УРОВЕНЬ ТЕХНИКИ
Семейство белков TRAF включает семь членов, т.е. TRAF1, TRAF2, TRAF3, TRAF4, TRAF5, TRAF6 и TRAF7, которые обладают значительной степенью гомологии своих доменных структур (Xie, P., TRAF molecules in cell signaling and in human diseases. J Mol Signal, 2013. 8(1): p. 7 и Zotti, Т., P. Vito, and R. Stilo, The seventh ring: exploring TRAF7 functions. J Cell Physiol, 2012. 227(3): p. 1280-4). Исследования, проведенные в последние годы, выявили большое количество семейств сигнальных рецепторов, которые включают белки TRAF, для распространения сигнала. Эти пути включают Toll-подобные рецепторы (TLR), семейство рецепторов ИЛ-1, рецептор Т-клеток, рецепторы ИЛ-17, рецепторы ФНО и другие. TRAF-зависимые пути передачи сигналов в основном стимулируют активацию (i) фактора транскрипции NF-κВ и (ii) активируемых митогеном протеинкиназ (МАРК), участвуя тем самым в контроле различных клеточных процессов, таких как выживание, пролиферация, дифференцировка, активация, выработка цитокинов и аутофагия (Xie, P., TRAF molecules in cell signaling and in human diseases. J Mol Signal, 2013. 8(1): p. 7). Таким образом, было показано, что различные изменения в членах семейства TRAF вовлечены в патогенез отдельных заболеваний человека, включая аутоиммунность, иммунодефицит и рак (Hildebrand, J.M., Z. Yi, С.М. Buchta, et al., Roles of tumor necrosis factor receptor associated factor 3 (TRAF3) and TRAF5 in immune cell functions. Immunol Rev, 2011. 244(1): p. 55-74; Namjou, В., С.В. Choi, I.T. Harley, et al., Evaluation of TRAF 6 in a large multiancestral lupus cohort. Arthritis Rheum, 2012. 64(6): p. 1960-9 и Netea, M.G., С. Wijmenga, and L.A. O'Neill, Genetic variation in Toll-like receptors and disease susceptibility. Nat Immunol, 2012. 13(6): p. 535-42).
Белок TRAF6 действует как адаптерный белок, а также как убиквитинлигаза (Е3 ubiquitin ligase) для активации фактора транскрипции NF-κВ. В частности, TRAF6 содержит RING-домен и четыре домена цинковых пальцев на своем N-конце, тогда как C-концевой домен TRAF содержит суперспираль и консервативный домен MATH (Yin, Q., S.C Lin, В. Lamothe, et al., E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol, 2009. 16(6): p. 658-66). N-конец (RING и цинковый палец-1) TRAF6 взаимодействует непосредственно с гетеродимерным ферментом Е2, Ubc13/Uev1a, для присоединения K63-соединенных цепей убиквитина к своим белкам-субстратам. Этот процесс имеет решающее значение для распространения передачи сигнала в нескольких клеточных сигнальных процессах, таких как активация NF-κВ, опосредуемая Т-клеточными рецепторами (Oeckinghaus, А., Е. Wegener, V. Welteke, et al., Maltl ubiquitination triggers NF-kappaB signaling upon T-cell activation. EMBO J, 2007. 26(22): p. 4634-45) или врожденный иммунный ответ, включая передачу сигналов с участием ИЛ-1β и Toll-подобного рецептора (Bhoj, V.G. and Z.J. Chen, Ubiquitylation in innate and adaptive immunity. Nature, 2009. 458(7237): p. 430-7). Более того, TRAF6 вовлечен в передачу сигналов с участием рецептора ИЛ-17, а также в ответ на повреждение ДНК (Xie, P., TRAF molecules in cell signaling and in human diseases. J Mol Signal, 2013. 8(1): p. 7 и Walsh, M.C., J. Lee, and Y. Choi, Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev, 2015. 266(1): p. 72-92). Было показано, что гиперэкспрессия TRAF6, а также повышенная активность TRAF6 способствуют хронической иммунной стимуляции и секреции цитокинов, что вызывает широкий спектр нарушений, включая аутоиммунные заболевания, воспаление и рак. Подытоживая, нацеливание на активность TRAF6 за счет нарушения связывания TRAF6-Ubc13 представляет собой очень перспективную новую стратегию противодействия развитию заболеваний.
Предыдущие исследования в основном были сосредоточены на ингибировании взаимодействия белок-белок (PPI) между лигазой Е3 и ее субстратом для того чтобы уменьшить убиквитинирование; при этом наиболее известным примером является р53-Mdm2 (Vassilev, L.T., В.Т. Vu, В. Graves, et al., In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science, 2004. 303(5659): p. 844-8). Международная заявка на патент WO 2011/160016 А2 относится к выделенному фрагменту белка, который содержит связывающий карман или активный сайт на лигазе Е3, который модулирует поверхность контакта Е2-Е3, и к агенту, который взаимодействует с таким связывающим карманом. Однако в WO 2011/160016 А2 упоминаются два агента для этой цели, обозначенные как CRIN-1 и CRLN-2.
Соответственно, существует значительная потребность в новых, наилучших в своем классе, ингибиторных каркасах, которые могут ингибировать TRAF6, в частности, Е3-лигазную активность TRAF6 за счет нарушения его взаимодействия с ферментом Е2, Ubc13.
Авторы настоящего изобретения провели интенсивные исследования и обнаружили неожиданный факт, что соединения в соответствии с Формулой I, Формулой II, Формулой III и Формулой IV, которые более подробно описаны ниже, удовлетворяют эту потребность. Не желая быть связанными соответствием какой-либо теории, авторы настоящего изобретения полагают, что соединения в соответствии с настоящим изобретением нацелены на взаимодействие белок-белок TRAF6 (Е3-лигаза)-Ubc13 (фермент Е2) и тем самым прерывают этот биологический путь (ингибиторы Е2-Е3). В частности, полагают, что соединения противодействуют каталитической активности TRAF6 и тем самым приводят к снижению активации NF-κB.
Соединения в соответствии с настоящим изобретением эффективны при уменьшении активации NF-κB в клеточных линиях (см. Фиг. 4 и Фиг. 6), а также в первичных Т-клетках, полученных от мышей линии BALB/c (см. Фиг. 8). Помимо этого авторы настоящего изобретения обнаружили, что соединения в соответствии с настоящим изобретением улучшают исход при ревматоидном артрите в модели коллаген-индуцированного артрита (CIA) на мышах (см. Фиг. 13), улучшают исход при псориазе в модели IMQ-индуцированного псориаза на мышах (см. Фиг. 12), селективно уничтожают ABC-DLBCL с длительной MYD88-зависимой передачей сигналов (см. Фиг. 14), уменьшают прибавку массы тела в модели T2D на мышах и дополнительно уменьшают экспрессию ИЛ-1β (см. Фиг. 11), уменьшают пролиферацию клеток линии U2OS в степени, сходной с таковой при облучении в дозе 2 Гр, при этом применение соединений вместе с облучением приводит к синергическим эффектам (см. Фиг. 15), также указанные соединения влияют на передачу сигналов с участием иммунных рецепторов в первичных мононуклеарных клетках периферической крови человека (МКПК) (см. Фиг. 7). Безотносительно к какой-либо теории, полагают, что на молекулярном уровне соединения согласно настоящему изобретению нарушают связывание TRAF6-Ubc13 (см. раздел «Анализ» ниже).
КРАТКОЕ ОПИСАНИЕ
Соответственно, согласно первому аспекту настоящее изобретение относится к соединению для применения в лечении рака, иммунного заболевания, болезни Паркинсона, гипертрофии сердца или диабета 2 типа, причем указанное соединение имеет структуру в соответствии с Формулой I
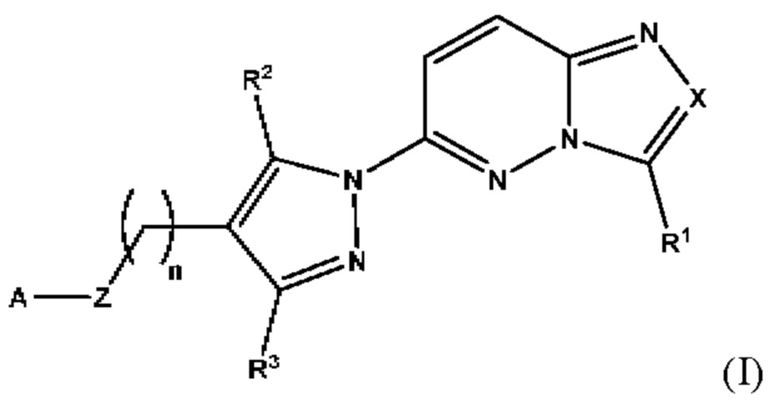
в которой
X выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила;
n представляет собой целое число от 0 до 3;
Z выбран из группы, состоящей из С=O, C=S и СН2;
А выбран из группы, состоящей из -N(R4)(R5) и 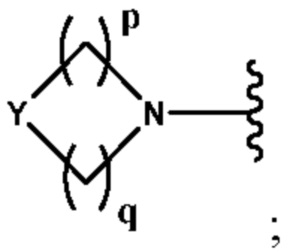
R4 представляет собой водород или (С1-С6)алкил;
R5 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)гетероалкила и (С6-С10)арил(С1-С6)гетероалкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-С4)алкила, (C1-С4)галогеналкила и -OR6;
R6 представляет собой (С1-С4)алкил или (С1-С4)галогеналкил;
Y выбран из группы, состоящей из N-B, СН-В и О;
В выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (C1-С6)алкила и N(R7)(R8), которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR6;
R7 представляет собой водород или (С1-С6)алкил;
R8 выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С1-С6)алкила;
р представляет собой целое число от 1 до 2;
q представляет собой целое число от 1 до 3;
или к его фармацевтически приемлемой соли, сольвату или гидрату.
Согласно второму аспекту настоящее изобретение относится к фармацевтической композиции для применения в лечении рака, иммунного заболевания, болезни Паркинсона, гипертрофии сердца или диабета 2 типа, причем указанная композиция содержит соединение, имеющее структуру в соответствии с Формулой I
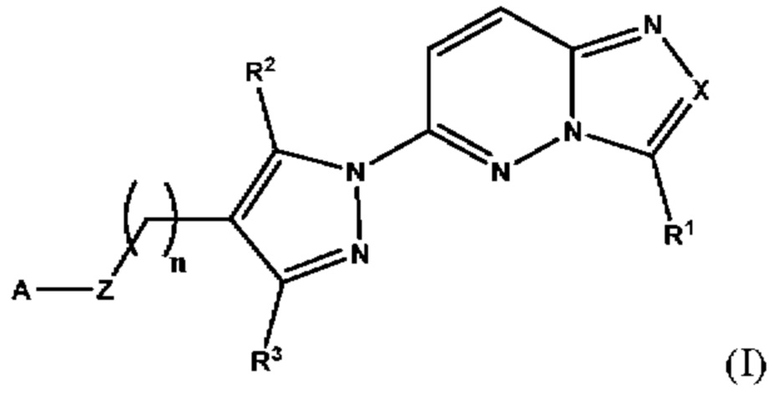
в которой
X выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила;
n представляет собой целое число от 0 до 3;
Z выбран из группы, состоящей из С=O, C=S и CH2;
А выбран из группы, состоящей из N(R4)(R5) и 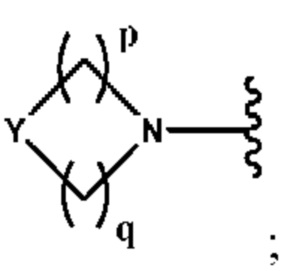
R4 представляет собой водород или (С1-С6)алкил;
R5 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)гетероалкила и (С6-С10)арил(С1-С6)гетероалкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-С4)алкила, (С1-С4)галогеналкила и -OR6;
R6 представляет собой (С1-С4)алкил или (С1-С4)галогеналкил;
Y выбран из группы, состоящей из N-B, СН-В и О;
В выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С1-С6)алкила и N(R7)(R8), которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR6;
R7 представляет собой водород или (С1-С6)алкил;
R8 выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-C10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (C1-С6)алкила;
р представляет собой целое число от 1 до 2;
q представляет собой целое число от 1 до 3;
или его фармацевтически приемлемую соль, сольват или гидрат.
Согласно третьему аспекту настоящее изобретение относится к соединению для применения в медицине, причем указанное соединение имеет структуру в соответствии с Формулой I
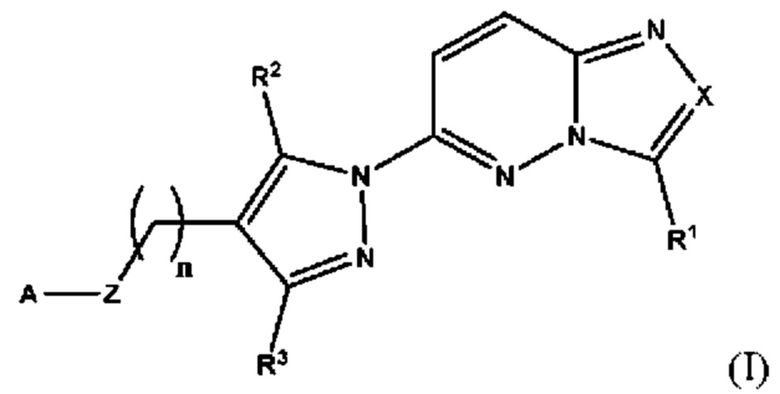
в которой
X выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила;
n представляет собой целое число от 0 до 3;
Z выбран из группы, состоящей из С=O, C=S и СН2;
А выбран из группы, состоящей из -N(R4)(R5) и 
R4 представляет собой водород или (С1-С6)алкил;
R5 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)гетероалкила и (С6-С10)арил(C1-С6)гетероалкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-С4)алкила, (C1-С4)галогеналкила и -OR6;
R6 представляет собой (С1-С4)алкил или (C1-С4)галогеналкил;
Y выбран из группы, состоящей из N-B, СН-В и О;
В выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (C1-С6)алкила и N(R7)(R8), которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR6;
R7 представляет собой водород или (С1-С6)алкил;
R8 выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С1-С6)алкила;
p представляет собой целое число от 1 до 2;
q представляет собой целое число от 1 до 3;
или к его фармацевтически приемлемой соли, сольвату или гидрату.
Согласно некоторым вариантам реализации настоящего изобретения указанное соединение, имеющее структуру в соответствии с Формулой I, представляет собой соединение, имеющее структуру в соответствии с Формулой (II)
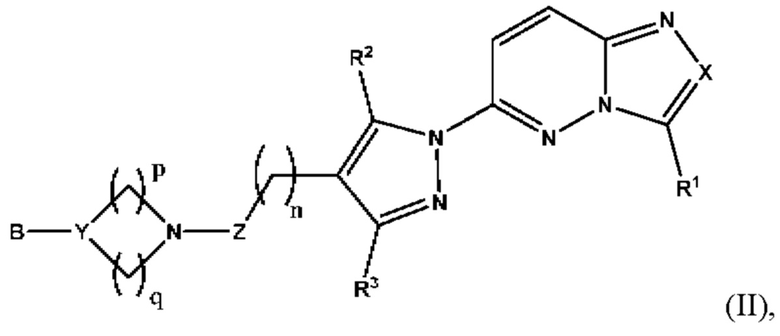
в которой
X выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила, предпочтительно водорода, (С1-С6)алкила, (С3-С8)циклоалкила, (С6-С10)арила и (С6-С10)арил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила, предпочтительно (С1-С6)алкила и (C1-С6)галогеналкила;
n представляет собой целое число от 0 до 3, предпочтительно 1-3, более предпочтительно 1-2, еще более предпочтительно 2;
Z выбран из группы, состоящей из С=O, C=S и СН2, предпочтительно С=O;
Y представляет собой N или СН;
В выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (C1-С6)алкила и N(R7)(R8), которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и OR6, предпочтительно галогена и OR6;
R7 представляет собой водород или (С1-С6)алкил, предпочтительно водород;
R8 выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (C1-С6)алкила, предпочтительно (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, более предпочтительно (С6-С10)арил(С1-С6)алкила;
р представляет собой целое число от 1 до 2;
q представляет собой целое число от 1 до 3;
или его фармацевтически приемлемую соль, сольват или гидрат.
Согласно некоторым вариантам реализации настоящего изобретения указанное соединение, имеющее структуру в соответствии с Формулой (I) или Формулой (II), характеризуется тем, что X представляет собой N.
Согласно некоторым вариантам реализации настоящего изобретения указанное соединение, имеющее структуру в соответствии с Формулой (I) или Формулой (II), характеризуется тем, что Z представляет собой С=O.
Согласно некоторым вариантам реализации настоящего изобретения указанное соединение, имеющее структуру в соответствии с Формулой (I) или Формулой (II), характеризуется тем, что,
R1 выбран из группы, состоящей из водорода, (С1-С4)алкила, (С3-С6)циклоалкила, (С6-С10)арила и (С6-С10)арил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из (С1-С4)алкила и (C1-С4)галогеналкила;
n представляет собой целое число от 1 до 3, предпочтительно 1-2, более предпочтительно 2; и
R5 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(C1-С6)алкила и (С3-С10)гетероарил(C1-С6)гетероалкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и OR6, предпочтительно галогена.
Согласно некоторым вариантам реализации настоящего изобретения указанное соединение, имеющее структуру в соответствии с Формулой (I), представляет собой соединение, имеющее структуру в соответствии с Формулой (III)
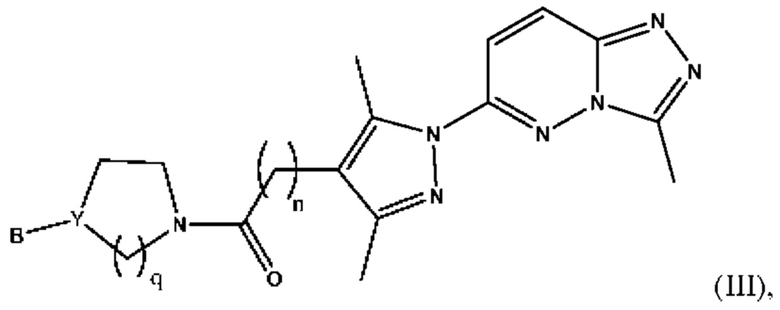
в которой
n представляет собой целое число от 1 до 3, предпочтительно 1-2, более предпочтительно 2;
Y представляет собой N или СН;
В выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С1-С6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и OR6, предпочтительно галогена и -OR6;
q представляет собой целое число от 1 до 3, предпочтительно 1-2, более предпочтительно 2;
или его фармацевтически приемлемую соль, сольват или гидрат.
Согласно некоторым вариантам реализации настоящего изобретения указанное соединение, имеющее структуру в соответствии с Формулой (I), представляет собой соединение, имеющее структуру в соответствии с Формулой (IV)
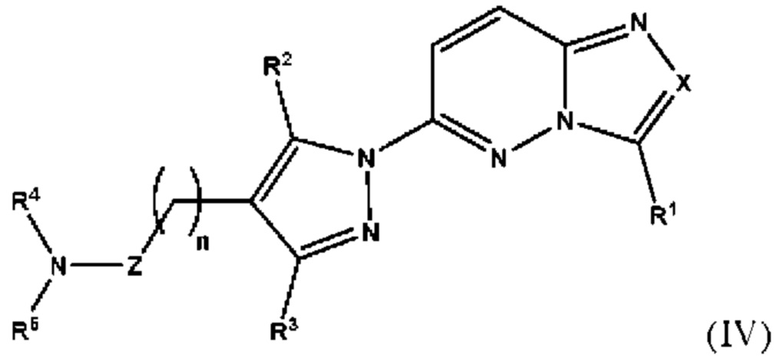
в которой
X выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила, предпочтительно водорода и (С1-С6)алкила, более предпочтительно (С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила, предпочтительно (С1-С6)алкила и (C1-С6)галогеналкила, более предпочтительно (С1-С6)алкила;
n представляет собой целое число от 0 до 3, предпочтительно 1-3, более предпочтительно 1-2, еще более предпочтительно 2;
Z выбран из группы, состоящей из С=O, C=S и CH2, предпочтительно С=O;
R4 представляет собой водород или (С1-С6)алкил, предпочтительно водород;
R5 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)гетероалкила и (С6-С10)арил(С1-С6)гетероалкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-С4)алкила, (С1-С4)галогеналкила и OR6, предпочтительно галогена;
R6 представляет собой (С1-С4)алкил или (С1-С4)галогеналкил;
или его фармацевтически приемлемую соль, сольват или гидрат.
Согласно некоторым вариантам реализации настоящего изобретения указанный рак выбран из группы, состоящей из лимфомы, такой как диффузная крупноклеточная В-клеточная лимфома (DLBCL) и лимфома MALT-типа, множественной миеломы (ММ), рака легких, аденокарциномы легких, рака толстой кишки, рака простаты, рака молочной железы, остеосаркомы, рака поджелудочной железы и плоскоклеточной карциномы пищевода (ESCC).
Согласно некоторым вариантам реализации настоящего изобретения указанное иммунное заболевание представляет собой аутоиммунное заболевание, предпочтительно аутоиммунное заболевание, выбранное из группы, состоящей из псориаза, ревматоидного артрита, целиакии, воспалительного заболевания кишечника, такого как болезнь Крона и язвенный колит, рассеянного склероза и сахарного диабета 1 типа.
Согласно четвертому аспекту настоящее изобретение относится к соединению, имеющему структуру в соответствии с Формулой I,
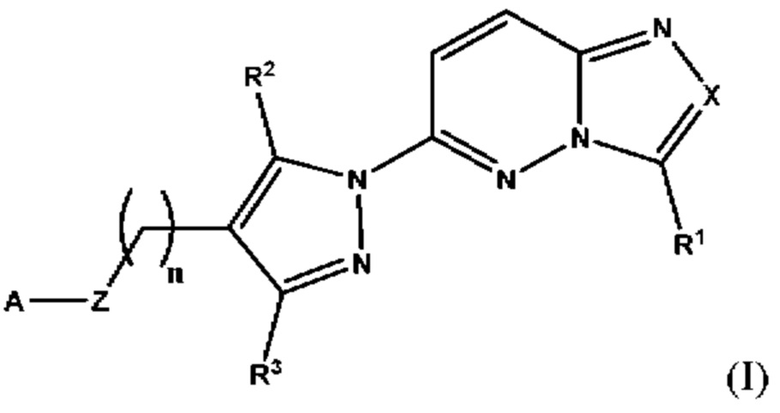
в которой
X выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила;
n представляет собой целое число от 0 до 3;
Z выбран из группы, состоящей из С=O, C=S и СН2;
А выбран из группы, состоящей из 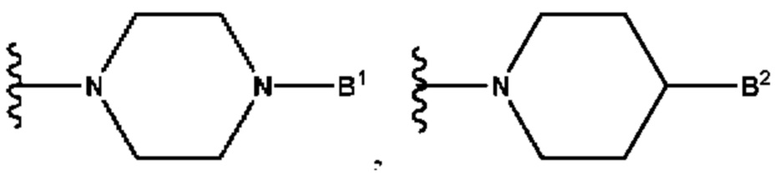 и
и 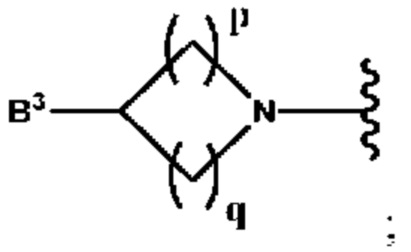
B1 представляет собой (С6-С10)арил(С1-С6)алкил или (С3-С10)гетероарил(С1-С6)алкил, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR6;
B2 представляет собой (С6-С10)арил(С1-С6)алкил, замещенный одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6, или
B2 представляет собой (С3-С10)гетероарил(С1-С6)алкил, который необязательно замещен одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6;
B3 выбран из группы, состоящей из водорода, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С1-С6)алкила и N(R7)(R8), которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6;
R6 представляет собой (С1-С4)алкил или (С1-С4)галогеналкил;
R7 представляет собой водород или (С1-С6)алкил;
R8 выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С1-С6)алкила;
p равно 1 и
q представляет собой целое число от 1 до 2, или
p равно 2 и
q равно 3;
или к его фармацевтически приемлемой соли, сольвату или гидрату.
Согласно некоторым вариантам реализации настоящего изобретения указанное соединение выбрано из группы, включающей:
1-(4-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперазин-1-ил)пропан-1-он,
(R,S)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперазин-1-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
(R)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
(S)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-циклопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-фенил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-(3-бензил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазо[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)этан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-тион,
6-(4-(3-(4-бензилпиперидин-1-ил)пропил)-3,5-диметил-1Н-пиразол-1-ил)-3-метил-[1,2,4]триазол[4,3-b]пиридазин,
1-(азепан-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-морфолинопропан-1-он,
N-(3-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-метилпропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(фуран-2-ил-метил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(2-фенилпропан-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-(3,4-диметоксибензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(нафтален-2-ил-метил)пиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилазетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-(бензиламино)азетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
N-бензил-3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-карбоксамид,
(3-бензилпирролидин-1-ил)(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)метанон,
1-(3-бензилпирролидин-1-ил)-3-(5-изопропил-3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он, и
1-(3-бензилпирролидин-1-ил)-3-(3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-5-(трифторметил)-1Н-пиразол-4-ил)пропан-1-он.
Согласно некоторым вариантам реализации настоящего изобретения указанное соединение выбрано из группы, включающей:
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенилпиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-фенилпиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-((5-метилфуран-2-ил)метил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(2-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(3-фенилпропил)пропанамид,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(2,5-диметилфенил)пиперазин-1-ил)пропан-1-он,
N-(4-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(тиофен-2-ил-метил)пропанамид,
N-(2-(1Н-индол-3-ил)этил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
N-бензил-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1H-(3-фенилпропил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-этилпиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-метилпиперидин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(2-фторбензил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(2-((фуран-2-ил-метил)тио)этил)пропанамид,
1-(4-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперазин-1-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперазин-1-ил)пропан-1-он,
(R)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
(S)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-циклопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-фенил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-(3-бензил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазол[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)этан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-тион,
6-(4-(3-(4-бензилпиперидин-1-ил)пропил)-3,5-диметил-1Н-пиразол-1-ил)-3-метил-[1,2,4]триазол[4,3-b]пиридазин,
1-(азепан-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-морфолинопропан-1-он,
N-(3-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-метилпропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(фуран-2-ил-метил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(2-фенилпропан-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-(3,4-диметоксибензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(нафтален-2-ил-метил)пиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилазетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-(бензиламино)азетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
N-бензил-3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-карбоксамид,
(3-бензилпирролидин-1-ил)(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)метанон,
1-(3-бензилпирролидин-1-ил)-3-(5-изопропил-3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-5-(трифторметил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
1-бензил-4-(3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропаноил)пиперазин-2-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(2,2-диметилпирролидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(3,3-диметилпирролидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(3,3-диметилпирролидин-1-ил)пропан-1-он,
1-((1S)-2-азабицикло[2.2.1]гептан-2-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
(1S,5R)-3-(3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропаноил)-2,3,4,5-тетрагидро-1,5-метанопиридо[1,2-с1][1,4]диазепин-7(1Н)-он,
1-((1S)-2-азабицикло[2.2.1]гептан-2-ил)-3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-(трифторметил)-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(3,3-диметилпирролидин-1-ил)пропан-1-он,
3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(3,3-диметилпирролидин-1-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диэтил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензил-1,4-диазепан-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3-изопропил-5-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-(3-бензил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-циклопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперазин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метилимидазол[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпиперазин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазол[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазол[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенэтилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метилимидазол[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенэтилпиперидин-1-ил)пропан-1-он.
Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, содержащей указанное соединение, имеющее структуру в соответствии с Формулой (I), или указанное соединение, выбранное из группы выше, и по меньшей мере один фармацевтически приемлемый носитель.
Согласно другому аспекту настоящее изобретение относится к набору, содержащему указанное соединение, имеющее структуру в соответствии с Формулой (I), указанное соединение, выбранное из группы выше, или указанную фармацевтическую композицию и по меньшей мере один фармацевтически приемлемый носитель.
Другие признаки и преимущества настоящего изобретения будут очевидны из следующего подробного описания, фигур и формулы изобретения.
ФИГУРЫ
Фигура 1. Идентификация соединения и исследование зависимости активности от структуры (SAR) С25. (А) Способ идентификации ингибиторов TRAF6-Ubc13. 25000 небольших молекул из трех собственных библиотек подвергали скринингу с использованием технологии ALPHAScreen. 520 молекул ингибировали связывание TRAF6-Ubc13 более чем на 25%. После устранения ложноположительных соединений в анализе ALPHA и ложноположительных соединений в анализе His-метки 178 соединений повторно испытывали в количественных исследованиях с 5-точечным титрованием ALPHAScreen. Оставшиеся 27 небольших молекул были перегруппированы и испытаны в количественных исследованиях ALPHAScreen (10-точечное титрование), а также в экспериментах по оценке сдвига электрофоретической подвижности (EMSA). С25 действительно показало лучшие результаты в количественных исследованиях в условиях in vitro и на клеточных системах. (В) С25-140, очевидно, имеет сходную эффективность в AlphaScreen, но (С) более активно в экспериментах на основе клеток ALPHA Surefire. С25 соответствует соединению 1, С25-140 соответствует соединению 3, С25-167 соответствует соединению 61 и С25-189 соответствует соединению 2 описания.
Фигура 2: Биохимическая характеристика и характеристика в клеточных системах С25-140. (А) Представлена химическая структура С25-140. (В) С25-140 дозозависимо снижает взаимодействие эндогенного Ubc13 с эктопически экспрессированным НА-меченым TRAF6 в клетках линии HEK 293Т. (С) С25-140 непосредственно связывается с белком RZ1 TRAF6 в экспериментах с применением ЯМР, как выявлено на основании смещения или исчезновения различных пиков. (D) Ингибитор TRAF6, С25-140, дозозависимо нарушает способность TRAF6 и Ubc13 образовывать свободные полиубиквитиновые цепи в количественном исследовании убиквитинирования в условиях in vitro. (Е) С25-140 не оказывало влияния на взаимодействие Ubc13 с Uevla. С25-140 соответствует соединению 3 описания.
Фигура 3: Влияние С25-140 на жизнеспособность клеток и клеточный цикл. (А) С25-140 не оказывает существенного влияния на метаболическую активность и жизнеспособность клеток до концентрации 50 мкМ через 24 часа инкубации, согласно результатам наблюдения в трех различных клеточных линиях (клетки линий MEF, HepG2 и Jurkat). Токсичность обнаруживается только при самой высокой дозе 100 мкМ. (В-С) Прогрессирование клеточного цикла в Т-клетках линии Jurkat и (D-E) клетках линии U2OS не нарушается после 24 часов и 72 часов обработки С25-140. С25-140 соответствует соединению 3 описания.
Фигура 4: С25-140 противодействует передаче сигналов с участием ИЛ-1β и ФНО-α в клетках линии MEF. (А) Индуцированное ИЛ-1β аутоубиквитинирование TRAF6 уменьшается после обработки С25-140. (В) В количественном исследовании активности IKK обработка С25-140 вызывает снижение фосфорилирования IκВα после стимуляции ИЛ-1β. (С) Экспрессия мРНК ИЛ-1β-индуцированных генов-мишеней (например, ICAM-1 и А20) нарушается в результате обработки С25-140. (D) С25-140 также влияет на ФНО-α-индуцированное фосфорилирование IκВα в количественном исследовании активности IKK и (Е) уровни экспрессии мРНК генов-мишеней ICAM-1 и А20. IKK KA означает киназную активность IKK. С25-140 соответствует соединению 3 описания.
Фигура 5: (А) С помощью количественных исследований убиквитинирования в условиях in vitro можно продемонстрировать, что аутоубиквитинирование cIAP1 (RING Е3-лигаза; образование цепи за счет K63) дозозависимо уменьшается после обработки С25-140. Напротив, С25-140 не оказывало влияния на (В) убиквитинирование р53 под действием MDM2 и убиквитинирование S5A по действием MURF/TRIM63 (обе RING Е3-лигазы; образование цепи за счет K48), (С) аутоубиквитинирование RNF4 (нацеленная на SUMO убиквитинлигаза), а также (D) аутоубиквитинирование ITCH и убиквитинирование S5A под действием Е6АР (обе НЕСТ Е3-лигазы; образование цепи за счет K48). (Е) Были выполнены эксперименты с панелью UbiquitinProfiler™. Различные Е3-лигазы в комбинации с различными Е2-лигазами и субстратами инкубировали с 30 мкМ соединения С25-140. Ни одна из реакций не ингибируется в значительной степени соединением С25-140. С25-140 соответствует соединению 3 описания.
Фигура 6: С25-140 нарушает передачу сигналов с участием Т-клеточных рецепторов в Т-клетках линии Jurkat. (А) Аутоубиквитинирование TRAF6, индуцированное при стимуляции Т-клеточного рецептора (ТКР) с помощью РМА/иономицина (P/I), дозозависимо уменьшается в результате обработки С25-140. (В) Киназная активность IKK уменьшается после обработки С25-140, что, в свою очередь, приводит к (С) меньшему разрушению IκВα и ослабленной активации NF-κВ. (D-E) Уровни мРНК и секреция цитокинов ИЛ-2 и ФНО-α, генов-мишеней ТКР, дозозависимо снижаются. IKK KA означает киназную активность IKK. С25-140 соответствует соединению 3 описания.
Фигура 7: С25-140 влияет на передачу сигналов с участием иммунных рецепторов в первичных МКПК человека. (А) Секреция воспалительных цитокинов (например, ИЛ-6 и ФНО-α) мононуклеарными клетками периферической крови человека (МКПК), которые были стимулированы ИЛ-1β, может быть ослаблена с помощью С25-140. (В) МКПК человека, которые были стимулированы ЛПС, секретировали более низкие уровни цитокинов ИЛ-1β и ФНО-α. (С) МКПК человека, стимулированные CD3/CD28 для индукции передачи сигналов с участием ТКР, имеют сниженные уровни секреции ИЛ-2 и ФНО-α после обработки С25-140. С25-140 соответствует соединению 3 описания.
Фигура 8: С25-140 препятствует передаче сигналов с участием ТКР в первичных мышиных Т-клетках. Выделенные первичные наивные CD4+ Т-клетки проявляют нарушенную экспрессию мРНК ИЛ-2 и секрецию белка ИЛ-2 в присутствии С25-140 при стимуляции с применением CD3/CD28. С25-140 соответствует соединению 3 описания.
Фигура 9: Исследования ADME и фармакокинетики С25-140. (А) Параметры всасывания, распределения, метаболизма и выведения (ADME) С25-140 выявили стабильную и приемлемую небольшую молекулу для дальнейшей разработки лекарственного препарата. (В-С) Фармакокинетические исследования С25-140, введенного внутривенным (в/в), пероральным (п/о) и внутрибрюшинным (в/б) путем, выявили умеренный период полужизни в условиях in vivo, фазу быстрого начального распределения и хорошую биодоступность. С25-140 соответствует соединению 3 описания.
Фигура 10: Оценка безопасности соединения С25-140 в условиях in vivo с помощью SafetyScreen87. 87 количественных исследований были проведены в Eurofins Cerep Panlabs. 87 первичных молекулярных мишеней включали киназы, некиназные ферменты, ядерные рецепторы, GPCR, транспортеры и ионные каналы, которые были испытаны при концентрации соединения 10 мкМ С25-140. С25-140 не оказывало существенного влияния ни на одну из 87 молекулярных мишеней. Представлены данные о количественном исследовании, лиганде/субстрате, видам и тканям. Величины ответа указаны в процентах. С25-140 соответствует соединению 3 описания.
Фигура 11: Обработка соединением С25-140 мышей, страдающих алиментарным ожирением (DIO), влияет на прибавку массы тела и воспаление эпидермальной белой жировой ткани. (А) Мыши, обработанные С25-140, имели меньшую прибавку массы тела по сравнению с контрольными мышами, (В) несмотря на то, что потребление пищи остается неизменным в обеих группах. (С) У мышей, обработанных С25-140, снизились уровни мРНК провоспалительного цитокина ИЛ-1β и гена-мишени VCAM, зависящего от NF-kB. С25-140 соответствует соединению 3 описания. Vhcl означает носитель.
Фигура 12: С25-140 улучшает симптомы псориаза, вызванного IMQ. (А) Дизайн исследования псориаза на мышах. IMQ применяли один раз в сутки, и С25-140 наносили местно два раза в сутки на бритую спину и правое ухо. Параметры оценивали каждый день, и образцы для измерения цитокина ИЛ-17 собирали на 6 день. (В) С25-140 уменьшает совокупную оценку, которая состоит из (С) оценки толщины спины, (D) оценки шелушения и (Е) оценки эритемы спины. (F) Толщина уха значительно уменьшается при обработке С25-140. (G) Содержание цитокина ИЛ-17 снижается в присутствии С25-140. С25-140 соответствует соединению 3 описания.
Фигура 13: С25-140 облегчает симптомы коллаген-индуцированного артрита. (А) Дизайн исследования коллаген-индуцированного артрита на мышах. Ревматоидный артрит (РА) индуцировали путем инъекции коллагена. На 21 день мыши получали стимулирующую инъекцию коллагена, индуцирующую развитие симптомов артрита. С25-140 вводили спустя 7 дней два раза в сутки в течение 14 дней. Мышей ежедневно оценивали для определения артритного индекса (AI) и подвергали эвтаназии на 42 день. Конечности собирали для гистопатологического исследования. (В) В ходе данного исследования регулярно измеряли массу тела, при этом не было обнаружено каких-либо признаков потери массы тела. (С) AI мышей, обработанных С25-140, дозозависимо уменьшается по сравнению с контрольными мышами. (D-E) Общая гистологическая оценка конечностей, включая воспаление, образование паннуса, повреждение хряща и резорбцию кости, может быть уменьшена с помощью обработки С25-140. (F) Гистопатологические срезы с лодыжки и задней лапы каждой отдельной группы подтверждают результаты для оцениваемых параметров. С25-140 соответствует соединению 3 описания.
Фигура 14: С25-140 снижает выживаемость MYD88-зависимых клеток ABC-DLBCL. Жизнеспособность MYD88-зависимых клеток ABC-DLBCL (HBL1) уменьшается в результате обработки С25-140, при концентрации С25-140 30 мкМ, тогда как обработка С25-140 не оказывает влияния на ABC-DLBCL с MYD88-зависимостью дикого типа (U2932) и GCB (В-клетки зародышевого центра)-DLBCL (BJAB). Соединение добавляли на 0, 4 и 8 день. С25-140 соответствует соединению 3 описания.
Фигура 15: Комбинированная терапия, включающая облучение и обработку С25-140, оказывает синергическое действие на размножение клеток остеосаркомы. Размножение клеток остеосаркомы уменьшается в результате как обработки 30 мкМ С25-140, так и при облучении в дозе 2 Гр. Эта комбинация (30 мкМ С25-140+2 Гр) уменьшает размножение клеток в той же степени, как и более высокие дозы облучения (4 Гр). С25-140 соответствует соединению 3 описания.
Фигура 16: Исследование влияния С25-140 на панель онкологических клеточных линий. Исследовали потерю жизнеспособности 78 онкологических клеточных линий различного происхождения после обработки С25-140. Использовали две контрольные клеточные линии (HS-5 и MCF 10А). Различные раковые клеточные линии погибали после обработки С25-140, но не контрольные клеточные линии. Основные эффекты были видны в раковых клетках гемопоэтического происхождения, хотя наблюдали влияние и на другие раковые клеточные линии из других источников. С25-140 соответствует соединению 3 описания.
Фигура 17: Исследование 9 структурных аналогов С25-140 в количественных исследованиях ALPHAScreen. 9 структурных аналогов С25-140, недоступных коммерчески, испытывали в экспериментах с последовательными разведениями с использованием количественного исследования ALPHAScreen. Все исследованные соединения проявляют ингибиторную активность, причем HZM06-04 проявляет наиболее выраженный эффект. Важно отметить, что ни одна из испытанных небольших молекул не вызывала неспецифических помех в технологии ALPHAScreen в экспериментах TruHits. С25 соответствует соединению 1, С25-140 соответствует соединению 3, HZM06-1 соответствует соединению 28, HZM06-2 соответствует соединению 29, HZM06-3 соответствует соединению 30, HZM06-4 соответствует соединению 31, HZM06-6 соответствует соединению 32, HZM06-7 соответствует соединению 33, HZM06-8 соответствует соединению 34, HZM06-9 соответствует соединению 35 и HZM06-10 соответствует соединению 36 описания. Коэффициент Z равен 0,77, и окно сигнала равно 10,29.
Фигура 18: Измерение цитотоксичности 9 структурных аналогов С25-140 в различных клеточных линиях. Исследовали жизнеспособность клеток линий Jurkat, HepG2 и MEF после обработки 9 аналогами в течение 6 или 24 часов. Все небольшие молекулы проявляют меньшую цитотоксичность по сравнению с С25-140. С25 соответствует соединению 1, С25-140 соответствует соединению 3, HZM06-1 соответствует соединению 28, HZM06-2 соответствует соединению 29, HZM06-3 соответствует соединению 30, HZM06-4 соответствует соединению 31, HZM06-6 соответствует соединению 32, HZM06-7 соответствует соединению 33, HZM06-8 соответствует соединению 34, HZM06-9 соответствует соединению 35 и HZM06-10 соответствует соединению 36 описания.
Фигура 19: Исследования коиммунопреципитации 9 структурных аналогов С25-140 выявили ингибиторный потенциал отдельных аналогов в клетках. В исследованиях коиммунопреципитации отдельные соединения, включая С25-140, HZM06-04, HZM06-07, HZM06-08 и HZM06-09, ингибировали связывание TRAF6-Ubcl3 в клетках линии НЕК293Т. С25 соответствует соединению 1, С25-140 соответствует соединению 3, HZM06-1 соответствует соединению 28, HZM06-2 соответствует соединению 29, HZM06-3 соответствует соединению 30, HZM06-4 соответствует соединению 31, HZM06-6 соответствует соединению 32, HZM06-7 соответствует соединению 33, HZM06-8 соответствует соединению 34, HZM06-9 соответствует соединению 35 и HZM06-10 соответствует соединению 36 описания.
Фигура 20: Аналогичные соединения. Все соединения испытывали в количественном исследовании ALPHAScreen. Уровень ингибирования указан.
ПОДРОБНОЕ ОПИСАНИЕ
Несмотря на то, что настоящее изобретение подробно описано ниже, следует понимать, что настоящее изобретение не ограничено конкретными методологиями, протоколами и реагентами, описанными в настоящем документе, поскольку они могут изменяться. Также следует понимать, что используемая в настоящем документе терминология предназначена только для описания конкретных вариантов реализации и не предназначена для ограничения объема настоящего изобретения, который будет ограничен только прилагаемой формулой изобретения. Если не указано иное, все технические и научные термины используются в настоящем документе в значении, соответствующем обычному пониманию специалиста в соответствующей области техники.
Далее будут описаны элементы настоящего изобретения. Эти элементы перечислены с конкретными вариантами реализации, однако следует понимать, что они могут быть комбинированы любым способом и в любом количестве для создания дополнительных вариантов реализации. Описанные различным образом примеры и предпочтительные варианты реализации, изложенные по всему описанию, не должны рассматриваться как ограничивающие настоящее изобретение только явно описанными вариантами реализации. Следует понимать, что это описание поддерживает и включает варианты реализации, которые объединяют явно описанные варианты реализации с любым количеством раскрытых и/или предпочтительных элементов. Кроме того, любые перестановки и комбинации всех элементов, описанных в настоящем документе, следует рассматривать как раскрытые с помощью описания настоящей заявки, если из контекста не следует иное. Например, если в одном варианте реализации R1 соединений согласно настоящему изобретению представляет собой гетероарил (такой как пиразолил), и в другом варианте реализации соединений согласно настоящему изобретению В представляет собой замещенное или незамещенное фенильное кольцо, тогда в предпочтительном варианте реализации R1 соединений согласно настоящему изобретению представляет собой гетероарил (такой как пиразолил) и В представляет собой замещенное или незамещенное фенильное кольцо.
Следует понимать, что по всему тексту настоящего описания и в формуле изобретения после него, если контекст не требует иного, слово «содержат» и такие варианты как «содержит» и «содержащий» подразумевают включение указанного члена, целого числа или этапа или группы членов, целых чисел или этапов, но не исключение какого-либо другого члена, целого числа или этапа или группы членов, целых чисел или этапов, несмотря на то, что в некоторых вариантах реализации такой другой член, целое число или этап или группа членов, целых чисел или этапов могут быть исключены, т.е. рассматриваемый объект заключается во включении указанного члена, целого числа или этапа или группы членов, целых чисел или этапов. Термины, обозначающие элемент в единственном числе (соотв. «а» и «an» или «the» в исходном тексте на английском языке) и аналогичная ссылка, используемая в контексте описания настоящего изобретения (особенно применительно к формуле изобретения), должны рассматриваться как включающие как единственное, так и множественное число, если иное не указано в настоящем документе или явно не противоречит контексту. Перечисление диапазонов значений в настоящем документе предназначено исключительно для того чтобы служить в качестве краткого способа индивидуальной ссылки на каждое отдельное значение, которое находится в пределах диапазона. Если в настоящем документе не указано иное, каждое отдельное значение включено в описание, как если бы оно было отдельно перечислено в настоящем документе.
Все способы, описанные в настоящем документе, могут быть выполнены в любом подходящем порядке, если иное не указано в настоящем документе или иное явно не противоречит контексту. Применение любого и всех примеров, или иллюстративных формулировок (например, «такой как»), приведенных в настоящем документе, предназначено только для лучшей иллюстрации настоящего изобретения и не ограничивает объем настоящего изобретения, заявленный иным образом. Ни одна из формулировок в описании не должна быть истолкована как указывающая на любой не заявленный элемент, существенный для практического применения настоящего изобретения.
Если не указано иное, термин «по меньшей мере», предшествующий ряду элементов, следует понимать как относящийся к каждому элементу в ряду. Специалисты в данной области техники распознают, или смогут установить с использованием только обычных экспериментов, множество эквивалентов конкретных вариантов реализации настоящего изобретения, описанных в настоящем документе. Подразумевается, что такие эквиваленты включены в объем настоящего изобретения.
В настоящем документе термин «состоящий из» исключает любой элемент, этап или ингредиент, не указанный в элементе формулы изобретения. В настоящем документе термин «состоящий по существу из» не исключает материалы или этапы, которые не оказывают существенного влияния на основные и новые характеристики формулы изобретения. В каждом конкретном случае в настоящем документе любой из терминов «содержащий», «состоящий по существу из» и «состоящий из» может быть заменен любым из двух других терминов.
Несколько документов процитированы по всему тексту настоящего описания. Каждый из документов, процитированных в настоящем документе (включая все патенты, заявки на патенты, научные публикации, спецификации производителя, инструкции и т.д.), приведенный выше или ниже, настоящим полностью включен посредством ссылки. Ничто в настоящем документе не должно быть истолковано как допущение того, что изобретение не имеет права предшествовать такому раскрытию на основании предшествующего изобретения.
Соединения
Для лучшего понимания настоящего изобретения сначала определены некоторые термины. Дополнительные определения приведены по всему описанию.
В настоящем документе и по всему описанию термин «алкил» относится к монорадикалу насыщенного линейного или разветвленного углеводорода. Предпочтительно алкильная группа содержит от 1 до 6 атомов углерода, т.е. 1, 2, 3, 4, 5 или 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода. Согласно другому варианту реализации настоящего изобретения применяемая алкильная группа содержит 1-5 атомов углерода (С1-5 алкил). Согласно другому варианту реализации настоящего изобретения применяемая алкильная группа содержит 1-4 атома углерода (С1-4 алкил). Согласно другому варианту реализации настоящего изобретения применяемая алкильная группа содержит 1-3 атома углерода (C1-3 алкил). Согласно другому варианту реализации настоящего изобретения применяемая алкильная группа содержит 1-2 атома углерода (С1-2 алкил). Согласно другому варианту реализации настоящего изобретения применяемая алкильная группа представляет собой метил. Примеры алкильных радикалов включают, но не ограничиваются ими, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, н-пентил, изопентил, втор-пентил, неопентил, 1,2-диметилпропил, изоамил, н-гексил, изогексил, втор-гексил и тому подобное, которые могут содержать один или более заместителей. Заместители алкильной группы включают, но не ограничиваются ими, любой из заместителей, описанных в настоящем документе, которые приводят к образованию стабильного фрагмента. Замещенная алкильная группа может быть замещена в любых положениях, при условии, что полученное соединение является достаточно стабильным и подходит в качестве фармацевтически активного соединения. Предварительное условие о том, чтобы конкретная группа и соединение согласно любой из Формул I, II, III и IV были достаточно стабильными и подходящими в качестве фармацевтически активного соединения, применимо в общем по отношению ко всем группам в соединениях согласно любой из Формул I, II, III и IV. Если алкильная группа может быть монозамещена или полизамещена фтором, она может быть незамещенной, т.е. не содержит атомы фтора, или замещенной, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11 атомами фтора, предпочтительно 1, 2, 3, 4 или 5 атомами фтора, которые могут присутствовать в любых положениях. Например, одна или более метальных групп во фторзамещенной алкильной группе могут нести по три атома фтора каждая и присутствовать в виде трифторметильных групп, и/или одна или более метиленовых групп (СН2) могут нести по два атома фтора каждая и присутствовать в виде дифторметиле новых групп. Разъяснения в отношении замены группы фтором также применимы, если группа дополнительно содержит другие заместители и/или является частью другой группы, например, алкил-О-группы. Примеры фторзамещенных алкильных групп включают трифторметил, 2-фторэтил, 1,1-дифторэтил, 2,2,2-трифторэтил, пентафторэтил, 3,3,3-трифторпропил, 2,2,3,3,3-пентафторпропил, 4,4,4-трифторбутил и гептафторизопропил. Примеры фторзамещенных алкил-О-групп включают трифторметокси, 2,2,2-трифторэтокси, пентафторэтокси и 3,3,3-трифторпропокси. Примеры фторзамещенных алкил-S(O)m- групп включают трифторметансульфанил- (CF3-S-, трифтор метил сульфанил-), трифторметансульфинил- (CF3-S(O)-) и трифторметансуль фонил- (CF3-S(O)2-). Согласно некоторым вариантам реализации настоящего изобретения алкильная группа может быть замещена одним или более идентичными или различными заместителями, выбранными из галогена, гидроксила, циано, (С1-С6)алкил-O- и (С1-С6)алкил-S(O)m-. Примеры (С1-С6)алкил-O- включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси и н-пентокси. Примеры алкил-S(O)m- включают метансульфанил- (СН3-S-, метилсульфанил), метансульфинил- (СН3-S(O)-), метанеульфонил- (СН3-S(O)2-), этансульфанил- (СН3-СН2-S-, этилсульфанил-), этансульфинил- (СН3-СН2-S(O)-), этансульфонил- (СН3-СН2-S(O)2-), 1-метилэтансульфанил- ((СН3)2СН-S-, 1-метилэтилсульфанил-), 1-метилэтансульфинил-((СН3)2CH-S(O)-) и 1-метилэтансульфонил- ((СН3)2СН-S(O)2-). Согласно одному варианту реализации настоящего изобретения число m выбрано из 0 и 2, причем все числа m не зависят друг от друга и могут быть идентичными или различными. Например, если алкильная группа замещена группой CF3-S-, группа CF3-S- связана с этой алкильной группой за счет серы, как это обозначено концевой линией (дефисом) рядом с атомом серы, представляющей свободную связь. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является линейной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является разветвленной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является замещенной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является незамещенной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является линейной и замещенной или незамещенной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является разветвленной и замещенной или незамещенной.
В настоящем документе и по всему описанию термин «алкилен» относится к дирадикалу насыщенного линейного или разветвленного углеводорода. Предпочтительно алкилен содержит от 1 до 6 атомов углерода, т.е. 1, 2, 3, 4, 5 или 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода. Примерные алкиленовые группы включают метилен, этилен (т.е. 1,1-этилен, 1,2-этилен), пропилен (т.е. 1,1-пропилен, 1,2-пропилен (-СН(СН3)СН2-), 2,2-пропилен (-С(СН3)2-) и 1,3-пропилен), изомеры бутилена (например, 1,1-бутилен, 1,2-бутилен, 2,2-бутилен, 1,3-бутилен, 2,3-бутилен (цис или транс или их смесь), 1,4-бутилен, 1,1-изобутилен, 1,2-изобутилен и 1,3-изобутилен), изомеры пентилена (например, 1,1-пентилен, 1,2-пентилен, 1,3-пентилен, 1,4-пентилен, 1,5-пентилен, 1,1-изопентилен, 1,1-втор-пентил, 1,1-неопентил), изомеры гексилена (например, 1,1-гексилен, 1,2-гексилен, 1,3-гексилен, 1,4-гексилен, 1,5-гексилен, 1,6-гексилен и 1,1-изогексилен) и т.п. Алкиленовые группы могут быть циклическими или ациклическими, разветвленными или неразветвленными, замещенными или незамещенными. Заместители алкиленовой группы включают, но не ограничиваются ими, любой из заместителей, описанных в настоящем документе, которые приводят к образованию стабильного фрагмента.
В настоящем документе и по всему описанию термин «галогеналкил» относится к алкильной группе, замещенной одним галогеновым заместителем до пер-галоген-замещения. Галогеновый заместитель предпочтительно представляет собой фтор. Галогеналкил предпочтительно представляет собой перфторалкил. Согласно некоторым вариантам реализации настоящего изобретения галогеналкильная группа, применяемая в настоящем изобретении, содержит 1-6 атомов углерода (C1-6 галогеналкил). Согласно другому варианту реализации настоящего изобретения применяемая галогеналкильная группа содержит 1-5 атомов углерода (С1-5 галогеналкил). Согласно другому варианту реализации настоящего изобретения применяемая галогеналкильная группа содержит 1-4 атома углерода (С1-4 галогеналкил). Согласно другому варианту реализации настоящего изобретения применяемая галогеналкильная группа содержит 1-3 атома углерода (С1-3 галогеналкил). Согласно другому варианту реализации настоящего изобретения применяемая галогеналкильная группа содержит 1-2 атома углерода (С1-2 галогеналкил). Согласно другому варианту реализации настоящего изобретения применяемая галогеналкильная группа содержит 1 атом углерода (C1 галогеналкил). Согласно другому варианту реализации настоящего изобретения применяемая галогеналкильная группа представляет собой трифторметил. Примерный фторзамещенный С1-С2 алкил включает -CFH2, -CF2H, -CF3, CH2CH2F, -CH2CHF2, -CHFCH3, -CHFCH3, -CF2CHF2. Перфторзамещенный С1-С2 галогеналкил, например, включает -CF3 и CF2CF3.
В настоящем документе и по всему описанию термин «гетероалкил», применяемый в настоящем документе, относится к алкильному фрагменту, определенному в настоящем документе, который содержит один или более гетероатомов (например, атомов кислорода, серы, азота, фосфора или кремния) между атомами углерода. Гетероалкил может быть замещенным или незамещенным. Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная группа содержит 1-6 атомов углерода и 1-3 гетероатома (C1-6 гетероалкил). Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная группа содержит 1-5 атомов углерода и 1-3 гетероатома (С1-5 гетероалкил). Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная группа содержит 1-4 атома углерода и 1-2 гетероатома (С1-4 гетероалкил). Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная группа содержит 1-3 атома углерода и 1 гетероатом (C1-3 гетероалкил). Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная группа содержит 1-2 атома углерода и 1 гетероатом (С1-2 гетероалкил). В настоящем документе термин «гетероалкилен» относится к бирадикалу, полученному из гетероалкильной группы, определенной в настоящем документе, путем удаления двух атомов водорода. Гетероалкиленовые группы могут быть циклическими или ациклическими, разветвленными или неразветвленными, замещенными или незамещенными. Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная группа представляет собой замещенную гетероалкильную группу, содержащую 1-6 атомов углерода и 1-3 гетероатома (С1-6 гетероалкил). Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная группа представляет собой незамещенную гетероалкильную группу, содержащую 1-6 атомов углерода и 1-3 гетероатома (C1-6 гетероалкил). Согласно некоторым вариантам реализации настоящего изобретения гетероалкил представляет собой алкильный фрагмент, в котором на метиленовой группе присутствует заместитель S. Согласно некоторым вариантам реализации настоящего изобретения гетероалкил представляет собой алкильный фрагмент, в котором на метиленовой группе присутствует заместитель О. Согласно некоторым вариантам реализации настоящего изобретения гетероалкил представляет собой алкильный фрагмент, в котором на метиленовой группе присутствует заместитель NRA, причем RA выбран из группы, состоящей из водорода, замещенного или незамещенного (C1-6)алкила, замещенного или незамещенного (С3-С8)циклоалкила, замещенного или незамещенного (С6-С14)арила и замещенного или незамещенного (С3-С14)гетероарила. Согласно некоторым вариантам реализации настоящего изобретения гетероалкил представляет собой -CH2SCH3. Согласно некоторым вариантам реализации настоящего изобретения гетероалкил представляет собой -СН2ОСН3.
В настоящем документе и по всему описанию термин «алкенил» относится к монорадикалу ненасыщенного линейного или разветвленного углеводорода, имеющего по меньшей мере одну двойную связь углерод-углерод. Обычно максимальное количество двойных связей углерод-углерод в алкенильной группе может быть равно целому числу, которое вычисляется путем деления количества атомов углерода в алкенильной группе на 2 и, если количество атомов углерода в алкенильной группе нечетное, округляя результат деления до следующего целого числа. Например, для алкенильной группы, имеющей 9 атомов углерода, максимальное количество двойных связей углерод-углерод составляет 4. Предпочтительно алкенильная группа имеет от 1 до 4, т.е. 1, 2, 3 или 4 двойные связи углерод-углерод. Предпочтительно алкенильная группа содержит от 2 до 10 атомов углерода, т.е. 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода, более предпочтительно от 2 до 8 атомов углерода, например, от 2 до 6 атомов углерода или от 2 до 4 атомов углерода. Соответственно, согласно предпочтительному варианту реализации алкенильная группа содержит от 2 до 10 атомов углерода и 1, 2, 3, 4 или 5 двойных связей углерод-углерод, более предпочтительно она содержит от 2 до 8 атомов углерода и 1, 2, 3 или 4 двойные связи углерод-углерод, например, 2-6 атомов углерода и 1, 2 или 3 двойные связи углерод-углерод или 2-4 атома углерода и 1 или 2 двойные связи углерод-углерод. Согласно определенным вариантам реализации настоящего изобретения алкенильная группа, применяемая в настоящем изобретении, содержит 2-20 атомов углерода (С2-20 алкенил). Согласно некоторым вариантам реализации настоящего изобретения алкенильная группа, применяемая в настоящем изобретении, содержит 2-15 атомов углерода (С2-15 алкенил). Согласно другому варианту реализации настоящего изобретения алкенильная группа содержит 2-10 атомов углерода (С2-10 алкенил). Согласно другим вариантам реализации настоящего изобретения алкенильная группа содержит 2-8 атомов углерода (С2-8 алкенил). Согласно дополнительным вариантам реализации настоящего изобретения алкенильная группа содержит 2-6 атомов углерода (С2-6 алкенил). Согласно дополнительным вариантам реализации настоящего изобретения алкенильная группа содержит 2-5 атомов углерода (С2-5 алкенил). Согласно дополнительным вариантам реализации настоящего изобретения алкенильная группа содержит 2-4 атома углерода (С2-4 алкенил). Согласно дополнительным вариантам реализации настоящего изобретения алкенильная группа содержит 2-3 атома углерода (С2-3 алкенил). Согласно дополнительным вариантам реализации настоящего изобретения алкенильная группа содержит 2 атома углерода (С2 алкенил). Двойная(ые) связь (и) углерод-углерод может (могут) иметь цис (Z) или транс (Е) конфигурацию. Примерные алкенильные группы включают винил, 1-пропенил, 2-пропенил (т.е. аллил), 1-бутенил, 2-бутенил, 3-бутенил, 1-пентенил, 2-пентенил, 3-пентенил, 4-пентенил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил, 5-гексенил, 1-гептенил, 2-гептенил, 3-гептенил, 4-гептенил, 5-гептенил, 6-гептенил, 1-октенил, 2-октенил, 3-октенил, 4-октенил, 5-октенил, 6-октенил, 7-октенил, 1-ноненил, 2-ноненил, 3-ноненил, 4-ноненил, 5-ноненил, 6-ноненил, 7-ноненил, 8-ноненил 1-деценил, 2-деценил, 3-деценил, 4-деценил, 5-деценил, 6-деценил, 7-деценил, 8-деценил, 9-деценил и т.п. Если алкенильная группа присоединена к атому азота, двойная связь не может быть в альфа-положении относительно атома азота. Заместители алкенильной группы включают, но не ограничиваются ими, любой из заместителей, описанных в настоящем документе, которые приводят к образованию стабильного фрагмента. Замещенная алкенильная группа может быть замещена в любых положениях, при условии, что полученное соединение является достаточно стабильным и подходит в качестве фармацевтически активного соединения. Предварительное условие о том, что конкретная группа и соединение согласно любой из Формул I, II, III и IV является достаточно стабильным и пригодным в качестве фармацевтически активного соединения, применимо в общем в отношении всех групп в соединениях согласно любой из Формул I, II, III и IV. Согласно некоторым вариантам реализации настоящего изобретения алкенильная группа может быть замещена одним или более идентичными или различными заместителями, выбранными из галогена, гидроксила, циано, (С1-С6)алкил-O- и (С1-С6)алкил-S(O)m-. Согласно одному варианту реализации настоящего изобретения число m выбрано из 0 и 2, причем все числа m не зависят друг от друга и могут быть идентичными или различными. Согласно некоторым вариантам реализации настоящего изобретения алкенильная цепь является линейной. Согласно некоторым вариантам реализации настоящего изобретения алкенильная цепь является разветвленной. Согласно некоторым вариантам реализации настоящего изобретения алкенильная цепь является замещенной. Согласно некоторым вариантам реализации настоящего изобретения алкенильная цепь является незамещенной. Согласно некоторым вариантам реализации настоящего изобретения алкенильная цепь является линейной и замещенной или незамещенной. Согласно некоторым вариантам реализации настоящего изобретения алкенильная цепь является разветвленной и замещенной или незамещенной. Заместители алкенильной группы включают, но не ограничиваются ими, любой из заместителей, описанных в настоящем документе, которые приводят к образованию стабильного фрагмента.
