Область техники
[0001] Настоящее изобретение относится к областям химии и медицины. Более конкретно, настоящее изобретение относится к немакроциклическим α-кетоамидным соединениям в качестве модуляторов кальпаина, являющихся малыми молекулами, композициям, их получению и их применению в качестве терапевтических агентов. Уровень техники
[0002] По оценкам, в развитых странах фиброзное заболевание является причиной 45% летальных исходов, однако разработка терапии таких заболеваний до сих пор находится на этапе становления. Современные способы лечения фиброзных заболеваний, таких как идиопатический фиброз легких, фиброз почки, системный склероз и цирроз печени, немногочисленны и обеспечивают лишь ослабление некоторых симптомов фиброза, но не могут устранить первопричину.
[0003] Несмотря на имеющееся в настоящее время ограниченное понимание различных этиологических факторов, обуславливающих эти состояния, сходство в фенотипе пораженных органов при различных фиброзных заболеваниях всецело подтверждает существование общих путей патогенеза. В настоящее время признано, что основной движущей силой фиброзного заболевания является сигнальный путь с высоким уровнем трансформирующего фактора роста-бета (TGFβ), который может способствовать трансформации нормально функционирующих клеток в клетки, способствующие фиброзу. Эти трансформированные клетки, называемые «миофибробластами», могут секретировать большие количества белков внеклеточного матрикса и разрушающих матрикс ферментов, что приводит к образованию рубцовой ткани и возможной недостаточности органов. Этот клеточный процесс является трансформирующим и называется «дифференцировкой миофибробластов» (которая включает эпителиально-мезенхимальный переход (ЕрМТ) и его вариации, такие как эндотелиально-мезенхимальный переход (EnMT) и переход фибробластов в миофибробласты (FMT)). Этот процесс является основной мишенью при лечении фиброзных заболеваний. Также было показано, что дифференцировка миофибробластов происходит внутри раковых клеток, которые хронически подвергаются воздействию высокого уровня TGFβ, и вызывает превращение покоящихся эпителиальных клеток в лабильные, инвазивные и метастазирующие. Таким образом, в отношении рака было документально подтверждено, что указанный путь передачи сигнала связан с приобретением устойчивости к лекарственным средствам, ускользанием от действия иммунной системы и развитием свойств стволовых клеток.
[0004] Несмотря на колоссальные потенциальные возможности лекарственных средств, подавляющих дифференцировку миофибробластов, и многочисленные попытки разработки эффективного лечения, полученным на настоящий момент данным еще только предстоит найти применение в практической терапии. Отчасти это связано с отсутствием идеального белка-мишени. Исходные стратегии нацеливания на процесс дифференцировки миофибробластов были направлены на проксимальное ингибирование сигнального пути TGFβ с помощью различных способов, включая нацеливание на активаторы лиганда (например, интегрины альфа-v), взаимодействия лиганда с рецептором (например, с применением нейтрализующих антител) или активность киназы рецептора TGFβ (например, лекарственные средства для блокирования трансдукции сигнала, представляющие собой химические соединения, являющиеся малыми молекулами). К сожалению, TGFβ является плейотропным цитокином, обладающим многочисленными физиологическими функциями, в связи с чем полное подавление сигналинга TGFβ также было связано с тяжелыми побочными эффектами. Кроме того, имеющиеся данные указывают на то, что такое проксимальное ингибирование может поддаваться патологическим стратегиям обхода (например, вследствие избыточности или компенсации), что ограничило бы полезность таких лекарственных средств. Дополнительные сложности связаны с тем, что при раке на ранних стадиях сигналинг TGFβ действует в качестве противоопухолевого ингибитора роста, однако затем начинает способствовать развитию опухоли, и это является еще одной причиной, по которой селективное ингибирование патогенных звеньев сигналинга является в столь высокой степени желательным. В связи с этими неотъемлемыми ограничениями современные стратегии лечения были переориентированы на выявление и подавление критически важных дистальных процессов сигналинга TGFβ, что в теории должно обеспечивать нацеливание преимущественно на патологические, но не физиологические функции сигналинга TGFβ.
Краткое описание изобретения
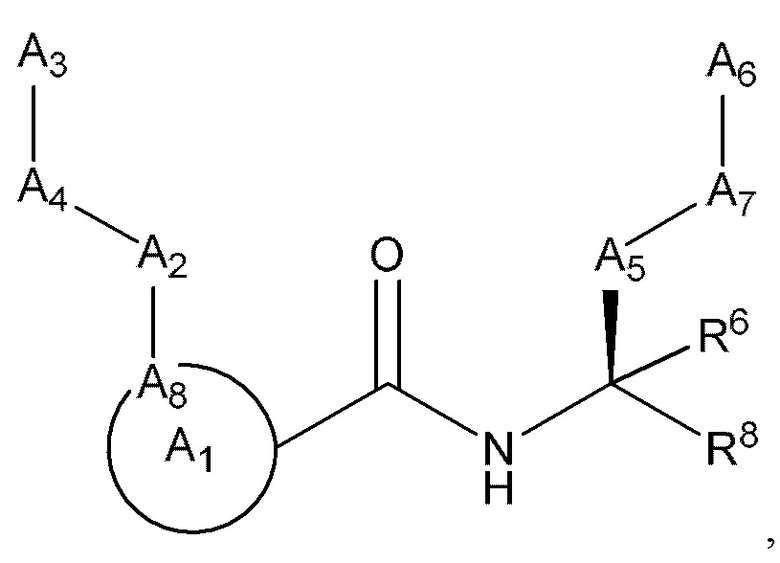
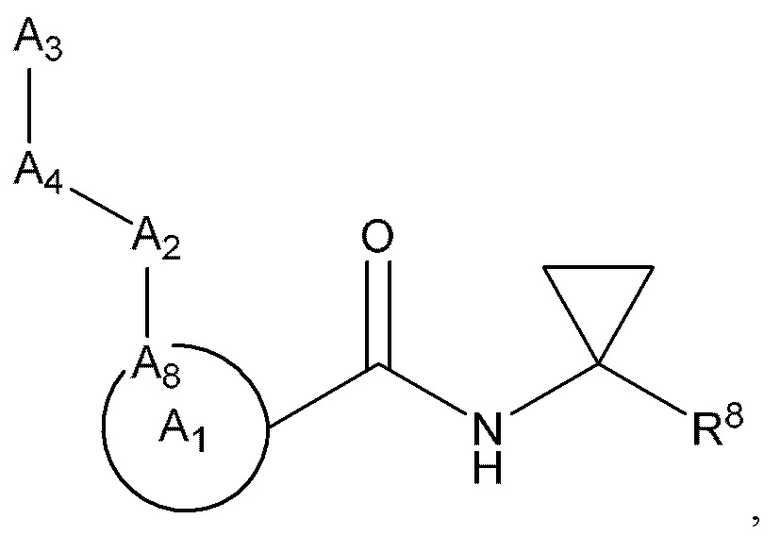
[0005] Соединение, имеющее структуру формулы I:
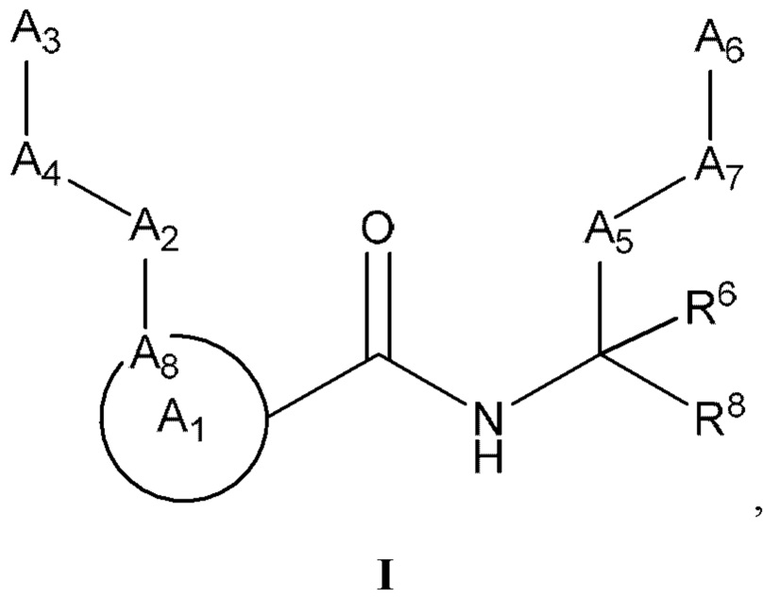
или его фармацевтически приемлемая соль, где:
A1 выбран из группы, состоящей из необязательно замещенного 5-10-членного гетероциклила, при условии, что указанный 5-10-членный гетероциклил не замещен оксо, необязательно замещенного 5-, 8- или 9-членного гетероарила и необязательно замещенного С3-10 карбоциклила;
А2 выбран из группы, состоящей из необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила и необязательно замещенного С3-10 карбоциклила, -CR2-, -S-, -S(=O)-, -SO2-, -О-, -C(=S)-, -С(=O)-, -NR-, -СН=СН-, -С≡С-, -OC(O)NH-, -NHC(O)NH-, -NHC(O)O-, -NHC(O)-, -NHC(S)NH-, -NHC(S)O-, -NHC(S)- и одинарной связи;
A4 выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С3-10 карбоциклила, необязательно замещенного C1-4 алкила, -(CR2)n-S-(CR2)n-, -(CR2)n-S(=O)-(CR2)n-, -(CR2)n-SO2-(CR2)n-, -(CR2)n-O-(CR2)n-, -(CR2)n-C(=S)-(CR2)n-, -(CR2)n-C(=O)-(CR2)n-, -(CR2)n-NR-(CR2)n-, -(CR2)n-CH=CH-(CR2)n-, -(CR2)n-OC(O)NH-(CR2)n-, -(CR2)n-NHC(O)NH-(CR2)n-, -(CR2)n-NHC(O)O-(CR2)n-, -(CR2)n-NHC(O)-(CR2)n-, -(CR2)n-NHC(S)NH-(CR2)n-, -(CR2)n-NHC(S)O-(CR2)n-, -(CR2)n-NHC(S)-(CR2)n- и одинарной связи;
в случае, если А2 и А4 представляют собой одинарную связь, А3 непосредственно присоединен к A8;
A3 выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила и необязательно замещенного С3-10 карбоциклила, или, если А2 выбран из необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила и необязательно замещенного С3-10 карбоциклила, то А3 выбран из группы, состоящей из атома водорода, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С3-10 карбоциклила, -С≡СН и необязательно замещенного 2-5-членного полиэтиленгликоля;
А5 выбран из группы, состоящей из необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного С3-10 карбоциклила, необязательно замещенного C1-8 алкила, -S-, -S(=O)-, -SO2-, -О-, -C(=S)-, -С(=O)-, -NR-, -СН=СН-, -OC(O)NH-, -NHC(O)NH-, -NHC(O)O-, -NHC(O)-, -NHC(S)NH-, -NHC(S)O-, -NHC(S)- и одинарной связи;
А6 выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила и необязательно замещенного С3-10 карбоциклила, необязательно замещенного C1-8 алкила, необязательно замещенного С2-8 алкенила, необязательно замещенного -O-C1-6 алкила, необязательно замещенного -О-С2-6 алкенила, -OSO2CF3 и боковой цепи любой природной или неприродной аминокислоты;
А7 выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С3-10 карбоциклила, необязательно замещенного C1-8 алкила, -S-, S(=O)-, -SO2-, -О-, -C(=S)-, -С(=O)-, -NR-, -СН=СН-, -OC(O)NH-, -NHC(O)NH-, -NHC(O)O-, -NHC(O)-, -NHC(S)NH-, -NHC(S)O-, -NHC(S)- и одинарной связи;
в случае, если A5 и А7 представляют собой одинарную связь, А6 непосредственно связан с атомом углерода, к которому присоединен R8;
A8 является членом кольца A1 и выбран из группы, состоящей из С, СН и N;
R8 выбран из группы, состоящей из -COR1, -CN, -CH=CHSO2R и -CH2NO2;
R1 выбран из группы, состоящей из Н, -ОН, С1-4 галогеналкила, -СООН, -CH2NO2, -C(=O)NOR, -NH2, -CONR2R3, -CH(CH3)=CH2, -CH(CF3)NR2R3,
-C(F)=CHCH2CH3, 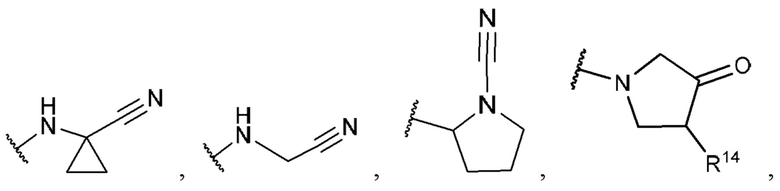
R14 представляет собой галоген;
каждый R, R2 и R3 независимо выбран из -Н, необязательно замещенного
С1-4 алкила, необязательно замещенного C1-8 алкоксиалкила, необязательно замещенного 2-5-членного полиэтиленгликоля, необязательно замещенного С3-7 карбоциклила, необязательно замещенного 5-10-членного гетероциклила, необязательно замещенного С6-10 арила и необязательно замещенного 5-10-членного гетероарила; и
R6 независимо выбран из -Н и необязательно замещенного С1-4 алкила.
[0006] Другие варианты реализации, раскрытые в настоящем описании, включают фармацевтическую композицию, содержащую терапевтически эффективное количество соединения, раскрытого в настоящем описании, и фармацевтически приемлемое вспомогательное вещество.
[0007] Другие варианты реализации, раскрытые в настоящем описании, включают способ лечения заболеваний и состояний, опосредованных, по меньшей мере частично, физиологическими эффектами CAPN1, CAPN2 или САР9 или их комбинаций, включающий введение нуждающемуся в этом субъекту соединения, раскрытого в настоящем описании.
[0008] В некоторых вариантах реализации соединения, раскрытые в настоящем описании, являются специфическими ингибиторами одного из: CAPN1, CAPN2 или CAPN9.
[0009] В некоторых вариантах реализации соединения, раскрытые в настоящем описании, являются селективными ингибиторами одного из: CAPN1, CAPN2 или CAPN9.
[0010] В некоторых вариантах реализации соединения, раскрытые в настоящем описании, являются селективными ингибиторами: CAPN1 и CAPN2, или CAPN1 и CAPN9, или CAPN2 и CAPN9.
[0011] В некоторых вариантах реализации соединения, раскрытые в настоящем описании, являются эффективными ингибиторами CAPN1, CAPN2 и/или CAPN9.
[0012] В некоторых вариантах реализации немакроциклические α-кетоамидные соединения, раскрытые в настоящем описании, в целом являются эффективными для лечения множества состояний, обусловленных фиброзом или воспалением, и, в частности, включая состояния, связанные с дифференцировкой миофибробластов. Соответственно, соединения, раскрытые в настоящем описании, являются терапевтическими средствами, активными в отношении ряда различных заболеваний или расстройств, которые включают или которые вызывают симптомы, которые включают, но не ограничиваются перечисленными: фиброз печени, фиброз почки, фиброз легкого, пневмонит гиперчувствительности, интерстициальный фиброз, системную склеродермию, макулярную дегенерацию, фиброз поджелудочной железы, фиброз селезенки, фиброз сердца, медиастинальный фиброз, миелофиброз, эндомиокардиальный фиброз, ретроперитонеальный фиброз, прогрессирующий массивный фиброз, нефрогенный системный фиброз, фиброзные послеоперационные осложнения, хроническую васкулопатию аллотрансплантата и/или хроническое отторжение трансплантированных органов, фиброз, связанный с ишемически-реперфузионным повреждением, фиброз после инъекции, цирроз, диффузное паренхиматозное заболевание легких, болевой синдром после вазэктомии и связанные с ревматоидным артритом заболевания или расстройства. В других вариантах реализации соединения, раскрытые в настоящем описании, можно применять в исследованиях обмена веществ и кинетики реакций, методиках обнаружения и визуализации и при лечении радиоактивным воздействием.
[0013] В некоторых вариантах реализации соединения, раскрытые в настоящем описании, применяют для лечения заболеваний или состояний, вызывающих у субъекта симптомы, которые включают, но не ограничиваются перечисленными: фиброз печени, фиброз почки, фиброз легкого, пневмонит гиперчувствительности, интерстициальный фиброз, системную склеродермию, макулярную дегенерацию, фиброз поджелудочной железы, фиброз селезенки, фиброз сердца, медиастинальный фиброз, миелофиброз, эндомиокардиальный фиброз, ретроперитонеальный фиброз, прогрессирующий массивный фиброз, нефрогенный системный фиброз, фиброзные послеоперационные осложнения, хроническую васкулопатию аллотрансплантата и/или хроническое отторжение трансплантированных органов, фиброз, связанный с ишемически-реперфузионным повреждением, фиброз после инъекции, цирроз, диффузное паренхиматозное заболевание легких, болевой синдром после вазэктомии и связанные с ревматоидным артритом заболевания.
[0014] В некоторых вариантах реализации предложены способы облегчения или улучшения состояния или расстройства, вызванного, по меньшей мере частично, ферментативной активностью кальпаина 1 (CAPN1), кальпаина 2 (CAPN2) и/или кальпаина 9 (CAPN9), или опосредованного, по меньшей мере частично, ферментативной активностью CAPN1, CAPN2 и/или CAPN9, где указанное состояние включает или вызывает симптом, который включает: фиброз печени, фиброз почки, фиброз легкого, пневмонит гиперчувствительности, интерстициальный фиброз, системную склеродермию, макулярную дегенерацию, фиброз поджелудочной железы, фиброз селезенки, фиброз сердца, медиастинальный фиброз, миелофиброз, эндомиокардиальный фиброз, ретроперитонеальный фиброз, прогрессирующий массивный фиброз, нефрогенный системный фиброз, фиброзные послеоперационные осложнения, хроническую васкулопатию аллотрансплантата и/или хроническое отторжение трансплантированных органов, фиброз, связанный с ишемически-реперфузионным повреждением, фиброз после инъекции, цирроз, диффузное паренхиматозное заболевание легких, болевой синдром после вазэктомии и/или ревматоидный артрит.
[0015] В некоторых вариантах реализации способы, соединения и/или композиции согласно настоящему изобретению применяют для профилактической терапии.
[0016] В некоторых вариантах реализации соединения, ингибирующие CAPN1, CAPN2 и/или CAPN9, проявляют эффективность в моделях заболевания человека у животных. В частности, лечение in vivo мышей, кроликов и других субъектов-млекопитающих с применением соединений, раскрытых в настоящем описании, демонстрирует пригодность этих соединений для применения в качестве терапевтических агентов для модулирования активности CAPN1, CAPN2 и/или CAPN9 у людей и, таким образом, улучшения соответствующих клинических состояний.
[0017] В некоторых вариантах реализации предложены соединения, фармацевтические композиции и способы их применения для подавления дифференцировки миофибробластов. В некоторых вариантах реализации предложены соединения, фармацевтические композиции и способы их применения для ингибирования CAPN1, CAPN2 и/или CAPN9 или комбинации этих ферментативных активностей, в частности CAPN1 и CAPN2, или CAPN1 и CAPN9, или CAPN2 и CAPN9. В некоторых вариантах реализации предложены способы лечения заболеваний и расстройств путем ингибирования CAPN1, CAPN2 и/или CAPN9 или комбинации этих ферментативных активностей.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
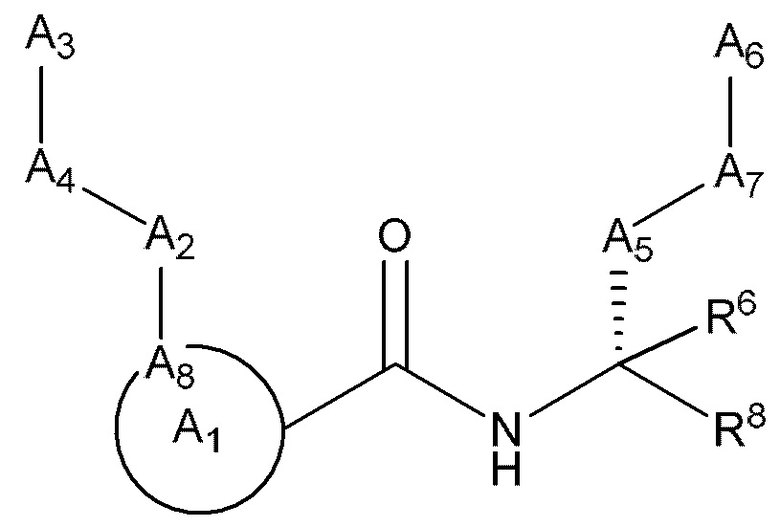
[0018] В некоторых вариантах реализации предложены соединения, являющиеся немакроциклическими α-кетоамидами, которые действуют в качестве модуляторов кальпаина. Различные варианты реализации этих соединений включают соединения, имеющие структуры формулы I, описанные выше, или их фармацевтически приемлемые соли. Структура формулы I охватывает все стереоизомеры и рацемические смеси, включая следующие структуры и их смеси:
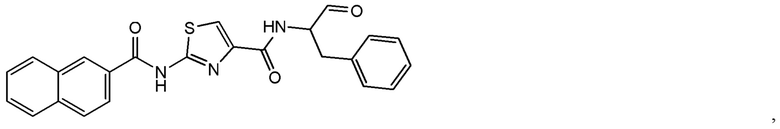
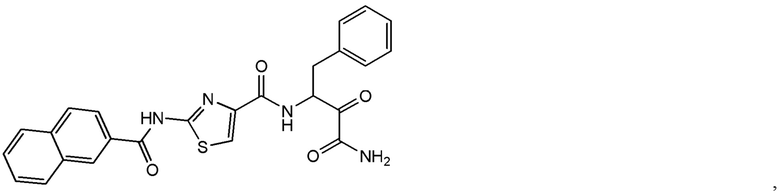
[0019] В некоторых вариантах реализации соединений формулы (I) соединение не является выбранным из группы, состоящей из:
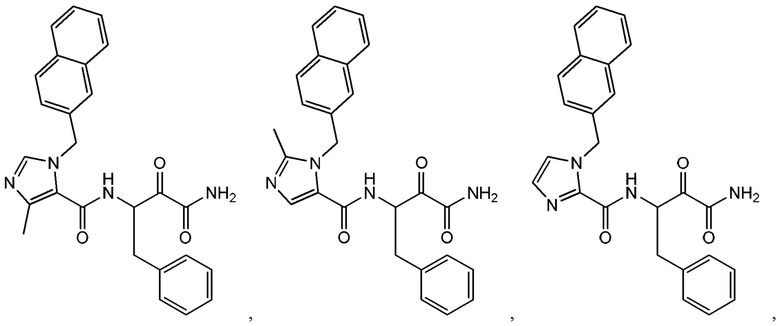
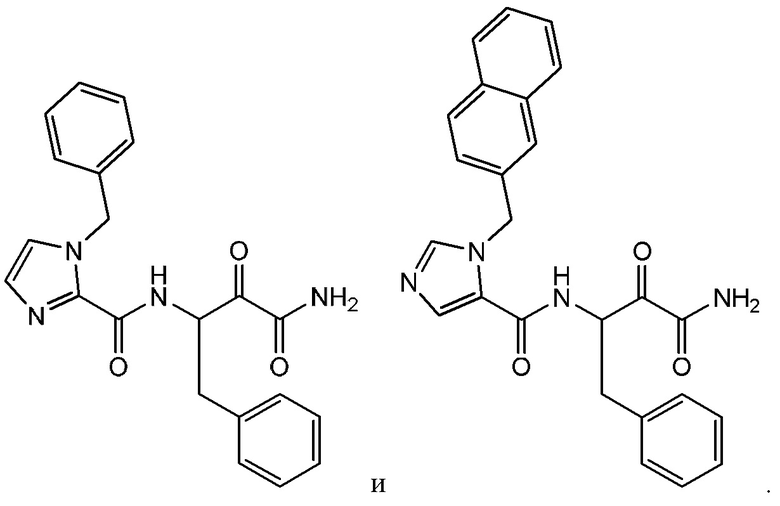
[0020] В некоторых вариантах реализации соединений формулы (I):
A1 выбран из группы, состоящей из необязательно замещенного 6-10-членного гетероциклила, при условии, что указанный 6-10-членный гетероциклил не замещен оксо; необязательно замещенного 5-, 8- или 9-членного гетероарила и необязательно замещенного С3-10 карбоциклила;
А2 выбран из группы, состоящей из необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного С3-10 карбоциклила, -CR2-, -S-, -S(=O)-, -SO2-, -О-, -C(=S)-, -С(=O)-, -NR-, -СН=СН-, -OC(O)NH-, -NHC(O)NH-, -NHC(O)O, -NHC(O)-, -NHC(S)NH-, -NHC(S)O-, -NHC(S)- и одинарной связи;
А4 выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С3-10 карбоциклила, необязательно замещенного C1-4 алкила, -S-, S(=O)-, -SO2-, -О-, -C(=S)-, -С(=O)-, -NR-, -СН=СН-, -OC(O)NH-, -NHC(O)NH-, -NHC(O)O-, -NHC(O)-, -NHC(S)NH-, -NHC(S)O-, -NHC(S)- и одинарной связи;
А3 выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила и необязательно замещенного С3-10 карбоциклила;
А6 выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С3-10 карбоциклила, необязательно замещенного C1-8 алкила, необязательно замещенного -O-C1-6 алкила, необязательно замещенного -О-С2-6 алкенила и боковой цепи любой природной или неприродной аминокислоты; и
каждый R, R2 и R3 независимо выбран из -Н, необязательно замещенного С1-4 алкила, необязательно замещенного С3-7 карбоциклила, необязательно замещенного 5-10-членного гетероциклила, необязательно замещенного С6-10 арила и необязательно замещенного 5-10-членного гетероарила.
[0021] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-а):
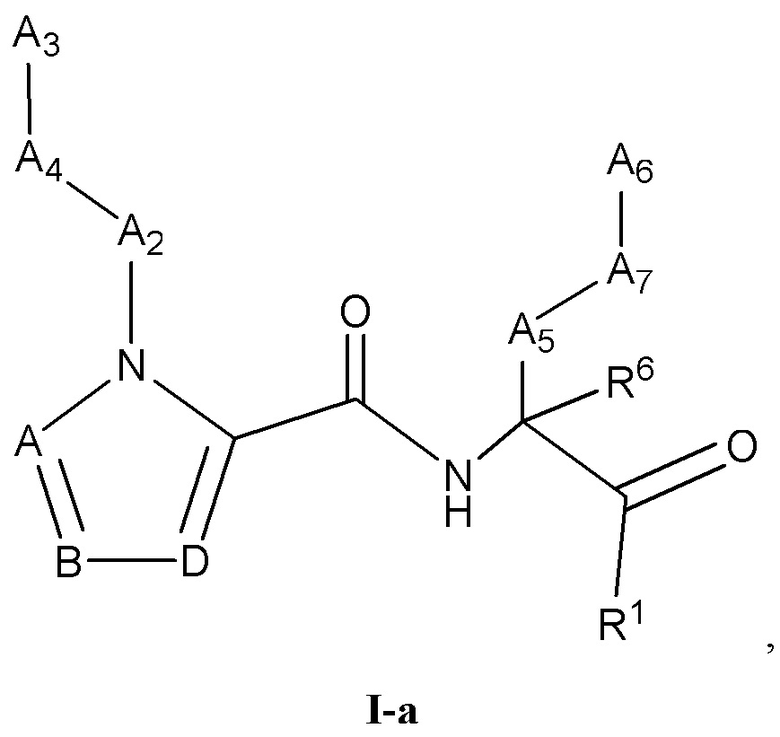
или их фармацевтически приемлемую соль, где:
каждый из А, В и D независимо выбран из группы, состоящей из C(R4) и N; и каждый R4 независимо выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
[0022] В некоторых вариантах реализации соединений формулы (I-а) или их фармацевтически приемлемых солей А, В и D независимо выбраны из группы, состоящей из СН и N. В некоторых вариантах реализации А представляет собой N, В представляет собой СН, и D представляет собой СН. В некоторых вариантах реализации А представляет собой СН, В представляет собой N, и D представляет собой СН.
[0023] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-b):
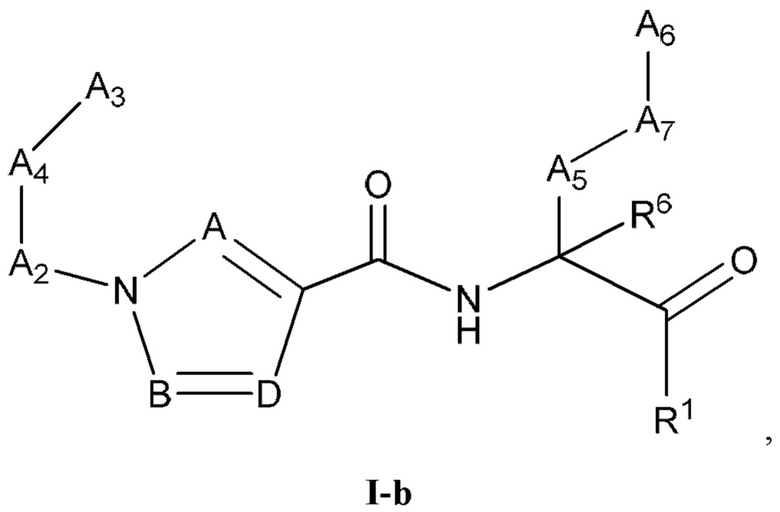
или их фармацевтически приемлемую соль, где:
каждый из А, В и D независимо выбран из группы, состоящей из C(R4) и N; и каждый R4 независимо выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
[0024] В некоторых вариантах реализации соединений формулы (I-b) или их фармацевтически приемлемых солей А, В и D независимо выбраны из группы, состоящей из СН и N.
[0025] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-c):
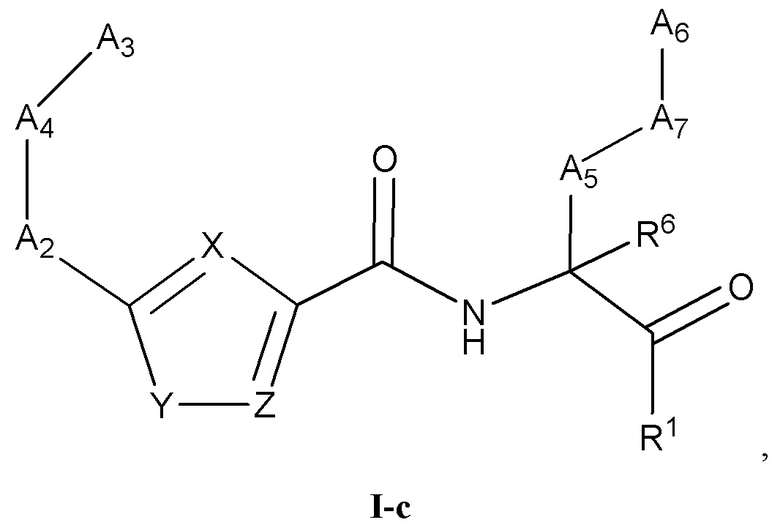
или их фармацевтически приемлемую соль, где:
Y выбран из группы, состоящей из NR5, О, S и SO2; каждый из X и Z независимо выбран из группы, состоящей из C(R4) и N; каждый R4 независимо выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси; и R5 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила и С3-7 карбоциклила.
[0026] В некоторых вариантах реализации соединений формулы (I-c) или их фармацевтически приемлемых солей X и Z независимо выбраны из группы, состоящей из СН и N.
[0027] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-d):
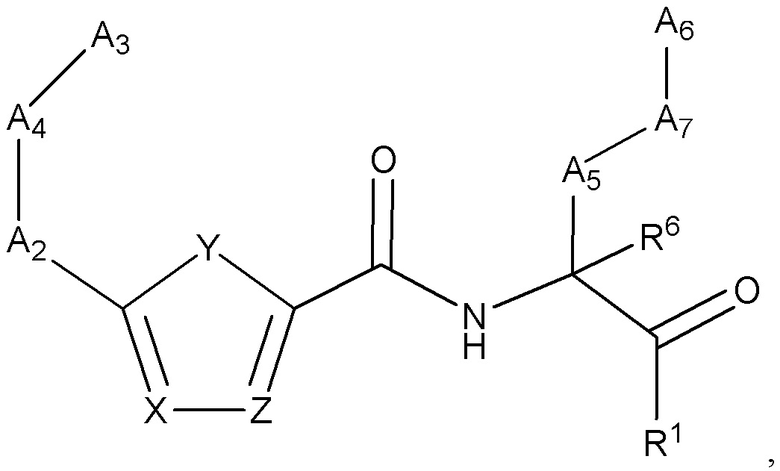
или их фармацевтически приемлемую соль, где:
Y выбран из группы, состоящей из NR5, О, S и SO2; каждый из X и Z независимо выбран из группы, состоящей из C(R4) и N; каждый R4 независимо выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси; и R5 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила и С3-7 карбоциклила.
[0028] В некоторых вариантах реализации соединений формулы (I-d) или их фармацевтически приемлемых солей X и Z независимо выбраны из группы, состоящей из СН и N.
[0029] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-e):
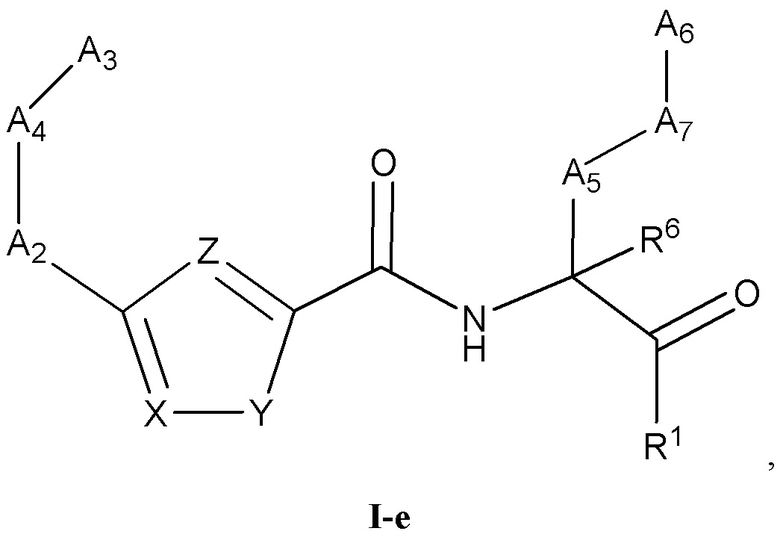
или их фармацевтически приемлемую соль, где:
Y выбран из группы, состоящей из NR5, О, S и SO2; каждый из X и Z независимо выбран из группы, состоящей из C(R4) и N; каждый R4 независимо выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси; и R5 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила и С3-7 карбоциклила.
[0030] В некоторых вариантах реализации соединений формулы (I-е) или их фармацевтически приемлемых солей X и Z независимо выбраны из группы, состоящей из СН и N.
[0031] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-f):
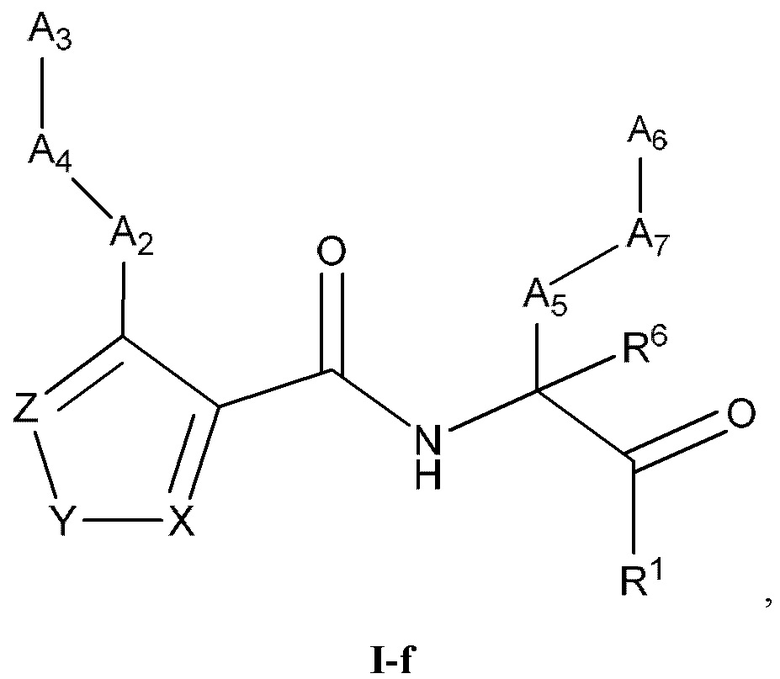
или их фармацевтически приемлемую соль, где:
Y выбран из группы, состоящей из NR5, О, S и SO2; каждый из X и Z независимо выбран из группы, состоящей из C(R4) и N; каждый R4 независимо выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси; и R5 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила и С3-7 карбоциклила.
[0032] В некоторых вариантах реализации соединений формулы (I-f) Z представляет собой N, Y представляет собой NR5, и X представляет собой СН.
[0033] В некоторых вариантах реализации соединений формулы (I-f) R5 выбран из группы, состоящей из -Н, C1-4 алкила, C1-C4 галогеналкила и циклопропила.
[0034] В некоторых вариантах реализации соединений формулы (I-f) Z представляет собой N, Y представляет собой О, и X представляет собой C(R4). В некоторых вариантах реализации соединений формулы (I-f) Z представляет собой N, Y представляет собой S, и X представляет собой C(R4). В некоторых вариантах реализации соединений формулы (I-f) Z представляет собой C(R4), Y представляет собой S, и X представляет собой C(R4).
[0035] В некоторых вариантах реализации соединений формулы (I-f) Z представляет собой C(R4), Y представляет собой О, и X представляет собой C(R4).
[0036] В некоторых вариантах реализации соединений формулы (I-f) Z представляет собой N, Y представляет собой S, и X представляет собой N. В некоторых вариантах реализации соединений формулы (I-f) Z представляет собой N, Y представляет собой О, и X представляет собой N.
[0037] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-g):
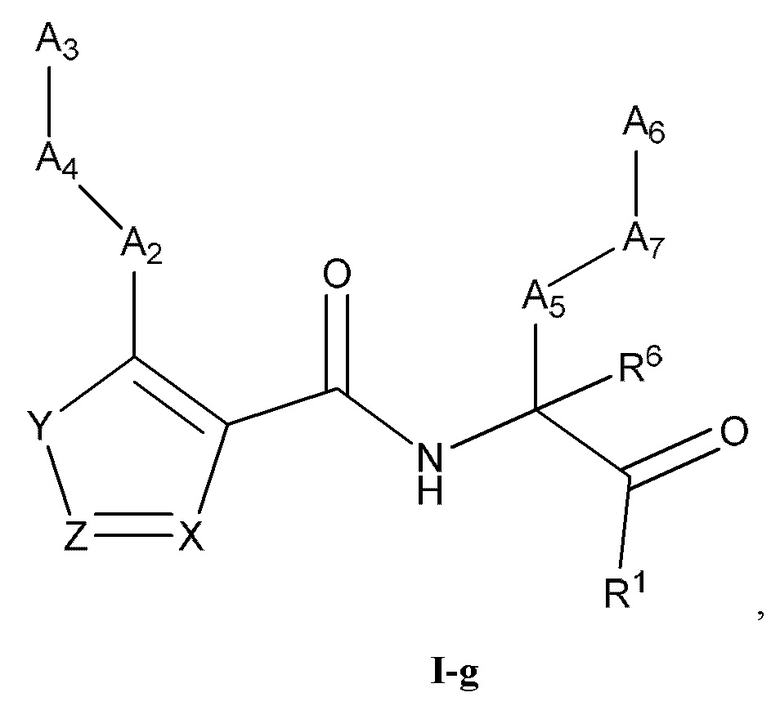
или их фармацевтически приемлемую соль, где:
Y выбран из группы, состоящей из NR5, О, S и SO2; каждый из X и Z независимо выбран из группы, состоящей из C(R4) и N; каждый R4 независимо выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси; и R5 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила и С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси).
[0038] В некоторых вариантах реализации соединений формулы (I-g) или их фармацевтически приемлемых солей X и Z независимо выбраны из группы, состоящей из СН и N. В некоторых вариантах реализации соединений формулы (I-g) Y представляет собой NR5, Z представляет собой N, и X представляет собой СН.
[0039] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-h):
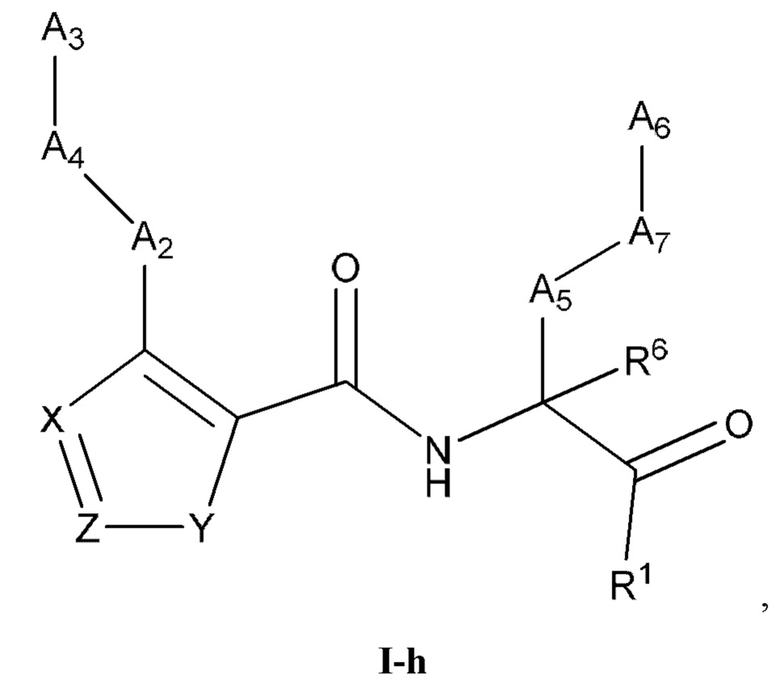
или их фармацевтически приемлемую соль, где:
Y выбран из группы, состоящей из NR5, О, S и SO2; каждый из X и Z независимо выбран из группы, состоящей из C(R4) и N; каждый R4 независимо выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси; и R5 выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила и С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси).
[0040] В некоторых вариантах реализации соединений формулы (I-h) или их фармацевтически приемлемых солей X и Z независимо выбраны из группы, состоящей из СН и N. В некоторых вариантах реализации соединений формулы (I-h) X представляет собой СН, Z представляет собой N, и Y представляет собой NR5.
[0041] В некоторых вариантах реализации соединений формулы (I-h) X представляет собой СН, Z представляет собой N, и Y представляет собой NR5. В некоторых вариантах реализации соединений формулы (I-h) X представляет собой N, Z представляет собой C(R4), и Y представляет собой О.
[0042] В некоторых вариантах реализации соединений формулы (I-h) R4 выбран из -Н и C1-4 алкила.
[0043] В некоторых вариантах реализации соединений формулы (I-h) X представляет собой N, Z представляет собой C(R4), и Y представляет собой S. В некоторых вариантах реализации соединений формулы (I-h) X представляет собой N, Z представляет собой N, и Y представляет собой S.
[0044] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-j):
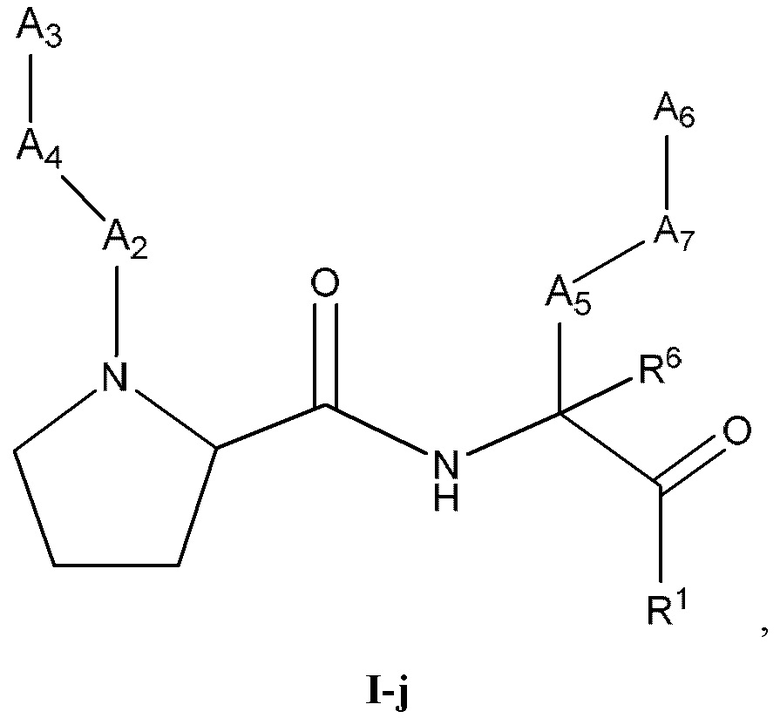
или их фармацевтически приемлемую соль.
[0045] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-k):
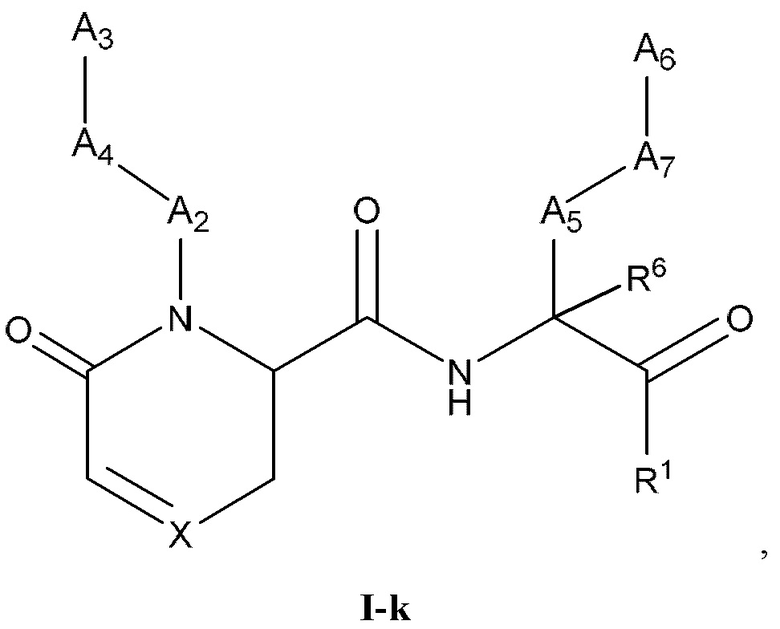
или их фармацевтически приемлемую соль, где:
X выбран из группы, состоящей из C(OR5), -C(R4) и N; R4 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси; и R5 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила и С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси).
[0046] В некоторых вариантах реализации соединений формулы (I-k) или их фармацевтически приемлемых солей X и Z независимо выбраны из группы, состоящей из СН и N.
[0047] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-m):

или их фармацевтически приемлемую соль, где:
X и Z независимо выбраны из группы, состоящей из C(R4) и N; Е выбран из группы, состоящей из необязательно замещенного C5-6 карбоциклила и необязательно замещенного 5-6-членного гетероциклила; и каждый R4 независимо выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
[0048] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-n):
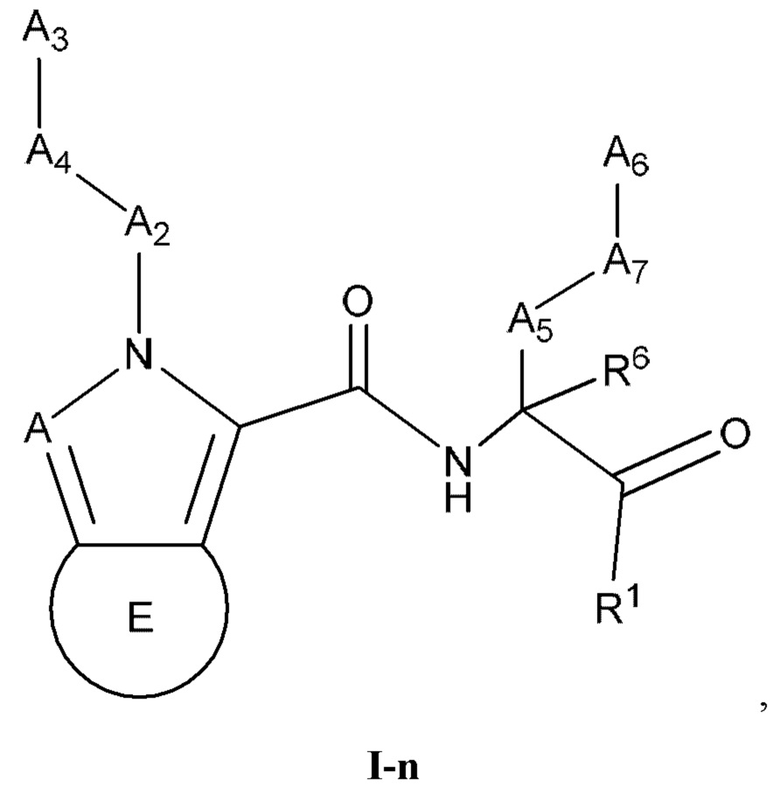
или их фармацевтически приемлемую соль, где:
А выбран из группы, состоящей из C(R4) и N; Е выбран из группы, состоящей из необязательно замещенного C5-6 карбоциклила, необязательно замещенного 5-6-членного гетероциклила, необязательно замещенного 5-6-членного гетероарила и необязательно замещенного фенила; и каждый R4 независимо выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
[0049] Некоторые варианты реализации включают соединения формулы (III)
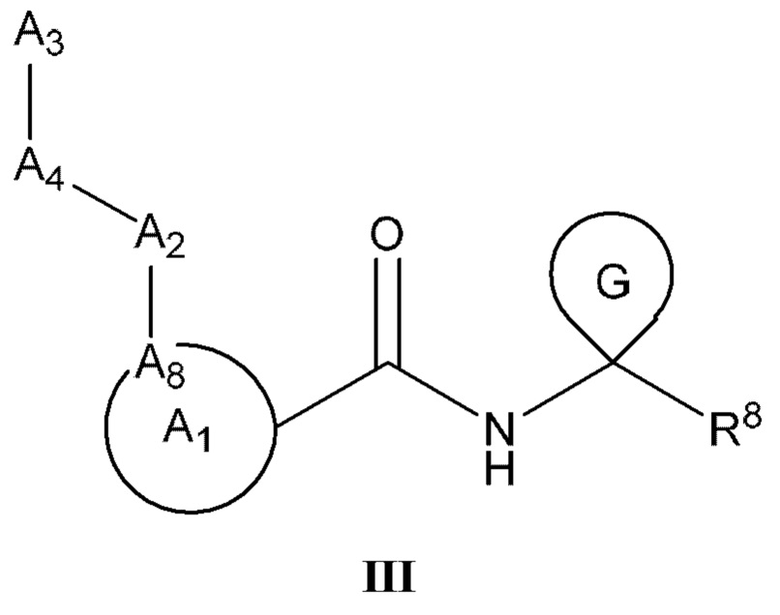
или их фармацевтически приемлемую соль, где:
A1 выбран из группы, состоящей из необязательно замещенного 5-10-членного гетероциклила, при условии, что 6-10-членный гетероциклил не замещен оксо; необязательно замещенного 5-, 8- или 9-членного гетероарила и необязательно замещенного С3-10 карбоциклила;
А2 выбран из группы, состоящей из необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного С3-10 карбоциклила, -CR2-, -S-, -S(=O)-, -SO2-, -О-, -C(=S)-, -С(=O)-, -NR-, -СН=СН-, -С≡С-, -OC(O)NH-, -NHC(O)NH-, -NHC(O)O-, -NHC(O)-, -NHC(S)NH-, -NHC(S)O-, -NHC(S)- и одинарной связи;
A4 выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С3-10 карбоциклила, необязательно замещенного C1-4 алкила, -(CR2)n-S-(CR2)n-, -(CR2)n-S(=O)-(CR2)n-, -(CR2)n-SO2-(CR2)n-, -(CR2)n-O-(CR2)n-, -(CR2)n-C(=S)-(CR2)n-, -(CR2)n-C(=O)-(CR2)n-, -(CR2)n-NR-(CR2)n-, -(CR2)n-CH=CH-(CR2)n-, -(CR2)n-OC(O)NH-(CR2)n-, -(CR2)n-NHC(O)NH-(CR2)n-, -(CR2)n-NHC(O)O-(CR2)n-, -(CR2)n-NHC(O)-(CR2)n-, -(CR2)n-NHC(S)NH-(CR2)n-, -(CR2)n-NHC(S)O-(CR2)n-, -(CR2)n-NHC(S)-(CR2)n- и одинарной связи;
в случае, если А2 и А4 представляют собой одинарную связь, А3 непосредственно присоединен к А8;
А3 выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила и необязательно замещенного С3-10 карбоциклила, или, если А2 выбран из необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила и необязательно замещенного С3-10 карбоциклила, то А3 выбран из группы, состоящей из атома водорода, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С3-10 карбоциклила, -С≡СН и необязательно замещенного 2-5-членного полиэтиленгликоля;
G представляет собой необязательно замещенный С3-С7 карбоциклил или необязательно замещенный 4-7-членный гетероциклил;
A8 является членом кольца A1 и выбран из группы, состоящей из С и N;
R8 выбран из группы, состоящей из -COR1, -CN, -CH=CHSO2R, -CH2NO2;
R1 выбран из группы, состоящей из Н, -ОН, С1-4 галогеналкила, -СООН, -CH2NO2, -C(=O)NOR, -NH2, -CONR2R3, -СН(СН3)=СН2, -CH(CF3)NR2R3,
-C(F)=CHCH2CH3, 
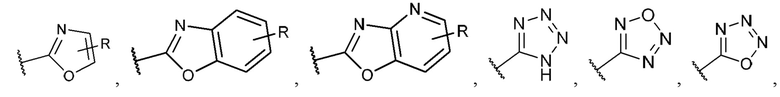
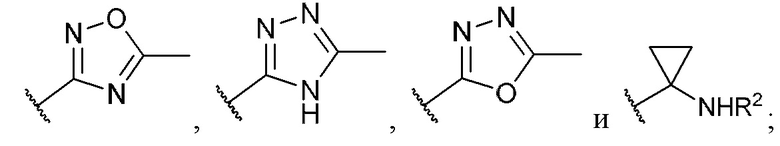
R14 представляет собой галоген; и
каждый R, R2 и R3 независимо выбран из -Н, необязательно замещенного С1-4 алкила, необязательно замещенного C1-8 алкоксиалкила, необязательно замещенного 2-5-членного полиэтиленгликоля, необязательно замещенного С3-7 карбоциклила, необязательно замещенного 5-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного С6-10 арил(С1-С6)алкила и необязательно замещенного 5-10-членного гетероарила; R6 независимо выбран из -Н и необязательно замещенного С1-4 алкила; и каждый п независимо выбран из целых чисел от 0 до 3.
[0050] Некоторые варианты реализации соединений формул (III) включают соединения, имеющие структуру формулы (III-а):
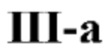
или их фармацевтически приемлемую соль.
[0051] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) по меньшей мере один из необязательно замещенных фрагментов А2, А4 и А3 замещен 18F.
[0052] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) по меньшей мере один из необязательно замещенных фрагментов А2, А4 и А3 замещен C1-С6 алкилом, содержащим один или более 11С.
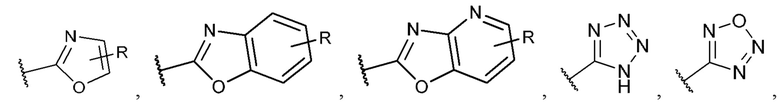
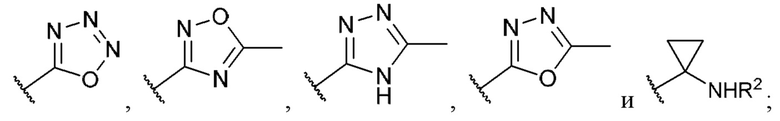
[0053] В некоторых вариантах реализации соединений формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) или их фармацевтически приемлемых солей А3 выбран из группы, состоящей из 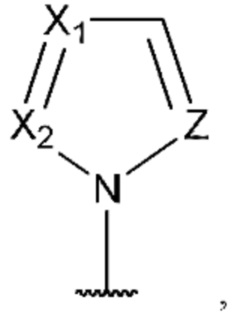
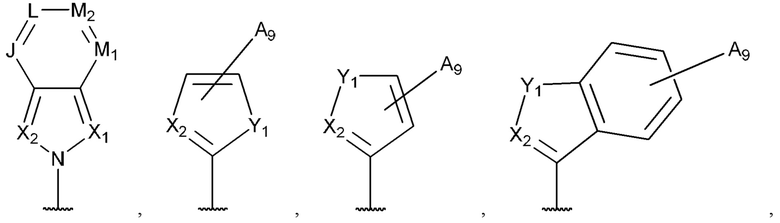
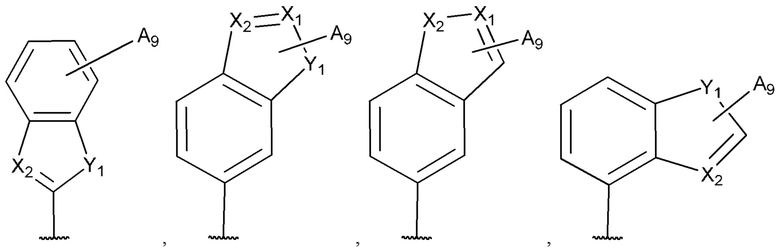
и 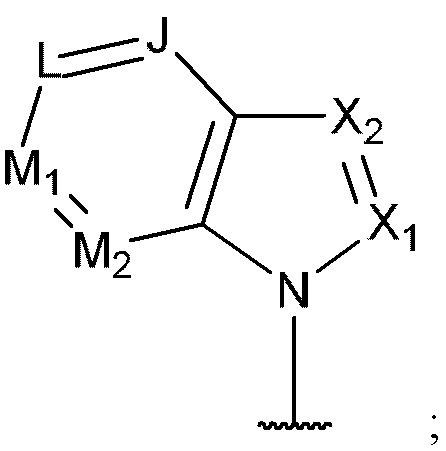 и А9 выбран из группы, состоящей из Н, С6-10 арила, 5-10-членного гетероарила, 3-10-членного гетероциклила и С3-10 карбоциклила, C1-4 алкила; каждый из Х2, X1 и Z независимо выбран из группы, состоящей из C(R4) и N; Y1 выбран из группы, состоящей из NR5, О и S; каждый из J, L, M1 и М2 независимо выбран из группы, состоящей из C(R4) и N; R4 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила, галогена, гидрокси и C1-С6 алкокси; R5 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила и С3-7 карбоциклила.
и А9 выбран из группы, состоящей из Н, С6-10 арила, 5-10-членного гетероарила, 3-10-членного гетероциклила и С3-10 карбоциклила, C1-4 алкила; каждый из Х2, X1 и Z независимо выбран из группы, состоящей из C(R4) и N; Y1 выбран из группы, состоящей из NR5, О и S; каждый из J, L, M1 и М2 независимо выбран из группы, состоящей из C(R4) и N; R4 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила, С3-7 карбоциклила, галогена, гидрокси и C1-С6 алкокси; R5 выбран из группы, состоящей из -Н, C1-4 алкила, C1-4 галогеналкила и С3-7 карбоциклила.
[0054] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A2 представляет собой -CH2-.
[0055] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A2 представляет собой -СН=СН-.
[0056] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A2 представляет собой -О-.
[0057] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A2 представляет собой S.
[0058] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A2 представляет собой одинарную связь.
[0059] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A2 представляет собой фенил.
[0060] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) А3 представляет собой необязательно замещенный С6-10 арил.
[0061] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A2 выбран из группы, состоящей из необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного 5- или 7-10-членного гетероарила, необязательно замещенного С3-10 карбоциклила, -S-, -S(=O)-, -SO2-, -C(=S)-, -С(=O)-, -NR-, -СН=СН-, -С≡С-, -OC(O)NH-, -NHC(O)NH-, -NHC(O)O-, -NHC(S)NH-, -NHC(S)O- и -NHC(S)-.
[0062] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A2 выбран из группы, состоящей из необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного С3-10 карбоциклила и -С≡С-.
[0063] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A2 выбран из группы, состоящей из необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила и необязательно замещенного С3-10 карбоциклила.
[0064] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A4 представляет собой одинарную связь.
[0065] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A3 выбран из группы, состоящей из фенила, 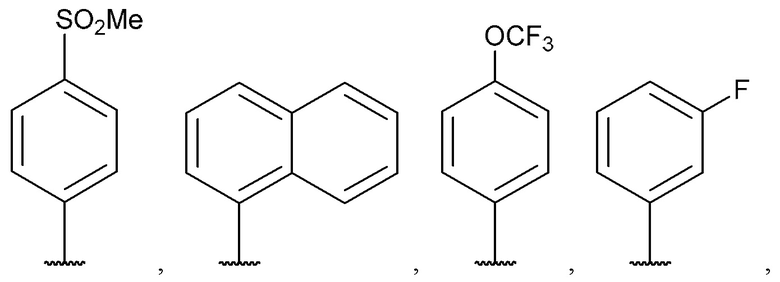

[0066] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) А3 представляет собой необязательно замещенный 5-10-членный гетероарил.
[0067] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p) A3 выбран из группы, состоящей из 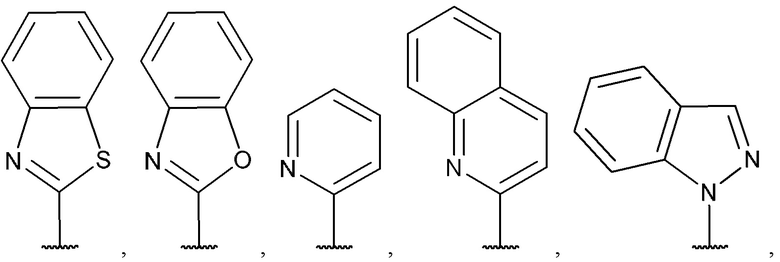
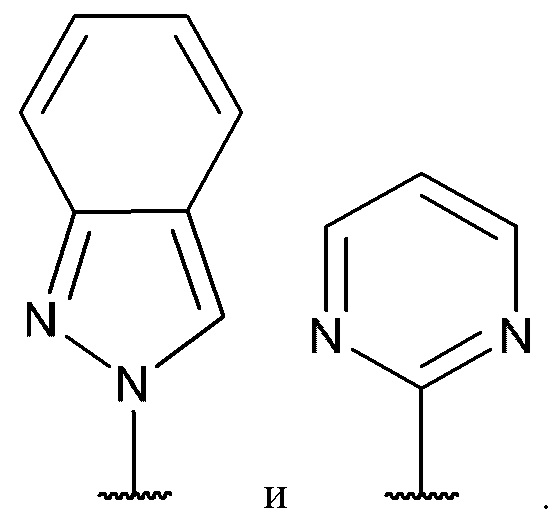
[0068] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p), где A2 представляет собой одинарную связь, А4 представляет собой одинарную связь, и А3 представляет собой необязательно замещенный С6-10 арил или необязательно замещенный 5-10-членный гетероарил.
[0069] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p), где A3 имеет структуру:
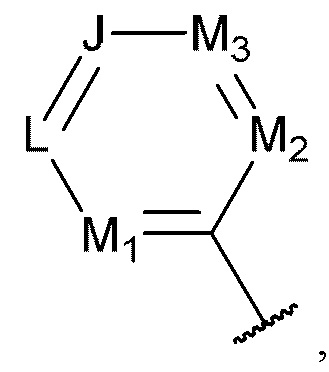 где каждый из J, L, M1, M2 и М3 независимо выбран из группы, состоящей из C(R4) и N; и каждый R4 независимо выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
где каждый из J, L, M1, M2 и М3 независимо выбран из группы, состоящей из C(R4) и N; и каждый R4 независимо выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
[0070] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p), где каждый из J, L, M1, M2 и М3 представляет собой C(R4).
[0071] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p), где каждый R4 независимо выбран из -Н и галогена.
[0072] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p), где M1 представляет собой галоген, и каждый из J, L, М2 и М3 представляет собой СН.
[0073] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p), где L представляет собой галоген, и каждый из J, M1, М2 и М3 представляет собой СН.
[0074] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p), где A3 имеет структуру, выбранную из группы, состоящей из:
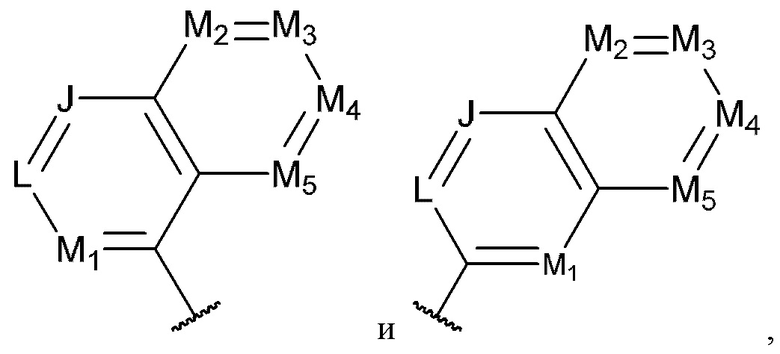 где каждый из J, L, M1, М2, М3, М4 и M5 независимо выбран из группы, состоящей из C(R4) и N; и каждый R4 независимо выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
где каждый из J, L, M1, М2, М3, М4 и M5 независимо выбран из группы, состоящей из C(R4) и N; и каждый R4 независимо выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
[0075] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n) или (I-p), где A3 имеет структуру:
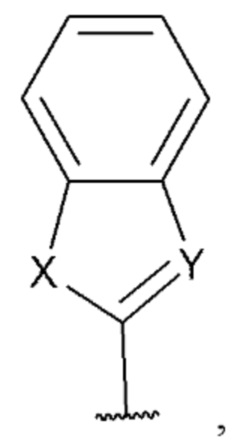 где X выбран из группы, состоящей из C(R4) и N; Y выбран из О и S; и R4 выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
где X выбран из группы, состоящей из C(R4) и N; Y выбран из О и S; и R4 выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила, С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, гидрокси и C1-С6 алкокси.
[0076] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-о):
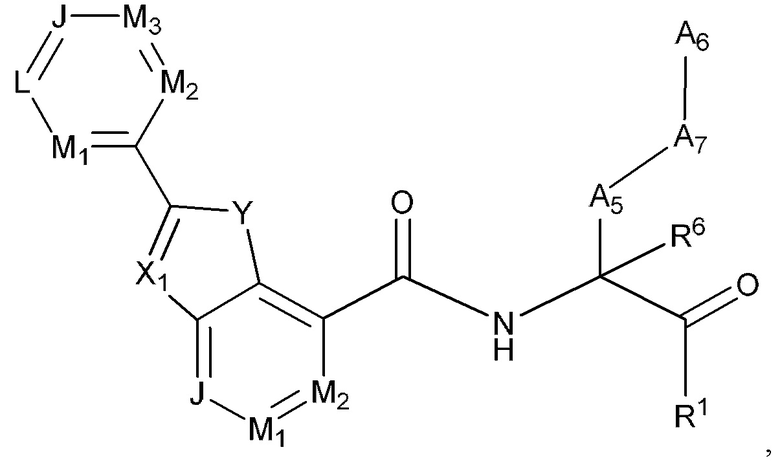
или их фармацевтически приемлемую соль, где:
Y выбран из группы, состоящей из NR5, О, S и SO2; X1 выбран из группы, состоящей из C(R4) и N; каждый из J, L, M1, М2 и М3 независимо выбран из группы, состоящей из C(R4) и N; R4 выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила, С3-7 карбоциклила, галогена, гидрокси и C1-С6 алкокси; R5 выбран из группы, состоящей из -Н, С1-4 алкила, С1-4 галогеналкила и С3-7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси).
[0077] В некоторых вариантах реализации соединений формулы (I-о) или их фармацевтически приемлемых солей J, L, M1, М2 и М3 независимо выбраны из группы, состоящей из СН и N.
[0078] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p), где по меньшей мере один из необязательно замещенных фрагментов А5, А7 и Аб замещен 18F.
[0079] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p), где по меньшей мере один из необязательно замещенных фрагментов А5, А7 и Аб замещен Ci-Сб алкилом, содержащим один или более 11С.
[0080] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) А6 представляет собой фенил.
[0081] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) Аб выбран из группы, состоящей из необязательно замещенного С6-10 арила, необязательно замещенного 5-10-членного гетероарила, необязательно замещенного 3-10-членного гетероциклила, необязательно замещенного С3-10 карбоциклила, необязательно замещенного C1-8 алкила, необязательно замещенного -O-C1-6 алкила и необязательно замещенного -О-С2-6 алкенила.
[0082] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) A7 представляет собой -CH2-.
[0083] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) А7 представляет собой -CH=CH-.
[0084] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) A7 представляет собой -О-.
[0085] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) A7 представляет собой S.
[0086] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) A7 представляет собой одинарную связь.
[0087] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) A7 представляет собой необязательно замещенный С6-10 арил.
[0088] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) A7 представляет собой фенил.
[0089] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) A5 представляет собой -CH2-.
[0090] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p), где As представляет собой -CH2-или -СН2СН2-; А7 представляет собой одинарную связь; и A6 выбран из группы, состоящей из С1-С4 алкила, необязательно замещенного фенила, необязательно замещенного 5-10-членного гетероарила.
[0091] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) А6 представляет собой необязательно замещенный фенил.
[0092] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p), где А6 представляет собой незамещенный фенил.
[0093] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p), где А6 представляет собой фенил, необязательно замещенный одним или более С1-4 алкилом, С3-7 карбоциклилом, галогеном, гидрокси и C1-С6 алкокси.
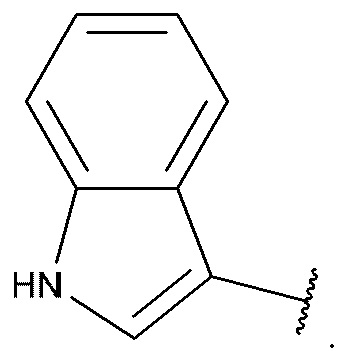
[0094] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) A6 имеет структуру:
[0095] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p), где A5 представляет собой одинарную связь, А7 представляет собой одинарную связь; и А6 представляет собой С1-С5 алкил.
[0096] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) А6 выбран из группы, состоящей из этила, н-пропила, изопропила, изобутила, 2,2-диметилпропила и 1,2-диметилпропила.
[0097] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-o) или (I-p) R1 представляет собой CONR2R3.
[0098] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-o) или (I-p) R2 представляет собой -H, и R3 представляет собой необязательно замещенный C1-4 алкил.
[0099] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-o) или (I-p), где R2 представляет собой -H, и R3 выбран из группы, состоящей из -Н, С1-С4 алкила, необязательно замещенного посредством С-амидо, и С3-С6 циклоалкила.
[0100] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-o) или (I-p) R3 выбран из этила или циклопропила.
[0101] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-o) или (I-p) R3 представляет собой метил, замещенный посредством С-амидо.
[0102] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-o) или (I-p) R3 представляет собой H.
[0103] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-o) или (I-p) R3 представляет собой необязательно замещенный C1-4 алкил.
[0104] В некоторых вариантах реализации формул (I), (III), (III-а), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-o) или (I-p) R3 представляет собой бензил.
[0105] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) R6 представляет собой -H и необязательно замещенный C1-4 алкил.
[0106] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) R6 представляет собой необязательно замещенный C1-4 алкил.
[0107] В некоторых вариантах реализации формул (I), (I-a), (I-b), (I-c), (I-d), (I-е), (I-f), (I-g), (I-h), (I-j), (I-k), (I-m), (I-n), (I-o) или (I-p) R6 представляет собой метил.
[0108] В некоторых вариантах реализации формулы (I) A1 выбран из группы, состоящей из необязательно замещенного 6-10-членного гетероциклила; 5-членного гетероциклила, необязательно замещенного одним или более C1-4 алкилом, С3-7 карбоциклилом, галогеном, гидрокси или C1-С6 алкокси; необязательно замещенного 5-, 8-или 9-членного гетероарила и необязательно замещенного С3-10 карбоциклила.
[0109] В некоторых вариантах реализации формулы (I) A1 выбран из группы, состоящей из 5-членного гетероциклила, необязательно замещенного одним или более C1-4 алкилом, С3-7 карбоциклилом, галогеном, гидрокси или C1-С6 алкокси, и необязательно замещенного 5-членного гетероарила.
[0110] В некоторых вариантах реализации формулы (I) A1 представляет собой необязательно замещенный 5-членный гетероарил.
[0111] Некоторые варианты реализации соединений формулы (I) включают соединения, имеющие структуру формулы (I-р):
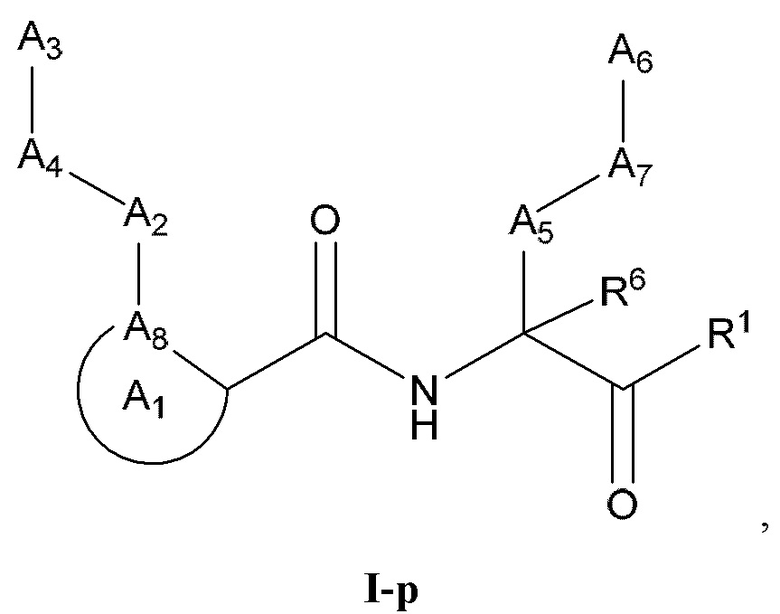
или их фармацевтически приемлемую соль.
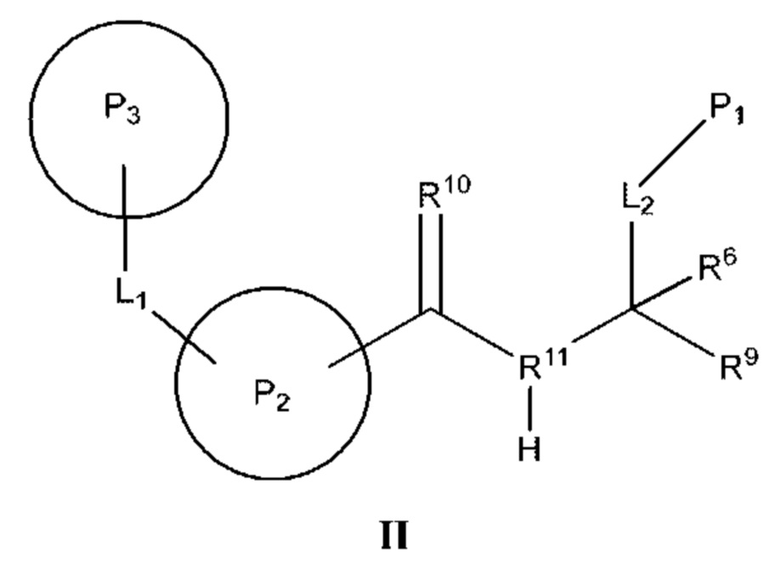
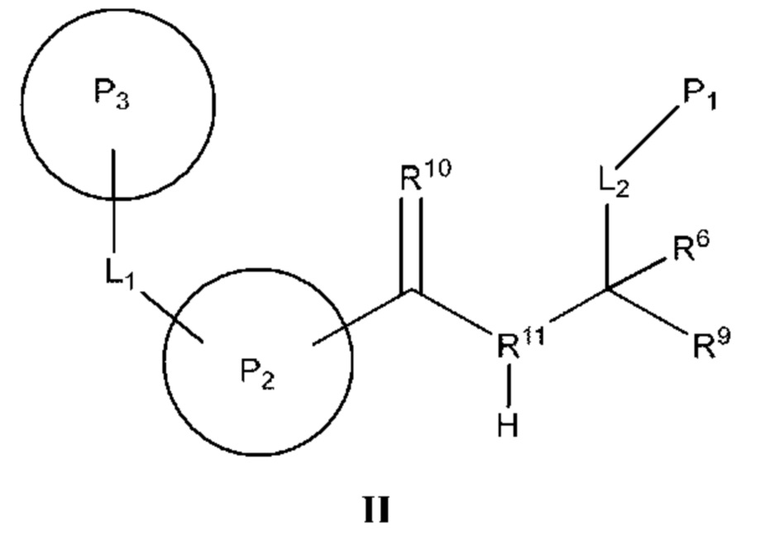
[0112] В некоторых вариантах реализации предложено соединение формулы (II)
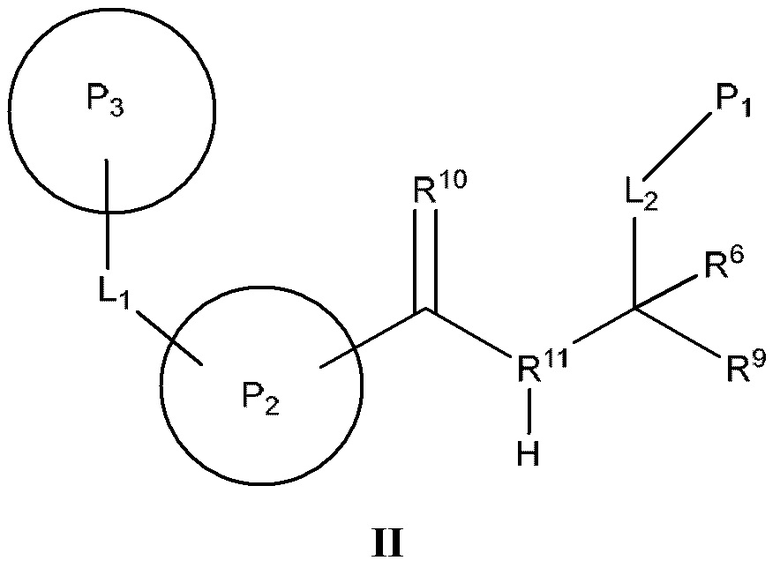
или его фармацевтически приемлемая соль, где:
[0113] Р2 представляет собой необязательно замещенный циклический фрагмент, имеющий такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р2 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р2 кальпаина 9, выбранным из группы, состоящей из Gly190, Phe233, Gly253, His254 и Ala255, и находится в пределах 5  или менее от него;
или менее от него;
[0114] L1 представляет собой связь или фрагмент, состоящий из 1-25 атомов, выбранных из группы, состоящей из атома углерода, атома кислорода, атома азота, атома водорода и атома серы;
[0115] Р3 представляет собой необязательно замещенный циклический фрагмент, расположенный вблизи L1 и имеющий такие размер и конфигурацию, что при связывании соединения с кальпаином 9 по меньшей мере один атом Р3 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р3 кальпаина 9, выбранным из группы, состоящей из Gly189, Gly190, Ser191, Thr236 и Gly253, и находится в пределах 5  или менее от него;
или менее от него;
[0116] R10 представляет собой оксо и расположен вблизи Р2 таким образом, что при связывании соединения с кальпаином 9 R10 образует полярное взаимодействие с амидом Gly190 кальпаина 9 и находится в пределах 4 или менее от него;
[0117] R11 представляет собой атом азота и расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании соединения с кальпаином 9 R11 образует полярное взаимодействие с карбонилом Gly253 кальпаина 9 и находится в пределах 4 или менее от него;
[0118] L2 представляет собой связь или фрагмент, состоящий из 1-25 атомов, выбранных из группы, состоящей из атома углерода, атома кислорода, атома азота, атома водорода и атома серы;
[0119] P1 представляет собой фрагмент, расположенный вблизи L2 и имеющий такие размер и конфигурацию, что при связывании соединения с кальпаином 9 по меньшей мере один атом P1 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р1 кальпаина 9, выбранным из группы, состоящей из Gly95, Lys188, Gly189 и Ser242, и находится в пределах 5 или менее от него;
[0120] R9 представляет собой фрагмент, расположенный вблизи атома углерода, к которому он присоединен, таким образом, что при связывании соединения с кальпаином 9 по меньшей мере один атом R9 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 9, выбранным из группы, состоящей из Gln91, Cys97 и His254, и находится в пределах 4 или менее от него; и
[0121] R6 выбран из -Н и необязательно замещенного C1-4 алкила.
[0122] Некоторые варианты реализации соединений формулы (II) включают соединения, где R9 представляет собой -(C=R12)(C=R13)NR2R3;
[0123] R12 представляет собой оксо и расположен таким образом, что при связывании соединения с кальпаином 9 R12 образует полярное взаимодействие с имидазолом His254 кальпаина 9 и находится в пределах 4 или менее от него;
[0124] R13 представляет собой оксо и расположен таким образом, что при связывании соединения с кальпаином 9 R13 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 9, выбранным из группы, состоящей из карбоксамида боковой цепи Gln91 и амида основной цепи Cys97, и находится в пределах 4 или менее от него; и
[0125] R2 и R3 независимо выбраны из -Н, необязательно замещенного C1-4 алкила, необязательно замещенного С3-7 карбоциклила, необязательно замещенного 5-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного С6-10арил(С1-С6)алкила и необязательно замещенного 5-10-членного гетероарила.
[0126] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R12 расположен таким образом, что при связывании указанного соединения с кальпаином 9 R12 находится в пределах 2,6-3,2 или менее от имидазола His254 кальпаина 9.
[0127] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R12 расположен таким образом, что при связывании указанного соединения с кальпаином 9 R12 находится в пределах 2,6-3,0 или менее от имидазола His254 кальпаина 9.
[0128] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R13 расположен таким образом, что при связывании указанного соединения с кальпаином 9 R13 находится в пределах 2,6-3,5 от фрагментов кальпаина 9, включая и карбоксамид боковой цепи Gln91, и амид основной цепи Cys97.
[0129] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R13 расположен таким образом, что при связывании указанного соединения с кальпаином 9 R13 находится в пределах 2,6-3,2 от фрагментов кальпаина 9, включая и карбоксамид боковой цепи Gln91, и амид основной цепи Cys97.
[0130] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R9 расположен вблизи атома углерода, к которому он присоединен, таким образом, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом R9 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 9, выбранным из группы, состоящей из Gln91, Cys97 и His254, и находится в пределах 3,6 или менее от него.
[0131] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R9 расположен вблизи атома углерода, к которому он присоединен, таким образом, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом R9 находится в пределах 2,6-3,6 от фрагментов кальпаина 9, включая и карбоксамид боковой цепи Gln91, и амид основной цепи Cys97.
[0132] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R9 расположен вблизи атома углерода, к которому он присоединен, таким образом, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом R9 находится в пределах 2,9-3,2 от фрагментов кальпаина 9, включая и карбоксамид боковой цепи Gln91, и амид основной цепи Cys97.
[0133] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором атом углерода в R9 в точке его присоединения образует ковалентную связь с Cys97.
[0134] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором длина указанной ковалентной связи составляет от 1,7 до 1,9 .
[0135] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Рг представляет собой необязательно замещенный 5-членный гетероарил.
[0136] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R11 расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании указанного соединения с кальпаином 9 R11 образует полярное взаимодействие с карбонилом Gly253 кальпаина 9 и находится в пределах 3,6  или менее от него.
или менее от него.
[0137] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 1 по меньшей мере один атом Р2 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р2 кальпаина 1, выбранным из группы, состоящей из Gly208, Ser251, Gly271, His272 и Ala273, и находится в пределах 5  или менее от него;
или менее от него;
[0138] Р3 расположен вблизи L1 и имеет такие размер и конфигурацию, что при связывании соединения с кальпаином 1 по меньшей мере один атом Р3 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р3 кальпаина 1, выбранным из группы, состоящей из Gly207, Gly208, Ser209, Ile254 и Gly271, и находится в пределах 5 или менее от него;
[0139] R10 расположен вблизи Р2 таким образом, что при связывании соединения с кальпаином 1 R10 образует полярное взаимодействие с амидом Gly208 кальпаина 1 и находится в пределах 4 или менее от него;
[0140] R11 расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании соединения с кальпаином 1 R11 образует полярное взаимодействие с карбонилом Gly271 кальпаина 1 и находится в пределах 4 или менее от него;
[0141] P1 расположен вблизи L2 и имеет такие размер и конфигурацию, что при связывании соединения с кальпаином 1 по меньшей мере один атом P1 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р1 кальпаина 1, выбранным из группы, состоящей из Gly113, Ser206, Gly207 и Met260, и находится в пределах 5 или менее от него; и
[0142] R9 расположен вблизи атома углерода, к которому он присоединен, таким образом, что при связывании соединения с кальпаином 1 по меньшей мере один атом R9 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 1, выбранным из группы, состоящей из Gln109, Cys115 и His272, и находится в пределах 4 или менее от него.
[0143] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором:
[0144] Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 2 по меньшей мере один атом Р2 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р2 кальпаина 2, выбранным из группы, состоящей из Gly198, Ser241, Gly261, His262 и Ala263, и находится в пределах 5 или менее от него;
[0145] Р3 расположен вблизи L1 и имеет такие размер и конфигурацию, что при связывании соединения с кальпаином 2 по меньшей мере один атом Р3 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р3 кальпаина 2, выбранным из группы, состоящей из Gly197, Gly198, Ala199, Ile244 и Gly261, и находится в пределах 5  или менее от него;
или менее от него;
[0146] R10 расположен вблизи Р2 таким образом, что при связывании соединения с кальпаином 2 R10 образует полярное взаимодействие с амидом Gly198 кальпаина 2 и находится в пределах 4 или менее от него;
[0147] R11 расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании соединения с кальпаином 2 R11 образует полярное взаимодействие с карбонилом Gly261 кальпаина 2 и находится в пределах 4 или менее от него;
[0148] P1 расположен вблизи L2 и имеет такие размер и конфигурацию, что при связывании соединения с кальпаином 2 по меньшей мере один атом P1 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р1 кальпаина 2, выбранным из группы, состоящей из Gly103, Ser196, Gly197 и Ser250, и находится в пределах 5 или менее от него; и
[0149] R9 расположен вблизи атома углерода, к которому он присоединен, таким образом, что при связывании соединения с кальпаином 2 по меньшей мере один атом R9 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 2, выбранным из группы, состоящей из Gln99, Cys105 и His262, и находится в пределах 4 или менее от него.
[0150] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р2 находится в пределах 2,6-3,6 от атома кислорода карбонила Gly190.
[0151] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р2 находится в пределах 2,9-3,3 от атома кислорода карбонила Gly190.
[0152] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р2 находится в пределах 2,8-4,8 от атома углерода в Phe233.
[0153] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р2 находится в пределах 2,9-3,3 от атома углерода в Phe233.
[0154] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р2 находится в пределах 2,6-3,7 от атома кислорода карбонила Gly253.
[0155] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р2 находится в пределах 2,9-3,3 от атома кислорода карбонила Gly253.
[0156] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р2 находится в пределах 2,9-4,8 от атома азота Ala255.
[0157] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р2 находится в пределах 3,2-4,0 от атома азота Ala255.
[0158] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р3 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р3 находится в пределах 3,1-4,3 от С-альфа Gly189.
[0159] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р3 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р3 находится в пределах 3,2-4,0 от С-альфа Gly189.
[0160] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р3 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р3 находится в пределах 3,0-4,3 от атома кислорода карбонила Gly190.
[0161] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р3 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р3 находится в пределах 3,2-4,0 от атома кислорода карбонила Gly190.
[0162] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р3 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р3 находится в пределах 3,2-4,8 от атома азота Ser191.
[0163] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р3 имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом Р3 находится в пределах 3,2-4,0 от атома азота Ser191.
[0164] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R10 расположен вблизи Р2 таким образом, что при связывании указанного соединения с кальпаином 9 R10 находится в пределах 2,6-3,5 от амида Gly190 кальпаина 9.
[0165] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R10 расположен вблизи Р2 таким образом, что при связывании указанного соединения с кальпаином 9 R10 находится в пределах 2,9-3,3 от амида Gly190 кальпаина 9.
[0166] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R11 расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании указанного соединения с кальпаином 9 R11 находится в пределах 2,6-3,6 или менее от карбонила Gly253 кальпаина 9.
[0167] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R11 расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании указанного соединения с кальпаином 9 R11 находится в пределах 2,9-3,3 или менее от карбонила Gly253 кальпаина 9.
[0168] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором P1 расположен вблизи L2 и имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом P1 находится в пределах 3,2-4,4 от атома кислорода карбонила Gly95.
[0169] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором P1 расположен вблизи L2 и имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом P1 находится в пределах 3,2-4,0 от атома кислорода карбонила Gly95.
[0170] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором P1 расположен вблизи L2 и имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом P1 находится в пределах 3,2-4,7 от атома углерода карбонила Lys188.
[0171] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором P1 расположен вблизи L2 и имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом P1 находится в пределах 2,6-4,0 от атома углерода карбонила Lys188.
[0172] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором P1 расположен вблизи L2 и имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом P1 находится в пределах 3,0-4,1 от С-альфа Gly189.
[0173] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором P1 расположен вблизи L2 и имеет такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 9 по меньшей мере один атом P1 находится в пределах 3,2-4,0 от С-альфа Gly189.
[0174] В некоторых вариантах реализации предложено соединение формулы (II)
или его фармацевтически приемлемая соль, где:
[0175] Р2 представляет собой необязательно замещенный циклический фрагмент, имеющий такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 1 по меньшей мере один атом Р2 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р2 кальпаина 1, выбранным из группы, состоящей из Gly208, Ser251, Gly271, His272 и Ala273, и находится в пределах 5 или менее от него;
[0176] L1 представляет собой связь или фрагмент, состоящий из 1-25 атомов, выбранных из группы, состоящей из атома углерода, атома кислорода, атома азота, атома водорода и атома серы;
[0177] Р3 представляет собой необязательно замещенный циклический фрагмент, расположенный вблизи L1 и имеющий такие размер и конфигурацию, что при связывании соединения с кальпаином 1 по меньшей мере один атом Р3 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р3 кальпаина 1, выбранным из группы, состоящей из Gly207, Gly208, Ser209, Ile254 и Gly271, и находится в пределах 5 или менее от него;
[0178] R10 представляет собой оксо и расположен вблизи Р2 таким образом, что при связывании соединения с кальпаином 1 R10 образует полярное взаимодействие с амидом Gly208 кальпаина 1 и находится в пределах 4 или менее от него;
[0179] R11 представляет собой атом азота и расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании соединения с кальпаином 1 R11 образует полярное взаимодействие с карбонилом Gly271 кальпаина 1 и находится в пределах 4 или менее от него;
[0180] L2 представляет собой связь или фрагмент, состоящий из 1-25 атомов, выбранных из группы, состоящей из атома углерода, атома кислорода, атома азота, атома водорода и атома серы;
[0181] P1 представляет собой фрагмент, расположенный вблизи L2 и имеющий такие размер и конфигурацию, что при связывании соединения с кальпаином 1 по меньшей мере один атом P1 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р1 кальпаина 1, выбранным из группы, состоящей из Gly113, Ser206, Gly207 и Met260, и находится в пределах 5 или менее от него;
[0182] R9 представляет собой фрагмент, расположенный вблизи атома углерода, к которому он присоединен, таким образом, что при связывании соединения с кальпаином 1 по меньшей мере один атом R9 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 1, выбранным из группы, состоящей из Gln109, Cys115 и His272, и находится в пределах 4 или менее от него; и R6 выбран из -Н и необязательно замещенного C1-4 алкила.
[0183] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R9 представляет собой -(C=R12)(C=R13)NR2R3;
[0184] R12 представляет собой оксо и расположен таким образом, что при связывании указанного соединения с кальпаином 1 R12 образует полярное взаимодействие с имидазолом His272 кальпаина 1 и находится в пределах 4 или менее от него;
[0185] R13 представляет собой оксо и расположен таким образом, что при связывании соединения с кальпаином 1 R13 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 1, выбранным из группы, состоящей из карбоксамида боковой цепи Gln109 и амида основной цепи Cys115, и находится в пределах 4 или менее от него; и
[0186] R2 и R3 независимо выбраны из -Н, необязательно замещенного C1-4 алкила, необязательно замещенного С3-7 карбоциклила, необязательно замещенного 5-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного С6-10арил(С1-С6)алкила и необязательно замещенного 5-10-членного гетероарила.
[0187] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R9 расположен вблизи атома углерода, к которому он присоединен, таким образом, что при связывании указанного соединения с кальпаином 1 по меньшей мере один атом R9 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 1, выбранным из группы, состоящей из Gln109, Cys115 и His272, и находится в пределах 3,5 или менее от него.
[0188] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором атом углерода в R9 в точке его присоединения образует ковалентную связь с Cys115.
[0189] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором длина указанной ковалентной связи составляет от 1,7 до 1,9 .
[0190] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 представляет собой необязательно замещенный 5-членный гетероарил.
[0191] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R11 расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании указанного соединения с кальпаином 1 R11 образует полярное взаимодействие с карбонилом Gly271 кальпаина 1 и находится в пределах 3,5 или менее от него.
[0192] В некоторых вариантах реализации предложено соединение формулы (II)
или его фармацевтически приемлемая соль, где
[0193] Р2 представляет собой необязательно замещенный циклический фрагмент, имеющий такие размер и конфигурацию, что при связывании указанного соединения с кальпаином 2 по меньшей мере один атом Р2 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р2 кальпаина 2, выбранным из группы, состоящей из Gly198, Ser241, Gly261, His262 и Ala263, и находится в пределах 5 или менее от него;
[0194] L1 представляет собой связь или фрагмент, состоящий из 1-25 атомов, выбранных из группы, состоящей из атома углерода, атома кислорода, атома азота, атома водорода и атома серы;
[0195] Р3 представляет собой необязательно замещенный циклический фрагмент, расположенный вблизи L1 и имеющий такие размер и конфигурацию, что при связывании соединения с кальпаином 2 по меньшей мере один атом Р3 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р3 кальпаина 2, выбранным из группы, состоящей из Gly197, Gly198, Ala199, Ile244 и Gly261, и находится в пределах 5 или менее от него;
[0196] R10 представляет собой оксо и расположен вблизи Р2 таким образом, что при связывании соединения с кальпаином 2 R10 образует полярное взаимодействие с амидом Gly198 кальпаина 2 и находится в пределах 4 или менее от него;
[0197] R11 представляет собой атом азота и расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании соединения с кальпаином 2 R11 образует полярное взаимодействие с карбонилом Gly261 кальпаина 2 и находится в пределах 4 или менее от него;
[0198] L2 представляет собой связь или фрагмент, состоящий из 1-25 атомов, выбранных из группы, состоящей из атома углерода, атома кислорода, атома азота, атома водорода и атома серы;
[0199] P1 представляет собой фрагмент, расположенный вблизи L2 и имеющий такие размер и конфигурацию, что при связывании соединения с кальпаином 2 по меньшей мере один атом P1 образует неполярное взаимодействие с по меньшей мере одним фрагментом кармана Р1 кальпаина 2, выбранным из группы, состоящей из Gly103, Ser196, Gly197 и Ser250, и находится в пределах 5 или менее от него;
[0200] R9 представляет собой фрагмент, расположенный вблизи атома углерода, к которому он присоединен, таким образом, что при связывании соединения с кальпаином 2 по меньшей мере один атом R9 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 2, выбранным из группы, состоящей из Gln99, Cys105 и His262, и находится в пределах 4 или менее от него; и R6 выбран из -Н и необязательно замещенного C1-4 алкила.
[0201] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R9 представляет собой -(C=R12)(C=R13)NR2R3;
[0202] R12 представляет собой оксо и расположен таким образом, что при связывании указанного соединения с кальпаином 2 R12 образует полярное взаимодействие с имидазолом His262 кальпаина 2 и находится в пределах 4 или менее от него;
[0203] R13 представляет собой оксо и расположен таким образом, что при связывании соединения с кальпаином 2 R13 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 2, выбранным из группы, состоящей из карбоксамида боковой цепи Gln99 и амида основной цепи Cys105, и находится в пределах 4 или менее от него; и
[0204] R2 и R3 независимо выбраны из -Н, необязательно замещенного C1-4 алкила, необязательно замещенного С3-7 карбоциклила, необязательно замещенного 5-10-членного гетероциклила, необязательно замещенного С6-10 арила, необязательно замещенного С6-10арил(С1-С6)алкила и необязательно замещенного 5-10-членного гетероарила.
[0205] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R9 расположен вблизи атома углерода, к которому он присоединен, таким образом, что при связывании указанного соединения с кальпаином 2 по меньшей мере один атом R9 образует полярное взаимодействие с по меньшей мере одним фрагментом кальпаина 2, выбранным из группы, состоящей из Gln99, Cys105 и His262, и находится в пределах 3,5 или менее от него.
[0206] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором атом углерода в R9 в точке его присоединения образует ковалентную связь с Cys195.
[0207] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором длина указанной ковалентной связи составляет от 1,7 до 1,9 .
[0208] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором Р2 представляет собой необязательно замещенный 5-членный гетероарил.
[0209] Некоторые варианты реализации соединений формулы (II) включают соединение, в котором R11 расположен вблизи атомов углерода, с которыми он связан, таким образом, что при связывании указанного соединения с кальпаином 2 R11 образует полярное взаимодействие с карбонилом Gly261 кальпаина 2 и находится в пределах 3,5 или менее от него.
[0210] Некоторые варианты реализации включают соединение, выбранное из группы, состоящей из соединений 1-90, соединений 92-94, соединения 195, соединений 197-235, соединений 238-273, соединений 276-281, соединений 283-299, соединений 303-309, соединений 313-363, соединения 365, соединений 367-410, соединений 413-424, соединений 428-445, соединений 447-448, соединений 454-532, соединения 540, соединений 546-588, соединений 591-605, соединений 607-611, соединений 613-630 и их фармацевтически приемлемых солей, указанные соединения по существу описаны в настоящем описании.
[0211] Некоторые варианты реализации включают соединение, выбранное из группы, состоящей из соединений 91, 196, 274, 282, 310-312, 364, 366, 411, 536, 541 и их фармацевтически приемлемых солей, указанные соединения по существу описаны в настоящем описании.
[0212] Некоторые варианты реализации включают соединение, выбранное из группы, состоящей из:
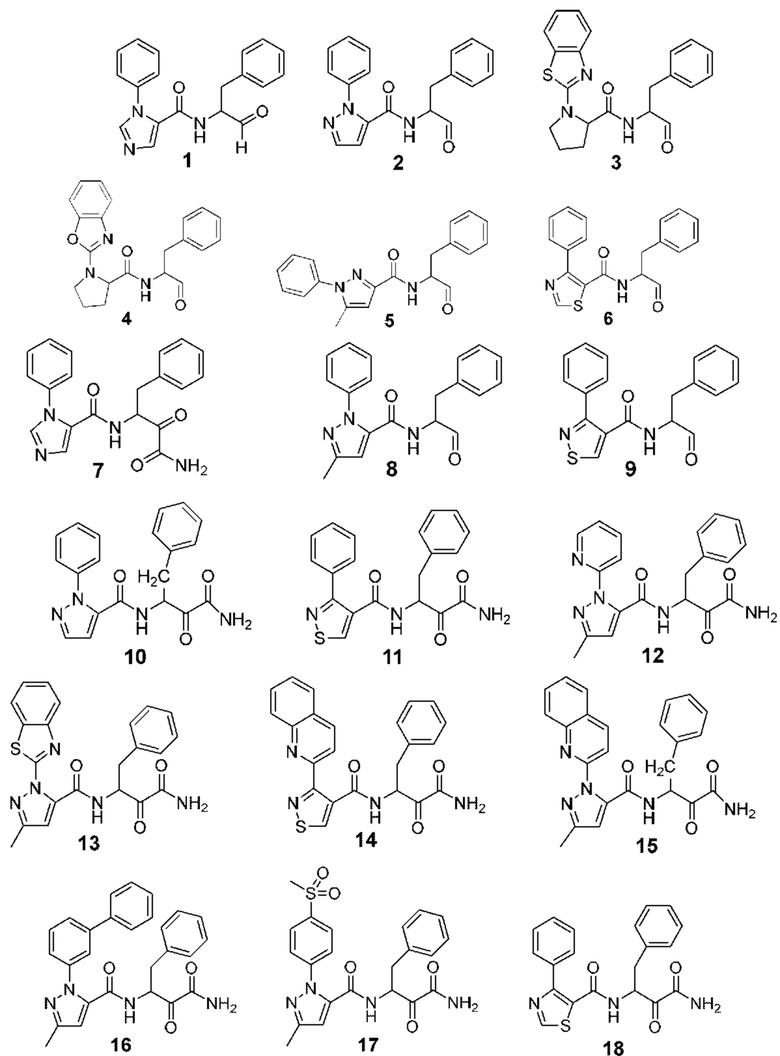
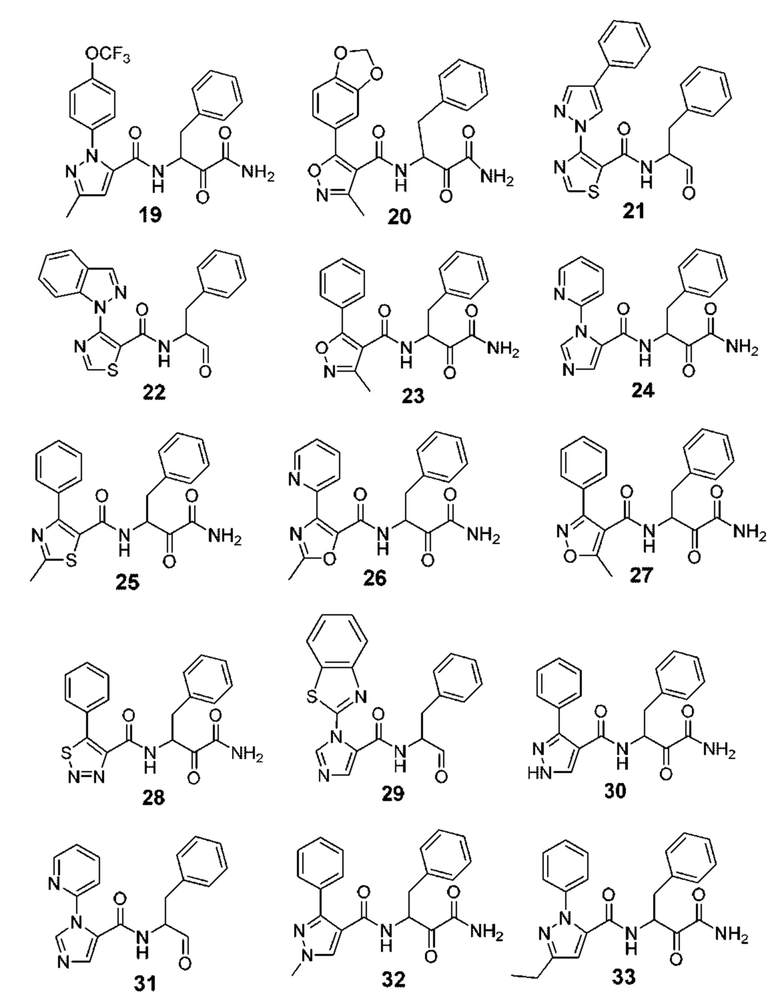
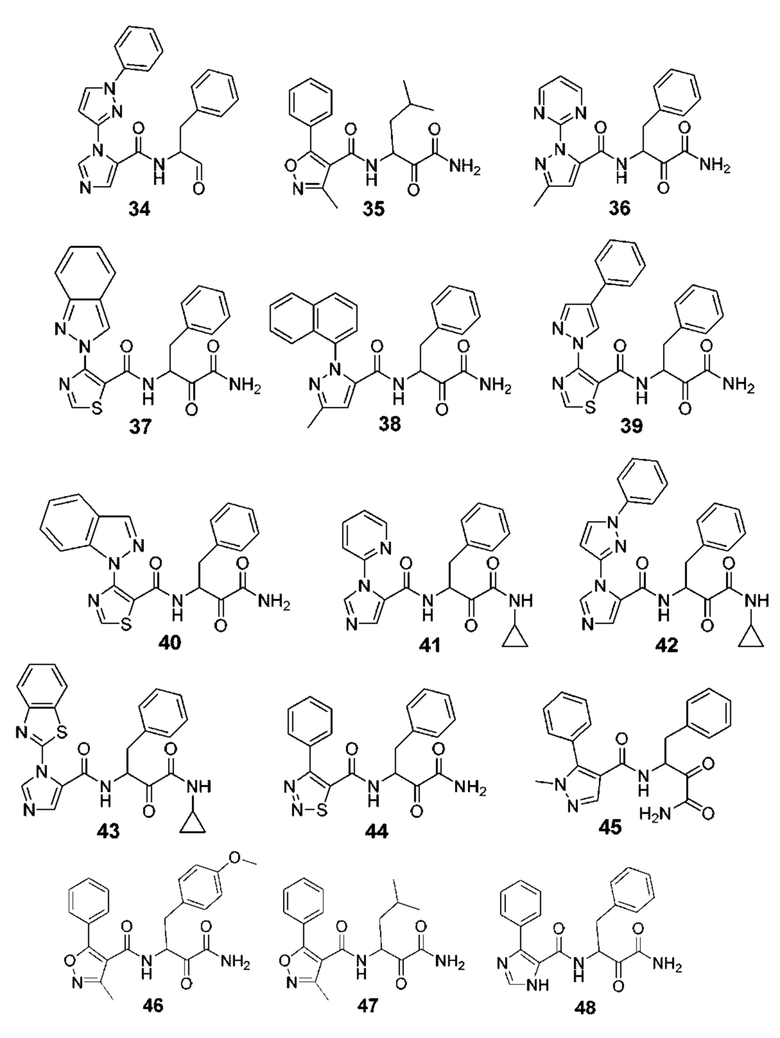
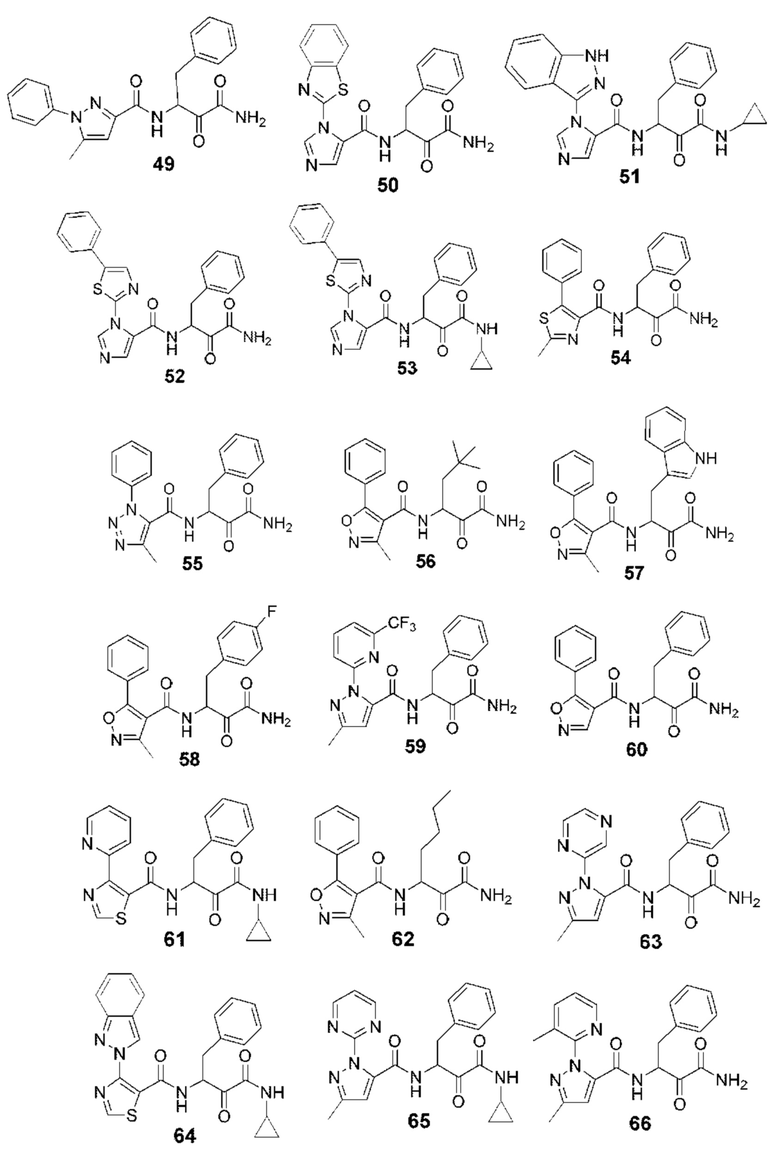
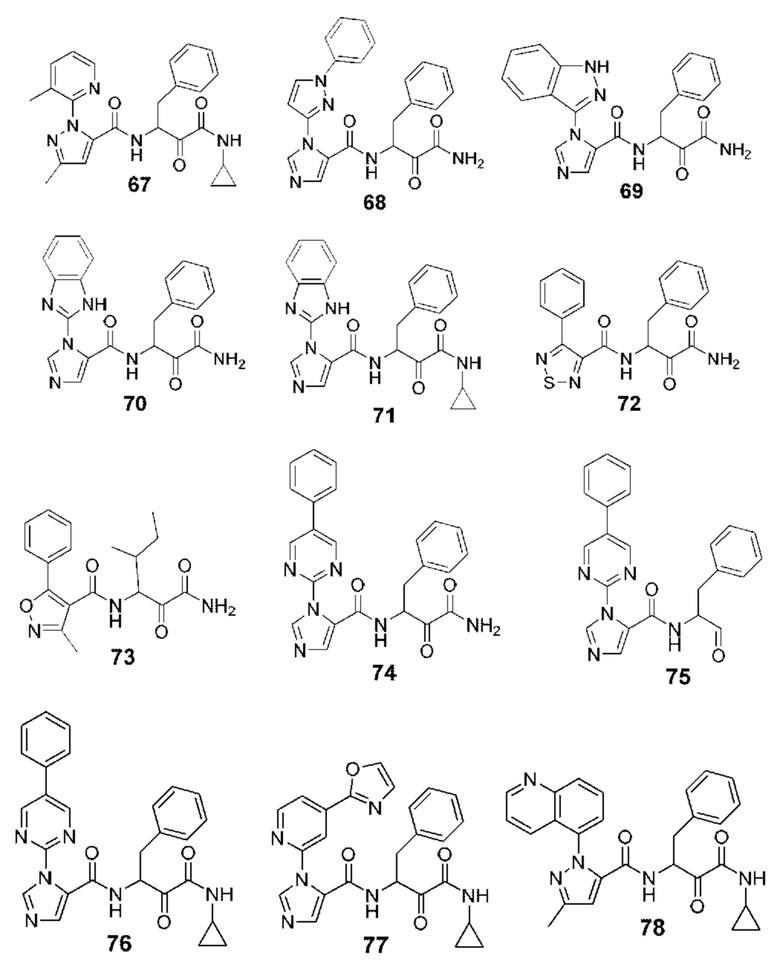
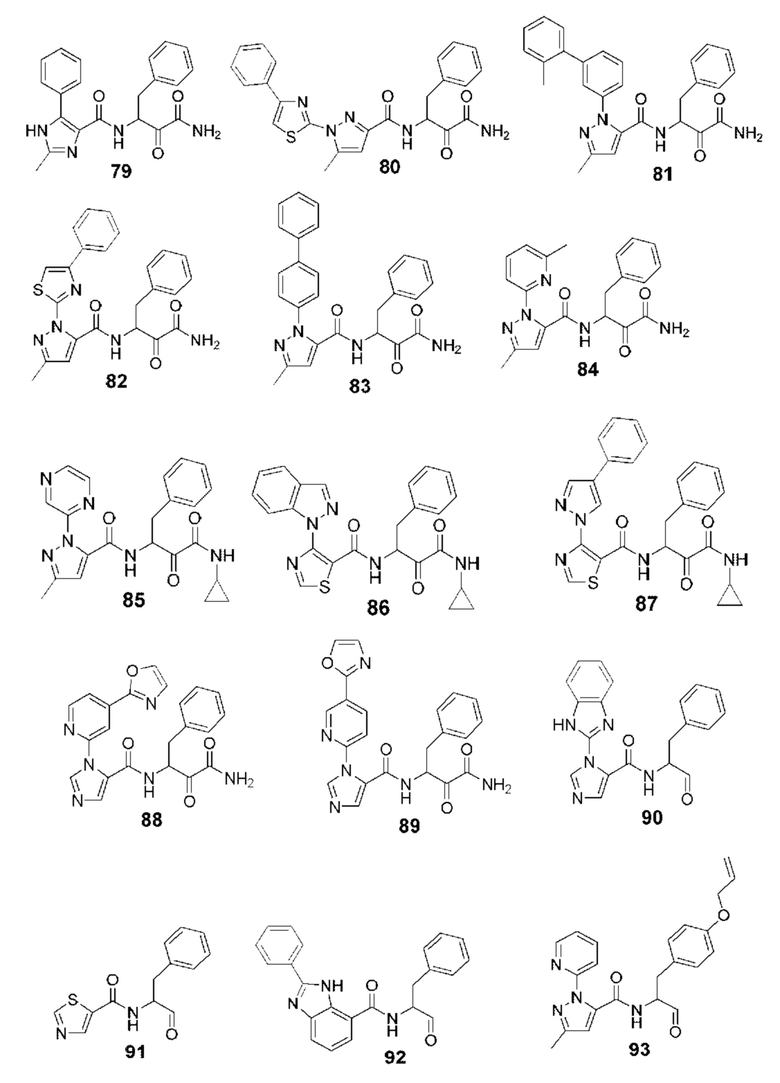
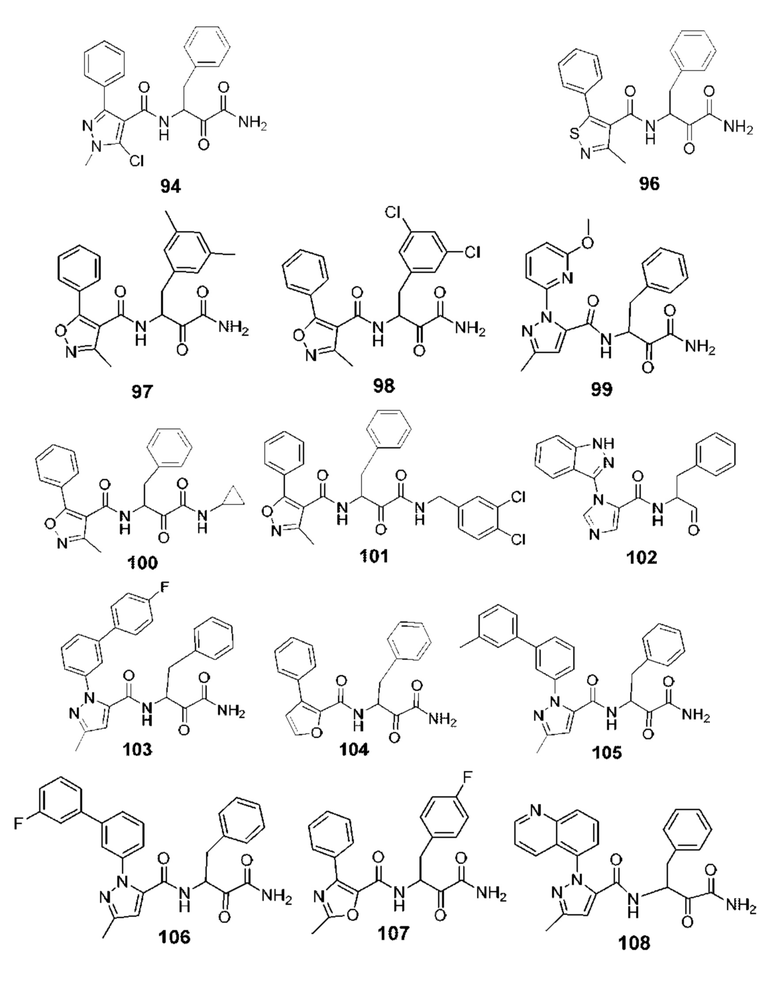

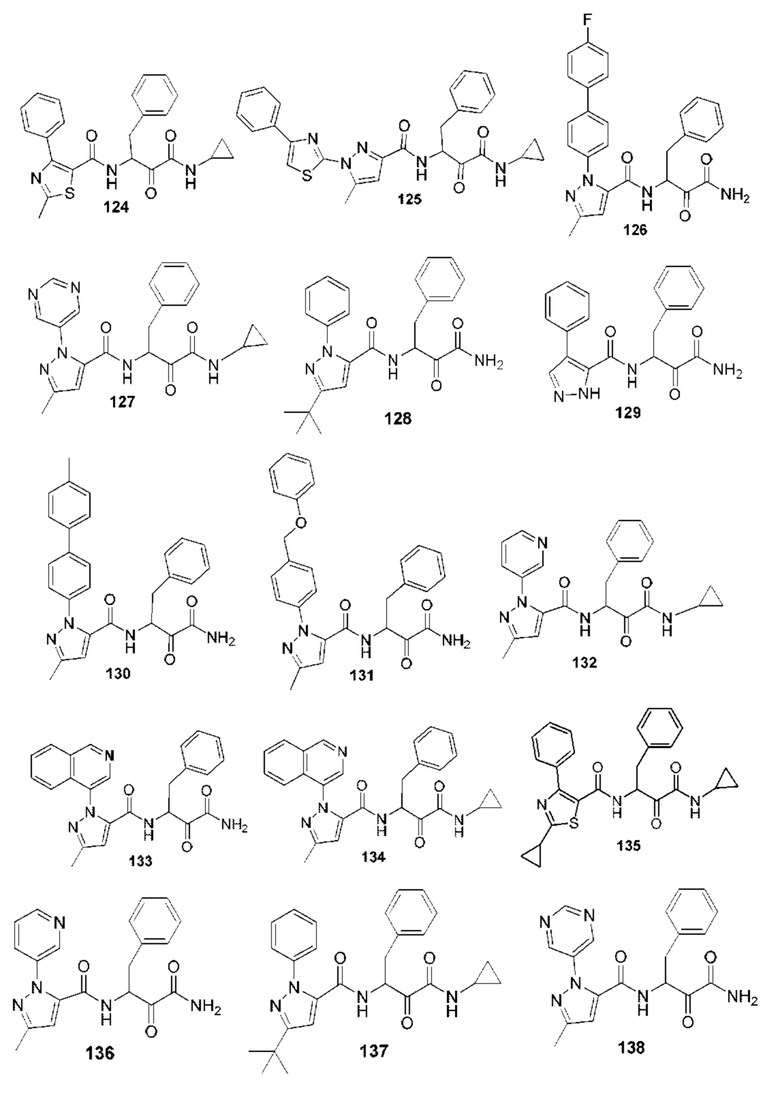
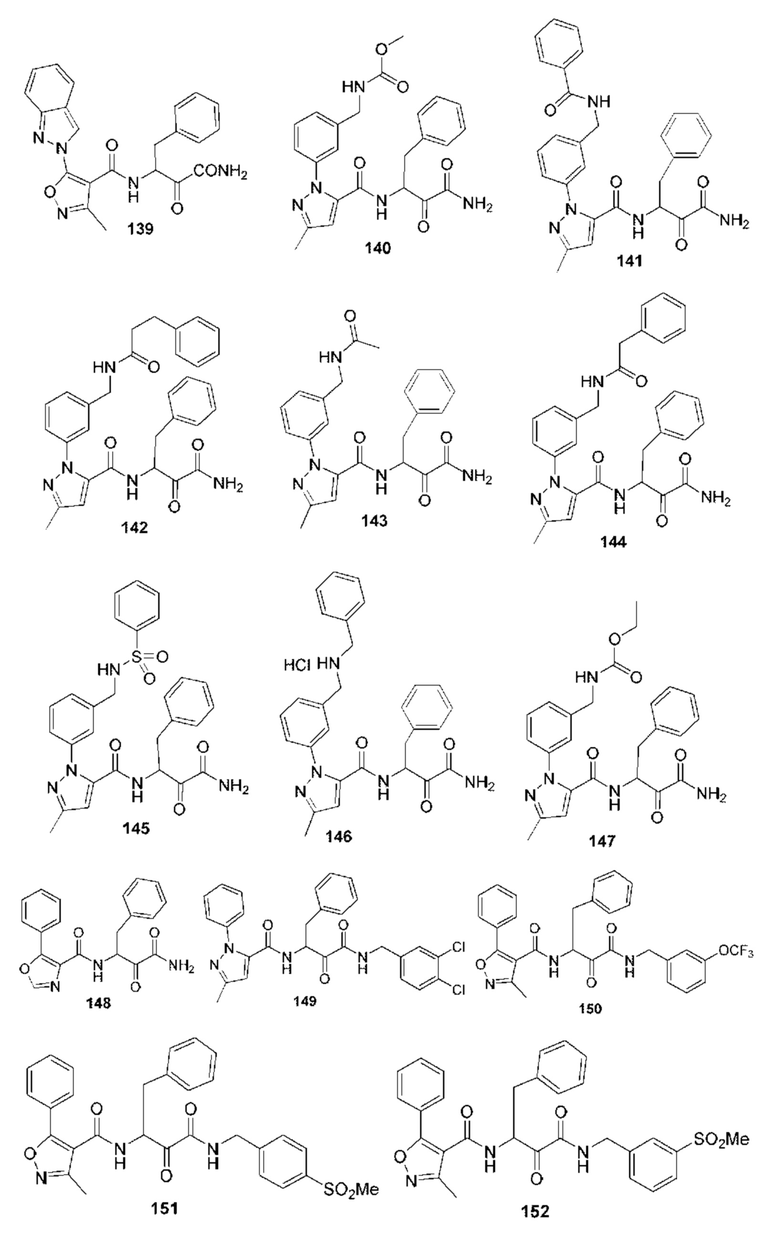
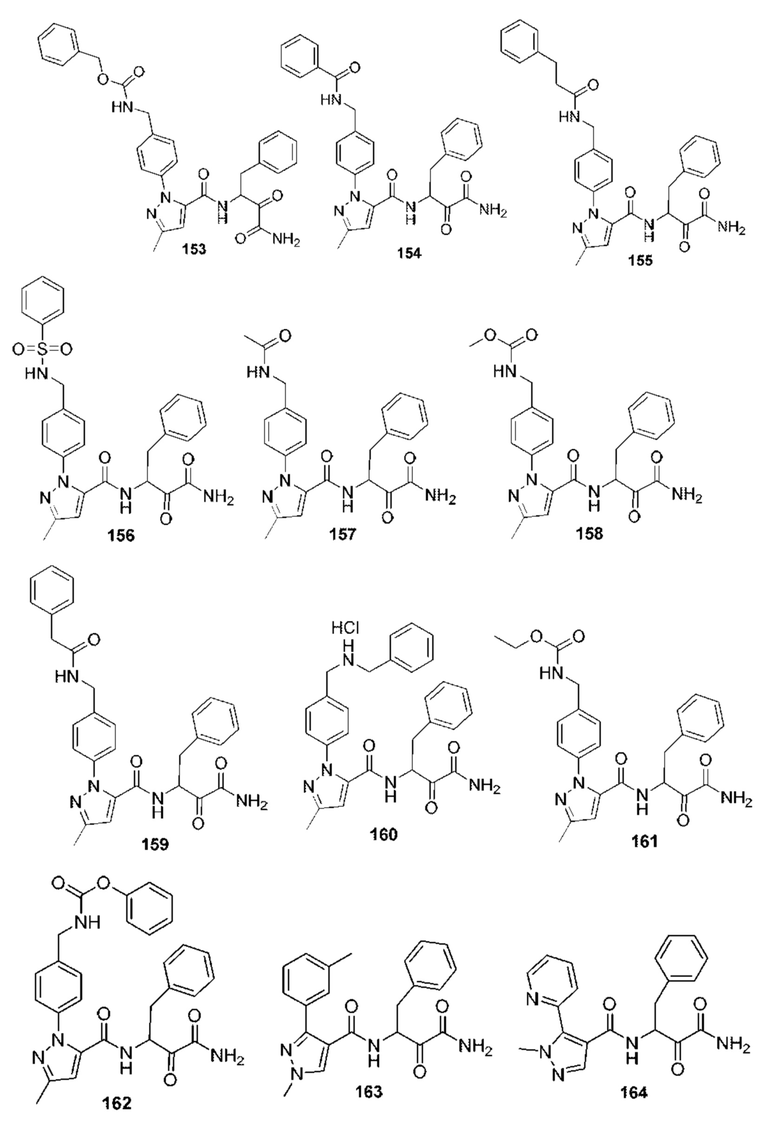
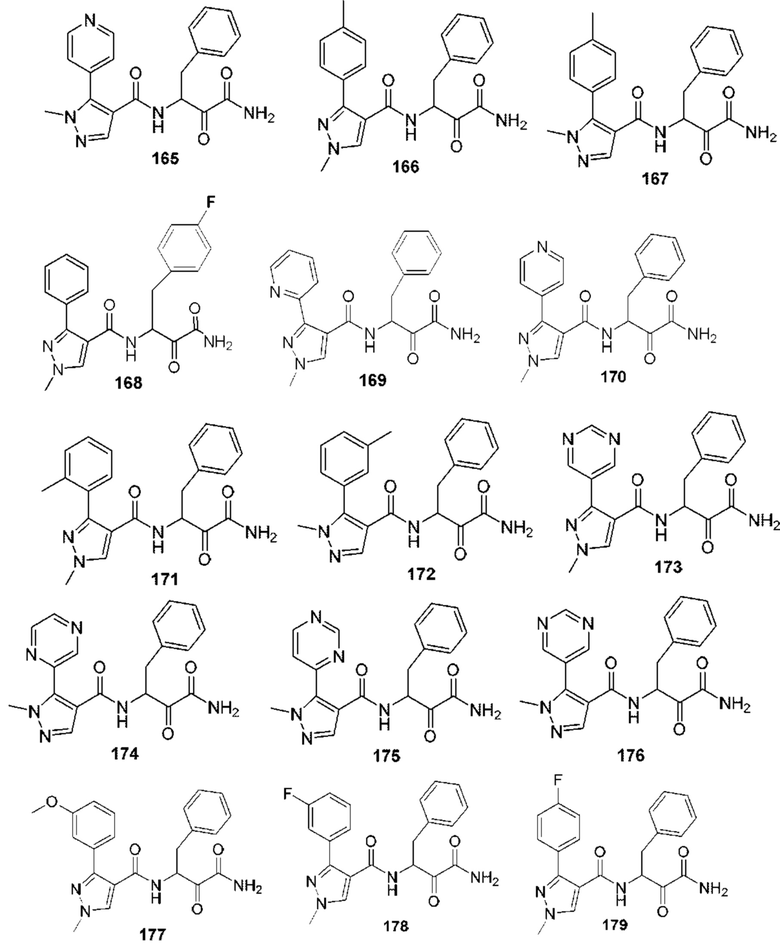
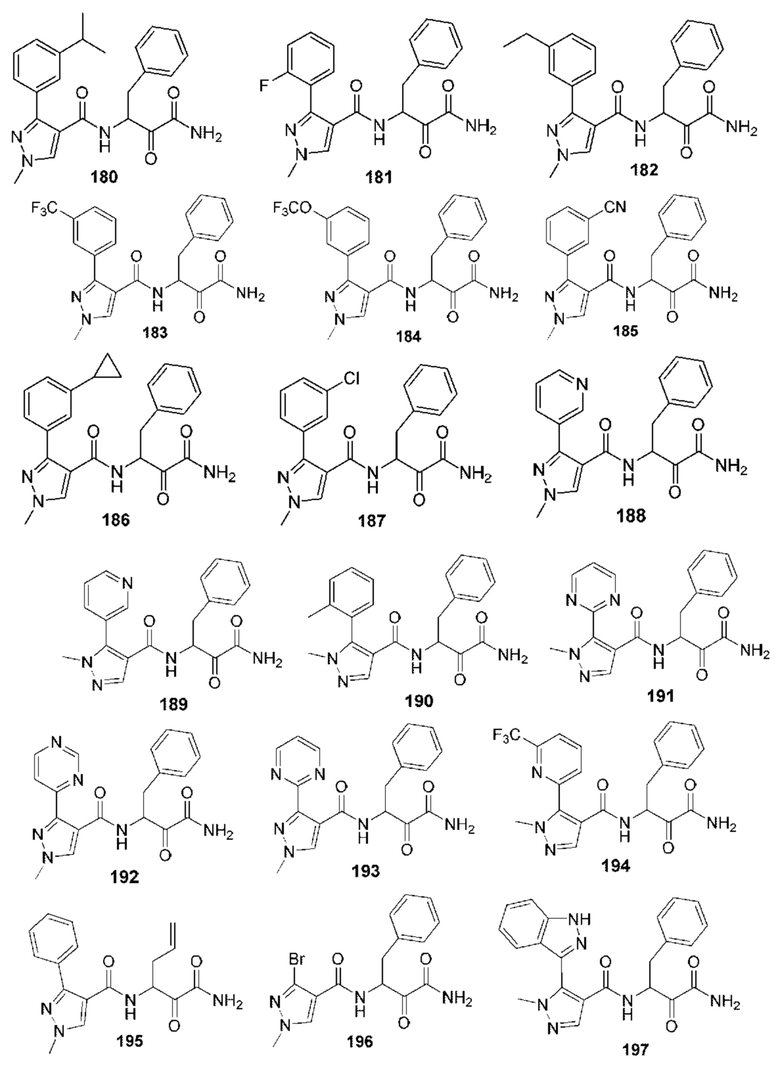

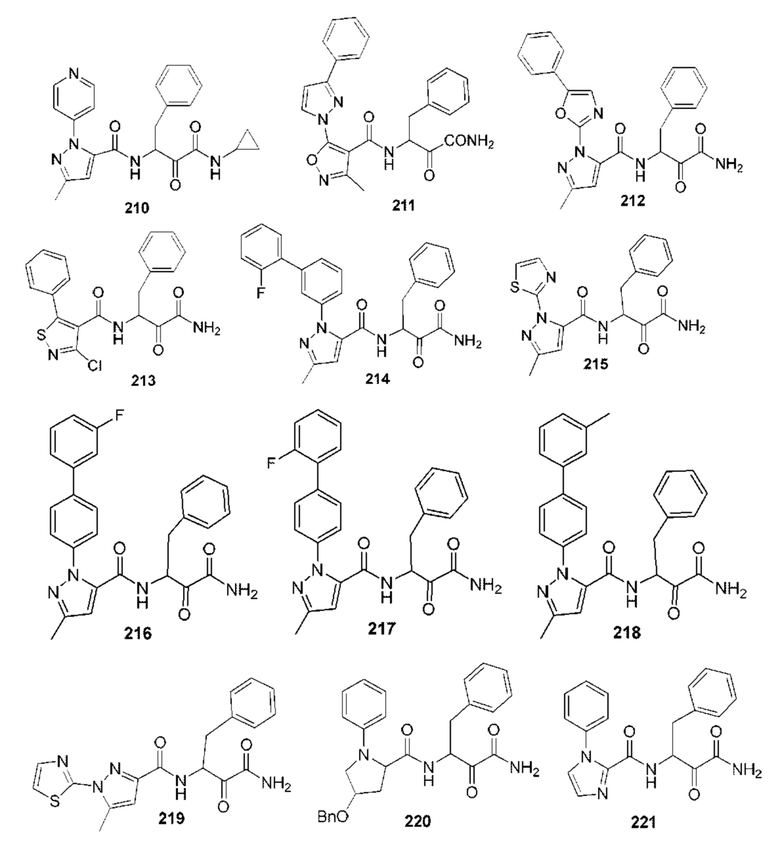
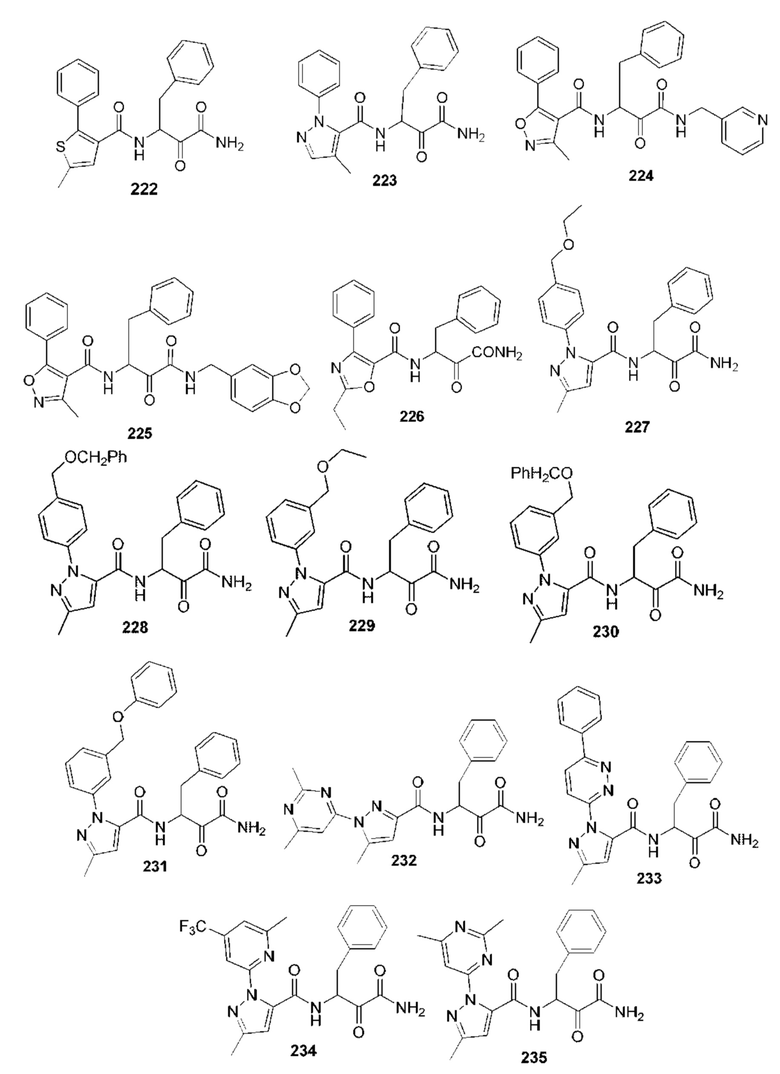
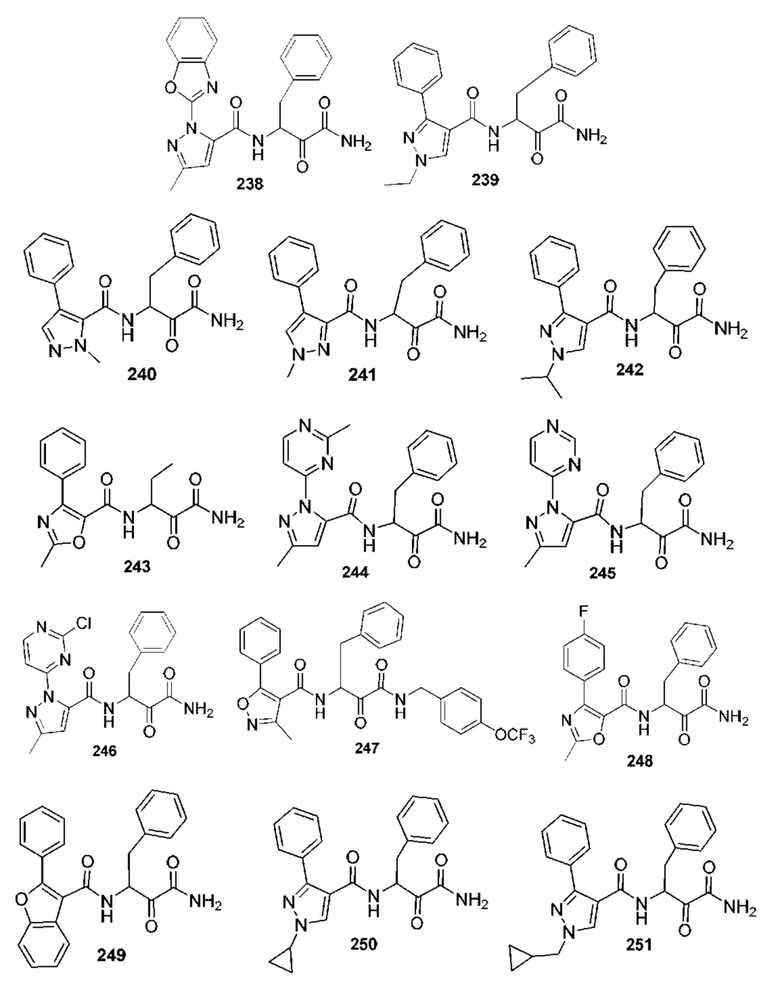
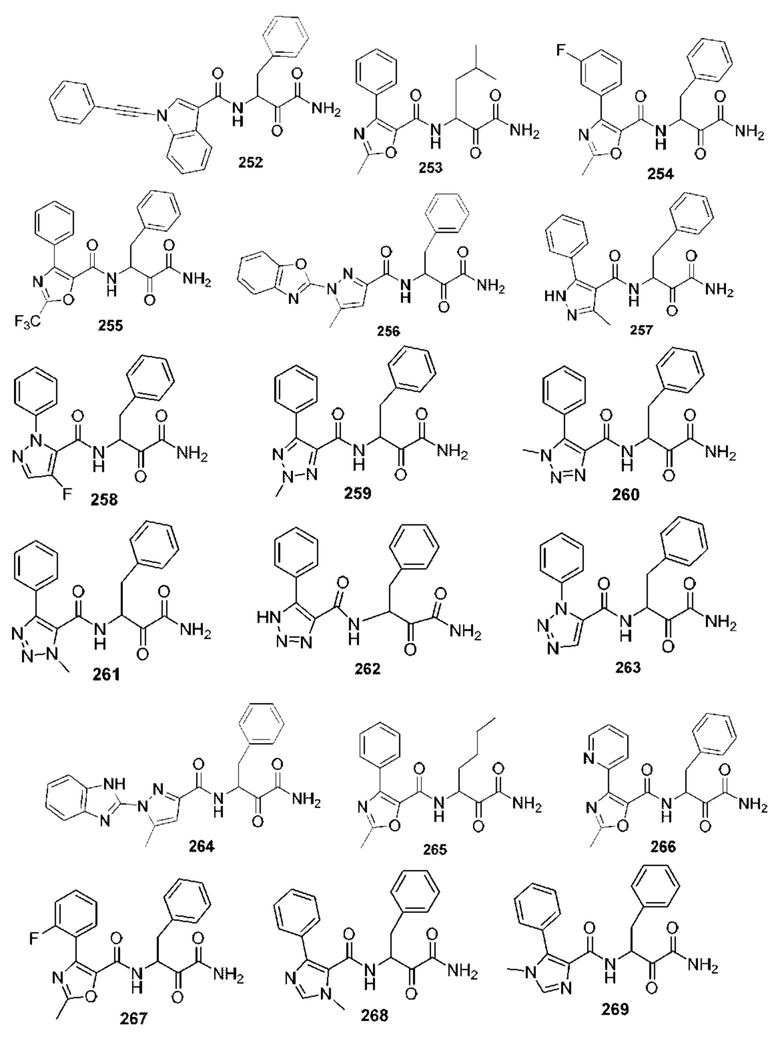
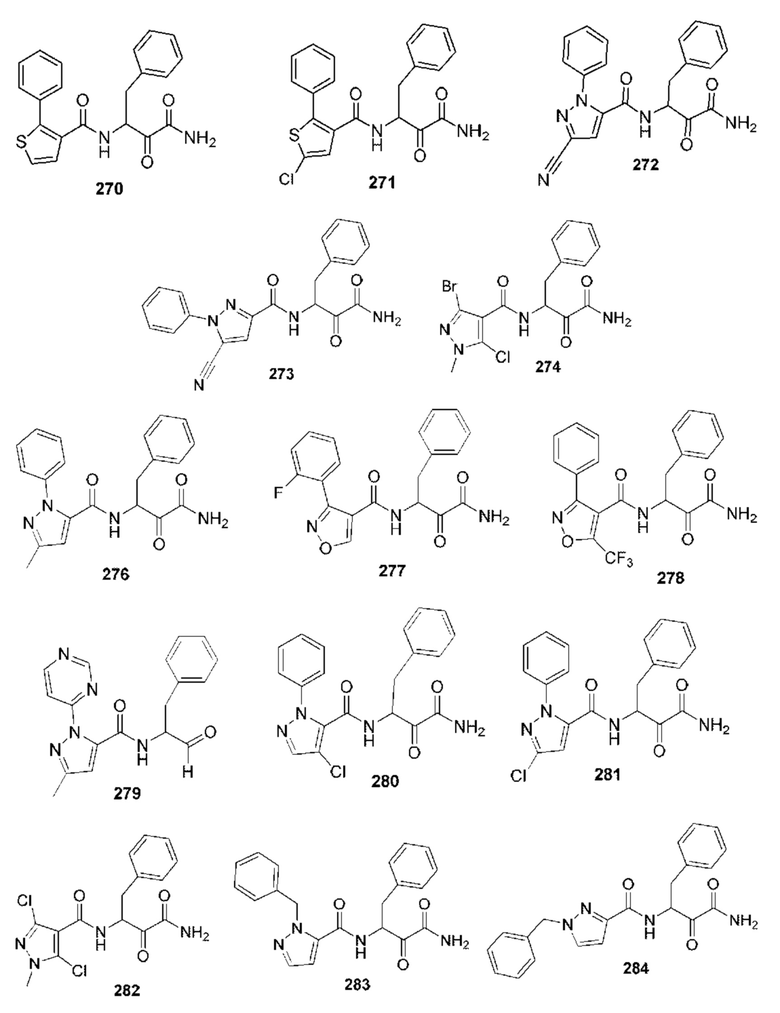
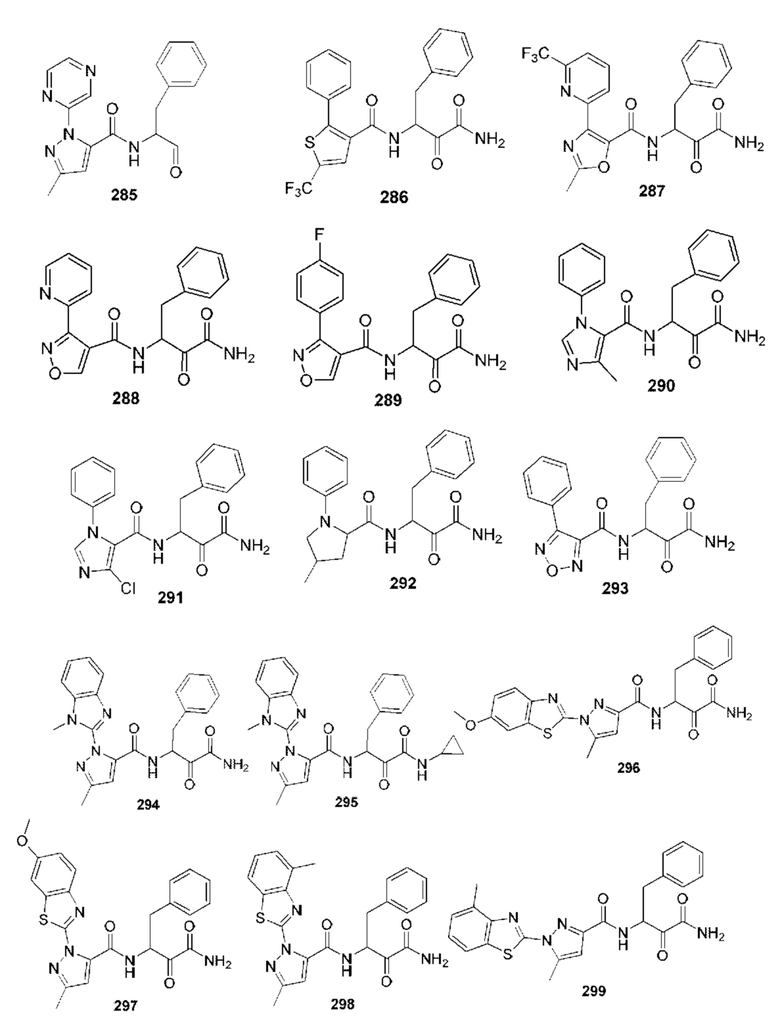
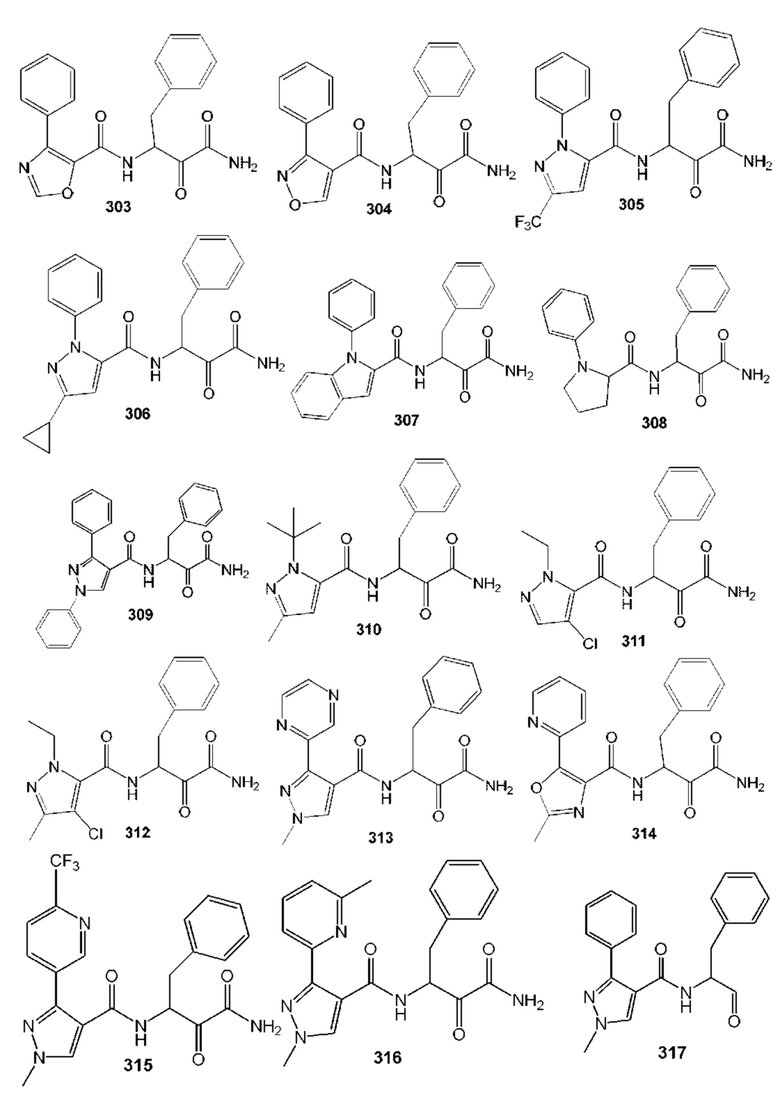
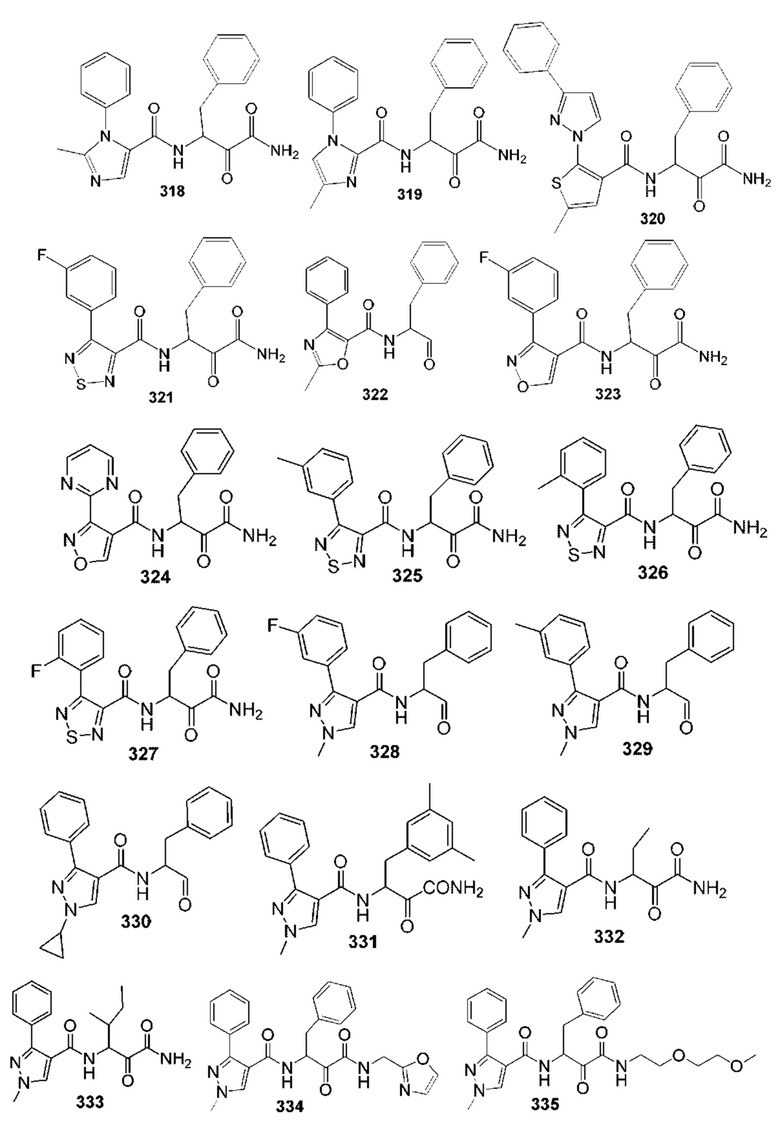
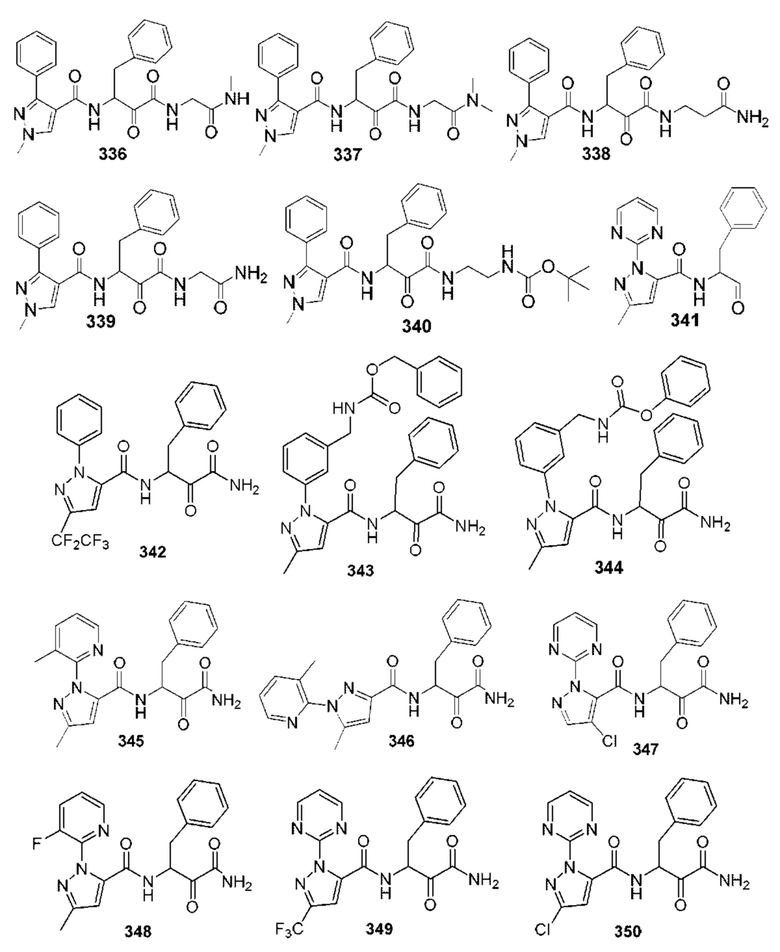
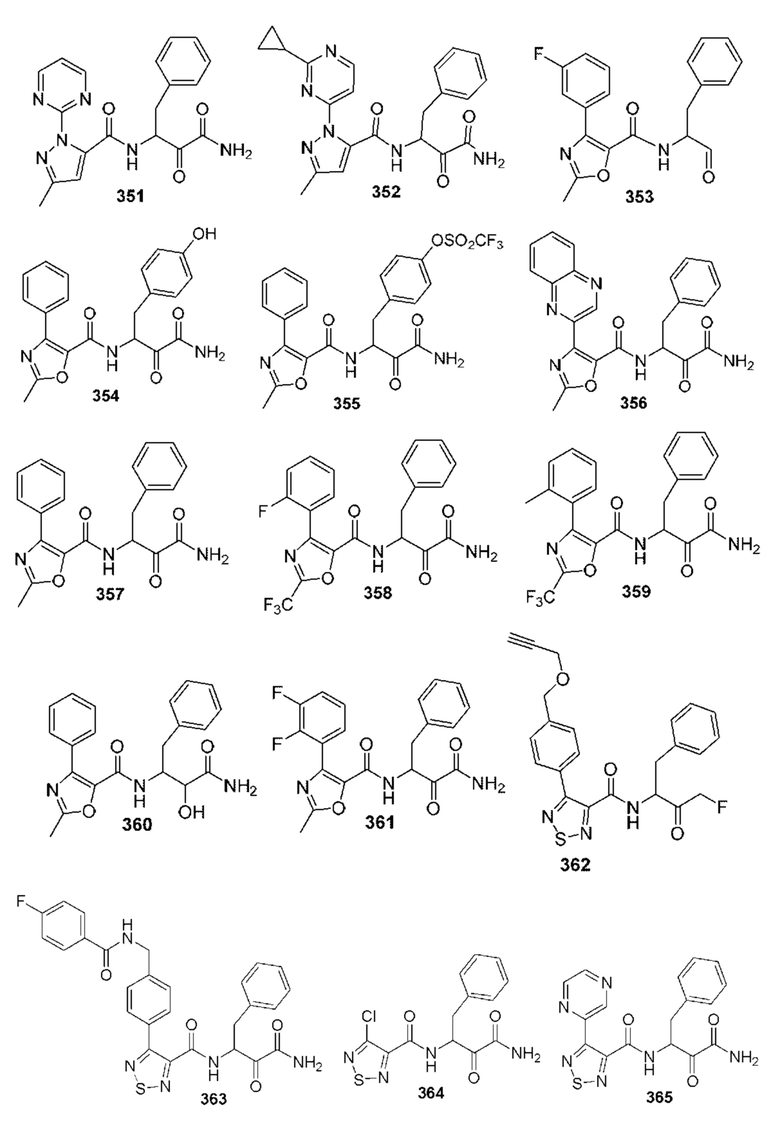
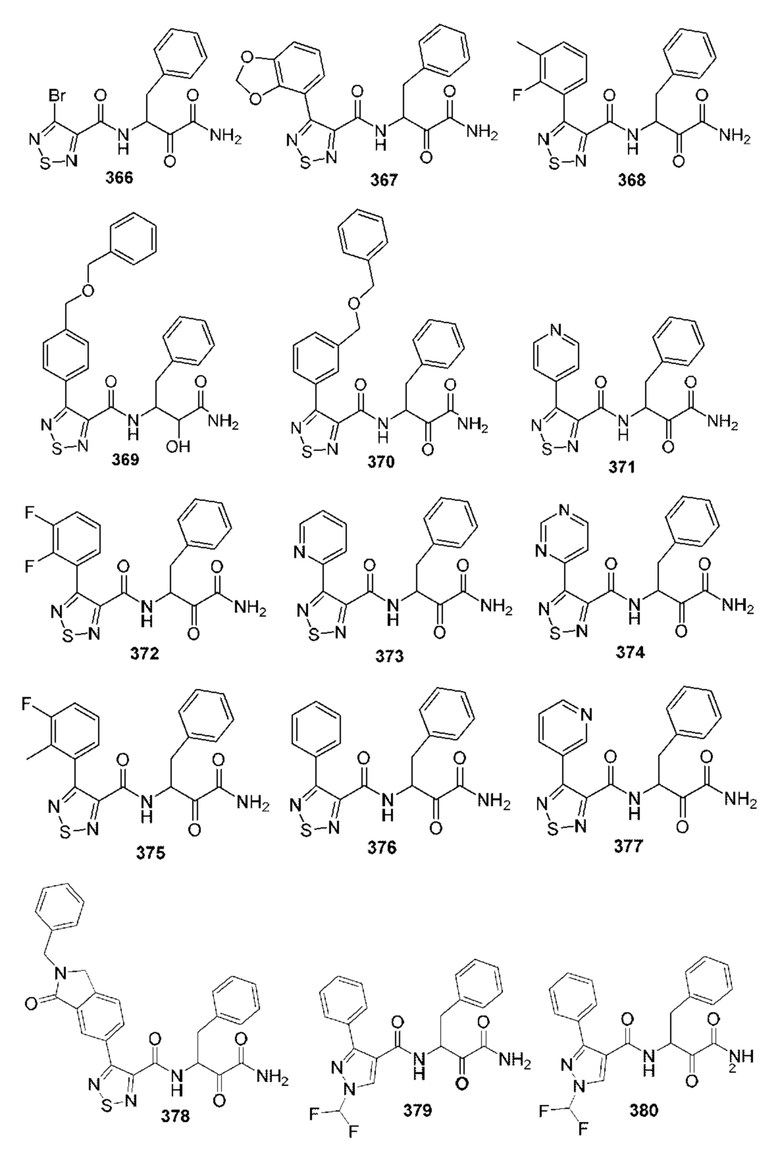
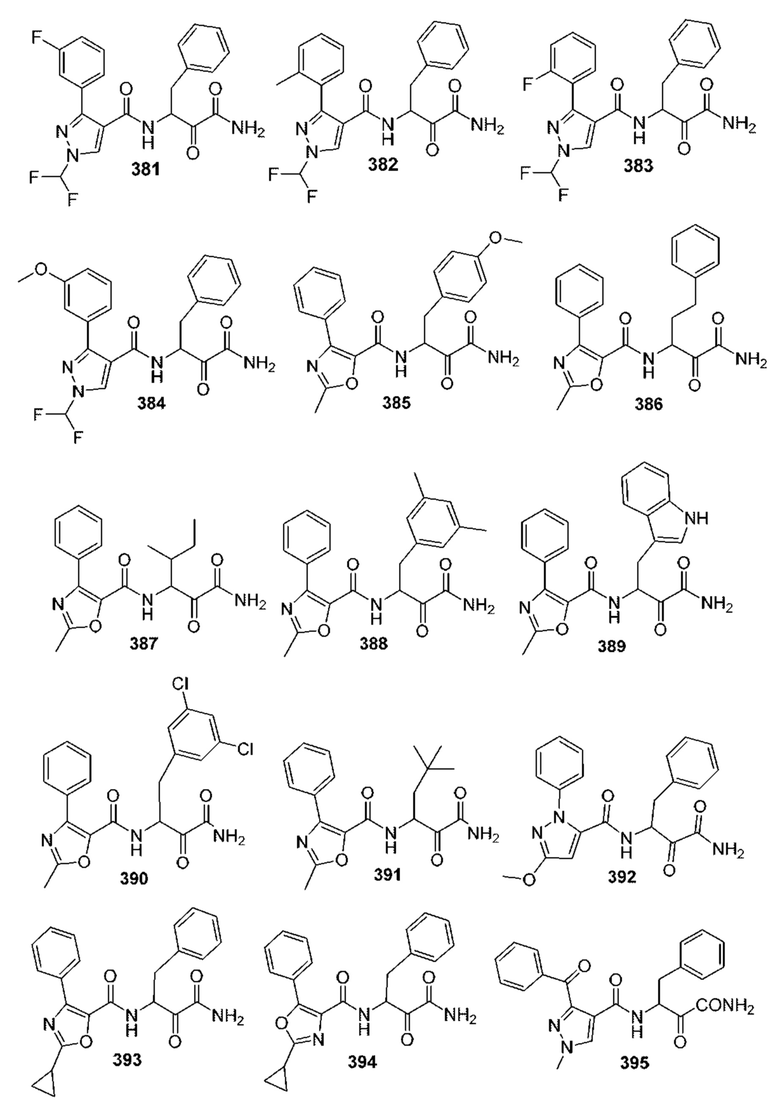
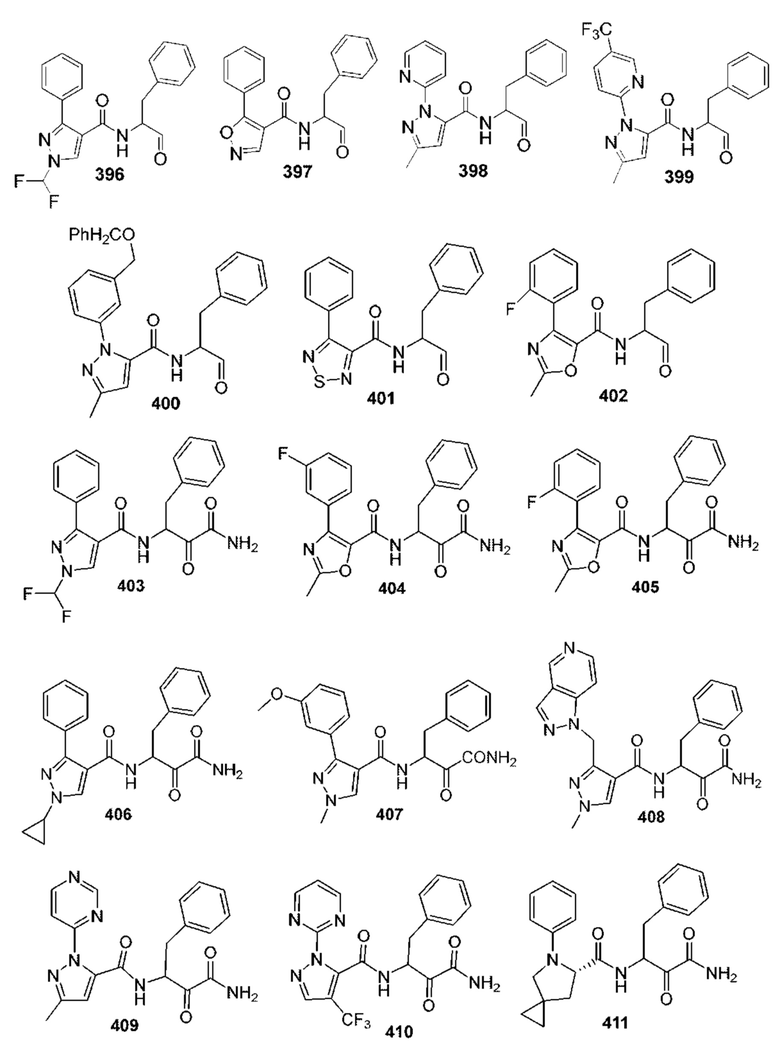
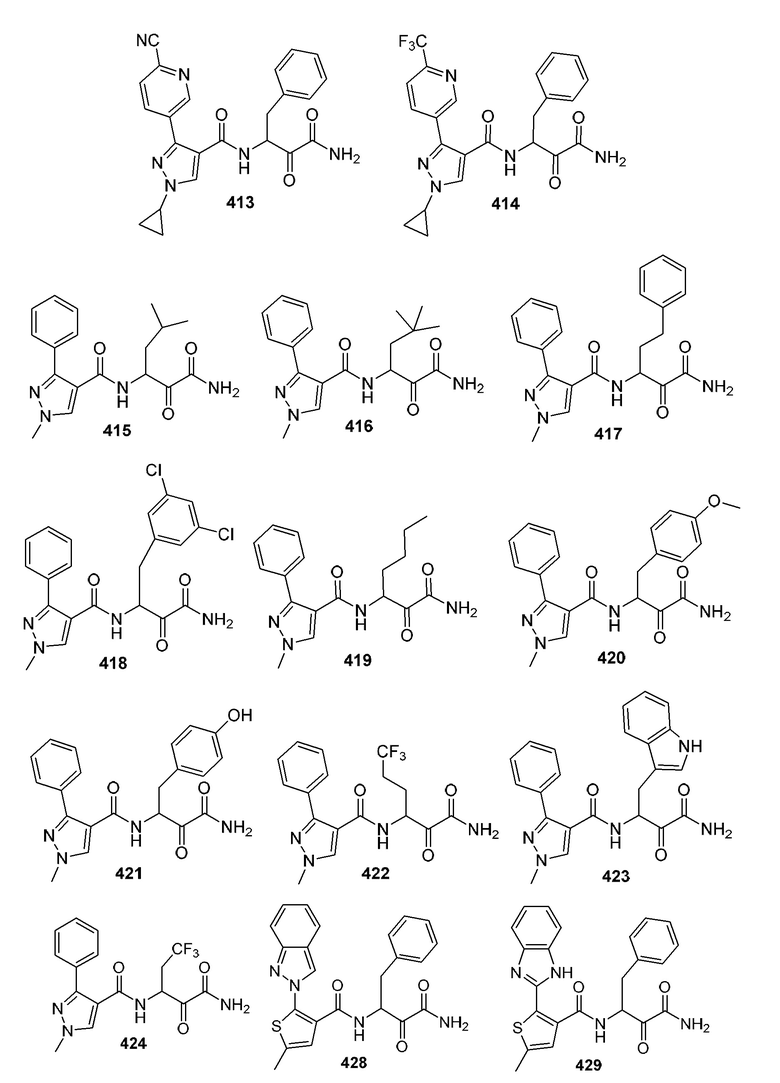
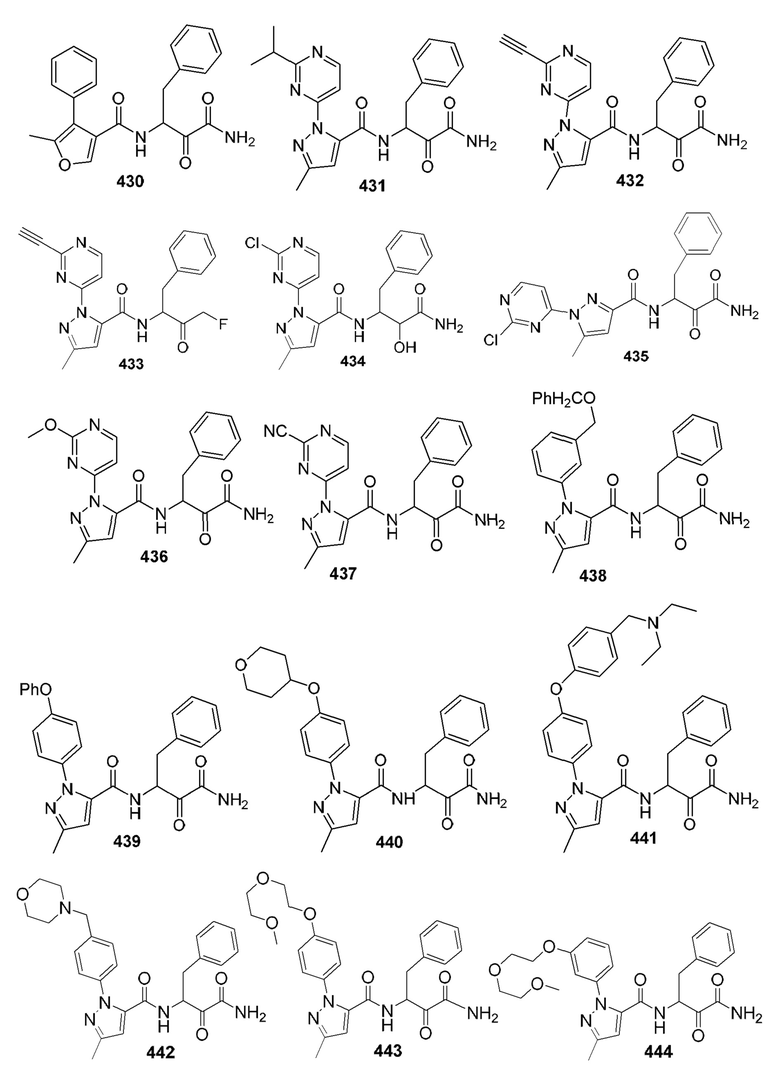
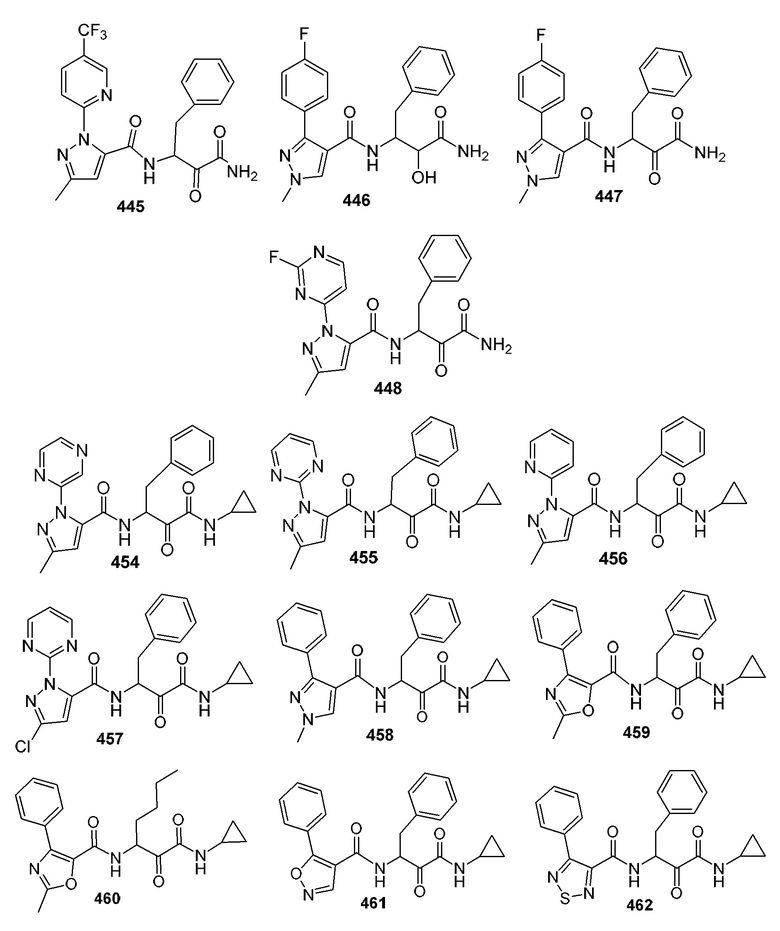
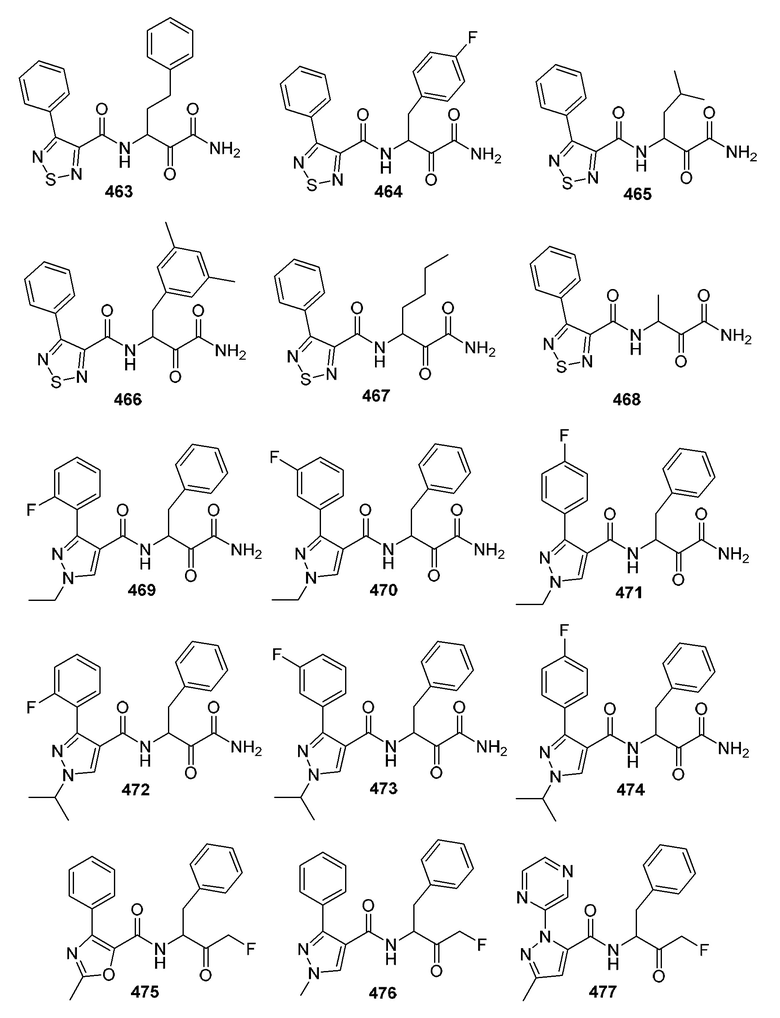
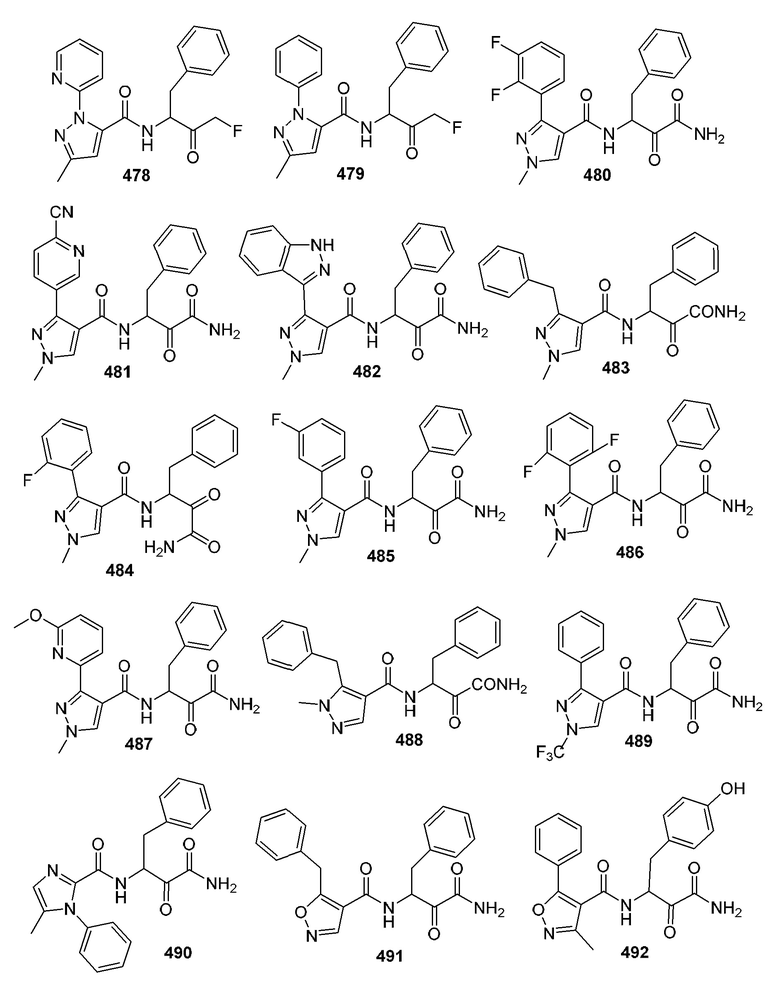
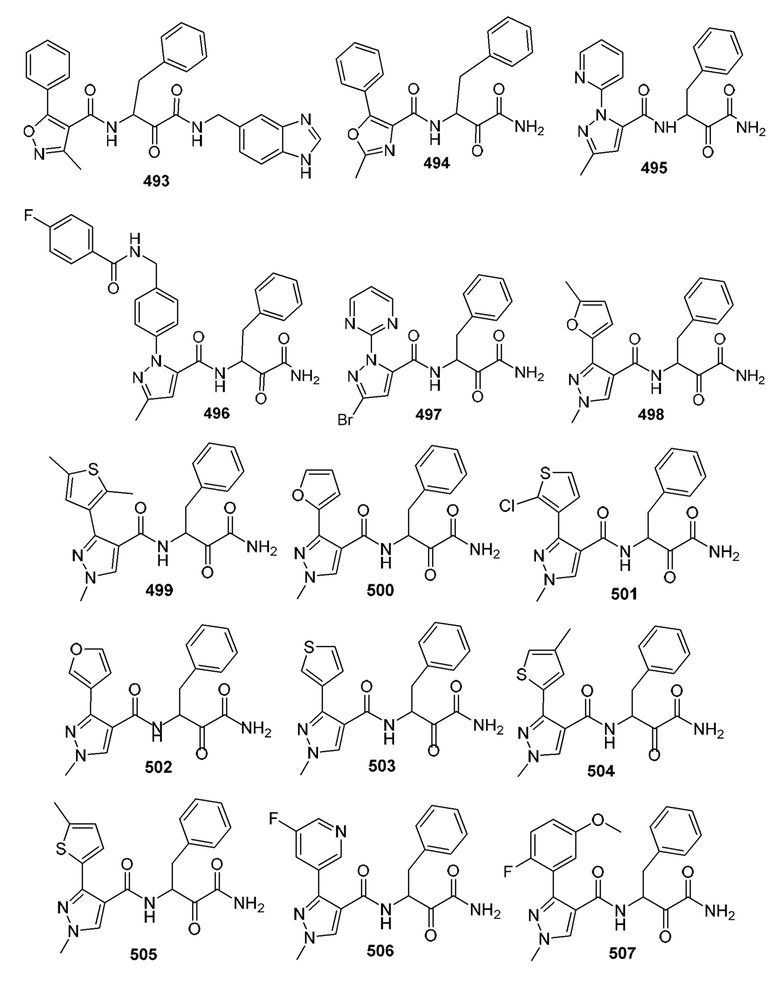
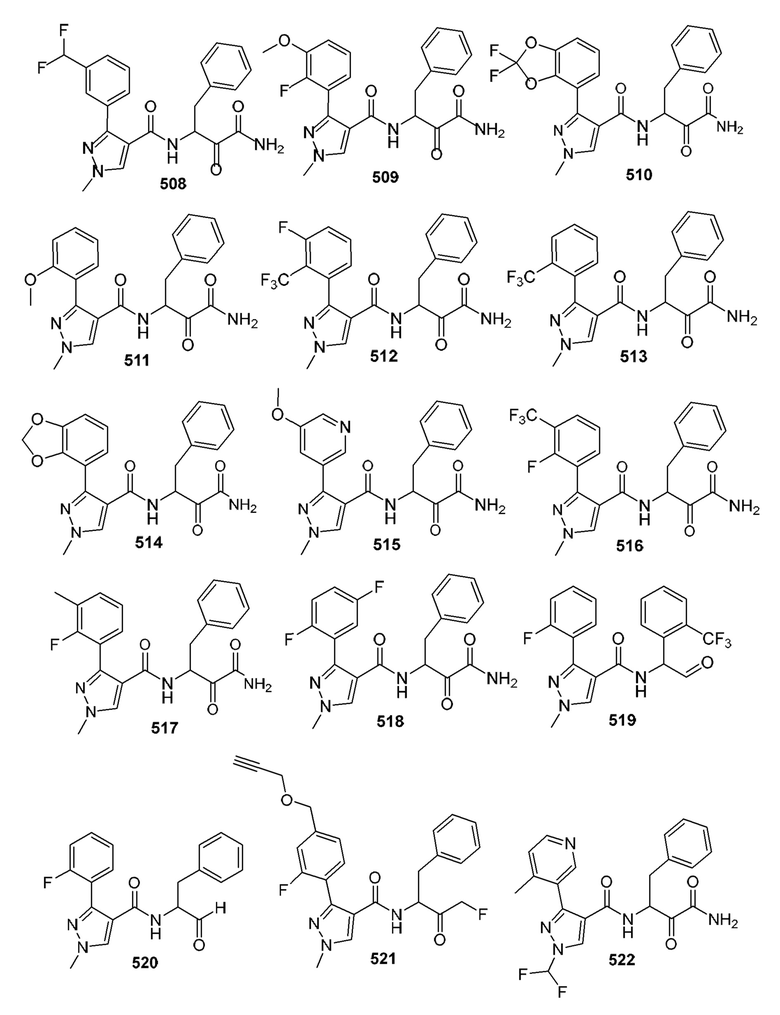
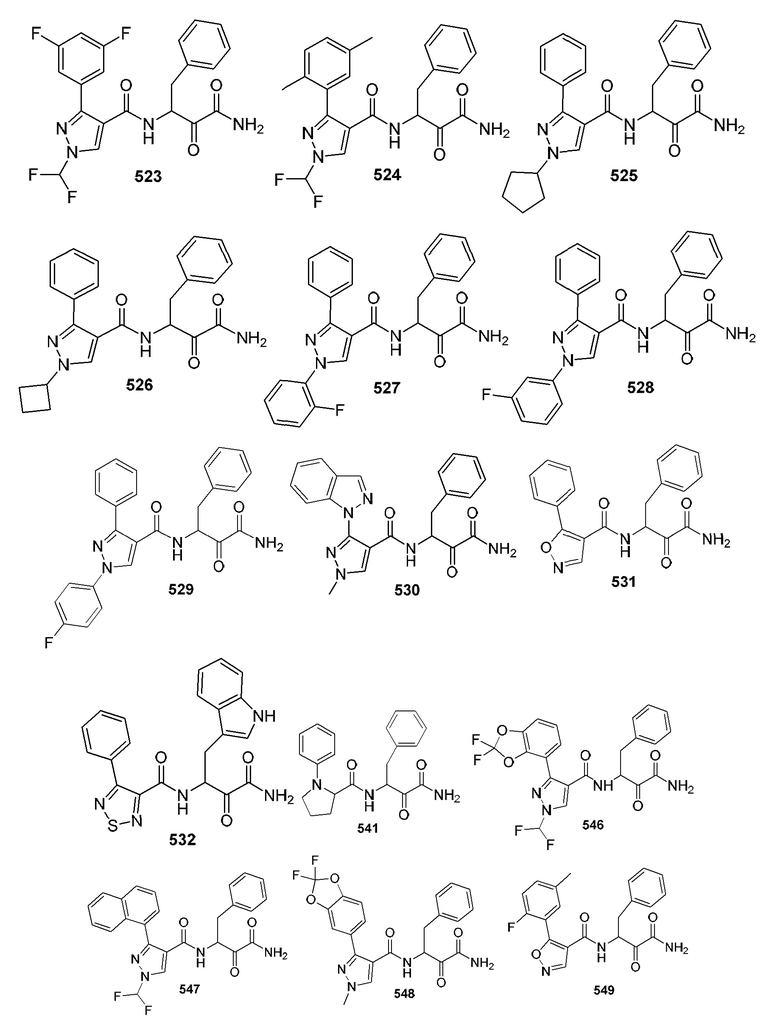
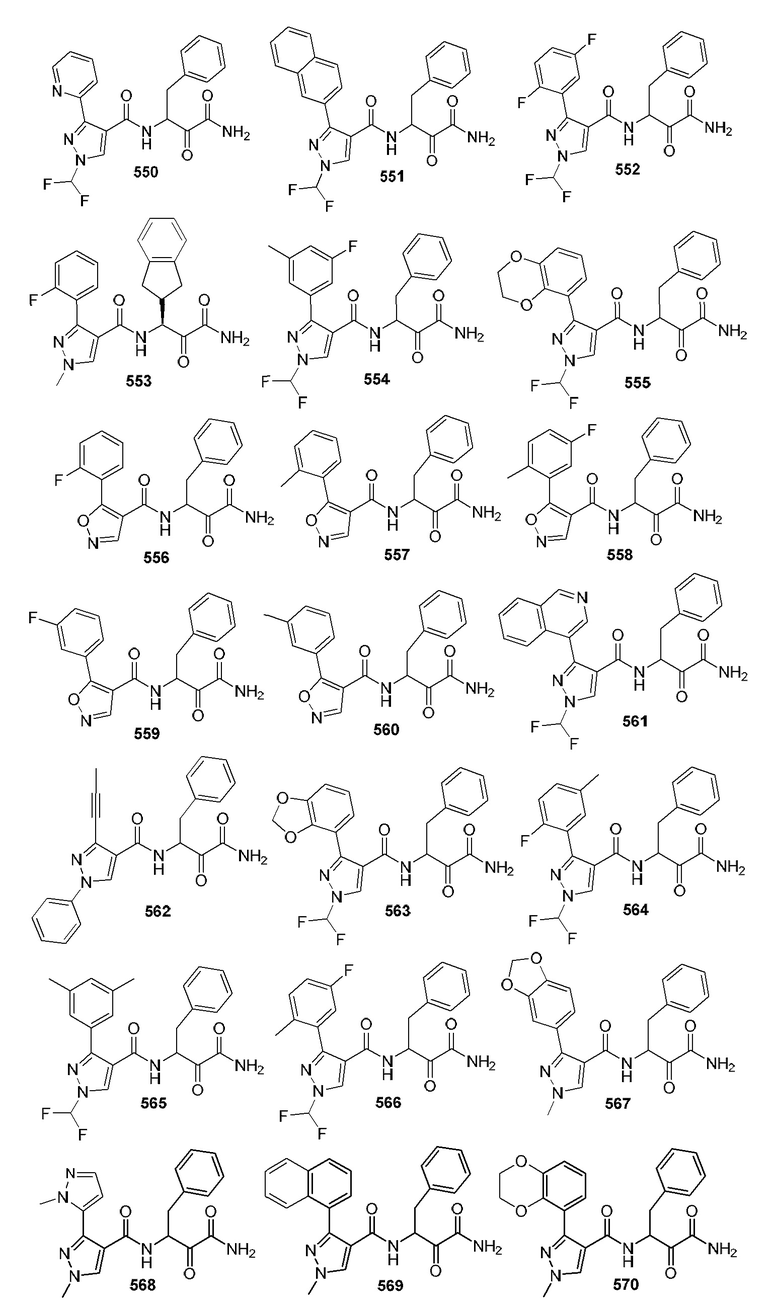
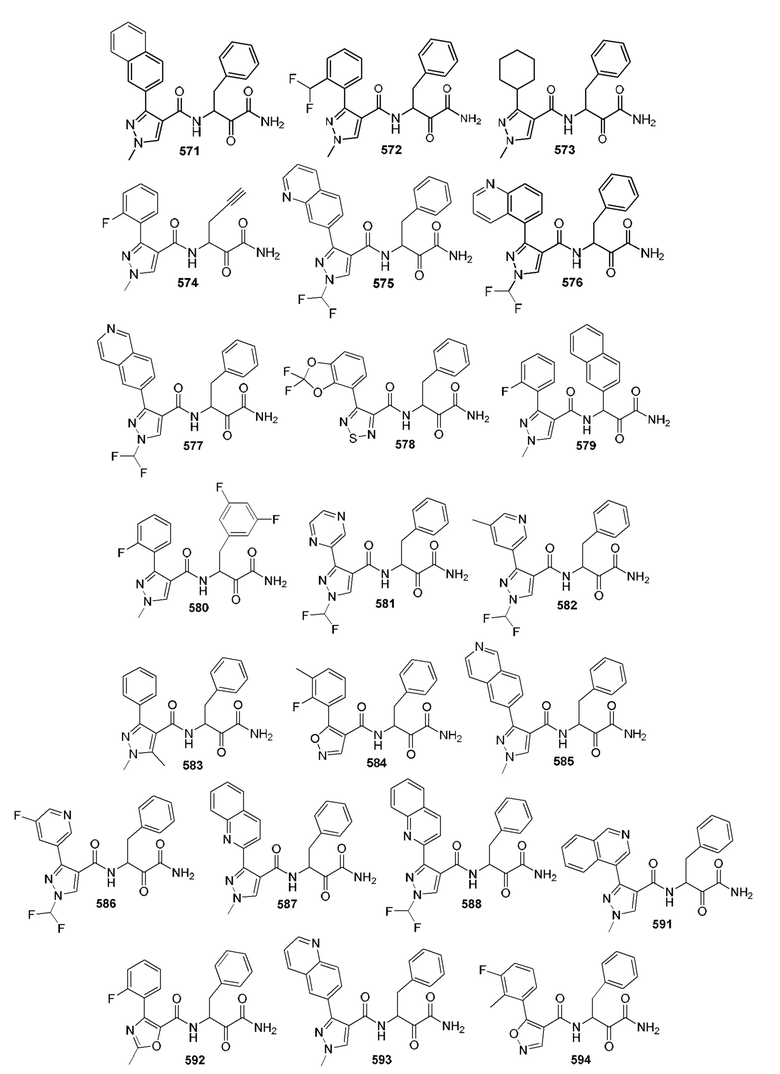
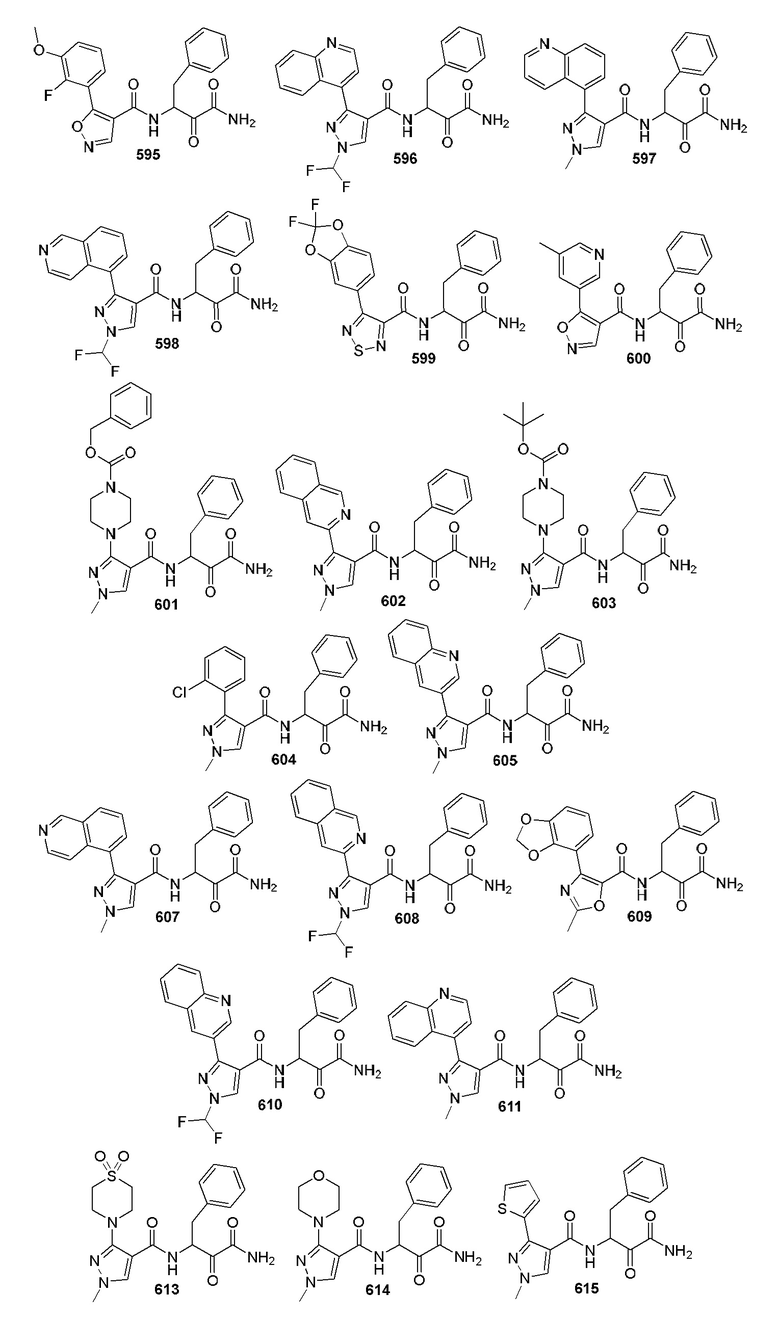
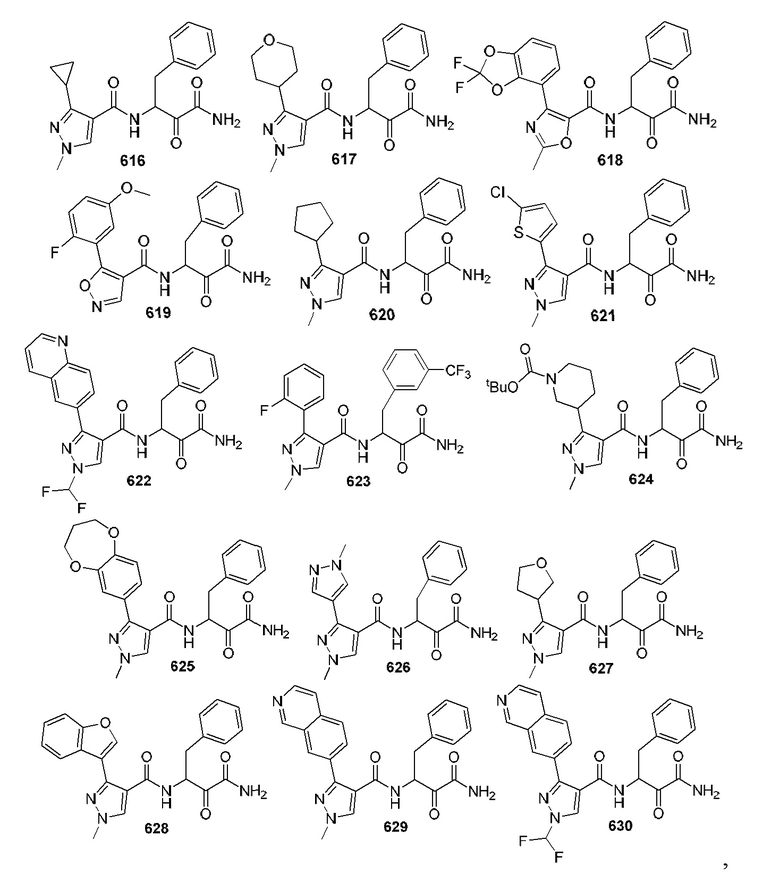
[0213] или его фармацевтически приемлемую соль. Различные варианты реализации включают S-энантиомер, R-энантиомер или рацемат указанных выше соединений.
[0214] Дополнительные соединения, подходящие для применения согласно настоящему описанию, которые также могут быть получены с применением способов, описанных в настоящем описании, представлены в таблице 1.
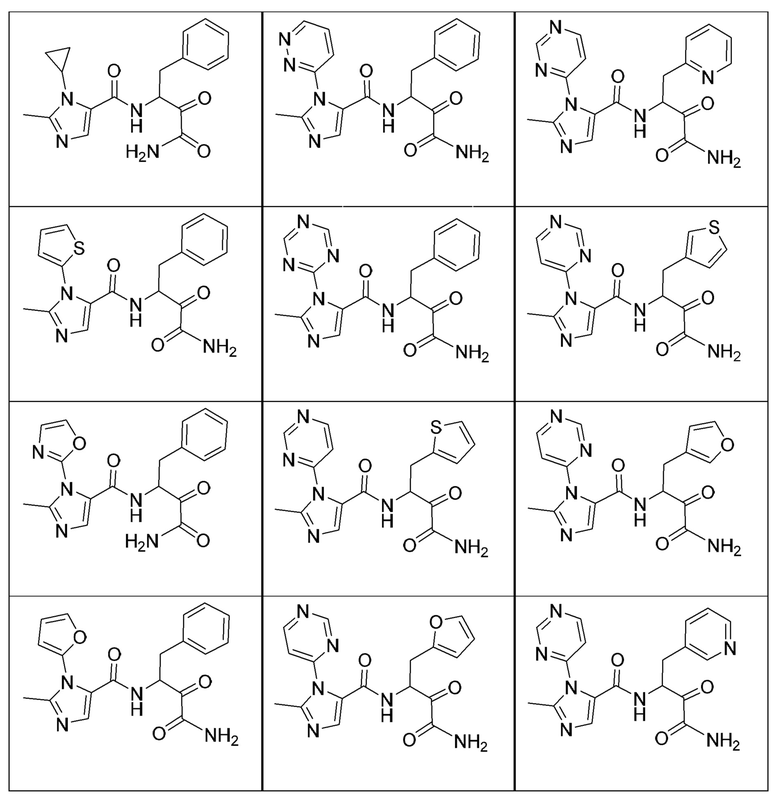
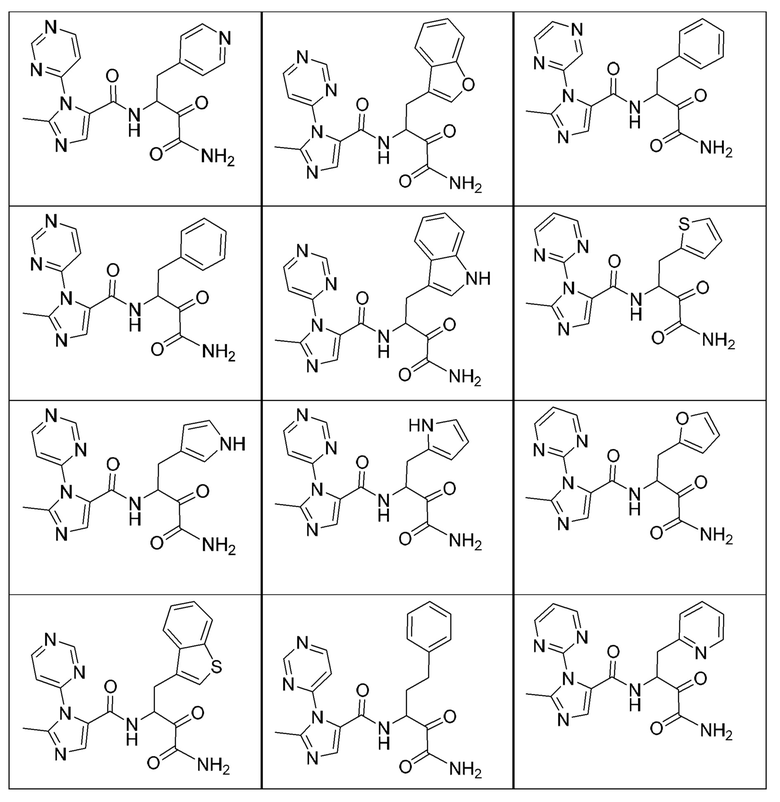
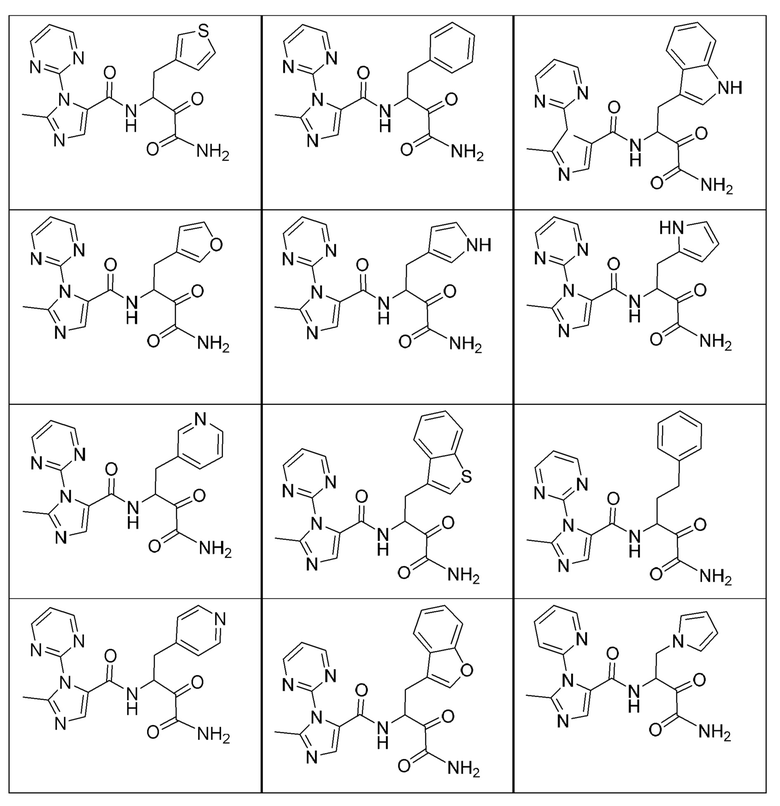
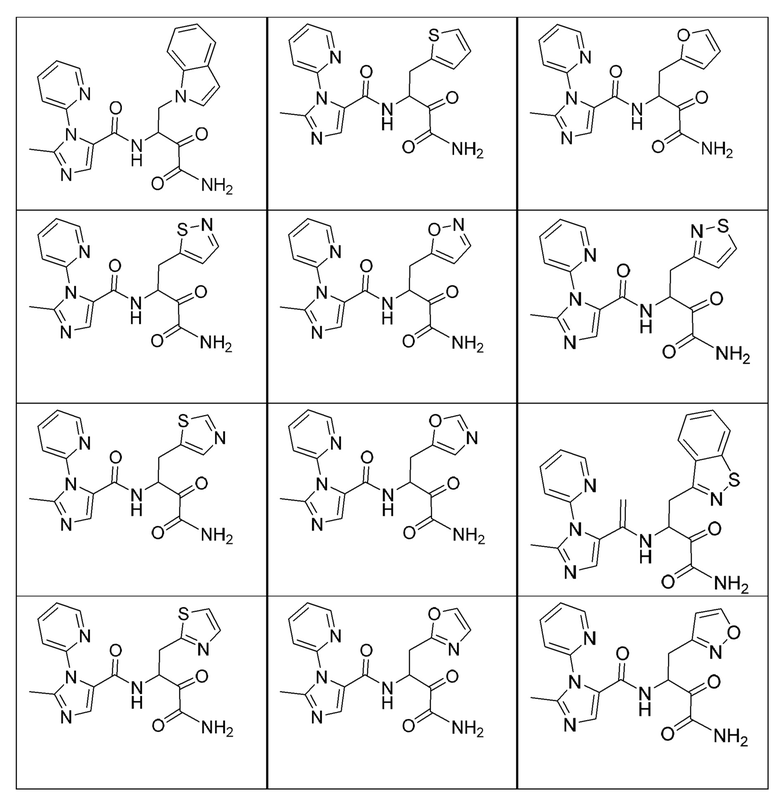
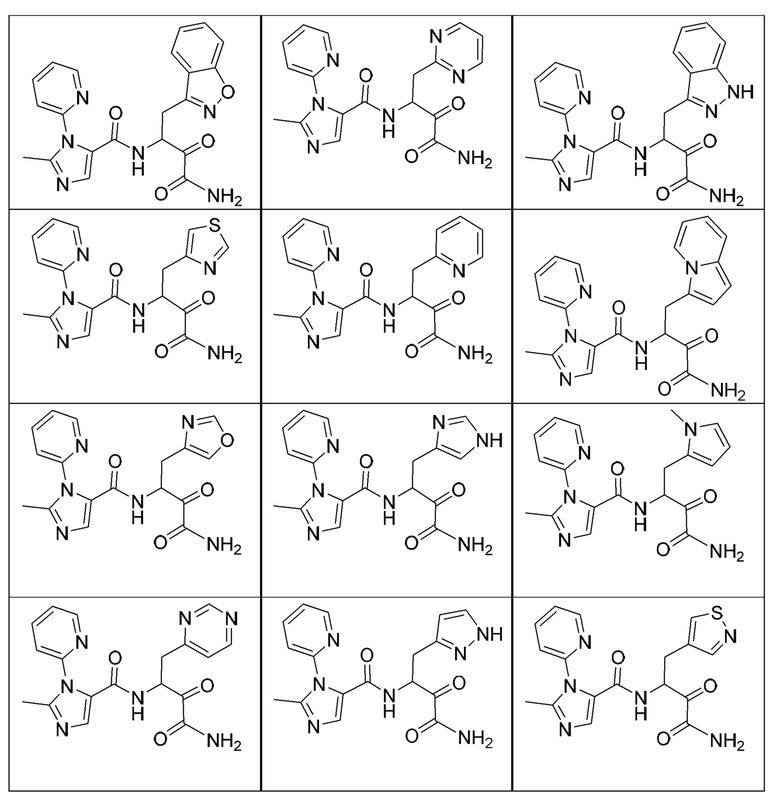
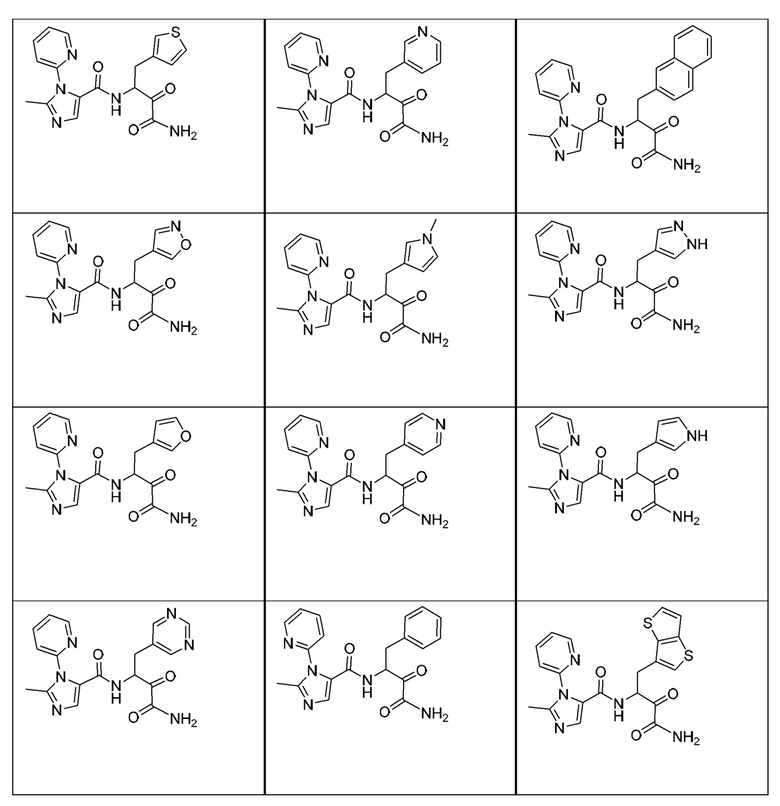
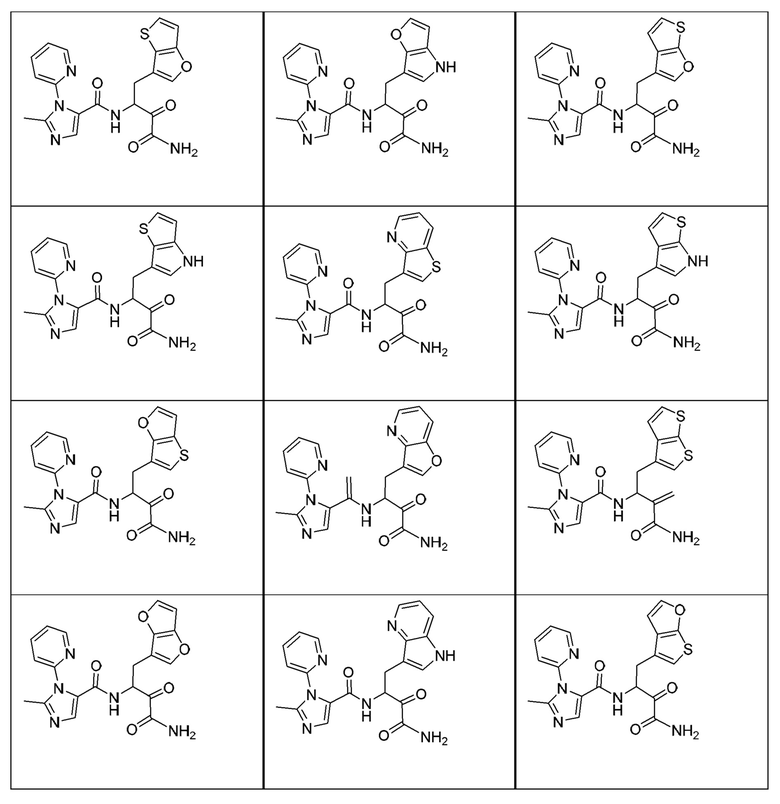
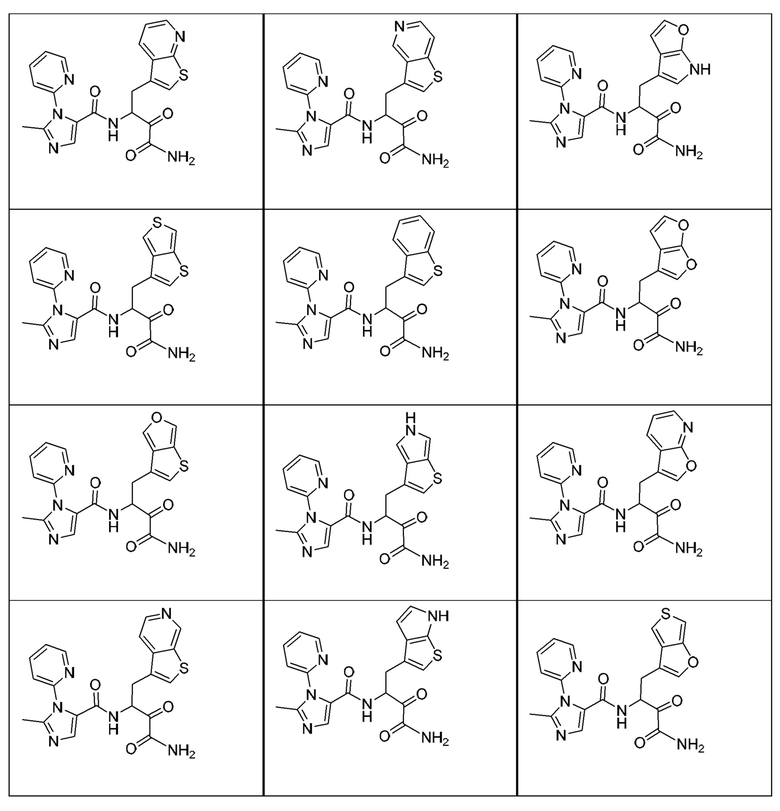
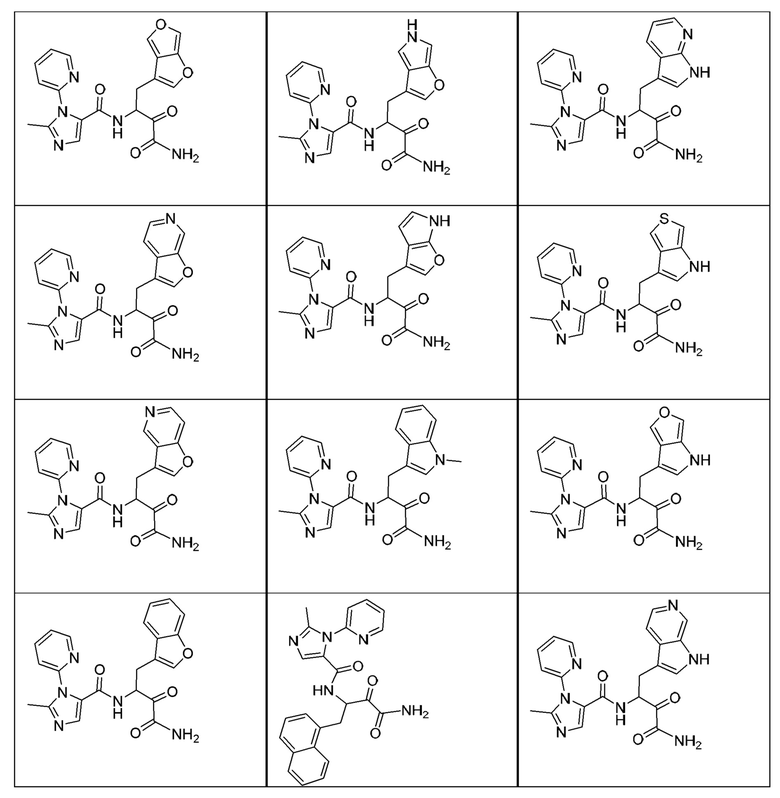
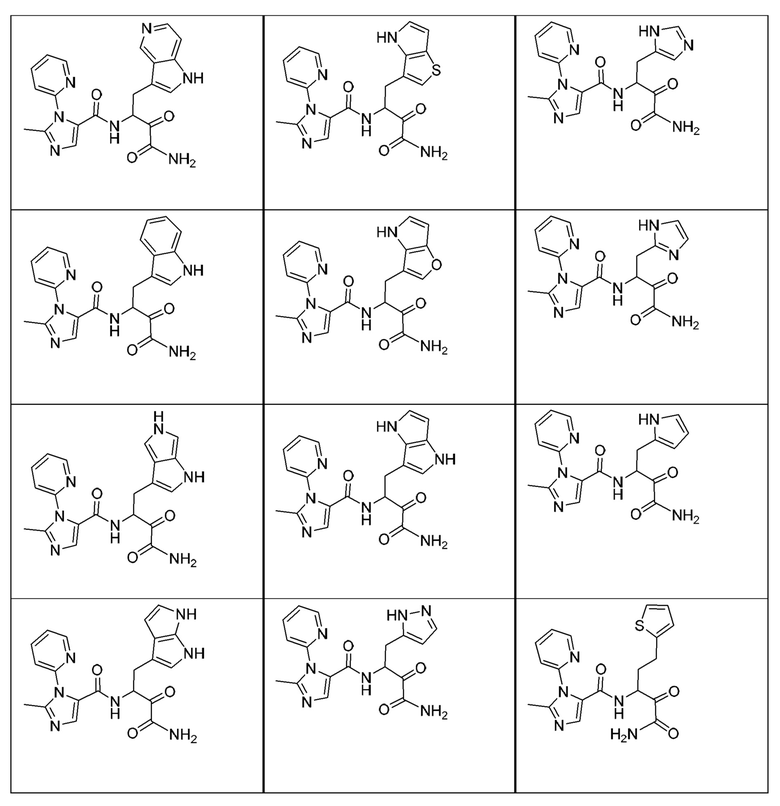
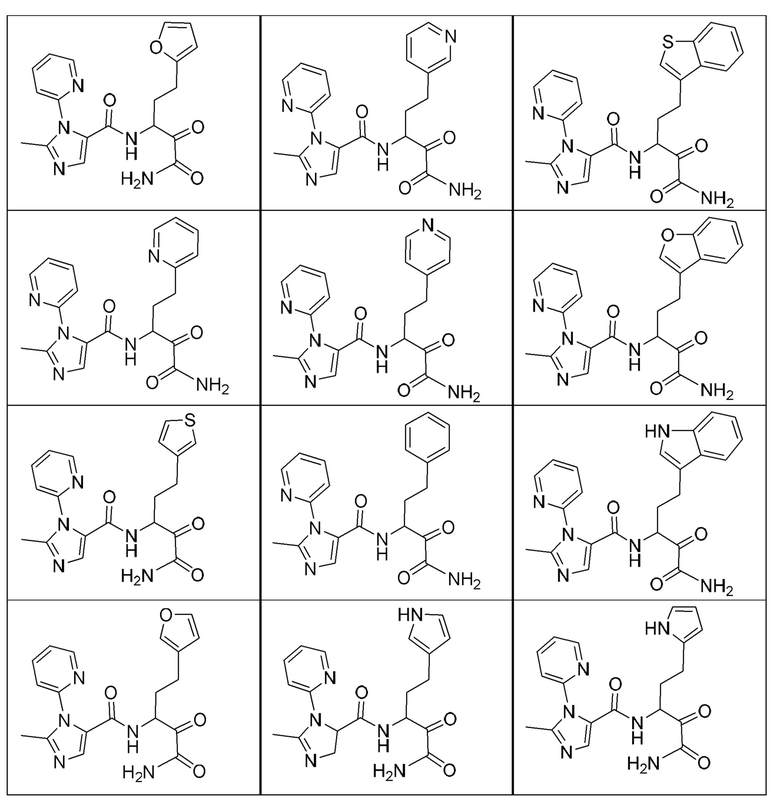
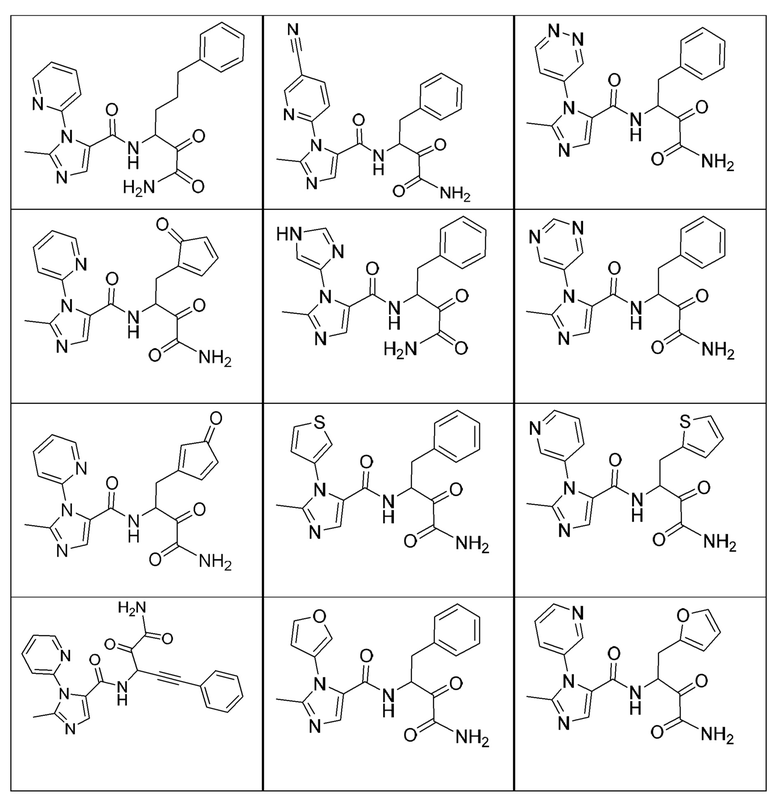
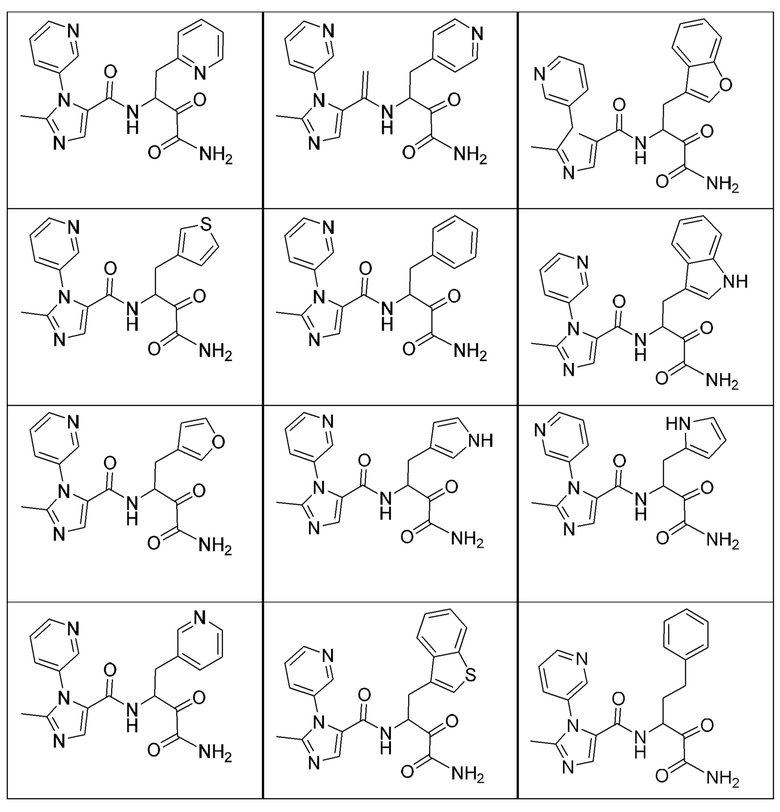
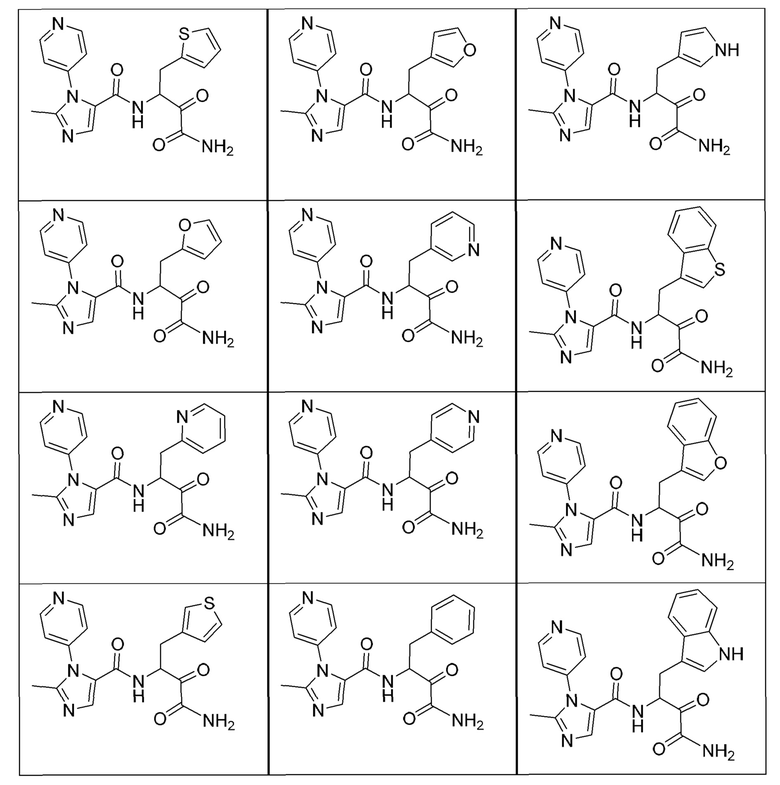
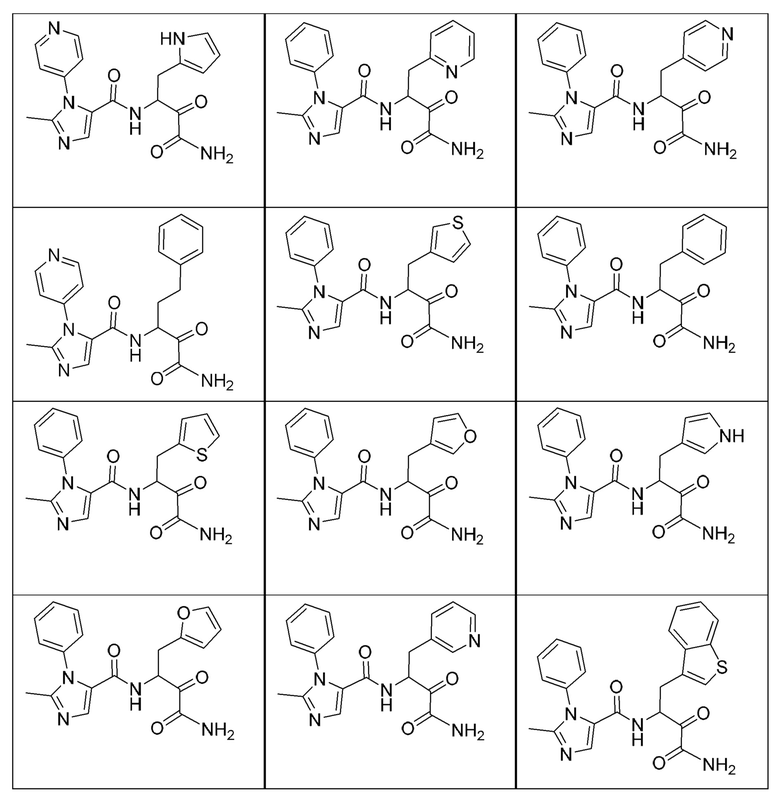
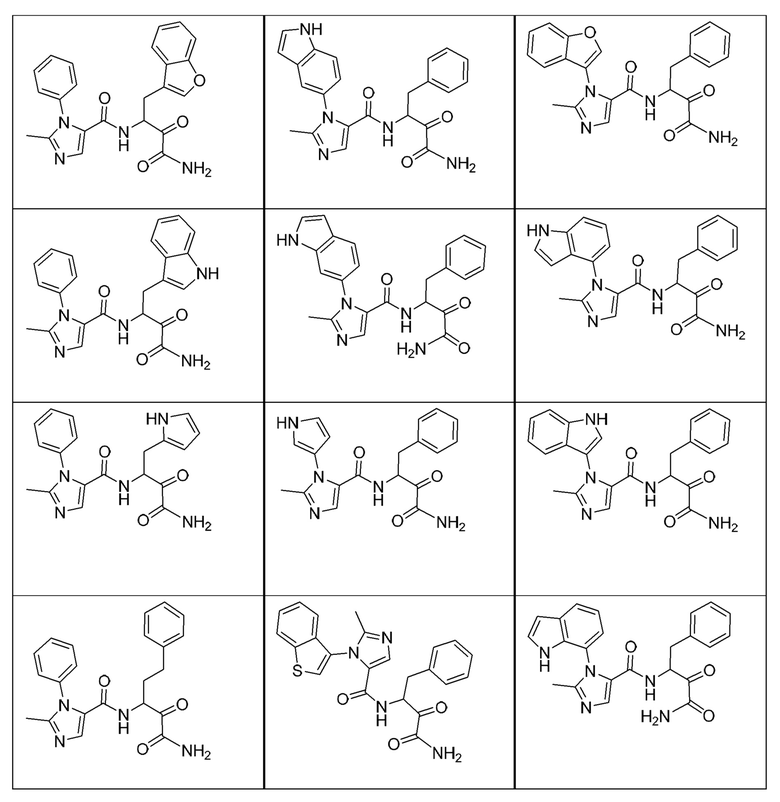
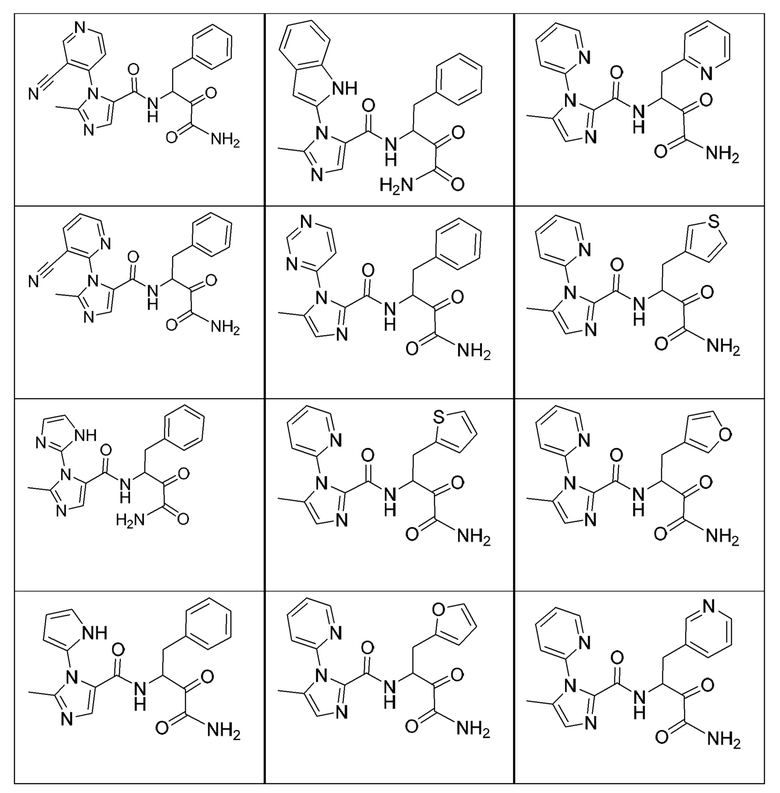
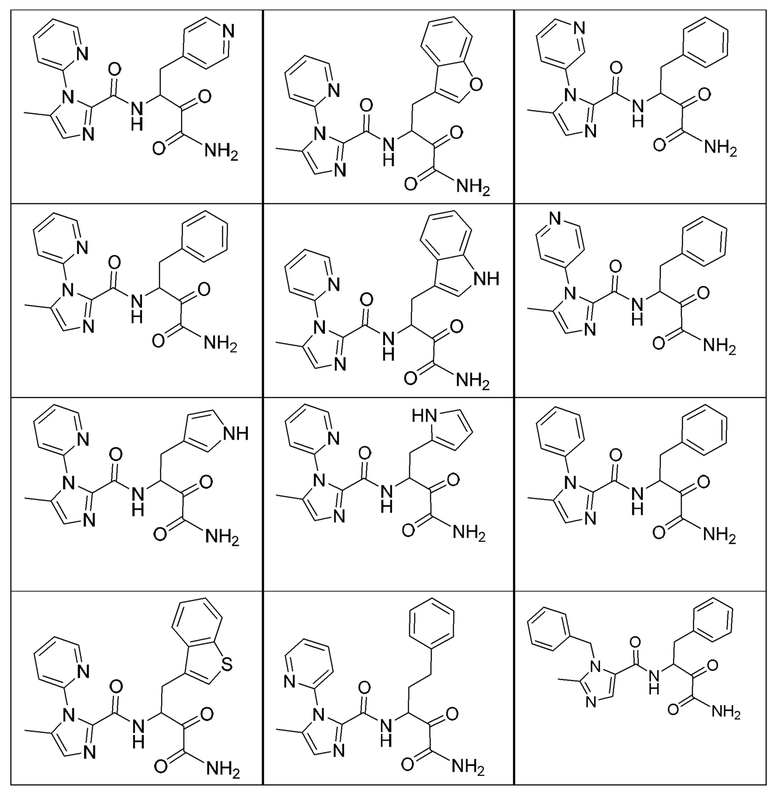
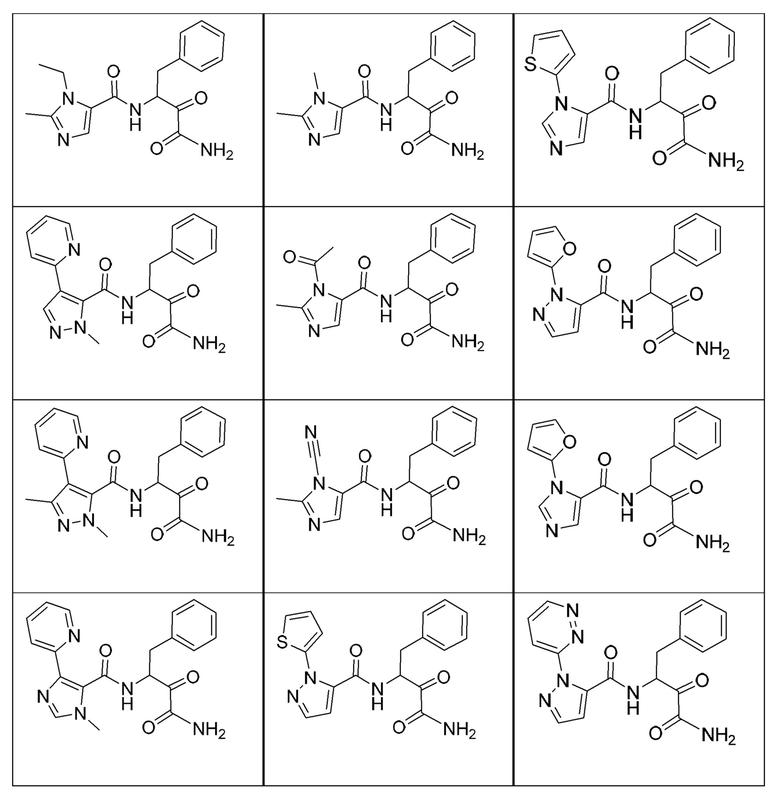
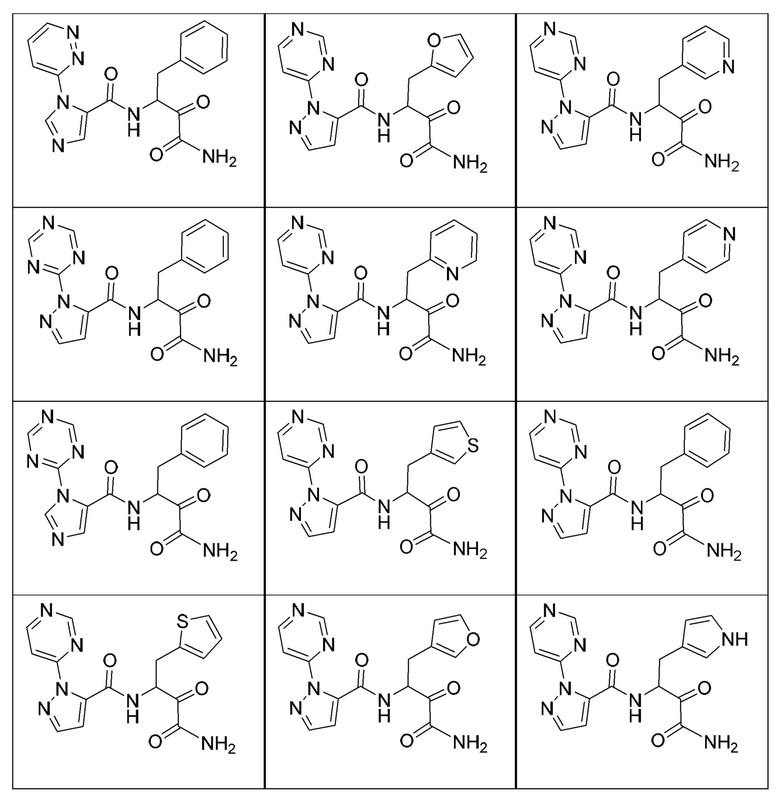
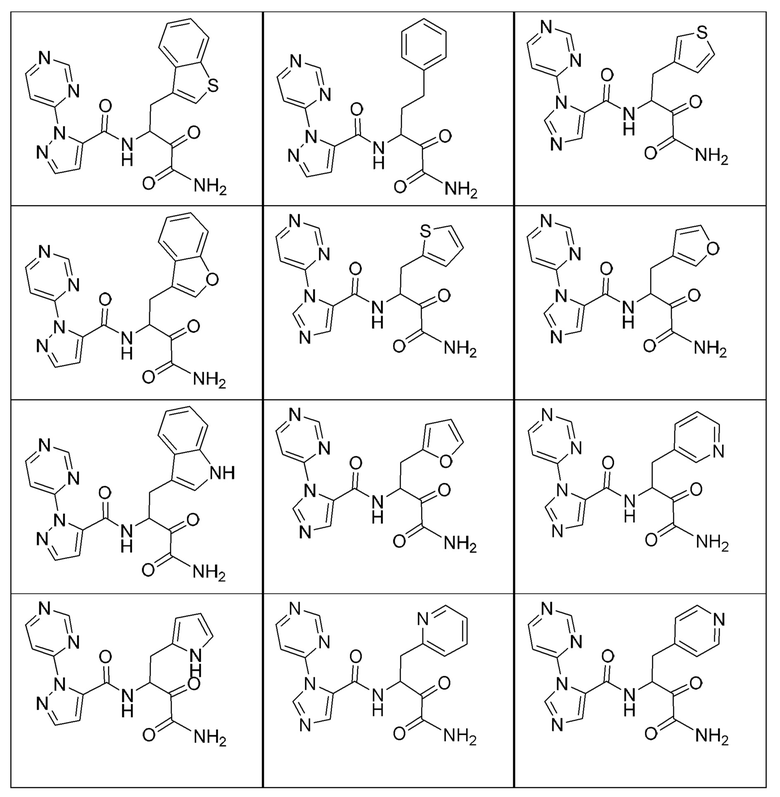
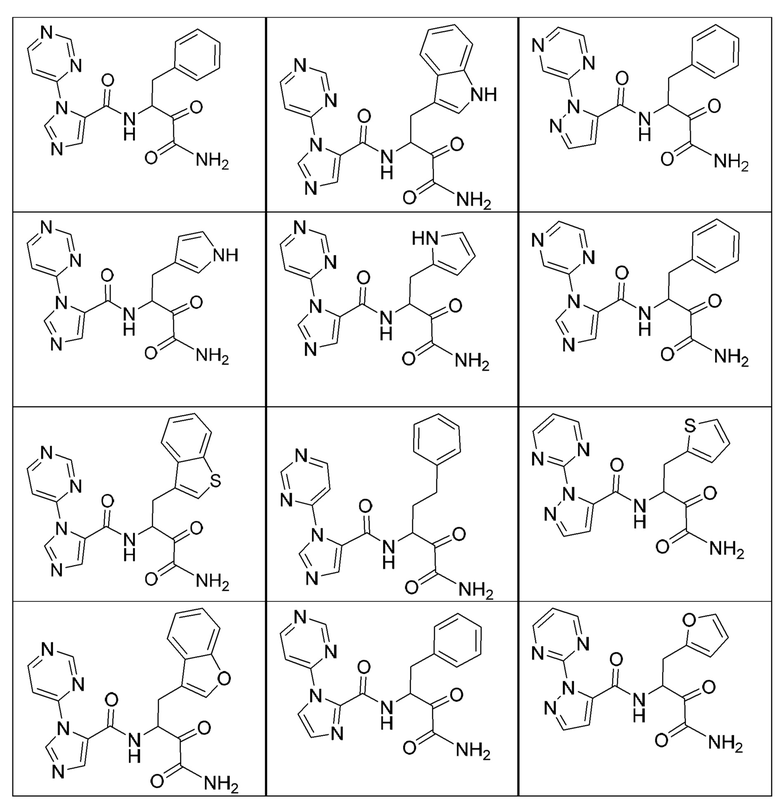
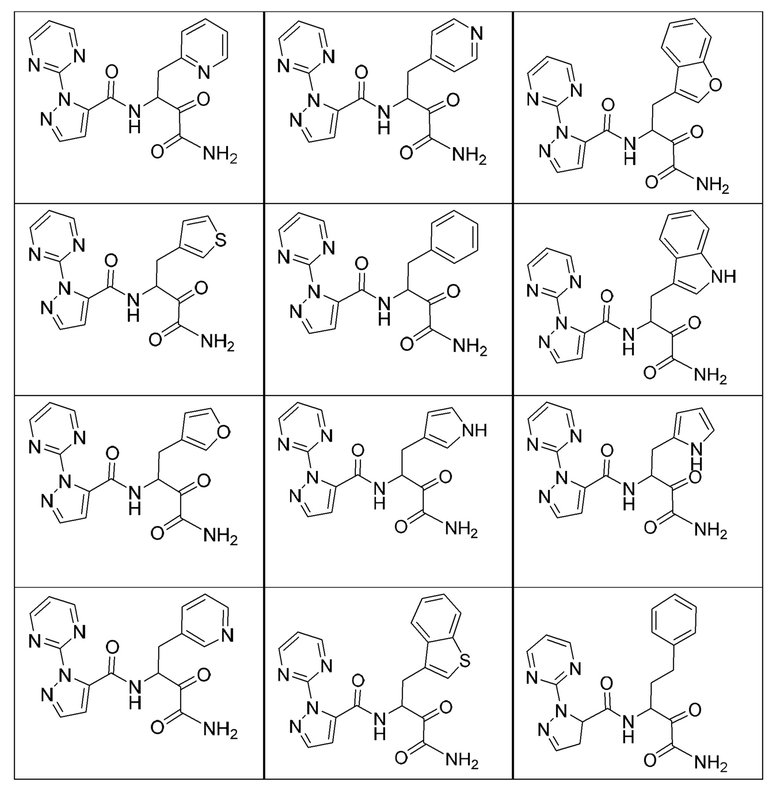
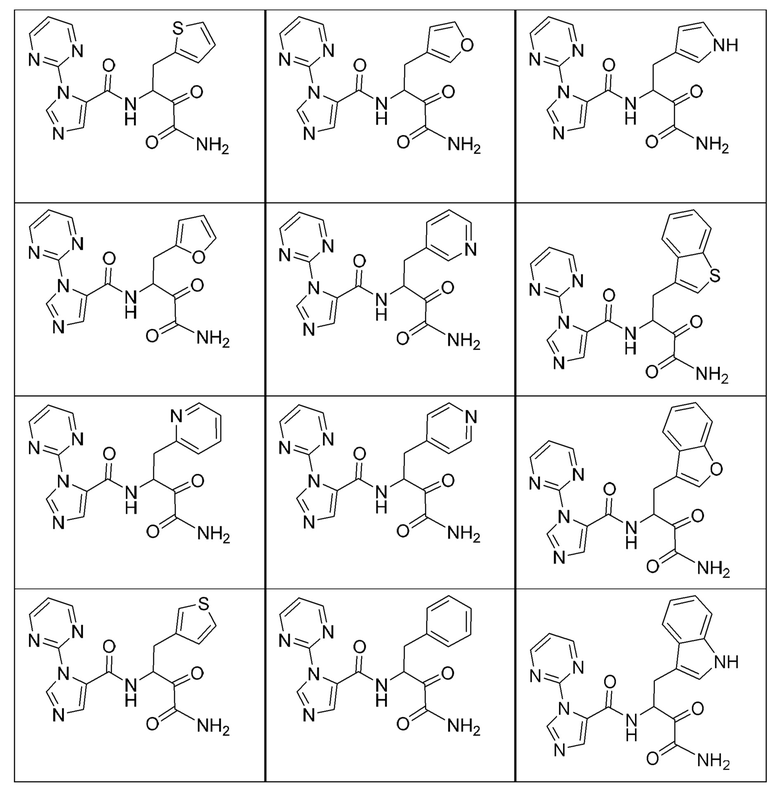
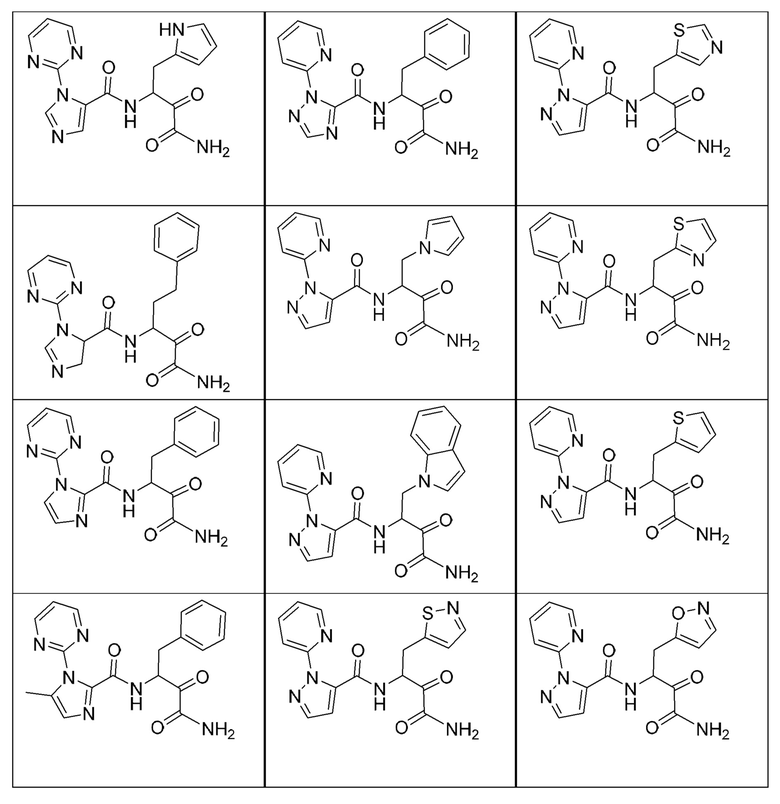
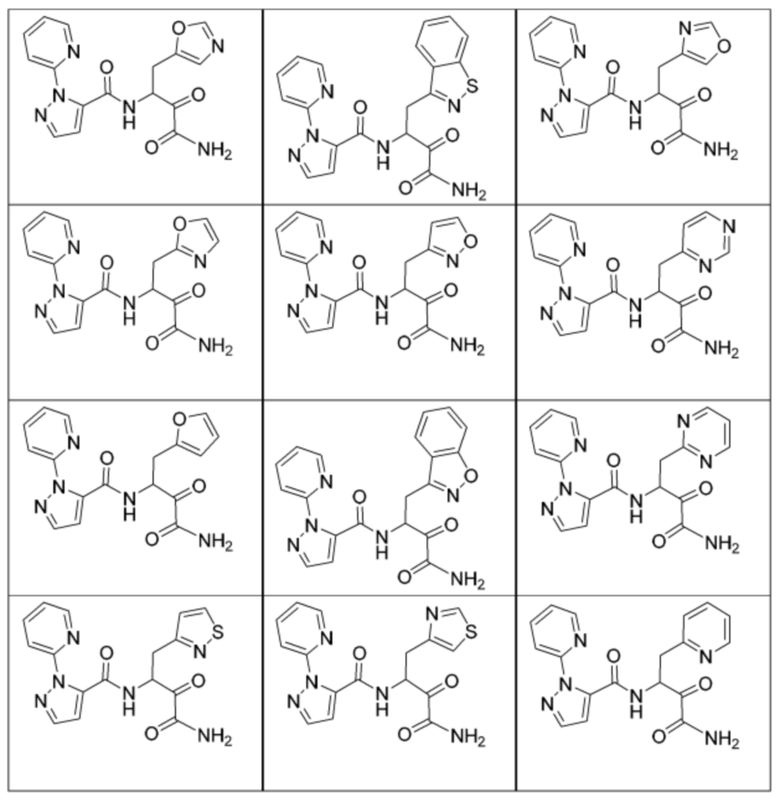
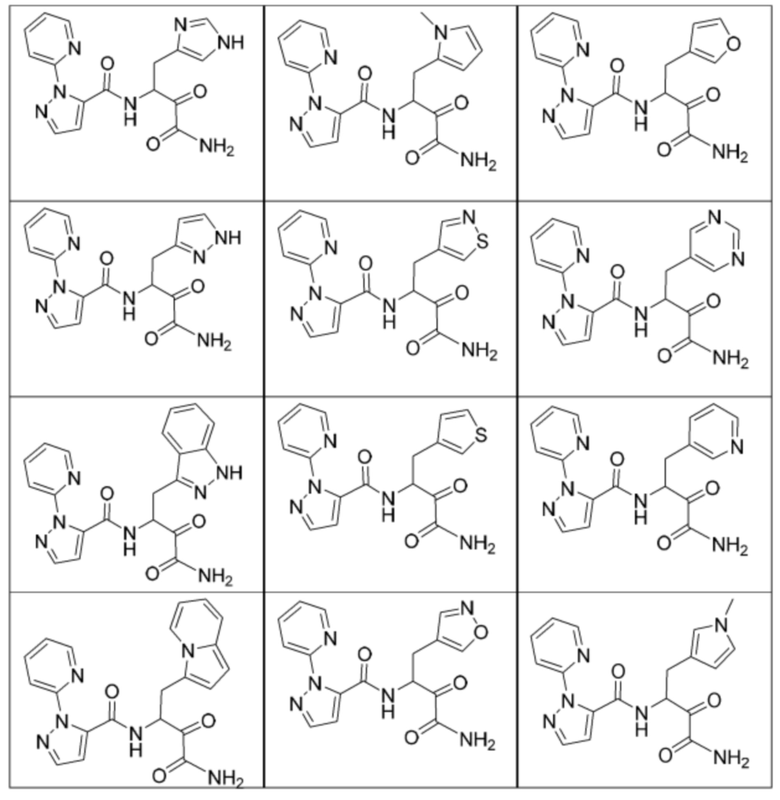
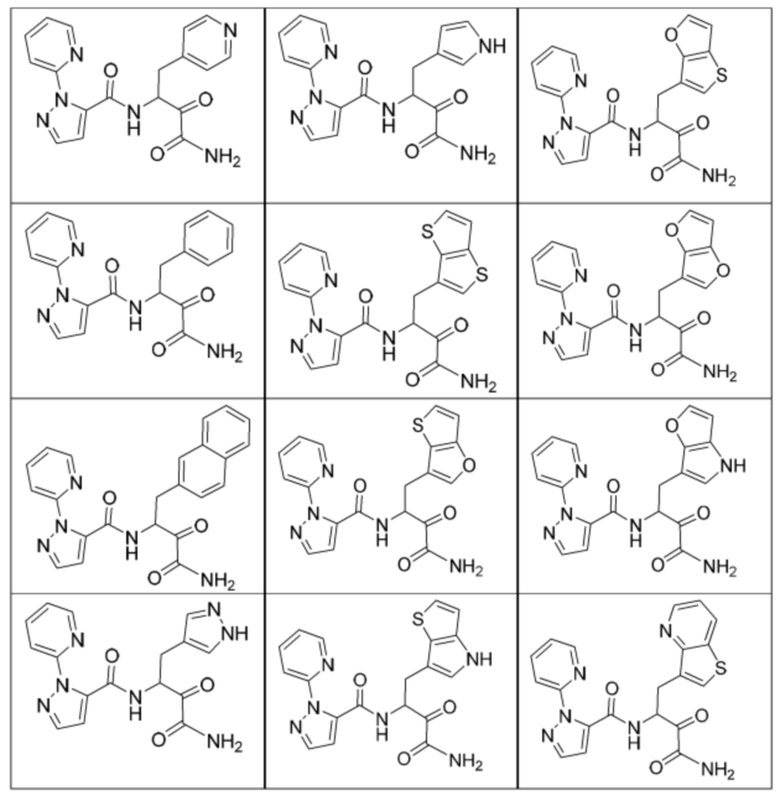
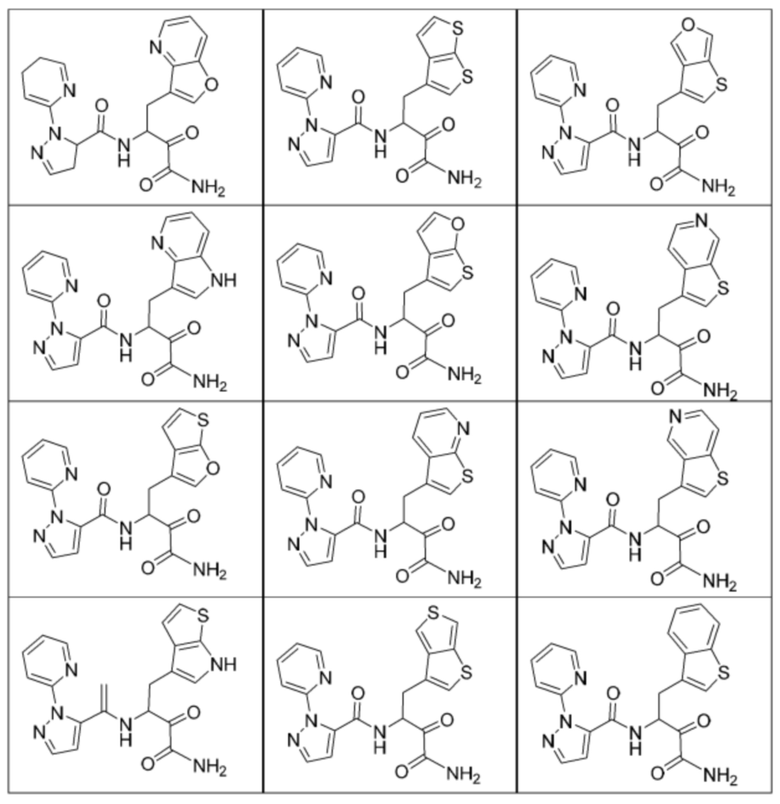
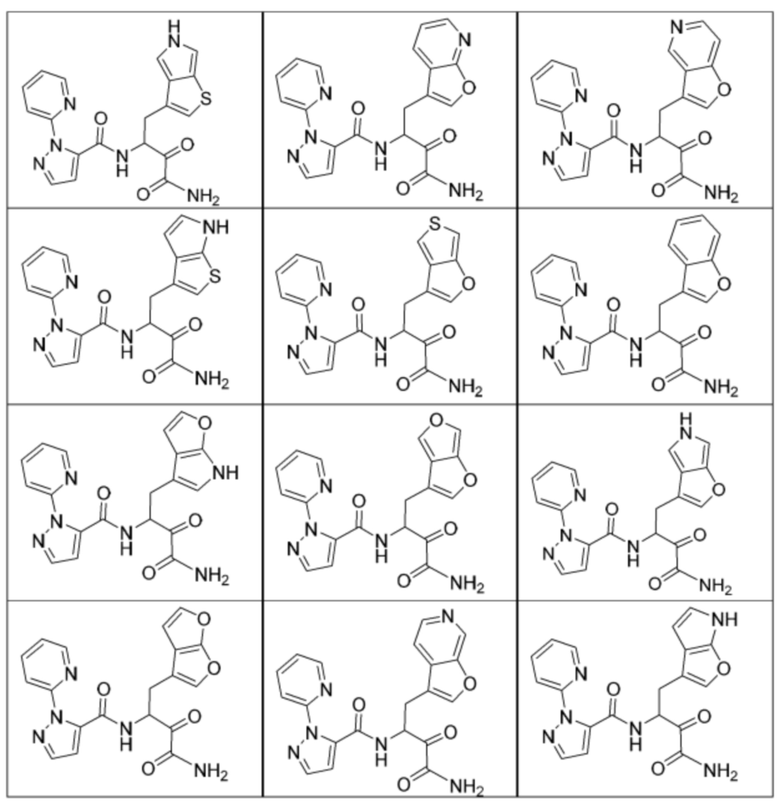
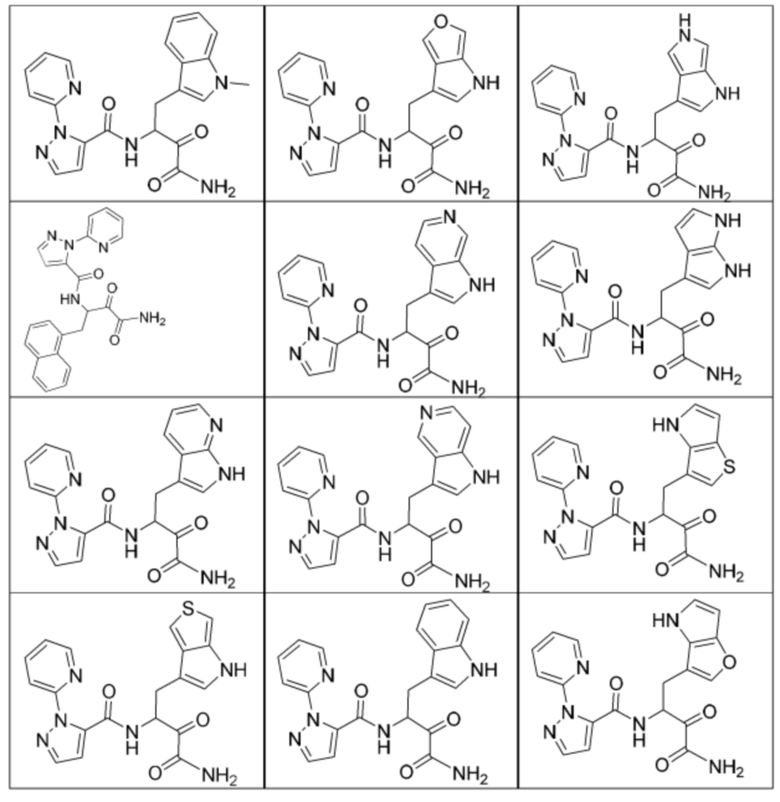
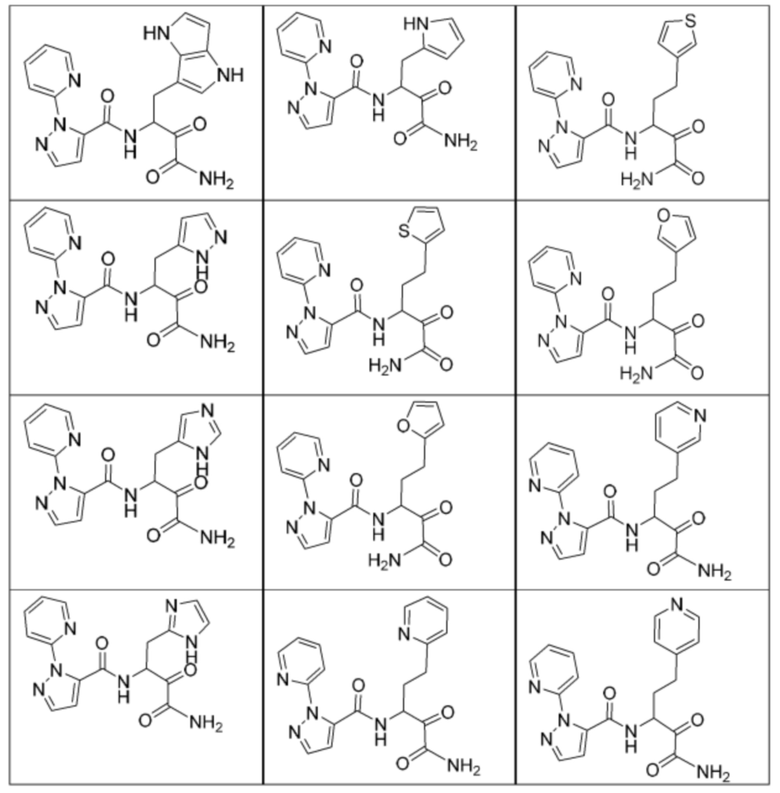
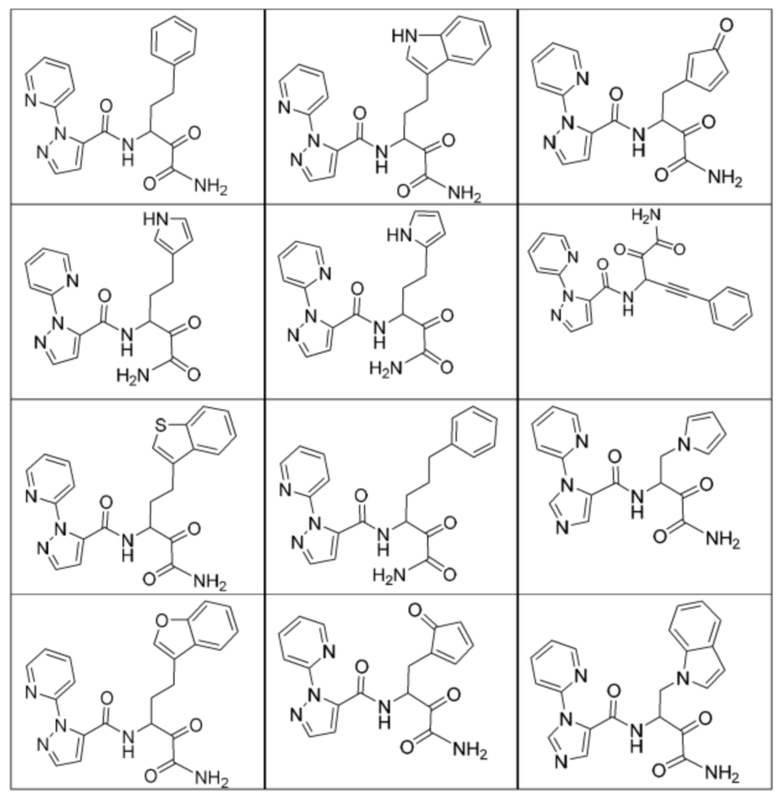
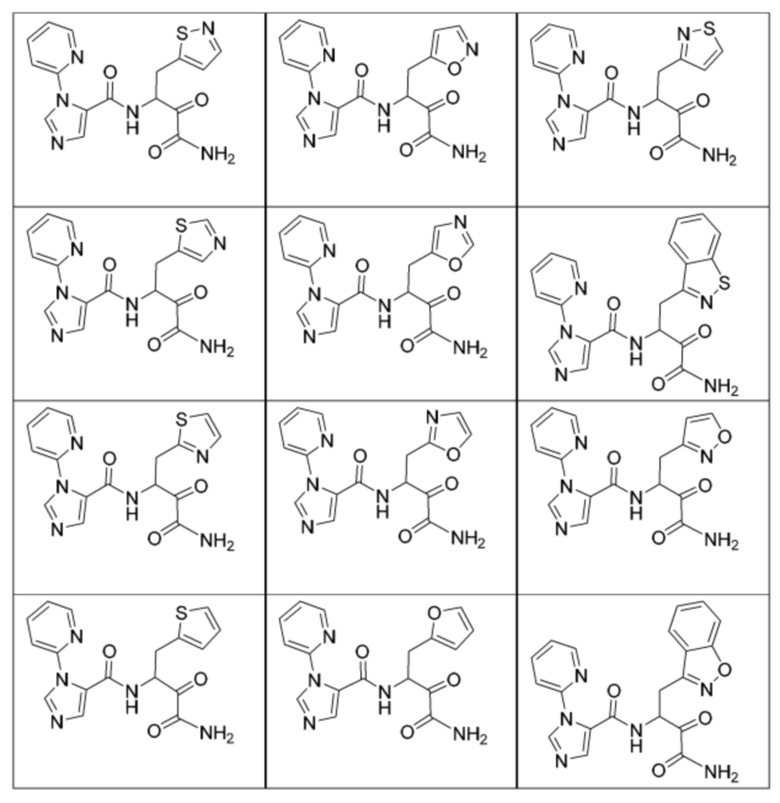
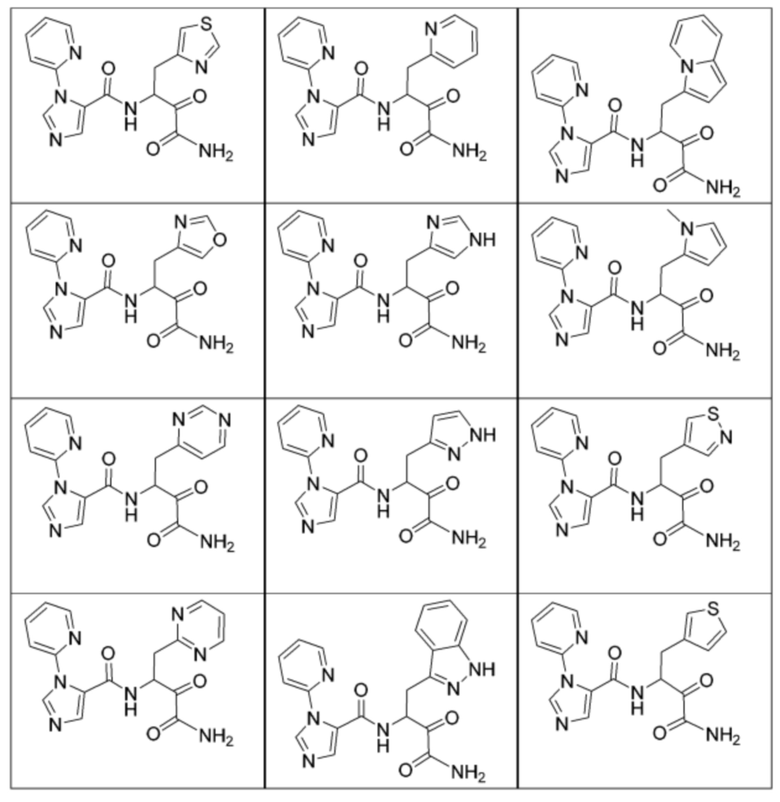
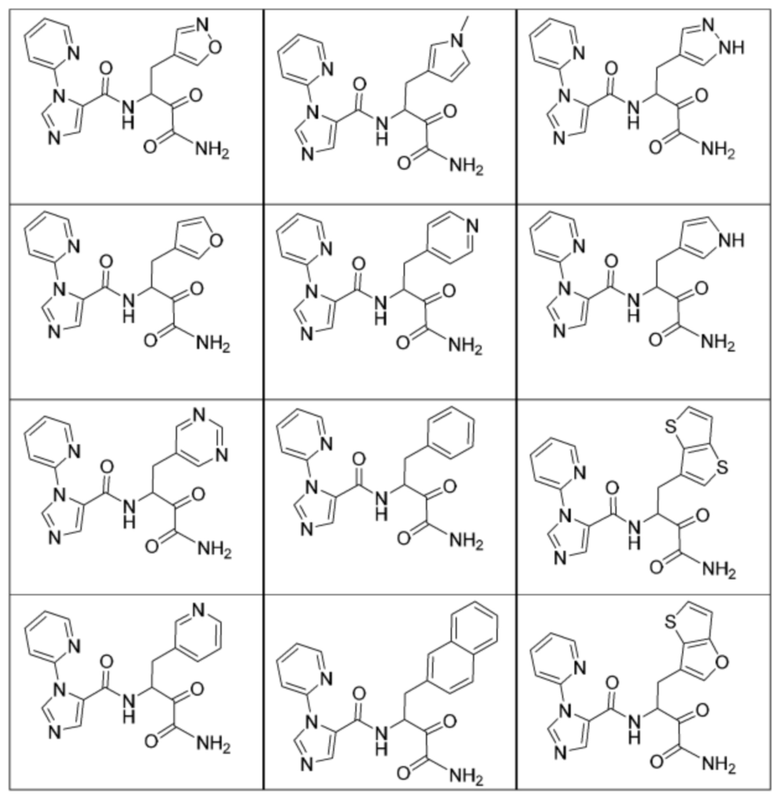
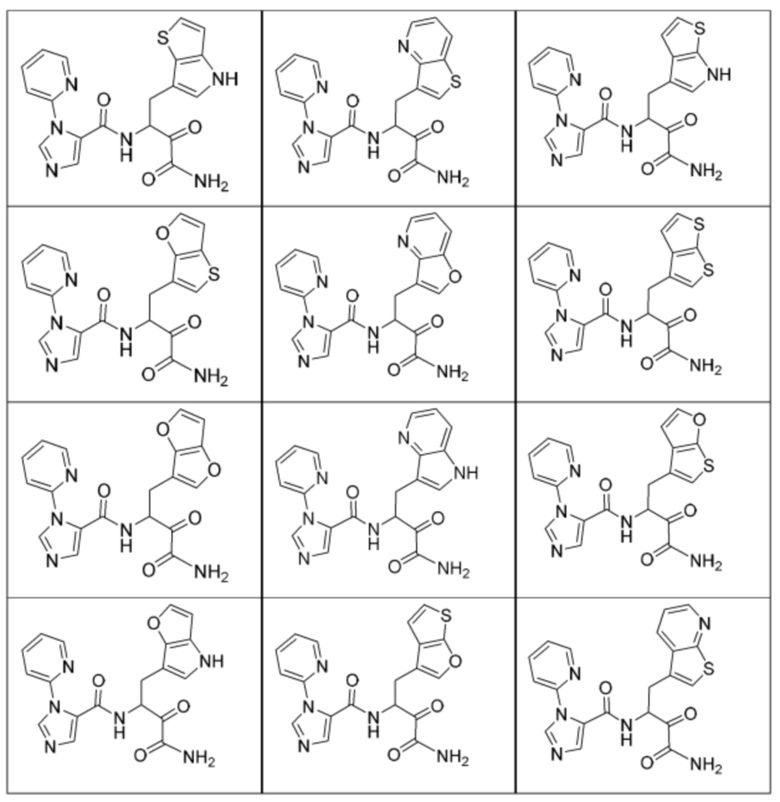
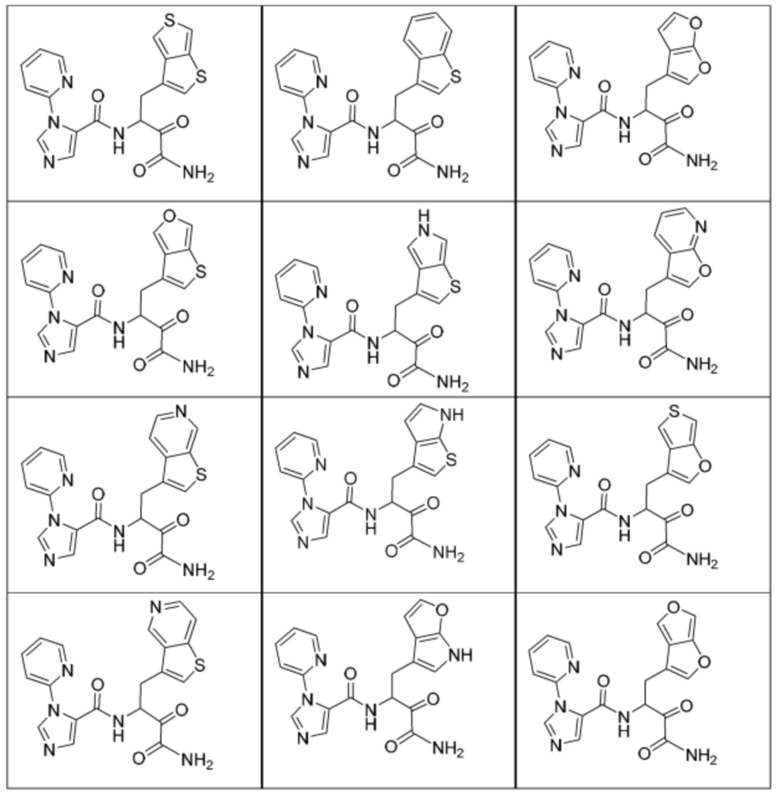
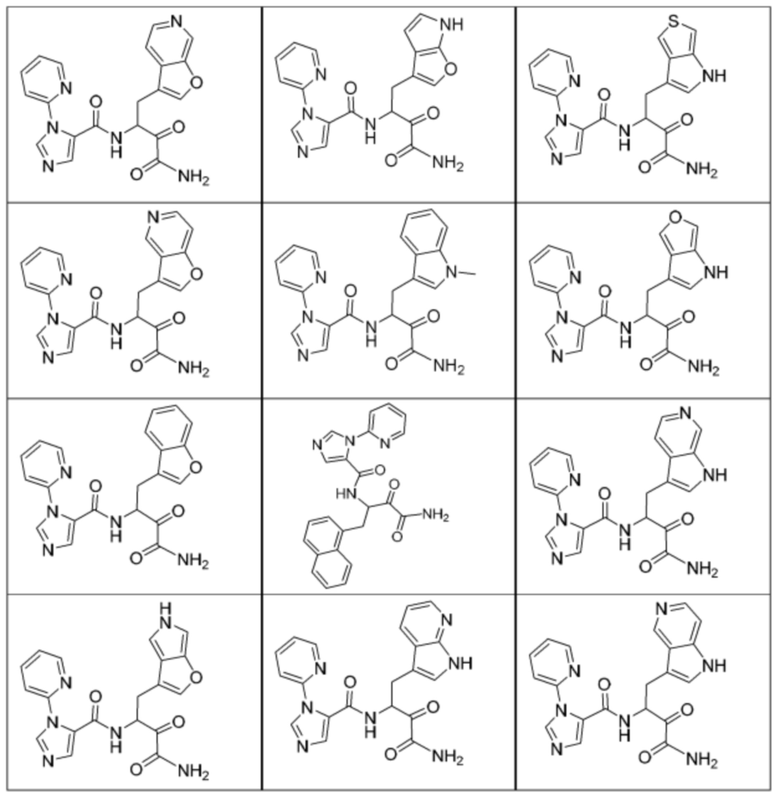
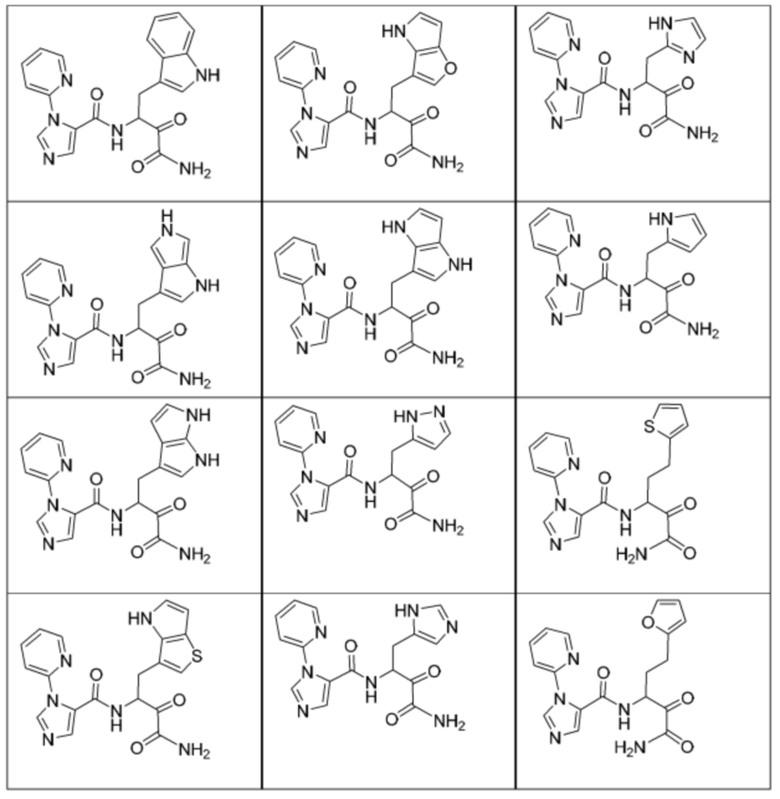
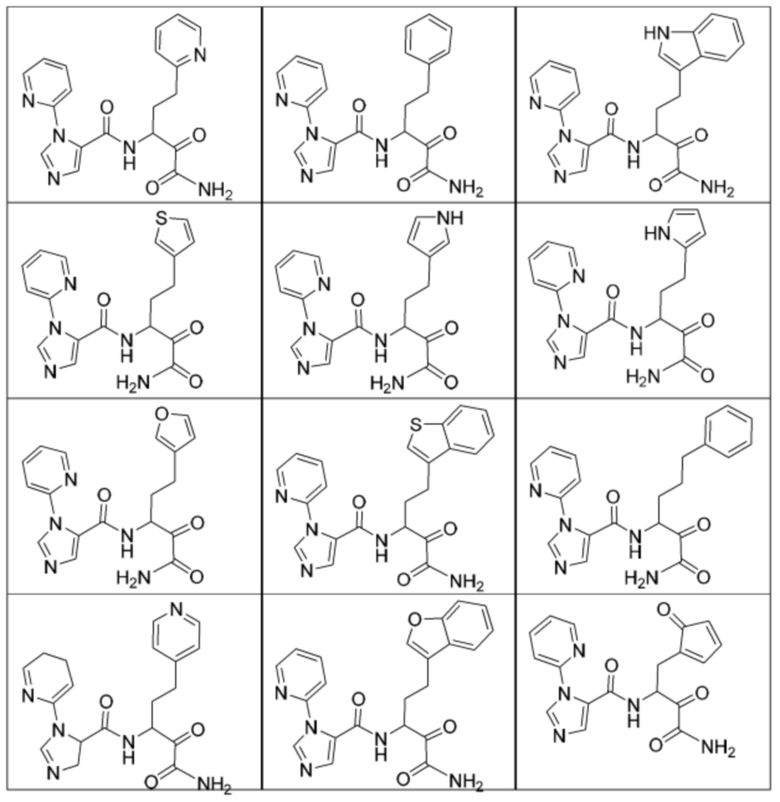

