ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
В настоящем описании представлены 6-карбоновые кислоты бензимидазолов и 4-аза-, 5-аза-, 7-аза-, и 4,7-диазабензимидазолов в качестве агонистов GLP-1R, способы получения указанных соединений и способы, включающие введение указанных соединений нуждающемуся в этом млекопитающему.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Диабет является серьезной угрозой здоровью общества из-за его растущей распространенности и сопутствующих рисков для здоровья. Заболевание характеризуется высокими уровнями глюкозы в крови в результате нарушений выработки инсулина, действия инсулина или и того и другого. Выделяют две основные формы диабета: тип 1 и тип 2. Диабет 1 типа (T1D) развивается, когда иммунная система организма разрушает панкреатические бета-клетки, единственные клетки в организме, которые вырабатывают гормон инсулин, который регулирует уровень глюкозы в крови. Для выживания людям с диабетом 1 типа нужно вводить инсулин с помощью инъекции или насоса. Сахарный диабет 2 типа (обычно называемый T2DM) обычно начинается или с инсулиновой резистентности, или если существует недостаточная выработка инсулина для поддержания приемлемого уровня глюкозы.
В настоящее время существуют различные фармакологические подходы для лечения гипергликемии и впоследствии T2DM (Hampp, C. et al. Use of Antidiabetic Drugs in the U.S., 2003-2012, Diabetes Care 2014, 37, 1367-1374). Они могут быть сгруппированы в шесть основных классов, каждый из которых действует через различный первичный механизм: (A) Стимуляторы секреции инсулина, включающие сульфонилмочевины (например, глипизид, глимепирид, глибурид), меглитиниды (например, натеглидин, репаглинид), ингибиторы дипептидилпептидазы IV (DPP-IV) (например, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин, саксоглиптин) и агонисты рецептора глюкагоноподобного пептида-1 (GLP-1R) (например, лираглутид, албиглутид, эксенатид, ликсисенатид, дулаглутид, семаглутид), которые усиливают секрецию инсулина, воздействуя на панкреатические бета-клетки. Сульфонилмочевины и меглитиниды имеют ограниченную эффективность и переносимость, вызывают увеличение веса и часто индуцируют развитие гипогликемии. Ингибиторы DPP-IV имеют ограниченную эффективность. Имеющиеся в продаже агонисты GLP-1R представляют собой пептиды, вводимые путем подкожной инъекции. Лираглутид дополнительно одобрен для лечения ожирения. (B) Бигуаниды (например, метформин), как полагают, действуют главным образом за счет уменьшения образования глюкозы в печени. Бигуаниды часто вызывают желудочно-кишечные расстройства и лактоацидоз, что еще больше ограничивает их применение. (C) Ингибиторы альфа-глюкозидазы (например, акарбоза) снижают всасывание глюкозы в кишечнике. Эти вещества часто вызывают желудочно-кишечные расстройства. (D) Тиазолидиндионы (например, пиоглитазон, росиглитазон) действуют на специфический рецептор (гамма-рецептор, активируемый пролифератором пероксисомы) в печени, мышцах и жировых тканях. Они регулируют липидный обмен, впоследствии усиливая реакцию этих тканей на действие инсулина. Частое применение этих лекарственных средств может привести к увеличению веса и может индуцировать отек и анемию. (E) Инсулин используется в более тяжелых случаях, отдельно или в сочетании с вышеуказанными агентами, и частое использование также может привести к увеличению веса и несет риск гипогликемии. (F) Ингибиторы натрий-зависимого переносчика глюкозы 2 типа (SGLT2) (например, дапаглифлозин, эмпаглифлозин, канаглифлозин, эртуглифлозин) ингибируют реабсорбцию глюкозы в почках и тем самым снижают уровень глюкозы в крови. Этот новый класс лекарственных средств может быть связан с кетоацидозом и инфекциями мочевыводящих путей.
Однако, за исключением агонистов GLP-1R и ингибиторов SGLT2, лекарственные средства имеют ограниченную эффективность и не решают наиболее важные проблемы, снижение функции β-клеток и связанное с этим ожирение.
Ожирение является хроническим заболеванием, которое широко распространено в современном обществе и связано с многочисленными медицинскими проблемами, включая гипертензию, гиперхолестеринемию и ишемическую болезнь сердца. Кроме того, оно сильно коррелирует с T2DM и инсулинорезистентностью, последняя обычно сопровождается гиперинсулинемией или гипергликемией, или и тем и другим. Кроме того, T2DM связан с увеличением риска ишемической болезни сердца в 2-4 раза. В настоящее время единственным методом лечения ожирения с высокой эффективностью является бариатрическая хирургия, но это лечение является дорогостоящим и рискованным. Фармакологическое воздействие обычно менее эффективно и сопровождается побочными эффектами. Соответственно, существует очевидная потребность в более эффективном фармакологическом воздействии с меньшим количеством побочных эффектов и удобным введением.
Хотя T2DM чаще всего ассоциируется с гипергликемией и инсулинорезистентностью, другие заболевания, связанные с T2DM, включают инсулинорезистентность печени, нарушенную толерантность к глюкозе, диабетическую невропатию, диабетическую нефропатию, диабетическую ретинопатию, ожирение, дислипидемию, гипертензию, гиперинсулинемию и неалкогольную жировую болезнь печени (NAFLD).
NAFLD является печеночным проявлением метаболического синдрома и представляет собой спектр состояний печени, включающих стеатоз, неалкогольный стеатогепатит (NASH), фиброз, цирроз и, наконец, гепатоцеллюлярную карциному. NAFLD и NASH считаются основными жировыми заболеваниями печени, поскольку они характерны для наибольшей доли лиц с повышенным уровнем липидов в печени. Степень тяжести NAFLD/NASH зависит от наличия липидов, воспалительного клеточного инфильтрата, увеличения гепатоцитов и степени фиброза. Хотя не у всех индивидуумов стеатоз прогрессирует до NASH, у существенной части это происходит.
GLP-1 представляет собой гормон инкретин длиной 30 аминокислот, секретируемый L-клетками в кишечнике в ответ на прием пищи. Было показано, что GLP-1 стимулирует секрецию инсулина физиологическим и глюкозозависимым образом, уменьшает секрецию глюкагона, ингибирует опорожнение желудка, снижает аппетит и стимулирует пролиферацию бета-клеток. В неклинических экспериментах GLP-1 способствует сохранению компетентности бета-клеток путем стимуляции транскрипции генов, важных для глюкозозависимой секреции инсулина, и путем стимулирования неогенеза бета-клеток (Meier, et al. Biodrugs. 2003; 17 (2): 93-102).
У здорового индивидуума GLP-1 играет важную роль в регуляции уровня глюкозы в крови после приема пищи, стимулируя глюкозозависимую секрецию инсулина поджелудочной железой, что приводит к увеличению абсорбции глюкозы на периферии. GLP-1 также подавляет секрецию глюкагона, что приводит к снижению выработки глюкозы в печени. Кроме того, GLP-1 задерживает опорожнение желудка и замедляет моторику тонкого кишечника, замедляя всасывание пищи. У людей с T2DM нормальное повышение уровня GLP-1 после приема пищи отсутствует или снижено (Vilsboll T, et al. Diabetes. 2001. 50; 609-613).
Holst (Physiol. Rev. 2007, 87, 1409) и Meier (Nat. Rev. Endocrinol. 2012, 8, 728) описывают, что агонисты рецептора GLP-1, такие как GLP-1, лираглутид и эксендин-4, обладают тремя основными фармакологическими активностями для улучшения гликемического контроля у пациентов с T2DM за счет снижения уровня глюкозы натощак и после приема пищи (FPG и PPG): (i) повышенной глюкозозависимой секрецией инсулина (улучшенная первая и вторая фаза), (ii) активностью подавления глюкагона при гипергликемических состояниях, (iii) задержкой скорости опорожнения желудка, приводящей к замедленному всасыванию глюкозы, полученной из пищи.
Остается потребность в легко проводимой профилактике и/или лечении кардиометаболических и сопутствующих заболеваний.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям формулы I
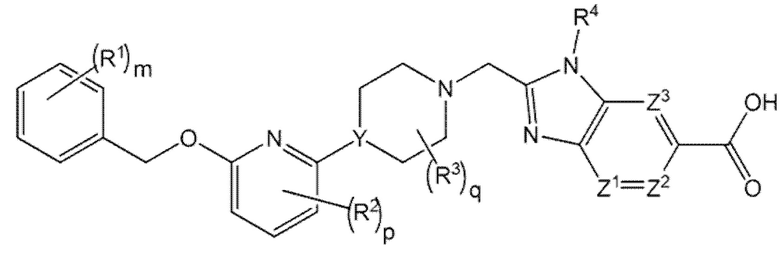
I
или к их фармацевтически приемлемой соли, где
каждый R1 независимо представляет собой галоген, -CN, -C1-3алкил или -OC1-3алкил, при этом алкил C1 3алкила и OC1-3алкила замещен 0-3 атомами F;
m равно 0, 1, 2 или 3;
каждый R2 независимо представляет собой F, Cl или -CN;
p равно 0, 1 или 2;
каждый R3 независимо представляет собой F, -OH, -CN, -C1-3алкил, -OC1-3алкил или -C3-4циклоалкил, или 2 R3 могут циклизоваться вместе с образованием -C3-4 спироциклоалкила, где алкил С1-3алкила и OC1-3алкила, циклоалкил или спироциклоалкил могут быть замещены в зависимости от валентности 0-3 атомами F и 0-1 -OH;
q равно 0, 1 или 2;
Y представляет собой CH или N;
R4 представляет собой -C1-3алкил, -C0-3алкилен-C3 6циклоалкил, -C0-3алкилен-R5, или C1 3алкилен R6, где указанный алкил может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из 0-3 атомов F и 0-1 заместителя, выбранного из C0-1алкилен CN, C0-1алкилен ORO и -N(RN)2, и
где указанные алкилен и циклоалкил могут быть независимо замещены в зависимости от валентности 0-2 заместителями, независимо выбранными из 0-2 атомов F и 0-1 заместителя, выбранного из C0-1алкилен CN, C0-1алкилен ORO, и -N(RN)2;
R5 представляет собой 4-6-членный гетероциклоалкил, где указанный гетероциклоалкил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-1 оксо (=O),
0-1 -CN,
0-2 атомов F, и
0-2 заместителями, независимо выбранными из -C1-3алкила и -OC1-3алкила, при этом алкил С1-3алкила и -OC1-3алкила может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-3 атомов F,
0-1 -CN и
0-1 -ORO;
R6 представляет собой 5-6-членный гетероарил, где указанный гетероарил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-2 галогенов,
0-1 заместителя, выбранного из -ORO и -N(RN)2, и
0-2 -C1-3алкилов, где алкил может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-3 атомов F, и
0-1 -ORO;
каждый RO независимо представляет собой H или -C1-3алкил, где C1-3алкил может быть замещен 0-3 атомами F;
каждый RN независимо представляет собой H или -C1-3 алкил;
Z1 представляет собой СН или N;
Z2 и Z3 каждый независимо представляет собой -CRZ или N, при условии, что когда Z1 или Z3 представляет собой N, Z2 представляет собой -CRZ; и
каждый RZ независимо представляет собой H, F, Cl или -CH3.
Другой вариант осуществления относится к соединениям формулы II
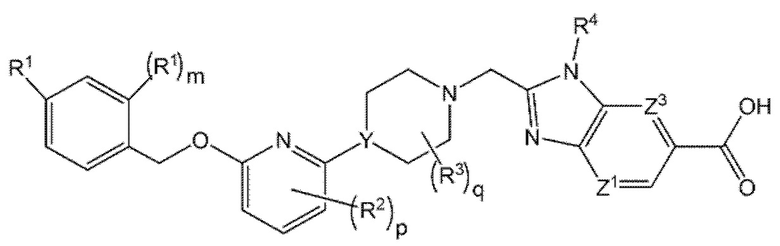
II
или к их фармацевтически приемлемой соли, где
m равно 0 или 1;
R2 представляет собой F;
р равно 0 или 1; и
q равно 0 или 1.
Другой вариант осуществления относится к соединениям формул I или II, где
m равно 0 или 1;
q равно 0 или 1; и
R3 представляет собой -F, -CH3, -CH2CH3, -CH2OH, -CF3, изопропил или циклопропил, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формулы III
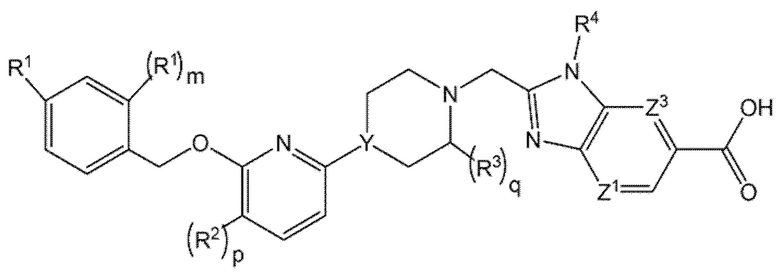
III
или к их фармацевтически приемлемой соли, где
m равно 0 или 1;
R2 представляет собой F;
р равно 0 или 1;
R3 представляет собой -C1-2алкил, где -C1-2алкил может быть замещен в зависимости от валентности 0-3 атомами F; и
q равно 0 или 1.
Другой вариант осуществления относится к соединениям формул I, II или III, где каждый R1 независимо представляет собой F, Cl, -CN, -CH3 или -CF3, или к их фармацевтически приемлемой соли.Другой вариант осуществления относится к соединениям формул I, II или III, где
R3 представляет собой -CH3;
q равно 0 или 1; и
R4 представляет собой -CH2CH2OCH3, C1-3алкилен-R5 или C1-3алкилен-R6, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II или III, где
R4 представляет собой -CH2-R5, где R5 представляет собой 4-5-членный гетероциклоалкил, где указанный гетероциклоалкил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-2 атомов F, и
0-1 заместителя, выбранного из -OCH3 и -CH2OCH3;
или к их фармацевтически приемлемой соли.
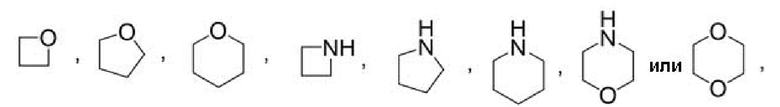
Другой вариант осуществления относится к соединениям формул I, II или III, где гетероциклоалкил представляет собой
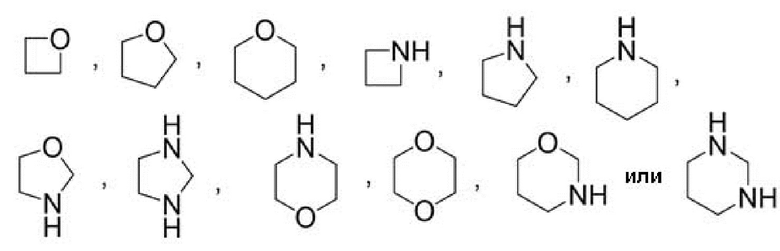
при этом гетероциклоалкил может быть замещен в зависимости от валентности 0-2 заместителями, например, замещающими водород, независимо выбранными из:
0-1 оксо (O=),
0-1 -CN,
0-2 атомов F, и
0-2 заместителями, независимо выбранными из -C1-3алкила и -OC1-3алкила, при этом алкил С1-3алкила и -OC1-3алкила могут быть независимо замещены в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-3 атомов F,
0-1 -CN и
0-1 -ORO,
или к их фармацевтически приемлемой соли.
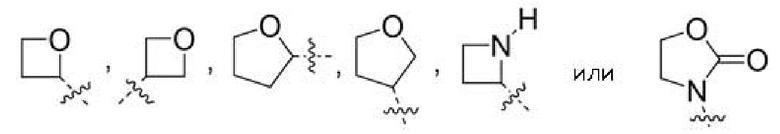
Другой вариант осуществления относится к соединениям формул I, II или III, где гетероциклоалкил представляет собой
при этом гетероциклоалкил может быть замещен в зависимости от валентности 0-2 заместителями, например, замещающими водород, независимо выбранными из:
0-1 -CN,
0-2 атомов F, и
0-2 заместителями, независимо выбранными из -C1-3алкила и -OC1-3алкила, при этом алкил С1-3алкила и -OC1-3алкила могут быть независимо замещены в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-3 атомов F,
0-1 -CN и
0-1 -ORO, или к их фармацевтически приемлемой соли.
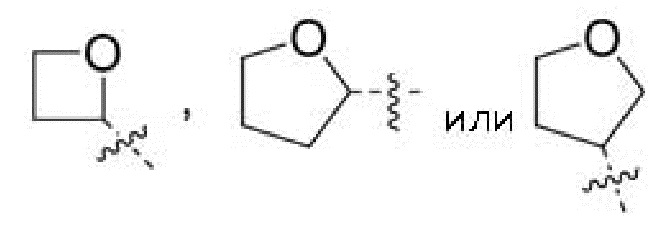
Другой вариант осуществления относится к соединениям формул I, II или III, где гетероциклоалкил представляет собой
при этом гетероциклоалкил может быть замещен в зависимости от валентности 0-1 заместителем, например, замещающим водород, выбранным из:
-CN,
атома F, и
0-1 заместителя, независимо выбранного из -C1-3алкила и -OC1-3алкила, при этом алкил С1-3алкила и -OC1-3алкила может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-3 атомов F,
0-1 -CN и
0-1 -ORO, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II или III, где гетероциклоалкил представляет собой
и при этом гетероциклоалкил может быть замещен в зависимости от валентности 0-1 заместителем, например, замещающим водород, выбранным из:
-CN,
атома F, и
0-1 заместителя, независимо выбранного из -C1-3алкила и -OC1-3алкила, при этом алкил С1-3алкила и -OC1-3алкила может быть замещен в зависимости от валентности 0-3 заместителями, включающими:
0-3 атомов F,
0-1 -CN или
0-1 -ORO, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II или III, где гетероциклоалкил представляет собой
и при этом гетероциклоалкил может быть замещен в зависимости от валентности 0-1 метилом, при этом указанный метил может быть замещен 0-3 атомами F, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, независимо выбранным из одного или любой комбинации следующего:
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2R)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-{[4-(6-{[(4-цианo-2-фторфенил)(метил-d2)]окси}пиридин-2-ил)пиперидин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота;
2-{[(2S)-4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(3R)-тетрагидрофуран-3-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(3R)-тетрагидрофуран-3-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(3S)-тетрагидрофуран-3-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианoбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианoбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианoбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2R)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2R)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(2,4-дифторбензил)окси]-5-фторпиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота;
2-{[(2S)-4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота; или
2-{[(2S)-4-{6-[(4-цианoбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединению, которое представляет собой 2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту, или к его фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединению, которое представляет собой 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту, или к его фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединению, которое представляет собой 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту, или к его фармацевтически приемлемой соли.
Другой вариант осуществления представляет собой трис-соль 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты.
Другой вариант осуществления представляет собой свободную кислоту 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты.
Другой вариант осуществления относится к соединению, которое представляет собой 2-{[4-(6-{[(4-цианo-2-фторфенил)(метил-d2)]окси}пиридин-2-ил)пиперидин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту, или к его фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединению, которое представляет собой 2-[(4-{6-[(4-цианo-2-фторбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту, или к его фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II или III, где
R4 представляет собой -CH2-R6, где R6 представляет собой 5-членный гетероарил, при этом указанный гетероарил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-2 галогенов, где галоген независимо выбран из F и Cl,
0-1 -OCH3, и
0-1 -CH3, -CH2CH3, -CF3 или -CH2CH2OCH3;
или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II или III, где гетероарил представляет собой
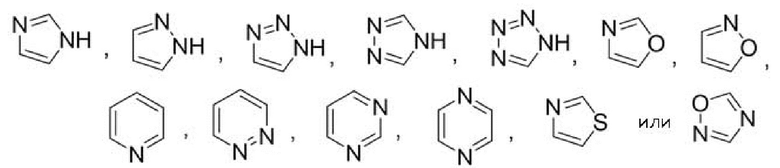
при этом указанный гетероарил может быть замещен в зависимости от валентности 0-2 заместителями, например, замещающими водород, независимо выбранными из:
0-2 галогенов, где галоген независимо выбран из F и Cl,
0-1 заместителя, выбранного из -ORO и -N(RN)2, или
0-2 -C1-3алкилов, где алкил может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-3 атомов F, и
0-1 -ORO;
или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II или III, где гетероарил представляет собой
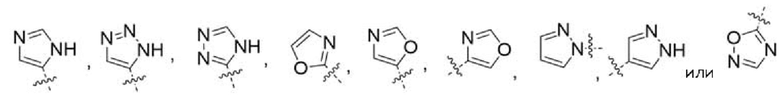
при этом указанный гетероарил может быть замещен в зависимости от валентности 0-2 заместителями, например, замещающими водород, независимо выбранными из:
0-2 галогенов, где галоген независимо выбран из F и Cl,
0-1 заместителя, выбранного из -ORO и -N(RN)2, или
0-2 -C1-3алкилов, где алкил может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-3 атомов F, и
0-1 -ORO;
или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям формул I, II или III, где гетероарил представляет собой
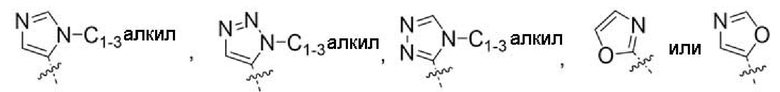
при этом C1-3 алкил на указанном гетероариле может быть замещен в зависимости от валентности 0-3 заместителями, например, замещающими водород, независимо выбранными из:
0-3 атомов F, и
0-1 -ORO;
или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, независимо выбранным из одного или любой комбинации следующего:
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-метил-1H-1,2,3-триазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-4-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(1-этил-1H-1,2,3-триазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,2-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,2-оксазол-3-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-этил-1H-1,2,3-триазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-(1,3-оксазол-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота; или
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-(1,3-оксазол-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота;
или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, независимо выбранным из одного или любой комбинации следующего:
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота; или
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям, независимо выбранным из одного или любой комбинации следующего:
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-7-фтор-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-7-фтор-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-цианoбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]-5-фторпиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{6-[(4-цианoбензил)окси]-5-фторпиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота; или
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-метоксициклобутил)метил]-1H-бензимидазол-6-карбоновая кислота;
или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления в данном описании, например, к соединениям формул I, II или III, где каждый из Z1, Z2 и Z3 представляет собой CRZ, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления в данном описании, например, к соединениям формул I, II или III, где RZ представляет собой H, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления в данном описании, например, к соединениям формул I, II или III, где каждый Z1, Z2 и Z3 представляет собой CH, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления в данном описании, например, к соединениям формул I, II или III, где р равно 0 или 1; и R2 представляет собой F.
Другой вариант осуществления относится к соединениям других вариантов осуществления в данном описании, например, к соединениям формул I, II или III, где R3 представляет собой CH3 или -CF3; и q равно 1, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления в данном описании, например, к соединениям формул I, II или III, где каждый R1 независимо представляет собой F, Cl или -CN; или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления в данном описании, например, к соединениям формул I, II или III, где R4 представляет собой -CH2-R5, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления в данном описании, например, к соединениям формул I, II или III, где R4 представляет собой -CH2-R6, или к их фармацевтически приемлемой соли.
Другой вариант осуществления относится к соединениям других вариантов осуществления в данном описании, например, к соединениям формул I, II или III, где соединение представляет собой свободную кислоту.
В другом варианте осуществления изобретение предлагает фармацевтическую композицию, содержащую соединение формул I, II, или III, или его фармацевтически приемлемую соль, как определено в любом из описанных здесь вариантов осуществления, в смеси с по меньшей мере одним фармацевтически приемлемым эксципиентом.
Изобретение также включает в себя следующие варианты осуществления:
соединение формул I, II или III или его фармацевтически приемлемая соль, как определено в любом из описанных здесь вариантов осуществления, для применения в качестве лекарственного средства;
соединение формул I, II или III или его фармацевтически приемлемая соль, как определено в любом из описанных здесь вариантов осуществления, для использования в профилактике и/или лечении кардиометаболических и сопутствующих заболеваний, обсуждаемых в настоящей заявке, включая T2DM, преддиабет, NASH и сердечно-сосудистое заболевание;
способ лечения заболевания, для которого показан агонист GLP-1R, у субъекта, нуждающегося в такой профилактике и/или лечении, включающий введение субъекту терапевтически эффективного количества соединения формул I, II или III, или его фармацевтически приемлемой соли, как определено в любом из описанных здесь вариантов осуществления;
применение соединения формул I, II или III, или его фармацевтически приемлемой соли, как определено в любом из описанных здесь вариантов осуществления, для производства лекарственного средства для лечения заболевания или состояния, для которых показан агонист GLP-1R;
соединение формул I, II или III или его фармацевтически приемлемая соль, как определено в любом из описанных здесь вариантов осуществления, для применения при лечении заболевания или состояния, для которых показан агонист GLP-1R; или же
фармацевтическая композиция для лечения заболевания или состояния, для которого показан агонист GLP-1R, содержащая соединение формул I, II или III или его фармацевтически приемлемую соль, как определено в любом из описанных здесь вариантов осуществления.
Соединение каждого примера или его фармацевтически приемлемая соль могут быть заявлены отдельно или сгруппированы вместе в любой комбинации с любым числом всех без исключения описанных здесь вариантов осуществления.
Изобретение также относится к фармацевтической композиции, содержащей соединение формул I, II или III или его фармацевтически приемлемую соль, как определено в любом из описанных здесь вариантов осуществления, для использования в профилактике и/или лечении кардиометаболических и сопутствующих заболеваний, обсуждаемых в настоящей заявке, включая T2DM, преддиабет, NASH и сердечно-сосудистое заболевание.
Другой вариант осуществления изобретения относится к соединению формул I, II, или III, или к его фармацевтически приемлемой соли, как определено в любом из описанных здесь вариантов осуществления, для использования в лечении и/или профилактике кардиометаболических и сопутствующих заболеваний, включая диабет (T1D и/или T2DM, включая преддиабет), идиопатический T1D (тип 1b), латентный аутоиммунный диабет у взрослых (LADA), T2DM с ранним началом (EOD), атипичный диабет молодого возраста (YOAD), диабет взрослого типа у молодых (MODY), диабет, связанный с недостаточностью питания, гестационный диабет, гипергликемию, инсулинорезистентность, инсулинорезистентность печени, нарушенную толерантность к глюкозе, диабетическую невропатию, диабетическую нефропатию, заболевание почек (например, острая почечная недостаточность, дисфункция канальцев, провоспалительные изменения проксимальных канальцев), диабетическую ретинопатию, дисфункцию адипоцитов, висцеральное жироотложение, апноэ во сне, ожирение (включая гипоталамическое ожирение и моногенное ожирение) и связанные с ним сопутствующие заболевания (например, остеоартрит и недержание мочи), расстройства пищевого поведения (включая синдром компульсивного переедания, нервную булимию и синдромное ожирение, например, синдромы Прадера-Вилли и Бардета-Бидля), увеличение массы тела от применения других средств (например, от применения стероидов и антипсихотических средств), чрезмерную тягу к сахару, дислипидемию (включая гиперлипидемию, гипертриглицеридемию, повышение уровня общего холестерина, высокий уровень холестерина ЛПНП и низкий уровень холестерина ЛПВП), гиперинсулинемию, NAFLD (включая сопутствующие заболевания, такие как стеатоз, NASH, фиброз, цирроз и гепатоцеллюлярную карциному),сердечно-сосудистые заболевания, атеросклероз (включая ишемическую болезнь сердца), заболевание периферических сосудов, гипертензию, эндотелиальную дисфункцию, уменьшенную эластичность сосудов, застойную сердечную недостаточность, инфаркт миокарда (например, некроз и апоптоз), инсульт, геморрагический инсульт, ишемический инсульт, травматическое повреждение головного мозга, легочную гипертензию, рестеноз после ангиопластики, перемежающуюся хромоту, постпрандиальную липемию, метаболический ацидоз, кетоз, артрит, остеопороз, болезнь Паркинсона, гипертрофию левого желудочка, периферическую артериальную болезнь, дегенерацию желтого пятна, катаракту, гломерулосклероз, хроническую почечную недостаточность, метаболический синдром, синдром X, предменструальный синдром, стенокардию, тромбоз, атеросклероз, преходящие ишемические приступы, сосудистый рестеноз, нарушенный метаболизм глюкозы, состояния нарушенного уровня глюкозы натощак, гиперурикемию, подагру, эректильную дисфункцию, заболевания кожи и соединительных тканей, псориаз, изъязвление стопы, язвенный колит, гипер-апо-B-липопротеинемию, болезнь Альцгеймера, шизофрению, ухудшение когнитивной деятельности, воспалительное заболевание кишечника, синдром укороченной тонкой кишки, болезнь Крона, колит, синдром раздраженного кишечника, профилактику или лечение синдрома поликистоза яичников и лечение зависимости (например, злоупотребления алкоголем и/или наркотиками).
В данном описании используются следующие сокращения:
Термин «алкил», используемый в настоящем документе, означает одновалентную углеводородную группу формулы -CnH(2n+1) с прямой или разветвленной цепью. Неограничивающие примеры включают метил, этил, пропил, бутил, 2-метилпропил, 1,1- диметилэтил, пентил и гексил.
Термин «алкилен», используемый в настоящем документе, означает двухвалентную углеводородную группу формулы -CnH2n- с прямой или разветвленной цепью. Неограничивающие примеры включают этилен и пропилен.
Термин «циклоалкил», используемый в настоящем документе, означает циклическую одновалентную углеводородную группу формулы -CnH(2n-1), содержащую по меньшей мере три атома углерода. Неограничивающие примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
Термин «галоген», используемый в настоящем документе, относится к, фториду, хлориду, бромиду или йодиду.
Термин «гетероциклоалкил», используемый в настоящем документе, относится к циклоалкильной группе, в которой одна или более кольцевых метиленовых групп (-CH2-) были замещены группой, выбранной из -O-, -S- или азота, при этом азот может обеспечивать место присоединения или может быть замещен, как предусмотрено в каждом варианте осуществления. Когда азот обеспечивает место присоединения, структурный рисунок гетероциклоалкила будет содержать водород на указанном азоте. Как правило, гетероциклоалкил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из оксо, -CN, галогена, алкила и -Oалкила, и алкил может быть дополнительно замещен. Следует отметить, что когда имеет место 0 замещение, гетероциклоалкил является незамещенным.
Термин «гетероарил», используемый в настоящем документе, относится к моноциклическому ароматическому углеводороду, содержащему от 5 до 6 атомов углерода, в котором по меньшей мере один из атомов углерода кольца замещен гетероатомом, выбранным из кислорода, азота и серы. Такая гетероарильная группа может быть присоединена через атом углерода кольца или, где позволяет валентность, через атом азота кольца. Как правило, гетероарил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из галогена, OH, алкила, O-алкила и амино (например, NH2, NHалкила, N(алкил)2), и алкил может быть дополнительно замещен. Следует отметить, что когда имеет место 0 замещение, гетероарил является незамещенным.
Комнатная температура: КТ.
Метанол: MeOH.
Этанол: EtOH.
Изопропанол: iPrOH.
Этилацетат: EtOAc.
Тетрагидрофуран: THF.
Толуол: PhCH3.
Карбонат цезия: Cs2CO3.
Бис(триметилсилил)амид лития: LiHMDS.
Трет-бутоксид натрия: NaOtBu.
Трет-бутоксид калия: KOtBu.
Диизопропиламид лития: LDA.
Триэтиламин: Et3N.
N,N-диизопропилэтиламин: DIPEA.
Карбонат калия: K2CO3.
Диметилформамид: DMF.
Диметилацетамид: DMAc.
Диметилсульфоксид: ДМСО.
N-Метил-2-пирролидинон: NMP.
Гидрид натрия: NaH.
Трифторуксусная кислота: TFA.
Трифторуксусный ангидрид: TFAA.
Уксусный ангидрид: Ac2O.
Дихлорметан: DCM.
1,2-Дихлорэтан: DCE.
Соляная кислота: HCl.
1,8-Диазабицикло[5.4.0]ундец-7-ен: DBU.
Комплекс боран-диметилсульфид: BH3-DMS.
Комплекс боран-тетрагидрофуран: BH3-THF.
Алюмогидрид лития: LAH.
Уксусная кислота: AcOH.
Ацетонитрил: MeCN.
п-Толуолсульфоновая кислота: pTSA.
Дибензилиденацетон: DBA.
2,2′-Бис(дифенилфосфино)-1,1′-бинафталин: BINAP.
1,1′-Ферроцендиил-бис(дифенилфосфин): dppf.
1,3-Бис(дифенилфосфино)пропан: DPPP.
3-Хлорпербензойная кислота: m-CPBA.
Трет-бутилметиловый простой эфир: MTBE.
Метансульфонил: Ms.
N-метилпирролидинон: NMP.
Тонкослойная хроматография: ТСХ.
Сверхкритическая флюидная хроматография: СФХ.
4-(Диметиламино)пиридин: DMAP.
Трет-бутилоксикарбонил: Boc.
1-[Бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний-3-оксидгексафторфосфат: HATU.
Простой петролейный эфир: PE.
2-(1H-Бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат: HBTU.
2-Амино-2-(гидроксиметил)пропан-1,3-диол: трис.
Трис(дибензилиденацетон)дипалладий: Pd2(dba)3
Спектры 1Н ядерного магнитного резонанса (ЯМР) во всех случаях соответствовали предложенным структурам. Характерные химические сдвиги (δ) приведены в миллионных долях относительно остаточного сигнала протона в дейтерированном растворителе (CHCl3 при 7,27 м.д.; CD2HOD при 3,31 м.д.; MeCN при 1,94 м.д.; ДМСО при 2,50 м.д.) и представлены с использованием обычных сокращений для обозначения главных пиков: например, с - синглет; д - дублет; т - триплет; кв - квартет; м - мультиплет; ушир. - уширенный. 1H ЯМР спектры были получены при напряженности поля 400 МГц или 600 МГц, если не указано иное.
В настоящем описании волнистая линия 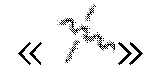 обозначает место присоединения заместителя к другой группе.
обозначает место присоединения заместителя к другой группе.
Описанные ниже соединения и промежуточные соединения были названы с использованием соглашения о названиях в программе ChemBioDraw Ultra, версия 13.0 (CambridgeSoft Corp., Кембридж, Массачусетс) или ACD/Labs, версия 12 (Advanced Chemistry Development, Inc., Торонто, Онтарио). Соглашения о названиях, используемые в ChemBioDraw Ultra версии 13.0 и ACD/Labs версии 12, хорошо известны специалистам в данной области техники и считается, что соглашения о названиях, предусмотренные в ChemBioDraw Ultra версии 13.0 и ACD/Labs версии 12, в целом соответствуют рекомендациям IUPAC (Международного союза теоретической и прикладной химии), как определено правилами Nomenclature of Organic Chemistry и CAS Index. Следует отметить, что химические названия могут иметь только круглые скобки или могут иметь круглые скобки и квадратные скобки. Стереохимические дескрипторы также могут располагаться в разных местах внутри самого наименования, в зависимости от соглашения о названиях. Специалисту в данной области известны эти варианты форматирования и должно быть понятно, что они относятся к одной и той же химической структуре.
Фармацевтически приемлемые соли соединений формулы I включают кислотно-аддитивные и основные соли.
Подходящие кислотно-аддитивные соли образуются из кислот, которые образуют нетоксичные соли. Примеры солей включают соли ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, цикламат, эдизилат, эзилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат, соли 1,5-нафталиндисульфоновой кислоты и ксинафоат.
Подходящие основные соли образуются из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, бис-(2-гидроксиэтил)амина (диоламина), глицина, лизина, магния, меглумина, 2-аминоэтанола (оламина), калия, натрия, 2-амино-2-(гидроксиметил)пропан-1,3-диола (трис или трометамин) и цинка.
Могут также образовываться полусоли кислот и оснований, например, полусульфаты и полукальциевые соли. Для обзора подходящих солей см. Handbook of Pharmaceutical Salts: Properties, Selection, and Use, by Stahl and Wermuth (Wiley-VCH, 2002).
Фармацевтически приемлемые соли соединений формулы (I) можно получить одним или более из трех способов:
(i) взаимодействием соединения формулы I с желаемой кислотой или основанием;
(ii) путем удаления лабильной в кислотных или щелочных условиях защитной группы из подходящего предшественника соединения формулы I или путем раскрытия кольца подходящего циклического предшественника, например лактона или лактама, с использованием желаемой кислоты или основания; или же
(iii) путем превращения одной соли соединения формулы I в другую реакцией с подходящей кислотой или основанием или с помощью подходящей ионообменной колонки.
Все три реакции обычно проводятся в растворе. Полученная соль может выпадать в осадок и может быть собрана фильтрованием или может быть выделена выпариванием растворителя. Степень ионизации полученной соли может меняться от полностью ионизированной до почти неионизированной.
Соединения формулы I и их фармацевтически приемлемые соли могут существовать в несольватированных и сольватированных формах. Термин «сольват» употребляется в настоящем документе для описания молекулярного комплекса, содержащего соединение формулы I или его фармацевтически приемлемую соль и молекулы одного или более фармацевтически приемлемых растворителей, например, этанола. Термин «гидрат» используется, когда указанным растворителем является вода.
Принятая в настоящее время система классификации для органических гидратов представляет собой систему, которая определяет гидраты с изолированным участком, канальные гидраты или гидраты, координируемые ионом металла - см. Polymorphism in Pharmaceutical Solids, K. R. Morris (Ed. H. G. Brittain, Marcel Dekker, 1995). Гидраты с изолированным участком представляют собой гидраты, в которых молекулы воды изолированы от прямого контакта друг с другом посредством промежуточных органических молекул. В канальных гидратах молекулы воды лежат в каналах решетки, где они находятся рядом с другими молекулами воды. В гидратах, координированных ионами металлов, молекулы воды связаны с ионом металла.
Когда растворитель или вода прочно связаны, комплекс может иметь хорошо определенную стехиометрию, независимо от влажности. Однако, когда растворитель или вода слабо связаны, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя может зависеть от влажности и условий сушки. В таких случаях отсутствие стехиометрии будет нормой.
Также в объем изобретения включены многокомпонентные комплексы (отличные от солей и сольватов), где лекарственное средство и по меньшей мере один другой компонент представлены в стехиометрических или нестехиометрических количествах. Комплексы этого типа включают клатраты (комплексы включения лекарственное средство-хозяин) и сокристаллы. Последние обычно определяют как кристаллические комплексы нейтральных молекулярных компонентов, которые связаны друг с другом через нековалентные взаимодействия, но они также могут быть комплексом нейтральной молекулы с солью. Сокристаллы могут быть получены кристаллизацией из расплава или перекристаллизацией из растворителей или физическим измельчением компонентов друг с другом - см. в Chem Commun, 17, 1889-1896, О.Almarsson и M. J. Zaworotko (2004). Что касается общего обзора многокомпонентных комплексов, - см. J Pharm Sci, 64 (8), 1269-1288, Haleblian (август 1975).
