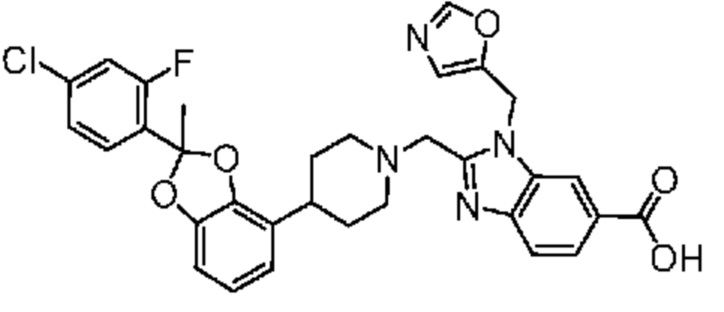
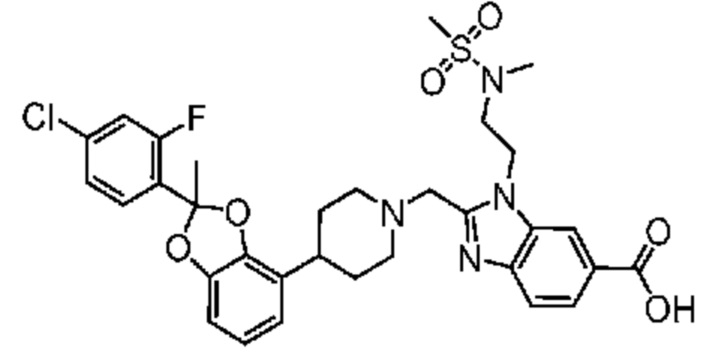
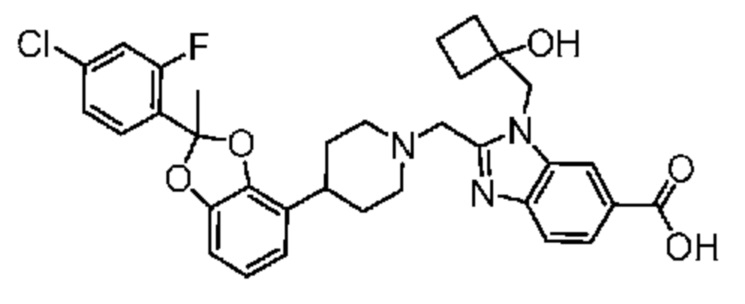
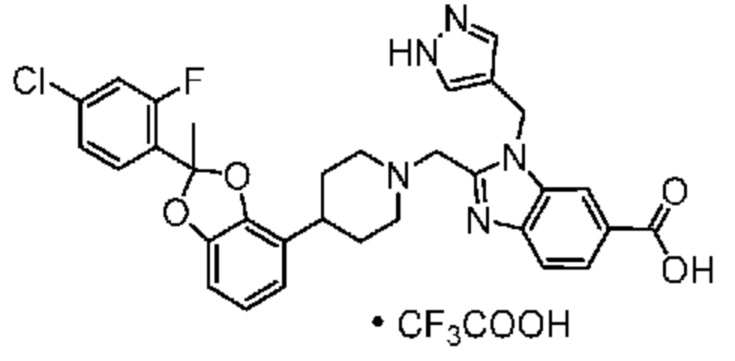
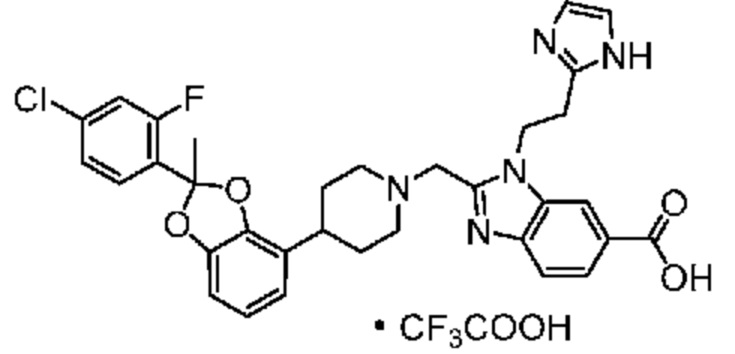
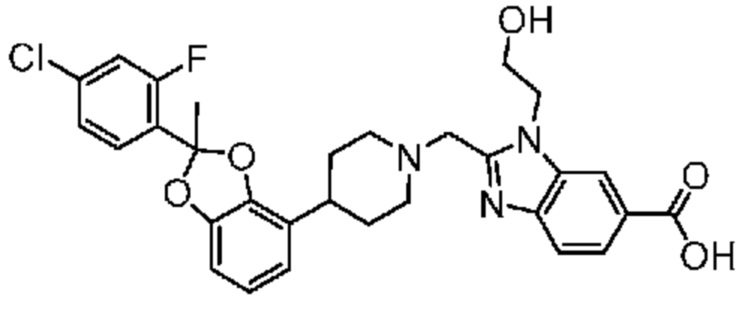
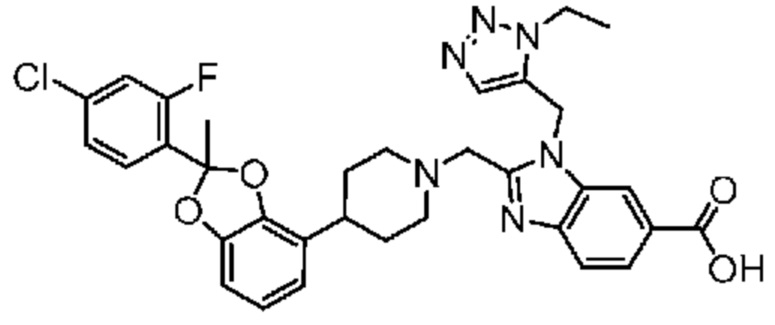
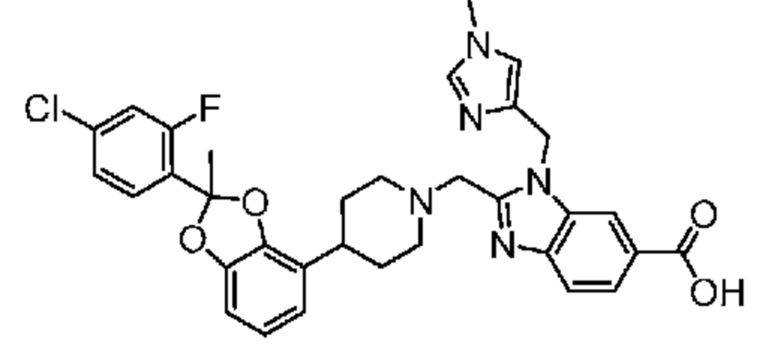
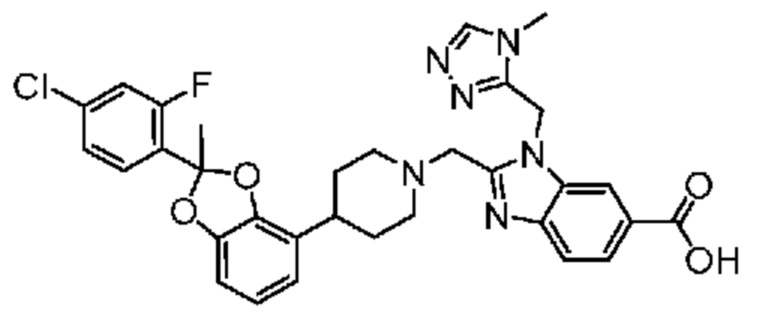
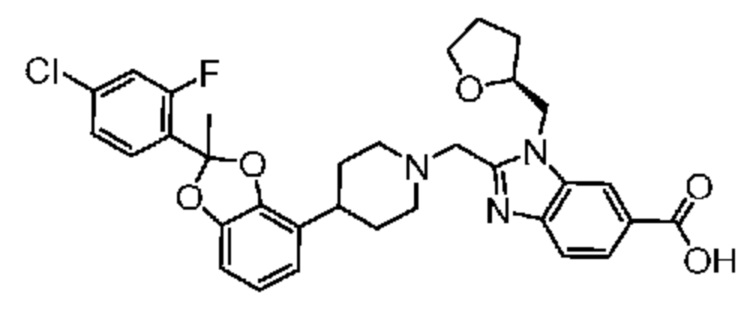
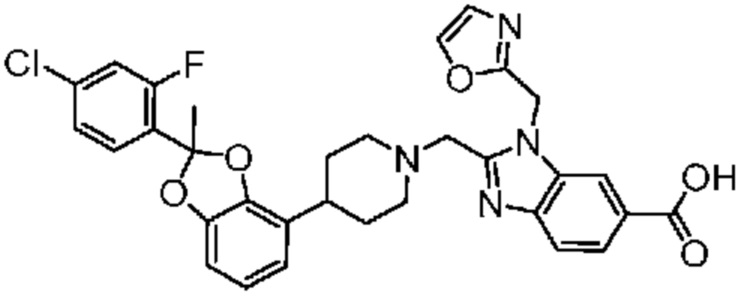
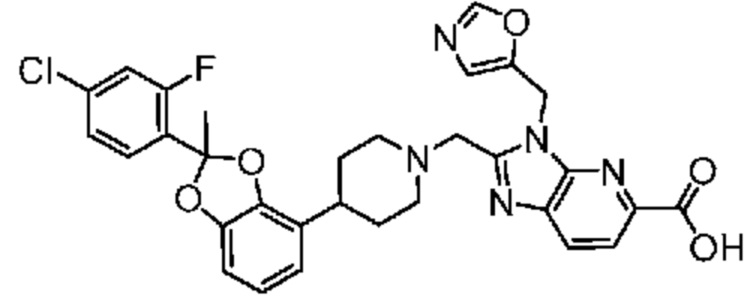

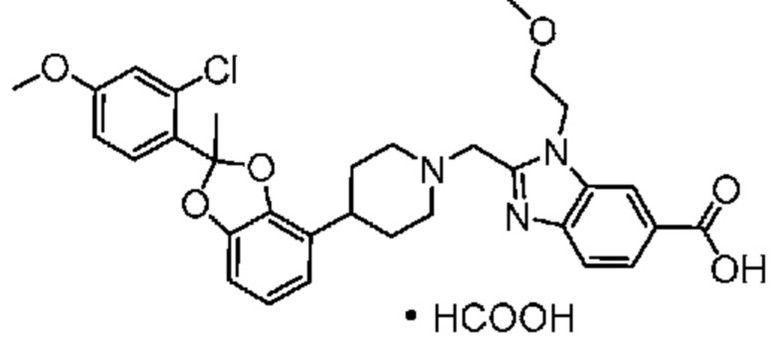

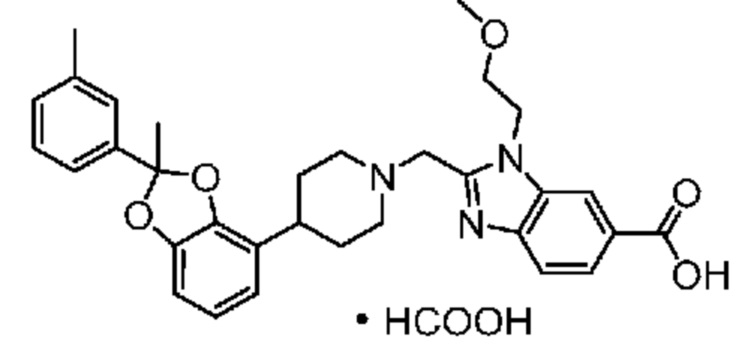
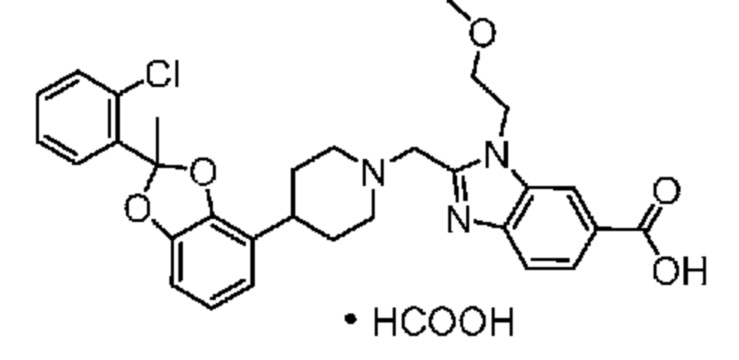
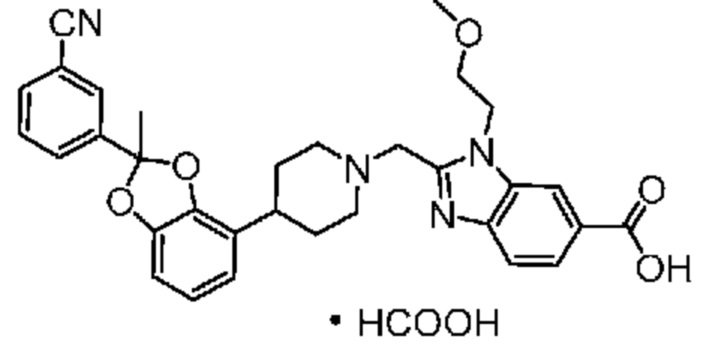
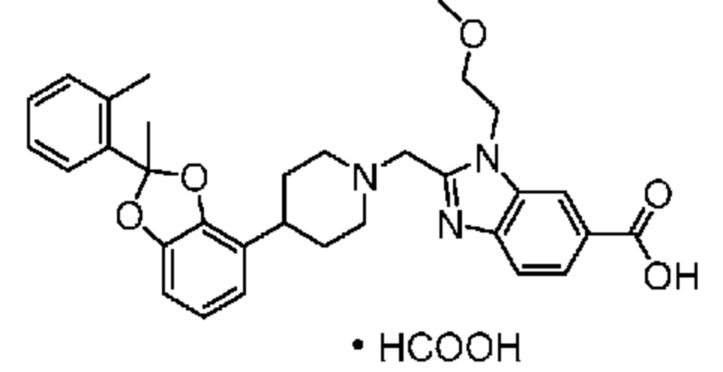

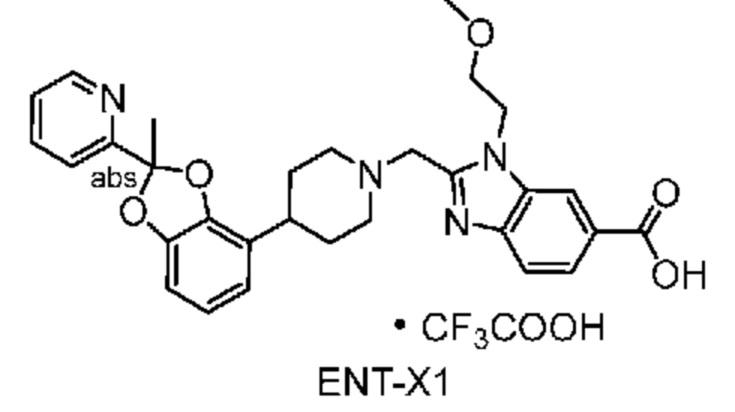
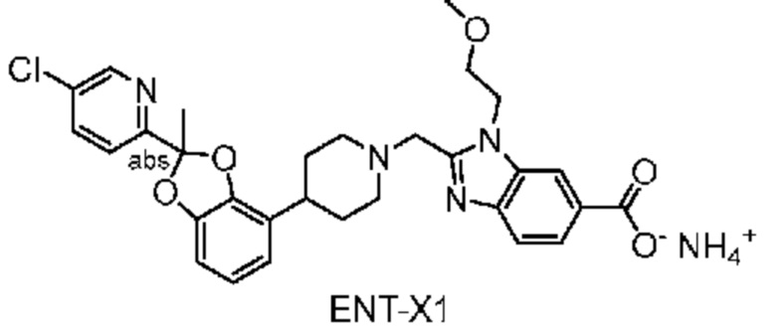
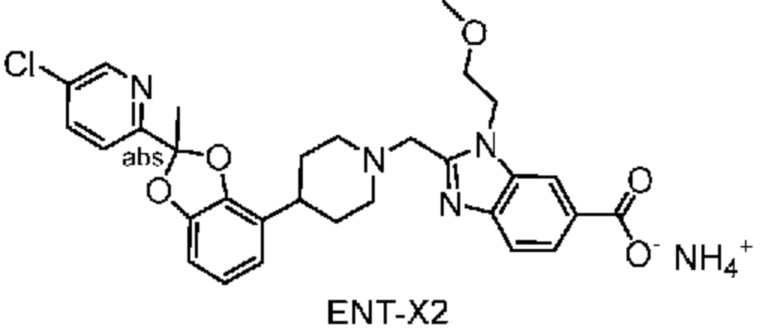

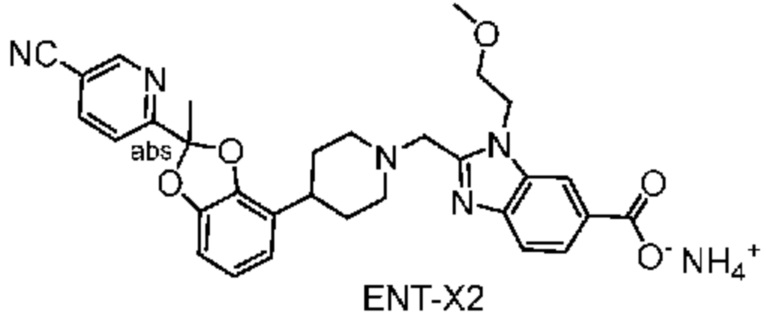
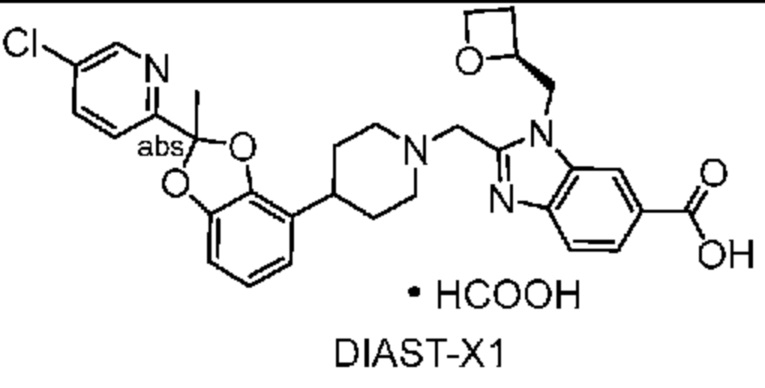



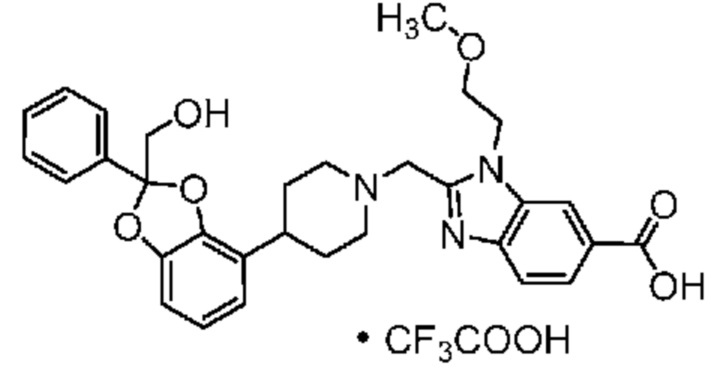
Таблица 2. Методика получения и физико-химические данные соединений примеров 19-102.
^ площадь является предположительной, пик частично закрыт пиком воды
^^ площадь является предположительной, пик частично закрыт пиком растворителя
♦ наблюдали сигналы изотопа хлора