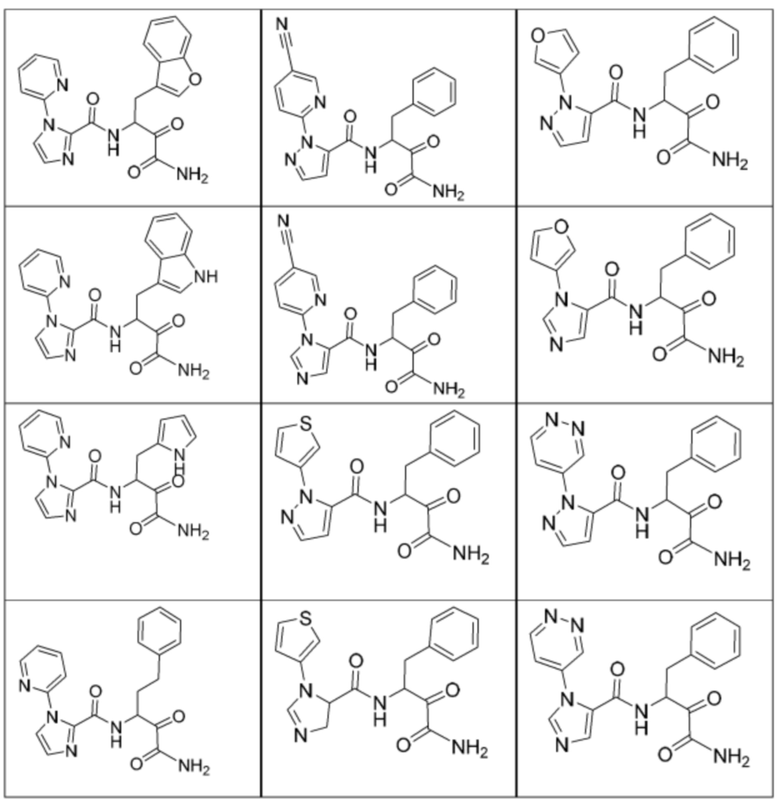
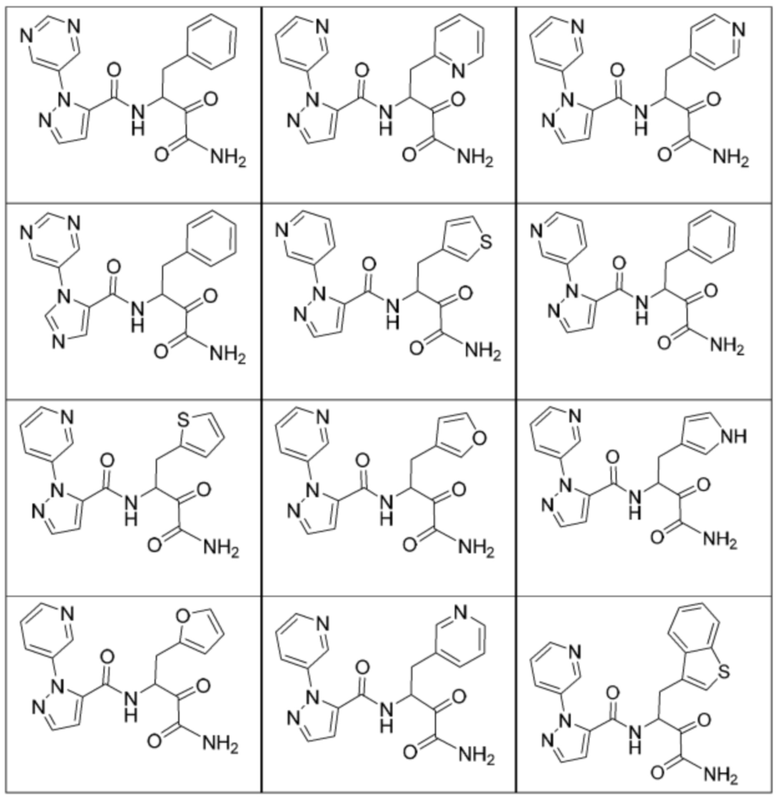
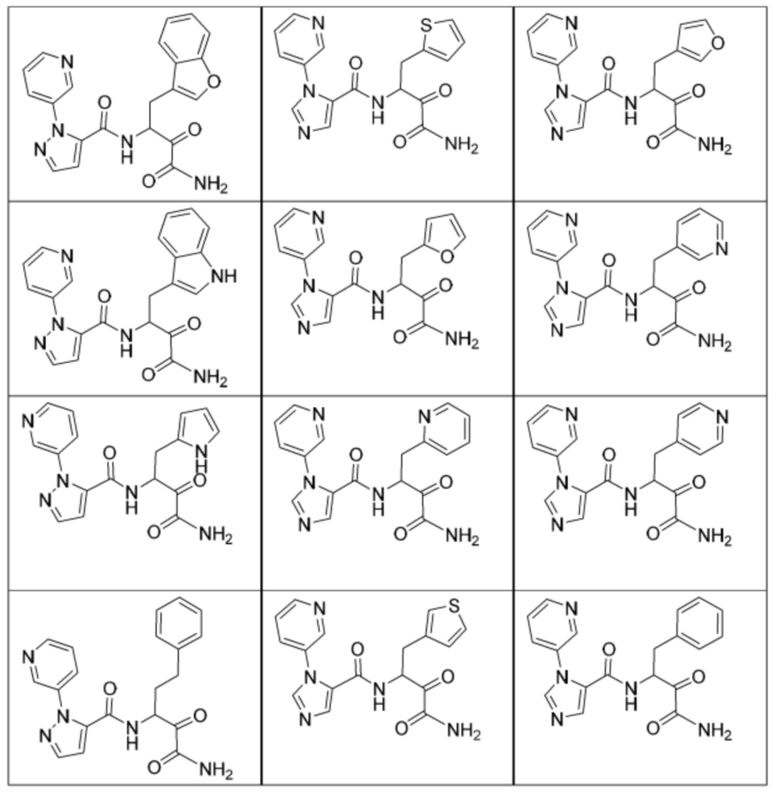
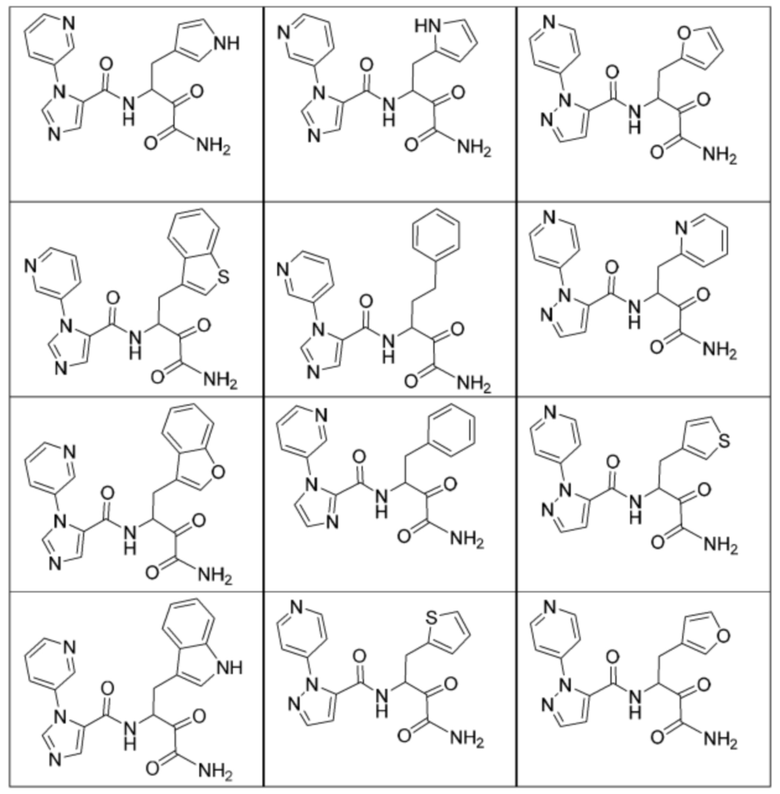
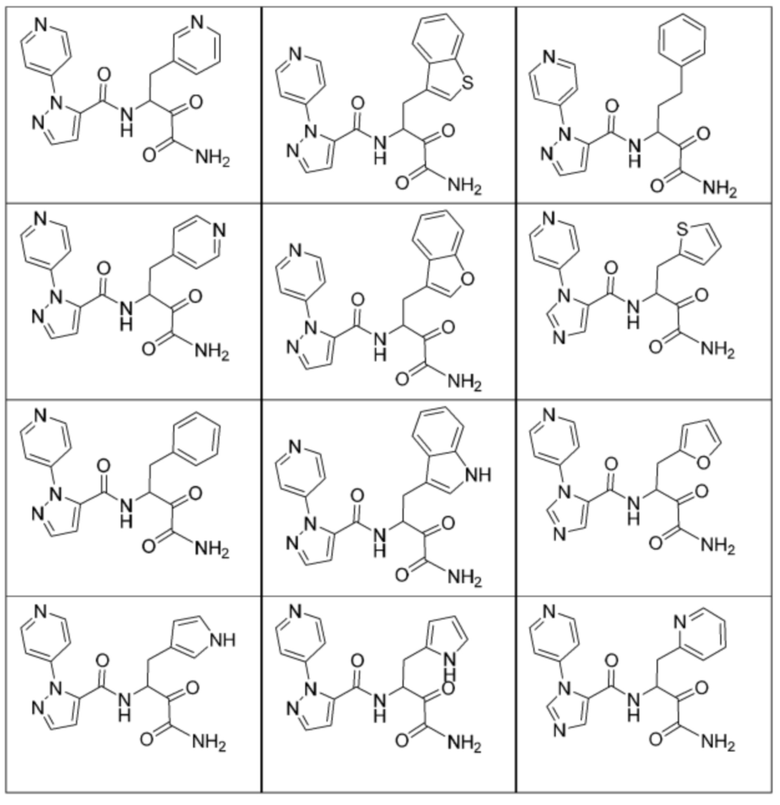
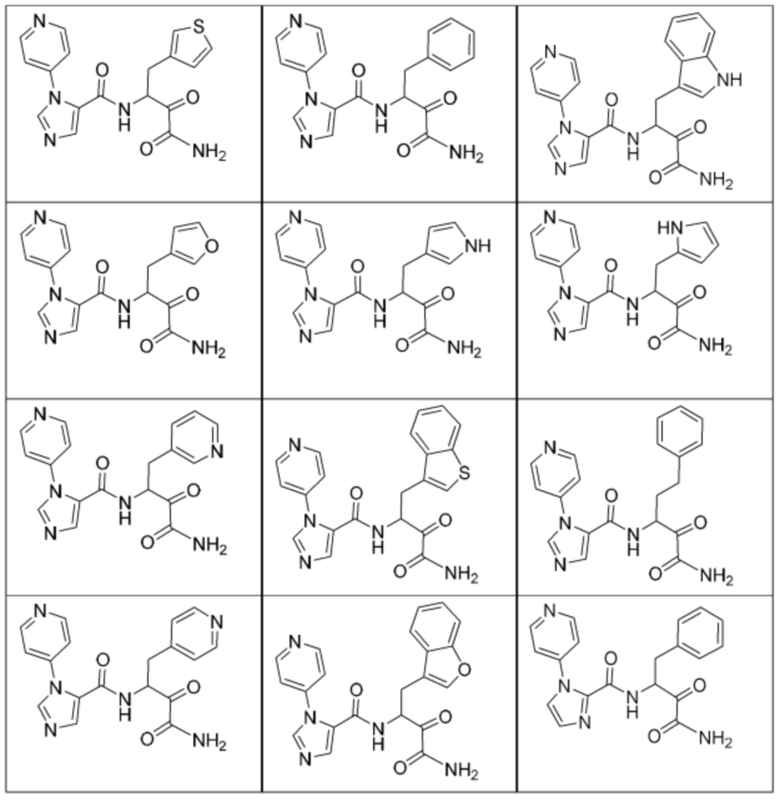
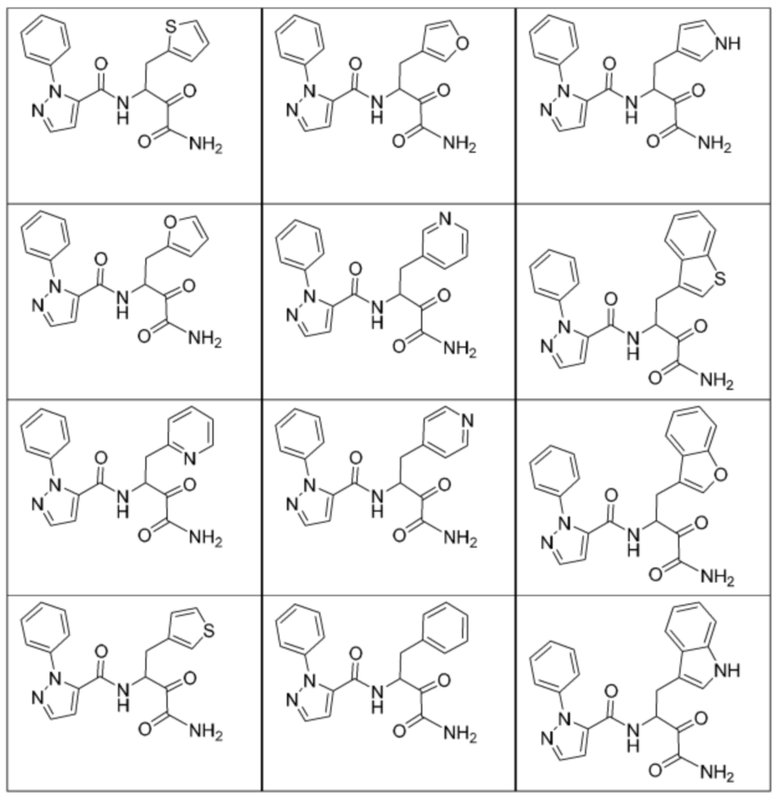
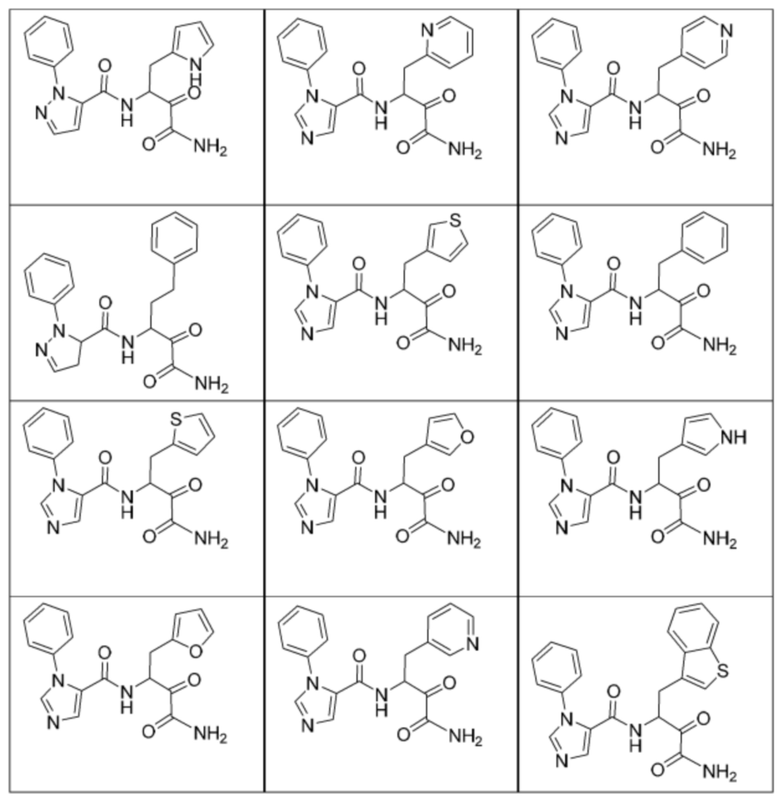
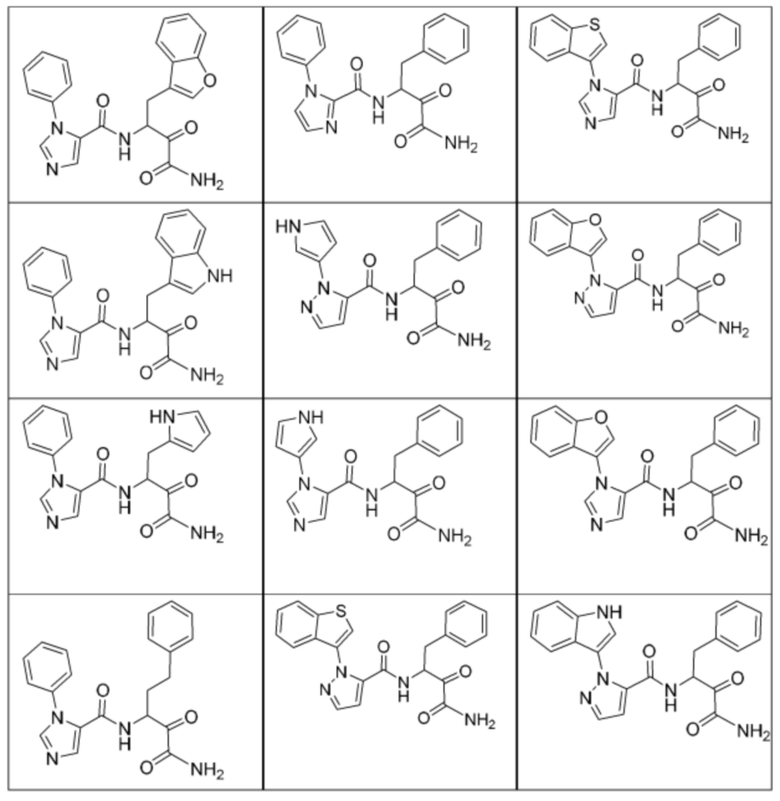
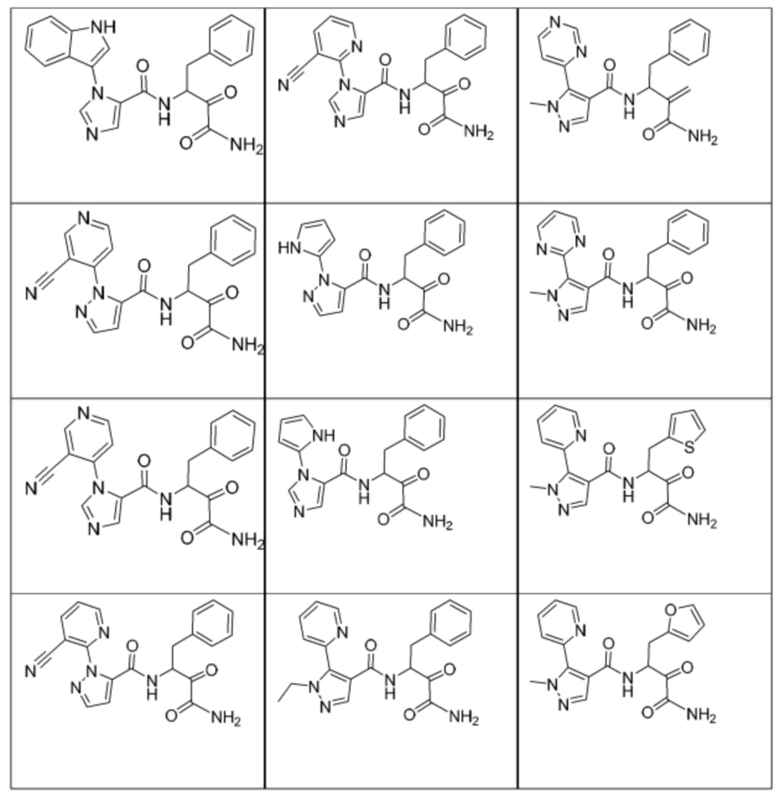
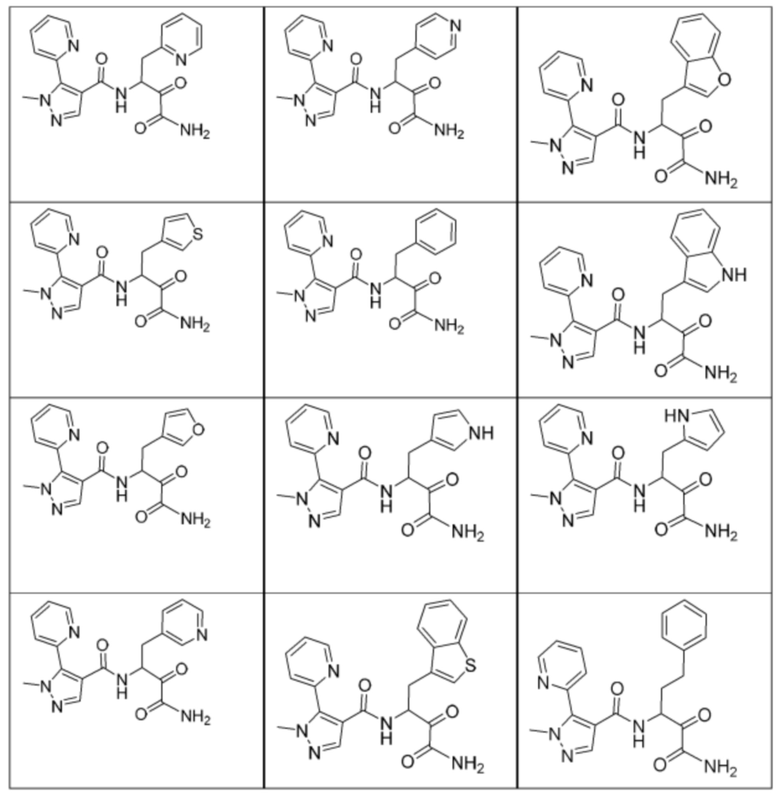
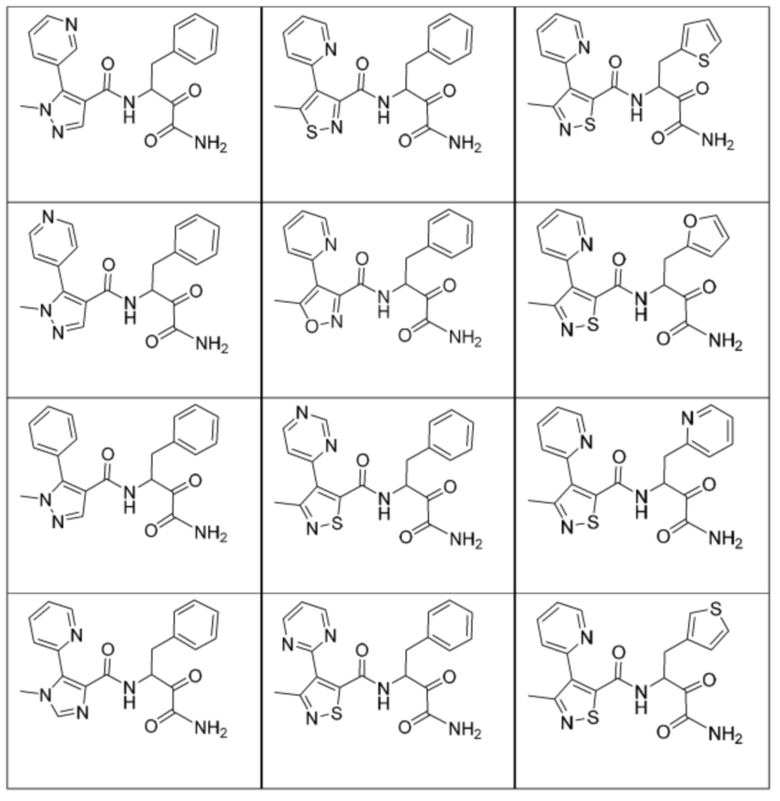
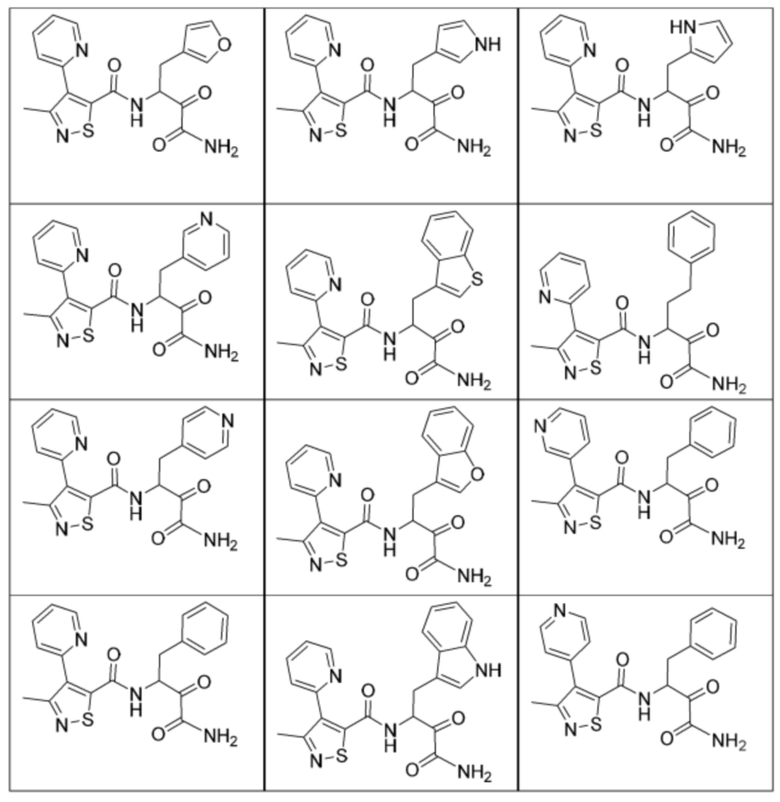
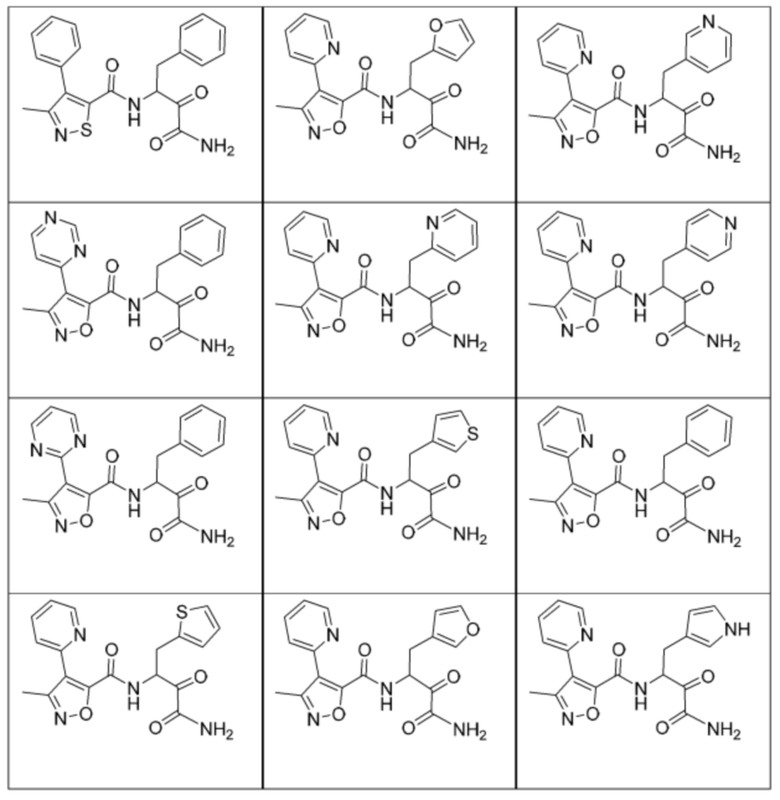
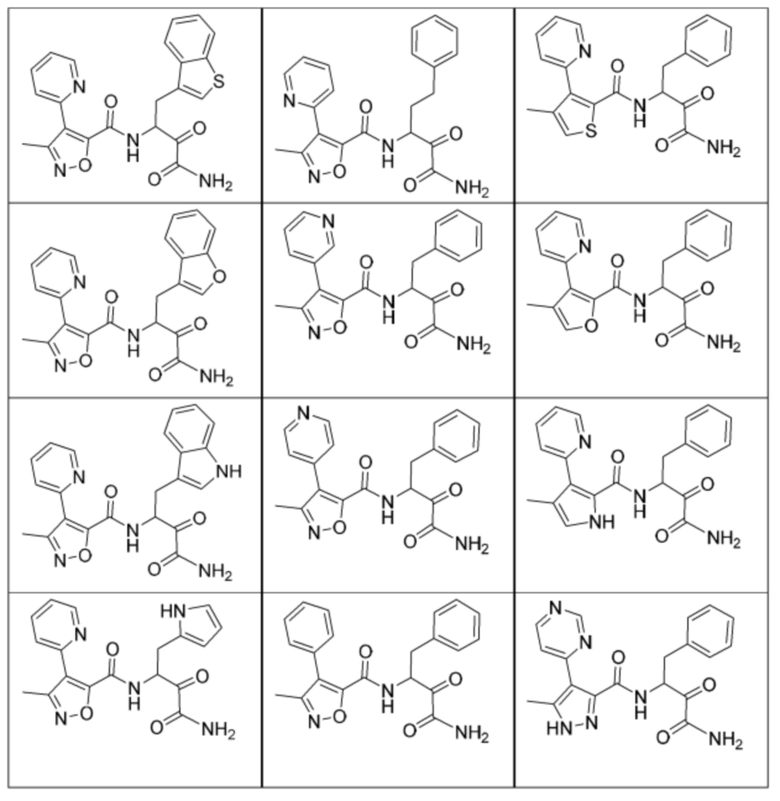
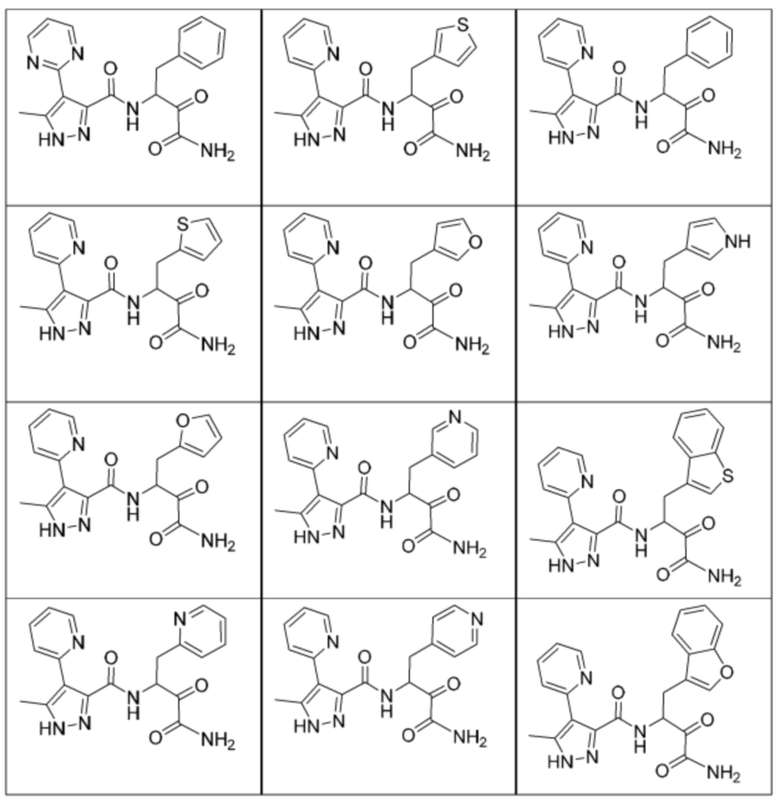
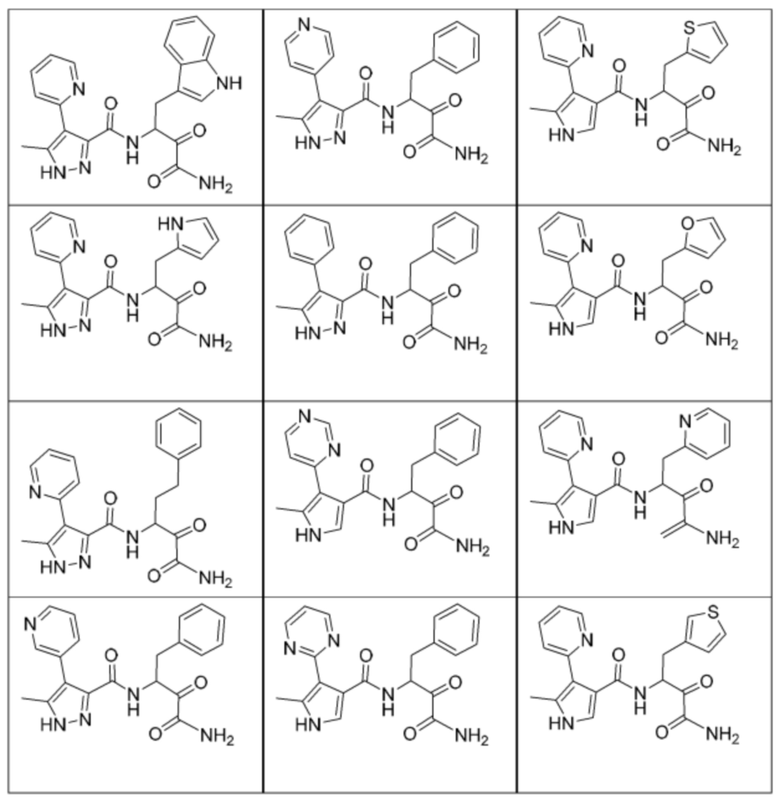
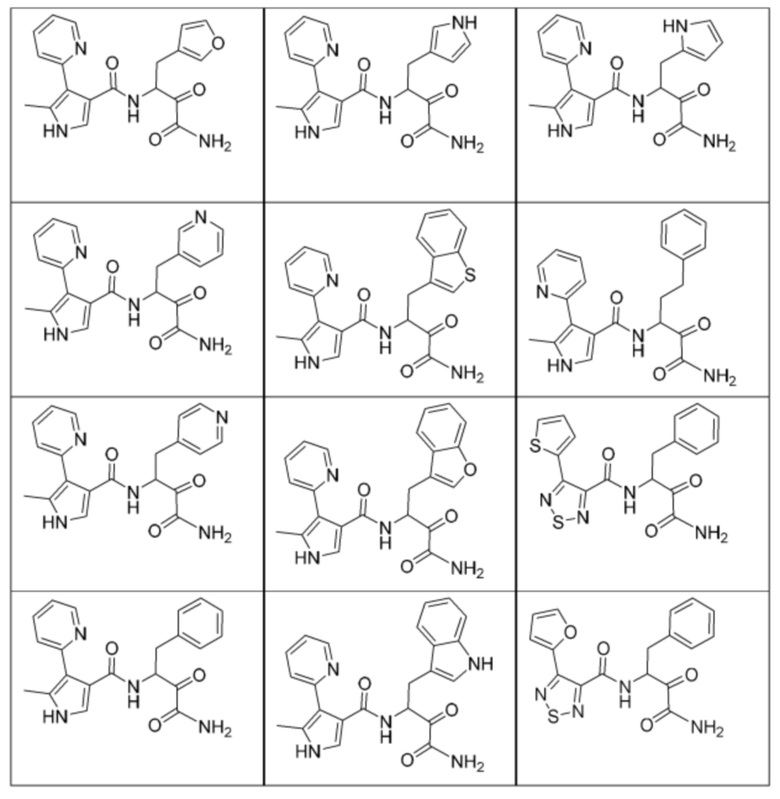
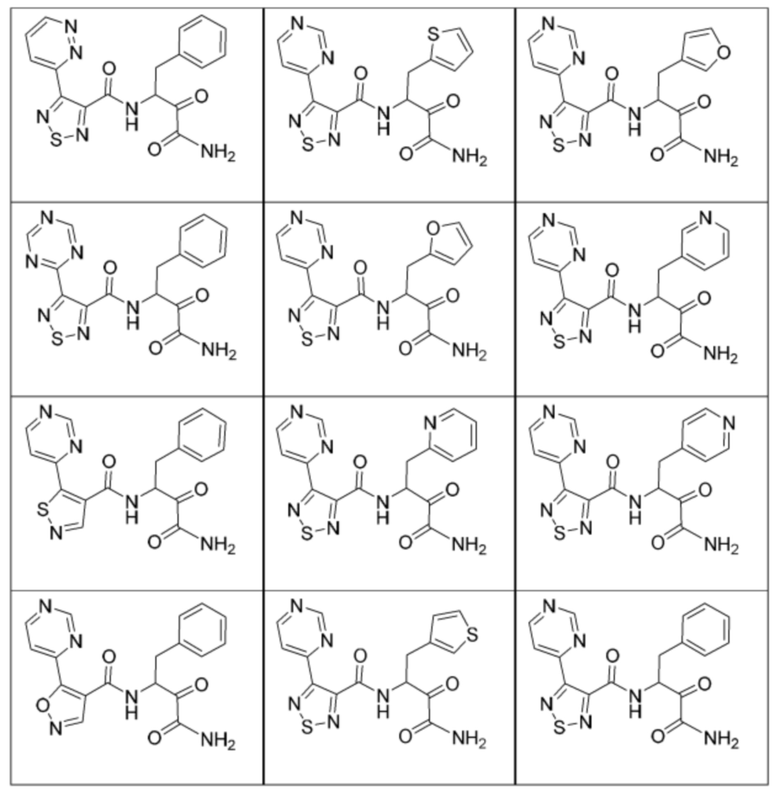
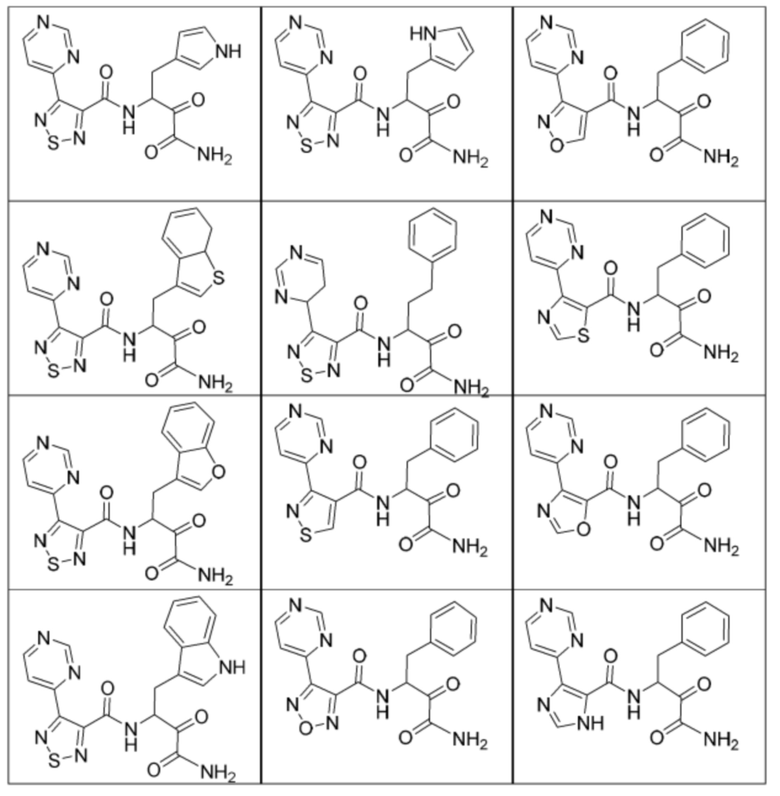
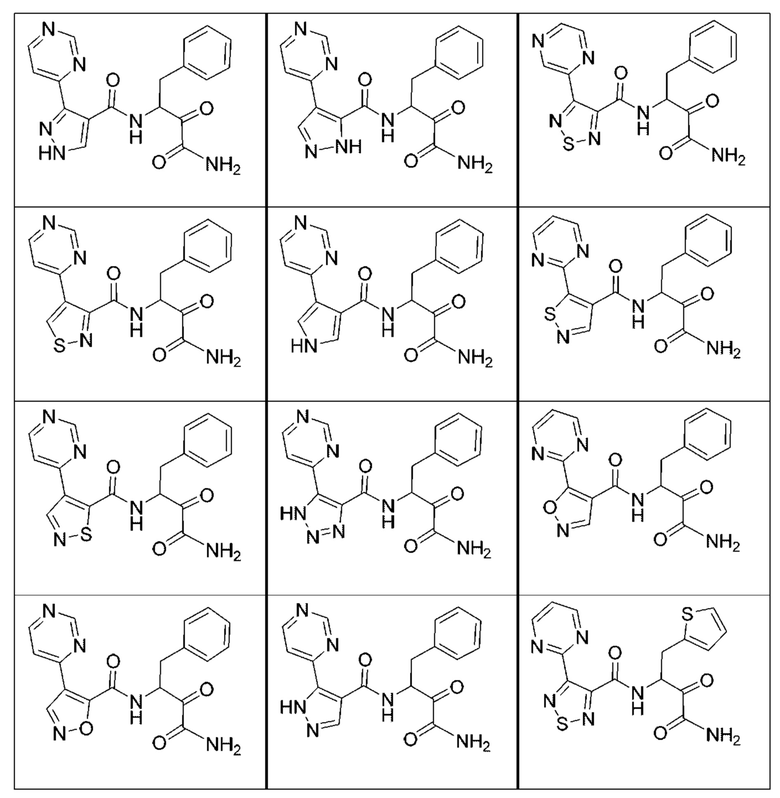
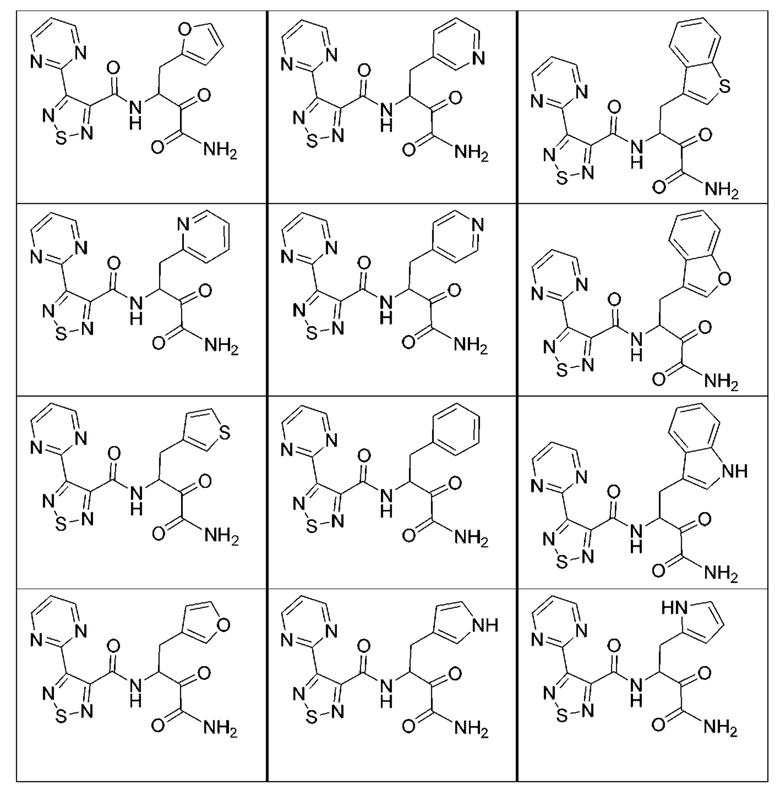
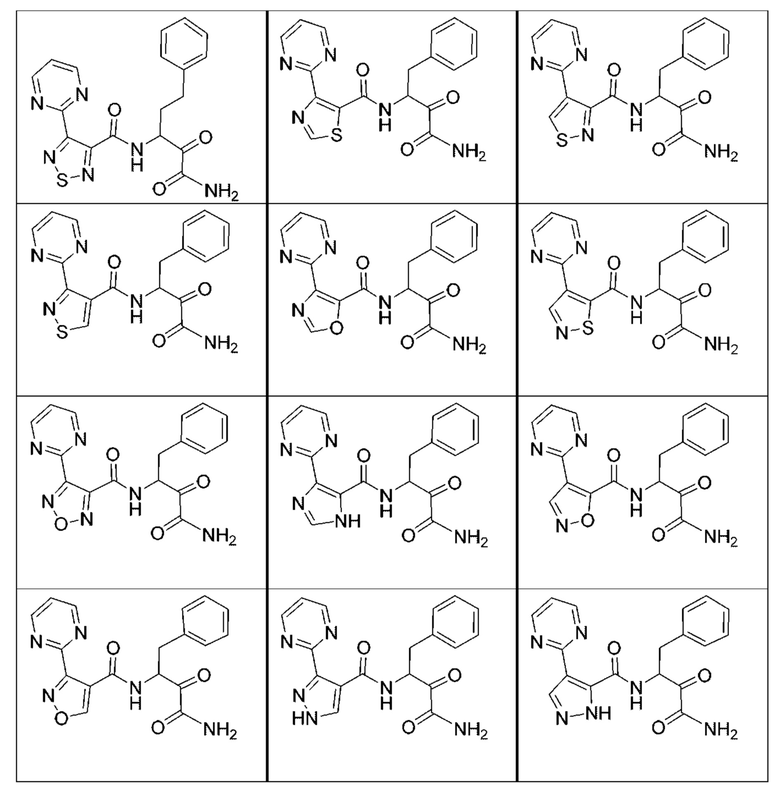
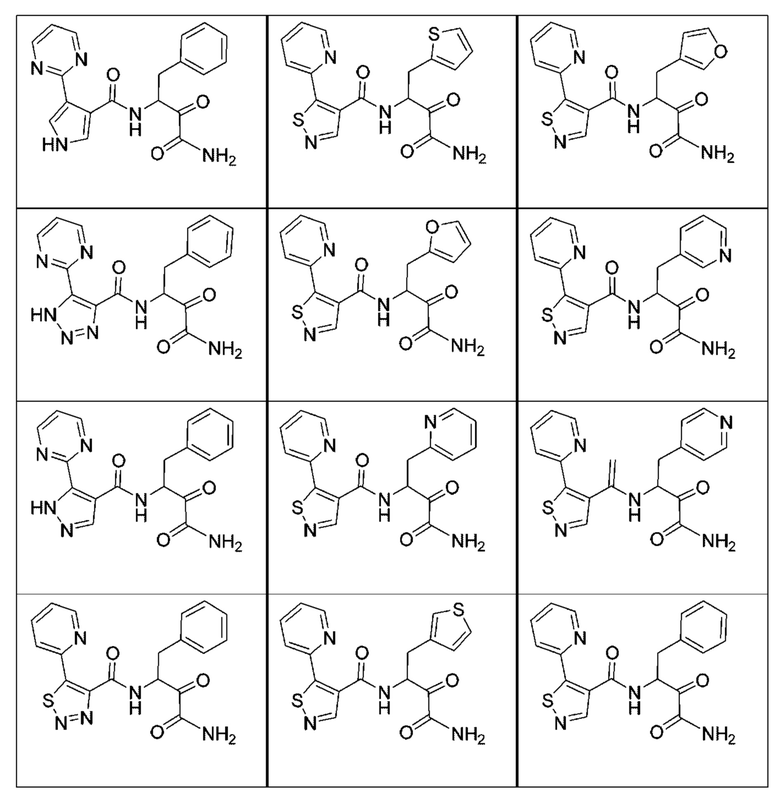
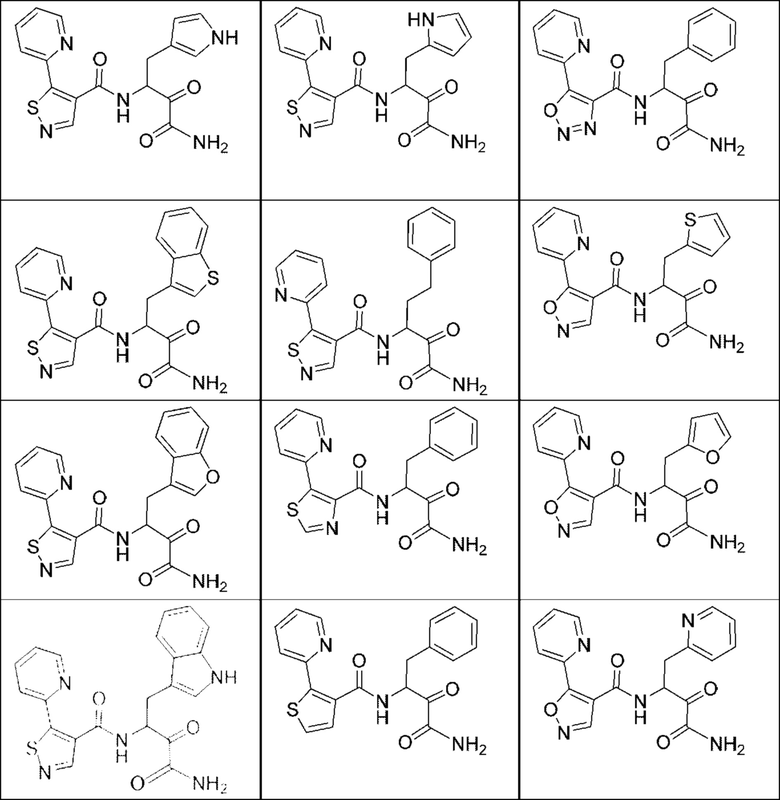
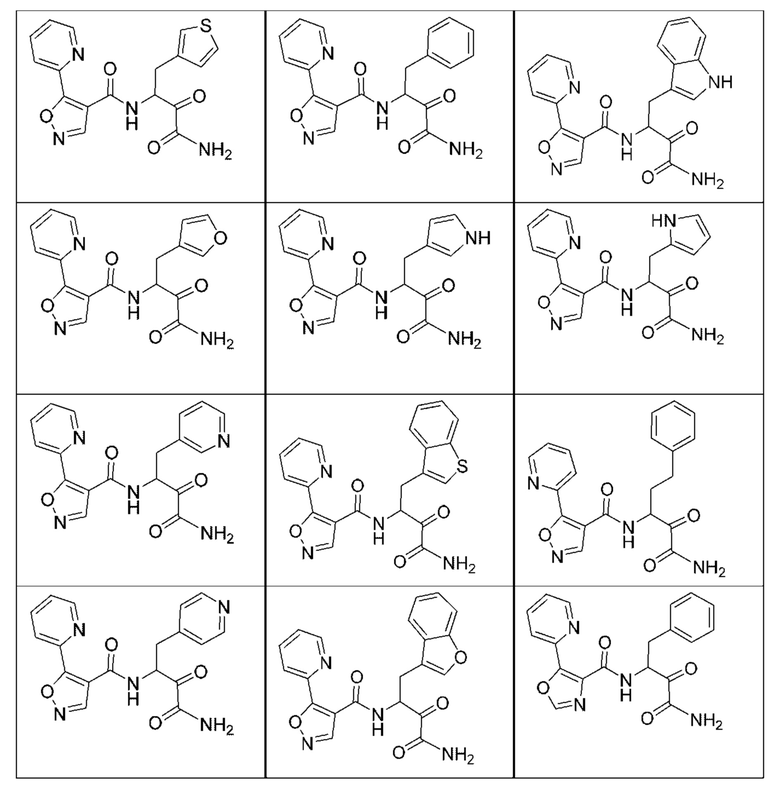
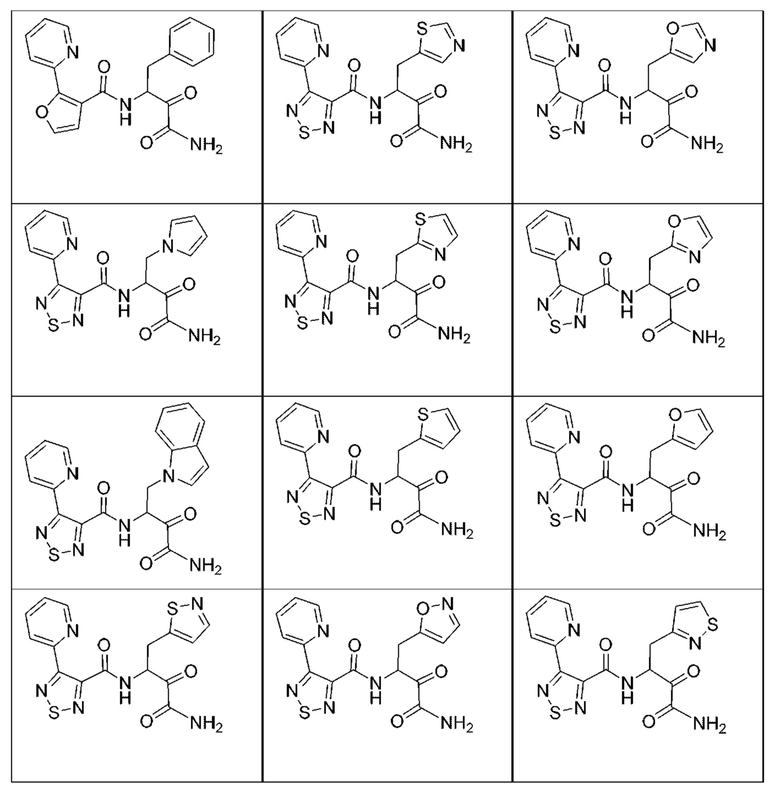
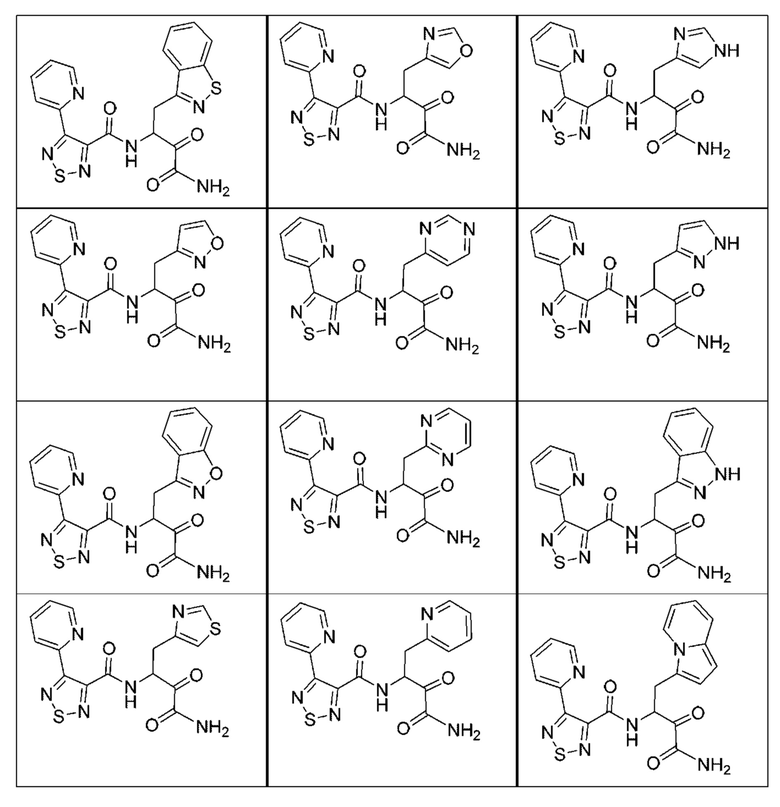
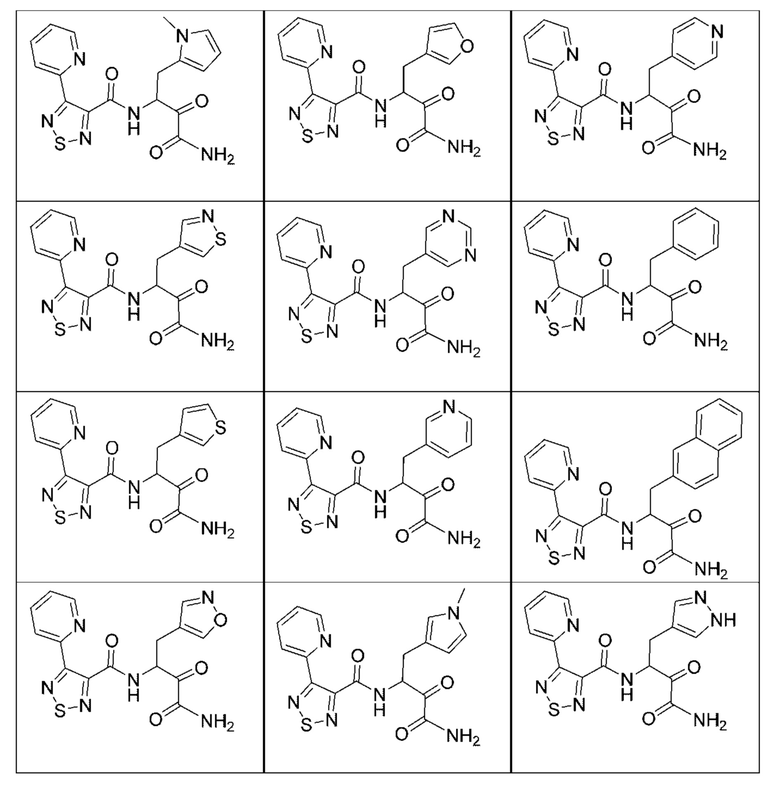
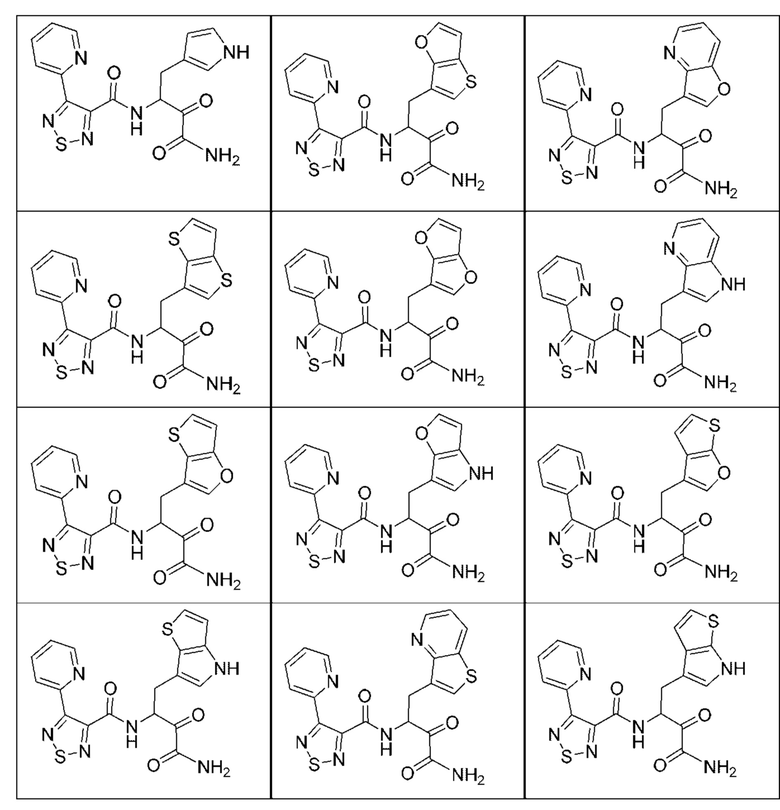
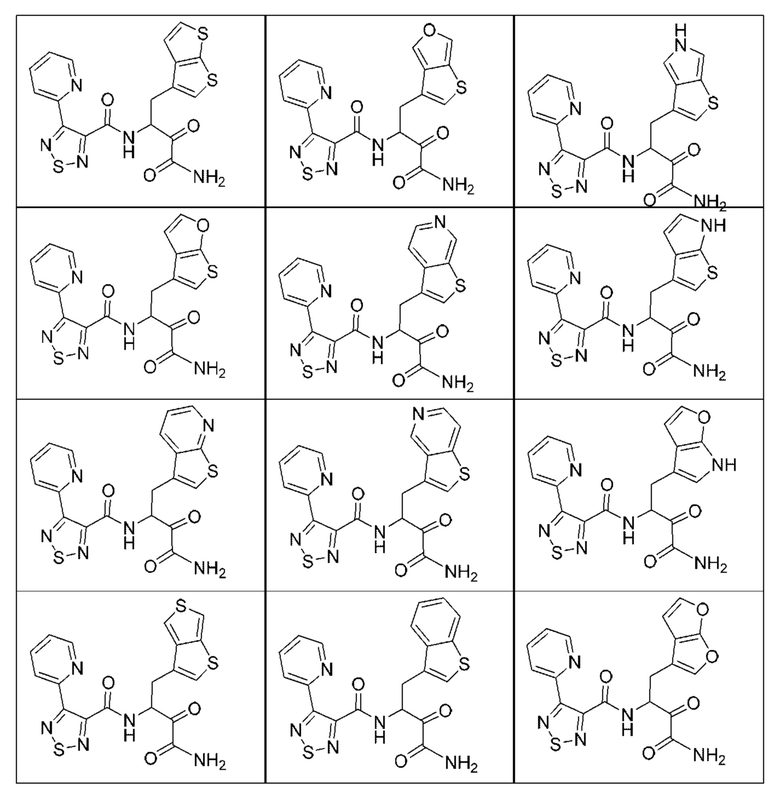
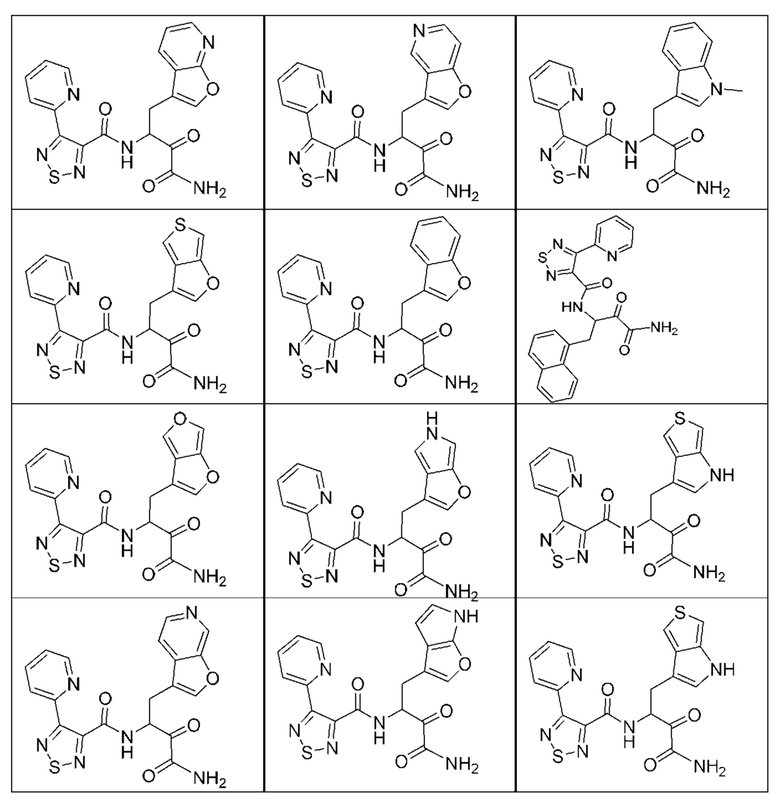
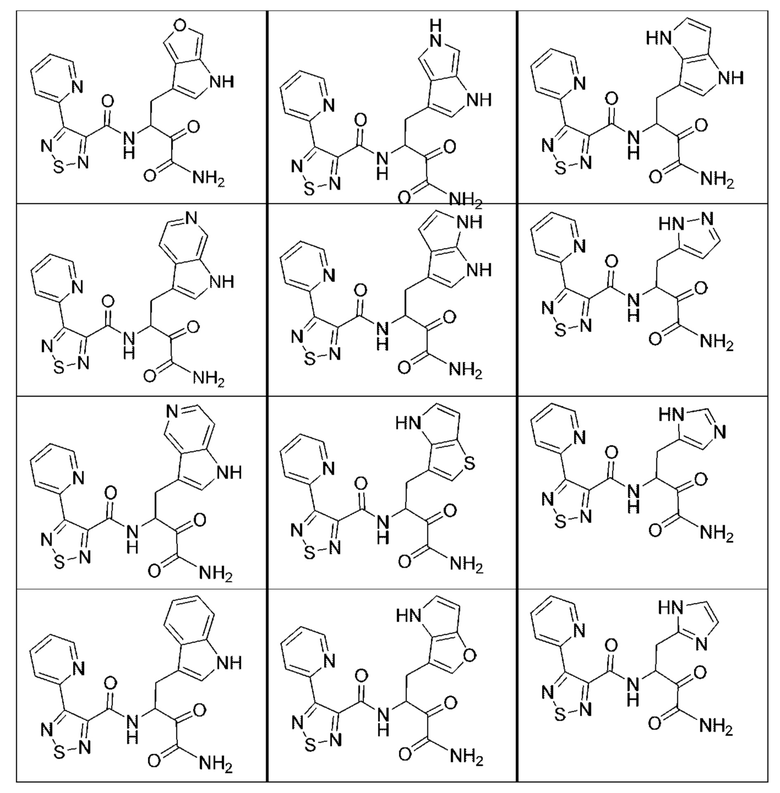
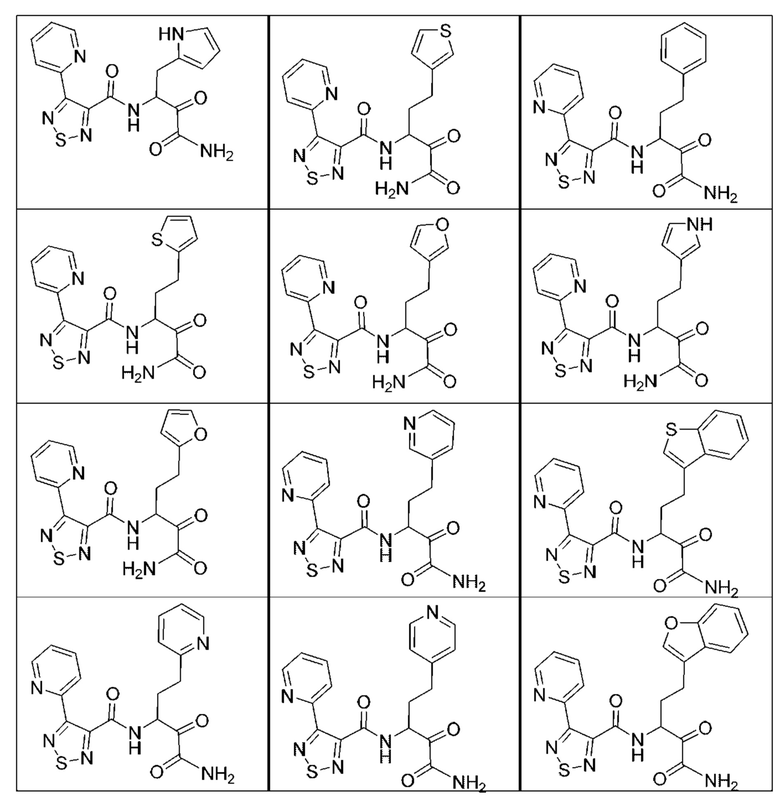
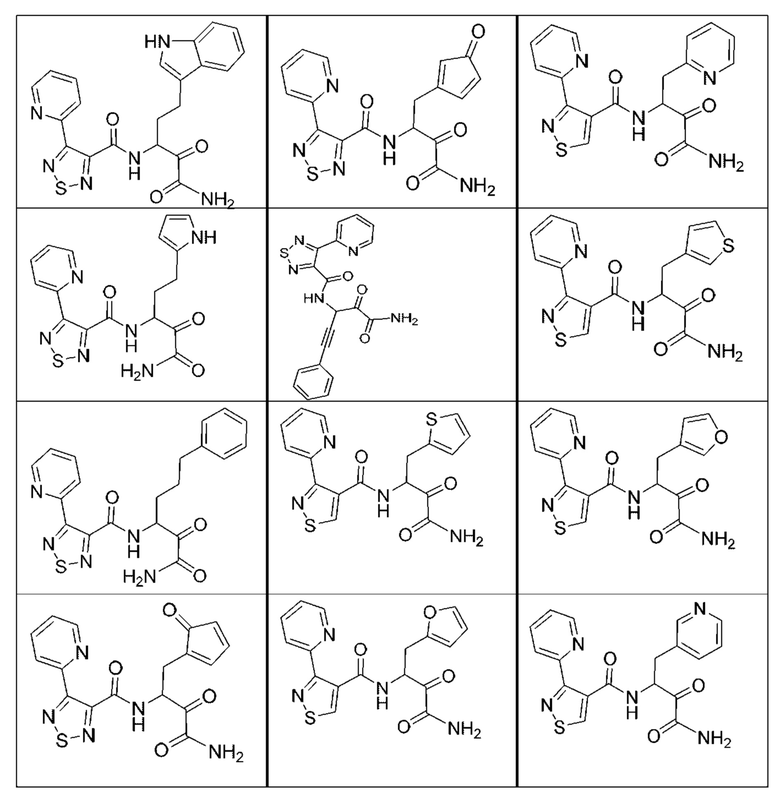
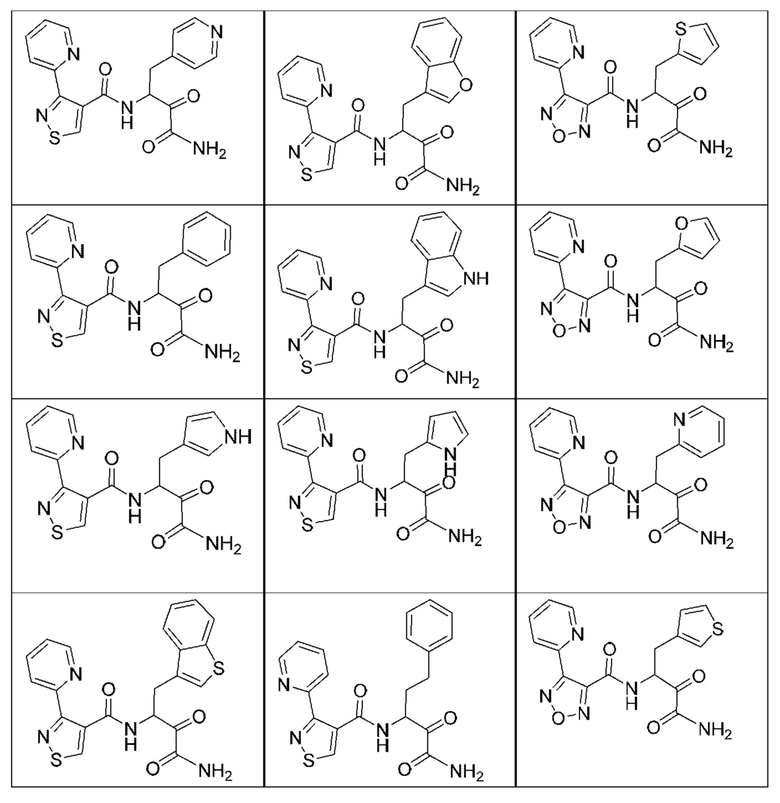
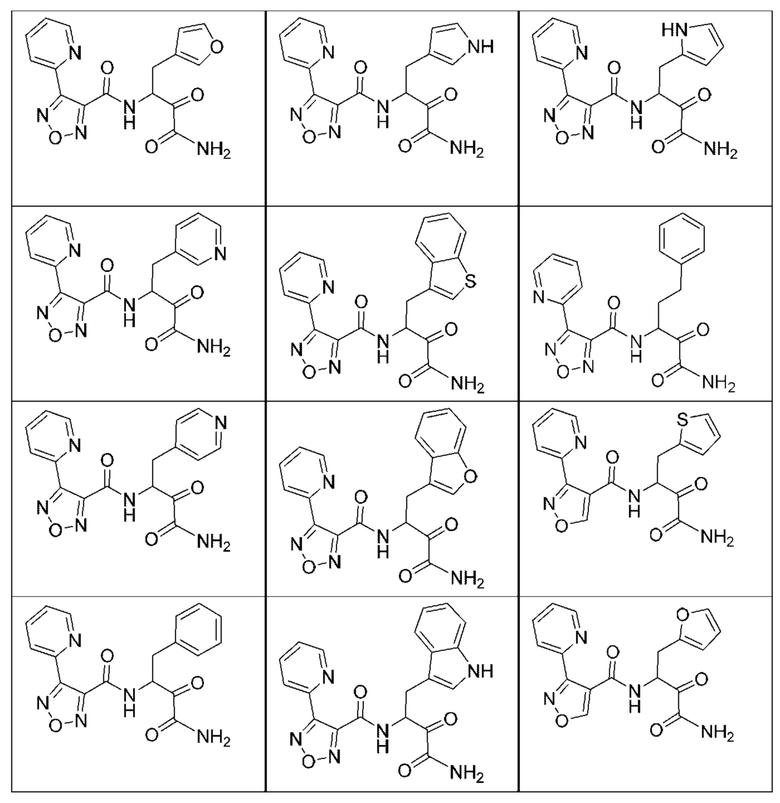
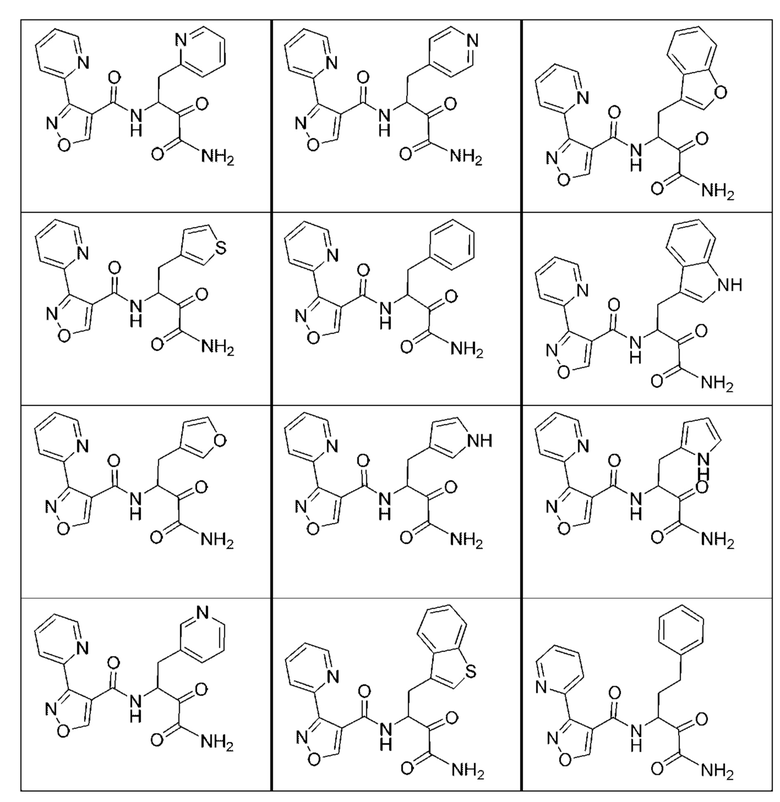
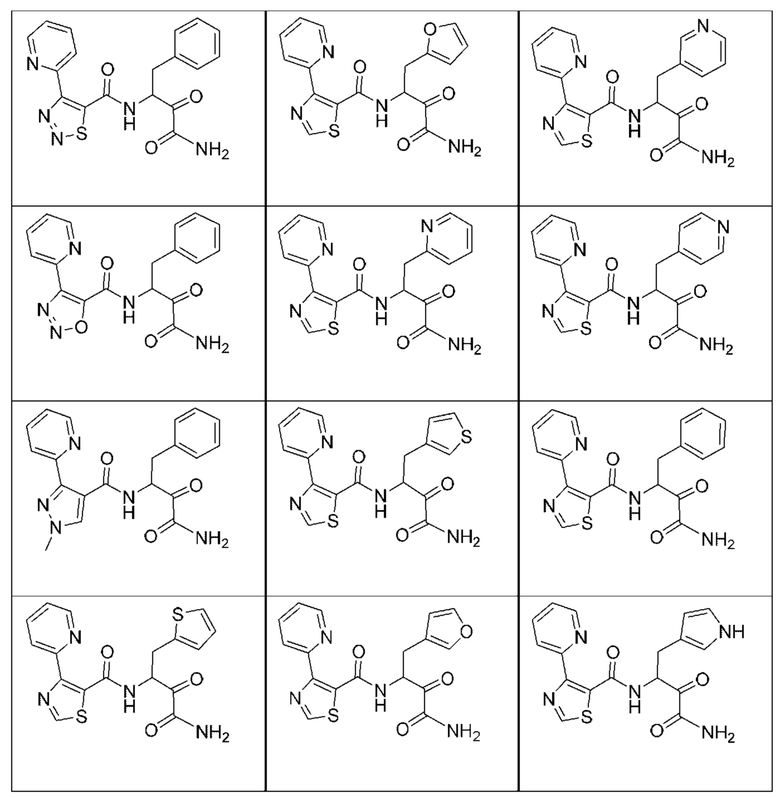
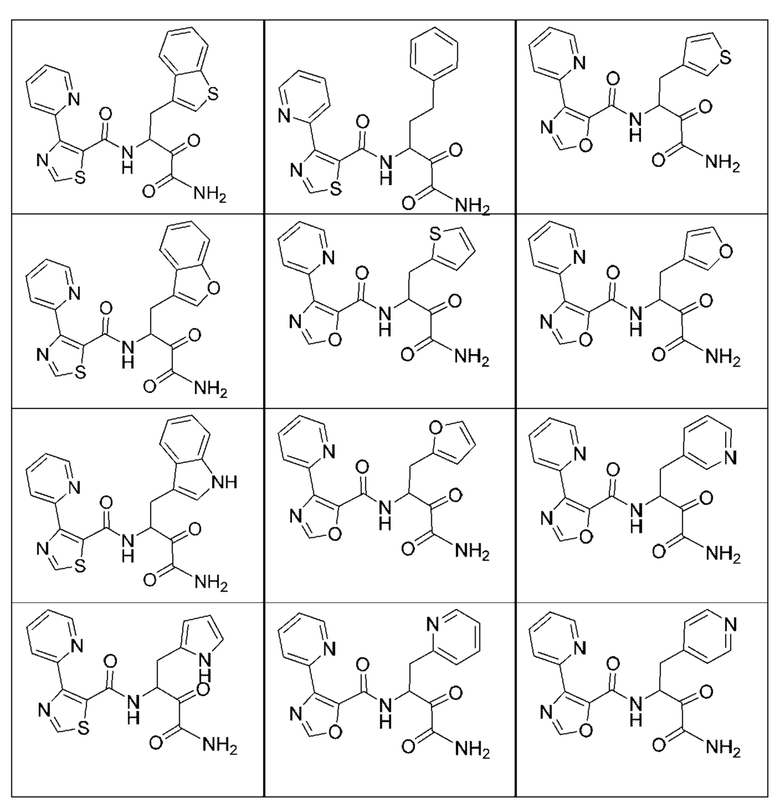
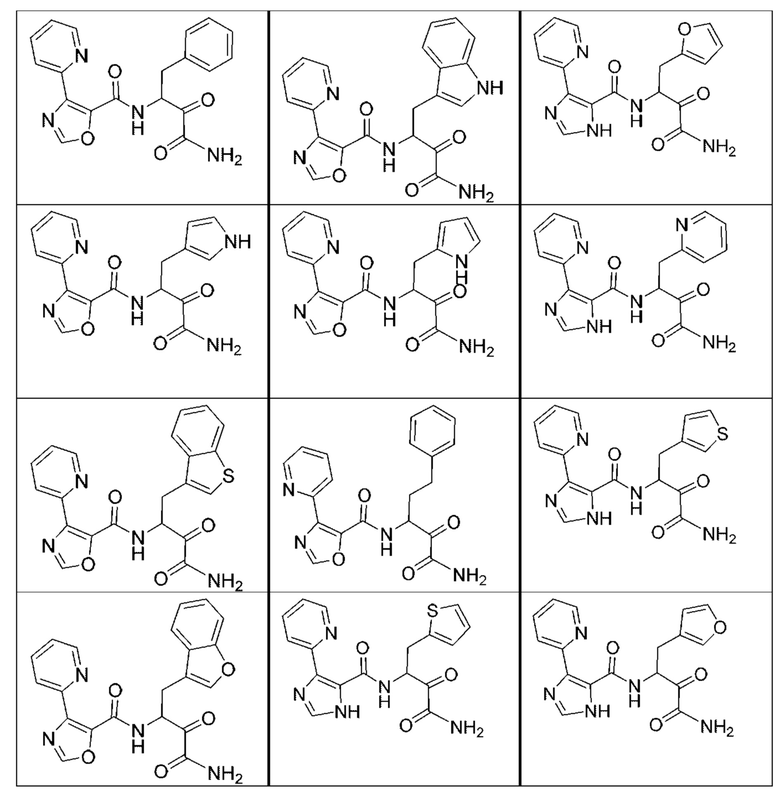

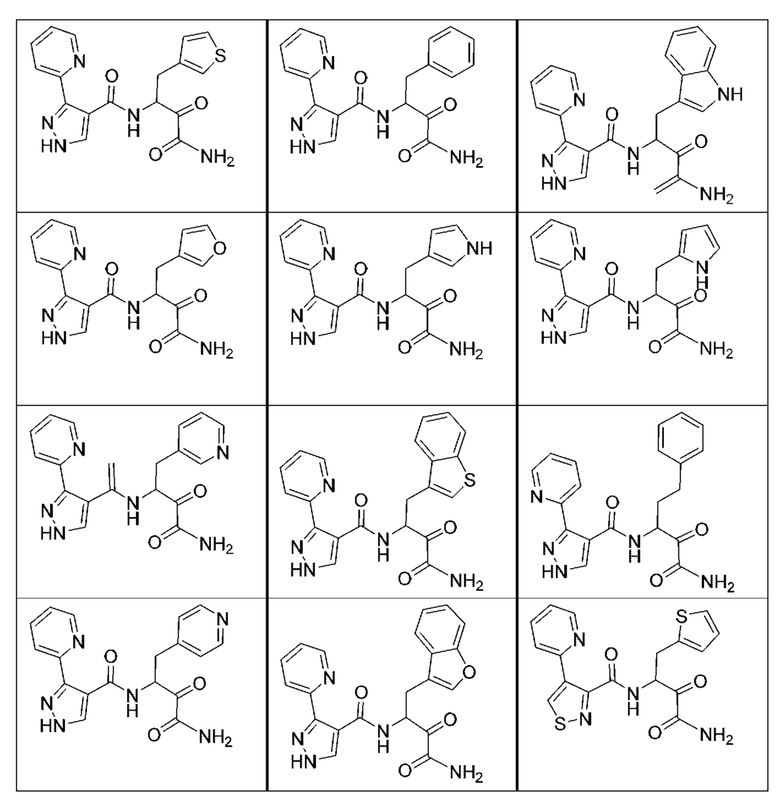
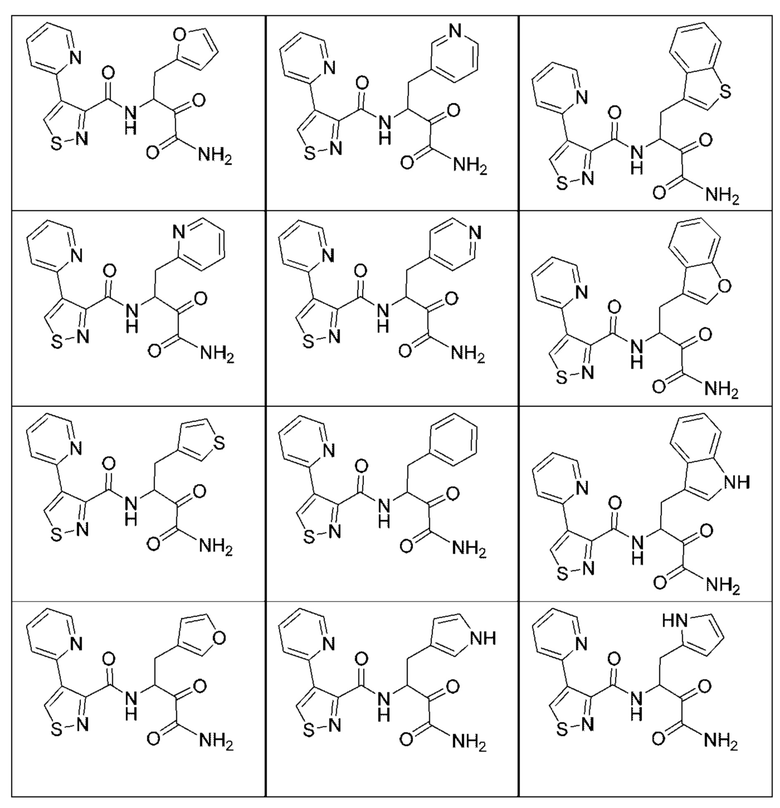
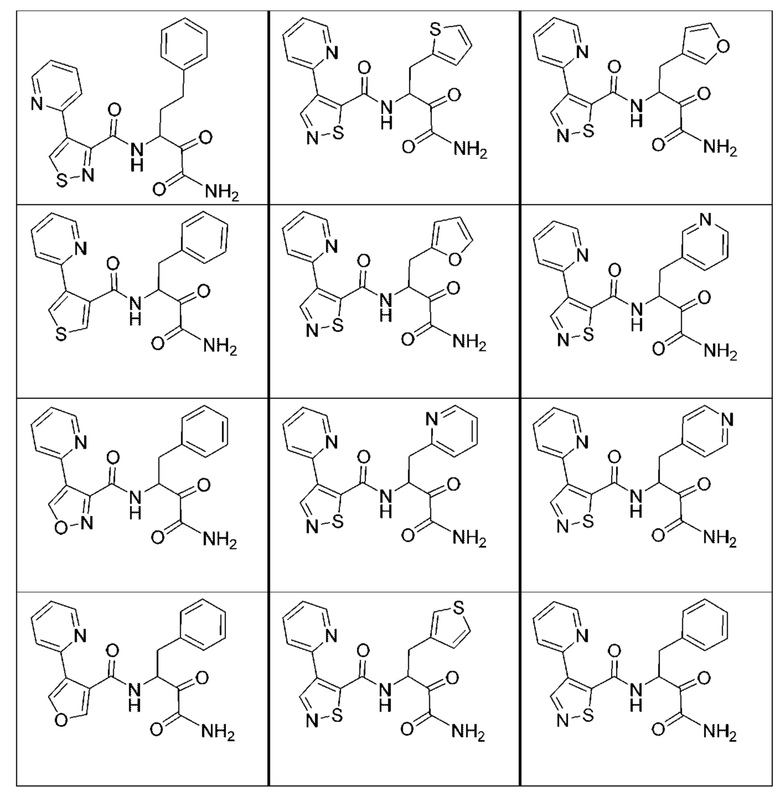
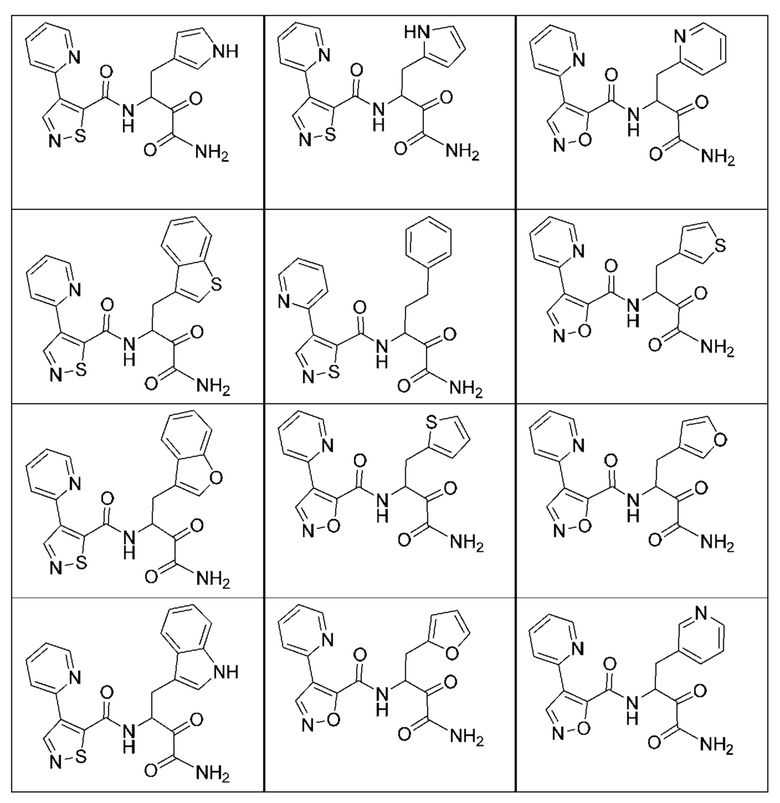
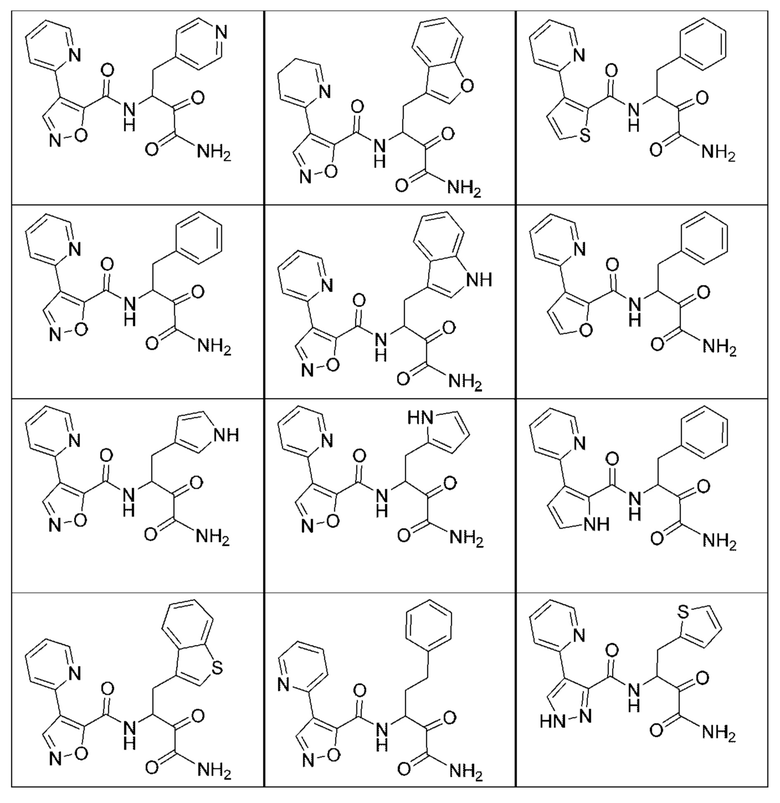
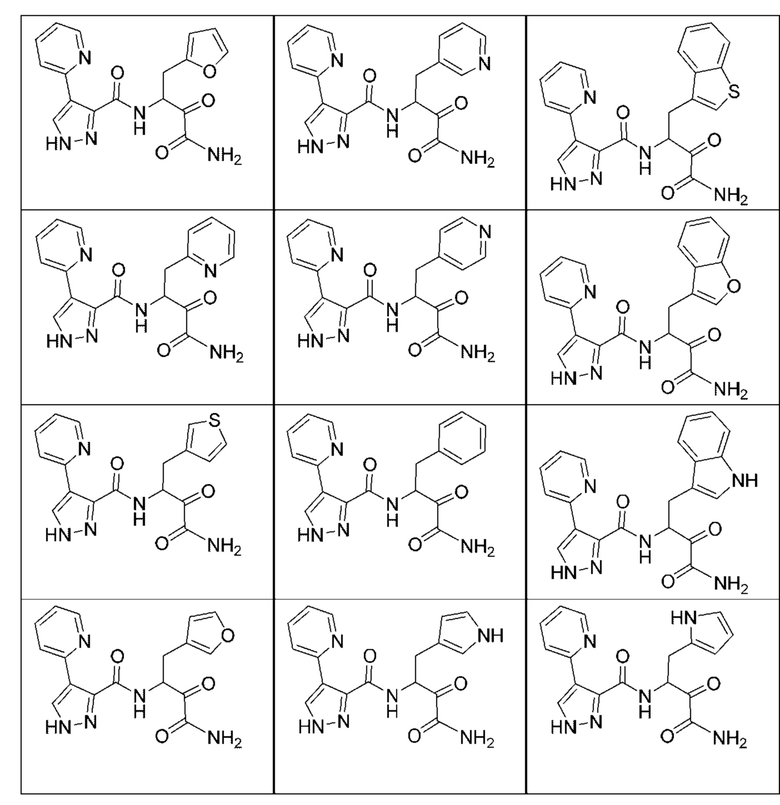
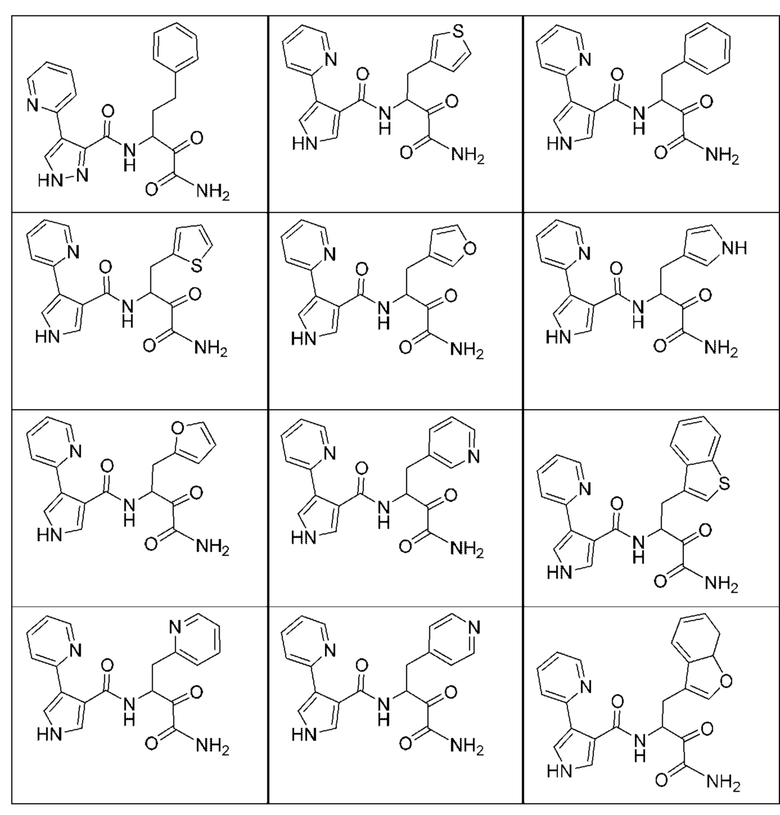
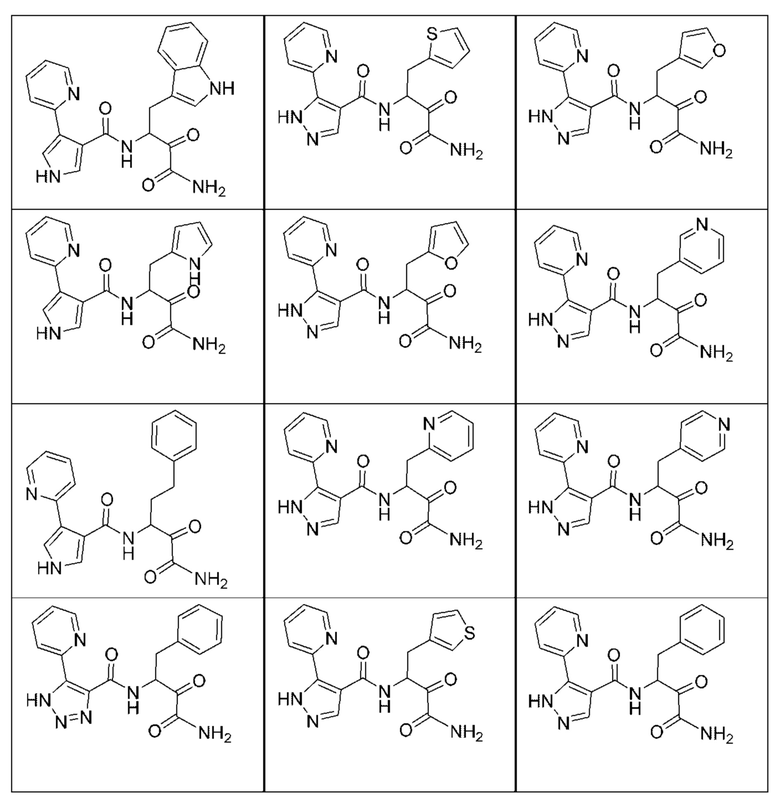
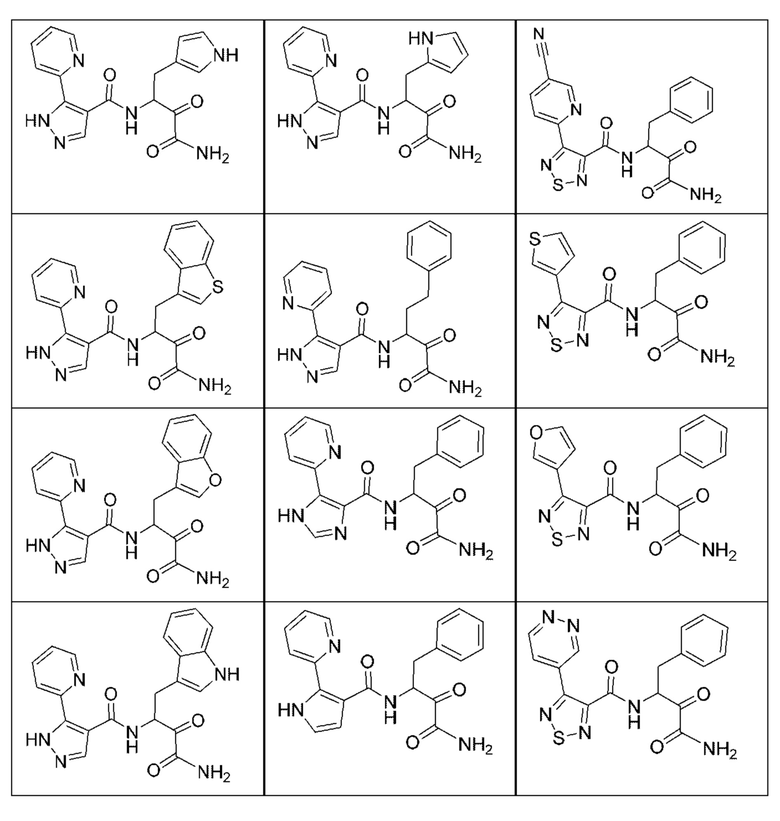
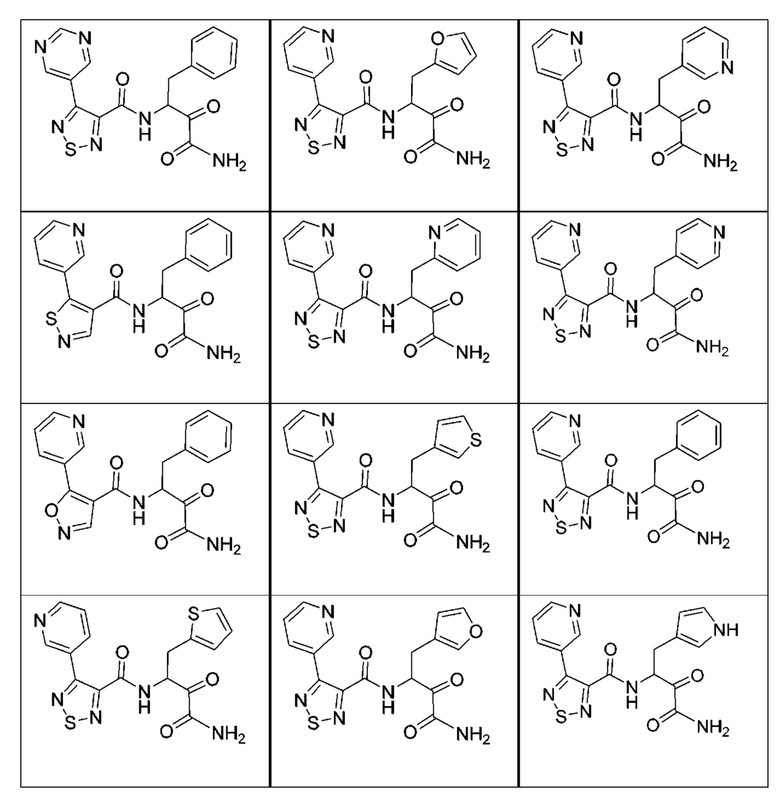
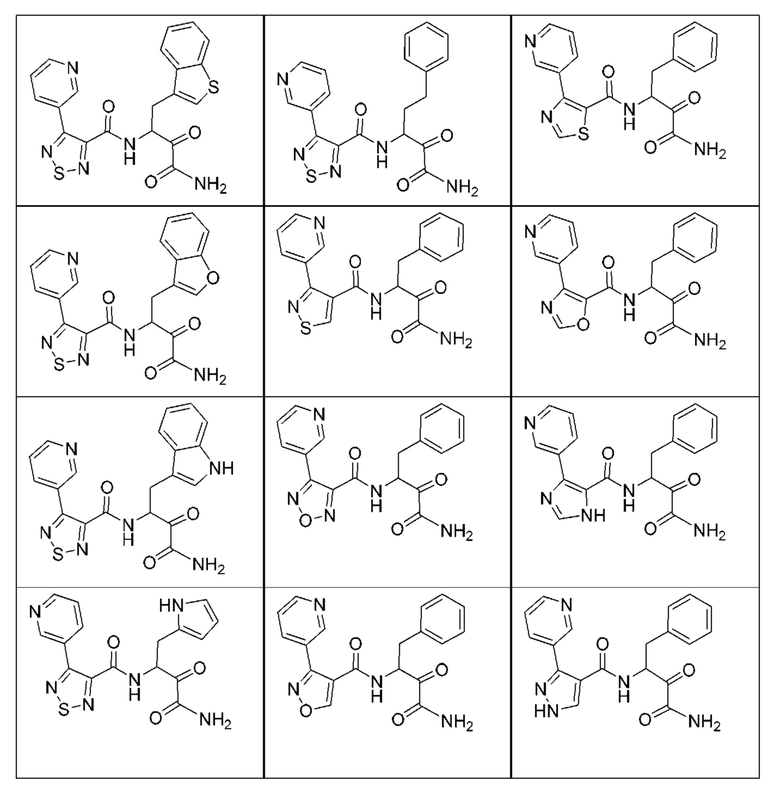
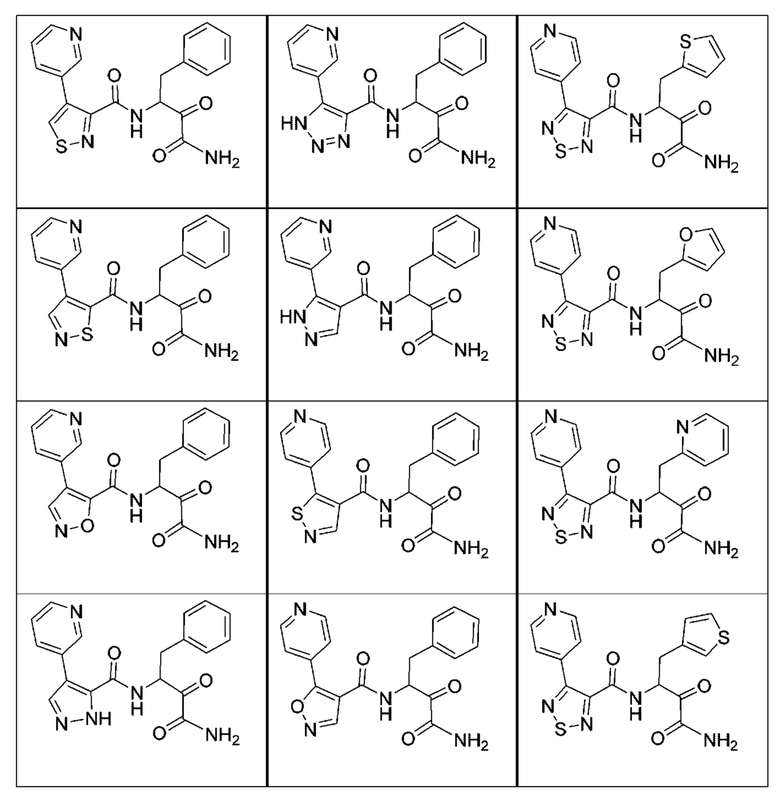
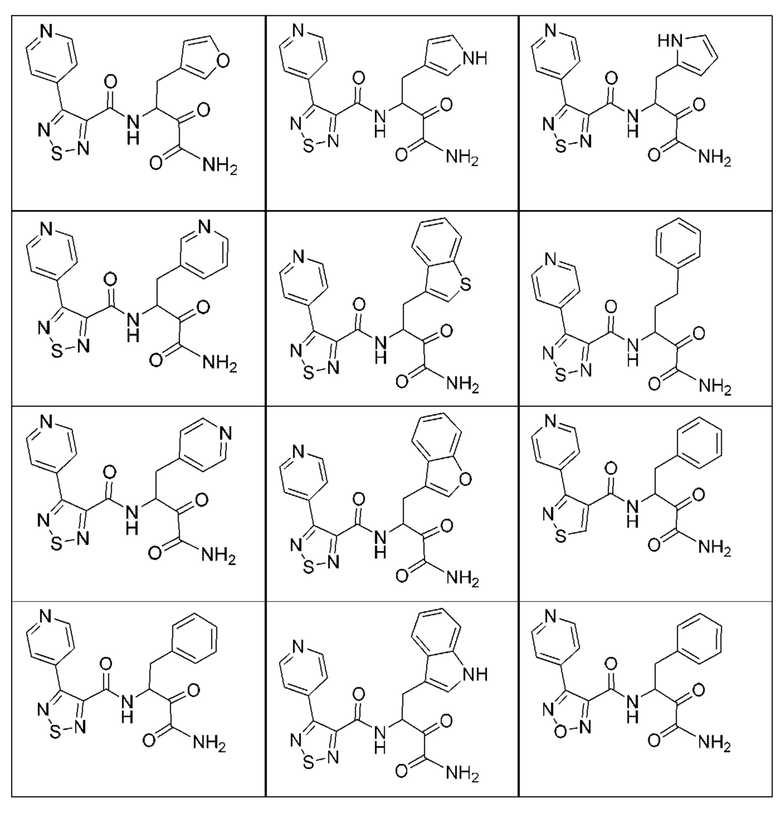
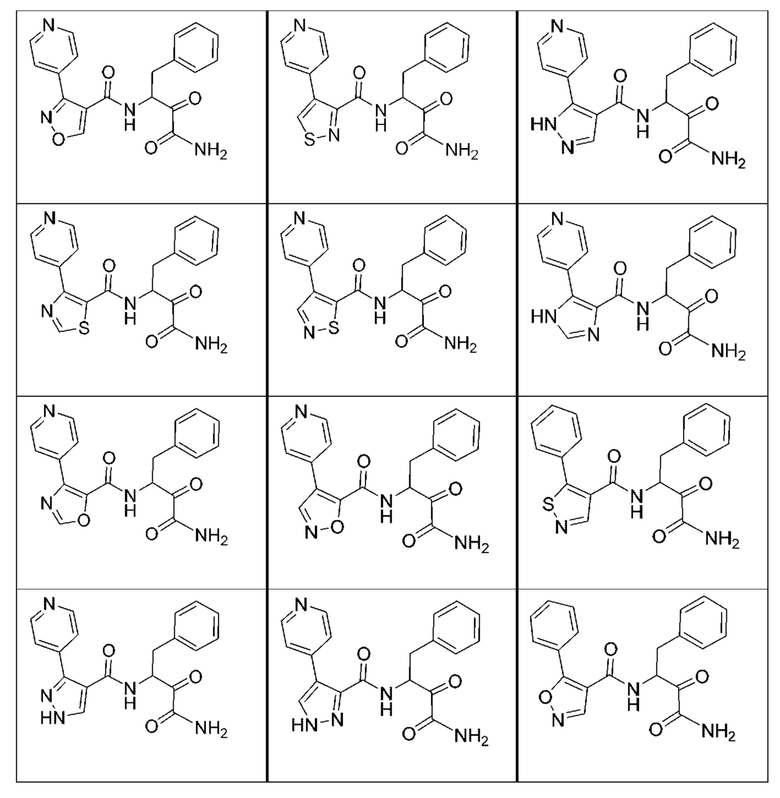
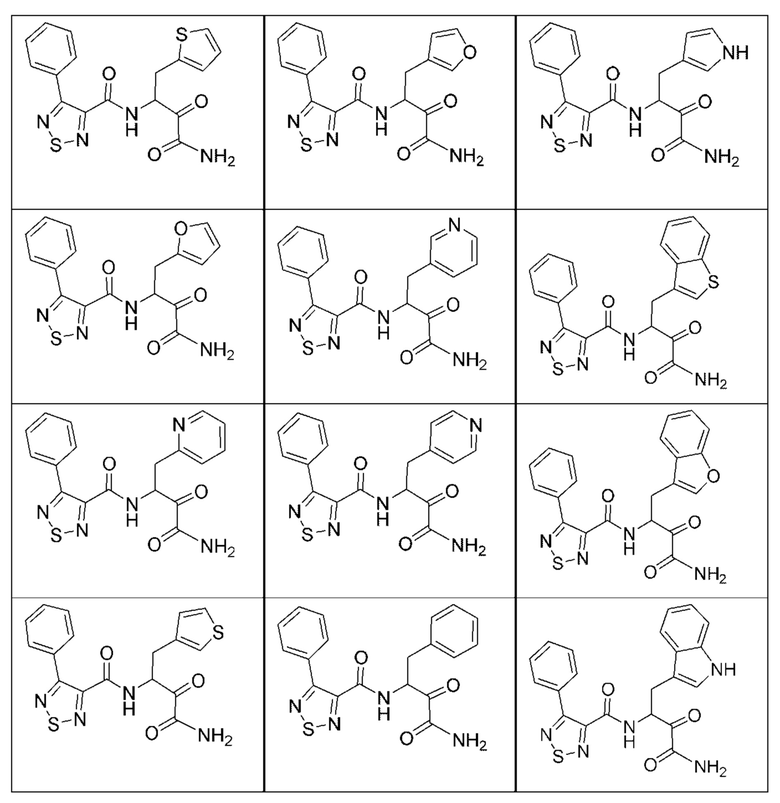
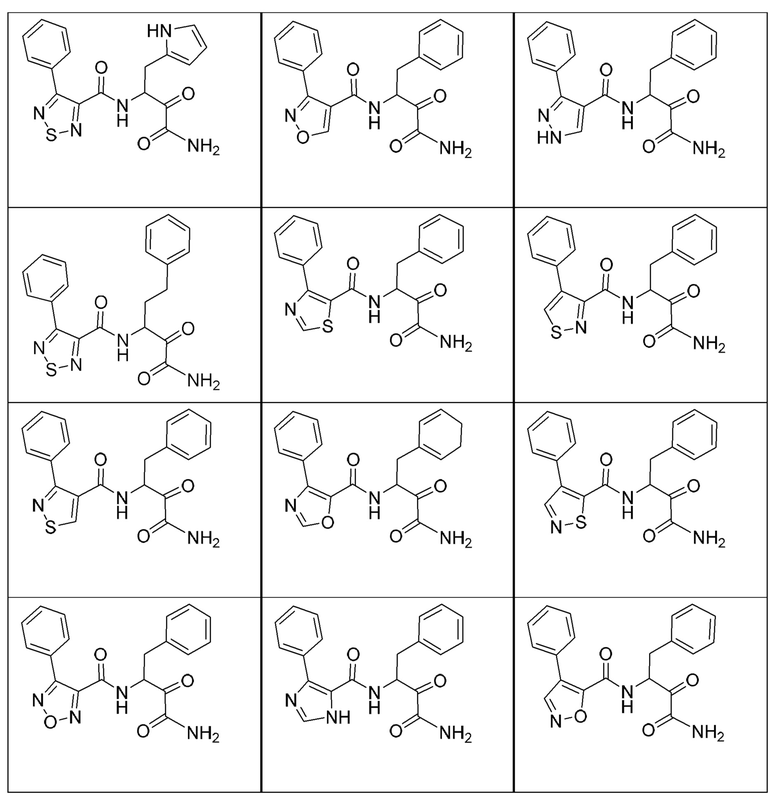
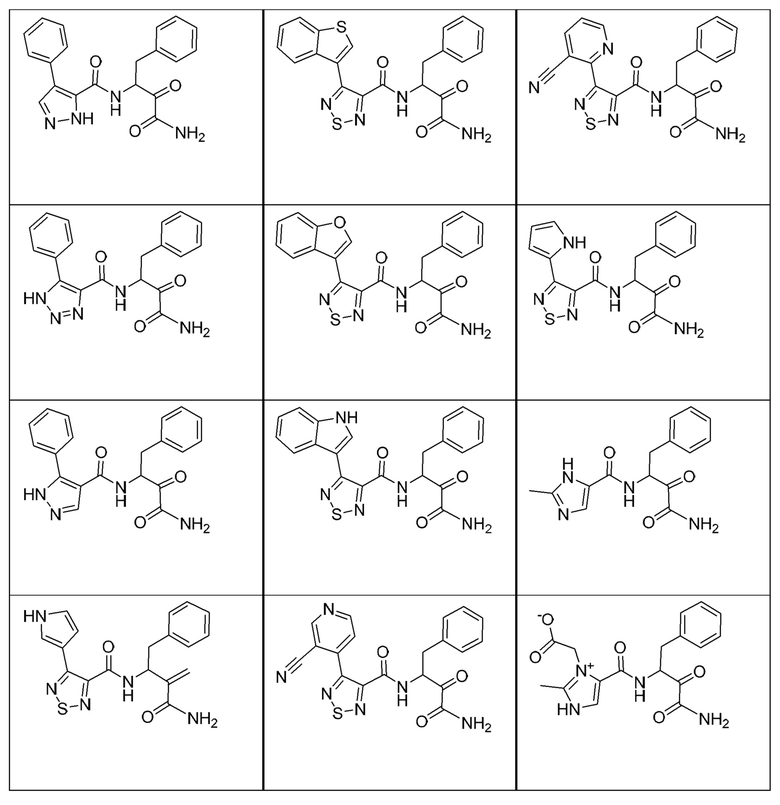
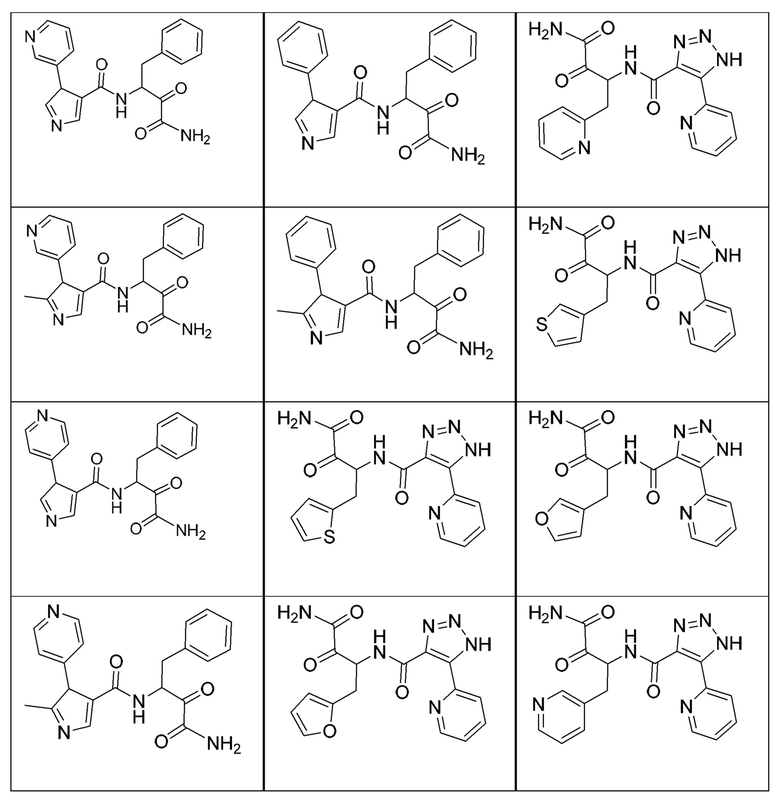
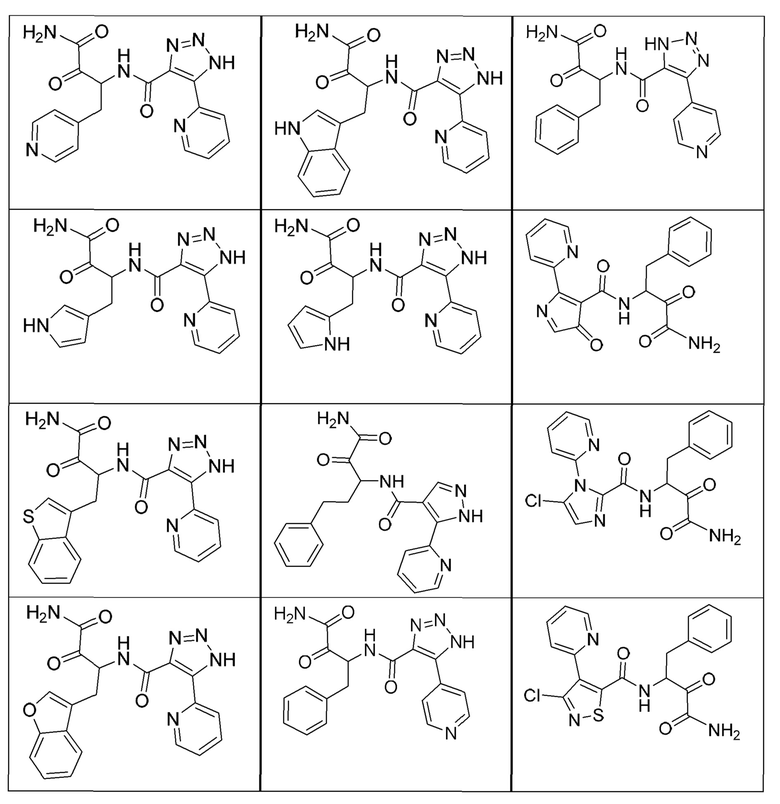
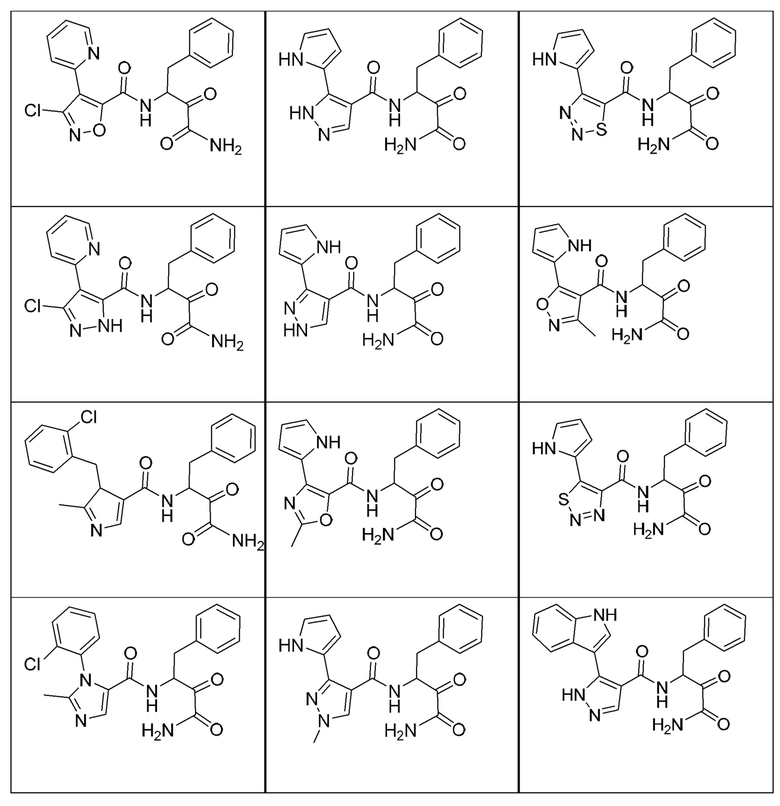
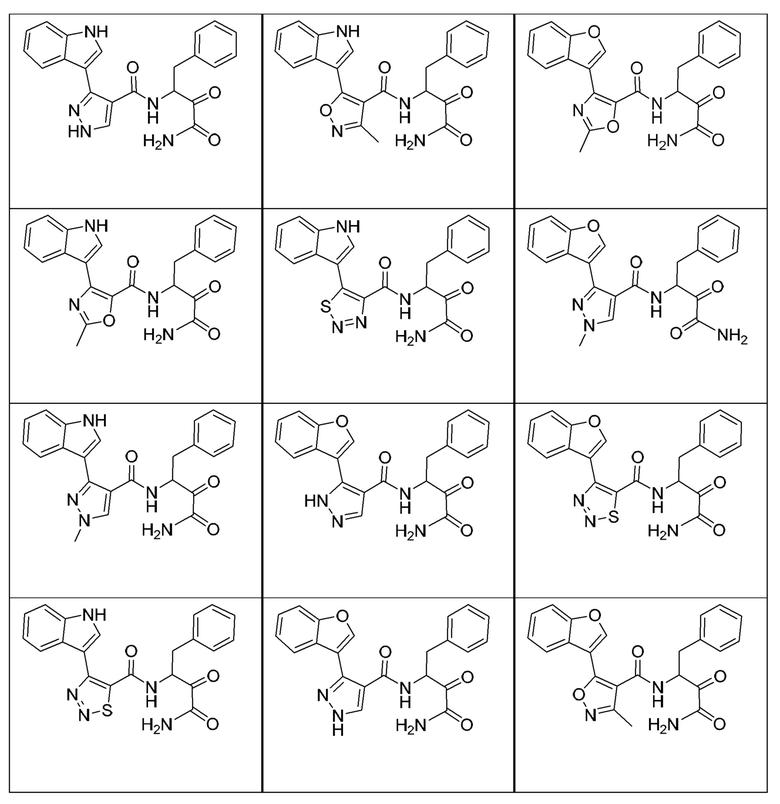
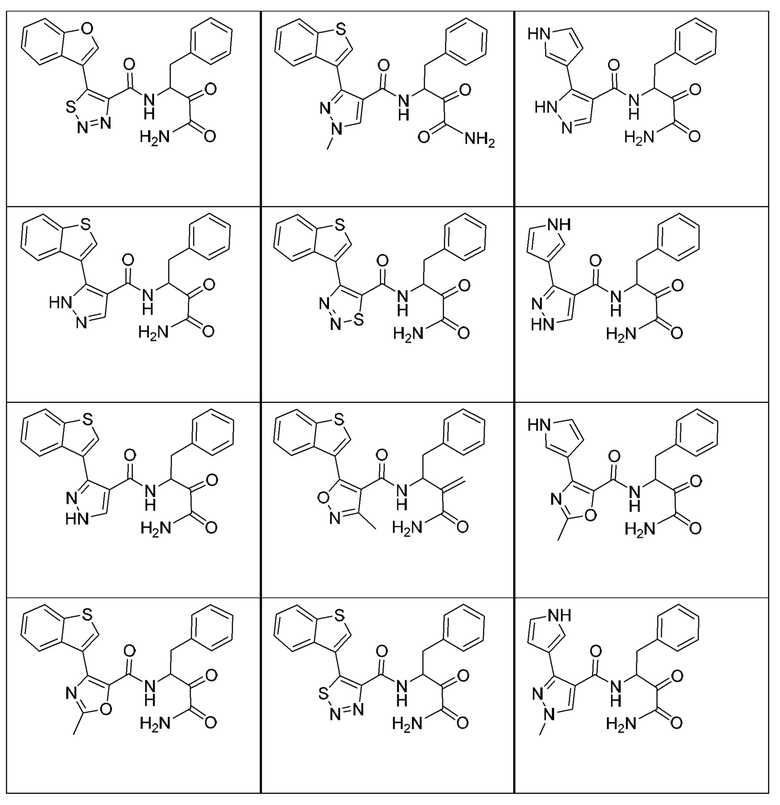
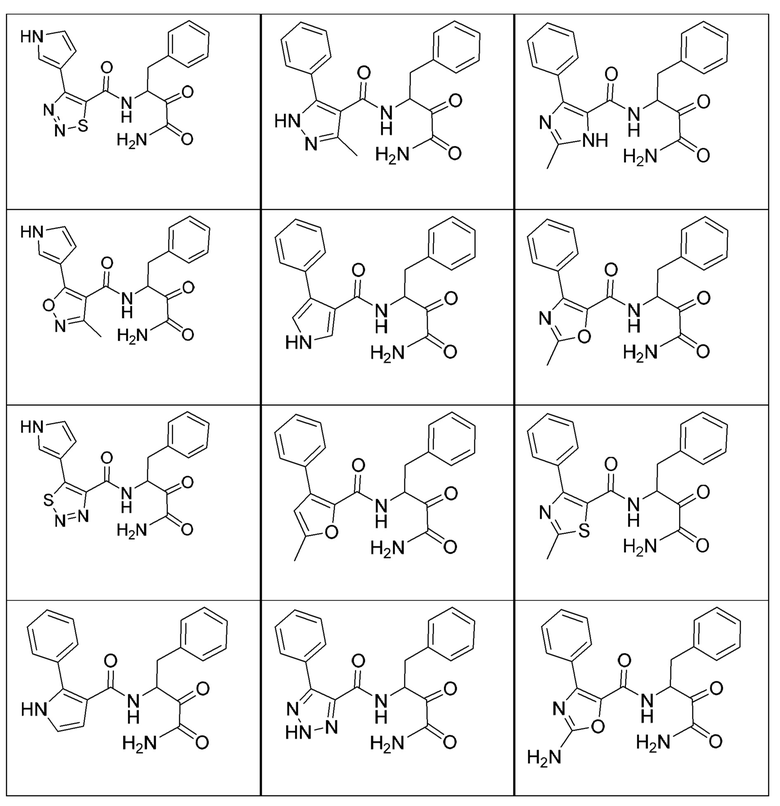
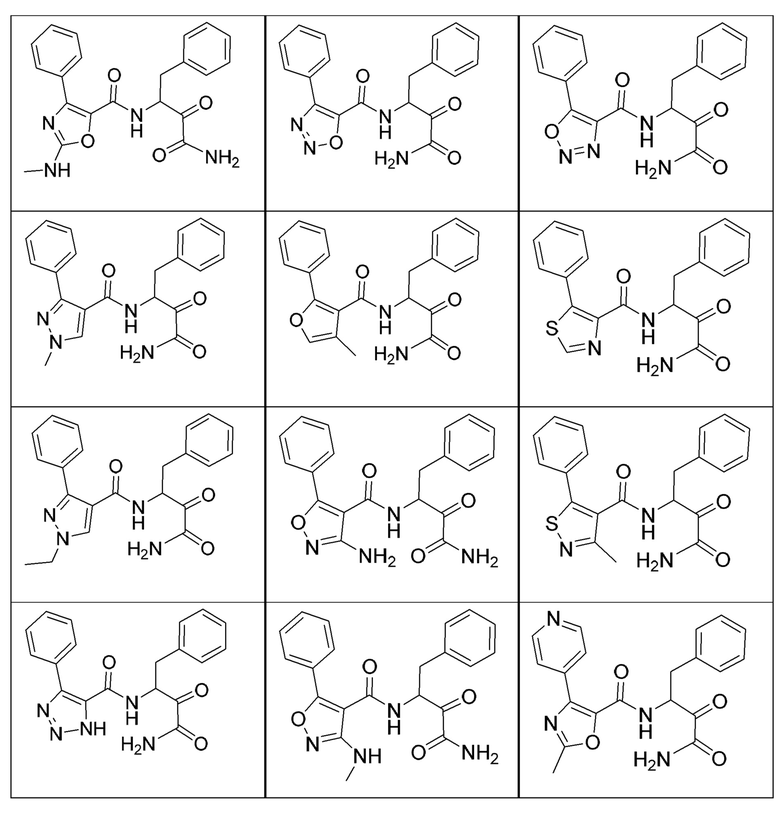
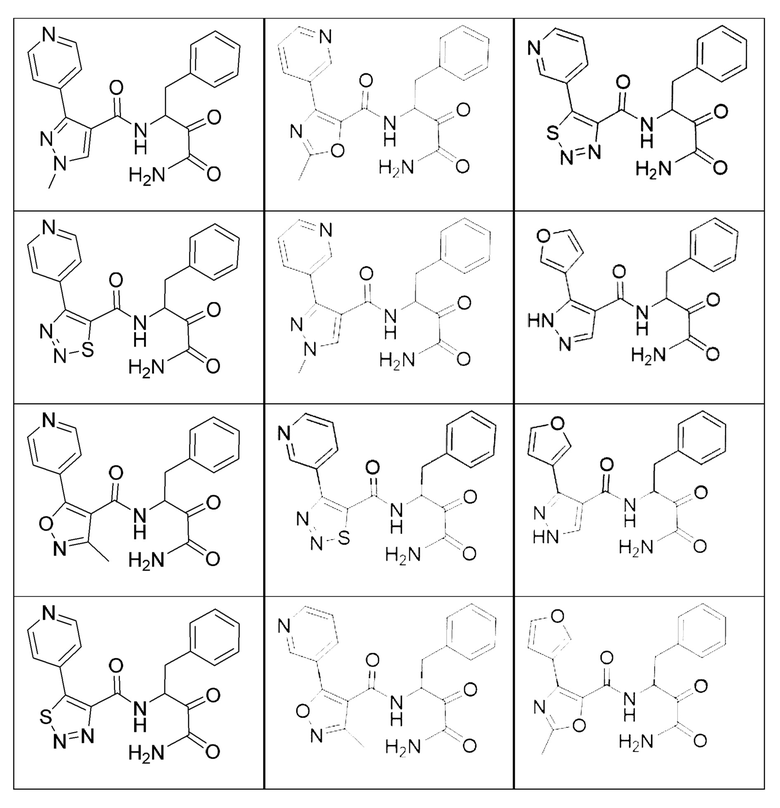
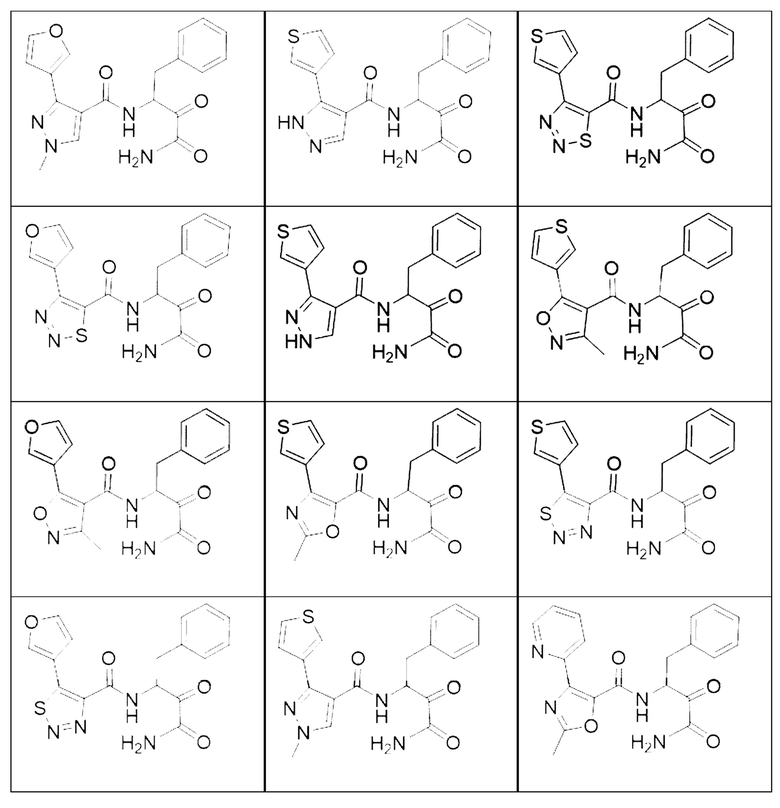
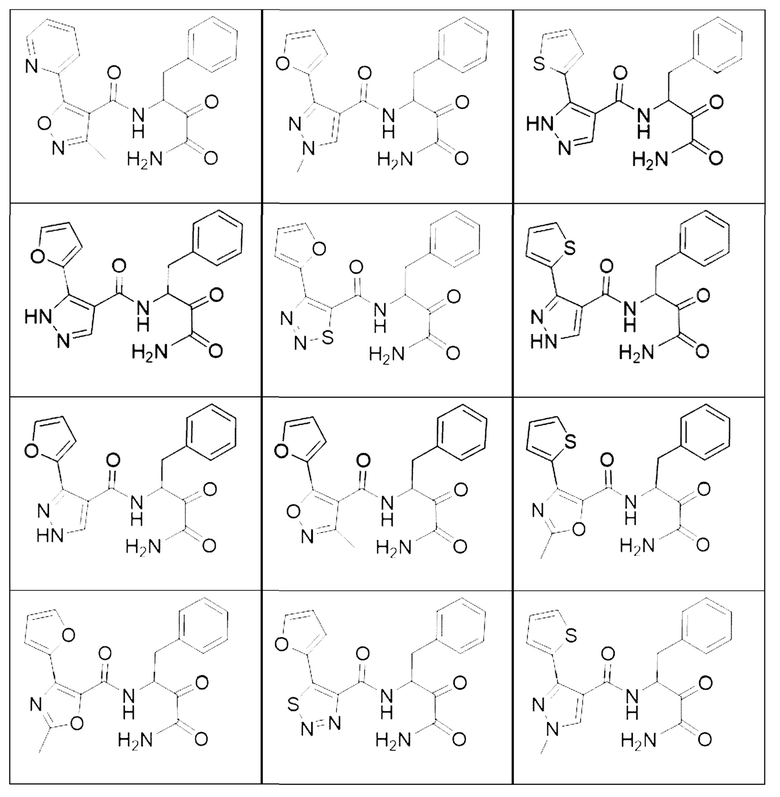
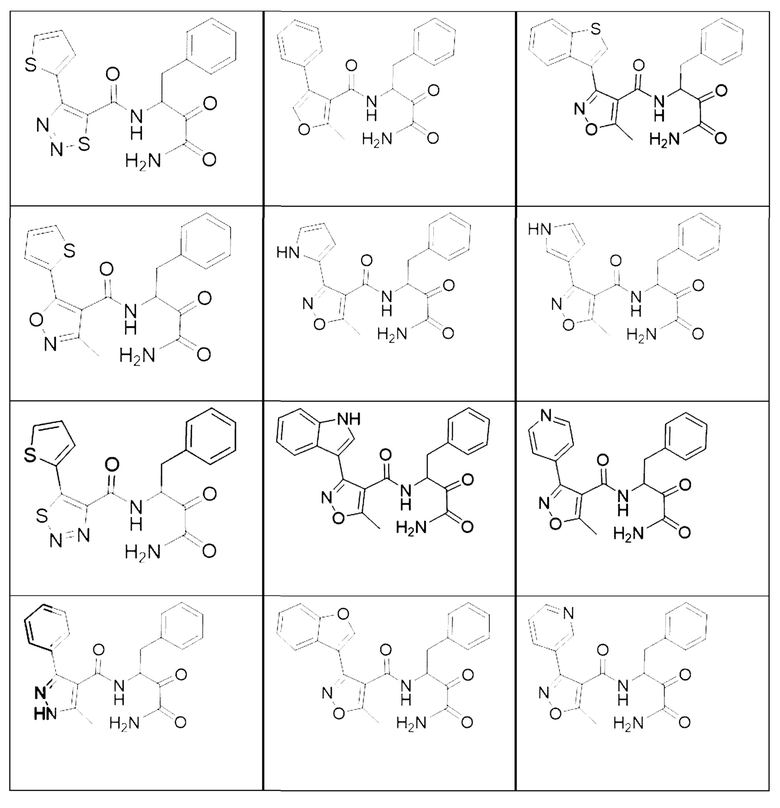
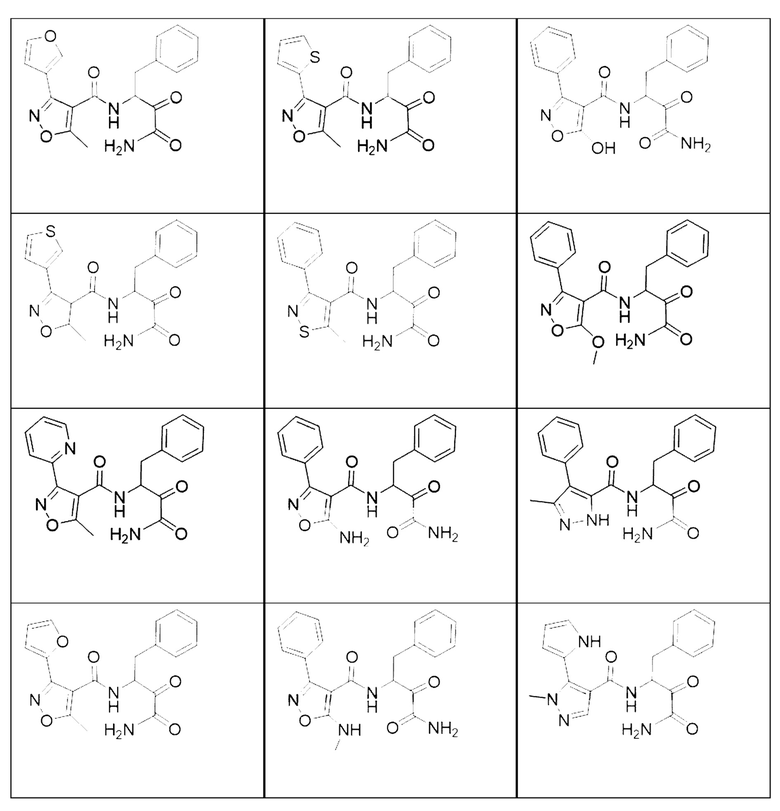
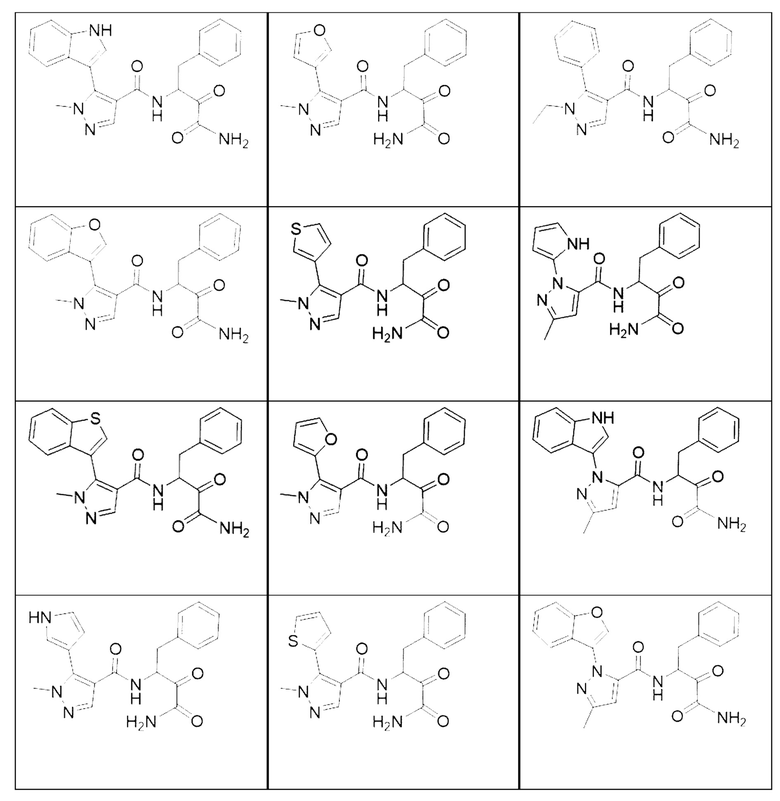
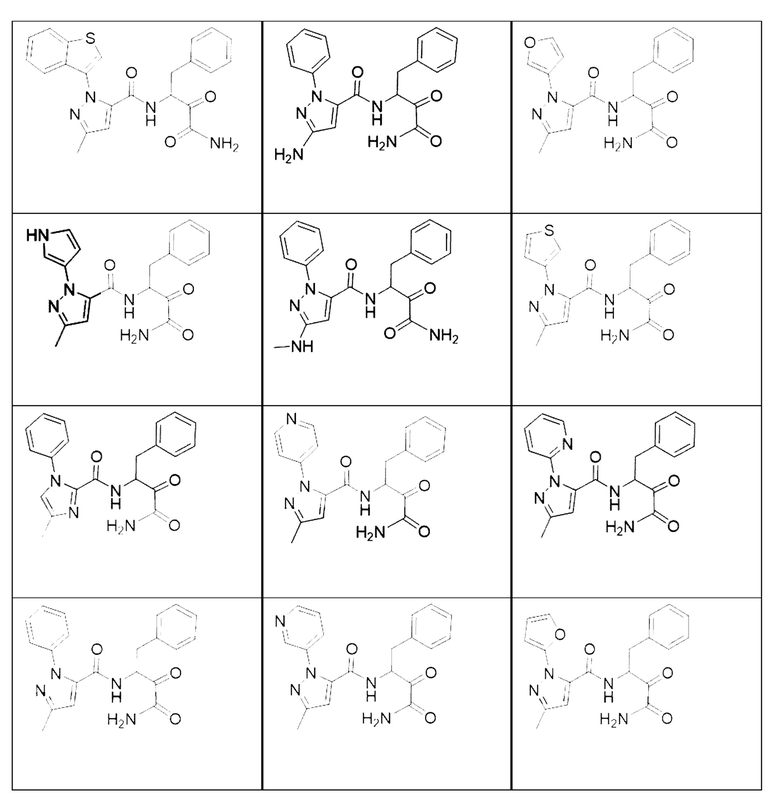
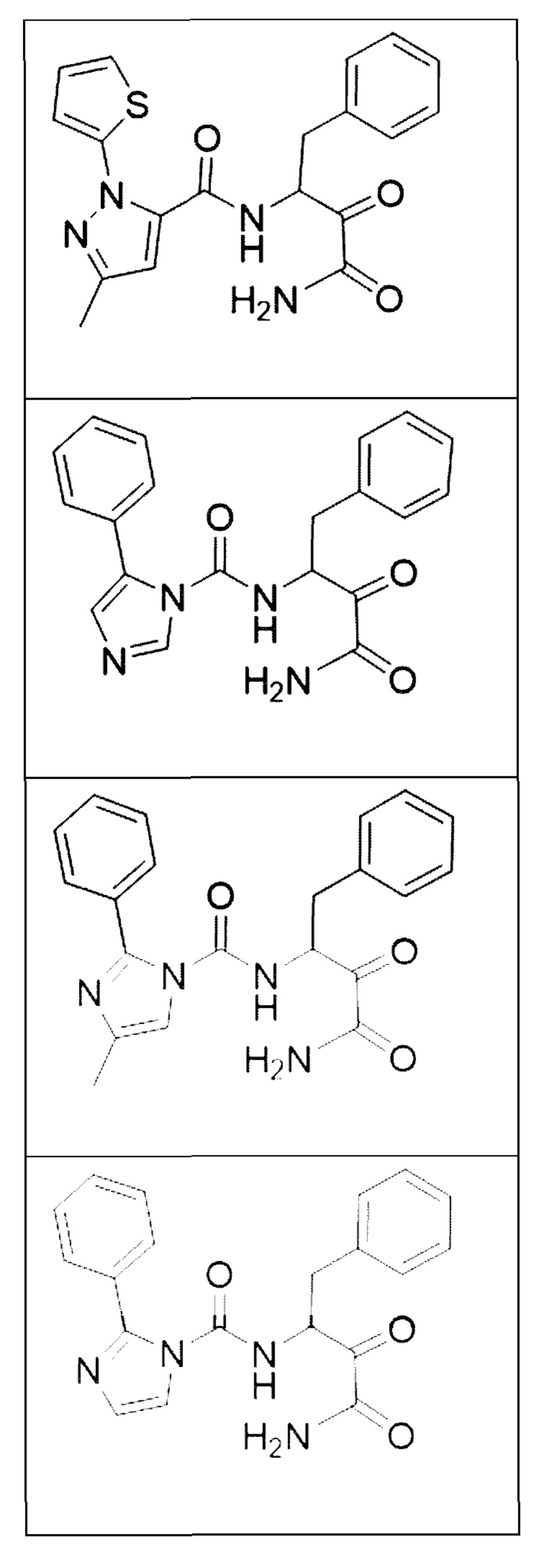
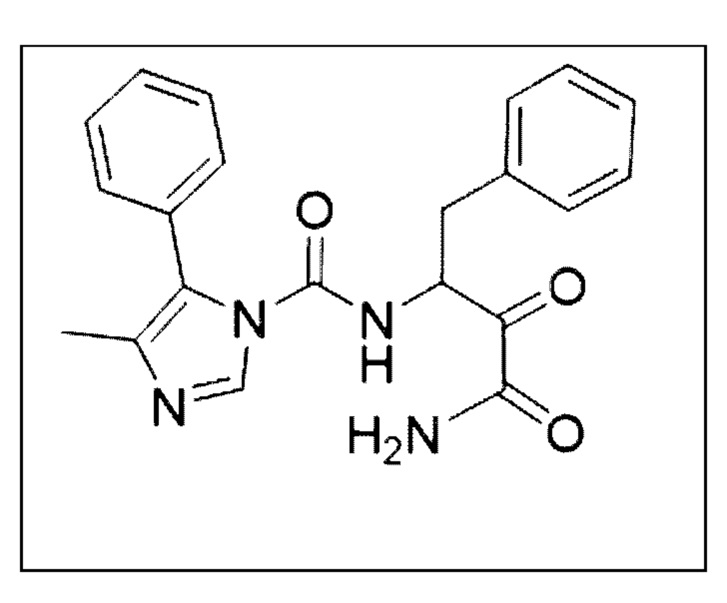
[0215] В случае, если соединения, раскрытые в настоящем описании, имеют по меньшей мере один хиральный центр, они могут существовать в виде отдельных энантиомеров и диастереомеров или в виде смесей таких изомеров, включая рацематы. Разделение отдельных изомеров или селективный синтез отдельных изомеров осуществляют путем применения различных способов, которые хорошо известны практикующим специалистам в данной области техники. Если не указано иное, все такие изомеры и их смеси включены в объем соединений, раскрытых в настоящем описании. Кроме того, соединения, раскрытые в настоящем описании, могут существовать в одной или более кристаллических или аморфных формах. Если не указано иное, все такие формы включены в объем соединений, раскрытых в настоящем описании, включая любые полиморфные формы. Помимо этого, некоторые из соединений, раскрытых в настоящем описании, могут образовывать сольваты с водой (то есть гидраты) или обычными органическими растворителями. Если не указано иное, такие сольваты включены в объем соединений, раскрытых в настоящем описании.
[0216] Специалисту в данной области техники будет понятно, что некоторые структуры, описанные в настоящем описании, могут представлять собой резонансные формы или таутомеры соединений, которые могут быть достаточно точно представлены другими химическими структурами даже с кинетической точки зрения; специалисту понятно, что такие структуры могут соответствовать лишь очень небольшой части образца такого соединения (соединений). Предполагается, что такие соединения включены в объем изображенных структур, хотя такие резонансные формы или таутомеры не представлены в настоящем описании.
Изотопно-меченные соединения
[0217] В описанных соединениях могут присутствовать изотопы. Каждый химический элемент, представленный в структуре соединения, может включать любой изотоп указанного элемента. Указанные изотопы могут представлять собой изотопы углерода, хлора, фтора, водорода, иода, азота, кислорода, фосфора, серы и технеция, включая 11С, 13С, 14С, 36Cl, 18F, 2Н, 3Н, 123I, 125I, 13N, 15N, 15О, 17О, 18О, 31Р, 32Р, 35S и 99mTc. Например, в структуре соединения атом водорода может быть явным образом указан или подразумеваться как присутствующий в соединении. В любом положении соединения, в котором может присутствовать атом водорода, указанный атом водорода может представлять собой любой изотоп водорода, включая, но не ограничиваясь перечисленными, водород-1 (протий) и водород-2 (дейтерий). Таким образом, в настоящем описании ссылка на соединение охватывает все потенциальные изотопные формы, если контекстом явным образом не продиктовано иное.
Изотопно-меченные соединения согласно настоящим вариантам реализации пригодны для применения в исследованиях распределения лекарственного вещества и субстрата в тканях и определении занятости мишени. Например, изотопно-меченные соединения в частности пригодны для применения в ОФЭКТ (однофотонной эмиссионной компьютерной томографии) и ПЭТ (позитронно-эмиссионной томографии), что обсуждается далее в настоящем описании.
Определения
[0218] Если не указано иное, все технические и научные термины в контексте настоящего описания имеют значения, обычно понимаемые средним специалистом в области техники, к которой относится настоящее описание. Содержание всех патентов, заявок, опубликованных заявок и других публикаций полностью включено в настоящее описание посредством ссылки. В случае, если существует множество определений термина, используемого в настоящем описании, приведенные в этом разделе определения имеют преимущественную силу, если не указано иное.
[0219] Термин «пролекарство» относится к агенту, который превращается в исходное лекарственное средство in vivo. Пролекарства часто являются пригодными для применения, поскольку в некоторых ситуациях их может быть легче вводить, чем исходное лекарственное средство. Например, они могут быть биодоступными при пероральном введении, тогда как для исходного лекарственного средства это не так. Пролекарство также может иметь улучшенную растворимость в фармацевтических композициях по сравнению с исходным лекарственным средством. Неограничивающим примером пролекарства может являться соединение, которое вводят в виде сложного эфира («пролекарства») для облегчения проникновения через клеточную мембрану, где растворимость в воде отрицательно влияет на подвижность, но которое далее метаболически гидролизуется до карбоновой кислоты, представляющей собой активное соединение, как только оказывается внутри клетки, где растворимость в воде обеспечивает положительный эффект Другим примером пролекарства может являться короткий пептид (полиаминокислота), связанный с кислотной группой, при этом указанный пептид метаболизируется с высвобождением активного фрагмента. Традиционные процедуры выбора и получения подходящих пролекарственных производных описаны, например, в источнике Design of Prodrugs (ed. H. Bundgaard, Elsevier, 1985), содержание которого полностью включено в настоящее описание посредством ссылки.
[0220] Термин «Пролекарство, представляющее собой сложный эфир» относится к производным соединений, раскрытых в настоящем описании, полученным путем добавления любой из нескольких образующих сложные эфиры групп, которые гидролизуются в физиологических условиях. Примеры сложноэфирных групп пролекарств включают пивоилоксиметил, ацетоксиметил, фталидил, инданил и метоксиметил, а также другие такие группы, известные в данной области техники, включая (5-R-2-оксо-1,3-диоксолен-4-ил)метильную группу. Другие примеры сложноэфирных групп пролекарств можно найти, например, в источниках Т. Higuchi and V. Stella, «Pro-drugs as Novel Delivery Systems», Vol. 14, A.C.S. Symposium Series, American Chemical Society (1975) и «Bioreversible Carriers in Drug Design: Theory and Application», edited by E.B. Roche, Pergamon Press: New York, 14-21 (1987) (где приведены примеры сложных эфиров, подходящих для применения в качестве пролекарств для соединений, содержащих карбоксильные группы). Содержание каждого из указанных выше источников полностью включено в настоящее описание посредством ссылки.
[0221] «Метаболиты» соединений, раскрытых в настоящем описании, включают активные вещества, которые образуются при введении указанных соединений в биологическую среду.
[0222] «Сольват» относится к соединению, полученному путем взаимодействия растворителя и соединения, описанного в настоящем описании, метаболита или соли указанного соединения. Подходящими сольватами являются фармацевтически приемлемые сольваты, включая гидраты.
[0223] Термин «фармацевтически приемлемая соль» относится к солям, которые сохраняют биологическую эффективность и свойства соединения, и которые не являются биологически или иным образом нежелательными для применения в фармацевтическом средстве. Во многих случаях соединения согласно настоящему описанию способны образовывать кислотные и/или основные соли благодаря присутствию амино- и/или карбоксильных групп или групп, подобных им. Фармацевтически приемлемые кислотно-аддитивные соли могут быть получены с применением неорганических кислот и органических кислот. Неорганические кислоты, из которых могут быть получены соли, включают, например, хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и тому подобное. Органические кислоты, из которых могут быть получены соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, пировиноградную кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, коричную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, n-толуолсульфоновую кислоту, салициловую кислоту и тому подобное. Фармацевтически приемлемые основно-аддитивные соли могут быть получены с применением неорганических и органических оснований. Неорганические основания, из которых могут быть получены соли, включают, например, основания, содержащие натрий, калий, литий, аммоний, кальций, магний, железо, цинк, медь, марганец, алюминий и тому подобное; особенно предпочтительными являются соли аммония, калия, натрия, кальция и магния. Органические основания, из которых могут быть получены соли, включают, например, первичные, вторичные и третичные амины, замещенные амины, включая встречающиеся в природе замещенные амины, циклические амины, основные ионообменные смолы и тому подобное, в частности, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин и этаноламин. Многие такие соли известны в данной области техники, как описано в источнике WO 87/05297, Johnston et al., опубликованном 11 сентября 1987 года (содержание которого полностью включено в настоящее описание посредством ссылки).
[0224] В контексте настоящего описания «Са-Cb» или «Ca-b», где «а» и «b» представляют собой целые числа, относится к числу атомов углерода в указанной группе. То есть указанная группа может содержать от «а» до «b» атомов углерода включительно. Таким образом, например, «C1-C4 алкильная» или «C1-4 алкильная» группа относится ко всем алкильным группам, содержащим от 1 до 4 атомов углерода, то есть СН3-, CH3CH2-, СН3СН2СН2-, (СН3)2СН-, СН3СН2СН2СН2-, СН3СН2СН(СН3)- и (СН3)3С-.
[0225] Термин «галоген» в контексте настоящего описания означает любой из нерадиоактивных атомов главной подгруппы 7 группы периодической таблицы элементов, например, фтор, хлор, бром или иод, при этом фтор и хлор являются предпочтительными.
[0226] В контексте настоящего описания «алкил» относится к неразветвленной или разветвленной углеводородной цепи, которая является полностью насыщенной (то есть не содержит двойных или тройных связей). Алкильная группа может содержать от 1 до 20 атомов углерода (при каждом появлении в настоящем описании числовой диапазон, такой как «от 1 до 20», относится к каждому целому числу в данном диапазоне; например «от 1 до 20 атомов углерода» означает, что алкильная группа может состоять из 1 атома углерода, 2 атомов углерода, 3 атомов углерода и т.д. до 20 атомов углерода включительно, хотя настоящее определение также охватывает употребление термина «алкил», когда числовой диапазон не обозначен). Алкильная группа также может представлять собой алкил среднего размера, содержащий от 1 до 9 атомов углерода. Алкильная группа также может представлять собой низший алкил, содержащий от 1 до 4 атомов углерода. Алкильная группа соединений может быть обозначена как «C1-4 алкил» или сходными обозначениями. Исключительно для примера, «C1-4 алкил» указывает на то, что в алкильной цепи содержится от одного до четырех атомов углерода, то есть указанная алкильная цепь выбрана из группы, состоящей из метила, этила, пропила, изопропила, н-бутила, изобутила, втор-бутила. и трет-бутила. Типичные алкильные группы включают, но не ограничиваются перечисленными, метил, этил, пропил, изопропил, бутил, изобутил, третичный бутил, пентил, гексил и тому подобное.
[0227] В контексте настоящего описания «галогеналкил» относится к алкильной группе с неразветвленной или разветвленной цепью, содержащий от 1 до 12 атомов углерода в цепи, в которой один или более атомов водорода заменены на атомы галогена. Примеры галогеналкильных групп включают, но не ограничиваются перечисленными, -CF3, -CHF2, -CH2F, -CH2CF3, -CH2CHF2, -CH2CH2F, -CH2CH2Cl, -CH2CH2CF3 и другие группы, которые, в соответствии со средним уровнем компетентности в данной области техники и принципами, представленными в настоящем описании, будут считаться эквивалентными любому из приведенных выше примеров.
[0228] В контексте настоящего описания «алкокси» относится к группе формулы -OR, где R представляет собой алкил, определенный выше, такой как «C1-9 алкокси», включая, но не ограничиваясь перечисленными, метокси, этокси, н-пропокси, 1-метилэтокси (изопропокси), н-бутокси, изобутокси, втор-бутокси и трет-бутокси и тому подобное.
[0229] В контексте настоящего описания «полиэтиленгликоль» относится к группе формулы 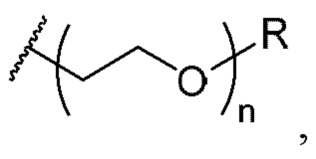 где n представляет собой целое число больше единицы, и R представляет собой атом водорода или алкил. Количество повторяющихся звеньев «n» может быть обозначено путем указания на количество членов. Так, например, «2-5-членный полиэтиленгликоль» относится к n, представляющему собой целое число, выбранное из чисел от двух до пяти. В некоторых вариантах реализации R выбран из метокси, этокси, н-пропокси, 1-метилэтокси (изопропокси), н-бутокси, изобутокси, втор-бутокси и трет-бутокси.
где n представляет собой целое число больше единицы, и R представляет собой атом водорода или алкил. Количество повторяющихся звеньев «n» может быть обозначено путем указания на количество членов. Так, например, «2-5-членный полиэтиленгликоль» относится к n, представляющему собой целое число, выбранное из чисел от двух до пяти. В некоторых вариантах реализации R выбран из метокси, этокси, н-пропокси, 1-метилэтокси (изопропокси), н-бутокси, изобутокси, втор-бутокси и трет-бутокси.
[0230] В контексте настоящего описания «гетероалкил» относится к неразветвленной или разветвленной углеводородной цепи, содержащей в основной цепи один или более гетероатомов, то есть элементов, отличных от углерода, включая, но не ограничиваясь перечисленными, азот, кислород и серу. Гетероалкильная группа может содержать от 1 до 20 атомов углерода, хотя настоящее определение также охватывает употребление термина «гетероалкил», когда числовой диапазон не обозначен. Гетероалкильная группа также может представлять собой гетероалкил среднего размера, содержащий от 1 до 9 атомов углерода. Гетероалкильная группа также может представлять собой низший гетероалкил, содержащий от 1 до 4 атомов углерода. В различных вариантах реализации гетероалкил может содержать от 1 до 4 гетероатомов, от 1 до 3 гетероатомов, 1 или 2 гетероатома или 1 гетероатом. Гетероалкильная группа в соединениях может быть обозначена как «C1-4 гетероалкил» или сходными обозначениями. Гетероалкильная группа может содержать один или более гетероатомов. Исключительно для примера, «C1-4 гетероалкил» указывает на то, что в гетероалкильной цепи содержится от одного до четырех атомов углерода, и в основной цепи дополнительно содержится один или более гетероатомов.
[0231] Термин «ароматический» относится к кольцу или кольцевой системе, содержащей сопряженную пи-электронную систему, и включает как карбоциклические ароматические группы (например, фенил), так и гетероциклические ароматические группы (например, пиридин). Указанный термин включает моноциклические группы или полициклические группы с конденсированными кольцами (то есть кольцами, которые имеют общие соседние пары атомов) при условии, что кольцевая система в целом является ароматической.
[0232] В контексте настоящего описания «арил» относится к ароматическому кольцу или кольцевой системе (то есть двум или более конденсированным кольцам, которые имеют два общих соседних атома углерода), содержащей в основной цепи кольца только атомы углерода. В случае, если арил представляет собой кольцевую систему, каждое кольцо в указанной системе является ароматическим. Арильная группа может содержать от 6 до 18 атомов углерода, хотя настоящее определение также охватывает употребление термина «арил», когда числовой диапазон не обозначен. В некоторых вариантах реализации арильная группа содержит от 6 до 10 атомов углерода. Арильная группа может быть обозначена как «С6-10 арил», «С6 или С10 арил» или сходными обозначениями. Примеры арильных групп включают, но не ограничиваются перечисленными, фенил, нафтил, азуленил и антраценил.
[0233] В контексте настоящего описания «арилокси» и «арилтио» относятся к группам RO- и RS-, в которых R представляет собой арил, определенный выше, таким как «С6-10 арилокси» или «С6-10 арилтио» и тому подобное, включая фенилокси, но не ограничиваясь им.
[0234] «Аралкил» или «арилалкил» представляет собой арильную группу, присоединяемую в качестве заместителя через алкиленовую группу, такую как «C7-14 аралкил» и тому подобное, включая, но не ограничиваясь перечисленными, бензил, 2-фенилэтил, 3-фенилпропил и нафтилалкил. В некоторых случаях указанная алкиленовая группа представляет собой низшую алкиленовую группу (то есть C1-4 алкиленовую группу).
[0235] В контексте настоящего описания «гетероарил» относится к ароматическому кольцу или кольцевой системе (то есть двум или более конденсированным кольцам, которые имеют два общих соседних атома), которая содержит в основной цепи кольца один или более гетероатомов, то есть элементов, отличных от углерода, включая, но не ограничиваясь перечисленными, азот, кислород и серу. В случае, если гетероарил представляет собой кольцевую систему, каждое кольцо в указанной системе является ароматическим. Гетероарильная группа может содержать 5-18 членов кольца (то есть указанное число атомов, составляющих основную цепь кольца, включая атомы углерода и гетероатомы), хотя настоящее определение также охватывает употребление термина «гетероарил», когда числовой диапазон не обозначен. В некоторых вариантах реализации гетероарильная группа содержит от 5 до 10 членов кольца или от 5 до 7 членов кольца. Гетероарильная группа может быть обозначена как «5-7-членный гетероарил», «5-10-членный гетероарил» или сходными обозначениями. В различных вариантах реализации гетероарил содержит от 1 до 4 гетероатомов, от 1 до 3 гетероатомов, от 1 до 2 гетероатомов или 1 гетероатом. Например, в различных вариантах реализации гетероарил содержит от 1 до 4 атомов азота, от 1 до 3 атомов азота, от 1 до 2 атомов азота, 2 атома азота и 1 атом серы или кислорода, 1 атом азота и 1 атом серы или кислорода, или 1 атом серы или кислорода. Примеры гетероарильных колец включают, но не ограничиваются перечисленными, фурил, тиенил, фталазинил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, триазолил, тиадиазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, триазинил, хинолинил, изохинолинил, бензимидазолил, бензоксазолил, бензотиазолил, индолил, изоиндолил и бензотиенил.
[0236] «Гетероаралкил» или «гетероарилалкил» представляет собой гетероарильную группу, присоединяемую в качестве заместителя через алкиленовую группу. Примеры включают, но не ограничиваются перечисленными, 2-тиенилметил, 3-тиенилметил, фурилметил, тиенилэтил, пирролилалкил, пиридилалкил, изоксазолилалкил и имидазолилалкил. В некоторых случаях указанная алкиленовая группа представляет собой низшую алкиленовую группу (то есть C1-4 алкиленовую группу).
[0237] В контексте настоящего описания «карбоциклил» означает неароматическое циклическое кольцо или кольцевую систему, содержащую в основной цепи кольцевой системы только атомы углерода. В случае, если карбоциклил представляет собой кольцевую систему, два или более колец могут быть соединены друг с другом таким образом, что образуются конденсированные, мостиковые или спиросоединения. Карбоциклилы могут иметь любую степень насыщения, при условии, что по меньшей мере одно кольцо в кольцевой системе не является ароматическим. Таким образом, карбоциклилы включают циклоалкилы, циклоалкенилы и циклоалкинилы. Карбоциклильная группа может содержать от 3 до 20 атомов углерода, хотя настоящее определение также охватывает употребление термина «карбоциклил», когда числовой диапазон не обозначен. Карбоциклильная группа также может представлять собой карбоциклил среднего размера, содержащий от 3 до 10 атомов углерода. Карбоциклильная группа также может представлять собой карбоциклил, содержащий от 3 до 6 атомов углерода. Карбоциклильная группа может быть обозначена как «С3-6 карбоциклил» или сходными обозначениями. Примеры карбоциклильных колец включают, но не ограничиваются перечисленными, циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, 2,3-дигидроинден, бицикло[2.2.2]октанил, адамантил и спиро[4.4]нонанил.
[0238] «(Карбоциклил)алкил» представляет собой карбоциклильную группу, присоединяемую в качестве заместителя через алкиленовую группу, такую как «С4-10 (карбоциклил)алкил» и тому подобное, включая, но не ограничиваясь перечисленными, циклопропилметил, циклобутилметил, циклопропилэтил, циклопропилбутил, циклобутилэтил, циклопропилизопропил, циклопентилметил, циклопентилэтил, циклогексилметил, циклогексилэтил, циклогептилметил и тому подобное. В некоторых случаях указанная алкиленовая группа представляет собой низшую алкиленовую группу.
[0239] В контексте настоящего описания «циклоалкил» означает полностью насыщенное карбоциклильное кольцо или кольцевую систему. Примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
[0240] В контексте настоящего описания «циклоалкенил» означает карбоциклильное кольцо или кольцевую систему, содержащую по меньшей мере одну двойную связь, при этом ни одно кольцо в указанной кольцевой системе не является ароматическим. Примером является циклогексенил.
[0241] В контексте настоящего описания «гетероциклил» означает неароматическое циклическое кольцо или кольцевую систему, содержащую в основной цепи кольца по меньшей мере один гетероатом. Гетероциклилы могут быть соединены друг с другом таким образом, что образуются конденсированные, мостиковые или спиросоединения. Гетероциклилы могут иметь любую степень насыщения, при условии, что по меньшей мере одно кольцо в кольцевой системе не является ароматическим. Гетероатом (гетероатомы) может присутствовать в неароматическом или ароматическом кольце кольцевой системы. Гетероциклильная группа может содержать от 3 до 20 членов кольца (то есть указанное число атомов, составляющих кольцевой остов, включая атомы углерода и гетероатомы), хотя настоящее определение также охватывает употребление термина «гетероциклил», когда числовой диапазон не обозначен. Гетероциклильная группа также может представлять собой гетероциклил среднего размера, содержащий от 3 до 10 членов кольца. Гетероциклильная группа также может представлять собой гетероциклил, содержащий от 3 до 6 членов кольца. Гетероциклильная группа может быть обозначена как «3-6-членный гетероциклил» или сходными обозначениями.
[0242] В различных вариантах реализации гетероциклил содержит от 1 до 4 гетероатомов, от 1 до 3 гетероатомов, от 1 до 2 гетероатомов или 1 гетероатом. Например, в различных вариантах реализации гетероциклил содержит от 1 до 4 атомов азота, от 1 до 3 атомов азота, от 1 до 2 атомов азота, 2 атома азота и 1 атом серы или кислорода, 1 атом азота и 1 атом серы или кислорода, или 1 атом серы или кислорода. В предпочтительных шестичленных моноциклических гетероциклилах гетероатом (гетероатомы) выбран из от одного до трех из О, N или S, и в предпочтительных пятичленных моноциклических гетероциклилах гетероатом (гетероатомы) выбран из одного или двух гетероатомов, выбранных из О, N или S. Примеры гетероциклильных колец включают, но не ограничиваются перечисленными, азепинил, акридинил, карбазолил, циннолинил, диоксоланил, имидазолинил, имидазолидинил, морфолинил, оксиранил, оксепанил, тиепанил, пиперидинил, пиперазинил, диоксопиперазинил, пирролидинил, пирролидонил, пирролидионил, 4-пиперидонил, пиразолинил, пиразолидинил, 1,3-диоксинил, 1,3-диоксанил, 1,4-диоксинил, 1,4-диоксанил, 1,3-оксатианил, 1,4-оксатиинил, 1,4-оксатианил, 2H-1,2-оксазинил, триоксанил, гексагидро-1,3,5-триазинил, 1,3-диоксолил, 1,3-диоксоланил, 1,3-дитиолил, 1,3-дитиоланил, изоксазолинил, изоксазолидинил, оксазолинил, оксазолидинил, оксазолидинонил, тиазолинил, тиазолидинил, 1,3-оксатиоланил, индолинил, изоиндолинил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиофенил, тетрагидротиопиранил, тетрагидро-1,4-тиазинил, тиаморфолинил, дигидробензофуранил, бензимидазолидинил и тетрагидрохинолин.
[0243] «(Гетероциклил)алкил» представляет собой гетероциклильную группу, присоединяемую в качестве заместителя через алкиленовую группу. Примеры включают, но не ограничиваются перечисленными, имидазолинилметил и индолинилэтил.
[0244] В контексте настоящего описания «ацил» относится к -C(=O)R, где R представляет собой атом водорода, C1-6 алкил, C2-6 алкенил, C2-6 алкинил, С3-7 карбоциклил, арил, 5-10-членный гетероарил и 5-10-членный гетероциклил, определенные в настоящем описании. Неограничивающие примеры включают формил, ацетил, пропаноил, бензоил и акрил.
[0245] Группа «O-карбокси» относится к группе «-OC(=O)R», в которой R выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0246] Группа «С-карбокси» относится к группе «-C(=O)OR», в которой R выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании. Неограничивающий пример включает карбоксил (то есть -С(=O)ОН).
[0247] Группа «циано» относится к группе «-CN».
[0248] Группа «цианато» относится к группе «-OCN».
[0249] Группа «изоцианато» относится к группе «-NCO».
[0250] Группа «тиоцианато» относится к группе «-SCN».
[0251] Группа «изотиоцианато» относится к группе «-NCS».
[0252] Группа «сульфинил» относится к группе «-S(=O)R», в которой R выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0253] Группа «сульфонил» относится к группе «-SO2R», в которой R выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0254] Группа «S-сульфонамидо» относится к группе «-SO2NRARB», в которой каждый из RA и RB независимо выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0255] Группа «N-сульфонамидо» относится к группе «-N(RA)SO2RB», в которой каждый из RA и RB независимо выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0256] Группа «O-карбамил» относится к группе «-OC(=O)NRARB», в которой каждый из RA и RB независимо выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0257] Группа «N-карбамил» относится к группе «-N(RA)OC(=O)RB», в которой каждый из RA и RB независимо выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0258] Группа «O-тиокарбамил» относится к группе «-OC(=S)NRARB», в которой каждый из RA и RB независимо выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0259] Группа «N-тиокарбамил» относится к группе «-N(RA)OC(=S)RB», в которой каждый из RA и RB независимо выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0260] Группа «С-амидо» относится к группе «-C(=O)NRARB», в которой каждый из RA и RB независимо выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0261] Группа «N-амидо» относится к группе «-N(RA)C(=O)RB», в которой каждый из RA и RB независимо выбран из атома водорода, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила 5-10-членного гетероциклила, определенных в настоящем описании.
[0262] Группа «амино» относится к группе «-NRARB», в которой каждый из RA и RB независимо выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 5-10-членного гетероциклила, определенных в настоящем описании.
[0263] Группа «аминоалкил» относится к аминогруппе, связанной через алкиленовую группу.
[0264] Группа «алкоксиалкил» относится к алкоксигруппе, связанной через алкиленовую группу, такой как «С2-8 алкоксиалкил» и тому подобное.
[0265] В контексте настоящего описания «боковая цепь природной аминокислоты» относится к заместителю боковой цепи встречающейся в природе аминокислоты. Встречающиеся в природе аминокислоты содержат заместитель, присоединенный к α-атому углерода. Встречающиеся в природе аминокислоты включают аргинин, лизин, аспарагиновую кислоту, глутаминовую кислоту, глутамин, аспарагин, гистидин, серии, треонин, тирозин, цистеин, метионин, триптофан, аланин, изолейцин, лейцин, фенилаланин, валин, пролин и глицин.
[0266] В контексте настоящего описания «боковая цепь неприродной аминокислоты» относится к заместителю боковой цепи не встречающейся в природе аминокислоты. Неприродные аминокислоты включают β-аминокислоты (β3 и β2), гомоаминокислоты, производные пролина и пировиноградной кислоты, 3-замещенные производные аланина, производные глицина, содержащие заместитель в кольце производные фенилаланина и тирозина, аминокислоты с линейной основной цепью и N-метиламинокислоты. Иллюстративные неприродные аминокислоты доступны в Sigma-Aldridge, перечислены в разделе «неприродные аминокислоты и производные». См. также источник Travis S. Young and Peter G. Schultz, «Beyond the Canonical 20 Amino Acids: Expanding the Genetic Lexicon», J. Biol. Chem. 2010 285: 11039-11044, содержание которого полностью включено в настоящее описание посредством ссылки.
[0267] В контексте настоящего описания замещенную группу получают из незамещенной исходной группы, в которой была произведена замена одного или более атомов водорода на другой атом или группу. Если не указано иное, когда считается, что группа является «замещенной», это означает, что указанная группа замещена одним или более заместителями, независимо выбранными из C1-С6 алкила, C1-С6 алкенила, C1-С6 алкинила, C1-С6 гетероалкила, С3-С7 карбоциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), С3-С7-карбоциклил-С1-С6-алкила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), 5-10-членного гетероциклила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), 5-10-членного гетероциклил-C1-С6-алкила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), арила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), арил(С1-С6)алкила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), 5-10-членного гетероарила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), 5-10-членного гетероарил(С1-С6)алкила (необязательно замещенного галогеном, C1-С6 алкилом, C1-С6 алкокси, C1-С6 галогеналкилом и C1-С6 галогеналкокси), галогена, циано, гидрокси, C1-С6 алкокси, C1-С6 алкокси(С1-С6)алкила (то есть простого эфира), арилокси, сульфгидрила (меркапто), галоген(С1-С6)алкила (например, -CF3), галоген(С1-С6)алкокси (например, -OCF3), C1-С6 алкилтио, арилтио, амино, амино(С1-С6)алкила, нитро, O-карбамила, N-карбамила, O-тиокарбамила, N-тиокарбамила, С-амидо, N-амидо, S-сульфонамидо, N-сульфонамидо, С-карбокси, O-карбокси, ацила, цианато, изоцианато, тиоцианато, изотиоцианато, сульфинила, сульфонила и оксо (=O). Во всех случаях, когда группа описана как «необязательно замещенная», указанная группа может быть замещена указанными выше заместителями.
[0268] В некоторых вариантах реализации замещенная группа (группы) замещена (замещены) одним или более заместителями, отдельно и независимо выбранными из C1-C4 алкила, амино, гидрокси и галогена.
[0269] Следует понимать, что некоторые способы наименования радикалов могут включать или монорадикал, или бирадикал в зависимости от контекста. Например, в случае, если для заместителя требуются две точки присоединения к остальной части молекулы, подразумевается, что указанный заместитель представляет собой бирадикал. Например, заместитель, определяемый как алкил, для которого требуются две точки присоединения, включает бирадикалы, такие как -CH2-, -СН2СН2-, -СН2СН(СН3)СН2- и тому подобное. Другие способы наименования радикалов явно указывают на то, что указанный радикал представляет собой бирадикал, как то «алкилен» или «алкенилен».
[0270] В случае, если сообщается, что две группы R образуют кольцо (например, карбоциклильное, гетероциклильное, арильное или гетероарильное кольцо) «совместно с атомом, к которому они присоединены», это означает, что полученное звено, образованное указанным атомом и указанными двумя группами R, представляет собой упомянутое кольцо. Указанное кольцо иным образом не ограничено определением каждой группы R, взятой в отдельности. Например, в случае, если присутствует следующая подструктура:
и R1 и R2 определены как выбранные из группы, состоящей из атома водорода и алкила, или R1 и R2 совместно с атомом азота, к которому они присоединены, образуют гетероциклил, это означает, что R1 и R2 могут быть выбраны из атома водорода или алкила, или в качестве альтернативы указанная подструктура имеет структуру:
где кольцо А представляет собой гетероциклильное кольцо, содержащее изображенный атом азота.
[0271] Аналогичным образом, в случае, если сообщается, что две «соседние» группы R образуют кольцо «совместно с атомами, к которым они присоединены», это означает, что полученное звено, образованное указанными атомами, промежуточными связями и указанными двумя группами R, представляет собой упомянутое кольцо. Например, в случае, если присутствует следующая подструктура:
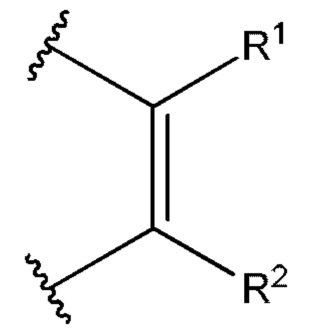
и R1 и R2 определены как выбранные из группы, состоящей из атома водорода и алкила, или R1 и R2 совместно с атомами, к которым они присоединены, образуют арил или карбоциклил, это означает, что R1 и R2 могут быть выбраны из атома водорода или алкила, или в качестве альтернативы указанная подструктура имеет структуру:
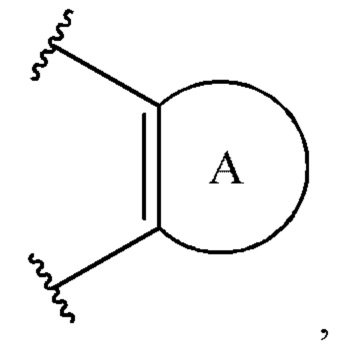
где А представляет собой арильное кольцо или карбоциклил, содержащий изображенную двойную связь.
[0272] Во всех случаях, когда заместитель изображен как бирадикал (то есть имеет две точки присоединения к остальной части молекулы), следует понимать, что указанный заместитель может быть присоединен в любом направлении, если не указано иное. Таким образом, например, заместитель, изображенный как -АЕ- или 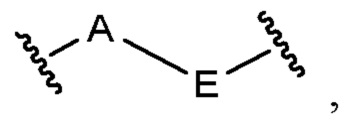 включает заместитель, ориентированный так, что А присоединен к крайней левой точке присоединения молекулы, а также случай, когда А присоединен к крайней правой точке присоединения указанной молекулы.
включает заместитель, ориентированный так, что А присоединен к крайней левой точке присоединения молекулы, а также случай, когда А присоединен к крайней правой точке присоединения указанной молекулы.
[0273] В контексте настоящего описания подструктура:
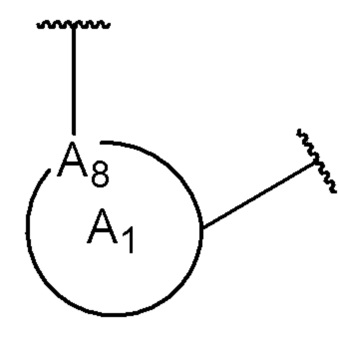 означает, что атом A8 может находиться в любом положении атома кольца внутри кольца или кольцевой системы A1. Подструктура:
означает, что атом A8 может находиться в любом положении атома кольца внутри кольца или кольцевой системы A1. Подструктура: 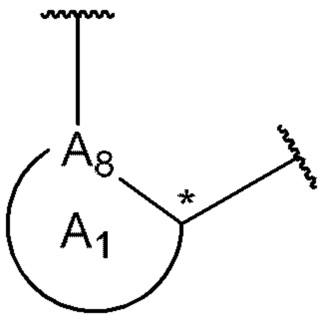 означает, что атом A8 находится в положении атома кольца, непосредственно соседнем (то есть альфа) по отношению к точке присоединения, обозначенной с помощью*.
означает, что атом A8 находится в положении атома кольца, непосредственно соседнем (то есть альфа) по отношению к точке присоединения, обозначенной с помощью*.
[0274] В контексте настоящего описания «изостеры» химической группы представляют собой другие химические группы, которые проявляют такие же или сходные свойства. Например, тетразол представляет собой изостер карбоновой кислоты, поскольку он имитирует свойства карбоновой кислоты даже несмотря на то, что они имеют очень разные молекулярные формулы. Тетразол представляет собой один из многих возможных изостерических заменителей карбоновой кислоты. Другие предполагаемые изостеры карбоновой кислоты включают -SO3H, -SO2HNR, -PO2(R)2, -РО3(R)2, -CONHNHSO2R, -COHNSO2R и -CONRCN, где R выбран из атома водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, С3-7 карбоциклила, С6-10 арила, 5-10-членного гетероарила и 3-10-членного гетероциклила, определенных в настоящем описании. Кроме того, изостеры карбоновой кислоты могут включать 5-7-членные карбоциклы или гетероциклы, содержащие любую комбинацию CH2, О, S или N в любой химически стабильной степени окисления, где любые из атомов указанной кольцевой структуры необязательно замещены в одном или более положениях. Следующие структуры представляют собой неограничивающие примеры предполагаемых карбоциклических и гетероциклических изостеров. Атомы указанной кольцевой структуры могут быть необязательно замещены R, определенным выше, в одном или более положениях.
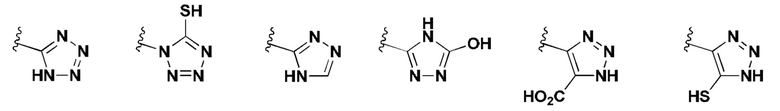
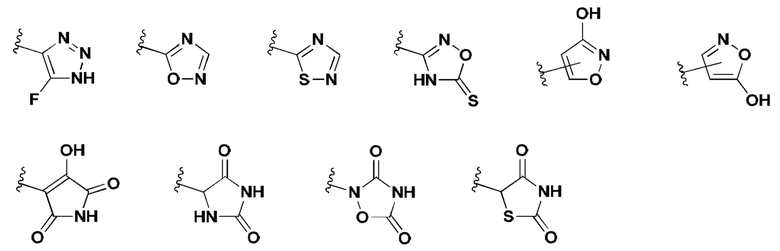
[0275] Также предполагается, что в случае, если к изостеру карбоновой кислоты добавляют химические заместители, полученное соединение сохраняет свойства изостера карбоновой кислоты. Предполагается, что в случае, если изостер карбоновой кислоты необязательно замещен одним или более фрагментами, выбранными из R, определенного выше, то заместитель и положение замещения выбраны таким образом, что они не лишают указанное соединение свойств изостера карбоновой кислоты. Аналогичным образом, также предполагается, что введение одного или более заместителей R в карбоциклический или гетероциклический изостер карбоновой кислоты не является замещением по одному или более атомам, которые сохраняют или имеют важное значение для свойств изостера карбоновой кислоты в случае указанного соединения, если такой заместитель (заместители) лишил бы указанное соединение свойств изостера карбоновой кислоты.
[0276] Также предполагаются другие изостеры карбоновой кислоты, конкретно не представленные в настоящем описании.
[0277] Термин «агент» или «исследуемый агент» включает любое вещество, молекулу, элемент, соединение, объект или их комбинацию. Он включает, но не ограничивается перечисленными, например, белок, полипептид, пептид или миметик, малые органические молекулы, полисахарид, полинуклеотид и тому подобное. Он может представлять собой природный продукт, синтетическое соединение или химическое соединение, или комбинацию двух или более веществ. Если не указано иное, термины «агент», «вещество» и «соединение» в настоящем описании используются взаимозаменяемо.
[0278] Термин «аналог» в контексте настоящего описания относится к молекуле, которая имеет структурное сходство с эталонной молекулой, но которая была целенаправленно и контролируемо модифицирована путем замены конкретного заместителя эталонной молекулы альтернативным заместителем. Специалист в данной области техники может ожидать, что в сравнении с эталонной молекулой аналог будет демонстрировать равную, близкую или улучшенную полезность. Синтез и скрининг аналогов для выявления вариантов известных соединений, обладающих улучшенными характеристиками (такими как более высокая аффинность связывания с молекулой-мишенью), является подходом, хорошо известным в области фармацевтической химии.
[0279] Термин «млекопитающее» применяют в его обычном биологическом значении. Таким образом, он, в частности, включает, но не ограничивается перечисленными, приматов, включая обезьян (шимпанзе, человекообразных обезьян (apes), нечеловекообразных обезьян (monkeys)) и людей, крупный рогатый скот, лошадей, овец, коз, свиней, кроликов, собак, кошек, крыс и мышей, но также включает и многие другие виды.
[0280] Термин «микробная инфекция» относится к инвазии организма-хозяина, независимо от того, является ли указанный организм позвоночным, беспозвоночным, рыбой, растением, птицей или млекопитающим, патогенными микроорганизмами. Это включает чрезмерный рост микроорганизмов, которые в норме присутствуют в теле или на теле млекопитающего или другого организма. В более общем случае микробная инфекция может представлять собой любую ситуацию, в которой присутствие популяции (популяций) микроорганизмов наносит вред млекопитающему-хозяину. Таким образом, млекопитающее «страдает» от микробной инфекции в случае, когда избыточное количество популяции микроорганизмов присутствует в теле или на теле млекопитающего, или когда воздействие присутствия популяции (популяций) микроорганизмов повреждает клетки или другую ткань млекопитающего. В частности, это описание относится к бактериальной инфекции. Следует отметить, что соединения согласно предпочтительным вариантам реализации также пригодны для применения для устранения микробного роста или загрязнения культур клеток или других сред, или обработки неодушевленных объектов или поверхностей, и ничто из содержащегося в настоящем описании не предназначено для ограничения предпочтительных вариантов реализации только лечением высших организмов, за исключением случаев, когда это явным образом указано в формуле изобретения.
[0281] Термин «фармацевтически приемлемый носитель» или «фармацевтически приемлемое вспомогательное вещество» включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические и замедляющие всасывание агенты и тому подобное. Применение таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области техники. За исключением случаев, когда какая-либо традиционная среда или агент несовместимы с активным ингредиентом, предполагается их применение в терапевтических композициях. Кроме того, могут быть включены различные адъюванты, такие как адъюванты, широко применяемые в данной области техники. Соображения относительно включения различных компонентов в фармацевтические композиции описаны, например, в источнике Oilman et al. (Eds.) (1990); Goodman and Gilman's: The Pharmacological Basis of Therapeutics, 8th Ed., Pergamon Press, содержание которого полностью включено в настоящее описание посредством ссылки.
[0282] «Субъект» в контексте настоящего описания означает человека или отличное от человека млекопитающее, например, собаку, кошку, мышь, крысу, корову, овцу, свинью, козу, отличного от человека примата, или птицу, например, курицу, а также любое другое позвоночное или беспозвоночное.
[0283] «Эффективное количество» или «терапевтически эффективное количество» в контексте настоящего описания относится к количеству терапевтического агента, которое является эффективным для ослабления, до некоторой степени, или уменьшения вероятности возникновения одного или более симптомов заболевания или состояния и включает излечение заболевания или состояния. «Излечение» означает, что симптомы заболевания или состояния устранены; однако, некоторые долговременные или постоянные эффекты могут сохраняться даже после получения курса лечения (такие как обширное повреждение тканей).
[0284] «Лечить», «лечение» или «осуществление лечения» в контексте настоящего описания относится к введению соединения или фармацевтической композиции субъекту для профилактических и/или терапевтических целей. Термин «профилактическое лечение» относится к лечению субъекта, у которого еще не проявляются симптомы заболевания или состояния, но который восприимчив или иным образом подвержен риску развития конкретного заболевания или состояния, при этом указанное лечение снижает вероятность того, что у пациента разовьется указанное заболевание или состояние. Термин «терапевтическое лечение» относится к проведению лечения в отношении субъекта, уже страдающего от заболевания или состояния.
Способы получения
[0285] Соединения, раскрытые в настоящем описании, могут быть синтезированы способами, описанными ниже, или путем модификации этих способов. Способы модификации указанной методологии включают, среди прочего, температуру, растворитель, реагенты и т.д., известные специалистам в данной области техники. В целом, во время любого из способов получения соединений, раскрытых в настоящем описании, может быть необходимо и/или желательно защитить чувствительные или реакционноспособные группы в любых из рассматриваемых молекул. Это может быть достигнуто с помощью обычных защитных групп, таких как группы, описанные в источниках Protective Groups in Organic Chemistry (ed. J.F.W. McOmie, Plenum Press, 1973) и P.G.M. Green, T.W. Wutts, Protecting Groups in Organic Synthesis (3rd ed.) Wiley, New York (1999), оба из которых полностью включены в настоящее описание посредством ссылки. Защитные группы могут быть удалены на любой удобной последующей стадии с использованием способов, известных в данной области техники. Синтетические химические преобразования, используемые при синтезе подходящих соединений, известны в данной области техники и включают, например, те, что описаны в источниках R. Larock, Comprehensive Organic Transformations, VCH Publishers, 1989, или L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons, 1995, оба из которых полностью включены в настоящее описание посредством ссылки. Пути, показанные и описанные в настоящем описании, являются только иллюстративными и не предназначены для ограничения, и не должны рассматриваться как ограничивающие каким-либо образом объем формулы изобретения. Специалисты в данной области техники смогут распознать модификации раскрытых синтезов и разработать альтернативные пути на основе раскрытых в настоящем описании сведений; все такие модификации и альтернативные пути входят в объем формулы изобретения.
[0286] В следующих схемах защитные группы для атомов кислорода выбраны исходя из их совместимости с необходимыми стадиями синтеза, а также совместимости стадий введения и снятия защиты с общими схемами синтеза (P.G.M. Green, T.W. Wutts, Protecting Groups in Organic Synthesis (3rd ed.) Wiley, New York (1999)).
[0287] Если соединения согласно настоящей технологии содержат один или более хиральных центров, такие соединения могут быть получены или выделены в виде чистых стереоизомеров, то есть в виде отдельных энантиомеров или d(l) стереоизомеров, или в виде обогащенных стереоизомерами смесей. Все такие стереоизомеры (и обогащенные смеси) включены в объем настоящей технологии, если не указано иное. Чистые стереоизомеры (или обогащенные смеси) могут быть получены с использованием, например, оптически активных исходных веществ или стереоселективных реагентов, хорошо известных в данной области техники. В качестве альтернативы, рацемические смеси таких соединений могут быть разделены с использованием, например, хиральной колоночной хроматографии, хиральных разделяющих агентов и тому подобного.
[0288] Исходные вещества для следующих реакций являются общеизвестными соединениями или могут быть получены известными способами или их очевидными модификациями. Например, многие исходные вещества доступны от коммерческих поставщиков, таких как Aldrich Chemical Co. (Milwaukee, Wisconsin, USA), Bachem (Torrance, California, USA), Emka-Chemce или Sigma (St. Louis, Missouri, USA). Другие могут быть получены с применением способов или их очевидных модификаций, описанных в соответствующих стандартных источниках, таких как Fieser and Fieser's Reagents for Organic Synthesis, Volumes 1-15 (John Wiley, and Sons, 1991), Rodd's Chemistry of Carbon Compounds, Volumes 1-5, и Supplemental (Elsevier Science Publishers, 1989), Organic Reactions, Volumes 1-40 (John Wiley, and Sons, 1991), March's Advanced Organic Chemistry, (John Wiley, and Sons, 5th Edition, 2001), и Larock's Comprehensive Organic Transformations (VCH Publishers Inc., 1989).
Синтез соединений формулы I
[0289] В одном варианте реализации способ включает взаимодействие подходящим образом замещенного промежуточного соединения с кислотным водородом (IV) со сложным эфиром (V) в условиях основного катализа с получением сложноэфирного производного (VI). Полученный продукт затем подвергали гидролизу в основных условиях с получением производного карбоновой кислоты (VII), которое затем подвергали условиям амидного сочетания с производным аминокислоты (VIII), где группа карбоновой кислоты замещена группой R1 (Схема 1). В качестве альтернативы, продукт карбоновой кислоты (VII) затем подвергают условиям амидного сочетания с производным аминоспирта (VIII-а) с получением соответствующего аддукта (IX). Полученный аддукт (IX) подвергают окислительным условиям с DMP-окислением (гипервалентным йодом) или окислителем, таким как РСС (хлорхромат пиридиния), с получением α-кетоамидного продукта (X). В качестве альтернативы, аддукт (IX) подвергали окислительным условиям с использованием EDC и дихлоруксусной кислоты или с использованием IBX в качестве окислителя с получением α-кетоамидного продукта (X). Специалисту в данной области техники также будет понятно, что существует множество других окислительных условий и агентов, которые входят в объем данного раскрытия, для окисления гидроксильной группы. Этот путь синтеза в целом показан на Схеме 2.
Схема 1:
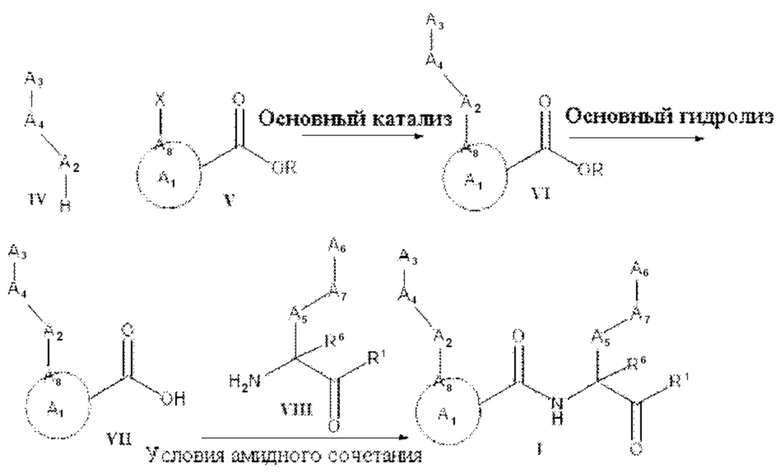
Схема 2;
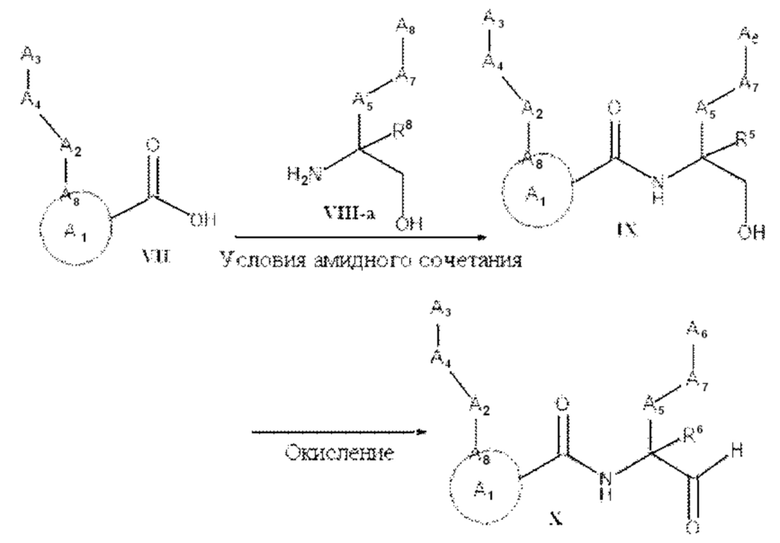
[0290] Следующие иллюстративные схемы представлены для руководства читателя и вместе представляют иллюстративный способ получения соединений, включенных в настоящее описание. Кроме того, другие способы получения соединений, описанных в настоящем описании, будут легко понятны специалисту в данной области в свете следующих схем реакций и примеров. Если не указано иное, все переменные имеют значения, указанные выше.
Применение изотопно-меченных соединений
[0291] Некоторые варианты реализации предусматривают способ применения изотопно-меченных соединений и пролекарств согласно настоящему описанию в: (i) метаболических исследованиях (предпочтительно с 14С), исследованиях кинетики реакции (например, с 2Н или 3Н); (ii) методах обнаружения или визуализации [таких как позитронно-эмиссионная томография (ПЭТ) или однофотонная эмиссионная компьютерная томография (ОФЭКТ)], включая анализы распределения лекарственного средства или субстрата в тканях; или (III) при радиоактивном лечении пациентов.
[0292] Изотопно-меченные соединения и пролекарства в соответствии с их вариантами реализации, как правило, могут быть получены путем реализации процедур, раскрытых в схемах или в примерах и способах получения, описанных ниже, путем замены неизотопно-меченого реагента на легко доступный изотопно-меченый реагент. Соединение, меченное 18F или 11С, может быть особенно предпочтительным для ПЭТ, а соединение, меченное 123I, может быть особенно предпочтительным для исследований ОФЭКТ. Дальнейшее замещение более тяжелыми изотопами, такими как дейтерий (то есть 2Н), может дать определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличение периода полувыведения in vivo или снижение требуемой дозы.
Синтез изотопно-меченных соединений
[0293] Соединения, меченные 18F, синтезируют, как показано на схемах ниже. В одном варианте реализации способ включает взаимодействие промежуточного соединения 450 с меченным 18F агентом с использованием условий, описанных в источниках Rotstein, et al., Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics, Nature Communications, 2014, Vol. 5, 4365-4371 и Rotstein, et al., Mechanistic Studies and Radiofluorination of Structurally Diverse Pharmaceuticals with Spirocyclic Iodonium(III) Ylides, Chemical Science, 2016, Vol. 7, 4407-4417, оба из которых полностью включены в настоящее описание посредством ссылки, чтобы получить меченный 18F промежуточный метил-2-((этоксикарбонил)амино)-3-(4-(фтор-18F)фенил)пропаноат (631), который затем превращается в конечный α-кетоамидный продукт, представленный общей структурой XI (Схема 3).
Схема 3:
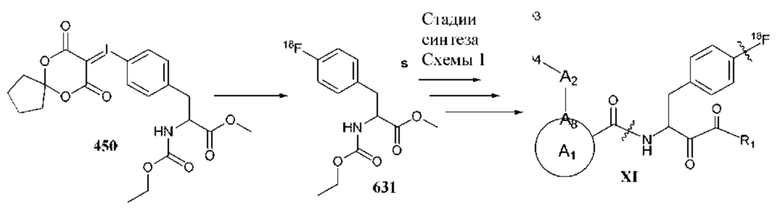
[0294] В качестве альтернативы, меченное 18F соединение XV синтезируют, как показано на схеме 4. В одном варианте реализации йоданилиденовое промежуточное соединение XII используют для введения метки 18F с использованием условий, описанных в источниках Rotstein, et al., Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics, Nature Communications, 2014, Vol. 5, 4365-4371 и Rotstein, et al., Mechanistic Studies and Radiofluorination of Structurally Diverse Pharmaceuticals with Spirocyclic Iodonium(III) Ylides, Chemical Science, 2016, Vol. 7, 4407-4417, чтобы получить меченый α-кетоамидный продукт XV. В другом варианте реализации йоданилиденовое промежуточное соединение (XIV) (схема 4) подвергается окислительным условиям с окислением DMP (с помощью гипервалентного йода) или окислителем, таким как РСС (хлорхромат пиридиния), с получением α-кетоамидного продукта (XV). В еще одном варианте реализации йоданилиденовое промежуточное соединение (XIII) (схема 4) подвергают условиям реакции 18F-мечения, как описано ранее, с последующим гидролизом сложного эфира в основных условиях с получением производного карбоновой кислоты, которое затем подвергают условиям амидного сочетания с производным аминокислоты, где группа карбоновой кислоты замещена группой R1 с получением меченого α-кетоамидного продукта XV.
Схема 4:
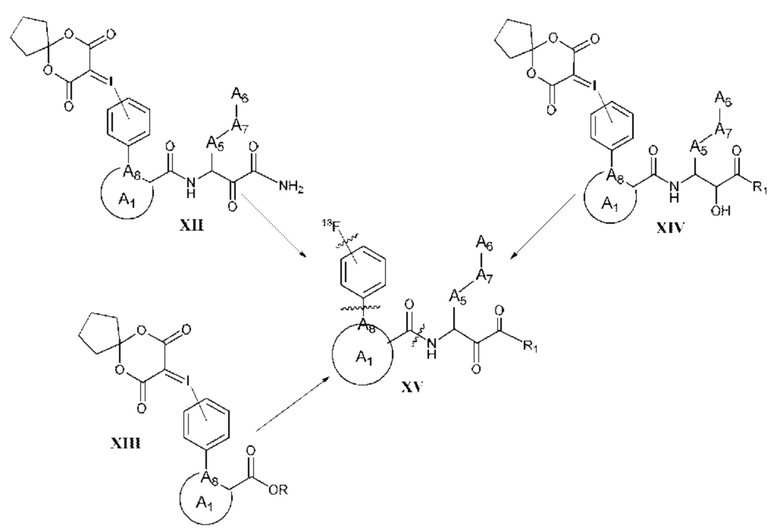
Введение и фармацевтические композиции
[0295] Указанные соединения вводят в терапевтически эффективной дозе. Несмотря на то, что величины доз для человека еще требуют оптимизации, для соединений, описанных в настоящем описании, суточная доза, как правило, может составлять от примерно 0,25 мг/кг до примерно 120 мг/кг массы тела или более, от примерно 0,5 мг/кг или менее до примерно 70 мг/кг, от примерно 1,0 мг/кг до примерно 50 мг/кг массы тела или от примерно 1,5 мг/кг до примерно 10 мг/кг массы тела. Таким образом, для введения человеку массой 70 кг диапазон доз составлял бы от примерно 17 мг в сутки до примерно 8000 мг в сутки, от примерно 35 мг в сутки или менее до примерно 7000 мг в сутки или более, от примерно 70 мг в сутки до примерно 6000 мг в сутки, от примерно 100 мг в сутки до примерно 5000 мг в сутки или от примерно 200 мг до примерно 3000 мг в сутки. Количество вводимого активного соединения, конечно, будет зависеть от субъекта и патологического состояния, подвергаемого лечению, тяжести болезни, способа и схемы введения и решения лечащего врача.
[0296] Введение соединений, раскрытых в настоящем описании, или их фармацевтически приемлемых солей может осуществляться посредством любого из принятых способов введения для агентов, которые служат аналогичными средствами, включая, но не ограничиваясь следующими: перорально, подкожно, внутривенно, интраназально, местно, трансдермально, внутрибрюшинно, внутримышечно, внутрипульмонарно, вагинально, ректально или внутриглазно. Пероральный и парентеральный пути введения являются обычными при лечении показаний, которые являются предметом предпочтительных вариантов реализации.
[0297] Соединения, применяемые, как описано выше, могут быть включены в состав фармацевтических композиций для применения при лечении этих состояний. Используют стандартные способы получения фармацевтических составов, такие как описанные в источнике Remington's The Science and Practice of Pharmacy, 21st Ed., Lippincott Williams & Wilkins (2005), полностью включенном в настоящее описание посредством ссылки. Соответственно, некоторые варианты реализации включают фармацевтические композиции, содержащие: (а) безопасное и терапевтически эффективное количество соединения, описанного в настоящем описании (включая энантиомеры, диастереоизомеры, таутомеры, полиморфы и их сольваты), или его фармацевтически приемлемых солей; и (b) фармацевтически приемлемый носитель, разбавитель, наполнитель или их комбинацию.
[0298] В дополнение к выбранному соединению, используемому, как описано выше, некоторые варианты реализации включают композиции, содержащие фармацевтически приемлемый носитель. Термин «фармацевтически приемлемый носитель» или «фармацевтически приемлемое вспомогательное вещество» включает любые растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические агенты и агенты, замедляющие абсорбцию, и тому подобное. Использование таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области техники. За исключением случаев, когда какая-либо обычная среда или агент несовместимы с активным ингредиентом, предполагается их использование в терапевтических композициях. Кроме того, могут быть включены различные адъюванты, такие как обычно используемые в данной области техники. Соображения относительно включения различных компонентов в фармацевтические композиции описаны, например, в источнике Gilman et al. (Eds.) (1990); Goodman and Gilman's: The Pharmacological Basis of Therapeutics, 8th Ed., Pergamon Press, который полностью включен в настоящее описание посредством ссылки.
[0299] Некоторыми примерами веществ, которые могут служить в качестве фармацевтически приемлемых носителей или их компонентов, являются сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натрий карбоксиметилцеллюлоза, этилцеллюлоза и метилцеллюлоза; порошкообразный трагакант; солод; желатин; тальк; твердые смазывающие вещества, такие как стеариновая кислота и стеарат магния; сульфат кальция; растительные масла, такие как арахисовое масло, хлопковое масло, кунжутное масло, оливковое масло, кукурузное масло и масло какао; полиолы, такие как пропиленгликоль, глицерин, сорбит, маннит и полиэтиленгликоль; альгиновая кислота; эмульгаторы, такие как Твины; смачивающие агенты, такие как лаурилсульфат натрия; окрашивающие агенты; ароматизирующие агенты; агенты для таблетирования, стабилизаторы; антиоксиданты; консерванты; апирогенная вода; изотонический солевой раствор и фосфатные буферные растворы.
[0300] Выбор фармацевтически приемлемого носителя для применения совместно с целевым соединением, как правило, определяется способом введения соединения.
[0301] Композиции, описанные в настоящем описании, предпочтительно представлены в виде единичной лекарственной формы. В контексте настоящего описания «единичная лекарственная форма» представляет собой композицию, содержащую количество соединения, которое подходит для введения субъекту-животному, предпочтительно субъекту-млекопитающему, в однократной дозе, в соответствии с надлежащей медицинской практикой (good medical practice). Получение однократной или единичной лекарственной формы, однако, не означает, что указанную лекарственную форму вводят один раз в сутки или один раз на курс терапии. Такие лекарственные формы рассматриваются как предполагаемые к введению один, два, три или более раз в сутки, и могут быть введены в виде инфузии в течение некоторого периода времени (например, от примерно 30 минут до примерно 2-6 часов), или введены в виде непрерывной инфузии, и могут быть введены более одного раза в ходе курса терапии, хотя и однократное введение конкретно не исключено. Специалисту в данной области техники будет понятно, что указанный состав не включает конкретно полный курс терапии, и такие решения остаются на усмотрение специалистов в области лечения, а не в области получения составов.
[0302] Композиции, применяемые описанным выше образом, могут находиться в любой из множества подходящих форм для различных путей введения, например, для перорального, назального, ректального, местного (включая трансдермальное), глазного, внутримозгового, внутричерепного, интратекального, внутриартериального, внутривенного, внутримышечного или других парентеральных путей введения. Специалисту в данной области техники будет понятно, что композиции для перорального и назального введения включают композиции, которые вводят путем ингаляции, и они получены с использованием доступных методик. В зависимости от конкретного желаемого пути введения могут быть использованы разнообразные фармацевтически приемлемые носители, хорошо известные в данной области техники. Фармацевтически приемлемые носители включают, например, твердые или жидкие наполнители, разбавители, гидротропные агенты, поверхностно-активные агенты и инкапсулирующие вещества. Могут быть включены необязательные фармацевтически активные вещества, которые по существу не мешают ингибирующей активности соединения. Количество носителя, используемое в сочетании с соединением, является достаточным для обеспечения практически применимого количества материала для введения на единичную дозу соединения. Способы и композиции для получения лекарственных форм, подходящих для применения в способах, описанных в настоящем описании, описаны в следующих источниках: Modern Pharmaceutics, 4th Ed., Chapters 9 and 10 (Banker & Rhodes, editors, 2002); Lieberman et al., Pharmaceutical Dosage Forms: Tablets (1989) и Ansel, Introduction to Pharmaceutical Dosage Forms 8th Edition (2004), содержание всех из указанных источников включено в настоящее описание посредством ссылки.
[0303] Могут быть применены различные лекарственные формы для перорального введения, включая такие твердые формы, как таблетки, капсулы, гранулы и сыпучие порошки. Таблетки могут быть спрессованы, могут быть предназначены для растирания перед употреблением, могут быть покрыты энтеросолюбильным покрытием, покрыты сахаром, покрыты пленкой или могут быть спрессованы по несколько, могут содержать подходящие связующие, смазывающие агенты, разбавители, разрыхляющие агенты, окрашивающие агенты, ароматизирующие агенты, агенты, способствующие текучести, и агенты, способствующие плавлению. Жидкие лекарственные формы для перорального введения включают водные растворы, эмульсии, суспензии, растворы и/или суспензии, разведенные из нешипучих гранул, и шипучие растворы, разведенные из шипучих гранул, содержащие подходящие растворители, консерванты, эмульгирующие агенты, суспендирующие агенты, разбавители, подсластители, агенты, способствующие плавлению, окрашивающие агенты и ароматизирующие агенты.
[0304] Фармацевтически приемлемый носитель, подходящий для получения единичных лекарственных форм для перорального введения, хорошо известен в данной области техники. Таблетки обычно содержат традиционные фармацевтически совместимые адъюванты в качестве инертных разбавителей, такие как карбонат кальция, карбонат натрия, маннит, лактоза и целлюлоза; связующие агенты, такие как крахмал, желатин и сахароза; разрыхлители, такие как крахмал, альгиновая кислота и кроскармеллоза; смазывающие агенты, такие как стеарат магния, стеариновая кислота и тальк. Для улучшения характеристик текучести порошковой смеси могут быть использованы вещества, способствующие скольжению, такие как диоксид кремния. Для обеспечения внешнего вида могут быть добавлены окрашивающие агенты, такие как красители для пищевых продуктов, лекарств и косметики (FD&C, англ.: food, drugs and cosmetic). Подсластители и ароматизирующие агенты, такие как аспартам, сахарин, ментол, перечная мята и фруктовые отдушки, являются пригодными для применения адъювантами для жевательных таблеток. Капсулы обычно содержат один или более твердых разбавителей, раскрытых выше. Выбор компонентов носителей зависит от второстепенных соображений, таких как вкус, стоимость и стабильность при хранении, которые не являются критическими, и может быть легко сделан специалистом в данной области техники.
[0305] Композиции для перорального введения также включают жидкие растворы, эмульсии, суспензии и т.п. Фармацевтически приемлемые носители, подходящие для получения таких композиций, хорошо известны в данной области техники. Типичные компоненты носителей для сиропов, эликсиров, эмульсий и суспензий включают этанол, глицерин, пропиленгликоль, полиэтиленгликоль, жидкую сахарозу, сорбит и воду. Для суспензий типичные суспендирующие агенты включают метилцеллюлозу, натрий карбоксиметилцеллюлозу, АВИЦЕЛ RC-591 (AVICEL RC-591), трагакант и альгинат натрия; типичные смачивающие агенты включают лецитин и полисорбат 80; и типичные консерванты включают метилпарабен и бензоат натрия. Жидкие композиции для перорального введения могут также содержать один или более компонентов, таких как подсластители, ароматизирующие агенты и красители, описанные выше.
[0306] Такие композиции могут быть также покрыты с помощью традиционных методов, обычно зависимыми от рН или времени покрытиями, такими что целевое соединение высвобождается в желудочно-кишечном тракте в непосредственной близости от желаемого местного применения или в разное время, чтобы пролонгировать желаемое действие. Такие лекарственные формы обычно содержат, но не ограничиваются ими, один или более из фталата ацетата целлюлозы, фталата поливинилацетата, фталата гидроксипропилметилцеллюлозы, этилцеллюлозы, покрытий Эудрагита (Eudragit), восков и шеллака.
[0307] Композиции, описанные в настоящем описании, могут необязательно содержать другие активные лекарственные вещества.
[0308] Другие композиции, подходящие для достижения системной доставки целевых соединений, включают лекарственные формы для сублингвального, буккального и назального введения. Такие композиции обычно содержат одно или более растворимых веществ-наполнителей, таких как сахароза, сорбит и маннит, и связующие вещества, такие как аравийская камедь (acacia), микрокристаллическая целлюлоза, карбоксиметилцеллюлоза и гидроксипропилметилцеллюлоза. Также могут быть включены вещества, способствующие скольжению, смазывающие вещества, подсластители, красители, антиоксиданты и ароматизирующие агенты, раскрытые выше.
[0309] Жидкую композицию, которая разработана для местного офтальмологического применения, готовят таким образом, чтобы ее можно было вводить местно в глаз. Комфорт должен быть максимальным, насколько это возможно, хотя иногда из соображений получения (например, стабильности лекарственного средства) может быть необходимо, чтобы комфорт оказался меньше оптимального. В том случае, если комфорт не может быть максимизирован, жидкость должна быть приготовлена таким образом, чтобы такая жидкость переносилась пациентом при местном офтальмологическом применении. Кроме того, офтальмологически приемлемая жидкость должна быть либо упакована для однократного применения, либо может содержать консервант для предотвращения загрязнения в течение нескольких применений.
[0310] Растворы или лекарственные средства для офтальмологического применения часто готовят с использованием физиологического раствора в качестве основного носителя. Офтальмологические растворы предпочтительно должны поддерживаться при комфортном рН с помощью подходящей буферной системы. Составы могут также содержать обычные фармацевтически приемлемые консерванты, стабилизаторы и поверхностно-активные вещества.
[0311] Консерванты, которые могут быть использованы в фармацевтических композициях, раскрытых в настоящем описании, включают, но не ограничиваются ими, бензалкония хлорид, полигексаметиленбигуанид (РНМВ), хлорбутанол, тимеросал, ацетат фенилртути и нитрат фенилртути. Подходящим поверхностно-активным веществом является, например, Твин 80. Кроме того, в офтальмологических препаратах, раскрытых в настоящем описании, могут быть использованы различные подходящие переносящие среды (vehicles). Эти переносящие среды включают, но не ограничиваются ими: поливиниловый спирт, повидон, гидроксипропилметилцеллюлозу, полоксамеры, карбоксиметилцеллюлозу, гидроксиэтилцеллюлозу и очищенную воду.
[0312] При необходимости или для удобства могут быть добавлены регуляторы тоничности. Они включают, но не ограничиваются ими, соли, в частности хлорид натрия, хлорид калия, маннит и глицерин или любой другой офтальмологически подходящий регулятор тоничности.
[0313] Могут быть использованы различные буферы и средства для регулирования рН, при условии, что полученный в результате препарат будет офтальмологически приемлемым. Для многих композиций рН будет составлять от 4 до 9. Соответственно, буферы включают ацетатные буферы, цитратные буферы, фосфатные буферы и боратные буферы. Для регулирования рН этих составов при необходимости могут быть использованы кислоты или основания.
[0314] В том же ключе офтальмологически приемлемые антиоксиданты включают, но не ограничиваются ими, метабисульфит натрия, тиосульфат натрия, ацетилцистеин, бутилированный гидроксианизол и бутилированный гидрокситолуол.
[0315] Другими вспомогательными компонентами, которые могут быть включены в офтальмологические препараты, являются хелатирующие агенты. Подходящим хелатирующим агентом является этилендиаминтетраацетат динатрия, хотя вместо него или в сочетании с ним могут быть также использованы и другие хелатирующие агенты.
[0316] Для местного применения используют кремы, мази, гели, растворы или суспензии и т.д., содержащие соединение, раскрытое в настоящем описании. Составы для местного применения обычно могут содержать фармацевтический носитель, сорастворитель, эмульгатор, усилитель проникновения, консервирующую систему и смягчающее средство.
[0317] Для внутривенного введения соединения и композиции, описанные в настоящем описании, могут быть растворены или диспергированы в фармацевтически приемлемом разбавителе, таком как солевой раствор или раствор декстрозы. Подходящие вспомогательные вещества могут быть включены для достижения желаемого рН, включая, но не ограничиваясь ими, NaOH, карбонат натрия, ацетат натрия, HCl и лимонную кислоту. В различных вариантах реализации рН конечной композиции составляет от 2 до 8 или предпочтительно от 4 до 7. Вспомогательные вещества-антиоксиданты, могут включать бисульфит натрия, ацетон-бисульфит натрия, формальдегид натрия, сульфоксилат, тиомочевину и ЭДТА. Другие неограничивающие примеры подходящих вспомогательных веществ, которые можно обнаружить в готовой композиции для внутривенного введения, могут включать фосфаты натрия или калия, лимонную кислоту, винную кислоту, желатин и углеводы, такие как декстроза, маннит и декстран. Другие приемлемые вспомогательные вещества описаны в источниках Powell, et al., Compendium of Excipients for Parenteral Formulations, PDA J Pharm Sci and Tech 1998, 52 238-311, и Nema et al., Excipients and Their Role in Approved Injectable Products: Current Usage and Future Directions, PDA J Pharm Sci and Tech 2011, 65 287-332, содержание обоих из указанных источников полностью включено в настоящее описание посредством ссылки. Для обеспечения бактериостатического или фунгистатического раствора также могут быть включены антимикробные агенты, включая, но не ограничиваясь ими, нитрат фенилртути, тимеросал, бензетония хлорид, бензалкония хлорид, фенол, крезол и хлорбутанол.
[0318] Композиции для внутривенного введения могут быть предоставлены для лиц, осуществляющих уход за пациентом, в форме одного или более твердых веществ, которые восстанавливают с помощью подходящего разбавителя, такого как стерильная вода, солевой раствор или декстроза в воде, незадолго до введения. В других вариантах реализации указанные композиции представлены в растворе, готовом для парентерального введения. В других вариантах реализации указанные композиции представлены в растворе, который дополнительно разбавляют и затем осуществляют введение. В вариантах реализации, которые включают введение комбинации соединения, описанного в настоящем описании, и другого агента, указанная комбинация может быть предоставлена лицам, осуществляющим уход, в виде смеси, или лица, осуществляющие уход, могут смешать указанные два агента и затем осуществить введение, или указанные два агента можно вводить отдельно.
[0319] Фактическая доза активных соединений, описанных в настоящем описании, зависит от конкретного соединения и от подлежащего лечению состояния; выбор подходящей дозы находится в сфере компетентности специалиста в данной области техники.
[0320] Соединения и композиции, описанные в настоящем описании, если это желательно, могут быть представлены в упаковке или дозирующем устройстве, содержащем одну или более единичных лекарственных форм, содержащих активный ингредиент. Такая упаковка или устройство может, например, содержать металлическую или пластиковую фольгу, такую, как блистерная упаковка, или стеклянный контейнер и резиновые пробки, такие как во флаконах. Упаковка или дозирующее устройство могут сопровождаться инструкциями по применению. Соединения и композиции, описанные в настоящем описании, входят в готовую смесь с фармацевтически совместимым носителем, а также могут быть приготовлены, помещены в соответствующий контейнер и маркированы для лечения указанного состояния.
[0321] Количество соединения в составе может варьировать в пределах полного диапазона, используемого специалистами в данной области техники. Как правило, состав будет содержать, в массовых процентах (мас. %), от примерно 0,01 до 99,99 мас. % соединения согласно настоящей технологии в расчете на весь состав, причем остаток составляют один или более подходящих фармацевтических наполнителей. Предпочтительно соединение присутствует в количестве около 1 80 мас. %. Типичные фармацевтические составы описаны ниже.
Примеры составов
[0322] Ниже представлены типичные фармацевтические составы, содержащие соединение формулы I.
Пример состава 1 - Состав таблетки
[0323] Следующие ингредиенты тщательно перемешивают и прессуют в таблетки с одной линией разлома.

Пример состава 2 - Состав капсулы
[0324] Следующие ингредиенты тщательно перемешивают и помещают в желатиновую капсулу с твердой оболочкой.


Пример состава 3 - Состав суспензии
[0325] Следующие ингредиенты смешивают для получения суспензии для перорального введения.

Пример состава 4 - Состав для инъекционного введения
[0326] Следующие ингредиенты смешивают для образования композиции для инъекционного введения.

