ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к 6-карбоновым кислотам бензимидазолов и 4-аза-, 5-аза- и 7-аза-бензимидазолов в качестве агонистов GLP-1R, способам получения указанных соединений и способам, включающим введение указанных соединений млекопитающему, нуждающемуся в этом.
УРОВЕНЬ ТЕХНИКИ
Диабет представляет собой серьезную проблему для общественного здравоохранения вследствие его растущей распространенности и связанных с этим рисков для здоровья. Это заболевание характеризуется высокими уровнями содержания глюкозы в крови в результате нарушения выработки инсулина, нарушения действия инсулина или того и другого. Различают две основные формы диабета: диабет 1 типа и диабет 2 типа. Диабет 1 типа (T1D) развивается, когда иммунная система организма разрушает бета-клетки поджелудочной железы - единственные клетки в организме, которые производят гормон инсулин, регулирующий уровень глюкозы в крови. Для выживания людям с диабетом 1 типа необходимо вводить инсулин инъекцией или с помощью помпы. Сахарный диабет 2 типа (обычно обозначаемый как T2DM) обычно начинается либо с инсулинорезистентности, либо с недостаточной для поддержания приемлемого уровня глюкозы выработки инсулина.
В настоящее время доступны различные фармакологические подходы к лечению гипергликемии и, как следствие, T2DM (Hampp, C. et al. Use of Antidiabetic Drugs in the U.S., 2003-2012, Diabetes Care 2014, 37, 1367-1374). Их можно сгруппировать в шесть основных классов, действующих посредством разных основных механизмов: (A) стимуляторы секреции инсулина, в том числе сульфонилмочевины (например, глипизид, глимепирид, глибурид), меглитиниды (например, натеглидин, репаглинид), ингибиторы дипептидилпептидазы IV (DPP-IV) (например, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин, саксоглиптин) и агонисты рецепторов глюкагоноподобного пептида-1 (GLP-1R) (например, лираглутид, альбиглутид, эксенатид, ликсисенатид, дилаглутид, семаглутид), которые усиливают секрецию инсулина, воздействуя на бета-клетки поджелудочной железы. Сульфонилмочевины и меглитиниды обладают ограниченной эффективностью и переносимостью, вызывают увеличение массы тела и часто вызывают гипогликемию. Ингибиторы DPP-IV обладают ограниченной эффективностью. Зарегистрированные агонисты GLP-1R представляют собой пептиды, вводимые подкожной инъекцией. Лираглутид одобрен также для лечения ожирения. (B) Считается, что бигуаниды (например, метформин) действуют в основном за счет снижения выработки глюкозы в печени. Бигуаниды часто вызывают желудочно-кишечные расстройства и лактоацидоз, что является дополнительным ограничением их использования. (C) Ингибиторы альфа-глюкозидазы (например, акарбоза) снижают всасывание глюкозы в кишечнике. Эти лекарственные средства часто вызывают желудочно-кишечные расстройства. (D) Тиазолидиндионы (например, пиоглитазон, розиглитазон) воздействуют на специфический рецептор (гамма-рецептор, активируемый пролифератором пероксисом) в печени, мышцах и жировых тканях. Они регулируют метаболизм липидов, впоследствии усиливая реакцию этих тканей на действие инсулина. Частое употребление этих препаратов может приводить к увеличению массы тела и вызывать отек и анемию. (E) Инсулин используется в более тяжелых случаях отдельно либо в сочетании с вышеуказанными лекарственными средствами, и его частое использование также может привести к увеличению массы тела и несет риск гипогликемии. (F) Ингибиторы натрий-зависимого переносчика глюкозы 2 типа (SGLT2) (например, дапаглифлозин, эмпаглифлозин, канаглифлозин, эртуглифлозин) подавляют ресорбцию глюкозы в почках и тем самым снижают уровни глюкозы в крови. Этот перспективный класс лекарственных средств может быть связан с кетоацидозом и инфекциями мочевыводящих путей.
Однако указанные лекарственные средства, за исключением агонистов GLP-1R и ингибиторов SGLT2, обладают ограниченной эффективностью и не решают наиболее важных проблем, таких как снижение функции β-клеток и связанное с этим ожирение.
Ожирение представляет собой хроническое заболевание, которое широко распространено в современном обществе и связано с многочисленными медицинскими проблемами, включая гипертонию, гиперхолестеринемию и ишемическую болезнь сердца. Кроме того, оно в значительной степени сильно коррелирует с T2DM и инсулинорезистентностью, а последняя обычно сопровождается гиперинсулинемией или гипергликемией либо обоими этими заболеваниями. Кроме того, T2DM ассоциируется с повышенным в два-четыре раза риском ишемической болезни сердца. В настоящее время единственным лечением, которое устраняет ожирение с высокой эффективностью, является бариатрическая хирургия, но это лечение является дорогостоящим и рискованным. Фармакологическое вмешательство обычно менее эффективно и связано с побочными эффектами. Следовательно, существует очевидная потребность в более эффективном фармакологическом вмешательстве с меньшим количеством побочных эффектов и удобным введением.
Хотя T2DM чаще всего ассоциируется с гипергликемией и инсулинорезистентностью, другие заболевания, связанные с T2DM, включают печеночную инсулинорезистентность, нарушение толерантности к глюкозе, диабетическую невропатию, диабетическую нефропатию, диабетическую ретинопатию, ожирение, дислипидемию, гипертензию, гиперинсулинемию и неалкогольную жировую болезнь печени (НАЖБП).
НАЖБП представляет собой печеночное проявление метаболического синдрома и включает различные патологические состояния печени, в том числе стеатоз, неалкогольный стеатогепатит (НАСГ), фиброз, цирроз и, в конечном итоге, гепатоцеллюлярную карциному. НАЖБП и НАСГ считаются первичными жировыми заболеваниями печени, поскольку пациенты с этими заболеваниями составляют наибольшую долю среди пациентов с повышенным уровнем липидов печени. Тяжесть НАЖБП/НАСГ зависит от наличия липидов, инфильтрата воспалительных клеток, баллонной дистрофии гепатоцитов и степени фиброза. Хотя стеатоз не у всех пациентов прогрессирует до НАСГ, это происходит у значительной их части.
GLP-1 представляет собой инкретиновый гормон длиной 30 аминокислот, секретируемый L-клетками в кишечнике в ответ на прием пищи. Было показано, что GLP-1 стимулирует секрецию инсулина физиологическим и глюкозозависимым образом, снижает секрецию глюкагона, ингибирует опорожнение желудка, снижает аппетит и стимулирует пролиферацию бета-клеток. В неклинических экспериментах GLP-1 способствует непрерывной компетенции бета-клеток, стимулируя транскрипцию генов, важных для секреции инсулина, и способствуя неогнезу бета-клеток (Meier, et al. Biodrugs. 2003; 17 (2): 93-102).
У здорового человека GLP-1 играет важную роль в регулировании постпрандиальных уровней глюкозы в крови стимулированием глюкозозависимой секреции инсулина поджелудочной железой, что приводит к повышению поглощения глюкозы на периферии. GLP-1 также подавляет секрецию глюкагона, что приводит к снижению выхода глюкозы из печени. Кроме того, GLP-1 задерживает опорожнение желудка и замедляет моторику тонкого кишечника, задерживая всасывание пищи. У пациентов с T2DM нормальное постпрандиальное повышение содержания GLP-1 отсутствует или снижено (Vilsboll T., et al. Diabetes. 2001. 50; 609-613).
Холст (Holst) (Physiol. Rev. 2007, 87, 1409) и Мейер (Meier) (Nat. Rev. Endocrinol. 2012, 8, 728) отмечают, что агонисты GLP-1 рецептора, такие как GLP-1, лираглутид и эксендин-4, обладают тремя основными фармакологическими активностями для улучшения гликемического контроля у пациентов с T2DM посредством снижения уровня глюкозы натощак (FPG) и после приема пищи (PPG): (i) повышение глюкозозависимой секреции инсулина (улучшенная первая и вторая фаза), (ii) активность подавления глюкагона в условиях гипергликемии, (iii) замедление скорости опорожнения желудка, приводящее к замедленному всасыванию глюкозы, полученной из пищи.
Таким образом, сохраняется потребность в легковводимых лекарственных средствах для предотвращения и/или лечения кардиометаболических и связанных с ними заболеваний.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
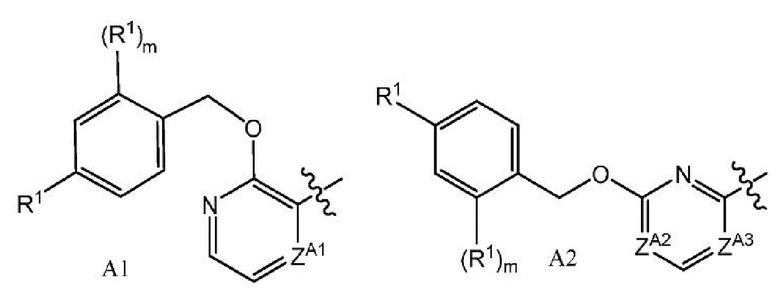
Настоящее изобретение относится к соединениям формулы I:
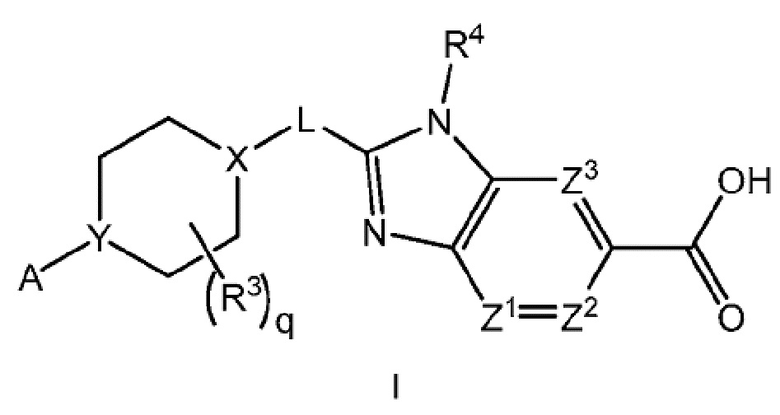
или их фармацевтически приемлемым солям, где A представляет собой A1 или A2
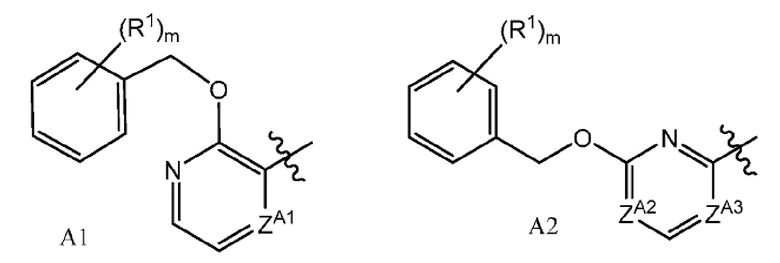
и где
каждый R1 независимо представляет собой галоген, -CN, -C1-3алкил или -OC1-3алкил, где алкил C1-3алкила и OC1-3алкила является замещенным 0-3 атомами F;
m равно 0, 1, 2 или 3;
Х-L представляет собой N-CH2, CHCH2 или циклопропил;
Y представляет собой CH или N;
ZA1 представляет собой CH, CR2 или N;
ZA2 представляет собой CH, CR2 или N;
ZA3 представляет собой CH, CR2 или N; при условии, что ZA2 и ZA3 одновременно не являются N, и дополнительно при условии, что один из ZA2 и ZA3 представляет собой N, когда Х-L представляет собой N-CH2;
каждый R2 независимо представляет собой F, Cl или -CN;
каждый R3 независимо представляет собой F, -OH, -CN, -C1-3алкил, -OC1-3алкил или -C3-4циклоалкил, или два R3 вместе могут циклизоваться с образованием -C3-4спироциклоалкила, где алкил C1-3алкила и OC1-3алкила, циклоалкила или спироциклоалкила может быть замещенным, как допускает валентность, 0-3 атомами F и 0-1 -OH;
q равно 0, 1 или 2;
R4 представляет собой -C1-3алкил, -C0-3алкилен-C3-6циклоалкил, -C0-3алкилен-R5 или -C1-3алкилен-R6, где указанный алкил может быть замещенным, как допускает валентность, 0-3 заместителями, независимо выбранными из 0-3 атомов F, и 0-1 заместителем, выбранным из -C0-1алкилен-CN, -C0-1алкилен-ORO и -N(RN)2, и
где указанные алкилен и циклоалкил могут быть независимо замещенными, как допускает валентность, 0-2 заместителями, независимо выбранными из 0-2 атомов F, и 0-1 заместителем, выбранным из -C0-1алкилен-CN, -C0-1алкилен-ORO и -N(RN)2;
R5 представляет собой 4-6-членный гетероциклоалкил, где указанный гетероциклоалкил может быть замещенным 0-2 заместителями, как допускает валентность, независимо выбранными из:
0-1 оксо (=O),
0-1 -CN,
0-2 атомов F и
0-2 заместителей, независимо выбранных из -C1-3алкила и -OC1-3 алкила, где алкил C1-3алкила и OC1-3алкила может быть замещенным 0-3 заместителями, как допускает валентность, независимо выбранными из:
0-3 атомов F,
0-1 -CN и
0-1 -ORO;
R6 представляет собой 5-6-членный гетероарил, где указанный гетероарил может быть замещенным 0-2 заместителями, как допускает валентность, независимо выбранными из:
0-2 атомов галогенов,
0-1 заместителя, выбранного из -ORO и -N(RN)2, и
0-2 -C1-3алкилов, где алкил может быть замещенным 0-3 заместителями, как допускает валентность, независимо выбранными из:
0-3 атомов F и
0-1 -ORO;
каждый RO независимо представляет собой H или -C1-3алкил, где C1-3алкил может быть замещенным 0-3 атомами F;
каждый RN независимо представляет собой H или -C1-3алкил;
каждый из Z1, Z2 и Z3 представляет собой -CRZ, или
один из Z1, Z2 и Z3 представляет собой N, а два другие представляют собой -CRZ; и
каждый RZ независимо представляет собой H, F, Cl или -CH3.
Другой вариант осуществления настоящего изобретения относится к соединению формулы II
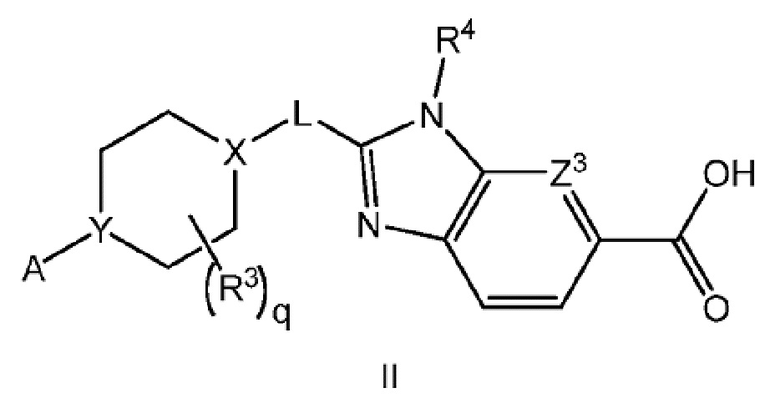
или его фармацевтически приемлемой соли, где А представляет собой A1 или A2
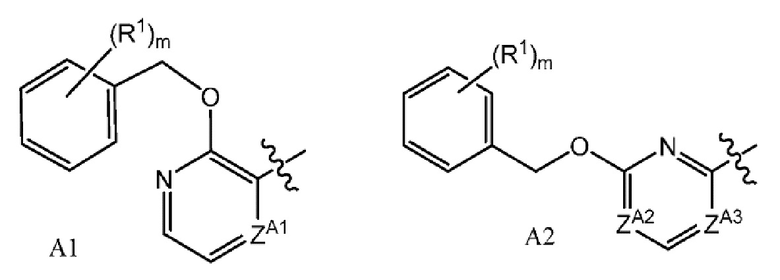
и где
каждый R1 независимо представляет собой F, Cl или -CN; и
m равно 0, 1 или 2.
Другой вариант осуществления настоящего изобретения относится к соединению формулы III
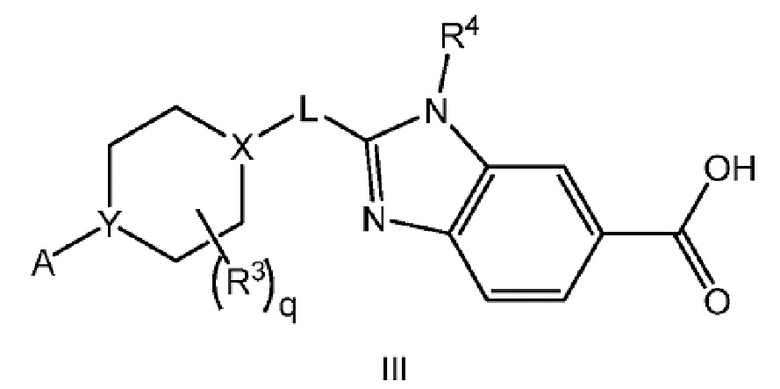
или его фармацевтически приемлемой соли, где А представляет собой A1 или A2
и где
каждый R1 независимо представляет собой F, Cl или -CN; и
m равно 0, 1 или 2.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I, II или III, где A представляет собой A1, или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к соединению формулы IV
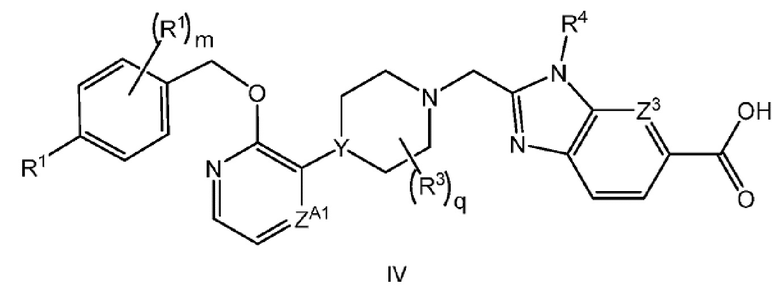
или его фармацевтически приемлемой соли, где
каждый R1 независимо представляет собой F, Cl или -CN;
m равно 0 или 1;
q равно 0 или 1; и
R3 представляет собой -CH3.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I, II, III или IV, где ZA1 представляет собой CH или CR2; и
R2 представляет собой F;
или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I, II, III или IV, где ZA1 представляет собой N, или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I, II или OII, где A представляет собой A2,
q равно 0 или 1; и
R3 представляет собой -CH3;
или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I, II или III, где ZA2 представляет собой CH или CR2; и ZA3 представляет собой N, или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I, II или III, где ZA2 представляет собой N, ZA3 представляет собой CH или CR2, или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I
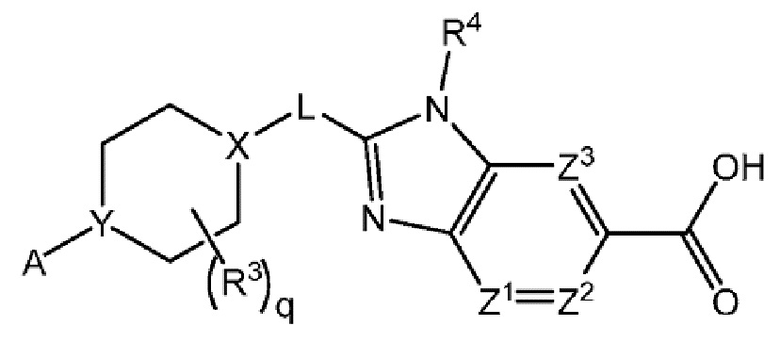
или его фармацевтически приемлемой соли, где A представляет собой A1 или A2
и где
каждый R1 независимо представляет собой F, Cl или -CN; и
m равно 0 или 1.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществлений, например соединениям формулы I, II или III, или их фармацевтически приемлемым солям, где Х-L представляет собой N-CH2; и Y представляет собой CH или N. В вариантах осуществления, описанных в настоящем документе, в этом случае X представляет собой N, L представляет собой CH2.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления, например соединениям формул I, II или III, или их фармацевтически приемлемым солям, где Х-L представляет собой CHCH2; и Y представляет собой N. В вариантах осуществления, описанных в настоящем документе, в этом случае X представляет собой CH, L представляет собой CH2.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II или III, или их фармацевтически приемлемым солям, где Х-L представляет собой CHCH2; и Y представляет собой CH. В вариантах осуществления, описанных в настоящем документе, в этом случае X представляет собой CH, и L представляет собой CH2.
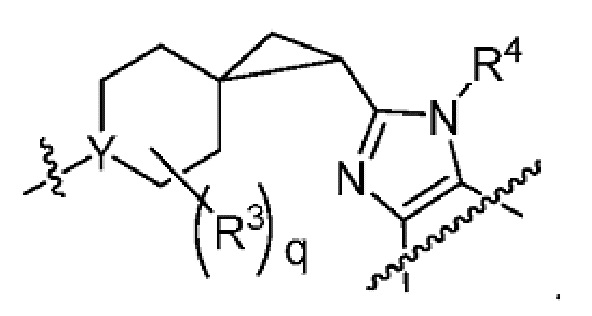
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например к соединениям формулы I или II, или их фармацевтически приемлемым солям, где Х-L представляет собой циклопропил; Y представляет собой N.
В вариантах осуществления, где Х-L представляет собой циклопропил, Х-L, являющийся циклопропилом, может обеспечить группу
Другой вариант осуществления настоящего изобретения относится к соединению формулы V
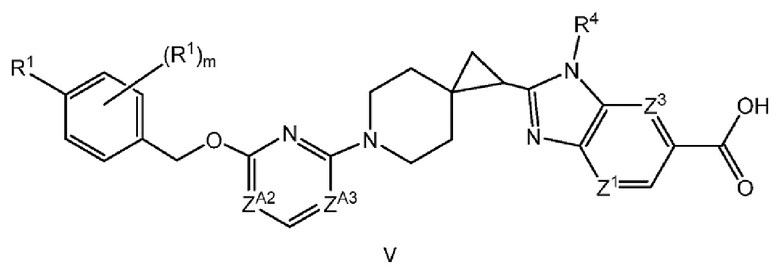
или его фармацевтически приемлемой соли, где
каждый R1 независимо представляет собой F, Cl или -CN;
m равно 0 или 1;
ZA2 представляет собой CH, CR2 или N;
ZA3 представляет собой CH, CR2 или N; при условии, что ZA2 и ZA3 одновременно не являются N; и
каждый R2 представляет собой F.
Другой вариант осуществления настоящего изобретения относится к соединению формулы VI
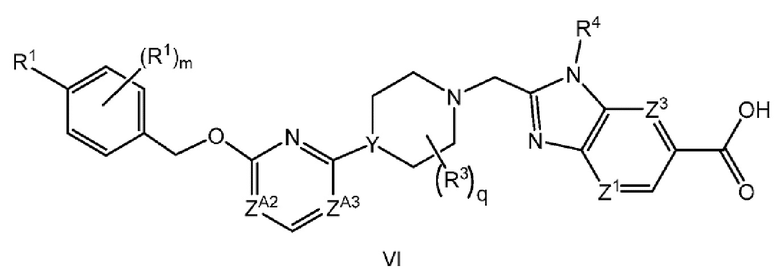
или его фармацевтически приемлемой соли, где
каждый R1 независимо представляет собой F, Cl или -CN;
m равно 0 или 1;
R3 представляет собой -CH3;
q равно 0, 1 или 2;
ZA2 представляет собой CH, CR2 или N;
ZA3 представляет собой CH, CR2 или N; при условии, что ZA2 и ZA3 одновременно не являются N;
R2 представляет собой F; и
Y представляет собой CH или N.
Другой вариант осуществления настоящего изобретения относится к соединениям любой из формул I, II, III, IV, V или IV и любым их вариантам осуществления, где R4 представляет собой -CH2-R5, R5 представляет собой 4-5-членный гетероциклоалкил, где указанный гетероциклоалкил может быть замещенным 0-2 заместителями, как допускает валентность, независимо выбранными из:
0-2 атомов F и
0-1 заместителя, выбранного из оксо (=O), -OCH3 и -CH2OCH3;
или их фармацевтически приемлемым солям.
В вариантах осуществления настоящего изобретения, относящихся к соединению формулы II, III, IV, V или VI, в котором переменная не определена, указанная переменная принимает значения, определенные для формулы I.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где R4 представляет собой -CH2CH2OCH3, C1-3алкилен-R5 или C1-3алкилен-R6, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где R4 представляет собой -C1-3алкил, где указанный алкил может быть замещенным, как допускает валентность, 0-1 заместителем, выбранным из -C0-1алкилен-ORO и -N(RN)2, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где R4 представляет собой -(CH2)2OCH3 или -(CH2)2N(CH3)2, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где R4 представляет собой -CH2-R5, R5 представляет собой 4-5-членный гетероциклоалкил, где указанный гетероциклоалкил может быть замещенным 0-1 заместителем, который представляет собой оксогруппу (=O), или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где R4 представляет собой -CH2-R5, R5 представляет собой 4-5-членный гетероциклоалкил, где указанный гетероциклоалкил может быть замещенным 0-2 заместителями, как допускает валентность, независимо выбранными из:
0-2 атомов F и
0-1 заместителя, выбранного из -OCH3 и -CH2OCH3;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям любой из формул I, II, III, IV, V или IV и их любым вариантам осуществления, где R4 представляет собой -CH2-R6, R6 представляет собой 5-членный гетероарил, где указанный гетероарил может быть замещенным 0-2 заместителями, как допускает валентность, независимо выбранными из:
0-2 атомов галогенов, где галоген независимо выбран из F и Cl,
0-1 -OCH3 и
0-1 -CH3, -CH2CH3, -CF3 или -CH2CH2OCH3;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям любой из формул I, II, III, IV, V или IV и их любым вариантам осуществления, где R4 представляет собой -C1-3алкил, где указанный алкил может быть замещенным, как допускает валентность, 0-3 заместителями, независимо выбранными из 0-3 атомов F, и 0-1 заместителем, который представляет собой -C0-1алкилен-ORO.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где гетероциклоалкил представляет собой
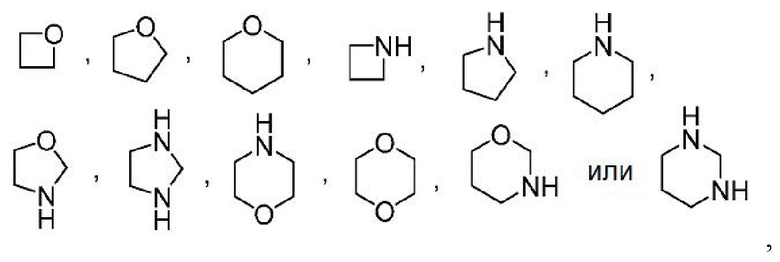
где гетероциклоалкил может быть замещенным, как допускает валентность, например замещением атома(ов) водорода, 0-2 заместителями, независимо выбранными из:
0-1 оксогруппы (=O),
0-1 -CN,
0-2 атомов F и
0-2 заместителей, независимо выбранных из -C1-3алкила и -OC1-3алкила, где алкил C1-3алкила и OC1-3алкила может быть независимо замещенным 0-3 заместителями, как допускает валентность, независимо выбранными из:
0-3 атомов F,
0-1 -CN и
0-1 -ORO,
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где гетероциклоалкил представляет собой
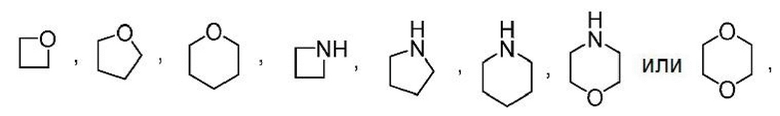
где гетероциклоалкил может быть замещенным, как допускает валентность, например замещением атома(ов) водорода, 0-2 заместителями, независимо выбранными из:
0-1 -CN,
0-2 атомов F и
0-2 заместителей, независимо выбранных из -C1-3алкила и -OC1-3алкила, где алкил C1-3алкила и OC1-3алкила может быть независимо замещенным 0-3 заместителями, как допускает валентность, независимо выбранными из:
0-3 атомов F,
0-1 -CN и
0-1 -ORO, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где гетероциклоалкил представляет собой
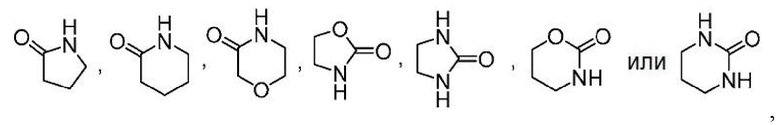
где гетероциклоалкил может быть замещенным, как допускает валентность, например замещением атома водорода, 0-1 заместителем, выбранным из:
-CN,
атома F и
0-1 заместителя, независимо выбранного из -C1-3алкила и -OC1-3алкила, где алкил C1-3алкила и OC1-3алкила может быть замещенным 0-3 заместителями, как допускает валентность, независимо выбранными из:
0-3 атомов F,
0-1 -CN и
0-1 -ORO,
или их фармацевтически приемлемым солям.
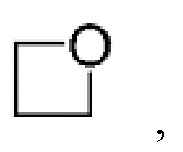
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где гетероциклоалкил представляет собой
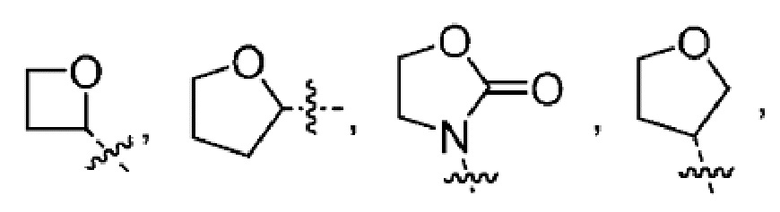
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где гетероциклоалкил представляет собой
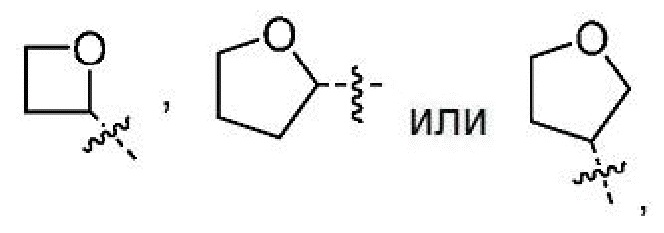
где гетероциклоалкил может быть замещенным, как допускает валентность, 0-1 метилом, где указанный метил может быть замещенным 0-3 атомами F, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где гетероциклоалкил представляет собой
где гетероциклоалкил является незамещенным, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где -CH2-R5 и атом азота, к которому присоединен R4, образуют:
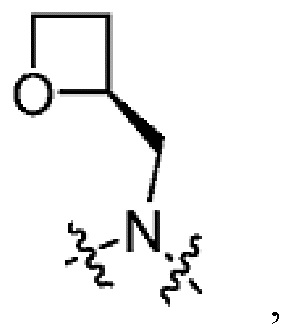
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где гетероарил представляет собой
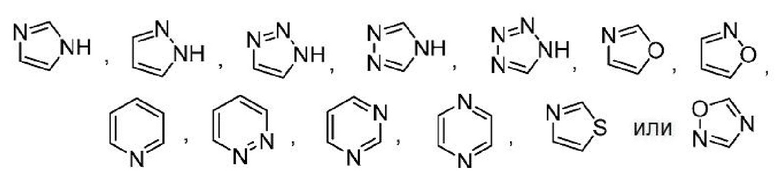 ,
,
где указанный гетероарил может быть замещенным, как допускает валентность, например замещением атома(ов) водорода, 0-2 заместителями, независимо выбранными из:
0-2 атомов галогенов, где галоген независимо выбран из F и Cl,
0-1 заместителя, выбранного из -ORO и -N(RN)2, или
0-2 -C1-3алкилов, где алкил может быть замещенным 0-3 заместителями, как допускает валентность, независимо выбранными из:
0-3 атомов F и
0-1 -ORO;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где гетероарил представляет собой
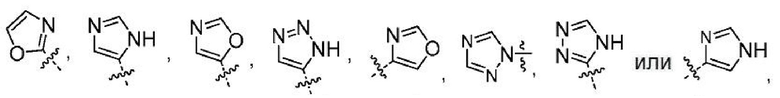
где указанный гетероарил может быть замещенным 0-1 заместителем, как допускает валентность, который представляет собой -C1-2алкил, где алкил может быть замещенным 0-3 заместителями, как допускает валентность, независимо выбранными из:
0-3 атомов F и
0-1 -ORO; и
каждый RO независимо представляет собой H или -C1-3алкил;
или их фармацевтически приемлемым солям. Понятно, что любой заместитель будет замещать атом Н на атоме углерода или азота, на котором происходит замещение. Неограничивающими примерами замещаемых гетероарилов являются
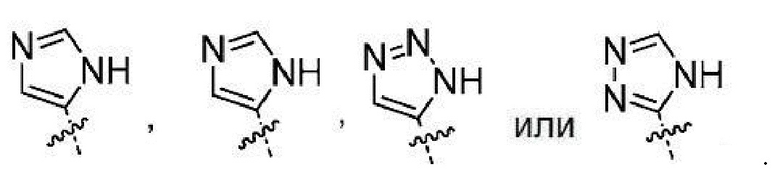
Понятно, что атом Н замещается заместителем, например R6s (заместителем, допустимым на любом гетероариле R6), с получением:
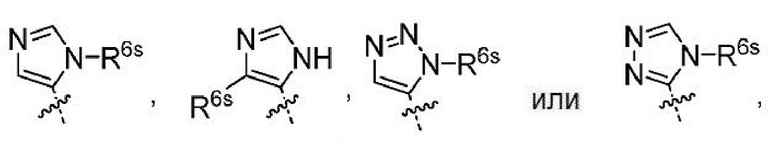
где R6s представляет собой -C1-2алкил, где алкил может быть замещенным 0-3 заместителями, как допускает валентность, независимо выбранными из:
0-3 атомов F и
0-1 -ORO; и
каждый RO независимо представляет собой H или -C1-3алкил;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям формул I, II, III, IV, V или VI, где гетероарил представляет собой
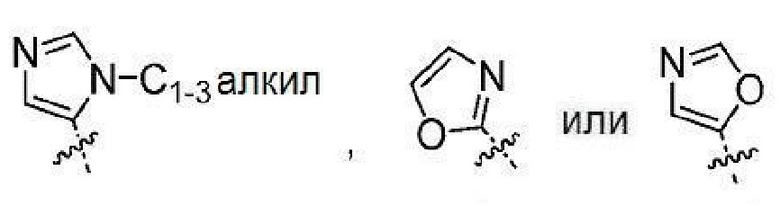 ,
,
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к любому одному или нескольким соединиям, которые представляют собой следующие соединения:
2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{2-[(4-циано-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{2-[(4-циано-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемым солям.
Другой вариант осуществления относится к любому одному или нескольким соединениям, которые представляют собой следующие соединения:
2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к любому одному или нескольким соединениям, которые представляют собой следующие соединения:
2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям, которые представляют собой следующие соединения:
2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, хиральность которой происходит из C79;
2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, хиральность которой происходит из P7;
2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота, хиральность которой происходит из P7;
2-(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота, хиральность которой происходит из C93;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-[(4-{2-[(4-циано-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-[(4-{2-[(4-циано-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям, которые представляют собой следующие соединения:
2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(1S)-6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота; или
2-[(1R)-6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям, которые представляют собой следующие соединения:
2-(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(1S)6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-[(1R)6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям, которые представляют собой следующие соединения:
2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(1S)6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота; или
2-[(1R)6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям, где соединение представляет собой 2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту или ее фармацевтически приемлемую соль.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где каждый из Z1, Z2 и Z3 представляет собой CRZ, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где RZ представляет собой H, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где каждый из Z1, Z2 и Z3 представляет собой CH, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где каждый R2 представляет собой F, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где q равно 0, или их фармацевтически приемлемым солям. Понятно, что когда q равно 0, R3 отсутствует.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где R3 представляет собой -CH3 или -CF3; и q равно 1, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где R3 представляет собой -CH3; и q равно 1, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где каждый R1 независимо представляет собой F, Cl или -CN, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где R4 представляет собой -CH2-R5, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где R4 представляет собой -CH2-R6, или их фармацевтически приемлемым солям.
Другой вариант осуществления настоящего изобретения относится к соединениям других вариантов осуществления настоящего изобретения, например соединениям формул I, II, III, IV, V или VI, где соединение представляет собой свободную кислоту.
В другом варианте осуществление изобретение относится к фармацевтической композиции, включающей соединение формул I, II, III, IV, V или VI или его фармацевтически приемлемую соль, как определено в любом из вариантов осуществлений, описанных в настоящем документе, в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом. Он включает фармацевтическую композицию, включающую соединение формул I, II, III, IV, V или VI или его фармацевтически приемлемую соль, которое определено в любом из вариантов осуществления, описанных в настоящем документе, в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом и одним или несколькими другими терапевтическими средствами, описанными в настоящем документе.
Настоящее изобретение также включает следующие варианты осуществления:
соединение формул I, II, III, IV, V или VI или его фармацевтически приемлемая соль, как определено в любом из вариантов осуществления, описанных в настоящем документе, для применения в качестве лекарственного средства;
соединение формул I, II, III, IV, V или VI или его фармацевтически приемлемая соль, как определено в любом из вариантов осуществления, описанных в настоящем документе, для применения в предотвращении и/или лечении кардиометаболических и связанных с ними заболеваний, описанных в настоящем документе, включая T2DM, предиабет, НАСГ и сердечно-сосудистое заболевание;
способ лечения заболевания, для которого показан агонист GLP-1R, у субъекта, нуждающегося в таком предотвращении и/или лечении, включающий введение субъекту терапевтически эффективного количества соединения формул I, II, III, IV, V или VI или его фармацевтически приемлемой соли, как определено в любом из вариантов осуществления, описанных в настоящем документе;
применение соединения формул I, II, III, IV, V или VI или его фармацевтически приемлемой соли, как определено в любом из вариантов осуществления, описанных в настоящем документе, для производства лекарственного средства для лечения заболевания или состояния, для которого показан агонист GLP-1R;
соединение формул I, II, III, IV, V или VI или его фармацевтически приемлемая соль, как определено в любом из вариантов осуществления, описанных в настоящем документе, для применения в лечении заболевания или состояния, для которого показан агонист GLP-1R; или
фармацевтическая композиция для лечения заболевания или состояния, для которого показан агонист GLP-1R, включающая соединение формул I, II, III, IV, V или VI или его фармацевтически приемлемую соль, как определено в любом из вариантов осуществления, описанных в настоящем документе.
Каждое соединение примера или его фармацевтически приемлемая соль может заявляться отдельно или в сочетании в любой комбинации.
Изобретение также относится к фармацевтической композиции, включающей соединение формул I, II, III, IV, V или VI или его фармацевтически приемлемую соль, как определено в любом из вариантов осуществления, описанных в настоящем документе, для применения в лечении и/или предотвращении кардиометаболических или связанных с ними заболеваний, которые обсуждены в настоящем документе, включая T2DM, предиабет, НАСГ и сердечно-сосудистое заболевание.
Другой вариант осуществления настоящего изобретения относится к соединению формул I, II, III, IV, V или VI или его фармацевтически приемлемой соли, как определено в любом из вариантов осуществления, описанных в настоящем документе, для применения в лечении и/или предотвращения кардиометаболических и связанных с ними заболеваний, включая диабет (T1D и/или T2DM, в том числе предиабет), идиопатический T1D (типа 1b), латентный аутоиммунный диабет у взрослых (LADA), юношеский T2DM (EOD), атипичный диабет в молодом возрасте (YOAD), юношеский диабет взрослого типа (MODY), диабет, связанный с недоеданием, гестационный диабет, гипергликемию, инсулинорезистентность, печеночную инсулинорезистентность, нарушение толерантности к глюкозе, диабетическую невропатию, диабетическую нефропатию, заболевания почек (например, острую почечную недостаточность, трубчатую дисфункцию, провоспалительные изменения проксимальных канальцев), диабетическую ретинопатию, дисфункцию адипоцитов, отложение висцеральной жировой ткани, апноэ во сне, ожирение (включая гипоталамическое ожирение и моногенное ожирение) и сопутствующие заболевания (например, остеоартрит и недержание мочи), расстройства пищевого поведения (включая синдром переедания, нервную булимию и синдромное ожирение, такое как синдромы Прайдера-Вилли и Барде-Бидля), увеличение массы тела при использовании других лекарственных средств (например, при использовании стероидов и антипсихотических лекарственных средств), чрезмерное пристрастие к сахару, дислипидемию (включая гиперлипидемию, гипертриглицеридемию, повышение общего холестерина, высокий уровень холестерина липопротеинов низкой плотности (холестерина ЛПНП) и низкий уровень холестерина липопротеинов высокой плотности (холестерина ЛПВП)), гиперинсулинемию, НАЖБП (включая связанные заболевания, такие как стеатоз, НАСГ, фиброз, цирроз и гепатоцеллюлярную карциному), сердечно-сосудистые заболевания, атеросклероз (включая ишемическую болезнь сердца), заболевания периферических сосудов, гипертензию, эндотелиальную дисфункцию, нарушение эластичности сосудов, застойную сердечную недостаточность, инфаркт миокарда (например, некроз и апоптоз), инсульт, геморрагический инсульт, ишемический инсульт, черепно-мозговую травму, легочную гипертензию, рестеноз после ангиопластики, перемежающуюся хромоту, постпрандиальную липемию, метаболический ацидоз, кетоз, артрит, остеопороз, болезнь Паркинсона, гипертрофию левого желудочка, заболевание периферических артерий, дегенерацию желтого пятна, катаракту, гломерулосклероз, хроническую почечную недостаточность, метаболический синдром, синдром X, предменструальный синдром, стенокардию, тромбоз, атеросклероз, преходящие ишемические атаки, рестеноз сосудов, нарушение метаболизма глюкозы, состояния нарушения уровня глюкозы в плазме крови натощак, гиперурикемию, подагру, эректильную дисфункцию, кожные заболевания и заболевания соединительной ткани, псориаз, изъязвления стопы, язвенный колит, гипер-апо В-липопротеинемию, болезнь Альцгеймера, шизофрению, нарушение когнитивных функций, воспалительное заболевание кишечника, синдром короткого кишечника, болезнь Крона, колит, синдром раздраженного кишечника, профилактику или лечение синдрома поликистозного яичника и лечение зависимости (например, алкоголизма и/или наркомании).
Используемые в описании аббревиатуры имеют следующие значения.
Термин «алкил», когда используется в настоящем описании, означает прямую или разветвленную моновалентную группу формулы -CnH(2n+1). Неограничивающие примеры включают метил, этил, пропил, бутил, 2-метилпропил, 1,1-диметилэтил, пентил и гексил.
Термин «алкилен», когда используется в настоящем описании, означает двухвалентную углеводородную группу формулы -CnH2n- м прямой или разветвленной цепью. Неогранирующие примеры примеры включают этилен и пропилен.
Термин «циклоалкил», когда используется в настоящем описании, означает циклическую моновалентную углеводородную группу формулы-CnH(2n-1), содержащую меньшей мере три атома углерода. Неограничивающие примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
Термин «галоген», когда используется в настоящем описании, относится к фториду, хлориду, бромиду или йодиду.
Термин «гетероциклоалкил», когда используется в настоящем описании, относится к циклоалкильной группе, в которой один или несколько метиленовых групп в кольце (-CH2-) замещены группой, выбранной из из -O-, -S- или атома азота, где на атоме азот может находиться точка присоединения или атом азота может быть замещенным, как это показано в каждом варианте осуществления. Когда точка присоединения находится на атоме азота, структурная формула гетероциклоалкила должна содержать атом водорода на указанном атоме азота. Обычно гетероциклоалкил может быть замещенным 0-2 заместителями, как допускает валентность, независимо выбранными из оксо, -CN, галогена, алкила и -Oалкила, где алкил может быть дополнительно замещенным. Следует отметить, что когда заместитель отсутствует, гетероциклоалкил является незамещенным.
Термин «гетероарил», когда используется в настоящем описании, относится к моноциклической ароматической углеводородной группе, содержащей от 5 до 6 атомов углерода, в которой по меньшей мере один из атомов углерода кольца замещен гетероатомом, выбранным из атома кислорода, атома азота и атома серы. Такая гетероарильная группа может присоединяться через атом углерода кольца или, когда допускает валентность, через атом азота кольца. Обычно гетероарил может быть замещенным 0-2 заместелями, как допускает валентность, независимо выбранными из галогена, OH, алкила, O-алкила и аминогруппы (например, NH2, NH-алкил, N(алкил)2), и алкил может быть дополнительно замещенным. Следует отметить, что когда замещение равно 0, гетероарил является незамещенным.
Комнатная температура: КТ.
Метанол: MeOH.
Этанол: EtOH.
Изопропанол: iPrOH.
Этилацетат: EtOAc.
Тетрагидрофуран: ТГФ.
Толуол: PhCH3.
Карбонат цезия: Cs2CO3.
бис(Триметилсилил)амид лития: LiHMDS.
трет-Бутоксид натрия: NaOtBu.
трет-Бутоксид калия: KOtBu.
Диизопропиламид лития: LDA.
Триэтиламин: Et3N.
N, N-диизопропилэтиламин: DIPEA.
Карбонат калия: K2CO3.
Диметилформамид: ДМФА.
Диметилацетамид: DMAc.
Диметилсульфоксид: ДМСО.
N-Метил-2-пирролидинон: NMP.
Гидрид натрия: NaH.
Трифторуксусная кислота: ТФУК.
Трифторуксусный ангидрид: ТФУА.
Уксусный ангидрид: Ac2O.
Дихлорметан: DCM.
1,2-Дихлорэтан: DCE.
Соляная кислота: HCl.
1,8-Диазабицикло[5.4.0]ундец-7-ен: DBU.
Комплекс боран-диметилсульфид: BH3-DMS.
Комплекс боран-тетрагидрофуран: BH3-ТГФ.
Алюмогидрид лития: LAH.
Уксусная кислота: AcOH.
Ацетонитрил: MeCN.
п-Толуолсульфоновая кислота: pTSA.
Дибензилидинацетон: DBA.
2,2′-Бис(дифенилфосфино)-1,1′-бинафталин: BINAP.
1,1′-Ферроцендиил-бис(дифенилфосфин): dppf.
1,3-Бис(дифенилфосфино)пропан: DPPP.
3-Хлорпербензойная кислота: m-CPBA.
трет-Бутилметиловый эфир: MTBE.
Метансульфонил: Ms.
N-Метилпирролидинон: NMP.
Тонкослойная хроматография: ТСХ.
Сверхкритическая жидкостная хроматография: SFC.
4-(Диметиламино)пиридин: DMAP.
трет-Бутилоксикарбонил: Boc.
Гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния: HATU.
Петролейный эфир: PE.
Гексафторфосфат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония: HBTU.
2-Амино-2-(гидроксиметил)пропат-1,3-диол: tris.
Трис(дибензилиденацетон)дипалладий: Pd2(dba)3.
Спектры 1H ядерного магнитного резонанса (ЯМР) во всех случаях соответствовали предложенным структурам. Характерные химические сдвиги (δ даны в частях на миллион (миллионных долях) относительно остаточного протонного сигнала в дейтерированном растворителе (CHCl3 при 7,27 м.д.; CD2HOD при 3,31 м.д.; MeCN при 1,94 м.д.; ДМСО при 2,50 м.д.) и представлены с использованием обычных сокращений для обозначения основных пиков: например, с - синглет; д - дублет; т - триплет; кв - квартет; м - мультиплет; уш. - уширенный.
Волнистая линия «  », когда используется в настоящем описании, означает точку присоединения заместителя к другой группе.
», когда используется в настоящем описании, означает точку присоединения заместителя к другой группе.
Названия соединений и промежуточных соединений, представленные ниже, получены с использованием способа присвоения названий, предоставленного ACD/ChemSketch 2012, File Version C10H41, Build 69045 (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada). Способ присвоения названий, предоставленный ACD/ChemSketch 2012, хорошо известен специалистам в данной области техники, и считается, что он в целом согласуется с рекомендациями IUPAC (International Union for Pure и Applied Chemistry) по номенклатуре органических химических соединений и с правилами CAS Index. Следует отметить, что химические названия могут содержать только круглые скобки или круглые и квадратные скобки. Стереохимические дискрипторы также могут располагаться в различных местах внутри самого названия в зависимости от способа составления названия. Специалистам в данной области техники будут понятны эти варианты названий, и они поймут, что эти варианты описывают одну и ту же химическую структуру.
Фармацевтически приемлемые соли соединений формулы I включают кислотно-аддитивные и соли, полученные из оснований.
Подходящие кислотно-аддитивные соли представляют собой соли, полученные из кислот, которые образуют нетоксичные соли. Примеры включают ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, цикламат, эдисилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гиксафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглютамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат, соль 1,5-нафталиндисульфоновой кислоты и ксинафоат.
Подходящие соли оснований представляют собой соли, полученные из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, бис(2-гидроксиэтил)амина (диоламина), глицина, лизина, магния, меглумина, 2-аминоэтанола (оламина), калия, натрия, 2-амино-2-(гидроксиметил)пропан-1,3-диола и (трис- или трометамина) и цинка.
Также могут быть получены полусоли (гемисоли) кислот и оснований, например гемисульфатные и гемикальциевые соли. Обзор подходящих солей см. в Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002).
Фармацевтически приемлемые соли соединений формулы I могут быть получены одним или несколькими из трех способов получения:
(i) взаимодействием соединения формулы I с подходящей кислотой или подходящим основанием;
(ii) удалением кислотно- или основно-лабильной защитной группы из подходящего предшественника соединения формулы I или раскрытием цикла подходящего циклического предшественника, например лактона или лактама, с использованием подходящей кислоты или подходящего основания; или
(iii) превращением одной соли соединения формулы I в другую взаимодействием с подходящей кислотой или подходящим основанием или с помощью подходящей ионообменной колонки.
Реакции всех указанных трех видов обычно проводятся в растворе. Полученная соль может осаждаться и собираться фильтрацией или может выделяться выпариванием растворителя. Степень ионизации полученной соли может изменяться от полностью ионизированной до почти неионизированной.
Соединения формулы I и их фармацевтически приемлемые соли могут существовать в несольватированной и сольватированной форме. Термин «сольват», когда используется в настоящем описании, относится к молекулярному комплексу, включающему соединение формулы I или его фармацевтически приемлемую соль и одну или несколько молекул фармацевтически приемлемого растворителя, например этанола. Термин «гидрат» используется, когда указанным растворителем является вода.
Общепринятая в настоящее время система классификации органических гидратов определяет гидраты, координированные с отдельным сайтом, каналом или ионом металла - см. Polymorphism в Pharmaceutical Solids by K. R. Morris (Ed. H.G. Brittain, Marcel Dekker, 1995). Гидраты изолированных сайтов представляют собой гидраты, в которых молекулы воды изолированы от прямого контакта друг с другом вследствие внедрения органических молекул. В канальных гидратах молекулы воды располагаются в каналах решетки, они находятся рядом с другими молекулами воды. В гидратах, координированных с ионами металлов, молекулы воды связаны с ионом металла.
Когда растворитель или вода прочно связаны, комплекс может иметь четко определенную стехиометрию, не зависящую от влажности. Однако когда растворитель или вода слабо связаны, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя может зависеть от влажности и условий сушки. В таких случаях нормой будет нестехиометрическое соотношение.
Настоящее изобретение включает также многокомпонентные комплексы (отличные от солей и сольватов), в которых лекарственное средство и по меньшей мере один другой компонент присутствуют в стехиометрических или нестехиометрических количествах. Комплексы такого типа включают клатраты (комплексы включения лекарственного средства-хозяина) и сокристаллы. Последние обычно определяются как кристаллические комплексы нейтральных составляющих молекул, которые связаны вместе посредством нековалентного взаимодействия, но также могут представлять собой комплекс нейтральной молекулы с солью. Сокристаллы могут быть получены кристаллизацией из расплава, перекристаллизацией из растворителя или физическим совместным измельчением компонентов - см. Chem. Commun., 17, 1889-1896, by O. Almarsson и M. J. Zaworotko (2004). Обзор многокомпонентных комплексов представлен в публикации J. Pharm. Sci., 64 (8), 1269-1288, by Haleblian (August 1975).
Соединения по настоящему изобретению могут существовать в ряде твердых состояний от полностью аморфного до полностью кристаллического. Термин «аморфное» относится к состоянию, при котором в материале отсутствует дальняя упорядоченность на молекулярном уровне и в зависимости от температуры материал может проявлять физические свойства твердого вещества или жидкости. Обычно такие материалы не дают характерных дифрактограмм и, проявляя свойства твердого вещества, более формально описываются как жидкость. При нагревании свойства твердого вещества изменяются до свойств жидкости, что характеризуется изменением состояния, обычно второго порядка («стеклование»). Термин «кристаллическое» относится к твердой фазе, в которой материал имеет регулярную упорядоченную внутреннюю структуру на молекулярном уровне и дает характерную дифрактограмму с определенными пиками. Такие материалы при достаточном нагревании также будут проявлять свойства жидкости, но переход от твердого состояния к жидкому характеризуется фазовым переходом, обычно первого порядка («температура плавление»).