В настоящем документе и по всему описанию термин «циклоалкил» или «циклоалифатический», или «карбоциклический», или «карбоцикл» представляет циклические неароматические варианты «алкила» и «алкенила», предпочтительно с 3-8 атомами углерода, т.е. 3, 4, 5, 6, 7 или 8 атомами углерода, более предпочтительно с 3-7 атомами углерода. Согласно другому варианту реализации настоящего изобретения циклоалкильная группа, применяемая в настоящем изобретении, содержит 3-7 атомов углерода (С3-7 циклоалкил). Согласно другому варианту реализации настоящего изобретения циклоалкильная группа, применяемая в настоящем изобретении, содержит 3-6 атомов углерода (С3-6 циклоалкил). Согласно другому варианту реализации настоящего изобретения циклоалкильная группа, применяемая в настоящем изобретении, содержит 3-5 атомов углерода (С3-5 циклоалкил). Согласно другому варианту реализации настоящего изобретения циклоалкильная группа, применяемая в настоящем изобретении, содержит 3-4 атома углерода (С3-4 циклоалкил). Согласно другому варианту реализации настоящего изобретения циклоалкильная группа, применяемая в настоящем изобретении, содержит 3 атома углерода (С3 циклоалкил). Примерные циклоалкильные группы включают циклопропил, циклопропенил, циклобутил, циклобутенил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогептил, циклогептенил, циклооктил и циклооктенил. Двойная связь в циклоалкенильной группе может присутствовать в любом положении по отношению к атому углерода в положении 1, за счет которого группа связана с остальной частью молекулы, т.е., например, с атомом азота в соединениях, имеющих структуру согласно Формуле I, и циклоалкенил может, соответственно, представлять собой, например, циклопент-1-енил, циклопент-2-енил, циклопент-3-енил, циклогекс-1-енил, циклогекс-2-енил, циклогекс-3-енил, циклогепт-1-енил, циклогепт-2-енил, циклогепт-3-енил, циклогепт-4-енил. Согласно предпочтительным вариантам реализации настоящего изобретения циклоалкильная группа, такая как (С3-С7)-циклоалкил, в определении любой группы, выбрана из подгруппы любых двух или более из указанных конкретных циклоалкильных групп, например, из циклопропила и циклобутила, или из циклопропила, циклобутила и циклопентила, или из циклопропила, циклопентила и циклогексила, или из циклопентила и циклогексила, или из циклопентила, циклогексила и циклогептила. Аналогичным образом, согласно предпочтительным вариантам реализации циклоалкенильная группа выбрана из подгруппы любых двух или более из указанных конкретных циклоалкенильных групп, например, из циклопентенила и циклогексенила, или из циклогексенила и циклогептенила, или из циклопент-1-енила, циклопент-2-енила, циклогекс-1-енила, циклогекс-2-енила, циклогепт-1-енила и циклогепт-2-енила, или из циклопент-2-енила, циклопент-3-енила, циклогекс-2-енила, циклогекс-3-енила, циклогепт-2-енила, циклогепт-3-енила и циклогепт-4-енила, или из циклопент-2-енила и циклогекс-2-енила, или из циклопент-2-енила, циклогекс-2-енила и циклогепт-2-енила. Циклоалкильные группы и циклоалкенильные группы обычно необязательно замещены одним или более (С1-С4)-алкильными заместителями. Т.е. они незамещены, т.е. не несут алкильные заместители, или замещены, например, 1, 2, 3 или 4 идентичными или различными (С1-С4)-алкильными заместителями, например, метальными группами и/или этильными группами и/или изопропильными группами и/или трет-бутильными группами, в частности, метальными группами, причем заместители могут присутствовать в любых положениях. Примеры алкилзамещенных циклоалкильных групп включают 1-метилциклопропил, 2,2-диметилциклопропил, 1-метилциклопентил, 2,3-диметилциклопентил, 1-метилциклогексил, 4-метилциклогексил, 4-изопропилциклогексил, 4-трет-бутилциклогексил и 3,3,5,5-тетраметилциклогексил. Примеры алкилзамещенных циклоалкенильных групп включают 1-метилциклопент-2-енил, 2-метилциклопент-2-енил, 3-метилциклопент-2-енил, 3,4-диметилциклопент-3-енил, 1-метилциклогекс-2-енил, 2-метилциклогекс-2-енил, 3-метилциклогекс-2-енил, 4-метилциклогекс-2-енил, 2-метилциклогекс-3-енил, 3-метилциклогекс-3-енил, 4-метилциклогекс-3-енил, 2,3-диметилциклогекс-2-енил, 4,4-диметилциклогекс-2-енил, 3,4-диметилциклогекс-3-енил. Циклоалкильные группы и циклоалкенильные группы обычно также необязательно замещены одним или более атомами фтора. Т.е. они являются незамещенными, т.е. не несут атомы фтора, или замещены, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11 атомами фтора, предпочтительно 1, 2, 3, 4, 5 или 6 атомами фтора. Циклоалкильные группы и циклоалкенильные группы также могут быть замещены одновременно фтором и алкилом. Атомы фтора могут присутствовать в любых положениях, а также могут присутствовать в алкильном заместителе. Примеры фторзамещенных циклоалкильных групп включают 1-фторцикло пропил, 2,2-дифторциклопропил, 3,3-дифторциклобутил, 1-фторциклогексил, 4,4-дифторциклогексил и 3,3,4,4,5,5-гексафторциклогексил. Примеры фторзамещенных циклоалкенильных групп включают 1-фторциклопент-2-енил, 1-фторциклогекс-2-енил, 4-фторциклогекс-2-енил, 4,4-дифторциклогекс-2-енил. Согласно одному варианту реализации настоящего изобретения циклоалкильные группы необязательно замещены заместителями, выбранными из фтора и (С1-С4)-алкила. Если цикло алкильная группа или циклоалкенильная группа может быть замещена дополнительными заместителями, такими как гидрокси, она может быть замещена одним или более такими дополнительными заместителями, такими как только гидрокси, но не заместителями, выбранными из фтора и (С1-С4)-алкила, или одним или более такими дополнительными заместителями и одновременно одним или более заместителями, выбранными из фтора и (С1-С4)-алкила. Количество таких дополнительных заместителей, таких как гидрокси, которые могут присутствовать в циклоалкильной или циклоалкенильной группе, предпочтительно составляет 1, 2 или 3, более предпочтительно 1 или 2, например 1. Общее количество всех заместителей в циклоалкильной группе или циклоалкенильной группе предпочтительно составляет 1, 2, 3, 4, 5, 6, 7 или 8, более предпочтительно 1, 2, 3, 4 или 5, например, 1, 2 или 3. Такие дополнительные заместители, такие как гидрокси, могут присутствовать в любых положениях, при условии, что полученное соединение является достаточно стабильным и подходит в качестве подгруппы в фармацевтически активном соединении. Предпочтительно гидроксильный заместитель не присутствует в положении 1 циклоалкенильной группы или циклоалкильной группы, и в циклоалкенильной группе гидроксильный заместитель не присутствует на атоме углерода, который является частью двойной связи. Примеры гидроксизамещенных циклоалкильных групп включают 3-гидроксициклобутил, 2-гидроксициклопентил, 3-гидроксициклопентил, 3,4-дигидроксициклопентил, 2-гидроксициклогексил, 3-гидроксициклогексил, 4-гидроксициклогексил, 2,3-дигидроксициклогексил, 2,4-дигидроксициклогексил, 3,4-дигидроксициклогексил, 3,5-дигидроксициклогексил, 3,4,5-тригидроксициклогексил, 2-гидроксициклогептил, 3-гидроксициклогептил, 4-гидроксициклогептил. Примеры гидроксизамещенных циклоалкенильных групп включают 5-гидроксициклопент-2-енил, 4-гидроксициклогекс-2-енил, 5-гидроксициклогекс-2-енил, 6-гидроксициклогекс-2-енил, 6-гидроксициклогексен-3-енил. Термин «циклоалкил» также подразумевает включение его бициклических и трициклических вариантов. Если образуются бициклические кольца, предпочтительно соответствующие кольца соединены друг с другом у двух соседних атомов углерода, однако, в другом варианте, два кольца соединены за счет одного и того же атома углерода, т.е. они образуют спиро-кольцевую систему или они образуют «мостиковые» кольцевые системы. Предпочтительные примеры циклоалкила включают С3-С8 циклоалкил, в частности, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, спиро[3,3]гептил, спиро[3,4]октил, спиро[4,3]октил, бицикло[4.1.0]гептил, бицикло[3.2.0]гептил, бицикло[2.2.1]гептил, бицикло[2.2.2]октил, бицикло[5.1.0]октил и бицикло[4.2.0]октил. Заместители циклоалкильной группы включают, но не ограничиваются ими, любой из заместителей, описанных в настоящем документе, которые приводят к образованию стабильного фрагмента.
В настоящем документе и по всему описанию термин «циклопропилен» означает циклопропильную группу, определенную выше, в которой один атом водорода был удален с получением дирадикала. Циклопропилен может соединять два атома или фрагмента за счет одного и того же атом углерода (1,1-циклопропилен, т.е. геминальный дирадикал) или за счет двух атомов углерода (1,2-циклопропилен).
В настоящем документе и по всему описанию термин «арил» или «ароматическое кольцо» относится к ароматической моно- или полициклической кольцевой системе, имеющей 6-10 кольцевых атомов, в которой все атомы кольца являются атомами углерода, и которая может быть замещенной или незамещенной. Согласно некоторым вариантам реализации настоящего изобретения «арил» относится к моно- или бициклической С6-С10 ароматической циклической системе, имеющей одно или два ароматических кольца, которые включают, но не ограничиваются ими, фенил, бифенил, нафтил и т.п., которые могут содержать один или более заместителей. Предпочтительно арильная группа содержит от 6 до 10 атомов углерода, которые могут быть расположены в одном кольце (например, фениле) или двух или более конденсированных кольцах (например, нафтиле). Примерные арильные группы включают циклопропенилий, циклопентадиенил, фенил, инденил, нафтил и азуленил. Предпочтительно «арил» относится к моноциклическому кольцу, содержащему 6 атомов углерода, или к ароматической бициклической кольцевой системе, содержащей 10 атомов углерода. Предпочтительными примерами являются замещенный или незамещенный фенил и нафтил. Еще более предпочтительным является замещенный или незамещенный фенил. Согласно некоторым вариантам реализации настоящего изобретения арильная группа, применяемая в настоящем изобретении, содержит 7-10 атомов углерода (С7-10 арил). Согласно другому варианту реализации настоящего изобретения арильная группа, применяемая в настоящем изобретении, содержит 6-8 атомов углерода (С6-8 арил). Согласно другому варианту реализации настоящего изобретения арильная группа, применяемая в настоящем изобретении, содержит 6 атомов углерода (С6 арил). Согласно другому варианту реализации настоящего изобретения арильная группа, применяемая в настоящем изобретении, содержит 10 атомов углерода (С10 арил).
В замещенных арильных группах заместители могут присутствовать в любых положениях. В монозамещенных фенильных группах заместитель может присутствовать в 2-положении, 3-положении или 4-положении. В дизамещенных фенильных группах заместители могут присутствовать в 2,3-положении, 2,4-положении, 2,5-положении, 2,6-положении, 3,4-положении или 3,5-положении. В тризамещенных фенильных группах заместители могут присутствовать в 2,3,4-положении, 2,3,5-положении, 2,3,6-положении, 2,4,5-положении, 2,4,6-положении или 3,4,5-положении. Если фенильная группа несет четыре заместителя, из которых один, два, три или четыре заместителя могут быть атомами фтора, например, незамещенный атом углерода кольца может присутствовать в 2-положении, 3-положении или 4-положении. Если полизамещенная фенильная группа или гетероарильная группа несет различные заместители, то каждый заместитель может присутствовать в любом подходящем положении, и настоящее изобретение включает все позиционные изомеры. Количество заместителей в замещенной фенильной группе может составлять 1, 2, 3, 4 или 5. Предпочтительно замещенная фенильная группа, а также замещенная гетероарильная группа, несет 1, 2 или 3, в частности, 1 или 2, идентичных или различных заместителей. Согласно предпочтительному варианту реализации настоящего изобретения замещенная фенильная группа несет 1 заместитель во 2, 3 или 4-положении. Согласно предпочтительным вариантам реализации настоящего изобретения заместители в замещенных фенильных и гетероарильных группах выбраны из любой подгруппы заместителей, перечисленных в соответствующем определении, например, заместителей, выбранных из галогена, замещенного или незамещенного (С1-С6)-алкила, (С1-С4)-галогеналкила, -NO2, циано, галогена, замещенного или незамещенного (С1-С6)алкил-O-, замещенного или незамещенного (С6-С14)арил(С1-С6)алкил-O-, C(O)NH2, -C(O)NH((C1-С6)-алкил), -С(O)N((С1-С6)-алкил)2, -NH((С1-С6)алкил-O(С1-С6)алкил)), -NH((С1-С6)алкил-ОН), азидо, -NH(С(=O)(С1-С6)алкил) и -N((С1-С6)алкил)(С(=O)(С1-С6)алкил).
В настоящем документе и по всему описанию термин «арилен» относится к бирадикалу арила, полученному из арильной группы, определенной в настоящем документе, путем удаления двух атомов водорода. Ариленовые группы могут быть замещенными или незамещенными. Заместители ариленовой группы включают, но не ограничиваются ими, любой из заместителей, описанных в настоящем документе, которые приводят к образованию стабильного фрагмента.
В настоящем документе и по всему описанию термин «гетероарил» или «гетероароматическое кольцо» означает арильную группу, определенную выше, в которой один или более атомов углерода в арильной группе замещены гетероатомами О, S или N. Предпочтительно гетероарильная группа содержит от 3 до 10 атомов углерода. Предпочтительно гетероарил относится к пяти- или шестичленому ароматическому моноциклическому кольцу, в котором 1, 2 или 3 атома углерода замещены идентичными или различными гетероатомами О, N или S. Согласно другому варианту это означает ароматическую бициклическую или трициклическую кольцевую систему, в которой 1, 2, 3, 4 или 5 атомов углерода замещены идентичными или различными гетероатомами О, N или S. Предпочтительно в каждом кольце гетероарильной группы максимальное количество атомов О составляет 1, максимальное количество атомов S составляет 1, и максимальное общее количество атомов О и S составляет 2. Согласно некоторым вариантам реализации настоящего изобретения гетероарильная группа, применяемая в настоящем изобретении, представляет собой пятичленное ароматическое моноциклическое кольцо, в котором 1, 2 или 3 атома углерода замещены идентичными или различными гетероатомами О, N или S. Согласно некоторым вариантам реализации настоящего изобретения гетероарильная группа, применяемая в настоящем изобретении, представляет собой пятичленное ароматическое моноциклическое кольцо, в котором 1, 2 или 3 атома углерода замещены идентичными или различными гетероатомами О. Согласно некоторым вариантам реализации настоящего изобретения гетероарильная группа, применяемая в настоящем изобретении, представляет собой пятичленное ароматическое моноциклическое кольцо, в котором 1, 2 или 3 атома углерода замещены идентичными или различными гетероатомами О и N. Согласно некоторым вариантам реализации настоящего изобретения гетероарильная группа, применяемая в настоящем изобретении, представляет собой пятичленное ароматическое моноциклическое кольцо, в котором 1, 2 или 3 атома углерода замещены идентичными или различными гетероатомами О и S. Согласно определенным вариантам реализации настоящего изобретения гетероарильная группа, применяемая в настоящем изобретении, представляет собой пятичленное ароматическое моноциклическое кольцо, в котором 1, 2 или 3 атома углерода замещены идентичными или различными гетероатомами N и S. Согласно некоторым вариантам реализации настоящего изобретения гетероарильная группа, применяемая в настоящем изобретении, представляет собой шестичленное ароматическое моноциклическое кольцо, в котором 1, 2 или 3 атома углерода замещены идентичными или различными гетероатомами О, S или N. Согласно некоторым вариантам реализации настоящего изобретения гетероарильная группа, применяемая в настоящем изобретении, представляет собой шестичленное ароматическое моноциклическое кольцо, в котором 1, 2 или 3 атома углерода замещены N. Согласно некоторым вариантам реализации настоящего изобретения гетероарильная группа, применяемая в настоящем изобретении, представляет собой ароматическую бициклическую систему, в которой 1, 2, 3, 4 или 5 атомов углерода замещены идентичными или различными гетероатомами О, N или S. Примерные гетероарильные группы включают фуранил, тиенил, оксазолил, изоксазолил, оксадиазолил (1,2,5- и 1,2,3-), пирролил, имидазолил, пиразолил, триазолил (1,2,3- и 1,2,4-), тетразолил, тиазолил, изотиазолил, тиадиазолил (1,2,3- и 1,2,5-), пиридил, пиримидинил, пиразинил, триазинил (1,2,3-, 1,2,4- и 1,3,5-), бензофуранил (1- и 2-), индолил, азаиндолил (4-, 5-, 6- и 7-), диазиндолил, изоиндолил, бензотиенил (1- и 2-), 1H-индазолил, бензимидазолил, бензоксазолил, индоксазинил, бензизоксазолил, бензотиазолил, бензизотиазолил, бензотриазолил, хинолил, изохинолинил, бензодиазинил, хиноксалинил, хиназолинил, бензотриазинил (1,2,3- и 1,2,4-бензотриазинил), пиридазинил, феноксазинил, тиазолпиридинил, пирролтиазолил, фенотиазинил, изобензофуранил, хроменил, ксантенил, феноксатиинил, пирролизинил, индолизинил, индазолил, пуринил, хинолизинил, фталазинил, нафтиридинил (1,5-, 1,6-, 1,7-, 1,8- и 2,6-), циннолинил, птеридинил, карбазолил, фенантридинил, акридинил, перимидинил, фенантролинил (1,7-, 1,8-, 1,10-, 3,8- и 4,7-), феназинил, оксазолопиридинил, изоксазолопиридинил, пирролоксазолил, пирролпирролил и т.п., который может нести один или более заместителей. Примерные 5- или 6-членные гетероарильные группы включают фуранил, тиенил, оксазолил, изоксазолил, оксадиазолил (1,2,5- и 1,2,3-), пирролил, имидазолил, пиразолил, триазолил (1,2,3- и 1,2,4-), тиазолил, изотиазолил, тиадиазолил (1,2,3- и 1,2,5-), пиридил, пиримидинил, пиразинил, триазинил (1,2,3-, 1,2,4- и 1,3,5-) и пиридазинил. Примерные бициклические гетероарильные группы включают 7-азаиндолил, 6-азаиндолил, 5-азаиндолил, 4-азаиндолил, бензофуранил и индолил. Гетероарильные заместители включают, но не ограничиваются ими, любые заместители, описанные в настоящем документе, которые приводят к образованию стабильного фрагмента.
В замещенных гетероарильных группах заместители могут присутствовать в любых положениях, например, в тиофен-2-ильной группе или фуран-2-ильной группе в 3-положении и/или в 4-положении и/или в 5-положении, в тиофен-3-ильной группе или фуран-3-ильной группе в 2-положении и/или в 4-положении и/или в 5-положении, в пиридин-2-ильной группе в 3-положении и/или в 4-положении и/или в 5-положении и/или в 6-положении, в пиридин-3-ильной группе в 2-положении и/или в 4-положении и/или в 5-положении и/или в 6-положении, в пир ид ин-4-ильной группе в 2-положении и/или в 3-положении и/или в 5-положении и/или в 6-положении, в 1H-пиразол-4-ильной группе в 3-положении и/или в 5-положении и/или в 1-положении, в 1H-пиразол-1-ильной группе в 3-положении и/или в 5-положении и/или в 4-положении, в 1H-индол-1-ильной группе в 2-положении и/или в 3-положении и/или в 4-положении и/или в 5-положении и/или в 6-положении и/или в 7-положении, в 1Н-индол-3-ильной группе в 1-положении и/или в 2-положении и/или в 4-положении и/или в 5-положении и/или в 6-положении и/или в 7-положении. Замещенные гетероарильные группы могут быть монозамещенными или полизамещенными, т.е. могут нести более одного заместителя. Согласно предпочтительным вариантам реализации 1Н-индол-1-ильная группа замещена в 2-положении и 3-положении и/или в 6-положении и 7-положении. Согласно другим предпочтительным вариантам реализации настоящего изобретения 1Н-индол-3-ильная группа замещена в 1-положении и 7-положении и/или в 6-положении и 7-положении. Предпочтительно замещенная гетероарильная группа замещена одним, двумя или тремя, в частности, одним или двумя, например, одним, идентичными или различными заместителями. Если присутствует кольцевой атом азота, который может нести атом водорода или заместитель, заместитель на этом атоме азота может быть, например, метальной группой, этильной группой, пропильной группой или трет-бутильной группой, причем группы также могут быть монозамещенными или полизамещенными фтором. Обычно подходящие кольцевые атомы азота в ароматическом кольце гетероарильной группы, например, атом азота в группе пиридинила, также могут нести оксидо заместитель -О, и соединения Формулы I, Формулы II, Формулы III и Формулы IV, соответственно, присутствуют в форме N-оксида.
В настоящем документе и по всему описанию термин «гетероарилен» относится к бирадикалу, полученному из гетероарильной группы, определенной в настоящем документе, путем удаления двух атомов водорода. Гетероариленовые группы могут быть замещенными или незамещенными.
В настоящем документе и по всему описанию термины «арилалкил» и «гетероарилалкил» подразумевают включение тех радикалов, в которых арильная группа и гетероарильная группа, соответственно, присоединены к алкильной группе (например, бензилу, фенетилу, пиридилметилу и т.п.), включая те алкильные группы, в которых атом углерода (например, метиленовая группа) замещен, например, атомом кислорода (например, феноксиметил, 2-пиридилоксиметил, 3-(1-нафтилокси)пропил и т.п.). Предпочтительно арилалкил представляет собой замещенный или незамещенный (С6-C10)арил(С1-С6)алкил. Предпочтительно гетероарилалкил представляет собой замещенный или незамещенный (С3-С10)гетероарил(С1-С6)алкил. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является линейной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является разветвленной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является замещенной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является незамещенной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является линейной и замещенной или незамещенной. Согласно некоторым вариантам реализации настоящего изобретения алкильная цепь является разветвленной и замещенной или незамещенной.
В настоящем документе и по всему описанию термины «арилгетероалкил» и «гетероарилгетероалкил» подразумевают включение тех радикалов, в которых арильная группа и гетероарильная группа, соответственно, присоединены к гетероалкильной группе. Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная цепь является линейной. Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная цепь является разветвленной. Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная цепь является замещенной. Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная цепь является незамещенной. Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная цепь является линейной и замещенной или незамещенной. Согласно некоторым вариантам реализации настоящего изобретения гетероалкильная цепь является разветвленной и замещенной или незамещенной.
В настоящем документе и по всему описанию термин «галоген» или «гало» означает фтор, хлор, бром или йод.
В настоящем документе и по всему описанию термин «циано» означает -CN.
В настоящем документе и по всему описанию термин «азидо» означает N3.
В настоящем документе и по всему описанию термин «необязательно замещенный» или «замещенный» указывает на то, что один или более (например, от 1 до максимального количества атомов водорода, связанных с группой, например, 1, 2, 3, 4, 5, 6, 7, 8, 9 или до 10, например, от 1 до 5, от 1 до 4, или от 1 до 3, или 1 или 2) атомов водорода могут быть замещены группой, отличной от водорода, такой как (С1-С6)алкил, (С1-С6)гетероалкил, (С1-С6)галогеналкил; (С2-С6)алкенил, (С3-С8)циклоалкил, (С6-С10)арил, (С6-С10)арил(C1-С6)алкил, (С3-С10)гетероарил, (С3-С10)гетероарил(С1-С6)алкил, галоген, -CN, -NO2, -OR161, -N(R162)(R163), -N(R161)(OR161), -S(O)o-2R161, -S(O)1-2OR161, -OS(O)1-2R161, -OS(O)1-2OR161, -S(O)1-2N(R162)(R163), -OS(O)1-2N(R162)(R163), -N(R161)S(O)1-2R161, -NR161S(O)1-2OR161, -NR161S(O)1-2N(R162)(R163), -C(=W)R161, -C(=W)WR161, -WC(=W)R161 и -WC(=W)WR161; где R161, R162 и R163 независимо выбраны из группы, состоящей из -Н, алкила, алкенила, алкинила, циклоалкила, арила и гетероарила, предпочтительно, где R161, R162 и R163 независимо выбраны из группы, состоящей из -Н, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, 3-7-членного циклоалкила, 5-6-членного арила, 5-6-членного гетероарила и 3-7-членного гетероциклила; R164 независимо выбран из группы, состоящей из -Н, алкила, алкенила, алкинила, циклоалкила, арила, гетероарила и -OR161; W независимо выбран из О, S и N(R164).
В настоящем документе и по всему описанию термин «необязательный» или «необязательно», применяемый в настоящем документе, означает, что впоследствии описанное событие, обстоятельство или условие может возникнуть, но необязательно, и что описание включает случаи, когда указанное событие, обстоятельство или условие возникает и случаи, в которых оно не возникает.
В настоящем документе и по всему описанию термин «сольват», применяемый в настоящем документе, относится к комплексу присоединения растворенного материала в растворителе (таком как органический растворитель (например, алифатический спирт (такой как метанол, этанол, н-пропанол, изопропанол), ацетон, ацетонитрил, простой эфир и т.п.), вода или смесь двух или более из этих жидкостей), причем указанный комплекс присоединения существует в форме кристалла или смешанного кристалла. Количество растворителя, содержащегося в комплексе присоединения, может быть стехиометрическим или нестехиометрическим. «Гидрат» представляет собой сольват, в котором растворителем является вода.
В настоящем документе и по всему описанию термин «фармацевтически приемлемая соль» относится к соли, которая сохраняет желаемую биологическую активность исходного соединения и не оказывает каких-либо нежелательных токсикологических эффектов (см., например, Berge, S. М., et al. (1977) J. Pharm. Sci. 66: 1-19). Физиологически приемлемые соли соединений согласно настоящему изобретению представляют собой, в частности, соли с нетоксичным солевым компонентом и предпочтительно представляют собой фармацевтически применимые соли. Они могут содержать неорганические или органические солевые компоненты. Такие соли могут быть получены, например, из соединений согласно настоящему изобретению, которые содержат кислотную группу, например, группу карбоновой кислоты (НО-СО-) или группу сульфоновой кислоты (НО-S(O)2-), и нетоксичные неорганические или органические основания. Подходящими основаниями являются, например, соединения щелочных металлов или соединения щелочноземельных металлов, такие как гидроксид натрия, гидроксид калия, карбонат натрия или гидрокарбонат натрия, или аммиак, органические аминосоединения и гидроксиды четвертичного аммония. Реакции соединений согласно настоящему изобретению с основаниями для получения солей обычно осуществляют в соответствии с обычными процедурами в растворителе или разбавителе. Вследствие физиологической и химической стабильности предпочтительными солями кислотных групп во многих случаях являются соли натрия, калия, магния или кальция или соли аммония, которые также могут нести одну или более органических групп на атоме азота. Соединения согласно настоящему изобретению, которые содержат основную, т.е. протонируемую группу, например, аминогруппу или другой основный гетероцикл, могут присутствовать в форме их кислотно-аддитивных солей с физиологически приемлемыми кислотами, например, в виде соли с хлорводородом, бромводородом, фосфорной кислотой, серной кислотой, уксусной кислотой, бензойной кислотой, метансульфоновой кислотой, п-толуолсульфокислотой, которая в общем случае может быть получена из соединений согласно настоящему изобретению путем взаимодействия с кислотой в растворителе или разбавителе в соответствии с обычными процедурами. Обычно, в частности, в случае кислотно-аддитивных солей соединения, содержащего две или более основных групп, в полученной соли соотношение компонентов соли может увеличиваться или уменьшаться относительно стехиометрического соотношения, такого как молярное соотношение 1:1 или 1:2 в случае кислотно-аддитивной соли соединения согласно настоящему изобретению, содержащего одну или две основные группы, с одновалентной кислотой, и варьируется в зависимости от применяемых условий. Настоящее изобретение также включает соли, содержащие компоненты в нестехиометрическом соотношении, и указание на то, что кислотно-аддитивная соль соединения согласно настоящему изобретению содержит кислоту в эквимолярном количестве, например, также допускает меньшее или большее количество кислоты в полученной соли, например, приблизительно 0,8 или приблизительно 1,1 моль кислоты на моль соединения согласно настоящему изобретению. Если соединения согласно настоящему изобретению одновременно содержат кислотную и основную группу в молекуле, настоящее изобретение также включает внутренние соли (бетаины, цвиттерионы) в дополнение к упомянутым солевым формам. Настоящее изобретение также включает все соли соединений согласно настоящему изобретению, которые из-за низкой физиологической переносимости не являются непосредственно подходящими для применения в качестве фармацевтического препарата, но пригодны в качестве промежуточных соединений для химических реакций или для получения физиологически приемлемых солей, например, посредством анионного обмена или катионного обмена. Объектом настоящего изобретения также являются сольваты соединений согласно настоящему изобретению и их соли, такие как гидраты и аддукты со спиртами, такими как (С1-С4)-алканолы, в частности, физиологически приемлемые сольваты, а также активные метаболиты соединений согласно настоящему изобретению.
В настоящем документе и по всему описанию термин «фармацевтически приемлемый», в частности, может означать одобрение регулирующего органа или другой общепризнанной фармакопеи для применения у животных и, в частности, у человека.
Настоящее изобретение включает все стереоизомерные формы соединений Формулы I, например, все возможные энантиомеры и диастереомеры, включая цис/транс изомеры. Настоящее изобретение также включает смеси двух или более стереоизомерных форм, например, смеси энантиомеров и/или диастереомеров, включая цис/транс изомеры, во всех соотношениях.
Асимметричные центры, содержащиеся в соединениях Формулы I, например, в незамещенных или замещенных алкильных группах или в стереогенном углероде СН-В, в случае если р и q не равны, изображенные в Формуле I, все могут независимо друг от друга иметь S-конфигурацию или R-конфигурацию. Настоящее изобретение относится к энантиомерам, как к левовращающему, так и к правовращающему антиподу, в энантиомерно чистой форме и по существу энантиомерно чистой форме, а также в форме рацематов и в форме смесей двух энантиомеров во всех соотношениях. Настоящее изобретение также относится к диастереомерам в форме чистых и по существу чистых диастереомеров и в форме смесей двух или более диастереомеров во всех соотношениях. Настоящее изобретение также включает все цис/транс-изомеры соединений Формулы I, например, в чистой форме и по существу в чистой форме и в виде смесей цис-изомера и транс-изомера во всех соотношениях. Цис/транс-изомерия может происходить, например, в замещенных циклоалкановых кольцах. Получение отдельных стереоизомеров, если необходимо, можно осуществлять путем разделения смеси в соответствии с обычными способами, например, с помощью хроматографии или кристаллизации, или с использованием в синтезе стереохимически однородных исходных соединений или стереоселективных реакций. Необязательно перед разделением стереоизомеров может быть проведена дериватизация. Разделение смеси стереоизомеров можно осуществлять на стадии соединения Формулы I или на стадии промежуточного соединения в ходе синтеза. Настоящее изобретение также включает все таутомерные формы соединений согласно настоящему изобретению.
Согласно одному аспекту настоящее изобретение относится к соединениям, имеющим структуру в соответствии с Формулой I, в любой из их стереоизомерных форм или к смеси стереоизомерных форм в любом соотношении, и их физиологически приемлемым солям, а также физиологически приемлемым сольватам или гидратам любого из них,
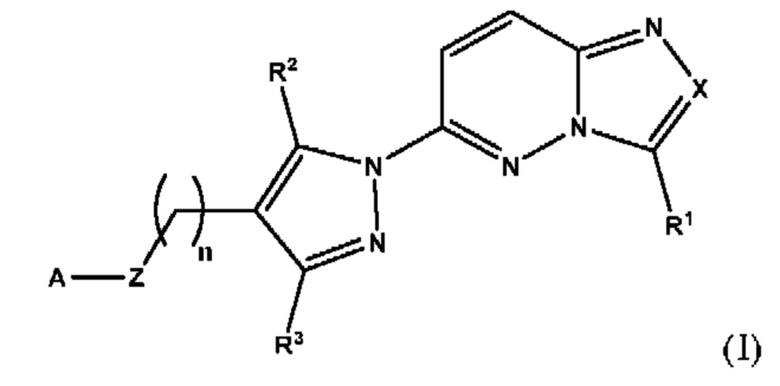
в которой
X выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила;
n представляет собой целое число от 0 до 3;
Z выбран из группы, состоящей из С=O, C=S и СН2;
А выбран из группы, состоящей из -N(R4)(R5) и 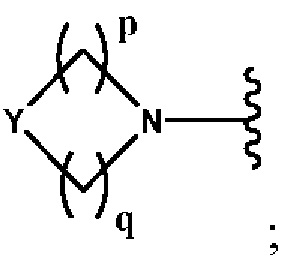
R4 представляет собой водород или (С1-С6)алкил;
R5 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)гетероалкила и (С6-С10)арил(C1-С6)гетероалкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-С4)алкила, (С1-С4)галогеналкила и -OR6;
R6 представляет собой (С1-С4)алкил или (С1-С4)галогеналкил;
Y выбран из группы, состоящей из N-B, СН-В и О;
В выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (C1-С6)алкила и N(R7)(R8), которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR6;
R7 представляет собой водород или (С1-С6)алкил;
R8 выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (C1-С6)алкила;
р представляет собой целое число от 1 до 2; и
q представляет собой целое число от 1 до 3;
Согласно некоторым вариантам реализации настоящего изобретения X представляет собой N. Согласно некоторым вариантам реализации настоящего изобретения X представляет собой CH2.
Согласно некоторым вариантам реализации настоящего изобретения R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила, согласно другим вариантам реализации из водорода, (С1-С4)алкила, (C1-С4)галогеналкила, (С3-С6)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С4)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С4)алкила, согласно другим вариантам реализации из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (Се-С10)арила и (С6-С10)арил(С1-С6)алкила, согласно другим вариантам реализации из водорода, (С1-С6)алкила, (С3-С8)циклоалкила, (С6-С10)арила и (С6-С10)арил(С1-С6)алкила, согласно другим вариантам реализации из водорода, (С1-С4)алкила, (С3-С6)циклоалкила, (С6-С10)арила и (С6-С10)арил(С1-С4)алкила, согласно другим вариантам реализации из водорода, (С1-С4)алкила, (С3-С6)циклоалкила и (С6)арила, (С6)арил(С1-С4)алкила, согласно другим вариантам реализации из водорода, (С1-С6)алкила и (С1-С6)галогеналкила, согласно другим вариантам реализации из водорода, (С1-С4)алкила и (C1-С4)галогеналкила, согласно другим вариантам реализации из водорода, (С2-С6)алкила и (С2-С6)галогеналкила, согласно другим вариантам реализации из водорода, (С3-С6)алкила и (С3-С6)галогеналкила, согласно другим вариантам реализации из водорода и (C1-С6)алкила, согласно другим вариантам реализации из водорода и (С1-С4)алкила, согласно другим вариантам реализации из водорода и (С3-С4)алкила, согласно другим вариантам реализации настоящего изобретения R1 представляет собой (C1-С6)алкил, предпочтительно (С1-С4)алкил, более предпочтительно (C1-С2)алкил, еще более предпочтительно метил. Согласно некоторым вариантам реализации настоящего изобретения R1 представляет собой (С6-С10)арил, при этом предпочтительно арил выбран из группы замещенного или незамещенного нафтила и замещенного или незамещенного фенила, при этом более предпочтительно арил представляет собой замещенный или незамещенный фенил. Согласно некоторым вариантам реализации настоящего изобретения R1 представляет собой (С1-С6)галогеналкил, предпочтительно -CF3, согласно другим вариантам реализации (С3-С8)циклоалкил, предпочтительно замещенный или незамещенный циклопропил, согласно другим вариантам реализации (С6-С10)арил(С1-С6)алкил, предпочтительно (С6)арил(С1-С6)алкил, более предпочтительно замещенный или незамещенный бензил. Алкил, циклоалкил, арил, арилалкил, гетероарил и гетероарилалкил могут быть замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из замещенного или незамещенного (С1-С6)алкила, замещенного или незамещенного (С1-С6)галогеналкила, -OR26, галогена и циано, предпочтительно галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR26, где R26 выбран из группы, состоящей из водорода, замещенного или незамещенного (C1-С6)алкила и замещенного или незамещенного (С2-С6)алкенила, предпочтительно из водорода и замещенного или незамещенного (С1-С6)алкила. Предпочтительно количество заместителей в замещенной группе R1 составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе R1 могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенных алкильных, циклоалкильных, арильных, арилалкильных, гетероарильных и гетероарилалкильных групп.
Согласно некоторым вариантам реализации настоящего изобретения R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила, согласно другим вариантам реализации из водорода, (С1-С6)алкила и (С1-С6)галогеналкила, согласно другим вариантам реализации из (С1-С6)алкила, (С1-С6)галогеналкила и (С3-С8)циклоалкила, согласно другим вариантам реализации из (С1-С6)алкила и (С1-С6)галогеналкила, согласно другим вариантам реализации настоящего изобретения R2 и R3 представляют собой водород, согласно другим вариантам реализации настоящего изобретения R2 и R3 представляют собой (C1-С6)алкил, предпочтительно (С1-С4)алкил, более предпочтительно (С1-С2)алкил, еще более предпочтительно метил, согласно другим вариантам реализации настоящего изобретения R2 и R3 представляют собой (С1-С6)галогеналкил, предпочтительно (С1-С4)галогеналкил, более предпочтительно (С1-С2)галогеналкил, еще более предпочтительно -CF3, согласно другим вариантам реализации настоящего изобретения один из R2 и R3 представляет собой водород, а другой представляет собой (С1-С6)алкил, предпочтительно метил, согласно другим вариантам реализации настоящего изобретения один из R2 и R3 представляет собой водород, а другой представляет собой (С1-С6)галогеналкил, предпочтительно -CF3, согласно другим вариантам реализации настоящего изобретения один из R2 и R3 представляет собой водород, а другой представляет собой (С3-С8)циклоалкил, предпочтительно циклопропил, согласно другим вариантам реализации настоящего изобретения один из R2 и R3 представляет собой (С1-С6)алкил, предпочтительно (С1-С4)алкил, более предпочтительно (С1-С2)алкил, еще более предпочтительно метил и другой представляет собой (C1-С6)галогеналкил, предпочтительно (С1-С4)галогеналкил, более предпочтительно (C1-С2)галогеналкил, еще более предпочтительно -CF3, согласно другим вариантам реализации настоящего изобретения один из R2 и R3 представляет собой (С1-С6)алкил, предпочтительно (C1-С4)алкил, более предпочтительно (С1-С2)алкил, еще более предпочтительно метил, а другой представляет собой (С3-С8)циклоалкил, предпочтительно (С3-С6)циклоалкил, более предпочтительно циклоалкил, согласно другим вариантам реализации настоящего изобретения один из R2 и R3 представляет собой метил, а другой представляет собой изопропил или этил, согласно другим вариантам реализации настоящего изобретения один из R2 и R3 выбран из группы, состоящей из метила, этила, пропила и изопропила, а другой представляет собой (С1-С6)галогеналкил, предпочтительно выбранный из -CF3, -CH2CF3, -CF2CF3, более предпочтительно -CF3. Алкил и циклоалкил могут быть замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из замещенного или незамещенного (С1-С6)алкила, замещенного или незамещенного (С1-С6)галогеналкила, -OR26, галогена и циано, предпочтительно галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR26, где R26 выбран из группы, состоящей из водорода, замещенного или незамещенного (С1-С6)алкила и замещенного или незамещенного (С2-С6)алкенила, предпочтительно из водорода и замещенного или незамещенного (С1-С6)алкила. Предпочтительно количество заместителей в замещенной группе R2 и/или R3 составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе R2 и/или R3 могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенных алкильных и циклоалкильных групп.
Согласно некоторым вариантам реализации настоящего изобретения n представляет собой целое число от 0 до 3, согласно другим вариантам реализации настоящего изобретения n представляет собой 0, 1, 2 или 3, согласно другим вариантам реализации настоящего изобретения n представляет собой целое число от 1 до 3, согласно другим вариантам реализации настоящего изобретения n представляет собой целое число от 1 до 2, согласно другим вариантам реализации настоящего изобретения n равно 1, согласно другим вариантам реализации настоящего изобретения n равно 2, согласно другим вариантам реализации настоящего изобретения n равно 3, согласно другим вариантам реализации настоящего изобретения n равно 0.
Согласно некоторым вариантам реализации настоящего изобретения Z выбран из группы, состоящей из С=O, C=S и CH2, согласно другим вариантам реализации из С=O и C=S, согласно другим вариантам реализации из С=O и СН2, согласно другим вариантам реализации из C=S и СН2, согласно другим вариантам реализации настоящего изобретения Z представляет собой С=O, согласно другим вариантам реализации настоящего изобретения Z представляет собой C=S, согласно другим вариантам реализации настоящего изобретения Z представляет собой CH2.
Согласно некоторым вариантам реализации настоящего изобретения А выбран из группы, состоящей из -N(R4)(R5), 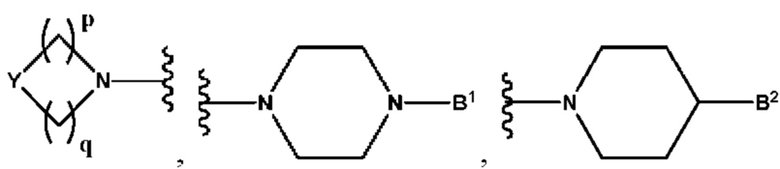 и
и 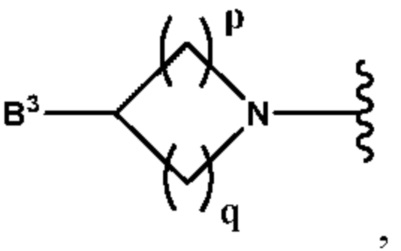
и согласно другим вариантам реализации из -N(R4)(R5) и 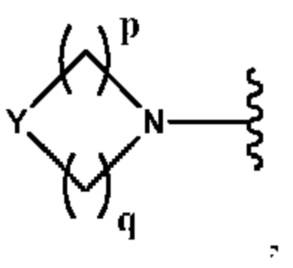
и согласно другим вариантам реализации из 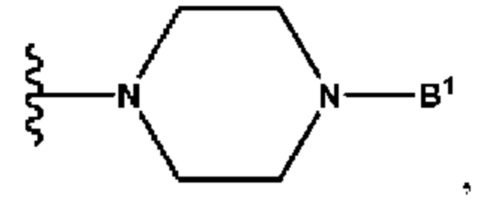
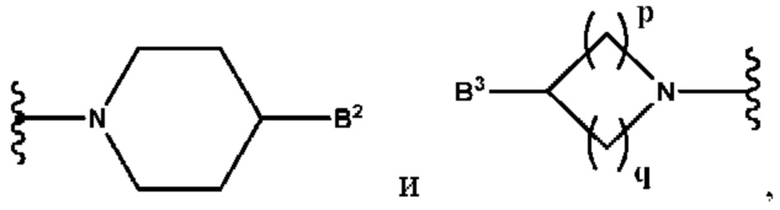 и согласно другим вариантам реализации настоящего изобретения А представляет собой -N(R4)(R5), согласно другим вариантам реализации настоящего изобретения А представляет собой
и согласно другим вариантам реализации настоящего изобретения А представляет собой -N(R4)(R5), согласно другим вариантам реализации настоящего изобретения А представляет собой 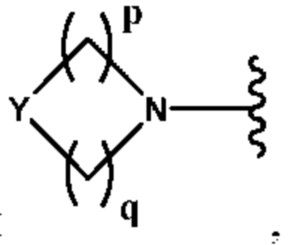 согласно другим вариантам реализации настоящего изобретения А представляет собой
согласно другим вариантам реализации настоящего изобретения А представляет собой 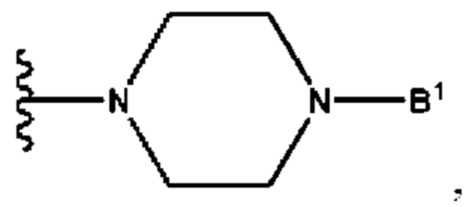 согласно другим вариантам реализации настоящего изобретения А представляет собой
согласно другим вариантам реализации настоящего изобретения А представляет собой 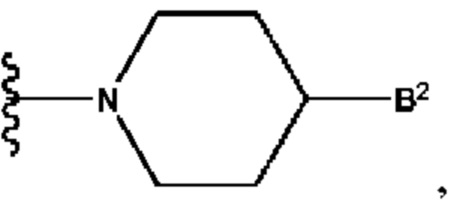 согласно другим вариантам реализации настоящего изобретения А представляет собой
согласно другим вариантам реализации настоящего изобретения А представляет собой 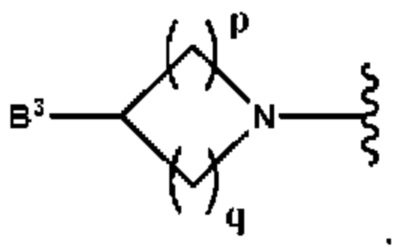
Согласно некоторым вариантам реализации настоящего изобретения R4 представляет собой водород или (С1-С6)алкил, согласно другим вариантам реализации настоящего изобретения R4 представляет собой водород или (С1-С4)алкил, согласно другим вариантам реализации настоящего изобретения R4 представляет собой водород или (С3-С4)алкил, согласно другим вариантам реализации настоящего изобретения R4 представляет собой водород или (С1-С2)алкил, согласно другим вариантам реализации настоящего изобретения R4 представляет собой водород, согласно другим вариантам реализации настоящего изобретения R4 представляет собой (С1-С6)алкил, предпочтительно (C1-С4)алкил, более предпочтительно (C1-С2)алкил, еще более предпочтительно метил. Алкил может быть замещен одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена и циано, предпочтительно галогена. Предпочтительно количество заместителей в замещенной группе R4 составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе R4 могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенной алкильной группы.
Согласно некоторым вариантам реализации настоящего изобретения R5 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)гетероалкила и (С6-С10)арил(С1-С6)гетероалкила, согласно другим вариантам реализации из (С6)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)гетероалкила и (С6)арил(С1-С6)гетероалкила, согласно другим вариантам реализации из (С6-С10)арил(C1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С3-С10)гетероарил(С1-С6)гетероалкила, согласно другим вариантам реализации из (С6-С10)арил(С1-С6)алкила и (С3-С10)гетероарил(С1-С6)алкила, предпочтительно (С6)арил(С1-С6)алкила и (С3-С10)гетероарил(С1-С6)алкила. Согласно некоторым вариантам реализации настоящего изобретения R5 представляет собой (С6-С10)арил(С1-С6)алкил, при этом предпочтительно арил выбран из группы замещенного или незамещенного нафтила и замещенного или незамещенного фенила, причем более предпочтительно арил представляет собой замещенный или незамещенный фенил, согласно другим вариантам реализации R5 представляет собой (С6)арил(С1-С6)алкил, согласно другим вариантам реализации R5 представляет собой замещенный или незамещенный бензил, согласно другим вариантам реализации R5 выбран из группы, состоящей из бензила, 2-фторбензила, 3-фторбензила, 4-фторбензила, 2-хлорбензила, 3-хлорбензила, 4-хлорбензила, 3-фенилпропила и фенетила, предпочтительно бензила, 3-хлорбензила, 4-хлорбензила и 3-фенилпропила. Согласно некоторым вариантам реализации настоящего изобретения R5 представляет собой (С3-С10)гетероарил(С1-С6)алкил, согласно другим вариантам реализации настоящего изобретения он выбран из группы, состоящей из 5-метилфуран-2-ил-метила, тиофен-2-ил-метила и 2-(1H-индол-3-ил)этила, предпочтительно 5-метилфуран-2-ил-метила и 2-(1H-индол-3-ил)этила. Согласно некоторым вариантам реализации настоящего изобретения R5 представляет собой (С3-С10)гетероарил(С1-С6)гетероалкил, согласно другим вариантам реализации настоящего изобретения он выбран из группы, состоящей из фуран-2-ил-метилтиометила, тиофен-2-ил-метилтиометила, фуран-2-ил-метоксиметила, тиофен-2-ил-метоксиметила, предпочтительно фуран-2-ил-метилтиометила. Согласно некоторым вариантам реализации настоящего изобретения R5 представляет собой (С6-С10)арил(С1-С6)гетероалкил, предпочтительно (С6)арил(С1-С6)гетероалкил, при этом предпочтительно арил выбран из группы замещенного или незамещенного нафтила и замещенного или незамещенного фенила, при этом более предпочтительно арил представляет собой замещенный или незамещенный фенил. Арилалкил, гетероарилалкил, гетероарилгетероалкил и арилгетероалкил могут быть замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6, предпочтительно галогена, более предпочтительно хлора или фтора, еще более предпочтительно хлора, еще более предпочтительно хлора, предпочтительно метила. Предпочтительно количество заместителей в замещенной группе R5 составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе R5 могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенной алкильной группы.
Согласно некоторым вариантам реализации настоящего изобретения R6 представляет собой (С1-С4)алкил или (С1-С4)галогеналкил, согласно другим вариантам реализации (C1-С2)алкил или (С1-С2)галогеналкил, согласно другим вариантам реализации (С1-С4)алкил, предпочтительно метил, согласно другим вариантам реализации настоящего изобретения (С1-С4)галогеналкил, предпочтительно -CF3. Алкил может быть замещен одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена и циано, предпочтительно галогена. Предпочтительно количество заместителей в замещенной группе R6 составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе R6 могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенной алкильной группы.
Согласно некоторым вариантам реализации настоящего изобретения Y1 выбран из группы, состоящей из N-B, СН-В и О, предпочтительно из N-B и СН-В. Согласно некоторым вариантам реализации настоящего изобретения Y1 представляет собой N-B, согласно другим вариантам реализации настоящего изобретения Y1 представляет собой СН-В, согласно другим вариантам реализации настоящего изобретения Y1 представляет собой О.
Согласно некоторым вариантам реализации настоящего изобретения В выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С1о)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С1-С6)алкила и N(R7)(R8), согласно другим вариантам реализации из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила и (С3-С10)гетероарил(С1-С6)алкила, согласно другим вариантам реализации из (С6-С10)арила, (С3-С10)гетероарила и (С6-С10)арил(С1-С6)алкила, согласно другим вариантам реализации из (С6)арила, (С3-С10)гетероарила и (С6)арил(С1-С6)алкила. Согласно некоторым вариантам реализации настоящего изобретения В представляет собой (С6-С10)арил, при этом предпочтительно арил выбран из группы замещенного или незамещенного нафтила и замещенного или незамещенного фенила, при этом более предпочтительно арил представляет собой замещенный или незамещенный фенил. Согласно некоторым вариантам реализации настоящего изобретения В выбран из группы, состоящей из фенила, 3-метоксифенила, 4-фторфенила, 4-метоксифенила, 2-фторфенила, 2,5-диметилфенила, предпочтительно фенила, 4-фторфенила и 3-метоксифенила, более предпочтительно фенила и 3-метоксифенила. Согласно некоторым вариантам реализации настоящего изобретения В представляет собой (С3-С10)гетероарил, согласно другим вариантам реализации В представляет собой (С3-С10)гетероарил, содержащий 1-3 атома азота, предпочтительно 1-2 атома азота, более предпочтительно 1 атом азота, согласно другим вариантам реализации настоящего изобретения В выбран из группы, состоящей из пиридин-2-ила, пиридин-3-ила, пиридин-4-ила, предпочтительно пиридин-2-ила. Согласно некоторым вариантам реализации настоящего изобретения В представляет собой (С6-С10)арил(С1-С6)алкил, согласно другим вариантам реализации (С6)арил(С1-С6)алкил, согласно другим вариантам реализации замещенный или незамещенный бензил, согласно другим вариантам реализации он выбран из группы, состоящей из бензила, 2-хлорбензила, 2-фторбензила, 3-хлорбензила, 3-фторбензила, 4-хлорбензила, 4-фтор бензила, 2-метоксибензила, 3-метоксибензила, 4-метоксибензила, 2,3-диметоксибензила, 3,4-диметоксибензила, 4-диметоксибензила и 3,5-диметоксибензила, предпочтительно бензила и 3-хлорбензила, более предпочтительно бензила. Согласно некоторым вариантам реализации настоящего изобретения В представляет собой (С3-С10)гетероарил(С1-С6)алкил, при этом предпочтительно гетероарильная часть представляет собой кислородсодержащий гетероарил, такой как фуранил, или серосодержащий гетероарил, такой как тиофен, или азотсодержащий гетероарил, такой как индол или диазаиндол. Согласно другим вариантам реализации настоящего изобретения (С3-С10)гетероарил(С1-С6)алкил выбран из пиридин-2-ил-метила, пиридин-3-ил-метила, пиридин-4-ил-метила, 5-метилфуран-2-ил-метила, фуран-2-ил-метила, фуран-3-ил-метила, тиофен-2-ил-метила и тиофен-3-ил-метила, предпочтительно пиридин-2-ил-метила и фуран-2-ил-метила, более предпочтительно пиридин-2-ил-метила. Согласно некоторым вариантам реализации настоящего изобретения В представляет собой (С1-С6)алкил, предпочтительно (C1-С4)алкил, согласно другим вариантам реализации настоящего изобретения В выбран из группы, состоящей из метила, этила, пропила, изопропила, бутила, втор-бутила и трет-бутила, предпочтительно метила или этила. Согласно некоторым вариантам реализации настоящего изобретения В представляет собой N(R7)(R8), при этом предпочтительно R7 представляет собой водород и R8 определен ниже, при этом предпочтительно R7 представляет собой (С1-С6)алкил, предпочтительно метил, и R8 определен ниже. Арильная, гетероарильная, арилалкильная, гетероарилалкильная, алкильная группы и группа N(R7)(R8) могут быть замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR6, предпочтительно галогена и -OR6. Предпочтительно количество заместителей в замещенной группе В составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе В могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенной арильной, гетероарильной, арилалкильной, гетероарилалкильной, алкильной группы и группы N(R7)(R8).
В1 представляет собой (С6-С10)арил(С1-С6)алкил или (С3-С10)гетероарил(С1-С6)алкил. Согласно некоторым вариантам реализации настоящего изобретения В1 представляет собой (С6-С10)арил(С1-С6)алкил, согласно другим вариантам реализации (С6)арил(С1-С6)алкил, согласно другим вариантам реализации (С10)арил(С1-С6)алкил, согласно другим вариантам реализации замещенный или незамещенный бензил, согласно другим вариантам реализации замещенный или незамещенный нафтален-2-ил-метил, согласно другим вариантам реализации он выбран из группы, состоящей из нафтален-2-ил-метила, бензила, 2-хлорбензила, 2-фторбензила, 3-хлорбензила, 3-фторбензила, 4-хлорбензила, 4-фторбензила, 2-метоксибензила, 3-метоксибензила, 4-метоксибензила, 2,3-диметоксибензила, 3,4-диметоксибензила, 2,4-диметоксибензила, нафтален-2-ил-метила и 3,5-диметоксибензила, предпочтительно бензила, 4-фторбензила, 2,4-диметоксибензила и 3-метоксибензила, более предпочтительно бензила. Согласно некоторым вариантам реализации настоящего изобретения В1 представляет собой (С3-С10)гетероарил(С1-С6)алкил, при этом предпочтительно гетеро арильная часть представляет собой кислородсодержащий гетероарил, такой как фуранил, или серосодержащий гетероарил, такой как тиофен, или азотсодержащий гетероарил, такой как пиридин, индол или диазаиндол. Согласно другим вариантам реализации настоящего изобретения (С3-С10)гетероарил(С1-С6)алкил выбран из пиридин-2-ил-метила, пиридин-3-ил-метила, пиридин-4-ил-метила, 5-метилфуран-2-ил-метила, фуран-2-ил-метила, фуран-3-ил-метила, тиофен-2-ил-метила и тиофен-3-ил-метила, предпочтительно пиридин-2-ил-метила и фуран-2-ил-метила, более предпочтительно пиридин-2-ил-метила. Арилалкильная и гетероарилалкильная группы могут быть замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-С4)алкила, (C1-С4)галогеналкила и -OR6, предпочтительно галогена и -OR6. Предпочтительно количество заместителей в замещенной группе В1 составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе В1 могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенной арилалкильной и гетероарил ал киль ной групп.
Согласно некоторым вариантам реализации настоящего изобретения В2 представляет собой (С6-С10)арил(С1-С6)алкил, замещенный одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-С4)алкила, (С1-С4)галогеналкила и -OR6, предпочтительно галогена и -OR6. Согласно некоторым вариантам реализации настоящего изобретения В2 представляет собой (С6)арил(С1-С6)алкил, согласно другим вариантам реализации настоящего изобретения (С10)арил(С1-С6)алкил, согласно другим вариантам реализации замещенный или незамещенный бензил, согласно другим вариантам реализации замещенный или незамещенный нафтален-2-ил-метил, согласно другим вариантам реализации выбранный из группы, состоящей из 2-хлорбензила, 2-фторбензила, 3-хлорбензила, 3-фторбензила, 4-хлорбензила, 4-фторбензила, 2-метоксибензила, 3-метоксибензила, 4-метоксибензила, 2,3-диметоксибензила, 3,4-диметоксибензила, 2,4-диметоксибензила и 3,5-диметоксибензила, предпочтительно 4-фторбензила и 3-метоксибензила. Согласно некоторым вариантам реализации настоящего изобретения В2 представляет собой (С3-С10)гетероарил(С1-С6)алкил, при этом предпочтительно гетероарильная часть представляет собой кислородсодержащий гетероарил, такой как фуранил, или серосодержащий гетероарил, такой как тиофен, или азотсодержащий гетероарил, такой как пиридин, индол или диазаиндол. Согласно другим вариантам реализации настоящего изобретения (С3-C10)гетероарил(С1-С6)алкил выбран из пиридин-2-ил-метила, пиридин-3-ил-метила, пиридин-4-ил-метила, 5-метилфуран-2-ил-метила, фуран-2-ил-метила, фуран-3-ил-метила, тиофен-2-ил-метила и тиофен-3-ил-метила, предпочтительно пиридин-2-ил-метила и фуран-2-ил-метила, более предпочтительно пиридин-2-ил-метила. Гетероарилалкильная группа может быть замещена одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR6, предпочтительно галогена и -OR6. Предпочтительно количество заместителей в замещенной группе В2 составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе В2 могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенной арилалкильной и гетероарилалкильной групп.
В3 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С1-С6)алкила и N(R7)(R8). Согласно некоторым вариантам реализации настоящего изобретения В3 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила и (С3-С10)гетероарил(С1-С6)алкила, согласно другим вариантам реализации настоящего изобретения В3 представляет собой (С6-С10)арил(С1-С6)алкил. Согласно некоторым вариантам реализации настоящего изобретения В3 представляет собой (С6-C10)арил(С1-С6)алкил, согласно другим вариантам реализации настоящего изобретения (С6)арил(С1-С6)алкил, согласно другим вариантам реализации замещенный или незамещенный бензил, согласно другим вариантам реализации замещенный или незамещенный бензил, согласно другим вариантам реализации замещенный или незамещенный нафтален-2-ил-метил, согласно другим вариантам реализации он выбран из группы, состоящей из бензила, нафтален-2-ил-метила, 2-хлорбензила, 2-фторбензила, 3-хлорбензила, 3-фторбензила, 4-хлорбензила, 4-фторбензила, 2-метоксибензила, 3-метоксибензила, 4-метоксибензила, 2,3-диметоксибензила, 3,4-диметоксибензила, 2,4-диметоксибензила и 3,5-диметоксибензила, предпочтительно бензила и 3-хлорбензила, более предпочтительно бензила. Согласно некоторым вариантам реализации настоящего изобретения В3 представляет собой (С3-С10)гетероарил(C1-С6)алкил, при этом предпочтительно гетероарильная часть представляет собой кислородсодержащий гетероарил, такой как фуранил, или серосодержащий гетероарил, такой как тиофен, или азотсодержащий гетероарил, такой как пиридин, индол или диазаиндол. Согласно другим вариантам реализации настоящего изобретения (С3-С10)гетероарил(С1-С6)алкил выбран из пиридин-2-ил-метила, пиридин-3-ил-метила, пиридин-4-ил-метила, 5-метилфуран-2-ил-метила, фуран-2-ил-метила, фуран-3-ил-метила, тиофен-2-ил-метила и тиофен-3-ил-метила, предпочтительно пиридин-2-ил-метила и фуран-2-ил-метила, более предпочтительно пиридин-2-ил-метила. Согласно некоторым вариантам реализации настоящего изобретения В3 представляет собой (С1-С6)алкил, предпочтительно (C1-С4)алкил, согласно другим вариантам реализации настоящего изобретения он выбран из группы, состоящей из метила, этила, пропила, изопропила, бутила, втор-бутила и трет-бутила, предпочтительно метила или этила. Согласно некоторым вариантам реализации настоящего изобретения В3 представляет собой N(R7)(R8), при этом предпочтительно R7 представляет собой водород и R8 определен ниже, при этом предпочтительно R7 представляет собой (С1-С6)алкил, предпочтительно метил, и R8 определен ниже. Арилалкильная, гетероарилалкильная, алкильная группы и группа N(R7)(R8) могут быть замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6, предпочтительно галогена и -OR6. Предпочтительно количество заместителей в замещенной группе В3 составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе В3 могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенной арилалкильной, гетероарилалкильной, алкильной группы и группы N(R7)(R8).