Пример состава 5 - Состав суппозитория
[0327] Суппозиторий общей массой 2,5 г готовят путем смешивания соединения согласно настоящей технологии с Witepsol® Н 15 (триглицериды насыщенной растительной жирной кислоты; Riches Nelson, Inc., New York), и он имеет следующий состав:

Способы лечения
[0328] Раскрытые в настоящем описании соединения или их таутомеры и/или их фармацевтически приемлемые соли могут эффективно действовать в качестве ингибиторов CAPN1, CAPN2 и/или CAPN9 и обеспечивать лечение состояний, на которые, по меньшей мере частично, влияют CAPN1, CAPN2 и/или CAPN9. В некоторых вариантах реализации предложены фармацевтические композиции, содержащие одно или более соединений, раскрытых в настоящем описании, и фармацевтически приемлемое вспомогательное вещество. Некоторые варианты реализации обеспечивают способ лечения фиброзного заболевания с помощью эффективного количества одного или более соединений, описанных в настоящем описании.
[0329] В некоторых вариантах реализации субъектом является человек.
[0330] Другие варианты реализации включают введение комбинации соединений субъекту, нуждающемуся в этом. Комбинация может содержать соединение, композицию, фармацевтическую композицию, описанные в настоящем описании, совместно с дополнительным лекарственным средством.
[0331] Некоторые варианты реализации включают совместное введение соединения, композиции и/или фармацевтической композиции, описанных в настоящем описании, с дополнительным лекарственным средством. Под термином «совместное введение» понимается, что два или более агентов могут быть обнаружены в кровотоке пациента в одно время вне зависимости от того, когда и как они были фактически введены. В одном из вариантов реализации указанные агенты вводят одновременно. В одном таком варианте реализации введение в комбинации осуществляют путем объединения агентов в одной лекарственной форме. В другом варианте реализации указанные агенты вводят последовательно. В одном из вариантов реализации указанные агенты вводят одним путем, например, перорально. В другом варианте реализации указанные агенты вводят разными путями, например, один вводят перорально, а другой вводят внутривенно (в/в).
[0332] Некоторые варианты реализации включают комбинации соединения, композиции или фармацевтической композиции, описанных в настоящем описании, с любым другим фармацевтическим соединением, которое одобрено для лечения заболеваний или расстройств, обусловленных фиброзом или связанных с дифференцировкой миофибробластов.
[0333] В некоторых вариантах реализации предложен способ ингибирования CAPN1, CAPN2 и/или CAPN9 и/или способ лечения заболевания, на которое, по меньшей мере частично, влияют CAPN1, CAPN2 и/или CAPN9, с помощью эффективного количества одного или более соединений, раскрытых в настоящем описании.
[0334] Соединения, раскрытые в настоящем описании, подходят для ингибирования ферментов CAPN1, CAPN2 и/или CAPN9 и/или лечения расстройств, связанных с фиброзом или дифференцировкой миофибробластов.
[0335] В некоторых вариантах реализации предложен способ ингибирования CAPN1, CAPN2 и/или CAPN9, включающий приведение клеток (включая нейроны/клетки микроглии/поступающие макрофаги) в контакт с эффективным количеством одного или более соединений, раскрытых в настоящем описании.
[0336] В некоторых вариантах реализации предложен способ лечения фиброзного заболевания, включающий введение субъекту эффективного количества одного или более соединений или фармацевтической композиции, содержащей фармацевтически приемлемое вспомогательное вещество, раскрытых в настоящем описании.
[0337] В некоторых вариантах реализации предложен способ лечения заболевания, на которое, по меньшей мере частично, влияют CAPN1, CAPN2 и/или CAPN9, включающий введение субъекту эффективного количества одного или более соединений или фармацевтической композиции, содержащей фармацевтически приемлемое вспомогательное вещество, раскрытых в настоящем описании.
[0338] В некоторых вариантах реализации предложен способ ингибирования CAPN1, CAPN2 и/или CAPN9, включающий приведение клеток в контакт с эффективным количеством одного или более соединений, раскрытых в настоящем описании. В некоторых вариантах реализации способ ингибирования CAPN1, CAPN2 и/или CAPN9 осуществляют in vitro или in vivo.
[0339] Кальпаины также экспрессируются в клетках, отличных от нейронов, клеток микроглии и поступающих макрофагов. В частности, они важны для скелетных мышц, и в настоящем описании ингибирование кальпаинов также относится к ингибированию в этих клетках.
Селективное ингибирование
[0340] Некоторые варианты реализации предусматривают способ конкурентного связывания с кальпастатином (CAST), включающий приведение соединения, раскрытого в настоящем описании, в контакт с ферментами CAPN1, CAPN2 и/или CAPN9, находящимися внутри субъекта. В таком способе соединение специфически ингибирует один или более ферментов, выбранных из группы, состоящей из: CAPN1, CAPN2 и CAPN9, по меньшей мере в 2 раза, по меньшей мере в 3 раза, по меньшей мере в 4 раза, по меньшей мере в 5 раз, по меньшей мере в 10 раз, по меньшей мере в 15 раз, по меньшей мере в 20 раз, по меньшей мере в 50 раз, по меньшей мере в 100 раз, по меньшей мере в 150 раз, по меньшей мере в 200 раз, по меньшей мере в 400 раз или по меньшей мере в 500 раз.
[0341] Некоторые варианты реализации предусматривают способ селективного ингибирования CAPN1 в присутствии CAPN2 и CAPN9, который включает приведение клеток (включая нейроны/клетки микроглии/поступающие макрофаги) в контакт с эффективным количеством одного или более соединений, раскрытых в настоящем описании.
[0342] Некоторые варианты реализации предусматривают способ селективного ингибирования CAPN2 в присутствии CAPN1 и CAPN9, который включает приведение клеток (включая нейроны/клетки микроглии/поступающие макрофаги) в контакт с эффективным количеством одного или более соединений, раскрытых в настоящем описании.
[0343] Некоторые варианты реализации предусматривают способ селективного ингибирования CAPN9 в присутствии CAPN2 и CAPN1, который включает приведение клеток (включая нейроны/клетки микроглии/поступающие макрофаги) в контакт с эффективным количеством одного или более соединений, раскрытых в настоящем описании.
[0344] Некоторые варианты реализации предусматривают способ селективного ингибирования CAPN1 и CAPN2 в присутствии CAPN9, который включает приведение клеток (включая нейроны/клетки микроглии/поступающие макрофаги) в контакт с эффективным количеством одного или более соединений, раскрытых в настоящем описании.
[0345] Некоторые варианты реализации предусматривают способ селективного ингибирования CAPN1 и CAPN9 в присутствии CAPN2, который включает приведение клеток (включая нейроны/клетки микроглии/поступающие макрофаги) в контакт с эффективным количеством одного или более соединений, раскрытых в настоящем описании.
[0346] Некоторые варианты реализации предусматривают способ селективного ингибирования CAPN2 и CAPN9 в присутствии CAPN1, который включает приведение клеток (включая нейроны/клетки микроглии/поступающие макрофаги) в контакт с эффективным количеством одного или более соединений, раскрытых в настоящем описании.
[0347] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют CAPN1, CAPN2 и/или CAPN9, указанных соединений или фармацевтической композиции, содержащей одно или более соединений, раскрытых в настоящем описании, и фармацевтически приемлемое вспомогательное вещество.
[0348] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем этот способ включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют CAPN1, CAPN2 и/или CAPN9, причем указанные соединения выбраны из соединений, раскрытых в настоящем описании, или фармацевтической композиции, содержащей одно или более соединений, раскрытых в настоящем описании, и фармацевтически приемлемое вспомогательное вещество.
[0349] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют CAPN1, CAPN2 и/или CAPN9, причем указанные соединения выбраны из соединений, раскрытых в настоящем описании, или фармацевтической композиции, содержащей одно или более соединений, описанных в настоящем описании, и фармацевтически приемлемое вспомогательное вещество.
[0350] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем указанный способ включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют CAPN1, CAPN2 и/или CAPN9, причем указанные соединения выбраны из соединений, раскрытых в настоящем описании, или фармацевтической композиции, содержащей одно или более соединений, раскрытых в настоящем описании, и фармацевтически приемлемое вспомогательное вещество.
[0351] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:5.
[0352] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:10.
[0353] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:20.
[0354] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:50.
[0355] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:100.
[0356] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:200.
[0357] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:250.
[0358] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:500.
[0359] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:5.
[0360] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:10.
[0361] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:20.
[0362] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:50.
[0363] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:100.
[0364] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9. в отношении не менее 1:1:200.
[0365] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:250.
[0366] Некоторые варианты реализации предусматривают способ лечения фиброзного заболевания, который включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более ферментов, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении не менее 1:1:500.
[0367] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:5.
[0368] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:10.
[0369] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:20.
[0370] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:50.
[0371] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем указанный способ включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:100.
[0372] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем этот способ включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:200.
[0373] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:250.
[0374] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, который включает введение субъекту эффективного количества одного или более соединений, которые специфически ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:500.
[0375] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем этот способ включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:5.
[0376] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем этот способ включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:10.
[0377] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем указанный способ включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:20.
[0378] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем этот способ включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:50.
[0379] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем этот способ включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:100.
[0380] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем этот способ включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:200.
[0381] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем этот способ включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:250.
[0382] Некоторые варианты реализации предусматривают способ лечения заболевания, обусловленного по меньшей мере частично CAPN1, CAPN2 и/или CAPN9, причем указанный способ включает введение субъекту эффективного количества одного или более соединений, которые селективно ингибируют два или более фермента, выбранных из группы, состоящей из CAPN1, CAPN2 и CAPN9, в отношении по меньшей мере 1:1:500.
[0383] Некоторые варианты реализации предусматривают способ для профилактической терапии или лечения субъекта, имеющего фиброзное расстройство, где указанный способ включает введение эффективного количества одного или более соединений, раскрытых в настоящем описании, субъекту, нуждающемуся в этом.
[0384] Некоторые варианты реализации предусматривают способ для профилактической терапии или лечения субъекта, имеющего расстройство, обусловленное CAPN1, CAPN2 и/или CAPN9, где указанный способ включает введение эффективного количества одного или более соединений, раскрытых в настоящем описании, субъекту, нуждающемуся в этом.
[0385] Некоторые варианты реализации предусматривают способ ингибирования дифференцировки миофибробластов (например, эпителиально/эндотелиально-мезенхимального перехода (ЕрМТ/EnMT)), который включает приведение клеток в контакт с эффективным количеством одного или более соединений, раскрытых в настоящем описании. В одном аспекте способ ингибирования дифференцировки миофибробластов, например, эпителиально/эндотелиально-мезенхимального перехода (ЕрМТ/EnMT)), выполняют in vitro или in vivo.
[0386] Некоторые варианты реализации предусматривают способ лечения заболевания или состояния, выбранного из группы, состоящей из или вызывающей симптом, выбранный из группы, состоящей из: фиброза печени, фиброза почки, фиброза легких, гиперчувствительного пневмонита, интерстициального фиброза, системной склеродермии, дегенерация желтого пятна, фиброза поджелудочной железы, фиброза селезенки, фиброза сердца, фиброза средостения, миелофиброза, фиброза эндомиокарда, ретроперитонеального фиброза, прогрессирующего массивного фиброза, нефрогенного системного фиброза, фиброзных осложнения после хирургических вмешательств, хронической васкулопатии аллотрансплантата и/или хронического отторжения аллотрансплантата, ишемического реперфузионного повреждения, связанного с фиброзом, фиброза вследствие инъекций, цирроза, диффузного заболевания паренхимы легкого, болевого синдрома после вазэктомии и ревматоидного артрита, при которых указанный способ включает введение субъекту эффективного количества одного или более соединений, раскрытых в настоящем описании, если субъекту это необходимо.
[0387] Некоторые варианты реализации предусматривают способ лечения фиброза печени.
[0388] Некоторые варианты реализации предусматривают способ лечения фиброза миокарда.
[0389] Некоторые варианты реализации предусматривают способ лечения фиброза при заболеваниях, связанных с ревматоидным артритом.
[0390] Некоторые варианты реализации предусматривают способ лечения состояния, обусловленного CAPN1, CAPN2 и/или CAPN9, который применяют как в терапевтических, так и в профилактических целях для субъектов. Оба способа включают введение одного или более соединений, раскрытых в настоящем описании, субъекту, нуждающемуся в этом.
[0391] Некоторые варианты реализации предусматривают способ лечения синдрома жесткой кожи.
[0392] Предпочтительные варианты реализации включают комбинации соединения, композиции или фармацевтической композиции, описанных в настоящем описании, с другими ингибиторами CAPN1, CAPN2 и/или CAPN9, такими как антитела против CAPN1, CAPN2 и/или CAPN9 или фрагменты антител, антисмысловые соединения против CAPN1, CAPN2 и/или CAPN9, иРНК или другие ингибиторы CAPN1, CAPN2 и/или CAPN9, представляющие собой малые молекулы.
[0393] Некоторые варианты реализации включают комбинации соединения, композиции или фармацевтической композиции, описанных в настоящем описании, для ингибирования дифференцировки миофибробластов (например, эпителиально/эндотелиально-мезенхимального перехода (ЕрМТ/EnMT)). Некоторые варианты реализации включают комбинации одного или более из этих соединений, которые являются ингибиторами одного или более (или всех трех) из группы, состоящей из CAPN1, CAPN2 и/или CAPN9, отдельно или в комбинации с другими ингибиторами пути сигналинга TGFβ, могут быть использованы для лечения или профилактики или облегчения симптомов фиброзного, склеротического или поствоспалительного заболевания или состояния, включая: фиброз печени, фиброз почки, фиброз легких, гиперчувствительный пневмонит, интерстициальный фиброз, системную склеродермию, макулярную дегенерацию, фиброз поджелудочной железы, фиброз селезенки, фиброз сердца, фиброз средостения, миелофиброз, эндомиокардиальный фиброз, забрюшинный фиброз, прогрессирующий массивный фиброз, нефрогенный системный фиброз, фиброзные осложнения после хирургического вмешательства, хроническую васкулопатию аллотрансплантата и/или хроническое отторжение трансплантированных органов, ишемически-реперфузионный травматический фиброз, фиброз вследствие инъекций, цирроз, диффузную паренхиматозную болезнь легких, болевой синдром после вазэктомии и ревматоидный артрит.
[0394] Некоторые варианты реализации включают комбинацию соединений, композиций и/или фармацевтических композиций, описанных в настоящем описании, с дополнительным агентом, таким как противовоспалительные средства, включая глюкокортикоиды, анальгетики (например, ибупрофен), аспирин, и агенты, которые модулируют Th2-иммунный ответ, иммунодепрессанты, включая метотрексат, микофенолат, циклофосфамид, циклоспорин, талидомид, помалидомид, лефлуномид, гидроксихлорохин, азатиоприн, растворимый бычий хрящ, вазодилататоры, включая антагонисты рецепторов эндотелина, аналоги простациклина, нифедипин, и силденафил, селективный ингибитор IL-6, селективные и неселективные ингибиторы тирозинкиназ, модуляторы Wnt-пути, активаторы PPAR, ингибиторы каспазы-3, антагонисты рецепторов LPA, агенты, разрушающие В-клетки, антагонисты CCR2, пирфенидон, агонисты каннабиноидных рецепторов, ингибиторы ROCK, агенты, нацеленные на миРНК, антагонисты толл-подобных рецепторов, агенты, нацеленные на CTGF, ингибиторы НАДФН-оксидазы, ингибиторы триптазы, ингибиторы TGFD, агонисты рецептора релаксина и регенеративные клетки, полученные из аутологичной жировой ткани.
Показания к применению
[0395] В некоторых вариантах реализации соединения и композиции, содержащие соединения, описанные в настоящем описании, можно применять для лечения множества состояний, возникающих в результате фиброза или воспаления, и, в частности, включая те, которые связаны с дифференцировкой миофибробластов. Примеры состояний включают фиброз печени (алкогольное, вирусное, аутоиммунное, метаболическое и наследственное хроническое заболевание), фиброз почки (например, в результате хронического воспаления, инфекций или диабета типа II), фиброз легких (идиопатический или в результате воздействия окружающей среды, включая токсические частицы, саркоидоз, асбестоз, гиперчувствительный пневмонит, бактериальные инфекции, включая туберкулез, лекарственные средства и т.д.), интерстициальный фиброз, системную склеродермию (аутоиммунное заболевание, при котором многие органы становятся фиброзными), дегенерацию желтого пятна (фиброзная болезнь глаза), фиброз поджелудочной железы (возникающий в результате, например, злоупотребления алкоголем и хронических воспалительных заболеваний поджелудочной железы), фиброз селезенки (вследствие серповидно-клеточной анемии, других заболеваний крови), фиброз сердца (в результате инфекции, воспаления и гипертрофии), фиброз средостения, миелофиброз, фиброз эндомиокарда, ретроперитонеальный фиброз, прогрессирующий массивный фиброз, нефрогенный системный фиброз, фиброзные осложнения после хирургических вмешательств, хроническую васкулопатию аллотрансплантата и/или хроническое отторжение в пересаженных органах, ишемически-реперфузионное повреждение, связанное с фиброзом, фиброз вследствие инъекций, цирроз печени, диффузные заболевания паренхимы легких, болевой синдромом после вазэктомии и заболевания или нарушения, связанные с ревматоидным артритом.
[0396] Чтобы дополнительно проиллюстрировать это изобретение, в описание включены следующие примеры. Эти примеры, конечно, не должны рассматриваться как конкретно ограничивающие изобретение. Варианты этих примеров в пределах объема формулы изобретения находятся в компетенции специалиста в данной области техники и считаются находящимися в пределах объема изобретения, как описано и заявлено в настоящем описании. Читатель поймет, что специалист в данной области техники, применяющий настоящее описание и квалифицированный в данной области техники, способен получить и применить настоящее изобретение без исчерпывающих примеров. Следующие примеры будут дополнительно описывать настоящее изобретение и используются только в целях иллюстрации и не должны рассматриваться как ограничивающие.
ПРИМЕРЫ
Общие методики
[0397] Для специалиста в данной области техники очевидно, что способы получения предшественников и функциональных групп, соответствующих заявленным соединениям, в целом описаны в литературе. В указанных реакциях также возможно применение вариантов, которые сами по себе известны специалистам в данной области техники, но не указываются в больших подробностях. Специалист в данной области техники с учетом сведений из литературы и настоящего описания хорошо подготовлен для того, чтобы получить любые предложенные соединения.
[0398] Очевидно, что специалист в области органической химии может легко провести манипуляции без дополнительных инструкций, а это означает, что осуществление данных манипуляций находится в сфере компетенции и практической деятельности специалиста в данной области техники. Такие манипуляции включают восстановление карбонильных соединений до их соответствующих спиртов, процессы окисления, ацилирования, ароматического замещения, как электрофильного, так и нуклеофильного, этерификации с получением простых эфиров, этерификации с получением сложных эфиров и омыления и тому подобное. Указанные манипуляции обсуждаются в стандартных руководствах, таких как March Advanced Organic Chemistry (Wiley), Carey and Sundberg, Advanced Organic Chemistry (содержание данной публикации полностью включено в настоящее описание посредством ссылки) и тому подобные. Все промежуточные соединения согласно настоящему изобретению использовали без дополнительной очистки, если не указано иное.
[0399] Специалисту в данной области техники будет очевидно, что некоторые реакции лучше всего проводить, когда другие функциональные группы в молекуле замаскированы или защищены, что позволяет избежать каких-либо нежелательных побочных реакций и/или увеличить выход реакции. Как правило, для обеспечения таких повышенных выходов или предотвращения нежелательных реакций специалист в данной области техники использует защитные группы. Указанные реакции могут быть найдены в литературе и также находятся в сфере компетенции специалиста в данной области техники. Примеры большинства указанных манипуляций могут быть найдены, например, в источнике Т. Greene and P. Wuts Protecting Groups in Organic Synthesis, 4th Ed., John Wiley & Sons (2007), содержание которого полностью включено в настоящее описание посредством ссылки.
[0400] Следующие иллюстративные схемы представлены для того, чтобы проинструктировать читателя, и отображают предпочтительные способы получения соединений, приведенных в качестве примеров в настоящем описании. Указанные способы являются неограничивающими, и будет очевидно, что для получения указанных соединений могут быть применены и другие способы. Такие способы конкретно включают химию твердых фаз, включая комбинаторную химию. Специалист в данной области техники основательно подготовлен для того, чтобы получить указанные соединения способами, приведенными в литературе и в настоящем описании. Нумерация соединений, используемая в схемах синтеза, изображенных ниже, относится только к конкретным схемам и не должна быть истолкована и спутана с номерами из других разделов настоящей заявки.
[0401] Товарные знаки, используемые в настоящем описании, приведены исключительно для примера и отражают иллюстративные материалы, использованные во время изобретения. Специалисту в данной области техники будет понятно, что вариации в серийных производственных и подобных процессах ожидаемы. Следовательно, примеры и использованные в них товарные знаки являются неограничивающими и не предполагаются для ограничения, а являются исключительно иллюстрацией того, как специалист в данной области техники может выбрать способ осуществления одного или более вариантов реализации настоящего изобретения.
[0402] Следующие сокращения имеют указанные значения:


[0403] Следующие иллюстративные схемы представлены для того, чтобы проинструктировать читателя, и в совокупности отображают иллюстративные способы получения соединений, предложенных согласно настоящему изобретению. Кроме того, другие способы получения соединений, описанных в настоящем описании, будут легко понятны специалисту в данной области техники в свете приведенных ниже схем реакций и примеров. Если не указано иное, все переменные имеют значения, определенные выше.
ПРИМЕР 1
СОЕДИНЕНИЯ 1-2, 5-6, 8, 91-92
(S)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-ФЕНИЛ-1Н-ИМИДАЗОЛ-5-КАРБОКСАМИД (1)
[0404] Смесь соединения 1А (102 мг, 1,0 экв.), соединения 1В (160 мг, 1,2 экв.) и HBTU (250 мг, 1,25 экв.) в ДМФА (8 мл) перемешивали при комнатной температуре в течение 5 мин и затем добавляли DIEA (0,3 мл, 3,0 экв.). После перемешивания при комнатной температуре в течение 30 мин реакционную смесь разбавляли 50 мл этилацетата и 20 мл гексана, промывали водой, насыщенным NaHCO3 и солевым раствором и концентрировали под вакуумом с получением промежуточного соединения 1С (190 мг, выход 92%).
[0405] Раствор соединения 1С (190 мг, 1,0 экв.) в сухом ТГФ (15 мл) охлаждали до -50°С в атмосфере N2 и затем добавляли по каплям раствор 1 н. LAH в ТГФ (0,55 мл, 1,1 экв.) при -50°С. Реакционную смесь перемешивали при температуре от -30°С до -10°С в течение 2 ч, добавляли в нее для гашения реакции насыщенный NaHCO3 при -20°С, и затем смесь подвергали экстракции 3×30 мл этилацетата. Объединенную органическую фазу сушили над Na2SO4 с получением неочищенной смеси, которую очищали посредством колоночной хроматографии на силикагеле. Соединение 1 (105 мг, 65%): МС (ИЭР) m/z (М+Н)+: 320,3; 1Н ЯМР (400 МГц, CDCl3): δ 9,64 (s, 1H), 7,65 (s, 1H), 7,56 (s, 1H), 7,46 (m, 3Н), 7,26-7,33 (m, 5Н), 7,09 (m, 2Н), 6,29 (d, 1H), 4,81 (m, 1H), 3,19 (d, 2Н) ppm.
(S)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-ФЕНИЛ-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (2)
((S)-5-МЕТИЛ-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-ФЕНИЛ-1Н-ПИРАЗОЛ-3-КАРБОКСАМИД (5)
(S)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-4-ФЕНИЛТИАЗОЛ-5-1САРБОКСАМИД (6)
(S)-3-МЕТИЛ-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-ФЕНИЛ-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (8)
[0406] Соединения 2, 5, 6 и 8 были получены, как описано в примере 1, с использованием подходящей карбоновой кислоты, соответственно. Соединение 2: МС (ИЭР) m/z (М+Н)+: 320,2; 1Н ЯМР (400 МГц, ДМСО): δ 9,6 (s, 1H), 9,15 (d, 1H), 7,73 (s, 1H), 7,4-7,2 (m, 10Н), 6,8 (s, 1H), 4,53 (m, 1H), 3,25 (dd, 1H), 2,8 (dd, 1H) ppm.
[0407] Соединение 5: МС (ИЭР) m/z (М+Н)+: 334,3; 1H ЯМР (400 МГц, CDCl3): δ 9,67 (s, 1H), 7,54-7,4 (m, 6Н), 7,3-7,2 (m, 5Н), 6,73 (s, 1H), 4,82 (m, 1H), 3,21 (d, 2Н), 2,33 (s, 3Н) ppm.
[0408] Соединение 6: МС (ИЭР) m/z (М+Н)+: 337,5; 1H ЯМР (400 МГц, CDCl3): δ 9,56 (s, 1H), 8,88 (s, 1H), 7,5-7,34 (m, 5Н), 7,27-7,2 (m, 3Н), 6,94 (m, 2Н), 6,35 (d, 1H), 4,73 (m, 1H), 3,1 (dd, 1H), 3,08 (dd, 1Н) ppm.
[0409] Соединение 8: МС (ИЭР) m/z (М+Н)+: 334,3; 1H ЯМР (400 МГц, ДМСО): δ 9,59 (s, 0,6Н), 9,01 (d, 0,6Н), 8,35 (d, 0,4Н), 7,38-7,06 (m, 10Н), 6,58 (s, 0,6Н), 6,48 (s, 0,4Н), 4,82 (m, 0,2Н), 4,54 (m, 0,6Н), 3,98 (m, 0,4Н), 3,25 (dd, 0,6Н), 2,98 (dd, 0,4Н), 2,78 (dd, 0,6Н), 2,7 (dd, 0,4Н), 2,48 (s, 1,8Н), 2,21 (s, 1,2Н) ppm.
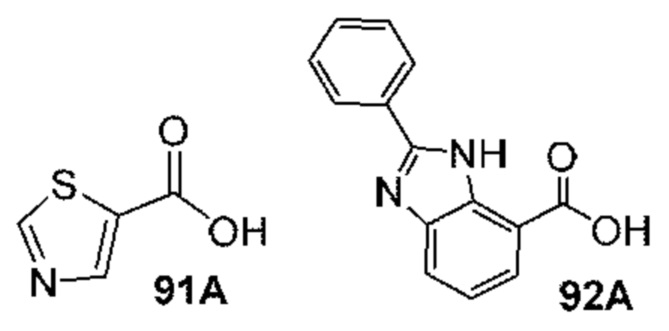
(S)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (91)
[0410] Соединение 91 было получено, как описано в примере 1, из соответствующих исходных веществ, соединений 91А и 1В. Соединение 91: 1Н ЯМР (400 МГц, CDCl3): δ 9,69 (s, 1H), 8,85 (s, 1H), 8,21 (s, 1H), 7,06 (d, 1H), 7,32-7,18 (m, 8Н), 4,88 (m, 1H), 3,26 (m, 2Н) ppm. МС (ИЭР) m/z (М+Н)+ 261,3.
(S)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-2-ФЕНИЛ-1H-БЕНЗО[d]ИМИДАЗОЛ-7-КАРБОКСАМИД (92)
[0411] Соединение 92 было получено, как описано в примере 1, с использованием соответствующей карбоновой кислоты. 1Н ЯМР (400 МГц, CDCl3): δ 12,07 (s, 1H), 11,92 (d, 1H), 9,84 (s, 1H), 8,1-8,0 (m, 3Н), 7,5-7,46 (m, 10Н), 7,32-7,18 (m, 8Н), 5,04 (m, 1H), 3,34 (d, 2Н) ppm. МС (ИЭР) m/z (М+Н)+ 370,4.
ПРИМЕР 2
СОЕДИНЕНИЯ 3-4
(S)-1-(БЕНЗО[D]ТИАЗОЛ-2-ИЛ)-N-((S)-1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)ПИРРОЛИДИН-2-КАРБОКСАМИД (3)
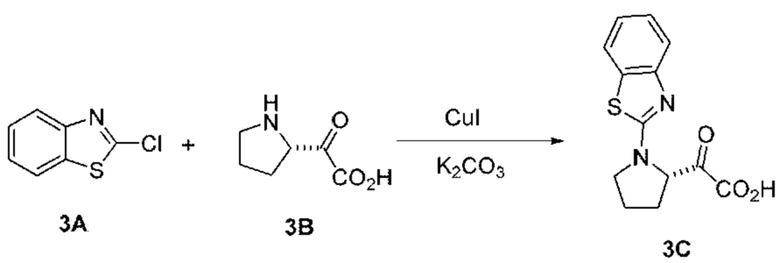
[0412] Смесь соединения 3А (500 мг, 1,0 экв.), соединения 3В (738 мг, 1,0 экв.), CuI (124 мг, 0,15 экв.) и K2CO3 (1,8 г, 3,0 экв.) в DMA (15 мл) нагревали при 100°С в течение 18 ч, и затем неорганические соединения удаляли путем фильтрования. Смесь разбавляли водой (50 мл), доводили рН до ~6 и затем подвергали экстракции 3×50 мл ацетата с получением промежуточного соединения 3С. Соединение 3 было получено, как описано в примере 1, с использованием соответствующей карбоновой кислоты, промежуточного соединения 3С. Соединение 3: МС (ИЭР) m/z (М+Н)+: 380,2; 1Н ЯМР (400 МГц, CDCl3): δ 9,66 (s, 1H), 8,32 (d, 1H), 7,63 (d, 1H), 7,52 (d, 1H), 7,30 (t, 1H), 7,10 (t, 1H), 6,92-7,01 (m, 5Н), 4,69 (m, 2Н), 3,45 (m, 1H), 3,36 (m, 1H), 3,17 (dd, 1H), 2,90 (dd, 1H), 2,55 (m, 1H), 2,03 (m, 3Н) ppm.
(S)-1-(БЕНЗО[D]ОКСАЗОЛ-2-ИЛ)-N-((S)-1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)ПИРРОЛИДИН-2-КАРБОКСАМИД (4)
[0413] Соединение 4 было получено, как описано в примере 2, с использованием соответствующих исходных веществ. МС (ИЭР) m/z (М+Н)+: 364,3; 1H ЯМР (400 МГц, CDCl3): δ 9,65 (s, 1H), 7,69 (ушир. d, 1H), 7,37 (d, 1H), 7,33 (t, 1H), 7,18 (t, 1H), 6,98-7,10 (m, 6Н), 4,71 (m, 2Н), 4,59 (m, 1H), 3,61 (m, 2Н), 3,19 (dd, 1H), 2,97 (dd, 1H), 2,41 (m, 1H), 1,91-2,12 (m, 3Н) ppm.
ПРИМЕР 3
(S)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-3-ФЕНИЛИЗОТИАЗОЛ-4-КАРБОКСАМИД (9)
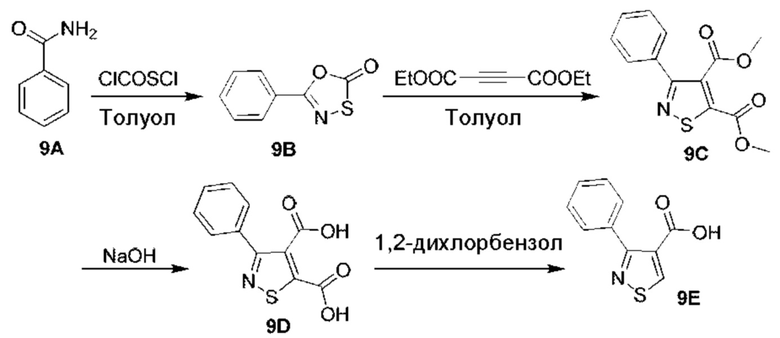
[0414] К суспензии соединения 9А (1,2 г) в толуоле (15 мл) добавляли хлоркарбонилсульфенилхлорид (1,3 мл). Смесь нагревали при 100°С в течение 2 ч с получением прозрачного раствора (наблюдали выделение газа). Когда анализ посредством ТСХ свидетельствовал о завершении превращения, реакционную смесь концентрировали, и твердый остаток растирали с гексаном, отфильтровывали и сушили с получением соединения 9В.
[0415] К раствору соединения 9В (1,4 г) в α,α,α-трифтортолуоле (10 мл) добавляли диэтилацетилендикарбоксилат (2,0 мл). После нагревания в микроволновом реакторе при 170°С в течение 1 ч реакционную смесь концентрировали, и маслянистый остаток очищали посредством колоночной флэш-хроматографии. Фракции, содержащие продукт, объединяли, концентрировали, и остаток растирали с гексаном, отфильтровывали и сушили с получением соединения 9С.
[0416] Раствор соединения 9С (2,1 г) и NaOH (1,4 г) в воде (20 мл) нагревали с обратным холодильником в течение 2,5 ч. Реакционную смесь охлаждали, разбавляли водой (150 мл) и подкисляли с помощью концентрированной HCl (водного раствора). Происходило образование осадка. Водный слой подвергали экстракции EtOAc (2×200 мл; осадок медленно растворялся). Объединенные органические слои промывали солевым раствором, сушили (Na2SO4) и концентрировали с получением соединения 9D.
[0417] Суспензию соединения 9D (1,8 г) в 1,2-дихлорбензоле (20 мл) нагревали с обратным холодильником в течение 20 мин (наблюдали образование газа). Реакционную смесь охлаждали, разбавляли гексаном (50 мл) и фильтровали с осаждением продукта. К суспензии неочищенного продукта в воде (40 мл) добавляли 1 н. NaOH (10 мл). Водный слой подвергали экстракции этилацетатом (2×100 мл) и подкисляли с помощью концентрированной HCl до рН ~3. Продукт экстрагировали EtOAc (2×100 мл). Объединенные органические слои промывали солевым раствором, сушили (Na2SO4) и концентрировали с получением промежуточного соединения 9Е.
[0418] Соединение 9 было получено, как описано в примере 1, с использованием соответствующей карбоновой кислоты, промежуточного соединения 9Е. МС (ПЭР) m/z (М+Н)+: 359,1; 1Н ЯМР (400 МГц, CDCl3): δ 9,59 (s, 1H), 9,16 (s, 1H), 7,56-7,5 (m, 2Н), 7,48-7,4 (m, 3Н), 7,27-7,22 (m, 3Н), 6,94 (m, 2Н), 6,15 (d, 1H), 4,79 (m, 1H), 3,1 (d, 2Н) ppm.
ПРИМЕР 4
СОЕДИНЕНИЯ 7, 10-11, 14, 18, 20
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ФЕНИЛ-1Н-ИМИДАЗОЛ-5-КАРБОКСАМИД (7)
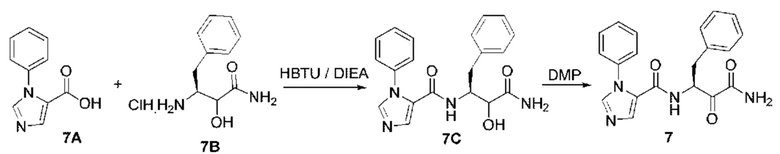
[0419] Смесь соединения 7А (50 мг, 1,0 экв.), соединения 7В (74 мг, 1,2 экв.) и HBTU (126 мг, 1,25 экв.) в ДМФА (3 мл) перемешивали при комнатной температуре в течение 5 мин и затем добавляли DIEA (0,15 мл, 3,0 экв.). После перемешивания при комнатной температуре в течение 30 мин реакционную смесь разбавляли этилацетатом (30 мл) и гексаном (10 мл), промывали 1 н. HCl, водой, насыщенным NaHCO3 и солевым раствором и концентрировали под вакуумом с получением промежуточного соединения 7С (65 мг, выход 67%) в виде белого твердого вещества.
[0420] К раствору соединения 7С (65 мг, 1,0 экв.) в сухом ДХМ (10 мл) и ДМСО (2 мл) добавляли DMP (305 мг, 4,0 экв.). После перемешивания при комнатной температуре в течение 1 ч смесь разбавляли ДХМ (30 мл), добавляли в нее для гашения реакции 10% водный Na2S2O3/насыщенный водный NaHCO3 (об./об. = 1/1, ~ 10 мл). Органический слой отделяли путем подвергания водного слоя экстракции ДХМ (30 мл × 5). Объединенный органический слой промывали H2O (10 мл), солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали с получением белого твердого вещества, которое затем растирали в смеси CH2Cl2/гексан с получением чистого продукта соединения 7 (29 мг, выход 45%). МС (ПЭР) m/z (М+Н)+: 363,4; 1H ЯМР (400 МГц, CDCl3): δ 7,66 (s, 1H), 7,57 (s, 1H), 7,45 (m, 3Н), 7,26-7,35 (m, 5Н), 7,05 (m, 2Н), 6,72 (s, 1H), 6,24 (d, 1H), 5,58 (m, 2Н), 4,81 (m, 1H), 3,38 (dd, 1H), 3,14 (dd, 1H) ppm.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ФЕНИЛ-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (10)
[0421] Указанное соединение было получено, как описано в примере 4, с использованием соответствующей карбоновой кислоты. МС (ИЭР) m/z (М+Н)+: 363,3; 1Н ЯМР (400 МГц, ДМСО): δ 9,15 (d, 1H), 8,11 (s, 1H), 7,71 (s, 1H), 7,4-7,2 (m, 10Н), 7,07 (d, 1H), 6,72 (s, 1H), 5,26 (m, 1H), 2,81 (dd, 1H), 2,64 (dd, 1H) ppm.
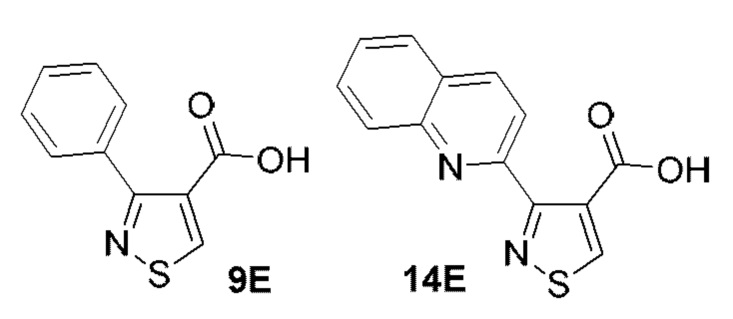
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ФЕНИЛИЗОТИАЗОЛ-4-КАРБОКСАМИД (11)
[0422] Указанное соединение было получено, как описано в примере 4, с использованием соответствующей карбоновой кислоты, промежуточного соединения 9Е. МС (ИЭР) m/z (М+Н)+ 380,2; 1Н ЯМР (400 МГц, ДМСО): δ 9,15 (d, 1H), 9,05 (s, 1H), 8,14 (s, 1H), 7,89 (s, 1H), 7,5-7,4 (m, 2Н), 7,3-7,2 (m, 8Н), 5,34 (m, 1H), 3,2 (d, 2Н) ppm.
(S)-1-(БЕНЗО[D]ОКСАЗОЛ-2-ИЛ)-N-((S)-1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)ПИРРОЛИДИН-2-КАРБОКСАМИД (14)
[0423] Промежуточное соединение 14Е было получено, как описано в примере 3. Соединение 14 затем было получено, как описано в примере 4, с использованием соответствующей карбоновой кислоты, промежуточного соединения 14Е. Соединение 14: МС (ИЭР) m/z (М+Н)+: 431,5; 1Н ЯМР (400 МГц, ДМСО): δ 9,14 (s, 1H), 9,05 (d, 1H), 8,16 (d, 1H), 7,9 (s, 1H), 7,62-7,56 (m, 2Н), 7,3-7,2 (m, 8Н), 5,36 (m, 1H), 3,17 (dd, 1H), 2,78 (dd, 1H) ppm.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФЕНИЛТИАЗОЛ-5-КАРБОКСАМИД (18)
[0424] Указанное соединение было получено, как описано в примере 4, с использованием соответствующей карбоновой кислоты. МС (ИЭР) m/z (М+Н)+: 380,1; 1Н ЯМР (400 МГц, ДМСО): δ 10,11 (d, 1H), 9,33 (s, 1H), 8,49 (d, 1H), 8,13 (s, 1H), 8,07 (d, 1H), 8,03 (d, 1H), 7,85 (s, 1H), 7,74 (m, 2Н), 7,65 (m, 1H), 7,12-7 (m, 5Н), 5,51 (m, 1H), 3,18 (dd, 1H) 2,89 (dd, 1H) ppm.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-(БЕНЗО[D][1,3]ДИОКСОЛ-5-ИЛ)-3-МЕТИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (20)
[0425] Указанное соединение было получено, как описано в примере 4, с использованием соответствующей карбоновой кислоты. МС (ИЭР) m/z (М+Н)+: 422,1; 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,88 (d, 1H), 8,19 (s, 1H), 7,91 (s, 1H), 7,17-7,30 (m, 7Н), 6,94 (d, 1H), 6,11 (s, 2Н), 5,45 (m, 1H), 3,22 (dd, 1H), 2,72 (dd, 1H), 2,03 (s, 3Н) ppm.
ПРИМЕР 5
СОЕДИНЕНИЯ 12-13, 15-17, 19, 27, 44, 47, 54, 60, 94, 117-118,128, 148, 207, 235, 303-305, 309-312, 23, 39, 456, 461, 492
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИДИН-2-ИЛ)-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (12)
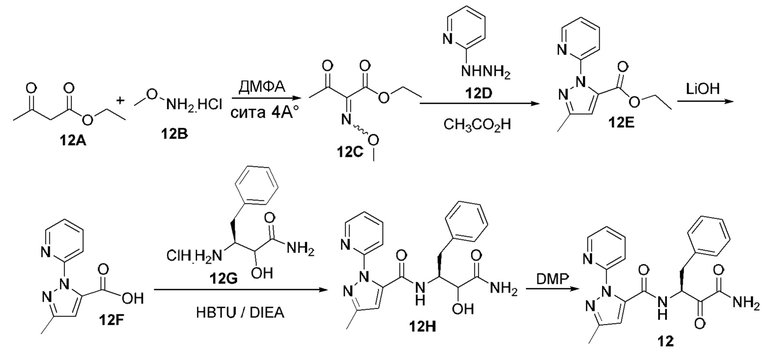
[0426] К раствору соединения 12А (5,0 г, 1,0 экв.) и соединения 12В (2,64 г, 1,0 экв.) в сухом ДМФА (20 мл) добавляли молекулярное сито с размером ячейки 4А° (5,0 г, порошок). Полученную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 20 ч, фильтровали для удаления молекулярных сит, разбавляли гексаном (80 мл) и этилацетатом (80 мл) и затем промывали 3×50 мл воды, 50 мл насыщенного NaHCO3 и солевым раствором. Неочищенную смесь очищали посредством колоночной хроматографии на силикагеле с получением соединения 12С (3,2 г, выход 48%) в виде прозрачного масла.
[0427] Смесь соединения 12С (350 мг, 1,0 экв.) и соединения 12D (190 мг, 1,0 экв.) в уксусной кислоте (8 мл) нагревали при 100°С в течение 1 ч. Остаток, после удаления растворителя под вакуумом, суспендировали в этилацетате (80 мл), промывали насыщенным NaHCO3 и солевым раствором. Неочищенную смесь очищали посредством колоночной хроматографии на силикагеле с получением соединения 12Е (100 мг, выход 25%). Соединение 12Е (100 мг) обрабатывали LiOH в смеси МеОН/вода с получением соединения 12F (87 мг, выход 100%).
[0428] Смесь соединения 12F (85 мг, 1,0 экв.), соединения 12G (116 мг, 1,2 экв.) и HBTU (190 мг, 1,2 экв.) в ДМФА (5 мл) перемешивали при комнатной температуре в течение 5 мин и затем добавляли DIEA (0,3 мл, 4,0 экв.). После перемешивания при комнатной температуре в течение 30 мин смесь разбавляли 50 мл этилацетата и 20 мл гексана, промывали 1 н. HCl, водой, насыщенным NaHCO3 и солевым раствором и концентрировали под вакуумом с получением промежуточного соединения 12Н (150 мг, выход 94%) в виде белого твердого вещества.
[0429] К раствору соединения 12Н (150 мг, 1,0 экв.) в сухом ДХМ (20 мл) и ДМСО (2,5 мл) добавляли DMP (673 мг, 4,0 экв.). После перемешивания при комнатной температуре в течение 1 ч смесь разбавляли ДХМ (80 мл), добавляли в нее для гашения реакции 10% Na2S2O3/насыщенный NaHCO3 (об./об. = 1/1, ~ 20 мл). Органический слой отделяли. Водный слой подвергали экстракции ДХМ (30 мл × 2). Объединенный органический слой промывали H2O (10 мл), солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали с получением белого твердого вещества. Твердое вещество растирали в смеси CH2Cl2/гексан с получением чистого соединения 12 (95 мг, выход 64%). МС (ПЭР) m/z (М+Н)+: 378,3; 1H ЯМР (400 МГц, ДМСО-d6): δ 9,15 (d, 1H), 8,21 (d, 1H), 7,91 (t, 1H), 7,82 (s, 1H), 7,54 (d, 1H), 7,17-7,32 (m, 6Н), 6,49 (s, 1H), 5,29 (m, 1H), 3,15 (dd, 1H), 2,84 (dd, 1H), 2,23 (s, 3Н) ppm.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(БЕНЗО[D]ТИАЗОЛ-2-ИЛ)-3-МЕТИЛ-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (13)
(S)-N(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ХИНОЛИН-2-ИЛ)-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (15)
(S)-1-([1,1'-БИФЕНИЛ]-3-ИЛ)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (16)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-(МЕТИЛСУЛЬФОНИЛ)ФЕНИЛ)-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД(17)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-(ТРИФТОРМЕТОКСИ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД(19)
[0430] Соединения 13, 15-17 и 19 были получены, соответственно, как описано в примере 5, с использованием соответствующего производного гидразина.
[0431] Соединение 13: МС (ИЭР) m/z (М+Н)+: 434,3; 1H ЯМР (400 МГц, ДМСО-d6): δ 10,09 (d, 1H), 8,10 (d, 1H), 7,99 (d, 1H), 7,83 (s, 1H), 7,62 (d, 1H), 7,40 (m, 2Н), 7,04-7,24 (m, 5Н), 6,68 (s, 1H), 5,51 (m, 1H), 3,16 (dd, 1H), 2,95 (dd, 1H), 2,24 (s, 3Н) ppm.
[0432] Соединение 15: МС (ИЭР) m/z (М+Н)+: 428,4; 1H ЯМР (400 МГц, ДМСО): δ 9,28 (d, 0,5Н), 8,77 (d, 0,5Н), 8,45 (d, 1H), 8 (d, 1H), 7,9-7,5 (6Н), 7,2-7,1 (6Н), 5,4 (m, 0,5Н), 4,44 (m, 0,5Н), 3,2-2,7 (m, 2Н) ppm.
[0433] Соединение 16: МС (ИЭР) m/z (М+Н)+: 453,3; 1H ЯМР (400 МГц, ДМСО-d6): δ 9,10 (d, 1H), 8,09 (s, 1H), 7,87 (s, 1H), 7,35-7,62 (m, 8Н), 7,19-7,29 (m, 5Н), 7,09 (d, 1H), 6,61 (s, 1H), 5,30 (m, 1H), 3,17 (dd, 1H), 2,81 (dd, 1H), 2,25 (s, 3Н) ppm.
[0434] Соединение 17: МС (ИЭР) m/z (М+Н)+: 455,3; 1H ЯМР (400 МГц, ДМСО-d6): δ 9,27 (d, 1H), 8,14 (s, 1H), 7,88 (m, 2Н), 7,82 (d, 1H), 7,35-7,62 (m, 8Н), 7,19-7,45 (m, 7Н), 6,62 (s, 1H), 5,25 (m, 1H), 3,19 (m, 4Н), 2,82 (dd, 1H), 2,25 (s, 3Н) ppm.
[0435] Соединение 19: МС (ИЭР) m/z (М+Н)+: 461,3; 1H ЯМР (400 МГц, ДМСО-d6): δ 9,15 (d, 1H), 8,11 (s, 1H), 7,87 (s, 1H), 7,82 (d, 1H), 7,21-7,35 (m, 9Н), 6,61 (s, 1H), 5,23 (m, 1H), 3,20 (dd, 1H), 2,82 (dd, 1H), 2,24 (s, 3Н) ppm.
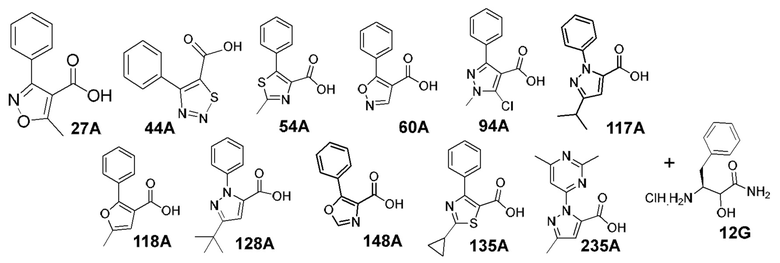
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-3-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (27)
[0436] Соединение 27 (30,0 мг, выход 43,0%, белое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, соединения 27А. Соединение 27: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,94 (d, J=7,6 Гц, 1H), 8,17 (s, 1H), 7,90 (s, 1H), 7,49-7,41 (m, 3Н), 7,41-7,34 (m, 2Н), 7,33-7,21 (m, 5Н), 5,42-5,35 (m, 1H), 3,29-3,21 (m, 1H), 2,80-2,70 (m, 1H), 2,35-2,27 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 378,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФЕНИЛ-1,2,3-ТИАДИАЗОЛ-5-КАРБОКСАМИД (44)
[0437] Соединение 44 (42,4 мг, выход: 47,7%, белое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 44А. Соединение 44: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,60 (ушир. d, J=7,5 Гц, 1H), 8,19 (s, 1H), 7,93 (s, 1H), 7,79-7,67 (m, 2Н), 7,52-7,39 (m, 3Н), 7,34-7,21 (m, 5Н), 5,52-5,39 (m, 1H), 3,23 (dd, J=14,0, 3,5 Гц, 1H), 2,78 (dd, J=13,6, 10,5 Гц, 1H). МС (ИЭР) m/z (М+Н)+381,0.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-5-ФЕНИЛТИАЗОЛ-4-КАРБОКСАМИД (54)
[0438] Соединение 54 (75 мг, выход: 75,4%, белое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 54А. 1Н ЯМР (400 МГц, ДМСО-d6) S 8,46 (ушир. d, J=7,7 Гц, 1H) 8,05 (ушир. s, 1H) 7,81 (ушир. s, 1H) 7,43-7,29 (m, 1H) 7,41-7,29 (m, 1H) 7,29-7,29 (m, 1H) 7,41-7,29 (m, 1H) 7,30-7,28 (m, 1H) 7,28-7,08 (m, 5Н) 5,37 (td, J=8,1, 4,5 Гц, 1H), 3,22-3,09 (m, 1H), 3,17 (ушир. dd, J=14,0, 4,1 Гц, 1H) 3,06-2,92 (m, 1H) 2,72-2,60 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 394,0.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (60)
[0439] Соединение 60 (40 мг, выход 36,20%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, соединения 60А. Соединение 60: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,02 (d, J=7,5 Гц, 1H), 8,83 (s, 1H), 8.16 (s, 1H), 7,92-7,78 (m, 3Н), 7,59-7,42 (m, 3Н), 7,35-7,17 (m, 4Н), 5,43-5,34 (m, 1H), 3,27-3,17 (m, 1H), 2,90-2,79 (m, 1H). МС (ИЭР) m/z (М+Н)+ 364,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ХЛОР-1-МЕТИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (94)
[0440] Соединение 94 было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 94А. Соединение 94: 1Н ЯМР (400 МГц, ДМСО): δ 8,8 (d, 1H), 8,16 (s, 1H), 7,89 (s, 1H), 7,5-7,46 (m, 2Н), 7,32-7.18 (m, 8Н), 5,41 (m, 1H), 3,82 (s, 3Н), 3,17 (dd, 1H), 2,76 (dd, 1H) ppm. МС (ИЭР) m/z (М+Н)+ 410,9.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ИЗОПРОПИЛ-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (117)
[0441] Соединение 117 (10 мг, выход 18,29%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 117А. Соединение 117: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,36 (d, J=7,8 Гц, 1H), 8,09 (s, 1H), 7,84 (s, 1H), 7,63-7,47 (m, 5Н), 7,32-7,14 (m, 5Н), 6,66 (s, 1H), 5,50-5,39 (m, 1H), 3,23-3,13 (m, 1H), 3,09-2,89 (m, 2Н), 1,12 (d, J=6,8 Гц, 6Н). МС (ИЭР) m/z (М+Н)+ 405,2.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-2-ФЕНИЛФУРАН-3-КАРБОКСАМИД (118)
[0442] Соединение 118 (58 мг, выход: 55,4%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 118А. Соединение 118: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,54 (d, J=7,5 Гц, 1H), 8,08 (s, 1H), 7,81 (s, 1H), 7,68 (d, J=7,0 Гц, 2Н), 7,35-7,26 (m, 7Н), 7,23-7,17 (m, 1H), 6,39 (d, J=0,9 Гц, 1H), 5,30 (ушир. d, J=0,7 Гц, 1H), 3,19-3,12 (m, 1H), 2,88-2,79 (m, 1H), 2,33-2,29 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 377,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ТРЕТ-БУТИЛ)-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (128)
[0443] Соединение 128 (101,7 мг, выход 68,04%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 128А. Соединение 128: 1Н ЯМР (400 МГц, CDCl3) δ 7,55-7,44 (m, 3Н), 7,42-7,35 (m, 2Н), 7,34-7,28 (m, 1H), 7,25-7,14 (m, 5Н), 6,74 (ушир. s, 1H), 6,70 (s, 1H), 5,73-5,64 (m, 1H), 5,53 (ушир. s, 1H), 3,44-3,35 (m, 1H), 3,18-3,09 (m, 1H), 1,16 (s, 9Н). МС (ИЭР) m/z (М+1)+419,3.
(S)-N-(4-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛОКСАЗОЛ-4-КАРБОКСАМИД (148)
[0444] Соединение 148 (10 мг, выход: 30,8%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 148А. Соединение 148: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,57 (s, 1H), 8,44 (d, J=7,7 Гц, 1H), 8,14-8,03 (m, 3Н), 7,85 (s, 1H), 7,49-7,42 (m, 3Н), 7,30-7,15 (m, 5H), 5,49-5,40 (m, 1H), 3,26-3,17 (m, 1H), 3,12-3,02 (m, 1H). MC (ИЭР) m/z (M+H)+ 364,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-ЦИКЛОПРОПИЛ-4-ФЕНИЛТИАЗОЛ-5-КАРБОКСАМИД (207)
[0445] Соединение 207 (54,0 мг, выход 44,09%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 135А. Соединение 207: 1Н ЯМР (400 МГц, CDCl3) δ 7,52-7,45 (m, 2Н), 7,43-7,38 (m, 3Н), 7,21-7,17 (m, 3Н), 6,79-6,77 (m, 2Н), 6,70 (s, 1H), 6,19-6,17 (d, J=6,0 Гц, 1H), 5,53 (s, 1H), 5,50-5,45 (m, 1H), 3,25-3,21 (m, 1H), 2,90-2,85 (m, 1H), 2,33-2,27 (m, 1H), 1,19-1,16 (m, 2Н), 1,13-1,10 (m, 2Н). МС (ИЭР) m/z (М+1)+ 420,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2,6-ДИМЕТИЛПИРИМИДИН-4-ИЛ)-3-МЕТИЛ-1Н-ПИРАЗОЛ-5КАРБОКСАМИД (235)
[0446] Соединение 235 (61,6 мг, выход 51,11%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 235А. Соединение 235: 1Н ЯМР (400 МГц, CDCl3) δ 9,83 (d, J=7,2 Гц, 1H), 7,57 (s, 1H), 7,21-7,16 (m, 3Н), 7,11-7,06 (m, 2Н), 6,87 (s, 1H), 6,75 (ушир. s, 1H), 5,84-5,76 (m, 1H), 5,56 (ушир. s, 1H), 3,49-3,31 (m, 2Н), 2,55 (s, 3Н), 2,34-2,32 (m, 6Н). МС (ИЭР) m/z (М+1)+ 407,1.
((S)-N-(1-АМИНО-1,2-ДИОКСОПЕНТАН-3-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (47)
[0447] Соединение 47 (90,00 мг, выход 60,4%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, 23А и 47А. Соединение 47: !H ЯМР (400 МГц, ДМСО-d6) δ 8,98 (d, J=6,6 Гц, 1H), 8,12 (ушир. s, 1H), 7,88-7,79 (m, 3Н), 7,57-7,50 (m, 3Н), 5,12-5,02 (m, 1H), 2,32 (s, 3Н), 1,95-1,77 (m, 1H), 1,65-1,48 (m, 1H), 0,93 (t, J=7,4 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 316,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (303)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (304)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ФЕНИЛ-3-(ТРИФТОРМЕТИЛ)-1H-ПИРАЗОЛ-5-КАРБОКС АМИД (305)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1,3-ДИФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (309)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(ТРЕТ-БУТИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (310)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ХЛОР-1-ЭТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (311)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ХЛОР-1-ЭТИЛ-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (312)
[0448] Соединения 303-305 и 309-312 были получены, как описано в примере 5, из соответствующей карбоновой кислоты с использованием соединения 12G, соответственно.
[0449] Соединение 303: 1H ЯМР (400 МГц, ДМСО-d6) δ 1H ЯМР (400 МГц, ДМСО): δ 8,51 (s, 1H), 7,9-7,85 (m, 2Н), 7,81 (d, 1H), 7,4-7,0 (m, 10Н), 4,53 (m, 1H), 2,98 (dd, 1H), 2,57 (dd, 1H) ppm. МС (ИЭР) m/z (М+Н)+ 364,3.
[0450] Соединение 304: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,2-8,9 (m, 1H), 8,11 (m, 1H), 7,7-7,1 (m, 12Н), 5,3 (m, 0,5Н), 4,4 (m, 0,5Н), 2,85-2,55 (m, 2Н) ppm. МС (ИЭР) m/z (М+Н)+ 364,3.
[0451] Соединение 305: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,3 (d, 1H), 8,07 (s, 1H), 7,83 (s, 1H), 7,4-7,1 (m, 10Н), 5,24 (m, 1H), 3,14 (dd, 1H), 2,74 (dd, 1H) ppm. МС (ИЭР) m/z (М+Н)+ 431,3.
[0452] Соединение 309: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,7 (m, 1H), 8,49 (d, 1H), 8,1-7,1 (m, 17Н), 5,31 (m, 0,5Н), 4,6-4,4 (m, 0,5Н), 3,1-2,7 (m, 2Н) ppm. МС (ИЭР) m/z (М+Н)+ 439,3.
[0453] Соединение 310: 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,75 (d, 1H), 7,4-7,1 (m, 5Н), 6,38 (s, 1H), 6,1 (d, 2Н), 4,48 (m, 1H), 3,02 (dd, 1H), 2,52 (dd, 1H) 2,08 (s, 3Н), 1,31 (s, 9Н) ppm. МС (ИЭР) m/z (М+Н)+ 379,3.
[0454] Соединение 311: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,89 (d, 1H), 8,13 (d, 1H), 7,86 (s, 1H), 7,33 (s, 1H), 7,3-7,1 (m, 5Н), 6,8 (s, 1H), 5,38 (m, 1H), 3,99 (q, 2Н), 3,21 (dd, 1H), 2,78 (dd, 1H) 1,11 (t, 3Н) ppm. МС (ИЭР) m/z (М+Н)+ 349,2.
[0455] Соединение 312: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,74 (d, 1H), 8,1 (s, 1H), 7,83 (s, 1H), 7,4-7,2 (m, 5Н), 6,58 (s, 1H), 5,29 (m, 1H), 4,25 (q, 2Н), 3,18 (dd, 1H), 2,87 (dd, 1H), 2,13 (s, 3Н), 1,15 (t, 3Н) ppm. МС (ИЭР) m/z (М+Н)+ 329,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (23)

[0456] К раствору соединения 23А (500 мг, 2,46 ммоль) в ТГФ (10 мл) добавляли 23 В (311 мг, 2,71 ммоль) и EDCI (566 мг, 2,95 ммоль) с ДХМ (10 мл). Смесь перемешивали при 25°С в течение 3 ч. Реакционную смесь концентрировали и разбавляли ЭА (20 мл). Затем смесь промывали HCl (1 М, 20 мл), насыщенным водным NaHCO3 (20 мл), сушили над Na2SO4 и концентрировали. Соединение 23С (800 мг, неочищенное, желтое масло): 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,93-7,88 (m, 2Н), 7,69-7,63 (m, 1H), 7,62-7,56 (m, 2Н), 2,87 (s, 4Н), 2,54-2,51 (m, 3Н).
[0457] Соединение 23 (30,0 мг, выход 35,0%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих промежуточных соединений 23С и 12G. Соединение 23: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,08 (ушир. d, J=8,0 Гц, 1H), 8,22 (s, 1H), 7,94 (s, 1H), 7,64 (ушир. d, J=7,2 Гц, 2Н), 7,55-7,41 (m, 3Н), 7,35-7,21 (m, 5Н), 5,54-5,45 (m, 1H), 3,29-3,23 (m, 1H), 2,81-2,71 (m, 1H), 2,09 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 378,0.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(4-ФЕНИЛ-1H-ПИРАЗОЛ-1-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (39)
[0458] Соединение 39 (5,20 мг, выход 26,12%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединений 21F и 12G. Соединение 39: 1Н ЯМР (CDCl3, 400 МГц): δ 11,75 (d, J=4,8 Гц, 1H), 8,73-8,71 (m, 1H), 8,73 (s, 1H), 8,66 (s, 1H), 7,69 (s, 1H), 7,54 (d, J=7,6 Гц, 2Н), 7,45-7,41 (m, 2Н), 7,35-7,31 (m, 1H), 7,29-7,27 (m, 1H), 7,25-7,21 (m, 4Н), 6,78 (ушир. s, 1H), 5,82-5,74 (m, 1H), 5,48 (ушир. s, 1H), 3,46-3,41 (m, 1H), 3,27-3,20 (m, 1H). МС (ИЭР) m/z (М+Н)+ 446,1.
N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (456)
[0459] Соединение 456 (240 мг, выход 86,0%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующих исходных веществ, соединений 12F и гидрохлорида 3-амино-N-циклопропил-2-гидрокси-4-фенилбутанамида. Соединение 456: 1Н ЯМР (CDCl3, 400 МГц): δ 9,15 (d, J=7,2 Гц, 1H), 8,80 (d, J=5,2 Гц, 1H), 8,21-8,17 (m, 1H), 7,92-7,86 (m, 1H), 7,55 (d, J=8,4 Гц, 1H), 7,33-7,16 (m, 6Н), 6,50 (s, 1H), 5,36-5,27 (m, 1H), 3,17-3,09 (m, 1H), 2,88-2,79 (m, 1H), 2,79-2,70 (m, 1H), 2,24 (s, 3Н), 0,69-0,53 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 418,2.
N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (461)
[0460] Соединение 461 (270 мг, выход 68,77%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующих исходных веществ, соединений 60А и гидрохлорида 3-амино-N-циклопропил-2-гидрокси-4-фенилбутанамида. Соединение 461: 1Н ЯМР (CDCl3, 400 МГц): δ 9,04 (d, J=7,5 Гц, 1H), 8,87 (d, J=4,8 Гц, 1H), 8,82 (s, 1H), 7,85 (d, J=7,3 Гц, 2Н), 7,59-7,44 (m, 3Н), 7,36-7,19 (m, 5Н), 5,37 (ушир. s, 1H), 3,27-3,17 (m, 1H), 2,90-2,73 (m, 2Н), 0,72-0,51 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 404,1.
(S)-N-(4-АМИНО-1-(4-ГИДРОКСИФЕНИЛ)-3,4-ДИОКСОБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (492)
[0461] Соединение 492 (35 мг, выход 60,9%, белое твердое вещество) получали, как в случае соединения 12, из соответствующих исходных веществ, соединений 23А и (3S)-3-амино-4-(4-(трет-бутокси)фенил)-2-гидроксибутанамида, с последующим удалением трет-бутилъной группы с получением конечного соединения 492. Соединение 492: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,29 (s, 1H), 9,01 (d, J=7,5 Гц, 1H), 8,19 (s, 1H), 7,92 (s, 1H), 7,66-7,41 (m, 5Н), 7,07 (d, J=8,4 Гц, 2Н), 6,68 (d, J=8,6 Гц, 2Н), 5,46-5,29 (m, 1H), 3,13 (ушир. d, J=10,8 Гц, 1H), 2,63 (ушир. d, J=2,9 Гц, 1H), 2,13 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 394,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (495)
[0462] Соединение 495 (4,0 г, выход 44,68%, белое твердое вещество) получали, как в случае соединения 12, из соответствующих исходных веществ, соединений 12F и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с получением конечного соединения 495. Соединение 495: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 7,74 (ушир. d, J=9,0 Гц, 1H), 7,31 (d, J=8,5 Гц, 2Н), 7,20-7,08 (m, 5Н), 7,04 (d, J=9,0 Гц, 1H), 6,97 (d, J=8,5 Гц, 2Н), 5,40 (d, J=6,3 Гц, 1H), 4,96 (s, 2Н), 4,79 (d, J=2,3 Гц, 2Н), 4,48-4,15 (m, 2Н), 3,97-3,86 (m, 1H), 3,68 (t, J=8,2 Гц, 1H), 3,63-3,49 (m, 2Н), 2,98 (dd, J=3,4, 13,9 Гц, 1H), 2,70-2,59 (m, 1H), 1,78 (qd, J=6,8, 13,6 Гц, 1H), 0,72 (d, J=6,8 Гц, 3Н), 0,69-0,62 (m, 1H), 0,67 (d, J=6,8 Гц, 2Н). МС (ИЭР) m/z (M+Na)+ 493,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (531)
[0463] Соединение 531 (4,0 г, выход 44,68%, белое твердое вещество) получали, как в случае соединения 12, из соответствующих исходных веществ, соединений 60А и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с получением конечного соединения 531. Соединение 531: 1Н ЯМР (CD3CN, 400 МГц): δ 8,52 (s, 1H), 7,84-7,75 (m, 2Н), 7,57-7,51 (m, 1H), 7,51-7,43 (m, 2Н), 7,32-7,23 (m, 3Н), 7,23-7,17 (m, 2Н), 7,17-7,07 (m, 1H), 7,06-6,93 (m, 1H), 6,23 (s, 1H), 5,55-5,47 (m, 1H), 3,29 (dd, J=4,9, 14,1 Гц, 1H), 2,92 (dd, J=8,9, 14,0 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 364,1.
ПРИМЕР 6
Соединения 21-22, 322, 29, 31, 75, 90, 279
(S)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-4-(4-ФЕНИЛ-1Н-ПИРАЗОЛ-1-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (21)
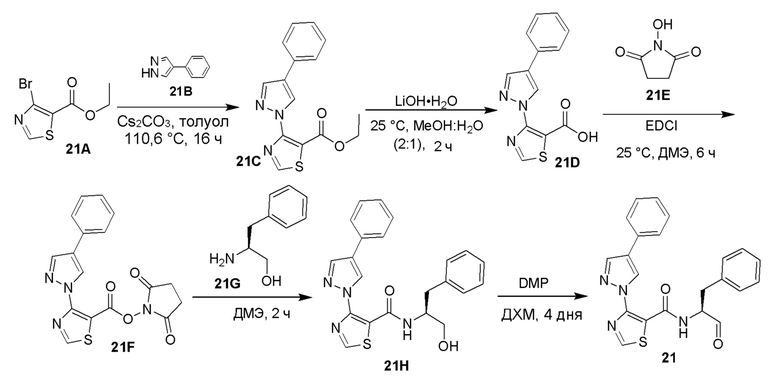
[0464] Смесь, состоящую из соединения 21А (500 мг, 2,12 ммоль), соединения 21 В (306 мг, 2,12 ммоль) и Cs2CO3 (2,07 г, 6,36 ммоль), перемешивали при 110,6°С в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (петролейный эфир : этилацетат = 3:2 и затем уксусная кислота : этилацетат = 1:100) с получением соединения 21С (80 мг, выход 12,61%) в виде светло-желтого твердого вещества и соединения 21D (125 мг, выход 21,73%) в виде желтого твердого вещества.
[0465] Соединение 21С: 1Н ЯМР (CDCl3, 400 МГц): δ 8,85 (s, 1H), 8,49 (s, 1H), 8,07 (s, 1H), 7,57 (d, J=7,6 Гц, 2Н), 7,40 (t, J=7,6 Гц, 2Н), 7,30-7,26 (m, 1H), 4,41-4,32 (m, 2Н), 1,34 (t, J=7,2 Гц, 3Н).
[0466] Соединение 21D: 1H ЯМР (CDCl3, 400 МГц): δ 14,70 (ушир. s, 1H), 9,33 (s, 1H), 8,87 (d, J=0,8 Гц, 1H), 8,41 (d, J=0,8 Гц, 1H), 7,75-7,71 (m, 2Н), 7,43-7,38 (m, 2Н), 7,30-7,24 (m, 1H). МС (ИЭР) m/z (М+Н)+ 271,8.
[0467] К раствору соединения 21С (80 мг, 267,25 мкмоль) в МеОН (5 мл) и H2O (2,5 мл) добавляли LiOH (19,20 мг, 801,75 мкмоль) одной порцией при 25°С в атмосфере N2. Смесь перемешивали при 25°С в течение 2 ч. Смесь концентрировали при пониженном давлении с получением остатка. Остаток разбавляли водой (10 мл) и с помощью 1 н. HCl доводили рН до ~ 3, подвергали экстракции 90 мл этилацетата (30 мл × 3). Объединенные органические слои промывали 30 мл солевого раствора, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 21D (71,1 мг, выход 98,07%) в виде белого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц): δ 8,84 (s, 1H), 8,78 (d, J=0,8 Гц, 1H), 8,13 (d, J=0,8 Гц, 1H), 7,60-7,56 (m, 2Н), 7,45 (t, J=7,6 Гц, 2Н), 7,39-7,34 (m, 1H). МС (ИЭР) m/z (M+H)+ 271,8.
[0468] К раствору соединения 21D (80 мг, 294,89 мкмоль) и 1-гидроксипирролидин-2,5-диона (21Е) (35,6 мг, 309,63 мкмоль) в ДМЭ (3,50 мл) добавляли EDCI (84,8 мг, 442,34 мкмоль) одной порцией при 25°С в атмосфере N2. Полученную смесь перемешивали при 25°С в течение 6 ч. Смесь концентрировали при пониженном давлении, разбавляли EtOAc (100 мл), промывали 1 н. HCl (10 мл) и насыщенным водным NaHCO3 (10 мл × 3) и затем промывали солевым раствором (20 мл). Органическую фазу сушили с помощью безводного Na2SO4, фильтровали и концентрировали под вакуумом с получением промежуточного соединения 21F (100 мг, неочищенного) в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 368,9.
[0469] Смесь, состоящую из соединения 21F (100 мг, 271,47 мкмоль) и соединения 21G (41,1 мг, 271,47 мкмоль) в ДМЭ (3 мл), перемешивали при 25°С в течение 2 ч. Смесь концентрировали под вакуумом, разбавляли этилацетатом (100 мл), промывали 1 н. HCl (10 мл) и насыщенным водным NaHCO3 (10 мл × 3) и затем промывали солевым раствором (20 мл). Органическую фазу сушили с помощью безводного Na2SO4, фильтровали и концентрировали под вакуумом с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (петролейный эфир : этилацетат = 3:2) с получением соединения 21Н (65 мг, выход 59,20%). 1Н ЯМР (CDCl3, 400 МГц): δ 10,54 (d, J=7,6 Гц, 1H), 9,21 (d, J=0,4 Гц, 1H), 8,93 (s, 1H), 8,40 (s, 1H), 7,78 (d, J=7,6 Гц, 2Н), 7,45-7,38 (m, 2Н), 7,31-7,26 (m, 1H), 7,24-7,18 (m, 4Н), 7,15-7,10 (m, 1H), 4,96-4,89 (m, 1H), 4,13-4,02 (m, 1H), 3,52-3,43 (m, 2Н), 2,97-2,91 (m, 1H), 2,82-2,74 (m, 1H). МС (ИЭР) m/z (М+Н)+ 405,0.
[0470] DMP (63 мг, 148,34 мкмоль) добавляли к раствору соединения 21Н (30 мг, 74,17 мкмоль) в дихлорметане (6 мл). Смесь перемешивали при 25°С в течение 12 ч. Добавляли дополнительное количество DMP (63 мг, 148,34 мкмоль), и смесь дополнительно перемешивали в течение 6 ч при 25°С. Добавляли дополнительное количество DMP (157 мг, 0,37 ммоль). После дополнительного перемешивания в течение 39 ч смесь разбавляли дихлорметаном (35 мл), добавляли в нее для гашения реакции 10% Na2S2O3/насыщенный водный NaHCO3 (об./об. = 1/1, ~35 мл). Органический слой отделяли, и водный слой подвергали экстракции ДХМ (20 мл × 2). Объединенный органический слой промывали Н2О (10 мл), солевым раствором (10 мл), сушили над безводным MgSO4, фильтровали и концентрировали. Остаток растирали с i-Pr2O (3 мл). Нерастворимое вещество собирали и сушили под вакуумом. Соединение 21 (20 мг, выход 67%, бледно-желтое твердое вещество): 1Н ЯМР (CDCl3, 400 МГц): δ 11,71 (ушир. d, J=6,0 Гц, 1H), 9,67 (s, 1H), 8,68 (s, 1H), 8,60 (s, 1H), 7,66 (s, 1H), 7,48-7,46 (m, 2Н), 7,36-7,34 (m, 3Н), 7,24-7,22 (m, 2Н), 7,20-7,16 (m, 3Н), 4,86-4,81 (m, 1H), 3,21-3,18 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 403,1.
(S)-4-(1Н-ИНДАЗОЛ-1-ИЛ)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (22)
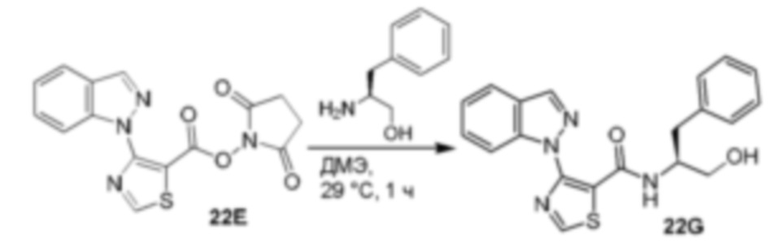
[0471] Соединение 22 (4,70 мг, выход 16,87%, желтое твердое вещество) было получено, как описано в примере 6, из соответствующих исходных веществ через стадии получения промежуточного соединения 22Е и далее соединения 22G. Соединение 22: 1Н ЯМР (CD3CN, 400 МГц): δ 10,41 (ушир. s, 1H), 9,67 (s, 1H), 9,04 (s, 1H), 8,21-8,19 (m, 1H), 8,15 (s, 1H), 7,91-7,89 (m, 1H), 7,61-7,58 (m, 1H), 7,40-7,37 (m, 1H) 7,12-7,10 (m, 5Н) 4,77-4,72 (m, 1H), 3,29-3,24 (m, 1H), 3,11-3,05 (m, 1H). МС (ИЭР) m/z (М+Н)+ 377,0.
(S)-2-МЕТИЛ-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (322)
[0472] Соединение 322 (102,9 мг, выход 36,1%, грязно-белое твердое вещество) было получено, как описано в примере 6, из соответствующих промежуточных соединений 107В и 21G ((S)-2-амино-3-фенилпропан-1-ола). Соединение 322: 1Н ЯМР (400 МГц, CDCl3) δ 9,70 (s, 1H), 8,17-8,09 (m, 2Н), 7,47-7,36 (m, 3Н), 7,35-7,26 (m, 3Н), 7,19 (d, J=6,84 Гц, 2H), 6,82 (d, J=6,00 Гц, 1H), 4,96-4,86 (m, 1H), 3,40-3,28 (m, 1H), 3,26-3,19 (m, 1H), 2,55 (s, 3Н). МС (ИЭР) m/z (M+1)+ 335,1.
(S)-1-(БЕНЗО[D]ТИАЗОЛ-2-ИЛ)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1Н-ИМИДАЗОЛ-5-КАРБОКСАМИД (29)
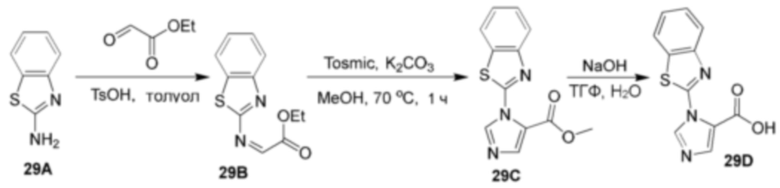
[0473] Смесь соединения 29А (20 г, 133 ммоль), этил-2-оксоацетата (136 г, 665 ммоль), TsOH⋅Н2О (2,5 г, 13,3 ммоль) в толуоле (200 мл) перемешивали при 120°С в течение 1 часа. Анализ посредством ТСХ (петролейный эфир : этилацетат = 3:1, Rf=0,5) свидетельствовал о том, что реагент 29А практически полностью израсходовался, и на хроматограмме появилось одно новое пятно. В анализе посредством ЖХМС был обнаружен один пик, которому соответствовал МС целевого соединения. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = от 20:1 до 5:1) с получением соединения 29В (30,0 г, неочищенного) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 9,40 (s, 1H), 7,98-7,78 (m, 1H), 7,77-7,57 (m, 1H), 7,55-7,31 (m, 1H), 7,30-7,07 (m, 1H), 5,38-5,26 (m, 1H), 4,33-4,21 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 234,9.
[0474] Смесь метального производного 29 В (10 г, 45,4 ммоль), толуолсульфонилметилизоцианида (Tosmic) (17,7 г, 90,8 ммоль), K2CO3 (9,4 г, 68,1 ммоль) в МеОН (200 мл) перемешивали при 70°С в течение 0,5 часа. Анализ посредством ТСХ (петролейный эфир : этилацетат = 3:1, Rf=0,4) свидетельствовал о том, что 29 В полностью израсходовалось, и на хроматограмме появилось несколько новых пятен. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = от 20:1 до 3:1) с получением соединения 29С (1,2 г, выход: 10,2%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,22 (d, J=0,9 Гц, 1H), 8,06 (d, J=7,9 Гц, 1H), 7,93-7,88 (m, 3Н), 7,58 (dt, J=1,3, 7,7 Гц, 1H), 7,52-7,49 (m, 1H), 7,49-7,43 (m, 1H), 4,58 (s, 1H), 3,87 (s, 3Н), 2,51 (s, 1Н). МС (ИЭР) m/z (М+Н)+ 259,9.
[0475] К раствору 29С (1,1 г, 4,24 ммоль) в ТГФ (30 мл), H2O (5 мл) добавляли NaOH (339 мг, 8,48 ммоль). Реакционную смесь перемешивали при 25°С в течение 3 ч. Анализ посредством ЖХМС свидетельствовал о том, что 29С полностью израсходовалось, и был обнаружен один основной пик, которому соответствовал МС целевого соединения. Реакционную смесь концентрировали с получением остатка. Остаток растворяли в воде (10 мл), с помощью водного раствора HCl (2 М) доводили рН до ~ 5, фильтровали, и осадок на фильтре концентрировали с получением продукта 29D (600 мг, выход: 57,7%) в виде серого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,48 (d, J=1,1 Гц, 1H), 8,22-8,18 (m, 1H), 8,06 (dd, J=0,8, 8,0 Гц, 1H), 7,81 (d, J=0,9 Гц, 1H), 7,64-7,53 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 245,9.
[0476] Соединение 29 (55,00 мг, выход: 76,12%, грязно-белое твердое вещество) было получено, как описано в примере 21, из соответствующих промежуточных соединений 29D и 21G. Соединение 29: 1Н ЯМР (400 МГц, CDCl3) δ 9,75 (s, 1H), 9,01 (d, J=6,2 Гц, 1H), 8,14 (s, 1H), 7,90-7,75 (m, 3Н), 7,57-7,42 (m, 2Н), 7,19-7,00 (m, 5Н), 5,04-4,94 (m, 1H), 3,37-3,21 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 377,2.
(S)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-(ПИРИДИН-2-ИЛ)-1Н-ИМИДАЗОЛ-5-КАРБОКСАМИД (31)
[0477] Соединение 31 (25 мг, выход: 57,86%, светло-желтое твердое вещество) было получено, как описано в примере 6, из соответствующих промежуточных соединений 24Е и 21G. Соединение 31: 1H ЯМР (400 МГц, CDCl3) δ 9,70 (s, 1H), 8,45-8,39 (m, 1H), 7,94 (d, J=1,1 Гц, 1H), 7,89-7,82 (m, 1H), 7,67-7,59 (m, 2Н), 7,39-7,32 (m, 2Н), 7,30-7,27 (m, 1H), 7,26-7,21 (m, 2Н), 7,17-7,11 (m, 2Н), 4,87 (q, J=6,6 Гц, 1H), 3,25 (dd, J=2,5, 6,5 Гц, 2Н). МС (ИЭР) m/z (М+Н)+ 321,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(5-ФЕНИЛПИРИМИДИН-2-ИЛ)-1Н-ИМИДАЗОЛ-5-КАРБОКСАМИД (75)
[0478] Соединение 75 (43,1 мг, выход: 66,6%, белое твердое вещество) было получено, как описано в примере 6, из соответствующих промежуточных соединений 74Е и 21G. Соединение 75: 1Н ЯМР (CDCl3, 400 МГц) δ 9,79 (s, 1H), 9,61 (ушир. d, J=6,0 Гц, 1H), 8,75 (s, 2Н), 8,65 (s, 1H), 7,84 (s, 1H), 7,58-7,53 (m, 5Н), 7,25-7,14 (m, 5Н), 5,06-5,01 (m, 1H), 3,43-3,38 (m, 1H), 3,33-3,28 (m, 1H). МС (ИЭР) m/z (М+Н)+ 398,1.
(S)-1-(1Н-БЕНЗО[d]ИМИДАЗОЛ-2-ИЛ)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (90)
[0479] Соединение 90 (20 мг, выход: 44,4%, белое твердое вещество) было получено, как описано в примере 6, из соответствующих промежуточных соединений 70D и 21G ((S)-2-амино-3-фенилпропан-1-ола). Соединение 90: 1Н ЯМР (400 МГц, CDCl3) δ 12,77 (ушир. s, 1H), 12,87-12,63 (m, 1H), 9,75 (s, 1H), 8,85 (s, 1H), 7,71 (ушир. s, 1H), 7,57 (s, 1H), 7,50 (ушир. s, 1H), 7,36-7,27 (m, 4Н), 7,19 (ушир. d, J=6,8 Гц, 2Н), 6,99 (ушир. d, J=5,3 Гц, 1H), 4,93 (q, J=6,7 Гц, 1H), 3,32 (d, J=6,4 Гц, 2Н). МС (ИЭР) m/z (М+Н)+ 360,1
(S)-3-МЕТИЛ-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-(ПИРИМИДИН-4-ИЛ)-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (279)
[0480] Соединение 279 (102,0 мг, 304,15 мкмоль, выход 55,26%, белое твердое вещество) было получено, как описано в примере 6, из соответствующих промежуточных соединений 245D и 21G ((S)-2-амино-3-фенилпропан-1-ола). Соединение 279: 1Н ЯМР (400 МГц, CDCl3): δ 9,88 (d, J=5,6 Гц, 1H), 9,76 (s, 1H), 8,74 (d, J=5,6 Гц, 1H), 8,61 (s, 1H), 7,91-7,87 (m, 1H), 7,27-7,23 (m, 3Н), 7,19-7,17 (m, 2Н), 8,89 (s, 1H), 5,02-4,97 (m, 1H), 3,40-3,35 (m, 1H), 3,30-3,25 (m, 1H), 2,34 (s, 1Н). МС (ИЭР) m/z (M+1)+ 336,1.
ПРИМЕР 7
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(ПИРИДИН-2-ИЛ)-1Н-ИМИДАЗОЛ-5-КАРБОКСАМИД (24)
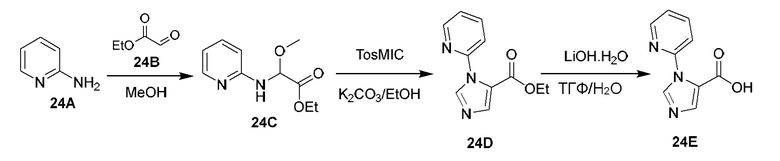
[0481] К раствору соединения 24 В (16,2 г, 79,7 ммоль) в МеОН (25 мл) добавляли соединение 24А (5 г, 53,1 ммоль). Смесь перемешивали при 80°С в течение 6 ч. Анализ посредством ТСХ (петролейный эфир : этилацетат = 2:1, Rf=0,24) свидетельствовал о том, что соединение 24А оставалось, и на хроматограмме было обнаружено одно основное новое пятно с меньшей полярностью. Затем реакционную смесь концентрировали при пониженном давлении с получением остатка. Далее остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 30:1 до 15:1) с получением соединения 24С (6 г, выход: 53,7%) в виде желтого масла.
[0482] К смеси соединения 24С (6 г, 28,5 ммоль) в EtOH (15 мл) добавляли TosMIC (8,3 г, 42,8 ммоль), K2CO3 (11,8 г, 85,6 ммоль). Смесь перемешивали при 80°С в течение 12 ч. Анализ посредством ТСХ (петролейный эфир : этилацетат = 1:1, Rf=0,70) свидетельствовал о том, что соединение 24С оставалось, и на хроматограмме было обнаружено одно основное новое пятно с большей полярностью. Затем реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 30:1 до 3:1) с получением соединения 24D (1,60 г, выход: 25,8%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,61-8,54 (m, 1H), 7,98 (s, 1H), 7,92-7,83 (m, 2Н), 7,45-7,38 (m, 2Н), 4,25 (q, J=7,1 Гц, 2Н), 1,28 (t, J=7,2 Гц, 4Н).
[0483] К раствору соединения 24D (1,6 г, 7,37 ммоль) в ТГФ (15 мл) и Н2О (5 мл) добавляли LiOH⋅H2O (618 мг, 14,7 ммоль). Реакционную смесь перемешивали при 25°С в течение 12 ч. Анализ посредством ЖХМС свидетельствовал о том, что соединение 24D полностью израсходовалось, и был обнаружен один основной пик, которому соответствовал МС целевого соединения. Затем рН смеси доводили до ~ 5 путем добавления HCl (1 М), и затем белое твердое вещество выпадало в осадок. Это белое твердое вещество отфильтровывали и сушили над с получением соединения 24Е (1 г, выход: 71,7%) в виде белого твердого вещества. МС (ИЭР) m/z (М+Н)+ 189,1.
[0484] Соединение 24 (20 мг, выход: 33,5%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 24Е, и соединения 12G. Соединение 24: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,95 (d, J=7,6 Гц, 1H), 8,50-8,38 (m, 1H), 8,12 (s, 1H), 8,03 (ушир. s, 1H), 7,89-7,85 (m, 1H), 7,80 (ушир. s, 1H), 7,53 (s, 1H), 7,43-7,40 (m, 1H), 7,33-7,25 (m, 4Н), 7,24-7,19 (m, 1H), 7,15 (d, J=8,4 Гц, 1H), 5,25-5,20 (m, 1H), 3,17 (dd, J=4,0, 14,4 Гц, 1H), 2,83 (dd, J=10,0, 13,6 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 364,1.
ПРИМЕР 8
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-4-ФЕНИЛТИАЗОЛ-5-КАРБОКСАМИД (25)
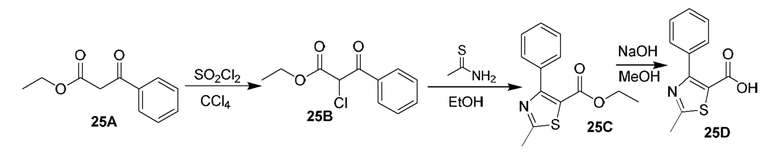
[0485] К раствору соединения 25А (20 г, 104 ммоль) в CCl4 (200 мл) добавляли SO2Cl2 (14 г, 104 ммоль) при 45-50°С в течение 0,3 ч. Затем смесь перемешивали при 45-50°С в течение 1 ч. Реакционную смесь разбавляли ледяной водой (200 мл). Органический слой отделяли, промывали Н2О (200 мл × 2), солевым раствором (200 мл) сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением соединения 25 В (25 г, неочищенного) в виде бледно-желтого масла, которое непосредственно использовали на следующей стадии. 1Н ЯМР (CDCl3, 400 МГц): δ 8,03-7,98 (m, 2Н), 7,67-7,62 (m, 1H), 7,54-7,50 (m, 2Н), 5,62 (s, 1H), 4,32-4,26 (q, J=7,2 Гц, 2Н), 1,25 (t, J=7,2 Гц, 3Н).
[0486] Смесь соединения 25 В (6,8 г, 30 ммоль) и тиоацетамида (2,25 г, 30,0 ммоль) в EtOH (75 мл) нагревали до 80°С и перемешивали в течение 6 ч. Растворитель отгоняли при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: петролейный эфир / этилацетат = 50/1) с получением соединения 25С (3,0 г, выход 40,4%) в виде желтого масла. 1Н ЯМР (CDCl3, 400 МГц): δ 7,77-7,71 (m, 2Н), 7,47-7,40 (m, 3Н), 4,28 (q, J=7,2 Гц, 2Н), 2,77 (s, 3Н), 1,29 (t, J=7,2 Гц, 3Н).
[0487] Раствор NaOH (2 н., 12 мл, 24 ммоль) добавляли к раствору соединения 25С (1,24 г, 5,01 ммоль) в смеси МеОН/Н2О (39 мл/13 мл). Смесь перемешивали при 25°С в течение 3 ч. Смесь разбавляли Н2О (5 мл). Летучий растворитель удаляли путем выпаривания. Остаток обрабатывали HCl (1 н.) до рН ~ 3. Осадок собирали путем фильтрования, сушили при пониженном давлении с получением соединения 25D (650 мг, выход 59,2%) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 7,72-7,65 (m, 2Н), 7,43-7,36 (m, 3Н), 2,68 (s, 3Н).
[0488] Соединение 25 (25 мг, выход 42%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 25D. Соединение 25: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,81 (d, J=7,6 Гц, 1H), 8,12 (s, 1H), 7,86 (s, 1H), 7,55-7,53 (m, 2Н), 7,31-7,19 (m, 8Н), 5,35-5,31 (m, 1H), 3,15 (dd, J=3,6, 13,8 Гц, 1H), 2,77 (dd, J=9,9, 13,8 Гц, 1H), 2,67 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 394,1.
ПРИМЕР 9
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (26)
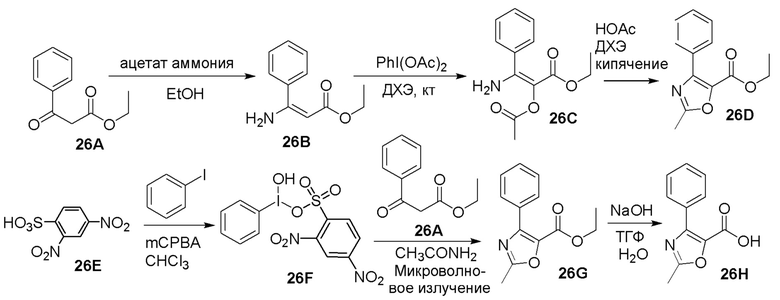
[0489] К смеси соединения 26А (7,2 г, 37,46 ммоль, 6,5 мл) и ацетата аммония (5,8 г, 74,92 ммоль) смешивали в EtOH (70 мл) и кипятили с обратным холодильником при 80°С в течение 16 ч. После удаления растворителя остаток растворяли в воде (50 мл), подвергали экстракции EtOAc (100 мл × 2). Эту объединенную органическую фазу промывали насыщ. NaHCO3 (50 мл × 2) и солевым раствором (50 мл), сушили над Na2SO4, фильтровали, и растворитель удаляли под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир / этилацетат = от 20:1 до 10:1) с получением соединения 26 В (3,5 г, выход: 48,9%) в виде желтого масла. 1Н ЯМР (CDCl3, 400 МГц): δ 7,57-7,53 (m, 2Н), 7,48-7,37 (m, 3Н), 5,07-4,89 (m, 1H), 4,22-4,11 (m, 2Н), 1,33-1,25 (m, 3Н).
[0490] К смеси соединения 26 В (2 г, 10,46 ммоль) в ДХЭ (4 мл) добавляли PhI(ОАс)2 (4,4 г, 13,60 ммоль) одной порцией при 25°С в атмосфере N2. Смесь перемешивали при 25°С в течение 16 ч. В реакционную смесь добавляли для гашения реакции насыщенный водный NaHCO3 (50 мл), и смесь подвергали экстракции ДХМ (50 мл × 3). Органические слои объединяли и сушили над безводным Na2SO4. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир / этилацетат = от 20:1 до 5:1) с получением соединения 26С (400 мг, выход: 15,3%) в виде желтого масла. 1Н ЯМР (CDCl3, 400 МГц): δ 7,58 (dd, J=1,1, 7,7 Гц, 1H), 7,44-7,39 (m, 4Н), 4,28-4,21 (m, 2Н), 1,94 (s, 3Н), 1,66-1,60 (m, 1H), 1,62 (ушир. s, 1H), 1,30-1,26 (m, 3Н).
[0491] К смеси соединения 26С (400 мг, 1,60 ммоль) в ДХЭ (5 мл) и АсОН (10 мл) перемешивали при 90°С в течение 16 ч. Растворитель удаляли. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир / этилацетат = от 10:1 до 5:1) с получением соединения 26D (220 мг, выход: 53,0%) в виде желтого масла. 1Н ЯМР (CDCl3, 400 МГц): δ 8,11-7,89 (m, 2Н), 7,56-7,32 (m, 3Н), 4,40 (t, J=7,3 Гц, 2Н), 2,59 (s, 3Н), 1,39 (q, J=7,1 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 231,8.
[0492] К смеси иодбензола (2,5 г, 12,25 ммоль, 1,4 мл) и соединения 26Е (3,59 г, 13,48 ммоль) в CHCl3 (25 мл) добавляли м-СРВА (2,33 г, 13,48 ммоль). Смесь перемешивали в течение 2 ч при 25°С в атмосфере N2. После завершения реакции в реакционную смесь добавляли МТБЭ (20 мл), и полученную смесь фильтровали и твердое вещество промывали МТБЭ (30 мл). Соединение 26F получали в виде белого твердого вещества.
[0493] Этил-3-оксо-3-фенил-пропаноат 26А (500 мг, 2,60 ммоль) и соединение 26F (1,58 г, 3,38 ммоль) в CH3CN (30 мл) нагревали до кипения с обратным холодильником в течение 1 ч при 80°С, и к смеси добавляли ацетамид (461 мг, 7,80 ммоль). Реакционную смесь нагревали при 120°С в течение 0,1 ч под действием микроволнового излучения. После охлаждения до комнатной температуры суспензию разбавляли насыщенным раствором NaHCO3 (30 мл), подвергали экстракции EtOAc (10 мл × 2), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир / этилацетат = 10/1) и посредством препаративной ТСХ (SiO2, петролейный эфир / этилацетат = 5/1). Соединение 26G (50 мг, выход: 8,32%) получали в виде белого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц): δ 8,06-7,93 (m, 2Н), 7,48-7,39 (m, 3Н), 4,38 (q, J=7,1 Гц, 2H), 2,58 (s, 3Н), 1,37 (t, J=7,1 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 231,8.
[0494] К смеси соединения 26G (60 мг, 259,46 мкмоль) в ТГФ (2 мл) и H2O (2 мл) добавляли NaOH (1 М, 778 мкл) одной порцией при 0°С. Смесь перемешивали при 25°С в течение 16 ч. Смесь подвергали экстракции МТБЭ (2 × 30 мл) и промывали водой (3 × 30 мл). Водные слои подкисляли до рН ~ 4 с помощью 1 н. HCl, затем раствор подвергали экстракции EtOAc (3 × 30 мл). Органические слои сушили над Na2SO4 и концентрировали с получением соединения 5 (50 мг, выход: 86,6%) в виде желтого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (CDCl3, 400 МГц): δ 8,05-8,01 (m, 2Н), 7,48-7,43 (m, 3Н), 2,62 (s, 3Н), МС (ИЭР) m/z (М+Н)+ 203,8.
[0495] Соединение 26 (15 мг, выход: 23,0%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 26Н. Соединение 26: 1Н ЯМР (CDCl3, 400 МГц): δ 8,08 (ушир. d, J=6,6 Гц, 2Н), 7,41 (ушир. d, J=6,8 Гц, 2Н), 7,32-7,24 (m, 4Н), 7,13 (ушир. d, J=6,6 Гц, 2Н), 6,77 (ушир. s, 2Н), 5,76-5,68 (m, 1H), 5,55 (ушир. s, 1H), 3,45 (ушир. dd, J=5,3, 14,3 Гц, 1H), 3,24 (ушир. dd, J=7,3, 14,1 Гц, 1H), 2,56 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 378,1.
ПРИМЕР 10
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛ-1,2,3-ТИАДИАЗОЛ-4-КАРБОКСАМИД (28)
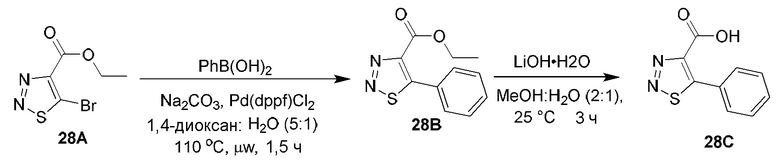
[0496] Смесь, состоящую из соединения 28А (200 мг, 843,63 мкмоль), фенилбороновой кислоты (23 мг, 110,98 мкмоль) и Na2CO3 (22,4 мг, 2,11 ммоль), перемешивали при 110°С в течение 1,5 ч под действием микроволнового излучения. Реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством препаративной ВЭЖХ (в основных условиях) с получением соединения 28В (23 мг, выход 11,64%) в виде светло-желого масла. 1Н ЯМР (CDCl3, 400 МГц): δ 7,44-7,58 (m, 5Н), 4,44 (q, J=7,20 Гц, 2Н), 1,32 (t, J=7,17 Гц, 3Н).
[0497] К смеси соединения 28В (50 мг, 213,43 мкмоль) в МеОН (3 мл) и H2O (1,50 мл) добавляли LiOH-Н2О (26,9 мг, 640,29 мкмоль) одной порцией, и смесь перемешивали при 25°С в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли Н2О (8 мл), доводили рН до ~ 3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (30 мл × 3). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 28С (38 мг, неочищенного) в виде коричневого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц): δ 7,63-7,57 (m, 2Н), 7,54-7,45 (m, 3Н). МС (ИЭР) m/z (М+1)+ 206,7.
[0498] Соединение 28 (18,9 мг, выход 38,00%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 28С. Соединение 28: 1H ЯМР (CDCl3, 400 МГц): δ 8,07 (d, J=7,2 Гц, 1H), 7,59-7,53 (m, 2Н), 7,49-7,41 (m, 3Н), 7,33-7,27 (m, 3Н), 7,23-7,19 (m, 2Н), 6,77 (ушир. s, 1H), 5,84-5,76 (m, 1H), 5,52 (ушир. s, 1H), 3,51-3,45 (m, 1H), 3,25-3,18 (m, 1H). МС (ИЭР) m/z (М+1)+ 381,1.
ПРИМЕР 11
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (30)

[04991 К раствору m-BuONO (3,8 мл, 30,94 ммоль) в CH3CN (60 мл) добавляли CuBr2 (6,91 г, 30,94 ммоль). Смесь перемешивали при 25°С в течение 1 ч в атмосфере N2. Затем добавляли порциями соединение 30А (4 г, 25,78 ммоль). Далее смесь нагревали до 70°С и перемешивали в течение 12 ч. Реакционную смесь промывали Н2О (100 мл), подвергали экстракции EtOAc (100 мл × 2). Органические фазы собирали, сушили с помощью Na2SO4, фильтровали и концентрировали с получением промежуточного соединения 30В (6 г, неочищенного) в виде черно-коричневого масла. МС (ИЭР) m/z (М+Н)+ 218,9, 220,9.
[05001 К раствору NaH (1,64 г, 41,09 ммоль, чистота 60%) в ТГФ (80 мл) при 0°С добавляли раствор соединения 30В (6 г, 27,39 ммоль) в ТГФ (20 мл). После добавления смесь нагревали до 25°С и перемешивали в течение 2 ч. Затем раствор охлаждали до 0°С и добавляли раствор SEM-Cl (5,34 мл, 30,13 ммоль) в ТГФ (100 мл) при 0°С. Далее смесь нагревали до 25°С и перемешивали в течение 12 ч. В реакционную смесь добавляли по каплям Н2О (100 мл) для гашения реакции. Смесь подвергали экстракции EtOAc (100 мл × 2). Органические фазы собирали и концентрировали. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = 10:1) с получением соединения 30С (3 г, выход: 31,14%) в виде желтого масла.
[0501] К раствору соединения 30С (2,60 г, 7,44 ммоль) и PhB(OH)2 (1,09 г, 8,93 ммоль) в диоксане (36 мл) и H2O (12 мл) добавляли Pd(dtbpf)Cl2 (485 мг, 0,74 ммоль) и K3PO4 (4,74 г, 22,32 ммоль). Смесь перемешивали при 70°С в атмосфере N2 в течение 2 ч. Реакционную смесь разбавляли Н2О (20 мл), подвергали экстракции EtOAc (20 мл × 2). Органические фазы собирали и концентрировали. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = 10:1) с получением соединения 30D (2,40 г, выход: 93,1%) в виде бесцветного масла. МС (ИЭР) m/z (М+Н)+ 347,1. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,60 (s, 1H), 7,78-7,72 (m, 2Н), 7,56-7,34 (m, 3Н), 5,52 (s, 2Н), 4,25-4,16 (m, 2Н), 3,70-3,62 (m, 2Н), 1,26 (t, J=7,1 Гц, 3Н), 0,94-0,85 (m, 2Н), 0,03-0,02 (m, 9Н).
[0502] Раствор соединения 30D (200 мг, 577,20 мкмоль) в МеОН (4 мл), и затем добавляли по каплям NaOH (230 мг, 5,77 ммоль) в Н2О (4 мл). Смесь перемешивали при 25°С в течение 19 ч. Реакционную смесь разбавляли путем добавления Н2О (10 мл) и затем подвергали экстракции МТБЭ (10 мл × 2). Водные слои нейтрализовали путем добавления 1 н. HCl до рН ~ 3 и подвергали экстракции EtOAc (10 мл × 3). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 30Е (140 мг, выход: 76,17%) в виде желтого масла. 1Н-ЯМР (400 МГц, ДМСО-d6) δ 12,41 (ушир. s, 1H), 8,54 (s, 1H), 7,81-7,74 (m, 2Н), 7,53-7,36 (m, 3Н), 5,50 (s, 2Н), 3,66 (t, J=8,0 Гц, 2Н), 0,90 (t, J=7,9 Гц, 2Н), 0,01-0,04 (m, 9Н).
[0503] Соединение 30G (140 мг, выход: 93,72%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 30Е. Соединение 30G: 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,54 (d, J=7,3 Гц, 1H), 8,25 (s, 1H), 8,15-8,00 (m, 1H), 7,87 (s, 1H), 7,66-7,52 (m, 2Н), 7,43-7,23 (m, 9Н), 5,46 (ушир. d, J=6,8 Гц, 1H), 5,38-5,30 (m, 1H), 3,76-3,57 (m, 2Н), 3,27-3,12 (m, 1H), 2,96-2,76 (m, 1H), 0,94-0,89 (m, 2Н), 0,03-0,00 (m, 9Н).
[0504] К раствору соединения 30G (18,00 мг, 36,54 мкмоль) в этилацетате (1,00 мл) добавляли 4 М HCl/EtOAc (5,00 мл). Затем реакционную смесь перемешивали при 30°С в течение 4 ч. В реакционную смесь добавляли петролейный эфир (50 мл), смесь перемешивали в течение 3 мин, фильтровали, и целевое твердое вещество сушили под вакуумом с получением соединения 30 (6,00 мг, выход: 45,31%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,34 (d, J=7,5 Гц, 1H), 8,02 (s, 1H), 7,99-7,95 (m, 1H), 7,77 (s, 1H), 7,59-7,53 (m, 2Н), 7,35-7,28 (m, 4Н), 7,28-7,23 (m, 5Н), 7,23-7,16 (m, 2Н), 5,30-5,21 (m, 1H), 3,19-3,10 (m, 1H), 2,83 (dd, J=9,8, 13,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 363,1.
ПРИМЕР 12
СОЕДИНЕНИЯ 32, 458, 476-479, 521
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (32)
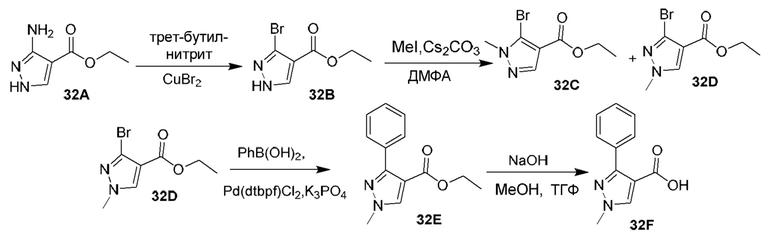
[0505] К раствору т-BuONO (3,19 г, 30,94 ммоль, 3,67 мл) в CH3CN (60 мл) добавляли CuBr2 (6,91 г, 30,94 ммоль). Смесь перемешивали при 25°С в течение 1 часа в атмосфере N2. Затем добавляли порциями соединение 32А (4,00 г, 25,78 ммоль). После нагревания до 70°С и перемешивания в течение 12 ч смесь концентрировали и разбавляли этилацетатом (100 мл). Затем смесь промывали HCl (1 М, 100 мл), насыщенным NaHCO3 (100 мл), солевым раствором (100 мл), сушили над Na2SO4 и концентрировали с получением промежуточного соединения 32 В (5,6 г, неочищенного) в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 220,9.
[0506] К раствору соединения 32В (5,6 г, 25,57 ммоль) в ДМФА (200 мл) добавляли MeI (14,52 г, 102,28 ммоль, 6,37 мл) и Cs2CO3 (33,32 г, 102,28 ммоль). Смесь перемешивали при 25°С в течение 12 ч. Смесь разбавляли Н2О (1000 мл) и подвергали экстракции этилацетатом (500 мл), затем органический слой промывали солевым раствором (500 мл × 3), сушили над Na2SO4 и концентрировали. Остаток (4 г) очищали посредством препаративной ВЭЖХ (в основных условиях). Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир / этилацетат = от 10/1 до 5:1). Соединение 32С (1 г, выход: 16,8%) получали в виде белого твердого вещества. Соединение 32D (2 г, выход: 33,6%) получали в виде белого твердого вещества.
[0507] Соединение 32С: 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,93 (s, 1H), 4,24-4,18 (m, 2Н), 3,85 (s, 3Н), 1,28-1,23 (m, 3Н).
[0508] Соединение 32D: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,33 (s, 1H), 4,22-4,16 (m, 2Н), 3,83 (s, 3Н), 1,26-1,22 (m, 3Н).
[0509] Смесь этильного производного соединения 32D (500,0 мг, 2,15 ммоль), фенилбороновой кислоты (314,6 мг, 2,58 ммоль), Pd(dtbpf)Cl2 (140,1 мг, 215,00 мкмоль), K3PO4 (1,37 г, 6,45 ммоль) в диоксане (30 мл) и Н2О (10 мл) дегазировали и продували N2 3 раза. После перемешивания при 70°С в течение 1 часа в атмосфере N2 смесь концентрировали и разбавляли этилацетатом (30 мл). Затем смесь промывали HCl (1 М, 50 мл), насыщенным водным NaHCO3 (50 мл), солевым раствором (50 мл), сушили над Na2SO4 и концентрировали с получением промежуточного соединения 32Е (480 мг, неочищенного) в виде коричневого масла. МС (ИЭР) m/z (М+Н)+ 230,9.
[0510] К раствору соединения 32Е (380,0 мг, 1,65 ммоль) в МеОН (5 мл) и ТГФ (5 мл) добавляли NaOH (2 М, 16,5 мл). Смесь перемешивали при 60°С в течение 1 часа. Смесь концентрировали и разбавляли Н2О (10 мл), смесь подвергали экстракции этилацетатом (10 мл), в водную фазу добавляли HCl (1 М) до рН ~ 3, затем смесь подвергали экстракции этилацетатом (20 мл), органический слой промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали. Соединение 32F (320 мг, выход: 95,9%) было получено в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,03-11,85 (m, 1H), 8,27 (s, 1H), 7,74-7,68 (m, 2Н), 7,40-7,30 (m, 3Н), 3,87 (s, 3Н).
[0511] Соединение 32 (40,0 мг, выход: 64,7%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 32F. Соединение 32: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,33 (d, J=7,2 Гц, 1H), 8,05 (s, 2Н), 7,81 (ушир. s, 1H), 7,63-7,53 (m, 2Н), 7,39-7,20 (m, 8Н), 5,33-5,26 (m, 1H), 3,93-3,86 (m, 3Н), 3,21-3,13 (m, 1H), 2,88-2,79 (m, 1H). МС (ИЭР) m/z (М+Н)+ 377,1.
N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (458)
[0512] Соединение 458 (270 мг, выход: 67,4%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей карбоновой кислоты, промежуточного соединения 32F, и гидрохлорида 3-амино-N-циклопропил-2-гидрокси-4-фенилбутанамида. Соединение 458: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,80 (d, J=4,4 Гц, 1H), 8,38 (d, J=7,3 Гц, 1H), 8,05 (s, 1H), 7,56 (s, 2Н), 7,36-7,17 (m, 8Н), 5,28 (s, 1H), 3,89 (s, 3Н), 3,16 (d, J=11,2 Гц, 1H), 2,89-2,73 (m, 2Н), 0,71-0,52 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 417,1.
(S)-N-(4-ФТОР-3-OKCO-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (476)
[0513] Соединение 476 (36,8 мг, выход: 34,22%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей карбоновой кислоты, промежуточного соединения 32F, и гидрохлорида (2S,3S)-3-амино-1-фтор-4-фенилбутан-2-ола. Соединение 476: 1Н ЯМР (400 МГц, CDCl3) δ 7,91 (s, 1H), 7,52-7,47 (m, 2Н), 7,46-7,37 (m, 3Н), 7,25-7,19 (m, 3Н), 6,94-6,84 (m, 2Н), 6,06 (d, J=6,4 Гц, 1H), 5,03-4,71 (m, 3Н), 3,93 (s, 3Н), 3,09-3,01 (m, 1H), 2,85-2,76 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 366,1.
(S)-N-(4-ФТОР-3-ОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРАЗИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (477)
[0514] Соединение 477 (110 мг, выход: 90,33%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей карбоновой кислоты, промежуточного соединения 85В, и гидрохлорида (2S,3S)-3-амино-1-фтор-4-фенилбутан-2-ола. Соединение 477: 1Н ЯМР (400 МГц, CDCl3) δ 9,20 (s, 1H), 9,08-9,00 (m, 1H), 8,50-8,48 (m, 1H), 8,11 (s, 1H), 7,27-7,24 (m, 3Н), 7,17-7,12 (m, 2Н), 6,79 (s, 1H), 5,33-5,24 (m, 1H), 5,11-4,79 (m, 2Н), 3,45-3,33 (m, 1H), 3,15-3,11 (m, 1H), 2,38 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 368,1.
(S)-N-(4-ФТОР-3-ОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (478)
[0515] Соединение 478 (82 мг, выход: 54,97%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей карбоновой кислоты, промежуточного соединения 12F, и гидрохлорида (2S,3S)-3-амино-1-фтор-4-фенилбутан-2-ола. Соединение 478: 1H ЯМР (400 МГц, CDCl3) δ 10,70 (d, J=6,40 Гц, 1H), 8,14 (d, J=4,40 Гц, 1H), 7,92-7,83 (m, 2Н), 7,26-7,14 (m, 6Н), 6,88 (s, 1H), 5,24-5,20 (m, 1H), 5,05-4,74 (m, 2Н), 3,29-3,18 (m, 2Н), 2,35 (s, 3Н). МС (ИЭР) m/z (М+1)+ 367,2.
(S)-N-(4-ФТОР-3-ОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (479)
[0516] Соединение 479 (100 мг, выход: 82,06%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей промежуточной карбоновой кислоты, 3-метил-1-фенил-1H-пиразол-5-карбоновой кислоты и гидрохлорида (2S,3S)-3-амино-1-фтор-4-фенилбутан-2-ола. Соединение 479: 1Н ЯМР (400 МГц, CDCl3) δ 7,46-7,28 (m, 8Н), 7,08-7,03 (m, 2Н), 6,51 (s, 1H), 6,28 (ушир. d, J=7,0 Гц, 1H), 5,20-5,13 (m, 1H), 5,03-4,73 (m, 2Н), 3,22-3,15 (m, 1H), 3,02-2,95 (m, 1H), 2,35 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 366,1.
(S)-N-(4-ФТОР-3-ОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ФТОР-4-((ПРОП-2-ИН-1-ИЛОКСИ)МЕТИЛ)ФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (521)
[0517] Соединение 521 (250 мг, выход: 78,40%, белое твердое вещество) получали в условиях сочетания, используемых для соединения 476, из соответствующего промежуточного соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2-фтор-4-(гидроксиметил)фенил)бороновой кислоты с последующим алкилированием с помощью 3-бромпроп-1-ина, и затем полученное промежуточное соединение подвергали гидролизу и сочетанию с гидрохлоридом (2S,3S)-3-амино-1-фтор-4-фенилбутан-2-ола, как в случае соединения 12, с получением соединения 521. Соединение 521: 1Н ЯМР (400 МГц, CDCl3) δ 7,89 (s, 1H), 7,40 (t, J=7,6 Гц, 1H), 7,24-7,13 (m, 5Н), 6,94-6,92 (m, 2Н), 5,97 (d, J=6,4 Гц, 1H), 5,09-5,01 (m, 1H), 4,96-4,83 (m, 1H), 4,83-4,70 (m, 1H), 4,66 (s, 2Н), 4,19 (d, J=2,4 Гц, 2Н), 3,95 (s, 3Н), 3,03 (d, J=6,4 Гц, 1H), 2,95-2,88 (m, 1H), 2,49 (t, J=2,3 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 452,2.
ПРИМЕР 13
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ЭТИЛ-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (33)
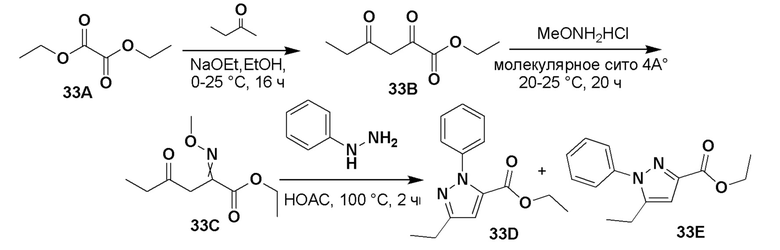
[0518] Смесь соединения 33А (46,73 мл, 342,14 ммоль) и бутан-2-она (30,46 мл, 342,14 ммоль) добавляли по каплям к раствору NaOEt (полученному путем растворения Na (9,5 г) в EtOH (200 мл)) при 0°С. Затем реакционную смесь перемешивали при 20-25°С в течение 16 ч. рН реакционной смеси доводили до ~ 6-7 с помощью HCl (2 М) и затем удаляли растворитель с получением остатка, который разбавляли этилацетатом (500 мл), промывали солевым раствором (150 мл), сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали посредством колоночной флэш-хроматографии (петролейный эфир : этилацетат = от 1:0 до 10:1) с получением соединения 33В (23,0 г, выход: 39,0%) в виде желтого масла. 1Н ЯМР (CDCl3, 400 МГц) δ 14,34 (ушир. s, 1H), 6,31 (s, 1H), 4,28 (q, J=7,2 Гц, 2Н), 2,47 (q, J=7,3 Гц, 2Н), 1,34-1,27 (m, 3Н), 1,11 (t, J=7,5 Гц, 3Н).
[0519] Смесь соединения 33 В (10 г, 58,08 ммоль), О-метилгидроксиламина (4,85 г, 58,08 ммоль, HCl) и молекулярного сита с размером ячейки 4А° (10 г) в ДМФА (100 мл) перемешивали при 20-25°С в течение 20 ч. Смесь фильтровали для удаления молекулярного сита с размером ячейки 4А°, и фильтрат разбавляли Н2О (800 мл), подвергали экстракции этилацетатом (300 мл × 3). Органическую фазу объединяли и промывали солевым раствором (300 мл × 3) и концентрировали с получением неочищенного продукта, который очищали посредством колоночной флэш-хроматографии (петролейный эфир : этилацетат = от 1:0 до 5:1) с получением соединения 33С (3,5 г, выход: 29,95%) в виде желтого масла. 1Н ЯМР (CDCl3, 400 МГц) δ 4,41-4,29 (m, 2H), 4,06 (s, 3Н), 3,71 (s, 2H), 2,60-2,46 (m, 2H), 1,42-1,32 (m, 3Н), 1,08 (t, J=7,3 Гц, 3Н).
[0520] Смесь соединения 33С (3,5 г, 17,39 ммоль) и фенилгидразина (1,88 г, 17,39 ммоль) в АсОН (20 мл) перемешивали при 100°С в течение 2 ч. Растворитель удаляли, и рН остатка доводили до ~ 7-8 с помощью насыщенного водного NaHCO3, и остаток подвергали экстракции этилацетатом (60 мл × 2). Органическую фазу объединяли и промывали солевым раствором (50 мл), концентрировали с получением остатка, который очищали посредством колоночной флэш-хроматографии (петролейный эфир : этилацетат = от 1:0 до 5:1) с получением соединения 33D (0,3 г, 1,23 ммоль, выход 7,05%) в виде желтого твердого вещества и соединения 33Е (3,0 г, 12,21 ммоль, выход 70,19%) в виде желтого масла.
[0521] Соединение 33D: 1H ЯМР (CDCl3, 400 МГц) δ 7,50-7,37 (m, 5Н), 6,87 (s, 1H), 4,24 (q, J=7,0 Гц, 2Н), 2,76 (q, J=7,5 Гц, 2Н), 1,32 (t, J=7,5 Гц, 3Н), 1,25 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 245,0. Соединение 33Е: 1H ЯМР (CDCl3, 400 МГц) δ 7,46-7,31 (m, 1H), 6,70 (s, 1H), 4,35 (q, J=7,1 Гц, 1H), 2,58 (q, J=7,4 Гц, 1H), 1,33 (t, J=7,2 Гц, 1H), 1,16 (t, J=7,5 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 245,0.
[0522] Смесь соединения 33D (3,0 г, 12,28 ммоль) и LiOH⋅H2O (3,09 г, 73,68 ммоль) в МеОН (10 мл) и H2O (3 мл) перемешивали при 25°С в течение 16 ч. рН реакционной смеси доводили до ~ 3-4 с помощью HCl (2 М) и удаляли растворитель. Остаток подвергали экстракции этилацетатом (100 мл × 3) и объединяли, промывали солевым раствором (100 мл), сушили над Na2SO4. Фильтровали, и фильтрат концентрировали с получением соединения 33F (2,7 г, неочищенного) в виде желтого твердого вещества. 1H ЯМР (CDCl3, 400 МГц) δ 8,58 (ушир. s, 1H), 7,46-7,34 (m, 5Н), 6,89 (s, 1H), 2,73 (q, J=7,6 Гц, 2Н), 1,28 (t, J=7,6 Гц, 3Н), МС (ИЭР) m/z (М+Н)+ 216,9.
[0523] Смесь соединения 33F (2,7 г, 12,49 ммоль) и 1-гидроксипирролидин-2,5-диона (1,44 г, 12,49 ммоль) в ТГФ (20 мл) перемешивали при 0°С в течение 15 мин, затем добавляли по каплям раствор DCC (2,6 г, 12,61 ммоль, 2,55 мл) в ТГФ (10 мл) при 0°С и перемешивали при 25-30°С в течение 16 ч. Затем фильтровали, и фильтрат концентрировали с получением соединения 33G (4,0 г, неочищенного) в виде желтого твердого вещества. Продукт непосредственно использовали на следующей стадии.
[0524] Смесь соединения 33G (0,2 г, 638,35 мкмоль), соединения 12G (147,3 мг, 638,35 мкмоль, HCl) и DIEA (0,25 мл, 1,28 ммоль) в ДМФА (10 мл) перемешивали при 20-25°С в течение 16 ч. Реакционную смесь разбавляли Н2О (60 мл) и этилацетатом (30 мл) и перемешивали при 20-25°С в течение 0,5 ч. Белое твердое вещество выпадало в осадок, и его отфильтровывали, осадок на фильтре промывали Н2О (10 мл × 2) и сушили над при пониженном давлении с получением соединения 33Н (100,0 мг, выход: 39,24%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,53-8,11 (m, 1H), 7,40-7,20 (m, 10Н), 7,16-7,01 (m, 2Н), 6,59 (s, 1H), 5,96-5,69 (m, 1H), 4,49-4,36 (m, 1H), 4,03-3,90 (m, 1H), 2,96-2,70 (m, 2Н), 2,62 (q, J=7,7 Гц, 2Н), 1,22 (t, J=7,5 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 393,0.
[0525] Смесь соединения 33Н (100 мг, 254,81 мкмоль) и DMP (540,4 мг, 1,27 ммоль, 394,44 мкл) в ДМСО (5,0 мл) перемешивали при 25-30°С в течение 16 ч. Реакционную смесь разбавляли ДХМ (20 мл) и добавляли в нее для гашения реакции смесь насыщенного водного NaHCO3 и водного (10%) Na2S2O3 (80 мл, 1:1) и перемешивали при 20-25°С в течение 0,5 часа. Белое твердое вещество выпадало в осадок, и его отфильтровывали, осадок на фильтре промывали Н2О (3 мл × 2) и сушили при пониженном давлении с получением соединения 33 (20,0 мг, выход: 20%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,10 (d, J=7,9 Гц, 1H), 8,10 (s, 1H), 7,85 (s, 1H), 7,36-7,20 (m, 8Н), 7,18-7,10 (m, 2Н), 6,58 (s, 1H), 5,30-5,21 (m, 1H), 3,18 (dd, J=3,5, 13,9 Гц, 1H), 2,80 (dd, J=10,6, 13,7 Гц, 1H), 2,60 (q, J=7,7 Гц, 2Н), 1,20 (t, J=7,6 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 391,1.
ПРИМЕР 14
(S)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-(1-ФЕНИЛ-1H-ПИРАЗОЛ-3-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (34)
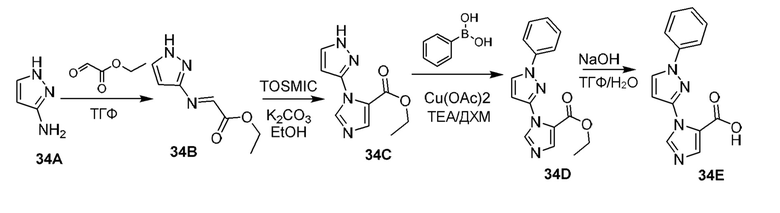
[0526] К раствору 34А (15 г, 180,53 ммоль) в ТГФ (200 мл) добавляли этил-2-оксоацетат (47,9 г, 234,69 ммоль). Смесь перемешивали при 25°С в течение 0,5 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 34 В (55,3 г, неочищенного) в виде коричневого твердого вещества. МС (ИЭР) m/z (М+Н)+ 167,8.
[0527] К раствору 34 В (40 г, 239 ммоль) в EtOH (400 мл) добавляли K2CO3 (50 г, 362 моль) и 1-(изоцианометилсульфонил)-4-метилбензол (40 г, 204,88 ммоль). Смесь перемешивали при 90°С в течение 0,5 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 1:0 до 5:2) с получением соединения 34С (12 г, выход: 24,3%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 11,80-11,35 (m, 1H), 7,87 (d, J=1,10 Гц, 1H), 7,84 (d, J=1,10 Гц, 1H), 7,58 (d, J=2,43 Гц, 1H), 6,45 (d, J=2,43 Гц, 1H), 4,25 (q, J=7,06 Гц, 2Н), 1,29 (t, J=7,17 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 207,0.
[0528] Смесь 34С (5 г, 24,3 ммоль), фенилбороновой кислоты (4,4 г, 36,4 ммоль), Cu(ОАс)2 (4,4 г, 24,3 ммоль), триэтиламина (7,4 г, 72,8 ммоль) в ДХМ (200 мл) дегазировали и продували О2 3 раза, и затем смесь перемешивали при 25°С в течение 10 ч в атмосфере О2. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир:этилацетат = от 1:0 до 2:1). Соединение 34D (2,3 г, выход: 33,6%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,04-7,94 (m, 2Н), 7,87 (s, 1H), 7,71 (ушир. d, J=7,7 Гц, 2Н), 7,49 (ушир. t, J=7,1 Гц, 2Н), 7,36 (ушир. d, J=7,1 Гц, 1H), 7,27 (d, J=2,0 Гц, 2Н), 6,70-6,61 (m, 1H), 4,29 (dd, J=2,1, 7,2 Гц, 2Н), 1,38-1,22 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 282,9.
[0529] К раствору 34D (2,5 г, 8,86 ммоль) в ТГФ (30 мл) и H2O (6 мл) добавляли NaOH (708 мг, 17,7 ммоль). Смесь перемешивали при 80°С в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении для удаления ТГФ и затем промывали EtOAc (20 мл). Водный слой подкисляли с помощью 1 М HCl (до рН ~ 5) и затем подвергали экстракции EtOAc (30 мл × 3). Объединенный органический слой промывали H2O (40 мл), солевым раствором (40 мл), сушили над Na2SO4, фильтровали и концентрировали с получением промежуточного соединения 34Е (1,90 г, выход: 84,31%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,62 (d, J=2,6 Гц, 1H), 8,19 (s, 1H), 7,86 (d, J=7,9 Гц, 2Н), 7,76 (s, 1H), 7,53 (t, J=7,9 Гц, 2Н), 7,39-7,31 (m, 1H), 6,77 (d, J=2,6 Гц, 1H). МС (ИЭР) (М+Н)+ 254,9.
[0530] Соединение 34 (50 мг, выход: 62,8%, белое твердое вещество) было получено, как описано в примере 6, из соответствующих промежуточных соединений 34Е и 21G. Соединение 34: 1H ЯМР (400 МГц, CDCl3) δ 9,66 (s, 1H), 7,95 (d, J=2,4 Гц, 1H), 7,89 (s, 1H), 7,67 (s, 1H), 7,58 (d, J=8,2 Гц, 2Н), 7,45 (t, J=7,8 Гц, 2Н), 7,36-7,31 (m, 1H), 7,26-7,19 (m, 4Н), 7,08 (d, J=6,4 Гц, 2Н), 6,55 (d, J=2,4 Гц, 1H), 4,84 (q, J=6,4 Гц, 1H), 3,21 (d, J=6,4 Гц, 2Н). МС (ИЭР) m/z (М+H2O+Н)+ 404,1.
ПРИМЕР 15
СОЕДИНЕНИЯ 35, 205
(S)-N-(1-AMHHO-5-МЕТИЛ-1,2-ДИОКСОГЕКСАН-3-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (35)
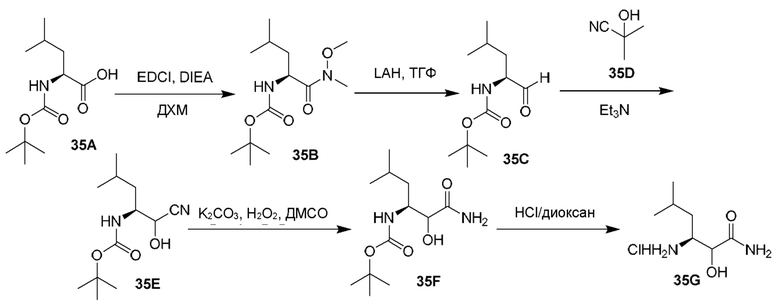
[0531] К раствору соединения 35А (20 г, 86,47 ммоль), N-метоксиметанамина (12,65 г, 129,71 ммоль, HCl), HOBt (11,68 г, 86,47 ммоль) в ДХМ (400 мл) добавляли DIEA (33,53 г, 259,41 ммоль, 45,31 мл) при 0°С. После этого реакционную смесь перемешивали при 0°С в течение 0,1 ч и затем добавляли EDCI (19,89 г, 103,76 ммоль), после добавления реакционную смесь перемешивали при 25°С в течение 16 ч. Реакционную смесь концентрировали с получением остатка, и остаток растворяли в EtOAc (400 мл), промывали 1 н. HCl (400 мл × 2), насыщ. NaHCO3 (400 мл × 2) и солевым раствором (400 мл). Органическую фазу сушили над Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир~петролейный эфир : EtOAc = 10:1). Соединение 35 В (40,32 г, выход: 84,98%) получали в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 5,04 (ушир. d, J=9,0 Гц, 1H), 4,71 (ушир. s, 1H), 3,77 (s, 3Н), 3,18 (s, 3Н), 1,80-1,63 (m, 2Н), 1,42 (s, 10Н), 0,93 (dd, J=6,5, 14,2 Гц, 6Н).
[0532] К смеси LAH (1,53 г, 40,41 ммоль) в ТГФ (200 мл) добавляли по каплям раствор соединения 35В (10,08 г, 36,74 ммоль) в ТГФ (100 мл) при 0°С в атмосфере N2. После добавления реакционную смесь перемешивали при 0°С в течение 2 ч. В реакционную смесь добавляли по каплям EtOAc (150 мл) при 0°С и подкисляли до рН ~ 1~2 с помощью 1 н. HCl, затем добавляли насыщенный водный NaHCO3 (150 мл × 3) и солевой раствор (150 мл). Органический слой сушили над Na2SO4 и концентрировали. Соединение 35С (27,89 г, выход: 88,15%) получали в виде желтого масла, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (400 МГц, CDCl3): δ 9,71-9,32 (m, 1H), 4,99 (ушир. s, 1H), 4,20 (ушир. d, J=2,9 Гц, 1H), 1,79-1,69 (m, 1H), 1,67-1,57 (m, 1H), 1,43-1,40 (m, 10Н), 0,93 (dd, J=1,4, 6,5 Гц, 6Н).
[0533] К раствору соединения 35С (4 г, 18,58 ммоль), соединения 35D (3,16 г, 37,16 ммоль, 3,40 мл) и Ef3N (2,26 г, 22,30 ммоль, 3,09 мл) в сухом ДХМ (40 мл) перемешивали при 25°С в течение 16 ч. Реакционную смесь разбавляли 50 мл ДХМ, промывали 0,5 н. HCl (100 мл), водой (100 мл) и солевым раствором (100 мл). Органическую фазу сушили над Na2SO4, концентрировали. Затем остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : EtOAc = 10:1). Соединение 35Е (3,9 г, выход: 86,63%) получали в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3): δ 4,80 (ушир. s, 1H), 4,58-4,36 (m, 1H), 4,02-3,91 (m, 0,5Н), 3,77 (ушир. s, 0,5Н), 1,75-1,60 (m, 2Н), 1,51-1,33 (m, 10Н), 1,03-0,89 (m, 6Н).
[0534] К раствору соединения 35Е (15 г, 61,90 ммоль) и K2CO3 (17,11 г, 123,80 ммоль) в ДМСО (300 мл) добавляли Н2О2 (70,17 г, 2,15 моль, 60 мл) в атмосфере N2 при 0°С. После добавления реакционную смесь перемешивали при 0°С в течение 1 ч. Затем реакционную смесь разбавляли водой (150 мл) и медленно добавляли в нее для гашения реакции насыщенный водный Na2S2O3 (300 мл) при охлаждении ледяной водой. Смесь подвергали экстракции EtOAc (300 мл × 3), и объединенные экстракты промывали насыщенным водным Na2S2O3 (300 мл × 3). Органический слой сушили над Na2SO4 и концентрировали. Остаток разбавляли EtOAc (20 мл) и МТБЭ (200 мл), твердое вещество собирали и сушили под вакуумом. Соединение 35F (15,15 г, выход: 47,01%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,31-6,96 (m, 2Н), 6,33 (ушир. d, J=9,0 Гц, 0,6Н), 5,95 (d, J=9,5 Гц, 0,4Н), 5,44 (ушир. d, J=5,1 Гц, 1H), 3,93-3,65 (m, 2Н), 1,57-1,47 (m, 1H), 1,41-1,23 (m, 10Н), 0,95-0,70 (m, 7Н).
[0535] К раствору соединения 35F (5,42 г, 20,82 ммоль) в диоксане (10 мл) добавляли HCl/диоксан (4 М, 55 мл) при 25°С. После добавления реакционную смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь концентрировали и добавляли в нее 40 мл МТБЭ, и смесь перемешивали в течение 5 мин. Затем смесь фильтровали с получением целевого соединения. Соединение 35G (3,8 г, выход: 92,80%) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,12 (ушир. s, 1,5Н), 7,87 (ушир. s, 0,5Н), 7,57-7,35 (m, 2Н), 4,22 (d, J=2,5 Гц, 0,7Н), 4,02 (d, J=3,8 Гц, 0,ЗН), 3,57 (s, 1H), 3,45 (ушир. d, J=3,5 Гц, 1H), 1,81-1,58 (m, 1H), 1,54-1,33 (m, 1, 3Н), 1,21 (ddd, J=4,3, 9,5, 14,1 Гц, 0,7Н), 0,93-0,67 (m, 6Н).
[0536] Соединение 35 (48 мг, выход: 44,55%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих промежуточных соединений 23А и 35G. Соединение 35: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,99 (ушир. d, J=7,1 Гц, 1H), 8,13 (s, 1H), 7,89-7,77 (m, 3Н), 7,54-7,48 (m, 3Н), 5,20 (ddd, J=3,3, 7,0, 10,6 Гц, 1H), 2,29 (s, 3Н), 1,74-1,62 (m, 1H), 1,56-1,36 (m, 2Н), 0,92 (d, J=6,4 Гц, 3Н), 0,89-0,84 (m, 3Н).
(S)-N-(1-AMHHO-1,2-ДИОКСО-5-ФЕНИЛПЕНТАН-3-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (205)
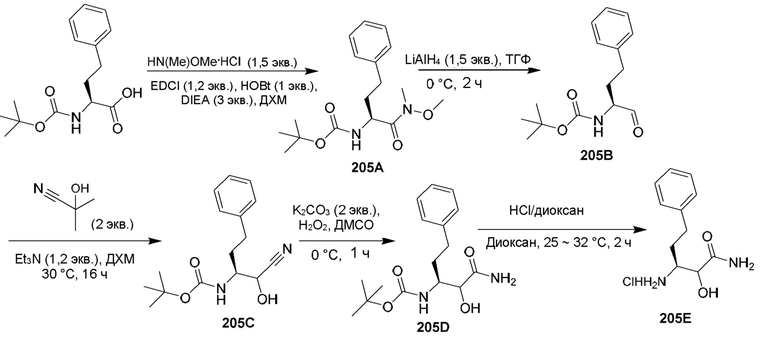
[0537] К смеси (S)-2-((трет-бутоксикарбонил)амино)-4-фенилбутановой кислоты (5 г, 17,90 ммоль) и N-метоксиметанамина (2,76 г, 28,26 ммоль, HCl), HOBt (2,55 г, 18,84 ммоль) в ДХМ (100,00 мл) добавляли по каплям DIEA (9,88 мл, 56,53 ммоль) и EDCI (4,33 г, 22,61 ммоль) порциями при 0°С в атмосфере N2. Смесь перемешивали при 0°С в течение 30 мин, затем смесь перемешивали при 25°С в течение 16 часов. Реакционную смесь разбавляли Н2О (200 мл). Два слоя разделяли, и водную фазу подвергали экстракции этилацетатом (2×150 мл). Объединенные органические слои промывали 0,5 н. HCl (2×150 мл) и NaHCO3 (2×150 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10:1 до 3:1) с получением соединения 205А (4,15 г, выход 68,32%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,35-7,23 (m, 2H), 7,22-7,04 (m, 4H), 4,45-4,21 (m, 1H), 3,59 (s, 3Н), 3,06 (s, 3Н), 2,81-2,68 (m, 1H), 2,61-2,54 (m, 1H), 1,86-1,65 (m, 2H), 1,45-1,29 (s, 9H).
[0538] К раствору LiAlH4 (88,3 мг, 2,32 ммоль) в ТГФ (15 мл) добавляли по каплям раствор соединения 205А (500 мг, 1,55 ммоль) в ТГФ (15 мл) при 0°С в атмосфере N2. После добавления реакционную смесь перемешивали при 0°С в течение 2 часов. Смесь разбавляли этилацетатом (100 мл) и промывали 1 н. HCl (20 мл), насыщенным NaHCO3 (2×20 мл), солевым раствором (15 мл). Органический слой сушили над Na2SO4 и концентрировали с получением соединения 205В (400 мг, 1,52 ммоль) в виде желтого масла. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,4 (s, 1H), 7,33-7,05 (m, 5H), 3,82-3,72 (m, 1H), 2,71-2,51 (m, 2H), 1,97-1,9 (m, 1H), 1,81-1,66 (m, 1H), 1,51-1,25 (m, 10Н).
[0539] Раствор соединения 205В (1,86 г, 7,06 ммоль), 2-гидрокси-2-метилпропаннитрила (1,29 мл, 14,12 ммоль) и Et3N (1,17 мл, 8,47 ммоль) в сухом ДХМ (60 мл) перемешивали при 30°С в течение 16 часов. Реакционную смесь разбавляли ДХМ (50 мл), промывали 0,5 н. HCl (20 мл), водой (20 мл) и солевым раствором (20 мл). Органическую фазу сушили над Na2SO4, концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 3:1) с получением соединения 205С (900 мг, выход 43,90%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,44-7,21 (m, 5H), 6,75-6,58 (m, 1H), 4,70-4,29 (m, 1H), 3,80-3,51 (m, 1H), 2,86-2,68 (m, 1H), 2,62-2,59 (m, 1H), 2,04-1,64 (m, 2H), 1,53-1,43 (m, 9H).
[0540] К раствору соединения 205С (900 мг, 3,1 ммоль) и K2CO3 (856,9 мг, 6,2 ммоль) в ДМСО (18 мл) добавляли H2O2 (3,06 мл, 106,14 ммоль) при 0°С. После добавления реакционную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь разбавляли водой (200 мл) и медленно добавляли в нее для гашения реакции насыщенный водный Na2S2O3 (500 мл) при охлаждении ледяной водой. Смесь подвергали экстракции EtOAc (3×500 мл), и объединенные экстракты промывали насыщенным водным Na2S2O3 (2×300 мл). Органический слой сушили над Na2SO4 и концентрировали. Смесь обрабатывали МТБЭ и затем фильтровали ее с получением соединения 205D (500 мг, выход 52,30%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 7,34-7,11 (m, 1Н), 6,81-6,67 (m, 1H), 5,54-5,35 (m, 1H), 5,19-5,05 (m, 2H), 4,28-4,12 (m, 1H), 3,85-3,72 (s, 1H), 2,81-2,54 (m, 2H), 2,24-1,99 (m, 2H), 2,98-2,78 (m, 1H), 1,65-1,41 (m, 9H).
[0541] К раствору соединения 205D (250 мг, 810,71 мкмоль) в диоксане (2 мл) добавляли HCl/диоксан (4 M, 1,06 мл) при 25°С. После добавления реакционную смесь перемешивали при 32°С в течение 2 часов и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10:1 до 1:2) с получением соединения 205Е (180 мг, выход 90,64%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,15-8,02 (m, 1H), 8,01-7,75 (m, 1H), 7,65-7,48 (m, 2H), 7,37-7,26 (m, 2H), 7,25-7,05 (m, 5H), 6,52-6,24 (s, 1H), 4,16-4,05 (m, 1H), 3,45-3,39 (m, 1H), 1,95-1,61 (m, 2H), 1,41-1,28 (m, 2H).
[0542] Соединение 205 (23,5 мг, выход 31,69%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующих промежуточных соединений 23А и 205Е. Соединение 205: 1H ЯМР (400 МГц, CDCl3) δ 7,84-7,72 (m, 2H), 7,60-7,50 (m, 3Н), 7,26-7,12 (m, 4H), 7,07-7,00 (m, 2H), 6,66 (ушир. s, 1H), 6,19 (ушир. s, 1H), 5,51-5,34 (m, 2H), 2,66-2,54 (m, 2H), 2,47 (s, 3Н), 2,38-2,25 (m, 1H), 1,99-1,85 (m, 1H). MC (ИЭР) m/z (M+H)+ 392,1.
ПРИМЕР 16
СОЕДИНЕНИЯ 36, 49, 409, 455
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИМИДИН-2-ИЛ)-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (36)
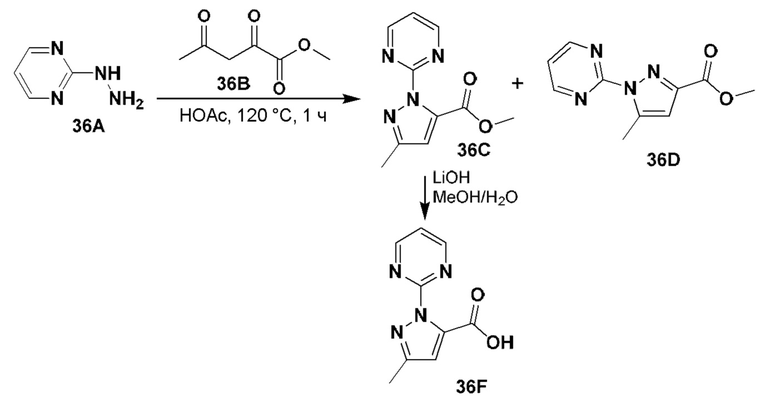
[0543] К раствору соединения 36В (1,31 г, 9,08 ммоль) в АсОН (50 мл) добавляли соединение 36А (1 г, 9,08 ммоль). Смесь перемешивали при 120°С в течение 1 ч. Смесь выливали в ДХМ (50 мл). Органический слой промывали водой (10 мл), NaHCO3 до рН ~ 8~9 и сушили над Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10:1 до 5:1) с получением соединений 36С и 36D. Соединение 36С (500 мг, 2,29 ммоль, выход 25,24%, белое твердое вещество): 1H ЯМР (400 МГц, ДМСО-d6) δ 9,03-8,79 (m, 2H), 7,67-7,45 (m, 1Н), 6,87 (s, 1H), 3,73 (s, 3H), 2,29 (s, 3Н). Соединение 36D (1 г, выход 50,47%, белое твердое вещество): 1H ЯМР (400 МГц, ДМСО-d6) δ 9,03-8,89 (m, J=4,9 Гц, 2H), 7,67-7,55 (m, 1H), 6,81 (s, 1H), 3,84 (s, 3Н), 2,60 (s, 3H).
[0544] Промежуточное соединение 36F (39,6 мг, выход 90%, белое твердое вещество) было получено, как описано в примере 85, из соединения 36С. МС (ИЭР) m/z (М+1)+ 205. Соединение 36 (15,5 мг, выход 43,77%, коричневое твердое вещество) было получено, как описано в примере 5, из соответствующего промежуточного соединения 36F. Соединение 36: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,99-8,95 (m, 2H), 8,50-8,39 (m, 1H), 8,11 (s, 1H), 7,85 (s, 1H), 7,65-7,57 (m, 1H), 7,31-7,17 (m, 5H), 6,68 (s, 1H), 5,51-5,45 (m, 1H), 3,26-3,18 (m, 1H), 3,13-3,03 (m, 1H), 2,58 (s, 3Н).
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИМИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (49)
[0545] Следуя методике получения соединения 36, соединение 49 (21 мг, выход 38,4%, белое твердое вещество) получали из соответствующего промежуточного соединения 36D. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,08-8,98 (m, 1H), 8,76-8,70 (m, 2H), 8,05 (s, 1H), 7,82 (s, 1H), 7,49-7,44 (m, 1H), 7,35-7,26 (m, 4H), 7,26-7,19 (m, 1H), 6,58 (s, 1H), 5,31-5,25 (m, 1H), 3,19-3,09 (m, 1H), 2,90-2,78 (m, 1H), 2,27 (s, 3H). МС (ИЭР) m/z (M+Na)+ 379.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИМИДИН-4-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (409)
[0546] Следуя методике получения соединения 36, соединение 409 (4225,7 мг, выход 80,2%, белое твердое вещество) получали из соответствующих исходных веществ, а именно 4-гидразинилпиримидина и промежуточного соединения 274D. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,22 (d, J=7,2 Гц, 1H), 8,84 (d, J=5,6 Гц, 1H), 8,75 (s, 1H), 8,14 (s, 1H), 7,89 (s, 1H), 7,78-7,74 (m, 1H), 7,31-7,21 (m, 5H), 6,52 (s, 1H), 5,41-5,33 (m, 1H), 3,22-3,12 (m, 1H), 2,90-2,78 (m, 1H), 2,28 (s, 3H). МС (ИЭР) m/z (M+1)+ 379,0.
N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИМИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (455)
[0547] Следуя методике получения соединения 36, соединение 455 (180 мг, выход 71,6%, белое твердое вещество) получали из соответствующих исходных веществ, а именно 36F и 3-амино-N-циклопропил-2-гидрокси-4-фенилбутанамида. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,05-9,00 (m, 1H), 9,03 (d, J=7,3 Гц, 1H), 8,78 (d, J=5,1 Гц, 1H), 8,69 (d, J=4,9 Гц, 2H), 7,44 (t, J=4,9 Гц, 1H), 7,29-7,18 (m, 5H), 6,56 (s, 1H), 5,31-5,24 (m, 1H), 3,12 (dd, J=3,7, 13,9 Гц, 1H), 2,84-2,71 (m, 2H), 2,24 (s, 2H), 2,27-2,19 (m, 1H), 0,67-0,54 (m, 4H). МС (ИЭР) m/z (M+H)+ 419,2.
ПРИМЕР 17
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(2H-ИНДАЗОЛ-2-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (37)
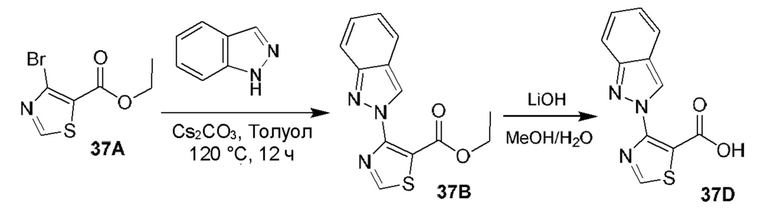
[0548] Смесь соединения 37А (250 мг, 2,12 ммоль), Cs2CO3 (2,07 г, 6,36 ммоль) в толуоле (40 мл) перемешивали при 110°С в течение 13 ч. Смесь концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10/1 до 5:1) с получением соединения 37В (43,75 мг, выход 7,55%) в виде белого твердого вещества. МС (ИЭР) m/z (М+1)+ 274. 1H ЯМР (400 МГц, CDCl3) δ 8,93 (s, 1Н), 8,68 (s, 1H), 7,85-7,65 (m, 2H), 7,39-7,30 (m, 1H), 7,17-7,05 (m, 1H), 4,44-4,24 (m, 2H), 1,28 (m, 3H).
[0549] Смесь соединения 37В (35 мг, 128,06 мкмоль), LiOH (9,2 мг, 384,18 мкмоль) в воде (1 мл) и МеОН (5 мл) перемешивали при 27°С в течение 2 ч. МеОН выпаривали. К остатку добавляли воду (10 мл). Смесь подвергали экстракции МТБЭ (5 мл) и разделяли. Водный слой подкисляли до рН ~ 3 с помощью 1 н. HCl и подвергали экстракции этилацетатом (3×20 мл). Объединенные органические слои сушили и концентрировали с получением соединения 37D (25,3 мг, выход 80,55%) в виде коричневого твердого вещества.
[0550] Соединение 37 (4 мг, 8,47 мкмоль, выход 5,95%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 37D. Соединение 37: 1H ЯМР (400 МГц, CDCl3) δ 12,72-12,49 (m, 1H), 8,97 (s, 1H), 8,80 (s, 1H), 7,80-7,64 (m, 1H), 7,46-7,29 (m, 2H), 7,20-6,98 (m, 6H), 6,83-6,72 (m, 1H), 5,97-5,84 (m, 1H), 5,52-5,40 (m, 1H), 3,56-3,33 (m, 2H).
ПРИМЕР 18
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(НАФТАЛИН-1-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (38)
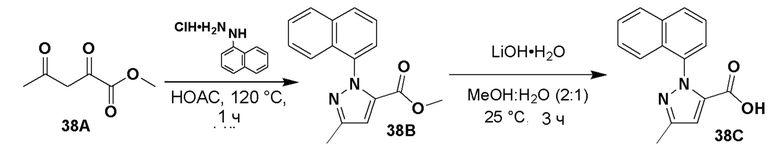
[0551] Смесь, состоящую из соединения гидрохлорида нафталин-1-илгидразина (4,05 г, 20,81 ммоль) и соединения 38А (3,0 г, 20,81 ммоль) в АсОН (30 мл), перемешивали при 120°С в течение 1 часа. Реакционную смесь охлаждали до 25°С, концентрировали при пониженном давлении и разбавляли CH2Cl2 (100 мл). Органическую фазу промывали насыщ. NaHCO3 (20 мл × 2), сушили над безводным Na2SO4, фильтровали, и фильтрат концентрировали при пониженном давлении с получением остатка, который очищали посредством флэш-хроматографии на силикагеле (петролейный эфир : этилацетат = 4:1) с получением соединения 38В (154,3 мг, выход 2,79%) в виде коричневого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 7,96 (d, J=8,0 Гц, 1Н), 7,91 (d, J=8,4 Гц, 1Н), 7,57-7,52 (m, 1Н), 7,51-7,47 (m, 2H), 7,46-7,41 (m, 1Н), 7,22 (d, J=8,4 Гц, 1Н), 6,90 (s, 1Н), 3,62 (s, 3Н), 2,43 (s, 3Н). МС (ИЭР) m/z (M+1)+ 267,1.
[0552] К смеси соединения 38 В (160 мг, 570,78 мкмоль) в МеОН (10 мл) и H2O (5 мл) добавляли LiOH⋅H2O (71,9 мг, 1,71 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли H2O (10 мл), доводили рН до ~ 3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (40 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 38С (140 мг, выход 97,23%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 7,96-7,83 (m, 2H), 7,52-7,44 (m, 2H), 7,44-7,36 (m, 2H), 7,15 (d, J=8,4 Гц, 1Н), 6,87 (s, 1Н), 2,37 (s, 3Н). МС (ИЭР) m/z (M+1)+ 252,9.
[0553] Соединение 38 (10,6 мг, выход 13,31%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 38С. Соединение 38: 1H ЯМР (400 МГц, CDCl3) δ 8,00-7,89 (m, 2H), 7,57-7,43 (m, 4H), 7,28 (d, J=8,4 Гц, 1Н), 7,22-7,10 (m, 3Н), 6,82-6,72 (m, 2H), 6,69 (s, 1Н), 6,54 (ушир. s, 1Н), 6,16 (d, J=6,8 Гц, 1Н), 5,48-5,33 (m, 2H), 3,19-3,09 (m, 1Н), 2,94-2,84 (m, 1Н), 2,40 (s, 3Н). МС (ИЭР) m/z (M+1)+ 427,2.
ПРИМЕР 19
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(1H-ИНДАЗОЛ-1-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (40)
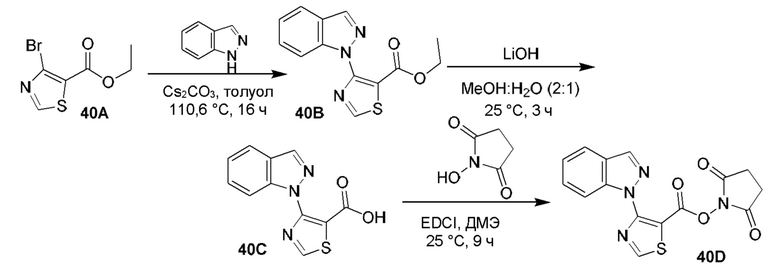
[0554] Смесь, состоящую из соединения 40А (500 мг, 2,12 ммоль), индазола (250,5 мг, 2,12 ммоль), Cs2CO3 (2,07 г, 6,36 ммоль) в толуоле (40 мл), перемешивали при 110,6°С в течение 16 ч. Реакционную смесь охлаждали до 25°С, фильтровали, концентрировали при пониженном давлении. Полученный остаток очищали посредством препаративной ВЭЖХ (в условиях HCl) с получением соединения 40В (56 мг, выход 9,67%) в виде светло-желтого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,94 (s, 1H), 8,26 (s, 1Н), 7,80 (d, J=8,0 Гц, 1Н), 7,62 (d, J=8,4 Гц, 1Н), 7,48-7,43 (m, 1Н), 7,28-7,24 (m, 1Н), 4,24 (q, J=7,2 Гц, 2H), 1,17-1,12 (m, 1Н), 1,14 (t, J=7,2 Гц, 2H).
[0555] К смеси соединения 40В (50 мг, 182,94 мкмоль) в МеОН (2 мл) добавляли LiOH (13,1 мг, 548,83 мкмоль) одной порцией, и смесь перемешивали при 25°С в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли H2O (8 мл), доводили рН до ~ 3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (30 мл × 3). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 40С (42 мг, выход 93,61%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,95 (s, 1Н), 8,73 (d, J=8,4 Гц, 1Н), 8,38 (s, 1Н), 7,89 (d, J=8,0 Гц, 1Н), 7,67 (t, J=7,6 Гц, 1Н), 7,43(t, J=7,6 Гц, 1Н).
[0556] К раствору, состоящему из соединения 40С (42 мг, 171,25 мкмоль) и 1-гидроксипирролидин-2,5-диона (20,7 мг, 179,81 мкмоль) в ДМЭ (5 мл), добавляли EDCI (49,24 мг, 256,87 мкмоль) одной порцией при 25°С в атмосфере N2. Смесь перемешивали при 25°С в течение 9 ч. Реакционную смесь концентрировали при пониженном давлении для удаления ДМЭ. Остаток разбавляли EtOAc (60 мл), промывали 1 н. HCl (10 мл) и насыщенным водным NaHCO3 (10 мл × 3). Органические слои промывали солевым раствором (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 40D (60 мг, неочищенного) в виде светло-желого масла. МС (ИЭР) m/z (М+1)+ 342,8.
[0557] Соединение 40 (15,10 мг, выход 50,57%, белое твердое вещество) было получено, как описано в примере 6, из соответствующих исходных веществ, соединения 40D и 12G. Соединение 40: 1H ЯМР (CDCl3, 400 МГц): δ 11,07 (d, J=6,0 Гц, 1Н), 8,85 (s, 1H), 8,33-8,28 (m, 1Н), 7,86 (d, J=0,8 Гц, 1Н), 7,77 (d, J=8,0 Гц, 1Н), 7,57-7,52 (m, 1Н), 7,35-7,31 (m, 1Н), 7,16-7,08 (m, 5H), 6,77 (ушир. s, 1Н), 5,81-5,75 (m, 1Н), 5,55 (ушир. s, 1Н), 3,43-3,37 (m, 1Н), 3,26-3,19 (m, 1Н). МС (ИЭР) m/z (М+1)+ 420,1.
ПРИМЕР 20
СОЕДИНЕНИЯ 41-43, 64-65, 67, 71, 76, 87, 100, 116, 132, 134-135, 137, 203-204
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(ПИРИДИН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (41)

[0558] К смеси соединения 24Е (100 мг, 528 мкмоль) и соединения 41В (148 мг, 634 мкмоль) в ДМФА (1,5 мл) добавляли HBTU (240 мг, 634 мкмоль) одной порцией при 25°С и перемешивали в течение 5 мин, и затем добавляли DIEA (273 мг, 2,1 ммоль). Смесь перемешивали при 25°С в течение 30 мин. Анализ посредством ЖХМС свидетельствовал о том, что соединение 24Е оставалось, и был обнаружен продукт, которому соответствовал МС целевого соединения. Затем остаток очищали посредством препаративной ВЭЖХ (в условиях ТФУ) с получением соединения 41А (130 мг, выход: 60,6%) в виде белого твердого вещества. 1H ЯМР (400 МГц, Метанол-d4) δ 9,12 (ушир. s, 1Н), 8,49 (ушир. d, J=3,8 Гц, 1H), 7,95-7,74 (m, 2H), 7,50 (ушир. s, 1H), 7,37-7,16 (m, 5H), 7,13-7,01 (m, 1H), 4,69-4,52 (m, 1H), 4,22-4,03 (m, 1H), 3,29 (ушир. s, 1H), 3,11-2,74 (m, 2H), 2,69-2,51 (m, 1H), 0,77-0,59 (m, 2H), 0,56-0,38 (m, 2H). MC (ИЭР) m/z (M+H)+ 405,2.
[0559] К раствору соединения 41А (130 мг, 320 мкмоль) в ДХМ (10 мл) добавляли DMP (543 мг, 1,3 ммоль, 397 мкл) одной порцией при 0°С. Смесь перемешивали при 25°С в течение 10 мин. Анализ посредством ЖХМС свидетельствовал о том, что соединение 41А полностью израсходовалось, и был обнаружен один основной пик, которому соответствовал MC целевого соединения. Затем смесь разбавляли ДХМ (80 мл), добавляли в нее для гашения реакции 10% Na2S2O3/насыщенный водный NaHCO3 (об./об. = 1/1, 20 мл). Органический слой отделяли, и водный слой подвергали экстракции ДХМ (30 мл × 2). Объединенный органический слой промывали H2O (10 мл), солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали с получением белого твердого вещества. Затем остаток очищали посредством рекристаллизации из простого изопропилового эфира (20 мл) с получением соединения 41 (20,6 мг, выход: 30,9%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,96 (d, J=7,8 Гц, 1H), 8,79 (ушир. d, J=5,0 Гц, 1H), 8,45 (ушир. d, J=4,6 Гц, 1H), 8,14 (s, 1H), 7,88 (t, J=7,8 Гц, 1H), 7,57 (s, 1H), 7,44 (dd, J=7,0, 5,2 Гц, 1H), 7,34-7,28 (m, 4H), 7,24 (ушир. d, J=4,2 Гц, 1H), 7,18 (d, J=8,2 Гц, 1H), 5,32-5,22 (m, 1H), 3,31 (s, 1H), 3,19 (dd, J=13,8, 3,6 Гц, 1H), 2,89-2,71 (m, 2H), 0,70-0,64 (m, 2H), 0,60-0,55 (m, 2H). MC (ИЭР) m/z (M+H)+ 403,2.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(1-ФЕНИЛ-1H-ПИРАЗОЛ-3-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (42)
[0560] Соединение 42 (19,3 мг, выход: 55,2%, желтое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 34Е. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,91 (d, J=7,7 Гц, 1H), 8,82 (d, J=5,1 Гц, 1H), 8,57 (d, J=2,6 Гц, 1H), 8,14 (s, 1H), 7,83 (d, J=7,9 Гц, 2H), 7,63 (s, 1H), 7,53 (t, J=8,0 Гц, 2H), 7,36 (t, J=7,4 Гц, 1H), 7,32 (d, J=4,4 Гц, 4H), 7,27-7,20 (m, 1H), 6,48 (d, J=2,6 Гц, 1H), 5,34-5,26 (m, 1H), 3,21 (dd, J=13,8, 3,6 Гц, 1H), 2,86 (dd, J=13,8, 10,3 Гц, 1H), 2,81-2,72 (m, 1H), 0,71-0,63 (m, 2H), 0,62-0,53 (m, 2H). MC (ИЭР) m/z (М+H)+ 469,1.
(S)-1-(БЕНЗО[D]ТИАЗОЛ-2-ИЛ)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (43)
[0561] Соединение 43 (24,4 мг, выход: 43%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 29D. Соединение 43: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,13 (d, J=7,7 Гц, 1Н), 8,77 (ушир. d, J=4,9 Гц, 1H), 8,39 (s, 1H), 8,08 (d, J=7,9 Гц, 1H), 7,96 (d, J=8,2 Гц, 1H), 7,67 (s, 1H), 7,59-7,43 (m, 2H), 7,27 (d, J=4,0 Гц, 4Н), 7,22-7,12 (m, 1H), 5,34-5,16 (m, 1H), 3,18 (dd, J=13,9, 3,3 Гц, 1H), 2,83 (dd, J=13,7, 10,1 Гц, 1H), 2,72 (ушир. d, J=4,2 Гц, 1H), 0,69-0,58 (m, 2H), 0,54 (ушир. d, J=2,9 Гц, 2H). МС (ИЭР) m/z (M+H)+ 460,1.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИМИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (65)
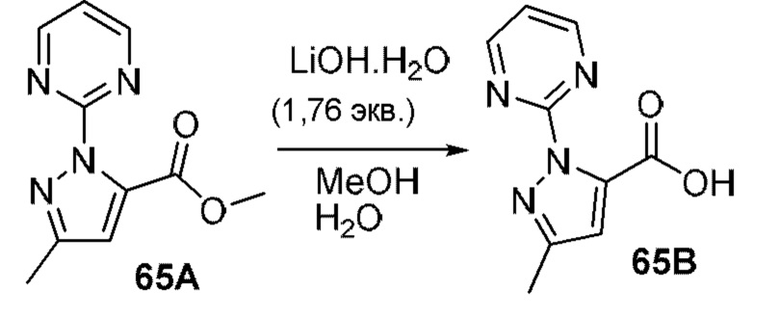
[0562] К смеси соединения 65А (80 мг, 366,62 мкмоль) в МеОН (5 мл) и H2O (1 мл) добавляли LiOH⋅H2O (27,1 мг, 645,87 мкмоль). Смесь перемешивали при 31°С в течение 1 ч. Смесь упаривали для удаления МеОН, затем ее промывали водой (3×50 мл) и подвергали экстракции МТБЭ (2×50 мл). Водные слои подкисляли до рН ~ 4 с помощью 1 н. HCl, затем раствор подвергали экстракции этилацетатом (3×100 мл). Органические слои сушили над Na2SO4 и концентрировали с получением соединения 65В (50 мг, выход 66,79%), которое было получено в виде белого твердого вещества.
[0563] Соединение 65 (11,8 мг, выход 87,93%, белое твердое вещество) было получено, как описано в примере 20, из соответствующих промежуточных соединений 65В и 41В. Соединение 65: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,94 (s, 2H), 8,87-8,79 (m, 1H), 8,51-8,44 (m, 1H), 7,62-7,55 (m, 1H), 7,34-7,09 (m, 5H), 6,65 (s, 1H), 5,53-5,39 (m, 1H), 3,26-3,12 (m, 4Н), 3,10-3,00 (m, 1H), 2,81-2,71 (m, 1H), 2,56 (s, 3H), 0,71-0,54 (m, 4Н). МС (ИЭР) m/z (M+H)+ 419,2.
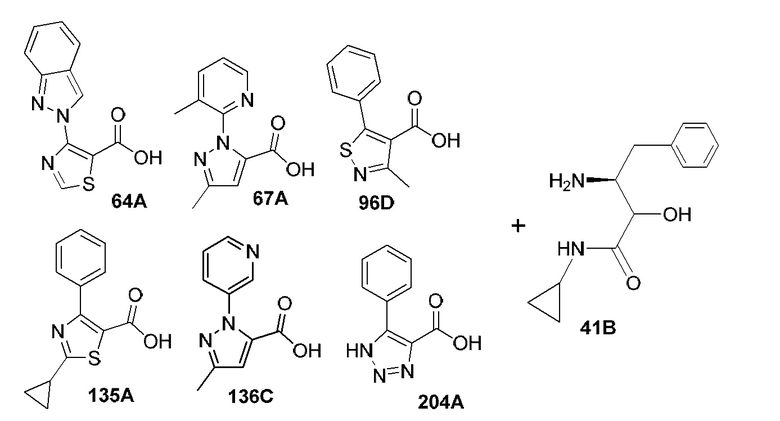
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(2Н-ИНДАЗОЛ-2-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (64)
[0564] Соединение 64 (48,2 мг выход 87,93%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 64А. Соединение 64: 1H ЯМР (400 МГц, ДМСО-d6) δ 11,99-11,83 (m, 1Н), 9,33 (s, 1H), 9,16 (s, 1H), 8,95-8,84 (m, 1H), 7,93-7,80 (m, 1H), 7,56-7,50 (m, 1H), 7,44-7,33 (m, 1H), 7,27-7,15 (m, 1H), 7,12-6,99 (m, 5H), 5,71-5,60 (m, 1H), 3,34-3,24 (m, 3H), 3,19-3,10 (m, 1H), 2,84-2,74 (m, 1H), 0,73-0,54 (m, 4H). MC (ИЭР) m/z (M+H)+ 460,1.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(3-МЕТИЛПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (67)
[0565] Соединение 67 (30,8 мг, выход 38,6%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 67А. Соединение 67: 1H ЯМР (CDCl3, 400 МГц) δ 8,24 (d, J=4,4 Гц, 1H), 7,67 (d, J=7,6 Гц, 1H), 7,2 (d, J=7,2 Гц, 1H), 7,34-7,26 (m, 2H), 7,25-7,20 (m, 3H), 6,99 (d, J=4,4 Гц, 2H), 6,86 (s, 1H), 6,56 (s, 1H), 5,66-5,58 (m, 1H), 3,38-3,29 (m, 1H), 3,21-3,13 (m, 1H), 2,82-2,74 (m, 1H), 2,34 (s, 3H), 2,16 (s, 3H), 0,91-0,84 (m, 2H), 0,64-0,57 (m, 2H). MC (ИЭР) m/z (М+H)+ 432,1.
(S)-1-(1H-БЕНЗО[d]ИМИДАЗОЛ-2-ИЛ)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (71)
[0566] Соединение 71 (75 мг, выход: 78,1%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 70D. Соединение 71: 1H ЯМР (400 МГц, ДМСО-d6) δ 12,93 (ушир. s, 1H), 9,25 (ушир. s, 1H), 8,74 (d, J=4,9 Гц, 1H), 8,29 (s, 1H), 7,83 (s, 1H), 7,52 (ушир. s, 2H), 7,30-7,18 (m, 6H), 7,18-7,13 (m, 1H), 5,42-5,25 (m, 1H), 3,17 (dd, J=3,5, 13,7 Гц, 1H), 2,83 (dd, J=10,0, 13,8 Гц, 1H), 2,74-2,64 (m, 1H), 0,70-0,42 (m, 4H). МС (ИЭР) m/z (M+H)+ 443,0.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(5-ФЕНИЛПИРИМИДИН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (76)
[0567] Соединение 76 (24,7 мг, выход: 44,7%, белое твердое вещество) было получено, как описано в примере 20, из соответствующего промежуточного соединения 74Е. Соединение 76: 1H ЯМР (CDCl3, 400 МГц) δ 9,46 (ушир. d, J=6,4 Гц, 1H), 8,66 (s, 2H), 8,62 (s, 1H), 7,80 (s, 1H), 7,57-7,49 (m, 5H), 7,22-7,16 (m, 2H), 7,16-7,07 (m, 3Н), 6,93 (ушир. s, 1H), 5,86-5,82 (m, 1H), 3,53-3,46 (m, 1H), 3,41-3,32 (m, 1H), 2,86-2,82 (m, 1H), 0,92-0,86 (m, 2H), 0,64-0,62 (m, 2H). МС (ИЭР) m/z (М+Н)+ 481,0.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(4-ФЕНИЛ-1H-ПИРАЗОЛ-1-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (87)
[0568] Соединение 87 (60,0 мг, выход 75,3%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 21D. 1H ЯМР (400 МГц, CDCl3) δ 11,72 (ушир. d, J=6,0 Гц, 1H), 8,73 (s, 1H), 8,68-8,64 (m, 1H), 7,76-7,72 (m, 1H), 7,58-7,52 (m, 2H), 7,48-7,40 (m, 2H), 7,37-7,30 (m, 1H), 7,29-7,21 (m, 5H), 6,97-6,91 (m, 1H), 5,86-5,74 (m, 1H), 3,53-3,41 (m, 1H), 3,29-3,17 (m, 1H), 2,88-2,75 (m, 1H), 0,93-0,82 (m, 2H), 0,68-0,58 (m, 2H). МС (ИЭР) m/z (М+1)+ 486,1.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (100)
[0569] Соединение 100 (85 мг, выход: 83,27%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 23А. Соединение 100: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,09 (d, J=7,5 Гц, 1H), 8,94 (ушир. d, J=5,1 Гц, 1H), 7,62 (d, J=7,1 Гц, 2H), 7,53-7,46 (m, 1H), 7,44-7,39 (m, 2H), 7,32-7,20 (m, 5H), 5,48 (ddd, J=3,3, 7,6, 10,7 Гц, 1H), 3,25 (ушир. dd, J=3,2, 14,0 Гц, 1H), 2,85-2,67 (m, 2H), 2,07 (s, 3Н), 0,73-0,56 (m, 4H). МС (ИЭР) m/z (M+H)+ 418,1.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОТИАЗОЛ-4-КАРБОКСАМИД (116)
[0570] Соединение 116 (88,00 мг, выход 87,41%, грязно-белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 96D. Соединение 116: 1H ЯМР (400 МГц, CDCl3) δ 7,44 (s, 5Н), 7,20-7,09 (m, 3Н), 6,86 (ушир. s, 1Н), 6,77-6,68 (m, 2H), 5,93 (ушир. d, J=6,6 Гц, 1Н), 5,68-5,57 (m, 1Н), 3,24-3,14 (m, 1Н), 2,99-2,89 (m, 1Н), 2,83-2,73 (m, 1Н), 2,46 (s, 3Н), 0,93-0,81 (m, 2H), 0,69-0,53 (m, 2H). MC (ИЭР) m/z (M+H)+ 434,1.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИДИН-3-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (132)
[0571] Соединение 132 (72,8 мг, выход 60,40%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 136С. Соединение 132: 1H ЯМР (400 МГц, CDCl3): δ 8,61 (d, J=2,4 Гц, 1Н), 8,57-8,54 (m, 1Н), 7,72-7,66 (m, 1Н), 7,34-7,26 (m, 4H), 7,10-7,05 (m, 2H), 7,03-6,94 (m, 1Н), 6,64-6,56 (m, 1Н), 6,44 (s, 1Н), 5,62-5,54 (m, 1Н), 3,44-3,36 (m, 1Н), 3,18-3,10 (m, 1Н), 2,85-2,76 (m, 1Н), 2,33 (s, 3Н), 0,92-0,85 (m, 2H), 0,66-0,59 (m, 2H). MC (ИЭР) m/z (M+1)+ 418,1.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(ИЗОХИНОЛИН-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (134)
[0572] Соединение 134 (57,4 мг, выход 62,9%, желтое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 133D. Соединение 134: 1H ЯМР (400 МГц, CDCl3) δ 9,31 (ушир. s, 1Н), 8,47 (ушир. s, 1Н), 8,06 (d, J=7,6 Гц, 1Н), 7,74-7,61 (m, 2H), 7,43 (d, J=8,0 Гц, 1Н), 7,25-7,20 (m, 3Н), 6,92 (ушир. s, 2H), 6,84 (ушир. s, 1Н), 6,60 (s, 1Н), 6,46 (d, J=7,2 Гц, 1Н), 5,50-5,41 (m, 1Н), 3,30-3,22 (m, 1Н), 3,14-3,04 (m, 1Н), 2,79-2,70 (m, 1Н), 2,40 (s, 3Н), 0,87-0,82 (m, 2H), 0,63-0,53 (m, 2H). MC (ИЭР) m/z (M+H)+ 468,1.
(S)-2-ЦИКЛОПРОПИЛ-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФЕНИЛТИАЗОЛ-5-КАРБОКСАМИД (135)
[0573] Соединение 135 (52,8 мг, выход 53,03%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 135А. Соединение 135: 1H ЯМР (CDCl3, 400 МГц) δ 1H ЯМР (400 МГц, CDCl3) δ 7,53-7,45 (m, 2H), 7,45-7,35 (m, 3H), 7,22-7,11 (m, 3H), 6,85 (ушир. s, 1Н), 6,80-6,70 (m, 2H), 6,17 (d, J=6,4 Гц, 1H), 5,54-5,45 (m, 1H), 3,27-3,22 (m, 1H), 2,89-2,84 (m, 1H),2,80-2,75 (m, 1H), 2,33-2,26 (m, 1H), 1,20-1,14 (m, 2H), 1,13-1,08 (m, 2H), 0,91-0,79 (m, 2H), 0,64-0,54 (m, 2H). MC (ИЭР) m/z (M+H)+ 460,1.
(S)-3-(трет-БУТИЛ)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (137)
[0574] Соединение 137 (96,70 мг, выход 64,74%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 128А. Соединение 137: 1H ЯМР (400 МГц, CDCl3) δ 7,54-7,45 (m, 3H), 7,41-7,36 (m, 2H), 7,31-7,27 (m, 1H), 7,25-7,13 (m, 5H), 6,87 (ушир. s, 1H), 6,69 (s, 1H), 5,77-5,68 (m, 1H), 3,44-3,36 (m, 1H), 3,17-3,09 (m, 1H), 2,82-2,74 (m, 1H), 1,16 (s, 9H), 0,89-0,81 (m, 2H), 0,64-0,54 (m, 2H). MC (ИЭР) m/z (M+H)+ 459,2.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-ФЕНИЛТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (203)
[0575] Соединение 203 (30 мг, выход 60,18%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 82D. Соединение 203: 1H ЯМР (CDCl3, 400 МГц) δ 7,69-7,64 (m, 2H), 7,41-7,33 (m, 3H), 7,22 (s, 1H), 7,12-7,06 (m, 3H), 7,01-6,94 (m, 3H), 6,84 (ушир. s, 1H), 5,65-5,58 (m, 1H), 3,41-3,34 (m, 1H), 2,97-2,89 (m, 1H), 2,79-2,71 (m, 1H), 2,32 (s, 3H), 0,86-0,80 (m, 2H), 0,61-0,53 (m, 2H). MC (ИЭР) m/z (М+H)+ 500,1.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛ-1H-1,2,3-ТРИАЗОЛ-4-КАРБОКСАМИД (204)
[0576] Соединение 204 (4 мг, выход 8,9%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 204А. Соединение 204: 1H ЯМР (CDCl3, 400 МГц) Т=80: δ 8,55 (ушир. s, 1H), 8,41 (d, J=7,2 Гц, 1H), 7,82-7,78 (m, 2H), 7,50-7,35 (m, 4H), 7,32-7,20 (m, 5H), 5,51-5,45 (m, 1H), 3,30-3,22 (m, 1H), 3,05 (ушир. s, 1H), 2,81-2,74 (m, 1H), 0,71-0,66 (m, 2H), 0,64-0,59 (m, 2H). MC (ИЭР) m/z (M+H)+ 404,1.
ПРИМЕР 21
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (45)
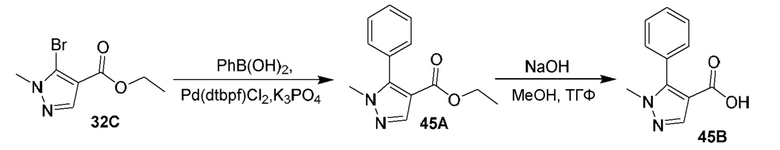
[0577] Смесь этильного производного соединения 32С (500,0 мг, 2,15 ммоль), фенилбороновой кислоты (262,1 мг, 2,15 ммоль), Pd(dtbpf)Cl2 (140,1 мг, 215,00 мкмоль), K3PO4 (1,37 г, 6,45 ммоль) в диоксане (30 мл) и H2O (10 мл) дегазировали и продували N2 3 раза, и затем смесь перемешивали при 70°С в течение 1 часа в атмосфере N2. Смесь концентрировали и разбавляли этилацетатом (30 мл), промывали HCl (1 М, 50 мл), насыщенным водным NaHCO3 (50 мл), солевым раствором (50 мл), сушили над Na2SO4 и концентрировали с получением промежуточного соединения 45А (490 мг, неочищенного) в виде коричневого масла. МС (ИЭР) m/z (М+H)+ 230,9.
[0578] К раствору соединения 45А (490,0 мг, 2,13 ммоль) в МеОН (5 мл) и ТГФ (5 мл) добавляли NaOH (2 М, 21,28 мл). Смесь перемешивали при 60°С в течение 1 часа. Смесь концентрировали и разбавляли H2O (10 мл), смесь подвергали экстракции этилацетатом (10 мл), в водную фазу добавляли HCl (1 М) до рН ~ 3, затем смесь подвергали экстракции этилацетатом (20 мл), органический слой промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали. Соединение 45В (400 мг, выход: 93,0%) было получено в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 12,09 (ушир. s, 1Н), 7,87 (s, 1H), 7,50-7,40 (m, 5H), 3,63 (s, 3H).
[0579] Соединение 45 (50,0 мг, выход: 71,4%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 45 В. Соединение 45: 1H ЯМР (400 МГц, CDCl3) δ 7,97 (s, 1H), 7,55-7,43 (m, 3H), 7,32-7,27 (m, 2H), 7,23-7,15 (m, 3H), 6,85-6,65 (m, 3H), 5,80-5,71 (m, 1H), 5,55-5,40 (m, 2H), 3,71-3,60 (m, 3H), 3,29-3,19 (m, 1H), 2,94-2,84 (m, 1H), 2,94-2,84 (m, 1H). МС (ИЭР) m/z (M+H)+ 377,1.
ПРИМЕР 22
(S)-N-(4-АМИНО-1-(4-МЕТОКСИФЕНИЛ)-3,4-ДИОКСОБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (46)
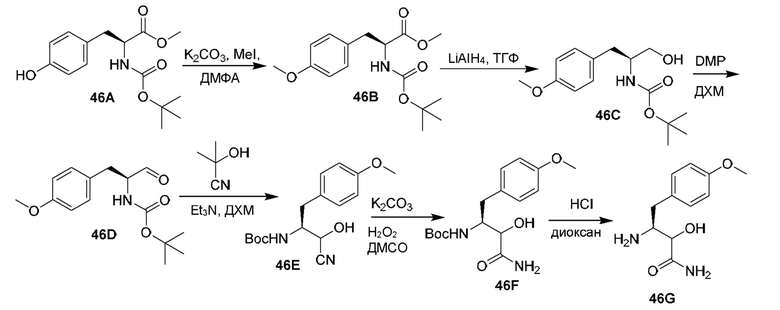
[0580] К раствору соединения 46А (13 г, 44,02 ммоль, 1 экв.) в ДМФА (150 мл) добавляли K2CO3 (12,17 г, 88,04 ммоль, 2 экв.) при 0°С. После добавления смесь перемешивали при этой температуре в течение 0,2 ч и затем добавляли CH3I (8,97 г, 63,20 ммоль, 3,93 мл) по каплям при 0°С. Полученную смесь перемешивали при 25°С в течение 18,8 часов. Реакционную смесь разбавляли EtOAc (50 мл). Объединенные органические слои промывали солевым раствором (100 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 46В (13,4 г, выход: 98,4%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,04 (d, J=8,6 Гц, 2Н), 6,84 (d, J=7,7 Гц, 2H). 4,96 (ушир. d, J=7,3 Гц, 1Н), 4,55 (ушир. d, J=7,1 Гц, 1H), 3,79 (s, 3Н), 3,72 (s, 3Н), 3,09-2,94 (m, 2H), 1,43 (s, 9H).
[0581] К раствору LAH (490 мг, 12,92 ммоль, 2 экв.) в ТГФ (10 мл), который дегазировали и продували N2 3 раза при 0°С, добавляли по каплям смесь соединения 46В (2 г, 6,46 ммоль, 1 экв.) в ТГФ (30 мл), и затем смесь перемешивали при 0°С в течение 2 ч в атмосфере N2. В реакционную смесь добавляли для гашения реакции Н2О (0,5 мл), затем добавляли NaOH (15% в H2O, 0,5 мл), H2O (1,5 мл), и затем смесь разбавляли EtOAc (20 мл), сушили над Na2SO4 и перемешивали в течение 30 мин, затем фильтровали с получением органических слоев. Объединенные органические слои промывали солевым раствором (20 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 46С (1,48 г, выход: 81,4%), которое было получено в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,12 (d, J=8,4 Гц, 2Н), 6,89-6,78 (m, 2H), 4,69 (ушир. s, 1H), 3,88-3,80 (m, 1H), 3,79 (s, 3H), 3,69-3,48 (m, 2H), 2,77 (d, J=7,1 Гц, 2H), 1,41 (s, 9H).
[0582] Раствор DMP (1,51 г, 3,56 ммоль) в ДХМ (10 мл) дегазировали и продували N2 3 раза и затем добавляли по каплям соединение 46С (500 мг, 1,78 ммоль) в ДХМ (10 мл), и затем смесь перемешивали при 25°С в течение 20 ч в атмосфере N2. В реакционную смесь добавляли для гашения реакции насыщенный водный Na2S2O3 (15 мл) и насыщенный водный NaHCO3 (15 мл), и затем смесь разбавляли ДХМ (10 мл) и подвергали экстракции H2O (20 мл × 3). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 46D (430 мг, выход: 86,48%), которое было получено в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,62 (s, 1H), 7,13-7,02 (m, 2H), 6,84 (ушир. d, J=8,6 Гц, 2H), 5,05 (ушир. d, J=5,5 Гц, 1H), 4,46-4,32 (m, 1H), 3,78 (s, 3H), 3,06 (ушир. d, J=6,4 Гц, 2H), 1,43 (s, 9H).
[0583] К раствору соединения 46D (1,53 г, 5,48 ммоль) в ДХМ (20 мл) добавляли соединение 2-гидрокси-2-метилпропаннитрил (3,30 г, 38,78 ммоль, 3,55 мл) и Et3N (832 мг, 8,22 ммоль, 1,14 мл). Смесь перемешивали при 25°С в течение 2 ч. В реакционную смесь добавляли для гашения реакции 1 н. HCl (20 мл), и затем смесь разбавляли H2O (20 мл) и подвергали экстракции ДХМ (20 мл × 2). Объединенные органические слои промывали солевым раствором (20 мл × 3), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10/1 до 4:1) с получением соединения 46Е (980 мг, выход: 58,37%), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,16-6,97 (m, 2H), 6,90-6,71 (m, 2H), 4,96-4,72 (m, 1H), 4,52-4,37 (m, 1H), 3,74-3,72 (m, 3H), 3,07-2,66 (m, 2H), 1,37 (s, 9H).
[0584] К раствору соединения 46Е (980 мг, 3,20 ммоль) и K2CO3 (885 мг, 6,40 ммоль) в ДМСО (15 мл) добавляли H2O2 (9,3 мл, чистота: 30%). Смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь разбавляли H2O (100 мл) и подвергали экстракции EtOAc (30 мл × 3). Объединенные органические слои промывали солевым раствором (50 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 46F (560 мг, выход: 53,95%), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,16-6,97 (m, 2H), 6,90-6,71 (m, 2Н), 4,96-4,72 (m, 1H), 4,52-4,37 (m, 1H), 3,74-3,72 (m, 3H), 3,07-2,66 (m, 2H), 1,37 (s, 9H).
[0585] К раствору соединения 46F (500 мг, 1,54 ммоль) в EtOAc (5 мл) добавляли HCl/EtOAc (4 M, 5 мл). Смесь перемешивали при 25°С в течение 1 ч. Реакционную смесь разбавляли МТБЭ (20 мл) и фильтровали с получением соединения 46G (300 мг, выход: 73,97%, HCl), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,06-7,81 (m, 3H), 7,51 (ушир. s, 2H), 7,26-7,07 (m, 2H), 6,95-6,79 (m, 2H), 6,65-6,35 (m, 1H), 4,21-3,78 (m, 1H), 3,71 (d, J=1,5 Гц, 3H), 3,53 (ушир. s, 1H), 2,87-2,62 (m, 2H).
[0586] Соединение 46 (65 мг, выход: 65,3%, белое твердое вещество) было получено, как описано в примере 15, из соответствующих промежуточных соединений, 23А и 46G. Соединение 46: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,05-8,64 (m, 1H), 8,18 (s, 1H), 7,90 (s, 1H), 7,67-7,55 (m, 2H), 7,53-7,32 (m, 3H), 7,24-7,10 (m, 2H), 6,89-6,76 (m, 2H), 5,48-5,36 (m, 1H), 3,74-3,65 (m, 3H), 3,23-2,95 (m, 1H), 2,76-2,58 (m, 1H), 2,17-2,00 (m, 3H). MC (ИЭР) m/z (M+H)+ 408,1.
ПРИМЕР 23
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФЕНИЛ-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (48)
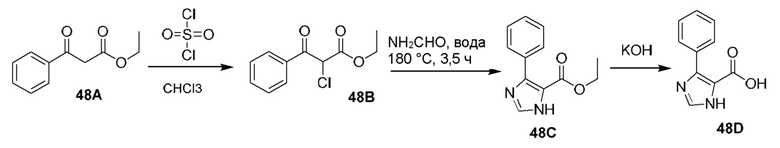
[0587] К раствору соединения 48А (40 г) в CHCl3 (200 мл), охлажденному до 0°С, добавляли по каплям сульфурилдихлорид (34 г). Смесь нагревали до 30°С в течение 0,5 ч и нагревали при 70°С в течение 5 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли хлороформом (40 мл), последовательно промывали водным NaHCO3 (40 мл × 2), водой (20 мл) и затем солевым раствором (30 мл). Органическую фазу сушили над Na2SO4 и упаривали с получением соединения 48В (47 г, неочищенного), которое было получено в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,06-7,87 (m, 2H), 7,67-7,56 (m, 1H), 7,52-7,42 (m, 1H), 7,48-7,39 (m, 1H), 7,48-7,39 (m, 1H), 7,67-7,38 (m, 1H), 7,26 (s, 1H), 5,61 (s, 1H), 5,29-5,26 (m, 1H), 4,39-4,21 (m, 2H), 1,70 (s, 1H), 1,40-1,14 (m, 3H).
[0588] Раствор соединения 48В (20 г) в NH2CHO (40 г, 882,40 ммоль, 35 мл) и воде (3,2 г, 176,48 ммоль) нагревали при 180°С в течение 3,5 ч. Смесь оставляли для охлаждения до комнатной температуры, затем добавляли воду (50 мл), и смесь подвергали экстракции ДХМ (100 мл × 3). Объединенные органические слои промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (элюент 0 ~ 100% этилацетат/петролейный эфир, градиент со скоростью потока 40 мл/мин) с получением соединения 48С (1,3 г) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,09 (d, J=7,3 Гц, 7Н), 4,28-4,08 (m, 2H), 1,24 (ушир. t, J=6,8 Гц, 1H), 1,29-1,10 (m, 1H).
[0589] К раствору этильного производного соединения 48С (800 мг, 3,70 ммоль) в EtOH (20 мл) добавляли раствор KOH (2,1 г, 37,00 ммоль) в H2O (20 мл) при 0°С. После добавления реакционную смесь перемешивали при 70°С в течение 16 ч. В реакционную смесь добавляли 20 мл воды, и смесь подвергали экстракции МТБЭ (20 мл). Водный слой подкисляли с помощью 1 н. HCl до рН ~ 4 и фильтровали с получением целевого соединения. Фильтрат подвергали экстракции EtOAc (50 мл × 3). Объединенные экстракты промывали солевым раствором (50 мл) и сушили над Na2SO4, смесь концентрировали под вакуумом с получением целевого соединения 48D (500 мг, выход 71,81%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,42-12,33 (m, 1H), 7,97-7,67 (m, 3H), 7,48-7,21 (m, 3H).
[0590] Соединение 48 (10 мг, выход 25,1%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 48D. Соединение 48: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,30-8,17 (m, 1H), 8,00-7,53 (m, 5H), 7,46-7,13 (m, 8H), 5,50-5,30 (m, 1H), 4,31-4,05 (m, 1H), 3,32-3,21 (m, 1H), 2,71-2,61 (m, 1H). МС (ИЭР) m/z (M+H)+ 363,2.
ПРИМЕР 24
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(БЕНЗО[d]ТИАЗОЛ-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (50)
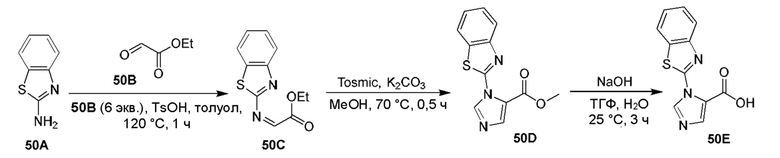
[0591] Смесь соединения 50А (20 г, 133 ммоль), соединения 50В (136 г, 665 ммоль), TsOH⋅H2O (2,5 г, 13,3 ммоль) в толуоле (200 мл) перемешивали при 120°С в течение 1 часа. Анализ посредством ТСХ (петролейный эфир : этилацетат = 3:1, Rf ~ 0,5) свидетельствовал о том, что 50А практически полностью израсходовалось, и на хроматограмме появилось одно новое пятно. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = от 20:1 до 5:1) с получением соединения 50С (30 г, неочищенного) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 9,40 (s, 1H), 7,98-7,78 (m, 1H), 7,77-7,57 (m, 1H), 7,55-7,31 (m, 1H), 7,30-7,07 (m, 1H), 5,38-5,26 (m, 1H), 4,33-4,21 (m, 3H). МС (ИЭР) m/z (M+H)+ 234,9.
[0592] Смесь метильного производного 50С (10 г, 45,4 ммоль), TosMIC (17,7 г, 90,8 ммоль), K2CO3 (9,4 г, 68,1 ммоль) в МеОН (200 мл) перемешивали при 70°С в течение 0,5 часа. Анализ посредством ТСХ (петролейный эфир : этилацетат = 3:1, Rf=0,4) свидетельствовал о том, что 50С полностью израсходовалось, и на хроматограмме появилось несколько новых пятен. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = от 20:1 до 3:1) с получением соединения 50D (1,2 г, выход: 10,2%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,22 (d, J=0,9 Гц, 1H), 8,06 (d, J=7,9 Гц, 1H), 7,93-7,88 (m, 3H), 7,58 (dt, J=1,3, 7,7 Гц, 1H), 7,52-7,49 (m, 1H), 7,49-7,43 (m, 1H), 4,58 (s, 1H), 3,87 (s, 3H), 2,51 (s, 1H). МС (ИЭР) m/z (М+Н)+ 259,9.
[0593] К раствору 50D (1,1 г, 4,24 ммоль в ТГФ (30 мл), H2O (5 мл) добавляли NaOH (339 мг, 8,48 ммоль). Реакционную смесь перемешивали при 25°С в течение 3 ч. Анализ посредством ЖХМС свидетельствовал о том, что 50D полностью израсходовалось, и был обнаружен один основной пик, которому соответствовал МС целевого соединения. Реакционную смесь концентрировали с получением остатка. Остаток растворяли в воде (10 мл), доводили рН до ~ 5 путем добавления водного раствора HCl, фильтровали, и осадок на фильтре концентрировали с получением продукта 50Е (0,6 г, выход: 57,7%) в виде серого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,48 (d, J=1,1 Гц, 1Н), 8,22-8,18 (m, 1H), 8,06 (dd, J=0,8, 8,0 Гц, 1Н), 7,81 (d, J=0,9 Гц, 1H), 7,64-7,53 (m, 2H). МС (ИЭР) m/z (М+Н)+ 245,9.
[0594] Соединение 50 (12,9 мг, выход: 18,8%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 50Е. Соединение 50: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,11 (ушир. d, J=7,7 Гц, 1H) 8,39 (s, 1H) 8,12-8,03 (m, 2H) 7,96 (d, J=8,2 Гц, 1H) 7,81 (s, 1H) 7,66 (s, 1H) 7,57-7,45 (m, 2H) 7,26 (d, J=4,2 Гц, 4Н) 7,20-7,16 (m, 1H) 5,33-5,20 (m, 1H) 3,18 (ушир. dd, J=13,9, 3,5 Гц, 1H) 2,90-2,76 (m, 1H). МС (ИЭР) m/z (M+H)+ 420,0.
ПРИМЕР 25
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(1H-ИНДАЗОЛ-3-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (51)
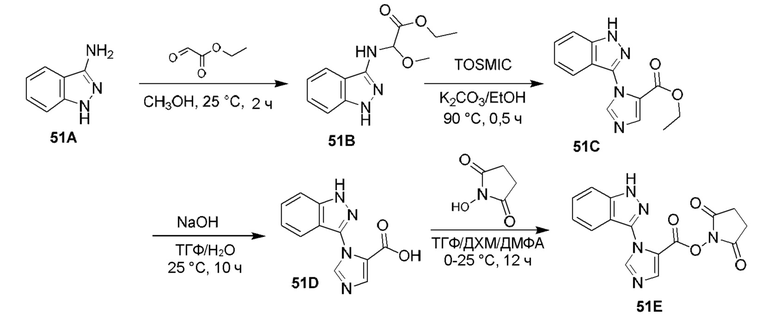
[0595] К раствору 51А (8,7 г, 65,3 ммоль) в МеОН (90 мл) добавляли этил-2-оксоацетат (20 г, 98,01 ммоль). После перемешивания при 25°С в течение 2 часов смесь фильтровали и концентрировали с получением неочищенного продукта 51В (15 г, неочищенного) в виде коричневого твердого вещества, который использовали на следующей стадии без очистки.
[0596] К раствору 51В (15 г, 69,1 ммоль) в EtOH (400 мл) добавляли K2CO3 (14,5 г, 104 ммоль) и TosMIC (11,6 г 59,4 ммоль). После перемешивания при 90°С в течение 0,5 часа реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1:0 до 1:1) с получением соединения 51С (2,9 г, выход: 16,4%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 11,04 (ушир. s, 1Н), 7,98 (d, J=0,7 Гц, 1Н), 7,91 (s, 1H), 7,48-7,41 (m, 3H), 7,25-7,19 (m, 1Н), 4,24-4,14 (m, 2H), 1,14 (t, J=7,1 Гц, 3Н).
[0597] К раствору 51С (2,9 г, 11,3 ммоль) в ТГФ (40 мл) и H2O (8 мл) добавляли NaOH (905 мг, 22,6 ммоль). Смесь перемешивали при 25°С в течение 10 часов. Смесь концентрировали при пониженном давлении для удаления органического растворителя и подвергали экстракции EtOAc (20 мл). Водный слой подкисляли с помощью 1 M HCl до рН ~ 5 и затем подвергали экстракции EtOAc (30 мл × 3). Объединенный органический слой промывали H2O (40 мл), солевым раствором (40 мл), сушили над Na2SO4, фильтровали и концентрировали с получением 51D (1,5 г, выход: 58,1%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,35 (s, 1Н), 8,16 (s, 1Н), 7,83 (s, 1Н), 7,61 (d, J=8,3 Гц, 1Н), 7,47-7,41 (m, 2H), 7,20-7,15 (m, 1Н). MC (ИЭР) m/z (M+H)+ 228,9.
[0598] К раствору 51D (500 мг, 2,19 ммоль) и 1-гидроксипирролидин-2,5-диона (252 мг, 2,19 ммоль) в ТГФ (10 мл), ДХМ (5 мл) и ДМФА (10 мл) добавляли EDCI (420 мг, 2,19 ммоль) при 0°С. После добавления смесь перемешивали при 25°С в течение 12 ч. Растворитель удаляли под вакуумом. Остаток разбавляли EtOAc (50 мл), промывали 1 н. HCl (20 мл), насыщенным NaHCO3 (20 мл) и солевым раствором (20 мл). Органические фазы объединяли, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного 51Е (476 мг, выход: 66,8%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,52 (s, 1Н), 8,57 (s, 1Н), 8,32 (s, 1Н), 7,63 (d, J=8,6 Гц, 1Н), 7,49-7,43 (m, 2H), 7,24-7,17 (m, 1Н), 2,77 (s, 5H).
[0599] Соединение 51 (28,5 мг, выход: 29,1%, желтое твердое вещество) было получено, как описано в примере 20, из соответствующих промежуточных соединений 51Е и 41В. Соединение 51: 1H ЯМР (400 МГц, CDCl3) δ 10,66 (ушир. s, 1Н), 7,86 (s, 1Н), 7,73 (s, 1Н), 7,49-7,34 (m, 3Н), 7,34-7,28 (m, 1Н), 7,23-7,10 (m, 4H), 7,09-6,90 (m, 3H), 5,65-5,53 (m, 1Н), 3,33 (dd, J=14,1, 5,1 Гц, 1Н), 3,15 (dd, J=14,1, 7,3 Гц, 1Н), 2,75 (td, J=7,2, 3,6 Гц, 1Н), 0,76-0,86 (m, 2H), 0,55 (ушир. d, J=2,6 Гц, 2Н). МС (ИЭР) m/z (M+H)+ 443,1.
ПРИМЕР 26
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(5-ФЕНИЛТИАЗОЛ-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (52)
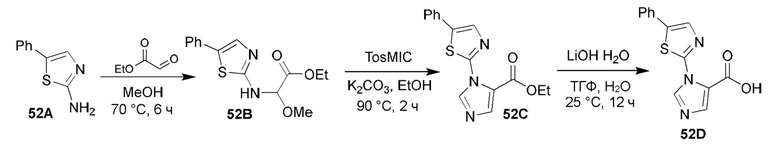
[0600] Смесь соединения 52А (4,2 г, 23,8 ммоль) и этил-2-оксоацетата (14,6 г, 71,4 ммоль) в МеОН (40 мл) перемешивали при 70°С в течение 6 часов. Анализ посредством ТСХ (петролейный эфир : этилацетат = 2:1, Rf ~ 0,7) свидетельствовал о том, что соединение 52А полностью израсходовалось, и на хроматограмме было обнаружено одно основное новое пятно с меньшей полярностью. Реакционную смесь концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 20:1 до 10:1) с получением соединения 52В (7 г, неочищенного) в виде желтого масла.
[0601] К смеси соединения 52В (7 г, 23,9 ммоль) и K2CO3 (6,6 г, 47,8 ммоль) в EtOH (15 мл) добавляли TosMIC (6,9 г, 35,9 ммоль). Смесь перемешивали при 90°С в течение 2 часов. Анализ посредством ТСХ (петролейный эфир : этилацетат = 2:1, Rf ~ 0,55) свидетельствовал о том, что соединение 52В полностью израсходовалось, и на хроматограмме было обнаружено одно основное новое пятно с большей полярностью. Реакционную смесь концентрировали с получением остатка. Неочищенный продукт очищали посредством хроматографии на силикагеле, элюируя смесью петролейный эфир : этилацетат = от 15:1 до 5:1, с получением соединения 52С (6 г, неочищенного) в виде желтого твердого вещества. МС (ИЭР) m/z (М+H)+ 299,9.
[0602] К раствору соединения 52С (3,5 г, 11,69 ммоль) в ТГФ (20 мл) и H2O (6 мл) добавляли LiOH⋅H2O (981 мг, 23,3 ммоль) одной порцией. Смесь перемешивали при 25°С в течение 12 часов. Анализ посредством ТСХ (петролейный эфир : этилацетат = 1:1, Rf ~ 0,25) свидетельствовал о том, что соединение 52С полностью израсходовалось, и на хроматограмме появилось одно новое пятно. рН смеси доводили до ~ 5 путем добавления HCl (2 М), и затем белое твердое вещество выпадало в осадок, который отфильтровывали и сушили при пониженном давлении с получением соединения 52D (1,5 г, выход: 47,3%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,37 (s, 1H), 8,19 (s, 1H), 7,76-7,70 (m, 3Н), 7,53-7,46 (m, 2H), 7,45-7,38 (m, 1H).
[0603] Соединение 52 (50,9 мг, выход: 43%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 52D. Соединение 52: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,05 (d, J=7,5 Гц, 1H), 8,32 (s, 1H), 8,11 (s, 2H), 7,85 (s, 1H), 7,71-7,66 (m, 3Н), 7,48 (t, J=7,5 Гц, 2H), 7,44-7,39 (m, 1H), 7,30 (d, J=4,4 Гц, 4Н), 7,23-7,19 (m, 1H), 5,35-5,25 (m, 1H), 3,21 (dd, J=3,7, 13,8 Гц, 1H), 2,85 (dd, J=10,3, 13,8 Гц, 1H). МС (ИЭР) m/z (M+H)+ 446,0.
ПРИМЕР 27
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(5-ФЕНИЛТИАЗОЛ-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (53)
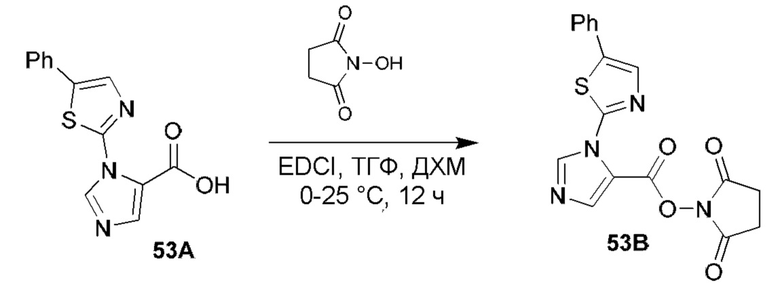
[0604] К смеси соединения 53А (600 мг, 2,21 ммоль) и соединения 1-гидроксипирролидин-2,5-диона (254 мг, 2,21 ммоль) в ТГФ (10 мл) при 0°С добавляли по каплям раствор EDCI (423 мг, 2,21 ммоль) в ДХМ (5 мл). Смесь перемешивали при 25°С в течение 12 часов. Анализ посредством ТСХ (петролейный эфир : этилацетат = 1:1, Rf ~ 0,4) свидетельствовал о том, что соединение 53А полностью израсходовалось, и на хроматограмме было обнаружено одно основное новое пятно с меньшей полярностью. Реакционную смесь концентрировали для удаления растворителя. Остаток разбавляли EtOAc (50 мл), промывали H2O (20 мл), насыщенным NaHCO3 (20 мл), солевым раствором (20 мл). Органические фазы собирали, сушили с помощью Na2SO4, фильтровали и концентрировали с получением целевого промежуточного соединения 53В (700 мг, выход: 85,9%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,29 (s, 1H), 8,20 (s, 1H), 7,86 (s, 1H), 7,57 (d, J=6,5 Гц, 2H), 7,48-7,36 (m, 3H), 7,27 (s, 1H), 2,88 (s, 4H).
[0605] Соединение 53 (41 мг, выход: 34,3%, белое твердое вещество) было получено, как описано в примере 20, из соответствующего промежуточного соединения 53В. Соединение 53: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,08 (d, J=7,7 Гц, 1H), 8,82 (d, J=5,0 Гц, 1H), 8,32 (d, J=0,8 Гц, 1H), 8,11 (s, 1H), 7,72-7,64 (m, 3H), 7,52-7,46 (m, 2H), 7,44-7,39 (m, 1H), 7,30 (d, J=4,4 Гц, 4H), 7,24-7,18 (m, 1H), 5,31-5,22 (m, 1H), 3,21 (dd, J=13,7, 3,6 Гц, 1H), 2,90-2,70 (m, 2H), 0,66-0,54 (m, 4H). МС (ИЭР) m/z (М+Н)+ 486,1.
ПРИМЕР 28
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-МЕТИЛ-1-ФЕНИЛ-1H-1,2,3-ТРИАЗОЛ-5-КАРБОКСАМИД (55)
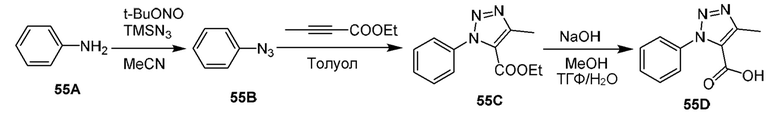
[0606] К раствору соединения 55А (2,5 г, 26,8 ммоль) в MeCN (50 мл) добавляли т-BuONO (4,15 г, 40,3 ммоль) при 0°С и затем добавляли TMSN3 (4,64 г, 40,3 ммоль). Реакционную смесь перемешивали при 20°С в течение 1 ч. Растворитель выпаривали с получением промежуточного соединения 55В (4 г, неочищенного) в виде желтого масла.
[0607] Смесь соединения 55В (4 г, неочищенного) и соединения этилбут-2-иноата (1 г, 8,92 ммоль) в толуоле (20 мл) перемешивали при 110°С в течение 5 ч. Растворитель выпаривали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 20:1 ~ 5:1) с получением соединения 55С (150 мг, выход: 7,27%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,54-7,49 (m, 2H), 7,44-7,40 (m, 2H), 7,37 (d, J=5,1 Гц, 1H), 4,25 (q, J=7,1 Гц, 2H), 1,21 (t, J=7,1 Гц, 3H).
[0608] К раствору соединения 55С (150 мг, 649 мкмоль) в ТГФ (2 мл) и H2O (2 мл) добавляли NaOH (51,9 мг). Реакционную смесь перемешивали при 20°С в течение 30 мин. Анализ посредством ТСХ свидетельствовал об образовании нового пика с большей полярностью. Растворитель выпаривали и добавляли 1 M HCl до рН ~ 6. Смесь фильтровали, и осадок на фильтре сушили с получением соединения 55D (120 мг, выход: 91,0%) в виде желтого твердого вещества.
[0609] Соединение 55 (46 мг, 121 мкмоль, выход: 42,0%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 55D. Соединение 55: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,36 (ушир. d, J=7,9 Гц, 1Н), 8,20 (s, 1H), 7,94 (s, 1H), 7,51-7,43 (m, 3H), 7,36-7,28 (m, 7H), 5,38 (ушир. t, J=7,7 Гц, 1H), 3,26 (ушир. s, 1H), 2,82-2,73 (m, 1H), 2,21 (s, 3H). MC (ИЭР) m/z (М+Н)+ 378,1.
ПРИМЕР 29
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-МЕТИЛ-1-ФЕНИЛ-1H-1,2,3-ТРИАЗОЛ-5-КАРБОКСАМИД (56)
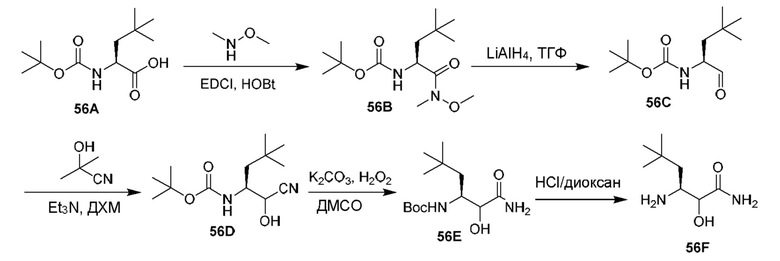
[0610] Смесь соединения 56А (1,0 г, 4,08 ммоль), соединения N,O-диметилгидроксиламина (478 мг, 4,90 ммоль, HCl), HOBt (552 мг, 4,08 ммоль) и NMM (1,24 г, 12,24 ммоль, 1,35 мл) в CHCl3 (20 мл) дегазировали и продували N2 3 раза при 0°С, затем добавляли порциями EDCI (1,17 г, 6,12 ммоль). Смесь перемешивали при 25°С в течение 20 ч в атмосфере N2. В реакционную смесь добавляли H2O (20 мл) для гашения реакции, и затем смесь разбавляли ДХМ (10 мл). Объединенные органические слои промывали 1 н. HCl (15 мл × 2), насыщенным водным NaHCO3 (15 мл × 2) и солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 56В (1,15 г, выход: 97,7%), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 6,97 (ушир. d, J=8,8 Гц, 1H), 4,47 (ушир. t, J=8,4 Гц, 1H), 3,73-3,64 (m, 3H), 3,06 (s, 3H), 1,51-1,27 (m, 11H), 0,87 (s, 9H).
[0611] К раствору LAH (303 мг, 7,98 ммоль) в ТГФ (10 мл), который дегазировали и продували N2 3 раза при 0°С, добавляли по каплям смесь соединения 56В (1,15 г, 3,99 ммоль) в ТГФ (20 мл), и затем смесь перемешивали при 0°С в течение 2 ч в атмосфере N2. В реакционную смесь добавляли для гашения реакции EtOAc (10 мл), затем добавляли 1 н. HCl (50 мл), и затем смесь разбавляли EtOAc (20 мл), сушили над Na2SO4 и перемешивали в течение 30 мин, затем фильтровали с получением органических слоев. Объединенные органические слои промывали солевым раствором (20 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 56С (900 мг, выход: 98,4%), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,55 (s, 1Н), 4,83 (ушир. s, 1H), 4,24 (ушир. s, 1H), 1,86-1,55 (m, 2H), 1,44 (s, 9H), 1,03-0,91 (m, 9H).
[0612] К раствору соединения 56С (900 мг, 3,92 ммоль) в ДХМ (20 мл) добавляли соединение 2-гидрокси-2-метилпропаннитрил (2,33 г, 27,32 ммоль, 2,50 мл) и Et3N (595 мг, 5,88 ммоль, 815 мкл). Смесь перемешивали при 25°С в течение 2 ч. В реакционную смесь добавляли для гашения реакции 1 н. HCl (20 мл), и затем смесь разбавляли H2O (20 мл) и подвергали экстракции ДХМ (20 мл × 2). Объединенные органические слои промывали солевым раствором (20 мл × 3), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 56D (930 мг, выход: 92,55%), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 5,06-4,66 (m, 1H), 4,55-4,35 (m, 1H), 4,05-3,73 (m, 1H), 1,80-1,65 (m, 2H), 1,45 (ушир. d, J=6,8 Гц, 9H), 1,10-0,80 (m, 9H).
[0613] К раствору соединения 56D (930 мг, 3,63 ммоль) и K2CO3 (1,00 г, 7,26 ммоль) в ДМСО (15 мл) добавляли H2O2 (4,12 г, 36,30 ммоль, 3,49 мл, чистота: 30%). Смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь разбавляли H2O (50 мл) и подвергали экстракции EtOAc (20 мл × 2). Объединенные органические слои промывали солевым раствором (30 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток перемешивали в ДХМ (0,1 мл) и ПЭ (5 мл) в течение 30 мин и фильтровали с получением соединения 56Е (480 мг, выход: 48,20%), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 6,83 (ушир. s, 1H), 5,65 (ушир. s, 1H), 5,27-5,06 (m, 1H), 4,99-4,82 (m, 1H), 4,23-4,00 (m, 1H), 3,88 (ушир. t, J=8,6 Гц, 1Н), 1,77 (ушир. s, 1H), 1,60-1,51 (m, 1H), 1,42 (d, J=9,3 Гц, 9H), 0,94 (d, J=10,1 Гц, 9Н).
[0614] К раствору соединения 56Е (480 мг, 1,75 ммоль) в EtOAc (5 мл) добавляли HCl/EtOAc (4 M, 5 мл). Смесь перемешивали при 25°С в течение 1 ч. Реакционную смесь разбавляли ПЭ (20 мл), фильтровали и концентрировали при пониженном давлении с получением соединения 56F (360 мг, выход: 97,63%, HCl), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,00 (ушир. s, 1H), 7,92-7,70 (m, 1H), 7,58-7,41 (m, 2Н), 4,21-3,93 (m, 1H), 3,33 (ушир. d, J=3,5 Гц, 2Н), 1,76-1,24 (m, 2Н), 0,86 (s, 9H).
[0615] Соединение 56 (94,20 мг, выход: 85,26%, белое твердое вещество) было получено, как описано в примере 35, из соответствующих промежуточных соединений, 23А и 56F. Соединение 56: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,98-8,61 (m, 1H), 8,20-7,95 (m, 1H), 7,85-7,71 (m, 2Н), 7,57-7,37 (m, 3Н), 5,25 (ушир. t, J=6,8 Гц, 1H), 2,35-2,20 (m, 3H), 1,63-1,28 (m, 2Н), 0,98-0,76 (m, 9H). МС (ИЭР) m/z (М+Н)+ 358,2.
ПРИМЕР 30
(S)-N-(4-АМИНО-1-(1H-ИНДОЛ-3-ИЛ)-3,4-ДИОКСОБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (57)
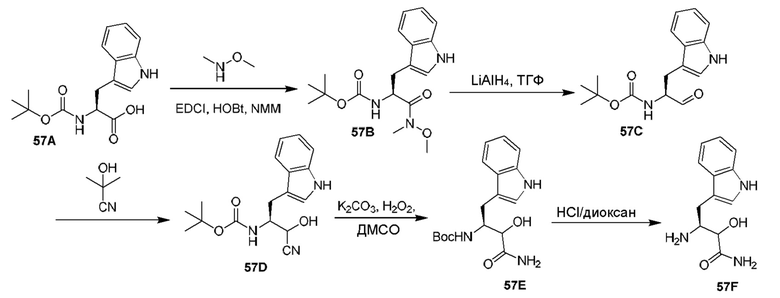
[0616] Смесь соединения 57А (5,00 г, 16,43 ммоль), соединения N,O-диметилгидроксиламина (1,76 г, 18,07 ммоль, HCl), HOBt (2,22 г, 16,43 ммоль) и NMM (4,99 г, 49,29 ммоль, 5,42 мл) в CHCl3 (150 мл) дегазировали и продували N2 3 раза при 0°С, затем добавляли порциями EDCI (4,72 г, 24,65 ммоль), и затем смесь перемешивали при 25°С в течение 23 ч в атмосфере N2. В реакционную смесь добавляли H2O (100 мл) для гашения реакции, и затем смесь разбавляли 1 н. HCl (200 мл) и подвергали экстракции NaHCO3 (50 мл × 2). Объединенные органические слои промывали солевым раствором (200 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 6/1 до 1/1) с получением соединения 57В (5,94 г), которое было получено в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,79 (ушир. s, 1Н), 7,50 (d, J=7,7 Гц, 1Н), 7,32 (d, J=8,2 Гц, 1H), 7,19-7,11 (m, 1H), 7,07-6,96 (m, 3Н), 4,59 (ушир. s, 1H), 3,70 (ушир. s, 3Н), 3,10 (s, 3Н), 3,03-2,94 (m, 1H), 2,89-2,77 (m. 1H). 1.29 (s. 9H).
[0617] К раствору LAH (330 мг, 8,64 ммоль) в ТГФ (10 мл) и затем добавляли по каплям соединение 57В (2,00 г, 5,76 ммоль) в ТГФ (20 мл). Смесь перемешивали при 0°С в течение 2 ч. В реакционную смесь добавляли для гашения реакции EtOAc (10 мл) при 0°С, и затем смесь разбавляли 1 н. HCl (40 мл) и подвергали экстракции EtOAc (20 мл × 2). Объединенные органические слои промывали 1 н. HCl (40 мл) и NaHCO3 (30 мл × 2) и солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 57С (1,55 г, выход: 93,33%), которое было получено в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,02-10,75 (m, 1H), 9,52 (s, 1H), 7,50 (ушир. d, J=7,7 Гц, 1H), 7,32 (d, J=7,9 Гц, 1H), 7,25 (ушир. d, J=7,3 Гц, 1H), 7,14 (d, J=1,8 Гц, 1H), 7,05 (t, J=7,2 Гц, 1H), 7,00-6,92 (m, 1H), 4,14-4,05 (m, 1H), 3,19-3,10 (m, 1H), 2,95-2,85 (m, 1H), 2,52-2,45 (m, 4H), 1,39-1,23 (m, 9H).
[0618] К раствору соединения 57С (1,50 г, 5,20 ммоль) в ДХМ (30,00 мл) добавляли соединение N,O-диметилгидроксиламин (885 мг, 10,40 ммоль, 960 мкл) и Et3N (790 мг, 7,80 ммоль, 1,08 мл). После перемешивания при 25°С в течение 20 ч в реакционную смесь добавляли для гашения реакции 30 мл 0,5 н. HCl, и затем смесь подвергали экстракции ДХМ (30 мл × 2). Объединенные органические слои промывали солевым раствором (30 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Соединение 57D (1,74 г, желтое твердое вещество): 1H ЯМР (400 МГц, CDCl3) δ 9,64 (s, 1H), 8,14 (ушир. s, 1H), 7,60 (d, J=7,7 Гц, 1H), 7,37 (d, J=8,2 Гц, 1H), 7,24-7,18 (m, 1H), 7,17-7,11 (m, 1H), 7,03 (d, J=2,2 Гц, 1H), 5,14 (ушир. s, 1H), 4,51 (ушир. d, J=6,6 Гц, 1H), 3,41-3,16 (m, 2H), 1,44 (s, 9H).
[0619] К раствору соединения 57D (1,74 г, 5,52 ммоль) и K2CO3 (1,53 г, 11,04 ммоль) в ДМСО (25,00 мл) добавляли H2O2 (6,43 г, 189,00 ммоль, 5,45 мл) при 0°С. Смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь разбавляли H2O (50 мл) и затем добавляли в нее для гашения реакции Na2S2O3 (50 мл), и смесь подвергали экстракции EtOAc (50 мл × 3) и Na2S2O3 (50 мл × 3). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1/2 до 0:1) с получением соединения 57Е (689,60 мг, выход: 37,47%), которое было получено в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,06 (ушир. s, 1Н), 7,70 (d, J=8,3 Гц, 1Н), 7,38 (d, J=7,8 Гц, 1Н), 7,25-7,06 (m, 4H), 5,42 (ушир. s, 1Н), 5,19-5,04 (m, 1Н), 4,21-4,08 (m, 3H), 3,30-3,12 (m, 2H), 1,41 (s, 9H).
[0620] К раствору соединения 57Е (680,00 мг, 2,04 ммоль) в EtOAc (5,00 мл) добавляли HCl/EtOAc (5,00 мл). Смесь перемешивали при 25°С в течение 2,5 ч. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя с получением соединения 57F (400,00 мг, выход: 72,69%, HCl), которое было получено в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,01 (ушир. s, 1Н), 7,92 (ушир. s, 2H), 7,70-7,46 (m, 3H), 7,39-7,26 (m, 2H), 7,12-6,95 (m, 2H), 4,01-3,89 (m, 1Н), 3,81-3,64 (m, 1Н), 3,14 (s, 2H), 3,08-2,80 (m, 2H).
[0621] Соединение 57 (11,20 мг, выход: 29,41%, белое твердое вещество) было получено, как описано в примере 15, из соответствующих промежуточных соединений, 23А и 57F. Соединение 57: 1H ЯМР (400 МГц, ДМСО-d6) δ 10,89 (ушир. s, 1Н), 9,03 (d, J=7,3 Гц, 1Н), 8,23 (s, 1Н), 7,95 (s, 1Н), 7,70 (d, J=7,8 Гц, 1Н), 7,63 (d, J=7,5 Гц, 2H), 7,48 (ушир. d, J=7,5 Гц, 1Н), 7,39 (t, J=7,4 Гц, 3H), 7,17 (d, J=1,8 Гц, 1Н), 7,13-6,96 (m, 2H), 5,56 (ушир. s, 1Н), 2,97-2,87 (m, 1Н), 2,70-2,54 (m, 1Н), 2,11 (s, 3H). МС (ИЭР) m/z (M+H)+ 417,1.
ПРИМЕР 31
(S)-N-(4-АМИНО-1-(1H-ИНДОЛ-3-ИЛ)-3,4-ДИОКСОБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (58)
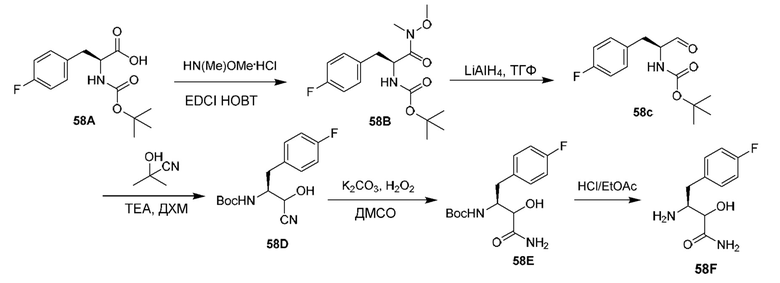
[0622] Раствор N-метоксиметанамина (1,89 г, 19,42 ммоль), соединения 58А (5,0 г, 17,65 ммоль), HOBt (2,38 г, 17,65 ммоль) и NMM (52,95 ммоль, 5,8 мл) в CHCl3 (100 мл) дегазировали и продували N2 3 раза при 0°С, затем добавляли порциями EDCI (5,1 г, 26,48 ммоль). Смесь перемешивали при 25°С в течение 16 ч в атмосфере N2. Реакционную смесь промывали H2O (100 мл). Органические слои промывали 1 М HCl (100 мл × 2), насыщенным NaHCO3 (100 мл × 2) и насыщенным солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® Silica Flash массой 80 г, элюент 0 ~ 30% этилацетат/петролейный эфир, градиент со скоростью потока 40 мл/мин) с получением соединения 58В (4,00 г, выход 69,4%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,11 (dd, J=5,6, 8,3 Гц, 2Н), 6,94 (t, J=8,7 Гц, 2Н), 5,18 (ушир. d, J=7,9 Гц, 1Н), 4,98-4,80 (m, 1H), 4,13-4,07 (m, 2Н), 3,72-3,64 (m, 4H), 3,14 (s, 3H), 3,08-2,94 (m, 1H), 2,91-2,70 (m, 1H), 2,02 (s, 2Н), 1,78 (ушир. s, 1H), 1,37 (s, 10Н), 1,28-1,20 (m, 3H).
[0623] К LiAlH4 (128 мг, 3,37 ммоль) в сухой колбе объемом 100 мл добавляли по каплям ТГФ (15 мл) при 0°С. После добавления смесь перемешивали при этой температуре, и затем к описанной выше смеси добавляли по каплям раствор соединения 58В (1,0 г, 3,06 ммоль) в ТГФ (15 мл) при 0°С. Полученную смесь перемешивали при 0°С в течение 1,5 ч. В реакционную смесь медленно добавляли для гашения реакции EtOAc (20 мл) при 0°С и затем добавляли 1 н. HCl (20 мл), и смесь подвергали экстракции EtOAc (30 мл × 2). Объединенные органические слои промывали NaHCO3 (30 мл × 2) и солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 58С (810 мг, выход 99,0%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,65 (ушир. s, 1H), 9,63 (ушир. s, 1H), 7,21-7,08 (m, 2Н), 7,00 (ушир. d, J=8,6 Гц, 2Н), 5,05 (ушир. s, 1H), 4,42 (ушир. s, 1H), 3,09-3,02 (m, 1H), 3,11 (ушир. d, J=6,2 Гц, 1H), 1,51-1,38 (m, 9H).
[0624] К раствору соединения 58С (3,2 г, 11,86 ммоль) и 2-гидрокси-2-метил-пропаннитрила (2,2 мл, 23,72 ммоль) в ДХМ (30 мл) добавляли TEA (2 мл, 14,23 ммоль). После добавления реакционную смесь перемешивали при 28°С в течение 14 ч. Реакционную смесь разбавляли 30 мл ДХМ и добавляли в нее для гашения реакции 30 мл 0,5 н. HCl. Органический слой промывали H2O (30 мл) и солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 58D (3,4 г, выход 89,2%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,15-7,07 (m, 2Н), 7,01-6,90 (m, 2Н), 4,94-4,70 (m, 1H), 4,52-4,36 (m, 1H), 4,16-3,67 (m, 1H), 3,11-2,78 (m, 2Н), 1,57-1,47 (m, 2Н).
[0625] К раствору соединения 58D (3,42 г, 11,62 ммоль) и K2CO3 (3,21 г, 23,24 ммоль) в ДМСО (30 мл) добавляли H2O2 (395,08 ммоль, 12 мл) при 0°С. После добавления реакционную смесь перемешивали при 0°С в течение 1,5 ч. Реакционную смесь разбавляли водой (100 мл) и медленно добавляли в нее для гашения реакции насыщенный водный Na2S2O3 при охлаждении ледяной водой. Смесь подвергали экстракции EtOAc (200 мл × 3), и объединенные экстракты промывали насыщенным водным Na2S2O3 (100 мл × 3). Органический слой сушили над Na2SO4 и концентрировали с получением остатка. Остаток разбавляли EtOAc (10 мл) и фильтровали с получением соединения 58Е (2,25 г, выход 61,99%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,25 (ушир. s, 6H), 6,62-6,03 (m, 1H), 5,75-5,55 (m, 1H), 4,02-3,67 (m, 2Н), 2,80-2,52 (m, 2Н), 2,52-2,51 (m, 1H), 1,26 (d, J=3,7 Гц, 9Н). МС (ПЭР) m/z (M+Na)+ 334,9.
[0626] К раствору соединения 58Е (1 г, 3,20 ммоль) в EtOAc (10 мл) добавляли HCl/EtOAc (4 ммоль, 20 мл). Смесь перемешивали при 28°С в течение 2 ч. Реакционную смесь разбавляли МТБЭ и фильтровали с получением соединения 58F (750 мг, выход 94,25%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,25-7,94 (m, 3Н), 7,58-7,43 (m, 2Н), 7,41-7,33 (m, 1H), 7,30-7,23 (m, 1H), 7,41-7,23 (m, 1H), 7,20-7,05 (m, 2Н), 6,90-6,37 (m, 1H), 6,80-6,25 (m, 1H), 4,24 (ушир. s, 1H), 3,88-3,81 (m, 1H), 3,85 (ушир. s, 1H), 3,68-3,50 (m, 1H), 2,96-2,76 (m, 2Н). MC (ИЭР) m/z (M+H)+ 213,1.
[0627] Соединение 58 (130 мг, выход 78,40%, светло-желтое твердое вещество) было получено, как описано в примере 15, из соответствующих промежуточных соединений, 23А и 58F. Соединение 58: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,05 (d, J=7,7 Гц, 1H), 8,20 (ушир. s, 1H), 7,93 (ушир. s, 1H), 7,66-7,59 (m, 2Н), 7,55-7,49 (m, 1H), 7,48-7,41 (m, 2Н), 7,35-7,26 (m, 2Н), 7,17-7,06 (m, 2Н), 5,51-5,40 (m, 1H), 3,28-3,19 (m, 1H), 2,81-2,69 (m, 1H), 2,11 (s, 3H). MC (ИЭР) m/z (M+H)+ 396,1.
ПРИМЕР 32
(S)-N-(4-АМИНО-1-(1H-ИНДОЛ-3-ИЛ)-3,4-ДИОКСОБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (59)
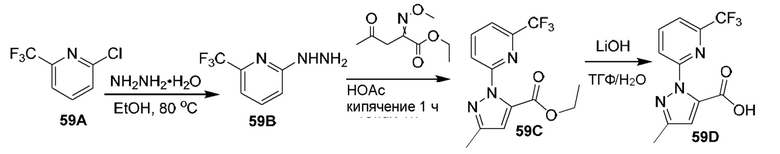
[0628] К раствору соединения 59А (10 г, 55,08 ммоль) в EtOH (30 мл) добавляли NH2NH2.H2O (32 мл, 550,80 ммоль). После добавления реакционную смесь перемешивали при 80°С в течение 14 ч. Реакционную смесь концентрировали, и остаток растворяли в 150 мл EtOAc, смесь промывали водой (50 мл) и солевым раствором (50 мл), затем сушили над Na2SO4 и концентрировали под вакуумом с получением соединения 59В (9,7 г, выход 99,4%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,02 (s, 1H), 7,71-7,53 (m, 1H), 7,03-6,79 (m, 2Н), 4,23 (s, 2Н).
[0629] К раствору соединения 59В (1 г, 5,65 ммоль) в АсОН (10 мл) добавляли этил-2-метоксиимино-4-оксо-пентаноат (1,1 г, 5,65 ммоль). После добавления реакционную смесь перемешивали при 120°С в течение 14 ч. Смесь концентрировали под вакуумом, и остаток растворяли в 80 мл EtOAc, смесь промывали 30 мл насыщенного водного NaHCO3 и солевым раствором (30 мл). Смесь сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 20/1 до 10:1) с получением целевого соединения 59С (1,2 г, выход: 71%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,34-8,25 (m, 1Н), 8,05 (d, J=8,2 Гц, 1H), 7,93 (d, J=7,5 Гц, 1Н), 6,86 (s, 1Н), 4,18 (q, J=7,1 Гц, 2H), 2,29 (s, 3H), 1,11 (t, J=7,1 Гц, 3Н). МС (ИЭР) m/z (М+H)+ 299,9.
[0630] К раствору соединения 59С (700 мг, 2,34 ммоль) в ТГФ (10 мл) добавляли раствор LiOH⋅H2O (393 мг, 9,36 ммоль) в H2O (10 мл) при 0°С. После добавления реакционную смесь перемешивали при 28°С в течение 16 ч, в реакционную смесь добавляли 20 мл МТБЭ, затем смесь разделяли и водный слой подкисляли с помощью 1 н. HCl до рН ~ 4, смесь фильтровали с получением белого твердого вещества, которое сушили с получением соединения 59D (330 мг, выход 51,95%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,48 (ушир. s, 1Н), 8,31-8,22 (m, 1Н), 8,02-7,89 (m, 2H), 6,81 (s, 1Н), 2,27 (s, 3Н). МС (ИЭР) m/z (M+H)+ 271,8.
[0631] Соединение 59 (50 мг, выход: 37,9%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 59D. Соединение 59: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,07 (d, J=7,3 Гц, 1Н), 8,27-8,16 (m, 1Н), 8,04 (s, 1Н), 7,89-7,77 (m, 3Н), 7,30-7,19 (m, 5H), 6,59 (s, 1Н), 5,40-5,28 (m, 1Н), 3,15 (dd, J=4,0, 13,8 Гц, 1Н), 2,82 (dd, J=9,5, 13,8 Гц, 1Н), 2,30 (s, 3Н). МС (ИЭР) m/z (M+H)+ 446,1.
ПРИМЕР 33
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (61)
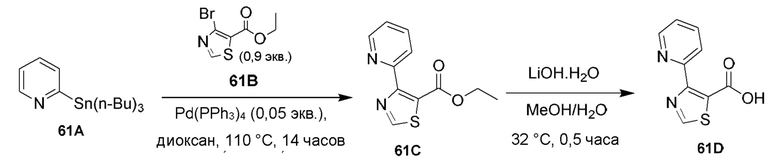
[0632] Смесь соединения 61В (500 мг, 2,12 ммоль), соединения 61А (859 мг, 2,33 ммоль), Pd(PPh3)4 (122 мг, 106 мкмоль) перемешивали при 105°С в течение 14 часов. Смесь концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 3/1 до 1/1) с получением соединения 61С (376 мг, выход 74,95%) в виде желтого масла. МС (ИЭР) m/z (M+H)+ 234,9.
[0633] К раствору соединения 61С (320 мг, 1,37 ммоль) в МеОН (20 мл) добавляли LiOH⋅H2O (144 мг, 3,43 ммоль). Смесь перемешивали при 32°С в течение 0,5 ч. МеОН выпаривали. К остатку добавляли воду (20 мл). Смесь подвергали экстракции МТБЭ (5 мл) и разделяли. Водный слой подкисляли до рН ~ 3 с помощью 1 н. HCl и подвергали экстракции этилацетатом (3×20 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением соединения 61D (220 мг, выход 77,9%) в виде белого твердого вещества.
[0634] Соединение 61 (21,8 мг, выход 21,78%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 61D. Соединение 61: 1H ЯМР (400 МГц, ДМСО-d6) δ 13,06-12,79 (m, 1H), 9,27 (s, 1H), 8,95-8,84 (m, 1H), 8,42-8,32 (m, 1H), 8,32-8,24 (m, 1H), 8,13-8,00 (m, 1H), 7,55-7,45 (m, 1H), 7,21-7,10 (m, 3H), 7,22-7,03 (m, 2H), 5,70-5,59 (m, 1H), 3,32-3,25 (m, 1H), 3,20-3,12 (m, 1H), 2,85-2,74 (m, 1H), 0,72-0,63 (m, 2H), 0,63-0,54 (m, 2H). MC (ИЭР) m/z (M+H)+ 421,1.
ПРИМЕР 34
(S)-N-(1-амино-1,2-диоксогептан-3-ил)-3-метил-5-фенилизоксазол-4-карбоксамид (62)
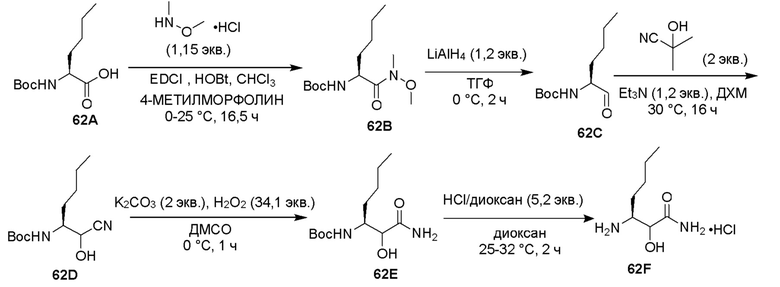
[0635] К смеси соединения 62А (2 г, 8,65 ммоль) и соединения гидрохлорида N,O-диметилгидроксиламина (970,3 мг, 9,95 ммоль), HOBt (1,34 г, 9,95 ммоль) в CHCl3 (40 мл) добавляли по каплям 4-метилморфолин (2,62 г, 25,95 ммоль) и EDCI (2,32 г, 12,11 ммоль) порциями при 0°С в атмосфере N2. Смесь перемешивали при 0°С в течение 30 мин, и затем смесь перемешивали при 25°С в течение 16 часов. Реакционную смесь разбавляли H2O (5 мл). Два слоя разделяли, и водную фазу подвергали экстракции ЭА (5 мл × 2). Объединенные органические слои промывали 0,5 н. HCl (5 мл × 2) и NaHCO3 (5 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 62В (1,7 г, выход 71,7%) в виде бесцветного масла. 1H ЯМР (CDCl3, 400 МГц): δ 5,19-5,06 (m, 1Н), 4,66 (ушир. s, 1H), 3,77 (s, 3H), 3,20 (s, 3H), 1,76-1,66 (m, 1H), 1,55-1,39 (m, 10Н), 1,37-1,28 (m, 4H), 0,93-0,83 (m, 3H). МС (ИЭР) m/z (М-Boc+H)+ 175,0.
[0636] К раствору LiAlH4 (258,7 мг, 6,82 ммоль) в ТГФ (36 мл) добавляли по каплям раствор соединения 62В (1,7 г, 6,2 ммоль) в ТГФ (18 мл) при 0°С в атмосфере N2. После добавления реакционную смесь перемешивали при 0°С в течение 2 часов. Смесь разбавляли этилацетатом (100 мл), промывали 1 н. HCl (20 мл), насыщенным NaHCO3 (20 мл × 2), солевым раствором (15 мл). Органический слой сушили над безводным Na2SO4 и концентрировали с получением соединения 62С (1,5 г, неочищенного) в виде желтого масла. 1H ЯМР (CDCl3, 400 МГц) δ 9,58 (s, 1H), 5,03 (ушир. s, 1H), 4,28-4,16 (m, 1H), 1,58-1,19 (m, 15H), 1,01-0,80 (m, 3H).
[0637] Раствор соединения 62С (1,5 г, 6,97 ммоль), соединения 2-гидрокси-2-метилпропаннитрила (1,3 мл, 13,94 ммоль) и Et3N (1,16 мл, 8,36 ммоль) в сухом ДХМ (30 мл) перемешивали при 30°С в течение 16 часов. Реакционную смесь разбавляли ДХМ (50 мл), промывали 0,5 н. HCl (20 мл), водой (20 мл) и солевым раствором (20 мл). Органическую фазу сушили над Na2SO4, концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 3:1) с получением соединения 62D (1,12 г, выход 66,32%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 5,44-4,34 (m, 3H), 3,94-3,83 (m, 1H), 3,74-3,61 (m, 1H), 3,98-3,55 (m, 1H), 1,66-1,28 (m, 14H), 0,99-0,90 (m, 3H).
[0638] К смеси соединения 62D (1,12 г, 4,62 ммоль) и K2CO3 (1,28 г, 9,24 ммоль) в ДМСО (18 мл) добавляли H2O2 (4,6 мл, 158,19 ммоль) при 0°С. После добавления реакционную смесь перемешивали при 0°С в течение 1 ч. После завершения реакции в реакционную смесь добавляли МТБЭ (20 мл), и полученную смесь фильтровали и твердое вещество промывали МТБЭ (30 мл) с получением соединения 62Е (1,1 г, выход 91,46%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,32-7,08 (m, 2H), 6,42-5,86 (m, 1H), 5,54-5,30 (m, 1H), 3,88-3,59 (m, 2H), 1,42-1,21 (m, 15H), 0,92-0,78 (m, 3H).
[0639] К раствору соединения 62Е (600 мг, 20,82 ммоль) в диоксане (10 мл) добавляли HCl/диоксан (3 мл, 4 M) при 25°С. Реакционную смесь перемешивали при 25°С в течение 2 часов. Смесь фильтровали с получением соединения 62F (320 мг, выход 70,7%, HCl), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,90 (ушир. s, 2H), 7,54-7,35 (m, 2H), 6,26-6,17 (m, 1H), 4,09 (ушир. s, 1H), 1,66-1,37 (m, 2H), 1,37-1,12 (m, 5H), 0,93-0,72 (m, 3H).
[0640] Соединение 62 (9,1 мг, выход: 38,6%, белое твердое вещество) было получено, как описано в примере 15, из соответствующих промежуточных соединений, 23А и 62F. Соединение 62: 1H ЯМР (CDCl3, 400 МГц): δ 7,84-7,67 (m, 2H), 7,57-7,43 (m, 3H), 6,73 (s, 1H), 6,17-6,03 (m, 1H), 5,52-5,29 (m, 2H), 2,46 (s, 3H), 1,99-1,84 (m, 1H), 1,41-1,04 (m, 5H), 0,90-0,78 (m, 3H). МС (ИЭР) m/z (M+H)+ 344,1.
ПРИМЕР 35 СОЕДИНЕНИЯ 63, 454
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРАЗИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (63)
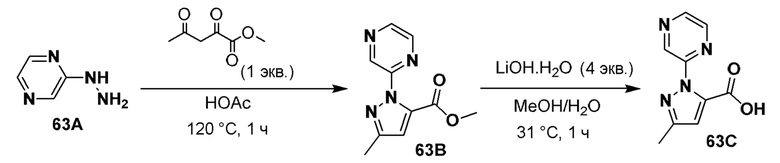
[0641] К раствору соединения метил-2,4-диоксопентаноата (100 мг, 693,82 мкмоль) в АсОН (20 мл) добавляли соединение 63А (76,4 мг, 693,82 мкмоль). Смесь перемешивали при 120°С в течение 1 часа. Смесь выливали в ДХМ (5 мл). Органический слой промывали водой (10 мл), NaHCO3 до рН ~ 8~9 и сушили над Na2SO4 и концентрировали с получением соединения 63 В (500 мг, выход 25,24%) в виде белого твердого вещества.
[0642] К раствору соединения 63 В (61 мг, 279,55 мкмоль) в МеОН (6 мл) и H2O (1 мл) добавляли LiOH⋅H2O (46,9 мг, 1,12 ммоль). Смесь перемешивали при 31°С в течение 1 ч. МеОН выпаривали. К остатку добавляли воду (10 мл), и смесь подвергали экстракции МТБЭ (5 мл) и разделяли. Водный слой подкисляли до рН ~ 3 с помощью 1 н. HCl и подвергали экстракции этилацетатом (3×20 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного продукта (50 мг, выход 87,59%) в виде белого твердого вещества.
[0643] Соединение 63 (25,1 мг, выход 63,1%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 63С.Соединение 63: 1Н ЯМР (400 МГц, CDCl3) δ 9,16 (s, 2Н), 9,08-8,97 (m, 1H), 8,44 (s, 1H), 7,95 (s, 1H), 7,20 (s, 3Н), 7,08 (s, 2Н), 5,78 (m, 1H), 5,54 (s, 1H), 3,45 (m, 1H), 3,38-3,24 (m, 1H), 2,36 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 379,1.
N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРАЗИН-2-ИЛ)-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (454)
[0644] Соединение 454 (210 мг, выход 91,7%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей карбоновой кислоты, промежуточного соединения 63С, и гидрохлорида 3-амино-N-циклопропил-2-гидрокси-4-фенилбутанамида. Соединение 454: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,07 (d, J=7,2 Гц, 1H), 8,84-8,77 (m, 2Н), 8,57 (d, J=2,8 Гц, 1H), 8,31-8,27 (m, 1H), 7,30-7,17 (m, 5Н), 6,64 (s, 1H), 5,33-5,25 (m, 1H), 3,17-3,09 (m, 1H), 2,83-2,70 (m, 2Н), 2,27 (s, 3Н), 0,69-0,53 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 419,2.
ПРИМЕР 36
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(3-МЕТИЛПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД(66)
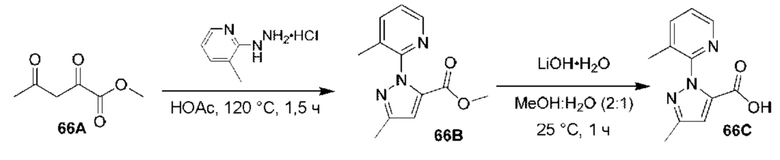
[0645] Смесь соединения гидрохлорида 2-гидразинил-3-метилпиридина (2 г, 12,53 ммоль) и соединения 66А (1,81 г, 12,53 ммоль) в АсОН (30 мл) дегазировали и продували N2 3 раза и затем перемешивали при 120°С в течение 1,5 ч в атмосфере N2. Полученную смесь концентрировали при пониженном давлении для удаления АсОН и разбавляли ДХМ (10 мл), нейтрализовали с помощью насыщенного водного NaHCO3. Смесь подвергали экстракции ДХМ (20 мл × 3), и объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством флэш-хроматографии на силикагеле (петролейный эфир : этилацетат = от 1:0 до 0:1) с получением соединения 66В (800,0 мг, выход 27,6%) в виде белого твердого вещества и соединения 66В-1 (110,0 мг, выход 4,04%) в виде белого твердого вещества и неочищенного 66В-1 (~800,0 мг).
[0646] Соединение 66В: Метил-3-метил-1-(3-метилпиридин-2-ил)-1Н-пиразол-5-карбоксилат: 1Н ЯМР (CDCl3, 400 МГц) δ 8,42-8,37 (m, 1H), 7,70 (d, J=7,6 Гц, 1H), 7,33 (d, J=7,6 Гц, 1H), 6,79 (s, 1H), 3,74 (s, 3Н), 2,38 (s, 3Н), 2,14 (s, 3Н).
[0647] Соединение 66В-1: Метил-5-метил-1-(3-метилпиридин-2-ил)-1Н-пиразол-3-карбоксилат: 1Н ЯМР (CDCl3, 400 МГц) δ 8,42 (d, J=3,6 Гц, 1H), 7,72 (d, J=6,8 Гц, 1H), 7,34 (d, J=7,6 Гц, 1H), 6,74 (s, 1H), 3,92 (s, 3Н), 2,26 (s, 3Н), 2,20 (s, 3Н).
[0648] К смеси соединения 66В (200,0 мг, 864,86 мкмоль) в МеОН (10 мл) и Н2О (5 мл) добавляли LiOH⋅Н2О (145,2 мг, 3,46 ммоль) одной порцией. После перемешивания при 25°С в течение 1 часа реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли Н2О (10 мл), доводили рН до ~3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (40 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 66С (150 мг, выход 79,84%, белое твердое вещество). Соединение 66С: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 13,11 (ушир. s, 1H), 8,31 (d, J=3,7 Гц, 1H), 7,84 (d, J=7,3 Гц, 1H), 7,47-7,40 (m, 1H), 6,78 (s, 1H), 2,25 (s, 3Н), 2,03 (s, 3Н). МС (ИЭР) m/z (М+1)+ 218,1.
[0649] Соединение 66 (24,5 мг, выход 54,7%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 66С. Соединение 66: 1Н ЯМР (CDCl3, 400 МГц) δ 8,23 (d, J=3,6 Гц, 1H), 7,67 (d, J=7,2 Гц, 1H), 7,34 (d, J=7,2 Гц, 1H), 7,27 (ушир. s, 1H), 7,25-7,21 (m, 3Н), 7,04-6,99 (m, 2Н), 6,70 (ушир. s, 1H), 6,57 (s, 1H), 5,65-5,6 (m, 1H), 5,57 (ушир. s, 1H), 3,37-3,29 (m, 1H), 3,2-3,14 (m, 1H), 2,34 (s, 3Н), 2,17 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 392,2.
ПРИМЕР 37
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(1-ФЕНИЛ-1H-ПИРАЗОЛ-3-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (68)
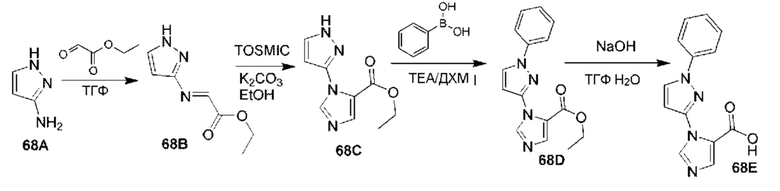
[0650] К раствору 68А (15 г, 181 ммоль) в ТГФ (200 мл) добавляли этил-2-оксоацетат (47,9 г, 235 ммоль). Смесь перемешивали при 25°С в течение 0,5 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 68В (55,3 г, неочищенного) в виде коричневого твердого вещества. МС (ИЭР) m/z (М+Н)+ 167,8.
[0651] К раствору 68В (40 г, 239 ммоль) в EtOH (400 мл) добавляли K2CO3 (50 г, 362 ммоль) и TosMIC (40 г, 204,88 ммоль). Смесь перемешивали при 90°С в течение 0,5 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 1:0 до 5:2) с получением соединения 68С (12 г, выход: 24,3%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 11,80-11,35 (m, 1H), 7,87 (d, J=1,10 Гц, 1H), 7,84 (d, J=1,10 Гц, 1H), 7,58 (d, J=2,43 Гц, 1H), 6,45 (d, J=2,43 Гц, 1H), 4,25 (q, J=7,06 Гц, 2Н), 1,29 (t, J=7,17 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 207,0.
[0652] Смесь 68С (5 г, 24,3 ммоль), фенилбороновой кислоты (4,4 г, 36,4 ммоль), Cu(ОАс)2 (4,4 г, 24,3 ммоль), TEA (7,4 г, 72,8 ммоль) в ДХМ (200 мл) дегазировали и продували О2 3 раза, и затем смесь перемешивали при 25°С в течение 10 часов в атмосфере О2. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 1:0 до 2:1). Соединение 68D (2,3 г, выход: 33,6%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,04-7,94 (m, 2Н), 7,87 (s, 1H), 7,71 (ушир. d, J=7,7 Гц, 2Н), 7,49 (ушир. t, J=7,1 Гц, 2Н), 7,36 (ушир. d, J=7,1 Гц, 1H), 7,27 (d, J=2,0 Гц, 2H), 6,70-6,61 (m, 1H), 4,29 (dd, J=2,1, 7,2 Гц, 2H), 1,38-1,22 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 282,9.
[0653] К раствору 68D (2,5 г, 8,86 ммоль) в ТГФ (30 мл) и H2O (6 мл) добавляли NaOH (708 мг, 17,7 ммоль). Смесь перемешивали при 80°С в течение 1,5 часа. Реакционную смесь концентрировали при пониженном давлении для удаления ТГФ и затем промывали EtOAc (20 мл). Водный слой подкисляли с помощью 1 М HCl до рН ~5 и затем подвергали экстракции EtOAc (30 мл × 3). Объединенный органический слой промывали H2O (40 мл), солевым раствором (40 мл), сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного промежуточного соединения 68Е (1,90 г, выход: 84,3%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,62 (d, J=2,6 Гц, 1H), 8,19 (s, 1H), 7,86 (d, J=7,9 Гц, 2Н), 7,76 (s, 1H), 7,53 (t, J=7,9 Гц, 2Н), 7,39-7,31 (m, 1H), 6,77 (d, J=2,6 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 254,9.
[0654] Соединение 68 (33,5 мг, выход: 42,1%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 68Е. Соединение 68: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,86 (d, J=7,7 Гц, 1H), 8,51 (d, J=2,6 Гц, 1H), 8,10 (d, J=0,9 Гц, 1H), 8,06 (s, 1H), 7,79 (dd, J=8,7, 1,0 Гц, 3Н), 7,60 (d, J=0,9 Гц, 1H), 7,46-7,53 (m, 2Н), 7,30-7,36 (m, 1H), 7,24-7,29 (m, 4Н), 7,16-7,23 (m, 1H), 6,44 (d, J=2,6 Гц, 1H), 5,23-5,32 (m, 1H), 3,17 (dd, J=13,8, 3,9 Гц, 1H), 2,83 (dd, J=13,9, 10,4 Гц, 1 Н). МС (ИЭР) m/z (М+Н)+ 429,1.
ПРИМЕР 38
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(1H-ИНДАЗОЛ-3-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (69)
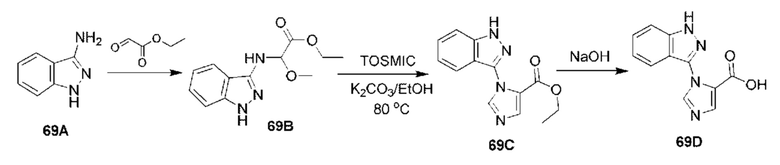
[0655] К раствору 69А (8,7 г, 65,3 ммоль) в МеОН (90 мл) добавляли этил-2-оксоацетат (20 г, 98,01 ммоль). Смесь перемешивали при 25°С в течение 2 часов. Смесь фильтровали и концентрировали с получением промежуточного соединения 69В (15 г, неочищенного) в виде коричневого твердого вещества.
[0656] К раствору 69В (15 г, 69,1 ммоль) в EtOH (400 мл) добавляли K2CO3 (14,5 г, 104 ммоль) и TosMIC (11,6 г 59,4 ммоль). Смесь перемешивали при 90°С в течение 0,5 часа. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 1:0 до 1:1) с получением соединения 69С (2,9 г, выход: 16,4%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 11,04 (ушир. s, 1H), 7,98 (d, J=0,7 Гц, 1H), 7,91 (s, 1H), 7,48-7,41 (m, 3Н), 7,25-7,19 (m, 1H), 4,24-4,14 (m, 2Н), 1,14 (t, J=7,1 Гц, 3Н).
[0657] К раствору 69С (2,9 г, 11,3 ммоль) в ТГФ (40 мл) и H2O (8 мл) добавляли NaOH (905 мг, 22,6 ммоль). Смесь перемешивали при 25°С в течение 10 часов. Смесь концентрировали при пониженном давлении для удаления органического растворителя и подвергали экстракции EtOAc (20 мл). Водный слой подкисляли с помощью 1 М HCl до рН ~5 и затем подвергали экстракции EtOAc (30 мл × 3). Объединенный органический слой промывали H2O (40 мл), солевым раствором (40 мл), сушили над Na2SO4, фильтровали и концентрировали с получением 69D (1,5 г, выход: 58,1%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,35 (s, 1H), 8,16 (s, 1H), 7,83 (s, 1H), 7,61 (d, J=8,3 Гц, 1H), 7,47-7,41 (m, 2Н), 7,20-7,15 (m, 1H). МС (ИЭР) m/z (М+Н)+ 228,9.
[0658] Соединение 69 (16,7 мг, выход: 20,9%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 69D. Соединение 69: 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,16 (ушир. s, 1H), 8,84 (ушир. d, J=7,7 Гц, 1H), 8,08-7,95 (m, 2Н), 7,80-7,70 (m, 2Н), 7,52 (d, J=8,4 Гц, 1H), 7,36 (ушир. t, J=7,5 Гц, 1H), 7,30-7,23 (m, 4Н), 7,22-7,13 (m, 3Н), 7,08-7,01 (m, 1H), 5,21-5,12 (m, 1H), 3,17-3,09 (m, 1H), 2,84-2,75 (m, 1H). МС (ИЭР) m/z (М+Н)+ 403,1.
ПРИМЕР 39
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(1H-БЕНЗО[d]ИМИДАЗОЛ-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (70)
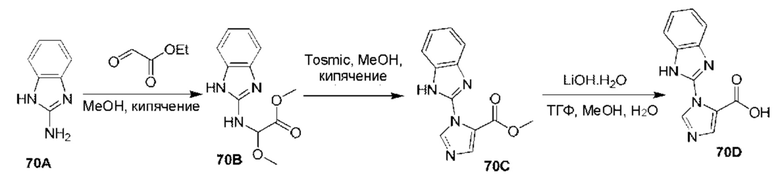
[0659] Смесь 70А (10 г, 75,1 ммоль), этил-2-оксоацетата (30,6 г, 150 ммоль) в МеОН (300 мл) перемешивали при 70°С в течение 12 часов в атмосфере N2. Анализ посредством ЖХМС свидетельствовал о том, что 70А полностью израсходовалось, и был обнаружен один пик, которому соответствовал МС целевого соединения. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта 70В (15 г, неочищенного) в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 235,9.
[0660] Смесь 70В (15 г, 63,7 ммоль), K2CO3 (13,2 г, 95,6 ммоль), TosMIC (24,9 г, 127 ммоль) в МеОН (300 мл) перемешивали при 70°С в течение 1 часа. Анализ посредством ЖХМС свидетельствовал о том, что 70В полностью израсходовалось, и был обнаружен один небольшой пик, которому соответствовал МС целевого соединения. Анализ посредством ТСХ (петролейный эфир : этилацетат = 1:1, Rf ~ 0,3) свидетельствовал о том, что 70В полностью израсходовалось, и на хроматограмме появилось одно новое пятно. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (петролейный эфир/этилацетат = от 10/1 до 1:1) с получением 70С (350 мг, выход: 2,3%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 12,52 (ушир. s, 1H) 8,96 (s, 1H) 7,98 (s, 1H) 7,69 (ушир. d, J=4,4 Гц, 1H) 7,48 (ушир. s, 1H) 7,28 (ушир. dd, J=5,8, 2,8 Гц, 2Н) 3,99 (s, 3 Н). МС (ИЭР) m/z (М+Н)+ 243,1.
[0661] Смесь 70С (350 мг, 1,44 ммоль), LiOH⋅H2O (120 мг, 2,88 ммоль) в ТГФ (5 мл), Н2О (2 мл) перемешивали при 25°С в течение 4 часов. Анализ посредством ЖХМС свидетельствовал о том, что 70С полностью израсходовалось, и был обнаружен один пик, которому соответствовал МС целевого соединения. В реакционную смесь добавляли водный раствор HCl (1 М) для доведения рН до ~5, фильтровали, и осадок на фильтре концентрировали при пониженном давлении. Осадок на фильтре промывали водой. Соединение 70D (230 мг, выход: 70,1%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,57 (d, J=1,3 Гц, 1H) 7,38-7,63 (m, 3Н) 7,03-7,22 (m, 1H) 7,03-7,17 (m, 1 Н). МС (ИЭР) m/z (М+Н)+ 229,0.
[0662] Соединение 70 (40 мг, выход: 46,7%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 70D. Соединение 70: 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,94 (ушир. s, 1H), 9,31 (ушир. s, 1H), 8,38-8,23 (m, 1H), 8,04 (ушир. s, 1H), 7,87-7,74 (m, 2Н), 7,60-7,43 (m, 2H), 7,29-7,10 (m, 6H), 5,33 (ушир. t, J=6,6 Гц, 1H), 3,17 (ушир. dd, J=3,1, 13,9 Гц, 1H), 3,22-3,09 (m, 1H), 2,84 (ушир. dd, J=10,3, 13,8 Гц, 1H), 2,91-2,74 (m, 1H). МС (ИЭР) m/z (М+Н)+ 403,1.
ПРИМЕР 40
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФЕНИЛ-1,2,5-ТИАДИАЗОЛ-3-КАРБОКСАМИД (72)
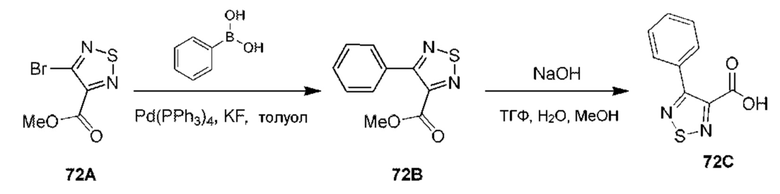
[0663] К смеси соединения 72А (800 мг, 3,59 ммоль) и фенилбороновой кислоты (1,31 г, 10,8 ммоль) в толуоле (10 мл) и Н2О (500 мкл) добавляли KF (417 мг, 7,18 ммоль) и Pd(PPh3)4 (414 мг, 359 мкмоль) в атмосфере N2. Затем реакционную смесь перемешивали при 100°С в атмосфере N2 в течение 16 ч. Растворитель выпаривали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 20:1 до 5:1) с получением соединения 72В (400 мг, неочищенного) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 7,70-7,68 (m, 2Н), 7,48-7,46 (m, 3Н), 3,94 (s, 3Н).
[0664] К раствору соединения 72В (500 мг, 2,27 ммоль) в ТГФ (5 мл), Н2О (5 мл) и МеОН (5 мл) добавляли NaOH (182 мг, 4,54 ммоль). Реакционную смесь перемешивали при 20°С в течение 1 ч. В реакционную смесь добавляли 1 М HCl до рН ~4. Растворитель выпаривали с получением неочищенного продукта 72С (500 мг, неочищенного) в виде белого твердого вещества. Неочищенный продукт использовали на следующей стадии без очистки.
[0665] Соединение 72 (50,5 мг, выход: 43,9%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 72С. Соединение 72: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,42 (d, J=7,7 Гц, 1H), 8,18 (s, 1H), 7,91 (s, 1H), 7,60-7,55 (m, 2Н), 7,48-7,43 (m, 1H), 7,42-7,36 (m, 2Н), 7,31-7,22 (m, 5Н), 5,51 (ddd, J=3,6, 7,7, 10,0 Гц, 1H), 3,23 (dd, J=3,6, 14,0 Гц, 1H), 2,87 (dd, J=10,1, 14,1 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 381,0.
ПРИМЕР 41
N-((3S,4R)-1-АМИНО-4-МЕТИЛ-1,2-ДИОКСОГЕКСАН-3-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (73)
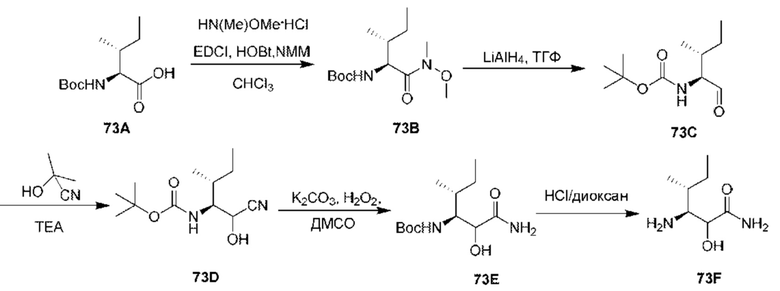
[0666] Смесь N-метоксиметанамина (2,32 г, 23,78 ммоль), соединения 73А (5,00 г, 21,62 ммоль), HOBt (2,92 г, 21,62 ммоль) и NMM (6,56 г, 64,86 ммоль) в CHCl3 (100 мл) дегазировали и продували N2 3 раза при 0°С, затем добавляли порциями EDCI (6,22 г, 32,43 ммоль). Смесь перемешивали при 25°С в течение 16 ч в атмосфере N2. В реакционную смесь добавляли для гашения реакции Н2О (100 мл). Органические слои промывали HCl (1 н., 100 мл × 2), и насыщенным NaHCO3 (100 мл × 2), и насыщенным солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (элюент градиент 0~10% этилацетат/петролейный эфир) с получением соединения 73В (5,0 г, выход: 84,3%) в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 5,10 (d, J=9,5 Гц, 1H), 4,67-4,53 (m, 1H), 3,76 (s, 3Н), 3,20 (s, 3Н), 1,70 (qt, J=6,8, 9,9 Гц, 1H), 1,54-1,51 (m, 1H), 1,41 (s, 9Н), 1,15-1,07 (m, 1H), 0,91-0,85 (m, 6Н). МС (ИЭР) m/z (M+Na)+ 296,9.
[0667] К раствору LiAlH4 (350 мг, 9,22 ммоль) в ТГФ (30 мл) добавляли раствор соединения 73В (2,30 г, 8,38 ммоль) в ТГФ (30 мл) при 0°С. После добавления реакционную смесь перемешивали в течение 1 ч при 5°С. В реакционную смесь добавляли для гашения реакции этилацетат (10 мл) и HCl (1 н., 10 мл), и смесь подвергали экстракции EtOAc (100 мл × 2). Объединенные органические слои промывали HCl (1 н., 30 мл × 2), насыщ. NaHCO3 (30 мл × 3) и солевым раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 73С (1,50 г, выход: 83,2%) в виде бесцветного масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,45 (d, J=1,3 Гц, 1H), 7,22 (ушир. d, J=7,5 Гц, 1H), 3,79 (ушир. t, J=6,4 Гц, 1H), 1,89-1,75 (m, 1H), 1,42-1,32 (m, 10Н), 1,25-1,10 (m, 1H), 0,86 (d, J=6,8 Гц, 3Н), 0,81 (t, J=7,4 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 216,0.
[0668] К раствору соединения 73С (1,5 г, 6,97 ммоль) в ДХМ (10 мл) добавляли 2-гидрокси-2-метилпропаннитрил (1,28 мл, 13,93 ммоль) и TEA (1,16 мл, 8,36 ммоль) и затем перемешивали при 25°С в течение 14 часов. Реакционную смесь разбавляли ДХМ (25 мл), промывали HCl (1 н., 20 мл × 2), Н2О (30 мл) и солевым раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 73D (1,5 г, выход: 88,8%) в виде бесцветной жидкости. 1Н ЯМР (400 МГц, CDCl3) δ 5,24-5,08 (m, 1H), 4,92-4,56 (m, 1H), 3,90-3,25 (m, 1H), 2,04-1,80 (m, 1H), 1,66-1,52 (m, 1H), 1,50-1,40 (m, 9Н), 1,33-1,09 (m, 2Н), 1,02-0,75 (m, 6Н).
[0669] К раствору соединения 73D (1,50 г, 6,19 ммоль) и K2CO3 (1,71 г, 12,38 ммоль) в ДМСО (15 мл) добавляли Н2О2 (7,21 г, 211,95 ммоль) при 0°С. После добавления реакционную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь разбавляли водой (20 мл) и медленно добавляли в нее для гашения реакции насыщенный водный Na2S2O3 при охлаждении ледяной водой. Смесь подвергали экстракции EtOAc (50 мл × 3), и объединенные экстракты промывали насыщенным водным Na2S2O3 (30 мл × 3). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (элюент градиент 0~20% этилацетат/петролейный эфир) с получением соединения 73Е (870 мг, выход: 54,0%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,31-6,97 (m, 2Н), 6,29-5,87 (m, 1H), 5,44-5,12 (m, 1H), 3,99-3,80 (m, 1H), 3,71-3,50 (m, 1H), 1,67-1,41 (m, 2Н), 1,39-1,30 (m, 9Н), 1,11-0,92 (m, 1H), 0,89-0,75 (m, 6Н). МС (ИЭР) m/z (M+Na)+ 282,9.
[0670] К раствору соединения 73Е (870 мг, 3,34 ммоль) в EtOAc (10 мл) добавляли HCl/EtOAc (4 М, 16,70 мл) при 0°С. После добавления реакционную смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток промывали МТБЭ (30 мл), фильтровали с получением соединения 73F (620 мг, выход: 94,4%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,14-7,71 (m, 3Н), 7,64-7,37 (m, 2Н), 6,57-6,28 (m, 1H), 4,32-3,99 (m, 1H), 3,21 (ушир. s, 1H), 1,82-1,43 (m, 2Н), 1,30-1,03 (m, 1H), 0,99-0,71 (m, 6Н). МС (ИЭР) m/z (М+Н)+ 161,1.
[0671] Соединение 73 (100 мг, выход: 63,6%, белое твердое вещество) было получено, как описано в примере 15, из соответствующих промежуточных соединений, 23А и 73F. Соединение 73: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,92 (d, J=7,0 Гц, 1H), 8,15 (s, 1H), 7,96-7,65 (m, 3Н), 7,62-7,44 (m, 3Н), 5,19 (t, J=6,5 Гц, 1H), 3,33 (ушир. s, 1H), 2,30 (s, 3Н), 2,10-1,94 (m, 1H), 1,36-1,13 (m, 2Н), 0,92 (d, J=6,8 Гц, 3Н), 0,81 (t, J=7,4 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 344,1.
ПРИМЕР 42
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(5-ФЕНИЛПИРИМИДИН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (74)