Соединения формулы I в подходящих условиях могут также существовать в мезоморфном состоянии (мезофаза или жидкий кристалл). Мезоморфное состояние представляет собой промежуточное состояние между истинным кристаллическим состояние и истинным жидким состоянием (расплав или раствор). Мезоморфизм, возникающий в результате изменения температуры, описывается как «термотропный», а возникающий в результате наличия второго компонента, такого как вода или другой растворитель, описывается как «лиотропный». Соединения, которые могут образовывать лиотропные мезофазы, описываются как «амфифильные» и состоят из молекул, которые содержат ионную (такую как -COO-Na+, -COO-K+ или -SO3-Na+) или неионную (таую как -N-N+(CH3)3) группу «полярной головки». Для более полной информации см. Crystals and the Polarizing Microscope by N.H. Hartshorne and A. Stuart, 4th Edition (Edward Arnold, 1970).
Соединения формулы I могут проявлять полиморфизм и/или одну или несколько разновидностей изомеризма (например, оптический, геометрический или таутомерный изомеризм). Соединения формулы I также могут быть изотопно-мечеными. Такой вариант подразумевается для соединений формулы I, определенных в соответствии с их структурными отличительными признаками, и, следовательно, включен в объем настоящего изобретения.
Соединения формулы I, содержащие один или большее количество асимметрических атомов углерода, могут существовать в виде двух или большего количества стереоизомеров. Когда соединение формулы I содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс (или Z/E) изомеры. Когда структурные изомеры являются взаимопревращающимися с низкоэнергетическим барьером превращения, может возникнуть таутомерная изомерия («таутомерия»). Она может принимать форму протонной таутомерии в соединениях формулы I, содержащих, например, имино, кето- или оксимную группу, или так называемой валентной таутомерии в соединениях, содержащих ароматический фрагмент. Таким образом, одно соединение может проявлять изомерию нескольких типов.
Фирмацевтически приемлемые соли соединений формулы I могут также содержать противоион, который является оптически активным (например, d-лактат или l-лизин) или рацемическим (например dl-тартрат или dl-аргинин).
Цис/транс изомеры могут быть разделены стандартными методами, хорошо известными специалистам в данной области техники, например хроматографией и фракционной кристаллизацией.
Стандартные методы разделения/выделения отдельных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (либо рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ЖХВД). Альтернативно, рацемический предшественник, содержащий хиральный эфир, может быть разделен ферментативным разделением (см., например, Int. J. Mol. Sci. 29682-29716 by A. C. L. M. Carvaho et. al. (2015)). В случае, когда соединение формулы I содержит кислотный или основной фрагмент, может быть получена соль с оптически чистым основанием или кислотой, такой как 1-фенилэтиламин или винная кислота. Полученная диастереомерная смесь может быть разделена фракционной кристаллизацией, и одна или обе диастереомерные соли могут быть преобразованы в соответствующий(е) чистый(е) энантиомер(ы) способами, хорошо известными специалистам в данной области техники. В качестве альтернативы, рацемат (или рацемический предшественник) может подвергаться ковалентному взаимодействию с подходящим оптически активным соединением, например спиртом, амином или бензилхлоридом. Полученная диастереомерная смесь может разделяться хроматографией и/или фракционной кристаллизацией способами, хорошо известными специалистам в данной области техники, для получения разделенных диастереомеров в виде отдельных энантиомеров с двумя или большим количеством хиральных центров. Хиральные соединения формулы I (и их хиральные предшественнки) могут быть получены в энантиомерно обогащенной форме с использованием хроматографии, обычно ЖХВД, на асимметричной смоле с подвижной фазой, содержащей углеводород, обычно гептан или гексан, содержащий от 0 до 50% по объему изопропанола, обычно от 2% до 20%, и от 0 до 5% по объему алкиламина, обычно 0,1% диэтиламина. Концентрирование элюата приводит к получению обогащенной смеси. Может применяться хиральная хроматография с использованием субкритических и сверкритических жидкостей. Методы хиральной хроматографии, применяемые в некоторых вариантах осуществления настоящего изобретения, известны в данной области техники (см., например, Smith, Roger M., Loughborough University, Loughborough, UK; Chromatographic Science Series (1998), 75 (SFC with Packed Columns), pp. 223-249 и ссылки, указанные в публикации). В некоторых конкретных примерах в настоящем документе колонки были получены от Chiral Technologies, Inc., West Chester, Pennsylvania, USA, дочерней организацией Daicel® Chemical Industries, Ltd., Tokyo, Japan.
При кристаллизации любого рацемата возможны кристаллы двух различных типов. Первый тип представляет собой рацемическое соединение (истинный рацемат), упомянутое выше, когда получают одну гомогенную форму кристалла, содержащую оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, когда получают две формы кристалла в эквимолярных количествах, каждая из которых включает один энантиомер. Хотя обе кристаллические формы, присутствующие в рацемической смеси, обладают идентичными физическими свойствами, их свойства могут отличаться от физических свойств истинного рацемата. Рацемические смеси могут быть разделены обычными методами, известными специалистам в данной области - см., например, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, 1994).
Следует подчеркнуть, что соединения формулы I представлены здесь в единственной таутомерной форме, все возможные таутомерные формы включены в объем изобретения.
Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченые соединения формулы I, в которых один или несколько атомов заменены атомами, имеющими такой же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, которые преобладают в природе.
Примеры изотопов, подходящих для введения в соединения по настоящему изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, и серы, такие как 35S.
Некоторые изотопно-меченые соединения формулы I, например соединения, содержащие радиоактивный изотоп, могут использоваться в исследованиях распределения лекарственного средства и/или субстратов в тканях. Радиоактивные изотопы: тритий, то есть 3H, и углерод-14, то есть 14C - особенно полезны для этой цели благодаря легкости введения и наличию средств обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например увеличенный период полураспада in vivo, или меньшие необходимые дозы.
Замещение позитронно-эмиссионными изотопами, такими как 11C, 18F, 15O и 13N, может быть полезно в исследованиях позитронно-эмиссионной томографии (ПЭТ) для изучения степени занятости рецептора субстратом.
Изотопно-меченые соединения формулы I могут быть получены обычными способами, которые известны специалистам в данной области техники, или способами, аналогичными описанным в прилагаемых примерах и примерах получения, с использованием соответствующего изотопно-меченого реагента вместо ранее применяемого немеченого реагента.
Фармацевтически приемлемые сольваты по настоящему изобретению включают сольваты, в которых растворитель кристаллизации может содержать изотопное замещение, например D2O, d6-ацетон, d6-ДМСО.
Одним из способов осуществления изобретения является введение соединения формулы I в форме пролекарства. Таким образом, некоторые производные соединений формулы I, которые сами по себе могут обладать небольшой фармакологической активностью или вообще не обладать ею, при введении в организм или при нанесении на тело могут превращаться в соединения формулы I, обладающие желаемой активностью, например посредством гидролитического расщепления, стимулируемого ферментом эстеразы или пептидазы. Такие производные называются «пролекарствами». Дополнительную информацию о применении пролекарств можно найти в публикации ‘Pro-drugs as Novel Delivery Systems’, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella); ‘Bioreversible Carriers in Drug Design’, Pergamon Press, 1987 (Ed. E. B. Roche, American Pharmaceutical Association). В качестве ссылки можно также привести Nature Reviews/Drug Discovery, 2008, 7, 355 и Current Opinion в Drug Discovery и Development, 2007, 10, 550.
Пролекарства в соответствии с изобретением можно получить, например, заменой соответствующих функциональных групп, присутствующих в соединениях формулы I, определенными фрагментами, известными специалистам в данной области как «профрагменты» - см., например, в ‘Design of Prodrugs’ by H. Bundgaard (Elsevier, 1985) and Y. M. Choi-Sledeski and C. G. Wermuth, ‘Designing Prodrugs and Bioprecursors’ in Practice of Medicinal Chemistry, (Fourth Edition), Chapter 28, 657-696 (Elsevier, 2015).
Таким образом, пролекарство в соответствии с изобретением представляет собой (а) сложноэфирное или амидное производное по группе карбоновой кислоты в соединении формулы I; (b) сложноэфирное, карбонатное, карбаматное, фосфатное или эфирное производное по гидроксильной группе в соединении формулы I; (c) амидное, иминное, карбаматное или аминное производные по аминогруппе в соединении формулы I; (d) оксимное или иминное производное по карбонильной группе в соединении формулы I; или (e) соединение, в котором метильная группа, группа первичного спирта или альдегидная группа может метаболически окисляться до группы карбоновой кислоты с получением соединения формулы I.
Некоторые конкретные примеры пролекарств в соответствии с изобретением включают:
(i) когда соединение формулы содержит функциональную группу карбоновой кислоты (-COOH), сложный эфир указанной кислоты, такой как соединение, в котором атом водорода функциональной группы карбоновой кислоты соединения формулы I замещен на C1-C8 алкил (например этил) или (C1-C8 алкил)C(=O)OCH2- (например tBuC(=O)OCH2-);
(ii) когда соединение формулы I содержит спиртовую функциональную группу (-OH), сложный эфир указанного соединения, такой как соединение, в котором атом водорода спиртовой функциональной группы соединения формулы I замещен на группу -CO(C1-C8 алкил) (например, метилкарбонил), или спирт, этерифицированный аминокислотой;
(iii) когда соединение формулы I содержит спиртовую функциональную группу (-OH), простой эфир указанного соединения, такой как соединение, в котором атом водорода спиртовой функциональной группы соединения формулы I замещен на группу (C1-C8 алкил)C(=O)OCH2- или -CH2OP(=O)(OH)2;
(iv) когда соединение формулы I содержит спиртовую функциональную группу (-OH), его фосфат, такой как соединение, в котором атом водорода спиртовой функциональной группы соединения формулы I замещен на группу -P(=O)(OH)2, -P(=O)(ONa)2 или -P(=O)(O-)2Ca2+;
(v) когда соединение формулы I содержит функциональную первичную или вторичную аминогруппу (-NH2 или -NHR, где R ≠ H), его амид, например соединение, в котором один или оба атома водорода функциональной аминогруппы соединения формулы I замещен/замещены на (C1-C10)алканоил, -COCH2NH2 или аминогруппу, полученную из аминокислоты;
(vi) когда соединение формулы I содержит функциональную первичную или вторичную аминогруппу (-NH2 или -NHR, где R≠H), его амин, например соединение, в котором один или оба атома водорода функциональной аминогруппы соединения формулы I замещен/замещены на группу -CH2OP(=O)(OH)2;
(vii) соединение, в котором группа карбоновой кислоты в соединении формулы I замещена метильной группой, группой -CH2OH или aльдегидной группой.
Некоторые соединения формулы I сами могут выступать в качестве пролекарств других соединений формулы I. Два соединения формулы I также могут соединяться с образованием пролекарства. В некоторых случаях пролекарство соединения формулы I может быть получено посредством внутримолекулярного связывания двух функциональлных групп в соединении формулы I, например, посредством образования лактона.
Ссылки на соединения формулы I включают сами соединения и их пролекарства. Изобретение включает такие соединения формулы I, а также фармацевтически приемлемые соли таких соединений и фармацевтически приемлемые сольваты указанных соединений и их солей.
Введение и дозировка
Обычно соединение по настоящему изобретению вводится в количестве, эффективном для лечения состояния, которое определено в описании. Соединения по настоящему изобретению могут вводиться сами по себе или, в качестве альтернативы, в виде фармацевтически приемлемой соли. Для определения способа введения и дозировки соединение само по себе или его фармацевтически приемлемая соль будет называться соединением по настоящему изобретению.
Соединения по настоящему изобретению вводятся любым подходящим способом в форме фармацевтической композиции, адаптированной к такому способу введения, и в дозе, эффективной для предполагаемого лечения. Соединения по настоящему изобретению могут вводиться перорально, ректально, вагинально, парентерально или местно.
Соединения по настоящему изобретению могут вводиться перорально. Пероральное введение может включать проглатывание, так что соединение поступает в желудочно-кишечный тракт, или может применяться буккальное или сублингвальное введение, при котором соединение поступает в кровоток непосредственно из ротовой полости.
В другом варианте осуществления соединения по настоящему изобретению могут также вводиться непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенное, внутриартериальное, интраперитонеальное, интратекальное, внутрижелудочковое, внутриуретальное, внутригрудинное, внутричерепное, внутримышечное и подкожное введение. Подходящие устройства для парентерального введения включают игольчатые (включая микроигольчатые) инъекторы, безыгольные инъекторы и устройства для инфузии.
В другом варианте осуществления соединения по настоящему изобретению могут также вводиться местно на кожу или слизистую оболочку, то есть дермально или трансдермально. В другом варианте осуществления соединения по настоящему изобретению могут также вводиться интраназально или ингаляцией. В другом варианте осуществления соединения по настоящему изобретению могут вводиться ректально или вагинально. В другом варианте осуществления соединения по настоящему изобретению могут также вводиться непосредственно в глаз или в ухо.
Схема приема соединений по настоящему изобретению и/или композиций, содержащих указанные соединения, основана на различных факторах, таких как тип, возраст, масса тела, пол и состояние здоровья пациента; тяжесть состояния; способ введения; и активность конкретного применяемого соединения. Таким образом, схема приема может меняться в широких пределах. В одном варианте осуществления общая суточная доза соединения по настоящему изобретению обычно составляет от примерно 0,001 до примерно 100 мг/кг (т.е. количество мг соединения по изобретению на кг массы тела пациента) для лечения состояний, обсужденных в настоящем документе. В другом варианте осуществления общая суточная доза соединения по настоящему изобретению составляет от примерно 0,01 до примерно 30 мг/кг, в еще одном варианте осуществления - от примерно 0,03 до примерно 10 мг/кг, и в еще одном варианте осуществления - от примерно 0,1 до примерно 3 мг/кг. Нередко введение соединений по настоящему изобретению будет повторяться несколько раз в день (обычно не более 4 раз). При желании для увеличения общей суточной дозы обычно можно использовать несколько доз в день.
Композиции для перорального введения могут быть представлены в форме таблеток, содержащих 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 30,0 50,0, 75,0, 100, 125, 150, 175, 200, 250 и 500 миллиграммов активного ингредиента для симптоматического корректирования дозы для пациента. Лекарственное средство обычно включает от примерно 0,01 мг до примерно 500 мг активного ингредиента или, в другом варианте осуществления, от примерно 1 мг до примерно 100 мг активного ингредиента. Дозы для внутривенного введения могут составлять от примерно 0,01 до примерно 10 мг/кг/минута при вливании с постоянной скоростью.
Подходящие пациенты по настоящему изобретению включают млекопитающих пациентов. В одном варианте осуществления подходящими пациентами являются люди. Люди могут быть любого пола и любой стадии развития.
Фармацевтические композиции
В другом варианте осуществления изобретение включает фармацевтические композиции. Такие фармацевтические композиции включают соединение по настоящему изобретению с фармацевтически приемлемым носителем. Могут присутствовать и другие фармацевтически активные соединения. Тармин «фармацевтически приемлемый носитель», когда используется в настоящем описании, включает любой и все растворители, дисперсионные среды, покрытия, бактерицидные и фунгицидные средства, изотонические добавки, а также вещества, замедляющие абсорбцию, и т.п., которые являются физиологически совместимыми. Примеры фармацевтически приемлемых носителей включают один или несколько носителей, выбранных из воды, физиологического раствора, фосфатно-буферного раствора, декстрозы, глицерина, этанола и т.п., а также их комбинации и могут включать изотонические добавки, например сахара, хлорид натрия или полиспирты, такие как маннит или сорбит. Могут добавляться фармацевтически приемлемые вещества, такие как смачивающие агенты, или незначительные количества вспомогательных веществ, таких как смачивающие агенты или эмульгаторы, консерванты или буферы, которые увеличивают срок хранения или эффективность антитела или фрагмента антитела.
Композиции по настоящему изобретению могут быть представлены в различных формах. Они включают, например, жидкие, полутвердые и твердые лекарственные формы, такие как жидкие растворы (например, растворы для инъекций или инфузий), дисперсии или суспензии, таблетки, пилюли, порошки, липосомы и суппозитории. Форма зависит от предполагаемого способа введения и терапевтического применения.
Типичные композиции представлены в форме растворов для инъекций или инфузий, аналогичных композициям, обычно применяемым для пассивной иммунизации человека антителами. Одним из способов введения является парентеральное введение (например, внутривенное, подкожное, внутрибрюшинное, внутримышечное введение). В другом варианте осуществления антитело вводят внутривенной инфузией или инъекцией. В еще одном варианте осуществления антитело вводится внутримышечной или подкожной инъекцией.
Лекарственная форма для перорального введения может быть представлена, например, в твердой лекарственной форме, полученной в дискретный единицах, таких как твердые или мягкие капсулы, пилюли, саше, пастилки или таблетки, каждая из которых содержит заданное количество по меньшей мере одного соединения по настоящему изобретению. В другом варианте осуществления лекарственная форма для перорального введения может быть представлена в форме порошка или гранул. В другом варианте осуществления лекарственная форма для перорального введения представляет собой форму для сублингвального введения, такую как, например, пастилка. В таких твердых лекарственных формах соединения формулы I обычно объединяются с одним или несколькими адъювантами. Такие капсулы или таблетки могут содержать препарат с контролируемым высвобождением действующего вещества. В случае капсул, таблеток и пилюль лекарственные формы также могут содержать буферные агенты или могут быть изготовлены с энтеросолюбильными покрытиями.
В другом варианте осуществления лекарственная форма для перорального введения может быть представлена в жидкой лекарственной форме. Примеры жидких лекарственных форм для перорального введения включают, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и элексиры, содержащие инертные разбавители, обычно используемые в данной области техники (например, воду). Такие композиции также могут включать адъюванты, такие как смачивающие, эмульгирующие, суспендирующие, вкусовые (например, подсластители) добавки и/или отдушки.
В другом варианте осуществления изобретение включает лекарственную форму для парентерального введения. Термин «парентеральное введение» включает, например, подкожные инъекции, внутривенные инъекции, внутрибрюшинное введение, внутримышечные инъекции, интраназальные инъекции и инфузию. Препараты для инъекций (т.е. стерильные водные или масляные суспензии для инъекций) могут быть получены с использованием традиционных в данной области техники подходящих дисперсантов, смачивающих агентов и/или суспендирующих агентов.
В другом варианте осуществления изобретение включает лекарственную форму для местного введения. Термин «местное введение» включает, например, чрескожное (трансдермальное) введение, такое как введение с помощью трансдермальных пластырей или устройства для ионтофореза, внутриглазное введение или интраназальное или ингаляционное введение. Композиции для местного введения также включают, например, гели для местного применения, спреи, мази и кремы. Препарат для местного введения может включать соединение, которое повышает абсорбцию или проникновение активного ингредиента через кожу или другие пораженные участки. Когда соединения по настоящему изобретению вводятся с помощью устройства для чрескожного введения, введение будет осуществляться с использованием пластыря резервуарного типа с пористой мембраной или пластыря с твердой матрицей. Типичные примеры препаратов для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пенки, пленки, кожные пластыри, накладки, импланты, спонжи, волокна, перевязочные материалы и микроэмульсии. Также могут использоваться липосомы. Типичные носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут также включаться добавки, повышающие проникновение - см., например, B. C. Finnin and T. M. Morgan, J. Pharm. Sci., vol. 88, pp. 955-958, 1999.
Препараты, подходящие для местного введения в глаз, включают, например, глазные капли, в которых соединение по настоящему изобретению растворено или суспендировано в подходящем носителе. Типичный препарат, подходящий для введения в глаз или в ухо, может быть представлен в форме капель микронизированной суспензии или раствора в изотоническом стерильном физиологическом растворе с заданным значением pH. Другие препараты, подходящие для введения в глаз или в ухо, включают мази, биологически разлагаемые (т.е абсорбируемые гелевые губки, коллаген) и биологически не разлагаемые (т.е. силиконовые) импланты, накладки, линзы и дисперсные или везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как сшитая полиакриловая килота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например гелановая камедь, может быть включен в препарат вместе с консервантом, таким как хлорид бензалкония. Такие препараты также могут доставляться с помощью ионофореза.
Для интраназального введения или введения ингаляцией соединения по настоящему изобретению удобно доставлять в форме раствора или суспензии из помпового аэрозольного баллончика, который сжимается или накачивается пациентом, или в виде аэрозольного спрея из баллона под давлением или набулайзера с использованием подходящего пропеллента. Препараты, подходящие для интраназального введения, обычно вводятся в форме сухого порошка (отдельно либо в виде смеси, например в сухой смеси с лактозой, или в виде частиц смешанного компонента, например в смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора сухого порошка или в виде аэрозольного спрея из контейнера под давлением, с помощью насоса, пульверизатора, аэрозольного ингалятора (предпочтительно распылителя, использующего электрогидродинамику для получения мелкодисперсного тумана) или небулайзера с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Порошшок для интраназального применения может содержать биоадгезивную добавку, например хитозан или циклодекстрин.
В другом варианте осуществления изобретение включает лекарственную форму для ректального введения. Лекарственная форма для ректального введения может представлять собой, например, суппозиторий. Традиционной основой для суппозиториев является какао-масло, но при необходимости могут использоваться и другие основы.
Также могут использоваться другие материалы носителей и способы введения, известные в фармацевтике. Фармацевтические композиции по настоящему изобретению могут быть получены любым из хорошо известных фармацевтических методов, таких как эффективное получение препарата и методики введения. Упомянутые выше способы эффективного получения препарата и методики введения хорошо известны в данной области и описаны в стандартных учебниках. Получение препаратов лекарственных средств описано, например, в Hoover, John E., Remington’s Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999.
Совместное введение
Соединения по настоящему изобретению могут использоваться отдельно или в комбинации с другими терапевтическими средствами. Изобретение включают любое применение, любой способ введения или любую композицию, определенную в настоящем документе, где соединение любого варианта осуществления формулы I или его фармацевтически приемлемая соль или фармацевтически приемлемый сольват указанного соединения или указанной соли используется в комбинации с одним или несколькими другими терапевтическими средствами, обсужденными в настоящем документе. Такое применение может включать фармацевтическую композицию, содержащую соединение формул I, II, III, IV, V или VI или его фармацевтически приемлемую соль, как определено в любом из вариантов осуществления, описанных в настоящем документе, в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом и одним или несколькими другими терапевтическими средствами, описанными в настоящем документе.
Введение двух или более соединений «в комбинации» означает, что все соединения вводятся достаточно близко по времени, чтобы каждое из них могло вызвать биологический эффект в одних и тех же временных рамках. Присутствие одного средства может изменить биологическое действие другого(их) соединения(й). Два или более соединений могут вводиться одновременно, с использованием «параллельного» введения или последовательно. Кроме того, совместное введение может осуществляться смешиванием соединений перед введением или введение соединений в один и тот же момент времени, но в отдельных лекарственных формах в один и тот же или в разные сайты введения.
Фразы «параллельное введение», «совместное введение», «одновременное введение» и «введение одновременно» означают, что соединения вводятся в комбинации.
В другом варианте осуществления изобретение относится к способам лечения, которые включают введение соединений по настоящему изобретению в комбинации с одним или несколькими другими фармацевтическими средствами, где одно или несколько других фармацевтических средств могут выбираться из средств, которые обсуждены в настоящем документе.
В одном варианте осуществления настоящего изобретения соединения по настоящему изобретению вводятся с противодиабетическим средством, включая, но без ограничения, бигуанид (например, метформин), сульфонилмочевину (например, толбутамид, глибенкламид, гликлазид, хлорпропамид, толазамид, ацетогексамид, гликлопирамид, глимепирид или глипизид), тиазолидиндион (например, пиоглитазон, росиглитазон или лобеглитазон), глитазар (например, сароглитазар, алеглитазар, мураглитазар или тесаглитазар), меглитинид (например, натегдилинид, репаглинид), ингибитор дипептидилпептидазы 4 (ДПП-4) (например, ситаглиптин, вилдаглиптин, саксаглиптин, линаглиптин, гемиглиптин, анаглиптин, тенелиглиптин, алоглиптин, трелаглиптин, дутоглиптин или омариглиптин), глитазон (например, пиоглитазон, росиглитазон, балаглитазон, ривоглитазон или лобеглитазон), ингибитор натрий-зависимого переносчика глюкозы 2 (sodium-glucose linked transporter 2 - SGLT2) (например, эмпаглифлозин, канаглифлозин, дапаглифлозин, ипраглифлозин, ипраглифлозин, тофоглифлозин, серглифлозина этабонат, ремоглифлозина этабонат или эртуглифлозин), ингибитор SGLTL1, агонист GPR40 (агонист FFAR1/FFA1, например фасиглифам), глюкозозависимый инсулинотропный пептид (GIP) и его аналоги, ингибитор альфа-глюкозидазы (например, воглибоза, акарбоза или миглитол) или инсулин или аналог инсулина, включая фармацевтически приемлемые соли указанных лекарственных средств и фармацевтически приемлемые сольваты указанных лекарственных средств и их солей.
В другом варианте осуществления соединения по настоящему изобретению вводятся со средством против ожирения, включая, но без ограничения, пептид YY или его аналог, агонист рецептора нейропептида Y типа 2 (NPYR2), антагонист NPYR1 или NPYR5, антагонист рецептора каннабиноидов 1 типа (CB1R), ингибитор липазы (например, орлистат), проостровковый пептид человека (human proislet peptide - HIP), агонист рецептора меланокортина 4 (например, сетмеланотид), антагонист рецептора-1 меланин-концентрирующего гормона 1, агонист фарнезоидного Х рецептора (FXR) (такой как обетихолевая кислота), зонисамид, фентермин (отдельно или в комбинации с топираматом), ингибитор обратного захвата норэпинефрина/дофамина (например, бупроприон), антагонист опиодного рецептора (например, налтрексон), комбинацию ингибитора обратного захвата норэпинефтина/дофамина и антагониста опиодного рецептора (такую как комбинация бупропиона и налтрексона), aналог GDF-15, сибутрамин, агонист холецистокинина, амилин и его аналоги (например, прамлинтид), лептин и его аналоги (например, метролептин), серотонинергическое средство (например, лоркасерин), ингибитор метионинаминопептидазы 2 (MetAP2) (например, белораниб или ZGN-1061), фендиметразин, диэтилпропион, бензфетамин, ингибитор SGLT2 (например, эмпаглифлозин, канаглифлозин, дапаглифлозин, ипраглифлозин, Ипраглифлозин, тофоглифлозин, серглифлозина этабонат, ремоглифлозина этабонат или эртуглифлозин), ингибитор SGLTL1, двойной ингибитор SGLT2/SGLT1, модулятор рецептора фактора роста фибробластов (FGFR), активатор AMФ-активируемой протеинкиназы (AMPK), биотин, модулятор MAS-рецептора или агонист рецептора глюкагона (отдельно или в комбинации с другим агонистом GLP-1R, например лираглютид, эксенатид, дулаглутид, албиглутид, ликсисенатид или семаглутид), включая фармацевтически приемлемые соли конкретно указанных лекарственных средств и фармацевтически приемлемые сольваты указанных лекарственных средств и их солей.
В другом варианте осуществления соединения по настоящему изобретения вводятся со средством для лечения НАСГ, включая, но без ограничения, PF-05221304, агонист FXR (например, обетихолевую кислоту, агонист PPAR α/δ (например, элафибранор), конъюгат синтетическая жирная кислота-желчная кислота (например, арамхол), ингибитор каспазы (например, эмрикасан), моноклональное антитело против гомолога лизилоксидазы 2 (LOXL2) (например, симтузумаб), ингибитор галектина 3 (например, GR-MD-02), ингибитор MAPK5 (например, GS-4997), двойной антагонист хемокинового рецептора 2 (CCR2) и CCR5 (например, ценикривирос), агонист фактора роста фибробласта 21 (FGF21) (например, BMS-986036), антагонист рецептора лейкотриена D4 (LTD4) (например, типелукаст), аналог ниацина (например, ARI 3037MO), ингибитор ASBT (например, воликсибат), ингибитор ацетил-КoA-карбоксилазы (AКК) (например, NDI 010976), ингибитор кетогексокиназы (KГK), ингибитор диацилглицерилацилтрансферазы 2 (DGAT2), антагонист рецептора CB1, антитело против CB1R или ингибитор регулирующей апопторические сигналы киназы 1 (ASK1), включая фармацевтически приемлемые соли конкретно указанных лекарственных средств и фармацевтически приемлемые сольваты указанных лекарственных средств и их солей.
Указанные лекарственные средства и соединения по настоящему изобретению могут объединяться с фармацевтически приемлемыми носителями, такими как физиологический раствор, раствор Рингера, раствор декстрозы и т.п. Конкретная схема лечения, то есть доза, время введения и количество повторных введений, будет зависеть от конкретного пациента и его истории болезни.
Подходящими являются носители, эксципиенты или стабилизаторы, нетоксичные для реципиентов в применяемых дозах и концентрациях, и могут включать буферы, такие как фосфатный, цитратный буферы и буферы других органических кислот; соли, такие как хлорид натрия; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как хлорид октадецилдиметилбензиламмония; хлорид гексаметония; хлорид бензалкония, хлорид бензетония; фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метил- или пропилпарабен; катехол; резорцин; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее примерно 10 остатоков) полипептиды; белки, такие как сывороточный альбумин, желатин или Igs; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатообразующие добавки, такие как EDTA; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как ион натрия; комплексы металлов (например, комплекс Zn-белок); и/или неионогенные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (PEG).
Липосомы, содержащие указанные лекарственные средства и/или соединения по настоящему изобретению, получают способами, известными в данной области техники, такими как описанные, например, в Патентах США № 4485045 и 4544545. Липосомы с увеличенным временем кругооборота в крови описаны в Патенте США № 5013556. Особенно подходящие липосомы могут быть получены методом обращенно-фазового выпаривания с липидной композицией, включающей фосфатидилхолин, холестерин и PEG-производный фосфатидилэтаноламин (PEG-PE). Липосомы подвергают экструзии через фильтры с определенным размером пор с получением липосом нужного диаметра.
Эти лекарственные средства и/или соединения по настоящему изобретению также могут вводиться в микрокапсулы, полученные, например, методами коацервации или межфазной полимеризации, например микрокапсулы гидроксиметилцеллюлозы или желатиновые микрокапсулы и поли(метилметакрилатныe) микрокапсулы, соответственно, в коллоидных системах доставки лекарственного средства (например, липосомах, альбуминовых микросферах, микроэмульсиях, наночастицах и нанокапсулах) или в макроэмульвиях. Такие методы описаны в Remington, The Science и Practice of Pharmacy, 20th Ed., Mack Publishing (2000).
Могут использоваться препараты с замедленным высвобождением действующего вещества. Примеры подходящих препаратов с замедленным высвобождением действующего вещества включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащие соединение формул I, II, III, IV, V или VI, причем указанные матрицы имеют определенную форму, например форму пленок или микрокапсул. Примеры матриц с замедленным высвобождением действующего вещества включают полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт)), полилактиды (Патент США № 3773919), сополимеры L-глутаминовой кислоты и 7-этил-L-глутамата, неразлагаемый этиленвинилацетат, разлагаемые сополимеры молочной кислоты и гликолевой кислоты, такие как сополимеры, используемые в LUPRON DEPOT™ (микросферы для инъекций, состоящие из сополимера молочной кислоты и гликолевой кислоты и ацетата лейпролида), изобутирата ацетата сахарозы и поли-D-(-)-3-оксимасляной кислоты.
Препараты, предназначенные для внутривенного введения, должны быть стерильными. Стерильность может быть легко достигнута, например, фильтрацией через стерильные фильтрующие мембраны. Соединения по настоящему изобретению обычно помещают в контейнер со стерильным отверстием для доступа, например в пакет с раствором для внутривенного введения или во флакон с пробкой, которую можно проткнуть иглой для подкожных инъекций.
Подходящие эмульсии могут быть получены с использованием коммерчески доступных жировых эмульсий, таких как Intralipid™, Liposyn™, Infonutrol™, Lipofundin™ и Lipiphysan™. Активный ингредиент может растворяться в предварительно полученной смешением эмульсионной композиции или, альтернативно, растворяться в масле (например, соевом масле, сафлоровом масле, хлопковом масле, кунжутном масле, кукурузном масле или миндальном масле) и эмульсии, полученной смешиванием с фосфолипидом (например, фосфолипидами яиц, соевыми фосфолипидами или соевым лецитином) и водой. Следует иметь в виду, что могут добавляться и другие ингредиенты, например глицерин или глюкоза, для регулирования тоничности эмульсии. Подходящие эмульсии обычно будут содержать до 20% масла, например от 5 до 20%. Жировая эмульсия может включать жировые капли диаметром от 0,1 до 1,0 мкм, в частности от 0,1 до 0,5 мкм, и ее значение рН может находиться в интервале от 5,5 до 8,0.
Композиции эмульсий могут быть получены смешиванием соединения по настоящему изобретению с Intralipid™ или его компонентами (соевым маслом, фосфолипидами яиц, глицерином и водой).
Композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях или их смесях, а также порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые эксципиенты, которые описаны выше. В некоторых вариантах осуществления композиции вводятся перорально или назально респираторным путем для местного или системного действия. Композиции в предпочтительно стерильных фармацевтически приемлемых растворителях могут вводиться опрыскиванием с использованием газов. Распределяемые опрыскиванием растворы могут вдыхаться непосредственно из распылительного устройства, или распыляющее устройство может прикрепляться к лицевой маске, тенту или дыхательному аппарату с периодическим нагнетанием. Композиции в виде раствора, суспензии или порошка могут вводиться, предпочтительно перорально или назально, из устройств, которые доставляют композицию соответствующим образом.
Наборы
Другой аспект настоящего изобретения относится к наборам, содержащим соединение формул I, II или III или фармацевтические композиции, включающие соединение формул I, II или III по настоящему изобретению. Набор может включать помимо соединения формул I, II или III по настоящему изобретению или его фармацевтической композиции диагностические или терапевтические средства. Набор также может включать инструкции по применению при диагностике или терапевтическом лечении. В некоторых вариантах осуществления набор включает соединение формул I, II, III, IV, V или VI или его фармацевтическую композицию и диагностическое средство. В других вариантах осуществления набор включает соединение формул I, II, III, IV, V или VI или его фармацевтическую композицию.
В еще одном варианте осуществления изобретение включает наборы, которые подходят для применения в способах лечения, описанных в настоящем документе. В одном варианте осуществления набор содержит первую лекарственную форму, включающую одно или несколько соединений по настоящему изобретению в количествах, достаточных для осуществления способа лечения по изобретению. В другом варианте осуществления набор включает одно или несколько соединений по настоящему изобретению в количествах, достаточных для осуществления способов лечения по настоящему изобретению, и контейнер для дозирования.
ПОЛУЧЕНИЕ
Соединения формул I, II, III, IV, V или VI могут быть получены общими и специфическими способами, описанными ниже, в соответствии с общей практикой органического синтеза. Такие общие методики можно найти в стандартных справочниках, таких как Comprehensive Organic Chemistry, Ed. Barton и Ollis, Elsevier; Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Larock, John Wiley and Sons; Compendium of Organic Synthetic Methods, Vol. I-XII (published by Wiley-Interscience). Исходные вещества, используемые в данных способах, являются коммерчески доступными или могут быть получены стандартными способами, известными в данной области техники.
Следует отметить, что при получении соединений формул I, II, III, IV, V или VI в соответствии с некоторыми способами получения может потребоваться защита удаленной функциональности (например, первичной аминогруппы, вторичной аминогруппы, карбоксильной группы в предшественниках соединений формулы I). Потребность в такой защите будет изменяться в зависимости от природы удаленной функциональности и условий синтеза. Необходимость такой защиты легко определяется специалистом в данной области техники. Применение методов введения/удаления защиты также опредляется специалистом в данной области техники. Общее описание защитных групп и их применения представлено в монографии T.W. Greene, Protective Groups в Organic Synthesis, John Wiley & Sons, New York, 1991.
Например, некоторые соединения содержат функциональные группы первичных аминов и карбоновой кислоты, которые могут оказывать неблагоприятное воздействие на реакции, протекающие в других сайтах молекулы, если остаются незащищенными. Соответственно, такие функциональности могут защищаться подходящей защитной группой, которая может удаляться на более поздней стадии. Подходящие защитные группы для аминогруппы и группы карбоновой кислоты включают защитные группы, обычно используемые в пептидном синтезе (такие как N-трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz) и 9-флуоренилметиленоксикарбонил (Fmoc) для аминов и низшие алкиловые или бензиловые эфиры для карбоновых кислот), которые обычно являются нереакционноспособными в описанных условиях реакции и могут удаляться без химического воздействия на другие функциональные группы соединений формулы I.
Схемы, описанные ниже, предназначены для общего описания методологии, применяемой при получении соединений по настоящему изобретению. Некоторые из соединений по настоящему изобретению могут содержать один или несколько хиральных центров со стереохимическим обозначением (R) или (S). Специалисту в данной области техники понятно, что все синтетические превращения могут осуществляться аналогичным образом, независимо от того, являются ли исходные вещества энантиообогащенными или рацемическими. Кроме того, разделение на желаемый оптически активный материал может осуществляться на любюбой стадии синтеза с использованием хорошо известных способов, которые описаны в настоящем документе и в химической литературе. Например, промежуточные соединения (например, S29, S32, S37 и S48) и конечные соединения (например, S49) могут разделяться с использованием методов хиральной хроматографии. Альтернативно, хиральные соли могут использоваться для выделения энантиомерно обогащенных промежуточных продуктов и конечных соединений.
На представленных далее схемах переменные A, A1, A2, X, Y, L, Z1, Z2, Z3, ZA1, ZA2, ZA3, R1, R2, R3, R4, R5, R6, m и q принимают значения, определенные для соединений формул I, II, III, IV, V или VI, если не указано иное. Для схем, представленных ниже, каждое p независимо равно 0 и 1. Для схем, представленных ниже, каждый X1, X2, X3 и X4 может независимо представлять собой удаляемую группу, такую как любой алкил или арилсульфонат (например, мезилат, тозилат или трифлат), галоген или любая другая группа, которая может замещаться амином или использоваться в реакции сочетания, опосредуемой металлом. X4 также может представлять собой защищенную группу карбоновой кислоты (т.e. сложный эфир). Когда защитная группа обозначена как Pg1, она может представлять собой защитную группу алкиламина, такую как бензил, бензгидрил или т.п.; защитную группу карбаматной группы, такую как Boc, Cbz, или т.п.; или защитную группу амидной группы, такую как трифторацетамид. Когда защитная группа обозначена как Pg2, она может представлять собой защитную группу кислоты, такую как метил, этил, бензил, трет-бутил или т.п. R4a представляет собой C1-2алкил, C0-2алкилен-C3-6циклоалкил, C0-2алкилен-R5 или C1-2алкилен-R6, где указанный алкил, алкилен или циклоалкил может быть независимо замещенным, как допускает валентность, 0-3 атомами F и 0-1 заместителем, независимо выбранным из C0-1алкилен-ORO и -N(RN)2.
Замещенный пиперидин общей структуры S6, где Y=CH, может быть получен как показано на схеме 1. Гетероцикл общей структуры S1 может подвергаться взаимодействию с бороновой кислотой или сложным боронатным эфиром (S2) в присутствии палладиевого катализатора и комплекса лиганда в соответствии с реакцией Сузуки (Maluenda и Navarro, Molecules, 2015, 20, 7528-7557) с получением соединений общей формулы S3. Когда ZA1 представляет собой CH или CR2, предпочтительная удаляемая группа X1 представляет собой F или SO2Me (полученная в результате окисления SMe, как показано на схеме 3), и предпочтительные заместители X2 включают Br и I. Когда ZA1 представляет собой N, X1 и X2 предпочтительно представляют собой Cl. Восстановление олефина для получения соединений общей структуры S4 может проводиться в атмосфере водорода (15-100 фунтов на кв. дюйм (103,42-689,48 кПа) Н2) в спиртовом растворителе, таком как MeOH или EtOH, или альтернативно, апротонном органическом растворителе, таком как EtOAc или ТГФ, в присутствии подходящего катализатора, такого как палладий на угле, Pd(OH)2 на угле (катализатор Перлмана), PtO2 (катализатор Адамса), или хлорид трис(трифенилфосфин)родия(I) (катализатор Вилкинсона). Реагенты трансферной гидрогенизации, например формиат аммония или дигидробензол или т.п., могут применяться с использованием подходящего катализатора. Альтернативно, восстановление может осуществляться альтернативными способами, известными специалисту в данной области техники, с использованием таких реагентов, как триэтилсилан или другие силаны, с кислотным или металлическим катализатором или металлическими восстановителями, такими как магний или т.п. Альтернативно, введение функциональной группы в олефин может осуществляться методами введения групп R3, известными специалистам в данной области техники. Например, олефин может подвергаться гидроборированию для получения спирта, который может подвергаться алкилированию или далее превращению в нитрильную группу, группу F или алкильную группу. Превращение в соединения общей структуры S6 может осуществляться таким способом, как реакция сочетания С-О Бухвальда-Хартвига (Lundgren and Stradiotto, Aldrich Chimica Acta, 2012, 45, 59-65) между соединениями общей структуры S4 и подходящим образом замещенного бензилового спирта S5 в присутствии палладиевого или медного катализатора и лигандного комплекса. Предпочтительным галогеном X1 является Cl. Данные реакции обычно проводятся при температуре в интервалев от 0 до 110°С в апротонных органических растворителях, таких как, но без ограничения, 1,4-диоксан и PhCH3, с добавленным основанием, такого как Cs2CO3, LiHMDS или NaOtBu. Альтернативно, взаимодействие S4 с соответственно замещенным бензиловым спиртом S5 в апротонном растворителе, таком как ДМФА или ТГФ, в присутствии сильного основания, такого как NaH, KOtBu или LiHMDS, может приводить к получению соединений общей структуры S6. Предпочтительные заместители X1 для данной реакции включают F и Cl или сульфоны (например, SO2Me).
Схема 1
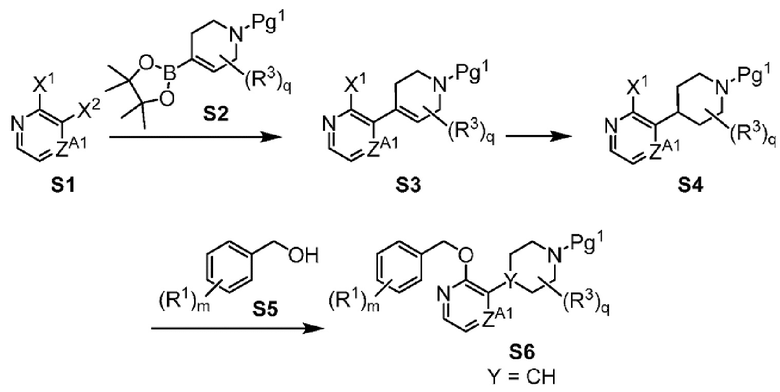
Замещенный пиперизин общей структуры S6, где Y=N, может быть получен как показано на схеме 2. Превращение S1 в соединения общей структуры S7 может осуществляться взаимодействием S1 с соответственно замещенным бензиловым спиртом S5 в апротонном растворителе, таком как ДМФА или ТГФ, в присутствии сильного основания, такого как NaH, KOtBu или LiHMDS, с получением соединений общей структуры S7. Когда ZA1 представляет собой CH или CR2, предпочтительная удаляемая группа X1 представляет собой F, и предпочтительными заместителями X2 являются Br и I. Когда ZA1 представляет собой N, предпочтительные группы Х1 и X2 представляют собой Cl. Превращение S7 в соединения общей структуры S6, где Y=N, может осуществляться в соответствии с реакцией сочетания С-О Бухвальда-Хартвига между соединениями общей структуры S7 и соответственно замещенным и защищенным пиперазином S8 в присутствии палладиевого или медного катализатора и лигандного комплекса. Данные реакции обычно проводятся при температуре в интервале от 0 до 110°C в апротонных растворителях, таких как, но без ограничения, 1,4-диоксан и PhCH3, с добавлением основания, такого как Cs2CO3, LiHMDS или NaOtBu.
Схема 2
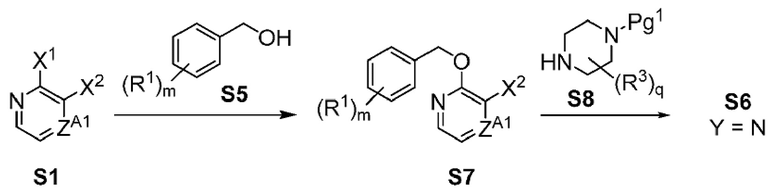
Как показано на схеме 3, соответственно замещенные сложные эфиры пиперидина общей структуры S9 могут подвергаться взаимодействию с пиримидинами S11 в присутствии сильного основания, такого как LiHMDS или LDA или другое подходящее основание, в апротонном органическом растворителе, таком как ТГФ, для получения соединений общей структуры S12. В реакциях S9 с S11 X1 предпочтительно представляет собой Cl, более предпочтительно SMe, X2 предпочтительно представляет собой Cl. Удаление Pg2 гидролизом сложных эфиров и декарбоксилированием полученных карбоновых кислота может приводить к получению пиперидинов S13 (X1=SMe) или S14 (X1=Cl). Когда X1 представляет собой SMe, окисление простого тиоэфира пероксидом, таким как мета-хлорпербензойная кислота, может приводить к получению сульфоксидов или сульфонов S14 (X1=S(O)Me, SO2Me). Взаимодействие пиримидина S14 (X1=Cl, S(O)Me, SO2Me) с соответственно замещенным бензиловым спиртом S5 в апротонном растворителе, таком как ДМФА или ТГФ, в присутствии сильного основания, такого как NaH, KOtBu, LiHMDS или NaHMDS, может приводить к получению соединений общей структуры S15.
Схема 3
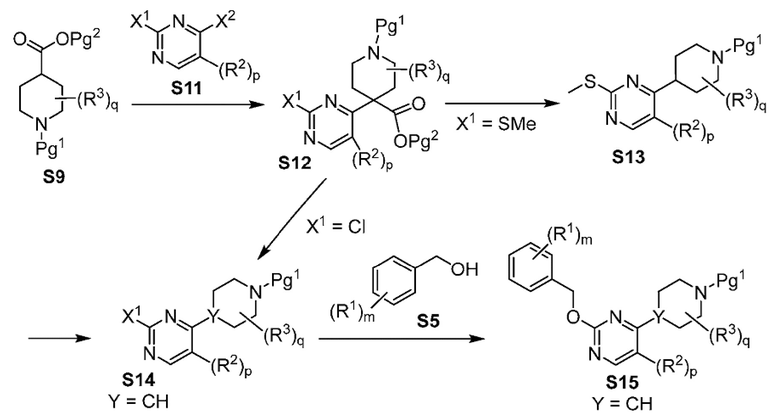
Соединения общей структуры S14, где Y=N, также могут быть получены как показано на схеме 4. Замещенный пиримидин S11 может подвергаться взаимодействию с пиперидинами общей структуры S8 в присутствии слабого основания, такого как триэтиламин или диизопропилэтиламин, в растворителе, таком как метанол, этанол, вода, ДМФА или ТГФ, при температуре примерно 30°C с получением соединений общей структуры S14. Предпочтительный заместитель X2 представляет собой Cl. Соединения S14 могут затем использоваться для получения S15, где Y=N, как показано на схеме 1, для получения S6 из S4, где предпочтительные заместители X1 включают Cl, Br или I.
Схема 4
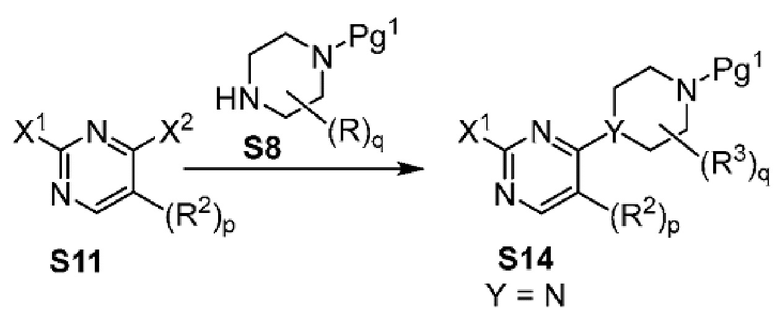
Как показано на схеме 5, взаимодействие S17 с соответственно замещенным бензиловым спиртом S5 в апротонном растворителе, таком как ДМФА или ТГФ, в присутствии сильного основания, такого как NaH, KOtBu или LiHMDS, может приводить к получению соединений общей структуры S18. Когда присутствуют электроноакцепторные заместители R2, могут использоваться такие основания как K2CO3. Предпочтительные заместители X1 для данной реакции включают F, Cl и Br, в то время как заместители X2 могут включать Cl, Br или I. Альтернативно, условия реакции сочетания С-О Бухвальда-Хартвига, аналогичные условиям получения S6, могут использоваться для получения S18 с предпочтительными X1 заместителями Cl, Br или I, как показано на схеме 1.
Схема 5
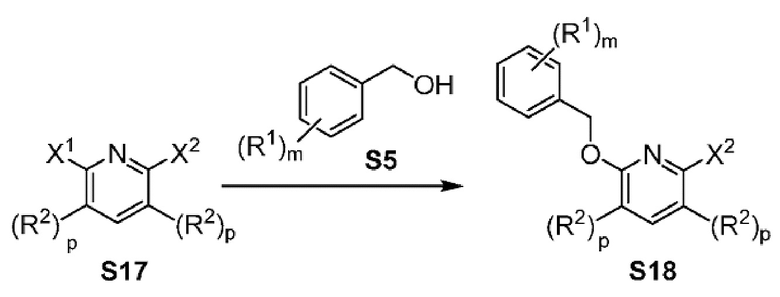
Как показано на схеме 6, взаимодействие пиримидина S19 с соответственно замещенным бензиловым спиртом S5 в апротонном растворителе, таком как ДМФА или ТГФ, в присутствии сильного основания, такого как NaH, KOtBu или LiHMDS, может приводить к получению соединений общей структуры S20. Предпочтительный заместитель X2 для данной реакции представляет собой Cl.
Схема 6
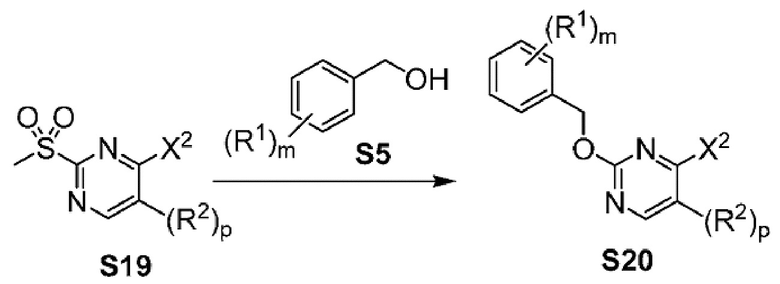
Как показано на схеме 7, взаимодействие пиримидина S21 с соответственно замещенным бензиловым спиртом S5 в апротонном растворителе, таком как ДМФА или ТГФ, в присутствии сильного основания, такого как NaH, KOtBu или LiHMDS, может приводить к получению соединений общей структуры S22. Предпочтительные заместители X1 и X2 для данной реакции представляют собой атомы Cl.
Схема 7
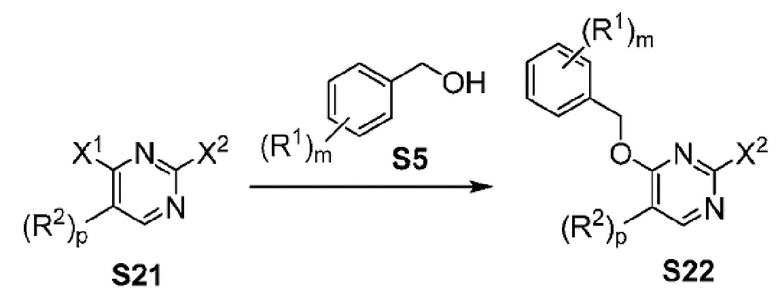
Замещенный пиперидин общей структуры S25, где Y=CH, может быть получен как показано на схеме 8. Соединения общей структуры S18, S20 и S22, обозначенные как S23, могут подвергаться взаимодействию с замещенной бороновой кислотой или сложным боронатным эфиром (S2), как показано для S1 на схеме 1. В реакции Сузуки галоген X2 предпочтительно представляет собой Cl, Br или I. Восстановление олефина, как показано на схеме 1, после этого может приводить к получению соединений общей структуры S25, где Y=CH.