Согласно некоторым вариантам реализации настоящего изобретения R7 представляет собой водород или (С1-С6)алкил, согласно другим вариантам реализации настоящего изобретения водород или (С1-С4)алкил, согласно другим вариантам реализации настоящего изобретения водород или (С3-С4)алкил, согласно другим вариантам реализации настоящего изобретения водород или (C1-С2)алкил, согласно другим вариантам реализации настоящего изобретения R7 представляет собой водород, согласно другим вариантам реализации настоящего изобретения R7 представляет собой (C1-С6)алкил, предпочтительно (С1-С4)алкил, более предпочтительно (С1-С2)алкил, еще более предпочтительно метил. Алкил может быть замещен одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена и циано, предпочтительно галогена. Предпочтительно количество заместителей в замещенной группе R7 составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе R7 могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенной алкильной группы.
Согласно некоторым вариантам реализации настоящего изобретения R8 выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С1-С6)алкила, согласно другим вариантам реализации из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила и (С3-С10)гетероарил(С1-С6)алкила, согласно другим вариантам реализации из (С6-С10)арила и (С3-С10)гетероарила, согласно другим вариантам реализации из (С6-С10)арил(С1-С6)алкила и (С3-С10)гетероарил(С1-С6)алкила, согласно другим вариантам реализации из (С6)арила, (С3-С10)гетероарила, (С6)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С1-С6)алкила. Согласно некоторым вариантам реализации настоящего изобретения R8 представляет собой (С6-С10)арил, при этом предпочтительно арил выбран из группы замещенного или незамещенного нафтила и замещенного или незамещенного фенила, при этом более предпочтительно арил представляет собой замещенный или незамещенный фенил. Согласно некоторым вариантам реализации настоящего изобретения R8 представляет собой (С3-С10)гетероарил, согласно другим вариантам реализации настоящего изобретения (С3-С10)гетероарил содержит 1-3 атома азота, предпочтительно 1-2 атома азота, более предпочтительно 1 атом азота, такой как пиридин-2-ил, пиридин-3-ил, пиридин-4-ил. Согласно некоторым вариантам реализации настоящего изобретения R8 представляет собой (С6-С10)арил(С1-С6)алкил, согласно другим вариантам реализации настоящего изобретения (С6)арил(С1-С6)алкил, согласно другим вариантам реализации замещенный или незамещенный бензил, согласно другим вариантам реализации он выбран из группы, состоящей из бензила, 2-хлорбензила, 2-фторбензила, 3-хлорбензила, 3-фторбензила, 4-хлорбензила, 4-фторбензила, 2-метоксибензила, 3-метоксибензила, 4-метоксибензила, 2,3-диметоксибензила, 3,4-диметоксибензила, 2,4-диметоксибензила и 3,5-диметоксибензила, предпочтительно бензила и 3-хлорбензила, более предпочтительно бензила. Согласно некоторым вариантам реализации настоящего изобретения R8 представляет собой (С3-С10)гетероарил(С1-С6)алкил, при этом предпочтительно гетероарильная часть представляет собой кислородсодержащий гетероарил, такой как фуранил, или серосодержащий гетероарил, такой как тиофен, или азотсодержащий гетероарил, такой как индол или диазаиндол. Согласно другим вариантам реализации настоящего изобретения (С3-С10)гетероарил(С1-С6)алкил выбран из пиридин-2-ил-метила, пиридин-3-ил-метила, пиридин-4-ил-метила, 5-метилфуран-2-ил-метила, фуран-2-ил-метила, фуран-3-ил-метила, тиофен-2-ил-метила и тиофен-3-ил-метила, предпочтительно пиридин-2-ил-метила и фуран-2-ил-метила, более предпочтительно пиридин-2-ил-метила. Согласно некоторым вариантам реализации настоящего изобретения R8 представляет собой (C1-С6)алкил, предпочтительно (C1-С4)алкил, согласно другим вариантам реализации настоящего изобретения он выбран из группы, состоящей из метила, этила, пропила, изопропила, бутила, втор-бутила и трет-бутила, предпочтительно метила или этила. Арильная, гетероарильная, арилалкильная, гетероарильная и алкильная группа может быть замещена одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6, предпочтительно галогена и -OR6. Предпочтительно количество заместителей в замещенной группе В составляет один, два, три или четыре, более предпочтительно один, два или три, например, один или два. Заместители в замещенной группе В могут присутствовать на атомах углерода в любых положениях, как указано выше в целом в отношении замещенной арильной, гетероарильной, арилалкильной, гетероарил алкильной и алкильной группы.
Согласно некоторым вариантам реализации настоящего изобретения р представляет собой целое число от 1 до 2, согласно другим вариантам реализации настоящего изобретения р равно 1, согласно другим вариантам реализации настоящего изобретения р равно 2.
Согласно некоторым вариантам реализации настоящего изобретения q представляет собой целое число от 1 до 3, согласно другим вариантам реализации настоящего изобретения q равно 1, 2 или 3, согласно другим вариантам реализации настоящего изобретения q представляет собой целое число от 1 до 2, согласно другим вариантам реализации настоящего изобретения q равно 1, согласно другим вариантам реализации настоящего изобретения q равно 2, согласно другим вариантам реализации настоящего изобретения q равно 3.
Согласно некоторым вариантам реализации настоящего изобретения р равно 1 и q равно 1, согласно другим вариантам реализации настоящего изобретения р равно 1 и q равно 2, согласно другим вариантам реализации настоящего изобретения р равно 1 и q равно 3, согласно другим вариантам реализации настоящего изобретения р равно 2 и q равно 2, согласно другим вариантам реализации настоящего изобретения р равно 2 и q равно 3.
Согласно некоторым вариантам реализации настоящего изобретения соединение, имеющее структуру в соответствии с Формулой (I), представляет собой соединение, имеющее структуру в соответствии с Формулой (II)
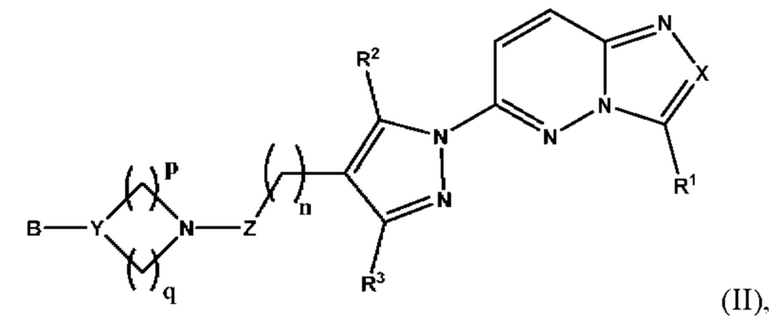
в которой R1, R2, R3, X, Y, Z, В, n, р и q определены выше и ниже.
Y представляет собой СН или N. Согласно некоторым вариантам реализации настоящего изобретения Y представляет собой СН, согласно другим вариантам реализации настоящего изобретения Y представляет собой N.
Согласно некоторым вариантам реализации настоящего изобретения соединение, имеющее структуру в соответствии с Формулой (I), представляет собой соединение, имеющее структуру в соответствии с Формулой (III)
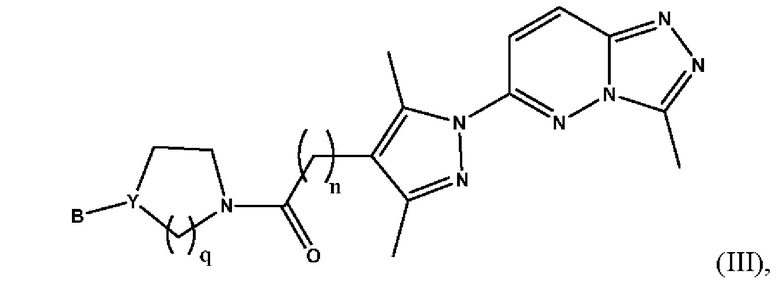
где Y, В, n и q определены выше и ниже.
Согласно некоторым вариантам реализации настоящего изобретения соединение, имеющее структуру в соответствии с Формулой (I), представляет собой соединение, имеющее структуру в соответствии с Формулой (IV)
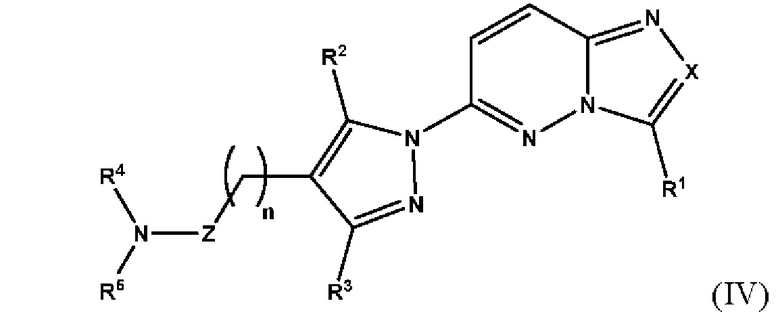
где R1, R2, R3, R4, R5, X, Z и n определены выше и ниже.
Согласно некоторым вариантам реализации настоящего изобретения А представляет собой 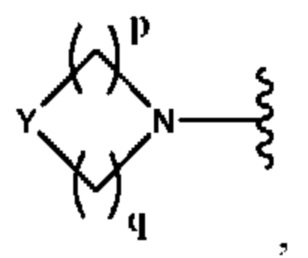 р равно 1 и q равно 1, согласно другим вариантам реализации настоящего изобретения А представляет собой
р равно 1 и q равно 1, согласно другим вариантам реализации настоящего изобретения А представляет собой 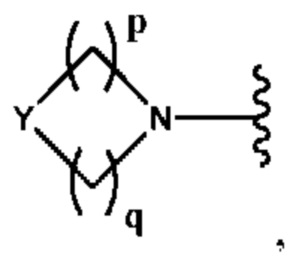 р равно 2 и q равно 1, согласно другим вариантам реализации настоящего изобретения А представляет собой
р равно 2 и q равно 1, согласно другим вариантам реализации настоящего изобретения А представляет собой 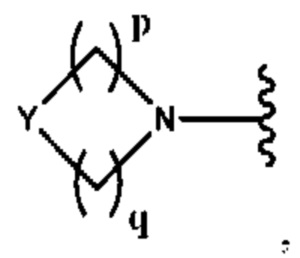 р равно 2 и q равно 2, согласно другим вариантам реализации настоящего изобретения А представляет собой
р равно 2 и q равно 2, согласно другим вариантам реализации настоящего изобретения А представляет собой 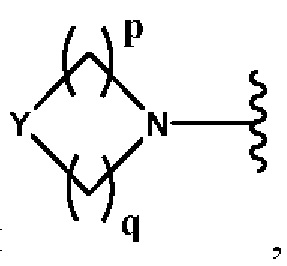 р равно 2 и q равно 3.
р равно 2 и q равно 3.
Согласно некоторым вариантам реализации настоящего изобретения соединение, имеющее структуру в соответствии с Формулой (I), характеризуется тем, что X выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила;
n представляет собой целое число от 0 до 3;
Z выбран из группы, состоящей из С=O, C=S и CH2;
А выбран из группы, состоящей из 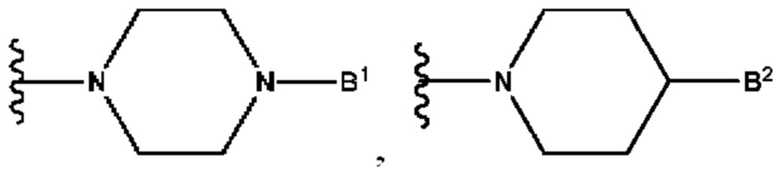 и
и 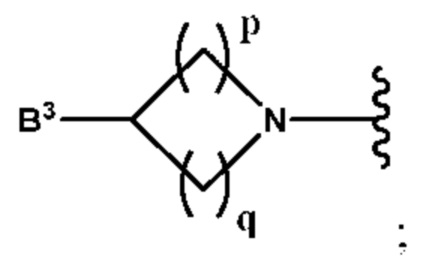
В1 представляет собой (С6-С10)арил(С1-С6)алкил или (С3-С10)гетероарил(С1-С6)алкил, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR6;
В2 представляет собой (С6-С10)арил(С1-С6)алкил, замещенный одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6, или
В2 представляет собой (С3-С10)гетероарил(С1-С6)алкил, который необязательно замещен одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6;
В3 выбран из группы, состоящей из водорода, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С1-С6)алкила и N(R7)(R8), которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6;
R6 представляет собой (С1-С4)алкил или (С1-С4)галогеналкил;
R7 представляет собой водород или (С1-С6)алкил;
R8 выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С1-С6)алкила;
р равно 1 и
q представляет собой целое число от 1 до 2, или
р равно 2 и
q равно 3;
или его фармацевтически приемлемая соль, сольват или гидрат.
Согласно некоторым вариантам реализации настоящего изобретения соединение, имеющее структуру в соответствии с Формулой (I), представляет собой соединение, имеющее структуру в соответствии с Формулой (II),
в которой
X выбран из группы, состоящей из N и СН, предпочтительно N, предпочтительно СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила, предпочтительно водорода, (С1-С6)алкила, (С3-С8)циклоалкила, (С6-С10)арила и (С6-С10)арил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила и (С3-С8)циклоалкила, предпочтительно (С1-С6)алкила и (C1-С6)галогеналкила;
n представляет собой целое число от 0 до 3, предпочтительно 1-3, более предпочтительно 1-2, еще более предпочтительно 2;
Z выбран из группы, состоящей из С=O, C=S и CH2, предпочтительно С=O;
Y представляет собой N или СН;
В выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (C1-С6)алкила и N(R7)(R8), которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (C1-С4)галогеналкила и -OR6, предпочтительно галогена и -OR6;
R7 представляет собой водород или (С1-С6)алкил, предпочтительно водород;
R8 выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (C1-С6)алкила, предпочтительно (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, более предпочтительно (С6-С10)арил(С1-С6)алкила;
р представляет собой целое число от 1 до 2;
q представляет собой целое число от 1 до 3;
или его фармацевтически приемлемую соль, сольват или гидрат.
Согласно некоторым вариантам реализации настоящего изобретения соединение, имеющее структуру согласно Формуле (I) или Формуле (II), характеризуется тем, что
R1 выбран из группы, состоящей из водорода, (С1-С4)алкила, (С3-С6)циклоалкила, (С6-С10)арила и (С6-С10)арил(С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из (С1-С4)алкила и (C1-С4)галогеналкила;
n представляет собой целое число от 1 до 3, предпочтительно 1-2, более предпочтительно 2; и
R5 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С3-С10)гетероарил(С1-С6)гетероалкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и OR6, предпочтительно галогена.
Согласно некоторым вариантам реализации настоящего изобретения соединение, имеющее структуру в соответствии с Формулой (I), представляет собой соединение, имеющее структуру в соответствии с Формулой (III)
в которой
n представляет собой целое число от 1 до 3, предпочтительно 1-2, более предпочтительно 2;
Y представляет собой N или СН;
В выбран из группы, состоящей из (С6-С10)арила, (С3-С10)гетероарила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила и (С1-С6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)галогеналкила и -OR6, предпочтительно галогена и -OR6;
q представляет собой целое число от 1 до 3, предпочтительно 1-2, более предпочтительно 2;
или его фармацевтически приемлемую соль, сольват или гидрат.
Согласно некоторым вариантам реализации настоящего изобретения соединение, имеющее структуру в соответствии с Формулой (I), представляет собой соединение, имеющее структуру в соответствии с Формулой (IV),
в которой
X выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из водорода, (С1-С6)алкила, (С1-С6)галогеналкила, (С3-С8)циклоалкила, (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарила и (С3-С10)гетероарил(С1-С6)алкила, предпочтительно водорода и (С1-С6)алкила, более предпочтительно (С1-С6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, (С1-С6)алкила, (C1-С6)галогеналкила и (С3-С8)циклоалкила, предпочтительно (С1-С6)алкила и (C1-С6)галогеналкила, более предпочтительно (С1-С6)алкила;
n представляет собой целое число от 0 до 3, предпочтительно 1-3, более предпочтительно 1-2, еще более предпочтительно 2;
Z выбран из группы, состоящей из С=O, C=S и СН2, предпочтительно С=O;
R4 представляет собой водород или (С1-С6)алкил, предпочтительно водород;
R5 выбран из группы, состоящей из (С6-С10)арил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)алкила, (С3-С10)гетероарил(С1-С6)гетероалкила и (С6-С10)арил(С1-С6)гетероалкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-С4)алкила, (С1-С4)галогеналкила и -OR6, предпочтительно галогена;
R6 представляет собой (С1-С4)алкил или (C1-С4)галогеналкил;
или его фармацевтически приемлемую соль, сольват или гидрат.
Согласно некоторым вариантам реализации настоящего изобретения соединение Формулы (I) выбрано из перечня, включающего:
1-(4-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперазин-1-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперазин-1-ил)пропан-1-он,
(R)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
(S)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол [4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-циклопропил-[1,2,4]триазол [4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-фенил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-(3-бензил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1 -он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазо[1,2-b]пиридазин-6-ил)-1Н-пир азол-4- ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)этан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-тион,
6-(4-(3-(4-бензилпиперидин-1-ил)пропил)-3,5-диметил-1Н-пиразол-1-ил)-3-метил-[1,2,4]триазол[4,3-b]пиридазин,
1-(азепан-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пир азол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-морфолинопропан-1-он,
N-(3-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-метилпропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол [4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(фуран-2-ил-метил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(2-фенилпропан-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-(3,4-диметоксибензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(нафтален-2-ил-метил)пиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилазетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-(бензиламино)азетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
Н-бензил-3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-карбоксамид,
(3-бензилпирролидин-1-ил)(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)метанон,
1-(3-бензилпирролидин-1-ил)-3-(5-изопропил-3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-5-(трифторметил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенилпиперазин- 1-ил)пропан- 1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-фенилпиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-((5-метилфуран-2-ил)метил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(2-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(3-фенилпропил)пропанамид,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(2,5-диметилфенил)пиперазин-1-ил)пропан-1-он,
N-(4-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(тиофен-2-ил-метил)пропанамид,
N-(2-(1Н-индол-3-ил)этил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
N-бензил-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-N-(3-фенилпропил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-этилпиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-метилпиперидин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(2-фторбензил)пропанамид, и
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(2-((фуран-2-ил-метил)тио)этил)пропанамид.
Согласно некоторым вариантам реализации настоящего изобретения соединение Формулы (I) выбрано из перечня, включающего:
1-(4-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперазин-1-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперазин-1-ил)пропан-1-он,
(N)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
(S)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-циклопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-фенил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-(3-бензил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазо[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)этан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-тион,
6-(4-(3-(4-бензилпиперидин-1-ил)пропил)-3,5-диметил-1Н-пиразол-1-ил)-3-метил-[1,2,4]триазол[4,3-b]пиридазин,
1-(азепан-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-морфолинопропан-1-он,
N-(3-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-метилпропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(фуран-2-ил-метил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(2-фенилпропан-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-(3,4-диметоксибензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(нафтален-2-ил-метил)пиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилазетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-(бензиламино)азетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
Н-бензил-3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-карбоксамид,
(3-бензилпирролидин-1-ил)(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)метанон,
1-(3-бензилпирролидин-1-ил)-3-(5-изопропил-3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он, и
1-(3-бензилпирролидин-1-ил)-3-(3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-5-(трифторметил)-1Н-пиразол-4-ил)пропан-1-он.
Согласно некоторым вариантам реализации настоящего изобретения соединение Формулы (I) выбрано из перечня, включающего:
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенилпиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-фенилпиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-((5-метилфуран-2-ил)метил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(2-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(3-фенилпропил)пропанамид,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(2,5-диметилфенил)пиперазин-1-ил)пропан-1-он,
N-(4-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(тиофен-2-ил-метил)пропанамид,
N-(2-(1Н-индол-3-ил)этил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
N-бензил-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-N-(3-фенилпропил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-этилпиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-метилпиперидин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(2-фторбензил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(2-((фуран-2-ил-метил)тио)этил)пропанамид.
Согласно некоторым вариантам реализации настоящего изобретения соединение Формулы (I) выбрано из перечня, включающего:
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенилпиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-фенилпиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-((5-метилфуран-2-ил)метил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(2-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(3-фенилпропил)пропанамид,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(2,5-диметилфенил)пиперазин-1-ил)пропан-1-он,
N-(4-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(тиофен-2-ил-метил)пропанамид,
N-(2-(1Н-индол-3-ил)этил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
N-бензил-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропанамид,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1H-(3-фенилпропил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-этилпиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-метилпиперидин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(2-фторбензил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-(2-((фуран-2-ил-метил)тио)этил)пропанамид,
1-(4-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперазин-1-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(4-фторбензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперазин-1-ил)пропан-1-он,
(R)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
(S)-1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-циклопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-фенил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-(3-бензил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазо[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)этан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-тион,
6-(4-(3-(4-бензилпиперидин-1-ил)пропил)-3,5-диметил-1Н-пиразол-1-ил)-3-метил-[1,2,4]триазол[4,3-b]пиридазин,
1-(азепан-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-морфолинопропан-1-он,
N-(3-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-N-метилпропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(фуран-2-ил-метил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(2-фенилпропан-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-(3,4-диметоксибензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(нафтален-2-ил-метил)пиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилазетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-(бензиламино)азетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
N-бензил-3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-карбоксамид,
(3-бензилпирролидин-1-ил)(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)метанон,
1-(3-бензилпирролидин-1-ил)-3-(5-изопропил-3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-5-(трифторметил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
1-бензил-4-(3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропаноил)пиперазин-2-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(2,2-диметилпирролидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(3,3-диметилпирролидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(3,3-диметилпирролидин-1-ил)пропан-1-он,
1-((1S)-2-азабицикло[2.2.1]гептан-2-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
(1S,5R)-3-(3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропаноил)-2,3,4,5-тетрагидро-1,5-метанпиридо[1,2-d][1,4]диазепин-7(1Н)-он,
1-((1S)-2-азабицикло[2.2.1]гептан-2-ил)-3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-(трифторметил)-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(3,3-диметилпирролидин-1-ил)пропан-1-он,
3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(3,3-диметилпирролидин-1-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диэтил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензил-1,4-диазепан-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3-изопропил-5-метил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
3-(1-(3-бензил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-циклопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперазин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метилимидазо[1,2-Ь]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпиперазин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазо[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазо[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)пропан-1-он,
2-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенэтилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метилимидазо[1,2-b]пиридазин-6-ил)-1Н-пиразол-4-ил)-1-(4-фенэтилпиперидин-1-ил)пропан-1-он.
Ряд соединений, включенных в объем настоящего изобретения или пригодных для применения в способах согласно настоящему изобретению, приведен в таблице 1 ниже.
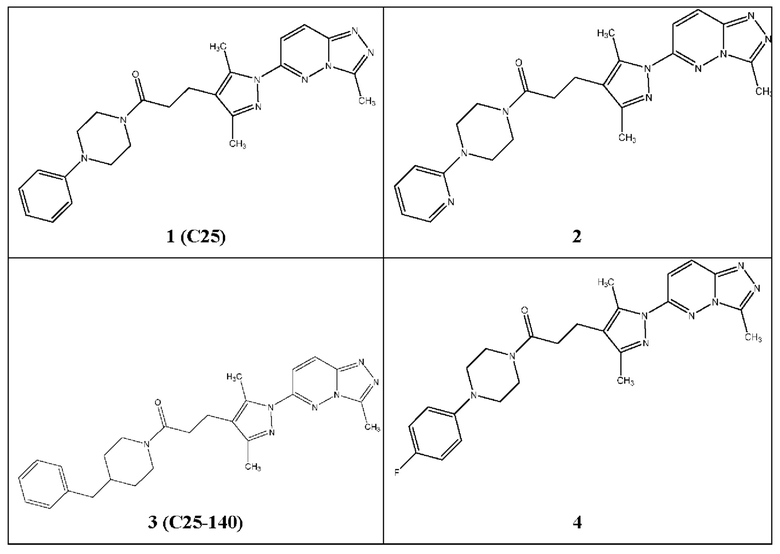
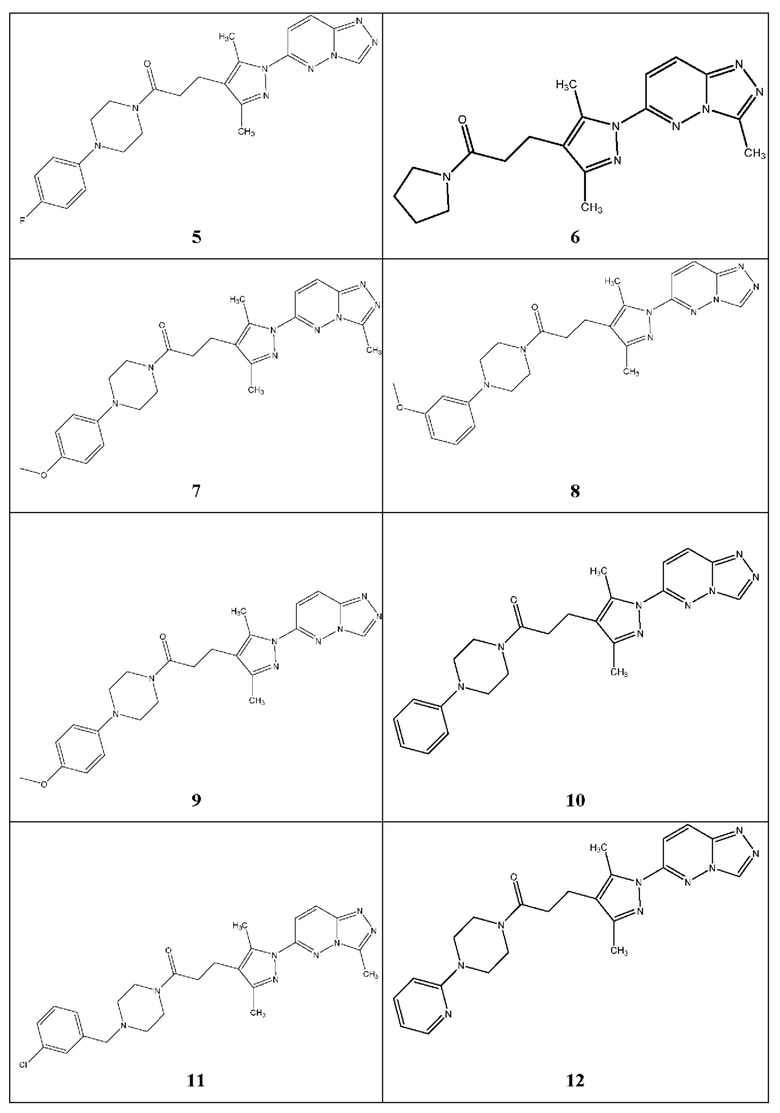
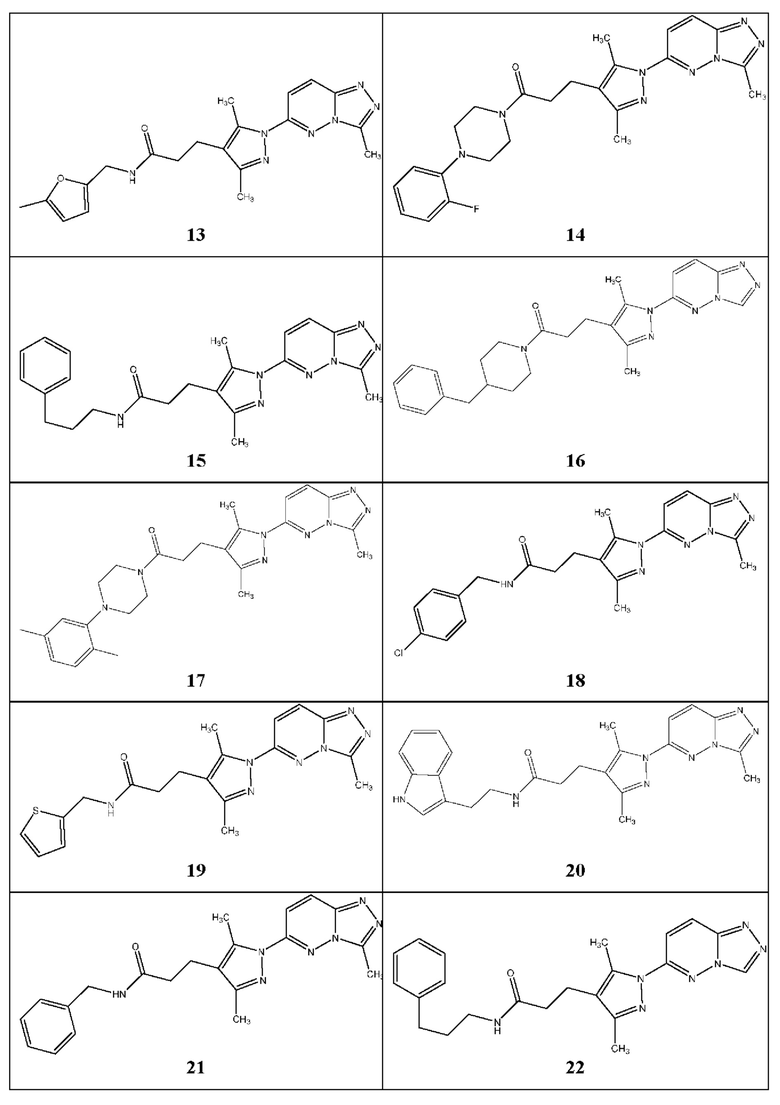
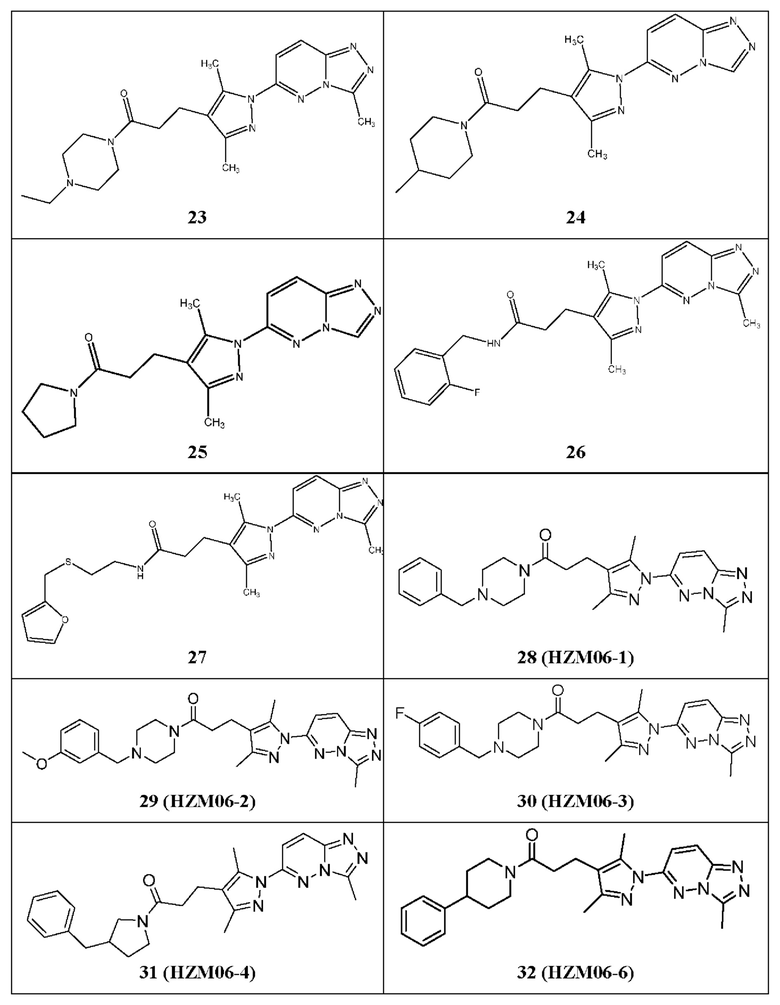
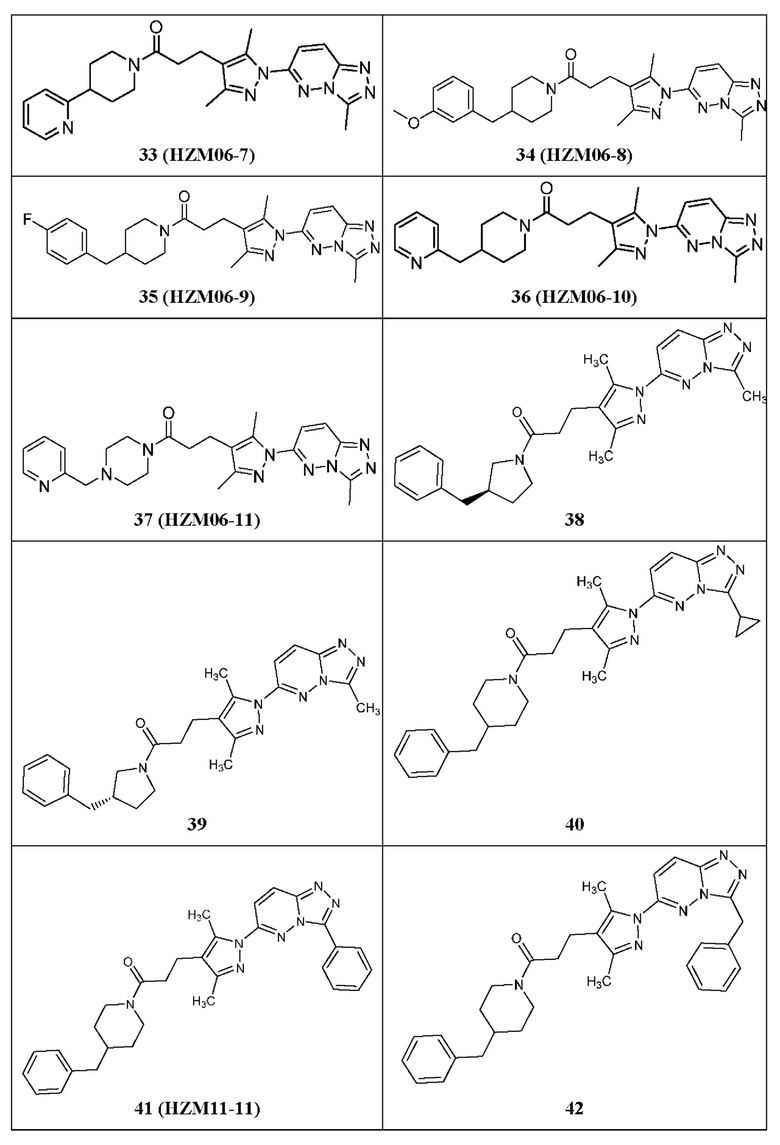
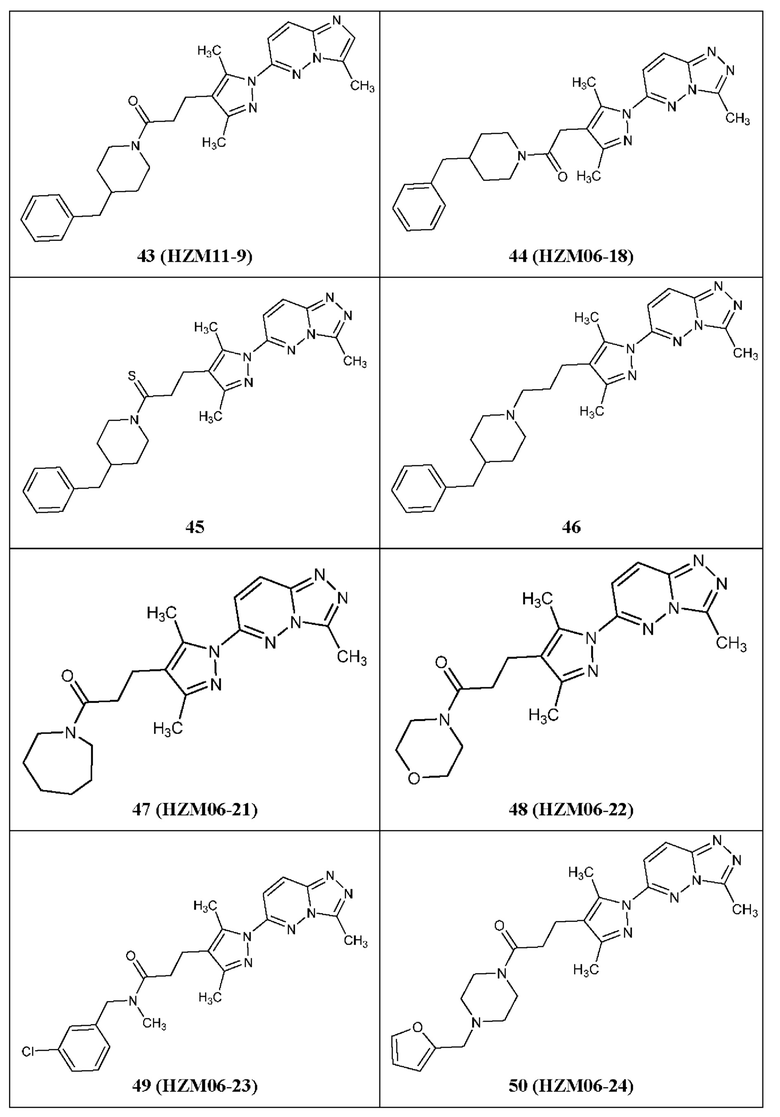
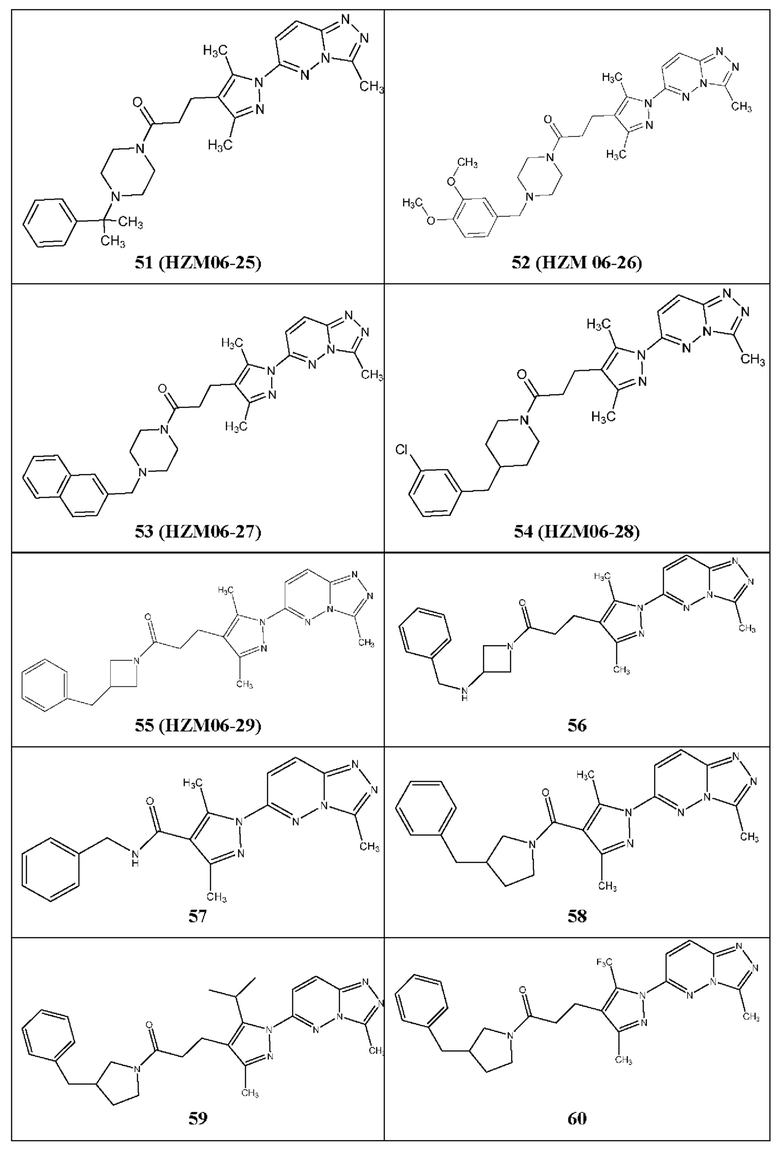
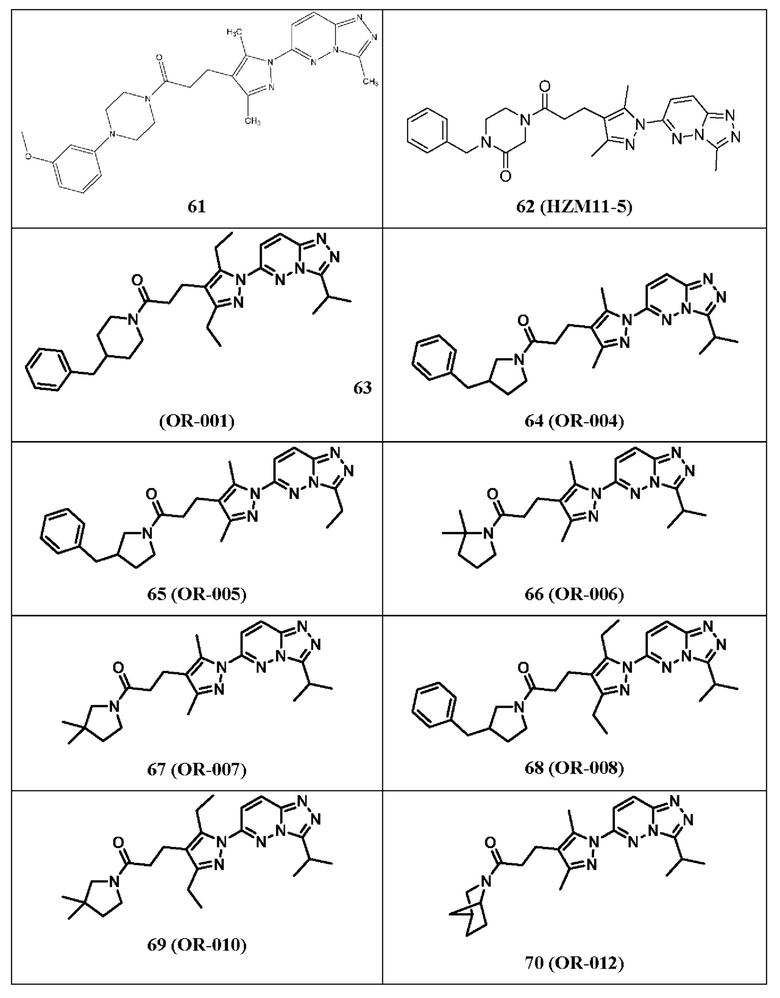
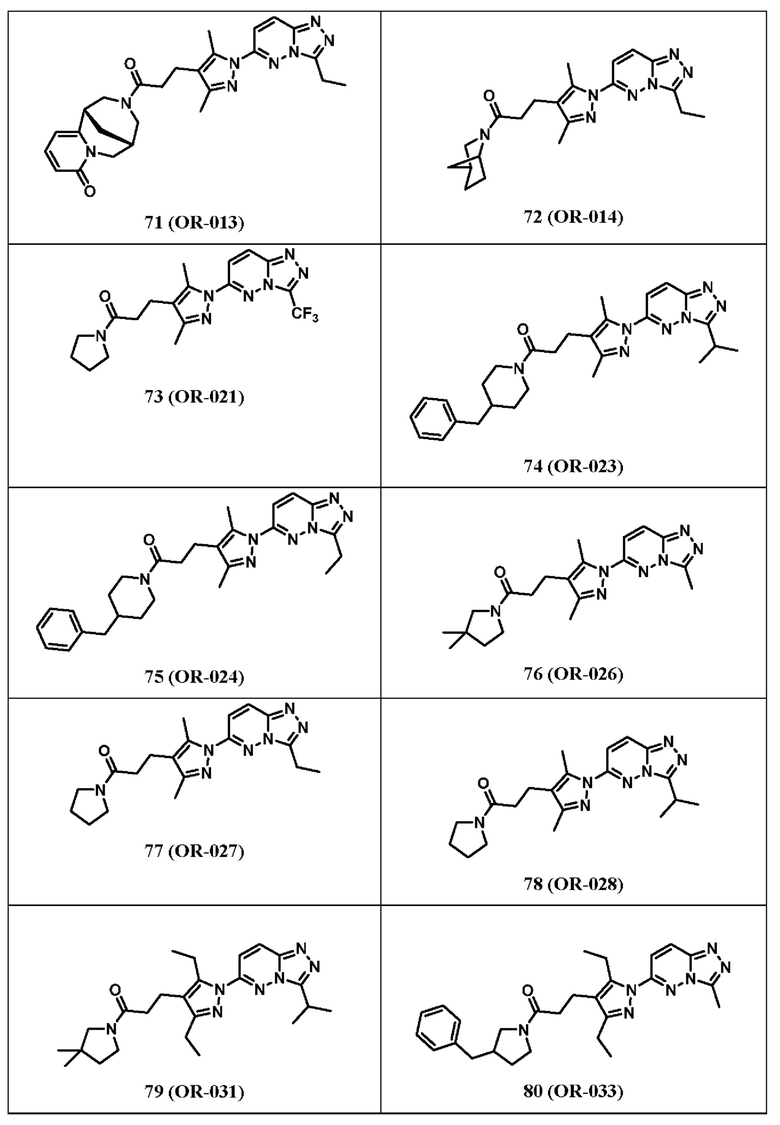
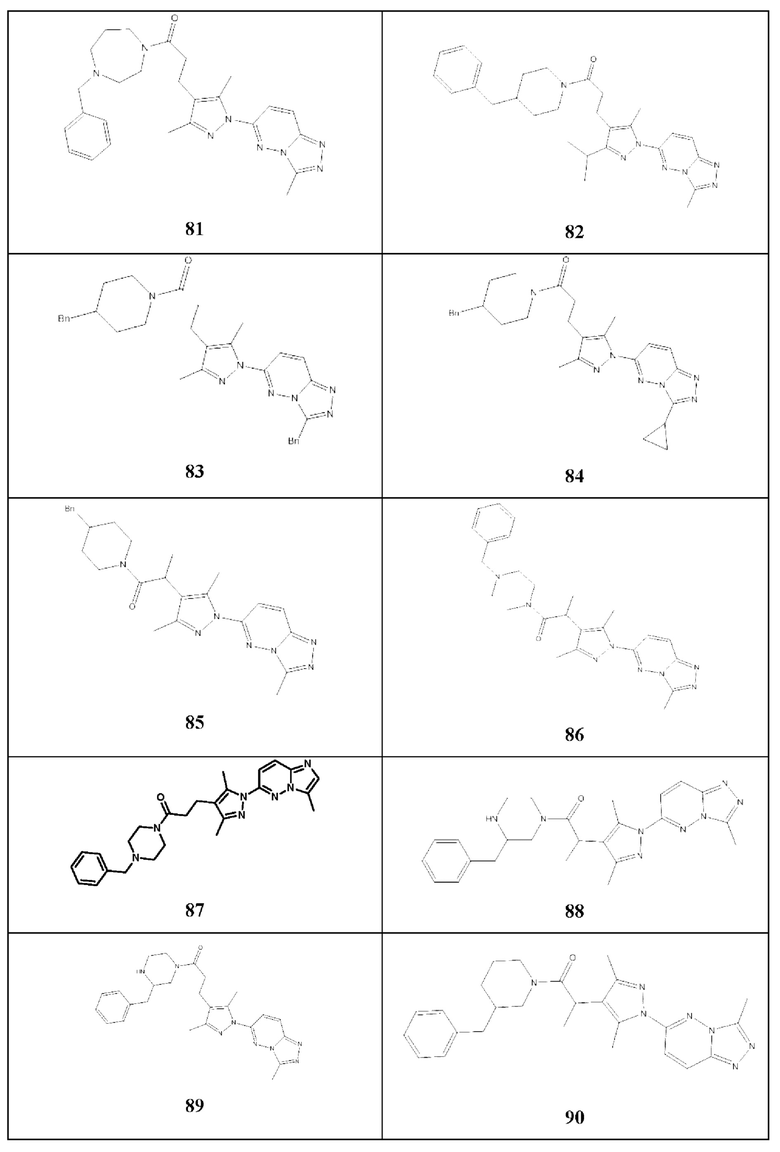
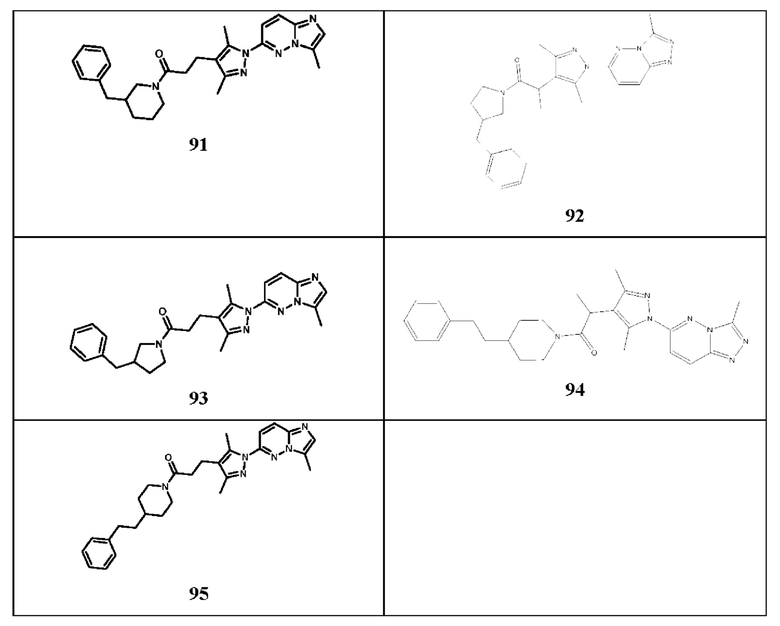
Согласно дополнительному аспекту настоящее изобретение относится к соединению согласно настоящему изобретению (в частности, к соединениям, указанным выше в отношении любой из Формул (I), (II), (III) и (IV)), а также к соединениям, представленным в таблице 1, для применения в качестве лекарственного средства.
Как следует из примеров, авторы настоящего изобретения обнаружили, что соединения согласно настоящему изобретению ингибируют или уменьшают взаимодействие TRAF6-Ubc13, в частности, соединения согласно настоящему изобретению ингибируют или уменьшают ферментативную активность TRAF6. Согласно некоторым вариантам реализации настоящего изобретения соединения согласно настоящему изобретению являются селективными ингибиторами TRAF6. Согласно некоторым вариантам реализации настоящего изобретения соединения согласно настоящему изобретению проявляют фармакологические свойства (биодоступность, токсичность, побочные эффекты, дозирование, приверженность пациента, совместимость, стабильность, период полужизни и т.д.), которые согласно по меньшей мере одному аспекту превосходят фармакологические свойства известных ингибиторов TRAF6.
В предпочтительных соединениях согласно настоящему изобретению любой один или более структурных элементов, таких как группы, заместители и числа, определены как в любом из предпочтительных определений элементов или в любом конкретном варианте реализации и/или могут иметь одно или более конкретных значений, которые упоминаются в качестве примеров элементов, причем все комбинации одного или более предпочтительных определений и вариантов реализации и/или конкретных значений являются объектом настоящего изобретения. Также в отношении всех предпочтительных соединений Формулы I, Формулы II, Формулы III и Формулы IV все их стереоизомерные формы и смеси стереоизомерных форм во всех соотношениях, а также физиологически приемлемые соли и физиологически приемлемые сольваты любого из них являются объектом настоящего изобретения. Аналогичным образом, также в отношении всех конкретных соединений, раскрытых в настоящем документе, таких как примеры соединений, которые представляют варианты реализации настоящего изобретения, где различные группы и числа в общем определении соединений Формулы I, Формулы II, Формулы III и Формулы IV имеют конкретные значения, присутствующие в соответствующем конкретном соединении, все их стереоизомерные формы и смеси стереоизомерных форм во всех соотношениях и их физиологически приемлемые соли, а также физиологически приемлемые сольваты любого из них являются объектом настоящего изобретения. В частности, объектом настоящего изобретения являются все конкретные соединения, раскрытые в настоящем документе, независимо от того, раскрыты ли они в форме свободного соединения и/или в форме конкретной соли, как в форме свободного соединения, так и в форме всех его физиологически приемлемых солей, и, если раскрыта конкретная соль, дополнительно в форме этой конкретной соли и ее физиологически приемлемых сольватов.
Соединения согласно настоящему изобретению могут быть получены, как описано ниже, или получены аналогичными способами, которые хорошо известны и доступны для специалиста в области органического синтеза. Синтетический подход к соединениям в соответствии с настоящим изобретением, т.е. соединениям Формул (I), (II), (III) и (IV), дополнительно иллюстрируется примерами соединений в описании.
В ходе синтеза соединений Формулы I гетероциклы Формулы V могут быть обработаны гидразином с получением соответствующих соединений Формулы VI с последующей конденсацией с соединением Формулы VII с получением соединения Формулы VIII, которое можно омылить с получением соединения Формулы IX, где группы X, R1, R2 и R3, а также число n определены, как в соединении Формулы I. LG1 представляет собой любую замещаемую группу, подходящую для химического превращения соединения V в соединение VI, такую как галогенид, тозилат и т.п. RL представляет собой любой остаток, подходящий для химической реакции соединения VI с соединением VII с образованием соединения VIII, т.е. стабильный в условиях реакции и отщепляемый в различных условиях реакции, что позволяет получить соединение IX, такой как алкильная группа, такая как метил или этил. Дополнительные функциональные группы могут присутствовать в защищенной форме или в форме группы-предшественника, которая позднее превращается в конечную группу. Применяемый гидразин для превращения соединения V в соединение VI может представлять собой гидразин или эквивалент гидразина, такой как гидрат гидразина или соль гидразина. Реакцию гидразина или эквивалента гидразина с соединением V обычно проводят при температуре от 0 до 150°С, предпочтительно от 30 до 100°С. Гидролитическая обработка реакционной смеси или выделение продукта с помощью фильтрации, которые как и обработку всех реакций при получении соединений Формулы I обычно можно осуществлять в стандартных условиях, затем приводят к получению соединения Формулы VI.
Реакцию конденсации соединения VI с соединением VII предпочтительно проводят в кислых условиях, например, в аквакислоте в качестве растворителя, с получением соединения Формулы VIII. Реакцию обычно проводят при температурах от приблизительно 30°С до 180°С, предпочтительно от 50°С до 150°С.
Соединение Формулы VIII превращают в соединение Формулы IX в кислых или основных условиях, предпочтительно в основных условиях. Омыление можно осуществлять в кислых условиях путем обработки соединения Формулы VIII сильными кислотами, такими как HBr, в подходящем растворителе. Омыление можно осуществлять в основных условиях путем обработки соединения Формулы VIII основанием, таким как NaOH, LiOH или KOH, в подходящем растворителе. Реакцию обычно осуществляют при температуре от приблизительно 0°С до 150°С, предпочтительно от 30°С до 130°С.
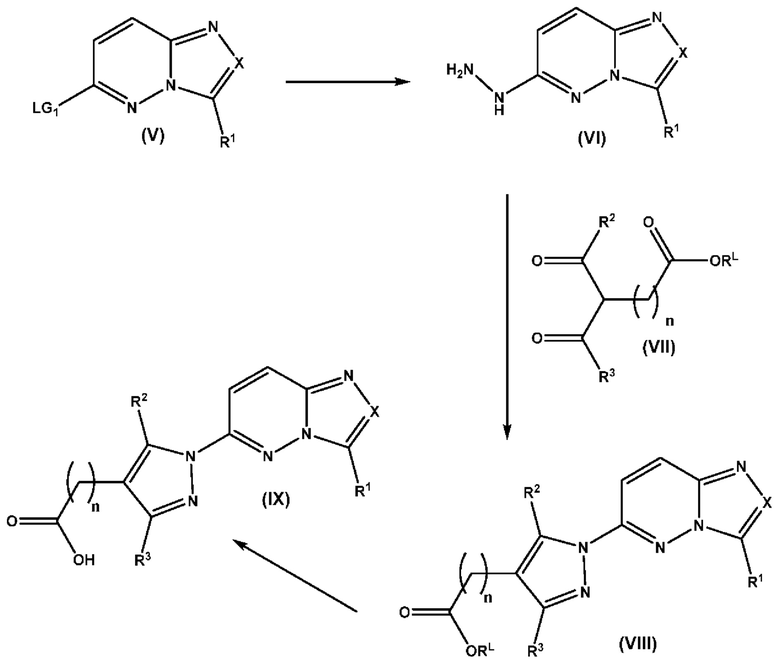
Соединения Формулы IX затем могут быть связаны в стандартных условиях для образования амидной связи с соединениями Формулы X с получением соединения Формулы XI. Группы R1, R2, R3, X и Y и числа n, р и q определены как и в соединениях Формулы I, и дополнительные функциональные группы могут присутствовать в защищенной форме или в форме группы-предшественника, которая позже превращается в конечную группу. Соединения Формулы IX определены выше. Для образования амидной связи карбоновую кислоту Формулы IX обычно превращают в реакционноспособное производное, которое может быть выделено или получено in situ или активировано in situ с помощью обычного реагента для связывания амидов.
Например, соединение Формулы IX может быть превращено в хлорид карбоновой кислоты с помощью обработки тионилхлоридом, оксалилхлоридом или (1-хлор-2-метилпропенил)диметиламином, в реакционноспособный сложный эфир или в смешанный ангидрид с помощью обработки алкилхлорформиатом, таким как этилхлорформиат или изобутилхлорформиат. Согласно другому варианту соединение Формулы IX может быть активировано с помощью реагента, такого как ангидрид пропанфосфоновой кислоты, N,N'-карбонилдиазола, такого как N,N'-карбонилдиимидазол (CDI), карбодиимида, такого как N,N'-диизопропилкарбодиимид (DIC), N,N'-дициклогексилкарбодиимид (DCC) или гидрохлорид N-этил-N'-(3-диметиламинопропил)карбодиимида (EDC), карбодиимида вместе с добавкой, такой как 1-гидроксибензотриазол (НОВТ) или 1-гидрокси-7-азабензотриазол, связывающего реагента на основе урония, такого как тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-N'-тетраметилурония (HBTU) или тетрафторборат О-(циано(этоксикарбонил)метиленамино)-N,N,N',N'-тетраметилурония (TOTU), или связывающего реагента на основе фосфония, такого как гексафторфосфат (бензотриазол-1-илокси)трис(диметиламино)фосфония (ВОР), гексафторфосфат (бензотриазол-1-илокси)трипирролидинфосфония (РуВОР) или гексафторфосфат бромтрипирролидинфосфония (PyBroP). Активацию соединения Формулы IX и реакцию активированного соединения Формулы IX или реакционноспособного производного карбоновой кислоты с соединением Формулы X обычно осуществляют в инертном растворителе, таком как простой эфир, такой как тетрагидрофуран (ТГФ), диоксан или 1,2-диметоксиэтан (ДМЭ), или углеводороде, таком как толуол, или хлорированном углеводороде, таком как дихлорметан или хлороформ, или амиде, таком как диметилформамид (ДМФА) или N-метилпирролидин-2-он (NMP), например, или в смеси растворителей, при температуре от приблизительно 0°С до приблизительно 60°С в присутствии подходящего основания, такого как третичный амин, такой как триэтиламин, этилдиизопропиламин, N-метилморфолин или пиридин, или соединения основного щелочного металла, например, такого как карбонат щелочного металла, такой как карбонат натрия, карбонат калия или карбонат цезия. Карбоновые кислоты Формулы IX, или соединения, которые вместо группы карбоновой кислоты, изображенной в Формуле IX, содержат производную группу карбоновой кислоты, например, хлоридную группу карбоновой кислоты, могут быть получены с помощью омыления из соответствующих сложных эфиров, таких как 5-этил-4-гидрокси-1-метил-2-оксо-1,2-дигидрохинолин-3-карбоновая кислота.