Соединения по изобретению могут существовать в континууме твердых состояний от полностью аморфного до полностью кристаллического. Термин «аморфный» относится к состоянию, в котором вещество не имеет дальнего порядка на молекулярном уровне и, в зависимости от температуры, может проявлять физические свойства твердого вещества или жидкости. Обычно такие материалы не дают четких рентгендифракционных картин, и, несмотря на то, что они демонстрируют свойства твердого вещества, их более формально описывают как жидкость. При нагревании происходит изменение от свойств твердого вещества к свойствам жидкости, что обычно характеризуется фазовым переходом второго рода («стеклование»). Термин «кристаллический» относится к твердой фазе, в которой вещество имеет регулярную упорядоченную внутреннюю структуру на молекулярном уровне и дает характерную рентгеновскую дифрактограмму с определенными пиками. Такие материалы при достаточном нагревании также будут демонстрировать свойства жидкости, но изменение от твердого состояния к жидкому характеризуется фазовым переходом первого рода («температура плавления»).
Соединения формулы (I) также могут существовать в мезоморфном состоянии (мезофаза или жидкий кристалл) при воздействии подходящих условий. Мезоморфное состояние является промежуточным между истинным кристаллическим состоянием и истинным жидким состоянием (либо расплав, либо раствор). Мезоморфизм, появляющийся в результате изменения температуры, описывают как «термотропный», и мезоморфизм, появляющийся в результате добавления второго компонента, такого как вода или другой растворитель, описывают как «лиотропный». Соединения, которые имеют возможность образовывать лиотропные мезофазы, описываются как «амфифильные» и содержат молекулы, которые имеют ионную (например, -COO-Na+, -COO-K+ или -SO3-Na+) или неионную (например, -N-N+(CH3)3) полярную головную группу. Дополнительную информацию см. в Crystals and the Polarizing Microscope by N. H. Hartshorne and A. Stuart, 4th Edition (Edward Arnold, 1970).
Соединения формулы I могут проявлять полиморфизм и/или один или более видов изомерии (например, оптическую, геометрическую или таутомерную изомерию). Соединения формулы I также могут быть изотопно-меченными. Такое изменение неявно относится к соединениям формулы I, определенным со ссылкой на их структурные особенности и, следовательно, находящимся в пределах объема изобретения.
Соединения формулы I, содержащие один или более асимметричных атомов углерода, могут существовать в виде двух или более стереоизомеров. Если соединение формулы I содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс (или Z/E) изомеры. Когда структурные изомеры являются взаимопревращаемыми за счет низкого энергетического барьера, может иметь место таутомерная изомерия («таутомерия»). Она может принимать форму протонной таутомерии в соединениях формулы I, содержащих, например, имино, кето или оксимную группу; или так называемой валентной таутомерии в соединениях, которые содержат ароматический фрагмент. Это означает, что одно соединение может проявлять более одного типа изомерии.
Фармацевтически приемлемые соли соединений формулы I также могут содержать противоион, который является оптически активным (например, d-лактат или l-лизин) или рацемическим (например, dl-тартрат или dl-аргинин).
Цис/транс изомеры могут быть разделены обычными методами, хорошо известными специалистам в данной области, например хроматографией и фракционной кристаллизацией.
Традиционные методы получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) используя, например, хиральную жидкостную хроматографию высокого давления (HPLC, ВЭЖХ). В качестве альтернативы, рацемат (или рацемический предшественник) может взаимодействовать с подходящим оптически активным соединением, например, спиртом, или в случае, когда соединение формулы I содержит кислотный или основной фрагмент, основанием или кислотой, такими как 1-фенилэтиламин или винная кислота. Полученная диастереомерная смесь может быть разделена хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера могут быть превращены в соответствующий чистый энантиомер (энантиомеры) с помощью методов, хорошо известных специалисту. Хиральные соединения формулы I (и их хиральные предшественники) могут быть получены в энантиомерно обогащенной форме с использованием хроматографии, обычно ВЭЖХ, на асимметрической смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащей 0-50 об.% изопропанола, обычно 2-20%, и 0-5 об.% алкиламина, обычно 0,1% диэтиламина. Концентрирование элюата позволяет получить обогащенную смесь. Может быть использована хиральная хроматография с использованием суб- и сверхкритических текучих сред. Способы хиральной хроматографии, используемые в некоторых вариантах осуществления настоящего изобретения, известны в данной области (см., например, Smith, Roger M., Loughborough University, Loughborough, UK; Chromatographic Science Series (1998), 75 (Supercritical Fluid Chromatography with Packed Columns), pp. 223-249 и приведенные там ссылки). В некоторых приведенных соответствующих примерах колонки получали от Chiral Technologies, Inc, Вест Честер, Пенсильвания, США, дочерней компании Daicel® Chemical Industries, Ltd., Токио, Япония.
При кристаллизации любого рацемата возможны кристаллы двух различных типов. Первый тип представляет собой указанное выше рацемическое соединение (истинный рацемат), в котором образуется одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, в котором две формы кристаллов образуются в эквимолярных количествах, каждая из которых содержит единственный энантиомер. Хотя обе кристаллические формы, присутствующие в рацемической смеси, имеют идентичные физические свойства, они могут иметь отличающиеся физические свойства по сравнению с истинным рацематом. Рацемические смеси могут быть разделены традиционными методами, известными специалистам в данной области - см., например, Stereochemistry of Organic Compounds, E. L. Eliel и S. H. Wilen (Wiley, 1994).
Следует подчеркнуть, что хотя соединения формулы I представлены в настоящем описании в виде единственной таутомерной формы, все возможные таутомерные формы находятся в пределах объема данного изобретения.
Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченные соединения формулы I, в которых один или более атомов замещены атомами, имеющими тот же самый атомный номер, но атомную массу или массовое число, отличающиеся от атомной массы или массового числа, которое преобладает в природе.
Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2H и 3H, углерод, такой как 11C, 13C и 14C, хлор, такой как 36Cl, фтор, такой как 18F, йод, такой как 123I и 125I, азот, такой как 13N и 15N, кислород, такой как 15O, 17O и 18O, фосфор, такой как 32P, и серу, такую как 35S.
Некоторые изотопно-меченные соединения формулы I, например, соединения, которые включают радиоактивный изотоп, можно использовать при исследованиях распределения лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы трития, т.е. 3Н, и углерод-14, т.е. 14C, особенно подходят для этой цели из-за простоты их включения и легкости способов обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2Н, может дать определенные терапевтические преимущества в результате большей метаболической стабильности, например, увеличенного периода полураспада in vivo или снижение требуемых дозировок.
Замещение позитрон-излучающими изотопами, такими как 11C, 18F, 15O и 13N, может быть полезным для позитронно-эмиссионной томографии (PET) для исследования заполнения рецептора субстратом.
Изотопно-меченные соединения формулы (I) обычно могут быть получены традиционными методами, известными специалистам в данной области техники, или способами, аналогичными описанным в прилагаемых примерах и способах получения с использованием подходящего изотопно-меченного реагента вместо применявшегося ранее немеченного реагента.
Фармацевтически приемлемые сольваты в соответствии с изобретением включают сольваты, в которых растворитель кристаллизации может быть может быть изотопно-замещенным, например, D2O, d6-ацетон, d6-ДМСО.
Одним из способов осуществления изобретения является введение соединения формулы I в форме пролекарства. Таким образом, некоторые производные соединения формулы I, которые могут иметь небольшую фармакологическую активность или вообще не иметь ее, при введении внутрь или применении на теле могут превращаться в соединение формулы I, обладающее желаемой активностью, например, путем гидролитического расщепления, особенно гидролитического расщепления, стимулируемого ферментом эстеразой или пептидазой. Такие производные называются «пролекарствами». Дополнительную информацию об использовании пролекарств можно найти в «Pro-drugs as Novel Delivery Systems», Vol. 14, ACS Symposium Series (T. Higuchi, W. Stella) и «Bioreversible Carriers in Drug Design», Pergamon Press, 1987 (Ed. E. B. Roche, American Pharmaceutical Association). Также можно сделать ссылку на Nature Reviews/Drug Discovery, 2008, 7, 355 и Current Opinion in Drug Discovery and Development, 2007, 10, 550.
Пролекарства в соответствии с изобретением могут, например, быть получены замещением соответствующих функциональных групп, присутствующих в соединениях формулы I, на определенные фрагменты, известные специалистам в данной области как «профрагменты», как описано, например, H. Bundgaard в «Design of Prodrugs» (Elsevier, 1985), а также Y. M. Choi-Sledeski, C. G. Wermuth, «Designing Prodrugs and Bioprecursors» в Practice of Medicinal Chemistry, (Fourth Edition), Chapter 28, 657-696 (Elsevier, 2015).
Таким образом, пролекарство в соответствии с изобретением представляет собой (а) сложноэфирное или амидное производное карбоновой кислоты в соединении формулы I; (b) сложноэфирное, карбонатное, карбаматное, фосфатное или простое эфирное производное гидроксильной группы в соединении формулы I; (c) амидное, иминовое, карбаматное или аминовое производное аминогруппы в соединении формулы I; (d) оксимное или иминовое производное карбонильной группы в соединении формулы I; или (е) метильную, первичную спиртовую или альдегидную группу, которая может быть метаболически окислена до карбоновой кислоты в соединении формулы I.
Некоторые конкретные примеры пролекарств в соответствии с изобретением включают:
(i) когда соединение формулы I содержит функциональную группу карбоновой кислоты (-COOH), - ее сложный эфир, как, например, соединение, в котором водород функциональной группы карбоновой кислоты соединения формулы I замещен C1-C8-алкилом (например, этилом) или (C1-C8-алкилом)C (=O)OCH2- (например, tBuC(=O) OCH2-);
(ii) когда соединение формулы I содержит спиртовую функциональную группу (-ОН), - ее сложный эфир, как, например, соединение, в котором водород спиртовой функциональной группы соединения формулы I замещен -CO(C1-C8 алкилом) (например, метилкарбонилом) или спирт этерифицирован аминокислотой;
(iii) когда соединение формулы I содержит спиртовую функциональную группу (-ОН), - ее простой эфир, как, например, соединение, в котором водород спиртовой функциональной группы соединения формулы I замещен (C1-C8 алкилом)C(=O)OCH2- или -CH2OP(=O)(OH)2;
(iv) когда соединение формулы I содержит спиртовую функциональную группу (-ОН), - ее фосфат, как, например, соединение, в котором водород спиртовой функциональной группы соединения формулы I замещен -P(=O)(OH)2 или -P(=O)(ONa)2 или -P(=O)(O-)2Ca2+;
(v) когда соединение формулы I содержит первичную или вторичную аминогруппу (-NH2 или -NHR, где R ≠ H), - ее амид, например, соединение, в котором, в соответствующих случаях, один или оба атома водорода аминогруппы соединения формулы I замещены (C1-C10) алканоилом, -COCH2NH2 или аминогруппа дериватизирована аминокислотой;
(vi) когда соединение формулы I содержит первичную или вторичную аминогруппу (-NH2 или -NHR, где R ≠ H), - ее амин, например, соединение, в котором, в соответствующих случаях, один или оба атома водорода аминогруппы соединения формулы I замещены -CH2OP(=O)(OH)2;
(vii) когда группа карбоновой кислоты в соединении формулы I замещена метильной группой, -CH2OH группой или альдегидной группой.
Некоторые соединения формулы I могут сами выступать в качестве пролекарств других соединений формулы I. Также возможно, чтобы два соединения формулы I были соединены вместе в форме пролекарства. В определенных обстоятельствах пролекарство соединения формулы I может быть создано путем внутреннего связывания двух функциональных групп в соединении формулы I, например, путем образования лактона.
Ссылки на соединения формулы I следует понимать как включающие в себя сами соединения и их пролекарства. Изобретение включает такие соединения формулы I, а также фармацевтически приемлемые соли таких соединений и фармацевтически приемлемые сольваты указанных соединений и солей.
Введение и дозирование
Как правило, соединение по изобретению вводится в количестве, эффективном для лечения состояния, как описано в настоящем документе. Соединения по изобретению можно вводить как соединение per se или, в качестве альтернативы, в виде фармацевтически приемлемой соли. Для целей введения и дозирования, соединение per se или его фармацевтически приемлемая соль будут просто называться соединениями по изобретению.
Соединения по изобретению вводят любым подходящим путем в форме фармацевтической композиции, адаптированной к такому пути, и в дозе, эффективной для предполагаемого лечения. Соединения по изобретению можно вводить перорально, ректально, вагинально, парентерально или местно.
Соединения по изобретению можно вводить перорально. Пероральное введение может включать глотание, благодаря чему соединение попадает в желудочно-кишечный тракт, или может применяться трансбуккальное или сублингвальное введение, при котором соединение поступает в кровоток непосредственно из полости рта.
В другом варианте осуществления соединения настоящего изобретения можно также вводить непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенное, внутриартериальное, интраперитонеальное, интратекальное, интравентрикулярное, интрауретральное, интрастернальное, интракраниальное, внутримышечное и подкожное введение. Подходящие устройства для парентерального введения включают игольные (включая микроигольные) инъекторы, безыгольные инъекторы и инфузионное оборудование.
В другом варианте осуществления соединения настоящего изобретения можно также вводить местно на кожу или слизистую оболочку, т.е. дермально или трансдермально. В другом варианте осуществления соединения по изобретению можно также вводить интраназально или с помощью ингаляции. В другом варианте осуществления соединения по изобретению можно вводить ректально или вагинально. В другом варианте осуществления соединения по изобретению можно также вводить непосредственно в глаз или ухо.
Схема дозирования соединений по изобретению и/или композиций, содержащих указанные соединения, основана на ряде факторов, включающих тип, возраст, вес, пол и медицинское состояние пациента; тяжесть состояния; способ введения; и активность конкретного применяемого соединения. Таким образом, режим дозирования может варьировать в широких пределах. В одном варианте осуществления общая суточная доза соединения по изобретению обычно составляет от примерно 0,001 мг/кг до примерно 100 мг/кг (т.е. мг соединения по изобретению на кг массы тела) для лечения указанных в настоящем описании состояний. В другом варианте осуществления общая суточная доза соединения по изобретению составляет от примерно 0,01 мг/кг до примерно 30 мг/кг, и в другом варианте осуществления от примерно 0,03 мг/кг до примерно 10 мг/кг, и в еще одном варианте осуществления, от примерно 0,1 мг/кг до примерно 3 мг/кг. Нередко введение соединений по изобретению повторяют несколько раз в день (обычно не более 4 раз). Для увеличения общей суточной дозы, если требуется, можно использовать многократные суточные дозы.
Для перорального введения композиции могут быть представлены в форме таблеток, содержащих 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 30,0 50,0, 75,0, 100, 125, 150, 175, 200, 250 и 500 мг активного ингредиента, для симптоматического регулирования дозирования для пациента. Лекарственное средство обычно содержит от примерно 0,01 мг до примерно 500 мг активного ингредиента или, в другом варианте осуществления, от примерно 1 мг до примерно 100 мг активного ингредиента. Для внутривенного введения дозы могут находиться в диапазоне от примерно 0,01 до примерно 10 мг/кг/мин при постоянной скорости инфузии.
Подходящие субъекты в соответствии с изобретением включают в себя субъекты млекопитающих. В одном варианте осуществления подходящими субъектами являются люди. Субъекты-люди могут быть любого пола и на любой стадии развития.
Фармацевтические композиции
В другом варианте осуществления изобретение включает фармацевтические композиции. Такие фармацевтические композиции содержат соединение по изобретению, представленное фармацевтически приемлемым носителем. Другие фармакологически активные вещества также могут присутствовать. Используемый в настоящем документе термин «фармацевтически приемлемый носитель» включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства, средства для поддержания изотоничности и средства, задерживающие поглощение, и тому подобное, которые являются физиологически совместимыми. Примеры фармацевтически приемлемых носителей включают одно или более из воды, физиологического раствора, фосфатно-солевого буферного раствора, декстрозы, глицерина, этанола и тому подобного, а также их комбинации, и могут включать изотонические агенты, например, сахара, хлорид натрия или многоатомные спирты, такие как маннит или сорбит в композиции. Фармацевтически приемлемые вещества, такие как увлажняющие вещества или незначительные количества вспомогательных веществ, таких как увлажняющие или эмульгирующие вещества, консерванты или буферы, которые увеличивают срок годности или эффективность антитела или части антитела.
Композиции данного изобретения могут быть в различных формах. Они включают, например, жидкие, полутвердые и твердые лекарственные формы, такие как жидкие растворы (например, растворы для инъекций и инфузий), дисперсии или суспензии, таблетки, пилюли, порошки, липосомы и суппозитории. Форма зависит от предполагаемого способа введения и терапевтического применения.
Типичные композиции находятся в форме растворов для инъекций или инфузий, таких как композиции, аналогичные тем, которые используются для пассивной иммунизации человека антителами в целом. Одним из способов введения является парентеральный (например, внутривенный, подкожный, интраперитонеальный, внутримышечный). В другом варианте осуществления антитело вводят посредством внутривенной инфузии или инъекции. В еще одном варианте осуществления антитело вводят путем внутримышечной или подкожной инъекции.
Пероральное введение твердой дозированной формы может быть, например, представлено в виде дискретных единиц, таких как твердые или мягкие капсулы, пилюли, крахмальные капсулы, пастилки или таблетки, каждая из которых содержит заранее определенное количество по меньшей мере одного соединения по изобретению. В другом варианте осуществления пероральное введение может осуществляться в виде порошка или гранул. В другом варианте осуществления пероральная дозированная форма является сублингвальной, как, например, пастилка. В таких твердых лекарственных формах соединения формулы I обычно объединяют с одним или несколькими вспомогательными веществами. Такие капсулы или таблетки могут содержать лекарственную форму с контролируемым высвобождением. В случае капсул, таблеток и пилюль, лекарственные формы также могут содержать буферные вещества или могут быть получены с энтеросолюбильными покрытиями.
В другом варианте осуществления пероральное введение может осуществляться в жидкой дозированной форме. Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно используемые в данной области (например, воду). Такие композиции также могут содержать вспомогательные вещества, такие как увлажняющие, эмульгирующие, суспендирующие, вкусо-ароматические (например, подстастители) и/или ароматизирующие добавки.
В другом варианте осуществления изобретение включает парентеральную дозированную форму. «Парентеральное введение» включает, например, подкожные инъекции, внутривенные инъекции, интраперитонеальные инъекции, внутримышечные инъекции, интрастернальные инъекции и инфузию. Препараты для инъекций (т.е. вводимые посредством инъекций стерильные водные или масляные суспензии), можно приготовить известным способом, используя подходящие диспергирующие, увлажняющие и/или суспендирующие вещества.
В другом варианте осуществления изобретение включает дозированную форму для местного применения. «Местное применение» включает, например, трансдермальное введение, например, посредством трансдермальных пластырей или устройств для ионтофореза, внутриглазное введение или интраназальное или ингаляционное введение. Композиции для местного применения также включают, например, гели для местного применения, спреи, мази и кремы. Лекарственная форма для местного применения может включать соединение, которое усиливает абсорбцию или проникновение активного ингредиента через кожу или другие области воздействия. Когда соединения данного изобретения вводят с помощью трансдермального устройства, введение будет осуществляться с использованием пластыря в виде резервуара и пористой мембраны или в виде различных твердых матриц. Типичные лекарственные формы для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, кожные пластыри, капсулы, имплантаты, губки, волокна, бандажи и микроэмульсии. Также можно использовать липосомы. Типичные носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены усилители проникновения - см., например, B. C. Finnin, T. M. Morgan, J. Pharm. Sci., vol. 88, pp. 955-958, 1999.
Лекарственные формы, подходящие для местного введения в глаза, включают, например, глазные капли, при этом соединение настоящего изобретения растворено или суспендировано в подходящем носителе. Типичная лекарственная форма, подходящая для глазного или ушного введения, может быть в виде капель микронизированной суспензии или раствора в изотоническом стерильном физиологическом растворе с отрегулированным рН. Другие лекарственные формы, подходящие для глазного и ушного введения, включают мази, биоразлагаемые (т.е. абсорбируемые гелевые губки, коллаген) и небиоразлагаемые (т.е. силикон) имплантаты, капсулы, линзы и системы микрочастиц или везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например, геллановая камедь, может быть включен вместе с консервантом, таким как бензалконийхлорид. Такие лекарственные формы также могут доставляться ионтофорезом.
Для интраназального введения или введения путем ингаляции, соединения по изобретению обычно доставляются в форме раствора или суспензии из баллончика-пульверизатора, который впрыскивается или нагнетается пациентом, или в виде аэрозольного спрея из контейнера под давлением или небулайзера, с использованием подходящего пропеллента. Лекарственные формы, подходящие для интраназального введения, обычно вводят в виде сухого порошка (или отдельно, в виде смеси, например, сухой смеси с лактозой, или как смесь частиц, например, смешанных с фосфолипидами, такими как фосфатидилхолин) из порошкового ингалятора, или в виде аэрозольного спрея из контейнера под давлением, насоса, разбрызгивателя, аэроионизатора (предпочтительно, аэроионизатора с использованием электрогидродинамики для получения мелкодисперсного тумана), или небулайзера, с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Для интраназального применения порошок может содержать биоадгезивный агент, например, хитозан или циклодекстрин.
В другом варианте осуществления изобретение включает дозированную форму для ректального введения. Такая дозированная форма для ректального введения может быть в виде, например, суппозитория. Масло какао является традиционной основой суппозиториев, но в соответствующих случаях можно использовать различные альтернативы.
Также могут использоваться и другие материалы носителя и способы введения, известные в области фармацевтики. Фармацевтические композиции по изобретению могут быть получены с помощью любой из хорошо известных фармацевтических методик, таких как эффективные способы получения и введения. Приведенные выше соображения относительно эффективных способов получения и введения хорошо известны в данной области и описаны в стандартных учебниках. Технология получения лекарственных средств рассмотрена, например, в Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999.
Совместное введение
Соединения по изобретению могут использоваться отдельно или в сочетании с другими терапевтическими средствами. Изобретение касается любого из применений, способов или составов, определенных в настоящем документе, в котором соединение любого приведенного здесь варианта осуществления формулы I или его фармацевтически приемлемая соль, или фармацевтически приемлемый сольват указанного соединения или соли используются в сочетании с одним или несколькими рассматриваемыми здесь другими терапевтическими средствами.
Введение двух или более соединений «в сочетании» означает, что все соединения вводятся достаточно близко по времени, так что каждое из них может вызвать биологический эффект в течение одного и того же интервала времени. Присутствие одного агента может изменять биологические эффекты другого соединения (соединений). Два или более соединения могут вводиться одновременно, совместно или последовательно. В дополнение к этому, одновременное введение может осуществляться путем смешивания соединений перед введением или с помощью введения соединений в один и тот же момент времени, но в виде разных лекарственных форм в одном и том же или разных местах введения.
Выражения «сопутствующее введение», «совместное введение», «одновременное введение» и «вводится одновременно» означают, что соединения вводятся в сочетании.
В другом варианте осуществления изобретение относится к способам лечения, которые включают введение соединений настоящего изобретения в сочетании с одним или более другими фармацевтическими средствами, где одно или более других фармацевтических средств могут быть выбраны из рассматриваемых в данном описании средств.
В одном варианте осуществления соединения данного изобретения вводят с противодиабетическим средством, включающим, без ограничения, бигуанид (например, метформин), сульфонилмочевину (например, толбутамид, глибенкламид, гликлазид, хлорпропамид, толазамид, ацетогексамид, гликлопирамид, глимепирид или глипизид), тиазолидиндион (например, пиоглитазон, росиглитазон или лобеглитазон), глитазар (например, сароглитазар, алеглитазар, мураглитазар или тезаглитазар), меглитинид (например, натеглинид, репаглинид), ингибитор дипептидилпептидазы 4 (DPP-4) (например, ситаглиптин, вилдаглиптин, саксаглиптин, линаглиптин, гемиглиптин, анаглиптин, тенелиглиптин, алоглиптин, трелаглиптин, дутоглиптин или омариглиптин), глитазон (например, пиоглитазон, росиглитазон, балаглитазон, ривоглитазон или лобеглитазон), ингибитор натрий-зависимого переносчика глюкозы 2 типа (SGLT2) (например, эмпаглифлозин, канаглифлозин, дапаглифлозин, ипраглифлозин, лпраглифлозин, тофоглифлозин, этабонат серглифлозина, этабонат ремоглифлозина или эртуглифлозин), ингибитор SGLTL1, агонист GPR40 (агонист FFAR1/FFA1, например, фасиглифам), глюкозозависимый инсулинотропный пептид (GIP) и его аналоги, ингибитор альфа-глюкозидазы (например, воглибозу, акарбозу или миглитол) или инсулин или аналог инсулина, включая фармацевтически приемлемые соли специально названных средств и фармацевтически приемлемые сольваты указанных средств и солей.
В другом варианте осуществления соединения настоящего изобретения вводят со средством против ожирения, включающим, без ограничения, пептид YY или его аналог, агонист рецептора нейропептида Y 2 типа (NPYR2), антагонист NPYR1 или NPYR5, антагонист каннабиноидного рецептора 1 типа (CB1R), ингибитор липазы (например, орлистат), проостровковый пептид человека (HIP), агонист рецептора меланокортина 4 (например, сетмеланотид), антагонист рецептора меланин-концентрирующего гормона 1, агонист фарнезоидного X рецептора (FXR) (например, обетихолевая кислота), зонизамид, фентермин (отдельно или в сочетании с топираматом), ингибитор обратного захвата норэпинефрина/дофамина (например, бупропион), антагонист опиоидного рецептора (например, налтрексон), сочетание ингибитора обратного захвата норэпинефрина/дофамина и антагониста опиоидного рецептора (например, сочетание бупропиона и налтрексона), аналог GDF-15, сибутрамин, агонист холецистокинина, амилин и его аналоги (например, прамлинтид), лептин и его аналоги (например, метролептин), серотонинергическое средство (например, лоркасерин), ингибитор метионинаминопептидазы 2 (MetAP2) (например, белораниб или ZGN-1061), фендиметразин, диэтилпропион, бензфетамин, ингибитор SGLT2 (например, эмпаглифлозин, канаглифлозин, дапаглифлозин, ипраглифлозин, лпраглифлозин, тофоглифлозин, этабонат серглифлозина, этабонат ремоглифлозина или эртуглифлозин), ингибитор SGLTL1, двойной ингибитор SGLT2/SGLT1, модулятор рецептора фактора роста фибробластов (FGFR), активатор AMP-активированной протеинкиназы (AMPK), биотин, модулятор рецептора MAS или агонист рецептора глюкагона (один или в сочетании с другим агонистом GLP-1R, например, лираглутидом, эксенатидом, дулаглутидом, албиглутидом, ликсисенатидом или семаглутидом), включая фармацевтически приемлемые соли специально названных средств и фармацевтически приемлемые сольваты указанных средств и солей.
В другом варианте осуществления соединения настоящего изобретения вводят со средством для лечения NASH, включающим, без ограничения, PF-05221304, агонист FXR (например, обетихолевую кислоту), агонист PPAR α/δ (например, элафибранор), конъюгат синтетических жирных кислот-желчной кислоты (например, арамхол), ингибитор каспазы (например, эмриказан), моноклональное антитело против гомолога лизилоксидазы 2 (LOXL2) (например, симтузумаб), ингибитор галектина 3 (например, GR-MD-02), ингибитор MAPK5 (например, GS-4997), двойной антагонист хемокинового рецептора 2 (CCR2) и CCR5 (например, ценикривирок), агонист фактора роста фибробластов 21 (FGF21) (например, BMS-986036), антагонист рецепторов лейкотриенов D4 (LTD4) (например, типелукаст), аналог ниацина (например, ARI 3037MO), ингибитор ASBT (например, воликсибат), ингибитор ацетил-КоА-карбоксилазы (ACC) (например, NDI 010976), ингибитор кетогексокиназы (KHK), ингибитор диацилглицерилацилтрансферазы 2 (DGAT2), антагонист рецептора CB1, антитело против CB1R или ингибитор киназы-1, регулирующей апоптозный сигнал (ASK1), включая фармацевтически приемлемые соли специально названных средств и фармацевтически приемлемые сольваты указанных средств и солей.
Эти агенты и соединения по изобретению можно комбинировать с фармацевтически приемлемыми носителями, такими как физиологический раствор, раствор Рингера, раствор декстрозы и тому подобное. Конкретный режим дозирования, т.е. доза, время и кратность, будет зависеть от конкретного индивидуума и его истории болезни.
Приемлемые носители, эксципиенты или стабилизаторы являются нетоксичными для реципиентов в используемых дозах и концентрациях, и могут включать буферы, такие как фосфатный, цитратный и другие буферы органических кислот; соли, такие как хлорид натрия; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензилхлорид аммония; гексаметонийхлорид; бензалконийхлорид, бензэтонийхлорид; феноловый, бутиловый или бензиловый спирт; алкилпарабены, такие как метилпарабен или пропилпарабен; катехин; резорцин; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее чем из приблизительно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как ЭДТА; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы металлов (например, Zn-белковые комплексы); и/или неионогенные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (PEG).
Липосомы, содержащие эти агенты и/или соединения по изобретению, получают способами, известными в данной области, например, описанными в патентах US №№ 4485045 и 4544545. Липосомы с увеличенным временем циркуляции описаны в патенте US № 5013556. Особенно пригодные липосомы можно получить методом обращенно-фазового выпаривания со смесью липидов, содержащей фосфатидилхолин, холестерин и PEG-дериватизированный фосфатидилэтаноламин (PEG-PE). Липосомы экструдируют через фильтры с определенным размером пор для получения липосом желаемого диаметра.
Эти вещества и/или соединения данного изобретения также могут быть заключены в микрокапсулы, приготовленные, например, способами коацервации или полимеризации на границе фаз, например, микрокапсулы из гидроксиметилцеллюлозы или желатиновые микрокапсулы и микрокапсулы из поли(метилметакрилата), соответственно, в коллоидные системы доставки лекарственных веществ (например, липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие способы описаны в Remington, The Science and Practice of Pharmacy, 20th Ed., Mack Publishing (2000).
Можно использовать препараты с замедленным высвобождением. Подходящие примеры препаратов с замедленным высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие соединение формул I, II или III, где матрицы находятся в форме профилированных изделий, например, пленок или микрокапсул. Примеры матриц для замедленного высвобождения включают сложные полиэфиры, гидрогели (например, поли-(2-гидроксиэтилметакрилат) или поливиниловый спирт)), полилактиды (патент US 3773919), сополимеры L-глутаминовой кислоты и 7-этил-L-глутамата, недеградируемый этиленвинилацетат, деградируемые сополимеры молочной кислоты и гликолевой кислоты, такие как используются в LUPRON DEPOT™ (инъецируемые микросферы, состоящие из сополимера молочной кислоты и гликолевой кислоты и ацетата леупролида), ацетат-изобутират сахарозы и поли-D-(-)-3-гидроксимасляную кислоту.
Лекарственные формы, используемые для внутривенного введения, должны быть стерильными. Это легко осуществляется, например, фильтрованием через мембраны для стерилизующего фильтрования. Соединения данного изобретения обычно помещают в емкость, имеющую стерильное входное отверстие, например, пакет для внутривенного раствора или флакон, имеющий пробку, прокалываемую иглой для подкожной инъекции.
Подходящие эмульсии могут быть получены с использованием коммерчески доступных жировых эмульсий, таких как Intralipid™, Liposyn™, Infonutrol™, Lipofundin™ и Lipiphysan™. Активный ингредиент может быть либо растворен в предварительно смешанной эмульсионной композиции, либо, в качестве альтернативы, его можно растворить в масле (например, соевом масле, сафлоровом масле, хлопковом масле, кунжутном масле, кукурузном масле или миндальном масле) и образованную эмульсию затем смешать с фосфолипидом (например, яичными фосфолипидами, соевыми фосфолипидами или соевым лецитином) и водой. Следует иметь в виду, что могут быть добавлены и другие ингредиенты, например, глицерин или глюкоза, для регулирования тоничности эмульсии. Подходящие эмульсии обычно содержат до 20% масла, например, 5-20%. Эмульсия жира может содержать капельки жира, имеющие размер 0,1-1,0 мкм, в частности 0,1-0,5 мкм, и иметь рН в диапазоне 5,5-8,0.
Эмульсионные композиции могут быть композициями, полученными путем смешивания соединения по изобретению с Intralipid™ или его компонентами (соевое масло, яичные фосфолипиды, глицерин и вода).
Композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях или их смесях, а также порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые эксципиенты, указанные выше. В некоторых вариантах осуществления композиции вводят пероральным или назальным респираторным путем для местного или системного действия. Композиции в предпочтительно стерильных фармацевтически приемлемых растворителях можно распылять при помощи газов. Распыляемые растворы могут вдыхаться непосредственно из распыляющего устройства, или распыляющее устройство может быть присоединено к надеваемой на лицо маске, палатке или аппарату с перемежающимся положительным давлением. Композиции в виде растворов, суспензий или порошков можно вводить предпочтительно перорально или назально из устройств, которые доставляют готовую лекарственную форму подходящим образом.
Наборы
Другой аспект изобретения относится к наборам, содержащим соединение формул I, II, или III, или фармацевтические композиции, содержащие соединение формул I, II или III изобретения. Набор может включать, в дополнение к соединению формул I, II или III по изобретению или его фармацевтической композиции, диагностические или терапевтические средства. Набор может также включать инструкции по применению в диагностическом или терапевтическом способе. В некоторых вариантах осуществления набор включает соединение формул I, II или III или его фармацевтическую композицию и диагностическое средство. В других вариантах осуществления набор включает соединение формул I, II или III или его фармацевтическую композицию.
В еще одном варианте осуществления изобретение включает наборы, которые подходят для использования при осуществлении способов лечения, описанных в настоящей заявке. В одном варианте осуществления набор содержит первую лекарственную форму, содержащую одно или более соединений по изобретению в количествах, достаточных для осуществления способов изобретения. В другом варианте осуществления набор включает одно или более соединений по изобретению в количествах, достаточных для осуществления способов изобретения, а также контейнер для дозирования.
ПОЛУЧЕНИЕ
Соединения формул I, II или III могут быть получены общими и специальными способами, описанными ниже, используя общеизвестные сведения для специалиста в области химии органического синтеза. Такие общеизвестные сведения можно найти в стандартных справочниках, таких как Comprehensive Organic Chemistry, Ed. Barton and Ollis, Elsevier; Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Larock, John Wiley and Sons; и Compendium of Organic Synthetic Methods, Vol. I-XII (опубликовано Wiley-Interscience). Используемые здесь исходные материалы являются коммерчески доступными или могут быть получены обычными способами, известными в данной области.
При получении соединений формул I, II или III следует отметить, что в некоторых из описанных здесь способов получения может требоваться защита отдаленной функциональной группы (например, первичного амина, вторичного амина, карбоксила в предшественниках формулы I). Потребность в такой защите будет меняться в зависимости от природы отдаленной функциональной группы и условий способов получения. Необходимость такой защиты легко определяется специалистом в данной области. Применение таких способов защиты/снятия защиты также известно специалистам в данной области. Общее описание защитных групп и их использования см. в T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Например, некоторые соединения содержат функциональные группы первичных аминов или карбоновых кислот, которые могут мешать реакциям в других местах молекулы, если они остаются незащищенными. Соответственно, такие функциональные группы могут быть защищены соответствующей защитной группой, которая может быть удалена на последующей стадии. Подходящие защитные группы для защиты амина и карбоновой кислоты включают защитные группы, обычно используемые в синтезе пептидов (такие как N-трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz) и 9-флуоренилметиленоксикарбонил (Fmoc) для аминов и сложные эфиры низших алкилов или бензилов для карбоновых кислот), которые обычно не являются химически реакционноспособными в описанных условиях реакции и обычно могут быть удалены без химического изменения других функциональных групп в соединениях формулы I.
Описанные ниже схемы предназначены для общего описания методологии, применяемой при получении соединений настоящего изобретения. Некоторые из соединений настоящего изобретения могут содержать один или несколько хиральных центров со стереохимическим обозначением (R) или (S). Специалисту в данной области техники будет очевидно, что все синтетические превращения могут быть проведены одинаковым образом, независимо от того, являются ли материалы энантиообогащенными или рацемическими. Кроме того, разделение на желаемый оптически активный материал может происходить в любой желаемой точке последовательности с использованием хорошо известных способов, описанных здесь и в химической литературе.
На следующих схемах переменные Y, Z1, Z2, Z3, R1, R2, R3, R4, m, p и q являются такими, как описано в настоящем документе для соединений формул I, II или III, если не указано иное. Для схем, представленных ниже, каждый X1, X2, X3 и X4 может быть независимо удаляемой группой, такой как любой алкил или арилсульфонат (например, мезилат, тозилат или трифлат), или галогеном, или любой другой группой, которая может быть замещена амином или использована в реакции сочетания, опосредованной металлом. Х4 также может представлять собой защищенную карбоновую кислоту (т.е. сложный эфир). Когда защитная группа идентифицируется как Pg1, она может быть алкиламиновой защитной группой, такой как бензил, бензгидрил или тому подобное; карбаматной защитной группой, такой как Boc, Cbz или тому подобное; или амидной защитной группой, такой как трифторацетамид. Когда защитная группа идентифицируется как Pg2, она может быть кислотной защитной группой, такой как метил, этил, бензил, трет-бутил или тому подобное. R4a представляет собой C1-2алкил, C0-2алкилен-C3 6циклоалкил, C0-2алкилен-R5 или C1-2алкилен-R6, где указанный алкил, алкилен или циклоалкил может быть независимо замещен, в зависимости от валентности, 0-3 атомами F, и 0-1 заместителем, независимо выбранным из C0-1алкилен ORO и -N(RN)2.