1. Рацемический метиловый сложный эфир [метил-2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат] разделяли на его компоненты-энантиомеры с помощью SFC [колонка: Chiral Technologies ChiralCel OD-H, 5 мкм; подвижная фаза: 7:3 диоксид углерода/(2-пропанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся энантиомер, ENT-1 (C76), использовали для синтеза соединения примера 21 и второй элюирующийся энантиомер, ENT-2 (C77), превращали в соединение примера 20. C76 время удерживания: 5,72 мин (колонка: Chiral Technologies Chiralpak OD-3, 4,6×150 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: 2-пропанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 5,5 мин, затем непрерывно при 40% B в течение 3,0 мин; скорость потока: 2,5 мл/мин). C77 время удерживания: 6,01 мин (условия проведения анализа с помощью SFC такие же, как использованные для C76).
2. Метиловый сложный эфир (метил-2-{6-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]-6-азаспиро[2.5]окт-1-ил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат), полученный сочетанием C4 и P17, разделяли на его компоненты-стереоизомеры по диоксолану с помощью SFC [колонка: Chiral Technologies Chiralpak AD, 10 мкм; подвижная фаза: 65:35 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся изомер, DIAST-1 (C78), превращали в соединение примера 26; на основании изучения данных 1H NMR заключили, что это вещество является энантиомером соединения примера 15. Второй элюирующийся изомер, DIAST-2 (C79), использовали для синтеза соединения