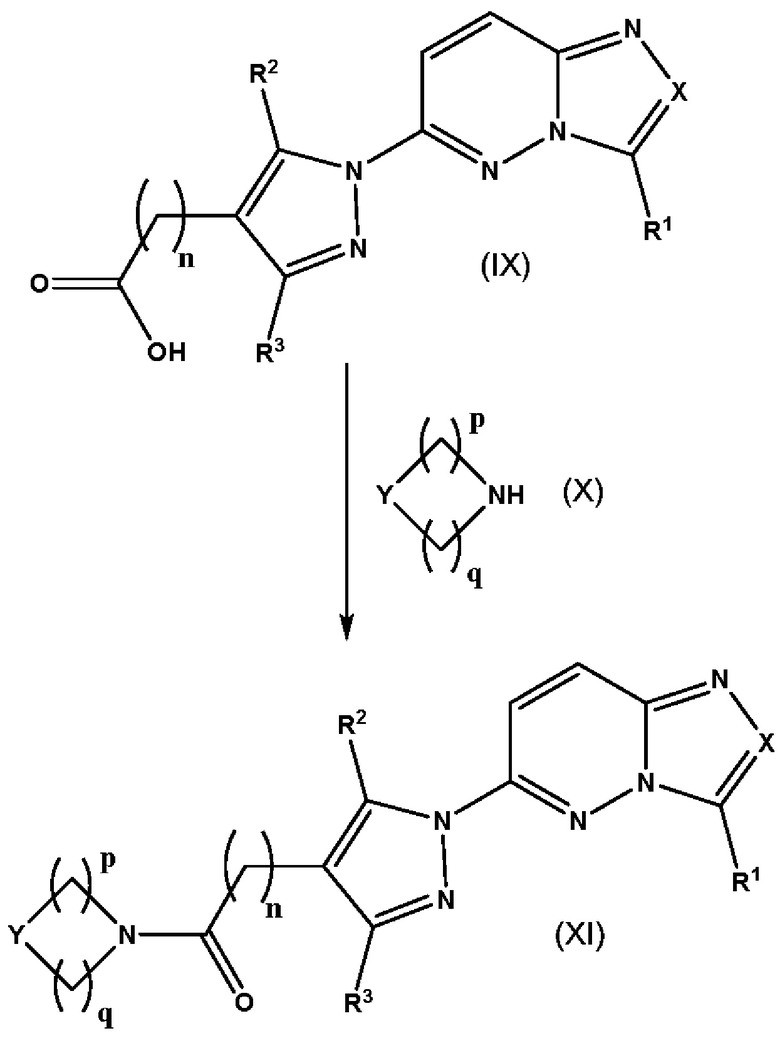
В том случае, если все группы Формулы XI имеют желаемые значения, включенные в определение соединений Формулы I, полученное таким способом соединение Формулы XI уже является готовым соединением Формулы I. В том случае, если любая из групп Формулы XI присутствует в защищенной форме или в форме группы-предшественника, полученное таким способом соединение Формулы XI может быть превращено в конечном итоге в желаемое соединение Формулы I путем удаления защитных групп и/или превращения любых других групп. Как указано выше, для того чтобы избежать нежелательного протекания реакции или побочных реакций на любой одной или более стадиях синтеза соединений Формулы I, функциональные группы могут присутствовать в защищенной форме или в форме группы-предшественника. Кроме того, на заключительной стадии синтеза соединения Формулы I защитные группы также могут быть удалены, и группы-предшественники могут быть превращены, на других стадиях синтеза. Соответствующие стратегии синтеза и подробная информация о подходящих защитных группах и их введении и удалении хорошо известны специалисту в данной области техники и могут быть найдены, например, в P. G. М. Wuts and Т. W. Greene, Greene's Protective Groups in Organic Synthesis, 4. ed. (2007), John Wiley & Sons.
Помимо этого, чтобы получить дополнительные соединения Формулы I, в соединениях Формулы I или соединениях Формулы XI или других соединениях, встречающихся при синтезе соединений Формулы I, можно осуществлять различные другие превращения функциональной группы.
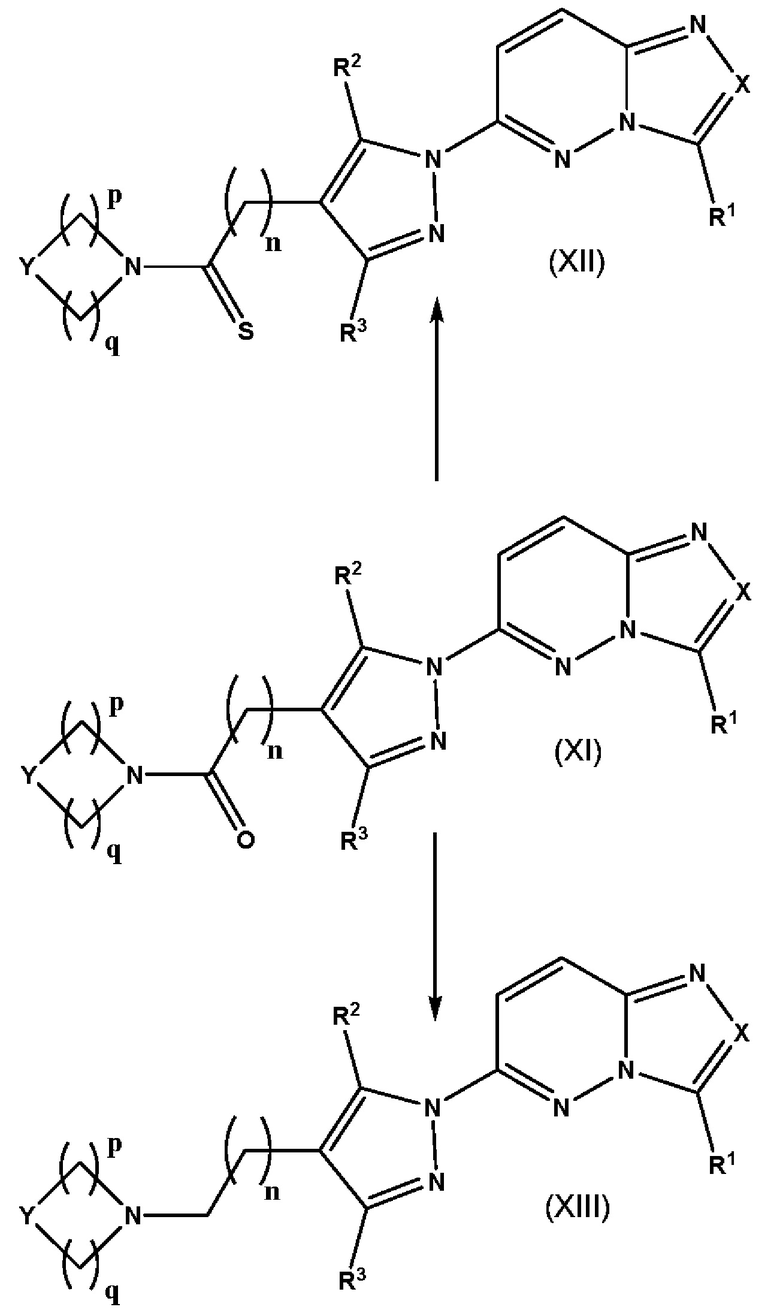
Соединения Формулы XI также могут быть превращены в соединение Формулы XII или XIII. Группы R1, R2, R3, X и Y и числа n, р и q определены как и в соединениях Формулы I, и дополнительные функциональные группы могут присутствовать в защищенной форме или в форме группы-предшественника, которую затем превращают в конечную группу. Соединения Формулы XI определены выше.
Превращение карбонильной группы соединения Формулы XI в тиокарбонильную группу соединения Формулы XII можно осуществлять с помощью различных реагентов, известных в данной области техники, таких как реагент Лавессона, фосфодитионат аммония или система PSCl3/H2O/Et3N. Применяемый растворитель и время реакции зависят от используемого реагента или комбинации реагентов.
Восстановление карбонильной группы соединения Формулы XI с получением соединения Формулы XII можно осуществлять с помощью различных реагентов, известных в данной области техники, таких как опосредуемое металлом восстановление, такое как катализируемое платиной восстановление, катализируемое цинком восстановление, восстановление, опосредуемое комплексами, такими как трис(пентафторфенил)бор В(C6F5)3 или [Ir(СОЕ)2Cl]2, или восстановление за счет переноса гидрида с помощью реагентов, таких как гидрат лития-алюминия (LiAlH4).
В том случае, если все группы Формул XII и XIII имеют желаемые значения, включенные в определение соединений Формулы I, полученное таким способом соединение Формул XII и XIII уже является конечным соединением Формулы I. В том случае, если любая из групп Формул XII и XIII присутствует в защищенной форме или в форме группы-предшественника, полученное таким способом соединение Формул XII и XIII может быть в конечном итоге превращено в желаемое соединение Формулы I путем удаления защитных групп и/или превращения любых других групп.
Исходные соединения и структурные звенья для синтеза соединений Формулы I являются коммерчески доступными или могут быть получены в соответствии с описанными в литературе методиками или аналогичными им методиками.
Порядок введения групп в ходе синтеза соединения Формулы I также может отличаться от указанных выше. Например, тиокарбонильная группа соединения Формулы XII может быть введена путем превращения карбонильной группы соединения Формулы VIII в соответствующий тиоэфир. В частности, другие пути синтеза можно фактически применять к определенным вариантам реализации соединений, раскрытых в настоящем документе. Специалист с обычной квалификацией может обратиться к общим руководствам, таким как March's Advanced Organic Chemistry (Michael В. Smith & Jerry March, Wiley-Interscience, 2000), The Practice of Medicinal Chemistry (Camile G. Wermuth, Academia Press, 2003) и Protective Groups in Organic Synthesis (Theosora W. Greene & Peter G.M. Wuts; John Wiley & Sons Inc, 1999).
Фармацевтические композиции
Согласно дополнительному аспекту настоящее изобретение относится к фармацевтической композиции, содержащей соединение, указанное выше под заголовком «Соединения», и одно или более фармацевтически приемлемых вспомогательных веществ.
Соединения, описанные в настоящем изобретении (в частности, те, которые указаны выше, такие как соединения Формулы (I), (II), (III) и/или (IV), а также соединения, представленные в таблице 1), предпочтительно вводят пациенту, нуждающемуся в этом, с помощью фармацевтической композиции. Согласно одному варианту реализации настоящего изобретения фармацевтическая композиция содержит соединение, описанное выше (например, соединение, имеющее общую Формулу (I), (II), (III) и/или (IV), а также соединения, представленные в таблице 1, или их гидрат, сольват, соль, комплекс, рацемическую смесь, диастереомер, энантиомер или таутомер или обогащенную изотопами форму любого из вышеперечисленных), и одно или более фармацевтически приемлемых вспомогательных веществ.
Фармацевтическую композицию можно вводить индивидууму любым путем, таким как энтеральный или парентеральный путь.
В настоящем документе выражения «энтеральное введение» и «введенный энтерально» означают, что введенный лекарственный препарат поглощается желудком и/или кишечником. Примеры энтерального введения включают пероральное и ректальное введение. В настоящем документе выражения «парентеральное введение» и «введенный парентерально» означают способы введения, отличные от энтерального введения, обычно путем инъекции или местного применения, и включают, но не ограничиваются ими, внутривенное, внутримышечное, внутриартериальное, интратекальное, внутрикапсулярное, внутрикостное, внутриорбитальное, внутрисердечное, внутрикожное, внутрибрюшинное, транстрахеальное, подкожное, подкутикулярное, внутрисуставное, подкапсулярное, внутримозговое, интрацеребровентрикулярное, субарахноидальное, внутрипозвоночное, эпидуральное и внутригрудинное введение (например, путем инъекции и/или инфузии), а также местное введение (например, накожное, путем ингаляции или через слизистые оболочки (такое как буккальное, сублингвальное или вагинальное введение)).
Соединения, применяемые в настоящем изобретении, обычно применяют в «фармацевтически приемлемых количествах» и в «фармацевтически приемлемых препаратах». Такие композиции могут содержать соли, буферы, консерванты, носители и необязательно другие терапевтические агенты.
В настоящем документе термин «вспомогательное вещество» предназначен для обозначения всех веществ в фармацевтической композиции, которые не являются активными ингредиентами (например, которые являются терапевтически неактивными ингредиентами, которые не проявляют какое-либо терапевтическое действие в применяемом количестве/концентрации), таких как, например, носители, связывающие вещества, смазывающие вещества, загустители, поверхностно-активные агенты, консерванты, эмульгаторы, буферы, ароматизаторы, красители или антиоксиданты.
Композиции, описанные в настоящем изобретении, могут содержать фармацевтически приемлемый носитель. В настоящем документе термин «фармацевтически приемлемый носитель» включает любые и все растворители, дисперсионные среды, покрытия, изотонические агенты и агенты, замедляющие всасывание, и аналогичные агенты, которые являются физиологически совместимыми. «Фармацевтически приемлемый носитель» может быть твердым, полутвердым, жидким или может представлять собой комбинацию указанных форм. Предпочтительно носитель подходит для энтерального (такого как пероральное) или парентерального введения (такого как внутривенное, внутримышечное, подкожное, спинальное или эпидермальное введение (например, путем инъекции или инфузии)). В зависимости от пути введения активное вещество, т.е. соединение, применяемое в настоящем изобретении, как по отдельности, так и в комбинации с одним или более дополнительными активными соединениями, может быть покрыто материалом для защиты активного(ых) соединения (й) от действия кислот и других природных условий, которые могут инактивировать активное соединение.
Примеры подходящих водных и неводных носителей, которые можно применять в фармацевтических композициях, применяемых в соответствии с настоящим изобретением, включают воду (например, воду для инъекций), этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.), водные растворы соли, углевода, сахарного спирта или аминокислоты (такие как физиологический солевой раствор или водный раствор аминокислоты) и их подходящие смеси и/или забуференные формы, растительные масла (такие как оливковое масло) и инъецируемые органические сложные эфиры (такие как этилолеат). Надлежащую текучесть можно поддерживать, например, за счет применения материалов для покрытия, таких как лецитин, за счет поддержания требуемого размера частиц в случае дисперсий и за счет применения поверхностно-активных веществ.
Фармацевтически приемлемые носители включают стерильные водные растворы или дисперсии и стерильные порошки для приготовления стерильных инъекционных растворов или дисперсий для немедленного применения. Применение таких сред и агентов для фармацевтически активных соединений известно в данной области техники. За исключением случаев, когда какая-либо обычная среда или агент несовместимы с активным соединением, предусмотрено их применение в фармацевтических композициях, применяемых в соответствии с настоящим изобретением.
Дополнительные активные соединения могут быть введены вместе, до или после соединения, применяемого в настоящем изобретении (в частности, соединения, указанного выше, такого как соединения Формул (I), (II), (III) и/или (IV), а также соединений, представленных в таблице 1, или включенных в композиции). Согласно одному варианту реализации настоящего изобретения фармацевтическая композиция, описанная в настоящем документе, содержит описанное выше соединение (например, имеющее общую Формулу (I), (II), (III) и/или (IV)), а также соединения, представленные в таблице 1, или их гидрат сольват, соль, комплекс, рацемическую смесь, диастереомер, энантиомер или таутомер или обогащенную изотопами форму любого из перечисленных выше), по меньшей мере одно дополнительное активное соединение и одно или более фармацевтически приемлемых вспомогательных веществ.
«Дополнительное активное соединение» (которое не является соединением, имеющим Формулы (I), (II), (III) и/или (IV), а также соединениями, представленными в таблице 1, как указано в настоящем документе), может быть выбрано из любого соединения, которое можно применять в лечении рака и/или иммунных заболеваний. Дополнительное активное соединение может индуцировать аддитивный или синергетический терапевтический эффект.
Фармацевтическая композиция, описанная в настоящем документе, может содержать, в дополнение к соединению, имеющему структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединениям, представленным в таблице 1, как описано выше, по меньшей мере одно, например, 1, 2, 3, 4, 5, 6, 7 или 8 дополнительных активных соединений. В соответствии с идеей настоящего изобретения по меньшей мере дополнительное активное соединение, например, противораковый лекарственный препарат, и/или агент для лечения иммунных заболеваний, может быть изготовлено вместе с соединением, имеющим структуру в соответствии с Формулами (I), (II)), (III) и/или (IV), или соединениями, представленными в таблице 1, описанными выше, в одной фармацевтической композиции. Согласно другому варианту фармацевтическая композиция может быть структурирована как набор частей, в котором соединение, имеющее структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленные в таблице 1, обеспечены в первом составе и по меньшей мере одно дополнительное активное соединение, например, противораковый лекарственный препарат и/или агент для лечения иммунных заболеваний, обеспечено во втором составе, т.е. во второй фармацевтической композиции. Первую и вторую фармацевтические композиции можно комбинировать перед применением. Другими словами, перед введением фармацевтической композиции состав, содержащий дополнительное активное соединение, может быть добавлен к первой фармацевтической композиции, содержащей соединение, имеющее структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленные в таблице 1, описанные выше. В другом варианте согласно идее настоящего изобретения предусмотрено введение соединения, имеющего структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединений, представленных в таблице 1, описанных выше, изготовленных в виде первой фармацевтической композиции, и введение по меньшей мере одного дополнительного активного соединения, изготовленного в виде второй фармацевтической композиции. Фармацевтические композиции могут быть введены одновременно или последовательно. Например, первую фармацевтическую композицию можно вводить в первый момент времени, и вторую фармацевтическую композицию можно вводить во второй момент времени, причем моменты времени могут быть разделены, например, интервалом в 0 или до 1, 2, 3, 4, 5 или 10 минут, до 1, 2, 3, 4, 5 или 10 часов, до 1, 2, 3, 4, 5 или 10 дней, до 1, 2, 3, 4, 5 или 10 недель, до 1, 2, 3, 4, 5 или 10 месяцев или до 1, 2, 3, 4, 5 или 10 лет. Согласно некоторым вариантам реализации настоящего изобретения набор частей содержит соединение, имеющее структуру в соответствии с Формулой (I), (II), (III) и/или (IV), или соединения, представленные в таблице 1, описанные выше, или фармацевтическую композицию, содержащую соединение, имеющее структуру в соответствии с Формулой (I), (II), (III) и/или (IV), или соединения, представленные в таблице 1, описанные выше, и по меньшей мере один фармацевтически приемлемый носитель.
Композиции также могут содержать адъюванты, такие как консерванты, смачивающие агенты, эмульгирующие агенты, забуферивающие рН агенты и диспергирующие агенты. Предотвращение присутствия микроорганизмов может быть обеспечено с помощью методик стерилизации и/или с помощью включения различных антибактериальных и противогрибковых агентов, например, парабена, хлорбутанола, фенолсорбиновой кислоты и т.п. Желательным также может быть включение в композиции изотонических агентов, таких как сахара, хлорид натрия и т.п. Помимо этого пролонгированное всасывание инъецируемой фармацевтической формы может быть достигнуто за счет включения агентов, которые замедляют всасывание, таких как моностеарат алюминия и желатин.
Независимо от выбранного пути введения активные соединения, которые можно применять в подходящей гидратированной форме, и/или фармацевтические композиции, применяемые в соответствии с настоящим изобретением, изготовлены в виде фармацевтически приемлемых лекарственных форм с помощью обычных способов, известных специалистам в данной области техники (см., например, Remington, «The Science and Practice of Pharmacy» под ред. Allen, Loyd V., Jr., 22 изд., Pharmaceutical Sciences, September 2012; Ansel et al., «Pharmaceutical Dosage Forms and Drug Delivery Systems», 7 изд., Lippincott Williams & Wilkins Publishers, 1999).
Фармацевтическая композиция может быть введена различными способами, известными в данной области техники. Специалист в данной области техники поймет, что путь и/или способ введения будут изменяться в зависимости от желаемых результатов. Фармацевтические композиции, содержащие одно или более активных соединений, могут быть получены с носителями, которые будут защищать одно или более активных соединений от быстрого высвобождения, например, состав с контролируемым высвобождением, включая имплантаты, трансдермальные пластыри и микроинкапсулированные системы доставки. Можно применять биоразлагаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Способы получения таких композиций обычно известны специалистам в данной области техники. См., например, Sustained and Controlled Release Drug Delivery Systems, J.R. Robinson, ed., Marcel Dekker, Inc., New York, 1978.
Для введения соединения, применяемого в настоящем изобретении, с помощью определенных путей введения может быть необходимо покрыть соединение материалом или ввести соединение совместно с ним, чтобы предотвратить инактивацию соединения. Например, соединение может быть введено индивидууму в подходящем носителе, например, в липосомах или разбавителе. Фармацевтически приемлемые разбавители включают физиологический солевой раствор и водные буферные растворы. Липосомы включают эмульсии CGF вода-в-масле-в-воде, а также обычные липосомы (Strejan et al., J. Neuroimmunol. 7: 27(1984)).
Фармацевтические композиции обычно являются стерильными и стабильными в условиях изготовления и хранения. Композиция может быть изготовлена в виде раствора, микроэмульсии, липосомы или другой упорядоченной структуры, подходящей для высокой концентрации лекарственного препарата. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль и т.п.) и их подходящие смеси. Надлежащую текучесть можно поддерживать, например, за счет применения покрытия, такого как лецитин, за счет поддержания требуемого размера частиц в случае дисперсии и за счет применения поверхностно-активных веществ. Во многих случаях предпочтительным является включение в композицию изотонических агентов, например, сахаров, полиспиртов, таких как маннит, сорбит, или хлорида натрия. Пролонгированное всасывание инъецируемых композиций может быть достигнуто за счет включения в композицию агента, который задерживает всасывание, например, моностеаратных солей и желатина.
Инъецируемая композиция должна быть стерильной и жидкой в такой степени, чтобы композиция могла быть доставлена с помощью шприца. В дополнение к воде носитель может представлять собой изотонический забуференный солевой раствор, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль и т.п.) и их подходящие смеси. Стерильные инъецируемые растворы могут быть получены с помощью включения активного соединения в необходимом количестве в подходящем растворителе с одним или комбинацией ингредиентов, перечисленных выше, по мере необходимости, с последующей стерилизацией с помощью микрофильтрации.
Обычно дисперсии получают с помощью включения активного соединения в стерильный носитель, который содержит основную дисперсионную среду и другие необходимые ингредиенты, выбранные из тех, которые перечислены выше. В случае стерильных порошков для получения стерильных инъецируемых растворов предпочтительными способами получения являются вакуумная сушка и сублимационная сушка (лиофилизация), которые позволяют получать порошок активного ингредиента с любым дополнительным желаемым ингредиентом из его раствора, предварительно стерилизованного с помощью фильтрации.
Схемы дозирования корректируют для обеспечения оптимального желаемого ответа (например, терапевтического ответа). Например, может быть введен один болюс, несколько разделенных доз могут быть введены с течением времени, или доза может быть пропорционально уменьшена или увеличена, если этого требует терапевтическая ситуация. Особенно предпочтительным является изготовление парентеральных композиций в виде стандартной лекарственной формы для простоты введения и единообразия дозировки. В настоящем документе стандартная лекарственная форма относится к физически дискретным единицам, пригодным в качестве единичных доз для индивидуумов, подлежащих лечению; каждая единица содержит заранее определенное количество активного соединения, рассчитанное для получения желаемого терапевтического действия, в сочетании с необходимым фармацевтическим носителем. Спецификация для стандартных лекарственных форм, применяемых в соответствии с настоящим изобретением, обусловлена и напрямую зависит от (а) уникальных характеристик активного соединения и конкретного терапевтического действия, которое должно быть достигнуто, и (и) ограничений, присущих уровню техники для составления смесей такого активного соединения для лечения чувствительности у людей.
Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и т.п.; (2) маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (ВНА), бутилированный гидрокситолуол (ВНТ), лецитин, пропилгаллат, альфа-токоферол и т.п.; и (3) хелатирующие металлы агенты, такие как лимонная кислота, этилендиаминтетрауксусная кислота (ЭДТА), сорбит, винная кислота, фосфорная кислота и т.п.
В случае терапевтических/фармацевтических составов композиции, применяемые в соответствии с настоящим изобретением, включают композиции, подходящие для энтерального введения (такого как пероральное или ректальное) или парентерального введения (такого как назальное, местное (включая вагинальное, буккальное и сублингвальное)). Композиции могут быть удобно представлены в стандартной лекарственной форме и могут быть получены любыми способами, известными в области фармации. Количество активного ингредиента (в частности, количество соединения, применяемого в соответствии с настоящим изобретением), которое можно комбинировать с материалом носителя для получения фармацевтической композиции (такой как единичная лекарственная форма), будет варьироваться в зависимости от индивидуума, которого лечат, и конкретного способа введения. Количество активного ингредиента, которое можно комбинировать с материалом носителя для получения единичной лекарственной формы, обычно будет представлять собой количество композиции, которое оказывает терапевтическое действие.
Обычно из 100% (для фармацевтических составов/композиций) количество активного ингредиента (в частности, количество соединения, применяемого в соответствии с настоящим изобретением, необязательно вместе с другими терапевтически активными агентами, если они присутствуют в фармацевтических составах/композиции) будет находиться в пределах диапазона от приблизительно 0,01% до приблизительно 99%, предпочтительно от приблизительно 0,1% до приблизительно 70%, наиболее предпочтительно от приблизительно 1% до приблизительно 30%, причем оставшаяся часть предпочтительно состоит из одного или более фармацевтически приемлемых вспомогательных веществ.
Количество активного ингредиента, например, соединения, применяемого в соответствии с настоящим изобретением, в стандартной лекарственной форме и/или при введении индивидууму или при применении в терапии, может находиться в пределах диапазона от приблизительно 0,1 до приблизительно 1000 мг (например, от приблизительно 1 до приблизительно 500 мг (например, от приблизительно 10 мг до приблизительно 200 мг) на единицу, введение или терапию. Согласно некоторым вариантам реализации настоящего изобретения подходящее количество такого активного ингредиента может быть рассчитано с использованием массы или площади поверхности тела индивидуума, включая количества от приблизительно 1 мг/кг до 10 мг/кг (например, от приблизительно 2 мг/кг до 5 мг/кг) или от приблизительно 1 мг/м2 до приблизительно 400 мг/м2 (например, от приблизительно 3 мг/м2 до приблизительно 350 мг/м2 или от приблизительно 10 мг/м2 до приблизительно 200 мг/м2).
Фактические уровни дозировки активных ингредиентов в фармацевтических композициях, применяемых в соответствии с настоящим изобретением, могут быть изменены так, чтобы получить количество активного ингредиента, которое эффективно для достижения желаемого терапевтического ответа для конкретного пациента, композиции и способа введения, без токсичности для пациента. Выбранный уровень дозировки будет зависеть от множества фармакокинетических факторов, включая активность конкретных примененных композиций, пути введения, времени введения, скорости выведения конкретного применяемого соединения, продолжительности лечения, других лекарственных препаратов, соединений и/или материалов, применяемых в комбинации с конкретными примененными композициями, возраста, пола, массы, состояния, общего состояния здоровья и предшествующей истории болезни пациента, которого лечат, и сходных факторов, хорошо известных в области медицины.
Врач или ветеринар, имеющий обычную квалификацию в данной области техники, может легко определить и назначить эффективное количество требуемой фармацевтической композиции. Например, врач или ветеринар может начать с доз соединений, применяемых в соответствии с настоящим изобретением, на уровнях ниже уровней, требуемых для достижения желаемого терапевтического действия, и постепенно увеличивать дозировку до тех пор, пока не будет достигнуто желаемое действие. Обычно подходящая суточная доза композиции, применяемой в соответствии с настоящим изобретением, будет представлять собой количество соединения, которое является самой низкой дозой, эффективной для получения терапевтического действия. Такая эффективная доза обычно зависит от описанных выше факторов. Предпочтительно введение является пероральным, внутривенным, внутримышечным, внутрибрюшинным или подкожным, предпочтительно введение осуществляют проксимально относительно целевого места. Если необходимо, эффективную суточную дозу фармацевтической композиции можно вводить в виде двух, трех, четырех, пяти, шести или более субдоз, вводимых по отдельности с соответствующими интервалами в течение дня, необязательно в виде единичных лекарственных форм. Несмотря на то, что соединение, применяемое в соответствии с настоящим изобретением, можно вводить по отдельности, предпочтительно соединение вводят в виде фармацевтического состава/композиции.
Для перорального введения фармацевтическая композиция, применяемая в соответствии с настоящим изобретением, может принимать форму, например, таблеток или капсул, полученных обычными способами с фармацевтически приемлемыми вспомогательными веществами, такими как связывающие агенты (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон, гидроксипропилметилцеллюлоза), наполнителями (например, лактозой, микрокристаллической целлюлозой, гидрофосфатом кальция), смазывающими веществами (например, стеаратом магния, тальком, диоксидом кремния), разрыхлителями (например, картофельным крахмалом, гликолятом крахмала натрия) или смачивающими агентами (например, лаурилсульфатом натрия). Жидкие препараты для перорального введения могут быть в форме, например, растворов, сиропов или суспензий, или могут быть представлены в виде сухого продукта для восстановления водой или другим подходящим носителем перед применением. Такой жидкий препарат может быть получен с помощью обычных способов с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сорбит, сироп, производные целлюлозы, гидрогенизированные пищевые жиры), эмульгирующие агенты (например, лецитин, гуммиарабик), неводные носители (например, миндальное масло, сложные эфиры жирных кислот, этиловый спирт, фракционированные растительные масла), консерванты (например, метил или пропил-п-гидроксикарбонаты, сорбиновые кислоты). Препараты также могут содержать буферные соли, ароматизаторы, красители и подсластители, если это будет считаться целесообразным. Препараты для перорального введения могут быть изготовлены соответствующим образом, чтобы обеспечить контролируемое высвобождение фармацевтической композиции согласно настоящему изобретению.
Согласно одному варианту реализации настоящего изобретения соединение вводят перорально в концентрации не более 100 мг/кг массы тела (например, не более 50 мг/кг массы тела, не более 40 мг/кг массы тела, не более 30 мг/кг массы тела, не более 20 мг/кг массы тела, не более 10 мг/кг массы тела, не более 5 мг/кг массы тела, не более 4 мг/кг массы тела, не более 3 мг/кг массы тела, не более 2 мг/кг массы тела, не более 1 мг/кг массы тела).
Согласно одному варианту реализации настоящего изобретения соединение вводят парентерально (например, внутривенно, внутримышечно или подкожно) в концентрации не более 10 мг/кг массы тела (например, не более 5 мг/кг массы тела, не более 4 мг/кг масса тела, не более 3 мг/кг массы тела, не более 2 мг/кг массы тела, не более 1 мг/кг массы тела, не более 0,5 мг/кг массы тела, не более 0,4 мг/кг массы тела, не более 0,3 мг/кг массы тела, не более 0,2 мг/кг массы тела, не более 0,1 мг/кг массы тела).
Фармацевтическая композиция может быть изготовлена в виде суппозитория с обычными связывающими веществами и носителями, такими как триглицериды. Пероральный состав может содержать стандартные носители, такие как маннит, лактоза, крахмал, стеарат магния, сахарин натрия, целлюлоза, карбонат магния и т.д. фармацевтического качества.
Фармацевтическая композиция, применяемая в соответствии с настоящим изобретением, может быть изготовлена для парентерального введения путем инъекции, например, путем болюсной инъекции или непрерывной инфузии. Согласно одному варианту реализации настоящего изобретения соединения или композиции, применяемые в соответствии с настоящим изобретением, могут быть введены путем медленной непрерывной инфузии в течение длительного периода времени, такого как более 24 часов, для уменьшения побочных токсических эффектов. Введение также может быть выполнено путем непрерывной инфузии в течение периода времени от 2 до 24 часов, например, от 2 до 12 часов. Такую схему можно повторять один или более раз, если необходимо, например, через 6 месяцев или 12 месяцев.
Согласно еще одному варианту реализации настоящего изобретения соединения или композиции, применяемые в соответствии с настоящим изобретением, вводят в виде поддерживающей терапии, такой как, например, один раз в неделю в течение периода 6 месяцев или более.
Составы для инъекции могут быть представлены в виде единичной лекарственной формы (например, во флаконе, в многодозовом контейнере) и с добавленным консервантом. Фармацевтическая композиция, применяемая в соответствии с настоящим изобретением, может быть в таких формах как суспензии, растворы или эмульсии в масляных или водных носителях, и может содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие или диспергирующие агенты. Согласно другому варианту агент может быть в форме порошка для восстановления подходящим носителем (например, стерильной апирогенной водой) перед применением. Как правило, композиции для внутривенного введения представляют собой растворы в стерильном изотоническом водном буфере. При необходимости композиция также может содержать солюбилизирующий агент и местный анестетик, такой как лигнокаин, для облегчения боли в месте инъекции. Обычно ингредиенты поставляются либо по отдельности, либо в смешанном виде в стандартной лекарственной форме, например, в виде сухого лиофилизированного порошка или безводного концентрата в герметически закрытом контейнере, таком как ампула или саше, с указанием количества активного агента. В случае если композиция должна быть введена путем инфузии, она может быть распределена с бутылкой для инфузии, содержащей стерильную воду или физиологический солевой раствор фармацевтического качества. В случае если композицию вводят путем инъекции, может быть обеспечена ампула стерильной воды для инъекций или физиологического солевого раствора так, чтобы ингредиенты могли быть смешаны перед введением.
Композиции, применяемые в соответствии с настоящим изобретением, которые подходят для вагинального введения, также включают пессарии, тампоны, кремы, гели, пасты, пены или аэрозольные составы, содержащие носители, которые известны в данной области техники как подходящие. Лекарственные формы для местного или трансдермального введения композиций, применяемых в соответствии с настоящим изобретением, включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и лекарственные формы для ингаляций. Активное соединение может быть смешано в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферами или пропеллентами, которые могут потребоваться.
Согласно одному варианту реализации настоящего изобретения соединения, применяемые в соответствии с настоящим изобретением, изготовлены в липосомах. Согласно более предпочтительному варианту реализации настоящего изобретения липосомы включают нацеливающий фрагмент. Согласно наиболее предпочтительному варианту реализации настоящего изобретения соединения в липосомах доставляют путем болюсной инъекции в место, расположенное проксимально относительно желаемой области. Такая композиция на основе липосом должна быть жидкой до такой степени, чтобы обеспечить легкость забора в шприц, должна быть стабильной в условиях изготовления и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибки.
«Терапевтически эффективная доза» для терапии/лечения может быть измерена на основании объективных ответов, которые могут быть как полными, так и частичными. Полный ответ (CR) определяется как отсутствие клинических, рентгенологических или других признаков состояния, нарушения или заболевания. Частичный ответ (PR) возникает в результате уменьшения заболевания более чем на 50%. Среднее время до прогрессирования является мерой, которая характеризует устойчивость объективного ответа опухоли.
«Терапевтически эффективная дозировка» для терапии/лечения также может быть измерена на основании ее способности стабилизировать прогрессирование состояния, нарушения или заболевания. Способность соединения ингибировать TRAF6 можно оценить с применением любого из количественных исследований в условиях in vitro или на основе клеток, описанных в настоящем документе, которые позволяют измерить активность/ингибирование TRAF6. Согласно другому варианту свойства соединения, описанного в настоящем изобретении, могут быть оценены с помощью исследования способности соединения в подходящих модельных системах на животных, известных специалисту в данной области техники. Терапевтически эффективное количество соединения, применяемого в соответствии с настоящим изобретением, может излечивать, заживлять, снижать, облегчать, изменять, исправлять, улучшать, нормализовать или влиять на состояние, нарушение или заболевание или симптомы состояния, нарушения или заболевания или предрасположенность к состоянию, нарушению или заболеванию у человека. Специалист в данной области техники сможет определить такие количества на основе таких факторов, как размер индивидуума, тяжесть симптомов индивидуума и конкретная композиция или выбранный путь введения.
Фармацевтическая композиция, применяемая в соответствии с настоящим изобретением, также может быть представлена, при необходимости, в упаковке или в дозирующем устройстве, которое может содержать одну или более стандартных лекарственных форм, содержащих активное соединение. Упаковка может содержать, например, металлическую или пластиковую фольгу, например, блистерная упаковка. Упаковка или дозирующее устройство могут сопровождаться инструкцией по введению.
Фармацевтическая композиция, применяемая в соответствии с настоящим изобретением, может быть введена в качестве единственного активного агента или может быть введена в комбинации с другими терапевтически и/или косметически активными агентами. Согласно одному варианту реализации настоящего изобретения фармацевтическая композиция, применяемая в соответствии с настоящим изобретением, содержит, или вводится с ними, один или более других терапевтически активных агентов, выбранных из группы, состоящей из противовирусных агентов, антител (которые направлены против антигена патогенного вируса животных или другого микроорганизма (например, патогенной бактерии или грибков) или против ракового антигена), агентов, стимулирующих иммунную систему субъекта (например, интерферонов, таких как интерферон-альфа или интерферон-бета, имиквимода и резиквимода), и противомикробных агентов.
Терапевтическая и другие сферы применения
Согласно дополнительным аспектам настоящая заявка относится к соединению, указанному выше под заголовком «Соединения», или фармацевтической композиции, указанной выше под заголовком «Фармацевтические композиции», для применения в терапии.
В целом, настоящее изобретение демонстрирует, что соединения, имеющие структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленные в таблице 1, описанные в настоящем документе под заголовком «Соединения», или фармацевтическая композиция, указанная выше под заголовком «Фармацевтические композиции», способны ингибировать или уменьшать взаимодействие TRAF6-Ubc13, в частности, способны ингибировать или уменьшать ферментативную активность TRAF6.
Соответственно, согласно одному аспекту настоящее изобретение относится к соединению, имеющему структуру в соответствии с Формулой (I), (II), (III) и/или (IV), или соединению, представленному в таблице 1, и их гидратам, сольватам, солям, комплексам, рацемическим смесям, диастереомерам, энантиомерам и таутомерам и обогащенным изотопами формам любых из вышеперечисленных, для применения в лечении связанного с TRAF6 заболевания, в частности, для применения в лечении рака, иммунного заболевания, болезни Паркинсона, гипертрофии сердца или диабета 2 типа, предпочтительно рака, иммунного заболевания и диабета 2 типа, более предпочтительно рака или иммунного заболевания. Необязательно способ включает стадию введения индивидууму по меньшей мере одного дополнительного активного соединения. По меньшей мере одно дополнительное активное соединение может быть введено совместно, до или после соединения, имеющего структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленного в таблице 1.
В настоящем документе и по всему описанию термин «заболевание, связанное с TRAF6» означает любое патологическое состояние или заболевание, которое возникает в совокупности в результате гиперэкспрессии TRAF6 или коррелирует с гиперэкспрессией или активностью TRAF6.
Было показано, что активность или гиперэкспрессия TRAF6 связана со следующими патологиями:
Ревматоидный артрит (РА): начало заболевания и прогрессирование РА в значительной степени зависит от различных путей передачи сигналов врожденного и адаптивного иммунного ответа (McInnes, I.B., C.D. Buckley, and J.D. Isaacs, Cytokines in rheumatoid arthritis - shaping the immunological landscape. Nat Rev Rheumatol, 2016. 12(1): p. 63-8; McInnes, I.B. and G. Schett, Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol, 2007. 7(6): p. 429-42; Schett, G., J.M. Dayer, and B. Manger, Interleukin-1 function and role in rheumatic disease. Nat Rev Rheumatol, 2016. 12(1): p. 14-24; Choy, E., Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford), 2012. 51 Suppl 5: p. v3-11; McInnes, I.B. and G. Schett, The pathogenesis of rheumatoid arthritis. N Engl J Med, 2011. 365(23): p. 2205-19; Brzustewicz, E. and E. Bryl, The role of cytokines in the pathogenesis of rheumatoid arthritis-Practical and potential application of cytokines as biomarkers and targets of personalized therapy. Cytokine, 2015. 76(2): p. 527-36 и Joosten, L.A., S. Abdollahi-Roodsaz, C.A. Dinarello, et al., Toll-like receptors and chronic inflammation in rheumatic diseases: new developments. Nat Rev Rheumatol, 2016. 12(6): p. 344-57). Интересно отметить, что во всех этих путях (ИЛ-1β, TLR, Th1, Th17 и других) Е3-лигазная активность TRAF6 применяется для корректной активации пути, распространения сигнала и секреции цитокинов (ИЛ-1β, ФНО-α, ИЛ-6 и других). Более того, подразумевается, что TRAF6 участвует в дифференцировке остеокластов (Lamothe, В., W.K. Webster, A. Gopinathan, et al., TRAF6 ubiquitin ligase is essential for RANKL signaling and osteoclast differentiation. Biochem Biophys Res Commun, 2007. 359(4): p. 1044-9). Наконец, исследования однонуклеотидных полиморфизмов связывали TRAF6 с PA (Namjou, В., С.В. Choi, I.T. Harley, et al., Evaluation of TRAF6 in a large multiancestral lupus cohort. Arthritis Rheum, 2012. 64(6): p. 1960-9). Соответственно, ингибирование взаимодействия TRAF6-Ubc13 является новой стратегией для противодействия воспалению и резорбции кости при РА. Исследование авторов настоящего изобретения для подтверждения концепции на доклинической модели CIA-индуцированного РА на мышах четко подтверждает эту гипотезу о том, что ингибирование взаимодействия TRAF6-Ubc13 с помощью С25-140 (соединение 3) действительно улучшает исходы заболевания РА (см. Фиг. 11). Кроме того, было показано, что экспрессия TRAF6 существенно повышена в суставах мышей с CIA-индуцированным РА и что обработка siTRAF6 мышей с CIA-индуцированным РА улучшает артритный индекс, а также воспаление и разрушение кости (Wang, Н., W. Chen, L. Wang, et al., Tumor necrosis factor receptor-associated factor 6 promotes migration of rheumatoid arthritis fibroblast-like synoviocytes. Mol Med Rep, 2015. 11(4): p. 2761-6). Интересно отметить, что результаты генетического истощения TRAF6 очень хорошо коррелируют с результатами исследования ингибитора активности TRAF6 авторами настоящего изобретения. Наконец, исследование, в котором наблюдали за 44 пациентами с РА и 9 пациентами с остеоартритом, выявило, что экспрессия TRAF6 увеличивается только в группе пациентов с РА, что еще раз подчеркивает аргумент о том, что TRAF6 может быть преимущественно вовлечен в синовиальное воспаление и резорбцию кости за счет дифференцировки остеокластов (Zhu, L.J., L. Dai, D.H. Zheng, et al., Upregulation of tumor necrosis factor receptor-associated factor 6 correlated with synovitis severity in rheumatoid arthritis. Arthritis Res Ther, 2012. 14(3): p. R133) и поэтому является интересной мишенью для лечения РА.
Псориаз: патогенез псориаза во многом зависит от передачи сигналов с участием ИЛ-17, которая включает TRAF6 как важный компонент в распространении сигнала до стадии экспрессии цитокинов (Baliwag, J., D.H. Barnes, and A. Johnston, Cytokines in psoriasis. Cytokine, 2015. 73(2): p. 342-50; Grine, L., L. Dejager, C. Libert, et al., An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev, 2015. 26(1): p. 25-33 и Gu, C., L. Wu, and X. Li, IL-17 family: cytokines, receptors and signaling. Cytokine, 2013. 64(2): p. 477-85). Более того, исследования активации TLR7 имиквимодом (IMQ) показали, что это стимулирует псориаз, что также подтверждает участие TLR и врожденного иммунного ответа в развитии заболевания (Gilliet, М., С. Conrad, М. Geiges, et al., Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol, 2004. 140(12): p. 1490-5). Bee TLR, за исключением TLR3, применяют TRAF6 в качестве центрального регулятора для передачи сигналов. В соответствии с этим настоящая заявка обеспечивает доказательство из исследования IMQ-индуцированного псориаза на мышах, что ингибирование взаимодействия TRAF6-Ubc13 с помощью С25-140 улучшает исходы заболевания (см. Фиг. 10).
Воспалительное заболевание кишечника (ВЗК): Воспаление при ВЗК (болезнь Крона и язвенный колит) зависит от событий K63-убиквитинирования, стимулированных TRAF6. Было показано, что Е3-лигазная активность TRAF6 имеет решающее значение для развития заболевания (Watanabe, Т., N. Asano, G. Meng, et al., NOD2 downregulates colonic inflammation by IRF4-mediated inhibition of K63-linked polyubiquitination of RICK and TRAF6. Mucosal Immunol, 2014. 7(6): p. 1312-25 и Wei, J., C. Wei, M. Wang, et al., The GTPase-activating protein GIT2 protects against colitis by negatively regulating Toll-like receptor signaling. Proc Natl Acad Sci U S A, 2014. 111(24): p. 8883-8). Более того, недавнее исследование доказало, что передача сигналов с участием TLR2 вовлечена в возникновение колита, поскольку пептидный ингибитор рецептора TLR2 уменьшал DSS-индуцированный колит (Shmuel-Galia, L., Т. Aychek, A. Fink, et al., Neutralization of proinflammatory monocytes by targeting TLR2 dimerization ameliorates colitis. EMBO J, 2016. 35(6): p. 685-98). При передаче сигналов с участием TLR2 TRAF6 является ключевым регулятором распространения сигнала до стадии экспрессии NF-κВ и цитокинов. Следовательно, ингибитор активности TRAF6 должен быть очень подходящим для лечения ВЗК. Помимо этого в ходе клинического исследования образцов пациентов с ВЗК было обнаружено, что уровни TRAF6 повышены как при болезни Крона, так и при язвенном колите (Shen, J., Y. Qiao, Z. Ran, et al., Different activation of TRAF4 and TRAF6 in inflammatory bowel disease. Mediators Inflamm, 2013. 2013: p. 647936), это еще раз подчеркивает перспективность нацеливания на TRAF6 для лечения ВЗК.
Диабет 1 типа (TIP): ИЛ-1-опосредуемая активация NF-κВ при синергическом действии ФНО-α и ИФН-γ является основной движущей силой воспаления и апоптоза в β-клетках (Bending, D., P. Zaccone, and A. Cooke, Inflammation and type one diabetes. Int Immunol, 2012. 24(6): p. 339-46; Csorba, T.R., A.W. Lyon, and M.D. Hollenberg, Autoimmunity and the pathogenesis of type 1 diabetes. Crit Rev Clin Lab Sci, 2010. 47(2): p. 51-71 и Mandrup-Poulsen, Т., L. Pickersgill, and M.Y. Donath, Blockade of interleukin 1 in type 1 diabetes mellitus. Nat Rev Endocrinol, 2010. 6(3): p. 158-66). Соответственно, ингибирование передачи сигналов с участием ИЛ-1 в моделях на животных уменьшало последствия T1D и улучшало гликемию, а также функцию β-клеток (Mandrup-Poulsen, Т., L. Pickersgill, and M.Y. Donath, Blockade of interleukin 1 in type 1 diabetes mellitus. Nat Rev Endocrinol, 2010. 6(3): p. 158-66). До настоящего времени в клинических исследованиях применяли антагонисты ИЛ-1 на основе антител, при этом они влияли на начало заболевания (Baumann, В., Н.Н. Salem, and В.О. Boehm, Anti-inflammatory therapy in type 1 diabetes. Curr Diab Rep, 2012. 12(5): p. 499-509). Также было показано, что TLR участвуют в воспалительных процессах при инсулите (Eizirik, D.L., M.L. Colli, and F. Ortis, The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol, 2009. 5(4): p. 219-26). TRAF6 является центральным регулятором передачи сигналов с участием ИЛ-1 и TLR и, соответственно, может представлять новую стратегию для уменьшения воспаления в предиабетической фазе инсулита, чтобы предотвратить гибель β-клеток.
Рассеянный склероз (PC). Предыдущие исследования показали, что экспериментальный аутоиммунный энцефалит (ЕАЕ), модель PC на мышах, усиливается под действием иммунных ответов Th1 и Th17 (Jager, А., V. Dardalhon, R.A. Sobel, et al., Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol, 2009. 183(11): p. 7169-77 и Zepp, J., L. Wu, and X. Li, IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol, 2011. 32(5): p. 232-9). В этом случае описано, что ИЛ-17 является основным стимулятором развития PC (Zepp, J., L. Wu, and X. Li, IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol, 2011. 32(5): p. 232-9 и Petermann, F. and T. Korn, Cytokines and effector T cell subsets causing autoimmune CNS disease. FEBS Lett, 2011. 585(23): p. 3747-57). Аналогично вышеуказанному, активность TRAF6 играет основную роль в передаче сигналов с участием Th1 и Th17 и может быть превосходной мишенью для лечения PC.
Целиакия: Употребление пшеницы, ячменя или ржи вызывает воспаление тонкого кишечника у пациентов с целиакией. В этом случае глютен вызывает адаптивный иммунный Th1-ответ у индивидуумов с определенной генетической предрасположенностью. Более того, было продемонстрировано, что ингибиторы α-амилазы/трипсина (ATI), молекулы устойчивости к вредителям пшеницы, выступают в качестве активаторов врожденных иммунных ответов. ATI связывают комплекс TLR4-MD2-CD14 и стимулируют активацию и секрецию провоспалительных цитокинов (Junker, Y., S. Zeissig, S.J. Kim, et al., Wheat amylase trypsin inhibitors drive intestinal inflammation via activation of toll-like receptor 4. J Exp Med, 2012. 209(13): p. 2395-408). TRAF6 играет решающую роль в иммунном Th1-ответе и передаче сигналов с участием TLR4 и может служить новой мишенью для лечения целиакии.
Аутоиммунные заболевания в целом: аутоиммунные заболевания возникают в результате сочетанного усиления иммунных и воспалительных процессов передачи сигналов, таких как Th1, Th17, TLR, ИЛ-1 и ФНО. Поскольку TRAF6 является основным регулятором почти во всех этих путях передачи сигналов до стадии активации NF-κB, очень вероятно, что ингибирование активности TRAF6 является приемлемой стратегией для общего нацеливания на аутоиммунитет. Результаты экспериментов на клетках в настоящей заявке демонстрируют, что ингибитор TRAF6-Ubc13 противодействует этим процессам передачи сигналов.
Диффузная крупноклеточная В-клеточная лимфома (DLBCL): DLBCL подразделяются на две основные группы: тип активированных В-клеток (ABC) и тип зародышевых В-клеток (GCB). Выживание ABC DLBCL основано на множестве различных мутаций, приводящих к конститутивной активации передачи сигналов с участием NF-κВ. Основные мутации происходят в В-клеточном рецепторе (BCR) и пути передачи сигналов с участием MYD88 (Nagel, D., М. Vincendeau, А.С. Eitelhuber, et al., Mechanisms and consequences of constitutive NF-kappaB activation in B-cell lymphoid malignancies. Oncogene, 2014. 33(50): p. 5655-65 и Ngo, V.N., R.M. Young, R. Schmitz, et al., Oncogenically active MYD88 mutations in human lymphoma. Nature, 2011. 470(7332): p. 115-9). Современные виды терапии в основном направлены на передачу сигналов с участием BCR с применением соединений, нацеленных, например, на ВТК, MALT1, cIAP (Krappmann, D. and M. Vincendeau, Mechanisms of NF-kappaB deregulation in lymphoid malignancies. Semin Cancer Biol, 2016 и Wilson, W.H., Treatment strategies for aggressive lymphomas: what works? Hematology Am Soc Hematol Educ Program, 2013. 2013: p. 584-90). Однако к настоящему моменту ингибиторы IRAK4 являются единственным доступным средством для уменьшения передачи сигналов с участием MYD88 (Kelly, P.N., D.L. Romero, Y. Yang, et al., Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med, 2015. 212(13): p. 2189-201). Данные настоящей заявки касательно DLBCL и С25-140 (соединение 3) подтверждают, что это соединение селективно уничтожает ABC-DLBCL с длительной передачей сигналов с участием MYD88 (см. Фиг. 12). В соответствии с этим ингибитор Ubc13 также успешно уменьшал передачу сигналов с участием NF-κВ и выживание клеток ABC-DLBCL (Pulvino, М., Y. Liang, D. Oleksyn, et al., Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood, 2012. 120(8): p. 1668-77). Однако ингибирование Ubc13 как фермента E2 оказывает слишком общее влияние на передачу сигналов в клетке.
Лимфома MALT-типа: лимфома MALT-типа содержит гибридный белок API2-MALT1 из-за хромосомной транслокации. Помимо других, этот гибридный белок взаимодействует с TRAF6, чтобы инициировать активацию NF-κВ, ведущую к развитию рака (Krappmann, D. and М. Vincendeau, Mechanisms of NF-kappaB deregulation in lymphoid malignancies. Semin Cancer Biol, 2016 и Staudt, L.M., Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol, 2010. 2(6): p. a000109). Эти пути ведут к пролиферации, выживанию и устойчивости к разным видам терапии. Следовательно, ингибирование активности TRAF6 может быть благоприятным при лимфоме MALT-типа.
Множественная миелома (ММ). Основными запускающими факторами злокачественного роста ММ являются нестабильность генома в опухоли и взаимодействие между клетками миеломы и микроокружением костного мозга. В данном случае активированы различные пути передачи сигналов, включая пути передачи сигналов с участием МАРК, JAK-STAT3, (PI3K)-АКТ, NF-κВ, Wnt, Notch, IGF и плейотрофина, приводящие к повышенной секреции цитокинов и факторов роста, пролиферации клеток и активности остеокластов. Клинически ММ характеризуется остеолитическими поражениями, анемией, почечной недостаточностью, гиперкальциемией, периферическими невропатиями и т.д. Однако заболевание костей является наиболее клинически значимым фактором заболеваемости. TRAF6 участвует в остеокластогенезе при ММ за счет его решающей роли в активации NF-κВ посредством передачи сигналов с участием RANK. В настоящем документе активность ТИАР6-лигазы в комбинации с Ubc13 важна для распространения сигнала. Соответственно, ингибитор активности TRAF6 является чрезвычайно перспективным для ингибирования передачи сигналов с участием RANK и, соответственно, уменьшения образования метастазов в кости и остеокластогенеза при ММ. Важно отметить, что пептиды-приманки, отсоединяющие TRAF6 от RANK-рецептора или миРНК против TRAF6 уже показали обнадеживающие результаты при остеокластогенезе и ингибировании роста, соответственно (Liu, Н., S. Tamashiro, S. Baritaki, et al., TRAF6 activation in multiple myeloma: a potential therapeutic target. Clin Lymphoma Myeloma Leuk, 2012. 12(3): p. 155-63). Соответственно, очень вероятно, что ММ можно лечить с помощью ингибирования TRAF6.
Рак легких: было показано, что при различных видах рака легких (линии NSCLC и SCLC) амплифицирован локус 11p13. Эта амплификация связана с активацией TRAF6, что характеризует TRAF6 как амплифицированный онкоген при раке легких. Амплифицированный TRAF6 стимулирует запускаемую Ras активацию провоспалительного фактора транскрипции NF-κВ и последующие стимулирующие опухоль ответы (Starczynowski, D.T., W.W. Lockwood, S. Delehouzee, et al., TRAF6 is an amplified oncogene bridging the RAS and NF-kappaB pathways in human lung cancer. J Clin Invest, 2011. 121(10): p. 4095-105). Также было показано, что Е3-лигазная активность TRAF6 необходима для запускаемой EGFR активации NF-κВ и прогрессирования рака легких (Pan, D., С.Jiang, Z. Ma, et al., MALT1 is required for EGFR-induced NF-kappaB activation and contributes to EGFR-driven lung cancer progression. Oncogene, 2016. 35(7): p.919-28). Соответственно, ингибирование TRAF6 может быть хорошей стратегией для комбинированной терапии при разных видах рака легких, запускаемых Ras, особенно при видах рака, устойчивых к текущей терапии первой линии.
Аденокатинома легких: уровень экспрессии TRAF6 значительно выше в клетках аденокарциномы легких, и TRAF6 вовлечен в пролиферацию и инвазию потенциальных клеток аденокарциномы легких (Zhong, L., F. Cao, and Q. You, Effect of TRAF6 on the biological behavior of human lung adenocarcinoma cell. Tumour Biol, 2013. 34(1): p. 231-9), это указывает на то, что его ингибирование может быть целесообразным в качестве терапевтического подхода для этого типа рака.
Рак толстой кишки: экспрессия TRAF6 повышена в тканях рака толстой кишки по сравнению с нормальными параканкрозными тканями. Уровень экспрессии TRAF6 отрицательно коррелирует с 5-летней выживаемостью, что ведет к снижению выживаемости пациентов с высокими уровнями экспрессии TRAF6 (Zhang, Т., Н. Wang, and L. Han, Expression and Clinical Significance of Tumor Necrosis Factor Receptor-Associated Factor 6 in Patients With Colon Cancer. Iran Red Crescent Med J, 2016. 18(1): p.e23931). Это свидетельствует о том, что TRAF6 играет важную роль в возникновении рака толстой кишки, и лекарственные препараты, нацеленные на TRAF6, могут быть перспективными для лечения этого заболевания. Другое исследование также показало, что экспрессия TRAF6 повышена при разных видах рака толстой кишки, что коррелирует со степенью/стадией опухоли. Эксперименты по РНК-интерференции (РНК-и) с применением миРНК TRAF6 уменьшали пролиферацию клеток рака толстой кишки в комбинации с лечением обычными противораковыми лекарственными препаратами, 5-фторурацилом и этопозидом (Sun, Н., X. Li, L. Fan, et al., TRAF6 is upregulated in colon cancer and promotes proliferation of colon cancer cells. Int J Biochem Cell Biol, 2014. 53: p. 195-201). Следовательно, ингибирование TRAF6 может усиливать эффекты этих лекарственных препаратов при комбинированном лечении.
Рак предстательной железы: было показано, что TRAF6 убиквитинирует рецептор ФРО-β I (TβRI) и презенилин 1 (PS1), чтобы стимулировать отщепление N-концевого внутриклеточного домена (ВКД) TβRI под действием ТАСЕ и PS1. Впоследствии ВКД транслоцируется в ядро, где он действует как регулятор транскрипции генов, вовлеченных в инвазивность опухолевых клеток. Важно отметить, что Е3-лигазная активность TRAF6 имеет существенное значение для этого процесса (Gudey, S.K., R. Sundar, Y. Mu, et al., TRAF6 stimulates the tumor-promoting effects of TGFbeta type I receptor through polyubiquitination and activation of presenilin 1. Sci Signal, 2014. 7(307): p.ra2 и Mu, Y., R. Sundar, N. Thakur, et al., TRAF6 ubiquitinates TGFbeta type I receptor to promote its cleavage and nuclear translocation in cancer. Nat Commun, 2011. 2: p. 330). Более того, ФРО-β индуцирует передачу сигналов с участием МАРК способом, который зависит от TRAF6, для выработки Snail 1. Этот процесс индуцирует инвазивность при агрессивном раке предстательной железы (Thakur, N., S.K. Gudey, A. Marcusson, et al., TGFbeta-induced invasion of prostate cancer cells is promoted by c-Jun-dependent transcriptional activation of SnaiU. Cell Cycle, 2014. 13(15): p.2400-14). Соответственно, TRAF6, по-видимому, является хорошей мишенью при раке предстательной железы человека.
Рак молочной железы: в недавнем исследовании было показано, что передача сигналов с участием TLR2 способствует развитию рака молочной железы. Соответственно, истощение TLR2 или подавление его регуляторов на последующих стадиях (MYD88 и IRAKI) уменьшало рост рака молочной железы (Scheeren, F.A., А.Н. Kuo, L.J. van Weele, et al., A cell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia and oncogenesis. Nat Cell Biol, 2014. 16(12): p. 1238-48). Другое исследование выявило, что ось TLR9-TRAF6 значительно вовлечена в инвазию клеток рака молочной железы. В нем также было показано, что образцы от пациентов с раком молочной железы содержат повышенные уровни TLR9 (Ilvesaro, J.M., М.А. Merrell, L. Li, et al., Toll-like receptor 9 mediates CpG oligonucleotide-induced cellular invasion. Mol Cancer Res, 2008. 6(10): p. 1534-43). Оба исследования предполагают, что нацеливание на TRAF6 в качестве основного регулятора передачи сигналов с участием TLR2 и TLR9 может быть благоприятным для лечения рака молочной железы.
Остеосаркома: уровень экспрессии TRAF6 значительно выше в тканях остеосаркомы. Более того, уровень экспрессии был еще выше, если остеосаркома была основана на метастазировании в легкие. Это исследование также может продемонстрировать, что TRAF6 положительно регулирует способность к пролиферации и инвазии линии клеток остеосаркомы (Meng, Q., М. Zheng, Н. Liu, et al., TRAF6 regulates proliferation, apoptosis, and invasion of osteosarcoma cell. Mol Cell Biochem, 2012. 371(1-2): p. 177-86). Данные в настоящей заявке свидетельствуют о том, что С25-140 (соединение 3) снижает пролиферацию клеток U20S в той же степени, как и облучение в дозе 2 Гр, и что С25-140 совместно с облучением оказывает синергические эффекты (см. Фиг. 13). Соответственно, ингибирование TRAF6 представляет собой стратегию противодействия остеосаркоме при разных видах комбинированной лекарственной/лучевой терапии.
Рак поджелудочной железы: в клиническом исследовании было показано, что уровни TRAF6 повышены в тканях 53 пациентов с раком поджелудочной железы. Более того, гиперэкспрессия TRAF6 в раковых клетках поджелудочной железы индуцировала пролиферацию и миграцию клеток. Напротив, подавление TRAF6 с использованием РНК-и снижало онкогенность клеток рака поджелудочной железы. Еще более важным является то, что миРНК против TRAF6 уменьшали рост опухоли в условиях in vivo (Rong, Y., D. Wang, W. Wu, et al., TRAF6 is over-expressed in pancreatic cancer and promotes the tumorigenicity of pancreatic cancer cells. Med Oncol, 2014. 31(11): p. 260). В заключение нужно отметить, что TRAF6 представляет собой новую мишень для разработки разных видов терапии рака поджелудочной железы.
Плоскоклеточная карцинома пищевода (ESCC): на основании данных исследования 38 образцов пациентов экспрессия TRAF6 повышена при ESCC. Более того, уровень TRAF6 был повышен в клеточных линиях ESCC. В соответствии с этим гиперэкспрессия TRAF6 способствовала размножению клеток ESCC, в то время как миРНК против TRAF6 оказывали нежелательные эффекты. Наконец, миРНК против TRAF6 эффективно уменьшала рост опухоли в условиях in vivo (Yao, F., Q. Han, C. Zhong, et al., TRAF6 promoted the tumorigenicity of esophageal squamous cell carcinoma. Tumour Biol, 2013. 34(5): p. 3201-7). Следовательно, TRAF6 может представлять собой привлекательную мишень для разработки разных видов терапии ESCC.
Болезнь Паркинсона (БП): считается, что хроническое воспаление влияет на развитие БП (Block, M.L. and J.S. Hong, Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol, 2005. 76(2): p. 77-98). В недавнем исследовании было продемонстрировано, что паркин стимулирует разрушение TRAF2 и TRAF6 в протеасомах, чтобы защитить от воспалительного ответа и индуцированной цитокинами гибели клеток в результате действия путей с участием ФНО и ИЛ-1. В случае потери паркина или мутаций паркина эта защита исчезает. Соответственно, большое количество тканей людей с БП имеют повышенные уровни TRAF6 (Chung, J.Y., H.R. Park, S.J. Lee, et al., Elevated TRAF2/6 expression in Parkinson's disease is caused by the loss of Parkin E3 ligase activity. Lab Invest, 2013. 93(6): p.663-76). Соответственно, ингибирование TRAF6 может представлять собой потенциальную терапевтическую стратегию у пациентов с паркин-зависимой БП.