[0672] Смесь соединения 74А (10,0 г, 57,47 ммоль), фенилбороновой кислоты (10,5 г, 86,21 ммоль), K3PO4 (24,4 г, 114,94 ммоль), Pd(OAc)2 (1,3 г, 5,75 ммоль) в этиленгликоле (200 мл) перемешивали при 80°С в течение 12 ч. Реакционную смесь добавляли в Н2О (200 мл), нерастворимое вещество удаляли путем фильтрования; фильтрат подвергали экстракции этилацетатом (200 мл × 3). Объединенный органический слой промывали насыщенным водным NaHCO3 (150 мл × 3), насыщенным водным NaCl (150 мл × 3), сушили над Na2SO4 и концентрировали под вакуумом. Полученное твердое вещество обрабатывали этилацетатом (10 мл). Осадок фильтровали и сушили под вакуумом с получением соединения 74В (4,97 г, выход: 50,5%) в виде светло-желтого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,55 (s, 2Н), 7,62-7,58 (m, 2Н), 7,43-7,40 (m, 2Н), 7,33-7,27 (m, 1H), 6,76 (ушир. s, 2Н).
[0673] Смесь соединения 74В (3,0 г, 17,35 ммоль) и соединения этил-2-оксоацетата (2,3 г, 22,55 ммоль) в МеОН (50 мл) перемешивали при 80°С в течение 12 ч. Реакционную смесь концентрировали, и твердое вещество отфильтровывали. Полученное твердое вещество обрабатывали МеОН (10 мл), отфильтровывали и сушили под вакуумом с получением соединения 74С (3,23 г, выход: 64,8%) в виде светло-желтого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,71 (s, 2Н), 8,24 (d, J=8,8 Гц, 1H), 7,67-7,63 (m, 2Н), 7,47-7,41 (m, 2Н), 7,37-7,31 (m, 1H), 5,64 (d, J=8,8 Гц, 1H), 4,19-4,10 (m, 2Н), 3,33 (s, 3Н), 1,21 (t, J=7,2 Гц, 3Н).
[0674] Смесь соединения 74С (500 мг, 1,74 ммоль), Tosmic (680 мг, 3,48 ммоль), K2CO3 (720 мг, 5,22 ммоль) в абсолютном EtOH (50 мл) перемешивали при 65°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении; полученный остаток добавляли в воду (30 мл) и подвергали экстракции этилацетатом (30 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 15:1 до 8:1) с получением соединения 4 (293 мг, выход: 52,2%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,30 (s, 2Н), 8,46 (s, 1H), 7,89 (t, J=7,2 Гц, 2Н), 7,76 (s, 1H), 7,59-7,49 (m, 3Н), 4,23-4,13 (m, 2Н), 1,17 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 295,1.
[0675] К смеси соединения 74D (1,15 г, 3,91 ммоль) в ТГФ (10 мл) и МеОН (10 мл) добавляли KOH (2 М, 1,96 мл, 3,92 ммоль) по каплям при 25°С. Смесь перемешивали при 25°С в течение 23 ч и затем концентрировали при пониженном давлении с получением промежуточного соединения 74Е (2 г, неочищенного).
[0676] Соединение 74 (14,9 мг, выход 28,2%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих промежуточных соединений 74Е и 12G. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,60 (ушир. d, J=6,4 Гц, 1H), 8,64 (ушир. s, 3Н), 7,82 (s, 1H), 7,59-7,50 (m, 6Н), 7,22-7,11 (m, 5Н), 5,83 (m, 1H), 5,61 (ушир. s, 1H), 3,52-3,44 (m, 1H), 3,40-3,31 (m, 1H). МС (ИЭР) m/z (М+Н)+ 441,0.
ПРИМЕР 43
СОЕДИНЕНИЯ 77, 88
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4-(ОКСАЗОЛ-2-ИЛ)ПИРИДИН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (77)
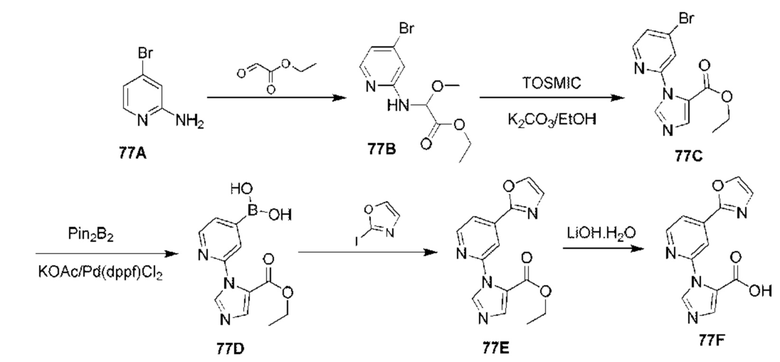
[0677] Смесь 77А (20 г, 115,60 ммоль) и этил-2-оксоацетата (30,7 г, 150,28 ммоль) в МеОН (300 мл) нагревали до 80°С в течение 3 ч. В анализе посредством ЖХМС был обнаружен МС целевого соединения. В анализе посредством ТСХ (петролейный эфир : этилацетат = 3:1, Rf ~ 0,8) было обнаружено новое пятно, смесь концентрировали и остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 20:1). Соединение 77В (28,9 г, выход 86,5%, желтое твердое вещество): 1Н ЯМР (400 МГц, CDCl3) δ 7,96 (d, J=5,2 Гц, 1H), 6,86 (dd, J=5,2, 1,75 Гц, 1H), 6,77 (d, J=1,3 Гц, 1H), 5,75 (ушир. s, 1H), 5,61 (d, J=8,3 Гц, 1H), 4,29 (q, J=7,0 Гц, 2Н), 3,41 (s, 3Н), 1,37-1,31 (m, 3Н).
[0678] Смесь 77В (15 г, 51,9 ммоль) и K2CO3 (21,5 г, 156 ммоль) в EtOH (300 мл) перемешивали при 80°С в течение 0,5 ч, затем добавляли TosMIC (15,2 г, 77,82 ммоль), полученную смесь перемешивали при 80°С в течение еще 2 ч. В анализе посредством ЖХМС был обнаружен МС целевого соединения, большую часть этанола удаляли, и происходило образование осадка, твердое вещество отфильтровывали и промывали водой (100 мл × 2), твердое вещество сушили и концентрировали с получением 77С (6,4 г, выход: 41,7%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,38 (d, J=5,2 Гц, 1H), 7,97 (s, 1H), 7,85 (s, 1H), 7,61 (s, 1H), 7,56 (dd, J=5,26 1,3 Гц, 1H), 4,27 (q, J=7,02 Гц, 2Н), 1,29 (t, J=7,02 Гц, 3Н).
[0679] Соединение 77С (3 г, 10,13 ммоль), Pin2B2 (2,57 г, 10,13 ммоль), KOAc (2,98 г, 30,4 ммоль) и Pd(dppf)Cl2 (741 мг, 1,01 ммоль) в диоксане (100 мл) дегазировали и затем нагревали при 70°С в течение 4 часов в атмосфере N2. В анализе посредством ЖХМС был обнаружен МС целевого соединения. Анализ посредством ТСХ (этилацетат : метанол = 10:1, Rf ~ 0), смесь фильтровали, и фильтрат концентрировали, остаток очищали посредством хроматографии на силикагеле (ДХМ : метанол = 5:1) с получением 77D (1,70 г, неочищенного) в виде черного твердого вещества.
[0680] Соединение 77D (300 мг, 1,15 ммоль), 2-иодоксазол (157 мг, 805,00 мкмоль), Pd(dppf)Cl2 (84,1 мг, 115,00 мкмоль) и Na2CO3 (244 мг, 2,30 ммоль) в толуоле (2 мл), EtOH (2 мл), Н2О (1 мл) дегазировали и затем нагревали до 120°С в течение 1 ч в условиях микроволнового излучения. В анализе посредством ЖХМС был обнаружен МС целевого соединения, смесь добавляли в воду (5 мл) и подвергали экстракции этилацетатом (10 мл × 2), органические фазы сушили и концентрировали, остаток очищали посредством препаративной ТСХ (петролейный эфир : этилацетат = 1:1) с получением 77Е (80 мг, выход: 24,5%) в виде желтого твердого вещества.
[0681] Смесь 77Е (80 мг, 281,42 мкмоль) и LiOH⋅H2O (17,7 мг, 422,13 мкмоль) в ТГФ (5 мл), Н2О (1 мл) перемешивали при 25°С в течение 12 ч. В анализе посредством ЖХМС был обнаружен МС целевого соединения, ТГФ удаляли под вакуумом, водный слой подвергали экстракции этилацетатом (10 мл × 2), рН водного слоя доводили до ~6 с помощью 1 н. HCl, и слой лиофилизировали, остаток очищали посредством препаративной ВЭЖХ (ТФУ) с получением 77F (35 мг, выход: 48,5%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, метанол-d4) δ 8,70 (d, J=5,3 Гц, 1H), 8,25 (s, 1H), 8,16 (s, 1H), 8,11 (s, 1H), 8,08 (d, J=5,1 Гц, 1H), 7,81 (s, 1H), 7,46 (s, 1H).
[0682] Соединение 77 (38,4 мг, выход: 64,3%, белое твердое вещество) было получено, как описано в примере 41, из соответствующей карбоновой кислоты, промежуточного соединения 77F. Соединение 77: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,93 (d, J=7,5 Гц, 1H), 8,74 (d, J=5,1 Гц, 1H), 8,60 (d, J=5,7 Гц, 1H), 8,39 (d, J=0,7 Гц, 1H), 8,22 (d, J=0,7 Гц, 1H), 7,94 (dd, J=1,4, 5,2 Гц, 1H), 7,82 (s, 1H), 7,65 (d, J=0,7 Гц, 1H), 7,54 (d, J=0,7 Гц, 1H), 7,28 (d, J=4,4 Гц, 4Н), 7,20 (qd, J=4,2, 8,5 Гц, 1H), 5,30-5,22 (m, 1H), 3,16 (dd, J=3,9, 13,8 Гц, 1H), 2,85 (dd, J=10,1, 13,9 Гц, 1H), 2,77-2,68 (m, 1H), 0,67-0,59 (m, 2Н), 0,58-0,50 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 471,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4-(ОКСАЗОЛ-2-ИЛ)ПИРИДИН-2-ИЛ)-1Н-ИМИДАЗОЛ-5-КАРБОКСАМИД (88)
[0683] Соединение 88 (18,5 мг, выход: 46,5%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 77F. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,93 (d, J=7,5 Гц, 1H), 8,58 (d, J=5,3 Гц, 1H), 8,37 (s, 1H), 8,21 (s, 1H), 8,01 (ушир. s, 1H), 7,91 (d, J=5,1 Гц, 1H), 7,79 (s, 2Н), 7,62 (s, 1H), 7,52 (s, 1H), 7,25 (d, J=4,2 Гц, 4Н), 7,18 (ушир. dd, J=4,5, 8,7 Гц, 1H), 5,25-5,17 (m, 1H), 3,14 (dd, J=3,6, 14,0 Гц, 1H), 2,83 (dd, J=10,5, 13,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 431,1.
ПРИМЕР 44
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ХИНОЛИН-5-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (78)
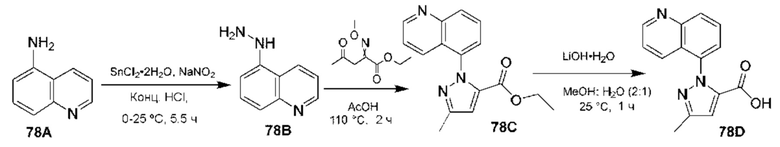
[0684] К смеси, состоящей из соединения 78А (1,0 г, 6,94 ммоль) в конц. HCl (4,00 мл) при 0°С добавляли по каплям NaNO2 (526,8 мг, 7,63 ммоль), и полученную смесь перемешивали при 0°С в течение 0,5 часа. Реакционную смесь нагревали до 25°С в течение более 0,5 часа и затем охлаждали до 0°С. В реакционную смесь добавляли по каплям SnCl2⋅2H2O (3,13 г, 13,88 ммоль, в 1,2 мл конц. HCl) и перемешивали при 0°С в течение 0,5 часа. Полученную смесь оставляли нагреваться до комнатной температуры с интенсивным перемешиванием в течение более 4 часов и затем концентрировали при пониженном давлении для удаления растворителя. Остаток фильтровали, и осадок на фильтре промывали этанолом (30 мл × 3) и затем сушили при пониженном давлении с получением соединения 78В (700,0 мг, выход 51,55%) в виде желтого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,95 (ушир. s, 1H), 9,25-9,13 (m, 2Н), 8,04-7,95 (m, 2Н), 7,88 (d, J=8,8 Гц, 1H), 7,26 (d, J=7,6 Гц, 1H), 7,28-7,23 (m, 1H).
[0685] Смесь соединения 78В (500 мг, 3,14 ммоль) и соединения этил-2-(метоксиимино)-4-оксопентаноата (588 мг, 3,14 ммоль) в АсОН (1 мл) дегазировали и продували N2 3 раза, и затем смесь перемешивали при 110°С в течение 2 ч в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении для удаления АсОН. Остаток разбавляли CH2Cl2 (100 мл), доводили рН до ~7-8 с помощью насыщенного водного NaHCO3 и затем подвергали экстракции CH2Cl2 (40 мл × 2). Органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1:0 до 1:0) с получением соединения 78С (200 мг, выход 22,6%) в виде желтого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц) δ 8,93 (d, J=4,0 Гц, 1H), 8,23 (d, J=8,4 Гц, 1H), 7,78 (t, J=8,0 Гц, 1H), 7,64 (d, J=8,4 Гц, 1H), 7,56 (d, J=7,2 Гц, 1H), 7,38 (d, J=8,8 Гц, 1H), 6,93 (s, 1H), 4,05 (q, J=7,2 Гц, 2Н), 2,41 (s, 3Н), 1,00 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+1)+ 282,0.
[0686] Промежуточное соединение 78D (135 мг, выход 74,98%, белое твердое вещество) было получено, как описано в примере 85, из соединения 78С. Соединение 78D: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,97 (d, J=4,0 Гц, 1H), 8,17 (d, J=8,4 Гц, 1H), 7,89-7,82 (m, 1H), 7,67-7,52 (m, 3Н), 6,97 (s, 1H), 2,32 (s, 3Н). МС (ИЭР) m/z (М+1)+ 253,9.
[0687] Соединение 78 (8,8 мг, выход 16,77%, желтое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 78D. Соединение 78: 1Н ЯМР (CDCl3, 400 МГц): δ 8,95 (d, J=4,0 Гц, 1H), 8,22 (d, J=8,4 Гц, 1H), 7,76-7,66 (m, 2Н), 7,49 (d, J=6,4 Гц, 1H), 7,38 (d, J=8,4 Гц, 1H), 7,24-7,16 (m, 3Н), 6,87 (d, J=7,6 Гц, 2Н), 6,79 (ушир. s, 1H), 6,64 (s, 1H), 6,33 (d, J=7,2 Гц, 1H), 5,49-5,42 (m, 1H), 3,27-3,19 (m, 1H), 3,08-2,98 (m, 1H), 2,78-2,69 (m, 1H), 0,90-0,83 (m, 2Н), 0,61-0,50 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 468,1.
ПРИМЕР 45
СОЕДИНЕНИЯ 79, 146, 160, 264
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-5-ФЕНИЛ-1H-ИМИДАЗОЛ-4-КАРБОКСАМИД (79)