Схема 8
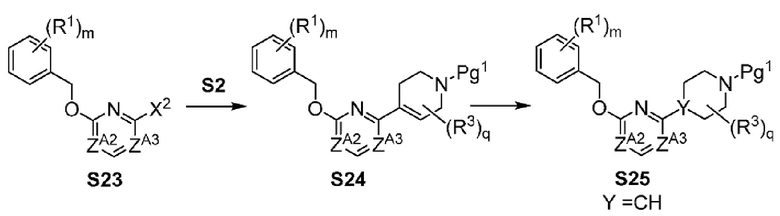
Как показано на схеме 9, превращение S23 в соединения общей структуры S25, где Y=N, может осуществляться таким способом, как реакция сочетания С-О Бухвальда-Хартвига между соединениями общей структуры S23 и соответственно замещенным и защищенным пиперазином в присутствии палладиевого или медного катализатора и лигандного комплекса, как показано на схеме 2 при превращении S7 в S6. Когда X2 представляет собой Cl и ZA3 представляет собой N, S23 и S8 могут взаимодействовать с получением S25 в присутствии слабого основания, такого как триэтиламин или диизопропилэтиламин, в растворителе, таком как метанол, этанол, вода, ДМФА или ТГФ, при температуре около 30°С.
Схема 9
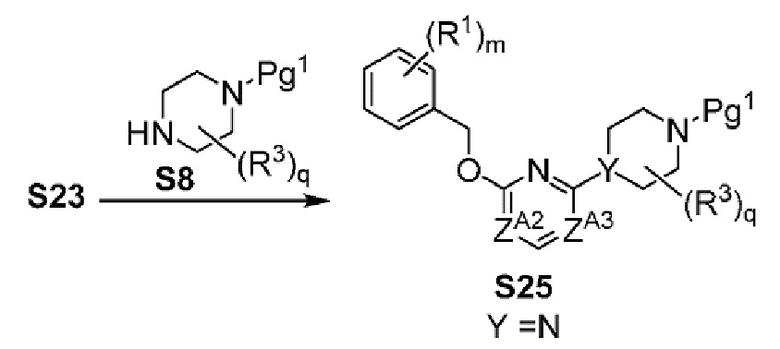
Карбоновые кислоты общей структуры S29, где X=N и L=CH2, могут быть получены как показано на схеме 10. Из соединений общих структур S6, S15 и S25, полученных способами, показанными на схемах 1-3, 8 и 9, и в совокупности обозначенных как S26, защитные группы могут быть удалены способами, описанными в литературе, с получением аминов общей структуры S27. Амины S27 могут подвергаться алкилированию защищенным 2-бромацетатом в присутствии подходящего основания, такого как K2CO3, Et3N, NaH или LiHMDS, в полярном апротонном растворителе, таком как, но без ограничения, ДМФА, DMAc, ДМСО или NMP, с получением соединений общей структуры S28, где X=N и L=CH2. Если Pg2 представляет собой трет-бутил, для получения кислот S29 могут использоваться стандартные способы удаления защитных групп кислот, такие как ТФУК/ДХМ, HCl/1,4-диоксан, HCl/EtOAc или другие подходящие условия. Если Pg2 представляет собой метил или этил, для получения кислот S29 могут использоваться стандартные способы удаления защитных групп кислот, такие как водный раствор NaOH в метаноле или этаноле или другие подходящие условия.
Схема 10
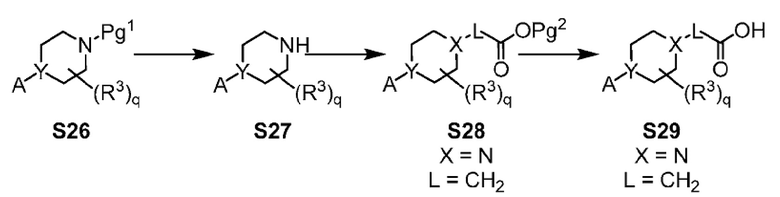
Соединения общей структуры S29, где Y=N и Х-L представляет собой циклопропил, могут быть получены как показано на схеме 11. Защищенный пиперидинон S30 может подвергаться гомологенированию до ненасыщенного сложного эфира 31 с использованием способов, хорошо известных специалистам в данной области техники. Например, олефинирование в соответствии с реакцией Хорнера-Вадсворта-Эммонса S30 с помощью фосфоната, такого как этил(диэтоксифосфорил)ацетат, который был депротонирован с помощью сильного основания, такого как гидрид натрия или трет-бутоксид калия, может приводить к получению S31. Реакция обычно проводится в апротонном растворителе, таком как ТГФ или DME, при температуре в интервале примерно от 0 до -50°С. Превращение S31 в циклопропановое производное S32 может осуществляться обработкой илидом сульфоксония, полученным из йодида триметилсульфоксония и основания, такого как трет-бутоксид калия или гидрид натрия. Удаление Pg1 из S32 будет приводить к получению аминов общей структуры S33, где Х-L представляет собой циклопропил. Арилгалогениды S7, S18, S20 и S22 обозначаются общей структурой S34. Связывание S33 с соединениями общей структуры S34 способом, аналогичным показанному на схеме 2 для получения S6 из S7 и S8, приводит к получении S35, где Y=N, и Х-L представляет собой циклопропил. Удаление Pg2 затем может приводить к получению соединений общей структуры S29, где Y=N и Х-L представляет собой циклопропил.
Схема 11
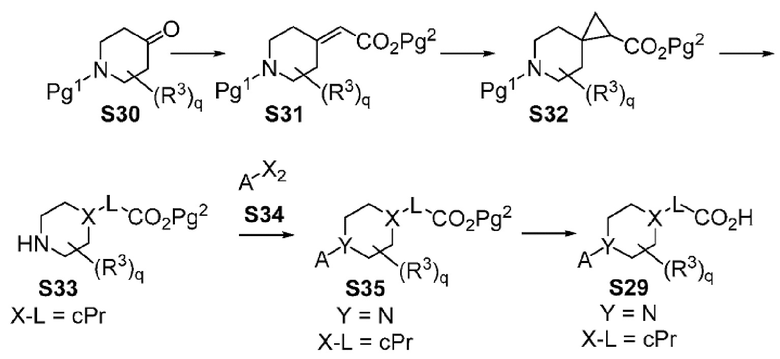
Альтернативно, защищенные карбоновые кислоты общей структуры S33, где X=N и L=CH2, могут быть получены как показано на схеме 12. Соответственно защищенные пиперазины S8 могут подвергаться алкилированию с помощью защищенного 2-бромацетата, как показано в способе получения S28 на схеме 10, с получением соединений общей структуры S36. Удаление Pg1 может затем приводить к получению соединений общей структуры S33, которые далее могут использоваться для получения S29, где X=N и L=CH2, как показано на схеме 11.
Схема 12
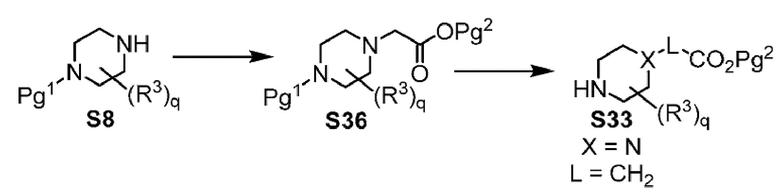
Соединения общей структуры S29, где Y=N, A=A2, ZA2=N и ZA3=CH или CR2, также могут быть получены как показано на схеме 13. Замещенный пиримидин S16 может подвергаться взаимодействию с аминами общей структуры S33 в присутствии слабого основания, такого как триэтиламин или диизопропилэтиламин, в растворителе, таком как метанол, этанол, вода, ДМФА или ТГФ, при температуре около 30°С для получения соединений общей структуры S37. Взаимодействие S37 с соответственно замещенным бензиловым спиртом в апротонном растворителе, таком как ДМФА или ТГФ, в присутствии сильного основания, такого как NaH, KOtBu или LiHMDS, может приводить к получению соединений общей структуры S38. Предпочтительными заместителями X1 и X2 для данных реакций являются атомы Cl. Альтернативно, условия реакция сочетания С-О Бухвальда-Хартвига, аналогичные реакции получения S6, могут использоваться для получения S38 с предпочтительным X1 заместителем Cl. Удаление Pg2 может затем приводить к получению соединений общей структуры S29, где Y=N, A=A2, ZA2=N и ZA3=CH или CR2.
Схема 13
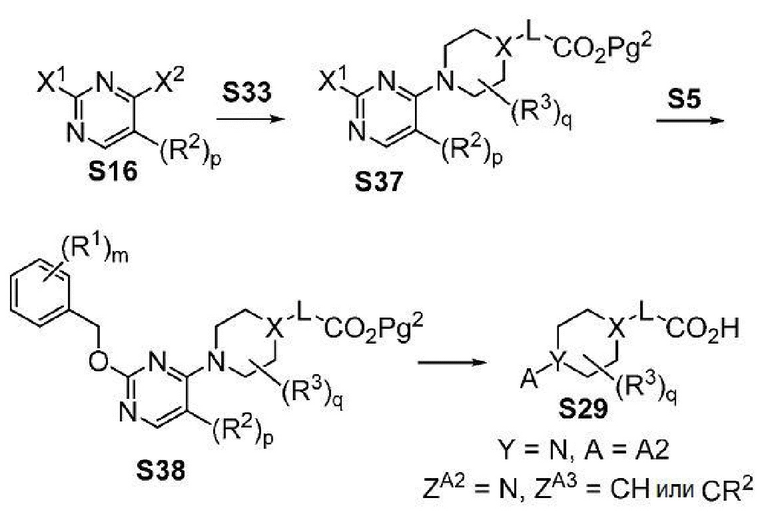
Как показано на схеме 14, соединения общей структуры S39 могут подвергаться взаимодействию с аминами R4NH2 в присутствии оснований, таких как карбонат, бикарбонат, гидроксид, ацетат натрия, калия или цезия, или органического аминного основания, такого как Et3N, DIPEA, DBU и т.п., в полярном апротонном растворителе, таком как, но без ограничения, ТГФ, ДМФА, DMAc, ДМСО или NMP, или протонном растворителе, таком как вода, MeOH, EtOH или iPrOH, или в смеси указанных растворителей с получением соединений общей структуры S40. Следует отметить, что если пример обеспечивает получение R4 с разделенным энантиомерным центром, другой энантиомер или их рацемическую смесь можно получить с помощью подбора подходящего исходного материала. Предпочтительные X3 заместители включают F, Cl и Br, предпочтительные группы X4 включают Cl, Br, -CO2-Pg2. Восстановление нитрогруппы может осуществляться гидрированием H2 с давлением 1-6 атм (101,32-607,95 кПа) с металлическим катализатором, таким как палладий на угле или никель Ренея, в протонном растворителе, таком как MeOH или EtOH, или апротонном растворителе, таком как ДМФА, ТГФ или EtOAc. Альтернативно, нитрогруппа может подвергаться восстановлению железом, цинком, SnCl2 или другим подходящим металлом в кислой среде, такой как 1N HCl, AcOH или водный NH4Cl в ТГФ, с получением соединений общей структуры S41 (схема 8a). Соединения S42 могут подвергаться ацилированию ацилгалогенидами стандартным способом или карбоксилатами в соответствии со стандартными методиками амидного связывания с получением соединений S43. Восстановление до соединений S44 может осуществляться в стандартных условиях с помощью восстановителей, таких как LAH или BH3-ТГФ или BH3-DMS (схема 14b).
Схема 14
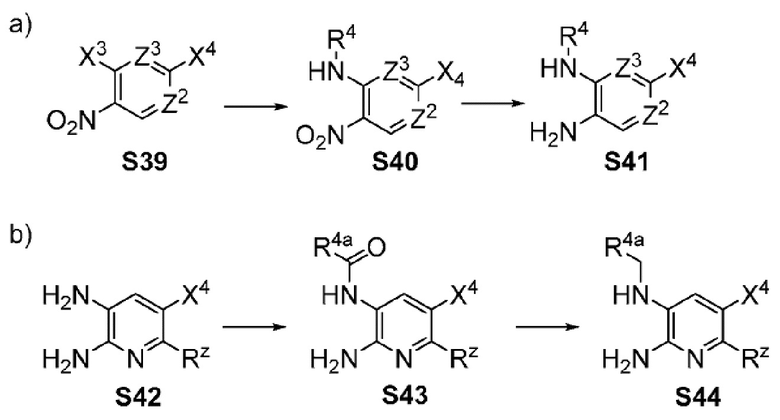
Диамины S41 и S44, полученные способами, представленными на схемах 14a и 14b, и совокупно обозначенные как диамин S45 (схема 15), могут подвергаться ацилированию кислотами общей структуры S29 в соответствии со стандартными методиками связывания амидов с получением аминов S46, которые будут представлены в виде смеси с содержанием от 100% S46a до 100% S46b. Данная смесь аминов S46 может подвергаться циклизации с получением соединений общей структуры S47 различными способами. Амины S46 могут подвергаться нагреванию с дегидратирующим агентом, таким как T3P® или алкиловый спирт, такой как н-бутанол, в условиях микроволнового облучения (10-60 мин. при 120-180°C) с получением соединений S47. Альтернативно, смесь аминов S46 может подвергаться нагреванию в кислотных условиях, таких как AcOH, до 60-100°C или в основных условиях, таких как водный NaOH или KOH в 1,4-диоксане, до температуры 60-100°C с получением S47. Соединения общей структуры S47 (X4=Cl, Br или I) могут подвергаться превращению в сложные эфиры структуры S48 карбонилированием в присутствии палладиевого катализатора в атмосфере моноксида углерода с давлением 15-100 фунтов на кв. дюйм (103,42-689,48 кПа) при температуре 20-100°С с подходящим спиртом, таким как MeOH или EtOH или другой алкиловый спирт. Гидролиз сложного эфира S48 может осуществляться как показано на схеме 10 с получением кислот S49. Для соединений S46, где X4=CO2-Pg2, превращение в сложный эфир S48 проводится в аналогичных условиях, как описано выше, но с тем отличием, что применяется метод циклизации в основных условиях, где соединение S49 может выделяться непосредственно из реакционной смеси. Для соединений S48, где X4 представляет собой CO2tBu, удаление защитной группы с получением кислоты 49 может осуществляться в кислотных условиях, показанных на схеме 10. Альтернативно, для соединений S48, где Pg2 представляет собой C1-C8 алкил, такой как метил, этил, гексил или октил, удаление защитной группы из сложного эфира может осуществляться в присутствии различных ферментов, включая эстеразы, протеазы, пептидазы, липазы и гликозидазы, которые хорошо известны специалистам в данной области техники. Гидролиз также может осуществляться обработкой сложного эфира водным раствором 1,5,7-триазабицикло[4.4.0]дец-5-ена при комнатной температуре.
Схема 15
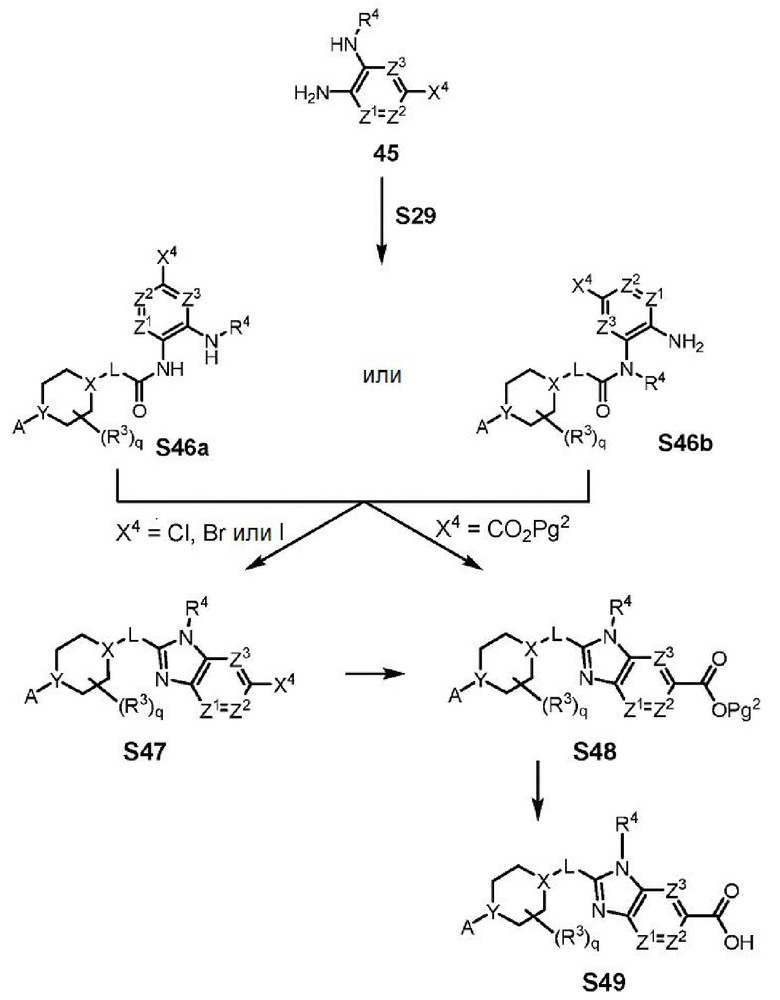
Кроме того, диамин S45 может подвергаться превращению в 2-хлорметилбензимидазол S50 (схема 16) несколькими способами. Обработка 2-хлорацетилхлоридом в апротонном растворителе, таком как 1,4-диоксан, с последующим нагреванием до 40-100°С и выдерживанием при указанной температуре в течение 2-18 часов может приводить к получению целевого бензимидазола S50, где Z1, Z2 и Z3 представляют собой CH. В случае, когда Z1, Z2 и Z3 не являются одновременно CRz, после обработки 2-хлорацетилхлоридом в апротонном растворителе, таком как 1,4-диоксан, в течение от 30 минут до 4 часов растворитель заменяют на кислотную среду, такую как AcOH или ТФУК, с последующим нагреванием до 40-100°С и выдерживанием при указанной температуре в течение 2-18 часов с получением целевого соединения S50. Диамин S45 может также подвергаться обработке хлоруксусным ангидридом при температуре в интервале от 0 до 80°С в апротонном растворителе, таком как, но без ограничения, 1,4-диоксан, ТГФ или MeCN, и последующему выдерживанию в течение от 2 до 18 часов при температуре 60-100°C с получением целевого соединения S50. Кроме того, диамин S45 может подвергаться обработке 2-хлор-1,1,1-триметоксиэтаном в апротонном растворителе, таком как, но без ограничения, 1,4-диоксан, ТГФ или MeCN, или протонном растворителе, например, MeOH или EtOH, в присутствии кислотного катализатора, например pTSA, при 20-100°C. Альтернативно, диамины S45 могут подвергаться нагреванию до 100-180°C с 2-оксиуксусной кислотой в апротонном растворителе, таком как, но без ограничения, мезитилен, с получением гидроксиметилового промежуточного соединения. Превращение гидроксиметильной группы в хлорметильную группу в соединении S50 может осуществляться стандартными способами, включая обработку SOCl2 в апротонном растворителе. Соединения общей структуры S50 могут подвергаться взаимодействию с соединениями S27 в присутствии оснований, таких как карбонат, бикарбонат натрия, калия или цезия, NaH, или органического аминного основания, такого как Et3N, DIPEA, DBU и т.п., в полярном апротонном растворителе, таком как, но без ограничения, ТГФ, MeCN, ДМФА, DMAc, ДМСО или NMP, с получением соединений S47 (X4=Cl, Br, I) или соединений S48 (X4=CO2-Pg2), которые затем используются для получения соединений S49 способами, представленными на схеме 15.
Схема 16
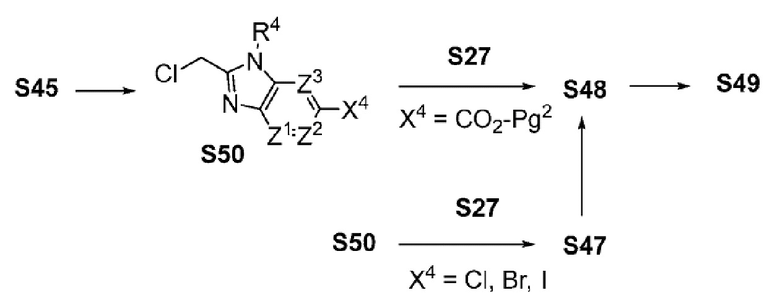
Альтернативно, соединения общей структуры S50 могут подвергаться взаимодействию с соответственно замещенными и защищенными пиперазинами S8 с получением соединений S51 (схема 17). Удаление Pg1 может осуществляться рядом способов, описанных в литературе, с получением аминов S52. Превращение в соединения общей структуры S47 (X4=Cl, Br или I) или S48 (X4=CO2-Pg2) может осуществляться таким способом, как реакция сочетания С-О Бухвальда-Хартвига между соединениями общей структуры S7 и как показано ранее на схеме 2. Соединения общей структуры S47 или S48 затем могут использоваться для получения соединений структуры S49 способами, представленными на схеме 15.
Схема 17
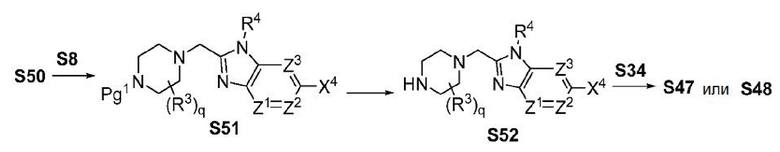
Способом, аналогичным представленному на схеме 1 для получения S4, соединения общей структуры S29, где X=Y=CH, L=CH2, A=A2, ZA2=CH или CR2 и ZA3=CH или CR2, также могут быть получены как показано на схеме 18a. Соединения общей структуры S17 могут подвергаться взаимодействию со сложными боронатными эфирами S53 в присутствии палладиевого катализатора и комплекса лиганда способом, аналогичным описанному для получения S3 из S1 и S2, с получением промежуточного олефина S54. В этом взаимодействии предпочтительные заместители X2 представляют собой Cl и Br. Восстановление олефина будет приводить к получению циклогексильных производных S55 со стереоселективностью восстановления, обеспечивающей цис- или транс-изомер или их смесь в зависимости от условий и специфической природы заместителей. Взаимодействие S55 со спиртами S5, которые описаны выше, будет приводить к получению соединений общей структуры S56. Удаление Pg2 будет обеспечивать получение соединений S29 (X=Y=CH, L=CH2, A=A2, ZA2=CH или CR2 и ZA3=CH или CR2). Альтернативно, соединения общей структуры S29, где X=Y=CH, L=CH2, могут быть получены из замещенных гетероарилгалогенидов общей структуры S34 взаимодействием с S53 в присутствии палладиевого катализатора и комплекса лиганда (схема 18b). Восстановление полученного олефина (S57) до S58 с последующим удалением Pg2 будет обеспечивать получение соединений S29 (X=Y=CH, L=CH2). Соединения S29, полученные данным способом, могут подвергаться превращению в соединения общей структуры S49 способами, представленными выше на схеме 15.
Схема 18
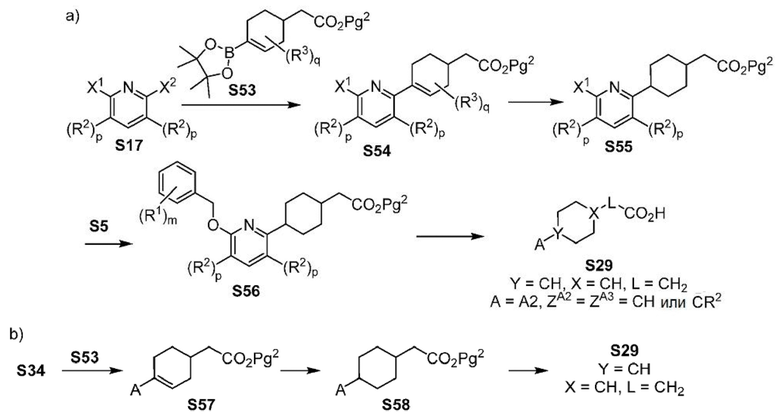
Примеры
Далее представлены примеры синтеза различных соединений по настоящему изобретению. Дополнительные соединения, относящиеся к настоящему изобретению, могут быть получены с использованием методов, проиллюстрированных в этих примерах, отдельно либо в сочетании с методами, широко известными в данной области техники.
Эксперименты обычно проводятся в инертной атмосфере (азот или аргон), особенно в тех случаях, когда используются чувствительные к кислороду или влаге реагенты или промежуточные соединения. Коммерчески доступные растворители и реагенты обычно используются без дополнительной очистки. При необходимости используются безводные растворители, как правило продукты AcroSeal® от Acros Organics, Aldrich® Sure/Seal™ от Sigma-Aldrich или продукты DriSolv® от EMD Chemicals. В других случаях коммерческие растворители пропускают через колонки, заполненные молекулярными ситами 4Å, до достижения содержания воды, соответствующего следующим стандартам контроля качества (КК): а) <100 м.д. для дихлорметана, толуола, N, N-диметилформамида и ТГФ; b) <180 м.д. для метанола, этанола, 1,4-диоксана и диизопропиламина. Для очень чувствительных реакций растворители дополнительно обрабатывают металлическим натрием, гидридом кальция или молекулярными ситами и дистиллируют непосредственно перед использованием. Вещества, как правило, сушат в вакууме, прежде чем передают их в последующие реакции или на биологические испытания. Данные масс-спектрометрии получают либо с помощью приборов жидкостной хроматографии-масс-спектрометрии (ЖХМС), химической ионизации при атмосферном давлении (atmospheric pressure chemical ionization - APCI), либо с помощью приборов газовой хроматографии-масс-спектрометрии (ГХМС). Символ ♦ означает, что в масс-спектре наблюдается характерная картина изотопа хлора.
Для разделения отдельных энантиомеров используют хиральное разделение. Полученные после разделения энантиомеры обозначают как ENT-1 или ENT-2 в соответствии с порядном их элюирования. Для соединений с двумя хиральными центрами стереоизомеры при каждом стереоцентром разделяют в разное время. Обозначение ENT-1 или ENT-2 промежуточного соединения или соединения примера относится к хирального центру, энтиомеры которого разделяяются на данной стадии. Понятно, что когда разделяются стереоизомеры при хиральном центре в соединении с двумя или более центрами, разделенные энантиомеры являются дистереомерами относительно друг друга. Обозначение ENT-1 или ENT-2 используется в настоящем описании для единообразия и относится к конкретному хиральному центру. Например, но без ограничения, соединения примеров 35 и 36 содержат два хиральных центра. Энантиомеры по хиральному центру циклопропильного фрагмента разделяют при разделении промежуточного соединения C77 на ENT-1, который представляет собой промежуточное соединение C78, и ENT-2, которое представляет собой промежуточное соединение C79. После этого C78 используют для получения из соединения С78 соединения примера 35, которое идентифицируется названием как 2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония. В этих способах получения после осуществления методик разделения хиральный центр идентифицируется символом “abs” около этого центра, подразумевая, что разделенные энантиомеры могут не быть энантиомерно чистыми. Обычно содержание обогащенного энантиомера при каждом хиральном центре составляет >90% в выделенном материале. Предпочтительно, содержание обобщенного энантиомера при каждом хиральном центре составляет >98%.
В некоторых примерах оптическое вращение энантиомера измеряют с использованием поляриметра. В соответствии с данными наблюдаемого вращения (или данными специфического вращения), энантиомер с вращением по часовой стрелке обозначается как (+)-энантиомер, энантиомер с вращением против часовой стрелки обозначается как (-)-энантиомер. У рацемических соединений отсутствует обозначение значками стереохимии, или они обозначаются значком (+/-) рядом со структурой; в последнем случае указанная стереохимия представляет относительную (а не абсолютную) конфигурацию заместителей соединения.
Реакции, протекающие через получение обнаруживаемых промежуточных соединения, обычно отслеживают с помощью ЖХМС и проводят до полной конверсии перед добавлением последующих реагентов. Условия реакций (продолжительность и температура реакции) синтеза, в котором сделала ссылка на другие примеры или способы, могут изменяться. Обычно после реакция проводится анализ тонкослойной хроматографией или масс-спектрометрией и при необходимости реакционная смесь подвергается дополнительной обработке. Очистка может изменяться в зависимости от эксперимента: обычно растворители и соотношения растворителей, используемые для элюентов/градиентов, подбираются для обеспечения подходящих значений Rf или времени удерживания. Все исходные материалы в этих способах получения и примерах либо являются коммерчески доступными, либо могут быть получены способами, известными в данной области техники, или как описано в настоящем документе.
Получение P1
(4-{2-[(4-Хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)уксусная кислота (P1)
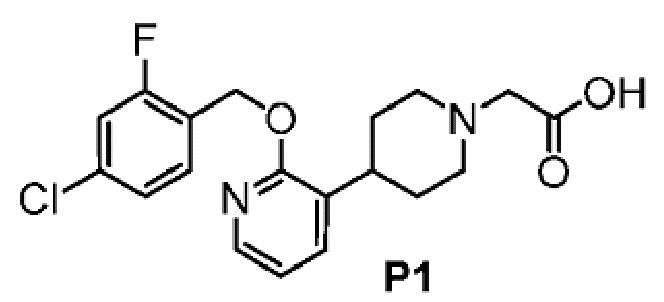
Стадия 1. Синтез трет-бутил-2-фтор-3',6'-дигидро-3,4'-бипиридин-1'(2'H)-карбоксилата (C1).
Смесь 3-бром-2-фторпиридина (14,5 г, 82,4 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилата (28,0 г, 90,6 ммоль), [1,1’-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) (3,80 г, 5,19 ммоль) и карбоната калия (25,0 г, 181 ммоль) в 1,4-диоксане (200 мл) и воде (50 мл) перемешивают при 100°C в течение 16 часов. После этого реакционную смесь распределяют между насыщенным водным раствором хлорида натрия (200 мл) и EtOAc (300 мл); органический слой сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 0% до 9% EtOAc в петролейном эфире) приводит к получению С1 в виде масла желтого цвета. Выход: 20,0 г, 71,9 ммоль, 87%.
Стадия 2. Синтез трет-бутил-4-(2-фторпиридин-3-ил)пиперидин-1-карбоксилата (C2).
Смесь C1 (20,0 г, 71,9 ммоль) и оксида платины (IV) (1,97 г) в MeOH (500 мл) гидрируют при 15°C в течение 16 часов, после чего реакционную смесь фильтруют. Фильтрат концентрируют в вакууме и остаток очищают хроматографией (силикагель, градиент: от 9% до 17% EtOAc в петролейном эфире) с получением C2 в виде масла бледно-желтого цвета. Выход: 20,0 г, 71,3 ммоль, 99%.
Стадия 3. Синтез трет-бутил-4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-карбоксилата (C3).
Гидрид натрия (60% дисперсия в минеральном масле; 9,0 г, 220 ммоль) добавляют порциями к смеси C2 (20,0 г, 71,3 ммоль) и (4-хлор-2-фторфенил)метанола (12,0 г, 74,7 ммоль) в N, N-диметилформамиде (150 мл) при 0°C. После этого реакционной смеси дают возможность нагреться до 15°C, смесь перемешивают при 15°C в течение 1 часа, после чего охлаждают до 0°C и медлденно обрабатывают насыщенным водным раствором хлорида аммония (100 мл). Полученную смесь экстрагируют EtOAc (300 мл), органический слой промывают насыщенным водным раствором хлорида натрия (200 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, элюент: 20:1 петролейный эфир/EtOAc) приводит к получению C3 в виде твердого белого вещества. Выход: 21,0 г, 49,9 ммоль, 70%.
Стадия 4. Синтез 2-[(4-хлор-2-фторбензил)окси]-3-(пиперидин-4-ил)пиридина (C4)
К раствору C3 (14,0 г, 33,3 ммоль) в дихлорметане (25 мл) добавляют трифторуксусную кислоту (25 мл). Реакционную смесь перемешивают при 20°C в течение 1,5 часа, после чего концентрируют в вакууме с получением C4 в виде масла желтого цвета. Полученный продукт используют непосредственно в следующей стадии.
Стадия 5. Синтез этил-(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)ацетата (C5)
Карбонат калия (30,0 г, 217 ммоль) добавляют к раствору C4 (полученного на предыдущей стадии; ≤33,3 ммоль) и этилбромацетата (6,00 г, 35,9 ммоль) в N, N-диметилформамидe (150 мл). Реакционную смесь перемешивают при 50°C в течение 2 часов и затем фильтруют. Фильтрат разбавляют EtOAc (250 мл), промывают последовательно водой (250 мл) и насыщенным раствором хлорида натрия (250 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 9% до 25% EtOAc в петролейном эфире) приводит к получению C5 в виде масла желтого цвета. Выход: 9,80 г, 24,1 ммоль, 72% для 2 стадий.
Стадия 6. Синтез (4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)уксусной кислоты (P1)
К раствору C5 (8,80 г, 21,6 ммоль) в ТГФ (45 мл) и MeOH (45 мл) добавляют водный раствор гидроксида натрия (3 M; 90 мл, 270 ммоль). Реакционную смесь перемешивают при 20°C в течение 2 часов, после чего значение pH доводят до 4 добавлением 1 M соляной кислоты. Полученную смесь фильтруют, собранный твердый продукт промывают три раза водой и затем переносят в смесь MeOH (20 мл) и воды (60 мл). Полученную смесь концентрируют в вакууме для удаления органических растворителей и затем лиофилизуют с получением P1 в виде твердого белого вещества. Выход: 5,90 г, 15,6 ммоль, 72%. ЖХМС m/z 378.9♦ [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,03 (уш. д, 1H), 7,60 (уш. д, 1H), 7,55 (дд, 1H), 7,49 (уш. д, 1H), 7,32 (дд, 1H), 7,01 (дд, 1H), 5,41 (с, 2H), 3,26-3,17 (м, 4H), 2,90-2,79 (м, 1H), 2,66-2,55 (м, 2H), 1,85-1,73 (м, 4H).
Получение P2
(4-{3-[(4-Хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)ацетат аммония (P2)
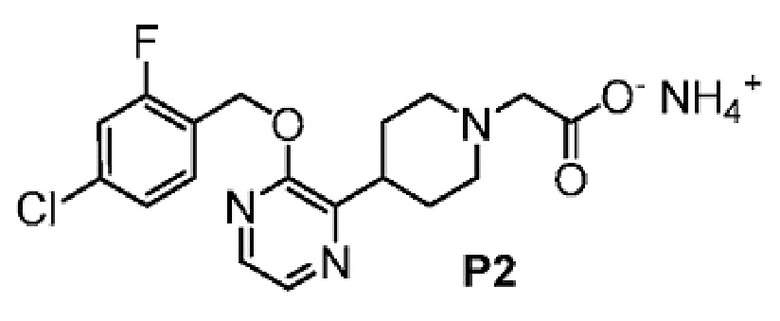
Стадия 1. Синтез трет-бутил-4-(3-хлорпиразин-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилата (C6
Смесь трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилата (20,0 г, 64,7 ммоль), 2,3-дихлорпиразина (14,5 г, 97,3 ммоль), [1,1’-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) (2,60 г, 3,55 ммоль) и карбоната натрия (15,1 г, 142 ммоль) в смеси 1,4-диоксана (120 мл) и воды (50 мл) перемешивают при 95°C в течение 4 часов. После этого реакционную смесь разбавляют EtOAc (500 мл) и промывают последовательно водой (100 мл) и насыщенным раствором хлорида натрия (300 мл). Органический слой сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и остаток очищают хроматографией (силикагель, градиент: от 5% до 17% EtOAc в петролейном эфире) с получением C6 в виде масла бледно-желтого цвета. Выход: 15,7 г, 53,1 ммоль, 82%.
Стадия 2. Синтез трет-бутил-4-(3-хлорпиразин-2-ил)пиперидин-1-карбоксилата (C7)
К раствору C6 (12,6 г, 42,6 ммоль) в MeOH (400 мл) добавляют хлорид трис(трифенилфосфин)родия (I) (2,50 г, 2,70 ммоль) и перемешивают полученную реакционную смесь в течение 2 часов при 30°C в атмосфере водорода из баллона. Полученную смесь фильтруют, фильтрат концентрируют в вакууме и остаток объединяют с продуктом аналогичной реакции гидрирования, проведенной с использованием C6 (1,08 г, 3,65 ммоль); очистка хроматографией (силикагель, градиент: от 9% до 17% EtOAc в петролейном эфире) приводит к получению C7 в виде твердого вещества бледно-желтого цвета. Общий выход: 12,3 г, 41,3 ммоль, 89%.
Стадия 3. Синтез трет-бутил-4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-карбоксилата (C8)
Ацетат палладия (II) (271 мг, 1,21 ммоль), 1,1'-бинафталин-2,2'-диилбис(дифенилфосфан) (BINAP; 1,51 г, 2,42 ммоль) и карбонат цезия (13,1 мг, 40,3 ммоль) добавляют к раствору C7 (6,00 г, 20,1 ммоль) и (4-хлор-2-фторфенил)метанола (3,56 г, 22,2 ммоль) в 1,4-диоксане (60 мл). Через суспензию барботируют азот, после чего реакционную смесь перемешивают при 90°C в течение 18 часов и затем фильтруют. Фильтрат концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 3% до 20% EtOAc в петролейном эфире) с получением C8 в виде смолы желтого цвета. Выход: 7,96 г, 18,9 ммоль, 94%.
Стадия 4. Синтез 2-[(4-хлор-2-фторбензил)окси]-3-(пиперидин-4-ил)пиразина (C9)
К раствору C8 (7,00 г, 16,6 ммоль) в дихлорметане (80 мл) добавляют трифторуксусную кислоту (20 мл). Реакционную смесь перемешивают при 15°C в течение 1 часа, после чего концентрируют и осторожно обрабатывают остаток насыщенным водным раствором бикарбоната натрия (100 мл). Полученную смесь экстрагируют EtOAc (3 × 100 мл), объединенные органические слои промывают насыщенным водным раствором хлорида натрия (300 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 10% до 25% MeOH в дихлорметане) приводит к получению C9 в виде твердого вещества желтого цвета. Выход: 5,2 г, 16 ммоль, 96%.
Стадия 5. Синтез этил-(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)ацетата (C10)
Этилбромацетат (2,55 г, 15,3 ммоль) и карбонат калия (6,03 г, 43,6 ммоль) добавляют к раствору C9 (4,68 мг, 14,5 ммоль) в N, N-диметилформамиде (50 мл). Реакционную смесь перемешивают при 15°C в течение 18 часов, затем разбавляют водой (100 мл) и экстрагируют EtOAc (3 × 100 мл). Объединенные органические слои промывают последовательно водой (200 мл) и насыщенным раствором хлорида натрия (200 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением C10 в виде смолы желтого цвета. Выход: 4,75 г, 11,6 ммоль, 80%.
Стадия 6. Синтез (4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)ацетата аммония (P2)
К раствору C10 (4,70 г, 11,5 ммоль) в MeOH (40 мл) добавляют водный раствор гидроксида натрия (3 M; 20 мл, 60 ммоль). Реакционную смесь перемешивают при 15°C в течение 2 часов, после чего разбавляют водой (80 мл) и промывают EtOAc (3 × 80 мл). Значение рН водного слоя затем доводят до 6 добавлением 1 М соляной кислоты и концентрируют смесь в вакууме. Остаток обрабатывают смесью дихлорметана и MeOH (10:1, 50 мл), смесь перемешивают при 15°C в течение 30 минут и фильтруют через диатомовую землю. Фильтрат концентрируют при пониженном давлении и очищают ВЭЖХ с обращенной фазой (колонка: Phenomenex Gemini C18, 10 мкм; подвижная фаза A: 10 мМ бикарбонат аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 10% до 33% B) с получением P2 в виде твердого белого вещества. Выход: 2,30 г, 5,80 ммоль, 50%. ЖХМС m/z 380,1♦ [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,13 (д, 1H), 8,06 (д, 1H), 7,55 (дд, 1H), 7,31-7,21 (м, 2H), 5,49 (с, 2H), 3,69 (уш. д, 2H), 3,61 (с, 2H), 3,43-3,33 (м, 1H), 3,25-3,11 (м, 2H), 2,25-2,06 (м, 4H).
Получение P3
(4-{2-[(4-Хлор-2-фторбензил)окси]пиридин-3-ил}пиперазин-1-ил)уксусная кислота(P3)
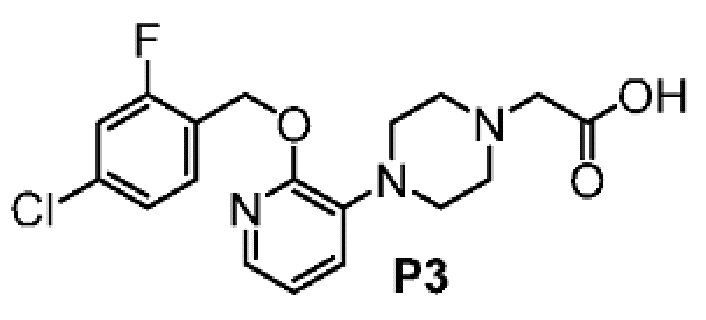
Стадия 1. Синтез 3-бром-2-[(4-хлор-2-фторбензил)окси]пиридина (C11)
(4-Хлор-2-фторфенил)метанол (2,21 г, 13,8 ммоль) по каплям через шприц добавляют к охлажденной до 0°C суспензии гидрида натрия (60% дисперсия в минеральном масле; 0,80 г, 20,0 ммоль) в ТГФ (50 мл). Ледяную баню удаляют и перемешивают полученную реакционную смесь при комнатной температуре (14°C) в течение 40 минут, после чего к смеси по каплям с помощью шприца добавляют 3-бром-2-фторпиридин (2,00 г, 11,4 ммоль). Перемешивание продолжают при комнатной температуре (14°C) в течение 1 часа, после чего реакционную смеь выливают в насыщенный водный раствор хлорида аммония (20 мл) и экстрагируют EtOAc (3 × 20 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (50 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 0% до 5% дихлорметан в петролейном эфире) приводит к получению С11 в виде твердого белого вещества. Выход: 1,77 г, 49%.
Стадия 2. Синтез трет-бутил-4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперазин-1-карбоксилата (C12)
Суспензию C11 (1,00 г, 3,16 ммоль), трет-бутил-пиперазин-1-карбоксилата (650 мг, 3,49 ммоль), 1,1'-бинафталин-2,2'-диилбис(дифенилфосфана) (236 мг, 0,379 ммоль), трис(дибензилиденацетон)дипалладия (0) (202 мг, 0,221 ммоль) и карбоната цезия (2,06 г, 6,32 ммоль) в 1,4-диоксане (10 мл) дегазируют азотом в течение 1 минуты и затем перемешивают при 85°C в течение 16 часов. Реакционную смесь фильтруют и концентрируют фильтрат в вакууме; очистка остатка хроматографией (силикагель, элюент: 20:1 петролейный эфир/EtOAc) приводит к получению C12 в виде твердого вещества бледно-желтого цвета. Выход: 920 мг, 69%. ЖХМС m/z 422,1♦ [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,81 (дд, 1H), 7,46 (дд, 1H), 7,18-7,07 (м, 3H), 6,88 (дд, 1H), 5,47 (с, 2H), 3,61-3,52 (м, 4H), 3,07-2,97 (м, 4H), 1,48 (с, 9H).
Стадия 3. Синтез 1-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперазина (C13)
Трифторуксусную кислоту (1 мл) добавляют к раствору C12 (160 мг, 0,379 ммоль) в дихлорметане (2 мл). Реакционную смесь перемешивают при 20°C в течение 30 минут, после чего концентрируют в вакууме с получением C13 в виде масла желтого цвета, которое используют непосредственно в следующей стадии.
Стадия 4. Синтез этил-(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперазин-1-ил)ацетата (C14)
К суспензии C13 (с предыдущей стадии; ≤0,379 ммоль) и карбоната калия (262 мг, 1,90 ммоль) в N, N-диметилформамидe (1 мл) добавляют этилбромацетат (82,3 мг, 0,493 ммоль). Реакционную смесь перемешивают при 15°C в течение 1,5 часа, затем разбавляют EtOAc (50 мл) и промывают водой (50 мл). Органический слой промывают насыщенным водным раствором хлорида натрия (50 мл), сушат над сульфатом магния, фильтруют, концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 17% до 50% EtOAc в петролейном эфире) с получением C14 в виде масла желтого цвета. Выход: 102 мг, 66% для 2 стадий. ЖХМС m/z 408,0♦ [M+H]+.
Стадия 5. Синтез (4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперазин-1-ил)уксусной кислоты (P3).
Раствор C14 (102 мг, 0,250 ммоль) и водный раствор гидроксида натрия (3 M; 0,3 мл, 0,9 ммоль) в смеси MeOH (1 мл) и ТГФ (1 мл) перемешивают при 20°C в течение 16 часов. После этого значение рН реакционной смеси доводят до 7 добавлением 1 М соляной кислоты и экстрагируют полученную смесь смесью дихлорметана и MeOH (10:1, 3 × 30 мл). Объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют в вакууме с получением P3 в виде масла желтого цвета. Выход: 95,0 мг, 100%.
Получение P4
2-Хлор-3-[(4-хлор-2-фторбензил)окси]пиразин(P4)
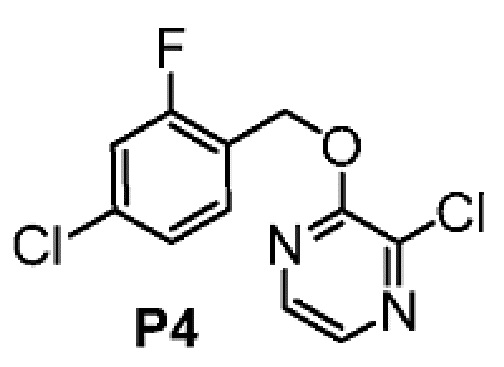
К раствору (4-хлор-2-фторфенил)метанола (7,17 г, 44,65 ммоль) и 2,3-дихлорпиразина (7,09 г, 47,59 ммоль) в 1,4-диоксане (50 мл) при 19°C добавляют трет-бутоксид натрия (5,42 г, 56,40 ммоль). Реакционную смесь перемешивают при 19°C в течение 4 часов, после чего выливают в петролейный эфир (150 мл) и затем фильтруют через слой диатомовой земли. Фильтрат концентрируют в вакууме с получением остатка, который очищают хроматографией (силикагель, градиент: от 0% до 17% дихлорметана в петролейном эфире); P4 выделяют в виде твердого белого вещества. Выход: 8,93 г, 73%. ЖХМС m/z 272,7 (наблюдается картина масс-спектра дихлор-изотопа) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,03 (д, 1H), 7,98 (д, 1H), 7,48 (дд, 1H), 7,21-7,11 (м, 2H), 5,49 (с, 2H).
Получение P5
Метил-6-азаспиро[2.5]октан-1-карбоксилат (P5)
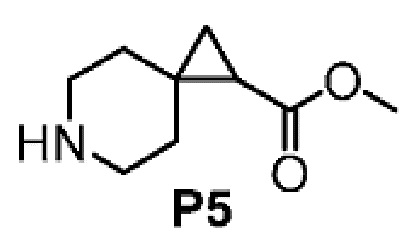
Стадия 1. Синтез трет-бутил-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилата (C15)
Раствор трет-бутоксида калия (65,9 г, 587 ммоль) в ТГФ (500 мл) добавляют к охлажденному до 0°C раствору этил(диэтоксифосфорил)ацетата (132 г, 589 ммоль) в ТГФ (500 мл), полученную суспензию перемешивают при 0°C в течение 1 часа, после чего охлаждают до −50°C. К смеси по каплям добавляют раствор трет-бутил-4-оксопиперидин-1-карбоксилата (90,0 г, 452 ммоль) в ТГФ (1,5 л) при −50°C, реакционной смеси дают возможность медленно нагреться до 20°C и затем перемешивают смесь в течение 16 часов при 20°C. После добавления воды (1 л) смесь концентрируют в вакууме для удаления ТГФ. Водный остаток экстрагируют EtOAc (2 × 800 мл), объединенные органические слои промывают насыщенным водным раствором хлорида натрия (500 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный продукт промывают несколько раз петролейным эфиром (200 мл) с получением C15 в виде твердого белого вещества. Выход: 95,0 г, 353 ммоль, 78%. 1H ЯМР (400 МГц, хлороформ-d) δ 5,71 (с, 1H), 4,16 (кв, 2H), 3,55-3,43 (м, 4H), 2,94 (уш. т, 2H), 2,28 (уш. т, 2H), 1,47 (с, 9H), 1,28 (т, 3H).
Стадия 2. Синтез 6-трет-бутил-1-этил-6-азаспиро[2.5]октан-1,6-дикарбоксилата (C16)
К раствору йодида триметилсульфоксония (140 г, 636 ммоль) в диметилсульфоксидe (800 мл) одной порцией добавляют трет-бутоксид калия (71,2 г, 634 ммоль) при 20°C. Реакционную смесь перемешивают при 20°C в течение 1,5 часа, затем по каплям добавляют раствор C15 (95,0 г, 353 ммоль) в диметилсульфоксиде (800 мл) и продолжают перемешивание при 20°C в течение 16 часов. К смеси добавляют насыщенный водный раствор хлорида натрия (2,0 л); полученную смесь нейтрализуют добавлением хлорида аммония и экстрагируют EtOAc (3,0 л). Объединенные органические слои промывают последовательно водой (2 × 1,0 л) и насыщенным раствором хлорида натрия (2,0 л), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, элюент: 10:1 петролейный эфир/EtOAc) приводит к получению С16 в виде масла желтого цвета. 1H ЯМР анализ показал, что присутствует алифатическая примесь. Выход: 80 г, 280 ммоль, 79%. 1H ЯМР (400 МГц, хлороформ-d): δ 4,19-4,09 (м, 2H), 3,55-3,39 (м, 3H), 3,27 (ддд, 1H), 1,76-1,64 (м, 2H), 1,56 (дд, 1H, предполагаемый; частично скрыт пиком воды), 1,47 (с, 9H), 1,47-1,37 (м, 2H), 1,27 (т, 3H), 1,17 (дд, 1H), 0,93 (дд, 1H).
Стадия 3. Синтез 6-(трет-бутоксикарбонил)-6-азаспиро[2.5]октан-1-карбоновой кислоты (C17)
К смеси C16 (80 г, 280 ммоль) в ТГФ (500 мл) и воды (500 мл) одной порцией добавляют моногидрат гидроксида лития (37,4 г, 891 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, после чего разбавляют водой (600 мл) и промывают EtOAc (3 × 300 мл). Органические слои утилизируют, водный слой подкисляют до pH 3-4 добавлением 6 M соляной кислоты. Полученную смесь экстрагируют EtOAc (3 × 600 мл) и объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток растирают с петролейным эфиром (300 мл) с получением C17 в виде твердого белого вещества. Выход: 42,0 г, 164 ммоль, 59%. ЖХМС m/z 278,2 [M+Na+]. 1H ЯМР (400 МГц, ДМСО-d6) δ 12,15-12,03 (уш. с, 1H), 3,43-3,25 (м, 3H, предполагаемый; частично скрыт пиком воды), 3,23-3,12 (м, 1H), 1,64-1,50 (м, 2H), 1,52 (дд, 1H), 1,39 (с, 9H), 1,39-1,28 (м, 2H), 0,96-0,88 (м, 2H).
Стадия 4. Синтез метил-6-азаспиро[2.5]октан-1-карбоксилата (P5)
Тионилхлорид (5 мл) добавляют к охлажденному до 15°C раствору C17 (5,00 г, 19,6 ммоль) в MeOH (50 мл) и перемешивают полученную реакционную смесь при 15°C в течение 16 часов. Затем смесь концентрируют в вакууме и выливают остаток в воду (20 мл). Значение рН полученной смеси доводят до 9 добавление водного раствора бикарбоната натрия, после чего экстрагируют смесь сначала EtOAc (3 × 100 мл), а затем смесью дихлорметана и MeOH (10:1; 5 × 100 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении с получением P5 в виде твердого вещества бледно-коричневого цвета. Выход: 3,0 г, 18 ммоль, 92%. 1H ЯМР (400 МГц, хлороформ-d) δ 3,68 (с, 3H), 2,92-2,88 (м, 2H), 2,88-2,82 (м, 1H), 2,74 (ддд, 1H), 1,76-1,62 (м, 2H), 1,51 (дд, 1H), 1,49-1,36 (м, 2H), 1,13 (дд, 1H), 0,90 (дд, 1H).