Гипертрофия сердца: Недавнее исследование продемонстрировало, что уровни TRAF6 увеличиваются при патологической гипертрофии сердца как следствие выработки АФК. Еще более важно, что авторы демонстрируют, что Е3-лигазная активность TRAF6, направленная на гиперактивацию ТАК1, имеет решающее значение для ответа на гипертрофию сердца (Ji, Y.X., P. Zhang, X.J. Zhang, et al., The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via ТАК 1-dependent signalling. Nat Commun, 2016. 7: p.11267). Эти данные свидетельствуют о том, что ингибирование активности TRAF6 может быть хорошей возможностью для лечения патологической гипертрофии сердца.
Диабет 2 типа (T2D): алиментарное ожирение может стимулировать воспаление и резистентность к инсулину за счет ИЛ-1β (Tack, С.J., R. Stienstra, L.A. Joosten, et al., Inflammation links excess fat to insulin resistance: the role of the interleukin-1 family. Immunol Rev, 2012. 249(1): p.239-52). Более того, было показано, что моноциты пациентов с ожирением содержат более высокие уровни TRAF6, это связывает TRAF6 с метаболическими нарушениями (Hulsmans, М., Е. Van Dooren, С.Mathieu, et al., Decrease of miR-146b-5p in monocytes during obesity is associated with loss of the anti-inflammatory but not insulin signaling action of adiponectin. PLoS One, 2012. 7(2): p. e32794). Несколько других исследований дополнительно продемонстрировали важность передачи сигналов с участием TLR4 для развития резистентности к инсулину (Pal, D., S. Dasgupta, R. Kundu, et al., Fetuin-A acts as an endogenous ligand ofTLR4 to promote lipid-induced insulin resistance. Nat Med, 2012. 18(8): p.1279-85 и Uchimura, K., M. Hayata, T. Mizumoto, et al., The serine protease prostasin regulates hepatic insulin sensitivity by modulating TLR4 signalling. Nat Commun, 2014. 5: p.3428). В завершение следует отметить, что у мышей с нарушением передачи сигналов с участием CD40-TRAF6 наблюдали уменьшение прироста массы тела и уменьшение жировой ткани в модели с рационом с высоким содержанием жиров (Chatzigeorgiou, А., Т. Seijkens, В. Zarzycka, et al., Blocking CD40-TRAF6 signaling is a therapeutic target in obesity-associated insulin resistance. Proc Natl Acad Sci USA, 2014. 111(7): p. 2686-91). В совокупности эти исследования позволяют предположить, что TRAF6 является терапевтической мишенью для индуцированной ожирением резистентности к инсулину. Данные настоящей заявки ясно демонстрируют, что С25-140 (соединение 3) уменьшает прирост массы тела в модели T2D на мышах и дополнительно уменьшает экспрессию ИЛ-1β (см. Фиг. 9).
Термины «заболевание», «нарушение» и «состояние», при применении в отношении лечения или терапии (включая профилактическую терапию), используются в настоящем документе взаимозаменяемо и относятся к любому патологическому состоянию, включая рак, в частности, к патологическим состояниям (включая формы рака), описанным в настоящем документе.
Согласно дополнительному аспекту настоящее изобретение относится к фармацевтической композиции для применения в лечении заболевания, связанного с TRAF6, в частности, для лечения рака, иммунного заболевания, болезни Паркинсона, гипертрофии сердца или диабета 2 типа, причем указанная композиция содержит соединение, имеющее структуру в соответствии с Формулой (I), (II), (III) и/или (IV), или соединение, представленное в таблице 1, и одно или более вспомогательных веществ, и необязательно по меньшей мере одно дополнительное активное соединение. По меньшей мере одно дополнительное активное соединение можно вводить вместе, до или после соединения, имеющего структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленного в таблице 1.
Согласно дополнительному аспекту настоящее изобретение относится к способу лечения и/или предотвращения состояния, нарушения или заболевания, которое связано с гиперэкспрессией TRAF6, причем указанный способ включает введение терапевтически эффективного количества соединения, описанного в настоящем документе (в частности, введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества соединения, имеющего структуру в соответствии с Формулой (I), (II), (III) и/или (IV), или соединения, представленного в таблице 1). Необязательно способ включает стадию введения индивидууму по меньшей мере одного дополнительного активного соединения. По меньшей мере одно дополнительное активное соединение можно вводить вместе, до или после соединения, имеющего структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленного в таблице 1.
В настоящем документе и по всему описанию термины «субъект» или «индивидуум» используются взаимозаменяемо и означают животных, включая теплокровных млекопитающих, таких как человек и приматы, птиц, семейных домашних или сельскохозяйственных животных, таких как кошки, собаки, овцы, козы, крупный рогатый скот, лошади и свиньи, лабораторных животных, таких как мыши, крысы и морские свинки, рыб, рептилий, зоопарковых и диких животных и т.д. Субъект предпочтительно представляет собой млекопитающее, более предпочтительно человека.
В настоящем документе и по всему описанию термин «эффективное количество», применительно к композиции или лекарственной форме для введения субъекту, относится к количеству композиции или лекарственной формы, достаточному для обеспечения пользы при лечении рака, иммунного заболевания, болезни Паркинсона, гипертрофии сердца или диабета 2 типа, чтобы замедлить или свести к минимуму симптомы, связанные с раком, иммунным заболеванием, болезнью Паркинсона, гипертрофией сердца или диабетом 2 типа, или вылечить или способствовать улучшению рака, иммунного заболевания, болезни Паркинсона, гипертрофии сердца или диабета 2 типа. В частности, терапевтически эффективное количество означает количество, достаточное для обеспечения терапевтической пользы в условиях in vivo. При использовании применительно к количеству соединения согласно настоящему изобретению, термин предпочтительно включает нетоксичное количество, которое улучшает общую терапию, уменьшает или позволяет избегать симптомов или причин заболевания или увеличивает терапевтическую эффективность или синергическое действие с другим терапевтическим агентом.
Как известно, эффективные количества будут зависеть от конкретного субъекта, которого лечат; тяжести состояния, заболевания или нарушения; индивидуальных параметров пациента, включая возраст, физическое состояние, размер и массу; продолжительность лечения; характер сопутствующей терапии (если имеется); конкретного пути ведения и аналогичных факторов в пределах знаний и опыта практикующего медицинского работника. Эти факторы хорошо известны специалистам в данной области техники и могут быть установлены с помощью всего лишь обычных экспериментов. Обычно предпочтительно применяют максимальную дозу, т.е. самую высокую безопасную дозу в соответствии с обоснованным медицинским заключением. Однако специалисты в данной области техники поймут, что пациент может настаивать на более низкой дозе или переносимой дозе по медицинским причинам, психологическим причинам или фактически по любой другой причине.
Согласно любому из вышеуказанных терапевтических аспектов по меньшей мере одно дополнительное активное соединение может быть выбрано из дополнительных активных соединений, описанных выше.
Согласно любому из вышеуказанных терапевтических аспектов, в которых способ включает стадии введения соединения, имеющего структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленного в таблице 1, как указано выше, и введение по меньшей мере одного дополнительного активного соединения, по меньшей мере дополнительное активное соединение может быть изготовлено вместе с соединениями согласно настоящему изобретению, как описано выше, в одной фармацевтической композиции. Согласно другому варианту фармацевтическая композиция может быть структурирована как набор частей, причем соединение, имеющее структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединение, представленное в таблице 1, обеспечено в первом составе, и по меньшей мере одно дополнительное активное соединение обеспечено во втором составе, т.е. во второй фармацевтической композиции. Первую и вторую фармацевтические композиции можно комбинировать перед применением. Иными словами, перед введением фармацевтической композиции состав, содержащий дополнительное активное соединение, может быть добавлен к первой фармацевтической композиции, содержащей соединение, имеющее структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединение, представленное в таблице 1. Согласно другому варианту идея настоящего изобретения предусматривает введение соединения, имеющего структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленного в таблице 1, изготовленного в виде первой фармацевтической композиции, и введение по меньшей мере одного дополнительного активного соединения, изготовленного в виде второй фармацевтической композиции. Фармацевтические композиции могут быть введены одновременно или последовательно. Например, первую фармацевтическую композицию можно вводить в первый момент времени, и вторую фармацевтическую композицию можно вводить во второй момент времени, причем моменты времени могут быть разделены, например, интервалами в 0 или до 1, 2, 3, 4, 5 или 10 минут, до 1, 2, 3, 4, 5 или 10 часов, до 1, 2, 3, 4, 5 или 10 дней, до 1, 2, 3, 4, 5 или 10 недель, до 1, 2, 3, 4, 5 или 10 месяцев или до 1, 2, 3, 4, 5 или 10 лет.
Согласно некоторым вариантам реализации настоящего изобретения заболевание, связанное с TRAF6, представляет собой рак, иммунное заболевание, болезнь Паркинсона, гипертрофию сердца или диабет 2 типа, предпочтительно рак, иммунное заболевание и диабет 2 типа, более предпочтительно рак или иммунное заболевание.
Рак предпочтительно выбран из группы, состоящей из лимфомы, такой как диффузная крупноклеточная В-клеточная лимфома (DLBCL) и лимфома MALT-типа, множественной миеломы (ММ), рака легких, аденокарциномы легких, рака толстой кишки, рака предстательной железы, рака молочной железы, остеосаркомы, рака поджелудочной железы и плоскоклеточной карциномы пищевода (ESCC), согласно другим вариантам реализации настоящего изобретения из диффузной крупноклеточной В-клеточной лимфомы (DLBCL) и остеосаркомы.
Иммунное заболевание предпочтительно представляет собой аутоиммунное заболевание. Аутоиммунное заболевание предпочтительно выбрано из группы, состоящей из псориаза, ревматоидного артрита, целиакии, воспалительного заболевания кишечника, такого как болезнь Крона и язвенный колит, рассеянного склероза и сахарного диабета 1 типа, согласно другим вариантам реализации из псориаза, ревматоидного артрита и рассеянного склероза, согласно другим вариантам реализации из псориаза и ревматоидного артрита.
В настоящем документе термин «раковое заболевание» или «рак» включает заболевание, характеризующееся абберантно регулируемым размножением, пролиферацией, дифференцировкой, адгезией и/или миграцией клеток. Под «раковой клеткой» подразумевается патологическая клетка, которая размножается в результате быстрой неконтролируемой клеточной пролиферации и продолжает размножаться после прекращения действия стимулов, которые инициировали новое размножение.
Обычно количество соединения согласно настоящему изобретению, описанного в настоящем документе (в частности, количество соединения, имеющего структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленного в таблице 1), вводимое индивидууму ежедневно, может составлять не более 100 мг/кг (например, не более 50 мг/кг, не более 40 мг/кг, не более 30 мг/кг, не более 20 мг/кг, не более 10 мг/кг, не более 9 мг/кг, не более 8 мг/кг, не более 7 мг/кг, не более 6 мг/кг, не более 5 мг/кг, не более 4 мг/кг, не более 3 мг/кг, не более 2 мг/кг или не более 1 мг/кг), в зависимости от таких факторов как состояние субъекта, подлежащего лечению, и способ введения. Например, количество соединения в соответствии с настоящим изобретением, описанного в настоящем документе (в частности, количество соединения, имеющего структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединения, представленного в таблице 1), вводимое индивидууму ежедневно, может находиться в пределах диапазона от приблизительно 0,01 мг/кг до 100 мг/кг (например, от приблизительно 0,05 мг/кг до 50 мг/кг, от приблизительно 0,1 мг/кг до 40 мг/кг, от приблизительно 0,2 мг/кг до 30 мг/кг, от приблизительно 0,3 мг/кг до 20 мг/кг, от приблизительно 0,4 мг/кг до 10 мг/кг, от приблизительно 0,5 мг/кг до 9 мг/кг, от приблизительно 0,6 мг/кг до 8 мг/кг, от приблизительно 0,7 мг/кг до 7 мг/кг, от приблизительно 0,8 мг/кг до 6 мг/кг, от приблизительно 0,9 мг/кг до 5 мг/кг, от приблизительно 1 мг/кг до 4 мг/кг, или от приблизительно 2 мг/кг до 3 мг/кг) в зависимости от таких факторов как состояние субъекта, подлежащего лечению, и способ введения.
Согласно одному варианту реализации настоящего изобретения соединение согласно настоящему изобретению, описанное в настоящем документе (в частности, соединение, имеющее структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединение, представленное в таблице 1), вводят перорально в концентрации не более 100 мг/кг массы тела (например, не более 50 мг/кг массы тела, не более 40 мг/кг массы тела, не более 30 мг/кг массы тела, не более 20 мг/кг масса тела, не более 10 мг/кг массы тела, не более 5 мг/кг массы тела, не более 4 мг/кг массы тела, не более 3 мг/кг массы тела, не более 2 мг/кг массы тела, не более 1 мг/кг массы тела).
Согласно одному варианту реализации настоящего изобретения соединение согласно настоящему изобретению, описанное в настоящем документе (в частности, соединение, имеющее структуру в соответствии с Формулами (I), (II), (III) и/или (IV), или соединение, представленное в таблице 1), вводят парентерально (например, внутривенно, внутримышечно или подкожно) в концентрации не более 10 мг/кг массы тела (например, не более 5 мг/кг массы тела, не более 4 мг/кг массы тела, не более 3 мг/кг массы тела, не более 2 мг/кг массы тела, не более 1 мг/кг массы тела, не более 0,5 мг/кг массы тела, не более 0,4 мг/кг массы тела, не более 0,3 мг/кг массы тела, не более 0,2 мг/кг массы тела, не более 0,1 мг/кг массы тела).
Варианты реализации и определения терминов, описанные применительно к средствам, таким как соединения в соответствии с Формулой (I), Формулой (II), Формулой (III), Формулой (IV) и/или таблицей 1 настоящего изобретения, в равной степени применимы к способам и вариантам применения, описанным выше, с соответствующими изменениями.
Согласно настоящему изобретению также предусмотрен способ лечения у субъекта заболевания, связанного с гиперэкспрессией или активностью TRAF6, включающий введение указанному субъекту эффективного количества соединения в соответствии с Формулой (I), Формулой (II), Формулой (III), Формулой (IV) и/или таблицей 1 или его фармацевтически приемлемой соли, сольвата или гидрата или фармацевтической композиции, содержащей указанное соединение. Указанный способ предпочтительно включает дополнительное введение по меньшей мере одного дополнительного фармацевтически активного соединения. Вышеописанные аспекты, варианты реализации, определения и т.д. также применимы к указанному способу лечения, с соответствующими изменениями.
Настоящее изобретение будет более подробно описано ниже с помощью следующих примеров.
1) Материалы и методы
1.1) ВЭЖХ
Система для ВЭЖХ представляла собой Agilent 1100 (Agilent, США), оснащенную насосом для четырехкомпонентных смесей, вакуумным дегазатором, детектором с изменяемой длиной волны (VWD), колоночным термостатом, ручным пробоотборником Rheodyne-7725i и колонкой для ВЭЖХ (CNW Athena C18-WP, 3 мкм, 2,1 × 50 мм). Получение образца: 1,0 мг образца растворяли в 0,8 мл растворителя А и 0,4 мл растворителя В и фильтровали через нейлоновую мембрану с размером пор 0,45 мкм перед исследованием. Подвижная фаза: растворитель А представлял собой ацетонитрил, и растворитель В представлял собой 0,1% фосфорную кислоту в воде.
Способ 1: Градиентное элюирование программировали следующим образом: А/В=5/95 (об./об., 0 мин) → 95/5 (об./об., 4 мин) → 95/5 (об./об., 5,9 мин) → 5/95 (об./об., 6,0 мин). Температура колонки: 25°С. Скорость потока: 0,40 мл/мин. Длина волны детектирования: 254 нм. Впрыскиваемый объем: 2 мкл.
Способ 2: Градиентное элюирование программировали следующим образом: А/В=20/80 (об./об., 0 мин) → 95/5 (об./об., 3 мин) → 95/5 (об./об., 5,9 мин) → 5/95 (об./об., 6,0 мин). Температура колонки: 35°С. Скорость потока: 0,45 мл/мин. Длина волны детектирования: 254 нм. Впрыскиваемый объем: 2 мкл.
2) Соединения
2.1) Получение 1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)гидразина (Р571-1)

К раствору 6-хлор-3-метил-[1,2,4]триазол[4,3-b]пиридазина (2,80 г, 16,6 ммоль) в EtOH (30,0 мл) добавляли гидрат гидразина (85%, 2,93 мл, 49,8 ммоль). Реакционную смесь нагревали до 80°С в течение 4 часов. Проверка методом ТСХ (EtOAc, УФ) показала, что исходный материал (R/=0,10) израсходован. Реакционную смесь охлаждали до 25°С. Появился осадок, и после фильтрации получали 2,50 г желаемого продукта. Выход: 2,50 г (91%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ (м.д.)=7,84 (d, 1H), 6,75 (d, 1H), 4,28 (s, 2Н), 2,52 (s, 3Н)
Ссылка: Polish Journal of Chemistry, 1996, 70, 10, 683-692.
2.2) Получение этил-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропаноата (Р571-2)
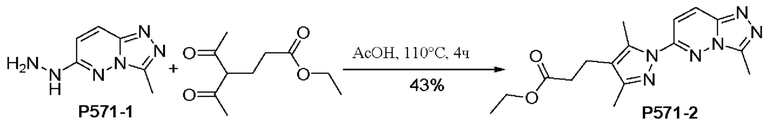
Смесь Р571-1 (1,50 г, 9,14 ммоль) и этил-4-ацетил-5-оксогексаноата (2,01 г, 10,1 ммоль) в АсОН (15,0 мл) нагревали до 110°С в течение 4 часов. Проверка методом ТСХ (EtOAc, УФ) показала, что можно наблюдать желаемый продукт в виде основного пятна (Rf=0,20). Реакционную смесь концентрировали для удаления АсОН. Остаток разбавляли EtOAc и промывали 5% раствором NaHCO3, солевым раствором и высушивали над Na2SO4. Остаток повторно кристаллизовали из Et2O с получением 1,30 г чистого желаемого продукта.
Выход: 1,30 г (43%).
1H-ЯМР: (400 МГц, метанол-d4): δ (м.д.)=8,36 (d, 1H), 7,84 (d, 1H), 4,05 (q, 2Н), 2,70 (t, 2Н), 2,68 (s, 3Н), 2,59 (s, 3Н), 2,23 (s, 3Н), 1,16 (t, 3Н).
Ссылка: European Journal of Medicinal Chemistry, 2013, 69, 701-710.
2.3) Получение этил-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4 ил)пропановой кислоты (Р571-3)
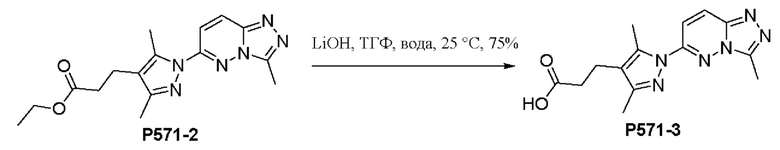
К раствору Р571-2 (1,30 г, 3,96 ммоль) в ТГФ (15,0 мл) и воде (5,00 мл) добавляли LiOH⋅H2O (498 мг, 11,9 ммоль). Реакционную смесь перемешивали при 25°С в течение 4 часов. Проверка методом ТСХ (EtOAc, УФ) показала, что исходный материал (Rf=0,20) был израсходован, и желаемый продукт можно наблюдать в виде основного пятна (Rf=0,05). Реакционную смесь концентрировали для удаления ТГФ. Остаток доводили до рН=3 с помощью 6 н HCl с получением осадка. Твердое вещество собирали с помощью фильтрации, промывали водой и высушивали с получением 900 мг чистого желаемого продукта.
Выход: 900 мг (75%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ (м.д.)=12,19 (s, 1Н), 8,36 (d, 1Н), 7,85 (d, 1Н), 2,68 (s, 3Н), 2,65 (d, 2Н), 2,60 (s, 3Н), 2,40 (t, 2Н), 2,23 (s, 3Н).
Ссылка: European Journal of Medicinal Chemistry, 2013, 69, 701-710.
2.4.1) Получение 1-(4-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин)-6-ил)-1H-пиразол-4-ил)пропан-1-она (HZM06-1)

Смесь Р571-3 (50,0 мг, 0,17 ммоль), HATU (69,6 мг, 0,18 ммоль), 1-бензилпиперазина (58,7 мг, 0,33 ммоль) и ТЭА (33,7 мг, 0,33 ммоль) в ДМ ФА (3,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ТСХ (10% МеОН в EtOAc, УФ) показала, что исходный материал (Rf=0,10) был израсходован, и желаемый продукт можно наблюдать в виде основного пятна (Rf=0,30). Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 млх5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью Chromatotron (силикагель, 50% МеОН в EtOAc) с получением 55,0 мг чистого целевого соединения.
Выход: 55,0 мг (72%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ (м.д.)=8,38 (d, 1H), 7,86 (d, 1H), 7,30 - 7,25 (m, 1H), 7,23 - 7,20 (m, 2Н), 3,44 (t, 2Н), 3,39 (s, 2Н), 2,66 (s, 3Н), 2,63 (d, 2Н), 2,58 (s, 3Н), 2,46 (d, 2Н), 2,26 (t, 2Н), 2,22 (s, 3Н), 2,17 (t, 2Н).
ЖХ/МС: m/z=459,2, R=3,57 мин (способ 1).
2.4.2 Получение 1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]-триазол- [4,3-b]-пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-она С25-140
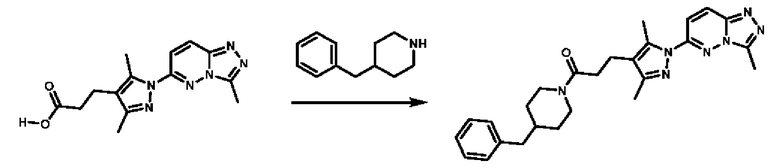
В колбу с круглым дном, заполненную раствором 3-(3,5-диметил-1-(3-метил-[1,2,4]-триазол-[4,3-b]-пиридазин-6-ил)-1H-пиразол-4-ил)пропановой кислоты (50 мг, 0,17 ммоль) в диметилформамиде (3 мл), добавляли 4-бензилпиперидин (0,06 мл, 0,33 ммоль), HATU (68,52 мг, 0,18 ммоль) и триэтиламин (0,04 мл, 0,306 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре до полного расходования исходного материала (17 ч). Затем ее разбавляли водой (15 мл) и экстрагировали этилацетатом (3 × 10 мл). Объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали в вакууме. Продукт очищали с помощью флэш-хроматографии, элюировали линейным градиентом этилацетата/метанола 100:00→50:50 в качестве элюента. Комбинация соответствующих фракций позволила получить 55 мг (70%) указанного в заголовке продукта: 1H ЯМР (400 МГц, ДМСО-d6): δ=8,4 (d, J=10 Гц, 1Н), 7,88 (d, J=10 Гц, 1H), 7,33-7,09 (br m, 3Н), 7,10-6,99 (br m, 2Н), 4,39 (br d, 1H), 3,76 (br d, 1Н), 2,86 (t, 1Н), 2,71-2,61 (br m, 6Н), 2,59 (s, 3Н), 2,47-2,14 (br m, 4Н), 2,24 (s, 3Н), 1,77-1,63 (br m, 1H), 1,59-1,43 (br m, 2Н), 0,99-0,73 (br m, 2Н); ЖХ/МС(ESI+) m/z: [М+Н]+ Вычислено для C26H32N7O - 458,58, установлено - 458,61, tR=1,16 мин.
2.5) Получение 1-(4-(3-метоксибензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-она (HZM06-2)

Смесь Р571-3 (50,0 мг, 0,17 ммоль), HATU (69,6 мг, 0,18 ммоль), 1-(3-метоксибензил)пиперазина (68,7 мг, 0,33 ммоль) и ТЭА (33,7 мг, 0,33 ммоль) в ДМФА (3,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ТСХ (10% МеОН в EtOAc, УФ) показала, что исходный материал (Rf=0,10) был израсходован, и желаемый продукт можно наблюдать в виде основного пятна (Rf=0,30). Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 мл × 5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью Chromatotron (силикагель, 50% МеОН в EtOAc) с получением 30,0 мг чистого целевого соединения.
Выход: 30,0 мг (37%).
1H-ЯМР: (400 МГц, D2O): δ (м.д.)=8,38 (d, 1H), 7,86 (d, 1H), 7,22-7,15 (m, 1H), 6,82-6,75 (m, 2H), 3,71 (s, 3H), 3,44 (d, 2H), 3,36 (s, 2H), 2,67 (s, 3H), 2,64 (d, 2H), 2,58 (s, 3H), 2,46 (d, 2H), 2,27 (s, 2H), 2,23 (s, 3H), 2,18 (d, 2H).
ЖХ/МС: m/z=489,1, Rt=3,64 мин (способ 1).
2.6) Получение 1-(4-(4-фторбензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-6]1 шридазин-6-ил)-1H-пиразол-4-ил)пропан-1-она (HZM06-3)

Смесь Р571-3 (50,0 мг, 0,17 ммоль), HATU (69,6 мг, 0,18 ммоль), 1-(4-фторбензил)пиперазина (64,7 мг, 0,33 ммоль) и ТЭА (33,7 мг, 0,33 ммоль) в ДМФА (3,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ТСХ (10% МеОН в EtOAc, УФ) показала, что исходный материал (Rf=0,10) был израсходован, и желаемый продукт можно наблюдать в виде основного пятна (Rf=0,30). Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 мл × 5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью Chromatotron (силикагель, 50% МеОН в EtOAc) с получением 50,0 мг целевого соединения.
Выход: 50,0 мг (63%).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ (м.д.)=8,38 (d, 1H), 7,85 (d, 1H), 7,28 - 7,23 (m, 1H), 7,13-7,08 (m, 2Н), 3,45 (d, 2Н), 3,38 (s, 2Н), 2,67 (s, 3Н), 2,63 (d, 2Н), 2,58 (s, 3Н), 2,46 (d, 2Н), 2,26 (t, 2Н), 2,22 (s, 3Н), 2,18 (d, 2Н).
ЖХ/МС: m/z=477,1, Rt=3,64 мин (способ 1).
2.7) Получение 1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-6]пиридазин)-6-ил)-1H-пиразол-4-ил)пропан-1-она (HZM06-4)
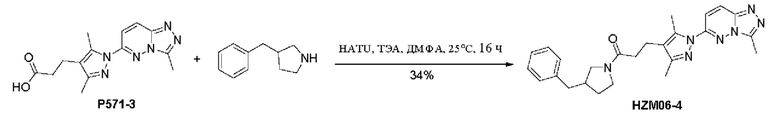
Смесь Р571-3 (200 мг, 0,67 ммоль), HATU (253 мг, 0,67 ммоль), 3-бензилпирролидина (107 мг, 0,67 ммоль) и ТЭА (135 мг, 1,33 ммоль) в ДМАА (5,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ВЭЖХ-УФ показала, что исходное вещество было израсходовано, и желаемый продукт можно наблюдать в виде основного пика. Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 млх5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью колонки с основным Al2O3 (10% МеОН в EtOAc) с получением 100 мг чистого целевого соединения. Выход: 100 мг (34%).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ (м.д.)=8,36 (dd, 1H), 7,84 (dd, 1H), 7,29-7,19 (m, 2Н), 7,16 (t, 3Н), 3,49-3,35 (m, 2Н), 3,23-3,13 (m, 1Н), 3,04-2,89 (m, 1Н), 2,67 (d, 3Н), 2,65-2,60 (m, 2Н), 2,57 (d, 3Н), 2,46-2,32 (m, 2Н), 2,22 (d, 3Н), 1,95-1,75 (m, 1H), 1,61-1,41 (m, 1H).
ЖХ/МС: m/z=444,3, Rt=3,55 мин (способ 2).
2.8) Получение 3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-фенилпиперидин-1-ил)пропан-1-она (HZM06-6)

Смесь Р571-3 (50,0 мг, 0,17 ммоль), HATU (69,6 мг, 0,18 ммоль), 4-фенилпиперидина (64,7 мг, 0,33 ммоль) и ТЭА (33,7 мг, 0,33 ммоль) в ДМФА (3,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ТСХ (10% МеОН в EtOAc, УФ) показала, что исходный материал (Rf=0,10) был израсходован, и желаемый продукт можно наблюдать в виде основного пятна (Rf=0,30). Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 млх5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью Chromatotron (силикагель, 50% МеОН в EtOAc) с получением 45,0 мг чистого целевого соединения.
Выход: 45,0 мг (61%).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ (м.д.)=8,36 (d, 1H), 7,84 (d, 1H), 7,15-7,02 (m, 5Н), 4,57 (d, 1H), 3,90 (d, 1H), 3,03 (t, 1H), 2,69-2,64 (m, 2Н), 2,62 (s, 3Н), 2,60 (s, 3Н), 2,47-2,37 (m, 1Н), 2,26 (s, 3Н), 1,71 (dd, 2Н), 1,32-1,13 (m, 2Н).
ЖХ/МС: m/z=444,3, Rt=4,59 мин (способ 1).
2.9) Получение 3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперидин-1-ил)пропан-1-она (HZM06-7)
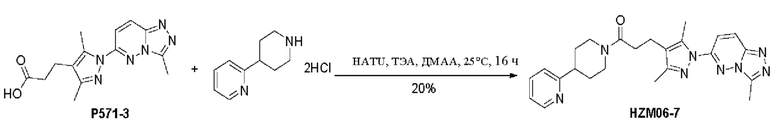
Смесь Р571-3 (128 мг, 0,43 ммоль), HATU (178 мг, 0,47 ммоль), дигидрохлорида 2-(пиперидин-4-ил)пиридина (100 мг, 0,43 ммоль) и ТЭА (172 мг, 1,70 ммоль) в ДМАА (3,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ВЭЖХ показала, что исходное вещество было израсходовано, и желаемый продукт можно наблюдать в виде основного пика. Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 мл × 5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью Chromatotron (основной Al2O3, 50% МеОН в EtOAc) с получением 37,0 мг чистого целевого соединения.
Выход: 37,0 мг (20%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ (м.д.)=8,34 (d, 1Н), 8,32 (d, 1H), 7,82 (d, 1H), 7,56 (td, 1Н), 7,14 (d, 1H), 7,09 (ddd, 1H), 4,55 (d, 1H), 3,90 (d, 1Н), 3,06 (t, 1Н), 2,73-2,65 (m, 2Н), 2,63 (s, 3Н), 2,59 (s, 3Н), 2,25 (s, 3Н), 1,80 (dd, 2Н), 1,47-1,23 (m, 2Н).
ЖХ/МС: m/z=445,2, Rt=3,44 мин (способ 1).
2.10) Получение 1-(4-(3-метоксибензил)пиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-она (HZM06-8)

Смесь Р571-3 (50,0 мг, 0,17 ммоль), HATU (69,6 мг, 0,18 ммоль), гидрохлорида 4-(3-метоксибензил)пиперидина (80,0 мг, 0,33 ммоль) и ТЭА (67,4 мг, 0,67 ммоль) в ДМФА (3,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ТСХ (10% МеОН в EtOAc, УФ) показала, что исходный материал (Rf=0,10) был израсходован, и желаемый продукт можно наблюдать в виде основного пятна (Rf=0,30). Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 мл × 5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью Chromatotron (силикагель, 50% МеОН в EtOAc) с последующей повторной кристаллизацией из Et2O с получением 30,0 мг чистого целевого соединения.
Выход: 30,0 мг (37%).
1H-ЯМР: (400 МГц, D2O: δ (м.д.)=8,38 (d, 1H), 7,87 (d, 1H), 7,11 (t, 1H), 6,74-6,69 (m, 1H), 6,65-6,57 (m, 2H), 4,38 (d, 1H), 3,76 (d, 1H), 3,69 (s, 3Н), 2,85 (t, 1H), 2,67 (s, 3Н), 2,59 (s, 3Н), 2,47-2,38 (m, 2H), 2,23 (s, 3Н), 1,69 (s, 1H), 1,51 (m, 2H), 0,99-0,70 (m, 2H).
ЖХ/МС: m/z=488,2, Rt=4,87 мин (способ 1).
2.11) Получение 1-(4-(4-фторбензил)пиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-она (HZM06-9)

Смесь Р571-3 (100 мг, 0,33 ммоль), HATU (133 мг, 0,35 ммоль), 4-(4-фторбензил)пиперидина (64,0 мг, 0,33 ммоль) и ТЭА (67,4 мг, 0,67 ммоль) в ДМАА (3,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ТСХ (10% МеОН в EtOAc, УФ) показала, что исходный материал (Rf=0,10) был израсходован, и желаемый продукт можно наблюдать в виде основного пятна (Rf=0,30). Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 мл × 5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью Chromatotron (силикагель, 50% МеОН в EtOAc) с получением 45,0 мг чистого целевого соединения.
Выход: 45,0 мг (28%).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ (м.д.)=8,36 (d, 1H), 7,84 (d, 1H), 7,14-7,05 (m, 2Н), 7,04-6,96 (m, 2Н), 4,36 (d, 1H), 3,75 (d, 1H), 2,83 (t, 1H), 2,65 (s, 3Н), 2,57 (s, 3Н), 2,41-2,36 (m, 2Н), 2,21 (s, 3Н), 1,72-1,58 (m, 1Н), 1,48 (dd, 2Н), 0,98-0,72 (m, 2Н).
ЖХ/МС: m/z=476,1, Rt=4,97 мин (способ 1).
2.12) Получение 3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-((пиридин-2-ил)метил)пиперидин-1-ил)пропан-1-она (HZM06-10)