[0688] Раствор соединения 79А (500 мг, 3,96 ммоль) в HCl/MeOH (4 М, 50 мл) перемешивали при 80°С в течение 12 ч. Растворитель удаляли под вакуумом. рН остатка доводили до ~8 с помощью насыщенного водного NaHCO3. Раствор подвергали экстракции EtOAc (100 мл × 3). Органические фазы собирали, сушили с помощью Na2SO4, фильтровали и концентрировали. Соединение 79В (360 мг, выход: 64,87%, светло-желтое твердое вещество): 1Н ЯМР (CDCl3, 400 МГц) δ 7,90-7,88 (m, 1H), 7,34-7,31 (m, 1H), 7,15-7,09 (m, 2Н), 5,72-5,63 (m, 1H), 4,87-4,76 (m, 2Н), 2,90-2,86 (m, 2Н), 1,92-1,87 (m, 2Н), 1,50-1,47 (m, 2Н), 1,26-1,14 (m, 8Н).
[0689] К раствору соединения 79В (360 мг, 2,57 ммоль) в ДМФА (5 мл) при 0°С добавляли NBS (550 мг, 3,08 ммоль). Затем смесь нагревали до 25°С и перемешивали в течение 12 ч. Реакционную смесь промывали H2O (10 мл), подвергали экстракции ДХМ (20 мл). Органические фазы собирали, сушили с помощью Na2SO4, фильтровали и концентрировали с получением промежуточного соединения 79С (550 мг, неочищенного) в виде желтого твердого вещества. МС (ИЭР) m/z (М+2)+ 220,7.
[0690] К раствору NaH (151 мг, 3,76 ммоль, чистота 60%) в ТГФ (8 мл) при 0°С добавляли по каплям раствор соединения 79С (550 мг, 2,51 ммоль) в ТГФ (2 мл). После добавления смесь нагревали до 25°С и перемешивали в течение 1 ч. Затем добавляли SEM-Cl (0,5 мл, 2,76 ммоль). Смесь перемешивали при 25°С в течение 12 ч. В реакционную смесь добавляли H2O (10 мл) для гашения реакции, смесь подвергали экстракции EtOAc (20 мл × 2). Органические фазы собирали и концентрировали. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = 5:1) с получением соединения 79D (180 мг, выход: 20,51%) в виде бесцветного масла. МС (ИЭР) m/z (М+2)+ 350,9.
[0691] К раствору соединения 79D (180 мг, 0,52 ммоль) и фенилбороновой кислоты (76 мг, 0,62 ммоль) в диоксане (12 мл) и H2O (4 мл) добавляли Pd(dtbpf)Cl2 (34 мг, 0,052 ммоль) и K3PO4 (330 мг, 1,55 ммоль). Смесь перемешивали при 80°С в атмосфере N2 в течение 2 ч. Реакционную смесь промывали Н2О (10 мл), подвергали экстракции EtOAc (15 мл × 2). Органические фазы собирали и концентрировали. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = 5:1) с получением соединения 79Е (150 мг, выход: 84,0%) в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 347,0.
[0692] К раствору соединения 79Е (180 мг, 0,52 ммоль) в ТГФ (5 мл), МеОН (5 мл) и Н2О (5 мл) добавляли LiOH⋅H2O (110 мг, 2,60 ммоль). Смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь подкисляли с помощью 1 н. HCl до рН ~3. Смесь подвергали экстракции EtOAc (10 мл × 2). Органические фазы собирали, сушили с помощью Na2SO4, фильтровали и концентрировали с получением соединения 79F (130 мг, неочищенного) в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 333,0. Промежуточное соединение 79Н (65 мг, неочищенное, желтое масло) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, соединения 79F. Соединение 79Н: МС (ИЭР) m/z (М+Н)+ 507,2.
[0693] К раствору соединения 79Н (65 мг, 0,13 ммоль) в EtOAc (5 мл) добавляли по каплям HCl/EtOAc (4 М, 10 мл). После добавления смесь перемешивали при 25°С в течение 12 ч. Растворитель удаляли под вакуумом. Остаток очищали посредством препаративной ВЭЖХ (HCl) с получением соединения 79 (10,00 мг, выход: 18,7%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, D2O) δ 7,45-7,28 (m, 3Н), 7,23-6,97 (m, 7Н), 4,46-4,38 (m, 1H), 3,02-2,93 (m, 1H), 2,48-2,41 (m, 3Н), 2,39-2,29 (m, 1H). МС (ИЭР) m/z (М+Н)+ 377,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(3-((БЕНЗИЛАМИНО)МЕТИЛ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (146)
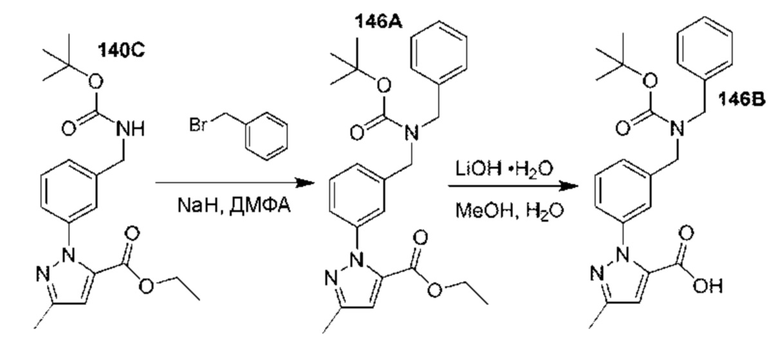
[0694] К смеси соединения 140С (250 мг, 0,72 ммоль) и бензилбромида (310 мг, 1,8 ммоль) в ДМФА (10 мл) добавляли NaH (87 мг, 2,2 ммоль, чистота 60%) порциями при 0°С в атмосфере N2. Смесь перемешивали при 25°С в течение 3 ч. В смесь добавляли NH4Cl (10 мл) для гашения реакции, смесь разбавляли Н2О (30 мл), подвергали экстракции этилацетатом (20 мл × 3). Органическую фазу объединяли и промывали солевым раствором (30 мл × 2), сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Остаток очищали посредством колоночной флэш-хроматографии (SiO2, петролейный эфир/этилацетат = от 1/0 до 5/1) с получением соединения 146А (182 мг, выход: 57,7%) в виде бесцветной прозрачной жидкости.
[0695] К смеси соединения 146А (180 мг, 0,41 ммоль) в МеОН (10 мл) и Н2О (2 мл) добавляли LiOH⋅Н2О (52 мг, 1,24 ммоль) одной порцией при 25°С. Смесь перемешивали при 25°С в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Затем остаток разбавляли водой (15 мл) и подвергали экстракции МТБЭ (20 мл), водную фазу подкисляли с помощью водного раствора HCl (1 М) до рН ~5~6 и подвергали экстракции этилацетатом (20 мл × 2). Объединенные органические слои промывали солевым раствором (40 мл) и сушили над Na2SO4, фильтровали и концентрировали с получением соединения 146В (158 мг, выход: 90,8%) в виде бесцветной жидкости, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 7,44-7,39 (m, 1H), 7,37-7,30 (m, 3Н), 7,26 (q, J=6,9 Гц, 5Н), 6,81 (s, 1H), 4,48-4,26 (m, 4Н), 2,25 (s, 3Н), 1,39 (s, 9Н).
[0696] Соединение 146 было получено, как описано в примере 45, из промежуточного соединения 146В. Соединение 146 (40,0 мг, выход 74,6%, белое твердое вещество): 1Н ЯМР (D2O, 400 МГц): δ 7,51-7,42 (m, 6Н), 7,41-7,36 (m, 1H), 7,36-7,27 (m, 5Н), 7,25 (s, 1H), 6,78 (d, J=8,6 Гц, 1H), 6,60 (s, 1H), 4,52-4,43 (m, 1H), 4,30-4,18 (m, 4Н), 3,24-3,15 (m, 1H), 2,82-2,71 (m, 1H), 2,29 (s, 3Н). МС (ИЭР) m/z (М-HCl+Н)+ 496,2.
ГИДРОХЛОРИД (S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4-((БЕНЗИЛАМИНО)МЕТИЛ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИДА (160)
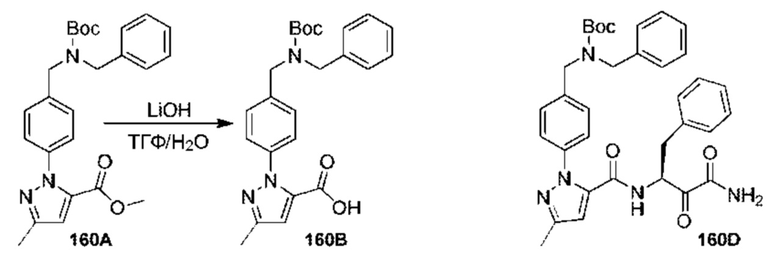
[0697] К раствору соединения 153Е (350 мг, 1,01 ммоль) и бензилбромида (432 мг, 2,53 ммоль, 0,3 мл) в ДМФА (10 мл) добавляли NaH (121 мг, 3,03 ммоль, чистота 60%) при 0°С. Смесь перемешивали при 25°С в течение 1 ч. В смесь добавляли NH4Cl (5 мл) для гашения реакции, смесь разбавляли H2O (20 мл), подвергали экстракции этилацетатом (20 мл × 3), органическую фазу объединяли и промывали NaCl (30 мл × 2), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 20/1 до 5/1) с получением соединения 160А (400 мг, выход: 41,47%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,02 (dd, J=1,1 Гц, 1H), 7,37-7,25 (m, 9Н), 6,88-6,75 (m, 1H), 4,49-4,28 (m, 4Н), 2,98-2,88 (m, 4Н), 2,46-2,30 (m, 3Н), 1,57-1,41 (m, 8Н). МС (ИЭР) m/z (М-56)+ 380,0.
[0698] К смеси соединения 160А (400 мг, 918,46 мкмоль) в ТГФ (10 мл) и H2O (10 мл) добавляли LiOH⋅H2O (116 мг, 2,76 ммоль) порциями при 25°С и перемешивали в течение 2,5 ч. Смесь разбавляли H2O (10 мл) и концентрировали для удаления ТГФ, затем водную фазу подвергали экстракции МТБЭ (30 мл × 2). Водные слои подкисляли до рН ~ 2 с помощью 1 н. HCl, затем раствор подвергали экстракции этилацетатом (30 мл × 3). Органические слои сушили над Na2SO4 и концентрировали с получением промежуточного соединения 160В (350 мг, выход: 86,82%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,40-7,20 (m, 9Н), 6,85 (s, 1H), 4,43 (s, 2Н), 4,33 (dd, J=13,9 Гц, 2H), 2,34 (s, 3Н), 1,47 (s, 8Н). МС (ИЭР) m/z (М-56)+ 366,1.
[0699] Соединение 160 было получено, как описано в примере 79, из соответствующей карбоновой кислоты, соединения 160В, и затем через стадию получения промежуточного соединения 160D. Соединение 160 (30 мг, выход: 53,02%, светло-желтое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,62-9,57 (m, 1H), 9,15 (d, J=7,9 Гц, 1H), 8,12 (s, 1H), 7,88 (s, 1H), 7,58-7,49 (m, 4Н), 7,44 (dd, J=6,8 Гц, 3Н), 7,36-7,27 (m, 5Н), 7,22 (d, J=8,4 Гц, 2Н), 6,61 (s, 1H), 5,35-5,28 (m, 1H), 4,18 (s, 4Н), 3,25-3,18 (m, 1H), 2,83 (dd, J=10,6, 13,9 Гц, 1H), 2,26 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 496,2.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(1H-БЕНЗО[d]ИМИДАЗОЛ-2-ИЛ)-5-МЕТИЛ-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (264)
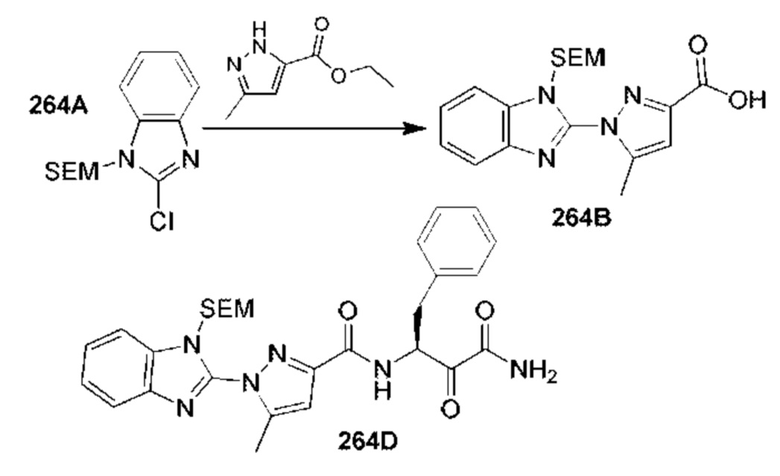
[0700] 2-хлор-1H-бензо[d]имидазол (5 г, 32,8 ммоль) добавляли к раствору NaH (1,31 г, 32,8 ммоль, 60%) в ДМФА (50 мл) при температуре ниже 10°С. После добавления реакционную смесь перемешивали при 20°С в течение 2 ч. Затем в реакционную смесь добавляли SEM-Cl (5,46 г, 32,8 ммоль). Реакционную смесь перемешивали при 20°С в течение 16 ч. Добавляли воду (150 мл) и EtOAc (150 мл). Органический слой отделяли и промывали солевым раствором (100 мл), концентрировали с получением остатка. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 20:1~4:1) с получением соединения 264А (3,50 г, выход: 37,8%) в виде масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,78-7,71 (m, 1H), 7,54-7,48 (m, 1H), 7,41-7,32 (m, 2Н), 5,62 (s, 2Н), 3,66-3,59 (m, 2Н), 0,99-0,93 (m, 2Н), 0,07 (d, J=2,0 Гц, 2Н), 0,00 (s, 9Н).
[0701] Соединение 264 было получено, как описано в примере 79, из соответствующего промежуточного соединения 264В, и затем через стадию получения промежуточного соединения 264D. Соединение 264 (31,8 мг, выход: 28,0%, грязно-белое твердое вещество): 1Н ЯМР (400 МГц, CDCl3) δ 13,04 (ушир. s, 1H), 8,39 (d, J=7,6 Гц, 1H), 8,13 (ушир. s, 1H), 7,88 (ушир. s, 1H), 7,67 (ушир. d, J=7,2 Гц, 1H), 7,53 (ушир. d, J=7,5 Гц, 1H), 7,34-7,19 (m, 7Н), 6,79 (s, 1H), 5,51 (dt, J=4,0, 8,2 Гц, 1H), 3,27 (ушир. d, J=4,0 Гц, 1H), 3,02 (dd, J=9,3, 13,9 Гц, 1H), 2,73 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 417,2.
ПРИМЕР 46
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-1-(4-ФЕНИЛТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (80)
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-1-(4-ФЕНИЛТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (125)
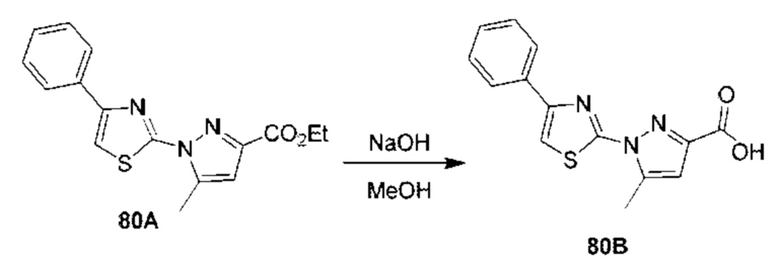
[0702] Промежуточное соединение 80В (182,00 мг, выход 99,95%, белое твердое вещество): 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,02 (s, 1H), 7,96 (ушир. d, J=1,5 Гц, 2Н), 7,47 (ушир. t, J=7,5 Гц, 2Н), 7,41-7,32 (m, 1H), 6,80 (s, 1H), 2,78 (s, 3Н).
[0703] Соединение 80 (44 мг, выход 64,6%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих промежуточных соединений 80В и 12G. Соединение 80: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,53 (ушир. d, J=7,3 Гц, 1H), 8,12 (ушир. s, 1H), 8,04-7,94 (m, 3Н), 7,86 (ушир. s, 1H), 7,52-7,44 (m, 2Н), 7,39 (ушир. d, J=6,4 Гц, 1H), 7,32-7,17 (m, 5Н), 6,76 (s, 1H), 5,43 (ушир. s, 1H), 3,24 (ушир. d, J=12,1 Гц, 1H), 3,12-3,03 (m, 1H), 2,78 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 460,1.
[0704] Соединение 125 (118 мг, выход 77,6%, белое твердое вещество) было получено, как описано в примере 20, из соответствующих промежуточных карбоксильных соединений 80В и 41В. Соединение 125: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,84 (ушир. s, 1H), 8,60-8,53 (m, 1H), 8,06-7,94 (m, 3Н), 7,52-7,44 (m, 2Н), 7,42-7,35 (m, 1H), 7,29 (ушир. s, 4Н), 7,21 (ушир. s, 1H), 6,76 (s, 1H), 5,44 (ушир. s, 1H), 3,27-3,19 (m, 1H), 3,11-3,02 (m, 1H), 2,78 (ушир. s, 4Н), 0,72-0,57 (m, 4Н), МС (ИЭР) m/z (М+Н)+ 500,1.
ПРИМЕР 47
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(2'-МЕТИЛ-[1,1'-БИФЕНИЛ]-3-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (81)
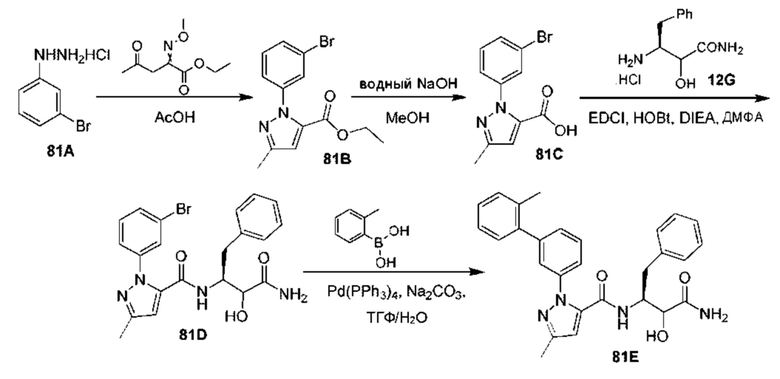
[0705] Смесь соединения 81А (25 г, 111,86 ммоль) и соединения этил-2-(метоксиимино)-4-оксопентаноата (22 г, 117,45 ммоль) в АсОН (150 мл) перемешивали при 110°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении для удаления большого количества АсОН. Остаток подкисляли с помощью насыщенного водного NaHCO3 до рН ~ 7-8. Осажденное вещество собирали путем фильтрования, и осадок на фильтре растирали с петролейным эфиром (20 мл), отфильтровывали и сушили под вакуумом с получением соединения 81В (26,41 г, выход: 74,0%) в виде серого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц) δ 7,71-7,68 (m, 1H), 7,65 (td, J=1,5, 7,7 Гц, 1H), 7,50-7,38 (m, 2Н), 6,95-6,84 (m, 1H), 4,18 (q, J=7,0 Гц, 2Н), 2,27 (s, 3Н), 1,17 (t, J=7,0 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 310,8.
[0706] К смеси соединения 81В (5 г, 16,17 ммоль) в МеОН (20,00 мл) добавляли NaOH (2 М, 40 мл) одной порцией при 25°С. Смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. К остатку добавляли H2O (10 мл) и этилацетат (20 мл) и затем смесь подкисляли с помощью 1 М HCl до рН водной фазы ~ 5-6. Отделенный водный слой подвергали экстракции этилацетатом (30×3 мл), объединенные органические слои промывали солевым раствором (60 мл), сушили над Na2SO4, фильтровали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт обрабатывали простым изопропиловым эфиром (15 мл), осадок фильтровали и сушили под вакуумом с получением соединения 81С (4,21 г, выход: 80,67%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,65-7,58 (m, 2Н), 7,45-7,36 (m, 2Н), 6,83 (s, 1H), 2,23 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 282,8.
[0707] К раствору соединения 81С (1 г, 3,56 ммоль) в ДМФА (50 мл) добавляли HOBt (144 мг, 1,07 ммоль), соединение 12G (903 мг, 3,92 ммоль, HCl) и DIEA (1,38 г, 10,68 ммоль). После перемешивания в течение 5 мин добавляли EDCI (682 мг, 3,56 ммоль) при 0°С. Затем реакционную смесь перемешивали при 25°С в течение 9 ч. Реакционную смесь концентрировали при пониженном давлении для удаления ДМФА, и к остатку добавляли этилацетат (100 мл) и, соответственно, промывали H2O (80 мл), насыщенным водным NaHCO3 (80 мл × 2), солевым раствором (80 мл × 3). Органическую фазу сушили над Na2SO4 и концентрировали. Неочищенный продукт обрабатывали простым i-пропиловым эфиром. Твердое вещество собирали и сушили под вакуумом с получением соединения 81D (1,3 г, выход: 79,85%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,55-8,18 (m, 1H), 7,54-7,45 (m, 2Н), 7,34 (ушир. d, J=18,5 Гц, 1H), 7,30-7,14 (m, 7Н), 7,00-6,89 (m, 1H), 6,58 (d, J=1,5 Гц, 1H), 5,98-5,73 (m, 1H), 4,44-4,33 (m, 1H), 4,00-3,89 (m, 1H), 2,93-2,87 (m, 0,5Н), 2,84-2,73 (m, 1H), 2,71 (ушир. s, 0,6Н), 2,22 (s, 3Н).
[0708] К смеси соединения 81D (150 мг, 328,00 мкмоль) и соединения о-толилбороновой кислоты (89,2 мг, 656,00 мкмоль) в ТГФ (50 мл) и H2O (10 мл) добавляли Na2CO3 (70 мг, 656,00 мкмоль) и Pd(PPh3)4 (38 мг, 32,80 мкмоль) одной порцией при 25°С в атмосфере N2. Смесь перемешивали при 80°С в течение 12 ч. Затем в реакционную смесь добавляли H2O (100 мл), и смесь подвергали экстракции этилацетатом (100 мл × 3). Объединенный органический слой промывали насыщенным водным NaHCO3 (150 мл × 3), солевым раствором (150 мл × 3), сушили над Na2SO4 и концентрировали с получением соединения 81Е (110 мг, выход: 71,58%), которое было получено в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,50-8,11 (m, 1H), 7,38-7,32 (m, 2Н), 7,31-7,22 (m, 6Н), 7,18 (ушир. s, 5Н), 7,10 (ушир. d, J=6,4 Гц, 1H), 7,00 (ушир. dd, J=8,0, 16,4 Гц, 1H), 6,57 (s, 1H), 5,96-5,69 (m, 1H), 4,51-4,30 (m, 1H), 4,03-3,85 (m, 1H), 2,92-2,63 (m, 2Н), 2,27-2,12 (m, 6Н). МС (ИЭР) m/z (М+Н)+ 469,2.
[0709] К смеси соединения 81Е (70 мг, 149,4 мкмоль) в ДХМ (10 мл) и ДМСО (0,5 мл) добавляли DMP (190 мг, 448,2 мкмоль) одной порцией при 0°С. Смесь перемешивали при 0°С в течение 5 мин, затем нагревали до 25°С и перемешивали в течение 1,5 часа. В реакционную смесь добавляли для гашения реакции 20 мл 10% водного раствора Na2S2O3 и 20 мл насыщенного водного раствора NaHCO3, и затем смесь подвергали экстракции ДХМ (30 мл × 3). Объединенную органическую фазу промывали солевым раствором (40 мл × 3), сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Остаток обрабатывали смесью простой i-пропиловый эфир/CH3CN (об./об. = 10/1, 10 мл). Твердое вещество собирали и сушили под вакуумом с получением соединения 81 (48,3 мг, выход: 66,3%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,11 (ушир. d, J=8,0 Гц, 1H), 8,18-7,79 (m, 2Н), 7,49-7,37 (m, 1H), 7,29-7,26 (m, 8Н), 7,21-7,12 (m, 4Н), 6,60 (s, 1H), 5,32 (ушир. s, 1H), 3,21-3,18 (m, 1H), 2,86-2,76 (m, 1H), 2,25 (ушир. s, 3Н), 2,18 (ушир. s, 3Н). МС (ИЭР) m/z (М+Н)+ 467,1.
ПРИМЕР 48
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-ФЕНИЛТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (82)
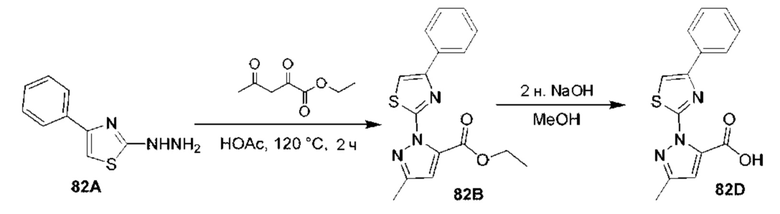
[0710] К раствору соединения 82А (2,0 г, 10,46 ммоль) в СН3СООН (30,0 мл) добавляли по каплям соединение этил-2,4-диоксопентаноат (1,65 г, 10,46 ммоль, 1,48 мл), затем смесь нагревали до 120°С и перемешивали в течение 2 ч и удаляли растворитель при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и обрабатывали NaHCO3 до рН ~ 8, и затем органический слой собирали и упаривали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии (петролейный эфир/этилацетат: от 0 до 10/1).
[0711] Соединение 82В (660,0 мг, 2,11 ммоль, выход 20,14%) получали в виде белого твердого вещества. Соединение 82В слабополярное (слабополярное): 1Н ЯМР (CDCl3, 400 МГц) δ 7,88-7,83 (m, 2H), 7,43-7,38 (m, 3Н), 7,36-7,31 (m, 1H), 6,71 (s, 1H), 4,33 (q, J=7,2 Гц, 2H), 2,37 (s, 3Н), 1,24 (t, J=7,2 Гц, 3Н).
[0712] К раствору соединения 82В (650,0 мг, 2,07 ммоль) в МеОН (10,00 мл) добавляли по каплям NaOH (2 М, 6,00 мл), и смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь разбавляли H2O (10 мл) и подвергали экстракции МТБЭ (10 мл × 2). Водную фазу обрабатывали HCl (1 М) до рН ~ 4, затем осадок отфильтровывали и сушили при пониженном давлении. Соединение 82D (540,0 мг, выход 91,3%) получали в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,04 (s, 1H), 7,88 (d, J=7,1 Гц, 2Н), 7,47-7,41 (m, 2Н), 7,38-7,33 (m, 1H), 6,83 (s, 1H), 2,27 (s, 3Н).
[0713] Соединение 82 (20,0 мг, выход 42,74%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 82D. Соединение 82: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,49 (d, J=7,6 Гц, 1H), 8,12 (s, 1H), 7,94 (s, 1H), 7,88 (ушир. s, 1H), 7,78 (ушир. d, J=7,2 Гц, 2Н), 7,42-7,37 (m, 2Н), 7,35-7,29 (m, 1H), 7,22-7,12 (m, 5Н), 6,55 (s, 1H), 5,55-5,47 (m, 1H), 3,16 (m, 1H), 2,80 (m, 1H), 2,27 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 403,1.
ПРИМЕР 49
СОЕДИНЕНИЯ 83, 126, 130
(S)-1-([1,1'-БИФЕНИЛ]-4-ИЛ)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (83)
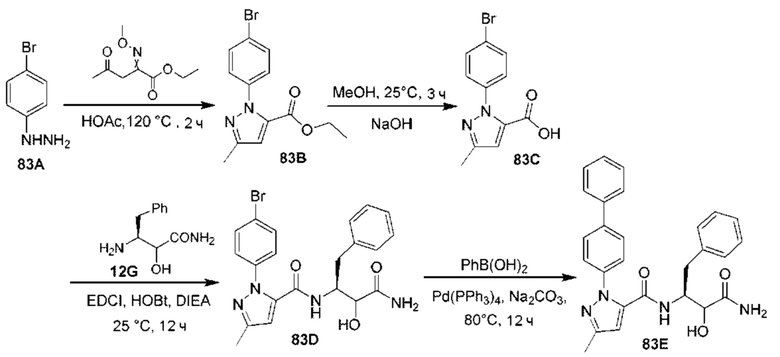
[0714] К раствору соединения 83А (20,0 г, 89,49 ммоль, HCl) в CH3COOH (150,0 мл) добавляли соединение этил-2-(метоксиимино)-4-оксопентаноат (14,0 г, 89,49 ммоль), затем смесь нагревали до 120°С и перемешивали в течение 2 ч и удаляли растворитель при пониженном давлении. Остаток растворяли в этилацетате (150 мл), обрабатывали NaHCO3 до рН ~ 7 и фильтровали. Твердое вещество обрабатывали петролейный эфиром. Соединение 83В (22,0 г, 71,16 ммоль, выход 79,52%) получали в виде желтого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц) δ 7,56 (d, J=8,4 Гц, 2Н), 7,31-7,24 (m, 2Н), 6,81 (s, 1H), 4,23 (q, J=7,2 Гц, 2Н), 2,34 (s, 3Н), 1,26 (t, J=7,2 Гц, 3Н).
[0715] К раствору соединения 83В (5,0 г, 16,17 ммоль) в МеОН (40,0 мл) добавляли по каплям NaOH (2 М, 45,0 мл), и смесь перемешивали при 25°С в течение 3 ч и удаляли растворитель при пониженном давлении, затем смесь разбавляли H2O (30 мл) и подвергали экстракции МТБЭ (60 мл × 2). Водную фазу обрабатывали HCl (1 М) до рН ~ 4, и затем осадок отфильтровывали и сушили при пониженном давлении. Водную фазу подвергали экстракции этилацетатом (50 мл × 2), органический слой (подвергнутый экстракции этилацетатом) упаривали при пониженном давлении. Собирали твердое вещество, которое представляло собой соединение 83С (3,75 г, выход 82,5%), полученное в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,62 (d, J=8,8 Гц, 2Н), 7,36 (d, J=8,8 Гц, 2Н), 6,81 (s, 1H), 2,23 (s, 3Н).
[0716] Соединение 83 (25,0 мг, выход 61,24%, белое твердое вещество) было получено, как описано в примере 47, из соответствующих промежуточных соединений 83С, 12G и фенилбороновой кислоты. Соединение 83: 1Н ЯМР (CDCl3, 400 МГц) δ 9,49 (d, J=7,3 Гц, 1H), 8,12 (s, 1H), 7,94 (s, 1H), 7,88 (ушир. s, 1H), 7,78 (ушир. d, J=7,3 Гц, 2Н), 7,48-7,27 (m, 4Н), 7,26-6,96 (m, 7Н), 6,55 (s, 1H), 5,55-5,47 (m, 1H), 3,16 (ушир. dd, J=4,2, 14,1 Гц, 1H), 2,80 (ушир. dd, J=9,7, 13,9 Гц, 1H), 2,27 (s, 3Н), 2,06 (s, 1H). МС (ИЭР) m/z (М+Н)+ 453,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4'-ФТОР-[1,1'-БИФЕНИЛ]-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (126)
[0717] Соединение 126 (32 мг, выход 25,5%, светло-желтое твердое вещество) было получено, как описано в примере 63, из соответствующих исходных веществ, соединения 83D и (4-фторфенил)бороновой кислоты. Соединение 126: 1Н ЯМР (CD3CN, 400 МГц) δ 7,73-7,67 (m, 2Н), 7,63-7,59 (m, 2Н), 7,37-7,22 (m, 10Н), 7,02 (ушир. s, 1H), 6,54 (s, 1H), 6,27 (ушир. s, 1H), 5,42 (ddd, J=4,5, 7,8, 9,5 Гц, 1H), 3,32 (dd, J=4,5, 13,8 Гц, 1H), 2,93 (dd, J=9,6, 14,0 Гц, 1H), 2,31 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 471,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4'-ФТОР-[1,1'-БИФЕНИЛ]-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (130)
[0718] Соединение 130 (30 мг, выход 34,7%, светло-желтое твердое вещество) было получено, как описано в примере 105, из соответствующих исходных веществ, соединения 83D и п-толилбороновой кислоты. Соединение 130: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,14 (ушир. d, J=1,1 Гц, 1H), 8,11 (ушир. s, 1H), 7,87 (ушир. s, 1H), 7,60-7,55 (m, 3Н), 7,33-7,22 (m, 10Н), 6,56-6,50 (m, 1H), 5,24 (ушир. s, 1H), 3,20 (ушир. d, J=13,5 Гц, 1H), 2,87-2,78 (m, 1H), 2,33 (s, 3Н), 2,24 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 467,1.
ПРИМЕР 50
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(6-МЕТИЛПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (84)
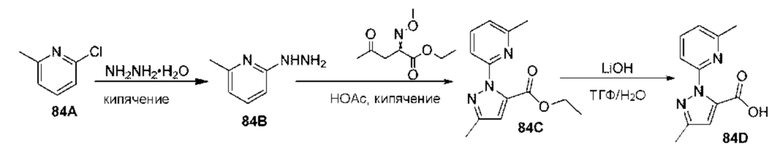
[0719] Смесь соединения 84А (5 г, 39,19 ммоль) и NH2NH2⋅H2O (20 г, 391,94 ммоль) нагревали с обратным холодильником (119°С) в течение 36 часов. Реакционную смесь концентрировали при пониженном давлении для удаления не вступившего в реакцию гидразингидрата. Остаток разбавляли H2O (30 мл) и подвергали экстракции ДХМ (30 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством рекристаллизации из петролейного эфира (15 мл) при -10°С с получением соединения 84В (2,40 г, выход: 49,35%) в виде черно-коричневого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,39-7,29 (m, 1H), 7,25 (s, 1H), 6,51 (d, J=8,4 Гц, 1H), 6,39 (d, J=7,3 Гц, 1H), 4,06 (s, 2Н), 2,26 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 127,8.
[0720] К раствору соединения 84В (970 мг, 7,88 ммоль) в АсОН (20 мл) добавляли соединение этил-2-(метоксиимино)-4-оксопентаноат (1,36 г, 7,88 ммоль). Смесь перемешивали при 120°С в течение 20 ч. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли H2O (30 мл) и подвергали экстракции этилацетатом (10 мл × 3). Объединенные органические слои промывали насыщенным водным NaHCO3 (15 мл × 3), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле и затем посредством препаративной ВЭЖХ (в условиях HCl) с получением соединения 84С (160 мг, выход: 8,22%), которое было получено в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,88 (t, J=7,8 Гц, 1H), 7,50 (d, J=8,0 Гц, 1H), 7,28 (d, J=7,5 Гц, 1H), 6,76 (s, 1H), 4,20 (d, J=7,3 Гц, 2Н), 2,43 (s, 3Н), 2,28 (s, 3Н), 1,14 (t, J=7,0 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 246,0.
[0721] К раствору соединения 84С (100 мг, 432,43 мкмоль) в ТГФ (5 мл) добавляли раствор LiOH⋅H2O (91 мг, 2,16 ммоль) в H2O (5 мл) при 0°С. После добавления реакционную смесь перемешивали в течение 14 ч при 25°С. Реакционную смесь разбавляли H2O (10 мл) и подвергали экстракции МТБЭ (30 мл). Водную фазу нейтрализовали 1 н. HCl до рН ~ 4 и затем подвергали экстракции EtOAc (30 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 84D (50 мг, выход: 53,2%) в виде красного твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,06 (d, J=8,4 Гц, 1H), 7,89 (t, J=8,0 Гц, 1H), 7,20-7,14 (m, 2Н), 2,64 (s, 3Н), 2,36 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 217,9.
[0722] Соединение 84 (70 мг, выход: 54,12%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 84D. Соединение 84: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,09 (d, J=7,3 Гц, 1H), 8,04 (s, 1H), 7,81 (s, 1H), 7,76 (t, J=7,7 Гц, 1H), 7,32 (d, J=7,9 Гц, 1H), 7,29-7,17 (m, 5Н), 7,15 (d, J=7,5 Гц, 1H), 6,44 (s, 1H), 5,37-5,25 (m, 1H), 3,13 (dd, J=4,0, 13,9 Гц, 1H), 2,82 (dd, J=9,7, 13,9 Гц, 1H), 2,24 (s, 6Н). МС (ИЭР) m/z (М+Н)+ 392,1.
ПРИМЕР 51
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРАЗИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (85)
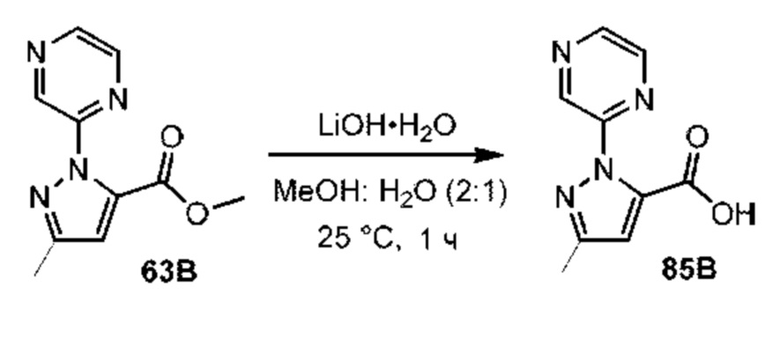
[0723] К смеси соединения 63В (200 мг, 916,55 мкмоль) в МеОН (10 мл) и H2O (5 мл) добавляли LiOH⋅H2O (153,8 мг, 3,67 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли H2O (10 мл), доводили рН до ~ 3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (40 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 85В (160 мг, выход 85,49%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,60 (ушир. s, 1H), 8,67 (ушир. s, 1H), 8,32 (ушир. s, 1H), 7,25 (ушир. s, 1H), 2,41 (ушир. s, 3Н). МС (ИЭР) m/z (М+1)+ 205,0.
[0724] Соединение 85 (30,7 мг, выход 59,7%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 85В. Соединение 85: 1H ЯМР (400 МГц, CDCl3) δ 8,95 (d, J=4,0 Гц, 1H), 8,22 (d, J=8,4 Гц, 1H), 7,76-7,66 (m, 2Н), 7,49 (d, J=6,4 Гц, 1H), 7,38 (d, J=8,4 Гц, 1H), 7,24-7,16 (m, 3Н), 6,87 (d, J=7,6 Гц, 2Н), 6,79 (ушир. s, 1H), 6,64 (s, 1H), 6,33 (d, J=7,2 Гц, 1H), 5,49-5,42 (m, 1H), 3,27-3,19 (m, 1H), 3,08-2,98 (m, 1H), 2,78-2,69 (m, 1H), 0,90-0,83 (m, 2Н), 0,61-0,50 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 419,1.
ПРИМЕР 52
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(1Н-ИНДАЗОЛ-1-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (86)
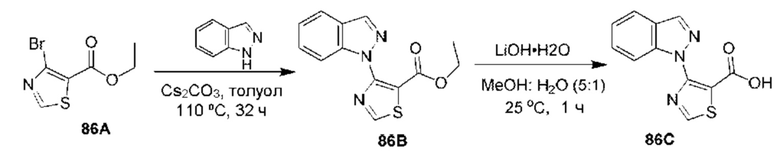
[0725] Смесь, состоящую из соединения 86А (250 мг, 1,06 ммоль), 1H-индазола (125,2 мг, 1,06 ммоль) и Cs2CO3 (1,04 г, 3,18 ммоль) в толуоле (15 мл), перемешивали при 110°С в течение 32 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством препаративной ВЭЖХ (в условиях HCl) с получением соединения 86В (20 мг, выход 6,90%) в виде белого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц) δ 8,87 (s, 1H), 8,19 (d, J=0,8 Гц, 1H), 7,75-7,70 (m, 1H), 7,54 (d, J=8,4 Гц, 1H), 7,41-7,35 (m, 1H), 7,22-7,17 (m, 2Н), 4,17 (q, J=7,2 Гц, 2H), 1,07 (t, J=7,2 Гц, 3Н).
[0726] К смеси 86В (40 мг, 146,35 мкмоль) в МеОН (5 мл) и H2O (1 мл) добавляли LiOH⋅Н2О (24,6 мг, 585,40 мкмоль) одной порцией, и смесь перемешивали при 25°С в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН, и остаток разбавляли Н2О (10 мл), доводили рН до ~ 3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (40 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 86С (30 мг, выход 83,58%) в виде белого твердого вещества. МС (ИЭР) m/z (М+1)+ 245,8.
[0727] Соединение 86 (5,4 мг, выход 10,0%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 86С. Соединение 86: 1Н ЯМР (400 МГц, CDCl3) δ 10,97 (d, J=5,2 Гц, 1H), 8,78 (s, 1H), 8,23 (d, J=6,4 Гц, 1H), 7,82 (s, 1H), 7,71 (d, J=3,2 Гц, 1H), 7,54-7,44 (m, 1H), 7,32-7,23 (m, 1H), 7,11-6,96 (m, 5Н), 6,85 (ушир. s, 1H), 5,79-5,66 (m, 1H), 3,40-3,29 (m, 1H), 3,21-3,09 (m, 1H), 2,78-2,69 (m, 1H), 0,80 (d, J=6,2 Гц, 2Н), 0,55 (ушир. s, 2Н). МС (ИЭР) m/z (М+Н)+ 460,1.
ПРИМЕР 53
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(5-(ОКСАЗОЛ-2-ИЛ)ПИРИДИН-2-ИЛ)-1Н-ИМИДАЗОЛ-5-КАРБОКСАМИД (89)
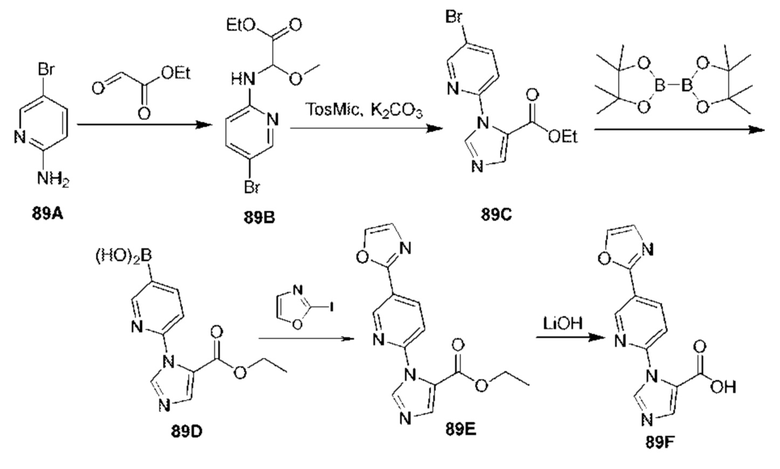
[0728] К раствору соединения 89А (40 г, 231 ммоль) в МеОН (500 мл) добавляли этил-2-оксоацетат (188 г, 924 ммоль) при 25°С. Реакционную смесь перемешивали при 70°С в течение 1 ч. Реакционную смесь концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 50:1 до 30:1). Соединение 89В (70 г, неочищенное) получали в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 258,8.
[0729] К раствору соединения 89В (35 г, 136 ммоль) в EtOH (300 мл) добавляли TosMIC (66,4 г, 340 ммоль) и K2CO3 (28,2 г, 204 ммоль) при 25°С. Реакционную смесь перемешивали при 70°С в течение 1 ч. Реакционную смесь фильтровали, и фильтрат концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 20:1 до 3:1). Соединение 89С (17 г) получали в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,62 (d, J=2,4 Гц, 1H), 8,00-7,96 (m, 2Н), 7,86 (d, J=0,9 Гц, 1H), 7,36-7,32 (m, 1H), 4,27 (q, J=7,1 Гц, 2Н), 1,31 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 297,0.
[0730] К раствору соединения 89С (5 г, 16,8 ммоль) в диоксане (100 мл) добавляли бис(пинаколато)дибор (8,58 г, 33,7 ммоль), KOAc (16,5 г, 168 ммоль) при 25°С. Смесь дегазировали и продували N2 3 раза, затем добавляли Pd(dppf)Cl2 (617 мг, 844 мкмоль). Реакционную смесь дегазировали и продували N2 3 раза и перемешивали при 75°С в течение 4 ч. Реакционную смесь концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, от петролейный эфир : этилацетат = 20:1 до дихлорметан : метанол = 5:1). Соединение 89D (2,7 г, выход: 61,2%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, метанол-d4) δ 8,63 (ушир. s, 1H), 8,06 (s, 2Н), 7,76 (s, 1H), 7,36 (ушир. d, J=7,1 Гц, 1H), 4,14 (q, J=7,1 Гц, 2Н), 1,14 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 261,9.
[0731] К раствору соединения 89D (500 мг, 1,92 ммоль) в диоксане (4 мл) добавляли 2-иодоксазол (561,48 мг, 2,88 ммоль), K2CO3 (796,09 мг, 5,76 ммоль), Н2О (1 мл) при 25°С. Реакционную смесь дегазировали и продували N2. Затем добавляли Pd(dppf)Cl2 (140 мг, 192 мкмоль). Смесь дегазировали и продували N2 и перемешивали при 150°С в течение 1 ч в условиях микроволнового излучения. Реакционную смесь концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = 10:1-1,5:1). Соединение 89Е (300 мг, неочищенное) было получено в виде серого твердого вещества. МС (ИЭР) m/z (М+Н)+ 285,0.
[0732] К раствору соединения 89Е (200 мг, 703 мкмоль) в ТГФ (2 мл), H2O (500 мкл) добавляли LiOH⋅Н2О (59 мг, 1,41 ммоль) и перемешивали при 25°С в течение 12 ч. Реакционную смесь подкисляли путем добавления HCl (1 н.) до рН ~ 5, и осадок отфильтровывали с получением неочищенного продукта. Соединение 89F (60 мг, неочищенное) было получено в виде серого твердого вещества. МС (ИЭР) m/z (М+Н)+ 257,0.
[0733] Соединение 89 (35 мг, выход: 65,4%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 89F. Соединение 89: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,04-8,93 (m, 2Н), 8,77 (ушир. d, J=5,1 Гц, 1H), 8,38-8,28 (m, 2Н), 8,20 (s, 1H), 7,56 (s, 1H), 7,46 (s, 1H), 7,34 (d, J=8,6 Гц, 1H), 7,29-7,22 (m, 4Н), 5,32-5,14 (m, 1H), 3,28 (ушир. s, 1H), 3,20-3,10 (m, 1H), 2,81 (ушир. dd, J=10,1, 13,7 Гц, 1H), 2,77-2,69 (m, 1H), 0,73-0,42 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 471,1.
ПРИМЕР 54
(S)-N-(1-(4-(АЛЛИЛОКСИ)ФЕНИЛ)-3-ОКСОПРОПАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (93)
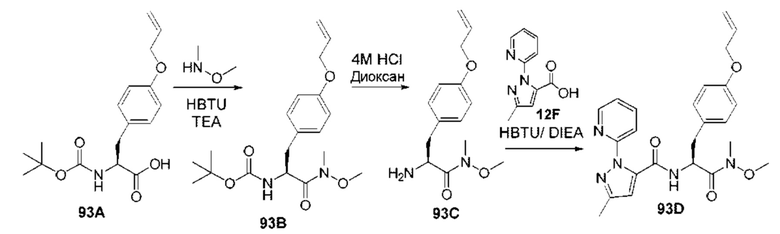
[0734] Соединение 93А (1 г, 1,0 экв.), N,O-диметилгидроксиламин (607 мг, 2 экв.) и HBTU (1,36 г, 1,15 экв.) объединяли в 10 мл ДМФА, смесь перемешивали при комнатной температуре в течение 5 мин и затем добавляли TEA (1,3 мл, 3,0 экв.). Полученную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разбавляли 100 мл этилацетата и 20 мл гексана, промывали 0,25 н. HCl, водой, насыщенным водным NaHCO3 и солевым раствором и концентрировали под вакуумом с получением промежуточного соединения 93В (1 г, выход 88%) в виде белого твердого вещества.
[0735] К раствору соединения 93В (1 г, 1,0 экв.) в 6 мл сухого ДХМ добавляли 3 мл 4 М HCl в диоксане. Полученную смесь перемешивали при комнатной температуре в течение 2 ч. ДХМ и диоксан удаляли под вакуумом, остаток разбавляли EtOAc, промывали насыщенным водным NaHCO3 и солевым раствором и концентрировали под вакуумом с получением промежуточного соединения 93С (650 мг, выход 90%) в виде белого твердого вещества.
[0736] Соединение 93С (125 мг, 1,0 экв.), соединение 12F (115 мг, 1,2 экв.) и HBTU (226 мг, 1,25 экв.) объединяли в 5 мл ДМФА, смесь перемешивали при комнатной температуре в течение 5 мин и затем добавляли DIEA (0,23 мл, 3,0 экв.). Полученную смесь перемешивали при комнатной температуре в течение 30 мин. Смесь разбавляли 50 мл этилацетата и 20 мл гексана, промывали водой, насыщенным водным NaHCO3 и солевым раствором и концентрировали под вакуумом с получением промежуточного соединения 93D (180 мг, выход 85%).
[0737] Соединение 6 (90 мг, 1,0 экв.) растворяли в 8 мл сухого ТГФ, охлаждали до -50°С в атмосфере N2. Раствор 1 н. LAH в ТГФ (0,22 мл, 1,1 экв.) добавляли по каплям при -50°С. Полученную смесь перемешивали при температуре от -30 до -10°С в течение 2 ч. В реакционную смесь добавляли для гашения реакции насыщенный водный NaHCO3 при -20°С, и затем смесь подвергали экстракции 3×15 мл ацетата. Объединенную органическую фазу сушили над Na2SO4. Неочищенную смесь очищали посредством колоночной хроматографии на силикагеле с получением соединения 93 (40 мг, 51%).
ПРИМЕР 55
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОТИАЗОЛ-4-КАРБОКСАМИД (96)
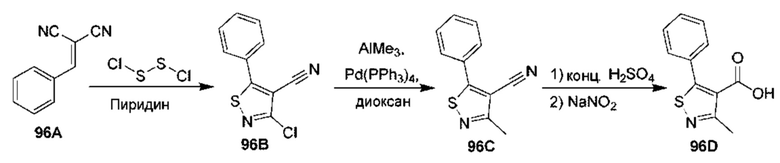
[0738] К раствору бензальдегида (10,00 г, 94,23 ммоль) и малононитрила (6,54 г, 98,94 ммоль) в EtOH (75,00 мл) добавляли катализатор пиперидин (80,24 мг, 942,30 мкмоль). Затем реакционную смесь перемешивали при 90°С в течение 2 ч. Когда реакционную смесь охлаждали до комнатной температуры, желтое твердое вещество выпадало в осадок, смесь фильтровали, целевое желтое твердое вещество промывали EtOH (20 мл) и сушили под вакуумом с получением промежуточного соединения 96А (23,00 г, выход 79,2%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,91 (d, J=7,7 Гц, 2Н), 7,79 (s, 1H), 7,67-7,60 (m, 1H), 7,58-7,50 (m, 2Н).
[0739] К смеси соединения 96А (17,50 г, 113,51 ммоль) и хлорсульфанилтиогипохлорита (70,00 г, 518,36 ммоль, 41,42 мл) добавляли пиридин (900,00 мг, 11,38 ммоль). Затем реакционную смесь перемешивали при 140°С в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли в нее для гашения реакции смесь лед/Н2О (200 мл) и EtOAc (500 мл), желтое твердое вещество выпадало в осадок, фильтровали, и фильтрат подвергали экстракции EtOAc (100 мл × 2), объединенную органическую фазу промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали, и фильтрат концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® Silica Flash массой 120 г, элюент 0~10% этилацетат/петролейный эфир, градиент со скоростью потока 50 мл/мин) с получением соединения 96В (21,00 г, выход 70,4%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,77 (ушир. d, J=7,1 Гц, 2Н), 7,65-7,53 (m, 3Н).
[0740] К смеси соединения 96В (2,00 г, 9,06 ммоль) в диоксане (150,00 мл) добавляли AlMe3 (2 М, 20,00 мл) и Pd(PPh3)4 (1,05 г, 906,00 мкмоль) в атмосфере N2, затем реакционную смесь перемешивали при 110°С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли в нее для гашения реакции смесь лед/Н2О (100 мл) и EtOAc (150 мл), желтое твердое вещество выпадало в осадок, фильтровали, и фильтрат подвергали экстракции EtOAc (60 мл × 2), объединенную органическую фазу промывали солевым раствором (70 мл), сушили над Na2SO4, фильтровали, и фильтрат концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® Silica Flash массой 40 г, элюент 0~10% этилацетат/петролейный эфир, градиент со скоростью потока 40 мл/мин) с получением соединения 96С (700,00 мг, выход 16,59%, чистота 43%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,79-7,75 (m, 2Н), 7,56-7,51 (m, 3Н), 2,67 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 200,9.
[0741] К соединению 96С (490,00 мг, 2,45 ммоль) добавляли H2SO4 (9,20 г, 93,81 ммоль, 5,00 мл), и реакционную смесь перемешивали при 135°С в течение 1,5 ч. Затем реакционную смесь охлаждали до 0°С и к описанной выше смеси добавляли раствор NaNO2 (339,79 мг, 4,92 ммоль) в Н2О (2,00 мл), и реакционную смесь перемешивали при 70°С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в смесь лед/Н2О (40 мл) и EtOAc (40 мл), подвергали экстракции EtOAc (50 мл × 2), объединенную органическую фазу подвергали экстракции 0,1 н. NaOH (40 мл × 2), к целевой основной водной фазе затем добавляли 1 н. HCl до рН < 4, затем ее подвергали экстракции EtOAc (40 мл × 3) и промывали солевым раствором (40 мл), сушили над Na2SO4, фильтровали, и фильтрат концентрировали под вакуумом с получением соединения 96D (410,00 мг, выход 76,25%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,49 (s, 5Н), 2,57 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 219,9.
[0742] Соединение 96 (35 мг, выход: 65,86%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 96D. Соединение 96: 1Н ЯМР (400 МГц, CD3CN) δ 7,48-7,33 (m, 5Н), 7,29-7,17 (m, 3Н), 7,15-7,06 (m, 3Н), 7,01 (ушир. s, 1H), 6,26 (ушир. s, 1H), 5,56 (ddd, J=4,4, 7,5, 9,5 Гц, 1H), 3,23 (dd, J=4,3, 14,2 Гц, 1H), 2,77 (dd, J=9,5, 14,3 Гц, 1H), 2,28 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 394,1.
ПРИМЕР 56
(S)-N-(4-АМИНО-1-(3,5-ДИМЕТИЛФЕНИЛ)-3,4-ДИОКСОБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (97)
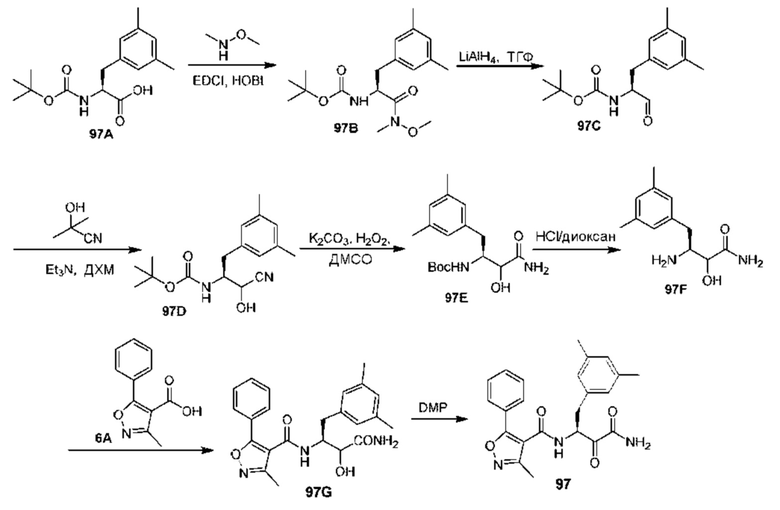
[0743] Смесь соединения 97А (1,0 г, 3,41 ммоль), соединения N,O-диметилгидроксиламина (400 мг, 4,09 ммоль, HCl), HOBt (460 мг, 3,41 ммоль) и NMM (1,03 г, 10,23 ммоль, 1,12 мл) в CHCl3 (20 мл) дегазировали и продували N2 3 раза при 0°С, затем добавляли порциями EDCI (980 мг, 5,12 ммоль). Смесь перемешивали при 25°С в течение 20 ч в атмосфере N2. В реакционную смесь добавляли Н2О (20 мл) для гашения реакции, и затем смесь разбавляли ДХМ (10 мл). Объединенные органические слои промывали 1 н. HCl (15 мл × 2), насыщенным водным NaHCO3 (15 мл × 2) и солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 97В (1,13 г, выход: 98,5%), которое было получено в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,08 (ушир. d, J=8,2 Гц, 1H), 6,82 (s, 3Н), 4,55 (ушир. s, 1H), 3,71 (ушир. s, 3Н), 3,09 (s, 3Н), 2,82-2,72 (m, 1H), 2,68-2,58 (m, 1H), 2,22 (s, 6Н), 1,32 (s, 9Н).
[0744] К раствору LAH (255 мг, 6,72 ммоль) в ТГФ (10 мл), который дегазировали и продували N2 3 раза при 0°С, добавляли по каплям смесь соединения 97В (1,13 г, 3,36 ммоль) в ТГФ (20 мл), и затем смесь перемешивали при 0°С в течение 2 ч в атмосфере N2. В реакционную смесь добавляли для гашения реакции EtOAc (10 мл), затем добавляли 1 н. HCl (50 мл), и затем смесь разбавляли EtOAc (20 мл), сушили над Na2SO4 и перемешивали в течение 30 мин, затем фильтровали с получением органических слоев. Объединенные органические слои промывали солевым раствором (20 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 97С (860 мг, выход: 92,3%), которое было получено в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,48 (s, 1H), 7,24 (ушир. d, J=7,7 Гц, 1H), 6,85-6,73 (m, 3Н), 4,08-3,94 (m, 1H), 3,04-2,91 (m, 1H), 2,70-2,57 (m, 1H), 2,20 (s, 6Н), 1,39-1,19 (m, 9Н).
[0745] К раствору соединения 97С (860 мг, 3,10 ммоль) в ДХМ (10 мл) добавляли соединение 2-гидрокси-2-метилпропаннитрил (530 мг, 6,20 ммоль, 570 мкл) и Et3N (470 мг, 4,65 ммоль, 650 мкл). Смесь перемешивали при 25°С в течение 22 ч. В реакционную смесь добавляли 1 н. HCl (20 мл) для гашения реакции, и затем смесь разбавляли H2O (20 мл) и подвергали экстракции ДХМ (20 мл × 2). Объединенные органические слои промывали солевым раствором (20 мл × 3), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 97D (930,00 мг, выход: 98,6%), которое было получено в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,19-6,99 (m, 1H), 6,91-6,78 (m, 3Н), 6,77-6,51 (m, 1H), 4,66-4,34 (m, 1H), 3,84 (ушир. s, 1H), 2,99-2,81 (m, 1H), 2,75-2,60 (m, 1H), 2,27 (ушир. s, 6Н), 1,40-1,20 (m, 9Н).
[0746] К раствору соединения 97D (930 мг, 3,63 ммоль) и K2CO3 (850 мг, 6,11 ммоль) в ДМСО (10 мл) добавляли Н2О2 (3,46 г, 30,55 ммоль, 2,94 мл, чистота: 30%). Смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь разбавляли H2O (100 мл) и подвергали экстракции EtOAc (30 мл × 3). Объединенные органические слои промывали солевым раствором (50 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток перемешивали в ДХМ (3 мл) и ПЭ (25 мл) в течение 30 мин и фильтровали с получением соединения 5 (970 мг, выход: 98,32%), которое было получено в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,42-7,08 (m, 1H), 6,86-6,45 (m, 3Н), 6,21-5,49 (m, 1H), 4,06-3,82 (m, 1H), 3,31 (s, 1H), 2,72-2,52 (m, 2Н), 2,26-2,13 (m, 6Н), 1,40-1,18 (m, 9Н).
[0747] К раствору соединения 97Е (970 мг, 3,01 ммоль) в EtOAc (5 мл) добавляли HCl/EtOAc (4 М, 5 мл). Смесь перемешивали при 25°С в течение 3 ч.
Реакционную смесь разбавляли ПЭ (20 мл), фильтровали и концентрировали при пониженном давлении с получением соединения 97F (370 мг, выход: 43,7%, HCl), которое было получено в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,09-7,89 (m, 3Н), 7,58 (ушир. s, 2Н), 6,96 (ушир. s, 2Н), 6,89 (s, 1H), 4,26 (ушир. s, 1H), 3,89 (ушир. s, 1H), 3,69-3,57 (m, 1H), 2,91-2,73 (m, 2Н), 2,30 (ушир. s, 6Н). МС (ИЭР) m/z (М+Н)+ 223,1.
[0748] Смесь соединения 97F (310 мг, 1,18 ммоль, HCl), соединения 6А (200 мг, 984,30 мкмоль), НОВТ (133 мг, 984,30 мкмоль) и DIEA (520 мкл, 2,95 ммоль) в ДХМ (15 мл) добавляли EDCI (285 мг, 1,48 ммоль), и затем смесь перемешивали при 25°С в течение 18 ч. Реакционную смесь промывали H2O (20 мл × 2). Объединенные органические слои промывали HCl (1 н., 30 мл), насыщенным водным NaHCO3 (30 мл) и солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток перемешивали в петролейном эфире (5 мл) и ДХМ (1 мл) в течение 30 мин и фильтровали с получением соединения 97G (270 мг, выход: 61,9%), которое было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,59-8,24 (m, 1H), 7,64-7,51 (m, 2Н), 7,49-7,28 (m, 5Н), 6,89-6,77 (m, 3Н), 5,98-5,63 (m, 1H), 4,61-4,49 (m, 1H), 4,11-3,86 (m, 1H), 2,86-2,59 (m, 2Н), 2,21-2,03 (m, 9Н). МС (ИЭР) m/z (М+Н)+ 408,1.
[0749] К раствору соединения 97G (100 мг, 245,42 мкмоль) в ДХМ (10 мл) добавляли DMP (320 мг, 736,26 мкмоль) при 0°С. Смесь перемешивали при 25°С в течение 7 ч. В реакционную смесь добавляли для гашения реакции насыщенный водный Na2S2O3 (15 мл) и насыщенный водный NaHCO3 (15 мл), смесь перемешивали в течение 0,2 ч и затем разбавляли ДХМ (10 мл) и подвергали экстракции H2O (20 мл × 3). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток перемешивали в петролейном эфире (15 мл) и EtOAc (1 мл) в течение 30 мин и фильтровали с получением соединения 97 (60 мг, выход: 60,3%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,01 (ушир. d, J=7,5 Гц, 1H), 8,18 (ушир. s, 1H), 7,90 (ушир. s, 1H), 7,64 (ушир. d, J=7,3 Гц, 2Н), 7,53-7,46 (m, 1H), 7,45-7,38 (m, 2Н), 6,92-6,81 (m, 3Н), 5,40 (ушир. t, J=7,3 Гц, 1H), 3,15 (ушир. d, J=10,6 Гц, 1H), 2,72-2,58 (m, 1H), 2,18 (s, 6Н), 2,11 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 406,1.
ПРИМЕР 57
(S)-N-(4-АМИНО-1-(2,5-ДИМЕТИЛФЕНИЛ)-3,4-ДИОКСОБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (98)
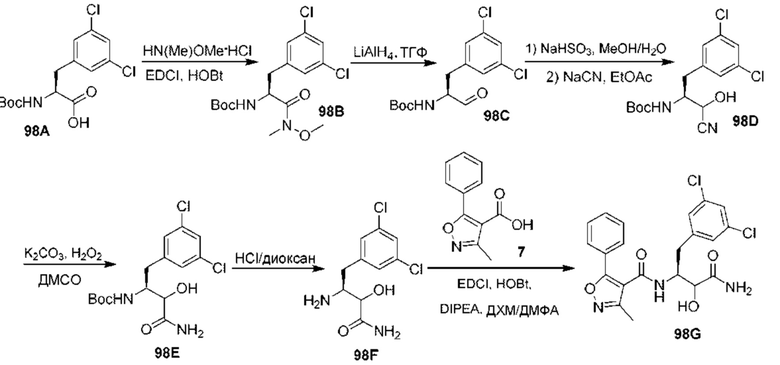
[0750] К раствору соединения 98А (1,0 г, 2,99 ммоль) и N-метоксиметанамина (321 мг, 3,29 ммоль, HCl) в CHCl3 (30 мл) добавляли HOBt (404 мг, 2,99 ммоль) и EDCI (803 мг, 4,19 ммоль). Затем в реакционную смесь добавляли NMM (1,3 мл, 11,96 ммоль). После добавления реакционную смесь перемешивали при 28°С в течение 14 ч. Реакционную смесь концентрировали под вакуумом, и остаток растворяли в 80 мл EtOAc. Смесь промывали 1 н. HCl (30 мл × 2) и насыщенным водным NaHCO3 (30 мл × 2), затем солевым раствором (30 мл). Смесь сушили над Na2SO4 и концентрировали под вакуумом с получением соединения 98В (1,1 г, выход 82,9%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,44 (s, 1H), 7,34-7,19 (m, 3Н), 4,55 (ушир. s, 1H), 3,71 (ушир. s, 3Н), 3,17-3,00 (m, 3Н), 2,90-2,80 (m, 1H), 2,76-2,67 (m, 1H), 1,29 (s, 8Н). МС (ИЭР) m/z (М-56)+ 320,9.
[0751] К раствору LiAlH4 (122 мг, 3,21 ммоль) в ТГФ (10 мл) добавляли раствор соединения 98В (1,1 г, 2,92 ммоль) в ТГФ (20 мл) при 0°С в атмосфере N2. После добавления реакционную смесь перемешивали при 0°С в течение 1 ч. В реакционную смесь добавляли 2 мл EtOAc при 0°С, и смесь перемешивали в течение 10 мин. Затем в реакционную смесь медленно добавляли 2 мл 1 н. HCl. После добавления смесь разбавляли 80 мл EtOAc, и смесь промывали 1 н. HCl (30 мл × 2), солевым раствором (30 мл). Затем смесь сушили над Na2SO4 и концентрировали под вакуумом с получением соединения 98С (800 мг, выход 80,9%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,52 (s, 1H), 7,53-7,17 (m, 4Н), 4,20-4,08 (m, 1H), 3,19-3,08 (m, 1H), 2,72-2,63 (m, 1H), 1,37-1,27 (m, 9Н).
[0752] К раствору соединения 98С (800 мг, 2,51 ммоль) в МеОН (10 мл) добавляли по каплям раствор NaHSO3 (261 мг, 2,51 ммоль) в H2O (15 мл) при 0-5°С. После этого реакционную смесь перемешивали при 25°С в течение 5 ч. В реакционную смесь добавляли NaCN (129 мг, 2,64 ммоль) в H2O (20 мл), затем EtOAc (40 мл). После этого реакционную смесь перемешивали при 25°C в течение 14 ч. Органический слой отделяли и промывали солевым раствором (30 мл), затем сушили над Na2SO4. Смесь концентрировали с получением соединения 98D (800 мг, выход 92,33%) в виде светло-желтой смолы. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,46-7,22 (m, 3Н), 7,16-7,02 (m, 1H), 6,89-6,70 (m, 1H), 4,65-4,30 (m, 1H), 3,95-3,76 (m, 1H), 3,07-2,87 (m, 1H), 2,76-2,55 (m, 1H), 1,32-1,20 (m, 8Н).
[0753] К раствору соединения 98D (800 мг, 2,32 ммоль) и K2CO3 (641 мг, 4,64 ммоль) в ДМСО (8 мл) добавляли Н2О2 (2 мл, 22,25 ммоль, чистота 30%) при 0°С. После добавления реакционную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь разбавляли ледяной водой (20 мл) и 50 мл насыщенного водного Na2SO3. Смесь подвергали экстракции EtOAc (50 мл × 3), и объединенные экстракты промывали насыщенным водным Na2SO3 (50 мл × 2). Органический слой сушили над Na2SO4 и концентрировали с получением неочищенного соединения. Неочищенное соединение разбавляли МТБЭ (5 мл) и фильтровали с получением соединения 98Е (800 мг, выход 94,9%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,48-7,12 (m, 5Н), 6,73-6,20 (m, 1H), 5,86-5,63 (m, 1H), 4,04-3,71 (m, 2Н), 2,86-2,54 (m, 1H), 1,34-1,19 (m, 9Н). МС (ИЭР) m/z (М+23)+ 384,9.
[0754] К раствору соединения 98Е (800 мг, 2,20 ммоль) в EtOAc (10 мл) добавляли HCl/EtOAc (4 М, 55 мл). После добавления реакционную смесь перемешивали при 26°С в течение 1 ч. В реакционную смесь добавляли 20 мл петролейного эфира, и смесь фильтровали с получением соединения 98F (400 мг, выход 58,87%, HCl) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,35 (ушир. s, 1H), 8,14 (ушир. s, 1H), 7,62-7,41 (m, 3H), 7,33 (d, J=1,8 Гц, 1H), 6,90-6,50 (m, 1H), 4,28 (ушир. s, 1H), 3,94-3,84 (m, 1H), 3,77-3,56 (m, 1H), 3,03-2,80 (m, 2Н).
[0755] К раствору соединения 7 (100 мг, 492,15 мкмоль) и соединения 98F (162 мг, 541,37 мкмоль, HCl) в ДМФА (10 мл) добавляли НОВТ (67 мг, 492,15 мкмоль) и DIEA (340 мкл, 1,97 ммоль), затем добавляли EDCI (133 мг, 689,01 мкмоль). После добавления реакционную смесь перемешивали при 26°С в течение 14 ч. Смесь разбавляли 30 мл EtOAc. Смесь промывали 1 н. HCl (15 мл × 2) и насыщенным водным NaHCO3 (15 мл × 3), затем солевым раствором (20 мл). Остаток сушили над Na2SO4 и концентрировали под вакуумом. Остаток разбавляли 4 мл EtOAc и фильтровали с получением соединения 98G (110 мг, выход 45,87%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,65-8,26 (m, 1H), 7,60-7,30 (m, 9Н), 7,28-7,17 (m, 1H), 6,04-5,65 (m, 1H), 4,73-4,56 (m, 1H), 4,11-4,06 (m, 0,5Н), 4,01-3,95 (m, 0,5Н), 3,01-2,70 (m, 2Н), 2,18-2,09 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 448,1.
[0756] К раствору соединения 98G (110 мг, 245,37 мкмоль) в ДХМ (30 мл) и ДМСО (4 мл) добавляли DMP (416 мг, 981,48 мкмоль). После добавления реакционную смесь перемешивали при 26°С в течение 2 ч. В реакционную смесь добавляли 10 мл насыщенного водного Na2S2O3 и 10 мл насыщенного водного NaHCO3, и смесь перемешивали в течение 20 мин. Затем смесь разделяли, органический слой промывали 10 мл насыщенного водного Na2S2O3 и 10 мл насыщенного водного NaHCO3, затем водой (20 мл) и солевым раствором (20 мл). Смесь сушили над Na2SO4 и концентрировали под вакуумом с получением соединения 98 (30 мг, выход 24,66%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,08 (d, J=7,5 Гц, 1H), 8,21 (s, 1H), 7,94 (s, 1H), 7,68-7,58 (m, 2Н), 7,54-7,42 (m, 4Н), 7,34 (d, J=1,8 Гц, 2Н), 5,45-5,33 (m, 1H), 3,28-3,19 (m, 1H), 2,83-2,73 (m, 1H), 2,12 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 446,0.
ПРИМЕР 58
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(6-МЕТОКСИПИРИДИН-2-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (99)
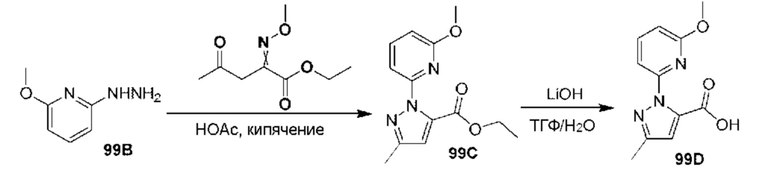
[0757] Смесь соединения 2-хлор-6-метоксипиридина (5,0 г, 34,83 ммоль) в NH2NH2⋅H2O (17,44 г, 348,30 ммоль, 16,93 мл) перемешивали при 120°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении с получением остатка, затем разбавляли H2O (30 мл) и подвергали экстракции этилацетатом (40 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 5:1 до 1:1) с получением соединения 99В (1,06 г, выход 21,87%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,41 (t, J=7,8 Гц, 1H), 6,24-6,08 (m, 2Н), 5,73 (ушир. s, 1H), 3,86 (s, 3Н), 3,83-2,75 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 140,1.
[0758] Смесь соединения 99В (1,00 г, 7,19 ммоль) и этил-2-(метоксиимино)-4-оксопентаноата (1,35 г, 7,19 ммоль) в НОАс (20,00 мл) перемешивали при 120°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении для удаления НОАс. Остаток разбавляли H2O (20 мл) и подвергали экстракции этилацетатом (20 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4, концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1:0 до 5:1) и дополнительно очищали посредством препаративной ВЭЖХ (в условиях ТФУ) с получением соединения 99С (487,00 мг, выход 25,87%), которое было получено в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,70 (t, J=7,9 Гц, 1H), 7,21 (d, J=7,5 Гц, 1H), 6,71 (d, J=8,2 Гц, 1H), 6,66 (s, 1H), 4,27 (q, J=7,2 Гц, 2Н), 3,85 (s, 3Н), 2,36 (s, 3Н), 1,25 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 261,9.
[0759] К раствору соединения 99С (487,00 мг, 1,99 ммоль) в ТГФ (15,00 мл) добавляли LiOH⋅H2O (417,50 мг, 9,95 ммоль) в H2O (5,00 мл). Смесь перемешивали при 28°С в течение 16 ч. Реакционную смесь разбавляли H2O (10 мл) и подвергали экстракции МТБЭ (15 мл × 2), в водную фазу добавляли 1 н. HCl до рН=3~4, подвергали ее экстракции ЭА (15 мл × 2). Объединенные органические слои промывали солевым раствором (15 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 99D (396 мг, выход 91,61%) в виде белого твердого вещества. Соединение 99D: 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,86 (t, J=7,8 Гц, 1H), 7,26 (d, J=7,5 Гц, 1H), 6,80 (d, J=8,2 Гц, 1H), 6,67 (s, 1H), 3,80 (s, 3Н), 2,26 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 234,1.
[0760] Соединение 99 (10,00 мг, выход 13,78%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 99D. Соединение 99: 1Н ЯМР (400 МГц, CDCl3) δ 7,68 (ушир. t, J=7,9 Гц, 1H), 7,26-7,18 (m, 4Н), 7,12-7,01 (m, 3Н), 6,73 (ушир. s, 1H), 6,68-6,60 (m, 1H), 6,65 (ушир. d, J=8,2 Гц, 1H), 6,50 (s, 1H), 5,73-5,64 (m, 1H), 5,50 (ушир. s, 1H), 3,67 (s, 3Н), 3,45-3,35 (m, 1H), 3,25-3,11 (m, 1H), 2,33 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 408,1.
ПРИМЕР 59
СОЕДИНЕНИЯ 101, 493
(S)-N-(4-((3,4-ДИХЛОРБЕНЗИЛ)АМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (101)
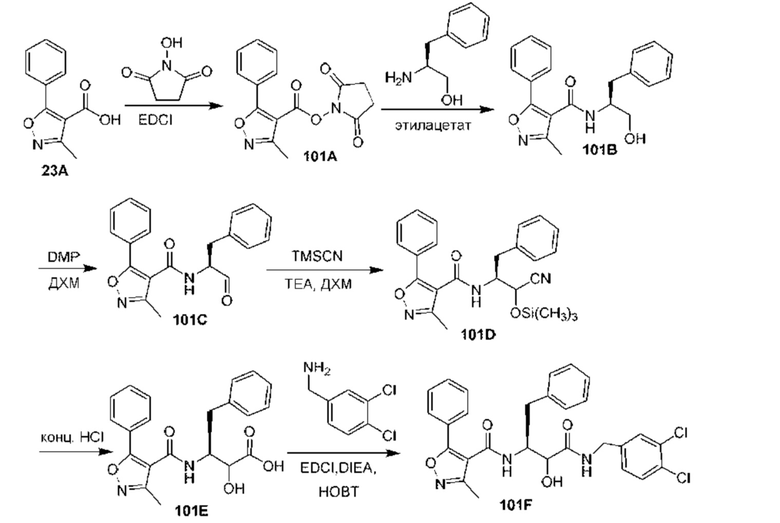
[0761] К раствору соединения 23А (20,00 г, 98,43 ммоль) в ТГФ (300 мл) добавляли 1-гидроксипирролидин-2,5-дион (12,46 г, 108,27 ммоль) и EDCI (22,64 г, 118,12 ммоль) с ДХМ (200 мл). Смесь перемешивали при 25°С в течение 12 часов. Реакционную смесь концентрировали и разбавляли этилацетатом (200 мл). Затем смесь промывали HCl (1 М, 200 мл), насыщенным водным NaHCO3 (200 мл), сушили над Na2SO4 и концентрировали. Соединение 101А (28,00 г, неочищенное) получали в виде желтого масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,94-7,88 (m, 2Н), 7,69-7,63 (m, 1H), 7,62-7,56 (m, 2Н), 2,87 (ушир. s, 4Н), 2,50-2,48 (m, 3H).
[07621 К раствору соединения 101А (28,00 г, 93,25 ммоль) в ДМФА (200 мл) добавляли (2S)-2-амино-3-фенилпропан-1-ол (15,51 г, 102,57 ммоль). Смесь перемешивали при 25°С в течение 12 часов. Смесь разбавляли H2O (1000 мл), подвергали экстракции этилацетатом (1000 мл), органический слой промывали HCl (водный раствор, 1000 мл), NaHCO3 (водный, 1000 мл), сушили над Na2SO4 и концентрировали. Соединение 3 (20,00 г, выход 63,8%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,45 (ушир. d, J=8,8 Гц, 1H), 7,66-7,61 (m, 2H), 7,53-7,40 (m, 3H), 7,32-7,18 (m, 5H), 4,97-4,92 (m, 1H), 4,33-4,23 (m, 1H), 3,54-3,41 (m, 2H), 3,01-2,97 (m, 1H), 2,69-2,57 (m, 1H), 2,06 (s, 3H).
[0763] К раствору соединения 101В (3,00 г, 8,92 ммоль) в ДХМ (100 мл) добавляли DMP (5,67 г, 13,38 ммоль). Смесь перемешивали при 25°С в течение 3 часов. В смесь добавляли для гашения реакции 10% Na2S2O3 (водный) : насыщенный NaHCO3 (водный) (1:1, 200 мл), смесь подвергали экстракции ДХМ (200 мл) и промывали солевым раствором (200 мл × 3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Соединение 101С (2,70 г, выход 90,5%) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,66 (s, 1H), 8,91 (d, J=8,4 Гц, 1H), 7,67-7,63 (m, 2Н), 7,53-7,47 (m, 1H), 7,46-7,40 (m, 2Н), 7,29-7,19 (m, 5Н), 4,79-4,72 (m, 1H), 3,37-3,32 (m, 1H), 2,81-2,72 (m, 1H), 2,09 (s, 3H).
[0764] К раствору соединения 101С (500,0 мг, 1,50 ммоль) в ДХМ (20 мл) добавляли TMSCN (223,2 мг, 2,25 ммоль, 280 мкл) и TEA (15,2 мг, 150,00 мкмоль, 20 мкл). Смесь перемешивали при 0°С в течение 3 часов. Смесь концентрировали, разбавляли этилацетатом (20 мл), промывали водой (20 мл), солевым раствором (20 мл), сушили над Na2SO4 и концентрировали с получением соединения 101D (600,0 мг, неочищенного) в виде бесцветного масла.
[0765] К раствору соединения 101D (600,0 мг, 1,41 ммоль) в ТГФ (10 мл) добавляли HCl (10 мл). Смесь перемешивали при 60°С в течение 12 часов. Смесь разбавляли H2O (200 мл), подвергали экстракции этилацетатом (100 мл), органический слой промывали NaHCO3 (водный, 100 мл), в водную фазу добавляли HCl (1 М) до рН~1, затем подвергали ее экстракции этилацетатом (100 мл), органический слой промывали солевым раствором (100 мл), сушили над Na2SO4 и концентрировали. Соединение 101Е (240,0 мг, неочищенное) получали в виде бесцветного масла и непосредственно использовали на следующей стадии.
[0766] К раствору соединения 101Е (200,0 мг, 526 мкмоль) в ТГФ (10,00 мл) добавляли (3,4-дихлорфенил)метанамин (92,6 мг, 525,78 мкмоль, 70 мкл), DIEA (203,85 мг, 1,58 ммоль, 275,48 мкл), HOBt (71,04 мг, 525,78 мкмоль) и EDCI (120,95 мг, 630,93 мкмоль). Смесь перемешивали при 25°С в течение 4 часов. Смесь концентрировали и разбавляли этилацетатом (50 мл), промывали HCl (1 М, 50 мл), насыщенным NaHCO3 (водный, 50 мл), солевым раствором (50 мл × 3), сушили над Na2SO4 и концентрировали. Смесь растирали с CH3CN (5 мл) и фильтровали. Соединение 101F (70,0 мг, выход 24,7%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,63-8,53 (m, 1H), 8,30 (d, J=9,2 Гц, 1H), 7,58-7,10 (m, 13Н), 6,20-5,94 (m, 1H), 4,68-4,57 (m, 1H), 4,32-4,16 (m, 2Н), 4,08-3,99 (m, 1H), 2,97-2,67 (m, 2Н), 2,07-1,96 (m, 1H), 2,07-1,96 (m, 2Н).
[0767] К раствору соединения 101F (60,0 мг, 111,44 мкмоль) в ДХМ (10 мл) и ДМСО (1,00 мл) добавляли DMP (141,8 мг, 334,32 мкмоль). Смесь перемешивали при 25°С в течение 3 часов. В смесь добавляли для гашения реакции 10% Na2S2O3 (водный) : насыщенный NaHCO3 (водный) (1:1, 20 мл), смесь подвергали экстракции ДХМ (10 мл) и промывали солевым раствором (20 мл × 3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Соединение 101 (33,2 мг, выход 55,5%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,52-9,43 (m, 1H), 9,12 (d, J=7,6 Гц, 1H), 7,69-7,38 (m, 7Н), 7,35-7,20 (m, 6Н), 5,53-5,42 (m, 1H), 4,40-4,32 (m, 2Н), 3,31-3,19 (m, 1H), 2,93-2,71 (m, 1H), 2,12-2,00 (m, 3H). МС (ИЭР) m/z (М+Н)+ 536,1.
(S)-N-(4-(((1H-БЕНЗО[d]ИМИДАЗОЛ-5-ИЛ)МЕТИЛ)АМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-1САРБОКСАМИД (493)
[0768] Соединение 493 (20 мг, выход 23,4%, желтое твердое вещество) было получено, как в случае соединения 101, из соответствующей карбоновой кислоты, промежуточного соединения 101Е и (1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-5-ил)метанамина с последующим удалением 2-(триметилсилил)этокси)метильной группы с получением соединения 493. Соединение 493: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,07 (s, 1H), 7,67-7,59 (m, 3H), 7,56 (ушир. s, 1H), 7,52-7,38 (m, 4Н), 7,30 (ушир. s, 1H), 7,25-7,14 (m, 4Н), 6,89 (ушир. d, J=6,2 Гц, 2Н), 6,12 (ушир. d, J=6,8 Гц, 1H), 5,72-5,63 (m, 1H), 4,62 (ушир. d, J=5,5 Гц, 2Н), 3,37 (ушир. dd, J=4,7, 14,0 Гц, 1H), 2,99 (ушир. dd, J=7,9, 14,3 Гц, 1H), 2,33 (s, 3H). МС (ИЭР) m/z (М+Н)+ 378,1.
ПРИМЕР 60
(S)-1-(1H-ИНДАЗОЛ-3-ИЛ)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (102)
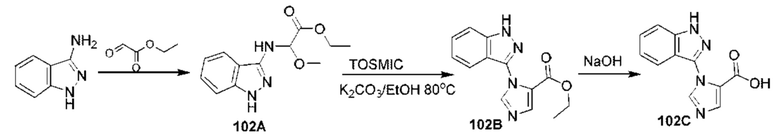
[0769] К раствору 1H-индазол-3-амина (8,7 г, 65,3 ммоль) в МеОН (90 мл) добавляли этил-2-оксоацетат (20 г, 98,01 ммоль). Смесь перемешивали при 25°С в течение 2 часов. Смесь фильтровали и концентрировали с получением неочищенного продукта 102А (15 г, неочищенного) в виде коричневого твердого вещества, которое использовали на следующей стадии без очистки.
[0770] К раствору 102А (15 г, 69,1 ммоль) в EtOH (400 мл) добавляли K2CO3 (14,5 г, 104 ммоль) и TosMIC (11,6 г 59,4 ммоль). Смесь перемешивали при 90°С в течение 0,5 часа. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1:0 до 1:1) с получением соединения 102В (2,9 г, выход: 16,4%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 11,04 (ушир. s, 1H), 7,98 (d, J=0,7 Гц, 1H), 7,91 (s, 1H), 7,48-7,41 (m, 3H), 7,25-7,19 (m, 1H), 4,24-4,14 (m, 2Н), 1,14 (t, J=7,1 Гц, 3H).
[0771] К раствору 102В (2,9 г, 11,3 ммоль) в ТГФ (40 мл) и H2O (8 мл) добавляли NaOH (905 мг, 22,6 ммоль). Смесь перемешивали при 25°С в течение 10 часов. Смесь концентрировали при пониженном давлении для удаления органического растворителя и подвергали экстракции EtOAc (20 мл). Водный слой подкисляли с помощью 1 М HCl до рН~5 и затем подвергали экстракции EtOAc (30 мл × 3). Объединенный органический слой промывали H2O (40 мл), солевым раствором (40 мл), сушили над Na2SO4, фильтровали и концентрировали с получением 102С (1,5 г, выход: 58,1%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,35 (s, 1H), 8,16 (s, 1H), 7,83 (s, 1H), 7,61 (d, J=8,3 Гц, 1H), 7,47-7,41 (m, 2Н), 7,20-7,15 (m, 1H). МС (ИЭР) m/z (М+Н)+ 228,9.
[0772] Соединение 102 (20 мг, выход 52,9%, бледно-желтое твердое вещество) было получено, как описано в примере 6, из соответствующих промежуточных соединений 102С и 21G ((S)-2-амино-3-фенилпропан-1-ола). Соединение 102: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 13,22 (s, 1H), 9,48 (s, 1H), 8,95 (d, J=8,0 Гц, 1H), 8,07 (s, 1H), 7,72 (s, 1H), 7,59-7,51 (m, 1H), 7,42-7,40 (m, 1H), 7,31-7,24 (m, 2Н), 7,24-7,18 (m, 4Н), 7,12-7,06 (m, 1H), 7,06-7,06 (m, 1H), 4,34-4,23 (m, 1H), 3,19-3,15 (m, 1H), 2,77-2,74 (m, 1H). МС (ИЭР) m/z (М+Н)+ 360,1.
ПРИМЕР 61
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(3'-МЕТИЛ-[1,1'-БИФЕНИЛ]-3-ИЛ)-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (105)
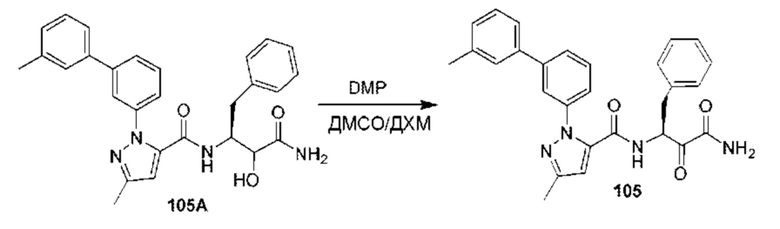
[0773] К смеси соединения 103А (150 мг, 0,33 ммоль) и м-толилбороновой кислоты (89 мг, 0,66 ммоль) в ТГФ (50 мл) добавляли H2O (10 мл), Na2CO3 (70 мг, 0,66 ммоль) и Pd(PPh3)4 (38 мг, 0,033 ммоль) одной порцией при 25°С в атмосфере N2. Смесь перемешивали при 80°С в течение 12 ч. В реакционную смесь добавляли H2O (100 мл), и смесь подвергали экстракции ЭА (100 мл × 3). Объединенный органический слой промывали насыщенным водным NaHCO3 (150 мл × 3), солевым раствором (150 мл × 3), сушили над Na2SO4 и концентрировали. Неочищенный продукт обрабатывали смесью простой i-пропиловый эфир/CH3CN (10/1, 10 мл). Твердое вещество собирали и сушили под вакуумом с получением соединения 2А (72,7 мг, выход 42,70%) в виде серого твердого вещества. Соединение 105А: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,51-8,10 (m, 1H), 7,60-7,52 (m, 2Н), 7,47-7,38 (m, 2Н), 7,36-7,28 (m, 3H), 7,26-7,12 (m, 7Н), 7,03-6,93 (m, 1H), 6,57 (d, J=3,3 Гц, 1H), 5,93-5,73 (m, 1H), 4,49-4,29 (m, 1H), 4,04-3,86 (m, 1H), 2,90-2,81 (m, 1H), 2,81-2,72 (m, 1H), 2,35 (s, 3H), 2,24 (s, 3H). МС (ИЭР) m/z (М+Н)+ 469,2.
[0774] К смеси соединения 105А (65 мг, 0,14 ммоль) в ДХМ (10 мл) и ДМСО (1 мл) добавляли DMP (177 мг, 0,42 ммоль) одной порцией при 0°С. Смесь перемешивали при 0°С в течение 10 мин, затем поднимали температуру до 25°С и перемешивали в течение 2 часов. В реакционную смесь добавляли для гашения реакции 20 мл 10% водного раствора Na2S2O3 и 20 мл насыщенного водного раствора NaHCO3, и затем смесь подвергали экстракции ДХМ (30 мл × 3). Объединенную органическую фазу промывали солевым раствором (40 мл × 3), сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством препаративной ВЭЖХ (в основных условиях) с получением соединения 105 (35,0 мг, выход 53,6%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц) δ 9,08 (d, J=1,1 Гц, 1H), 8,08-8,00 (m, 1H), 7,84 (ушир. s, 1H), 7,60-7,52 (m, 2Н), 7,45-7,37 (m, 3H), 7,36-7,30 (m, 1H), 7,28-7,25 (m, 3H), 7,23-7,16 (m, 3H), 7,12-7,06 (m, 1H), 6,60 (ушир. s, 1H), 5,29 (ушир. s, 1H), 3,22-3,14 (m, 1H), 2,86-2,76 (m, 1H), 2,35 (s, 3H), 2,28-2,22 (m, 3H). МС (ИЭР) m/z (М+Н)+ 467,2.
ПРИМЕР 62
СОЕДИНЕНИЯ 103, 106, 216-218, 214
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4,-ФТОР-[1,1'-БИФЕНИЛ]-3-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (103)
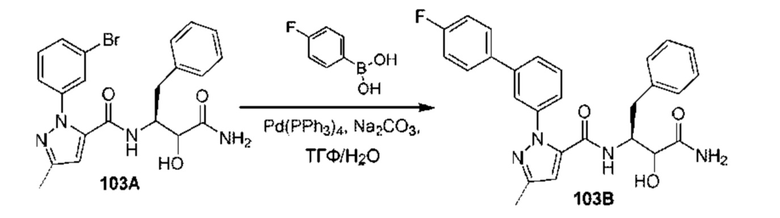
[0775] Соединение 103В (110 мг, выход 70,98%, грязно-белое твердое вещество) было получено, как описано в примере 49, из соответствующих промежуточных соединений 103А и (4-фторфенил)бороновой кислоты. Соединение 103В: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,45 (d, J=9,0 Гц, 0,5Н), 8,15 (d, J=9,0 Гц, 0,5Н), 7,68-7,52 (m, 4Н), 7,40-7,13 (m, 10Н), 7,02 (ушир. d, J=8,4 Гц, 0,5Н), 6,94 (ушир. d, J=7,9 Гц, 0,5Н), 6,59 (d, J=2,4 Гц, 1H), 5,93-5,74 (m, 1H), 4,49-4,32 (m, 1H), 4,02-3,88 (m, 1H), 2,95-2,66 (m, 2Н), 2,26-2,19 (m, 3H).
[0776] Соединение 103 (78 мг, выход 68,93%, бледно-желтое твердое вещество) было получено, как описано в примере 61, из соответствующих промежуточных соединений 103В. Соединение 103: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,09 (d, J=1,1 Гц, 1H), 8,09 (s, 1H), 7,86 (s, 1H), 7,65 (dd, J=5,4, 8,7 Гц, 2Н), 7,58 (ушир. d, J=1,1 Гц, 1H), 7,52 (s, 1H), 7,41 (t, J=7,8 Гц, 1H), 7,32-7,25 (m, 6Н), 7,23-7,19 (m, 1H), 7,11 (ушир. d, J=1,9 Гц, 1H), 6,60 (s, 1H), 5,35-5,25 (m, 1H), 3,18 (dd, J=3,5, 13,7 Гц, 1H), 2,81 (dd, J=10,4, 13,7 Гц, 1H), 2,25 (s, 3H). МС (ИЭР) m/z (М+Н)+ 471,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(3'-ФТОР-[1,1'-БИФЕНИЛ]-3-ИЛ)-3-МЕТИЛ-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (106)
[0777] Соединение 106 (18,7 мг, выход 46,2%, светло-желтое твердое вещество) было получено, как описано в примере 61, из соответствующих исходных веществ, соединения 103А и (3-фторфенил)бороновой кислоты. Соединение 106: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,08 (ушир. d, J=7,7 Гц, 1H), 8,05 (ушир. s, 1H), 7,85 (ушир. s, 1H), 7,69-7,62 (m, 1H), 7,60-7,56 (m, 1H), 7,52-7,43 (m, 3H), 7,43-7,37 (m, 1H), 7,33-7,24 (m, 4Н), 7,24-7,15 (m, 2Н), 7,13-7,05 (m, 1H), 6,62 (s, 1H), 5,35-5,25 (m, 1H), 3,20-3,15 (m, 1H), 2,86-2,76 (m, 1H), 2,28-2,21 (m, 3H). МС (ИЭР) m/z (М+Н)+ 471,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(3'-ФТОР-[1,1'-БИФЕНИЛ]-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (216)
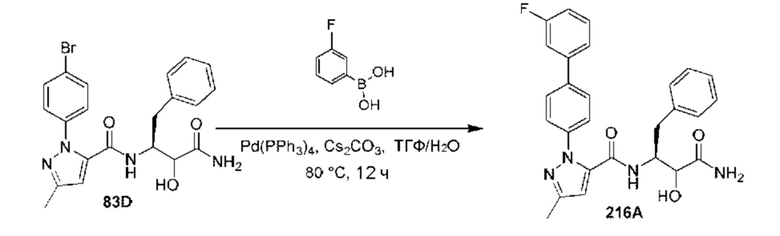
[0778] Соединение 216 (26 мг, выход 16,7%, желтое твердое вещество) было получено, как описано в примере 62, из соответствующих исходных веществ, соединения 83D и (3-фторфенил)бороновой кислоты. Соединение 216: 1Н ЯМР (CDCl3, 400 МГц) δ 7,59 (ушир. d, J=7,7 Гц, 2Н), 7,47-7,35 (m, 4Н), 7,33-7,27 (m, 4Н), 7,05 (ушир. d, J=6,4 Гц, 3H), 6,75 (ушир. s, 1H), 6,49 (s, 1H), 6,42-6,33 (m, 1H), 5,66-5,50 (m, 2Н), 3,39 (ушир. dd, J=5,0, 13,8 Гц, 1H), 3,16 (ушир. dd, J=7,4, 14,0 Гц, 1H), 2,40-2,29 (m, 3H). МС (ИЭР) m/z (М+Н)+ 471,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2'-ФТОР-[1,1'-БИФЕНИЛ]-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (217)
[0779] Соединение 217 было получено, как описано в примере 62, из соответствующих исходных веществ, соединения 83D и (2-фторфенил)бороновой кислоты. Соединение 217 (13 мг, выход 14,49%, желтое твердое вещество): 1Н ЯМР (CDCl3, 400 МГц) δ 7,59 (d, J=7,3 Гц, 2Н), 7,49-7,39 (m, 3H), 7,37-7,26 (m, 3H), 7,24-7,13 (m, 3H), 7,02 (ушир. d, J=6,0 Гц, 2H), 6,72 (ушир. s, 1H), 6,49 (s, 1H), 6,37-6,29 (m, 1H), 5,62-5,49 (m, 2H), 3,36 (dd, J=5,3, 14,1 Гц, 1H), 3,11 (dd, J=7,4, 14,0 Гц, 1H), 2,38-2,29 (m, 3H). МС (ИЭР) m/z (M+H)+ 471,2.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(3'-МЕТИЛ-[1,1'-БИФЕНИЛ]-4-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (218)
[0780] Соединение 218 было получено, как описано в примере 62, из соответствующих исходных веществ, соединения 83D и м-толилбороновой кислоты. Соединение 218 (выход 36,1%, желтое твердое вещество): 1Н ЯМР (CDCl3, 400 МГц) δ 9,16 (d, J=7,7 Гц, 1H), 8,11 (s, 1H), 7,87 (s, 1H), 7,60 (d, J=8,6 Гц, 2Н), 7,50-7,43 (m, 2Н), 7,38-7,28 (m, 5Н), 7,26-7,21 (m, 3H), 7,18 (ушир. d, J=7,5 Гц, 1H), 6,54 (s, 1H), 5,28-5,18 (m, 1H), 3,20 (ушир. dd, J=3,6, 13,8 Гц, 1H), 2,83 (dd, J=10,6, 13,7 Гц, 1H), 2,37 (s, 3H), 2,24 (s, 3H).
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2'-ФТОР-[1,1'-БИФЕНИЛ]-3-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (214)
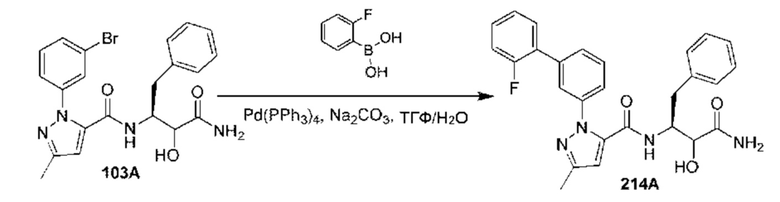
[0781] Соединение 214 было получено, как описано в примере 62, из соответствующих исходных веществ, соединения 103А и (2-фторфенил)бороновой кислоты. Соединение 214 (20 мг, выход 29,5%, белое твердое вещество): 1Н ЯМР (CDCl3, 400 МГц) δ 7,62-7,55 (m, 2Н), 7,52-7,43 (m, 2Н), 7,41-7,30 (m, 2Н), 7,26-7,22 (m, 3H), 7,21-7,13 (m, 2Н), 7,03-6,94 (m, 2Н), 6,65 (ушир. s, 1H), 6,49 (s, 1H), 6,33-6,26 (m, 1H), 5,56-5,52 (m, 1H), 5,37 (ушир. s, 1H), 3,38-3,31 (m, 1H), 3,17-3,09 (m, 1H), 2,36-2,30 (m, 3H). МС (ИЭР) m/z (М+Н)+ 471,1.
ПРИМЕР 63
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ФЕНИЛФУРАН-2-КАРБОКСАМИД (104)
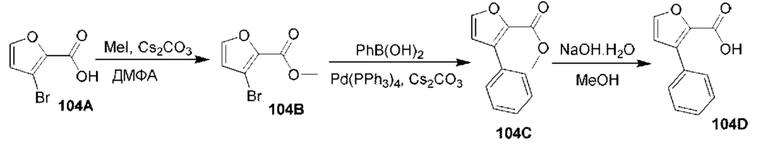
[0782] К раствору i-Pr2NH (3 мл, 18,71 ммоль) в безводном ТГФ (13 мл) добавляли n-BuLi (7 мл, 18,71 ммоль) по каплям при -78°С и перемешивали при 0°С в течение 30 мин. Затем к смеси добавляли по каплям раствор 3-бромфурана (2,5 г, 17,01 ммоль) в ТГФ (13 мл) при -78°С, и смесь перемешивали при -78°С в течение 30 минут. В раствор выливали безводный СО2 при -78°С в течение 30 минут. В реакционную смесь добавляли H2O (20 мл) для гашения реакции, и смесь подвергали экстракции этилацетатом (20 мл), затем водную фазу обрабатывали HCl до рН~3. Осадок отфильтровывали и сушили при пониженном давлении. Соединение 104А (1,8 г, неочищенное) получали в виде желтого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,96 (d, J=1,8 Гц, 1H), 6,89 (d, J=1,8 Гц, 1H).
[0783] Cs2CO3 (2,13 г, 6,55 ммоль) добавляли к раствору соединения 104А (500 мг, 2,62 ммоль) в ДМФА (10 мл). Затем к смеси добавляли MeI (652,43 мкл, 10,48 ммоль). Смесь перемешивали при 25°С в течение 13 ч. Смесь разбавляли этилацетатом (35 мл) и H2O (30 мл). Органический слой отделяли, и водный слой промывали, подвергали экстракции этилацетатом (20 мл × 2). Объединенный органический слой промывали солевым раствором (30 мл), сушили над MgSO4, фильтровали и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии (петролейный эфир/этилацетат = 15/1). Соединение 104В (250 мг, выход 46,54%) получали в виде белого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц) δ 7,50 (d, J=2,0 Гц, 1H), 6,61 (d, J=2,0 Гц, 1H), 3,94-3,92 (m, 3H).
[0784] К раствору соединения 104В (221 мг, 1,08 ммоль) в ТГФ (4 мл) и H2O (2 мл) добавляли фенилбороновую кислоту (263 мг, 2,16 ммоль) и Cs2CO3 (553 мг, 1,70 ммоль), затем Pd(PPh3)4 (125 мг, 108,00 мкмоль), затем смесь нагревали до 80°С и перемешивали в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли в нее для гашения реакции H2O (6 мл). Смесь подвергали экстракции этилацетатом (10 мл × 2). Объединенный органический слой промывали H2O (10 мл), солевым раствором (10 мл), сушили над Na2SO4, фильтровали, упаривали при пониженном давлении. Остаток очищали посредством КФХ (ПЭ/ЭА: от 0 до 10/1). Соединение 104С (180 мг, выход 82,42%) получали в виде желтого масла. 1Н ЯМР (CDCl3, 400 МГц) δ 7,61-7,55 (m, 3H), 7,45-7,35 (m, 3H), 6,64 (d, J=1,8 Гц, 1H), 3,86 (s, 3H).
[0785] К раствору соединения 104С (170 мг, 840,71 мкмоль) в МеОН (5 мл) добавляли по каплям NaOH (2 М, 2 мл), затем смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь разбавляли H2O (5 мл) и удаляли растворитель при пониженном давлении, затем смесь подвергали экстракции МТБЭ (5 мл). Водную фазу обрабатывали HCl (1 М) до рН~3, затем водную фазу подвергали экстракции этилацетатом (5 мл × 3). Органический слой промывали солевым раствором, сушили над безводным Na2SO4, упаривали при пониженном давлении. Соединение 104D (120 мг, выход 75,85%) получали в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1H ЯМР (ДМСО-d6, 400 МГц) δ 7,90 (d, J=1,8 Гц, 1H), 7,59-7,53 (m, 2Н), 7,39-7,28 (m, 3H), 6,80 (d, J=1,8 Гц, 1H).
[0786] Соединение 104 (35 мг, выход 44,0%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 104D. Соединение 104: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,56 (d, J=7,5 Гц, 1H), 8,07 (s, 1H), 7,88 (d, J=1,8 Гц, 1H), 7,81 (s, 1H), 7,61-7,54 (m, 2Н), 7,36-7,29 (m, 3H), 7,29-7,25 (m, 4Н), 7,21-7,17 (m, 1H), 6,90-6,83 (m, 1H), 5,39-5,29 (m, 1H), 3,21-3,12 (m, 1H), 3,01-2,92 (m, 1H). МС (ИЭР) m/z (М+Н)+ 363,1.
ПРИМЕР 64
СОЕДИНЕНИЯ 107, 243, 253, 265, 168, 459, 460, 475
(S)-N-(4-АМИНО-1-(4-ФТОРФЕНИЛ)-3,4-ДИОКСОБУТАН-2-ИЛ)-2-МЕТИЛ-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (107)
(S)-N-(1-АМИНО-1,2-ДИОКСОПЕНТАН-3-ИЛ)-2-МЕТИЛ-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (243)
(S)-N-(1-АМИНО-5-МЕТИЛ-1,2-ДИОКСОГЕКСАН-3-ИЛ)-2-МЕТИЛ-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (253)
(S)-N-(1-АМИНО-1,2-ДИОКСОГЕПТАН-3-ИЛ)-2-МЕТИЛ-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (265)
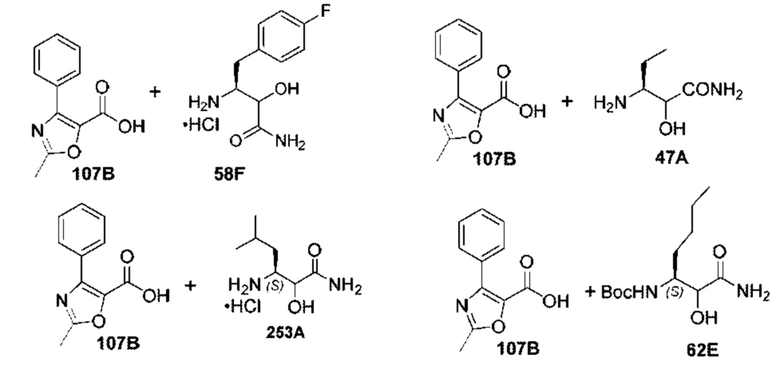
[0787] Соединения 107, 243, 253 и 265 были получены, как описано в примере 5, из соответствующих исходных веществ соответственно, соединения 107В и соединения 58F, 47А, 253А или 62Е.
[0788] Соединение 107 (77,3 мг, выход 51,80%, белое твердое вещество): 1Н ЯМР (400 МГц, CDCl3) δ 8,12-8,05 (m, 2Н), 7,46-7,36 (m, 3H), 7,12-7,05 (m, 2Н), 7,00-6,94 (m, 2Н), 6,79-6,70 (m, 2Н), 5,72-5,64 (m, 1H), 5,53 (ушир. s, 1H), 5,57-5,47 (m, 1H), 3,46-3,38 (m, 1H), 3,24-3,16 (m, 1H), 2,56 (s, 3H). МС (ИЭР) m/z (М+Н)+ 396,1.
[0789] Соединение 243 (52,8 мг, выход 42,87%, желтое твердое вещество): 1Н ЯМР (400 МГц, CDCl3): δ 8,13 (d, J=6,8 Гц, 2Н), 7,47-7,33 (m, 3H), 6,91-6,81 (m, 1H), 6,75 (ушир. s, 1H), 5,53-5,36 (m, 2Н), 2,58 (s, 3H), 2,20-2,08 (m, 1Н),1,88-1,76 (m, 1H), 0,99 (t, J=7,2 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 316,1.
[0790] Соединение 253 (6,5 мг, выход 6,42%, белое твердое вещество): 1Н ЯМР (CDCl3, 400 МГц): δ 8,19-8,09 (m, 2Н), 7,50-7,34 (m, 3H), 6,84-6,68 (m, 2Н), 5,55-5,38 (m, 2Н), 2,60 (s, 3H), 1,87-1,74 (m, 2Н), 1,63-1,58 (m, 1H), 1,04 (d, J=6,4 Гц, 3H), 0,98 (d, J=6,4 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 344,1.
[0791] Соединение 265 (79,7 мг, чистота 94,04%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,73 (d, J=6,8 Гц, 1H), 8,13-8,03 (m, 3H), 7,79 (s, 1H), 7,45-7,35 (m, 3H), 5,17-5,10 (m, 1H), 2,56 (s, 3H), 1,87-1,76 (m, 1H), 1,73-1,60 (m, 1H), 1,45-1,26 (m, 4Н), 0,93-0,83 (m, 3H). МС (ИЭР) m/z (М+Н)+ 344,1.
(S)-N-(4-АМИНО-1-(4-ФТОРФЕНИЛ)-3,4-ДИОКСОБУТАН-2-ИЛ)-1-МЕТИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (168)
[0792] Указанное соединение было получено, как описано в примере 64, из соответствующих исходных веществ, соединений 32F и 58F. Соединение 168 (21,3 мг, выход: 45,1%, светло-желтое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,38 (ушир. d, J=7,7 Гц, 1H), 8,12-7,99 (m, 2Н), 7,81 (s, 1H), 7,54 (ушир. d, J=3,7 Гц, 2Н), 7,36-7,24 (m, 5Н), 7,12 (ушир. t, J=8,7 Гц, 2Н), 5,30-5,20 (m, 1H), 3,89 (s, 3H), 3,19-3,09 (m, 1H), 2,87-2,74 (m, 1H). МС (ИЭР) m/z (М+Н)+ 395,1.
N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (459)
[0793] Указанное соединение было получено, как в случае соединения 107, из соответствующих исходных веществ, соединения 107В и гидрохлорида 3-амино-N-циклопропил-2-гидрокси-4-фенилбутанамида. Соединение 459 (210 мг, выход: 65,2%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,88-8,79 (m, 2Н), 8,06-7,99 (m, 2Н), 7,43-7,34 (m, 3H), 7,33-7,26 (m, 4Н), 7,25-7,18 (m, 1H), 5,48-5,35 (m, 1H), 3,26-3,17 (m, 1H), 3,05-2,94 (m, 1H), 2,82-2,71 (m, 1H), 2,55 (s, 3H), 0,70-0,52 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 418,2.
N-(1-(ЦИКЛОПРОПИЛАМИНО)-1,2-ДИОКСОГЕПТАН-3-ИЛ)-2-МЕТИЛ-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (460)
[0794] Указанное соединение было получено, как в случае соединения 107, из соответствующих исходных веществ, соединения 107В и гидрохлорида 3-амино-N-циклопропил-2-гидроксигептанамида. Соединение 460 (180 мг, выход: 53,3%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,76 (ушир. s, 2Н), 8,10 (ушир. s, 2Н), 7,40 (ушир. s, 3H), 5,12 (ушир. s, 1H), 2,77 (ушир. s, 1Н), 2,56 (ушир. s, 3H), 1,81 (ушир. s, 1H), 1,68 (ушир. s, 1H), 1,32 (ушир. s, 4Н), 0,88 (ушир. s, 3H), 0,70-0,52 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 384,2.
(S)-N-(4-ФТОР-3-ОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (475)
[0795] Указанное соединение было получено, как в случае соединения 107, из соответствующих исходных веществ, соединения 107В и гидрохлорида (2S,3S)-3-амино-1-фтор-4-фенилбутан-2-ола. Соединение 475 (75 мг, выход: 50,28%, белое твердое вещество): 1Н ЯМР (400 МГц, CDCl3) δ 8,13-8,08 (m, 2Н), 7,47-7,38 (m, 3H), 7,36-7,28 (m, 3H), 7,18 (d, J=6,6 Гц, 2Н), 6,81-6,76 (m, 1H), 5,31-5,22 (m, 1H), 5,05-4,89 (m, 1H), 4,88-4,72 (m, 1H), 3,29-3,22 (m, 1H), 3,17-3,10 (m, 1H), 2,57 (s, 3H). МС (ИЭР) m/z (М+Н)+ 367,1.
ПРИМЕР 65
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ХИНОЛИН-5-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (108)
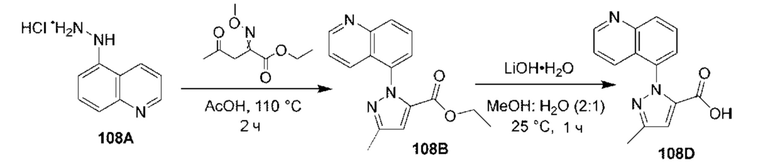
[0796] К смеси, состоящей из хинолин-5-амина (5 г, 34,68 ммоль) в конц. HCl (20 мл), при 0°С добавляли по каплям NaNO2 (2,63 г, 38,15 ммоль), и полученную смесь перемешивали при 0°С в течение 0,5 часа. Реакционную смесь нагревали до 25°С в течение более 0,5 часа и затем охлаждали до 0°С. В реакционную смесь добавляли по каплям SnCl2⋅2H2O (15,65 г, 68.36 ммоль, в 20 мл конц. HCl) и перемешивали при 0°С в течение 0,5 часа. Полученную смесь оставляли нагреваться до 25°С с интенсивным перемешиванием в течение более 4 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли 90 мл этанола (30 мл × 3), фильтровали и концентрировали при пониженном давлении с получением соединения 108А (5,2 г, выход 76,64%, HCl) в виде желтого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,98 (ушир. s, 1H), 9,26-9,15 (m, 2Н), 8,07-7,97 (m, 2Н), 7,89 (d, J=8,8 Гц, 1H), 7,27 (d, J=7,8 Гц, 1H).
[0797] Смесь соединения 108А (2 г, 12,56 ммоль, HCl) и этил-2-(метоксиимино)-4-оксопентаноата (1,91 г, 10,22 ммоль) в АсОН (20 мл) дегазировали и продували N2 3 раза, и затем смесь перемешивали при 110°С в течение 2 ч в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении для удаления АсОН. Остаток разбавляли CH2Cl2 (100 мл), доводили рН до ~ 7-8 с помощью насыщенного водного NaHCO3 и затем подвергали экстракции CH2Cl2 (40 мл × 2). Органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1:0 до 0:1) с получением соединения 108В (1,2 г, выход 41,78%) в виде желтого твердого вещества и соединения 108С (150 мг, выход 5,22%) в виде желтого твердого вещества. Соединение 108В: 1Н ЯМР (CDCl3, 400 МГц): δ 8,93 (d, J=4,0 Гц, 1H), 8,23 (d, J=8,4 Гц, 1H), 7,78 (t, J=8,0 Гц, 1H), 7,64 (d, J=8,4 Гц, 1H), 7,56 (d, J=7,2 Гц, 1H), 7,38 (d, J=8,8 Гц, 1H), 6,93 (s, 1H), 4,05 (q, J=7,2 Гц, 2Н), 2,41 (s, 3H), 1,00 (t, J=7,2 Гц, 3H).
[0798] Соединение 108С: 1Н ЯМР (CDCl3, 400 МГц): δ 1H ЯМР (400 МГц, ДМСО-d6) δ 8,98-8,87 (m, 1H), 8,27 (d, J=8,8 Гц, 1H), 7,83-8,77 (m, 1H), 7,68-7,56 (m, 2Н), 7,45-7,40 (m, 1H), 6,85 (s, 1H), 4,42 (q, J=7,2 Гц, 2Н), 2,13 (s, 3H), 1,40 (t, J=7,2 Гц, 3H).
[0799] К смеси 108В (250 мг, 888,7 мкмоль) в МеОН (10 мл) и H2O (5 мл) добавляли LiOH⋅Н2О (149,2 мг, 3,55 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли Н2О (10 мл), доводили рН до ~ 3 с помощью 1 н. HCl и затем подвергали экстракции ДХМ (40 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 108D (200 мг, выход 88,03%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,97 (d, J=4,0 Гц, 1H), 8,17 (d, J=8,8 Гц, 1H), 7,89-7,79 (m, 1H), 7,67-7,62 (m, 1H), 7,61-7,52 (m, 2Н), 6,96 (s, 1H), 5,76 (s, 1H), 2,32 (s, 3H). МС (ИЭР) m/z (М+1)+ 253,9.
[0800] Соединение 108 (21,2 мг, выход 23,11%, белое твердое вещество) было получено, как описано в примере 107, из соответствующих промежуточных соединений 108D и 12G. Соединение 108: 1Н ЯМР (CDCl3, 400 МГц): δ 8,95 (d, J=2,8 Гц, 1H), 8,22 (d, J=8,8 Гц, 1H), 7,77-7,66 (m, 2Н), 7,50 (d, J=7,2 Гц, 1H), 7,39 (d, J=8,4 Гц, 1H), 7,24-7,18 (m, 3H), 6,89 (d, J=5,6 Гц, 2H), 6,63 (s, 2H), 6,28 (d, J=7,2 Гц, 1H), 5,53-5,39 (m, 2H), 3,24 (d, J=14,4 Гц, 1H), 3,03 (d, J=14,4 Гц, 1H), 2,39 (s, 3H). МС (ИЭР) m/z (М+Н)+ 428,1.
ПРИМЕР 66
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(ПИРИДИН-2-ИЛ)ТИАЗОЛ-5-КАРБОКСАМИД (109)
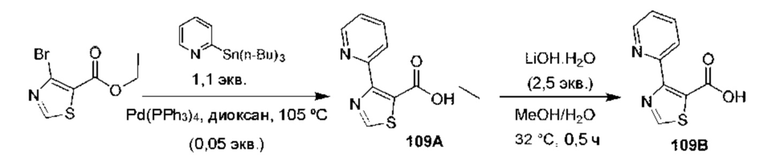
[0801] Смесь этил-4-бромтиазол-5-карбоксилата (500 мг, 2,12 ммоль), 2-(трибутилстаннил)пиридина (858,5 мг, 2,33 ммоль), Pd(PPh3)4 (122,5 мг, 106 мкмоль) в диоксане (15 мл) перемешивали при 105°С в течение 14 ч. Смесь концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 3:1 до 1:1) с получением соединения 109А (221 мг, выход 44,05%) в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 235,0.
[0802] К раствору соединения 109А (221 мг, 943,36 мкмоль) в МеОН (10 мл) и воде (2 мл) добавляли LiOH⋅H2O (99 мг, 2,36 ммоль, 2,5 экв.). Смесь перемешивали при 32°С в течение 0,5 ч. МеОН выпаривали. К остатку добавляли воду (20 мл). Смесь подвергали экстракции МТБЭ (5 мл) и разделяли. Водный слой подкисляли до рН~3 с помощью 1 н. HCl и подвергали экстракции этилацетатом (3×20 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением соединения 109В (155 мг, выход 79,68%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,37 (s, 1H), 8,84 (ушир. d, J=4,8 Гц, 1H), 8,57 (d, J=8,0 Гц, 1H), 8,34 (ушир. t, J=7,4 Гц, 1H), 7,78 (t, J=6,2 Гц, 1H).
[0803] Соединение 109 (5,7 мг, 13,91 выход %, желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 109В. Соединение 109: МС (ИЭР) m/z (М+Н)+ 381,0. 1Н ЯМР (400 МГц, CDCl3) δ 13,47-13,34 (m, 1H), 8,85 (s, 1H), 8,43 (d, J=8,0 Гц, 1H), 8,03 (d, J=5,2 Гц, 1H), 7,96-7,79 (m, 1H), 7,26-7,22 (m, 1H), 7,20-7,06 (m, 5Н), 6,81 (ушир. s, 1H), 5,94-5,86 (m, 1H), 5,68 (ушир. s, 1H), 3,49-3,33 (m, 2Н).
ПРИМЕР 67
(S)-1-(4-(ОКСАЗОЛ-2-ИЛ)ПИРИДИН-2-ИЛ)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (110)
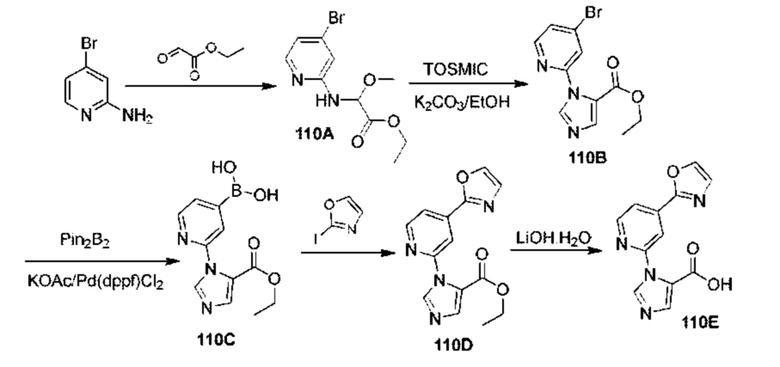
[0804] Смесь 4-бромпиридин-2-амина (20 г, 115,60 ммоль) и этил-2-оксоацетата (30,7 г, 150,28 ммоль) в МеОН (300 мл) нагревали до 80°С в течение 3 ч. Смесь концентрировали, остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 20:1). Соединение 110А (28,9 г, выход: 86,5%, желтое твердое вещество): 1Н ЯМР (400 МГц, CDCl3) δ 7,96 (d, J=5,2 Гц, 1H), 6,86 (dd, J=5,2, 1,75 Гц, 1H), 6,77 (d, J=1,3 Гц, 1H), 5,75 (ушир. s, 1H), 5,61 (d, J=8,3 Гц, 1H), 4,29 (q, J=7,0 Гц, 2Н), 3,41 (s, 3H), 1,37-1,31 (m, 3H).
[0805] Смесь 110А (15 г, 51,9 ммоль) и K2CO3 (21,5 г, 156 ммоль) в EtOH (300 мл) перемешивали при 80°С в течение 0,5 ч, затем добавляли 1-(изоцианометилсульфонил)-4-метилбензол (15,2 г, 77,82 ммоль), полученную смесь перемешивали при 80°С в течение еще 2 ч. Большую часть этанола удаляли, и происходило образование осадка, твердое вещество отфильтровывали и промывали водой (100 мл × 2), твердое вещество сушили и концентрировали с получением 110В (6,4 г, выход: 41,7%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,38 (d, J=5,2 Гц, 1H), 7,97 (s, 1H), 7,85 (s, 1H), 7,61 (s, 1H), 7,56 (dd, J=5,26, 1,3 Гц, 1H), 4,27 (q, J=7,02 Гц, 2Н), 1,29 (t, J=7,02 Гц, 3H).
[0806] 110В (3 г, 10,13 ммоль), Pin2B2 (2,57 г, 10,13 ммоль), KOAc (2,98 г, 30,4 ммоль) и Pd(dppf)Cl2 (741 мг, 1,01 ммоль) в диоксане (100 мл) дегазировали и затем нагревали при 70°С в течение 4 часов в атмосфере N2. Смесь фильтровали, и фильтрат концентрировали, остаток очищали посредством хроматографии на силикагеле (ДХМ : метанол = 5:1) с получением 110C (1,70 г, неочищенного) в виде черного твердого вещества.
[0807] 110С (300 мг, 1,15 ммоль), 2-иодоксазол (157 мг, 805,00 мкмоль), Pd(dppf)Cl2 (84,1 мг, 115,00 мкмоль) и Na2CO3 (244 мг, 2,30 ммоль) в толуоле (2 мл), EtOH (2 мл), H2O (1 мл) дегазировали и затем нагревали до 120°С в течение 1 часа в условиях микроволнового излучения. В анализе посредством ЖХМС был обнаружен МС целевого соединения, в смесь добавляли воду (5 мл) и подвергали ее экстракции этилацетатом (10 мл × 2), органические фазы сушили и концентрировали, остаток очищали посредством препаративной ТСХ (петролейный эфир : этилацетат = 1:1) с получением 110D (80 мг, выход: 24,5%) в виде желтого твердого вещества.
[0808] Смесь 110D (80 мг, 281,42 мкмоль) и LiOH⋅H2O (17,7 мг, 422,13 мкмоль) в ТГФ (5 мл), H2O (1 мл) перемешивали при 25°С в течение 12 ч. В анализе посредством ЖХМС был обнаружен МС целевого соединения, ТГФ удаляли под вакуумом, водный слой подвергали экстракции этилацетатом (10 мл × 2), рН водного слоя доводили до ~ 6 с помощью 1 н. HCl, и слой лиофилизировали, остаток очищали посредством препаративной ВЭЖХ (ТФУ) с получением 110Е (35 мг, выход: 48,5%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, метанол-d4) δ 8,70 (d, J=5,3 Гц, 1H), 8,25 (s, 1H), 8,16 (s, 1H), 8,11 (s, 1H), 8,08 (d, J=5,1 Гц, 1H), 7,81 (s, 1H), 7,46 (s, 1H).
[0809] Соединение 110 (38 мг, выход: 58,8%, белое твердое вещество) было получено, как описано в примере 6, из соответствующих промежуточных соединений 110Е и 21G ((S)-2-амино-3-фенилпропан-1-ола). Соединение 110: 1Н ЯМР (400 МГц, CDCl3) δ 9,69 (s, 1H), 8,52 (d, J=5,1 Гц, 1H), 7,98 (ушир. d, J=9,9 Гц, 2Н), 7,92 (ушир. d, J=5,1 Гц, 1H), 7,81 (s, 1H), 7,59 (s, 1H), 7,52 (ушир. d, J=5,3 Гц, 1H), 7,34 (s, 1H), 7,31-7,17 (m, 4Н), 7,13 (ушир. d, J=7,1 Гц, 2Н), 4,84 (q, J=6,5 Гц, 1H), 3,33-3,18 (m, 2Н). МС (ИЭР) m/z (M+H)+ 388,1.
ПРИМЕР 68
СОЕДИНЕНИЯ 111-112
(S)-1-(5-(ОКСАЗОЛ-2-ИЛ)ПИРИДИН-2-ИЛ)-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (111)
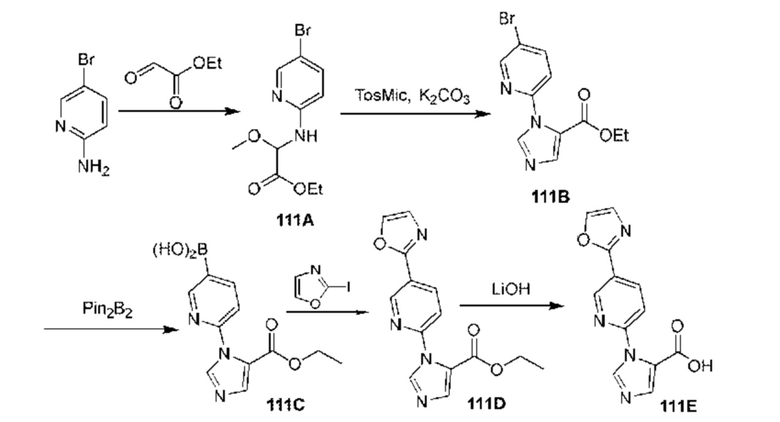
[0810] Соединение 111Е (60 мг, неочищенное, серое твердое вещество) было получено, как описано в примере 110, из соответствующих исходных веществ, 5-бромпиридин-2-амина. Соединение 111Е: МС (ИЭР) m/z (М+Н)+ 257,0. Соединение 111 (55 мг, выход: 76,9%, белое твердое вещество) было получено, как описано в примере 21, из соответствующих промежуточных соединений 111Е и 21G ((S)-2-амино-3-фенилпропан-1-ола). Соединение 111: 1Н ЯМР (400 МГц, CDCl3) δ 9,71 (s, 1H), 9,05 (d, J=1,5 Гц, 1H), 8,42 (dd, J=2,2, 8,4 Гц, 1H), 8,02 (s, 1H), 7,79 (s, 1H), 7,57 (s, 1H), 7,39 (d, J=8,4 Гц, 1H), 7,34 (ушир. d, J=6,4 Гц, 1H), 7,31 (s, 1H), 7,28-7,24 (m, 2Н), 7,22-7,17 (m, 1H), 7,14 (ушир. d, J=7,1 Гц, 2Н), 4,87 (q, J=6,5 Гц, 1H), 3,24 (d, J=6,4 Гц, 2Н). МС (ИЭР) m/z (М+Н)+ 388,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(5-(ОКСАЗОЛ-2-ИЛ)ПИРИДИН-2-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (112)
[0811] Соединение 112 (20 мг, выход: 48,2%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединений 111Е и 12G. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,06 (d, J=7,5 Гц, 1H), 8,99 (d, J=1,8 Гц, 1H), 8,39 (dd, J=2,4, 8,4 Гц, 1H), 8,34 (s, 1H), 8,26-8,21 (m, 1H), 8,08 (s, 1H), 7,84 (s, 1H), 7,57 (s, 1H), 7,48 (s, 1H), 7,34 (d, J=8,4 Гц, 1H), 7,31-7,25 (m, 4Н), 7,24-7,16 (m, 1H), 7,24-7,16 (m, 1H), 5,28-5,13 (m, 1H), 3,18 (dd, J=3,7, 13,9 Гц, 1H), 2,85 (dd, J=10,3, 13,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 431,1.
ПРИМЕР 69
СОЕДИНЕНИЯ 113, 115
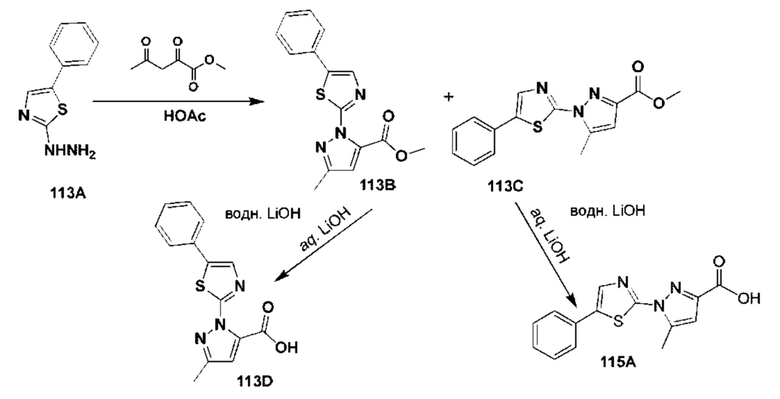
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(5-ФЕНИЛТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (113)
[0812] 5-фенилтиазол-2-амин (850 мг, 4,82 ммоль) добавляли в концентрированную соляную кислоту (5 мл). В смесь медленно добавляли по каплям водный раствор NaNO2 (998 мг, 14,5 ммоль) в H2O (3 мл) при перемешивании при 0°С, и смесь перемешивали в течение 1 ч. Затем медленно добавляли по каплям раствор SnCl2⋅2H2O (3,26 г, 14,4 ммоль) в соляной кислоте, и смесь перемешивали при 25°С в течение 3 ч. Анализ посредством ЖХМС свидетельствовал о том, что 5-фенилтиазол-2-амин полностью израсходовался, и был обнаружен один пик, которому соответствовал МС целевого соединения. Реакционную смесь фильтровали. Осадок на фильтре промывали водой (20 мл) и концентрировали при пониженном давлении с получением продукта 113А (1 г, неочищенного) в виде желтого твердого вещества. МС (ИЭР) m/z (М+Н)+ 191,9.
[0813] Смесь соединения 113А (1 г, 5,23 ммоль), метил-2,4-диоксопентаноата (754 мг, 5,23 ммоль) в НОАс (15 мл) перемешивали при 120°С в течение 1 ч. Реакционную смесь фильтровали, фильтрат концентрировали при пониженном давлении для удаления растворителя и доводили рН до 8~9 с помощью насыщенного водного NaHO3. Затем смесь подвергали экстракции этилацетатом (60 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток сначала очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = от 50:1 до 10:1) с получением чистого соединения 113С (300 мг) и смеси 113В и 113С (300 мг), и затем смесь 113В и 113С (300 мг) очищали посредством препаративной ВЭЖХ (в условиях ТФУ) с получением 113В (30 мг) и 113С (120 мг), каждое соединение в виде желтого твердого вещества.
[0814] Соединение 113В: 1H ЯМР (400 МГц, CDCl3) δ 7,79 (ушир. s, 1H), 7,60-7,51 (m, 2Н), 7,46-7,38 (m, 2Н), 7,38-7,29 (m, 1H), 6,75 (ушир. s, 1H), 4,05-3,71 (m, 3H), 2,54-2,16 (m, 3H). МС (ИЭР) m/z (М+Н)+ 300,0.
[0815] Соединение 113С: 1Н ЯМР (400 МГц, CDCl3) δ 7,74 (s, 1H), 7,58-7,53 (m, 2Н), 7,46-7,40 (m, 2Н), 7,38-7,32 (m, 1H), 7,26 (s, 1H), 6,72 (d, J=0,7 Гц, 1H), 3,96 (s, 3H), 2,75 (s, 3H). МС (ИЭР) m/z (М+Н)+ 300,0.
[0816] К раствору 113В (30 мг, 100 мкмоль) в ТГФ (5 мл), H2O (1 мл) добавляли LiOH⋅H2O (6,31 мг, 150 мкмоль). Смесь перемешивали при 25°С в течение 12 часов. В реакционную смесь добавляли водный раствор HCl для доведения рН до ~ 5. Затем смесь замораживали. Соединение 113D (35 мг, неочищенное) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,07 (s, 1H), 7,68 (d, J=7,3 Гц, 2Н), 7,49-7,41 (m, 2Н), 7,40-7,32 (m, 1H), 6,79 (s, 1H), 2,25 (s, 3H).
[0817] Соединение 113 (20 мг, выход: 66,4%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих промежуточных соединений 113D и 12G. Соединение 113: 1H ЯМР (400 МГц, CDCl3) δ 11,73 (ушир. d, J=5,5 Гц, 1H), 7,55-7,45 (m, 2Н), 7,42 (ушир. t, J=7,3 Гц, 2Н), 7,38-7,31 (m, 1H), 7,23 (ушир. dd, J=3,9, 8,0 Гц, 6Н), 7,03 (s, 1H), 6,80 (ушир. s, 1H), 5,87-5,70 (m, 1H), 5,58 (ушир. s, 1H), 3,43 (ушир. dd, J=4,5, 14,2 Гц, 1H), 3,22 (ушир. dd, J=8,2, 14,3 Гц, 1H), 2,31 (s, 3H). МС (ИЭР) m/z (M+H)+ 460,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-1-(5-ФЕНИЛТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (115)
[0818] Следуя методике получения соединения 113, соединение 115 (62,0 мг, выход: 68,3%, белое твердое вещество) получали из соответствующей карбоновой кислоты, промежуточного соединения 115А. Соединение 115: 1H ЯМР (400 МГц, CDCl3) δ 7,71 (s, 1H), 7,56 (d, J=7,3 Гц, 2H), 7,48-7,40 (m, 2H), 7,39-7,33 (m, 2H), 7,33-7,27 (m, 2H), 7,26 (s, 1H), 7,19 (ушир. d, J=6,8 Гц, 2H), 6,76 (ушир. s, 1H), 6,65 (s, 1H), 5,77-5,62 (m, 1H), 5,52 (ушир. s, 1H), 3,43 (dd, J=5,5, 13,9 Гц, 1H), 3,26 (dd, J=7,1, 13,9 Гц, 1H), 2,71 (s, 3H). МС (ИЭР) m/z (М+23)+ 460,1.
ПРИМЕР 70
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-1-(5-ФЕНИЛОКСАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (114)
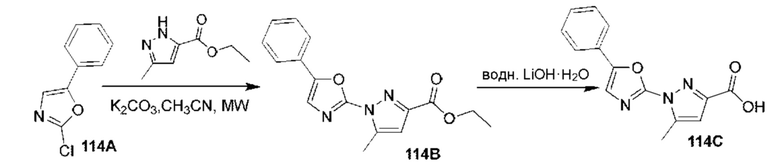
[0819] К раствору 5-фенилоксазола (800 мг, 5,51 ммоль) в ТГФ (10 мл) добавляли n-BuLi (2,5 М, 2,76 мл) по каплям при -78°С и перемешивали в течение 30 мин, затем добавляли гексахлорэтан (1,96 г, 8,27 ммоль) в ТГФ (2 мл), реакционную смесь медленно нагревали до 25°С и перемешивали в течение 12 ч. Смесь выливали в ледяную воду (20 мл) и подвергали экстракции этилацетатом (10 мл × 2), органическую фазу промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали, остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 10:1) с получением 114А (900 мг, выход: 90,9%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3-d) δ 7,58 (d, J=7,3 Гц, 2Н), 7,45-7,38 (m, 2Н), 7,38-7,31 (m, 1H), 7,27 (s, 1Н).
[0820] Смесь 114А (90 мг, 501 мкмоль), этил-3-метил-1Н-пиразол-5-карбоксилата (92,7 мг, 601 мкмоль) и K2CO3 (103 мг, 752 мкмоль) в CH3CN (3 мл) перемешивали при 120°С в течение 2 ч в условиях микроволнового излучения. Смесь разбавляли этилацетатом (20 мл) и водой (20 мл), органическую фазу сушили над Na2SO4, фильтровали и концентрировали, остаток очищали посредством препаративной ТСХ (петролейный эфир : этилацетат = 5:1) с получением 114В (0,14 г, выход: 60,4%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,76-7,67 (m, 2Н), 7,47-7,41 (m, 2Н), 7,39-7,32 (m, 2Н), 6,75 (d, J=0,9 Гц, 1H), 4,44 (q, J=7,2 Гц, 2Н), 2,67 (d, J=0,7 Гц, 3H), 1,43 (t, J=7,2 Гц, 3H).
[0821] Смесь 114В (140 мг, 471 мкмоль) и LiOH⋅H2O (39,5 мг, 942 мкмоль) в ТГФ (5 мл), H2O (1 мл) перемешивали при 25°С в течение 2 ч. Органический растворитель удаляли под вакуумом, рН водного слоя доводили до ~5 с помощью 1 н. HCl и фильтровали, водный слой подвергали экстракции ДХМ (10 мл × 3), органическую фазу промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали, остаток объединяли с осадком на фильтре с получением 114С (120 мг, неочищенного) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,88 (s, 1H), 7,81-7,71 (m, 2Н), 7,52 (t, J=7,7 Гц, 2Н), 7,46-7,40 (m, 1H), 6,81 (d, J=0,7 Гц, 1H), 2,62 (s, 3H).
[0822] Соединение 114 (53 мг, выход: 66,5%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, соединения 114С. Соединение 114: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,59 (d, J=7,7 Гц, 1H), 8,10 (s, 1H), 7,87 (s, 1H), 7,84 (s, 1H), 7,76 (d, J=7,3 Гц, 2Н), 7,52 (t, J=7,7 Гц, 2Н), 7,45-7,38 (m, 1H), 7,31-7,22 (m, 4Н), 7,19 (qd, J=4,3, 8,8 Гц, 1H), 6,72 (d, J=0,7 Гц, 1H), 5,49-5,40 (m, 1H), 3,25-3,17 (m, 1H), 3,06 (dd, J=9,4, 14,0 Гц, 1H), 2,57 (s, 3H). МС (ИЭР) m/z (М+Н)+ 444,1.
ПРИМЕР 71
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(ИЗОХИНОЛИН-1-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (119)
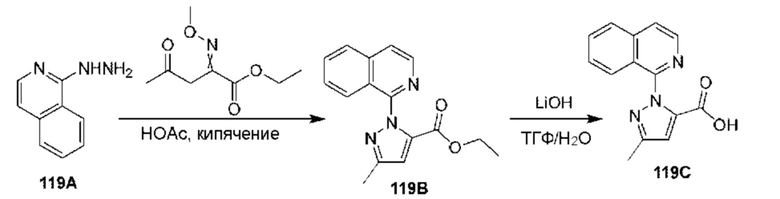
[0823] К смеси 1-хлоризохинолина (5,0 г, 30,56 ммоль) в диоксане (10,00 мл) добавляли NH2NH2⋅H2O (305,62 ммоль, 15 мл). Смесь перемешивали при 80°С в течение 16 ч. Реакционную смесь промывали H2O (100 мл). Реакционную смесь разбавляли МТБЭ и фильтровали с получением соединения 119А (4,27 г, выход 87,77%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,16 (d, J=8,3 Гц, 1H), 7,87 (d, J=5,8 Гц, 1H), 7,72-7,65 (m, 1H), 7,64-7,56 (m, 1H), 7,48-7,41 (m, 1H), 6,90 (d, J=5,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 160,1.
[0824] Смесь соединения 119А (4,20 г, 26,38 ммоль) и этил-2-(метоксиимино)-4-оксопентаноата (4,94 г, 26,38 ммоль) в НОАс (40,00 мл) перемешивали при 120°С в течение 48 ч. Реакционную смесь концентрировали при пониженном давлении для удаления НОАс. Остаток разбавляли H2O (20 мл) и подвергали экстракции этилацетатом (40 мл). Объединенные органические слои промывали солевым раствором (40 мл), сушили над Na2SO4, концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10/1 до 0:1) с получением соединения 119В (238,00 мг, выход 3,08%), которое было получено в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,46 (ушир. d, J=5,5 Гц, 1H), 7,92 (d, J=8,2 Гц, 1H), 7,79 (d, J=5,5 Гц, 1H), 7,72 (t, J=7,4 Гц, 1H), 7,67-7,53 (m, 2Н), 6,92 (s, 1H), 4,06 (q, J=7,1 Гц, 2Н), 2,43 (s, 3H), 0,99 (t, J=7,2 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 282.
[0825] К раствору соединения 119В (238,00 мг, 846,04 мкмоль) в ТГФ (6,00 мл) добавляли LiOH⋅H2O (177,50 мг, 4,23 ммоль) в H2O (2,00 мл). Смесь перемешивали при 28°С в течение 16 ч. Реакционную смесь разбавляли H2O (10 мл) и подвергали экстракции МТБЭ (15 мл × 2), в водную фазу добавляли 1 н. HCl до рН ~ 3~4, подвергали ее экстракции этилацетатом (15 мл × 2). Объединенные органические слои промывали солевым раствором (15 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 119С (201,00 мг, выход 92,87%) в виде желтого твердого вещества. Соединение 119С: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,42 (d, J=5,5 Гц, 1H), 8,12 (d, J=8,2 Гц, 1H), 8,01 (d, J=5,7 Гц, 1H), 7,85 (t, J=7,2 Гц, 1H), 7,69 (t, J=7,4 Гц, 1H), 7,56 (d, J=8,4 Гц, 1H), 6,92 (s, 1H), 2,32 (s, 3H). МС (ИЭР) m/z (M+H)+ 254,1.
[0826] Соединение 119 (20,00 мг, выход 35,64%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 119С. Соединение 119: 1Н ЯМР (400 МГц, CDCl3) δ 8,23 (ушир. d, J=6,2 Гц, 1H), 7,94-7,85 (m, 3H), 7,75 (ушир. t, J=7,8 Гц, 1H), 7,69 (ушир. d, J=5,3 Гц, 1H), 7,62 (ушир. t, J=7,7 Гц, 1H), 7,12 (ушир. d, J=7,1 Гц, 1H), 7,09-7,03 (m, 2Н), 6,92 (ушир. d, J=7,1 Гц, 2Н), 6,73 (s, 1H), 6,67 (ушир. s, 1H), 5,65-5,59 (m, 1H), 5,51 (ушир. s, 1H), 3,36-3,28 (m, 1H), 3,21-3,14 (m, 1H), 2,41 (s, 3H). МС (ИЭР) m/z (М+Н)+ 428,1.
ПРИМЕР 72
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(5-МЕТИЛПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (120)
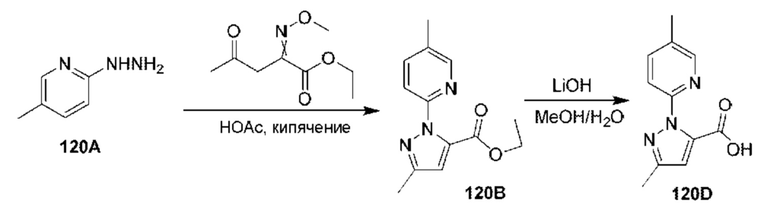
[0827] Смесь 2-фтор-5-метилпиридина (10.00 г, 89,99 ммоль, 9,35 мл) в NH2NH2⋅H2O (53,00 г, 899,93 ммоль, 51,5 мл) дегазировали и продували N2 3 раза, и затем смесь перемешивали при 120°С в течение 15 ч в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли Н2О (30 мл) и подвергали экстракции этилацетатом (20 мл × 2). Объединенные органические слои промывали солевым раствором (20 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 120А (6,09 г, выход: 54,9%), которое было получено в виде светло-розового твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,95 (s, 1H), 7,32 (dd, J=2,0, 8,4 Гц, 1H), 6,63 (d, J=8,4 Гц, 1H), 5,68 (ушир. s, 1H), 2,20 (s, 3H).
[0828] Смесь соединения 120А (2 г, 16,24 ммоль), этил-2-(метоксиимино)-4-оксопентаноата (3,04 г, 16,24 ммоль) в НОАс (20 мл) перемешивали при 120°С в течение 20 ч. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли Н2О (15 мл) и подвергали экстракции этилацетатом (20 мл × 2). Объединенные органические слои промывали NaHCO3 (20 мл × 3) и затем промывали солевым раствором (20 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством препаративной ВЭЖХ (в условиях HCl) с получением соединения 120В (340 мг, выход: 8,5%), которое было получено в виде белого твердого вещества. Соединение 120В: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,27 (s, 1H), 7,82 (dd, J=1,8, 8,3 Гц, 1H), 7,58 (d, J=8,3 Гц, 1H), 6,77 (s, 1H), 4,19 (q, J=7,0 Гц, 2Н), 2,35 (s, 3H), 2,28 (s, 3H), 1,14 (t, J=7,0 Гц, 3H).
[0829] К раствору соединения 120В (340 мг, 1,39 ммоль) в ТГФ (10 мл) добавляли LiOH⋅H2O (291 мг, 6,95 ммоль) в H2O (3 мл). Смесь перемешивали при 25°С в течение 30 ч. Реакционную смесь разбавляли H2O (15 мл) и подвергали экстракции МТБЭ (10 мл). рН объединенных водных слоев доводили до ~ 6 путем добавления 1 н. HCl, и затем эти слои подвергали экстракции этилацетатом (20 мл × 2). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 120D (300 мг, выход: 99,4%), которое было получено в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,50 (ушир. s, 1H), 8,26 (s, 1H), 7,79 (dd, J=1,8, 8,2 Гц, 1H), 7,54 (d, J=8,2 Гц, 1H), 6,73 (s, 1H), 2,33 (s, 3H), 2,24 (s, 3H).
[0830] Соединение 120 (15 мг, выход: 54,1% светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 120D. Соединение 120: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,21 (d, J=7,3 Гц, 1H), 8,09 (s, 1H), 8,04 (s, 1H), 7,85 (s, 1H), 7,73 (dd, J=1,6, 8,2 Гц, 1H), 7,44 (d, J=8,3 Гц, 1H), 7,31-7,23 (m, 5Н), 6,53 (s, 1H), 5,35-5,26 (m, 1H), 3,16 (dd, J=4,0, 14,1 Гц, 1H), 2,87 (dd, J=9,8, 14,1 Гц, 1H), 2,31 (s, 3H), 2,26 (s, 3H).
ПРИМЕР 73
СОЕДИНЕНИЯ 121-122, 445
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(5-МЕТИЛПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД(121)
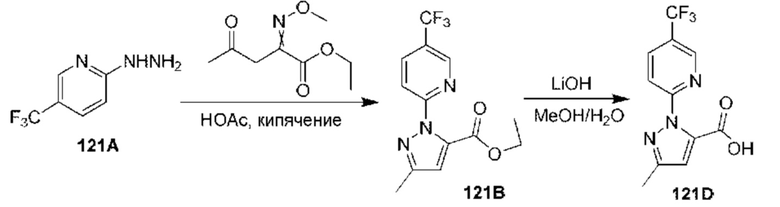
[0831] Промежуточного соединения 121D (650 мг, выход: 89,8%, белое твердое вещество) было получено, как описано в примере 120, из соответствующих исходных веществ, соединения 121А и 2-хлор-5-(трифторметил)пиридина. Соединение 121А: 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,55 (ушир. s, 1H), 8,86 (s, 1H), 8,39 (dd, J=2,3, 8,5 Гц, 1H), 7,91 (d, J=8,4 Гц, 1H), 6,81 (s, 1H), 2,27 (s, 3H).
[0832] Соединение 121 (35,9 мг, выход: 55,2%, белое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 121D. Соединение 121: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,15 (d, J=7,3 Гц, 1H), 8,45 (s, 1H), 8,29 (dd, J=2,1, 8,7 Гц, 1H), 8,11 (s, 1H), 7,88-7,80 (m, 2Н), 7,28-7,24 (m, 4Н), 7,22-7,17 (m, 1H), 6,51 (s, 1H), 5,36-5,28 (m, 1H), 3,14 (dd, J=3,6, 14,0 Гц, 1H), 2,81 (dd, J=9,9, 14,1 Гц, 1H), 2,27 (s, 3H).
(S)-N-(4-ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(5-ТРИФТОРМЕТИЛ)ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (122)
[0833] Соединение 122 (54,1 мг, выход: 87,9%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 121D. Соединение 122: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,16 (d, J=7,3 Гц, 1H), 8,85 (d, J=5,1 Гц, 1H), 8,42 (s, 1H), 8,30 (dd, J=2,1, 8,7 Гц, 1H), 7,83 (d, J=8,6 Гц, 1H), 7,29-7,23 (m, 4Н), 7,22-7,16 (m, 1H), 6,51 (s, 1H), 5,38-5,30 (m, 1H), 3,14 (dd, J=3,7, 14,1 Гц, 1H), 2,86-2,72 (m, 2Н), 2,27 (s, 3H), 0,68-0,55 (m, 4Н).
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(5-(ТРИФТОРМЕТИЛ)ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (445)
[0834] Соединение 445 (140 мг, выход: 47,4%, белое твердое вещество) было получено, как в случае соединения 121, из соответствующих промежуточных соединений 121D и 274D. Соединение 445: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,16 (d, J=7,5 Гц, 1H), 8,50-8,43 (m, 1H), 8,31 (dd, J=2,2, 8,8 Гц, 1H), 8,12 (s, 1H), 7,90-7,81 (m, 2Н), 7,29-7,18 (m, 4Н), 6,53 (s, 1H), 5,38-5,29 (m, 1H), 3,16 (dd, J=4,0, 14,1 Гц, 1H), 2,83 (dd, J=9,9, 14,1 Гц, 1H), 2,28 (s, 3H). МС (ИЭР) m/z (М+Н)+ 446,1.
ПРИМЕР 74
СОЕДИНЕНИЯ 123-124
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4,6-ДИМЕТИЛПИРИДИН-2-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (123)
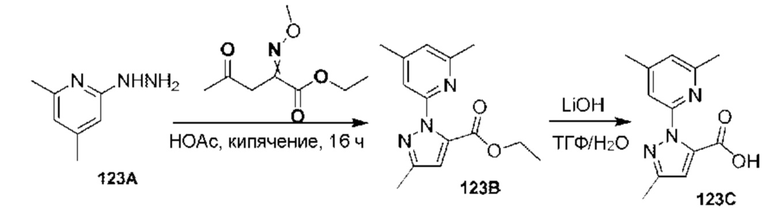
[0835] Промежуточное соединение 123С (210 мг, выход: 78,29%, белое твердое вещество) было получено, как описано в примере 120, из соответствующих исходных веществ, соединения 123А и 2-хлор-5-(трифторметил)пиридина. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,38 (s, 1H), 7,14 (s, 1H), 6,77 (s, 1H), 2,40 (s, 3H), 2,37 (s, 3H), 2,26 (s, 3H). МС (ИЭР) m/z (М+Н)+ 232,0.
[0836] Соединение 123 (40 мг, выход: 38,78%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 123С. Соединение 123: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,13 (d, J=7,3 Гц, 1H), 8,04 (s, 1H), 7,81 (s, 1H), 7,28-7,18 (m, 6Н), 6,99 (s, 1H), 6,44 (s, 1H), 5,41-5,21 (m, 1H), 3,12 (dd, J=4,0, 13,9 Гц, 1H), 2,82 (dd, J=9,7, 13,9 Гц, 1H), 2,31 (s, 3H), 2,23 (s, 3H), 2,19 (s, 3H). МС (ИЭР) m/z (М+Н)+ 406,1.
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-4-ФЕНИЛТИАЗОЛ-5-КАРБОКСАМИД (124)
[0837] Соединение 124 (40 мг, выход: 57,35%, белое твердое вещество) было получено, как описано в примере 41, из соответствующей карбоновой кислоты, 2-метил-4-фенилтиазол-5-карбоновой кислоты. 1Н ЯМР (400 МГц, CDCl3) δ 7,58-7,50 (m, 2Н), 7,49-7,36 (m, 3H), 7,22-7,13 (m, 3H), 6,84 (ушир. s, 1H), 6,80-6,69 (m, 2Н), 6,22 (ушир. d, J=6,3 Гц, 1H), 5,58-5,46 (m, 1H), 3,26 (dd, J=4,9, 14,2 Гц, 1H), 2,89 (dd, J=7,5, 14,1 Гц, 1H), 2,79 (qt, J=3,8, 7,4 Гц, 1H), 2,71 (s, 3H), 0,94-0,82 (m, 2Н), 0,66-0,55 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 434,1.
ПРИМЕР 75
(S)-N-(4-ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-1-(4-ФЕНИЛТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (127)
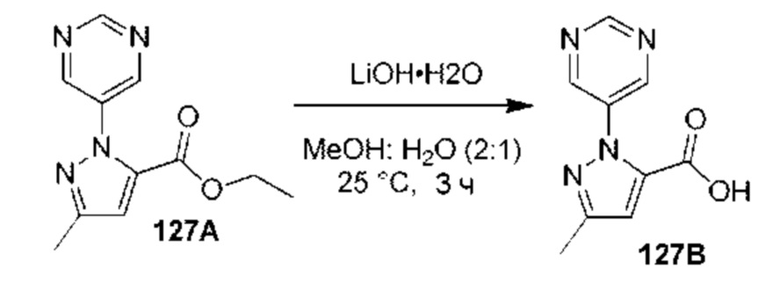
[0838] Промежуточное соединение 127В (150 мг, выход 94,78%, белое твердое вещество) было получено, как описано в примере 85, из соединения 127А. Соединение 127В: 1Н ЯМР (400 МГц, ДМСО-d6): δ 9,20 (s, 1H), 8,99 (s, 2Н), 6,94 (s, 1H), 2,27 (s, 3H).
[0839] Соединение 127 (55,3 мг, выход 45,18%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 127В. Соединение 127: 1Н ЯМР (400 МГц, CDCl3): δ 9,15 (s, 1H), 8,76 (s, 2Н), 7,35-7,28 (m, 3H), 7,13-7,09 (m, 2Н), 6,95 (ушир. s, 1H), 6,66-6,60 (m, 1H), 6,47 (s, 1H), 5,60-5,54 (m, 1H), 3,46-3,38 (m, 1H), 3,20-3,13 (m, 1H), 2,87-2,77 (m, 1H), 2,35 (s, 3H), 0,92-0,87 (m, 2Н), 0,66-0,61 (m, 2Н). МС (ИЭР) m/z (М+1)+ 419,1.
ПРИМЕР 76
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (129)
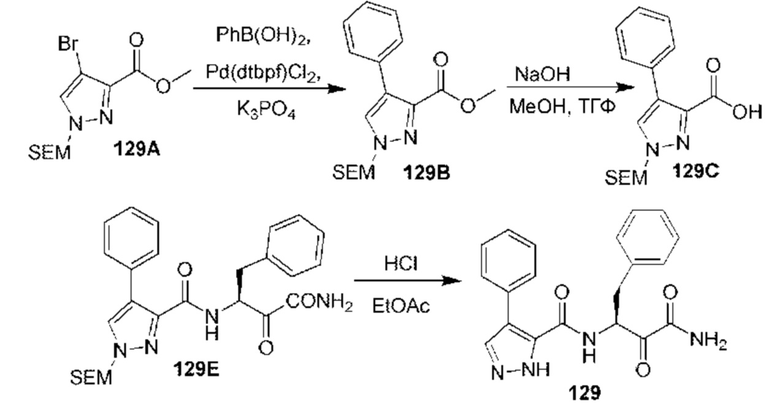
[0840] К раствору NaH (1,46 г, 36,59 ммоль, чистота 60%) в ТГФ (80 мл) добавляли метил-4-бром-1H-пиразол-3-карбоксилат (5,00 г, 24,39 ммоль) в ТГФ (20 мл) при 0°С. После добавления смесь нагревали до 25°С и перемешивали в течение 2 ч. Затем смесь охлаждали до 0°С и добавляли раствор SEM-Cl (4,47 г, 26,83 ммоль, 4,8 мл) в ТГФ (100 мл). Смесь перемешивали при 25°С в течение 12 ч. Смесь разбавляли H2O (200 мл), органический слой промывали HCl (1 М, 100 мл), насыщенным NaHCO3 (100 мл), солевым раствором (100 мл), сушили над Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10/1 до 3:1). Соединение 129А (3,40 г, выход 41,6%) получали в виде бесцветного масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,39 (s, 1H), 5,51 (s, 2Н), 3,87 (s, 3H), 3,62-3,56 (m, 2Н), 0,95-0,81 (m, 2Н), 0,07-0,07 (m, 9Н).
[0841] Смесь соединения 2 (3,40 г, 10,14 ммоль), фенилбороновой кислоты (1,48 г, 12,17 ммоль), ди-трет-бутил(циклопентил)фосфан; дихлорпалладий; железо (660,9 мг, 1,01 ммоль), K3PO4 (6,46 г, 30,42 ммоль) в диоксане (30 мл) и H2O (10 мл) дегазировали и продували N2 3 раза, и затем смесь перемешивали при 70°С в течение 1 часа в атмосфере N2. Смесь концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10/1 до 3:1). Соединение 129В (3,00 г, неочищенное) было получено в виде коричневого масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,26 (s, 1H), 7,51-7,32 (m, 5Н), 5,53 (s, 2Н), 3,78 (s, 3H), 3,67-3,59 (m, 2Н), 0,94-0,82 (m, 2Н), 0,06-0,07 (m, 9Н).
[0842] К раствору соединения 129В (3,00 г, 9,02 ммоль) в МеОН (100 мл) и ТГФ (100 мл) добавляли NaOH (2 М, 90 мл). Смесь перемешивали при 60°С в течение 1 часа. Смесь концентрировали и разбавляли H2O (200 мл), смесь подвергали экстракции этилацетатом (200 мл), в водную фазу добавляли HCl (1 М) до рН~3, затем смесь подвергали экстракции этилацетатом (200 мл), органический слой промывали солевым раствором (200 мл), сушили над Na2SO4 и концентрировали. Соединение 129С (300,0 мг, выход 10,4%) было получено в виде коричневого масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,20 (s, 1H), 7,51-7,47 (m, 2Н), 7,43-7,37 (m, 2Н), 7,35-7,30 (m, 1H), 5,50 (s, 2Н), 3,67-3,61 (m, 2Н), 0,92-0,87 (m, 2Н), 0,03-0,03 (m, 9Н).
[0843] Промежуточное соединение 129Е (70,0 мг, неочищенное, бесцветное масло) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, соединения 129С. Соединение 129Е: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,45 (d, J=7,6 Гц, 1H), 8,25-8,21 (m, 1H), 8,20-8,13 (m, 1H), 7,90 (s, 1H), 7,44-7,38 (m, 2Н), 7,36-7,19 (m, 8Н), 5,51-5,43 (m, 3H), 3,68-3,60 (m, 2Н), 3,26-3,18 (m, 1H), 3,08-2,99 (m, 1H), 0,95-0,87 (m, 2Н), 0,06-0,05 (m, 9Н).
[0844] К раствору соединения 129Е (70,0 мг, 142,09 мкмоль) в этилацетате (10 мл) добавляли HCl/EtOAc (4 М, 710 мкл). Смесь перемешивали при 25°С в течение 3 часов. Смесь концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в условиях HCl). Соединение 129 (20,0 мг, HCl, выход 34,4%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, D2O) δ 7,74-7,61 (m, 1H), 7,41-7,33 (m, 2Н), 7,30-7,09 (m, 10Н), 4,54-4,53 (m, 1H), 3,00-2,92 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 363,1.
ПРИМЕР 77
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-(ФЕНОКСИМЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (131)
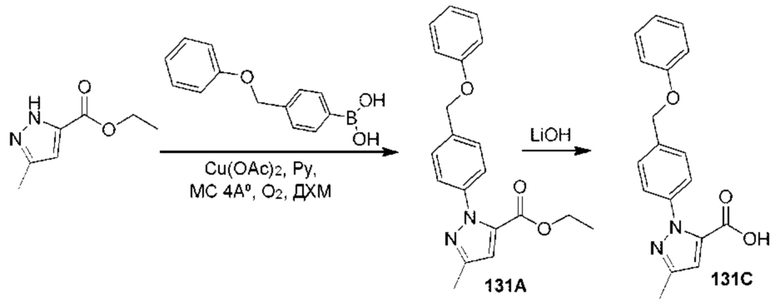
[0845] К смеси этил-3-метил-1H-пиразол-5-карбоксилата (250 мг, 1,62 ммоль), [4-(феноксиметил)фенил]бороновой кислоты (554,7 мг, 2,43 ммоль), молекулярного сита с размером ячейки 4А° (МС 4А°) (8 г) и пиридина (141 мг, 1,78 ммоль, 0,15 мл) в ДХМ (50 мл) добавляли Cu(OAc)2 (383 мг, 2,11 ммоль), смесь перемешивали при 25°С в течение 16 ч в атмосфере О2 из баллона (15 фунтов на кв. дюйм). Реакционную смесь фильтровали для удаления МС 4А° и катализатора, и затем фильтрат концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1) и посредством препаративной ТСХ (SiO2, петролейный эфир/этилацетат = 5/1).
[0846] Соединение 131А (69,3 мг, выход: 13,03%) получали в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,99-7,93 (m, 2Н), 7,91-7,82 (m, 2Н), 7,80-7,68 (m, 2Н), 7,46-7,40 (m, 3H), 5,59 (s, 2Н), 4,68 (q, J=7,1 Гц, 2Н), 2,81 (s, 3H), 2,05 (s, 1H), 1,69 (t, J=7,2 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 337,1.
[0847] К раствору соединения 131А (69,3 мг, 206,02 мкмоль), в ТГФ (5 мл) и H2O (3 мл) добавляли LiOH⋅H2O (26 мг, 618,06 мкмоль). После перемешивания при 25°С в течение 3 ч в реакционную смесь добавляли H2O (10 мл) и подвергали экстракции МТБЭ (20 мл). Органический слой промывали H2O (10 мл). Объединенный водный слой подкисляли до рН ~1~2 с помощью 1 н. HCl, подвергали экстракции этилацетатом (20 мл × 3), сушили над Na2SO4, фильтровали и концентрировали. Соединение 131С (70 мг, неочищенное, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,23 (ушир. s, 1H), 7,57-7,51 (m, 2Н), 7,43 (d, J=8,3 Гц, 2Н), 7,35-7,27 (m, 2Н), 7,05 (d, J=7,8 Гц, 2Н), 6,96 (t, J=7,3 Гц, 1H), 6,83 (s, 1H), 5,17 (s, 2Н), 2,26 (s, 3H).
[0848] Соединение 131 (37,2 мг, выход: 45,9%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 131С. Соединение 131: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,12 (ушир. d, J=7,5 Гц, 1H), 8,11 (ушир. s, 1H), 7,86 (s, 1H), 7,39 (ушир. d, J=8,2 Гц, 2Н), 7,33-7,26 (m, 6Н), 7,23 (ушир. d, J=6,4 Гц, 1H), 7,17 (ушир. d, J=8,2 Гц, 2Н), 7,01 (ушир. d, J=8,2 Гц, 2Н), 6,93 (t, J=7,4 Гц, 1H), 6,56 (s, 1H), 5,31-5,22 (m, 1H), 5,09 (s, 2Н), 3,19 (ушир. dd, J=3,2, 13,8 Гц, 1H), 2,82 (ушир. dd, J=10,9, 13,6 Гц, 1H), 2,23 (s, 3H). МС (ИЭР) m/z (М+Н)+ 483,1.
ПРИМЕР 78
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(ИЗОХИНОЛИН-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (133)
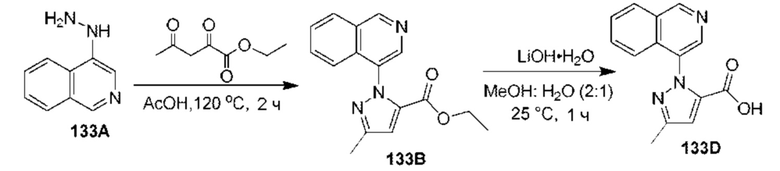
[0849] К раствору изохинолин-4-амина (1,4 г, 9,71 ммоль) в 5 н. водном растворе соляной кислоты (12 мл) при 0°С добавляли раствор NaNO2 (670 мг, 9,71 ммоль) в деионизированной воде (1 мл). Реакционную смесь перемешивали при 0°С в течение 0,5 ч и добавляли по каплям раствор SnCl2⋅H2O (5,48 г, 24,28 ммоль), растворенный концентрированной соляной кислоте (5 мл). Смесь перемешивали при 25°С в течение 2 ч. рН смеси доводили до ~12-14 с помощью 20% водного NaOH. Смесь подвергали экстракции смесью CHCl3/iPrOH 2:1 (200 мл). Органический слой сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1/0 до 0:1) и затем сушили при пониженном давлении с получением соединения 133А (720 мг, выход 46,55 %) в виде коричневого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,95 (ушир. s, 1H), 9,25-9,13 (m, 2Н), 8,04-7,95 (m, 2Н), 7,88 (d, J=8,8 Гц, 1H), 7,26 (d, J=7,6 Гц, 1H), 7,28-7,23 (m, 1H).
[0850] Смесь соединения 133А (620 мг, 3,89 ммоль) и этил-2,4-диоксопентаноата (615,9 мг, 3,89 ммоль) в АсОН (5 мл) дегазировали и продували N2 3 раза, и затем смесь перемешивали при 120°C в течение 2 часов в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении для удаления АсОН. Остаток разбавляли 10 мл EtOAc и доводили с помощью насыщенного NaHCO3 и затем, наконец, подвергали экстракции EtOAc (30 мл × 3). Объединенные органические слои сушили с помощью Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Реакционный раствор очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1/0 до 0/1) с получением соединения 133В (600,00 мг, выход 45,16%) в виде желтого масла. Соединение 133В: 1Н ЯМР (CDCl3, 400 МГц): δ 9,34 (s, 1H), 8,54 (s, 1H), 8,07 (d, J=7,6 Гц, 1H), 7,72-7,61 (m, 2Н), 7,37 (d, J=8,0 Гц, 1H), 6,96 (s, 1H), 4,05 (q, J=7,2 Гц, 2Н), 2,42 (s, 3Н), 0,98 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+1)+ 282,1.
[0851] К смеси 133В (200 мг, 711 мкмоль) в МеОН (10 мл) и H2O (5 мл) добавляли LiOH⋅H2O (119,3 мг, 2,84 ммоль) одной порцией, и смесь перемешивали при 25°C в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли Н2О (5 мл), доводили рН до ~3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (40 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 133D (150 мг, выход 75,43%) в виде желтого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,45 (s, 1H), 8,54 (s, 1H), 8,28 (d, J=7,6 Гц, 1H), 7,86-7,73 (m, 2Н), 7,27 (d, J=8,4 Гц, 1H), 6,99 (s, 1H), 2,33 (s, 3Н). МС (ИЭР) m/z (М+1)+ 254,0.
[0852] Соединение 133 (22,2 мг, выход 31,49%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 133D. Соединение 133: 1Н ЯМР (CDCl3, 400 МГц): δ 9,30 (s, 1H), 8,46 (s, 1H), 8,06 (d, J=8,0 Гц, 1H), 7,73-7,63 (m, 2Н), 7,47 (d, J=8,4 Гц, 1H), 7,26-7,24 (m, 3Н), 6,97-6,95 (m, 2Н), 6,66 (ушир. s, 1H), 6,59 (s, 1H), 6,48 (d, J=7,2 Гц, 1H), 5,65 (ушир. s, 1H), 5,41-5,36 (m, 1H), 3,28-3,24 (m, 1H), 3,11-3,06 (m, 1H), 2,40 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 428,1.
ПРИМЕР 79
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИДИН-3-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (136)
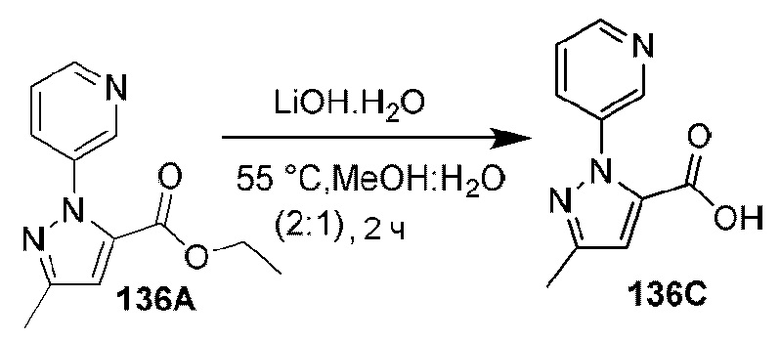
[0853] К раствору этил-3-метил-1-(пиридин-3-ил)-1Н-пиразол-5-карбоксилата (2,0 г, 12,97 ммоль) и пиридин-3-илбороновой кислоты (1,59 г, 12,97 ммоль) в пиридине (30 мл) добавляли Cu(ОАс)2 (1,18 г, 6,49 ммоль). Смесь перемешивали при 55°C в течение 18 ч. Смесь фильтровали и концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® Silica Flash массой 80 г, элюент 0~50% этилацетат/петролейный эфир, градиент со скоростью потока 40 мл/мин). Соединение 136А (850 мг, выход 28,34%, белое твердое вещество): 1Н ЯМР (400 МГц, CDCl3): δ 8,69 (d, J=2,0 Гц, 1H), 8,65-8,62 (m, 1H), 7,80-7,77 (m, 1H), 7,42-7,38 (m, 1H), 6,86 (s, 1H), 4,27-4,21 (m, 2Н), 2,37 (s, 3Н), 1,27-1,23 (m, 3Н).
[0854] Соединение 136С (160 мг, выход 60,57%, белое твердое вещество) было получено, как описано в примере 85. Соединение 136С: 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,65-8,55 (m, 2Н), 7,91-7,85 (m, 1H), 7,53-7,47 (m, 1H), 6,88 (s, 1H), 2,26 (s, 3Н).
[0855] Соединение 136 (46,2 мг, выход 54,66%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующих промежуточных соединений 136С и 12G. Соединение 136: 1H ЯМР (400 МГц, CDCl3) δ 8,61 (d, J=2,4 Гц, 1H), 8,58-8,55 (m, 1H), 7,74-7,69 (m, 1H), 7,36-7,28 (m, 4Н), 7,12-7,07 (m, 2Н), 6,79 (ушир. s, 1H), 6,55-6,48 (m, 1H), 6,43 (s, 1H), 5,69 (ушир. s, 1H), 5,56-5,49 (m, 1H), 3,43-3,36 (m, 1H), 3,20-3,13 (m, 1H), 2,33 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 378,1.
ПРИМЕР 80
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИМИДИН-5-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (138)
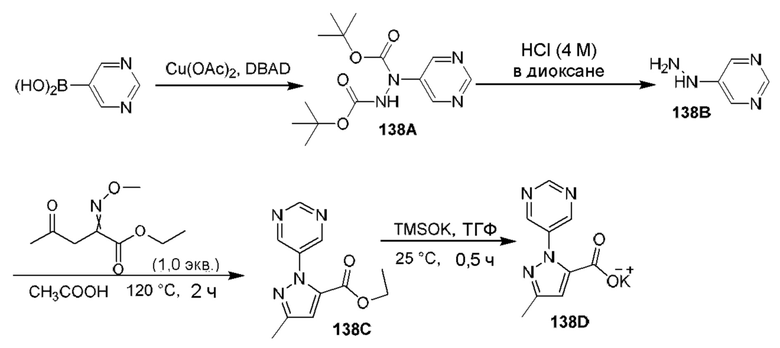
[0856] К раствору пиримидин-5-илбороновой кислоты (5,00 г, 40,35 ммоль) в МеОН (32 мл) добавляли Cu(ОАс)2 (732,8 мг, 4,04 ммоль) и DBAD (9,29 г, 40,35 ммоль). Полученную смесь перемешивали при 60°C в течение 1 часа. Реакционную смесь охлаждали до 25°C, концентрировали при пониженном давлении, разбавляли водой (50 мл) и подвергали экстракции этилацетатом (80 мл × 3). Экстракт сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде желтого масла, которое использовали на следующей стадии без очистки. Соединение 138А (9,00 г, выход 71,87%) получали в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,96 (s, 1H), 8,89 (ушир. s, 1H), 1,53-1,50 (m, 18Н).
[0857] К раствору соединения 138А (9,00 г, 25,00 ммоль) в 1,4-диоксане (60 мл) добавляли 4 М HCl 1,4-диокеан (60 мл), и смесь перемешивали при комнатной температуре в течение 30 часов. Суспензию фильтровали, и остаток промывали этилацетатом (100 мл × 2) и сушили при пониженном давлении с получением указанного в заголовке соединения (3,45 г, неочищенного), которое использовали на следующей стадии без очистки. Соединение 138В (3,45 г, 81,17 выход %, HCl) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,80 (s, 1H), 8,61 (s, 2Н).
[08581 К раствору соединения 138В (800,0 мг, 5,46 ммоль, HCl) в СН3СООН (12 мл) добавляли этил-2-(метоксиимино)-4-оксопентаноат (1,02 г, 5,46 ммоль), затем смесь перемешивали при 120°C в течение 2 часов. Смесь разбавляли CH2Cl2 (70 мл) и промывали насыщенным бикарбонатом натрия (20 мл × 2) и насыщенным солевым раствором (20 мл × 2), органический слой сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством колоночной флэш-хроматографии (SiO2, петролейный эфир : этилацетат = от 10/1 до 3/1). Соединение 138С (250,0 мг, выход 19,72%) получали в виде белого твердого вещества.
[0859] К раствору соединения 138С (50,0 мг, 215,29 мкмоль) в ТГФ (3,00 мл) добавляли TMSOK (55,2 мг, 430,58 мкмоль), затем смесь перемешивали при 25°C в течение 0,5 часа. Смесь разбавляли петролейным эфиром (20 мл), и осадок фильтровали с получением промежуточного соединения 138D (45,0 мг, выход 86,27%) в виде белого твердого вещества. МС (ИЭР) m/z (М+1)+ 204,9.
[0860] Соединение 138 (10,0 мг, выход 14,63%) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 138D. Соединение 138: 1Н ЯМР (400 МГц, CDCl3): δ 9,15 (s, 1H), 8,78 (s, 2Н), 7,35-7,31 (m, 3Н), 7,13 (d, J=6,4 Гц, 2Н), 6,82 (s, 1H), 6,58 (d, J=7,2 Гц, 1H), 6,47 (s, 1H), 5,57-5,52 (m, 1H), 5,46-5,58 (m, 1H), 3,45-3,40 (m, 1H), 3,21-3,15 (m, 1H), 2,35 (s, 3Н). МС (ИЭР) m/z (М+1)+ 379,1.
ПРИМЕР 81
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-(2H-ИНДАЗОЛ-2-ИЛ)-3-МЕТИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (139)
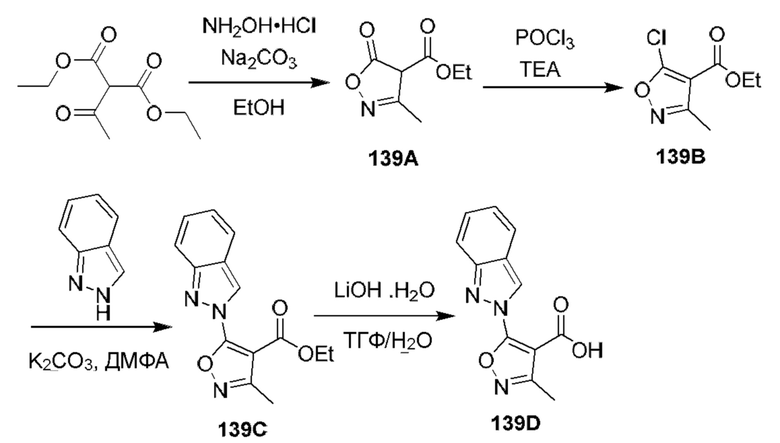
[0861] К раствору диэтил-2-ацетилмалоната (5 г, 24,7 ммоль) в EtOH (50 мл) добавляли NH2OH.HCl (1,9 г, 27,2 ммоль) и Na2CO3 (1,3 г, 12,4 ммоль) одной порцией, смесь перемешивали при 90°C в течение 2 часов. Затем содержимое смеси выливали в ледяную воду (6 мл), и затем фильтровали с получением промежуточного соединения 139А (3,2 г, выход: 75,6%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 4,37 (q, J=7,0 Гц, 2H), 2,43-2,37 (m, 3Н), 1,38 (t, J=7,2 Гц, 3Н). MC (ИЭР) m/z (М+Н)+ 126,2.
[0862] К соединению 139А (3 г, 17,5 ммоль) добавляли POCl3 (21,5 г, 140,2 ммоль, 13 мл) одной порцией. Затем добавляли TEA (1,8 г, 17,5 ммоль). Смесь перемешивали при 110°C в течение 24 часов в атмосфере N2. Затем к смеси добавляли ледяную воду (15 мл), и водную фазу подвергали экстракции EtOAc (25 мл х 3), объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и концентрировали с получением промежуточного соединения 139В (2,6 г, 13,7 ммоль, выход: 78,2%) в виде коричневого масла. 1Н ЯМР (400 МГц, CDCl3) δ 4,36 (q, J=7,1 Гц, 2Н), 2,48 (s, 3Н), 1,38 (t, J=7,2 Гц, 5Н).
[0863] К смеси соединения 139В (400 мг, 2,1 ммоль) и 2H-индазола (299 мг, 2,5 ммоль) в ДМФА (3 мл) добавляли K2CO3 (1,2 г, 8,4 ммоль) одной порцией. Смесь перемешивали при 80°C в течение 12 часов. Затем к смеси добавляли Н2О (9 мл), и водную фазу подвергали экстракции EtOAc (15 мл × 3), и объединенный органический слой концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 300:1 до 40:1) с получением соединения 139С (340 мг, выход: 59,4%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,33 (s, 1H), 7,82 (d, J=7,9 Гц, 1H), 7,68 (d, J=8,8 Гц, 1H), 7,57-7,51 (m, 1H), 7,35 (t, J=7,7 Гц, 1H), 4,24 (q, J=7,0 Гц, 2Н), 2,56 (s, 3Н), 1,13 (t, J=7,2 Гц, 3Н).
[0864] К раствору соединения 139С (100 мг, 368,6 мкмоль) в ТГФ (2 мл) и Н2О (500 мкл) добавляли LiOH⋅H2O (15,5 мг, 368,6 мкмоль) одной порцией. Смесь перемешивали при 25°C в течение 12 часов. Затем рН водной фазы доводили до примерно 5 путем добавления HCl (1 М), и остаток концентрировали на роторном испарителе с получением промежуточного соединения 139D (83 мг, выход: 92,6%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,62 (s, 1H), 7,94 (d, J=7,9 Гц, 1H), 7,59 (d, J=3,5 Гц, 2Н), 7,41-7,35 (m, 1H), 3,30 (ушир. s, 3Н). МС (ИЭР) m/z (М+Н)+ 243,9.
[0865] Соединение 139 (18 мг, выход: 24,8%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 139D. Соединение 139: 1H ЯМР (400 МГц, CDCl3) δ 10,32 (ушир. d, J=5,7 Гц, 1H), 8,17 (d, J=8,6 Гц, 1H), 7,93 (s, 1H), 7,81 (d, J=7,9 Гц, 1H), 7,65 (t, J=7,4 Гц, 1H), 7,43 (t, J=7,5 Гц, 1H), 7,19-7,12 (m, 5Н), 6,77 (ушир. s, 1H), 5,77-5,69 (m, 1H), 5,49 (ушир. s, 1H), 3,42 (dd, J=5,1, 14,3 Гц, 1H), 3,20 (dd, J=7,9, 14,3 Гц, 1H), 2,57 (s, 3Н). МС (ИЭР) m/z (М+Н)+418,0.
ПРИМЕР 82
МЕТИЛ (S)-(3-(5-((4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)КАРБАМОИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-1-ИЛ)БЕНЗИЛ)КАРБАМАТ (140)
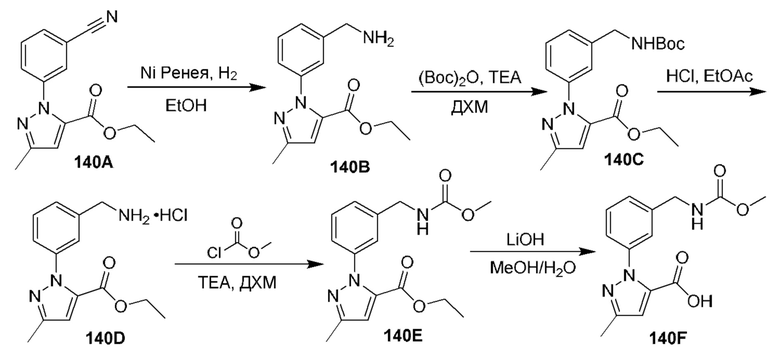
[0866] К раствору 3-гидразинилбензонитрила (30,0 г, 176,9 ммоль, гидрохлорид) в НОАс (500 мл) добавляли этил-2-метоксиимино-4-оксо-пентаноат (33,1 г, 176,9 ммоль). Смесь перемешивали при 100°C в течение 12 часов. Смесь концентрировали, разбавляли этилацетатом (200 мл), промывали NaHCO3 (водный, 200 мл), солевым раствором (200 мл), сушили над Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1:0 до 5:1). Полученный продукт растирали со смесью петролейный эфир/этилацетат = 10:1 (100 мл) и отфильтровывали. Соединение 140А (20,0 г, выход 44,3%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,05-8,02 (m, 1H), 7,93-7,88 (m, 1H), 7,83-7,78 (m, 1H), 7,70-7,63 (m, 1H), 6,94 (s, 1H), 4,21-4,13 (m, 2Н), 2,26 (s, 3Н), 1,19-1,11 (m, 3Н).
[0867] К раствору соединения 140А (9,00 г, 35,26 ммоль) в МеОН (500 мл) добавляли Ni Ренея (1,51 г) и NH3⋅H2O (4 мл). Смесь перемешивали при 25°C в атмосфере Нг при давлении 40 фунтов на кв. дюйм в течение 12 часов. Смесь концентрировали, разбавляли этилацетатом (500 мл), промывали HCl (500 мл), в водную фазу добавляли NaHCO3 (водный) до рН ~ 11. Затем смесь подвергали экстракции этилацетатом (500 мл), промывали солевым раствором (500 мл), сушили над Na2SO4 и концентрировали с получением промежуточного соединения 140В (15 г, неочищенного) в виде желтого масла.
[0868] К раствору соединения 140В (9,6 г, 37,06 ммоль) в ДХМ (100 мл) добавляли TEA (7 мл, 55,6 ммоль), затем к смеси добавляли Вос2О (9 мл, 40,77 ммоль), и смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь промывали лимонной кислотой (10%, 100 мл), подвергали экстракции ДХМ (100 мл × 2), промывали H2O (100 мл), сушили над безводным Na2SO4, фильтровали, упаривали при пониженном давлении. Неочищенный продукт очищали посредством колоночной флэш-хроматографии (петролейный эфир/этилацетат = 5/1) с получением соединения 140С (8,5 г, выход 63,8%) в виде желтого масла. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,50-7,44 (m, 1H), 7,43-7,37 (m, 1H), 7,32-7,24 (m, 3Н), 6,88 (s, 1H), 4,20-4,13 (m, 4Н), 2,27 (s, 3Н), 1,37 (s, 9Н), 1,20-1,14 (m, 3Н).
[0869] К суспензии соединение 140С (4,5 г, 13,03 ммоль) в ЭА (350 мл) добавляли HCl/EtOAc (4 М, 35 мл), и смесь перемешивали при 25°C в течение 2 ч. Реакционную смесь упаривали при пониженном давлении с получением соединения 140D (3,3 г, выход 89,9%, HCl) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии.
[0870] К раствору соединения 140D (1 г, 3,4 ммоль, HCl) в ДХМ (20 мл) добавляли TEA (1,4 мл, 10,1 ммоль), затем соединение метилхлорформиат (1,6 мл, 20,1 ммоль), затем смесь перемешивали при 25°C в течение 1 ч. Реакционную смесь разбавляли H2O (10 мл), смесь подвергали экстракции ДХМ (20 мл × 2). Органический слой собирали, промывали солевым раствором (20 мл × 3), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали посредством колоночной флэш-хроматографии (петролейный эфир/этилацетат, от 0 до 10/1) с получением соединения 140Е (400 мг, выход 37,3%), которое было получено в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,77 (ушир. t, J=6,2 Гц, 1H), 7,43-7,38 (m, 1H), 7,33-7,25 (m, 3Н), 6,88 (s, 1H), 4,23 (d, J=6,2 Гц, 2Н), 4,16 (q, J=7,1 Гц, 2Н), 3,55 (s, 3Н), 2,26 (s, 3Н), 1,14 (t, J=7,1 Гц, 3Н).
[0871] Соединение 140F (230 мг, выход 64,6%, белое твердое вещество) было получено, как описано в примере 85, из промежуточного соединения 140Е. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,76 (ушир. t, J=6,1 Гц, 1H), 7,42-7,35 (m, 1H), 7,31-7,22 (m, 3Н), 6,82 (s, 1H), 4,23 (ушир. d, J=6,2 Гц, 2Н), 3,55 (s, 3Н), 2,25 (s, 3Н).
[0872] Соединение 140 (35 мг, выход 21,1%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 140F. Соединение 140: 1Н ЯМР (CD3CN, 400 МГц) δ 7,40-7,17 (m, 10Н), 7,07 (ушир. d, J=18,3 Гц, 2Н), 6,47 (ушир. s, 1H), 6,26 (ушир. s, 1H), 6,09 (ушир. s, 1H), 5,34 (ушир. s, 1H), 4,29 (ушир. s, 2Н), 3,60 (ушир. s, 3Н), 3,27 (ушир. d, J=9,5 Гц, 1H), 2,99-2,85 (m, 1H), 2,27 (ушир. s, 3Н). МС (ИЭР) m/z (М+Н)+ 464,2.
ПРИМЕР 83
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(3-(БЕНЗАМИДОМЕТИЛ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (141)
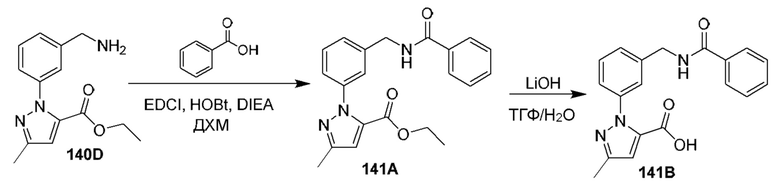
[0873] К раствору соединения 140D (300 мг, 1,22 ммоль) и бензойной кислоты (150 мг, 1,22 ммоль) в ДХМ (10 мл) добавляли HOBt (330 мг, 2,44 ммоль), DIEA (0,5 мл, 3,05 ммоль) и EDCI (470 мг, 2,44 ммоль). Смесь перемешивали при 25°C в течение 12 ч. Растворитель удаляли под вакуумом. Остаток растворяли в EtOAc (20 мл), промывали 1 н. HCl (20 мл). Органические фазы собирали, промывали насыщенным NaHCO3 (20 мл). Органические фазы собирали, промывали солевым раствором (20 мл), сушили с помощью Na2SO4, фильтровали и концентрировали с получением соединения 141А (400 мг, неочищенного) в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 364,0.
[0874] К раствору соединения 141А (400 мг, 1,14 ммоль) в ТГФ (5 мл) и H2O (5 мл) добавляли LiOH-H2O (241 мг, 5,72 ммоль). Смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь подкисляли с помощью 1 н. HCl до рН ~ 4, подвергали экстракции EtOAc (15 мл × 2). Органические фазы собирали и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в нейтральных условиях) с получением соединения 141 В (100 мг, выход: 26,16%) в виде белого твердого вещества. МС (ИЭР) m/z (M+Na)+ 358,0.
[0875] Соединение 141 (4,4 мг, выход: 14,40%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 141В. Соединение 141: МС (ИЭР) m/z (М+Н)+ 510,0. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,86-8,68 (m, 2Н), 7,94-7,88 (m, 2Н), 7,84-7,57 (m, 2Н), 7,54-7,20 (m, 11Н), 7,11-6,99 (m, 1H), 6,55 (s, 1H), 5,33-5,24 (m, 1H), 4,56-4,48 (m, 2Н), 3,26-3,18 (m, 1H), 2,95-2,86 (m, 1H), 2,25 (s, 3Н).
ПРИМЕР 84
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(3-((3-ФЕНИЛПРОПАНАМИДО)МЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (142)
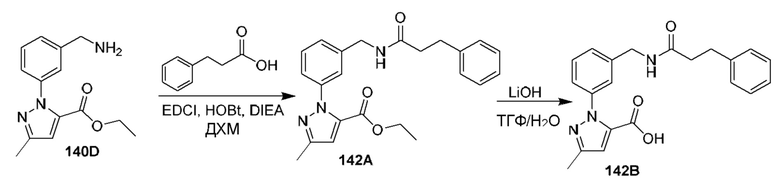
[0876] К раствору соединения 140D (500 мг, 2,04 ммоль) и 3-фенилпропановой кислоты (310 мг, 2,04 ммоль) в ДХМ (20 мл) добавляли DIEA (0,9 мл, 5,10 ммоль), HOBt (552 мг, 4,08 ммоль) и EDCI (783 мг, 4,08 ммоль). Смесь перемешивали при 25°C в течение 12 ч. Растворитель удаляли под вакуумом. Остаток растворяли в EtOAc (30 мл), промывали 1 н. HCl (30 мл). Органические фазы собирали, промывали насыщенным (30 мл), солевым раствором (30 мл), сушили с помощью Na2SO4, фильтровали, собирали и сушили под вакуумом с получением промежуточного соединения 22 (700 мг, неочищенного) в виде желтого масла. МС (ИЭР) m/z (M+Na)+ 414,0.
[0877] К раствору соединения 142А (700 мг, 1,85 ммоль) в ТГФ (10 мл) и H2O (10 мл) добавляли LiOH⋅H2O (390 мг, 9,27 ммоль). Смесь перемешивали при 25°C в течение 12 ч. Остаток подкисляли с помощью 1 н. HCl до рН ~ 4. Раствор подвергали экстракции EtOAc (20 мл × 2). Органические фазы собирали и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в нейтральных условиях) с получением соединения 142В (210 мг, выход: 31,24%) в виде белого твердого вещества. МС (ИЭР) m/z (M+Na)+ 386,0.
[0878] Соединение 142 (49,5 мг, выход: 37,88%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 142В. Соединение 142: МС (ИЭР) m/z (М+Н)+ 538,2. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,80-8,68 (m, 1H), 8,18-8,08 (m, 1H), 7,88-7,54 (m, 2Н), 7,33-7,02 (m, 15Н), 6,59-6,49 (m, 1H), 5,33-5,26 (m, 1H), 4,32-4,25 (m, 2Н), 3,26-3,20 (m, 1H), 2,95-2,90 (m, 1H), 2,90-2,85 (m, 2Н), 2,49-2,45 (m, 2Н), 2,28-2,22 (m, 3Н).
ПРИМЕР 85
(S)-1-(3-(АЦЕТАМИДОМЕТИЛ)ФЕНИЛ)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (143)
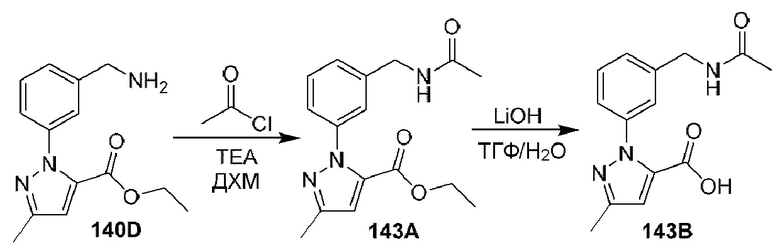
[0879] К раствору соединения 140D (500 мг, 2,04 ммоль) и ацетилхлорида (160 мг, 2,04 ммоль) в ДХМ (20 мл) добавляли TEA (0,7 мл, 5,10 ммоль). Смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь промывали 1 н. HCl (10 мл). Органические фазы собирали, сушили с помощью Na2SO4, фильтровали и концентрировали с получением промежуточного соединения 143А (580 мг, неочищенного) в виде желтого масла. МС (ИЭР) m/z (M+Na)+ 323,9.
[0880] К раствору соединения 143А (580 мг, 2,02 ммоль) в ТГФ (10 мл) и H2O (10 мл) добавляли LiOH⋅H2O (424 мг, 10,09 ммоль). Смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь подкисляли с помощью 1 н. HCl до рН ~ 4. Раствор подвергали экстракции EtOAc (20 мл × 2). Органические фазы собирали и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в нейтральных условиях) с получением соединения 26 (100 мг, выход: 18,11%) в виде белого твердого вещества. МС (ИЭР) m/z (M+Na)+ 295,9.
[0881] Соединение 143 (6,2 мг, выход: 12,26%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 143В. Соединение 143: МС (ИЭР) m/z (М+Н)+ 448,1. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,78-8,67 (m, 0,6Н), 8,19-8,05 (m, 1H), 7,85-7,72 (m, 1H), 7,67-7,53 (m, 0,6Н), 7,36-6,87 (m, 10Н), 6,59-6,46 (m, 1H), 6,30-5,89 (m, 1H), 5,33-5,23 (m, 0,6Н), 4,52-4,40 (m, 0,6Н), 4,32-4,22 (m, 2Н), 3,27-3,19 (m, 0,5Н), 2,96-2,85 (m, 0,6Н), 2,77-2,66 (m, 1H), 2,29-2,19 (m, 3Н), 1,89 (s, 3Н).
ПРИМЕР 86
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(3-((2-ФЕНИЛАЦЕТАМИДО)МЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (144)
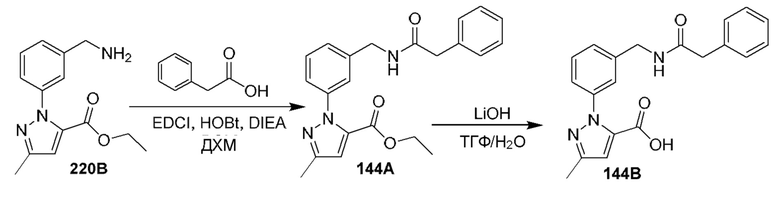
[0882] К раствору соединения 140D (500 мг, 2,04 ммоль) и 2-фенилуксусной кислоты (278 мг, 2,04 ммоль) в ДХМ (20 мл) добавляли DIEA (0,9 мл, 5,10 ммоль), HOBt (552 мг, 4,08 ммоль) и EDCI (783 мг, 4,08 ммоль). Смесь перемешивали при 25°C в течение 12 ч. Растворитель удаляли под вакуумом. Остаток растворяли в EtOAc (30 мл), промывали 1 н. HCl (30 мл). Органические фазы собирали, промывали насыщенным NaHCO3 (30 мл). Органические фазы собирали, промывали солевым раствором (30 мл). Органические фазы собирали, сушили с помощью Na2SO4, фильтровали и концентрировали с получением промежуточного соединения 144А (700,00 мг, неочищенного) в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 378,0.
[0883] К раствору соединения 144А1 (700 мг, 1,93 ммоль) в ТГФ (10 мл) и H2O (10 мл) добавляли LiOH⋅H2O (405 мг, 9,63 ммоль). Смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь подкисляли с помощью 1 н. HCl до рН ~ 4. Раствор подвергали экстракции EtOAc (15 мл × 2). Органические фазы собирали и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в нейтральных условиях) с получением соединения 144В (260 мг, выход: 38,56%) в виде желтого масла. МС (ИЭР) m/z (М+Н)+ 349,9.
[0884] Соединение 144 (36 мг, выход: 45,17%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединения 144В и 12G. Соединение 144: МС (ИЭР) m/z (М+Н)+ 524,2. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,07 (d, J=7,6 Гц, 1H), 8,66-8,49 (m, 1H), 8,08 (ушир. s, 1H), 7,84 (ушир. s, 1H), 7,34-7,10 (m, 13Н), 6,94-6,86 (m, 1H), 6,53 (s, 1H), 5,27-5,16 (m, 1H), 4,32-4,16 (m, 2Н), 3,44 (s, 2Н), 3,22-3,10 (m, 1H), 2,85-2,73 (m, 1H), 2,22 (s, 3Н).
ПРИМЕР 87
(S)-N-(4-(АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(3-(ФЕНИЛСУЛЬФОНАМИДОМЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (145)
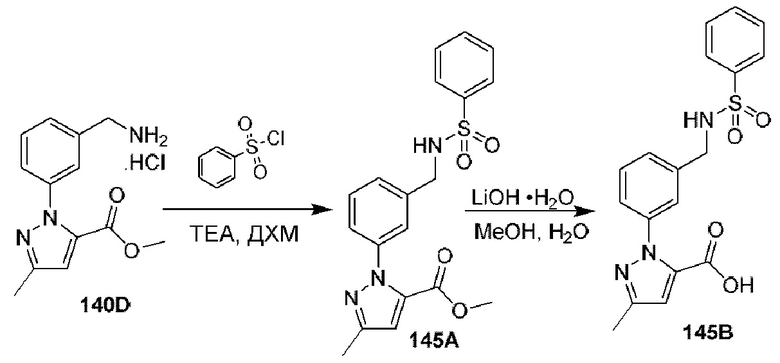
[0885] К смеси соединения 140D (300 мг, 1,1 ммоль, гидрохлорид) в ДХМ (20 мл) добавляли TEA (0,44 мл, 3,2 ммоль) одной порцией. К смеси добавляли по каплям бензолсульфонилхлорид (0,15 мл, 1,2 ммоль) при 0°C в течение 30 мин и затем перемешивали при 25°C в течение 1 ч. Реакционную смесь промывали 0,5 н. HCl (10 мл), насыщенным водным NaHCO3 (10 мл) и солевым раствором (10 мл). Отделенный органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток растирали с CH3CN (2 мл). Твердое вещество собирали и сушили под вакуумом с получением соединения 145А (330 мг, выход 79,2%) в виде белого твердого вещества. МС (ИЭР) m/z (М+Н)+ 386,0.
[0886] К смеси соединения 145А (150 мг, 0,39 ммоль) в МеОН (10 мл) и H2O (0,5 мл) добавляли LiOH⋅H2O (81,6 мг, 1,9 ммоль) одной порцией. Смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Затем остаток разбавляли водой (15 мл) и подвергали экстракции этилацетатом (10 мл), водную фазу подкисляли с помощью водного раствора HCl (1 М) до рН ~ 6~7 и подвергали экстракции этилацетатом (20 мл × 3). Объединенные органические слои промывали солевым раствором (40 мл) и сушили над Na2SO4, фильтровали и концентрировали с получением промежуточного соединения 145В (140 мг, неочищенного) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,29-8,22 (m, 1H), 7,85-7,78 (m, 2Н), 7,65-7,54 (m, 3Н), 7,38-7,23 (m, 4Н), 6,82 (s, 1H), 4,04 (d, J=6,0 Гц, 2Н), 2,26 (s, 3Н).
[0887] Соединение 145 (30 мг, выход 46,8%, белое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 145В. Соединение 145: 1Н ЯМР (CDCl3, 400 МГц): δ 7,86-7,81 (m, 2Н), 7,58-7,46 (m, 3Н), 7,37-7,26 (m, 5Н), 7,15-7,06 (m, 5Н), 6,41 (s, 1H), 6,24-6,18 (m, 1H), 6,16-6,10 (m, 2Н), 5,38-5,31 (m, 1H), 4,20-4,08 (m, 2Н), 3,34-3,27 (m, 1H), 3,10-3,03 (m, 1H), 2,30 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 546,1.
ПРИМЕР 88
ЭТИЛ-(S)-(3-(5-((4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)КАРБАМОИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-1-ИЛ)БЕНЗИЛ)КАРБАМАТ (147)
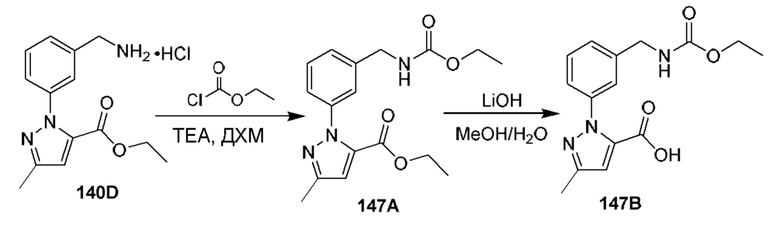
[0888] К раствору соединения 140D (1 г, 3,38 ммоль, гидрохлорид) в ДХМ (20 мл) добавляли по каплям TEA (1,4 мл, 10,14 ммоль), этилхлорформиат (1,9 мл, 20,27 ммоль), затем смесь перемешивали при 25°C в течение 1 ч. Реакционную смесь разбавляли H2O (10 мл), смесь подвергали экстракции ДХМ (20 мл × 2). Объединенный органический слой промывали солевым раствором (20 мл × 3), сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении. Продукт очищали посредством колоночной флэш-хроматографии (петролейный эфир/этилацетат: от 0 до 10/1) с получением соединения 147А (570 мг, выход 50,9%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,72 (ушир. t, J=6,1 Гц, 1H), 7,43-7,37 (m, 1H), 7,32-7,25 (m, 3Н), 6,88 (s, 1H), 4,23 (ушир. d, J=6,2 Гц, 2Н), 4,15 (q, J=7,1 Гц, 2Н), 4,03-3,97 (m, 2Н), 2,26 (s, 3Н), 1,16-1,12 (m, 6Н).
[0889] К раствору соединения 147А (560 мг, 1,69 ммоль) в МеОН (15 мл) добавляли по каплям LiOH (2 М, 5 мл), и затем смесь перемешивали при 25°C в течение 1 ч. Реакционную смесь разбавляли H2O (10 мл) и концентрировали при пониженном давлении. Смесь подвергали экстракции МТБЭ (10 мл), и водную фазу обрабатывали HCl (1 М) до рН ~ 5. Смесь подвергали экстракции этилацетатом (15 мл × 3), объединенный органический слой промывали солевым раствором (20 мл), сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении с получением соединения 147В (420 мг, выход 81,9%), которое было получено в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1H ЯМР (ДМСО-d6, 400 МГц) δ 7,69 (ушир. t, J=6,2 Гц, 1H), 7,38-7,33 (m, 1H), 7,27-7,20 (m, 3Н), 6,78 (s, 1H), 4,19 (ушир. d, J=6,2 Гц, 2Н), 3,97 (q, J=7,1 Гц, 2Н), 2,22 (s, 3Н), 1,16-1,10 (m, 3Н).
[0890] Соединение 147 (45 мг, выход 27,4%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 147В. Соединение 147: 1Н ЯМР (CD3CN, 400 МГц) δ 7,37-7,22 (m, 9Н), 7,10 (ушир. d, J=7,7 Гц, 2Н), 6,49 (s, 1H), 6,33 (ушир. s, 1H), 6,10 (ушир. s, 1H), 5,40-5,31 (m, 1H), 4,31 (ушир. d, J=6,2 Гц, 2Н), 4,08 (q, J=7,1 Гц, 2Н), 3,29 (dd, J=4,5, 14,0 Гц, 1H), 2,93 (dd, J=9,4, 14,0 Гц, 1H), 2,29 (s, 3Н), 1,21 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 478,2.
ПРИМЕР 89
(S)-N-((3,4-((3,4-ДИХЛОРБЕНЗИЛ)АМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (149)
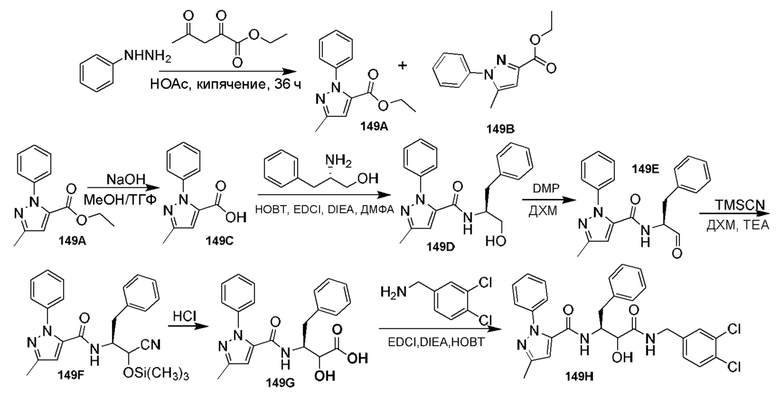
[0891] К раствору фенилгидразина (1,00 г, 9,25 ммоль, 910 мкл) в НОАс (20 мл) добавляли этил-2,4-диоксопентаноат (1,46 г, 9,25 ммоль, 1,3 мл). Смесь перемешивали при 100°C в течение 12 часов. Смесь концентрировали и разбавляли этилацетатом (50 мл), промывали NaHCO3 (водный, 50 мл × 3), солевым раствором (50 мл), сушили над Na2SO4 и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в условиях ТФУ). Соединение 149А (700,0 мг, выход 32,9%) получали в виде желтого масла. Соединение 149В (1,00 г, выход 46,9%) получали в виде желтого масла.
[0892] Соединение 149А: 1H ЯМР (400 МГц, ДМСО-d6) δ 7,48-7,36 (m, 5Н), 6,87 (s, 1H), 4,18-4,10 (m, 2Н), 2,25 (s, 3Н), 1,16-1,11 (m, 3Н).
[0893] Соединение 149В: 1H ЯМР (400 МГц, ДМСО-d6) δ 7,61-7,44 (m, 5Н), 6,75 (s, 1H), 4,31-4,23 (m, 2Н), 2,31 (s, 3Н), 1,30-1,24 (m, 3Н).
[0894] К раствору соединения 149А (700,0 мг, 3,04 ммоль) в ТГФ (20 мл) и МеОН (20 мл) добавляли NaOH (2 М, 30). Смесь перемешивали при 25°C в течение 12 часов. Смесь концентрировали, разбавляли H2O (20 мл), подвергали экстракции этилацетатом (20 мл), в водную фазу добавляли HCl (1 М) до рН ~ 1, затем смесь подвергали экстракции этилацетатом (20 мл), органический слой промывали солевым раствором (20 мл), сушили над Na2SO4 и концентрировали. Соединение 149С (600,0 мг, выход 97,6%) получали в виде бесцветного масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,21 (ушир. s, 1H), 7,49-7,31 (m, 5Н), 6,81 (s, 1H), 2,24 (s, 3Н).
[0895] К раствору соединения 149С (600,0 мг, 2,97 ммоль) в ТГФ (20 мл) добавляли DIEA (1,54 г, 11,88 ммоль, 2 мл), (2S)-2-амино-3-фенилпропан-1-ол (448,7 мг, 2,97 ммоль), HOBt (401,3 мг, 2,97 ммоль) и EDCI (683,2 мг, 3,56 ммоль). Смесь перемешивали при 25°C в течение 12 часов. Смесь концентрировали и разбавляли этилацетатом (50 мл), промывали HCl (1 М, 50 мл), насыщенным NaHCO3 (водный, 50 мл), солевым раствором (50 мл × 3), сушили над Na2SO4 и концентрировали. Соединение 149D (600,0 мг, выход 60,2%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,38 (d, J=8,8 Гц, 1H), 7,32-7,17 (m, 8Н), 7,12-7,07 (m, 2Н), 6,50 (s, 1H), 4,89-4,83 (m, 1H), 4,10-3,99 (m, 1H), 3,48-3,35 (m, 2Н), 2,95-2,87 (m, 1H), 2,68-2,59 (m, 1H), 2,21 (s, 3Н).
[0896] К раствору соединения 149D (600,0 мг, 1,79 ммоль) в ДХМ (200 мл) добавляли DMP (1,14 г, 2,69 ммоль). Смесь перемешивали при 25°C в течение 2 часов. В смесь добавляли для гашения реакции 10% Na2S2O3 (водный) : насыщенный NaHCO3 (водный) (1:1, 200 мл), смесь подвергали экстракции ДХМ (100 мл) и промывали солевым раствором (20 мл × 3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Соединение 149Е (500,0 мг, выход 83,8%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,58 (s, 1H), 9,03 (d, J=8,0 Гц, 1H), 7,36-7,13 (m, 10Н), 6,57 (s, 1H), 4,56-4,49 (m, 1H), 3,28-3,21 (m, 1H), 2,81-2,72 (m, 1H), 2,25-2,17 (m, 3Н).
[0897] К раствору соединения 149Е (500,0 мг, 1,50 ммоль) в ДХМ (10 мл) добавляли TEA (15,2 мг, 150,00 мкмоль, 20 мкл) и TMSCN (223,2 мг, 2,25 ммоль, 280 мкл). Смесь перемешивали при 0°C в течение 3 часов. Смесь промывали H2O (100 мл), солевым раствором (100 мл), сушили над Na2SO4 и концентрировали с получением промежуточного соединения 149F (600,0 мг, неочищенного) в виде бесцветного масла.
[0898] К раствору соединения 149F (600,0 мг, 1,39 ммоль) в ТГФ (30 мл) добавляли HCl (10 мл). После перемешивания при 60°C в течение 12 часов смесь разбавляли H2O (100 мл), подвергали экстракции этилацетатом (50 мл). Органический слой промывали NaHCO3 (водн., 50 мл). В водную фазу добавляли HCl (1 М) до рН ~ 1 и затем подвергали ее экстракции этилацетатом (500 мл). Органический слой промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали с получением промежуточного соединения 149G (500,0 мг, неочищенного) в виде бесцветного масла.
[0899] К раствору соединения 149G (500,0 мг, 1,32 ммоль) в ТГФ (10 мл) добавляли (3, 4-дихлорфенил)метанамин (255,6 мг, 1,45 ммоль, 190 мкл), HOBt (178,4 мг, 1,32 ммоль), DIEA (682,4 мг, 5,28 ммоль, 920 мкл) и EDCI (303,7 мг, 1,58 ммоль) с ДХМ (10 мл). Смесь перемешивали при 25°C в течение 12 часов. Смесь концентрировали и разбавляли этилацетатом (30 мл), промывали HCl (1 М, 30 мл), насыщенным NaHCO3 (водный, 30 мл), солевым раствором (30 мл), сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ (в условиях ТФУ). Полученный продукт (70 мг) растирали с CH3CN (5 мл) и отфильтровывали. Соединение 149Н (30,0 мг, 4,23%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,67-8,50 (m, 1H), 8,11 (d, J=9,6 Гц, 1H), 7,50-7,41 (m, 1H), 7,39-7,34 (m, 1H), 7,32-7,14 (m, 9Н), 7,07-6,96 (m, 2Н), 6,47-6,36 (m, 1H), 4,46-4,36 (m, 1H), 4,34-4,10 (m, 2Н), 4,06-3,99 (m, 1H), 2,95-2,71 (m, 2Н), 2,26-2,13 (m, 2Н), 2,26-2,13 (m, 1H).
[0900] К раствору соединения 149Н (30,0 мг, 55,82 мкмоль) в ДХМ (10 мл) и ДМСО (1 мл) добавляли DMP (47,4 мг, 111,64 мкмоль). Смесь перемешивали при 25°C в течение 48 часов. В смесь добавляли для гашения реакции 10% Na2S2O3 (водный) : насыщенный NaHCO3 (водный) (1:1, 20 мл), смесь подвергали экстракции ДХМ (10 мл) и промывали солевым раствором (20 мл × 3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Неочищенный продукт растирали с CH3CN (3 мл) и отфильтровывали. Соединение 149 (15,0 мг, выход 40,0%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,36-9,30 (m, 1H), 9,11 (ушир. A, J=1,6 Гц, 1H), 7,54-7,48 (m, 2Н), 7,33-7,19 (m, 9Н), 7,13 (ушир. d, J=6,6 Гц, 2Н), 6,52 (s, 1H), 5,29-5,22 (m, 1H), 4,34-4,28 (m, 2Н), 3,22-3,15 (m, 1H), 2,89-2,80 (m, 1H), 2,26-2,18 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 535,1.
ПРИМЕР 90
СОЕДИНЕНИЯ 150-152
(S)-N-(3,4-ДИОКСО-1-ФЕНИЛ-4-((3-(ТРИФТОРМЕТОКСИ)БЕНЗИЛ)АМИНО)БУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (150)
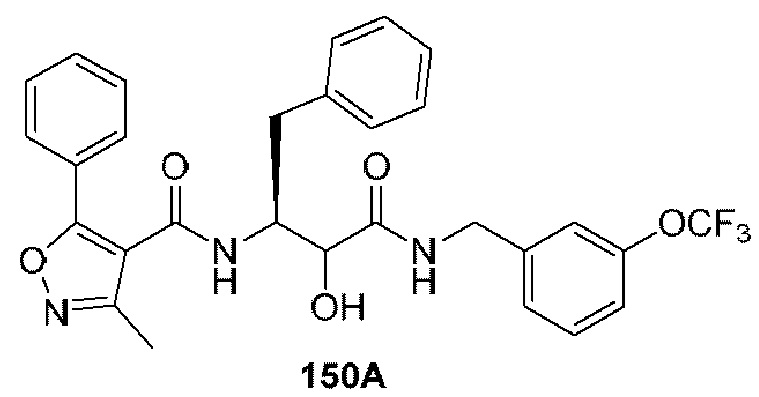
[0901] К раствору соединения 101Е (500,0 мг, 1,31 ммоль) в ТГФ (10 мл) добавляли [3-(трифторметокси)фенил]метанамин (251,3 мг, 1,31 ммоль), DIEA (509,6 мг, 3,94 ммоль, 690 мкл), HOBt (177,6 мг, 1,31 ммоль) и EDCI (302,4 мг, 1,58 ммоль) с ДХМ (5 мл). После перемешивания при 25°C в течение 12 часов смесь концентрировали и разбавляли этилацетатом (50 мл), промывали HCl (1 М, 50 мл), насыщенным водным NaHCO3 (50 мл), солевым раствором (50 мл × 3), сушили над Na2SO4 и концентрировали. Неочищенный продукт (0,30 г) растирали с CH3CN (5 мл) и отфильтровывали. Соединение 150А (140,0 мг, выход 19,3%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,75-8,53 (m, 1H), 8,31 (d, J=9,6 Гц, 1H), 7,59-7,08 (m, 14Н), 6,21-5,91 (m, 1H), 4,71-4,56 (m, 1H), 4,40-4,24 (m, 2Н), 4,22-4,01 (m, 1H), 2,98-2,67 (m, 2Н), 2,09-1,96 (m, 3Н).
[0902] К раствору соединения 150А (60,0 мг, 108,40 мкмоль) в ДХМ (10 мл) и ДМСО (1 мл) добавляли DMP (137,9 мг, 325,20 мкмоль). После перемешивания при 25°C в течение 4 часов в смесь добавляли для гашения реакции 10% Na2S2O3 (водный) : насыщенный водн. NaHCO3 (1:1, 20 мл), смесь подвергали экстракции ДХМ (10 мл) и промывали солевым раствором (20 мл × 3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Неочищенный продукт растирали с CH3CN (3 мл) и отфильтровывали. Соединение 150 (50,0 мг, выход 82,8%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,54-9,45 (m, 1H), 9,11 (d, J=7,6 Гц, 1H), 7,67-7,57 (m, 2Н), 7,54-7,36 (m, 4Н), 7,34-7,18 (m, 8Н), 5,52-5,43 (m, 1H), 4,40 (ушир. d, J=6,0 Гц, 2Н), 3,27-3,18 (m, 1H), 2,84-2,72 (m, 1H), 2,04 (s, 3Н). МС (ИЭР) m/z (М+Н)+552,1.
(S)-3-МЕТИЛ-N-(4-((4-((4ЧМЕТИЛСУЛЬФОНИЛ)БЕНЗИЛ)АМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (151)
(S)-3-МЕТИЛ-N-(4-((3-(МЕТИЛСУЛЬФОНИЛ)БЕНЗИЛ)АМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (152)
[0903] Соединения 151 и 152 были получены, как описано в примере 150, из соединения 101Е и соответствующего амина, соответственно. Соединение 151 (40,0 мг, выход 63,6%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,58-9,51 (m, 1H), 9,12 (d, J=7,2 Гц, 1H), 7,86 (d, J=8,4 Гц, 2Н), 7,65-7,59 (m, 2Н), 7,56-7,38 (m, 5Н), 7,31-7,19 (m, 5Н), 5,53-5,44 (m, 1H), 4,48-4,42 (m, 2Н), 3,29-3,21 (m, 1H), 3,20-3,10 (m, 3Н), 2,83-2,73 (m, 1H), 2,08-1,96 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 546,1.
[0904] Соединение 152 (42,0 мг, выход 68,8%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,59-9,51 (m, 1H), 9,11 (d, J=7,6 Гц, 1H), 7,91-7,78 (m, 2Н), 7,67-7,57 (m, 4Н), 7,53-7,37 (m, 3Н), 7,34-7,17 (m, 5Н), 5,53-5,45 (m, 1H), 4,46 (ушир. d, J=6,4 Гц, 2Н), 3,29-3,21 (m, 1H), 3,20-3,10 (m, 3Н), 2,83-2,72 (m, 1H), 2,09-1,98 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 546,1.
ПРИМЕР 91
БЕНЗИЛ (S)-(4-(5-((4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)КАРБАМОИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-1-ИЛ)БЕНЗИЛ)КАРБАМАТ (153)
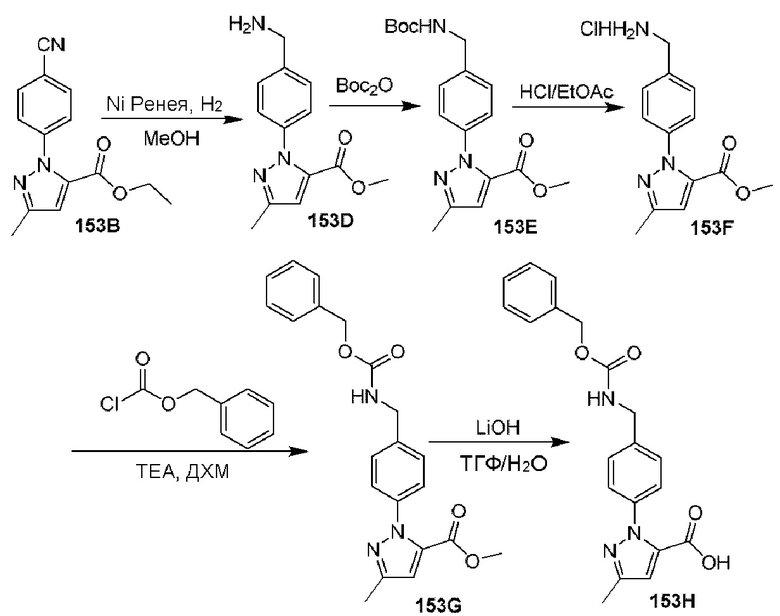
[0905] К раствору 4-гидразинилбензонитрила (20 г, 117,92 ммоль, HCl) в НОАс (200 мл) добавляли этил-2-метоксиимино-4-оксо-пентаноат (23,18 г, 123,82 ммоль), затем смесь нагревали до 110°C и перемешивали в течение 12 ч и затем удаляли растворитель при пониженном давлении. Остаток растворяли в этилацетате (200 мл) и обрабатывали NaHCO3 до рН ~ 8, и затем органический слой собирали и упаривали при пониженном давлении. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 15/1 до 3/1) с получением соединения 153В (5 г, выход: 16,61%) в виде белого твердого вещества. Соединение 153В: 1Н ЯМР (400 МГц, CDCl3) δ 7,71 (dd, J=7,9 Гц, 2Н), 7,53 (ушир. d, J=7,5 Гц, 2Н), 6,84 (s, 1H), 4,23 (q, J=7,0 Гц, 2Н), 2,33 (s, 3Н), 1,25 (t, J=7,1 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 255,9.
[0906] К раствору соединения 153В (6,5 г, 25,46 ммоль) в МеОН (70 мл) добавляли Ni Ренея (1,09 г, 12,73 ммоль) и NH3⋅H2O (2,68 г, 76,38 ммоль, 3 мл) в атмосфере аргона. Суспензию дегазировали под вакуумом и продували Н2 3 раза. Смесь перемешивали при 30°C в течение 16 ч в атмосфере H2 (40 фунтов на кв. дюйм). Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением промежуточного соединения 153D (6,6 г, неочищенного) в виде желтого масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,46-7,36 (m, 2Н), 7,35-7,29 (m, 2Н), 6,87 (s, 1H), 3,77 (s, 2Н), 3,71 (s, 3Н), 2,26 (s, 3Н).
[0907] К смеси соединения 153D (3,3 г, 13,45 ммоль) в ДХМ (40 мл) добавляли Et3N (2,04 г, 20,17 ммоль, 2,8 мл) и Вос2О (3,52 г, 16,14 ммоль, 3,7 мл) порциями при 25°C в атмосфере N2. Смесь перемешивали при 25°C в течение 1,5 ч. Реакционную смесь разбавляли ДХМ (20 мл) и промывали H2O (50 мл). Органический слой отделяли, и водный слой подвергали экстракции ДХМ (20 мл × 2). Объединенные органические слои промывали солевым раствором (30 мл × 2), сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 2/1) с получением соединения 153Е (3,3 г, выход: 64,86%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,37 (s, 4Н), 6,80 (s, 1H), 4,38 (dd, J=5,1 Гц, 2Н), 3,78 (s, 3Н), 2,36 (s, 3Н), 1,47 (s, 9Н). МС (ИЭР) m/z (М+Н)+ 346,1.
[0908] К смеси соединения 153Е (3,3 г, 9,55 ммоль) в этилацетате (20 мл) добавляли HCl/EtOAc (4 М, 20 мл) по каплям при 0°C. Реакционную смесь перемешивали при 25°C в течение 2 ч. Смесь концентрировали с получением промежуточного соединения 153F (2,7 г, неочищенного, HCl) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,62 (s, 1H), 7,63 (dd, J=7,9 Гц, 2Н), 7,40 (dd, J=7,5 Гц, 2Н), 6,80 (s, 1H), 4,16 (s, 2Н), 3,74 (s, 3Н), 2,34 (s, 3Н).
[0909] К смеси соединения 153F (300 мг, 1,06 ммоль, HCl) в ДХМ (20 мл) добавляли Et3N (268,15 мг, 2,65 ммоль, 0,4 мл) и бензилхлорформиат (181 мг, 1,06 ммоль, 0,2 мл) порциями при 25°C и перемешивали в течение 1,5 ч. Реакционную смесь обрабатывали ДХМ (20 мл), добавляли в нее H2O (30 мл). Органический слой отделяли и промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 1/1) с получением соединения 153G (350 мг, выход: 87,03%) в виде грязно-белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,42-7,29 (m, 9Н), 6,82-6,78 (m, 1H), 5,16 (s, 2Н), 4,45 (dd, J=6,2 Гц, 2Н), 3,80-3,77 (m, 3Н), 2,39-2,34 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 380,0.
[0910] К смеси соединения 153G (350 мг, 922,48 мкмоль) в ТГФ (10 мл) и H2O (10 мл) добавляли LiOH⋅H2O (116 мг, 2,77 ммоль) порциями при 25°C и перемешивали в течение 1,5 ч. Смесь разбавляли H2O (10 мл) и концентрировали для удаления ТГФ, затем водную фазу подвергали экстракции МТБЭ (30 мл × 2). Водные слои подкисляли до рН ~ 2 с помощью 1 н. HCl, затем раствор подвергали экстракции этилацетатом (30 мл × 3). Органические слои сушили над Na2SO4 и концентрировали с получением промежуточного соединения 153Н (300 мг, выход: 89,04%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,35 (dd, J=5,3, 8,2 Гц, 6Н), 7,30-7,16 (m, 2Н), 7,14-6,98 (m, 1H), 6,90-6,82 (m, 1H), 5,15 (s, 2Н), 4,47-4,30 (m, 2Н), 2,46-2,28 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 366,1.
[0911] Соединение 153 (35 мг, выход: 50,19%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 153Н. Соединение 153: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,04 (d, J=7,7 Гц, 1H), 8,09 (s, 1H), 7,84 (ушир. s, 2Н), 7,38-7,17 (m, 12Н), 7,09 (d, J=8,2 Гц, 2Н), 6,53 (s, 1H), 5,27 (t, J=7,5 Гц, 1H), 5,04 (s, 2Н), 4,20 (d, J=6,0 Гц, 2Н), 3,19 (dd, J=3,3, 14,1 Гц, 1H), 2,86-2,75 (m, 1H), 2,22 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 540,2.
ПРИМЕР 92
СОЕДИНЕНИЯ 154-159, 496
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4-(БЕНЗАМИДОМЕТИЛ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (154)
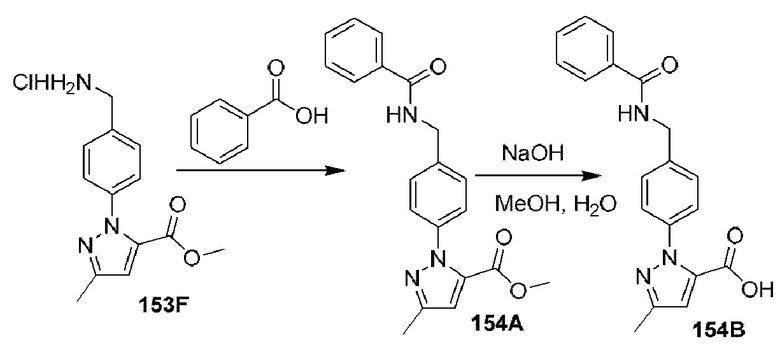
[0912] К смеси соединения 153F (300 мг, 1,06 ммоль, HCl), бензойной кислоты (155 мг, 1,27 ммоль, 0,2 мл), HOBt (286 мг, 2,12 ммоль) и DIEA (343 мг, 2,65 ммоль, 0,5 мл) в ДХМ (20 мл) добавляли EDCI (406 мг, 2,12 ммоль) порциями при 25°C и перемешивали в течение 4 ч. Реакционную смесь обрабатывали ДХМ (10 мл), промывали H2O (20 мл). Органический слой отделяли, и водный слой подвергали экстракции ДХМ (10 мл × 2). Объединенный органический слой промывали 0,5 н. HCl (20 мл × 2), NaHCO3 (20 мл × 2) и солевым раствором (30 мл), сушили над Na2SO4, фильтровали, и растворитель удаляли под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 1/1) с получением соединения 154А (280 мг, выход: 75,61%) в виде грязно-белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,81 (dd, J=6,4 Гц, 2Н), 7,58-7,40 (m, 7Н), 6,85-6,78 (m, 1H), 6,48 (ушир. s, 1H), 4,72 (ушир. d, J=5,1 Гц, 2Н), 3,84-3,77 (m, 3Н), 2,41-2,34 (m, 4Н). МС (ИЭР) m/z (M+Na)+ 372,0.
[0913] К смеси соединения 154А (280 мг, 801,42 мкмоль) в МеОН (10 мл) и H2O (10 мл) добавляли NaOH (2 М, 2 мл) порциями при 25°C и перемешивали в течение 3 ч. Смесь концентрировали для удаления МеОН, и затем водную фазу подвергали экстракции МТБЭ (30 мл × 2). Водный слой подкисляли до рН ~ 2 с помощью 1 н. HCl, затем раствор подвергали экстракции этилацетатом (20 мл × 2). Органические слои сушили над Na2SO4 и концентрировали с получением промежуточного соединения 154В (200 мг, выход: 74,41%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,79 (dd, J=8,2 Гц, 2Н), 7,50-7,31 (m, 7Н), 6,78 (s, 1H), 4,62 (s, 2Н), 2,31 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 336,0.
[0914] Соединение 154 (20 мг, выход: 29,53%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 154В. Соединение 154: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,11-9,04 (m, 2Н), 8,11 (s, 1H), 7,96-7,91 (m, 2Н), 7,86 (s, 1H), 7,56-7,47 (m, 3Н), 7,30 (d, J=4,4 Гц, 3Н), 7,27 (d, J=8,6 Гц, 2Н), 7,23-7,19 (m, 1H), 7,12 (d, J=8,4 Гц, 2Н), 6,55 (s, 1H), 5,32-5,26 (m, 1H), 4,51 (ушир. d, J=5,7 Гц, 2Н), 3,21 (dd, J=3,5, 13,9 Гц, 1H), 2,86-2,78 (m, 1H), 2,25 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 510,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-((3-ФЕНИЛПРОПАНАМИДО)МЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (155)
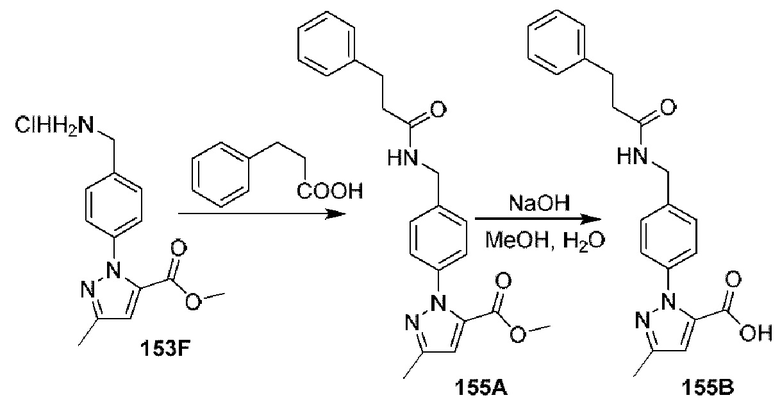
[0915] Следуя методике получения соединения 154В, промежуточное соединение 155В (200 мг, выход: 74,41%, белое твердое вещество) получали из соединения 153F через стадию получения 155А. Соединение 155 В: 1Н ЯМР (400 МГц, CDCl3) δ 7,28-7,12 (m, 8Н), 6,94 (s, 1H), 6,78 (s, 1H), 4,42-4,31 (m, 2Н), 2,94 (t, J=7,5 Гц, 2Н), 2,50 (t, J=7,2 Гц, 2Н), 2,31 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 364,1.
[0916] Соединение 155 (20 мг, выход: 27,16%, светло-желтое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 155В. Соединение 155: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,06 (d, J=7,7 Гц, 1H), 8,38 (t, J=5,8 Гц, 1H), 8,11 (s, 1H), 7,87 (s, 1H), 7,33-7,29 (m, 4Н), 7,27 (d, J=7,5 Гц, 2Н), 7,24-7,18 (m, 3Н), 7,13-7,07 (m, 4Н), 6,56 (s, 1H), 5,30 (dd, J=2,6 Гц, 1H), 4,27 (d, J=5,7 Гц, 2Н), 3,25-3,19 (m, 1H), 2,87-2,83 (m, 2Н), 2,53 (d, J=2,0 Гц, 1H), 2,49-2,44 (m, 2Н), 2,25 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 538,2.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-(ФЕНИЛСУЛЬФОНАМИДОМЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (156)
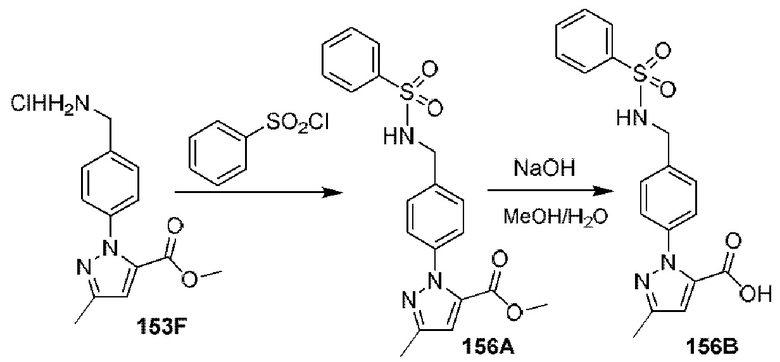
[0917] Следуя методике получения соединения 154В, промежуточное соединение 156В (250 мг, выход: 86,48%, белое твердое вещество) получали из соединения 153F через стадию получения 156А. Соединение 156В: 1Н ЯМР (400 МГц, CDCl3) δ 7,85 (dd, J=7,7 Гц, 2Н), 7,61-7,43 (m, 3Н), 7,29-7,20 (m, 4Н), 6,78 (s, 1H), 4,09 (s, 2Н), 2,30 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 372,0.
[0918] Соединение 156 (45 мг, выход: 78,05%, белое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 156В. Соединение 156: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,10-9,01 (m, 1H), 8,19 (ушир. s, 1H), 8,09 (s, 1H), 7,86-7,79 (m, 3Н), 7,63-7,56 (m, 3Н), 7,32-7,25 (m, 5Н), 7,19 (d, J=8,4 Гц, 2Н), 7,07 (d, J=8,4 Гц, 2Н), 6,52 (s, 1H), 5,26 (ушир. s, 1H), 3,97 (d, J=5,3 Гц, 2Н), 3,23-3,13 (m, 1H), 2,87-2,75 (m, 1H), 2,22 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 546,1.
(S)-1-(4-(АЦЕТАМИДОМЕТИЛ)ФЕНИЛ)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (157)
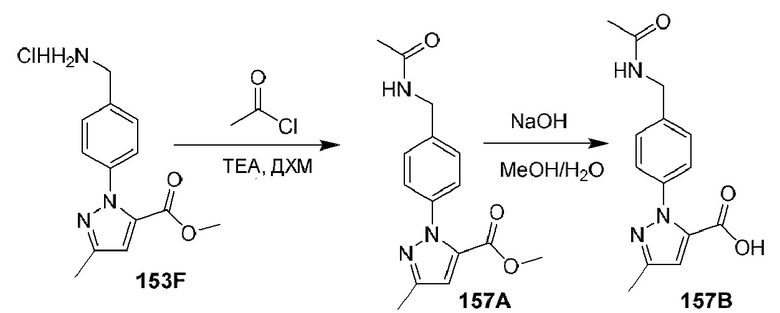
[0919] Следуя методике получения соединения 154В, промежуточное соединение 157В (162 мг, выход: 94,62%, белое твердое вещество) получали из соединения 153F через стадию получения 157А. Соединение 157В: 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,20 (ушир. s, 1H), 8,43 (ушир. t, J=5,8 Гц, 1H), 7,37-7,25 (m, 4Н), 6,79 (s, 1H), 4,29 (d, J=6,0 Гц, 2Н), 2,23 (s, 3Н), 1,88 (s, 3Н).
[0920] Соединение 157 (17 мг, выход: 33,13%, серое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 157В. Соединение 157: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,71 (ушир. d, J=7,5 Гц, 1H), 8,12 (ушир. s, 1H), 7,83-7,54 (m, 2Н), 7,31-7,18 (m, 9Н), 6,55 (s, 1H), 5,35-5,27 (m, 1H), 4,28 (d, J=6,0 Гц, 2Н), 3,25 (d, J=4,3 Гц, 0,5Н), 3,21 (d, J=4,0 Гц, 0,5Н), 2,94 (s, 0,5Н), 2,91 (d, J=4,3 Гц, 0,5Н), 2,25 (s, 3Н), 1,91 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 448,1.
МЕТИЛ-(S)-(4-(5-((4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)КАРБАМОИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-1-ИЛ)БЕНЗИЛ)КАРБАМАТ (158)
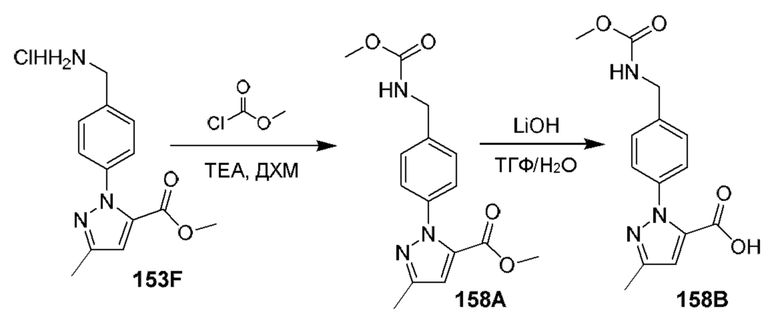
[0921] Следуя методике получения соединения 154В, промежуточное соединение 158В (150 мг, выход: 62,91%, белое твердое вещество) получали из соединения 153F через стадию получения 158А. Соединение 158В: 1Н ЯМР (400 МГц, CDCl3) δ 7,36-7,28 (m, 4Н), 6,78 (s, 1H), 4,36 (s, 2Н), 3,67 (s, 3Н), 2,34-2,30 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 289,9.
[0922] Соединение 158 (12 мг, выход: 22,68%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 158В. Соединение 158: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,08 (dd, J=7,5 Гц, 1H), 8,13 (s, 1H), 7,87 (ушир. s, 1H), 7,74 (s, 1H), 7,35-7,17 (m, 7Н), 7,16-7,07 (m, 2Н), 6,54 (s, 1H), 5,34-5,24 (m, 1H), 4,19 (dd, J=6,0 Гц, 2Н), 3,57 (s, 3Н), 3,28-3,18 (m, 1H), 2,82 (dd, J=10,9, 13,3 Гц, 1H), 2,24 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 464,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-((2-ФЕНИЛАЦЕТАМИДО)МЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (159)
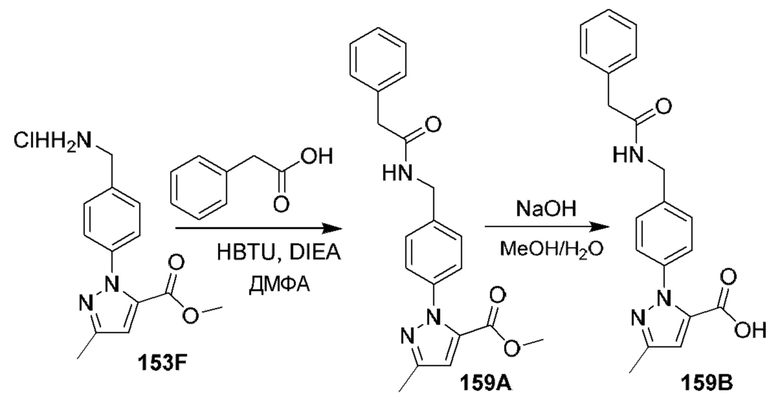
[0923] К смеси соединения 153F (300 мг, 1,06 ммоль, HCl) и 2-фенилуксусной кислоты (173 мг, 1,27 ммоль, 0,16 мл) в ДМФА (10 мл) добавляли DIEA (548 мг, 4,24 ммоль, 0,75 мл) и HBTU (603 мг, 1,59 ммоль) одной порцией при 25°C. Смесь перемешивали при 25°C в течение 1,5 ч. Смесь разбавляли 30 мл этилацетата и 20 мл H2O, органический слой отделяли и промывали 1 н. HCl (20 мл × 2), насыщенным NaHCO3 (20 мл × 2) и солевым раствором (20 мл), органический слой сушили над Na2SO4 и фильтровали, и органический слой концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 4/1). Соединение 159А (190 мг, выход: 49,32%) получали в виде желтого масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,62 (ушир. t, J=5,8 Гц, 1H), 7,36-7,14 (m, 9Н), 6,91-6,78 (m, 1H), 4,31 (d, J=6,0 Гц, 2Н), 3,69 (s, 3Н), 3,48 (s, 2Н), 2,24 (s, 3Н).
[0924] К раствору соединения 159А (190 мг, 522,83 мкмоль) в МеОН (8 мл) и H2O (5 мл) добавляли NaOH (84 мг, 2,09 ммоль). Смесь перемешивали при 25°C в течение 2 ч. Реакционную смесь концентрировали и добавляли 10 мл воды, и смесь подвергали экстракции МТБЭ (10 мл × 2), водный слой подкисляли с помощью 1 н. HCl до рН ~ 2~3 при 0°C и подвергали экстракции EtOAc (10 мл × 2), органическую фазу сушили над Na2SO4 и концентрировали с получением остатка. Соединение 159В (143 мг, выход: 78,28%) получали в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,62 (ушир. t, J=5,7 Гц, 1H), 7,34-7,24 (m, 8Н), 7,24-7,19 (m, 1H), 6,76 (s, 1H), 4,30 (d, J=5,7 Гц, 2Н), 3,48 (s, 2Н), 2,22 (s, 3Н).
[0925] Соединение 159 (25 мг, выход: 33,34%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 159В. Соединение 159: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,31 (ушир. s, 1H), 7,40-7,05 (m, 17Н), 6,56 (s, 1H), 5,31 (dd, J=4,3, 9,8 Гц, 1H), 4,31 (d, J=4,3 Гц, 2Н), 3,52 (s, 2Н), 3,23 (dd, J=4,3, 14,1 Гц, 1H), 2,91 (dd, J=10,0, 13,8 Гц, 1H), 2,25 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 524,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4-((4-ФТОРБЕНЗАМИДО)МЕТИЛ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (496)
[0926] Соединение 496 (246,9 мг, выход: 80,2%, белое твердое вещество) было получено, как в случае соединения 154, с использованием промежуточного соединения 153F и 4-фторбензоилхлорида, и полученный продукт вводили в реакции, как в случае соединения 12, с получением соединения 496. Соединение 496: 1Н ЯМР (400 МГц, ДМСО-d6) δ 99,20-9,05 (m, 2Н), 8,13 (s, 1H), 8,05-7,96 (m, 2Н), 7,87 (s, 1H), 7,40-7,16 (m, 9Н), 7,16-7,08 (m, 2Н), 6,54 (s, 1H), 5,32-5,22 (m, 1H), 4,55-4,45 (m, 2Н), 3,22-3,14 (m, 1H), 2,83-2,73 (m, 1H), 2,24 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 526,2.
ПРИМЕР 93
МЕТИЛ-(S)-(4-(5-((4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)КАРБАМОИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-1-ИЛ)БЕНЗИЛ)КАРБАМАТ (161)
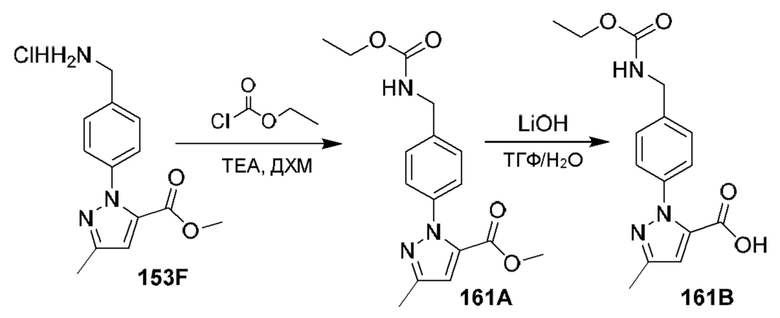
[0927] К смеси соединения 153F (450 мг, 1,60 ммоль, HCl) в ДХМ (15,00 мл) добавляли TEA (485 мг, 4,79 ммоль, 0,7 мл) и этилхлорформиат (452 мг, 4,17 ммоль, 0,4 мл) порциями при 25°C и перемешивали в течение 2 ч. Реакционную смесь обрабатывали ДХМ (20 мл), промывали H2O (30 мл). Органический слой отделяли и промывали солевым раствором (30 мл), сушили над безводным NaSO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 1/1) с получением соединения 161А (400 мг, выход: 52,81%) в виде грязно-белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,37 (ушир. s, 4Н), 6,80 (dd, J=3,5 Гц, 1H), 4,43 (s, 2Н), 4,20-4,09 (m, 2Н), 3,79 (d, J=3,7 Гц, 3Н), 2,36 (dd, J=3,5 Гц, 3Н), 1,27 (td, J=3,5, 7,1 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 318,0.
[0928] К смеси соединения 161А (400 мг, 1,26 ммоль) в ТГФ (10 мл) и H2O (10 мл) добавляли LiOH⋅H2O (159 мг, 3,78 ммоль) порциями при 25°C и перемешивали в течение 0,5 ч. Смесь разбавляли H2O (10 мл) и концентрировали для удаления ТГФ, затем водную фазу подвергали экстракции МТБЭ (30 мл × 2). Водные слои подкисляли до рН ~ 2 с помощью 1 н. HCl, затем раствор подвергали экстракции этилацетатом (30 мл × 3). Органические слои сушили над Na2SO4 и концентрировали с получением промежуточного соединения 161В (300 мг, выход: 78,50%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,33 (s, 4Н), 6,79 (s, 1H), 4,35 (s, 2Н), 4,23-4,03 (m, 2Н), 2,38-2,27 (m, 3Н), 1,41-1,19 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 304,0.
[0929] Соединение 161 (25 мг, выход: 22,8%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 161В. Соединение 161: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,07 (dd, J=8,2 Гц, 1H), 8,12 (s, 1H), 7,87 (s, 1H), 7,69 (s, 1H), 7,42-7,27 (m, 5Н), 7,20 (dd, J=7,9 Гц, 2Н), 7,16-7,06 (m, 2Н), 6,54 (s, 1H), 5,34-5,24 (m, 1H), 4,18 (dd, J=5,5 Гц, 2Н), 4,02 (q, J=7,1 Гц, 2Н), 3,21 (dd, J=2,9, 13,2 Гц, 1H), 2,92-2,77 (m, 1H), 2,24 (s, 3Н), 1,18 (ушир. t, J=7,1 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 478,1.
ПРИМЕР 94
ФЕНИЛ-(S)-(4-(5-((4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)КАРБАМОИЛ)-3-МЕТИЛ-1Н-ПИРАЗОЛ-1-ИЛ)БЕНЗИЛ)КАРБАМАТ (162)
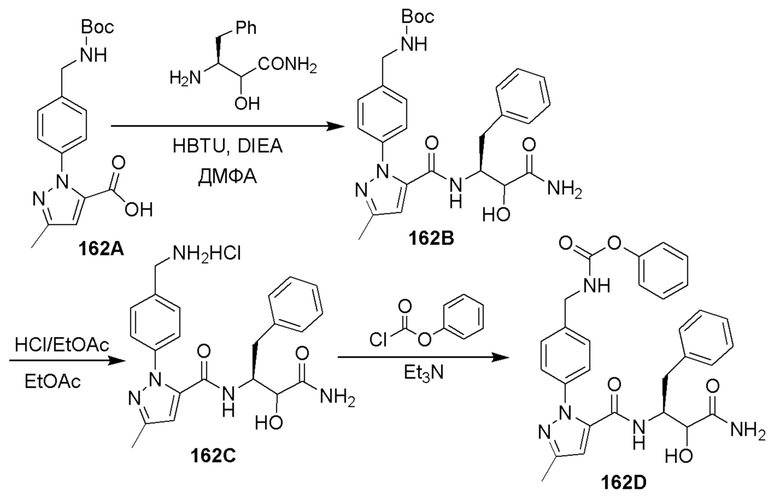
[0930] К смеси соединения 153Е (300 мг, 868,58 мкмоль) в ТГФ (10 мл) и H2O (10 мл) добавляли LiOH⋅Н2О (109 мг, 2,61 ммоль) порциями при 25°C и перемешивали в течение 12 ч. Смесь разбавляли H2O (10 мл) и концентрировали для удаления ТГФ, затем водную фазу подвергали экстракции МТБЭ (30 мл × 2). Водные слои подкисляли до рН ~ 2 с помощью 1 н. HCl, затем раствор подвергали экстракции этилацетатом (30 мл × 3). Органические слои сушили над Na2SO4 и концентрировали с получением промежуточного соединения 162А (250 мг, выход: 86,86%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,41-7,30 (m, 2Н), 7,27-7,04 (m, 2Н), 6,86 (s, 1H), 4,43-4,26 (m, 2Н), 2,46-2,32 (m, 3Н), 1,60-1,40 (m, 9Н). МС (ИЭР) m/z (М+Н)+ 332,0.
[0931] К смеси соединения 12G (209 мг, 905,33 мкмоль, HCl) и соединения 162А (250 мг, 754,44 мкмоль) в ДМФА (10 мл) добавляли DIEA (244 мг, 1,89 ммоль, 0,3 мл) и HBTU (343 мг, 905,33 мкмоль) порциями при 25°C и перемешивали в течение 1,5 ч. Реакционную смесь обрабатывали этилацетатом (40 мл), промывали H2O (50 мл × 2). Органический слой промывали солевым раствором (30 мл), сушили над Na2SO4, фильтровали, и растворитель удаляли под вакуумом. Остаток растирали в ДХМ (2 мл) и петролейном эфире (10 мл), твердое вещество собирали и сушили под вакуумом с получением соединения 162В (300 мг, выход: 75,76%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,72-8,41 (m, 1H), 7,95 (s, 1H), 7,45-7,20 (m, 7Н), 7,12 (dd, J=8,4 Гц, 2Н), 7,02-6,91 (m, 2Н), 6,70-6,48 (m, 1H), 6,08 (d, J=5,5 Гц, 1H), 4,45 (s, 1H), 4,12-3,98 (m, 2Н), 2,89 (s, 1H), 2,87-2,80 (m, 1H), 2,73 (s, 1H), 2,28-2,14 (m, 3Н), 1,52-1,33 (m, 9Н). МС (ИЭР) m/z (М-56)+ 452,1.
[0932] К смеси соединения 162В (300 мг, 591,04 мкмоль) в ЭА (10 мл) добавляли HCl/EtOAc (4 М, 10 мл) по каплям при 0°C. Реакционную смесь перемешивали при 25°C в течение 2 ч. Смесь концентрировали с получением промежуточного соединения 162С (250 мг, выход: 95,28%, HCl) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,71-8,64 (m, 2Н), 7,53-7,35 (m, 3Н), 7,34-7,17 (m, 7Н), 7,03 (dd, J=8,4 Гц, 2Н), 6,65 (s, 1H), 4,59-4,30 (m, 1H), 4,15 (s, 2Н), 2,88 (s, 1H), 2,83 (dd, J=11,9 Гц, 1H), 2,72 (s, 1H), 2,22 (s, 3Н).
[0933] К смеси соединения 162С (120 мг, 270,31 мкмоль, HCl) в ДХМ (10 мл) добавляли Et3N (68 мг, 675,78 мкмоль, 0,1 мл) и фенилхлорформиат (51 мг, 324,38 мкмоль, 0,1 мл) порциями при 25°C и перемешивали в течение 1 ч. Реакционную смесь обрабатывали ДХМ (20 мл), добавляли H2O (30 мл). Органический слой отделяли и промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в условиях HCl) с получением соединения 162D (70 мг, выход: 47,71%) в виде грязно-белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,36 (s, 1H), 8,50 (dd, J=9,0 Гц, 1H), 7,41-7,11 (m, 14Н), 7,08 (s, 1H), 7,02 (dd, J=8,4 Гц, 1H), 6,99 (dd, J=8,4 Гц, 1H), 6,78-6,72 (m, 2Н), 6,57-6,50 (m, 1H), 4,43 (s, 1H), 4,31-4,22 (m, 2Н), 3,99 (s, 1H), 2,87-2,75 (m, 2Н), 2,74-2,65 (m, 2Н), 2,27-2,20 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 528,1.
[0934] К смеси соединения 162D (40 мг, 75,82 мкмоль) в ДМСО (3 мл) и ДХМ (15 мл) добавляли DMP (96 мг, 227,46 мкмоль) одной порцией при 25°C в атмосфере N2. Смесь перемешивали при 25°C в течение 1,5 ч. Реакционную смесь разбавляли ДХМ (10 мл), NaHCO3 (5 мл) и Na2S2O3 (10 мл), затем перемешивали в течение 10 мин, и слои разделяли. Органические слои промывали водой (50 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток растирали в ДХМ (2 мл) и петролейном эфире (10 мл), твердое вещество собирали и сушили под вакуумом с получением соединения 162 (25 мг, выход: 51,13%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,15 (d, J=8,0 Гц, 1H), 8,39 (t, J=6,0 Гц, 1H), 8,14 (s, 1H), 7,88 (s, 1H), 7,41-7,19 (m, 11Н), 7,14 (dd, J=8,0 Гц, 3Н), 6,68-6,51 (m, 1H), 5,41-5,22 (m, 1H), 4,29 (dd, J=6,0 Гц, 2H), 3,21 (dd, J=3,5, 13,6 Гц, 1H), 2,85 (dd, J=10,8, 13,8 Гц, 1H), 2,25 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 526,1.
ПРИМЕР 95
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(м-ТОЛИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (163)
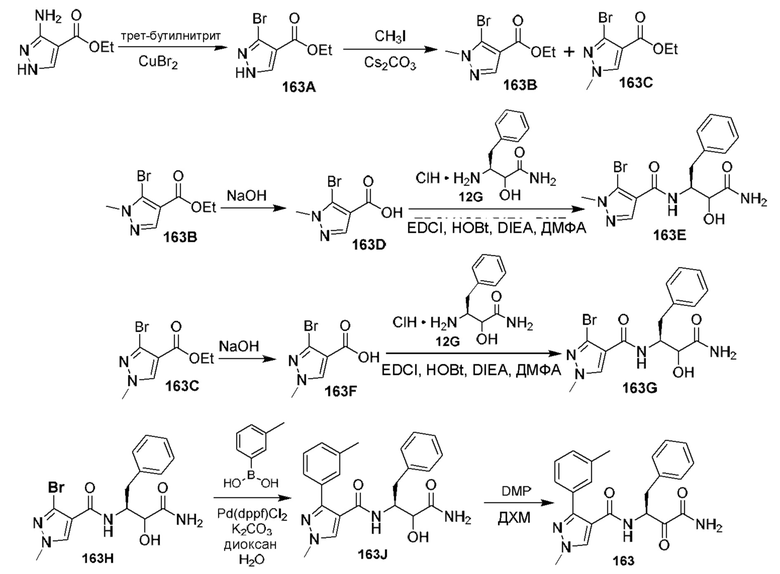
[0935] К раствору т-BuONO (3,8 мл, 30,94 ммоль) в CH3CN (60 мл) добавляли CuBr2 (6,91 г, 30,94 ммоль). Смесь перемешивали при 25°C в течение 1 ч в атмосфере N2. Затем добавляли порциями этил-3-амино-1Н-пиразол-4-карбоксилат (4 г, 25,78 ммоль). Затем смесь нагревали до 70°C и перемешивали в течение 12 ч. Реакционную смесь промывали Н2О (100 мл), подвергали экстракции EtOAc (100 мл × 2). Органические фазы собирали, сушили с помощью Na2SO4, фильтровали и концентрировали с получением промежуточного соединения 163А (6 г, неочищенного) в виде черного масла. МС (ИЭР) m/z (М+2)+ 220,9.
[0936] К раствору соединения 163А (10 г, 45,65 ммоль) и Cs2CO3 (29,75 г, 91,30 ммоль) в ДМФА (250 мл) добавляли Mel (19,44 г, 136,95 ммоль, 8,53 мл). Смесь перемешивали при 25°C в течение 16 ч. Смесь фильтровали, фильтрат разбавляли Н2О (500 мл) и подвергали экстракции этилацетатом (100 мл × 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1). Соединение 163В (2,5 г, выход: 23,50%) получали в виде желтого масла, и соединение 163С (5,5 г, выход: 51,70%) получали в виде белого твердого вещества.
[0937] Соединение 163В: 1Н ЯМР (400 МГц, CDCl3) δ 7,93 (s, 1H), 4,31 (q, J=7,1 Гц, 2Н), 3,95-3,87 (m, 3Н), 1,36 (t, J=7,1 Гц, 3Н).
[0938] Соединение 163С: 1Н ЯМР (400 МГц, CDCl3) δ 7,82 (s, 1H), 4,30 (q, J=7,1 Гц, 2Н), 3,99-3,77 (m, 3Н), 1,35 (t, J=7,2 Гц, 3Н).
[0939] К раствору соединения 163В (600 мг, 2,57 ммоль) в МеОН (10 мл) и H2O (10 мл) добавляли NaOH (514 мг, 12,85 ммоль). Смесь перемешивали при 25°C в течение 3 ч. Реакционную смесь концентрировали и добавляли 20 мл воды, смесь подвергали экстракции МТБЭ (10 мл × 2), водный слой подкисляли с помощью 1 н. HCl до рН ~ 2~3 при 0°C и подвергали экстракции EtOAc (20 мл × 2), органическую фазу сушили над Na2SO4, концентрировали с получением остатка. Соединение 163D (480 мг, выход: 91,05%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,65 (s, 1H), 8,13 (s, 1H), 3,82 (s, 3Н).
[0940] К раствору соединения 163D (450 мг, 2,20 ммоль), (3S)-3-амино-2-гидрокси-4-фенил-бутанамида 12G (761 мг, 3,30 ммоль, HCl) и НОВТ (445 мг, 3,30 ммоль) в ДХМ (20 мл) добавляли DIEA (1,14 г, 8,80 ммоль, 1,54 мл) и EDCI (843 мг, 4,40 ммоль). Смесь перемешивали при 25°C в течение 16 ч. Смесь разбавляли CHCl3:iPrOH=3:1 (50 мл), промывали 1 н. HCl (30 мл), насыщенным водным NaHCO3 (30 мл) и солевым раствором (30 мл). Органический слой сушили над безводным Na2SO4 и концентрировали под вакуумом. Твердое вещество растирали в этилацетате (30 мл), фильтровали. Соединение 163Е (550 мг, выход: 61,64%) получали в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,09-7,95 (m, 1H), 7,78 (d, J=8,8 Гц, 0,6Н), 7,46 (d, J=9,0 Гц, 0,4Н), 7,38-7,07 (m, 6Н), 6,01-5,86 (m, 1H), 4,54-4,33 (m, 1H), 4,00 (dd, J=3,4, 5,2 Гц, 1H), 3,85-3,74 (m, 4Н), 2,93-2,67 (m, 1H), 2,62 (dd, J=2,3, 13,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 381,0.
[0941] К раствору соединения 163С (2,6 г, 11,16 ммоль) в МеОН (10 мл) и H2O (10 мл) добавляли LiOH⋅H2O (2,34 г, 55,80 ммоль). Смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь концентрировали и добавляли 20 мл воды, и смесь подвергали экстракции МТБЭ (20 мл × 2), водный слой подкисляли с помощью 1 н. HCl до рН ~ 2-3 при 0°C и подвергали экстракции EtOAc (30 мл × 2), органическую фазу сушили над Na2SO4 и концентрировали с получением остатка. Соединение 163F (2,2 г, выход: 96,16%) было получено в виде серого твердого вещества, которое непосредственно использовали на следующей стадии. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,55 (ушир. s, 1H), 8,24 (s, 1H), 3,81 (s, 3Н).
[0942] К смеси соединения 163F (2,2 г, 10,73 ммоль) и соединения 12G (2,97 г, 12,88 ммоль HCl) в ДМФА (20 мл) и HOBt (2,17 г, 16,10 ммоль) и DIEA (4,16 г, 32,19 ммоль, 5,62 мл) и EDCI (4,11 г, 21,46 ммоль) одной порцией при 25°C. Смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь разбавляли H2O (40 мл) и подвергали экстракции смесью CHCl3 : изопропанол (об./об.=3:1; 30 × 3 мл), затем органическую фазу промывали 1 н. HCl (20 мл × 2) и насыщенным водным NaHCO3 (20 мл × 2). Смесь сушили над Na2SO4 и концентрировали. Остаток разбавляли EtOAc (15 мл), твердое вещество собирали и сушили под вакуумом. Соединение 163G (2,9 г, выход: 68,06%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,18 (s, 1H), 7,61 (ушир. d, J=8,8 Гц, 1H), 7,31 (ушир. d, J=2,4 Гц, 2Н), 7,24-7,13 (m, 5Н), 5,89 (d, J=5,7 Гц, 1H), 4,51-4,40 (m, 1H), 4,00-3,97 (m, 1H), 3,79 (s, 3Н), 2,80-2,76 (m, 1H), 2,65-2,58 (m, 1H). МС (ИЭР) m/z (М+Н)+ 381,0.
[0943] Соединение 163G (200 мг, 525 мкмоль), м-толилбороновую кислоту (85,6 мг, 629 мкмоль), Pd(dppf)Cl2 (38,4 мг, 52,5 мкмоль) и K2CO3 (145 мг, 1,05 ммоль) в диоксане (5 мл) дегазировали и затем нагревали до 100°C в течение 12 часов в атмосфере N2. Смесь фильтровали и концентрировали, остаток очищали посредством препаративной ТСХ (дихлорметан : метанол = 10:1) с получением соединения 163Н (100 мг, выход: 48,6%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,99 (d, J=19,4 Гц, 1H), 7,37 (ушир. s, 1H), 7,31-7,06 (m, 11Н), 5,76 (ушир. s, 1H), 4,52-4,31 (m, 1H), 3,98 (ушир. s, 1H), 3,83 (s, 3Н), 3,80 (ушир. s, 1H), 2,87-2,72 (m, 1H), 2,71-2,56 (m, 1H), 2,26 (d, J=6,8 Гц, 3Н).
[0944] Смесь соединения 163Н (100 мг, 255 мкмоль) и DMP (432 мг, 1,02 ммоль) в ДХМ (10 мл), ДМСО (2 мл) перемешивали при 25°C в течение 1 ч. Смесь разбавляли ДХМ (20 мл), добавляли в нее для гашения реакции насыщенный водный NaHCO3 (20 мл), насыщенный водный Na2S2O3 (20 мл) и перемешивали в течение 20 мин, смесь подвергали экстракции ДХМ (20 мл × 2), объединенную органическую фазу промывали водой (20 мл), солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали, остаток перемешивали в ДХМ и н-гексане в течение 20 мин, твердое вещество отфильтровывали и сушили с получением 163 (43,5 мг, выход: 43,7%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,23 (ушир. d, J=7,3 Гц, 1H), 8,08-7,97 (m, 2Н), 7,78 (s, 1H), 7,38 (s, 1H), 7,31 (ушир. d, J=7,5 Гц, 1H), 7,28-7,13 (m, 6Н), 7,11-7,05 (m, 1H), 5,31-5,21 (m, 1H), 3,85 (s, 3Н), 3,12 (dd, J=3,7, 13,9 Гц, 1H), 2,79 (dd, J=9,7, 13,9 Гц, 1H), 2,25 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 391,1.
ПРИМЕР 96
СОЕДИНЕНИЯ 164,169, 480-488, 498-518, 530, 548, 567-573, 585, 587, 591, 593, 597, 601-605, 607, 611, 613-617, 620-621, 624-629
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-(ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (164)
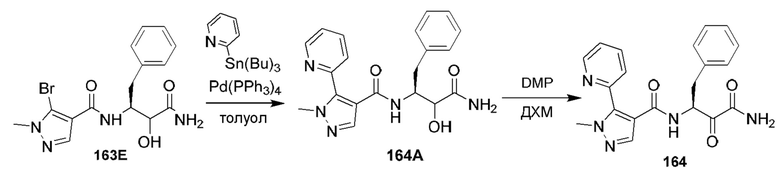
[0945] К смеси 163Е (200 мг, 527 мкмоль) и трибутил(2-пиридил)станнана (388 мг, 1,05 ммоль) в толуоле (5 мл) добавляли Pd(PPh3)4 (60,9 мг, 52,7 мкмоль) в атмосфере N2 (15 фунтов на кв. дюйм). После перемешивания при 110°C в течение 10 ч смесь концентрировали под вакуумом с получением остатка. Остаток очищали посредством препаративной ВЭЖХ (в кислых условиях) с получением соединения 164А (85 мг, выход: 42,5%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3-d) δ 8,97 (ушир. d, J=7,06 Гц, 1H), 8,72 (ушир. d, J=7,28 Гц, 1H), 8,55 (dd, J=17,64, 4,85 Гц, 1H), 8,03-7,95 (m, 1H), 7,88-7,79 (m, 1H), 7,43-7,31 (m, 2Н), 7,13-6,99 (m, 6Н), 5,49 (ушир. d, J=10,14 Гц, 1H), 4,27-4,17 (m, 2Н), 3,89 (d, J=3,53 Гц, 3Н), 3,26-2,91 (m, 2Н).
[0946] Соединение 164 (31 мг, выход: 41,6%, белое твердое вещество) было получено, как описано в примере 105, из промежуточного соединения 164А. Соединение 164: 1H ЯМР (400 МГц, CDCl3-d) δ 8,51 (ушир. d, J=6,4 Гц, 1H), 8,43 (d, J=5,1 Гц, 1H), 8,01 (s, 1H), 7,84 (ушир. t, J=7,8 Гц, 1H), 7,49 (d, J=7,9 Гц, 1H), 7,32 (dd, J=5,3, 7,1 Гц, 1H), 7,21-7,09 (m, 3Н), 6,96 (ушир. d, J=5,7 Гц, 2H), 6,75 (ушир. s, 1H), 5,70-5,60 (m, 2Н), 3,89 (s, 3Н), 3,33 (dd, J=5,1, 14,3 Гц, 1H), 3,15 (dd, J=7,1, 14,1 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 378,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (169)
[0947] Соединение 169 (20 мг, выход: 48,2%, белое твердое вещество) было получено, как в случае соединения 163, из соответствующих исходных веществ, соединения 163G и трибутил(2-пиридил)станнана. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,88 (ушир. d, J=7,9 Гц, 1H), 8,32 (ушир. d, J=5,3 Гц, 1H), 8,20 (s, 1H), 8,08 (d, J=8,3 Гц, 1H), 7,91 (t, J=7,7 Гц, 1H), 7,74 (ушир. s, 1H), 7,52 (ушир. s, 1H), 7,40-7,32 (m, 1H), 7,20-7,05 (m, 5Н), 5,64-5,47 (m, 1H), 3,91 (s, 3Н), 3,27 (dd, J=4,8, 14,5 Гц, 1H), 3,12-3,07 (m, 1H). МС (ИЭР) m/z (М+Н)+ 378,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2,3-ДИФТОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (480)
[0948] Соединение 480 (60 мг, выход: 48,13%, белое твердое вещество) было получено, как в случае соединения 163, из соответствующих исходных веществ, соединения 163G и (2,3-дифторфенил)бороновой кислоты. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,39 (d, J=7,3 Гц, 1H), 8,30 (s, 1H), 8,03 (s, 1H), 7,78 (s, 1H), 7,45-7,34 (m, 1H), 7,33-7,13 (m, 7Н), 5,28-5,22 (m, 1H), 3,92 (s, 3Н), 3,13 (dd, J=3,6, 13,8 Гц, 1H), 2,83 (dd, J=10,1, 13,9 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 413,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(6-ЦИАНОПИРИДИН-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (481)
[0949] Соединение 481 (15 мг, выход: 37,47%, белое твердое вещество) было получено, как в случае соединения 163, из соответствующих исходных веществ, соединения 163G и (6-цианопиридин-3-ил)бороновой кислоты. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,91 (d, J=1,8 Гц, 1H), 8,67 (d, J=7,3 Гц, 1H), 8,25 (s, 1H), 8,15 (dd, J=2,0, 8,2 Гц, 1H), 8,08 (s, 1H), 7,97 (d, J=8,2 Гц, 1H), 7,81 (s, 1H), 7,27 (d, J=4,4 Гц, 4Н), 7,24-7,17 (m, 1H), 5,27 (t, J=3,0 Гц, 1H), 3,94 (s, 3Н), 3,15 (dd, J=3,7, 13,9 Гц, 1H), 2,86-2,74 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 403,2.
N-4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(1H-ИНДАЗОЛ-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (482)
[0950] Соединение 482 (18 мг, выход: 14,77%, белое твердое вещество) было получено, как в случае соединения 163, из соответствующих исходных веществ, соединения 163С и 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-((2-(триметилсилил)этокси)метил)-1H-индазола, и конечное соединение 482 было получено путем удаления 2-(триметилсилил)этокси)метильной группы. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. s, 1H), 9,57 (ушир. s, 1H), 8,44 (d, J=8,0 Гц, 1H), 8,08 (s, 1H), 7,46-7,38 (m, 2Н), 7,24 (s, 6Н), 6,84 (ушир. s, 1H), 5,70 (ушир. s, 1H), 5,50 (ушир. s, 1H), 3,97 (s, 3Н), 3,42-3,32 (m, 1H), 3,26-3,20 (m, 1H). МС (ИЭР) m/z (М+Н)+ 417,0.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-БЕНЗИЛ-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (483)
[0951] Соединение 483 (65 мг, выход: 66,57%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и 2-бензил-4,4,5,5-тетраметил-1,3,2-диоксаборолана, и конечное соединение 483 было получено путем удаления 2-(триметилсилил)этокси)метильной группы. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,33 (d, J=7,1 Гц, 1H), 8,19 (s, 1H), 8,05 (s, 1H), 7,79 (s, 1H), 7,31-7,23 (m, 4Н), 7,20-7,07 (m, 6Н), 5,31-5,23 (m, 1H), 4,05 (s, 2Н), 3,77 (s, 3Н), 3,20-3,09 (m, 1H), 2,91-2,80 (m, 1H). МС (ИЭР) m/z (М+Н)+ 391,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ФТОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (484)
[0952] Соединение 484 (3,35 г, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения 163С и (2-фторфенил)бороновой кислоты, и было получено конечное соединение 484. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,30 (d, J=7,3 Гц, 1H), 8,26 (s, 1H), 8,04 (s, 1H), 7,79 (s, 1H), 7,41-7,11 (m, 9Н), 5,30-5,18 (m, 1H), 3,90 (s, 3Н), 3,12 (dd, J=3,6, 14,0 Гц, 1H), 2,83 (dd, J=9,8, 13,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 395,0.
ГИДРОХЛОРИД N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ФТОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИДА(485)
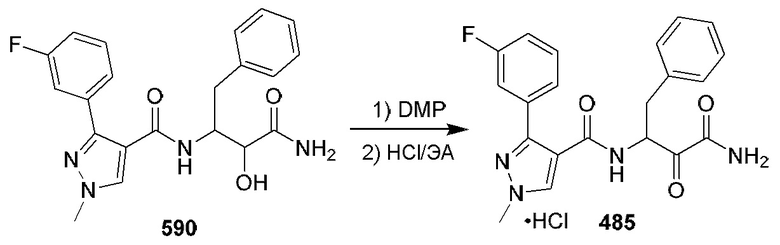
[0953] Соединение 485 синтезировали из промежуточного соединения 590, которое было синтезировано с использованием 163Н. Соединение 485 (3,1 г, выход: 79,85%, белое твердое вещество) получали из промежуточного соединения 590 с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, и было получено конечное соединение 485. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,51 (d, J=7,3 Гц, 1H), 8,12 (s, 2Н), 7,85 (ушир. s, 1H), 7,46 (d, J=8,3 Гц, 2Н), 7,39-7,28 (m, 5Н), 7,25-7,20 (m, 1H), 7,14 (t, J=8,0 Гц, 1H), 5,43-5,25 (m, 1H), 3,18 (dd, J=3,4, 13,7 Гц, 1H), 2,84 (dd, J=10,3, 13,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 395,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2,6-ДИФТОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (486)
[0954] Соединение 486 (60 мг, выход: 43,0%, светло-желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения 163С и (2,6-дифторфенил)бороновой кислоты, и было получено конечное соединение 486. 1Н ЯМР (400 МГц, CDCl3) δ 7,94 (s, 1H), 7,41-7,33 (m, 1H), 7,22-7,17 (m, 3Н), 6,99-6,93 (m, 2Н), 6,88 (dd, J=3,0, 6,3 Гц, 2Н), 6,67 (ушир. s, 1H), 6,01 (ушир. d, J=7,1 Гц, 1H), 5,63-5,57 (m, 1H), 5,53 (ушир. s, 1H), 3,97 (s, 3Н), 3,27 (dd, J=5,3, 14,1 Гц, 1H), 3,09 (dd, J=6,6, 14,1 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 413,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(6-МЕТОКСИПИРИДИН-2-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (487)
[0955] Соединение 487 (10 мг, выход: 20,06%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения 163С и 2-метокси-6-(трибутилстаннил)пиридина, и было получено конечное соединение 487. 1Н ЯМР (400 МГц, CD3CN) δ 11,02 (d, J=5,5 Гц, 1Н), 8,01 (s, 1Н), 7,81 (t, J=7,9 Гц, 1Н), 7,60 (d, J=7,5 Гц, 1Н), 7,14-7,06 (m, 5Н), 7,00-6,88 (m, 1Н), 6,81 (d, J=8,2 Гц, 1Н), 6,23-6,01 (m, 1Н), 5,21-5,15 (m, 1Н), 3,85 (s, 3Н), 3,73 (s, 3Н), 3,27 (dd, J=4,4, 13,9 Гц, 1Н), 2,97 (dd, J=9,9, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 408,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-БЕНЗИЛ-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (488)
[0956] Соединение 488 (35,6 мг, выход: 35,57%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-5-иод-1-метил-1H-пиразол-4-карбоксилата и 2-бензил-4,4,5,5-тетраметил-1,3,2-диоксаборолана, и было получено конечное соединение 488. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,40 (d, J=7,8 Гц, 1Н), 8,03 (ушир. s, 1Н), 7,92 (s, 1Н), 7,77 (ушир. s, 1Н), 7,31-7,03 (m, 10Н), 5,32-5,23 (m, 1Н), 4,36-4,24 (m, 2Н), 3,58 (s, 3Н), 3,19-3,10 (m, 1Н), 2,89-2,79 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 391,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(5-МЕТИЛФУРАН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (498)
[0957] Соединение 498 (70 мг, выход: 54,1%, светло-желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (5-метилфуран-2-ил)бороновой кислоты, и было получено конечное соединение 498. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,38-8,22 (m, 1Н), 8,20-8,01 (m, 2Н), 7,91-7,71 (m, 1Н), 7,31-7,17 (m, 5Н), 6,90 (d, J=3,1 Гц, 1Н), 6,08 (d, J=2,2 Гц, 1Н), 5,47-5,20 (m, 1Н), 3,87 (s, 3Н), 3,17 (dd, J=3,9, 13,8 Гц, 1Н), 2,88 (dd, J=9,7, 14,1 Гц, 1Н), 2,29-2,19 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 381,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2,5-ДИМЕТИЛТИОФЕН-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (499)
[0958] Соединение 499 (110 мг, выход: 64,9%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2,5-диметил-3-тиенил)бороновой кислоты, и было получено конечное соединение 499. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,14-8,00 (m, 2Н), 7,87-7,73 (m, 2Н), 7,34-7,12 (m, 5Н), 6,58 (d, J=1,1 Гц, 1Н), 5,28 (ddd, J=4,0, 7,3, 9,5 Гц, 1Н), 3,91-3,79 (m, 3Н), 3,14 (dd, J=4,0, 13,9 Гц, 1Н), 2,77 (dd, J=9,4, 14,0 Гц, 1Н), 2,33 (s, 3Н), 2,22-2,12 (m, 3Н). MC (ИЭР) m/z (М+Н)+ 411,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ФУРАН-2-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (500)
[0959] Соединение 500 (55 мг, выход: 91,2%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и 2-фурилбороновой кислоты, и было получено конечное соединение 500. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,37 (d, J=7,1 Гц, 1Н), 8,14 (s, 1Н), 8,07 (ушир. s, 1Н), 7,81 (ушир. s, 1Н), 7,61 (s, 1Н), 7,28 (s, 4Н), 7,20 (ушир. s, 1Н), 6,99 (d, J=2,9 Гц, 1Н), 6,48 (ушир. s, 1Н), 5,37-5,28 (m, 1Н), 3,88 (s, 3Н), 3,18 (dd, J=3,5, 13,7 Гц, 1Н), 2,86 (dd, J=9,7, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 367,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ХЛОРТИОФЕН-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (501)
[0960] Соединение 501 (110 мг, выход: 68,3%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2-хлортиофен-3-ил)бороновой кислоты, и было получено конечное соединение 501. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,13 (s, 1Н), 7,80-7,46 (m, 3Н), 7,35 (d, J=5,8 Гц, 1Н), 7,30-7,24 (m, 2Н), 7,23-7,16 (m, 3Н), 6,98 (d, J=5,8 Гц, 1Н), 5,31 (m, 1Н), 3,89 (s, 3Н), 3,17 (dd, J=4,4, 13,9 Гц, 1Н), 2,88 (dd, J=8,9, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 417,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ФУРАН-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (502)
[0961] Соединение 502 (160 мг, выход: 63,78%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и фуран-3-илбороновой кислоты, и было получено конечное соединение 502. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,35 (d, J=7,6 Гц, 1Н), 8,22-8,18 (m, 1Н), 8,13 (s, 1Н), 8,04 (ушир. s, 1Н), 7,76 (ушир. s, 1Н), 7,61-7,57 (m, 1Н), 7,31-7,14 (m, 5Н), 6,80-6,75 (m, 1Н), 5,32-5,23 (m, 1Н), 3,85 (s, 3Н), 3,19-3,11 (m, 1Н), 2,88-2,78 (m, 1Н). MC (ИЭР) m/z (М+Н)+ 367,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ТИОФЕН-3-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (503)
[0962] Соединение 503 (65 мг, выход: 47,7%, светло-желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и тиофен-3-илбороновой кислоты, и было получено конечное соединение 503. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,37 (d, J=7,3 Гц, 1Н), 8,04 (s, 2Н), 7,92 (s, 1Н), 7,77 (ушир. s, 1Н), 7,50-7,34 (m, 2Н), 7,30-7,22 (m, 4Н), 7,18 (dd, J=4,4, 8,6 Гц, 1Н), 5,38-5,18 (m, 1Н), 3,84 (s, 3Н), 3,22-3,08 (m, 1Н), 2,81 (dd, J=9,9, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 383,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(4-МЕТИЛТИОФЕН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (504)
[0963] Соединение 504 (100 мг, выход: 65,93%, светло-желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (4-метилтиофен-2-ил)бороновой кислоты, и было получено конечное соединение 504. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,37 (d, J=7,2 Гц, 1Н), 8,09 (s, 1Н), 8,06 (ушир. s, 1Н), 7,78 (ушир. s, 1Н), 7,57-7,54 (m, 1Н), 7,29-7,23 (m, 4Н), 7,22-7,15 (m, 1Н), 6,99-6,95 (m, 1Н), 5,34-5,27 (m, 1Н), 3,83 (s, 3Н), 3,19-3,10 (m, 1Н), 2,88-2,77 (m, 1Н), 2,14 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 397,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(5-МЕТИЛТИОФЕН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (505)
[0964] Соединение 505 (130 мг, выход: 61%, светло-желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (5-метилтиофен-2-ил)бороновой кислоты, и было получено конечное соединение 505. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,68 (s, 1Н), 8,57 (d, J=7,5 Гц, 1Н), 8,53-8,48 (m, 1Н), 8,23 (s, 1Н), 8,08 (s, 1Н), 7,92-7,86 (m, 1Н), 7,81 (s, 1Н), 7,34-7,25 (m, 4Н), 7,23-7,18 (m, 1Н), 5,36-5,28 (m, 1Н), 3,94 (s, 3Н), 3,17 (dd, J=3,9, 14,0 Гц, 1Н), 2,83 (dd, J=10,0, 14,0 Гц, 1Н). MC (ИЭР) m/z (М+Н)+ 396,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(5-ФТОРПИРИДИН-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (506)
[0965] Соединение 506 (35 мг, выход: 20,4%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (5-фторпиридин-3-ил)бороновой кислоты, и было получено конечное соединение 506. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,40 (d, J=7,3 Гц, 1Н), 8,16-8,03 (m, 2Н), 7,81 (s, 1Н), 7,54 (d, J=3,5 Гц, 1Н), 7,29 (d, J=4,2 Гц, 4Н), 7,24-7,19 (m, 1Н), 6,66 (dd, J=1,0, 3,6 Гц, 1Н), 5,34-5,29 (m, 1Н), 3,85 (s, 3Н), 3,17 (dd, J=3,7, 13,9 Гц, 1Н), 2,84 (dd, J=9,9, 13,9 Гц, 1Н), 2,40 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 397,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ФТОР-5-МЕТОКСИФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (507)
[0966] Соединение 507 (34 мг, выход: 15,75%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2-фтор-5-метоксифенил)бороновой кислоты, и было получено конечное соединение 507. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,14 (s, 1Н), 7,68 (ушир. d, J=7,0 Гц, 2Н), 7,51 (ушир. s, 1Н), 7,30-7,15 (m, 5Н), 7,10-7,01 (m, 1Н), 6,98-6,87 (m, 2Н), 5,28 (ddd, J=4,6, 7,3, 8,8 Гц, 1Н), 3,90 (s, 3Н), 3,75 (s, 3Н), 3,15 (dd, J=4,5, 14,1 Гц, 1Н), 2,87 (dd, J=8,9, 14,2 Гц, 1Н). 19F ЯМР (376 МГц, ДМСО-d6) δ -124,53 (ушир. d, J=88,69 Гц, 1F). МС (ИЭР) m/z (M+Na)+ 447,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-(ДИФТОРМЕТИЛ)ФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (508)
[0967] Соединение 508 (140 мг, выход: 69,98%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (3-(дифторметил)фенил)бороновой кислоты, и было получено конечное соединение 508. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,43 (d, J=7,6 Гц, 1Н), 8,09 (s, 1Н), 8,02 (ушир. s, 1Н), 7,85-7,81 (m, 1Н), 7,78 (ушир. s, 1Н), 7,73-7,67 (m, 1Н), 7,51-7,46 (m, 1Н), 7,44-7,39 (m, 1Н), 7,30-7,16 (m, 5H), 7,15-6,84 (m, 1Н), 5,32-5,23 (m, 1Н), 3,89 (s, 3Н), 3,19-3,11 (m, 1Н), 2,87-2,76 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 427,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ФТОР-3-МЕТОКСИФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (509)
[0968] Соединение 509 (190 мг, выход: 66,92%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2-фтор-3-метоксифенил)бороновой кислоты, и было получено конечное соединение 509. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,18 (s, 1Н), 8,13 (d, J=7,6 Гц, 1Н), 7,96 (ушир. s, 1Н), 7,73 (ушир. s, 1Н), 7,30-7,16 (m, 5Н), 7,14-7,01 (m, 2Н), 6,87-6,79 (m, 1Н), 5,27-5,16 (m, 1Н), 3,88 (s, 3Н), 3,80 (s, 3Н), 3,14-3,06 (m, 1Н), 2,84-2,74 (m, 1Н). МС (ИЭР) m/z (М+H)+ 425,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2,2-ДИФТОРБЕНЗО[d][1,3]ДИОКСОЛ-4-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (510)
[0969] Соединение 510 (70 мг, выход: 34%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2,2-дифторбензо[d][1,3]диоксол-4-ил)бороновой кислоты, и было получено конечное соединение 510. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,44 (d, J=7,5 Гц, 1Н), 8,19 (s, 1Н), 7,98 (s, 1Н), 7,76 (s, 1Н), 7,35 (d, J=7,8 Гц, 1Н), 7,31-7,12 (m, 7Н), 5,34-5,21 (m, 1Н), 3,93 (s, 3Н), 3,14 (dd, J=3,6, 13,9 Гц, 1Н), 2,82 (dd, J=9,9, 14,2 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 457,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-МЕТОКСИФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (511)
[0970] Соединение 511 (23 мг, выход: 8,04%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2-метоксифенил)бороновой кислоты, и было получено конечное соединение 511. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,04 (s, 1Н), 7,72 (ушир. s, 1Н), 7,55 (ушир. s, 1Н), 7,37 (ушир. t, J=7,9 Гц, 1Н), 7,28-7,10 (m, 5Н), 7,08-6,90 (m, 4Н), 5,30 (ушир. d, J=4,3 Гц, 1Н), 3,91-3,81 (m, 3Н), 3,66-3,54 (m, 3Н), 3,12 (ушир. d, J=4,3 Гц, 1Н), 2,76 (dd, J=8,8, 14,1 Гц, 1Н). МС (ИЭР) m/z (M+Na)+ 407,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ФТОР-2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (512)
[0971] Соединение 512 (60 мг, выход: 21,65%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и 2-(3-фтор-2-(трифторметил)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана, и было получено конечное соединение 512. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,28 (s, 1Н), 8,21 (d, J=7,5 Гц, 1Н), 7,99 (s, 1Н), 7,76 (s, 1Н), 7,67-7,59 (m, 1Н), 7,52-7,43 (m, 1Н), 7,31-7,17 (m, 5Н), 7,05 (d, J=7,7 Гц, 1Н), 5,25-5,16 (m, 1Н), 3,90 (s, 3Н), 3,11 (dd, J=3,6, 14,0 Гц, 1Н), 2,79 (dd, J=10,0, 14,0 Гц, 1Н). 19F ЯМР (376 МГц, ДМСО-d6) δ -55,03 (d, J=18,3 Гц, 3F), -114,43 (tdd, J=6,1, 12,3, 18,7 Гц, 1F). МС (ИЭР) m/z (M+Na)+ 463,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (513)
[0972] Соединение 513 (45 мг, выход: 54,58%, белое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2-(трифторметил)фенил)бороновой кислоты, и было получено конечное соединение 513. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,25 (s, 1Н), 7,99 (ушир. d, J=7,1 Гц, 2Н), 7,80-7,69 (m, 2Н), 7,62-7,52 (m, 2Н), 7,30-7,17 (m, 6Н), 5,20 (ddd, J=3,9, 7,4, 9,7 Гц, 1Н), 3,89 (s, 3Н), 3,17-3,03 (m, 1Н), 2,78 (dd, J=9,7, 13,9 Гц, 1Н). 19F ЯМР (376 МГц, ДМСО-d6) δ -57,15 (s, 3F). МС (ИЭР) m/z (М+Н)+ 445,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(БЕНЗО[d][1,3]ДИОКСОЛ-4-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (514)
[0973] Соединение 514 (92 мг, выход: 34,71%, светло-желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и бензо[d][1,3]диоксол-4-илбороновой кислоты, и было получено конечное соединение 514. 1Н ЯМР (400 МГц, ДМСО-d6) δ 2,82 (dd, J=13,89, 9,48 Гц, 1Н) 3,12 (dd, J=14,00, 4,08 Гц, 1Н) 3,88 (s, 3Н) 5,16-5,34 (m, 1Н) 5,77 (s, 1Н) 5,87 (s, 1Н) 6,76-6,90 (m, 3Н) 7,18-7,31 (m, 5H) 7,77 (s, 1Н) 8,00 (s, 1Н) 8,07 (s, 1Н) 8,11 (d, J=7,28 Гц, 1 H). МС (ИЭР) m/z (М+Н)+ 421,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(5-МЕТОКСИПИРИДИН-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (515)
[0974] Соединение 515 (60 мг, выход: 20,6%, желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (5-метоксипиридин-3-ил)бороновой кислоты, и было получено конечное соединение 515. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,50 (d, J=7,5 Гц, 1Н), 8,39 (d, J=1,5 Гц, 1Н), 8,22 (d, J=2,6 Гц, 1Н), 8,17 (s, 1Н), 8,06 (s, 1Н), 7,80 (s, 1Н), 7,59 (dd, J=1,8, 2,9 Гц, 1Н), 7,29 (d, J=4,4 Гц, 4Н), 7,24-7,18 (m, 1Н), 5,35-5,26 (m, 1Н), 3,93 (s, 3Н), 3,79 (s, 3Н), 3,16 (dd, J=3,6, 13,8 Гц, 1Н), 2,83 (dd, J=9,9, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 408,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ФТОР-3-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (516)
[0975] Соединение 516 (85 мг, выход: 78,65%, светло-желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2-фтор-3-(трифторметил)фенил)бороновой кислоты, и было получено конечное соединение 516. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,42 (d, J=7,5 Гц, 1Н), 8,29 (s, 1Н), 8,00 (s, 1Н), 7,79-7,72 (m, 2Н), 7,63 (ушир. t, J=7,1 Гц, 1Н), 7,37 (t, J=7,7 Гц, 1Н), 7,31-7,18 (m, 5Н), 5,29-5,19 (m, 1Н), 3,93 (s, 3Н), 3,13 (dd, J=3,5, 14,1 Гц, 1Н), 2,82 (dd, J=10,1, 13,9 Гц, 1Н). 19F ЯМР (376 МГц, ДМСО-d6) δ -59,92 (s, 1F), -59,96 (s, 1F), -116,72 -116,79 (m, 1F), -116,80-116,86 (m, 1F). МС (ИЭР) m/z (M+H)+ 463,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ФТОР-3-МЕТИЛФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (517)
[0976] Соединение 517 (170 мг, выход: 53,97%, светло-желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2-фтор-3-метилфенил)бороновой кислоты, и было получено конечное соединение 517. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,20 (s, 1Н), 8,13 (d, J=7,5 Гц, 1Н), 8,02 (s, 1Н), 7,78 (s, 1Н), 7,31-7,20 (m, 6Н), 7,15-7,10 (m, 1Н), 7,07-7,02 (m, 1Н), 5,25 (ddd, J=3,9, 7,3, 9,5 Гц, 1Н), 3,91 (s, 3Н), 3,75 (s, 3Н), 3,13 (dd, J=4,0, 13,9 Гц, 1Н), 2,82 (dd, J=9,7, 13,9 Гц, 1Н), 2,21 (d, J=1,5 Гц, 3Н). 19F ЯМР (376 МГц, ДМСО-d6) δ -116,59 -120,74 (m, 1F). МС (ИЭР) m/z (М+Н)+ 409,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2,5-ДИФТОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (518)
[0977] Соединение 518 (35,9 мг, выход: 24,08%, светло-желтое твердое вещество) получали с использованием методик, аналогичных методикам получения соединения 163, из соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и (2,5-дифторфенил)бороновой кислоты, и было получено конечное соединение 518. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,33 (d, J=7,3 Гц, 1Н), 8,23 (s, 1Н), 8,02 (s, 1Н), 7,78 (s, 1Н), 7,31-7,17 (m, 8Н), 5,29-5,21 (m, 1Н), 3,92 (s, 3Н), 3,13 (dd, J=3,6, 14,0 Гц, 1Н), 2,82 (dd, J=9,8, 13,8 Гц, 1Н). 19F ЯМР (376 МГц, ДМСО-d6) δ -119,41 (tdd, J=4,8, 8,7, 17,7 Гц, 1F), -119,69 -120,11 (m, 1F). МС (ИЭР) m/z (М+Н)+ 413,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(1H-ИНДАЗОЛ-1-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (530)
[0978] Соединение 530 (85 мг, выход: 39,2%, светло-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата и 1H-индазола, и алкилировали с использованием K3РО4, CuI, и (1R,2R)-N1,N2-диметилциклогексан-1,2-диамина, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 530. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,41 (d, J=5,8 Гц, 1Н), 8,28 (d, J=2,3 Гц, 2Н), 8,00 (d, J=8,5 Гц, 1Н), 7,91 (d, J=8,0 Гц, 1Н), 7,79 (ушир. s, 1Н), 7,68-7,48 (m, 2Н), 7,33 (t, J=7,4 Гц, 1Н), 7,19-7,04 (m, 5Н), 5,47 (dt, J=4,8, 7,7 Гц, 1Н), 3,95 (s, 3Н), 3,23 (dd, J=4,6, 14,2 Гц, 1Н), 2,94 (dd, J=8,4, 14,2 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 417,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2,2-ДИФТОРБЕНЗО[d][1,3]ДИОКСОЛ-5-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (548)
[0979] Соединение 548 (130 мг, выход: 53,1%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и (2,2-дифторбензо[d][1,3]диоксол-5-ил)бороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 548. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,48 (d, J=7,5 Гц, 1Н), 8,14-8,04 (m, 2Н), 7,82 (s, 1Н), 7,57 (d, J=1,5 Гц, 1Н), 7,46 (dd, J=1,8, 8,4 Гц, 1Н), 7,36-7,17 (m, 5Н), 5,38-5,19 (m, 1Н), 3,90 (s, 3Н), 3,17 (dd, J=4,0, 13,9 Гц, 1Н), 2,83 (dd, J=9,9, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 457,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(БЕНЗО[d][1,3]ДИОКСОЛ-5-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (567)
[0980] Соединение 567 (125 мг, выход: 72,7%, желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и бензо[d][1,3]диоксол-5-илбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 567. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,29 (d, J=7,5 Гц, 1Н), 8,08-7,99 (m, 2Н), 7,80 (s, 1Н), 7,32-7,19 (m, 5Н), 7,16-7,08 (m, 2Н), 6,83 (d, J=8,2 Гц, 1Н), 6,01 (s, 2Н), 5,33-5,24 (m, 1Н), 3,86 (s, 3Н), 3,16 (dd, J=4,1, 13,8 Гц, 1Н), 2,82 (dd, J=9,9, 14,1 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 421,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1,2'-ДИМЕТИЛ-1H,2'H-[3,3'-БИПИРАЗОЛ]-4-КАРБОКСАМИД (568)
[0981] Соединение 568 (24 мг, выход: 12,0%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и (1-метил-1H-пиразол-5-ил)бороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 568. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,28-8,10 (m, 1Н), 7,99-7,69 (m, 2Н), 7,59 (ушир. s, 1Н), 7,40-7,10 (m, 6Н), 6,42-6,29 (m, 1Н), 5,31 (ушир. s, 1Н), 3,98-3,85 (m, 3Н), 3,77-3,57 (m, 3Н), 3,19 (ушир. d, J=13,6 Гц, 1Н), 2,92-2,81 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 381,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(НАФТАЛИН-1-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (569)
[0982] Соединение 569 (125 мг, выход: 72,0%, грязно-белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и нафталин-1-илбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 569. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,32 (s, 1Н), 8.02 (s, 1Н) 8,03-7,89 (m, 3Н), 7,82 (d, J=7,3 Гц, 1Н), 7,76 (s, 1Н), 7,68 (d, J=8,4 Гц, 1Н), 7,53-7,44 (m, 2Н), 7,40 (dt, J=1,1, 7,6 Гц, 1Н), 7,35 (dd, J=1,1, 7,1 Гц, 1Н), 7,28-7,15 (m, 3Н), 7,08-7,00 (m, 2Н), 5,15 (ddd, J=4,0, 7,4, 9,4 Гц, 1Н), 3,96 (s, 3Н), 3,04 (dd, J=3,5, 13,9 Гц, 1Н), 2,67 (dd, J=9,7, 13,7 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 427,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2,3-ДИГИДРОБЕНЗО[b][1,4]ДИОКСИН-5-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (570)
[0983] Соединение 570 (41,8 мг, выход: 45,3%, грязно-белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и (2,3-дигидробензо[b][1,4]диоксин-5-ил)бороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 570. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,09-7,93 (m, 2Н), 7,83-7,69 (m, 2Н), 7,31-7,10 (m, 5Н), 6,88-6,73 (m, 3Н), 5,34-5,22 (m, 1Н), 4,15-4,06 (m, 2Н), 4,02-3,93 (m, 1Н), 3,91-3,82 (m, 4Н), 3,11 (dd, J=4,0, 13,9 Гц, 1Н), 2,79 (dd, J=9,3, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 435,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(НАФТАЛИН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (571)
[0984] Соединение 571 (41,8 мг, выход: 45,3%, грязно-белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и нафталин-2-илбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 571. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,45 (d, J=7,3 Гц, 1Н), 8,18-8,06 (m, 3Н), 7,92-7,81 (m, 4Н), 7,72 (dd, J=1,4, 8,5 Гц, 1Н), 7,55-7,45 (m, 2Н), 7,33-7,19 (m, 5Н), 5,36-5,27 (m, 1Н), 3,94 (s, 3Н), 3,17 (dd, J=3,9, 14,0 Гц, 1Н), 2,84 (dd, J=10,0, 13,8 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 427,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-(ДИФТОРМЕТИЛ)ФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (572)
[0985] Соединение 572 (110 мг, выход: 40,9%, светло-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и 1-бром-2-(дифторметил)бензола и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана), в присутствии палладиевого катализатора, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 572. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,31-8,21 (m, 2Н), 8,03 (s, 1Н), 7,79 (s, 1Н), 7,64 (d, J=7,5 Гц, 1Н), 7,55-7,42 (m, 2Н), 7,33-7,17 (m, 6Н), 7,00-6,66 (m, 1Н), 5,30-5,18 (m, 1Н), 3,92 (s, 3Н), 3,13 (dd, J=3,6, 13,7 Гц, 1Н), 2,79 (dd, J=10,0, 13,8 Гц, 1Н). 19F ЯМР (376 МГц, ДМСО-d6) δ -107,64 -110,93 (m, 2F). МС (ИЭР) m/z (М+Н)+ 396.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ЦИКЛОГЕКСИЛ-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (573)
[0986] Соединение 573 (172 мг, выход: 81,1%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и 2-(циклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана с последующим гидрогенолизом полученного продукта, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 573. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,11 (d, J=7,3 Гц, 1Н), 8,02 (s, 2Н), 7,76 (s, 1Н), 7,28-7,21 (m, 4Н), 7,20-7,11 (m, 1Н), 5,24 (ddd, J=3,6, 7,1, 10,3 Гц, 1Н), 3,74 (s, 3Н), 3,11 (dd, J=3,7, 13,9 Гц, 1Н), 3,05-2,96 (m, 1Н), 2,79 (dd, J=10,0, 13,8 Гц, 1Н), 1,76-1,55 (m, 5Н), 1,39-1,05 (m, 5Н). МС (ИЭР) m/z (М+Н)+ 383,3.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ИЗОХИНОЛИН-6-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (585)
[0987] Соединение 585 (120 мг, выход: 60,0%, светло-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и изохинолин-7-илбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 585. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,28 (s, 1Н), 8,56 (d, J=7,3 Гц, 1Н), 8,48 (d, J=5,8 Гц, 1Н), 8,20 (s, 1Н), 8,16-8,08 (m, 2Н), 8,04 (d, J=8,8 Гц, 1Н), 7,90-7,77 (m, 3Н), 7,29 (d, J=4,3 Гц, 4Н), 7,25-7,18 (m, 1Н), 5,38-5,25 (m, 1Н), 3,95 (s, 3Н), 3,18 (dd, J=3,8, 14,1 Гц, 1Н), 2,85 (dd, J=10,0, 13,8 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 428,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ХИНОЛИН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (587)
[0988] Соединение 587 (70 мг, выход: 49,7%, светло-розовое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и 2-бромхинолина и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана), в присутствии палладиевого катализатора, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 587. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,08 (ушир. d, J=8,0 Гц, 1Н), 8,50 (ушир. d, J=8,8 Гц, 1Н), 8,36 (s, 1Н), 8,26 (ушир. d, J=8,8 Гц, 1Н), 8,14 (ушир. s, 1Н), 8,02 (ушир. d, J=7,5 Гц, 1Н), 7,84 (ушир. s, 1Н), 7,74-7,59 (m, 3Н), 7,07-6,90 (m, 5Н), 5,75-5,66 (m, 1Н), 3,98 (s, 3Н), 3,31-3,26 (m, 1Н), 3,18-3,08 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 428,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ИЗОХИНОЛИН-4-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (591)
[0989] Соединение 591 (35 мг, выход: 56,3%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и 4-изохинолилбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 591. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,26 (s, 1Н), 8,36-8,30 (m, 2Н), 8,25-8,21 (m, 1Н), 8,14-8,09 (m, 1Н), 7,93 (s, 1Н), 7,71 (ушир. s, 1Н), 7,68-7,60 (m, 3Н), 7,27-7,20 (m, 2Н), 7,19-7,13 (m, 3Н), 5,17-5,09 (m, 1Н), 3,95 (s, 3Н), 3,09-3,02 (m, 1Н), 2,77-2,69 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 428,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ХИНОЛИН-6-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (593)
[0990] Соединение 593 (30 мг, выход 29,9%; бледно-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и хинолин-6-илбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 593. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,86 (ушир. s, 1Н), 8,50 (ушир. d, J=6,6 Гц, 1Н), 8,31 (ушир. d, J=7,9 Гц, 1Н), 8,20 (ушир. s, 1Н), 8,09 (ушир. d, J=9,5 Гц, 2Н), 7,93 (ушир. s, 2Н), 7,81 (ушир. s, 1Н), 7,53-7,47 (m, 1Н), 7,30-7,15 (m, 5H), 5,29 (ушир. s, 1Н), 3,92 (s, 3Н), 3,16 (ушир. d, J=11,5 Гц, 1Н), 2,88-2,78 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 428,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ХИНОЛИН-5-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (597)
[0991] Соединение 597 (20 мг, выход: 25,1%, желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и хинолин-5-илбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 597. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,88 (dd, J=1,8, 4,0 Гц, 1Н), 8,34-8,19 (m, 1Н), 8,17-7,97 (m, 2Н), 7,81-7,35 (m, 6Н), 7,27-7,03 (m, 5Н), 5,29-5,11 (m, 1Н), 4,02-3,90 (m, 3Н), 3,07-3,0 (m, 1Н), 2,80-2,74 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 428,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ИЗОХИНОЛИН-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (602)
[0992] Соединение 602 (55 мг, выход: 36,3%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и 3-бромизохинолина и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана), в присутствии палладиевого катализатора, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 602. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,07 (d, J=7,1 Гц, 1Н), 8,98 (s, 1Н), 8,51 (s, 1Н), 8,28 (s, 1Н), 8,10 (d, J=7,5 Гц, 3Н), 7,89-7,79 (m, 2Н), 7,75-7,67 (m, 1Н), 7,15-7,06 (m, 4Н), 7,04-6,97 (m, 1Н), 5,64-5,53 (m, 1Н), 4,03-3,84 (m, 3Н), 3,27 (dd, J=5,0, 14,0 Гц, 1Н), 3,10 (dd, J=7,7, 14,1 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 428,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ХЛОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (604)
[0993] Соединение 604 (120 мг, выход: 58,6%, бледно-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и (2-хлорфенил)бороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 604. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,19 (s, 1Н), 8,03 (d, J=7,3 Гц, 1Н), 7,97 (s, 1Н), 7,73 (s, 1Н), 7,39-7,15 (m, 9Н), 5,24-5,19 (m, 1Н), 3,87 (s, 3Н), 3,08 (dd, J=3,6, 14,0 Гц, 1Н), 2,76 (dd, J=9,8, 14,0 Гц, 1H). МС (ИЭР) m/z (M+H)+ 411,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ХИНОЛИН-3-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (605)
[0994] Соединение 605 (140 мг, выход: 55,6%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и хинолин-3-илбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 605. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,08 (s, 1Н), 8,65-8,45 (m, 2Н), 8,23 (s, 1Н), 8,08 (ушир. s, 1Н),7,99 (dd, J=8,3, 12,7 Гц, 2Н), 7,85-7,70 (m, 2Н), 7,60 (t, J=7,4 Гц, 1Н), 7,34-7,13 (m, 5Н), 5,36-5,20 (m, 1Н), 4,05-3,89 (m, 3Н), 3,18-2,78 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 428,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ИЗОХИНОЛИН-5-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (607)
[0995] Соединение 607 (50 мг, выход: 34,6%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и изохинолин-5-илбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 607. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,41-9,21 (m, 1Н), 8,52-8,27 (m, 2Н), 8,23-7,93 (m, 3Н), 7,81-7,48 (m, 4Н), 7,32-7,09 (m, 5Н), 5,22-5,10 (m, 1Н), 4,06-3,90 (m, 3Н), 3,09 (dd, J=3,2, 13,8 Гц, 1Н), 2,76 (dd, J=9,8, 13,8 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 428,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ХИНОЛИН-4-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (611)
[0996] Соединение 611 (55 мг, выход: 78,2%, светло-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и хинолин-4-илбороновой кислоты, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 611. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,86-8,76 (m, 1Н), 8,41-8,34 (m, 1Н), 8,07-7,88 (m, 2Н), 7,85-7,58 (m, 3Н), 7,49 (t, J=7,8 Гц, 1Н), 7,33-7,11 (m, 7H), 5,21-5,11 (m, 1Н), 4,30 (ушир. s, 1Н), 4,04-3,93 (m, 3Н), 3,10 (dd, J=3,5, 14,1 Гц, 1Н), 2,78 (dd, J=10,1, 13,9 Гц, 1Н). МС (ИЭР) (М+Н)+ 428,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ТИОФЕН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (615)
[0997] Соединение 615 (122 мг, выход: 73,0%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 1-метил-3-(тиофен-2-ил)-1H-пиразол-4-карбоновой кислоты и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 615. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,42 (d, J=7,3 Гц, 1Н), 8,10 (s, 1Н), 8,06 (s, 1Н), 7,79 (s, 1Н), 7,73 (d, J=3,5 Гц, 1Н), 7,41 (dd, J=1,1, 5,1 Гц, 1Н), 7,28 (d, J=4,2 Гц, 4Н), 7,24-7,18 (m, 1Н), 6,97 (dd, J=3,6, 5,0 Гц, 1Н), 5,49-5,20 (m, 1Н), 3,85 (s, 3Н), 3,16 (dd, J=3,5, 13,9 Гц, 1Н), 2,83 (dd, J=10,1, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 383,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ЦИКЛОПРОПИЛ-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (616)
[0998] Соединение 616 (120 мг, выход: 71,5%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 3-циклопропил-1-метил-1H-пиразол-4-карбоновой кислоты и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 616. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,08 (d, J=7,5 Гц, 1Н), 8,04 (s, 1Н), 8,03 (ушир. s, 1Н), 7,77 (s, 1Н), 7,27 (d, J=4,4 Гц, 4Н), 7,20-7,16 (m, 1Н), 5,31-5,26 (m, 1Н), 3,71 (s, 3Н), 3,15 (dd, J=3,9, 13,8 Гц, 1Н), 2,84 (dd, J=9,9, 13,9 Гц, 1Н), 2,41-2,35 (m, 1Н), 0,77-0,70 (m, 2Н), 0,69-0,65 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 341,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ТЕТРАГИДРО-2H-ПИРАН-4-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (617)
[0999] Соединение 617 (55 мг, выход: 34,4%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 1-метил-3-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоновой кислоты и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 617. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,20 (d, J=7,5 Гц, 1Н), 8,07 (s, 1Н), 8,01 (s, 1Н), 7,75 (s, 1Н), 7,30-7,21 (m, 4Н), 7,19-7,14 (m, 1Н), 5,31-5,15 (m, 1Н), 3,80 (d, J=9,5 Гц, 2H), 3,76 (s, 3Н), 3,29-3,20 (m, 3Н), 3,11 (dd, J=3,5, 13,9 Гц, 1Н), 2,79 (dd, J=10,0, 13,8 Гц, 1Н), 1,65-1,52 (m, 4H). МС (ИЭР) m/z (М+Н)+ 385,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ЦИКЛОПЕНТИЛ-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (620)
[1000] Соединение 620 (70 мг, выход: 53,1%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 3-циклопентил-1-метил-1H-пиразол-4-карбоновой кислоты и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 620. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,14 (ушир. d, J=7,3 Гц, 1Н), 8,06-8,02 (m, 2Н), 7,77 (ушир. s, 1Н), 7,29 (d, J=4,3 Гц, 4Н), 7,24-7,17 (m, 1Н), 5,29-5,21 (m, 1Н), 3,77 (s, 3Н), 3,42 (ушир. t, J=8,0 Гц, 1Н), 3,14 (ушир. dd, J=3,5, 13,8 Гц, 1Н), 2,83 (ушир. dd, J=10,2, 13,4 Гц, 1Н), 1,80 (ушир. d, J=7,8 Гц, 2Н), 1,69-1,45 (m, 6Н). МС (ИЭР) m/z (М+Н)+ 369,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(5-ХЛОРТИОФЕН-2-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (621)
[1001] Соединение 621 (25 мг, выход: 25,1%, бледно-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 3-(5-хлортиофен-2-ил)-1-метил-1H-пиразол-4-карбоновой кислоты и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 621. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,53 (ушир. d, J=6,8 Гц, 1Н), 8,22-8,04 (m, 2Н), 7,87-7,61 (m, 2Н), 7,35-6,97 (m, 6Н), 5,33 (ушир. s, 1Н), 3,88 (ушир. s, 3Н), 3,19 (ушир. d, J=14,1 Гц, 1Н), 2,92-2,79 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 417,0.
ТРЕТ-БУТИЛ 3-(4-((4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)КАРБАМОИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-3-ИЛ)ПИПЕРИДИН-1-КАРБОКСИЛАТ (624)
[1002] Соединение 624 (100 мг, выход: 33,04%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 3-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-1-метил-1H-пиразол-4-карбоновой кислоты и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 624. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,27-8,18 (m, 1Н), 8,12 (s, 1Н), 8,02 (d, J=2,9 Гц, 1Н), 7,78 (s, 1Н), 7,32-7,13 (m, 5H), 5,29 (s, 1Н), 4,02-3,85 (m, 2H), 3,82-3,75 (m, 3Н), 3,20-3,09 (m, 2H), 3,19-2,99 (m, 1Н), 2,99-2,78 (m, 2H), 2,73-2,64 (m, 1Н), 1,84 (s, 1Н), 1,62 (s, 1Н), 1,48 (d, J=11,9 Гц, 1Н), 1,39-1,27 (m, 10Н). МС (ИЭР) m/z (М+Н)+ 484,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3,4-ДИГИДРО-2H-БЕНЗО[b][1,4]ДИОКСЕПИН-7-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (625)
[1003] Соединение 625 (70 мг, выход: 58,6%, желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 3-(3,4-дигидро-2H-бензо[b][1,4]диоксепин-7-ил)-1-метил-1H-пиразол-4-карбоновой кислоты, и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 625. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,29 (d, J=7,3 Гц, 1Н), 8,09-7,94 (m, 2Н), 7,80 (s, 1Н), 7,32-7,14 (m, 7Н), 6,90-6,83 (m, 1Н), 5,39-5,22 (m, 1Н), 4,16-4,07 (m, 4Н), 3,88-3,82 (m, 3Н), 3,16 (dd, J=4,0, 13,7 Гц, 1Н), 2,82 (dd, J=9,8, 13,8 Гц, 1Н), 2,13-2,06 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 449,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1,1'-ДИМЕТИЛ-1H,1'Н-[3,4'-БИПИРАЗОЛ]-4-КАРБОКСАМИД (626)
[1004] Соединение 626 (55 мг, выход: 34,4%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 1,1'-диметил-1H,1'H-[3,4'-бипиразол]-4-карбоновой кислоты и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 626. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,06 (d, J=17,6 Гц, 2Н), 7,94 (s, 1Н), 7,76-7,45 (m, 3Н), 7,31-7,23 (m, 4Н), 7,23-7,15 (m, 1Н), 5,37-5,28 (m, 1Н), 3,83 (d, J=8,3 Гц, 6Н), 3,22 (dd, J=4,1, 13,7 Гц, 1Н), 2,95 (dd, J=8,9, 14,4 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 381,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ТЕТРАГИДРОФУРАН-3-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (627)
[1005] Соединение 627 (40 мг, выход: 25,1%, бледно-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 1-метил-3-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоновой кислоты и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 627. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,06 (s, 1Н), 8,00-7,87 (m, 1Н), 7,69 (s, 0,5Н), 7,57-7,44 (m, 0,7Н), 7,30-7,03 (m, 6Н), 5,33-5,23 (m, 1Н), 3,92 (dt, J=2,6, 7,7 Гц, 1Н), 3,86-3,81 (m, 1Н), 3,79 (s, 3Н), 3,77-3,73 (m, 1Н), 3,71 (d, J=7,8 Гц, 1Н), 3,62-3,54 (m, 1Н), 3,20 (dd, J=4,3, 14,1 Гц, 1Н), 2,96-2,88 (m, 1Н), 2,09 (q, J=7,3 Гц, 2Н). МС (ИЭР) m/z (М+Н)+ 371,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(БЕНЗОФУРАН-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (628)
[1006] Соединение 628 (143 мг, выход: 98,6%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения 3-(бензофуран-3-ил)-1-метил-1H-пиразол-4-карбоновой кислоты и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 628. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,56 (d, J=7,3 Гц, 1Н), 8,24 (s, 1Н), 8,10 (s, 1Н), 7,82 (s, 1Н), 7,63 (d, J=6,8 Гц, 1Н), 7,56-7,50 (m, 2Н), 7,33-7,15 (m, 7Н), 5,36 (ddd, J=3,9, 7,3, 9,8 Гц, 1Н), 3,94 (s, 3Н), 3,24-3,14 (m, 1Н), 2,87 (dd, J=10,0, 14,0 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 417,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ИЗОХИНОЛИН-3-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (629)
[1007] Соединение 629 (25 мг, выход: 50,14%, бледно-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (198А) и 7-бромизохинолина и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана), в присутствии палладиевого катализатора, затем с полученным промежуточным соединением выполняли такие же процедуры, как в случае соединения 12, с получением соединения 629. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,27 (s, 1Н), 8,54 (d, J=7,5 Гц, 1Н), 8,49 (d, J=5,7 Гц, 1Н), 8,35 (s, 1Н), 8,15 (s, 1Н), 8,11 (s, 1Н), 8,00-7,94 (m, 1Н), 7,92-7,87 (m, 1Н), 7,85-7,78 (m, 2Н), 7,30-7,17 (m, 5Н), 5,36-5,26 (m, 1Н), 3,95 (s, 3Н), 3,18 (dd, J=4,0, 13,9 Гц, 1Н), 2,85 (dd, J=9,9, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 428,2.
БЕНЗИЛ-4-(4-((4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)КАРБАМОИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-3-ИЛ)ПИПЕРАЗИН-1-КАРБОКСИЛАТ (601)
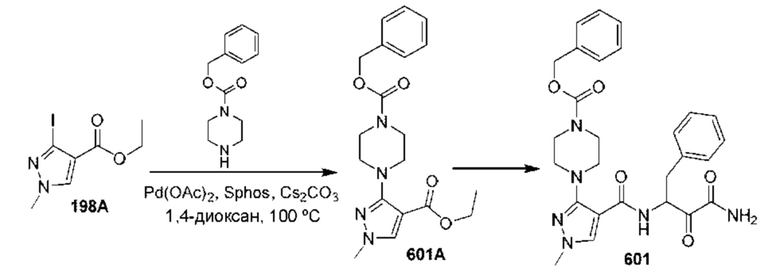
[1008] К раствору соединения 198А (500 мг, 1,79 ммоль) и бензилпиперазин-1-карбоксилата (590 мг, 2,68 ммоль) в 1,4-диоксане (20 мл) добавляли Pd(OAc)2 (40 мг, 0,18 ммоль), Cs2СО3 (116 мг, 3,57 ммоль) и Sphos (147 мг, 0,36 ммоль) в атмосфере N2. Реакционную смесь перемешивали при 100°С в течение 16 ч. Реакционную смесь фильтровали и промывали EtOAc (10 мл). Органическую фазу концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью системы для хроматографии CombiFlash (элюент: ПЭ~10% ~30% EtOAc/ПЭ) с получением соединения 601А (183 мг, выход 25,7%) в виде желтого масла.
[1009] Соединение 601 (55 мг, выход: 78,8%, розовое твердое вещество) получали с использованием соответствующих исходных веществ, соединения бензил-4-(4-(этоксикарбонил)-1-метил-1H-пиразол-3-ил)пиперазин-1-карбоксилата (601А) и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 601. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,28-8,15 (m, 2Н), 8,03 (s, 1Н), 7,92 (s, 1Н), 7,43-7,33 (m, 5Н), 7,28-7,21 (m, 2Н), 7,19-7,13 (m, 1Н), 7,06 (d, J=7,0 Гц, 2Н), 5,53 (q, J=6,3 Гц, 1Н), 5,13-5,04 (m, 2Н), 3,79-3,68 (m, 3Н), 3,33-3,18 (m, 5Н), 3,17-3,08 (m, 1Н), 2,86 (ушир. s, 2Н), 2,81-2,75 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 519,2.
ТРЕТ-БУТИЛ-4-(4-((4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)КАРБАМОИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-3-ИЛ)ПИПЕРАЗИН-1-КАРБОКСИЛАТ (603)
[1010] Промежуточное соединение 603А получали с помощью той же методики, как в случае промежуточного соединения 601А, с использованием трет-бутилпиперазин-1-карбоксилата.
[1011] Соединение 603 (18 мг, выход: 41,6%, белое твердое вещество) получали с использованием соответствующих исходных веществ, соединения трет-бутил-4-(4-(этоксикарбонил)-1-метил-1H-пиразол-3-ил)пиперазин-1-карбоксилата (603А) и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 603. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,07 (d, J=7,5 Гц, 1Н), 7,98-7,81 (m, 2Н), 7,80-7,61 (m, 1Н), 7,30-7,24 (m, 2Н), 7,22-7,19 (m, 1Н), 7,13 (d, J=7,5 Гц, 2Н), 5,56-5,45 (m, 1Н), 3,76-3,67 (m, 3Н), 3,42 (ушир. s, 1Н), 3,35-3,28 (m, 2Н), 3,27-3,21 (m, 2Н), 3,15-3,13 (m, 1Н), 2,88 (d, J=6,3 Гц, 2Н), 2,84-2,77 (m, 2Н), 1,44 (s, 9Н). МС (ИЭР) m/z (М+Н)+ 485,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(1,1-ДИОКСИДОТИОМОРФОЛИНО)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (613)
[1012] Промежуточное соединение 613А получали с помощью той же методики, как в случае промежуточного соединения 601А, с использованием тиоморфолин-1,1-диоксида.
[1013] Соединение 613 (70 мг, выход: 33,7%, светло-желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-3-(1,1-диоксидотиоморфолино)-1-метил-1H-пиразол-4-карбоксилата (613А) и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 613. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,15 (d, J=7,0 Гц, 2Н), 8,05 (s, 1Н), 7,87 (s, 1Н), 7,31-7,26 (m, 2Н), 7,23 (ушир. d, J=7,0 Гц, 1Н), 7,16 (d, J=6,8 Гц, 2Н), 5,48-5,31 (m, 1Н), 3,75 (s, 3Н), 3,42 (m, 4Н), 3,25-3,20 (m, 1Н), 3,14-3,07 (m, 2Н), 3,06-2,98 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 434,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-МОРФОЛИНО-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (614)
[1014] Промежуточное соединение 614А получали с помощью той же методики, как в случае промежуточного соединения 601А, с использованием морфолина.
[1015] Соединение 614 (45 мг, выход: 86,3%, желтое твердое вещество) получали с использованием соответствующих исходных веществ, соединения этил-1-метил-3-морфолино-1H-пиразол-4-карбоксилата (614А) и гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида, с помощью таких же методик, как в случае соединения 12, с получением соединения 614. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,21-8,11 (m, 2Н), 7,98 (s, 1Н), 7,91-7,84 (m, 1Н), 7,26-7,21 (m, 2Н), 7,18 (s, 1Н), 7,07 (d, J=6,8 Гц, 2Н), 5,52-5,44 (m, 1Н), 3,71 (s, 3Н), 3,55-3,47 (m, 2Н), 3,46-3,39 (m, 2Н), 3,23 (dd, J=5,3, 14,1 Гц, 1Н), 3,06 (dd, J=7,1, 14,1 Гц, 1Н), 2,89-2,82 (m, 2Н), 2,80-2,75 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 386,2.
ПРИМЕР 97
СОЕДИНЕНИЯ 165-167, 170-173,176-190, 315, 407, 408, 446, 447 (S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-(ПИРИДИН-4-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (165)
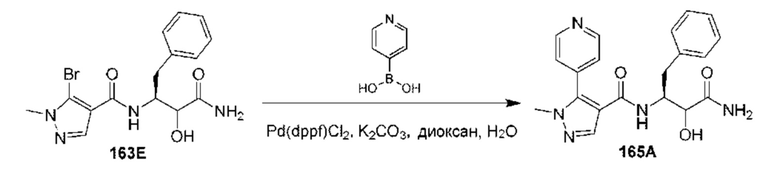
[1016] К смеси соединения 163Е (200 мг, 525 мкмоль), 4-пиридилбороновой кислоты (129 мг, 1,05 ммоль) и K2CO3 (218 мг, 1,57 ммоль) в диоксане (9 мл) и Н2О (1 мл) добавляли Pd(dppf)Cl2 (76,8 мг, 105 мкмоль) в атмосфере N2. Смесь перемешивали при 130°С в условиях микроволнового излучения в течение 2 ч. Растворитель удаляли под вакуумом. Остаток очищали посредством препаративной ТСХ (дихлорметан : метанол = 10:1, Rf=0,5) с получением 165А (90 мг, выход: 45,2%) в виде белого твердого вещества. Соединение 165А: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,55-8,70 (m, 2Н), 7,96 (ушир. dd, J=15,55, 5,40 Гц, 1Н), 7,79 (ушир. d, J=7,94 Гц, 1Н), 7,03-7,44 (m, 10Н), 5,74-5,87 (m, 1Н), 4,25-4,45 (m, 1Н), 3,75-4,02 (m, 1Н), 3,68 (ушир. t, J=5,40 Гц, 3Н), 2,57-2,91 (m, 3Н).
[1017] Соединение 165 было получено, как описано в примере 61, из соответствующего промежуточного соединения 165А. Соединение 165: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,60-8,65 (m, 2Н), 8,40 (d, J=7,50 Гц, 1Н), 8,00 (s, 2Н), 7,77 (s, 1Н), 7,32-7,36 (m, 2Н), 7,18-7,31 (m, 5Н), 5,14-5,25 (m, 1Н), 3,68 (s, 3Н), 3,14 (dd, J=13,89, 3,75 Гц, 1Н), 2,83 (dd, J=13,67, 10,14 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 378,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(n-ТОЛИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (166)
[1018] Соединение 166 (17,5 мг, выход: 27,9%, грязно-белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163G и n-толилбороновой кислоты. Соединение 166: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,28 (d, J=7,3 Гц, 1Н), 8,09-7,99 (m, 2Н), 7,81 (ушир. s, 1Н), 7,46 (d, J=8,2 Гц, 2Н), 7,33-7,19 (m, 5Н), 7,11 (d, J=7,9 Гц, 2Н), 5,33-5,23 (m, 1Н), 3,93-3,81 (m, 3Н), 3,15 (dd, J=3,6, 13,8 Гц, 1Н), 2,82 (dd, J=10,0, 13,8 Гц, 1Н), 2,30 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 391,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-(n-ТОЛИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (167)
[1019] Соединение 167 (40,0 мг, выход: 66,9%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163Е и n-толилбороновой кислоты. Соединение 167: 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,98-7,91 (m, 2Н), 7,89 (s, 1Н), 7,73 (s, 1Н), 7,30-7,08 (m, 9Н), 5,24-5,11 (m, 1Н), 3,58 (s, 3Н), 3,08 (dd, J=3,6, 14,0 Гц, 1Н), 2,77 (dd, J=9,8, 13,6 Гц, 1Н), 2,32 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 391,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ПИРИДИН-4-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (170)
[1020] Соединение 170 (15,6 мг, выход: 17,3%, грязно-белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163G и 4-пиридилбороновой кислоты. Соединение 170: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,61 (ушир. d, J=7,50 Гц, 1Н), 8,49 (d, J=4,85 Гц, 2Н), 8,03-8,15 (m, 2Н), 7,82 (ушир. s, 1Н), 7,56 (d, J=4,85 Гц, 2Н), 7,19-7,34 (m, 5Н), 5,22-5,36 (m, 1Н), 3,87-3,96 (m, 3Н), 3,18 (ушир. dd, J=14,00, 3,42 Гц, 1Н), 3,12-3,22 (m, 1Н), 2,83 (ушир. dd, J=13,56, 10,25 Гц, 1Н). МС (ИЭР) m/z (М+Н2O+Н)+ 396,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(о-ТОЛИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (171)
[1021] Соединение 171 (16 мг, 41 мкмоль, выход: 14,6%, чистота: 100,0%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163G и о-толилбороновой кислоты. Соединение 171: 1H ЯМР (400 МГц, CDCl3) δ 7,95 (s, 1Н), 7,38-7,33 (m, 1Н), 7,30-7,26 (m, 1Н), 7,25-7,21 (m, 2Н), 7,19-7,12 (m, 3Н), 6,75-6,71 (m, 2Н), 6,68 (ушир. s, 1Н), 5,81 (ушир. d, J=5,7 Гц, 1Н), 5,57 (ушир. s, 1Н), 5,47-5,39 (m, 1Н), 3,94-3,87 (m, 3Н), 3,15 (dd, J=4,4, 14,1 Гц, 1Н), 2,62 (dd, J=8,5, 14,2 Гц, 1Н), 2,11 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 391,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-(м-ТОЛИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (172)
[1022] Соединение 172 (25,3 мг, выход: 21,2%, желтое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163Е и м-толилбороновой кислоты. Соединение 172: 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,98 (ушир. s, 1Н), 7,91 (s, 1Н), 7,88 (ушир. d, J=7,3 Гц, 1Н), 7,75 (ушир. s, 1Н), 7,33-7,21 (m, 4Н), 7,20-7,10 (m, 4Н), 7,07 (ушир. d, J=7,3 Гц, 1Н), 5,19 (ушир. s, 1Н), 3,58 (s, 3Н), 3,09 (ушир. dd, J=3,3, 13,7 Гц, 1Н), 2,77 (ушир. dd, J=9,7, 13,7 Гц, 1Н), 2,29 (s, 3Н). МС (ИЭР) m/z (М+H)+ 391,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ПИРИМИДИН-5-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (173)
[1023] Соединение 173 (6,9 мг, выход: 18,6%, светло-желтое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163G и пиримидин-5-илбороновой кислоты. Соединение 173: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,15-9,06 (m, 1Н), 8,97-8,77 (m, 1Н), 8,62 (d, J=7,5 Гц, 1Н), 8,34-8,22 (m, 1Н), 8,09 (s, 1Н), 7,86-7,62 (m, 1Н), 7,40-7,07 (m, 6Н), 5,36-5,25 (m, 1Н), 3,98-3,90 (m, 3Н), 3,17 (dd, J=4,0, 13,7 Гц, 1Н), 2,83 (ушир. dd, J=10,3, 13,8 Гц, 1Н). МС (ИЭР) (М+Н)+ 379,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-(ПИРИМИДИН-5-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (176)
[1024] Соединение 176 (15 мг, выход: 12.19%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163Е и пиримидин-5-илбороновой кислоты. 1Н ЯМР (400 МГц, CD3CN) δ 9,24-9,17 (m, 1Н), 8,77-8,68 (m, 1Н), 7,87 (s, 1Н), 7,61 (s, 1Н), 7,34-7,18 (m, 5Н), 6,97 (ушир. s, 2Н), 6,24-6,09 (m, 1Н), 5,38-5,33 (m, 1Н), 3,73 (s, 3Н), 3,25 (dd, J=5,0, 14,0 Гц, 1Н), 2,92 (dd, J=9,0, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 379,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-МЕТОКСИФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (177)
[1025] Соединение 177 (46 мг, выход: 74,2%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163G и (3-метоксифенил)бороновой кислоты. Соединение 177: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,02 (s, 1Н), 7,85 (ушир. d, J=7,3 Гц, 1Н), 7,78-7,65 (m, 1Н), 7,54 (ушир. s, 1Н), 7,30-7,25 (m, 3Н), 7,24-7,18 (m, 5Н), 6,92-6,87 (m, 1Н), 5,38-5,31 (m, 1Н), 3,89 (s, 3Н), 3,78-3,74 (m, 3Н), 3,20 (dd, J=4,5, 14,1 Гц, 1Н), 2,90 (dd, J=9,0, 14,1 Гц, 1Н).
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ФТОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (178)
[1026] Соединение 178 (26 мг, выход: 49,4% белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,46 (ушир. d, J=7,3 Гц, 1Н), 8,07 (s, 2Н), 7,80 (ушир. s, 1Н), 7,42 (d, J=8,2 Гц, 2Н), 7,36-7,31 (m, 1Н), 7,30-7,25 (m, 4Н), 7,22-7,18 (m, 1Н), 7,11 (ушир. t, J=8,3 Гц, 1Н), 5,33-5,25 (m, 1Н), 3,88 (s, 3Н), 3,15 (ушир. dd, J=3,5, 13,7 Гц, 1Н), 2,80 (dd, J=10,1, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 395,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(4-ФТОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (179)
[1027] Соединение 179 (15 мг, выход: 36,7%, светло-желтое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,42 (d, J=7,5 Гц, 1Н), 8,07 (s, 2Н), 7,81 (s, 1Н), 7,61 (dd, J=5,7, 8,8 Гц, 2Н), 7,34-7,19 (m, 5Н), 7,12 (ушир. t, J=8,9 Гц, 2Н), 5,37-5,22 (m, 1Н), 3,89 (s, 3Н), 3,16 (ушир. dd, J=3,5, 14,1 Гц, 1Н), 2,82 (ушир. dd, J=10,1, 13,7 Гц, 1Н). МС (ИЭР) m/z (М+H)+ 395,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ИЗОПРОПИЛФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (180)
[1028] Соединение 180 (58 мг, выход: 60,95%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,29 (d, J=7,5 Гц, 1Н), 8,11-7,98 (m, 2Н), 7,81 (s, 1Н), 7,48 (s, 1Н), 7,37-7,16 (m, 9Н), 5,38-5,23 (m, 1Н), 3,89 (s, 3Н), 3,15 (dd, J=3,6, 14,0 Гц, 1Н), 2,91-2,77 (m, 3Н), 1,18 (d, J=6,8 Гц, 6Н). МС (ИЭР) m/z (М+Н)+ 419,2.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ФТОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (181)
[1029] Соединение 181 (46 мг, выход: 70,7%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,26-8,16 (m, 2Н), 8,01 (s, 1Н), 7,77 (s, 1Н), 7,43-7,07 (m, 9Н), 5,28-5,18 (m, 1Н), 3,96-3,85 (m, 3Н), 3,12 (dd, J=3,6, 14,0 Гц, 1Н), 2,81 (dd, J=9,7, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 395,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ЭТИЛФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (182)
[1030] Соединение 182 (24 мг, выход: 65,4%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,13 (s, 1Н), 7,94 (ушир. d, J=7,0 Гц, 1Н), 7,60 (s, 1Н), 7,52 (d, J=7,8 Гц, 1Н), 7,40-7,36 (m, 2Н), 7,35-7,26 (m, 6Н), 5,48-5,43 (m, 1Н), 4,01 (s, 3Н), 3,30 (dd, J=4,5, 14,1 Гц, 1Н), 3,00 (dd, J=9,0, 14,1 Гц, 1Н), 2,74 (q, J=7,5 Гц, 3Н), 1,32 (t, J=7,7 Гц, 4Н).
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(3-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (183)
[1031] Соединение 183 (38 мг, выход: 61,9%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,51 (d, J=7,5 Гц, 1Н), 8,12 (s, 1Н), 8,03 (s, 1Н), 7,97 (s, 1Н), 7,84 (ушир. d, J=7,9 Гц, 1Н), 7,80 (s, 1Н), 7,63 (ушир. d, J=7,7 Гц, 1Н), 7,55-7,48 (m, 1Н), 7,26 (d, J=4,2 Гц, 4Н), 7,21-7,16 (m, 1Н), 5,32-5,25 (m, 1Н), 3,89 (s, 3Н), 3,17-3,11 (m, 1Н), 2,83-2,76 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 445,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(3-(ТРИФТОРМЕТОКСИ)ФЕНИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (184)
[1032] Соединение 184 (18 мг, выход: 53,5%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,48 (d, J=7,7 Гц, 1Н), 8,10 (s, 1Н), 8,03 (s, 1Н), 7,79 (s, 1Н), 7,64-7,56 (m, 2Н), 7,41 (t, J=8,2 Гц, 1Н), 7,30-7,24 (m, 5Н), 7,12-7,16 (m, 1Н), 5,33-5,27 (m, 1Н), 3,89 (s, 3Н), 3,15 (dd, J=3,9, 13,8 Гц, 1Н), 2,81 (dd, J=10,1, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 461,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ЦИАНОФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (185)
[1033] Соединение 185 (20 мг, выход: 43,2%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,65 (dd, J=7,3 Гц, 1Н), 8,28 (s, 1Н), 8,07 (s, 1Н), 8,00 (s, 1Н), 7,89 (dd, J=7,5 Гц, 1Н), 7,82 (s, 1Н), 7,77 (dd, J=8,2 Гц, 1Н), 7,52 (t, J=7,9 Гц, 1Н), 7,31-7,19 (m, 5Н), 5,31 (dd, J=6,8 Гц, 1Н), 3,91 (s, 3Н), 3,16 (dd, J=9,9 Гц, 1Н), 2,91-2,81 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 402,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ЦИКЛОПРОПИЛФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (186)
[1034] Соединение 186 (30 мг, выход: 53,72%, желтое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,36 (dd, J=6,8 Гц, 1Н), 8,14-8,01 (m, 2Н), 7,82 (s, 1Н), 7,30-7,14 (m, 8Н), 6,99 (dd, J=7,5 Гц, 1Н), 5,28 (s, 1Н), 3,88 (s, 3Н), 3,14 (dd, J=11,0 Гц, 1Н), 2,87-2,80 (m, 1Н), 1,89 (s, 1Н), 0,92 (dd, J=6,6 Гц, 2Н), 0,62 (s, 2Н). МС (ИЭР) m/z (М+Н)+ 417,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ХЛОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (187)
[1035] Соединение 187 (35 мг, выход: 53,0%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,52-8,44 (m, 1Н), 8,11 (s, 1Н), 8,06 (s, 1Н), 7,81 (s, 1Н), 7,74-7,65 (m, 1Н), 7,60-7,50 (m, 1Н), 7,39-7,33 (m, 2Н), 7,30-7,28 (m, 3Н), 7,25-7,18 (m, 2Н), 5,38-5,25 (m, 1Н), 3,90 (s, 3Н), 3,17 (dd, J=3,6, 14,0 Гц, 1Н), 2,89-2,80 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 411,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ПИРИДИН-3-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (188)
[1036] Соединение 188 (15,8 мг, выход: 26,5%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163G и пиридин-3-илбороновой кислоты. Соединение 188: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,77 (d, J=1,6 Гц, 1Н), 8,53-8,47 (m, 2Н), 8,19 (s, 1Н), 8,06 (ушир. s, 1Н), 7,92 (td, J=1,9, 7,9 Гц, 1Н), 7,80 (ушир. s, 1Н), 7,34 (dd, J=5,1, 7,6 Гц, 1Н), 7,32-7,28 (m, 4Н), 7,26-7,18 (m, 1Н), 5,35-5,26 (m, 1Н), 3,93 (s, 3Н), 3,18 (dd, J=3,8, 13,8 Гц, 1Н), 2,84 (dd, J=10,1, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 378,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ)-5-(ПИРИДИН-3-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (189)
[1037] Соединение 189 (16 мг, 42,4 мкмоль, выход: 30,9%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163Е и пиридин-3-илбороновой кислоты. Соединение 189: 1Н ЯМР (400 МГц, CDCl3) δ 8,74 (ушир. d, J=4,2 Гц, 1Н), 8,60 (s, 1Н), 7,86 (s, 1Н), 7,69 (ушир. d, J=7,9 Гц, 1Н), 7,42-7,36 (m, 1Н), 7,25 (ушир. d, J=3,3 Гц, 2Н), 6,94 (ушир. d, J=3,5 Гц, 2Н), 6,74 (ушир. s, 1Н), 5,93 (ушир. d, J=6,6 Гц, 1Н), 5,63 (ушир. s, 1Н), 5,52 (q, J=6,5 Гц, 1Н), 3,73 (s, 3Н), 3,32 (dd, J=5,3, 13,9 Гц, 1Н), 3,08 (dd, J=6,8, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 378,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-(о-ТОЛИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (190)
[1038] Соединение 190 (28,4 мг, выход: 30,7%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163Е и о-толилбороновой кислоты. Соединение 190: 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,94 (d, J=4,8 Гц, 1Н), 7,43-7,31 (m, 3Н), 7,30-7,15 (m, 5Н), 7,15-7,05 (m, 4Н), 5,30-5,19 (m, 1Н), 3,48 (s, 3Н), 3,12 (dd, J=4,4, 14,0 Гц, 1Н), 2,76 (ddd, J=4,8, 9,0, 13,8 Гц, 1Н), 1,96 (d, J=10,1 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 391,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(6-(ТРИФТОРМЕТИЛ)ПИРИДИН-3-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (315)
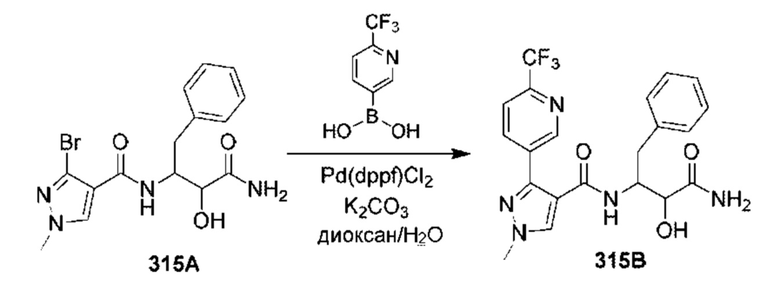
[1039] К смеси гидрохлорида 3-амино-2-гидрокси-4-фенилбутанамида (1,35 г, 5,86 ммоль, HCl) и соединения 163F (1 г, 4,88 ммоль), HOBt (659 мг, 4,88 ммоль) в ДМФА (20 мл) добавляли DIEA (1,58 г, 12,20 ммоль, 2 мл) и EDCI (1,4 г, 7,32 ммоль) порциями при 20°С и перемешивали в течение 16 ч. Реакционную смесь обрабатывали ЭА (40 мл), промывали Н2О (50 мл × 2). Органический слой промывали 0,5 н. HCl (40 мл), насыщенным водным NaHCO3 (40 мл) и солевым раствором (30 мл), сушили над Na2SO4, фильтровали, и растворитель удаляли под вакуумом. Остаток растирали с ДХМ (2 мл) и ПЭ (10 мл), образовывался осадок, твердое вещество собирали и сушили под вакуумом с получением соединения 315А (900 мг, выход: 45,38%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,25 (s, 1Н), 7,85-7,42 (m, 1Н), 7,35 (dd, J=7,3 Гц, 1Н), 7,28-7,18 (m, 5Н), 6,15 (d, J=6,2 Гц, 1Н), 5,95 (d, J=5,5 Гц, 1Н), 4,56-4,32 (m, 1Н), 3,86-3,78 (m, 4Н), 2,95-2,84 (m, 1Н), 2,82-2,73 (m, 1Н), 2,69-2,58 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 381,0.
[1040] Соединение 315 (30 мг, выход: 28,27%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих промежуточных соединений, 315А и (6-(трифторметил)пиридин-3-ил)бороновой кислоты, и через стадию получения промежуточного соединения 315В. Соединение 315: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,92 (s, 1Н), 8,65 (dd, J=7,3 Гц, 1Н), 8,26 (s, 1Н), 8,20 (dd, J=8,4 Гц, 1Н), 8,05 (s, 1Н), 7,88-7,76 (m, 2Н), 7,28 (d, J=3,7 Гц, 4Н), 7,24-7,16 (m, 1Н), 5,27 (t, J=3,1 Гц, 1Н), 3,94 (s, 3Н), 3,16 (dd, J=3,7, 14,1 Гц, 1Н), 2,83 (dd, J=10,4, 13,7 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 446,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-МЕТОКСИФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (407)
[1041] Соединение 407 (3,9 г, выход: 94,15%, белое твердое вещество) было получено, как описано в примере 97, из соответствующих исходных веществ, соединения 163С, (3-метоксифенил)бороновой кислоты и 274D. Соединение 407: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,44-8,25 (m, 1Н), 8,15-7,97 (m, 2Н), 7,84 (ушир. s, 1Н), 7,37-7,11 (m, 8Н), 6,96-6,80 (m, 1Н), 5,44-5,19 (m, 1Н), 3,90 (ушир. s, 3Н), 3,73 (ушир. s, 3Н), 3,26-3,07 (m, 1Н), 2,92-2,72 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 407,1.
3-((1H-ПИРАЗОЛО[4,3-с]ПИРИДИН-1-ИЛ)МЕТИЛ)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (408)
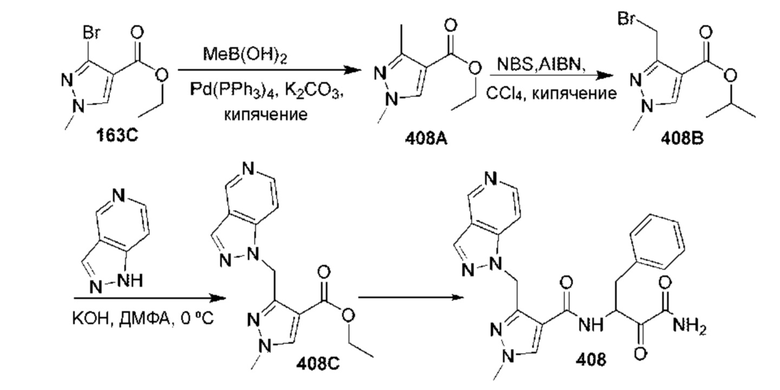
[1042] Смесь соединения 163С (2 г, 8,58 ммоль), МеВ(ОН)2 (2,05 г, 34,3 ммоль), Pd(PPh3)4 (793 мг, 687 мкмоль), K2СО3 (2,37 г, 17,2 ммоль) в диоксане (50 мл) и Н2O (10 мл) дегазировали и продували N2 3 раза, и затем смесь перемешивали при 120°С в течение 12 часов в атмосфере N2. Реакционную смесь подвергали экстракции этилацетатом 50 мл (50 мл × 2). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 100:1 до 80:1). Соединение 408А (1,4 г, выход: 97,0%) получали в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,87-7,73 (m, 1Н), 4,27 (q, J=7,1 Гц, 2Н), 3,91-3,75 (m, 3Н), 2,51-2,40 (m, 3Н), 1,33 (t, J=7,1 Гц, 3Н).
[1043] Смесь соединения 408А (1,2 г, 7,13 ммоль), NBS (1,9 г, 10,7 ммоль), AIBN (586 мг, 3,57 ммоль) в CCl4 (30 мл) перемешивали при 80°С в течение 12 часов. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 98:1 до 90:1). Соединение 408В (600 мг, выход: 34,1%) получали в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,76 (s, 1Н), 4,66 (s, 2Н), 4,30-4,21 (m, 2Н), 3,90-3,79 (m, 3Н), 1,38-1,22 (m, 3Н).
[1044] К смеси 1Н-пиразоло[4,3-с]пиридина (797 мг, 6,69 ммоль) в ДМФА (20 мл) добавляли KOH (501 мг, 8,92 ммоль). Смесь перемешивали при 15°С в течение 30 мин, и затем к смеси добавляли соединение 408В (550 мг, 2,23 ммоль) в ДМФА (10 мл) по каплям при 0°С в течение более 10 мин, и смесь перемешивали при 0°С в течение 2 ч. Структуру целевого продукта подтверждали посредством NOE (спектроскопии ЯМР с использованием ядерного эффекта Оверхаузера). Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством препаративной ВЭЖХ (в основных условиях). Соединение 408С (300 мг, выход: 47,2%) получали в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 9,14-8,99 (m, 1Н), 8,40 (d, J=6,2 Гц, 1Н), 8,15 (s, 1Н), 7,92-7,75 (m, 1Н), 7,51 (d, J=6,0 Гц, 1Н), 5,85 (s, 2Н), 4,26 (q, J=7,2 Гц, 2Н), 3,93-3,75 (m, 3Н), 1,29 (t, J=7,1 Гц, 3Н).
[1045] Соединение 408 (24,1 мг, выход: 40,4%, белое твердое вещество) было получено, как описано в примере 95, из соответствующих исходных веществ, соединения 408С и 274D
[1046] Соединение 408: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,07 (s, 1Н), 8,91 (ушир. d, J=5,5 Гц, 1Н), 8,46 (ушир. d, J=5,5 Гц, 1Н), 7,93 (s, 1Н), 7,69 (s, 1Н), 7,59 (ушир. d, J=5,5 Гц, 1Н), 7,24-7,17 (m, 1Н), 7,23 (ушир. s, 2Н), 7,25-7,15 (m, 1Н), 6,79 (ушир. s, 1Н), 5,69-5,51 (m, 1Н), 5,73-5,38 (m, 1Н), 5,49-5,37 (m, 1Н), 5,50-5,37 (m, 1Н), 5,74-5,33 (m, 1Н), 3,79 (s, 3Н), 3,56-3,42 (m, 1Н), 3,17 (ушир. dd, J=9,5, 14,1 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 432,3.
N-(4-АМИНО-3-ГИДРОКСИ-4-ОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(4-ФТОРФЕНИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (446) и N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(4-ФТОРФЕНИЛ)-1-МЕТИЛ-1Н-ПИРАЗОЛ-4-КАРБОКСАМИД (447)
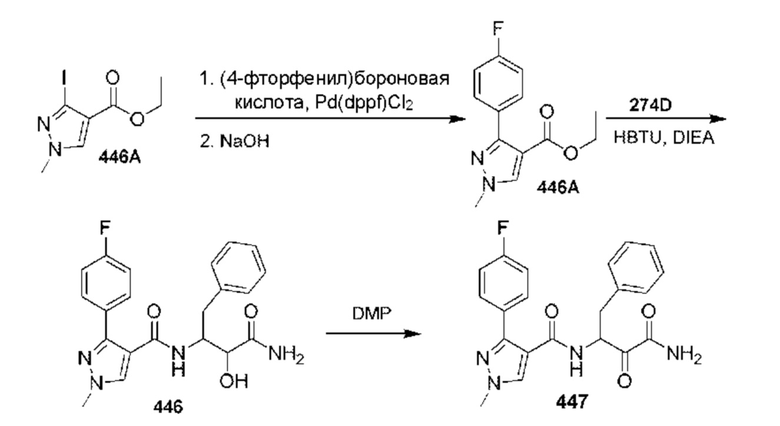
[1047] Соединение 446А обрабатывали (4-фторфенил)бороновой кислотой с помощью методики, как в случае соединения 163Е, затем полученный продукт подвергали сложноэфирному гидролизу с использованием гидроксида натрия и сочетанию с промежуточным соединением 274D с помощью методик, как в случае соединения 12, с получением соединения 446 и соединения 447. Соединение 446 (550 мг, выход: 75,9%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,08-8,00 (m, 1Н), 7,75 (d, J=9,0 Гц, 1Н), 7,56 (dd, J=5,6, 8,7 Гц, 2Н), 7,33-7,08 (m, 8Н), 5,83 (d, J=5,7 Гц, 1Н), 4,59-4,37 (m, 1Н), 4,00 (d, J=1,8 Гц, 1Н), 3,86 (s, 3Н), 2,80-2,73 (m, 1Н), 2,68 (s, 1Н), 2,66 (d, J=5,1 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 397,1. Соединение 447 (80 мг, выход: 39,9%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,42 (d, J=7,3 Гц, 1Н), 8,11-8,03 (m, 2Н), 7,81 (s, 1Н), 7,65-7,57 (m, 2Н), 7,32-7,18 (m, 5Н), 7,12 (t, J=8,8 Гц, 2Н), 5,27 (d, J=3,1 Гц, 1Н), 3,89 (s, 3Н), 3,30-3,12 (m, 1Н), 2,83 (dd, J=9,9, 13,7 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 395,1.
ПРИМЕР 98
СОЕДИНЕНИЯ 174-175, 191-193, 313, 293 (S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ПИРИМИДИН-5-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (174)
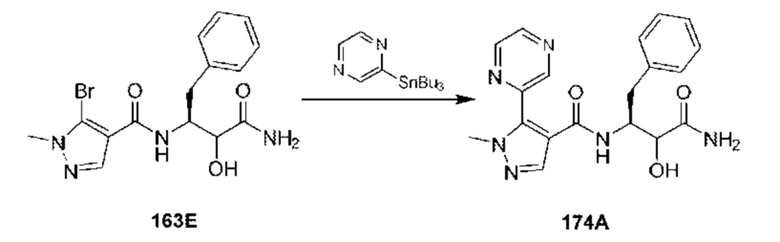
[1048] К раствору соединения 163Е (150 мг, 393,47 мкмоль) и трибутил(пиразин-2-ил)станнана (217 мг, 590,21 мкмоль) в диоксане (15 мл) добавляли палладий; тритрет-бутилфосфан (100 мг, 196,74 мкмоль). Смесь перемешивали при 90°С в течение 16 ч. В смесь добавляли водный KF (20 мл) для гашения реакции, смесь фильтровали, промывали этилацетатом (20 мл), фильтрат подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали посредством препаративной ВЭЖХ (в основных условиях) и посредством препаративной ТСХ (SiO2, ДХМ : МеОН = 10:1). Соединение 174А (70 мг, выход: 45,6%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ 8,77-8,69 (m, 1Н), 8,66-8,54 (m, 2Н), 7,94-7,78 (m, 1Н), 7,25-7,12 (m, 5Н), 4,63-4,58 (m, 1Н), 4,26-4,02 (m, 1Н), 3,91-3,84 (m, 3Н), 3,01-2,83 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 381,1.
[1049] Соединение 174 (25 мг, выход: 34,93%, белое твердое вещество) было получено, как описано в примере 61, из промежуточного соединения 174А. Соединение 174А: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,75 (d, J=1,5 Гц, 1Н), 8,71 (dd, J=1,6, 2,6 Гц, 1Н), 8,66-8,64 (m, 1Н), 8,31 (d, J=7,5 Гц, 1Н), 7,99 (s, 1Н), 7,70 (ушир. s, 1Н), 7,51 (ушир. s, 1Н), 7,28-7,23 (m, 4Н), 7,22-7,17 (m, 1Н), 5,33-5,28 (m, 1Н), 3,83 (s, 3Н), 3,20 (dd, J=4,5, 14,1 Гц, 1Н), 2,93 (dd, J=9,3, 14,1 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 379,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-(ПИРИМИДИН-4-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (175)
[1050] Соединение 175 (35 мг, выход: 47,75%, белое твердое вещество) было получено, как описано в примере 98, из соответствующих исходных веществ, соединения 163Е и трибутил(пиримидин-4-ил)станнана. Соединение 175: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,24 (d, J=1,1 Гц, 1Н), 8,82 (d, J=5,3 Гц, 1Н), 8,71 (d, J=7,5 Гц, 1Н), 8,03 (s, 1Н), 7,93 (s, 1Н), 7,78 (s, 1Н), 7,54 (dd, J=1,3, 5,3 Гц, 1Н), 7,27-7,17 (m, 6Н), 5,28-5,19 (m, 1Н), 3,85 (s, 3Н), 3,15 (dd, J=4,0, 13,9 Гц, 1Н), 2,83 (dd, J=10,1, 13,9 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 379,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-(ПИРИМИДИН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (191)
[1051] Соединение 191 (20 мг, выход: 38,77%, белое твердое вещество) было получено, как описано в примере 98, из соответствующих исходных веществ, соединения 163Е и трибутил(пиримидин-2-ил)станнана. Соединение 191: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,63 (d, J=6,3 Гц, 1Н), 8,86 (d, J=5,0 Гц, 2Н), 7,90 (s, 1Н), 7,81-7,56 (m, 2Н), 7,55 (t, J=4,9 Гц, 1Н), 7,21-7,14 (m, 3Н), 7,12-7,06 (m, 2Н), 5,47 (t, J=5,1, 7,5 Гц, 1Н), 4,05 (s, 3Н), 3,24 (dd, J=5,1, 14,2 Гц, 1Н), 3,05-3,00 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 379,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ПИРИМИДИН-4-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (192)
[1052] Соединение 192 (15 мг, выход: 29,82%, белое твердое вещество) было получено, как описано в примере 98, из соответствующих исходных веществ, соединения 163G и 4-(трибутилстаннил)пиримидина. Соединение 192: 1Н ЯМР (400 МГц, CD3CN) δ 11,76-11,65 (m, 1Н), 8,81-8,73 (m, 2Н), 8,14 (s, 1Н), 8,07 (dd, J=1,1, 5,3 Гц, 1Н), 7,59 (s, 1Н), 7,15 (s, 4Н), 7,04 (s, 1Н), 6,24 (s, 1Н), 5,65-5,59 (m, 1Н), 3,93 (s, 3Н), 3,33 (dd, J=5,1, 14,1 Гц, 1Н), 3,14 (dd, J=7,7, 14,1 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 379,1.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ПИРИМИДИН-4-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (193)
[1053] Соединение 193 (25 мг, выход: 54,29%, белое твердое вещество) было получено, как описано в примере 98, из соответствующих исходных веществ, соединения 163G и трибутил(пиримидин-2-ил)станнана. Соединение 193: 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,24 (d, J=7,3 Гц, 1Н), 8,69 (d, J=4,9 Гц, 2Н), 8,29 (s, 1Н), 8,08 (s, 1Н), 7,82 (s, 1Н), 7,47 (t, J=4,9 Гц, 1Н), 7,16-7,09 (m, 3Н), 7,07-7,03 (m, 2Н), 5,61-5,49 (m, 1Н), 3,92 (s, 3Н), 3,23 (dd, J=5,1, 14,1 Гц, 1Н), 3,07 (dd, J=7,3, 14,1 Гц, 1Н). МС (ИЭР) m/z (М+Н)+ 379,0.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(ПИРАЗИН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (313)
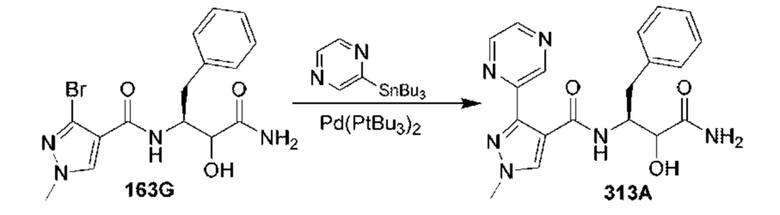
[1054] Соединение 313 было получено, как описано в примере 98, из соответствующих исходных веществ, соединения 163G и 2-(трибутилстаннил)пиразина, через стадию получения промежуточного соединения 313А. Соединение 313 (70 мг, выход: 69,7%, белое твердое вещество): 1Н ЯМР (400 МГц, CD3CN) δ 11,26 (d, J=6,0 Гц, 1Н), 9,34 (d, J=1,3 Гц, 1H), 8,54 (d, J=2,6 Гц, 1H), 8,23-8,10 (m, 2H), 7,17-7,11 (m, 5H), 7,01 (ушир. s, 1H), 6,19 (ушир. s, 1H), 5,67-5,55 (m, 1H), 3,94 (s, 3H), 3,33 (dd, J=5,3, 14,1 Гц, 1H), 3,13 (dd, J=7,7, 14,1 Гц, 1H). МС (ИЭР) m/z (M+H)+ 379,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(6-МЕТИЛПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (316)
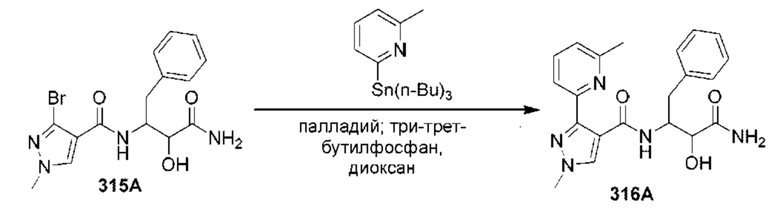
[1055] Соединение 316 было получено, как описано в примере 98, из соответствующих исходных веществ, соединения 315А и 2-метил-6-(трибутилстаннил)пиридина, через стадию получения промежуточного соединения 316А. Соединение 316 (20 мг, выход: 21,64%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,79 (d, J=7,7 Гц, 1Н), 8,24 (s, 1Н), 8,02 (s, 1Н), 7,92-7,86 (m, 1Н), 7,84-7,78 (m, 1Н), 7,75 (s, 1Н), 7,24 (d, J=7,7 Гц, 1Н), 7,15-7,08 (m, 3Н), 7,08-7,02 (m, 2Н), 5,52 (t, J=5,2, 8,1 Гц, 1Н), 3,89 (s, 3Н), 3,22 (dd, J=4,7, 13,8 Гц, 1Н), 2,99 (dd, J=8,4, 13,7 Гц, 1Н), 2,28 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 392,2.
ПРИМЕР 99
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-(6-(ТРИФТОРМЕТИЛ)ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (194)
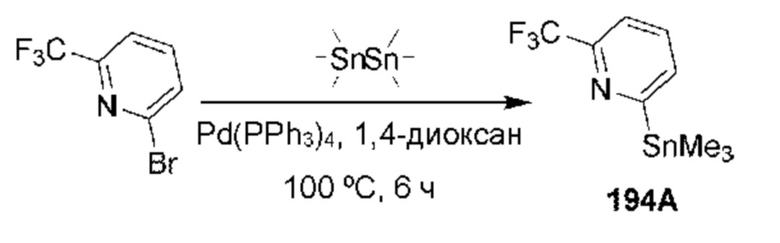
[1056] К смеси 2-бром-6-(трифторметил)пиридина (3 г, 13,27 ммоль) и 1,1,1,2,2,2-гексаметилдистаннана (5,7 г, 17,40 ммоль) в 1,4-диоксане (96 мл) добавляли Pd(PPh3)4 (3,07 г, 2,65 ммоль) при 25°С в атмосфере азота, и полученную смесь перемешивали при 100°С в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через тонкий слой целита. Осадок на фильтре промывали ДХМ. Фильтрат концентрировали под вакуумом с получением темного твердого соединения, которое очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 100:1 до 10:1) с получением неочищенного соединения 194А (3,2 г, чистота в анализе ЖХМС 26,46%) в виде белого твердого вещества. МС (ИЭР) m/z (М+Н)+ 311,8.
[1057] Соединение 194 (32,1 мг, выход 62,15%, белое твердое вещество) было получено, как описано в примере 98, из соответствующих исходных веществ, промежуточных соединений 163Е и 194А. Соединение 194: 1Н ЯМР (400 МГц, CDCl3): δ 7,98-7,91 (m, 1Н), 7,85 (d, J=7,6 Гц, 1Н), 7,75 (s, 1Н), 7,72 (d, J=7,6 Гц, 1Н), 7,26-7,21 (m, 3Н), 7,07-7,02 (m, 2Н), 6,74 (ушир. s, 1Н), 6,63-6,56 (m, 1Н), 5,60-5,54 (m, 1Н), 5,51 (ушир. s, 1Н), 3,94 (s, 3Н), 3,41-3,31 (m, 1Н), 3,14-3,06 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 446,0.
ПРИМЕР 100
N-(1-АМИНО-1,2-ДИОКСОГЕКС-5-ЕН-3-ИЛ)-1-МЕТИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (195)
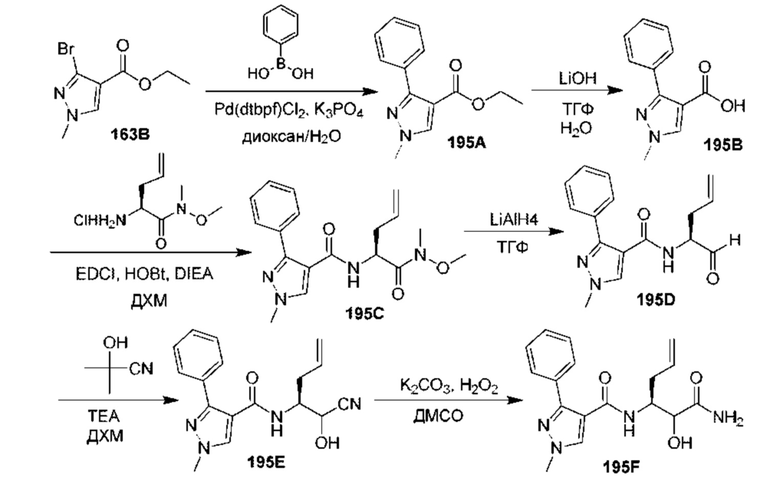
[1058] К раствору соединения 163В (2 г, 8,58 ммоль) и фенилбороновой кислоты (1,26 г, 10,30 ммоль) в диоксане (30 мл) и Н2О (10 мл) добавляли Pd(dtbpf)Cl2 (280 мг, 0,43 ммоль) и K3РО4 (5,46 г, 25,74 ммоль). Смесь перемешивали при 70°С в атмосфере N2 в течение 3 ч. Реакционную смесь промывали Н2О (20 мл), подвергали экстракции EtOAc (15 мл × 2). Органические фазы собирали и концентрировали. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = 10:1) с получением соединения 195А (1,9 г, выход: 96,17%) в виде желтого масла.
[1059] К раствору соединения 195А (1,9 г, 8,25 ммоль) в ТГФ (20 мл) и Н2O (20 мл) добавляли LiOH⋅Н2О (1,73 г, 41,25 ммоль). Смесь перемешивали при 25°С в течение 24 ч. Реакционную смесь подкисляли с помощью 1 н. HCl до рН ~ 4. Смесь подвергали экстракции EtOAc (25 мл × 2). Органические фазы собирали, промывали солевым раствором (30 мл), сушили с помощью Na2SO4, фильтровали и концентрировали с получением соединения 195В (1,3 г, выход: 77,93%) в виде светло-желтого твердого вещества. МС (ИЭР) m/z (М+1)+ 202,9.
[1060] К раствору соединения 195 В (800 мг, 3,96 ммоль) и гидрохлорида (S)-2-амино-N-метокси-N-метилпент-4-енамида (926 мг, 4,75 ммоль) в ДХМ (20 мл) добавляли DIEA (1,73 мл, 9,90 ммоль), HOBt (1,07 г, 7,92 ммоль) и EDCI (1,52 г, 7,92 ммоль). Смесь перемешивали при 25°С в течение 12 ч. Растворитель удаляли под вакуумом. Реакционную смесь разбавляли EtOAc (30 мл), промывали 1 н. HCl (20 мл). Органические фазы собирали и концентрировали. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = 1:1) с получением соединения 195С (1,2 г, выход: 88,50%) в виде бесцветного масла. МС (ИЭР) m/z (М+1)+ 343,1.
[1061] К раствору соединения 195С (1 г, 2,92 ммоль) в ТГФ (10 мл) добавляли по каплям при -40°С LiAlH4 (1 М, 3,1 мл). После добавления смесь перемешивали при 0°С в течение 1 ч. В реакционную смесь добавляли по каплям 1 н. HCl (30 мл) для гашения реакции, смесь подвергали экстракции EtOАс (20 мл × 3). Органические фазы собирали, промывали солевым раствором (30 мл), сушили с помощью Na2SO4, фильтровали и концентрировали с получением промежуточного соединения 195D (800 мг, неочищенного) в виде белого твердого вещества. МС (ИЭР) m/z (М+1)+ 284,0.
[1062] К раствору соединения 195D (280 мг, 0,99 ммоль) и 2-гидрокси-2-метилпропаннитрила (0,54 мл, 5,88 ммоль) в ДХМ (20 мл) добавляли TEA (0,17 мл, 1,19 ммоль). Смесь перемешивали при 25°С в течение 12 ч. Растворитель удаляли под вакуумом. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = 1:1) с получением соединения 195Е (200 мг, выход: 65,21%) в виде желтого масла. МС (ИЭР) m/z (М+1)+ 311,0.
[1063] К раствору соединения 195Е (736 мг, 2,37 ммоль) в ДМСО (5 мл) при 0°С добавляли K2CO3 (656 мг, 4,74 ммоль). Затем добавляли по каплям Н2О2 (2,28 мл, 23,72 ммоль, чистота 30%). Смесь перемешивали при 25°С в течение 1 ч. В реакционную смесь добавляли по каплям 10% водн. Na2S2О3 (30 мл) для гашения реакции. Смесь подвергали экстракции EtOAc (20 мл × 3). Органические фазы собирали, промывали солевым раствором (30 мл), сушили с помощью Na2SO4, фильтровали и концентрировали. Остаток промывали CH3CN (5 мл). Твердое вещество отфильтровывали, собирали и сушили под вакуумом с получением соединения 195F (200 мг, выход: 25,60%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,09 (s, 1Н), 7,72-7,61 (m, 2Н), 7,48-7,42 (m, 1Н), 7,40-7,26 (m, 3Н), 7,25-7,12 (m, 2Н), 5,81-5,60 (m, 2Н), 5,12-4,93 (m, 2Н), 4,33-4,17 (m, 1Н), 3,96-3,93 (m, 1Н), 3,86 (s, 3Н), 2,34-2,05 (m, 2Н). МС (ИЭР) m/z (М+1)+ 329,0.
[1064] К раствору соединения 195F (200 мг, 609,07 мкмоль) в ДХМ (15 мл) и ДМСО (5 мл) добавляли DMP (775 мг, 1,83 ммоль). Смесь перемешивали при 25°С в течение 30 мин. Реакционную смесь разбавляли ДХМ (20 мл), добавляли в нее для гашения реакции раствор 10% водного Na2S2О3 и насыщенного NaHCO3 (об./об. = 1/1) (40 мл). Органические фазы собирали, промывали солевым раствором (30 мл × 3). Органические фазы собирали, сушили с помощью Na2SO4, фильтровали и концентрировали. Остаток промывали CH3CN (5 мл). Твердое вещество отфильтровывали, собирали и сушили под вакуумом с получением 195 (38 мг, выход: 18,72%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,23 (d, J=6,8 Гц, 1Н), 8,13 (s, 1Н), 7,98 (ушир. s, 1Н), 7,73 (ушир. s, 1Н), 7,68-7,61 (m, 2Н), 7,38-7,25 (m, 3Н), 5,89-5,71 (m, 1Н), 5,11-4,98 (m, 3Н), 3,88 (s, 3Н), 2,57-2,50 (m, 1Н), 2,40-2,31 (m, 1Н). МС (ИЭР) m/z (М+1)+ 327,1.
ПРИМЕР 101
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-БРОМ-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (196)
[1065] Соединение 196 (54 мг, выход: 50,44%, белое твердое вещество) было получено, как описано в примере 61, из соответствующего промежуточного соединения 163G. Соединение 196: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,21-8,15 (m, 2Н), 8,06 (ушир. s, 1Н), 7,80 (ушир. s, 1Н), 7,28-7,23 (m, 4Н), 7,20-7,16 (m, 1Н), 5,33-5,26 (m, 1Н), 3,82 (s, 3Н), 3,15 (ушир. dd, J=3,7, 13,9 Гц, 1Н), 2,83 (ушир. dd, J=9,8, 13,8 Гц, 1Н). МС (ИЭР) m/z (M+H)+ 379,0 и 381,0.
ПРИМЕР 102
N-(1-АМИНО-1,2-ДИОКСОГЕКС-5-ЕН-3-ИЛ)-1-МЕТИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (197)
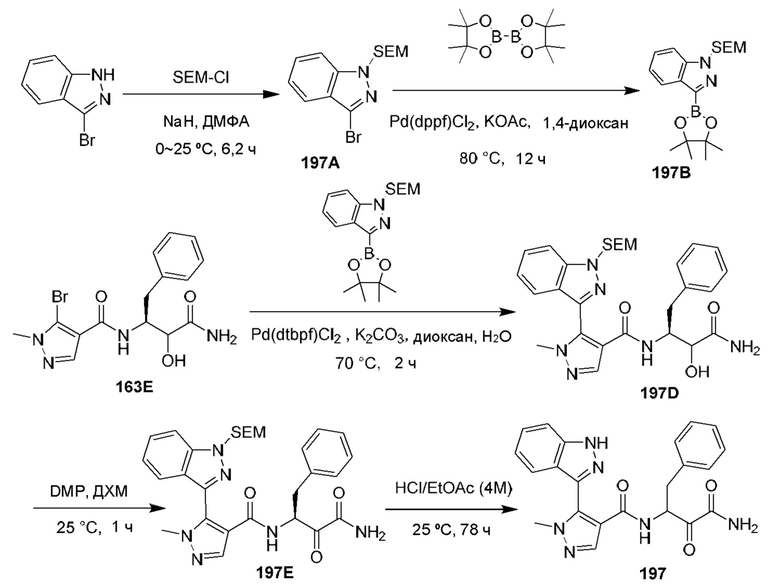
[1066] К холодному (0°С), перемешиваемому раствору 3-бром-1H-индазола (5 г, 25,38 ммоль) в ДМФА (130 мл) добавляли порциями NaH (1,22 г, 50,76 ммоль). Через 0,2 ч добавляли SEM-Cl (5,08 г, 30,46 ммоль), и затем смесь перемешивали при 25°С в течение 6 часов в атмосфере N2. В реакционную смесь добавляли насыщенный водный раствор NH4Cl для гашения реакции, и полученный слой подвергали экстракции EtOAc (100 мл × 2). Объединенные органические слои сушили, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (элюирование с помощью смеси петролейный эфир : EtOAc от 100:1 до 10:1) с получением соединения 197А (5,5 г, выход 66,21%) в виде масла. 1H ЯМР (CDCl3, 400 МГц): δ 7,71-7,69 (m, 1Н), 7,65-7,60 (m, 1Н), 7,57-7,51 (m, 1Н), 7,37-7,29 (m, 1Н), 5,76 (d, J=3,2 Гц, 2H), 3,70-3,55 (m, 2H), 1,02-0,87 (m, 2H), 0,00 (d, J=3,2 Гц, 9Н). МС (ИЭР) m/z (M+H)+ 338,3.
[1067] К смеси соединения 197А (2 г, 6,11 ммоль), соединения 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (1,86 г, 7,33 ммоль), КОАс (2,4 г, 24,44 ммоль), Pd(dppf)Cl2 (894,3 мг, 1,22 ммоль) в диоксане (80 мл) нагревали при 80°С в течение 12 часов в атмосфере N2. Соединение 197В (неочищенное) было получено в виде раствора (15,8 мг/мл в диоксане).
[1068] К смеси соединения 197В (461,6 мг, 1,23 ммоль, 30 мл в диоксане), соединения 163Е (100 мг, 262,3 мкмоль), Pd(dtbpf)Cl2 (34,19 мг, 52,46 мкмоль) и K3PO4 (111,4 мг, 524,6 мкмоль) в H2O (2 мл) дегазировали и продували N2 3 раза, и затем смесь перемешивали при 70°С в течение 2 часов в атмосфере N2. Реакционный раствор очищали посредством препаративной ВЭЖХ (в основных условиях) с получением соединения 197D (50 мг, выход 16,54%) в виде коричневого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,14-8,02 (m, 1Н), 8,01-7,82 (m, 1H), 7,70-7,63 (m, 1H), 7,58 (t, J=7,6 Гц, 1Н), 7,51 (d, J=8,4 Гц, 1H), 7,39-7,32 (m, 1H), 7,02-6,89 (m, 1H), 6,88-6,69 (m, 5H), 6,42 (ушир. s, 1H), 5,70-5,48 (m, 2H), 5,34 (ушир. s, 1H), 4,39-4,27 (m, 1H), 4,24-4,08 (m, 1H), 3,84 (s, 3Н), 3,58 (t, J=8,0 Гц, 2H), 3,04-2,74 (m, 2H), 0,92-0,87 (m, 2H), -0,07 (s, 9H). МС (ИЭР) m/z (M+H)+ 549,2.
[1069] К смеси соединения 197D (50 мг, 91,12 мкмоль) в ДХМ (10 мл) добавляли DMP (116 мг, 273,4 мкмоль) одной порцией при 0°С в атмосфере N2, и затем смесь перемешивали при 0°С в течение 1 часа в атмосфере N2. В смесь добавляли насыщенный водный NaHCO3 (15 мл) и насыщенный водный Na2S2O3 (15 мл) для гашения реакции, и смесь перемешивали в течение 20 мин, затем разбавляли дихлорметаном (100 мл). Смесь перемешивали в течение 20 мин и промывали водой (2×20 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении с получением неочищенного продукта. Остаток очищали посредством препаративной ВЭЖХ (в основных условиях) с получением соединения 197Е (30 мг, выход 55,67%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,23 (s, 1H), 7,98 (d, J=6,8 Гц, 1H), 7,74 (d, J=8,8 Гц. 1H), 7,70-7,60 (m, 2H), 7,48-7,38 (m, 1H), 7,03 (t, J=7,2 Гц, 1H), 6,89-6,75 (m, 3Н), 6,74-6,67 (m, 2H), 5,84-5,75 (m, 1H), 5,63 (d, J=11,6 Гц, 2H), 5,45 (d, J=11,2 Гц, 1H), 4,01-3,87 (m, 3Н), 3,78-3,57 (m, 2H), 3,39-3,12 (m, 2H), 1,02-0,92 (m, 2H), 0,16-0,07 (m, 9H). МС (ИЭР) m/z (М+Н)+ 547,2.
[1070] Смесь соединения 197Е (30 мг, 54,88 мкмоль) в HCl/EtOAc (4 M, 1,50 мл) перемешивали при 25°С в течение 78 часов. Реакционную смесь концентрировали при пониженном давлении с получением остатка, который очищали посредством препаративной ВЭЖХ (в основных условиях) с получением соединения 197 (1,6 мг, выход 6,68%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 10,35 (ушир. s, 1H), 8,12 (s, 1H), 7,77 (ушир. s, 1H), 7,61-7,50 (m, 3Н), 7,34-7,28 (m, 1H), 7,10-7,04 (m, 1H), 6,98-6,90 (m, 2H), 6,84-6,68 (m, 3Н), 5,62-5,42 (m, 2H), 3,83 (s, 3Н), 3,29-3,19 (m, 1H), 3,08-2,97 (m, 1H). МС (ИЭР) m/z (M+H)+ 417,2.
ПРИМЕР 103
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-3-(6-(ТРИФТОРМЕТИЛ)ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (198)
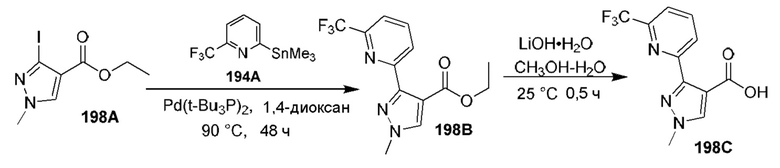
[1071] К смеси соединения этил-3-иод-1-метил-1H-пиразол-4-карбоксилата (200 мг, 714,13 мкмоль) и соединения 194А (1,91 г, 856,96 мкмоль) в диоксане (3 мл) добавляли Pd(t-Bu3P)2 (110 мг, 214,24 мкмоль) в атмосфере азота. Смесь перемешивали при 90°С в течение 48 часов. Смесь разбавляли CH2Cl2 (30 мл), фильтровали для удаления осадка, и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной флэш-хроматографии (петролейный эфир : этилацетат = от 10/1 до 5/1) с получением соединения (175,00 мг, выход 77,93%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,01-7,91 (m, 3Н), 7,72-7,67 (m, 1H), 4,26-4,20 (m, 2H), 4,00 (s, 3Н), 1,25-1,19 (s, 3Н).
[1072] К смеси соединения 198В (170 мг, 568,09 мкмоль) в МеОН (8 мл) и H2O (4 мл) добавляли LiOH⋅H2O (191 мг, 4,54 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли H2O (20 мл), доводили рН до ~ 3 с помощью 1 н. НСl и затем подвергали экстракции EtOAc (20 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 198С (150 мг, выход 97,36%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,44 (s 1H), 8,25 (d, J=4 Гц, 1Н), 8,00-7,98 (m, 2H). 3.95 (s, 3Н).
[1073] Соединение 198 (94,2 мг, 63,09 выход %, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 198С. Соединение 198: 1H ЯМР (400 МГц, CDCl3) δ 10,92 (d, J=6,80 Гц, 1H), 8,39 (d, J=8,40 Гц, 1H), 8,08 (s, 1 H) 7,92-8,02 (m, 1 H), 7,65 (d, J=8,00 Гц, 1 H), 7,06-7,23 (m, 5 H), 6,72 (s, 1 H), 5,51 (ушир. s, 1 H), 5,31-5,43 (m, 1 H), 3,93 (s, 3 H), 3,38-3,49 (m, 1 H), 2,96-3,11 (m, 1 H). MC (ИЭР) m/z (M+1)+ 446,1.
ПРИМЕР 104
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(3'-МЕТИЛ-[1,1'-БИФЕНИЛ]-3-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (199)
[1074] Соединение 199 (35,0 мг, выход 53,6%, белое твердое вещество) было получено, как описано в примере 61, из соответствующих исходных веществ, соединения 103А и м-толилбороновой кислоты. Соединение 199: 1H ЯМР (ДМСО-d6, 400 МГц) δ 9,08 (d, J=7,7 Гц, 1H), 8,08-8,00 (m, 1H), 7,84 (ушир. s, 1H), 7,60-7,52 (m, 2H), 7,45-7,37 (m, 3Н), 7,36-7,30 (m, 1H), 7,28-7,25 (m, 3Н), 7,23-7,16 (m, 3Н), 7,12-7,06 (m, 1H), 6,60 (ушир. s, 1H), 5,29 (ушир. s, 1H), 3,22-3,14 (m, 1H), 2,86-2,76 (m, 1H), 2,35 (s, 3Н), 2,28-2,22 (m, 3Н). MC (ИЭР) m/z (M+H)+ 467,2.
ПРИМЕР 105
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(6-ФЕНИЛПИРИДАЗИН-3-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (200)
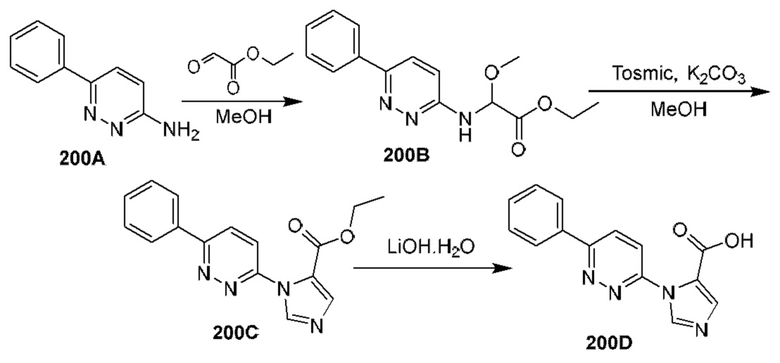
[1075] К смеси 6-бромпиридазин-3-амина (10 г, 57 ммоль) и фенилбороновой кислоты (10,5 г, 86 ммоль) в толуоле (150 мл) и EtOH (150 мл) добавляли LiCl (7,3 г, 172,4 ммоль). Затем добавляли Na2CO3 (1 M, 155 мл), после чего добавляли Pd(PPh3)2Cl2 (403 мг, 0,57 ммоль). Смесь нагревали до кипения с обратным холодильником в течение 16 ч. Смесь фильтровали через целит. Фильтрат разбавляли H2O (15 мл), подвергали экстракции этилацетатом (15 мл × 3). Объединенный органический слой промывали солевым раствором (15 мл), сушили над MgSO4, фильтровали и концентрировали. Остаток растирали со смесью МТБЭ/этилацетат (об./об. = 1/1, 50 мл). Осадок на фильтре сушили под вакуумом с получением соединения 2 (4,4 г, выход 44,7%) в виде грязно-белого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц) δ 7,94 (d, J=7,2 Гц, 2Н), 7,79 (d, J=9,2 Гц, 1H), 7,46-7,42 (m, 2Н), 7,38-7,36 (m, 1H), 6,86 (d, J=9,2 Гц, 2Н), 6,52 (s, 2Н). MC (ИЭР) m/z (M+H)+ 172,0.
[1076] Этил-2-оксоацетат (26 мл, 128,50 ммоль) добавляли к раствору соединения 200А (4,4 г, 25,7 ммоль) в МеОН (100 мл). Смесь нагревали до 65°С и перемешивали в течение 15 ч. Смесь концентрировали. Остаток очищали посредством колоночной флэш-хроматографии (петролейный эфир/этилацетат = 3/1) с получением соединения 200В (5,5 г, выход 52%, чистота 69,8%) в виде коричневого масла. MC (ИЭР) m/z (M+H)+ 288,l.
[1077] K2CO3 (6,61 г, 47,85 ммоль) добавляли к раствору соединения 200В (5,5 г, 19,14 ммоль) и тозилметилизоцианида (5,04 г, 25,84 ммоль) в EtOH (190 мл). Смесь нагревали до 65°С и перемешивали в течение 3 ч. Смесь концентрировали под вакуумом, и остаток обрабатывали этилацетатом (100 мл) и H2O (75 мл). Органический слой отделяли, и водный слой подвергали экстракции этилацетатом (50 мл × 2). Объединенный органический слой промывали солевым раствором (100 мл), сушили над MgSO4, фильтровали и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии (петролейный эфир/этилацетат = от 5/1 до 1/1) с получением (1,80 г, выход 30%) в виде желтого твердого вещества. 1H ЯМР (CDCl3, 400 МГц) δ 8,22 (s, 1H), 8,12-8,10 (m, 2Н), 8,01 (d, J=7,2 Гц, 1H), 7,94, (s, 1H), 7,76-7,74 (m, 1H), 7,56-7,54 (m, 3H), 4,29 (q, J=6,8 Гц, 2Н), 1,33 (t, J=6,8 Гц, 3H). MC (ИЭР) m/z (M+H)+ 295,0.
[1078] LiOH⋅H2O (114 мг, 2,72 ммоль) добавляли к раствору соединения 200С (100 мг, 0,34 ммоль) в МеОН (5 мл). Затем добавляли H2O (0,5 мл). Смесь перемешивали при 25°С в течение 3 ч. Смесь разбавляли H2O (25 мл), и летучий растворитель выпаривали под вакуумом. Остаток подкисляли до рН ~ 3 с помощью 1 н. НСl. Смесь подвергали экстракции этилацетатом (25 мл × 3). Объединенный органический слой промывали солевым раствором (20 мл), сушили над MgSO4, фильтровали и концентрировали с получением соединения 200D (90 мг, выход 99,5%) в виде желтого твердого вещества, которое непосредственно использовали на следующей стадии. 1H ЯМР (ДМСО-d6, 400 МГц) δ 8,50 (d, J=9,3 Гц, 1Н), 8,38 (d, J=1,0 Гц, 1Н), 8,24 (dd, J=1,9, 7,7 Гц, 2Н), 8,13 (d, J=9,0 Гц, 1Н), 7,85 (d, J=1,0 Гц, 1Н), 7,66-7,55 (m, 3H).
[1079] Соединение 200 (35 мг, выход 35,2%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 200D. В анализе посредством 1H ЯМР был обнаружен гидрат. Соединение 200: 1H ЯМР (ДМСО-d6, 400 МГц) δ 8,83 (ушир. s, 1Н), 8,34-8,25 (m, 2Н), 8,19 (dd, J=1,8, 7,8 Гц, 2Н), 7,81 (ушир. s, 1Н), 7,75 (s, 1Н), 7,66-7,48 (m, 5H), 7,34-7,20 (m, 5H), 5,32-5,23 (m, 1Н), 3,24 (dd, J=4,3, 14,1 Гц, 1Н), 2,94 (dd, J=9,7, 13,9 Гц, 1Н). МС (ИЭР) m/z (M+H)+ 441,1.
ПРИМЕР 106
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(6-ФЕНИЛПИРИДАЗИН-3-ИЛ)-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (201)
[1080] Соединение 201 (89 мг, выход 54,5%, желтое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 200D. В анализе посредством 1H ЯМР был обнаружен гидрат. Соединение 201: 1H ЯМР (ДМСО-d6, 400 МГц) δ 8,82 (ушир. s, 0,8Н), 8,51 (ушир. s, 0,9Н), 8,31-8,22 (m, 2,7Н), 8,20-8,14 (m, 2,3Н), 7,93 (s, 0,9Н), 7,79-7,70 (m, 1,7H), 7,69-7,64 (m, 1,7H), 7,63-7,54 (m, 5,7H), 7,37 (ушир. d, J=8,5 Гц, 0,9Н), 7,31 (d, J=4,5 Гц, 3,6Н), 7,28-7,17 (m, 5,2H), 5,35-5,28 (m, 1Н), 4,42-4,35 (m, 0,9H), 3,23 (dd, J=4,0, 14,1 Гц, 0,7Н), 3,07 (ушир. s, 1,7Н), 2,97-2,89 (m, 1,1Н), 2,80-2,74 (m, 1,7Н), 2,68-2,64 (m, 1,0H), 0,71-0,63 (m, 2,0Н), 0,62-0,55 (m, 3,2Н), 0,48 (ушир. s, 1,6Н). МС (ИЭР) m/z (M+H)+ 481,1.
ПРИМЕР 107
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(2'-МЕТИЛ-[1,1'-БИФЕНИЛ]-4-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (202)
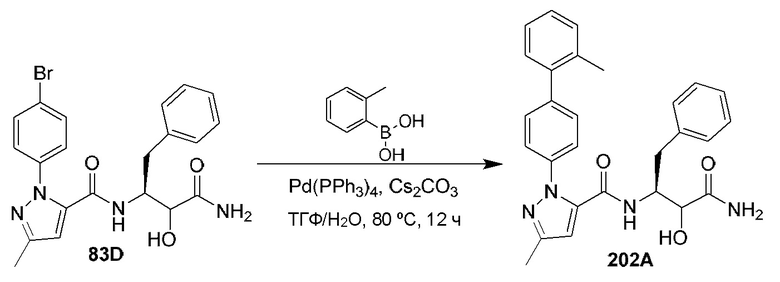
[1081] К раствору соединения 83D (150 мг, 0,33 ммоль) в ТГФ (6 мл) и H2O (3 мл) добавляли Cs2CO3 (168 мг, 0,51 ммоль) и о-толилбороновую кислоту (89 мг, 0,66 ммоль), затем Pd(PPh3)4 (38 мг, 0,033 ммоль). Затем смесь нагревали до 80°С и перемешивали в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли Н2О (10 мл) для гашения реакции, и затем смесь упаривали при пониженном давлении. Водную фазу подвергали экстракции этилацетатом (10 мл × 3). Объединенный органический слой промывали NaHCO3 (10 мл), H2O (10 мл), солевым раствором (10 мл), сушили над Na2SO4, фильтровали, упаривали при пониженном давлении. Неочищенный продукт растирали со смесью простой изопропиловый эфир/ацетонитрил (10/1, 5 мл) с получением соединения 202А (50 мг, выход 32,5%) в виде желтого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц) δ 8,54 (ушир. d, J=9,3 Гц, 1Н), 8,20 (ушир. d, J=9,7 Гц, 1Н), 7,66-7,46 (m, 1Н), 7,38 (ушир. s, 1Н), 7,35-7,19 (m, 12H), 7,14 (ушир. d, J=8,4 Гц, 1Н), 6,61-6,51 (m, 1Н), 6,01-5,66 (m, 1Н), 4,45 (ушир. s, 1Н), 4,04-3,90 (m, 1Н), 3,00-2,62 (m, 2H), 2,37-2,17 (m, 6H).
[1082] К раствору соединения 202А (46 мг, 98,2 мкмоль) в ДХМ (10 мл) добавляли DMP (125 мг, 294,5 мкмоль), и смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь разбавляли ДХМ (10 мл) и добавляли в нее для гашения реакции NaHCO3/Na2S2O3 (1/1, 20 мл), затем смесь перемешивали в течение 0,25 ч. Смесь подвергали экстракции ДХМ (10 мл × 2), объединенный органический слой промывали NaHCO3 (10 мл × 3) и солевым раствором (10 мл × 3), сушили над безводным Na2SO4, фильтровали, упаривали при пониженном давлении. Неочищенный продукт очищали посредством препаративной ВЭЖХ (в основных условиях) с получением 202 (30 мг, выход 60,65%), которое было получено в виде белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц) δ 7,43-7,39 (m, 2Н), 7,39-7,35 (m, 2Н), 7,30-7,28 (m, 3H), 7,26-7,21 (m, 4H), 7,03 (dd, J=1,9, 7,6 Гц, 2Н), 6,72 (ушир. s, 1H), 6,52 (s, 1H), 6,37 (ушир. d, J=7,5 Гц, 1Н), 5,66-5,60 (m, 1H), 5,55 (ушир. s, 1H), 3,38 (dd, J=5,4, 14,2 Гц, 1H), 3,16 (dd, J=7,3, 14,3 Гц, 1H), 2,36 (s, 3H), 2,29-2,27 (m, 3H). МС (ИЭР) m/z (M+H)+ 467,1.
ПРИМЕР 108
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(6-МЕТИЛ-4-(ТРИФТОРМЕТИЛ)ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (206)
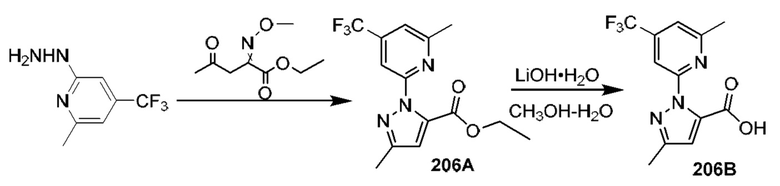
[1083] К раствору 2-гидразинил-6-метил-4-(трифторметил)пиридина (500 мг, 2,62 ммоль) в СН3СООН (10 мл) добавляли этил-2-(метоксиимино)-4-оксопентаноат (490,4 мг, 2,62 ммоль), затем смесь перемешивали при 120°С в течение 2 часов. Смесь разбавляли CH2Cl2 (100 мл) и промывали насыщенным бикарбонатом натрия (30 мл × 2) и насыщенным солевым раствором (30 мл × 2), сушили с помощью безводного Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством колоночной флэш-хроматографии (SiO2, петролейный эфир : этилацетат = от 10/1 до 3/1). Соединение 206А (200,0 мг, выход 24,37%) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,72 (s, 1H), 7,33 (s, 1H), 6,68 (s, 1H), 4,33-4,28 (m, 2Н), 2,60 (s, 3H), 3,23 (s, 3H), 1,31-1,20 (m, 3H).
[1084] К смеси соединения 206А (180,0 мг, 574,58 мкмоль) в МеОН (6 мл) и H2O (3 мл) добавляли LiOH⋅H2O (96,4 мг, 2,30 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 6 часов. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Затем смесь разбавляли H2O (10 мл) и доводили рН до ~ 3 с помощью 1 н. HCl, и затем смесь подвергали экстракции EtOAc (40 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 206В (145,0 мг, выход 88,48%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,80 (s, 1H), 7,73 (s, 1H), 6,79 (d, J=4 Гц, 1H), 2,56 (s, 3H), 2,29 (s, 3H).
[1085] Соединение 206 (52,0 мг, выход 52,16%, бледно-желтое твердое вещество) было получено, как описано в примере 41, из соответствующей карбоновой кислоты, промежуточного соединения 206В. Соединение 206: 1H ЯМР (400 МГц, CDCl3) δ 8,65 (J=7,6 Гц, 1H), 7,86 (s, 1H), 7,22 (s, 1H), 7,18-7,16 (m, 3H), 7,04-7,03 (m, 2H), 6,88 (s, 1H), 6,70 (s, 1H), 5,77-5,72 (m, 1H), 3,46-3,41 (m, 1H), 3,35-3,30 (m, 1H), 2,83-2,79 (m, 1H), 2,34 (s, 3H), 2,32 (s, 3H), 0,91-0,86 (m, 2H), 0,63-0,60 (m, 2H). МС (ИЭР) m/z (M+1)+ 500,2.
ПРИМЕР 109
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(6-МЕТИЛ-4-(ТРИФТОРМЕТИЛ)ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (208)
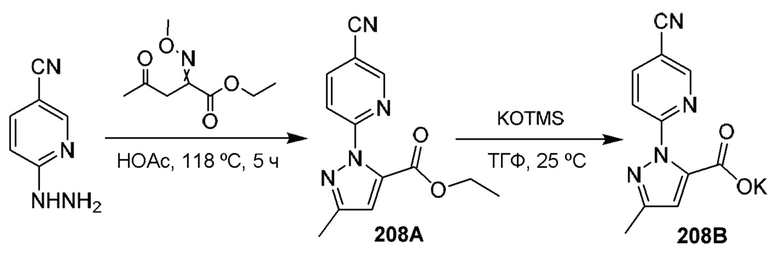
[1086] Смесь этил-2-(метоксиимино)-4-оксопентаноата (558,2 мг, 2,98 ммоль) и 6-гидразинилникотинонитрила (400 мг, 2,98 ммоль) в АсОН (5 мл) перемешивали при 118°С в течение 5 часов. Реакционную смесь охлаждали до 25°С и концентрировали при пониженном давлении с получением остатка, который разбавляли CH2Cl2 (100 мл). Органическую фазу промывали насыщенным водным NaHCO3 (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1:0 до 5:1) с получением соединения 208А (160 мг, выход 19,09%) в виде белого твердого вещества, но его структура (предполагаемая структура) не могла быть подтверждена посредством N-H НМВС (гетероядерной корреляционной спектроскопии с корреляцией через несколько связей). 1H ЯМР (400 МГц, CDCl3): δ 8,66 (dd, J=0,8, 2,4 Гц, 1H), 8,07 (dd, J=2,4, 8,4 Гц, 1Н), 7,88 (dd, J=0,8, 8,4 Гц, 1Н), 6,67 (s, 1H), 4,35 (q, J=6,8 Гц, 2H), 2,37 (s, 3H), 1,32 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (M+1)+ 257,1.
[1087] К раствору соединения 208А (100 мг, 390,23 мкмоль) в ТГФ (4 мл) добавляли KOTMS (100 мг, 780,46 мкмоль), затем смесь перемешивали при 25°С в течение 0,3 часа. Смесь разбавляли петролейным эфиром (20 мл), осадок отфильтровывали с получением остатка. Смесь разбавляли петролейным эфиром (20 мл), осадок отфильтровывали с получением промежуточного соединения 208B (80 мг, выход 76,98%) в виде белого твердого вещества.
[1088] Соединение 208 (13,5 мг, выход 27,13%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединений 208В и 12G. Соединение 208: 1H ЯМР (400 МГц, ДМСО-d6): δ 9,09 (d, J=7,2 Гц, 1H), 8,49 (d, J=1,6 Гц, 1H), 8,40-8,36 (m, 1H), 8,10 (ушир. s, 1H), 7,86 (ушир. s, 1H), 7,82 (d, J=8,4 Гц, 1H), 7,31-7,23 (m, 5H), 6,51 (s, 1H), 5,35-5,29 (m, 1H), 3,19-3,13 (m, 1H), 2,84-2,76 (m, 1H). МС (ИЭР) m/z (M+1)+ 403,1.
ПРИМЕР 110
СОЕДИНЕНИЯ 209, 439-441, 443-444
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИДИН-4-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (209)
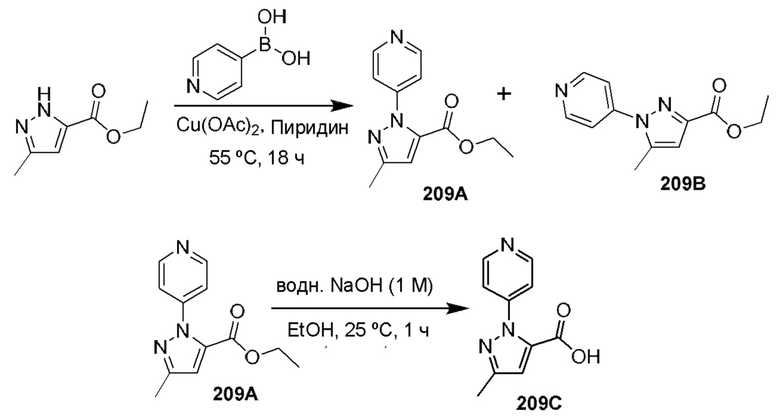
[1089] К раствору этил-3-метил-1H-пиразол-5-карбоксилата (3,0 г, 19,46 ммоль), пиридин-4-илбороновой кислоты (5,98 г, 48,65 ммоль) в пиридине (40 мл) добавляли Cu(ОАс)2 (1,8 г, 9,73 ммоль). Смесь перемешивали при 55°С в течение 18 ч. Смесь фильтровали и концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® Silica Flash массой 120 г, элюент 0~50% этилацетат/петролейный эфир, градиент со скоростью потока 40 мл/мин). Соединение 209А (850 мг, выход 18,91%) получали в виде белого твердого вещества. Соединение 209В (850 мг, выход 18,91%) получали в виде белого твердого вещества. Соединение 209А (850 мг, выход 18,91%, белое твердое вещество): 1H ЯМР (400 МГц, CDCl3) δ 8,75-8,65 (m, 2Н), 7,42 (d, J=6,0 Гц, 2Н), 6,87 (s, 1H), 4,33-4,25 (m, 2H), 2,36 (s, 3Н), 1,32-1,28 (m, 3H).
[1090] К смеси соединения 209А (600 мг, 2,59 ммоль) в EtOH (5 мл) добавляли водн. NaOH (1 M, 2,59 мл) одной порцией, и смесь перемешивали при 25°С в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления EtOH. рН остатка доводили до ~ 3 с помощью 1 н. HCl, и затем остаток подвергали экстракции EtOAc (200 мл × 4). Объединенные органические слои промывали солевым раствором (80 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 209С (390 мг, выход 74,10%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,63 (ушир. s, 2Н), 7,50 (ушир. d, J=5,2 Гц, 2Н), 6,90 (s, 1H), 2,26 (s, 3Н).
[1091] Соединение 209 (27,1 мг, выход 25,46%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 209С. Соединение 209: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,33 (d, J=7,8 Гц, 1H), 8,55-8,49 (m, 2Н), 8,17 (s, 1H), 7,92 (s, 1H), 7,37-7,27 (m, 5H), 7,22-7,18 (m, 2Н), 6,60 (s, 1H), 5,33 (s, 1H), 3,28-3,21 (m, 1H), 2,88-2,79 (m, 1H), 2,28 (s, 3Н). МС (ИЭР) m/z (M+H)+ 378,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-ФЕНОКСИФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (439)
[1092] Соединение 439 (65 мг, выход: 61,4%, желтое твердое вещество) получали из этил-3-метил-1H-пиразол-5-карбоксилата, который подвергали сочетанию с (4-феноксифенил)бороновой кислотой, как в случае соединения 209, с последующим сложноэфирным гидролизом и сочетанием с промежуточным соединением 274D с помощью методик, как в случае соединения 12, с получением соединения 439. Соединение 439: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,08 (ушир. d, J=7,3 Гц, 1H), 8,12 (ушир. s, 1H), 7,87 (ушир. s, 1H), 7,44 (ушир. t, J=7,4 Гц, 2Н), 7,31-7,13 (m, 8H), 7,06 (ушир. d, J=7,9 Гц, 2Н), 6,93 (ушир. d, J=8,4 Гц, 2Н), 6,57 (s, 1H), 5,26 (ушир. s, 1H), 3,20 (ушир. d, J=14,6 Гц, 1H), 2,81 (ушир. t, J=12,3 Гц, 1H), 2,24 (s, 3H). MC (ИЭР) m/z (M+H)+ 469,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-((ТЕТРАГИДРО-2H-ПИРАН-4-ИЛ)ОКСИ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (440)
[1093] Соединение 440 (90 мг, выход: 58%, белое твердое вещество) получали из этил-1-(4-гидроксифенил)-3-метил-1Н-пиразол-5-карбоксилата, который подвергали сочетанию Мицунобу с тетрагидро-2H-пиран-4-олом с последующим сложноэфирным гидролизом и сочетанием с промежуточным соединением 274D с помощью методик, как в случае соединения 12, с получением соединения 440. Соединение 440: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,03 (d, J=7,8 Гц, 1H), 8,11 (s, 1H), 7,87 (s, 1H), 7,35-7,23 (m, 5H), 7,07 (d, J=8,8 Гц, 2Н), 6,92 (d, J=9,0 Гц, 2Н), 6,53 (s, 1H), 5,27-5,14 (m, 1H), 4,59-4,54 (m, 1H), 3,89-3,82 (m, 2H), 3,49 (t, J=9,3 Гц, 2Н), 3,19 (dd, J=3,1, 13,9 Гц, 1Н), 2,81 (dd, J=10,8, 13,8 Гц, 1H), 2,23 (s, 3H), 1,97 (d, J=12,0 Гц, 2Н), 1,58 (d, J=7,8 Гц, 2Н). MC (ИЭР) m/z (M+H)+ 477,2.
ГИДРОХЛОРИД N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4-(4-((ДИЭТИЛАМИНО)МЕТИЛ)ФЕНОКСИ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИДА (441)
[1094] Соединение 441 (14 мг, выход: 26,21%, белое твердое вещество) получали из этил-1-(4-гидроксифенил)-3-метил-1Н-пиразол-5-карбоксилата, который подвергали сочетанию с (4-((диэтиламино)метил)фенил)бороновой кислотой, как в случае соединения 209, с последующим сложноэфирным гидролизом и сочетанием с промежуточным соединением 274D с помощью методик, как в случае соединения 12, с получением соединения 441. Соединение 441: 1H ЯМР (400 МГц, ДМСО-d6) δ 11,55 (ушир. s, 1H), 7,77-7,71 (m, 2Н), 7,37-7,15 (m, 8H), 7,12-7,06 (m, 2Н), 6,99 (d, J=9,0 Гц, 2Н), 6,57 (s, 2Н), 6,13-5,96 (m, 1H), 4,72-4,63 (m, 1H), 4,23-4,13 (m, 2Н), 3,27-3,18 (m, 1H), 3,13-2,92 (m, 5H), 2,28 (s, 3H), 1,34 (t, J=3,5, 7,3 Гц, 6Н). MC (ИЭР) m/z (M+H)+ 554,3.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4-(2-(2-МЕТОКСИЭТОКСИ)ЭТОКСИ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (443)
[1095] Соединение 443 (75 мг, выход: 61,3%, белое твердое вещество) получали из этил-3-метил-1H-пиразол-5-карбоксилата, который подвергали сочетанию с (4-(бензилокси)фенил)бороновой кислотой, как в случае соединения 209, с последующим гидрогенолизом с получением фенольного производного, которое подвергали сочетанию Мицунобу с 2-(2-метоксиэтокси)этан-1-олом, с последующим сложноэфирным гидролизом и сочетанием с промежуточным соединением 274D с помощью методик, как в случае соединения 12, с получением соединения 443. Соединение 443: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,02 (ушир. d, J=7,8 Гц, 1Н), 8,10 (s, 1H), 7,86 (s, 1H), 7,37-7,20 (m, 5H), 7,08 (ушир. d, J=8,8 Гц, 2H), 6,89 (ушир. d, J=9,0 Гц, 2Н), 6,53 (s, 1H), 5,28-5,17 (m, 1H), 4,16-4,06 (m, 2H), 3,80-3,71 (m, 2H), 3,65-3,56 (m, 2H), 3,51-3,44 (m, 2H), 3,25 (s, 3H), 3,21-3,14 (m, 1H), 2,81 (ушир. dd, J=10,7, 13,7 Гц, 1H), 2,23 (s, 3H). МС (ИЭР) m/z (M+H)+ 495,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(3-(2-(2-МЕТОКСИЭТОКСИ)ЭТОКСИ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (444)
[1096] Соединение 444 (60 мг, выход: 39,58%, белое твердое вещество) получали из этил-3-метил-1H-пиразол-5-карбоксилата, который подвергали сочетанию с (3-(бензилокси)фенил)бороновой кислотой, как в случае соединения 209, с последующим гидрогенолизом с получением фенольного производного, которое подвергали сочетанию Мицунобу с 2-(2-метоксиэтокси)этан-1-олом, с последующим сложноэфирным гидролизом и сочетанием с промежуточным соединением 274D с помощью методик, как в случае соединения 12, с получением соединения 444. Соединение 444: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,07 (d, J=7,3 Гц, 1H), 8,09 (s, 1H), 7,85 (s, 1H), 7,35-7,18 (m, 6H), 6,93-6,81 (m, 2H), 6,71 (d, J=8,2 Гц, 1H), 6,54 (s, 1H), 5,28 (s, 1H), 4,05 (s, 2H), 3,72 (s, 2H), 3,57 (d, J=4,2 Гц, 2H), 3,46 (d, J=4,2 Гц, 2H), 3,24 (s, 3H), 3,18 (s, 1H), 2,88-2,77 (m, 1H), 2,24 (s, 3H). МС (ИЭР) m/z (М+Н)+ 495,2.
ПРИМЕР 111
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-5-(3-ФЕНИЛ-1Н-ПИРАЗОЛ-1-ИЛ)ИЗОКСАЗОЛ-4-КАРБОКСАМИД (211)
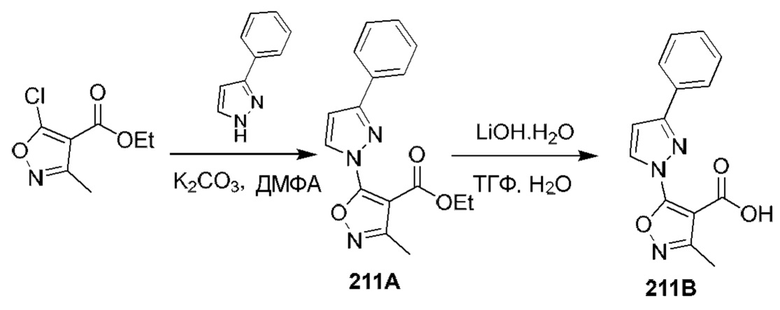
[1097] К смеси этил-5-хлор-3-метилизоксазол-4-карбоксилата (400 мг, 2,1 ммоль) и 3-фенил-1H-пиразола (365 мг, 2,5 ммоль) в ДМФА (3 мл) добавляли K2CO3 (1,2 г, 8,4 ммоль) одной порцией. Затем смесь перемешивали при 80°С в течение 12 часов. Затем к смеси добавляли H2O (9 мл), и водную фазу подвергали экстракции EtOAc (15 мл × 3), и объединенный органический слой концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 300:1 до 30:1). Соединение 211А (256 мг, выход: 40,8%, бледно-желтое твердое вещество): 1H ЯМР (400 МГц, CDCl3) δ 8,62 (d, J=2,6 Гц, 1Н), 7,93 (d, J=6,8 Гц, 2Н), 7,49-7,37 (m, 3H), 6,86 (d, J=2,6 Гц, 1Н), 4,37 (q, J=7,1 Гц, 2Н), 2,53 (s, 3H), 1,40-1,34 (m, 2Н), 1,41-1,33 (m, 1Н).
[1098] К раствору соединения 211А (150 мг, 504,5 мкмоль) в ТГФ (2 мл) и H2O (500 мкл) добавляли LiOH⋅H2O (31,8 мг, 756,8 мкмоль) одной порцией. Затем смесь перемешивали при 25°С в течение 3 часов. Анализ посредством ТСХ (петролейный эфир : этилацетат = 1:1, Rf ~ 0,45) свидетельствовал о том, что соединение 211А полностью израсходовалось, и на хроматограмме появилось одно новое основное пятно. рН водной фазы доводили примерно до 5 путем добавления НСl (1 M), и затем остаток концентрировали на роторном испарителе с получением промежуточного соединения 211В (92 мг, выход: 67,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,39 (ушир. s, 1Н), 7,92 (d, J=7,0 Гц, 2Н), 7,50-7,43 (m, 2Н), 7,42-7,36 (m, 1Н), 7,02 (s, 1Н), 2,51-2,51 (m, 3H). MC (ИЭР) m/z (M+H)+ 269,9.
[1099] Соединение 211 (20 мг, выход: 44,7%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 211В. Соединение 211: 1H ЯМР (400 МГц, CDCl3) δ 10,59 (ушир. d, J=6,8 Гц, 1Н), 8,19 (d, J=2,6 Гц, 1Н), 7,74-7,65 (m, 2H), 7,45-7,38 (m, 3H), 7,12-7,05 (m, 3H), 7,03-6,98 (m, 2H), 6,89 (d, J=2,4 Гц, 1Н), 6,72 (ушир. s, 1H), 5,73-5,64 (m, 1Н), 5,47 (ушир. s, 1Н), 3,38 (dd, J=5,3, 14,1 Гц, 1Н), 3,06 (dd, J=8,4, 13,9 Гц, 1Н), 2,56 (s, 3H). МС (ИЭР) m/z (M+H)+ 444,1.
ПРИМЕР 112
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(5-ФЕНИЛОКСАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (212)
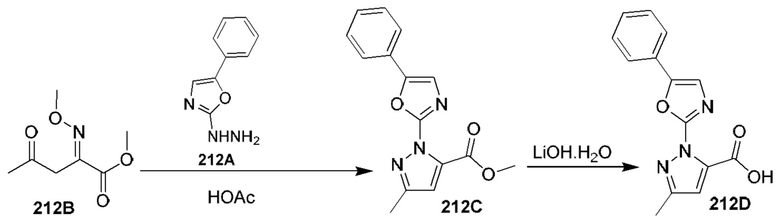
[1100] Раствор 2-хлор-5-фенилоксазола (соединение 114А) (560 мг, 3,12 ммоль) и NH2NH2⋅H2O (468 мг, 9,35 ммоль) в диоксане (10 мл) нагревали до кипения с обратным холодильником в течение 3 ч. Смесь концентрировали с получением соединения 212А (610 мг, неочищенного) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,27 (ушир. s, 1Н), 7,48 (ушир. d, J=7,9 Гц, 1Н), 7,36 (ушир. t, J=7,7 Гц, 2H), 7,29 (s, 1Н), 7,19 (ушир. t, J=7,4 Гц, 2H), 4,53-4,12 (m, 1Н), 4,35 (ушир. s, 1Н).
[1101] Раствор O-метилгидроксиламина (1,74 г, 20,8 ммоль) в H2O (20 мл) добавляли по каплям к раствору метил-2,4-диоксопентаноата (5 г, 34,7 ммоль) в этаноле (45 мл), H2O (25 мл), смесь перемешивали при 25°С в течение 12 ч. Органический растворитель удаляли под вакуумом, водный слой подвергали экстракции этилацетатом (20 мл × 2), объединенный органический слой промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали, остаток очищали посредством хроматографии на силикагеле с получением соединения 212В (3,3 г, выход: 54,9%), в виде желтого масла. 1H ЯМР (400 МГц, CDCl3-d) δ 4,06 (s, 3H), 3,87 (s, 3H), 3,72 (s, 2H), 2.21 (s. 2H).
[1102] Смесь соединения 212В (360 мг, 2,05 ммоль) и соединение 212А (355 мг, 2,05 ммоль) в диоксане (5 мл) нагревали до 110°С в течение 12 ч. Смесь концентрировали, остаток очищали посредством ТСХ (петролейный эфир : этилацетат = 5:1) с получением соединения 212С (140 мг, выход: 24,1%) в виде желтого масла.
[1103] Смесь соединения 212С (140 мг, 494 мкмоль) и LiOH⋅H2O (31,1 мг, 741 мкмоль) в ТГФ (5 мл), H2O (1 мл) перемешивали при 25°С в течение 2 ч. Анализ посредством ТСХ (петролейный эфир : этилацетат = 1:1, Rf ~ 0) свидетельствовал о завершении реакции. Органический растворитель удаляли при пониженном давлении, водный слой подвергали экстракции этилацетатом (3 мл), затем рН водного слоя доводили до ~ 3 с помощью 1 н. HCl с получением осадка, твердое вещество отфильтровывали и сушили с получением соединения 212D (100 мг, выход: 75,2%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,85 (s, 1H), 7,74-7,69 (m, 2H), 7,48 (t, J=7,7 Гц, 2H), 7,44-7,37 (m, 1H), 6,91 (s, 1H), 2,28 (s, 3Н).
[1104] Соединение 212 (34 мг, выход: 52,6%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 212D. Соединение 212: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,17 (ушир. d, J=7,5 Гц, 1H), 8,08 (ушир. s, 1H), 7,83 (ушир. s, 1H), 7,73 (s, 1H), 7,62 (ушир. d, J=7,5 Гц, 2H), 7,48-7,41 (m, 2H), 7,40-7,34 (m, 1H), 7,25 (s, 4H), 7,19 (ушир. d, J=4,0 Гц, 1H), 6,92 (s, 1H), 5,28 (ушир. d, J=7,7 Гц, 1H), 3,16 (ушир. dd, J=3,0, 14,0 Гц, 1H), 2,81 (ушир. dd, J=10,6, 13,7 Гц, 1H), 2,27 (s, 3Н). MC (ИЭР) m/z (M+H)+ 444,1.
ПРИМЕР 113
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ХЛОР-5-ФЕНИЛИЗОТИАЗОЛ-4-КАРБОКСАМИД (213)
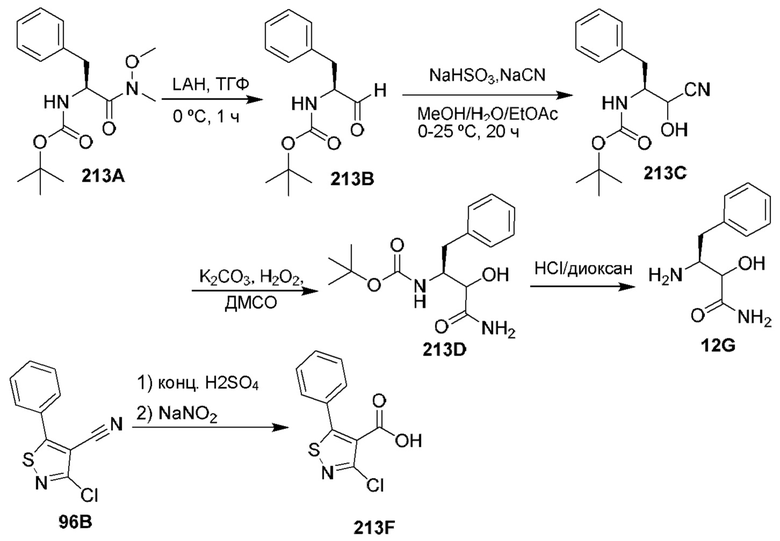
[1105] Смесь (трет-бутоксикарбонил)-L-фенилаланина (50 г, 188,47 ммоль), N-метоксиметанамина (20 г, 207,32 ммоль, HCl), NMM (57 г, 565,41 ммоль) и НОВТ (25 г, 188,47 ммоль) в CHCl3 (700 мл) дегазировали и продували N2 3 раза при 0°С, затем добавляли порциями EDCI (54 г, 282,71 ммоль). Смесь перемешивали при 25°С в течение 16 ч в атмосфере N2. В реакционную смесь добавляли H2O (500 мл) для гашения реакции. Органический слой отделяли, промывали 1 н. НСl (300 мл × 2), насыщенным водным NaHCO3 (300 мл × 3) и солевым раствором (300 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток растворяли в петролейном эфире (300 мл) и перемешивали в течение 2 ч, раствор фильтровали и концентрировали при пониженном давлении с получением соединения 213А (46 г, выход: 79,15%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,27-7,13 (m, 5H), 5,26-4,85 (m, 2H), 3,64 (s, 3H), 3,15 (ушир. s, 3Н), 3,04 (ушир. dd, J=5,8, 13,6 Гц, 1Н), 2,91-2,81 (m, 1Н), 1,37 (s, 9H). MC (ИЭР) m/z (M+23)+ 331,0.
[1106] К раствору LiAlH4 (5,32 г, 140,09 ммоль) в ТГФ (1 л) добавляли раствор соединения 213А (36 г, 116,74 ммоль) в ТГФ (500 мл) при 0°С. После добавления реакционную смесь перемешивали в течение 1 ч при 0°С. В реакционную смесь добавляли для гашения реакции этилацетат (200 мл) и 1 н. HCl (200 мл), и затем смесь подвергали экстракции EtOAc (300 мл × 3). Объединенные органические слои промывали 1 н. HCl (300 мл × 2), насыщенным водным NaHCO3 (300 мл × 3) и солевым раствором (300 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 213В (25,3 г, выход: 86,93%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,61 (s, 1H), 7,33-7,24 (m, 3H), 7,16 (ушир. d, J=7,1 Гц, 2Н), 5,16-5,03 (m, 1H), 4,40 (q, J=6,6 Гц, 1H), 3,10 (ушир. d, J=5,3 Гц, 2Н), 1,42 (s, 9H).
[1107] К раствору соединения 213В (32 г, 128,36 ммоль) в МеОН (250 мл) добавляли по каплям раствор NaHSO3 (13,5 г) в H2O (400 мл) при 0-5°С. После этого реакционную смесь перемешивали при 28°С в течение 5 ч. В реакционную смесь добавляли NaCN (6,6 г, 134,78 ммоль) в H2O (600 мл), затем EtOAc (1,2 л). После этого реакционную смесь перемешивали при 28°С в течение 14 ч. Смесь разделяли, и органический слой промывали солевым раствором (500 мл). Смесь сушили над Na2SO4 и концентрировали с получением соединения 213С (35 г, выход: 98,68%) в виде светло-желтой смолы. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,26-7,14 (m, 6H), 6,82-6,70 (m, 1H), 4,57-4,28 (m, 1H), 3,88-3,72 (m, 1H), 3,01-2,58 (m, 2Н), 1,31-1,22 (m, 9H). MC (ИЭР) m/z (M-55)+ 220,9.
[1108] К раствору соединения 213С (35 г, 126,66 ммоль) и K2CO3 (35 г, 253,32 ммоль) в ДМСО (400 мл) добавляли H2O2 (148 г, 4,35 моль) при 0°С.После добавления реакционную смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь фильтровали с получением остатка. Остаток промывали насыщенным водным Na2S2O3 (100 мл × 2) и H2O (100 мл), растворяли в толуоле (200 мл), концентрировали при пониженном давлении для удаления H2O. К фильтрату медленно добавляли для гашения реакции насыщенный водный Na2SO3 при 0°С. Смесь подвергали экстракции EtOAc (200 мл × 3), и объединенные экстракты промывали насыщенным водным Na2SO3 (300 мл × 3), солевым раствором (200 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 213D (37,5 г, выход: 88,51%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,29-7,16 (m, 7H), 6,64-6,09 (m, 1H), 5,74-5,61 (m, 1H), 4,09-3,67 (m, 2Н), 2,86-2,56 (m, 2Н), 1,36-1,22 (m, 9H). MC (ИЭР) m/z (M-100+H)+ 194,9.
[1109] К раствору соединения 213D (41 г, 139,29 ммоль) в EtOAc (300 мл) добавляли HCl/EtOAc (4 М, 696,45 мл) при 0°С. После добавления реакционную смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь фильтровали с получением остатка. Остаток промывали этилацетатом (30 мл), концентрировали при пониженном давлении с получением соединения 12G (26 г, выход: 66,35%, НС1) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,42-7,95 (m, 3Н), 7,60-7,43 (m, 2H), 7,37-7,12 (m, 6H), 4,38-3,79 (m, 2H), 3,73-3,62 (m, 1H), 3,03-2,73 (m, 2H). MC (ИЭР) m/z (М+Н)+ 195,1.
[1110] К соединению 96В (2,00 г, 9,06 ммоль), помещенному в круглодонную колбу объемом 100 мл, добавляли по каплям H2SO4 (27,60 г, 281,40 ммоль) и перемешивали при 135°С в течение 1 ч. Затем в смесь добавляли NaNO2 (907 мг, 13,14 ммоль) в H2O (10 мл) по каплям при 0°С. Полученный раствор перемешивали при 50°С в течение 0,5 ч. Реакционную смесь разбавляли H2O (50 мл) и подвергали экстракции этилацетатом (50 мл × 3). Объединенные органические слои were подвергали экстракции 10% NaOH (50 мл × 2). рН водной фазы доводили до 3 с помощью 1 н. HCl, водную фазу подвергали экстракции этилацетатом (50 мл × 3). Объединенные органические слои промывали солевым раствором (50 мл), сушили с помощью Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 213F (1,17 г, выход: 49,13%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,55 (d, J=1,3 Гц, 5Н). MC (ИЭР) m/z (M+H)+ 239,9.
[1111] Соединение 213 (65 мг, выход: 40,23%, серое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединения 12G и 213F. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,35 (d, J=7,3 Гц, 1H), 8,19 (ушир. s, 1H), 7,91 (ушир. s, 1H), 7,51-7,32 (m, 5Н), 7,27-7,17 (m, 5Н), 5,71-5,21 (m, 1H), 3,17 (dd, J=3,4, 14,2 Гц, 1H), 2,72 (dd, J=10,4, 14,1 Гц, 1H). MC (ИЭР) m/z (М+Н)+ 414,0.
ПРИМЕР 114
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (215)
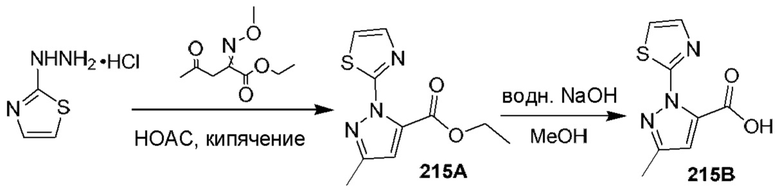
[1112] Смесь гидрохлорида 2-гидразинилтиазола (600 мг, 3,9 ммоль, гидрохлорид) и этил-2-(метоксиимино)-4-оксопентаноата (815 мг, 4,35 ммоль) в АсОН (15 мл) перемешивали при 110°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении для удаления АсОН. Остаток обрабатывали H2O (50 мл) и этилацетатом (50 мл), и затем смесь подкисляли с помощью насыщенного водного NaHCO3 до рН водной фазы ~ 7-8. Отделенный водный слой подвергали экстракции этилацетатом (80 мл × 3), объединенные органические слои промывали насыщенным водным NaCl (100 мл), сушили над Na2SO4, фильтровали при пониженном давлении с получением неочищенного продукта, который очищали посредством колоночной флэш-хроматографии (петролейный эфир : этилацетат = 1~9) с получением соединения 215А (138 мг, выход 12,6%) в виде белого твердого вещества. Соединение 215А: 1H ЯМР (ДМСО-d6, 400 МГц) δ 7,71-7,69 (m, 1Н), 7,66 (d, J=3,5 Гц, 1Н), 6,87 (s, 1H), 4,23 (q, J=7,1 Гц, 2Н), 2,26 (s, 3Н), 1,20-1,15 (m, 3Н). МС (ИЭР) m/z (M+H)+ 237,9.
[1113] К смеси соединения 215А (130 мг, 0,55 ммоль) в МеОН (10 мл) добавляли NaOH (2 M, 1,4 мл) одной порцией при 25°С. После перемешивания при 25°С в течение 1,5 ч реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток добавляли H2O (10 мл) и подвергали экстракции этилацетатом (10 мл), отделенную водную фазу подкисляли с помощью 1 M HCl до рН ~ 5-6. Твердое вещество отделяли и отфильтровывали при пониженном давлении с получением соединения 215В (70 мг, неочищенного) в виде белого твердого вещества. Соединение 215В: 1H ЯМР (CDCl3, 400 МГц) δ 7,56 (d, J=3,7 Гц, 1Н), 7,21-7,17 (m, 2Н), 2,37 (s, 3Н).
[1114] Соединение 215 (10 мг, выход 53,7%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 215В. Соединение 215: 1H ЯМР (CDCl3, 400 МГц) δ 11,76 (s, 1H), 7,27-7,18 (m, 5H), 7,17-7,12 (m, 1H), 7,10-7,00 (m, 2H), 6,78 (s, 1H), 5,83-5,74 (m, 1H), 5,50 (s, 1H), 3,48-3,40 (m, 1H), 3,28-3,18 (m, 1H), 2,32 (s, 3Н). МС (ИЭР) m/z (M+H)+ 384,0.
ПРИМЕР 115
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-1-(ТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (219)
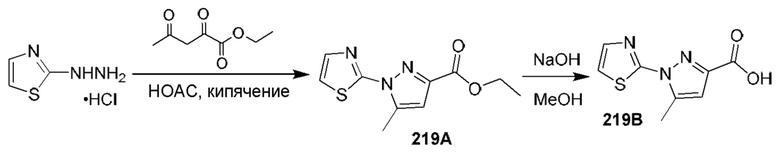
[1115] К раствору гидрохлорида 2-гидразинилтиазола (700 мг, 4,39 ммоль, гидрохлорид) в СН3СООН (15 мл) добавляли по каплям этил-2,4-диоксопентаноат (632 мкл, 4,48 ммоль), затем смесь нагревали до 120°С и перемешивали в течение 2 ч. Растворитель удаляли при пониженном давлении, остаток растворяли в этилацетате (5 мл) и обрабатывали NaHCO3 до рН ~ 8. Органический слой собирали и упаривали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии (петролейный эфир/этилацетат = от 1/0 до 10/1) с получением соединения 219А (160 мг, выход 15,4%) в виде белого твердого вещества. Соединение 219А: 1H ЯМР (CDCl3, 400 МГц) δ 7,60 (d. J=3,5 Гц, 1H), 7,18 (d, J=3,5 Гц, 1H), 6,70 (d, J=0,9 Гц, 1H), 4,42 (q, J=7,1 Гц, 2H), 2,74 (d, J=0,9 Гц, 3Н), 1,42 (t, J=7,1 Гц, 3Н).
[1116] К раствору соединения 219А (160 мг, 674,31 мкмоль) в МеОН (10 мл) добавляли по каплям NaOH (2 M, 2,00 мл), и затем смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь разбавляли H2O (5 мл), упаривали при пониженном давлении, и затем водную фазу подвергали экстракции МТБЭ (5 мл). Водную фазу (подкисленную с помощью HCl, рН ~ 3) подвергали экстракции этилацетатом (10 мл × 3), затем органический слой (этилацетат) собирали, промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и фильтровали, концентрировали при пониженном давлении. Соединение 219В (110 мг, выход 78%, белое твердое вещество): 1H ЯМР (ДМСО-d6, 400 МГц) δ 7,72 (d, J=3,5 Гц, 1H), 7,64 (d, J=3,5 Гц, 1H), 6,77 (d, J=0,7 Гц, 1H), 2,67 (s, 3Н).
[1117] Соединение 219 (15 мг, выход 31,8%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 219В. Соединение 219: 1H ЯМР (ДМСО-d6, 400 МГц) δ 7,58 (d, J=3,5 Гц, 1Н), 7,37-7,26 (m, 3Н), 7,26-7,24 (m, 1H), 7,22-7,15 (m, 3Н), 6,78 (ушир. s, 1H), 6,65 (s, 1H), 5,73-5,64 (m, 1H), 5,58 (ушир. s, 1H), 3,43 (dd, J=5,4, 14,0 Гц, 1H), 3,26 (dd, J=6,9, 14,2 Гц, 1H), 2,71 (s, 3Н). MC (ИЭР) m/z (M+H)+ 384,1.
ПРИМЕР 116
(2S,4R)-N-((S)-4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(БЕНЗИЛОКСИ)-1-ФЕНИЛПИРРОЛИДИН-2-КАРБОКСАМИД (220)
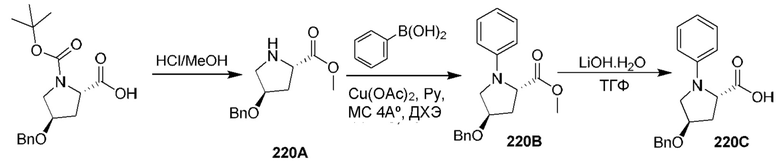
[1118] Смесь (2S,4R)-4-(бензилокси)-1-(трет-бутоксикарбонил)пирролидин-2-карбоновой кислоты (500 мг, 1,56 ммоль) в МеОН (3 мл), HCl/MeOH (15 мл) перемешивали при 20°С в течение 12 часов. Анализ посредством ЖХМС свидетельствовал о том, что исходный материал полностью израсходовался, и был обнаружен один пик, которому соответствовал MC целевого соединения. Реакционную смесь разбавляли водным NaHCO3, доводили рН до ~ 7 и подвергали экстракции ДХМ (30 мл × 2). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Соединение 220А (340 мг, неочищенное, желтое масло): 1H ЯМР (400 МГц, CDCl3) δ 7,37-7,20 (m, 5H), 4,52-4,41 (m, 2H), 4,12 (ушир. s, 1H), 4,00 (ушир. t, J=7,6 Гц, 1H), 3,77-3,62 (m, 3Н), 3,11 (ушир. s, 2H), 2,66 (ушир. s, 1H), 2,29 (ушир. dd, J=7,8, 13,1 Гц, 1H), 1,98 (td, J=6,6, 13,4 Гц, 1H). MC (ИЭР) m/z (М+Н)+ 236,1.
[1119] Смесь соединения 220А (240 мг, 1,02 ммоль), фенилбороновой кислоты (249 мг, 2,04 ммоль), Cu(ОАс)2 (278 мг, 1,53 ммоль), пиридина (161 мг, 2,04 ммоль) и MC 4А° (400 мг) в ДХЭ (20 мл) дегазировали и продували O2 3 раза, и затем смесь перемешивали при 60°С в течение 12 часов в атмосфере O2. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 50:1 до 10:1). Соединение 220В (130 мг, желтое масло): 1H ЯМР (400 МГц, CDCl3) δ 7,36-7,16 (m, 5H), 7,16-7,06 (m, 2H), 6,68-6,57 (m, 1H), 6,42 (d, J=8,1 Гц, 2H), 4,45 (s, 2H), 4,39-4,25 (m, 2H), 3,77-3,67 (m, 1H), 3,66-3,56 (m, 3H), 3,34 (dd, J=4,4, 9,5 Гц, 1H), 2,42-2,28 (m, 1H), 2,28-2,15 (m, 1H). MC (ИЭР) m/z (M+H)+ 312,0.
[1120] Смесь соединения 220В (130 мг, 418 мкмоль), LiOH⋅H2O (26,3 мг, 626 мкмоль) в ТГФ (5 мл), H2O (2 мл) перемешивали при 20°С в течение 12 часов. В реакционную смесь добавляли водный раствор HCl для доведения рН до ~ 5, и затем смесь фильтровали, и осадок на фильтре сушили путем лиофилизации. Соединение 220С (130 мг, неочищенное, белое твердое вещество): 1H ЯМР (400 МГц, ДМСО-d6) δ 7,36-7,15 (m, 6H), 7,13-6,98 (m, 2H), 6,57-6,47 (m, 1H), 6,40 (ушир. d, J=7,9 Гц, 2H), 4,48-4,40 (m, 2H), 4,28 (ушир. s, 1H), 4,15-4,05 (m, 1H), 3,29-3,26 (m, 1H), 3,27-3,17 (m, 1H), 2,33-2,22 (m, 1H), 2,21-2,09 (m, 1H). MC (ИЭР) m/z (M+H)+ 298,2.
[1121] Соединение 220 (18,6 мг, выход: 70,3%, коричневое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 220С. Соединение 220: 1H ЯМР (400 МГц, CDCl3) δ 7,33-7,07 (m, 11H), 6,93 (ушир. s, 2H), 6,84-6,70 (m, 2H), 6,59 (ушир. s, 1H), 6,49 (ушир. d, J=8,4 Гц, 2H), 5,35 (ушир. s, 1H), 5,30-5,13 (m, 1H), 4,50-4,29 (m, 2H), 4,01 (ушир. dd, J=4,2, 9,0 Гц, 1H), 3,95-3,85 (m, 1H), 3,62 (dd, J=5,8, 8,9 Гц, 1H), 3,30 (ушир. dd, J=5,0, 14,0 Гц, 1H), 3,17 (dd, J=6,4, 9,0 Гц, 1H), 2,80 (dd, J=9,0, 13,9 Гц, 1H), 2,31-2,17 (m, 1H), 2,16-2,03 (m, 1H). MC (ИЭР) m/z (М+Н)+ 472,2.
ПРИМЕР 117
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ФЕНИЛ-1H-ИМИДАЗОЛ-2-КАРБОКСАМИД (221)
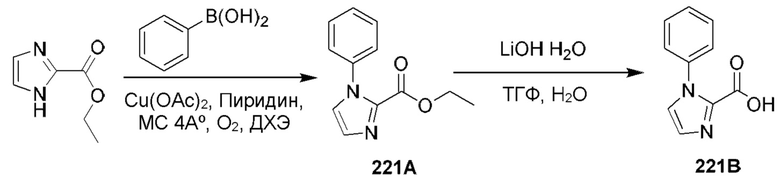
[1122] К смеси этил-1H-имидазол-2-карбоксилата (5 г, 35,7 ммоль) и фенилбороновой кислоты (8,7 г, 71,4 ммоль) в ДХЭ (150 мл) добавляли Cu(ОАс)2 (7,13 г, 39.25 ммоль), пиридин (5,64 г, 71,36 ммоль, 5,76 мл), MC 4А° (3 г). Смесь перемешивали при 60°С в течение 16 часов в атмосфере O2. Реакционную смесь фильтровали, и фильтрат концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле, элюируя с помощью смеси петролейный эфир : этилацетат = 10:1, 4:1, с получением соединения 221А (3 г, 13,9 ммоль, выход: 38,9%) в виде желтого твердого вещества. Соединение 221А: 1H ЯМР (400 МГц, CDCl3-d) δ 7,43-7,39 (m, 3H), 7,27-7,23 (m, 2Н), 7,22-7,19 (m, 1H), 7,11 (d, J=1,0 Гц, 1Н), 4,22 (q, J=7,2 Гц, 2Н), 1,24 (t, J=7,2 Гц, 3H).
[1123] К смеси соединения 221А (300 мг, 1,39 ммоль) в ТГФ (3 мл) и H2O (1 мл) добавляли LiOH⋅H2O (52 мг, 1,25 ммоль). Смесь перемешивали при 25°С в течение 12 часов. Остаток подвергали экстракции этилацетатом (5 мл × 2). рН смеси доводили до ~ 5 с помощью водного раствора HCl (1 M), и смесь концентрировали путем лиофилизации с получением промежуточного соединения 221В (550 мг, неочищенного) в виде белого твердого вещества.
[1124] Соединение 221 (17,1 мг, выход: 30,9%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 221В.
[1125] Соединение 221: 1H ЯМР (400 МГц, CDCl3) δ 7,76 (ушир. d, J=7,9 Гц, 1H), 7,35 (ушир. d, J=2,6 Гц, 3H), 7,24-7,16 (m, 5H), 7,12 (ушир. d, J=7,1 Гц, 2Н), 7,08 (s, 1H), 7,04 (s, 1H), 6,67 (ушир. s, 1H), 5,60-5,47 (m, 2Н), 3,32 (dd, J=4,9, 13,9 Гц, 1H), 3,08 (dd, J=7,4, 14,0 Гц, 1H). МС (ИЭР) m/z (M+H)+ 363,1.
ПРИМЕР 118
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-2-ФЕНИЛТИОФЕН-3-КАРБОКСАМИД (222)
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-(2H-ИНДАЗОЛ-2-ИЛ)-5-МЕТИЛТИОФЕН-3-КАРБОКСАМИД (428) и N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-(1H-БЕНЗО[d]ИМИДАЗОЛ-2-ИЛ)-5-МЕТИЛТИОФЕН-3-КАРБОКСАМИД (429)
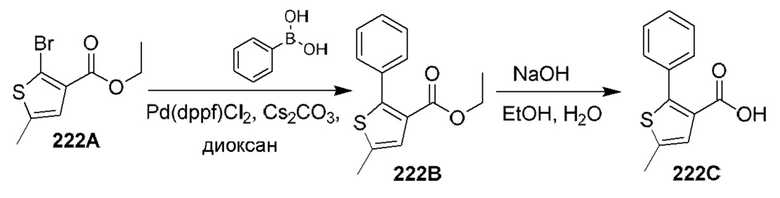
[1126] Смесь этил-2-амино-5-метилтиофен-3-карбоксилата (9 г, 48,6 ммоль) и CuBr2 (13 г, 58,3 ммоль) в MeCN (150 мл) перемешивали при 0°С-5°С. Добавляли по каплям т-BuONO (5,5 г, 53,5 ммоль). Реакционную смесь перемешивали в течение 0,5 часа при 0-5°С и 2 часа при 20°С. Реакционную смесь разбавляли EtOAc (400 мл), промывали водой (100 мл) и солевым раствором (100 мл), сушили над MgSO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = 100:1) с получением соединения 222А (2 г, выход: 16,5%) в виде желтого масла. Соединение 222А: 1H ЯМР (400 МГц, CDCl3-d) δ 7,01 (s, 1Н), 4,30 (q, J=7,1 Гц, 2Н), 2,38 (s, 3Н), 1,40-1,29 (m, 3H).
[1127] К смеси соединения 222А (400 мг, 1,61 ммоль) и фенилбороновой кислоты (393 мг, 3,22 ммоль), Cs2CO3 (1,05 г, 3,22 ммоль) в диоксане (20 мл) и H2O (2 мл) добавляли Pd(dppf)Cl2 (118 мг, 161 мкмоль) в атмосфере N2. Смесь перемешивали при 110°С в течение 12 часов в атмосфере N2. Реакционную смесь фильтровали, и фильтрат концентрировали. Остаток очищали посредством препаративной ТСХ (SiO2, петролейный эфир : этилацетат = 5:1) с получением соединения 222В (350 мг, выход: 88,3%) в виде белого твердого вещества. Соединение 222В: 1H ЯМР (400 МГц, CDCl3) δ 7,48 (ушир. s, 2Н), 7,38 (ушир. s, 3Н), 7,19 (ушир. s, 1Н), 4,19 (q, J=6,9 Гц, 2Н), 2,50 (ушир. s, 3Н), 1,23-l,15 (m, 3H).
[1128] К смеси соединения 222В (350 мг, 1,42 ммоль) в EtOH (10 мл) и H2O (5 мл) добавляли NaOH (142 мг, 3,55 ммоль). Смесь перемешивали при 80°С в течение 3 часов. Смесь концентрировали для удаления растворителя и доводили рН до ~ 5 с помощью водного раствора HCl (1 M). Смесь фильтровали, и твердое вещество промывали H2O (3 мл) с получением промежуточного соединения 222С (250 мг, выход: 81,0%) в виде белого твердого вещества. Соединение 222С: 1H ЯМР (400 МГц, ДМСО-d6) δ 12,47 (ушир. s, 1Н), 7,43-7,38 (m, 2Н), 7,37-7,32 (m, 3Н), 7,10 (d, J=0,9 Гц, 1Н), 2,41 (s, 3Н).
[1129] Соединение 222 (15,7 мг, выход: 29,6%, белое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 222С. Соединение 222: 1H ЯМР (400 МГц, CDCl3) δ 7,36-7,30 (m, 4H), 7,19 (s, 1Н), 7,13-7,07 (m, 3Н), 6,97 (s, 1Н), 6,71-6,60 (m, 3Н), 5,89 (ушир. d, J=5,3 Гц, 1Н), 5,49-5,34 (m, 2Н), 3,10 (dd, J=5,0, 14,0 Гц, 1Н), 2,81 (dd, J=7,9, 13,9 Гц, 1Н), 2,37 (s, 3Н). МС (ИЭР) m/z (М+H)+ 393,1.
[1130] Соединение 428 (44,7 мг, выход: 40,5%, белое твердое вещество) получали с использованием промежуточного соединения 222А для синтеза промежуточной карбоновой кислоты, 2-(2H-индазол-2-ил)-5-метилтиофен-3-карбоновой кислоты, которую превращали в соединение 428 с помощью методик, как в случае соединения 12. Соединение 428: 1H ЯМР (400 МГц, CDCl3) δ 9,17 (ушир. d, J=5,6 Гц, 1Н), 8,25 (s, 1Н), 7,70 (ушир. d, J=8,3 Гц, 1Н), 7,61 (ушир. d, J=8,7 Гц, 1Н), 7,34 (ушир. t, J=7,5 Гц, 1Н), 7,21-7,13 (m, 2H), 7,00 (ушир. s, 3Н), 6,85 (ушир. s, 2Н), 6,69 (ушир. s, 1Н), 5,69-5,58 (m, 1Н), 5,43 (ушир. s, 1Н), 3,27 (ушир. dd, J=4,8, 14,0 Гц, 1Н), 2,95 (ушир. dd, J=7,3, 14,1 Гц, 1Н), 2,48 (s, 3Н). MC (ИЭР) m/z (M+H)+ 433,1.
[1131] Соединение 429 (31,6 мг, выход: 35,7%, желтое твердое вещество) получали с использованием промежуточного соединения 222А для синтеза промежуточной карбоновой кислоты, 5-метил-2-(1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-2-ил)тиофен-3-карбоновой кислоты, которую превращали в соединение 429 с помощью методик, как в случае соединения 12. Соединение 429: 1H ЯМР (400 МГц, CDCl3) δ 12,80 (ушир. s, 1Н), 10,05 (ушир. d, J=6,8 Гц, 1Н), 8,18 (ушир. s, 1H), 7,85 (ушир. s, 1Н), 7,66-7,57 (m, 2H), 7,32-7,19 (m, 7H), 7,17-7,11 (m, 1Н), 5,44 (ушир. s, 1Н), 3,27 (ушир. s, 1Н), 3,03-2,93 (m, 1Н). MC (ИЭР) m/z (M+H)+ 433,1.
ПРИМЕР 119
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-МЕТИЛ-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (223)
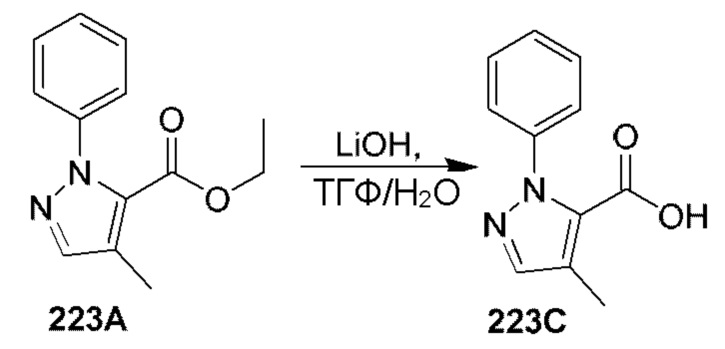
[1132] К смеси этил 4-метил-1Н-пиразол-5-карбоксилата (2,00 г, 12,97 ммоль), фенилбороновой кислоты (2,37 г, 19,45 ммоль), Ру (1,13 г, 14,27 ммоль, 1,2 мл) в ДХМ (40,00 мл) добавляли MC 4А° (10,00 г) (активированное MC 4А°) и Cu(ОАс)2 (2,59 г, 14,27 ммоль), смесь перемешивали при 40°С в течение 63 ч. Реакционную смесь фильтровали, фильтрат концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле (ПЭ:ЭА=1:0 до 5:1) с получением соединения 223А (725 мг, выход: 24,3%) получали в виде бесцветного масла. Соединение 223А: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,44 (s, 1Н), 7,83 (d, J=7,7 Гц, 2Н), 7,52 (t, J=7,9 Гц, 2Н), 7,43-7,32 (m, 1H), 4,31 (q, J=7,1 Гц, 2Н), 2,27 (s, 3Н), 1,32 (t, J=7,1 Гц, 3Н).
[1133] К раствору соединения 223А (720 мг, 3,13 ммоль) в ТГФ (20,00 мл) добавляли LiOH⋅H2O (700 мг, 16,68 ммоль) в H2O (6,00 мл). Реакционную смесь перемешивали при 25°С в течение 27 ч и затем раствор NaOH (626 мг, 15,65 ммоль) в H2O (5,00 мл) и МеОН (4,00 мл) добавляли к смеси. Смесь перемешивали при 25°С в течение 3,5 ч. Реакционную смесь разбавляли H2O (25 мл) и подвергали экстракции МТБЭ (15 мл). рН водных слоев доводили до ~ 3 путем добавления 1 н. HCl, и затем водный слой подвергали экстракции ЭА (20 мл × 3). Объединенный органический слой промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 223С (526 мг, выход: 83,1%) получали в виде белого твердого вещества. Соединение 223С: 1H ЯМР (400 МГц, ДМСО-d6) δ 7,63 (s, 1H), 7,51-7,28 (m, 5H), 2,25 (s, 3Н).
[1134] Соединение 223 (34 мг, выход: 68,3%, белое твердое вещество) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 223С. Соединение 223: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,18 (d, J=7,8 Гц, 1H), 8,21 (s, 1H), 7,94 (s, 1H), 7,54 (s, 1H), 7,37-7,22 (m, 10Н), 5,46-5,36 (m, 1H), 3,26 (ушир. dd, J=3,0, 13,8 Гц, 1H), 2,78 (dd, J=11,2, 13,9 Гц, 1H), 2,00-1,93 (m, 3Н).
ПРИМЕР 120
СОЕДИНЕНИЯ 224-225
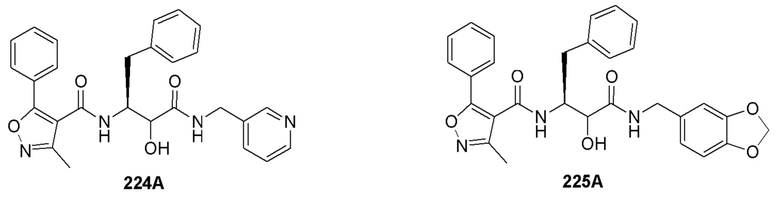
(S)-N-(3,4-ДИОКСО-1-ФЕНИЛ-4-((ПИРИДИН-3-ИЛМЕТИЛ)АМИНО)БУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (224)
(S)-N-(4-((БЕНЗО[D][1,3]ДИОКСОЛ-5-ИЛМЕТИЛ)АМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (225)
[1135] К раствору соединения 101Е (350,0 мг, 920,11 мкмоль) в ДМФА (10 мл) добавляли 3-пиридилметанамин (119,4 мг, 1,10 ммоль, 110 мкл), DIEA (0,5 мл), HOBt (124,33 мг, 920,11 мкмоль) и EDCI (211,66 мг, 1,10 ммоль). После перемешивания при 25°С в течение 48 ч, смесь добавляли HBTU (418,7 мг, 1,10 ммоль) и DIEA (0,5 мл), и затем перемешивали при 25°С в течение 12 ч. Смесь разбавляли H2O (100 мл), подвергали экстракции ЭА (30 мл), промывали HCl (1 М, 30 мл), насыщенным NaHCO3 (водн, 30 мл), солевым раствором (30 мл), сушили над Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1 до 0:1) с получением соединения 224А.
[1136] Соединение 224А (60,0 мг, выход 13,9%, белое твердое вещество): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,67-8,50 (m, 1Н), 8,48-8,45 (m, 1H), 8,42-8,37 (m, 1H), 8,30-8,24 (m, 1H), 7,73-7,33 (m, 6H), 7,31-7,09 (m, 6H), 6,14-5,86 (m, 1H), 4,69-4,55 (m, 1H), 4,36-4,14 (m, 2H), 4,08-4,01 (m, 1H), 2,97-2,87 (m, 1H), 2,77-2,66 (m, 1H), 2,08-1,96 (m, 3H). МС (ИЭР) m/z (М+Н)+ 471,2.
[1137] Соединение 225А (130,0 мг, 27,5% выход, белое твердое вещество) было синтезировано, как описано выше для 224А, из соответствующего амина. Соединение 225А: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,35-8,28 (m, 1H), 8,26-8,21 (m, 1H), 7,57-7,51 (m, 2H), 7,48-7,42 (m, 1H), 7,41-7,35 (m, 2H), 7,29-7,12 (m, 5H), 6,89-6,79 (m, 1H), 6,79-6,68 (m, 2H), 5,94-5,88 (m, 2H), 5,87-5,81 (m, 1H), 4,66-4,57 (m, 1H), 4,23-4,09 (m, 2H), 4,04-3,99 (m, 1H), 4,04-3,99 (m, 1H), 2,95-2,86 (m, 1H), 2,78-2,66 (m, 1H), 2,07-1,98 (m, 3H). МС (ИЭР) m/z (M+H)+ 514,2.
[1138] К раствору соединения 225А (120,0 мг, 233,67 мкмоль) в ДМСО (10 мл) и ДХМ (1 мл) добавляли DMP (297,3 мг, 701,01 мкмоль). После перемешивания при 25°С в течение 1 часа в смесь добавляли для гашения реакции 10% Na2S2O3 (водный): насыщенный водный NaHCO3 (1:1, 50 мл), органический слой промывали солевым раствором (50 мл × 3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Неочищенный продукт растирали с CH3CN (5 мл) и фильтровали с получением соединения 225 (62,0 мг, выход 51,9%) в виде желтого твердого вещества. Соединение 225: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,41-9,32 (m, 1H), 9,10 (d, J=7,6 Гц, 1H), 7,66-7,62 (m, 2H), 7,53-7,48 (m, 1H), 7,46-7,40 (m, 2H), 7,33-7,23 (m, 5H), 6,89-6,83 (m, 2H), 6,79-6,76 (m, 1H), 5,98 (s, 2H), 5,54-5,47 (m, 1H), 4,27 (d, J=6,4 Гц, 2Н), 3,30-3,23 (m, 1H), 2,83-2,74 (m, 1H), 2,09-2,06 (m, 3Н). МС (ИЭР) m/z (М+H)+ 512,2.
[1139] Соединение 224 было синтезировано из соответствующего промежуточного соединения 224А, как описано выше для соединения 225. Соединение 224 (25,0 мг, выход 50,2%) было получено в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,53-9,45 (m, 1H), 9,11 (d, J=7,6 Гц, 1H), 8,54 (s, 1H), 8,49-8,46 (m, 1H), 7,72-7,67 (m, 1H), 7,67-7,62 (m, 2H), 7,54-7,48 (m, 1H), 7,46-7,40 (m, 2H), 7,38-7,33 (m, 1H), 7,32-7,23 (m, 5H), 5,54-5,47 (m, 1H), 4,40 (d, J=6,3 Гц, 2H), 3,30-3,23 (m, 1H), 2,83-2,75 (m, 1H), 2,09-2,05 (m, 3Н). МС (ИЭР) m/z (M+H)+ 469,1.
ПРИМЕР 121
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-ЭТИЛ-4-ФЕНИЛОКСАЗОЛ-5-КАРБОКСАМИД (226)
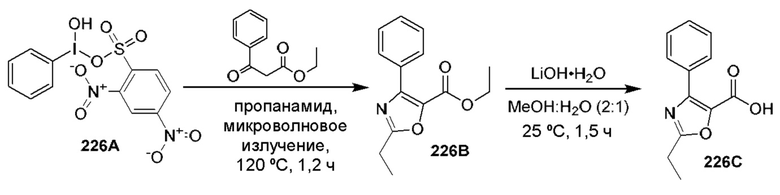
[1140] К смеси иодбензола (5 г, 24,51 ммоль) и 2,4-динитробензолсульфоновая кислота (7,83 г, 29,41 ммоль, H2O) в CHCl3 (20 мл), добавляли m-СРВА (4,23 г, 24,51 ммоль). Смесь перемешивали в течение 2 часов при 25°С в атмосфере N2. После завершения реакции в реакционную смесь добавляли МТБЭ (20 мл), и полученную смесь фильтровали и твердое вещество промывали МТБЭ (30 мл) и соединение 226А (8,9 г, выход 77,6%) получали в виде белого твердого вещества. Соединение 226А: 1H ЯМР (CDCl3, 400 МГц): δ 9,76 (ушир. s, 1H), 8,57 (d, J=2,4 Гц, 1H), 8,43-8,40 (m, 1H), 8,23 (d, J=7,6 Гц, 2H), 8,10 (d, J=8,4 Гц, 1H), 7,75-7,69 (m, 1H), 7,66-7,59 (m, 2H).
[1141] Этил-3-оксо-3-фенилпропаноат (1,3 г, 6,76 ммоль) и соединение 226А (4,12 г, 8,79 ммоль) в CH3CN (50 мл) перемешивали при 80°С в течение 1 ч, и пропанамид (5,93 г, 81,1 ммоль) добавляли к смеси, затем смесь перемешивали при 120°С в течение 0,2 часа под действием микроволнового излучения. После охлаждения до 25°С, суспензию разбавляли насыщенным NaHCO3 раствор (30 мл), подвергали экстракции EtOAc (100 мл × 2), сушили над Na2SO4, фильтровали, и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 10/1) с получением соединения 226В (200 мг, 11,22% выход) в виде белого твердого вещества. Соединение 226В: 1H ЯМР (CDCl3, 400 МГц): δ 8,03-7,96 (m, 2Н), 7,46-7,35 (m, 3Н), 4,36 (q, J=7,2 Гц, 2Н), 2,88 (q, J=7,6 Гц, 2Н), 1,40 (t, J=7,6 Гц, 3Н), 1,35 (t, J=7,2 Гц, 3Н).
[1142] Соединение 226С (170 мг, 93,71% выход, желтое твердое вещество) было получено, как описано в примере 51, из соответствующего промежуточного соединения 226В. Соединение 226С: 1H ЯМР (CDCl3, 400 МГц): δ 7,99-7,97 (m, 2Н), 7,47-7,35 (m, 3Н), 2,83 (q, J=7,6 Гц, 2Н), 1,27 (t, J=7,6 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 217,9.
[1143] Соединение 226 (59,7 мг, 58,17% выход, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, соединение 226С. Соединение 226: 1H ЯМР (CDCl3, 400 МГц): δ 8,12-8,04 (m, 2Н), 7,45-7,35 (m, 3Н), 7,32-7,23 (m, 3Н), 7,13 (d, J=6,4 Гц, 2Н), 6,81-6,68 (m, 2Н), 5,74-5,59 (m, 2Н), 3,46-3,41 (m, 1H), 3,26-3,21 (m, 1H), 2,91-2,80 (m, 2Н), 1,40 (t, J=7,6 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 392,1.
ПРИМЕР 122
СОЕДИНЕНИЯ 268-269
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-4-ФЕНИЛ-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (268)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-ФЕНИЛ-1H-ИМИДАЗОЛ-4-КАРБОКСАМИД (269)
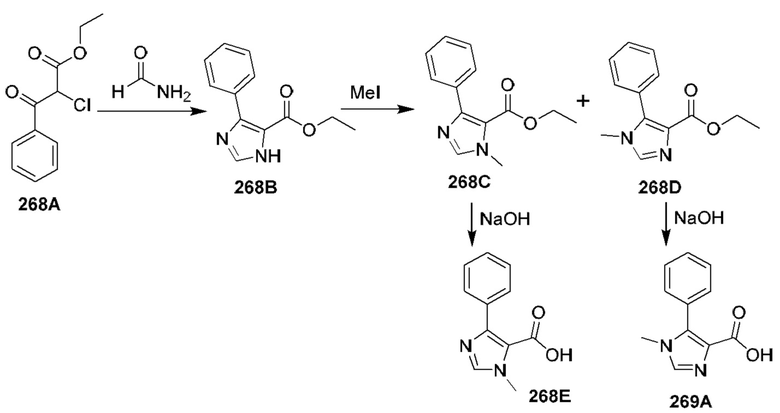
[1144] Cepayl хлорид (33,7 г, 250 ммоль) добавляли по каплям до этил-3-оксо-3-фенилпропаноат (40 г, 208 ммоль) в СНСl3 (200 мл) при 0°С. Смесь нагревали до 25°С в течение 30 мин, и затем нагревали до 80°С в течение 3,5 ч. Анализ посредством ТСХ (петролейный эфир : этилацетат = 10:1, Rf ~ 0,45) свидетельствовал о завершении реакции. Анализ посредством ЖХМС свидетельствовал о том, что целевого МС после охлаждения до комнатной температуры, реакционную смесь разбавляли хлороформ (40 мл), промывали NaHCO3 (водный; 40 мл × 2), вода (40 мл) и затем солевым раствором (30 мл) последовательно. Органическую фазу сушили над Na2SO4, фильтровали и упаривали с получением неочищенный продукт соединение 268А (48 г, неочищенного), в виде желтого масла. Соединение 268А: 1H ЯМР (400 МГц, CDCl3-d) δ 8,10-7,96 (m, 2H), 7,73-7,58 (m, 1Н), 7,57-7,39 (m, 2H), 5,62 (s, 1H), 4,39-4,25 (m, 2H), 1,32-1,13 (m, 3H).
[1145] Раствор соединения 268А (20 г, 88,2 ммоль), формамид (39,7 г, 882 ммоль) и H2O (3,18 г, 176 ммоль) нагревали до 180°С в течение 3,5 ч. После охлаждения в смесь добавляли воду (100 мл) и подвергали экстракции ДХМ (50 мл × 3), органическую фазу осаждали, твердое вещество отфильтровывали и сушили с получением соединения 268В (1,45 г, выход: 7,6%) в виде грязно-белого твердого вещества. Соединение 268В: 1H ЯМР (400 МГц, ДМСО-d6) δ 13,26-12,66 (m, 1H), 7,96-7,78 (m, 1H), 7,81 (s, 1H), 7,63 (ушир. d, J=7,2 Гц, 1H), 7,53-7,26 (m, 3H), 4,33-4,09 (m, 2H), 1,33-1,13 (m, 3H).
[1146] К раствору NaH (277 мг, 6,94 ммоль, 60% чистота) в ДМФА (5 мл) добавляли соединение 268В (1,25 г, 5,78 ммоль) порциями и перемешивали в течение 30 мин, затем CH3I (903 мг, 6,36 ммоль) добавляли, смесь перемешивали при 15°С в течение 2 ч. В смесь добавляли для гашения реакции воду (15 мл) и подвергали экстракции этилацетатом (20 мл × 3), органическую фазу сушили над Na2SO4, фильтровали и концентрировали, остаток очищали посредством препаративной ВЭЖХ (neutral) с получением соединения 268С и 268D. Соединение 268С (430 мг, выход: 64,7%, желтое твердое вещество): 1H ЯМР (400 МГц, CDCl3-d) δ 7,67 (ушир. d, J=7,1 Гц, 2H), 7,57 (s, 1H), 7,43-7,30 (m, 3H), 4,25 (q, J=7,1 Гц, 2H), 3,93 (s, 3H), 1,22 (t, J=7,2 Гц, 3H).
[1147] Соединение 268D (380 мг, выход: 57,1%, желтое твердое вещество): 1H ЯМР (400 МГц, CDCl3-d) δ 7,54 (s, 1H), 7,52-7,44 (m, 3H), 7,41-7,34 (m, 2H), 4,24 (q, J=7,1 Гц, 2H), 3,50 (s, 3H), 1,25 (t, J=7,2 Гц, 3H).
[1148] Смесь соединения 268С (200 мг, 869 мкмоль) и NaOH (69,5 мг, 1,74 ммоль) в ТГФ (5 мл), H2O (1 мл) перемешивали при 15°С в течение 12 ч. Анализ посредством ТСХ (этилацетат, Rf ~ 0) свидетельствовал о завершении реакции. Органический растворитель удаляли под вакуумом, рН водного слоя доводили до ~5 с помощью 1 н. HCl с получением осадка, твердое вещество отфильтровывали и сушили с получением соединения 268Е (120 мг, выход: 68,3%) в виде белого твердого вещества. Соединение 268Е: 1H ЯМР (400 МГц, ДМСО-d6) δ 12,90 (ушир. s, 1H), 7,86 (s, 1H), 7,62 (ушир. d, J=7,1 Гц, 2Н), 7,38-7,21 (m, 3H), 3,80 (s, 3H).
[1149] Соединение 268 (40,2 мг, выход: 27,2%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 268Е. Соединение 268: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,96 (d, J=7,5 Гц, 1H), 8,19 (s, 1H), 7,92 (s, 1H), 7,66 (s, 1H), 7,55 (d, J=6,8 Гц, 2Н), 7,31-7,18 (m, 8H), 5,52 (ddd, J=3,5, 7,4, 10,5 Гц, 1H), 3,40 (s, 3H), 3,21 (dd, J=3,5, 14,1 Гц, 1H), 2,75 (dd, J=10,6, 14,1 Гц, 1H). МС (ИЭР) m/z (M+H)+ 377,1.
[1150] Следуя методике, использованной для промежуточного соединения 268Е и соединения 268, промежуточного соединения 269А и соединения 269 получали. Соединение 269А (130 мг, выход: 74%, белое твердое вещество): 1H ЯМР (400 МГц, ДМСО-d6) δ 7,76 (s, 1H), 7,47-7,30 (m, 5H), 3,40 (s, 3H). Соединение 269 (32,4 мг, выход: 37,5%, белое твердое вещество): 1H ЯМР (400 МГц, CDCl3-d) е 7,67 (ушир. d, J=7,2 Гц, 1H), 7,51-7,43 (m, 4H), 7,42-7,35 (m, 2Н), 7,32-7,29 (m, 1H), 7,28 (s, 1H), 7,26-7,23 (m, 1H), 7,22-7,18 (m, 2Н), 6,72 (ушир. s, 1H), 5,64 (dt, J=5,3, 7,5 Гц, 1H), 5,44 (ушир. s, 1H), 3,51 (s, 3H), 3,41 (dd, J=5,3, 14,1 Гц, 1H), 3,20 (dd, J=7,4, 14,0 Гц, 1H). МС (ИЭР) m/z (M+H)+ 377,1.
ПРИМЕР 123
СОЕДИНЕНИЯ 227-228
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4-(ЭТОКСИМЕТИЛ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (227)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(4-((БЕНЗИЛОКСИ)МЕТИЛ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (228)
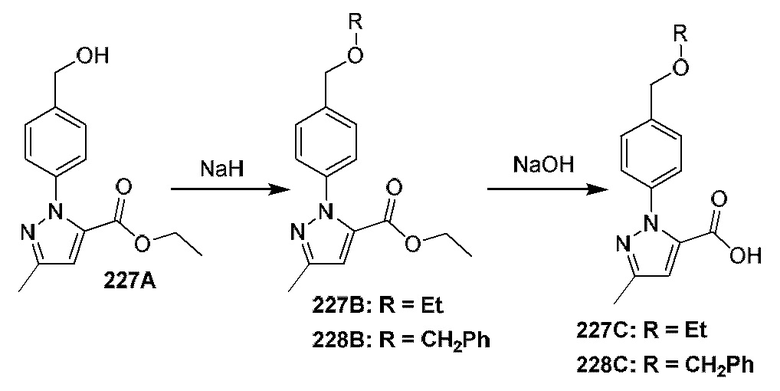
[1151] К раствору этил-3-метил-1H-пиразол-5-карбоксилата (2 г, 12,97 ммоль), [4-(гидроксиметил)фенил]бороновой кислоты (3,94 г, 25,94 ммоль) в NMP (200 мл) добавляли пиридин (2,05 г, 25,94 ммоль, 2,09 мл), Cu(ОАс)2 (3,53 г, 19,45 ммоль), МС 4А° (20 г, 12,97 ммоль). После перемешивания при 25°С в течение 24 ч, смесь фильтровали. Фильтрат промывали H2O (500 мл), подвергали экстракции этилацетатом (50 мл × 3). Органическую фазу промывали солевым раствором (500 мл), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1 до 3/1) с получением промежуточного соединения 227А (1 г, выход: 29,62%) в виде белого твердого вещества. Соединение 227А: 1H ЯМР (400 МГц, CDCl3) δ 7,41-7,34 (m, 4Н), 6,80 (s, 1Н), 4,71 (s, 2H), 4,22 (q, J=7,1 Гц, 2H), 2,35 (s, 3H), 1,24 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 261,0.
[1152] К раствору соединения 227А (350 мг, 1,34 ммоль) и бензилбромид (458 мг, 2,68 ммоль, 318 мкл) в ДМФА (10 мл) добавляли NaH (160 мг, 4,02 ммоль, 60% чистота) при 0°С. Смесь перемешивали при 25°С в течение 1 ч. В смесь добавляли для гашения реакции NH4Cl (1 мл), разбавляли H2O (30 мл), подвергали экстракции этилацетатом (20 мл × 3), Органическую фазу объединяли, промывали NaCl (50 мл × 2), сушили над Na2SO4, и концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10:1 до 5:1) с получением соединения 228В (350 мг, выход: 64,85%, желтое масло). Соединение 228В: 1H ЯМР (400 МГц, CDCl3) δ 7,47-7,28 (m, 9Н), 6,87-6,78 (m, 1Н), 4,66-4,60 (m, 2H), 4,56 (s, 2H), 4,23 (q, J=7,1 Гц, 2H), 2,36 (s, 3H), 1,24 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 351,0.
[1153] К раствору соединения 228В (350 мг, 998,83 мкмоль) в МеОН (10 мл) и H2O (10 мл) добавляли NaOH (119 мг, 3,00 ммоль). Смесь перемешивали при 25°С в течение 3 ч. Реакционную смесь концентрировали и добавляли 20 мл воды, смесь подвергали экстракции МТБЭ (10 мл × 2), Водный слой подкисляли с помощью 1 н. НС1 до рН ~ 2-3 при 0°С, и подвергали экстракции EtOAc (20 мл × 2), органическую фазу сушили над Na2SO4, концентрировали с получением остатка. Соединение 228С (270 мг, выход: 83,86%, белое твердое вещество): 1H ЯМР (400 МГц, ДМСО-d6) δ 13,58-12,95 (m, 1Н), 7,45-7,24 (m, 9Н), 6,79 (s, 1Н), 4,57 (d, J=6,8 Гц, 4Н), 2,23 (s, 3Н). МС (ИЭР) m/z (M+H)+ 323,0.
[1154] Соединение 228 (37,6 мг, выход: 51,78%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 228С. Соединение 228: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,09 (d, J=7,9 Гц, 1Н), 8,11 (s, 1Н), 7,86 (s, 1Н), 7,39-7,21 (m, 12H), 7,14 (d, J=8,2 Гц, 2H), 6,58-6,50 (m, 1Н), 5,33-5,17 (m, 1Н), 4,53 (d, J=6,0 Гц, 4Н), 3,18 (dd, J=3,2, 13,6 Гц, 1Н), 2,80 (dd, J=10,8, 13,7 Гц, 1Н), 2,23 (s, 3Н). МС (ИЭР) m/z (M+H)+ 497,2.
[1155] Промежуточного соединения 227С (300 мг, выход: 95,04%) получали в виде белого твердого вещества с использованием той же методики, как для соединения 228С. Соединение 227С: 1H ЯМР (400 МГц, ДМСО-d6) δ 13,20 (ушир. s, 1Н), 7,47-7,24 (m, 4Н), 6,79 (s, 1Н), 4,49 (s, 2H), 3,50 (q, J=7,0 Гц, 2H), 2,23 (s, 3Н), 1,16 (t, J=7,1 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 261,0.
[1156] Соединение 227 (31 мг, выход: 54,75%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 227С. Соединение 227: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,07 (d, J=7,7 Гц, 1Н), 8,11 (d, J=3,1 Гц, 1Н), 7,87-7,84 (m, 1Н), 7,31-7,24 (m, 7H), 7,13 (ушир. d, J=8.2 Гц, 2H), 6,53 (s, 1Н), 5,25 (t, J=7,4 Гц, 1Н), 4,44 (s, 2H), 3,52-3,44 (m, 2H), 3,19 (dd, J=3,0, 13,6 Гц, 1Н), 2,84-2,76 (m, 1Н), 2,23 (s, 3Н), 1,15 (t, J=6,9 Гц, 3Н). МС (ИЭР) m/z (М+H)+ 435,1.
ПРИМЕР 124
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(3-(ЭТОКСИМЕТИЛ)ФЕНИЛ)-3-МЕТИЛ- 1H-ПИРАЗОЛ-5-КАРБОКСАМИД (229)
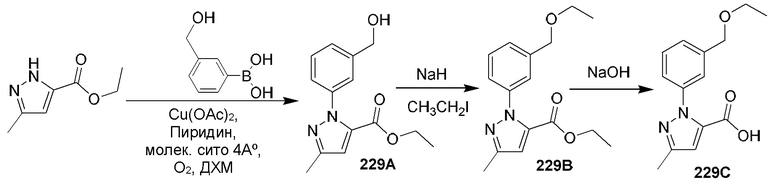
[1157] То этил-3-метил-1H-пиразол-5-карбоксилата (3 г, 19,46 ммоль), [3-(гидроксиметил)фенил]бороновой кислоты (4,44 г, 29,19 ммоль), МС 4А° (8 г) и Пиридин (1,69 г, 21,41 ммоль, 1,8 мл) в ДХМ (70 мл) добавляли Cu(ОАс)2 (4,59 г, 25,30 ммоль), смесь перемешивали при 25°С в течение 16 ч в атмосфере O2 из баллона (15 фунтов на кв. дюйм). Реакционную смесь фильтровали для удаления МС 4А° и катализатора, и затем фильтрат концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в условиях ТФУ). Соединение 229А (1,8 г, выход: 35,54%) получали в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,48-7,35 (m, 3Н), 7,32-7,28 (m, 1Н), 6,81 (s, 1H), 4,71 (s, 2H), 4,22 (q, J=7,0 Гц, 2Н), 2,35 (s, 3Н), 1,24 (t, J=7,0 Гц, 3Н).
[1158] К раствору соединения 229А (465 мг, 1,79 ммоль) и иодэтан (1,4 г, 8,95 ммоль, 0,75 мл) в сухом ДМФА (10 мл) добавляли NaH (214,8 мг, 5,37 ммоль, 60% чистота) при 0°С, затем смесь подвергали реакции при 25°С в течение 2 ч. Реакционную в смесь добавляли для гашения реакции 50 мл насыщенное NH4Cl при 0°С, подвергали экстракции этилацетатом (30 мл × 2), органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1). Соединение 229В (443 мг, выход: 85,83%) получали в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,43-7,36 (m, 3Н), 7,33-7,28 (m, 1H), 6,80 (s, 1H), 4,56 (s, 2H), 4,21 (q, J=7,3 Гц, 2H), 3,54 (q, J=7,0 Гц, 2H), 2,35 (s, 3Н), 1,23 (t, J=2,4, 7,1 Гц, 6Н).
[1159] К раствору соединения 229В (443 мг, 1,54 ммоль) в МеОН (10 мл) и H2O (6 мл) добавляли NaOH (184,8 мг, 4,62 ммоль). Смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь концентрировали и добавляли 20 мл воды и смесь подвергали экстракции МТБЭ (10 мл × 2), Водный слой подкисляли с помощью 1 н. НС1 до рН ~ 2 до 3 при 0°С, и подвергали экстракции EtOAc (10 мл × 2), органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Соединение 229С (400 мг, выход: 99,79%) получали в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,43-7,37 (m, 1H), 7,35-7,26 (m, 3Н), 6,79 (s, 1H), 4,48 (s, 2H), 3,48 (q, J=6,9 Гц, 2Н), 2,23 (s, 3H), 1,16-1,12 (m, 3Н).
[1160] Соединение 229 (30,1 мг, выход: 47,21%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 229С. Соединение 229: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,07 (ушир. d, J=7,7 Гц, 1H), 8,08 (s, 1H), 7,84 (s, 1H), 7,32-7,22 (m, 8H), 6,99 (ушир. d, J=7,5 Гц, 1H), 6,54 (s, 1H), 5,31-5,20 (m, 1H), 4,42 (s, 2H), 3,47-3,43 (m, 2H), 3,17 (ушир. dd, J=3,4, 13,8 Гц, 1H), 2,80 (ушир. dd, J=10,6, 13,7 Гц, 1H), 2,23 (s, 3Н), 1,11 (t, J=6,9 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 435,1.
ПРИМЕР 125
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(3-((БЕНЗИЛОКСИ)МЕТИЛ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (230)
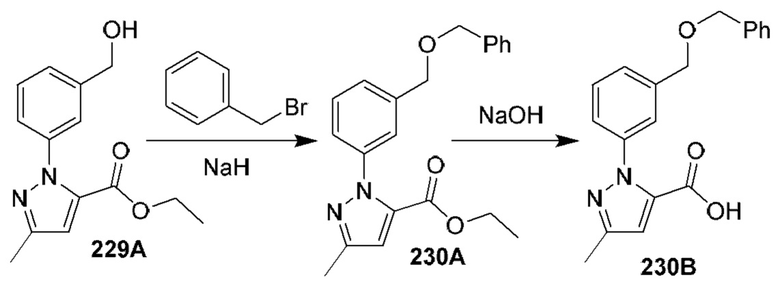
[1161] К раствору соединения 229А (472 мг, 1,81 ммоль) и бромметилбензол (1,55 г, 9,05 ммоль, 1,1 мл) в сухом ДМФА (15 мл) добавляли NaH (218 мг, 5,43 ммоль, 60% чистота) при 0°С и затем смесь подвергали реакции при 25°С в течение 2 ч. Реакционную в смесь добавляли для гашения реакции 50 мл насыщенное NH4Cl при 0°С, подвергали экстракции этилацетатом (30 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1). Соединение 230А (623 мг, выход: 98,23%) получали в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,43-7,41 (m, 3Н), 7,37-7,34 (m, 5H), 7,31 (d, J=2,0 Гц, 1Н), 6,81 (s, 1H), 4,62 (s, 2H), 4,57 (s, 2H), 4,23-4,18 (m, 2H), 2,36 (s, 3Н), 1,22 (t, J=7,1 Гц, 3Н).
[1162] К раствору соединения 230А (623 мг, 1,78 ммоль) в МеОН (15 мл) и H2O (8 мл) добавляли NaOH (214 мг, 5,34 ммоль). Смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь концентрировали и добавляли 20 мл воды, смесь подвергали экстракции МТБЭ (10 мл × 2), Водный слой подкисляли с помощью 1 н. НС1 до рН ~ 2~3 при 0°С, и подвергали экстракции EtOAc (10 мл × 2), органическую фазу сушили над Na2SO4, концентрировали с получением остатка. Соединение 230В (569 мг, выход: 99,16%) получали в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,45-7,23 (m, 10Н), 6,80 (s, 1H), 4,57 (s, 2H), 4,54 (s, 2H), 2,24 (s, 3Н).
[1163] Соединение 230 (33,3 мг, выход: 54,97%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 230В. Соединение 230: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,08 (d, J=7,7 Гц, 1H), 8,08 (s, 1H), 7,85 (s, 1H), 7,35-7,25 (m, 12H), 7,23-7,20 (m, 1H), 7,03-6,98 (m, 1H), 6,55 (s, 1H), 5,30-5,21 (m, 1H), 4,51 (d, J=2,9 Гц, 4Н), 3,17 (dd, J=3,4, 13,8 Гц. 1H), 2,80 (dd, J=10,6, 13,7 Гц, 1H), 2,23 (s, 3Н). MC (ИЭР) m/z (M+H)+ 497,1.
ПРИМЕР 126
СОЕДИНЕНИЯ 231, 438, 442
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(3-(ФЕНОКСИМЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (231)

[1164] К суспендированному раствору соединения 229А (400 мг, 1,54 ммоль) и фенол (174 мг, 1,85 ммоль) в сухом ТГФ (10 мл) добавляли PPh3 (605 мг, 2,31 ммоль) и затем медленно добавляли DIAD (467 мг, 2,31 ммоль, 449 мкл) в атмосфере N2. Смесь подвергали реакции при 25°С в течение 12 ч в атмосфере N2. Реакционную смесь растворяли в ДХМ (30 мл) и Н2О (20 мл), затем подвергали экстракции ДХМ (20 мл × 2), объединяли органический слой и смесь сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 4/1). Соединение 231А (489,7 мг, выход: 94,53%) получали в виде бесцветного масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,51-7,43 (m, 3Н), 7,36 (d, J=7,7 Гц, 1H), 7,31-7,24 (m, 2Н), 7,13 (t, J=7,8 Гц, 1H), 7,00 (d, J=7,7 Гц, 2Н), 6,87 (s, 1H), 5,14 (s, 2Н), 4,13 (q, J=7,1 Гц, 2Н), 2,25 (s, 3Н), 1,11 (t, J=7,1 Гц, 3Н).
[1165] К раствору соединения 231А (551 мг, 1,64 ммоль) в МеОН (5 мл) и Н2О (5 мл) добавляли NaOH (262 мг, 6,56 ммоль). Смесь перемешивали при 25°С в течение 1 ч. Реакционную смесь концентрировали и добавляли 10 мл воды и смесь подвергали экстракции МТБЭ (10 мл × 2), Водный слой подкисляли с помощью 1 н. HCl до рН ~ 2~3 при 0°С, и подвергали экстракции EtOAc (10 мл × 2), органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Соединение 231В (490 мг, выход: 96,90%) получали в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,50-7,42 (m, 3Н), 7,36-7,32 (m, 1H), 7,31-7,25 (m, 2Н), 7,01 (dd, J=1,0, 8,7 Гц, 2Н), 6,95-6,90 (m, 1H), 6,81 (s, 1H), 5,13 (s, 2Н), 2,24 (s, 3Н).
[1166] Соединение 231 (68 мг, выход: 65,87%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 231В. Соединение 231: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,08 (d, J=7,7 Гц, 1H), 8,07 (s, 1H), 7,84 (s, 1H), 7,40-7,23 (m, 10Н), 7,21-7,17 (m, 1H), 7,04-6,95 (m, 3Н), 6,91 (ушир. t, J=7,3 Гц, 1H), 6,55 (s, 1H), 5,28-5,20 (m, 1H), 5,08 (s, 2Н), 3,17 (dd, J=3,3, 13,9 Гц, 1H), 2,80 (ушир. dd, J=10,5, 13,8 Гц, 1H), 2,22 (s, 3Н).
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(3-((БЕНЗИЛОКСИ)МЕТИЛ)ФЕНИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (438)
[1167] Соединение 438 (2,9 г, выход: 86,54%, белое твердое вещество) получали из соответствующего промежуточного соединения 229А путем алкилирования бензилбромидом с последующим сложноэфирным гидролизом и сочетанием с промежуточным соединением 274D, как в случае соединения 12. Соединение 438: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,10 (d, J=7,7 Гц, 1H), 8,11 (s, 1H), 7,87 (s, 1H), 7,39-7,23 (m, 13Н), 7,10-6,99 (m, 1H), 6,58 (s, 1H), 5,28 (s, 1H), 4,53 (d, J=3,1 Гц, 4Н), 3,32-3,16 (m, 1H), 2,83 (dd, J=10,6, 13,7 Гц, 1H), 2,25 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 497,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-(МОРФОЛИНОМЕТИЛ)ФЕНИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (442)
[1168] Соединение 442 (50 мг, выход: 50,01%, желтое твердое вещество) получали из соответствующего промежуточного соединения 229А путем превращения его до морфолинового производного через мезилат. Морфолиновое производное подвергали сложный эфир гидролизу и сочетанию с промежуточным соединением 274D, как в случае соединения 12. Соединение 442: 1H ЯМР (400 МГц, CD3CN) δ 7,34-7,15 (m, 11Н), 7,07-6,96 (m, 1H), 6,50 (s, 1H), 6,24 (s, 1H), 5,39 (dd, J=4,6, 8,0, 9,4 Гц, 1H), 3,64-3,60 (m, 4Н), 3,50 (s, 2Н), 3,30-3,25 (m, 1H), 2,89 (dd, J=9,4, 14,0 Гц, 1H), 2,40 (s, 4Н), 2,26 (s, 3H). МС (ИЭР) m/z (М+Н)+ 476,2.
ПРИМЕР 127
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2,6-ДИМЕТИЛПИРИМИДИН-4-ИЛ)-5-МЕТИЛ-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (232)
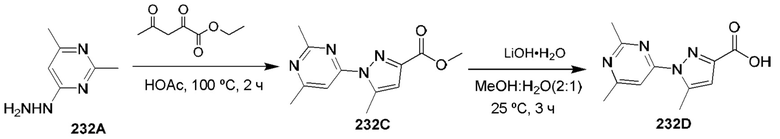
[1169] К раствору 4-хлор-2,6-диметилпиримидин (3,0 г, 21,04 ммоль) и NH2NH2⋅H2O (10,5 г, 210,40 ммоль) в EtOH (40 мл). Смесь перемешивали при 70°С в течение 2 часов. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением промежуточного соединения 232А (2,30 г, 62,60% выход) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6): δ 6,33 (ушир. s, 1H), 2,24 (s, 3H), 2,15 (s, 3H).
[1170] К раствору соединения 232А (2,30 г, 13,17 ммоль, HCl) и этил-2,4-диоксопентаноат (2,08 г, 13,17 ммоль) в АсОН (30 мл). Смесь перемешивали при 100°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении для удаления АсОН, затем разбавляли Н2О, доводили рН до примерно 9 путем постепенного добавления NaHCO3, затем распределяли между EtOAc (20 мл × 3), сушили над Na2SO4. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; 40 g SepaFlash® Silica Flash Колонка, элюент 0~50% этилацетат/петролейный эфир, градиент со скоростью потока 30 мл/мин), затем остаток очищали посредством препаративной ВЭЖХ (в основных условиях). Соединение 232С (50 мг, 1,46% выход) получали в виде белого твердого вещества. Соединение 232С: 1Н ЯМР (400 МГц, CDCl3) δ 7,76-7,71 (m, 1H), 6,72-6,66 (m, 1H), 4,48-4,37 (m, 2Н), 2,82-2,74 (m, 3H), 2,73-2,66 (m, 3H), 2,58-2,52 (m, 3H), 1,47-1,38 (m, 3H). Соединение 232D (38 мг, 85,18% выход, белое твердое вещество) было получено, как описано в примере 85, из соответствующего промежуточного соединения 232С. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,63 (s, 1H), 6,74 (s, 1H), 2,68 (s, 3H), 2,60 (s, 3H), 2,50 (s, 3H).
[1171] Соединение 232 (48,4 мг, 57,40% выход, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 232D. Соединение 232: 1Н ЯМР (400 МГц, CDCl3) δ 7,53 (s, 1H), 7,40 (ушир. d, J=7,2 Гц, 1H), 7,33-7,23 (m, 3H), 7,21-7,15 (m, 2Н), 6,80 (ушир. s, 1H), 6,66 (s, 1H), 5,77-5,69 (m, 2Н), 3,49-3,40 (m, 1H), 3,34-3,24 (m, 1H), 2,75 (s, 3H), 2,69 (s, 3H), 2,58 (s, 3H). МС (ИЭР) m/z (М+Н)+ 407,1.
ПРИМЕР 128
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(6-ФЕНИЛПИРИДАЗИН-3-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (233)
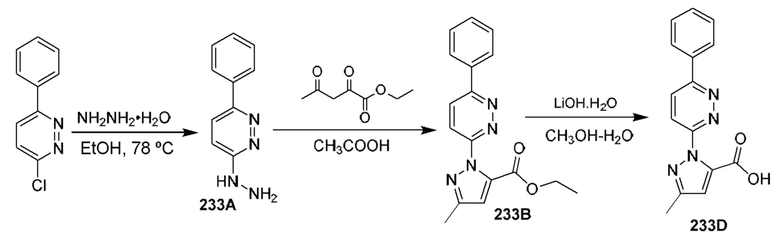
[1172] К раствору 3-хлор-6-фенилпиридазина (1,00 г, 5,25 ммоль) в EtOH (20 мл) добавляли N2H4⋅H2O (2,63 г, 52,46 ммоль, 2,55 мл). После перемешивания при 78°С в течение 10 часов реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли 30 мл петролейного эфира, перемешивали в течение 30 мин и затем фильтровали с получением неочищенного продукта промежуточного соединения 233А в виде серого остатка. 1Н ЯМР (400 МГц, ДМСО-d6): δ 7,96 (d, J=7,2 Гц, 1H), 7,85 (d, J=9,6 Гц, 1H), 7,47-7,43 (m, 2H), 7,39-7,35 (m, 1H), 7,09 (d, J=9,2 Гц, 1H).
[1173] К раствору соединения 233А (1,30 г, 6,98 ммоль) в СН3СООН (12 мл) добавляли этил-2,4-диоксопентаноат (1,10 г, 6,98 ммоль, 985,61 мкл), затем смесь перемешивали при 120°С в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли растворителем этилацетатом (70 мл), и промывали раствор насыщенным водным раствором NHCO3 (20 мл × 3), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (петролейный эфир:этилацетат = от 30/1 до 10/1) с получением остатка. Неочищенный остаток дополнительно разделяли посредством препаративной ВЭЖХ (в кислых условиях). Соединение 233В получали в виде белого твердого вещества (270,00 мг, 875,69 мкмоль, выход 12,55%).
[1174] К смеси соединения 233В (180,0 мг, 583,79 мкмоль) в МеОН (6 мл) и Н2О (3,00 мл) добавляли LiOH⋅Н2О (73,5 мг, 1,75 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 6 часов. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли H2O (20 мл), доводили рН до ~ 3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (60 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 233D (160,00 мг, выход 97,78%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,52 (d, J=9,2 Гц, 1H), 8,27-8,22 (m, 2Н), 7,63-7,56 (m, 2Н), 6,85 (s, 1H), 2,73 (s, 3H).
[1175] Соединение 233 (67,0 мг, выход 63,93%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 233D. Соединение 233: 1Н ЯМР (400 МГц, CDCl3) δ 8,16 (d, J=9,2 Гц, 1H), 8,13-8,11 (m, 2Н), 8,03 (d, J=9,2 Гц, 1H), 7,59-7,54 (m, 3H), 7,42 (d, J=7,2 Гц, 1H), 7,32-7,27 (m, 3H), 7,20-7,18 (m, 1H), 6,77 (s, 1H), 6,75 (d, J=0,4 Гц, 1H), 5,76-5,71 (m, 1H), 5,55 (s, 1H), 3,49-3,44 (m, 1H), 3,32-3,27 (m, 1H), 2,85 (s, 3H). МС (ИЭР) m/z (М+1)+ 455,1.
ПРИМЕР 129
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(6-МЕТИЛ-4-(ТРИФТОРМЕТИЛ)ПИРИДИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (234)
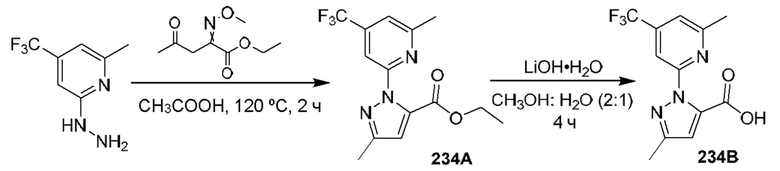
[1176] К раствору 2-гидразинил-6-метил-4-(трифторметил)пиридина (400 мг, 2,09 ммоль) в СН3СООН (4 мл) добавляли этил-2-(метоксиимино)-4-оксопентаноат (391 мг, 2,09 ммоль), затем смесь перемешивали при 120°С в течение 2 часов. Смесь разбавляли CH2Cl2 (70 мл) и промывали насыщенным бикарбонатом натрия (20 мл × 2) и насыщенным солевым раствором (20 мл × 2), сушили с помощью безводного Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством колоночной флэш-хроматографии (SiO2, петролейный эфир:этилацетат = от 10:1 до 3:1) с получением предполагаемого соединения 3 (150 мг, выход 22,91%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3): δ 8,08 (s, 1H), 7,32 (s, 1H), 6,72 (s, 1H), 4,46-4,36 (m, 2Н), 2,72 (s, 3H), 2,65 (s, 3H), 1,44-1,41 (m, 3H).
[1177] К раствору соединения 234А (100 мг, 319,21 мкмоль) в МеОН (4 мл) и Н2О (2 мл) добавляли LiOH⋅Н2О (53 мг, 1,28 ммоль), затем смесь перемешивали при 25°С в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли H2O (10 мл), доводили рН до ~ 3 с помощью 1 н. HCl и подвергали экстракции EtOAc (40 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 234В (80 мг, выход 87,87%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6): δ 7,79 (s, 1H), 7,72 (s, 1H), 6,79 (s, 1H), 2,56 (s, 3H), 2,28 (s, 3H).
[1178] Соединение 234 (24,1 мг, выход 34,58%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, соединение 234В. Соединение 234: 1H ЯМР (400 МГц, ДМСО-d6, t=80°С) δ 8,81 (d, J=6,4 Гц, 1H), 7,75 (ушир. s, 1H), 7,67 (s, 1H), 7,59 (ушир. s, 1H), 7,52 (s, 1H), 7,27-7,19 (m, 5Н), 6,53 (s, 1H), 5,43-5,35 (m, 1H), 3,23-3,16 (m, 1H), 2,96-2,87 (m, 1H), 2,39 (s, 3H), 2,30 (s, 3H). МС (ИЭР) m/z (М+1)+ 460,1.
ПРИМЕР 130
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(БЕНЗО[D]ОКСАЗОЛ-2-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (238)

[1179] Раствор 2-хлорбензо[d]оксазола (2,5 г, 16,3 ммоль) в диоксане (4 мл) добавляли по каплям к раствору N2H4⋅H2O (4,07 г, 81,4 ммоль) в диоксане (20 мл), поддерживая температуру реакционной смеси ниже 30°С. Реакционную смесь перемешивали при 20°С в течение 1 ч. Растворитель выпаривали. Добавляли воду (50 мл), и смесь перемешивали в течение 10 мин. Твердое вещество собирали путем фильтрования, и осадок на фильтре промывали водой (50 мл). Осадок на фильтре сушили с получением чистого продукта 238А (2 г, выход: 82,4%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,76 (ушир. s, 1H), 7,31 (d, J=7,7 Гц, 1H), 7,22 (d, J=7,3 Гц, 1H), 7,09 (dt, J=1,0, 7,7 Гц, 1H), 6,95 (dt, J=1,1, 7,7 Гц, 1H), 4,47 (ушир. s, 2Н).
[1180] Смесь соединения 238А (1 г, 6,70 ммоль) и метил-2,4-диоксопентаноата (966 мг, 6,70 ммоль) в АсОН (5 мл) перемешивали при 120°С в течение 16 ч. Растворитель выпаривали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 20:1~3:1) с получением соединения 238В (900 мг, неочищенного) в виде грязно-белого твердого вещества.
[1181] К раствору соединения 238 В (200 мг, 777 мкмоль) в толуоле (5 мл) добавляли TMSOK (199 мг, 1,55 ммоль). Реакционную смесь перемешивали при 80°С в течение 5 ч. Реакционную смесь выливали в насыщенный NH4Cl (5 мл). Продукт экстрагировали EtOAc (10 мл × 3). Объединенный органический слой очищали посредством препаративной ВЭЖХ (НСООН) с получением соединения 238С (30 мг, выход: 15,9%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,30 (ушир. s, 1H), 7,80 (td, J=4,2, 8,4 Гц, 2H), 7,52-7,37 (m, 2Н), 6,83 (s, 1H), 5,72 (s, 1H), 2,69 (s, 3H).
[1182] Соединение 238 (21 мг, выход: 70%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, соединения 238С. Структура возможного изомера не могла быть подтверждена посредством 2D ЯМР. Соединение 238: 1Н ЯМР (400 МГц, CDCl3) δ 7,78-7,69 (m, 1H), 7,61 (dd, J=3,2, 5,8 Гц, 1H), 7,51 (ушир. d, J=7,1 Гц, 1H), 7,43-7,35 (m, 2H), 7,32-7,16 (m, 6H), 6,80-6,70 (m, 2H), 5,81-5,71 (m, 1H), 5,55 (ушир. s, 1H), 3,45 (dd, J=5,4, 14,0 Гц, 1H), 3,22 (dd, J=7,3, 14,1 Гц, 1H), 2,75 (s, 3H). MC (ИЭР) m/z (M+H)+ 418,1.
ПРИМЕР 131
СОЕДИНЕНИЯ 239-242, 469-474
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ЭТИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (239)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ИЗОПРОПИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (242)
[1183] Раствор п-TsOH⋅Н2О (61,3 г, 322,27 ммоль) в H2O (20 мл) добавляли к суспензии соединения 1 (20,0 г, 128,91 ммоль) в CH3CN (400 мл) при 0°С. Смесь становилась прозрачной. Смесь перемешивали при 0°С в течение 30 мин. Затем к смеси добавляли по каплям раствор NaNO2 (13,3 г, 193,4 ммоль) и KI (32,1 г, 193,4 ммоль) в H2O (20 мл) при 0°С. После добавления смесь перемешивали при 20°С в течение 1 ч. В смесь добавляли для гашения реакции насыщенный Na2SO3 (~100 мл) при 0°С. Черная смесь становилась желтой. Смесь концентрировали до 200 мл и затем подвергали экстракции ДХМ (75 мл × 3). Объединенный органический слой промывали солевым раствором (75 мл × 2), сушили над MgSO4, фильтровали и концентрировали. Остаток обрабатывали 100 мл этилацетата. Нерастворимое вещество удаляли путем фильтрования. Фильтрат концентрировали и очищали посредством КФХ (ПЭ/ЭА = 1/1) с получением соединения 239А (17,50 г, выход 48,4%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 13,01-12,65 (m, 1H), 8,13-8,11 (m, 1H), 4,38-4,32 (m, 2Н), 1,41-1,37 (ш, 3H). МС (ИЭР) m/z (М+Н)+ 266,8.
[1184] Cs2CO3 (7,35 г, 22,56 ммоль) добавляли к раствору соединения 239А (2,0 г, 7,52 ммоль) в ДМФА (15 мл). Затем добавляли EtI (1,50 мл, 18,8 ммоль). Смесь перемешивали при 25°С в течение 2,5 ч. Смесь обрабатывали ЭА (50 мл) и H2O (50 мл). Органический слой отделяли, и водный слой подвергали экстракции ЭА (25 мл × 2). Объединенный органический слой промывали солевым раствором (30 мл × 3), сушили над MgSO4, фильтровали и концентрировали. Остаток очищали посредством КФХ (ПЭ/ЭА = 8/1) с получением соединения 239В (1,31 г, выход 59,2%) в виде бесцветного масла. Соединение 239В (Rf=0,24, ПЭ/ЭА = 8/1): 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,28 (s, 1H), 4,21-4,12 (m, 4Н), 1,34 (t, J=7,3 Гц, 3H), 1,25 (t, J=7,1 Гц, 3H).
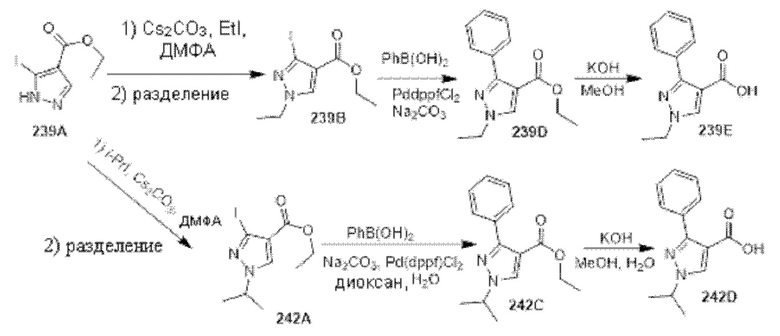
[1185] Na2CO3 (360 мг, 3,4 ммоль) добавляли к раствору соединения 239В (500 мг, 1,7 ммоль) и фенилбороновой кислоты (311 мг, 2,6 ммоль) в диоксане (10 мл). Затем добавляли H2O (2 мл), затем Pd(dppf)Cl2 (124 мг, 0,17 ммоль). Смесь дегазировали 3 раза и нагревали до 80°С и перемешивали в течение 22 ч при 80°С. Смесь фильтровали через слой целита, твердое вещество промывали ЭА (25 мл × 3). Органический слой отделяли от фильтрата и затем промывали солевым раствором (30 мл × 2), сушили над MgSO4, фильтровали и концентрировали. Остаток очищали посредством КФХ (ПЭ/ЭА = 10/1) с получением соединения 239D (380 мг, выход 91,5%) в виде бледно-желтого масла. 1Н ЯМР (CDCl3, 400 МГц): δ 7,99 (s, 1H), 7,76 (dd, J=1,5, 7,9 Гц, 2Н), 7,44-7,32 (m, 3H), 4,28-4,17 (m, 4Н), 1,55 (t, J=7,3 Гц, 3H), 1,27 (t, J=7,2 Гц, 3H).
[1186] К раствору соединения 239D (380 мг, 1,56 ммоль) в МеОН (15 мл) добавляли раствор KOH (875 мг, 15,6 ммоль) в H2O (3 мл). Смесь перемешивали при 70°С в течение 2 ч. Смесь разбавляли H2O (15 мл), и затем летучий растворитель удаляли путем выпаривания. Остаток подкисляли до рН ~ 2 с помощью 1 н. HCl. Осадок собирали путем фильтрования и сушили под вакуумом с получением соединения 239Е (250 мг, выход 74,1%), которое было получено в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,19 (ушир. s, 1H), 8,32 (s, 1H), 7,72 (dd, J=1,4, 7,8 Гц, 2Н), 7,41-7,24 (m, 3H), 4,17 (q, J=7,3 Гц, 2Н), 1,39 (t, J=7,3 Гц, 3H).
[1187] Соединение 239 (80 мг, выход 51,3%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 239Е. Соединение 239: 1Н ЯМР (CDCl3, 400 МГц): δ 8,32 (d, J=7,2 Гц, 1H), 8,07 (s, 1H), 8,05 (ушир. s, 1H), 7,79 (ушир. s, 1H), 7,62-7,51 (m, 2Н), 7,32-7,17 (m, 8Н), 5,32-5,22 (m, 1H), 4,16 (q, J=7,2 Гц, 2Н), 3,14 (dd, J=4,0, 14,0 Гц, 1H), 2,81 (dd, J=10,0, 14,0 Гц, 1H), 1,40 (t, J=7,2 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 391,1.
[1188] Следуя методике, использованной для получения соединения 239, последовательно получали промежуточные соединения 242А, 242С и 242D. Соединение 242А (1,41 г, выход 60,9%, бесцветное масло): 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,31 (s, 1H), 4,60-4,53 (m, 1H), 4,22 (q, J=7,2 Гц, 2Н), 1,41 (d, J=6,4 Гц, 6Н), 1,28 (t, J=7,2 Гц, 3H).
[1189] Соединение 242С (318 мг, выход 76,0%, бесцветная жидкость): 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,39 (s, 1H), 7,73-7,67 (m, 2Н), 7,42-7,34 (m, 3H), 4,64-4,52 (m, 1H), 4,16 (q, J=7,0 Гц, 2Н), 1,46 (d, J=6,8 Гц, 6Н), 1,21 (t, J=7,2 Гц, 3H). Соединение 242D (119 мг, неочищенное, белое твердое вещество): 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,19 (s, 1H), 8,33 (s, 1H), 7,76-7,71 (m, 2Н), 7,41-7,32 (m, 3H), 4,61-4,51 (m, 1H), 1,46 (d, J=6,8 Гц, 3H).
[1190] Соединение 242 (47 мг, выход 46,7%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 242D. Соединение 242: 1Н ЯМР (CDCl3, 400 МГц): δ 7,99 (s, 1H), 7,55-7,49 (m, 2Н), 7,47-7,37 (m, 3H), 7,23-7,14 (m, 3H), 6,85-6,78 (m, 2Н), 6,73 (s, 1H), 6,13-6,05 (m, J=6,2 Гц, 1H), 5,57-5,42 (m, 2Н), 4,51 (spt, J=6,7 Гц, 1H), 3,25 (dd, J=4,7, 14,0 Гц, 1H), 2,90 (dd, J=8,0, 14,2 Гц, 1H), 1,53 (d, J=6,6 Гц, 6Н). МС (ИЭР) m/z (М+Н)+ 405,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ЭТИЛ-3-(2-ФТОРФЕНИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (469)
[1191] Соединение 469 (130 мг, выход 49%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей промежуточной карбоновой кислоты, 1-этил-3-(2-фторфенил)-1H-пиразол-4-карбоновой кислоты, которую получали с помощью методики, аналогичной методике получения соединения 239Е. Соединение 469: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,21 (s, 1H), 7,89-7,45 (m, 3H), 7,43-7,31 (m, 2Н), 7,30-7,10 (m, 7Н), 5,33-5,22 (m, 1H), 4,20 (q, J=7,1 Гц, 2Н), 3,22-3,15 (m, 1H), 2,92-2,82 (m, 1H), 1,45 (ушир. t, J=7,2 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 409,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ЭТИЛ-3-(3-ФТОРФЕНИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (470)
[1192] Соединение 470 (140 мг, выход 66,8%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей промежуточной карбоновой кислоты, 1-этил-3-(3-фторфенил)-1H-пиразол-4-карбоновой кислоты, которую получали с помощью методики, аналогичной методике получения соединения 239Е. Соединение 470: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,44 (d, J=7,5 Гц, 1H), 8,11 (s, 1H), 8,04 (s, 1H), 7,78 (s, 1H), 7,47-7,39 (m, 2Н), 7,36-7,29 (m, 1H), 7,28-7,23 (m, 4Н), 7,22-7,16 (m, 1H), 7,14-7,07 (ш, 1H), 5,35-5,25 (m, 1H), 4,21-4,11 (m, 2Н), 3,15 (dd, J=3,9, 14,0 Гц, 1H), 2,81 (dd, J=9,9, 13,9 Гц, 1H), 1,40 (t, J=7,3 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 409,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ЭТИЛ-3-(4-ФТОРФЕНИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (471)
[1193] Соединение 471 (90 мг, выход 32,9%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей промежуточной карбоновой кислоты, 1-этил-3-(4-фторфенил)-1H-пиразол-4-карбоновой кислоты, которую получали с помощью методики, аналогичной методике получения соединения 239Е. Соединение 471: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,37 (d, J=7,5 Гц, 1H), 8,08 (s, 1H), 8,05-7,96 (m, 1H), 7,77 (s, 1H), 7,63-7,52 (m, 2Н), 7,31-7,14 (m, 5Н), 7,13-7,01 (m, 2Н), 5,31-5,16 (m, 1H), 4,24-4,03 (m, 2Н), 3,18-3,06 (m, 1H), 2,87-2,75 (m, 1H), 1,38 (t, J=7,3 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 409,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ФТОРФЕНИЛ)-1-ИЗОПРОПИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (472)
[1194] Соединение 472 (88 мг, выход 45,8%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей промежуточной карбоновой кислоты, 3-(2-фторфенил)-1-изопропил-1H-пиразол-4-карбоновой кислоты, которую получали с помощью методики, аналогичной методике получения соединения 242D. Соединение 472: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,25 (s, 1H), 8,17 (d, J=7,5 Гц, 1H), 7,96 (s, 1H), 7,73 (s, 1H), 7,40-7,05 (m, 9Н), 5,26-5,16 (m, 1H), 4,52 (td, J=6,8, 13,2 Гц, 1H), 3,09 (br, dd, J=3,9, 14,2 Гц, 1H), 2,78 (ушир. dd, J=9,7, 13,9 Гц, 1H), 1,48-1,39 (m, 6Н). МС (ИЭР) m/z (М+Н)+ 423,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ФТОРФЕНИЛ)-1-ИЗОПРОПИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (473)
[1195] Соединение 473 (51 мг, выход 41,2%, бледно-желтое твердое вещество) было получено, как в случае соединения 12, из соответствующей промежуточной карбоновой кислоты, 3-(3-фторфенил)-1-изопропил-1H-пиразол-4-карбоновой кислоты, которую получали с помощью методики, аналогичной методике получения соединения 242D. Соединение 473: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,42 (d, J=7,3 Гц, 1H), 8,14 (s, 1H), 8,07-8,01 (m, 1H), 7,78 (s, 1H), 7,51-7,42 (m, 2Н), 7,36-7,29 (m, 1H), 7,28-7,24 (m, 4Н), 7,22-7,16 (m, 1H), 7,14-7,05 (m, 1H), 5,34-5,25 (m, 1H), 4,60-4,48 (m, 1H), 3,15 (dd, J=4,0, 14,1 Гц, 1H), 2,82 (dd, J=9,8, 14,0 Гц, 1H), 1,44 (d, J=6,4 Гц, 6Н). МС (ИЭР) m/z (М+Н)+ 423,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(4-ФТОРФЕНИЛ)-1-ИЗОПРОПИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (474)
[1196] Соединение 474 (80 мг, выход 52,6%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующей промежуточной карбоновой кислоты, 3-(4-фторфенил)-1-изопропил-1H-пиразол-4-карбоновой кислоты, которую получали с помощью методики, аналогичной методике получения соединения 242D. Соединение 474: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,10 (s, 1H), 7,97 (s, 1H), 7,85-7,39 (m, 4Н), 7,34-7,17 (m, 5Н), 7,15-7,03 (m, 2Н), 5,37-5,27 (m, 1H), 4,59-4,47 (m, 1H), 3,26-3,16 (m, 1H), 2,97-2,87 (m, 1H), 1,49 (d, J=6,8 Гц, 6Н). МС (ИЭР) m/z (М+Н)+ 423,2.
ПРИМЕР 132
СОЕДИНЕНИЯ 240-241
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ЭТИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (240)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-4-ФЕНИЛ-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (241)
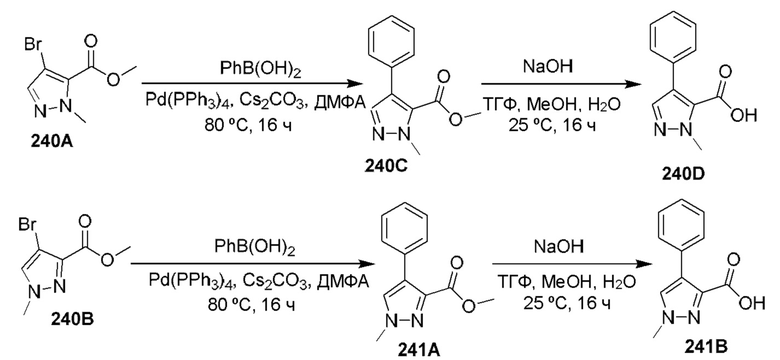
[1197] К смеси метил-4-бром-1H-пиразол-3-карбоксилата (15,0 г, 73,2 ммоль) и Cs2CO3 (59,6 г, 182,9 ммоль) в ДМФА (150 мл) добавляли MeI (14,7 мл, 236,0 ммоль) по каплям при 0°С в атмосфере N2. Смесь перемешивали при 25°С в течение 16 часов. Реакционную смесь фильтровали, осадок на фильтре промывали этилацетатом (200 мл × 2). Фильтрат промывали водой (70 мл × 4), и водную фазу подвергали экстракции этилацетатом (150 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали до сухого состояния при пониженном давлении до сухого состояния. Неочищенный продукт, который очищали посредством КФХ (элюция градиентом: петролейный эфир/этилацетат от 100/0 до 50/50) с получением указанного в заголовке соединения 240А (8,3 г, выход 51,8%) в виде белого твердого вещества и указанного в заголовке соединения 240В (7,0 г, выход 43,7%) в виде белого твердого вещества. Соединение 240А: 1Н ЯМР (400 МГц, CDCl3): δ 7,48 (s, 1H), 4,15 (s, 3H), 3,93 (s, 3H). МС (ИЭР) m/z (М+Н)+ 219,0. Соединение 240В: 1H ЯМР (400 МГц, CDCl3): δ 7,47 (s, 1H), 3,95 (s, 3H), 3,92 (s, 3H). МС (ИЭР) m/z (М+Н)+ 219,0.
[1198] Соединение 240А (2,0 г, 9,1 ммоль), фенилбороновую кислоту (1,3 г, 11,0 ммоль), Cs2CO3 (8,9 г, 27,4 ммоль) и Pd(PPh3)4 (211 мг, 183 мкмоль) в ДМФА (30 мл) дегазировали и затем нагревали до 80°С в течение 16 часов в атмосфере N2. Реакционную смесь фильтровали, осадок на фильтре промывали этилацетатом (30 мл × 2). Фильтрат промывали водой (20 мл × 4), и водную фазу подвергали экстракции этилацетатом (50 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали до сухого состояния при пониженном давлении до сухого состояния. Неочищенный продукт, который очищали посредством КФХ (элюция градиентом: петролейный эфир/этилацетат от 100/0 до 50/50) с получением указанного в заголовке соединения 240С (1,0 г, выход 47,3%) в виде светло-желтого твердого вещества. Соединение 240С: 1Н ЯМР (400 МГц, CDCl3): δ 7,52 (s, 1H), 7,40-7,36 (m, 4Н), 7,35-7,31 (m, 1H), 4,20 (s, 3H), 3,76 (s, 3H). МС (ИЭР) m/z (М+Н)+ 217,0.
[1199] Раствор NaOH (370 мг, 9,2 ммоль) в H2O (10 мл) добавляли к раствору соединения 240С (1,0 г, 4,6 ммоль) в ТГФ (10 мл) и МеОН (10 мл) при 25°С. Смесь перемешивали при 25°С в течение 16 часов. рН смеси доводили до ~ 6 с помощью 1 н. HCl (10 мл) при 25°С, и смесь подвергали экстракции этилацетатом (30 мл × 3). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали с получением соединения 240D (900 мг, выход 94,1%) в виде светло-желтого твердого вещества. Соединение 240D: 1Н ЯМР (400 МГц, CD3OD): δ 7,54-7,49 (m, 1H), 7,44-7,38 (m, 2Н), 7,37-7,27 (m, 3H), 4,14 (s, 3H). МС (ИЭР) m/z (М+Н)+ 202,9.
[1200] Соединение 240 (68,6 мг, выход 41,4%) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 240D. Соединение 240: 1Н ЯМР (400 МГц, ДМСО-d6): δ 7,46-7,39 (m, 4Н), 7,39-7,34 (m, 2Н), 7,20-7,13 (m, 3H), 6,73-6,68 (m, 2Н), 6,14-6,07 (m, 1H), 5,57-5,46 (m, 2Н), 4,09 (s, 3H), 3,19 (dd, J=4,8, 14,0 Гц, 1H), 2,80 (dd, J=8,0, 14,0 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 377,1.
[1201] Следуя методике, использованной для получения соединения 240, соединение 241 (90 мг, выход 58,5%, белое твердое вещество) получали из промежуточного соединения 241 В. Соединение 241: 1Н ЯМР (400 МГц, CDCl3): δ 7,53-7,47 (m, 2Н), 7,41 (s, 1H), 7,37-7,27 (m, 6Н), 7,19-7,13 (m, 2Н), 6,72 (ушир. s, 1H), 5,69-5,62 (m, 1H), 5,43 (ушир. s, 1H), 3,94 (s, 3H), 3,42 (dd, J=5,2, 14,0 Гц, 1H), 3,20 (dd, J=7,6, 14,0 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 377,1.
ПРИМЕР 133
СОЕДИНЕНИЯ 244-245
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(2-МЕТИЛПИРИМИДИН-4-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (244)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(ПИРИМИДИН-4-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (245)
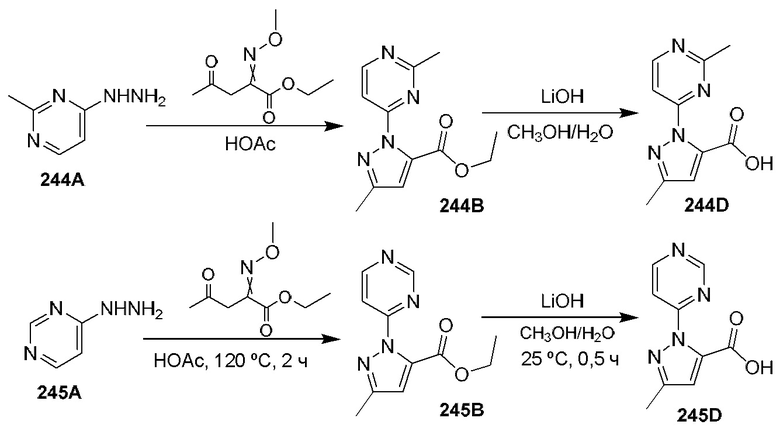
[1202] Смесь 4-хлор-2-метилпиримидина (5,00 г, 38,89 ммоль) и N2H4⋅H2O (22,91 г, 388,90 ммоль, 22,24 мл, чистота 85%) в EtOH (100 мл) дегазировали и продували N2 3 раза, затем смесь перемешивали при 70°С в течение 2 часов в атмосфере N2. Смесь концентрировали при пониженном давлении с получением неочищенного вещества, неочищенное вещество промывали ПЭ (50 мл) и фильтровали, остаток очищали посредством колоночной хроматографии (ДХМ:СН3ОН = 10:1) с получением соединения 244А (1,60 г, выход 33,14%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,21 (s, 1H), 7,99 (d, J=5,6 Гц, 1H), 6,48 (ушир. s, 1H), 4,31 (ушир. s, 2Н), 2,30 (s, 3H).
[1203] К раствору соединения 244А (900,0 мг, 7,25 ммоль) в СН3СООН (12 мл) добавляли этил-2-(метоксиимино)-4-оксопентаноат (1,36 г, 7,25 ммоль), затем смесь перемешивали при 120°С в течение 2 часов. Остаток разбавляли растворителем EtOAc (70 мл), и растворитель промывали насыщенным раствором NaHCO3 (20 мл × 3), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (ПЭ:ЭА = от 30/1 до 10/1) с получением неочищенного вещества. Неочищенное вещество дополнительно разделяли посредством препаративной ВЭЖХ (в основных условиях) с получением соединения 244В (158,0 мг, выход 8,85%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,66 (d, J=5,6 Гц, 1H), 7,53 (d, J=3,6 Гц, 1H), 6,59 (s, 1H), 4,39-4,33 (m, 2Н), 2,66 (s, 3H), 2,35 (s, 3H), 1,35-1,32 (m, 3H). МС (ИЭР) m/z (М+1)+ 247,1.
[1204] К смеси соединения 244В (130,0 мг, 527,90 мкмоль) в МеОН (8 мл) и H2O (4 мл) добавляли LiOH⋅Н2О (88,6 мг, 2,11 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 0,5 часа. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли H2O (10 мл), доводили рН до ~ 3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (40 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 244D (110,00 мг, выход 95,49%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,77 (d, J=5,6 Гц, 1H), 7,56 (d, J=3,6 Гц, 1H), 6,76 (s, 1H), 2,65 (s, 3H), 2,27 (s, 3H). МС (ИЭР) m/z (М+1)+ 219,1.
[1205] Соединение 244 (40,0 мг, выход 33,17%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 244D. Соединение 244: 1Н ЯМР (400 МГц, CDCl3) δ 9,48 (d, J=5,2 Гц, 1H), 8,66 (d, J=4,8 Гц, 1H), 7,70 (d, J=5,2 Гц, 1H), 7,26-7,20 (m, 3H), 7,10 (s, 2Н), 6,84 (s, 1H), 6,78 (s, 1H), 5,81 (d, J=6,0 Гц, 1H), 5,58 (s, 1H), 3,47-3,38 (m, 1H), 3,39-3,34 (m, 1H), 2,37 (s, 3H), 2,34 (s, 3H). МС (ИЭР) m/z (М+1)+ 393,1.
[1206] Следуя методике, использованной для получения соединения 244А, соединение 245А (1,80 г, выход 49,37%, коричневое твердое вещество) получали из 4-хлорпиримидина и NH2NH2⋅H2O. Соединение 245А: 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,32 (s, 2Н), 8,06 (d, J=5,2 Гц, 1H), 6,65 (s, 1H), 4,32 (m, 2Н).
[1207] Следуя методике, использованной для получения соединения 244В, соединение 245В (586,0 мг, 2,52 ммоль, выход 17,39%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,92 (s, 1H), 8,71 (d, J=5,6 Гц, 1H), 7,70-7,69 (m, 1H), 6,56 (s, 1H), 4,34-4,38 (m, 2Н), 2,29 (s, 1H), 1,31-1,24 (m, 3H). МС (ИЭР) m/z (М+1)+ 233,1.
[1208] Следуя методике, использованной для получения соединения 244D, соединение 245D (476,0 мг, 2,33 ммоль, выход 93,30%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,04 (s, 1H), 8,90 (d, J=5,2 Гц, 1H), 7,81 (d, J=5,6 Гц, 1H), 6,79 (s, 1H), 2,27 (s, 3H).
[1209] Соединение 245 (35,0 мг, выход 28,33%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, соединения 245D. Соединение 245: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,18 (d, J=7,2 Гц, 1H), 8,83 (d, J=5,6 Гц, 1H), 8,74 (s, 1H), 8,11 (s, 1H), 7,87 (s, 1H), 7,73 (d, J=4,8 Гц, 1H), 7,28-7,23 (m, 5Н), 6,50 (s, 1H), 5,38 (s, 1H), 3,19-3,16 (m, 1H), 2,88-2,82 (m, 1H), 2,29 (s, 3H). МС (ИЭР) m/z (М+1)+ 379,1.
ПРИМЕР 134
Соединения 246, 431-437, 448
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2-ХЛОРПИРИМИДИН-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (246)
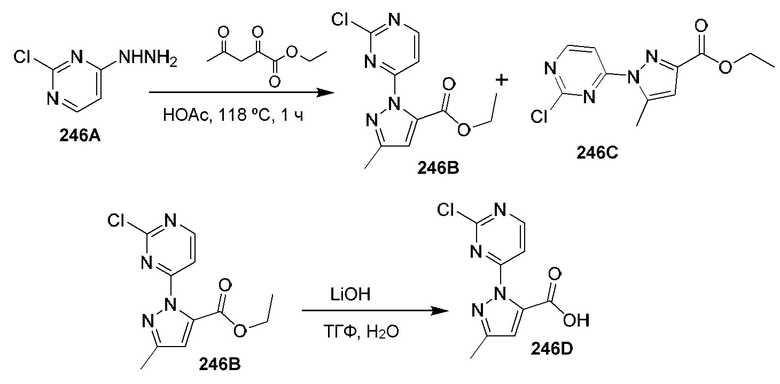
[1210] К раствору 2,4-дихлорпиримидина (10 г, 67,12 ммоль) и Et3N (10,2 мл, 73,83 ммоль) в EtOH (120 мл) добавляли NH2NH2⋅H2O (4,6 мл, 80,54 ммоль) при 0~5°С. Смесь перемешивали при 5°С в течение 1,5 ч. Смесь концентрировали. Остаток растирали в EtOH (15 мл) и воде (15 мл) с получением соединения 246А (4 г, выход 41,22%) в виде желтого масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,00-8,79 (m, 1H), 8,09-7,63 (m, 1H), 6,82-6,59 (m, 1H), 4,82-4,30 (m, 2Н).
[1211] Смесь этил-2,4-диоксопентаноата (4,38 г, 27,67 ммоль), соединения 246А (4 г, 27,67 ммоль) в АсОН (60 мл) перемешивали при 118°С в течение 1 ч. Смесь выливали в ДХМ (50 мл). Органический слой промывали водой (10 мл), NaHCO3 до рН ~ 8~9 и сушили над Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10/1 до 5:1). Соединение 246В (650 мг, выход 8,81%) получали в виде белого твердого вещества. Соединение 246С (240 мг, выход 3,25%) получали в виде белого твердого вещества. Соединение 246В: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,87 (d, J=5,6 Гц, 1H), 7,98 (d, J=5,6 Гц, 1H), 6,89 (s, 1H), 4,34 (q, J=7,2 Гц, 2Н), 2,71 (s, 3H), 1,32 (t, J=7,2 Гц, 3H). Соединение 246С: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,84 (d, J=5,6 Гц, 1H), 7,86 (d, J=5,6 Гц, 1H), 6,91 (s, 1H), 4,32 (q, J=7,2 Гц, 2Н), 2,31 (s, 3H), 1,30-1,20 (m, 3H).
[1212] К смеси соединения 246В (300 мг, 1,12 ммоль) в ТГФ (36 мл) и H2O (12 мл) добавляли LiOH⋅H2O (27,1 мг, 645,87 мкмоль). Смесь перемешивали при 31°С в течение 1 ч. Смесь концентрировали и подкисляли до рН ~ 5 с помощью 1 М HCl, затем подвергали экстракции смесью хлороформ:изопропиловый спирт = 10:1 (10 мл × 2). Эту объединенную органическую фазу промывали насыщенным водным NaCl и сушили над Na2SO4, фильтровали, и растворитель удаляли под вакуумом с получением соединения 246D (200 мг, выход 74,83%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6): δ 14,19-13,39 (m, 1H), 8,83 (d, J=5,4 Гц, 1H), 7,83 (d, J=5,6 Гц, 1H), 6,84 (s, 1H), 2,28 (s, 3H).
[1213] Соединение 246 (2,7 мг, выход 12,35%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, соединения 246D. Соединение 246: 1H ЯМР (400 МГц, ДМСО-d6): δ 9,16-9,11 (m, 1H), 8,77-8,72 (m, 1H), 8,03 (ушир. s, 1H), 7,80 (ушир. s, 1H), 7,73-7,68 (m, 1H), 7,23 (ушир. s, 4Н), 7,21-7,17 (m, 1H), 6,53 (s, 1H), 5,42 (ушир. s, 1H), 3,17-3,13 (m, 1H), 2,88-2,84 (m, 1H), 2,27 (s, 3H). МС (ИЭР) m/z (М+Н)+ 413,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2-ИЗОПРОПИЛПИРИМИДИН-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМ (431)
[1214] Соединение 431 (65 мг, выход 87,0%, белое твердое вещество) получали с использованием промежуточного соединения 246В, которое подвергали сочетанию Сузуки с использованием 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2-диоксаборолана с последующим сложноэфирным гидролизом с помощью методики, использованной для получения соединения 12, и гидрированию и сочетанию с промежуточным соединением 274D, как в случае соединения 12, с получением соединения 431. Соединение 431: 1H ЯМР (400 МГц, ДМСО-d6): δ 9,11 (d, J=7,5 Гц, 1H), 8,74 (d, J=5,5 Гц, 1H), 8,12 (s, 1H), 7,87 (s, 1H), 7,52 (d, J=5,5 Гц, 1H), 7,32-7,20 (m, 5Н), 6,47 (s, 1H), 5,52-5,41 (m, 1H), 3,16 (dd, J=3,6, 13,8 Гц, 1H), 2,80-2,75 (m, 1H), 2,29 (s, 3H), 1,04 (dd, J=6,9, 12,2 Гц, 6Н). МС (ИЭР) m/z (М+H)+ 421,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2-ЭТИНИЛПИРИМИДИН-4-ИЛ)-3-МЕТИЛ-1Н-ПИРАЗОЛ-5-КАРБОКСАМИД (432)
[1215] Соединение 432 (35 мг, выход 43,5%, белое твердое вещество) получали с использованием промежуточного соединения 246В, которое подвергали сочетанию с 4-этинилтриметилсиланом с последующим удалением триметилсилильной группы и далее сложноэфирным гидролизом с помощью методики, использованной для получения соединения 12, и сочетанию с промежуточным соединением 274D, как в случае соединения 12, с получением соединения 432. Соединение 432: 1H ЯМР (400 МГц, ДМСО-d6): δ 9,14 (d, J=7,0 Гц, 1H), 8,82 (d, J=5,5 Гц, 1H), 8,02 (s, 1H), 7,80 (s, 1H), 7,70 (d,J=5,5 Гц, 1H), 7,31-7,17 (m, 5Н), 6,55 (s, 1H), 5,46-5,35 (m, 1H), 4,39 (s, 1H), 3,19 (dd, J=4,8, 14,1 Гц, 1H), 2,93 (dd, J=9,0, 14,1 Гц, 1H), 2,29 (s, 3H). МС (ИЭР) m/z (М+Н)+ 403,1.
(S)-1-(2-ЭТИНИЛПРИМИДИН-4-ИЛ)-N-(4-ФТОР-3-ОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (433)
[1216] Соединение 433 (35 мг, выход 43,5%, белое твердое вещество) получали с использованием промежуточного соединения 246В, которое подвергали сочетанию с 4-этинилтриметилсиланом с последующим удалением триметилсилильной группы и далее сложноэфирным гидролизом с помощью методики, использованной для получения соединения 12, и сочетанию с гидрохлоридом (2S,3S)-3-амино-1-фтор-4-фенилбутан-2-ола с помощью методики, использованной для получения соединения 12, с получением соединения 433. Соединение 433: 1Н ЯМР (400 МГц, ДМСО-d6): δ 9,24 (d, J=7,8 Гц, 1H), 8,85 (d, J=5,5 Гц, 1H), 7,78 (d, J=5,5 Гц, 1H), 7,34-7,29 (m, 2Н), 7,29-7,21 (m, 3H), 6,38 s, 1H), 5,50-5,21 (m, 2Н), 4,73-4,64 (m, 1H), 4,45 (s, 1H), 3,19 (dd, J=4,9, 13,9 Гц, 1H), 2,93 (dd, J=9,8, 14,1 Гц, 1H), 2,28 (s, 3H). МС (ИЭР) m/z (М+Н)+ 392,1.
N-(4-АМИНО-3-ГИДРОКСИ-4-ОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2-ХЛОРПИРИМИДИН-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (434)
[1217] Соединение 434 (80 мг, выход 15,2%, белое твердое вещество) получали с использованием промежуточного соединения 246В, которое подвергали сочетанию с промежуточным соединением 274D с помощью методики, использованной для получения соединения 12, с получением соединения 434. Соединение 434: 1H ЯМР (400 МГц, ДМСО-d6): δ 8,76 (t, J=5,6 Гц, 1H), 8,63 (d, J=8,8 Гц, 1H), 8,36 (d, J=9,0 Гц, 1H), 7,68 (dd, J=5,5, 19,6 Гц, 1H), 7,39-7,14 (m, 7Н), 6,55-6,41 (m, 1H), 4,57-4,31 (m, 1H), 4,19 (d, J=3,3 Гц, 1H), 3,85 (d, J=2,4 Гц, 1H), 3,04-2,66 (m, 2Н), 2,28 (d, J=5,5 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 415,0.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2-ХЛОРПИРИМИДИН-4-ИЛ)-5-МЕТИЛ-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (435)
[1218] Соединение 435 (160 мг, выход 46,02%, белое твердое вещество) получали с использованием промежуточного соединения 246С, которое подвергали сочетанию с промежуточным соединением 12G с помощью методики, использованной для получения соединения 12, с получением соединения 435. Соединение 435: 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,88 (d, J=5,7 Гц, 1H), 8,09 (d, J=5,5 Гц, 1H), 7,82 (d, J=9,3 Гц, 1H), 7,42 (s, 1H), 7,27 (d, J=4,4 Гц, 4Н), 7,23-7,13 (m, 1H), 6,72 (s, 1H), 6,21 (s, 1H), 4,49 (d, J=7,3 Гц, 1H), 3,88 (d, J=2,4 Гц, 1H), 3,02-2,87 (m, 1H), 2,86-2,74 (m, 1H), 2,68 (s, 3H). МС (ИЭР) m/z (М+Н)+ 415,0.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2-МЕТОКСИПИРИМИДИН-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (436)
[1219] Соединение 436 (25 мг, выход 57,7%, белое твердое вещество) получали с использованием промежуточного соединения 246D, которое подвергали обработке метоксидом натрия и сочетанию с промежуточным соединением 274D с помощью методики, использованной для получения соединения 12, с получением соединения 436. Соединение 436: 1Н ЯМР (400 МГц, ДМСО-d6): δ 9,14 (d, J=7,5 Гц, 1H), 8,60 (d, J=5,3 Гц, 1H), 8,15 (s, 1H), 7,91 (s, 1H), 7,34 (d, J=5,3 Гц, 1H), 7,30-7,19 (m, 5Н), 6,50 (s, 1H), 5,45-5,36 (m, 1H), 3,48 (s, 3H), 3,16 (dd, J=3,3, 13,9 Гц, 1H), 2,77 (dd, J=10,1, 13,9 Гц, 1H), 2,29 (s, 3H). МС (ИЭР) m/z (М+Н)+ 409,2.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2-ЦИАНОПИРИМИДИН-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (437)
[1220] Соединение 437 (30 мг, выход 81,32%, белое твердое вещество) получали с использованием промежуточного соединения 246D, которое подвергали обработке цианидом цинка с использованием условий сочетания в присутствии палладиевого катализатора с последующим сочетанием с промежуточным соединением 274D с помощью методики, использованной для получения соединения 12, с получением соединения 437. Соединение 437: 1Н ЯМР (400 МГц, ДМСО-d6): δ 9,13 (d, J=7,3 Гц, 1H), 8,96 (d, J=5,5 Гц, 1H), 8,05-7,93 (m, 2Н), 7,84 (s, 1H), 7,27-7,15 (m, 5Н), 6,57 (s, 1H), 5,51-5,31 (m, 1H), 3,15 (dd, J=4,2, 13,9 Гц, 1H), 2,79 (dd, J=9,4, 14,0 Гц, 1H), 2,30-2,24 (m, 3H). МС (ИЭР) m/z (М+Н)+ 404,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(2-ЦИАНОПИРИМИДИН-4-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (448)
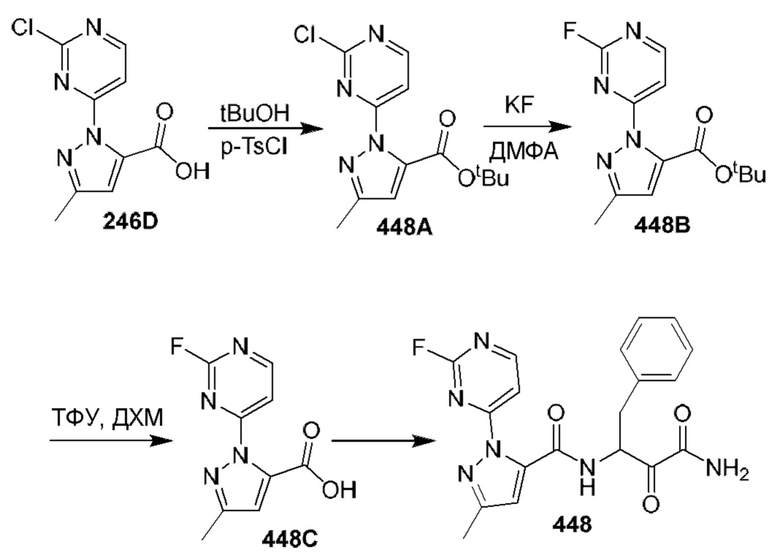
[1221] К раствору соединения 246D (400 мг, 1,68 ммоль) в 2-метилпропан-2-оле (6,2 г, 83,65 ммоль, 8,00 мл) и ТГФ (10 мл) добавляли пиридин (928 мг, 11,73 ммоль, 947 мкл) и затем добавляли п-TsCl (799 мг, 4,19 ммоль) одной порцией при 0°С. Смесь перемешивали при 25°С в течение 48 ч. В реакционную смесь добавляли для гашения реакции насыщ. NaHCO3 при 0°С, смесь подвергали экстракции ЭА (20 мл × 2), сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® Silica Flash массой 24 г, элюция градиентом 0~10%~20% этилацетат/петролейный эфир, скорость потока 35 мл/мин). Соединение 448А (400 мг, выход 81,0%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,63 (d, J=5,3 Гц, 1Н), 7,80-7,63 (m, 1H), 6,59 (s, 1H), 2,34 (m, 3H), 1,59 (m, 9H). МС (ИЭР) m/z (M+H)+ 295,1.
[1222] К раствору соединения 448A (400 мг, 1,36 ммоль) в ДМФА (13 мл) добавляли КГ (788 мг, 13,57 ммоль) и дициклогексапо-18-краун-6 (51 мг, 135,71 мкмоль). Смесь перемешивали при 120°С в течение 3 ч в атмосфере N2. Реакционную смесь охлаждали до комнатной температуры и добавляли ледяную воду (80 мл), образовывался белый осадок. Твердое вещество собирали путем фильтрования. Остаток очищали посредством препаративной ВЭЖХ (в условиях HCl). Колонка: YMC-Actus Triart C18 100 * 30 мм * 5 мкм; подвижная фаза: [вода (0,05% HCl)-ACN]; В %: 55%-85%, 9,5 мин. Соединение 448В (130 мг, выход: 34,4%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,63 (dd, J=2,0, 5,3 Гц, 1H), 7,72 (dd, J=3,2, 5,4 Гц, 1H), 6,57 (s, 1H), 2,36 (s, 3H), 1,59 (s, 9Н). МС (ИЭР) m/z (М+Н)+ 279,1.
[1223] К раствору соединения 448В (130 мг, 467,15 мкмоль) в ДХМ (15 мл) добавляли ТФУ (2,31 г, 20,26 ммоль, 1,5 мл). Смесь перемешивали при 25°С в течение 5 ч. Реакционную смесь концентрировали с получением остатка. Остаток использовали на следующей стадии без очистки. Соединение 448С (105 мг, неочищенное) получали в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,84 (dd, J=2,1, 5,4 Гц, 1H), 7,81 (dd, J=3,5, 5,5 Гц, 1H), 6,89-6,78 (m, 1H), 2,25 (s, 3H). МС (ИЭР) m/z (М+Н)+ 222,9.
[1224] Соединение 448 (35 мг, выход 69,7%, белое твердое вещество) получали с использованием промежуточного соединения 246D и 448С с помощью методики, использованной для получения соединения 12, с получением соединения 448. Соединение 448: 1Н ЯМР (400 МГц, ДМСО-d6): δ 9,14 (d, J=7,3 Гц, 1H), 8,76 (dd, J=2,0, 5,5 Гц, 1H), 8,06 (s, 1H), 7,83 (s, 1H), 7,70 (dd, J=3,5, 5,5 Гц, 1H), 7,27-7,17 (m, 5Н), 6,53 (s, 1H), 5,42-5,37 (m, 1H), 3,14 (dd, J=4,0, 14,1 Гц, 1H), 2,82 (dd, J=9,3, 14,1 Гц, 1H), 2,28 (s, 3H). МС (ИЭР) m/z (М+Н)+ 397,1.
ПРИМЕР 135
(S)-N-(3,4-ДИОКСО-1-ФЕНИЛ-4-((4-(ТРИФТОРМЕТОКСИ)БЕНЗИЛ)АМИНО)БУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛИЗОКСАЗОЛ-4-КАРБОКСАМИД (247)
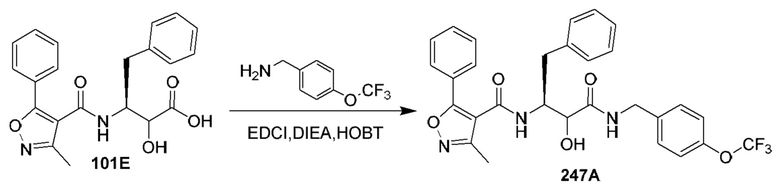
[1225] К раствору соединения 101Е (500,0 мг, 1,31 ммоль) в ДМФА (10 мл) добавляли [4-(трифторметокси)фенил]метанамин (250,4 мг, 1,31 ммоль, 200 мкл), DIEA (507,9 мг, 3,93 ммоль, 690 мкл), HOBt (53,1 мг, 393,00 мкмоль) и EDCI (301,4 мг, 1,57 ммоль). Смесь перемешивали при 25°С в течение 12 часов. Смесь разбавляли H2O (100 мл), подвергали экстракции ЭА (30 мл), промывали HCl (1 М, 30 мл), насыщенным NaHCO3 (водн., 30 мл), солевым раствором (30 мл), сушили над Na2SO4 и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в основных условиях). Соединение 247А (80,0 мг, неочищенное) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,46-8,20 (m, 1H), 7,57-7,11 (m, 15Н), 5,90-5,64 (m, 1H), 4,71-4,56 (m, 1H), 4,10-3,90 (m, 2Н), 2,99-2,89 (m, 1H), 2,86-2,74 (m, 1H), 2,11-2,01 (m, 3H).
[1226] К раствору соединения 247А (80,0 мг, 144,53 мкмоль) в ДХМ (10 мл) и ДМСО (1 мл) добавляли DMP (183,9 мг, 433,59 мкмоль). Смесь перемешивали при 25°С в течение 3 часов. В смесь добавляли для гашения реакции 10% Na2S2O3 (водный):насыщенный NaHCO3 (водный) (1:1, 20 мл), смесь подвергали экстракции ДХМ (10 мл) и промывали солевым раствором (20 мл × 3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Неочищенный продукт растирали с CH3CN (5 мл) и отфильтровывали. Соединение 247 (20,0 мг, выход 25,1%) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,50-9,43 (m, 1H), 9,12-9,06 (m, 1H), 7,66-7,57 (m, 2Н), 7,51-7,36 (m, 5Н), 7,34-7,19 (m, 7Н), 5,53-5,45 (m, 1H), 4,41-4,16 (m, 2Н), 3,27-3,20 (m, 1H), 2,82-2,72 (m, 1H), 2,10-2,01 (m, 1H), 2,05 (s, 2Н). МС (ИЭР) m/z (М+Н)+ 552,1.
ПРИМЕР 136
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(4-ФТОРФЕНИЛ)-2-МЕТИЛОКСАЗОЛ-5-КАРБОКСАМИД (248)
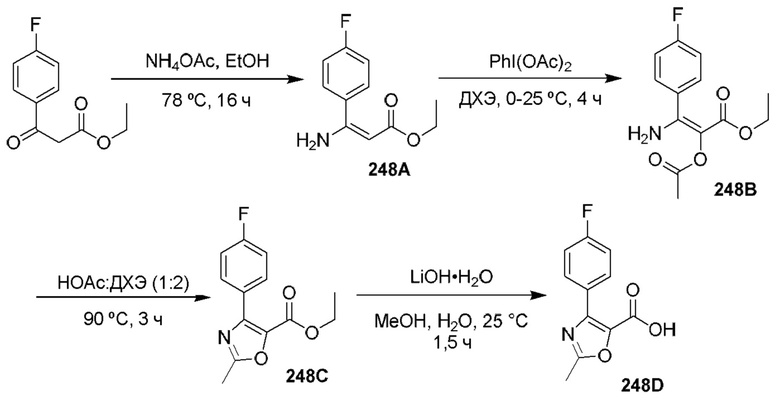
[1227] Смесь этил-3-(4-фторфенил)-3-оксопропаноата (8 г, 38,06 ммоль) и NH4OAC (5,87 г, 76,12 ммоль) в EtOH (450 мл) перемешивали при 78°С в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток разбавляли водой (60 мл) и затем подвергали экстракции EtOAc (100 мл × 3). Объединенную органическую фазу промывали насыщ. NHCO3 (50 мл × 3) и солевым раствором (50 мл), сушили над безводным Na2SO4, фильтровали, и растворитель удаляли при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 100:1 до 10:1) с получением соединения 248А (7,10 г, выход 89,16%) в виде светло-желого масла. 1Н ЯМР (400 МГц, CDCl3): 0 7,56-7,48 (m, 2Н), 7,12-7,04 (m, 2Н), 4,90 (s, 1H), 4,16 (q, J=7,2 Гц, 2Н), 1,32-1,24 (m, 3H).
[1228] К смеси соединения 248А (9 г, 43,02 ммоль) в ДХЭ (90 мл) добавляли PhI(ОАс)2 (18,0 г, 55,93 ммоль) тремя порциями при 0°С в атмосфере N2, смесь перемешивали при 0°С в течение 3 часов и затем медленно нагревали до 25°С. Далее смесь перемешивали при 25°С в течение 1 часа. В реакционную смесь добавляли для гашения реакции насыщенный водный NaHCO3 (200 мл), и смесь подвергали экстракции ДХМ (200 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 20:1 до 5:1) с получением соединения 248В (6,0 г, выход 52,19%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3): δ 7,49-7,35 (m, 2Н), 7,12-7,00 (m, 2Н), 4,27-4,16 (m, 2Н), 1,93 (s, 3H), 1,27 (t, J=7,2 Гц, 3H).
[1229] Смесь этильного производного соединения 248В (6,0 г, 22,45 ммоль) в ДХЭ (30 мл) и АсОН (15 мл) перемешивали при 90°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали до сухого состояния при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 100:1 до 10:1) с получением соединения 248С (2,80 г, выход 50,04%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3): δ 8,10-8,00 (m, 2Н), 7,11 (t, J=8,4 Гц, 2Н), 4,38 (q, J=7,2 Гц, 2Н), 2,57 (s, 3H), 1,37 (t, J=7,2 Гц, 3H).
[1230] К смеси соединения 248С (1 г, 4,01 ммоль) в МеОН (30 мл) и H2O (15 мл) добавляли LiOH⋅H2O (505,1 мг, 12,03 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 1,5 часов. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. рН остатка доводили до ~ 3 с помощью 1 н. HCl, остаток разбавляли водой (30 мл) и затем подвергали экстракции EtOAc (100 мл × 4). Объединенные органические слои промывали солевым раствором (80 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 248D (820 мг, выход 92,45%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3): δ 8,10-8,00 (m, 2Н), 7,17-7,07 (m, 2Н), 2,60 (s, 3H).
[1231] Соединение 248 (36,1 мг, выход 24,19%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 248D. Соединение 248: 1Н ЯМР (400 МГц, CDCl3): δ 8,19-8,12 (m, 2Н), 7,34-7,26 (m, 3H), 7,17-7,11 (m, 2Н), 7,10-7,03 (m, 2Н), 6,80-6,71 (m, 2Н), 5,74-5,68 (m, 1H), 5,58 (ушир. s, 1H), 3,48-3,40 (m, 1H), 3,28-3,19 (m, 1H), 2,53 (s, 3H). МС (ИЭР) m/z (М+Н)+ 396,0.
ПРИМЕР 137
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-ФЕНИЛБЕНЗОФУРАН-3-КАРБОКСАМИД (249)
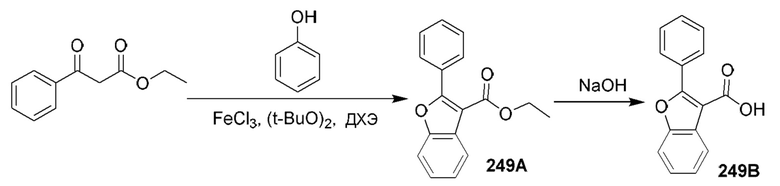
[1232] К смеси этил-3-оксо-3-фенилпропаноата (2 г, 10,41 ммоль, 1,8 мл), фенола (2,94 г, 31,23 ммоль, 2,75 мл), и FeCl3⋅6H2O (281 мг, 1,04 ммоль) добавляли ДХЭ (70 мл) в атмосфере азота при 25°С. Затем в смесь добавляли по каплям ди-трет-бутилпероксид (3,04 г, 20,82 ммоль, 3,85 мл) в атмосфере азота. Температуру реакционной смеси увеличивали до до 100°С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры. В полученный реакционный раствор добавляли для гашения реакции 30 мл насыщенного NaHCO3, и раствор подвергали экстракции ДХМ (20 мл × 3). Экстракт промывали 100 мл насыщенного NaHCO3 и 100 мл 10% Na2S2O3. Экстракт сушили над Na2SO4. Растворитель выпаривали под вакуумом с получением неочищенных продуктов. Остаток очищали посредством колоночной хроматографии (SiO2, ПЭ ~ петролейный эфир/этилацетат = 10/1) с получением соединения 249А (1,5 г, выход: 54,08%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,09-7,99 (m, 3H), 7,57-7,45 (m, 4Н), 7,39-7,32 (m, 2Н), 4,41 (q, J=7,1 Гц, 2Н), 1,43-1,39 (m, 3H). МС (ИЭР) m/z (М+Н)+ 267,0.
[1233] К раствору соединения 249А (600 мг, 2,25 ммоль) в МеОН (30 мл) и H2O (15 мл) добавляли NaOH (270 мг, 6,75 ммоль). Смесь перемешивали при 25°С в течение 16 ч. Реакционную смесь концентрировали и добавляли 20 мл воды, смесь подвергали экстракции МТБЭ (10 мл × 2), водный слой подкисляли с помощью 1 н. HCl до рН ~ 2~3 при 0°С и подвергали экстракции EtOAc (20 мл × 2), органическую фазу сушили над Na2SO4, концентрировали с получением остатка. Соединение 249В (330 мг, выход: 61,78%) получали в виде белого твердого вещества, которое использовали на следующей стадии без очистки. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,10 (ушир. s, 1H), 8,05-7,92 (m, 3H), 7,72-7,62 (m, 1H), 7,57-7,47 (m, 3H), 7,41-7,34 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 239,0.
[1234] Соединение 249 (65 мг, выход: 60,09%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 249В. Соединение 249: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,08 (d, J=7,3 Гц, 1H), 8,23 (ушир. s, 1H), 7,94 (ушир. s, 1H), 7,69 (ушир. s, 2Н), 7,63 (d, J=7,9 Гц, 1H), 7,40 (ушир. s, 3H), 7,39-7,20 (m, 8Н), 5,56 (ушир. s, 1H), 3,26 (ушир. s, 1H), 2,79 (t, J=12,2 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 413,1.
ПРИМЕР 138
СОЕДИНЕНИЯ 250-251
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ЦИКЛОПРОПИЛ-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (250)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(ЦИКЛОПРОПИЛМЕТИЛ)-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (251)
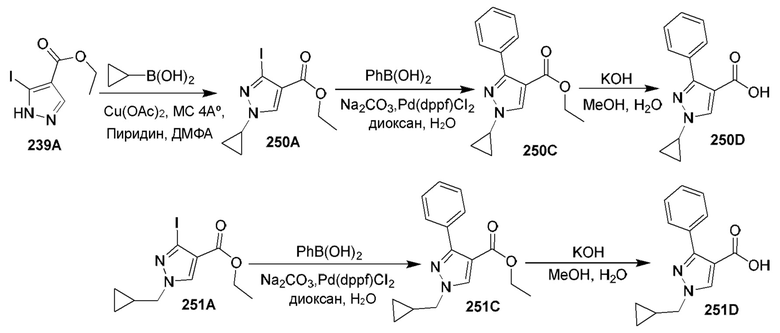
[1235] К смеси соединения 239А (2,0 г, 7,5 ммоль) и циклопропилбороновой кислоты (1,29 г, 15,0 ммоль) в ДМФА (40 мл) добавляли Cu(ОАс)2 (2,05 г, 11,28 ммоль), МС 4А° (20 г) и пиридин (1,2 мл 15,0 ммоль) при 25°С в атмосфере О2 (15 фунтов на кв. дюйм). Смесь перемешивали при 25°С в течение 38 ч. К смеси добавляли дополнительное количество циклопропилбороновой кислоты (1,29 г, 15,04 ммоль), и смесь перемешивали при 70-80°С в течение 20 ч. В реакционную смесь добавляли Cu(ОАс)2 (2,05 г, 11,28 ммоль) и перемешивали при 70-80°С в течение 22 ч. Смесь фильтровали, фильтрат разбавляли H2O (200 мл), подвергали экстракции ЭА (150 мл × 3), объединенную органическую фазу промывали солевым раствором (100 мл), сушили над Na2SO4 и концентрировали с получением остатка. Остаток очищали посредством КФХ (SiO2, петролейный эфир/этилацетат = 1:0 до 10:1) с получением соединения 250А (552 мг, выход 24,0%) в виде белого твердого вещества. Соединение 250А: 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,32 (s, 1H), 4,20 (q, J=7,1 Гц, 2Н), 3,82 (tt, J=3,8, 7,4 Гц, 1H), 1,27 (t, J=7,1 Гц, 3H), 1,12-1,07 (m, 2Н), 1,00-0,94 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 307,0.
[1236] К смеси соединения 250А (544 мг, 1,7 ммоль) и фенилбороновой кислоты (434 мг, 3,5 ммоль) в диоксане (50 мл) и Н2О (10 мл) добавляли Pd(dppf)Cl2 (130 мг, 0,18 ммоль) и Na2CO3 (377 мг, 3,5 ммоль) при 25°С в атмосфере N2. Смесь перемешивали при 80°С в течение 12 ч. Смесь фильтровали через целит. К фильтрату добавляли ЭА (150 мл) и затем промывали Н2О (100 мл × 3). Органический слой сушили над Na2SO4 и концентрировали. Остаток очищали посредством КФХ (SiO2, петролейный эфир/этилацетат = от 1:0 до 5:1)с получением соединения 250С (431 мг, выход 94,5%) в виде светло-желтой жидкости. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,42 (s, 1H), 7,71-7,65 (m, 2Н), 7,42-7,36 (m, 3H), 4,16 (q, J=7,1 Гц, 2Н), 3,85 (tt, J=3,8, 7,4 Гц, 1H), 1,21 (t, J=7,1 Гц, 3H), 1,18-1,13 (m, 2Н), 1,04-0,96 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 257,0.
[1237] К смеси соединения 250С (425 мг, 1,7 ммоль) в МеОН (10 мл) добавляли смесь KOH (931 мг, 16,6 ммоль) и Н2О (2 мл) одной порцией при 25°С. Смесь перемешивали при 70°С в течение 1 ч 40 мин. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН, водную фазу подкисляли с помощью водного раствора HCl (0,5 М) до рН ~ 4-5. Осадок фильтровали и сушили с получением соединения 250D (333 мг, неочищенного) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,24 (s, 1H), 8,35 (s, 1H), 7,73-7,69 (m, 2Н), 7,41-7,34 (m, 3H), 3,83 (tt, J=3,7, 7,5 Гц, 1H), 1,17-1,12 (m, 2Н), 1,02-0,97 (m, 2Н).
[1238] Соединение 250 (70,0 мг, выход 46,3%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 250D. Соединение 250: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,09 (s, 1H), 7,95-7,86 (m, 1H), 7,81-7,46 (m, 4Н), 7,42-7,12 (m, 8Н), 5,33 (s, 1H), 3,79 (s, 1H), 3,25-3,16 (m, 1H), 2,96-2,84 (m, 1H), 1,16-0,97 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 403,1.
[1239] Следуя методике, использованной для получения соединения 250D, соединение 251D (150 мг, выход 95,64%, белое твердое вещество) получали из соответствующих исходных веществ, соединения 239А и бромметилциклопропана. Соединение 251D: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,33 (s, 1H), 7,73 (dd, J=1,5, 7,9 Гц, 2Н), 7,40-7,34 (m, 3H), 4,01 (d, J=7,1 Гц, 2Н), 1,31 (ушир. d, J=7,7 Гц, 1H), 0,57-0,52 (m, 2Н), 0,42-0,37 (m, 2Н).
[1240] Соединение 251 (70 мг, выход 43,27%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 251D. Соединение 251: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,40 (ушир. d, J=7,3 Гц, 1H), 8,15-8,05 (m, 2Н), 7,82 (ушир. s, 1H), 7,62-7,52 (m, 2Н), 7,30 (ушир. s, 4Н), 7,28-7,20 (m, 4Н), 5,33-5,24 (m, 1H), 4,06-3,95 (m, 2Н), 3,17 (ушир. dd, J=3,5, 13,9 Гц, 1H), 2,84 (ушир. dd, J=9,9, 13,7 Гц, 1H), 1,34-1,18 (m, 1H), 0,58 (ушир. d, J=6,8 Гц, 2Н), 0,42 (ушир. d, J=4,4 Гц, 2Н). МС (ИЭР) m/z (М+Н)+ 417,2.
ПРИМЕР 139
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(ФЕНИЛЭТИНИЛ)-1H-ИНДОЛ-3-КАРБОКСАМИД (252)
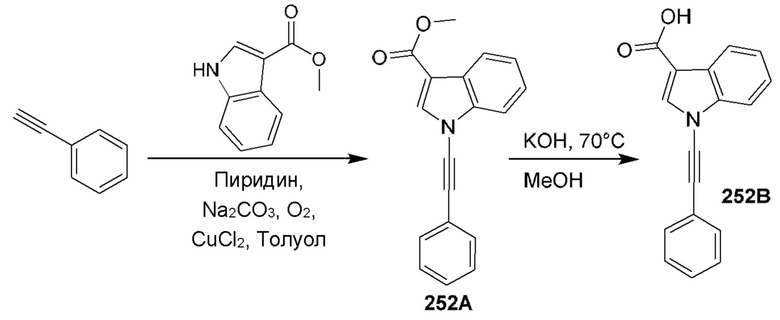
[1241] К раствору метил-1H-индол-3-карбоксилата (5,0 г, 28,55 ммоль) в толуоле (50 мл) добавляли N2CO3 (1,2 г, 11,42 ммоль), CuCl2 (153 мг, 1,14 ммоль), пиридин (922 мкл, 11,42 ммоль), затем к смеси добавляли этинилбензол (627 мкл, 5,71 ммоль). Смесь нагревали до 70°С и перемешивали в течение 4 ч в атмосфере О2. Реакционную смесь разбавляли H2O (15 мл) и ЭА (15 мл), фильтровали. Смесь подвергали экстракции ЭА (15 мл × 2), органический слой собирали и промывали NaHCO3 (25 мл × 2), промывали солевым раствором (20 мл), сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении. Продукт очищали посредством КФХ (ПЭ/ЭА: от 0 до 10/1) с получением соединения 252А (630 мг, выход 40,08%) в виде светло-красного твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,46 (s, 1H), 8,09 (d, J=1,1 Гц, 1H), 7,75 (d, J=7,9 Гц, 1H), 7,68-7,65 (m, 2Н), 7,49-7,44 (m, 4Н), 7,43-7,38 (m, 1H), 3,86 (s, 3H).
[1242] К раствору соединения 252А (300 мг, 1,09 ммоль) в МеОН (10 мл) добавляли KOH (611 мг, 10,90 ммоль), и затем смесь перемешивали при 70°С в течение 3 ч. Реакционную смесь разбавляли H2O (5 мл) и упаривали при пониженном давлении, водную фазу подвергали экстракции МТБЭ (5 мл), и затем водную фазу обрабатывали с помощью HCl (1 М) до рН ~ 4. Смесь подвергали экстракции ЭА (10 мл × 3), органический слой собирали, промывали солевым раствором (10 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Соединение 252В (240 мг, выход 84,27%) получали в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,37 (s, 1H), 8,11 (d, J=7,5 Гц, 1H), 7,75 (d, J=7,9 Гц, 1H), 7,69-7,65 (m, 2Н), 7,50-7,45 (m, 4Н), 7,45-7,36 (m, 2Н).
[1243] Соединение 252 (70 мг, выход 24,39%, желтое твердое вещество) было получено, как описано в примере 5, из промежуточного соединения 252В. Соединение 252: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,61 (d, J=7,5 Гц, 1H), 8,45 (s, 1H), 8,14-8,05 (m, 2Н), 7,85 (s, 1H), 7,73 (d, J=8,2 Гц, 1H), 7,67 (dd, J=2,4, 7,3 Гц, 2Н), 7,50-7,46 (m, 3H), 7,42 (t, J=7,7 Гц, 1H), 7,38-7,35 (m, 2Н), 7,33-7,29 (m, 3H), 7,23-7,19 (m, 1H), 5,47-5,36 (m, 1H), 3,23 (dd, J=3,6, 14,0 Гц, 1H), 2,91 (dd, J=10,0, 13,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 436,1.
ПРИМЕР 140
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(3-ФТОРФЕНИЛ)-2-МЕТИЛОКСАЗОЛ-5-КАРБОКСАМИД (254)
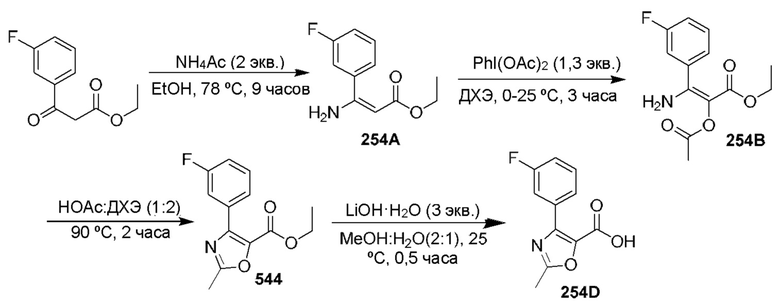
[1244] К раствору этил-3-(3-фторфенил)-3-оксопропаноата (3,00 г, 14,27 ммоль) в EtOH (40 мл) добавляли CH3COONH4 (2,20 г, 28,54 ммоль), затем смесь перемешивали при 78°С в течение 9 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли ЭА (100 мл) и промывали насыщ. раствором NaHCO3 (30 мл × 3) и насыщенным водным NaCl (30 мл × 3). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной флэш-хроматографии (ПЭ:ЭА = от 20/1 до 10:1). Соединение 254А (2,40 г, выход 80,39%) получали в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3): δ 7,38-7,35 (m, 1H), 7,35-7,32 (m, 1H), 7,22 (d, J=9,6 Гц, 1H), 7,13-7,09 (m, 1H), 4,93 (s, 1H), 4,19-4,13 (m, 2Н), 1,29-1,26 (m, 3H). МС (ИЭР) m/z (М+1)+ 210,1.
[1245] К смеси соединения 254А (2,00 г, 9,56 ммоль) в ДХЭ (25 мл) добавляли PhI(ОАс)2 (4,00 г, 12,43 ммоль) при 0°С в атмосфере N2 пятью порциями, смесь перемешивали при 0°С в течение 3 ч и затем медленно нагревали до 25°С. Затем смесь перемешивали при 25°С в течение 0,5 ч. В реакционную смесь добавляли для гашения реакции насыщенный водный NaHCO3 (150 мл) при 0°С, смесь медленно нагревали до 25°С и подвергали экстракции ДХМ (70 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 20:1 до 10:1). Соединение 254В (1,24 г, выход 48,53%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3): δ 7,37-7,34 (m, 1H), 7,22-7,00 (m, 1H), 7,17-7,13 (m, 1H), 7,12-7,09 (m, 1H), 4,23-4,18 (m, 2Н), 1,94 (s, 3H), 1,29-1,26 (m, 3H). МС (ИЭР) m/z (М+1)+ 268,1.
[1246] Смесь соединения 254В (1,20 г, 4,49 ммоль) в ДХЭ (20 мл) и СН3СООН (10 мл) перемешивали при 90°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и с получением остатка. Остаток очищали посредством колоночной флэш-хроматографии (ПЭ:ЭА = от 20/1 до 10/1). Соединение 544 (360,0 мг, выход 32,07%) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,87-7,86 (m, 1H), 7,83-7,80 (m, 1H), 7,41-7,39 (m, 1H), 7,13-7,09 (m, 1H), 4,43-4,37 (m, 2Н), 2,58 (s, 3H), 1,40-1,37 (m, 3H). МС (ИЭР) m/z (М+1)+ 250,1.
[1247] К смеси этильного производного соединения 544 (350,0 мг, 1,40 ммоль) в МеОН (10 мл) и Н2О (5 мл) добавляли LiOH⋅Н2О (235,0 мг, 5,60 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 0,5 часа. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли Н2О (20 мл), доводили рН до ~ 3 с помощью 1 н. HCl и затем подвергали экстракции EtOAc (50 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 254D (300,0 мг, выход 96,88%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,92-7,87 (m, 1H), 7,52-7,46 (m, 1H), 7,29-7,24 (m, 1H), 2,51 (s, 3H).
[1248] Соединение 254 (90,3 мг, выход 58,91%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 254D. Соединение 254: 1Н ЯМР (400 МГц, CDCl3) δ 7,98-7,95 (m, 1H), 7,94-7,91 (m, 1H), 7,39-7,35 (m, 1H), 7,33-7,30 (m, 1H), 7,29-7,26 (m, 2Н), 7,15-7,13 (m, 2Н), 7,09-7,04 (m, 1H), 6,79 (d, J=7,2 Гц, 1H), 6,76 (s, 1H), 5,74-5,69 (m, 1H), 5,53 (s, 1H), 3,47-3,42 (m, 1H), 3,27-3,22 (m, 1H), 2,54 (s, 3H). МС (ИЭР) m/z (М+1)+ 396,1.
ПРИМЕР 141
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФЕНИЛ-2-(ТРИФТОРМЕТИЛ)ОКСАЗОЛ-5-КАРБОКСАМИД (255)
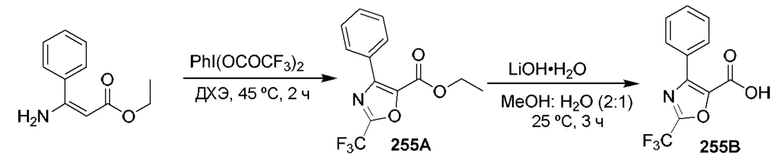
[1249] К смеси этил-(Е)-3-амино-3-фенилакрилата (1,5 г, 7,84 ммоль) в ДХЭ (400 мл) добавляли фенилиод-бис(2,2,2-трифторацетат) (4,38 г, 10,19 ммоль) тремя порциями при 45°С в атмосфере N2, смесь перемешивали при 45°С в течение 2 часов. Реакционную смесь охлаждали до 25°С и концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 100:1 до 10:1) с получением соединения 255А (650 мг, выход 28,43%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3): δ 8,11-8,05 (m, 2Н), 7,50-7,45 (m, 3H), 4,44 (q, J=7,2 Гц, 2Н), 1,40 (t, J=7,2 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 285,9.
[1250] Соединение 255В (200 мг, чистота 51,2%, желтое масло) было получено, как описано в примере 85, из соединения 255А. Соединение 255В: МС (ИЭР) m/z (М+Н)+ 258,0.
[1251] Соединение 255 (7,0 мг, выход 9,89%, грязно-белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 255В. Соединение 255: 1Н ЯМР (400 МГц, CDCl3): δ 8,17-8,08 (m, 2Н), 7,48-7,41 (m, 3H), 7,34-7,28 (m, 3H), 7,17-7,10 (m, 2Н), 6,88-6,80 (m, 1H), 6,77 (ушир. s, 1H), 5,78-5,71 (m, 1H), 5,55 (ушир. s, 1H), 3,50-3,42 (m, 1H), 3,27-3,20 (m, 1H). МС (ИЭР) m/z (М+Н)+ 432,2.
ПРИМЕР 142
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(БЕНЗО[d]ОКСАЗОЛ-2-ИЛ)-5-МЕТИЛ-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (256)
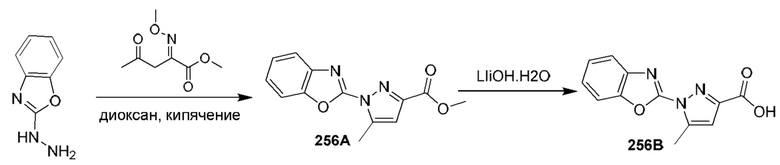
[1252] Смесь 2-гидразинилбензо[d]оксазола (320 мг, 2,15 ммоль) и метил-(Е)-2-(метоксиимино)-4-оксопентаноата (447 мг, 2,58 ммоль) в диоксане (10 мл) нагревали до 110°С в течение 12 ч. Смесь концентрировали, остаток очищали посредством препаративной ТСХ (петролейный эфир:этилацетат = 2:1) с получением соединения 256А (0,12 г, выход: 16,6%) в виде желтого масла.
[1253] Смесь соединения 256А (120 мг, 466 мкмоль) и LiOH⋅H2O (17,6 мг, 420 мкмоль) в ТГФ (5 мл), Н2О (1 мл) перемешивали при 25°С в течение 20 мин. Органический растворитель удаляли при пониженном давлении, водный слой подвергали экстракции этилацетатом (2 мл) и затем доводили рН до ~ 6 с помощью 1 н. HCl с получением осадка, твердое вещество отфильтровывали и сушили с получением соединения 256В (90 мг, выход: 79,3%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,02-7,87 (m, 2Н), 7,61 (dq, J=1,4, 7,7 Гц, 2Н), 7,11 (s, 1H), 2,43 (s, 3H).
[1254] Соединение 256 (22,2 мг, выход: 44,6%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 256 В. Соединение 256: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,97 (s, 1H), 7,82 (ушир. s, 1H), 7,64 (ушир. s, 1H), 7,55-7,44 (m, 2Н), 7,30-7,21 (m, 2Н), 6,94 (q, J=7,9 Гц, 4Н), 6,90-6,83 (m, 1H), 6,64 (s, 1H), 5,37 (dd, J=4,1, 8,0 Гц, 1H), 3,39 (dd, J=4,1, 14,2 Гц, 1H), 2,83 (dd, J=8,4, 14,1 Гц, 1H), 2,24 (s, 3H). МС (ИЭР) m/z (М+Н)+ 418,1.
ПРИМЕР 143
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-5-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (257)
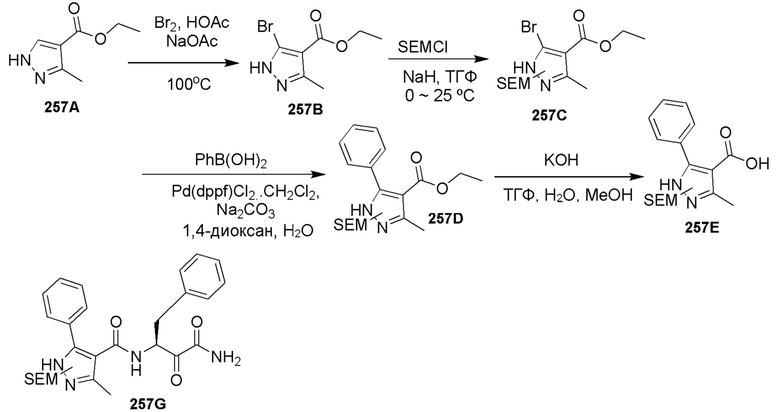
[1255] К раствору этил-3-оксобутаноата (20,0 г, 153,7 ммоль) в ТГФ (150 мл) добавляли ДМФА-DMA (19,2 г, 161,4 ммоль). Смесь перемешивали при 70°С в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении с получением остатка. К раствору неочищенного продукта, растворенного в EtOH (150 мл), добавляли по каплям NH2NH2⋅H2O (9,2 г, 184,4 ммоль). Смесь перемешивали при 80°С в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении для удаления EtOH. Остаток разбавляли 80 мл солевого раствора и подвергали экстракции этилацетатом (200 мл × 2). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением соединения 257А (21,0 г, выход 87,8%) в виде светло-зеленого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц): δ 7,96 (s, 1H), 4,29 (q, J=7,2 Гц, 2Н), 2,55 (s, 3H), 1,34 (t, J=7,2 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 154,8.
[1256] К раствору соединения 257А (12,0 г, 77,8 ммоль) в АсОН (120 мл) добавляли NaOAc (19,2 г, 233,5 ммоль) и Br2 (12 мл, 233,5 ммоль) при 25°С. Смесь перемешивали при 100°С в течение 16 часов в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли 150 мл воды и подвергали экстракции этилацетатом (200 мл × 2). Объединенные органические экстракты промывали насыщенным водным NaHCO3 (80 мл × 2), сушили над Na2SO4, фильтровали и концентрировали до сухого состояния при пониженном давлении до сухого состояния. Неочищенный продукт очищали посредством колоночной флэш-хроматографии (элюция градиентом: петролейный эфир/этилацетат от 100/0 до 50/50) с получением соединения 257В (8,2 г, выход 43,3%) в виде светло-желтого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц): δ 10,89 (ушир. s, 1H), 4,34 (q, J=7,2 Гц, 2Н), 2,64 (s, 3H), 1,38 (t, J=7,2 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 235,1.
[1257] К раствору соединения 257В (5,0 г, 21,5 ммоль) в ТГФ (50 мл) добавляли NaH (944 мг, 23,6 ммоль, чистота 60%) при 0°С, и реакционную смесь перемешивали в течение 30 минут. Добавляли SEM-Cl (3,9 г, 23,6 ммоль), и реакционную смесь перемешивали при 25°С в течение 16 часов. В реакционную смесь добавляли воду (30 мл) для гашения реакции, и смесь подвергали экстракции этилацетатом (50 мл × 3). Органический экстракт сушили над MgSO4 и концентрировали под вакуумом. Соединение 257С (6,2 г, выход 78,1%, бесцветное масло): 1H ЯМР (CDCl3, 400 МГц): δ 5,52-5,39 (m, 2Н), 4,35 (q, J=7,2 Гц, 2Н), 3,62 (td, J=8,4, 17,2 Гц, 2Н), 2,66-2,44 (m, 3H), 1,40 (dt, J=2,4, 7,2 Гц, 3H), 0,92 (td, J=8,4, 13,6 Гц, 2Н), 0,08-0,05 (m, 9Н). МС (ИЭР) m/z (М+Н)+ 364,9.
[1258] К раствору, состоящему из соединения 257С (2,0 г, 5,5 ммоль), фенилбороновой кислоты (871,8 мг, 7,2 ммоль), N2CO3 (1,8 г, 16,5 ммоль) в 1,4-диоксане (20 мл) и H2O (4 мл), добавляли Pd(dppf)Cl2. CH2Cl2 (89,9 мг, 110,0 мкмоль) при 25°С, и реакционную смесь перемешивали в течение 10 минут. Реакционную смесь нагревали до 80°С в течение 16 часов в атмосфере N2. Смесь концентрировали при пониженном давлении при 40°С. В реакционную смесь добавляли солевой раствор (40 мл) для гашения реакции, и смесь подвергали экстракции этилацетатом (30 мл × 3). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали до сухого состояния при пониженном давлении. Неочищенный продукт очищали посредством колоночной флэш-хроматографии (петролейный эфир:этилацетат = 100:0~1:1) с получением соединения 257D (1,6 г, выход 79,4%) в виде светло-желтого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц): δ 7,62-7,53 (m, 1H), 7,48-7,34 (m, 4Н), 5,48 (s, 1H), 5,18 (s, 1H), 4,25-4,06 (m, 2Н), 3,62 (td, J=8,0, 15,6 Гц, 2Н), 2,67-2,50 (m, 3H), 1,23-1,03 (m, 3H), 0,96-0,83 (m, 2Н), 0,03-0,07 (m, 9Н). МС (ИЭР) m/z (М+Н)+ 361,1.
[1259] KOH (2,5 г, 44,4 ммоль) добавляли к раствору, состоящему из соединения 257D (1,6 г, 4,4 ммоль), ТГФ (10 мл), H2O (5 мл) и МеОН (10 мл). Полученную смесь перемешивали при 25°С в течение 16 часов. Полученную смесь перемешивали при 75°С в течение 48 часов. Реакционный раствор концентрировали при пониженном давлении. Добавляли 2 н. HCl (30 мл) и подвергали экстракции EtOAc (30 мл × 3). Объединенные экстракты EtOAc промывали солевым раствором (20 мл), сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной флэш-хроматографии (петролейный эфир:этилацетат = 100:0~3:2) с получением соединения 257Е (1,0 г, выход 62,8%) в виде бесцветного масла. 1Н ЯМР (CDCl3, 400 МГц): δ 7,63-7,58 (m, 1H), 7,48-7,36 (m, 4Н), 5,49 (s, 1H), 5,17 (s, 1H), 3,62 (td, J=8,4, 18,4 Гц, 2Н), 2,66 (s, 1,5Н), 2,52 (s, 1,5Н), 0,95-0,83 (m, 2Н), 0,00-0,06 (m, 9Н). МС (ИЭР) m/z (М+Н)+ 333,2.
[1260] Промежуточное соединение 257G (150 мг, неочищенное, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 257Е. Соединение 257G: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,68-8,61 (m, 0,1Н), 8,26-7,88 (m, 1H), 7,59-7,09 (m, 10Н), 5,52-5,41 (m, 1H), 5,24 (s, 1H), 3,69-3,52 (m, 1H), 2,42-2,28 (m, 1H), 2,27-2,19 (m, 2Н), 0,96-0,79 (m, 2Н), 0,08-0,02 (m, 4,4Н), 0,02-0,06 (m, 4,3H). МС (ИЭР) m/z (М+Н)+ 507,2.
[1261] К раствору, состоящему из соединения 257G (100 мг, 0,20 ммоль) в ЭА (20 мл), добавляли HCl/EtOAc (4 М, 2 мл) при 25°С. Смесь перемешивали при 25°С в атмосфере N2 в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ВЭЖХ (0,05% гидроксид аммония) с получением 257 (15 мг, выход 19,3%) в виде белого твердого вещества. Соединение 257: 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,67 (ушир. s, 1H), 7,85-7,72 (m, 2Н), 7,64-7,57 (m, 1H), 7,57-7,49 (m, 2Н), 7,38-7,17 (m, 8Н), 5,45-5,32 (m, 1H), 3,21 (dd, J=4,0, 14,0 Гц, 1H), 2,85 (dd, J=9,2, 14,0 Гц, 1H), 2,19 (s, 3H). МС (ИЭР) m/z (М+Н)+ 377,1.
ПРИМЕР 144
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФТОР-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (258)
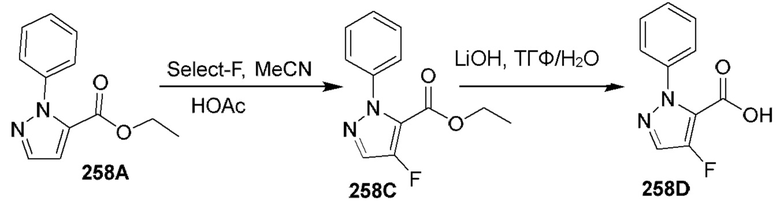
[1262] К смеси этил-1H-пиразол-5-карбоксилата (5,00 г, 35,68 ммоль), фенилбороновой кислоты (6,53 г, 53,52 ммоль), Ру (3,10 г, 39,25 ммоль, 3,17 мл) в ДХМ (70 мл) добавляли МС 4А° (20,0 г) и Cu(ОАс)2 (7,13 г, 39,25 ммоль), смесь перемешивали при 30°С в течение 20 ч. Реакционную смесь фильтровали, фильтрат концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле (ПЭ:ЭА = 1:0 до 0:1) с получением соединения 258А (1,10 г, выход: 14,3%), которое было получено в виде красновато-коричневого масла. Соединение 258А: 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,81 (d, J=2,0 Гц, 1H), 7,53-7,40 (m, 5Н), 7,09 (d, J=2,0 Гц, 1H), 4,17 (q, J=7,0 Гц, 2Н), 1,15 (t, J=7,0 Гц, 3H).
[1263] К раствору соединения 258А (1,00 г, 4,62 ммоль) в MeCN (60 мл) добавляли СН3СООН (20 мл), и затем в смесь добавляли Select F (4,91 г, 13,86 ммоль). Смесь перемешивали при 105°С в течение 21 ч в атмосфере N2. Смесь охлаждали до комнатной температуры, и летучие вещества удаляли под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле (ПЭ:ЭА = от 1:0 до 10:1) с получением соединения 258С (194 мг, выход: 17,9%), которое было получено в виде бесцветного масла. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,71-7,56 (m, 1H), 7,51-7,36 (m, 5Н), 4,29 (q, J=7,2 Гц, 2Н), 1,27 (t, J=7,1 Гц, 3H).
[1264] К раствору соединения 258С (190 мг, 811,17 мкмоль) в ТГФ (15 мл) добавляли LiOH⋅Н2О (170 мг, 4,06 ммоль) в Н2О (5 мл). Смесь перемешивали при 25°С в течение 20,3 ч. Реакционную смесь разбавляли МТБЭ (15 мл) и подвергали экстракции Н2О (15 мл × 3). рН объединенных водных слоев доводили до ~ 3 путем добавления 1 н. HCl, и затем водный слой подвергали экстракции ЭА (20 мл × 3). Объединенный органический слой промывали солевым раствором (15 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 258D (160 мг, выход: 95,7%), которое было получено в виде белого твердого вещества. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,76-7,60 (m, 1H), 7,55-7,33 (m, 5Н).
[1265] Соединение 258 (25 мг, выход: 44,3%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединений 258D и 12G. Соединение 258: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,33 (d, J=7,7 Гц, 1H), 8,18 (s, 1H), 8,00-7,85 (m, 2Н), 7,39-7,36 (m, 2Н), 7,33-7,27 (m, 5Н), 7,26-7,19 (m, 3H), 5,42-5,28 (m, 1H), 3,22 (ушир. dd, J=3,3, 14,1 Гц, 1H), 2,82 (ушир. dd, J=10,5, 13,8 Гц, 1H).
ПРИМЕР 145
СОЕДИНЕНИЯ 259-261
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-5-ФЕНИЛ-2H-1,2,3-ТРИАЗОЛ-4-КАРБОКСАМИД (259)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-5-ФЕНИЛ-1H-1,2,3-ТРИАЗОЛ-4-КАРБОКСАМИД (260)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-МЕТИЛ-4-ФЕНИЛ-1H-1,2,3-ТРИАЗОЛ-5-КАРБОКСАМИД (261)
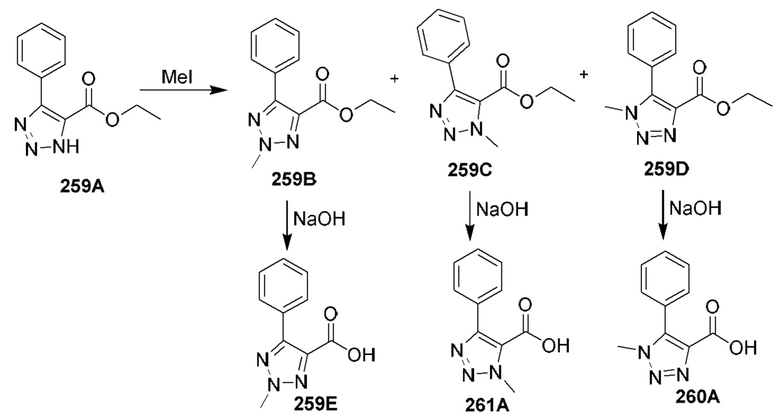
[1266] К раствору бензальдегида (5 г, 47,12 ммоль, 4,76 мл) в ДМФА (100 мл) добавляли гидрохлорид N,N-диэтилэтанамина (19,46 г, 141,36 ммоль), NaN3 (9,19 г, 141,36 ммоль) и этил-2-цианоацетат (5,33 г, 47,12 ммоль, 5,03 мл). Реакционную смесь нагревали при 70°С в течение 18 ч в защитной атмосфере азота. После завершения реакции смесь выливали в воду (500 мл) и подвергали экстракции CHl3:i-PrOH=3:1 (50 мл × 4). Органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством хроматографии (ПЭ:ЭА=5:1). Соединение 259А (4 г, выход: 38,3%) получали в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,81 (dd, J=2,9, 6,4 Гц, 2Н), 7,49-7,38 (m, 3Н), 4,38 (q, J=7,2 Гц, 2Н), 1,43-1,26 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 217,9.
[1267] MeI (6,53 г, 46,03 ммоль, 2,86 мл) добавляли к раствору соединения 259А (4 г, 18,41 ммоль) и K2CO3 (5,09 г, 36,82 ммоль) в CH3CN (50 мл) и ДМФА (50 мл). Реакционную смесь перемешивали при 25°С в течение 16 ч. Смесь фильтровали, в фильтрат добавляли H2O (200 мл), подвергали экстракции ЭА (50 мл × 3). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = ПЭ ~ 10/1 до 2/1).
[1268] Соединение 259В (1,8 г, выход: 41,9%, белое твердое вещество): 1Н ЯМР (400 МГц, CDCl3) δ 7,83-7,77 (m, 2Н), 7,45-7,36 (m, 3Н), 4,45-4,35 (m, 2Н), 4,27 (s, 3Н), 1,40-1,31 (m, 3Н). Соединение 259С (1,2 г, выход: 27,9%, белое твердое вещество): 1Н ЯМР (400 МГц, CDCl3) δ 7,75-7,67 (m, 2Н), 7,48-7,38 (m, 3Н), 4,37-4,30 (m, 5Н), 1,27 (t, J=7,1 Гц, 3Н). Соединение 259D (700 мг, выход: 16,3%, белое твердое вещество): 1Н ЯМР (400 МГц, CDCl3) δ 7,55-7,48 (m, 3Н), 7,40-7,35 (m, 2Н), 4,36-4,25 (m, 2Н), 3,95 (s, 3Н), 1,31-1,23 (m, 3Н).
[1269] К раствору соединения 259 В (400 мг, 1,73 ммоль) в МеОН (15 мл) и H2O (15 мл) добавляли NaOH (345,95 мг, 8,65 ммоль). Смесь перемешивали при 25°С в течение 1 ч. Реакционную смесь подкисляли с помощью 1 н. HCl до рН ~ 2~3 при 0°С, и образовывался белый осадок. Твердое вещество собирали путем фильтрования, фильтрат подвергали экстракции EtOAc (20 мл × 2), органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением остатка, остаток объединяли с твердым веществом с получением соединения 259Е (337 мг, выход: 95,9%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,82-7,69 (m, 2Н), 7,46-7,34 (m, 3Н), 4,22 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 204,0.
[1270] Соединение 259 (78 мг, выход: 77,2%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 259Е. Соединение 259: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,81 (ушир. d, J=7,5 Гц, 1H), 8,13 (ушир. s, 1H), 7,87 (ушир. s, 1H), 7,70 (ушир. s, 2Н), 7,38 (ушир. s, 3Н), 7,29 (ушир. d, J=4,0 Гц, 4Н), 7,23 (ушир. d, J=4,3 Гц, 1H), 5,47 (ушир. s, 1H), 4,25 (s, 3Н), 3,21 (ушир. d, J=10,8 Гц, 1H), 3,04-2,91 (m, 1H). МС (ИЭР) m/z (М+Н)+ 378,1.
[1271] Следуя методике, использованной для получения соединения 259Е, промежуточные соединения 261А и 260А получали из соединений 259С и 259D, соответственно. Соединение 261А (240 мг, выход: 91,6%, белое твердое вещество): 1Н ЯМР (400 МГц, CDCl3) δ 7,69-7,63 (m, 2Н), 7,38-7,29 (m, 3Н), 4,26 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 203,9. Соединение 260А (260 мг, выход: 98,5%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,81 (ушир. s, 1H), 7,60-7,42 (m, 5Н), 4,02-3,74 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 204,0.
[1272] Следуя методике, использованной для получения соединения 259, соединения 261 и 260 получали из соответствующей промежуточной карбоновой кислоты, соединений 261А и 260А, соответственно. Соединение 260 (75 мг, выход: 72,4%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,63 (d, J=7,5 Гц, 1H), 8,09 (s, 1H), 7,84 (s, 1H), 7,51-7,41 (m, 5Н), 7,32-7,20 (m, 5Н), 5,45-5,33 (m, 1H), 3,91 (s, 3Н), 3,23-3,14 (m, 1H), 3,11-2,97 (m, 1H). МС (ИЭР) m/z (М+Н)+ 378,1.
[1273] Соединение 261 (52 мг, выход: 59,5%, желтое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,54 (dd, J=7,9 Гц, 1H), 8,26 (s, 1H), 7,99 (s, 1H), 7,60 (dd, J=3,0, 6,5 Гц, 2Н), 7,36-7,25 (m, 8Н), 5,58 (dd, J=3,3, 7,7, 10,9 Гц, 1H), 3,83 (s, 3Н), 3,29 (dd, J=3,3 Гц, 1H), 2,77 (dd, J=10,9, 14,0 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 378,1.
ПРИМЕР 146
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ФЕНИЛ-1H-1,2,3-ТРИАЗОЛ-4-КАРБОКСАМИД (262)
[1274] Соединение 262 (8,1 мг, выход 20,36%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 204А. Соединение 262: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,38-7,89 (m, 2Н), 7,87-7,69 (m, 2Н), 7,63-7,34 (m, 4Н), 7,31-7,06 (m, 6Н), 5,55-5,42 (m, 1H), 3,32-3,24 (m, 1H), 3,12-3,06 (m, 1H). МС (ИЭР) m/z (М+Н)+ 364,1.
ПРИМЕР 147
(S)-N-(4-AMHHO-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ФЕНИЛ-1H-1,2,3-ТРИАЗОЛ-5-КАРБОКСАМИД (263)
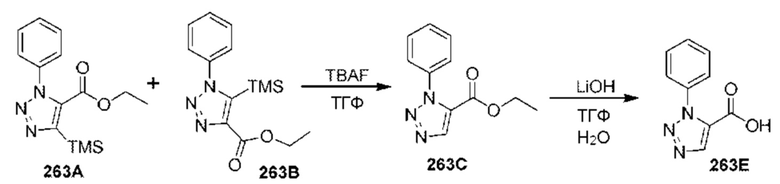
[1275] К раствору азидобензола (0,5 М, 11,8 мл) в толуоле (20 мл) добавляли этил-3-(триметилсилил)пропиолат (1 г, 5,87 ммоль). Смесь перемешивали при 100°С в течение 12 ч. Растворитель удаляли под вакуумом с получением смеси соединений 263А и 263В (1,7 г, неочищенной) в виде желтого масла, которую непосредственно использовали на следующей стадии без очистки. МС (ИЭР) m/z (М+Н)+ 290,1.
[1276] К смеси 263А и 263В (1,7 г, 5,87 ммоль) в ТГФ (20 мл) добавляли TBAF (1 М, 8,8 мл). Смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь промывали H2O (40 мл), подвергали экстракции EtOAc (20 мл × 3). Органические фазы собирали и концентрировали. Остаток очищали посредством колоночной хроматографии (ПЭ:ЭА=5:1) с получением соединения 263С (400 мг, выход: 31,37%) в виде желтого масла; 1Н ЯМР (CDCl3, 400 МГц): δ 8,26 (s, 1Н), 7,56-7,47 (m, 5Н), 4,29 (q, J=7,2 Гц, 2Н), 1,28 (t, J=7,2 Гц, 3Н).
[1277] К раствору соединения 263С (400 мг, 1,84 ммоль) в H2O (5 мл) и ТГФ (5 мл) добавляли LiOH⋅H2O (386 мг, 9,20 ммоль). Смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь подкисляли с помощью 1 н. HCl до рН ~ 3. Смесь подвергали экстракции EtOAc (20 мл × 2). Органические фазы собирали, промывали солевым раствором (20 мл), сушили с помощью Na2SO4, фильтровали и концентрировали с получением соединения 263Е (340 мг, выход: 97,68%) в виде желтого твердого вещества. МС (ИЭР) m/z (М+1)+ 189,9.
[1278] Соединение 263 (6,5 мг, выход: 6,10%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 263Е. Соединение 263: МС (ИЭР) m/z (М+1)+ 364,1. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,39 (d, J=8,0 Гц, 1H), 8,16 (s, 1H), 8,12 (ушир. s, 1H), 7,87 (ушир. s, 1H), 7,53-7,43 (m, 3Н), 7,34-7,20 (m, 7H), 5,35-5,26 (m, 1H), 3,23-3,14 (m, 1H), 2,85-2,75 (m, 1H).
ПРИМЕР 148
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-4-(ПИРИДИН-2-ИЛ)ОКСАЗОЛ-5-КАРБОКСАМИД (266)
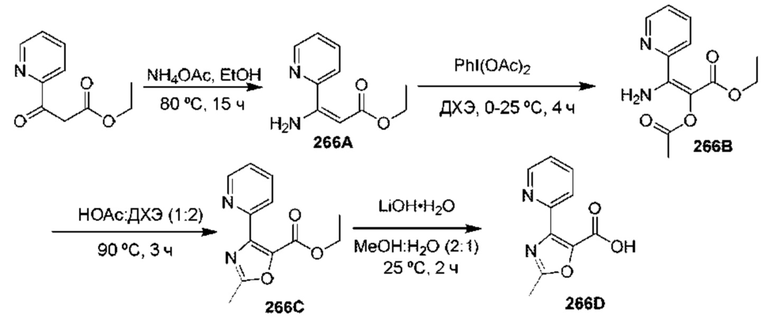
[1279] К смеси этил-3-оксо-3-(пиридин-2-ил)пропаноата (5 г, 25,88 ммоль) в EtOH (50 мл) добавляли NH4OAc (3,99 г, 51,76 ммоль) и продували N2 3 раза, и затем смесь перемешивали при 80°С в течение 15 часов в атмосфере N2. После удаления растворителя остаток растворяли в воде (50 мл), подвергали экстракции EtOAc (100 мл × 2). Эту объединенную органическую фазу промывали насыщенным водным NaHCO3 (50 мл × 2) и солевым раствором (50 мл), сушили над Na2SO4, фильтровали, и растворитель удаляли под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 0:1 до 10:1) с получением соединения 266А (3,80 г, выход 69,23%) в виде желтого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,62 (d, J=4,4 Гц, 1H), 7,78-7,71 (m, 2Н), 7,37-7,29 (m, 1H), 5,33 (s, 1H), 4,20 (q, J=7,2 Гц, 2Н), 1,31 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 193,1.
[1280] К смеси иодозобензолдиацетата (2 г, 10,41 ммоль) в ДХЭ (21 мл) добавляли соединение 266А (4,36 г, 13,53 ммоль) шестью порциями при 0°С в атмосфере N2, смесь перемешивали при 0°С в течение 3 часов и затем медленно нагревали до 25°С. Смесь перемешивали при 25°С в течение 1 ч. В реакционную смесь добавляли насыщенный водный NaHCO3 (60 мл) для гашения реакции, и смесь подвергали экстракции ДХМ (60 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 20:1 до 1:1) с получением соединения 266В (1,30 г, выход 49,90%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3): δ 8,72-8,61 (m, 1H), 7,81-7,69 (m, 2Н), 7,36-7,29 (m, 1H), 4,24 (q, J=7,2 Гц, 2Н), 2,13 (s, 3Н), 1,29 (t, J=7,2 Гц, 3Н).
[1281] Раствор соединения 266В (1,30 г, 5,19 ммоль) в ДХЭ (20 мл) и АсОН (10 мл) перемешивали при 90°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, и смесь концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10:1 до 0:1) с получением соединения 266С (300 мг, выход 23,06%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3): δ 8,78-8,73 (m, 1H), 8,13-8,08 (m, 1H), 7,80-7,74 (m, 1H), 7,33-7,28 (m, 1H), 4,40-4,34 (m, 2Н), 2,60 (s, 3Н), 1,37-1,33 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 233,1.
[1282] К смеси соединения 266С (300 мг, 1,29 ммоль) в МеОН (10 мл) и H2O (5 мл) добавляли LiOH⋅Н2О (162,4 мг, 3,87 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. рН остатка доводили до ~ 3 с помощью 1 н. HCl, остаток разбавляли водой (20 мл) и затем подвергали экстракции EtOAc (50 мл × 4). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 266D (170 мг, выход 64,54%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3: δ 8,59 (d, J=4,8 Гц, 1H), 8,31 (d, J=8,0 Гц, 1H), 8,11-8,03 (m, 1H), 7,54-7,49 (m, 1H), 2,61 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 204,8.
[1283] Соединение 266 (15,3 мг, выход 14,91%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 266D. Соединение 266: 1Н ЯМР (CDCl3, 400 МГц): δ 12,79 (d, J=6,0 Гц, 1H), 8,17 (d, J=8,0 Гц, 1H), 8,00 (d, J=4,4 Гц, 1H), 7,82 (t, J=7,6 Гц, 1H), 7,23-7,19 (m, 1H), 7,14-7,04 (m, 5Н), 6,76 (ушир. s, 1H), 5,89 (q, J=6,0 Гц, 1H), 5,56 (ушир. s, 1H), 3,47-3,30 (m, 2Н), 2,59-2,54 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 379,1.
ПРИМЕР 149
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(2-ФТОРФЕНИЛ)-2-МЕТИЛОКСАЗОЛ-5-КАРБОКСАМИД (267)
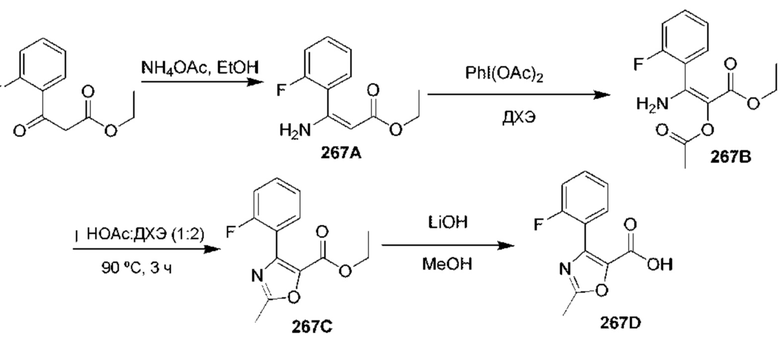
[1284] К смеси этил-3-оксо-3-(пиридин-2-ил)пропаноата (5 г, 25,88 ммоль) в EtOH (50 мл) добавляли NH4OAc (3,99 г, 51,76 ммоль) и продували N2 3 раза, и затем смесь перемешивали при 80°С в течение 15 часов в атмосфере N2. После удаления растворителя остаток растворяли в воде (50 мл), подвергали экстракции EtOAc (100 мл × 2). Эту объединенную органическую фазу промывали насыщенным водным NaHCO3 (50 мл × 2) и солевым раствором (50 мл), сушили над Na2SO4, фильтровали и растворитель удаляли под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 0:1 до 10:1) с получением соединения 266А (3,80 г, выход 69,23%) в виде желтого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц): δ 8,62 (d, J=4,4 Гц, 1H), 7,78-7,71 (m, 2Н), 7,37-7,29 (m, 1H), 5,33 (s, 1H), 4,20 (q, J=7,2 Гц, 2Н), 1,31 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 193,1.
[1285] К смеси иодозобензолдиацетата (2 г, 10,41 ммоль) в ДХЭ (21 мл) добавляли соединение 266А (4,36 г, 13,53 ммоль) шестью порциями при 0°С в атмосфере N2, смесь перемешивали при 0°С в течение 3 часов и затем медленно нагревали до 25°С. Смесь перемешивали при 25°С в течение 1 ч. В реакционную смесь добавляли насыщенный водный NaHCO3 (60 мл) для гашения реакции, и смесь подвергали экстракции ДХМ (60 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 20:1 до 1:1) с получением соединения 266В (1,30 г, выход 49,90%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3): δ 8,72-8,61 (m, 1H), 7,81-7,69 (m, 2Н), 7,36-7,29 (m, 1H), 4,24 (q, J=7,2 Гц, 2Н), 2,13 (s, 3Н), 1,29 (t, J=7,2 Гц, 3Н).
[1286] Раствор соединения 266В (1,30 г, 5,19 ммоль) в ДХЭ (20 мл) и АсОН (10 мл) перемешивали при 90°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, и смесь концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10:1 до 0:1) с получением соединения 266С (300 мг, выход 23,06%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3): δ 8,78-8,73 (m, 1H), 8,13-8,08 (m, 1H), 7,80-7,74 (m, 1H), 7,33-7,28 (m, 1H), 4,40-4,34 (m, 2Н), 2,60 (s, 3Н), 1,37-1,33 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 233,1.
[1287] К смеси соединения 266С (300 мг, 1,29 ммоль) в МеОН (10 мл) и H2O (5 мл) добавляли LiOH⋅Н2О (162,4 мг, 3,87 ммоль) одной порцией, и смесь перемешивали при 25°С в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. рН остатка доводили до ~ 3 с помощью 1 н. HCl, остаток разбавляли водой (20 мл) и затем подвергали экстракции EtOAc (50 мл × 4). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 266D (170 мг, выход 64,54%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3: δ 8,59 (d, J=4,8 Гц, 1H), 8,31 (d, J=8,0 Гц, 1H), 8,11-8,03 (m, 1H), 7,54-7,49 (m, 1H), 2,61 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 204,8.
[1288] Соединение 267 (15,3 мг, выход 14,91%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 267D. Соединение 267: 1Н ЯМР (CDCl3, 400 МГц): δ 12,79 (d, J=6,0 Гц, 1H), 8,17 (d, J=8,0 Гц, 1H), 8,00 (d, J=4,4 Гц, 1H), 7,82 (t, J=7,6 Гц, 1H), 7,23-7,19 (m, 1H), 7,14-7,04 (m, 5Н), 6,76 (ушир. s, 1H), 5,89 (q, J=6,0 Гц, 1H), 5,56 (ушир. s, 1H), 3,47-3,30 (m, 2Н), 2,59-2,54 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 379,1.
ПРИМЕР 150
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-ФЕНИЛТИОФЕН-3-КАРБОКСАМИД (270)
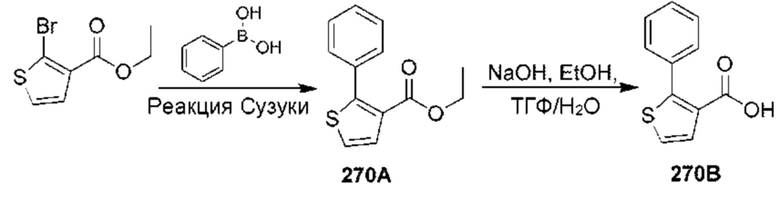
[1289] Этил-2-бромтиофен-3-карбоксилат (2 г, 8,51 ммоль), фенилбороновую кислоту (1,35 г, 11,1 ммоль), K2CO3 (2,35 г, 17 ммоль) и Pd(dppf)Cl2 (622 мг, 851 мкмоль) в диоксане (30 мл), H2O (3 мл) дегазировали и затем нагревали до 100°С в течение 6 часов в атмосфере N2. Смесь концентрировали, остаток очищали посредством хроматографии на силикагеле (от петролейного эфира до смеси петролейный эфир : этилацетат = 25:1) с получением соединения 270А (1,9 г, выход: 96,11%), в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3-d6) δ 7,53 (d, J=5,4 Гц, 1H), 7,52-7,47 (m, 2Н), 7,44-7,38 (m, 3Н), 7,25 (d, J=5,4 Гц, 1H), 4,20 (q, J=7,1 Гц, 2Н), 1,19 (t, J=7,1 Гц, 3Н).
[1290] Смесь 270А (150 мг, 646 мкмоль) и NaOH (51,7 мг, 1,29 ммоль) в ТГФ (5 мл), EtOH (3 мл), Н2О (2 мл) перемешивали при 15°С в течение 12 ч. Анализ посредством ТСХ (петролейный эфир/этилацетат = 10:1) свидетельствовал о присутствии непрореагировавшего исходного материала, и затем смесь нагревали до 60°С в течение еще 3 ч. Органический растворитель удаляли при пониженном давлении, рН водного слоя доводили до ~ 6 с помощью 1 н. HCl с получением осадка, твердое вещество отфильтровывали и сушили с получением 270В (100 мг, выход: 75,8%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,63 (ушир. s, 1H), 7,55 (d, J=5,3 Гц, 1H), 7,49-7,41 (m, 2Н), 7,41-7,31 (m, 4Н).
[1291] Соединение 270 (16,00 мг, выход: 20,1%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 270В. Соединение 270: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,73 (d, J=7,5 Гц, 1H), 8,12 (s, 1H), 7,87 (s, 1H), 7,58 (d, J=5,3 Гц, 1H), 7,36-7,20 (m, 10Н), 7,09 (d, J=5,3 Гц, 1H), 5,29 (ddd, J=3,6, 7,3, 10,4 Гц, 1H), 3,17 (dd, J=3,6, 13,8 Гц, 1H), 2,80 (dd, J=10,4, 13,9 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 379,1.
ПРИМЕР 151
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ХЛОР-2-ФЕНИЛТИОФЕН-3-КАРБОКСАМИД (271)
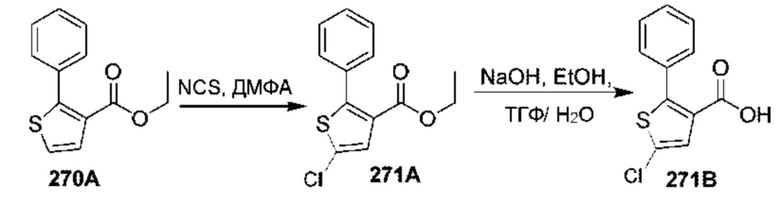
[1292] К раствору 270А (300 мг, 1,29 ммоль) в ДМФА (5 мл) добавляли NCS (345 мг, 2,58 ммоль) при 80°С, и смесь перемешивали при 80°С в течение 1,5 ч. Смесь выливали в воду (20 мл) и подвергали экстракции этилацетатом (10 мл × 2), объединенный органический слой промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством хроматографии на силикагеле (от петролейного эфира до смеси петролейный эфир : этилацетат = 20:1) с получением 271А (0,38 г, выход: 82,8%), в виде белого твердого вещества (см. стр. 158). 1Н ЯМР (400 МГц, CDCl3) δ 7,50-7,44 (m, 2H), 7,43-7,37 (m, 3Н), 7,34 (s, 1H), 4,19 (q, J=7,1 Гц, 2H), 1,18 (t, J=7,2 Гц, 3Н).
[1293] Смесь 271А (380 мг, 1,42 ммоль) и NaOH (114 мг, 2,84 ммоль) в ТГФ (5 мл), EtOH (3 мл), H2O (2 мл) перемешивали при 15°С в течение 12 ч. Органический растворитель удаляли под вакуумом, рН водного слоя доводили до ~ 3 с помощью 1 н. HCl, и слой подвергали экстракции этилацетатом (10 мл × 2), органический слой промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали с получением 271В (330 мг, выход: 97,4%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3-d) δ 7,43-7,36 (m, 2Н), 7,36-7,29 (m, 3Н), 7,28 (s, 1Н).
[1294] Соединение 271 (28,2 мг, выход: 30,4%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 271В. Соединение 271: 1Н ЯМР (400 МГц, CDCl3) δ 7,40 (ушир. s, 5Н), 7,19 (ушир. s, 4Н), 6,76 (ушир. s, 2Н), 6,66 (ушир. s, 1H), 5,93 (ушир. s, 1H), 5,44 (ушир. d, J=19,3 Гц, 2Н), 3,18 (ушир. d, J=16,7 Гц, 1H), 2,94-2,81 (m, 1H). МС (ИЭР) m/z (М+H)+ 413,0, 415,0.
ПРИМЕР 152
СОЕДИНЕНИЯ 272-273
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ЦИАНО-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (272)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-ЦИАНО-1-ФЕНИЛ-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (273)
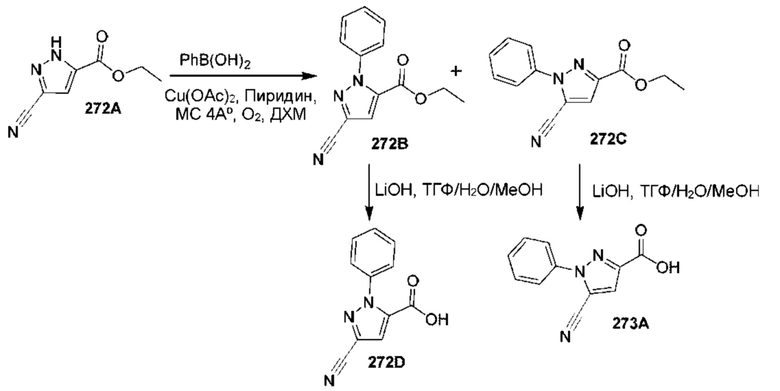
[1295] К раствору этилпропиолата (4,30 г, 43,83 ммоль) и гидрохлорида 2-аминоацетонитрила (8,11 г, 87,67 ммоль, HCl) в CHCl3 (250 мл) и H2O (10 мл) добавляли NaNO2 (9,07 г, 131,50 ммоль). Смесь перемешивали в течение 14 ч при 25°С. Затем реакционную смесь разбавляли ДХМ (50 мл) и фильтровали. Фильтрат промывали H2O (20 мл) и солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® Silica Flash массой 20 г, элюция градиентом 0~30% этилацетат/петролейный эфир) с получением 272А (1,40 г, выход: 19,34%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 12,19 (ушир. s, 1H), 7,21 (s, 1H), 4,45 (q, J=7,2 Гц, 2Н), 1,41 (t, J=7,2 Гц, 3Н).
[1296] К смеси 272А (1,40 г, 8,48 ммоль), фенилбороновой кислоты (1,55 г, 12,72 ммоль), пиридина (737,60 мг, 9,32 ммоль) в ДХМ (50 мл) добавляли МС 4А° (20 г) (активированное МС 4А°) и Cu(OAc)2 (1,69 г, 9,32 ммоль). После этого смесь перемешивали при 40°С в течение 72 часов в атмосфере O2 (15 фунтов на кв. дюйм). Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® Silica Flash массой 20 г, элюция градиентом 0~30% этилацетат/петролейный эфир) и затем посредством препаративной ВЭЖХ (в условиях HCl) с получением 272В (170 мг, выход: 8,23%) и 272С (264 мг, выход: 12,78%) в виде белого твердого вещества.
[1297] Соединение 272В: 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,73 (dd, J=1,3, 8,3 Гц, 2Н), 7,64-7,46 (m, 4Н), 4,46 (q, J=7,2 Гц, 2Н), 1,42 (t, J=7,0 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 241,9.
[1298] Соединение 272С: 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,55-7,47 (m, 3Н), 7,45-7,39 (m, 2Н), 7,37 (s, 1H), 4,27 (q, J=7,0 Гц, 2Н), 1,25 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+H)+ 241,9.
[1299] К раствору 272В (170 мг, 704,69 мкмоль) в ТГФ (5 мл) и МеОН (5 мл) добавляли раствор LiOH⋅Н2О (148 мг, 3,52 ммоль) в H2O (5 мл) при 0°С. После добавления реакционную смесь перемешивали в течение 3 ч при 25°С и затем разбавляли H2O (10 мл) и подвергали экстракции МТБЭ (30 мл). Водную фазу нейтрализовали 1 н. HCl до рН ~ 4 и затем подвергали экстракции EtOAc (30 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Соединение 272D (98 мг, выход: 64,58%, белое твердое вещество): 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,76-7,74 (m, 2Н), 7,61-7,52 (m, 4Н), 2,83 (ушир. s, 1H).
[1300] Соединение 272 (40 мг, выход: 53,56%, белое твердое вещество) было получено, как описано в примере 6, из соответствующих исходных веществ, соединений 272D и 12G. Соединение 272: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,88 (ушир. d, J=7,8 Гц, 1H), 8,12 (ушир. s, 1H), 7,91-7,74 (m, 4Н), 7,71-7,57 (m, 3Н), 7,31-7,15 (m, 5Н), 5,47 (ушир. s, 1H), 3,25-2,96 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 388,1.
[1301] Следуя методике, использованной для получения соединения 274, соединение 273 (19 мг, выход: 44,44%, белое твердое вещество) получали из соответствующей карбоновой кислоты, промежуточного соединения 273А. Соединение 273А (201 мг, выход: 55,94%, белое твердое вещество): 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,58-7,53 (m, 1H), 7,46-7,42 (m, 5Н), 4,05 (ушир. s, 1H). Соединение 273: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,43 (ушир. d, J=7,8 Гц, 1H), 8,15 (ушир. s, 1H), 7,90 (ушир. s, 1H), 7,44 (ушир. d, J=8,5 Гц, 4Н), 7,36-7,25 (m, 7Н), 5,42-5,22 (m, 1H), 3,21 (ушир. d, J=11,5 Гц, 1H), 2,90-2,75 (m, 1H). МС (ИЭР) m/z (М+Н)+ 388,1.
ПРИМЕР 153
СОЕДИНЕНИЯ 274, 320
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-БРОМ-5-ХЛОР-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (274)
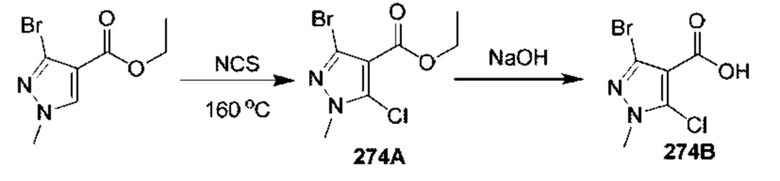
[1302] Раствор этил-3-бром-1-метил-1Н-пиразол-4-карбоксилата (500 мг, 2,15 ммоль) и NCS (574 мг, 4,30 ммоль) перемешивали. Смесь перемешивали при 160°С в течение 3 ч в атмосфере N2. Реакционную смесь разбавляли путем добавления CCl4 (20 мл) и затем разбавляли NaHCO3 (30 мл). Смесь подвергали экстракции ДХМ (20 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством препаративной ТСХ (SiO2, ПЭ:ЭА=5:1). Соединение 274А (180 мг, выход: 31,30%) получали в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 4,34 (q, J=7,1 Гц, 2Н), 3,85 (s, 3Н), 1,46-1,24 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 266,9.
[1303] К раствору соединения 274А (300 мг, 1,12 ммоль) в МеОН (10 мл) и Н2О (10 мл) добавляли NaOH (134 мг, 3,36 ммоль). Смесь перемешивали при 25°С в течение 3 ч. Реакционную смесь концентрировали и добавляли 20 мл воды, смесь подвергали экстракции МТБЭ (10 мл × 2), водный слой подкисляли с помощью 1 н. HCl до рН ~ 2~3 при 0°С и подвергали экстракции EtOAc (20 мл × 2), органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Соединение 274В (250 мг, выход: 93,22%) получали в виде белого твердого вещества, которое использовали на следующей стадии без очистки. 1Н ЯМР (400 МГц, CDCl3) δ 4,02-3,72 (m, 3Н). МС (ИЭР) m/z (М+H)+ 238,9.
[1304] Соединение 274 (70 мг, выход: 66,75%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединения 274В и 3-амино-2-гидрокси-4-фенилбутанамида (274D). Соединение 274: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,41-8,27 (m, 1H), 8,12 (s, 1H), 7,87 (s, 1H), 7,29 (dd, J=4,0 Гц, 4Н), 7,22 (dd, J=4,0 Гц, 1H), 5,36 (s, 1H), 3,78 (s, 3Н), 3,20 (dd, J=10,4 Гц, 1H), 2,90-2,81 (m, 1H). МС (ИЭР) m/z (М+Н)+ 415,0.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-2-(3-ФЕНИЛ-1H-ПИРАЗОЛ-1-ИЛ)ТИОФЕН-3-КАРБОКСАМИД (320)
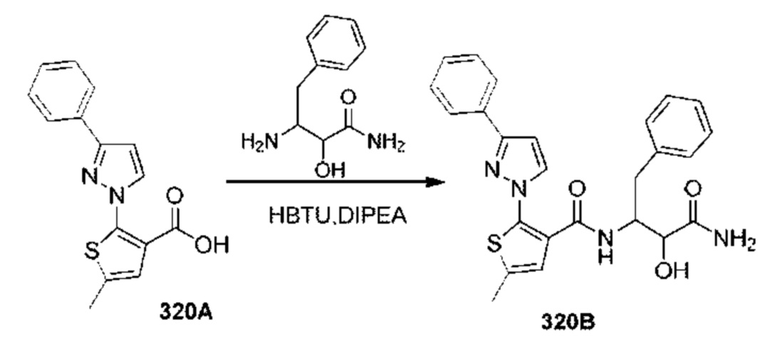
[1305] Соединение 320 (33,7 мг, выход: 51,5%, белое твердое вещество) было получено, как описано в примере 153, из соответствующей карбоновой кислоты, соединения 320А, и 3-амино-2-гидрокси-4-фенилбутанамида (274D). Соединение 320: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,88 (d, J=7,6 Гц, 1H), 8,12 (s, 1H), 7,97 (d, J=2,6 Гц, 1H), 7,85 (ушир. d, J=7,1 Гц, 3Н), 7,50-7,42 (m, 2Н), 7,41-7,34 (m, 1H), 7,33-7,19 (m, 5Н), 6,92 (d, J=2,6 Гц, 1H), 6,84 (d, J=1,0 Гц, 1H), 5,40-5,31 (m, 1H), 3,19 (dd, J=3,6, 14,0 Гц, 1H), 2,80 (dd, J=10,3, 13,8 Гц, 1H), 2,44 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 459,1.
ПРИМЕР 154
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (276)
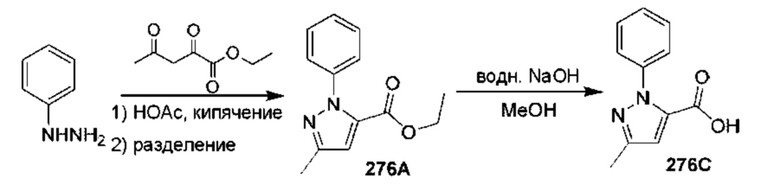
[1306] К смеси фенилгидразина (50 г, 462,3 ммоль) и этил-2,4-диоксопентаноата (76,8 г, 485,5 ммоль) в АсОН (600 мл) при 25°С. Смесь перемешивали при 100°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении для удаления АсОН. К остатку добавляли H2O (200 мл) и ЭА (200 мл), и затем смесь подщелачивали с помощью насыщенного водного NaHCO3 до рН водной фазы ~ 7-8. Отделенный водный слой подвергали экстракции ЭА (150 мл × 3), объединенные органические слои промывали насыщенным водным NaHCO3 (200 мл), насыщенным водным NaCl (200 мл), сушили над Na2SO4, фильтровали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали посредством КФХ (SiO2, петролейный эфир : этилацетат = 1:0~3:1). Соединение 276А (39,0 г, выход: 36,7%, желтое твердое вещество): 1Н ЯМР (ДМСО-d6, 400 МГц): δ 7,50-7,37 (m, 5Н), 6,88 (s, 1H), 4,16 (q, J=7,0 Гц, 2Н), 2,26 (s, 3Н), 1,14 (t, J=7,2 Гц, 3Н).
[1307] К смеси соединения 276А (250 мг, 1,1 ммоль) в МеОН (10 мл) добавляли NaOH (2 М, 3 мл) одной порцией при 25°С. Смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. К смеси добавляли H2O (10 мл), и смесь подкисляли с помощью водного раствора HCl (1 М) до рН ~ 3-4. Водную фазу подвергали экстракции ЭА (10 мл × 3). Объединенную органическую фазу промывали насыщенным водным NaCl (15 мл × 2), сушили над Na2SO4 и фильтровали при пониженном давлении с получением соединения 276С (170 мг, неочищенного) в виде желтого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 7,49-7,37 (m, 5Н), 6,82 (s, 1H), 2,25 (s, 3Н).
[1308] Соединение 276 (100 мг, выход: 65,68%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 276С. Соединение 276: 1Н ЯМР (CDCl3, 400 МГц): δ 9,14-9,00 (m, 1H), 8,09 (s, 1H), 7,85 (s, 1H), 7,44-7,11 (m, 10Н), 6,56 (s, 1H), 5,28 (s, 1H), 3,26-3,16 (m, 1H), 2,91-2,76 (m, 1H), 2,26 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 377,1.
ПРИМЕР 155
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(2-ФТОРФЕНИЛ)ИЗОКСАЗОЛ-4-КАРБОКСАМИД (277)
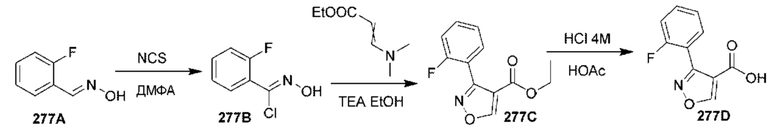
[1309] К суспензии 2-фторбензальдегида (10 г, 80,6 ммоль) и NH2OH⋅HCl (6,2 г, 88,6 ммоль) в EtOH (10 мл) и H2O (20 мл) добавляли лед (50 г). Затем добавляли по каплям водный раствор NaOH (8 г, 201 ммоль) в H2O (25 мл) в течение 10 мин, при этом большая часть твердого вещества растворялась. Затем смесь перемешивали в течение 2 часов при 16°С. Полученную смесь затем подкисляли с помощью HCl (5 н.). Далее смесь два раза подвергали экстракции дихлорметаном (100 мл). Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 1:0 до 200:1) с получением соединения 277А (10 г, выход: 89,2%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,68 (s, 1H), 8,39 (s, 1H), 7,74 (ушир. t, J=7,5 Гц, 1H), 7,39 (q, J=6,8 Гц, 1H), 7,18 (t, J=7,4 Гц, 1H), 7,14-7,06 (m, 1H).
[1310] К раствору соединения 277А (5 г, 35,9 ммоль) в ДМФА (20 мл) добавляли 1-хлорпирролидин-2,5-дион (5,3 г, 39,5 ммоль) с последующим перемешиванием при 20°С в течение 3 часов. Реакционную смесь разбавляли H2O (60 мл), и смесь подвергали экстракции этилацетатом (100 мл × 2). Органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 277В (5 г, выход: 80,2%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,15 (s, 1H), 7,68 (ушир. t, J=7,5 Гц, 1H), 7,50-7,39 (m, 1H), 7,23 (t, J=7,6 Гц, 1H), 7,20-7,13 (m, 1H).
[1311] К раствору этил-3-(диметиламино)акрилата (165 мг, 1,2 ммоль) и TEA (233 мг, 2,3 ммоль) в ТГФ (15 мл) добавляли по каплям раствор соединения 277В (400 мг, 2,3 ммоль) в ТГФ (5 мл) в течение более чем 30 мин. Смесь перемешивали при 20°С в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством препаративной ТСХ (SiO2, петролейный эфир : этилацетат = 1:1) с получением соединения 277С (240 мг, выход: 44,4%) в виде бледно-желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 8,94 (s, 1H), 7,51-7,36 (m, 2Н), 7,21-7,14 (m, 1H), 7,13-7,04 (m, 1H), 4,17 (q, J=7,1 Гц, 2Н), 1,15 (t, J=7,2 Гц, 3Н).
[1312] Смесь соединения 277С (230 мг, 977,9 мкмоль) в H2O (2,00 мл), НОАс (1,5 мл) и HCl (3 мл) нагревали до 130°С и перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с получением соединения 277D (120 мг, выход: 59,2%) в виде коричневого твердого вещества. Продукт использовали на следующей стадии без дополнительной очистки. МС (ИЭР) m/z (М+Н)+ 208,1.
[1313] Соединение 277 (33 мг, выход: 37,7%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 277D. Соединение 277: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,43 (ушир. s, 1H), 8,94 (ушир. d, J=6,8 Гц, 1H), 8,09 (ушир. s, 1H), 7,83 (ушир. s, 1H), 7,54 (ушир. d, J=5,3 Гц, 1H), 7,47-7,40 (m, 1H), 7,36-7,16 (m, 9Н), 5,31 (ушир. s, 1H), 3,17 (ушир. d, J=13,5 Гц, 1H), 2,89-2,75 (m, 1H), 2,89-2,75 (m, 1H). МС (ИЭР) m/z (М+Н)+ 382,1.
ПРИМЕР 156
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ФЕНИЛ-5-(ТРИФТОРМЕТИЛ)ИЗОКСАЗОЛ-4-КАРБОКСАМИД (278)
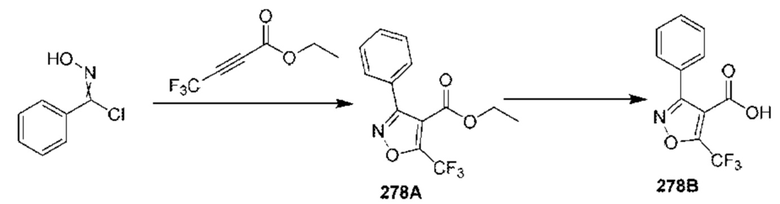
[1314] К смеси N-гидроксибензимидоилхлорида (1 г, 6,43 ммоль) и этил-4,4,4-трифторбут-2-иноата (1,28 г, 7,71 ммоль) в ТГФ (10 мл) добавляли TEA (1,3 г, 12,9 ммоль) при 25°С. Смесь перемешивали при 25°С в течение 1 ч, и к смеси добавляли H2O (10 мл), и смесь подвергали экстракции этилацетатом (10 мл × 2). Объединенную органическую фазу промывали солевым раствором (10 мл × 2), сушили с помощью безводного Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ с получением соединения 278А (1 г, выход: 54,5%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,68 (dd, J=1,3, 7,9 Гц, 2Н), 7,56-7,47 (m, 3Н), 4,39-4,31 (m, 2Н), 1,34-1,28 (m, 3Н).
[1315] К смеси соединения 278А (750 мг, 2,63 ммоль) в НОАс (2 мл) добавляли HCl (12 М, 939 мкл). Смесь перемешивали при 130°С в течение 48 ч. В анализе посредством ТСХ (дихлорметан:метанол = 10:1, Rf ~ 0,09) было обнаружено пятно, соответствующее целевому соединению. Смесь концентрировали с получением неочищенного соединения продукта 278В (450 мг, неочищенного) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (ушир. d, J=7,9 Гц, 2Н), 7,62-7,50 (m, 3Н).
[1316] Соединение 278 (15 мг, выход: 12,6%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 278В. Соединение 278: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,17 (ушир. d, J=9,2 Гц, 1H), 7,55-7,48 (m, 3Н), 7,43-7,36 (m, 4Н), 7,28-7,14 (m, 5Н), 5,88 (d, J=5,3 Гц, 1H), 4,60 (ушир. s, 1H), 4,04 (ушир. s, 1H), 3,31 (s, 12Н), 2,73-2,61 (m, 1H). МС (ИЭР) m/z (М+H)+ 432,1.
ПРИМЕР 157
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ХЛОР-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (280)
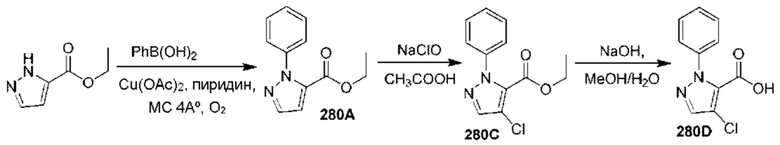
[1317] К смеси этил-1H-пиразол-5-карбоксилата (10,00 г, 71,36 ммоль), фенилбороновой кислоты (13,05 г, 107,04 ммоль), Ру (8,82 г, 111,50 ммоль, 9,0 мл) в ДХМ (120 мл) добавляли МС 4А° (40,00 г) и Cu(ОАс)2 (14,26 г, 78,50 ммоль), смесь перемешивали при 30°С в течение 154 ч. Реакционную смесь фильтровали, фильтрат концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле (ПЭ:ЭА=от 1:0 до 10:1) с получением соединения 280А (3,10 г, выход: 19,1%) в виде желтого масла. Соединение 280А: 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,81 (d, J=2,0 Гц, 1H), 7,57-7,37 (m, 5Н), 7,09 (d, J=2,0 Гц, 1H), 4,17 (q, J=7,0 Гц, 2Н), 1,15 (t, J=7,0 Гц, 3Н).
[1318] К раствору соединения 280А (1,0 г, 4,62 ммоль) в СН3СООН (15 мл) добавляли NaClO (24,2 г, 47,12 ммоль, 20,00 мл, чистота 14,5%). Смесь перемешивали при 25°С в течение 21 ч в атмосфере N2. В реакционную смесь добавляли Н2О (25 мл) для гашения реакции, и смесь разбавляли ЭА (20 мл) и перемешивали в течение 30 мин и затем подвергали экстракции ЭА (25 мл). Объединенные органические слои промывали Н2О (20 мл × 2), и затем промывали NaHCO3 (20 мл × 2), и затем промывали солевым раствором (15 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (ПЭ:ЭА = от 1:0 до 1:1) с получением соединения 280С (327 мг, выход: 28,1%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,69 (s, 1H), 7,53-7,31 (m, 5Н), 4,27 (q, J=7,1 Гц, 2Н), 1,22 (t, J=7,2 Гц, 3Н).
[1319] К раствору соединения 280С (300 мг, 1,20 ммоль) в МеОН (15 мл) добавляли NaOH (240 мг, 6,00 ммоль) в H2O (5 мл). Смесь перемешивали при 20°С в течение 2 ч. Реакционную смесь разбавляли МТБЭ (15 мл) и H2O (15 мл) и затем перемешивали в течение 10 мин. Водный слой отделяли, и органический слой подвергали экстракции H2O (15 мл × 2). рН объединенных водных слоев доводили до ~ 3 путем добавления 1 н. HCl, и затем водный слой подвергали экстракции ЭА (20 мл × 3). Объединенный органический слой промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 280D (252 мг, выход: 94,1%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, МЕТАНОЛ-d4) δ 7,76 (s, 1H), 7,52-7,38 (m, 5Н).
[1320] Соединение 280 (61 мг, выход: 61,2%, светло-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 280D. Соединение 280: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,46 (d, J=7,5 Гц, 1H), 8,20 (s, 1H), 7,97-7,86 (m, 2Н), 7,41-7,24 (m, 10Н), 5,48-5,35 (m, 1H), 3,21 (ушир. dd, J=3,2, 14,0 Гц, 1H), 2,80 (dd, J=10,5, 14,0 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 397,1.
ПРИМЕР 158
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ХЛОР-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (281)
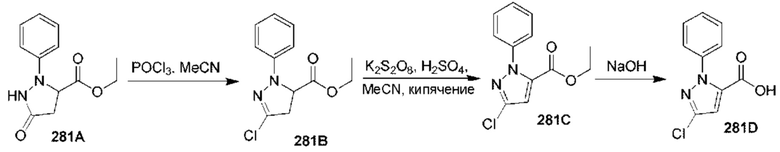
[13211 К EtOH (40 мл) добавляли Na (470 мг, 20,34 ммоль) при 20°С. После того, как весь натрий вступил в реакцию, смесь нагревали до 78°С и добавляли фенилгидразин (2,0 г, 18,49 ммоль, 1,82 мл) и перемешивали в течение 0,1 ч, и затем добавляли по каплям диэтилмалеат (3,5 г, 20,34 ммоль, 3,27 мл). Смесь перемешивали при 78°С в течение 4 ч. После охлаждения до 65°С реакционную смесь обрабатывали АсОН (2,0 г, 33,28 ммоль, 1,9 мл). Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли H2O (80 мл) и подвергали экстракции EtOAc (80 мл × 2). Объединенные органические слои промывали солевым раствором (50 мл × 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 1:1) с получением соединения 281А (2,72 г, выход: 58,40%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,24 (s, 1H), 7,34-7,24 (m, 2Н), 7,02-6,92 (m, 3Н), 4,59 (dd, J=2,0, 9,7 Гц, 1H), 4,24-4,13 (m, 2Н), 3,00-2,86 (m, 1H), 2,46-2,39 (m, 1H), 1,23 (t, J=7,1 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 235,0.
[1322] К раствору соединения 281А (2,7 г, 11,53 ммоль) в MeCN (50 мл) добавляли POCl3 (2,15 г, 14,02 ммоль). Смесь перемешивали при 85°С в течение 5 ч. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток нейтрализовали насыщ. NaHCO3 до рН~8, затем подвергали экстракции ДХМ (50 мл × 2). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 10/1 до 5:1) с получением соединения 281В (1,9 г, выход: 65,21%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,31-7,17 (m, 2Н), 6,98 (ушир. d, J=7,3 Гц, 2Н), 6,89 (ушир. t, J=7,2 Гц, 1H), 4,86-4,60 (m, 1H), 4,34-4,14 (m, 2Н), 3,62-3,37 (m, 1H), 3,33-3,17 (m, 1H), 1,37-1,11 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 252,9.
[1323] К раствору соединения 281В (800 мг, 3,17 ммоль) в MeCN (20 мл) добавляли H2SO4 (620 мг, 6,34 ммоль, 337,95 мкл) и K2S2O8 (1,29 г, 4,76 ммоль). Смесь перемешивали при 80°С в течение 6 ч. Реакционную смесь концентрировали при пониженном давлении для удаления большей части растворителя. Остаток добавляли по каплям в H2O (30 мл), фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток промывали 30% MeCN (5 мл × 2). Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 20/1 до 15:1) с получением соединения 281С (190 мг, выход: 21,52%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,47 (d, J=1,3 Гц, 4Н), 7,23-7,14 (m, 1H), 4,15 (q, J=7,1 Гц, 2Н), 1,12 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 250,9.
[1324] К раствору соединения 281С (150 мг, 598,37 мкмоль) в ТГФ/H2O (5 мл/5 мл) добавляли NaOH (120 мг, 2,99 ммоль). Смесь перемешивали при 25°С в течение 4 ч. Реакционную смесь разбавляли H2O (20 мл), затем смесь концентрировали при пониженном давлении для удаления ТГФ и подвергали экстракции МТБЭ (15 мл × 2). Водные слои нейтрализовали путем добавления 1 н. HCl до рН ~ 3 и затем подвергали экстракции ДХМ (20 мл × 2). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 281D (120 мг, выход: 90,08%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,50-7,34 (m, 5Н), 6,97 (s, 1Н).
[1325] Соединение 281 (20 мг, выход: 40,20%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 281D. Соединение 281: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,27 (d, J=7,7 Гц, 1H), 8,11 (s, 1H), 7,87 (s, 1H), 7,41-7,34 (m, 3Н), 7,30 (ушир. d, J=7,1 Гц, 2Н), 7,28-7,23 (m, 3Н), 7,21-7,16 (m, 2Н), 6,80 (s, 1H), 5,31-5,19 (m, 1H), 3,24-3.
ПРИМЕР 159
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3,5-ДИХЛОР-1-МЕТИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (282)
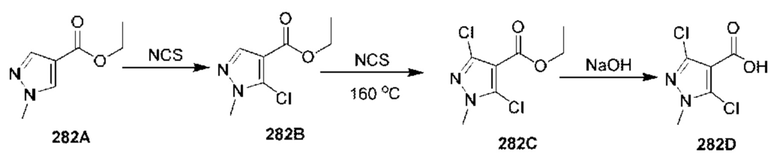
[1326] К раствору этил-1H-пиразол-4-карбоксилата (5 г, 35,68 ммоль) и Cs2CO3 (23,25 г, 71,36 ммоль) в ДМФА (100 мл) добавляли MeI (10,13 г, 71,36 ммоль, 4,44 мл). Смесь перемешивали при 25°С в течение 16 ч. Смесь фильтровали, фильтрат разбавляли H2O (500 мл), подвергали экстракции ЭА (50 мл × 3), сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1). Соединение 282А (4,5 г, выход: 81,81%) получали в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,84 (d, J=8,5 Гц, 2Н), 4,24 (q, J=7,3 Гц, 2Н), 4,03-3,70 (m, 3Н), 1,30 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 155,0.
[1327] К раствору соединения 282А (1,5 г, 9,73 ммоль) добавляли NCS (2,6 г, 19,46 ммоль) в атмосфере N2. Смесь перемешивали при 160°С в течение 3 ч. К реакционной смеси добавляли CCl4 (10 мл) и насыщенный водный NaHCO3 (10 мл). Смесь подвергали экстракции ДХМ (20 мл × 2), и затем объединенные органические слои и органическую фазу высушивали над Na2SO4, фильтровали, и фильтрат концентрировали под вакуумом. Соединение 282В (1,84 г, неочищенное) было получено в виде коричневого масла, которое непосредственно использовали на следующей стадии. МС (ИЭР) m/z (М+Н)+ 188,9.
[1328] К раствору соединения 282В (1,84 г, 9,76 ммоль) добавляли NCS (2,61 г, 19,52 ммоль) в атмосфере N2, и смесь перемешивали при 160°С в течение 4 ч в атмосфере N2. К реакционной смеси добавляли CCl4 (10 мл) и насыщенный водный NaHCO3 (10 мл), и смесь подвергали экстракции ДХМ (20 мл × 2), и затем объединенные органические слои и органическую фазу высушивали над Na2SO4, фильтровали, и фильтрат концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1). Соединение 282С (482 мг, выход: 22,14%) получали в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 4,35 (q, J=7,1 Гц, 2Н), 3,84 (s, 3Н), 1,38 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 222,9.
[13291 К смеси соединения 282С (480 мг, 2,15 ммоль) в H2O (5 мл) и МеОН (10 мл) добавляли NaOH (258 мг, 6,45 ммоль) порциями при 20°С и перемешивали в течение 2 ч. Смесь концентрировали для удаления МеОН, затем смесь разбавляли Н2О (20 мл) и подвергали экстракции МТБЭ (50 мл × 2). Водные слои подкисляли до рН ~ 2 с помощью 1 н. HCl, затем раствор подвергали экстракции ЭА (50 мл × 3). Органические слои сушили над Na2SO4 и концентрировали с получением промежуточного соединения 282D (390 мг, выход: 93,02%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,27 (s, 1H), 3,77 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 194,8 и 196,8.
[1330] Соединение 282 (50 мг, выход: 46,25%, грязно-белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 282D. Соединение 282: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,32 (dd, J=7,7 Гц, 1H), 8,21-8,07 (m, 1H), 7,87 (s, 1H), 7,36-7,16 (m, 5Н), 5,33 (s, 1H), 3,82-3,69 (m, 3Н), 3,19 (dd, J=13,2 Гц, 1H), 2,95-2,78 (m, 1H). МС (ИЭР) m/z (М+Н)+ 369,1 и 371,1.
ПРИМЕР 160
СОЕДИНЕНИЯ 283-284
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-БЕНЗИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (283)
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-БЕНЗИЛ-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (284)
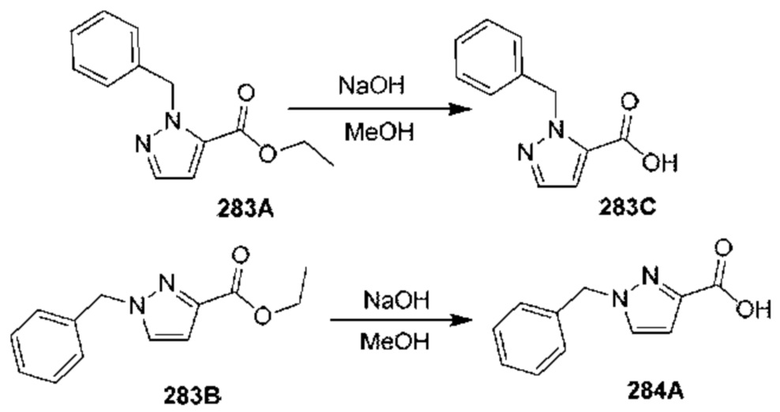
[1331] К смеси этил-1H-пиразол-5-карбоксилата (1 г, 7,14 ммоль) и бензилбромида (0,93 мл, 7,8 ммоль) в ДМФА (30 мл) добавляли K2CO3 (1,18 г, 8,6 ммоль) одной порцией при 25°С. Смесь перемешивали при 25°С в течение 14 ч. В реакционную смесь добавляли H2O (80 мл), и смесь подвергали экстракции ЭА (50 мл × 3). Объединенную органическую фазу промывали насыщ. NaCl (50 мл × 2). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством КФХ (SiO2, петролейный эфир/этилацетат = от 1/0 до 3/1) с получением соединения 283А (523 мг, выход 31,7%) в виде бесцветной жидкости и соединения 283В (989 мг, выход 60,1%) в виде белого твердого вещества.
[1332] Соединение 283А: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,65 (d, J=2,0 Гц, 1H), 7,36-7,23 (m, 3Н), 7,13 (d, J=7,0 Гц, 2Н), 6,95 (d, J=2,0 Гц, 1H), 5,72 (s, 2Н), 4,27 (q, J=7,2 Гц, 2Н), 1,26 (t, J=7,0 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 231,0.
[1333] Соединение 283В: 1H ЯМР (ДМСО-d6, 400 МГц) δ 7,99 (d, J=2,5 Гц, 1H), 7,40-7,29 (m, 3Н), 7,28-7,23 (m, 2Н), 6,77 (d, J=2,3 Гц, 1H), 5,43 (s, 2Н), 4,25 (q, J=7,0 Гц, 2Н), 1,27 (t, J=7,0 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 231,0.
[1334] К смеси соединения 283А (517 мг, 2,2 ммоль) в МеОН (10 мл) добавляли NaOH (2 М, 6 мл, 12,0 ммоль) одной порцией при 25°С. После перемешивания при 25°С в течение 2 ч реакционную смесь концентрировали при пониженном давлении для удаления МеОН, водную фазу подкисляли с помощью водного раствора HCl (1 М) до рН ~ 4-5. Твердое вещество отфильтровывали и сушили с получением соединения 283С (389 мг, неочищенного) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,60 (d, J=2,0 Гц, 1H), 7,34-7,22 (m, 3Н), 7,15-7,08 (m, 2Н), 6,88 (d, J=2,0 Гц, 1H), 5,73 (s, 2Н).
[1335] Соединение 283 (177 мг, выход 89,0%) было получено, как описано в примере 12, из соответствующей карбоновой кислоты, промежуточного соединения 283С. Соединение 283: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,91 (d, J=7,1 Гц, 1H), 8,11 (s, 1H), 7,84 (s, 1H), 7,52 (s, 1H), 7,26 (s, 8Н), 7,14-7,01 (m, 2Н), 6,89 (s, 1H), 5,60 (s, 2Н), 5,40-5,24 (m, 1H), 3,27-3,13 (m, 1H), 2,95-2,80 (m, 1H). МС (ИЭР) m/z (М+Н)+ 377,1.
[1336] Следуя той же методике, которую использовали для получения соединения 283, соединение 284 (35 мг, выход 35,7%, белое твердое вещество) получали из соответствующей карбоновой кислоты, промежуточного соединения 284А. Соединение 284: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,18 (ушир. d, J=7,5 Гц, 1H), 8,05 (ушир. s, 1H), 7,94-7,79 (m, 2Н), 7,40-7,30 (m 3Н), 7,28-7,18 (m, 7Н), 6,64 (s, 1H), 5,40 (s, 3Н), 3,19 (ушир. dd, J=3,7, 13,7 Гц, 1H), 3,04 (ушир. dd, J=8,9, 13,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 377,1.
ПРИМЕР 161
(S)-3-МЕТИЛ-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-(ПИРАЗИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (285)
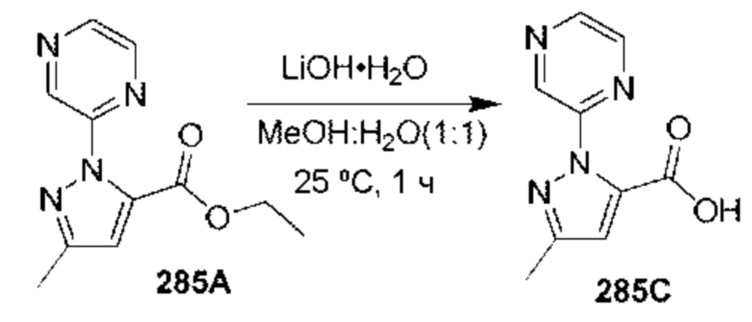
[1337] Смесь 2-гидразинилпиразина (2 г, 18,16 ммоль) и этил-2,4-диоксопентаноата (2,87 г, 18,16 ммоль) в АсОН (40 мл) перемешивали при 118°С в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления АсОН. Остаток разбавляли H2O (8 мл), доводили рН до ~ 7 с помощью Na2CO3 и затем подвергали экстракции ДХМ (200 мл × 3). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® Silica Flash массой 40 г, элюция градиентом 0~20% этилацетат/петролейный эфир, скорость потока 40 мл/мин). Соединение 285А (1,5 г, выход 35,57%) получали в виде белого твердого вещества. Соединение 285А: 1Н ЯМР (400 МГц, CDCl3) δ 8,98-8,93 (m, 1H), 8,60-8,54 (m, 1H), 8,46-8,41 (m, 1H), 6,78 (s, 1H), 4,34-4,25 (m, 2Н), 2,38 (s, 3Н), 1,27 (t, J=7,2 Гц, 3Н).
[1338] Промежуточное соединение 285С (1,2 г, выход 92,84%, белое твердое вещество) подвергали реакции снятия защиты, как описано в примере 85, из соединения 285А. Соединение 285С: 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,96-8,92 (m, 1H), 8,70-8,66 (m, 1H), 8,58-8,54 (m, 1H), 6,84 (s, 1H), 2,25-2,24 (m, 1H), 2,26 (s, 2Н).
[1339] Соединение 285 (143,0 мг, выход 53,28%, белое твердое вещество) было получено, как описано в примере 6, из соответствующих промежуточных соединений 285С и 21G ((S)-2-амино-3-фенилпропан-1-ола). Соединение 285: 1Н ЯМР (400 МГц, CDCl3) δ 9,74 (s, 1H), 9,19-9,16 (m, 1H), 9,03-8,96 (m, 1H), 8,48-8,43 (m, 1H), 8,06-8,03 (m, 1H), 7,27-7,25 (m, 1H), 7,24-7,20 (m, 3Н), 7,17-7,12 (m, 2Н), 6,80 (s, 1H), 5,00-4,91 (m, 1H), 3,39-3,31 (m, 1H), 3,30-3,23 (m, 1H), 2,37 (s, 3Н). МС (ИЭР) m/z (М+H2O+Н)+ 354,2.
ПРИМЕР 162
(S)-3-МЕТИЛ-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-(ПИРАЗИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (286)
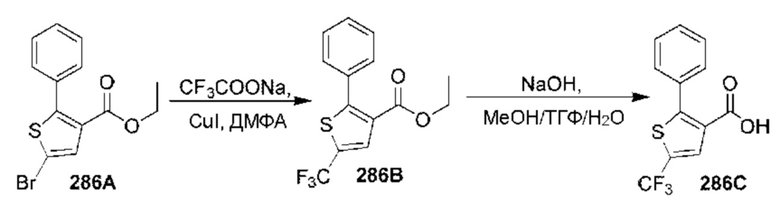
[1340] К раствору соединения 270А (1 г, 4,30 ммоль) в ДМФА (20 мл) добавляли NBS (1,53 г, 8,60 ммоль) при 80°С, и смесь перемешивали при 80°С в течение 1,5 ч. Смесь выливали в воду (40 мл) и подвергали экстракции этилацетатом (20 мл × 2), объединенный органический слой промывали насыщенным NaHCO3 (20 мл), солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством хроматографии на силикагеле (от петролейного эфира до смеси петролейный эфир : этилацетат = 20:1) с получением соединения 286А (1,3 г, выход: 97,2%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,49-7,41 (m, 3Н), 7,40-7,34 (m, 3Н), 4,16 (q, J=7,2 Гц, 2Н), 1,16 (t, J=7,1 Гц, 3Н).
[1341] Смесь соединения 286А (1 г, 3,21 ммоль), CuI (1,22 г, 6,42 ммоль) и 2,2,2-трифторацетата натрия (4,37 г, 32,1 ммоль) в DMA (20 мл) нагревали до 160°С в течение 5 ч. К смеси добавляли этилацетат (30 мл), воду (50 мл), 1 н. HCl (50 мл), смесь фильтровали, и фильтрат отделяли. Органический растворитель промывали водой (30 мл), солевым раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством ЖХСД (жидкостной хроматографии среднего давления) (от петролейного эфира до смеси петролейный эфир : этилацетат = 30:1) с получением соединения 286В (210 мг, выход: 21,8%), в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,85 (d, J=1,1 Гц, 1H), 7,53-7,47 (m, 2Н), 7,47-7,37 (m, 3Н), 4,21 (q, J=7,2 Гц, 2Н), 1,20 (t, J=7,1 Гц, 3Н).
[1342] Смесь соединения 286В (210 мг, 699 мкмоль) и NaOH (55,9 мг, 1,40 ммоль) в ТГФ (5 мл), EtOH (3 мл), Н2О (2 мл) перемешивали при 10°С в течение 12 ч. Органические растворители удаляли под вакуумом, рН водного слоя доводили до ~ 5 с помощью 1 н. HCl, и слой подвергали экстракции этилацетатом (20 мл), органический слой сушили над Na2SO4, фильтровали и концентрировали с получением соединения 286С (160 мг, выход: 84%), в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,81 (d, J=1,1 Гц, 1H), 7,46-7,41 (m, 2Н), 7,40-7,31 (m, 3Н).
[1343] Соединение 286 (28,3 мг, выход: 45,5%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединения 286С и 3-амино-2-гидрокси-4-фенилбутанамида (274D). Соединение 286: 1Н ЯМР (400 МГц, CDCl3) δ 7,76 (s, 1H), 7,61-7,38 (m, 5Н), 7,26-7,16 (m, 3Н), 6,80 (ушир. d, J=5,6 Гц, 2Н), 6,70 (ушир. s, 1H), 6,01 (ушир. d, J=5,9 Гц, 1H), 5,61-5,47 (m, 2Н), 3,24 (dd, J=5,0, 14,2 Гц, 1H), 2,93 (dd, J=7,6, 14,2 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 447,1.
ПРИМЕР 163
(S)-3-МЕТИЛ-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1-(ПИРАЗИН-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (287)
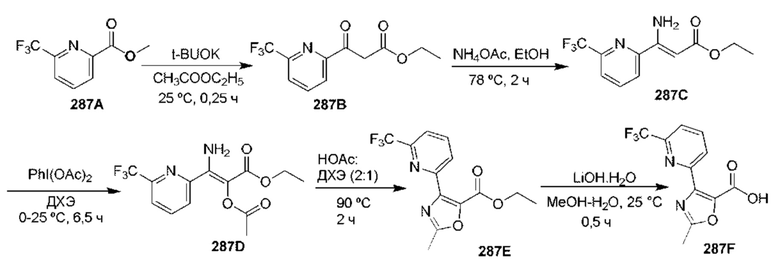
[1344] К раствору 6-(трифторметил)пиколиновой кислоты (10 г, 52,33 ммоль) в МеОН (150 мл) добавляли по каплям H2SO4 (1,03 г, 10,47 ммоль, 557,88 мкл). После перемешивания при 65°С в течение 10 часов смесь охлаждали до комнатной температуры, нейтрализовали с помощью насыщенного водного раствора NaHCO3 и подвергали экстракции CH2Cl2 (70 мл × 3). Органическую фазу объединяли, сушили с помощью безводного Na2SO4 и упаривали с получением неочищенного промежуточного соединения 287А (9,20 г, выход 85,71%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,32 (d, J=8,0 Гц, 1H), 8,09-8,05 (m, 1H), 7,88 (d, J=8,0 Гц, 1H), 4,03 (s, 3Н).
[1345] К раствору соединения 287А (4,5 г, 21,94 ммоль) в СН3СООС2Н5 (150 мл) добавляли t-BuOK (3,20 г, 28,52 ммоль). Смесь перемешивали в течение 0,25 часа при 25°С. В смесь добавляли Н2О (150 мл) для гашения реакции. Органический слой отделяли, и водный слой подвергали экстракции ЭА (70 мл × 3). Органическую фазу объединяли, сушили с помощью безводного Na2SO4, фильтровали и упаривали с получением промежуточного соединения 287В (3,95 г, выход 66,86%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,24 (d, J=8,0 Гц, 1H), 8,09-8,06 (m, 1H), 7,87 (d, J=7,2 Гц, 1H), 4,22-4,16 (m, 4Н), 1,26-1,23 (m, 3Н).
[1346] К раствору соединения 287В (3,90 г, 14,93 ммоль) в EtOH (80 мл) добавляли NH4OAc (5,75 г, 74,65 ммоль), затем смесь перемешивали при 78°С в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли ЭА (500 мл) и промывали насыщенным водным раствором NaHCO3 (30 мл × 3) и солевым раствором (30 мл × 3). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной флэш-хроматографии (ПЭ:ЭА=от 30/1 до 10/1) с получением соединения 287С (2,52 г, выход 64,87%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,94-7,93 (m, 2Н), 7,73-7,71 (m, 1H), 5,38 (s, 1H), 4,23-4,18 (m, 2Н), 1,33-1,29 (m, 3Н).
[1347] К смеси соединения 287С (2,5 г, 9,61 ммоль) в ДХЭ (60,00 мл) добавляли PhI(ОАс)2 (4,02 г, 12,49 ммоль) при 0°С в атмосфере N2 четырьмя порциями, смесь перемешивали при 0°С в течение 6 ч и затем медленно нагревали до 25°С. Затем смесь перемешивали при 25°С в течение 0,5 ч. В реакционную смесь добавляли для гашения реакции насыщенный водный NaHCO3 (150 мл) при 0°С, медленно нагревали до 25°С, и смесь подвергали экстракции ДХМ (100 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной флэш-хроматографии (ПЭ:ЭА = от 20/1 до 10/1) с получением соединения 287D (650 мг, выход 21,23%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,34-8,18 (m, 1H), 8,16-8,13 (s, 1H), 7,96-7,90 (m, 1H), 4,28-4,23 (m, 2Н), 2,13 (s, 3Н), 1,28-1,23 (m, 3Н).
[1348] Смесь соединения 287D (600 мг, 1,89 ммоль) в ДХЭ (5 мл) и СН3СООН (10 мл) перемешивали при 90°С в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и с получением остатка. Остаток очищали посредством колоночной флэш-хроматографии (ПЭ:ЭА = от 30/1 до 10/1) с получением соединения 287Е (120 мг, 221,31 мкмоль, выход 11,71%, чистота 55,37%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,42-8,33 (m, 1H), 8,17-8,15 (m, 1H), 7,99-7,26 (m, 1H), 4,40-4,37 (m, 2Н), 2,62 (s, 3Н), 1,35-1,32 (m, 3Н).
[1349] К раствору соединения 287Е (110 мг, 366,39 мкмоль) в МеОН (6 мл) и Н2О (3 мл) добавляли LiOH⋅Н2О (61 мг, 1,47 ммоль), затем смесь перемешивали при 25°С в течение 0,5 часа. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН. Остаток разбавляли Н2О (20 мл), доводили рН до ~ 3 с помощью 1 н. HCl, затем подвергали экстракции EtOAc (30 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 287F (86 мг, выход 59,17%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,45-8,26 (m, 2Н), 8,10-8,07 (m, 1H), 2,58 (s, 3Н).
[1350] Соединение 287 (12,1 мг, выход 20,26%, грязно-белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединения 287F и 3-амино-2-гидрокси-4-фенилбутанамида (274D). Соединение 287: 1Н ЯМР (400 МГц, CDCl3): δ 11,10 (d, J=7,2 Гц, 1H), 8,46 (d, J=8,0 Гц, 1H), 8,18-7,99 (m, 1H), 7,72 (d, J=8,0 Гц, 1H), 7,23-7,04 (m, 5Н), 6,70 (ушир. s, 1H), 5,64-5,52 (m, 1H), 5,45 (ушир. s, 1H), 3,57-3,47 (m, 1H), 3,10-2,94 (m, 1H), 2,55 (d, J=0,80 Гц, 3Н). МС (ИЭР) m/z (М+1)+ 447,2.
ПРИМЕР 164
СОЕДИНЕНИЯ 288-289
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ПИРИДИН-2-ИЛ)ИЗОКСАЗОЛ-4-КАРБОКСАМИД (288)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(4-ФТОРФЕНИЛ)ИЗОКСАЗОЛ-4-КАРБОКСАМИД (289)
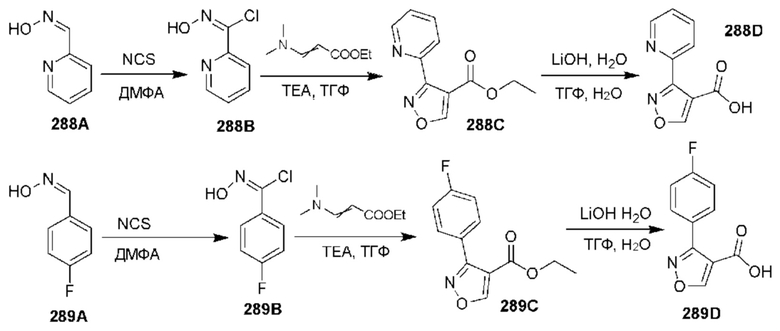
[1351] В раствор пиколинальдегида (200 мг, 1,87 ммоль) добавляли NH2OH⋅HCl (156 мг, 2,24 ммоль) и NaOAc (184 мг, 2,24 ммоль) в EtOH (15 мл). Смесь нагревали при 60°С в течение 2 часов. Реакционную смесь концентрировали. Добавляли дихлорметан (50 мл). Органическую фазу промывали Н2О (10 мл) и солевым раствором (10 мл), сушили с помощью безводного Na2SO4, фильтровали и концентрировали под вакуумом с получением промежуточного соединения 288А (180 мг, выход: 78,8%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,47 (ушир. s, 1H), 8,57 (ушир. d, J=4,2 Гц, 1H), 8,50 (ушир. d, J=4,4 Гц, 1H), 8,26 (s, 1H), 7,85 (t, J=7,7 Гц, 1H), 7,76 (d, J=7,9 Гц, 1H), 7,69-7,62 (m, 1H), 7,40-7,32 (m, 1H), 7,26-7,17 (m, 1Н).
[1352] К смеси соединения 288А (180 мг, 1,47 ммоль) в ДМФА (2 мл) добавляли NCS (216 мг, 1,62 ммоль) одной порцией при 20°С. Смесь перемешивали при 20°С в течение 12 часов. Реакционную смесь концентрировали. Остаток выливали в воду (20 мл). Водную фазу подвергали экстракции этилацетатом (50 мл × 3). Объединенную органическую фазу промывали солевым раствором (20 мл × 2), сушили с помощью безводного Na2SO4, фильтровали и концентрировали под вакуумом с получением промежуточного соединения 288В (200 мг, выход: 87,1%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 10,79 (ушир. s, 1H), 8,72 (d, J=4,2 Гц, 1H), 7,94 (d, J=8,2 Гц, 1H), 7,80 (dt, J=1,7, 7,8 Гц, 1H), 7,39 (ddd, J=0,8, 5,0, 7,4 Гц, 1H), 2,79 (s, 1H).
[1353] К смеси этил-3-(диметиламино)акрилата (92 мг, 639 мкмоль), TEA (129 мг, 1,28 ммоль) в ТГФ (10 мл) добавляли раствор соединения 288В (200 мг, 1,28 ммоль) в ТГФ (10 мл) в течение более чем 20 мин. Смесь перемешивали при 20°С и перемешивали в течение 12 часов. Реакционную смесь концентрировали. Остаток очищали посредством препаративной ТСХ (SiO2, петролейный эфир : этилацетат = 1:1) с получением соединения 288С (150 мг, выход: 53,8%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3-d) δ 9,02-8,97 (m, 1H), 8,75 (d, J=4,6 Гц, 1H), 7,87-7,76 (m, 2Н), 7,45-7,36 (m, 1H), 4,27 (q, J=7,3 Гц, 2Н), 1,26 (t, J=7,2 Гц, 3Н).
[1354] К смеси соединения 288С (150 мг, 687 мкмоль) в ТГФ (5 мл) и H2O (1 мл) добавляли LiOH⋅H2O (58 мг, 1,4 ммоль). Смесь перемешивали при 15°С в течение 12 часов. Смесь концентрировали для удаления растворителя. рН смеси доводили до ~ 5 с помощью водного раствора HCl (1 М), и смесь концентрировали с получением промежуточного соединения 288D (130 мг, выход: 99,5%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,57 (s, 1H), 8,76 (d, J=4,9 Гц, 1H), 8,11-8,05 (m, 2Н), 7,65 (dt, J=3,1, 5,3 Гц, 1H).
[1355] Соединение 288 (73,8 мг, выход: 81,7%, желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 288D. Соединение 288: 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. d, J=7,0 Гц, 1H), 9,44 (s, 1H), 8,46 (d, J=4,4 Гц, 1H), 8,11-8,01 (m, 2Н), 7,81 (ушир. s, 1H), 7,65-7,51 (m, 2H), 7,17-7,08 (m, 5H), 5,62-5,52 (m, 1H), 3,27 (dd, J=5,0, 13,8 Гц, 1H), 3,13-3,06 (m, 1H). МС (ИЭР) m/z (M+H)+ 365,1.
[1356] Следуя методике, использованной для получения соединения 288, соединение 289 (83,1 мг, выход: 70,9%, белое твердое вещество) получали из соответствующей карбоновой кислоты, промежуточного соединения 289D. Промежуточное соединение 289D (100 мг, выход: 56,8%, белое твердое вещество): 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,55 (s, 1H), 7,89-7,81 (m, 2Н), 7,39-7,28 (m, 2Н). Соединение 289: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,32 (s, 1H), 9,10 (d, J=7,6 Гц, 1H), 8,11 (s, 1H), 7,86 (s, 1H), 7,65-7,56 (m, 2Н), 7,33-7,21 (m, 7Н), 5,40-5,29 (m, 1H), 3,19 (dd, J=3,9, 13,9 Гц, 1H), 2,84 (dd, J=10,0, 13,9 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 382,1.
ПРИМЕР 165
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-МЕТИЛ-1-ФЕНИЛ-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (290)
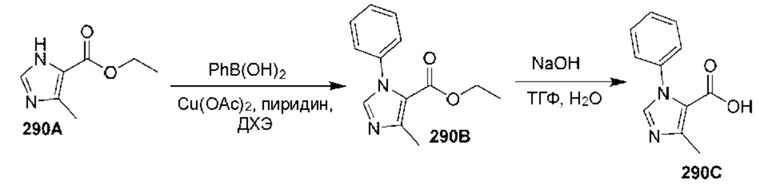
[1357] Смесь этил-2-хлор-3-оксобутаноата (16 г, 97,2 ммоль), формамида (43,8 г, 972 ммоль), H2O (3,50 г, 194 ммоль) в автоклаве перемешивали при 180°С в течение 3,5 часов. Реакционную смесь фильтровали, и осадок на фильтре растворяли в ДХМ (200 мл). Смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением остатка. Соединение 290А (2 г, неочищенное) получали в виде желтого твердого вещества. МС (ИЭР) m/z (М+Н)+ 154,8.
[1358] Смесь 290А (1,8 г, 11,7 ммоль), фенилбороновой кислоты (2,85 г, 23,4 ммоль), Cu(ОАс)2 (3,18 г, 17,5 ммоль), пиридина (1,85 г, 23,4 ммоль) и МС 4А° (2 г) в ДХЭ (60 мл) дегазировали и продували О2 3 раза, и затем смесь перемешивали при 60°С в течение 12 часов в атмосфере О2. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (петролейный эфир : этилацетат = от 50:1 до 2:1). Соединение 290В (1,15 г, выход: 42,7%) получали в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,53 (s, 1H), 7,45-7,35 (m, 3Н), 7,27-7,16 (m, 2H), 4,18-4,05 (m, 2H), 2,53 (s, 3Н), 1,10 (t, J=7,1 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 231,0.
[13591 Смесь 290В (300 мг, 1,30 ммоль), LiOH⋅H2O (109 мг, 2,60 ммоль) в ТГФ (10 мл), Н2О (10 мл) перемешивали при 15°С в течение 12 ч. Анализ посредством ЖХМС свидетельствовал о том, что большая часть 290В оставалась. К смеси добавляли NaOH (416 мг, 10,4 ммоль), и смесь перемешивали при 70°С в течение 12 ч. В реакционную смесь добавляли водн. HCl для доведения рН до ~5, и затем смесь фильтровали, и осадок на фильтре концентрировали с получением указанного продукта. Соединение 290С (300 мг, неочищенное) получали в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,90 (s, 1H), 7,50-7,37 (m, 3Н), 7,36-7,28 (m, 2Н), 2,39 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 203,1.
[13601 Соединение 290 (15,3 мг, выход: 11,5%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 290С.Соединение 290: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,23 (ушир. d, J=7,0 Гц, 1H), 7,73 (s, 1H), 7,80-7,56 (m, 1H), 7,41-7,13 (m, 11Н), 5,31 (ушир. s, 1H), 3,20 (dd, J=3,7, 13,8 Гц, 1H), 2,92-2,80 (m, 1H), 2,18-2,13 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 377,2.
ПРИМЕР 166
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ХЛОР-1-ФЕНИЛ-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (291)
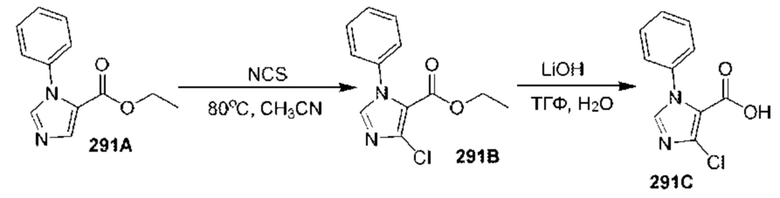
[1361] Смесь этил-1H-имидазол-5-карбоксилата (10 г, 71,4 ммоль), фенилбороновой кислоты (13,1 г, 107 ммоль), Cu(ОАс)2 (19,4 г, 107 ммоль), пиридина (11,3 г, 142,72 ммоль) и МС 4А° (4,0 г) в ДХЭ (200 мл) дегазировали и продували О2 3 раза, и затем смесь перемешивали при 60°С в течение 12 часов в атмосфере О2. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 20:1 до 5:1). Соединение 291А (2,8 г, выход: 18,2%) получали в виде желтого твердого вещества. (Примечание: структура была подтверждена посредством NOE). 1H ЯМР (400 МГц, CDCl3) δ 7,79 (s, 1H), 7,63 (s, 1H), 7,47-7,33 (m, 3Н), 7,32-7,22 (m, 2Н), 4,14 (q, J=7,2 Гц, 2H), 1,16 (t, J=7,1 Гц, 3Н).
[13621 К раствору 291А (1 г, 4,62 ммоль) в CH3CN (20 мл) добавляли NCS (925 мг, 6,93 ммоль) при 80°С. Смесь перемешивали при 80°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток растворяли в ДХМ (50 мл), фильтровали, и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 80:1 до 50:1) и затем очищали посредством препаративной ТСХ (петролейный эфир : этилацетат = 3:1). Соединение 291В (150 мг, выход: 13,0%) получали в виде желтого масла. (Примечание: Структура была подтверждена посредством НМВС). 1H ЯМР (400 МГц, CDCl3) δ 7,77-7,62 (m, 1H), 7,54-7,37 (m, 3Н), 7,29-7,10 (m, 2Н), 4,09 (q, J=7,1 Гц, 2Н), 1,11 (t, J=7,2 Гц, 3Н).
[1363] Смесь 291В (150 мг, 598 мкмоль), LiOH⋅H2O (50,2 мг, 1,20 ммоль) в ТГФ (5 мл), H2O (5 мл) перемешивали при 15°С в течение 12 часов. В реакционную смесь добавляли водн. HCl для доведения рН до ~ 5, и затем смесь фильтровали, и осадок на фильтре концентрировали с получением указанного продукта. Соединение 291С (130 мг, неочищенное) получали в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) 6 7,45 (ушир. d, J=2,2 Гц, 4Н), 7,29 (ушир. s, 2Н). МС (ИЭР) m/z (М+Н)+ 222,8.
[1364] Соединение 291 (47,1 мг, выход: 47,6%, коричневое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 291С.Соединение 291: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,74 (ушир. d, J=7,7 Гц, 1H), 8,02 (ушир. s, 1H), 7,79 (ушир. s, 1H), 7,66 (s, 1H), 7,43 (ушир. d, J=4,0 Гц, 3Н), 7,37-7,10 (m, 7Н), 5,16 (ушир. t, J=6,8 Гц, 1H), 3,13 (ушир. d, J=10,8 Гц, 1H), 2,86-2,68 (m, 1H). МС (ИЭР) m/z (М+Н)+ 397,1.
ПРИМЕР 167
(2S,4R)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-МЕТИЛ-1-ФЕНИЛПИРРОЛИДИН-2-КАРБОКСАМИД (292)
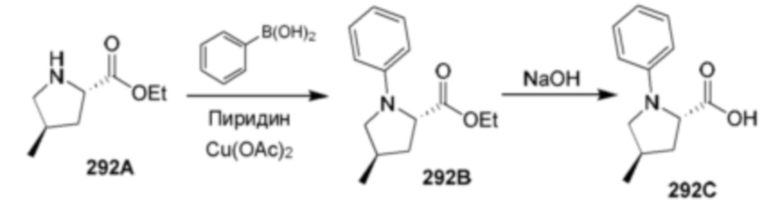
[1365] К смеси 1-(трет-бутил)-2-этил-(2S,4R)-4-метилпирролидин-1,2-дикарбоксилата (2 г, 7,77 ммоль) в EtOAc (5 мл) добавляли HCl/EtOAc (4 М, 20 мл) при 25°С. Смесь перемешивали при 25°С в течение 10 ч. Смесь концентрировали с получением остатка, и к остатку добавляли насыщенный водный Na2CO3 (1,5 мл), затем добавляли ДХМ (200 мл). Затем смесь сушили с помощью безводного Na2SO4, фильтровали и концентрировали под вакуумом с получением неочищенного соединения 292А (1,9 г, неочищенного) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,76 (ушир. s, 2Н), 5,29 (s, 1H), 4,48 (dd, J=4,1, 9,2 Гц, 1H), 4,27 (q, J=7,2 Гц, 2Н), 3,73 (dd, J=7,4, 11,3 Гц, 1H), 2,95 (dd, J=9,1, 11,3 Гц, 1H), 2,48-2,28 (m, 2Н), 2,05-1,95 (m, 1H), 1,31 (t, J=7,2 Гц, 3Н), 1,13 (d, J=6,6 Гц, 3Н).
[1366] К смеси соединения 292А (1,9 г, 12,1 ммоль) и фенилбороновой кислоты (2,95 г, 24,2 ммоль) в ДХЭ (15 мл) добавляли МС 4А° (4 г), пиридин (1,91 г, 24,2 ммоль), Cu(ОАс)2 (3,29 г, 18,1 ммоль) одной порцией при 25°С. Смесь перемешивали при 60°С в течение 10 ч в атмосфере О2 (15 фунтов на кв. дюйм). Реакционную смесь фильтровали, и фильтрат концентрировали под вакуумом. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 100/1, 50/1) с получением соединения 292В (900 мг, выход: 31,9%) в виде светлого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,42-7,32 (m, 2Н), 7,29-7,17 (m, 3Н), 4,28-4,08 (m, 3Н), 3,67 (t, J=8,0 Гц, 1H), 2,90 (t, J=8,7 Гц, 1H), 2,69-2,55 (m, 1H), 2,24-2,14 (m, 1H), 1,96-1,83 (m, 1H), 1,24 (t, J=7,1 Гц, 3Н), 1,12 (d, J=6,6 Гц, 3Н).
[1367] К смеси соединения 292В (300 мг, 1,29 ммоль) в EtOH (5 мл) и H2O (1 мл) добавляли NaOH (129 мг, 3,23 ммоль) при 25°С. Смесь перемешивали при 25°С в течение 10 ч. Реакционную смесь концентрировали, и водную фазу подвергали экстракции этилацетатом (15 мл × 2). Затем к водной фазе добавляли HCl (1 М) до рН ~ 3. Целевой продукт подвергали экстракции этилацетатом (15 мл × 2). Объединенную органическую фазу промывали солевым раствором (20 мл × 2), сушили с помощью безводного Na2SO4, фильтровали и концентрировали под вакуумом с получением неочищенного соединения 292С (100 мг, неочищенного) в виде желтого масла.
[1368] Соединение 292 (12,7 мг, выход: 25,5%, грязно-белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 292С.Соединение 292: 1H ЯМР (400 МГц, CDCl3) δ 7,30-7,19 (m, 4H), 7,16-7,09 (m, 1H), 7,09-6,97 (m, 3Н), 6,88-6,77 (m, 2H), 6,73 (ушир. s, 1H), 6,55 (dd, J=8,2, 18,3 Гц, 2H), 5,75-5,61 (m, 1H), 5,61-5,36 (m, 1H), 4,04-3,92 (m, 1H), 3,65-3,47 (m, 1H), 3,42 (dd, J=5,0, 14,0 Гц, 1H), 3,23-3,02 (m, 1H), 2,90 (dd, J=8,9, 14,0 Гц, 1H), 2,82-2,68 (m, 1H), 2,41-2,29 (m, 1H), 2,28-2,04 (m, 1H), 2,03-1,89 (m, 1H), 1,88-1,70 (m, 1H), 1,13-1,00 (m, 3Н). МС (ИЭР) m/z (М+Н)+ 380,2.
ПРИМЕР 168
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-ФЕНИЛ-1,2,5-ОКСАДИАЗОЛ-3-КАРБОКСАМИД (293)
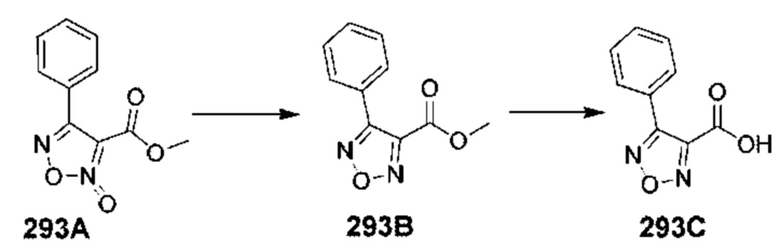
[1369] К раствору метилциннамата (1 г, 1 экв.) в пиридине (20 мл) добавляли NOBF4 (2,34 г, 3,2 экв.) при 0°С. Реакционную смесь перемешивали при 0°С в течение 2 days. Раствор вливали в ледяную воду и подвергали экстракции посредством EtOAc (3 раза). Объединенную органическую фазу промывали водой, сушили над NaSO4 и концентрировали при пониженном давлении. Остаток очищали на ISCO с получением соединения 293А.
[1370] Раствор соединения 293А (0,5 г) в триметилфосфите (5 мл) нагревали при 100°С в атмосфере N2 в течение ночи. Реакционную смесь охлаждали до комнатной температуры и добавляли в нее для гашения реакции 1 н. HCl (10 мл). Смесь подвергали экстракции EtOAc (3 раза). Объединенную органическую фазу промывали водой, сушили над NaSO4 и концентрировали при пониженном давлении. Остаток очищали на ISCO с получением соединения 293В.
[1371] Соединение 293 было получено, как описано в примере 5, из соответствующей кислоты, промежуточного соединения 293С, которое было получено путем обработки соединения 293В (720 мг) с помощью LiOH в МеОН и водой. 1Н ЯМР (400 МГц, ДМСО): δ 9,81 (d, 1H), 8,22 (s, 1H), 7,95 (s, 1H), 7,6-7,2 (m, 10Н), 5,53 (m, 1H), 3,25 (dd, 1H), 2,83 (dd, 1H) ppm. МС (ИЭР) m/z (M+Na)+ 387,2.
ПРИМЕР 169
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(1-МЕТИЛ-1H-БЕНЗО[d]ИМИДАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (294)
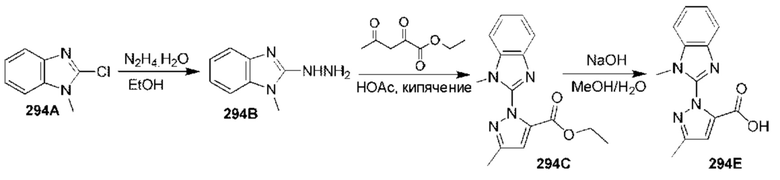
[1372] MeI (6,1 мл, 98,3 ммоль) добавляли к смеси 2-хлор-1H-бензо[d]имидазола (5,0 г, 32,8 ммоль) и K2CO3 (13,6 г, 98,3 ммоль) в ДМФА (20 мл). Смесь перемешивали при 25°С в течение 1 ч. Нерастворимое вещество удаляли путем фильтрования, и фильтрат обрабатывали посредством ЭА (50 мл), Н2О (50 мл). Отделяли органический слой и водный слой подвергали экстракции ЭА (35 мл × 3). Объединенный органический слой промывали Н2О (35 мл × 2), солевым раствором (35 мл × 2), сушили над MgSO4, фильтровали и концентрировали. Остаток растирали с МТБЭ/РЕ (об./об.=1/1, ~20 мл) с получением соединения 294А (3,3 г, выход 60,38%) в виде бледно-желтого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,60-7,56 (m, 2Н), 7,31-7,24 (m, 2Н), 3,80 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 167,0.
[1373] К смеси соединения 294А (3,3 г, 19,8 ммоль в EtOH (10 мл) добавляли N2H4.H2O (5,8 г, 99,1 ммоль, 85% чистота) одной порцией. Смесь перемешивали при 110°С в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток растирали МТБЭ (20 мл), осадок фильтровали и сушили под вакуумом с получением соединения 294В (2,4 г, выход 73,1%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,25-7,21 (m, 1H), 7,17-7,13 (m, 1H), 6,98-6,89 (m, 2Н), 3,45 (s, 3Н).
[1374] К смеси соединения 294В (1,0 г, 6,17 ммоль) и этил-2,4-диоксопентаноата (1,0 г, 6,48 ммоль) в АсОН (20 мл) перемешивали при 110°С в течение 5 ч. Реакционную смесь концентрировали при пониженном давлении для удаления АсОН. Остаток добавляли Н2О (50 мл) и ЭА (50 мл), и затем смесь подкисляли с помощью насыщенного водного NaHCO3 до рН водной фазы ~ 7-8. Отделенный водный слой подвергали экстракции ЭА (100 мл × 3), Объединенные органические слои промывали насыщенным водным NaCl (150 мл), сушили над Na2SO4, фильтровали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали посредством КФХ (SiO2, петролейный эфир : этилацетат = 1:0 ~ 3:1) с получением соединения 294С (494 мг, выход 27,9%) в виде желтой жидкости.
[13751 Соединение 294С: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,69-7,63 (m, 2Н), 7,41-7,35 (m, 1H), 7,33-7,28 (m, 1H), 7,05 (s, 1H), 4,10 (q, J=7,1 Гц, 2Н), 3,55 (s, 3Н), 2,32 (s, 3Н), 0,99 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 285,1.
[1376] К смеси соединения 294С (645 мг, 2,3 ммоль) в МеОН (10 мл) добавляли NaOH (2 М, 5,7 мл) одной порцией при 25°С. Смесь перемешивали при 25°С в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН, остаток добавляли H2O (10 мл) и подкисляли с помощью 1 н. HCl раствор до рН водной фазы ~ 6-7. Твердое вещество отделяли и фильтровали при пониженном давлении с получением соединения 294Е (482 мг, неочищенного) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 13,58 (s, 1H), 7,69-7,61 (m, 2Н), 7,40-7,34 (m, 1H), 7,33-7,26 (m, 1H), 6,96 (s, 1H), 3,54 (s, 3Н), 2,31 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 257,0.
[1377] Соединение 294 (27 мг, выход 57,7%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 294Е. Соединение 294: 1Н ЯМР (CDCl3, 400 МГц) δ 9,98 (d, J=6,0 Гц, 1H), 7,63 (d, J=7,9 Гц, 1H), 7,44-7,29 (m, 4Н), 7,04 (m, 3Н), 6,99 (m, 2Н), 6,90-6,85 (m, 1H), 6,78-6,71 (m, 1H), 5,74-5,65 (m, 1H), 3,81 (s, 3Н), 3,36 (m, 1H), 3,09 (m, 1H), 2,37 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 431,1.
ПРИМЕР 170
(S)-N-(4-(ЦИКЛОПРОПИЛАМИНО)-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(1-МЕТИЛ-1H-БЕНЗО[d]ИМИДАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (295)
[1378] Соединение 295 (50,0 мг, выход: 50,16%, белое твердое вещество) было получено, как описано в примере 20, из соответствующей карбоновой кислоты, промежуточного соединения 294Е. Соединение 295: 1Н ЯМР (CDCl3, 400 МГц) δ 9,89-9,76 (m, 1H), 7,66-7,62 (m, 1H), 7,43-7,37 (m, 2Н), 7,36-7,30 (m, 1H), 7,06-7,00 (m, 3Н), 7,00-6,94 (m, 2H), 6,92-6,85 (m, 2H), 5,77-5,65 (m, 1H), 3,79 (s, 3Н), 3,43-3,32 (m, 1H), 3,13-3,02 (m, 1H), 2,86-2,73 (m, 1H), 2,37 (s, 3Н), 0,91-0,81 (m, 2H), 0,65-0,52 (m, 2H). МС (ИЭР) m/z (М+Н)+ 471,1.
ПРИМЕР 171
СОЕДИНЕНИЯ 296-297
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(6-МЕТОКСИБЕНЗО[d]ТИАЗОЛ-2-ИЛ)-5-МЕТИЛ-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (296)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-(6-МЕТОКСИБЕНЗО[d]ТИАЗОЛ-2-ИЛ)-3-МЕТИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (297)

[1379] К NH2NH2.H2O (6,9 мл, 120 ммоль) добавляли HCl (12 М, 5,50 мл) при перемешивании при 0-5°С, а затем этиленгликоль (30 мл). Далее порциями добавляли 6-метоксибензо[d]тиазол-2-амин (3,6 г, 20,0 ммоль). Смесь нагревали до 125°С и перемешивали в течение 3 ч. После охлаждения до комнатной температуры осадок собирали путем фильтрования. Осадок на фильтре промывали EtOH (5 мл × 3) с получением соединения 296А (3,0 г, 15,37 ммоль, выход 76,8%) в виде бледно-зеленого твердого вещества.
[1380] Смесь соединения 296А (1,0 г, 5,1 ммоль) и этил-2,4-диоксопентаноата (0,7 мл, 5,1 ммоль) в НОАс (20 мл) нагревали до 120°С и перемешивали в течение 3 ч. Смесь концентрировали. Остаток обрабатывали МеОН (15 мл). Нерастворимое вещество удаляли путем фильтрования. Фильтрат концентрировали и остаток очищали посредством препаративной ВЭЖХ с получением соединения 296С (670 мг, выход 41,2%) в виде бледно-желтого твердого вещества и соединения 296В (74 мг, выход 4,6%) в виде розового твердого вещества. Нерастворимое вещество (0,5 г, нечистое) в виде розового твердого вещества обрабатывали ДХМ (50 мл). Нерастворимое вещество удаляли путем фильтрования. Фильтрат промывали насыщенным NaHCO3 (15 мл × 3), солевым раствором (15 мл × 2), сушили над MgSO4, фильтровали и концентрировали с получением соединения 296В (0,35 г, выход 21,5%) в виде розового твердого вещества.
[1381] Соединение 296В: 1H ЯМР (CDCl3, 400 МГц) δ 7,80 (d, J=9,2 Гц, 1H), 7,30 (d, J=2,4 Гц, 1H), 7,06 (dd, J=2,4, 8,8 Гц, 1H), 6,72 (s, 1H), 4,42 (q, J=6,8 Гц, 2Н), 3,89 (s, 3Н), 2,82 (s, 3Н), 1,42 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 317,9.
[1382] Соединение 296С: 1Н ЯМР (CDCl3, 400 МГц) δ 7,80 (d, J=8,8 Гц, 1H), 7,30 (d, J=2,4 Гц, 1H), 7,80 (dd, J=2,0, 8,8 Гц, 1H) 6,71 (s, 1H), 4,37 (q, J=7,2 Гц, 2Н), 3,88 (s, 3Н), 2,37 (s, 3Н), 1,30 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (М+Н)+ 317,9.
[1383] К раствору соединения 296С (400 мг, 1,26 ммоль) в МеОН (15 мл) добавляли NaOH (2 М, 3,15 мл, 6,3 ммоль). Смесь перемешивали при 25°С в течение 12 ч. Смесь разбавляли Н2О (50 мл) и летучий растворитель удаляли путем выпаривания. Полученный водный раствор подкисляли до рН~3 с помощью 1 н. HCl. Осадок собирали и подвергали азеотропной перегонке с толуолом с получением соединения 296D (320 мг, выход 87,8%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 7,78 (d, J=9,0 Гц, 1H), 7,70 (d, J=2,6 Гц, 1H), 7,11 (dd, J=2,5, 8,9 Гц, 1H), 6,86 (s, 1H), 3,82 (s, 3Н), 2,28 (s, 3Н).
[1384] Соединение 296 (100 мг, выход 43,9%, бледно-желтое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 296D. Соединение 296: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 10,20 (ушир. d, J=7,3 Гц, 1H), 8,12 (s, 1H), 7,86 (ушир. s, 1H), 7,64 (d, J=2,4 Гц, 1H), 7,56 (d, J=9,0 Гц, 1H), 7,18 (q, J=7,8 Гц, 1Н), 7,12 (ушир. d, J=6,8 Гц, 1H), 7,05 (dd, J=2,6, 9,0 Гц, 1H), 6,73 (s, 1H), 5,59-5,49 (m, 1H), 3,81 (s, 3H), 3,22 (ушир. dd, J=4,5, 14,0 Гц, 1H), 3,02 (ушир. dd, J=8,5, 14,4 Гц, 1H), 2,27 (s, 3Н). MC (ИЭР) m/z (M+H)+ 464,1.
[1385] Следуя той же методике, которую использовали для соединения 296, соединения 297 (30 мг, выход 54,3%, белое твердое вещество) получали из соответствующей карбоновой кислоты, промежуточного соединения 297А. Соединение 297:1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,55 (ушир. d, J=7,5 Гц, 1H), 8,12 (s, 1H), 7,88-7,80 (m, 2Н), 7,72 (d, J=2,6 Гц, 1H), 7,31-7,25 (m, 4Н), 7,22-7,17 (m, 1H), 7,11 (dd, J=2,6, 8,8 Гц, 1H), 6,76 (s, 1H), 5,45-5,35 (m, 1H), 3,83 (s, 3Н), 3,22 (ушир. dd, J=4,1, 13,8 Гц, 1H), 3,04 (dd, J=9,4, 13,8 Гц, 1H), 2,73 (s, 3H). МС (ИЭР) m/z (М+Н)+ 464,1.
ПРИМЕР 172
СОЕДИНЕНИЯ 298-299
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-МЕТИЛ-1-(4-МЕТИЛБЕНЗО[d]ТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (298)
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-5-МЕТИЛ-1-(4-МЕТИЛБЕНЗО[d]ТИАЗОЛ-2-ИЛ)-1H-ПИРАЗОЛ-3-КАРБОКСАМИД (299)
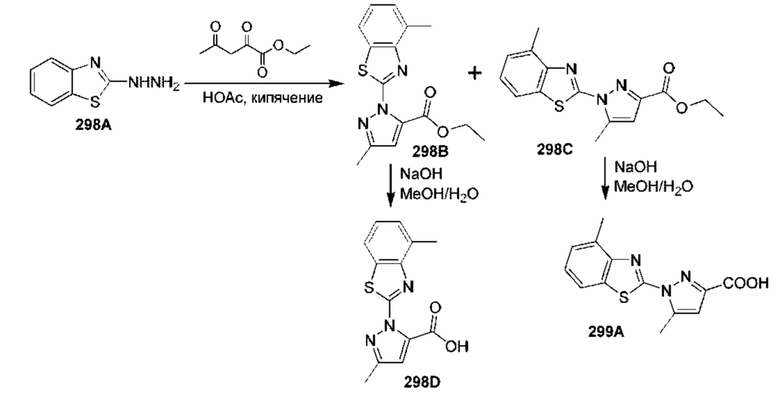
[1386] К смеси 4-метилбензо[d]тиазол-2-амина (5,0 г, 30, 5 ммоль) и NH2NH2.H2O (19,2 мл, 335 ммоль) в этиленгликоле (30 мл) добавляли HCl (12 М, 2,5 мл). Смесь нагревали 120°С и перемешивали в течение 5 ч. После охлаждения до комнатной температуры наблюдали осаждение. Осадок собирали путем фильтрования и промывали посредством EtOH (15 мл) с получением соединения 298А (2,3 г, выход 41,7%) в виде белого игольчатого кристалла. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,98 (ушир. s, 1H), 7,46 (d, J=8,0 Гц, 1H), 7,00 (d, J=7,6 Гц, 1H), 6,88-6,84 (m, 2Н), 4,98 (s, 2Н), 2,38 (s, 3H). МС (ИЭР) m/z (М+H)+ 179,8.
[1387] Смесь соединения 298А (1,7 г, 9,5 ммоль) и этил-2,4-диоксопентаноата (1,5 г, 9,5 ммоль) в АсОН (30 мл) нагревали до 125°С и перемешивали в течение 3 ч. Смесь концентрировали. Остаток обрабатывали МеОН (15 мл). Нерастворимое вещество удаляли путем фильтрования. Фильтрат концентрировали и остаток очищали посредством препаративной ВЭЖХ (FA) с получением соединения 298В (260 мг, 9,1% выход) получали в виде желтого твердого вещества и соединение 298С (1,12 г, выход 39,2%). Нерастворимое вещество (1,4 г, нечистое) обрабатывали ДХМ (50 мл) и насыщенным водным NaHCO3 (15 мл). Отделяли органический слой, и затем промывали насыщенным NaHCO3 (15 мл × 2), солевым раствором (15 мл × 3), сушили над MgSO4, фильтровали и концентрировали. Остаток очищали посредством КФХ (ПЭ/ЭА=10/1) с получением соединения 298В (620 мг, 21,7% выход) в виде желтого твердого вещества.
[1388] Соединение 298В: 1H ЯМР (CDCl3, 400 МГц) δ 7,71-7,65 (m, 1H), 7,28 (d, J=5,3 Гц, 2Н), 6,74 (d, J=0,9 Гц, 1H), 4,43 (q, J=7,3 Гц, 2Н), 2,87 (d, J=0,9 Гц, 3H), 2,69 (s, 3H), 1,43 (t, J=7,2 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 302,0.
[13891 Соединение 298С: 1H ЯМР (CDCl3, 400 МГц) δ 7,681-7,66 (m, 1H), 7,30-7,26 (m, 2Н), 6,66 (s, 1H), 4,39 (q, J=7,2 Гц, 2Н), 2,65 (s, 3H), 2,38 (s, 3H), 1,30 (t, J=6,8 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 301,9.
[1390] NaOH (2 М, 2,5 мл, 5,0 ммоль) добавляли к раствору этильного производного соединения 298В (300 мг, 1,0 ммоль) в МеОН (10 мл). Смесь перемешивали при 25°С в течение 2 ч. Наблюдали образование густого белого осадка. Смесь разбавляли H2O (30 мл). и летучий растворитель удаляли путем упаривания. Остаток подкисляли до рН~3 с помощью 1 н. HCl. Осадок собирали и подвергали азеотропной перегонке с толуолом с получением соединения 298D (190 мг, выход 69,8%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц) δ 13,32 (ушир. s, 1H), 7,93-7,87 (m, 1H), 7,37-7,31 (m, 2Н), 6,83 (s, 1H), 2,79 (s, 3H), 2,62 (s, 3H).
[1391] Соединение 298 (35 мг, выход 42,1%, белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 298D. Соединение 298: 1H ЯМР (ДМСО-d6, 400 МГц) δ 8,60 (ушир. d, J=7,0 Гц, 1H), 8,14 (ушир. s, 1H), 7,94 (ушир. s, 1H), 7,88 (ушир. s, 1H), 7,41-7,17 (m, 8Н), 6,81 (s, 1H), 5,44 (ушир. s, 1H), 3,23 (ушир. s, 1H), 3,27-3,23 (m, 1H), 3,13-3,02 (m, 1H), 2,81 (ушир. s, 3H), 2,65 (ушир. s, 3H). МС (ИЭР) m/z (М+Н)+ 448,1.
[1392] Следуя той же методике, которую использовали для соединения 298, соединение 299 (30 мг, выход 25,0%, белое твердое вещество) получали из соответствующей карбоновой кислоты, промежуточного соединения 299А. Соединение 299: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 11,49 (d, J=6,8 Гц, 1H), 7,68-7,62 (m, 1H), 7,32-7,28 (m, 1H), 7,26-7,21 (m, 1H), 7,20-7,04 (m, 6Н), 6,73 (s, 1H), 5,56-5,43 (m, 2Н), 3,55-3,47 (m, 1H), 3,25-3,16 (m, 1H), 2,39 (s, 3H), 2,36 (s, 3H). МС (ИЭР) m/z (М+Н)+ 448,1.
ПРИМЕР 173
СОЕДИНЕНИЯ 306-307
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-ЦИКЛОПРОПИЛ-1-ФЕНИЛ-1H-ПИРАЗОЛ-5-КАРБОКСАМИД (306)
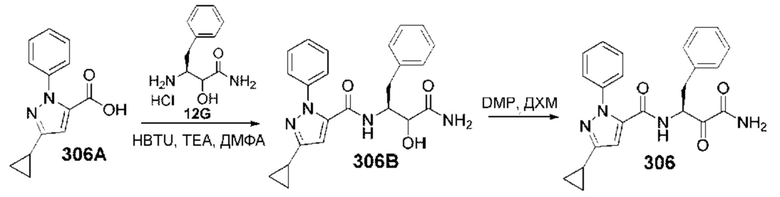
[1393] Соединение 306 (25 мг, 24%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединений 306А и 12G. Соединение 306: МС (ИЭР) m/z (М+Н)+ 403.
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ФЕНИЛ-1H-ИНДОЛ-2-КАРБОКСАМИД (307)
[1394] Соединение 307 синтезировали из соответствующих исходных веществ с использованием тех же методик, которые были описаны ранее для соединения 306.
ПРИМЕР 174
СОЕДИНЕНИЯ 314, 494
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-5-(ПИРИДИН-2-ИЛ)ОКСАЗОЛ-4-КАРБОКСАМИД (314)
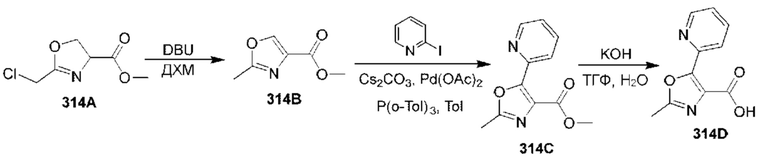
[1395] Na (29,55 мг, 1,29 ммоль) растворяли в МеОН (90 мг). Полученный раствор добавляли к смеси МеОН (10 мл) и ДХМ (90 мл) при 0-5°С, спустя 5 мин добавляли 2-хлорацетонитрил (10,2 мл, 160,7 ммоль), и полученную смесь перемешивали при 0-5°С в течение 1,5 ч. Затем добавляли этилацетимидата гидрохлорид (20 г, 128,55 ммоль, гидрохлорид) при 0-5°С. Полученную суспензию оставляли нагреваться до 20°С и перемешивали в течение 18 ч. Н2О (50 мл) добавляли к смеси и смесь перемешивали в течение 15 мин до ensure осадок растворяли. Отделяли органический слой, промывали солевым раствором (30 мл), сушили над MgSO4, фильтровали и концентрировали с получением соединения 314А (20,2 г, выход 88,5%) в виде прозрачного масла, которое непосредственно использовали на следующей стадии. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 4,87-4,82 (m, 1H), 4,55-4,46 (m, 2Н), 4,39 (s, 2Н), 3,71 (s, 3H).
[1396] К раствору соединения 314А (20,2 г, 113,75 ммоль) в ДХМ (100 мл) медленно добавляли DBU (17,2 мл, 113,75 ммоль). Смесь перемешивали при 25°С в течение 1 ч. Смесь обрабатывали 2 н. НСl (40 мл). Отделяли органический слой и затем промывали Н2О (30 мл), солевым раствором (30 мл), сушили над MgSO4, фильтровали и концентрировали с получением соединения 314В (13,5 г, выход 84,1%) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,68 (s, 1H), 3,76 (s, 3H), 2,42 (s, 3H).
[1397] Cs2CO3 (4,6 г, 14,2 ммоль) добавляли к смеси соединения 314В (1,0 г, 7,1 ммоль) и 2-иодпиридин (2,9 г, 14,2 ммоль) в толуоле (20,00 мл). Затем добавляли Р(о-толил)3 (216 мг, 0,71 ммоль) и Pd(OAc)2 (80 мг, 0,35 ммоль). Смесь дегазировали 3 раза. Затем смесь нагревали до 110°С и перемешивали в течение 18 ч. Смесь фильтровали через целит; осадок на фильтре промывали ЭА (15 мл × 2). Объединенный фильтрат концентрировали. Остаток очищали посредством колоночной флэш-хроматографии (ПЭ/ЭА=от 10/1 до 1/1) с получением соединения 314С (1,0 г, выход 64,6%) в виде бледно-желтого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,69-8,67 (m, 1H), 8,08-8,06 (m, 1H), 7,96-7,91 (m, 1H), 7,48-7,45 (m, 1H), 3,77 (s, 3H), 2,50 (s, 3H).
[1398] К смеси соединения 314С (500 мг, 2,3 ммоль) в ТГФ (10 мл) и Н2О (2 мл) добавляли KОН (1,28 г, 22,9 ммоль) одной порцией при 25°С. Смесь перемешивали при 25°С в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении для удаления ТГФ. Водную фазу подкисляли с помощью водного раствора HCl (1 М) до рН~4-5, и затем подвергали экстракции ДХМ (20 мл × 5). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением соединения 314D (600 мг, неочищенного) в виде светло-желтого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,84-8,75 (m, 1H), 8,25-8,19 (m, 1H), 8,06 (d, J=8,2 Гц, 1H), 7,71-7,66 (m, 1H), 2,56 (s, 3H).
[1399] Соединение 314 (30 мг, выход 26,2%, белое твердое вещество) было получено, как в случае соединения 12, из соответствующих исходных веществ, соединения 314D и 3-амино-2-гидрокси-4-фенил-бутанамид (274D). Соединение 314: 1Н ЯМР (CDCl3, 400 МГц) δ 11,59 (d, J=6,2 Гц, 1H), 8,14-8,09 (m, 1H), 8,03-7,98 (m, 1H), 7,87-7,80 (m, 1H), 7,26-7,22 (m, 1H), 7,18-7,05 (m, 5Н), 6,81 (s, 1H), 5,86-5,78 (m, 1H), 5,64 (s, 1H), 3,50-3,41 (m, 1H), 3,37-3,29 (m, 1H), 2,58 (s, 3H). МС (ИЭР) m/z (М+Н)+ 379,1.
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-5-ФЕНИЛОКСАЗОЛ-4-КАРБОКСАМИД (494)
[1400] Соединение 494 (40 мг, выход 25,1%, белое твердое вещество) было получено, как в случае соединения 314, из соответствующих исходных веществ, соединения 314В и иодбензола, следуя методикам, описанным для соединения 12, с получением соединения 494. Соединение 494: 1Н ЯМР (CDCl3, 400 МГц) δ 8,20-7,97 (m, 3H), 7,80 (ушир. s, 1H), 7,61 (ушир. d, J=10,0 Гц, 1H), 7,49-7,34 (m, 3H), 7,31-7,04 (m, 5Н), 6,22 (ушир. s, 0,22Н), 6,09 (ушир. s, 0,22Н), 5,46 (dt, J=4,8, 8,0 Гц, 0,75Н), 4,59 (dt, J=3,0, 10,3 Гц, 0,28Н), 3,27 (dd, J=5,0, 14,1 Гц, 1H), 3,11 (ушир. dd, J=8,3, 14,1 Гц, 1H), 2,54-2,50 (m, 3H). МС (ИЭР) m/z (М+Н)+ 378,1.
ПРИМЕР 175
(S)-1-МЕТИЛ-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-3-ФЕНИЛ-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (317)