примера 25; на основании изучения данных 1H NMR заключили, что это вещество является энантиомером соединения примера 16. C78 время удерживания: 3,60 мин (колонка: Chiral Technologies Chiralpak AD-3, 4,6×100 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: этанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 4,5 мин, затем непрерывно при 40% B в течение 2,5 мин; скорость потока: 2,8 мл/мин). C79 время удерживания: 3,82 мин (условия проведения анализа с помощью SFC такие же, как использованные для C78).
3. 4-(4-Бром-1,3-бензодиоксол-2-ил)-3-фторбензонитрил получали путем обработки 3-фтор-4-формилбензонитрила и 3-бромбензол-1,2-диола п-толуолсульфоновой кислотой в толуоле с удалением воды с использованием аппарата Дина-Штарка. Это вещество затем вводили в реакцию с [1-(трет-бутоксикарбонил)пиперидин-4-ил](йод)цинк в присутствии [1,1"-бис(дифенилфосфино)ферроцен]дихлорпалладием(II) и йодидом меди(I), затем сложный эфир расщепляли с помощью п-толуолсульфоновой кислоты и получали искомый 3-фтор-4-[4-(пиперидин-4-ил)-1,3-бензодиоксол-2-ил]бензонитрил.
4. Условия проведения аналитической HPLC. Колонка: Waters XBridge C18, 2,1×50 мм, 5 мкм; подвижная фаза A: 0,0375% трифторуксусной кислоты в воде; подвижная фаза B: 0,01875% трифторуксусной кислоты в ацетонитриле; градиентный режим: от 10% до 100% B за 4,0 мин; скорость потока: 0,8 мл/мин.