Смесь Р571-3 (120 мг, 0,40 ммоль), HATU (160 мг, 0,42 ммоль), дигидрохлорида 2-((пиперидин-4-ил)метил)пиридина (100 мг, 0,40 ммоль) и ТЭА (162 мг, 1,61 ммоль) в ДМАА (3,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ВЭЖХ показала, что исходное вещество было израсходовано, и желаемый продукт наблюдали в виде основного пика. Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 мл × 5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью Chromatotron (основной Al2O3, 50% МеОН в EtOAc) с получением 50,0 мг чистого целевого соединения. Выход: 50,0 мг (27%).
1H-ЯМР: (400 МГц, метанол-d4): δ (м.д.)=8,41 (dt, 1H), 8,37 (d, 1H), 7,86 (d, 1H), 7,62 (td, 1Н), 7,16 (ddd, 1H), 7,09 (d, 1H), 4,37 (d, 1H), 3,76 (d, 1Н), 2,86 (q, 1H), 2,67 (s, 3Н), 2,65-2,61 (m, 2Н), 2,58 (s, 3Н), 2,54 (dd, 2Н), 2,47-2,35 (m, 2Н), 2,23 (s, 3Н), 1,50 (dd, 2Н), 1,10-0,80 (m, 2Н).
ЖХ/МС: m/z=459,2, Rt=3,49 мин (способ 1).
2.13) Получение 3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-((пиридин-2-ил)метил)пиперазин-1-ил)пропан-1-она (HZM06-П)

Смесь Р571-3 (50,0 мг, 0,17 ммоль), HATU (69,6 мг, 0,18 ммоль), 1-((пиридин-2-ил)метил)пиперазина (59,0 мг, 0,33 ммоль) и ТЭА (33,7 мг, 0,33) ммоль) в ДМ ФА (3,00 мл) перемешивали при 25°С в течение 16 часов. Проверка методом ВЭЖХ показала, что исходное вещество было израсходовано, и желаемый продукт наблюдали в виде основного пика. Реакционную смесь разбавляли водой (15,0 мл) и экстрагировали EtOAc (10,0 мл × 5). Объединенную органическую фазу промывали водой и солевым раствором, высушивали над Na2SO4 и очищали с помощью Chromatotron (силикагель, МеОН), с получением 25,0 мг чистого целевого соединения.
Выход: 25,0 мг (33%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ (м.д.)=8,43 (dt, 1H), 8,37 (d, 1H), 7,85 (d, 1H), 7,73 (td, 1H), 7,41-7,37 (m, 1H), 7,23 (ddd, 1H), 3,52 (s, 2Н), 3,46 (t, 2Н), 3,39 (d, 2Н), 2,67 (s, 3Н), 2,64 (d, 2Н), 2,58 (s, 3Н), 2,47 (d, 2Н), 2,33 (t, 2Н), 2,26 (t, 2Н), 2,23 (s, 3Н).
ЖХ/МС: m/z=460,2, Rt=3,48 мин (способ 1).
Следующие соединения получали в соответствии с несинтетической методикой, аналогичной той, которая описана для примера:
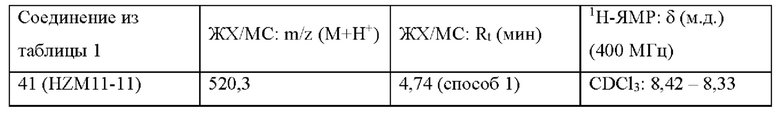
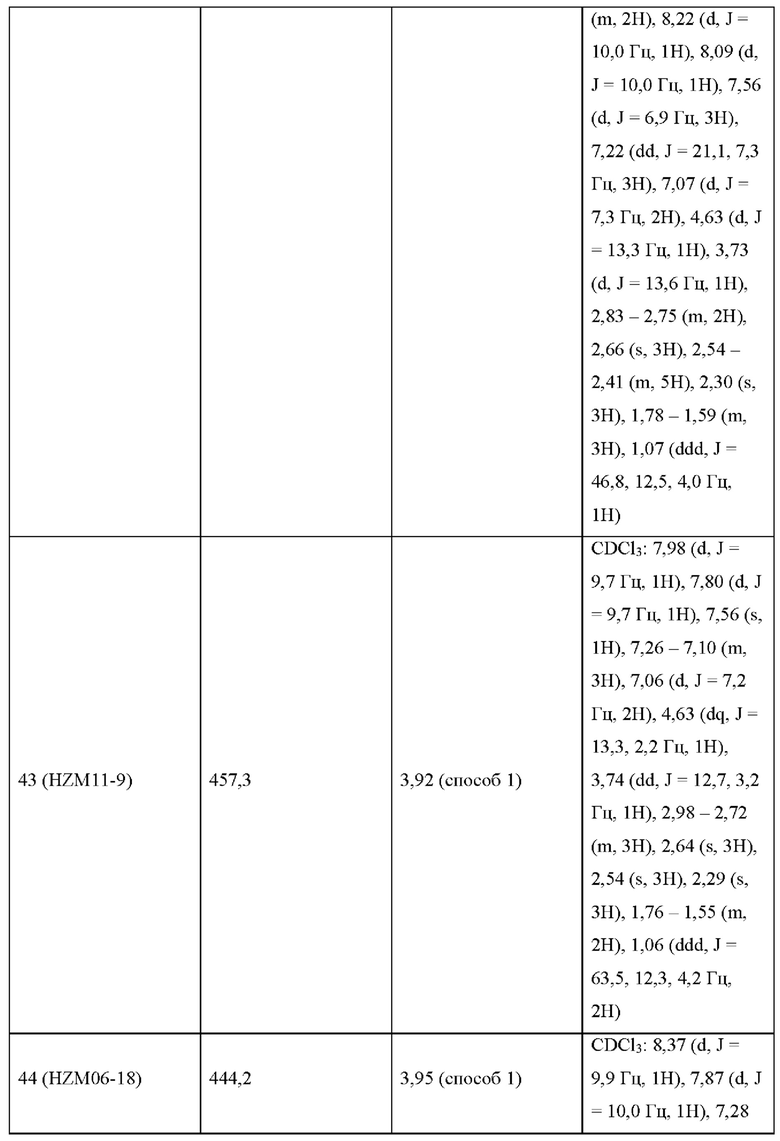
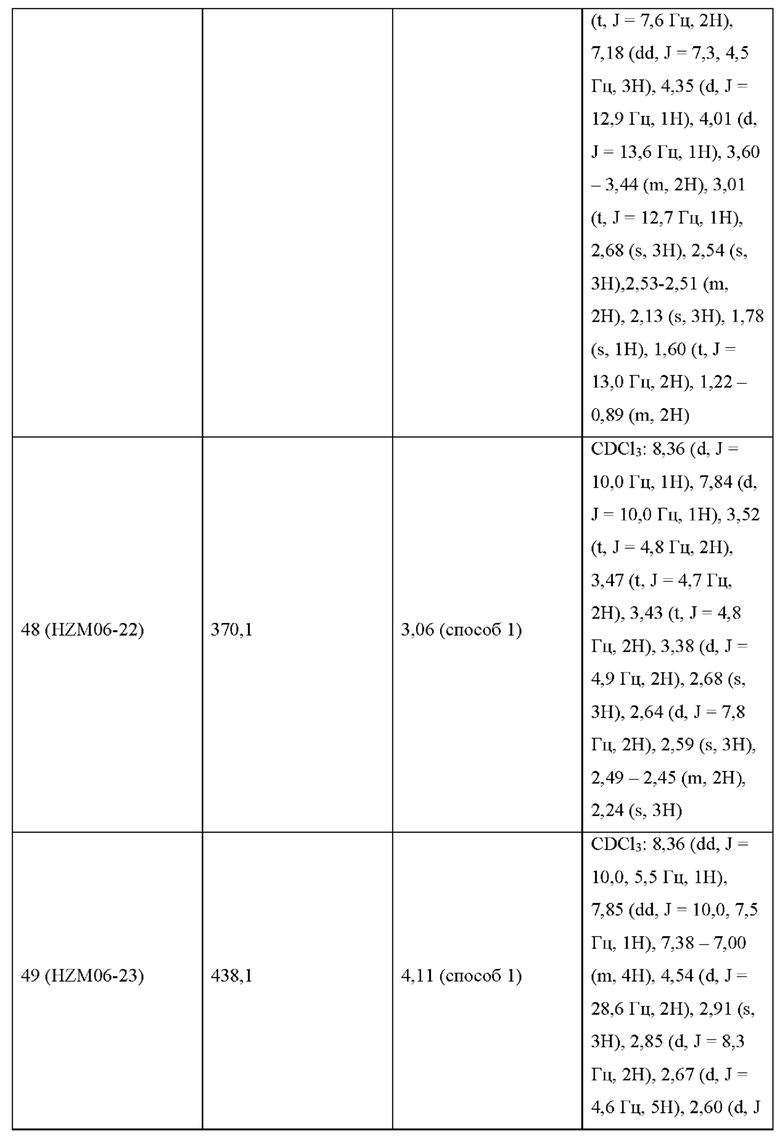
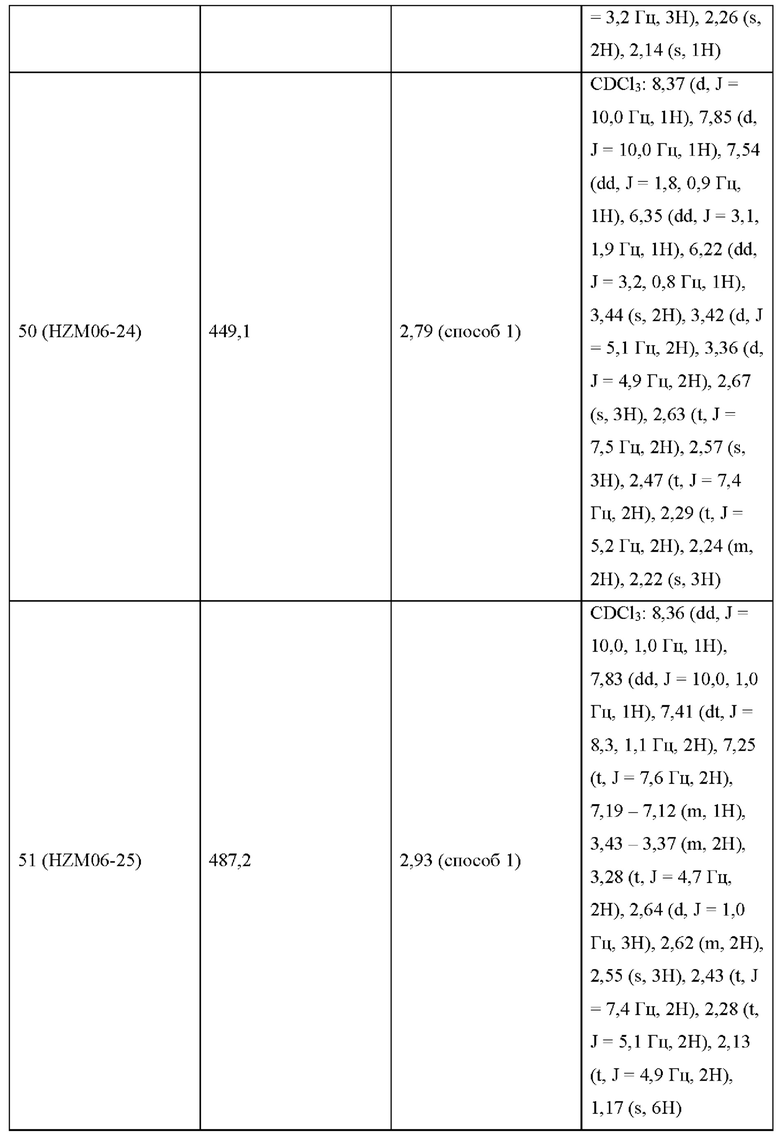
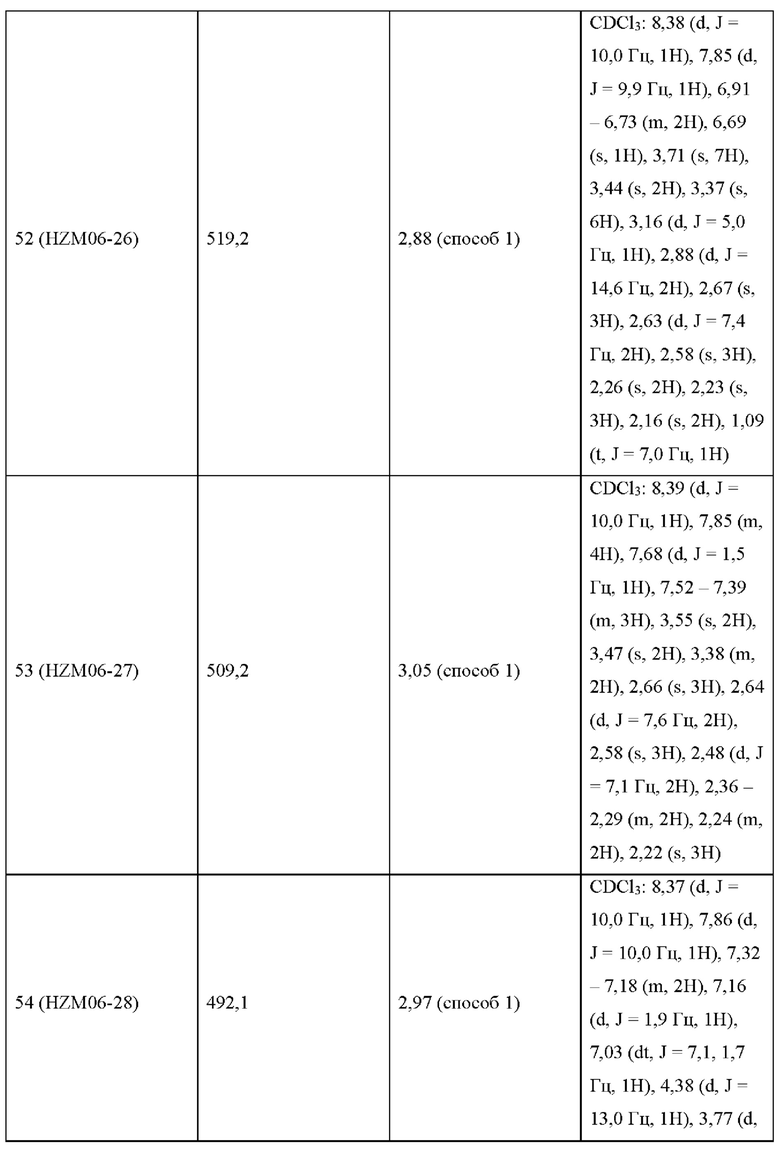
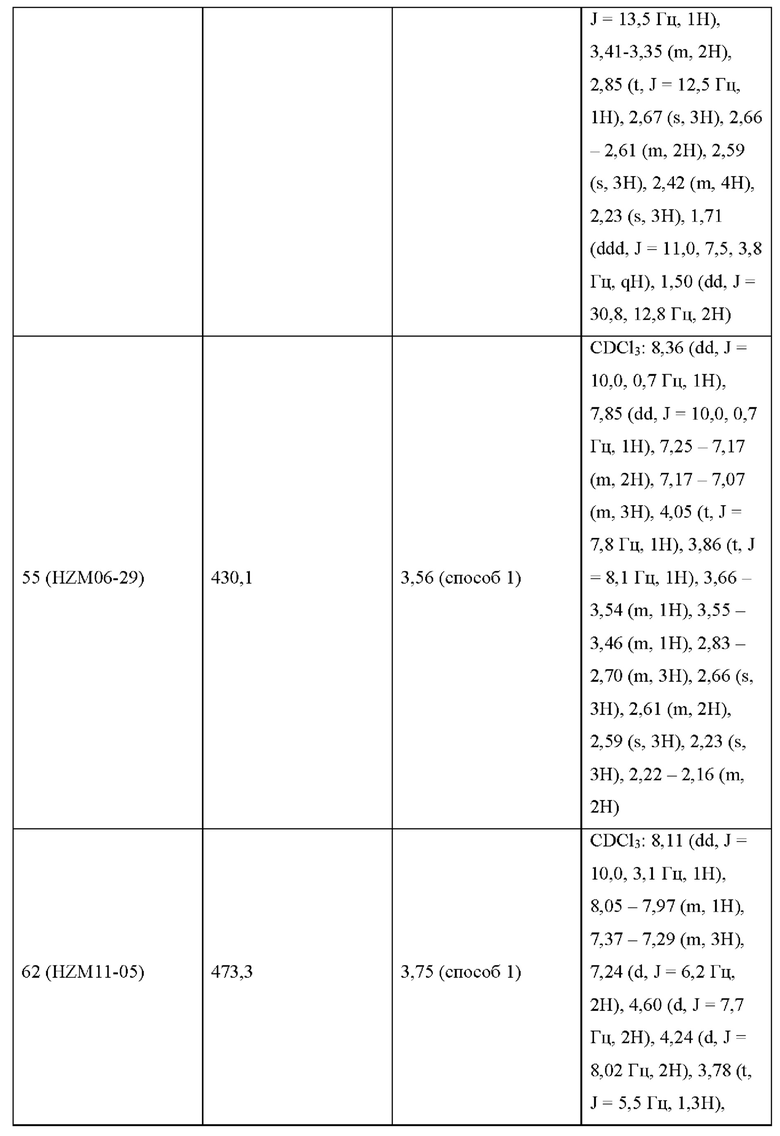
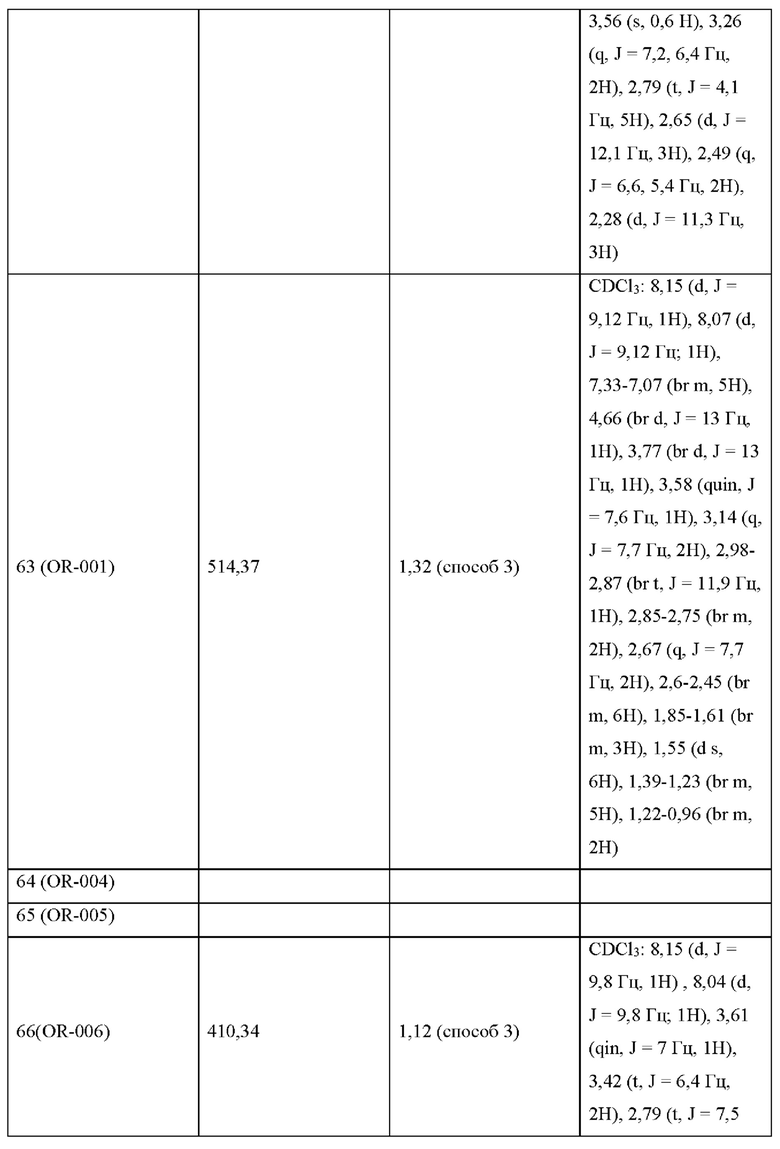
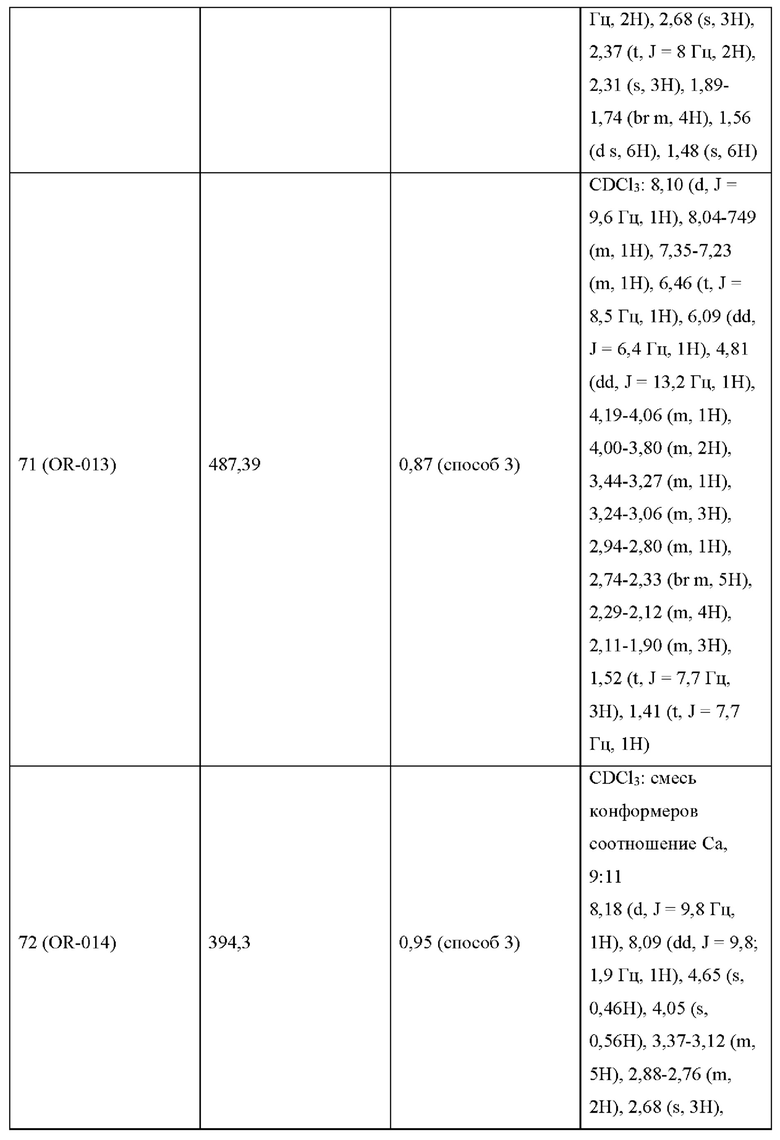
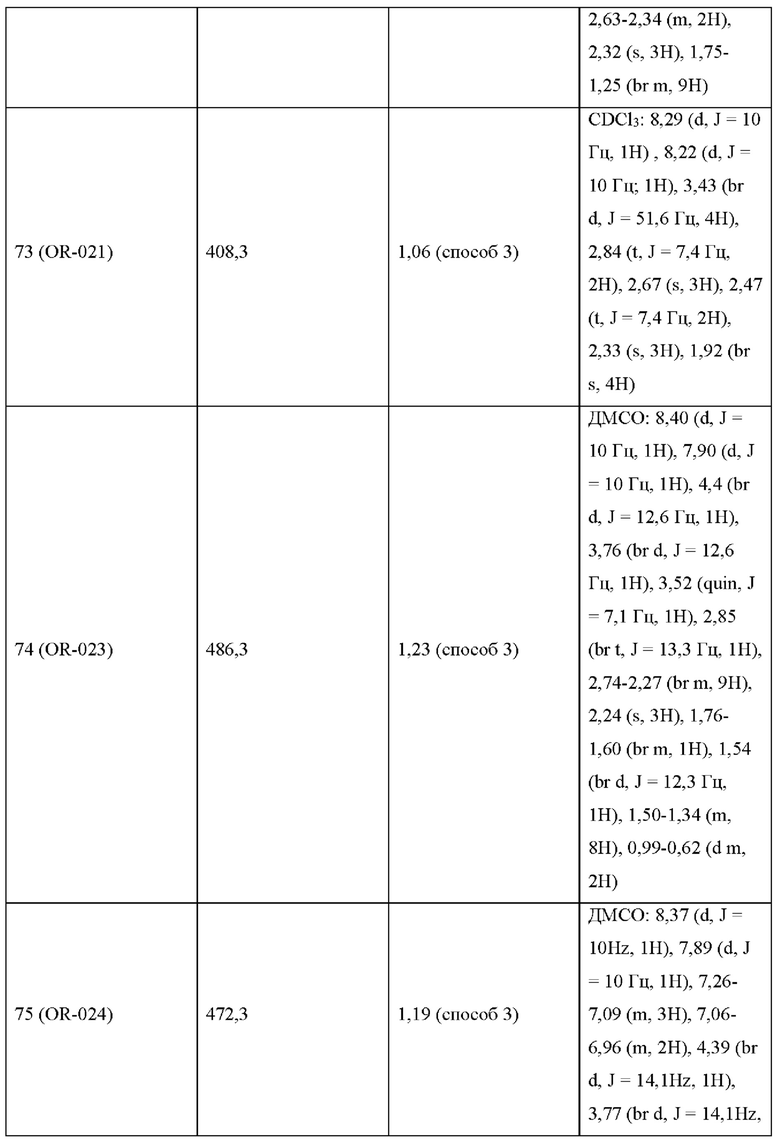
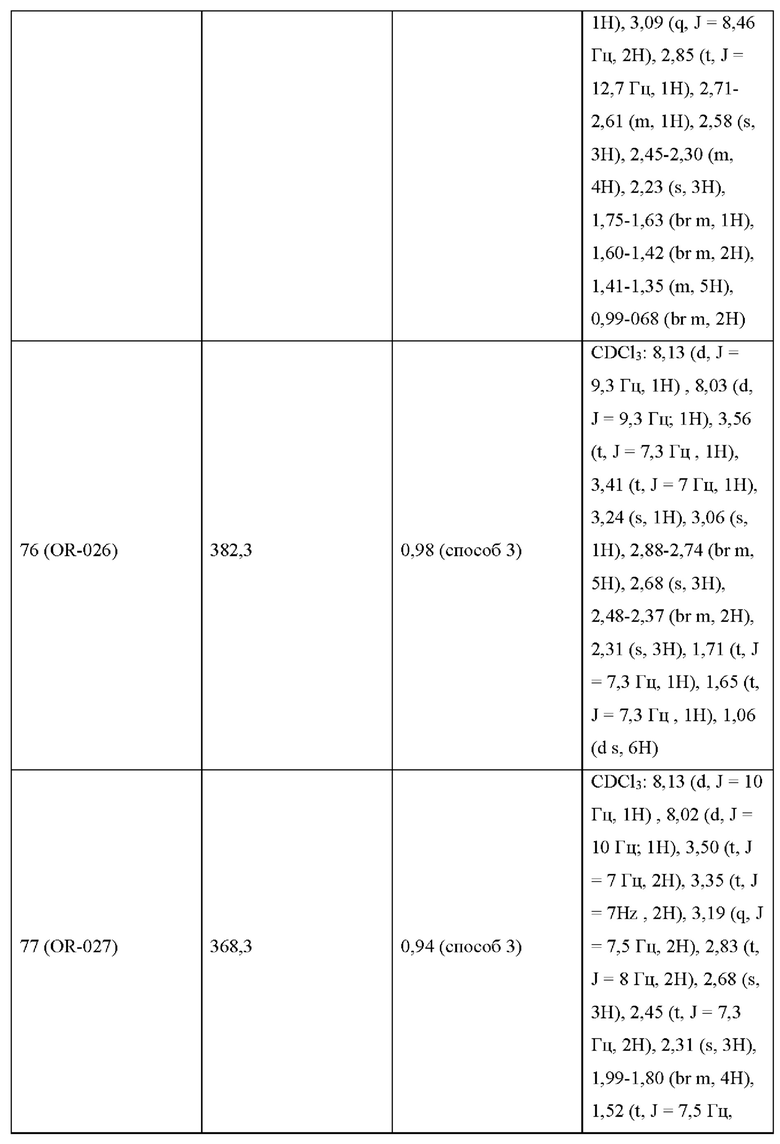
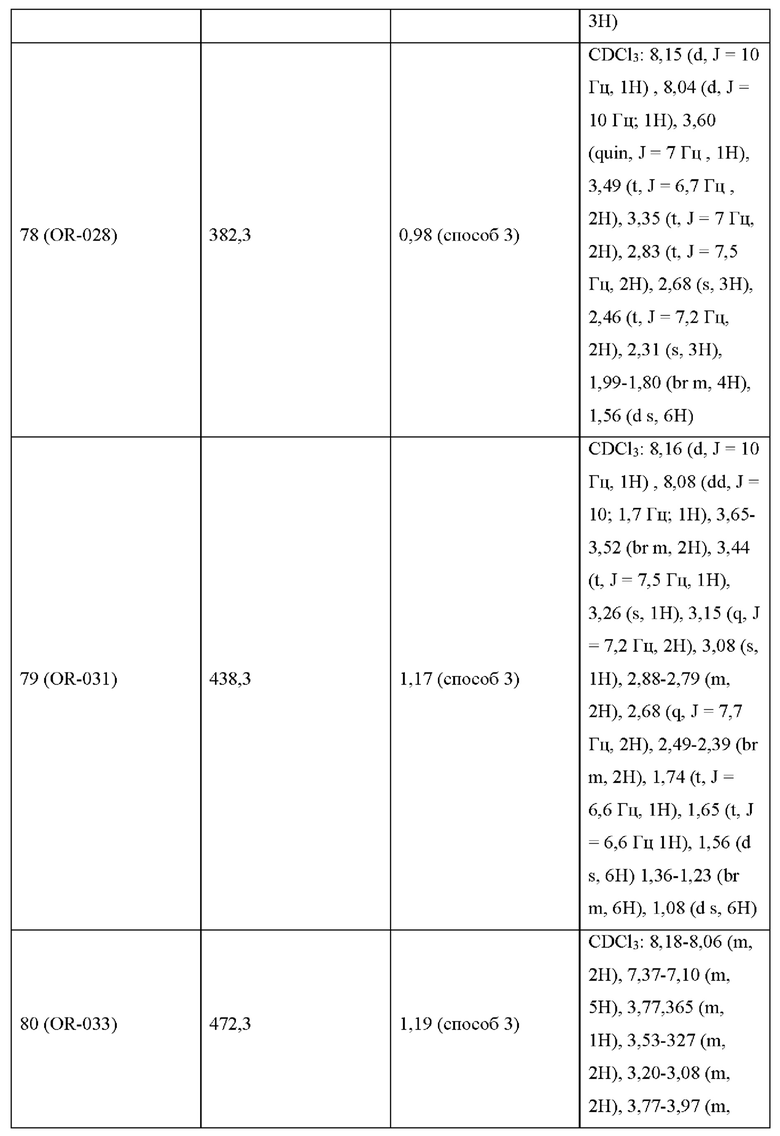
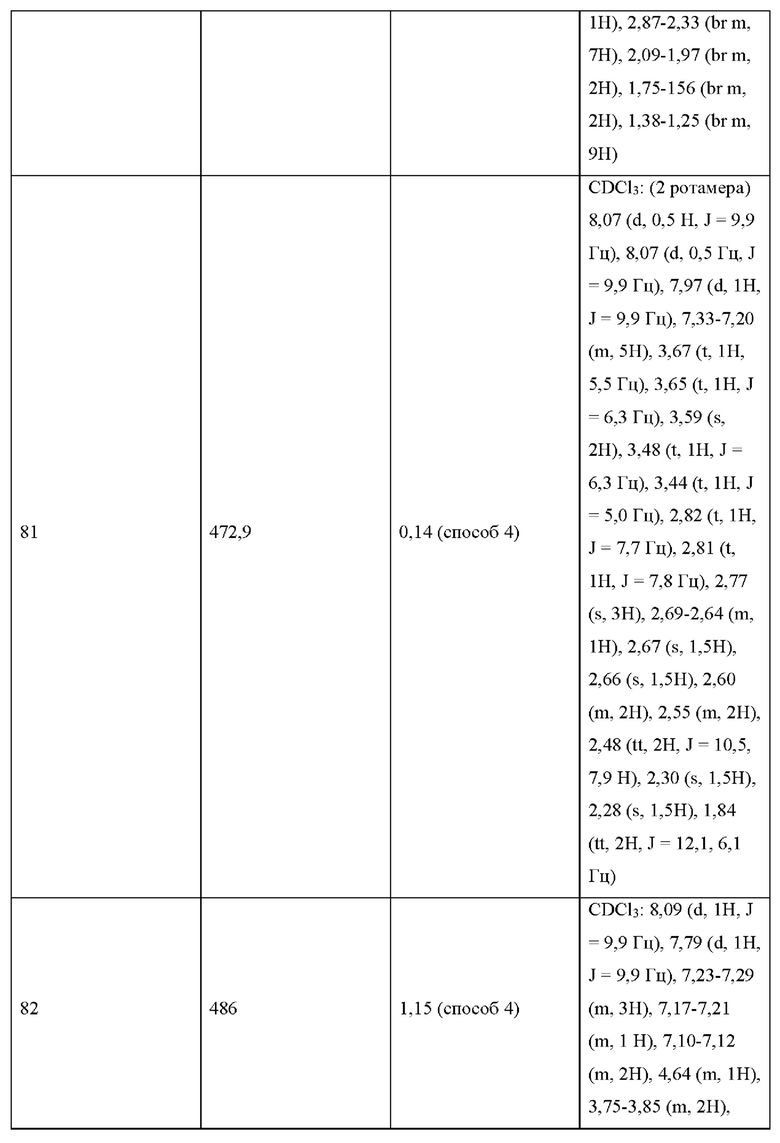
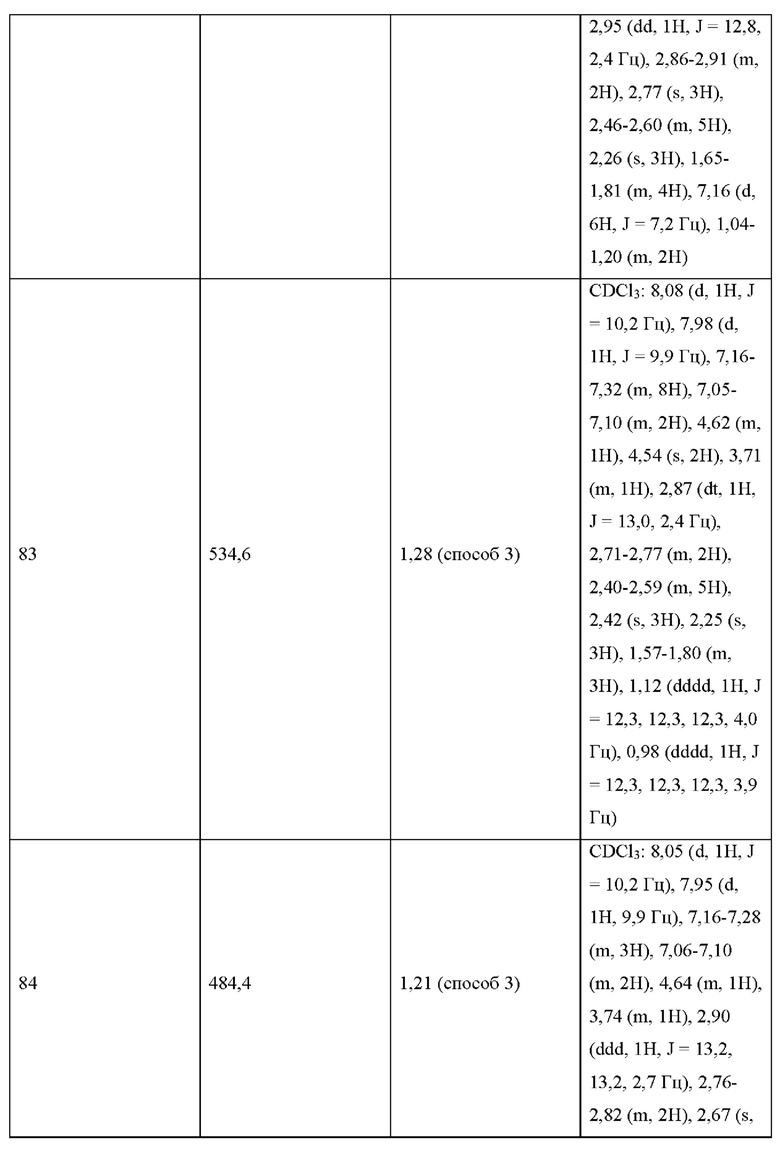
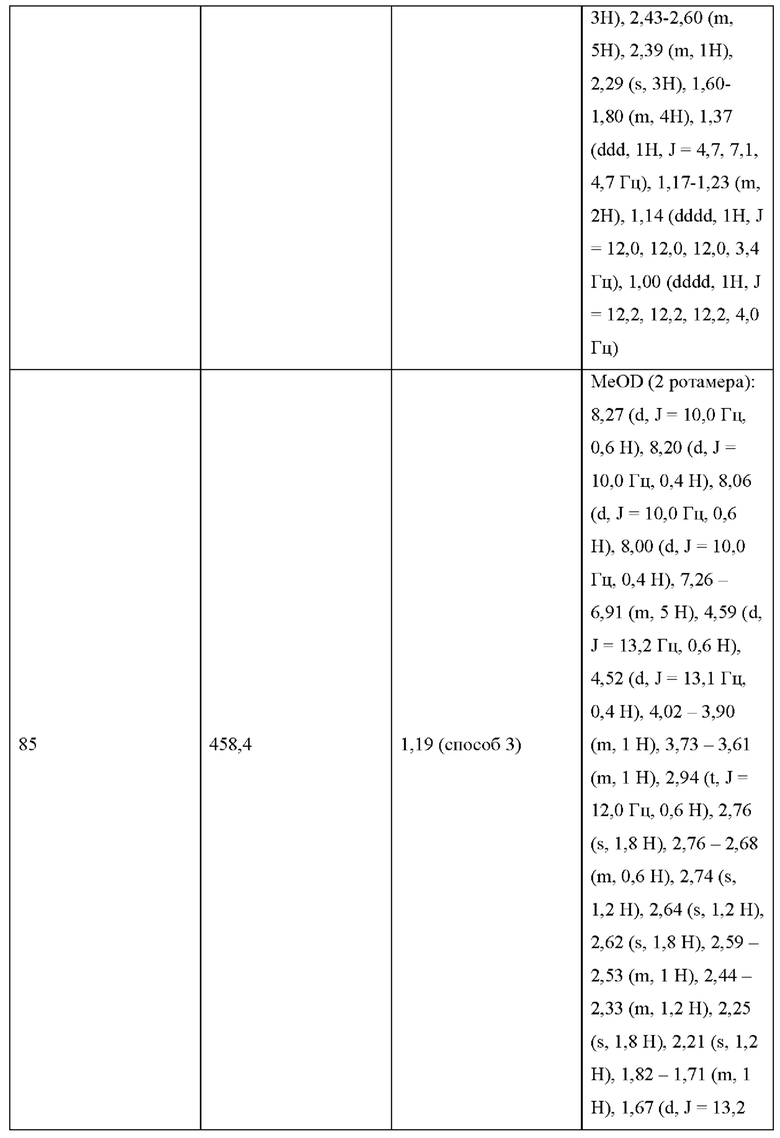
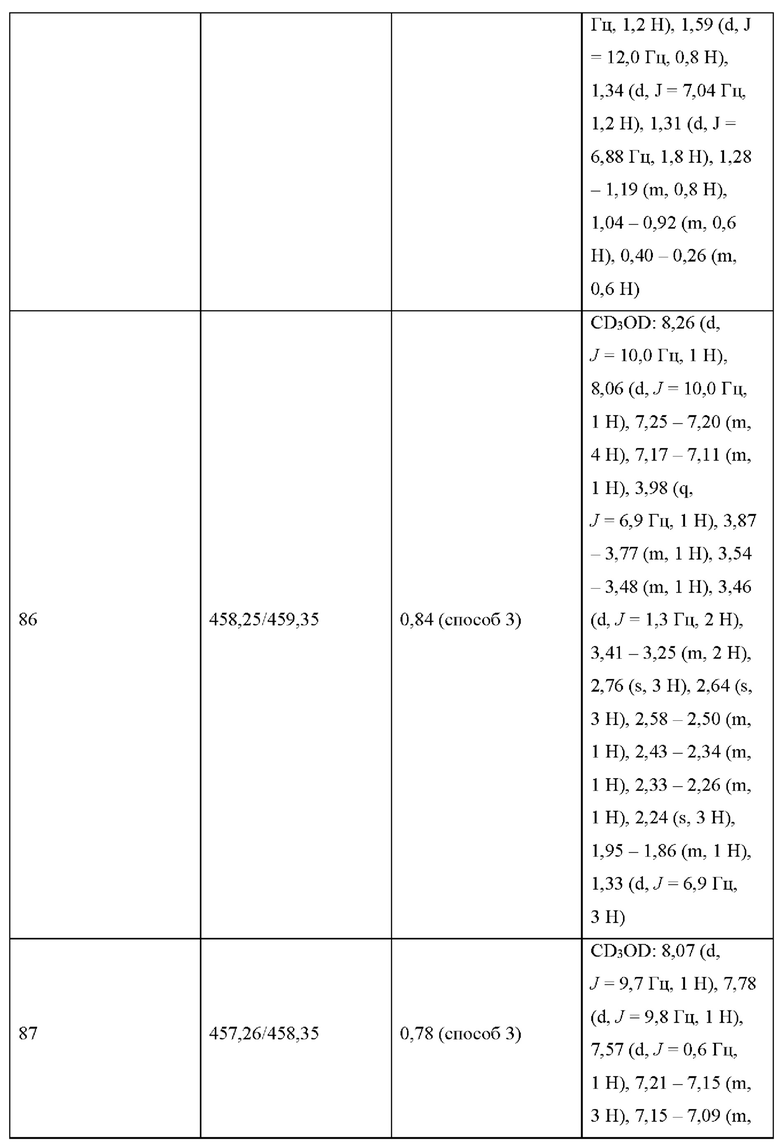
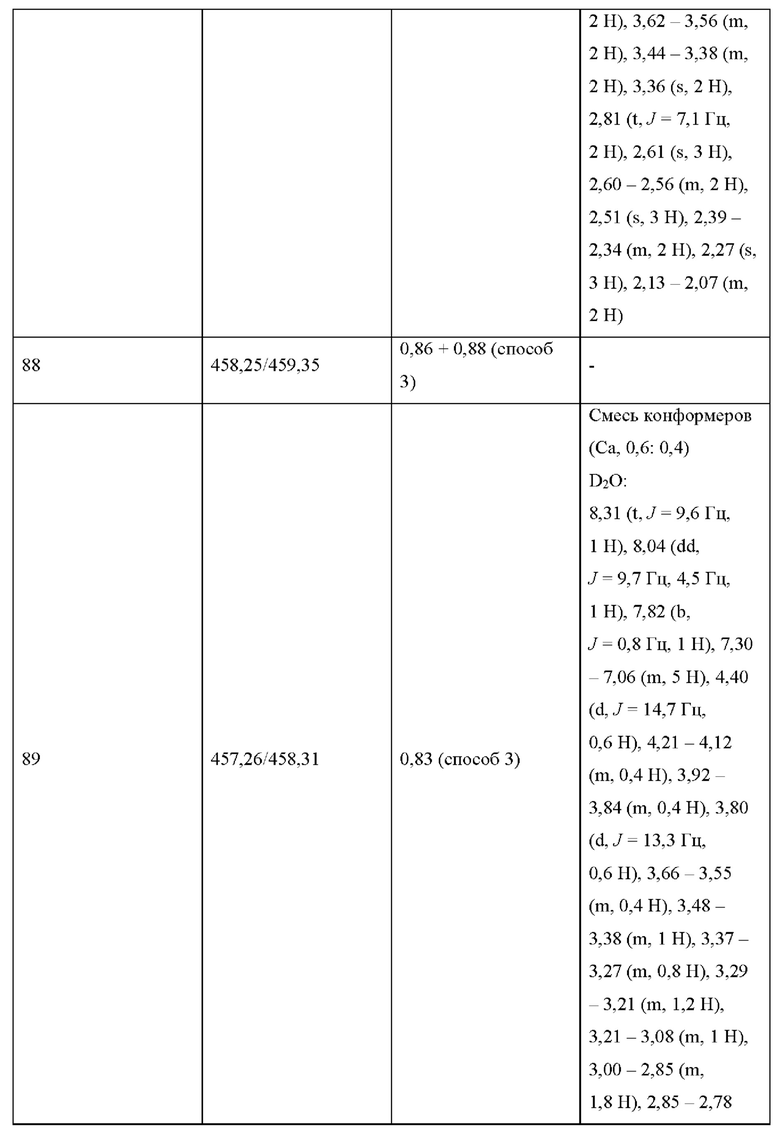
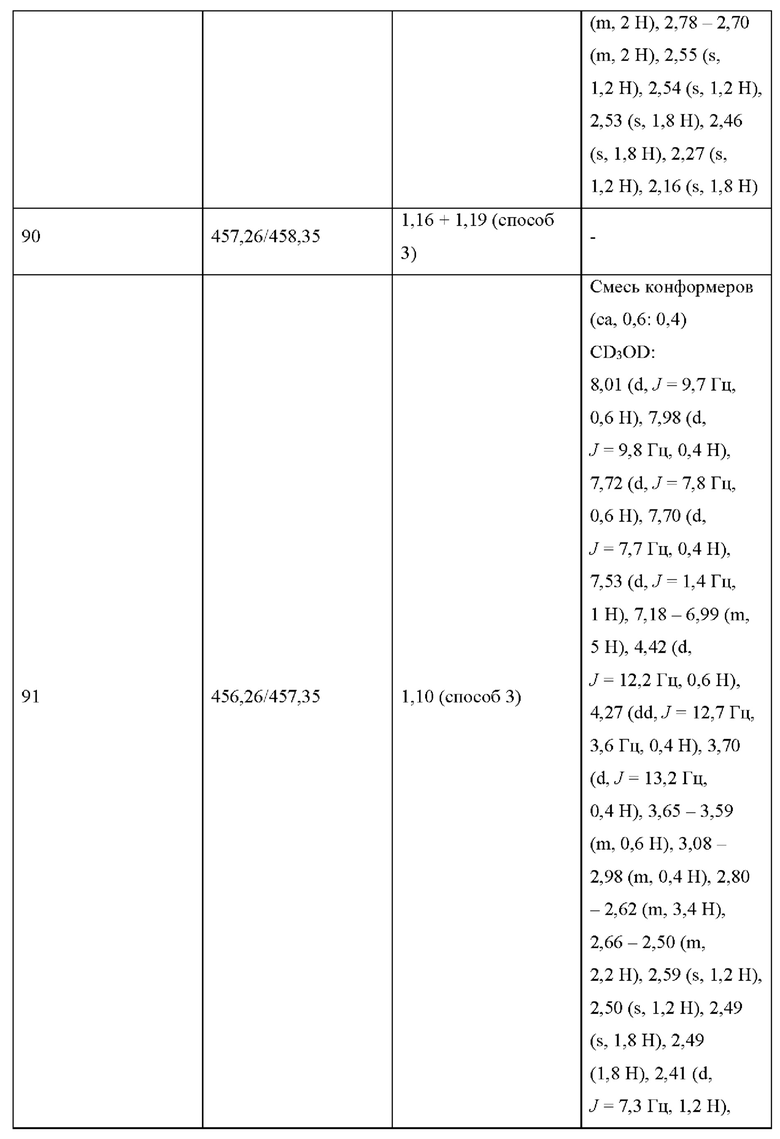
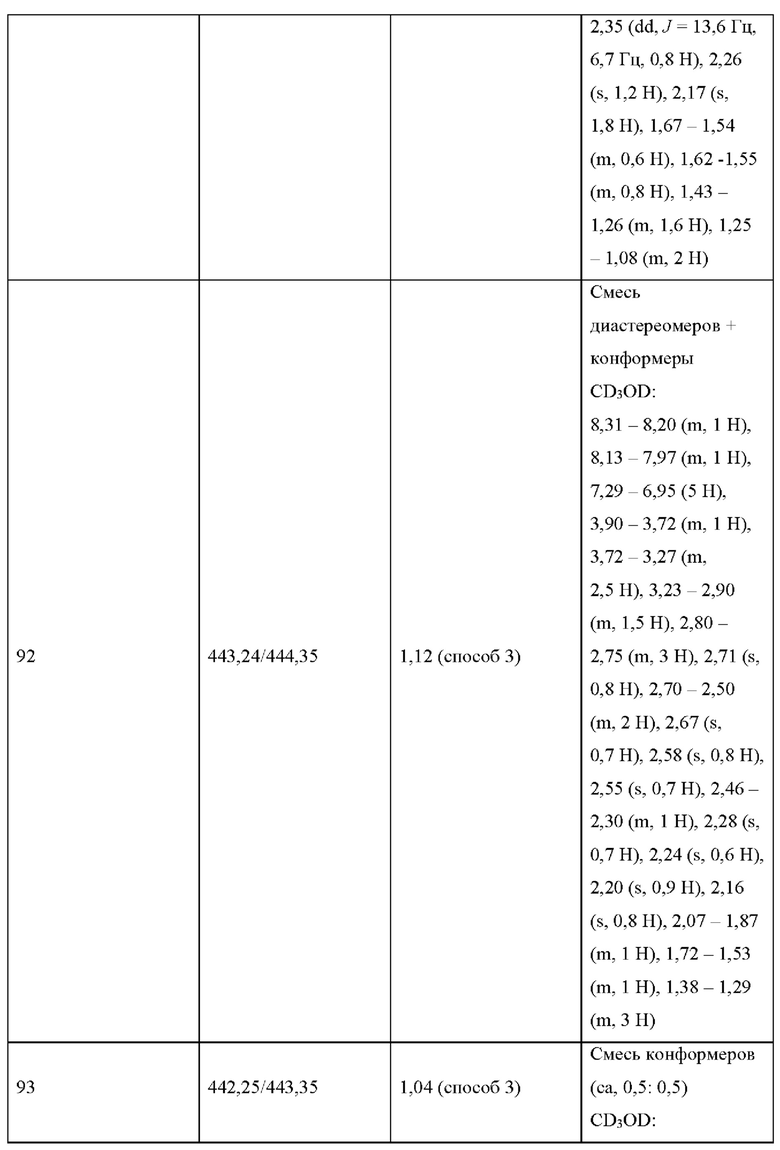
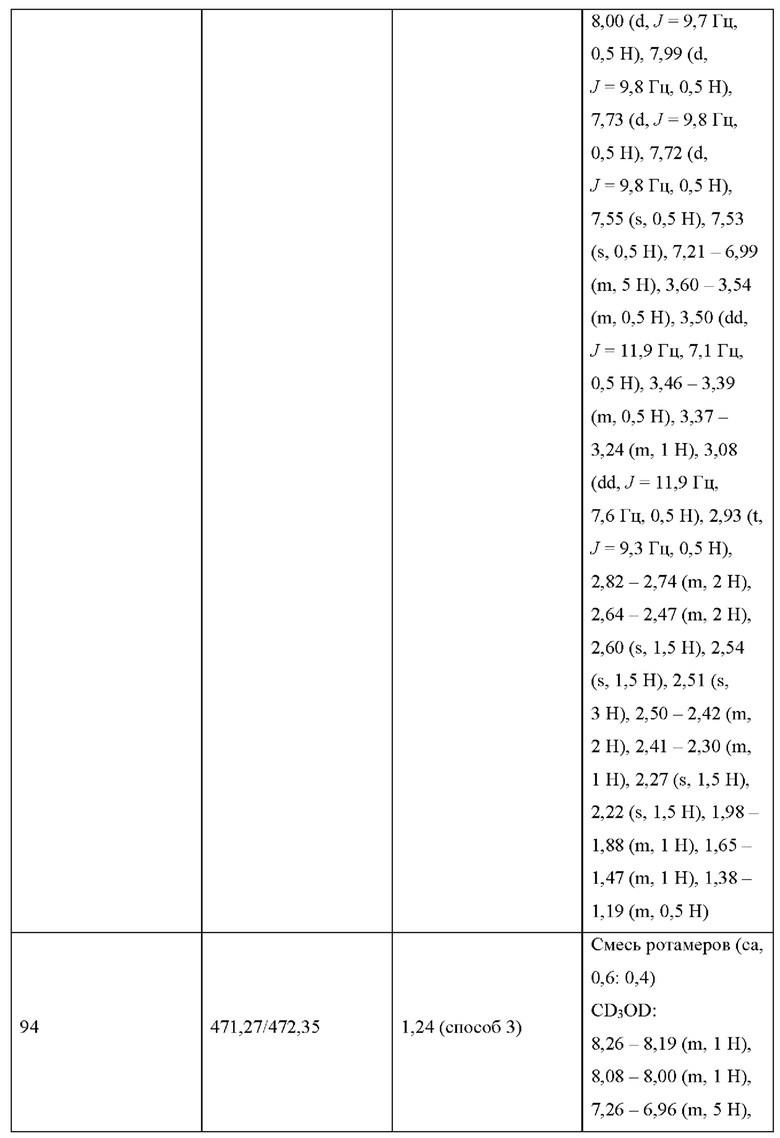
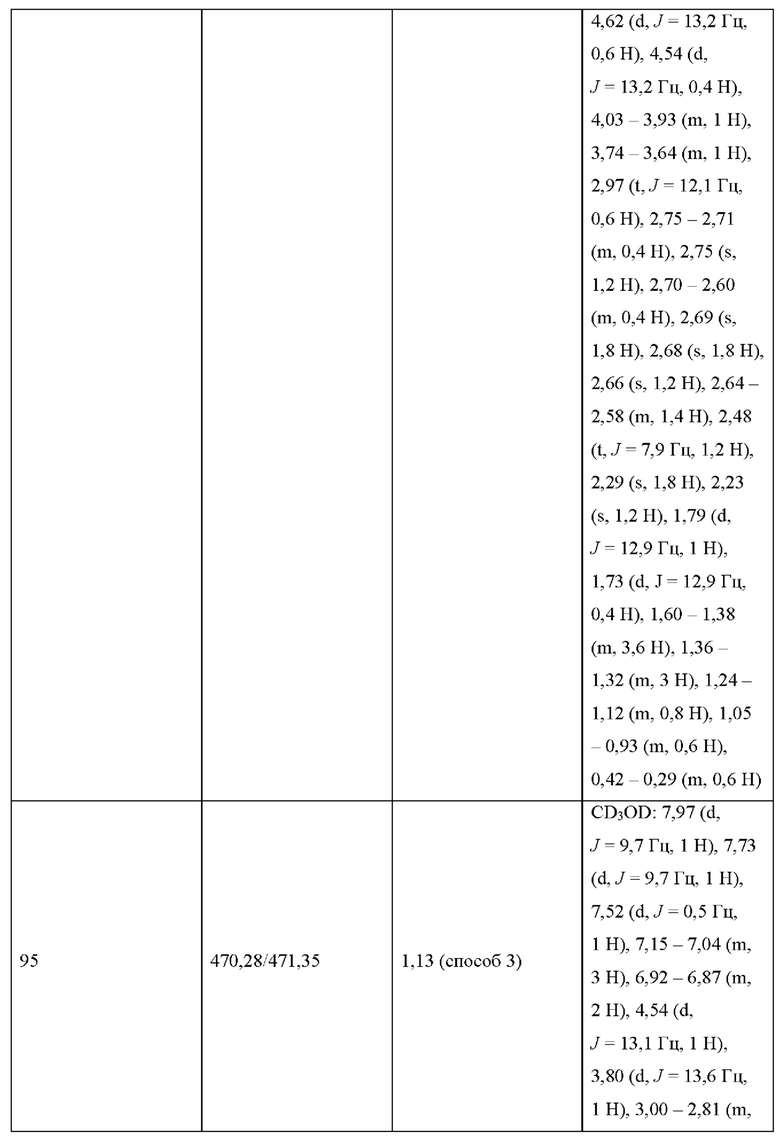
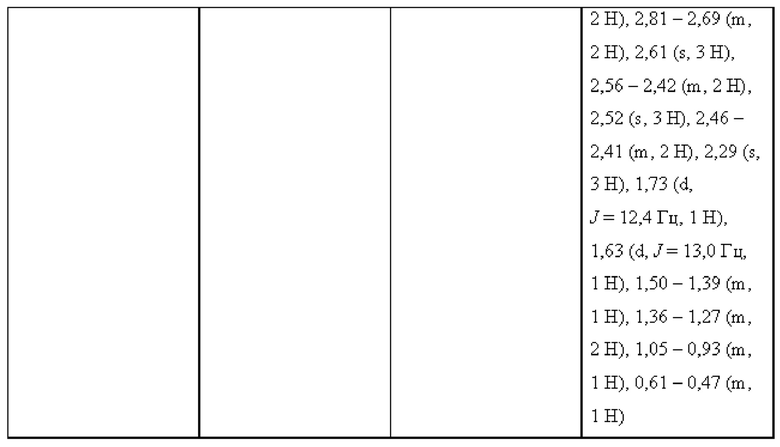
Способ ЖХ-МС 3: Система Waters Acquity Н UPLC CLASS с колонной Aqmty UPLC БЕН С18 (1,7 мкм, 2,1×50 мм), скорость потока 0,8 мл/мин, с использованием способа на основе 3-минутного градиента с подвижной фазой, состоящей из смеси вода/ацетонитрил (в каждый компонент было добавлено по 0,05% об./об. ТФУ): 95:5→5:95 (0-2,25 мин), 95:5 (2,27-3 мин).
Способ ЖХ-МС 4 (система Agilent для ЖХ, серия 1200; квадрупольный детектор Agilent 6130; колонка: колонка LUNA С18 (2) (3 мкм, 10×2 мм):

2.14) Получение 1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6)ил]-1H-пиразол-4-ил)пропан-1-тиона(45)
Примерный С25-140 растворяли в толуоле и добавляли реагент Лавессона (0,6 экв.). Реакционную смесь перемешивали при 110°С в закрытой пробирке в течение 3 часов. Неочищенную смесь концентрировали в вакууме и очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2/ProH) с получением желаемого продукта (выход 96%).
1H-ЯМР: (400 МГц, CDCl3): δ (м.д.)=8,08 (d, 1Н, J=9,9 Гц), 7,98 (d, 1H, J=9,9 Гц), 7,21-7,25 (m, 2Н), 7,16-7,21 (m, 1Н), 7,03-7,07 (m, 2Н), 5,67 (m, 1Н), 4,14 (rn, 1Н), 3,05 (ddd, 1Н, J=12,5, 12,5, 2,0 Гц), 2,85-3,00 (m, 5Н), 2,76 (s, 3Н), 2,66 (s, 3Н), 2,57 (dd, 1Н, J=13,5, 6,7 Гц), 2,44 (dd, 1H, J=13,3, 7,5 Гц), 2,29 (s, 3Н), 1,67-1,91 (m, 3Н), 1,35 (dddd, 1H, J=12,5, 12,5, 12,5, 3,3 Гц), 1,03 (dddd, 1H, J=12,3, 12,3, 12,3, 4.0 Гц).
ЖХМС: m/z=473,9, Rt=1,5 5 мин (способ 4).
2.15) Получение 6-(4-(3-(4-бегоилпиперидин-1-ил)-3,5-диметил-1Н-пиразол-1-ил)-3-метил-[1,2,4]триазол[4,3-6]пиридазина (46)
К раствору тиоамида 45 в ТГФ добавляли Mel (50 экв.). Реакционную смесь перемешивали при комнатной температуре до полного превращения, растворитель удаляли в вакууме. Неочищенную смесь растворяли в этилацетате и воде (1:1) и добавляли NaBH3CN (1,1 экв.). Смесь перемешивали до завершения реакции, реакцию останавливали добавлением 20% водного раствора гидроксида натрия и несколько раз экстрагировали с применением диэтилового эфира. Объединенные органические слои высушивали над Na2SO4, растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на диоксиде кремния (CH2Cl2/МеОН) с получением желаемого продукта (41%).
1H-ЯМР: (400 МГц, CDCl3): δ (м.д.)=8,06 (d, 1Н, 10,2 Гц), 7,99 (d, 1Н, J=9,9 Гц), 7,24-7,29 (m, 2Н), 7,16-7,21 (m, 1Н), 7,11-7,15 (m, 2Н) 2,97 (d, 1Н, J=12,3 Гц), 2,77 (s, 3Н), 2,63 (s, 3Н), 2,54 (d, 1Н, J=6,8 Гц), 2,37-2,47 (m, 5Н), 2,26 (s, 3Н), 1,95 (t, 2Н, J=11,3 Гц), 1,62-1,78 (m, 4Н), 1,48-1,61 (m, 1Н), 1,34-1,44 (m, 2Н).
ЖХ/МС: m/z=444,0, Rt=1,16 мин (способ 4).
3) Биологические и фармакологические исследования
3.1) Очистка рекомбинангного белка
3.1.1) Очистка Strep-меченого белка RZ1 TRAF6
Последовательности кДНК домена RING-Zincfinger1 (RZ1) (остатки 50-159) TRAF6 дикого типа и Ubc13-связывающего мутанта (TRAF6 D57K) клонировали в вектор pASK-IBA3+ (IBA Life Sciences). Эти белки экспрессировали в клетках BL21-CodonPlus-RIPL (Agilent Technologies) в среде Luria/Miller, дополненной 100 мкМ ZnCl2, и очищали с использованием системы Strep-Tag®-Strep-Tactin®, разработанной IBA Life Sciences, с использованием очистителя АКТА (GE Healthcare Life Sciences).
3.1.2) Очистка немеченого белка RZ1 TRAF6
Последовательность кДНК домена RZ1 TRAF6 дикого типа клонировали в вектор pGex 4Т1 (GE Healthcare Life Sciences). Для очистки немеченого RZ1 TRAF6 белок экспрессировали в клетках BL21-CodonPlus-RIPL в минеральной среде М9, дополненной 100 мкМ ZnCl2. Для 15N - меченого белка обычный NH4 заменяли 15NH4. Очистку белка проводили с использованием гранул Glutathione Sepharose® 4 Fast Flow (GE Healthcare Life Sciences), расщепления тромбином (Sigma-Aldrich) и эксклюзионной хроматографии (Superdex 75, GE Healthcare Life Sciences).
3.1.3) Очистка Flag-His-меченого белка Ubc13 (Ubc1 3-FH)
Последовательность кДНК для полноразмерного Ubc13 и метку Flag-His клонировали в вектор pGex 4Т1. GST-меченый белок экспрессировали в клетках BL21-CodonPlus-RIPL в среде LB и очищали с использованием гранул Glutathione Sepharose® 4 Fast Flow, расщепления тромбином и эксклюзионной хроматографии (Superdex 75, GE Healthcare Life Sciences).
3.1.4) Очистку белков GST (включая GST-OTUB1 и GST-Ubcl3) проводили, как описано ранее в Weber, Е., Rothenaigner, I., Brandner, S., Hadian, K. & Schorpp, K. A High-Throughput Screening Strategy for Development of RNF8-Ubcl3 Protein-Protein Interaction Inhibitors. SLASDiscov 22, 316-323 (2017).
3.1.5) His-меченый белок Uevla очищали в соответствии с протоколом GST-OTUB1 следующим буфером: буфер для лизиса (20 мМ NaH2PO4, 20 мМ NaCl, 50 мМ имидазола), буфер для элюирования (20 мМ NaH2PO4, 20 мМ NaCl, 500 мМ имидазола) и буфер для обессоливания (20 мМ NaH2PO4, 20 мМ NaCl).
3.2) Идентификация и характеристика ингибиторов PPI TRAF6-Ubcl3 в биохимических количественных исследованиях
3.2.1) ALPHAScreen
Чтобы идентифицировать небольшие молекулы, которые специфически нацелены на домен RZ1 TRAF6 и за счет этого предотвращают его связывание с Ubc13, выбрали технологию ALPHAScreen (PerkinElmer) для детектирования взаимодействия TRAF6wTStrepII-Ubc13FH при скрининге с высокой пропускной способностью (ВПС), а также анализ зависимости активности от структуры (SAR).
Для проведения автоматического количественного исследования ALPHAScreen все следующие компоненты предварительно разводили в буфере ALPHAScreen (1×ФСБ, 0,5% БСА, 0,01% твин-20). Сначала белок TRAF6 (конечная концентрация 100 нМ, объем 30 мкл) добавляли в 384-луночные оптические планшеты, включающие систему распределения MultiFlo (BioTek), с последующим переносом соединений с помощью станции для переноса Sciclone G3 (PerkinElmer). После добавления Ubcl3FH (конечная концентрация 75 нМ, объем 10 мкл) с помощью системы MultiFlo и инкубации в течение одного часа при комнатной температуре оба типа гранул (донорные гранулы Strep-Tactin Alpha и акцепторные гранулы Nickel-Chelate; конечные концентрации 4 мкг для каждого типа, по 10 мкл каждого типа) смешивали и добавляли с использованием системы MultiFlo при приглушенном свете. Считывание с планшетов происходило через еще один час инкубации при комнатной температуре. Статистические параметры, включая коэффициент изменчивости, фактор Z' и окно сигнала, рассчитывали для определения качества кампании ВПС. Чтобы оценить эффективность соединений, единицы сигнала ALPHAScreen образцов, обработанных соединением, нормировали к сигналам контрольных образцов, обработанных ДМСО. Соответственно, мутант TRAF6d57K служил контролем для минимального сигнала и был включен в каждый планшет для скрининга. Соединения, которые ингибировали TRAF6wTStrepII Ubcl3FH более чем на 25%, рассматривали в качестве активных агентов. После устранения ложноположительных соединений в ALPHAScreen и ложноположительных соединений при анализе His-метки (Schorpp, K., I. Rothenaigner, Е. Salmina, et al., Identification of Small-Molecule Frequent Hitters from AlphaScreen High-Throughput Screens. J Biomol Screen, 2014. 19(5): p. 715-26) 178 соединений определяли в качестве первичных пиков и затем испытывали в количественном исследовании с пятиточечным последовательным разведением (2,5-40 мкм) в экспериментах ALPHAScreen TRAF6wTStrepII Ubc13FH. Для дальнейших исследований отбирали только соединения с дозозависимыми эффектами на TRAF6wTStrepII-Ubc13FH (n=27). Для исследования аналогов все соединения испытывали в экспериментах ALPHAScreen TRAF6wTStrepII Ubc13FH с титрованием по десяти точкам (0,2-100 мкМ). Одновременно с этим соединения испытывали с помощью набора ALPHAScreen TruHits Kit (PerkinElmer), чтобы исключить неспецифические помехи в технологии ALPHAScreen.
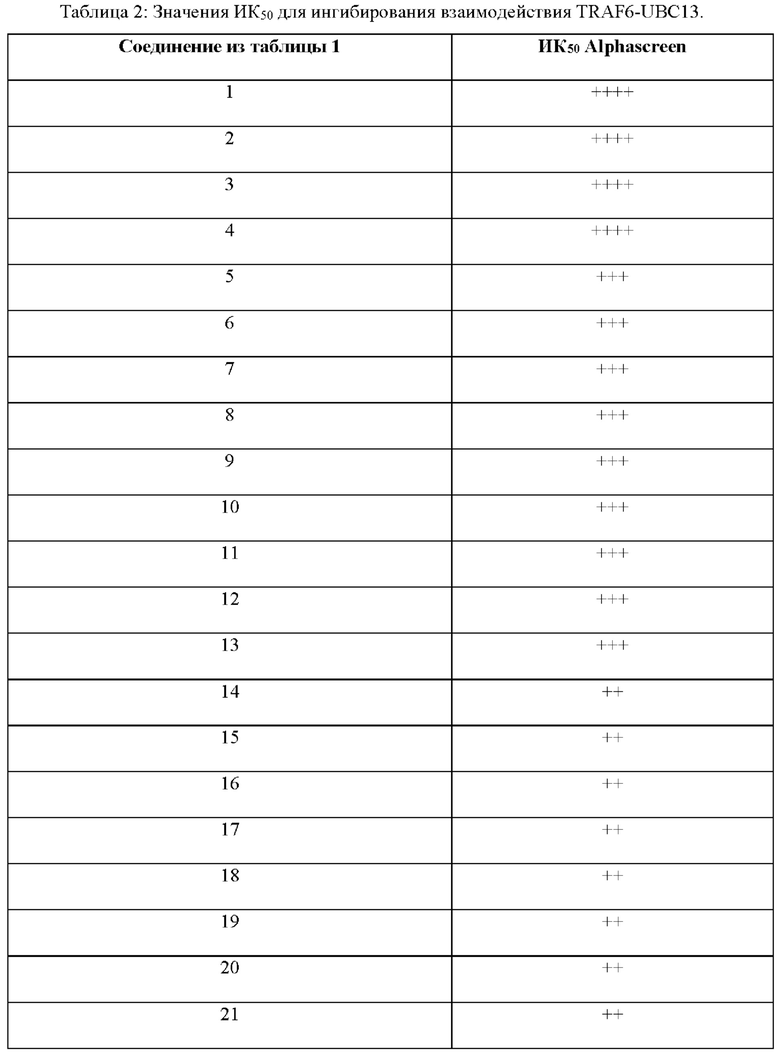

Дополнительные результаты AlphaScreen приведены на Фигуре 20.
3.2.2) Двумерный ядерный магнитный резонанс (2D-ЯМР)
Чтобы определить, могут ли ингибирующие соединения непосредственно связывать TRAF6, но не Ubc13, проводили эксперименты с помощью 2D-ЯМР. 15N-маркированный белок PvZl TRAF6 (с=120 мкМ), не содержащий метку, и соединение (с=20 мМ) инкубировали в соотношении 1:5 в 3 мм пробирке для ЯМР в течение 10 минут перед получением спектров. Двумерные спектры SOFAST-HMQC получали, чтобы обнаружить возмущения при химическом сдвиге после добавления соединения. Все измерения проводили с использованием спектрометра Bruker Avance 600 МГц.
3.2.3) Количественное исследование убиквитинирования в условиях in vitro
3.2.3.1) Количественные исследования на основе Вестерн-блоттинга
Способность TRAF6 образовывать K63-соединенные полиубиквитиновые цепи в сочетании с Ubc13 исследовали после обработки соединением. Соответственно, немеченый TRAF6WT и TRAF6D57K, соответственно, рекомбинантно очищали. По 0,125 мкМ белка TRAF6 предварительно инкубировали с ДМСО или соединением в общем объеме 100 мкл в буфере для количественного исследования K63 (25 нМ Трис-HCl, рН=7,6; 250 нМ MgCl2; 500 нМ креатинфосфата; 0,3 ед./мл неорганической пирофосфатазы; 0,3 ед./мл креатинфосфокиназы) в течение 30 минут при комнатной температуре. Брали небольшие аликвоты для вносимых образцов. Исходную смесь, содержащую 0,01 мкМ E1-активирующего фермента (UBE1) (Boston Biochem), 0,2 мкМ Е2-конъюгирующего ферментного комплекса (Ubc13/Uev1a) (Boston Biochem), 1 мМ ZnCl2, 2 мМ АТФ и 4 мкМ моноубиквитина (Boston Biochem), добавляли к белку TRAF6, и реакционную смесь инкубировали в течение 120 мин при 37°С. Реакцию останавливали добавлением 4× ДСН-содержащего буфера для загрузки и кипятили при 95°С в течение 5 минут. Вносимые образцы исследовали с использованием набора для окрашивания серебром Pierce Silver Stain (Thermo Fisher Scientific). Убиквитинирование детектировали методом Вестерн-блоттинга с использованием антитела к убиквитину.
Для исследования полиубиквитинирования, опосредованного cIAP1, MDM2, TRIM63, RNF4, ITCH и Е6АР, использовали соответствующий набор убиквитинлигаз от Boston Biochem (Boston Biochem (K-102, K-260, K-200 В, K-220, K-240 и K-270). В настоящем документе реакционный буфер, фермент E1, Е2 и Е3 (и субстрат р53 в случае MDM2) смешивали и предварительно инкубировали с соединением или ДМСО в течение 5 минут при комнатной температуре перед добавлением Mg2+-ATP и моноубиквитина, чтобы начать реакцию. Убиквитинирование визуализировали методом Вестерн-блоттинга.
3.2.3.2) Получение убиквитинового профиля (Eurofins Pharma Discovery Services, Великобритания)
C25-140 отправляли в Eurofins Pharma Discovery Services (Великобритания) для проведения одноточечного определения убиквитинового профиля при действии С25-140 в концентрации 30 мкМ на 24 различных каскада реакций убиквитинирования с использованием различных комбинаций Е2, Е3 и субстрата. Каждую реакцию измеряли с двумя повторами. Каскадную активность выражали в процентах от средней каскадной активности в образцах для положительного контроля. Значение положительного контроля считали равным 100%, и все испытанные образцы нормировали к этому значению.
3.3) Проверка ингибиторов PPI TRAF6-Ubc13 в клеточных количественных исследованиях
3.3.1) Количественное исследование изменения электрофоретической подвижности (EMSA) для измерения активации NF-κВ
Для обработки соединением клетки линии MEF высевали в 6-луночные планшеты в количестве 1,2×105 клеток/лунку за день до обработки. Т-клетки линии Jurkat высевали непосредственно перед обработкой в количестве 2×105 клеток/образец. Через шесть часов после обработки соединением или обработки ДМСО клетки симулировали, как указано. Соответственно, клетки линии MEF оставляли необработанными или стимулировали рекомбинантным мышиным ИЛ-1β (1 нг/мл) или ФНО-α (10 нг/мл) в течение 20 минут. Т-клетки линии Jurkat стимулировали форбол-12-миристат-13-ацететом (РМА) (400 нг/мл) и иономицином (600 нг/мл) в течение 30 минут. В завершение, клетки лизировали в буфере для лизиса с высоким содержанием соли (20 мМ HEPES, рН=7,9; 350 мМ NaCl; 1 мМ MgCl2; 0,5 мМ ЭДТА; 0,1 мМ ЭГТА; 20% глицерина; 1% NP-40; 10 мМ NaF; 8 мМ β-глицерофосфата; 1 мМ ДТТ; 300 мкМ ванадата натрия; полного ингибитора протеаз) и проводили количественные исследования EMSA с использованием зондов Н2Л (NF-κB) или Oct-1 (контроль загрузки). Для EMS А применяли следующие олигонуклеотиды:

3.2. Количественное исследование киназы IKK
Высевали 5,5×105 клеток линии MEF, затем обрабатывали соединением в течение 6 часов и в завершение стимулировали ИЛ-1β или ФНО-α в течение 8 минут. 1×106 Т-клеток линии Jurkat обрабатывали соединением в течение 6 часов и в завершение стимулировали РМА (400 нг/мл) и иономицином (600 нг/мл) в течение 8 минут. После лизиса клеток в буфере для лизиса Со-IP (150 мМ NaCl; 25 мМ HEPES (рН=7,5); 0,2% NP-40; 1 мМ глицерина; 10 мМ NaF; 8 мМ β-глицерофосфата; 1 мМ ДТТ; 300 мкМ ванадата натрия; полный ингибитор протеаз) отбирали вносимые образцы, и супернатант затем инкубировали с 6 мкл антитела к IKKγ, FL-419 (Santa Cruz Biotechnology). После инкубации с гранулами белок G-сефароза (GE Healthcare) гранулы промывали в промывочном буфере Co-IP (150 мМ NaCl; 25 мМ HEPES (рН=7,5); 0,2% NP-40; 1 мМ глицерина) и переносили в буфер для киназной реакции (20 мкМ HEPES, рН=7,9; 10 мМ MgCl2; 20 мкМ АТФ; 20 мМ β-глицерофосфата; 200 мкМ ванадата натрия; 1 мМ ДТТ) с добавлением 1,5 мг GST-IκBα (1-53) и 0,5 мкл 32Р-γ-АТФ. После 30 минут инкубации при 37°С реакцию останавливали и исследовали методом авторадиографии, а также Вестерн-блоттинга после разделения на ДСН-ПААГ.
3.3.3) Исследование экспрессии мРНК
Высевали 1,2×105 клеток линии MEF на образец, обрабатывали соединением в течение 6 часов и стимулировали либо ИЛ-1β, либо ФНО-α в течение 60 минут. Т-клетки линии Jurkat и первичные мышиные CD4+ Т-клетки высевали в количестве 2×105 клеток на образец, также обрабатывали соединением в течение 6 часов и стимулировали РМА/иономицином в течение 3 часов. РНК выделяли в соответствии с протоколом производителя набора Qiagen RNeasy (клетки линии MEF) или мини-набора STRATEC INVITRAP® SPIN UNIVERSAL RNA (Т-клетки линии Jurkat). Выделение мРНК из эпидермальной белой жировой ткани мышей DIO выполняли с использованием реагента TRIzol® в соответствии с инструкцией производителя. Для очистки выделенной РНК от геномной ДНК образцы (до 5 мкг РНК) обрабатывали ДНКазой I, не содержащей РНКаз, RQ1 (Promega), как описано в техническом руководстве. Для обратной транскрипции РНК в кДНК образцы РНК, расщепленные ДНКазой I, обрабатывали с использованием системы для синтеза первой цепи ДНК SuperScript III (Invitrogen/ThermoFisher Scientific) в соответствии с протоколом изготовителя. Соответственно, случайные гексамеры, поставляемые с набором, использовали для обратной транскрипции. Образцы кДНК хранили при -20°С. Для количественного определения кДНК в образцах использовали LightCylcer480 (Roche) и набор для кПЦР KAPA SYBR FAST qPCR. Для количественной оценки применяли способ ΔΔСр, впервые описанный Pfaff1 (Pfaff1, M.W., A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res, 2001. 29 (9): p. e45). Для кПЦР в реальном времени применяли следующие праймеры:

3.3.4) ALPHASurefire
Для детектирования уровней белка р-IκВα (Ser32/36), а также общего белка IκВα по 1×104 клеток линии MEF высевали в 96-луночные планшеты, обрабатывали соединением в течение 6 часов и стимулировали 1 нг/мл ИЛ-1β в течение 7 минут. Уровни белка р-IκВα и общего белка IκВα исследовали с использованием наборов ALPHASurefire (PerkinElmer) в соответствии с инструкциями производителя. Выполняли общий протокол в 2 планшетах.
3.3.5) Иммупопреципитация TRAF6 для детектирования эндогенного убиквитинирования
Высевали 2,5×106 клеток линии MEF, обрабатывали соединением в течение 6 часов и стимулировали ИЛ-1β (3,5 нг/мл) в течение 10 минут. Клетки лизировали в буфере для лизиса Co-IP, содержащем 1% ДСН, для устранения всех клеточных взаимодействий. После лизиса клеток и удаления внесенных образцов, образцы разбавляли до содержания 0,1% ДСН и инкубировали с 5 мкл антитела к TRAF6, EP591Y (Abeam), и вызывали иммунопреципитацию с использованием гранул белок G-сефароза. После промывки гранул в промывочном буфере Co-IP белок TRAF6 элюировали и уровни убиквитинирования TRAF6 определяли методом Вестерн-блоттинга.
3.3.6) Твердофазный иммуноферментный анализ (ИФА)
Для измерения секреции цитокинов высевали по 2×105 Т-клеток линии Jurkat или первичных CD4+ Т-клеток или МКПК человека на образец, обрабатывали соединением в течение 6 часов и стимулировали, как указано. Через 20 часов после стимуляции супернатанты собирали и исследовали с помощью наборов для ИФА ИЛ-2, ИЛ-1β и ФНО-α Ready-Set-Go! (2 поколения), поставляемых Affymatrix eBioscience.
3.3.7) Исследования коиммунопреципитации в клетках линии HEK 293Т
Для исследования взаимодействия TRAF6 и Ubc13 в клетках высевали 1×106 клеток линии 293Т, и через 18 часов клетки трансфецировали 1 мкг пустой плазмиды pEF-HA или плазмиды pEF-HA-TRAF6 RZ1 с использованием реагента для трансфекции ДНК X-tremeGENE HP (Roche). Через 6 часов после трансфекции клетки обрабатывали соединением или ДМСО. После 42 часов дополнительной инкубации клетки собирали в буфере для лизиса Co-IP, и HA-TRAF6 иммунопреципитировали с помощью аффинного матрикса против НА (Roche). Связывание Ubc13 с TRAF6 исследовали методом Вестерн-блоттинга.
3.4) Исследования жизнеспособности, клеточного цикла, ADME и ФК
3.4.1) Количественное исследование жизнеспособности
Для экспериментов по оценке жизнеспособности клеток с использованием системы CellTiter-Glo® 2.0 (Promega) клетки линии MEF (1,2×103 клеток/лунку), клетки линии HepG2 (4×103 клеток/лунку) и Т-клетки линии Jurkat (5×103 клеток/лунку) высевали в 384-луночные планшеты. Через 18 часов клетки обрабатывали соединениями с использованием автоматической системы Sciclone G3. Через 6 или 24 часа инкубации измерение люциферазы выполняли в соответствии с руководством производителя.
3.4.2) Оценка клеточного цикла
Для окрашивания клеток на разных стадиях клеточного цикла клетки высевали в количестве 2×105 клеток на образец и обрабатывали соединением или ДМСО. После указанного времени инкубации (24 или 72 часа) клетки фиксировали в 100% этаноле и затем окрашивали окрашивающим раствором иодида пропидия (0,1% тритона Х-100; 20 мкг/мл йодида пропидия; 200 мкг/мл РНКазы А в ФСБ) в течение 40 мин в темноте. Флуоресценцию йодида пропидия регистрировали с использованием проточного цитометра Attune (Thermo Fisher Scientific). Исследование проводили с использованием программного обеспечения для анализа отдельных клеток FlowJo.
3.4.3) Определение размножения линии клеток остеосаркомы U2OS
Для исследования размножения линии клеток остеосаркомы U2OS по 5×103 клеток высевали в 96-луночные планшеты. Через 18 часов клетки обрабатывали С25-140 (соединение 3) или ДМСО в течение 6 часов. Используя источник цезия-137 (HWM-D2000), отобранные образцы облучали в дозе 2 Гр или 4 Гр. Размножение клеток измеряли каждые 24 часа в течение 7 дней путем подсчета Hoechst33342-положительных клеток в системе визуализации Operetta High-Content (PerkinElmer) и с помощью программного обеспечения для анализа Harmony/Columbus (PerkinElmer).
3.4.4) Определение жизнеспособности клеток при DLBCL
Чтобы определить действие С25-140 на MYD88-зависимую ABC-DLBCL, были выбраны клеточные линии HBL1 (ABC-DLBCL - MYD88 L265P), U2932 (ABC DLBCL - MYD88 дикого типа) и BJAB (GCB DLBCL). По 1×106 клеток каждой клеточной линии высевали в 5 мл и обрабатывали 30 мкМ соединения С25-140 (соединение 3) или ДМСО. Весь эксперимент осуществляли в течение 9 дней. Каждый день отбирали по 500 мкл, и количество жизнеспособных клеток определяли с использованием счетчика клеток ViCEll (Beckmann Coulter). Ha 4 и 8 дни эксперимента объем снова доводили до 5 мл, и в образцы добавляли 30 мкМ соединения С25-140 или ДМСО. Этот процесс был необходим для обеспечения линейной пролиферации клеток, обработанных ДМСО. Все кривые для образцов, обработанных С25-140, соотносили с соответствующими контрольными образцами, обработанными ДМСО.
3.4.5) Исследования ADME и фармакокинетики
Все исследования ADME (стабильность в плазме, связывание с белками плазмы, микросомальная стабильность, LogD, количественное исследование Сасо-2, ингибирование CYP450 и количественное исследование Predictor™ hERG), а также фармакокинетики (Tmax, Cmax, AUC0-240мин, среднее время удержания, полупериод выведения, константа скорости выведения, объем распределения и клиренс) С25-140 (соединение 3) были проведены биологическим службами Bienta Enamine компании Bienta/Enamine Ltd.
3.4.6) SafetyScreen87
SafetyScreen87, в общей сложности 87 количественных исследований, проводили в Eurofins Cerep Panlabs. 87 первичных молекулярных мишеней, включая 13 ферментных количественных исследований (киназы и некиназные ферменты) и 74 количественных исследования связывания (ядерные рецепторы, GPCR, транспортеры и ионные каналы), выполняли при концентрации соединения С25-140 10 мкМ. Каждую из 87 реакций количественных исследований проводили в оптимизированных условиях, которые были доступны исходно в Eurofins Cerep Panlabs. Одновременно с С25-140 испытывали эталонное соединение для каждой из 87 реакций, чтобы обеспечить оптимальное условие количественных исследований. В данном исследовании эталонные соединения функционировали в том же диапазоне, как и исходные значения Ki.
3.4.7) Omniscreen
Оценку действия С25-140 на жизнеспособность клеток из 80 линий раковых клеток с использованием количественного люминесцентного исследования жизнеспособности клеток CellTiter-Glo проводили в CrownBioscience Inc. в исследовательском центре в Пекине. В данном исследовании 80 отобранных клеточных линий культивировали в среде, дополненной 10-15% ФБС, при температуре 37°С, 5% СО2 и 95% влажности. Культуральную среду приобретали у GIBCO или Sigma (США). Клетки обрабатывали С25-140 при 9 уровнях дозы, начиная с 80 мкМ с 3,16-кратными последовательными разведениями. Цисплатин служил эталонным контролем и его начинали применять в максимальной рабочей концентрации 100 мкМ. Исследуемые образцы инкубировали в течение 72 часов, и жизнеспособность клеток измеряли в количественном исследовании CellTiter-Glo.
3.5) Исследования на мышах
3.5.1) Выделение первичных мышиных С Неположительных Т-клеток
Выделение CD4+ Т-клеток из периферических лимфатических узлов мышей линии Balb/C проводили путем отбора с отрицательной магнитно-активированной сортировкой клеток (MACS) с использованием набора для выделения CD4+ Т-клеток II (Miltenyi). Первичные CD4+ Т-клетки культивировали в среде RPMI с добавлением 10% инактивированной нагреванием фетальной сыворотки теленка (ФСТ), 1% пенициллина/стрептомицина, 1% NEAA (Life Technologies), 1% HEPES, 1% L-глутамина, 1% пирувата натрия (Life Technologies) и 0,1% β-меркаптоэтанола. Для каждого образца высевали 2×105 клеток и обрабатывали соединением в течение 6 часов. Клетки стимулировали анти-CD3 (0,5 мг/мл) и анти-CD28 (1 мг/мл) на планшетах, которые были предварительно покрыты иммуноглобулином кролика к IgG хомяка или гранулами, покрытыми антителами к CD3/CD28 (3:1). Образцы для кПЦР в реальном времени собирали через 4 часа, тогда как супернатанты для исследования секреции цитокинов собирали через 20 часов после стимуляции.
3.5.2) Исследование на мышах с алиментарным ожирением (DIO)
Мыши получали рацион с высоким содержанием жиров до тех пор, пока их масса тела не достигала по меньшей мере 40 г и у них не развивалось ожирение, вызванное диабетом. Этих мышей разделяли на контрольную группу и группу для инъекции С25-140 (соединение 3). Мыши получали каждый день 14 мкмоль/кг (=7,4 мг/кг) С25-140 внутрибрюшинно в течение 20 дней. Соответственно, С25-140 растворяли в 2,9% ДМСО, 1,5% твин-80 и 1×ФСБ. Каждый день перед инъекцией измеряли массу тела и потребление пищи. На 20 день мышей умерщвляли через два часа после инъекции соединения и извлекали эпидермальную белую жировую ткань с обоих участков тела, замораживали в жидком азоте и хранили при -80°С.
3.5.3) Исследование псориаза на мышах
Чтобы оценить противовоспалительную активность соединения С25-140 (соединение 3), в Washington Biotechnology, Inc. (Baltimore, USA) была получена модель индуцированного имиквимодом (IMQ) псориаза на мышах. У 8 самцов мышей Balb/C на группу выбривали спину (1,5 см × 2 см) и IMQ и С25-140 наносили местно на бритую спину и правое ухо ежедневно в течение 6 дней. Соответственно, С25-140 растворяли в ацетоне и наносили в общей дозировке 500 мкг в сутки (2×250 мкг). За мышами наблюдали, и суточные оценки (эритема, шелушение и толщина кожи спины) независимо собирали по шкале от 0 до 4: 0 = нет; 1 = слабое; 2 = умеренное; 3 = заметное и 4 = очень заметное. Толщину уха измеряли электронным штангенциркулем как показатель отека. Секрецию цитокинов ИЛ-17 в ткань правого уха измеряли методом ИФА.
3.5.6) Модель артрита, индуцированного коллагеном
Чтобы исследовать способность С25-140 уменьшать симптомы РА, модель индуцированного коллагеном артрита (CIA) на мышах линии DBA1/J была создана Washington Biotechnology, Inc. (Baltimore, USA). Каждая группа из десяти мышей получала однократную подкожную инъекцию коллагена/эмульсии полного адъюванта Фрейнда (0,05 мл/мышь; 100 мкг коллагена/CFA). Через 20 дней (21 день) мышам вводили повторную инъекцию коллагена (0,05 мл/мышь; 100 мкг/мышиного коллагена в неполном адъюванте Фрейнда). На 28 день у мышей оценивали артритный индекс (AI) и вводили внутрибрюшинно дважды в сутки 6 мг/кг, 10 мг/кг и 14 мг/кг С25-140, соответственно, в течение 14 дней. У мышей ежедневно оценивали макроскопические признаки артрита. Соответственно, для каждой отдельной лапы применяли следующий оценочный индекс: 0 = нет видимых эффектов артрита; 1 = отек и/или эритема 1 пальца; 2 = отек и/или эритема 2 пальцев; 3 = отек и/или эритема более 2 пальцев; 4 = тяжелый артрит всей лапы и пальцев. Добавляли и записывали оценки для одной лапы. На 42 день животных подвергали эвтаназии, собирали конечности и проводили гистопатологическое исследование с использованием окрашивания толуидиновым синим с оценкой нескольких параметров.