[1401] К смеси этил-3-иод-1H-пиразол-4-карбоксилата (30 г, 112,7 ммоль) в ДМФА (200 мл) добавляли Cs2CO3 (110,22 г, 338,28 ммоль) одной порцией при 25°С. Затем добавляли иодметан (18,83 мл, 302,45 ммоль). Смесь перемешивали при 25°С в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении для удаления большей части ДМФА. Смесь обрабатывали ЭА (100 мл) и Н2О (100 мл). Отделяли органический слой и водный слой подвергали экстракции ЭА (50 мл × 3). Объединенный органический слой промывали солевым раствором (100 мл × 2), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством КФХ (SiO2, РЕ:ЭА=1:0-5:1) с получением соединения 317В (17,24 г, выход 54,59%) в виде белого твердого вещества. Соединение 317В: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,25 (s, 1H), 4,20 (q, J=7,1 Гц, 2Н), 3,87 (s, 3H), 1,29-1,23 (m, 3H).
[1402] К смеси соединения 317В (10,0 г, 35,7 ммоль) и фенилбороновой кислоты (8,71 г, 71,4 ммоль) в 1,4-диоксане (300 мл) и Н2О (80 мл) добавляли K2CO3 (9,87 г, 71,4 ммоль) и Pd(dppf)Cl2 (2,61 г, 3,57 ммоль). Смесь дегазировали и продували N2 3 раза и затем перемешивали при 80°С в течение 18 ч. Реакционную смесь концентрировали при пониженном давлении с удалением 1,4-диоксана. Смесь добавляли ЭА (150 мл), и затем промывали Н2О (100 мл × 3). Органический слой сушили над Na2SO4 и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии (SiO2, петролейный эфир:этилацетат = 1:0 до 3:1) с получением соединения 317С (8,30 г, неочищенного) в виде светло-красного твердого вещества, 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,34 (s, 1H), 7,71-7,64 (m, 2Н), 7,41-7,33 (m, 3H), 4,20-4,06 (m, 2Н), 3,88 (s, 3H), 1,26-1,12 (m, 3H). МС (ИЭР) m/z (М+H)+ 231,0.
[1403] К смеси соединения 317С (8,29 г, 36,0 ммоль) в ТГФ (15 мл) и МеОН (10 мл) добавляли смесь KОН (20,20 г, 360,0 ммоль) и Н2О (10 мл) при 25°С. Смесь перемешивали при 70°С в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН и ТГФ, водную фазу подкисляли с помощью концентрированной HCl (36-38%) до рН~3-4, осажденное твердое вещество фильтровали и сушили с получением соединения 317D (5,95 г, выход 81,72%) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,29 (s, 1H), 7,75-7,70 (m, 2Н), 7,42-7,30 (m, 3H), 3,89 (s, 3H).
[1404] Соединение 317 (1,29 г, выход 63,83%) было получено, как описано в примере 6, из соответствующих промежуточных соединений 317D и 21G ((S)-2-амино-3-фенилпропан-1-ол). Соединение 317: 1Н ЯМР (ДМСО-d6, 400 МГц) δ 9,56 (s, 1H), 8,38 (d, J=7,5 Гц, 1H), 8,05 (s, 1H), 7,60-7,48 (m, 2Н), 7,34-7,13 (m, 8Н), 4,51-4,37 (m, 1H), 3,87 (s, 3H), 3,26-3,15 (m, 1H), 2,90-2,76 (m, 1H). МС (ИЭР) m/z (М+Н)+ 334,1.
ПРИМЕР 176
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-2-МЕТИЛ-1-ФЕНИЛ-1H-ИМИДАЗОЛ-5-КАРБОКСАМИД (318)
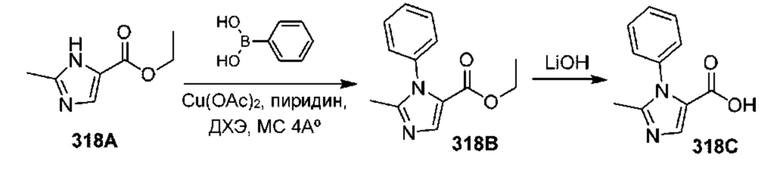
[1405] Смесь N'-гидроксиацетимидамида (5 г, 67,5 ммоль) и этил-проп-2-иноата (8,94 г, 91,1 ммоль) в МеОН (50 мл) перемешивали при 65°С в течение 4 ч. Затем растворитель выпаривали и добавляли PhO2 (25 мл). Реакционную смесь перемешивали при 250°С в течение 4 ч. Смесь охлаждали до 70°С и порциями выливали в МТБЭ (100 мл). Смесь перемешивали в течение 10 мин. Фильтровали и собирали осадок на фильтре. Твердое вещество растворяли в EtOAc (200 мл) и МеОН (50 мл). Фильтровали и собирали фильтрат. Фильтрат концентрировали с получением неочищенный продукт в виде коричневого масла. Остаток суспендировали в МТБЭ (100 мл) и перемешивали в течение 10 мин. Фильтровали и осадок на фильтре собирали с получением соединения 318А (2 г, выход: 19,2%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,32 (ушир. s, 1H), 7,72-7,49 (m, 1H), 4,25-4,10 (m, 2Н), 3,33 (ушир. d, J=8,8 Гц, 1H), 2,33-2,23 (m, 3H), 1,25 (t, J=7,2 Гц, 3H).
[1406] К смеси соединения 318А (2 г, 13,0 ммоль), фенилбороновой кислоты (3,16 г, 25,9 ммоль), пиридина (2,05 г, 25,9 ммоль) и молекулярного сита 4А° (2 г) в ДХЭ (50 мл) добавляли Cu(ОАс)2 (3,53 г, 19,5 ммоль). Смесь перемешивали при 60°С в течение 12 ч в атмосфере О2 (15 фунтов на кв. дюйм). Фильтровали и фильтрат очищали посредством хроматографии на силикагеле, элюируя смесью петролейный эфир:этилацетат = 4:1 с получением неочищенного продукта. Неочищенный продукт затем снова очищали посредством препаративной ТСХ (EtOAc, Rf~0,5) дважды с получением соединения 318В (160 мг, выход: 5,36%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,77 (s, 1H), 7,53-7,48 (m, 3H), 7,26-7,20 (m, 2Н), 4,15 (q, J=7,2 Гц, 2Н), 2,24 (s, 3H), 1,19 (t, J=7,1 Гц, 3H).
[1407] К раствору соединения 318В (100 мг, 434 мкмоль) в ТГФ (6 мл) и H2O (2 мл) добавляли LiOH⋅Н2О (36,5 мг, 869 мкмоль). Смесь перемешивали при 15°С в течение 24 ч. Анализ посредством ТСХ (EtOAc, Rf~0) свидетельствовал о завершении реакции. рН смеси доводили до ~7,0 с использованием 1 н. HCl. Затем растворитель удаляли под вакуумом с получением неочищенного соединения 318С (87,0 мг, неочищенного) в виде желтого масла.
[1408] Соединение 318 (30,3 мг, выход: 28,0%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединения 318С и 3-амино-2-гидрокси-4-фенил-бутанамида (274D). Соединение 318: 1Н ЯМР (400 МГц, CDCl3) δ 7,57-7,39 (m, 5Н), 7,30 (ушир. s, 2Н), 7,24-7,17 (m, 2Н), 7,11-6,96 (m, 2Н), 6,70 (ушир. s, 1H), 6,13 (ушир. s, 1H), 5,63-5,46 (m, 2Н), 3,34 (dd, J=5,5, 14,2 Гц, 1H), 3,12 (dd, J=6,8, 14,1 Гц, 1H), 2,22 (s, 3H). МС (ИЭР) m/z (М+Н)+ 377,1.
ПРИМЕР 177
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-МЕТИЛ-1-ФЕНИЛ-1H-ИМИДАЗОЛ-2-КАРБОКСАМИД (319)
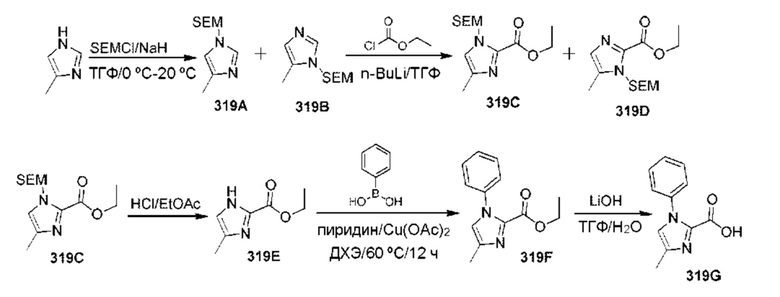
[1409] К раствору 4-метил-1H-имидазола (5 г, 60,9 ммоль) в ТГФ (100 мл) медленно добавляли NaH (2,68 г, 67 ммоль) при 0°С. Суспензию перемешивали при 0°С в течение 30 мин и затем добавляли SEM-Cl (12,2 г, 73,1 ммоль). Реакционную смесь перемешивали при 20°С в течение 12 часов. В смесь добавляли насыщенный водный раствор NaHCO3 (200 мл) для гашения реакции и проводили экстракцию посредством EtOAc (300 мл × 2). Объединенные органические слои сушили над Na2SO4, концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством хроматографии на силикагеле, элюируя посредством EtOAc с получением смеси соединения 319А и 319В (10 г, неочищенного) в виде желтого масла.
[1410] К раствору соединения 319А и 319В (10 г, 47,1 ммоль) в ТГФ (40 мл) добавляли n-BuLi (2,5 М, 28,3 мл) при -78°С. Смесь перемешивали при -78°С в течение 1 ч. К полученному раствору добавляли этилхлорформиат (7,67 г, 70,6 ммоль) и перемешивали при 20°С в течение 12 ч. В смесь добавляли NH4Cl (водный; 200 мл) для гашения реакции, проводили экстракцию посредством EtOAc (300 мл × 2). Объединенные органические слои сушили над Na2SO4, концентрировали с получением неочищенного продукта в виде оранжевого масла. Неочищенный продукт очищали посредством хроматографии на силикагеле, элюируя смесью петролейный эфир:этилацетат = 5:1 с получением смеси соединений 319С и 319D (2,9 г, неочищенного) в виде желтого масла.
[1411] Раствор 319С и 319D (2 г, 7,03 ммоль) в HCl/EtOAc (50 мл) перемешивали при 20°С в течение 24 ч. Анализ посредством ЖХМС свидетельствовал о том, что большая часть 319С и 319D была израсходована. Растворитель удаляли, и остаток подвергали экстракции посредством EtOAc (50 мл) и воды (50 мл). Затем рН водного слоя доводили до ~8,0 с использованием насыщенного водного NaHCO3 и остаток подвергали экстракции посредством EtOAc (50 мл × 6). Объединенные органические слои сушили над Na2SO4, концентрировали с получением 319Е (900 мг, неочищенного) в виде коричневого твердого вещества. Неочищенный продукт непосредственно применяли на следующей стадии. 1Н ЯМР (400 МГц, CDCl3) δ 10,42 (ушир. s, 1H), 10,62-10,25 (m, 1H), 6,96 (s, 1H), 4,42 (q, J=7,3 Гц, 2Н), 2,33 (s, 3H), 1,41 (t, J=7,2 Гц, 3H).
[1412] К смеси соединения 319Е (405 мг, 2,63 ммоль), фенилбороновой кислоты (480 мг, 3,94 ммоль), пиридина (416 мг, 5,25 ммоль) и молекулярного сита 4А° (500 мг) в ДХЭ (30 мл) добавляли Cu(ОАс)2 (716 мг, 3,94 ммоль). Смесь перемешивали при 60°С в течение 12 ч в атмосфере О2 (15 фунтов на кв. дюйм). Фильтровали и остаток очищали посредством хроматографии на силикагеле, элюируя смесью петролейный эфир:этилацетат = 5:1 с получением соединения 319F (400 мг, неочищенного) в виде чистого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,49-7,44 (m, 3H), 7,33-7,28 (m, 2Н), 6,93 (s, 1H), 4,30 (q, J=7,1 Гц, 2H), 2,35 (s, 3H), 1,31 (t, J=7,1 Гц, 4H).
[1413] Раствор соединения 319F (75 мг, 326 мкмоль) и LiOH⋅Н2О (13,7 мг, 326 мкмоль) в ТГФ (3 мл) и Н2О (1 мл) перемешивали при 15°С в течение 12 ч. Анализ посредством ТСХ (петролейный эфир:этилацетат = 1:1, Rf~0,01) и анализ посредством ЖХМС свидетельствовал о том, что реакция была завершена. рН смеси доводили до ~7,0 и ТГФ удаляли посредством N2. Затем остаток лиофилизировали с получением неочищенного соединения 319G (130 мг, неочищенного) в виде белого твердого вещества. Неочищенный продукт непосредственно применяли на следующей стадии. МС (ИЭР) m/z (М+Н)+ 202,8.
[1414] Соединение 319 (38,3 мг, выход: 42,3%, грязно-белое твердое вещество) было получено, как описано в примере 5, из соответствующей карбоновой кислоты, промежуточного соединения 319G. Соединение 319: 1Н ЯМР (400 МГц, CDCl3) δ 7,80 (ушир. s, 3H), 7,41 (ушир. s, 3H), 7,33-7,28 (m, 2Н), 7,21 (ушир. A, J=1,2 Гц, 4Н), 6,85 (s, 1H), 6,70 (ушир. s, 1H), 5,69-5,57 (m, 1H), 5,42 (ушир. s, 1H), 3,39 (ушир. dd, J=5,0, 14,2 Гц, 1H), 3,15 (ушир. dd, J=7,6, 14,1 Гц, 1H), 2,28 (s, 3H). МС (ИЭР) m/z (М+Н)+ 377,2.
ПРИМЕР 178
СОЕДИНЕНИЯ 321, 519-520
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-4-(3-ФТОРФЕНИЛ)-1,2,5-ТИАДИАЗОЛ-3-КАРБОКСАМИД (321)
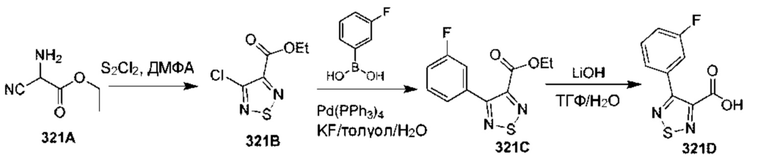
[1415] К смеси этил-(Е)-2-циано-2-(гидроксиимино)ацетата (25 г, 1,76 моль) в EtOH (100 мл) добавляли PtO2 (2 г, 8,8 ммоль). Смесь перемешивали при 25°С в течение 12 ч в атмосфере Н2 (50 фунтов на кв. дюйм). Фильтровали и фильтрат концентрировали с получением соединения 321А (44 г, неочищенного) в виде красного масла. Неочищенный продукт непосредственно применяли на следующей стадии.
[1416] К раствору соединения 321А (22 г, 172 ммоль) в ДМФА (500 мл) добавляли хлорсульфанилтиогипохлорит (69,6 г, 515 ммоль). Смесь перемешивали при 20°С в течение 12 ч. Смесь выливали в ледяную воду (1000 мл), проводили экстракцию посредством EtOAc (500 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир:этилацетат = 30:1. Соединение 321В (12 г, выход: 18,1%, желтое прозрачное масло): 1Н ЯМР (400 МГц, CDCl3) δ 4,50 (q, J=7,1 Гц, 2Н), 1,46 (t, J=7,2 Гц, 3H).
[1417] К смеси соединения 321В (1 г, 5,19 ммоль) и (3-фторфенил)бороновой кислоты (1,09 г, 7,79 ммоль) в Н2О (2 мл) и толуоле (20 мл) добавляли KF (603 мг, 10,38 ммоль) и Pd(PPh3)4 (300 мг, 260 мкмоль) в атмосфере N2. Затем реакционную смесь перемешивали при 100°С в атмосфере N2 в течение 16 ч. Растворитель выпаривали. Неочищенный продукт очищали посредством препаративной ТСХ (петролейный эфир:этилацетат = 5:1, Rf=0,69) с получением соединения 321С (100 мг, выход: 7,64%) в виде белого твердого вещества.
[1418] Смесь соединения 321С (120 мг, 476 мкмоль) в ТГФ (4 мл) и Н2О (2 мл) добавляли LiOH⋅Н2О (39,9 мг, 951 мкмоль). Затем реакционную смесь перемешивали при 20°С в течение 16 ч. 1 М НСl добавляли в реакционную смесь до рН~6. Растворитель удаляли под вакуумом с получением неочищенного соединение 321D (100 мг, неочищенного) в виде белого твердого вещества. Неочищенный продукт использовали на следующей стадии без очистки. 1Н ЯМР (400 МГц, CDCl3) δ 7,70-7,50 (m, 3H), 7,36-7,33 (m, 1H).
[1419] Соединение 321 (43,8 мг, выход: 61,6%, белое твердое вещество) было получено, как описано в примере 6, из соответствующих исходных веществ, соединения 321D и 3-амино-2-гидрокси-4-фенил-бутанамида (274D). Соединение 321: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,39 (d, J=7,7 Гц, 1H), 8,16 (s, 1H), 7,91 (s, 1H), 7,48-7,17 (m, 8Н), 5,73 (s, 1H), 5,55-5,43 (m, 1H), 3,21 (dd, J=3,6, 14,0 Гц, 1H), 2,85 (dd, J=10,0, 14,0 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 399,1.
3-(2-ФТОРФЕНИЛ)-1-МЕТИЛ-N-(2-ОКСО-1-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)ЭТИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (519)
[1420] Соединение 519 (50 мг, выход: 17,5%, белое твердое вещество) было получено, как в случае соединения 21, из соответствующих исходных веществ, 3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоновой кислоты и 2-амино-2-(2-(трифторметил)фенил)этан-1-ола. Соединение 519: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,61 (s, 1H), 8,98 (d, J=6,8 Гц, 1H), 8,37 (s, 1H), 7,80 (d, J=8,0 Гц, 1H), 7,75-7,70 (m, 1H), 7,62-7,56 (m, 2Н), 7,49-7,40 (m, 2Н), 7,26-7,17 (m, 2Н), 5,77 (d, J=6,8 Гц, 1H), 3,95 (s, 3H). МС (ИЭР) m/z (М+H)+ 406,1.
3-(2-ФТОРФЕНИЛ)-1-МЕТИЛ-N-(1-ОКСО-3-ФЕНИЛПРОПАН-2-ИЛ)-1H-ПИРАЗОЛ-4-КАРБОКСАМИД (520)
[1421] Соединение 520 (60 мг, выход: 39,6%, светло-желтое твердое вещество) было получено, как в случае соединения 21, из соответствующих исходных веществ, 3-(2-фторфенил)-1-метил-1Н-пиразол-4-карбоновой кислоты и 3-амино-2-гидрокси-4-фенил-бутанамида (274D). Соединение 520: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,56 (s, 1H), 7,93 (s, 1H), 7,47-7,33 (m, 2Н), 7,23-7,08 (m, 5Н), 6,95 (dd, J=2,9, 6,6 Гц, 2Н), 6,00 (d, J=6,2 Гц, 1H), 4,70 (q, J=6,7 Гц, 1H), 3,96 (s, 3H), 3,07 (d, J=6,4 Гц, 2Н). МС (ИЭР) m/z (М+Н)+ 392,0.
ПРИМЕР 179
(S)-N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(3-ФТОРФЕНИЛ)ИЗОКСАЗОЛ-4-КАРБОКСАМИД (323)
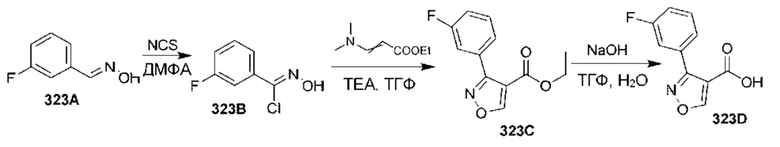
[1422] К суспензии 3-фторбензальдегида (10 г, 80,6 ммоль) и NH2OH.HCl (6,2 г, 88,6 ммоль) в EtOH (10 мл) и Н2О (20 мл) добавляли лед (50 г). Затем водный раствор NaOH (8,1 г, 201,4 ммоль) в Н2О (20 мл) по каплям добавляли в течение 10 мин, при этом растворялась большая часть твердого вещества. Затем смесь перемешивали 2 часа при 16°С. Полученную смесь затем подкисляли с помощью НСl (5 н.). Затем смесь три раза подвергали экстракции дихлорметаном (80 мл) с получением соединения 323А (10 г, выход: 89,2%) в виде светло-желтого твердого вещества. Продукт использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ 8,13 (s, 1H), 7,95 (ушир. s, 1H), 7,39-7,30 (m, 3H), 7,13-7,06 (m, 1H).
[1423] NCS (5,3 г, 39,5 ммоль) добавляли к раствору соединения 323А (5 г, 35,9 ммоль) в ДМФА (20 мл) с последующим перемешиванием при 20°С в течение 3 часов. Реакционную смесь разбавляли Н2О (60 мл) и подвергали экстракции этилацетатом (100 мл × 2). Органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 323В (5,7 г, выход: 91,4%) в виде желтого твердого вещества. Продукт использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,21 (s, 1H), 7,66 (d, J=7,9 Гц, 1H), 7,57 (ушир. d, J=10,1 Гц, 1H), 7,43-7,33 (m, 1H), 7,16 (tt, J=1,1, 8,3 Гц, 1H).
[1424] К раствору этил-3-(диметиламино)акрилата (825 мг, 5,8 ммоль) и TEA (583 мг, 5,8 ммоль) в ТГФ (15 мл) добавляли раствор соединения 323В (1 г, 5,8 ммоль) в ТГФ (35 мл) по каплям в течение 30 мин. Смесь перемешивали при 16°С в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир:этилацетат = 1:0 до 30:1) с получением соединения 323С (800 мг, выход: 59%) в виде бледно-желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 9,03 (s, 1H), 7,62-7,51 (m, 2Н), 7,45 (dt, J=5,8, 8,0 Гц, 1H), 7,25-7,17 (m, 1H), 4,32 (q, J=7,1 Гц, 2H), 1,33 (t, J=7,2 Гц, 3H).
[1425] К смеси соединения 323С (224 мг, 952 мкмоль) в ТГФ (5 мл) и Н2О (1 мл) добавляли LiOH⋅Н2О (80 мг, 1,90 ммоль). Смесь перемешивали при 15°С в течение 12 часов. Смесь концентрировали для удаления растворителя и доводили рН до ~5 с помощью водного раствора HCl (1 М). Смесь фильтровали и твердое вещество промывали Н2О (3 мл) с получением промежуточного соединения 323D (200 мг, неочищенного) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,40 (s, 1H), 7,74 (td, J=1,9, 10,3 Гц, 1H), 7,66 (d, J=7,7 Гц, 1H), 7,52 (dt, J=6,2, 7,9 Гц, 1H), 7,34 (dt, J=2,2, 8,4 Гц, 1H).
[1426] Соединение 323 (68,9 мг, выход: 66,0%, белое твердое вещество) было получено, как описано в примере 5, из соответствующих исходных веществ, соединения 323D и 12G. Соединение 323: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,31 (s, 1H), 9,05 (ушир. d, J=7,5 Гц, 1H), 8,13 (ушир. s, 1H), 7,87 (ушир. s, 1H), 7,54-7,17 (m, 9Н), 5,38 (ушир. s, 1H), 3,26-3,15 (m, 1H), 2,81 (ушир. dd, J=10,6, 13,2 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 382,1.
ПРИМЕР 180
N-(4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-3-(ПИРИМИДИН-2-ИЛ)ИЗОКСАЗОЛ-4-КАРБОКСАМИД (324)
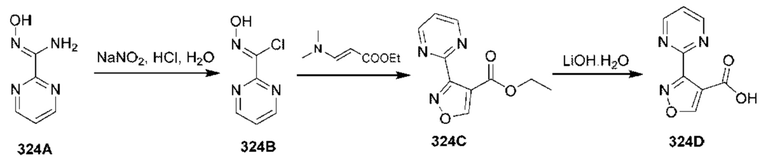
[1427] Смесь пиримидин-2-карбонитрила (10 г, 95,2 ммоль), NH2OH.HCl (6,94 г, 99,9 ммоль) и CH3ONa (5,40 г, 99,9 ммоль) в МеОН (100 мл) нагревали до 70°С в течение 2 ч. Смесь концентрировали, в остаток добавляли воду (50 мл) с получением осадка, твердое вещество отфильтровывали, промывали водой (5 мл × 2) и МТБЭ (10 мл) с получением соединения 324А (10,4 г, выход: 79,1%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,18 (s, 1H), 8,84 (d, J=4,9 Гц, 2Н), 7,51 (t, J=4,9 Гц, 1H), 5,84 (ушир. s, 2Н).
[1428] NaNO2 (1,25 г, 18,1 ммоль) в Н2О (7 мл) добавляли к раствору соединения 324А (2 г, 14,5 ммоль) в HCl (40 мл) при 0°С, смесь перемешивали при 0°С в течение 2 ч. рН смеси доводили до ~6 с помощью насыщенного водного NaHCO3 с получением осадка. Твердое вещество отфильтровывали, промывали водой (5 мл × 2) и сушили с получением соединения 324В (1,40 г, выход: 61,4%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,95 (s, 1H), 8,92 (d, J=4,9 Гц, 2Н), 7,60 (t, J=5,0 Гц, 1H).
[1429] Суспензию соединения 324В (500 мг, 3,17 ммоль) в ТГФ (4 мл) добавляли порциями к смеси 3-амино-2-гидрокси-4-фенилбутанамида (454 мг, 3,17 ммоль) и TEA (321 мг, 3,17 ммоль) в ТГФ (6 мл), смесь перемешивали при 10°С в течение 12 ч. Смесь фильтровали и фильтрат концентрировали, остаток очищали посредством препаративной ТСХ (петролейный эфир:этилацетат = 1:1) с получением соединения 324С (300 мг, выход: 43,2%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,46-7,39 (m, 1H), 7,39-7,32 (m, 2Н), 7,30-7,27 (m, 1H), 7,25-7,19 (m, 4Н), 7,05-7,00 (m, 2Н), 6,87 (ушир. s, 1H), 5,92-5,85 (m, 2Н), 5,40 (ушир. s, 1H), 4,22 (dd, J=1,3, 4,9 Гц, 1H), 4,17-4,09 (m, 1H), 2,92-2,81 (m, 2Н).
[1430] Смесь соединения 324С (150 мг, 684 мкмоль) и LiOH⋅H2O (43,1 мг, 1,03 ммоль) в ТГФ (5 мл), EtOH (3 мл), Н2О (2 мл) перемешивали при 10°С в течение 12 ч. В анализе посредством ЖХМС был обнаружен МС целевого соединения, органический растворитель удаляли под вакуумом, водный слой подвергали экстракции МТБЭ (5 мл) и затем доводили рН до ~4 с помощью 1 н. HCl, смесь концентрировали с получением неочищенного соединения 324D (130 мг, неочищенного) в виде черного твердого вещества.
[1431] Соединение 324 (27,9 мг, выход: 62,3%, желтое твердое вещество) было получено, как описано в примере 6, из соответствующих исходных веществ, соединения 324D и 3-амино-2-гидрокси-4-фенил-бутанамида (274D). Соединение 324: 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,16 (d, J=7,3 Гц, 1H), 9,56 (s, 1H), 8,84 (d, J=5,1 Гц, 2Н), 8,13 (s, 1H), 7,88 (s, 1H), 7,66 (t, J=5,0 Гц, 1H), 7,27-7,05 (m, 5Н), 5,51 (dt, J=5,0, 7,6 Гц, 1H), 3,21 (dd, J=4,9, 14,1 Гц, 1H), 3,00 (dd, J=7,8, 14,0 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 366,1.
ПРИМЕР 181
(S)-N-((S)-4-АМИНО-3,4-ДИОКСО-1-ФЕНИЛБУТАН-2-ИЛ)-1-ФЕНИЛПИРРОЛИДИН-2-КАРБОКСАМИД (308)
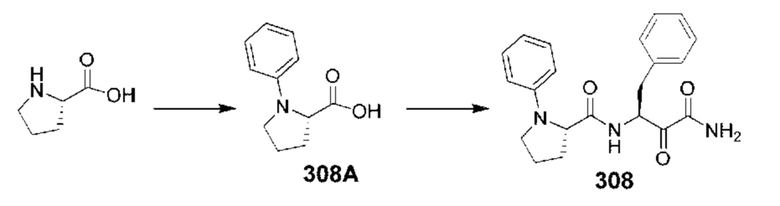
[1432] Смесь L-пролина (1,15 г, 1 экв.) и иодбензола (2,04 г, 1 экв.), K2CO3 (2,07 г, 1,5 экв.) и CuI (0,19 г, 0,1 экв.) в DMA (15 мл) нагревали до 90°С в атмосфере N2 в течение 48 часов. Реакционную смесь охлаждали до комнатной температуры, доводили рН до ~3 с помощью HCl и разбавляли этилацетатом и водой. Отделяли органический слой и водный слой подвергали экстракции этилацетатом (5 раз). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали на ISCO с получением соединения-кислоты 308А.
[1433] Соединение 308 было получено, как описано в примере 5, из кислоты, промежуточного соединения 308А. 1Н ЯМР (400 МГц, ДМСО): 1Н ЯМР (400 МГц, ДМСО): δ 8,3-7,5 (m, 2Н), 7,38-7 (m, 7Н), 6,7-6,2 (m, 4 Н), 5,2 (m, 0,5 Н), 4,35 (m, 0,5Н), 3,9-3,3 (m, 3H), 3,2-2,8 (m, 2Н) 2,2-1,7 (m, 4Н) ppm. МС (ИЭР) m/z (М+Н)+ 356,9.
ПРИМЕР 182
СОЕДИНЕНИЯ 325-327
[1434] Соединения 325-327 были синтезированы из соответствующих исходных веществ с использованием тех же методик, которые были описаны ранее для соединения 321
[1435] Соединение 325: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(м-толил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,42 (ушир. d, J=7,3 Гц, 1H), 8,17 (ушир. s, 1H), 7,92 (ушир. s, 1H), 7,49 (s, 1H), 7,39-7,13 (m, 8Н), 5,55-5,43 (m, 1H), 3,20 (ушир. dd, J=3,0, 14,2 Гц, 1H), 2,85 (ушир. dd, J=10,0, 14,0 Гц, 1H), 2,30 (s, 3H). МС (ИЭР) m/z (М+Н)+ 395,1.
[1436] Соединение 326: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(о-толил)-1,2,5-тиадиазол-3-карбоксамид: 1Н ЯМР (400 МГц, CDCl3) δ 7,52 (ушир. d, J=7,7 Гц, 1H), 7,39-7,32 (m, 1H), 7,31-7,19 (m, 6Н), 7,13 (ушир. d, J=6,6 Гц, 2Н), 6,71 (ушир. s, 1H), 5,66 (dt, J=5,3, 7,5 Гц, 1H), 5,59 (ушир. s, 1H), 3,41 (dd, J=5,1, 14,1 Гц, 1H), 3,18 (dd, J=7,3, 14,1 Гц, 1H), 2,17-2,01 (m, 3H). МС (ИЭР) m/z (M+H)+ 395,1.
[1437] Соединение 327: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2-фторфенил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, CDCl3) δ 7,58-7,41 (m, 3H), 7,33-7,21 (m, 4Н), 7,19-7,08 (m, 3H), 6,74 (ушир. s, 1H), 5,79-5,68 (m, 1H), 5,65 (ушир. s, 1H), 3,50-3,37 (m, 1H), 3,36-3,23 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 399,1.
ПРИМЕР 183
СОЕДИНЕНИЯ 328-329
[1438] Соединения 328-329 были синтезированы из соответствующих исходных веществ с использованием тех же методик, которые описаны ранее для соединения 317.
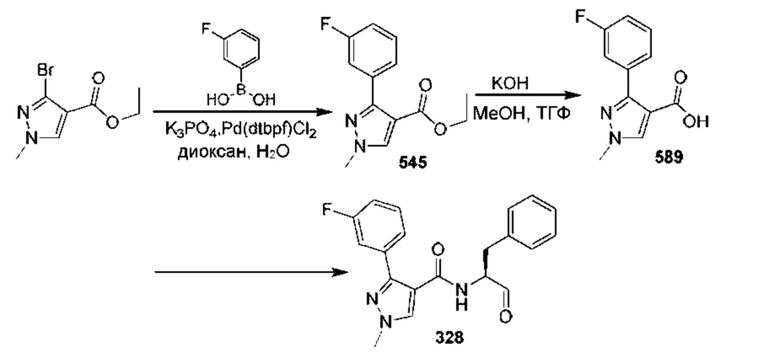
[1439] Соединение 328 синтезировали с использованием этил-3-бром-1-метил-1H-пиразол-4-карбоксилата и (3-фторфенил)бороновой кислоты через промежуточные соединения 545 и 589, используя те же методики, как в случае соединения 317. Соединение 328: (S)-3-(3-фторфенил)-1-метил-N-(1-оксо-3-фенилпропан-2-ил)-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,57 (s, 1H), 8,51 (d, J=8,0 Гц, 1H), 8,08 (s, 1H), 7,48-7,41 (m, 2Н), 7,37-7,29 (m, 1H), 7,29-7,21 (m, 4Н), 7,21-7,09 (m, 2Н), 4,50-4,42 (m, 1H), 3,88 (s, 3H), 3,24-3,18 (m, 1H), 2,85-2,77 (m, 1H). МС (ИЭР) m/z (М+Н)+ 352,1.
[1440] Соединение 329: (S)-1-метил-N-(1-оксо-3-фенилпропан-2-ил)-3-(М-ТОЛИЛ)-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,56 (s, 1H), 8,35 (d, J=7,2 Гц, 1H), 8,03 (s, 1H), 7,42 (s, 1H), 7,32 (d, J=7,6 Гц, 1H), 7,29-7,18 (m, 5Н), 7,17-7,14 (m, 1H), 7,12-7,07 (m, 1H), 4,47-4,40 (m, 1H), 3,86 (s, 3H), 3,22-3,16 (m, 1H), 2,85-2,78 (m, 1H), 2,29-2,25 (m, 1H), 2,27 (s, 2Н). МС (ИЭР) m/z (М+Н)+ 348,1.
ПРИМЕР 184
СОЕДИНЕНИЕ 330
[1441] Соединение 330 синтезировали из промежуточного соединения 250D и с использованием тех же методик, которые были описаны ранее для соединения 317.
[1442] Соединение 330: (S)-1-циклопропил-N-(1-оксо-3-фенилпропан-2-ил)-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, CD3CN) δ 9,59 (s, 1Н), 7,92 (s, 1H), 7,56-7,08 (m, 1Н), 6,65 (s, 1H), 4,58-4,44 (m, 1H), 3,77-3,58 (m, 1H), 3,30-3,15 (m, 1H), 2,95-2,86 (m, 1H), 1,15-0,99 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 360,1.
ПРИМЕР 185
СОЕДИНЕНИЯ 331-333, 415-424
[1443] Соединения 331-333 синтезировали промежуточного соединения 32F и с использованием тех же методик, которые были описаны ранее для соединения 168.
[1444] Соединение 331: (S)-N-(4-амино-3,4-диоксо-1-(м-толил)бутан-2-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,25 (d, J=7,3 Гц, 1H), 8,09-8,01 (m, 2Н), 7,80 (s, 1H), 7,61-7,56 (m, 2Н), 7,40-7,22 (m, 3H), 6,85 (s, 3H), 5,28 (ушир. s, 1H), 3,90 (s, 3H), 3,11-3,04 (m, 1H), 2,77-2,68 (m, 1H), 2,22 (s, 6Н). МС (ИЭР) m/z (М+Н)+ 405,2.
[1445] Соединение 332: (S)-N-(1-амино-1,2-диоксопентан-3-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,26 (ушир. d, J=6,8 Гц, 1H), 8,19 (s, 1H), 8,02 (ушир. s, 1H), 7,76 (ушир. s, 1H), 7,69 (ушир. d, J=7,0 Гц, 2Н), 7,39-7,28 (m, 3H), 4,95 (ушир. t, J=10,0 Гц, 1H), 3,91 (s, 3H), 1,90-1,75 (m, 1H), 1,66-1,50 (m, 1H), 0,94 (t, J=7,3 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 315,1.
[1446] Соединение 333: N-((3S,4R)-1-амино-4-метил-1,2-диоксогексан-3-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,19 (s, 1H), 8,07-7,94 (m, 2Н), 7,75-7,59 (m, 3H), 7,43-7,27 (m, 3H), 5,06 (t, J=6,9 Гц, 1H), 3,90 (s, 3H), 2,11-1,88 (m, 1H), 1,36 (ddd, J=3,9, 7,3, 13,7 Гц, 1H), 1,20-1,04 (m, 1H), 0,91-0,79 (m, 6Н). МС (ИЭР) m/z (М+Н)+ 343,2.
[1447] Соединение 415 (45 мг, выход: 60,98%): (S)-N-(1-амино-5-метил-1,2-диоксогексан-3-ил)-1-метил-3-фенил-H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,24 (d, J=6,8 Гц, 1H), 8,14 (s, 1H), 8,00 (s, 1H), 7,73 (s, 1H), 7,66 (d, J=7,4 Гц, 2Н), 7,35-7,26 (m, 3H), 5,13-5,07 (m, 1H), 3,88 (s, 3H), 1,75-1,65 (m, 1H), 1,51-1,42 (m, 2Н), 0,88 (d, J=6,6 Гц, 6H). МС (ИЭР) m/z (М+Н)+ 343,1.
[1448] Соединение 416 (25 мг, выход: 34,6%): (S)-N-(1-амино-5,5-диметил-1,2-диоксогексан-3-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,26 (d, J=7,5 Гц, 1H), 8,13-8,09 (m, 1H), 8,04 (s, 1H), 7,78-7,66 (m, 3H), 7,38-7,28 (m, 3H), 5,19 (ушир. t, J=6,9 Гц, 1H), 3,93-3,87 (m, 3H), 1,61-1,46 (m, 2Н), 0,95 (s, 9Н). МС (ИЭР) m/z (М+Н)+ 357,2.
[1449] Соединение 417 (25 мг, выход: 71,7%): N-(1-амино-1,2-диоксо-5-фенилпентан-3-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,44 (d, J=6,8 Гц, 1H), 8,19 (s, 1H), 8,00 (s, 1H), 7,74 (s, 1H), 7,68 (d, J=6,8 Гц, 2Н), 7,36-7,23 (m, 5Н), 7,22-7,14 (m, 3H), 4,99-4,91 (m, 1H), 3,90 (s, 3H), 2,79-2,69 (m, 1H), 2,67-2,59 (m, 1H), 2,10-1,99 (m, 1H), 1,87-1,76 (m, 1H). МС (ИЭР) m/z (М+Н)+ 391,2.
[1450] Соединение 418 (25,1 мг, выход: 22,25%): N-(4-амино-1-(3,5-дихлорфенил)-3,4-диоксобутан-2-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,47 (d, J=7,6 Гц, 1H), 8,05 (ушир. s, 1H), 7,98 (s, 1H), 7,79 (ушир. s, 1H), 7,55-7,44 (m, 3H), 7,32-7,21 (m, 5Н), 5,25-5,17 (m, 1H), 3,87 (s, 3H), 3,19-3,11 (m, 1H), 2,88-2,77 (m, 1H). МС (ИЭР) m/z (М+Н)+ 445,0.
[1451] Соединение 419 (10 мг, выход: 17,6%): (S)-N-(1-амино-1,2-диоксогептан-3-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,23 (ушир. d, J=6,6 Гц, 1H), 8,15 (s, 1H), 8,00 (s, 1H), 7,74 (s, 1H), 7,65 (ушир. d, J=6,6 Гц, 2Н), 7,36-7,25 (m, 3H), 5,07-4,93 (m, 1H), 3,88 (s, 3H), 1,79-1,67 (m, 1H), 1,57-1,44 (m, 1H), 1,37-1,20 (m, 4Н), 0,84 (ушир. t, J=6,9 Гц, 3H). МС (ИЭР) m/z (М+Н)+ 343,2.
[1452] Соединение 420 (25 мг, выход: 38,7%): (S)-N-(4-амино-1-(4-метоксифенил)-3,4-диоксобутан-2-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,04 (s, 1H), 7,85-7,67 (m, 2Н), 7,66-7,46 (m, 3H), 7,37-7,29 (m, 3H), 7,17-7,09 (m, 2Н), 6,89-6,77 (m, 2Н), 5,33-5,24 (m, 1H), 3,94-3,85 (m, 3H), 3,76-3,71 (m, 3H), 3,16-3,10 (m, 1H), 2,88-2,80 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 407,1.
[1453] Соединение 421 (10 мг, выход: 28,2%): (S)-N-(4-амино-1-(4-гидроксифенил)-3,4-диоксобутан-2-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,25 (s, 1H), 8,25 (ушир. d, J=7,3 Гц, 1H), 8,06 (s, 2Н), 7,80 (s, 1H), 7,60-7,52 (m, 2Н), 7,36-7,26 (m, 3H), 7,03 (d, J=8,6 Гц, 2Н), 6,67 (d, J=8,6 Гц, 2Н), 5,28-5,17 (m, 1H), 3,92-3,85 (m, 3H), 3,03 (dd, J=4,1, 13,8 Гц, 1H), 2,73-2,68 (m, 1H). MC (ИЭР) m/z (M+H)+ 393,1.
[1454] Соединение 422 (20,3 мг, выход: 27,2%): N-(1-амино-6,6,6-трифтор-1,2-диоксогексан-3-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,54 (ушир. d, J=6,8 Гц, 1H), 8,20 (s, 1H), 8,03 (ушир. s, 1H), 7,78 (ушир. s, 1H), 7,68 (ушир. d, J=7,6 Гц, 2Н), 7,39-7,27 (m, 3H), 5,00-4,90 (m, 1H), 3,91 (s, 3H), 2,43-2,36 (m, 2Н), 2,12-1,98 (m, 1H), 1,87-1,72 (m, 1Н). МС (ИЭР) m/z (М+1)+ 383,1.
[1455] Соединение 423 (23 мг, выход: 35,5%): (S)-N-(4-амино-1-(1H-индол-3-ил)-3,4-диоксобутан-2-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,81 (s, 1H), 8,18 (d, J=6,8 Гц, 1H), 8,11-8,01 (m, 2Н), 7,80 (s, 1H), 7,62 (d, J=7,6 Гц, 1H), 7,56-7,46 (m, 2Н), 7,32 (d, J=7,6 Гц, 1H), 7,28-7,21 (m, 3H), 7,14-7,08 (m, 1H), 7,08-7,02 (m, 1H), 6,99-6,93 (m, 1H), 5,39-5,31 (m, 1H), 3,85 (s, 3H), 3,30-3,23 (m, 1H), 2,96-2,87 (m, 1H). МС (ИЭР) m/z (М+Н)+ 416,2.
[1456] Соединение 424 (23,6 мг, выход: 24,48%): N-(5-амино-1,1,1-трифтор-4,5-диоксопентан-3-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,78 (d, J=7,2 Гц, 1H), 8,14-8,02 (m, 2Н), 7,81 (s, 1H), 7,70-7,63 (m, 2Н), 7,39-7,29 (m, 3H), 5,20-5,13 (m, 1H), 3,91 (s, 3H), 2,97-2,80 (m, 1H), 2,74-2,60 (m, 1H). МС (ИЭР) /и/z (М+1)+ 369,1.
ПРИМЕР 186
СОЕДИНЕНИЯ 334-340
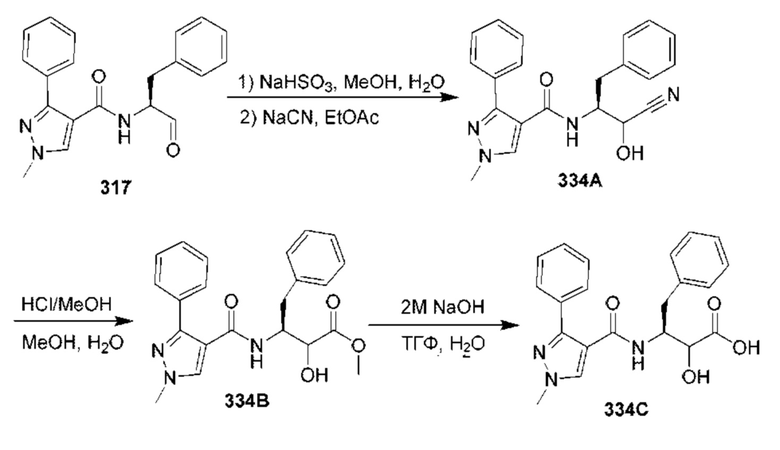
[1457] Соединение 317 подвергали действию условий реакции, используемых для превращения промежуточного соединения 98С в 98D с получением промежуточного соединения 334А (1,82 г, выход 89,9%) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,17-8,05 (m, 1H), 8,03-7,89 (m, 1H), 7,51-7,40 (m, 2Н), 7,29-7,13 (m, 8Н), 6,89-6,76 (m, 1H), 4,69-4,42 (m, 1H), 4,40-4,26 (m, 1H), 3,85 (s, 3H), 3,10-2,94 (m, 1H), 2,84-2,60 (m, 1H). МС (ИЭР) m/z (М+Н)+ 361,1.
[1458] К смеси соединения 334А (1,82 г, 5,1 ммоль) в МеОН (20 мл) добавляли НСl/МеОН (20 мл) при 25°С. Смесь перемешивали при 25°С в течение 15 ч. После удаления растворителя реакционной смеси при пониженном давлении добавляли МеОН (25 мл) и Н2О (25 мл), и затем смесь перемешивали при 25°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении для удаления растворителей с получением соединения 334В (2 г, неочищенного) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без очистки.
[1459] К смеси соединения 334В (2 г, 5,1 ммоль) в ТГФ (15 мл) и МеОН (15 мл) добавляли водный NaOH (2 М, 13 мл) при 25°С. Смесь перемешивали при 25°С в течение 6 ч. Реакционную смесь концентрировали при пониженном давлении для удаления МеОН и ТГФ. В смесь, промытую МТБЭ (10 мл × 2), добавляли Н2О (10 мл), и затем водную фазу подкисляли с помощью водного раствора HCl (1 М) до рН~4-5. Осадок фильтровали и сушили с получением соединения 334С (1,33 г, выход 69,1%) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 12,55 (s, 1H), 8,03-7,93 (m, 1H), 7,90-7,39 (m, 3H), 7,32-7,14 (m, 8Н), 5,74-5,25 (m, 1H), 4,54-4,36 (m, 1H), 4,10-3,94 (m, 1H), 3,85 (d, J=4,9 Гц, 3H), 2,92-2,71 (m, 2Н). МС (ИЭР) m/z (М+Н)+ 380,1.
[1460] Соединения 334-340 синтезировали из промежуточного соединения 334С и соответствующего амина с использованием тех же методик, которые были описаны ранее для соединения 168.
[1461] Соединение 334: (S)-1-метил-N-(4-((ОКСАЗОЛ-2-илметил)амино)-3,4-диоксо-1-фенилбутан-2-ил)-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (400 МГц, CDCl3) δ 7,90 (s, 1H), 7,64 (s, 1H), 7,54-7,33 (m, 6Н), 7,22-7,12 (m, 3H), 7,10 (s, 1H), 6,84-6,77 (m, 2H), 6,10 (d, J=6,0 Гц, 1H), 5,60-5,46 (m, 1H), 4,76-4,53 (m, 2H), 3,92 (s, 3H), 3,33-3,22 (m, 1H), 2,96-2,87 (m, 1H). МС (ИЭР) m/z (M+H)+ 458,2.
[1462] Соединение 335: (S)-N-(4-((2-(2-метоксиэтокси)этил)амино)-3,4-диоксо-1-фенилбутан-2-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (CDCl3, 400 МГц) δ 7,89 (s, 1H), 7,53-7,45 (m, 2Н), 7,44-7,29 (m, 4Н), 7,18 (s, 3H), 6,80 (s, 2Н), 6,09 (d, J=4,5 Гц, 1H), 5,58 (d, J=4,8 Гц, 1H), 3,91 (s, 3H), 3,68-3,48 (m, 8Н), 3,38 (s, 3H), 3,28 (d, J=10,3 Гц, 1H), 2,97-2,84 (m, 1H). МС (ИЭР) m/z (М+Н)+ 479,2.
[1463] Соединение 336: (S)-1-метил-N-(4-((2-(метиламино)-2-оксоэтил)амино)-3,4-диоксо-1-фенилбутан-2-ил)-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (CDCl3, 400 МГц) δ 7,83 (s, 1H), 7,58-7,32 (m, 6Н), 7,18 (s, 3H), 6,82 (s, 2Н), 6,32 (s, 1H), 6,11 (s, 1H), 5,31 (s, 1H), 4,15-3,80 (m, 5Н), 3,28-3,08 (m, 1H), 2,93-2,64 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 448,2.
[1464] Соединение 337: (S)-N-(4-((2-(диметиламино)-2-оксоэтил)амино)-3,4-диоксо-1-фенилбутан-2-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (CDCl3, 400 МГц) δ 7,92-7,79 (m, 2Н), 7,52-7,44 (m, 2Н), 7,44-7,33 (m, 3H), 7,21-7,11 (m, 3H), 6,85-6,74 (m, 2Н), 6,09 (d, J=6,3 Гц, 1H), 5,62 (q, J=6,5 Гц, 1H), 4,18-3,99 (m, 2Н), 3,91 (s, 3H), 3,31-3,22 (m, 1H), 3,01 (d, J=3,3 Гц, 6Н), 2,95-2,86 (m, 1H). МС (ИЭР) m/z (М+Н)+ 462,2.
[1465] Соединение 338: (S)-N-(4-((3-амино-3-оксопропил)амино)-3,4-диоксо-1-фенилбутан-2-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (CDCl3, 400 МГц) δ 7,86 (s, 1H), 7,52-7,46 (m, 2Н), 7,45-7,35 (m, 4Н), 7,21-7,13 (m, 3H), 6,86-6,74 (m, 2Н), 6,08 (d, J=5,8 Гц, 1H), 5,71 (s, 1H), 5,58-5,46 (m, 1H), 5,34 (s, 1H), 3,91 (s, 3H), 3,73-3,51 (m, 2Н), 3,24 (dd, J=4,8, 14,1 Гц, 1H), 2,86 (dd, J=7,8, 14,3 Гц, 1H), 2,49 (t, J=5,9 Гц, 2Н). МС (ИЭР) m/z (М+Н)+ 448,2.
[1466] Соединение 339: (S)-N-(4-((2-амино-2-оксоэтил)амино)-3,4-диоксо-1-фенилбутан-2-ил)-1-метил-3-фенил-1H-пиразол-4-карбоксамид: 1Н ЯМР (CDCl3, 400 МГц) δ 7,83 (s, 1H), 7,55-7,34 (m, 6Н), 7,22-7,14 (m, 3H), 6,89-6,77 (m, 2Н), 6,33 (s, 1H), 6,14 (d, J=4,6 Гц, 1H), 5,52 (s, 1H), 5,37-5,19 (m, 1H), 4,12-4,02 (m, 1H), 3,99-3,94 (m, 1H), 3,90 (s, 3H), 3,28-3,13 (m, 1H), 2,99-2,77 (m, 1H). МС (ИЭР) m/z (М+Н)+ 434,2.
[1467] Соединение 340: Трет-бутил-(S)-(2-(3-(1-метил-3-фенил-1H-пиразол-4-карбоксамидо)-2-оксо-4-фенилбутанамидо)этил)карбамат: 1Н ЯМР (CDCl3, 400 МГц) δ 7,89 (s, 1H), 7,50-7,35 (m, 5H), 7,26-7,23 (m, 1H), 7,20-7,13 (m, 3H), 6,86-6,70 (m, 2H), 6,09 (d, J=6,0 Гц, 1H), 5,58-5,42 (m, 1H), 4,97-4,82 (m, 1H), 3,91 (s, 3H), 3,48-3,39 (m, 2H), 3,35-3,20 (m, 3H), 2,93-2,85 (m, 1H), 1,44 (s, 9H). МС (ИЭР) m/z (M+H)+ 520,3.
ПРИМЕР 187
СОЕДИНЕНИЕ 341
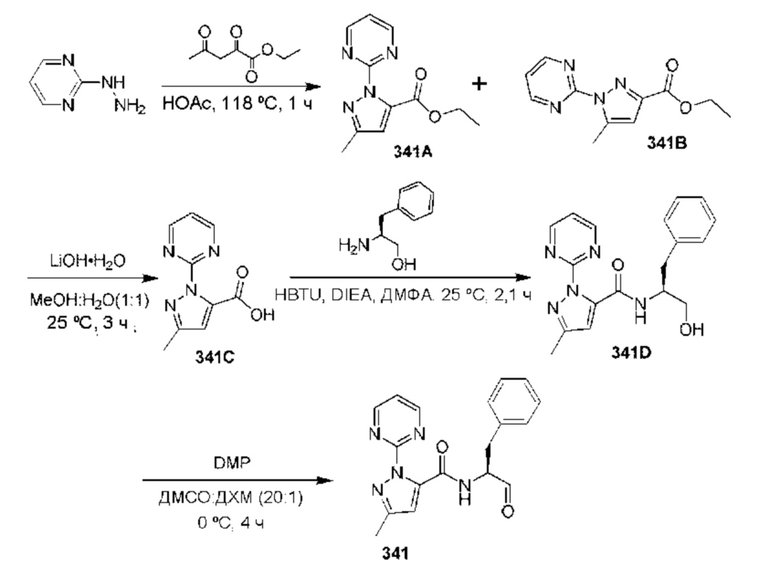
[1468] Соединение 341C синтезировали из 2-гидразинилпиримидина и с использованием тех же методик, которые описаны ранее для соединения 38.
[1469] Соединение 341 синтезировали из 341С с использованием тех же методик, которые описаны ранее для соединения 317. Соединение 341: (S)-3-метил-N-(1-оксо-3-фенилпропан-2-ил)-1-(пиримидин-2-ил)-1H-пиразол-5-карбоксамид: 1Н ЯМР (400 МГц, CDCl3) δ 9,75 (d, J=1,8 Гц, 1H), 8,70-8,60 (m, 3H), 7,28-7,26 (m, 1H), 7,25-7,16 (m, 5Н), 6,74 (s, 1H), 5,00-4,94 (m, 1H), 3,40-3,33 (m, 1H), 3,32-3,24 (m, 1H), 2,42-2,39 (m, 3H). МС (ИЭР) m/z (М+H2O+Н)+ 354,2.
ПРИМЕР 188
СОЕДИНЕНИЕ 342
[1470] Соединение 342 синтезировали из 2,2,3,3,3-пентафторпропан-1-амина гидрохлорида и с использованием тех же методик, которые описаны ранее для соединения 272.
[1471] Соединение 342: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-(перфторэтил)-1-фенил-1H-пиразол-5-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,36 (d, J=7,7 Гц, 1H), 8,10 (s, 1H), 7,85 (s, 1H), 7,48-7,34 (m, 4Н), 7,32-7,18 (m, 9Н), 5,31-5,24 (m, 1H), 3,24-3,13 (m, 1H), 2,85-2,69 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 481,1.
ПРИМЕР 189
СОЕДИНЕНИЕ 343
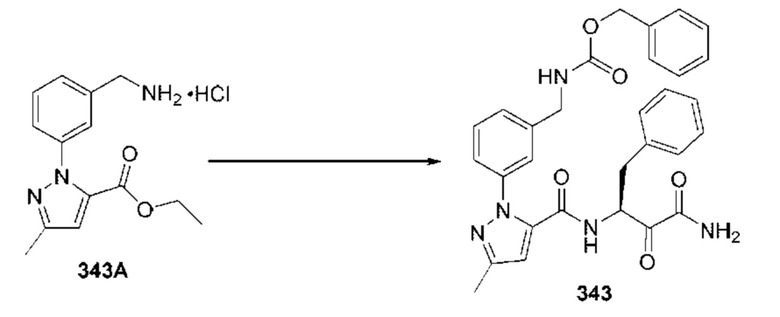
[1472] Соединение 343 синтезировали из этил-1-(3-(аминометил)фенил)-3-метил-1H-пиразол-5-карбоксилата гидрохлорида (343А) и с использованием тех же методик, которые описаны ранее для соединения 153. Соединение 343: Бензил (S)-(3-(5-((4-амино-3,4-диоксо-1-фенилбутан-2-ил)карбамоил)-3-метил-1H-пиразол-1-ил)бензил)карбамат: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,14-9,05 (m, 0,4Н), 8,16-8,00 (m, 0,9Н), 7,93-7,82 (m, 1,3H), 7,59-7,06 (m, 14,2Н), 7,00-6,87 (m, 0,5Н), 6,77-6,64 (m, 0,5Н), 6,31-6,50 (m, 0,9Н), 6,49-6,40 (m, 0,4Н), 6,29-6,17 (m, 0,4Н), 5,34-5,23 (m, 0,4Н), 5,04 (s, 1,9Н), 4,53-4,34 (m, 0,5Н), 4,31-4,09 (m, 1,9Н), 3,24-2,99 (m, 0,8Н), 2,90-2,63 (m, 1,4Н), 2,30-2,16 (m, 3H). МС (ИЭР) m/z (М+Н)+ 540,2.
ПРИМЕР 190
СОЕДИНЕНИЕ 344
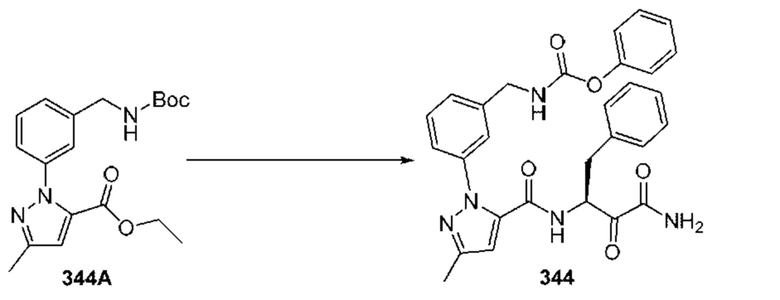
[1473] Соединение 344 синтезировали из этил-1-(3-(((трет-бутоксикарбонил)амино)метил)фенил)-3-метил-1H-пиразол-5-карбоксилата 344А и с использованием тех же методик, которые описаны ранее для соединения 162. Соединение 344: Фенил-(S)-(3-(5-((4-амино-3,4-диоксо-1-фенилбутан-2-ил)карбамоил)-3-метил-1H-пиразол-1-ил)бензил)карбамат: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,14-9,08 (m, 1Н), 8,40-8,33 (m, 1H), 8,13-8,04 (m, 1H), 7,87 (s, 1H), 7,45-7,09 (m, 13Н), 6,99-6,71 (m, 1H), 6,60-6,20 (m, 1H), 5,31-5,23 (m, 1H), 4,46-3,97 (m, 2Н), 3,26-3,01 (m, 1H), 2,90-2,68 (m, 1H), 2,31-2,19 (m, 3H). МС (ИЭР) m/z (М+Н)+ 526,2.
ПРИМЕР 191
СОЕДИНЕНИЯ 345-346
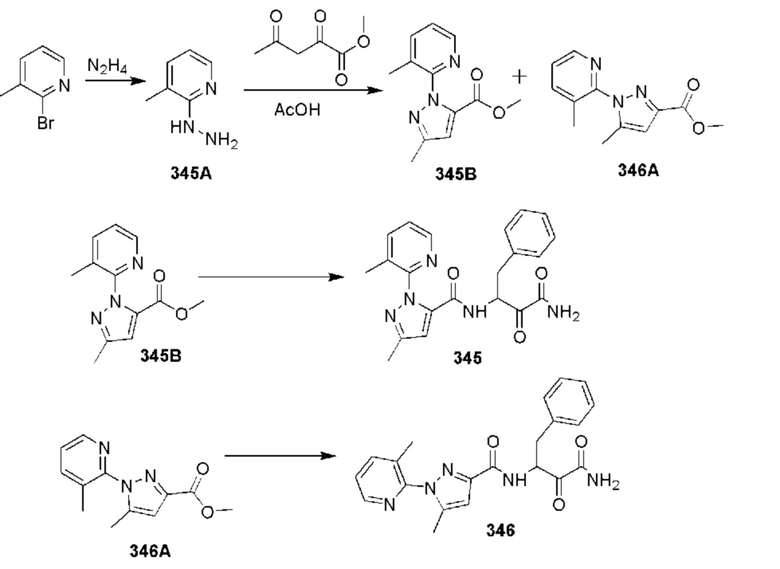
[1474] Соединения 345 и 346 получали из 2-гидразинил-3-метилпиридина и метил-2,4-диоксопентаноата с использованием методик, описанных для соединения 38 и 317
[1475] Соединение 345: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-метил-1-(3-метилпиридин-2-ил)-1H-пиразол-5-карбоксамид: 1Н ЯМР (400 МГц, CDCl3) δ 8,36 (ушир. d, J=4,0 Гц, 1H), 7,67 (ушир. d, J=7,6 Гц, 1H), 7,33-7,24 (m, 2Н), 7,18-7,05 (m, 5Н), 6,67 (ушир. s, 1H), 6,62 (s, 1H), 5,69-5,60 (m, 1H), 5,46 (ушир. s, 1H), 3,35 (dd, J=5,3, 14,0 Гц, 1H), 3,13 (dd, J=7,2, 14,0 Гц, 1H), 2,19 (s, 3H), 2,12 (s, 3H).
[1476] Соединение 346: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-метил-1-(3-метилпиридин-2-ил)-1H-пиразол-3-карбоксамид: 1Н ЯМР (400 МГц, CDCl3) δ 8,23 (ушир. d, J=4,6 Гц, 1H), 7,67 (d, J=7,5 Гц, 1H), 7,33 (ушир. d, J=7,3 Гц, 1H), 7,26-7,18 (m, 4Н), 7,01 (ушир. d, J=3,7 Гц, 2Н), 6,68 (ушир. s, 1H), 6,57 (s, 1H), 5,62 (q, J=6,5 Гц, 1H), 5,50 (ушир. s, 1H), 3,34 (dd, J=5,3, 14,1 Гц, 1H), 3,17 (dd, J=6,5, 14,2 Гц, 1H), 2,34 (s, 3H), 2,17 (s, 3H). МС (ИЭР) m/z (М+Н)+ 392,2.
ПРИМЕР 192
СОЕДИНЕНИЕ 347
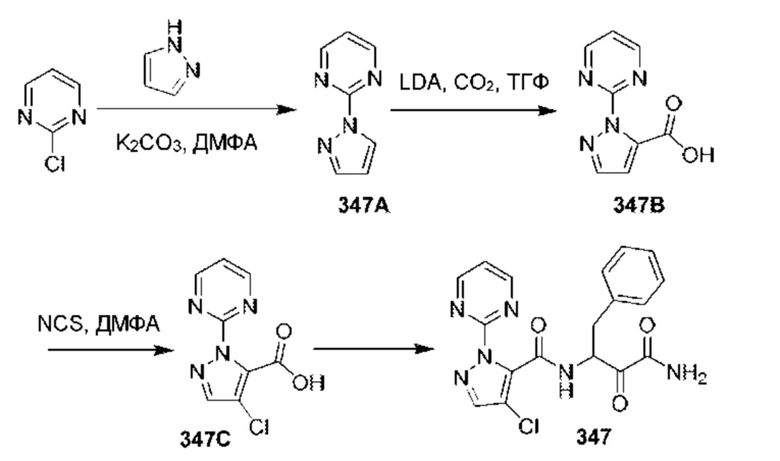
[1477] Смесь 2-хлорпиримидина (10 г, 87,3 ммоль), 1H-пиразола (7,73 г, 114 ммоль) и K2CO3 (24,1 г, 175 ммоль) в ДМФА (150 мл) нагревали до 120°С в течение 12 ч. В анализе посредством ЖХМС был обнаружен МС целевого соединения. В анализе посредством ТСХ (петролейный эфир:этилацетат = 3:1, Rf~0) было обнаружено новое пятно, после охлаждения смесь фильтровали и фильтрат концентрировали, остаток очищали посредством MPLC (петролейный эфир:этилацетат = 1:1) с получением соединения 347А (10,4 г, выход: 81,5%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,76 (d, J=4,8 Гц, 2Н), 8,60 (d, J=2,4 Гц, 1H), 7,84 (s, 1H), 7,21 (t, J=4,8 Гц, 1H), 6,51 (s, 1H).
[1478] К раствору соединения 347А (500 мг, 3,42 ммоль) в ТГФ (10 мл) добавляли LDA (1 М, 4,45 мл) по каплям при -70°С и перемешивали в течение 10 мин, затем через смесь пропускали диоксид углерода в течение 30 мин, смесь медленно нагревали до 15°С в течение 20 мин. К смеси добавляли МТБЭ (20 мл) и Н2О (20 мл), рН водного слоя доводили до ~4 с помощью 1 н. HCl и проводили экстракцию этилацетатом (20 мл × 4), органический слой сушили над Na2SO4, фильтровали и концентрировали с получением соединения 347В (480 мг, выход: 73,8%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,49 (ушир. s, 1H), 8,94 (d, J=4,9 Гц, 2Н), 7,83 (d, J=1,8 Гц, 1H), 7,63 (t, J=4,9 Гц, 1H), 6,98 (d, J=1,8 Гц, 1H).
[1479] Смесь соединения 347В (200 мг, 1,05 ммоль) и NCS (154 мг, 1,16 ммоль) в ДМФА (3 мл) нагревали до 90°С в течение 4 ч. В анализе посредством ЖХМС был обнаружен МС целевого соединения, смесь очищали посредством препаративной ВЭЖХ (ТФУ) с получением соединения 347С (0,2 г, выход: 56,5%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 14,05 (ушир. s, 1H), 8,96 (d, J=4,9 Гц, 2Н), 8,07 (s, 1H), 7,66 (t, J=4,9 Гц, 1H).
[1480] Соединение 347 синтезировали из 347С и с использованием тех же методик, которые описаны ранее для превращения соединения 321D в соединение 321. Соединение 347: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-хлор-1-(пиримидин-2-ил)-1H-пиразол-5-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,25 (d, J=7,3 Гц, 1H), 8,67 (d, J=4,9 Гц, 2Н), 8,12 (s, 1H), 7,96 (s, 1H), 7,86 (s, 1H), 7,46 (t, J=4,9 Гц, 1H), 7,32-7,15 (m, 5Н), 5,50-5,38 (m, 1H), 3,13 (dd, J=3,6, 14,2 Гц, 1H), 2,77 (dd, J=9,9, 14,1 Гц, 1H). МС (ИЭР) (М+Н)+ 399,1.
ПРИМЕР 193
СОЕДИНЕНИЕ 348
[1481] Соединение 348 синтезировали из 2,3-дифтор пиридина и с использованием тех же методик, которые описаны ранее, например, для 313. Соединение 348: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(3-фторпиридин-2-ил)-3-метил-1H-пиразол-5-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,97 (d, J=7,5 Гц, 1H), 8,29 (d, J=4,6 Гц, 1H), 8,07 (s, 1H), 7,92-7,80 (m, 2Н), 7,57 (td, J=4,2, 8,4 Гц, 1H), 7,32-7,25 (m, 4Н), 7,21 (dt, J=2,5, 6,1 Гц, 1H), 6,91 (s, 1H), 5,26-5,17 (m, 1H), 3,16 (dd, J=3,3, 13,9 Гц, 1H), 2,91-2,79 (m, 1H), 2,27 (s, 3H). МС (ИЭР) m/z (М+Н)+ 396,1.
ПРИМЕР 194
СОЕДИНЕНИЕ 349
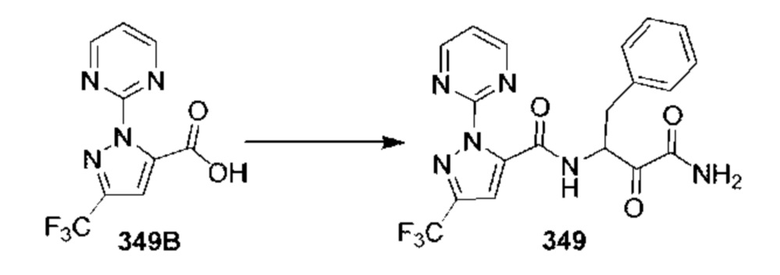
[1482] Соединение 349В получали из 3-(трифторметил)-1H-пиразола, используя ту же методику как описано для соединения 347В.
[1483] Соединение 349 синтезировали из 349В и с использованием тех же методик, которые описаны ранее для превращения соединений 347С в соединение 347. Соединение 349: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(пиримидин-2-ил)-3-(трифторметил)-1H-пиразол-5-карбоксамид: 1Н ЯМР (400 МГц, CDCl3) δ 8,69 (ушир. s, 2Н), 8,11 (ушир. s, 1H), 7,33 (ушир. s, 1H), 7,38-7,31 (m, 1H), 7,24 (ушир. s, 4Н), 7,15-7,08 (m, 1H), 7,15-7,08 (m, 1H), 7,15-7,01 (m, 1H), 6,77 (ушир. s, 1H), 5,82 (ушир. s, 1H), 5,63 (ушир. s, 1H), 3,47 (ушир. s, 1H), 3,35 (ушир. s, 1H). МС (ИЭР) m/z (М+Н)+ 433,1.
ПРИМЕР 195
СОЕДИНЕНИЯ 350, 457
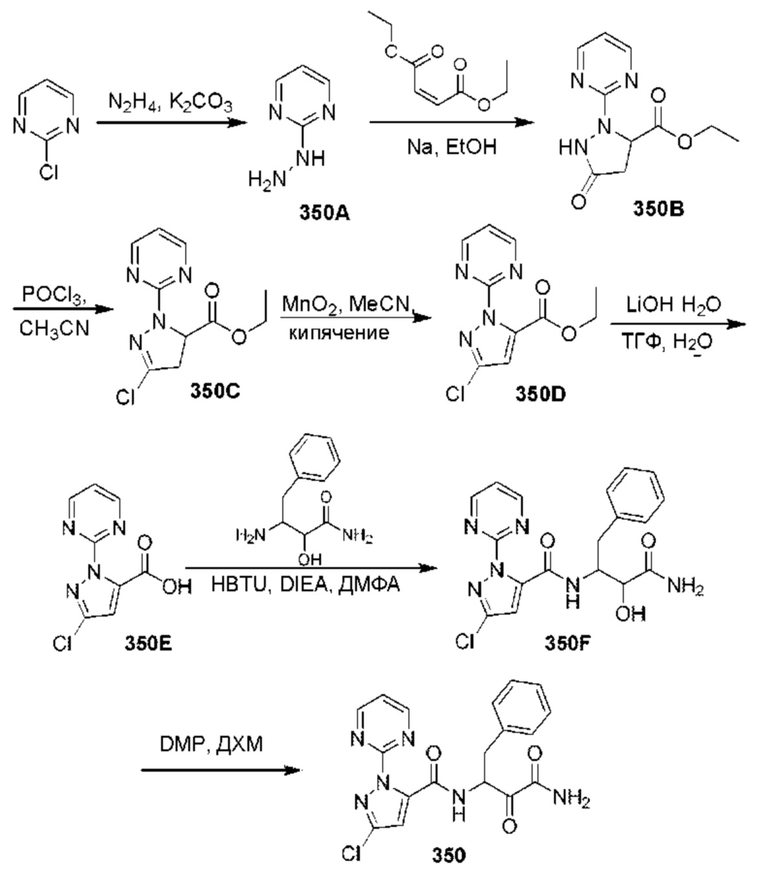
[1484] Смесь 2-хлорпиримидина (15 г, 131 ммоль), NH2NH2.H2O (30 мл), K2СО3 (15 г, 109 ммоль) перемешивали при 100°С в течение 30 мин. Смесь охлаждали с помощью льда, и полученные неочищенные кристаллы собирали путем фильтрования. Кристаллы промывали холодной водой, сушили на воздухе и перекристаллизовывали из петролейного эфира (150 мл) с получением соединения 350А (14,4 г, 131 ммоль, выход: 99,8%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,30 (d, J=4,8 Гц, 2Н), 8,12 (ушир. s, 1H), 6,59 (t, J=4,7 Гц, 1H), 4,13 (s, 2Н).
[1485] К смеси соединения 350А (2 г, 18,2 ммоль) и Na (1,46 г, 63,6 ммоль) в EtOH (60 мл) добавляли диэтилмалеат (3,75 г, 21,8 ммоль) при 15°С. Смесь перемешивали при 60°С в течение 3 часов. Реакционную смесь охлаждали до 15°С и добавляли в нее для гашения реакции уксусную кислоту до рН~7. Смесь концентрировали с получением остаток. Остаток очищали посредством препаративной ВЭЖХ (в условиях ТФУ) с получением соединения 350В (3,5 г, 14,8 ммоль, выход: 81,6%) в виде коричневого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,49 (d, J=4,9 Гц, 2Н), 6,85 (t, J=5,0 Гц, 1H), 5,23 (dd, J=4,2, 11,0 Гц, 1H), 4,33-4,20 (m, 2Н), 3,41-3,28 (m, 1H), 3,01 (dd, J=4,2, 17,6 Гц, 1H), 2,05-1,96 (m, 1H), 1,32-1,21 (m, 3H).
[1486] К смеси соединения 350В (3,5 г, 14,8 ммоль) в MeCN (40 мл) добавляли POCl3 (2,73 г, 17,8 ммоль, 1,65 мл). Смесь перемешивали при 80°С в течение 12 часов. Реакционную смесь концентрировали. Остаток вливали в насыщенный NaHCO3 (30 мл) и перемешивали в течение 10 мин. Водную фазу подвергали экстракции этилацетатом (50 мл × 4). Объединенную органическую фазу сушили с помощью безводном Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир:этилацетат = 0:1 до 1:1) с получением соединения 350С (900 мг, выход: 23,9%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,44 (d, J=4,9 Гц, 2Н), 6,75 (t, J=4,9 Гц, 1H), 5,11 (dd, J=6,5, 12,7 Гц, 1H), 4,27-4,16 (m, 2Н), 3,56 (dd, J=12,7, 18,0 Гц, 1H), 3,21 (dd, J=6,6, 18,1 Гц, 1H), 1,23 (t, J=7,2 Гц, 3H).
[1487] К смеси соединения 350С (900 мг, 3,53 ммоль) в MeCN (15 мл) добавляли MnO2 (3,07 г, 35,3 ммоль). Смесь перемешивали при 80°С в течение 12 часов. Реакционную смесь фильтровали и фильтрат концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир:этилацетат = 1:0 до 0:1) с получением соединения 350D (215 мг, выход: 24,1%) в виде коричневого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,81 (d, J=4,9 Гц, 2Н), 7,36 (t, J=4,9 Гц, 1H), 6,80 (s, 1H), 4,35 (q, J=7,2 Гц, 2H), 1,31 (t, J=7,2 Гц, 3H).
[1488] Соединения 350 синтезировали из промежуточного соединения 350D с использованием тех же методик, которые описаны ранее для превращения соединения 38В в соединение 38. Соединение 350: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-хлор-1-(пиримидин-2-ил)-1H-пиразол-5-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,23 (d, J=7,3 Гц, 1H), 8,78 (d, J=4,9 Гц, 2H), 8,09 (s, 1H), 7,85 (s, 1H), 7,55 (t, J=4,9 Гц, 1H), 7,33-7,19 (m, 5H), 6,84 (s, 1H), 5,35-5,25 (m, 1H), 3,16 (dd, J=3,6, 14,0 Гц, 1H), 2,82 (dd, J=10,0, 14,0 Гц, 1H). МС (ИЭР) m/z (M+H)+ 399,1.
[1489] Соединение 457 синтезировали из промежуточного соединения 350Е и 3-амино-N-циклопропил-2-гидрокси-4-фенилбутанамида гидрохлорида с использованием тех же методик, которые описаны ранее для соединения 350. Соединение 457 (70,21 г, выход: 70,32%): 3-хлор-N-(4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)-1-(пиримидин-2-ил)-1H-пиразол-5-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,29 (d, J=7,5 Гц, 1H), 8,84 (d, J=5,1 Гц, 1H), 8,77 (d, J=4,9 Гц, 2Н), 7,56 (t, J=4,7 Гц, 1H), 7,32-7,20 (m, 5Н), 6,90 (s, 1H), 5,36-5,29 (m, 1H), 3,16 (dd, J=3,6, 14,0 Гц, 1H), 2,83 (dd, J=9,9, 13,9 Гц, 1H), 2,79-2,72 (m, 1H), 0,71-0,56 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 439,1.
ПРИМЕР 196
СОЕДИНЕНИЕ 351
[1490] Соединение 351 синтезировали из 341С с использованием тех же методик, которые описаны ранее для превращения соединения 321D в соединение 321. Соединение 341: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-метил-1-(пиримидин-2-ил)-1H-пиразол-5-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,05 (d, J=7,2 Гц, 1H), 8,71 (d, J=4,8 Гц, 2Н), 8,07 (s, 1H), 7,83 (s, 1H), 7,47-7,42 (m, 1H), 7,32-7,17 (m, 5Н), 6,56 (s, 1H), 5,29-5,21 (m, 1H), 3,16-3,08 (m, 1H), 2,85-2,77 (m, 1H), 2,25 (s, 3H). МС (ИЭР) m/z (М+Н)+ 379,0.
ПРИМЕР 197
СОЕДИНЕНИЕ 352
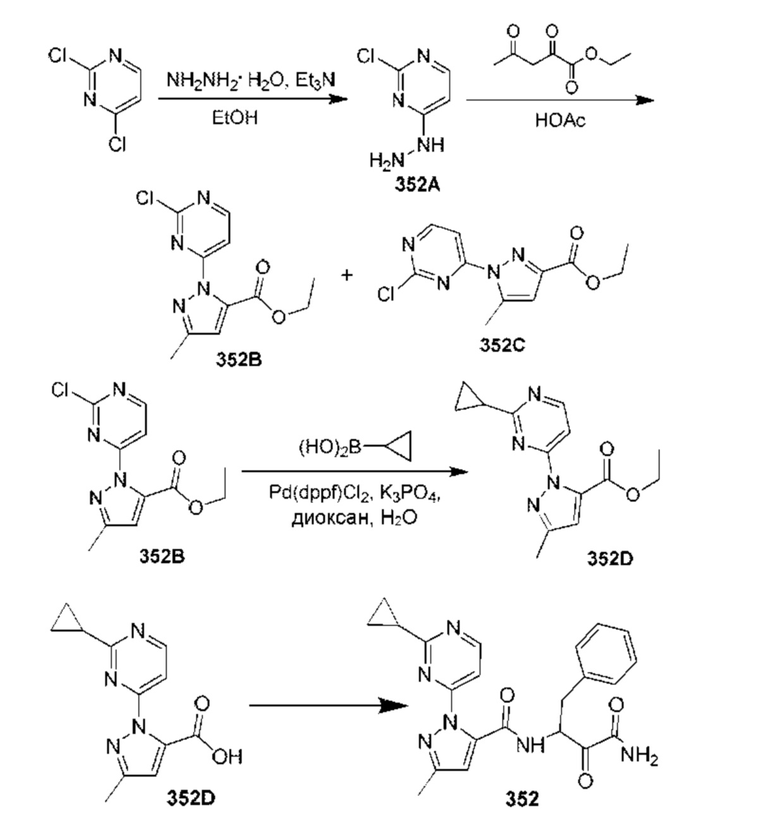
[1491] Соединение 352 получали из 2-хлор-4-гидразинилпиримидина и этил-2,4-диоксопентаноата с использованием методик, описанных для соединений 345 и 321. Соединение 352: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(2-циклопропилпиримидин-4-ил)-3-метил-1H-пиразол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,10 (ушир. d, J=7,0 Гц, 1H), 8,62 (d, J=5,5 Гц, 1H), 8,12 (ушир. s, 1H), 7,87 (ушир. s, 1H), 7,45 (d, J=5,5 Гц, 1H), 7,35-7,19 (m, 5Н), 6,44 (s, 1H), 5,56-5,48 (m, 1H), 3,18 (ушир. dd, J=3,8, 13,8 Гц, 1H), 2,81 (ушир. dd, J=9,8, 13,8 Гц, 1H), 2,28 (s, 3H), 2,00-1,90 (m, 1H), 0,94-0,67 (m, 4Н). МС (ИЭР) m/z (М+Н)+ 419,2.
ПРИМЕР 198
СОЕДИНЕНИЕ 353
[1492] Соединение 353 синтезировали из 254D с использованием тех же методик, которые описаны ранее для синтеза соединения 322. Соединение 353: (S)-4-(3-фторфенил)-2-метил-N-(1-оксо-3-фенилпропан-2-ил)оксазол-5-карбоксамид: 1Н ЯМР (400 МГц, CDCl3) δ 9,71 (s, 1H), 8,05-7,94 (m, 2H), 7,43-7,27 (m, 4H), 7,21 (ушир. d, J=6,8 Гц, 2H), 7,13-7,05 (m, 1H), 6,89-6,84 (m, 1H), 4,98-4,90 (m, 1H), 3,37-3,31 (m, 1H), 3,29-3,20 (m, 1H), 2,55 (s, 3H). МС (ИЭР) m/z (M+H2O+H)+ 353,1.
ПРИМЕР 199
СОЕДИНЕНИЕ 354
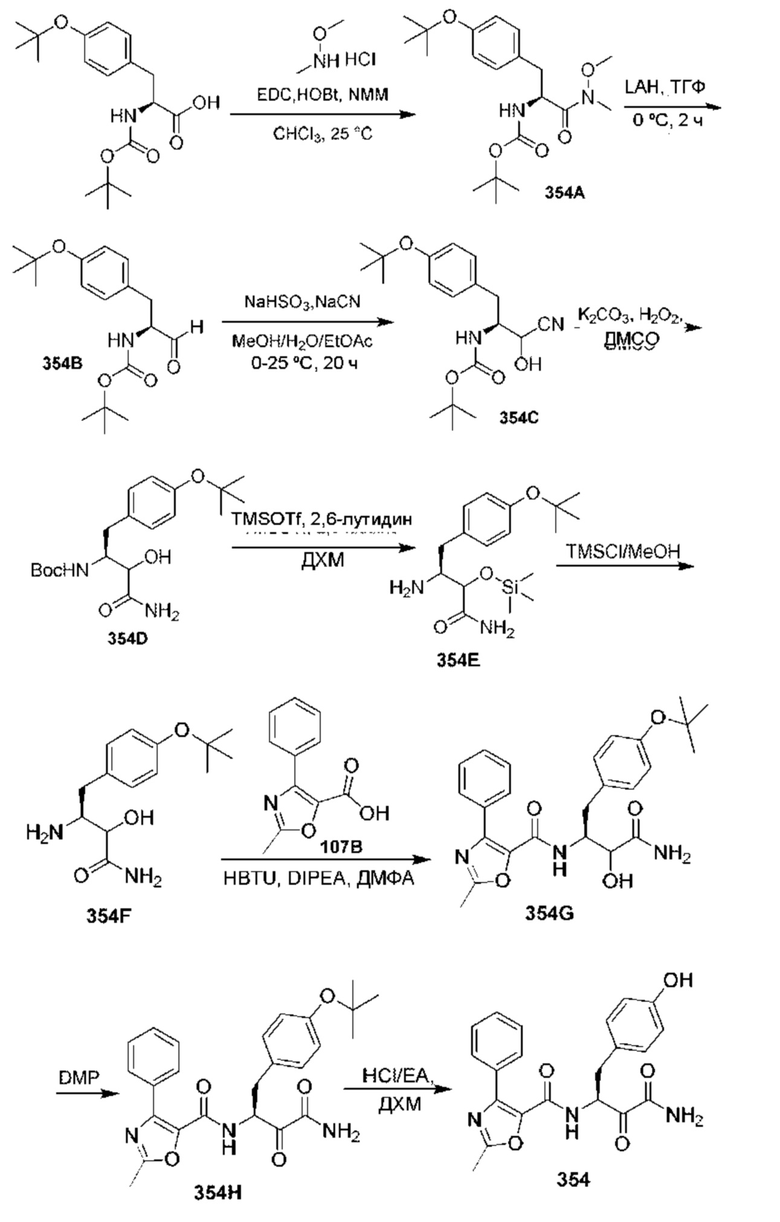
[1493] Соединение 354D получали с использованием методики, описанной для соединения 213D. К раствору соединения 354D (2,50 г, 6,82 ммоль) и 2,6-лутидина (6,36 мл, 54,56 ммоль) в ДХМ (25 мл) добавляли триметилсилилтрифторметансульфонат (7,40 мл, 40,92 ммоль) при 0°С в атмосфере N2. После добавления реакционную смесь перемешивали при 0°С в течение 0,25 ч, затем перемешивали при 20°С в течение 3 ч. Реакционную смесь охлаждали до 0°С и добавляли в охлажденный нас. NH4Cl (50 мл), затем смесь подвергали экстракции EtOAc (100 мл × 2), объединенные экстракты промывали нас. NaHCO3 (100 мл), затем смесь сушили над Na2SO4 и фильтровали, фильтрат концентрировали под вакуумом с получением соединения 354Е (2,50 г, неочищенного) в виде красного масла. МС (ИЭР) m/z (М+Н)+ 339,1.
[1494] К раствору соединения 354Е (2,50 г, 7,39 ммоль) в МеОН (40 мл) добавляли TMSCl (1,50 мл, 11,87 ммоль) при 0°С. После добавления реакционную смесь перемешивали при 0°С в течение 0,5 ч. TEA В реакционную смесь добавляли до рН~8, затем смесь концентрировали под вакуумом с получением неочищенного соединения 354F (2,50 г, неочищенного) в виде красного твердого вещества. МС (ИЭР) m/z (М+Н)+ 266,9.
[1495] К раствору соединения 107В (700 мг, 3,45 ммоль), соединения 354F (1,01 г, 3,80 ммоль) и HBTU (1,57 г, 4,13 ммоль) в ДМФА (40 мл) добавляли DIEA (2,41 мл, 13,78 ммоль) при 0-10°С. После добавления реакционную смесь перемешивали при 20°С в течение 2 ч. В реакционную смесь добавляли 5 мл воды, и смесь концентрировали под вакуумом для удаления большей части ДМФА. Затем в смесь добавляли 100 мл воды и 80 мл EtOAc и проводили перемешивание в течение 2 мин. Смесь отделяли и водный слой подвергали экстракции EtOAc (80 мл). Объединенные экстракты промывали 0,3 н. HCl (80 мл × 2), нас. NaHCO3 (80 мл × 2) и солевым раствором (80 мл). Затем смесь сушили над Na2SO4 и фильтровали, смесь концентрировали под вакуумом с получением неочищенного продукта. Остаток очищали посредством препаративной ВЭЖХ (нейтральные условия) с получением соединения 354G (1,0 г, выход 63,55%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,12-7,55 (m, 3H), 7,45-7,25 (m, 5Н), 7,22-7,05 (m, 2Н), 6,91-6,77 (m, 2Н), 6,10-5,84 (m, 1H), 4,64-4,46 (m, 1H), 4,11-3,99 (m, 1H), 2,94-2,80 (m, 1H), 2,79-2,61 (m, 1H), 2,56-2,52 (m, 3H), 1,23-1,17 (m, 9Н). МС (ИЭР) m/z (M+H)+ 452,1.
[1496] К раствору соединения 354G (863,1 мг, 1,91 ммоль) в ДХМ (200 мл) добавляли DMP (3,24 г, 7,65 ммоль) при 0°С в атмосфере N2. После добавления реакционную смесь перемешивали при 10°С в течение 1 ч. 50 мл sat. Na2S2O3 и 50 мл NaHCO3 В реакционную смесь добавляли, смесь перемешивали в течение 20 мин. Затем смесь отделяли, органический слой промывали 50 мл нас. Na2S2O3 и 50 мл NaHCO3, затем вода (80 мл), солевым раствором (80 мл). Смесь сушили над Na2SO4 и фильтровали, затем смесь концентрировали под вакуумом с получением соединения 354Н (640 мг, выход 74,45%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,77 (d, J=7,5 Гц, 1H), 8,17-7,94 (m, 3H), 7,87-7,74 (m, 1H), 7,45-7,31 (m, 3H), 7,16 (d, J=8,4 Гц, 2Н), 6,88-6,81 (m, 2Н), 5,45-5,31 (m, 1H), 3,18-3,05 (m, 1H), 2,99-2,86 (m, 1H), 2,55-2,49 (m, 3H), 1,23-1,18 (m, 9Н). МС (ИЭР) m/z (М+Н)+ 448,2.
[1497] К раствору соединения 354Н (440 мг, 978,87 мкмоль) в ДХМ (30 мл) добавляли HCl/EtOAc (4 М, 22 мл) при 0°С. После добавления реакционную смесь перемешивали при 10°С в течение 2 ч. Реакционную смесь концентрировали и остаток растворяли в 80 мл EtOAc, смесь промывали водой (80 мл), 0,1% NaHCO3 (80 мл) и солевым раствором (80 мл). Затем смесь сушили над Na2SO4 и фильтровали, затем концентрировали под вакуумом с получением соединения 354. Соединение 354 (650 мг, 1,49 ммоль) растворяли в 3 мл CH3CN и 15 мл 2-изопропоксипропан добавляли into the stirring mixture. После этого смесь фильтровали с получением чистого соединения 354 (450 мг, выход 71.40%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,25 (s, 1H), 8,70 (d, J=7,5 Гц, 1H), 8,11 (s, 1H), 8,07-8,00 (m, 2Н), 7,84 (s, 1H), 7,42-7,36 (m, 3H), 7,08 (d, J=8,5 Гц, 2Н), 6,67 (d, J=8,5 Гц, 2Н), 5,45-5,29 (m, 1H), 3,13-3,05 (m, 1H), 2,93-2,84 (m, 1H), 2,56 (s, 3H). МС (ИЭР) m/z (М+Н)+ 394,1.
ПРИМЕР 200
СОЕДИНЕНИЕ 355
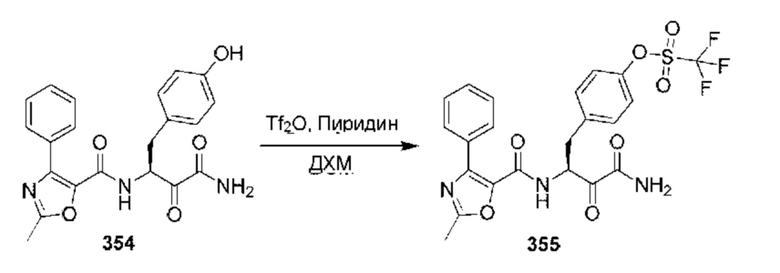
[1498] К раствору соединения 354 (40 мг, 101,68 мкмоль) и пиридина (19 мг, 233,86 мкмоль) в ДХМ (2 мл) добавляли Tf2O (34 мг, 122,02 мкмоль) в ДХМ (0,5 мл) при 0°С в атмосфере N2. После добавления реакционную смесь перемешивали при 0°С в течение 4 ч. Реакционную смесь разбавляли EtOAc (50 мл), смесь промывали 0,2 н. HCl (20 мл), NaHCO3 (20 мл) и солевым раствором (20 мл), затем смесь сушили над Na2SO4 и фильтровали, смесь концентрировали под вакуумом с получением соединения 355 (40 мг, выход 63,64%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,89 (d, J=7,5 Гц, 1H), 8,09 (s, 1H), 8,01-7,95 (m, 2Н), 7,82 (s, 1H), 7,47-7,42 (m, 2Н), 7,42-7,38 (m, 2Н), 7,37-7,32 (m, 3H), 5,44-5,28 (m, 1H), 3,26-3,21 (m, 1H), 3,06-2,96 (m, 1H), 2,52 (s, 3H). МС (ИЭР) m/z (М+Н)+ 526,1.
ПРИМЕР 201
СОЕДИНЕНИЕ 356
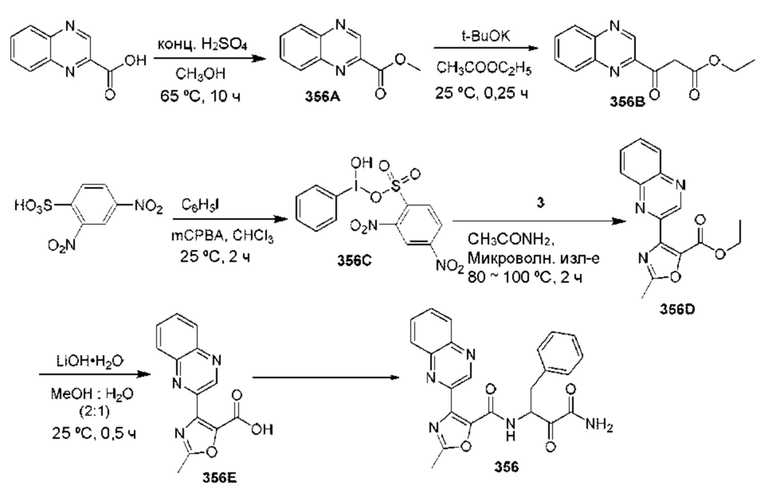
[1499] К раствору хиноксалин-2-карбоновой кислоты (6 г, 34,45 ммоль) в МеОН (80 мл) по каплям добавляли конц. H2SO4 (675,8 мг, 6,89 ммоль), затем полученную смесь перемешивали при 65°С в течение 10 часов. После охлаждения до комнатной температуры, смесь нейтрализовали с помощью нас. NaHCO3 и подвергали экстракции посредством ДХМ (60 мл × 3). Органическую фазу объединяли, сушили с помощью безводного Na2SO4, и упаривали с получением соединения 356А (5,80 г, выход: 89,47%) в виде коричневого твердого вещества. Неочищенный продукт непосредственно применяли на следующей стадии без дополнительной очистки. 1Н ЯМР (CDCl3, 400 МГц) δ 9,56 (s, 1H), 8,31 (d, J=7,6 Гц, 1H), 8,20 (d, J=8,0 Гц, 1H), 7,97-7,84 (m, 2Н), 4,13 (s, 3H).
[1500] Раствор соединения 356А (2,5 г, 13,29 ммоль) в СН3СООС2Н5 (60 мл) добавляли t-BuOK (1,94 г, 17,28 ммоль). Смесь перемешивали в течение 0,25 часа при 25°С. Смесь гасили посредством H2O (50 мл). Отделяли органический слой, и водный подвергали экстракции ЭА (50 мл × 3). Органическую фазу объединяли, сушили с помощью безводном Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии (ПЭ:ЭА=20/1 до 10/1) с получением соединения 356В (2,45 г, 75,48% выход) в виде бледно-желтого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц) δ 9,57-9,33 (m, 1H), 8,19-8,06 (m, 2Н), 7,96-7,74 (m, 2Н), 4,35-4,26 (m, 2Н), 4,24-4,13 (m, 2Н), 1,28-1,18 (m, 3H).
[1501] К смеси 2,4-динитробензолсульфоновой кислоты (7,83 г, 29,41 ммоль, Н2О) и иодбензола (5 г, 24,51 ммоль) в CHCl3 (20 мл) добавляли m-СРВА (4,23 г, 24,51 ммоль), смесь перемешивали при 25°С в течение 2 часов в атмосфере N2. После завершения реакции в реакционную смесь добавляли МТБЭ (20 мл), и полученную смесь фильтровали и твердое вещество промывали МТБЭ (30 мл). Полученную смесь фильтровали и твердое вещество промывали МТБЭ (30 мл) с получением соединения 356С (8,7 г, 75,82% выход) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,60 (ушир. s, 1H), 8,53 (d, J=2,0 Гц, 1H), 8,39-8,36 (m, 1H), 8,18 (d, J=7,6 Гц, 2Н), 8,07 (d, J=8,8 Гц, 1H), 7,71-7,64 (m, 1H), 7,63-7,55 (m, 2Н).
[1502] Смесь соединения 356С (3,49 г, 7,45 ммоль) и соединения 356В (1,4 г, 5,73 ммоль) перемешивали при 80°С в течение 1 ч, и к полученной смеси добавляли ацетамид (4,06 г, 68,76 ммоль), затем смесь перемешивали при 120°С в течение 1 ч под действием микроволнового излучения. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1/0 до 0:1) с получением соединения 356D (300 мг, неочищенного) в виде бледно-красного твердого вещества. МС (ИЭР) m/z (М+Н)+ 284,1.
[1503] Соединение 356 синтезировали из 356D и с использованием тех же методик, которые описаны ранее для превращения соединения 321С в соединение 321. Соединение 356: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-2-метил-4-(хиноксалин-2-ил)оксазол-5-карбоксамид: 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,39 (d, J=8,4 Гц, 1H), 9,58 (s, 1H), 8,24 (ушир. s, 1H), 8,12 (d, J=8,0 Гц, 1H), 7,95 (ушир. s, 1H), 7,89 (t, J=7,6 Гц, 1H), 7,76 (t, J=7,6 Гц, 1Н), 7,60 (d, J=8,4 Гц, 1Н), 6,98-6,86 (m, 4H), 6,85-6,78 (m, 1H), 5,90-5,78 (m, 1Н), 3,27-3,19 (m, 2H), 2,59 (s, 3H). МС (ИЭР) m/z (M+H)+ 430,1.
ПРИМЕР 202
СОЕДИНЕНИЕ 357
[1504] Соединение 357 синтезировали из 107В и с использованием тех же методик, которые были описаны ранее для превращения соединения 321D в соединение 321. Соединение 357: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-2-метил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, CDCl3) δ 8,12-8,05 (m, 2H), 7,43-7,37 (m, 3H), 7,32-7,26 (m, 3H), 7,15-7,10 (m, 2H), 6,78-6,71 (m, 2H), 5,75-5,68 (m, 1Н), 5,54 (ушир. s, 1Н), 3„50-3,38 (m, 1Н), 3,29-3,18 (m, 1Н), 2,55 (s, 3H). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,82 (d, J=7,2 Гц, 1Н), 8,15-7,92 (m, 3H), 7,86 (s, 1Н), 7,41-7,35 (m, 3H), 7,32-7,26 (m, 4H), 7,25-7,17 (m, 1Н), 5,48-5,38 (m, 1Н), 3,27-3,15 (m, 1Н), 3,06-2,93 (m, 1Н), 2,55 (s, 3H). МС (ИЭР) m/z (M+1)+ 378,1.
ПРИМЕР 203
СОЕДИНЕНИЯ 358-359
[1505] Соединения 358 и 359 синтезировали с использованием тех же методик, которые были описаны ранее для соединения 255. Соединение 358: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2-фторфенил)-2-(трифторметил)оксазол-5-карбоксамид: 1H ЯМР (CDCl3, 400 МГц) δ 7,52 (t, J=7,6 Гц, 1Н), 7,45-7,35 (m, 1Н), 7,28-7,20 (m, 3H), 7,19-7,07 (m, 2H), 7,02 (d, J=7,6 Гц, 2H), 6,68 (d, J=4,8 Гц, 2H), 5,70-5,60 (m, 1Н), 5,49 (ушир. s, 1Н), 3,42-3,32 (m, 1Н), 3,24-3,14 (m, 1Н). МС (ИЭР) m/z (M+H)+ 450,1. Соединение 359: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(о-толил)-2-(трифторметил)оксазол-5-карбоксамид: 1H ЯМР (CDCl3, 400 МГц) δ 7,43-7,34 (m, 1Н), 7,33-7,26 (m, 2H), 7,25-7,19 (m, 4H), 6,92 (ушир. s, 2H), 6,69 (ушир. s, 1Н), 6,44 (d, J=6,4 Гц, 1Н), 5,66-5,58 (m, 1Н), 5,50 (ушир. s, 1Н), 3,41-3,28 (m, 1Н), 3,08-2,97 (m, 1Н), 2,21 (s, 3H). МС (ИЭР) m/z (М+Н)+ 446,1.
ПРИМЕР 204
СОЕДИНЕНИЕ 360
[1506] Соединение 360 синтезировали с использованием тех же методик, которые описаны для соединения 26. Соединение 360: N-((2S)-4-амино-3-гидрокси-4-оксо-1-фенилбутан-2-ил)-2-метил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,19-7,58 (m, 3Н), 7,47-7,34 (m, 5Н), 7,32-7,26 (m, 2H), 7,25-7,06 (m, 3H), 6,16-5,82 (m, 1H), 4,73-4,39 (m, 1H), 4,06-3,88 (m, 1H), 3,04-2,65 (m, 2H), 2,59-2,53 (m, 3H). MC (ИЭР) m/z (M+1)+ 380,0.
ПРИМЕР 205
СОЕДИНЕНИЕ 361
[1507] Соединение 361 синтезировали из этил-3-(2,3-дифторфенил)-3-оксопропаноата с использованием тех же методик, которые описаны ранее для соединения 267. Соединение 361: N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2,3-дифторфенил)-2-метилоксазол-5-карбоксамид: 1H ЯМР (CDCl3, 400 МГц) δ 7,36-7,27 (m, 3H), 7,26-7,17 (m, 2H), 7,15-7,06 (m, 3H), 6,78-6,63 (m, 2H), 5,74-5,62 (m, 1H), 5,55 (ушир. s, 1H), 3,42 (dd, J=5,5, 14,3 Гц, 1H), 3,25 (dd, J=6,6, 14,3 Гц, 1H), 2,57 (s, 3H). MC (ИЭР) m/z (M+H)+ 414,1.
ПРИМЕР 206
СОЕДИНЕНИЯ 362-377, 462-468
[1508] Соединения 362-377, 462-468 синтезировали из соответствующего промежуточного соединения или промежуточного соединения 321 В и с использованием тех же методик, которые были описаны ранее для соединения 321.
[1509] Соединение 362 (35,2 мг, выход 47,49%, чистота 94%, ЭИ%: 97%): (S)-N-(4-фтор-3-оксо-1-фенилбутан-2-ил)-4-(4-((проп-2-ин-1-илокси)метил)фенил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,50-9,40 (m, 1H), 9,45 (d, J=7,7 Гц, 1H), 7,50 (d, J=8,2 Гц, 2H), 7,38-7,22 (m, 7H), 5,44-5,14 (m, 2H), 4,99-4,88 (m, 1H), 4,58 (s, 2H), 4,23 (d, J=2,4 Гц, 2H), 3,53 (t, J=2,3 Гц, 1H), 3,21 (dd, J=4,1, 14,2 Гц, 1H), 2,89 (dd, J=10,5, 14,0 Гц, 1H). MC (ИЭР) m/z (М+H)+ 438,1, (M+Na)+ 460,0.
[1510] Соединение 363 (39,2 мг, выход 36,85%, чистота 94%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(4-((4-фторБЕНЗАМИДО)метил)фенил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,44 (d, J=7,7 Гц, 1H), 9,14 (t, J=6,0 Гц, 1H), 8,21 (s, 1H), 8,06-7,97 (m, 2H), 7,93 (s, 1H), 7,51 (d, J=8,2 Гц, 2H), 7,37-7,23 (m, 8H), 7,20-7,12 (m, 1H), 5,60-5,44 (m, 1H), 4,52 (d, J=6,0 Гц, 2H), 3,24 (dd, J=3,2, 14,0 Гц, 1H), 2,85 (dd, J=10,3, 14,0 Гц, 1H). MC (ИЭР) m/z (M+H)+ 532,2.
[1511] Соединение 364 (27,5 мг, выход 26,56%, чистота 96%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-хлор-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,24 (d, J=7,5 Гц, 1Н), 8,19 (s, 1H), 7,92 (s, 1H), 7,49-7,19 (m, 5H), 5,47 (ddd, J=3,9, 7,7, 9,6 Гц, 1H), 3,24 (dd, J=3,9, 14,0 Гц, 1H), 2,95 (dd, J=9,8, 14,0 Гц, 1H). МС (ИЭР) m/z (M+H)+ 339,0.
[1512] Соединение 365 (60 мг, выход 39,4%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(пиразин-2-ил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,23 (d, J=7,3 Гц, 1H), 9,06 (d, J=1,5 Гц, 1H), 8,69 (d, J=2,4 Гц, 1H), 8,51 (dd, J=1,5, 2,4 Гц, 1H), 8,13 (s, 1H), 7,87 (s, 1H), 7,29-7,14 (m, 6H), 5,47 (ddd, J=4,1, 7,6, 9,3 Гц, 1H), 3,16 (dd, J=4,0, 14,1 Гц, 1H), 2,87 (dd, J=9,3, 14,3 Гц, 1H). МС (ИЭР) m/z (M+H)+ 383,1.
[1513] Соединение 366 (50 мг, выход 36,9%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-бром-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,23 (d, J=7,5 Гц, 1H), 8,18 (s, 1H), 7,91 (s, 1H), 7,35-7,17 (m, 4H), 5,54-5,41 (m, 1H), 3,23 (dd, J=3,9, 14,2 Гц, 1H), 2,95 (dd, J=9,7, 13,9 Гц, 1H). МС (ИЭР) m/z (M+H)+ 383,0.
[1514] Соединение 367 (50 мг, выход 18,43%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(бензо[d][1,3]ДИОКСОЛ-4-ил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,92 (d, J=8,0 Гц, 1H), 7,94-7,52 (m, 2H), 7,36-7,18 (m, 5H), 7,08-6,93 (m, 2H), 6,92-6,79 (m, 1H), 5,87 (d, J=10,0 Гц, 2H), 5,55-5,38 (m, 1H), 3,23 (dd, J=3,5, 14,1 Гц, 1H), 3,03-2,97 (m, 1H). МС (ИЭР) m/z (M+H)+ 425,1.
[1515] Соединение 368 (130 мг, выход 60,3%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2-фтор-3-метилфенил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,22 (d, J=7,7 Гц, 1H), 8,12 (s, 1H), 7,87 (s, 1H), 7,39 (t, J=6,9 Гц, 1H), 7,31-7,19 (m, 6H), 7,17-7,10 (m, 1H), 5,42 (ddd, J=3,7, 7,7, 9,7 Гц, 1H), 3,19 (dd, J=3,7, 14,1 Гц, 1H), 2,96 (dd, J=9,7, 13,9 Гц, 1H), 2,22 (d, J=2,0 Гц, 3Н). МС (ИЭР) m/z (М+H)+ 413,1.
[1516] Соединение 369 (55 мг, выход 27,8%): N-(4-амино-3-гидрокси-4-оксо-1-фенилбутан-2-ил)-4-(4-((бензилокси)метил)фенил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,45 (d, J=7,5 Гц, 1H), 8,21 (s, 1H), 7,94 (s, 1H), 7,63-7,52 (m, 2H), 7,44-7,16 (m, 12H), 5,54-5,49 (m, 1H), 4,62-4,56 (m, 4H), 3,24 (dd, J=3,6, 14,0 Гц, 1H), 2,86 (dd, J=10,1, 13,9 Гц, 1H). МС (ИЭР) m/z (М+H)+ 501,1.
[1517] Соединение 370 (55 мг, выход 20,1%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(3-((бензилокси)метил)фенил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,42 (d, J=7,7 Гц, 1Н), 8,18 (s, 1H), 7,93 (s, 1H), 7,71 (s, 1H), 7,49-7,43 (m, 2H), 7,42-7,33 (m, 5H), 7,32-7,24 (m, 5H), 7,24-7,19 (m, 1H), 6,49-6,39 (m, 1H), 5,55-5,48 (m, 1H), 4,55 (d, J=5,7 Гц, 4Н), 3,22 (dd, J=3,6, 14,0 Гц, 1H), 2,88 (dd, J=9,9, 13,9 Гц, 1H). МС (ИЭР) m/z (M+H2O)+ 518,2.
[1518] Соединение 371 (90 мг, выход 63,2%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(пиридин-4-ил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,47 (d, J=7,8 Гц, 1H), 8,65-8,55 (m, 2H), 8,21 (s, 1H), 7,95 (s, 1H), 7,52-7,38 (m, 2H), 7,33-7,22 (m, 5H), 6,53-6,39 (m, 1H), 5,55-5,47 (m, 1H), 3,28-3,22 (m, 1H), 2,92-2,83 (m, 1H). МС (ИЭР) m/z (M+H)+ 382,1.
[1519] Соединение 372 (50 мг, выход 22,8%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2,3-дифторфенил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,24 (d, J=7,6 Гц, 1H), 8,12 (s, 1H), 7,86 (s, 1H), 7,59-7,50 (m, 1H), 7,29-7,16 (m, 7H), 5,44-5,37 (m, 1H), 3,21-3,14 (m, 1H), 2,98-2,90 (m, 1H). МС (ИЭР) m/z (M+H)+ 417,1.
[1520] Соединение 373 (85 мг, выход 44,6%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(пиридин-2-ил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d) δ 9,36 (d, J=7,1 Гц, 1H), 8,42 (d, J=5,6 Гц, 1H), 8,10 (s, 1H), 7,97-7,79 (m, 3Н), 7,50-7,34 (m, 1H), 7,21 (s, 5H), 5,54-5,46 (m, 1H), 3,20-3,11 (m, 1H), 2,90 (dd, J=8,8, 14,1 Гц, 1H). МС (ИЭР) m/z (M+H)+ 382,1.
[1521] Соединение 374 (100 мг, выход 49,9%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(пиримидин-4-ил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,27 (d. J=7,5 Гц, 1H), 9,05 (d, J=1,3 Гц, 1H), 8,94 (d, J=5,1 Гц, 1H), 8,14 (s, 1H), 7,94-7,83 (m, 2H), 7,28-7,16 (m, 5H), 5,50 (ddd, J=4,2, 7,5, 9,3 Гц, 1H), 3,18 (dd, J=4,1, 14,2 Гц, 1H), 2,88 (dd, J=9,5, 14,1 Гц, 1H). МС (ИЭР) m/z (M+H)+ 383,1.
[1522] Соединение 375 (60 мг, выход 28,5%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(3-фтор-2-метилфенил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (d, J=8,0 Гц, 1H), 8,10 (s, 1H), 7,84 (s, 1H), 7,27-7,15 (m, 7H), 6,98-6,94 (m, 1H), 5,39-5,31 (m, 1H), 3,18-3,12 (m, 1H), 2,93-2,85 (m, 1H), 1,87-1,83 (m, 3Н). МС (ИЭР) m/z (M+H)+ 413,1.
[1523] Соединение 376 (300 мг, выход 47,9%): (S)-N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,46 (d, J=8,0 Гц, 1H), 8,22 (s, 1H), 7,95 (s, 1H), 7,58 (d, J=6,8 Гц, 2Н), 7,50-7,45 (m, 1H), 7,44-7,38 (m, 2Н), 7,35-7,24 (m, 5H), 5,56-5,49 (m, 1H), 3,29-3,21 (m, 1H), 2,93-2,83 (m, 1H). (ESI) m/z (M+H)+ 381,1.
[1524] Соединение 377 (80 мг, выход 39,9%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(пиридин-3-ил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,39 (d, J=7,7 Гц, 1H), 8,82 (d, J=2,2 Гц, 1H), 8,65 (dd, J=1,5, 4,9 Гц, 1H), 8,19 (s, 1H), 7,96-7,82 (m, 2Н), 7,43 (dd, J=4,9, 7,9 Гц, 1H), 7,33-7,20 (m, 5H), 5,53-5,45 (m, 1H), 3,23 (dd, J=3,6, 14,0 Гц, 1H), 2,90 (dd, J=10,0, 14,0 Гц, 1H). МС (ИЭР) m/z (M+H)+ 382,1.
[1525] Соединение 462 (150 мг, выход 73,8%): N-(4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,45 (d, J=7,7 Гц, 1H), 8,91 (ушир. d, J=5,1 Гц, 1H), 7,57 (d, J=7,1 Гц, 2Н), 7,50-7,43 (m, 1H), 7,42-7,34 (m, 2Н), 7,34-7,25 (m, 5H), 5,59-5,48 (m, 1H), 3,24 (dd, J=3,1, 13,9 Гц, 1H), 2,93-2,76 (m, 2Н), 0,72-0,59 (m, 4H). МС (ИЭР) m/z (M+H)+ 421,1.
[1526] Соединение 463 получали из соответствующих промежуточных соединений, 4-фенил-1,2,5-тиадиазол-3-карбоновой кислоты и 3-амино-2-гидрокси-5-фенилпентанамида гидрохлорида с использованием тех же методик, которые описаны для соединения 321. Соединение 463 (120 мг, выход 35%): N-(1-амино-1,2-диоксо-5-фенилпентан-3-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,49 (ушир. d, J=7,1 Гц, 1H), 8,14 (ушир. s, 1H), 7,91-7,78 (m, 3H), 7,56-7,45 (m, 3H), 7,33-7,25 (m, 2Н), 7,22-7,15 (m, 3H), 5,15 (ушир. t, J=6,6 Гц, 1H), 2,77-2,58 (m, 2Н), 2,21-2,08 (m, 1H), 1,96-1,81 (m, 1H). МС (ИЭР) m/z (M+H)+ 395,1.
[1527] Соединение 464 получали из соответствующих промежуточных соединений, 4-фенил-1,2,5-тиадиазол-3-карбоновой кислоты и 3-амино-4-(4-фторфенил)-2-гидроксибутанамида гидрохлорида с использованием тех же методик, которые описаны для соединения 321. Соединение 464 (120 мг, выход 47%): N-(4-амино-1-(4-фторфенил)-3,4-диоксобутан-2-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,45 (d, J=6,4 Гц, 1H), 8,20 (s, 1H), 7,94 (s, 1H), 7,56 (d, J=7,1 Гц, 2Н), 7,50-7,37 (m, 3Н), 7,31 (s, 2H), 7,18-7,05 (m, 2H), 5,47 (s, 1H), 3,29-3,15 (m, 1H), 2,91-2,78 (m, 1H). MC (ИЭР) m/z (M+H)+ 399,0.
[1528] Соединение 465 получали из соответствующих промежуточных соединений, 4-фенил-1,2,5-тиадиазол-3-карбоновой кислоты и 3-амино-2-гидрокси-5-метилгексанамида гидрохлорида с использованием тех же методик, которые описаны для соединения 321. Соединение 465 (110 мг, 38,2% выход): N-(1-амино-5-метил-1,2-диоксогексан-3-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,32 (d, J=6,6 Гц, 1H), 8,14 (s, 1H), 7,95-7,69 (m, 3Н), 7,51 (s, 3Н), 5,30 (s, 1H), 1,78-1,39 (m, 3Н), 0,90 (dd, J=5,8, 15,3 Гц, 6Н). MC (ИЭР) m/z (M+H)+ 347,1.
[1529] Соединение 466 получали из соответствующих промежуточных соединений, 4-фенил-1,2,5-тиадиазол-3-карбоновой кислоты и 3-амино-4-(3,5-диметилфенил)-2-гидроксибутанамида гидрохлорида с использованием тех же методик, которые описаны для соединения 321. Соединение 466 (70 мг, 39,2% выход): N-(4-амино-1-(3,5-диметилфенил)-3,4-диоксобутан-2-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,40 (d, J=7,5 Гц, 1H), 8,19 (s, 1H), 7,92 (s, 1H), 7,62 (d, J=7,3 Гц, 2H), 7,53-7,35 (m, 3Н), 6,89 (s, 3Н), 5,56-5,40 (m, 1H), 3,20-3,08 (m, 1H), 2,85-2,71 (m, 1H), 2,21 (s, 6Н). MC (ИЭР) m/z (M+H)+ 409,1.
[1530] Соединение 467 получали из соответствующих промежуточных соединений, 4-фенил-1,2,5-тиадиазол-3-карбоновой кислоты и 3-амино-2-гидроксигептанамида гидрохлорида с использованием тех же методик, которые описаны для соединения 321. Соединение 467 (80 мг, 73% выход): N-(1-амино-1,2-диоксогептан-3-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,31 (ушир. d, J=7,0 Гц, 1H), 8,14 (ушир. s, 1H), 7,94-7,69 (m, 3Н), 7,58-7,43 (m, 3Н), 5,30-5,15 (m, 1H). 1,82 (ушир. d, J=7,5 Гц, 1H), 1,57 (ушир. d, J=4,8 Гц, 1H), 1,40-1,39 (m, 1H), 1,36-1,22 (m, 1H), 1,36-1,20 (m, 3Н), 0,88-0,81 (m, 3Н). MC (ИЭР) m/z (М+H)+ 347,1.
[1531] Соединение 468 получали из соответствующих промежуточных соединений, 4-фенил-1,2,5-тиадиазол-3-карбоновой кислоты и 3-амино-2-гидроксибутанамида гидрохлорида с использованием тех же методик, которые описаны для соединения 321. Соединение 468 (70 мг, 54,2% выход): N-(4-амино-3,4-диоксобутан-2-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,40 (d, J =6,4 Гц, 1Н), 8,12 (ушир. s, 1Н), 7,92-7,75 (m, 3Н), 7,58-7,43 (m, 3H), 5,25-5,18 (m, 1H), 1,36 (d, J=7,3 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 305,1.
ПРИМЕР 207
СОЕДИНЕНИЯ 378, 578, 599
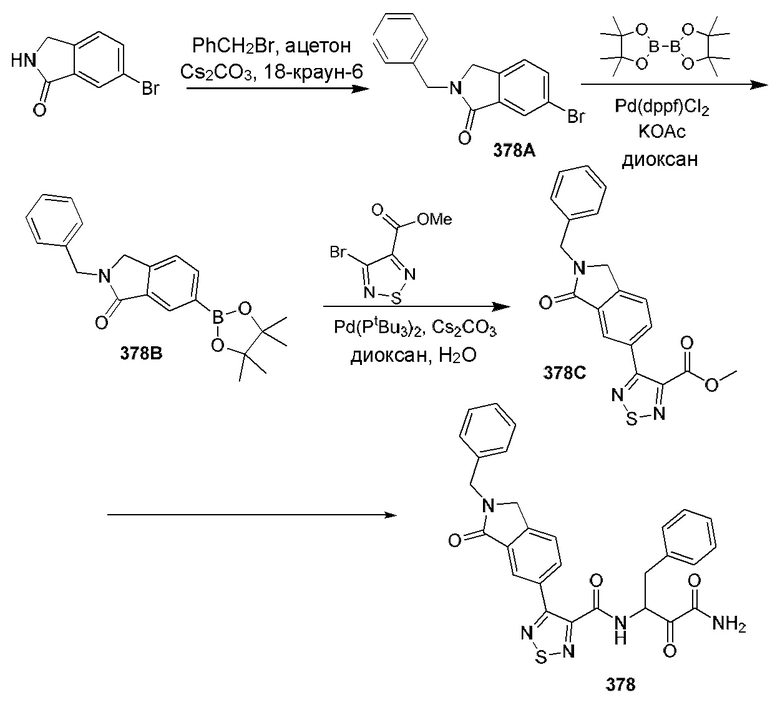
[1532] К раствору 6-бромизоиндолин-1-она (0,5 г, 2,36 ммоль) в ацетоне (20 мл) добавляли BnBr (605 мг, 3,54 ммоль, 0,420 мл), CS2CO3 (1,92 г, 5,90 ммоль) и 18-Краун-6 (62 мг, 235,80 мкмоль), затем смесь перемешивали при 70°С в течение 16 ч. Реакционную смесь концентрировали для удаления растворителя, затем разбавляли водой (50 мл) и подвергали экстракции посредством ЭА (40 мл × 2), органические слои сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® с диоксидом кремния 12 g, элюент 0~20% этилацетат/петролейный эфир, градиент со скоростью потока 30 мл/мин). Соединение 378А (0,46 г, выход: 59,5%) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,02 (d, J=1,8 Гц, 1Н), 7,63 (dd, J=1,9, 8,0 Гц, 1Н), 7,39-7,16 (m, 6H), 4,79 (s, 2H), 4,21 (s, 2H). МС (ИЭР) m/z (M+H)+ 302,0.
[1533] К раствору соединения 378А (0,46 г, 1,52 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (580 мг, 2,28 ммоль) в диоксане (20 мл) добавляли KOAc (299 мг, 3,04 ммоль), и в атмосфере затем N2 добавляли Pd(dppf)Cl2 (111 мг, 152.23 мкмоль), смесь перемешивали при 85°С в течение 16 ч. Реакционную смесь разбавляли ЭА (20 мл), затем фильтровали и промывали ЭА (20 мл × 3), фильтрат концентрировали с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® для флэш-хроматографии с диоксидом кремния 12 g, элюент 0 ~ 30% этилацетат/петролейный эфир, градиент со скоростью потока 30 мл/мин). Затем проводили дополнительную очистку посредством препаративной ТСХ (ПЭ:ЭА=2:1). Получали соединение 378В (0.35 г, выход: 46,1%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,98 (t, J=0,9 Гц, 1Н), 7,86 (dd, J=1,1, 7,5 Гц, 1Н), 7,57 (dd, J=0,9, 7,5 Гц, 1Н), 7,38-7,32 (m, 2H), 7,31-7,23 (m, 3H), 4,77-4,69 (m, 2H), 4,39 (s, 2H), 1,31 (s, 12H). МС (ИЭР) m/z (M+H)+ 349,9.
[1534] К раствору метил-4-бром-1,2,5-тиадиазол-3-карбоксилата (186 мг, 835,17 мкмоль) и соединения 378В (0,35 г, 1,00 ммоль) в диоксане (20 мл) и H2O (2 мл) добавляли K2CO3 (231 мг, 1,67 ммоль), затем Pd(dppf)Cl2 (61 мг, 83,52 мкмоль) добавляли в атмосфере N2, затем смесь перемешивали при 85°С в течение 16 ч в атмосфере N2. Реакционную смесь разбавляли ЭА (30 мл), затем фильтровали и концентрировали с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка SepaFlash® для флэш-хроматографии с диоксидом кремния 12 g, элюент 0~30% этилацетат/петролейный эфир, градиент со скоростью потока 30 мл/мин). Соединение 378С (0,13 г, выход: 37,3%) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,20 (d, J=1,8 Гц, 1Н), 7,87 (dd, J=1,8, 7,9 Гц, 1Н), 7,51 (d, J=7,7 Гц, 1Н), 7,41-7,28 (m, 5H), 4,84 (s, 2H), 4,35 (s, 2H), 4,03-3,95 (m, 3H). МС (ИЭР) m/z (М+Н)+366,0.
[1535] Соединение 378 синтезировали из 378С с использованием тех же методик, которые описаны ранее для соединения 321. Соединение 378 (20 мг, 13,5% выход): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2-бензил-3-оксоизоиндолин-5-ил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,07 (ушир. s, 1H), 8,20-8,00 (m, 1Н), 7,96-7,48 (m, 4H), 7,42-7,12 (m, 10Н), 5,58-5,38 (m, 1Н), 4,78 (s, 2H), 4,45 (s, 2H), 3,26 (dd, J=3,8, 14,3 Гц, 1Н), 2,98 (d, J=14,1 Гц, 1Н). МС (ИЭР) m/z (M+H)+ 526,1.
[1536] Соединение 578 синтезировали путем сочетания метил-4-бром-1,2,5-тиадиазол-3-карбоксилата и (2,2-дифторбензо[d][1,3]диоксол-4-ил)бороновой кислоты, после чего продукт обрабатывали по тем же методикам, которые описаны ранее для соединения 321. Соединение 578 (100 мг, 57,9% выход): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2,2-дифторбензо[d][1,3]ДИОКСОЛ-4-ил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,36-9,32 (m, 1Н), 8,12 (s, 1H), 7,88 (s, 1H), 7,52-7,49 (m, 1H), 7,28-7,17 (m, 7H), 5,50-5,43 (m, 1H), 3,22-3,15 (m, 1H), 2,94-2,86 (m, 1H). MC (ИЭР) m/z (M+H)+ 461,0.
[1537] Соединение 599 синтезировали путем сочетания метил-4-бром-1,2,5-тиадиазол-3-карбоксилата и (2,2-дифторбензо[d][1,3]диоксол-5-ил)бороновой кислоты, после чего полученный продукт обрабатывали по тем же методикам, которые описаны ранее для соединения 321. Соединение 599 (140 мг, 69,6% выход): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,40 (d, J=7,7 Гц, 1H), 8,19 (s, 1H), 7,93 (s, 1H), 7,59 (s, 1H), 7,42 (d, J=0,9 Гц, 2Н), 7,29-7,18 (m, 5H), 5,55-5,41 (m, 1H), 3,23 (dd, J=3,5, 13,9 Гц, 1H), 2,86 (dd, J=10,1, 14,1 Гц, 1H). MC (ИЭР) m/z (M+H)+ 461,0.
ПРИМЕР 208
СОЕДИНЕНИЯ 379-380
[1538] Соединения 379-380 синтезировали из соответствующего промежуточного соединения: 1-(дифторметил)-3-фенил-1H-пиразол-4-карбоновой кислоты - с использованием тех же методик, которые были описаны ранее в Примере 5.
[1539] Соединение 379 (240 мг, выход 76,2%): (S)-N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,79 (d, J=7,5 Гц, 1H), 8,48 (s, 1H), 8,14-7,72 (m, 3Н), 7,55 (dd, J=2,0, 7,3 Гц, 2Н), 7,37-7,20 (m, 8H), 5,40-5,26 (m, 1H), 3,18 (dd, J=3,6, 14,0 Гц, 1H), 2,80 (dd, J=10,1, 13,9 Гц, 1H). MC (ИЭР) m/z (M+H)+ 413,1.
[1540] Соединение 380 (50 мг, выход 61,7%): (R)-N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,79 (d, J=7,3 Гц, 1H), 8,47 (s, 1H), 8,15-7,71 (m, 3Н), 7,60-7,48 (m, 2Н), 7,39-7,21 (m, 8H), 5,40-5,25 (m, 1H), 3,17 (dd, J=3,7, 13,9 Гц, 1H), 2,80 (dd, J=10,0, 14,0 Гц, 1H). MC (ИЭР) m/z (М+H)+ 413,1.
ПРИМЕР 209
СОЕДИНЕНИЯ 381-384, 403, 522-524, 546-547, 550-552, 554-555, 575-577, 588, 596, 598, 608,610,622, 630
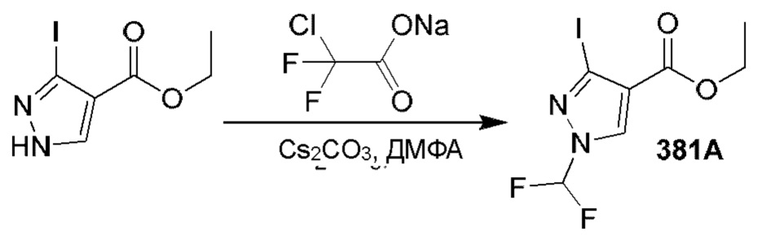
[1541] К раствору этил-3-иод-1H-пиразол-4-карбоксилата (20 г, 75,18 ммоль) в ДМФА (100 мл) добавляли 2-хлор-2,2-дифторацетат натрия (22,92 г, 150,36 ммоль) и CS2CO3 (48,99 г, 150,36 ммоль). Смесь перемешивали при 100°С в течение 16 ч. Реакционную смесь концентрировали, остаток разбавляли H2O (200 мл) и подвергали экстракции EtOAc (100 мл × 3). Объединенные органические слои промывали солевым раствором (200 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка для флэш-хроматографии SepaFlash® с диоксидом кремния Х g, элюент 0% ~ 10% ~20% этилацетат/петролейный эфир градиент). Соединение 381А (9,1 г, выход: 38,30%) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,47-7,95 (m, 1H), 7,44-6,95 (m, 1H), 4,53-4,17 (m, 2H), l,54-l,17(m, 3H).
[1542] Соединения 381-384, 403, 522-524, 546-547, 550-552, 554-555, 561, 563-566, 575-577, 581-582, 586, 588, 596, 598, 608, 610, 622 и 630 синтезировали из соответствующего промежуточного соединения этил-1-(дифторметил)-3-иод-1H-пиразол-4-карбоксилата (381А) с использованием тех же методик, которые были описаны ранее для соединения 242.
[1543] Соединение 381 (125 мг, выход 43,5%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(3-фторфенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,87 (ушир. d, J=7,3 Гц, 1H), 8,56 (s, 1H), 8,18-8,04 (m, 1H), 7,99-7,75 (m, 2H), 7,51-7,36 (m, 3H), 7,33-7,18 (m, 6H), 5,35 (ушир. s, 1H), 3,20 (ушир. dd, J=3,0, 13,8 Гц, 1H), 2,89-2,76 (m, 1H). МС (ИЭР) m/z (М+H)+ 431,1.
[1544] Соединение 382 (70 мг, выход 40,9%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(o-толил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,69 (s, 1H), 8,35 (ушир. d, J=7,3 Гц, 1Н), 8,06 (ушир. s, 1H), 7,95-7,75 (m, 2H), 7,27 (ушир. d, J=6,8 Гц, 3Н), 7,25-7,10 (m, 6H), 5,26 (ушир. s, 1H), 3,19-3,10 (m, 1H), 2,81-2,70 (m, 1H), 2,02 (s, 3Н). МС (ИЭР) m/z (M+H)+ 427,1.
[1545] Соединение 383 (150 мг, выход 58,2%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(2-фторфенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,77-8,62 (m, 2H), 8,16-7,76 (m, 3Н), 7,50-7,17 (m, 9H), 5,36-5,23 (m, 1H), 3,17 (dd, J=3,9, 14,0 Гц, 1H), 2,82 (dd, J=10,0, 13,8 Гц, 1H). МС (ИЭР) m/z (М+Н)+431,1.
[1546] Соединение 384 (130 мг, выход 39,4%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(3-метоксифенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,54-8,35 (m, 2H), 8,03-7,51 (m, 3Н), 7,35-7,14 (m, 8H), 6,96 (d, J=7,8 Гц, 1H), 5,37 (ушир. s, 1H), 3,76 (s, 3Н), 3,22 (d, J=14,3 Гц, 1H), 2,97-2,83 (m, 1H). МС (ИЭР) m/z (М+Н)+ 443,1.
[1547] Соединение 403 (3,1 г, выход 49,74%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,80 (d, J=7,5 Гц, 1H), 8,58-8,37 (m, 1H), 8,10 (s, 1H), 8,05-7,69 (m, 2H), 7,58-7,47 (m, 2H), 7,36-7,19 (m, 8H), 5,47-5,19 (m, 1H), 3,19-3,14 (m, 1H), 2,82-2,75 (m, 1H). МС (ИЭР) m/z (М+Н)+413,2.
[1548] Соединение 522 (25 мг, выход 16,6%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(4-метилпиридин-3-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,77 (s, 1H), 8,65 (d, J=7,3 Гц, 1H), 8,39 (d, J=5,1 Гц, 1H), 8,24 (s, 1H), 8,09-8,02 (m. 1H), 7,93 (s, 1H), 7,78 (d, J=6,2 Гц, 1H), 7,29-7,13 (m, 7H), 5,27-5,19 (m, 1H), 3,13 (dd, J=3,7, 13,9 Гц, 1H), 2,76 (dd, J=9,9, 13,9 Гц, 1H), 1,99 (s, 3Н). МС (ИЭР) m/z (M+H)+ 428,1.
[1549] Соединение 523 (63 мг, выход 63,6%; светло-желтое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(3,5-дифторфенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,89 (d, J=7,5 Гц, 1H), 8,62 (s, 1H), 8,14-8,08 (m, 1H), 7,97-7,79 (m, 2H), 7,39-7,25 (m, 7H), 7,23-7,19 (m, 1H), 5,42-5,33 (m, 1H), 3,20 (dd, J=3,7, 13,9 Гц, 1H), 2,82 (dd, J=10,3, 14,0 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 449,0.
[1550] Соединение 524 (50 мг, выход 31,5%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(2,5-диметилфенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,69 (s, 1H), 8,23 (d, J=7,3 Гц, 1Н), 8,05 (s, 1H), 7,92-7,73 (m, 2H), 7,31-7,24 (m, 2H), 7,24-7,15 (m, 3H), 7,13-7,09 (m, 2H), 6,97 (s, 1H), 5,28-5,24 (m, 1H), 3,14 (dd, J=3,6, 14,0 Гц, 1H), 2,75 (dd, J=9,7, 14,1 Гц, 1H), 2,25 (s, 3H), 1,96 (s, 3H). МС (ИЭР) m/z (M+H)+ 441,1.
[1551] Соединение 546 (120 мг, выход 47,9%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-(2,2-дифторбензо[d][1,3]диоксол-4-ил)-1-(дифторметил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 5 8,87 (d, J=7,5 Гц, 1H), 8,72 (s, 1H), 8,19-7,79 (m, 3H), 7,44 (d, J=7,7 Гц, 1H), 7,32-7,17 (m, 7H), 5,42-5,24 (m, 1H), 3,17 (dd, J=3,5, 13,7 Гц, 1H), 2,81 (dd, J=10,1, 13,9 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 493,1.
[1552] Соединение 547 (140 мг, выход 67,0%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(НАФТАЛИН-1-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,79 (s, 1H), 8,46 (d, J=7,5 Гц, 1H), 8,15-7,71 (m, 5H), 7,62 (d, J=7,9 Гц, 1H), 7,54-7,45 (m, 2H), 7,44-7,35 (m, 2H), 7,28-7,13 (m, 5H), 5,19-5,15 (m, 1H), 3,08 (dd, J=3,7, 13,9 Гц, 1H), 2,72 (dd, J=9,9, 13,9 Гц, 1H). МС (ИЭР) m/z (M+H)+ 463,1.
[1553] Соединение 550 (25 мг, выход 30,3%; светло-желтое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(пиридин-2-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 11,78 (s, 1H), 8,71 (s, 1H), 8,40 (d, J=4,5 Гц, 1H), 8,15(d, J=8,0 Гц, 1H), 8,03-7,99 (m, 1H), 7,91-7,56 (m, 3H), 7,55-7,46 (m, 1H), 7,23-7,08 (m, 5H), 5,67-5,53 (m, 1H), 3,31 (dd, J=5,0, 14,3 Гц, 1H), 3,18-3,11 (m, 1H). МС (ИЭР) m/z (M+H)+ 414,1.
[1554] Соединение 551 (65 мг, выход 82,3%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(НАФТАЛИН-2-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,90 (d, J=7,3 Гц, 1H), 8,53 (s, 1H), 8,16 (d, J=9,9 Гц, 2H), 8,09-7,79 (m, 5H), 7,67 (dd, J=1,7, 8,5 Гц, 1H), 7,56-7,50 (m, 2H), 7,30-7,25 (m, 4H), 7,23-7,18 (m, 1H), 5,39-5,29 (m, 1H), 3,18 (dd, J=3,9, 13,8 Гц, 1H), 2,82 (dd, J=10,3, 14,0 Гц, 1H). МС (ИЭР) m/z (M+H)+ 463,1.
[1555] Соединение 552 (60 мг, выход 53,6%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(2,5-дифторфенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,75-8,67 (m, 2H), 8,09-7,76 (m, 3Н), 7,32-7,15 (m, 8H), 5,30-5,22 (m, 1H), 3,16-3,09 (m, 1H), 2,82-2,73 (m, 1H). MC (ИЭР) m/z (М+H)+ 449,1.
[1556] Соединение 554 (174 мг, выход 86,6%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(3-фтор-5-метилфенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,83 (d, J=7,3 Гц, 1H), 8,53 (s, 1H), 8,21-7,72 (m, 3Н), 7,34-7,23 (m, 5H), 7,21 (ушир. dd, J=2,5, 8,5 Гц, 2H), 7,06 (ушир. d, J=9,9 Гц, 1H), 5,46-5,22 (m, 1H), 3,17 (dd, J=3,9, 14,0 Гц, 1H), 2,80 (dd, J=10,1, 13,9 Гц, 1H), 2,31 (s, 3Н). MC (ИЭР) m/z (М+H)+ 445,1.
[1557] Соединение 555 (85 мг, выход 57,0%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-(2,2-дифторбензо[d][1,3]диоксол-4-ил)-1-(дифторметил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,34 (s, 1H), 7,57-7,27 (m, 1H), 7,24 (ушир. s, 3Н), 7,05-6,97 (m, 4H), 6,96-6,92 (m, 1H), 6,90 (t, J=6,9 Гц, 1H), 6,71 (d, J=6,6 Гц, 1H), 6,26 (ушир. s, 1H), 5,55-5,42 (m, 1H), 4,16 (ушир. s, 2H), 4,11-4,05 (m, 1H), 4,11-4,05 (m, 1H), 4,00-3,92 (m, 1H), 3,22 (m, J=4,7, 14,2 Гц, 1H), 2,93-2,82 (m, 1H). MC (ИЭР) m/z (М-H)+ 471,1.
[1558] Соединение 561 (100 мг, выход 46,4%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(изохинолин-4-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,35 (s, 1H), 8,89 (s, 1H), 8,73 (d, J=7,5 Гц, 1H), 8,39 (s, 1H), 8,24-7,88 (s, 3Н), 7,78 (s, 1H), 7,74-7,66 (m, 2H), 7,66-7,59 (m, 1H), 7,33-7,15 (m, 5H), 5,25-5,14 (m, 1H), 3,21-3,05 (m, 1H), 2,83-2,72 (m, 1H). MC (ИЭР) m/z (M+H)+ 464,1.
[1559] Соединение 563 (110 мг, выход 84,1%; светло-желтое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-(бензо[d][1,3]диоксол-4-ил)-1-(дифторметил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,67 (d, J=7,3 Гц, 1H), 8,53 (s, 1H), 8,08 (s, 1H), 7,97-7,76 (m, 2H), 7,34-7,19 (m, 5H), 6,97-6,91 (m, 1H), 6,90-6,80 (m, 2H), 5,90 (s, 1H), 5,78 (s, 1H), 5,33-5,23 (m, 1H), 3,15 (dd, J=3,9, 14,0 Гц, 1H), 2,81 (dd, J=9,7, 13,9 Гц, 1H). MC (ИЭР) m/z (M+H)+ 457,1.
[1560] Соединение 564 (128 мг, выход 79,3%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(2-фтор-5-метилфенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,71-8,60 (m, 2H), 8,11-8,02 (m, 1H), 7,97-7,76 (m, 2H), 7,33-7,16 (m, 7H), 7,11-7,02 (m, 1H), 5,32-5,23 (m, 1H), 3,15 (dd, J=3,9, 14,0 Гц, 1H), 2,81 (dd, J=9,9, 13,9 Гц, 1H), 2,29 (s, 3H). МС (ИЭР) m/z (M+H)+ 445,1.
[1561] Соединение 565 (75 мг, выход 80,2%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(3,5-диметилфенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,42 (s, 1H), 8,34-8,23 (m, 1H), 7,98-7,47 (m, 3H), 7,28-7,20 (m, 7H), 7,03 (s, 1H), 5,39-5,33 (m, 1H), 3,23-3,19 (m, 1H), 2,90 (dd, J=9,2, 13,9 Гц, 1H), 2,29 (s, 6H). МС (ИЭР) m/z (M+H)+ 441,1.
[1562] Соединение 566 (135 мг, выход 78,7%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(5-фтор-2-метилфенил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,74 (s, 1H), 8,49 (d, J=7,7 Гц, 1H), 8,10-7,77 (m, 3H), 7,31-7,18 (m, 6H), 7,17-7,10 (m, 1H), 6,96 (dd, J=2,9, 9,5 Гц, 1H), 5,32-5,23 (m, 1H), 3,15 (dd, J=3,7, 13,9 Гц, 1H), 2,78 (dd, J=9,9, 13,9 Гц, 1H), 1,97 (s, 3H). МС (ИЭР) m/z (M+H)+ 445,1.
[1563] Соединение 575 (20 мг, выход 66,9%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(хинолин-7-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,94 (dd, J=1,6, 4,1 Гц, 1H), 8,67-8,48 (m, 2H), 8,43-8,33 (m, 2H), 7,98-7,71 (m, 4H), 7,58-7,53 (m, 1H), 7,31-7,17 (m, 6H), 5,48-5,28 (m, 1H), 3,24 (dd, J=4,5, 14,1 Гц, 1H), 2,93 (dd, J=9,3, 14,1 Гц, 1H). МС (ИЭР) m/z (M+H)+ 464,1.
[1564] Соединение 576 (125 мг, выход 66,2%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(хинолин-5-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,94-8,82 (m, 2H), 8,66 (ушир. d, J=7,5 Гц, 1H), 8,20-7,98 (m, 4H), 7,87-7,72 (m, 2H), 7,55-7,40 (m, 2H), 7,32-7,17 (m, 5H), 5,27-5,14 (m, 1H), 3,13 (ушир. dd, J=3,5, 13,9 Гц, 1H), 2,78 (ушир. dd, J=10,0, 13,8 Гц, 1H). МС (ИЭР) m/z(M+H)+ 471,1.
[1565] Соединение 577 (110 мг, выход 47,2%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(изохинолин-6-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,31 (s, 1H), 8,93 (d, J=7,5 Гц, 1H), 8,58 (s, 1H), 8,51 (d, J=5,7 Гц, 1H), 8,23-8,17 (m, 1H), 8,15-7,81 (m, 6H), 7,31-7,25 (m, 4H), 7,24-7,17 (m, 1H), 5,45-5,26 (m, 1H), 3,19 (dd, J=4,0, 13,9 Гц, 1H), 2,90-2,77 (m, 1H). МС (ИЭР) m/z (M+H)+ 471,1.
[1566] Соединение 581 (30 мг, выход 37,5%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(пиразин-2-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 10,55 (d, J=7,0 Гц, 1H), 9,40-9,27 (m, 1H), 8,91-8,83 (m, 2H), 8,73-8,15 (m, 1H), 8,06-7,67 (m, 3H), 7,30-7,23 (m, 5H), 5,75-5,62 (m, 1H), 3,28-3,16 (m, 2H). МС (ИЭР) m/z (M+H)+ 415,1.
[1567] Соединение 582 (11,4 мг, выход 13,6%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(5-метилпиридин-3-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,83 (d, J=7,5 Гц, 1H), 8,62 (s, 1H), 8,55 (d, J=2,0 Гц, 1H), 8,40 (d, J=1,3 Гц, 1H), 8,08-7,79 (m, 3H), 7,76 (s, 1H), 7,30-7,25 (m, 4H), 7,21 (td, J=4,5, 8,8 Гц, 1H), 5,36-5,27 (m, 1H), 3,17 (dd, J=3,7, 13,7 Гц, 1H), 2,81 (dd, J=10,0, 14,0 Гц, 1H), 2,29 (s, 3H). МС (ИЭР) m/z (M+H)+ 428,1.
[1568] Соединение 586 (56 мг, выход 32,4%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(5-фторпиридин-3-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,94-8,86 (m, 1H), 8,74 (s, 1H), 8,69-8,58 (m, 2H), 8,15-8,08 (m, 1H), 8,01-7,81 (m, 3H), 7,29 (ушир. d, J=4,2 Гц, 4H), 7,21 (ушир. d, J=4,4 Гц, 1H), 5,36 (ушир. s, 1H), 3,23-3,14 (m, 1H), 2,87-2,77 (m, 1H). МС (ИЭР) m/z (M+H)+ 431,0.
[1569] Соединение 588 (95 мг, выход 63,0%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(хинолин-2-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 11,90 (d, J=8,2 Гц, 1H), 8,87 (s, 1H), 8,59 (d, J=8,6 Гц, 1H), 8,27 (d, J=8,6 Гц, 1H), 8,20-8,04 (m, 2H), 7,98-7,79 (m, 2H), 7,76-7,64 (m, 3H), 7,02-6,91 (m, 5H), 5,78-5,70 (m, 1H), 3,32-3,27 (m, 1H), 3,14 (dd, J=8,0, 14,0 Гц, 1H). МС (ИЭР) m/z (M+H)+ 464,1.
[1570] Соединение 596 (110 мг, выход 58,1%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(хинолин-4-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,94-8,77 (m, 3H), 8,19-7,86 (m, 3H), 7,83-7,69 (m, 3H), 7,55-7,51 (m, 1H), 7,41-7,20 (m, 6H), 5,28-5,12 (m, 1H), 3,14 (dd, J=3,6, 14,0 Гц, 1H), 2,79 (dd, J=9,9, 14,1 Гц, 1H). МС (ИЭР) m/z (M+H)+ 464,1.
[1571] Соединение 598 (85 мг, выход 53,3%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(изохинолин-5-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,35 (s, 1Н), 8,90-8,63 (m, 2H), 8,41 (d, J=6,0 Гц, 1Н), 8,30-7,94 (m, 3Н), 7,90-7,64 (m, 3Н), 7,51 (d, J=6,0 Гц, 1Н), 7,32-7,17 (m, 5H), 5,29-5,12 (m, 1Н), 3,13 (dd, J=3,9, 14,0 Гц, 1Н), 2,77 (dd, J=10,0, 13,8 Гц, 1Н). МС (ИЭР) m/z (M+H)+ 464,1.
[1572] Соединение 608 (25 мг, выход 37,2%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(изохинолин-3-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 11,96 (d, J=7,1 Гц, 1Н), 9,03 (s, 1Н), 8,78 (s, 1Н), 8,60 (s, 1Н), 8,22-7,84 (m, 6H), 7,80-7,75 (m, 1Н), 7,12-7,05 (m, 4H), 7,03-6,96 (m, 1Н), 5,69-5,55 (m, 1Н), 3,30-3,24 (m, 1Н), 3,12 (dd, J=7,6, 14,0 Гц, 1Н). МС (ИЭР) m/z (M+H)+ 464,1.
[1573] Соединение 610 (42 мг, выход 15,5%; бледно-желтое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(хинолин-3-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,12 (s, 1Н), 8,73-8,55 (m, 3Н), 8,13-7,89 (m, 3Н), 7,79 (d, J=8,8 Гц, 2H), 7,65 (d, J=7,0 Гц, 2H), 7,28 (s, 5H), 5,39 (s, 1Н), 3,00-2,82 (m, 1Н), 3,3-3,15 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 464,1.
[1574] Соединение 622 (25 мг, выход 22,7%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(хинолин-6-ил)-1H-пиразол-4-карбоксамид гидрохлорид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,13 (d, J=3,7 Гц, 1Н), 8,99 (d, J=7,5 Гц, 1Н), 8,86-8,75 (m, 1Н), 8,67 (s, 1Н), 8,44 (s, 1Н), 8,19-8,12 (m, 2H), 8,12-8,07 (m, 1Н), 8,00-7,80 (m, 3Н), 7,32-7,25 (m, 4H), 7,24-7,18 (m, 1Н), 5,43-5,26 (m, 1Н), 3,26-3,12 (m, 1Н), 2,90-2,80 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 464,1.
[1575] Соединение 630 (20 мг, выход 42,58%; бледно-желтое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(дифторметил)-3-(изохинолин-7-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,31 (s, 1Н), 8,68-8,48 (m, 3Н), 8,41 (s, 1Н), 8,08-7,71 (m, 5H), 7,60 (ушир. s, 1Н), 7,31-7,16 (m, 5H), 5,39 (ушир. t, J=10,5 Гц, 1Н), 3,24 (ушир. dd, J=4,3, 14,1 Гц, 1Н), 2,99-2,86 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 464,2.
ПРИМЕР 210
СОЕДИНЕНИЯ 385-391, 532
[1576] Соединения 385-391, 532 синтезировали из соответствующих исходных веществ с использованием тех же методик, которые были описаны ранее для соединения 265.
[1577] Соединение 385 (25 мг, выход 38,7%): (S)-N-(4-амино-1-(4-метоксифенил)-3,4-диоксобутан-2-ил)-2-метил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,74 (ушир. d, J=7,6 Гц, 1Н), 8,10 (ушир. s, 1H), 8,06-7,95 (m, 2H), 7,83 (s, 1H), 7,43-7,29 (m, 3Н), 7,18 (ушир. d, J=8,4 Гц, 2Н), 6,83 (ушир. d, J=8,4 Гц, 2Н), 5,40-5,29 (m, 1H), 3,73-3,62 (m, 3Н), 3,15-3,06 (m, 1H), 2,97-2,86 (m, 1H), 2,53 (s, 3Н). MC (ИЭР) m/z (M+H)+ 408,1.
[1578] Соединение 386 (14 мг, выход 22,8%): (S)-N-(1-амино-1,2-диоксо-5-фенилпентан-3-ил)-2-метил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,87 (ушир. d, J=7,2 Гц, 1H), 8,13-8,04 (m, 3Н), 7,77 (ушир. s, 1H), 7,43-7,34 (m, 3Н), 7,30-7,13 (m, 5H), 5,09 (ушир. s, 1H), 2,81-2,70 (m, 1H), 2,67-2,61 (m, 1H), 2,56 (s, 3Н), 2,15-1,90 (m, 2Н). MC (ИЭР) m/z (M+H)+ 392,1.
[1579] Соединение 387 (18 мг, выход 29,4%): N-((3S,4R)-1-амино-4-метил-1,2-диоксогексан-3-ил)-2-метил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,48 (ушир. d, J=7,2 Гц, 1H), 8,09-8,03 (m, 3Н), 7,77 (ушир. s, 1H), 7,43-7,32 (m, 3Н), 5,17-5,09 (m, 1H), 2,53 (s, 3Н), 2,04 (ушир. s, 1H), 1,41 (ушир. s, 1H), 1,24-1,13 (m, 1H), 0,91-0,78 (m, 6H). MC (ИЭР) m/z (M+H)+ 344,2.
[1580] Соединение 388 (35 мг, выход 47,8%): (S)-N-(4-амино-1-(3,5-диметилфенил)-3,4-диоксобутан-2-ил)-2-метил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,71 (d, J=7,2 Гц, 1H), 8,10 (s, 1H), 8,06-7,98 (m, 2Н), 7,82 (s, 1H), 7,42-7,32 (m, 3Н), 6,87 (s, 2Н), 6,81 (s, 1H), 5,41-5,35 (m, 1H), 3,13-3,06 (m, 1H), 2,92-2,84 (m, 1H), 2,52 (s, 3Н), 2,18 (s, 6H). MC (ИЭР) m/z (M+H)+ 406,1.
[1581] Соединение 389 (55 мг, выход 15,9%): (S)-N-(4-амино-1-(1H-индол-3-ил)-3,4-диоксобутан-2-ил)-2-метил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 10,83 (s, 1H), 8,60 (d, J=7,2 Гц, 1H), 8,11 (s, 1H), 8,04-7,99 (m, 2Н), 7,85 (s, 1H), 7,66 (d, J=8,0 Гц, 1H), 7,38-7,28 (m, 4H), 7,19-7,16 (m, 1H), 7,08-7,01 (m, 1H), 7,00-6,94 (m, 1H), 5,51-5,44 (m, 1H), 3,36-3,32 (m, 1H), 3,15-3,06 (m, 1H), 2,50 (s, 3Н). МС (ИЭР) m/z (М+H)+ 417,1.
[1582] Соединение 390 (50,1 мг, выход 83,9%): N-(4-амино-1-(3,5-дихлорфенил)-3,4-диоксобутан-2-ил)-2-метил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,93 (d, J=7,6 Гц, 1H), 8,09 (ушир. s, 1H), 8,02-7,92 (m, 2H), 7,81 (ушир. s, 1H), 7,46-7,29 (m, 6H), 5,38-5,26 (m, 1H), 3,25-3,17 (m, 1H), 3,02-2,88 (m, 1H), 2,52 (s, 3Н). МС (ИЭР) m/z (M+H)+ 446,0.
[1583] Соединение 391 (60 мг, выход 45,63%): (S)-N-(1-амино-5,5-диметил-1,2-диоксогексан-3-ил)-2-метил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,06 (ушир. s, 0,23H), 7,78 (ушир. s, 0,23H), 7,65 (d, J=10,0 Гц, 0,73Н), 7,43-7,33 (m, 3Н), 7,32-7,26 (m, 1,4H), 6,39-6,14 (m, 1H), 5,31-5,24 (m, 0,25H), 4,35-4,28 (m, 0,74H), 2,55-2,50 (m, 3Н), 1,74-1,66 (m, 0,3H), 1,62-1,50 (m, 1H), 1,33-1,24 (m, 0,79H), 0,95-0,82 (m, 9H). МС (ИЭР) m/z (M+H)+ 358,1.
[1584] Соединение 532 (65 мг, выход 27,6%; светло-желтое твердое вещество): N-(4-амино-1-(1H-индол-3-ил)-3,4-диоксобутан-2-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 10,67 (ушир. s, 1H), 9,00 (ушир. d, J=7,3 Гц, 1H). 7,87 (ушир. s, 1H), 7,73-7,58 (m, 4H), 7,46-7,39 (m, 1H), 7,34 (q, J=7,5 Гц, 3Н), 7,15 (d, J=2,3 Гц, 1H), 7,09 (t, J=7,2 Гц, 1H), 7,02-6,96 (m, 1H), 5,59 (ddd, J=4,3, 7,3, 9,0 Гц, 1H), 3,40 (dd, J=4,1, 14,7 Гц, 1H), 3,16-3,10 (m, 1H). МС (ИЭР) m/z (M+H)+ 420,1.
ПРИМЕР 211
СОЕДИНЕНИЕ 392
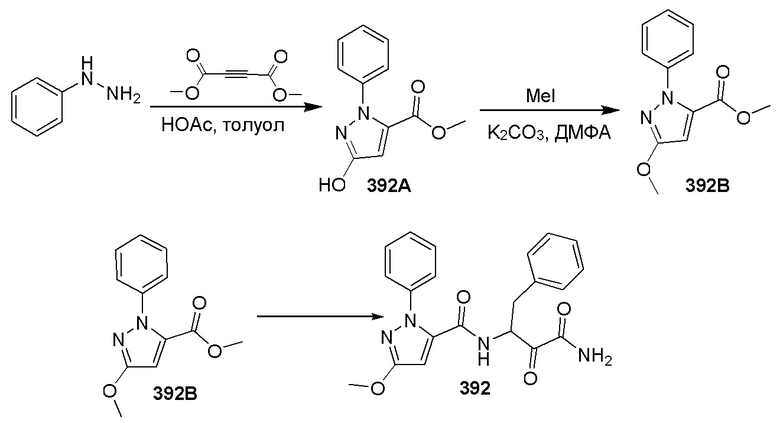
[1585] Перемешиваемый раствор диметил-бут-2-индиоата (5,4 мл, 44,7 ммоль) в толуоле (35 мл) и АсОН (35 мл, 612,0 ммоль) при 0°С аккуратно обрабатывали фенилгидразином (4 мл, 40,6 ммоль). Смесь перемешивали при 20°С в течение 1 ч. Затем смесь нагревали до 115°С и перемешивали в течение 4 ч. Реакционную смесь выдерживали при 20°С в течение 12 ч. Реакционную смесь фильтровали и осадок на фильтре промывали посредством EtOH (30 мл × 3). Осадок на фильтре сушили при пониженном давлении с получением соединения 392А (2,2 г, выход 24,7%) получали в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1H ЯМР (ДМСО-d6, 400 МГц): δ 12,16 (ушир. s, 1H), 7,77-7,70 (m, 2H), 7,54-7,47 (m, 2H), 7,40-7,37 (m, 1H), 5,97 (s, 1H), 3,80 (s, 3Н).
[1586] К раствору соединения 392А (1 г, 4,5 ммоль) в ДМФА (20 мл) добавляли K2CO3 (1,2 г, 9,1 ммоль) и MeI (855 мкл, 13,7 ммоль). Затем смеси перемешивали при 15°С в течение 12 ч. Смесь фильтровали и остаток промывали посредством ЭА (20 мл × 2). К смеси добавляли H2O (20 мл) и отделяли органический слой, водный слой подвергали экстракции посредством ЭА (20 мл × 2). Объединенный органический слой промывали солевым раствором, сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении с получением соединения 392В (0,515 г, выход 48,3%) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии. 1H ЯМР (ДМСО-d6, 400 МГц): δ 7,67 (d, J=7,9 Гц, 2H), 7,51 (t, J=7,8 Гц, 2H), 7,44-7,37 (m, 1H), 6,41 (s, 1H), 3,98 (s, 3Н), 3,83 (s, 3Н).
[1587] Соединение 392 синтезировали из 392В с использованием тех же методик, которые описаны ранее для соединения 66. Соединение 392 (40 мг, выход 21,8%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-метокси-1-фенил-1H-пиразол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,38 (ушир. d, J=7,8 Гц, 1H), 8,11 (ушир. s, 1H), 7,86 (ушир. s, 1H), 7,71 (ушир. d, J=8,0 Гц, 2H), 7,53 (ушир. t, J=7,7 Гц, 2H), 7,44-7,35 (m, 1H), 7,32-7,15 (m, 5H), 6,26 (s, 1H), 5,44 (ушир. d, J=3,3 Гц, 1H), 3,96 (s, 3Н), 3,21 (ушир. dd, J=3,8, 14,1 Гц, 1H), 3,11-3,00 (m, 1H). МС (ИЭР) m/z (M+H)+ 393,1.
ПРИМЕР 212
СОЕДИНЕНИЕ 393
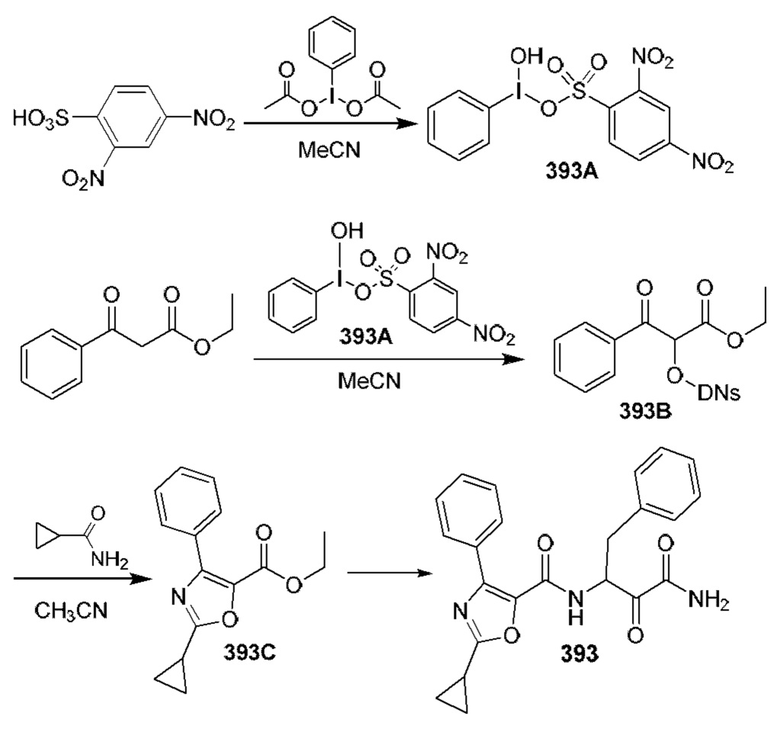
[1588] К раствору 2,4-динитробензолсульфоновой кислоты (10,0 г, 31,05 ммоль) в CH3CN (150 мл) добавляли фенилиодозобензолдиацетат (15,4 г, 62,09 ммоль). Смесь перемешивали при 25°С в течение 1 ч. В смесь добавляли МТБЭ (400 мл), охлаждали ледяной водой в течение 10 мин. Затем смесь фильтровали и собирали фильтрат. Соединение 393А (11 г, неочищенного) получали в виде желтого твердого вещества. Неочищенный продукт использовали на следующей стадии в том виде, в котором он был получен.
[1589] К раствору соединения 393А (11,3 г, 24,14 ммоль) в CH3CN (100 мл) добавляли этил-3-оксо-3-фенилпропаноат (4,23 г, 21,98 ммоль). Смесь перемешивали при 90°С в течение 1 ч. Реакционную смесь использовали на следующей стадии в том виде, в котором она была получена.
[1590] К раствору соединения 393В (9,69 г, 21,95 ммоль) в CH3CN (100 мл) добавляли циклопропанкарбоксамид (2,24 г, 26,34 ммоль). Смесь перемешивали при 90°С в течение 12 ч. Смесь концентрировали. Остаток очищали посредством колоночной хроматографии SiO2, петролейный эфир/этилацетат = 10/1. Соединение 393С (800,0 мг, неочищенного) получали в виде бесцветного масла. Неочищенный продукт использовали на следующей стадии в том виде, в котором он был получен.
[1591] Соединение 393 синтезировали из 393С и с использованием тех же методик, которые описаны ранее для соединения 66. Соединение 393 (60 мг, 29,0% выход): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-2-циклопропил-4-фенилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,71 (d, J=7,6 Гц, 1Н), 8,11 (s, 1H), 8,04-7,96 (m, 2H), 7,84 (s, 1H), 7,40-7,14 (m, 8H), 5,39-5,31 (m, 1H), 3,23-3,15 (m, 1H), 2,99-2,91 (m, 1H), 2,24-2,14 (m, 1H), 1,23-1,03 (m, 4H). MC (ИЭР) m/z (M+H)+ 404,1.
ПРИМЕР 213
СОЕДИНЕНИЕ 394
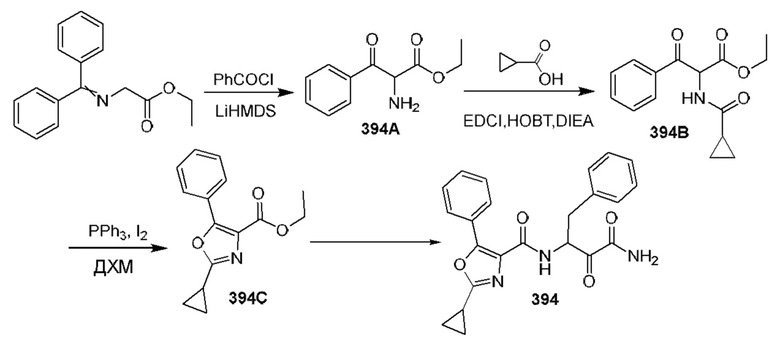
[1592] Смесь этил-2-((дифенилметилен)амино)ацетата (5,00 г, 18,70 ммоль) в ТГФ (100 мл) дегазировали и продували N2 3 раза, к ней добавляли LiHMDS (1 M, 22 мл) при -78°С, затем смесь перемешивали в течение 0,5 ч, затем добавляли бензоилхлорид (22,44 ммоль, 2,61 мл) при -78°С, и смесь перемешивали при 25°С в течение 2 ч в атмосфере N2. Смесь гасили посредством HCl (2 н., 240 мл) и перемешивали в течение 1 ч, затем промывали ЭА (50 мл). Водную фазу собирали, добавляли NaHCO3 (водный) до рН ~ 9, затем подвергали экстракции посредством ЭА (100 мл). Органический слой промывали солевым раствором (100 мл), сушили над Na2SO4 и концентрировали. Соединение 394А (2,6 г, неочищенного) получали в виде желтого масла. Неочищенный продукт использовали на следующей стадии в том виде, в котором он был получен.
[1593] К раствору соединения 394А (1,30 г, 6,27 ммоль) в ДМФА (20 мл) добавляли DIEA (25,09 ммоль, 4,4 мл), циклопропанкарбоновую кислоту (648,1 мг, 7,53 ммоль) и HBTU (2,62 г, 6,90 ммоль). Смесь перемешивали при 25°С в течение 1 ч. Смесь концентрировали, разбавляли ЭА (200 мл), промывали HCl (1 M, 200 мл), NaHCO3 (водный, 200 мл), солевым раствором (200 мл), сушили над Na2SO4 и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в условиях ТФУ). Соединение 394В (400,0 мг, 1,45 ммоль, 23,2% выход) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,13 (d, J=8,0 Гц, 1Н), 7,98-7,92 (m, 2H), 7,71-7,64 (m, 1H), 7,58-7,50 (m, 2H), 6,17 (d, J=8,0 Гц, 1H), 4,16-4,05 (m, 2H), 1,83-1,74 (m, 1H), 1,11-1,04 (m, 3Н), 0,74-0,60 (m, 4H).
[1594] К раствору соединения 394В (400,0 мг, 1,45 ммоль) в ДХМ (15 мл) добавляли Et3N (5,96 ммоль, 800 мкл), I2 (737,6 мг, 2,91 ммоль) и PPh3 (762,2 мг, 2,91 ммоль). Смесь перемешивали при 25°С в течение 2 ч. Смесь концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1 до 0:1). Соединение 394С (300,0 мг, неочищенного) получали в виде желтого масла. Неочищенный продукт использовали in next стадия без дополнительной очистки.
[1595] Соединение 394 синтезировали из 394С и с использованием тех же методик, которые описаны ранее для соединения 66. Соединение 394 (50 мг, выход 31,4%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-2-циклопропил-5-фенилоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,31 (d, J=7,6 Гц, 1H), 8,11 (s, 1H), 8,05-7,99 (m, 2H), 7,86 (s, 1H), 7,45-7,36 (m, 3Н), 7,29-7,16 (m, 5H), 5,45-5,38 (m, 1H), 3,23-3,16 (m, 1H), 3,11-3,03 (m, 1H), 2,22-2,13 (m, 1H), 1,13-1,04 (m, 4H). МС (ИЭР) m/z (M+H)+ 404,2.
ПРИМЕР 214
СОЕДИНЕНИЕ 395

[1596] К смеси этил-3-бром-1-метил-1H-пиразол-4-карбоксилата (1 г, 4,29 ммоль), 2-бензил-4,4,5,5-тетраметил-1,3,2-диоксаборолана (1,12 г, 5,15 ммоль), K2CO3 (1,19 г, 8,58 ммоль) в диоксане (15 мл) и H2O (5 мл) добавляли Pd(dppf)Cl2 (314 мг, 429,07 мкмоль) порциями при 15°С в атмосфере N2. Смесь перемешивали при 90°С в течение 16 ч. Реакционную смесь разбавляли H2O (20 мл), подвергали экстракции EtOAc (40 мл × 2), органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = 5:1 до 3:1) с получением соединения 395А (1 г, выход: 35,4%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,82 (s, 1H), 7,82 (s, 1H), 7,38-7,10 (m, 5H), 4,25-4,19 (m, 2H), 3,89 (s, 3H), 3,85 (s, 2H), 1,34 (t, J=7,1 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 245,0.
[1597] К смеси соединения 395А (0,85 г, 3,48 ммоль) в CHCl3 (30 мл) добавляли MnO2 (4,54 г, 52,19 ммоль) одной порцией при 15°С. Смесь перемешивали при 70°С в течение 36 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = от 5:1 до 1:1) с получением соединения 395В (0,25 г, выход: 24,12%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,97 (s, 1H), 7,95-7,91 (m, 2H), 7,59 (t, J=6,9 Гц, 1H), 7,46 (t, J=7,2 Гц, 2H), 4,12-4,06 (m, 2H), 4,01 (s, 3Н), 1,03 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 258,9.
[1598] Соединение 395 синтезировали из 395В и с использованием тех же методик, которые описаны ранее для соединения 66. Соединение 395 (20 мг, 18,61% выход): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-бензоил-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, CD3CN) δ 9,77 (d, J=5,3 Гц, 1H), 8,13 (s, 1H), 8,02 (d, J=7,9 Гц, 2H), 7,75-7,62 (m, 1H), 7,52 (t, J=7,3 Гц, 2H), 7,24-7,16 (m, 5H), 7,07-6,95 (m, 1H), 6,22 (s, 1H), 5,62-5,44 (m, 1H), 3,92 (s, 3Н), 3,33 (dd, J=4,5, 13,8 Гц, 1H), 3,03 (dd, J=8,4, 13,9 Гц, 1H). МС (ИЭР) m/z (М+Н)+ 405,1.
ПРИМЕР 215
СОЕДИНЕНИЯ 396-402
[1599] Соединения 396-402 синтезировали из соответствующего исходного материала с использованием тех же методик, которые описаны ранее для Соединения 21.
[1600] Соединение 396 (260 мг, выход 81,12%): 1-(дифторметил)-N-(1-оксо-3-фенилпропан-2-ил)-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, CD3CN) δ 9,61 (s, 1H), 8,27 (s, 1H), 7,59-7,51 (m, 2H), 7,44-7,35 (m, 3Н), 7,33-7,23 (m, 3Н), 7,22-7,16 (m, 2H), 6,93 (d, J=7,7 Гц, 1Н), 4,60 (ddd, J=5,0, 7,6, 9,1 Гц, 1Н), 3,26 (dd, J=5,1, 14,1 Гц, 1Н), 2,92 (dd, J=9,0, 14,1 Гц, 1Н). МС (ИЭР) m/z (M+H)+ 370,0.
[1601] Соединение 397 (153 мг, выход 50,5%): N-(1-оксо-3-фенилпропан-2-ил)-5-фенилизоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,61 (s, 1Н), 8,96 (d, J=7,3 Гц, 1Н), 8,83 (s, 1Н), 7,82 (d, J=7,3 Гц, 2H), 7,57-7,39 (m, 3Н), 7,31-7,13 (m, 5H), 4,55 (s, 1Н), 3,28-3,19 (m, 1Н), 2,92-2,76 (m, 1Н), МС (ИЭР) m/z (M+H)+ 321,0.
[1602] Соединение 398 (180 мг, выход 52,4%): 3-метил-N-(1-оксо-3-фенилпропан-2-ил)-1-(пиридин-2-ил)-1H-пиразол-5-карбоксамид: 1H ЯМР (400 МГц, CD3CN) δ 9,59 (s, 1Н), 9,17 (d, J=7,2 Гц, 1Н), 8,29-8,24 (m, 1Н), 7,94-7,87 (m, 1Н), 7,61 (d, J=8,0 Гц, 1Н), 7,34-7,23 (m, 5H), 7,22-7,16 (m, 1Н), 6,46 (s, 1Н), 4,37-4,29 (m, 1Н), 3,20-3,12 (m, 1Н), 2,89-2,80 (m, 1Н), 2,24 (s, 3Н). МС (ИЭР) m/z (M+H)+ 375,0.
[1603] Соединение 399 (580 мг, выход 85,67%): 3-метил-N-(1-оксо-3-фенилпропан-2-ил)-1-(5-(трифторметил)пиридин-2-ил)-1H-пиразол-5-карбоксамид: 1H ЯМР (400 МГц, CD3CN) δ 9,69 (s, 1Н), 8,52-8,40 (m, 1Н), 8,16 (dd, J=2,4, 8,6 Гц, 1Н), 7,99 (ушир. d, J=6,2 Гц, 1Н), 7,89 (d, J=8,6 Гц, 1Н), 7,34-7,16 (m, 5H), 6,55 (s, 1Н), 4,57 (ddd, J=5,1, 7,2, 8,9 Гц, 1Н), 3,28 (dd, J=5,1, 14,3 Гц, 1Н), 3,01 (dd, J=8,9, 14,2 Гц, 1Н), 2,31 (s, 3Н). МС (ИЭР) m/z (М+Н)+ 403,1.
[1604] Соединение 400 (135 мг, выход 46,3%): 1-(3-((бензилокси)метил)фенил)-3-метил-N-(1-оксо-3-фенилпропан-2-ил)-1H-пиразол-5-карбоксамид: 1H ЯМР (400 МГц, CDCl3) δ 9,60 (s, 1Н), 7,45 (s, 1Н), 7,42-7,28 (m, 10Н), 7,09-7,04 (m, 2H), 6,51 (s, 1Н), 6,32 (d, J=6,0 Гц, 1Н), 4,78 (q, J=6,6 Гц, 1Н), 4,59 (d, J=14,3 Гц, 5H), 3,22-3,10 (m, 2H), 2,35 (s, 3Н). МС (ИЭР) m/z (M+H)+ 454,2.
[1605] Соединение 401 (180 мг, выход 37,0%): N-(1-оксо-3-фенилпропан-2-ил)-4-фенил-1,2,5-тиадиазол-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,64 (s, 1Н), 9,35 (ушир. d, J=8,0 Гц, 1Н), 7,57-7,53 (m, 2H), 7,47-7,41 (m, 1Н), 7,40-7,34 (m, 2H), 7,29-7,18 (m, 5H), 4,75-4,68 (m, 1Н), 3,29-3,25 (m, 1Н), 2,89-2,81 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 338,0.
[1606] Соединение 402 (250 мг, выход 35,9%): 4-(2-фторфенил)-2-метил-N-(1-оксо-3-фенилпропан-2-ил)оксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,53 (s, 1Н), 8,85 (d, J=7,6 Гц, 1Н), 7,49-7,40 (m, 2H), 7,28-7,14 (m, 7H), 4,50-4,44 (m, 1Н), 3,25-3,18 (m, 1Н), 2,94-2,86 (m, 1H), 2,52 (s, 3Н). МС (ИЭР) m/z (M+H)+ 353,1.
ПРИМЕР 216
СОЕДИНЕНИЯ 404-405, 609, 618
[1607] Соединения 404-405, 609 и 618 синтезировали из соответствующих исходных веществ с использованием тех же методик, которые были описаны ранее, например, для 254.
[1608] Соединение 404 (3,69 г, выход 85,6%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(3-фторфенил)-2-метилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, CDCl3) δ 8,02-7,89 (m, 2H), 7,44-7,26 (m, 4H), 7,21-7,01 (m, 3H), 6,88-6,69 (m, 2H), 5,79-5,70 (m, 1H), 5,66 (ушир. s, 1H), 3,53-3,39 (m, 1H), 3,35-3,18 (m, 1H), 2,56 (s, 3H). MC (ИЭР) m/z (M+H)+ 396,1.
[1609] Соединение 405 получали путем окисления N-(4-амино-3-гидрокси-4-оксо-1-фенилбутан-2-ил)-4-(2-фторфенил)-2-метилоксазол-5-карбоксамида с использованием тех же условий, которые использовали для окисления промежуточногое соединения 12Н до 12. Соединение 405 (3,97 г, 34,6% выход): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2-фторфенил)-2-метилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,78 (d, J=7,2 Гц, 1H), 8,07 (ушир. s, 1H), 7,82 (ушир. s, 1H), 7,48-7,38 (m, 2H), 7,31-7,14 (m, 7H), 5,38-5,29 (m, 1H), 3,17-3,09 (m, 1H), 2,98-2,88 (m, 1H), 2,53 (s, 3H). MC (ИЭР) m/z (М+Н)+ 396,1.
[1610] В качестве альтернативы N-(4-амино-3-гидрокси-4-оксо-1-фенилбутан-2-ил)-4-(2-фторфенил)-2-метилоксазол-5-карбоксамид окисляли с использованием EDC и дихлоруксусную кислоту, как показано ниже, с получением соединения 405.
[1611] К раствору соединения N-(4-амино-3-гидрокси-4-оксо-1-фенилбутан-2-ил)-4-(2-фторфенил)-2-метилоксазол-5-карбоксамида (100 мг, 0,25 ммоль, 1,0 эквив.) в ДМСО (5 мл) добавляли N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (351,5 мг, 2,3 ммоль, 9 экв.) при комнатной температуре. После перемешивания в течение 10 минут добавляли дихлоруксусную кислоту (0,083 мл, 1 ммоль, 4 эквив.). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, при которой точка ЖХ-МС показала, что реакция была завершена. Смесь разбавляли дихлорметаном (10 мл) и последовательно промывали насыщенным бикарбонатом натрия (2×10 мл), 1 н. HCl (2 × 10 мл) и насыщенным солевым раствором (10 мл). Органический слой сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученное твердое вещество растирали с этилацетатом (2×1 мл) при комнатной температуре в течение 2 часов и сушили под вакуумом при 45°С в течение ночи с получением соединения 405 в виде белого твердого вещества (69 мг, 70% выход; (М+H)+ 396,1.
[1612] Соединение 609 (70 мг, выход 35,13%, белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(бензо[d][1,3]диоксол-4-ил)-2-метилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6,) δ 8,31-8,18 (m, 1H), 7,80 (ушир. s, 1H), 7,61 (ушир. s, 1H), 7,33-7,14 (m, 5H), 7,06-6,98 (m, 1H), 6,94-6,79 (m, 2H), 5,96-5,85 (m, 2H), 5,45-5,34 (m, 1H), 3,27-3,18 (m, 1H), 3,05-2,95 (m, 1H). MC (ИЭР) m/z (M+H)+ 422,1.
[1613] Соединение 618 (80 мг, выход 39,73%, белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2,2-дифторбензо[d][1,3]диоксол-4-ил)-2-метилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 7,52-7,45 (m, 1H), 7,32-7,26 (m, 2H), 7,25-7,18 (m, 1H), 7,13-7,05 (m, 4H), 6,79-6,68 (m, 2H), 5,71-5,63 (m, 1H), 5,56 (ушир. s, 1H), 3,48-3,40 (m, 1H), 3,30-3,21 (m, 1H), 2,57 (s, 3H). MC (ИЭР) m/z (M+H)+ 458,1.
ПРИМЕР 217
СОЕДИНЕНИЕ 406
[1614] Соединения 406 синтезировали из соответствующих исходных промежуточных соединений 274D и 250D с использованием тех же методик, которые были описаны ранее, например, для 306. Соединение 406 (3,3 г): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-циклопропил-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,34 (ушир. d, J=7,1 Гц, 1H), 8,12 (s, 1H), 8,06 (ушир. s, 1H), 7,80 (ушир. s, 1H), 7,53 (ушир. d, J=3,1 Гц, 2H), 7,35-7,09 (m, 8H), 5,26 (ушир. s, 1H), 3,78 (ушир. s, 1H), 3,14 (ушир. d, J=11,5 Гц, 1H), 2,87-2,73 (m, 1H), 2,05 (s, 1H), 1,11-0,92 (m, 4H). MC (ИЭР) m/z (M+H)+ 403,4.
ПРИМЕР 218
СОЕДИНЕНИЕ 410
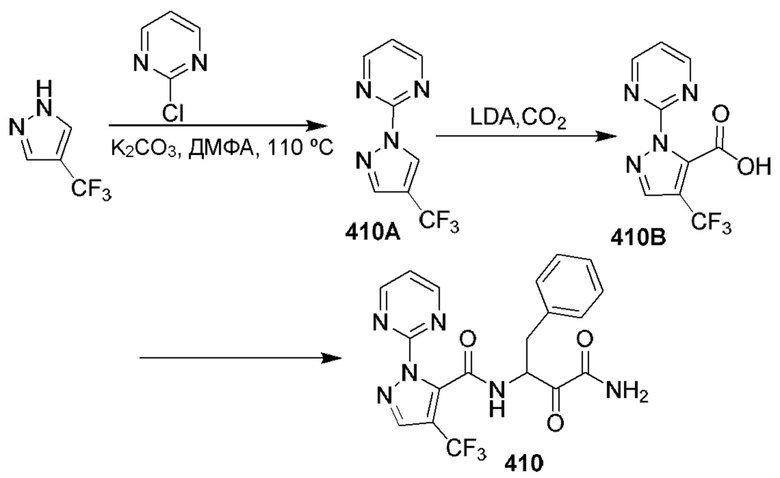
[1615] Смесь 4-(трифторметил)-1H-пиразольного соединения (1 г, 7,35 ммоль), 2-хлорпиримидина (926 мг, 8,09 ммоль) и K2CO3 (2,03 г, 14,7 ммоль) в ДМФА (15 мл) нагревали до 110°С в течение 12 ч. В смесь добавляли воду (20 мл) и проводили экстракцию этилацетатом (20 мл × 2), органическую фазу промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток перекристаллизовывали посредством МТБЭ с получением соединения 410А (800 мг, выход: 50,8%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,94 (s, 1H), 8,83 (d, J=4,8 Гц, 2Н), 8,03 (s, 1H), 7,34 (t, J=4,8 Гц, 1H).
[1616] К раствору соединения 410А (350 мг, 1,63 ммоль) в ТГФ (10 мл) по каплям добавляли LDA (2 М, 1,06 мл) при -78°С и перемешивали в течение 10 мин, затем смесь продували CO2 в течение 20 мин при -78°С, после чего медленно нагревали до 15°С в течение 1 ч. В смесь добавляли воду (20 мл) и проводили экстракцию этилацетатом (10 мл × 2), рН водного слоя доводили до ~ 3 с помощью 1 н. HCl и проводили экстракцию этилацетатом (10 мл × 2), органические фазы сушили и концентрировали с получением соединения 410В (140 мг, выход: 33,3%), в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,79 (d, J=4,9 Гц, 2Н), 7,93 (s, 1H), 7,34 (t, J=4,7 Гц, 1H).
[1617] Соединения 410 синтезировали из соответствующих исходных промежуточных соединений 274D и 410В с использованием тех же методик, которые были описаны ранее, например, для 305. Соединение 410 (3,3 г): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(пиримидин-2-ил)-4-(трифторметил)-1H-пиразол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,68 (d, J=4,6 Гц, 2Н), 7,94 (s, 1H), 7,32-7,26 (m, 2Н), 7,24-7,14 (m, 4H), 6,81 (ушир. d, J=7,1 Гц, 1H), 6,71 (ушир. s, 1H), 5,85 (q, J=6,4 Гц, 1H), 5,51 (ушир. s, 1H), 3,49-3,40 (m, 1H), 3,39-3,29 (m, 1H). МС (ИЭР) m/z (M+H)+ 433,1.
ПРИМЕР 219
СОЕДИНЕНИЕ 411
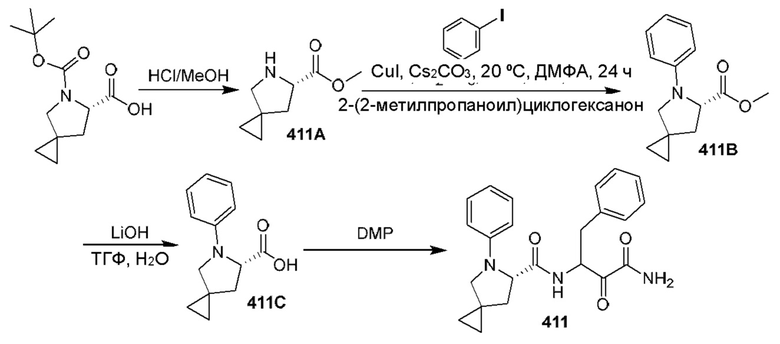
[1618] Смесь (S)-5-(трет-бутоксикарбонил)-5-азаспиро[2.4]гептан-6-карбоновой кислоты (2 г, 8,29 ммоль) в МеОН (5 мл), HCl/MeOH (50 мл) перемешивали при 1°С в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Соединение 411А (1,3 г, неочищенного) получали в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 11,09 (ушир. s, 1H), 9,14 (ушир. s, 1H), 4,64 (ушир. s, 1H), 3,85 (s, 3Н), 3,55-3,33 (m, 3Н), 2,87-2,64 (m, 2Н), 2,38 (ушир. dd, J=8,2, 13,0 Гц, 1H), 2,10 (ушир. dd, J=5,3, 13,0 Гц, 1H), 0,87-0,71 (m, 3Н), 0,71-0,61 (m, 1H).
[1619] Смесь соединения 411А (1 г, 6,44 ммоль), иодбензола (5,26 г, 25,8 ммоль), Cs2CO3 (6,30 г, 19,3 ммоль), CuI (981,76 мг, 5,15 ммоль) в ДМФА (40 мл) дегазировали и продували N2 3 раза, и затем полученную смесь перемешивали при 110°С в течение 12 часов в атмосфере N2. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = 95:1 до 90:1). и затем остаток очищали посредством препаративной ВЭЖХ (в условиях ТФУ). Соединение 411В (120 мг, выход: 8,06%) получали в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,32-7,12 (m, 2Н), 6,72 (t, J=7,3 Гц, 1H), 6,50 (d, J=7,9 Гц, 2Н), 4,40 (dd, J=2,4, 8,6 Гц, 1H), 3,80-3,62 (m, 3Н), 3,48 (d, J=8,6 Гц, 1H), 3,29 (d, J=8,6 Гц, 1H), 2,50 (dd, J=8,8, 12,6 Гц, 1H), 1,87 (dd, J=2,4, 12,6 Гц, 1H), 0,79-0,51 (m, 4H).
[1620] Соединения 411 синтезировали из соответствующих исходных промежуточных соединений 274D и 411В с использованием тех же методик, которые были описаны ранее, например, для 321. Соединение 411 (53,2 мг, выход: 50,5%): (6S)-N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-фенил-5-азаспиро[2.4]гептан-6-карбоксамид: 1H ЯМР (400 МГц, CDCl3) δ 7,16-7,06 (m, 4H), 7,04-6,87 (m, 3H), 6,77-6,66 (m, 2H), 6,60 (ушир. s, 1H), 6,54-6,38 (m, 2H), 5,58-5,21 (m, 2H), 4,05-3,93 (m, 1H), 3,39-3,22 (m, 1H), 3,09-2,90 (m, 2H), 2,82-2,69 (m, 1H), 2,45-2,30 (m, 1H), 1,68-1,53 (m, 1H), 0,56-0,34 (m, 3H), 0,17-0,05 (m, 1H). MC (ИЭР) m/z (M+H)+ 392,2.
ПРИМЕР 220
2-(1,1-ДИОКСИДО-1,2-ТИАЗИНАН-2-ИЛ)-3-МЕТИЛ-N-(4-МЕТИЛ-1-ОКСОПЕНТАН-2-ИЛ)БУТАНАМИД (412)
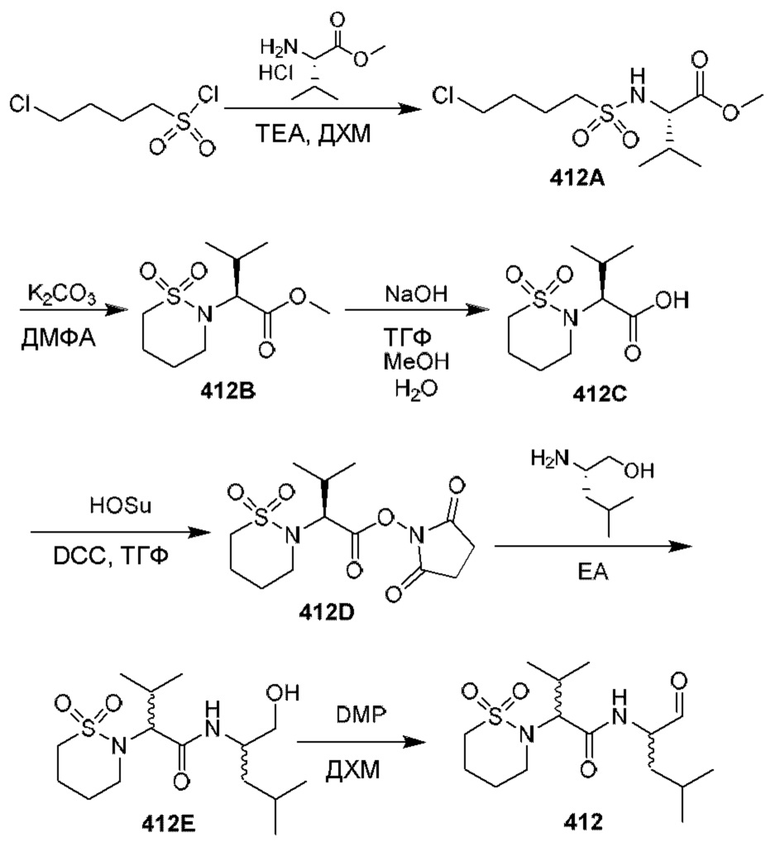
[1621] К смеси метил-L-валината (2,63 г, 15,7 ммоль, HCl) в ДХМ (50 мл) одной порцией добавляли TEA (4,5 мл, 32,3 ммоль). По каплям добавляли 4-хлорбутан-1-сульфонилхлорид (2,5 г, 13,1 ммоль) при 0°С, после чего смесь перемешивали в течение 30 мин при 0°С, затем перемешивали при 15°С в течение 1,5 ч. Реакционную смесь промывали 0,5 н. HCl (20 мл), насыщ. NaHCO3 (20 мл) и sat. NaCl (20 мл). Отделенный органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 412А (2,26 г, выход 60,4%) в виде светло-желтого вязкого масла, которое непосредственно использовали на следующей стадии без очистки. МС (ИЭР) m/z (M+H)+ 286,1.
[1622] К смеси соединения 412А (3,16 г, 11,1 ммоль) в ДМФА (150 мл) одной порцией добавляли K2CO3 (3,82 г, 27,6 ммоль). Смесь перемешивали при 15°С в течение 18 ч. К смеси добавляли H2O (200 мл) и проводили экстракцию посредством ЭА (150 мл × 3). Объединенную органическую фазу промывали нас. NaCl (200 мл × 2) и сушили над Na2SO4. Растворитель выпаривали при пониженном давлении с получением соединения 412В (2,7 г, неочищенного) в виде светло-желтой жидкости, которую непосредственно использовали на следующей стадии без очистки. 1H ЯМР (ДМСО-d6, 400 МГц): δ 3,94 (d, J=10,6 Гц, 1Н), 3,69-3,64 (m, 3H), 3,40-3,34 (m, 0,72H), 3,33-3,30 (m, 0,24H), 3,29-3,20 (m, 1H), 3,16-3,07 (m, 1Н), 3,06-2,97 (m, 1Н), 2,11-1,95 (m, 3H), 1,61-1,47 (m, 2H), 0,87 (dd, J=6,7, 14,0 Гц, 6Н). МС (ИЭР) m/z (M+H)+ 250,1.
[1623] К смеси соединения 412В (1 г, 4,0 ммоль) в ТГФ (10 мл) и МеОН (10 мл) одной порцией добавляли раствор NaOH (802,1 мг, 20,0 ммоль) в H2O (2 мл). Смесь перемешивали при 55°С в течение 1 ч. К реакционной смеси добавляли H2O (15 мл) и затем проводили концентрированно при пониженном давлении для удаления МеОН. Водную фазу промывали МТБЭ (10 мл). Отделенную водную фазу подкисляли с помощью водного раствора HCl (1 M) до рН ~ 5-6, после чего проводили экстракцию посредством ЭА (15 мл × 5). Затем органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом с получением соединения 412С (88,6 мг, неочищенного) в виде желтого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. 1H ЯМР (ДМСО-d6, 400 МГц): δ 12,83 (s, 1Н), 3,82 (d, J=10,4 Гц, 1Н), 3,29-3,23 (m, 2H), 3,14-2,90 (m, 2H), 2,10-1,89 (m, 3H), 1,65-1,44 (m, 2H), 0,85 (dd, J=4,2, 6,6 Гц, 6Н).
[1624] К смеси соединения 412С (88,5 мг, 3,7 ммоль) в ТГФ (20 мл) добавляли 1-гидроксипирролидин-2,5-дион (HOSu) (432,9 мг, 3,8 ммоль), а затем DCC (776,0 мг, 3,7 ммоль) одной порцией при 0°С. Смесь перемешивали при 15°С в течение 12 ч. Нерастворимое вещество удаляли посредством фильтрования. Фильтрат концентрировали под вакуумом. Остаток растирали с изопропанолом (10 мл). Твердое вещество собирали и сушили под вакуумом с получением соединения 412D (95,8 мг, выход 76,6%) в виде бесцветной жидкости. 1H ЯМР (ДМСО-d6, 400 МГц): δ 4,26 (d, J=11,0 Гц, 1Н), 3,45-3,37 (m, 2H), 3,28-3,22 (m, 1Н), 3,15-3,05 (m, 1Н), 2,81 (s, 4H), 2,19-2,09 (m, 1Н), 2,08-2,02 (m, 2H), 1,58-1,45 (m, 2H), 0,93 (dd, J=6,6, 18,7 Гц, 6Н).
[1625] К смеси соединения 412D (950 мг, 2,9 ммоль) в ЭА (25 мл) добавляли (S)-2-амино-4-метилпентан-1-ол (40,2 мг, 3,4 ммоль) одной порцией. Смесь перемешивали при 15°С в течение 12 ч. Смесь промывали водн. HCl (0.5 N, 10 мл), насыщ. NaHCO3 (10 мл), насыщ. NaCl (10 мл). Отделенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством препаративной ВЭЖХ (в основных условиях) с получением соединения 412Е (220 мг, выход 23,0%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц): δ 7,78 (d, J=8,8 Гц, 1Н), 4,57 (t, J=5,5 Гц, 1Н), 3,85-3,70 (m, 2H), 3,55-3,44 (m, 1Н), 3,41-3,37 (m, 0,52H), 3,32-3,25 (m, 1,45H), 3,20-3,06 (m, 2H), 2,88-2,75 (m, 1Н), 2,08-1,88 (m, 3H), 1,73-1,61 (m, 1Н), 1,60-1,41 (m, 2H), 1,38-1,21 (m, 2H), 0,84 (td, J=6,6, 17,4 Гц, 12Н).
[1626] К смеси соединения 412Е (0,1 г, 299 мкмоль) в ДХМ (15 мл) добавляли DMP (380,4 мг, 896,93 мкмоль) одной порцией. Смесь перемешивали при 15°С в течение 1,5 ч. В реакционную смесь добавляли для гашения реакции 30 мл 10% раствор Na2S2O3 и 30 мл насыщ. NaHCO3 раствор и перемешивали в течение 10 мин. После гашения реакции реакционную смесь вливали в разделительную воронку и разделяли. Отделенную водную фазу подвергали экстракции ДХМ (20 мл × 3). Объединенную органическую фазу промывали солевым раствором (30 мл × 2), сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Остаток растирали со смесью CH3CN : изопропиловый эфир (1:8, 2 мл). Твердое вещество собирали и сушили под вакуумом с получением соединения 412 (45 мг, выход 45,1%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц): δ 9,39 (s, 1Н), 8,64 (d, J=6,5 Гц, 1Н), 4,06 (s, 1Н), 3,88 (d, J=10,8 Гц, 1Н), 3,50-3,37 (m, 2H), 3,19-3,06 (m, 1Н), 2,96-2,83 (m, 1Н), 2,03 (s, 3H), 1,77-1,38 (m, 5H), 0,97-0,79 (m, 12Н). МС (ИЭР) m/z (M+H)+ 333,1.
ПРИМЕР 221
СОЕДИНЕНИЯ 413-414, 525-529
[1627] Соединения 413-414 синтезировали из соответствующих исходных промежуточных соединений 274D и 250А с использованием тех же методик, которые были описаны ранее, например, для 250.
[1628] Соединение 413 (55 мг, выход: 53,4%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-(6-цианопиридин-3-ил)-1-циклопропил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,04-8,86 (m, 1H), 8,36-8,06 (m, 3Н), 7,95-7,86 (m, 1H), 7,76 (s, 1H), 7,62-7,43 (m, 1H), 7,35-7,04 (m, 5H), 5,40-5,24 (m, 1H), 3,94-3,73 (m, 1H), 3,26-3,18 (m, 1H), 2,95-2,86 (m, 1H), 1,19-1,02 (m, 4H). МС (ИЭР) m/z (М+H)+ 429,1.
[1629] Соединение 414 (42 мг, выход: 26,9%): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-циклопропил-3-(6-(трифторметил)пиридин-3-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,98 (s, 1H), 8,34-8,11 (m, 3Н), 7,82 (d, J=8,0 Гц, 1H), 7,78-7,66 (m, 1H), 7,54 (s, 1H), 7,34-7,06 (m, 5H), 5,39-5,24 (m, 1H), 3,94-3,57 (m, 1H), 3,28-3,16 (m, 1H), 2,97-2,84 (m, 1H), 1,18-1,04 (m, 4H). МС (ИЭР) m/z (М+H)+ 472,1.
[1630] Соединения 525-526 синтезировали из соответствующего исходного промежуточного соединения 250А и соответствующего алкилирующего агента с последующим прохождением полученным промежуточным соединением стадий согласно методикам, описанным для соединения 12, с получением конечного соединения.
[1631] Соединение 525 (13 мг, выход: 5,84%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-циклопентил-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,33 (d, J=7,3 Гц, 1H), 8,09 (s, 1H), 8,05 (s, 1H), 7,79 (s, 1H), 7,59-7,54 (m, 2H), 7,33-7,18 (m, 8H), 5,27 (ddd, J=4,0, 7,4, 9,8 Гц, 1H), 4,73 (quin, J=6,8 Гц, 1H), 3,16 (dd, J=4,1, 14,0 Гц, 1H), 2,82 (dd, J=9,9, 13,9 Гц, 1H), 2,16-2,06 (m, 2H), 1,94 (td, J=6,1, 12,2 Гц, 2H), 1,85-1,77 (m, 2H), 1,72-1,60 (m, 2H). МС (ИЭР) m/z (M+H)+ 431,2.
[1632] Соединение 526 (69,7 мг, выход: 22,7%; бледно-желтое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-циклобутил-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,35 (d, J=7,3 Гц, 1H), 8,15 (s, 1H), 8,06 (s, 1H), 7,80 (s, 1H), 7,60-7,54 (m, 2H), 7,33-7,17 (m, 8H), 5,29 (ddd, J=4,0, 7,4, 9,8 Гц, 1H), 4,92-4,84 (m, 1H), 3,16 (dd, J=4,0, 13,9 Гц, 1H), 2,82 (dd, J=9,7,13,9 Гц, 1H), 2,48-2,30 (m, 4H), 1,86-1,75 (m, 2H). МС (ИЭР) m/z (M+Na)+ 441,1.
[1633] Соединения 527-529 синтезировали из соответствующего исходного промежуточного соединения 250А и соответствующего агента на основе бороновых кислот, как в случае соединения 250 с последующим прохождением полученными промежуточными соединениями стадий согласно методикам, описанным для соединения 12, с получением конечного соединения.
[1634] Соединение 527 (69,9 мг, выход: 23,4%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(2-фторфенил)-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,70 (d, J=7,5 Гц, 1Н), 8,53 (d, J=2,2 Гц, 1Н), 8,09 (s, 1Н), 7,91 (dt, J=1,7, 8,0 Гц, 1Н), 7,83 (s, 1Н), 7,68-7,63 (m, 2H), 7,58-7,46 (m, 2H), 7,43-7,33 (m, 4H), 7,31-7,27 (m, 4H), 7,25-7,20 (m, 1Н), 5,33 (ddd, J=4,2, 7,4, 10,0 Гц, 1Н), 3,20 (dd, J=4,0, 13,9 Гц, 1Н), 2,85 (dd, J=10,1, 13,9 Гц, 1Н). МС (ИЭР) m/z (M+H)+ 457,1.
[1635] Соединение 528 (50 мг, выход: 16,2%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(3-фторфенил)-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,87 (s, 1Н), 8,59 (d, J=7,3 Гц, 1Н), 8,10 (s, 1Н), 7,84 (s, 1Н), 7,80-7,72 (m, 2H), 7,70-7,57 (m, 3Н), 7,40-7,20 (m, 9H), 5,49-5,31 (m, 1Н), 3,24-3,16 (m, 1Н), 2,91-2,82 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 457,2.
[1636] Соединение 529 (17,5 мг, выход: 10%; белое твердое вещество): N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-(4-фторфенил)-3-фенил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,76 (s, 1Н), 8,54 (d, J=7,3 Гц, 1Н), 8,09 (s, 1Н), 7,95-7,81 (m, 3Н), 7,67 (dd, J=3,0, 6,5 Гц, 2H), 7,46-7,21 (m, 10Н), 5,43-5,31 (m, 1Н), 3,20 (dd, J=4,0, 14,1 Гц, 1Н), 2,90-2,82 (m, 1Н). МС (ИЭР) m/z (М+Н)+ 457,2.
ПРИМЕР 222
СОЕДИНЕНИЯ 425-427
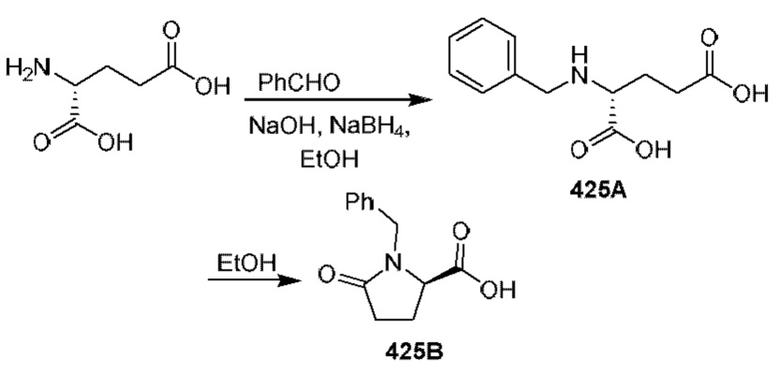
[1637] -D-глутаминовую кислоту (15,0 г, 101,9 ммоль) растворяли в водном NaOH (2 M, 100 мл) и перемешивали в течение 15 минут. К смеси добавляли раствор бензальдегида (11 мл, 108,84 ммоль) в EtOH (30 мл) и перемешивали при 15°С в течение 30 минут. Смесь охлаждали до 0°С. К полученной смеси добавляли NaBH4 (1.16 г, 30.6 ммоль), который оставляли нагреваться до 15°С при перемешивании в течение более 3 ч. Смесь промывали МТБЭ (30 мл × 2), после чего подкисляли концентрированной соляной кислотой до рН ~ 4-5. Полученный осадок отфильтровывали и сушили над с получением соединения 425А (10,26 г, неочищенного) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. МС (ИЭР) m/z (M+H)+ 238,0.
[1638] Суспензию соединения 425А (4,2 г, 17,7 ммоль) в EtOH (400 мл) нагревали до кипения 95°С в течение 10 часов. Реакционную смесь концентрировали при пониженном давлении для удаления EtOH. Остаток очищали посредством препаративной ВЭЖХ (условия ТФУ: колонка Phenomenex Synergi Max-RP 250 × 50 мм × 10 мкм; подвижная фаза: [вода (0,1%ТФУ)-ACN]; В%: 2% - 30%, 20 мин) с получением соединения 3 (2,7 г, выход 69,44%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц): δ 7,36-7,24 (m, 3Н), 7,22-7,16 (m, 2H), 4,88 (d, J=15,2 Гц, 1Н), 3,96-3,81 (m, 2H), 2,41-2,21 (m, 3Н), 2,01-1,89 (m, 1Н). МС (ИЭР) m/z (M+H)+ 219,9.
[1639] Соединение 425 синтезировали из соответствующего исходного промежуточного соединения 3-амино-N-циклопропил-2-гидрокси-4-фенилбутанамида гидрохлорида и 425В с использованием тех же методик, которые были описаны ранее для соединения 65, с последующим разделением с помощью SFC с получением соединения 426 и 427
[1640] Соединение 425: (2R)-1-бензил-N-(4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)-5-оксопирролидин-2-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,83 (s, 1Н), 8,66 (s, 1Н), 7,38-7,18 (m, 8H), 7,17-6,97 (m, 2H), 5,18 (s, 1Н), 4,90-4,67 (m, 1Н), 3,87 (s, 1Н), 3,56-3,41 (m, 1Н), 3,26-3,13 (m, 1Н), 2,85-2,65 (m, 2H), 2,30-1,97 (m, 3Н), 1,77-1,45 (m, 1Н), 0,73-0,52 (m, 4H). МС (ИЭР) m/z (M+H)+ 434,2.
[1641] Соединение 426 (129 мг, выход: 49,5%): (2R)-1-бензил-N-(4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)-5-оксопирролидин-2-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,85 (d, J=5,1 Гц, 1Н), 8,68 (d, J=7,5 Гц, 1Н), 7,36-7,18 (m, 8H), 7,13 (d, J=7,1 Гц, 2H), 5,24-5,11 (m, 1Н), 4,84 (d, J=15,0 Гц, 1Н), 3,92-3,83 (m, 1Н), 3,49 (d, J=15,0 Гц, 1Н), 3,18 (dd, J=3,5, 13,9 Гц, 1Н), 2,84-2,69 (m, 2H), 2,22 (t, J=7,9 Гц, 2H), 2,12-1,97 (m, 1Н), 1,59-1,47 (m, 1Н), 0,73-0,56 (m, 4H). МС (ИЭР) m/z (M+H)+ 434,2.
[1642] Соединение 427 (63,2 мг, выход: 25,2%): (2R)-1-бензил-N-(4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)-5-оксопирролидин-2-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,83 (d, J=5,1 Гц, 1Н), 8,67 (d, J=7,5 Гц, 1Н), 7,34-7,21 (m, 8H), 7,02 (d, J=6,6 Гц, 2Н), 5,27-5,16 (m, 1Н), 4,75 (d, J=15,0 Гц, 1Н), 3,86 (dd, J=3,3, 8,8 Гц, 1Н), 3,33 (s, 1Н), 3,19 (dd, J=4,0, 14,1 Гц, 1Н), 2,83-2,69 (m, 2Н), 2,32-2,06 (m, 3Н), 1,77-1,61 (m, 1Н), 0,73-0,50 (m, 4H). MC (ИЭР) m/z (M+H)+ 434,2.
ПРИМЕР 223
СОЕДИНЕНИЕ 430
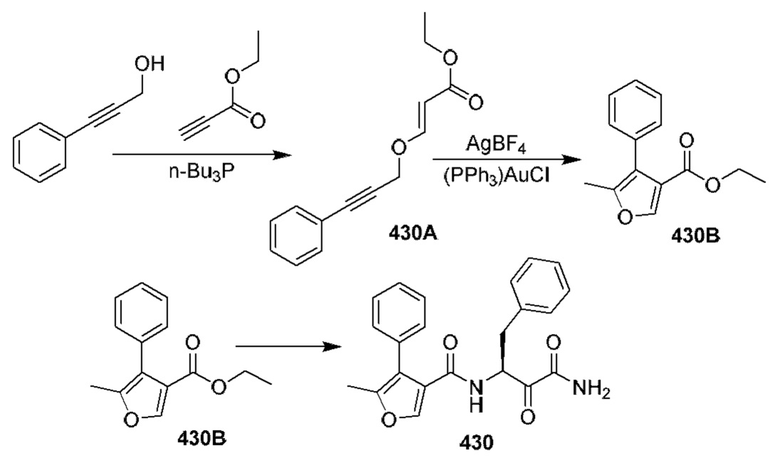
[1643] К раствору 3-фенилпроп-2-ин-1-ола (2 г, 15,13 ммоль) и этилпропиолата (1,48 г, 15,13 ммоль) в ДХМ (20 мл) по каплям добавляли n-Bu3P (307 мг, 1,51 ммоль). Смесь перемешивали при 25°С в течение 12 ч. Растворитель удаляли под вакуумом. Остаток очищали посредством колоночной хроматографии (ПЭ:ЭА=10:1) с получением соединения 425А (3,4 г, неочищенного) в виде светло-желтого масла. MC (ИЭР) m/z (M+H)+ 231,0.
[1644] К раствору соединения 425А (500 мг, 2,17 ммоль) в толуоле (10 мл) добавляли (PPh3)AuCl (22 мг, 43,40 мкмоль) и AgBF4 (9 мг, 43,40 мкмоль). Смесь перемешивали при 25°С в течение 12 ч. Растворитель удаляли под вакуумом. Остаток очищали посредством колоночной хроматографии (ПЭ:ЭА=10:1) с получением соединения 425В (120 мг, выход: 24,02%) в виде бесцветного масла. 1H ЯМР (CDCl3, 400 МГц): δ 7,95 (s, 1Н), 7,45-7,29 (m, 5Н), 4,20-4,14 (m, 2Н), 2,26 (s, 3Н), 1,22-1,17 (m, 3Н).
[1645] Соединение 430 синтезировали из промежуточного соединения 430В с использованием тех же методик, которые были описаны ранее для соединения 65 с получением соединения 430.
[1646] Соединение 430 (35 мг, выход: 33,25%) (S)-N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-метил-4-фенилфуран-3-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 7,82 (s, 1H), 7,38-7,14 (m, 11H), 7,04-6,94 (m, 2H), 6,35 (s, 1H), 6,11 (s, 1H), 4,43-4,30 (m, 1H), 3,04-2,85 (m, 1H), 2,61-2,53 (m, 1H), 2,21 (s, 3Н). МС (ИЭР) m/z (M+H)+ 377,1.
ПРИМЕР 224
СОЕДИНЕНИЕ 449
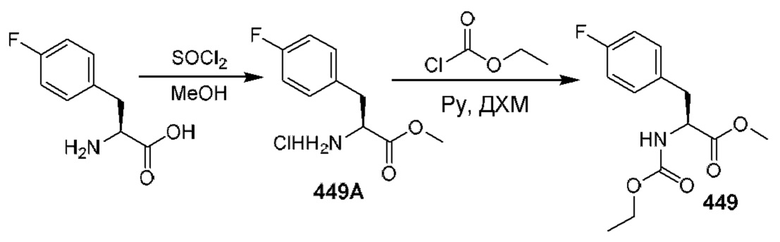
[1647] К смеси (S)-2-амино-3-(4-фторфенил)пропановой кислоты (1 г, 5,46 ммоль) в МеОН (10 мл) добавляли SOCl2 (2,60 г, 21,84 ммоль, 1,6 мл) порциями при 0°С в атмосфере N2. Смесь перемешивали при 60°С в течение 1,5 ч. Растворитель удаляли под вакуумом с получением соединения 449А (1,2 г, выход: 94,1%, HCl) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,59 (s, 3Н), 7,29 (t, J=5,9 Гц, 2H), 7,17 (t, J=8,2 Гц, 2H), 4,28 (s, 1H), 3,68 (s, 3Н), 3,19-3,07 (m, 2H). МС (ИЭР) m/z (M+H)+ 197,9.
[1648] К смеси соединения 449А (1,2 г, 5,46 ммоль, HCl) и этилхлорформиата (712 мг, 6,56 ммоль, 0,6 мл) в ДХМ (20 мл) добавляли пиридин (1,30 г, 16,39 ммоль, 1,3 мл) одной порцией при 0°С в атмосфере N2. Смесь перемешивали при 20°С в течение 2 ч. Реакционную смесь обрабатывали ДХМ (30 мл), промывали 0,5 н. HCl (50 мл × 2) и солевым раствором (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир : этилацетат = 10:1 до 4: 1) с получением соединения 449 (1,2 г, выход: 80,6%) в виде бесцветного масла. Метил-(S)-2-((этоксикарбонил)амино)-3-(4-фторфенил)пропаноат: 1H ЯМР (400 МГц, ДМСО-d6) δ 7,64 (d, J=8,0 Гц, 1H), 7,27 (dd, J=5,8, 8,3 Гц, 2H), 7,10 (t, J=8,8 Гц, 2H), 4,24-4,07 (m, 1H), 3,92 (dd, J=4,0, 7,0 Гц, 2H), 3,61 (s, 3Н), 3,00 (dd, J=5,0, 14,1 Гц, 1H), 2,83 (dd, J=10,5, 13,6 Гц, 1H), 1,14-0,97 (m, 3H). МС (ИЭР) m/z (M+H)+ 269,9.
ПРИМЕР 225
СОЕДИНЕНИЕ 450
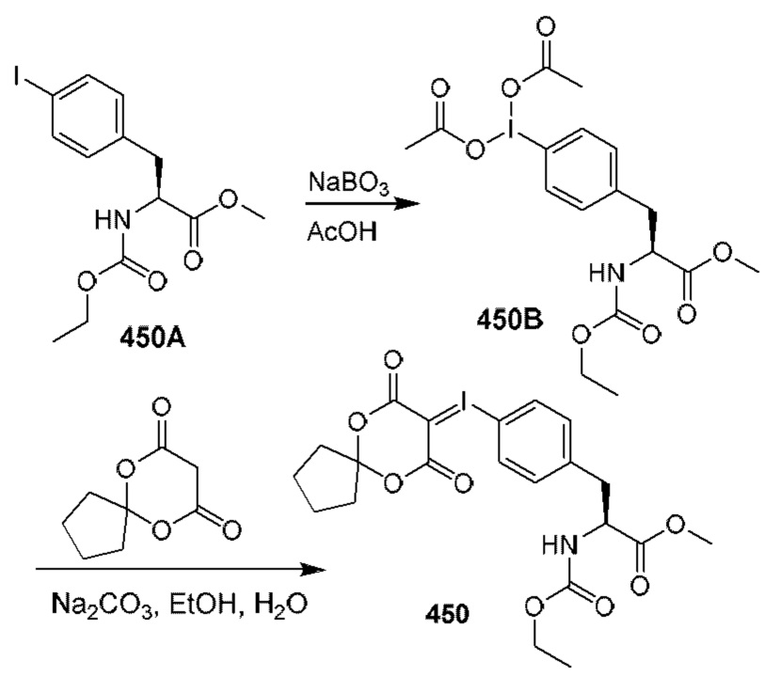
[1649] Соединение 450А получали из (S)-2-амино-3-(4-иодфенил)пропановой кислоты с помощью методик, описанных для соединения 449.
[1650] К раствору соединения 450А (400 мг, 1,06 ммоль) в АсОН (8,5 мл) порциями добавляли NaBO3 (1,63 г, 10,61 ммоль) и нагревали до 50°С. Реакционную смесь перемешивали при 50°С в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли ДХМ (20 мл), фильтровали, фильтрат разбавляли водой (30 мл), и подвергали экстракции ДХМ (10 мл × 2). Объединенные органические экстракты сушили с помощью безводного сульфата натрия, фильтровали и концентрировали. Остаток растирали в ДХМ: ПЭ = 1:10 (10 мл × 2), обеспечивая осаждение, и полученные твердые вещества собирали. Соединение 450В (380 мг, выход: 72,4%) получали в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,99 (d, J=8,4 Гц, 2H), 7,29-7,23 (m, 2H), 5,20 (d, J=7,7 Гц, 1Н), 4,72-4,64 (m, 1H), 4,11 (q, J=6,9 Гц, 2H), 3,76-3,72 (m, 3H), 3,25-3,08 (m, 2H), 2,00 (s, 6H), 1,23 (t, J=7,1 Гц, 3Н).
[1651] К раствору соединения 450В (380 мг, 767,27 мкмоль) в Na2CO3 (243,97 мг, 2,30 ммоль) в H2O (3 мл) добавляли EtOH (3 мл), после чего быстро добавляли 6,10-диоксаспиро[4.5]декан-7,9-дион (130,56 мг, 767,27 мкмоль). Реакционную смесь интенсивно перемешивали при 18°С в течение 4 ч. Реакционную смесь затем разбавляли водой (10 мл), и подвергали экстракции ДХМ (10 мл × 3). Объединенные органические экстракты сушили с помощью безводном Na2SO4, фильтровали и концентрировали. К остатку добавляли ДХМ (1 мл) и ПЭ (15 мл) для обеспечения осаждения, твердые вещества собирали. Соединение 450 (300 мг, выход: 71,7%) получали в виде белого твердого вещества. Метил-(S)-3-(4-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-2-((этоксикарбонил)амино)пропаноат 1H ЯМР (400 МГц, CDCl3) δ 7,78 (d, J=8,4 Гц, 2H), 7,18 (d, J=8,4 Гц, 2H), 5,18 (d, J=7,7 Гц, 1H), 4,67-4,55 (m, 1H), 4,12-4,02 (m, 2H), 3,71 (s, 3H), 3,26-3,13 (m, 1H), 3,11-2,99 (m, 1H), 2,13 (t, J=7,4 Гц, 4Н), 1,84-1,74 (m, 4Н), 1,21 (t, J=7,1 Гц, 3H). МС (ИЭР) m/z (М-H)+ 544,0.
ПРИМЕР 226
СОЕДИНЕНИЯ 451-453, 533-540
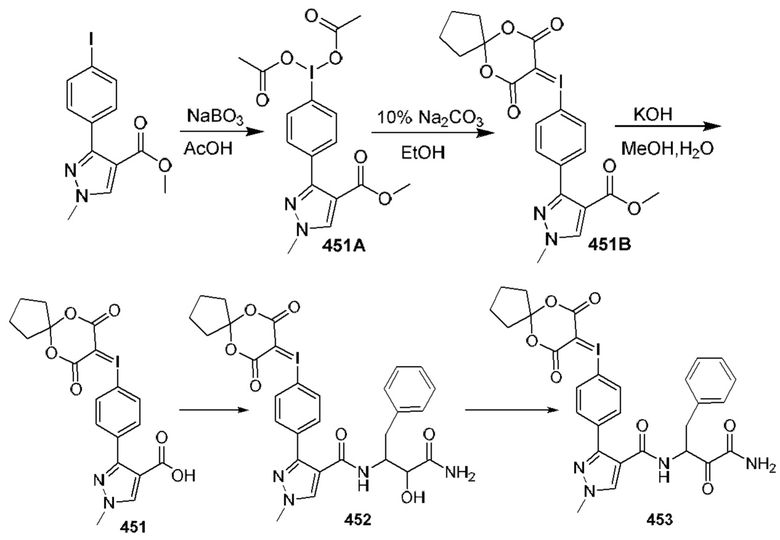
[1652] К раствору метил-3-(4-иодфенил)-1-метил-1H-пиразол-4-карбоксилата (2 г, 5,85 ммоль) в АсОН (40 мл) порциями добавляли натрия перборат тетрагидрат (8,99 г, 58,46 ммоль) и нагревали до 50°С. Реакционную смесь перемешивали при 50°С в течение 8 ч, охлаждали до комнатной температуры, разбавляли ДХМ (50 мл), фильтровали, фильтрат разбавляли водой (100 мл), и подвергали экстракции метиленхлорид (30 мл × 2). Объединенные органические экстракты сушили с помощью безводного сульфата натрия, фильтровали и концентрировали. Остаток растирали в ДХМ: ПЭ (1: 20) (100 мл), фильтровали с получением соединения 451А (2,1 г, выход: 78,1%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,30-8,01 (m, 2H), 7,98-7,84 (m, 3H), 4,10-3,90 (m, 3H), 3,86-3,70 (m, 3H), 2,17-1,83 (m, 6H).
[1653] К раствору соединения 451А (2,1 г, 4,56 ммоль) в EtOH (60 мл) добавляли Na2CO3 (1,93 г, 18,25 ммоль) в H2O (30 мл) и 6,10-диоксаспиро[4.5]декан-7,9-дион (932 мг, 5,48 ммоль). Смесь перемешивали при 25°С в течение 4 ч. Реакционную смесь затем разбавляли водой (50 мл) и подвергали экстракции ДХМ (20 мл × 3). Объединенные органические экстракты сушили с помощью безводного Na2SO4, фильтровали, и концентрировали. Остаток добавляли ДХМ (5 мл) и ПЭ (50 мл) для обеспечения осаждения. Твердые вещества собирали с получением соединения 451В (1,6 г, выход: 68,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,95 (s, 1H), 7,90-7,80 (m, 4H), 3,94 (s, 3H), 3,79-3,72 (m, 3H), 2,14 (t, J=7,5 Гц, 4Н), 1,81-1,75 (m, 4H).
[1654] Соединения 451-453 синтезировали из промежуточного соединения 451В с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 451-453.
[1655] Соединение 451 (220 мг, выход: 58,1%) 3-(4-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-1-метил-1H-пиразол-4-карбоновая кислота: 1H ЯМР (400 МГц, ДМСО-d6) δ 12,31 (ушир. s, 1H), 8,35-8,28 (m, 1H), 7,82-7,73 (m, 4H), 3,88 (s, 3H), 2,02-1,93 (m, 4H), 1,70-1,62 (m, 4H). МС (ИЭР) m/z (M-H)+ 495,01.
[1656] Соединение 452 (250 мг, выход: 35,1%) N-(4-амино-3-гидрокси-4-оксо-1-фенилбутан-2-ил)-3-(4-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,13-7,98 (m, 1H), 7,93-7,78 (m, 1H), 7,72-7,62 (m, 2H), 7,59-7,46 (m, 2H), 7,33 (d, J=7,8 Гц, 2Н), 7,28-7,17 (m, 6Н), 5,87-5,73 (m, 1H), 4,49 (d, J=9,5 Гц, 1H), 4,02 (ушир. s, 1H), 3,89 (s, 3H), 2,84-2,65 (m, 2H), 2,01 (ушир. s, 4H), 1,70 (ушир. s, 4H). МС (ИЭР) m/z (M+H)+ 673,11.
[1657] Соединение 453 (32 мг, выход: 64,2%) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-(4-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,52 (d, J=7,5 Гц, 1H), 8,11-8,02 (m, 2H), 7,81-7,76 (m, 1H), 7,72-7,65 (m, 2H), 7,56 (d, J=8,6 Гц, 2H), 7,29-7,24 (m, 4H), 7,21 (dd, J=4,5, 8,7 Гц, 1H), 5,34-5,19 (m, 1H), 3,91-3,86 (m, 3H), 3,18-3,14 (m, 1H), 2,79 (dd, J=10,3, 13,8 Гц, 1H), 1,97 (t, J=7,3 Гц, 4H), 1,68-1,64 (m, 4H). МС (ИЭР) m/z (М+Н)+ 671,09.
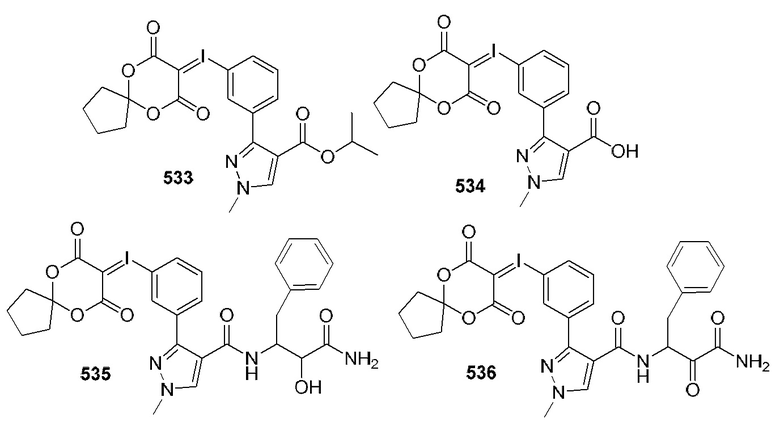
[1658] Соединения 533-536 синтезировали из соответствующего промежуточного соединения, этил-3-(3-иодфенил)-1-метил-1H-пиразол-4-карбоксилата, с использованием тех же методик, которые были описаны ранее для соединения 451В и 451-453 с получением соединения 533-536.
[1659] Соединение 533 (1,3 г, выход: 67,8%; желтое твердое вещество) Этил-3-(3-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-1-метил-1H-пиразол-4-карбоксилат: 1H ЯМР (400 МГц, CDCl3) δ 8,35 (s, 1H), 8,07 (d, J=8,2 Гц, 1Н), 7.94 (s, 1H), 7,85 (dd, J=0,9, 8,2 Гц, 1H). 7,43 (t, J=7,9 Гц, 1H), 4,24 (q, J=7,1 Гц. 2Н), 3,94 (s, 3Н), 2,14 (t, J=7,5 Гц, 4Н), 1,83-1,70 (m, 4Н), 1,28 (t, J=7,2 Гц, 3Н).
[1660] Соединение 534 (78 мг; белое твердое вещество) 3-(3-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-1-метил-1H-пиразол-4-карбоновая кислота: 1H ЯМР (400 МГц, ДМСО-d6) δ 12,46-12,28 (m, 1H), 8,31 (s, 1H), 8,11 (t, J=1,7 Гц, 1H), 7,94 (d, J=8,2 Гц, 1H), 7,75 (d, J=8,8 Гц, 1H), 7,46 (t, J=7,9 Гц, 1H), 3,88 (s, 3Н), 2,01-1,94 (m, 4Н), 1,69-1,63 (m, 4Н), MC (ИЭР) m/z (M-H)+ 495,0.
[1661] Соединение 535 (250 мг, выход: 48%; белое твердое вещество) N-(4-амино-3-гидрокси-4-оксо-1-фенилбутан-2-ил)-3-(3-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,14-8,00 (m, 2Н), 7,87 (d, J=8,8 Гц, 0,5Н), 7,66 (d, J=7,9 Гц, 1H), 7,60-7,55 (m, 1H), 7,53 (d, J=9,3 Гц, 0,5Н), 7,35-7,10 (m, 8H), 5,86-5,68 (m, 1H), 4,49-4,33 (m, 1H), 4,02-3,98 (m, 0,5H), 3,88-3,81 (m, 3,5H), 2,90-2,83 (m, 0,5H), 2,79-2,71 (m, 1H), 2,67-2,62 (m, 0,5H), 2,00-1,94 (m, 4Н), 1,69-1,62 (m, 4Н). MC (ИЭР) m/z (М+Н)+ 673,1.
[1662] Соединение 536 (200 мг, выход: 69,8%; бледно-желтое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-(3-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,49 (d, J=7,5 Гц, 1Н), 8,16-7,92 (m, 3H), 7,82-7,72 (m, 1Н), 7,71-7,54 (m, 2H), 7,33 (t, J=7,7 Гц, 1Н), 7,29-7,21 (m, 4H), 7,19 (d, J=3,7 Гц, 1Н), 5,24 (ушир. s, 1Н), 3,95-3,80 (m, 3H), 3,21-3,07 (m, 1Н), 2,80 (dd, J=10,0, 13,8 Гц, 1Н), 2,02-1,91 (m, 4H), 1,65 (ушир. s, 4H). MC (ИЭР) m/z (M+H)+ 671,1.
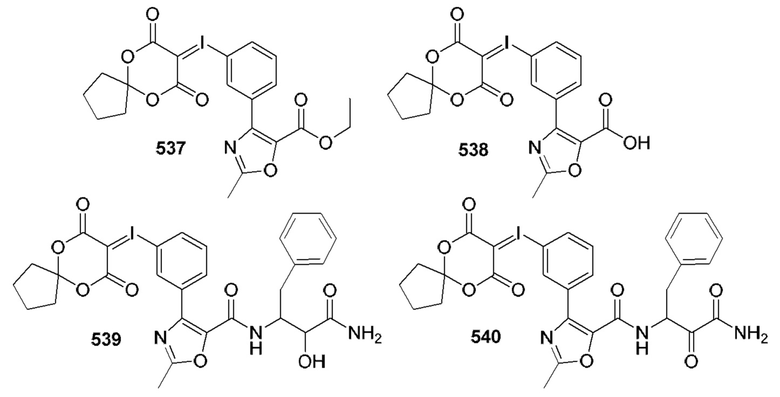
[1663] Соединения 537-540 синтезировали из соответствующего промежуточного соединения, этил-3-(3-иодфенил)-3-оксопропаноата, для превращения его в этил-4-(3-иодфенил)-2-метилоксазол-5-карбоксилата с использованием методик, описанных для соединения 248С, а затем этил-4-(3-иодфенил)-2-метилоксазол-5-карбоксилат превращали в соединения 537-540 с использованием тех же методик, которые были описаны ранее для соединения 451В и 451-453.
[1664] Соединение 537 (1,3 г, выход: 41,5%; белое твердое вещество) Этил 4-(3-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-2-метилоксазол-5-карбоксилат: 1H ЯМР (400 МГц, CDCl3) δ 8,64 (t, J=1,7 Гц, 1Н), 8,42-8,35 (m, 1Н), 7,98-7,80 (m, 1Н), 7,50 (t, J=7,9 Гц, 1Н), 4,42 (q, J=7,1 Гц, 2H), 2,59 (s, 3H), 2,21-2,14 (m, 4H), 1,83-1,76 (m, 4H), 1,41 (t, J=7,2 Гц, 3Н).
[1665] Соединение 538 (300 мг; выход: 63,1% белое твердое вещество) 4-(3-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-2-метилоксазол-5-карбоновая кислота: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,37 (t, J=1,7 Гц, 1H), 8,28-8,23 (m, 1H), 7,85-7,78 (m, 1H), 7,53 (t, J=7,9 Гц, 1H), 2,52 (s, 3H), 2,00-1,96 (m, 4H), 1,69-1,64 (m, 4H), MC (ИЭР) m/z (М-H)+ 495,99.
[1666] Соединение 539 (360 мг, выход: 68,7%; бледно-желтое твердое вещество) N-(4-амино-3-гидрокси-4-оксо-1-фенилбутан-2-ил)-4-(3-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-2-метилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,47-8,38 (m, 1H), 8,29 (d, J=8,2 Гц, 0,5Н), 8,22 (t, J=8,8 Гц, 1H), 7,83-7,68 (m, 1,5H), 7,44 (td, J=7,9, 13,0 Гц, 1H), 7,34 (d, J=9,3 Гц, 1H), 7,27-7,08 (m, 6H), 6,03 (d, J=6,0 Гц, 0,5Н), 5,87 (d, J=5,7 Гц, 0,5Н), 4,69-4,41 (m, 1H), 4,03 (t, J=4,6 Гц, 0,5Н), 3,90-3,83 (m, 0,5H), 2,98-2,85 (m, 1H), 2,82-2,71 (m, 1H), 2,52 (d, J=3,7 Гц, 3H), 2,01-1,96 (m, 4H), 1,69-1,64 (m, 4H). MC (ИЭР) m/z (М-H)+ 672,0.
[1667] Соединение 540 (150 мг, выход: 65,2%; белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(3-((7,9-диоксо-6,10-диоксаспиро[4.5]декан-8-илиден)-λ3-иоданил)фенил)-2-метилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,56 (ушир. s, 1H), 8,50-8,42 (m, 1H), 8,33-8,21 (m, 1H), 7,92-7,73 (m, 2H), 7,63 (ушир. s, 1H), 7,50-7,41 (m, 1H), 7,30-7,20 (m, 5H), 5,50-5,39 (m, 1H), 3,28 (dd, J=4,4, 14,2 Гц, 1H), 3,08-3,02 (m, 1H), 2,56-2,54 (m, 3H), 2,02 (t, J=7,4 Гц, 4H), 1,75-1,67 (m, 4H). MC (ИЭР) m/z (M+H)+ 672,0.
ПРИМЕР 227
СОЕДИНЕНИЕ 489
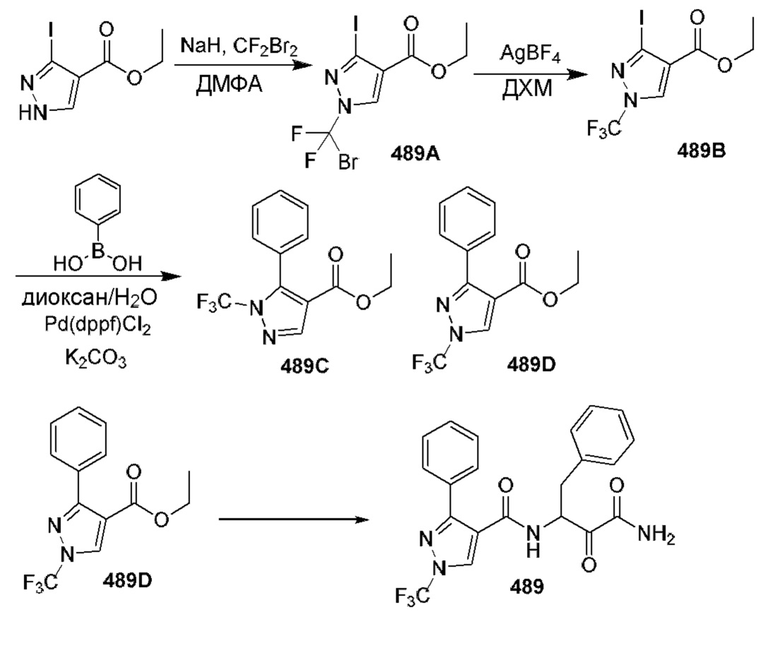
[1668] К раствору этил-3-иод-1H-пиразол-4-карбоксилата (4,3 г, 16,16 ммоль) в ДМФА (50 мл) добавляли NaH (1,29 г, 32,32 ммоль, чистота 60%) при 0°С, смесь перемешивали при 15°С в течение 30 мин, затем добавляли дибром(дифтор)метан (10,17 г, 48,48 ммоль, 4,5 мл) при 0°С. Смесь перемешивали при 15°С в течение 16 ч. Смесь гасили посредством NH4Cl (15 мл), разбавляли H2O (30 мл), подвергали экстракции ЭА (50 мл × 3), Органическую фазу объединяли, промывали NaCl (50 мл), сушили над Na2SO4, фильтровали и концентрировали с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир:этилацетат = 1:0 до 10:1) с получением соединения 489А (4,4 г, выход: 61,15%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 8,25-8,08 (m, 1Н), 4,42-4,31 (m, 2H), 1,39 (t, J=3,3, 7,2 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 394,9.
[1669] К смеси соединения 489А (2,6 г, 6,58 ммоль) в ДХМ (30 мл) добавляли AgBF4 (3,84 г, 19,74 ммоль) порциями при -78°С в атмосфере N2. Смесь перемешивали при 15°С в течение 16 ч. Реакционную смесь разбавляли ДХМ (50 мл), затем смесь промывали H2O (60 мл) и солевым раствором (60 мл), органическую фазу сушили над Na2SO4 и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир:этилацетат = 1:0 до 10:1) с получением соединения 489В (1,7 г, выход: 45,17%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 8,24-8,03 (m, 1Н), 4,44-4,31 (m, 2H), 1,46-1,34 (m, 3Н). МС (ИЭР) m/z (M+H)+ 334,9.
[1670] К смеси соединения 489В (200 мг, 598,75 мкмоль), фенилбороновой кислоты (110 мг, 898,13 мкмоль), K2CO3 (166 мг, 1,20 ммоль) в диоксане (10 мл) и H2O (0,5 мл) добавляли Pd(dppf)Cl2 (44 мг, 59,88 мкмоль) порциями при 15°С в атмосфере N2. Смесь перемешивали при 90°С в течение 16 ч. Реакционную смесь концентрировали. Остаток очищали посредством препаративной ТСХ (SiO2, ПЭ:ЭА=15:1) с получением соединения 489D (40 мг, выход: 9,31%) в виде желтого масла и соединение 489С (40 мг, выход: 20,97%) в виде желтого масла. Соединение 489D: 1H ЯМР (400 МГц, CDCl3) δ 8,43 (s, 1Н), 7,83-7,77 (m, 2H), 7,48-7,43 (m, 3Н), 4,30 (q, J=7,1 Гц, 2H), 1,31 (t, J=7,2 Гц, 4Н). МС (ИЭР) m/z (M+H)+ 285,0. Соединение 489С: 1H ЯМР (400 МГц, CDCl3) δ 8,16 (s, 1Н), 7,55-7,44 (m, 3Н), 7,38 (d, J=7,3 Гц, 2H), 4,15 (q, J=7,2 Гц, 2H), 1,14 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 285,0.
[1671] Соединение 489 синтезировали из промежуточного соединения 489D с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 489. Соединение 489 (55 мг, выход: 73,01%) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-фенил-1-(трифторметил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,93 (dd, J=7,3 Гц, 1Н), 8,76 (s, 1H), 8,11 (s, 1H), 7,85 (s, 1H), 7,54 (dd, J=7,1 Гц, 2H), 7,41-7,31 (m, 3H), 7,29-7,20 (m, 5H), 5,39-5,32 (m, 1H), 3,17 (dd, J=3,6, 14,0 Гц, 1H), 2,82 (dd, J=10,0, 14,0 Гц, 1H). МС (ИЭР) m/z (M+H)+ 431,1.
ПРИМЕР 228
СОЕДИНЕНИЕ 490
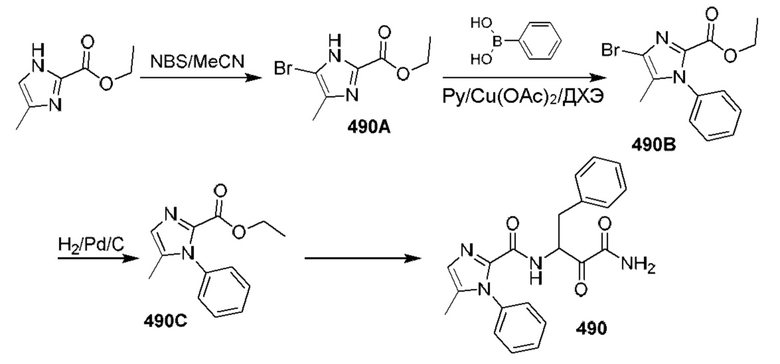
[1672] Раствор этил-4-метил-1H-имидазол-2-карбоксилата (800 мг, 5,19 ммоль) в MeCN (20 мл) добавляли NBS (970 мг, 5,45 ммоль). Реакционную смесь перемешивали при 20°С в течение 5 ч. Растворитель выпаривали под вакуумом. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 20:1 ~ 3:1) с получением соединения 490А (1,50 г, неочищенного) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 4,42 (q, J=7,2 Гц, 2H), 2,32 (s, 3H), 1,40 (t, J=7,1 Гц, 3H).
[1673] Смесь соединения 490А (1,5 г, 6,44 ммоль), фенилбороновой кислоты (1,57 г, 12,9 ммоль), Cu(ОАс)2 (2,34 г, 12,9 ммоль), пиридин (1,53 г, 19,3 ммоль) и МС 4А° in ДХЭ (20 мл) перемешивали при 70°С в атмосфере О2 в течение 12 ч. Смесь фильтровали и фильтрат концентрировали, остаток очищали посредством КФХ (петролейный эфир:этилацетат = 15:1) с получением соединения 490В (300 мг, выход: 15,1%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,57-7,47 (m, 3H), 7,23-7,17 (m, 2H), 4,24 (q, J=7,1 Гц, 2H), 2,02 (s, 3H), 1,25 (t, J=7,2 Гц, 3H).
[1674] К раствору соединения 490В (50 мг, 162 мкмоль) в EtOH (10 мл) добавляли Pd-C (0,1 г) в атмосфере N2. Суспензию дегазировали под вакуумом и несколько раз продували H2. Смесь перемешивали в H2 (15 фунтов на кв. дюйм) при 15°С в течение 12 часов. Смесь фильтровали и фильтрат концентрировали с получением соединения 490С (40 мг, неочищенного) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,72-7,56 (m, 4Н), 7,33 (ушир. d, J=7,1 Гц, 2Н), 4,40 (q, J=7,1 Гц, 2Н), 1,40 (t, J=7,2 Гц, 3Н).
[1675] Соединение 490 синтезировали из промежуточное соединение 490С с использованием тех же методик, которые были описаны ранее для соединения 12 с получением соединения 490. Соединение 490 (26 мг, выход: 52,3%) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-метил-1-фенил-1H-имидазол-2-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 7,74 (ушир. d, J=7,9 Гц, 1Н), 7,55-7,45 (m, 3Н), 7,35-7,29 (m, 1H), 7,27-7,14 (m, 6H), 6,93 (d, J=0,9 Гц, 1H), 6,70 (ушир. s, 1H), 5,63 (dt, J=5,2, 7,7 Гц, 1H), 5,44 (ушир. s, 1H), 3,38 (dd, J=5,3, 14,1 Гц, 1H), 3,17 (dd, J=7,3, 14,1 Гц, 1H), 2,02 (d, J=0,9 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 377,1.
ПРИМЕР 229
СОЕДИНЕНИЕ 491
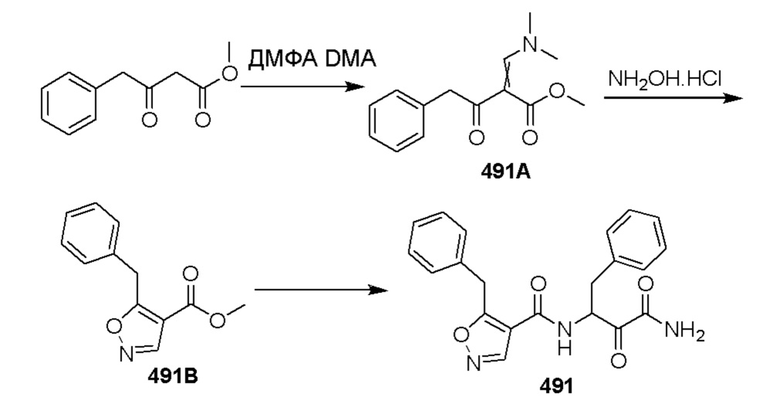
[1676] Смесь метил-3-оксо-4-фенилбутаноата (1 г, 5,20 ммоль) и ДМФА-DMA (682 мг, 5,72 ммоль) перемешивали при 20°С в течение 1 ч. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = от 20:1 до 5:1) с получением соединения 491А (900 мг, выход: 70,0%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,70 (s, 1H), 7,37-7,16 (m, 5Н), 4,06 (s, 2Н), 3,75 (s, 3Н), 3,33-2,57 (m, 6H).
[1677] Раствор соединения 491А (900 мг, 3,64 ммоль) в МеОН (20 мл) добавляли NH2OH.HCl (253 мг, 3,64 ммоль). Реакционную смесь перемешивали при 65°С в течение 1 ч. Растворитель выпаривали. Неочищенный остаток очищали посредством препаративной ТСХ (петролейный эфир:этилацетат = 3:1) с получением соединения 491В (500 мг, выход: 63,2%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 8,50 (s, 1H), 7,35-7,27 (m, 5H), 4,48 (s, 2H), 3,90 (s, 3Н).
[1678] Соединение 491 синтезировали из промежуточного соединения 491В с использованием тех же методик, которые были описаны ранее для соединения 12 с получением соединения 491. Соединение 491 (20,4 мг, выход: 41,0%) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-бензилизоксазол-4-карбоксамид: 1H ЯМР (400 МГц, CDCl3) δ 8,30 (s, 1H), 7,39-7,23 (m, 9H), 7,14-7,03 (m, 2H), 6,78 (ушир. s, 1H), 6,21 (ушир. d, J=7,0 Гц, 1H), 5,72-5,66 (m, 1H), 5,59 (ушир. s, 1H), 4,41 (s, 2H), 3,43 (dd, J=5,5, 14,2 Гц, 1H), 3,21 (dd, J=6,8, 14,2 Гц, 1H).
ПРИМЕР 230
СОЕДИНЕНИЕ 497
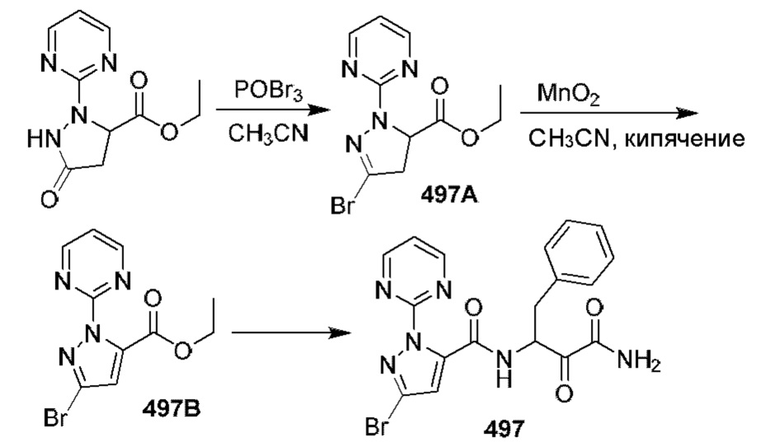
[1679] К раствору этил-5-оксо-2-(пиримидин-2-ил)пиразолидин-3-карбоксилата (300 мг, 1,27 ммоль) в CH3CN (10 мл) добавляли POBr3 (291,26 мг, 1,02 ммоль). Смесь перемешивали при 80°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении для удаления CH3CN. Остаток разбавляли H2O (10 мл) и подвергали экстракции ДХМ (10 мл × 2). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток использовали на следующей стадии без очистки. Соединение 497А (300 мг, неочищенного) получали в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 8,42 (d, J=4,9 Гц, 2H), 6,74 (t, J=4,9 Гц, 1H), 5,03 (dd, J=6,6, 12,6 Гц, 1Н), 4,27-4,10 (m, 2H), 3,60 (dd, J=12,7, 18,0 Гц, 1Н), 3,25 (dd, J=6,6, 18,1 Гц, 1Н), 1,20 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 298,7.
[1680] К раствору соединения 497А (300 мг, 1,00 ммоль) в CH3CN (10 мл) добавляли MnO2 (871,92 мг, 10,03 ммоль). Смесь перемешивали при 80°С в течение 48 ч. Смесь фильтровали. Фильтрат концентрировали с получением остатка. Остаток очищали посредством препаративной ТСХ (SiO2, ПЭ:ЭА=2:1). Соединение 497В (150 мг, выход 50,3%) получали в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 8,79 (d, J=4,9 Гц, 2H), 7,34 (t, J=4,9 Гц, 1Н), 6,95-6,78 (m, 1Н), 4,33 (q, J=7,1 Гц, 2H), 1,28 (t, J=7,2 Гц, 3Н), МС (ИЭР) m/z (M+H)+ 296,8.
[1681] Соединение 497 синтезировали из промежуточных соединений, 497В и 3-амино-2-гидрокси-4-фенилбутанамида гидрохлорида и с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 497. Соединение 497 (6 мг, выход: 41,0%) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-3-бром-1-(пиримидин-2-ил)-1H-пиразол-5-карбоксамид: 1H ЯМР (400 МГц, CDCl3) δ 8,72 (d, J=4,8 Гц, 2H), 7,70 (ушир. d, J=7,3 Гц, 1Н), 7,42 (t, J=4,8 Гц, 1Н), 7,35-7,20 (m, 5H), 7,03 (ушир. s, 1Н), 6,76 (s, 1Н), 6,27 (ушир. s, 1Н), 5,50 (dt, J=4,8, 8,0 Гц, 1Н), 3,31 (dd, J=4,9, 13,9 Гц, 1Н), 2,99 (dd, J=8,5, 14,1 Гц, 1Н). МС (ИЭР) m/z (M+H)+ 443,0.
ПРИМЕР 231
СОЕДИНЕНИЯ 549, 556-560, 584, 594, 595, 600, 619
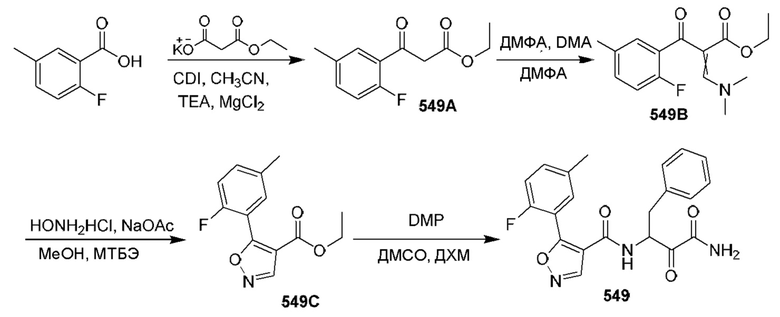
[1682] В круглодонной колбе к раствору 2-фтор-5-метилбензойной кислоты (10 г, 64,9 ммоль) в CH3CN (40 мл) добавляли CDI (11,8 г, 72,7 ммоль). Смесь перемешивали при 25°С в течение 4 ч. В другой колбе к раствору 3-этокси-3-оксопропаноата калия (14,6 г, 85,6 ммоль) в CH3CN (130 мл) порциями добавляли MgCl2 (6,2 г, 64,9 ммоль) в течение более 15 мин. Смесь перемешивали при 25°С в течение 0,5 ч, затем добавляли TEA (27 мл, 194,0 ммоль), и полученную суспензию перемешивали в течение 0,5 ч. Раствор из первой круглодонной колбы переносили в суспензию, содержавшуюся во второй колбе. Смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь гасили посредством HCl (3 н., 180 мл), и полученный раствор концентрировали при пониженном давлении. Полученный подвергали экстракции МТБЭ (200 мл × 2). Органический слой промывали H2O (200 мл), нас. NaHCO3 (200 мл × 2), sat. NaCl (200 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 549А (6,7 г, выход 45,5%) в виде бесцветной жидкости, который применяли на следующей стадии без дополнительной очистки. 1H ЯМР (ДМСО-d6, 400 МГц): δ 7,68-7,63 (m, 1H), 7,53-7,46 (m, 1H), 7,28-7,19 (m, 1H), 4,11 (q, J=7,1 Гц, 2Н), 4,03 (d, J=3,1 Гц, 2Н), 2,33 (s, 3Н), 1,16 (t, J=7,1 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 224,9.
[1683] Смесь соединения 549А (6,7 г, 29,9 ммоль) и ДМФА-DMA (16 мл, 120,4 ммоль) в ДМФА (60 мл) перемешивали при 80°С в течение 12 ч. Смесь концентрировали под вакуумом с получением соединения 549В (8,4 г, выход 97,6%) в виде красной жидкости, которую применяли на следующей стадии без дополнительной очистки. МС (ИЭР) m/z (М+H)+ 280,1.
[1684] К смеси соединения 549В (8,4 г, 30,1 ммоль) и гидроксиламина гидрохлорида (4,2 г, 60,2 ммоль) в МТБЭ (70 мл) и МеОН (70 мл) добавляли NaOAc (4,9 г, 60,2 ммоль) одной порцией. Смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь фильтровали и растворитель удаляли из фильтрата путем концентрирования под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле (элюент 0~10% этилацетат/петролейный эфир градиент) с получением соединения 549С (3,6 г, выход 46,7%) в виде бесцветной жидкости. 1H ЯМР (ДМСО-d6, 400 МГц): δ 9,11 (s, 1H), 7,60-7,54 (m, 1H), 7,52-7,44 (m, 1H), 7,37-7,28 (m, 1H), 4,20 (q, J=7,2 Гц, 2Н), 2,36 (s, 3Н), 1,17 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 250,1.
[1685] Соединение 549 синтезировали из промежуточных соединений, 549С и 3-амино-2-гидрокси-4-фенилбутанамида гидрохлорида с использованием тех же методик, которые описаны ранее для соединения 12, с получением соединения 549. Соединение 549 (100 мг, выход: 68,0%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(2-фтор-5-метилфенил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,96 (s, 1H), 8,92-8,81 (m, 1H), 8,08 (s, 1H), 7,83 (s, 1H), 7,40 (s, 2H), 7,33-7,11 (m, 6H), 5,33 (s, 1H), 3,24-3,05 (m, 1H), 2,93-2,75 (m, 1H), 2,31 (s, 3Н). МС (ИЭР) m/z (M+H)+ 396,1.
[1686] Соединение 556 синтезировали из этил-3-(2-фторфенил)-3-оксопропаноата с помощью методик, описанных для соединения 549, а затем с использованием тех же методик, которые описаны ранее для соединения 12, с получением соединения 556. Соединение 556 (60 мг, выход: 39,8%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(2-фторфенил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,00-8,89 (m, 2H), 8,11 (s, 1H), 7,85 (s, 1H), 7,65-7,55 (m, 2H), 7,38-7,19 (m, 7H), 5,37-5,28 (m, 1H), 3,18 (dd, J=3,6, 14,0 Гц, 1H), 2,91-2,63 (m, 1H). МС (ИЭР) m/z (М+Н)+ 382,1.
[1687] Соединение 557 синтезировали из этил-3-оксо-3-(о-толил)пропаноата с помощью методик, описанных для соединения 549, а затем с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 557. Соединение 557 (80 мг, выход: 52,5%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(о-отолил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,99 (s, 1H), 8,73 (d, J=7,3 Гц, 1H), 8,10 (s, 1H), 7,84 (s, 1H), 7,45-7,39 (m, 1H), 7,34-7,19 (m, 8H), 5,33-5,26 (m, 1H), 3,17 (dd, J=3,7, 13,9 Гц, 1H), 2,80 (dd, J=9,9, 13,9 Гц, 1H), 2,08 (s, 3Н). МС (ИЭР) m/z (М+H)+ 378,1.
[1688] Соединение 558 синтезировали из этил-3-(5-фтор-2-метилфенил)-3-оксопропаноата с помощью методик, описанных для соединения 549, затем с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 558. Соединение 558 (100 мг, выход: 48,0%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(5-фтор-2-метилфенил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,04 (s, 1H), 8,79 (d, J=7,5 Гц, 1H), 8,10 (s, 1H), 7,84 (s, 1H), 7,39-7,33 (m, 1H), 7,39-7,19 (m, 7H), 5,38-5,27 (m, 1H), 3,18 (dd, J=3,9, 13,9 Гц, 1H), 2,91-2,72 (m, 1H), 2,04 (s, 3Н). МС (ИЭР) m/z (M+H)+ 396,1.
[1689] Соединение 559 синтезировали из этил-3-(3-фторфенил)-3-оксопропаноат с помощью методик, описанных для соединения 549, затем с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 559. Соединение 559 (150 мг, выход: 74,3%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(3-фторфенил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,04 (d, J=7,3 Гц, 1Н), 8,88 (s, 1H), 8,14 (s, 1H), 7,86 (s, 1H), 7,76 (d, J=9,8 Гц, 1H), 7,70 (d, J=7,8 Гц, 1H), 7,59-7,48 (m, 1H), 7,46-7,37 (m, 1H), 7,36-7,25 (m, 4H), 7,25-7,17 (m, 1H), 5,46-5,32 (m, 1H), 3,27-3,15 (m, 1H), 2,92-2,77 (m, 1H), MC (ИЭР) m/z (М+H)+ 382,1.
[1690] Соединение 560 синтезировали из этил-3-оксо-3-(м-толил)пропаноата с помощью методик, описанных для соединения 549, а затем с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 560. Соединение 560 (160 мг, выход: 61,3%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(м-толил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,08-8,88 (m, 1H), 8,80 (s, 1H), 8,15 (s, 1H), 7,88 (s, 1H), 7,73-7,52 (m, 2H), 7,43-7,11 (m, 7H), 5,43-5,29 (m, 1H), 3,27-3,15 (m, 1H), 2,91-2,78 (m, 1H), 2,34 (ушир. s, 3Н). MC (ИЭР) m/z (M+H)+ 378,1.
[1691] Соединение 584 синтезировали из этил-3-(2-фтор-3-метилфенил)-3-оксопропаноата с помощью методик, описанных для соединения 549, а затем с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 584. Соединение 584 (130 мг, выход: 60,1%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(2-фтор-3-метилфенил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,93 (s, 1H), 8,84 (d, J=7,5 Гц, 1H), 8,06 (s, 1H), 7,81 (s, 1H), 7,46 (t, J=7,1 Гц, 1H), 7,40-7,33 (m, 1H), 7,30-7,15 (m, 6H), 5,31 (ddd, J=4,0, 7,4, 9,8 Гц, 1H), 3,15 (dd, J=4,0, 13,9 Гц, 1H), 2,80 (dd, J=9,9, 13,9 Гц, 1H), 2,23 (d, J=1,8 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 396,1.
[1692] Соединение 594 синтезировали из этил-3-(3-фтор-2-метилфенил)-3-оксопропаноата с помощью методик, описанных для соединения 549, а затем с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 594. Соединение 594 (30 мг, выход: 18,1%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(3-фтор-2-метилфенил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,00 (s, 1H), 8,77 (d, J=7,3 Гц, 1H), 8,06 (s, 1H), 7,80 (s, 1H), 7,36-7,13 (m, 8H), 5,32-5,24 (m, 1H), 3,16 (dd, J=4,1, 14,0 Гц, 1H), 2,79 (ушир. dd, J=10,0, 13,6 Гц, 1H), 1,93 (d, J=2,2 Гц, 3Н). 19F ЯМР (400 МГц, ДМСО-d6) δ -115,508-115,531 (s, 1F). МС (ИЭР) m/z (M+H)+ 396,0.
[1693] Соединение 595 синтезировали из этил-3-(2-фтор-3-метоксифенил)-3-оксопропаноата с помощью методик, описанных для соединения 549, а затем с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 595. Соединение 595 (86 мг, выход: 68,2%, светло-желтое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(2-фтор-3-метоксифенил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,97 (s, 1H), 8,89 (d, J=7,5 Гц, 1H), 8,09 (s, 1H), 7,84 (s, 1H), 7,40-7,33 (m, 1H), 7,32-7,17 (m, 6H), 7,12-7,04 (m, 1H), 5,43-5,25 (m, 1H), 3,87 (s, 3H), 3,22-3,13 (m, 1H), 2,89-2,75 (m, 1H). MC (ИЭР) m/z (M+H)+ 412,1.
[1694] Соединение 600 синтезировали из этил-3-(5-метилпиридин-3-ил)-3-оксопропаноата с помощью методик, описанных для соединения 549, а затем с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 600. Соединение 600 (90 мг, выход: 49,1%, желтое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(5-метилпиридин-3-ил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,04 (d, J=7,5 Гц, 1H), 8,96 (s, 1H), 8,81 (d, J=1,8 Гц, 1H), 8,56 (d, J=1,5 Гц, 1H), 8,15 (s, 1H), 8,06 (s, 1H), 7,86 (s, 1H), 7,29 (d, J=4,3 Гц, 3H), 7,27-7,17 (m, 2H), 5,42-5,34 (m, 1H), 3,21 (dd, J=3,8, 13,8 Гц, 1H), 2,85 (dd, J=10,0, 14,1 Гц, 1H), 2,35 (s, 3H). MC (ИЭР) m/z (M+H)+ 412,1.
[1695] Соединение 619 синтезировали из этил-3-(2-фтор-5-метоксифенил)-3-оксопропаноата с помощью методик, описанных для соединения 549, а затем с использованием тех же методик, которые были описаны ранее для соединения 12, с получением соединения 619. Соединение 619 (72 мг, выход: 71,5%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-5-(2-фтор-5-метоксифенил)изоксазол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,97 (s, 1H), 8,88 (d, J=7,5 Гц, 1H), 8,08 (s, 1H), 7,83 (s, 1H), 7,31-7,12 (m, 8H), 5,38-5,30 (m, 1H), 3,75 (s, 3H), 3,17 (dd, J=3,8, 14,1 Гц, 1H), 2,83 (dd, J=9,9, 13,9 Гц, 1H). MC (ИЭР) m/z (M+H)+ 412,1.
ПРИМЕР 232
СОЕДИНЕНИЯ 553, 574, 579, 580, 592, 623
[1696] Соединения 553, 574, 579, 580, 592, 623 синтезировали путем сочетания соответствующих промежуточных соединений, которые в свою очередь синтезировали с использованием методик, использованных для получения промежуточных соединений 62F и 32F, соответственно, и последующего воздействия на продукт сочетания условиями, как в случае соединения 107, с получением конечного продукта.
[1697] Соединение 553 (100 мг, 66,9% выход, белое твердое вещество) синтезировали из промежуточного соединения (3S)-3-амино-3-(2,3-дигидро-1H-инден-2-ил)-2-гидроксипропанамида и 3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоновой кислоты. Соединение 553: (S)-N-(3-амино-1-(2,3-дигидро-1H-инден-2-ил)-2,3-диоксопропил)-3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,24-8,19 (m, 2Н), 7,97-7,91 (m, 1Н), 7,70 (s, 1H), 7,40-7,33 (m, 2H), 7,19-7,05 (m, 6H), 5,16-5,10 (m, 1H), 3,87 (s, 3Н), 2,89-2,67 (m, 5H). MC (ИЭР) m/z (M+H)+ 421,1.
[1698] Соединение 574 (100 мг, 45,0% выход, светло-желтое твердое вещество) синтезировали из промежуточного соединения: (3S)-3-амино-2-гидроксигекс-5-упамид и 3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоновой кислоты. Соединение 574: (S)-N-(1-амино-1,2-диоксогекс-5-ин-3-ил)-3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,31-8,24 (m, 2H), 7,97 (s, 1H), 7,74 (s, 1H), 7,44-7,37 (m, 2H), 7,23-7,15 (m, 2H), 5,11-5,02 (m, 1H), 3,92 (s, 3Н), 2,88 (t, J=2,5 Гц, 1H), 2,75-2,57 (m, 2H). MC (ИЭР) m/z (M+H)+ 343,1.
[1699] Соединение 579 (52 мг, 33,85% выход, белое твердое вещество) синтезировали из промежуточного соединения: 3-амино-2-гидрокси-3-(НАФТАЛИН-2-ил)пропанамида гидрохлорида и 3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоновой кислоты. Соединение 579: N-(3-амино-1-(НАФТАЛИН-2-ил)-2,3-диоксопропил)-3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (d, J=6,0 Гц, 1H), 8,34 (s, 1H), 8,01 (ушир. s, 1H), 7,93-7,85 (m, 3Н), 7,81 (s, 1H), 7,68 (ушир. s, 1H), 7,55-7,36 (m, 5H), 7,23-7,13 (m, 2H), 6,45 (d, J=6,0 Гц, 1H), 3,87 (s, 3Н). MC (ИЭР) m/z (M+H)+ 431,1.
[1700] Соединение 580 (60 мг, 40,15% выход, белое твердое вещество) синтезировали из промежуточного соединения (3S)-3-амино-4-(3,5-дифторфенил)-2-гидроксибутанамида, гидрохлорида и 3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоновая кислота. Соединение 580: N-(4-амино-1-(3,5-дифторфенил)-3,4-диоксобутан-2-ил)-3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,31 (d, J=7,6 Гц, 1H), 8,13 (s, 1H), 7,95 (ушир. s, 1H), 7,72 (ушир. s, 1H). 7,39-7,27 (m, 2H), 7,17-7,00 (m, 3Н), 6,96-6,87 (m, 2H), 5,20-5,11 (m, 1H), 3,88 (s, 3Н), 3,17-3,09 (m, 1H), 2,89-2,79 (m, 1H). МС (ИЭР) m/z (M+H)+ 431,1.
[1701] Соединение 592 (320 мг, 60,69% выход, белое твердое вещество) синтезировали из промежуточного соединения (3R)-3-амино-2-гидрокси-4-фенилбутанамида, гидрохлорида и 3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоновая кислота. Соединение 592: (R)-N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-4-(2-фторфенил)-2-метилоксазол-5-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,78 (d, J=7,6 Гц, 1H), 8,06 (ушир. s, 1H), 7,81 (ушир. s, 1H), 7,47-7,37 (m, 2H), 7,30-7,13 (m, 7H), 5,37-5,28 (m, 1H), 3,16-3,09 (m, 1H), 2,97-2,88 (m, 1H), 2,52 (s, 3Н). МС (ИЭР) m/z (M+H)+ 396,1.
[1702] Соединение 623 (60 мг, 59,9% выход, белое твердое вещество) синтезировали из промежуточного соединения 3-амино-2-гидрокси-4-(3-(трифторметил)фенил)бутанамида, гидрохлорида и 3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоновая кислота. Соединение 623: N-(4-амино-3,4-диоксо-1-(3-(трифторметил)фенил)бутан-2-ил)-3-(2-фторфенил)-1-метил-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,35 (ушир. d, J=6,6 Гц, 1H), 8,15 (s, 1H), 8,00 (ушир. s, 1H), 7,76 (ушир. s, 1H), 7,64-7,48 (m, 4H), 7,33 (ушир. dd, J=7,3, 15,4 Гц, 2H), 7,20-7,08 (m, 2H), 5,22 (ушир. s, 1H), 3,90 (s, 3Н), 3,28-2,84 (m, 2H). МС (ИЭР) m/z (М+Н)+ 363,1.
ПРИМЕР 233
СОЕДИНЕНИЕ 562
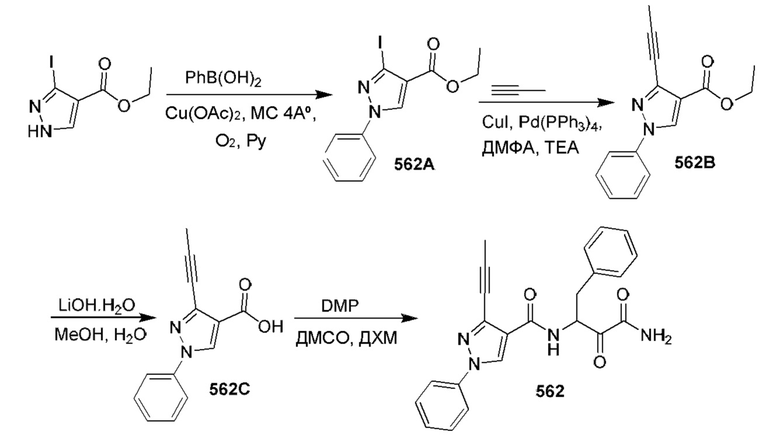
[1703] К смеси этил-3-иод-1H-пиразол-4-карбоксилатного соединения (1 г, 3,8 ммоль) и фенилбороновой кислоты (504,1 мг, 4,1 ммоль) в пиридине (20 мл) добавляли Cu(OAc)2 (751 мг, 4,1 ммоль, 1,1 экв.) и молекулярное сито 4А° (1 г). Смесь перемешивали при 80°С в течение 12 ч в атмосфере О2 (~15 фунтов на кв. дюйм). Смесь разбавляли ЭА (40 мл), фильтровали через целит. Фильтрат концентрировали при пониженном давлении для удаления растворителя, затем добавляли H2O (60 мл) и ЭА (50 мл). Полученное черное нерастворимое вещество отделяли и фильтровали через целит два раза. Осадок на фильтре промывали ЭА (40 мл × 2). Объединенный фильтрат отделяли и водную фазу подвергали экстракции посредством ЭА (40 мл × 2). Объединенную органическую фазу промывали солевым раствором (50 мл × 2), сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 20/1 до 5/1), затем отбирали фракцию и концентрировали. Остаток растирали с ПЭ (40 мл). Твердое вещество собирали и сушили под вакуумом с получением соединения 2 (0,8 г, выход 61,4%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц) δ 9,01 (s, 1H), 7,89 (d, J=8,4 Гц, 2Н), 7,58-7,49 (m, 2H), 7,44-7,36 (m, 1H), 4,28 (q, J=7,2 Гц, 2Н), 1,35-1,27 (m, 3Н). МС (ИЭР) m/z (M+H)+ 342,9.
[1704] К смеси соединения 562А (0,3 г, 876,9 мкмоль) и проп-1-ина (1М в ДМФА, 1,8 мл, 1,8 ммоль) в ДМФА (6 мл) добавляли TEA (2 мл), CuI (33,4 мг, 175,4 мкмоль), затем Pd(PPh3)4 (101 мг, 87,7 мкмоль). Смесь дегазировали и продували N2 3 раза, и затем перемешивали при 55°С в течение 12 ч. Реакционную смесь объединяли с реакционной смесью на странице ES5524-401-P1 для концентрирования при пониженном давлении. Остаток очищали посредством флэш-хроматографии на силикагеле (элюирование с градиентом 0-15% этилацетат/петролейный эфир) с получением соединения 562В (204,2 мг, выход 91,5%) в виде светло-желтого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц): δ 9,07 (s, 1H), 7,98-7,85 (m, 2Н), 7,59-7,47 (m, 2Н), 7,45-7,33 (m, 1H), 4,26 (q, J=7,0 Гц, 2Н), 2,11 (s, 3Н), 1,31 (t, J=7,2 Гц, 3Н). МС (ИЭР) m/z (M+H)+ 255,1.
[1705] Соединение 562 синтезировали из промежуточных соединений 562В путем превращения их в 562С и обработки (3S)-3-амино-2-гидрокси-4-фенилбутанамида гидрохлоридом и с использованием тех же методик, которые описаны ранее для соединения 12, с получением соединения 562. Соединение 549 (100 мг, выход: 35,7%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-фенил-3-(проп-1-ин-1-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,85 (s, 1H), 7,92-7,76 (m, 4H), 7,66 (s, 1H), 7,57-7,48 (m, 2H), 7,43-7,36 (m, 1H), 7,33-7,26 (m, 2H), 7,26-7,17 (m, 3H), 5,63-5,48 (m, 1H), 3,34-3,26 (m, 1H), 3,15-3,09 (m, 1H), 2,03 (s, 3H). МС (ИЭР) m/z (М+H)+ 401,2.
ПРИМЕР 234
СОЕДИНЕНИЕ 583
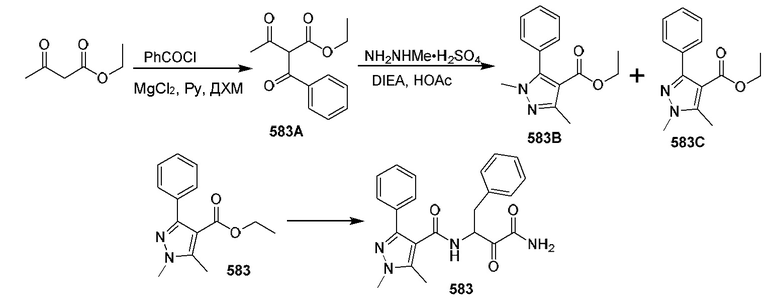
[1706] Смесь этил-3-оксобутаноата (5 г, 38,42 ммоль), MgCl2 (4,39 г, 46,10 ммоль) и пиридина (6,8 мл, 84,52 ммоль) в ДХМ (100 мл) перемешивали при 0°С в течение 30 мин. Затем в раствор медленно добавляли бензоилхлорид (4,9 мл, 42,26 ммоль) в ДХМ (20 мл). Смесь перемешивали при 25°С в течение 10 ч. Реакцию гасили путем добавления 6 н. HCl (~20 мл). Затем смесь разбавляли H2O (60 мл). Отделяли органический слой, и водный раствор подвергали экстракции посредством ДХМ (35 мл × 2). Объединенный органический слой промывали насыщенным NaHCO3 (50 мл), солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 8:1) с получением соединения 583А (6,2 г, выход 68,9%) в виде бесцветного масла.
[1707] К суспензии метилгидразина (14,6 г, 101,6 ммоль, соль H2SO4) в ДМФА (15 мл) добавляли DIEA (35,3 мл, 203,2 ммоль). Смесь перемешивали при 25°С в течение 15 мин. Затем смесь добавляли к смеси соединение 583А (11,9 г, 50,8 ммоль) в НОАс (150 мл). Смесь перемешивали при 25°С в течение 8 ч. Смесь концентрировали. Остаток обрабатывали H2O (300 мл) и ЭА (100 мл). Отделяли органический слой и водный слой подвергали экстракции ЭА (100 мл). Объединенный органический слой промывали насыщенным NaHCO3 (100 мл), солевым раствором (100 мл), сушили над MgSO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = от 10:1 до 5:1) с получением соединения 583В (2,3 г, выход 18,5%) в виде бесцветного масла и соединение 583С (6 г, выход 48,3%) в виде бледно-желтого масла. Соединение 583В: 1H ЯМР (ДМСО-d6, 400 МГц): δ 7,49-7,43 (m, 3H), 7,39-7,34 (m, 2H), 3,95 (q, J=7,1 Гц, 2Н), 3,56-3,51 (m, 3H), 2,34 (s, 3H), 0,96 (t, J=7,1 Гц, 3H). Соединение 583С: 1H ЯМР (ДМСО-d6, 400 МГц): δ 7,51-7,47 (m, 2H), 7,38-7,31 (m, 3H), 4,09 (q, J=7,1 Гц, 2H), 3,78 (s, 3H), 2,47 (ушир. s, 3H), 1,11 (t, J=7,1 Гц, 3H).
[1708] Соединение 583 синтезировали из промежуточных соединений, 583С и 3-амино-2-гидрокси-4-фенилбутанамида, гидрохлорида и с использованием тех же методик, которые описаны для соединения 12, с получением соединения 583. Соединение 583 (100 мг, выход: 46,2%, белое твердое вещество) N-(4-амино-3,4-диоксо-1-фенилбутан-2-ил)-1-фенил-3-(проп-1-ин-1-ил)-1H-пиразол-4-карбоксамид: 1H ЯМР (400 МГц, ДМСО-d6) δ 8,38 (d, J=7,5 Гц, 1Н), 8,12 (s, 1H), 7,86 (s, 1H), 7,47-7,41 (m, 2H), 7,31-7,21 (m, 8H), 5,38 (ddd, J=3,6, 7,4, 10,6 Гц, 1H), 3,72 (s, 3H), 3,19 (dd, J=3,4, 14,0 Гц, 1H), 2,74 (dd, J=10,6, 13,9 Гц, 1H), 2,12 (s, 3H). MC (ИЭР) m/z (M+H)+ 391,1.
БИОЛОГИЧЕСКИЕ ДАННЫЕ
ПРИМЕР 235
[1709] Активность кальпаинов 1, 2 и 9 и ее ингибирование оценивали с помощью непрерывного флуоресцентного анализа. Субстрат кальпаина SensoLyte 520 (Anaspec Inc) был оптимизирован для определения активности кальпаина. Этот субстрат содержит пару 5-FAM/QXLTM 520 FRET с внутримолекулярным тушением флуоресценции. Кальпаины 1, 2 и 9 расщепляют субстрат FRET на два отдельных фрагмента, что приводит к возрастанию флуоресценции 5-FAM, которая пропорциональна активности кальпаина.
[1710] Как правило, анализ проводили в черных 384-луночных планшетах с использованием автоматической системы дозирования жидкостей следующим образом. Основной буфер для анализа кальпаина, как правило, содержит 50 мМ Трис, рН 7,5, 100 мМ NaCl и 1 мМ ДТТ. Готовили серийные разведения ингибиторов в ДМСО и использовали для приготовления 2-кратных (2х) смесей с кальпаинами в указанном выше буфере. После инкубирования при температуре окружающей среды (25 С) реакцию инициировали путем добавления 2х смеси флуоресцентного пептидного субстрата и CaCl2 (необходимого для активации кальпаина in situ) в том же буфере. Данные кривой процесса реакции, как правило, регистрировали в течение 10 мин с использованием длин волн возбуждения/эмиссии 490 нм/520 нм на планшетных ридерах SpectraMax i3x или FLIPR-Tetra (Molecular Devices Inc). Скорости реакций рассчитывали по наклонам кривых процесса реакции, как правило, за 1-5 мин. Кривые зависимости ответа от дозы (скорость в зависимости от логарифма концентрации ингибитора), как правило, аппроксимировали с помощью 4-параметрической логистической функции для получения значений IC50.
[1711] Активность кальпаина в клетках SH-SY5Y и ее ингибирование оценивали с помощью гомогенного флуоресцентного анализа, в котором используется доступный для клеток и профлуоресцентный субстрат кальпаина Suc-LLVY-AMC (Sigma-Aldrich Inc). При внутриклеточном расщеплении Suc-LLVY-AMC кальпаином в среду высвобождается флуоресцентный аминометилкумарин (АМК), что приводит к непрерывному увеличению флуоресцентного сигнала, который пропорционален внутриклеточной активности кальпаина.
[1712] Как правило, анализ проводили путем высевания клеток SH-SY5Y в черные 384-лун очные планшеты в концентрации 40000/лунку в RPMI-1640, содержащей 1% сыворотки, с последующим инкубированием при 37 С в течение ночи. На следующее утро клетки предварительно инкубировали в течение 30 мин с серийными разведениями соединений с последующим добавлением 100 мкМ субстрата Suc-LLVY-AMC. Непрерывное увеличение флуоресценции АМК регистрируют с помощью планшетного ридера FLIPR Tetra (Molecular Devices Inc) и определяют наклоны кривых для определения активности кальпаина. Кривые зависимости ответа от дозы (наклоны кривых в зависимости от логарифма концентрации ингибитора), как правило, аппроксимировали с помощью 4-параметрической логистической функции для получения значений IC50.
[1713] Активность кальпаина в клетках SH-SY5Y и ее ингибирование также оценивали с помощью анализа на основе вестерн-блоттинга, в котором определяют специфический для кальпаина продукт деградации альфа-цепи неэритроцитарного спектрина (SBDP-150). Для индукции активности кальпаина и образования SBDP-150 применяли добавление кальциевого ионофора А23187.
[1714] Как правило, эти анализы проводили путем высевания клеток SH-SY5Y в 96-лун очные планшеты в концентрации 150000/лунку в DMEM, содержащей 10% сыворотки, с последующим инкубированием при 37 С в течение 24 часов. Затем клетки предварительно инкубировали в течение 60 мин с серийными разведениями соединений с последующим добавлением 25 мкМ А23187 и дальнейшим инкубированием в течение 90 мин. Общий клеточный белок экстрагировали в буфере RIPA, кипятили в буфере для загрузки в гель и проводили электрофорез в ПААГ с добавлением ДНС. Гель подвергали вестерн-блоттингу (сухому переносу) для количественного определения SBDP-150 (антитело АА6, Enzo Inc) и либо GAPDH, либо HSP90 в качестве контролей нанесения. Строили график зависимости нормированных количеств SBDP-150 от логарифма концентрации ингибитора для получения кривых зависимости ответа от дозы, которые, как правило, аппроксимировали с помощью 4-параметрической логистической функции для получения значений IC50.
ИНГИБИРОВАНИЕ КАЛЬПАИНА
Таблица 2. Анализ ингибирования кальпаина
Столбец А: IC50 кальпаин 1 человека/NS1
Столбец В: IC50 кальпаин 2 человека/NS1
Столбец С: IC50 кальпаин 9 человека/NS1
Столбец D: IC50 SH-SY5Y спектрин
Столбец Е: IC50 SH-SY5Y + АМК
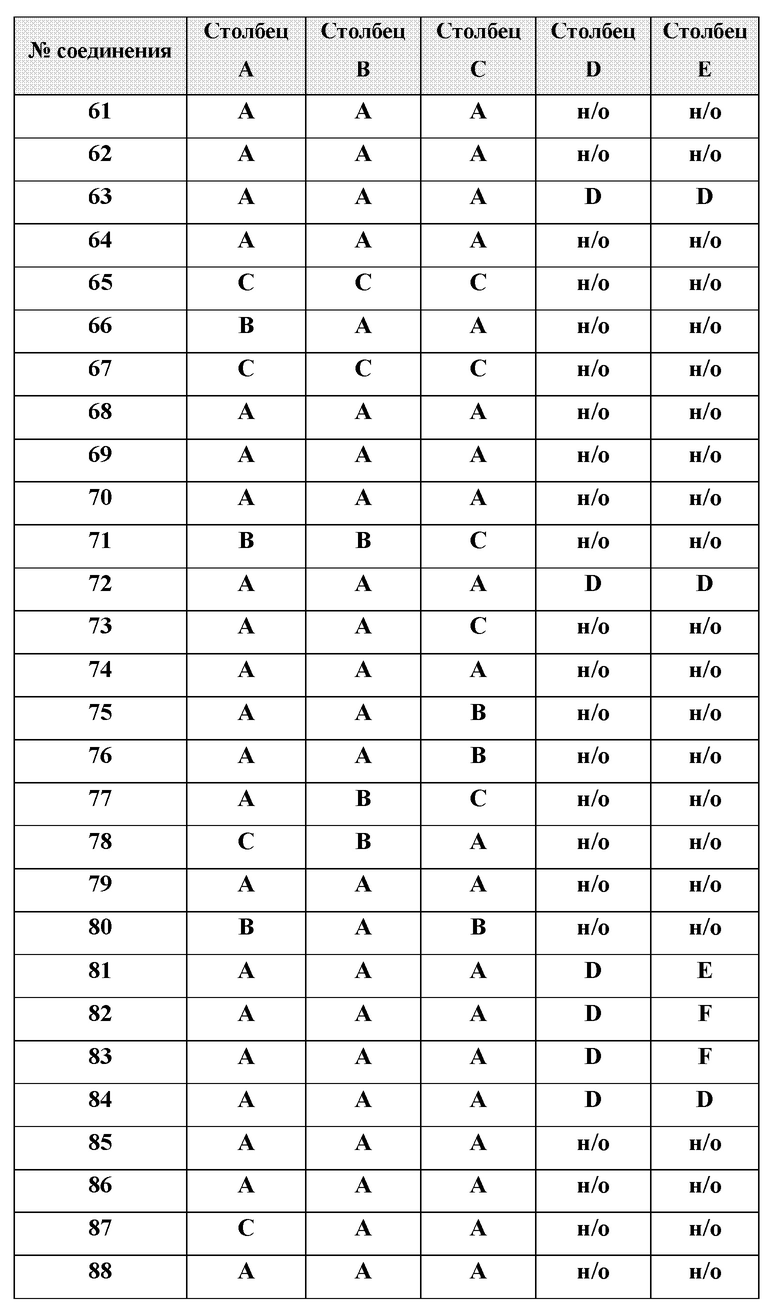
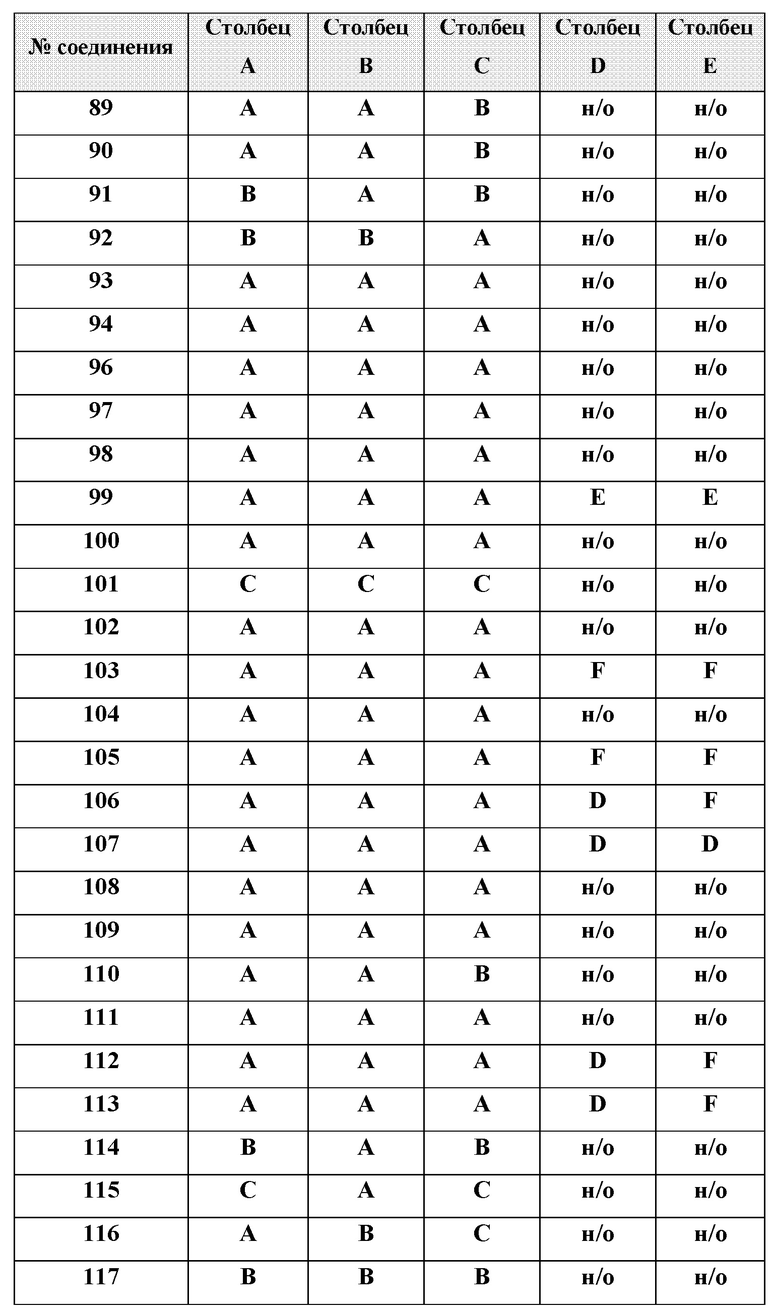
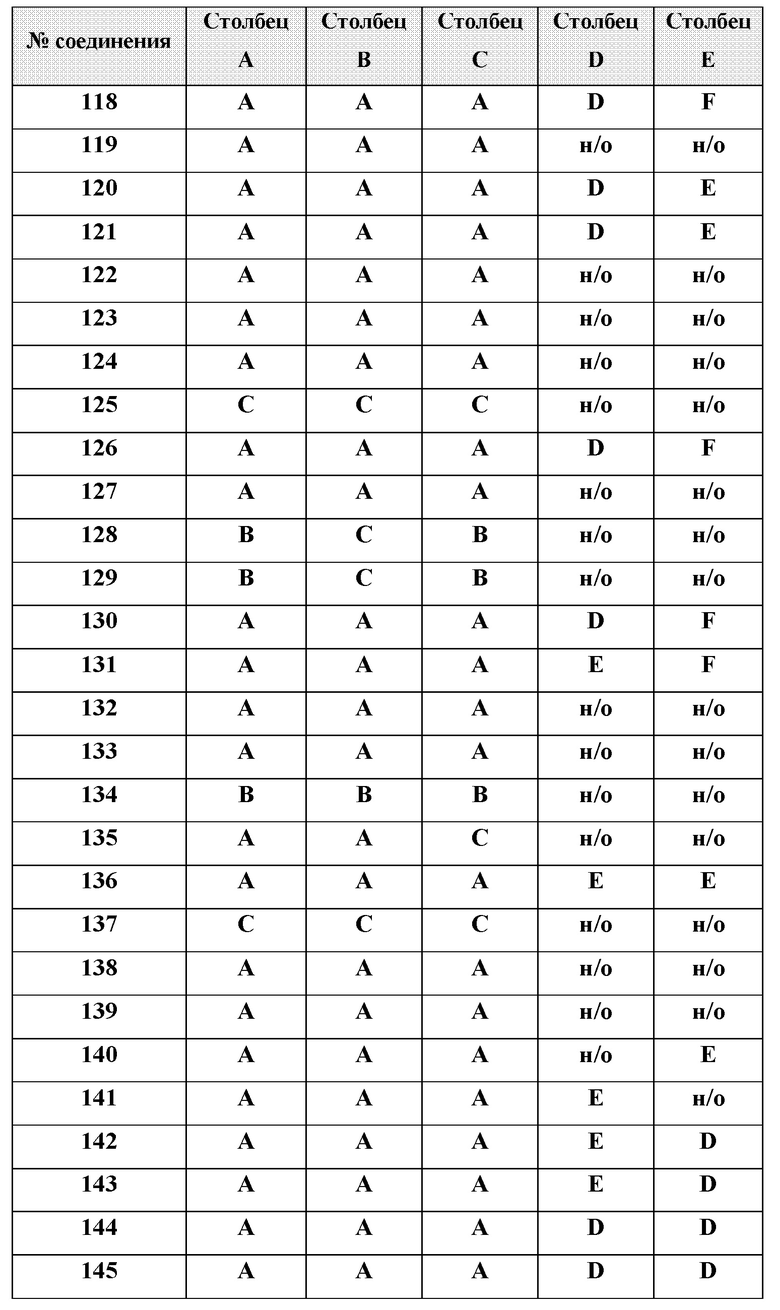
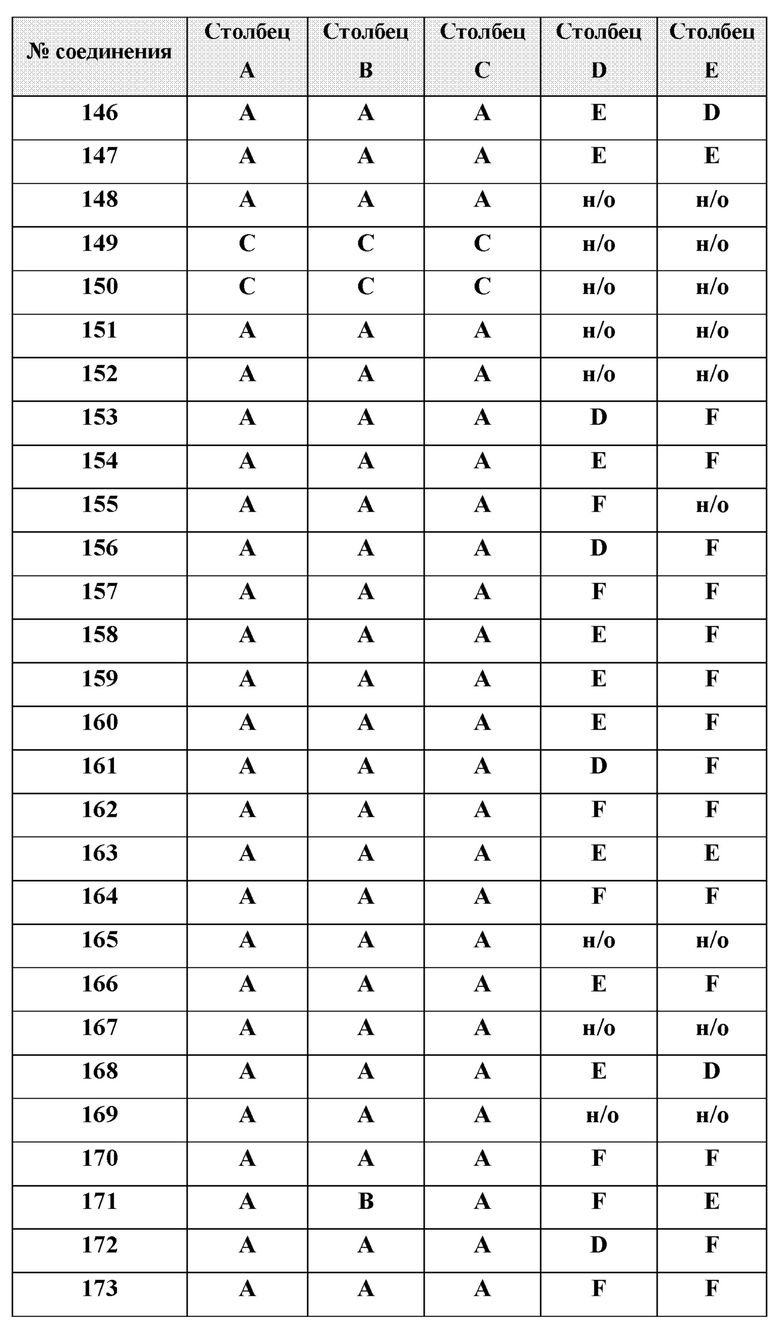
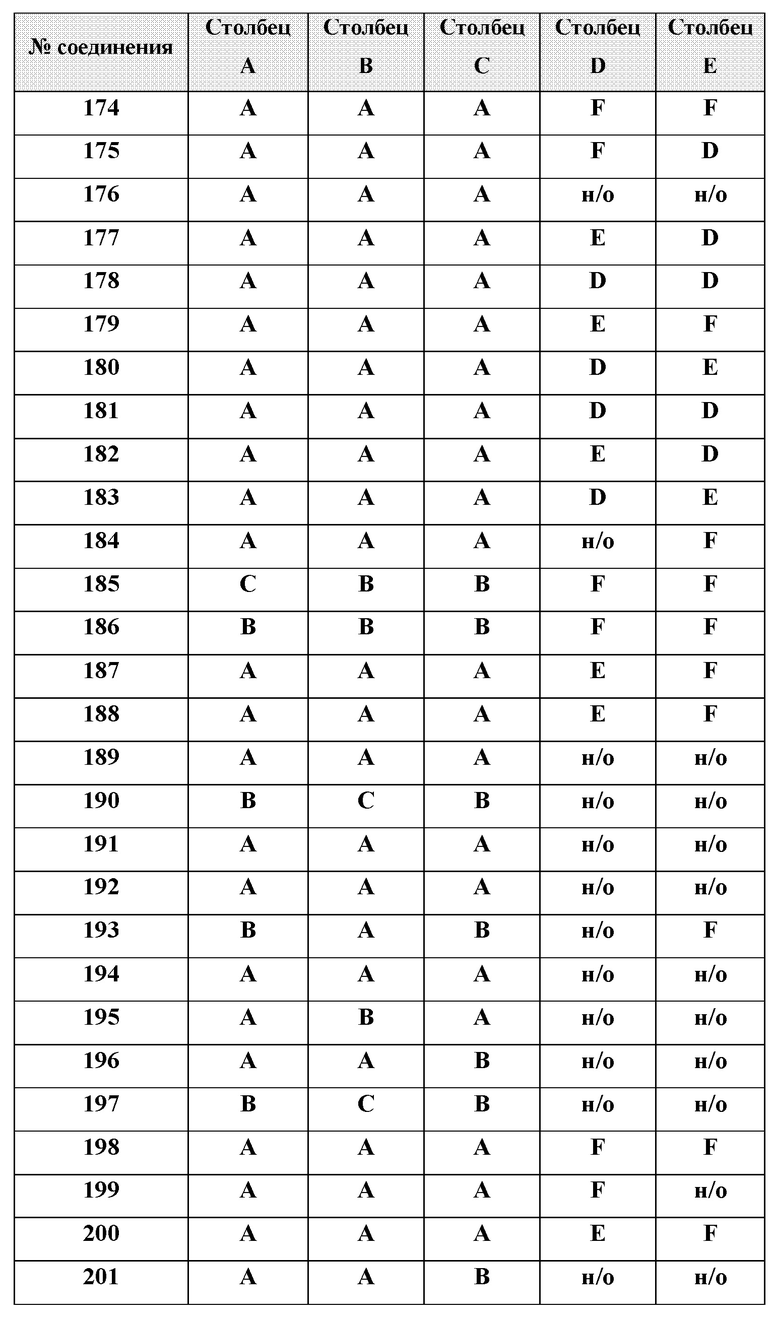
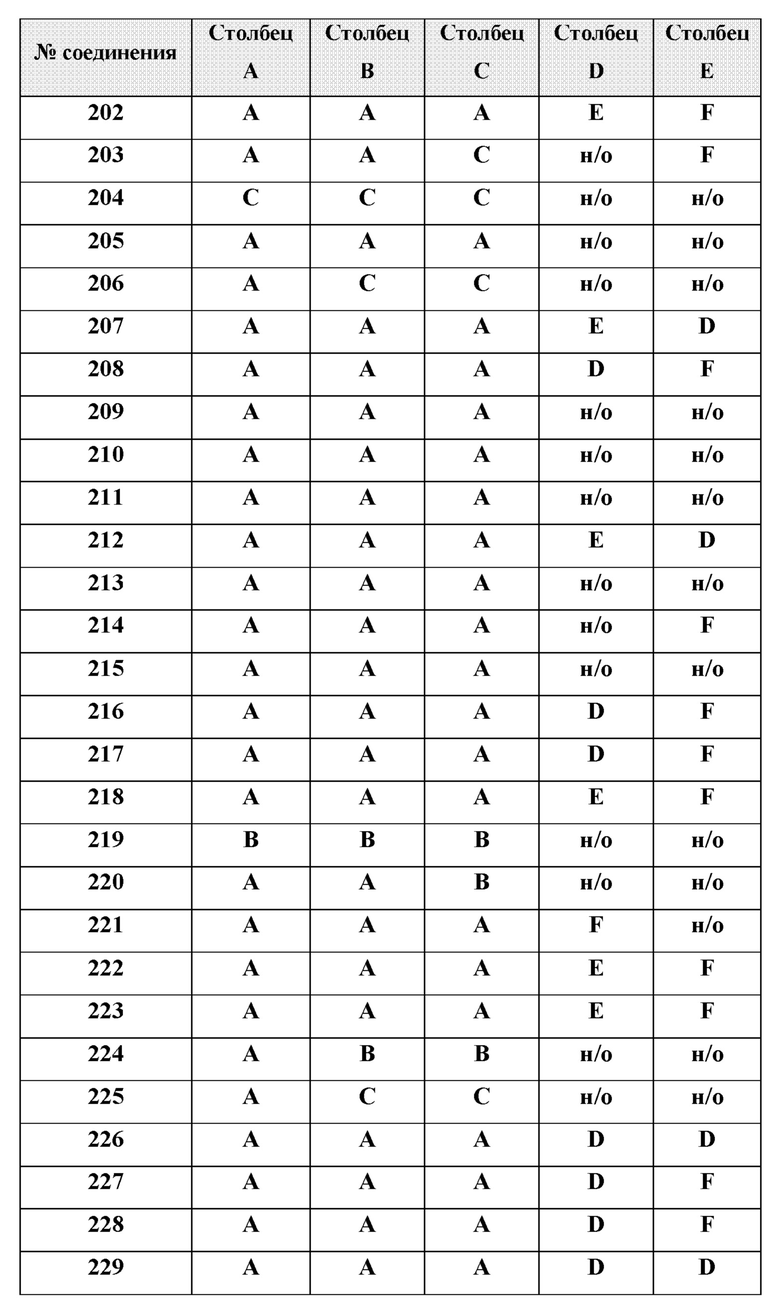
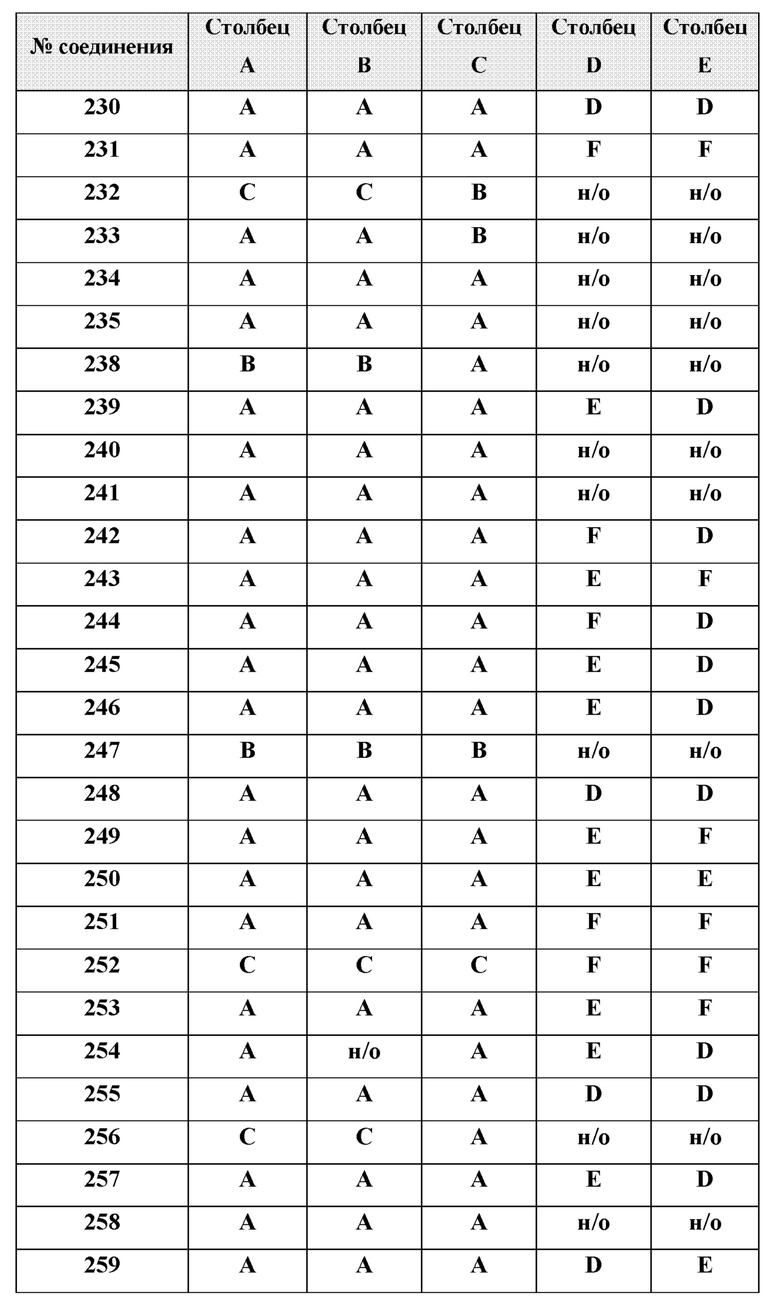
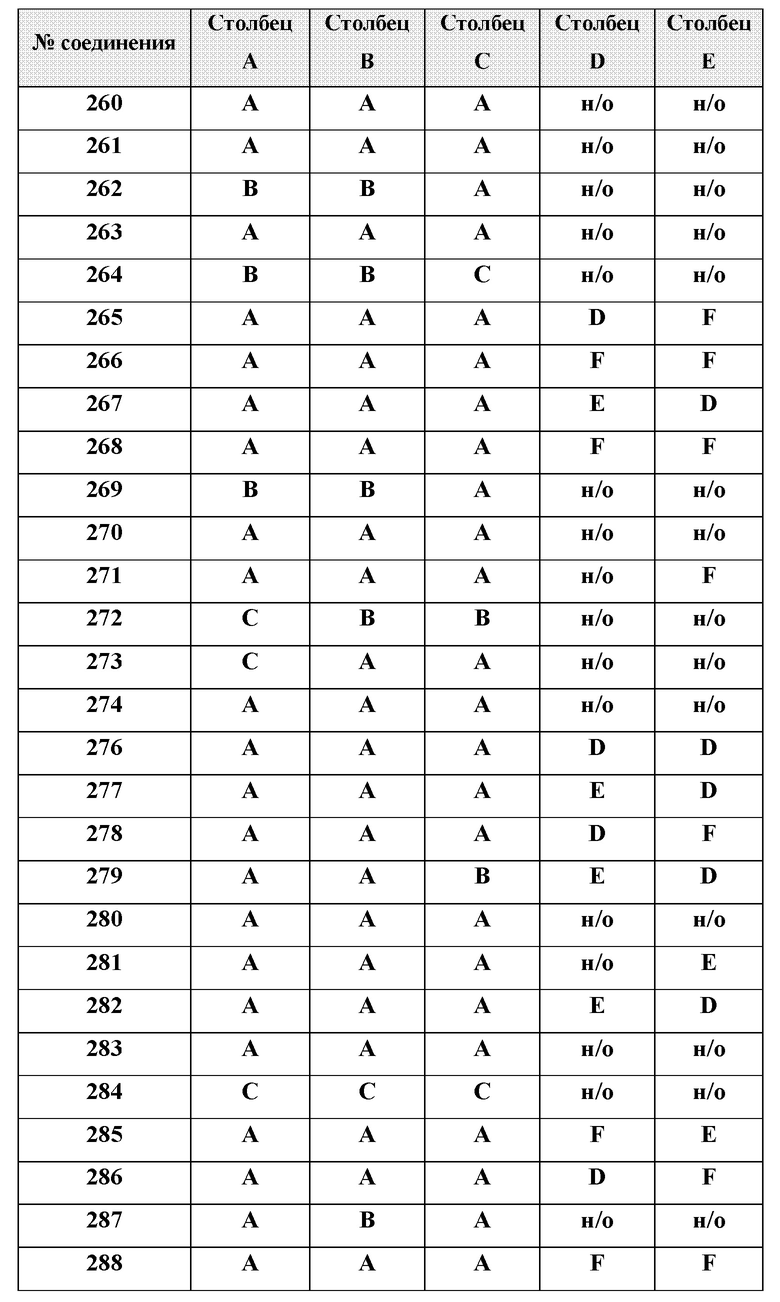
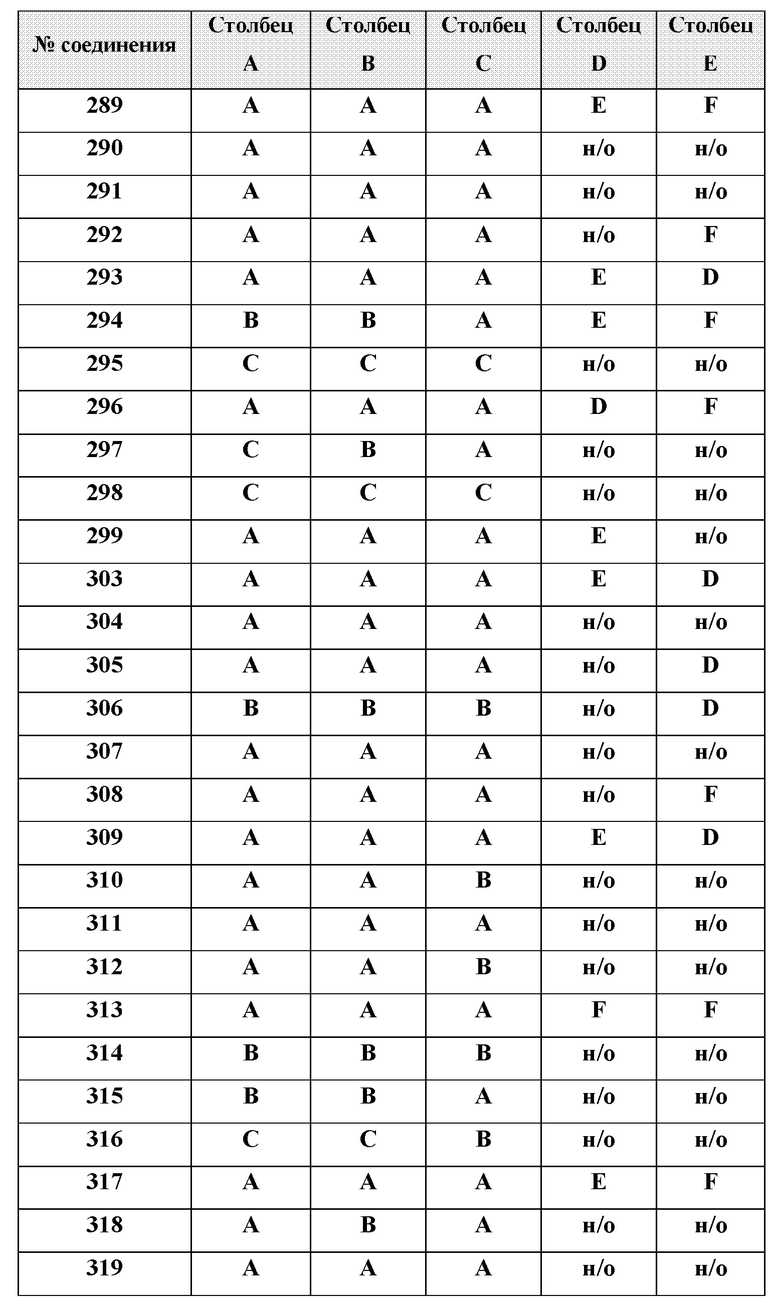
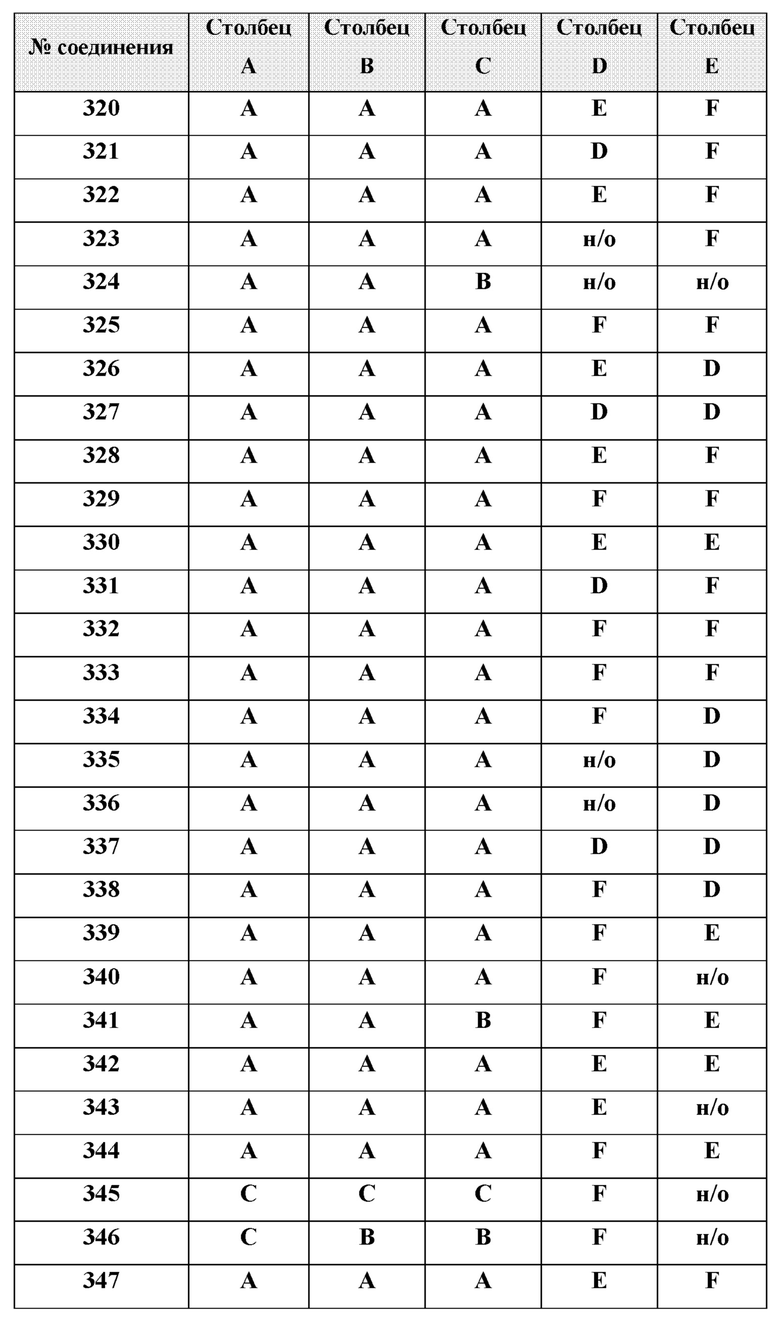
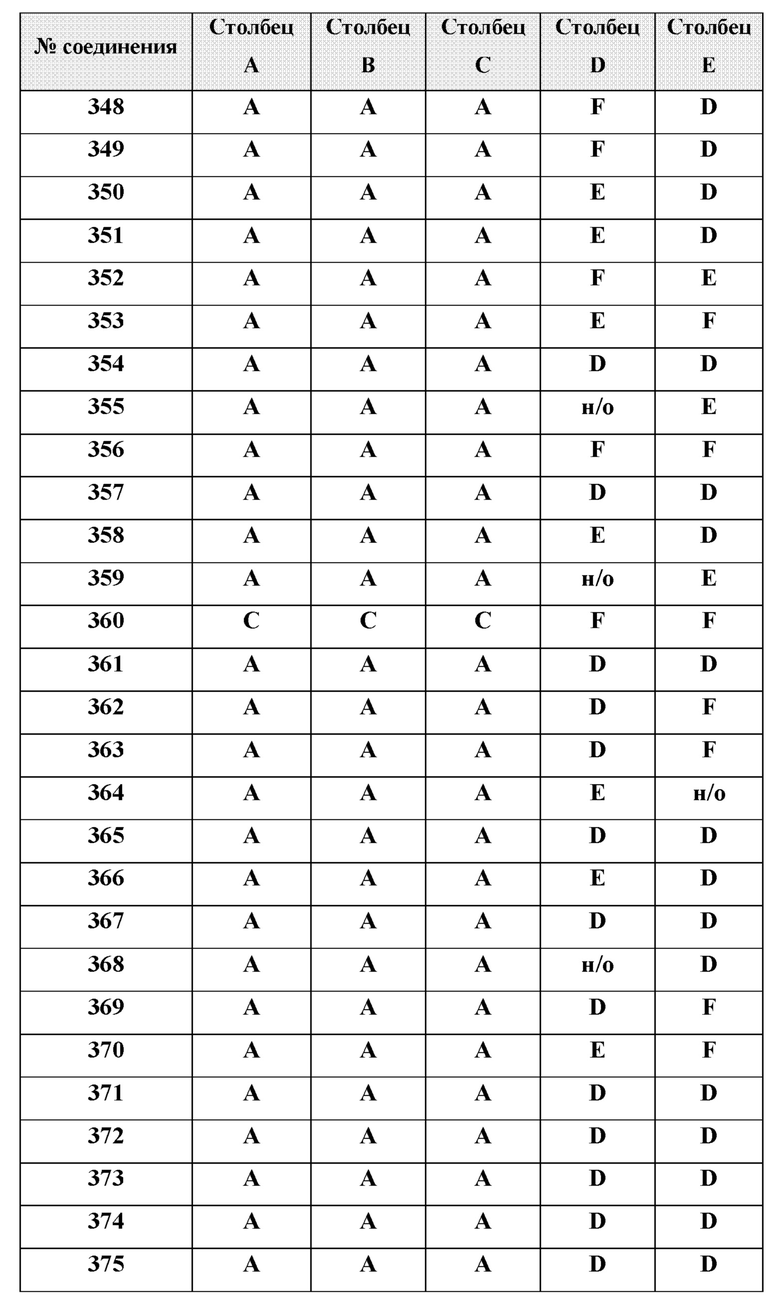
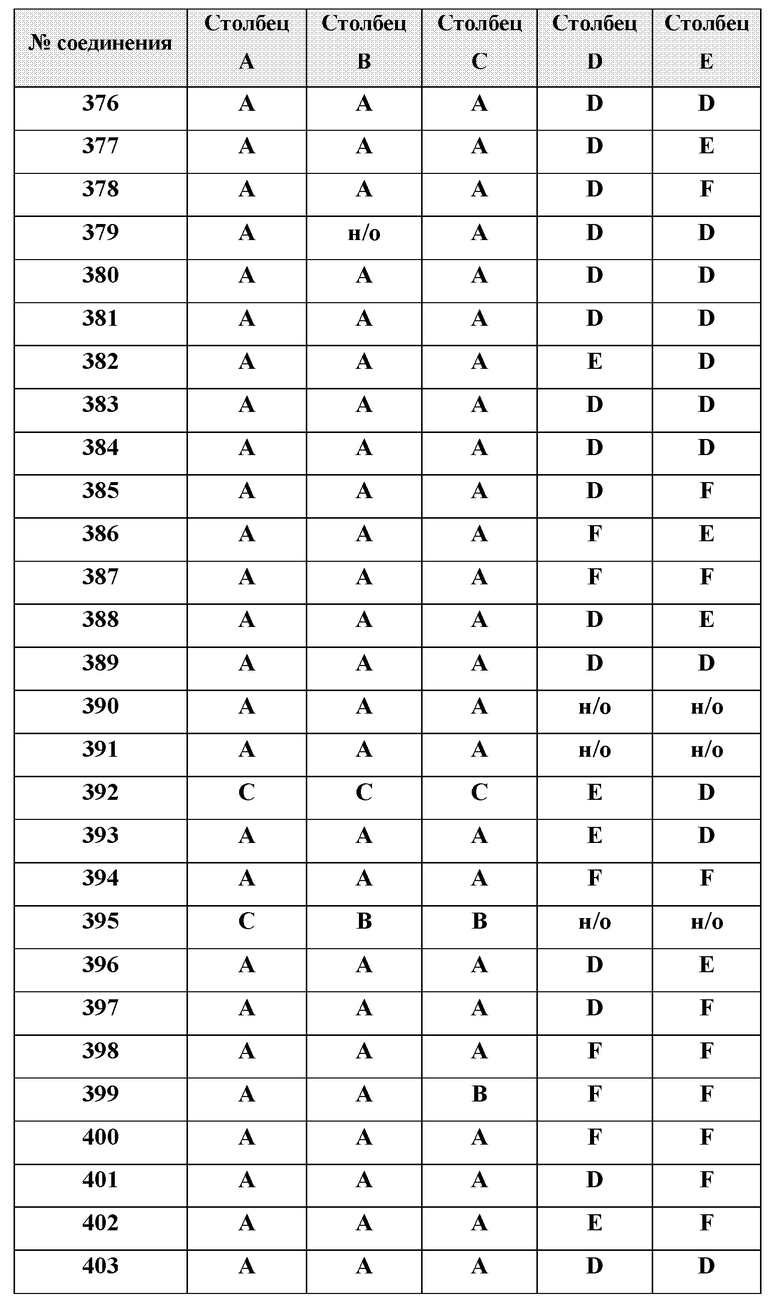
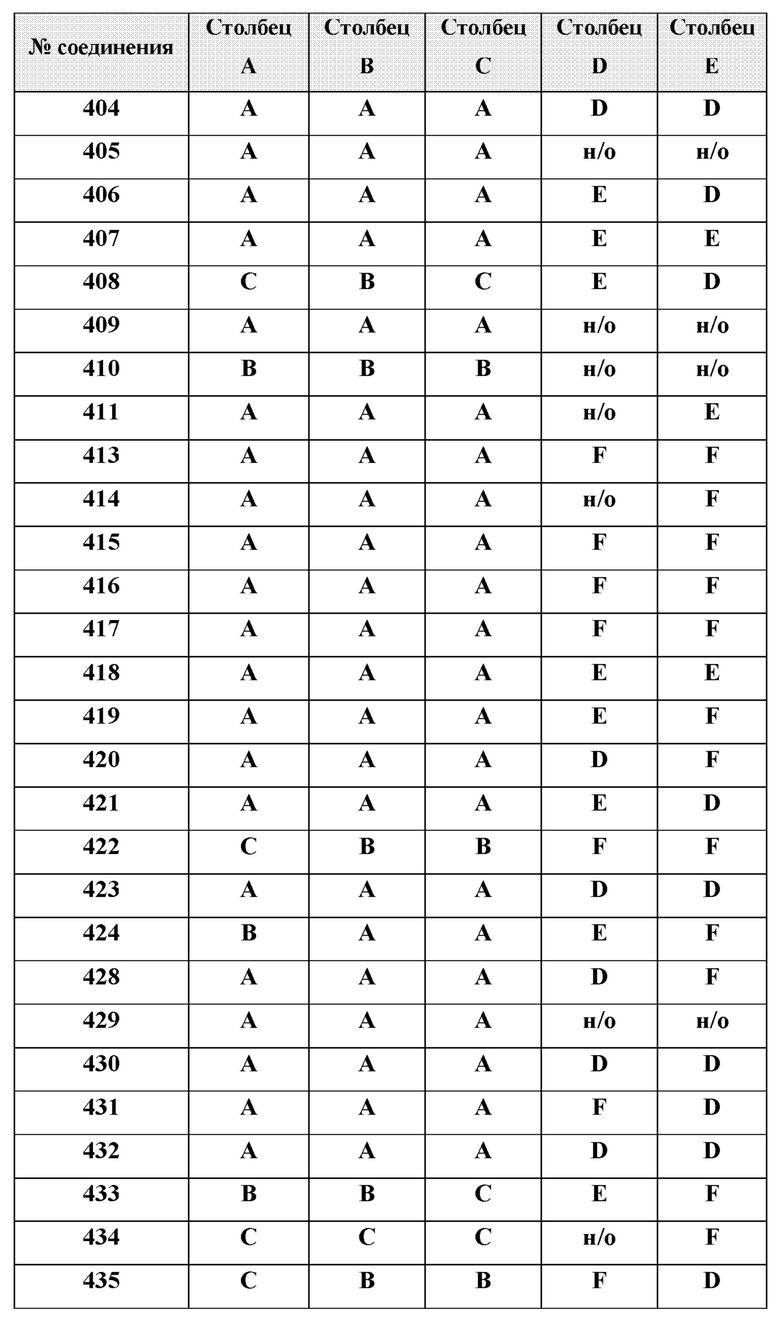
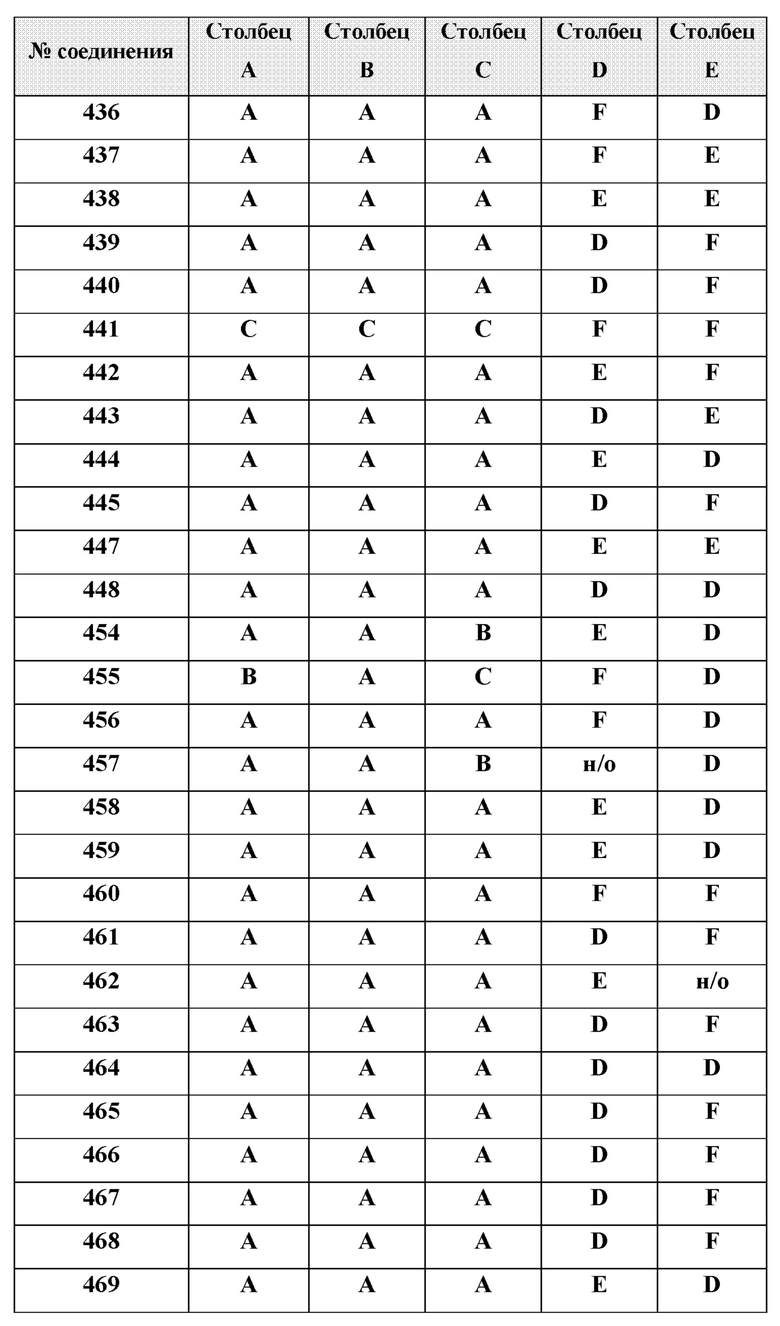
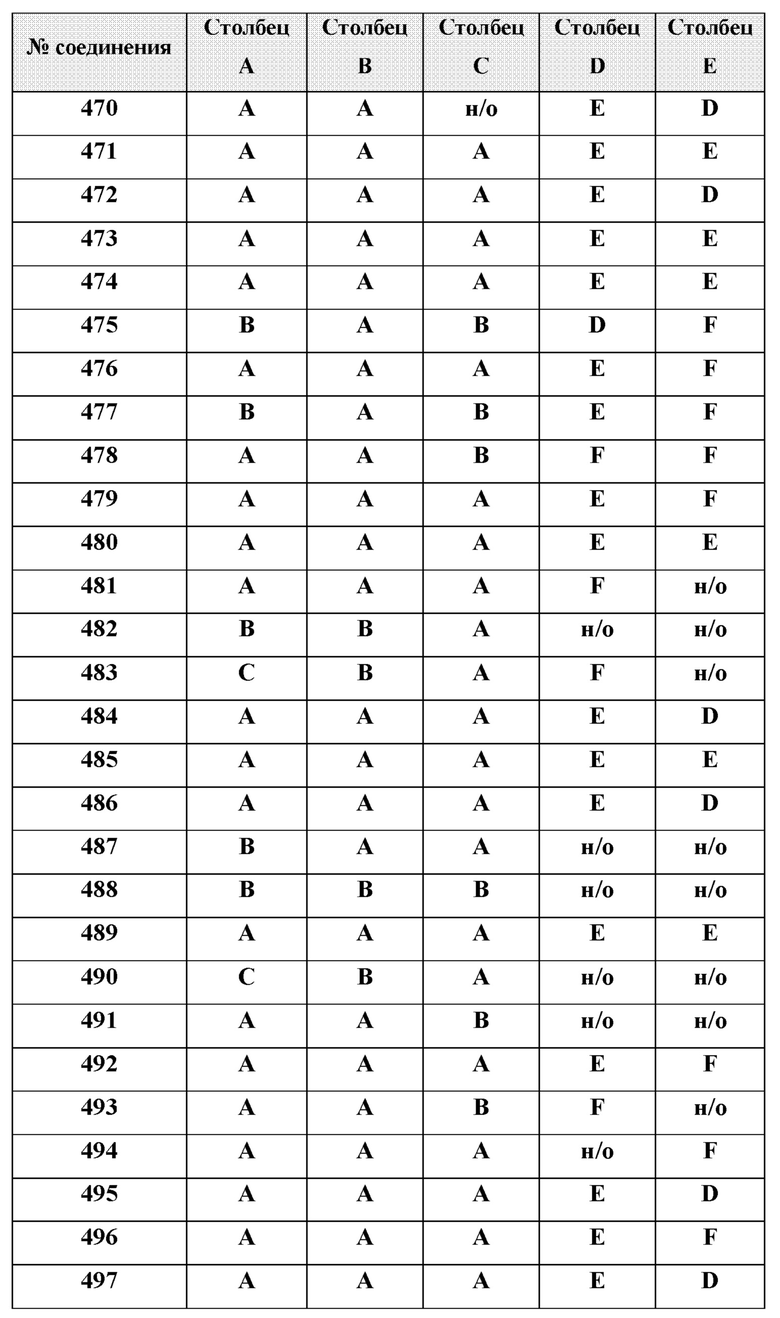
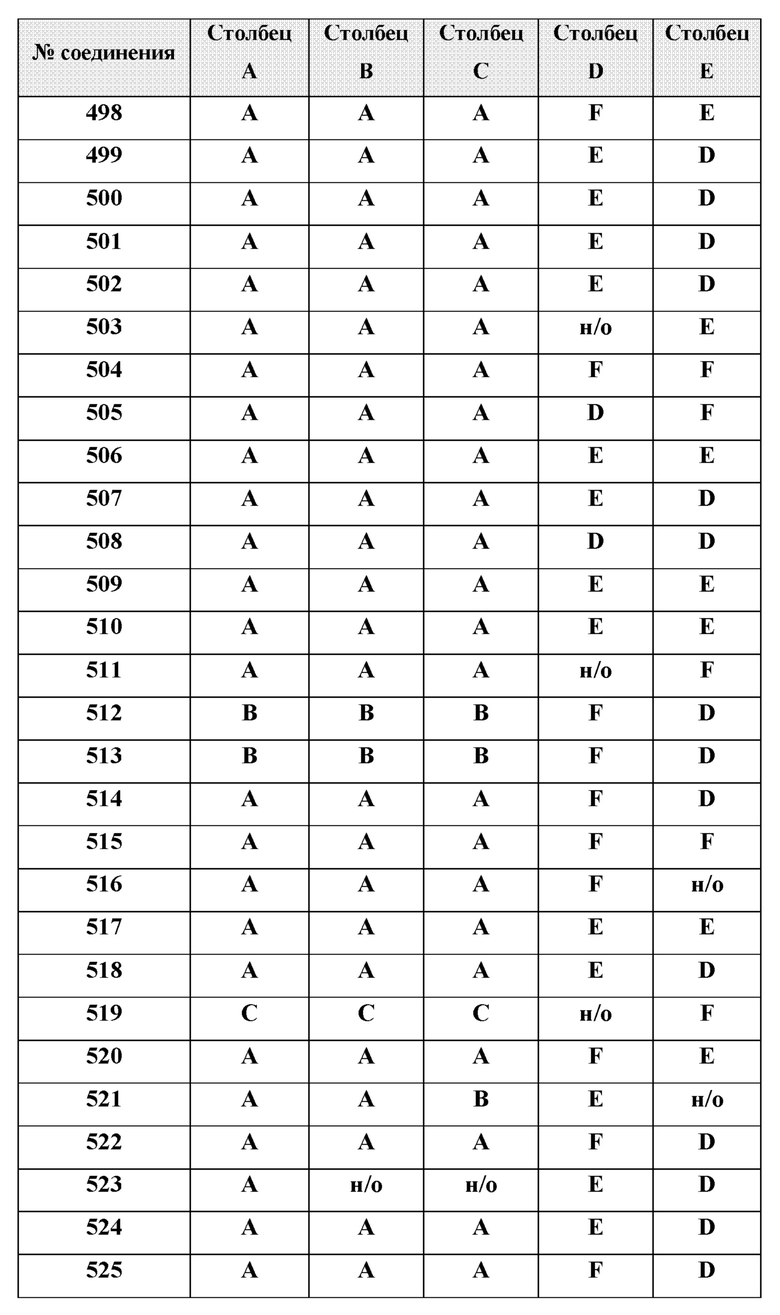
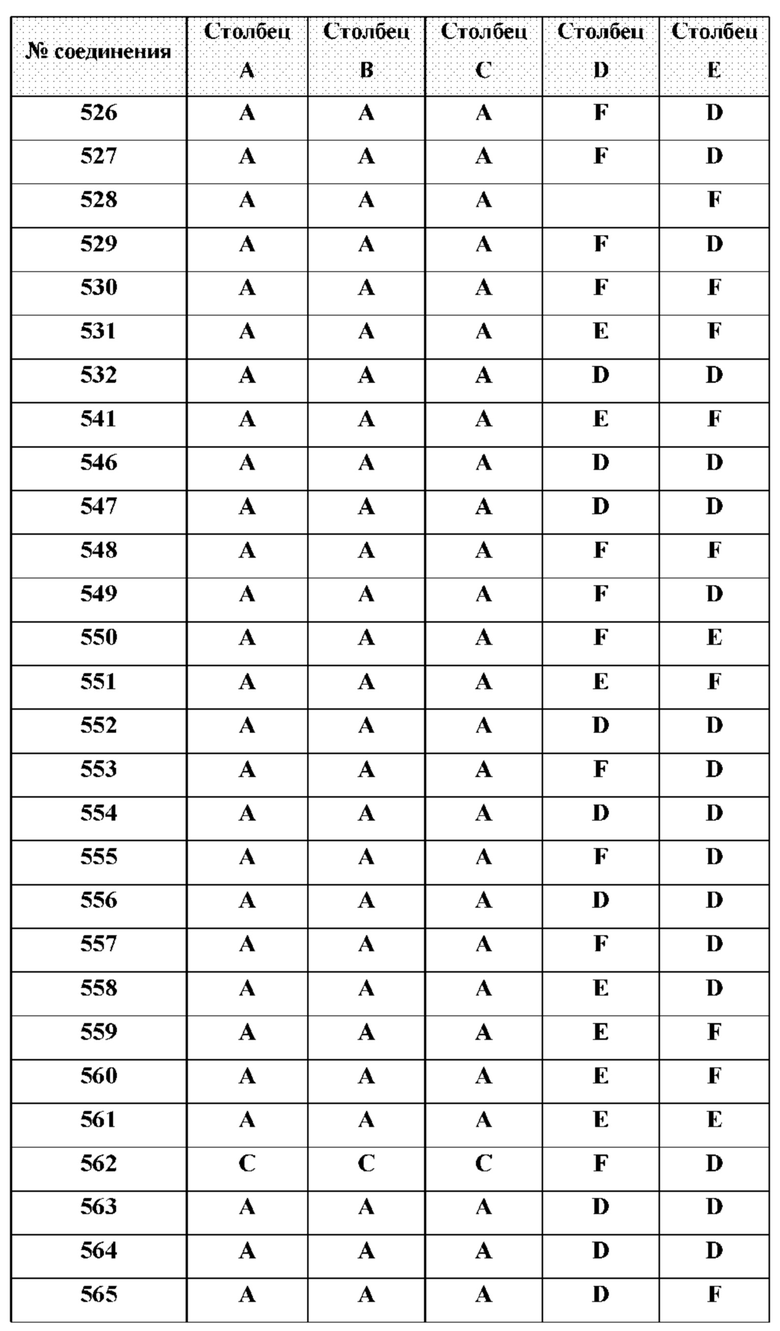
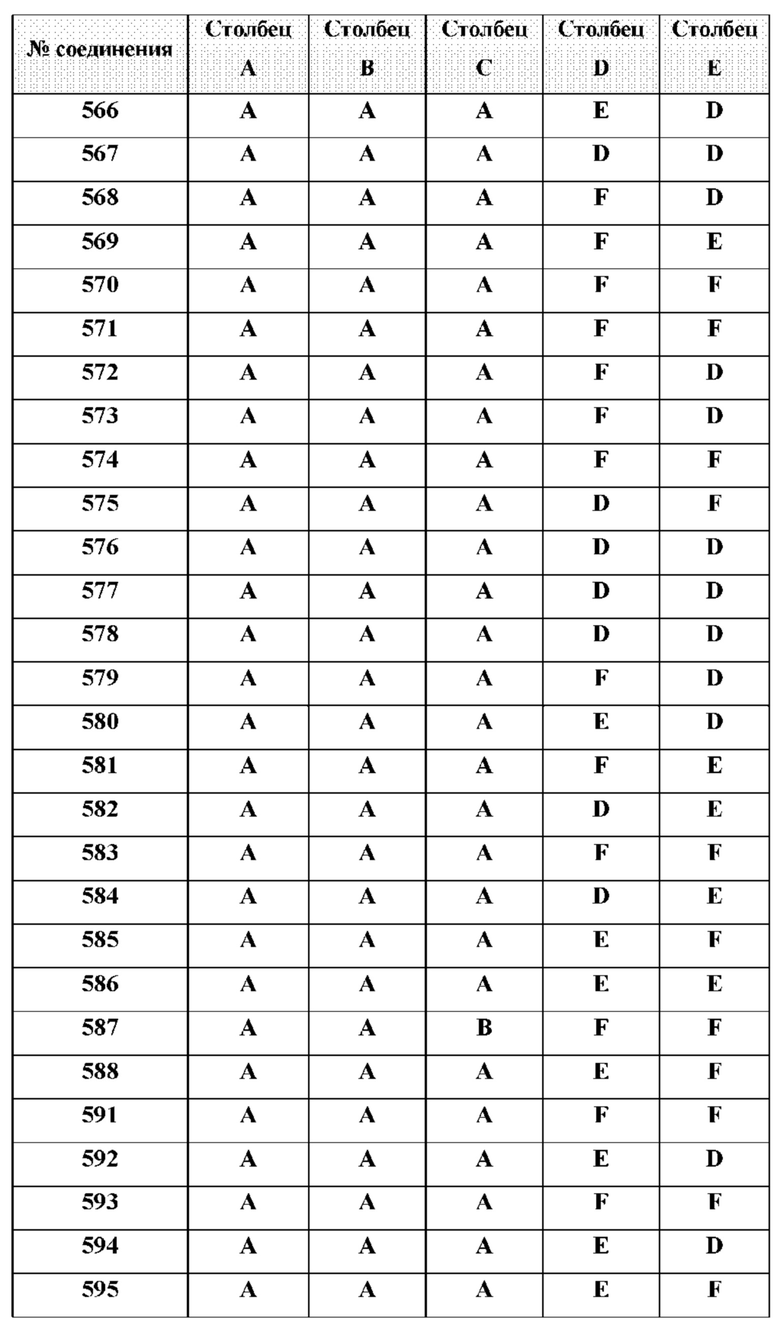
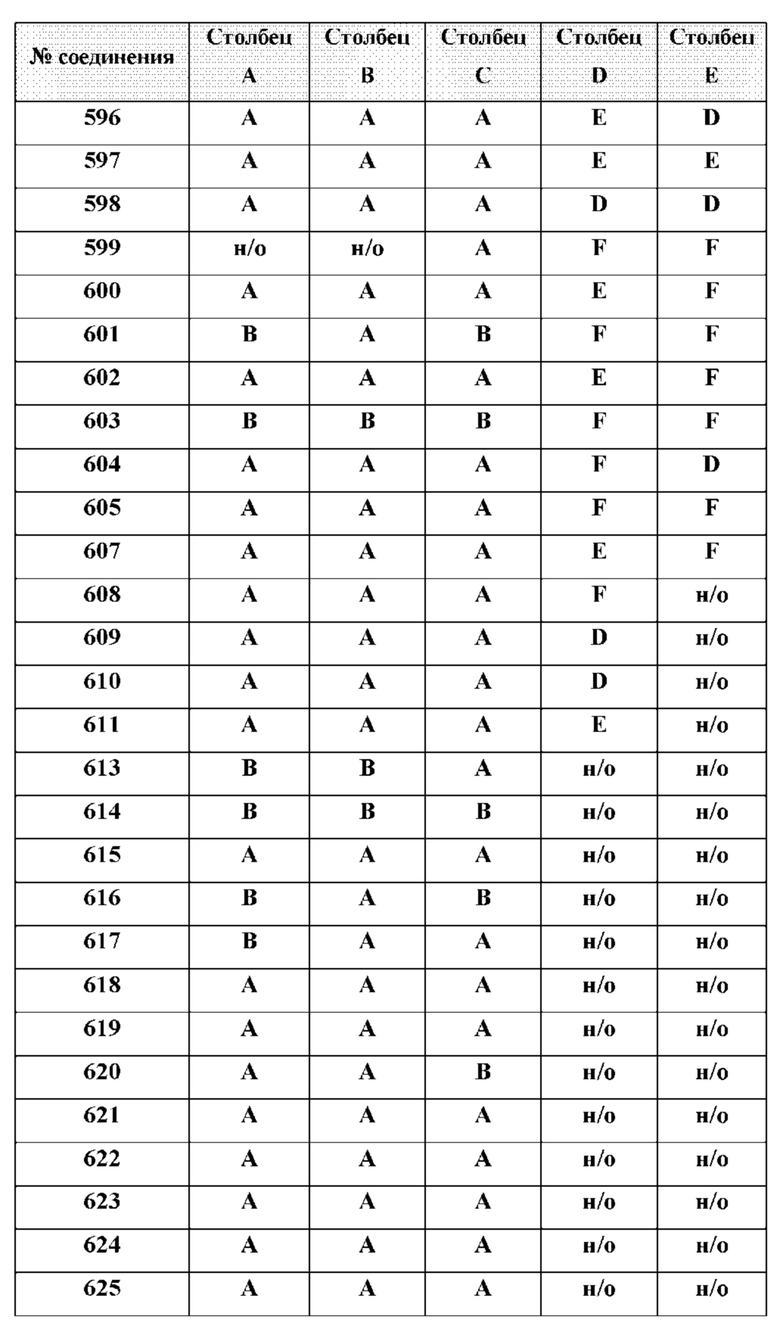
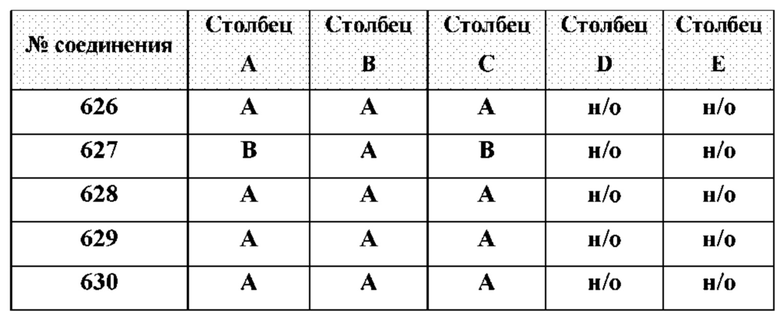
А: <3 мкМ;
В: 3-10 мкМ;
С: >10 мкМ;
D: <10 мкМ;
Е: 10-25 мкМ;
F: >25 мкМ
н/о: не определено
ПРИМЕР 236: МОДЕЛИ И ИССЛЕДОВАНИЯ НА ЖИВОТНЫХ
Индуцированный блеомицином фиброз легких у мышей или крыс
[1715] Способ индукции фиброза легких у мышей описан в источнике Current Protocols in Pharmacology: 5.46.1 под названием «Mouse Models of Bleomycin-induced Pulmonary Fibrosis)). Для индукции фиброза легких мышам С57В 1/6 или крысам Вистар в возрасте 6-8 недель однократно орофарингеально вводили ~1,5 МЕ/кг блеомицина сульфата (Calbiochem, Billerica, MA) в форме капель. Кратко, для орофарингеального введения блеомицина мышей или крыс подвергают анестезии с применением изофлурана и затем подвешивают на спине под углом ~60 градусов на наклонной поверхности с резиновой лентой, проходящей под верхними резцами. Дыхательные пути открывают, фиксируя язык с помощью одной бранши обернутого мягким материалом зажима, и вводят блеомицин с помощью шприца в заднюю часть полости рта. Язык и рот животного удерживали в открытом состоянии до тех пор, пока жидкость не исчезала из полости рта. Затем животное возвращали в клетку и наблюдали до полного восстановления после анестезии. Исследование прекращают на 14-28 день после орофарингеального введения блеомицина мышам и крысам.
ДАННЫЕ ОБ ЭФФЕКТИВНОСТИ IN VIVO
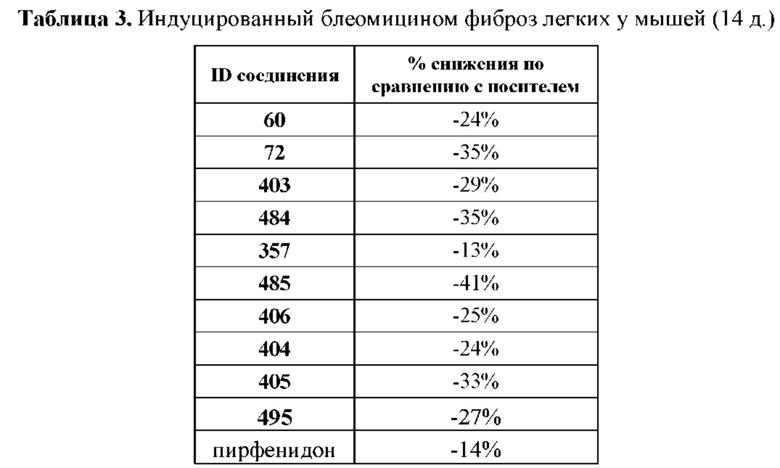
[1716] В качестве альтернативы, для системного введения блеомицина мышам с помощью осмотических насосов, указанные насосы наполняют блеомицином и имплантируют подкожно под изофлурановой анестезией, как описано в источнике Lee, Am J Physiol Lung Cell Mol Physiol, 2014. Кратко, мышам системно вводят ~50-100 МЕ/кг блеомицина (Blenoxane; Teva Pharma, North Wales, PA) через осмотические насосы в течение 7 дней. На 10 день осмотические насосы удаляют и продолжают исследование до 35 дня.
[1717] По окончании исследований всех животных умерщвляют путем смещения шейных позвонков для макроскопического исследования после вскрытия и собирают кровь путем пункции сердца. Легкие каждого животного отделяют от тела животного и взвешивают. Клетки BAL и жидкость собирают, дважды промывая легкие 0,5 мл сбалансированного солевого раствора Хенкса (HBSS; VWR, Radnor, РА). После сбора клеток BAL и жидкости легкие иссекают и удаляют у каждого животного. Целые легкие наполняют 10% NBF и затем фиксируют в 10% NBF для гистологии. Тяжесть фиброза в легких оценивают с использованием модифицированной шкалы Эшкрофта (Ashcroft score (Hubner, Biotechniques, 2008)) и субъективных показателей фиброза. Срезы легких оценивали путем усреднения 5 полей зрения микроскопа при 20-кратном увеличении по шкале Эшкрофта следующим образом: степень 0 = нормальное легкое; степень 1 = минимально определяемое утолщение альвеолярных стенок; степень 2 = умеренное утолщение альвеолярных стенок. Степень 3 = умеренное смежное утолщение стенок с фиброзными узелками; степень 4 = утолщенные септы и сплошные фиброзные образования, которые в целом составляют менее 10% поля зрения микроскопа. Степень 5 = повышенный фиброз с явным повреждением структуры легких и образованием фиброзных тяжей или небольших фиброзных образований, которые составляют от 10 до 50% поля зрения микроскопа; степень 6 = крупные сплошные фиброзные образования, затемняющие более 50% поля зрения микроскопа. Степень 7 = выраженная деформация структуры и крупные области фиброза; степень 8 = полная фиброзная облитерация легкого в пределах поля зрения микроскопа. Каждый срез исследовали при 20-кратном увеличении и оценку для 5 отдельных репрезентативных полей усредняли для каждого животного. Субъективные показатели (для окрашенных гематоксилином и эозином (Н&Е) срезов и срезов с трихромным окрашиванием) оценивали при 2-кратном увеличении для общей оценки патологического изменения. Оценка от 0 = отсутствие обнаруживаемых проявлений до 5 = полное вовлечение в консолидацию. Оценки в каждой группе усредняли и рассчитывали стандартную ошибку с помощью программного обеспечения Excel 2010. Плотные, организованные воспалительные экссудаты оценивали как фиброз. Другие ткани исследовали с помощью микроскопии и оценивали обычным образом.
Индуцированный четыреххлористым углеродом фиброз печени у мышей или крыс
[1718] Индуцированный четыреххлористым углеродом фиброз печени является широко используемой и общепринятой моделью для оценки новых способов лечения фиброза. Способы индукции фиброза печени путем введения четыреххлористого углерода описаны в источниках Lee, J Clin Invest, 1995 и Tsukamoto, Semin Liver Dis, 1990. Кратко, самцам мышей C57BL/6 вводят 1 мг/кг четыреххлористого углерода (Sigma Aldrich, разбавленный 1:7 в кукурузном или оливковом масле) путем внутрибрюшинной инъекции два раза в неделю в течение 4 недель. Мышей умерщвляют на 28-й день. В альтернативном варианте реализации крысам Вистар вводят четыреххлористый углерод путем внутрибрюшинной инъекции три раза в неделю в течение 8-12 недель. Крыс умерщвляют по окончании эксперимента, через 8-12 недель после начала исследования.
[1719] Кровь собирают путем пункции сердца и получают сыворотку для оценки ферментов печени (включая АЛТ, ACT, ЩФ и так далее) в несколько моментов времени в течение всего исследования и по окончании исследования. Ткани печени от всех животных собирают и фиксируют путем погружения в 10% нейтральный забуференный формалин, обрабатывают, заливают парафином, изготавливают срезы, получают препараты и окрашивают методом трихромного окрашивания по Массону (Tri) или пикросириусом красным (PSR) с использованием стандартных гистологических методов оценки тяжести фиброза.
Модель фиброза почки с односторонней обструкцией мочеточника у мышей
[1720] Самкам мышей C57BL/6 (Harlan, возраст 4-6 недель) предоставляют свободный доступ к пище и воде и оставляют для акклиматизации в течение по меньшей мере 7 дней, затем начинают исследование. После акклиматизации мышей подвергают анестезии и выполняют операцию по односторонней обструкции мочеточника (ООМ) или имитацию операции на левой почке. Кратко, выполняют продольный разрез в верхней левой части для обнажения левой почки. Определяют местонахождение почечной артерии и пропускают шелковую нить 6/0 между артерией и мочеточником. Нить обвивают вокруг мочеточника и трижды завязывают, обеспечивая полное лигирование мочеточника. Почку возвращают в брюшную полость, мышцу брюшной стенки ушивают и скрепляют кожу. Всех животных умерщвляют через 4, 8, 14, 21 или 28 дней после операции ООМ. После умерщвления кровь собирают путем пункции сердца, забирают почки, и одну половину почки замораживают при -80°С, а другую половину фиксируют в 10% нейтральном забуференном формалине для гистопатологической оценки фиброза почки.
Модель индуцированного блеомицином кожного фиброза
[1721] Блеомицин (Calbiochem, Billerica MA) растворяют в фосфатно-солевом буфере (ФСБ) в концентрации 10 мкг/мл и стерилизуют с помощью фильтрации. Блеомицин или контроль - ФСБ вводят путем подкожной инъекции в два участка выбритой поверхности спины мышей C57/BL6 или S129 (Charles River/Harlan Labs, 20-25 г) один раз в день в течение 28 дней под изофлурановой анестезией (5% в 100% 02). Через 28 дней мышей умерщвляют и из каждого места инъекции получают пункционные биопсии толщиной 6 мм. Кожный фиброз оценивают с помощью стандартного гистопатологического анализа и биохимического определения гидроксипролина.
ПРИМЕР 237: НАЦЕЛИВАНИЕ НА КАЛЬПАИНЫ
Подавление ЕрМТ
[1722] Для оценки ЕМТ in vitro клетки NMuMG (АТСС) выращивают до достижения конфлюэнтности в среде для роста (среда Игла в модификации Дульбекко с добавлением 10 мкг/мл инсулина) с добавлением 10% сыворотки (эмбриональной телячьей сыворотки) и затем проводят старвацию в течение 24 часов в среде с добавлением 0,5% сыворотки +/- лекарственные ингибиторы. Затем клетки обрабатывают рекомбинантным TGFb1 человека (R&D Systems, 5 нг/мл) +/- лекарственные ингибиторы в среде с добавлением 0,5% сыворотки. В моменты времени, превышающие 24 ч, упомянутую выше среду заменяют на свежую каждые 24 часа. Клеточные лизаты анализировали в отношении экспрессии белка aSMA с помощью вестерн-блоттинга.
[1723] Miettinen et al. (1994). «TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors.» J Cell Biol 127(6 Pt 2):2021-36.
[1724] Lamouille et al. (2014). «Molecular mechanisms of epithelial-mesenchymal transition.» Nat Rev Mol Cell Biol 15(3): 178-96.
[1725] Для оценки FMT in vitro клетки - нормальные фибробласты легких человека (NHLF) (Lonza) выращивали в среде для роста фибробластов-2 (Lonza СС-3131/с СС-4126 bullet kit) и затем проводили старвацию в течение 24 ч в основной среде для фибробластов-2 (Fibroblast Basal Media-2) (Lonza CC-3131) без добавления сыворотки/факторов роста +/- лекарственные ингибиторы. Затем клетки обрабатывали TGFb1 (5 нг/мл) в основной среде для фибробластов +/- лекарственные ингибиторы. Клеточные лизаты анализируют в отношении экспрессии белка aSMA с помощью вестерн-блоттинга.
[1726] Дополнительные подробности приведены в источнике Pegorier et al. (2010). «Вопе Morphogenetic Protein (BMP)-4 and BMP-7 regulate differentially Transforming Growth Factor (TGF)-B1 in normal human lung fibroblasts (NHLF)» Respir Res 11:85, полное содержание которого включено в настоящее описание посредством ссылки.
ПРИМЕР 238: ЛЕЧЕНИЕ ЧЕЛОВЕКА
[1727] Оценивают эффективность лечения с применением соединения согласно предпочтительному варианту реализации по сравнению с плацебо у пациентов с идиопатическим фиброзом легких (ИФЛ) и безопасность лечения с применением соединения согласно предпочтительному варианту реализации по сравнению с плацебо у пациентов с ИФЛ. Главной переменной результата (primary outcome variable) является абсолютное изменение прогнозируемой форсированной жизненной емкости легких (ФЖЕЛ) в процентах от исходного уровня к 52 неделе. Другие возможные конечные точки могут включать, но не ограничиваются перечисленными: смертность, выживаемость без прогрессирования, изменение скорости снижения ФЖЕЛ, изменение Sp02 и изменение биомаркеров (анализ изображений КТВР; молекулярные и клеточные маркеры активности заболевания). Вторичные показатели эффективности включают: комбинированные показатели важных событий, связанных с ИФЛ; выживаемость без прогрессирования; уровень смертности от любой причины; уровень смертности от ИФЛ; категориальную оценку абсолютного изменения прогнозируемой ФЖЕЛ в процентах от исходного уровня к 52 неделе; изменение одышки от исходного уровня к 52 неделе; изменение прогнозируемой диффузионной способности легких для монооксида углерода (DLco) с поправкой на концентрацию гемоглобина (Hb) в процентах от исходного уровня к 52 неделе; изменение насыщения кислородом во время теста с 6-минутной ходьбой (6MWT) от исходного уровня к 52 неделе; изменение оценки компьютерной томографии высокого разрешения (КТВР) от исходного уровня к 52 неделе; изменение пройденного расстояния в 6MWT от исходного уровня к 52 неделе. Пациенты, имеющие право на участие в этом исследовании, включают, но не ограничиваются перечисленными: пациенты, удовлетворяющие следующим критериям включения: диагноз ИФЛ; возраст от 40 до 80 лет; прогнозируемое значение ФЖЕЛ 50%; прогнозируемое значение DLco35%; прогнозируемое значение либо ФЖЕЛ, либо DLco90%; отсутствие улучшения в прошлом году; отношение объема форсированного выдоха за 1 секунду (FEV1) к ФЖЕЛ составляет 0,80 или более; способны пройти 150 метров за 6 минут и поддерживать насыщение 83% при дополнительном расходе кислорода не более 6 л/мин. Пациентов исключают из этого исследования, если они удовлетворяют любому из следующих критериев: неспособны пройти тестирование функции легких; проявления значительного обструктивного заболевания легких или гиперчувствительности дыхательных путей; согласно клиническому мнению исследователя ожидается, что пациент будет нуждаться в пересадке легкого и соответствовать ее критериям в течение 52 недель после рандомизации; активная инфекция; заболевание печени; рак или другое клиническое состояние, которое может привести к смерти в течение 2 лет; диабет; беременность или лактация; злоупотребление психоактивными веществами; личный или семейный анамнез синдрома удлинения интервала QT; другое лечение ИФЛ; неспособность принимать исследуемое лекарственное средство; досрочное исключение из других исследований ИФЛ. Пациентам перорально вводят либо плацебо, либо дозу соединения согласно предпочтительному варианту реализации (1 мг/день-1000 мг/день). Главной переменной результата будет являться абсолютное изменение прогнозируемой ФЖЕЛ в процентах от исходного уровня к 52 неделе. Пациенты будут получать лечение с применением замаскированного исследуемого лекарственного средства с момента рандомизации до тех пор, пока последний рандомизированный пациент не получит лечение в течение 52 недель. Физические и клинические лабораторные оценки будут проводиться через определенные промежутки времени во время лечения, например, на 2, 4, 8, 13, 26, 39 и 52 неделе. Функцию легких, толерантность к физической нагрузке и одышку будут оценивать через определенные промежутки времени во время лечения, например, на 13, 26, 39 и 52 неделе. Комитет по мониторингу данных (DMC) будет периодически анализировать данные о безопасности и эффективности для обеспечения безопасности пациентов.
50%; прогнозируемое значение DLco35%; прогнозируемое значение либо ФЖЕЛ, либо DLco90%; отсутствие улучшения в прошлом году; отношение объема форсированного выдоха за 1 секунду (FEV1) к ФЖЕЛ составляет 0,80 или более; способны пройти 150 метров за 6 минут и поддерживать насыщение 83% при дополнительном расходе кислорода не более 6 л/мин. Пациентов исключают из этого исследования, если они удовлетворяют любому из следующих критериев: неспособны пройти тестирование функции легких; проявления значительного обструктивного заболевания легких или гиперчувствительности дыхательных путей; согласно клиническому мнению исследователя ожидается, что пациент будет нуждаться в пересадке легкого и соответствовать ее критериям в течение 52 недель после рандомизации; активная инфекция; заболевание печени; рак или другое клиническое состояние, которое может привести к смерти в течение 2 лет; диабет; беременность или лактация; злоупотребление психоактивными веществами; личный или семейный анамнез синдрома удлинения интервала QT; другое лечение ИФЛ; неспособность принимать исследуемое лекарственное средство; досрочное исключение из других исследований ИФЛ. Пациентам перорально вводят либо плацебо, либо дозу соединения согласно предпочтительному варианту реализации (1 мг/день-1000 мг/день). Главной переменной результата будет являться абсолютное изменение прогнозируемой ФЖЕЛ в процентах от исходного уровня к 52 неделе. Пациенты будут получать лечение с применением замаскированного исследуемого лекарственного средства с момента рандомизации до тех пор, пока последний рандомизированный пациент не получит лечение в течение 52 недель. Физические и клинические лабораторные оценки будут проводиться через определенные промежутки времени во время лечения, например, на 2, 4, 8, 13, 26, 39 и 52 неделе. Функцию легких, толерантность к физической нагрузке и одышку будут оценивать через определенные промежутки времени во время лечения, например, на 13, 26, 39 и 52 неделе. Комитет по мониторингу данных (DMC) будет периодически анализировать данные о безопасности и эффективности для обеспечения безопасности пациентов.
Иллюстративное исследование при СС
[1728] Оценивают эффективность лечения с применением соединения согласно предпочтительному варианту реализации по сравнению с плацебо у пациентов с системным склерозом (СС) и безопасность лечения с применением соединения согласно предпочтительному варианту реализации по сравнению с плацебо у пациентов с СС. Главной переменной результата является абсолютное изменение модифицированной оценки кожи по Роднану (Modified Rodnan Skin Score (mRSS)) в процентах от исходного уровня к 48 неделе. Другие возможные конечные точки могут включать, но не ограничиваются перечисленными: смертность, процент пациентов с возникшими на фоне лечения нежелательными явлениями (НЯ) и серьезными нежелательными явлениями (СНЯ), комбинированная оценка прогрессирования заболевания и изменение биомаркеров (молекулярные и клеточные маркеры активности заболевания, такие как С-реактивный белок). Вторичные показатели эффективности включают, но не ограничиваются перечисленными: показатель согласно опроснику для оценки состояния здоровья больного склеродермией (Scleroderma Health Assessment Questionnaire (SHAQ)); индекс инвалидизации согласно опроснику для оценки состояния здоровья (Health Assessment Questionnaire Disability Index (HAQ-DI)); оценка по шкале функциональной оценки терапии хронического заболевания - подшкале утомляемости (Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT)); тяжесть зуда, определенная с помощью стандартной шкалы, такой как шкала оценки тяжести зуда по 5 показателям (5-D Itch Scale); оценка согласно опроснику госпиталя Святого Георга для оценки дыхательной функции (St. George's Respiratory Questionnaire (SGRQ)); число болезненных суставов из 28 (ЧБС28); показатели функции легких; стандартные показатели жизнедеятельности (включая кровяное давление, частоту сердечных сокращений и температуру); показатели электрокардиограммы (ЭКГ); лабораторные исследования (клиническая химия, гематология и анализ мочи); показатели фармакокинетики (ФК). Включенные в указанные исследования и дополнительные клинические образцы и образцы для определения биомаркеров, такие как биопсия кожи и кровь (или сыворотка и/или плазма), также будут собраны до начала лечения. Кроме того, пациенты, имеющие право на участие в этом исследовании, включают, но не ограничиваются перечисленными, пациентов, удовлетворяющих следующим критериям: пациенты в возрасте по меньшей мере 18 лет; имеющие диагноз СС в соответствии с критериями Американской коллегии ревматологов (American College of Rheumatology (ACR)) и Европейской лиги по борьбе с ревматизмом (European League Against Rheumatism (EULAR)), удовлетворяющие критериям активного заболевания и имеющие общую продолжительность заболевания 60 месяцев или менее; 10 mRSS35. Пациентов исключают из этого исследования, если они удовлетворяют любому из следующих критериев: крупное хирургическое вмешательство в период 8 недель до скрининга; склеродермия ограничена дистальной областью локтей или колен; ревматическое аутоиммунное заболевание, отличное от СС; применение любой исследуемой, биологической или иммуносупрессивной терапии, включая внутрисуставное или парентеральное введение кортикостероидов в течение 4 недель до скрининга. Пациентам перорально вводят либо плацебо, либо дозу соединения согласно предпочтительному варианту реализации (1 мг/день-1000 мг/день). Главной переменной результата будет являться абсолютное изменение mRSS \от исходного уровня к 48 неделе. Пациенты будут получать лечение с применением замаскированного исследуемого лекарственного средства с момента рандомизации до тех пор, пока последний рандомизированный пациент не получит лечение в течение 48 недель. Физические и клинические лабораторные оценки будут проводиться через определенные промежутки времени во время лечения, например, на 2, 4, 8, 12, 24, 36 и 48 неделе. Клинические образцы и образцы для определения биомаркеров также будут собраны на 48 неделе. Комитет по мониторингу данных (DMC) будет периодически анализировать данные о безопасности и эффективности для обеспечения безопасности пациентов.
mRSS35. Пациентов исключают из этого исследования, если они удовлетворяют любому из следующих критериев: крупное хирургическое вмешательство в период 8 недель до скрининга; склеродермия ограничена дистальной областью локтей или колен; ревматическое аутоиммунное заболевание, отличное от СС; применение любой исследуемой, биологической или иммуносупрессивной терапии, включая внутрисуставное или парентеральное введение кортикостероидов в течение 4 недель до скрининга. Пациентам перорально вводят либо плацебо, либо дозу соединения согласно предпочтительному варианту реализации (1 мг/день-1000 мг/день). Главной переменной результата будет являться абсолютное изменение mRSS \от исходного уровня к 48 неделе. Пациенты будут получать лечение с применением замаскированного исследуемого лекарственного средства с момента рандомизации до тех пор, пока последний рандомизированный пациент не получит лечение в течение 48 недель. Физические и клинические лабораторные оценки будут проводиться через определенные промежутки времени во время лечения, например, на 2, 4, 8, 12, 24, 36 и 48 неделе. Клинические образцы и образцы для определения биомаркеров также будут собраны на 48 неделе. Комитет по мониторингу данных (DMC) будет периодически анализировать данные о безопасности и эффективности для обеспечения безопасности пациентов.
ПРИМЕР 239: СВЯЗЫВАНИЕ С КАЛЫ1АИНОМ
[1729] Получение белка калъпаина 9: содержащий на N-конце гексагистидиновую метку кальпаин 9 человека, остатки 27-347, был сверхэкспрессирован в Е. coli BL21 (DE3) и очищен с помощью Ni-NTA и эксклюзионной хроматографии с выходом >10 мг/литр культуры и чистотой >99%. Последовательность кальпаина 9 имеет код O14815 в базе данных uniprot.org.
[1730] Кристаллизация калъпаина 9: раствор кальпаина 1 крысы или кальпаина 9 человека с концентрацией 10 мг/мл (полученных, как описано выше) готовили в 50 мМ Трис (рН 8), 0,5 М NaCl, 1 мМ ЭДТА, 1 мМ CaCl2 и 1 мМ бета-меркаптоэтаноле против среды для скрининга MCSG1 С2 (Anatrace, Maumee, ОН), содержащей 0,2 М LiSO4, 0,1 М Бис-трис (рН 5,5) и 25% ПЭГ 3350. Раствор содержал 2,5 мМ исследуемого соединения с добавлением 20% этиленгликоля в качестве криопротектора. Связанный белок кристаллизовали путем диффузии паров, методом сидячей капли, при примерно 15,85°С (289°К). Группа симметрии имела характеристики Р 41 21 2 с параметрами элементарной ячейки 97,2  , 97,2 , 173,4 .
, 97,2 , 173,4 .
[1731] Получение и уточнение кристаллографических данных кальпаина 9: Данные получали на синхротроне APS с пучком излучения 21-ID-F при примерно -173,15°С (100°K). Отражения получали в диапазоне 45,0-2,1 с полнотой 100%. Данные отражений анализировали с помощью XDS и Xscale (Heidelberg, Germany). Разрешение структуры проводили методом молекулярного замещения и уточняли с помощью PHENIX (Berkeley, СА) до значения R 0,168, Rfree=0,210.
[1732] Моделирование кальпаина 9: Экспериментальные расстояния, определенные на основе кристаллических структур, были дополнены расчетными моделями связывания кальпаина 9. Для получения начальных координат использовали структуру кристалла кальпаин 9/исследуемое соединение. Ее освобождали от влияния растворителя и минимизировали с помощью МОЕ 2016.08 (CCG, Montreal) с использованием стандартного протокола quickprep и силового поля Amber10:EHT. Различные исследуемые соединения затем подвергали минимизации в активном центре. Полученные расстояния продемонстрировали хорошее соответствие с доступными данными кристаллов.
[1733] Получение, кристаллизация кальпаина 1 и получение данных: Подходящие конструкции для экспрессии кальпаина 1 крысы были получены ранее. Экспрессию выполняли в соответствии с ранее разработанными протоколами. Был разработан протокол очистки и получен гомогенный белок в препаративных количествах. Проводили очистку белка кальпаина 1, включая стадии аффинной и гельфильтрационной хроматографии. Эта методика обеспечивала получение гомогенного белка с чистотой более 95% по данным окрашивания геля Кумасси после электрофореза в ПААГ с добавлением ДНС. Очищенный белок использовали в анализе кристаллизации с исследуемыми соединениями с применением как стандартного скрининга с примерно 1200 различными условиями, так и условий кристаллизации, определенных с использованием данных литературы. Первоначально полученные условия были оптимизированы с использованием стандартных стратегий, систематически изменяющих параметры, критически влияющие на кристаллизацию, такие как температура, концентрация белка, отношения капель и другие. Эти условия также были уточнены путем систематического изменения рН или концентраций осадителя. Кристаллы мгновенно замораживали и измеряли при температуре примерно -173,15°С (100°K). Данные рентгеновской дифракции получали на основе кристаллов комплексов с лигандами на источнике синхротронного излучения с применением криогенных условий. Данные обрабатывали с использованием программного обеспечения XDS и XSCALE.
[1734] Были определены ключевые взаимодействия между фрагментами исследуемого соединения (со ссылкой на переменные формулы II) и остатками кальпаина 1 или кальпаина 9, приведенные ниже в таблицах 4-7.
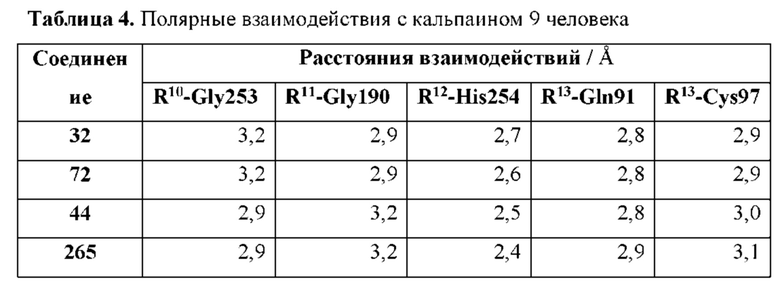
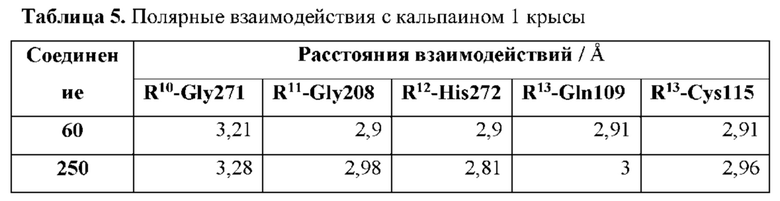
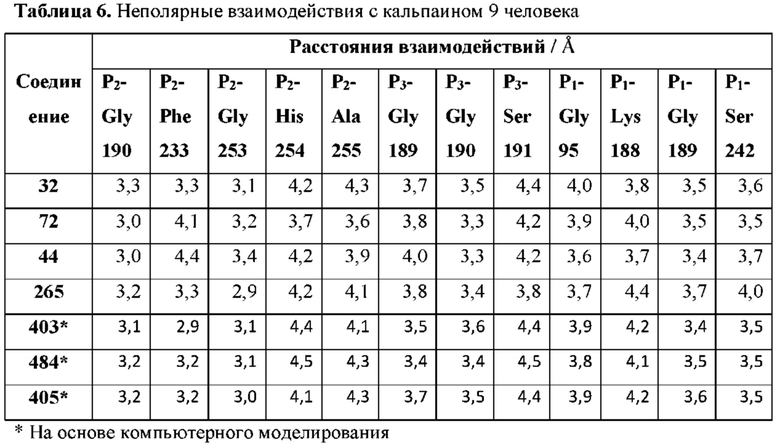
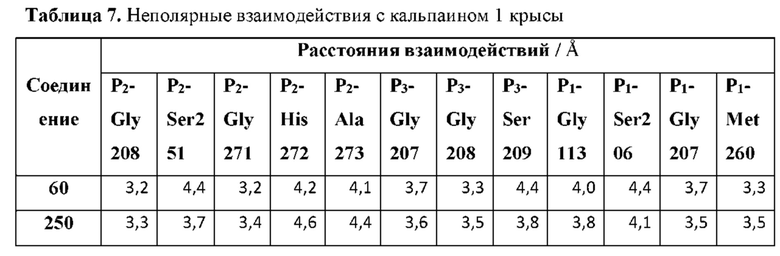
[1735] Несмотря на то что были проиллюстрированы и описаны некоторые варианты реализации, средний специалист в данной области техники после прочтения приведенного выше описания может произвести изменения, замены эквивалентов и другие типы изменений соединений согласно настоящей технологии или их солей, фармацевтических композиций, производных, пролекарств, метаболитов, таутомеров или рацемических смесей, указанных в настоящем описании. Каждый аспект и вариант реализации, описанный выше, также может включать или объединять такие вариации или аспекты, которые раскрыты в отношении любого или всех других аспектов и вариантов реализации.
[1736] Настоящая технология также не должна быть ограничена с точки зрения конкретных аспектов, описанных в настоящем описании, которые служат в качестве отдельных иллюстраций отдельных аспектов настоящей технологии. Многочисленные модификации и вариации этой настоящей технологии могут быть выполнены без отступления от ее сущности и объема, что будет очевидно для специалистов в данной области техники. Функционально эквивалентные способы в пределах объема настоящей технологии, помимо перечисленных в настоящем описании, будут очевидны для специалистов в данной области техники из приведенных выше описаний. Подразумевается, что такие модификации и вариации входят в объем прилагаемой формулы изобретения. Следует понимать, что настоящая технология не ограничивается конкретными способами, реагентами, соединениями, композициями, мечеными соединениями или биологическими системами, которые, безусловно, могут различаться. Также следует понимать, что используемая в контексте настоящего описания терминология предназначена только для описания конкретных аспектов и не предназначена для ограничения. Таким образом, предполагается, что настоящее описание будет рассматриваться как иллюстративное только с учетом широты, объема и сущности настоящей технологии, указанных только в прилагаемой формуле изобретения, ее определениях и любых их эквивалентах.
[1737] Варианты реализации, иллюстративно описанные в настоящем описании, могут подходящим образом применяться на практике в отсутствие какого-либо элемента или элементов, ограничения или ограничений, конкретно не обозначенных в настоящем описании. Таким образом, например, термины «включающий», «включая», «содержащий» и так далее следует понимать в широком смысле и без ограничения. Кроме того, используемые в настоящем описании термины и выражения использовались в качестве терминов описания, а не ограничения, и при использовании таких терминов и выражений нет намерения исключать какие-либо эквиваленты описанных и представленных признаков или их частей, но признается, что возможны различные модификации в пределах объема заявленной технологии. Кроме того, фраза «по существу состоящий из» будет пониматься как включающая те элементы, которые конкретно указаны, и те дополнительные элементы, которые не оказывают существенного влияния на основные и новые характеристики заявленной технологии. Фраза «состоящий из» исключает любой не указанный элемент.
[1738] Помимо этого, в случае, когда признаки или аспекты настоящего описания описаны посредством групп Маркуша, специалистам в данной области техники будет понятно, что настоящее описание также будет описано посредством любого отдельного элемента или подгруппы элементов группы Маркуша. Каждый из более узких видов и внутренних групп, подпадающих под общее описание, также является частью настоящей технологии. Это включает общее описание настоящей технологии с условием или отрицательным ограничением, исключающим что-либо из рода объектов, независимо от того, был ли исключенный объект специально изложен в настоящем описании.
[1739] Содержание всех публикаций, патентных заявок, выданных патентов и других документов (например, журналов, статей и/или пособий), упомянутых в настоящем описании, включено в настоящее описание посредством ссылки, как если бы полное содержание каждой отдельной публикации, патентной заявки, выданного патента или другого документа было конкретно и отдельно обозначено как включенное посредством ссылки. Определения, которые содержатся в источнике, включенном посредством ссылки, исключаются в той степени, в которой они противоречат определениям в настоящем описании.
[1740] Другие варианты реализации приведены в следующей формуле изобретения совместно с полным объемом эквивалентов, на которые распространяется действие указанной формулы изобретения.
[1741] Несмотря на то, что настоящее изобретение было конкретно представлено и описано со ссылкой на предпочтительный вариант реализации и различные альтернативные варианты реализации, специалистам в данной области техники будет понятно, что могут быть выполнены различные изменения в форме и деталях без отступления от сущности и объема настоящего изобретения.
[1742] Все источники, выданные патенты и патентные заявки, упомянутые в основной части настоящего описания, полностью включены в настоящее описание посредством ссылки для всех целей.
[1743] Несмотря на то что настоящее изобретение было описано со ссылкой на варианты реализации и примеры, следует понимать, что могут быть выполнены многочисленные и различные модификации без отступления от сущности настоящего изобретения. Соответственно, настоящее изобретение ограничено только лишь следующей формулой изобретения.
СПИСОК ЛИТЕРАТУРЫ
1. Патент США №5,145,684.
2. Goll et al. (2003). "The calpain system." Physiol Rev 83(3): 731-801.
3. Schad et al. (2002). "A novel human small subunit calpains." Biochem J 362(Pt 2): 383-8.
4. Ravulapalli et al. (2009). "Distinguishing between calpain heterodimerization and homodimerization." FEBS J 276(4): 973-82.
5. Dourdin et al. (2001). "Reduced cell migration and disruption of the actin cytoskeleton in calpain-deficient embryonic fibroblasts." J Biol Chem 276(51): 48382-8.
6. Leloup et al. (2006). "Involvement calpains in growth factor-mediated migration." Int J Biochem Cell Biol 38(12): 2049-63.
7. Janossy et al. (2004). "Calpain as a multi-site regulator of cell cycle." Biochem Pharmacol 67(8): 1513-21.
8. Santos et al. (2012). "Distinct regulatory functions of calpain 1 and 2 during neural stem cell self-renewal and differentiation." PLoS One 7(3): e33468.
9. Miettinen et al. (1994). "TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors." J Cell Biol 127(6 Pt 2): 2021-36.
10. Lamouille et al. (2014). "Molecular mechanisms of epithelial-mesenchymal transition." Nat Rev Mol Cell Biol 15(3): 178-96.
11. Pegorier et al. (2010). "Bone Morphogenetic Protein (BMP)-4 and BMP-7 regulate differentially Transforming Growth Factor (TGF)-B1 in normal human lung fibroblasts (NHLF)" Respir Res 11:85.
| название | год | авторы | номер документа |
|---|---|---|---|
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| 2-ЦИАНОПИРИМИДИН-4-ИЛКАРБАМАТ, ИЛИ ПРОИЗВОДНОЕ МОЧЕВИНЫ, ИЛИ ЕГО СОЛЬ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2788740C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| ПРОТИВОФИБРОЗНЫЕ ПИРИДИНОНЫ | 2015 |
|
RU2692485C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ИХ ПРИМЕНЕНИЕ ПРОТИВ МИКОБАКТЕРИЙ | 2015 |
|
RU2664587C1 |
| Соединения С,O-спиро-арил-гликозидов, их приготовление и их использование | 2016 |
|
RU2746858C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| 10H-ФЕНОТИАЗИНОВЫЕ ИНГИБИТОРЫ ФЕРРОПТОЗА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2787366C2 |
Изобретение относится к соединениям, имеющим структуру, выбранную из формул I-a, I-b, I-f, I-g и I-h, или их фармацевтически приемлемым солям, которые обладают модулирующей активностью в отношении кальпаинов CAPN1, CAPN2 и CAPN9. В указанных формулах A2 выбран из группы, состоящей из C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, и одинарной связи; A4 выбран из группы, состоящей из C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, -(CR2)n-S-(CR2)n-, -(CR2)n-S(=O)-(CR2)n-, -(CR2)n-SO2-(CR2)n-, -(CR2)n-O-(CR2)n-, -(CR2)n-C(=S)-(CR2)n-, -(CR2)n-C(=O)-(CR2)n-, -(CR2)n-CH=CH-(CR2)n- и т.д., где каждый R представляет собой -H; A3 выбран из группы, состоящей из необязательно замещенного C6-10 арила, необязательно замещенного 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, необязательно замещенного 3-10-членного гетероциклила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, и необязательно замещенного C3-10 карбоциклила и т.д.; A5 представляет собой C1-4 алкил; A6 выбран из группы, состоящей из C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, C1-4 алкила и C2-4 алкенила; A7 представляет собой одинарную связь; R1 выбран из группы, состоящей из H и -CONR2R3; каждый R2 и R3 независимо выбран из -H, необязательно замещенного C1-4 алкила, 2-5-членного полиэтиленгликоля, C3-7 карбоциклила и необязательно замещенного C6-10 арил(C1-C6)алкила; R6 представляет собой -H; каждый n независимо выбран из целых чисел от 0 до 3; каждый из A и Z независимо выбран из группы, состоящей из C(R4) и N; когда соединение имеет структуру формулы I-a, B и D представляют собой C(R4); когда соединение имеет структуру формулы I-b, B выбран из группы, состоящей из C(R4) и N, и D представляет собой C(R4); каждый R4 независимо выбран из группы, состоящей из -H, C1-4 алкила, C1-4 галогеналкила, C3-7 карбоциклила, галогена, гидрокси и C1-C3 алкокси; когда соединение имеет структуру формулы I-f, Y выбран из группы, состоящей из NR5, O, S и SO2, и X выбран из группы, состоящей из C(R4) и N; когда соединение имеет структуру формулы I-g или формулы I-h, Y выбран из группы, состоящей из NR5 и S, и X представляет собой C(R4), или Y выбран из O и SO2 и X выбран из группы, состоящей из C(R4) и N; R5 выбран из группы, состоящей из -H, C1-4 алкила, C1-4 галогеналкила и C3-7 карбоциклила. Изобретение относится также к конкретным соединениям, фармацевтической композиции, содержащей указанные соединения, способу лечения фиброзного заболевания с их использованием, а также к их применению для лечения фиброзного заболевания. 7 н. и 53 з.п. ф-лы, 7 табл., 239 пр.
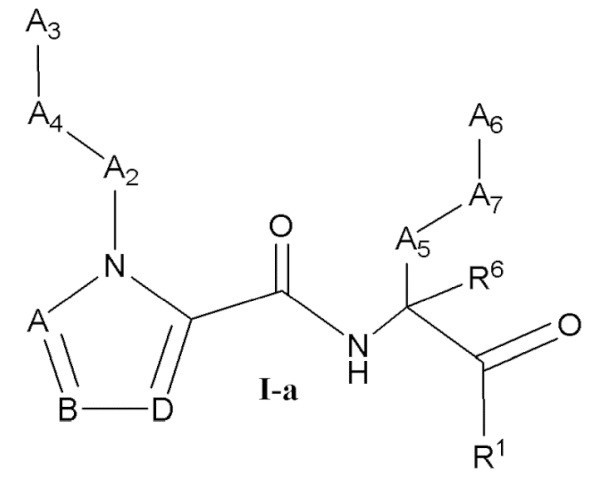
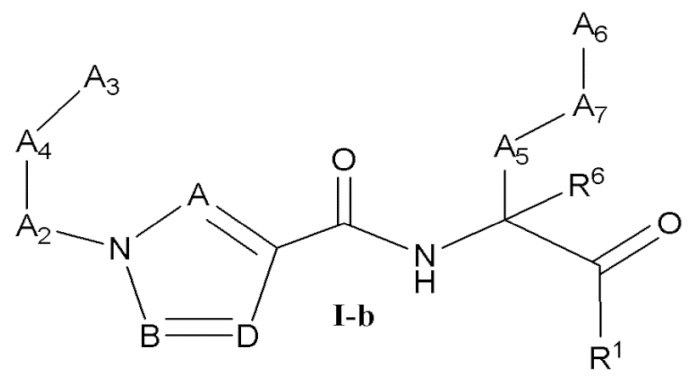
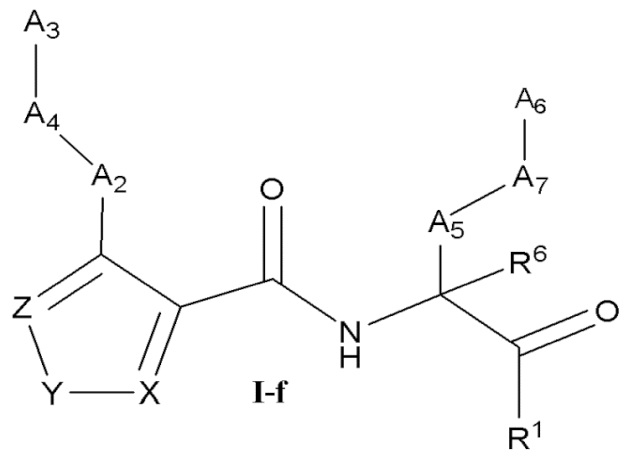
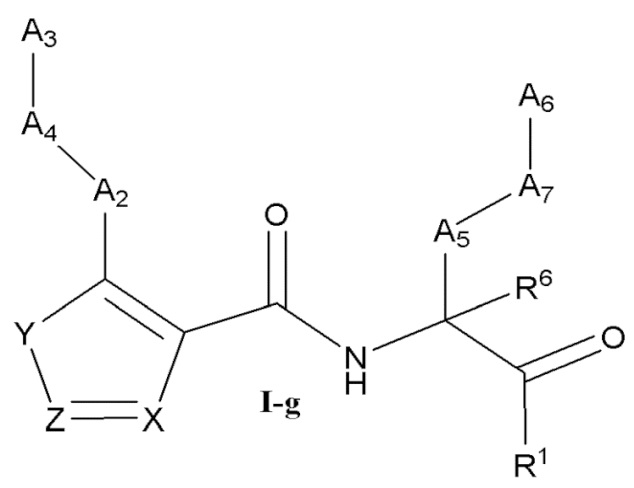
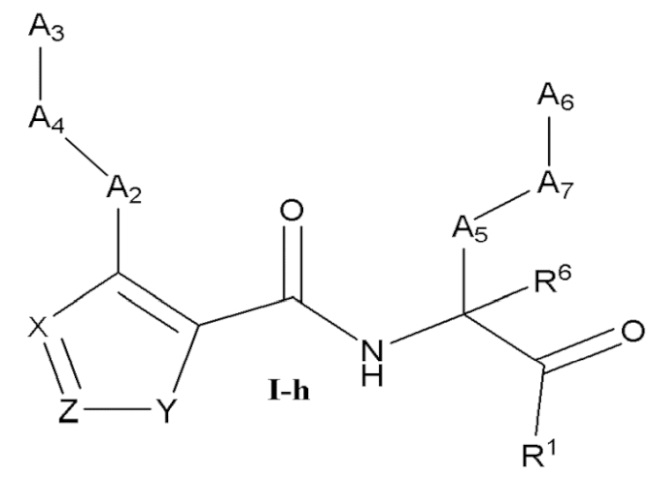
1. Соединение, имеющее структуру, выбранную из следующих формул:
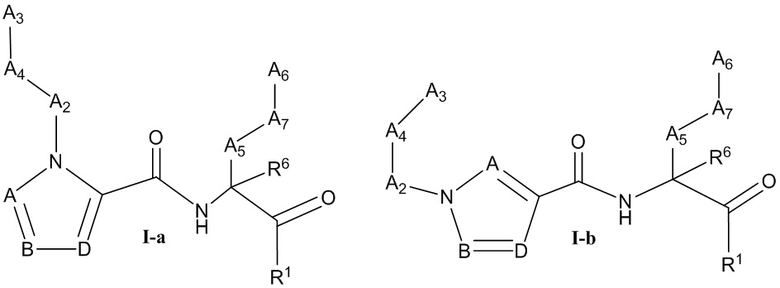
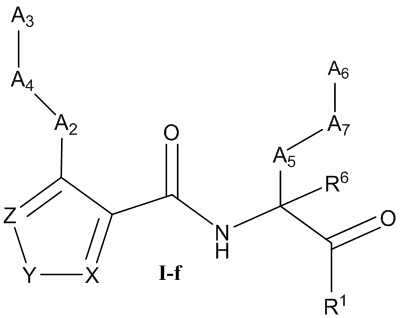
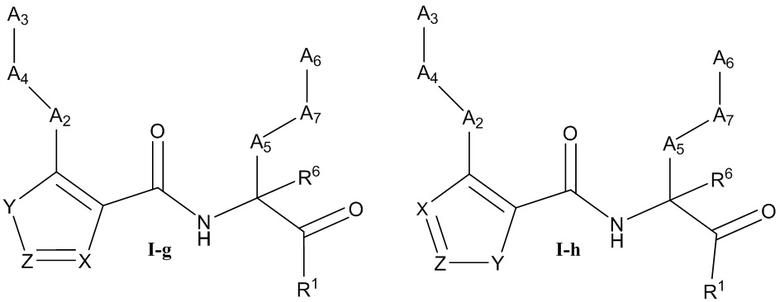 ,
,
или его фармацевтически приемлемая соль,
где A2 выбран из группы, состоящей из C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, и одинарной связи;
A4 выбран из группы, состоящей из C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, -(CR2)n-S-(CR2)n-, -(CR2)n-S(=O)-(CR2)n-, -(CR2)n-SO2-(CR2)n-, -(CR2)n-O-(CR2)n-, -(CR2)n-C(=S)-(CR2)n-, -(CR2)n-C(=O)-(CR2)n-, -(CR2)n-CH=CH-(CR2)n-, -(CR2)n-OC(O)NH-(CR2)n-, -(CR2)n-NHC(O)NH-(CR2)n-, -(CR2)n-NHC(O)O-(CR2)n-, -(CR2)n-NHC(S)NH-(CR2)n-, -(CR2)n-NHC(S)O-(CR2)n-, -(CR2)n-NHC(S)-(CR2)n- и одинарной связи, где каждый R представляет собой -H;
когда A2 и A4 представляют собой одинарную связь, A3 непосредственно присоединен к атому кольца, к которому присоединен A2;
A3 выбран из группы, состоящей из необязательно замещенного C6-10 арила, необязательно замещенного 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, необязательно замещенного 3-10-членного гетероциклила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, и необязательно замещенного C3-10 карбоциклила, или, если A2 выбран из C6-10 арила и 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, то A3 выбран из группы, состоящей из водорода, необязательно замещенного C6-10 арила, необязательно замещенного C3-10 карбоциклила, -C≡CH и 2-5-членного полиэтиленгликоля;
A5 представляет собой C1-4 алкил;
A6 выбран из группы, состоящей из C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, C1-4 алкила и C2-4 алкенила;
A7 представляет собой одинарную связь;
R1 выбран из группы, состоящей из H и -CONR2R3;
каждый R2 и R3 независимо выбран из -H, необязательно замещенного C1-4 алкила, 2-5-членного полиэтиленгликоля, C3-7 карбоциклила и необязательно замещенного C6-10 арил(C1-C6)алкила;
R6 представляет собой -H;
каждый n независимо выбран из целых чисел от 0 до 3;
каждый из A и Z независимо выбран из группы, состоящей из C(R4) и N;
когда соединение имеет структуру формулы I-a, B и D представляют собой C(R4);
когда соединение имеет структуру формулы I-b, B выбран из группы, состоящей из C(R4) и N, и D представляет собой C(R4);
каждый R4 независимо выбран из группы, состоящей из -H, C1-4 алкила, C1-4 галогеналкила, C3-7 карбоциклила, галогена, гидрокси и C1-C3 алкокси;
когда соединение имеет структуру формулы I-f, Y выбран из группы, состоящей из NR5, O, S и SO2, и X выбран из группы, состоящей из C(R4) и N;
когда соединение имеет структуру формулы I-g или формулы I-h, Y выбран из группы, состоящей из NR5 и S, и X представляет собой C(R4), или Y выбран из O и SO2 и X выбран из группы, состоящей из C(R4) и N;
R5 выбран из группы, состоящей из -H, C1-4 алкила, C1-4 галогеналкила и C3-7 карбоциклила;
при этом необязательно замещенная группа может быть замещена, если не указано иное, одним или более заместителями, независимо выбранными из C1-C6 алкила, C3-C7 карбоциклила, 5-10-членного гетероциклила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, C6-C10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, галогена, циано, гидрокси, C1-C6 алкокси, C1-C6 алкокси(C1-C6)алкила (то есть группы простого эфира), арилокси, галоген(C1-C6)алкила (например, -CF3), галоген(C1-C6)алкокси (например, -OCF3), амино, амино(C1-C6)алкила, C-амидо, N-амидо, N-сульфонамидо и сульфонила.
2. Соединение по п. 1, где
A2 выбран из группы, состоящей из C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, и одинарной связи;
A4 выбран из группы, состоящей из C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, -S-, S(=O)-, -SO2-, -O-, -C(=S)-, -C(=O)-, -CH=CH-, -OC(O)NH-, -NHC(O)NH-, -NHC(O)O-, -NHC(S)NH-, -NHC(S)O-, -NHC(S)- и одинарной связи;
A3 выбран из группы, состоящей из необязательно замещенного C6-10 арила, необязательно замещенного 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, необязательно замещенного 3-10-членного гетероциклила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, и необязательно замещенного C3-10 карбоциклила;
A6 выбран из группы, состоящей из C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, C1-4 алкила и C2-4 алкенила; и
каждый R2 и R3 независимо выбран из -H, необязательно замещенного C1-4 алкила и C3-7 карбоциклила.
3. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-a, A выбран из группы, состоящей из CH и N.
4. Соединение по п. 3, где A представляет собой N, B представляет собой CH и D представляет собой CH.
5. Соединение по п. 3, где A представляет собой CH, B представляет собой CH и D представляет собой CH.
6. Соединение по п. 3, где A представляет собой N и B представляет собой CH.
7. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-b, A и B независимо выбраны из группы, состоящей из CH и N.
8. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-f, Z представляет собой N, Y представляет собой NR5 и X представляет собой CH.
9. Соединение по п. 8, где R5 выбран из группы, состоящей из -H, C1-4 алкила, C1-C4 галогеналкила и циклопропила.
10. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-f, Z представляет собой N, Y представляет собой O и X представляет собой C(R4).
11. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-f, Z представляет собой N, Y представляет собой S и X представляет собой C(R4).
12. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-f, Z представляет собой C(R4), Y представляет собой S и X представляет собой C(R4).
13. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-f, Z представляет собой C(R4), Y представляет собой O и X представляет собой C(R4).
14. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-f, Z представляет собой N, Y представляет собой S и X представляет собой N.
15. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-f, Z представляет собой N, Y представляет собой O и X представляет собой N.
16. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-g, Z выбран из группы, состоящей из CH и N.
17. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-g, Z представляет собой N и X представляет собой CH.
18. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-h, Z выбран из группы, состоящей из CH и N.
19. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-h, X представляет собой CH, Z представляет собой N и Y представляет собой NR5.
20. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-h, X представляет собой CH, Z представляет собой C(R4) и Y представляет собой O.
21. Соединение по п. 20, где R4 выбран из -H и C1-4 алкила.
22. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-h, X представляет собой CH, Z представляет собой C(R4) и Y представляет собой S.
23. Соединение по любому из пп. 1 и 2, в котором, когда структура соединения представляет собой структуру формулы I-h, X представляет собой CH, Z представляет собой N и Y представляет собой S.
24. Соединение по любому из пп. 1-23, где по меньшей мере один из необязательно замещенных фрагментов A2, A4 и A3 замещен 18F.
25. Соединение по любому из пп. 1-24, где по меньшей мере один из необязательно замещенных фрагментов A2, A4 и A3 замещен C1-C6 алкилом, содержащим один или более 11C.
26. Соединение по любому из пп. 1-25, где A3 выбран из группы, состоящей из
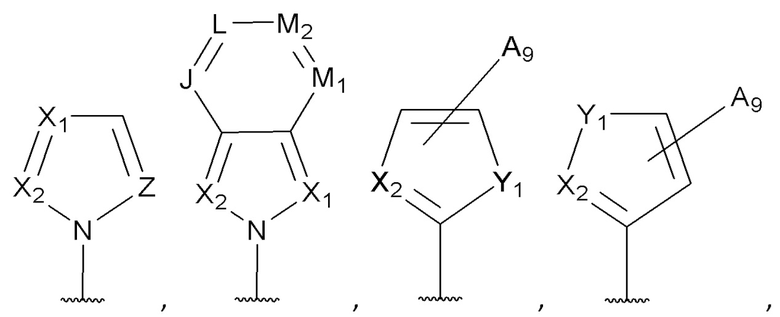
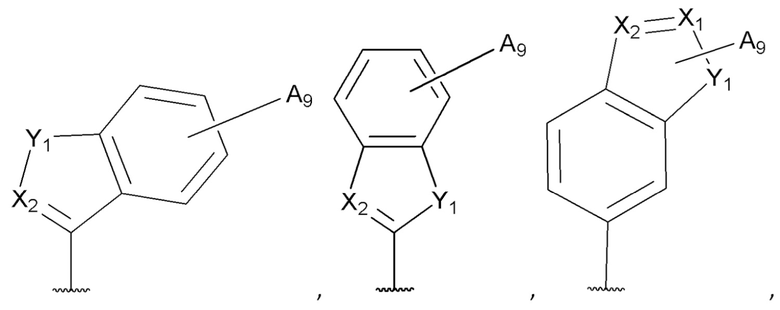
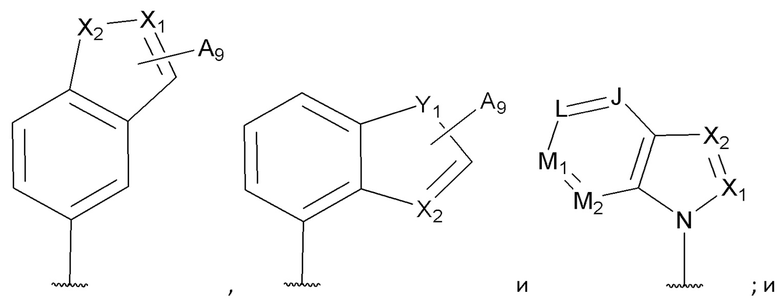
A9 выбран из группы, состоящей из H, C6-10 арила, 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, 3-10-членного гетероциклила, содержащего от 1 до 2 гетероатомов, выбранных из N, O и/или S, C3-10 карбоциклила и C1-4 алкила;
каждый из X2, X1 и Z независимо выбран из группы, состоящей из C(R4) и N;
Y1 выбран из группы, состоящей из NR5, O и S;
каждый из J, L, M1 и M2 независимо выбран из группы, состоящей из C(R4) и N;
R4 выбран из группы, состоящей из -H, C1-4 алкила, C1-4 галогеналкила, C3-7 карбоциклила, галогена, гидрокси и C1-C6 алкокси;
R5 выбран из группы, состоящей из -H, C1-4 алкила, C1-4 галогеналкила и C3-7 карбоциклила (необязательно замещенного галогеном, C1-C6 алкилом, C1-C6 алкокси, C1-C6 галогеналкилом и C1-C6 галогеналкокси).
27. Соединение по любому из пп. 1-25, где A3 выбран из группы, состоящей из
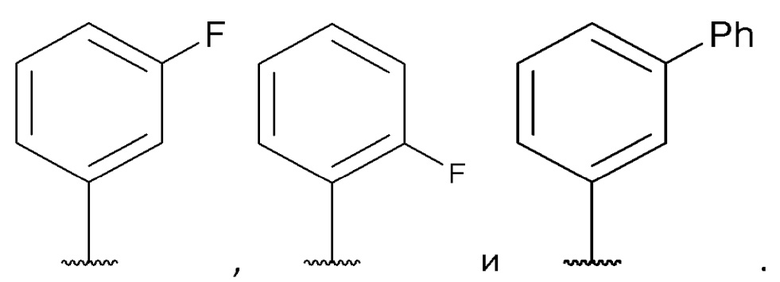
28. Соединение по любому из пп. 1-25, где A3 выбран из группы, состоящей из
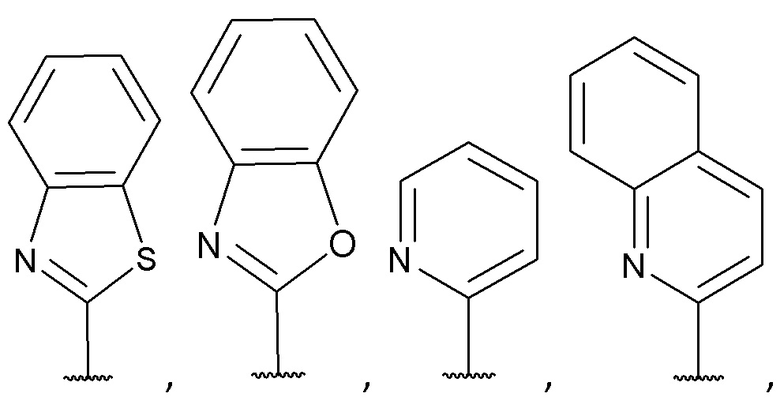
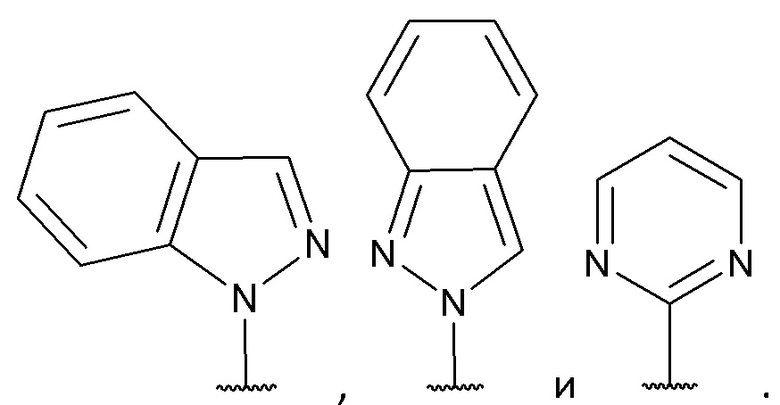
29. Соединение по любому из пп. 1-28, где A2 выбран из группы, состоящей из одинарной связи и фенила.
30. Соединение по любому из пп. 1-25, где A2 представляет собой одинарную связь, A4 представляет собой одинарную связь и A3 представляет собой необязательно замещенный C6-10 арил или необязательно замещенный 5-10-членный гетероарил, содержащий от 1 до 2 гетероатомов, выбранных из N, O и/или S.
31. Соединение по п. 30, где A3 имеет структуру
 ,
,
где каждый из J, L, M1, M2 и M3 независимо выбран из группы, состоящей из C(R4) и N; и
каждый R4 независимо выбран из группы, состоящей из -H, C1-4 алкила, C1-4 галогеналкила, C3-7 карбоциклила, галогена, гидрокси и C1-C6 алкокси.
32. Соединение по п. 31, где каждый из J, L, M1, M2 и M3 представляет собой C(R4).
33. Соединение по п. 32, где каждый R4 независимо выбран из -H и галогена.
34. Соединение по п. 31, где M1 представляет собой C(R4); R4 представляет собой галоген и каждый из J, L, M2 и M3 представляет собой CH.
35. Соединение по п. 31, где L представляет собой C(R4); R4 представляет собой галоген и каждый из J, M1, M2 и M3 представляет собой CH.
36. Соединение по любому из пп. 1-25 и 30, где A3 имеет структуру, выбранную из группы, состоящей из
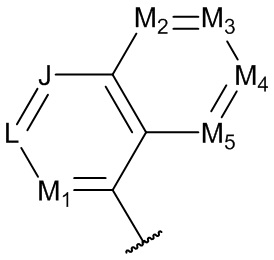 и
и  ,
,
где каждый из J, L, M1, M2, M3, M4 и M5 независимо выбран из группы, состоящей из C(R4) и N; и
каждый R4 независимо выбран из группы, состоящей из -H, C1-4 алкила, C1-4 галогеналкила, C3-7 карбоциклила, галогена, гидрокси и C1-C6 алкокси.
37. Соединение по любому из пп. 1-25 и 30, где A3 имеет структуру
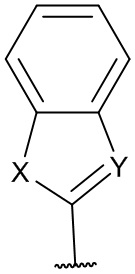 ,
,
где X выбран из группы, состоящей из C(R4) и N;
Y выбран из O и S и
R4 выбран из группы, состоящей из -H, C1-4 алкила, C1-4 галогеналкила, C3-7 карбоциклила, галогена, гидрокси и C1-C6 алкокси.
38. Соединение, имеющее структуру формулы I-o
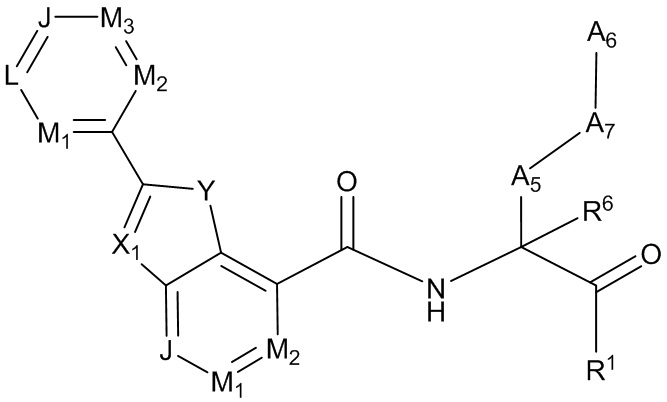 ,
,
I-o
или его фармацевтически приемлемая соль,
где Y выбран из группы, состоящей из NR5;
X1 представляет собой N;
каждый из J, L, M1, M2 и M3 представляет собой C(R4);
A5 представляет собой C1-4 алкил;
A6 представляет собой C6-10 арил;
A7 представляет собой одинарную связь и
каждый из R4, R5, R1 и R6 представляет собой -H.
39. Соединение по п. 38, где J, L, M1, M2 и M3 каждый представляет собой CH.
40. Соединение по любому из пп. 1-39, где по меньшей мере один из A5 и A6 замещен 18F.
41. Соединение по любому из пп. 1-39, где по меньшей мере один из A5 и A6 замещен C1-C6 алкилом, содержащим один или более 11C.
42. Соединение по любому из пп. 1-39, где A6 представляет собой фенил.
43. Соединение по любому из пп. 1-42, где A5 представляет собой -CH2-.
44. Соединение по любому из пп. 1-39, где A5 представляет собой -CH2- или -CH2CH2- и A6 выбран из группы, состоящей из C1-C4 алкила, фенила и 5-10-членного гетероарила.
45. Соединение по п. 44, где A6 представляет собой фенил.
46. Соединение по п. 44, где A6 имеет структуру
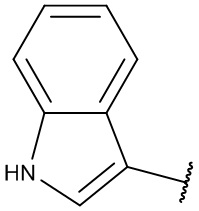 .
.
47. Соединение по любому из пп. 1-39, где A6 представляет собой C1-C4 алкил.
48. Соединение по п. 47, где A6 выбран из группы, состоящей из этила, н-пропила, изопропила и изобутила.
49. Соединение по любому из пп. 1-48, где R1 представляет собой CONR2R3.
50. Соединение по п. 49, где R2 представляет собой -H и R3 представляет собой необязательно замещенный C1-4 алкил.
51. Соединение по п. 49, где R2 представляет собой -H и R3 выбран из группы, состоящей из -H, C1-C4 алкила, необязательно замещенного посредством C-амидо, и C3-C6 циклоалкила.
52. Соединение по п. 51, где R3 выбран из группы, состоящей из -H, этила, циклопропила и метила, замещенного посредством C-амидо.
53. Соединение по п. 49, где R3 выбран из группы, состоящей из необязательно замещенного C1-4 алкила и бензила.
54. Соединение по любому из пп. 1-53, где A4 представляет собой одинарную связь.
55. Соединение, имеющее структуру, выбранную из группы, состоящей из:
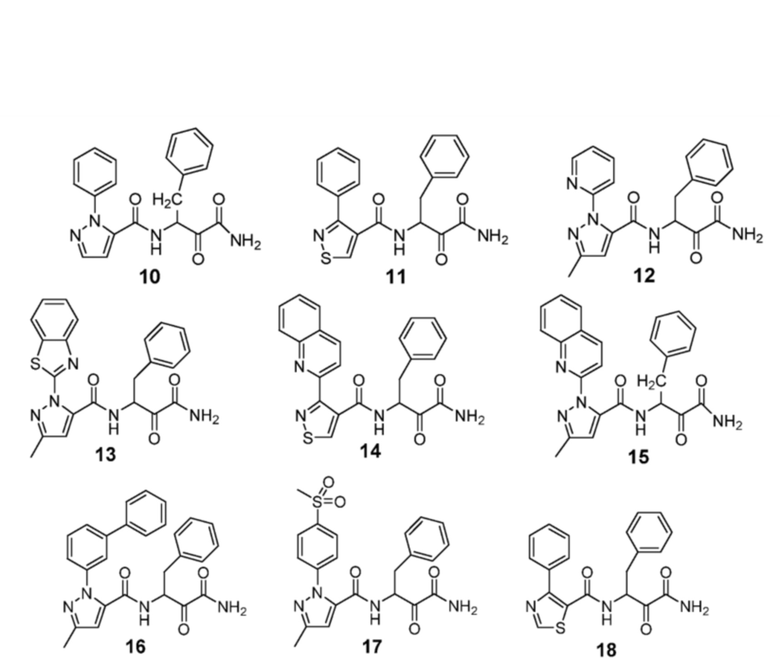

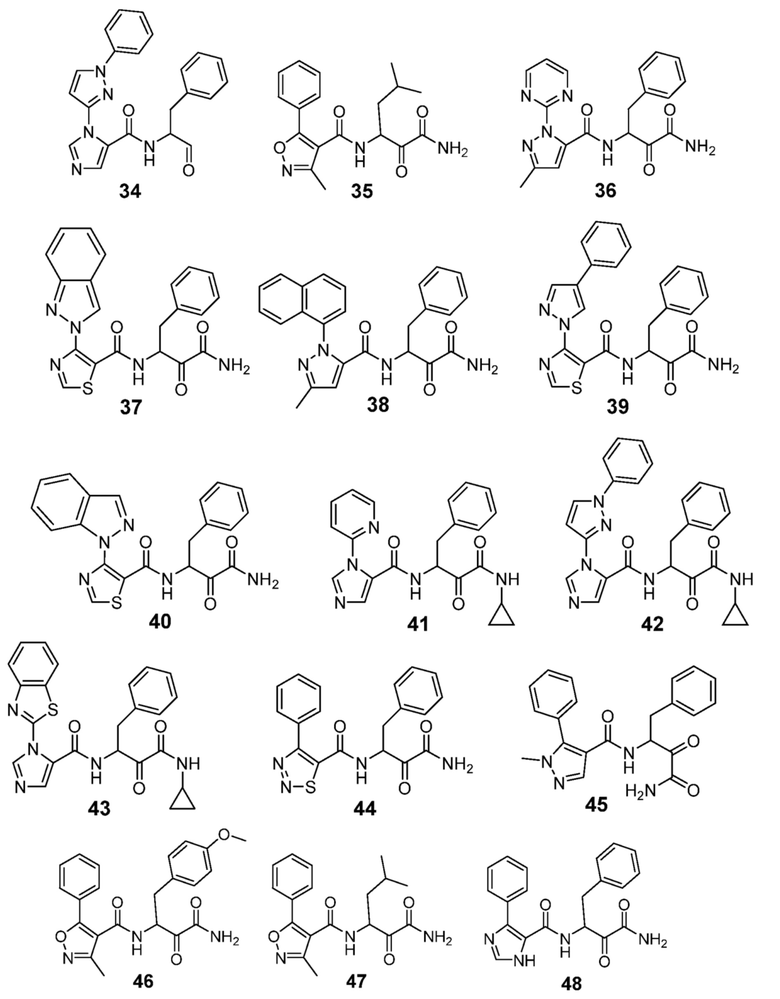
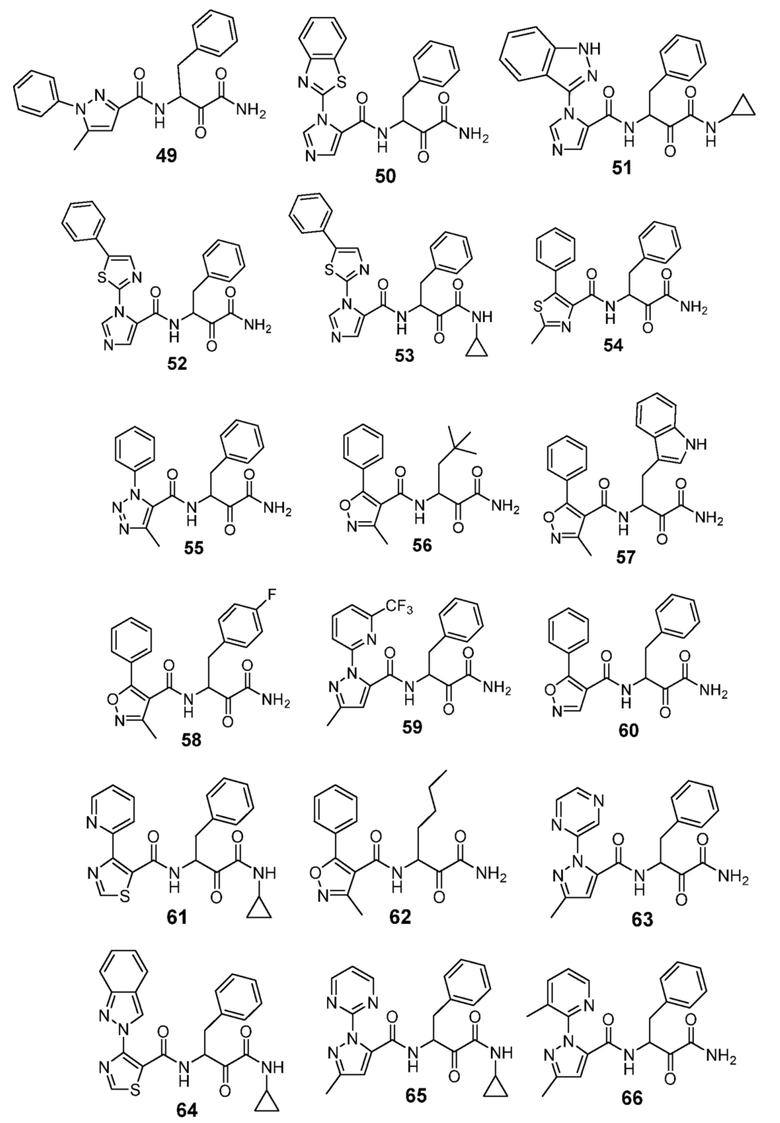
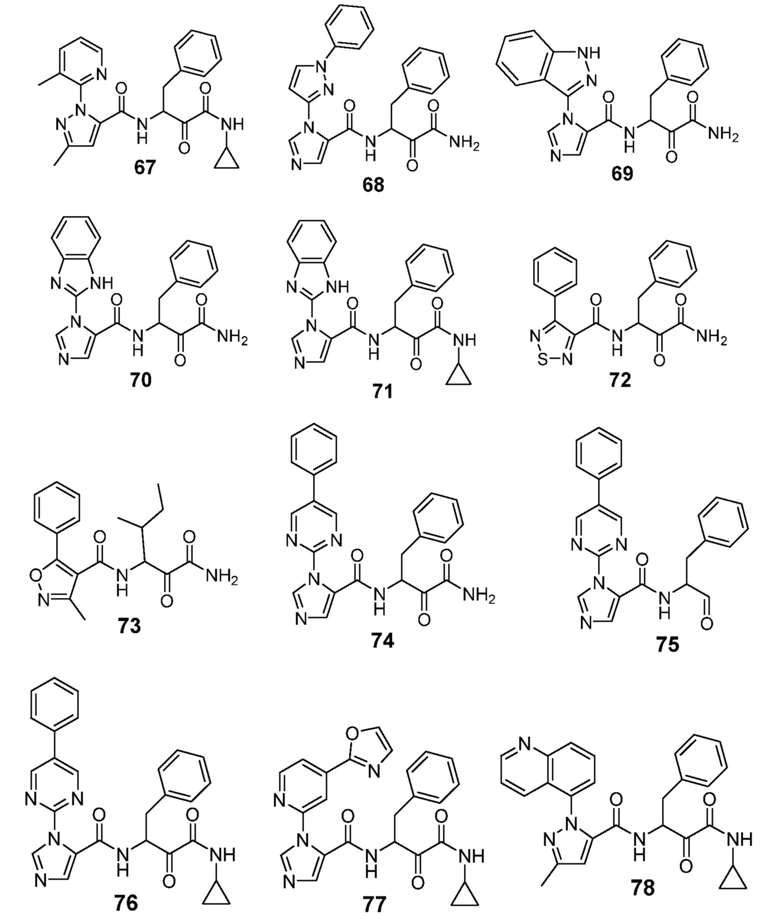
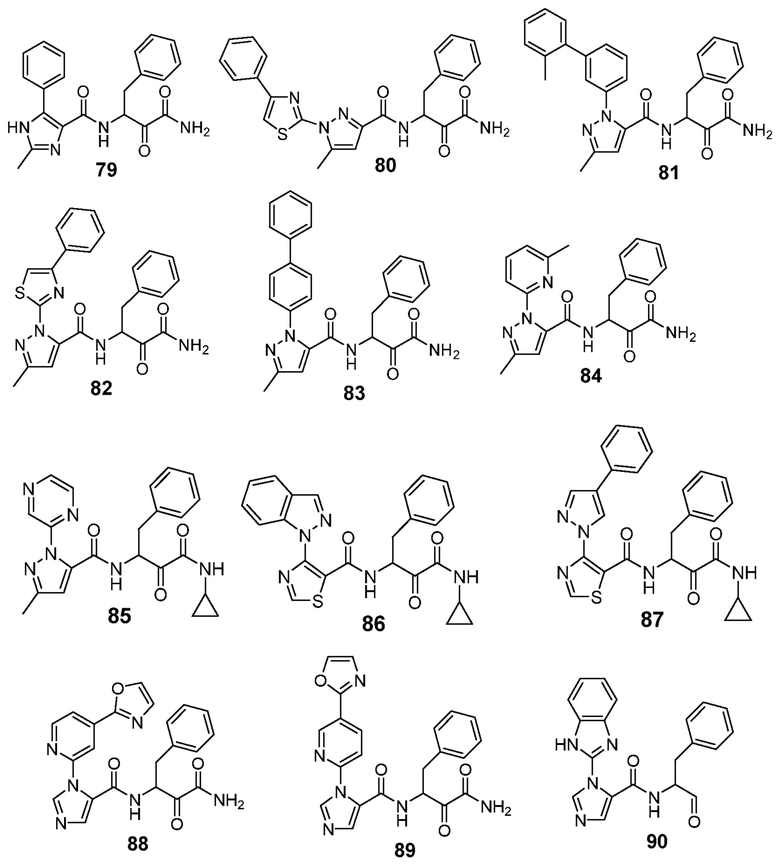

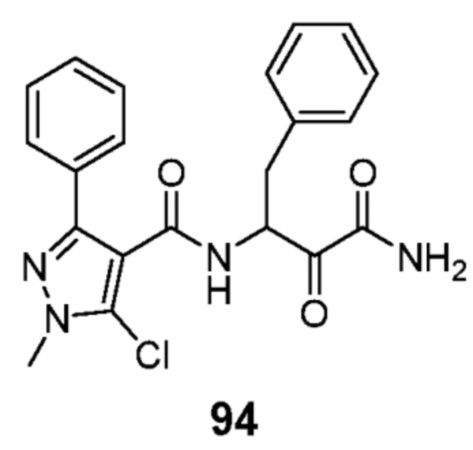
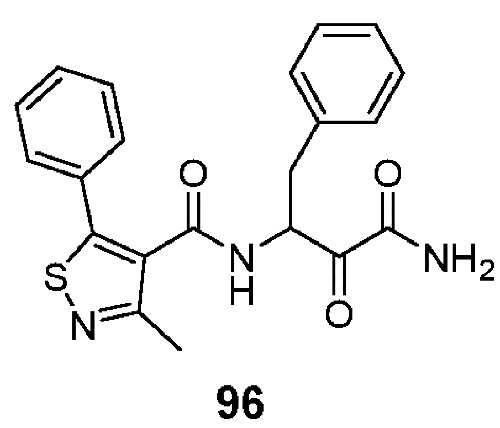
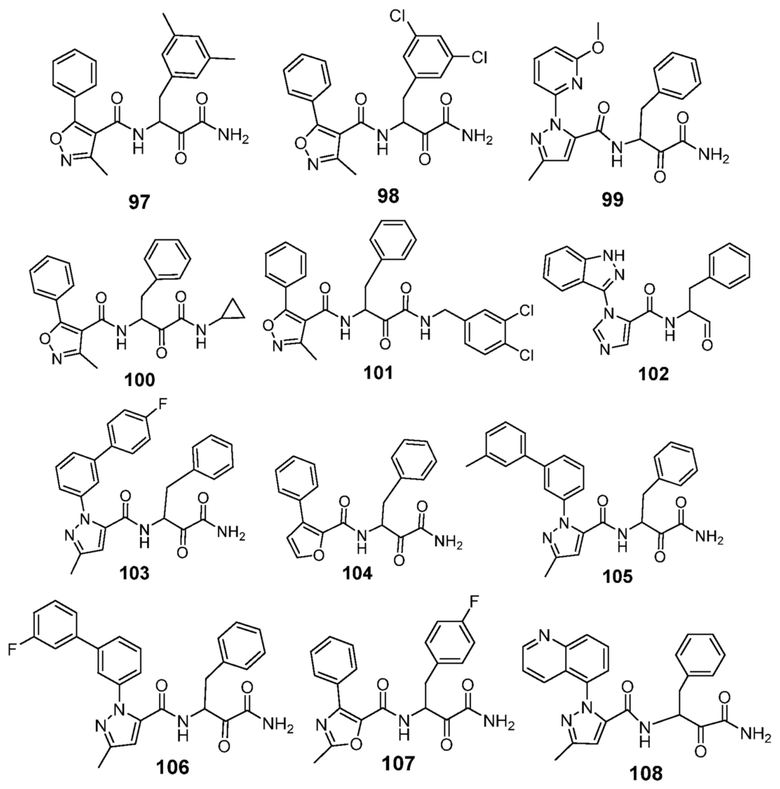
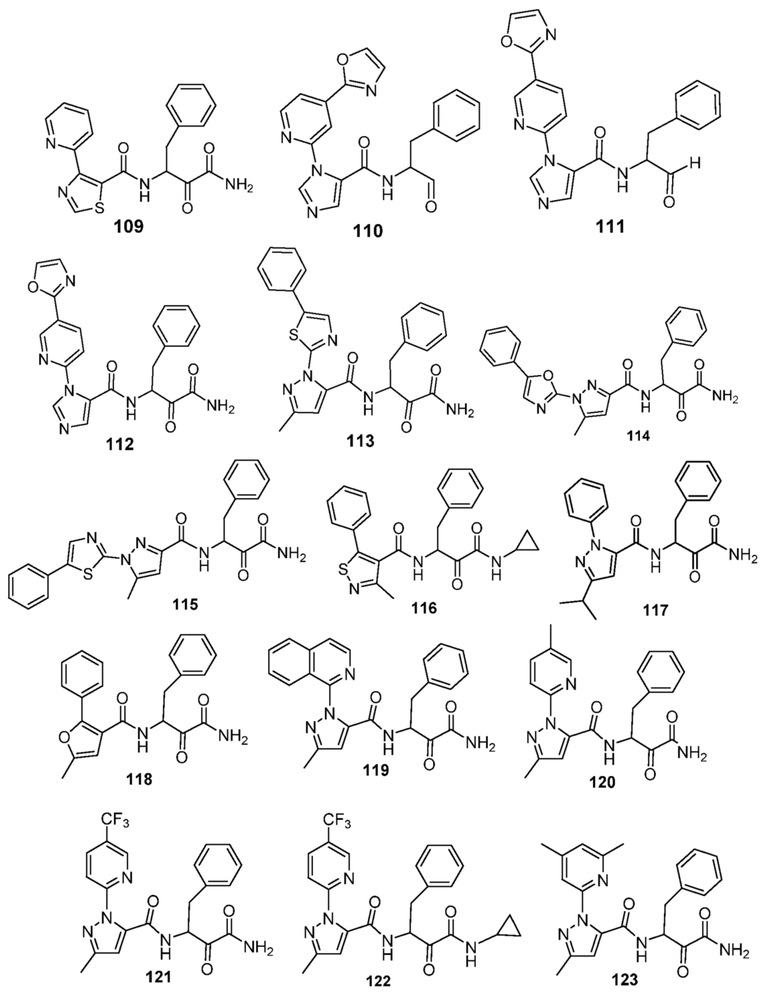
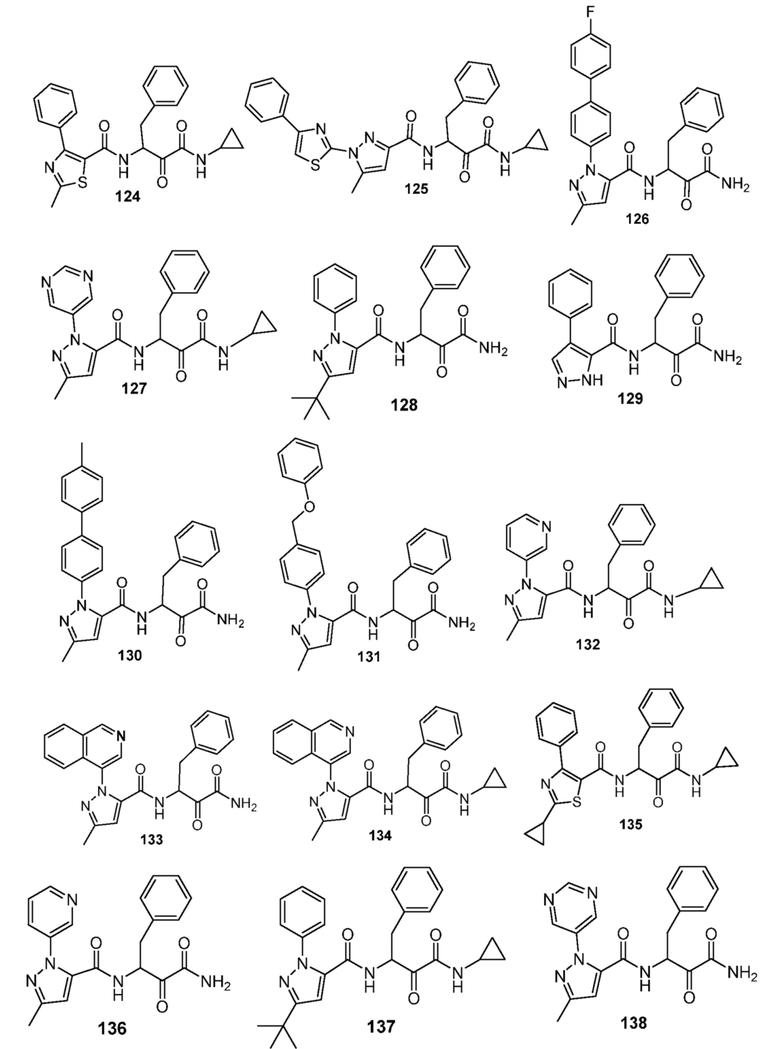
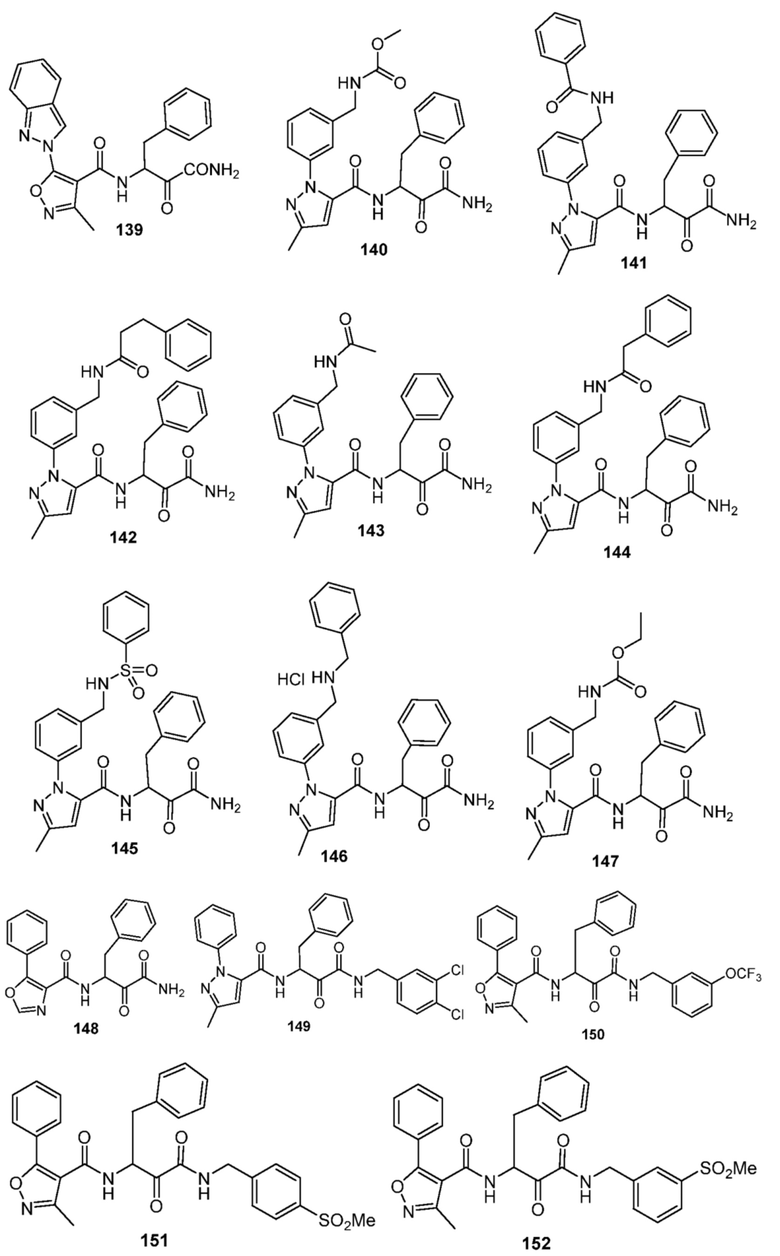
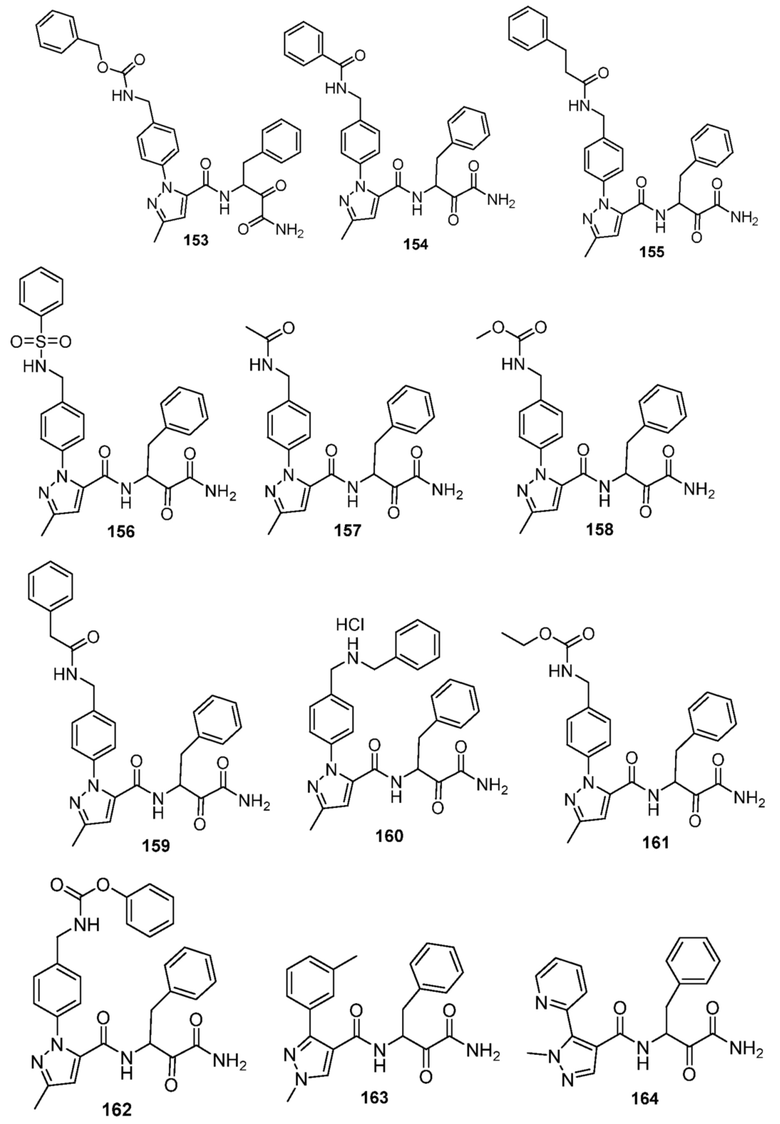
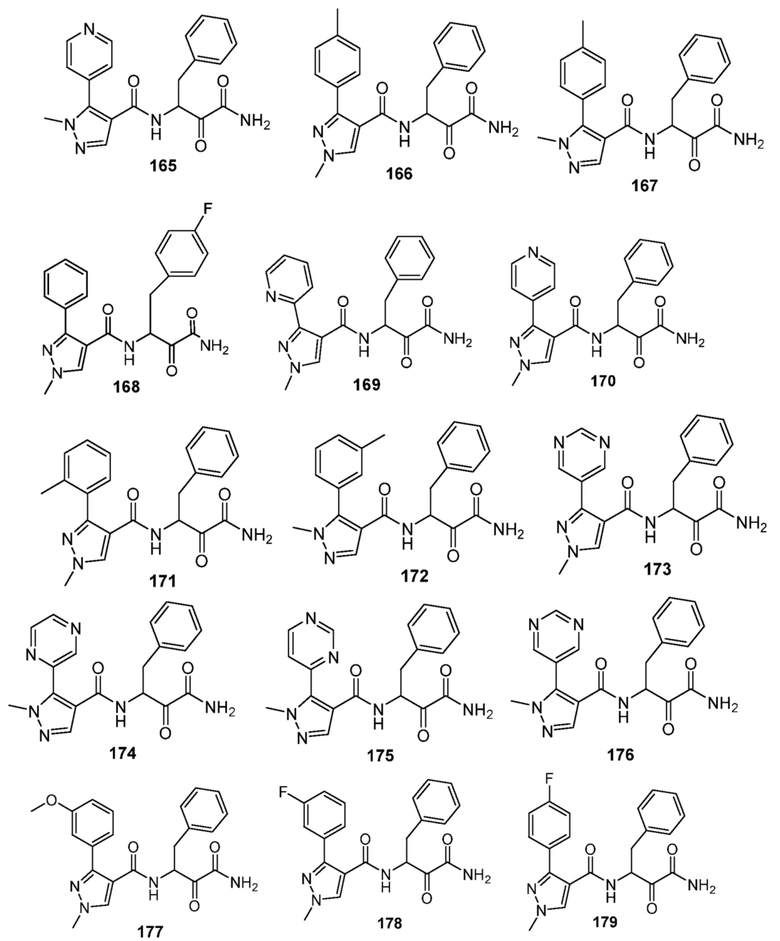
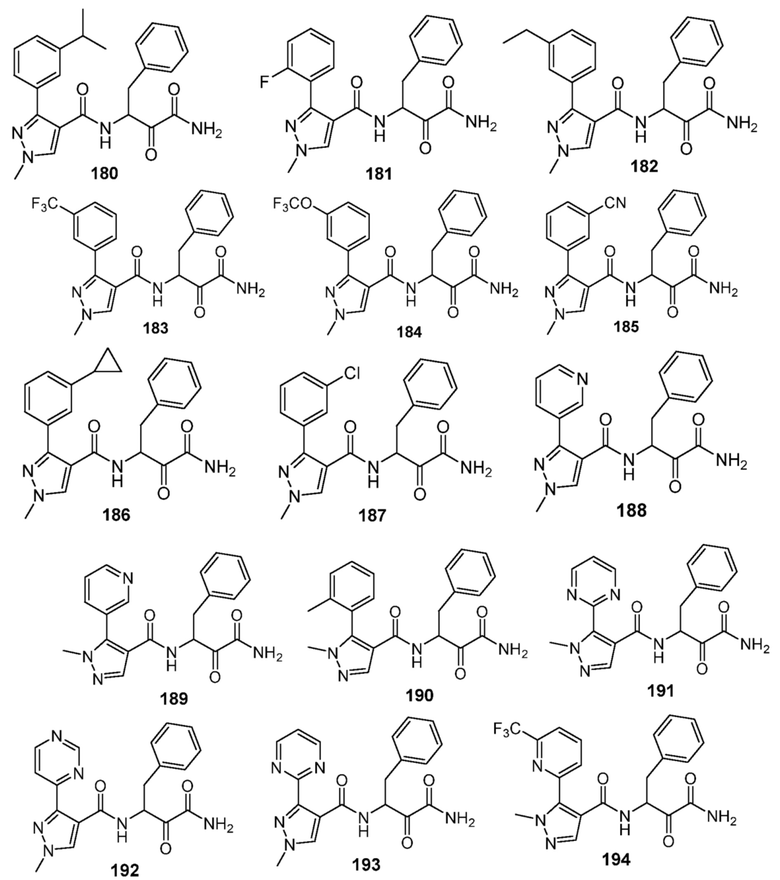
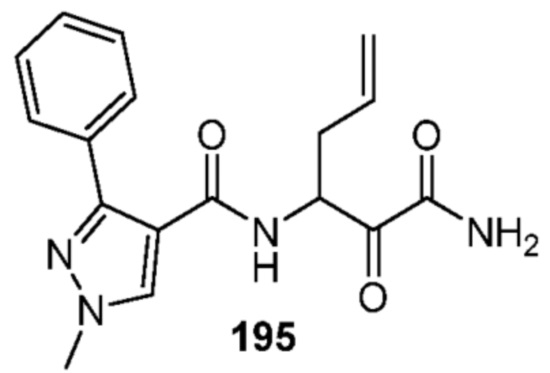
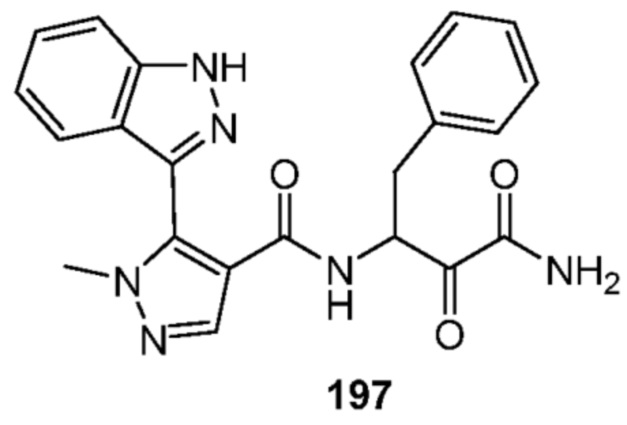
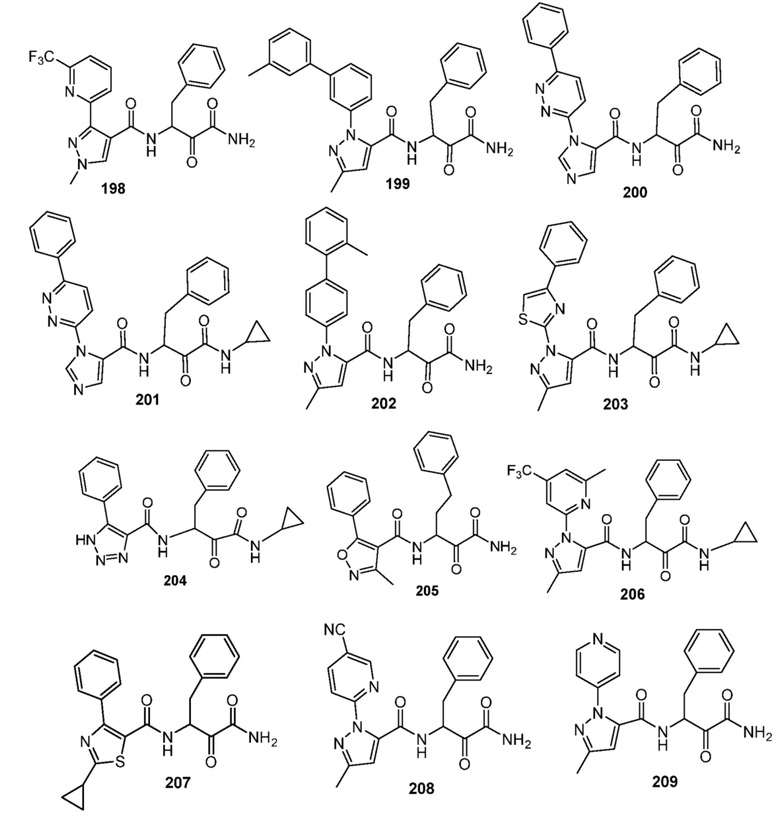
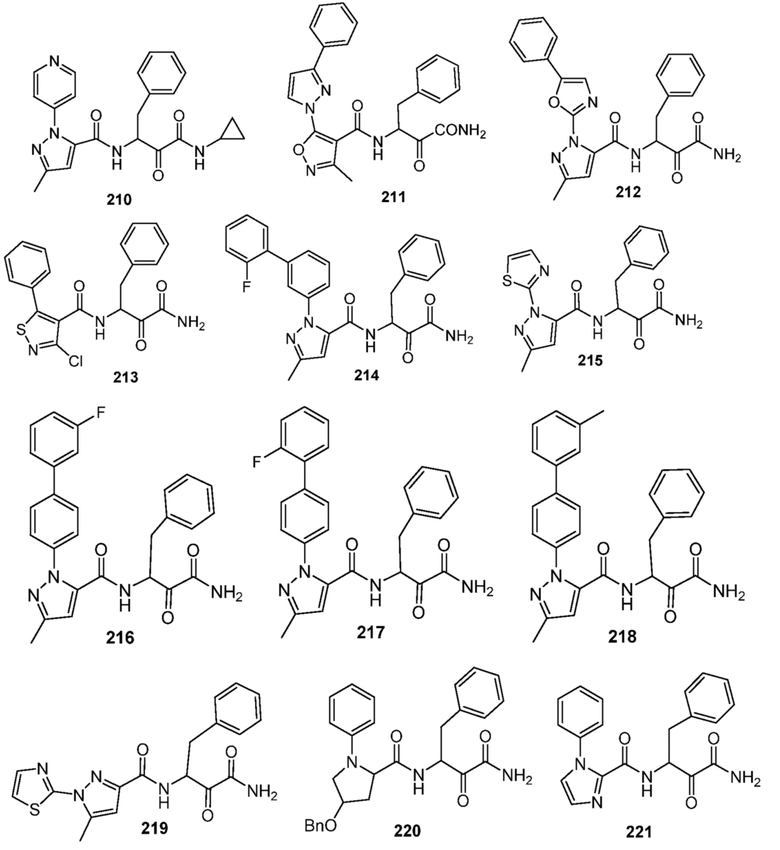
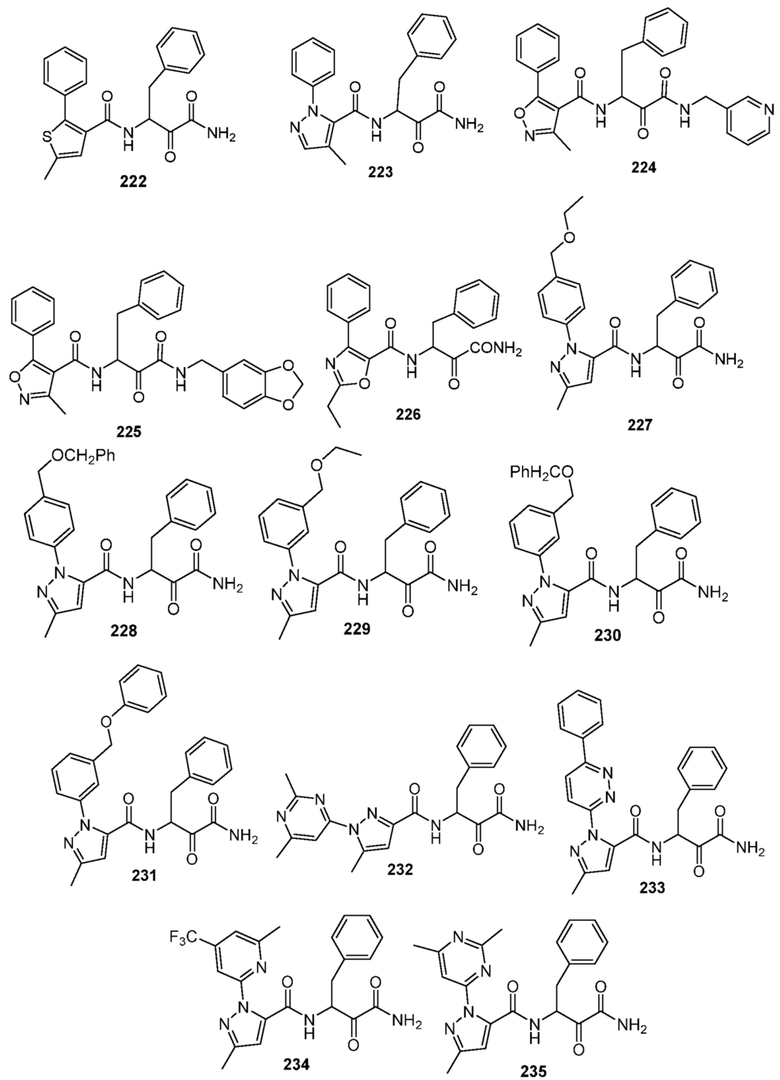
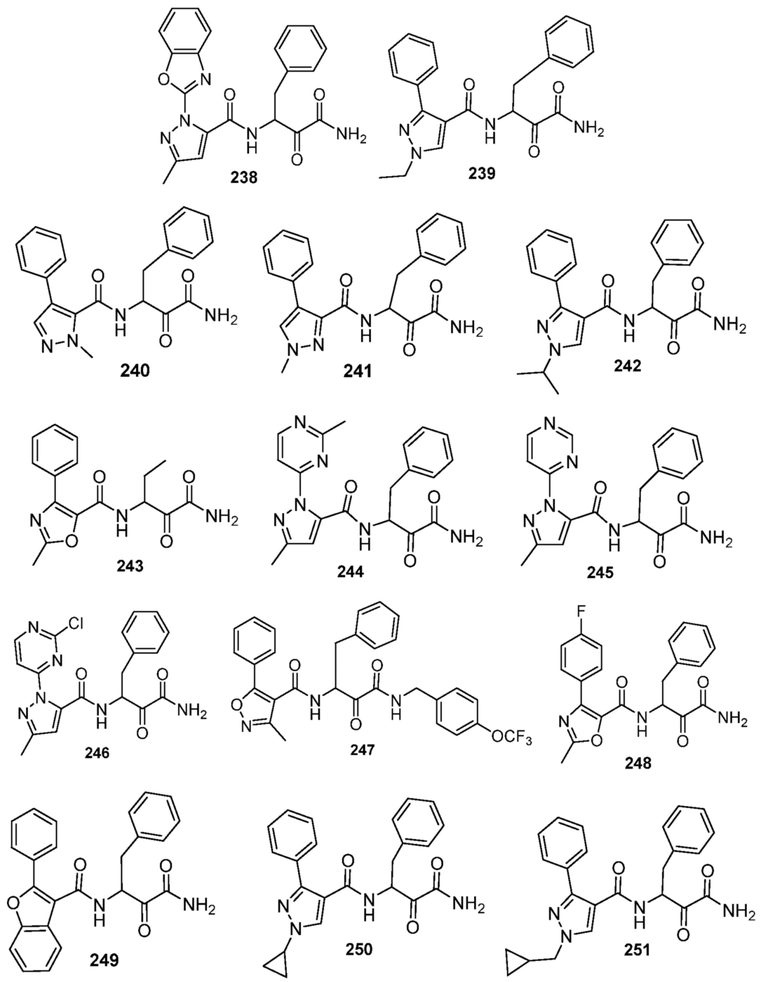
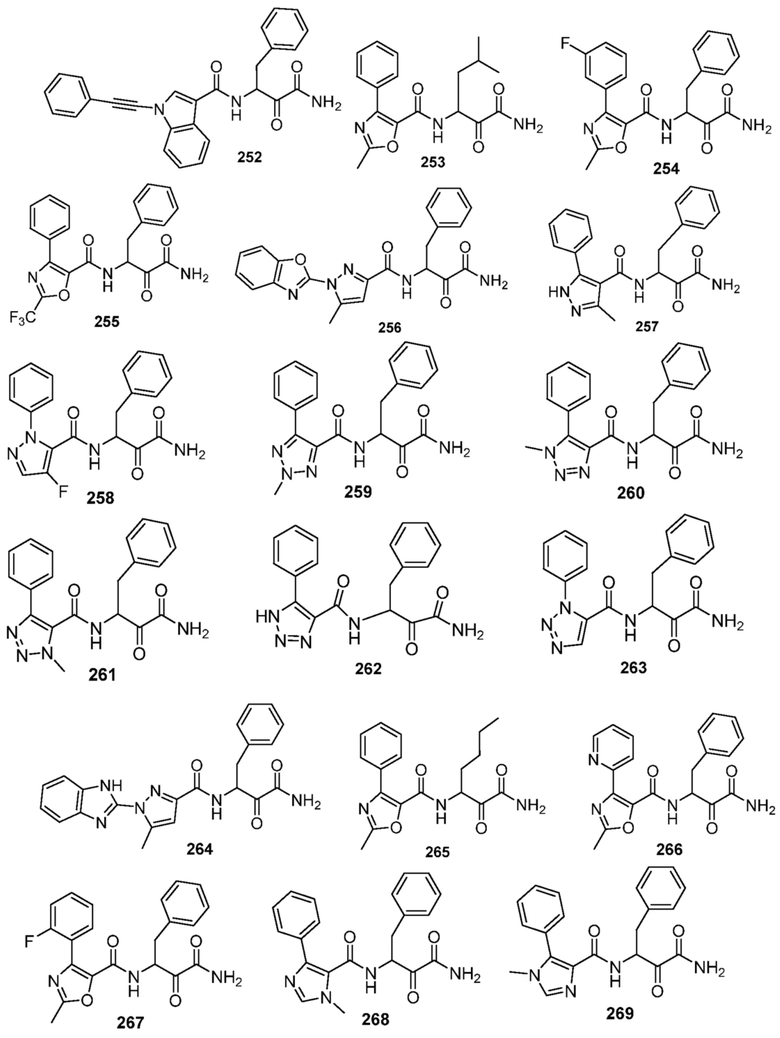
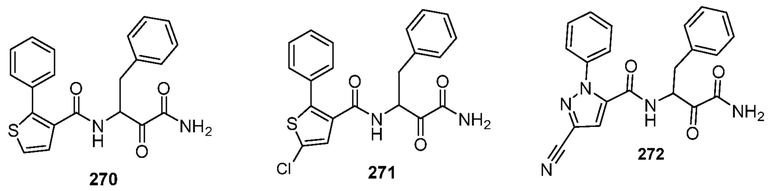
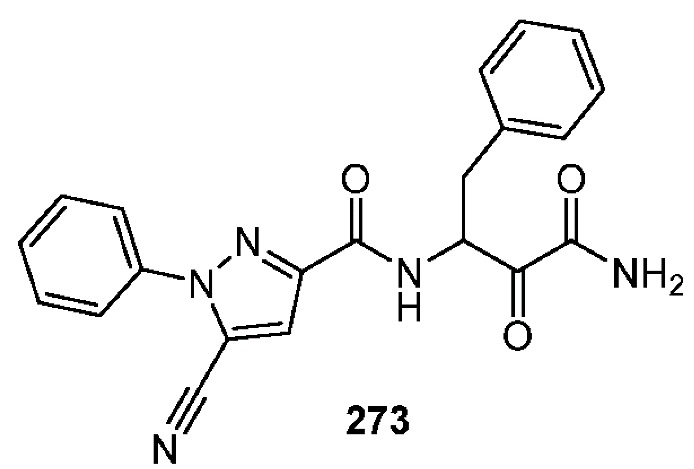
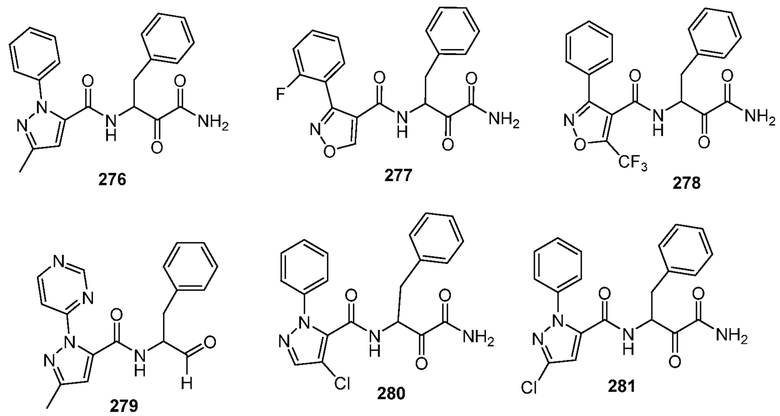
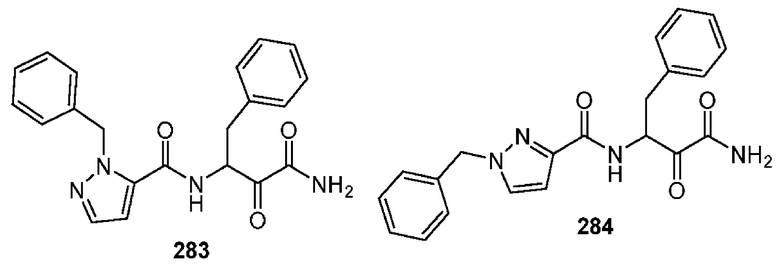
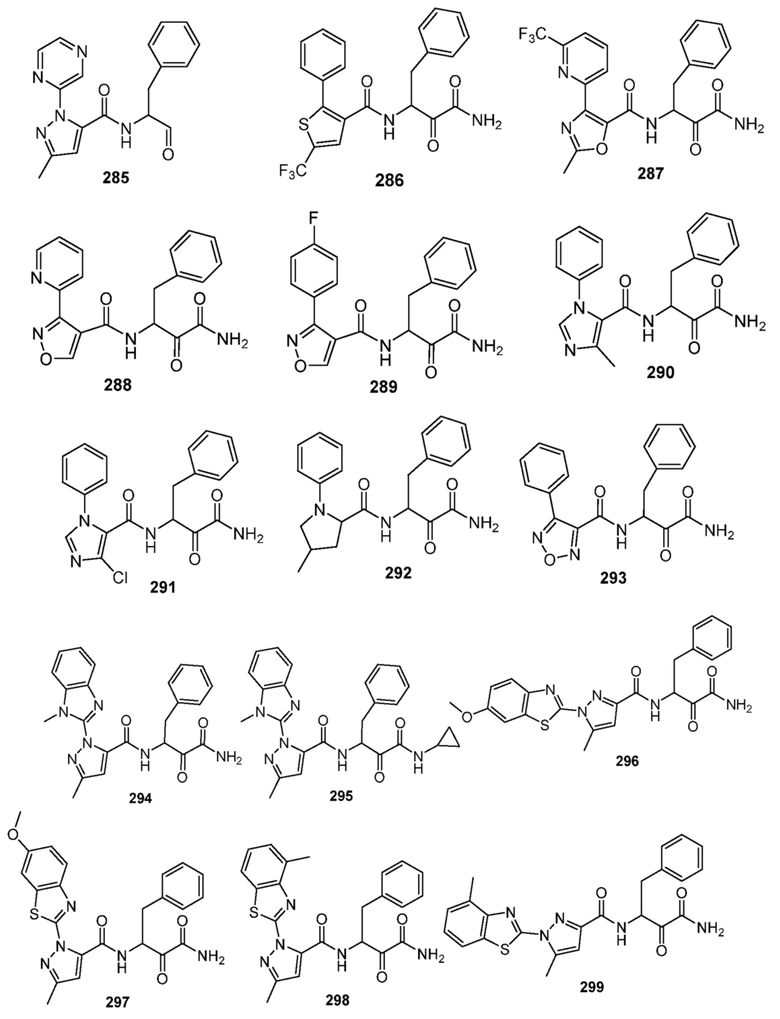
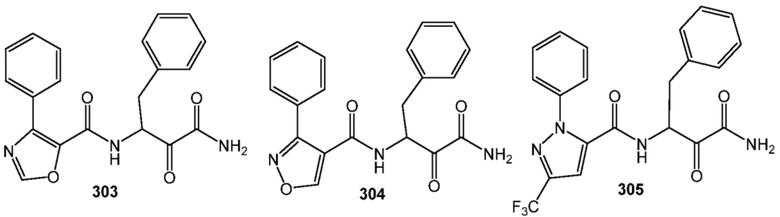
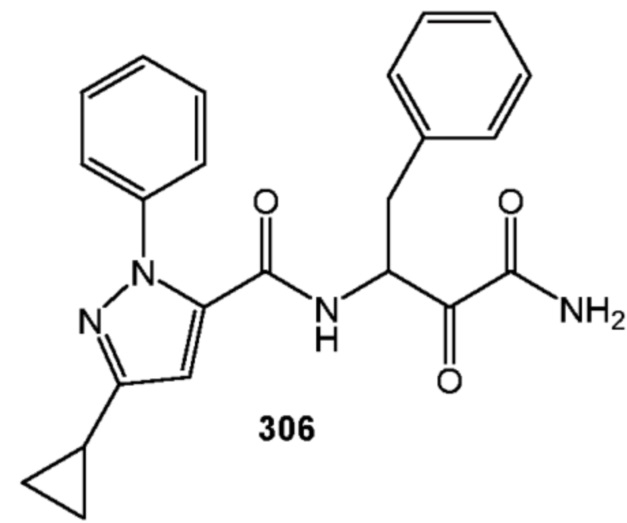
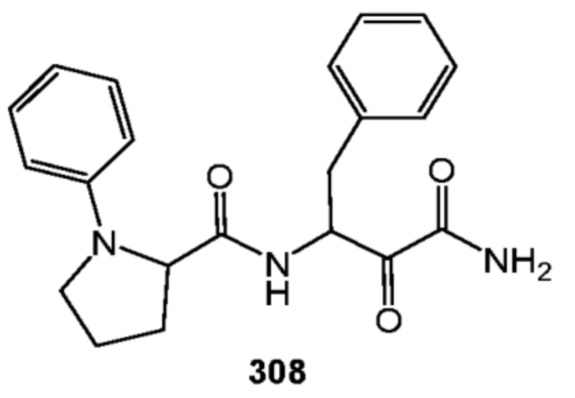
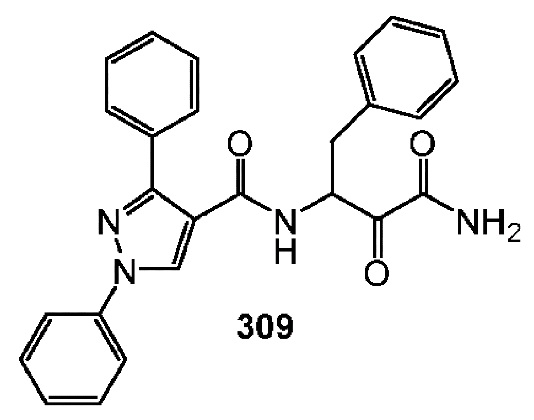
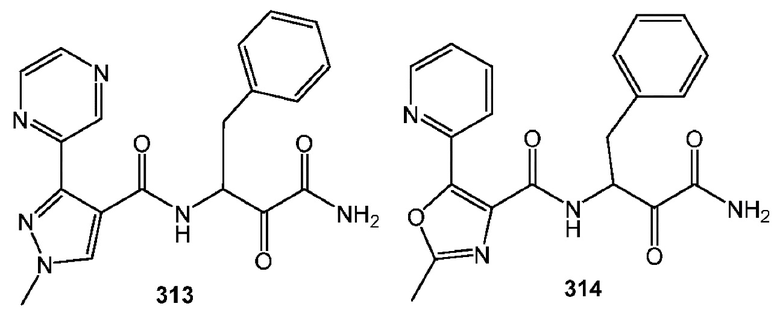
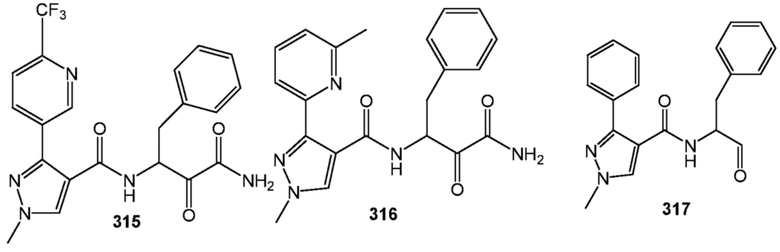
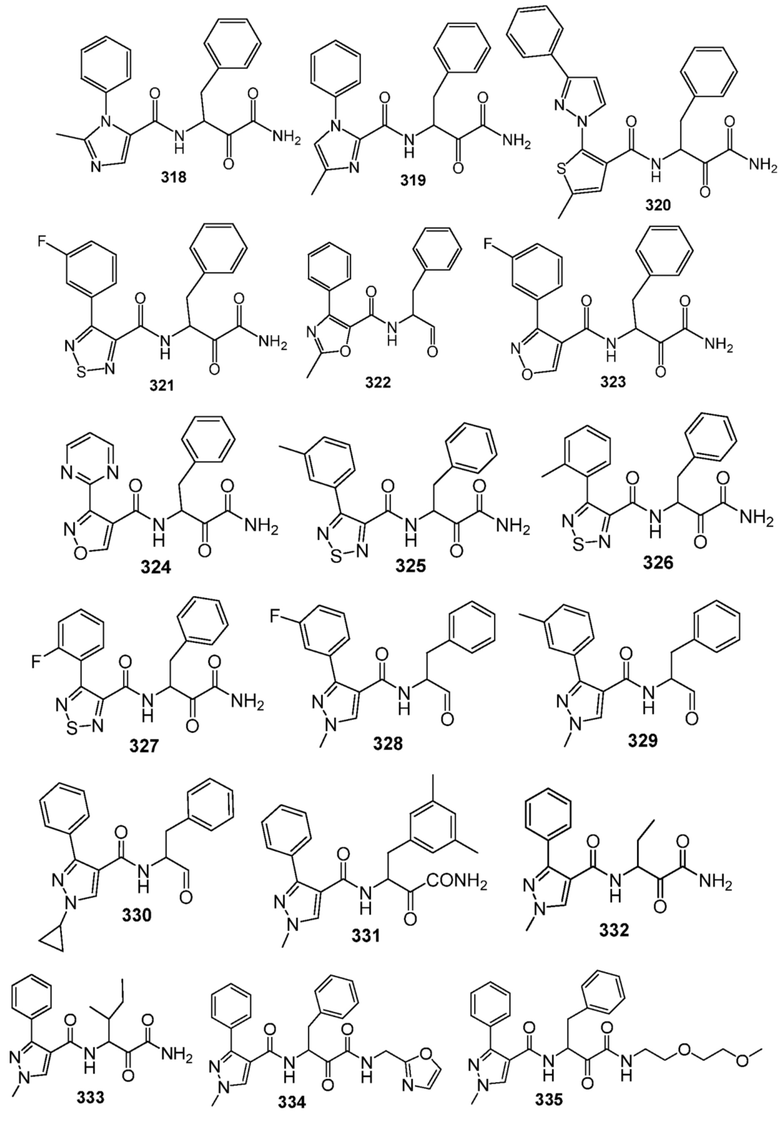
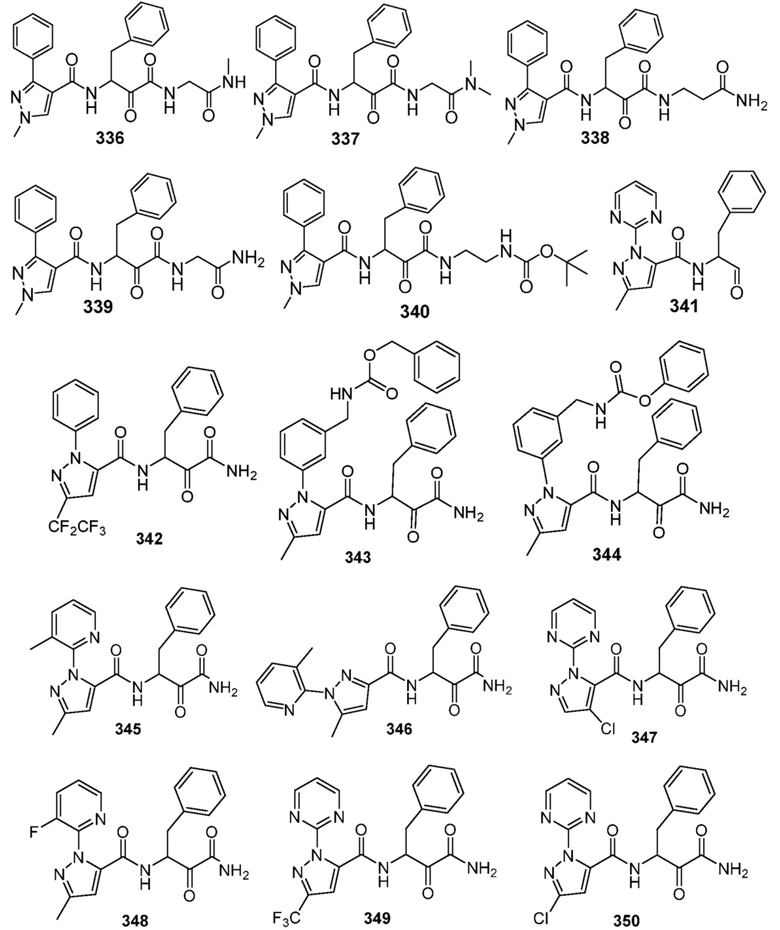
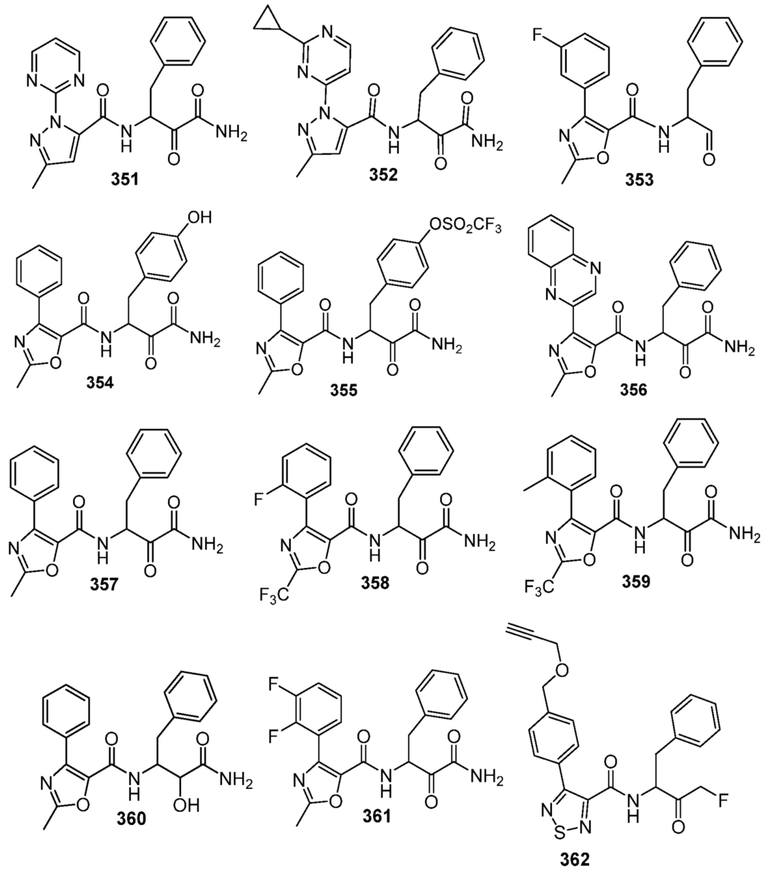
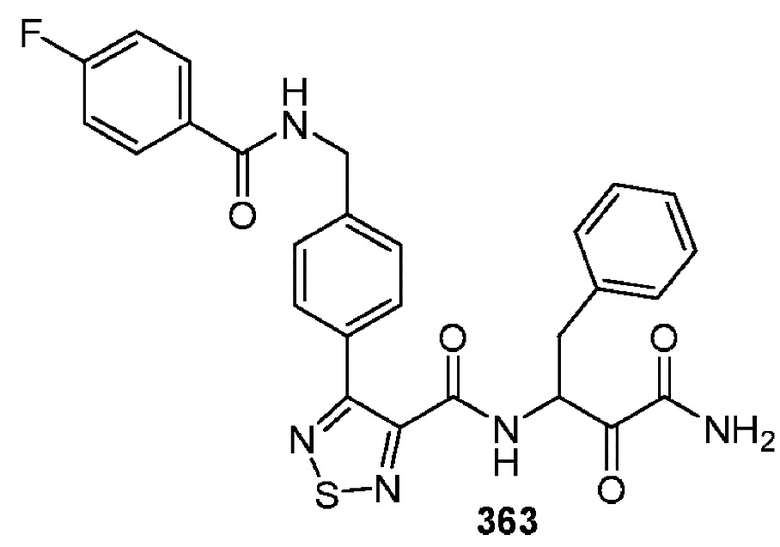
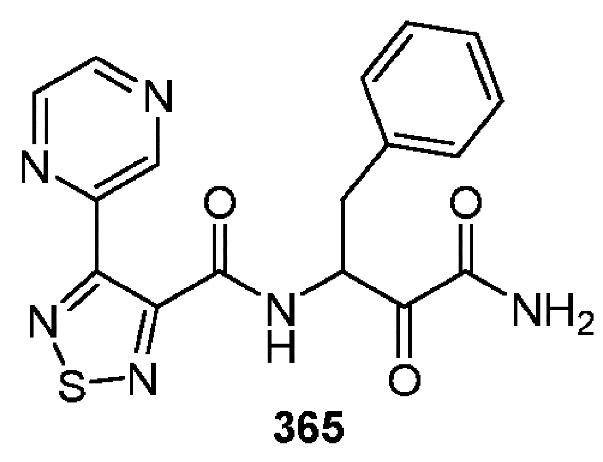

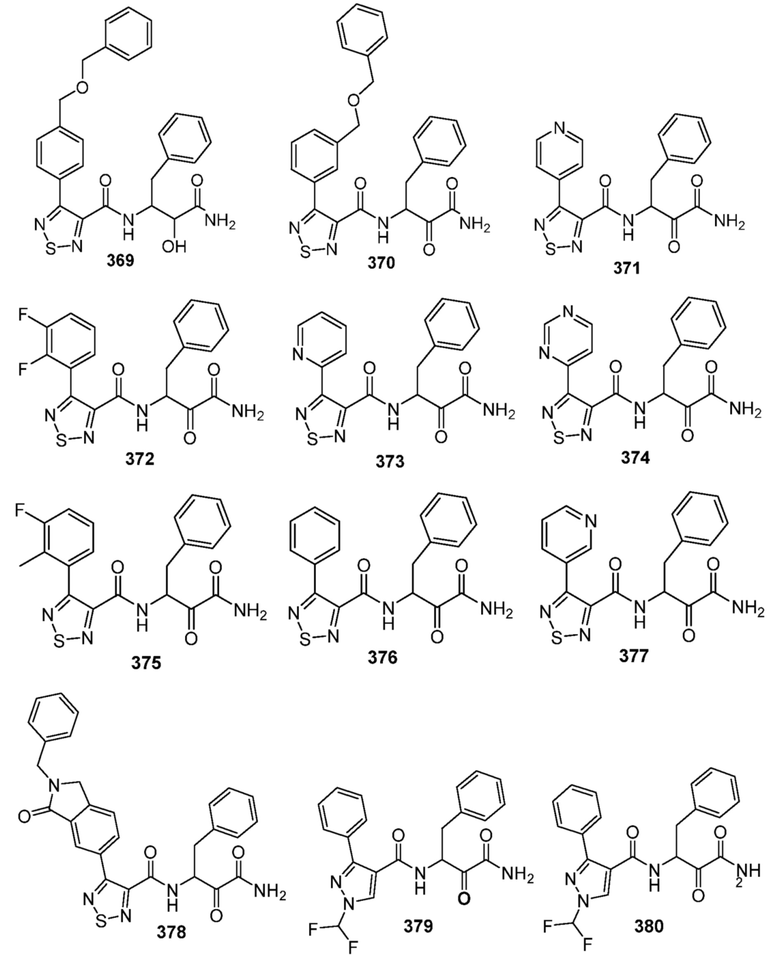
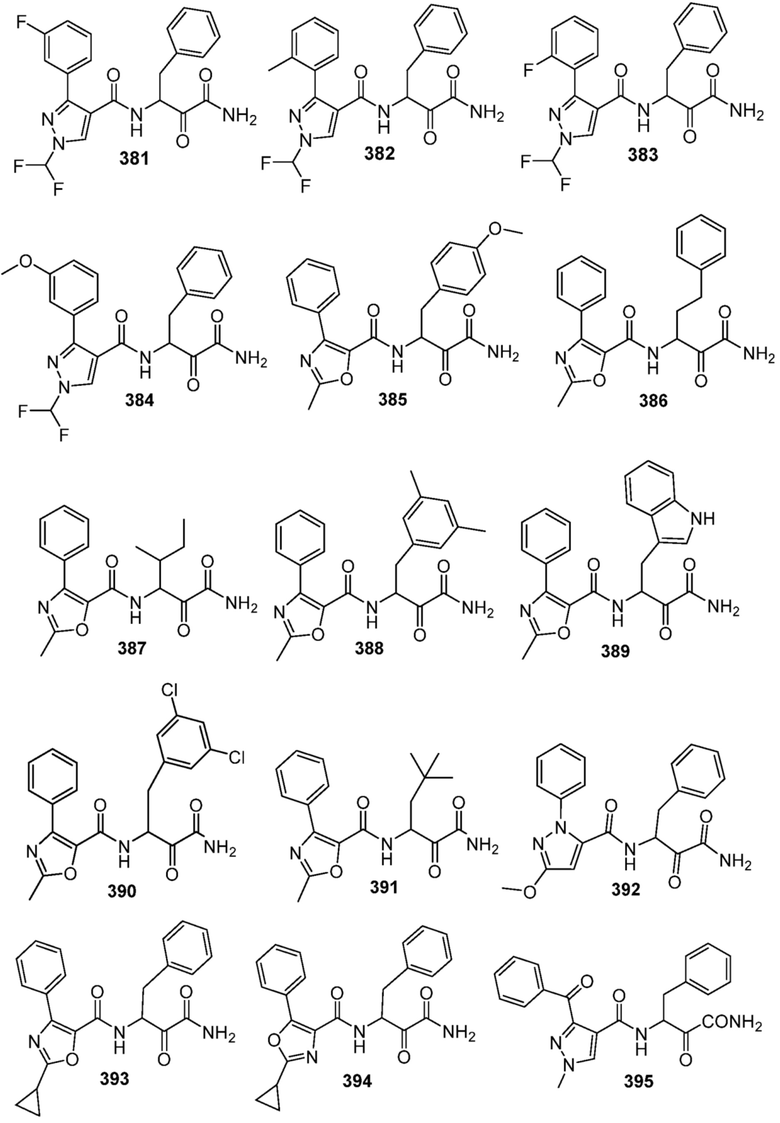
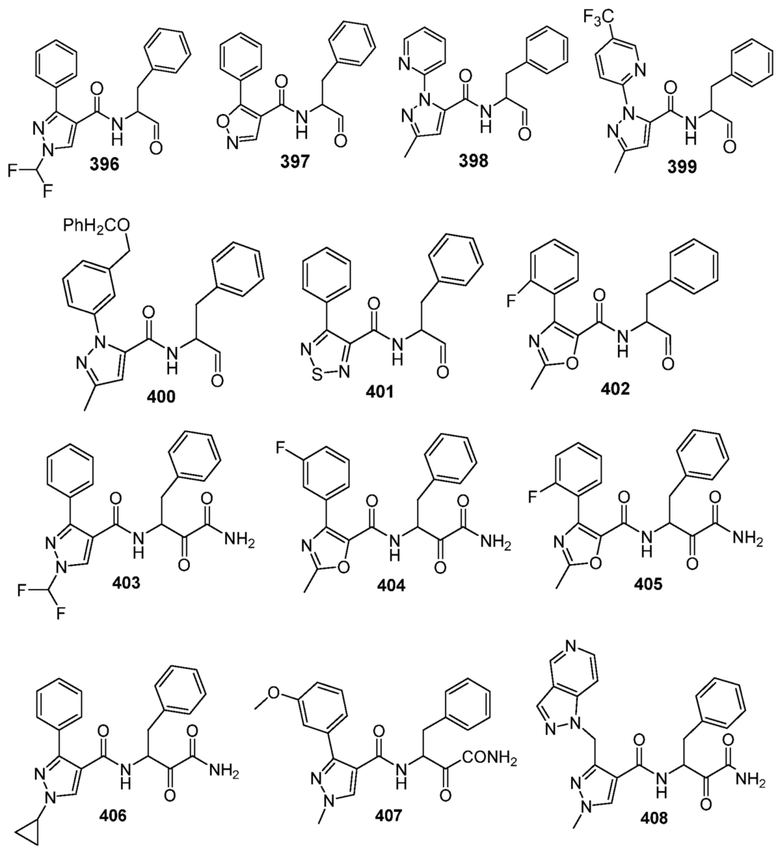
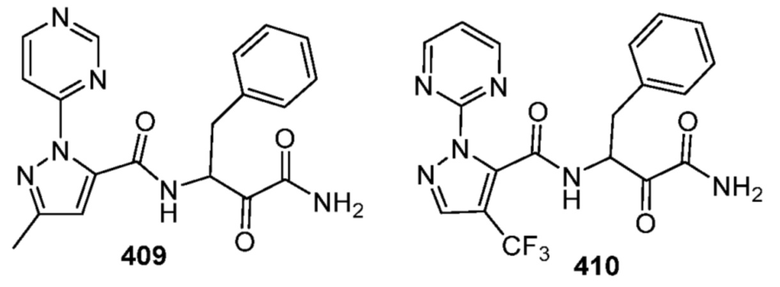
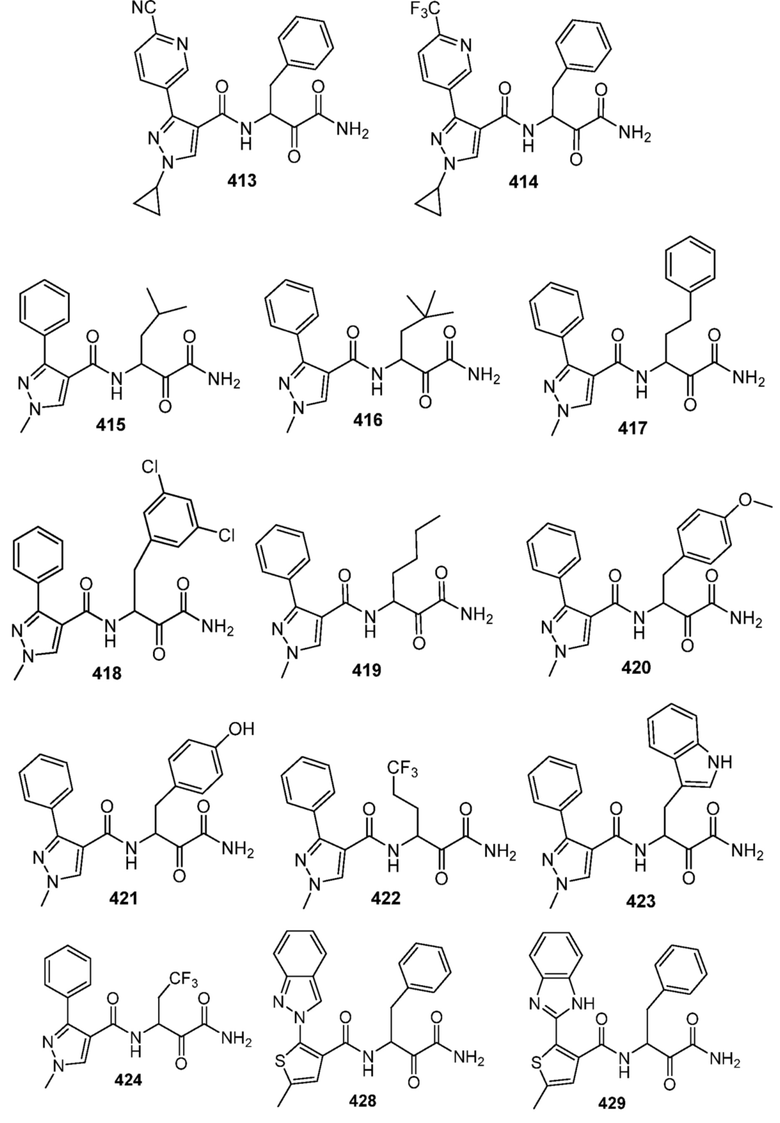
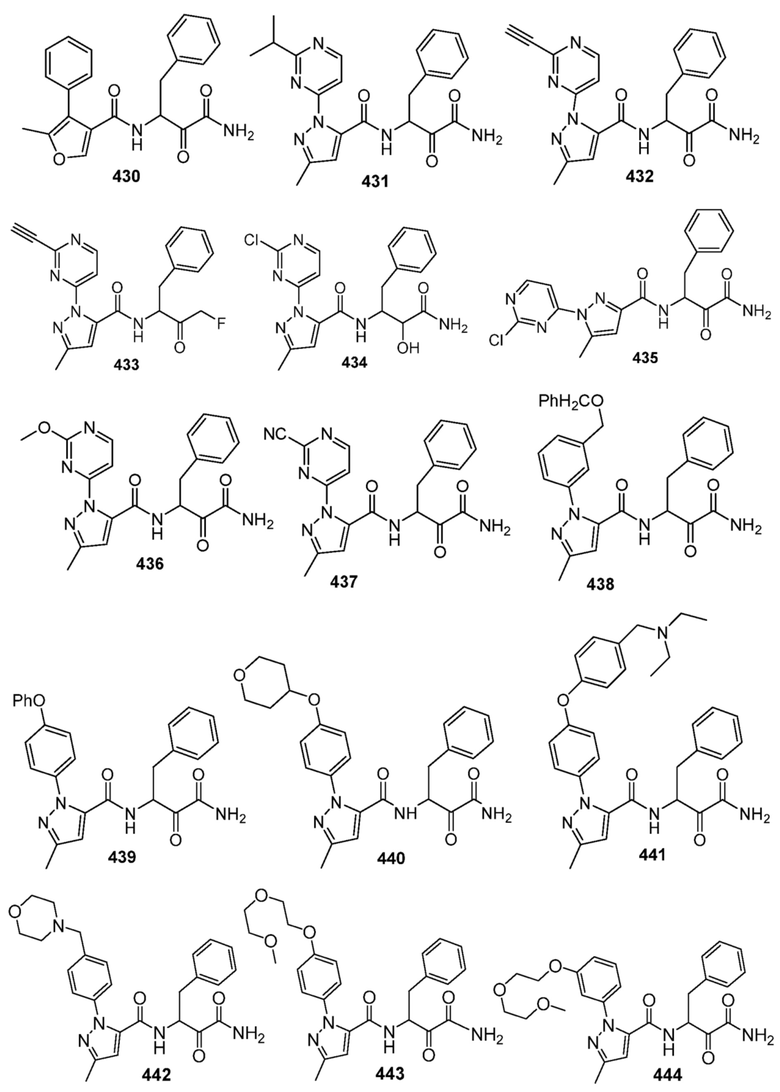
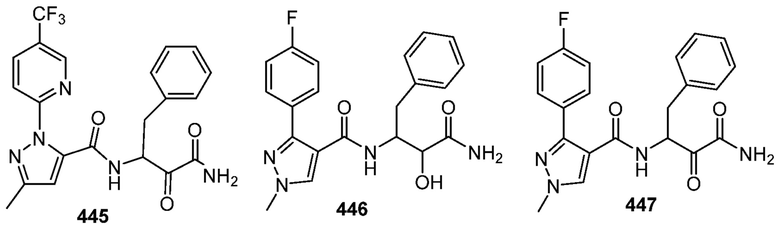
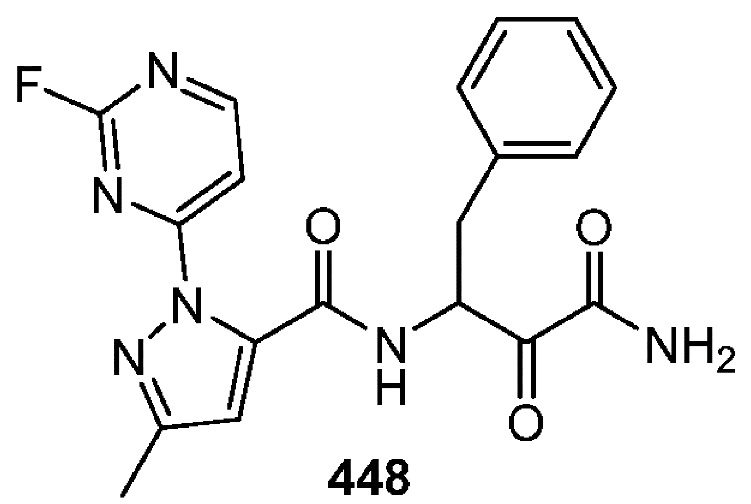
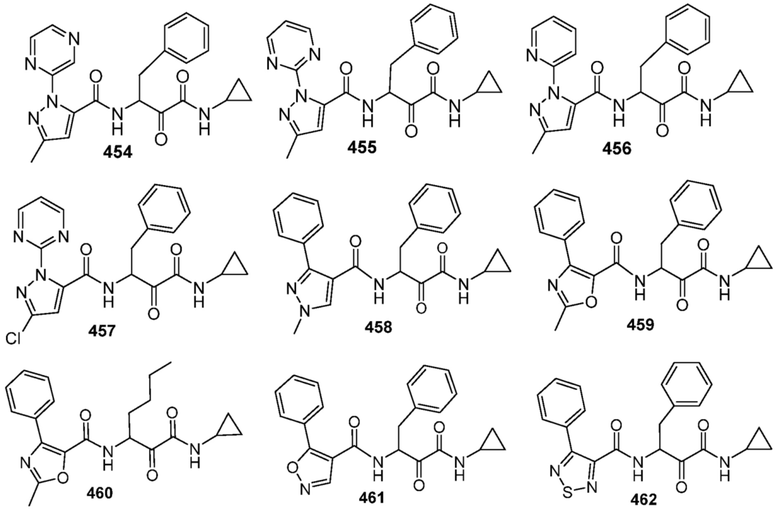
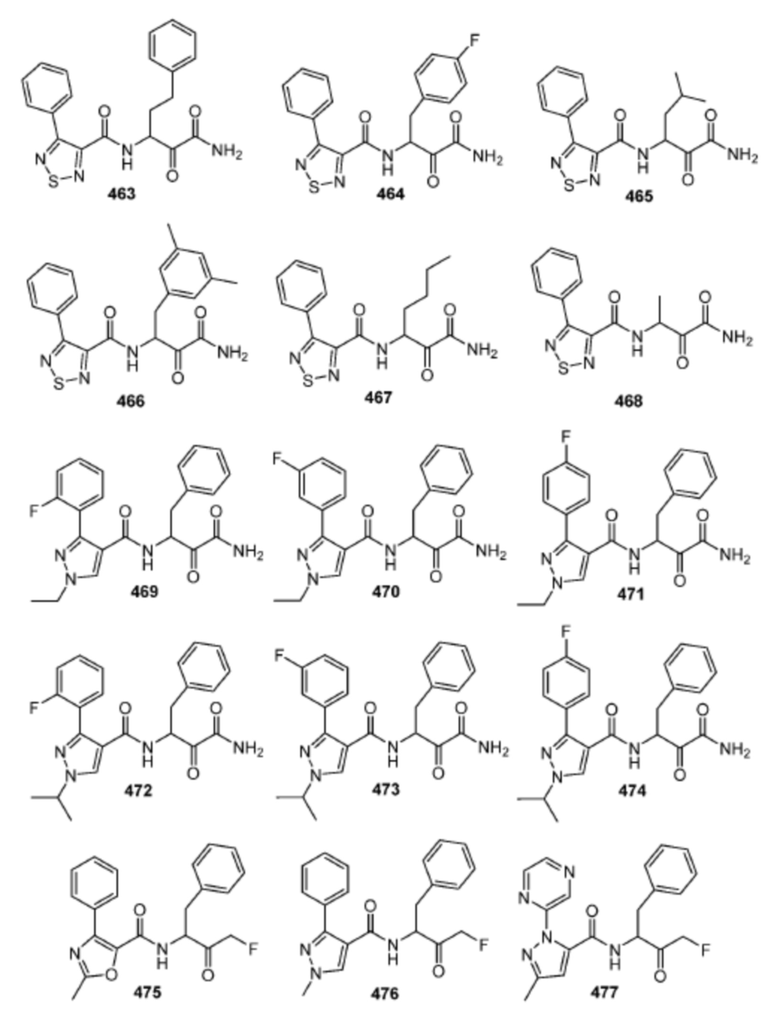
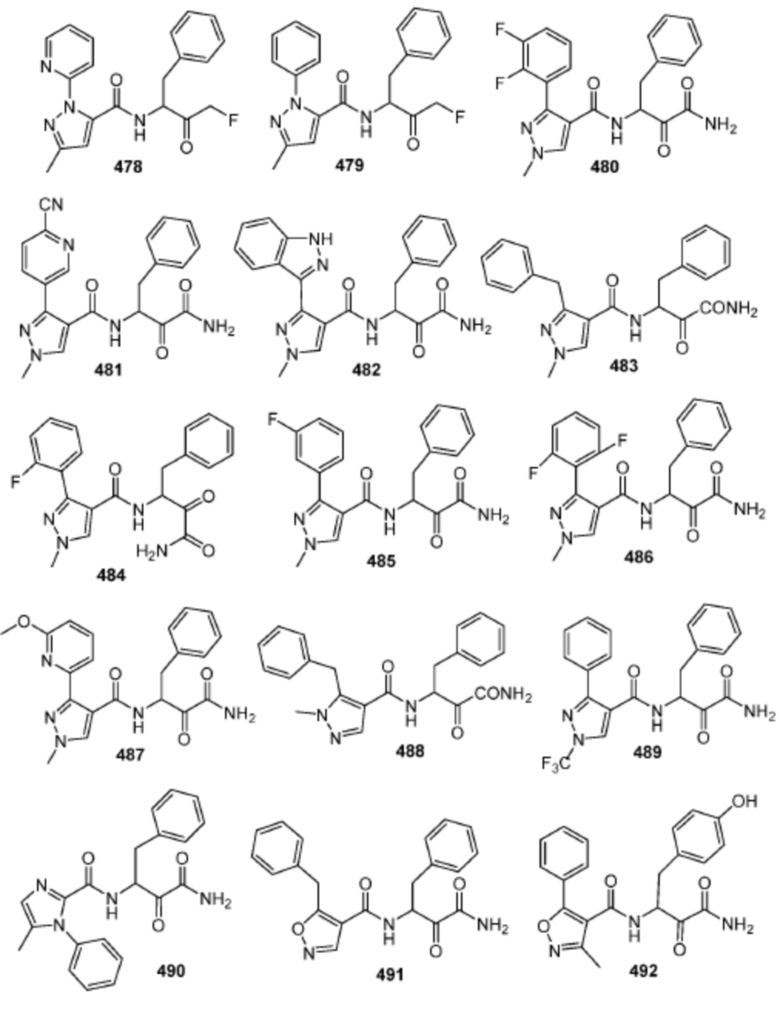
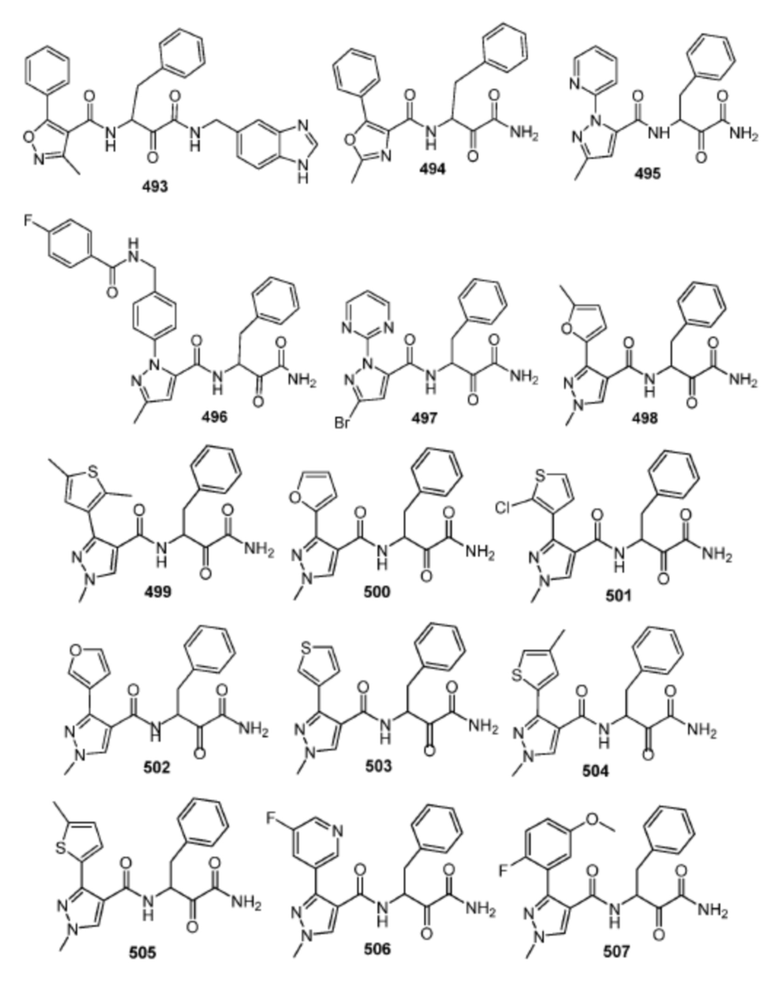
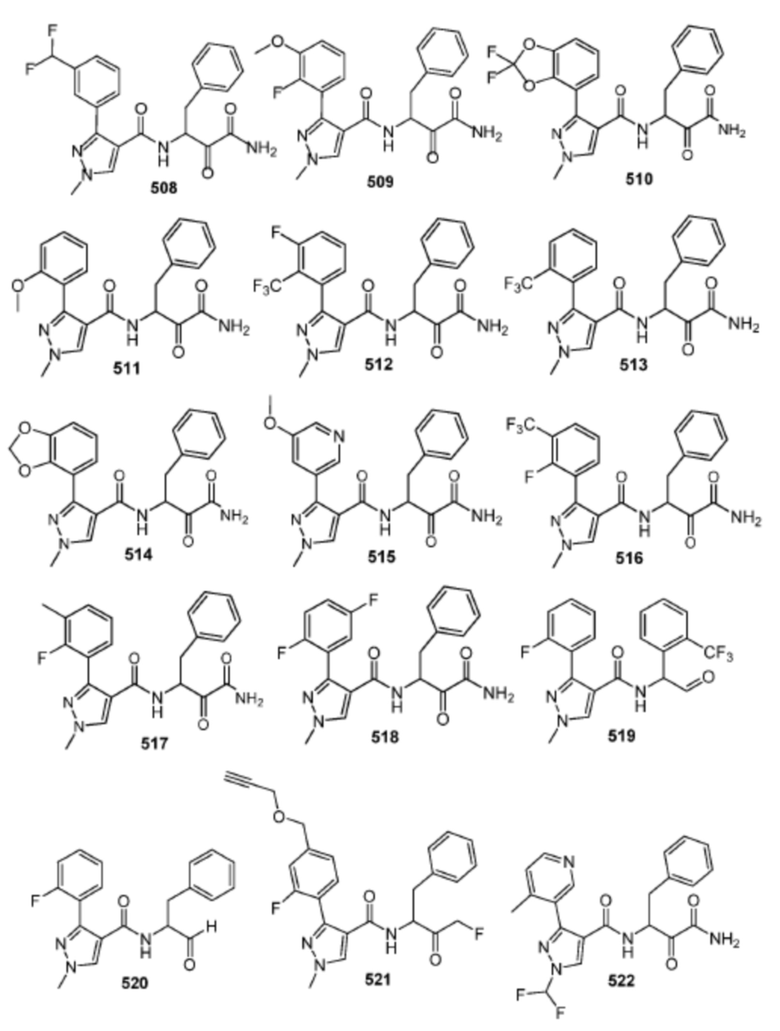
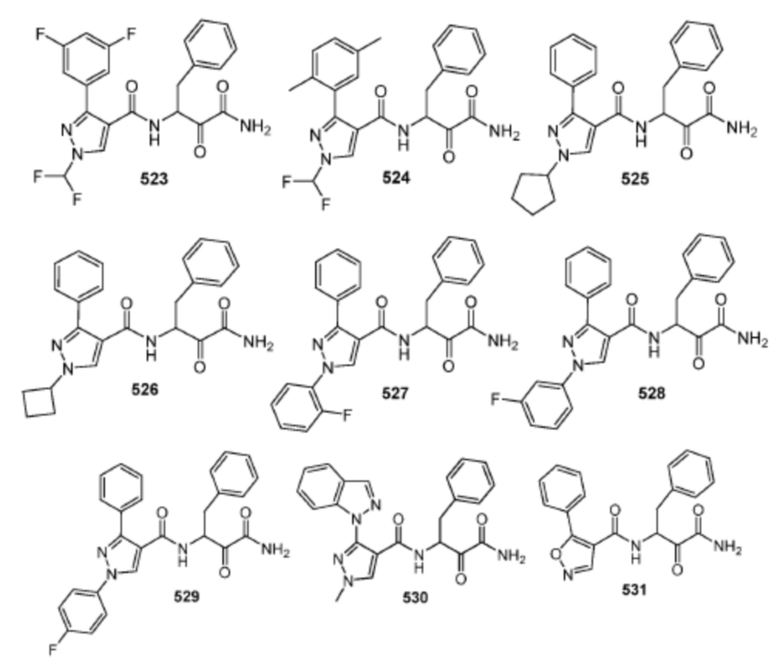
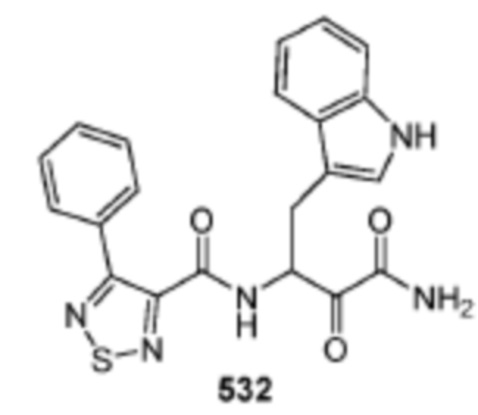
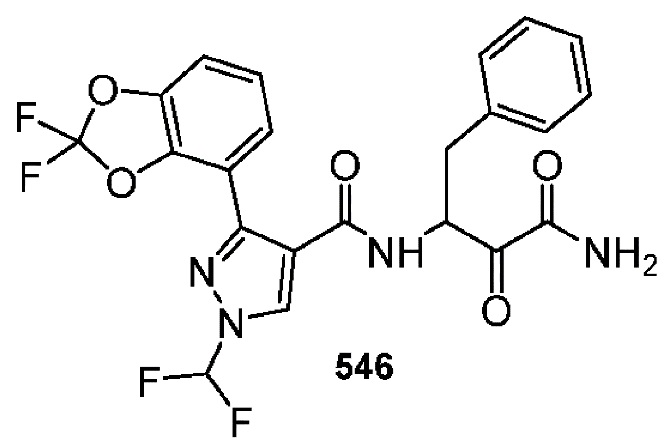
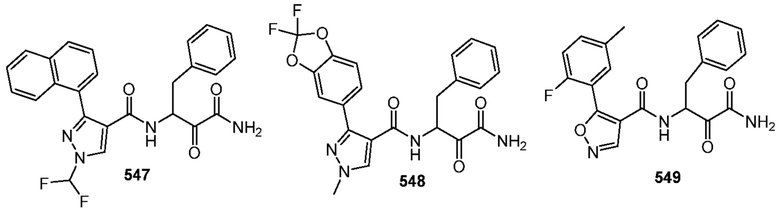
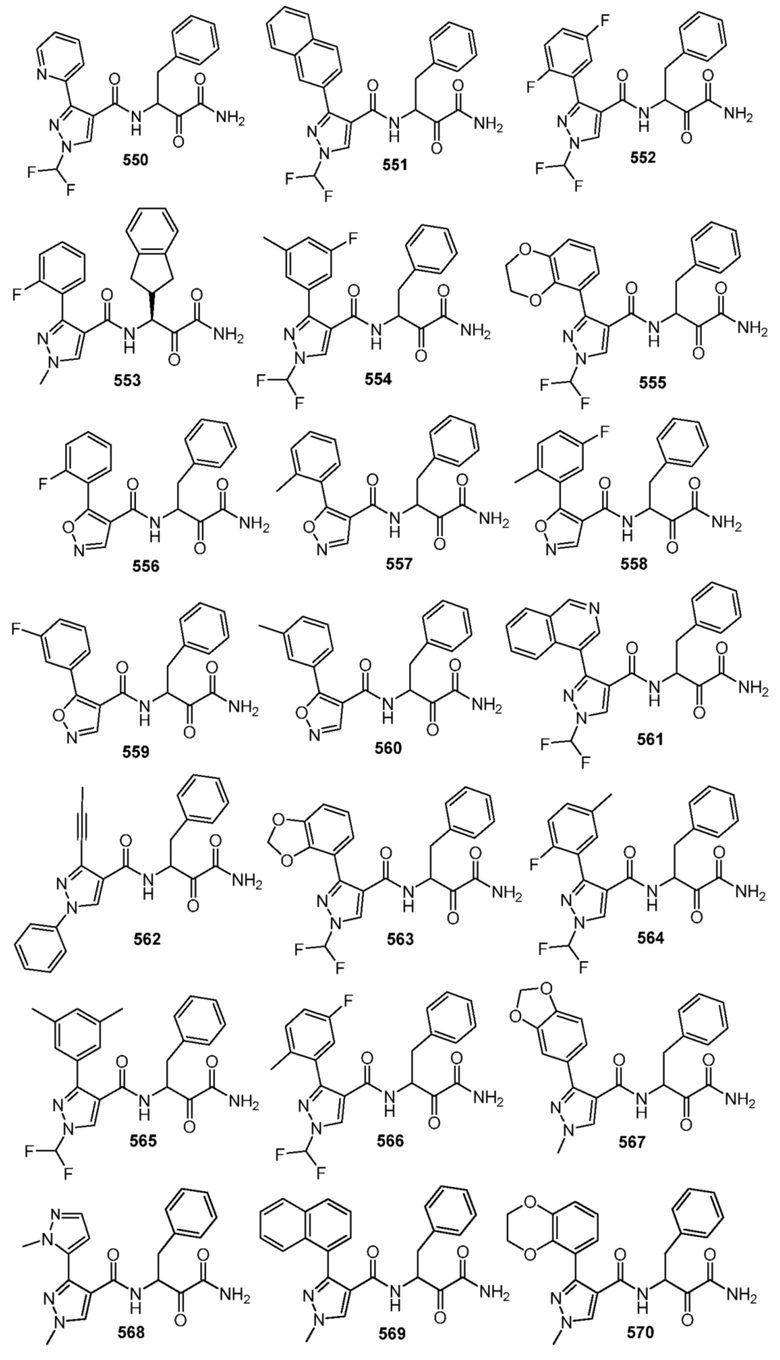
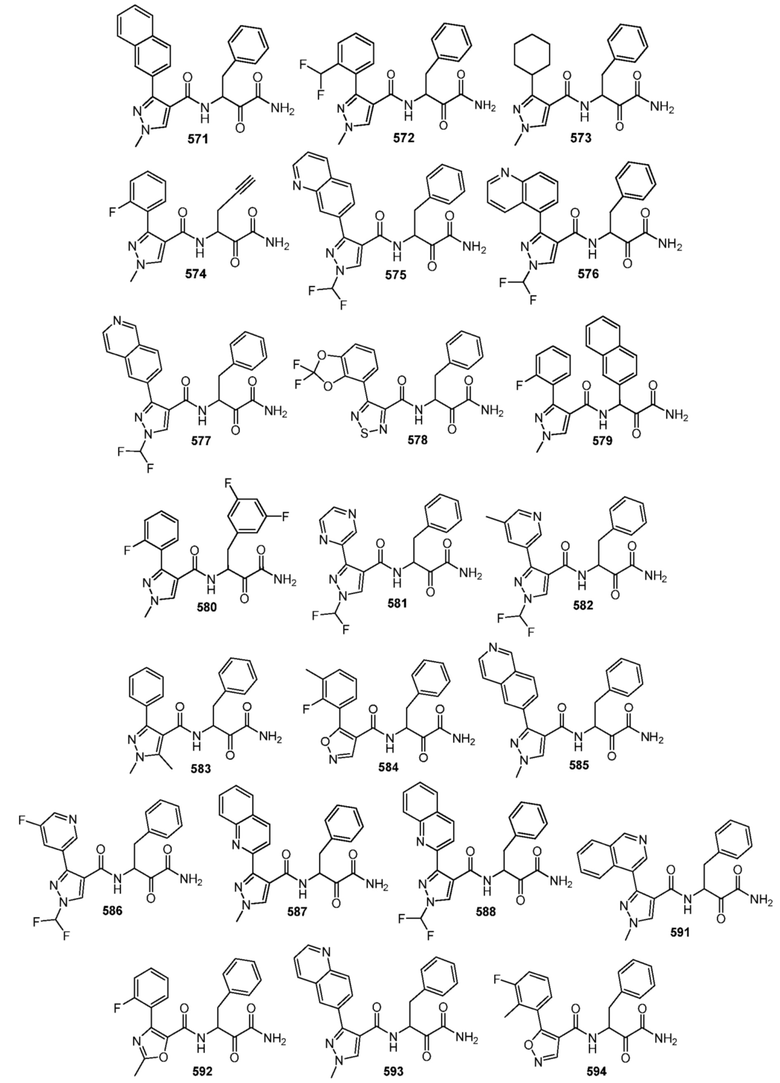
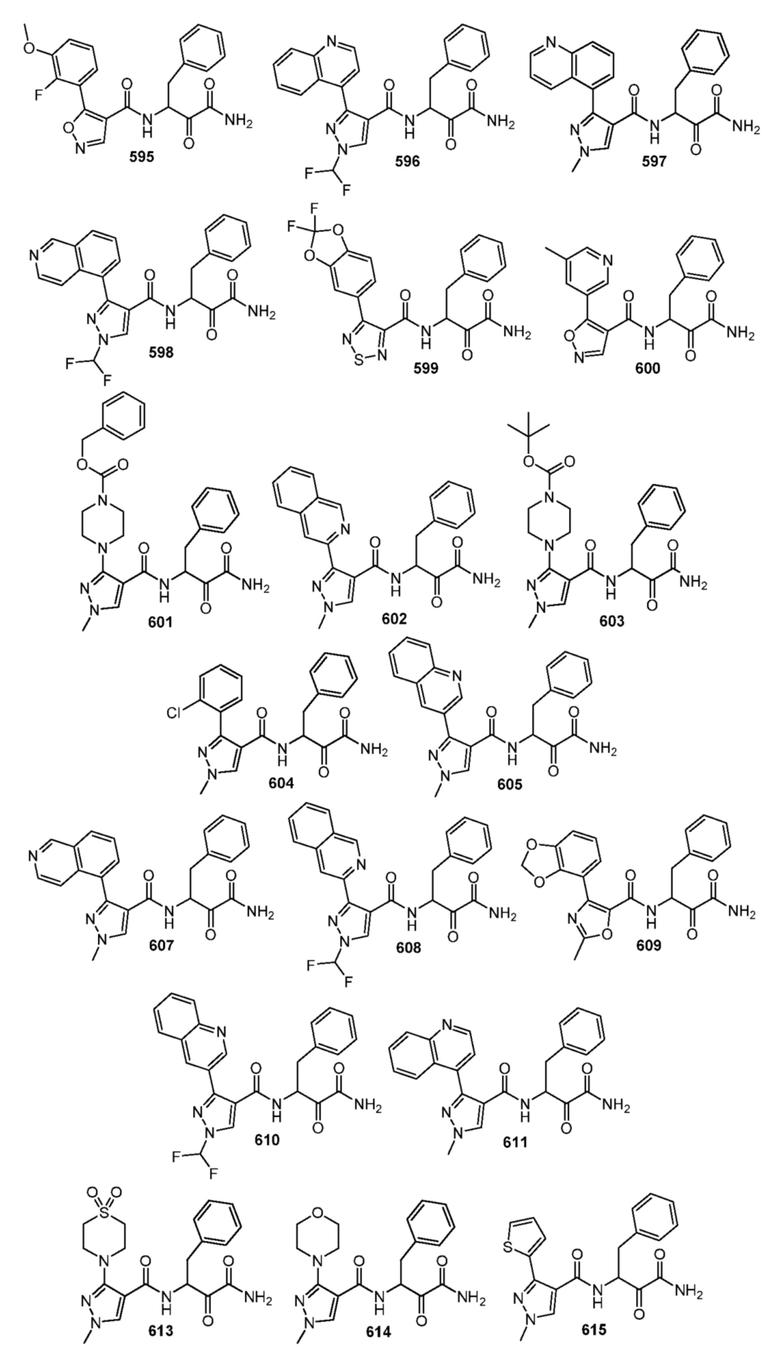
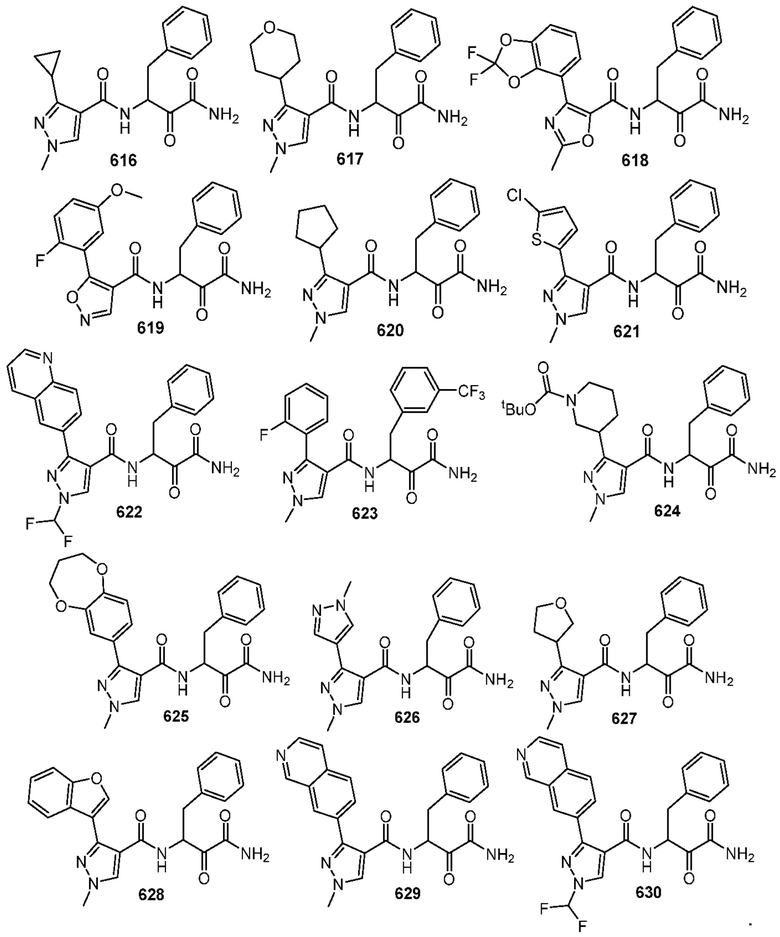
56. Соединение, имеющее структуру, выбранную из группы, состоящей из:
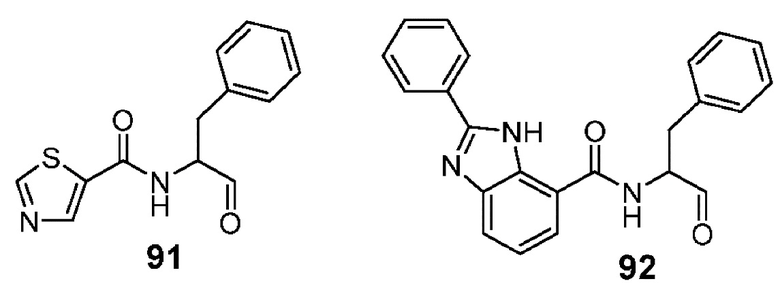
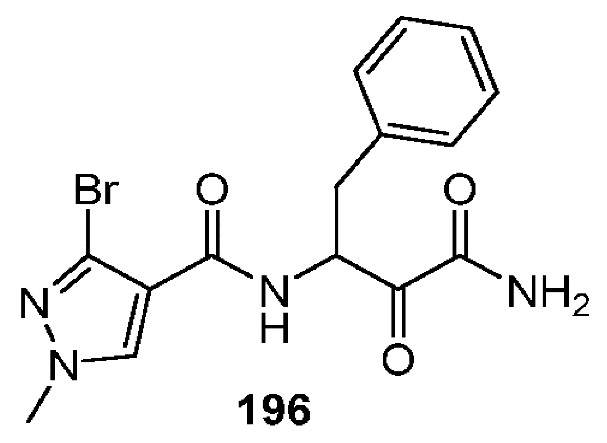
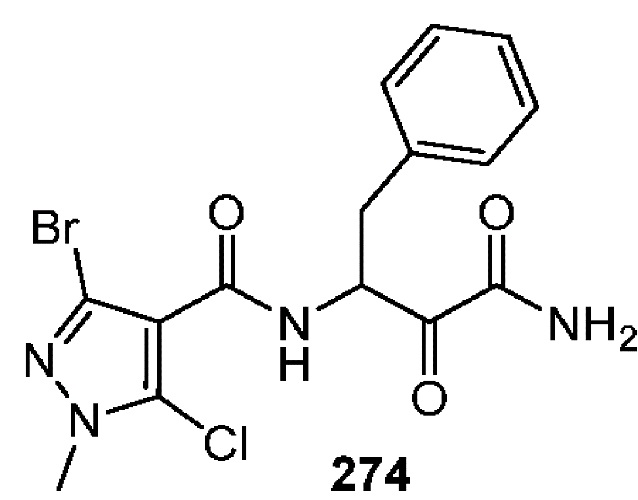
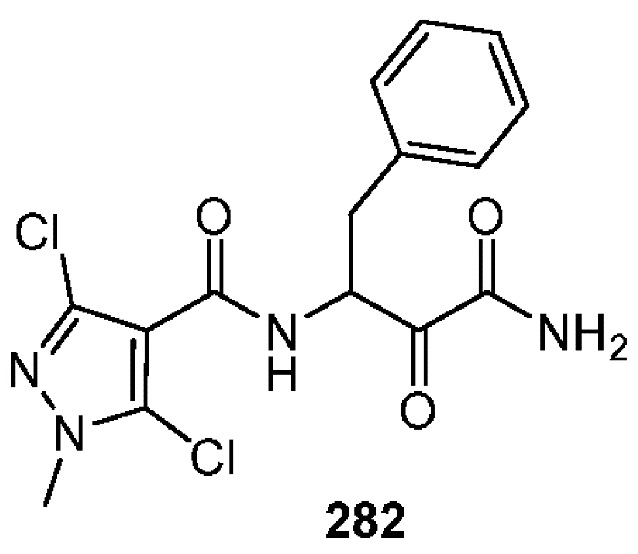
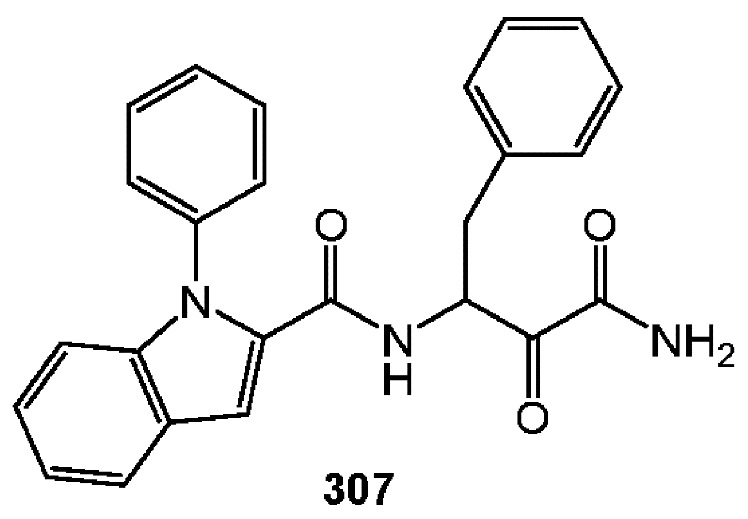
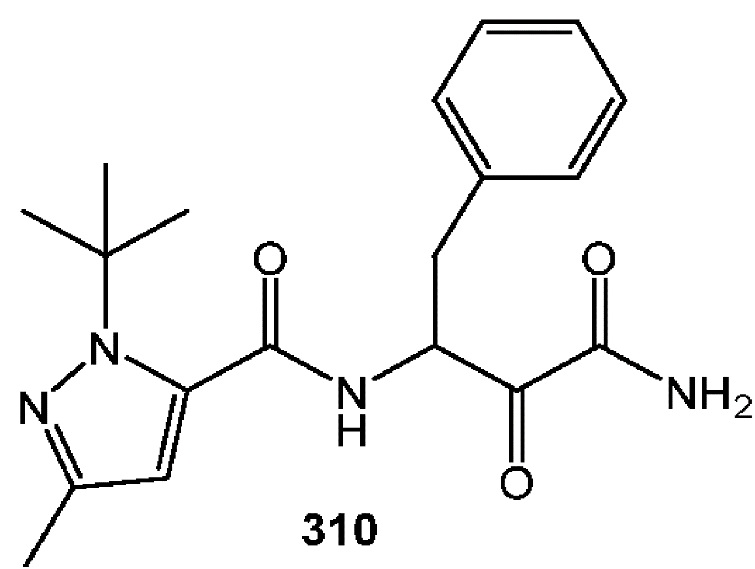
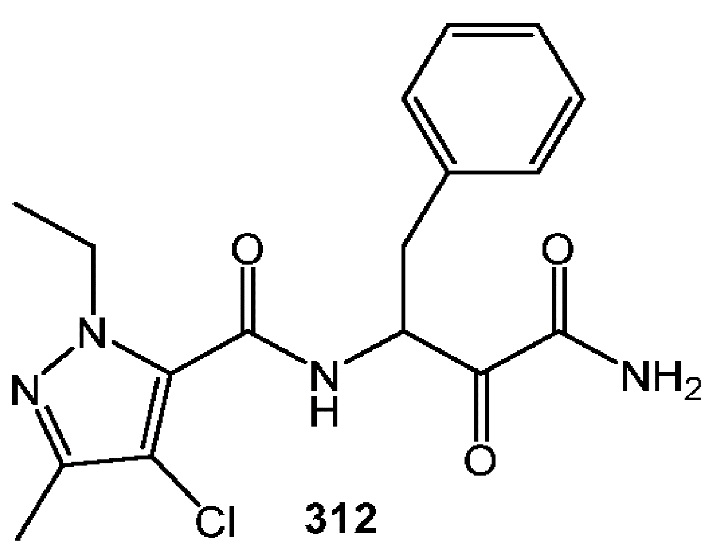
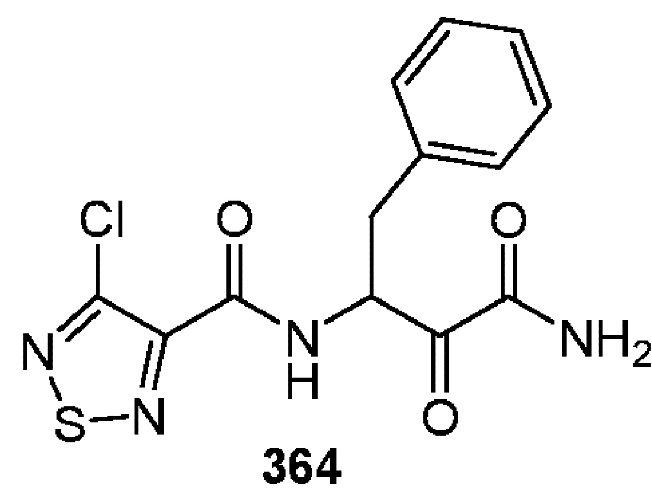
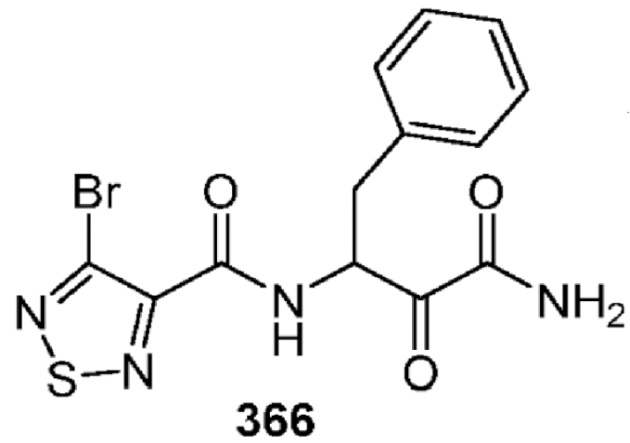
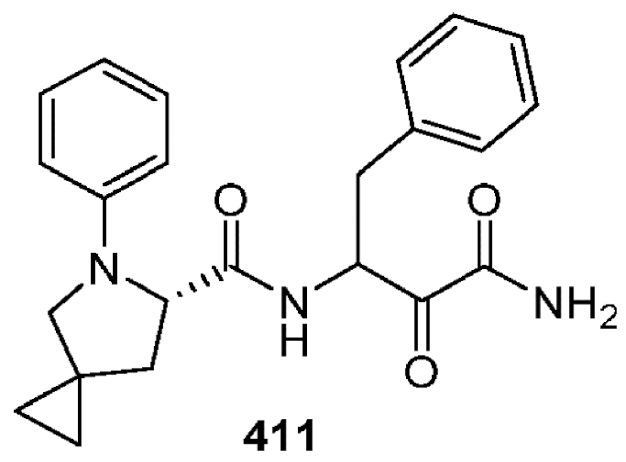
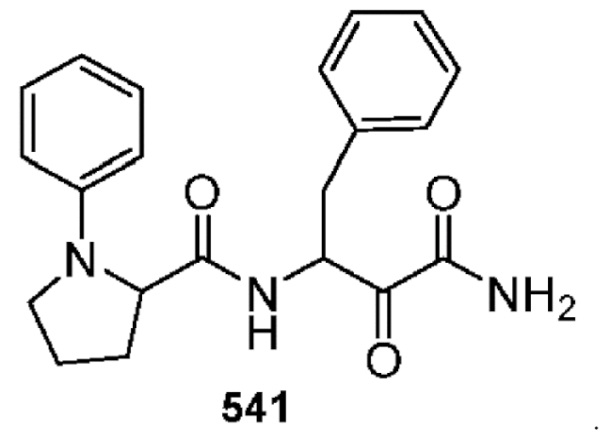
57. Фармацевтическая композиция, имеющая модулирующую активность в отношении кальпаинов CAPN1, CAPN2, CAPN9 и любой их комбинации, содержащая эффективное количество по меньшей мере одного соединения по любому из пп. 1-56 и фармацевтически приемлемое вспомогательное вещество.
58. Способ лечения фиброзного заболевания или его вторичного заболевания или состояния, включающий введение субъекту, нуждающемуся в этом, соединения по любому из пп. 1-56.
59. Способ по п. 58, где заболевание выбрано из группы, состоящей из фиброза печени, фиброза почки, фиброза легкого, пневмонита гиперчувствительности, интерстициального фиброза, системной склеродермии, макулярной дегенерации, фиброза поджелудочной железы, фиброза селезенки, фиброза сердца, медиастинального фиброза, миелофиброза, эндомиокардиального фиброза, ретроперитонеального фиброза, прогрессирующего массивного фиброза, нефрогенного системного фиброза, фиброзных послеоперационных осложнений, хронической васкулопатии аллотрансплантата и/или хронического отторжения трансплантированных органов, фиброза, связанного с ишемически-реперфузионным повреждением, фиброза после инъекции, цирроза, диффузного паренхиматозного заболевания легких, болевого синдрома после вазэктомии и ревматоидного артрита.
60. Применение соединения по любому из пп. 1-56 для лечения фиброзного заболевания или его вторичного заболевания или состояния, где заболевание выбрано из группы, состоящей из фиброза печени, фиброза почки, фиброза легкого, пневмонита гиперчувствительности, интерстициального фиброза, системной склеродермии, макулярной дегенерации, фиброза поджелудочной железы, фиброза селезенки, фиброза сердца, медиастинального фиброза, миелофиброза, эндомиокардиального фиброза, ретроперитонеального фиброза, прогрессирующего массивного фиброза, нефрогенного системного фиброза, фиброзных послеоперационных осложнений, хронической васкулопатии аллотрансплантата и/или хронического отторжения трансплантированных органов, фиброза, связанного с ишемически-реперфузионным повреждением, фиброза после инъекции, цирроза, диффузного паренхиматозного заболевания легких, болевого синдрома после вазэктомии и ревматоидного артрита.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| WO 00/55114 А1, 21.09.2000 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| WO 00/55125 А2, 21.09.2000 | |||
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| W | |||
| BLUM ET AL., Complementary use of ion trap/time-of-flight mass spectrometry in combination with capillary high-pressure liquid chromatography: early characterization of in vivo metabolites of | |||