5. Условия проведения аналитической HPLC. Колонка: Waters XBridge C18, 2,1×50 мм, 5 мкм; подвижная фаза A: 0,0375% трифторуксусной кислоты в воде; подвижная фаза B: 0,01875% трифторуксусной кислоты в ацетонитриле; градиентный режим: от 1% до 5% B за 0,6 мин; от 5% до 100% B за 3,4 мин; скорость потока: 0,8 мл/мин.
6. трет-Бутил-4-[2-метил-2-(пиридин-2-ил)-1,3-бензодиоксол-4-ил]-3,6-дигидропиридин-1(2H)-карбоксилат синтезировали из 3-бромбензол-1,2-диола и 2-этинилпиридина по методике, описанной для синтеза C12 в процедуре P7. Последующее гидрирование над палладием на угле с последующей обработкой хлоридом водорода в этилацетате, давало искомый 2-[2-метил-4-(пиперидин-4-ил)-1,3-бензодиоксол-2-ил]пиридингидрохлорид.
7. Рацемический метиловый сложный эфир [метил-1-(2-метоксиэтил)-2-({4-[2-метил-2-(пиридин-2-ил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1H-бензимидазол-6-карбоксилат] разделяли на его компоненты-энантиомеры с помощью SFC [колонка: Chiral Technologies Chiralpak AD, 10 мкм; подвижная фаза: 65:35 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся энантиомер ENT-1 (C80) использовали для синтеза соединения примера 90 и второй элюирующийся энантиомер ENT-2 (C81) превращали в соединение примера 89. C80 время удерживания: 4,11 мин (колонка: Chiral Technologies Chiralpak AD-3, 4,6×100 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: этанол, содержащий 0,05% диэтиламина; градиентный режим: от 5% до 40% B за 4,5 мин, затем непрерывно при 40% B в течение 2,5 мин; скорость потока: 2,8 мл/мин). C81 время удерживания: 4,62 мин (условия проведения анализа с помощью SFC такие же, как использованные для C80).
8. Превращение P8 и P9 в соответствующие цианозамещенные производные проводили по методике, описанной для синтеза P4 из P2 в процедуре P4.
9. Обработка 1-(4-хлор-2-фторфенил)этанона триметилортоформиатом и п-толуолсульфоновой кислотой давала 4-хлор-1-(1,1-диметоксиэтил)-2-фторбензол, который вводили в реакцию с 3-бром-6-фторбензол-1,2-диолом в присутствии п-толуолсульфоновой кислоты и получали 4-бром-2-(4-хлор-2-фторфенил)-7-фтор-2-метил-1,3-бензодиоксол. Это вещество превращали в искомый трет-бутил-4-[2-(4-хлор-2-фторфенил)-7-фтор-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-карбоксилат по методике, описанной в процедуре P1 для синтеза P1 из C2.
10. Разделение стереоизомеров по диоксолану для 96 и 97 проводили с помощью SFC [колонка: Chiral Technologies Chiralpak IG, 5 мкм; подвижная фаза: 3:1 диоксид углерода/(2-пропанол, содержащий 0,2% гидроксида аммония)]. Первый элюирующийся изомер обозначали, как DIAST-1 (96), и второй элюирующийся изомер, как DIAST-2 (97).
11. Условия проведения аналитической SFC. Колонка: Chiral Technologies Chiralpak IG, 4,6×100 мм, 5 мкм; подвижная фаза: 7:3 диоксид углерода/(2-пропанол, содержащий 0,2% гидроксида аммония); скорость потока: 1,5 мл/мин; противодавление: 150 бар.
12. трет-Бутил-2-(хлорметил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоксилат синтезировали из трет-бутил-3-фтор-4-нитробензоата и 1-(1,3-оксазол-2-ил)метанамина по методике, описанной для синтеза P11. Последующую реакцию с C54 проводили с использованием триэтиламина и получали трет-бутил-2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоксилат, который разделяли на его компоненты-энантиомеры с помощью SFC [колонка: Chiral Technologies ChiralCel OD-H, 5 мкм; подвижная фаза: 55:45 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Первый элюирующийся энантиомер ENT-1 (C82) использовали для синтеза 99 и второй элюирующийся энантиомер ENT-2 (C83) превращали в 98. C82 время удерживания: 1,47 мин (колонка: Chiral Technologies Chiralpak OD-3, 4,6×50 мм, 3 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: метанол, содержащий 0,05% диэтиламина; градиентный режим: 5% B в течение 0,2 мин, затем от 5% до 40% B за 1,4 мин, затем непрерывно при 40% B в течение 1,05 мин; скорость потока: 4 мл/мин). C83 время удерживания: 1,85 мин (условия проведения анализа с помощью SFC такие же, как использованные для C82).
13. Реакция 1-бром-2,3-дифтор-4-нитробензола с цианидом меди(I) в 1-метилпирролидин-2-оне при повышенной температуре давала 2,3-дифтор-4-нитробензонитрил, который обрабатывали тионилхлоридом и метанолом и получали метил-2,3-дифтор-4-нитробензоат. Это вещество с использованием C29 превращали в искомый метил-2-(хлорметил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат по методике, описанной в процедуре P11 для синтеза P11 из метил-3-фтор-4-нитробензоати.
14. Искомый [2-фенил-4-(пиперидин-4-ил)-1,3-бензодиоксол-2-ил]метанол синтезировали из 2-оксо-2-фенилэтилацетата по методике, аналогичной описанной для синтеза C13.
CHO GLP-1R клон H6 - анализ 1
Опосредуемую с помощью GLP-1R агонистическую активность определяли с помощью клеточного функционального анализа с использованием набора HTRF (гомогенная флуоресценция с разрешением по времени) для определения cAMP (cAMP HI Range Assay Kit; CisBio cat #62AM6PEJ), с помощью которого определяют содержания cAMP в клетке. Методикой является конкурентный иммуноанализ нативного cAMP, продуцируемого клетками, и экзогенного cAMP, меченого красителем d2. Связывание маркера визуализируют с помощью антител mAb к cAMP, меченых криптатом. Удельный сигнал (т. е. перенос энергии) обратно пропорционален концентрации cAMP в стандартном или экспериментальном образце.
Кодирующую последовательность GLP-1R человека (NCBI Reference Sequence NP_002053.3, включая природный вариант Gly168Ser) субклонировали в pcDNA3 (Invitrogen) и выделяли линию клеток, стабильно экспрессирующих рецептор (обозначена, как клон H6). Анализы насыщения связывания (процедура фильтрационного анализа) с использованием 125I-GLP-17-36 (Perkin Elmer) показали, что плазменные мембраны, образованные из клеток это линии, с высокой плотностью экспрессируют GLP-1R (Kd: 0,4 нМ, Bmax: 1900 фмоль/мг белка).
Клетки забирали из криоконсерватора, повторно суспендировали в 40 мл забуференного фосфатом физиологического раствора Дульбекко (DPBS - Lonza Cat # 17-512Q) и центрифугировали при 800×g в течение 5 мин при 22°C. Затем таблетку клеток повторно суспендировали в 10 мл среды для выращивания [DMEM/F12 1:1 смесь с HEPES, L-Gln, 500 мл (DMEM/F12 Lonza Cat # 12-719F), 10% инактивированной нагреванием фетальной бычьей сыворотки (Gibco Cat # 16140-071), 5 мл 100× Pen-Strep (Gibco Cat # 15140-122), 5 мл 100× L-глутамина (Gibco Cat # 25030-081) и 500 мкг/мл генетицина (G418) (Invitrogen #10131035)]. Для 1 мл образца суспензии клеток в средах для выращивания проводили подсчет на приборе Becton Dickinson ViCell для определения жизнеспособности клеток и количества клеток в 1 мл. Затем оставшуюся суспензию клеток пополняли средами для выращивания и готовили 2000 жизнеспособных клеток на лунку с использованием дозатора реагентов Matrix Combi Multidrop, и клетки дозировали в белый 384-луночный обработанный клеточной культурой планшет для анализа (Corning 3570). Затем планшет для анализа инкубировали в течение 48 ч при 37°C в увлажненной среде в 5% диоксиде углерода.