3.6) Эксперименты с участием доноров-людей
Для выделения мононуклеарных клеток периферической крови (МКПК) из 50 мл крови трех разных людей-доноров кровь смешивали с 800 единицами гепарина (Sigma-Aldrich) и центрифугировали при 300×g в течение 10 минут без остановки. После удаления плазматической фракции лейкоцитарную пленку разбавляли в 2 объемах ФСБ и добавляли поверх 15 мл Lymphoprep (STEMCELL technologies). После центрифугирования при 160×g в течение 20 минут при комнатной температуре без остановки тромбоциты удаляли, и суспензию клеток снова центрифугировали при 350×g в течение 20 минут без остановки. Мононуклеарные клетки из промежуточного слоя переносили в новый флакон и промывали 3 раза в ФСБ с добавлением 0,1% БСА и 2 мМ ЭДТА. В завершение клетки ресуспендировали в среде RPMI, содержащей 10% ФСТ, 1% пенициллина/стрептомицина и 50 мкМ бета-меркаптоэтанола. Для каждого образца высевали 2×105 клеток и обрабатывали соединением в течение 6 часов. Клетки стимулировали либо 1 мкг/мл липополисахарида (ЛПС), либо CD3/CD28 (1 мкг hCD3 (IgG2A), 4 мкг hCD28 (IgG1), 2 мкг анти-IgG2A и 2 мкг анти-IgG2A) или 20 нг ИЛ-1β. Супернатанты для исследования секреции цитокинов собирали через 20 часов после стимуляции.
3.7) Статистический анализ
Результаты экспериментов представлены как среднее значение ± стандартное отклонение среднего. Для статистического анализа применяли «непарный t-тест» и определяли статистическую значимость с использованием р-значения <0,05. В исследовании на мышах DIO статистическую значимость потери массы тела рассчитывали с помощью двухфакторного дисперсионного анализа. Чтобы определить достоверность влияния обработки С25-140 (соединение 3) на исходы симптомов псориаза, применяли двухфакторный дисперсионный анализ. Наконец, двухфакторный дисперсионный анализ применяли для экспериментов с DLBCL по изучению кривых выживаемости. Для определения статистических параметров использовали программное обеспечение Prism 6.0 (GraphPad).
4) Анализ
При проведении высокопроизводительного скрининга (ВПС) для идентификации небольших молекул, ингибирующих взаимодействие TRAF6-Ubc13, С25 (которое соответствует соединению 1 описания) было открыто как перспективное активное соединение в ступенчатом способе с применением исследований в условиях in vitro и на основе клеток (Фиг. 1А). Исследования зависимости активности от структуры (SAR) С25 выявили три соединения со сходной эффективностью в условиях in vitro (Фиг. 1В и 2А), но С25-140 (которое соответствует соединению 3 описания) проявило улучшенную ингибирующую активность в клеточных количественных исследованиях активации NF-κB (Фиг. 1С). Помимо этого С25-140 дозозависимо уменьшало взаимодействие между эндогенным Ubc13 и эктопически экспрессированным TRAF6 в клетках (Фиг. 2 В). Дальнейшая характеристика С25-140 в экспериментах в условиях in vitro показала, что это соединение непосредственно связывается с белком TRAF6, что доказано в исследованиях методом ЯМР (Фиг. 2С), и С25-140 способно дозозависимо ослаблять Е3-лигазную активность TRAF6 образовывать полиубиквитиновые цепи (Фиг. 2D). Важно отметить, что С25-140 не влияло на связывание Ubc13 с Uev1a (Фиг. 2Е). Следовательно, С25-140 способно связывать TRAF6, впоследствии нарушать его связывание с Ubc13 и в конечном итоге уменьшать активность TRAF6.
Перед проведением углубленных клеточных исследований С25-140 авторы настоящего изобретения сначала исследовали влияние этого соединения на жизнеспособность клеток и фазы клеточного цикла. В данном исследовании С25-140 не оказывало существенного влияния на жизнеспособность клеток в трех различных клеточных линиях (Jurkat, MEF и HepG2), за исключением самой высокой концентрации 100 мкМ (Фиг. 3А). Кроме того, при обработке С25-140 клеток линий Jurkat и U2OS не наблюдали влияние на фазы клеточного цикла (Фиг. 3В-С). В целом, можно продемонстрировать, что С25-140 не влияло на жизнеспособность клеток и прогрессирование клеточного цикла в концентрациях до 30 мкМ, которые использовали для последующих клеточных экспериментов.
В качестве первой клеточной модели выбрали клетки линии MEF, которые можно стимулировать с помощью ИЛ-1β и ФНО-α. Стимуляция этих клеток с помощью ИЛ-1β индуцирует аутоубиквитинирование TRAF6, которое необходимо для распространения сигнала до стадии фосфорилирования IκВα под действием комплекса IKK, активации NF-κВ и экспрессии NF-κВ-зависимых генов-мишеней. Обработка клеток линии MEF С25-140 привела к уменьшению аутоубиквитинирования TRAF6 (Фиг. 4А), отражающему результаты убиквитинирования в условиях in vitro с применением рекомбинантных белков (Фиг. 2D). После уменьшения аутоубиквитинирования TRAF6 имело место снижение киназной активности IKK (Фиг. 4В), что преобразовалось в ослабление экспрессии гена-мишени (Фиг. 4С). Эти данные указывают на то, что ингибирование активности TRAF6 на ранних этапах передачи сигналов с участием рецептора ИЛ-1 передается в ходе разных этапов передачи сигналов, и ингибирование активности TRAF6 можно визуализировать на всех этих отдельных этапах. Неожиданным фактом явилось то, что обработка С25-140 также влияла на ФНО-α-индуцированое фосфорилирование IκВα (Фиг. 4D) и NF-κВ-индуцированную экспрессию генов-мишеней (Фиг. 4Е). Это явилось неожиданным фактом, поскольку согласно имеющимся данным передача сигналов с участием рецептора ФНО-α действительно незначительно вовлекает TRAF6 для распространения сигнала. Чтобы изучить это открытие, исследовали влияние С25-140 на активность нескольких других Е3-лигаз, чтобы установить, существует ли перекрестная реактивность. Эти исследования выявили, что С25-140 действительно влияло на активность Е3-лигазы cIAP1, Е3-лигазы, которая образует K63-соединенные полиубиквитиновые цепи сходно с TRAF6 (Фиг. 5А). Напротив, С25-140 не оказывало влияния на несколько других Е3-лигаз (MDM2, TRIM63, ITCH, Е6АР и RNF4), которые образуют другие типы полиубиквитиновых цепей (в основном, K11 и K48) (Фиг. 5B-D). Кроме того, в исследовании с использованием панели Ubiquitin Profiler™ испытывали еще 24 реакции убиквитинирования, включающие различные комбинации Е3-Е2 и субстрата. Соединение С25-140 не ингибировало ни одну из этих реакций (Фиг. 5Е). Эти данные объясняют ингибирующие эффекты С25-140 на передачу сигналов с участием ФНО-α, поскольку Е3-лигаза cIAP1 является центральным регулятором передачи сигналов с участием ФНО-α, которая ингибируется С25-140. Важно отметить, что эти данные также иллюстрируют то, что С25-140 не является универсальным ингибитором Е3-лигазы и, соответственно, сохраняет хорошую селективность, несмотря на то, что наблюдали незначительные неспецифические эффекты на передачу сигналов с участием ФНО-α.
Авторы настоящего изобретения также исследовали влияние С25-140 на передачу сигналов с участием Т-клеточного рецептора (ТКР) на линии Т-клеток человека (Т-клетки линии Jurkat) при адаптивном иммунном ответе. В настоящем документе авторы могут показать, что указанное соединение также влияет на передачу сигналов с участием ТКР, которая основана на TRAF6 (Oeckinghaus, А., Е. Wegener, V. Welteke, et al., Maltl ubiquitination triggers NF-kappaB signaling upon T-cell activation. EMBO J, 2007. 26 (22): p. 4634-45). В Т-клетках линии Jurkat стимуляция ТКР приводила к аутоубиквитинированию TRAF6, которое ослаблялось под действием С25-140 дозозависимым образом (Фиг. 6А). Сниженное аутоубиквитинирование TRAF6 привело к подавлению последующих реакций, включая киназную активность IKK, активацию NF-κВ, а также экспрессию и секрецию соответствующих цитокинов (Фиг. 6 В-Е). Эти данные подчеркивают, что соединение согласно настоящему изобретению также влияет на опосредованный ТКР адаптивный иммунный ответ в линиях клеток человека.
После того, как авторы настоящего изобретения показали, что С25-140 успешно конкурирует с TRAF6-зависимой передачей сигналов с участием рецепторов в линиях клеток мыши и человека, были продолжены работы с более биологически значимыми клетками. С этой целью авторы настоящего изобретения выделили мононуклеарные клетки периферической крови человека (МКПК) от трех разных индивидуумов и стимулировали эти клетки ЛПС для активации врожденных иммунных ответов. В соответствии с предыдущими данными авторов настоящего изобретения соединение С25-140 уменьшало секрецию NF-κВ-запускаемых воспалительных цитокинов, таких как ФНО-α и ИЛ-6 после стимуляции ИЛ-1β (Фиг. 7А), или ИЛ-1β и ФНО-α после стимуляции ЛПС (Фиг. 7В), это свидетельствует о том, что С25-140 не только влияет на активность TRAF6 в иммортализованных клетках, но, что более важно, оно уменьшает врожденные иммунные процессы в первичных клетках крови человека. Сходные ингибирующие эффекты под действием С25-140 можно наблюдать в МКПК человека при стимуляции адаптивного иммунного ответа с помощью CD3/CD28 (Фиг. 7С). В завершение авторы настоящего изобретения также выделили первичные мышиные CD4+ Т-клетки для экспериментов с предшественниками в исследованиях на мышах в условиях in vivo. Аналогично вышеуказанному, соединение С25-140 противодействовало CD3/CD28-опосредуемой активации NF-κВ и выработке ИЛ-2 (Фиг. 8).
В совокупности данные из исследований в условиях in vitro, а также исследований на основе клеток с применением иммортализованных клеточных линий, а также первичных клеток человека и мыши демонстрируют, что С25-140 оказывает ингибирующее действие на различные иммунные и воспалительные пути передачи сигналов, что может быть благоприятным для лечения сильно воспаленной иммунной системы.
Чтобы перейти к исследованиям на мышах в условиях in vivo, авторы настоящего изобретения исследовали, обладает ли соединение С25-140 подходящими свойствами для применения в условиях in vivo. С этой целью авторы настоящего изобретения сначала провели исследование ADME, изучив следующий параметр: (i) стабильность в плазме, (ii) связывание с белками плазмы, (iii) микросомальная стабильность, (iv) logD, (v) проницаемость Сасо-2, (vi) ингибирование CYP450 и (vii) ингибирование hERG. Соединение С25-140 обладает довольно хорошим набором свойств во всех этих количественных исследованиях (Фиг. 9А). В целом, соединение стабильно в плазме и микросомах, хорошо проникает в слой Сасо-2, что указывает на хорошую пероральную биодоступность, довольно хорошо связывается с белками плазмы и, что очень важно, не оказывает существенного ингибирующего действия на hERG. Только в отношении СР450 оно имеет некоторую способность ингибировать отдельные CYP.
Авторы настоящего изобретения также исследовали фармакокинетику (ФК) С25-140 после внутривенной (в/в), пероральной (п/о) и внутрибрюшинной (в/б) инъекции 10 мг/кг соединения. Все проанализированные данные обобщены на Фиг. 9В, и график зависимости концентрации от времени для в/б введения представлен на Фиг. 9С. При в/б введении соединение С25-140 проявило более высокий рассчитанный период полужизни в условиях in vivo в кровотоке (184 минуты) по сравнению с в/в (80 минут) и п/о (127 мин) доставкой. В целом, наблюдали фазу быстрого начального распределения. Соединение обладает хорошей расчетной пероральной биодоступностью приблизительно 45%. Во время исследований ФК на животных не наблюдали явных нежелательных эффектов соединения.
Далее авторы настоящего изобретения исследовали безопасность С25-140, применяя панель Cerep SafetyScreen87, набор важнейших рецепторов, белков и ферментов, которые не должны подвергаться воздействию для обеспечения хорошей переносимости в условиях in vivo. Интересно отметить, что существенное связывание С25-140 с какой-либо из этих мишеней не детектировалось (Фиг. 10), это очевидным образом свидетельствует о высокой степени безопасности С25-140 для доклинического/клинического применения.
В первом исследовании на мышах авторы настоящего изобретения ставили целью изучить способности ингибитора активности TRAF6, С25-140, противодействовать воспалению и прибавке массы тела, которая обусловлена ожирением, индуцированным рационом с высоким содержанием жира (HFD). В сотрудничестве с Институтом диабета и ожирения при Helmholtz Zentrum Munchen GmbH - Deutsches Forschungszentrum fur Gesundheit und Umwelt (HMGU) выбрали 16 мышей, которых поддерживали на HFD по меньшей мере 12 месяцев. Эти мыши имели среднюю массу тела 55 г с очевидными характеристиками ожирения и диабета 2 типа (T2D). Мышей разделяли на две группы и вводили внутрибрюшинно один раз в сутки носитель или С25-140 в дозе 7,4 мг/кг в течение 20 дней. Каждый день перед инъекцией контролировали массу тела мышей и их потребление пищи. Обработанные контролем мыши непрерывно набирали массу тела в течение 20 дней в условиях HFD, в то время как мыши, обработанные С25-140, хуже набирали массу тела (Фиг. 11А). Важно отметить, что потребление пищи в двух группах не различалось (Фиг. 11В), это указывает на то, что лечение не оказывало общего нежелательного эффекта на мышей. Авторы настоящего изобретения также исследовали экспрессию ИЛ-1β в выделенных адипоцитах в качестве маркера воспаления, а также VCAM как гена-мишени NF-κВ. Экспрессия обоих генов была значительно уменьшена после лечения, обеспечивая молекулярное доказательство корректного функционирования ингибитора TRAF6. В целом, данное исследование предоставляет важную информацию о том, что ингибитор TRAF6, С25-140, может уменьшать воспаление в условиях in vivo и, соответственно, существенно способствует снижению прибавки массы тела в условиях рациона с высоким содержанием жиров.
Вторая модель на мышах в условиях in vivo была предназначена для изучения С25-140 в исследовании псориаза на мышах, как модели аутоиммунного заболевания, которое было проведено CRO Washington Biotechnology. В этой модели псориаз индуцирован имиквимодом (IMQ) за счет активации пути передачи сигналов с участием TLR7 (Gilliet, М., С. Conrad, М. Geiges, et al., Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol, 2004. 140 (12): p. 1490-5). Более конкретно, IMQ наносили один раз в сутки на бритую спину и правое ухо мышей. С25-140 наносили местно два раза в сутки на одни и те же области. Параметры, фиксирующие исход заболевания, оценивали каждый день, и образцы для измерения цитокина ИЛ-17 собирали на 6 день (Фиг. 12А). Соединение 25-140 значительно уменьшило «совокупную оценку» (Фиг. 12В) и «оценку толщины» (Фиг. 12С). Эти оценки отражали индивидуальные оценки, такие как «шелушение» (Фиг. 12D), «эритема» (Фиг. 12Е) и «толщина уха» (Фиг. 12F). Все эти параметры отражали улучшение симптомов заболевания. Наконец, исследование ткани правого уха для определения ИЛ-17 выявило, что секреция/экспрессия этого цитокина была значительно снижена (Фиг. 12G). Следовательно, данное исследование действительно продемонстрировало, что С25-140, как ингибитор активности TRAF6, способен улучшать симптомы модели аутоиммунного псориаза в условиях in vivo.
В третьем исследовании на мышах в условиях in vivo авторы настоящего изобретения исследовали эффективность соединения С25-140 в доклинической модели аутоиммунного ревматоидного артрита (РА) на мышах. В данном исследовании авторы настоящего изобретения испытывали С25-140 в модели коллаген-индуцированного артрита (CIA) на мышах, которая является наиболее часто используемой моделью для оценки эффективности при PA (Brand, D.D., K.А. Latham, and E.F. Rosloniec, Collagen-induced arthritis. Nat Protoc, 2007. 2 (5): p. 1269-75). Исследование также было проведено CRO Washington Biotechnology. В данном исследовании ревматоидный артрит (РА) индуцировали с помощью инъекции коллагена на 0 день. Через 21 день мыши получали инъекцию коллагена, вызывающую развитие симптомов артрита. На 28 день вводили С25-140 два раза в сутки в трех разных дозах (6 мг/кг, 10 мг/кг и 14 мг/кг) в течение 14 дней. Преднизолон служил положительным контролем. У мышей ежедневно оценивали артритный индекс (AI) и подвергали эвтаназии на 42 день. Конечности отбирали для гистопатологического анализа (Фиг. 13А). Массу тела контролировали на протяжении всего исследования, и не наблюдали каких-либо признаков ее снижения (Фиг. 13В). Важно отметить, что соединение согласно настоящему изобретению в дозах 10 мг/кг и 14 мг/кг практически полностью улучшало артритный индекс до исходных уровней в этой модели для оценки эффективности (Фиг. 13С). Еще более важно, что эти результаты были подтверждены гистологическим исследованием конечностей. Количественная оценка различных параметров (суммированные оценки, воспаление, паннус, повреждение хряща и резорбция кости) проявления заболевания (Фиг. 13D-E) и срезов тканей конечностей (Фиг. 13F) подтвердили, что С25-140 дозозависимо улучшало симптомы РА. Полный набор данных демонстрирует, что С25-140 успешно противодействовало клиническим симптомам РА и полностью обращало патологическое состояние. Посредством этого авторы настоящего изобретения представляют успешное исследование для подтверждения концепции в отношении соединения С25-140 в доклинической модели для оценки эффективности РА.
Авторы настоящего изобретения дополнительно исследовали способность С25-140 селективно уничтожать подгруппу агрессивной ABC DLBCL. Выживание ABC DLBCL опирается на ряд различных мутаций, ведущих к конститутивной активности пути передачи сигналов с участием NF-κВ. Основные мутации находятся в пределах В-клеточного рецептора (BCR) и пути передачи сигналов с участием MYD88 (Nagel, D., М. Vincendeau, A.C. Eitelhuber, et al., Mechanisms and consequences of constitutive NF-kappaB activation in B-cell lymphoid malignancies. Oncogene, 2014. 33 (50): p. 5655-65 и Ngo, V.N., R.M. Young, R. Schmitz, et al., Oncogenically active MYD88 mutations in human lymphoma. Nature, 2011. 470 (7332): p. 115-9). В настоящее время большинство доступных терапевтических средств сосредоточены на пути BCR. Поскольку TRAF6 является основным участником передачи сигналов с участием MYD88, авторы настоящего изобретения исследовали, будет ли ингибитор TRAF6 селективно уничтожать ABC DLBCL с конститутивно активной мутацией MYD88. Применяли клеточные линии MYD88-зависимой ABC DLBCL (HBL-1 с мутацией MYD88 L265P), ABC DLBCL с MYD88 дикого типа (U2932) и GCB DLBCL (BJAB). Все клеточные линии обрабатывали С25-140 на 0, 4 и 8 день. Исследование проводили в течение 9 дней, и жизнеспособность клеток (количество клеток) измеряли ежедневно. С25-140 значительно уменьшало жизнеспособность MYD88-зависимой клеточной линии HBL1 и оказывало незначительное влияние на MYD88-независимую клеточную линию U2932 (Фиг. 14). Неродственная GCB DLBCL клеточная линия BJAB не проявила различий в выживаемости после обработки С25-140 (Фиг. 14). Эти данные показывают, что ингибитор TRAF6, С25-140, является селективным ингибитором MYD88-зависимой клеточной линии ABC DLBCL и может служить в качестве важного ингибитора для разных видов комбинированного лечения с одновременным введением терапевтических средств, действующих на BCR.
Применительно ко второму типу рака авторы настоящего изобретения исследовали эффективность ингибитора TRAF6, С25-140, против клеток, происходящих из остеосаркомы, и возможность его применения для терапии остеосаркомы. Авторы настоящего изобретения обрабатывали клетки U2OS облучением (2 Гр или 4 Гр) или только С25-140, или комбинацией облучения в дозе 2 Гр и С25-140. Несмотря на то, что С25-140 по отдельности и облучение в дозе 2 Гр оказывали одинаковое ингибирующее действие на пролиферацию клеток U2OS, комбинация обоих факторов усиливала действие на область облучения в дозе 4 Гр (Фиг. 15). Эти данные указывают на то, что С25-140 может усиливать эффекты лучевой терапии так, что меньшее воздействие облучения в комбинации с С25-140 по-прежнему будет обеспечивать эффективную терапию.
Чтобы получить более полное представление о действии С25-140 на раковые клетки, авторы настоящего изобретения начали исследование рака, в котором применяли 78 линий раковых клеток различного органного происхождения. Эти линии раковых клеток испытывали против девяти концентраций С25-140. Результаты показывают, что основное действие было оказано на раковые клетки крови (Фиг. 16). Однако воздействию подверглись также линии раковых клеток из других тканей, включая мочевой пузырь, кишечник, печень, легкие, поджелудочную железу, глотку и простату (Фиг. 16). Важно отметить, что две контрольные клеточные линии, HS-5 и MCF 10А, вообще не подверглись воздействию, что свидетельствует об отсутствии признаков общей токсичности (Фиг. 16).