Замещенный пиридин 6 может быть получен, как показано на схеме 1. 2,6-дигалопиридин (1, синтезированный или приобретаемый коммерчески) может вступать в реакцию с замещенной бороновой кислотой или сложным боронатным эфиром (2) в присутствии палладиевого катализатора и лигандного комплекса по реакции Сузуки (Maluenda and Navarro, Molecules, 2015, 20, 7528-7557) с получением соединений общей формулы 3. Для достижения наилучших результатов в реакции Сузуки галоген Х2 предпочтительно представляет собой Cl, Br или I. Восстановление олефина с получением соединений общей структуры 4 будет проводиться в атмосфере водорода (15-100 фунт/кв. дюйм H2 (0,10-0,69 МПа)) в спиртовом растворителе, таком как MeOH или EtOH, или, в качестве альтернативы, апротонном органическом растворителе, таком как EtOAc или THF, в присутствии подходящего катализатора, такого как палладий на угле, Pd(OH)2 на угле (катализатор Перлмана) или PtO2 (катализатор Адамса). В качестве альтернативы, восстановление может осуществляться с альтернативными способами, известными специалистам в данной области техники, с использованием таких реагентов, как триэтилсилан или другие силаны, в условиях кислотного или металлического катализа, или металлических восстановителей, таких как магний или подобные. В качестве альтернативы, олефин может быть функционализирован известными специалисту способами для введения R3 групп. Например, олефин может быть гидроборирован с получением спирта, который может быть алкилирован или дополнительно превращен в нитрильную, F или алкильную группу. Превращение в соединения общей структуры 5 может быть осуществлено таким способом, как C-O сочетание Бухвальда-Хартвига (Lundgren and Stradiotto, Aldrich Chimica Acta, 2012, 45, 59-65) между соединениями общей структуры 4 и соответствующим образом замещенным бензиловым спиртом в присутствии палладиевого или медного катализатора и лигандного комплекса. Предпочтительным галогеном X1 является Cl. Эти реакции обычно проводятся при температуре 0-110 °C в апротонных органических растворителях, таких как, без ограничения, 1,4-диоксан и PhCH3, с добавлением основания, такого как Cs2CO3, LiHMDS или NaOtBu. В качестве альтернативы, реакция 4 с соответствующим образом замещенным бензиловым спиртом в апротонном растворителе, таком как DMF или THF, в присутствии сильного основания, такого как NaH, KOtBu или LiHMDS, может давать соединения общей структуры 5. Предпочтительные заместители X1 для этой реакции включают F и Cl или сульфоны (например, SO2Me). Удаление Pg1 может быть осуществлено многими способами, описанными в литературе, для получения аминов 6.
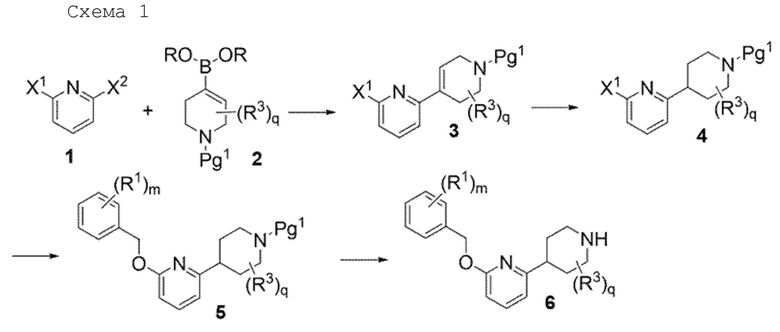
В качестве альтернативы, как показано на схеме 2, соответствующим образом замещенные сложные эфиры пиперидина общей структуры 7 могут взаимодействовать с 1 в присутствии сильного основания, такого как LiHMDS или LDA, или другого подходящего основания в апротонном органическом растворителе, таком как THF, с получением соединений общей структуры 8. Для достижения наилучших результатов при получении таких соединений, как 8, Х2 предпочтительно представляет собой F или Cl. Удаление Pg2 посредством гидролиза сложного эфира для получения карбоновых кислот 9 может быть выполнено традиционным способом, таким как реакция водного гидроксида лития, натрия или калия в смешивающимся с водой растворителе, таком как MeOH, EtOH, THF или тому подобное. Подвергание карбоновых кислот 9 нагреванию (60-120 °С) в подходящем растворителе, таком как DCE или PhCH3, приведет к декарбоксилированию с получением соединений общей формулы 4 для использования, как описано в схеме 1, для получения аминов 6.
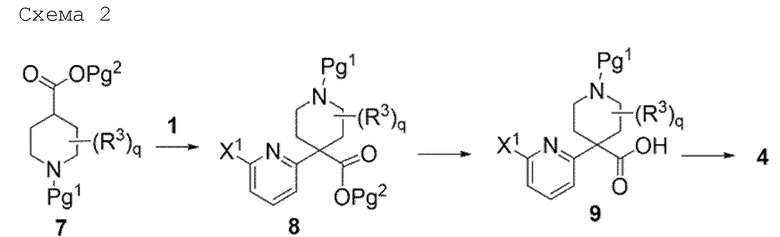
Схема 3 предусматривает альтернативный способ получения соединений 5. Реакция 1 с соответствующим образом замещенным бензиловым спиртом в апротонном растворителе, таком как DMF или THF, в присутствии сильного основания, такого как NaH, KOtBu или LiHMDS, может давать соединения общей структуры 10. Предпочтительные заместители X1 для этой реакции включают F и Cl, тогда как заместители X2 могут включать Cl, Br или I. В качестве альтернативы, условия реакции С-О сочетания Бухвальда-Хартвига, аналогичные получению 5, можно использовать для получения 10 с предпочтительными заместителями Х1 - Cl, Br или I. Условия реакции Сузуки, аналогичные получению общей структуры 3, можно использовать для получения соединений общей структуры 11 из 10. Предпочтительные заместители Х2 для использования в реакции сочетания включают Cl, Br или I. Олефин может быть восстановлен способами, ранее описанными на схеме 1, с получением соединений общей структуры 5, которые затем используются для получения аминов 6.
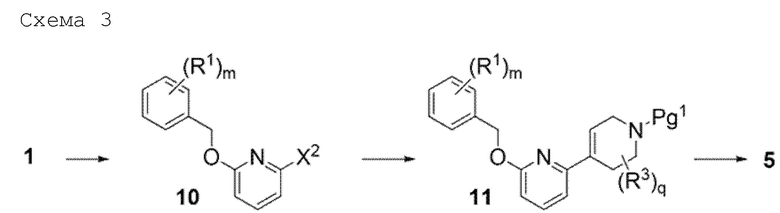
Как представлено на схеме 4, превращение 10 в соединения общей структуры 12 может быть осуществлено таким способом, как C-N сочетание Бухвальда-Хартвига между соединениями общей структуры 10 и соответствующим образом замещенным и защищенным пиперазином в присутствии палладиевого или медного катализатора и лигандного комплекса. Предпочтительные заместители Х2 для использования в реакции сочетания включают Cl, Br или I. Эти реакции обычно проводятся при температуре 0-110 °C в апротонных органических растворителях, таких как, без ограничения, 1,4-диоксан и PhCH3, с добавлением основания, такого как Cs2CO3, LiHMDS или NaOtBu. Удаление Pg1 может быть осуществлено многими способами, описанными в литературе, для получения аминов 13.
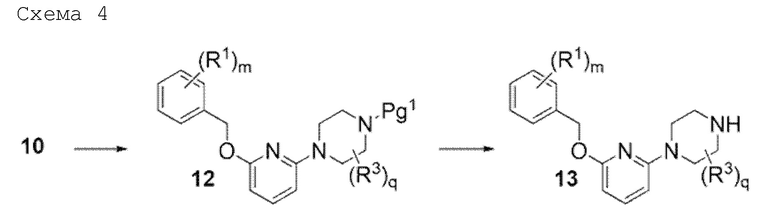
Соединения структуры 14 (схема 5) могут быть превращены в соединения общей структуры 15 способами, описанными ранее на схеме 1 или схеме 2. Предпочтительные заместители Х2 для использования в реакции сочетания включают Cl, Br или I. Превращение промежуточных соединений 15 в их соответствующие N-оксиды 16 можно проводить с окислителями, такими как 3-хлорпероксибензойная кислота, Oxone® или другой подходящий окислитель. На перегруппировку соединений структуры 17 может влиять обработка ангидридом органической кислоты, таким как Ac2O или TFAA, в апротонных растворителях с подходящим основанием органического амина, таким как Et3N, DIPEA или другим подходящим основанием. Получение простых бензиловых эфиров общей структуры 18 может быть достигнуто стандартными способами алкилирования с подходящим образом замещенными бензилбромидами или по стандартным протоколам алкилирования Мицунобу (Swamy et al., Chem. Rev. 2009, 109, 2551-2651) с соответствующим образом замещенными бензиловыми спиртами. Удаление Pg1 может быть осуществлено многими способами, описанными в литературе, для получения аминов 19.
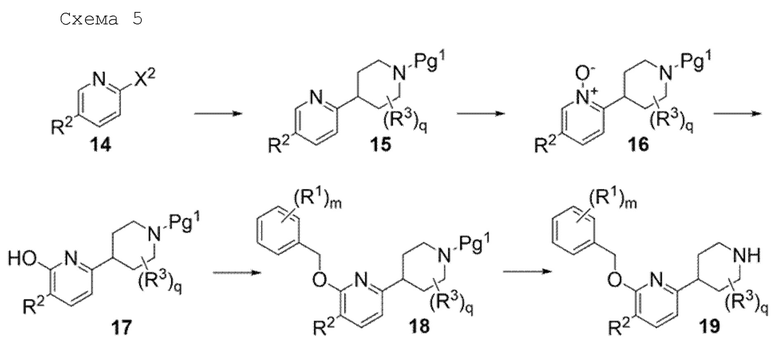
Соединение 20 (схема 6) может вступать в реакцию с соответствующим образом замещенным и защищенным пиперазином в присутствии подходящего основания, такого как Cs2CO3, K2CO3, NaH или LiHMDS, или органического основания, такого как Et3N, DIPEA или DBU, в полярном апротонном растворителе, таком как, без ограничения, DMF, DMAc, ДМСО или NMP, для получения соединений общей структуры 21. Предпочтительные заместители X1 и Х2 для использования в реакции сочетания включают F и Cl; F является наиболее предпочтительным. Простые бензиловые эфиры 22 могут быть получены аналогично соединениям 10 на схеме 3. В качестве альтернативы, выполняя вышеуказанные стадии в обратном порядке, соединения общей структуры 25 могут быть получены из того же исходного материала 20. Удаление Pg1 может быть осуществлено многими способами, описанными в литературе, для получения аминов 23 и 26.
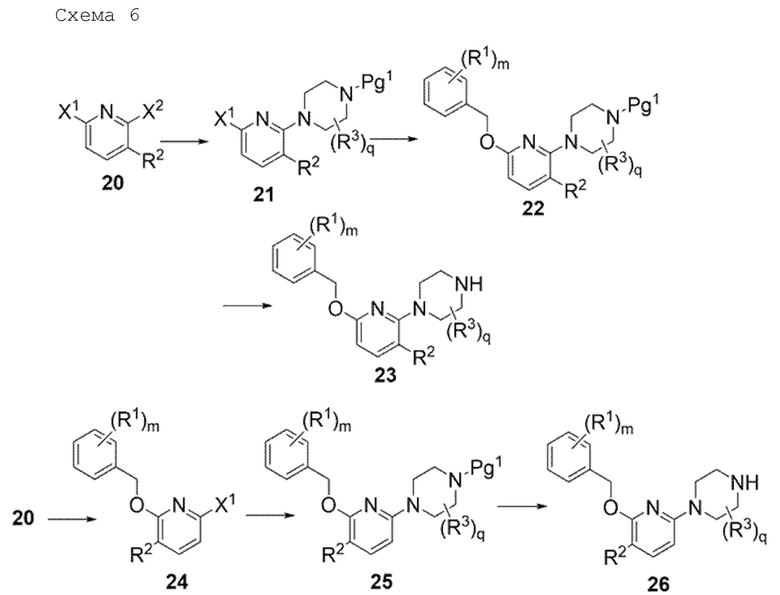
Аминные соединения, полученные способами, описанными на схемах 1-6, в совокупности обозначаемые как амины 27, могут быть алкилированы защищенным 2-бромацетатом в присутствии подходящего основания, такого как K2CO3, Et3N, NaH или LiHMDS в полярном апротонном растворителе, таком как, без ограничения, DMF, DMAc, ДМСО или NMP, для получения соединений общей структуры 28. Для получения кислот 29 может быть проведен стандартный гидролиз сложного эфира. Если Pg2 представляет собой трет-бутил, для получения кислот 29 могут быть использованы стандартные способы снятия защиты с помощью кислоты, например, TFA/DCM, HCl/1,4-диоксан, HCl/EtOAc или другие подходящие условия.
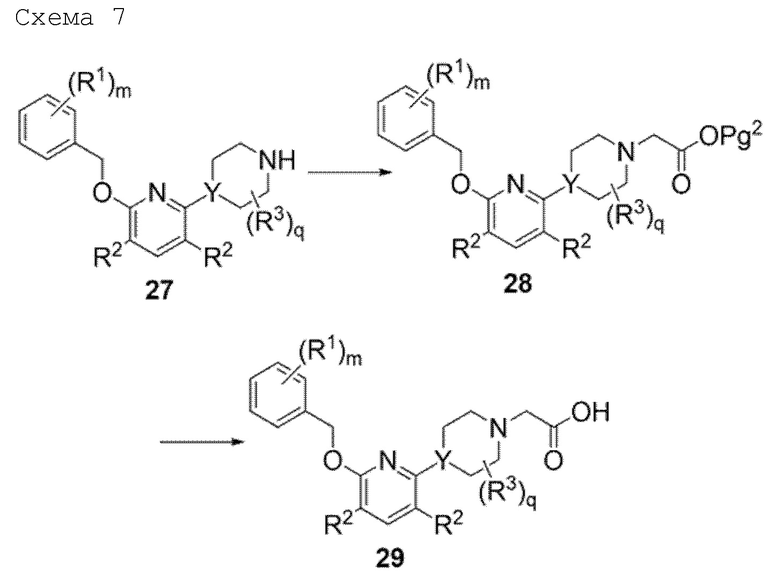
Соединения общей структуры 30 (схема 8) могут вступать в реакцию с аминами R4NH2 в присутствии оснований, таких как карбонат, бикарбонат, гидроксид, ацетат натрия, калия или цезия, или основания органического амина, такого как Et3N, DIPEA, DBU и тому подобное, в полярном апротонном растворителе, таком как, без ограничения, THF, DMF, DMAc, ДМСО или NMP, или протонном растворителе, таком как вода, MeOH, EtOH или iPrOH, или их смесь для получения соединений общей структуры 31. Следует отметить, что если в примере R4 имеет разделенный энантиомерный центр, другой энантиомер или его рацемическая смесь могут быть получены путем выбора подходящего исходного материала. Предпочтительные X3 заместители включают F, Cl и Br, предпочтительные X4 группы включают Cl, Br, -CO2, -Pg2. На восстановление нитрогруппы может влиять гидрирование при 1-6 атм (0,1-0,6 МПа) Н2 с использованием металлического катализатора, такого как палладий на угле или никель Ренея в протонном растворителе, таком как МЕОН или EtOH, или апротонном растворителе, таком как DMF, THF или EtOAc. В качестве альтернативы, нитрогруппа может быть восстановлена железом, цинком, SnCl2 или другим подходящим металлом в кислой среде, такой как 1 н. HCl, AcOH или водный NH4Cl в THF, с получением соединений общей структуры 32 (схема 8а). Соединения, такие как 33, могут быть ацилированы ацилгалогенидами стандартным способом или карбоксилатами по стандартным протоколам амидного сочетания с получением соединений 34. Восстановление до соединений 35 можно проводить в стандартных условиях с использованием восстановителей, таких как LAH или BH3-THF или BH3-DMS (схема 8b).
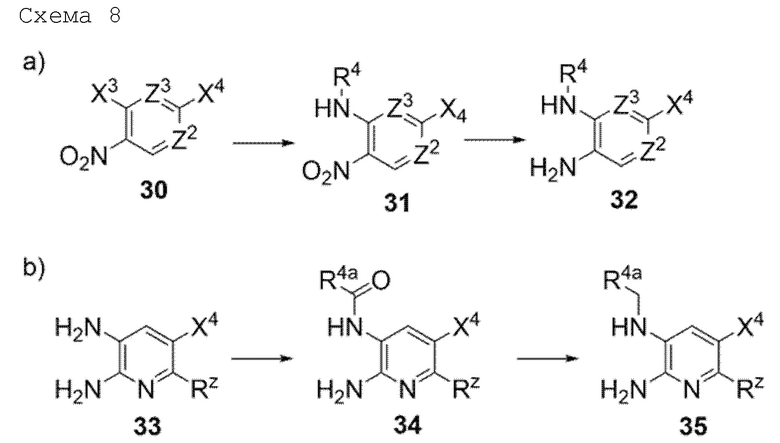
Диаминовые соединения 32 и 35, получаемые способами, описанными на схемах 8а и 8b, в совокупности обозначаемые как диамин 37 (схема 9), могут быть ацилированы кислотами общей структуры 29 в соответствии со стандартными протоколами амидного сочетания с получением аминов 38, которые будут существовать в виде смеси от 100% 38a до 100% 38b. Эта смесь аминов 38 может быть циклизована с получением соединений общей структуры 39 различными способами. Амины 38 могут быть нагреты в присутствии обезвоживающего агента, такого как T3P®, или алкилового спирта, такого как н-бутанол в условиях микроволнового излучения (10-60 мин при 120-180 °С) с получением соединений 39. В качестве альтернативы, смесь соединений 38 может быть нагрета в кислых условиях, таких как AcOH, при температуре 60-100 °C или в основных условиях, таких как водный раствор NaOH или KOH в 1,4-диоксане, при температуре 60-100 °C, для получения 39. Соединения общей структуры 39 (X4=Cl, Br или I) могут быть превращены в сложные эфиры структуры 40 с помощью катализируемого палладием карбонилирования, в атмосфере монооксида углерода при 15-100 фунт/кв. дюйм (0,10-0,69 МПа) при температуре 20-100 °C, с подходящим спиртом, таким как MeOH или EtOH или другой алкиловый спирт. Гидролиз сложного эфира 40 может быть осуществлен, как описано на схеме 7, с получением кислот 41. Для соединений 38, где X4=CO2-Pg2, превращение в сложный эфир 40 происходит в условиях, аналогичных описанным ранее, за исключением использования способа циклизации в основных условиях, где соединение 41 может быть выделено непосредственно из реакционной смеси. Для соединений 40, где X4 представляет собой CO2tBu, снятие защиты с получением кислоты 41 можно проводить в кислых условиях, описанных на схеме 7.
Схема 9
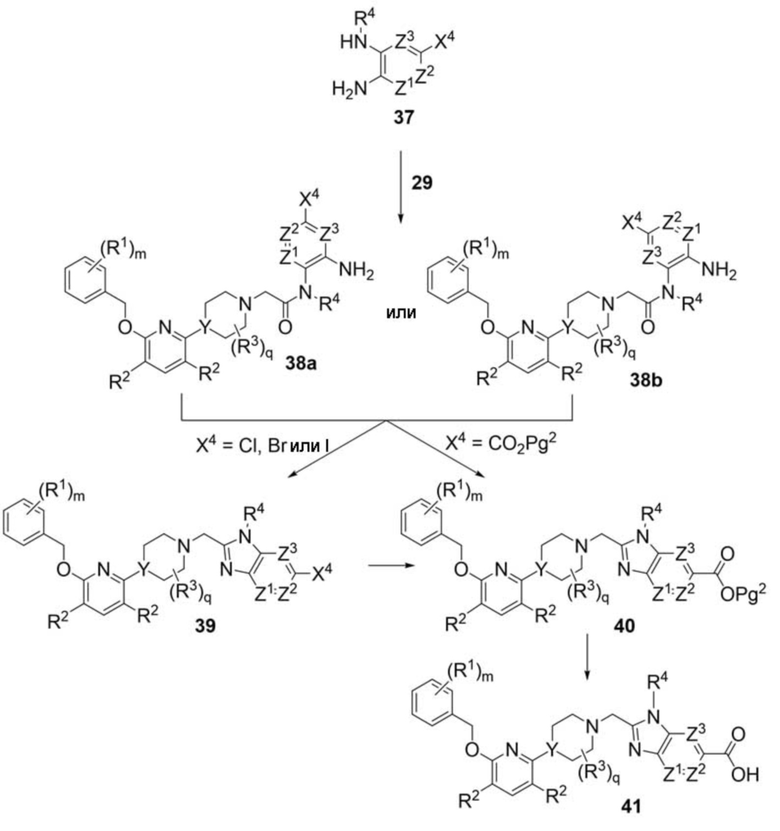
В дополнение к этому, диамин 37 может быть превращен в 2-хлорметилбензимидазол 42 (схема 10) несколькими способами. Обработка 2-хлорацетилхлоридом в апротонном растворителе, таком как 1,4-диоксан, с последующим нагреванием при 40-100 °С в течение 2-18 ч, позволяет получить желаемый бензимидазол 42, где Z1, Z2 и Z3 представляют собой СН. В случаях, когда не все из Z1, Z2 и Z3 являются CRz, после обработки 2-хлорацетилхлоридом в апротонном растворителе, таком как 1,4-диоксан, в течение периода от 30 мин до 4 ч, растворитель заменяют на кислую среду, такую как AcOH или TFA, с последующим нагреванием при 40-100 °C в течение 2-18 ч, для получения желаемого соединения 42. Диамин 37 также можно обработать хлоруксусным ангидридом при температуре 0-80 °С в апротонном растворителе, таком как, без ограничения, 1,4-диоксан, THF или MeCN, с последующим нагреванием в течение 2-18 ч при 60-100 °С для получения желаемого соединения 42. В дополнение к этому, диамин 37 может быть обработан 2-хлор-1,1,1-триметоксиэтаном в апротонном растворителе, таком как, без ограничения, 1,4-диоксан, THF или MeCN, или протонном растворителе, например, MeOH или EtOH, в присутствии кислотного катализатора, например, pTSA, при 20-100 °C. В качестве альтернативы, диамины 37 могут быть нагреты до 100-180 °С с 2-гидроксиуксусной кислотой в апротонном растворителе, таком как, без ограничения, мезитилен, с получением гидроксиметильного промежуточного соединения. Превращение гидроксиметильной группы в хлорметильное соединение 42 может быть осуществлено стандартными способами, включая обработку SOCl2 в апротонном растворителе. Соединения общей структуры 42 могут вступать в реакцию с соединениями 27 в присутствии оснований, таких как карбонат, бикарбонат натрия, калия или цезия, NaH или основания органического амина, такого как Et3N, DIPEA, DBU и тому подобное, в полярном апротонном растворителе, таком как, без ограничения, THF, MeCN, DMF, DMAc, ДМСО или NMP, с получением соединений 39 (X4=Cl, Br, I) или соединений 40 (X4=CO2-Pg2), которые затем используются для получения соединений 41 способами, описанными на схеме 9.
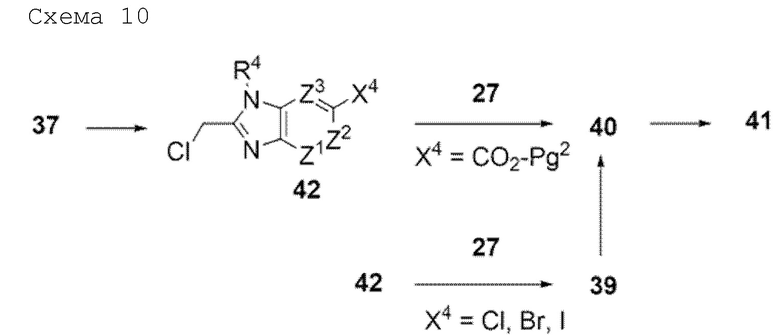
В качестве альтернативы, соединения общей структуры 42 могут вступать в реакцию с соответствующим образом замещенными и защищенными пиперазинами для получения соединений 43 (схема 11). Удаление Pg1 может быть осуществлено многими способами, описанными в литературе, для получения аминов 44. Превращение в соединения общей структуры 39 (X4=Cl, Br или I) или 40 (X4=CO2-Pg2) может быть осуществлено таким способом, как C-N сочетание Бухвальда-Хартвига между соединениями общей структуры 10 и как описано ранее на схеме 4. Соединения общей структуры 39 или 40 могут затем быть использованы для получения соединений структуры 41 с помощью способов, описанных на схеме 9.
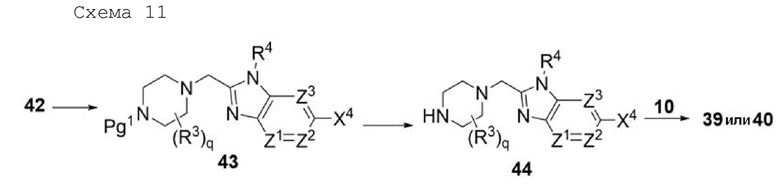
Примеры
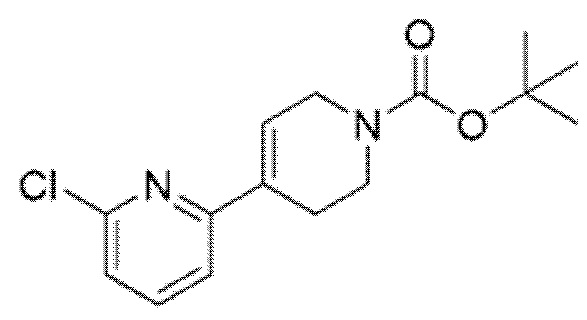
Промежуточное соединение 1
Трет-бутил-6-хлор-3',6'-дигидро-[2,4'-бипиридин]-1'(2'H)-карбоксилат
В реакционный сосуд, оснащенный обратным холодильником, загружали трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (1,5 г, 4,9 ммоль), 2,6-дихлорпиридин (1,4 г, 9,7 ммоль), Pd(dppf)Cl2 (0,34 г, 0,49 ммоль), и карбонат цезия (3,5 г, 11 ммоль). Добавляли барботируемый раствор 1,4-диоксана (15 мл) и воды (3 мл) и смесь нагревали до 90°C в атмосфере N2 (г). Через 7 ч смеси давали возможность остыть до КТ и фильтровали через слой Celite® с EtOAc (50 мл). Смесь разбавляли водой (20 мл), водный слой экстрагировали EtOAc (3 × 50 мл), и объединенные органические слои сушили над безводным Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Неочищенный материал очищали с помощью колоночной хроматографии, элюируя 10% EtOAc в гептане с получением промежуточного соединения 1 в виде бесцветного масла (1,1 г, 75%). 1H ЯМР (CDCl3) δ: 7,57 (т, 1H), 7,23 (д, 1H), 7,14 (д, 1H), 6,66 (ушир. с, 1H), 4,11 (ушир. с, 2H), 3,61 (ушир. с, 2H), 2,57 (ушир. с, 2H), 1,43-1,52 (м, 9H).
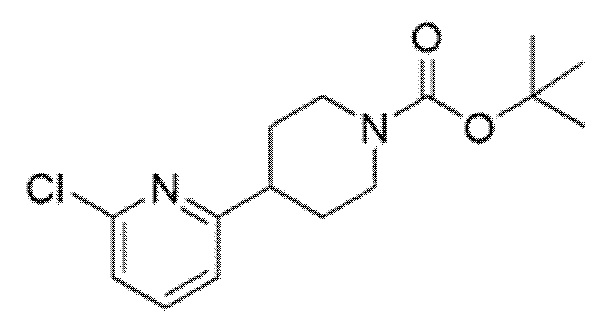
Промежуточное соединение 2
Трет-бутил-4-(6-хлорпиридин-2-ил)пиперидин-1-карбоксилат
К перемешиваемому раствору промежуточного соединения 1 (0,55 г, 1,9 ммоль) в MeOH (19 мл) добавляли PtO2 (0,042 г, 0,19 ммоль). Раствор подвергали воздействию атмосферы водорода (30 фунт/кв. дюйм (0,21 МПа) при КТ. Через 3 ч раствор фильтровали через слой Celite®, промывали МеОН (2 × 15 мл) и концентрировали при пониженном давлении. Неочищенный материал очищали с помощью колоночной хроматографии, элюируя 30% EtOAc в гептане с получением промежуточного соединения 2 (0,22 г, 40%) в виде бесцветного масла. 1H ЯМР (CDCl3) δ: 7,57 (т, 1H), 7,15 (д, 1H), 7,05 (д, 1H), 4,23 (ушир. с, 2H), 2,80 (д, 3H), 1,89 (д, 2H), 1,60-1,73 (м, 2H), 1,45 (с, 9H).
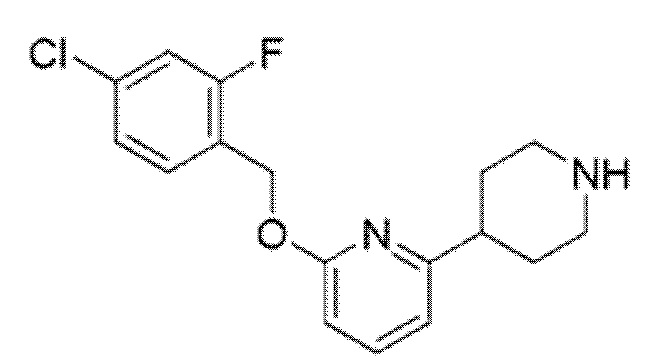
Промежуточное соединение 3
2-((4-Хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридин-бис(4-метилбензолсульфонат)
Стадия 1
В реакционный сосуд, оснащенный обратным холодильником, загружали промежуточное соединение 2 (6,5 г, 22 ммоль), (4-хлор-2-фторфенил)метанол (3,5 г, 22 ммоль), Pd2(dba)3 (1,0 г, 1,1 ммоль), BINAP (1,4 г, 2,2 ммоль) и карбонат цезия (14 г, 44 ммоль). Добавляли толуол (73 мл) и смесь нагревали до 100°С. Через 16 ч смеси давали возможность остыть до КТ, фильтровали через Celite® с EtOAc (100 мл) и концентрировали при пониженном давлении. Неочищенный материал очищали с помощью колоночной хроматографии, элюируя 10% EtOAc в PE с получением трет-бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата в виде желтого масла (7,6 г, 82%). 1H ЯМР (CDCl3) δ: 7,51 (дд, 1H), 7,39-7,47 (м, 1H), 7,06-7,18 (м, 2H), 6,73 (д, 1H), 6,62 (д, 1H), 5,40 (с, 2H), 4,22 (ушир. с, 2H), 2,83 (м, 2H), 2,73 (тт, 1H), 1,81-1,94 (м, 2H), 1,64-1,79 (м, 2H), 1,50 (с, 9H).
Стадия 2
К перемешиваемому раствору трет-бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата (50 г, 120 ммоль) в EtOAc (700 мл) добавляли pTSA·H2O (59 г, 310 ммоль). Смесь нагревали до 60°C. Через 30 мин раствору давали возможность остыть до КТ. Полученный твердый осадок суспендировали в течение 16 ч, собирали фильтрованием и затем сушили при пониженном давлении, с получением промежуточного соединения 3 в виде твердого вещества (81 г, колич. выход). 1H ЯМР (600 МГц, ДМСО-d6) δ: 8,55 (ушир. с, 1H), 8,28 (д, 1H), 7,68 (т, 1H), 7,60 (т, 1H), 7,48 (д, 4H), 7,32 (д, 1H), 7,12 (д, 4H), 6,89 (д, 1H), 6,74 (д, 1H), 5,38 (с, 2H), 3,37 (д, 2H), 2,98-3,09 (м, 2H), 2,87-2,96 (м, 1H), 2,29 (с, 6H), 1,96-2,01 (м, 2H), 1,80-1,94 (м, 2H).
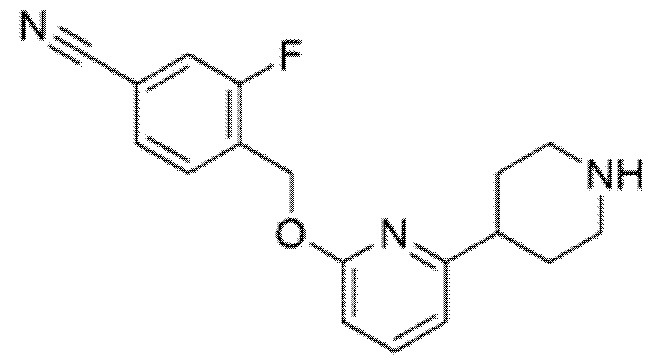
Промежуточное соединение 4
3-Фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрил-бис(4-метилбензолсульфонат)
Стадия 1
К раствору диизопропиламина (92 мл, 656 ммоль) в THF (350 мл) при -26°С добавляли н-бутиллитий в гептанах (2,6 M, 250 мл, 650 ммоль) в течение 15 мин. Смесь охлаждали до -30°С и раствор 1-(трет-бутил)-4-метилпиперидин-1,4-дикарбоксилата (156 г, 641 ммоль) в THF (150 мл) добавляли в течение 25 мин. Через 10 мин раствор 2,6-дихлорпиридина (94 г, 635 ммоль) в THF (150 мл) добавляли в течение 2 мин. Смесь нагревали до 25°С в течение 2,5 ч и затем охлаждали до 8°С и обрабатывали 6 M HCl (125 мл) в течение 20 мин, чтобы довести рН смеси до ~7-8. Смесь разбавляли водой (100 мл) и МТВЕ (150 мл) и слои разделяли. Водный слой экстрагировали МТВЕ (150 мл) и объединенные органические слои промывали насыщенным солевым раствором (150 мл), сушили над MgSO4. Растворитель удаляли при пониженном давлении с получением неочищенного 1-(трет-бутил)-4-метил-4-(6-хлорпиридин-2-ил)пиперидин-1,4-дикарбоксилата (241 г) в виде желтого масла, которое использовали на следующей стадии без очистки. 1H-ЯМР очищенного образца (400 МГц, CDCl3) δ: 7,62 (т, 1H), 7,21 (д, 2H), 3,83 (ушир. с, 2H), 3,71 (с, 3H), 3,14 (ушир. с, 2H), 2,41 (д, 2H), 2,08 (ддд, 2H), 1,45 (с, 9H).
Стадия 2
Неочищенный 1-(трет-бутил)-4-метил-4-(6-хлорпиридин-2-ил)пиперидин-1,4-дикарбоксилат (241 г, предположительно 635 ммоль) растворяли в MeOH (400 мл) при 43°С и обрабатывали 4 М водн. NaOH (300 мл) в течение 20 мин. Смесь нагревали до 50°С и перемешивали в течение 35 мин. Затем смесь охлаждали до 11°С и рН доводили до ~2 путем добавления 6 М HCl (200 мл) ) в течение 25 мин, продолжая охлаждение до 5°С, после чего образовывался твердый осадок. Суспензию разбавляли водой (300 мл) и перемешивали в течение 40 мин, после чего твердое вещество собирали фильтрованием, промывали водой и затем сушили под вакуумом при 50°С с получением белого твердого вещества (224 г). Твердое вещество растирали в гептане (750 мл) при 45°С в течение 45 мин. Смесь охлаждали до 16°С и твердое вещество собирали фильтрованием, промывали гептаном и сушили с получением 1-(трет-бутоксикарбонил)-4-(6-хлорпиридин-2-ил)пиперидин-4-карбоновой кислоты (187 г, 549 ммоль, 86% в две стадии) в виде белого твердого вещества.
Стадия 3
Раствор 1-(трет-бутоксикарбонил)-4-(6-хлорпиридин-2-ил)пиперидин-4-карбоновой кислоты (187 г, 549 ммоль) в DCE (900 мл) нагревали при 82°С в течение ночи и затем охлаждали до 20°С. Смесь обрабатывали Magnesol® (30 г) в течение 40 мин. Суспензию фильтровали через слой Magnesol® (30 г) и твердые вещества промывали 1:1 МТВЕ:гептан (300 мл). Фильтрат концентрировали при пониженном давлении с получением бледно-желтого твердого вещества, которое растирали в гептане (250 мл) при 50°С. Смесь охлаждали до 12°С и твердое вещество собирали фильтрованием, промывали гептаном и сушили под вакуумом при 45°С с получением трет-бутил-4-(6-хлорпиридин-2-ил)пиперидин-1-карбоксилата (143 г, 481 ммоль, 88%) в виде твердого вещества. 1H ЯМР (600 МГц, CDCl3) δ: 7,58 (т, 1H), 7,17 (д, 1H), 7,06 (д, 1H), 4,25 (ушир. с, 2H), 2,66-2,93 (м, 3H), 1,91 (д, 2H), 1,69 (кв,д, 2H), 1,47 (с, 9H).
Стадия 4
Смесь трет-бутил-4-(6-хлорпиридин-2-ил)пиперидин-1-карбоксилата (100 г, 337 ммоль), 3-фтор-4-(гидроксиметил)бензонитрила (53,9 г, 357 ммоль) и Cs2CO3 (170 г, 522 ммоль) в диоксане (900 мл) дезоксигенировали 5 циклами вакуумирования/заполнения азотом. Добавляли JohnPhos ([1,1'-бифенил]-2-ил-ди-трет-бутилфосфин, 2,02 г, 6,77 ммоль) и Pd2(dba)3 (3,10 г, 3,39 ммоль) и проводили еще 2 цикла вакуумирования/заполнения азотом. Затем смесь нагревали при 95°С в течение 3 ч. Дополнительно добавляли JohnPhos (660 мг, 2,21 ммоль) и Pd2(dba)3 (990 мг, 1,08 ммоль) и нагревание продолжали в течение ночи. Смесь охлаждали до 20°С и фильтровали через слой Celite®, промывая МТВЕ (250 мл). Фильтрат концентрировали при пониженном давлении с получением красно-оранжевого масла (174 г). Это вещество растворяли в 30% MTBE/гексан (600 мл), перемешивали с Magnesol® (20 г) и Darco® G-60 (10 г) в течение 70 мин и затем фильтровали через слой диоксида кремния (100 г), промывая 50% MTBE/гексан (600 мл). Фильтрат концентрировали при пониженном давлении и подвергали азеотропной перегонке с EtOAc (100 мл) с получением трет-бутил-4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата в виде масла (147 г), которое использовали без дальнейшей очистки. 1H-ЯМР очищенного образца (600 МГц, CDCl3) δ: 7,62 (т, 1H), 7,53 (т, 1H), 7,44 (д, 1H), 7,37 (д, 1H), 6,75 (д, 1H), 6,65 (д, 1H), 5,49 (с, 2H), 4,20 (ушир. с, 2H), 2,81 (ушир. с, 2H), 2,70 (тт, 1H), 1,82 (д, 2H), 1,67 (д, 2H), 1,49 (с, 9H).