Каждое исследуемое соединение в разных концентрациях (в DMSO) разводили в буфере для анализа (HBSS с кальцием/магнием (Lonza/BioWhittaker cat # 10-527F)/0,1% BSA (Sigma Aldrich cat # A7409-1L)/20 мМ HEPES (Lonza/BioWhittaker cat #17-737E), содержащем 100 мкМ 3-изобутил-1-метилксантина (IBMX; Sigma cat # I5879). Конечная концентрация DMSO равна 1%.
Через 48 ч среды для выращивания удаляли из лунок планшета для анализа и клетки обрабатывали с помощью 20 мкл серийно разведенного соединения(IBMX; Sigma cat # I5879 в течение 30 мин при 37°C в увлажненной среде в 5% диоксиде углерода. После инкубации в течение 30 мин в каждую лунку планшета для анализа добавляли 10 мкл меченого d2 cAMP и 10 мкл антител к cAMP (оба разводили в отношении 1:20 в буфере для лизиса клеток; как описано изготовителем в протоколе анализа). Затем планшеты инкубировали при комнатной температуре и через 60 мин изменения сигнала HTRF считывали с помощью многофункционального планшетного анализатора Envision 2104 с использованием возбуждения при 330 нм и испускания при 615 и 665 нм. Необработанные данные пересчитывали в количество нМ cAMP с помощью интерполяции калибровочной кривой для cAMP (как описано изготовителем в протоколе анализа) и выраженное в процентах воздействие определяли относительно всего агониста GLP-17-36 в насыщенной концентрации (1 мкМ), включенного в каждый планшет. Определения EC50 проводили по зависимостям доза-ответ для агониста, проанализированным с помощью программы аппроксимации зависимости с использованием 4-параметрического логистического уравнения доза-ответ.
CHO GLP-1R клон C6 - анализ 2
Опосредуемую с помощью GLP-1R агонистическую активность определяли с помощью клеточного функционального анализа с использованием набора HTRF (гомогенная флуоресценция с разрешением по времени) для определения cAMP (cAMP HI Range Assay Kit; CisBio cat #62AM6PEJ), с помощью которого определяют содержания cAMP в клетке. Методикой является конкурентный иммуноанализ нативного cAMP, продуцируемого клетками, и экзогенного cAMP, меченого красителем d2. Связывание маркера визуализируют с помощью антител mAb к cAMP, меченых криптатом. Удельный сигнал (т. е. перенос энергии) обратно пропорционален концентрации cAMP в стандартном или экспериментальном образце.
Кодирующую последовательность GLP-1R человека (NCBI Reference Sequence NP_002053.3, включая природный вариант Leu260Phe) субклонировали в pcDNA5-FRT-TO и клонированную линию клеток CHO, стабильно с низкой плотностью экспрессирующих рецептор, выделяли с использованием системы Flp-In™ T-Rex™, как описано изготовителем (ThermoFisher). Анализы насыщения связывания (процедура фильтрационного анализа) с использованием 125I-GLP-1 (Perkin Elmer) показали, что плазменные мембраны, образованные из клеток это линии (обозначена, как клон C6), с низкой плотностью экспрессируют GLP-1R (Kd: 0,3 нМ, Bmax: 240 фмоль/мг белка), по сравнению с клоном H6 линии клеток.
Клетки забирали из криоконсерватора, повторно суспендировали в 40 мл забуференного фосфатом физиологического раствора Дульбекко (DPBS - Lonza Cat # 17-512Q) и центрифугировали при 800×g в течение 5 мин при 22°C. DPBS отсасывали и таблетку клеток повторно суспендировали в 10 мл полной среды для выращивания (DMEM:F12 1:1 смесь с HEPES, L-Gln, 500 мл (DMEM/F12 Lonza Cat # 12-719F), 10% инактивированной нагреванием фетальной бычьей сыворотки (Gibco Cat # 16140-071), 5 мл 100× Pen-Strep (Gibco Cat # 15140-122), 5 мл 100× L-глутамина (Gibco Cat # 25030-081), 700 мкг/мл гигромицина (Invitrogen Cat # 10687010) и 15 мкг/мл бластицидина (Gibco Cat # R21001). Для 1 мл образца суспензии клеток в средах для выращивания проводили подсчет на приборе Becton Dickinson ViCell для определения жизнеспособности клеток и количества клеток в 1 мл. Затем оставшуюся суспензию клеток пополняли средами для выращивания и готовили 1600 жизнеспособных клеток на лунку с использованием дозатора реагентов Matrix Combi Multidrop, и клетки дозировали в белый 384-луночный обработанный клеточной культурой планшет для анализа (Corning 3570). Затем планшет для анализа инкубировали в течение 48 ч при 37°C в увлажненной среде (95% O2, 5% CO2).
Каждое исследуемое соединение в разных концентрациях (в DMSO) разводили в буфере для анализа (HBSS с кальцием/магнием (Lonza/BioWhittaker cat # 10-527F)/0,1% BSA (Sigma Aldrich cat # A7409-1L)/20 мМ HEPES (Lonza/BioWhittaker cat #17-737E)], содержащем 100 мкМ 3-изобутил-1-метилксантина (IBMX; Sigma cat # I5879). Конечная концентрация DMSO в соединении/смеси буфера для анализа равна 1%.
Через 48 ч среды для выращивания удаляли из лунок планшета для анализа и клетки обрабатывали с помощью 20 мкл серийно разведенного соединения(IBMX; Sigma cat # I5879 в течение 30 мин при 37°C в увлажненной среде (95% O2, 5% CO2). После инкубации в течение 30 мин в каждую лунку планшета для анализа добавляли 10 мкл меченого d2 cAMP и 10 мкл антител к cAMP (оба разводили в отношении 1:20 в буфере для лизиса клеток; как описано изготовителем в протоколе анализа). Затем планшеты инкубировали при комнатной температуре и через 60 мин изменения сигнала HTRF считывали с помощью многофункционального планшетного анализатора Envision 2104 с использованием возбуждения при 330 нм и испускания при 615 и 665 нм. Необработанные данные пересчитывали в количество нМ cAMP с помощью интерполяции калибровочной кривой для cAMP (как описано изготовителем в протоколе анализа) и выраженное в процентах воздействие определяли относительно всего агониста GLP-1 в насыщенной концентрации (1 мкМ), включенного в каждый планшет. Определения EC50 проводили по зависимостям доза-ответ для агониста, проанализированным с помощью программы аппроксимации зависимости с использованием 4-параметрического логистического уравнения доза-ответ.
В таблице 3 данные анализов приведены с точностью до двух (2) значащих цифр в виде средних геометрических (EC50s) и средних арифметических (Emax) значений на основе указанного количества повторов. Отсутствие числа в ячейке означает, что для этого примера не было данных или Emax не рассчитывали.
Таблица 3. Биологическая активность соединений примеров 1-102.