Во время идентификации соединений, которые не доступны коммерчески, и уникальных структур, соединения синтезировали и испытывали в количественном исследовании ALPHAScreen для определения их способность нарушать связывание TRAF6-Ubc13. Эти соединения одновременно испытывали с использованием набора TruHits, чтобы исключить соединения, которые вносят помехи в технологию ALPHAScreen. Все новые соединения ингибировали взаимодействие TRAF6-Ubc13 с различными ИК50, варьирующимися от 2,3 мкМ до 19,6 мкМ (Фиг. 17). Ни одно из соединений не препятствовало количественному исследованию TruHits. Эти данные подтверждают существование новых ингибиторов связывания TRAF6-Ubc13 с новыми структурами, которые не являются коммерчески доступными. Важно отметить, что все соединения проявляли сходные или лучшие токсические свойства по сравнению с С25-140. Все соединения в концентрации до 50 мкМ практически не оказывали токсического действия на три разные клеточные линии (Фиг. 18). Только для отдельных соединений при концентрации соединения 100 мкМ была обнаружена некоторая токсичность. Наконец, способность всех новых соединений препятствовать связыванию TRAF6-Ubc13 испытывали в клеточных количественных исследованиях коиммунопреципитации. Помимо С25-140 в клетках были активны другие новые соединения HZM06-1 (которое соответствует соединению 28), HZM06-2 (которое соответствует соединению 29), HZM06-3 (которое соответствует соединению 30), HZM06-4 (которое соответствует соединению 31), HZM06-6 (которое соответствует соединению 32), HZM06-7 (которое соответствует соединению 33), HZM06-8 (которое соответствует соединению 34), HZM06-9 (которое соответствует соединению 35) и HZM06-10 (которое соответствует соединению 36), причем наиболее активными аналогами были С25-140, HZM06-4, HZM06-7, HZM06-8 и HZM06-9 (Фиг. 19). В совокупности данные на Фиг. 14-16 представляют набор новых, коммерчески недоступных соединений, которые активны в условиях in vitro и в клетках и имеют незначительную токсичность.
Другие новые соединения синтезировали и испытывали в количественном исследовании ALPHAScreen. Эти соединения проявляли разную степень ингибиторного действия, как указано в таблице (Фиг. 20).
Следует понимать, что настоящее изобретение можно осуществлять способами, отличными от конкретно описанных в предшествующем описании и примерах. Многочисленные модификации и варианты настоящего изобретения возможны с учетом вышеизложенных идей и, следовательно, находятся в пределах объема прилагаемой формулы изобретения.
--->
Перечень последовательностей
<110> Helmholtz Zentrum München Deutsches Forschungszentrum für Gesundheit
Und Umwelt (GmbH)
<120> Ингибиторы TRAF 6
<130> HEL15785PCT
<150> 16189150.2
<151> 2016-09-16
<160> 22
<170> PatentIn version 3.5
<210> 1
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Зонд H2K (NF-каппа B)
<400> 1
gatccagggc tggggattcc ccatctccac agg 33
<210> 2
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Зонд H2K (NF-каппа B)
<400> 2
gatccctgtg gagatgggga atccccagcc ctg 33
<210> 3
<211> 26
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Зонд Oct-1
<400> 3
gatctgtcga atgcaaatca ctagaa 26
<210> 4
<211> 26
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Зонд Oct-1
<400> 4
gatcttctag tgatttgcat tcgaca 26
1
<210> 5
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Прямой праймер для мышиного A20
<400> 5
gctcaactgg tgtcgtgaag 20
<210> 6
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Обратный праймер для мышиного A20
<400> 6
atgaggcagt ttccatcacc 20
<210> 7
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Прямой праймер для мышиного бета-актина
<400> 7
cctctatgcc aacacagtgc 20
<210> 8
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Обратный праймер для мышиного бета-актина
<400> 8
gtactcctgc ttgctgatcc 20
<210> 9
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Прямой праймер для мышиного ICAM-1
<400> 9
cgctcagaag aaccaccttc 20
<210> 10
<211> 21
<212> ДНК
<213> Искусственная последовательность
2
<220>
<223> Обратный праймер для мышиного ICAM-1
<400> 10
ggagacgcag aggaccttaa c 21
<210> 11
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Прямой праймер для мышиного ИЛ-1бета
<400> 11
tcagcacctc acaagcagag 20
<210> 12
<211> 22
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Обратный праймер для мышиного ИЛ-1бета
<400> 12
gcccatactt taggaagaca cg 22
<210> 13
<211> 21
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Прямой праймер для мышиного ИЛ-2
<400> 13
gagtgccaat tcgatgatga g 21
<210> 14
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Обратный праймер для мышиного ИЛ-2
<400> 14
agggcttgtt gagatgatgc 20
<210> 15
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Прямой праймер для мышиного VCAM
3
<400> 15
cccctcattc cttaccaccc 20
<210> 16
<211> 21
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Обратный праймер для мышиного VCAM
<400> 16
agttggggat tcggttgttc t 21
<210> 17
<211> 24
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Прямой праймер для мышиного ИЛ-2
<400> 17
cacagctaca actggagcat ttac 24
<210> 18
<211> 21
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Обратный праймер для мышиного ИЛ-2
<400> 18
tgctgattaa gtccctgggt c 21
<210> 19
<211> 21
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Прямой праймер для мышиного ФНО-альфа
<400> 19
cccagggacc tctctctaat c 21
<210> 20
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Обратный праймер для мышиного ФНО-альфа
<400> 20
gctacaggct tgtcactcgg 20
4
<210> 21
<211> 18
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Прямой праймер для РНК полимеразы II человека
<400> 21
gcaccacgtc caatgaca 18
<210> 22
<211> 18
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Обратный праймер для РНК полимеразы II человека
<400> 22
gtgcggctgc ttccataa 18
5
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| КОНДЕНСИРОВАННЫЕ ТРИАЗОЛАМИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ Р2Х7 | 2010 |
|
RU2533122C2 |
| АЦИЛИРОВАННЫЕ ИНДАНИЛАМИНЫ И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2002 |
|
RU2339614C2 |
| ИНГИБИТОР, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ ПРОИЗВОДНОЕ ПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2807611C2 |
| КОНДЕНСИРОВАННЫЕ ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПИРИДИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПРОИЗВОДСТВА И ПРИМЕНЕНИЕ | 2014 |
|
RU2668074C2 |
| ПИРАЗОЛОПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ НАДФН-ОКСИДАЗЫ | 2009 |
|
RU2538041C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИММУНОЛОГИЧЕСКИХ И ОНКОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2009 |
|
RU2545023C9 |
| ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ-МОДУЛЯТОРЫ ФОСФОИНОЗИДИТ-3-КИНАЗЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2013 |
|
RU2665036C9 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И ИХ ПРОИЗВОДНЫЕ | 2020 |
|
RU2834236C2 |
| ПИРАЗОЛОПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ НАДФН-ОКСИДАЗЫ | 2009 |
|
RU2532161C2 |
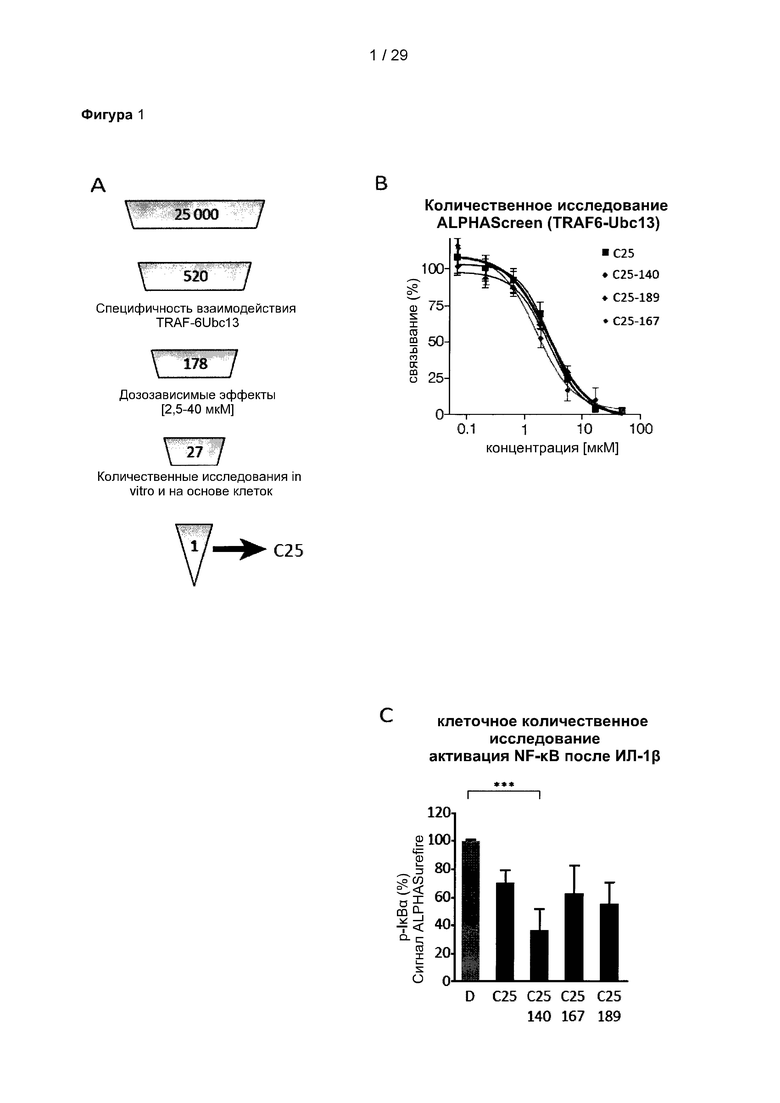
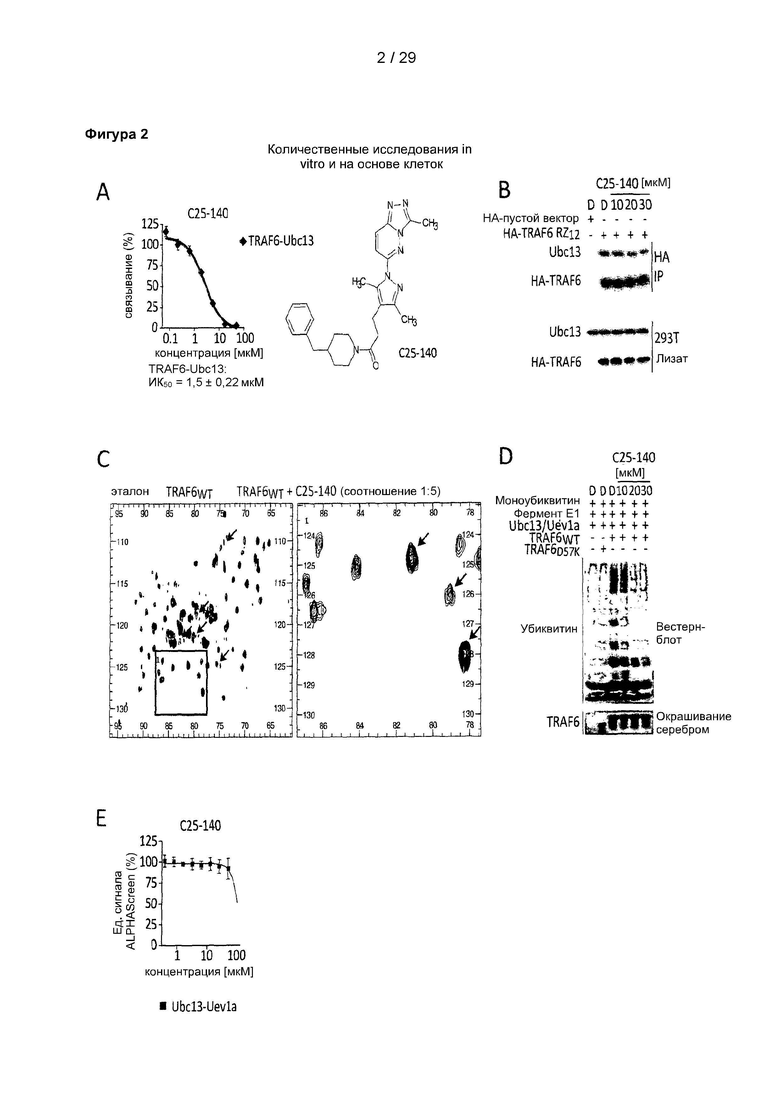
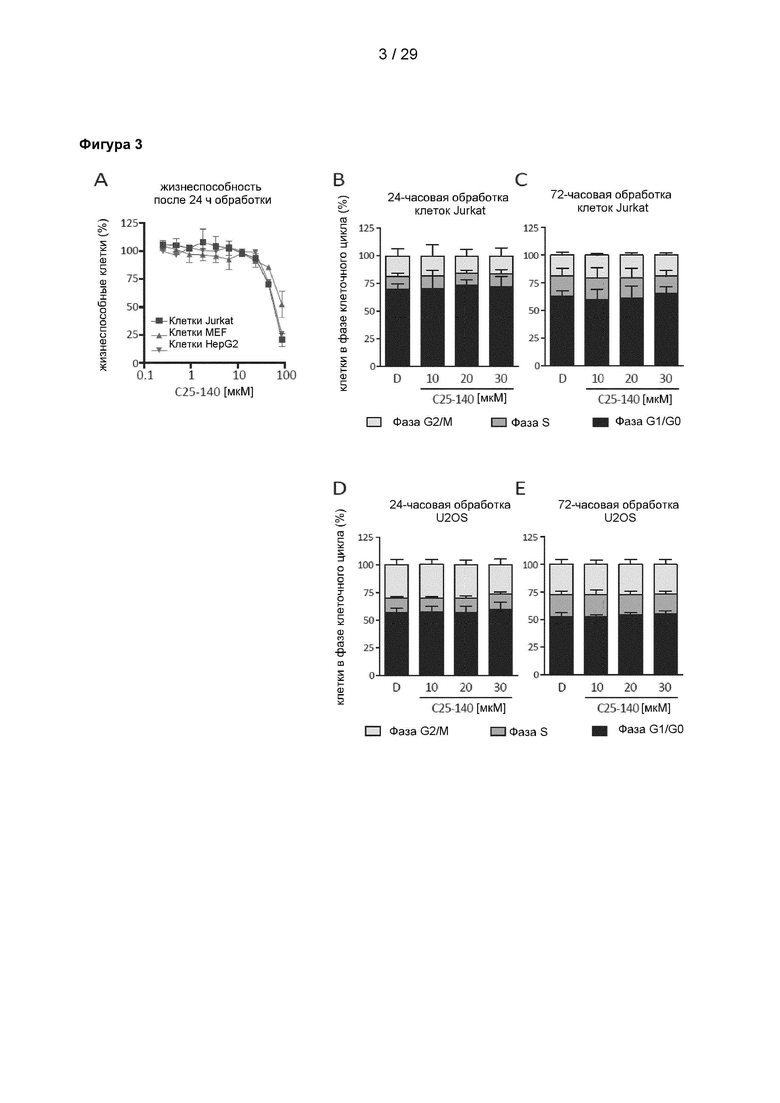
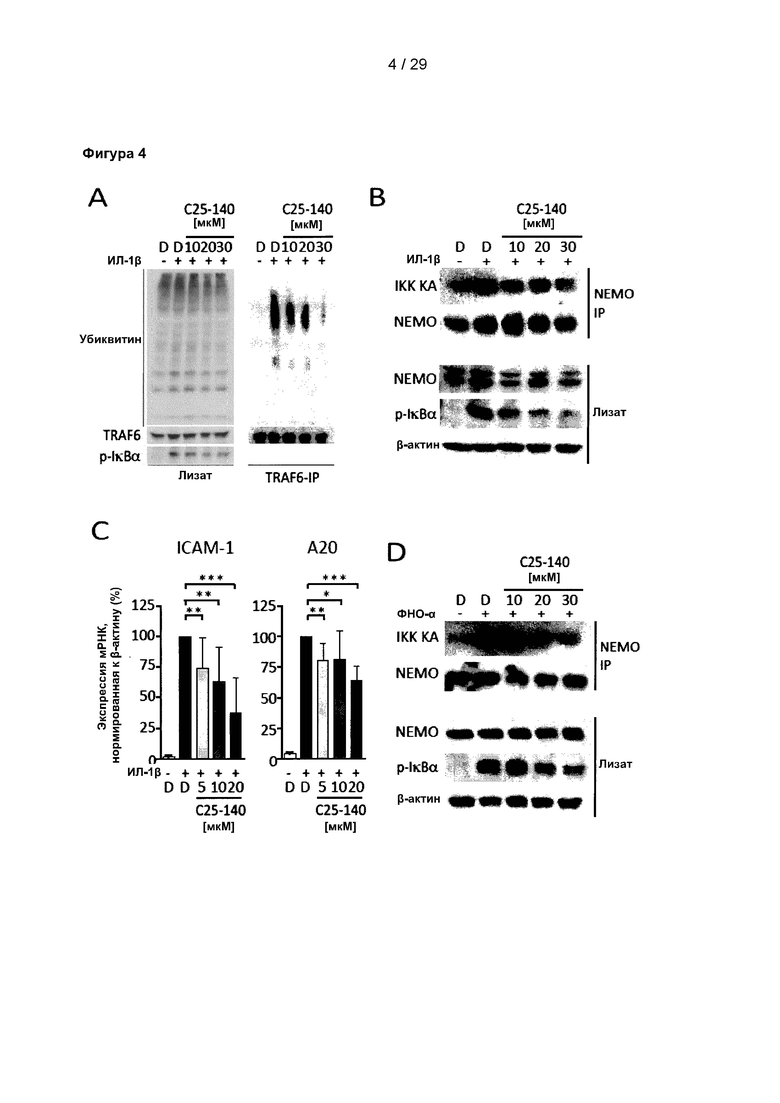
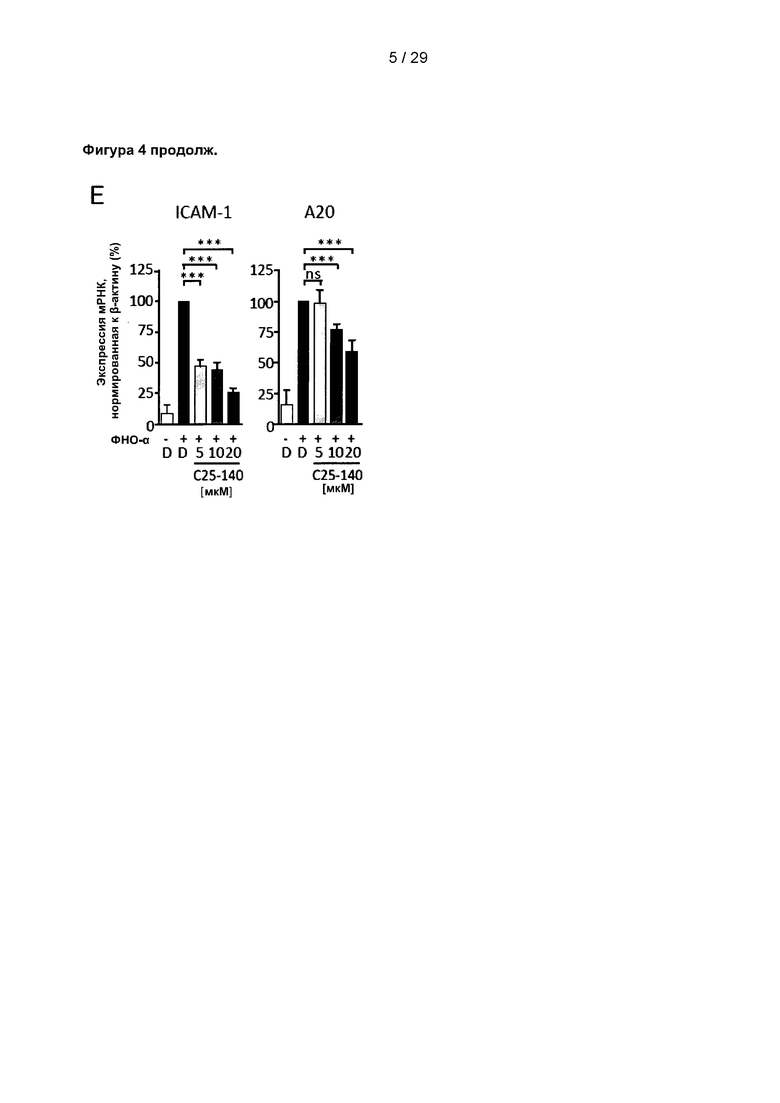
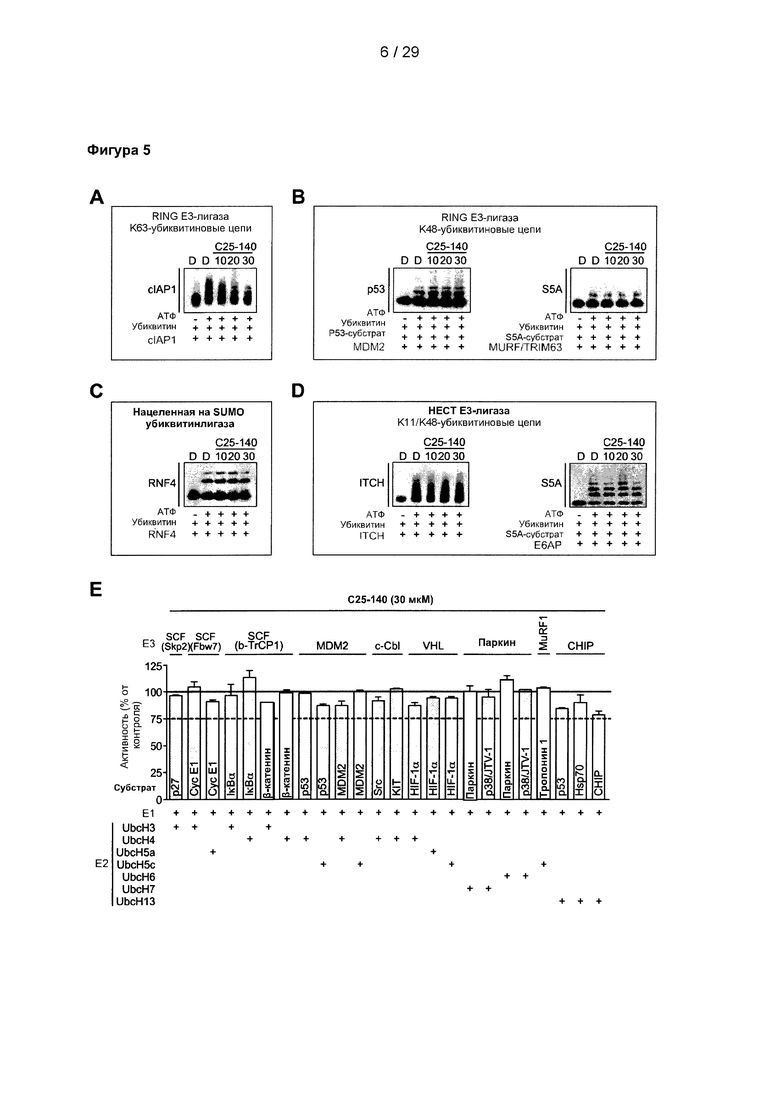
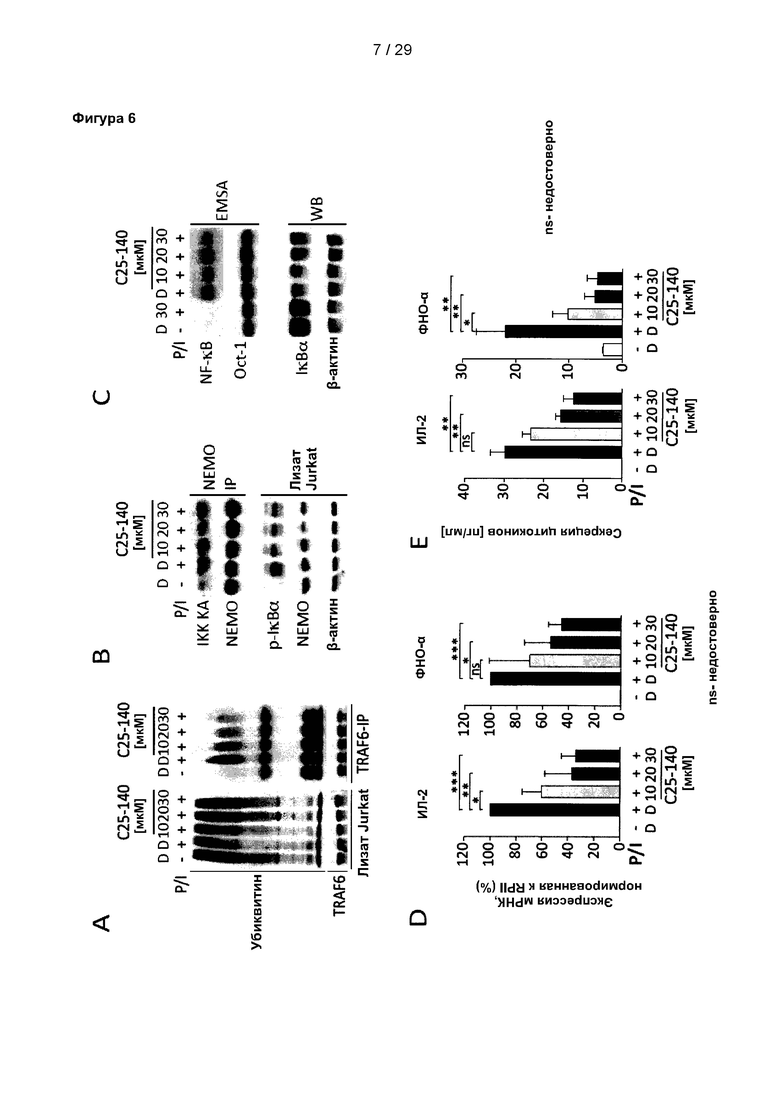
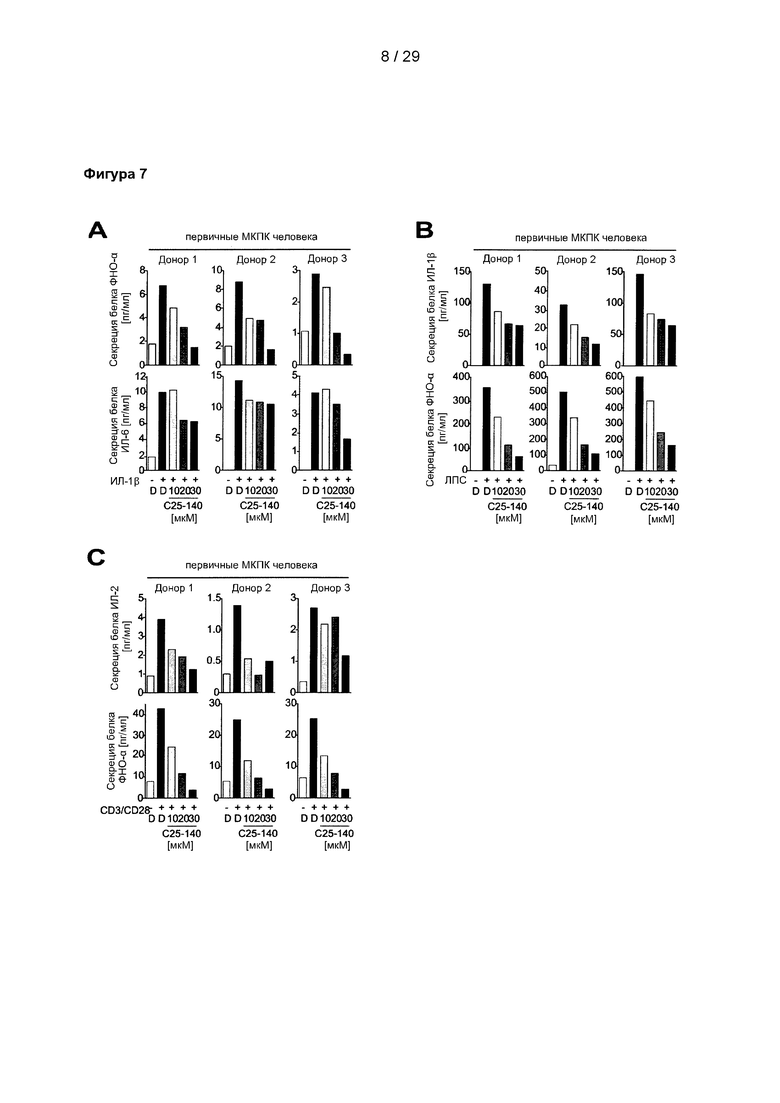
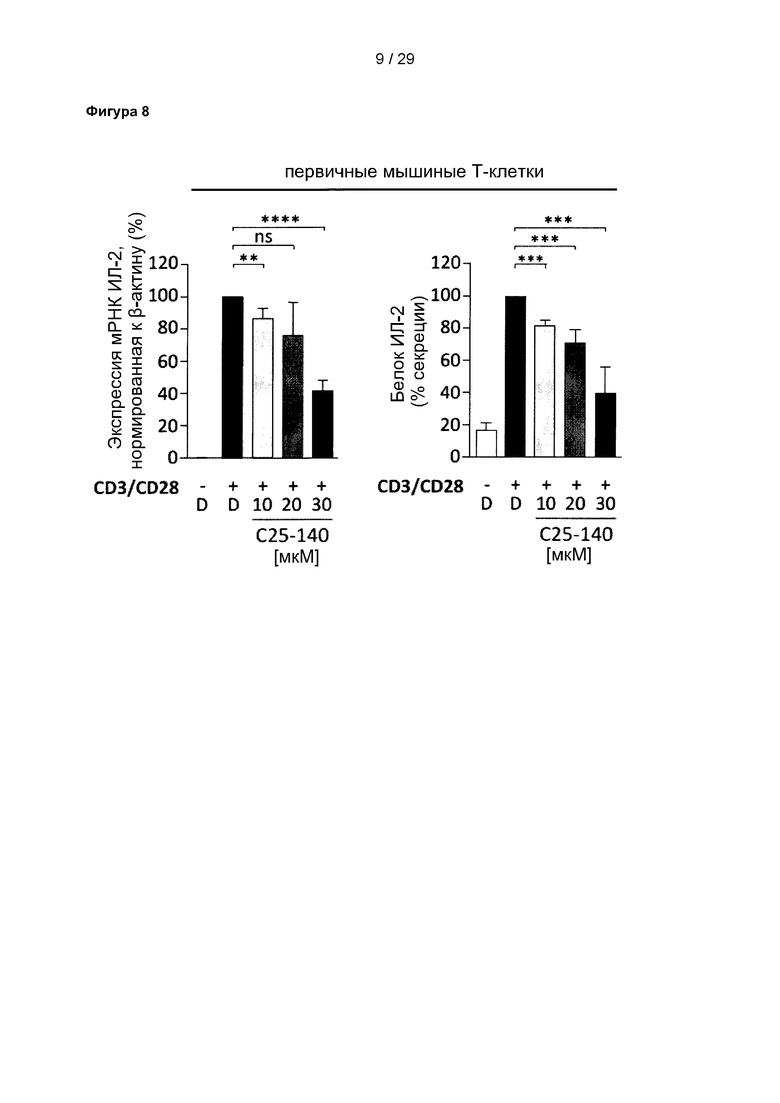
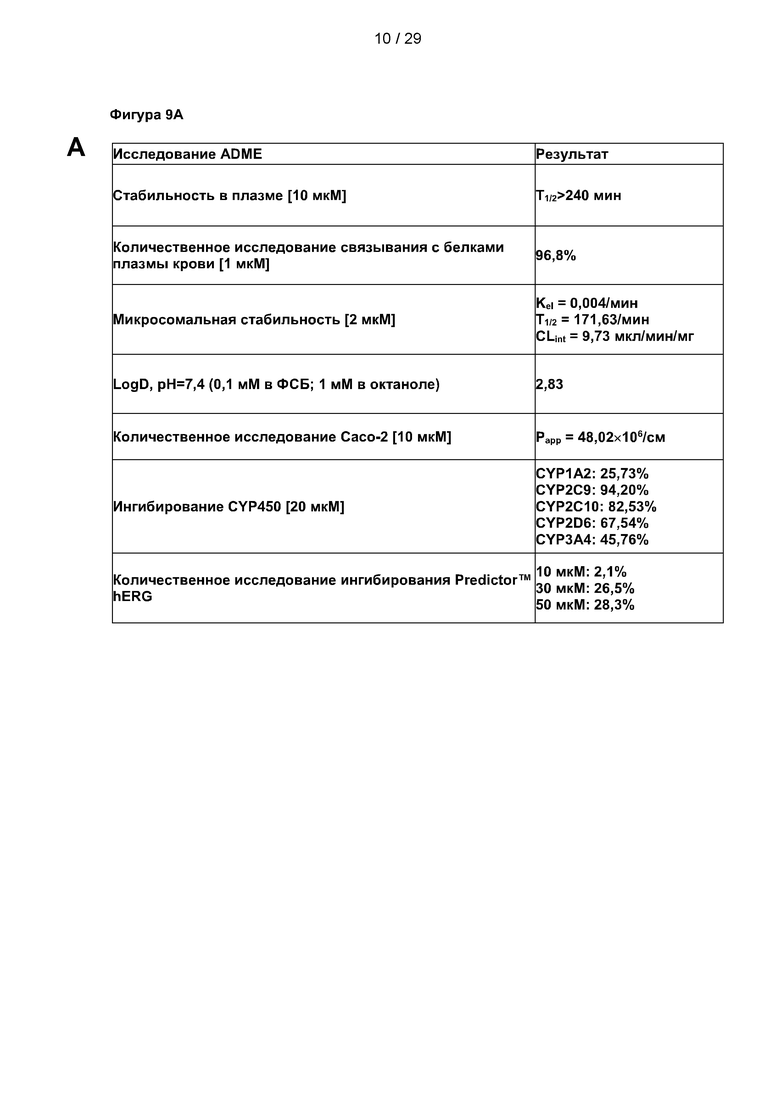
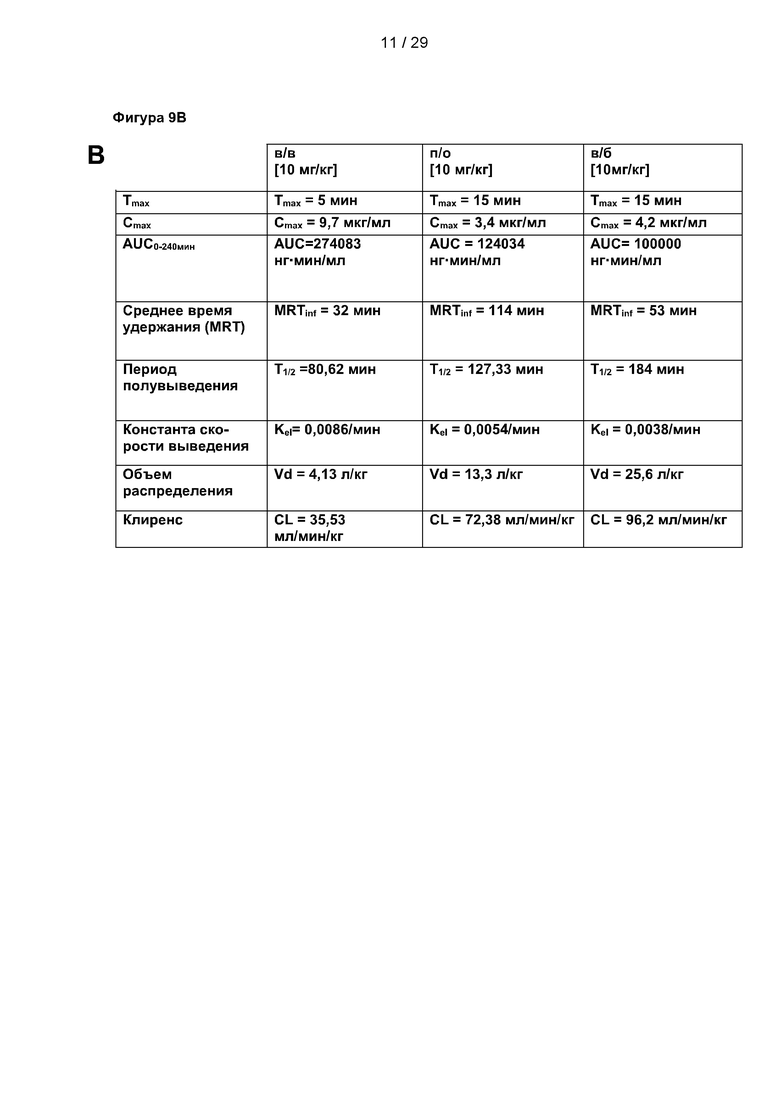
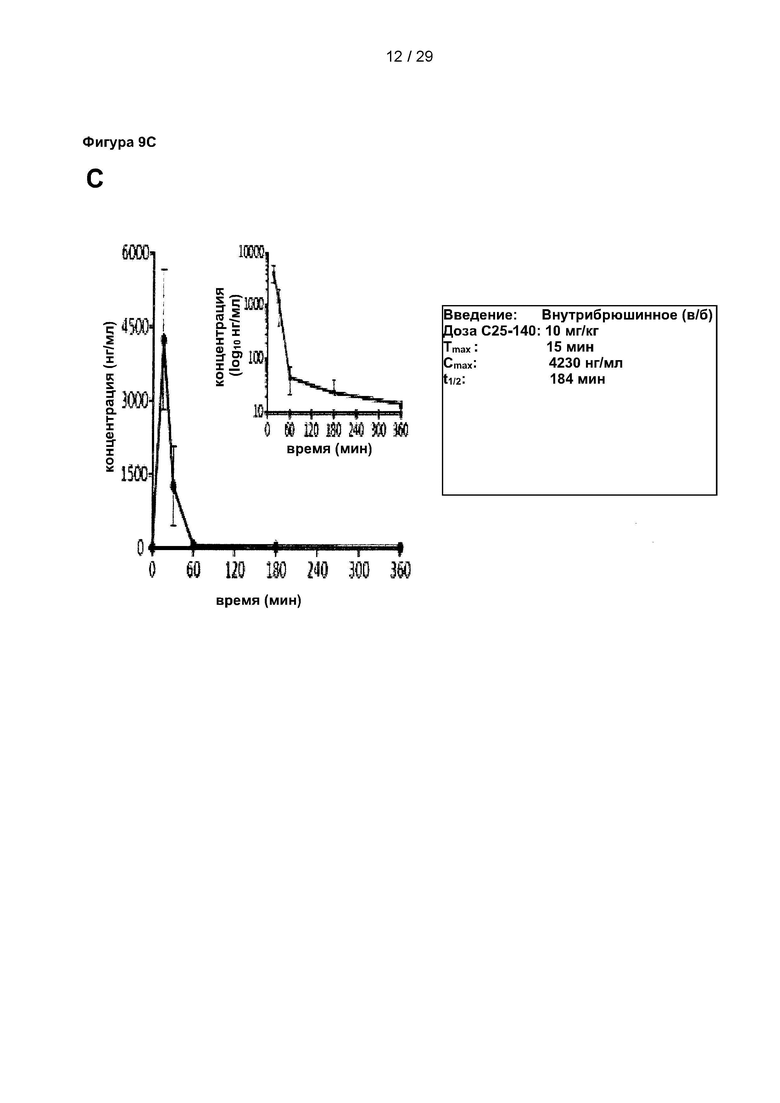
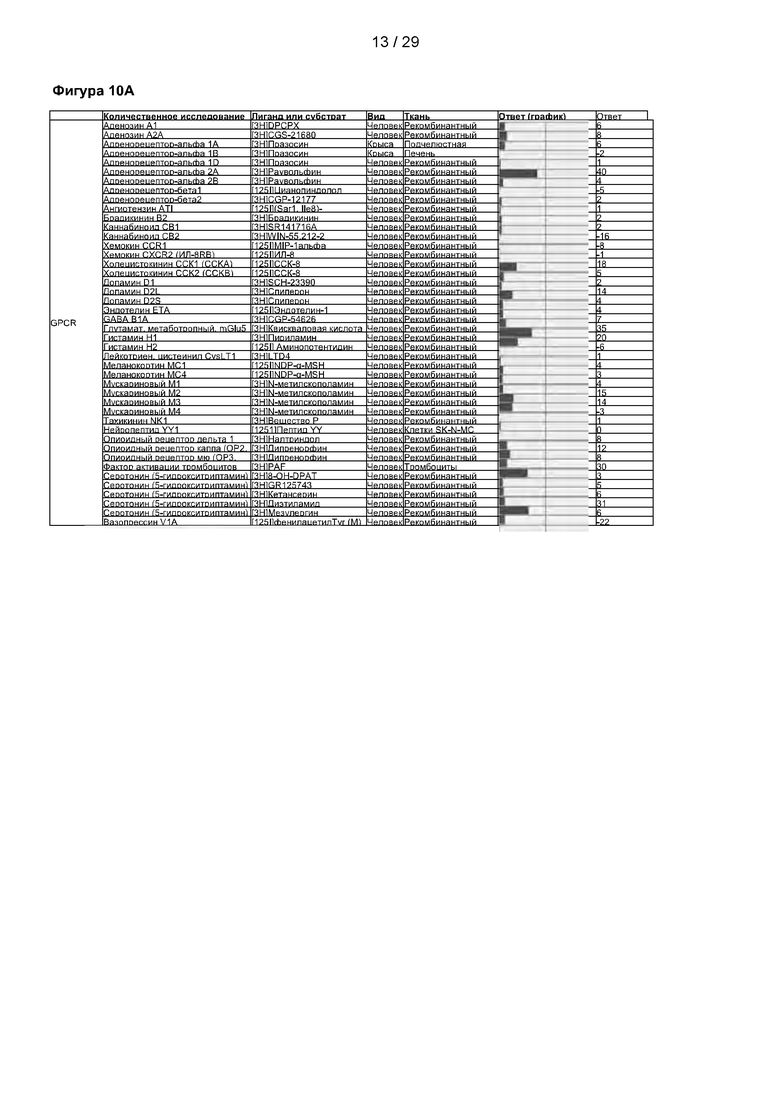

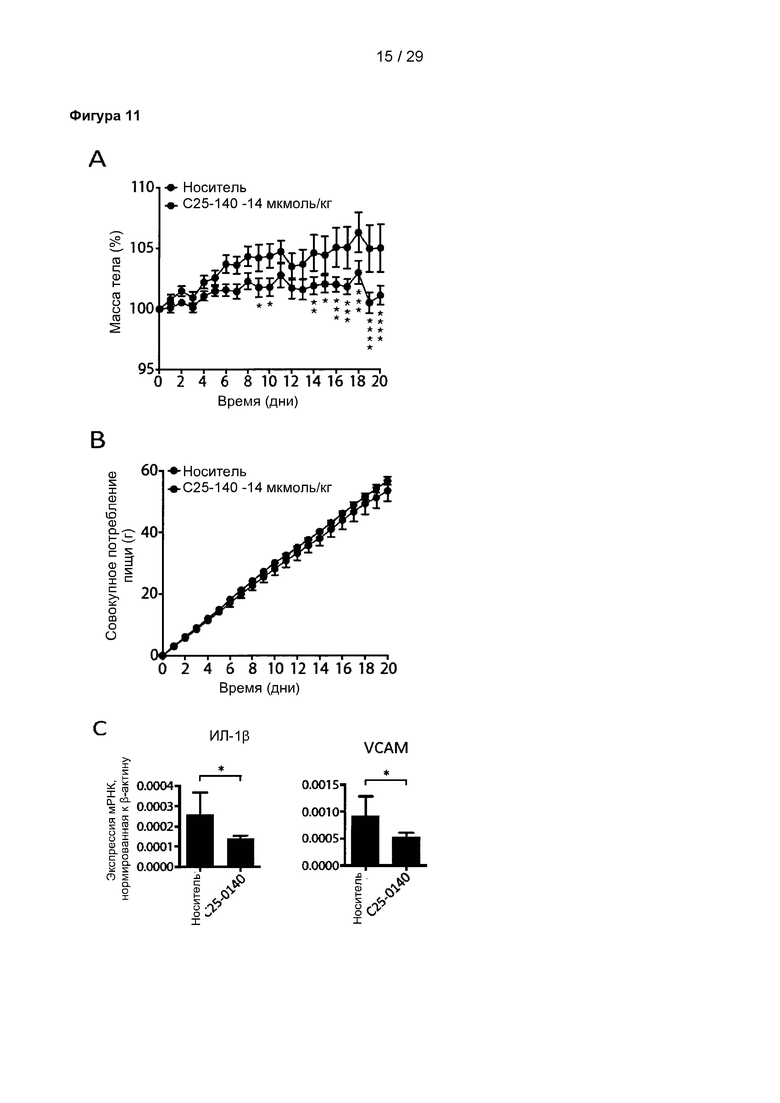
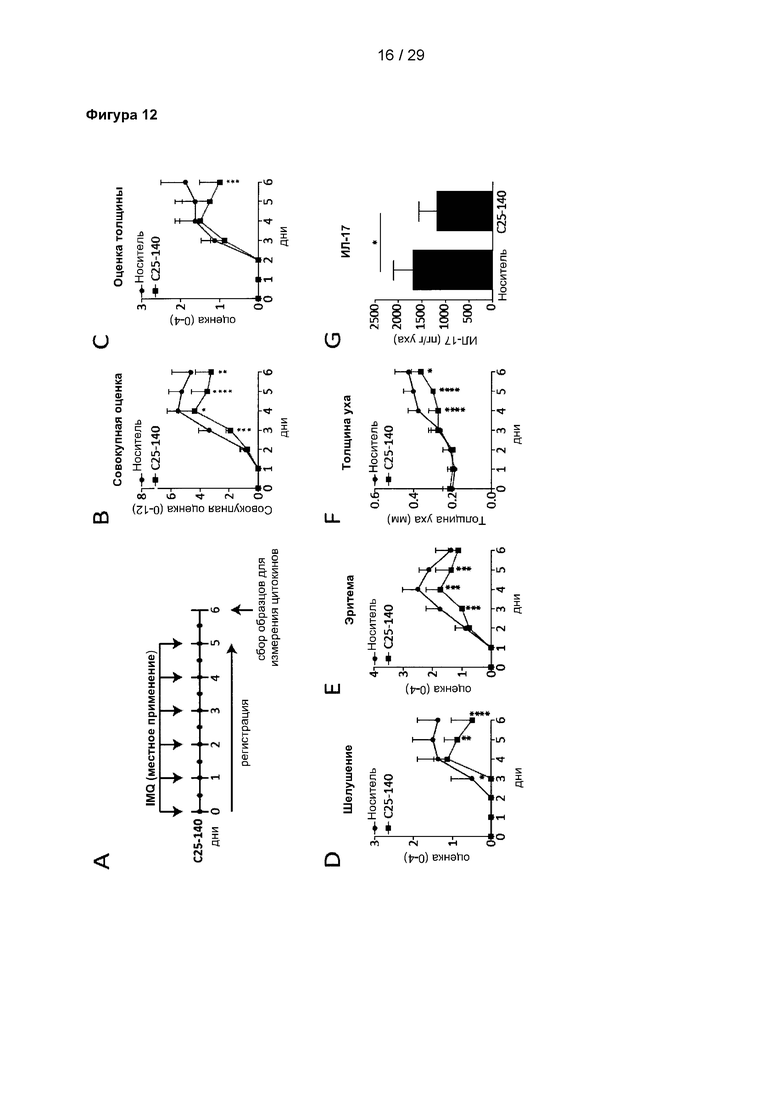
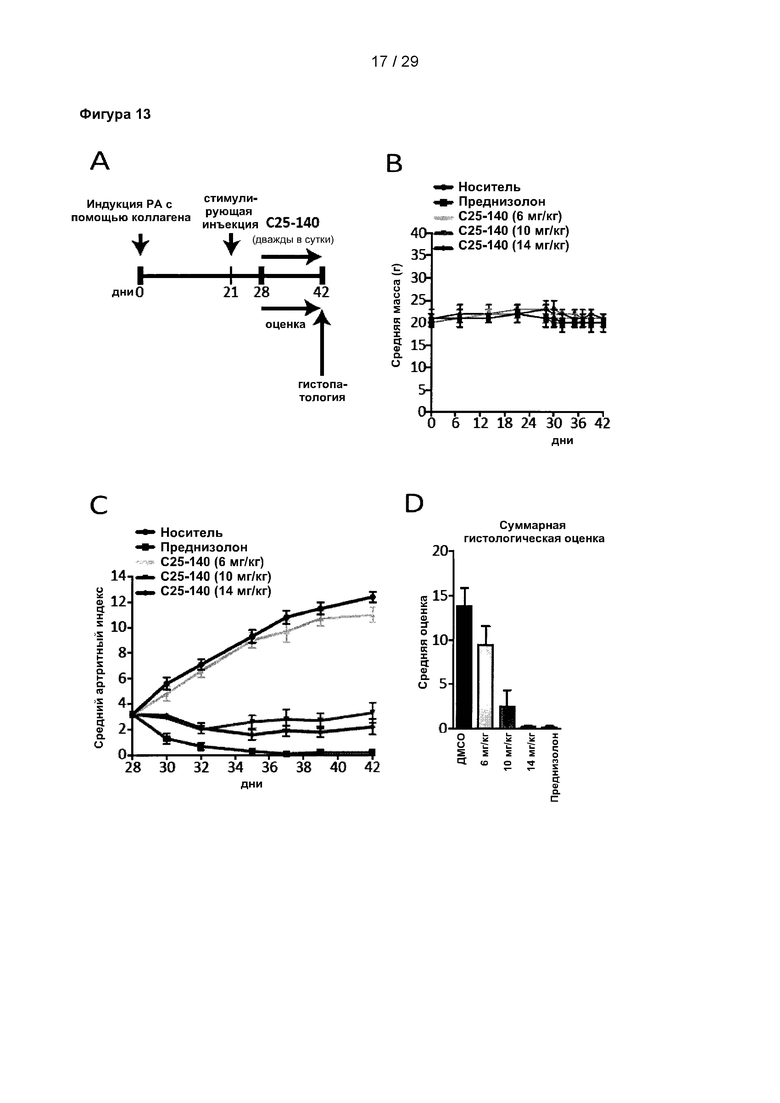
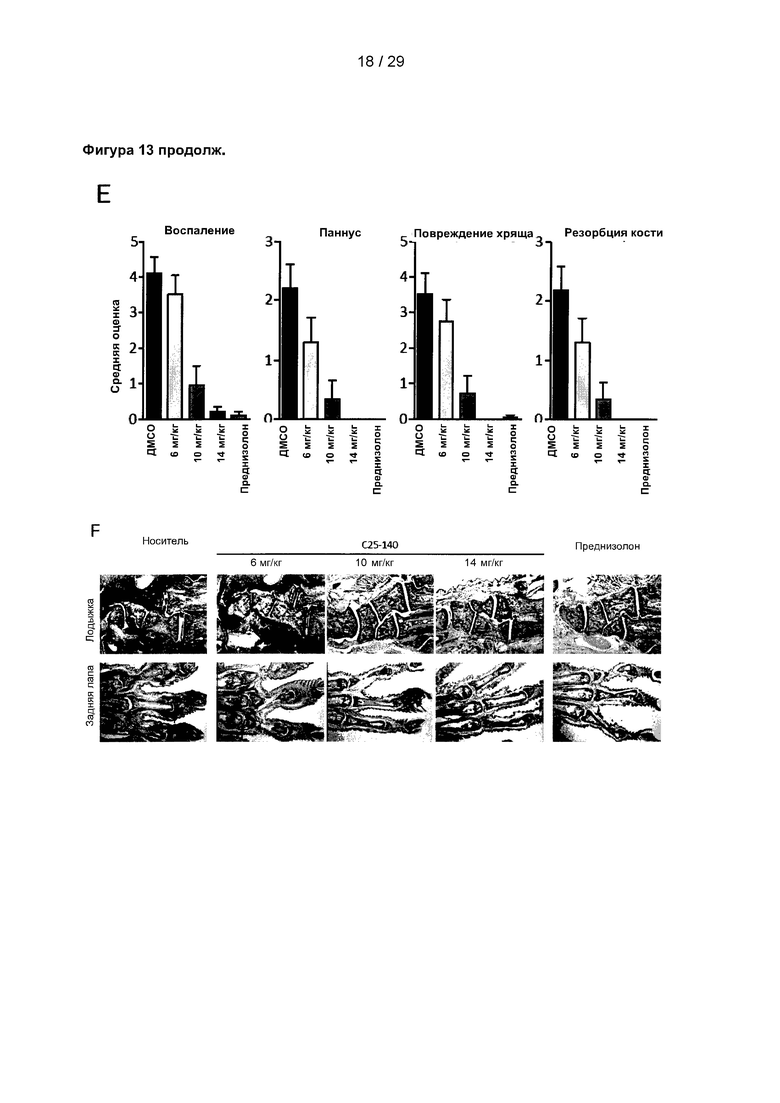
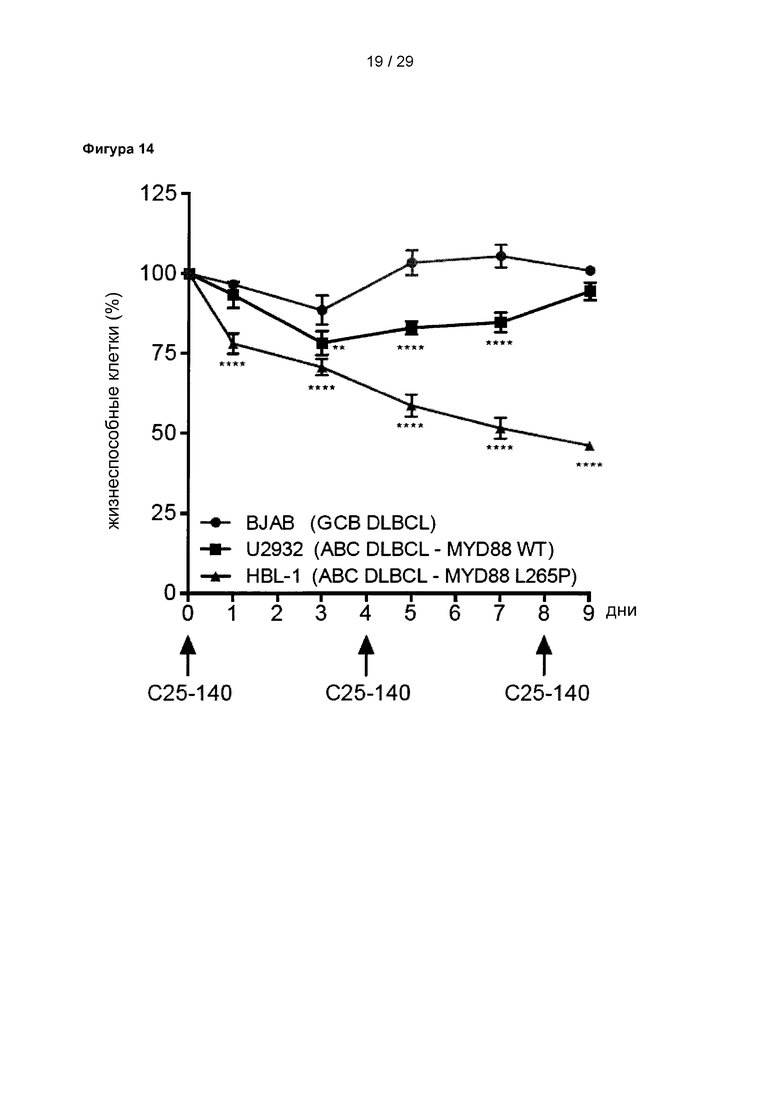
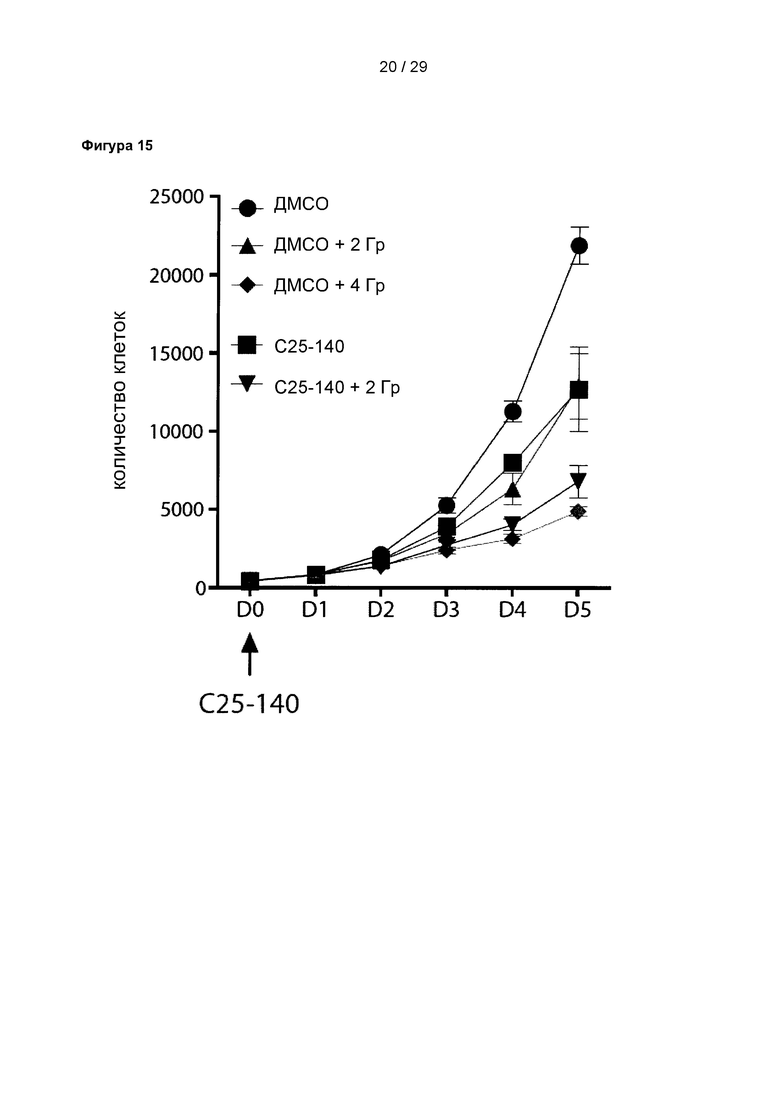
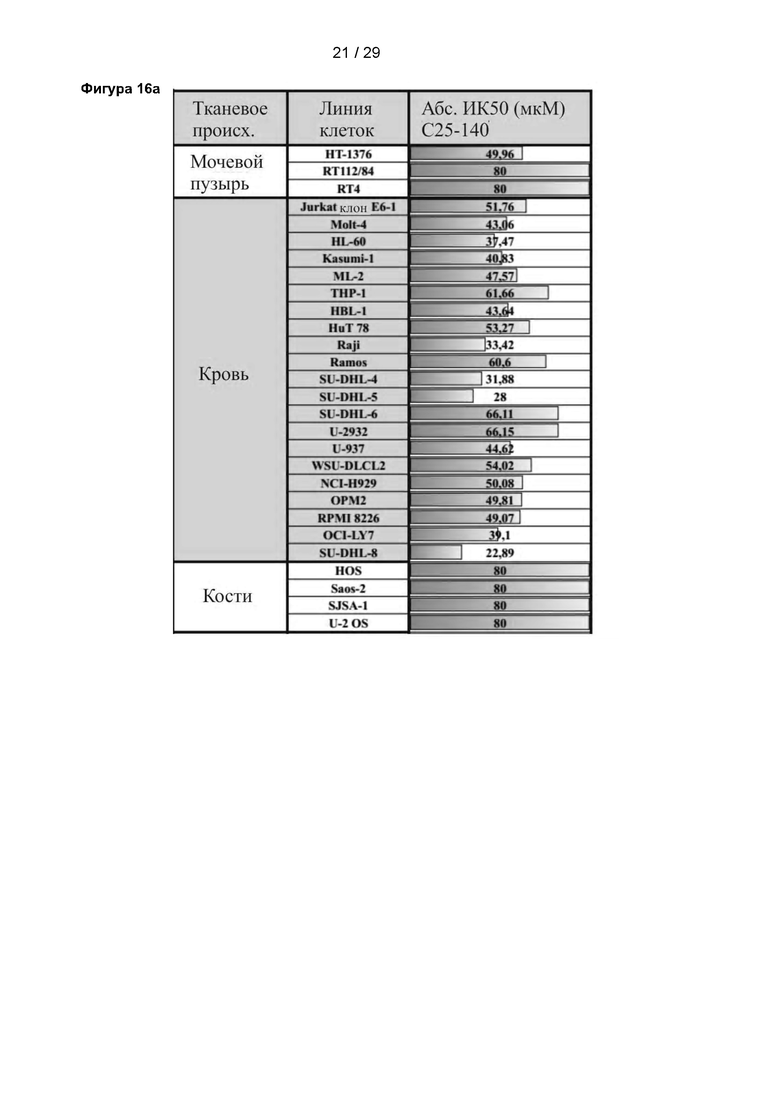
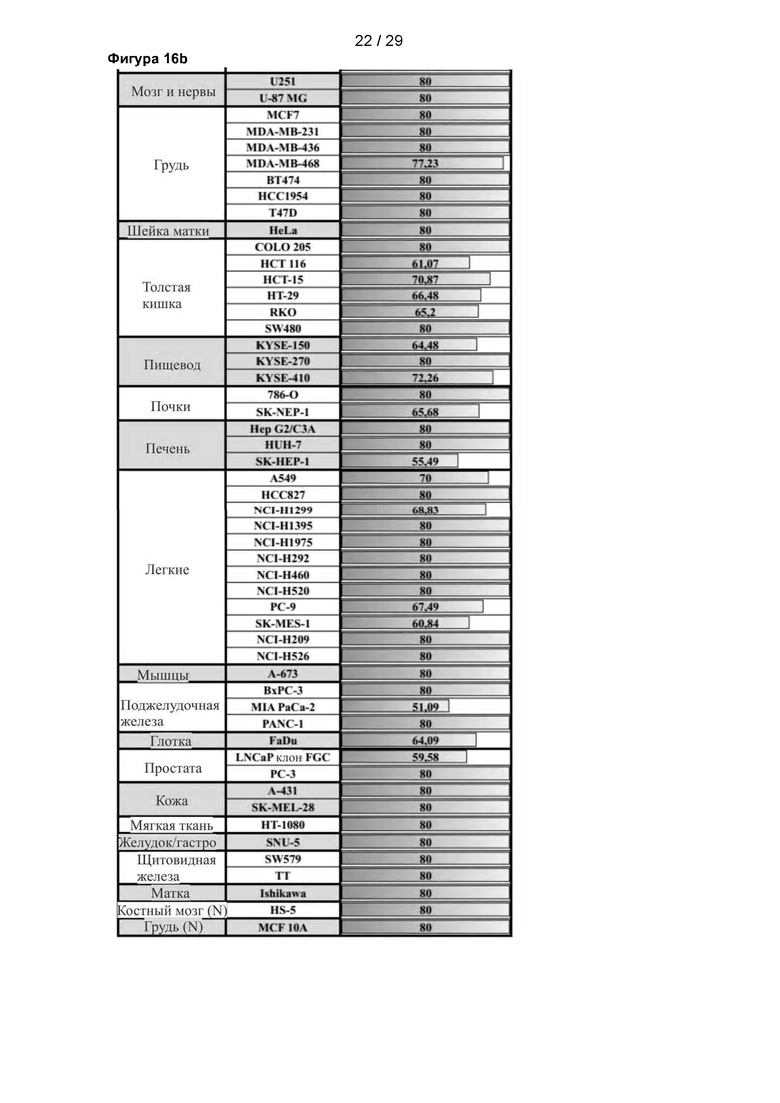
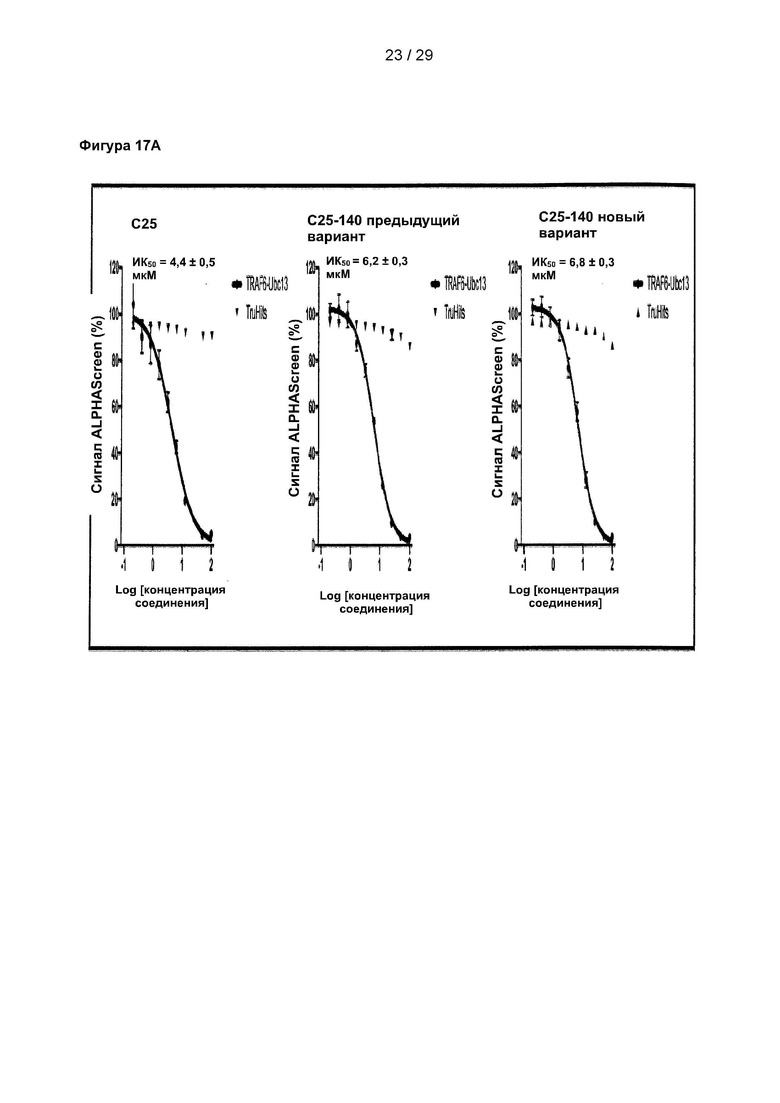
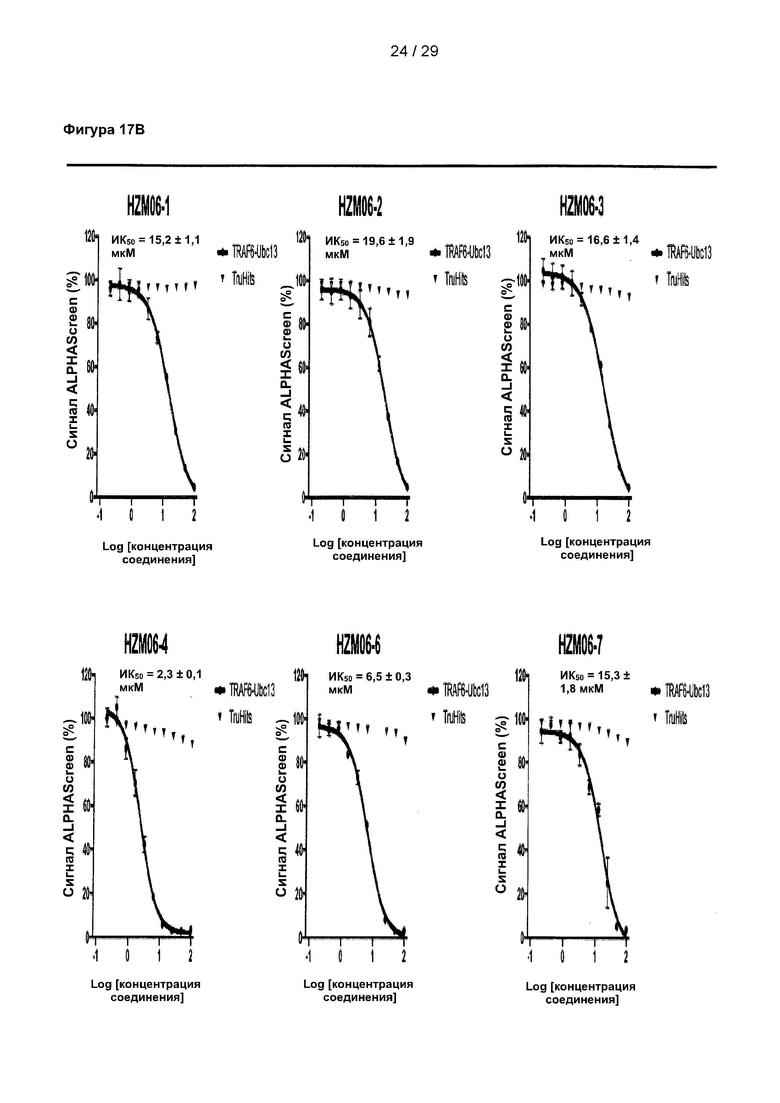
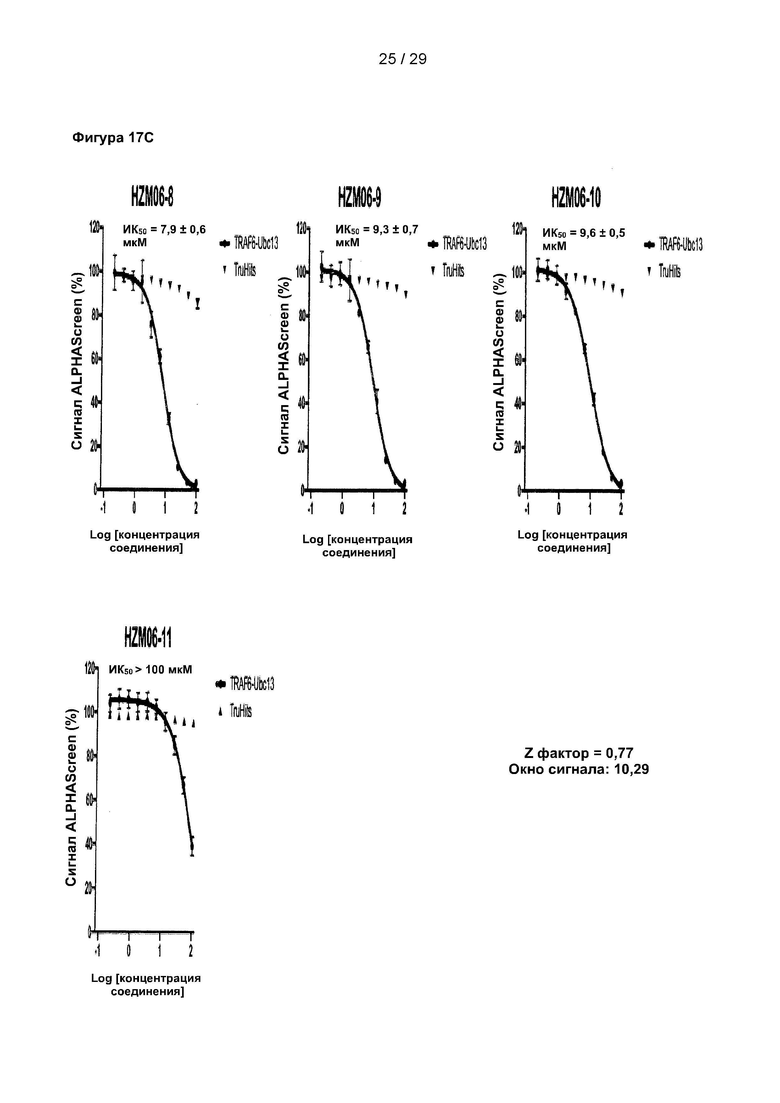
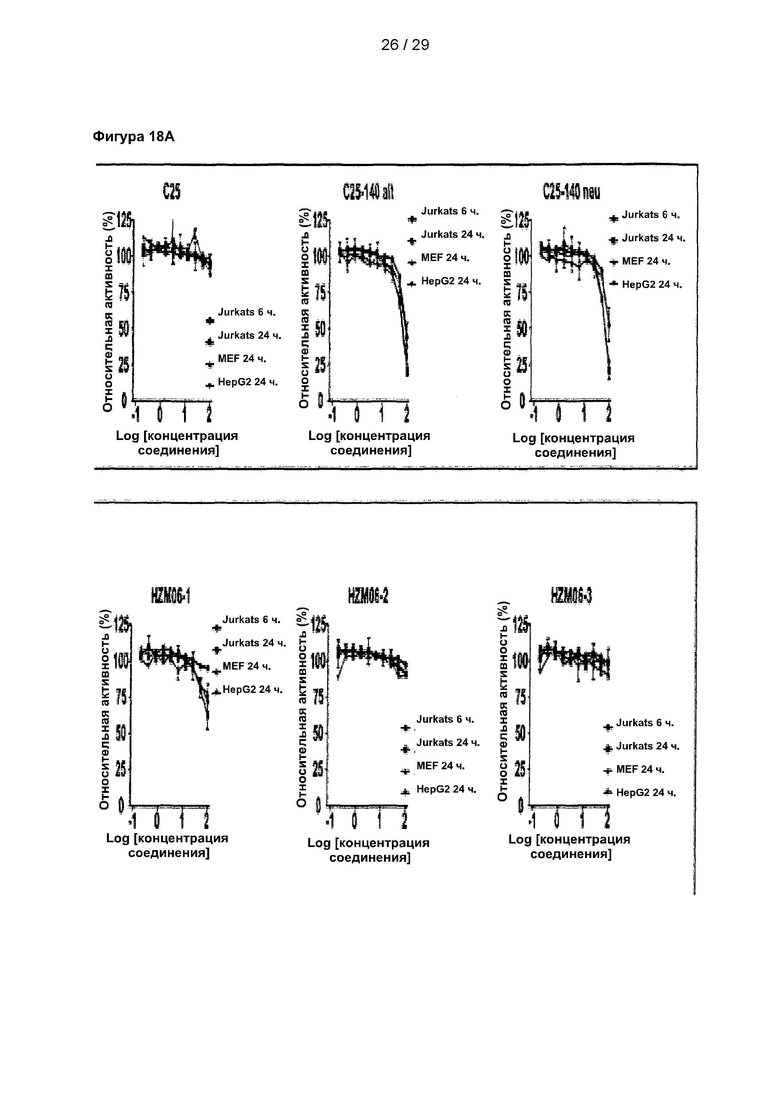
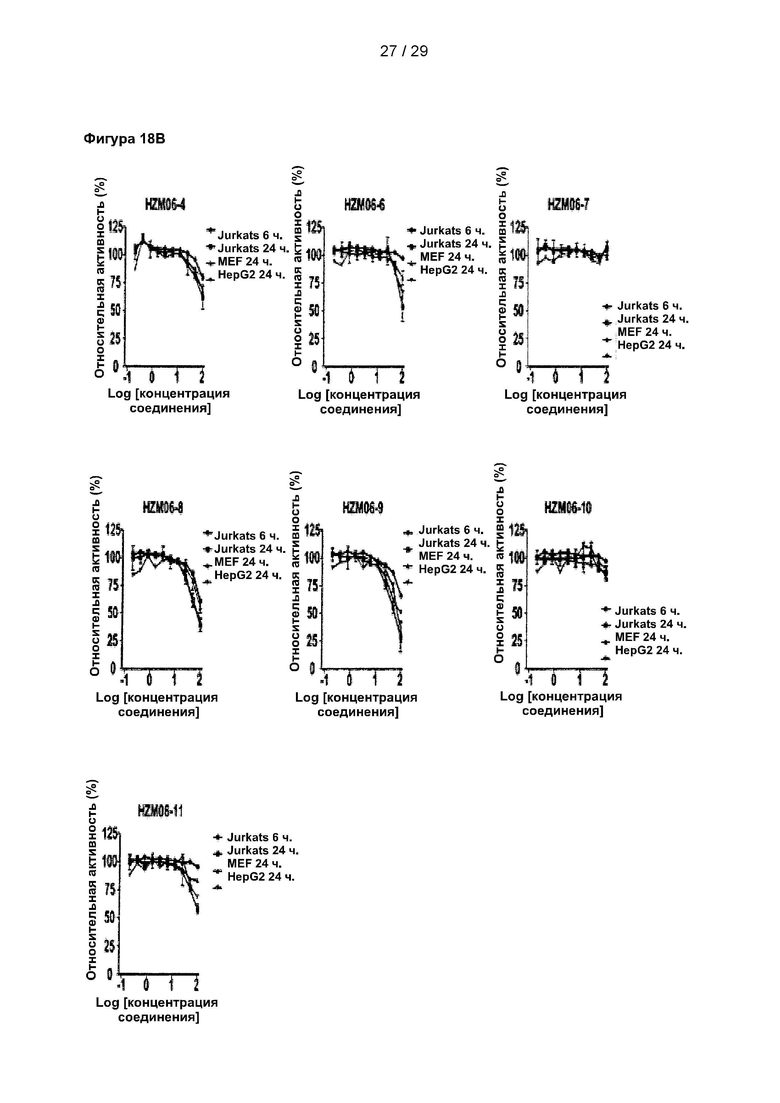
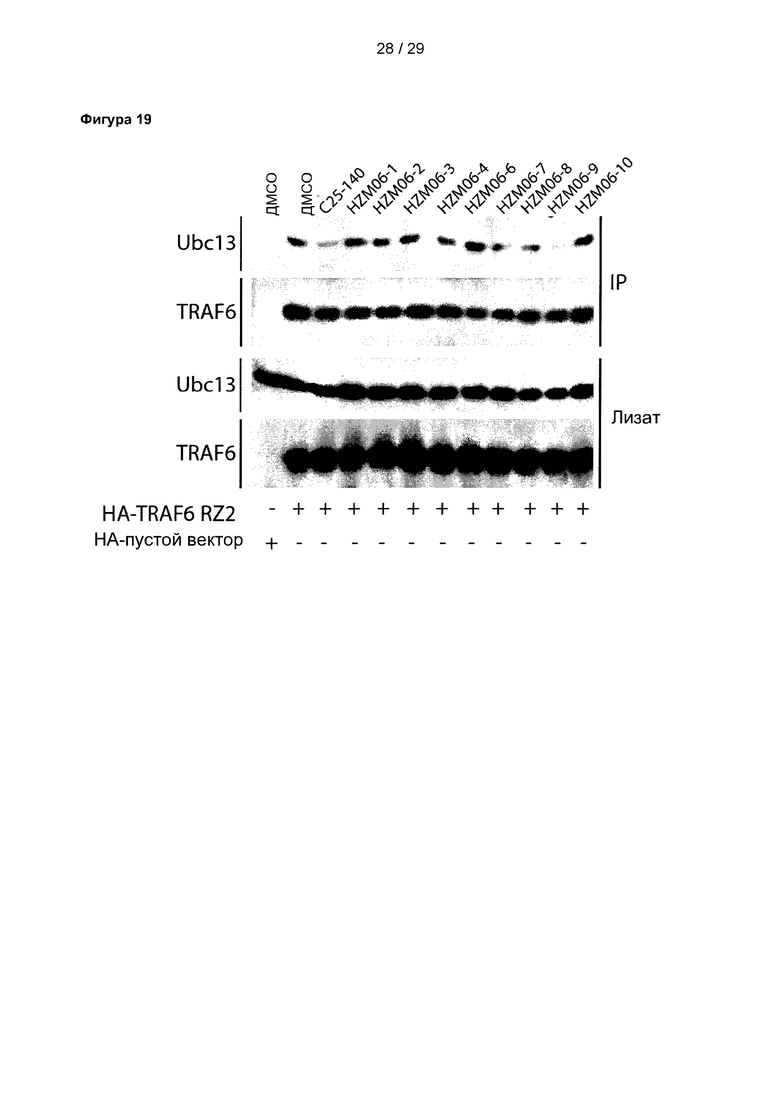
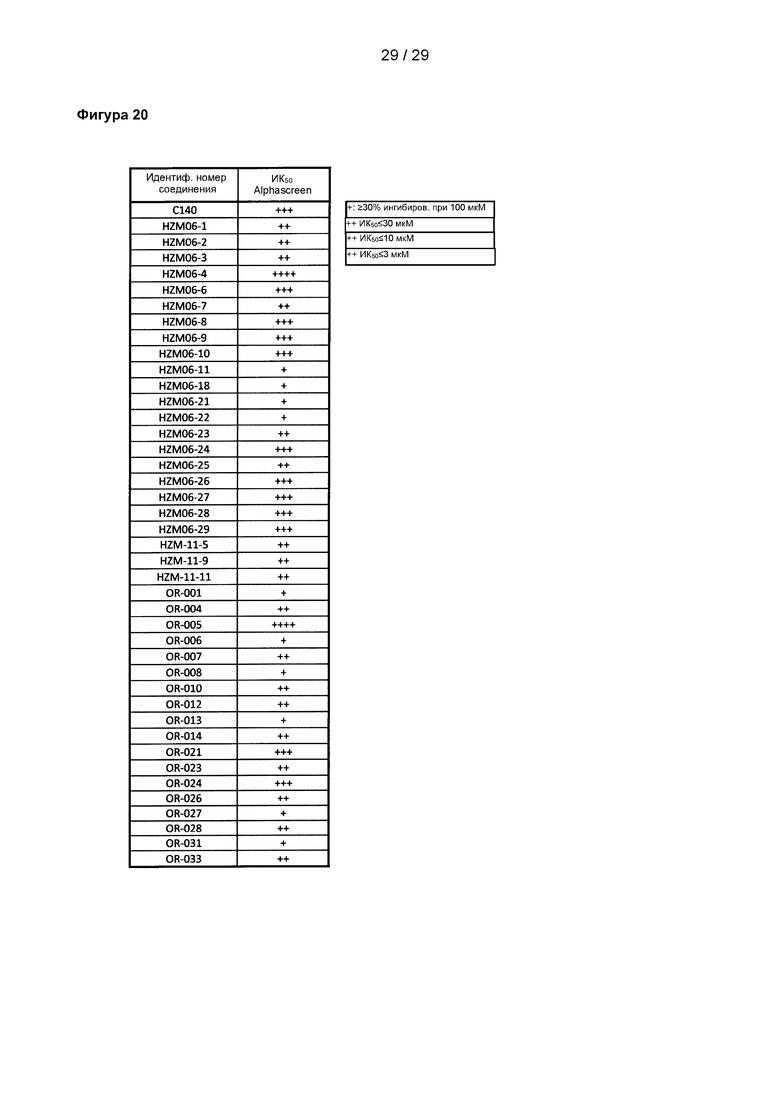
Настоящее изобретение относится к области органической химии, а именно к применению соединения формулы (I) для ингибирования взаимодействия TRAF6 с UBC13, где Х выбран из группы, состоящей из N и CH; R1 выбран из группы, состоящей из водорода, (C1-C6)алкила, (C1-C6)галогеналкила; R2 и R3 независимо выбраны из группы, состоящей из линейного (C1-C6)алкила; n представляет собой 2; Z представляет собой C=O; А выбран из группы, состоящей из –N(R4)(R5) и 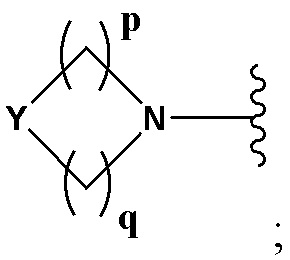 R4 представляет собой водород или (C1-C6)алкил; R5 выбран из группы, состоящей из (C6-C10)арил(C1-C6)алкила, фуранил(C1-C6)алкила, тиофенил(C1-C6)алкила и индолил(C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила; R6 представляет собой (C1-C4)алкил; Y выбран из группы, состоящей из N-B, CH-B и O; B выбран из группы, состоящей из (C6-C10)арила, пиридинила, (C6-C10)арил(C1-C6)алкила, пиридинил(C1-C6)алкила, фуранил(C1-C6)алкила и (C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила, (C1-C4)галогеналкила и -OR6; р и q представляют собой целое число от 1 до 2. Также изобретение относится к соединению формулы (I), где значения радикалов имеют указанные в формуле изобретения значения и конкретным соединениям. Технический результат: ингибирование взаимодействия TRAF6 с UBC13 соединением формулы (I) при лечении артрита, псориаза и некоторых видов рака. 3 н. и 9 з.п. ф-лы, 20 ил., 2 табл.
R4 представляет собой водород или (C1-C6)алкил; R5 выбран из группы, состоящей из (C6-C10)арил(C1-C6)алкила, фуранил(C1-C6)алкила, тиофенил(C1-C6)алкила и индолил(C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила; R6 представляет собой (C1-C4)алкил; Y выбран из группы, состоящей из N-B, CH-B и O; B выбран из группы, состоящей из (C6-C10)арила, пиридинила, (C6-C10)арил(C1-C6)алкила, пиридинил(C1-C6)алкила, фуранил(C1-C6)алкила и (C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила, (C1-C4)галогеналкила и -OR6; р и q представляют собой целое число от 1 до 2. Также изобретение относится к соединению формулы (I), где значения радикалов имеют указанные в формуле изобретения значения и конкретным соединениям. Технический результат: ингибирование взаимодействия TRAF6 с UBC13 соединением формулы (I) при лечении артрита, псориаза и некоторых видов рака. 3 н. и 9 з.п. ф-лы, 20 ил., 2 табл.
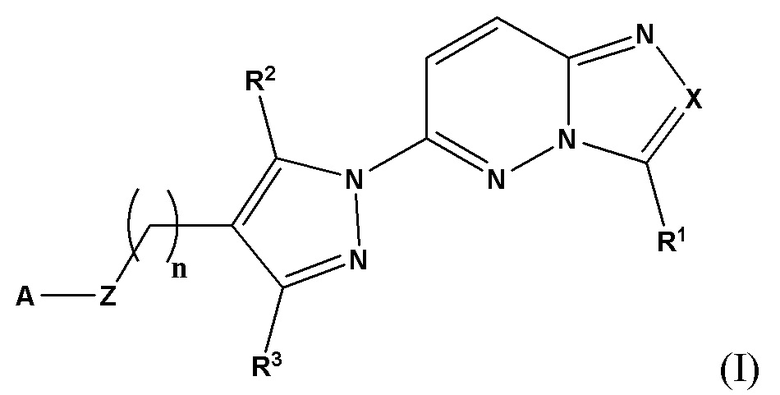
1. Применение соединения для ингибирования взаимодействия TRAF6 с UBC13, причем указанное соединение имеет структуру в соответствии с Формулой I
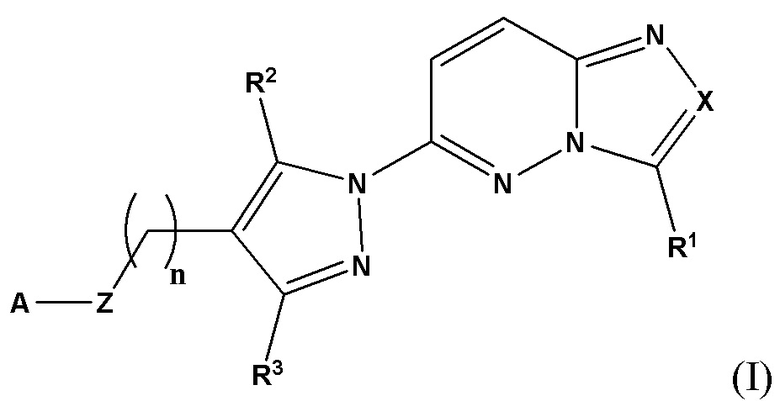
в которой
Х выбран из группы, состоящей из N и CH;
R1 выбран из группы, состоящей из водорода, (C1-C6)алкила, (C1-C6)галогеналкила;
R2 и R3 независимо выбраны из группы, состоящей из линейного (C1-C6)алкила;
n представляет собой 2;
Z представляет собой C=O;
А выбран из группы, состоящей из –N(R4)(R5) и 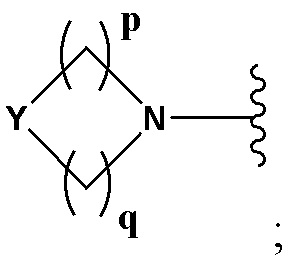
R4 представляет собой водород или (C1-C6)алкил;
R5 выбран из группы, состоящей из (C6-C10)арил(C1-C6)алкила, фуранил(C1-C6)алкила, тиофенил(C1-C6)алкила и индолил(C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила;
R6 представляет собой (C1-C4)алкил;
Y выбран из группы, состоящей из N-B, CH-B и O;
B выбран из группы, состоящей из (C6-C10)арила, пиридинила, (C6-C10)арил(C1-C6)алкила, пиридинил(C1-C6)алкила, фуранил(C1-C6)алкила и (C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила, (C1-C4)галогеналкила и -OR6;
p представляет собой целое число от 1 до 2;
q представляет собой целое число от 1 до 2.
2. Применение по п. 1, отличающееся тем, что указанное соединение Формулы (I) представляет собой соединение, имеющее структуру в соответствии с Формулой (II)
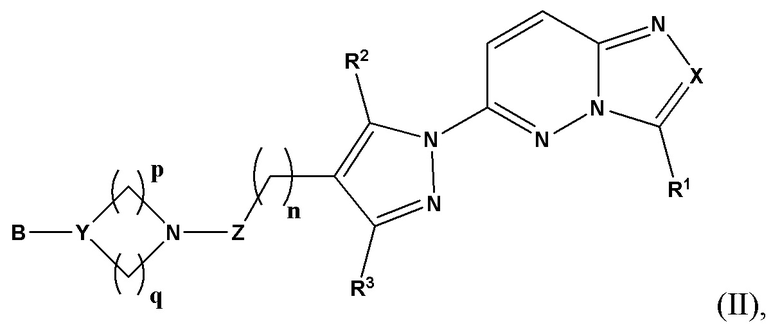
в которой
Х выбран из группы, состоящей из N и CH;
R1 выбран из группы, состоящей из водорода, (C1-C6)алкила, (C1-C6)галогеналкила;
R2 и R3 независимо выбраны из группы, состоящей из линейного (C1-C6)алкила;
n представляет собой 2;
Z представляет собой C=O;
Y представляет собой N или CH;
B выбран из группы, состоящей из (C6-C10)арила, пиридинила, (C6-C10)арил(C1-C6)алкила, пиридинил(C1-C6)алкила, фуранил(C1-C6)алкила и (C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила, (C1-C4)галогеналкила и -OR6, предпочтительно галогена и -OR6;
p представляет собой целое число от 1 до 2;
q представляет собой целое число от 1 до 2.
3. Применение по любому из предшествующих пунктов, отличающееся тем, что указанное соединение, имеющее структуру в соответствии с Формулой (I) или Формулой (II), характеризуется тем, что
Х представляет собой N.
4. Применение по любому из предшествующих пунктов, отличающееся тем, что указанное соединение, имеющее структуру в соответствии с Формулой (I) или Формулой (II), характеризуется тем, что
Z представляет собой C=O.
5. Применение по любому из предшествующих пунктов, отличающееся тем, что указанное соединение, имеющее структуру в соответствии с Формулой (I) или Формулой (II), характеризуется тем, что
R1 выбран из группы, состоящей из водорода, (C1-C4)алкила;
R2 и R3 независимо выбраны из группы, состоящей из линейного (C1-C4)алкила;
n представляет собой 2; и
R5 выбран из группы, состоящей из (C6-C10)арил(C1-C6)алкила, фуранил(C1-C6)алкила, тиофенил(C1-C6)алкила и индолил(C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила, предпочтительно галогена.
6. Применение по любому из предшествующих пунктов, отличающееся тем, что указанное соединение Формулы (I) представляет собой соединение, имеющее структуру в соответствии с Формулой (III)
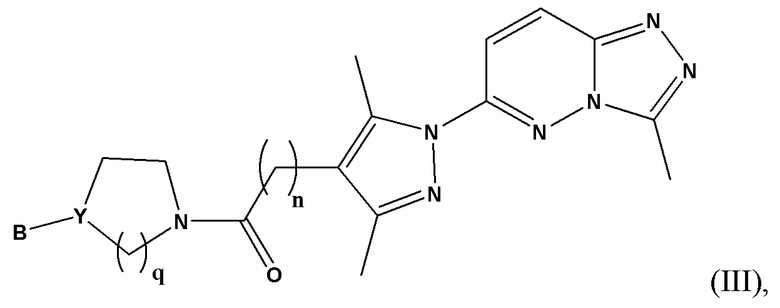
в которой
n представляет собой 2;
Y представляет собой N или CH;
B выбран из группы, состоящей из (C6-C10)арила, пиридинила, (C6-C10)арил(C1-C6)алкила, пиридинил(C1-C6)алкила, фуранил(C1-C6)алкила и (C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила, (C1-C4)галогеналкила и -OR6, предпочтительно галогена и -OR6;
q представляет собой целое число от 1 до 2, более предпочтительно 2.
7. Применение по любому из пп. 1-5, отличающееся тем, что указанное соединение Формулы (I) представляет собой соединение, имеющее структуру в соответствии с Формулой (IV)
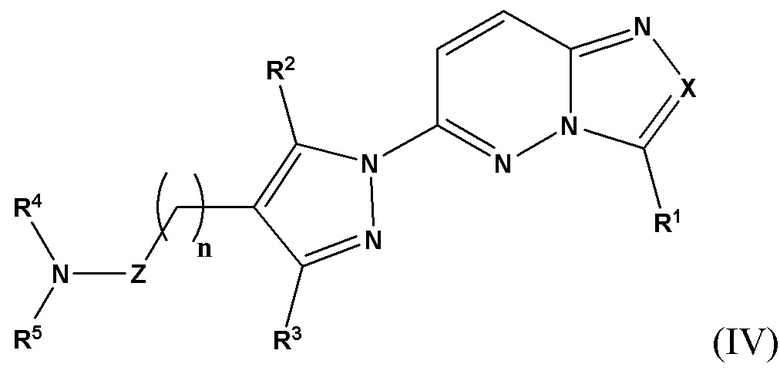
в которой
Х представляет собой N;
R1 выбран из группы, состоящей из водорода, (C1-C6)алкила, (C1-C6)галогеналкила, предпочтительно водорода и (C1-C6)алкила, более предпочтительно (C1-C6)алкила;
R2 и R3 независимо выбраны из группы, состоящей из линейного (C1-C6)алкила;
n представляет собой 2;
Z представляет собой C=O;
R4 представляет собой водород или (C1-C6)алкил, предпочтительно водород;
R5 выбран из группы, состоящей из (C6-C10)арил(C1-C6)алкила, фуранил(C1-C6)алкила, тиофенил(C1-C6)алкила и индолил(C1-C6)алкила, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила, предпочтительно галогена;
R6 представляет собой (C1-C4)алкил.
8. Применение соединения по любому из предшествующих пунктов для лечения псориаза и артрита.
9. Применение соединения по любому из пп. 1-7,
где X представляет собой N;
для лечения диффузной крупноклеточной B-клеточной лимфомы (DLBCL), рака мочевого пузыря, рака крови, рака толстой кишки, рака печени, рака легких, рака поджелудочной железы, рака глотки и рака простаты.
10. Применение по пп. 8 и 9, отличающееся тем, что соединение представляет собой
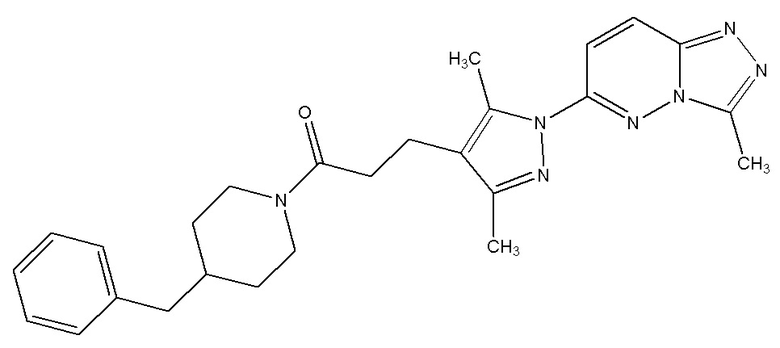
11. Соединение, имеющее структуру в соответствии с Формулой I,
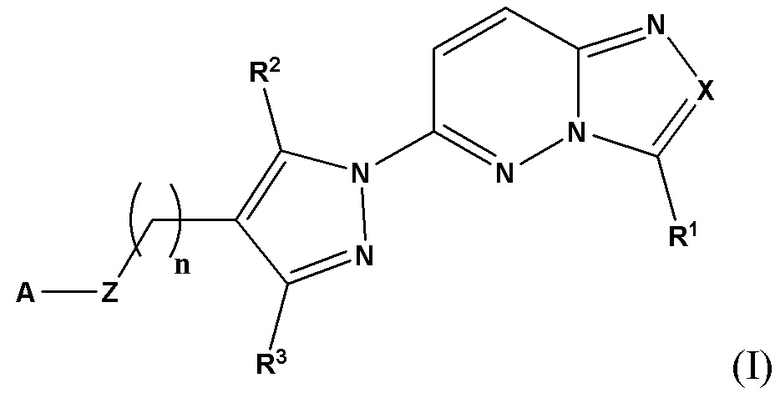
в которой
Х выбран из группы, состоящей из N и CH;
R1 представляет собой (C1-C6)алкил;
R2 и R3 независимо выбраны из группы, состоящей из (C1-C6)алкила;
n представляет собой 2;
Z представляет собой C=O;
А выбран из группы, состоящей из 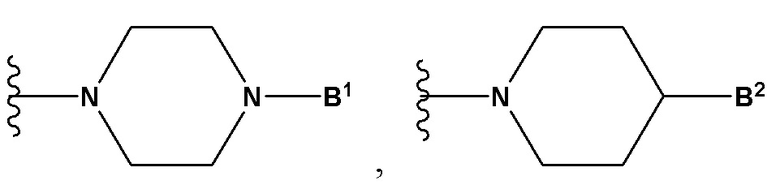 и
и 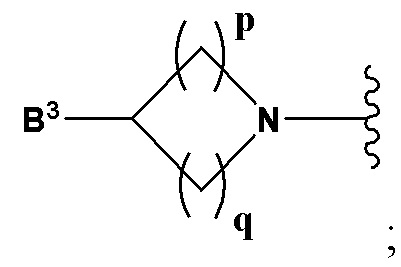
B1 представляет собой (C6-C10)арил(C1-C6)алкил, фуранил(C1-C6)алкил, которые необязательно замещены одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена и –OR6;
B2 представляет собой (C6-C10)арил(C1-C6)алкил, замещенный одним или более идентичными или различными заместителями, выбранными из группы, состоящей из галогена или
B2 представляет собой пиридинил(C1-C6)алкил;
B3 выбран из группы, состоящей из (C6-C10)арил(C1-C6)алкила;
R6 представляет собой (C1-C4)алкил;
p равно 1 и
q представляет собой целое число от 1 до 2.
12. Соединение, выбранное из группы, включающей:
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(4-фторбензил)пиперазин-1-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-фенилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(3-метоксибензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(4-фторбензил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(пиридин-2-ил-метил)пиперазин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-фенил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диметил-1-(3-метилимидазо[1,2-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)этан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-морфолинопропан-1-он,
N-(3-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-N-метилпропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(фуран-2-ил-метил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(2-фенилпропан-2-ил)пиперазин-1-ил)пропан-1-он,
1-(4-(3,4-диметоксибензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(нафтален-2-ил-метил)пиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(3-бензилазетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(4-(4-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(4-(4-метоксифенил)пиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(4-фенилпиперазин-1-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
3-(1-([1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(4-(пиридин-2-ил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-N-((5-метилфуран-2-ил)метил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(2-фторфенил)пиперазин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-N-(3-фенилпропил)пропанамид,
3-(1-([1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(4-бензилпиперидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(2,5-диметилфенил)пиперазин-1-ил)пропан-1-он,
N-(4-хлорбензил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-N-(тиофен-2-ил-метил)пропанамид,
N-(2-(1H-индол-3-ил)этил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропанамид,
N-бензил-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропанамид,
3-(1-([1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-N-(3-фенилпропил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-этилпиперазин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(4-метилпиперидин-1-ил)пропан-1-он,
3-(1-([1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-N-(2-фторбензил)пропанамид,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-N-(2-((фуран-2-ил-метил)тио)этил)пропанамид,
1-(4-(3,4-диметоксибензил)пиперазин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(4-(3-хлорбензил)пиперидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(3-бензилазетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(3-(бензиламино)азетидин-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
N-бензил-3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-карбоксамид,
(3-бензилпирролидин-1-ил)(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)метанон,
1-(3-бензилпирролидин-1-ил)-3-(5-изопропил-3-метил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3-метил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-5-(трифторметил)-1H-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(4-(3-метоксифенил)пиперазин-1-ил)пропан-1-он,
1-бензил-4-(3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропанoил)пиперазин-2-он,
1-(4-бензилпиперидин-1-ил)-3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(1-(3-этил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропан-1-он,
1-(2,2-диметилпирролидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропан-1-он,
1-(3,3-диметилпирролидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
3-(3,5-диэтил-1-(3-изопропил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(3,3-диметилпирролидин-1-ил)пропан-1-он,
1-((1S)-2-азабицикло[2.2.1]гептан-2-ил)-3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропан-1-он,
(1S,5R)-3-(3-(1-(3-этил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропанoил)-2,3,4,5-тетрагидро-1,5-метанпиридо[1,2-d][1,4]диазепин-7(1H)-он,
1-((1S)-2-азабицикло[2.2.1]гептан-2-ил)-3-(1-(3-этил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-(трифторметил)-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-изопропил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(1-(3-этил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)пропан-1-он,
3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)-1-(3,3-диметилпирролидин-1-ил)пропан-1-он,
3-(1-(3-этил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
3-(1-(3-изопропил-[1,2,4]триазол[4,3-b]пиридазин-6-ил)-3,5-диметил-1H-пиразол-4-ил)-1-(пирролидин-1-ил)пропан-1-он,
1-(3-бензилпирролидин-1-ил)-3-(3,5-диэтил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(4-бензил-1,4-диазепан-1-ил)-3-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-3-(3-изопропил-5-метил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперидин-1-ил)-2-(3,5-диметил-1-(3-метил-[1,2,4]триазоло[4,3-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он,
1-(4-бензилпиперазин-1-ил)-3-(3,5-диметил-1-(3-метилимидазо[1,2-b]пиридазин-6-ил)-1H-пиразол-4-ил)пропан-1-он.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Аппарат для определения плотности жидкостей | 1928 |
|
SU12393A1 |