Стадия 5
К перемешиваемому раствору трет-бутил-4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата (147 г, предположительно 337 ммоль) в EtOAc (1,8 л) при КТ добавляли pTSA·H2O (161 г, 846 ммоль). Смесь нагревали до 60°С, что приводило к выделению газа и образованию твердого вещества. Смесь перемешивали в течение 1,5 ч, после чего добавляли дополнительное количество pTSA·H2O (12 г, 63 ммоль) и перемешивание продолжали в течение 45 мин. Смесь охлаждали до 17°С и твердые вещества собирали фильтрованием, промывали EtOAc (200 мл) и сушили с получением 205 г твердого вещества. Это вещество растворяли в МеОН (500 мл) при 55°C и разбавляли EtOAc (1 л). Полученную суспензию охлаждали до 20°C, и твердые вещества собирали фильтрованием, промывали смесью 9:1 EtOAc:MeOH (100 мл) и EtOAc (250 мл) и сушили с получением промежуточного соединения 4 (176,6 г, 269 ммоль, 80% в 2 стадии) в виде белого твердого вещества. 1H ЯМР (600 МГц, ДМСО-d6) δ: 8,53 (ушир. с, 1H), 8,26 (ушир. с, 1H), 7,89 (д, 1H), 7,67-7,78 (м, 3H), 7,48 (д, 4H), 7,11 (д, 4H), 6,90 (д, 1H), 6,79 (д, 1H), 5,48 (с, 2H), 3,35 (д, 2H), 2,96-3,09 (м, 2H), 2,79-2,96 (м, 1H), 2,29 (с, 6H), 1,93-2,03 (м, 2H), 1,77-1,90 (м, 2H).
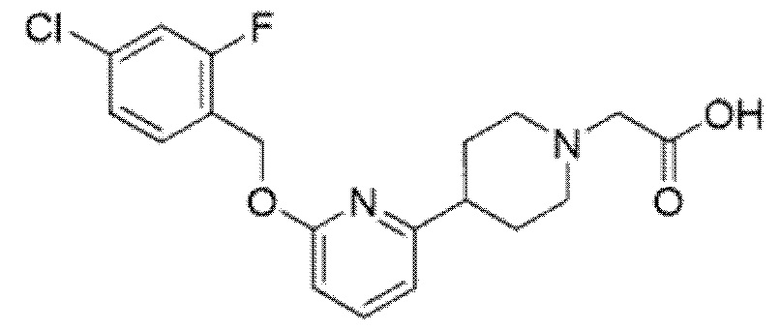
Промежуточное соединение 5
2-(4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)уксусная кислота
Стадия 1
К смеси промежуточного соединения 3 (70,0 г, 209 ммоль) и K2CO3 (118 г, 863 ммоль) в DMF (800 мл) добавляли этил-2-бромацетат (39,9 г, 236 ммоль) порциями. Смесь перемешивали при 30°С в течение 1 ч. Смесь разбавляли водой (500 мл) и экстрагировали EtOAc (400 мл × 3). Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (колонка с силикагелем, 10:1 PE/EtOAc) с получением 74 г этил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)ацетата (84%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,51 (т, 1H), 7,45 (т, 1H), 7,09-7,17 (м, 2H), 6,75 (д, 1H), 6,61 (д, 1H), 5,41 (с, 2H), 4,22 (кв, 2H), 3,27 (с, 2H), 3,07 (д, 2H), 2,54-2,65 (м, 1H), 2,32 (тд, 2H), 1,93-2,07 (м, 2H), 1,85-1,92 (м, 2H), 1,30 (т, 3H).
Стадия 2
К раствору этил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)ацетата (73 г, 179 ммоль) в EtOH (270 мл) добавляли 5 M NaOH (156 мл, 780 ммоль). Раствор перемешивали при 25°С в течение 2 ч. Смесь подкисляли до pH ~3,5 с помощью 1 М HCl. Полученный осадок собирали фильтрованием. Твердые вещества промывали водой и сушили под вакуумом с получением 54 г промежуточного соединения 5 (78%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,65-7,72 (м, 1H), 7,62 (т, 1H), 7,47 (дд, 1H), 7,32 (дд, 1H), 6,92 (д, 1H), 6,73 (д, 1H), 5,40 (с, 2H), 4,13 (с, 2H), 3,58 (д, 2H), 3,16-3,26 (м, 2H), 2,89 (ушир. с, 1H), 2,00-2,19 (м, 4H); ЖХМС=378,8.
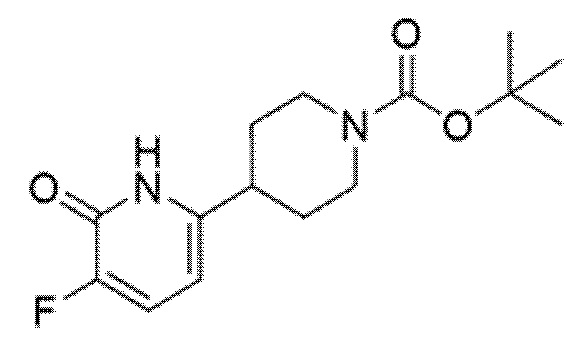
Промежуточное соединение 6
Трет-бутил-4-(5-фтор-6-оксо-1,6-дигидропиридин-2-ил)пиперидин-1-карбоксилат
Стадия 1
К раствору 2-бром-5-фторпиридина (20 г, 110 ммоль) и трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилата (35,1 г, 114 ммоль) в THF (240 мл) добавляли Pd(PPh3)4 (13,1 г, 11,4 ммоль) и Na2CO3 (24,1 г, 227 ммоль). Полученную желтую реакционную смесь перемешивали при 90°С в течение 48 ч. Реакционную смесь охлаждали до КТ, разбавляли водой (100 мл) и экстрагировали EtOAc (3 × 200 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (с градиентом 1-20% EtOAc/PE) с получением трет-бутил-5-фтор-3',6'-дигидро-[2,4'-бипиридин]-1'(2'H)-карбоксилата (31 г, 98%) в виде бесцветного масла. 1H ЯМР (CDCl3) δ 8,41 (т, 1H), 7,37 (дд, 2H), 6,52 (ушир. с, 1H), 4,13 (д, 2H), 3,65 (м, 2H), 2,57-2,70 (м, 2H), 1,49 (с, 9H).
Стадия 2
К бесцветному раствору трет-бутил-5-фтор-3',6'-дигидро-[2,4'-бипиридин]-1'(2'H)-карбоксилата (31 г, 110 ммоль) в EtOAc (300 мл) добавляли 10% влажный Pd/C (1,2 г, 5,6 ммоль). Черную смесь перемешивали при 25°C в атмосфере H2 (15 фунт/кв. дюйм (0,10 МПа)) в течение 16 ч. Смесь фильтровали через Celite® и концентрировали при пониженном давлении с получением трет-бутил-4-(5-фторпиридин-2-ил)пиперидин-1-карбоксилата (31 г, 99%) в виде бесцветного масла. 1H ЯМР (CDCl3) δ 8,39 (д, 1H), 7,34 (тд, 1H), 7,15 (дд, 1H), 4,25 (ушир. с, 2H), 2,74-2,93 (м, 3H), 1,89 (д, 2H), 1,69 (кв,д, 2H), 1,48 (с, 9H).
Стадия 3
К раствору полученного трет-бутил-4-(5-фторпиридин-2-ил)пиперидин-1-карбоксилата (31 г, 110 ммоль) в DCM (400 мл) добавляли m-CPBA (47,7 г, 276 ммоль) при 0°С. Полученную реакционную смесь перемешивали при КТ в течение 16 ч. Белую суспензию фильтровали и фильтрат затем гасили водн. Na2SO3 (200 мл). Водный слой отделяли и затем экстрагировали DCM (3 × 200). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (градиент 0,5-4% MeOH/DCM) с получением 2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-5-фторпиридин-1-оксида (20 г, 61%) в виде твердого вещества. 1H ЯМР (CDCl3) δ 8,21 (дд, 1H), 7,11-7,18 (м, 1H), 7,02-7,09 (м, 1H), 4,26 (ушир. с, 2H), 3,58 (м, 1H), 2,89 (ушир. с, 2H), 2,02 (д, 2H), 1,43-1,52 (м, 11H).
Стадия 4
К раствору 2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-5-фторпиридин-1-оксида (10 г, 34 ммоль) в THF (150 мл) при 0°С по каплям добавляли Et3N (6,83 г, 67,5 ммоль) и TFAA (70,9 г, 337 ммоль). Смесь перемешивали при 0°С в течение 1 ч, и при КТ в течение 16 ч. Светло-желтый раствор гасили водн. NaHCO3 (400 мл). Величину pH доводили до ~4 с помощью TFA, и смесь экстрагировали EtOAc (3 × 200 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (4-80% EtOAc в PE) с получением промежуточного соединения 6 (5,4 г, 54%) в виде твердого вещества. 1H ЯМР (CDCl3) δ 12,92 (ушир. с, 1H), 7,17 (дд, 1H), 5,97 (дд, 1H), 4,25 (ушир. с, 2H), 2,86 (ушир. с, 2H), 2,72 (т, 1H), 1,95 (д, 2H), 1,56 (кв,д, 2H), 1,48 (с, 9H).
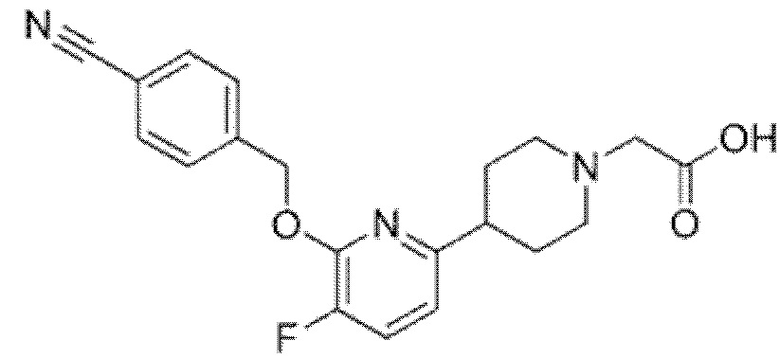
Промежуточное соединение 7
2-(4-(6-((4-Цианoбензил)окси)-5-фторпиридин-2-ил)пиперидин-1-ил)уксусная кислота
Стадия 1
К раствору промежуточного соединения 6 (2,0 г, 6,8 ммоль), 4-цианoбензилового спирта (1,35 г, 10,1 ммоль) и 1,1'-(азодикарбонил)дипиперидина (2,55 г, 10,1 ммоль ) в PhCH3 (30 мл) по каплям добавляли три-н-бутилфосфин (2,05 г, 10,1 ммоль) в атмосфере N2. Полученный светло-желтый раствор перемешивали при 80°C в атмосфере N2 в течение 48 ч. Смесь разбавляли EtOAc (100 мл) и промывали водой (100 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (0-15% EtOAc/PE) с получением трет-бутил-4-(6-((4-цианoбензил)окси)-5-фторпиридин-2-ил)пиперидин-1-карбоксилата (1,72 г, 62% выход) в виде бесцветного масла. 1H ЯМР (CDCl3) δ 7,67 (д, 2H), 7,58 (д, 2H), 7,29 (дд, 1H), 6,71 (дд, 1H), 5,51 (с, 2H), 4,20 (ушир. с, 2H), 2,81 (т, 2H), 2,69 (дт, 1H), 1,81 (д, 2H), 1,65 (ушир. с, 2H), 1,49 (с, 9H).
Стадия 2
К раствору трет-бутил-4-(6-((4-цианoбензил)окси)-5-фторпиридин-2-ил)пиперидин-1-карбоксилата (1,72 г, 4,18 ммоль) в DCM (15 мл) по каплям добавляли TFA (5 мл). Полученный светло-желтый раствор перемешивали при 25°С в течение 2 ч. Смесь концентрировали при пониженном давлении с получением 4-(((3-фтор-6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрилтрифторацетата (1,3 г, количественно) в виде светло-желтого твердого вещества. 1H ЯМР (CD3OD) δ 7,74 (д, 2H), 7,63 (д, 2H), 7,46 (дд, 1H), 6,89 (дд, 1H), 5,56 (с, 2H), 3,42-3,53 (м, 2H), 3,11 (тд, 2H), 2,96 (тт, 1H), 2,03-2,13 (м, 2H), 1,87-2,02 (м, 2H).
Стадия 3
К бесцветному раствору 4-(((3-фтор-6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрилтрифторацетата (1,3 г, 4,2 ммоль) и этил-2-бромацетата (767 мг, 4,59 ммоль) в MeCN (20 мл) добавляли K2CO3 (2,89 г, 20,9 ммоль). Полученную белую суспензию перемешивали при 60°С в течение 3 ч и оставляли при КТ на 16 ч. Смесь фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (0-33% EtOAc в PE) с получением этил-2-(4-(6-((4-цианoбензил)окси)-5-фторпиридин-2-ил)пиперидин-1-ил)ацетата (1,07 г, 65%) в виде светло-желтого твердого вещества. 1H ЯМР (CDCl3) δ 7,67 (д, 2H), 7,58 (д, 2H), 7,28 (дд, 1H), 6,72 (дд, 1H), 5,52 (с, 2H), 4,21 (м, 2H), 3,26 (с, 2H), 3,06 (д, 2H), 2,55 (тт, 1H), 2,29 (дт, 2H), 1,94 (дкв, 2H), 1,77-1,86 (м, 2H), 1,30 (м, 3H).
Стадия 4
К раствору этил-2-(4-(6-((4-цианoбензил)окси)-5-фторпиридин-2-ил)пиперидин-1-ил)ацетата (1,07 г, 2,69 ммоль) в MeOH (10 мл) по каплям добавляли раствор NaOH (162 мг, 4,04 ммоль) в воде (2 мл). Полученный бесцветный раствор перемешивали при 25°С в течение 3 ч. Смесь разбавляли водой (30 мл), экстрагировали МТВЕ (30 мл). Органическую фазу подкисляли до pH ~7 с помощью 2 М HCl и лиофилизировали в течение 16 ч. Неочищенный продукт очищали флэш-хроматографией (градиент 0-5% MeOH/DCM) с получением промежуточного соединения 7 (850 мг, 86% выход) в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7,75 (д, 2H), 7,67 (д, 2H), 7,46 (дд, 1H), 6,92 (дд, 1H), 5,59 (с, 2H), 3,71-3,80 (м, 2H), 3,35 (с, 2H), 3,10-3,27 (м, 2H), 2,90-3,06 (м, 1H), 2,11-2,29 (м, 2H), 2,01-2,10 (м, 2H), ЖХМС (ES+): 369,9 (M+H).
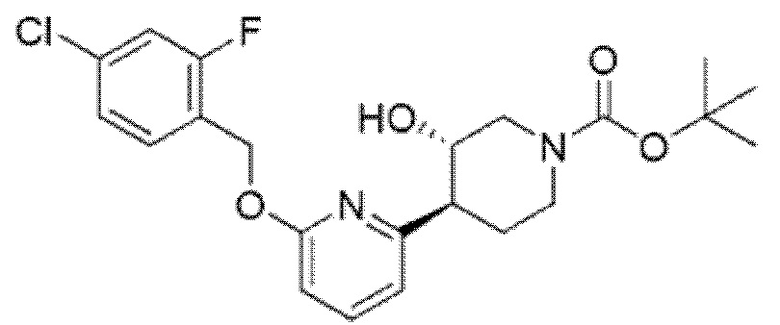
Промежуточное соединение 8
Рац-трет-бутил-(3R,4R)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-карбоксилат
К раствору промежуточного соединения 1 (800 мг, 1,9 ммоль) в THF (15 мл) при 0°С в атмосфере азота добавляли комплекс боран-THF (1 M в THF, 2,1 мл, 2,1 ммоль). Реакционную смесь перемешивали при 0°С в течение 10 мин и затем нагревали до 30°С в течение 30 мин. Реакционный сосуд затем охлаждали до 0°C, открывали для воздуха и медленно добавляли раствор NaOH (190 мг, 4,8 ммоль) в воде (5 мл) и перекиси водорода (30% масс. в воде, 0,86 мл, 9,6 ммоль). Затем смесь нагревали до 26°С и перемешивали в течение 16 ч. Водный Na2SO3 (15 мл) и NaHCO3 (15 мл) добавляли к полученной белой суспензии и смесь экстрагировали DCM (3 × 50 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный материал очищали с помощью колоночной хроматографии, элюируя EtOAc в PE (градиент от 10% до 30% до 60%), с получением промежуточного соединения 8 в виде бесцветного масла (320 мг, 38%). ЖХМС(ES+): 437 (M+H), 459 (M+Na).
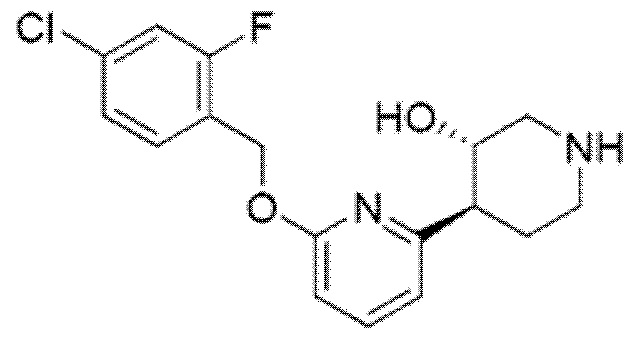
Промежуточное соединение 9
Рац-(3R,4R)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-3-олтрифторацетат
К раствору промежуточного соединения 8 (60 мг, 0,14 ммоль) в DCM (2 мл) добавляли TFA (0,5 мл) при КТ, и смесь перемешивали в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного промежуточного соединения 9 в виде светло-желтого масла, которое использовали без очистки. ЖХМС(ES+): 337 (M+H).
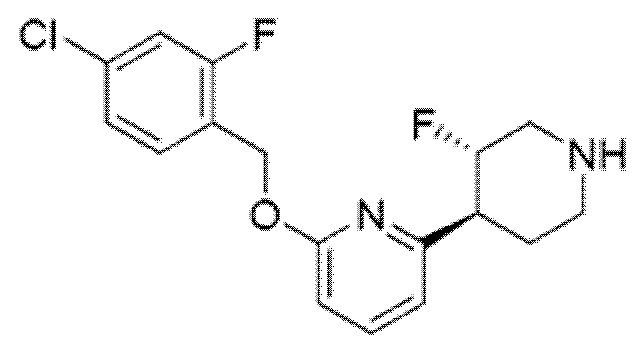
Промежуточное соединение 10
Рац-2-((4-хлор-2-фторбензил)окси)-6-((3R,4R)-3-фторпиперидин-4-ил)пиридингидрохлорид
К раствору промежуточного соединения 8 (60 мг, 0,14 ммоль) в DCM (6 мл) при 0°С в атмосфере азота добавляли DAST (трифторид диэтиламиносеры, 38 мг, 0,23 ммоль). Полученную смесь перемешивали при 0°С в течение 10 мин и затем при КТ в течение 2 ч. Затем к раствору добавляли воду и смесь экстрагировали EtOAc (3 × 100 мл). Объединенный органический раствор промывали насыщенным солевым раствором и затем концентрировали при пониженном давлении. Неочищенный продукт очищали преп-ТСХ (PE:EtOAc=10:1) с получением рац-трет-бутил-(3R,4R)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-карбоксилата (80 мг), который использовали без дальнейшей очистки. Вещество растворяли в DCM (2 мл) при КТ и по каплям добавляли 4 M HCl в EtOAc (1 мл). Смесь перемешивали в течение 1 ч, после чего концентрировали при пониженном давлении с получением промежуточного соединения 10 в виде белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7,65-7,77 (м, 1H), 7,51 (т, 1H), 7,18-7,32 (м, 2H), 7,00 (д, 1H), 6,81 (д, 1H), 5,45 (с, 2H), 5,09-5,33 (м, 1H), 3,72 (ддд, 1H), 3,40-3,49 (м, 1H), 3,40-3,49 (м, 1H), 3,32-3,38 (м, 1H), 3,13-3,25 (м, 1H), 2,08-2,39 (м, 2H). Примечание: стереохимия пиперидиновых заместителей была определена как транс по аналогии с опубликованным прецедентом (см., например, WO 2010/022055), но не была подтверждена экспериментально.
Промежуточное соединение 11
Рац-(3R,4S)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-3-олгидрохлорид
Стадия 1
К раствору промежуточного соединения 8 (170 мг, 0,39 ммоль) в DCM (5 мл) при 0°С добавляли Et3N (0,16 мл, 1,2 ммоль) и MsCl (58 мг, 0,51 ммоль) и смесь перемешивали в течение 2 ч. Смесь разбавляли DCM (30 мл), промывали насыщенным водн. NH4Cl и насыщенным солевым раствором, сушили над Na2SO4 и затем концентрировали при пониженном давлении с получением желтого масла. Это вещество растворяли в ДМСО (1,5 мл) и добавляли к суспензии формиата цезия (140 мг, 0,78 ммоль) в ДМСО (1 мл). Смесь перемешивали при 120°С в течение 4 ч и при 25°С в течение 14 ч. Смесь выливали в воду (15 мл) и экстрагировали EtOAc (3 × 15 мл). Объединенный органический раствор промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали преп-ТСХ (PE:EtOAc=4:1) с получением бесцветного масла (60 мг).
Масло растворяли в MeOH (2 мл) при КТ, добавляли K2CO3 и смесь перемешивали в течение 1 ч. Смесь разбавляли EtOAc, промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении с получением неочищенного продукта. Продукт очищали препаративной СФХ с получением рац-трет-бутил-(3R,4S)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-карбоксилата в виде желтой смолы (20 мг, 12%). ЖХМС(ES+): 437 (M+H), 459 (M+Na)
Способ СФХ: Колонка: OJ (250 мм × 30 мм, 5 мкм); Подвижная фаза: CO2 c 15% iPrOH (0,1% NH4OH); Скорость потока: 60 мл/мин; Длина волны: 220 нм. Время удерживания=3,65 мин.
Стадия 2
К раствору рац-трет-бутил (3R,4S)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-карбоксилата (20 мг, 0,046 ммоль) в EtOAc (4 мл) при 0°С добавляли 4 M HCl в EtOAc (4 мл) и смесь перемешивали в течение 2 ч. Затем смесь концентрировали при пониженном давлении с получением неочищенного примера промежуточного соединения 11 в виде светло-желтого масла, которое использовали без очистки. ЖХМС(ES+): 337 (M+H).
Промежуточное соединение 12a
Рац-2-((4-хлор-2-фторбензил)окси)-6-((3R,4S)-3-метилпиперидин-4-ил)пиридинтрифторацетат
Промежуточное соединение 12b
Рац-2-((4-хлор-2-фторбензил)окси)-6-((3R,4R)-3-метилпиперидин-4-ил)пиридинтрифторацетат
Стадия 1
Рац-трет-бутил-(3R,4S)-4-(6-хлорпиридин-2-ил)-3-метилпиперидин-1-карбоксилат и рац-трет-бутил-(3R,4R)-4-(6-хлорпиридин-2-ил)-3-метилпиперидин-1-карбоксилат получали аналогично способу, который был описан для промежуточных соединений 1 и 2, используя трет-бутил-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат в реакции Сузуки. Смесь цис- и транс-изомеров разделяли колоночной хроматографией, элюируя EtOAc в РЕ (градиент 0-15%). Транс (рац-3R,4S)-изомер был элюирован первым.
Рац-трет-бутил-(3R,4S)-4-(6-хлорпиридин-2-ил)-3-метилпиперидин-1-карбоксилат: 1H ЯМР (400 МГц, CDCl3) δ 7,56 (т, 1H), 7,71 (д, 1H), 7,04 (д, 1H), 4,22 (ушир. с, 2H), 2,76 (ушир. с, 1H), 2,43-2,39 (м, 2H), 2,02-1,92 (м, 1H), 1,79-1,71 (м, 2H), 1,48 (с, 9H), 0,70 (д, 3H).
Рац-трет-бутил-(3R,4R)-4-(6-хлорпиридин-2-ил)-3-метилпиперидин-1-карбоксилат: 1H ЯМР (400 МГц, CDCl3) δ 7,59 (т, 1H), 7,16 (д, 1H), 6,99 (д, 1H), 4,36 (ушир. с, 1H), 4,01 (ушир. с, 1H), 3,05 (дт, 2H), 2,79 (ушир. с, 1H), 2,33 (кв, 1H), 2,07-2,01 (м, 1H), 1,71 (д, 1H), 1,46 (с, 9H), 0,66 (д, 3H).
Стадия 2
Промежуточные соединения 12a и 12b получали из соответствующих отделенных изомеров хлорпиридина путем этерификации способом, аналогичным промежуточному соединению 3, стадия 1, и снимали защиту способом, аналогичным промежуточному соединению 9, и использовали без очистки.
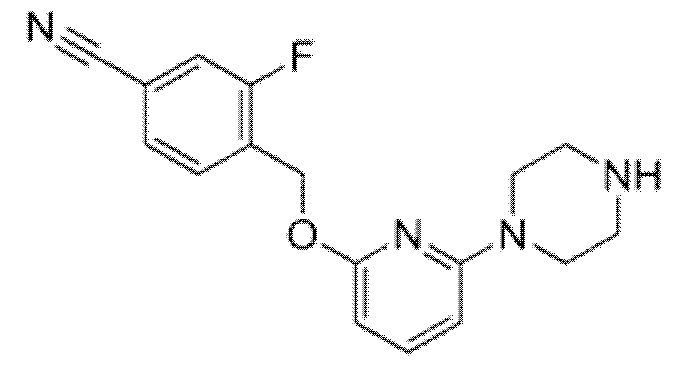
Промежуточное соединение 13
3-фтор-4-(((6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)бензoнитрил-бис-гидрохлорид
Стадия 1
Реакцию проводили в двух параллельных загрузках; пример приготовления загрузки приведен ниже: В перемешиваемую суспензию KOtBu (313 г, 2,79 моль) в THF (4,0 л) добавляли 4-цианo-2-фторбензиловый спирт (281 г, 1,86 моль) порциями при 10-15 °С. Смесь перемешивали при 15°С в течение 45 мин и 2,6-дихлорпиридин (230 г, 1,55 моль) добавляли несколькими порциями в реакционную смесь при 15°С, и смесь перемешивали при 15°С в течение 18 ч. Смесь выливали в насыщ. водн. NH4Cl (10 л). Добавляли EtOAc (10 л) и смесь перемешивали в течение 15 мин. Смесь фильтровали через слой Celite®. Органический слой отделяли и водный слой экстрагировали EtOAc (2 × 6,0 л). Объединенные органические слои промывали насыщенным солевым раствором (5,0 л), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (градиент PE/EtOAc 10-15%) с получением 4-(((6-хлорпиридин-2-ил)окси)метил)-3-фторбензонитрила в виде светло-желтого твердого вещества. Объединенные загрузки давали 550 г (67%). 1H ЯМР (CDCl3) δ 7,67 (т, 1H), 7,58 (т, 1H), 7,48 (дд, 1H), 7,40 (дд, 1H), 6,97 (д, 1H), 6,75 (д, 1H), 5,49 (с, 2H).
Стадия 2
К перемешиваемому раствору 4-(((6-хлорпиридин-2-ил)окси)метил)-3-фторбензонитрила (180 г, 0,685 моль) и трет-бутил пиперазин-1-карбоксилата (140 г, 0,754 моль) в PhCH3 (2,0 л) добавляли Cs2CO3 (446 г, 1,37 моль), BINAP (42,6 г, 0,0685 моль) и Pd2(dba)3 (31,4 г, 0,0343 моль) в атмосфере N2 при 15°С. Смесь дегазировали и повторно заполняли N2 три раза. Полученную смесь нагревали до 120°С в атмосфере N2 в течение 18 ч. Реакционную смесь охлаждали до 80°С и фильтровали через слой Celite®. Фильтровальный осадок промывали EtOAc (4×1,0 л) и объединенные органические слои концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (градиент PE/EtOAc 10-15%). Продукт растирали с PE (1,0 л) при перемешивании при 10°C в течение 2 ч. Твердые вещества собирали фильтрованием с получением трет-бутил-4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата (168 г, 76%) в виде почти белого твердого вещества. 1H ЯМР (CDCl3) δ 7,62 (т, 1H), 7,41-7,49 (м, 2H), 7,38 (дд, 1H), 6,20 (дд, 2H), 5,45 (с, 2H), 3,37-3,57 (м, 8H), 1,49 (с, 9H).
Стадия 3
К раствору EtOH (2,8 мл, 48 ммоль) в EtOAc (20 мл) по каплям добавляли ацетилхлорид (2,0 мл, 28 ммоль). После перемешивания в течение 1 ч при 40°С добавляли трет-бутил-4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилат (1,75 г, 4,24 ммоль) одной порцией и затем смесь перемешивали при 40°С в течение 2 ч. Реакционной смеси давали возможность остыть до КТ и перемешивали в течение 1 ч. EtOAc (10 мл) добавляли к белой суспензии и полученную суспензию энергично перемешивали при КТ в течение 1 ч. Твердое вещество собирали фильтрованием с получением бис-HCl соли целевого продукта промежуточного соединения 13 (1,45 г, 89%) в виде твердого вещества. 1H ЯМР (600 МГц, ДМСО-d6) δ 9,45 (ушир. с, 2H), 7,89 (д, 1H), 7,65-7,73 (м, 2H), 7,55 (м, 1H), 6,44 (д, 1H), 6,22 (д, 1H), 5,42 (с, 2H), 3,61-3,74 (м, 4H), 3,09 (ушир. с, 4H).
Промежуточное соединение 14
(S)-1-(6-((4-Хлор-2-фторбензил)окси)-5-фторпиридин-2-ил)-3-метилпиперазингидрохлорид
Стадия 1
К раствору 2,3,5-трифторпиридина (1,5 г, 11 ммоль) и 4-хлор-2-фторбензилового спирта (1,81 г,11,3 ммоль) в NMP (20 мл) добавляли K2CO3 (4,67 г, 33,8 ммоль) при 25°С, и смесь перемешивали при 100°С в течение 16 ч. Смесь выливали в воду (30 мл) и затем экстрагировали EtOAc (3 × 50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (3×40 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (градиент 0-5% EtOAc/PE) с получением 2-((4-хлор-2-фторбензил)окси)-3,6-дифторпиридина (2,45 г, 80%) в виде бесцветного масла. 1H ЯМР (CDCl3) δ 7,41-7,54 (м, 2H), 7,11-7,20 (м, 2H), 6,47 (ддд, 1H), 5,44 (с, 2H).
Стадия 2
К раствору 2-((4-хлор-2-фторбензил)окси)-3,6-дифторпиридина (200 мг, 0,731 ммоль) и трет-бутил-(S)-2-метилпиперазин-1-карбоксилата (161 мг, 0,804 ммоль) в ДМСО (3 мл) добавляли K2CO3 (303 мг, 2,19 ммоль) при КТ. Реакционную смесь перемешивали при 120°С в течение 18 ч. Смесь выливали в воду (10 мл) и затем экстрагировали EtOAc (3 × 20 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (3×20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали преп-ТСХ (15% EtOAc/PE) с получением трет-бутил-(S)-4-(6-((4-хлор-2-фторбензил)окси)-5-фторпиридин-2-ил)-2-метилпиперазин-1-карбоксилата (60 мг, 18%) в виде бесцветного масла. ЖХМС(ES+): 397,9 (M+H-tBu).
Стадия 3
К раствору (S)-4-(6-((4-хлор-2-фторбензил)окси)-5-фторпиридин-2-ил)-2-метилпиперазин-1-карбоксилата (60 мг, 0,13 ммоль) в DCM (3 мл) добавляли HCl/EtOAc (3 мл). Раствор перемешивали при 30°С в течение 0,5 ч. Суспензию концентрировали при пониженном давлении с получением промежуточного соединения 14 (50 мг, 89%) в виде твердого вещества. ЖХМС(ES+): 353,9 (M+H).
Промежуточное соединение 15
(S)-1-(6-((4-хлор-2-фторбензил)окси)-3-фторпиридин-2-ил)-3-метилпиперазингидрохлорид
Стадия 1
К раствору 2,3,5-трифторпиридина (500 мг, 3,76 ммоль) и трет-бутилпиперазин-1-карбоксилата (753 мг, 3,76 ммоль) в MeCN (8 мл) добавляли Et3N (1,14 г, 11,3 ммоль) при 30°С и реакционную смесь нагревали и затем перемешивали при 70°С в течение 16 ч. Реакционную смесь выливали в воду (20 мл) и экстрагировали EtOAc (3 × 30 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (2×20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (0-5% EtOAc/PE) с получением трет-бутил-(S)-4-(3,6-дифторпиридин-2-ил)-2-метилпиперазин-1-карбоксилата (870 мг, 74%) в виде бледно-коричневого масла. 1H ЯМР (CDCl3) δ 7,24-7,34 (м, 1H), 6,22 (ддд, 1H), 4,31 (ушир. с, 1H), 4,09 (ддт, 1H), 3,92 (дт, 2H), 3,22 (тд, 1H), 3,13 (дд, 1H), 2,93 (тд, 1H), 1,49 (с, 9H), 1,23 (д, 3H).
Стадия 2
К раствору 4-хлор-2-фторбензилового спирта (102 мг, 0,638 ммоль) в DMF (2 мл) добавляли NaH (44,7 мг, 1,12 ммоль, 60% в минеральном масле). Желтую смесь перемешивали при 30°С в течение 15 мин. Затем раствор трет-бутил-(S)-4-(3,6-дифторпиридин-2-ил)-2-метилпиперазин-1-карбоксилата (100 мг, 0,319 ммоль) в DMF (2 мл) добавляли при КТ. Реакционную смесь перемешивали при 90°С в течение 18 ч. Реакционную смесь выливали в воду (30 мл) и затем экстрагировали EtOAc (3 × 30 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (3×20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали преп-ТСХ (EtOAc:PE 5:1) с получением трет-бутил-(S)-4-(6-((4-хлор-2-фторбензил)окси)-3-фторпиридин-2-ил)-2-метилпиперазин-1-карбоксилата (136 мг, 47%) в виде бесцветного масла. ЖХМС(ES+): 397,9 (M+H - tBu).
Стадия 3
К раствору трет-бутил-(S)-4-(6-((4-хлор-2-фторбензил)окси)-3-фторпиридин-2-ил)-2-метилпиперазин-1-карбоксилата (136 мг, 0,300 ммоль) в DCM (4 мл) добавляли HCl/EtOAc (4 мл). Раствор перемешивали при 30°С в течение 2 ч. Суспензию концентрировали при пониженном давлении с получением промежуточного соединения 15 (132 мг, количественно) в виде бледно-желтого твердого вещества. ЖХМС(ES+): 354,1 (M+H).
Промежуточное соединение 16
Метил-4-амино-3-(метиламино)бензоат
Стадия 1
К раствору метил-3-фтор-4-нитробензоата (5,10 г, 25,6 ммоль) в THF (60 мл) по каплям добавляли метиламин (38,4 мл, 76,8 ммоль, 2 M в THF) в течение 10 мин. Бледно-желтый раствор становился насыщенно-оранжевым сразу после добавления, и его перемешивали 2 ч при КТ. Смесь разбавляли Et2O (100 мл) и отделенный органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 5,26 г метил-3-(метиламино)-4-нитробензоата (98%) в виде твердого вещества насыщенно-оранжевого цвета. 1H ЯМР (400 МГц, CDCl3) δ 8,21 (д, 1H), 7,99 (ушир. с, 1H), 7,54 (д, 1H), 7,23 (дд, 1H), 3,94 (с, 3H), 3,08 (д, 3H); ЖХМС(ES+): 211,1 (M+H).
Стадия 2
Метил-3-(метиламино)-4-нитробензоат (5,26 г, 25,0 ммоль) растворяли в EtOH (150 мл). Раствор добавляли в 500 мл колбу Parr®, в которую предварительно загружали 1 г 10% Pd/C (50% воды). Смесь встряхивали в атмосфере H2 при 50 фунт/кв.дюйм (0,34 МПа) в течение 1 ч при КТ. Смесь фильтровали и осадок на фильтре промывали EtOH (100 мл). Бесцветный фильтрат концентрировали при пониженном давлении с получением 4,38 г промежуточного соединения 16 (97%) в виде почти белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,47 (дд, 1H), 7,35 (д, 1H), 6,69 (д, 1H), 3,88 (с, 3H), 3,75 (ушир. с, 2H), 3,22 (ушир. с, 1H), 2,92 (с, 3H); МС(APCI+): 181,1 (M+H).
Промежуточное соединение 17
Метил-2-(хлорметил)-1-метил-1H-бензo[d]имидазол-6-карбоксилат
Промежуточное соединение 16 (206 мг, 1,14 ммоль) растворяли в диоксане (11,5 мл) и обрабатывали хлорацетилхлоридом (109 мкл, 1,37 ммоль). Смесь перемешивали при 100°С в течение 3 ч и охлаждали до КТ. Добавляли Et3N (0,8 мл, 7 ммоль) и гептан (10 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенный продукт очищали флэш-хроматографией (колонка с силикагелем, 40% EtOAc/гептаны) с получением 120 мг промежуточного соединения 17 (44%). 1H ЯМР (400 МГц, CDCl3) δ 8,14 (с, 1H), 8,01 (д, 1H), 7,78 (д, 1H), 4,87 (с, 2H), 3,97 (с, 3H), 3,94 (с, 3H); ЖХМС(ES+): 239,1 (M+H).
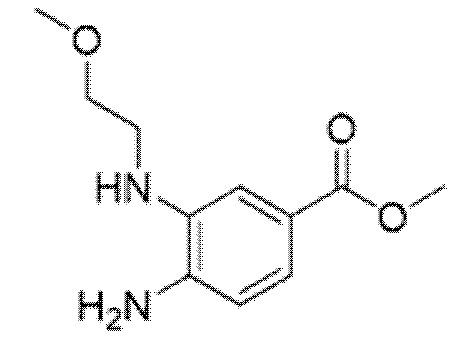
Промежуточное соединение 18
Метил-4-амино-3-((2-метоксиэтил)амино)бензоат
Стадия 1
К бесцветному раствору метил-3-фтор-4-нитробензоата (50 г, 250 ммоль) в THF (400 мл) добавляли Et3N (40,7 г, 402 ммоль, 55,8 мл), затем по каплям добавляли 2-метоксиэтиламин (30,2 г, 402 ммоль) в THF (100 мл) при КТ. Полученный желтый раствор перемешивали при 55°С в течение 18 ч. Раствор охлаждали до КТ и концентрировали при пониженном давлении для удаления THF. Полученное желтое твердое вещество растворяли в EtOAc (800 мл) и промывали насыщ. водн. NH4Cl (250 мл). Водную фазу отделяли и экстрагировали EtOAc (200 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×250 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением метил-3-((2-метоксиэтил)амино)-4-нитробензоата (60,2 г, 94%) в виде желтого твердого вещества. 1H ЯМР (CDCl3) δ 8,23 (д, 1H), 8,17 (ушир. с, 1H), 7,58 (д, 1H), 7,25(dd, 1H), 3,95 (с, 3H), 3,69-3,73 (м, 2H), 3,56 (м, 2H), 3,45 (с, 3H); ЖХМС(ES+): 255,4 (M+H).