*Исследовали в виде соли аммония и трифторацетатов
**Исследовали в виде солей аммония и 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминия (Tris) и свободной кислоты
***Исследовали в виде соли аммония и свободной кислоты
****Исследовали в виде формиата и свободной кислоты
Все патенты, заявки на патенты и литература, цитированные в настоящем изобретении во всей их полноте включены в настоящее изобретение в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| АГОНИСТЫ РЕЦЕПТОРА GLP-1 И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2740135C1 |
| АГОНИСТЫ GLP-1 РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2765721C1 |
| ПРОИЗВОДНЫЕ ПРОПИОНОВОЙ КИСЛОТЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА | 2000 |
|
RU2255933C9 |
| ТИАЗОЛИНО-2-ПИРИДОНЫ С КОНДЕНСИРОВАННЫМИ ЦИКЛАМИ В КОМБИНАЦИИ С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ ПРОТИВ ТУБЕРКУЛЕЗА | 2018 |
|
RU2791467C2 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2597364C2 |
| НОВЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2607788C2 |
| 6-СУЛЬФАМОИЛХИНОЛИН-4-КАРБОНОВЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И КОМБИНАТОРНАЯ БИБЛИОТЕКА | 2003 |
|
RU2229475C1 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2589878C2 |
| МОЛЕКУЛЫ С ПЕСТИЦИДНОЙ ФУНКЦИЕЙ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ, СВЯЗАННЫЕ С НИМИ | 2016 |
|
RU2735602C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2016 |
|
RU2744766C2 |
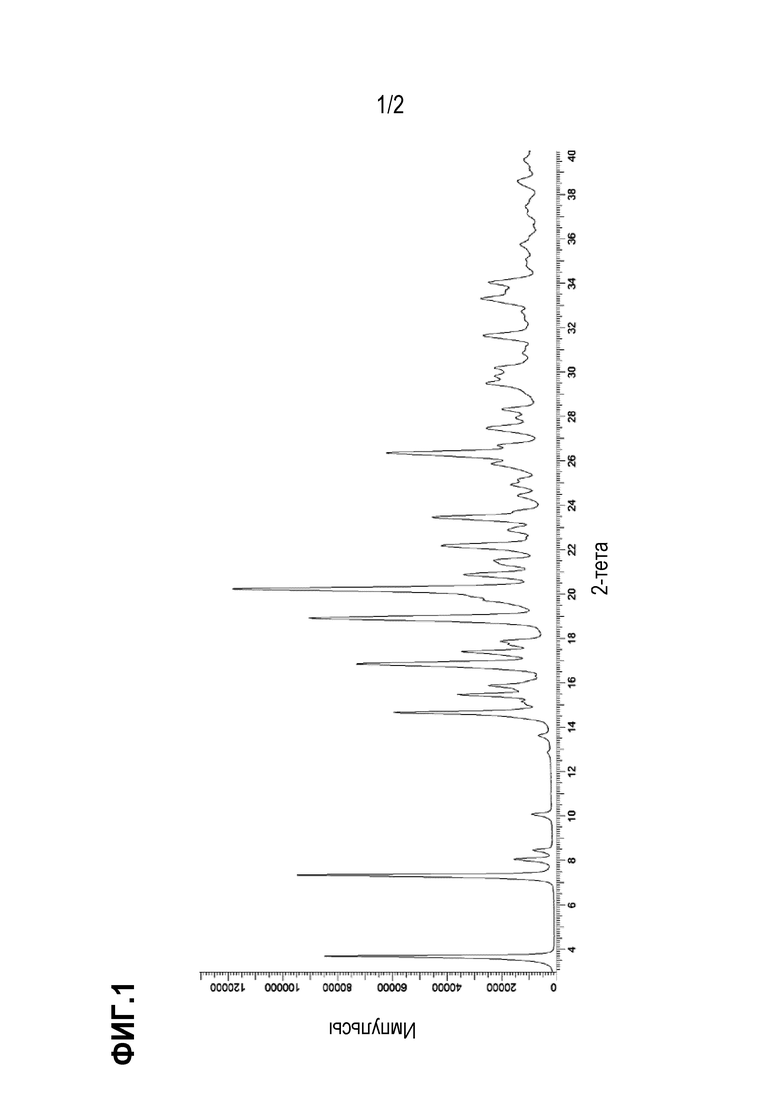
Изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R означает F; p равно 0 или 1; кольцо A представляет собой фенил или пиридинил; m равно 0, 1, 2 или 3; каждый R1 независимо выбран из группы, включающей галоген, -CN, -C1-3алкил или -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила замещен 0-3 атомами F; R2 означает H или -C1-3алкил, где указанный алкил замещен с помощью от 0 до 1 OH; X-L означает N-CH2 или циклопропил; Y означает CH или N; R4 означает -C1-3алкил, -C0-3алкилен-C3-6циклоалкил, -C0-3алкилен-R5 или -C1-3алкилен-R6, где указанный алкил может быть замещен, насколько допускает валентность, 0-3 заместителями, независимо выбранными из группы, включающей 0-3 атома F, и 0-1 заместителем, выбранным из группы, включающей -C0-1алкилен-ORO, -SO2-N(RN)2, -C(O)-N(RN)2, -N(C=O)(RN) и -N(RN)2; R5 означает 4-6-членный гетероциклоалкил, выбранный из оксетанила, тетрагидрофуранила, морфолинила, 1,3-оксазолидинила и пирролидинила, где указанный гетероциклоалкил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей 0-1 оксогруппу (=O); R6 означает 5-6-членный гетероарил, выбранный из оксазолила, имидазолила, пиридинила, пиразолила и триазолила, где указанный гетероарил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей: 0-2 -C1-3алкила, где алкил может быть замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей 0-1 -ORO; каждый RO независимо означает H или -C1-3алкил, где C1-3алкил может быть замещен 0-3 атомами F; каждый RN независимо означает H или -C1-3алкил; Z1, Z2 и Z3 все означают -CRZ или один из Z1, Z2 и Z3 означает N и два других означают -CRZ; и каждый RZ независимо означает H, F, Cl или -CH3; или 2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-{2-[метил(метилсульфонил)амино]этил}-1H-бензимидазол-6-карбоновой кислоте или ее фармацевтически приемлемой соли. Также изобретение относится к фармацевтической композиции, обладающей агонистической активностью GLP-1R, содержащей терапевтически эффективное количество соединения по изобретению и инертный наполнитель. Технический результат - 6-карбоксипроизводные бензимидазолов и 4-аза-, 5-аза- и 7-азабензимидазолов в качестве агонистов GLP-1R. 16 н. и 17 з.п. ф-лы, 2 ил., 13 табл., 102 пр.

1. Соединение формулы I
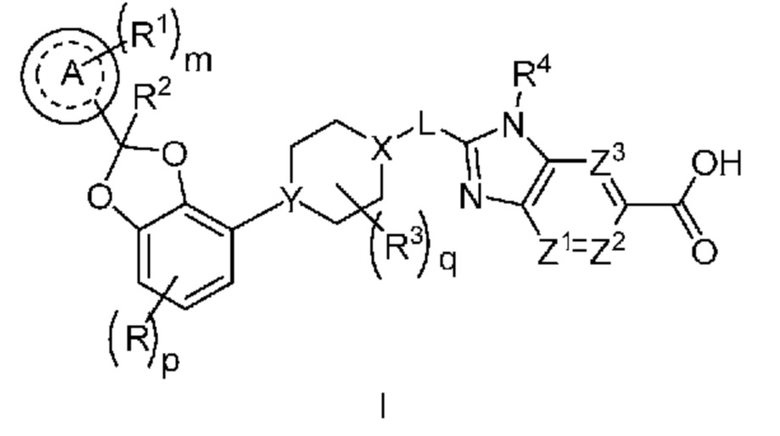
или его фармацевтически приемлемая соль, где
R означает F;
p равно 0 или 1;
кольцо A представляет собой фенил или пиридинил;
m равно 0, 1, 2 или 3;
каждый R1 независимо выбран из группы, включающей галоген, -CN, -C1-3алкил или -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила замещен 0-3 атомами F;
R2 означает H или -C1-3алкил, где указанный алкил замещен с помощью от 0 до 1 OH;
X-L означает N-CH2 или циклопропил;
Y означает CH или N;
R4 означает -C1-3алкил, -C0-3алкилен-C3-6циклоалкил, -C0-3алкилен-R5 или -C1-3алкилен-R6, где указанный алкил может быть замещен, насколько допускает валентность, 0-3 заместителями, независимо выбранными из группы, включающей 0-3 атома F, и 0-1 заместителем, выбранным из группы, включающей -C0-1алкилен-ORO, -SO2-N(RN)2, -C(O)-N(RN)2, -N(C=O)(RN) и -N(RN)2;
R5 означает 4-6-членный гетероциклоалкил, выбранный из оксетанила, тетрагидрофуранила, морфолинила, 1,3-оксазолидинила и пирролидинила, где указанный гетероциклоалкил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-1 оксогруппу (=O),
R6 означает 5-6-членный гетероарил, выбранный из оксазолила, имидазолила, пиридинила, пиразолила и триазолила, где указанный гетероарил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-2 -C1-3алкила, где алкил может быть замещен 0-3 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-1 -ORO;
каждый RO независимо означает H или -C1-3алкил, где C1-3алкил может быть замещен 0-3 атомами F;
каждый RN независимо означает H или -C1-3алкил;
Z1, Z2 и Z3 все означают -CRZ, или
один из Z1, Z2 и Z3 означает N и два других означают -CRZ; и
каждый RZ независимо означает H, F, Cl или -CH3; или
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-{2-[метил(метилсульфонил)амино]этил}-1H-бензимидазол-6-карбоновая кислота или ее фармацевтически приемлемая соль.