Получение P6
6-{6-[(4-Циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]октан-1-карбоновая кислота (P6)
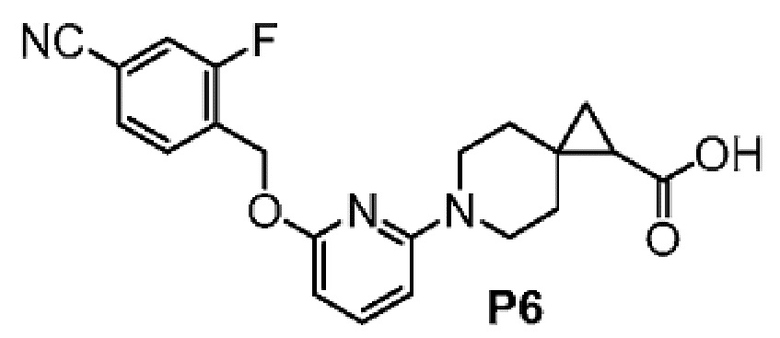
Стадия 1. Синтез 4-{[(6-хлорпиридин-2-ил)окси]метил}-3-фторбензонитрила (C18)
Данную реакцию проводят в двух параллельных загрузках. К перемешиваемой суспензии трет-бутоксида калия (313 г, 2,79 моль) в ТГФ (4,0 л) при температуре в интервале от 10°C до 15°C небольшими порциями добавляют 3-фтор-4-(гидроксиметил)бензонитрил (281 г, 1,86 моль). Реакционную смесь перемешивают при 15°C в течение 45 минут, после чего несколькими порциями добавляют 2,6-дихлорпиридин (230 г, 1,55 моль), поддерживая температуру реакционной смеси на уровне 15°C. Реакционную смесь выдерживают в течение дополнительных 18 часов при 15°C и затем выливают в насыщенный водный раствор хлорида аммония (10 л). К смеси добавляют EtOAc (10 л), перемешивают полученную смесь в течение 15 минут и затем фильтруют через слой диатомовой земли. Водный слой фильтрата экстрагируют EtOAc (2 × 6 л), объединенные органические слои промывают насыщенным водным раствором хлорида натрия (5 л), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 10% до 15% EtOAc в петролейном эфире) приводит к получению С18 в виде твердого вещества светло-желтого цвета. Общий выход: 550 г, 2,09 ммоль, 67%. 1H ЯМР (400 МГц, CDCl3) δ 7,67 (дд, 1H), 7,58 (дд, 1H), 7,48 (дд, 1H), 7,40 (дд, 1H), 6,97 (д, 1H), 6,75 (д, 1H), 5,49 (с, 2H).
Стадия 2. Синтез метил-6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]октан-1-карбоксилата (C19)
К раствору C18 (3,00 г, 11,4 ммоль) в толуоле (80 мл) добавляют P5 (1,93 г, 11,4 ммоль), трис(дибензилиденацетон)дипалладия (0) (523 мг, 0,571 ммоль), 1,1'-бинафталин-2,2'-диилбис(дифенилфосфан) (711 мг, 1,14 ммоль) и карбонат цезия (11,2 г, 34,3 ммоль). Реакционную смесь перемешивают при 100°C в течение 16 часов, после чего фильтруют через слой диатомовой земли. Фильтрат концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 0% до 15% EtOAc в петролейном эфире) с получением C19 в виде масла бледно-желтого цвета. Выход: 2,30 г, 5,82 ммоль, 51%. ЖХМС m/z 395,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,61 (дд, 1H), 7,44 (дд, 1H), 7,42 (дд, 1H), 7,36 (дд, 1H), 6,21 (д, 1H), 6,13 (д, 1H), 5,45 (с, 2H), 3,69 (с, 3H), 3,63-3,49 (м, 3H), 3,38 (ддд, 1H), 1,83-1,70 (м, 2H), 1,60 (дд, 1H), 1,51-1,45 (м, 2H), 1,21 (дд, 1H), 0,99 (дд, 1H).
Стадия 3. Синтез 6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]октан-1-карбоновой кислоты (P6)
К раствору C19 (770 мг, 1,95 ммоль) в смеси ТГФ (8,0 мл) и MeOH (8,0 мл) добавляют водный раствор гидроксида лития(2 M; 5,8 мл, 12 ммоль) и перемешивают полученную реакционную смесь при 15°C в течение 60 часов. Затем смесь концентрируют для удаления органических растворителей и доводят значение рН водного остатка до 6 добавлением 1 М соляной кислоты. Полученную смесь экстрагируют дихлорметаном (3 × 30 мл), объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 0% до 1% MeOH в дихлорметане) с последующей тонкослойной хроматографией (элюент: 20:1 дихлорметан/MeOH) приводит к получению P6 в виде смолы красного цвета. Выход: 530 мг, 1,39 ммоль, 71%. ЖХМС m/z 382,1 [M+H]+.
Получение P7
6-{2-[(4-Хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]октан-1-карбоксилат аммония, ENT-2 (P7)
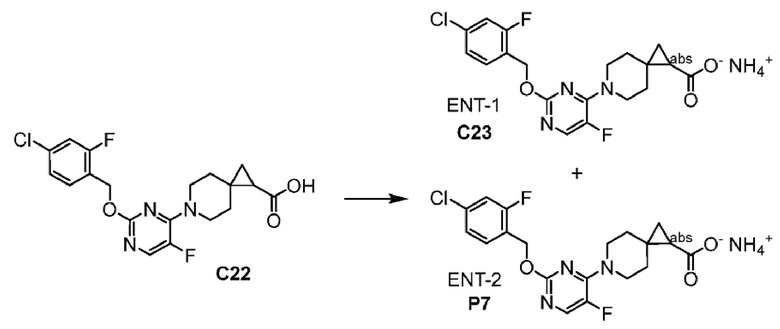
Стадия 1. Синтез гидрохлоридной соли метил-6-азаспиро[2.5]октан-1-карбоксилата (P5, HCl соль)
Тионилхлорид (8 мл) добавляют к раствору C17 (12,4 г, 48,6 ммоль) в MeOH (200 мл) и перемешивают полученную реакционную смесь при 30°C в течение 16 часов. Смесь концентрируют в вакууме с получением P5 (HCl соль) в виде твердого вещества бежевого цвета. Выход: 10,0 г, 48,6 ммоль, количественный выход. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,29 (уш. с, 2H), 3,61 (с, 3H), 3,09-2,97 (м, 3H), 2,92-2,81 (м, 1H), 1,91-1,76 (м, 2H), 1,74 (дд, 1H), 1,73-1,57 (м, 2H), 1,06 (дд, 1H), 1,02 (дд, 1H).
Стадия 2. Синтез метил-6-(2-хлор-5-фторпиримидин-4-ил)-6-азаспиро[2.5]октан-1-карбоксилата (C20)
К раствору P5 (HCl соль) (8,0 г, 39 ммоль) и 2,4-дихлор-5-фторпиримидина (7,80 г, 46,7 ммоль) в MeOH (150 мл) добавляют триэтиламин (16,5 мл, 118 ммоль). Реакционную смесь перемешивают при комнатной температуре (30°C) в течение 16 часов и затем концентрируют в вакууме. Полученную смолу распределяют между EtOAc (80 мл) и водой (80 мл); водный слой затем дополнительно экстрагируют EtOAc (3 × 80 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (2 × 50 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Очистка хроматографией (силикагель, градиент: от 0% до 21% EtOAc в петролейном эфире) приводит к получению C20 в виде масла желтого цвета. Выход: 11,5 г, 38,4 ммоль, 98%. 1H ЯМР (400 МГц, CDCl3) δ 7,91 (д, 1H), 3,88-3,78 (м, 3H), 3,74-3,65 (м, 1H), 3,70 (с, 3H), 1,92-1,79 (м, 2H), 1,65 (дд, 1H, предполагаемый; частично скрыт пиком воды), 1,59-1,54 (м, 2H), 1,25 (дд, 1H), 1,02 (дд, 1H).
Стадия 3. Синтез метил-6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]октан-1-карбоксилата (C21)
Ацетат палладия (II) (888 мг, 3,96 ммоль), ди-трет-бутил[2',4',6'-три(пропан-2-ил)бифенил-2-ил]фосфан (t-Bu XPhos; 3,27 г, 7,70 ммоль) и карбонат цезия (31,6 г, 96,9 ммоль) добавляют к раствору C20 (11,5 г, 38,4 ммоль) и (4-хлор-2-фторфенил)метанола (7,47 г, 46,5 ммоль) в толуоле (200 мл), после этого из реактора удаляют воздух и заполняют реактор азотом. Данную операцию повторяют дважды и затем реакционную смесь перемешивают при 110°C в течение 16 часов. Смесь фильтруют, фильтрат концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 0% до 22% EtOAc в петролейном эфире) с получением C21 в виде масла бледно-желтого цвета. Выход: 13,4 г, 31,6 ммоль, 82%. ЖХМС m/z 424,0♦ [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,87 (д, 1H), 7,45 (дд, 1H), 7,15-7,07 (м, 2H), 5,33 (с, 2H), 3,84-3,73 (м, 3H), 3,70 (с, 3H), 3,70-3,63 (м, 1H), 1,88-1,75 (м, 2H), 1,63 (дд, 1H), 1,56-1,50 (м, 2H), 1,23 (дд, 1H), 1,00 (дд, 1H).
Стадия 4. Синтез 6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]октан-1-карбоновой кислоты (C22)
К раствору C21 (21 г, 50 ммоль) в смеси MeOH (80 мл) и ТГФ (80 мл) добавляют водный раствор гидроксида натрия (5 M; 30 мл, 150 ммоль). Реакционную смесь перемешивают при 30°C в течение 15 часов, после чего концентрируют в вакууме, доводят значение pH до 6-7 добавлением 1 М соляной кислоты и экстрагируют полученную смесь смесью дихлорметана и MeOH (10:1, 4 × 100 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют, концентрируют при пониженном давлении и объединяют с продуктом аналогичной реакции с использованием C21 (13,4 г, 31,6 ммоль). Очистка хроматографией (силикагель, градиент: от 0% до 10% MeOH в дихлорметане) приводит к получению C22 в виде смолы желтого цвета. Общий выход: 27,0 г, 65,9 ммоль, 81%. ЖХМС m/z 410,1♦ [M+H]+.
Стадия 5. Выделение 6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]октан-1-карбоксилата аммония, ENT-1 (C23), и 6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]октан-1-карбоксилата аммония, ENT-2 (P7)
Разделение C22 (27,0 г, 65,9 ммоль) на энантиомеры проводят с помощью сверхкритической жидкостной хроматографии [колонка: Chiral Technologies Chiralpak AD, 10 мкм; подвижная фаза: 7:3 диоксид углерода/(MeOH, содержащий 0,1% гидроксида аммония)]. Энантиомер первого элюирования, выделенный в виде твердого белого вещества, обозначают как ENT-1 (C23). Выход: 10,27 г, 25,06 ммоль, 38%. ЖХМС m/z 410,0♦ [M+H]+. 1H ЯМР (400 МГц, CDCl3) д 7,88 (д, 1H), 7,44 (дд, 1H), 7,12 (дд, 1H), 7,09 (дд, 1H), 5,33 (с, 2H), 3,89-3,69 (м, 4H), 1,86 (т, 2H), 1,63 (дд, 1H), 1,60-1,48 (м, 2H), 1,25 (дд, 1H), 1,05 (дд, 1H).
Энантиомер второго элюирования, ENT-2 (P7), дополнительно очищают с использованием ВЭЖХ с обращенной фазой (колонка: Phenomenex Gemini C18, 10 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 15% до 38% B); это соединение получают также в виде твердого белого вещества. Выход: 9,86 г, 24,1 ммоль, 37%. ЖХМС m/z 410.0♦ [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,88 (д, 1H), 7,45 (дд, 1H), 7,12 (дд, 1H), 7,09 (дд, 1H), 5,33 (с, 2H), 3,88-3,69 (м, 4H), 1,86 (т, 2H), 1,64 (дд, 1H), 1,61-1,49 (м, 2H), 1,26 (дд, 1H), 1,07 (дд, 1H).
Получение P8
4-Хлор-2-[(4-хлор-2-фторбензил)окси]пиримидин (P8)
Данную реакцию проводят в двух идентичных загрузках. Раствор (4-хлор-2-фторфенил)метанола (5,42 г, 33,8 ммоль) в ТГФ (60 мл) по каплям в течение 25 минут добавляют к охлажденной до −25°C суспензии 4-хлор-2-(метилсульфонил)пиримидина (13,0 г, 67,5 ммоль) и гидрида натрия (60% дисперсия в минеральном масле; 2,43 г, 60,8 ммоль) в ТГФ (180 мл). Реакционную смесь перемешивают при комнатной температуре (25°C) в течение 18 часов, после чего две загрузки объединяют и концентрируют в вакууме. Остаток разбавляют насыщенным водным раствором хлорида аммония (120 мл) и экстрагируют EtOAc (3 × 100 мл); объединенные органические слои сушат над сульфатом натрия, фильтруют, концентрируют при пониженном давлении и очищают хроматографией (силикагель, градиент: от 0% до 6% EtOAc в петролейном эфире) с получением P8 в виде твердого белого вещества. Общий выход: 9,93 г, 36,4 ммоль, 54%. ЖХМС m/z 272,7 (картина масс-спектра дихлор-изотопа) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,41 (д, 1H), 7,48 (дд, 1H), 7,17-7,10 (м, 2H), 7,02 (д, 1H), 5,46 (с, 2H).
Получение P9
6-{4-[(4-Хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]октан-1-карбоновая кислота (P9)
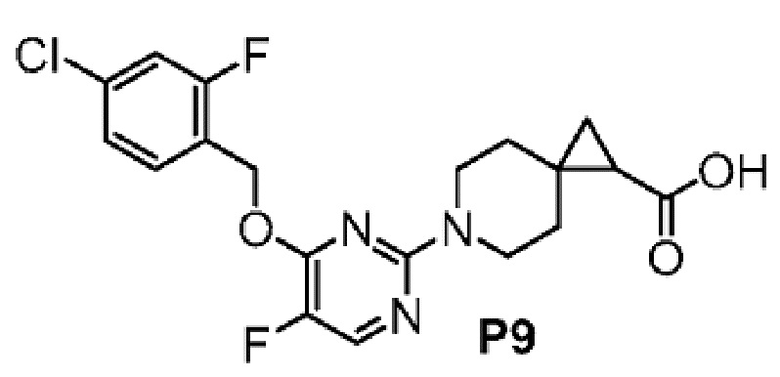
Стадия 1. Синтез 2-хлор-4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидина (C24)
Гидрид натрия (60% дисперсия в минеральном масле; 264 мг, 6,59 ммоль) добавляют порциями к охлажденному до 0°C раствору (4-хлор-2-фторфенил)метанола (1,06 г, 6,59 ммоль) в ТГФ (15 мл) и перемешивают полученную смесь при 10°C в течение 30 минут. Затем порциями добавляют раствор 2,4-дихлор-5-фторпиримидина (1,00 мг, 5,99 ммоль) в ТГФ (5 мл) и перемешивают полученную реакционную смесь при 10°C в течение 3 часов, после чего выливают в водный раствор хлорида аммония (100 мл) и экстрагируют EtOAc (2 × 50 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Очистка хроматографией (силикагель, градиент: от 0% до 5% EtOAc в петролейном эфире) приводит к получению C24 в виде твердого белого вещества. Выход: 1,21 г, 69%.
Стадия 2. Синтез метил-6-{4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]октан-1-карбоксилата (C25)
Раствор C24 (1,10 г, 3,78 ммоль), P5 (HCl соль) (855 мг, 4,16 ммоль) и триэтиламина (1,15 г, 11,3 ммоль) в N, N-диметилформамидe (20 мл) перемешивают при 100°C в течение 6 часов, после чего объединяют с продуктом аналогичной реакции с использованием C24 (100 мг, 0,344 ммоль), выливают в воду (300 мл) и экстрагируют смесь EtOAc (2 × 60 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме; очистка остатка хроматографией (силикагель, градиент: от 0% до 10% EtOAc в петролейном эфире) приводит к получению С25 в виде масла оранжевого цвета. Общий выход: 813 мг, 1,92 ммоль, 47%.
Стадия 3. Синтез 6-{4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]октан-1-карбоновой кислоты (P9)
К раствору C25 (1,79 г, 4,22 ммоль) в MeOH (50 мл) добавляют водный раствор гидроксида натрия (2 M; 21,1 мл, 42,2 ммоль). Реакционную смесь перемешивают при 20°C в течение 3 часов, затем при 40°C в течение 4 часов, после чего смесь подкисляют до pH 5 добавлением 12 M соляной кислоты, разбавляют водой (300 мл) и экстрагируют дихлорметаном (2 × 200 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистку остатка проводят хроматографией (силикагель, градиент: от 0% до 5% MeOH в дихлорметане) и затем ВЭЖХ с обращенной фазой (колонка: Phenomenex Synergi C18, 30 × 150 мм, 4 мкм; подвижная фаза A: 0,225% муравьиная кислота в воде; подвижная фаза B: ацетонитрил; градиент: от 50% до 80% B). Фракции, полученные разделением с помощью ВЭЖХ, концентрируют при пониженном давлении до половины исходного объема и затем экстрагируют дихлорметаном (2 × 100 мл); объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением P9 в виде вспененного твердого белого вещества. Выход: 1,04 г, 60%. ЖХМС m/z 409,8♦ [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,97 (д, 1H), 7,42 (дд, 1H), 7,15 (уш. дд,1H), 7,12 (дд, 1H), 5,43 (с, 2H), 3,86-3,74 (м, 3H), 3,73-3,64 (м, 1H), 1,79 (т, 2H), 1,63 (дд, 1H), 1,49 (т, 2H), 1,26 (дд, 1H), 1,07 (дд, 1H).
Получение P10
6-{6-[(4-Циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]октан-1-карбоновая кислота (P10)
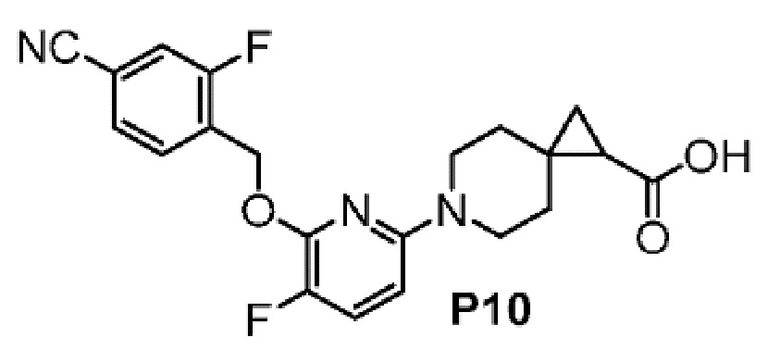
Стадия 1. Синтез 4-{[(3,6-дифторпиридин-2-ил)окси]метил}-3-фторбензонитрила (C26)
К раствору 2,3,6-трифторпиридина (4,40 г, 33,1 ммоль) и 3-фтор-4-(гидроксиметил)бензонитрила (5,00 г, 33,1 ммоль) в 1-метилпирролидин-2-оне (60 мл) добавляют карбоната калия (13,7 г, 99,2 ммоль). Реакционную смесь перемешивают при 100°C в течение 16 часов, после чего выливают в воду (100 мл) и экстрагируют полученную смесь EtOAc (3 × 300 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (4 × 200 мл), сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и объединяют с продуктом аналогичной реакции, проведенной с использованием 2,3,6-трифторпиридина (200 мг, 1,50 ммоль). Очистка хроматографией (силикагель, градиент: от 0% до 5% EtOAc в петролейном эфире) приводит к получению C26 виде твердого белого вещества. Общий выход: 6,93 г, 26,2 ммоль, 76%. МС (ESI) m/z 265,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,69 (дд, 1H), 7,53-7,45 (м, 2H), 7,41 (дд, 1H), 6,50 (ддд, 1H), 5,52 (с, 2H).
Стадия 2. Синтез метил-6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]октан-1-карбоксилата (C27)
Данную реакцию проводят в двух идентичных загрузках. Триэтиламин (766 мг, 7,57 ммоль) добавляют к раствору C26 (1,00 г, 3,78 ммоль) и P5 (640 мг, 3,78 ммоль) в диметилсульфоксидe (9 мл) и перемешивают полученную реакционную смесь при 140°C в течение 14 часов в микроволновом реакторе. Две реакционные смеси затем объединяют, выливают в воду (50 мл) и экстрагируют дихлорметаном (3 × 50 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и остаток очищают хроматографией (силикагель, элюент: 20% EtOAc в петролейном эфире) с получением C27 в виде масла желтого цвета. Общий выход: 696 мг, 1,68 ммоль, 22%. ЖХМС m/z 413.9 [M+H]+. 1H ЯМР (400 МГц, CDCl3): δ 7,65 (дд, 1H), 7,46 (дд, 1H), 7,37 (дд, 1H), 7,24 (дд, 1H), 6,12 (дд, 1H), 5,52 (с, 2H), 3,70 (с, 3H), 3,56-3,42 (м, 3H), 3,30 (ддд, 1H), 1,85-1,72 (м, 2H), 1,60 (дд, 1H), 1,53-1,46 (м, 2H), 1,21 (дд, 1H), 0,99 (дд, 1H).
Стадия 3. Синтез 6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]октан-1-карбоновой кислоты (P10)
К раствору C27 (646 мг, 1,56 ммоль) в смеси ТГФ (10 мл) и MeOH (1 мл) добавляют водный раствор гидроксида лития (2 M; 4,7 мл, 9,4 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, объединяют с продуктом аналогичной реакции с использованием C27 (50 мг, 0,12 ммоль) и упаривают досуха в вакууме. Остаток растворяют в воде (5 мл) и доводят значение pH до 6 добавлением 1 М соляной кислоты; осадок собирают фильтрацией и промывают водой (5 мл) с получением P10 в виде твердого вещества желтого цвета. Общий выход: 645 мг, 1,61 ммоль, 96%. ЖХМС m/z 400,1 [M+H]+.
Получение P11
6-(6-Бром-3-фторпиридин-2-ил)-6-азаспиро[2.5]октан-1-карбоновая кислота (P11)
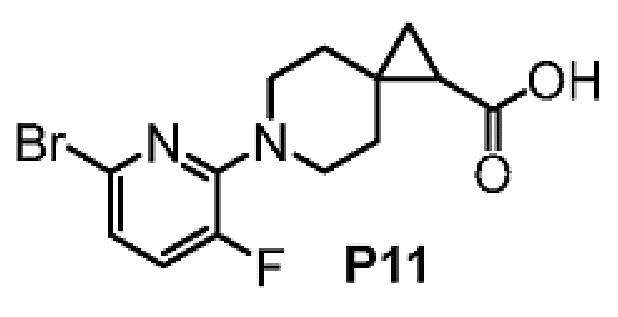
Стадия 1. Синтез метил-6-(6-бром-3-фторпиридин-2-ил)-6-азаспиро[2.5]октан-1-карбоксилата (C28)
Карбонат калия (2,44 г, 17,7 ммоль) добавляют к раствору 2,6-дибром-3-фторпиридина (1,50 г, 5,88 ммоль) и P5 (1,10 г, 6,50 ммоль) в N, N-диметилформамидe (25 мл) и перемешивают полученную суспензию при 100°C в течение 16 часов. Реакционную смесь объединяют с двумя продуктами аналогичных реакций, проводимых с использованием 2,6-дибром-3-фторпиридина (1,50 г, 5,88 ммоль и 1,0 г, 3,9 ммоль) и выливают в воду (300 мл). После экстракции трет-бутилметиловым эфиром (2 × 200 мл) объединенные органические слои промывают насыщенным водным раствором хлорида натрия (200 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 0% до 10% EtOAc в петролейном эфире) и затем ВЭЖХ с обращенной фазой (колонка: Phenomenex Gemini C18, 10 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 60% до 88% B) приводит к получению С28 в виде твердого белого вещества. Общий выход: 2,25 г, 6,56 ммоль, 42%. ЖХМС m/z 344,7 (масс-спектр изотопа брома) [M+H]+. δ (400 МГц, CDCl3) δ 7,06 (дд, 1H), 6,80 (дд, 1H), 3,69 (с, 3H), 3,63-3,50 (м, 3H), 3,40 (ддд, 1H), 1,91-1,78 (м, 2H), 1,64-1,50 (м, 3H, предполагаемый; частично скрыт пиком воды), 1,22 (дд, 1H), 0,99 (дд, 1H).
Стадия 2. Синтез 6-(6-бром-3-фторпиридин-2-ил)-6-азаспиро[2.5]октан-1-карбоновой кислоты (P11)
Водный раствор гидроксида натрия (2 M; 6,56 мл, 13,1 ммоль) добавляют к раствору C28 (2,25 г, 6,56 ммоль) в ТГФ (35 мл) и MeOH (25 мл), перемешивают полученную реакционную смесь при комнатной температуре в течение 73 часов, после чего концентрируют при пониженном давлении до двух третей исходного объема и подкисляют до pH 6-7 осторожным добавлением концентрированной соляной кислоты. Полученную смесь разбавляют насыщенным водным раствором хлорида натрия (100 мл) и экстрагируют дихлорметаном (2 × 80 мл); объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением P11 в виде твердого не совсем белого вещества. Выход: 2,15 г, 6,53 ммоль, количественный выход. ЖХМС m/z 330,7 (масс-спектр изотопа брома) [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,06 (дд, 1H), 6,80 (дд, 1H), 3,62-3,44 (м, 4H), 1,90 (дд, 2H), 1,65-1,56 (м, 3H), 1,26 (дд, 1H), 1,07 (дд, 1H).
Получение P12
6-{6-[(4-Циано-2-фторбензил)окси]-3,5-дифторпиридин-2-ил}-6-азаспиро[2.5]октан-1-карбоновая кислота P12)
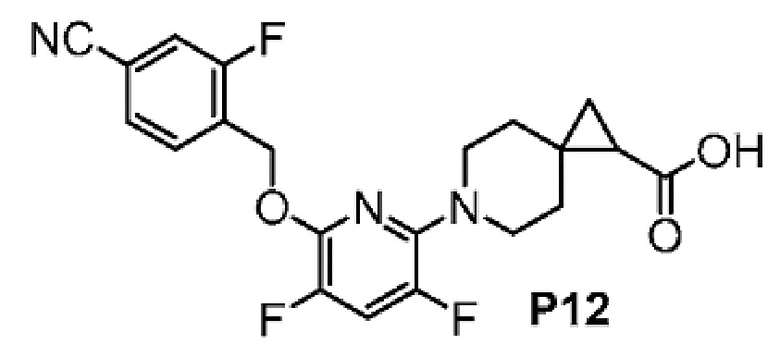
Стадия 1. Синтез 3-фтор-4-{[(3,5,6-triфторпиридин-2-ил)окси]метил}бензонитрил (C29)
Взаимодействие 3-фтор-4-(гидроксиметил)бензонитрила с 2,3,5,6-тетрафторпиридином проводят с использованием способа, описанного для синтеза C26 в примере получения P10. Соединение C29 получают в виде твердого белого вещества. Выход: 1,15 г, 40%. 1H ЯМР (400 МГц, хлороформ-d) δ 7,66 (дд, 1H), 7,49 (дд, 1H), 7,46-7,38 (м, 2H), 5,49 (с, 2H).
Стадия 2. Синтез метил-6-{6-[(4-циано-2-фторбензил)окси]-3,5-дифторпиридин-2-ил}-6-азаспиро[2.5]октан-1-карбоксилата (C30)
К раствору C29 (522 мг, 1,85 ммоль) в N, N-диметилформамидe (10 мл) добавляют P5 (348 мг, 2,06 ммоль) и карбонат калия (281 мг, 2,04 ммоль). Реакционную смесь перемешивают при 110°C в течение 10 часов, после чего разбавляют водой (30 мл) и экстрагируют EtOAc (3 × 25 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (2 × 20 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 0% до 4% EtOAc в петролейном эфире) приводит к получению С30 в виде смолы бледно-желтого цвета. Выход: 400 мг, 50%. ЖХМС m/z 431,9 [M+H]+.
Стадия 3. Синтез 6-{6-[(4-циано-2-фторбензил)окси]-3,5-дифторпиридин-2-ил}-6-азаспиро[2.5]октан-1-карбоновой кислоты (P12)
К раствору C30 (540 мг, 1,25 ммоль) в ТГФ (16 мл) добавляют водный раствор гидроксида лития (2 M; 1,9 мл, 3,8 ммоль) и MeOH (1,8 мл). Реакционную смесь перемешивают при 25°C в течение 16 часов, затем снова добавляют водный раствор гидроксида лития (2 M; 1,9 мл, 3,8 ммоль) и продолжают перемешивание при 25°C в течение 20 часов, после значение pH доводят до 5-6 добавлением 1 М соляной кислоты и экстрагируют смесь дихлорметаном (2 × 25 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением P12 (540 мг, предполагаемый количественный выход) в виде твердого белого вещества. Полученный продукт используют в последующих химических реакциях без дополнительной очистки. ЖХМС m/z 417,9 [M+H]+.
Получение P13
Соль бис(п-толуолсульфонат) 2-[(4-хлор-2-фторбензил)окси]-4-(пиперидин-4-ил)пиримидина (P13)
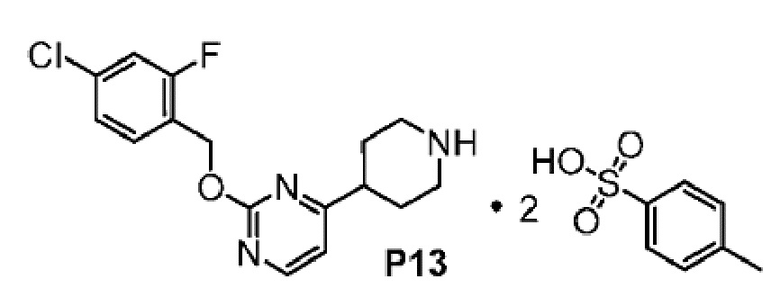
Стадия 1. Синтез 1-трет-бутил-4-этил-4-[2-(метилтио)пиримидин-4-ил]пиперидин-1,4-дикарбоксилата (C31)
Раствор бис(триметилсилил)амида лития в ТГФ (1 M; 2,3 л, 2,3 моль) добавляют к охлажденному до −60°C раствору 1-трет-бутил-4-этилпиперидин-1,4-дикарбоксилата (500 г, 1,93 моль) в ТГФ (5 л). Реакционную смесь перемешивают при −60°C в течение 30 минут и затем к смеси по каплям добавляют раствор 4-хлор-2-(метилтио)пиримидина (300 г, 1,87 моль) в ТГФ (1,5 л). Реакционной смеси дают возможность нагреться до комнатной температуры в течение 1,5 часов, смесь перемешивают при комнатной температуре в течение 1 часа и затем охлаждают до 0°C. К смеси добавляют раствор лимонной кислоты (386 г, 2,01 моль) в воде (5 л) и насыщенный водный раствор хлорида натрия (5 л) и экстрагируют полученную смесь EtOAc (2 × 10 л). Объединенные органические слои сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 75% до 100% EtOAc в петролейном эфире) с получением C31 в виде масла желтого цвета. Выход: 690 г, 1,81 моль, 97%. ЖХМС m/z 382,2 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,61 (д, 1H), 7,25 (д, 1H), 4,13 (кв, 2H), 3,69 (ддд, 2H), 3,16-2,96 (м, 2H), 2,48 (с, 3H), 2,27-2,18 (м, 2H), 1,96 (ддд, 2H), 1,39 (с, 9H), 1,13 (т, 3H).
Стадия 2. Синтез трет-бутил-4-[2-(метилтио)пиримидин-4-ил]пиперидин-1-карбоксилата (C32)
Раствор C31 (690 г, 1,81 моль) в MeOH (4,6 л) и ТГФ (2,3 л) нагревают до 40°C и затем добавляют водный раствор гидроксида натрия (2,0 M, 2 эквивалента). Реакционную смесь перемешивают при 40°C в течение 8 часов, после этого ей дают возможность охладиться до комнатной температуры и затем доводят значение рН до 4 добавлением 1 M водного раствора лимонной кислоты. Полученную смесь разбавляют насыщенным водным раствором хлорида натрия (5 л) и экстрагируют EtOAc (3 × 5 л); объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением C32, который непосредственно используют в следующе стадии. Выход: 550 г, 1,78 моль, 98%. ЖХМС m/z 310,2 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,52 (д, 1H), 7,12 (д, 1H), 4,10-3,98 (м, 2H), 2,90-2,74 (м, 2H), 2,80 (тт, 1H), 2,49 (с, 3H), 1,82 (уш. д, 2H), 1,60-1,47 (м, 2H), 1,41 (с, 9H).
Стадия 3. Синтез трет-бутил-4-[2-(метилсульфонил)пиримидин-4-ил]пиперидин-1-карбоксилата (C33)
3-Хлорпероксибензойную кислоту (80%; 765 г, 3,55 моль) добавляют к охлажденному до 0°C раствору C32 (550 г, 1,78 моль) в дихлорметане (14 л). Реакционной смеси дают возможность нагреться до комнатной температуры в течение 1,5 часов, затем перемешивают в течение дополнительных 15 часов и фильтруют через слой диатомовой земли. Фильтр промывают дихлорметаном (3 × 5 л), объединенные фильтраты промывают последовательно насыщенным водным раствором бикарбоната натрия (2 × 1,1 л) и насыщенным раствором хлорида натрия (1,5 л), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, элюент: 50:1 дихлорметан/MeOH) приводит к получению С33 в виде твердого вещества желтого цвета. Выход: 420 г, 1,23 моль, 69%. ЖХМС m/z 364,2 [M+Na+]. 1H ЯМР (400 МГц, CDCl3) δ 8,83 (д, 1H), 7,40 (д, 1H), 4,28 (уш. д, 2H), 3,37 (с, 3H), 2,99 (тт, 1H), 2,85 (ддд, 2H), 1,98 (уш. д, 2H), 1,81-1,67 (м, 2H), 1,48 (с, 9H).
Стадия 4. Синтез трет-бутил-4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}пиперидин-1-карбоксилата (C34)
К раствору (4-хлор-2-фторфенил)метанола (201 г, 1,25 моль) в ТГФ (12 л) добавляют раствор бис(триметилсилил)амида натрия в ТГФ (2 M; 1,2 эквивалента). Реакционную смесь перемешивают в течение 15 минут, затем добавляют раствор C33 (410 г, 1,20 моль) в ТГФ (1 л) и продолжают перемешивание при комнатной температуре в течение 1 часа. Затем добавляют воду (5 л) и экстрагируют полученную смесь EtOAc (2 × 5 л); объединенные органические слои промывают насыщенным водным раствором хлорида натрия (5 л), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка остатка хроматографией (силикагель, элюент: 5:1 петролейный эфир/EtOAc) приводит к получению C34. Выход: 252 г, 597 ммоль, 50%. ЖХМС m/z 422,1♦ [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 8,43 (д, 1H), 7,48 (дд, 1H), 7,15-7,08 (м, 2H), 6,82 (д, 1H), 5,44 (c, 2H), 4,32-4,15 (м, 2H), 2,88-2,74 (м, 2H), 2,75 (тт, 1H), 1,88 (уш. д, 2H), 1,77-1,63 (м, 2H), 1,47 (c, 9H).
Стадия 5. Синтез бис(п-толуолсульфоната) 2-[(4-хлор-2-фторбензил)окси]-4-(пиперидин-4-ил)пиримидина (P13)
Моногидрат п-толуолсульфоновой кислоты (17,1 г, 89,9 ммоль) одной порцией добавляют к раствору C34 (16,2 г, 38,4 ммоль) в EtOAc (220 мл). Реакционную смесь нагревают до внутренней температуры 60°C и выдерживают при указанной температуры в течение 35 минут, после чего дают возможность смеси охладиться до комнатной температуры в течение ночи при перемешивании на масляной бане. ЖХМС анализ в это время показывает конверсию в P13: ЖХМС m/z 322,2♦ [M+H]+. Твердый продукт собирают фильтрацией и промывают EtOAc (100 мл) с получением P13 в виде твердого вещества бледно-розового цвета. Выход: 23,7 г, 35,6 ммоль, 93%. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,64-8,53 (уш. м, 1H), 8,56 (д, 1H), 8,37-8,23 (уш. м, 1H), 7,60 (дд, 1H), 7,53-7,48 (м, 1H), 7,49 (д, 4H), 7,34 (уш. д, 1H), 7,12 (д, 4H), 7,09 (д, 1H), 5,41 (с, 2H), 3,37 (уш. д, 2H), 3,08-2,93 (м, 3H), 2,29 (с, 6H), 2,01 (уш. д, 2H), 1,92-1,77 (м, 2H).
Получение P14
трет-Бутил-(2S)-4-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-2-метилпиперазин-1-карбоксилат (P14)
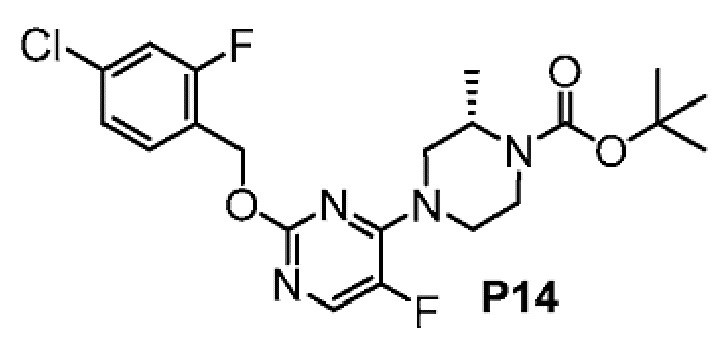
Стадия 1. Синтез трет-бутил-(2S)-4-(2-хлор-5-фторпиримидин-4-ил)-2-метилпиперазин-1-карбоксилата (C35)
К охлажденному до 0°C раствору 2,4-дихлор-5-фторпиримидина (12,0 г, 71,9 ммоль) в дихлорметане (130 мл) добавляют триэтиламин (20 мл, 140 ммоль) и затем раствор трет-бутил-(2S)-2-метилпиперазин-1-карбоксилата (15,0 г, 74,9 ммоль) в дихлорметане (70 мл). Реакционную смесь перемешивают при 30°C в течение 15 часов, после чего промывают последовательно водой (150 мл) и насыщенным раствором хлорида натрия (100 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 20% до 50% EtOAc в петролейном эфире) приводит к получению С35 в виде твердого белого вещества. Выход: 21,4 г, 64,7 ммоль, 90%. ЖХМС m/z 330,9♦ [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,94 (д, 1H), 4,54 4,45 (м, 1H), 4,39-4,29 (м, 1H), 4,28 (уш. д, 1H), 3,94 (уш. д, 1H), 3,35 (дд, 1H), 3,25-3,06 (м, 2H), 1,48 (с, 9H), 1,17 (д, 3H).
Стадия 2. Синтез трет-бутил-(2S)-4-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-2-метилпиперазин-1-карбоксилата (P14)
Превращение C35 в P14 проводят способом, описанным для синтеза C21 из C20 в примере получения P7. В этом случае сырой продукт очищают хроматографией (силикагель, градиент: от 0% до 20% EtOAc в петролейном эфире) с получением P14 в виде масла бледно-желтого цвета. Выход: 10,7 г, 23,5 ммоль, 98%. 1H ЯМР (400 МГц, хлороформ-d) δ 7,88 (д, 1H), 7,44(дд, 1H), 7,15-7,06 (м, 2H), 5,33 (с, 2H), 4,50-4,42 (м, 1H), 4,35-4,20 (м, 2H), 3,90 (уш. д, 1H), 3,28 (дд, 1H), 3,16 (ддд, 1H), 3,05 (ддд, 1H), 1,47 (с, 9H), 1,14 (д, 3H).
Получение P15
Соль бис(п-толуолсульфонат) 2-[(4-хлор-2-фторбензил)окси]-4-[(3S)-3-метилпиперазин-1-ил]пиримидина (P15)
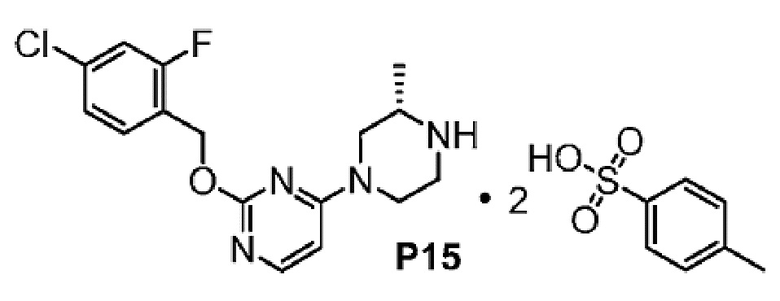
Стадия 1. Синтез трет-бутил-(2S)-4-(2-хлорпиримидин-4-ил)-2-метилпиперазин-1-карбоксилата (C36)
Раствор трет-бутил-(2S)-2-метилпиперазин-1-карбоксилата (22,2 г, 111 ммоль), 2,4-дихлорпиримидина (15,0 г, 101 ммоль) и триэтиламина (30 мл) в дихлорметане (200 мл) перемешивают при 30°C в течение 15 часов, затем смесь промывают последовательно водой (150 мл) и насыщенным раствором хлорида натрия (100 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка остатка хроматографией (силикагель, градиент: от 20% до 50% EtOAc в петролейном эфире) приводит к получению С36 в виде твердого белого вещества. Выход: 25,0 г, 79,9 ммоль, 79%.
Стадия 2. Синтез трет-бутил-(2S)-4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-2-метилпиперазин-1-карбоксилата (C37)
Раствор бис(триметилсилил)амида натрия в ТГФ (1,0 M; 90,0 мл, 90,0 ммоль) по каплям добавляют к раствору (4-хлор-2-фторфенил)метанола (13,3 г, 82,8 ммоль) в ТГФ (50 мл), полученную смесь перемешивают при 60°C в течение 15 минут и затем добавляют к раствору C36 (20,0 г, 63,9 ммоль) в ТГФ (120 мл). Перемешивание продолжают при 60°C в течение 1 часа, затем реакционную смесь распределяют между EtOAc и водой и объединяют с продуктом аналогичной реакции, проведенной с использованием C36 (5,00 г, 16,0 ммоль). Водный слой экстрагируют EtOAc (2 × 120 мл), объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 0% до 30% EtOAc в петролейном эфире) приводит к получению C37 в виде смолы желтого цвета. Общий выход: 33,6 г, 76,9 ммоль, 96%.
Стадия 3. Синтез соли бис(п-толуолсульфоната) 2-[(4-хлор-2-фторбензил)окси]-4-[(3S)-3-метилпиперазин-1-ил]пиримидина (P15).
Моногидрат п-толуолсульфоновой кислоты (17,8 г, 93,6 ммоль) добавляют одной порцией к смеси C37 (15,7 г, 35,9 ммоль) и EtOAc (220 мл). Реакционную смесь нагревают до внутренней температуры 60°C и выдерживают при указанной температуры в течение 35 минут, после чего смеси дают возможность охладиться до комнатной температуры при перемешивании на масляной бане в течение ночи. Полученный твердый продукт собирают фильтрацией и промывают EtOAc (100 мл) с получением P15 в виде твердого не совсем белого вещества. Выход: 24,5 г, количественный выход. ЖХМС m/z 337,2♦ [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6), характеристические пики δ 9,21-9,06 (м, 1H), 8,89-8,73 (м, 1H), 8,22 (д, 1H), 7,62 (дд, 1H), 7,55 (дд, 1H), 7,48 (д, 4H), 7,37 (дд, 1H), 7,11 (д, 4H), 6,87 (д, 1H), 5,53 (с, 2H), 3,22 (дд, 1H), 3,18-3,05 (м, 1H), 2,29 (с, 6H), 1,27 (д, 3H).
Получение P16
Метил-4-амино-3-{[(2S)-оксетан-2-илметил]амино}бензоат (P16)
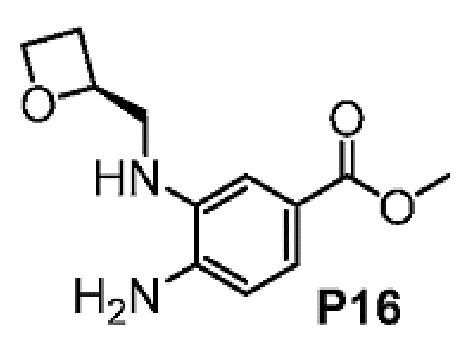
Данную последовательность реакций выполняют с использованием крупномасштабных загрузок. Обычно перед реакциями, а также после добавления реагентов реакторы вакуумируют до −0,08 −0,05 МПа и затем заполняют азотом до нормального давления. Этот процесс обычно повторяют 3 раза и затем оценивают содержание кислорода, чтобы убедиться, что оно составляет ≤1,0%. Для экстракции и промывки органических слоев смесь обычно перемешивают в течение 15-60 минут, а затем оставляют осаждаться в течение 15-60 минут перед разделением слоев.
Стадия 1. Синтез (2S)-2-[(бензиокси)метил]оксетана (C38)
Данную реакцию проводят в трех примерно одинаковых загрузках. В стеклянный реактор объемом 2000 л загружают 2-метилпропан-2-ол (774,7 кг). трет-Бутоксид калия (157,3 кг, 1402 моль) добавляют с помощью загрузочной воронки для твердых веществ и перемешивают полученную смесь в течение 30 минут. Таким же образом добавляют йодид метилсульфоксония (308,2 кг, 1400 моль), полученную реакционную смесь нагревают до 55-65°C и выдерживают при указанной температуре в течение 2-3 часов, после чего добавляют (2S)-2-[(бензиокси)метил]оксиран (92,1 кг, 561 моль) со скоростью от 5 до 20 кг/час. Реакционную смесь выдерживают при 55-65°C в течение 25 часов, затем охлаждают до 25-35°C и фильтруют через диатомовую землю (18,4 кг). Осадок на фильтре промывают трет-бутилметиловым эфиром (3 × 340 кг), объединенные фильтраты переносят к реактор объемом 5000 л, обрабатывают очищенной водой (921 кг) и перемешивают в течение от 15 до 30 минут при 15-30°C. Органический слой дважды промывают раствором хлорида натрия (230,4 кг) в очищенной воде (920,5 кг) и концентрируют при пониженном давлении (≤−0,08 MПа) при ≤45°C. К остатку добавляют н-гептан (187 кг) и концентрируют полученную смесь при пониженном давлении (≤−0,08 мПа) при ≤45°C; органическую фазу очищают хроматографией (силикагель, 280 кг) с хлоридом натрия в верхней части колонки (18,5 кг). Сырой продукт загружают в колонку с использованием н-гептана (513 кг) и затем элюируют смесью н-гептана (688,7 кг) и EtOAc (64,4 кг). Продукт, полученный в трех загрузках, объединяют, получая C38 с чистотой 85% в виде масла светло-желтого цвета (189,7 кг, 906 ммоль, 54%). 1H ЯМР (400 МГц, хлороформ-d), только пики C38: δ 7,40-7,32 (м, 4H), 7,32-7,27 (м, 1H), 4,98 (дддд, 1H), 4,72-4,55 (м, 4H), 3,67 (дд,1H), 3,62 (дд,1H), 2,72-2,53 (м, 2H).
Стадия 2. Синтез (2S)-оксетан-2-илметанола (C39)
10% Палладий на угле (30,7 кг) добавляют с помощью воронки для загрузки твердых веществ при температуре от 10°C до 30°C в раствор C38 с чистотой 85% (с предыдущей стадии; 185,3 кг, 884,8 моль) в ТГФ (1270 кг) в автоклавный реактор из нержавеющей стали объемом 3000 л. Загрузочную воронку промывают очищенной водой и в реакционную смесь добавяют ТГФ (143 кг). Реактор продувают азотом и затем наполняют водородом, повышая давление до 0,3-0,5 МПа и затем сбрасывая по 0,05 МПа. Эту водородную продувку повторяют 5 раз, после чего давление водорода повышают до 0,3-0,4 МПа. Реакционную смесь затем нагревают до 35-45°C. В течение 13 часов давление водорода поддерживают на уровне 0,3-0,5 МПа, после чего смесь дренируют до 0,05 МПа и пять раз продувают азотом, повышая давление до 0,15-0,2 МПа и затем сбрасывая до 0,05 МПа. Смесь охлаждают до температуры в интервале от 10°C до 25°C, фильтруют и реактор промывают ТГФ (2 × 321 кг). Осадок на фильтре дважды смачивают промывной жидкостью и затем фильтруют; фильтрат концентрируют при пониженном давлении (≤−0,06 MПa) при ≤40°C с получением C39 (62,2 кг, 706 моль, 80%) в ТГФ (251 кг)
Стадия 3. Синтез (2S)-оксетан-2-илметил-4-метилбензолсульфоната (C40)
4-(Диметиламино)пиридин (17,5 кг, 143 моль) добавляют при температуре в интервале от 10°C до 25°C к смеси раствора C39 (с предыдущей стадии; 62,2 кг, 706 моль) в ТГФ (251 кг) и триэтиламина (92,7 кг, 916 моль) в дихлорметане (1240 кг). Спустя 30 минут небольшими порциями в течение от 20 до 40 минут добавляют п-толуолсульфонилхлорид (174,8 кг, 916,9 моль) и перемешивают полученную реакционную смесь при температуре в интервале от 15°C до 25°C в течение 16 часов 20 минут. Добавляют очищенную воду (190 кг); после перемешивания органический слой промывают водным раствором бикарбоната натрия (полученным с использованием 53,8 кг бикарбоната натрия и 622 кг очищенной воды) и затем промывают водным раствором хлорида аммония (полученного с использованием 230 кг хлорида аммония и 624 кг очищенной воды). Органический слой промывают очищенной водой (311 кг), затем органический слой фильтруют через нуч-фильтр из нержавеющей стали, в который предварительно загружают силикагель (60,2 кг). Осадок на фильтре промывают дихлорметаном (311 кг) в течение 20 минут и затем фильтруют; объединенные фильтраты концентрируют при пониженном давлении (≤−0,05 МПа) и температуре ≤40°C до конечного объема 330-400 л. Затем добавляют ТГФ (311 кг) при температуре в интервале от 15°C до 30°C и концентрируют смесь в указанных условиях до конечного объема 330-400 л. Добавление ТГФ и упаривание растворитля повторяют снова до объема 330-400 л с получением раствора С40 светло-желтого цвета (167,6 кг, 692 ммоль, 98%) в ТГФ (251,8 кг). 1H ЯМР (400 МГц, хлороформ-d), только пики C40: δ 7,81 (д, 2H), 7,34 (д, 2H), 4,91 (ддт, 1H), 4,62-4,55 (м, 1H), 4,53-4,45 (м, 1H), 4,14 (д, 2H), 2,75-2,63 (м, 1H), 2,60-2,49 (м, 1H), 2,44 (с, 3H).
Стадия 4. Синтез (2S)-2-(азидометил)оксетана (C41)
N, N-Диметилформамид (473 кг), азид натрия (34,7 кг, 534 моль) и йодид калия (5,2 кг, 31 моль) объединяют в реакторе из нержавеющей стали объемом 3000 л при температуре в интервале от 10°C до 25°C. После добавления C40 (83,5 кг, 344,6 моль) в ТГФ (125,4 кг) реакционную смесь нагревают до температуры в интервале от 55°C до 65°C и выдерживают при указанной температуре в течение 17 часов 40 минут, после чего охлаждают до температуры в интервале от 25°C до 35°C и барботируют через смесь азот из донного вентиля в течение 15 минут. Затем добавляют трет-бутилметиловый эфир (623 кг) и очищенную воду (840 кг) и образующийся водный слой дважды экстрагируют трет-бутилметиловым эфиром (312 кг и 294 кг). Объединенные органические слои промывают очищенной водой (2 × 419 кг), поддерживая температуру на уровне от 10°C до 25°C, с получением C41 (31,2 кг, 276 моль, 80%) в растворе указанного выше органического слоя (1236,8 кг).
Стадия 5. Синтез 1-[(2S)-оксетан-2-ил]метанамина (C42)
10% Палладий на угле (3,7 кг) добавляют через загрузочную воронку в раствор C41 (с предыдущей стадии; 1264 кг, 31,1 кг, 275 моль) в ТГФ (328 кг) при температуре в интеврале от 10°C до 30°C в реакторе из нержавеющей стали объемом 3000 л. Загрузочную воронку промывают ТГФ (32 кг) и смыв добавляют в реакционную смесь. Содержимое реактора продувают азотом, затем аналогично продувают водородом, повышая давление до 0,05-0,15 МПа и затем сбрасывая давления до 0,03-0,04 МПа. Такую продувку водородом повторяют 5 раз, после чего давление водорода повышают до 0,05-0,07 МПа. Температуру реакции повышают до 25°C - 33°C, давление водорода поддерживают на уровне 0,05-0,15 МПа в течение 22 часов, заменяя водород каждые 3-5 часов. Смесь после этого реактор продувают пять раз азотом, повышая давление до 0,15-0,2 МПа и затем сбрасывая давления до 0,05 МПа. После фильтрации смеси реактор и осадок на фильтре промывают ТГФ (92 кг и 93 кг). Объединенные фильтраты концентрируют при пониженном давлении (≤−0,07 МПа) и температуре ≤45°C с получением C42 (18,0 кг, 207 моль, 75%) в ТГФ (57,8 кг). 1H ЯМР (400 МГц, ДМСО-d6), только пики C42: δ 4,62 (ддт, 1H), 4,49 (ддд, 1H), 4,37 (дт, 1H), 2,69 (д, 2H), 2,55-2,49 (м, 1H), 2,39 (м, 1H).
Стадия 6. Синтез метил-4-нитро-3-{[(2S)-оксетан-2-илметил]амино}бензоата (C43).
Карбонат калия (58,1 кг, 420 моль) добавляют к раствору метил 3-фтор-4-нитробензоата (54,8 кг, 275 моль) в ТГФ (148 кг) в эмалированном с внутренней стороны реакторе объемом 100 л и перемешивают полученную смесь в течение 10 минут. В реактор добавляют раствор C42 (29,3 кг, 336 моль) в ТГФ (212,9 кг) и перемешивают полученную реакционную смесь при температуре в интервале от 20°C до 30°C в течение 12 часов, после чего добавляют EtOAc (151 кг) и фильтруют смесь через силикагель (29 кг). Осадок на фильтре промывают EtOAc (150 кг и 151 кг) и объединенные фильтраты концентрируют при пониженном давлении (≤−0,08 МПа) и температуре ≤45°C до объема от 222 до 281 л. Смесь охлаждают до температуры в интервале от 10°C до 30°C, добавляют н-гептан (189 кг), полученную смесь перемешивают в течение 20 минут и концентрируют при пониженном давлении (≤−0,08 МПа) и температуре ≤45°C до объема 222 л. Снова добавляют н-гептан (181 кг) с контрольной скоростью от 100 до 300 кг/час и продолжают перемешивание в течение 20 минут. Смесь анализируют до остаточного содержания ТГФ ≤5% и остаточного содржания EtOAc 10-13%. Смесь нагревают до температуры в интервале от 40°C до 45°C и перемешивают в течение 1 часа, после чего охлаждают до температуры в интервале от 15°C до 25°C со скоростью от 5°C до 10°C в час и затем перемешивают при температуре в интервале от 15°C до 25°C в течение 1 часа. Фильтрация с использованием центрифуги из нержавеющей стали приводит к получению осадка на фильтре, который промывают смесью EtOAc (5,0 кг) и н-гептана (34 кг) и затем смешивают с ТГФ (724 кг) при температуре в интервале 10°C до 30°C в течение 15 минут; фильтрация приводит к получению C43 в виде твердого вещества желтого цвета (57,3 кг, 210 моль, 76%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,34 (т, 1H), 8,14 (д, 1H), 7,63 (д, 1H), 7,13 (дд, 1H), 4,99 (дддд, 1H), 4,55 (ддд, 1H), 4,43 (дт, 1H), 3,87 (с, 3H), 3,67-3,61 (м, 2H), 2,67 (дддд, 1H), 2,57-2,47 (м, 1H).
Стадия 7. Синтез метил-4-амино-3-{[(2S)-оксетан-2-илметил]амино}бензоата (P16)
К раствору C43 (5,00 г, 18,8 ммоль) в MeOH (150 мл) добавляют увлажненный палладий на угле (500 мг) и перемешивают полученную смесь при 15°C в течение 3 часов в атмосфере водорода из баллона. Реакционную смесь фильтруют; фильтрат концентрируют в вакууме с получением P16 в виде бесцветного масла. Выход: 4,40 г, 18,6 ммоль, 99%. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,16 (дд, 1H), 7,02 (д, 1H), 6,55 (д, 1H), 5,48 (c, 2H), 4,92-4,87 (м, 1H), 4,70 (т, 1H), 4,54 (ддд, 1H), 4,47 (ддд, 1H), 3,72 (c, 3H), 3,39-3,23 (м, 2H, предполагаемый; частично скрыт пиком воды), 2,72-2,61 (м, 1H), 2,5-2,40 (м, 1H, предполагаемый; частично скрыт пиком растворителя).
Получение P17
Метил-2-(хлорметил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат (P17)
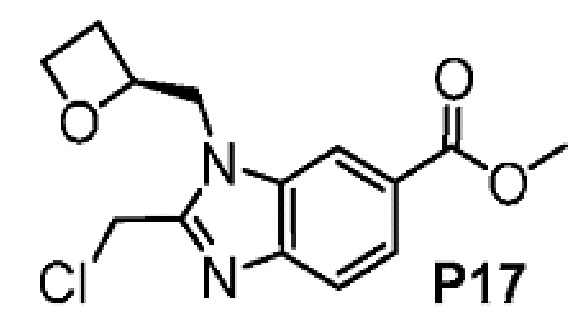
Раствор C43 (со стадии 6 получения P16; 51,8 кг, 190 моль) в ТГФ (678 кг) в автоклавном реакторе объемом 3000 л обрабатывают 10% палладием на угле (5,2 кг) при температуре в интервале от 10°C до 30°C. Трубку добавления промывают ТГФ (46 кг) и смывную жидкость добавляют к реакционной смеси. Содержимое реактора продувают азотом, затем аналогично продувают водородом, повышая давление до 0,1-0,2 МПа и затем сбрасывая до 0,02-0,05 МПа. Такую продувку водородом повторяют 5 раз, после чего давление водорода повышают до 0,1-0,25 МПа. Реакционную смесь перемешивают при температуре в интервале от 20°C до 30°C, и каждые 2-3 часа смесь продувают азотом три раза, и затем продувают водородом пять раз; после каждой конечной замены водорода давление водорода повышают до 0,1-0,25 МПа. Спустя 11,25 часов общего времени реакции реакционную смесь дренируют до нормального давления и продувают пять раз азотом, повышая давление до 0,15-0,2 МПа и затем сбрасывая давления до 0,05 МПа. Смесь затем фильтруют и осадок на фильтре дважды промывают ТГФ (64 кг и 63 кг); смывную жидкость объединяют с фильтратом и концентрируют при пониженном давлении (≤−0,08 МПа) и тепературе ≤40°C до объема 128-160 л. Добавляют ТГФ (169 кг) и смесь снова концентрируют до объема 128-160 л; эту процедуру повторяют 4 раза с получением раствора промежуточного соединения P16.
К полученному раствору добавляют ТГФ (150 кг) с последующим добавлением 2-хлор-1,1,1-триметоксиэтана (35,1 кг, 227 моль) и моногидрата п-толуолсульфоновой кислоты (1,8 кг, 9,5 моль). Реакционную смесь перемешивают в течение 25 минут, затем нагревают до температуры в интервале от 40°C до 45°C и выдерживают при указанной температуре в течение 5 часов, после чего концентрируют при пониженном давлении до объема 135-181 л. К раствору добавляют 2-пропанол (142 кг) и смесь снова концентрируют до объема 135-181 л, после чего добавляют 2-пропанол (36,5 кг) и очищенную воду (90 кг) и продолжают перемешивание до получения раствора. Полученный раствор фильтруют через подключенный жидкостной фильтр и затем обрабатывают очищенной водой (447 кг) в контрольной скоростью от 150 до 400 кг/час при температуры от 20°C до 40°C. Смесь охлаждают до темпемпературы в интервале от 20°C до 30°C, затем перемешивают в течение 2 часов и собирают твердый продукт фильтрацией в центрифуге. Осадок на фильтре промывают 2-пропанолом (20,5 кг) и очищенной водой (154 кг); после сушки получают P17 в виде твердого белого вещества (32,1 кг, 109 моль, 57%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,14-8,11 (м, 1H), 8,01 (дд, 1H), 7,79 (уш. д, 1H), 5,26-5,18 (м, 1H), 5,04 (с, 2H), 4,66-4,58 (м, 2H), 4,53 (дд,1H), 4,34 (дт, 1H), 3,96 (с, 3H), 2,82-2,71 (м, 1H), 2,48-2,37 (м, 1H).
Получение P18
Метил-3-метил-2-{[(2S)-2-метилпиперазин-1-ил]метил}-3H-имидазо[4,5-b]пиридин-5-карбоксилат (P18)
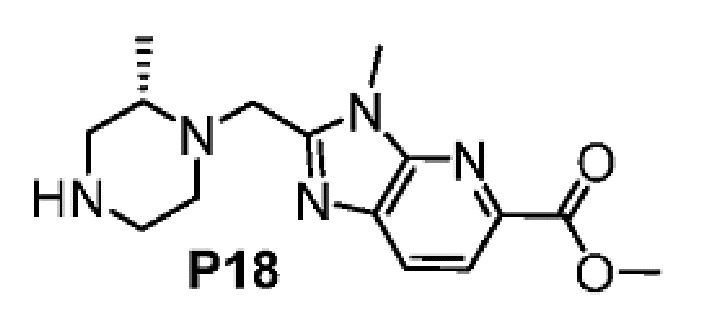
Стадия 1. Синтез трет-бутил-(3S)-4-(2-этокси-2-оксоэтил)-3-метилпиперазин-1-карбоксилата (C44)
К раствору трет-бутил-(3S)-3-метилпиперазин-1-карбоксилата (1,20 г, 5,99 ммоль) в N, N-диметилформамидe (15 мл) добавляют этилбромацетат (1,20 г, 7,19 ммоль) и карбонат калия (2,48 г, 18,0 ммоль). Реакционную смесь перемешивают при 50°C в течение 2 часов, затем охлаждают до комнатной температуры, разбавляют EtOAc (25 мл) и промывают водой (25 мл). Водный слой экстрагируют EtOAc (2 × 30 мл), объединенные органические слои промывают насыщенным водным раствором хлорида натрия (3 × 20 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 0% до 10% MeOH в дихлорметане) приводит к получению С44 в виде масла желтого цвета. Выход: 1,6 г, 5,6 ммоль, 93%.
Стадия 2. Синтез [(2S)-4-(трет-бутоксикарбонил)-2-метилпиперазин-1-ил]уксусной кислоты (C45)
Водный раствор гидроксида натрия (2 M; 5,34 мл, 10,7 ммоль) и воду (1 мл) добавляют к раствору C44 (612 мг, 2,14 ммоль) в MeOH (5 мл) и перемешивают полученную реакционную смесь при комнатной температуре (10°C) в течение 2 часов. После удаления органического растворителя в вакууме остаток подкисляют до pH 7 добавлением 1 M соляной кислоты и концентрируют полученную смесь при пониженном давлении. Полученный твердый остаток после этого добавляют к смеси дихлорметана и MeOH (10:1, 45 мл), смесь перемешивают при комнатной температуре (10°C) в течение 20 часов, после чего фильтруют и затем фильтрат концентрируют в вакууме с получением C45 в виде смолы бледно-желтого цвета. Выход: 300 мг, 1,16 ммоль, 54%.
Стадия 3. Синтез трет-бутил-(3S)-4-(2-{[6-хлор-2-(метиламино)пиридин-3-ил]амино}-2-оксоэтил)-3-метилпиперазин-1-карбоксилата (C46)
К раствору C45 (270 мг, 1,04 ммоль) и 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50% раствор в EtOAc; 1,27 г, 2,00 ммоль) в N, N-диметилформамидe (3 мл) добавляют 6-хлор-N2-метилпиридин-2,3-диамин (182 мг, 1,15 ммоль) и N, N-диизопропилэтиламин (465 мг, 3,60 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, после чего объединяют с продуктом аналогичной реакции с использованием C45 (25,5 мг, 98,8 мкмоль), разбавляют водой (20 мл) и экстрагируют EtOAc (3 × 20 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (2 × 20 мл), сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 0% до 44% EtOAc в петролейном эфире) с получением C46 в виде черной смолы. Общий выход: 320 мг, 0,804 ммоль, 70%.
Стадия 4. Синтез трет-бутил-(3S)-4-[(5-хлор-3-метил-3H-имидазо[4,5-b]пиридин-2-ил)метил]-3-метилпиперазин-1-карбоксилата (C47)
Смесь C46 (320 мг, 0,804 ммоль) и уксусной кислоты (4 мл) перемешивают при 100°C в течение 16 часов и затем концентрируют в вакууме. Остаток смешивают с дихлорметаном (10 мл) и затем добавляют ди-трет-бутилдикарбонат (287 мг, 1,32 ммоль) и триэтиламин (200 мг, 1,97 ммоль), перемешивают полученную реакционную смесь при комнатной температуре (15°C) в течение 2 часов, после чего промывают водой (20 мл) и экстрагируют дихлорметаном (3 × 20 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и остаток очищают хроматографией (силикагель, градиент: от 0% до 45% EtOAc в петролейном эфире) с получением C47 в виде смолы желтого цвета. Выход: 180 мг, 0,474 ммоль, 59%.
Стадия 5. Синтез метил-2-{[(2S)-4-(трет-бутоксикарбонил)-2-метилпиперазин-1-ил]метил}-3-метил-3H-имидазо[4,5-b]пиридин-5-карбоксилата (C48)
1,3-Бис(дифенилфосфино)пропан (43,0 мг, 0,104 ммоль), ацетат палладия (II) (12,7 мг, 56,6 мкмоль) и триэтиламин (528 мг, 5,22 ммоль) добавляют к раствору C47 (180 мг, 0,474 ммоль) в смеси MeOH (7 мл) и N, N-диметилформамида и перемешивают полученную реакционную смесь при 80°C в атмосфере монооксида углерода (50 фунтов на кв. дюйм (344,74 кПа)) в течение 20 часов. Затем смесь концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 0% до 78% EtOAc в петролейном эфире) с получением C48 в виде смолы желтого цвета. Выход: 150 мг, 0,372 ммоль, 78%.
Стадия 6. Синтез метил-3-метил-2-{[(2S)-2-метилпиперазин-1-ил]метил}-3H-имидазо[4,5-b]пиридин-5-карбоксилата (P18)
К раствору C48 (100 мг, 0,248 ммоль) в дихлорметане (4 мл) добавляют трифторуксусную кислоту (1 мл) и перемешивают полученную реакционную смесь при комнатной температуре (15°C) в течение 2 часов. Затем смесь концентрируют в вакууме и подвергают катионному обмену твердофазной экстракцией (колонка Agela Cleanert SCX) с получением P18 в виде смолы коричневого цвета. Выход: 70 мг, 0,23 ммоль, 93%. ЖХМС m/z 304,1 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,08 (AB квартет, 2H), 4,34 (д, 1H), 4,06 (с, 3H), 4,03 (с, 3H), 3,59 (д, 1H), 2,99-2,91 (м, 1H), 2,89-2,82 (м, 1H), 2,80-2,71 (м, 1H), 2,65-2,55 (м, 2H), 2,53-2,43 (м, 1H), 2,32-2,22 (м, 1H), 1,19 (д, 3H).
Получение P19
Метил-2-(6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат (P19)
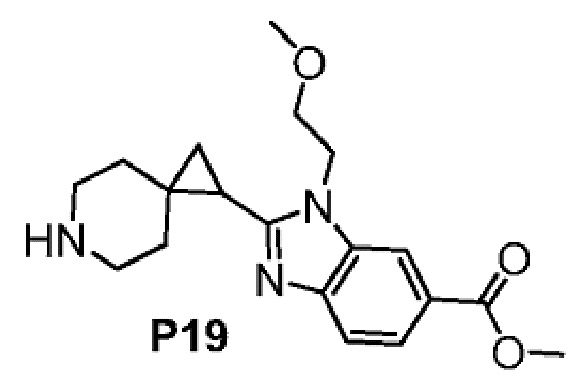
Стадия 1. Синтез метил-3-[(2-метоксиэтил)амино]-4-нитробензоата (C49)
К бесцветному раствору метил-3-фтор-4-нитробензоата (50 г, 250 ммоль) в ТГФ (400 мл) добавляют триэтиламин (40,7 г, 402 ммоль, 55,8 мл) с последующим добавлением по каплям при комнатной температуре 2-метоксиэтанамина (30,2 г, 402 ммоль) в ТГФ (100 мл). Полученный раствор желтого цвета перемешивают при 55°C в течение 18 часов. Раствор охлаждают до комнатной температуры и концентрируют при пониженном давлении для удаления ТГФ. Полученный твердый продукт желтого цвета растворяют в EtOAc (800 мл) и промывают насыщенным водным раствором хлорида аммония (250 мл). Водную фазу отделяют и экстрагируют EtOAc (200 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (3 × 250 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении с получением C49 (60,2 г, 94%) в виде твердого вещества желтого цвета. 1H ЯМР (CDCl3) δ 8,23 (д, 1H), 8,17 (уш. с, 1H), 7,58 (д, 1H), 7,25 (дд, 1H), 3,95 (с, 3H), 3,69-3,73 (м, 2H), 3,56 (м, 2H), 3,45 (с, 3H); ЖХМС m/z 255,4 [M+H]+.
Стадия 2. Синтез метил-4-амино-3-[(2-метоксиэтил)амино]бензоата (C50)
К раствору C49 (30 г, 118 ммоль) в MeOH (500 мл) добавляют палладий на угле (10 г, 94 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в атмосфере водорода с давлением 15 фунтов на кв. дюйм (103,42 кПа) в течение 18 часов. Суспензию черного цвета фильтруют через диатомовую землю и осадок на фильтре промывают MeOH (500 мл). Объединенные фильтраты концентрируют в вакууме с получением C50 (26,5 г, количественный выход) в виде масла коричневого цвета, которое затвердевает при стоянии. 1H ЯМР (400 МГц, CDCl3) δ 7,48 (дд, 1H), 7,36 (д, 1H), 6,69 (д, 1H), 3,87 (с, 3H), 3,77 (уш. с, 2H), 3,68 (т, 2H), 3,41 (с, 3H), 3,32 (т, 2H); ЖХМС m/z 224,7 [M+H]+.
Стадия 3. Синтез трет-бутил-1-({4-(метоксикарбонил)-2-[(2-метоксиэтил)амино]фенил}карбонил)-6-азаспиро[2.5]октан-6-карбоксилата (C51)
К раствору C17 (1,50 г, 5,88 ммоль) и C50 (1,49 г, 6,64 ммоль) в N, N-диметилформамидe (30 мл) при комнатной температуре (15°C) добавляют гексафторфосфат О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (HATU; 3,35 г, 8,81 ммоль). Реакционную смесь перемешивают при 15°C в течение 20 минут, после чего добавляют триэтиламин (1,19 г, 11,8 ммоль) и перемешивают полученную реакционную смесь при 50°C в течение 4 часов. Смесь выливают в воду (160 мл) и экстрагируют EtOAc (3 × 100 мл); объединенные органические слои промывают насыщенным водным раствором хлорида натрия (3 × 100 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 0% до 5% MeOH в дихлорметане) приводит к получению С51 в виде масла желтого цвета. Выход: 2,70 г, 5,85 ммоль, 99%.
Стадия 4. Синтез метил-2-[6-(трет-бутоксикарбонил)-6-азаспиро[2.5]окт-1-ил]-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (C52)
Раствор C51 (2,70 г, 5,85 ммоль) в уксусной кислоте (25 мл) перемешивают при 50°C в течение 16 часов, после чего осторожно подщелачивают добавлением насыщенного водного раствора карбоната калия. Полученную смесь экстрагируют EtOAc (2 × 100 мл), объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка остатка хроматографией (силикагель, градиент: от 50% до 100% EtOAc в петролейном эфире) приводит к получению C52 в виде твердого вещества желтого цвета. Выход: 1,45 г, 3,27 ммоль, 56%.
Стадия 5. Синтез метил-2-(6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (P19).
К раствору C52 (550 мг, 1,24 ммоль) в дихлорметане (10 мл) добавляют раствор соляной кислоты в EtOAc (4 M; 10 мл). Реакционную смесь перемешивают при 20°C°C в течение 2 часов, после чего смесь концентрируют при пониженном давлении. Остаток обрабатывают насыщенным водным раствором карбоната калия (10 мл) и экстрагируют дихлорметаном (3 × 40 мл); объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют в вакууме с получением P19 в виде твердого вещества желтого цвета. Выход: 400 мг, 1,16 ммоль, 94%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,06-8,02 (м, 1H), 7,93 (дд, 1H), 7,66 (д, 1H), 4,51 (ддд, компонент ABXY шаблона; 1H), 4,38 (ддд, компонент ABXY шаблона; 1H), 3,93 (с, 3H), 3,81-3,69 (м, 2H), 3,30 (с, 3H), 3,08-3,00 (м, 1H), 2,93 (ддд, 1H), 2,80-2,66 (м, 2H), 2,09 (дд, 1H), 1,88-1,77 (м, 1H), 1,67 (дд, 1H), 1,51-1,41 (м, 2H), 1,40-1,30 (м, 1H), 1,12 (дд, 1H).
Получение P20
Метил-2-(хлорметил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат (P20)

Стадия 1. Синтез метил-2-(хлорметил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (P20)
К раствору C50 (5,00 г, 22,3 ммоль) в ТГФ (100 мл) добавляют 2-хлор-1,1,1-триметоксиэтан (3,31 мл, 24,6 ммоль) и затем моногидрат п-толуолсульфоновой кислоты (84,8 мг, 0,446 ммоль). Реакционную смесь нагревают до 45°C и выдерживают при указанной температуре в течение 5 часов, после чего концентрируют в вакууме; к маслянистому остатку добавляют в EtOAc (10 мл) и нагревают смесь до получения раствора. Раствор медленно перемешивают в течение ночи, охлаждая до комнатной температуры. Осадок собирают фильтрацией и промывают гептаном с получением P20 в виде твердого вещества серого цвета. Выход: 5,73 г, 20,3 ммоль, 91%. 1H ЯМР (600 МГц, хлороформ-d) δ 8,12 (уш. с, 1H), 8,01 (уш. д, 1H), 7,79 (д, 1H), 4,96 (с, 2H), 4,52 (т, 2H), 3,96 (с, 3H), 3,74 (т, 2H), 3,28 (с, 3H).
Стадия 2. Синтез гидрохлоридой соли метил-2-(хлорметил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (P20, HCl соль)
Раствор C50 (5,0 г, 24 ммоль) в 1,4-диоксане (100 мл) нагревают до 100°C, к раствору с помощью капельной воронки в течение 10 часов добавляют раствор хлоруксусного ангидрида (4,1 г, 24,5 ммоль) в 1,4-диоксане (60 мл) и перемешивают полученную реакционную смесь в течение дополнительных 12 часов при 100°C. На следующий день реакционную смесь охлаждают до комнатной температуры и 1,4-диоксан удаляют при пониженном давлении. Сырую реакционную смесь растворяют в EtOAc и промывают насыщенным водным раствором бикарбоната натрия. EtOAc слой отделяют, сушат над сульфатом натрия и фильтруют. Раствор 4 M соляной кислоты в 1,4-диоксане (1,1 экв.) добавляют к раствору продукта в EtOAc при постоянном перемешивании. HCl соль целевого продукта осаждается в виде твердого вещества бледно-желтого цвета. Суспензию перемешивают в течение 1 часа и продукт собирают фильтрацией с получением HCl соли P20 в виде твердого вещества желтого цвета (6,1 г, 86%). 1H ЯМР (600 МГц, CD3OD) δ 8,64 (с, 1H), 8,30 (д, 1H), 7,92 (д, 1H), 5,32 (с, 2H), 4,84 (м, 2H), 3,99 (с, 3H), 3,83 (т, 2H), 3,31 (с, 3H). ЖХМС m/z 283,2 [M+H]+.
Получение P21
Метил-1-(2-метоксиэтил)-2-(пиперазин-1-илметил)-1H-бензимидазол-6-карбоксилат (P21)
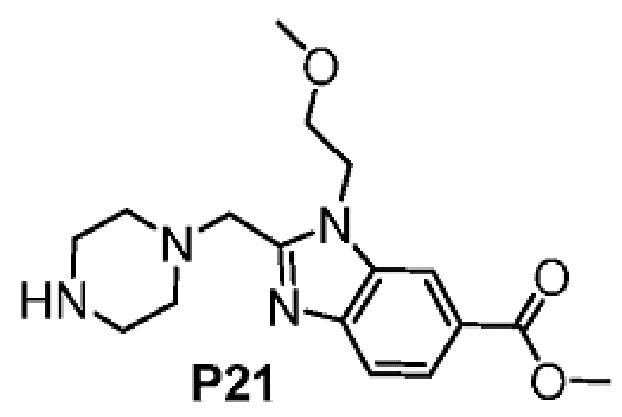
Стадия 1. Синтез метил-2-{[4-(трет-бутоксикарбонил)пиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (C53)
Соединение P20 (1,59 г, 5,62 ммоль) добавляют к смеси трет-бутилпиперазин-1-карбоксилата (1,00 г, 5,37 ммоль) и карбоната калия (2,97 г, 21,5 ммоль) в ацетонитриле (15 мл) при 15°C и перемешивают полученную реакционную смесь при 55°C в течение 12 часов. Полученную смесь затем объединяют с продуктом аналогичной реакции с использованием P20 и трет-бутил-пиперазин-1-карбоксилата (200 мг, 1,07 ммоль) и фильтруют полученную смесь. Фильтрат концентрируют в вакууме, остаток очищают хроматографией (силикагель, градиент: от 0% до 60% EtOAc в петролейном эфире) с получением C53 в виде твердого вещества бледно-желтого цвета. Общий выход: 2,30 г, 5,32 ммоль, 83%. ЖХМС m/z 433,0 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,12 (д, 1H), 7,96 (дд, 1H), 7,73 (д, 1H), 4,58 (т, 2H), 3,95 (с, 3H), 3,89 (с, 2H), 3,73 (т, 2H), 3,46-3,37 (уш. м, 4H), 3,28 (с, 3H), 2,54-2,44 (уш. м, 4H), 1,45 (с, 9H).
Стадия 2. Синтез метил-1-(2-метоксиэтил)-2-(пиперазин-1-илметил)-1H-бензимидазол-6-карбоксилата (P21)
К раствору C53 (2,30 г, 5,32 ммоль) в дихлорметане (80 мл) добавляют раствор соляной кислоты в EtOAc (20 мл). Реакционную смесь перемешивают при 20°C в течение 2 часов и затем концентрируют в вакууме. Остаток разбавляют водой (20 мл), значение pH раствора доводят до 9-10 добавлением насыщенного водного раствора бикарбоната натрия и экстрагируют раствор смесью EtOAc и MeOH (10:1, 15 × 50 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением P21 в виде твердого вещества бледно-желтого цвета. Выход: 1,68 г, 5,05 ммоль, 95%. ЖХМС m/z 332,8 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,13 (уш. с, 1H), 7,96 (уш. д, 1H), 7,72 (д, 1H), 4,59 (т, 2H), 3,95 (с, 3H), 3,86 (с, 2H), 3,75 (т, 2H), 3,29 (с, 3H), 2,87 (т, 4H), 2,50 (уш. м, 4H).
Получение P22
Метил-2-(хлорметил)-1-метил-1H-бензимидазол-6-карбоксилат (P22)
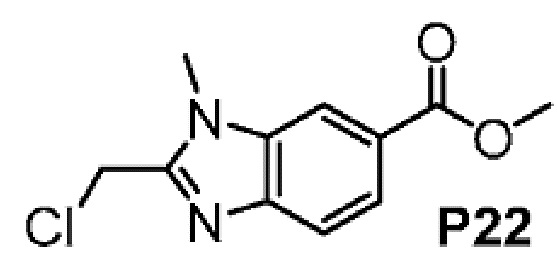
Метил-4-амино-3-(метиламино)бензоат (206 мг, 1,14 ммоль) растворяют в 1,4-диоксане (11,5 мл) и обрабатывают хлорацетилхлоридом (109 мкл, 1,37 ммоль). Смесь перемешивают при 100°C в течение 3 часов и охлаждают до комнатной температуры. К смеси добавляют триэтиламин (0,8 мл, 7 ммоль) и гептан (10 мл) и полученную смесь фильтруют. Фильтрат концентрируют при пониженном давлении, сырой продукт очищают хроматографией (силикагель, элюент: 40% EtOAc в гептане) с получением 120 мг P22 (44%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,14 (с, 1H), 8,01 (д, 1H), 7,78 (д, 1H), 4,87 (с, 2H), 3,97 (с, 3H), 3,94 (с, 3H); ЖХМС m/z 239,1 [M+H]+.
Получение P23
Метил-4-амино-3-{[(1-этил-1H-имидазол-5-ил)метил]амино}бензоат (P23)
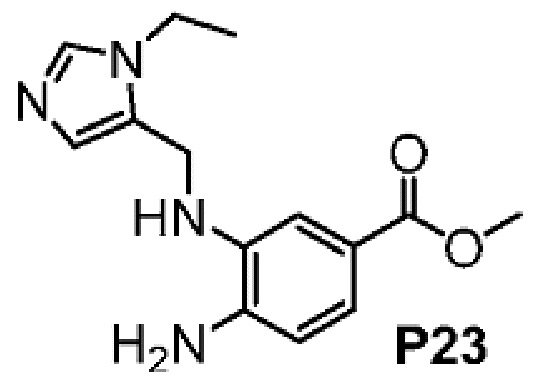
Стадия 1. Синтез метил-3-{[(1-этил-1H-имидазол-5-ил)метил]амино}-4-нитробензоата (C54)
Триэтиламин (3,65 мл, 26,2 ммоль) добавляют к раствору метил-3-фтор-4-нитробензоата (1,00 г, 5,02 ммоль) и дигидрохлоридной соли 1-(1-этил-1H-имидазол-5-ил)метанамина (1,00 г, 5,05 ммоль) в смеси ТГФ (12 мл) и MeOH (8 мл). Реакционную смесь перемешивают при 60°C в течение 40 часов, после чего смесь концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 0% до 2% MeOH в дихлорметане). Соединение C54 получают в виде твердого вещества оранжевого цвета. Выход: 1,27 г, 4,17 ммоль, 83%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,24 (д, 1H), 7,98-7,91 (м, 1H), 7,68 (д, 1H), 7,57 (уш. с, 1H), 7,33 (дд, 1H), 7,11 (уш. с, 1H), 4,53 (д, 2H), 3,99 (кв, 2H), 3,95 (с, 3H), 1,47 (т, 3H).
Стадия 2. Синтез метил-4-амино-3-{[(1-этил-1H-имидазол-5-ил)метил]амино}бензоата (P23)
Смесь влажного палладия на угле (144 мг) и C54 (412 мг, 1,35 ммоль) в MeOH (13 мл) перемешивают в атмосфере водорода из баллона в течение 16 часов при 25°C. Реакционную смесь фильтруют через слой диатомовой земли и концентрируют фильтрат в вакууме с получением P23 в виде твердого вещества серого цвета. Выход: 340 мг, 1,24 ммоль, 92%. 1H ЯМР (400 МГц, метанол-d4) δ 7,66 (уш. с, 1H), 7,38-7,29 (м, 2H), 6,97 (уш. с, 1H), 6,67 (д, 1H), 4,35 (с, 2H), 4,11 (кв, 2H), 3,81 (с, 3H), 1,44 (т, 3H).
Получение P24
Гидрохлоридная соль метил-2-(хлорметил)-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилата (P24)
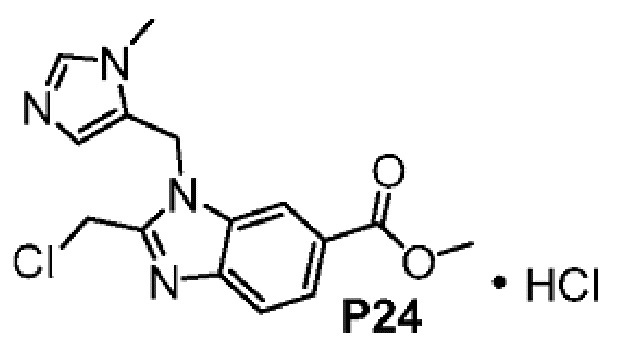
Стадия 1. Синтез метил-3-{[(1-метил-1H-имидазол-5-ил)метил]амино}-4-нитробензоата (C55)
К бесцветному раствор метил-3-фтор-4-нитробензоата (1,0 г, 5,0 ммоль) в N, N-диметилформамидe (10 мл) медленно добавляют 1-(1-метил-1H-имидазол-5-ил)метанамин (670 мг, 6,0 ммоль) и триэтиламин (762 мг, 7,53 ммоль). Раствор перемешивают при 60°C в течение 16 часов. Реакционную смесь выливают в воду (30 мл) и экстрагируют дихлорметаном (3 × 30 мл). Объединенные органические экстракты сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают флэш-хроматографией (20% MeOH/дихлорметан). Полученное твердое вещество желтого цвета растирают в смеси (30:1) петролейный эфир/EtOAc с получением C55 (1,2 г, 82%) в виде твердого вещества желтого цвета. 1H ЯМР (CDCl3) δ 8,26 (д, 1H), 7,96 (уш. с, 1H), 7,71 (д, 1H), 7,50 (с, 1H), 7,35 (дд, 1H), 7,13 (с, 1H), 4,55 (д, 2H), 3,97 (с, 3H), 3,68 (с, 3H).
Стадия 2. Синтез метил-4-амино-3-{[(1-метил-1H-имидазол-5-ил)метил]амино}бензоата (C56)
К суспензии C55 (5,46 г, 18,8 ммоль) желтого цвета в MeOH (160 мл) добавляют влажный 10% палладий на угле (1 г). Смесь перемешивают в атмосфере водорода с давлением 1 атмосфера (101,32 кПа) в течение 36 часов при 20°C. Реакционную смесь фильтруют и промывают осадок на фильтре MeOH (200 мл). Фильтрат концентрируют при пониженном давлении с получением C56 (4,8 г, 98%) в виде твердого вещества коричневого цвета. 1H ЯМР (ДМСО-d6) δ 7,56 (с, 1H), 7,18 (д, 1H), 7,13 (с, 1H), 6,87 (с, 1H), 6,55 (д, 1H), 5,50 (с, 2H), 4,84 (т, 1H), 4,23 (д, 2H), 3,73 (с, 3H), 3,63 (с, 3H).
Стадия 3. Синтез метил-2-(гидроксиметил)-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилата (C57)
Смесь C56 (780 мг, 3,00 ммоль) и гидроксиуксусной кислоты (342 мг, 4,49 ммоль) в 1,3,5-триметилбензоле (8 мл) перемешивают при 140°C в атмосфере N2 в течение 14 часов и затем при 25°C в течение 48 часов. Прозрачный раствор желтого цвета декантируют с получением остатка коричневого цвета, который растворяют в MeOH (50 мл) и концентрируют при пониженном давлении. Сырой продукт очищают флэш-хроматографией (20% MeOH/дихлорметан) с получением C57 (318 мг, 35%) в виде пены желтого цвета. 1H ЯМР (ДМСО-d6) δ 8,13 (д, 1H), 7,83 (дд, 1H), 7,71 (д, 1H), 7,60 (с, 1H), 6,59 (с, 1H), 5,69 (с, 2H), 4,76 (с, 2H), 3,91 (с, 1H), 3,84 (с, 3H), 3,53 (с, 3H).
Стадия 4. Синтез гидрохлоридной соли метил-2-(хлорметил)-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоксилата (P24)
К суспензии желтого цвета C57 (500 мг, 1,66 ммоль) в дихлорметане (10 мл) и N, N-диметилформамидe (3 мл) при комнатной температуре по каплям добавляют SOCl2 (990 мг, 0,60 мл, 8,32 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, концентрируют при пониженном давлении и растирают полученный остаток коричневого цвета с дихлорметаном (10 мл). Твердый продукт собирают фильтрацией, промывают дихлорметаном (5 мл) и сушат в вакууме с получением P24 (431 мг, 73%) в виде твердого не совсем белого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,17 (с, 1H), 8,31 (с, 1H), 7,91-7,99 (м, 1H), 7,77-7,87 (м, 1H), 7,11 (с, 1H), 5,92 (с, 2H), 5,13 (с, 2H), 3,87 (с, 3H), 3,86 (с, 3H); MS(ES+): 319,0 (M+H).
Получение P25
трет-Бутил-4-нитро-3-[(1,3-оксазол-2-илметил)амино]бензоат (P25)
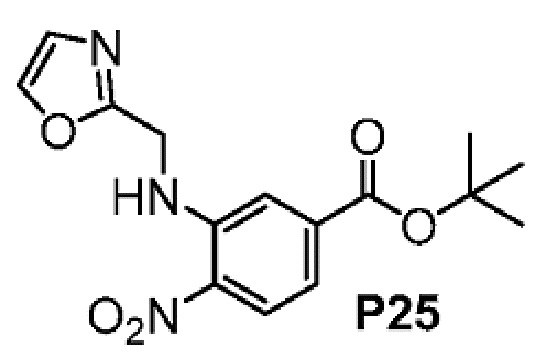
К суспензии гидрохлоридной соли 1-(1,3-оксазол-2-ил)метанамина (491 мг, 3,65 ммоль) и трет-бутил-3-фтор-4-нитробензоата (800 мг, 3,32 ммоль) в N, N-диметилформамидe (5 мл) добавляют карбонат калия (1,04 г, 6,63 ммоль). Реакционную смесь перемешивают при 60°C в течение 2 часов. Добавляют дополнительную порцию гидрохлоридной соли 1-(1,3-оксазол-2-ил)метанамина (100 мг, 1,0 ммоль) и реакционную смесь перемешивают в течение дополнительных 30 минут при 60°C. Реакционную смесь охлаждают до комнатной температуры, затем разбавляют водой (30 мл) и экстрагируют EtOAc (60 мл). Органический слой промывают водой, затем насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток оранжевого цвета остаток очищают флэш-хроматографией (12 г силикагель, элюирование с градиентом: 0-50% EtOAc/гептан) с получением P25 (764 мг, 75%) в виде твердого вещества оранжевого цвета. 1H ЯМР (CDCl3) δ 8,48 (уш. с, 1H), 8,23 (д, 1H), 7,68 (д, 1H), 7,61 (д, 1H), 7,28 (дд, 1H), 7,15 (с, 1H), 4,72 (д, 2H), 1,60 (с, 9H).
Получение P26
Метил-5-амино-6-{[(2S)-оксетан-2-илметил]амино}пиридин-2-карбоксилат (P26)
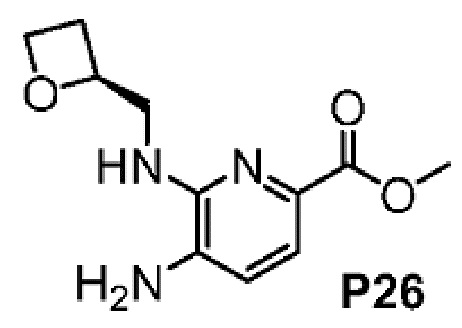
Стадия 1. Синтез метил-5-нитро-6-{[(2S)-оксетан-2-илметил]амино}пиридин-2-карбоксилата (C58)
Метил-6-хлор-5-нитропиридин-2-карбоксилат (270 г, 1,25 моль) и триэтиламин (500 г, 5,1 моль) добавляют к раствору C42 (152 г, 1,7 моль) в N, N-диметилформамидe (3 л) и ТГФ (3 л) при 25°C. Смесь перемешивают при 25°C в течение 16 часов. Смесь концентрируют при пониженном давлении для удаления ТГФ и добавляют воду (5 л). Смесь экстрагируют EtOAc (2 × 5 л) и объединенные органические растворы промывают насыщенным водным раствором хлорида натрия (2 ×), сушат и концентрируют при пониженном давлении. Сырой продукт объединяют с сырым продуктом второй загрузки из аналогичного эксперимента (70 г) и твердые вещества растирают со смесью петролейный эфир:EtOAc (4:1, 500 мл) в течение 2 часов. Твердый продукт собирают фильтрацией и сушат с получением C58 (304 г, 52%) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CDCl3) δ 8,58 (уш. с, 1H), 8,56 (д, 1H), 7,39 (д, 1H), 5,08-5,18 (м, 1H), 4,73 (ддд, 1H), 4,61 (td, 1H), 4,06-4,16 (м, 1H), 3,98 (с, 3H), 3,88-3,97 (м, 1H), 2,68-2,80 (м, 1H), 2,55 (тдд, 1H).
Стадия 2. Синтез метил-5-амино-6-{[(2S)-оксетан-2-илметил]амино}пиридин-2-карбоксилата (P26)
Соединение C58 (10 г, 37 ммоль) суспендируют в MeOH (150 мл), добавляют 10% палладий на угле (1,0 г) и перемешивают полученную смесь при комнатной температуре в атмосфере водорода с давлением 50 фунтов на кв. дюйм (344,74 кПа) в течение 4 часов. Смесь фильтруют через Celite® и фильтрат концентрируют при пониженном давлении с получением P26 (8,4 г, 95%) в виде масла желтого цвета, которое затвердевает при стоянии. 1H ЯМР (600 МГц, CDCl3) δ 7,49 (д, 1H), 6,86 (д, 1H), 5,06-5,15 (м, 1H), 4,68-4,77 (м, 1H), 4,53-4,63 (м, 2H), 3,91 (с, 3H), 3,80-3,86 (м, 2H), 3,72 (уш. с, 2H), 2,68-2,78 (м, 1H), 2,52-2,61 (м, 1H).
Получение P27
Метил-2-(хлорметил)-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоксилат (P27)
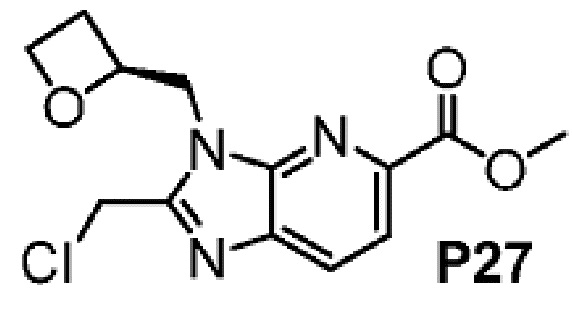
В трехгорлую колбу объемом 2 литра, снабженную механической мешалкой с верхним приводом, загружают P26 (43,0 г, 181 ммоль) в ТГФ (780 мл). Полученную суспензию бледно-розового цвета обрабатывают раствором хлоруксусного ангидрида (33,5 г, 190 ммоль в 100 мл ТГФ), добавляя его из капельной воронки в течение 30 минут. Полученный раствор светло-янтарного цвета перемешивают при комнатной температуре в течение 2 часов, затем нагревают до 60°C и выдерживают при указанной температуре в течение 7 часов. Реакционную смесь охлаждают до комнатной температуры. Примерно 400 мл растворителя удаляют из реакционной смеси на роторном испарителе при пониженном давлении. Полученный раствор разбавляют EtOAc (500 мл) и обрабатывают насыщенным водным раствором бикарбоната натрия (200 мл). Двухфазную смесь перемешивают при комнатной температуре в течение 30 минут. Органический слой отделяют, водный слой экстрагируют EtOAc (500 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (500 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении с получением P27 (52,5 г, 98%) в виде твердого вещества желтовато-коричневого цвета. 1H ЯМР (600 МГц, CDCl3) δ 8,14 (д, 2H), 5,19-5,28 (м, 1H), 4,99-5,16 (м, 2H), 4,70-4,88 (м, 2H), 4,55-4,67 (м, 1H), 4,24-4,44 (м, 1H), 4,01 (с, 3H), 2,70-2,88 (м, 1H), 2,37-2,53 (м, 1H); ЖХ-МС(ES+): 296,4 (M+H).
Получение P28
5-Хлор-2-(хлорметил)-3-метил-3H-имидазо[4,5-b]пиридин (P28)
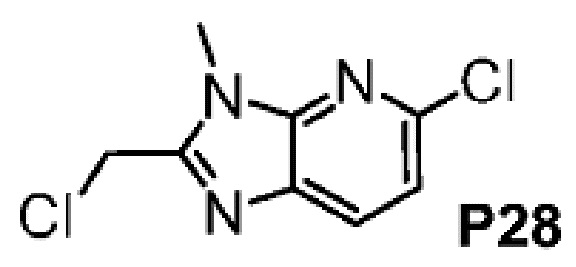
Стадия 1. Синтез 6-хлор-N-метил-3-нитропиридин-2-амина (C59)
К суспензии 2,6-дихлор-3-нитропиридина (200 г, 1,04 моль) и карбоната натрия (132 г, 1,24 моль) в этаноле (1 л) по каплям с помощью шприца добавляют 2,0 M раствор метиламина в ТГФ (622 мл, 1,24 моль) при 0°C. После добавления реакционную смесь перемешивают при 18°C в течение 6 часов. Смесь желтого цвета фильтруют и фильтрат концентрируют при пониженном давлении с получением твердого вещества желтого цвета. Сырой продукт очищают флэш-хроматографией (петролейный эфир/EtOAc 0-5%) с получением C59 (158 г, выход 81%) в виде твердого вещества желтого цвета. 1H ЯМР (ДМСО-d6) δ 8,72 (уш. с, 1H), 8,41 (д, 1H), 6,76 (д, 1H), 3,00 (д, 3H).
Стадия 2. Синтез 6-хлор-N2-метилпиридин-2,3-диамина (C60)
К смеси C59 (15,8 г, 84,2 ммоль) в уксусной кислоте (100 мл) добавляют железный порошок (15,4 г, 276 ммоль). Смесь желтого цвета перемешивают при 80°C в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры и фильтруют. Осадок на фильтре промывают EtOAc (2 × 100). Объединенные органические слои концентрируют при пониженном давлении и сырой продукт очищают флэш-хроматографией (120 г силикагеля, 50% EtOAc/петролейный эфир) с получением C60 (8,40 г, выход 63%) в виде твердого вещества коричневого цвета. 1H ЯМР (CDCl3) δ 6,80 (д, 1H), 6,50 (д, 1H), 3,39 (уш. с, 2H), 3,01 (с, 3H).
Стадия 3. Синтез 5-хлор-2-(хлорметил)-3-метил-3H-имидазо[4,5-b]пиридина (P28)
К раствору C60 (50,0 г, 317 ммоль) в 1,4-диоксане (1,2 л) добавляют хлорацетилхлорид (55,5 мл, 698 ммоль) и перемешивают полученную смесь при 15°C в течение 50 минут. Смесь коричневого цвета концентрируют при пониженном давлении с получением твердого вещества коричневого цвета, которое переносят в трифторуксусную кислоту (1,2 л) и перемешивают при 80°C в течение 60 часов. Смесь концентрируют при пониженном давлении с получением масла коричневого цвета. Масло разбавляют EtOAc (1 л) и нейтрализуют насыщенным водным раствором бикарбоната натрия. Когда выделение CO2 прекращается, слои разделяют и экстрагируют водный слой EtOAc (200 мл). Органические экстракты объединяют, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают флэш-хроматографией (элюирование с градиентом: 10-25% EtOAc/петролейный эфир) с получением P28 (61,0 г, 79%) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,13 (д, 1H), 7,37 (д, 1H), 5,11 (с, 2H), 3,84 (с, 3H).
Получение P29
5-Бром-N3,6-диметилпиридин-2,3-диамин (P29)
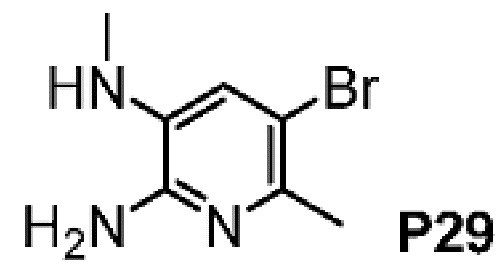
Соединение P29 синтезируют в соответствии с методикой, описанной в литературе (Choi, J. Y. et al., J. Med. Chem. 2012, 55, 852−870).
Получение P30
Гидрохлоридная соль метил-2-(6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин-6-карбоксилата (P30)
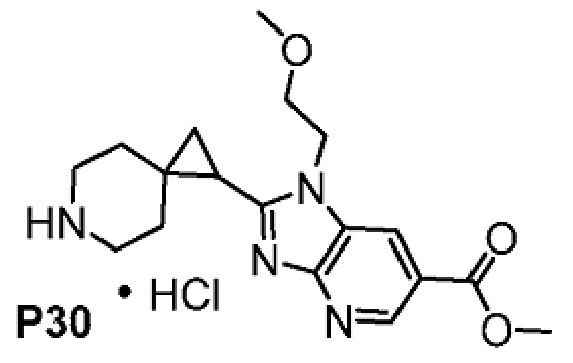
Стадия 1. Синтез трет-бутил-1-(6-бром-1H-имидазо[4,5-b]пиридин-2-ил)-6-азаспиро[2.5]октан-6-карбоксилата (C61)
Смесь C17 (1,00 г, 3,92 ммоль), гексафторфосфата О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (2,98 г, 7,83 ммоль), 5-бромпиридин-2,3-диамина (1,10 г, 5,85 ммоль) и триэтиламина (1,64 мл, 11,8 ммоль) растворяют в N, N-диметилформамидe и перемешивают полученную реакционную смесь при комнатной температуре в течение ночи, затем разбавляют водой и экстрагируют EtOAc. Органический слой концентрируют в вакууме и очищают хроматографией (силикагель). Полученный амид растворяют в 1,4-диоксане (8 мл), обрабатывают водным раствором гидроксида натрия (4 M; 2 мл, 8 ммоль) и перемешивают в течение 15 часов при 100°C. После стандартной обработки и хроматографии (силикагель, градиент: EtOAc в углеводородном растворителе) получают C61 (255 мг) и нециклизованный амид (255 мг). Амид подвергают действию гидроксида калия с получением дополнительного количества C61 (250 мг) в виде твердого вещества. Общий выход: 505 мг, 1,24 ммоль, 32%.
Стадия 2. Синтез трет-бутил-1-[6-бром-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин-2-ил]-6-азаспиро[2.5]октан-6-карбоксилата (C62)
Раствор C61 (225 мг, 0,552 ммоль) и трет-бутоксида калия (136 мг, 1,21 ммоль) в ТГФ перемешивают при комнатной температуре в течение 20 минут, после чего добавляют 2-бромэтилметиловый эфир (0,125 мл, 1,33 ммоль), затем реакционную смесь нагревают до 60°C и выдерживают при указанной температуре в течение 15 часов. ЖХМС анализ показывает, что получены два изомера. Хроматография (силикагель) приводит к получению очищенного C62 (120 мг) в виде смолы и C63 (69 мг). Указанные региоизомеры C62 и C63 определяют с использованием экспериментов ядерного эффекта Оверхаузера. Выход C62: 120 мг, 0,258 ммоль, 47%. ЖХМС m/z 465,3 (масс-спектр изотопа брома) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики δ 8,40 (с, 1H), 7,70 (с, 1H), 4,40-4,20 (м, 2H), 3,75-3,58 (м, 3H), 3,36-3,25 (м, 2H), 3,21 (с, 3H), 3,14-3,08 (м, 1H), 1,98 (дд, 1H), 1,74 (дд, 1H), 1,70-1,63 (м, 1H), 1,48-1,42 (м, 2H), 1,36 (с, 9H), 1,11 (дд, 1H).
Стадия 3. Синтез гидрохлоридной соли метил-2-(6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин-6-карбоксилата (P30)
Смесь C62 (120 мг, 0,258 ммоль), 1,3-бис(дифенилфосфино)пропана (31,9 мг, 77,4 мкмоль) и ацетата палладия (II) (11,6 мг, 51,7 мкмоль) в N, N-диметилформамидe (0,5 мл) обрабатывают MeOH (4 мл) и триэтиламином (0,36 мл, 2,6 ммоль), затем нагревают до 80°C и выдерживают при указанной температуре в атмосфере моноксида углерода (50 фунтов на кв. дюйм (344,74 кПа)) в течение 20 часов. После стандартной обработки очистка хроматографией (силикагель, элюент: 10:1 дихлорметан/MeOH) приводит к получению продукта, который затем для удаления защитной группы обрабатывают раствором соляной кислоты в 1,4-диоксане с получением P30 в виде твердого вещества. Выход: 40 мг, 0,105 ммоль, 41%. ЖХМС m/z 345,3 [M+H]+.
Получение P31
5-Бром-N3-(2-метоксиэтил)пиридин-2,3-диамин (P31)
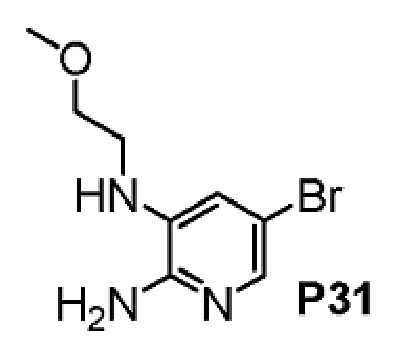
Стадия 1. Синтез N-(2-амино-5-бромпиридин-3-ил)-2-метоксиацетамида (C64)
В колбу, содержащую раствор метоксиуксусной кислоты (1,00 г, 11,1 ммоль) в N, N-диметилформамидe (30 мл) добавляют гексафторфосфат О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (6,33 г, 16,7 ммоль) и триэтиламин (3,37 г, 33,3 ммоль). Смесь перемешивают в течение 20 минут, затем небольшими порциями добавляют 5-бромпиридин-2,3-диамин (2,3 г, 12 ммоль) и перемешивают полученную реакционную смесь в течение ночи. Спустя 15 часов к смеси добавляют воду и экстрагируют раствор EtOAc. Объединенные органические слои сушат и растворитель удаляют при пониженном давлении. Сырой продукт очищают флэш-хроматографией (градиент: от 0% до 80% EtOAc в гептане) с получением C64 (2,3 г, 80%). 1H ЯМР (400 МГц, CDCl3) δ 8,09 (с, 1H), 8,06 (д, 1H), 8,03 (с, 1H), 7,83 (д, 1H), 4,08 (с, 2H), 3,53 (с, 3H); ЖХ-МС(ES+): 260,2 (M+H).
Стадия 2. Синтез 5-бром-N3-(2-метоксиэтил)пиридин-2,3-диамина (P31)
К раствору C64 (3,3 г, 13 ммоль) в ТГФ в течение 10 минут добавляют раствор BH3 в ТГФ (1 M; 14 мл, 14 ммоль) и перемешивают полученную смесь при комнатной температуре в течение ночи. Избыток борана гасят добавлением воды и затем смесь экстрагируют EtOAc. EtOAc слой сушат и концентрируют при пониженном давлении. Сырой продукт растворяют в MeOH, добавляют HCl в 1,4-диоксане (1,0 экв.) и перемешивают смесь в течение 2 часов. Избыток MeOH удаляют при пониженном давлении с получением сырого продукта. Соединение очищают флэш-хроматографией (градиент: от 0% до 70% EtOAc в гептане) с получением P31 в виде масла коричневого цвета (1,1 г, 35%). 1H ЯМР (600 МГц, CDCl3) δ 7,83 (д, 1H), 6,95 (д, 1H), 5,56 (с, 2H), 3,77 (т, 1H), 3,66 (т, 2H), 3,42 (с, 3H), 3,22 (кв, 2H); ЖХ-МС(ES+): 246,1.
Получение P32
6-Бром-2-(хлорметил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин (P32)
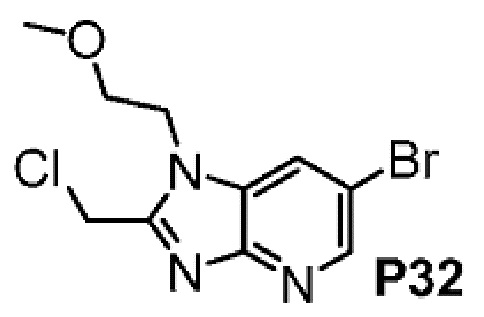
Соединение P31 (400 мг, 1,63 ммоль) переносят в 1,4-диоксан (8 мл) и обрабатывают хлорацетилхлоридом (0,284 мл, 3,58 ммоль). Смесь перемешивают при комнатной температуре. Растворитель удаляют при пониженном давлении, полученный остаток переносят в трифторуксусную кислоту (8 мл) и перемешивают при 80°C в течение 18 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении. Полученное масло коричневого цвета переносят в EtOAc (50 мл) и нейтрализуют насыщенным водным раствором бикарбоната натрия. После завершения выделения диоксида углерода слои разделяют и водный слой экстрагируют дополнительным количеством EtOAc (20 мл). Органические экстракты объединяют, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный сырой продукт очищают флэш-хроматографией (градиент: от 0% до 80% EtOAc в гептане) с получением P32 (176 мг, 36%) твердого вещества желтовато-коричневого цвета. 1H ЯМР (600 МГц, CDCl3) δ 8,59 (с, 1H), 7,90 (с, 1H), 4,93 (с, 2H), 4,45 (м, 2H), 3,72 (м, 2H), 3,29 (с, 3H); ЖХ-МС(ES+): 306,1 (M+H).
Получение P33
6-Бром-N4-(2-метоксиэтил)пиридин-3,4-диамин (P33)
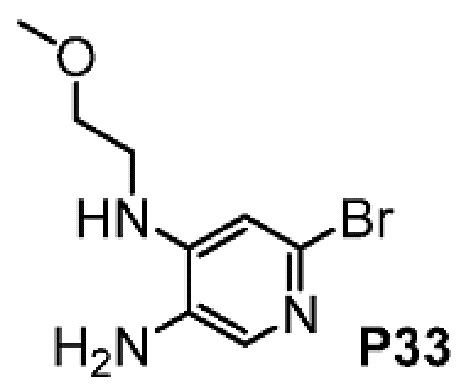
Стадия 1. Синтез 2-бром-N-(2-метоксиэтил)-5-нитропиридин-4-амина (C65)
Раствор 2,4-дибром-5-нитропиридина (8,65 г, 30,7 ммоль) в ТГФ (100 мл) обрабатывают триэтиламином (5,11 мл, 36,8 ммоль) и 2-метоксиэтанамином (2,77 г, 36,8 ммоль) и перемешивают полученную реакционную смесь при 20°C в течение 1 часа. После этого смесь выливают в воду (200 мл) и экстрагируют EtOAc (2 × 200 мл); объединенные органические слои сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и объединяют с продуктом аналогичной реакции с использованием 2,4-дибром-5-нитропиридина (600 мг, 2,13 ммоль). Очистка хроматографией (силикагель, градиент: от 0% до 35% EtOAc в петролейном эфире) приводит к получению С65 в виде твердого вещества желтого цвета. Общий выход: 9,02 г, 32,7 ммоль, 99%.
Стадия 2. Синтез 6-бром-N4-(2-метоксиэтил)пиридин-3,4-диамина (P33)
Восстановленный железный порошок (607 мг, 10,9 ммоль) и хлорид аммония (3,49 г, 65,2 ммоль) добавляют к раствору C65 (1,00 г, 3,62 ммоль) в смеси MeOH (10 мл) и воды (4 мл). Реакционную смесь перемешивают при 80°C в течение 4 часов, затем выливают в воду (30 мл) и экстрагируют EtOAc (4 × 50 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (50 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением P33 в виде твердого вещества бледно-коричневого цвета. Выход: 900 мг, предполагаемый количественный выход. 1H ЯМР (400 МГц, хлороформ-d) δ 7,63 (с, 1H), 6,59 (с, 1H), 4,58 (уш. с, 1H), 3,63 (t, 2H), 3,40 (с, 3H), 3,34-3,26 (м, 2H).
Получение P34
(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}циклогексил)уксусная кислота (P34)
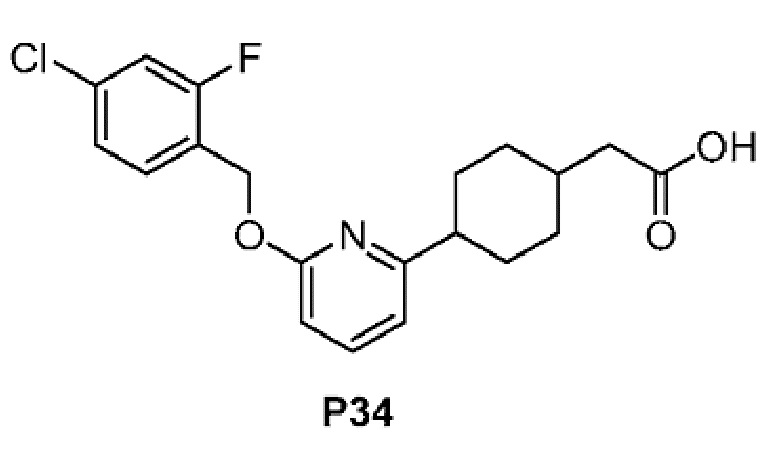
Соединение P34 получают в виде смеси цис- и транс-изомеров способом, аналогичным описанному в примере получения P2, исходя из 2,6-дихлорпиридина и метил-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклогекс-3-ен-1-ил]ацета реакцией сочетания Сузуки, хемоселективным восстановлением, получением простого эфира с использованием палладиевого катализатора и гидролизом сложного эфира. ЖХМС m/z 378,1 [M+H]+.
Пример 1
2-[(4-{2-[(4-Хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония(1)
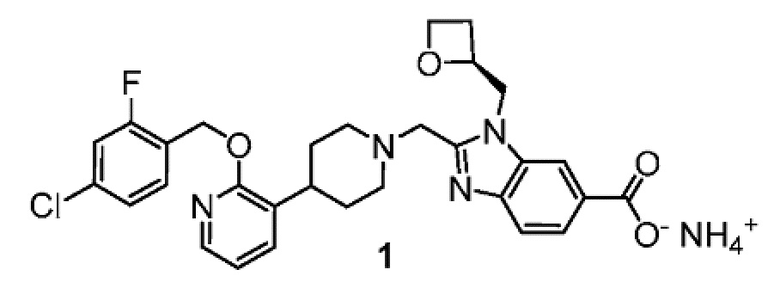
Стадия 1. Синтез метил-4-{[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)ацетил]амино}-3-{[(2S)-оксетан-2-илметил]амино}бензоата (C66)
К раствору P1 (100 мг, 0,264 ммоль) в N, N-диметилформамидe (5 мл) добавляют гексафторфосфат О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (151 мг, 0,396 ммоль). Смесь перемешивают при комнатной температуре (10°C) в течение 10 минут, добавляют P16 (62,4 мг, 0,264 ммоль) и N, N-диизопропилэтиламин (102 мг, 0,792 ммоль) и перемешивают полученную реакционную смесь при комнатной температуре (10°C) в течение 16 часов. Смесь обрабатывают насыщенным водным раствором хлоридом аммония (10 мл) и экстрагируют смесью дихлорметана и MeOH (10:1, 3 × 30 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме; очистка препаративной тонкослойной хроматографией (элюент: 20:1 дихлорметан/MeOH) приводит к получению С66 в виде бесцветного масла. Выход: 90 мг, 0,15 ммоль, 57%.
Стадия 2. Синтез метил-2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C67)
Раствор C66 (90 мг, 0,15 ммоль) в уксусной кислоте (5 мл) перемешивают при 50°C в течение 16 часов. Реакционную смесь концентрируют в вакууме, остаток растворяют в смеси дихлорметана и MeOH (10:1, 30 мл) и промывают насыщенным водным раствором карбоната натрия (20 мл). Водный слой экстрагируют смесью дихлорметана и MeOH (10:1, 3 × 30 мл), объединенные органические слои промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением C67 (90 мг) в виде бесцветного масла, часть которого используют непосредственно в следующей стадии.
Стадия 3. Синтез 2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (1)
К раствору C67 (полученного на предыдущей стадии; 80 мг, ≤0,13 ммоль) в MeOH (2 мл) добавляют гидроксид лития (4,96 мг, 0,209 ммоль) и воды (0,5 мл) и перемешивают реакционную смесь при 50°C в течение 16 часов. После этого смесь концентрируют в вакууме и остаток очищают ВЭЖХ с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 13% до 33% B) с получением 1 в виде твердого белого вещества. Выход: 28 мг, 48 мкмоль, 37% для 2 стадий. ЖХМС m/z 565,1♦ [M+H]+. δ (400 МГц, метанол-d4) δ 8,30 (д, 1H), 8,00-7,94 (м, 2H), 7,65 (д, 1H), 7,58 (дд, 1H), 7,51 (дд, 1H), 7,29-7,21 (м, 2H), 6,94 (дд, 1H), 5,42 (с, 2H), 5,29-5,21 (м, 1H), 4,9-4,83 (м, 1H, предполагаемый; частично скрыт пиком воды), 4,71 (дд, 1H), 4,67-4,60 (м, 1H), 4,45 (дт, 1H), 3,99 (AB квартет, 2H), 3,11-3,04 (м, 1H), 3,02-2,94 (м, 1H), 2,92-2,74 (м, 2H), 2,57-2,47 (м, 1H), 2,40-2,25 (м, 2H), 1,90-1,69 (м, 4H).
Соединения, представленные в таблице 1 ниже, синтезируют в соответствии с методиками, описанными в настоящем документе для синтеза соединений примеров и примеров получений, с использованием подходящих исходных соединений, которые являются коммерчески доступными либо получены с использованием способов, известных специалистам в данной области техники или аналогичных описанным для получения других промежуточных соединений. Соединения очищают с использованием методов, которые хорошо известны специалистам в данной области техники и могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 1. Структуры и названия в соответствии с номенклатурой ИЮПАК соединений примеров со 2 по 18
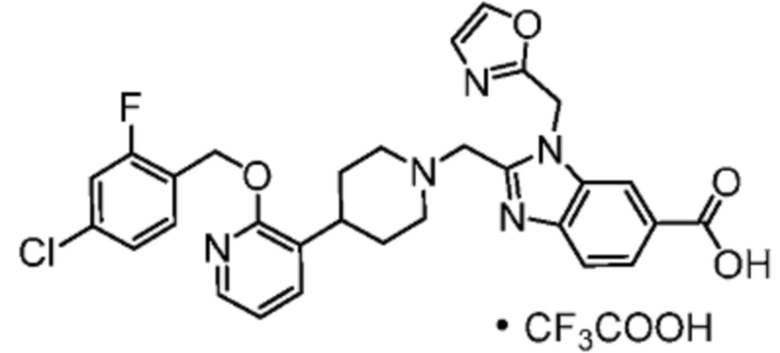
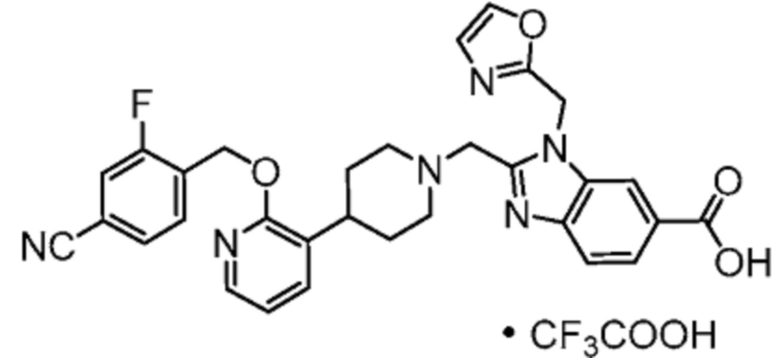
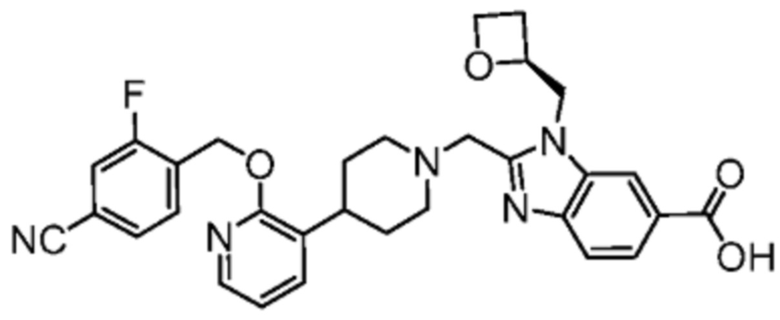
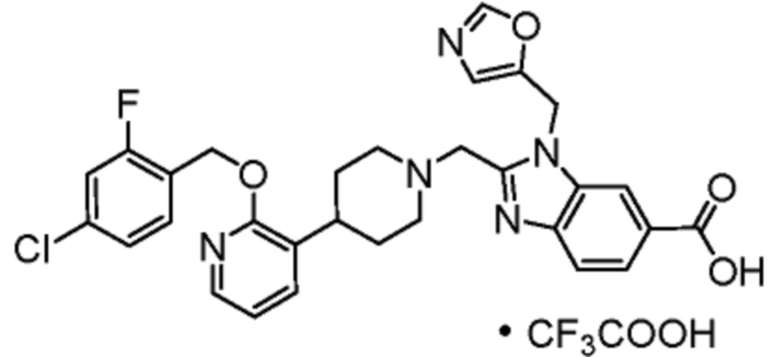
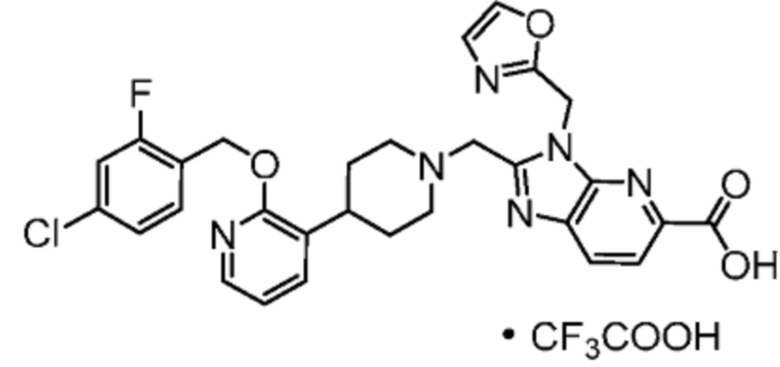
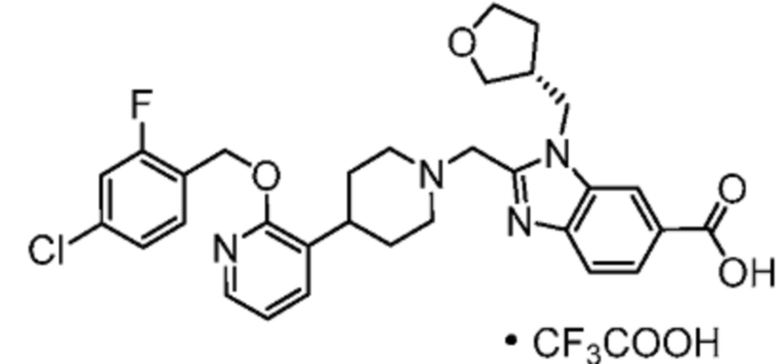
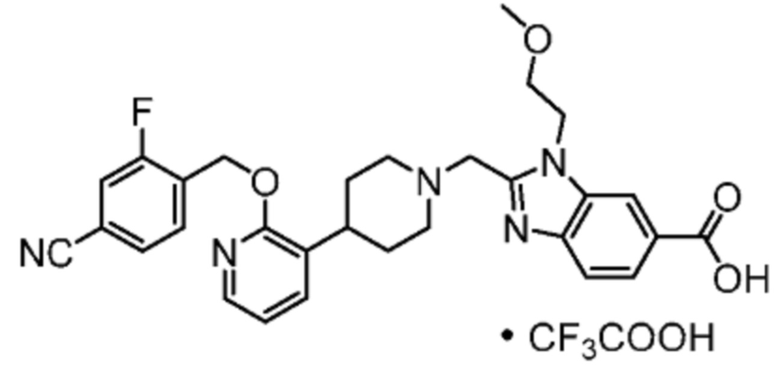
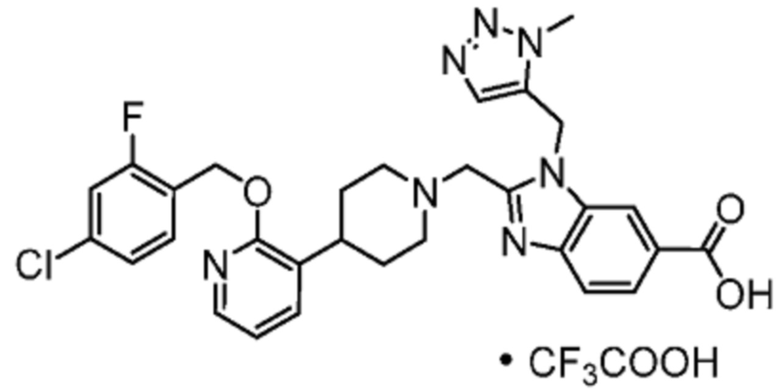
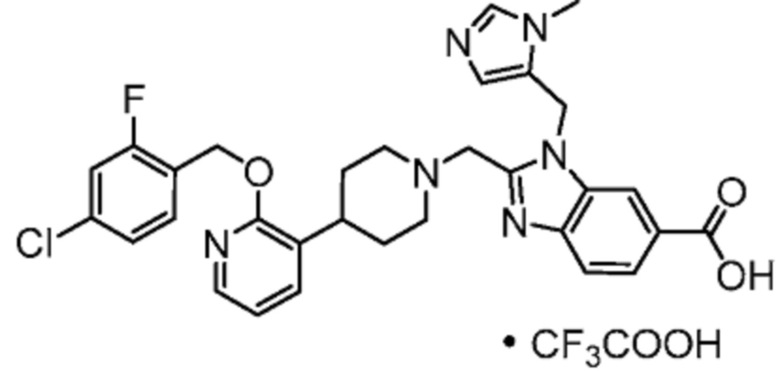
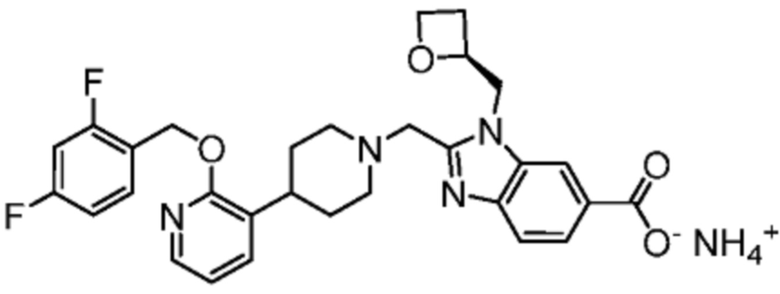
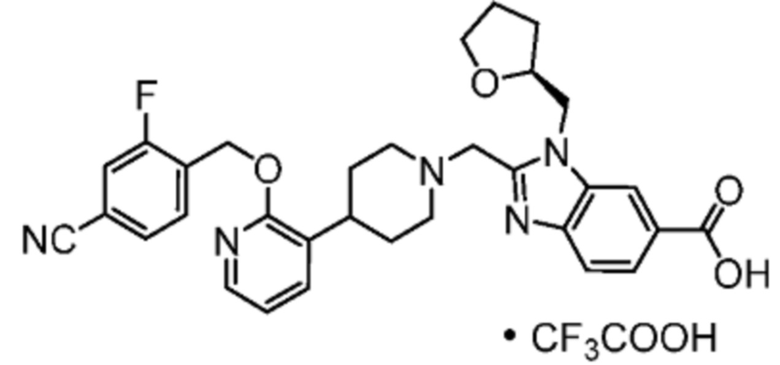
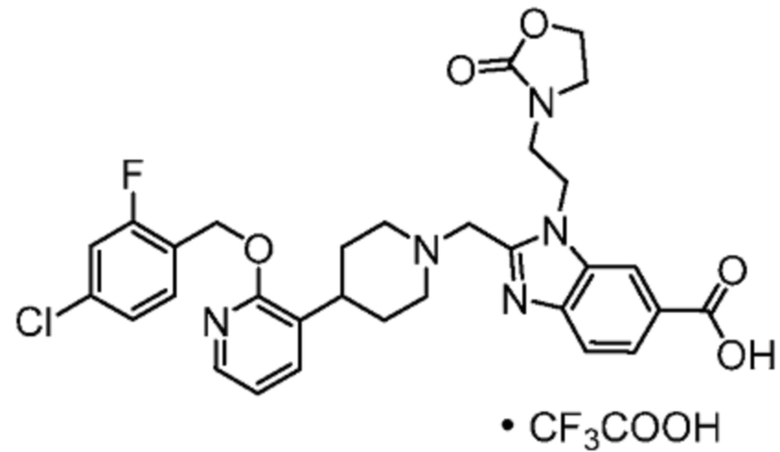
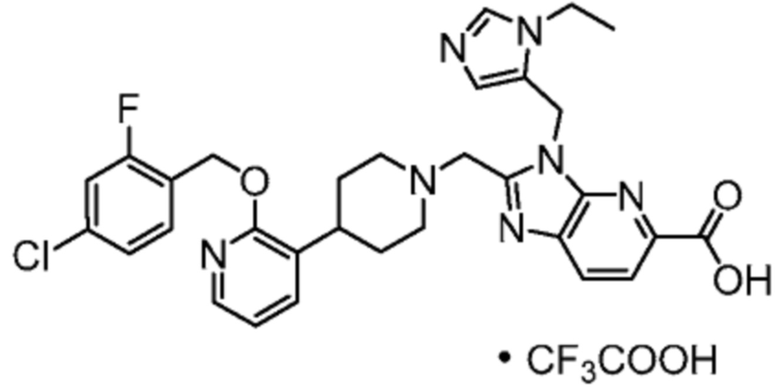
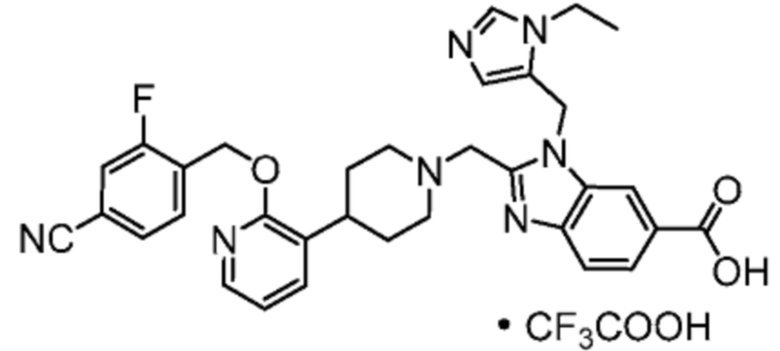
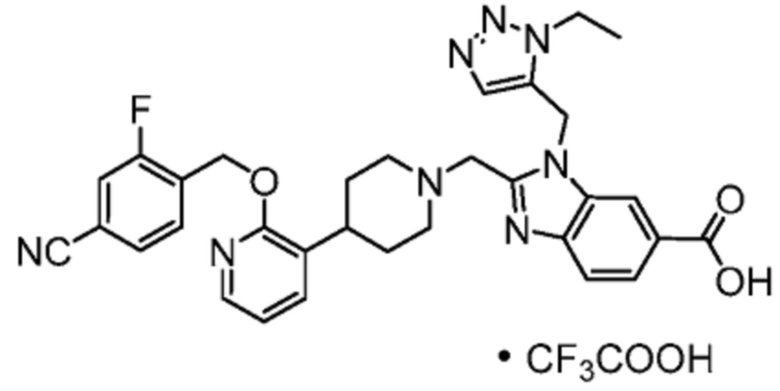
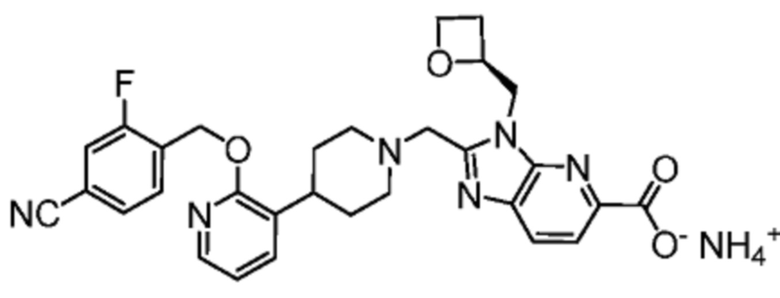
Таблица 1A. Физико-химические характеристики соединений примеров 2-18
Таблица 1/1A: 1. Обработка метил-5-{[(4-{2-[(4-циано-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)ацетил]амино}-6-{[(2S)-оксетан-2-илметил]амино}пиридин-2-карбоксилата водным раствором гидроксида натрия в 1,4-диоксане при повышенной температуре приводит к закрытию цикла и гидролизу сложного эфира с получением соединения примера 18 после очистки.
Пример 19
2-[(4-{3-[(4-Хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (19)
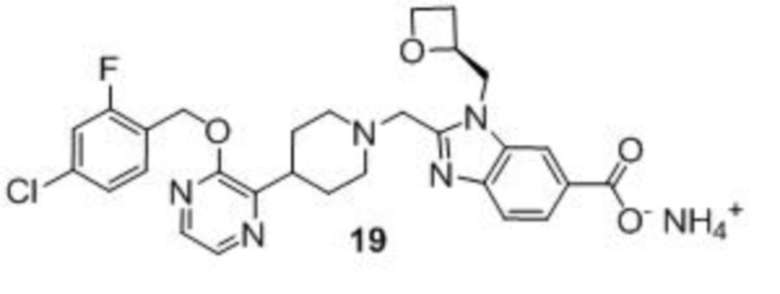
Стадия 1. Синтез метил-4-{[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)ацетил]амино}-3-{[(2S)-оксетан-2-илметил]амино}бензоата (C68)
К раствору P2 (800 мг, 2,02 ммоль) и P16 (476 мг, 2,02 ммоль) в N, N-диметилформамиде (12 мл) добавляют гексафторфосфат O-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (1,15 г, 3,02 ммоль) и триэтиламин (408 мг, 4,03 ммоль). Реакционную смесь перемешивают при 30°C в течение 18 часов, затем разбавляют насыщенным водным раствором хлорида аммония (20 мл) и экстрагируют EtOAc (3×20 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (40 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением C68 в виде смолы желтого цвета. Выход: 1,07 г, 1,79 ммоль, 89%.
Стадия 2. Синтез метил-2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C69)
Раствор C68 (1,08 г, 1,81 ммоль) в уксусной кислоте (8 мл) перемешивают при 50°C в течение 18 часов, затем концентрируют в вакууме, осторожно выливают в насыщенный водный раствор бикарбоната натрия (20 мл) и экстрагируют EtOAc (3 × 20 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (50 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка остатка хроматографией (силикагель, элюент: EtOAc) приводит к получению C69 в виде смолы желтого цвета. Выход: 550 мг, 0,948 ммоль, 52%.
Стадия 3. Синтез 2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (19)
К суспензии C69 (550 мг, 0,948 ммоль) в смеси MeOH (5 мл) и ТГФ (5 мл) добавляют водный раствор гидроксида натрия (3 M; 6,32 мл, 19,0 ммоль) и перемешивают реакционную смесь при 15°С в течение 3 часов. Смесь концентрируют в вакууме, разбавляют водой (8 мл) и промывают EtOAc (8 мл). Значение рН водного слоя доводят до 6-7 добавлением 1 M соляной кислоты и смесь экстрагируют EtOAc (3 × 10 мл). Объединенные органические экстракты промывают насыщенным водным раствором хлорида натрия (30 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Очистка ВЭЖХ с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 26% до 56% B) приводит к получению 19 в виде твердого белого вещества. Выход: 294 мг, 0,504 ммоль, 53%. ЖХМС m/z 566,1♦ [M+H]+. 1H ЯМР (400 MГц, метанол-d4) δ 8,25 (уш. с, 1H), 8,06 (д, 1H), 7,99 (д, 1H), 7,95 (дд, 1H), 7,61 (д, 1H), 7,52 (дд, 1H), 7,28-7,21 (м, 2H), 5,46 (с, 2H), 5,30-5,22 (м, 1H), 4,93-4,83 (м, 1H, предполагаемый; зачительно скрыт пиком воды), 4,70 (дд, 1H), 4,67-4,59 (м, 1H), 4,47 (дт, 1H), 3,96 (AB квартет, 2H), 3,13-3,02 (м, 2H), 2,95 (уш. д, 1H), 2,85-2,75 (м, 1H), 2,57-2,47 (м, 1H), 2,38-2,23 (м, 2H), 2,00-1,78 (м, 4H).
Соединения, представленные в таблице 2 ниже, синтезируют в соответствии с методиками, описанными в настоящем документе для синтеза соединений примеров и примеров получений, с использованием подходящих исходных соединений, которые являются коммерчески доступными либо получены с использованием способов, известных специалистам в данной области техники или аналогичных описанным для получения других промежуточных соединений. Соединения очищают с использованием методов, которые хорошо известны специалистам в данной области техники и могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 2. Структуры и названия в соответствии с номенклатурой ИЮПАК соединений примеров 20-28
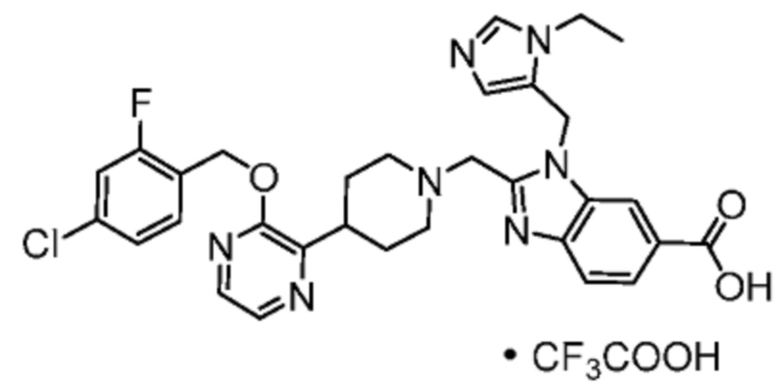
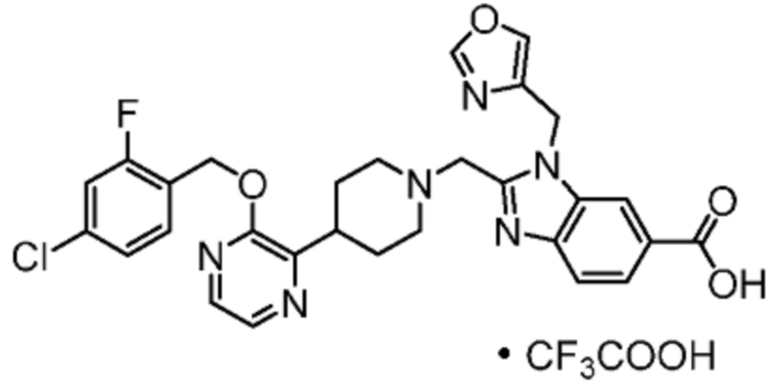
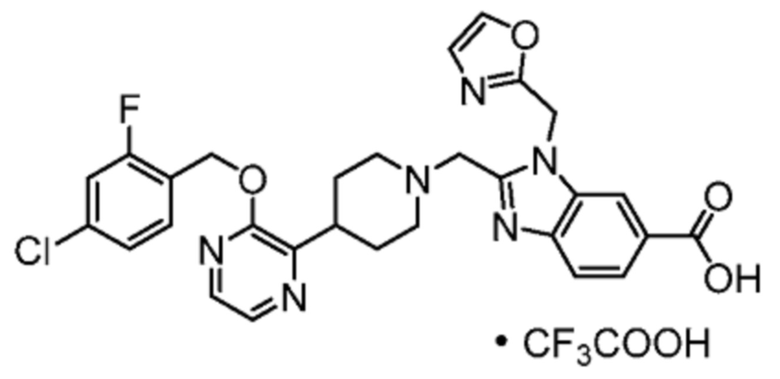
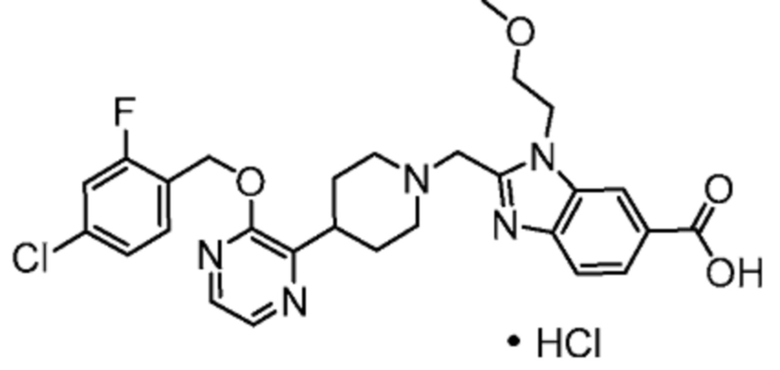
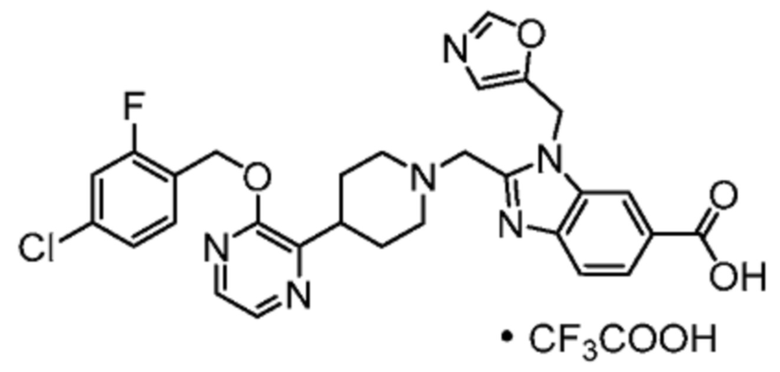
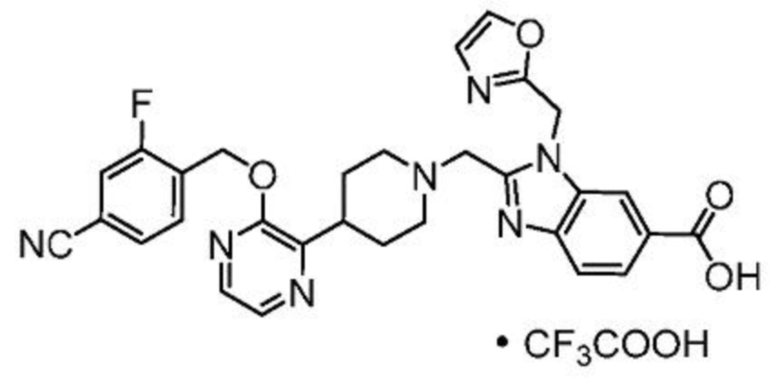
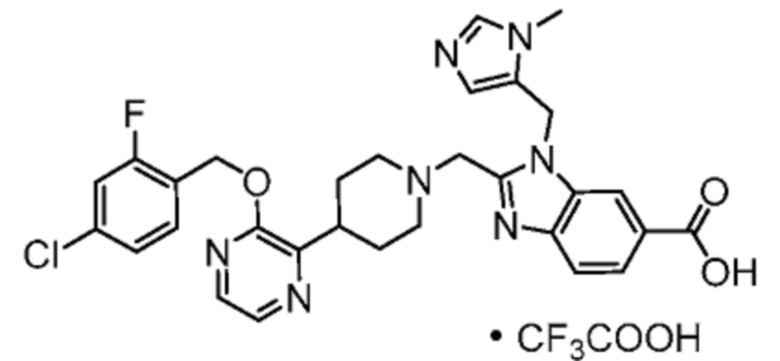
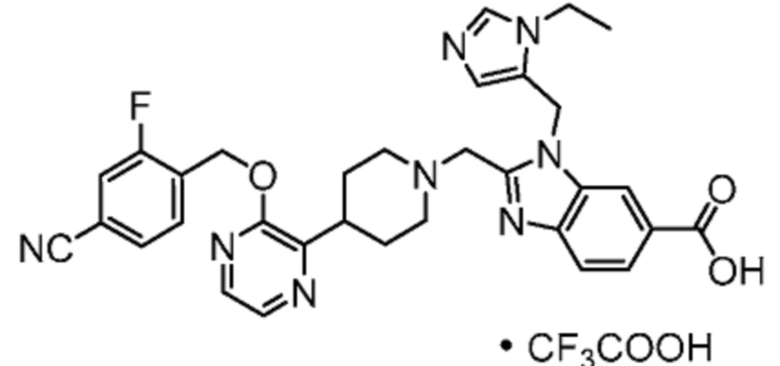
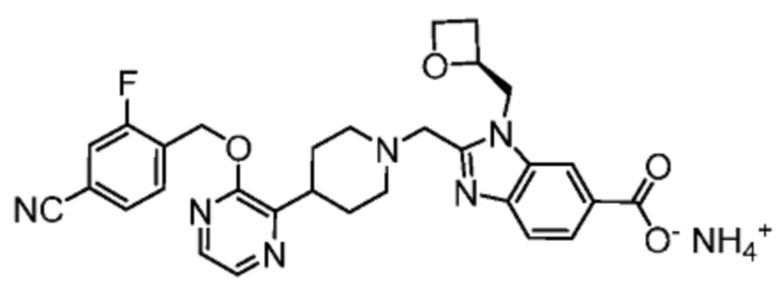
Таблица 2A. Физико-химические характеристики соединений примеров 20-28
Пример 29
Трифторацетатная соль 2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-метил-1H-имидазо[4,5-c]пиридин-6-карбоновой кислоты (29)
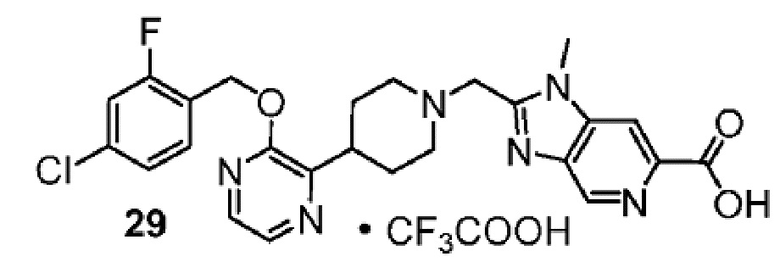
Стадия 1. Синтез N-[6-бром-4-(метиламино)пиридин-3-ил]-2-(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)ацетамида (C70)
К раствору HCl соли P2 (180 мг, 0,47 ммоль) в N, N-диметилформамидe (5 мл) добавляют 6-бром-N4 -метилпиридин-3,4-диамин (96 мг, 0,47 ммоль), гексафторфосфат О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (270 мг, 0,71 ммоль) и N, N-диизопропилэтиламин (0,25 мл, 1,4 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, после чего разбавляют водой (60 мл) и экстрагируют EtOAc (3 × 50 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают препаративной тонкослойной хроматографией (EtOAc) с получением C70 в виде смолы красного цвета. Выход: 200 мг, 0,36 ммоль, 75%.
Стадия 2. Синтез 6-бром-2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-метил-1H-имидазо[4,5-c]пиридина (C71)
К раствору C70 (200 мг, 0,36 ммоль) в 1,4-диоксане (9 мл) добавляют водный раствор гидроксида натрия (2 M; 1,8 мл, 4 ммоль). Раствор перемешивают при 100°C в течение 2 часов. Смесь разбавляют водой (30 мл) и экстрагируют EtOAc (3 × 40 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Полученный продукт объединяют с дополнительным сырым продуктом и очищают и очищают препаративной тонкослойной хроматографией (EtOAc) с получением C71 в виде твердого вещества желтого цвета (140 мг, 0,26 ммоль, 48% из расчета на общую массу исходых веществ).
Стадия 3. Синтез метил-2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-метил-1H-имидазо[4,5-c]пиридин-6-карбоксилата (C72)
1,3-Бис(дифенилфосфино)пропан (25 мг, 62 мкмоль), ацетат палладия (II) (26 мг, 0,12 ммоль) и триэтиламин (0,35 мл, 2,5 ммоль) добавляют к раствору C71 (140 мг, 0,26 ммоль) в смеси MeOH (3 мл) и N, N-диметилформамида (2 мл) и перемешивают полученную реакционную смесь при 80°C в атмосфере моноксида углерода (50 фунтов на кв. дюйм (344,74 кПа)) в течение 16 часов. После этого смесь концентрируют в вакууме и очищают препаративной тонкослойной хроматографией (EtOAc) с получением C72 в виде твердого вещества желтого цвета (110 мг, 0,21 ммоль, 82%).
Стадия 4. Синтез трифторацетатной соли 2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-метил-1H-имидазо[4,5-c]пиридин-6-карбоновой кислоты (29)
Водный раствор гидроксида лития (2 M; 0,4 мл, 0,8 ммоль) добавляют к раствору C72 (80 мг, 0,15 ммоль) в смеси ТГФ (2 мл) и MeOH (2 мл) и перемешивают полученную реакционную смесь при 30°C в течение 2 часов. Смесь концентрируют в вакууме, значение рН остатка доводят до 4 с помощью трифторуксусной кислоты и затем очищают ВЭЖХ с обращенной фазой (колонка: Waters XBridge C18 OBD, 5 мкм; подвижная фаза A: вода, содержащая 0,1% трифторуксусной кислоты; подвижная фаза B: ацетонитрил; градиент: от 5% до 95% B) с получением 29 в виде твердого белого вещества. Выход: 52,4 мг, 83,8 мкмоль, 56%. ЖХМС m/z 511,2♦ [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 9,11 (с, 1H), 8,60 (с, 1H), 8,16 (д, 1H), 8,09 (д, 1H), 7,56 (дд, 1H), 7,30-7,22 (м, 2H), 5,50 (с, 2H), 4,91 (с, 2H), 4,02-3,93 (м, 2H), 4,01 (с, 3H), 3,54-3,41 (м, 3H), 2,38-2,18 (м, 4H).
Соединения, представленные в таблице 3 ниже, синтезируют в соответствии с методиками, описанными в настоящем документе для синтеза соединений примеров и примеров получений, с использованием подходящих исходных соединений, которые являются коммерчески доступными либо получены с использованием способов, известных специалистам в данной области техники или аналогичных описанным для получения других промежуточных соединений. Соединения очищают с использованием методов, которые хорошо известны специалистам в данной области техники и могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 3. Структуры и названия в соответствии с номенклатурой ИЮПАК соединений примеров 30-32
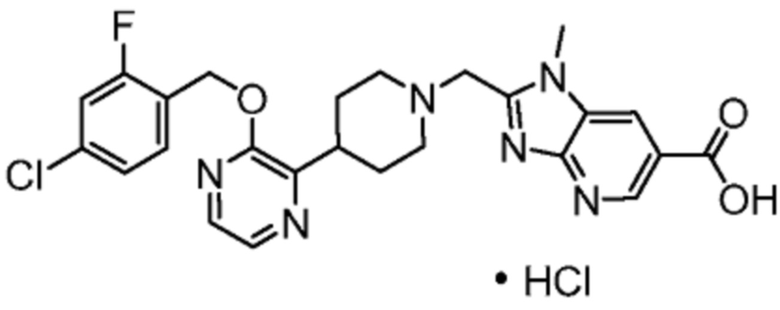
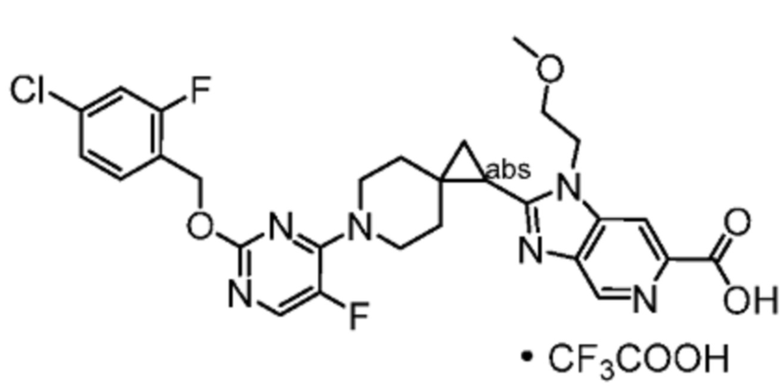
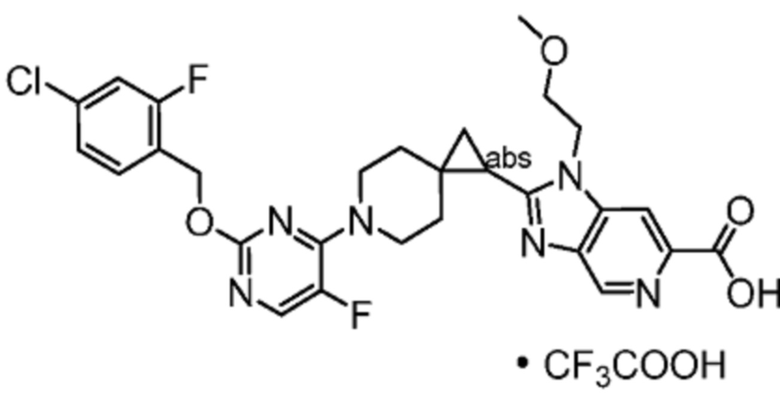
Таблица 3A. Физико-химические характеристики соединений примеров 30-32.
Пример 33
2-[(4-{2-[(4-Хлор-2-фторбензил)окси]пиридин-3-ил}пиперазин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (33)
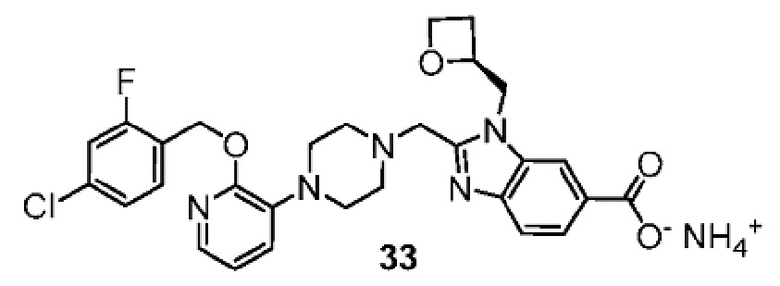
Стадия 1. Синтез метил-4-{[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперазин-1-ил)ацетил]амино}-3-{[(2S)-оксетан-2-илметил]амино}бензоата (C73)
Раствор P3 (95,0 мг, 0,250 ммоль), P16 (70,5 мг, 0,298 ммоль) и гексафторфосфата О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (143 мг, 0,376 ммоль) в N, N-диметилформамидe (1 мл) перемешивают при 20°C в течение 30 минут, после чего добавляют триэтиламин (50,6 мг, 0,500 ммоль). Реакционную смесь перемешивают при 15°C в течение 2 часов, затем разбавляют EtOAc (50 мл) и промывают водой (50 мл). Органический слой промывают насыщенным водным раствором хлорида натрия (50 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме; очистка остатка препаративной тонкослойной хроматографией (элюент: EtOAc) приводит к получению C73 в виде масла желтого цвета (150 мг), которое непосредственно используют в следующей стадии.
Стадия 2. Синтез метил-2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперазин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C74)
Раствор C73 (с предыдущей стадии; 150 мг, ≤0,250 ммоль) в уксусной кислоте (2 мл) перемешивают при 50°C в течение 16 часов, после чего упаривают досуха, остаток смешивают с EtOAc (60 мл) и промывают смесь насыщенным водным раствором карбоната натрия (50 мл). Органический слой сушат над сульфатом магния, фильтруют, концентрируют при пониженном давлении и очищают с использованием препаративной тонкослойной хроматографии (элюент: EtOAc) с получением C74 в виде масла желтого цвета. Выход: 80,5 мг, 0,139 ммоль, 56% для 2 стадий.
Стадия 3. Синтез 2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперазин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (33)
Раствор C74 (80,0 мг, 0,138 ммоль) и водный раствор гидроксида натрия (3 M; 0,3 мл, 0,9 ммоль) в MeOH (1 мл) и ТГФ (1 мл) перемешивают при 50°C в течение 1 часа. После этого значение рН реакционной смеси доводят до 7 добавлением 1 M соляной кислоты и экстрагируют полученную смесь смесью дихлорметана и MeOH (10:1, 3 × 30 мл). Объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют в вакууме; очистка с использованием ВЭЖХ с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 20% до 50% B) приводит к получению 33 в виде твердого белого вещества. Выход: 39,7 мг, 68,0 мкмоль, 49%. ЖХМС m/z 566,0♦ [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,32 (д, 1H), 7,97 (дд, 1H), 7,75 (дд, 1H), 7,67 (д, 1H), 7,55 (дд, 1H), 7,28-7,20 (м, 3H), 6,92 (дд, 1H), 5,42 (с, 2H), 5,29-5,21 (м, 1H), 4,9-4,81 (м, 1H, предполагаемый; частично скрыт пиком воды), 4,70 (дд, 1H), 4,66-4,59 (м, 1H), 4,46 (дт, 1H), 3,98 (AB квартет, 2H), 3,16-3,04 (уш. м, 4H), 2,84-2,74 (м, 1H), 2,74-2,61 (м, 4H), 2,56-2,45 (м, 1H).
Пример 34
Трифторацетатная соль 2-{[(2S)-4-{3-[(4-Хлор-2-фторбензил)окси]пиразин-2-ил}-2-метилпиперазин-1-ил]метил}-3-метил-3H-имидазо[4,5-b]пиридин-5-карбоновой кислоты (34)
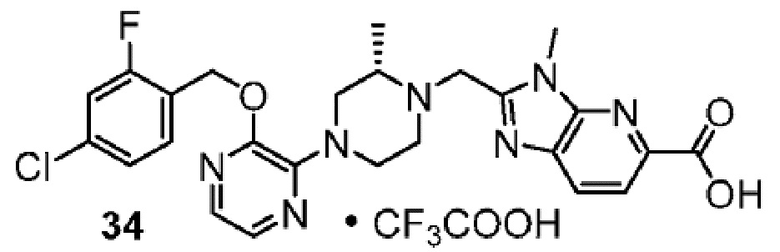
Стадия 1. Синтез метил-2-{[(2S)-4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}-2-метилпиперазин-1-ил]метил}-3-метил-3H-имидазо[4,5-b]пиридин-5-карбоксилата (C75)
К суспензии P18 (60,0 мг, 0,198 ммоль), P4 (64,8 мг, 0,237 ммоль), 1,1'-бинафталин-2,2'-диилбис(дифенилфосфан) (24,6 мг, 39,6 мкмоль) и трис(дибензилиденацетон)дипалладия (0) (18,1 мг, 19,8 мкмоль) в толуоле (2 мл) добавляют карбонат цезия (129 мг, 0,396 ммоль). Реакционную смесь продувают азотом в течение 30 секунд и перемешивают при 100°C в течение 16 часов, после чего разбавляют водой (10 мл) и экстрагируют дихлорметан (3 × 10 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка остатка препаративной тонкослойной хроматографией (элюент: 20:1 дихлорметан/MeOH) приводит к получению С75 в виде твердого вещества желтого цвета. Выход: 60 мг, 0,11 мкмоль, 56%.
Стадия 2. Синтез трифторацетатной соли 2-{[(2S)-4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}-2-метилпиперазин-1-ил]метил}-3-метил-3H-имидазо[4,5-b]пиридин-5-карбоновой кислоты (34)
К раствору C75 (60 мг, 0,11 ммоль) в MeOH (5 мл) добавляют ТГФ (1 мл) и раствор гидроксида натрия (22,2 мг, 0,556 ммоль) в воде (1 мл). Реакционную смесь перемешиваютпри 40°C в течение 2 часов, затем концентрируют в вакууме и подкисляют до pH 7 добавлением 1 M соляной кислоты. Полученную смесь экстрагируют дихлорметаном (3 × 20 мл), объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Очистка с использованием ВЭЖХ с обращенной фазой (колонка: Waters XBridge C18 OBD, 5 мкм; подвижная фаза A: вода, содержащая 0,1% трифторуксусной кислоты; подвижная фаза B: ацетонитрил; градиент: от 5% до 95% B) приводит к получению 34 в виде твердого вещества. Выход: 43,4 мг, 67, мкмоль, 62%. ЖХМС m/z 526,0♦ [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,20 (AB квартет, 2H), 7,80 (д, 1H), 7,70 (д, 1H), 7,54 (дд, 1H), 7,27 (дд, 1H), 7,24 (уш. дд, 1H), 5,49 (AB квартет, 2H), 5,01 (д, 1H), 4,68 (д, 1H), 4,14-4,01 (м, 2H), 3,99 (с, 3H), 3,87-3,69 (м, 2H), 3,67-3,40 (м, 3H), 1,49 (д, 3H).
Примеры 35 и 36
2-(6-{6-[(4-Циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (35) из C78 и
2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (36) из C79
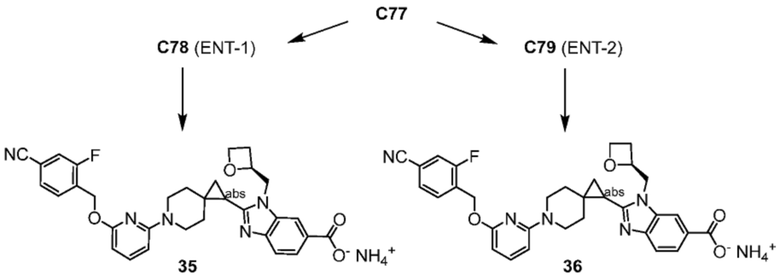
Стадия 1. Синтез метил-4-{[(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)карбонил]амино}-3-{[(2S)-оксетан-2-илметил]амино}бензоата (C76)
К раствору P6 (530 мг, 1,39 ммоль), гексафторфосфата О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (794 мг, 2,09 ммоль) и N, N-диизопропилэтиламина (0,70 мл, 4,1 ммоль) в N, N-диметилформамидe (4,0 мл) добавляют раствор P16 (324 мг, 1,37 ммоль) в N, N-диметилформамидe (4,0 мл). Реакционную смесь перемешивают при 25°C в течение 15 часов, затем концентрируют в вакууме для удаления N, N-диметилформамида. Полученную смолу промывают насыщенным водным раствором хлорида натрия (20 мл) и экстрагируют EtOAc (3 × 20 мл). Объединенные органические слои сушат над сульфатом магния, фильтруют, концентрируют при пониженном давлении и очищают хроматографией (силикагель, градиент: от 0% до 1,3% MeOH в дихлорметане) и затем препаративной тонкослойной хроматографией (элюент: 4:1 EtOAc/петролейный эфир). Соединение C76 выделяют в виде смолы красного цвета. Выход: 530 мг, 0,884 ммоль, 64%.
Стадия 2. Синтез метил-2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C77)
Раствор C76 (530 мг, 0,884 ммоль) в уксусной кислоте (5,0 мл) перемешивают при 60°C в течение 16 часов, после чего концентрируют в вакууме, промывают насыщенным водным раствором бикарбоната натрия (10 мл) и экстрагируют EtOAc (3 × 20 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Очистку проводят дважды с использованием препаративной тонкослойной хроматографии (элюент для #1: 1:1 петролейный эфир/EtOAc; элюент для #2: 10:1 дихлорметан/MeOH) и затем ВЭЖХ с обращенной фазой (колонка: Phenomenex Gemini C18, 10 мкм; подвижная фаза A: 0,05% гидроксида аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 60% до 90% B) с получением C77 в виде твердого вещества, который представляет собой смесь двух стереоизомеров относительно циклопропана. Выход: 140 мг, 0,241 ммоль, 27%.
Стадия 3. Выделение метил-2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата ENT-1 (C78) и метил-2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата ENT-2 (C79)
Разделение C77 (185 мг, 0,318 ммоль) на стереоизомеры по циклопропану проводят с использованием сверхкритической жидкостной хроматографии [колонка: Chiral Technologies ChiralcelOJ-H, 5 мкм; подвижная фаза: 3:2 диоксид углерода/(MeOH, содержащий 0,1% гидроксида аммония)]. Стереоизомер первого элюирования, полученный в виде смолы желтого цвета, обозначают как ENT-1 (C78). Выход: 80 мг, 0,14 ммоль, 44%. Стереоизомер второго элюирования, выделенный в виде смолы зеленого цвета, обозначают как ENT-2 (C79). Выход: 100 мг, 0,17 ммоль, предположительно 50%.
Стадия 4. Синтез 2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (35) из C78
Раствор C78 (75 мг, 0,13 ммоль) в смеси ТГФ (5,0 мл) и MeOH (2,0 мл) обрабатывают водным раствором гидроксида лития (2 M; 0,32 мл, 0,64 ммоль) и перемешивают полученную реакционную смесь при 30°C в течение 60 часов. Значение рН смеси доводят до 6-7 добавлением 1 M соляной кислоты и полученную смесь экстрагируют дихлорметаном (4 × 15 мл); объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка с использованием ВЭЖХ с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 20% до 40% B) приводит к получению 35 в виде твердого белого вещества. Выход: 11,3 мг, 18,8 мкмоль, 14%. ЖХМС m/z 568,1 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,22 (д, 1H), 7,94 (дд, 1H), 7,64-7,57 (м, 2H), 7,53-7,47 (м, 2H), 7,41 (дд, 1H), 6,27 (д, 1H), 6,09 (д, 1H), 5,40 (AB квартет, 2H), 5,23-5,15 (м, 1H), 4,69 (дд, 1H), 4,63-4,53 (м, 2H), 4,41 (дт, 1H), 3,98-3,90 (м, 1H), 3,61-3,52 (м, 1H), 3,42 (ддд, 1H), 3,23 (ддд, 1H), 2,84-2,73 (м, 1H), 2,58-2,48 (м, 1H), 2,38 (дд, 1H), 1,86 (ддд, 1H), 1,65 (дд, 1H), 1,54-1,44 (м, 2H), 1,36-1,27 (м, 1H), 1,24 (дд, 1H),
Стадия 5. Синтез 2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (36) из C79
Раствор C79 (95 мг, 0,16 ммоль) в смеси ТГФ (5,0 мл) и MeOH (2,0 мл) обрабатывают водным раствором гидроксида лития (2 M; 0,40 мл, 0,80 ммоль) и перемешивают полученную реакционную смесь при 30°C в течение 50 часов. Затем значение рН смеси доводят до 5-6 добавлением 1 M соляной кислоты и экстрагируют полученную смесь дихлорметаном (4 × 20 мл); объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка с использованием ВЭЖХ с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксида аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 21% до 41% B) приводит к получению 36 в виде твердого белого вещества. Выход: 21,9 мг, 38,5 мкмоль, 24%. ЖХМС m/z 568,1 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,21 (д, 1H), 7,95 (дд, 1H), 7,62 (д, 1H), 7,60 (дд, 1H), 7,53-7,47 (м, 2H), 7,40 (дд, 1H), 6,25 (д, 1H), 6,08 (д, 1H), 5,40 (AB квартет, 2H), 5,28-5,20 (м, 1H), 4,69 (дд, 1H), 4,58-4,48 (м, 2H), 4,31 (дт, 1H), 3,95-3,86 (м, 1H), 3,59-3,50 (м, 1H), 3,41 (ддд, 1H), 3,22 (ддд, 1H), 2,81-2,71 (м, 1H), 2,52-2,41 (м, 2H), 1,86-1,77 (м, 1H), 1,66 (дд, 1H), 1,56-1,42 (м, 2H), 1,36-1,27 (м, 1H), 1,23 (дд, 1H).
Соединение примера 37, представленное в таблице 4 ниже, синтезируют в соответствии в методиками, описанным в настоящем документе для синтеза соединений примеров и примеров получения, с использованием подходящих исходных веществ, которые являются коммерчески доступными либо могут быть получены с использованием способов получения, хорошо известных специалистам в данной области техники или аналогичных способам получения других промежуточных соединений. Соединение примера 37 очищают методами, которые хорошо известны специалистам в данной области техники и могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 4. Структура и название в соответствии с ИЮПАК соединения примера 37
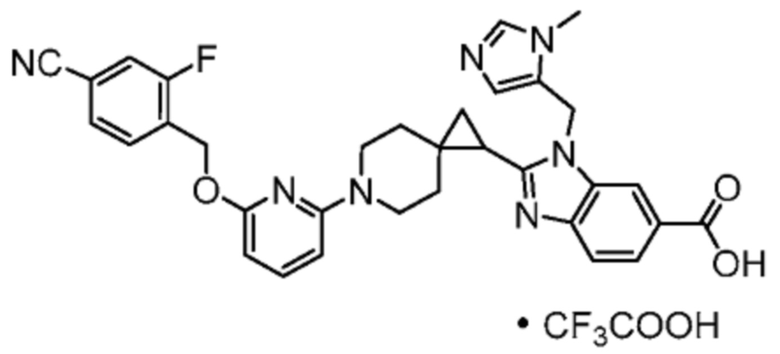
пиридин-2-ил}-6-азаспиро
[2.5]окт-1-ил)-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновой кислоты
Таблица 4A. Физико-химические характеристики соединения примера 37
Пример 38
2-(6-{2-[(4-Хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония из P7(38)
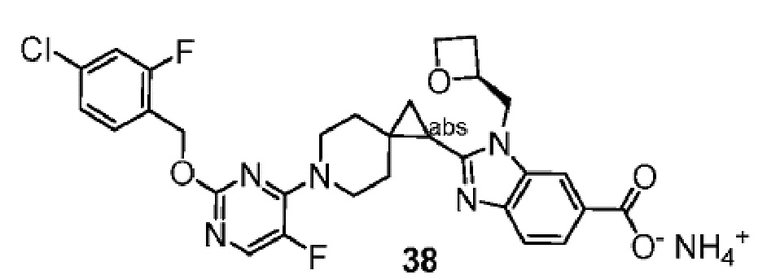
Стадия 1. Синтез метил-4-{[(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)карбонил]амино}-3-{[(2S)-оксетан-2-илметил]амино}бензоата (С80) из P7
К раствору P16 (540 мг, 2,29 ммоль) и P7 (1,03 г, 2,51 ммоль) в пиридине (10 мл) добавляют гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,31 г, 6,86 ммоль). Реакционную смесь перемешивают при 30°C в течение 16 часов, после чего выливают в воду (20 мл) и экстрагируют EtOAc (3 × 20 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме; очистка хроматографией (силикагель, элюент: 85% EtOAc в петролейном эфире) приводит к получению С80 в виде белой пены. Выход: 1,4 г, 2,23 ммоль, 97%.
Стадия 2. Синтез метил-2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата из P7(C81)
Раствор C80 (1,4 г, 2,2 ммоль) в уксусной кислоте (20 мл) перемешивают при 60°C в течение 21 часов, после чего реакционную смесь упаривают досуха в вакууме и нейтрализуют остаток добавлением водного раствора бикарбоната натрия (20 мл). Полученную смесь экстрагируют EtOAc (3 × 20 мл), объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, элюент: EtOAc) приводит к получению C81 в виде вспененного твердого белого вещества. Выход: 1,1 г, 1,8 ммоль, 82%.
Стадия 3. Синтез 2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (38) из P7
Водный раствор гидроксида натрия (2 M; 4,5 мл, 9,0 ммоль) добавляют к раствору C81 (1,1 г, 1,8 ммоль) в смеси ТГФ (11 мл) и MeOH (4,5 мл) и перемешивают полученную реакционную смесь при 25°C в течение 16 часов. Затем смесь объединяют с продуктом аналогичной реакции с использованием C81 (366 мг, 0,600 ммоль), упаривают досуха в вакууме, переносят в воду (10 мл) и доводят значение pH до 5 добавлением 1 M соляной кислоты. Полученную смесь экстрагируют дихлорметан (4 × 50 мл); объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Очистка хроматографией (силикагель, элюент: 20% MeOH в дихлорметане) приводит к получению твердого белого вещества, которое растворяют в ацетонитриле (5 мл), затем обрабатывают водой (20 мл) и концентрирированным гидроксидом аммония (1 мл). Смесь лиофилизуют с получением 38 в виде твердого белого вещества. Выход: 1,09 г, 1,78 ммоль, 74%. ЖХМС m/z 596,0♦ [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,20 (д, 1H), 7,95 (дд, 1H), 7,86 (д, 1H), 7,62 (д, 1H), 7,44 (дд, 1H), 7,18-7,11 (м, 2H), 5,30 (с, 2H), 5,28-5,20 (м, 1H), 4,73 (дд,1H), 4,59-4,49 (м, 2H), 4,34 (дт, 1H), 4,28-4,20 (м, 1H), 3,99-3,90 (м, 1H), 3,70-3,60 (м, 1H), 3,53-3,44 (м, 1H), 2,84-2,73 (м, 1H), 2,56-2,44 (м, 2H), 2,00-1,89 (м, 1H), 1,69 (дд, 1H), 1,65-1,50 (м, 2H), 1,43-1,34 (м, 1H), 1,28 (дд, 1H).
Соединения, представленные в таблице 5 ниже, синтезируют в соответствии с методиками, описанными в настоящем документе для синтеза соединений примеров и примеров получений, с использованием подходящих исходных соединений, которые являются коммерчески доступными либо получены с использованием способов, известных специалистам в данной области техники или аналогичных описанным для получения других промежуточных соединений. Соединения очищают методами, которые хорошо известны специалистам в данной области техники и могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 5. Структуры и названия в соответствии с номенклатурой ИЮПАК соединений примеров 39-45
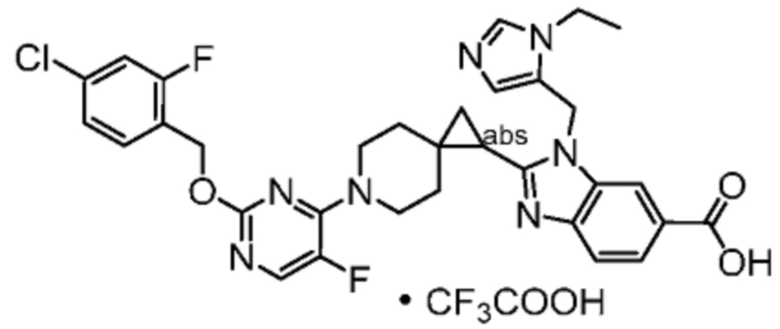
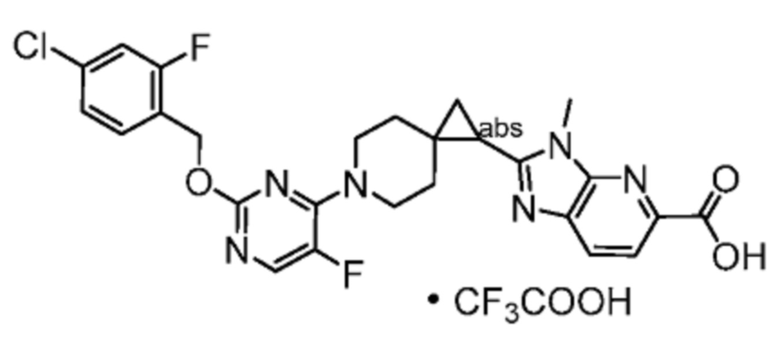
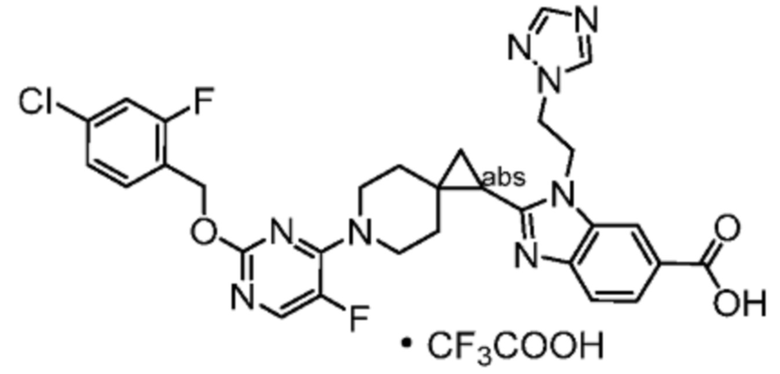
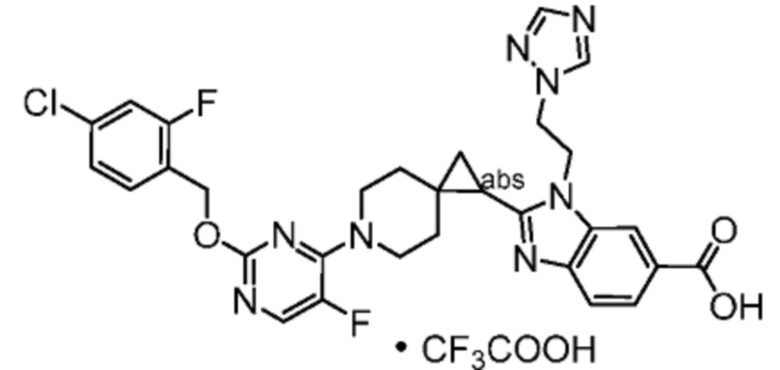
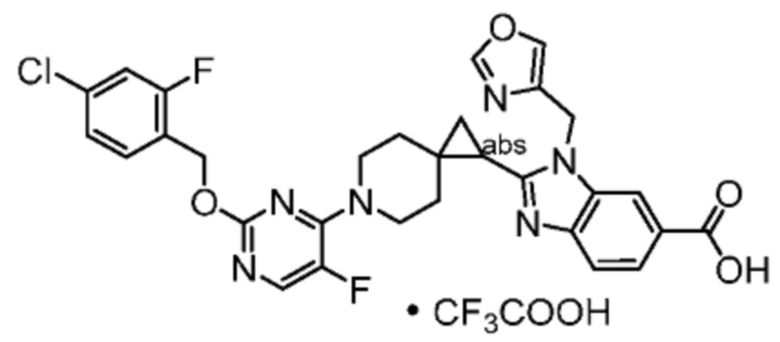
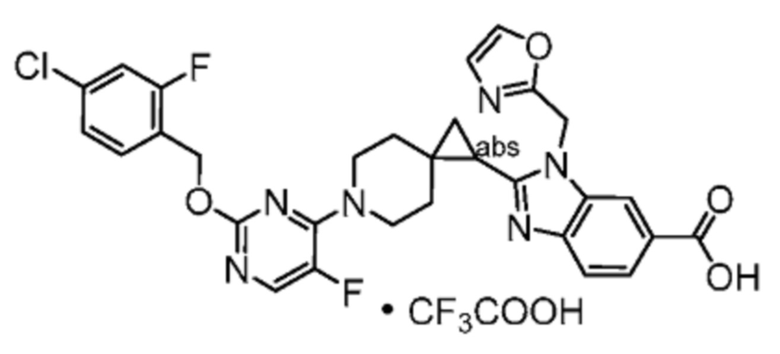
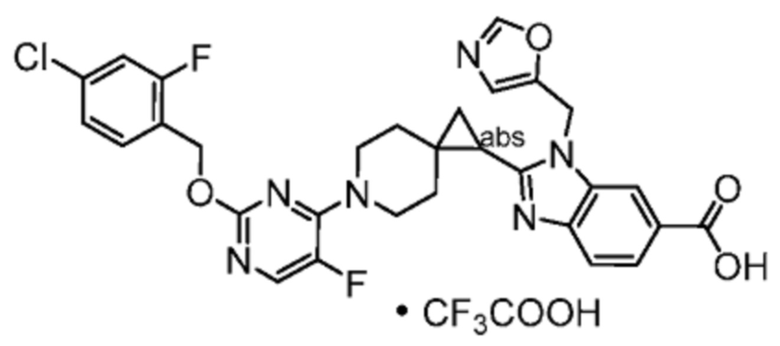
Таблица 5A. Физико-химические характеристики соединений примеров 39-45
Таблица 5/5A:
1. Амидный продукт реакции сочетания P7 с C60 подвергают циклизации нагреванием в триметилсилилполифосфoсфате при 120°C в течение 30 минут с получением единственного энантиомера 5-хлор-2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-3-метил-3H-имидазо[4,5-b]пиридина.
2. Амидный продукт реакции сочетания P7 с метил-4-амино-3-{[2-(1H-1,2,4-триазол-1-ил)этил]амино}бензоата циклизуют нагреванием при повышенной температуре с водным раствором гидроксида натрия в 1,4-диоксане; гидролиз сложного эфира с последующей очисткой приводит к получению соединения примера 41.
3. Амидный продукт реакции сочетания C23 с метил-4-амино-3-{[2-(1H-1,2,4-триазол-1-ил)этил]амино}бензоатом циклизуют нагреванием до повышенной температуры с водным раствором гидроксида натрия в 1,4-диоксане; гидролиз сложного эфира с последующей очисткой приводит к получению соединения примера 42.
Примеры 46 и 47
2-(6-{2-[(4-Хлор-2-фторбензил)окси]пиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат аммония, ENT-1 (46), и
2-(6-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилат аммония, ENT-2 (47)
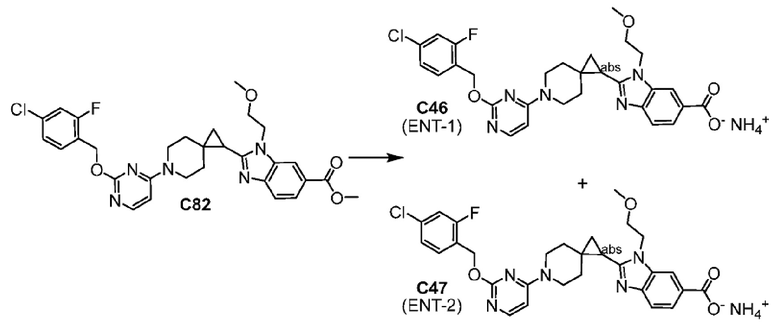
Стадия 1. Синтез метил-2-(6-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата (C82)
Из емкости, содержащей смесь P8 (61 мг, 0,22 ммоль), P19 (55 мг, 0,16 ммоль), трис(дибензилиденацетон)дипалладия (0) (14,7 мг, 16,0 мкмоль), 1,1'-бинафталин-2,2'-диилбис(дифенилфосфана) (20 мг, 32 мкмоль) и карбоната цезия (104 мг, 0,319 ммоль), откачивают воздух и затем заполняют емкость азотом в течение 10 минут. После добавления 1,4-диоксана (0,8 мл) реакционную смесь нагревают до 100°C и выдерживают при указанной температуре в течение 23 часов, после чего разбавляют дихлорметаном (0,5 мл) и очищают хроматографией (силикагель, градиент: от 15% до 100% EtOAc в гептане). Соединение C82 выделяют в виде твердого вещества желтого цвета (37,5 мг), которое непосредственно используют в следующей стадии.
Стадия 2. Синтез 2-(6-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата аммония, ENT-1 (46), и 2-(6-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоксилата аммония, ENT-2 (47)
Водный раствор гидроксида натрия (2 M; 150 мкл, 0,3 ммоль) добавляют к смеси C82 (с предыдущей стадии; 37,5 мг, ≤64,6 мкмоль) в MeOH (0,4 мл) и ТГФ (0,4 мл). Реакционную смесь перемешивают при 35°C в течение 2,25 часов, после чего добавляют соляную кислоту (1 M; 0,35 мл, 0,35 ммоль) и дают возможность реакционной смеси охладиться до комнатной температуры. Растворители удаляют в вакууме, остаток разделяют на энантиомеры с использованием сверхкритической жидкостной хроматографии: Chiral Technologies Chiralpak AD-H, 5 мкм; подвижная фаза: 65:35 диоксид углерода/(MeOH, содержащий 0,2% гидроксида аммония)]. Энантиомер первого элюирования обозначают как ENT-1 (46). Выход: 4,3 мг, 7,3 мкмоль, 5% для 2 стадий. ЖХМС m/z 566,4♦ [M+H]+. Время удерживания: 2,12 мин. [условия анализа: колонка Chiral Technologies Chiralpak AD-H, 4,6 × 100 мм, 5 мкм; подвижная фаза: 1:1 диоксид углерода/(MeOH, содержащий 0,2% гидроксида аммония); объемная скорость потока: 1,5 мл/мин.; обратное давление: 120 бар].
Энантиомер второго элюирования обозначают как ENT-2 (47). Выход: 4,1 мг, 7,0 мкмоль, 4% для 2 стадий. ЖХМС m/z 566,4♦ [M+H]+. Время удерживания: 2,74 мин. (условия анализа идентичны условиям, которые используют для анализа соединения 46).
Соединения, представленные в таблице 6 ниже, синтезируют в соответствии с методиками, описанными в настоящем документе для синтеза соединений примеров и примеров получений, с использованием подходящих исходных соединений, которые являются коммерчески доступными либо получены с использованием способов, известных специалистам в данной области техники или аналогичных описанным для получения других промежуточных соединений. Соединения очищают методами, которые хорошо известны специалистам в данной области техники и могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 6. Структуры и названия в соответствии с номенклатурой ИЮПАК соединений примеров 48 и 49
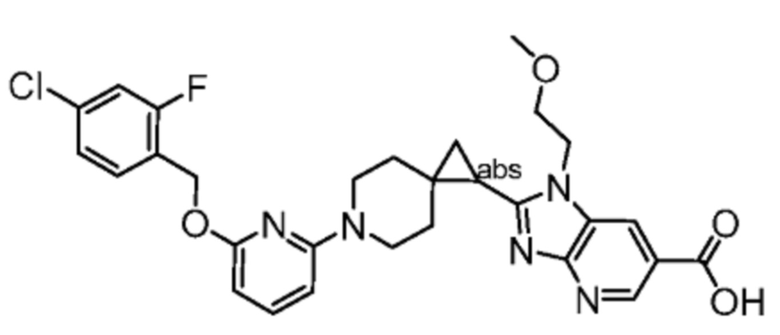
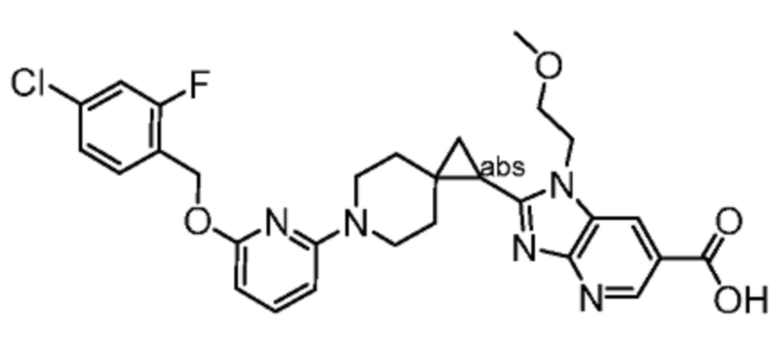
1. В этом случае реакцию сочетания P30 и 2-хлор-6-[(4-хлор-2-фторбензил)окси]пиридина катализируют хлор(2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладием (II) (RuPhos Pd G2) и 2-дициклогексилфосфино-2′,6′-диизопропоксибифенилом.
2. Рацемическую смесь соединений примеров 48 и 49 разделяют на энантиомеры с использованием сверхкритической жидкостной хроматографии [колонка: Phenomenex Lux Cellulose-2, 5 мкм; подвижная фаза: 7:3 диоксид углерода/(МеОН, содержащий 0,2% гидроксида аммония и 5% воды)]. Энантиомер первого элюирования обозначают как ENT-1(48), энантиомер второго элюирования обозначают как ENT-2(49).
Таблица 6A. Физико-химические характеристики соединений примеров 48 и 49
Примеры 50 и 51
2-(6-{4-[(4-Хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (50) из C85 и
2-(6-{4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (51) из C86
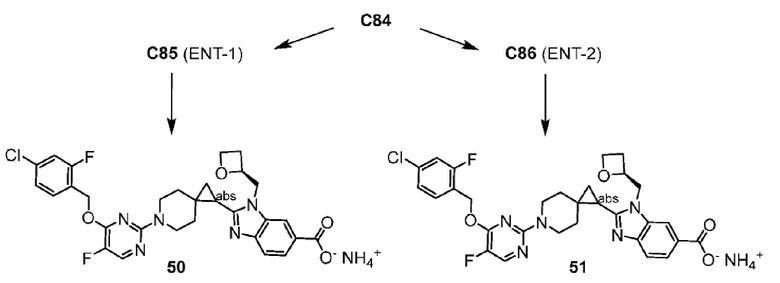
Стадия 1. Синтез метил-4-{[(6-{4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]окт-1-ил)карбонил]амино}-3-{[(2S)-оксетан-2-илметил]амино}бензоата (C83)
Взаимодействие P16 с P9 проводят с использованием способа, описанного для синтеза C80 из P16 и P7 в примере 38. В этом случае очистку остатка проводят хроматографией (силикагель, градиент: от 0% до 2,1% MeOH в дихлорметане) с получением C83, смеси стереоизомеров при циклопропане, в виде вспененного твердого вещества желтого цвета. Выход: 504 мг, 0,802 ммоль, 94%.
Стадия 2. Синтез метил-2-(6-{4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C84)
Раствор C83 (270 мг, 0,430 ммоль) в уксусной кислоте (4,5 мл) перемешивают при 60°C в течение 7 часов, после чего выливают в воду, подщелачивают до pH 8 добавлением карбоната натрия и экстрагируют EtOAc (3 × 30 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 0% до 80% EtOAc в петролейном эфире) с получением C84, которое представляет собой смесь стереоизомеров при циклопропане, в виде масла желтого цвета. Выход: 250 мг, 0,410 ммоль, 95%.
Стадия 3. Выделение метил-2-(6-{4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата, ENT-1 (C85), и метил-2-(6-{4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата, ENT-2 (C86)
Разделение C84 (250 мг, 0,410 ммоль) на стереоизомеры при стереоизомерном центре пропана проводят с помощью сверхкритической жидкостной хроматографии [колонка: Chiral Technologies ChiralcelOJ-H, 5 мкм; подвижная фаза: 3:2 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Стереоизомер первого элюирования, бесцветное масло, обозначают как ENT-1 (C85). Выход: 104 мг, 0,170 ммоль, 41%.
Стереоизомер второго элюирования, твердое белое вещество, обозначают как ENT-2 (C86). Выход: 109 мг, 0,179 ммоль, 44%.
Стадия 4. Синтез 2-(6-{4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (50) из C85
Водный раствор гидроксида натрия (0,767 мл, 2 M, 1,53 ммоль) добавляют к раствору C85 (104 мг, 0,170 ммоль) в MeOH (3 мл). Реакционную смесь перемешивают при 30°C в течение 6 часов, подкисляют осторожным добавлением 12 M соляной кислоты, разбавляют водой (30 мл) и экстрагируют дихлорметаном (3 × 30 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и очищают ВЭЖХ с обращенной фазой (колонка: Phenomenex Gemini C18, 10 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 25% до 55% B) с получением 50 в виде твердого белого вещества. Выход: 47,1 мг, 76,8 мкмоль, 45%. ЖХМС m/z 596,0♦ [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,20 (д, 1H), 7,97-7,92 (м, 2H), 7,61 (д, 1H), 7,45 (дд, 1H), 7,20-7,13 (м, 2H), 5,42 (с, 2H), 5,28-5,20 (м, 1H), 4,70 (дд, 1H), 4,58-4,49 (м, 2H), 4,33 (дт, 1H), 4,24-4,16 (м, 1H), 3,92-3,83 (м, 1H), 3,56 (ддд, 1H), 3,39 (ддд, 1H), 2,82-2,71 (м, 1H), 2,54-2,42 (м, 2H), 1,90-1,80 (м, 1H), 1,68 (дд, 1H), 1,54-1,43 (м, 2H), 1,35-1,28 (м, 1H), 1,25 (дд, 1H).
Стадия 5. Синтез 2-(6-{4-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (51) из C86
Синтез 51 из соединения C86 проводят с использованием способа, описанного для синтеза 50 из C85 в стадии 4 выше. Соединение 51 выделяют в виде твердого белого вещества. Выход: 46,0 мг, 75,0 мкмоль, 42%. ЖХМС m/z 596,0♦ [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,23 (д, 1H), 7,97 (д, 1H), 7,95 (дд, 1H), 7,62 (д, 1H), 7,45 (дд, 1H), 7,20-7,13 (м, 2H), 5,43 (с, 2H), 5,23-5,15 (м, 1H), 4,70 (дд,1H), 4,63-4,54 (м, 2H), 4,40 (дт, 1H), 4,29-4,21 (м, 1H), 3,96-3,87 (м, 1H), 3,57 (ддд, 1H), 3,41 (ддд, 1H), 2,85-2,74 (м, 1H), 2,59-2,48 (м, 1H), 2,42 (дд, 1H), 1,95-1,86 (м, 1H), 1,67 (дд, 1H), 1,54-1,42 (м, 2H), 1,36-1,24 (м, 2H).
Примеры 52 и 53
2-(6-{6-[(4-Циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония, ENT-1(52), и
2-(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония, ENT-2 (53)
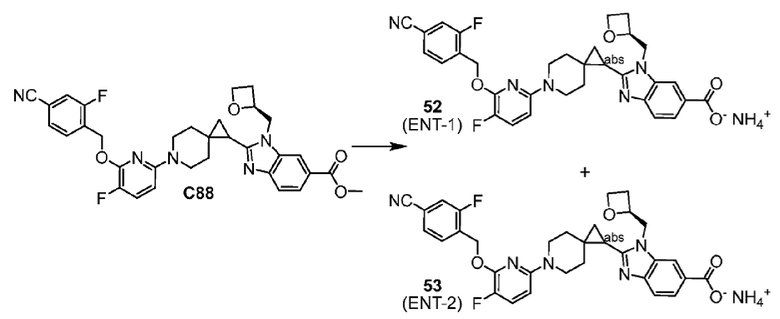
Стадия 1. Синтез метил-4-{[(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)карбонил]амино}-3-{[(2S)-оксетан-2-илметил]амино}бензоата (C87)
К раствору P10 (695 мг, 1,74 ммоль), P16 (411 мг, 1,74 ммоль) и гексафторфосфата О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (860 мг, 2,26 ммоль) в N, N-диметилформамидe (7 мл) добавляют триэтиламин (880 мг, 8,70 ммоль). Реакционную смесь перемешивают при 35°C в течение 16 часов, после чего объединяют с продуктом аналогичной реакции с использованием P10 (50 мг, 0,12 ммоль), выливают смесь в воду (20 мл) и экстрагируют EtOAc (3 × 20 мл). Объединенные органические слои промывают водным раствором хлоридом аммония (2 × 30 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, элюент: 80% EtOAc в петролейном эфире) и затем препаративной тонкослойной хроматографией (элюент: EtOAc) приводит к получению C87, смеси стереоизомеров при циклопропане, в виде пены желтого цвета. Общий выход: 525 мг, 0,850 ммоль, 46%.
Стадия 2. Синтез метил-2-(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C88)
Раствор C87 (351 мг, 0,568 ммоль) в уксусной кислоте (4 мл) перемешивают при 60°C в течение 16 часов, после чего объединяют с продуктом аналогичной реакции с использованием C87 (174 мг, 0,282 ммоль) и упаривают досуха в вакууме. Остаток нейтрализуют водным раствором бикарбоната натрия и экстрагируют дихлорметаном (3 × 10 мл); объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют фильтрат при пониженном давлении с получением C88, смеси изомеров при циклопропане, в виде вспененного твердого вещества желтого цвета. Общий выход: 510 мг, 0,850 ммоль, количественный выход.
Стадия 3. Синтез 2-(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония, ENT-1 (52), и 2-(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония, ENT-2 (53)
К раствору C88 (490 мг, 0,817 ммоль) в смеси ТГФ (8 мл) и MeOH (1 мл) добавляют водный раствор гидроксида лития (2 M; 1,63 мл, 3,26 ммоль) и перемешивают полученную реакционную смесь при 25°C в течение 16 часов. Смесь затем объединяют с продуктом аналогичной реакции с использованием C88(20 мг, 33 мкмоль), концентрируют в вакууме и разбавляют водой (5 мл). Значение рН полученной смеси доводят до 6 добавлением 1 M соляной кислоты и экстрагируют смесью дихлорметан и MeOH (9:1, 3 × 10 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют, концентрируют в вакууме и очищают с использованием с ВЭЖХ с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 17% до 47% B) с получением смеси стереоизомеров при циклопропановом центре 52 и 53 (161 мг, 0,275 ммоль, 32%) в виде твердого белого вещества.
Стереоизомеры при циклопропановом асимметрическом центре разделяют с использованием сверхкритической жидкостной хроматографии [колонка: Chiral Technologies ChiralcelOJ-H, 5 мкм; подвижная фаза: 3:2 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Стереоизомер первого элюирования, полученный в виде твердого белого вещества, обозначают как ENT-1 (52). Выход: 49,2 мг, 81,6 мкмоль, 30% для разделения. ЖХМС m/z 586,1 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,23 (с, 1H), 7,94 (д, 1H), 7,67-7,59 (м, 2H), 7,55-7,49 (м, 2H), 7,28 (дд, 1H), 6,22 (дд, 1H), 5,48 (AB квартет, 2H), 5,23-5,14 (м, 1H), 4,70 (дд, 1H), 4,63-4,54 (м, 2H), 4,41 (дт, 1H), 3,90-3,82 (м, 1H), 3,54-3,45 (м, 1H), 3,38 (ддд, 1H), 3,19 (ддд, 1H), 2,84-2,73 (м, 1H), 2,59-2,48 (м, 1H), 2,39 (дд, 1H), 1,88 (ддд, 1H), 1,69-1,62 (м, 1H), 1,56-1,46 (м, 2H), 1,38-1,30 (м, 1H), 1,24 (дд, 1H).
Стереоизомер второго элюирования, который также выделяют в виде твердого белого вещества, обозначают как ENT-2 (53). Выход: 37,9 мг, 62,9 мкмоль, 23% для разделения. ЖХМС m/z 586,1 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,21 (д, 1H), 7,94 (дд, 1H), 7,66-7,59 (м, 2H), 7,55-7,50 (м, 2H), 7,26 (дд, 1H), 6,20 (дд, 1H), 5,47 (AB квартет, 2H), 5,29-5,20 (м, 1H), 4,70 (дд, 1H), 4,58-4,48 (м, 2H), 4,32 (дт, 1H), 3,87-3,78 (м, 1H), 3,51-3,43 (м, 1H), 3,37 (ддд, 1H), 3,17 (ддд, 1H), 2,82-2,71 (м, 1H), 2,53-2,41 (м, 2H), 1,84 (ддд, 1H), 1,65 (дд, 1H), 1,59-1,44 (м, 2H), 1,38-1,27 (м, 1H), 1,23 (дд, 1H).
Примеры 54 и 55
2-(6-{6-[(4-Циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (54) из C92 и
2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (55) из C93
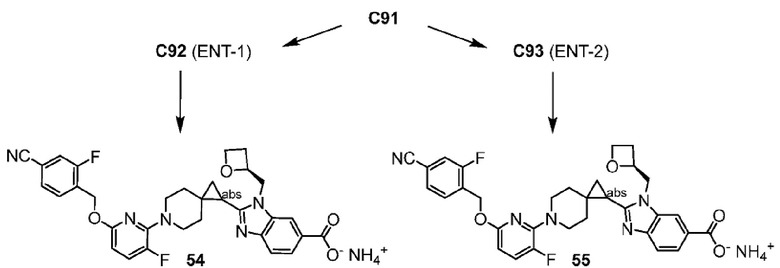
Стадия 1. Синтез метил-4-({[6-(6-бром-3-фторпиридин-2-ил)-6-азаспиро[2.5]окт-1-ил]карбонил}амино)-3-{[(2S)-оксетан-2-илметил]амино}бензоата (C89)
В соответствии с методикой, описанной для синтеза C73 в примере 33, P11 подвергают взаимодействию с P16. Хроматография на силикагеле приводит к получению С89, смеси стереоизомеров при циклопропане, в виде твердого не совсем белого вещества. Выход: 1,31 г, 2,39 ммоль, 79%.
Стадия 2. Синтез метил-2-[6-(6-бром-3-фторпиридин-2-ил)-6-азаспиро[2.5]окт-1-ил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C90)
Раствор C89 (900 мг, 1,64 ммоль) в уксусной кислоте (18 мл) перемешивают при 60°C в течение 16 часов, после чего разбавляют водой (200 мл), нейтрализуют осторожным добавлением карбоната натрия и экстрагируют EtOAc (3 × 100 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении; остаток объединяют с продуктом аналогичной реакции, проведенной с использованием C89 (400 мг, 0,731 ммоль), и очищают хроматографией (силикагель, градиент: от 0% до 60% EtOAc в петролейном эфире) с получением C90, смеси стереоизомеров при циклопропан, в виде твердого белого вещества. Общий выход: 823 мг, 1,55 ммоль, 65%.
Стадия 3. Синтез метил-2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C91)
Смесь C90 (400 мг, 0,756 ммоль), 3-фтор-4-(гидроксиметил)бензонитрила (228 мг, 1,51 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (Xantphos; 43,7 мг, 75,6 мкмоль), ацетата палладия (II) (8,48 мг, 37,8 мкмоль) и карбоната цезия (739 мг, 2,27 ммоль) в толуоле (8 мл) перемешивают при 100°C в течение 2 часов. Реакционную смесь объединяют с продуктом аналогичной реакции с использованием C90 (100 мг, 0,189 ммоль), разбавляют дихлорметанои (50 мл) и фильтруют. Фильтрат концентрируют в вакууме и очищают хроматографией (силикагель) (градиент: от 0% до 45% EtOAc в петролейном эфире) с получением C91, смеси стереоизомеров при циклопропан, в виде всперенного твердого вещества желтого цвета. Общий выход: 385 мг, 0,642 ммоль, 68%.
Стадия 4. Выделение метил-2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата, ENT-1 (C92), и метил-2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата, ENT-2 (C93).
Стереоизомеры при циклопропане, включающие C91 (385 мг, 0,642 ммоль), разделяют с использованием сверхкритической жидкостной хроматографии [колонка: Chiral Technologies Chiralpak AD, 5 мкм; подвижная фаза: 3:2 диоксид углерода/(этанол, содержащий 0,1% гидроксида аммония)]. Стереизомер первого элюирования, выделенный в виде белой пены, обозначают как ENT-1 (C92). Выход: 192 мг, 0,320 ммоль, 50%.
Стереоизомер второго элюирования, выделенный также в виде белой пены, обозначают как ENT-2 (C93). Выход: 193 мг, 0,322 ммоль, 50%.
Стадия 5. Синтез 2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (54) из C92
К раствору C92 (100 мг, 0,167 ммоль) в смеси ТГФ (1 мл) и MeOH (1 мл) добавляют водный раствор гидроксида лития(2 M; 0,834 мл, 1,67 ммоль). Реакционную смесь перемешивают при 30°C в течение 3 часов, затем нейтрализуют осторожным добавлением 12 M соляной кислоты, разбавляют водой (30 мл) и экстрагируют дихлорметаном (3 × 50 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме; остаток объединяют с продуктом аналогичной реакции, проведенной с использованием C92 (90 мг, 0,15 ммоль), и очищают ВЭЖХ с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 27% до 47% B) с получением 54 в виде твердого белого вещества. Общий выход: 39 мг, 65 мкмоль, 20%. ЖХМС m/z 586,1 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,24 (д, 1H), 7,95 (дд, 1H), 7,62 (д, 1H), 7,60 (дд, 1H), 7,51 (дд, 1H), 7,47 (дд, 1H), 7,28 (дд, 1H), 6,22 (дд, 1H), 5,39 (с, 2H), 5,26-5,17 (м, 1H), 4,72 (дд, компонент ABX шаблона, 1H), 4,66-4,55 (м, 2H), 4,43 (дт, 1H), 3,84-3,74 (м, 1H), 3,50-3,34 (м, 2H), 3,19 (ддд, 1H), 2,87-2,75 (м, 1H), 2,61-2,49 (м, 1H), 2,39 (дд, 1H), 1,95 (ддд, 1H), 1,65 (дд, 1H), 1,61-1,49 (м, 2H), 1,42-1,32 (м, 1H), 1,23 (дд, 1H).
Стадия 6. Синтез 2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (55) из C93
Синтез 55 из C93 проводят с использованием способа, описанного для синтеза 54 из C92 в стадии 5 выше. Соединение 55 получают в виде твердого белого вещества. Выход: 59,4 мг, 98,5 мкмоль, 31%. ЖХМС m/z 586,1 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,23 (д, 1H), 7,95 (дд, 1H), 7,62 (д, 1H), 7,59 (дд, 1H), 7,51 (дд,1H), 7,46 (дд,1H), 7,26 (дд, 1H), 6,20 (дд, 1H), 5,38 (с, 2H), 5,30-5,22 (м, 1H), 4,71 (дд,1H), 4,61-4,50 (м, 2H), 4,35 (дт, 1H), 3,80-3,72 (м, 1H), 3,47-3,33 (м, 2H), 3,17 (ддд, 1H), 2,83-2,72 (м, 1H), 2,54-2,46 (м, 1H), 2,44 (дд, 1H), 1,95-1,84 (м, 1H), 1,65 (дд, 1H), 1,62-1,47 (м, 2H), 1,39-1,31 (м, 1H), 1,22 (дд, 1H).
Примеры 56, 57, и 58
2-(6-{6-[(4-Циано-2-фторбензил)окси]-3,5-дифторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония (56),
2-(6-{6-[(4-циано-2-фторбензил)окси]-3,5-дифторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония, ENT-1(57), и
2-(6-{6-[(4-циано-2-фторбензил)окси]-3,5-дифторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония, ENT-2 (58)
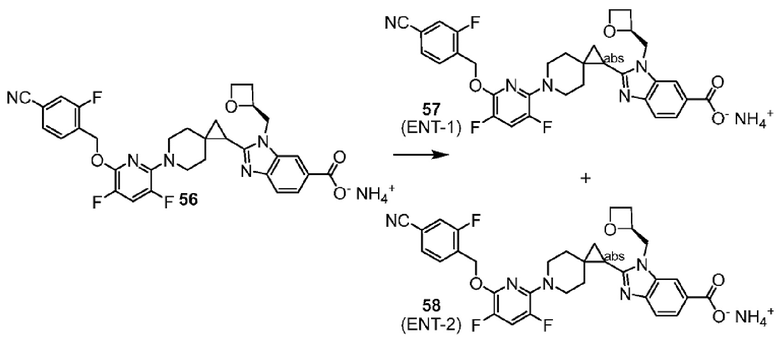
Стадия 1. Синтез метил-2-(6-{6-[(4-циано-2-фторбензил)окси]-3,5-дифторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C94)
К раствору P12 (488 мг, 1,17 ммоль) и гексафторфосфата О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилурония (522 мг, 1,37 ммоль) в N, N-диметилформамидe (8 мл) добавляют P16 (276 мг, 1,17 ммоль) и N, N-диизопропилэтиламин (515 мг, 3,99 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, после чего разбавляют водой (35 мл) и экстрагируют EtOAc (3 × 30 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (2 × 25 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме; очистка хроматографией (силикагель, градиент: от 0% до 60% EtOAc в петролейном эфире) приводит к получению промежуточного амида (500 мг, 0,787 ммоль, 67%) в виде бесцветной смолы. Полученное соединение растворяют в уксусной кислоте, нагревают до 60°C и выдерживают при указанной температуре в течение 16 часов, после чего объединяют с аналогичной реакционной смесью (полученной из P12, 52,2 мг, 0,125 ммоль) и концентрируют в вакууме. Остаток растворяют в дихлорметане (25 мл) и промывают насыщенным водным раствором бикарбоната натрия (30 мл). Водный слой экстрагируют дихлорметаном (2 × 20 мл), объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Препаративная тонкослойная хроматография (элюент: 1:1 EtOAc/петролейный эфир) приводит к получению С94, смеси стереоизомеров при циклопропане, в виде смолы желтого цвета. Общий выход: 280 мг, 0,453 ммоль, 35%.
Стадия 2. Cинтез 2-(6-{6-[(4-циано-2-фторбензил)окси]-3,5-дифторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (56)
Гидролиз C94 проводят с использованием способа, описанного для синтеза P12 из C30 в получении P12, с получением 56, смеси стереоизомеров при циклопропане, в виде твердого вещества. Сырой продукт очищают ВЭЖХ с обращенной фазой (колонка: Agela Durashell C18, 5 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 23% до 53% B). Стереоизомер первого элюирования, выделенный в виде твердого белого вещества, обозначают как ENT-1 (57). Выход: 11,0 мг, 17,7 мкмоль, 21%. ЖХМС m/z 604,1 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,22 (уш. с, 1H), 7,95 (дд, 1H), 7,66-7,60 (м, 2H), 7,54 (дд, 1H), 7,49 (дд, 1H), 7,38 (дд, 1H), 5,48 (с, 2H), 5,31-5,22 (м, 1H), 4,72 (дд, 1H), 4,62-4,52 (м, 2H), 4,36 (дт, 1H), 3,69-3,60 (м, 1H), 3,36-3,25 (м, 2H, предполагаемый; значительно скрыт пиком растворителя), 3,16-3,07 (м, 1H), 2,84-2,73 (м, 1H), 2,55-2,47 (м, 1H), 2,45 (дд, 1H), 1,99-1,86 (м, 1H), 1,65 (дд, 1H), 1,62-1,49 (м, 2H), 1,42-1,33 (м, 1H), 1,22 (дд, 1H).
Стереоизомер второго элюирования, также выделенный в виде твердого белого вещества, обозначают как ENT-2 (58). Выход: 23,5 мг, 39,0 мкмоль, 32%. ЖХМС m/z 604,1 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,25 (уш. с, 1H), 7,95 (дд, 1H), 7,67-7,60 (м, 2H), 7,54 (уш. д,1H), 7,49 (уш. д, 1H), 7,40 (дд, 1H), 5,49 (с, 2H), 5,26-5,18 (м, 1H), 4,74 (дд, компонент ABX системы; 1H), 4,66-4,57 (м, 2H), 4,44 (дт, 1H), 3,71-3,63 (м, 1H), 3,37-3,26 (м, 2H, предполагаемый; значительно скрыт пиком растворителя), 3,17-3,08 (м, 1H), 2,88-2,76 (м, 1H), 2,61-2,50 (м, 1H), 2,40 (дд, 1H), 2,03-1,93 (м, 1H), 1,65 (дд, 1H), 1,63-1,52 (м, 2H), 1,43-1,34 (м, 1H), 1,24 (дд, 1H).
Соединения, представленные в таблице 7 ниже, синтезируют в соответствии с методиками, описанными в настоящем документе для синтеза соединений примеров и примеров получений, с использованием подходящих исходных соединений, которые являются коммерчески доступными либо получены с использованием способов, известных специалистам в данной области техники или аналогичных описанным для получения других промежуточных соединений. Соединения очищают методами, хорошо известными специалистам в данной области техники, которые могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 7. Структуры и названия в соответствии с номенклатурой ИЮПАК соединений примеров 59-61
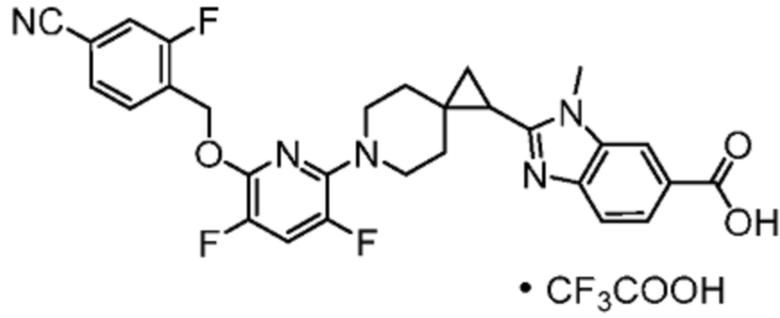
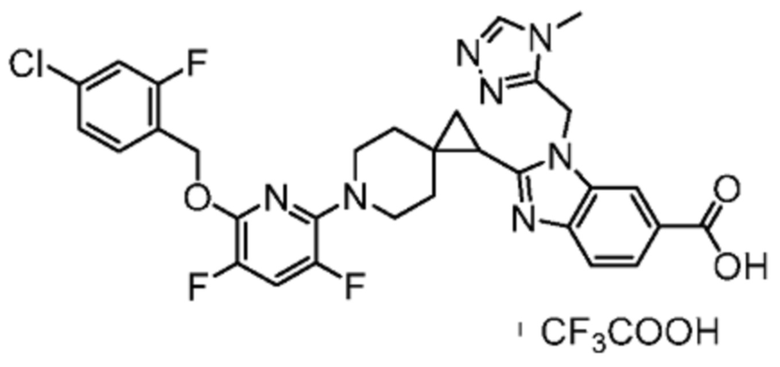
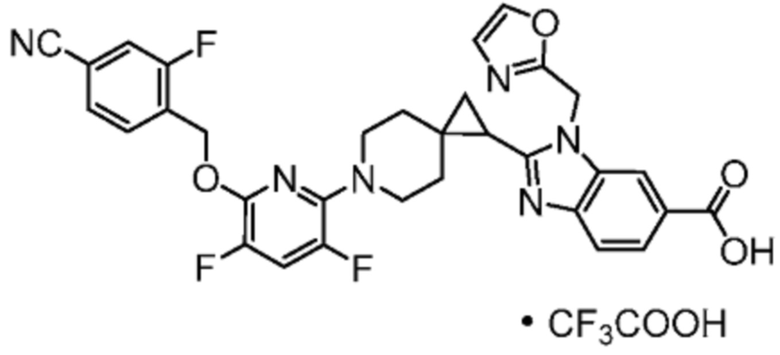
Таблица 7A. Физико-химические характеристики соединений примеров 59-61
Пример 62
2-[(4-{2-[(4-Хлор-2-фторбензил)окси]пиримидин-4-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота (62)
Стадия 1. Синтез метил-2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C95)
Смесь P13 (14,34 г, 21,5 ммоль), P17 (6,34 г, 21,5 ммоль) и карбоната калия (19,3 г, 140 ммоль) в 1,4-диоксане (144 мл) перемешивают при 58°C в течение 1,5 часа. После этого к смеси добавляют карбоната калия (5,0 г, 36 ммоль); спустя 30 минут добавляют ацетонитрил (50 мл) и еще через 50 минут добавляют еще одну порцию ацетонитрила (25 мл). Температуру смеси повышают до 63°C и выдерживают смесь при указанной температуре в течение ночи, после чего нагрев удаляют и медленно разбавляют смесь водой (300 мл). Полученную смесь экстрагируют EtOAc (3 × 200 мл), объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Очистка хроматографией (силикагель, градиент: от 0% до 10% MeOH в дихлорметане) приводит к получению C95 в виде твердого не совсем белого вещества. Выход: 10,7 г, 18,4 ммоль, 86%.
Стадия 2. Синтез 2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты (62)
Смесь C95 (9,33 г, 16,1 ммоль), MeOH (100 мл) и водного раствора гидроксида натрия (1,0 M; 40,2 мл, 40,2 ммоль) обрабатывают достаточным количеством ТГФ (12 мл) для получения раствора; реакционную смесь затем перемешивают при комнатной температуре в течение ночи. ЖХМС анализ в это время показывает конверсию исходных веществ в продукт: ЖХМС m/z 566,3 [M+H]+. Реакционную смесь концентрируют в вакууме до конечного объема приблизительно 50 мл и затем обрабатывают 10% водным раствором лимонной кислоты до pH 6. Смесь экстрагируют смесью 2-пропанола и дихлорметана (1:4; 3 × 75 мл), объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученное твердое белое вещество разбавляют циклогексаном и дихлорметаном и затем концентрируют в вакууме для удаления остаточного 2-пропанола с получением 62. Выход: 9,25 г, 16 ммоль, количественный выход. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,50 (д, 1H), 8,26 (уш. с, 1H), 7,80 (дд, 1H), 7,63 (д, 1H), 7,57 (дд, 1H), 7,48 (дд, 1H), 7,32 (дд, 1H), 7,09 (д, 1H), 5,39 (с, 2H), 5,13-5,04 (м, 1H), 4,80 (дд, 1H), 4,66 (дд, 1H), 4,52-4,44 (м, 1H), 4,37 (дт, 1H), 3,87 (AB квартет, 2H), 2,99 (уш. д, 1H), 2,86 (уш. д, 1H), 2,76-2,58 (м, 2H), 2,47-2,37 (м, 1H), 2,29-2,12 (м, 2H), 1,88-1,77 (м, 2H), 1,77-1,59 (м, 2H).
Соединения, представленные в таблице 8 ниже, синтезируют в соответствии с методиками, описанными в настоящем документе для синтеза соединений примеров и примеров получений, с использованием подходящих исходных соединений, которые являются коммерчески доступными либо получены с использованием способов, известных специалистам в данной области техники или аналогичных описанным для получения других промежуточных соединений. Соединения очищают методами, которые хорошо известны специалистам в данной области техники и могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 8. Структуры и названия в соответствии с номенклатурой ИЮПАК соединений примеров 63-68
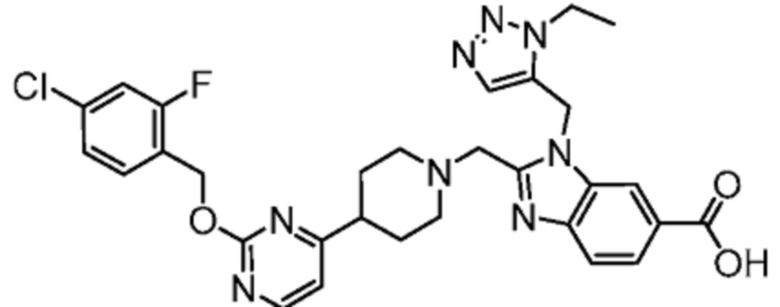
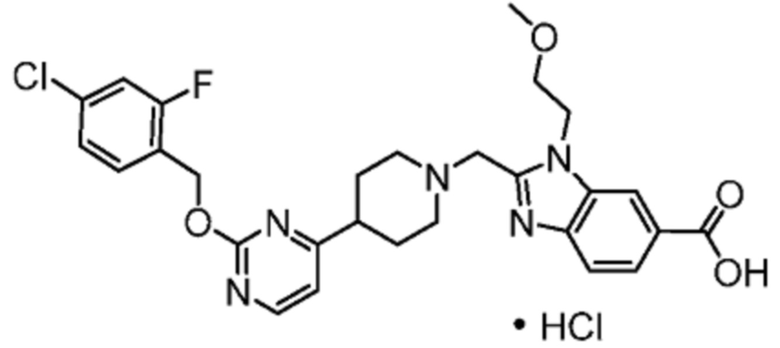
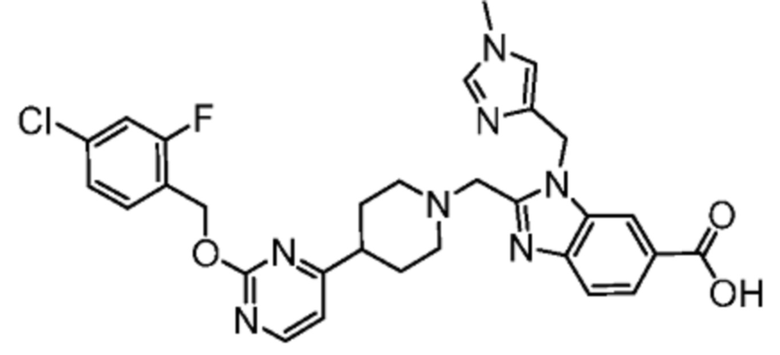
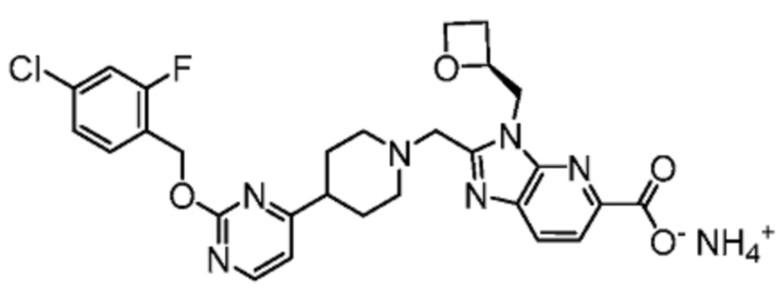
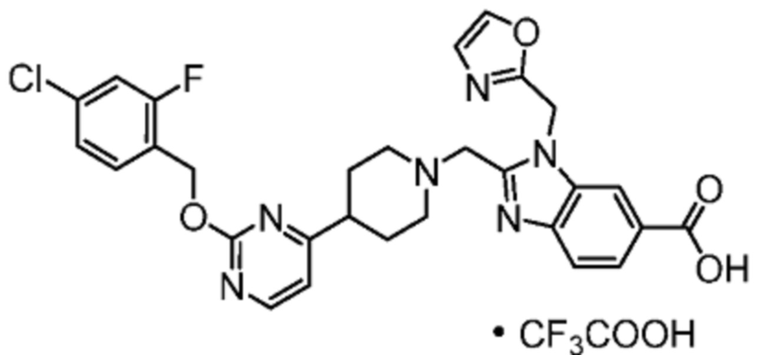
Таблица 8A. Физико-химические характеристики соединений примеров 63-68
Таблица 8/8A: 1. Обработка метил-5-{[(4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}пиперидин-1-ил)ацетил]амино}-6-{[(2S)-оксетан-2-илметил]амино}пиридин-2-карбоксилата водным раствором гидроксида натрия в 1,4-диоксане при повышенной температуре приводит к циклизации и гидролизу сложного эфира с получением после очистки соединения примера 67.
Пример 69
2-{[(2S)-4-{2-[(4-Хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония
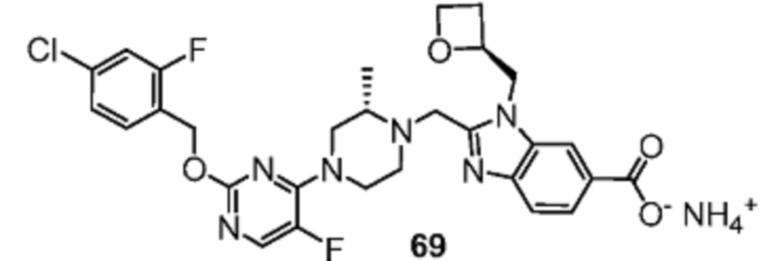
Стадия 1. Синтез трифторацетатной соли 2-[(4-хлор-2-фторбензил)окси]-5-фтор-4-[(3S)-3-метилпиперазин-1-ил]пиримидина (C96)
Трифторуксусную кислоту (36 мл) добавляют к раствору P14 (7,62 г, 16,8 ммоль) в дихлорметане (75 мл) и перемешивают полученную реакционную смесь при 18°C в течение 1 часа, после чего смесь объединяют с продуктом аналогичной реакции с использованием P14 (3,09 г, 6,79 ммоль). Смесь концентрируют в вакууме с получением С96 в виде масла коричневого цвета. Общий выход: 11,0 г, 23,5 ммоль, количественный выход.
Стадия 2. Синтез метил-2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C97)
Суспензию C96 (11,0 г, 23,5 ммоль), P17 (6,94 г, 23,5 ммоль) и карбоната калия (16,3 г, 118 ммоль) в ацетонитриле (150 мл) перемешивают при 50°C в течение 19 часов, после чего смесь распределяют между водой (300 мл) и EtOAc (300 мл). Водный слой экстрагируют EtOAc (2 × 200 мл), объединенные органические слои концентрируют в вакууме и очищают хроматографией (силикагель, градиент: от 35% до 60% EtOAc в петролейном эфире) с получением C97 в виде белой смолы. Выход: 11,1 г, 18,1 ммоль, 77%.
Стадия 3. Синтез 2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата аммония (69)
Водный раствор гидроксида натрия (2 M; 22 мл, 44 ммоль) добавляют к раствору C97 (5,00 г, 8,16 ммоль) в смеси MeOH (73 мл) и ТГФ (73 мл). Реакционную смесь перемешивают при 18°C в течение 16 часов, затем нагревают до 45°C и выдерживают при указанной температуре в течение 3 часов, после чего нейтрализуют до pH 7 добавлением 1 M соляной кислоты и концентрируют в вакууме. Остаток разбавляют водой (100 мл) и экстрагируют смесью дихлорметана и MeOH (10:1, 4 × 100 мл); объединенные органические слои концентрируют при пониженном давлении и затем очщают с использованием ВЭЖХ с обращенной фазой (колонка: Phenomenex Gemini C18, 10 мкм; подвижная фаза A: 0,05% гидроксид аммония в воде; подвижная фаза B: ацетонитрил; градиент: от 20% до 40% B) с получением 69 в виде твердого белого вещества. Выход: 2,75 г, 4,59 ммоль, 56%. ЖХМС m/z 598,9♦ [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,23 (дд,1H), 7,96 (дд,1H), 7,89 (д, 1H), 7,62 (дд, 1H), 7,46 (дд, 1H), 7,24-7,17 (м, 2H), 5,33 (с, 2H), 5,33-5,26 (м, 1H), 4,9-4,84 (м, 1H, предполагаемый; частично скрыт пиком воды), 4,75 (дд, 1H), 4,60 (ддд, 1H), 4,49 (д, 1H), 4,33 (дт, 1H), 4,14-4,07 (м, 1H), 4,02 (уш. д, 1H), 3,70 (д, 1H), 3,52 (ддд, 1H), 3,36-3,29 (м, 1H, предполагаемый; частично скрыт пиком растворителя), 2,81-2,70 (м, 2H), 2,72-2,63 (м, 1H), 2,54-2,44 (м, 1H), 2,39 (ддд, 1H), 1,18 (д, 3H).
Соединения, представленные в таблице 9 ниже, синтезируют в соответствии с методиками, описанными в настоящем документе для синтеза соединений примеров и примеров получений, с использованием подходящих исходных соединений, которые являются коммерчески доступными либо получены с использованием способов, известных специалистам в данной области техники или аналогичных описанным для получения других промежуточных соединений. Соединения очищают методами, которые хорошо известны специалистам в данной области техники и могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 9. Структуры и названия в соответствии с номенклатурой ИЮПАК соединений примеров 70-72
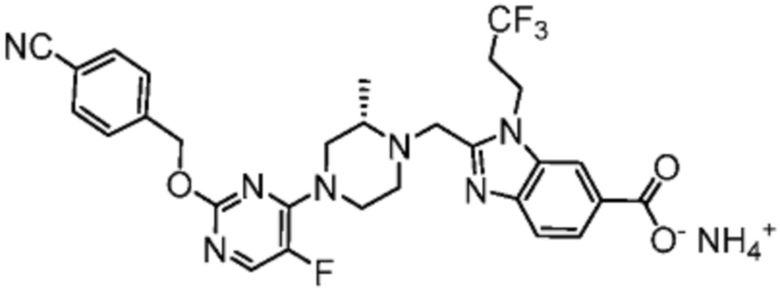
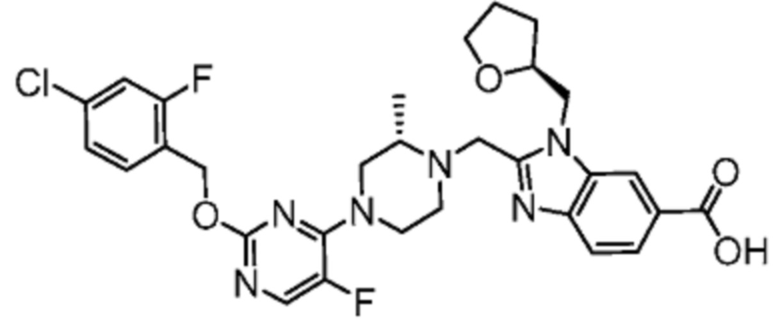
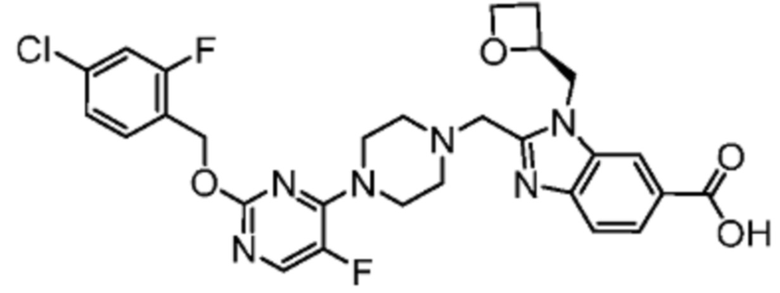
Таблица 9A. Физико-химические характеристики соединений примеров 70-72
Пример 73
2-{[(2S)-4-{2-[(4-Хлор-2-фторбензил)окси]пиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота (73)
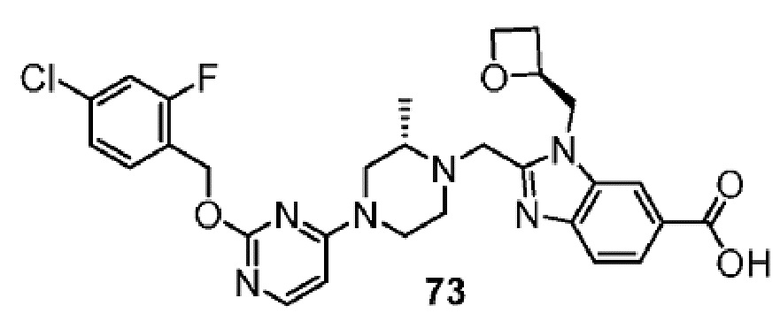
Стадия 1. Синтез метил-2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилата (C98)
Данную реакцию проводят в двух идентичных загрузках. Смесь P15 (26,3 г 38,6 ммоль) и карбоната калия (31,0 г, 225 ммоль) в ацетоне (190 мл) перемешивают при комнатной температуре в течение 5 минут, затем помещают на масляную баню при 70°C и выдерживают в течение 5 минут, затем удаляют нагрев и перемешивают в течение других 5 минут. К полученной смеси добавляют P17 (11,0 г, 37,3 ммоль) и кипятят реакционную смесь с обратным холодильником в течение 24 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют водой (350 мл), перемешивают при комнатной температуре в течение 1 часа и экстрагируют EtOAc (2 × 250 мл). EtOAc эстракты из обеих реакций объединяют, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме; повторная хроматография на силикагеле (градиент: от 0% до 10% MeOH в дихлорметане) приводит к получению C98 в виде твердого белого вещества. Общий выход: 29,2 г, 49,1 ммоль, 66%.
Стадия 2. Синтез 2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты (73)
Водный раствор гидроксида натрия (1 M; 110 мл, 110 ммоль) добавляют охлажденному до 0°C раствору C98 (18,8 г, 31,6 ммоль) в MeOH (220 мл); затем добавляют ТГФ (40 мл) до получения прозрачной смеси. Реакционную смесь перемешивают при комнатной температуре в течение ночи, ЖХМС анализ показывают конверсию исходных веществ продукт: ЖХМС m/z 581,2♦ [M+H]+. Реакционную смесь концентрируют в вакууме до объема примерно 125 мл, остаток разбавляют до 150 мл добавлением воды и медленно доводят значение pH до 7 с помощью 10% водного раствора лимонной кислоты. Полученную суспензию фильтруют, осадок на фильтре промывают большим количеством воды с получением 73 в виде твердого белого вещества. Выход: 18,2 г, 31,3 ммоль, 99%. 1H ЯМР (400 МГц, метанол-d4) δ 8,30 (уш. с, 1H), 7,98 (дд, 1H), 7,94 (д, 1H), 7,67 (д, 1H), 7,49 (дд, 1H), 7,26-7,18 (м, 2H), 6,40 (д, 1H), 5,38 (с, 2H), 5,34-5,26 (м, 1H), 4,95-4,85 (м, 1H, предполагаемый; частично скрыт пиком воды), 4,78 (дд, 1H), 4,62 (td, 1H), 4,50 (д, 1H), 4,35 (дт, 1H), 4,10-3,97 (уш. м, 1H), 3,97-3,83 (уш. м, 1H), 3,72 (д, 1H), 3,45-3,3 (м, 1H, предполагаемый; частично скрыт пиком растворителя), 3,23 (дд, 1H), 2,82-2,70 (м, 2H), 2,70-2,59 (м, 1H), 2,53-2,42 (м, 1H), 2,36 (ддд, 1H), 1,19 (д, 3H).
Соединения, представленные в таблице 10 ниже, синтезируют в соответствии с методиками, описанными в настоящем документе для синтеза соединений примеров и примеров получений, с использованием подходящих исходных соединений, которые являются коммерчески доступными либо получены с использованием способов, известных специалистам в данной области техники или аналогичных описанным для получения других промежуточных соединений. Соединения очищают методами, которые хорошо известны специалистам в данной области техники и могут включать хроматографию на силикагеле, ВЭЖХ или осаждение из реакционной смеси.
Таблица 10. Структуры и названия в соответствии с номенклатурой ИЮПАК соединений примеров 74-78
Таблица 10A. Физико-химические характеристики соединений примеров 74-78
Таблица 10/10A, 1. Взаимодействие P8 с трет-бутил-(3R)-3-метилпиперазин-1-карбоксилатом проводят с использованием фторида цезия и N, N-диизопропилэтиламина в ацетонитриле при температуре ≥100°C.
Пример 79 и 80
2-[(4-{6-[(4-Хлор-2-фторбензил)окси]пиридин-2-ил}циклогексил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония, изомер 1 (79), и
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}циклогексил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоксилат аммония, изомер 2 (80)
Соединение C99, смесь цис- и транс-изомеров при циклогексильной группе, получают из P34 и P16 способом, аналогичным описанному в Примере 1, из промежуточного соединения P1. Смесь изомеров разделяют с помощью ВЭЖХ, время удерживания показано ниже. Колонка: Chiral Technologies Chiralpak AD-H, 21 × 250 мм, 5 мкм; подвижная фаза A: диоксид углерода; подвижная фаза B: 0,2% гидроксида аммония в метаноле; элюент: 30% B; объемная скорость потока: 75 мл/мин.; температура колонки: 40°C).
Изомер первого элюирования обозначают как изомер 1 (79). Время удерживания 1,60 мин. ЖХМС m/z 564,5 [M+H]+.
Изомер второго элюирования обозначают как изомер 2 (80). Время удерживания 1,86 мин., ЖХМС m/z 564,4[M+H]+.
CHO GLP-1R клон H6 - анализ 1
GLP-1R-опосредуемую агонистическую активность определяют с помощью функционального клеточного анализа с использованием набора гомогенной флуоресценции с временным разрешением (HTRF) для определения цАМФ (cAMP HI Range Assay Kit; CisBio cat #62AM6PEJ), с помощью которого определяют уровни цАМФ в клетке. Метод представляет собой конкурентный иммуноанализ природного цАМФ, продуцируемого клетками, и экзогенного цАМФ, меченного красителем d2. Связывание меченого вещества визуализируют с помощью mAb против цАМФ, меченного криптатом. Специфический сигнал (т.е. передача энергии) обратно пропорционален концентрации цАМФ в стандартном или экспериментальном образце.
Кодирующую последовательность человеческого GLP-1R (NCBI Reference Sequence NP_002053.3, включающая существующий в природе вариант Gly168Ser) субклонируют в pcDNA3 (Invitrogen) и выделяют клеточную линию, стабильно экспрессирующую рецептор (обозначенный клоном H6). Анализы насыщения (методика аналитической фильтрации) с использованием 125I-GLP-17-36 (Perkin Elmer) показывают, что плазматические мембраны, полученные из этой линии клеток, экспрессируют высокую плотность GLP-1R (Kd: 0,4 нМ, Bmax: 1900 фмоль/мг белка).
Клетки удаляют из криоконсервации, повторно суспендируют в в 40 мл фосфатнoсолевого буфера Дульбекко (DPBS - Lonza Cat # 17-512Q) и центрифугируют со скоростью 800 × g в течение 5 мин. при 22°C. Клеточный пеллет снова суспендируют в 10 мл ростовой среды [DMEM/F12 1:1 смесь с HEPES, L-Gln, 500 мл (DMEM/F12 Lonza Cat # 12-719F), 10% инактивированной нагреванием фетальной бычьей сыворотки (Gibco Cat # 16140-071), 5 мл 100X Pen-Strep (Gibco Cat # 15140-122), 5 мл 100X L-глутамина (Gibco Cat # 25030-081) и 500 мкг/мл Geneticin (G418) (Invitrogen #10131035)]. Образец клеточной суспензии в ростовой среде объемом 1 мл обсчитывают на аппарате Becton Dickinson ViCell для определения жизнеспособности клеток и количества клеток на мл. Оставшуюся клеточную суспензию затем корректируют ростовой средой для доставки 2000 жизнеспособных клеток на лунку с использованием дозатора реагентов Matrix Combi Multidrop, и клетки распределяют в белый 384-луночный планшет для анализа, обработанный культурой ткани (Corning 3570). Затем аналитический планшет инкубируют в течение 48 часов при 37°С в увлажненной среде с 5% диоксида углерода.
Растворы различных концентраций каждого тестируемого соединения (в ДМСО) разводят аналитическим буфером (HBSS с кальцием/магнием (Lonza/BioWhittaker cat #10-527F)/0,1% BSA (Sigma Aldrich cat #A7409-1L)/20 мМ HEPES (Lonza/BioWhittaker cat #17-737E), содержащим 100 мкМ 3-изобутил-1-метилксантина (IBMX; Sigma cat #I5879). Конечная концентрация ДМСО составляет 1%.
Спустя 48 часов ростовую среду удаляют из лунок аналитического планшета и обрабатывают клетки 20 мкл последовательно разбавленного в аналитическом буфере раствора соединения в течение 30 минут при 37°C в увлажненной среде, содержащей 5% диоксида углерода. После 30 минутной инкубации в каждую лунку планшета для анализа добавляют 10 мкл меченого d2 цAMФ и 10 мкл антитела против цAMФ (оба резведены в соотношении 1:20 в буфере для клеточного лизиса; как описано в протоколе анализа производителя). После этого планшеты инкубируют при комнатной температуре и спустя 60 минут считывают изменения в HTRF сигнале с помощью планшет-ридера для планшетов с несколькими метками Envision 2104 с использованием возбуждения 330 нм и эмиссии 615 и 665 нм. Исходные данные преобразуют в нМ цAMФ интерполяцией из стандартной кривой цAMФ (как описано в протоколе анализа производителя), а эффект в процентах определяют относительно концентрации насыщения полного агониста GLP-17-36 (1 мкM), внесенного в каждый планшет. Значения EC50 определяют из кривых «доза-ответ» агониста, проанализированных с помощью программы аппроксимации кривой по точкам с использованием 4-параметрового логистического уравнение «доза-эффект».
CHO GLP-1R клон C6 - анализ 2
GLP-1R-опосредуемую агонистическую активность определяют с помощью функционального клеточного анализа с использованием набора гомогенной флуоресценции с временным разрешением (HTRF) для определения цАМФ (cAMP HI Range Assay Kit; CisBio cat #62AM6PEJ), с помощью которого определяют уровни цАМФ в клетке. Метод представляет собой конкурентный иммуноанализ природного цАМФ, продуцируемого клетками, и экзогенного цАМФ, меченного красителем d2. Связывание меченого вещества визуализируется с помощью mAb против цАМФ, меченного криптатом. Специфический сигнал (т.е. передача энергии) обратно пропорционален концентрации цАМФ в стандартном или экспериментальном образце.
Кодирующую последовательность человеческого GLP-1R (NCBI Reference Sequence NP_002053.3, включающая существующий в природе вариант Gly168Ser) субклонируют в pcDNA5-FRT-TO и выделяют клональную клеточную линию CHO, стабильно экспрессирующую низкую плотность рецепторов, с использованием Flp-In™ T-Rex™ System, как описано производителем (ThermoFisher). Анализы насыщения (процедура фильтрации) с использованием 125I-GLP-1 (Perkin Elmer) показали, что плазматические мембраны, полученные из этой линии клеток (обозначенной клоном C6), экспрессируют низкую плотность GLP-1R (Kd: 0,3 нM, Bmax: 240 240 фмоль/мг белка) относительно линии клеток клона H6.
Клетки удаляют из криоконсервации, повторно суспендируют в в 40 мл фосфатнoсолевого буфера Дульбекко (DPBS - Lonza Cat # 17-512Q) и центрифугируют со скоростью 800 × g в течение 5 мин. при 22°C. DPBS аспирируют, леточный пеллет снова суспендируют в 10 мл ростовой среды [DMEM/F12 1:1 смесь с HEPES, L-Gln, 500 мл (DMEM/F12 Lonza Cat # 12-719F), 10% инактивированной нагреванием фетальной бычьей сыворотки (Gibco Cat # 16140-071), 5 мл 100X Pen-Strep (Gibco Cat #15140-122), 5 мл 100X L-глутамина (Gibco Cat #25030-081), 700 мкг/мл Hygromycin (Invitrogen Cat #10687010) и 15 мкг/мл Blasticidin (Gibco Cat # R21001). Образец клеточной суспензии в ростовой среде объемом 1 мл обсчитывают на аппарате Becton Dickinson ViCell для определения жизнеспособности клеток и количества клеток на мл. Оставшуюся клеточную суспензию затем корректируют ростовой средой для доставки 1600 жизнеспособный клеток на лунку с использованием дозатора реагентов Matrix Combi Multidrop, и клетки распределяют в белый 384-луночный планшет для анализа, обработанный культурой ткани (Corning 3570). Затем аналитический планшет инкубируют в течение 48 часов при 37°С в увлажненной среде (95% O2, 5% CO2).
Растворы различных концентраций каждого тестируемого соединения (в ДМСО) разводят аналитическим буфером (HBSS с кальцием/магнием [Lonza/BioWhittaker cat #10-527F)/0,1% BSA (Sigma Aldrich cat #A7409-1L)/20 мМ HEPES (Lonza/BioWhittaker cat #17-737E), содержащим 100 мкМ 3-изобутил-1-метилксантина (IBMX; Sigma cat #I5879). Конечная концентрация ДМСО составляет 1%.
Спустя 48 часов ростовую среду удаляют из лунок аналитического планшета и обрабатывают клетки 20 мкл последовательно разбавленного в аналитическом буфере раствора соединения в течение 30 минут при 37°C в увлажненной среде (95% O2, 5% CO2). После 30 минутной инкубации в каждую лунку планшета для анализа добавляют 10 мкл меченого d2 цAMФ и 10 мкл антитела против цAMФ (оба разведены в соотношении 1:20 в буфере для клеточного лизиса; как описано в протоколе анализа производителя). После этого планшеты инкубируют при комнатной температуре и спустя 60 минут считывают изменения в HTRF сигнале с помощью планшет-ридера для планшетов с несколькими метками Envision 2104 с использованием возбуждения 330 нм и эмиссии 615 и 665 нм. Исходные данные преобразуют в нМ цAMФ интерполяцией из стандартной кривой цAMФ (как описано в протоколе анализа производителя), а эффект в процентах определяют относительно концентрации насыщения полного агониста GLP-17-36 (1 мкM), внесенного в каждый планшет. Значения EC50 определяют из кривых «доза-ответ» агониста, проанализированных с помощью программы аппроксимации кривой по точкам с использованием 4-параметрового логистического уравнение «доза-эффект».
В таблице 11 результаты анализа представлены двумя (2) значащими значениями среднего геометрического (EC50s) и среднего арифметического (Emax), полученных исходя из указанного числа повторов (число повторов). Пустая ячейка означает, что данные для этого примера отсутствуют или Emax не вычислено.
Таблица 11. Биологическая активность соединений примеров 1-80.
EC50 (нМ)
Emax (%)
Число повторов
EC50 (нМ)
Emax (%)
Число повторов
*Тестировалось в форме аммониевой соли и свободной кислоты
**Тестировалось в форме аммониевой и трис-солей
***Тестировалось в форме аммониевой и трис-солей и в форме свободной кислоты
Все патенты, заявки на патенты и ссылки, указанные в настоящем описании, полностью включены в настоящее изобретение в виде ссылок.
| название | год | авторы | номер документа |
|---|---|---|---|
| АГОНИСТЫ РЕЦЕПТОРА GLP-1 И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2740135C1 |
| АГОНИСТЫ РЕЦЕПТОРА GLP-1 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2769715C1 |
| АГОНИСТ РЕЦЕПТОРА GLP-1, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО, И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2021 |
|
RU2800290C1 |
| ИНГИБИТОРЫ ТРАНСГЛУТАМИНАЗЫ 2 (TG2) | 2019 |
|
RU2781370C2 |
| АМИНОПИРАЗИНОВЫЕ СОЕДИНЕНИЯ СО СВОЙСТВАМИ АНТАГОНИСТА A2A | 2015 |
|
RU2727805C2 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2019 |
|
RU2811601C1 |
| ПРОИЗВОДНЫЕ 5-ОКСА-2-АЗАСПИРО[3.4]ОКТАНА В КАЧЕСТВЕ АГОНИСТОВ M4 | 2020 |
|
RU2820477C1 |
| СУЛЬФОНАМИДНЫЕ ПЕРИ-ЗАМЕЩЕННЫЕ БИЦИКЛЫ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ПОРАЖЕНИЯ АРТЕРИЙ | 2005 |
|
RU2403240C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
Изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, которые обладают агонистической активностью в отношении GLP-1 рецептора и могут найти применение при лечении связанных с ним заболеваний. В формуле I А представляет собой A1 или A2, каждый R1 независимо представляет собой галоген или -CN; m равно 0, 1, 2 или 3; Х-L представляет собой N-CH2, CHCH2 или циклопропил; Y представляет собой CH или N; ZA1 представляет собой CH, CR2 или N; ZA2 представляет собой CH, CR2 или N; ZA3 представляет собой CH, CR2 или N; при условии, что ZA2 и ZA3 одновременно не являются N, а также дополнительно при условии, что один из ZA2 и ZA3 представляет собой N, когда Х-L представляет собой N-CH2; каждый R2 представляет собой F; каждый R3 независимо представляет собой -C1-3алкил; q равно 0, 1 или 2; R4 представляет собой -C1-3алкил, -C0-3алкилен-R5 или -C1-3алкилен-R6, где указанный алкил может быть замещенным, как допускает валентность, 0-3 заместителями, независимо выбранными из 0-3 атомов F, и 0-1 заместителем, выбранным из -C0-1алкилен-ORO; R5 представляет собой 4-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и O, где указанный гетероциклоалкил может быть замещенным 0-1 оксо (=O); R6 представляет собой 5-6-членный гетероарил, содержащий 2-3 гетероатома, независимо выбранных из N и O, где указанный гетероарил может быть замещенным 0-2 заместителями, как допускает валентность, независимо выбранными из 0-2 -C1-3алкилов; каждый RO независимо представляет собой H или -C1-3алкил; каждый из Z1, Z2 и Z3 представляет собой -CRZ или один из Z1, Z2 и Z3 представляет собой N, а два другие представляют собой -CRZ; и каждый RZ независимо представляет собой H. Изобретение относится также к конкретным соединениям, фармацевтической композиции и способу лечения кардиометаболического заболевания и связанных с ним заболеваний. 6 н. и 14 з.п. ф-лы, 11 табл., 80 пр.
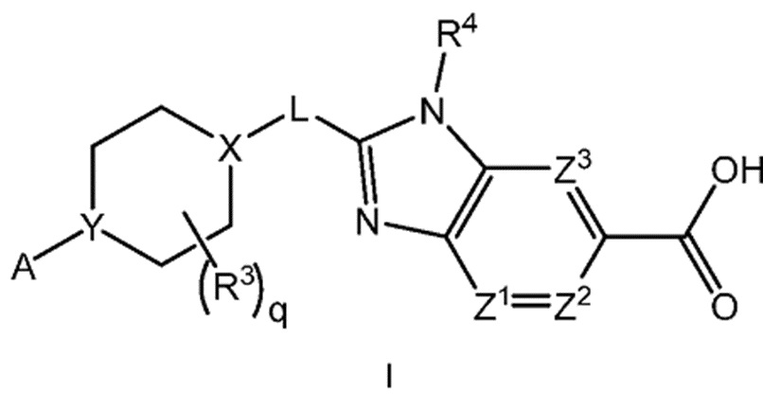
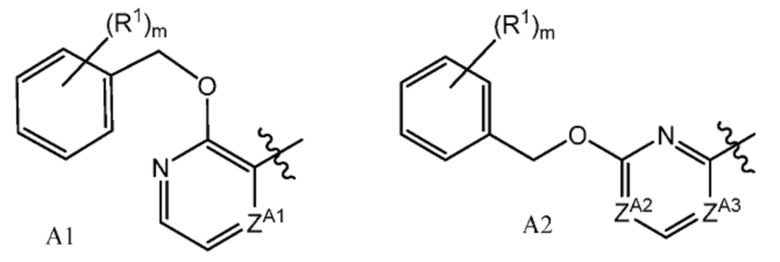
1. Соединение формулы I
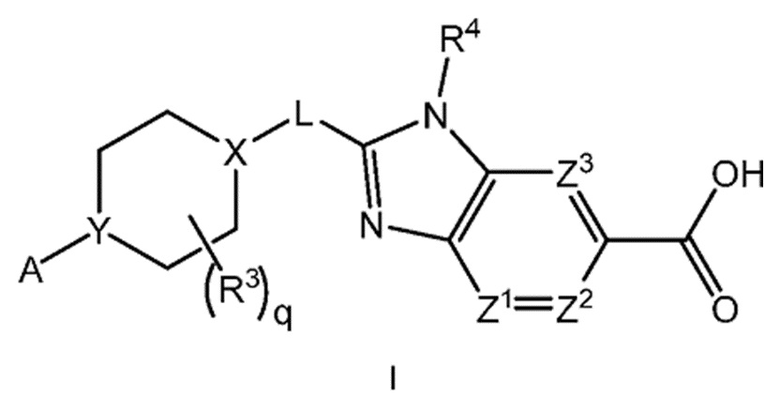
или его фармацевтически приемлемая соль, где А представляет собой A1 или A2
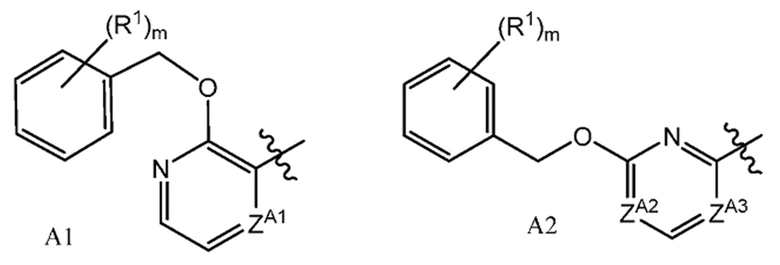
и где каждый R1 независимо представляет собой галоген или -CN;
m равно 0, 1, 2 или 3;
Х-L представляет собой N-CH2, CHCH2 или циклопропил;
Y представляет собой CH или N;
ZA1 представляет собой CH, CR2 или N;
ZA2 представляет собой CH, CR2 или N;
ZA3 представляет собой CH, CR2 или N; при условии, что ZA2 и ZA3 одновременно не являются N, а также дополнительно при условии, что один из ZA2 и ZA3 представляет собой N, когда Х-L представляет собой N-CH2;
каждый R2 представляет собой F;
каждый R3 независимо представляет собой -C1-3алкил;
q равно 0, 1 или 2;
R4 представляет собой -C1-3алкил, -C0-3алкилен-R5 или -C1-3алкилен-R6, где указанный алкил может быть замещенным, как допускает валентность, 0-3 заместителями, независимо выбранными из 0-3 атомов F, и 0-1 заместителем, выбранным из -C0-1алкилен-ORO;
R5 представляет собой 4-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и O, где указанный гетероциклоалкил может быть замещенным 0-1 оксо (=O);
R6 представляет собой 5-6-членный гетероарил, содержащий 2-3 гетероатома, независимо выбранных из N и O, где указанный гетероарил может быть замещенным 0-2 заместителями, как допускает валентность, независимо выбранными из 0-2 -C1-3алкилов;
каждый RO независимо представляет собой H или -C1-3алкил;
каждый из Z1, Z2 и Z3 представляет собой -CRZ или
один из Z1, Z2 и Z3 представляет собой N, а два другие представляют собой -CRZ; и
каждый RZ независимо представляет собой H.
2. Соединение по п. 1, которое представляет собой соединение формулы II
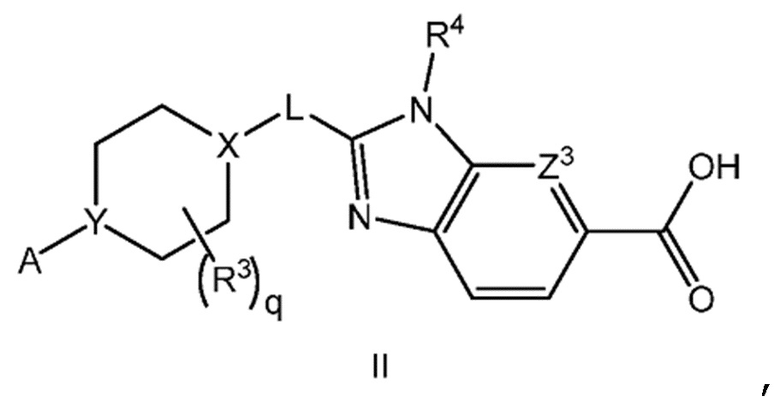
или его фармацевтически приемлемая соль, где A представляет собой A1 или A2
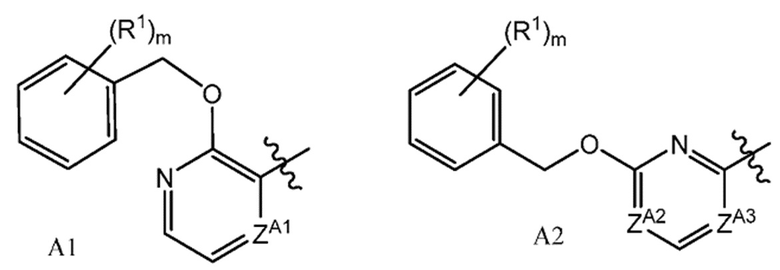
и где каждый R1 независимо представляет собой F, Cl или -CN; и
m равно 0, 1 или 2.
3. Соединение по п. 1, которое представляет собой соединение формулы III
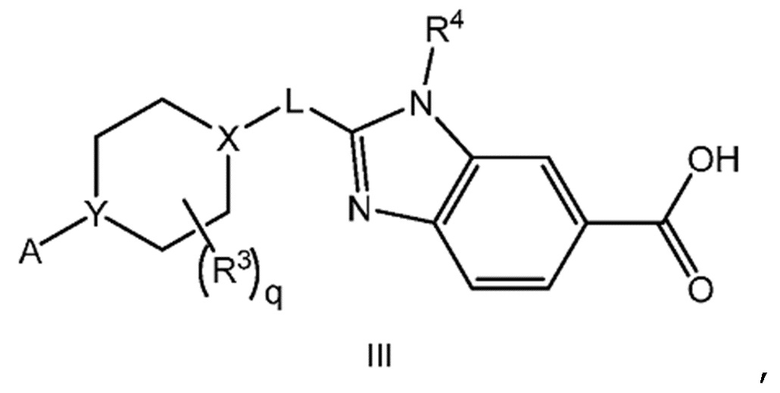
или его фармацевтически приемлемая соль, где A представляет собой A1 или A2
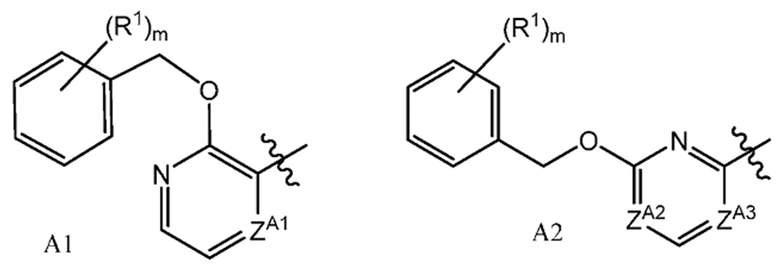
и где каждый R1 независимо представляет собой F, Cl или -CN; и
m равно 0, 1 или 2.
4. Соединение по любому из пп. 1-3, где A представляет собой A1, или его фармацевтически приемлемая соль.
5. Соединение по п. 4, которое представляет собой соединение формулы IV
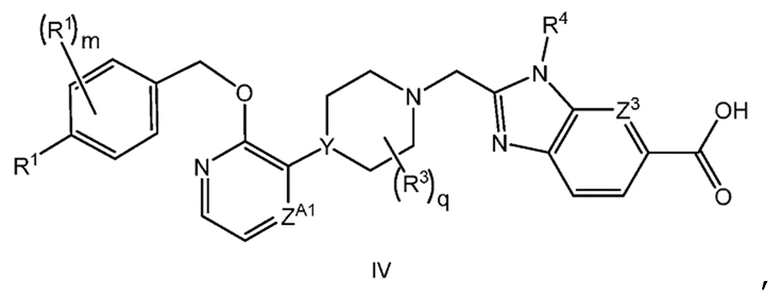
или его фармацевтически приемлемая соль, где
каждый R1 независимо представляет собой F, Cl или -CN;
m равно 0 или 1;
q равно 0 или 1; и
R3 представляет собой -CH3.
6. Соединение по п. 5, где ZA1 представляет собой CH или CR2; R2 представляет собой F; или его фармацевтически приемлемая соль.
7. Соединение по п. 5, где ZA1 представляет собой N, или его фармацевтически приемлемая соль.
8. Соединение по п. 1 или 2, где A представляет собой A2; q равно 0 или 1; и R3 представляет собой -CH3; или его фармацевтически приемлемая соль.
9. Соединение по п. 8, где ZA2 представляет собой CH или CR2; и ZA3 представляет собой N; или его фармацевтически приемлемая соль.
10. Соединение по п. 8, где ZA2 представляет собой N; и ZA3 представляет собой CH или CR2; или его фармацевтически приемлемая соль.
11. Соединение по любому из пп. 1, 9 или 10, которое представляет собой соединение формулы V
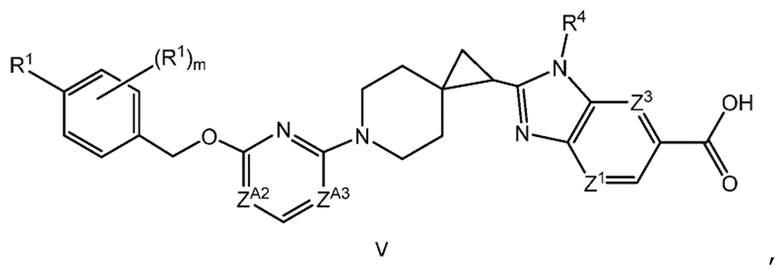
или его фармацевтически приемлемая соль, где каждый R1 независимо представляет собой F, Cl или -CN; m равно 0 или 1; ZA2 представляет собой CH, CR2 или N; ZA3 представляет собой CH, CR2 или N; при условии, что ZA2 и ZA3 одновременно не являются N; и каждый R2 представляет собой F.
12. Соединение по любому из пп. 1, 9 или 10, которое представляет собой соединение формулы VI
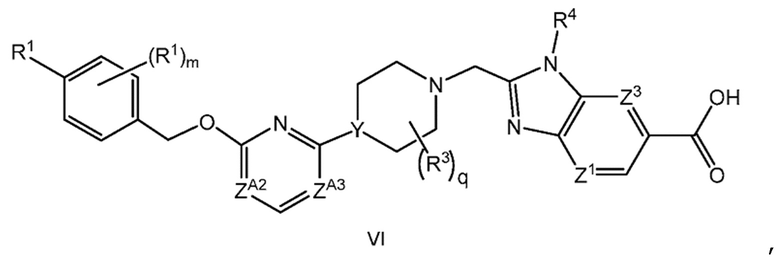
или его фармацевтически приемлемая соль, где
каждый R1 независимо представляет собой F, Cl или -CN; m равно 0 или 1; R3 представляет собой -CH3; q равно 0, 1 или 2; ZA2 представляет собой CH, CR2 или N; ZA3 представляет собой CH, CR2 или N; ZA2 и ZA3 одновременно не являются N; и дополнительно при условии, что один из ZA2 и ZA3 представляет собой N, когда Х-L представляет собой N-CH2; R2 представляет собой F; и Y представляет собой CH или N.
13. Соединение по любому из пп. 1-12, где R4 представляет собой -CH2-R5, где R5 представляет собой 4-5-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и O, где указанный гетероциклоалкил может быть замещенным 0-1 заместителем, выбранным из оксо (=O); или его фармацевтически приемлемая соль.
14. Соединение по любому из пп. 1-12, где R4 представляет собой -CH2-R6, где R6 представляет собой 5-членный гетероарил, содержащий 2-3 гетероатома, независимо выбранных из N и O, где указанный гетероарил может быть замещенным 0-2 заместителями, как допускает валентность, независимо выбранными из 0-1 -CH3 или -CH2CH3; или его фармацевтически приемлемая соль.
15. Соединение по любому из пп. 1-12, где R4 представляет собой -C1-3алкил, где указанный алкил может быть замещенным, как допускает валентность, 0-3 заместителями, независимо выбранными из 0-3 атомов F, и 0-1 заместителем, который представляет собой -C0-1алкилен-ORO.
16. Соединение, которое представляет собой
2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{2-[(4-циано-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{2-[(4-циано-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту; или
2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
или его фармацевтически приемлемая соль.
17. Соединение, которое представляет собой
2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту; или
2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
или его фармацевтически приемлемая соль.
18. Соединение, которое представляет собой
2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту; или
2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
или его фармацевтически приемлемая соль.
19. Фармацевтическая композиция, обладающая агонистической активностью в отношении GLP-1 рецептора, содержащая эффективное количество соединения по любому из пп. 1-18 или его фармацевтически приемлемой соли и фармацевтически приемлемый эксципиент.
20. Способ лечения кардиометаболического заболевания и связанных с ним заболеваний, для которых показан агонист GLP-1 рецептора, включающий введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по любому из пп. 1-18 или его фармацевтически приемлемой соли, где заболевание представляет собой диабет 1 типа (T1D), сахарный диабет 2 типа (T2DM), предиабет, идиопатический T1D, латентный аутоиммунный диабет у взрослых (LADA), юношеский Т2DM (EOD), атипичный диабет в молодом возрасте (YOAD), юношеский диабет взрослого типа (MODY), диабет, связанный с недоеданием, гестационный диабет, гипергликемию, инсулинорезистентность, печеночную инсулинорезистентность, нарушение толерантности к глюкозе, диабетическую невропатию, диабетическую нефропатию, заболевание почек, диабетическую ретинопатию, дисфункцию адипоцитов, отложение висцеральной жировой ткани, апноэ во сне, ожирение, расстройства пищевого поведения, увеличение массы тела при использовании других лекарственных средств, чрезмерное пристрастие к сахару, дислипидемию, гиперинсулинемию, неалкогольную жировую болезнь печени (НАЖБП), неалкогольный стеатогепатит (НАСГ), фиброз, цирроз, гепатоцеллюлярную карциному, сердечно-сосудистое заболевание, атеросклероз, ишемическую болезнь сердца, заболевание периферических сосудов, гипертензию, эндотелиальную дисфункцию, нарушение эластичности сосудов, застойную сердечную недостаточность, инфаркт миокарда, инсульт, геморрагический инсульт, ишемический инсульт, черепно-мозговую травму, легочную гипертензию, рестеноз после ангиопластики, перемежающуюся хромоту, постпрандиальную липемию, метаболический ацидоз, кетоз, артрит, остеопороз, болезнь Паркинсона, гипертрофию левого желудочка, заболевание периферических артерий, дегенерацию желтого пятна, катаракту, гломерулосклероз, хроническую почечную недостаточность, метаболический синдром, синдром X, предменструальный синдром, стенокардию, тромбоз, атеросклероз, преходящие ишемические атаки, рестеноз сосудов, нарушение метаболизма глюкозы, состояния нарушения уровня глюкозы в плазме крови натощак, гиперурикемию, подагру, эректильную дисфункцию, кожные заболевания и заболевания соединительной ткани, псориаз, изъязвления стопы, язвенный колит, гипер-апо В-липопротеинемию, болезнь Альцгеймера, шизофрению, нарушение когнитивных функций, воспалительное заболевание кишечника, синдром короткого кишечника, болезнь Крона, колит, синдром раздраженного кишечника, профилактику или лечение синдрома поликистозного яичника и лечение зависимости.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2006126122 A, 27.01.2008. | |||