Стадия 2
В раствор метил-3-((2-метоксиэтил)амино)-4-нитробензоата (30 г, 118 ммоль) в MeOH (500 мл) добавляли Pd/C (10 г, 94 ммоль). Данную реакционную смесь реакционную смесь перемешивали при КТ, под 15 фунт/кв. дюйм (0,10 МПа) H2 в течение 18 ч. Черную суспензию фильтровали через Celite® и осадок на фильтре промывали МеОН (500 мл). Объединенные фильтраты концентрировали под вакуумом с получением промежуточного соединения 18 (26,5 г, количественно) в виде коричневого масла, которое отверждалось при стоянии. 1H ЯМР (400 МГц, CDCl3) δ 7,48 (дд, 1H), 7,36 (д, 1H), 6,69 (д, 1H), 3,87 (с, 3H), 3,77 (ушир. с, 2H), 3,68 (т, 2H), 3,41 (с, 3H), 3,32 (т, 2H); ЖХМС(ES+): 224,7 (M+H).
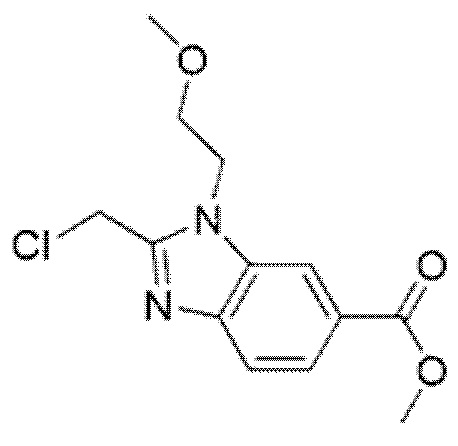
Промежуточное соединение 19
Метил-2-(хлорметил)-1-(2-метоксиэтил)-1H-бензo[d]имидазол-6-карбоксилатгидрохлорид
Раствор промежуточного соединения 18 (5,0 г, 24 ммоль) в диоксане (100 мл) нагревали до 100°С, раствор хлоруксусного ангидрида (4,1 г, 24,5 ммоль) в диоксане (60 мл) добавляли через капельную воронку в течение 10 ч и затем перемешивали в течение еще 12 ч при 100°C. На следующий день реакционную смесь охлаждали до КТ и диоксан удаляли при пониженном давлении. Неочищенную реакционную смесь растворяли в EtOAc и промывали насыщенным раствором NaHCO3. Слой EtOAc отделяли, сушили над Na2SO4 и фильтровали. Раствор 4 М HCl в диоксане (1,1 эквив.) добавляли к раствору продукта EtOAc при постоянном перемешивании. Соль HCl целевого продукта осаждалась в виде бледно-желтого твердого вещества. Суспензию перемешивали в течение 1 ч и затем продукт собирали фильтрованием с получением промежуточного соединения 19 в виде желтого твердого вещества (6,1 г, 86%).1H ЯМР (600 МГц, CD3OD) δ 8,64 (с, 1H), 8,30 (д, 1H), 7,92 (д, 1H), 5,32 (с, 2H), 4,84 (м, 2H), 3,99 (с, 3H), 3,83 (т, 2H), 3,31 (с, 3H). ЖХМС(ES+): 283,2 (M+H).
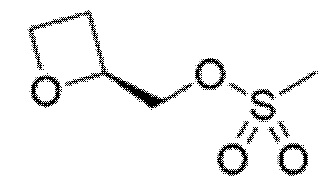
Промежуточное соединение 20
(S)-Оксетан-2-илметилметансульфонат
Стадия 1
К раствору трет-бутоксида калия (670 г, 5,98 моль) в t-BuOH (5 л) добавляли йодид триметилсульфоксония (1,32 кг, 5,98 моль) при 25°С. Смесь нагревали до 60°С и перемешивали в течение 30 мин, затем добавляли (S)-2-((бензилокси)метил)оксиран (500 г, 2,99 моль). Смесь нагревали до 80°C в течение 2 ч. Смесь охлаждали до 25°С и фильтровали через Celite®. Твердую фазу промывали PE (3×2 л). Фильтрат обрабатывали водой (10 л) и экстрагировали РЕ (2 × 5 л). Органический слой промывали насыщенным солевым раствором, сушили, фильтровали и концентрировали под вакуумом. Неочищенный продукт очищали колоночной хроматографией (PE/EtOAc с градиентом от 15:1 до 10:1) с получением (S)-2-((бензилокси)метил)оксетана (280 г, 52,6%) в виде прозрачного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,15-7,34 (м, 5H), 4,90 (тдд, 1H), 4,44-4,67 (м, 4H), 3,49-3,63 (м, 2H), 2,44-2,66 (м, 2H).
Стадия 2
Реакцию проводили в двух параллельных загрузках; пример загрузки приведен ниже: К раствору (S)-2-((бензилокси)метил)оксетана (140 г, 780 ммоль) в THF (1,4 л) добавляли Pd(OH)2 (14 г) в атмосфере азота. Смесь нагревали до 45°C и перемешивали в атмосфере H2 (50 фунт/кв. дюйм (0,34 МПа)) в течение 16 ч. Смесь охлаждали до 25°С и фильтровали через Celite® с получением целевого соединения (S)-оксетан-2-илметанола в виде раствора в THF. Небольшую аликвоту проверяли с помощью 1Н ЯМР, и оставшийся раствор использовали непосредственно на следующей стадии. 1H ЯМР (400 МГц, ДМСО-d6) δ 4,76-4,90 (м, 1H), 4,66 (тдд, 1H), 4,46 (ддд, 1H), 4,37 (тд, 1H), 3,47 (дд, 2H), 2,32-2,58 (м, 2H).
Стадия 3
Реакцию проводили в двух параллельных загрузках; пример загрузки приведен ниже: К раствору (S)-оксетан-2-илметанола (со стадии 2, предположительно 69 г, 780 ммоль) в THF (1,4 л) добавляли Et3N (197 г, 1,95 моль) при 0°С. Добавляли по каплям метансульфоновый ангидрид (204 г, 1,17 моль), поддерживая температуру содержимого ниже 10°С. Смесь перемешивали при 25°С в течение 2 ч. Две загрузки объединяли и смесь обрабатывали водой (1 л) и слои разделяли. Водную фазу экстрагировали DCM (3 × 2 л). Объединенный органический раствор сушили, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (градиент EtOAc/PE 50-100%) с получением промежуточного соединения 20 (250 г, 96% в две стадии) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 4,98-5,09 (м, 1H), 4,69 (ддд, 1H), 4,59 (тд, 1H), 4,37 (д, 2H), 3,11 (с, 3H), 2,72-2,82 (м, 1H), 2,64 (тдд, 1H).
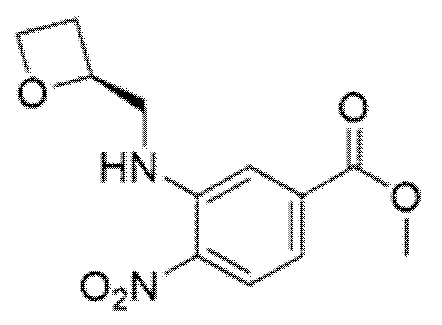
Промежуточное соединение 21
Метил-(S)-4-нитро-3-((оксетан-2-илметил)амино)бензоат
Стадия 1
К раствору (S)-оксетан-2-илметилметансульфоната (180 г, 1,08 моль) в DMF (1,2 л) добавляли азид натрия (105 г, 1,62 моль). Смесь нагревали до 80°С и перемешивали в течение 16 ч. Смесь охлаждали до 0°С и обрабатывали простым диэтиловым эфиром (1,5 л), и полученную суспензию перемешивали в течение 30 мин. Твердые вещества удаляли фильтрованием и осадок на фильтре промывали диэтиловым эфиром (2 × 200 мл). Диэтиловый эфир удаляли под вакуумом при 25°С с получением раствора (S)-2-(азидометил)оксетана в DMF (~1,2 л), который использовали непосредственно на следующей стадии.
Стадия 2
Реакцию проводили в трех параллельных загрузках; пример загрузки приведен ниже: К раствору (S)-2-(азидометил)оксетана (предположительно 41 г, 360 ммоль) в DMF (~400 мл) и THF (1 л) добавляли 10% Pd/C (50% масс. влажность, 13 г) в атмосфере азота. Смесь перемешивали при 25°C в атмосфере H2 (50 фунт/кв. дюйм (0,34 МПа)) в течение 16 ч. Раствор фильтровали через Celite®, добавляли 10% Pd/C (сухой, 4,0 г) и смесь перемешивали при 40°C в атмосфере H2 (50 фунт/кв. дюйм (0,34 МПа)) в течение 3 ч, после чего анализ ТСХ показывал, что реакция завершилась. Смесь охлаждали до 0°С и все три загрузки объединяли. Смесь фильтровали через Celite® с получением раствора (S)-2-(аминометил)оксетана в DMF (~1,4 л) и THF (~2,6 л), который использовали непосредственно на следующей стадии.
Стадия 3
К раствору (S)-2-(аминометил)оксетана (предположительно 94 г, 1,08 моль) в DMF (~1,4 л) и THF (~2,6 л) добавляли Et3N (327 г, 3,24 моль) и метил-3-фтор-4-нитробензоат (200 г, 1,0 моль) при 25°С. Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении для удаления THF и оставшийся раствор разбавляли водой (1 л). Смесь экстрагировали EtOAc (2 × 1,5 л). Объединенные органические экстракты промывали насыщенным солевым раствором (2 ×), сушили и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (градиент EtOAc/PE=10-50%) с получением промежуточного соединения 21 (158 г, 55%) в виде желтого твердого вещества. 1H ЯМР (600 МГц, CDCl3) δ 8,38 (ушир. с, 1H), 8,25 (д, 1H), 7,64 (с, 1H), 7,27 (д, 1H), 5,13-5,20 (м, 1H), 4,70-4,82 (м, 1H), 4,64 (тд, 1H), 3,95 (с, 3H), 3,57-3,71 (м, 2H), 2,71-2,86 (м, 1H), 2,55-2,70 (м, 1H); МС(ES+)=266,7.
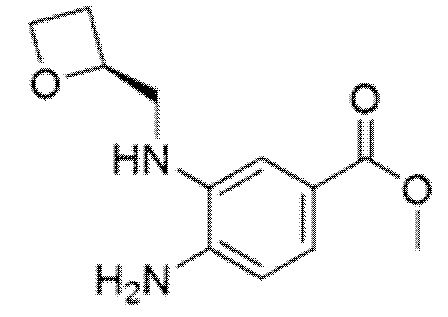
Промежуточное соединение 22
Метил-(S)-4-амино-3-((оксетан-2-илметил)амино)бензоат
Промежуточное соединение 21 (15 г, 56 ммоль) растворяли в THF (100 мл) в реакторе Parr®. В реактор добавляли Pd/C (10% мас./мас., 1,5 г) и смесь встряхивали при КТ в атмосфере H2 при 50 фунт/кв.дюйм (0,34 МПа) в течение 4 ч. Смесь фильтровали через Celite® и фильтрат концентрировали при пониженном давлении с получением промежуточного соединения 22 (12,3 г, 92%) в виде желтовато-коричневого твердого вещества. 1H ЯМР (600 МГц, CDCl3) δ 7,49 (дд, 1H), 7,39 (д, 1H), 6,70 (д, 1H), 5,05-5,18 (м, 1H), 4,76 (ддд, 1H), 4,62 (дт, 1H), 3,87 (с, 3H), 3,42-3,50 (м, 1H), 3,34-3,40 (м, 1H), 2,71-2,82 (м, 1H), 2,60 (ддт, 1H).
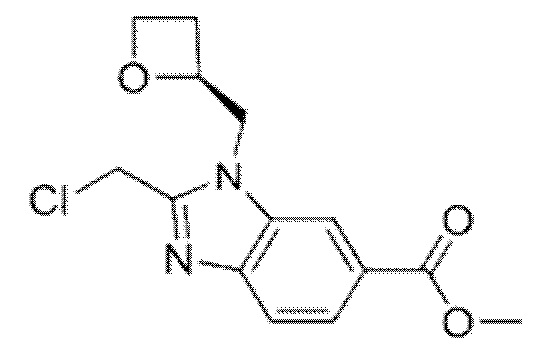
Промежуточное соединение 23
Метил-(S)-2-(хлорметил)-1-(оксетан-2-илметил)-1H-бензo[d]имидазол-6-карбоксилат
К раствору промежуточного соединения 22 (127 г, 0,54 моль) в MeCN (500 мл) добавляли 2-хлор-1,1,1-триметоксиэтан (76,2 мл, 0,57 моль) и pTSA·H2O (5,12 г, 26,9 ммоль). Смесь нагревали до 60°C в течение 1 ч. Реакционную смесь охлаждали до КТ и концентрировали при пониженном давлении. Неочищенный продукт растирали в смеси 50% EtOAc/гептан. Твердые вещества собирали фильтрованием с получением промежуточного соединения 23 (79 г, 50%) в виде желтовато-коричневого твердого вещества. 1H ЯМР (600 МГц, CDCl3) δ 8,12 (с, 1H), 8,00 (д, 1H), 7,79 (д, 1H), 5,16-5,26 (м, 1H), 5,03 (с, 2H), 4,57-4,66 (м, 2H), 4,48-4,56 (м, 1H), 4,33 (м, 1H), 3,95 (с, 3H), 2,71-2,81 (м, 1H), 2,36-2,47 (м, 1H).
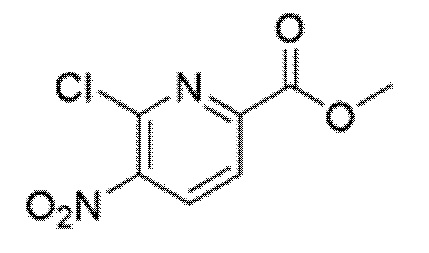
Промежуточное соединение 24
Метил-6-хлор-5-нитропиколинат
Стадия 1
2-Хлор-6-метил-3-нитропиридин (97 г, 560 ммоль) медленно добавляли в колбу, предварительно загруженную 18 М H2SO4 (400 мл), при перемешивании. Триоксид хрома (169 г, 1,69 моль) добавляли к реакционной смеси небольшими порциями, поддерживая температуру ниже 50°C. Реакционную смесь перемешивали при 15°С в течение 20 ч. Полученную зеленую смолу выливали в 2 кг льда, и полученные твердые вещества собирали фильтрованием и сушили под вакуумом, получая 6-хлор-5-нитропиколиновую кислоту (103 г, 90%) в виде бледного твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,70 (д, 1H), 8,24 (д, 1H).
Стадия 2
К суспензии 6-хлор-5-нитропиколиновой кислоты (103 г, 508,51 ммоль) в CH2Cl2 (1 л) добавляли оксалилхлорид (129 г, 1,02 моль) и DMF (6 мл) при 0°С. Реакционную смесь перемешивали при 15°С в течение 1 ч. К реакционной смеси добавляли МЕОН (60 мл) при 15°С. Раствор перемешивали при 15°С в течение дополнительных 10 мин. Желтый раствор концентрировали при пониженном давлении и полученный неочищенный продукт очищали колоночной хроматографией (градиент EtOAc/PE: 0-20%) с получением промежуточного соединения 24 (106 г, 96%) в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,55 (д, 1H), 8,27 (д, 1H), 4,01 (с, 3H).
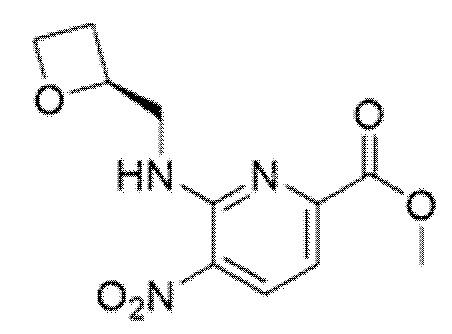
Промежуточное соединение 25
Метил-(S)-5-нитро-6-((оксетан-2-илметил)амино)пиколинат
Стадия 1
Раствор (S)-2-(аминометил)оксетана (предположительно 152 г, 1,7 моль) в DMF (3 л) и THF (3 л) получали из промежуточного соединения 20 как описано для промежуточного соединения 21 (стадии 1 и 2). Промежуточное соединение 24 (270 г, 1,25 моль) и Et3N (500 г, 5,1 моль) добавляли к раствору промежуточного соединения 20 (152 г, 1,7 моль) в DMF (3 л) и THF (3 л) при 25°С. Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении для удаления THF и добавляли воду (5 л). Смесь экстрагировали EtOAc (2 × 5 л) и объединенные органические растворы промывали насыщенным солевым раствором (2 ×), сушили и концентрировали при пониженном давлении. Неочищенный материал объединяли со второй загрузкой неочищенного продукта из аналогичного эксперимента (70 г) и твердые вещества растирали с РЕ:EtOAc (4:1, 500 мл) в течение 2 ч. Твердые вещества собирали фильтрованием и сушили с получением промежуточного продукта 25 (304 г, 52%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,58 (ушир. с, 1H), 8,56 (д, 1H), 7,39 (д, 1H), 5,08-5,18 (м, 1H), 4,73 (ддд, 1H), 4,61 (тд, 1H), 4,06-4,16 (м, 1H), 3,98 (с, 3H), 3,88-3,97 (м, 1H), 2,68-2,80 (м, 1H), 2,55 (тдд, 1H).
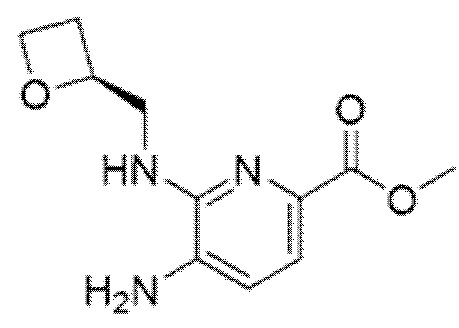
Промежуточное соединение 26
Метил-(S)-5-амино-6-((оксетан-2-илметил)амино)пиколинат
Промежуточное соединение 25 (10 г, 37 ммоль) суспендировали в MeOH (150 мл) и обрабатывали с 10% Pd/C (1,0 г), и смесь перемешивали при КТ при 50 фунт/кв. дюйм (0,34 МПа) H2 в течение 4 ч. Смесь фильтровали через Celite® и фильтрат концентрировали при пониженном давлении с получением промежуточного соединения 26 (8,4 г, 95%) в виде желтого масла, которое отверждалось при стоянии. 1H ЯМР (600 МГц, CDCl3) δ 7,49 (д, 1H), 6,86 (д, 1H), 5,06-5,15 (м, 1H), 4,68-4,77 (м, 1H), 4,53-4,63 (м, 2H), 3,91 (с, 3H), 3,80-3,86 (м, 2H), 3,72 (ушир. с, 2H), 2,68-2,78 (м, 1H), 2,52-2,61 (м, 1H).
Промежуточное соединение 27
Метил-(S)-2-(хлорметил)-3-(оксетан-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоксилат
В 2 л трехгорлой колбе, снабженной механической мешалкой с верхним приводом, промежуточное соединение 26 (43,0 г, 181 ммоль) растворяли в THF (780 мл). Полученную бледно-розовую суспензию обрабатывали раствором хлоруксусного ангидрида (33,5 г, 190 ммоль в 100 мл THF) через капельную воронку в течение 30 мин. Полученный светло-янтарный раствор перемешивали при КТ в течение 2 ч, и затем нагревали при 60°С в течение 7 ч. Реакционную смесь охлаждали до КТ. Приблизительно 400 мл растворителя из реакционной смеси удаляли при пониженном давлении на ротационном испарителе. Полученный раствор разбавляли EtOAc (500 мл) и обрабатывали насыщ. водн. NaHCO3 (200 мл). Двухфазную смесь перемешивали при КТ в течение 30 мин. Органический слой отделяли и водный слой экстрагировали EtOAc (500 мл). Объединенные органические слои промывали насыщенным солевым раствором (500 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 27 (52,5 г, 98%) в виде желтовато-коричневого твердого вещества. 1H ЯМР (600 МГц, CDCl3) δ 8,14 (д, 2H), 5,19-5,28 (м, 1H), 4,99-5,16 (м, 2H), 4,70-4,88 (м, 2H), 4,55-4,67 (м, 1H), 4,24-4,44 (м, 1H), 4,01 (с, 3H), 2,70-2,88 (м, 1H), 2,37-2,53 (м, 1H); ЖХМС(ES+): 296,4 (M+H).
Промежуточное соединение 28
цис(+/-)Метил-3-(((2-метоксициклопентил)метил)амино)-4-нитробензоат
Стадия 1
В колбу, содержащую цис(+/-)-2-(аминометил)циклопентан-1-ол (300 мг, 2,60 ммоль), метил-3-фтор-4-нитробензоат (571 мг, 2,87 ммоль) и Et3N (1,1 мл, 7,8 ммоль), добавляли DMF и смесь перемешивали в течение ночи. Реакционную смесь разбавляли водой и экстрагировали EtOAc. Органический экстракт концентрировали при пониженном давлении и неочищенный продукт очищали флэш-хроматографией (EtOAc/гептаны) с получением цис(+/-)метил-3-(((-2-гидроксициклопентил)метил)амино)-4-нитробензоата (493 мг, 64%). 1H ЯМР (CDCl3) δ 8,21 (ушир. с, 1H), 8,19 (д, 1H), 7,62 (с, 1H), 7,20 (д, 1H), 4,38 (ушир. с, 1H), 3,94 (с, 3H), 3,60 (ддд, 1H), 3,38-3,50 (м, 1H), 2,13-2,25 (м, 1H), 1,84-2,01 (м, 3H), 1,57-1,78 (м, 4H).
Стадия 2
В колбу, содержащую раствор цис(+/-)метил-3-(((2-гидроксициклопентил)метил)амино)-4-нитробензоата (0,48 г, 1,6 ммоль) в DCM ( 50 мл) добавляли 1,8-бис(диметиламино)нафталин (0,35 г, 1,6 ммоль). Раствор перемешивали в течение 5 мин и затем порциями добавляли триметилоксонийтетрафторборат (0,48 г, 3,3 ммоль) в течение 10 мин. Затем реакционную смесь перемешивали в течение дополнительных 18 ч при КТ. В колбу добавляли воду и полученную смесь экстрагировали DCM. Объединенные органические слои фильтровали и раствор концентрировали при пониженном давлении. Неочищенную смесь очищали флэш-хроматографией (EtOAc/гептаны) с получением промежуточного соединения 28 (0,4 г, 80%). 1H ЯМР (600 МГц, CDCl3) δ 8,19 (д, 1H), 7,61 (с, 1H), 7,20-7,12 (м, 1H), 3,93 (с, 3H), 3,57-3,50 (м, 1H), 3,43 (дд, 6,3 Гц, 1H), 3,30 (с, 3H), 2,22 (д, 1H), 1,89-1,43 (м, 7H).
Промежуточное соединение 29
Трет-бутил-3-фтор-4-нитробензоат
3-Фтор-4-нитробензойную кислоту (2,60 г, 14,0 ммоль) растворяли в THF (30 мл), смесь обрабатывали Boc-ангидридом (6,13 г, 28,1 ммоль) и DMAP (525 мг, 4,21 ммоль) и затем перемешивали при КТ. Густая суспензия быстро образовывалась и затем перемешивалась в течение 3 ч при 40°С, за это время суспензия становилась желтовато-коричневым раствором. После концентрирования реакционной смеси при пониженном давлении остаток растворяли в EtOAc, адсорбировали на силикагеле и затем элюировали через тонкий слой силикагеля 50% EtOAc/гептаном. Фильтрат концентрировали при пониженном давлении с получением промежуточного соединения 29 (8,88 г, 68%) в виде желтого масла. 1H ЯМР (600 МГц, CDCl3) δ 8,05-8,09 (м, 1H), 7,86-7,90 (м, 2H), 1,61 (с, 9H).
Промежуточное соединение 30
5-Бром-N3-метилпиридин-2,3-диамин
Промежуточное соединение 31
5-Бром-N3,6-диметилпиридин-2,3-диамин
Промежуточное соединение 30 синтезировали в соответствии с методикой, описанной в литературе (Choi, J. Y. et al. J. Med. Chem. 2012, 55, 852-870). Промежуточное соединение 31 синтезировали тем же методом.
Промежуточное соединение 32
Метил-2-(хлорметил)-1-((1-метил-1H-имидазол-5-ил)метил)-1H-бензo[d]имидазол-6-карбоксилат
Стадия 1
К бесцветному раствору метил-3-фтор-4-нитробензоата (1,0 г, 5,0 ммоль) в DMF (10 мл) медленно добавляли (1-метил-1H-имидазол-5-ил)метанамин (670 мг, 6,0 ммоль) и Et3N (762 мг, 7,53 ммоль). Раствор перемешивали при 60°С в течение 16 ч. Реакционную смесь выливали в H2O (30 мл) и экстрагировали DCM (3 × 30 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (20% MeOH/DCM). Полученное желтое твердое вещество растирали с 30:1 PE/EtOAc с получением метил-3-(((1-метил-1H-имидазол-5-ил)метил)амино)-4-нитробензоата (1,2 г, 82%) в виде желтого твердого вещества. 1H ЯМР (CDCl3) δ 8,26 (д, 1H), 7,96 (ушир. с, 1H), 7,71 (д, 1H), 7,50 (с, 1H), 7,35 (дд, 1H), 7,13 (с, 1H), 4,55 (д, 2H), 3,97 (с, 3H), 3,68 (с, 3H).
Стадия 2
К желтой суспензии метил-3-(((1-метил-1H-имидазол-5-ил)метил)амино)-4-нитробензоата (5,46 г, 18,8 ммоль) в MeOH (160 мл) добавляли влажный 10% Pd/C (1 г). Смесь перемешивали при 1 атм (0,10 МПа) Н2 в течение 36 ч при 20°С. Реакционную смесь фильтровали и осадок на фильтре промывали МеОН (200 мл). Фильтрат концентрировали при пониженном давлении с получением метил-4-амино-3-(((1-метил-1H-имидазол-5-ил)метил)амино)бензоата (4,8 г, 98%) в виде коричневого твердого вещества. 1H ЯМР (ДМСО-d6) δ 7,56 (с, 1H), 7,18 (д, 1H), 7,13 (с, 1H), 6,87 (с, 1H), 6,55 (д, 1H), 5,50 (с, 2H), 4,84 (т, 1H), 4,23 (д, 2H), 3,73 (с, 3H), 3,63 (с, 3H).
Стадия 3
Красную смесь метил-4-амино-3-(((1-метил-1H-имидазол-5-ил)метил)амино)бензоата (780 мг, 3,00 ммоль) и 2-гидроксиуксусной кислоты (342 мг, 4,49 ммоль) в мезитилене (8 мл) перемешивали при 140°С в атмосфере N2 в течение 14 ч и при 25°С в течение 48 ч. Прозрачный желтый раствор декантировали с получением коричневого остатка, который растворяли в MeOH (50 мл) и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (20% MeOH/DCM) с получением метил-2-(гидроксиметил)-1-((1-метил-1H-имидазол-5-ил)метил)-1H-бензo[d]имидазол-6-карбоксилат (318 мг, 35%) в виде желтой пены. 1H ЯМР (ДМСО-d6) δ 8,13 (д, 1H), 7,83 (дд, 1H), 7,71 (д, 1H), 7,60 (с, 1H), 6,59 (с, 1H), 5,69 (с, 2H), 4,76 (с, 2H), 3,91 (с, 1H), 3,84 (с, 3H), 3,53 (с, 3H).
Стадия 4
К желтой суспензии 2-(гидроксиметил)-1-((1-метил-1H-имидазол-5-ил)метил)-1H-бензo[d]имидазол-6-карбоксилата (500 мг, 1,66 ммоль) в DCM (10 мл) и DMF (3 мл) по каплям добавляли SOCl2 (990 мг, 0,60 мл, 8,32 ммоль) при КТ. Реакционную смесь перемешивали при КТ в течение 1 ч, концентрировали при пониженном давлении и полученный коричневый остаток растирали с DCM (10 мл). Твердые вещества собирали фильтрованием, промывали DCM (5 мл) и сушили под вакуумом с получением промежуточного соединения 32 (431 мг, 73%) в виде почти белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,17 (с, 1H), 8,31 (с, 1H), 7,91-7,99 (м, 1H), 7,77-7,87 (м, 1H), 7,11 (с, 1H), 5,92 (с, 2H), 5,13 (с, 2H),) 3,87 (с, 3H), 3,86 (с, 3H); МС(ES+): 319,0 (M+H).
Промежуточное соединение 33
5-Хлор-2-(хлорметил)-3-метил-3H-имидазо[4,5-b]пиридин
Стадия 1
К суспензии 2,6-дихлор-3-нитропиридина (200 г, 1,04 моль) и Na2CO3 (132 г, 1,24 моль) в EtOH (1 л) по каплям добавляли 2,0 M MeNH2 в THF (622 мл,1,24 моль) при 0°С при помощи шприца. После добавления реакционную смесь перемешивали при 18°С в течение 6 ч. Желтую смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением желтого твердого вещества. Неочищенный продукт очищали флэш-хроматографией (PE/EtOAc 0-5%) с получением 6-хлор-N-метил-3-нитропиридин-2-амина (158 г, 81% выход) в виде желтого твердого вещества. 1H ЯМР (ДМСО-d6) δ 8,72 (ушир. с, 1H), 8,41 (д, 1H), 6,76 (д, 1H), 3,00 (д, 3H).
Стадия 2
К смеси 6-хлор-N-метил-3-нитропиридин-2-амина (15,8 г, 84,2 ммоль) в AcOH (100 мл) добавляли железный порошок (15,4 г, 276 ммоль). Желтую смесь перемешивали при 80°С в течение 3 ч. Реакционную смесь охлаждали до КТ и фильтровали. Осадок на фильтре промывали EtOAc (2 × 100). Объединенные органические слои концентрировали при пониженном давлении и неочищенный продукт очищали флэш-хроматографией (120 г силикагеля, 50% EtOAc/PE) с получением 3-амино-6-хлор-2-метиламинопиридина (8,40 г, 63% выход) в виде коричневого твердого вещества. 1H ЯМР (CDCl3) δ 6,80 (д, 1H), 6,50 (д, 1H), 3,39 (ушир. с, 2H), 3,01 (с, 3H).
Стадия 3
К раствору 3-амино-6-хлор-2-метиламинопиридина (50,0 г, 317 ммоль) в диоксане (1,2 л) добавляли хлорацетилхлорид (55,5 мл, 698 ммоль) и смесь перемешивали при 15°С в течение 50 мин. Коричневую смесь концентрировали при пониженном давлении с получением коричневого твердого вещества, которое растворяли в TFA (1,2 л) и перемешивали при 80°С в течение 60 ч. Смесь концентрировали при пониженном давлении с получением коричневого масла. Масло разбавляли EtOAc (1 л) и нейтрализовали насыщ. водн. NaHCO3. Когда выделение CO2 уменьшалось, слои разделяли и водный слой экстрагировали EtOAc (200 мл). Органические экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (градиент 10-25% EtOAc/PE) с получением промежуточного соединения 33 (61,0 г, 79% выход) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,13 (д, 1H), 7,37 (д, 1H), 5,11 (с, 2H), 3,84 (с, 3H).
Промежуточное соединение 34
Метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензo[d]имидазол-6-карбоксилат
Смесь промежуточного соединения 3 (13,0 г, 23,8 ммоль) и промежуточного соединения 19 (6,72 г, 23,8 ммоль) и K2CO3 (16,4 г, 119 ммоль) в MeCN (200 мл) перемешивали при 50°С в течение 12 ч. Смесь охлаждали до КТ и выливали в воду (200 мл). Смесь экстрагировали EtOAc (3 × 500 мл) и объединенные органические слои промывали насыщенным солевым раствором (2×500 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (120 г, силикагель, градиент 0-2% MeOH/DCM) с получением промежуточного соединения 34 (12,5 г, 93%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,16 (с, 1H), 7,97 (д, 1H), 7,75 (д, 1H), 7,50 (т, 1H), 7,44 (т, 1H), 7,11 (м, 2H), 6,73 (д, 1H), 6,61 (д, 1H), 5,41 (с, 2H), 4,64 (т, 2H), 3,96 (с, 3H), 3,92 (с, 2H), 3,79 (т, 2H), 3,31 (с, 3H), 2,99 (д, 2H), 2,58-2,67 (м, 1H), 2,29 (т, 2H), 1,78-1,91 (м, 4H).
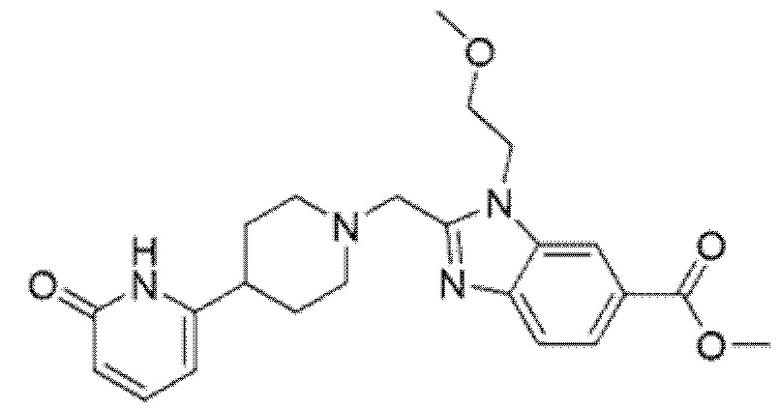
Промежуточное соединение 35
Метил-1-(2-метоксиэтил)-2-((4-(6-оксо-1,6-дигидропиридин-2-ил)пиперидин-1-ил)метил)-1H-бензo[d]имидазол-6-карбоксилат
В перемешиваемую суспензию промежуточного соединения 34 (500 мг, 0,88 ммоль) в MeOH (10 мл) добавляли 4 M HCl в диоксане (4,5 мл, 20 ммоль). Реакционную смесь нагревали до 70°С и перемешивали в течение 18 ч. Затем смесь охлаждали до КТ и концентрировали при пониженном давлении. Остаток растворяли в насыщ. водн. NaHCO3 и экстрагировали DCM (3×). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в минимальном количестве DCM и медленно добавляли PE до образования осадка. Смесь перемешивали для гранулирования твердых веществ в течение 2 ч. Твердые вещества отделяли фильтрованием и промывали РЕ с получением промежуточного соединения 35 (280 мг, 75%) в виде почти белого твердого вещества. 1H ЯМР (600 МГц, CDCl3) δ 11,43 (ушир. с, 1H), 8,13 (с, 1H), 7,96 (д, 1H), 7,73 (д, 1H), 7,36 (дд, 1H), 6,39 (д, 1H), 6,02 (д, 1H), 4,61 (т, 2H), 3,96 (с, 3H), 3,92 (с, 2H), 3,75 (т, 2H), 3,29 (с, 3H), 2,99 (д, 2H), 2,51 (т, 1H), 2,30 (т, 2H), 1,93 (д, 2H), 1,72 (кв.д, 2H).
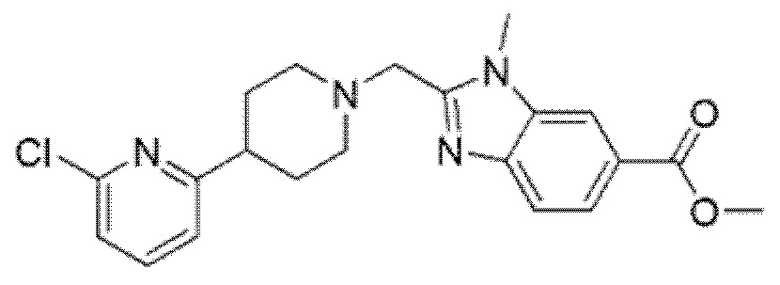
Промежуточное соединение 36
Метил-2-((4-(6-хлорпиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-бензo[d]имидазол-6-карбоксилат
Стадия 1
К бесцветному раствору промежуточного соединения 2 (6,00 г, 20,2 ммоль) в DCM (60 мл) добавляли 4 М HCl/EtOAc (60 мл) и раствор становился мутным. Суспензию перемешивали при 20°С в течение 2 ч и затем концентрировали при пониженном давлении с получением 2-хлор-6-(пиперидин-4-ил)пиридингидрохлорида (5,45 г, 99%) в виде твердого вещества. 1H ЯМР (ДМСО-d6) δ 9,32 (ушир. с, 1H), 8,95 (ушир. с, 1H), 7,83 (т, 1H), 7,37 (д, 1H), 7,31 (д, 1H), 3,31 (д, 2H), 2,89-3,06 (м, 3H), 1,85-2,04 (м, 4H).
Стадия 2
К смеси 2-хлор-6-(пиперидин-4-ил)пиридингидрохлорида (5,45 г, 20,2 ммоль) и K2CO3 (8,38 г, 60,6 ммоль) в DMF (50 мл) добавляли этил-2-бромацетат (4,05 г, 24,3 ммоль). Смесь перемешивали при 20°C в течение 2 ч и затем разбавляли EtOAc (300 мл) и промывали водой (100 мл). Органический слой промывали насыщенным солевым раствором (200 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (градиент EtOAc/PE 5-15%) с получением этил-2-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)ацетата (5,44 г, 95%) в виде желтого масла. 1H ЯМР (CDCl3) δ 7,59 (т, 1H), 7,16 (д, 1H), 7,11 (д, 1H), 4,21 (кв, 2H), 3,26 (с, 2H), 3,08 (д, 2H), 2,72 (тт, 1H), 2,31 (дт, 2H), 1,82-2,02 (м, 4H), 1,29 (т, 3H).
Стадия 3
К раствору этил-2-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)ацетата (5,44 г, 19,2 ммоль) в EtOH (50 мл) добавляли 5 M NaOH (11,5 мл, 57,5 ммоль). Раствор перемешивали при 25°С в течение 2 ч. Реакционную смесь гасили 1 М HCl и экстрагировали смесью DCM/MeOH (10:1, 5 × 80 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 2-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)уксусной кислоты (4,50 г, 92%) в виде желтого твердого вещества. 1H ЯМР (CD3OD) δ 7,71 (т, 1H), 7,24 (д, 2H), 3,20 (д, 2H), 3,13 (ушир. с, 2H), 2,70-2,83 (м, 1H), 2,29 (ушир. с, 2H), 1,83-2,06 (м, 4H).
Стадия 4
К желтому раствору 2-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)уксусной кислоты (4,50 г, 17,7 ммоль) и промежуточного соединения 16 (3,50 г, 19,4 ммоль) в DMF (50 мл) добавляли HATU (8,06 г, 21,2 ммоль) при КТ. Реакционную смесь перемешивали при 15°С в течение 20 мин, и затем добавляли Et3N (3,58 г, 35,3 ммоль). Желтую смесь перемешивали при 50°С в течение 2 ч. Полученную коричневую смесь выливали в воду (160 мл) и экстрагировали EtOAc (3 × 100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (3×100 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (градиент MeOH/DCM 0-5%) с получением метил-4-амино-3-(2-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)-N-метилацетамидо)бензоата (7,37 г, количественно) в виде желтого масла. ЖХМС(ES+): 417,1 (M+H).
Стадия 5
Смесь метил-4-амино-3-(2-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)-N-метилацетамидо)бензоата (7,37 г, 17,7 ммоль) в AcOH (100 мл) перемешивали при 60°С в течение 16 ч. Коричневую смесь концентрировали при пониженном давлении с получением коричневого масла, которое растворяли в EtOAc (300 мл) и промывали насыщ. водн. NaHCO3 (100 мл). Органический слой промывали насыщенным солевым раствором (3×100 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (градиент EtOAc/PE 0-50%) с получением промежуточного соединения 36 (3,51 г, 50%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,14 (с, 1H), 7,98 (д, 1H), 7,75 (д, 1H), 7,58 (т, 1H), 7,17 (д, 1H), 7,10 (д, 1H), 3,99 (с, 3H), 3,97 (с, 3H), 3,95 (ушир. с, 2H), 3,09 (д, 2H), 2,77 (ушир. с, 1H), 2,43 (ушир. с, 2H), 1,83-2,04 (м, 4H); ЖХМС(ES+): 399,1 (M+H).
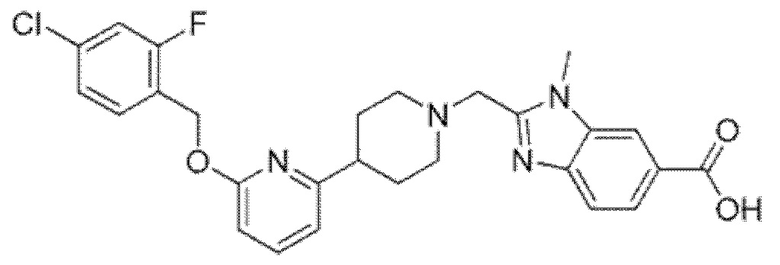
Пример 1A-01
Гидрохлорид 2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-бензo[d]имидазол-6-карбоновой кислоты
Стадия 1
Промежуточное соединение 17 (115 мг, 0,482 ммоль), промежуточное соединение 3 (178 мг, 0,554 ммоль) и K2CO3 (133 мг, 0,96 ммоль) объединяли в MeCN (4,8 мл) и смесь оставляли перемешиваться при 35°С в течение 3 ч. Реакционную смесь охлаждали до КТ, разбавляли EtOAc и экстрагировали водой. Органический слой сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали флэш-хроматографией (24 г силикагеля, 0-100% EtOAc/гептан) с получением 215 мг метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-бензo[d]имидазол-6-карбоксилата (85%) в виде белой пены. 1H ЯМР (400 МГц, CDCl3) δ 8,10 (ушир. с, 1H), 7,95 (д, 1H), 7,72 (д, 1H), 7,51-7,36 (м, 2H), 7,07 (ушир. с, 2H), 6,71 (ушир. с, 1H), 6,57 (д, 1H), 5,38 (ушир. с, 2H), 3,95 (с, 3H), 3,93 (с, 3H), 3,84 (ушир. с, 2H), 2,97 (ушир. с, 2H), 2,59 (ушир. с, 1H), 2,27 (ушир. с, 2H), 1,75-1,93 (м, 4H); ЖХМС(ES+): 523,3 (M+H).
Стадия 2
Метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-бензo[d]имидазол-6-карбоксилат (215 мг, 0,411 ммоль) суспендировали в MeOH (4 мл) и обрабатывали 2 M NaOH (820 мкл, 1,64 ммоль). Реакционную смесь оставляли перемешиваться при 40°C в течение 3 ч и в течение 14 ч при КТ. Реакционную смесь снова нагревали до 40°C и подкисляли 1 М HCl (2,50 мл, 2,50 ммоль). Смеси давали возможность остыть до КТ и, когда начинал образовываться осадок, через реакционную смесь продували пар N2 для удаления приблизительно половины МеОН. Затем твердое вещество собирали фильтрованием, промывали H2O (2 × 2 мл) и затем сушили под N2 с получением соединения примера 1A-01 (155 мг, 69%) в виде твердого вещества. 1H ЯМР (600 МГц, ДМСО-d6) δ 12,74 (ушир. с, 1H), 8,15 (с, 1H), 7,79 (д, 1H), 7,58-7,65 (м, 2H), 7,54 (т, 1H), 7,43 (д, 1H), 7,27 (д, 1H), 6,85 (д, 1H), 6,65 (д, 1H), 5,34 (с, 2H), 3,94 (с, 3H), 3,82 (с, 2H), 2,93 (д, 2H), 2,57 (т, 1H), 2,19 (т, 2H), 1,73-1,80 (м, 2H), 1,64-1,73 (м, 2H); ЖХМС(ES+): 509,2 (M+H).
Соединения, перечисленные в приведенной ниже таблице 1, были получены с использованием методик, аналогичных описанным выше для синтеза соединения 1А-01 с использованием соответствующих исходных веществ, которые имеются в продаже, были получены с использованием препаратов, хорошо известных специалистам в данной области, или получены способом, аналогичным описанным выше способам для других промежуточных соединений. Соединения очищали с использованием способов, хорошо известных специалистам в данной области, которые могут включать хроматографию на силикагеле, ВЭЖХ или кристаллизацию из реакционной смеси. Конечные соединения могут быть выделены в виде нейтральных или кислотных или основных солей.
Таблица 1
ЖХМС(ES+): 586,0 (M+H).
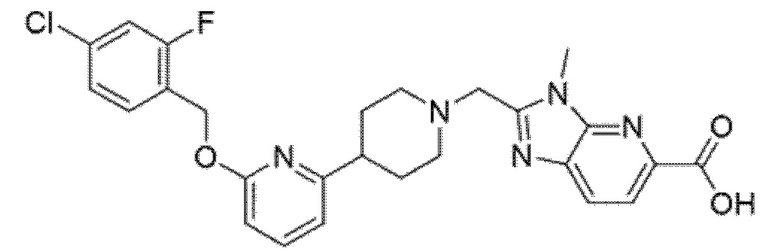
Пример 2A-01
Гидрохлорид 2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-3-метил-3H-имидазо[4,5-b]пиридин-5-карбоновой кислоты
Стадия 1
Желтую смесь промежуточного соединения 3 (92,3 г, 119 ммоль, 4 экв. соли TFA), промежуточного соединения 33 (25,9 г, 120 ммоль) и K2CO3 (98,5 г, 713 ммоль) в MeCN (300 мл) перемешивали при 50°С в течение 16 ч. Желтую смесь выливали в воду (300 мл) и экстрагировали EtOAc (3 × 500 мл). Объединенные органические слои промывали насыщенным солевым раствором (500 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (градиент MeOH/DCM 0-5%) с получением 5-хлор-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-3-метил-3H-имидазо[4,5-b]пиридина (59,0 г, 99%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,92 (д, 1H), 7,49 (м, 1H), 7,43 (м, 1H), 7,21 (д, 1H), 7,10-7,13 (м, 1H), 7,09 (д, 1H), 6,72 (д, 1H), 6,60 (д, 1H), 5,40 (с, 2H), 3,98 (с, 3H), 3,84 (с, 2H), 2,97 (д, 2H), 2,51-2,73 (м, 1H), 2,29 (м, 2H), 1,73-1,97 (м, 4H).
Стадия 2
Желтый раствор 5-хлор-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-3-метил-3H-имидазо[4,5-b]пиридина (59,0 г, 118 ммоль), DPPP (6,80 г, 16,5 ммоль), Pd(OAc)2 (3,65 г, 16,3 ммоль) и Et3N (125 г, 1240 ммоль) в MeOH (800 мл) и DMF (100 мл) перемешивали при 80°С в атмосфере 50 фунт/кв. дюйм (0,34 МПа) CO в течение 16 ч. Полученный оранжевый раствор концентрировали при пониженном давлении до коричневого масла, которое разбавляли EtOAc (300 мл) и промывали водой (200 мл). Органический слой промывали насыщенным солевым раствором (2×200 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт объединяли с продуктом из аналогичной реакции (масштабирование: 11 г) и очищали флэш-хроматографией (градиент 50-100% EtOAc/PE) с получением метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-3-метил-3H-имидазо[4,5-b]пиридин-5-карбоксилата (62,6 г, 85%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,13 (д, 1H), 8,07 (д, 1H), 7,49 (т, 1H), 7,43 (т, 1H), 7,12 (т, 1H), 7,08-7,11 (м, 1H), 6,73 (д, 1H), 6,60 (д, 1H), 5,40 (с, 2H), 4,09 (с, 3H), 4,03 (с, 3H), 3,90 (с, 2H), 2,93-3,05 (м, 2H), 2,55-2,69 (м, 1H), 2,31 (дт, 2H), 1,79-1,97 (м, 4H).
Стадия 3
Метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-3-метил-3H-имидазо[4,5-b]пиридин-5-карбоксилат (57,0 г, 109 ммоль) суспендировали в MeOH (1 л) и обрабатывали 2 M NaOH (218 мл). Суспензию перемешивали 5 мин при КТ и затем нагревали при 85°С в течение 3 ч. Смесь фильтровали через Celite® и прозрачный фильтрат снова нагревали до 70°C. Реакционную смесь подкисляли 2 М HCl (272 мл) и затем оставляли охлаждаться до КТ. Образовавшееся твердое вещество и суспензию перемешивали в течение 18 ч при КТ. Твердые вещества собирали фильтрованием с получением соединения примера 2A-01 (57,1 г, 96%) в виде кремово-белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,20 (ушир. с, 1H), 11,07 (ушир. с, 1H), 8,27 (д, 1H), 8,07 (д, 1H), 7,57-7,78 (м, 2H), 7,47 (м, 1H), 7,32 (м, 1H), 6,92 (д, 1H), 6,74 (д, 1H), 5,40 (с, 2H), 4,84 (ушир. с, 2H), 3,97 (с, 3H), 3,86 (ушир. с, 2H), 3,37 (ушир. с, 2H), 2,93 (ушир. с, 1H), 1,85-2,36 (м, 4H); ЖХМС(ES+): 510,2 (M+H).
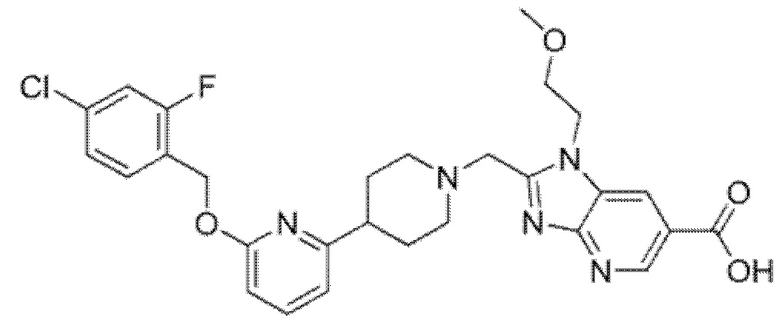
Гидрохлорид 2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин-6-карбоновой кислоты
Пример 2A-02
Стадия 1
В колбу, содержащую раствор метоксиуксусной кислоты (1,00 г, 11,1 ммоль) в DMF (30 мл), добавляли HATU (6,33 г, 16,7 ммоль) и Et3N (3,37 г, 33,3 ммоль). После перемешивания в течение 20 мин порциями добавляли 2,3-диамино-5-бромпиридин (2,3 г, 12 ммоль), и полученную реакционную смесь перемешивали в течение ночи. Через 15 ч добавляли воду и раствор экстрагировали EtOAc. Объединенные органические слои сушили и растворитель удаляли при пониженном давлении. Неочищенное соединение очищали флэш-хроматографией (градиент 0-80% EtOAc/гептан) с получением N-(2-амино-5-бромпиридин-3-ил)-2-метоксиацетамида (2,3 г, 80%). 1H ЯМР (400 МГц, CDCl3) δ 8,09 (с, 1H), 8,06 (д, 1H), 8,03 (с, 1H), 7,83 (д, 1H), 4,08 (с, 2H), 3,53 (с, 3H); ЖХМС(ES+): 260,2 (M+H).
Стадия 2
К раствору N-(2-амино-5-бромпиридин-3-ил)-2-метоксиацетамида (3,3 г, 13 ммоль) в THF добавляли 1 M раствор BH3 в THF (14 мл) в течение 10 мин, и перемешивали при КТ в течение ночи. В реакционную смесь медленно добавляли воду для гашения избытка борана, и затем смесь экстрагировали EtOAc. Слой EtOAc сушили и концентрировали при пониженном давлении. Неочищенный продукт растворяли в MeOH и добавляли HCl в диоксане (1,0 экв.) и перемешивали в течение 2 ч. Избыток метанола удаляли при пониженном давлении с получением неочищенного продукта. Соединение очищали флэш-хроматографией с градиентом от 0-70% EtOAc в гептанах с получением 5-бром-N3-(2-метоксиэтил)пиридин-2,3-диамина в виде коричневого масла (1,1 г, 35%). 1H ЯМР (600 МГц, CDCl3) δ 7,83 (д, 1H), 6,95 (д, 1H), 5,56 (с, 2H), 3,77 (т, 1H), 3,66 (т, 2H), 3,42 (с, 3H), 3,22 (кв, 2H); ЖХМС(ES+): 246,1.
Стадия 3
5-Бром-N3-(2-метоксиэтил)пиридин-2,3-диамин (400 мг, 1,63 ммоль) растворяли в 8 мл диоксана (8 мл) и обрабатывали хлорацетилхлоридом (0,284 мл, 3,58 ммоль). Смесь перемешивали при КТ. Растворитель удаляли при пониженном давлении и полученный остаток растворяли в TFA (8 мл) и перемешивали при 80°C в течение 18 ч. Реакционную смесь охлаждали до КТ и концентрировали при пониженном давлении. Полученное коричневое масло растворяли в EtOAc (50 мл) и нейтрализовали насыщ. водн. NaHCO3. Когда выделение CO2 уменьшалось, слои разделяли, и водный слой экстрагировали дополнительным EtOAc (20 мл). Органические экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали флэш-хроматографией (градиент 0-80% EtOAc/гептан) с получением 6-бром-2-(хлорметил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридина (176 мг, 36%) в виде желтовато-коричневого твердого вещества. 1H ЯМР (600 МГц, CDCl3) δ 8,59 (с, 1H), 7,90 (с, 1H), 4,93 (с, 2H), 4,45 (м, 2H), 3,72 (м, 2H), 3,29 (с, 3H); ЖХМС(ES+): 306,1 (M+H).
Стадия 4
Смесь промежуточного соединения 3 (294 мг, 0,97 ммоль, свободное основание), 6-бром-2-(хлорметил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридина (341 мг, 1,06 ммоль), KI (48 мг, 0,29 ммоль) и N,N-диизопропилэтиламина (0,51 мл, 0,97 ммоль) в MeCN (8 мл) перемешивали при 60°С в течение 16 ч. Смесь выливали в воду и экстрагировали EtOAc. Органический слой сушили, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (градиент 0-100% EtOAc/гептан) с получением 6-бром-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридина (406 мг, 71%) в виде желтовато-коричневого масла. 1H ЯМР (600 МГц, CDCl3) δ 8,54 (с, 1H), 7,91 (с, 1H), 7,50 (м, 1H), 7,43 (м, 1H), 7,11 (м, 2H), 6,73 (д, 1H), 6,60 (д, 1H), 5,41 (с, 2H), 4,54 (м, 2H), 3,92 (с, 2H), 3,76 (м, 2H), 3,30 (с, 3H), 2,97 (д, 2H), 2,58-2,67 (м, 1H), 2,31 (м, 2H), 1,76-1,93 (м, 4H).
Стадия 5
К смеси 6-бром-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридина (610 мг, 1,04 ммоль), ацетата палладия(II) (47 мг, 0,21 ммоль) и DPPP (128 мг, 0,31 ммоль) добавляли DMF (4 мл), MeOH (16 мл) и триметиламин (1,44 мл, 10,4 ммоль). Реакционную смесь нагревали при 80°С при перемешивании в атмосфере СО при давлении 50 фунт/кв. дюйм (0,34 МПа) в течение 20 ч. Реакционную смесь охлаждали до КТ и распределяли между водой и EtOAc. Органический слой отделяли и сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией (градиент 0-5% MeOH в DCM) с получением метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин-6-карбоксилата (540 мг, 92%) в виде желтовато-коричневой смолы. 1H ЯМР (600 МГц, CDCl3) δ 9,18 (с, 1H), 8,39 (с, 1H), 7,49 (т, 1H), 7,43 (т, 1H), 7,10 (т, 2H), 6,73 (д, 1H), 6,60 (д, 1H), 5,40 (с, 2H), 4,64 (т, 2H), 4,00-3,90 (м, 5H), 3,78 (т, 2H), 3,29 (с, 3H), 2,99 (д, 2H), 2,62 (м, 1H), 2,27-2,40 (м, 2H), 1,79-1,91 (м, 4H).
Стадия 6
К раствору метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-имидазо[4,5-b]пиридин-6-карбоксилата (2,0 г, 3,5 ммоль) в MeOH (60 мл) добавляли 2 M NaOH (8,9 мл) и смесь нагревали при 60°С в течение 1 ч. Реакционную смесь охлаждали до КТ и подкисляли 1 М HCl до рН ~4. Смесь концентрировали при пониженном давлении для удаления MeOH и твердое вещество собирали фильтрованием и сушили под вакуумом с получением соединения примера 2A-02 (1,7 г, 82%) в виде твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,35 (ушир. с, 1H), 10,90 (ушир. с, 1H), 9,01 (д, 1H), 8,70 (д, 1H), 7,69 (т, 1H), 7,63 (т, 1H), 7,47 (дд, 1H), 7,32 (дд, 1H), 6,93 (д, 1H), 6,73 (д, 1H), 5,40 (с, 2H), 4,86 (ушир. с, 2H), 4,70 (ушир. с, 2H), 3,81 (ушир. с, 2H), 3,65 (м, 2H), 3,20 (с, 3H), 2,94 (ушир. с, 1H), 2,08-2,25 (м, 4H); ЖХМС(ES+): 554,2 (M+H).
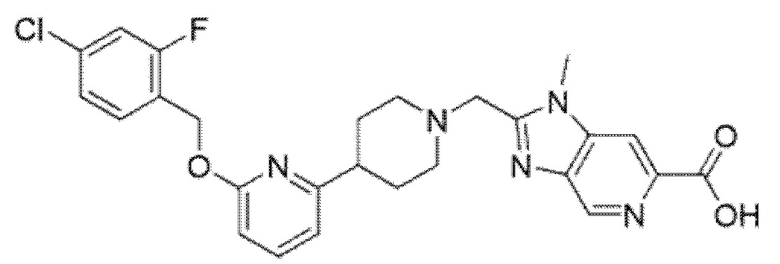
Пример 2A-03
2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-имидазо[4,5-c]пиридин-6-карбоновая кислота
Стадия 1
К перемешиваемому раствору 2,4-дибром-5-нитропиридина (0,21 г, 0,72 ммоль) в THF (4,1 мл) добавляли метиламин в THF (2 M, 1,2 мл, 2,5 ммоль). Через 0,5 ч раствор разбавляли водой (5 мл). Водную фазу экстрагировали EtOAc (3 × 15 мл), объединенные органические слои промывали насыщенным солевым раствором (20 мл), сушили над безводным Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с использованием колоночной хроматографии (50% EtOAc/гептан) с получением 2-бром-N-метил-5-нитропиридин-4-амина в виде желтого твердого вещества (0,15 г, 90%). 1H ЯМР (CDCl3) δ 8,99 (с, 1H), 6,95 (с, 1H), 3,08 (д, 3H).
Стадия 2
К перемешиваемому раствору 2-бром-N-метил-5-нитропиридин-4-амина (0,22 г, 0,96 ммоль) в AcOH (4,8 мл) добавляли Fe (0,053 г, 0,96 ммоль). Раствор нагревали до 75°С. Через 5 ч раствор фильтровали через слой Celite®, промывали EtOAc (10 мл) и затем гасили насыщ. Na2CO3. Водную фазу экстрагировали EtOAc (2 × 10 мл), объединенные органические слои сушили над безводным Na2SO4, фильтровали, обрабатывали HCl в диксоане (4 М, 2,4 мл, 9,6 ммоль) и растворитель удаляли при пониженном давлении. Неочищенное вещество перемешивали в Et2O/PE в течение 30 мин, и полученное твердое вещество затем собирали фильтрованием, промывали РЕ и сушили при пониженном давлении, получая 6-бром-N4-метилпиридин-3,4-диамингидрохлорид (0,20 г, 88%). 1H ЯМР (CD3OD) δ 7,48 (с, 1H), 6,95 (с, 1H), 3,04 (с, 3H).
Стадия 3
К перемешиваемому раствору 6-бром-N4-метилпиридин-3,4-диамингидрохлорида (0,15 г, 0,52 ммоль) в DMF (2,4 мл) добавляли промежуточное соединение 5 (0,18 г, 0,48 ммоль), затем DIPEA (0,25 мл, 1,4 ммоль) и HBTU (0,18 г, 0,57 ммоль). Через 2 ч раствор концентрировали при пониженном давлении, разбавляли EtOAc (20 мл) и промывали насыщ. Na2CO3. Органический слой сушили над безводным Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Неочищенный амид N-(6-бром-4-(метиламино)пиридин-3-ил)-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)ацетамид растворяли в 1,4-диоксане (5 мл), обрабатывали NaOH (2 M, 2,4 мл, 4,8 ммоль) и нагревали до 100°С. Через 0,5 ч раствор разбавляли водой (10 мл). Водную фазу экстрагировали CH2Cl2 (3 × 10 мл), объединенные органические слои сушили над безводным Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с использованием колоночной хроматографии, элюируя EtOAc с получением 6-бром-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-имидазо[4,5-c]пиридина в виде коричневого масла (0,19 г, 72%). 1H ЯМР (CDCl3) δ 8,74 (с, 1H), 7,44-7,51 (м, 2H), 7,41 (т, 1H), 7,08 (т, 2H), 6,71 (д, 1H), 6,59 (д, 1H), 5,39 (с, 2H), 3,89 (с, 3H), 3,83 (с, 2H), 2,94 (д, 2H), 2,60 (ддд, 1H), 2,28 (т, 2H), 1,85-1,90 (м, 2H), 1,75-1,84 (м, 2H).
Стадия 4
Во флакон, содержащий 6-бром-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-имидазо[4,5-c]пиридин (0,060 г, 0,11 ммоль), DPPP (0,011 г, 0,028 ммоль) и Pd(OAc)2 (0,035 г, 0,015 ммоль), добавляли DMF (0,4 мл), затем MeOH (2,6 мл) и Et3N (0,13 мл, 1,1 ммоль). Раствор нагревали до 80°С в атмосфере СО (50 фунт/кв. дюйм (0,34 МПа)). Через 16 ч раствор разбавляли насыщенным солевым раствором (5 мл). Водную фазу экстрагировали EtOAc (2 × 10 мл), объединенные органические слои сушили над безводным MgSO4, фильтровали и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с использованием колоночной хроматографии, элюируя 5% MeOH в CH2Cl2 с получением метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-имидазо[4,5-c]пиридин-6-карбоксилата в виде желтого масла (0,060 г, количественно). 1H ЯМР (600 МГц, CDCl3) δ 9,11 (с, 1H), 8,29 (с, 1H), 7,45-7,54 (м, 1H), 7,35-7,45 (м, 1H), 7,10 (т, 2H), 6,73 (д, 1H), 6,61 (д, 1H), 5,40 (с, 2H), 4,05 (с, 3H), 4,02 (с, 3H), 3,91 (с, 2H), 2,98 (д, 2H), 2,58-2,68 (м, 1H), 2,32 (т, 2H), 1,74-1,95 (м, 4H).
Стадия 5
К перемешиваемому раствору метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-имидазо[4,5-c]пиридин-6-карбоксилата (0,041 г, 0,078 ммоль) в MeOH (0,78 мл) добавляли раствор NaOH в воде (2 M, 0,14 мл) при перемешивании при 35°С. Через 2 ч раствор подкисляли до pH ~4 с помощью HCl в воде (1 М), охлаждали до 0°C, разбавляли водой (0,5 мл) и давали постоять в течение 2 ч. Полученный твердый осадок суспендировали в течение 1 ч, собирали фильтрованием, промывали водой (2×1 мл) и затем сушили при пониженном давлении с получением соединения примера 2А-03 в виде твердого вещества (21 мг, 48%). 1H ЯМР (400 МГц, CD3OD) δ: 9,10 (ушир. с, 1H), 8,57 (ушир. с, 1H), 7,67 (ушир. т, 1H), 7,52 (ушир. т, 1H), 7,13-7,33 (м, 2H), 6,95 (д, 1H), 6,75 (д, 1H), 5,46 (с, 2H), 4,92 (с, 2H), 3,92-4,18 (м, 5H), 3,45 (ушир. с, 2H), 3,08 (ушир. с, 1H), 2,11-2,46 (м, 4H). ЖХМС(ES+): 510,3 (M+H).
Соединения, перечисленные в приведенной ниже таблице 2, были получены с использованием методик, аналогичных описанным выше для синтеза соединений примеров 2A-01, 2A-02 и 2A-03 с использованием соответствующих исходных веществ, которые имеются в продаже, были получены с использованием препаратов, хорошо известных специалистам в данной области, или получены способом, аналогичным описанным выше способам для других промежуточных соединений. Соединения очищали с использованием способов, хорошо известных специалистам в данной области, которые могут включать хроматографию на силикагеле, ВЭЖХ или кристаллизацию из реакционной смеси. Конечные соединения могут быть выделены в виде нейтральных или кислотных или основных солей.
Таблица 2
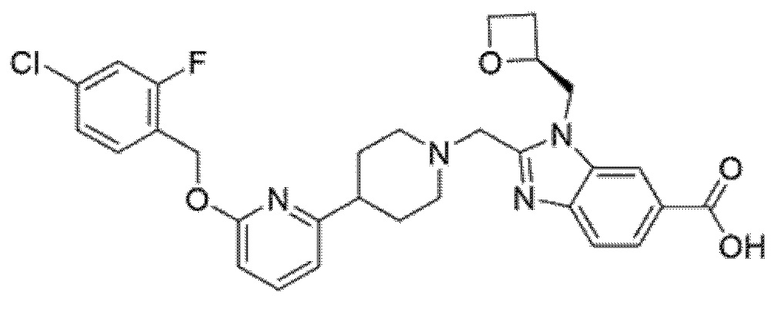
Пример 3A-01
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота
Стадия 1
К перемешиваемому раствору промежуточного соединения 22 (49,8 г, 211 ммоль) в MeCN (300 мл) добавляли 2-хлор-1,1,1-триметоксиэтан (30,0 мл, 223 ммоль), затем pTSA·H2O (2,0 г, 10 ммоль). Через 1 час при 60°C добавляли MeCN (400 мл), K2CO3 (116 г, 841 ммоль) и промежуточное соединение 3 (52,4 г, 90,2 ммоль). Через 2 ч раствор обрабатывали водой (1,6 л), оставляли охлаждаться до КТ и перемешивали в течение 2 ч. Полученный твердый осадок собирали фильтрованием, промывали водой (2×300 мл) и сушили при пониженном давлении с получением метил-(S)-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(оксетан-2-илметил)-1H-бензо[d]имидазол-6-карбоксилата в виде твердого вещества (102 г, 84%). 1H ЯМР (ДМСО-d6) δ 8,30 (с, 1H), 7,82 (д, 1H), 7,67 (д, 1H), 7,62 (т, 1H), 7,55 (т, 1H), 7,45 (д, 1H), 7,29 (д, 1H), 6,87 (д, 1H), 6,67 (д, 1H), 5,37 (с, 2H), 5,04-5,16 (м, 1H), 4,82 (дд, 1H), 4,62-4,73 (м, 1H), 4,44-4,52 (м, 1H), 4,37 (дт, 1H), 3,96 (д, 1H), 3,87 (с, 3H), 3,78 (д, 1H), 3,00 (д, 1H), 2,85 (д, 1H), 2,66-2,76 (м, 1H), 2,54-2,64 (м, 1H), 2,38-2,49 (м, 1H), 2,24 (т, 2,11-2,21 (м, 1H), 1,60-1,88 (м, 4H).
Стадия 2
К перемешиваемому раствору метил-(S)-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(оксетан-2-илметил)-1H-бензо[d]имидазол-6-карбоксилата (7,2 г, 12 ммоль) в MeOH (50 мл) и THF (50 мл) добавляли 2 M NaOH (25 мл, 50 ммоль). Через 2 ч при 45°С раствору давали возможность остыть до КТ, разбавляли водой (100 мл) и подкисляли до рН ~6 лимонной кислотой в воде (1 М, 20 мл). Полученный твердый осадок суспендировали в течение 1 ч, собирали фильтрованием, промывали водой (100 мл) и затем сушили при пониженном давлении с получением соединения примера 3A-01 в виде твердого вещества (6,4 г, 91%). 1H ЯМР (ДМСО-d6) δ 12,84 (ушир. с, 1H), 8,27 (с, 1H), 7,80 (д, 1H), 7,59-7,67 (м, 2H), 7,55 (т, 1H), 7,45 (дд, 1H), 7,29 (дд, 1H), 6,86 (д, 1H), 6,67 (д, 1H), 5,37 (с, 2H), 5,06-5,17 (м, 1H), 4,80 (дд, 1H), 4,66 (дд, 1H), 4,44-4,53 (м, 1H), 4,38 (дт, 1H), 3,95 (д, 1H), 3,78 (д, 1H), 3,00 (д, 1H), 2,85 (д, 1H), 2,64-2,77 (м, 1H), 2,54-2,64 (м, 1H), 2,40-2,48 (м, 1H), 2,20-2,29 (м, 1H), 2,17 (т, 1H), 1,61-1,85 (м, 4H). ЖХМС(ES+): 565,4 (M+H).
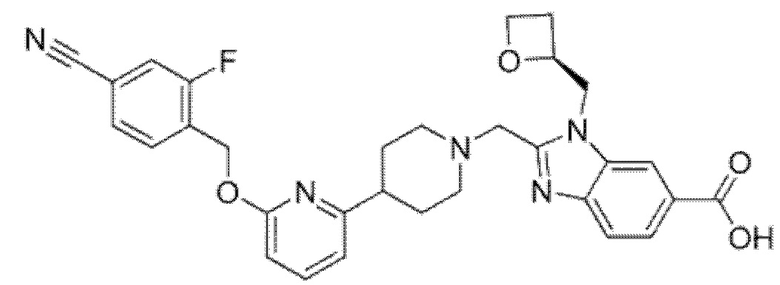
Пример 4A-01
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота
Стадия 1
К перемешиваемому раствору промежуточного соединения 22 (33,6 г, 142 ммоль) в MeCN (285 мл) добавляли 2-хлор-1,1,1-триметоксиэтан (20,1 мл, 149 ммоль), затем pTSA·H2O (1,35 г, 7,1 ммоль). Через 2 час при 50°C добавляли MeCN (280 мл), K2CO3 (79 г, 570 ммоль) и промежуточное соединение 4 (93,2 г, 142 ммоль). Через 2 ч раствор обрабатывали водой (800 мл), давали возможность остыть до КТ и перемешивали в течение 2 ч. Полученный твердый осадок собирали фильтрованием, промывали 10% MeCN в воде (150 мл), водой (2×200 мл) и затем сушили при пониженном давлении с получением метил-(S)-2-((4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(оксетан-2-илметил)-1H-бензo[d]имидазол-6-карбоксилата в виде бесцветного твердого вещества (77 г, 95%). 1H ЯМР (600 МГц, ДМСО-d6) δ 8,28 (с, 1H), 7,87 (д, 1H), 7,80 (д, 1H), 7,55-7,73 (м, 4H), 6,87 (д, 1H), 6,70 (д, 1H), 5,45 (с, 2H), 5,04-5,19 (м, 1H), 4,81 (дд, 1H), 4,66 (дд, 1H), 4,41-4,54 (м, 1H), 4,36 (дт, 1H), 3,94 (д, 1H), 3,86 (с, 3H), 3,76 (д, 1H), 2,97 (д, 1H), 2,82 (д, 1H), 2,63-2,77 (м, 1H), 2,49-2,63 (м, 1H), 2,37-2,46 (м, 1H), 2,18-2,29 (м, 1H), 2,05-2,18 (м, 1H), 1,47-1,82 (м, 4H).
Стадия 2
К перемешиваемому раствору метил-(S)-2-((4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(оксетан-2-илметил)-1H-бензo[d]имидазол-6-карбоксилата (4 г, 7 ммоль) в MeCN (70 мл) добавляли раствор 1,5,7-триазабицикло[4.4.0]дец-5-ена в воде (0,97 M, 14,7 мл). Через 20 ч раствор подкисляли до рН ~6 лимонной кислотой в воде (2 М, 7 мл) и разбавляли водой (50 мл). Водную фазу экстрагировали EtOAc (2 × 75 мл), объединенные органические слои сушили над безводным Na2SO4, фильтровали и растворитель удаляли при пониженном давлении с получением почти белого твердого вещества. Неочищенное вещество очищали с использованием колоночной хроматографии, элюируя MeOH/DCM (0:100 до 8:92) с получением соединения примера 4A-01 в виде твердого вещества (3,65 г, 90%). 1H ЯМР (400 МГц, ДМСО-d6) δ 12,75 (ушир. с, 1H), 8,27 (с, 1H), 7,89 (д, 1H), 7,80 (д, 1H), 7,68-7,72 (м, 2H), 7,60-7,67 (м, 2H), 6,89 (д, 1H), 6,72 (д, 1H), 5,47 (с, 2H), 5,11 (д, 1H), 4,74-4,86 (м, 1H), 4,62-4,72 (м, 1H), 4,43-4,53 (м, 1H), 4,35-4,42 (м, 1H), 3,95 (д, 1H), 3,77 (д, 1H), 2,98 (д, 1H), 2,84 (д, 1H), 2,65-2,77 (м, 1H), 2,53-2,64 (м, 1H), 2,37-2,45 (м, 1H), 2,10-2,28 (м, 2H), 1,57-1,84 (м, 4H). ЖХМС(ES+): 556,6 (M+H).
Трис-соль примера 4A-01
Трис-соль 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты
К перемешиваемому раствору соединения примера 4A-01 (6,5 г, 11,7 ммоль) в 1-пропаноле (275 мл) при 70°C по каплям добавляли водный раствор трис (2,0 М, 6,1 мл, 12,2 ммоль), при этом раствор оставался гомогенным. После перемешивания в течение 5 мин добавляли затравочные кристаллы и смеси давали возможность остыть до КТ в течение 2 ч. После перемешивания в течение ночи при КТ образовалось твердое вещество. Твердое вещество собирали фильтрованием, промывали 1-пропанoлом (2×30 мл) и сушили, сначала в потоке азота и затем в вакуумном сушильном шкафу при 45°C в течение 15 ч, с получением трис-соли примера 4A-01 (6,95 г, 88%) в виде кристаллического твердого вещества. 1H ЯМР (600 МГц, ДМСО-d6) δ: 8,20 (с, 1H), 7,89 (д, 1H), 7,79 (д, 1H), 7,70 (ушир. с, 2H), 7,64 (т, 1H), 7,56 (д, 1H), 6,89 (д, 1H), 6,72 (д, 1H), 5,47 (с, 2H), 5,11 (кв,д, 1H), 4,77 (дд, 1H), 4,64 (дд, 1H), 4,44-4,53 (м, 1H), 4,38 (дт, 1H), 3,93 (д, 1H), 3,76 (д, 1H), 3,35 (ушир. с, 9H), 2,98 (д, 1H), 2,85 (д, 1H), 2,64-2,75 (м, 1H), 2,54-2,64 (м, 1H), 2,40-2,49 (м, 1H), 2,08-2,26 (м, 2H), 1,56-1,83 (м, 4H), т. пл.:=194°С.
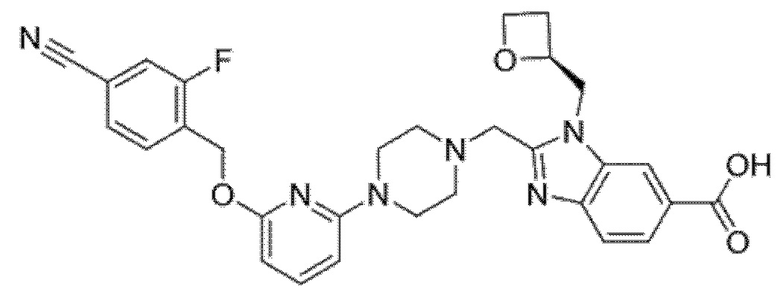
Пример 5A-01
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота
Стадия 1
Раствор промежуточного соединения 13 (5 г, 14,4 ммоль) в 5% MeOH:CH2Cl2 (60 мл) обрабатывали насыщ. водн. Na2CO3 (60 мл). Двухфазный раствор интенсивно перемешивали в течение 30 мин и органический слой отделяли. Органический слой сушили, фильтровали и концентрировали при пониженном давлении с получением 4-(((6-(4-пиперазин-1-ил)пиридин-2-ил)окси)метил)-3-фторбензoнитрила (4,4 г, количественно) в виде полутвердого вещества.
Стадия 2
В колбу, содержащую раствор 4-(((6-(4-пиперазин-1-ил)пиридин-2-ил)окси)метил)-3-фторбензoнитрила (1,58 г, 5,06 ммоль) в MeCN (15 мл) добавляли промежуточное соединение 23 (1,40 г, 5,06 ммоль) и K2CO3 (3,50 г, 25,3 ммоль). Полученную суспензию перемешивали в течение 2 ч при 50°С. Через 2 ч смесь обрабатывали водой (30 мл), давали возможность остыть до КТ и перемешивали в течение 2 ч. Твердое вещество собирали фильтрованием, промывали водой:MeCN (2:1) (2×30 мл) и сушили при пониженном давлении с получением метил-(S)-2-((4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)метил)-1-(оксетан-2-илметил)-1H-бензo[d]имидазол-6-карбоксилата (2,47 г, 86%) в виде твердого вещества. 1H ЯМР (600 МГц, CDCl3) δ 8,16 (с, 1H), 7,98 (д, 1H), 7,76 (д, 1H), 7,59 (т, 1H), 7,42 (дт, 2H), 7,34 (д, 1H), 6,17 (дд, 2H), 5,42 (с, 2H), 5,23 (дд, 1H), 4,77-4,58 (м, 3H), 4,38 (дт, 1H), 4,05-3,95 (м, 2H), 3,95 (с, 3H), 3,46 (д, 4H), 2,80-2,69 (м, 1H), 2,62 (т, 4H), 2,50-2,38 (м, 1H).
Стадия 3
В колбу, содержащую раствор метил-(S)-2-((4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)метил)-1-(оксетан-2-илметил)-1H-бензo[d]имидазол-6-карбоксилата (2,5 г, 4,3 ммоль) в 1:1 смеси iPrOH и THF (140 мл) добавляли 1,4 экв. LiOH (0,14 г, 6,1 ммоль) и полученный раствор нагревали при 45°С в течение 15 ч. Раствор оставляли охлаждаться до КТ, разбавляли водой (50 мл) и подкисляли до рН ~6 лимонной кислотой в воде. Полученный раствор экстрагировали EtOAc. Слой EtOAc сушили и растворитель удаляли при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали флэш-хроматографией (10% MeOH в CH2Cl2) с получением соединения примера 5A-01 (0,86 г, 35%) в виде твердого вещества. 1H ЯМР (600 МГц, CDCl3) δ 8,23 (с, 1H), 8,06 (д, 1H), 7,83 (д, 1H), 7,59 (т, 1H), 7,46-7,39 (м, 2H), 7,34 (д, 1H), 6,18 (дд, 2H), 5,43 (с, 2H), 5,28-5,20 (м, 1H), 4,81-4,58 (м, 3H), 4,44-4,33 (м, 1H), 4,04 (д, 2H), 3,48 (м, 4H), 2,82-2,71 (м, 1H), 2,65 (м, 4H), 2,46 (дд, 1H). ЖХМС(ES+): 557,2 (M+H).
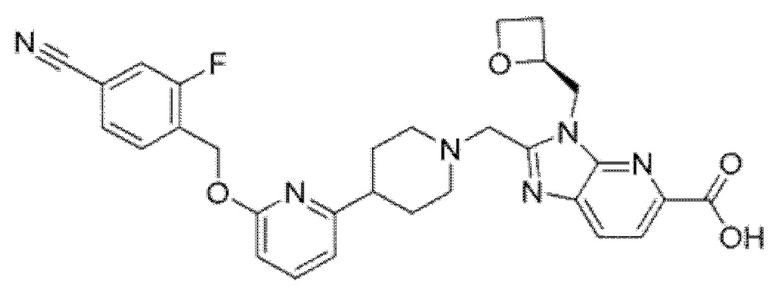
Пример 6A-01
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновая кислота
Стадия 1
В трехгорлую 3 л колбу, снабженную механической мешалкой, в которую загружали промежуточное соединение 4 (106 г, 161 ммоль), добавляли MeCN (886 мл), K2CO3 (89,0 г, 644 ммоль) и промежуточное соединение 27 (52,4 г, 177 ммоль). Смесь перемешивали при 60°С в течение 2 ч. Реакционную смесь выливали в 4 л колбу Эрленмейера и разбавляли 1,8 л воды. Полученную суспензию перемешивали при КТ в течение 4 ч с получением светло-желтой суспензии. Твердые вещества собирали фильтрованием и сушили в вакуумном сушильном шкафу при 45°С в течение ночи с получением целевого метил-(S)-2-((4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-3-(оксетан-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоксилата (88,6 г, 96%) в виде светло-желтого твердого вещества. 1H ЯМР (600 МГц, ДМСО-d6) δ 8,16 (д, 1H), 8,01 (д, 1H), 7,87 (д, 1H), 7,61-7,74 (м, 3H), 6,88 (д, 1H), 6,71 (д, 1H), 5,46 (с, 2H), 5,11-5,26 (м, 1H), 4,85 (дд, 1H), 4,73 (дд, 1H), 4,43-4,60 (м, 1H), 4,37 (дт, 1H), 3,96-4,04 (м, 1H), 3,89-3,95 (м, 3H), 2,87-3,01 (м, 2H), 2,66-2,81 (м, 1H), 2,55-2,64 (м, 1H), 2,52 (ушир. с, 3H), 2,24 (кв, 2H), 1,64-1,81 (м, 3H); ЖХМС(ES+): 571,5 (M+H).
Стадия 2
В трехгорлую колбу объемом 1 л, снабженную механической мешалкой с верхним приводом, загружали метил-(S)-2-((4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-3-(оксетан-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоксилат (35,5 г, 62,21 ммоль). В колбу добавляли MeCN (350 мл) и воду (70 мл). Полученную смесь перемешивали при КТ в течение 30 мин с образованием густой суспензии. LiOH·H2O (2,92 г, 68,4 ммоль) медленно добавляли в виде твердого вещества. Полученную суспензию перемешивали при 40°С в течение 1 ч. Реакционную смесь охлаждали до КТ и обрабатывали по каплям 1,0 М лимонной кислотой (15,5 мл) до достижения рН суспензии ~5. Полученную суспензию перемешивали при КТ в течение 4 ч. Полученные твердые вещества собирали фильтрованием, твердые вещества промывали ~20 мл воды и затем сушили в потоке N2 в течение 4 ч. Твердые вещества сушили в течение дополнительных 72 ч при 40°С в вакуумном сушильном шкафу с получением соединения примера 6А-01 (31,2 г, 90%) в виде твердого вещества. 1H ЯМР (600 МГц, ДМСО-d6) δ 13,03 (ушир. с, 1H), 8,15 (д, 1H), 8,00 (д, 1H), 7,87 (д, 1H), 7,67-7,73 (м, 2H), 7,64 (т, 1H), 6,88 (д, 1H), 6,71 (д, 1H), 5,45 (с, 2H), 4,93-5,03 (м, 1H), 4,87 (с, 1H), 4,70 (д, 1H), 4,36-4,45 (м, 1H), 4,23-4,35 (м, 1H), 4,05 (д, 1H), 3,79 (д, 1H), 2,93-3,06 (м, 1H), 2,76-2,88 (м, 1H), 2,54-2,69 (м, 1H), 2,34-2,46 (м, 1H), 2,25 (д, 2H), 2,05-2,21 (м, 1H), 1,73 (д, 3H), 1,47-1,67 (м, 1H); ЖХМС(ES+): 557,6 (M+H).
Пример 7A-01
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота
Стадия 1
К суспензии HCl-соли оксазол-2-илметанамина (491 мг, 3,65 ммоль) и промежуточного соединения 29 (800 мг, 3,32 ммоль) в DMF (5 мл) добавляли K2CO3 (1,04 г, 6,63 ммоль). Реакционную смесь перемешивали при 60°С в течение 2 ч. Добавляли дополнительное количество HCl-соли оксазол-2-илметанамина (100 мг, 1,0 ммоль) и реакционную смесь перемешивали в течение дополнительных 30 мин при 60°C. Реакционную смесь охлаждали до КТ, затем разбавляли водой (30 мл) и экстрагировали EtOAc (60 мл). Органический слой промывали водой, затем насыщенным солевым раствором, затем сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Оранжевый остаток очищали флэш-хроматографией (12 г силикагеля, градиент 0-50% EtOAc/гептан) с получением трет-бутил-4-нитро-3-((оксазол-2-илметил)амино)бензоата (764 мг, 75%) в виде оранжевого твердого вещества. 1H ЯМР (CDCl3) δ 8,48 (ушир. с, 1H), 8,23 (д, 1H), 7,68 (д, 1H), 7,61 (д, 1H), 7,28 (дд, 1H), 7,15 (с, 1H), 4,72 (д, 2H), 1,60 (с, 9H).
Стадия 2
К раствору трет-бутил-4-нитро-3-((оксазол-2-илметил)амино)бензоата (15 г, 47 ммоль) в THF (100 мл) добавляли 10% палладий на угле (1,5 г, 10% масс./масс.), и смесь затем перемешивали в атмосфере H2 при 50 фунт/кв.дюйм (0,34 МПа) при КТ в течение 6 ч. Затем реакционную смесь фильтровали через Celite® с получением темного раствора. Фильтрат фильтровали через второй слой Celite® и фильтрат концентрировали при пониженном давлении с получением трет-бутил-4-амино-3-((оксазол-2-илметил)амино)бензоата (13,1 г, 92%) в виде темной пены. 1H ЯМР (CDCl3) δ 7,62 (с, 1H), 7,43 (дд, 1H), 7,35 (д, 1H), 7,08 (с, 1H), 6,66 (д, 1H), 4,44 (с, 2H), 1,56 (с, 9H).
Стадия 3
К перемешиваемому раствору трет-бутил-4-амино-3-((оксазол-2-илметил)амино)бензоата (13 г, 45 ммоль) в MeCN (100 мл) добавляли 2-хлор-1,1,1-триметоксиэтан (9,0 мл, 65 ммоль) и pTSA·H2O (400 мг, 2,1 ммоль) и смесь нагревали при 60°С в течение 3 ч. Затем реакционную смесь охлаждали до КТ и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (120 г силикагеля, градиент 0-100% EtOAc/гептан) с получением трет-бутил-2-(хлорметил)-1-(оксазол-2-илметил)-1H-бензo[d]имидазол-6-карбоксилата (11,6 г, 74%) в виде светло-желтого твердого вещества. 1H ЯМР (CDCl3) δ 8,19 (д, 1H), 7,98 (дд, 1H), 7,77 (д, 1H), 7,64 (д, 1H), 7,12 (д, 1H), 5,64 (с, 2H), 5,00 (с, 2H), 1,62-1,66 (м, 9H).
Стадия 4
К суспензии трет-бутил-2-(хлорметил)-1-(оксазол-2-илметил)-1H-бензo[d]имидазол-6-карбоксилата (10,1 г, 29 ммоль) и промежуточного соединения 4 (11,2 г, 29,1 ммоль) в MeCN (100 мл) добавляли K2CO3 (16,1 г, 116 ммоль). Смесь перемешивали при 60°C в течение 2 ч и затем разбавляли водой (200 мл) и перемешивали в течение дополнительных 4 ч при КТ. Полученные твердые вещества собирали фильтрованием с получением трет-бутил-2-((4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(оксазол-2-илметил)-1H-бензo[d]имидазол-6-карбоксилата (16,23 г, 89%) в виде твердого вещества. 1H ЯМР (ДМСО-d6) δ 8,13 (с, 1H), 8,04 (с, 1H), 7,88 (д, 1H), 7,78 (дд, 1H), 7,70 (ушир. с, 2H), 7,66 (д, 1H), 7,62 (т, 1H), 7,13 (с, 1H), 6,79 (д, 1H), 6,69 (д, 1H), 5,91 (с, 2H), 5,44 (с, 2H), 3,84 (с, 2H), 2,80 (д, 2H), 2,46 (д, 1H), 2,05-2,13 (м, 2H), 1,64 (д, 2H), 1,55 (с, 9H), 1,35-1,43 (м, 2H).
Стадия 5
К раствору трет-бутил-2-((4-(6-((4-цианo-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(оксазол-2-илметил)-1H-бензo[d]имидазол-6-карбоксилата (31,1 г, 50,0 ммоль) в DCE (300 мл) добавляли TFA (40 мл, 530 ммоль). Смесь нагревали до 70°С в течение 4 ч и затем медленно охлаждали до КТ и перемешивали в течение ночи. Смесь концентрировали при пониженном давлении и остаток растворяли в MeOH (100 мл) и воде (300 мл). Насыщенный водн. NaHCO3 (85 мл) добавляли по каплям, чтобы довести pH раствора до ~7. Полученные твердые вещества перемешивали для гранулирования в течение 3 ч и затем собирали фильтрованием с получением соединения примера 7A-01 (27,3 г, 96%) в виде твердого вещества. 1H ЯМР (600 МГц, ДМСО-d6) δ 12,93 (ушир. с, 1H), 8,19 (с, 1H), 8,03 (с, 1H), 7,88 (д, 1H), 7,82 (д, 1H), 7,70 (ушир. с, 2H), 7,65 (д, 1H), 7,62 (т, 1H), 7,12 (с, 1H), 6,80 (д, 1H), 6,66-6,71 (м, 1H), 5,90 (с, 2H), 5,43 (с, 2H), 3,84 (с, 2H), 2,81 (д, 2H), 2,46 (м, 1H), 2,10 (т, 2H), 1,64 (д, 2H), 1,36-1,46 (м, 2H); ЖХМС(ES+): 568,3 (M+H).
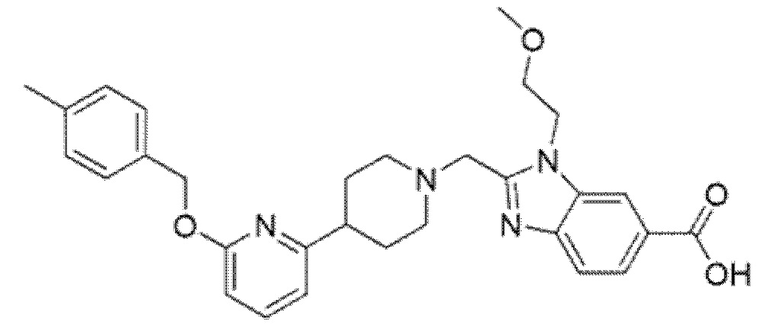
Пример 8A-01
Аммоний-2-((4-(6-((4-метилбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензo[d]имидазол-6-карбоксилат
Во флакон объемом 1 драхма (3,7 мл) добавляли промежуточное соединение 35 (20 мг, 47 мкмоль), а затем 4-метилбензиловый спирт (100 мкмоль). Добавляли THF (500 мкл), а затем реагент Тсунода (цианометилентрибутилфосфоран, 0,5 М в THF, 400 мкл, 0,20 ммоль), и смесь нагревали при 70°С в течение 3 ч. Реакционную смесь охлаждали до КТ и концентрировали при пониженном давлении. Остаток растворяли в MeOH (1 мл). Добавляли 1 М NaOH (0,15 мл, 150 мкмоль) и смесь нагревали при 60°С в течение 3 ч, и затем выдерживали при КТ в течение 48 ч. Смесь концентрировали при пониженном давлении и неочищенный продукт очищали препаративной СФХ с получением соединения примера 8A-01 (10,7 мг, 45%). Способ СФХ (Колонка: Phenomenex Biphenyl 4,6×150 мм), 5 мкм; Подвижная фаза А: CO2 (об./об.); Подвижная фаза В: Метанол с 0,2% NH4OH (об./об.) 85% CO2/15% Метанол с 0,2% NH4OH линейный градиент в течение 8 мин, выдерживали при 70% CO2/30% метанола с 0,2% NH4OH до 10 мин. Поток: 75 мл/мин. Противодавление: 120 бар (12 МПа); Время удерживания: 2,56 мин. ЖХМС(ES+): 515,4 (M+H).
Соединения, перечисленные в приведенной ниже таблице 3, были получены с использованием методик, аналогичных описанным выше для синтеза соединения примера 8А-01 с использованием соответствующих исходных веществ, которые имеются в продаже, были получены с использованием препаратов, хорошо известных специалистам в данной области, или получены способом, аналогичным описанным выше способам для других промежуточных соединений. Соединения очищали с использованием способов, хорошо известных специалистам в данной области, которые могут включать хроматографию на силикагеле, ВЭЖХ или кристаллизацию из реакционной смеси. Конечные соединения могут быть выделены в виде нейтральных или кислотных или основных солей.
Таблица 3
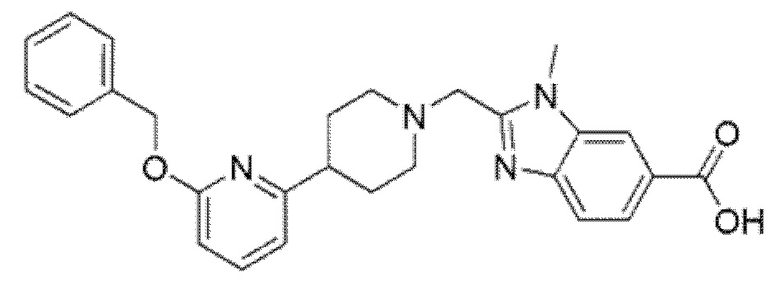
Пример 9A-01
2-((4-(6-(Бензилокси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-бензо[d]имидазол-6-карбоновая кислота
Стадия 1
Смесь промежуточного соединения 36 (100 мг, 0,251 ммоль), бензилового спирта (48,2 мг, 0,446 ммоль), BINAP (23,2 мг, 0,0373 ммоль), Pd2(dba)3 (15,2 мг, 0,0166 ммоль) и Cs2CO3 (123 мг, 0,378 ммоль) в PhMe (2 мл) перемешивали при 100°С в течение 14 ч. Коричневую смесь разбавляли DCM (50 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении с получением коричневого масла, которое очищали преп-ТСХ (DCM:MeOH=20:1) с получением метил-2-((4-(6-(бензилокси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-бензо[d]имидазол-6-карбоксилата (99,7 мг, 84%) в виде желтого твердого вещества. 1H ЯМР (CD3OD) δ 8,32 (с, 1H), 8,02 (дд, 1H), 7,79 (д, 1H), 7,59-7,70 (м, 1H), 7,40-7,48 (м, 2H), 7,35 (м, 2H), 7,23-7,32 (м, 1H), 6,90 (д, 1H), 6,73 (д, 1H), 5,40 (с, 2H), 4,79 (с, 2H), 3,96 (с, 6H), 3,91 (д, 2H), 3,40 (м, 2H), 3,05 (ушир. с, 1H), 2,14-2,38 (м, 4H).
Стадия 2
К раствору метил-2-((4-(6-(бензилокси)пиридин-2-ил)пиперидин-1-ил)метил)-1-метил-1H-бензо[d]имидазол-6-карбоксилата (90,0 мг, 0,191 ммоль) в MeOH (3 мл) добавляли 3,0 M NaOH (2,0 мл, 6,0 ммоль). Смесь перемешивали при 40°С в течение 4 ч. Реакционную смесь нейтрализовали 1 М HCl и полученную суспензию экстрагировали (DCM:MeOH 10:1, 2×40 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением желтого твердого вещества. Желтое твердое вещество очищали препаративной ВЭЖХ (Колонка: Waters Xbridge Prep OBD C18 100×19 мм × 5 мкм; Подвижная фаза: от 5% MeCN в воде (0,1% TFA) до 95% MeCN в воде (0,1% TFA); Длина волны: 220 нм; Скорость потока: 25 мл/мин), с получением соединения примера 9A-01 (33 мг, 28%) в виде твердого вещества. Из-за растворителя очистки конечное соединение, вероятно, представляло собой трифторацетатную соль. 1H ЯМР (400 МГц, CD3OD) δ 8,31 (с, 1H), 8,03 (дд, 1H), 7,78 (д, 1H), 7,65 (т, 1H), 7,40-7,46 (м, 2H), 7,35 (т, 2H), 7,25-7,31 (м, 1H), 6,90 (д, 1H), 6,73 (д, 1H), 5,40 (с, 2H), 4,79 (с, 2H), 3,96 (с, 3H), 3,90 (д, 2H), 3,40 (м, 2H), 3,05 (ушир. с, 1H), 2,14-2,37 (м, 4H); ЖХМС(ES+): 457,1 (M+H).
Соединения, перечисленные в приведенной ниже таблице 4, были получены с использованием методик, аналогичных описанным выше для синтеза соединения примера 9А-01 с использованием соответствующих исходных веществ, которые имеются в продаже, были получены с использованием препаратов, хорошо известных специалистам в данной области, или получены способом, аналогичным описанным выше способам для других промежуточных соединений. Соединения очищали с помощью ВЭЖХ. Из-за растворителя очистки конечные соединения, выделенные с использованием способов PF-AB01 и PF-AB10, были, вероятно, трифторацетатными солями, тогда как соединения, выделенные с использованием способа PF-CD05, являются, вероятно, солями аммония.
Таблица 4
*Найденный мол.вес (MW): МС(ES+): как (M+H)
**Способ очистки ВЭЖХ PF-AB01: Подвижная фаза А: 0,0375% TFA в H2O. Подвижная фаза В: 0,01875% TFA в MeCN. Исходные условия: B: 1%, A: 99% Градиент: B: 1%, A: 99% к B: 5%, A: 95% от t=0,00 мин до 0,60 мин, затем к B: 100% от t=0,60 мин до 4,00 мин, затем к B: 1%, A: 99% от t=4,00 мин до 4,30 мин, выдерживали до t=4,70 мин. Скорость потока=0,8 мл/мин, объем введенной пробы 2 мкл.
**Способ очистки ВЭЖХ PF-CD05: Подвижная фаза А: 0,05% NH4OH в H2O. Подвижная фаза В: 100% MeCN. Исходные условия: B: 5%, A: 95%. Градиент: B: 5%, A: 95% к B: 100%, от t=0,50 мин до 3,40 мин, выдерживали до t=4,20 мин, затем к В: 5%, A: 95% от t=4,21 мин до 4,70 мин, выдерживали до t=4,70 мин. Скорость потока=0,8 мл/мин, объем введенной пробы 2 мкл.
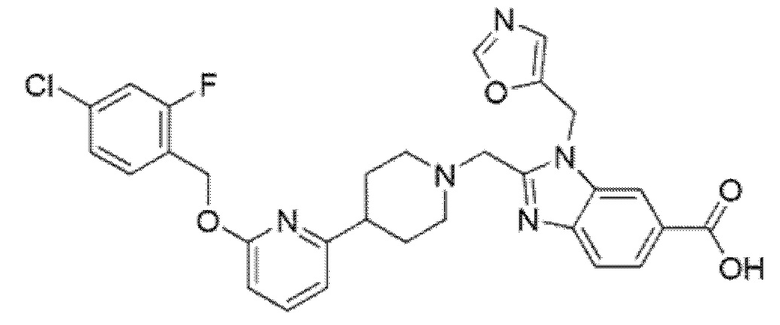
Пример 10А-01
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновая кислота
Стадия 1
К бесцветному раствору метил 3-фтор-4-нитробензоата (302 мг, 1,52 ммоль) и оксазол-5-илметанамина (164 мг, 1,67 ммоль) в DMF (5,0 мл) медленно добавляли Et3N (460 мг, 4,55 ммоль) при 20°С. Коричневый раствор перемешивали при 60°С в течение 36 ч. Смесь разбавляли EtOAc (50 мл) и промывали H2O (50 мл). Органическую фазу отделяли и водную фазу экстрагировали EtOAc (2 × 50 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (10-100% EtOAc/PE) с получением метил-4-нитро-3-((оксазол-5-илметил)амино)бензоата (320 мг, 76%) в виде оранжевого твердого вещества. 1H ЯМР (CDCl3) δ 8,26 (д, 2H), 7,89 (с, 1H), 7,66 (д, 1H), 7,35 (дд, 1H), 7,11 (с, 1H), 4,68 (д, 2H), 3,96 (с, 3H).
Стадия 2
К желтой суспензии метил-4-нитро-3-((оксазол-5-илметил)амино)бензоата (67 мг, 0,24 ммоль) в MeOH (8 мл) добавляли 10% Pd/C (10,3 мг). Смесь перемешивали при 1 атм (0,10 МПа) H2 при КТ в течение 1 ч. Твердые вещества удаляли фильтрованием и промывали MeOH (20 мл). Объединенные органические слои затем концентрировали при пониженном давлении с получением метил-4-амино-3-((оксазол-5-илметил)амино)бензоата (56 мг, 94%) в виде белого твердого вещества. ЖХМС(ES+): 247,9 (M+H).
Стадия 3
К желтому раствору промежуточного соединения 5 (85 мг, 0,22 ммоль), 4-амино-3-((оксазол-5-илметил)амино)бензоату (55,5 мг, 0,224 ммоль) и HATU (111 мг, 0,292 ммоль) в DMF (2 мл) добавляли Et3N (114 мг, 1,12 ммоль, 0,15 мл). Желтый раствор перемешивали при 25°С в течение 16 ч. Затем смесь выливали в H2O (8 мл) и экстрагировали EtOAc (3 × 10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали преп-ТСХ (EtOAc) с получением метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)ацетамидо)-3-((оксазол-5-илметил)амино)бензоата (58 мг, 43%) в виде желтого масла. ЖХМС(ES+): 630,0 (M+Na).
Стадия 4
Желтый раствор метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)ацетамидо)-3-((оксазол-5-илметил)амино)бензоата (58 мг, 0,095 ммоль) в AcOH (0,5 мл) перемешивали при 60°С в течение 3 ч и затем при КТ в течение 16 ч. Желтый остаток нейтрализовали насыщ. водн. Na2CO3 и экстрагировали DCM (3 × 10 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(оксазол-5-илметил)-1H-бензо[d]имидазол-6-карбоксилата (56 мг, 99%) в виде желтого масла. ЖХМС(ES+): 612,0 (M+Na).
Стадия 5
К раствору метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-(оксазол-5-илметил)-1H-бензо[d]имидазол-6-карбоксилата (56 мг, 0,095 ммоль) в THF (1 мл) и MeOH (0,2 мл) добавляли 2 M NaOH (0,0949 мл, 0,190 ммоль). Желтый раствор перемешивали при 25°C в течение 16 ч и затем выдерживали в течение 48 ч при 25°C. Желтый раствор концентрировали при пониженном давлении и остаток затем растворяли в H2O (5 мл), подкисляли до pH ~5 с помощью 1 М HCl и экстрагировали DCM (5 × 10 мл). Объединенные органические экстракты концентрировали при пониженном давлении и полученный неочищенный продукт очищали препаративной ВЭЖХ (Колонка: Waters Xbridge Prep OBD C18 150×30 мм × 5 мкм; Подвижная фаза: от 5% MeCN в воде (0,1% TFA) до 95% MeCN в воде (0,1% TFA); Длина волны: 220 нм; Скорость потока: 25 мл/мин), с получением соединения примера 10А-01 (22 мг, 33%) в виде твердого вещества. Из-за растворителя очистки конечное соединение, вероятно, было выделено в виде трифторацетатной соли.1H ЯМР (400 МГц, CD3OD) δ 8,41 (с, 1H), 8,18 (с, 1H), 8,03 (дд, 1H), 7,80 (д, 1H), 7,61-7,70 (м, 1H), 7,52 (т, 1H), 7,36 (с, 1H), 7,20-7,30 (м, 2H), 6,94 (д, 1H), 6,74 (д, 1H), 5,78 (с, 2H), 5,45 (с, 2H), 4,91 (ушир. с, 2H), 3,97 (д, 2H), 3,42 (ушир. с, 2H), 3,07 (ушир. с, 1H), 2,17-2,33 (м, 4H); ЖХМС(ES+): 576,1 (M+H).
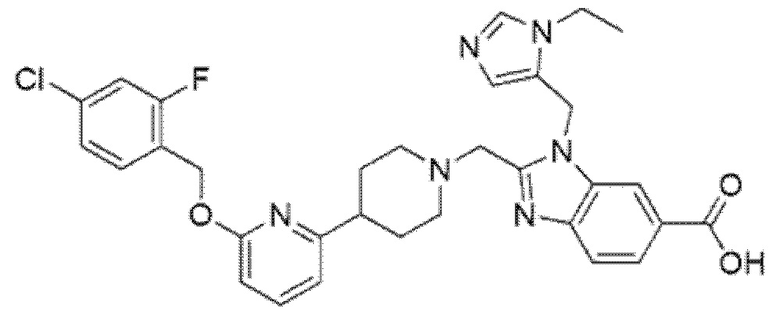
Пример 10A-02
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота
Стадия 1
К раствору промежуточного соединения 29 (200 мг, 0,829 ммоль) в DMF (8 мл) добавляли (1-этил-1H-имидазол-5-ил)метанамин (104 мг, 0,829 ммоль) и NaHCO3 (348 мг, 4,15 ммоль). Реакционную смесь перемешивали при 60°С в течение 16 ч. Реакционную смесь выливали в воду (10 мл) и затем экстрагировали EtOAc (2 × 30 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (2×20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (0-5% MeOH/DCM) с получением трет-бутил 3-(((1-этил-1H-имидазол-5-ил)метил)амино)-4-нитробензоата (105 мг, 37%) в виде бледно-красного масла. 1H ЯМР (CDCl3) δ 8,23 (д, 1H), 7,96 (ушир. с, 1H), 7,66 (д, 1H), 7,57 (с, 1H), 7,28 (дд, 1H), 7,12 (с, 1H), 4,54 (д, 2H), 4,00 (кв, 2H), 1,62 (с, 9H), 1,47 (т, 3H).
Стадия 2
К раствору трет-бутил-3-(((1-этил-1H-имидазол-5-ил)метил)амино)-4-нитробензоата (105 мг, 0,303 ммоль) в MeOH (3 мл) и H2O (1 мл) добавляли порошок Fe (59,2 мг, 1,06 ммоль) и NH4Cl (292 мг, 5,46 ммоль). Реакционную смесь перемешивали при 80°С в течение 50 мин. Реакционную смесь выливали в воду (10 мл) и экстрагировали EtOAc (3 × 15 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением трет-бутил-4-амино-3-(((1-этил-1H-имидазол-5-ил)метил)амино)бензоата (93 мг, 97%) в виде бледно-коричневого твердого вещества, которое использовали непосредственно на следующей стадии.
Стадия 3
К бледно-желтому раствору промежуточного соединения 5 (55 мг, 0,15 ммоль) и DMF (1 мл) добавляли HATU (66,2 мг, 0,174 ммоль). Смесь перемешивали при 30°С в течение 10 мин. Добавляли раствор трет-бутил-4-амино-3-(((1-этил-1H-имидазол-5-ил)метил)амино)бензоата (45,9 мг, 0,145 ммоль) и DIPEA (56,3 мг, 0,436 ммоль) в DMF (1 мл) и реакционную смесь перемешивали при 30°С в течение 16 ч. Смесь выливали в воду (10 мл) и затем экстрагировали EtOAc (3 × 20 мл). Объединенные органические экстракты промывали водн. NH4Cl (3×20 мл), насыщенным солевым раствором (2×20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали преп-ТСХ (5% MeOH/DCM) с получением трет-бутил-4-амино-3-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-N-((1-этил-1H-имидазол-5-ил)метил)ацетамидо)бензоата (60 мг, 61%) в виде бледно-коричневой смолы. ЖХМС(ES+): 699,4 (M+Na).
Стадия 4
Бледно-коричневый раствор полученного трет-бутил-4-амино-3-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-N-((1-этил-1H-имидазол-5-ил)метил)ацетамидо)бензоата (60 мг, 0,089 ммоль) в AcOH (2 мл) перемешивали при 60°С в течение 16 ч. Реакционную смесь концентрировали под вакуумом для удаления АсОН с получением трет-бутил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-((1-этил-1H-имидазол-5-ил)метил)-1H-бензо[d]имидазол-6-карбоксилата (56 мг, 96%) в виде бледно-коричневой смолы, которую использовали на следующей стадии без дополнительной очистки. ЖХМС(ES+): 681,3 (M+Na).
Стадия 5
К бледно-коричневому раствору трет-бутил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)метил)-1-((1-этил-1H-имидазол-5-ил)метил)-1H-бензо[d]имидазол-6-карбоксилата (56 мг, 0,085 ммоль) в DCM (2 мл) добавляли TFA (1 мл). Реакционную смесь перемешивали при КТ (10°С) в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и неочищенный продукт очищали препаративной ВЭЖХ (Колонка: Waters Xbridge Prep OBD C18 150×30 мм × 5 мкм; Подвижная фаза: от 5% MeCN в воде (0,1% TFA) до 95% MeCN в воде (0,1% TFA); Длина волны: 220 нм; Скорость потока: 25 мл/мин), с получением соединения примера 10А-02 (37 мг, 48%) в виде бежевого твердого вещества. Из-за растворителя очистки соединение, вероятно, было выделено в виде трифторацетатной соли. 1H ЯМР (400 МГц, CD3OD) δ 9,10 (д, 1H), 8,26 (с, 1H), 8,07 (дд, 1H), 7,88 (д, 1H), 7,66 (т, 1H), 7,52 (т, 1H), 7,19-7,28 (м, 2H), 7,08 (д, 1H), 6,93 (д, 1H), 6,73 (д, 1H), 5,88 (с, 2H), 5,45 (с, 2H), 4,84 (с, 2H), 4,36 (кв, 2H), 3,98 (д, 2H), 3,41 (т, 2H), 3,06 (т, 1H), 2,14-2,40 (м, 4H), 1,58 (т, 3H); ЖХМС(ES+): 603,1 (M+H).
Соединения, перечисленные в приведенной ниже таблице 5, были получены с использованием методик, аналогичных описанным выше для синтеза соединений примеров 10А-01 или 10А-02 с использованием соответствующих исходных веществ, которые имеются в продаже, были получены с использованием препаратов, хорошо известных специалистам в данной области, или получены способом, аналогичным описанным выше способам для других промежуточных соединений. Соединения очищали с использованием способов, хорошо известных специалистам в данной области, которые могут включать хроматографию на силикагеле, ВЭЖХ или кристаллизацию из реакционной смеси. Конечные соединения могут быть выделены в виде нейтральных или кислотных или основных солей.
Таблица 5
Соединения, перечисленные в приведенной ниже таблице 6, были получены с помощью параллельного синтеза с использованием методик, аналогичных описанным выше для синтеза соединения примера 10А-01 с использованием соответствующих исходных веществ, которые имеются в продаже, были получены с использованием препаратов, хорошо известных специалистам в данной области, или получены способом, аналогичным описанным выше способам для других промежуточных соединений. Соединения очищали с помощью ВЭЖХ. Из-за растворителя очистки конечные соединения, выделенные с использованием способов PF-AB01 и PF-AB10, были, вероятно, трифторацетатными солями, тогда как соединения, выделенные с использованием способа PF-CD05, являются, вероятно, солями аммония.
Таблица 6
*Найденный мол.вес (MW): МС(ES+): как (M+H)
**Способ очистки ВЭЖХ PF-AB01: Подвижная фаза А: 0,0375% TFA в H2O. Подвижная фаза В: 0,01875% TFA в MeCN. Исходные условия: B: 1%, A: 99% Градиент: B: 1%, A: 99% к B: 5%, A: 95% от t=0,00 мин до 0,60 мин, затем к B: 100% от t=0,60 мин до 4,00 мин, затем к B: 1%, A: 99% от t=4,00 мин до 4,30 мин, выдерживали до t=4,70 мин. Скорость потока=0,8 мл/мин, объем введенной пробы 2 мкл.
**Способ очистки ВЭЖХ PF-AB10: Подвижная фаза А: 0,0375% TFA в H2O. Подвижная фаза В: 0,01875% TFA в MeCN. Исходные условия: B: 10%, A: 90%. Градиент: B: 10%, A: 90% от t=0,00 мин до 0,50 мин, затем к B: 100% от t=0,60 мин до 4,00 мин, затем к B: 10%, A: 90% от t=4,00 мин до 4,30 мин, выдерживали до t=4,70 мин. Скорость потока=0,8 мл/мин, объем введенной пробы 2 мкл.
**Способ очистки ВЭЖХ PF-CD05: Подвижная фаза А: 0,05% NH4OH в H2O. Подвижная фаза В: 100% MeCN. Исходные условия: B: 5%, A: 95%. Градиент: B: 5%, A: 95% к B: 100%, от t=0,50 мин до 3,40 мин, выдерживали до t=4,20 мин, затем к В: 5%, A: 95% от t=4,21 мин до 4,70 мин, выдерживали до t=4,70 мин. Скорость потока=0,8 мл/мин, объем введенной пробы 2 мкл.
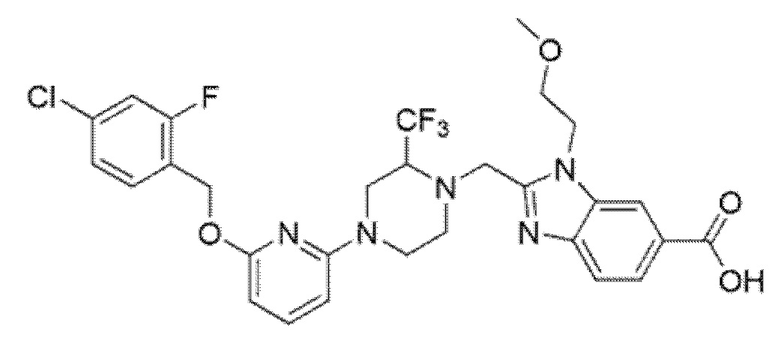
Пример 12A-01
2-((4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)-2-(трифторметил)пиперазин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоновая кислота, энантиомер 1
Пример 12A-02
2-((4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)-2-(трифторметил)пиперазин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоновая кислота, энантиомер 2
Стадия 1
К раствору 4-хлор-2-фторбензилового спирта (15,0 г, 93,4 ммоль) в DMF (250 мл) добавляли NaH (4,48 г, 112 ммоль, 60% сусп.) при 0°С. После перемешивания при 15°C в течение 40 мин добавляли 2,6-дихлорпиридин (16,6 г, 112 ммоль). Полученную смесь перемешивали при 15°С в течение 3 ч. Смесь выливали в воду (1 л) и экстрагировали EtOAc (2 × 200 мл). Объединенные органические слои промывали насыщ. NH4Cl (500 мл), насыщенным солевым раствором (500 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (PE) с получением 2-хлор-6-((4-хлор-2-фторбензил)окси)пиридина (19,2 г, 75%) в виде твердого вещества. 1H ЯМР (CDCl3) δ 7,55 (т, 1H), 7,47 (т, 1H), 7,15 (т, 2H), 6,94 (д, 1H), 6,71 (д, 1H), 5,40 (с, 2H).
Стадия 2
К раствору трет-бутил-3-(трифторметил)пиперазин-1-карбоксилата (100 мг, 0,393 ммоль) в MeCN (2 мл) добавляли промежуточное соединение 19 (111 мг, 0,393 ммоль), йодид тетрабутиламмония (145 мг, 0,39 ммоль) и N,N-диизопропилэтиламин (152 мг, 1,18 ммоль). Реакционную смесь перемешивали при 150°С в течение 1 ч в условиях микроволнового излучения. Реакционную смесь концентрировали при пониженном давлении и очищали преп-ТСХ (33% EtOAc/PE) с получением метил-2-((4-(трет-бутоксикарбонил)-2-(трифторметил)пиперазин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензo[d]имидазол-6-карбоксилата (100 мг, 51%) в виде желтого масла. МС(ES+): 501,1 (M+H).
Стадия 3
К раствору метил-2-((4-(трет-бутоксикарбонил)-2-(трифторметил)пиперазин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензo[d]имидазол-6-карбоксилата (100 мг, 0,2 ммоль) в EtOAc (5 мл) добавляли HCl-EtOAc (5 мл). Реакционную смесь перемешивали при КТ в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении. Неочищенный продукт растворяли в DCM (10 мл), промывали насыщ. водн. K2CO3, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением метил-1-(2-метоксиэтил)-2-((2-(трифторметил)пиперазин-1-ил)метил)-1H-бензo[d]имидазол-6-карбоксилата (42 мг, 53%) в виде желтого масла. МС(ES+): 401,0 (M+H).
Стадия 4
К раствору метил-1-(2-метоксиэтил)-2-((2-(трифторметил)пиперазин-1-ил)метил)-1H-бензо[d]имидазол-6-карбоксилата (90 мг, 0,22 ммоль) в PhCH3 (2 мл) добавляли 2-хлор-6-((4-хлор-2-фторбензил)окси)пиридин (61,2 мг, 0,225 ммоль), Pd2(dba)3 (20,6 мг, 0,1 ммоль), BINAP (28 мг, 0,045 ммоль) и Cs2CO3 (220 мг, 0,674 ммоль). Реакционную смесь перемешивали при 100°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и неочищенный продукт очищали препаративной ТСХ (33% EtOAc/PE) с получением рацемического продукта в виде желтого масла. Рацемическую смесь разделяли с помощью препаративной хиральной СФХ (Колонка: Whelk-01 250×30 мм × 10 мкм; Подвижная фаза: 45% изопропанол (1% NH4OH)/CO2; Скорость потока: 50 мл/мин) с получением выделенных энантиомеров метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-(трифторметил)пиперазин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилата:
Энантиомер 1: (28 мг, 20%); время удерживания (15,98 мин);
Энантиомер 2: (33 мг, 23%); время удерживания (20,92 мин).
Стадия 5
К раствору со стадии 4 энантиомера 1 (28 мг, 0,044 ммоль) в MeOH (5 мл) добавляли 2 M NaOH (1 мл). Реакционную смесь перемешивали при 50°С в течение 2 ч. Реакционную смесь охлаждали до 0°С и подкисляли 1 М HCl до рН ~4. Реакционную смесь экстрагировали EtOAc (3 × 10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной ВЭЖХ (Колонка: Waters Xbridge Prep OBD C18 150×30 мм × 5 мкм; Подвижная фаза: от 55% MeOH в воде (0,1% TFA) до 75% MeOH в воде (0,1% TFA); 10 мин градиент; Длина волны: 220 нм; Скорость потока: 25 мл/мин), с получением соединения примера 12А-01 (12,2 мг, 37%) в виде твердого вещества. Из-за растворителя очистки соединение, вероятно, было выделено в виде трифторацетатной соли. Аналитические данные ЖХМС: Xtimate C18 5×30 мм, 3 мкм; Подвижная фаза: от 1% MeCN в воде (0,1% TFA) до 5% MeCN в воде (0,1% TFA) в течение 1 мин; затем от 5% MeCN в воде (0,1% TFA) до 100% MeCN (0,1% TFA) в течение 5 мин; выдерживали при 100% MeCN (0,1% TFA) в течение 2 мин; снова до 1,0% MeCN в воде (0,1% TFA) при 8,01 мин и выдерживали 2 мин. Скорость потока: 1,2 мл/мин; Время удерживания: 4,465 мин, МС(ES+): 622,2 (M+H).
Соединение примера 12A-02 получали аналогичным образом из энантиомера 2 (33 мг, 0,052 ммоль) стадии 4 и очищали с использованием такого же метода препаративной ВЭЖХ с получением соединения примера 12A-02 (9,8 мг, 28%) в виде твердого вещества. Из-за растворителя очистки соединение, вероятно, было выделено в виде трифторацетатной соли. Аналитические данные ЖХМС: Время удерживания: 4,469 мин, МС(ES+): 622,2 (M+H).
Соединения, перечисленные в приведенной ниже таблице 7, были получены с использованием методик, аналогичных описанным выше для синтеза соединений примеров 12А-01 и 12А-02 с использованием соответствующих исходных веществ, которые имеются в продаже, были получены с использованием препаратов, хорошо известных специалистам в данной области, или получены способом, аналогичным описанным выше способам для других промежуточных соединений. Соединения очищали с использованием способов, хорошо известных специалистам в данной области, которые могут включать хроматографию на силикагеле, ВЭЖХ или кристаллизацию из реакционной смеси. Конечные соединения могут быть выделены в виде нейтральных или кислотных или основных солей.
Таблица 7
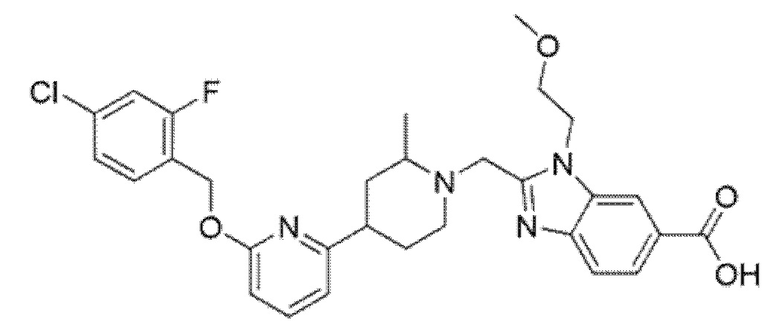
Примеры 13A-01 и 13А-02
Транс-2-{[4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперидин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота, энантиомеры 1 и 2
Примеры 13A-03 и 13A-04
Цис-2-{[4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперидин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновая кислота, энантиомеры 1 и 2
Стадия 1
Смесь 2-[(4-хлор-2-фторбензил)окси]-6-(2-метилпиперидин-4-ил)пиридина (350 мг, 0,86 ммоль) [полученного в виде смеси стереоизомеров способом, аналогичным используемому для промежуточного соединения 3], промежуточное соединение 19 (220 мг, 0,78 ммоль) и K2CO3 (540 мг, 3,9 ммоль) в MeCN (6 мл) перемешивали при 60°С в течение 16 ч. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали с использованием колоночной хроматографии, элюируя EtOAc/PE (1:1) с получением метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилата (250 мг, 55%, желтое масло) в виде смеси четырех стереоизомеров. Смесь стереоизомеров разделяли с помощью СФХ на хиральной колонке, используя условие 1 ниже, чтобы получить чистые пики 1, 3 и 4, а также пик 2, который не был чистым. Пик 2 повторно очищали СФХ с использованием условия 2. Времена удерживания относятся к условию 1 СФХ. Относительную стереохимию определяли с помощью 2D ЯМР. Абсолютная конфигурация каждого изомера не была определена.
Пик 1 (время удерживания 5,6 мин): транс-метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилат, энантиомер 1.
Пик 2 (время удерживания 5,8 мин): транс-метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилат, энантиомер 2.
Пик 3 (время удерживания 6,4 мин): цис-метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилат, энантиомер 1.
Пик 4 (время удерживания 6,9 мин): цис-метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилат, энантиомер 2.
СФХ условие 1: Колонка: AD (250 мм × 30 мм, 5 мкм); Подвижная фаза: CO2 с 35% EtOH (0,1% NH4OH); Скорость потока: 70 мл/мин; Длина волны: 220 нм.
СФХ условие 2: Колонка: AD (250 мм × 30 мм, 5 мкм); Подвижная фаза: CO2 c 40% iPrOH (0,1% NH4OH); Скорость потока: 60 мл/мин; Длина волны: 220 нм
Стадия 2
Сложные метиловые эфиры со стадии 1 превращали в свободные кислоты путем обработки NaOH в МеОН, как описано ранее, с получением четырех указанных в заголовке примеров соединений.
Пример 13A-01 (транс-метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилат, энантиомер 1): 1H ЯМР (400 МГц, CD3OD) δ: 8,32 (д, 1H), 8,01 (дд, 1H), 7,77 (д, 1H), 7,68 (дд, 1H), 7,49 (т, 1H), 7,17-7,31 (м, 2H), 6,97 (д, 1H), 6,76 (д, 1H), 5,37-5,50 (м, 2H), 4,75-4,86 (м, 2H), 4,62 (т, 2H), 4,19 (ушир. с, 1H), 3,76 (т, 2H), 3,66 (д, 1H), 2,49 (ддд, 1H), 2,30 (м, 1H), 2,16-2,24 (м, 1H), 2,10 (дт, 1H), 1,62 (д, 2H); ЖХМС(ES+): 567,1 (M+H).
Пример 13A-02 (транс-метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилат, энантиомер 2): 1H ЯМР (400 МГц, CD3OD) δ 8,31 (д, 1H), 8,00 (дд, 1H), 7,76 (д, 1H), 7,67 (дд, 1H), 7,48 (т, 1H), 7,22 (м, 2H), 6,96 (д, 1H), 6,75 (д, 1H), 5,47-5,38 (м, 2H), 4,80 (м, 1H), 4,61 (м, 2H), 4,18 (м, 1H), 3,80-3,59 (м, 3H), 2,48 (м, 1H), 2,29 (м, 1H), 2,19 (м, 1H), 2,09 (м, 1H), 1,61 (д, 3H); ЖХМС(ES+): 567,1 (M+H).
Пример 13A-03 (цис-метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилат, энантиомер 1): 1H ЯМР (400 МГц, CD3OD) δ 8,34 (д, 1H), 8,05 (дд, 1H), 7,81 (д, 1H), 7,73-7,64 (м, 1H), 7,53 (м, 1H), 7,26 (м, 2H), 6,94 (д, 1H), 6,76 (д, 1H), 5,48 (с, 2H), 5,12 (д, 1H), 4,77 (д, 1H), 4,64 (м, 2H), 4,01-3,82 (м, 2H), 3,78 (м, 2H), 3,52-3,42 (м, 1H), 3,15 (м, 1H), 2,35-2,05 (м, 4H), 1,57 (д, 3H); ЖХМС(ES+): 567,1 (M+H).
Пример 13A-04 (цис-метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперидин-1-ил)метил)-1-(2-метоксиэтил)-1H-бензо[d]имидазол-6-карбоксилат, энантиомер 2): 1H ЯМР (400 МГц, CD3OD) δ 8,34 (д, 1H), 8,05 (дд, 1H), 7,81 (д, 1H), 7,70-7,64 (м, 1H), 7,53 (т, 1H), 7,26 (м, 2H), 6,94 (д, 1H), 6,76 (д, 1H), 5,48 (с, 2H), 5,12 (д, 1H), 4,77 (д, 1H), 4,64 (т, 2H), 3,99-3,83 (м, 2H), 3,78 (т, 2H), 3,52-3,42 (м, 1H), 3,19-3,09 (м, 1H), 2,31-2,05 (м, 4H), 1,57 (д, 3H); ЖХМС(ES+): 567,1 (M+H).
Сложные метиловые эфиры соединений, перечисленные в таблице 8 ниже, были получены с использованием методик, аналогичных описанным выше для синтеза соединений примера 10А-01, с использованием рацемического 2-аминометилтетрагидрофурана или 3-аминометилтетрагидрофурана и других подходящих исходных веществ, которые имеются в продаже, были получены с использованием препаратов, хорошо известных специалистам в данной области, или получены способом, аналогичным описанным выше способам для других промежуточных соединений. Стереоизомеры THF разделяли с помощью СФХ с получением сложноэфирных промежуточных соединений в виде отдельных стереоизомеров. Сложные метиловые эфиры затем гидролизовали, как описано для примера 10А-01, с получением соединений, перечисленных в таблице 8. Время удерживания и способы хроматографии для промежуточных соединений сложного метилового эфира приведены в таблице. Стереохимия THF стереоцентра в каждом соединении не была определена.
Таблица 8
* Способы разделения и время удерживания сложных метиловых эфиров примеров:
Способ А: Препаративный способ: Колонка: AD (250 мм × 30 мм, 10 мкм); Подвижная фаза: CO2 с 50% MeOH (0,1% NH4OH); Скорость потока: 80 мл/мин; Длина волны: 220 нм. Аналитический метод: Колонка: AD (50 мм × 4,6 мм, 3 мкм); Подвижная фаза: CO2 с 40% EtOH (0,05% NHEt2); Скорость потока: 4 мл/мин; Длина волны: 220 нм
Способ B: Колонка: AD (250 мм × 30 мм, 10 мкм); Подвижная фаза: CO2 с 40% MeOH (0,1% NH4OH); Скорость потока: 80 мл/мин; Длина волны: 220 нм
Способ C: Колонка: IC (250 мм × 30 мм, 10 мкм); Подвижная фаза: CO2 с 50% MeOH (0,1% NH4OH); Скорость потока: 80 мл/мин; Длина волны: 220 нм
Способ D: Колонка: AD (250 мм × 30 мм, 10 мкм); Подвижная фаза: CO2 с 50% MeOH (0,1% NH4OH); Скорость потока: 80 мл/мин; Длина волны: 220 нм
Способ E: Колонка: OD (250 мм × 30 мм, 10 мкм); Подвижная фаза: CO2 с 45% EtOH (0,1% NH4OH); Скорость потока: 70 мл/мин; Длина волны: 220 нм
Способ F: Колонка: OJ (250 мм × 30 мм, 10 мкм); Подвижная фаза: CO2 с 30% EtOH (0,1% NH4OH); Скорость потока: 80 мл/мин; Длина волны: 220 нм
CHO GLP-1R клон H6 - Анализ 1
Опосредованную GLP-1R активность агониста определяли с помощью клеточного функционального анализа с использованием набора для определения cAMP HTRF (гомогенной флуоресценции с разрешением по времени) (cAMP HI Range Assay Kit; CisBio каталожный номер 62AM6PEJ), который измеряет уровни cAMP в клетке. Этот способ представляет собой конкурентный иммуноанализ между нативным cAMP, продуцируемым клетками, и экзогенным cAMP, меченным красителем d2. Связывание метки визуализируется с помощью mAb анти-cAMP, меченного Cryptate. Удельный сигнал (т.е. передача энергии) обратно пропорционален концентрации cAMP в стандартном или экспериментальном образце.
Кодирующая последовательность человеческого GLP-1R (эталонная последовательность NCBI NP_002053.3, включающая природный вариант Gly168Ser) была субклонирована в pcDNA3 (Invitrogen), и была выделена клеточная линия, стабильно экспрессирующая рецептор (обозначенная как клон H6). Анализы связывания с насыщением (методика фильтрации) с использованием 125I-GLP-17-36 (Perkin Elmer) показали, что плазматические мембраны, полученные из этой клеточной линии, экспрессируют высокую плотность GLP-1R (Kd: 0,4 нМ, Bmax: 1900 фмоль/мг белка).
Клетки извлекали из криоконсервации, повторно суспендировали в 40 мл фосфатно-солевого буферного раствора Дульбекко (DPBS - Lonza, каталожный номер 17-512Q) и центрифугировали при 800 × g в течение 5 мин при 22°C. Затем клеточный осадок ресуспендировали в 10 мл среды для выращивания [смесь 1:1 DMEM/F12 с HEPES, L-Gln, 500 мл (DMEM/F12 Lonza, каталожный номер 12-719F), 10% инактивированной нагреванием фетальной бычьей сыворотки (Gibco, каталожный номер 16140-071), 5 мл 100X Pen-Strep (Gibco, каталожный номер 15140-122), 5 мл 100X L-Глутамина (Gibco, каталожный номер 25030-081) и 500 мкг/мл Geneticin (G418) (Invitrogen, # 10131035)]. Образец 1 мл клеточной суспензии в среде для выращивания подсчитывали на Becton Dickinson ViCell для определения жизнеспособности клеток и количества клеток на мл. Затем оставшуюся клеточную суспензию доводили с помощью среды для выращивания для получения 2000 жизнеспособных клеток на лунку с использованием дозатора реагентов Matrix Combi Multidrop и клетки распределяли в белый 384-луночный аналитический планшет, обработанный для тканевого культивирования (Corning 3570). Затем аналитический планшет инкубировали в течение 48 ч при 37°С в увлажненной среде с 5% диоксида углерода.
Различные концентрации каждого тестируемого соединения (в ДМСО) разбавляли в аналитическом буфере (HBSS с кальцием/магнием (Lonza/BioWhittaker, каталожный номер 10-527F) /0,1% BSA (Sigma Aldrich, каталожный номер A7409-1L)/20 мM HEPES (Lonza/BioWhittaker, каталожный номер 17-737E), содержащем 100 мкМ 3-изобутил-1-метилксантин (IBMX; Sigma, каталожный номер I5879). Конечная концентрация ДМСО составляла 1%.
Через 48 ч среду для выращивания удаляли из лунок аналитического планшета, и клетки обрабатывали 20 мкл серийно разведенного соединения в аналитическом буфере в течение 30 мин при 37°С в увлажненной среде с 5% диоксида углерода. После 30 мин инкубации в каждую лунку аналитического планшета добавляли 10 мкл меченного d2 cAMP и 10 мкл анти-cAMP антитела (оба разбавлены 1:20 в буфере для лизиса клеток; как описано в протоколе анализа от производителя). Затем планшеты инкубировали при комнатной температуре и через 60 мин изменения в сигнале HTRF считывали помощью многоканального планшет-ридера Envision 2104 при возбуждении на волне 330 нм и эмиссии 615 нм и 665 нм. Исходные данные конвертировали в нМ сАМР путем интерполяции из стандартной кривой cAMP (как описано в протоколе анализа производителя) и процент эффекта определяли относительно насыщающей концентрации полного агониста GLP-17-36 (1 μМ) для каждого планшета. Определения EC50 были сделаны на основе кривых доза агониста-ответ, проанализированных с помощью программы аппроксимации кривых, с использованием 4-параметрического логистического уравнения доза-ответ.
CHO GLP-1R клон C6 - Анализ 2
Опосредованную GLP-1R активность агониста определяли с помощью клеточного функционального анализа с использованием набора для определения cAMP HTRF (гомогенной флуоресценции с разрешением по времени) (cAMP HI Range Assay Kit; CisBio каталожный номер 62AM6PEJ), который измеряет уровни cAMP в клетке. Этот способ представляет собой конкурентный иммуноанализ между нативным cAMP, продуцируемым клетками, и экзогенным cAMP, меченным красителем d2. Связывание метки визуализируется с помощью mAb анти-cAMP, меченного Cryptate. Удельный сигнал (т.е. передача энергии) обратно пропорционален концентрации cAMP в стандартном или экспериментальном образце.
Кодирующая последовательность человеческого GLP-1R (эталонная последовательность NCBI NP_002053.3, включающая природный вариант Leu260Phe) была субклонирована в pcDNA5-FRT-TO, и клональная клеточная линия CHO, стабильно экспрессирующая низкую плотность рецепторов, была выделена с использованием системы Flp-In™ T-Rex™, как описано производителем (ThermoFisher). Анализы связывания с насыщением (методика фильтрации) с использованием 125I-GLP-1 (Perkin Elmer) показали, что плазматические мембраны, полученные из этой клеточной линии (обозначаемой «клон С6»), экспрессируют низкую плотность GLP-1R (Kd: 0,3 нМ, Bmax: 240 фмоль/мг белка), относительно клеточной линии клона H6.
Клетки извлекали из криоконсервации, повторно суспендировали в 40 мл фосфатно-солевого буферного раствора Дульбекко (DPBS - Lonza, каталожный номер 17-512Q) и центрифугировали при 800 × g в течение 5 мин при 22°C. DPBS аспирировали и клеточный осадок ресуспендировали в 10 мл полной среды для выращивания (DMEМ:F12 1:1 смесь с HEPES, L-Gln, 500 мл (DMEM/F12 Lonza, каталожный номер 12-719F), 10% инактивированной нагреванием фетальной бычьей сыворотки (Gibco, каталожный номер 16140-071), 5 мл 100X Pen-Strep (Gibco, каталожный номер 15140-122), 5 мл 100X L-Глутамина (Gibco, каталожный номер 25030-081), 700 мкг/мл Hygromycin (Invitrogen, каталожный номер 10687010) и 15 мкг/мл Blasticidin (Gibco, каталожный номер R21001). Образец 1 мл клеточной суспензии в среде для выращивания подсчитывали на Becton Dickinson ViCell для определения жизнеспособности клеток и количества клеток на мл. Затем оставшуюся клеточную суспензию доводили с помощью среды для выращивания для получения 1600 жизнеспособных клеток на лунку с использованием дозатора реагентов Matrix Combi Multidrop и клетки распределяли в белый 384-луночный аналитический планшет, обработанный для тканевого культивирования (Corning 3570). Затем аналитический планшет инкубировали в течение 48 ч при 37°С в увлажненной среде (95% O2, 5% CO2).
Различные концентрации каждого тестируемого соединения (в ДМСО) разбавляли в аналитическом буфере [HBSS с кальцием/магнием (Lonza/BioWhittaker, каталожный номер 10-527F) /0,1% BSA (Sigma Aldrich, каталожный номер A7409-1L)/20 мM HEPES (Lonza/BioWhittaker, каталожный номер 17-737E)], содержащем 100 мкМ 3-изобутил-1-метилксантин (IBMX; Sigma, каталожный номер I5879). Конечная концентрация ДМСО в смеси соединение/аналитический буфер составляла 1%.
Через 48 ч среду для выращивания удаляли из лунок аналитического планшета, и клетки обрабатывали 20 мкл серийно разведенного соединения в аналитическом буфере в течение 30 мин при 37°С в увлажненной среде (95% O2, 5% CO2). После 30 мин инкубации в каждую лунку аналитического планшета добавляли 10 мкл меченного d2 cAMP и 10 мкл анти-cAMP антитела (оба разбавлены 1:20 в буфере для лизиса клеток; как описано в протоколе анализа от производителя). Затем планшеты инкубировали при комнатной температуре и через 60 мин изменения в сигнале HTRF считывали помощью многоканального планшет-ридера Envision 2104 при возбуждении на волне 330 нм и эмиссии 615 нм и 665 нм. Исходные данные конвертировали в нМ сАМР путем интерполяции из стандартной кривой cAMP (как описано в протоколе анализа производителя) и процент эффекта определяли относительно насыщающей концентрации полного агониста GLP-1 (1 μМ) для каждого планшета. Определения EC50 были сделаны на основе кривых доза агониста-ответ, проанализированных с помощью программы аппроксимации кривых, с использованием 4-параметрического логистического уравнения доза-ответ.
В таблице 9 данные анализа представлены двумя (2) значащими цифрами в виде геометрического среднего (EC50) и среднего арифметического (Emax) на основе приведенного количества повторов (Число). Пустая ячейка означает, что для этого примера не было данных или Emax не было рассчитано.
Таблица 9
EC50 (нM)
Число
Число
Все упомянутые здесь патенты, патентные заявки и источники включены в настоящее описание в полном объеме посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| АГОНИСТЫ GLP-1 РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2765721C1 |
| АГОНИСТ РЕЦЕПТОРА GLP-1, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО, И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2021 |
|
RU2800290C1 |
| АГОНИСТЫ РЕЦЕПТОРА GLP-1 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2769715C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ, ОПОСРЕДОВАННЫХ СХСR4 И CCR5 | 2001 |
|
RU2277092C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ПОВЫШЕННОЙ ЭФФЕКТИВНОСТЬЮ, СВЯЗЫВАЮЩИЕСЯ С РЕЦЕПТОРОМ ХЕМОКИНА | 2002 |
|
RU2325387C2 |
| Бициклические гетероциклические соединения и их применения в терапии | 2012 |
|
RU2662827C2 |
| ГЕТЕРОАРИЛ-БИФЕНИЛ АМИНЫ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2837843C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, МОДЕЛИРУЮЩИЕ АКТИВНОСТЬ ХЕМОКИНОВОГО РЕЦЕПТОРА, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2297413C2 |
| АГОНИСТ РЕЦЕПТОРА GPR40, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2013 |
|
RU2650506C2 |
| СОЕДИНЕНИЯ БЕНЗОЛСУЛЬФОНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ СРЕДСТВ | 2017 |
|
RU2769827C2 |
Настоящее изобретение относится к области органической химии, а именно к соединению формулы I
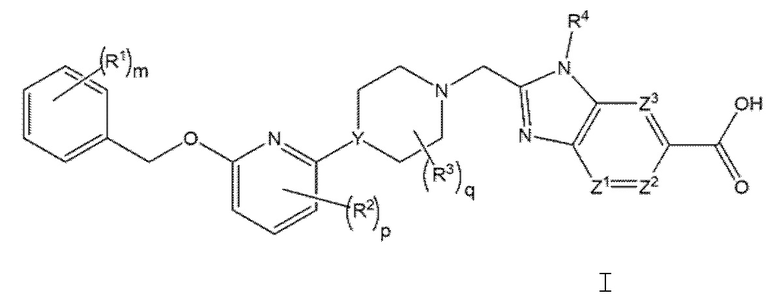 ,
,
или его фармацевтически приемлемой соли. В формуле I каждый R1 независимо представляет собой галоген, -CN, -C1-3алкил или -OC1-3алкил, при этом алкил C1-3алкила и OC1-3алкила замещен 0-3 атомами F; m равно 0, 1, 2 или 3; каждый R2 независимо представляет собой F или Cl; p равно 0 или 1; каждый R3 независимо представляет собой F, -OH, -C1-3алкил или -C3-4циклоалкил или 2 R3 могут циклизоваться вместе с образованием -C3-4спироциклоалкила, где указаный -С1-3алкил и -C3-4циклоалкил могут быть замещены в зависимости от валентности 0-3 атомами F и 0-1 -OH; q равно 0, 1 или 2; Y представляет собой CH или N; R4 представляет собой -C1-3алкил, -C0-3алкилен-C3-6циклоалкил, -C0-3алкилен-R5 или C1-3алкилен R6, где указанный алкил может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из 0-3 атомов F и 0-1 заместителя, выбранного из C0-1алкилен ORO, и где указанный циклоалкил может быть независимо замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из 0-2 атомов F и 0-1 заместителя, выбранного из C0-1алкилен ORO; R5 представляет собой 4-6-членный гетероциклоалкил (где указанный гетероциклоалкил может содержать от 1 до 2 гетероатомов, выбранных из О и/или N), где указанный гетероциклоалкил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из: 0-1 оксо (=O) и 0-2 заместителей, независимо выбранных из -C1-3алкила и -OC1-3алкила, при этом алкил С1-3алкила и -OC1-3алкила может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из: 0-1 -ORO; R6 представляет собой 5-6-членный гетероарил (где указанный гетероарил может содержать от 1 до 3 гетероатомов, выбранных из О и/или N), где указанный гетероарил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из: 0-2 галогенов и 0-2 -C1-3алкилов, где алкил может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из: 0-1 -ORO; каждый RO независимо представляет собой H или -C1-3алкил; Z1 представляет собой СН или N; Z2 и Z3 каждый независимо представляет собой -CRZ или N, при условии, что когда Z1 или Z3 представляет собой N, Z2 представляет собой -CRZ; и каждый RZ независимо представляет собой H, F, Cl или -CH3. Также предложена фармацевтическая композиция. Технический результат: предложены новые органические соединения, которые могут быть использованы в качестве агонистов GLP-1R. 2 н. и 21 з.п. ф-лы, 9 табл., 21 пр.
1. Соединение формулы I
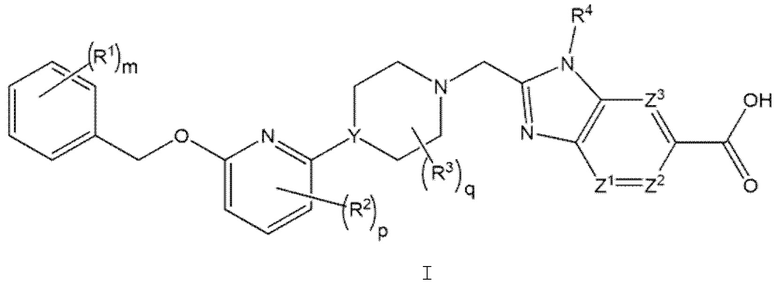
или его фармацевтически приемлемая соль, где
каждый R1 независимо представляет собой галоген, -CN, -C1-3алкил или -OC1-3алкил, при этом алкил C1-3алкила и OC1-3алкила замещен 0-3 атомами F;
m равно 0, 1, 2 или 3;
каждый R2 независимо представляет собой F или Cl;
p равно 0 или 1;
каждый R3 независимо представляет собой F, -OH, -C1-3алкил или -C3-4циклоалкил или 2 R3 могут циклизоваться вместе с образованием -C3-4спироциклоалкила, где указаный -С1-3алкил и -C3-4циклоалкил могут быть замещены в зависимости от валентности 0-3 атомами F и 0-1 -OH;
q равно 0, 1 или 2;
Y представляет собой CH или N;
R4 представляет собой -C1-3алкил, -C0-3алкилен-C3-6циклоалкил, -C0-3алкилен-R5 или C1-3алкилен R6, где указанный алкил может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из 0-3 атомов F и 0-1 заместителя, выбранного из C0-1алкилен ORO, и
где указанный циклоалкил может быть независимо замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из 0-2 атомов F и 0-1 заместителя, выбранного из C0- 1алкилен ORO;
R5 представляет собой 4-6-членный гетероциклоалкил (где указанный гетероциклоалкил может содержать от 1 до 2 гетероатомов, выбранных из О и/или N), где указанный гетероциклоалкил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-1 оксо (=O),
и
0-2 заместителей, независимо выбранных из -C1-3алкила и -OC1-3алкила, при этом алкил С1-3алкила и -OC1-3алкила может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-1 -ORO;
R6 представляет собой 5-6-членный гетероарил (где указанный гетероарил может содержать от 1 до 3 гетероатомов, выбранных из О и/или N), где указанный гетероарил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-2 галогенов,
и
0-2 -C1-3алкилов, где алкил может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-1 -ORO;
каждый RO независимо представляет собой H или -C1-3алкил;
Z1 представляет собой СН или N;
Z2 и Z3 каждый независимо представляет собой -CRZ или N, при условии, что когда Z1 или Z3 представляет собой N, Z2 представляет собой -CRZ; и
каждый RZ независимо представляет собой H, F, Cl или -CH3.
2. Соединение по п.1, где соединение представляет собой соединение формулы II
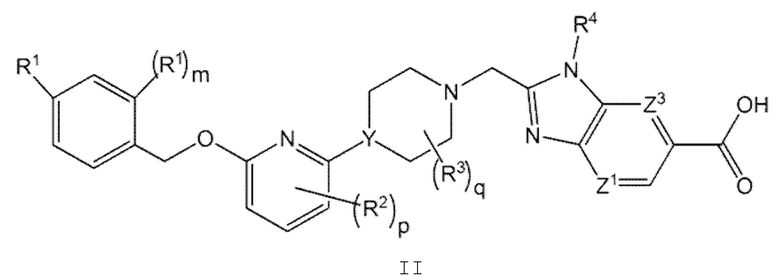
или его фармацевтически приемлемую соль, где
m равно 0 или 1;
R2 представляет собой F;
р равно 0 или 1; и
q равно 0 или 1.
3. Соединение по п.2, где соединение представляет собой соединение формулы III
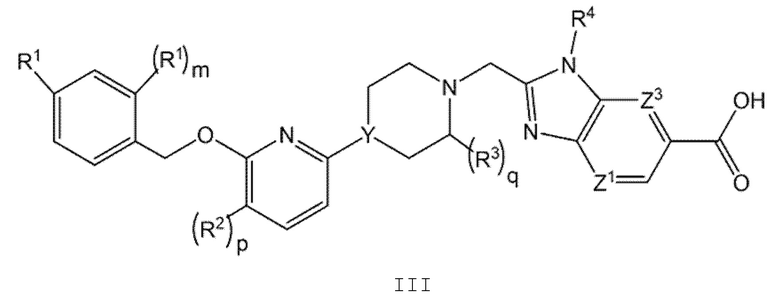
или его фармацевтически приемлемую соль, где
m равно 0 или 1;
R2 представляет собой F;
р равно 0 или 1;
R3 представляет собой -C1-2алкил, где -C1-2алкил может быть замещен в зависимости от валентности 0-3 атомами F; и
q равно 0 или 1.
4. Соединение по п.3, где каждый R1 независимо представляет собой F, Cl, -CN, -CH3 или -CF3, или его фармацевтически приемлемая соль.
5. Соединение по п.4, где гетероциклоалкил R5 представляет собой моновалентный радикал
где гетероциклоалкил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-1 оксо (O=),
0-1 -CN,
0-2 атомов F, и
0-2 заместителей, независимо выбранных из -C1-3алкила и -OC1-3алкила, при этом алкил С1-3алкила и -OC1-3алкила могут быть независимо замещены в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-3 атомов F,
0-1 -CN и
0-1 -ORO,
или его фармацевтически приемлемая соль.
6. Соединение по п.5, где
R4 представляет собой -CH2-R5, где R5 представляет собой 4-5-членный гетероциклоалкил, где указанный гетероциклоалкил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-2 атомов F, и
0-1 заместителя, выбранного из -OCH3 и -CH2OCH3;
или его фармацевтически приемлемая соль.
7. Соединение по п.1, где указанный гетероарил R6 представляет собой моновалентный радикал
и где указанный гетероарил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-3 галогенов,
0-1 заместителя, выбранного из -ORO и -N(RN)2, и
0-2 -C1-3алкилов, где алкил может быть замещен в зависимости от валентности 0-3 заместителями, независимо выбранными из:
0-3 атомов F, и
0-1 -ORO;
или его фармацевтически приемлемая соль.
8. Соединение по п.1, где
R4 представляет собой -CH2-R6, где R6 представляет собой 5-членный гетероарил, при этом указанный гетероарил может быть замещен в зависимости от валентности 0-2 заместителями, независимо выбранными из:
0-2 галогенов, где галоген независимо выбран из F и Cl,
0-1 -OCH3, и
0-1 -CH3, -CH2CH3, -CF3 или -CH2CH2OCH3;
или его фармацевтически приемлемая соль.
9. Соединение по п.1, которое представляет собой
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2R)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-{[4-(6-{[(4-цианo-2-фторфенил)(метил-d2)]окси}пиридин-2-ил)пиперидин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(3R)-тетрагидрофуран-3-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(3R)-тетрагидрофуран-3-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(3S)-тетрагидрофуран-3-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианoбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианoбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианoбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2R)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2R)-тетрагидрофуран-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(2,4-дифторбензил)окси]-5-фторпиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-3-[(2S)-оксетан-2-илметил]-3H-имидазо[4,5-b]пиридин-5-карбоновую кислоту; или
2-{[(2S)-4-{6-[(4-цианoбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту;
или его фармацевтически приемлемая соль.
10. Соединение по п.1, которое представляет собой
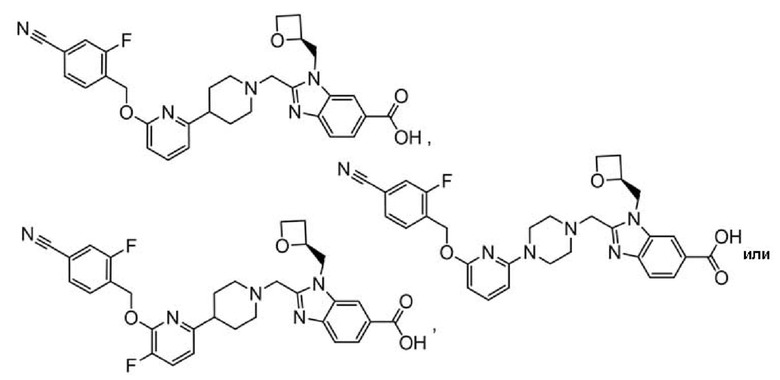
или его фармацевтически приемлемая соль.
11. Соединение по п.1, которое представляет собой 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту, или его фармацевтически приемлемая соль.
12. Соединение по п.11, которое представляет собой свободную кислоту 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты.
13. Соединение по п.11, которое представляет собой трис-соль 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты.
14. Соединение по п.11, которое представляет собой фармацевтически приемлемую соль 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты.
15. Соединение по п.1, которое представляет собой 2-[(4-{6-[(4-цианo-2-фторбензил)окси]-5-фторпиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту, или его фармацевтически приемлемая соль.
16. Соединение по п.1, которое представляет собой 2-{[4-(6-{[(4-цианo-2-фторфенил)(метил-d2)]окси}пиридин-2-ил)пиперидин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновую кислоту, или его фармацевтически приемлемая соль.
17. Соединение по п.1, которое представляет собой
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-метил-1H-1,2,3-триазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-4-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(2,4-дифторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-[(1-этил-1H-1,2,3-триазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,2-оксазол-5-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,2-оксазол-3-илметил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-этил-1H-1,2,3-триазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-метил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-(1,3-оксазол-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоновую кислоту; или
2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-3-(1,3-оксазол-2-илметил)-3H-имидазо[4,5-b]пиридин-5-карбоновую кислоту;
или его фармацевтически приемлемая соль.
18. Соединение по п.1, которое представляет собой
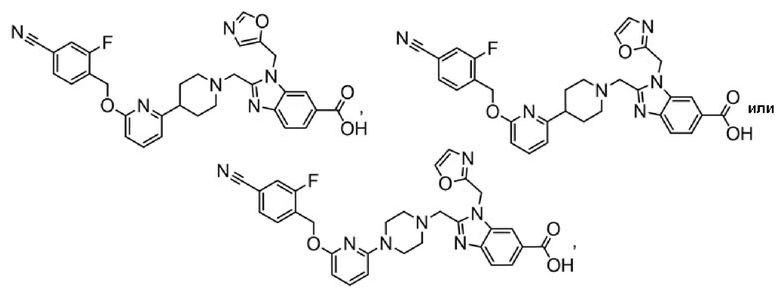
или его фармацевтически приемлемая соль.
19. Соединение по п.1, которое представляет собой 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-5-илметил)-1H-бензимидазол-6-карбоновую кислоту, или его фармацевтически приемлемая соль.
20. Соединение по п.1, которое представляет собой 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту, или его фармацевтически приемлемая соль.
21. Соединение по п.1, которое представляет собой 2-[(4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновую кислоту, или его фармацевтически приемлемая соль.
22. Соединение по п.1, которое представляет собой
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-7-фтор-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-7-фтор-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-цианoбензил)окси]пиридин-2-ил}пиперазин-1-ил)метил]-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-цианoбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-цианo-2-фторбензил)окси]пиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-хлор-2-фторбензил)окси]-5-фторпиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновую кислоту;
2-{[(2S)-4-{6-[(4-цианoбензил)окси]-5-фторпиридин-2-ил}-2-метилпиперазин-1-ил]метил}-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновую кислоту;
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-(2-метоксиэтил)-1H-бензимидазол-6-карбоновую кислоту; или
2-[(4-{6-[(4-хлор-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(1-метоксициклобутил)метил]-1H-бензимидазол-6-карбоновую кислоту;
или его фармацевтически приемлемая соль.
23. Фармацевтическая композиция, обладающая активностью агониста рецептора GLP-1, содержащая терапевтически эффективное количество соединения по любому из пп.1-22 или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
| WO 2011143365 A1, 17.11.2011 | |||
| US 2004127504 A1, 01.07.2004 | |||
| WO 2007029847 A1, 15.03.2005 | |||
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2293730C2 |