2. Соединение по п. 1, где соединением является соединение формулы II
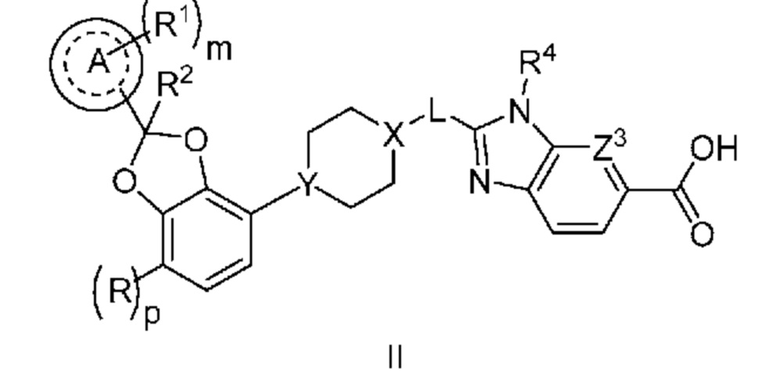
или его фармацевтически приемлемая соль, где
R означает F;
p равно 0 или 1;
кольцо A представляет собой фенил или пиридинил;
m равно 0, 1 или 2;
каждый R1 независимо выбран из группы, включающей галоген, -CN, -C1-3алкил или -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила замещен 0-3 атомами F;
R2 означает H или CH3;
X-L означает N-CH2 или циклопропил;
Y означает CH или N;
и
RZ означает H или -CH3.
3. Соединение по п. 1 или 2, где соединением является соединение формулы III
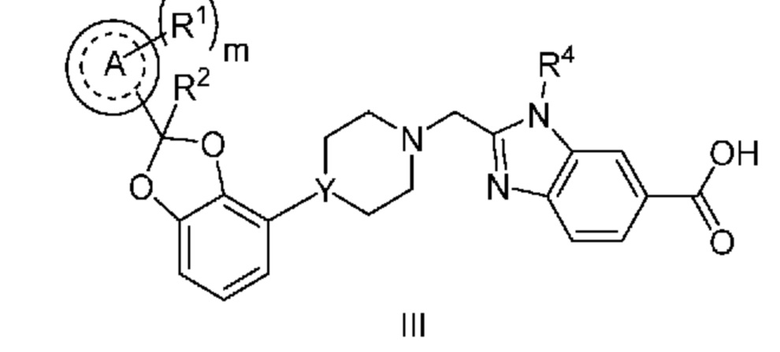
или его фармацевтически приемлемая соль, где
кольцо A представляет собой фенил или пиридинил;
m равно 0, 1 или 2;
каждый R1 независимо выбран из группы, включающей F, Cl или -CN;
R2 означает H или CH3; и
Y означает CH или N.
4. Соединение по любому из пп. 1-3, где R4 означает -CH2-R5, где R5 означает 4-5-членный гетероциклоалкил, выбранный из оксетанила, тетрагидрофуранила, 1,3-оксазолидинила и пирролидинила, где указанный гетероциклоалкил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-1 оксо (=О);
или его фармацевтически приемлемая соль.
5. Соединение по любому из пп. 1-3, где R4 означает -CH2-R6, где R6 означает 5-членный гетероарил, выбранный из оксазолила, имидазолила, пиразолила и триазолила, где указанный гетероарил может быть замещен 0-2 заместителями, насколько допускает валентность, независимо выбранными из группы, включающей:
0-1 -CH3, -CH2CH3 или -CH2CH2OCH3;
или его фармацевтически приемлемая соль.
6. Соединение по любому из пп. 1-5, где R2 означает H, или его фармацевтически приемлемая соль.
7. Соединение по п. 6, где соединением является
2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота,
или его фармацевтически приемлемая соль.
8. Соединение по п. 6, где соединением является
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, или
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота,
или его фармацевтически приемлемая соль.
9. Соединение по любому из пп. 1-5, где R2 означает CH3, или его фармацевтически приемлемая соль.
10. Соединение по п. 9, где соединением является
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-3-(1,3-оксазол-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-4-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(пиридин-3-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-1,2,3-триазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-7-фтор-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или его фармацевтически приемлемая соль.
11. Соединение по п. 9, где соединением является
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или его фармацевтически приемлемая соль.
12. Соединение по п. 9, где соединением является
2-({4-[(2S)-2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
или его фармацевтически приемлемая соль
13. Соединение по п. 9, где соединением является
2-({4-[(2R)-2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2R)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2R)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота,
или его фармацевтически приемлемая соль.
14. Соединение, которым является 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, или его фармацевтически приемлемая соль, где солью является соль с Tris.
15. Соединение, которым является 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота в виде свободной кислоты.
16. Соединение, которым является
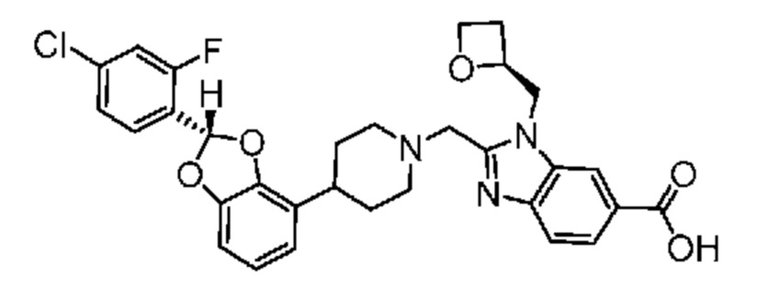
или его фармацевтически приемлемая соль.
17. Соединение, которым является 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота или фармацевтически приемлемая соль, где солью является соль с Tris.
18. Соединение, которым является 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота в виде свободной кислоты.
19. Соединение, которым является
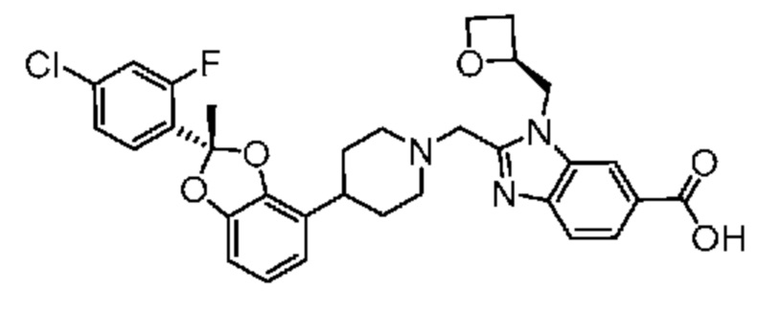
или его фармацевтически приемлемая соль.
20. Соединение, которым является
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2R)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота в виде свободной кислоты.
21. Соединение, которым является
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-({4-[(2R)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
его фармацевтически приемлемая соль, где солью является соль с Tris.
22. Соединение, которым является 2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, DIAST-X2:
 DIAST-X2,
DIAST-X2,
или его фармацевтически приемлемая соль.
23. Кристаллическая форма (форма I) безводной 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли 2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, обладающая порошковой рентгенограммой (излучение CuKα), содержащей характеристические пики с углами 2θ при 3,7±0,2° и 7,3±0,2°.
24. Кристаллическая форма по п. 23, обладающая порошковой рентгенограммой, содержащей пики с углами 2θ при 3,7±0,2°; 7,3±0,2° и 14,7±0,2°.
25. Кристаллическая форма по п. 24, обладающая порошковой рентгенограммой, содержащей пики с углами 2θ при 3,7±0,2°; 7,3±0,2°; 14,7±0,2° и 16,9±0,2°.
26. Кристаллическая форма по п. 25, обладающая порошковой рентгенограммой, содержащей пики с углами 2θ при 3,7±0,2°; 7,3±0,2°; 8,5±0,2°; 10,1±0,2°; 14,7±0,2° и 16,9±0,2°.
27. Кристаллическая форма (форма A) безводной 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминиевой соли 2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, DIAST-X2, обладающая порошковой рентгенограммой (излучение CuKα), содержащей характеристические пики с углами 2θ при 7,7±0,2° и 17,6±0,2°.
28. Кристаллическая форма по п. 27, обладающая порошковой рентгенограммой, содержащей пики с углами 2θ при 7,7±0,2°; 15,2±0,2° и 17,6±0,2°.
29. Кристаллическая форма по п. 27, обладающая порошковой рентгенограммой, содержащей пики с углами 2θ при 7,7±0,2°; 15,2±0,2°; 15,7±0,2° и 17,6±0,2°.
30. Фармацевтическая композиция, обладающая агонистической активностью GLP-1R, содержащая терапевтически эффективное количество соединения по любому из пп. 1-22 или его фармацевтически приемлемой соли и фармацевтически приемлемый инертный наполнитель.
31. Способ лечения кардиометаболического и сопутствующего заболевания или нарушения, включающий введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по любому из пп. 1-22 или его фармацевтически приемлемой соли, где заболевание или нарушение связано с агонистическим эффектом GLP-1R и выбрано из группы, включающей T1D, T2DM, преддиабет, идиопатический T1D, LADA, EOD, YOAD, MODY, диабет, связанный с недостаточным питанием, гестациозный диабет, гипергликемию, резистентность к инсулину, резистентность печени к инсулину, нарушенную переносимость глюкозы, диабетическую невропатию, диабетическую нефропатию, заболевание почек, диабетическую ретинопатию, дисфункцию адипоцитов, висцеральное отложение жира, апноэ во сне, ожирение, пищевые расстройства, увеличение массы тела вследствие применения других средств, чрезмерное пристрастие к сладкому, дислипидемию, гиперинсулинемию, NAFLD, NASH, фиброз, цирроз, печеночно-клеточную карциному, сердечно-сосудистое заболевание, атеросклероз, заболевание коронарной артерии, заболевание периферических сосудов, гипертензию, эндотелиальную дисфункцию, нарушенную эластичность сосудов, застойную сердечную недостаточность, инфаркт миокарда, удар, геморрагический удар, ишемический удар, травматическое повреждение головного мозга, легочную гипертензию, рестеноз после ангиопластики, перемежающуюся хромоту, липемию после еды, метаболический ацидоз, кетоз, артрит, остеопороз, болезнь Паркинсона, гипертрофию левого желудочка, заболевание периферической артерии, дегенерацию желтого пятна, катаракта, гломерулосклероз, хроническую почечную недостаточность, метаболический синдром, синдром X, предменструальный синдром, стенокардию, тромбоз, атеросклероз, преходящие ишемические нарушения, рестеноз сосудов, нарушенный метаболизм глюкозы, патологические состояния с нарушенным содержанием глюкозы в плазме натощак, гиперурикемию, подагру, эректильную дисфункцию, кожные нарушения и нарушения соединительной ткани, псориаз, изъязвления ног, язвенный колит, гипер-апо-B-липопротеинемию, болезнь Альцгеймера, шизофрению, нарушенную познавательную способность, воспалительную болезнь кишечника, синдром короткой кишки, болезнь Крона, колит, синдром раздраженной толстой кишки, предупреждения или лечения синдрома поликистоза яичников и лечения привыкания.
32. Фармацевтическая композиция, обладающая агонистической активностью GLP-1R, содержащая терапевтически эффективное количество кристаллической формы по любому из пп. 23-29 и фармацевтически приемлемый инертный наполнитель.
33. Способ лечения кардиометаболического и сопутствующего заболевания или нарушения, включающий введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллической формы по любому из пп. 23-29, где заболевание или нарушение выбрано из группы, включающей T1D, T2DM, преддиабет, идиопатический T1D, LADA, EOD, YOAD, MODY, диабет, связанный с недостаточным питанием, гестациозный диабет, гипергликемию, резистентность к инсулину, резистентность печени к инсулину, нарушенную переносимость глюкозы, диабетическую невропатию, диабетическую нефропатию, заболевание почек, диабетическую ретинопатию, дисфункцию адипоцитов, висцеральное отложение жира, апноэ во сне, ожирение, пищевые расстройства, увеличение массы тела вследствие применения других средств, чрезмерное пристрастие к сладкому, дислипидемия, гиперинсулинемию, NAFLD, NASH, фиброз, цирроз, печеночно-клеточную карциному, сердечно-сосудистое заболевание, атеросклероз, заболевание коронарной артерии, заболевание периферических сосудов, гипертензию, эндотелиальную дисфункцию, нарушенную эластичность сосудов, застойную сердечную недостаточность, инфаркт миокарда, удар, геморрагический удар, ишемический удар, травматическое повреждение головного мозга, легочную гипертензию, рестеноз после ангиопластики, перемежающуюся хромоту, липемию после еды, метаболический ацидоз, кетоз, артрит, остеопороз, болезнь Паркинсона, гипертрофию левого желудочка, заболевание периферической артерии, дегенерации желтого пятна, катаракту, гломерулосклероз, хроническую почечную недостаточность, метаболический синдром, синдром X, предменструальный синдром, стенокардию, тромбоз, атеросклероз, преходящие ишемические нарушения, рестеноз сосудов, нарушенный метаболизм глюкозы, патологические состояния с нарушенным содержанием глюкозы в плазме натощак, гиперурикемию, подагру, эректильную дисфункцию, кожные нарушения и нарушения соединительной ткани, псориаз, изъязвления ног, язвенный колит, гипер-апо-B-липопротеинемию, болезнь Альцгеймера, шизофрению, нарушенную познавательную способность, воспалительную болезнь кишечника, синдром короткой кишки, болезнь Крона, колит, синдром раздраженной толстой кишки, предупреждения или лечения синдрома поликистоза яичников и лечения привыкания.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| СА 3038479 А1, 29.03.2018 | |||
| НОВЫЕ БЕНЗОДИОКСОЛЫ | 2003 |
|
RU2304580C2 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |

