ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтической комбинации, содержащей (a) ингибитор Mdm2 и (b)(i) ингибитор MEK и/или (b)(ii) ингибитор Bcl2, в частности, для применения для лечения рака. Настоящее изобретение также относится к применениям такой комбинации для получения лекарственного средства для лечения рака; к способам лечения рака у индивидуума, нуждающегося в этом, включающим введение указанному индивидууму в совокупности терапевтически эффективного количества указанной комбинации; к фармацевтическим композициям, содержащим такую комбинацию, и к их коммерческим упаковкам.
УРОВЕНЬ ТЕХНИКИ, К КОТОРОМУ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Появление направленной терапии рака увеличило продолжительность жизни пациентов при различных злокачественных опухолях и помогло оценить комплексность опухолей посредством исследования механизмов резистентности лекарственных средств. Тот факт, что клинические ответы на направленные средства, как правило, являются неполными и/или временными, является следствием множества факторов, которые можно широко подразделить на два класса: токсичность, которая препятствует оптимальному дозированию лекарственных средств и, следовательно, ограничивают поражение цели (Brana and Siu 2012, Chapman, Solit et al. 2014), и способность раков к адаптации и поддержанию их пролиферативного потенциала в противодействие возмущающим воздействиям (Druker 2008, Chandarlapaty 2012, Doebele, Pilling et al. 2012, Duncan, Whittle et al. 2012, Katayama, Shaw et al. 2012, Lito, Rosen et al. 2013, Sullivan and Flaherty 2013, Solit and Rosen 2014). Комбинации лекарственных средств могут быть направлены на оба этих фактора посредством повышения общей эффективности и в то же время нацеливания на устойчивость и комплексность опухоли для противодействия резистентности (Robert, Karaszewska et al. 2015, Turner, Ro et al. 2015). Еще неясно, сколько требуется лекарственных средств и на какие процессы необходимо осуществлять нацеливание в комбинации для преодоления рака. Однако практически точно, что необходимо ингибировать различные каскады или движущие факторы, что, наиболее вероятно, требует двух или более лекарственных средств (Bozic, Reiter et al. 2013). Это подтверждается успехом комбинирования общепринятых химиотерапевтических средств для лечения раков (DeVita 1975), и комбинированных способов терапии инфекционных заболеваний, таких как ВИЧ (Porter, Babiker et al. 2003), а также теоретическими подходами, демонстрирующими, как биологическая устойчивость может быть преодолена путем увеличения порядка возмущающих воздействий (Lehar, Krueger et al. 2008).
Несмотря на многочисленные возможности лечения для пациентов с конкретными типами рака, остается потребность в эффективных и безопасных комбинированных способах терапии, которые можно проводить для длительного лечения рака.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Задачей настоящего изобретения является предоставление лекарственного средства для улучшения лечения рака, в частности, для улучшения лечения рака посредством ингибирования клеточного роста (пролиферации) и индукции апоптоза. Задачей настоящего изобретения является поиск новых комбинированных способов терапии, которые селективно оказывают синергичное действие, ингибируя пролиферацию и/или индуцируя апоптоз.
Такие ингибиторы, как ингибиторы MDM2, ингибиторы MEK и ингибиторы BCL2, в качестве монотерапии демонстрируют антипролиферативную (цитостатическую) и проапоптотическую (цитотоксическую) активность в доклинических анализах in vitro и in vivo. Неожиданно, было обнаружено, что фармацевтическая комбинация, содержащая
(a) ингибитор MDM2, выбранный из (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она или его фармацевтически приемлемой соли, и (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она или его фармацевтически приемлемой соли; и
(b)
(i) ингибитор MEK, выбранный из группы, состоящей из траметиниба, (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, (S)-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида, PD0325901, PD-184352, RDEA119, XL518, AS-701255, AS-701173, AS703026, RDEA436, E6201, RO4987655, RG7167 и RG7420 или их фармацевтически приемлемой соли; и/или
(ii) ингибитор Bcl2, выбранный из группы, состоящей из ABT-737, ABT-263 (навитоклакс) и ABT-199 или их фармацевтически приемлемой соли,
имеет благоприятное синергическое взаимодействие, улучшенную активность против рака, улучшенный антипролиферативный эффект и улучшенный проапоптотический эффект. Эти комбинации продемонстрировали синергичный эффект в отношении ингибирования роста клеток и индукции клеточной смерти посредством апоптоза.
Кроме того, было обнаружено, что комбинация
(a) ингибитора MDM2, выбранного из (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она или его фармацевтически приемлемой соли, и (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она или его фармацевтически приемлемой соли; и
(b)
(i) ингибитора MEK, выбранного из группы, состоящей из траметиниба, (2-гидроксиэтокси)-амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, (S)-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида, PD0325901, PD-184352, RDEA119, XL518, AS-701255, AS-701173, AS703026, RDEA436, E6201, RO4987655, RG7167 и RG7420 или их фармацевтически приемлемой соли;
и/или
(ii) ингибитора Bcl2, выбранного из группы, состоящей из ABT-737, ABT-263 (навитоклакс) и ABT-199 или их фармацевтически приемлемой соли,
преимущественно может содержать дополнительные ингибиторы, выбранные из ингибиторов EGFR, ингибиторов PI3K и ингибиторов BRAF. Кроме того, к комбинации ингибитора MDM2 ("MDM2i") и траметиниба можно добавлять ингибитор CDK4/6 или стандартное средство, такое как паклитаксел, что может приводить к дополнительному синергичному эффекту или мощной индукции апоптоза. Комбинация ингибитора MDM2 с ингибитором Bcl2 может быть дополнена ингибитором BRAF (например, дабрафениб) и ингибитором CMET (например, PF-04217903) с получением четырехкомпонентной комбинации. Было обнаружено, что последняя из комбинаций является слабо синергичной, но сильно индуцирует апоптоз.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтическую комбинацию по изобретению и по меньшей мере один фармацевтически приемлемый носитель.
В одном аспекте настоящее изобретение относится к фармацевтической комбинации или фармацевтической композиции по изобретению для применения в качестве лекарственного средства.
В другом аспекте настоящее изобретение относится к фармацевтической комбинации или фармацевтической композиции по изобретению для применения для лечения рака.
В другом аспекте изобретение относится к применению фармацевтической комбинации по изобретению для получения лекарственного средства для лечения рака.
В другом аспекте настоящее изобретение относится к способу лечения рака у индивидуума, нуждающегося в этом, включающему введение индивидууму терапевтически эффективного количества фармацевтической комбинации по настоящему изобретению или фармацевтической композиции по настоящему изобретению.
В частности, настоящее изобретение относится к приведенным ниже аспектам, преимущественным признакам и конкретным вариантам осуществления, соответственно, отдельно или в комбинации, как приведено в формуле изобретения ниже.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 Кривые доза-эффект для 8 клеточных линий рака ободочной и прямой кишки с TP53 дикого типа с ингибитором MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-оном (СОЕДИНЕНИЕ A) (круг) и ингибитором MEK траметинибом (треугольник) и их комбинацией (ромб). По оси x указан log10 длительности обработки; по оси y указано количество клеток после обработки относительно DMSO. Комбинации являются результатом комбинирования отдельных средств в фиксированных соотношениях (1:1). Жирная пунктирная линия указывает на количество клеток до начала обработки ("исходный уровень").
Фиг.2 Кривые доза-эффект для 8 клеточных линий рака ободочной и прямой кишки с TP53 дикого типа после обработки ингибитором MDM2 (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-оном (СОЕДИНЕНИЕ B) (круг) и ингибитором MEK траметинибом (треугольник) и их комбинацией (ромб). По оси x указан log10 длительности обработки; по оси y указано количество клеток после обработки относительно DMSO. Комбинации являются результатом комбинирования отдельных средств в фиксированных соотношениях (1:1). Жирная пунктирная линия указывает на количество клеток до начала обработки ("исходный уровень").
Фиг.3 Анализ с использованием изоболограммы на уровне ингибирования 75% для комбинаций ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) или ингибитора MDM2 (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она (СОЕДИНЕНИЕ B) (ось y) с ингибитором MEK траметинибом (ось x) для 8 клеточных линий рака ободочной и прямой кишки с TP53 дикого типа. Точки на диагональной кривой указывают на аддитивный эффект, точки справа от нее указывают на антагонизм и точки слева от нее указывают на синергию. Незакрашенным кругом показана комбинация с наиболее низким показателем аддитивности (наиболее высокая синергия) (см. таблицу 2 для значения).
Фиг.4 Максимальная индукция каспазы 3/7 для ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A), ингибитора MEK траметиниба и их комбинации в 5 клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа и через 24 ч, 48 ч и 72 ч (различные оттенки серого). По оси x указана обработка; по оси y указана максимальная индукция каспазы 3/7 (% клеток), наблюдаемая для каждой обработки.
Фиг.5 Долговременный анализ образования колоний для отдельных средств и комбинации ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) и ингибитора MEK траметиниба. "СОЕДИНЕНИЕ A (L)": 0,33 мкМ; "СОЕДИНЕНИЕ A (H)": 1 мкМ; "траметиниб (L)" для всех кроме LIM2405 и SW48: 4 нМ; "траметиниб (H)" для всех кроме LIM2405 и SW48: 12 нМ; "траметиниб (L)" для LIM2405 и SW48: 1 нМ, "траметиниб (H)" для LIM2405 и SW48: 3 нМ. (A) Репрезентативные изображения клеток после окрашивания кристаллическим фиолетовым. (B) Количественное определение сигнала кристаллического фиолетового с (A). Столбиками показано среднее значение±стандартное отклонение для n=3 повторений. Для теста значимости см. таблицу 3. RFU=относительная единица флуоресценции.
Фиг. 6 FACS-анализ для ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A), ингибитора MEK траметиниба и их комбинации после обработки в течение 24 ч. "СОЕДИНЕНИЕ A (L)": 0,33 мкМ; "СОЕДИНЕНИЕ A (H)": 1 мкМ; "траметиниб (L)" для всех кроме LIM2405 и SW48: 4 нМ; "траметиниб (H)" для всех кроме LIM2405 и SW48: 12 нМ; "траметиниб (L)" для LIM2405 и SW48: 1 нМ, "траметиниб (H)" для LIM2405 и SW48: 3 нМ. Столбики стопкой указывают на процент клеточной популяции на каждой из фаз клеточного цикла: subG1, G1, S и G2.
Фиг.7 Анализ с использованием вестерн-блоттинга ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A), ингибитора MEK траметиниба и их комбинации после обработки в течение 24 ч. "СОЕДИНЕНИЕ A (L)": 0,33 мкМ; "СОЕДИНЕНИЕ A (H)": 1 мкМ; "траметиниб (L)" для всех кроме LIM2405 и SW48: 4 нМ; "траметиниб (H)" для всех кроме LIM2405 и SW48: 12 нМ; "траметиниб (L)" для LIM2405 и SW48: 1 нМ, "траметиниб (H)" для LIM2405 и SW48: 3 нМ.
Фиг.8 Анализ посредством кОТ-ПЦР 5 генов-мишеней для ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A), ингибитора MEK траметиниба и их комбинации после обработки в течение 10 ч. "СОЕДИНЕНИЕ A (L)": 0,33 мкМ; "СОЕДИНЕНИЕ A (H)": 1 мкМ; "траметиниб (L)" для всех кроме LIM2405 и SW48: 4 нМ; "траметиниб (H)" для всех кроме LIM2405 и SW48: 12 нМ; "траметиниб (L)" для LIM2405 и SW48: 1 нМ, "траметиниб (H)" для LIM2405 и SW48: 3 нМ. Столбиками показана дифференциальная экспрессия в масштабе log2 по сравнению с обработкой DMSO, планками погрешности указано стандартное отклонение для n=2 реплик.
Фиг.9 Кривые доза-эффект для ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) (круги), ингибитора MEK траметиниба (СОЕДИНЕНИЕ B, треугольники), ингибитора BCL-2/-XL навитоклакса (ABT-263) (СОЕДИНЕНИЕ C, ромбы) и их комбинаций A+B (круги, пунктирная линия), A+C (треугольники), B+C (ромбы) и A+B+C (круги, сплошная линия) для 5 клеточных линий рака ободочной и прямой кишки с TP53 дикого типа. По оси x показан log10 разведения обработки; по y указано количество клеток после обработки относительно DMSO. Жирной пунктирной линией указано количество клеток до начала лечения ("исходный уровень").
Фиг.10 Максимальная индукция каспазы 3/7 для ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A), ингибитора MEK траметиниба (B) и ингибитора BCL-2/-XL навитоклакса (ABT-263) (C) и их комбинаций A+B, A+C, B+C и A+B+C в 5 клеточных линий рака ободочной и прямой кишки с TP53 дикого типа и через 24 ч, 48 ч и 72 ч (различные оттенки серого цвета). По оси x указана обработка; по оси y указана максимальная индукция каспазы 3/7 (% клеток), наблюдаемая для каждой обработки.
Фиг.11 Кривые доза-эффект для ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) (круги), ингибитора MEK траметиниба (СОЕДИНЕНИЕ B, треугольники), ингибитора EGFR эрлотиниба (СОЕДИНЕНИЕ C, ромбы) и их комбинаций A+B (круги, пунктирная линия), A+C (треугольники), B+C (ромбы) и A+B+C (круги, сплошная линия) для 5 клеточных линий рака ободочной и прямой кишки с TP53 дикого типа. По оси x показан log10 разведения обработки; по y указано количество клеток после обработки относительно DMSO. Жирной пунктирной линией указано количество клеток до начала лечения ("исходный уровень").
Фиг.12 Максимальная индукция каспазы 3/7 для ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A), ингибитора MEK траметиниба (СОЕДИНЕНИЕ B) и ингибитора EGFR эрлотиниба (СОЕДИНЕНИЕ C), и их комбинаций A+B, A+C, B+C и A+B+C в 5 клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа и через 24 ч, 48 ч и 72 ч (различные оттенки серого цвета). По оси x указана обработка; по оси y указана максимальная индукция каспазы 3/7 (% клеток), наблюдаемая для каждой обработки.
Фиг.13 Кривые доза-эффект для ингибитора PIK3CA 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амида) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты (СОЕДИНЕНИЕ A) (круги), ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ B) (треугольники), ингибитора BCL-2/-XL навитоклакса (ABT-263) (СОЕДИНЕНИЕ C) (ромбы) и их комбинаций A+B (круги, пунктирная линия), A+C (треугольники), B+C (ромбы) и A+B+C (круги, сплошная линия) для 5 клеточных линий рака ободочной и прямой кишки с TP53 дикого типа. По оси x показан log10 разведения обработки; по y указано количество клеток после обработки относительно DMSO. Жирной пунктирной линией указано количество клеток до начала лечения ("исходный уровень").
Фиг.14 Максимальная индукция каспазы 3/7 для ингибитора PIK3CA 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амида) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты (СОЕДИНЕНИЕ A), ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ B), ингибитора BCL-2/-XL навитоклакса (ABT-263) (СОЕДИНЕНИЕ C), A+B, A+C, B+C и A+B+C в 5 клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа и через 24 ч, 48 ч и 72 ч (различные оттенки серого цвета). По оси x указана обработка; по оси y указана максимальная индукция каспазы 3/7 (% клеток), наблюдаемая для каждой обработки.
Фиг.15 Кривые доза-эффект для ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) (круг) и ингибитора BCL-2/-XL навитоклакса (ABT-263) (треугольник) и комбинации СОЕДИНЕНИЯ A и ABT-263 (ромб) для 5 клеточных линий рака ободочной и прямой кишки с TP53 дикого типа. По оси x показан log10 разведения обработки; по y указано количество клеток после обработки относительно DMSO. Жирной пунктирной линией указано количество клеток до начала лечения ("исходный уровень").
Фиг.16 Максимальная индукция каспазы 3/7 для СОЕДИНЕНИЯ A и ABT-263 и комбинации ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) и ингибитора BCL-2/-XL навитоклакса (ABT-263) в 5 клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа и через 24 ч, 48 ч и 72 ч (различные оттенки серого цвета). По оси x указана обработка; по оси y указана максимальная индукция каспазы 3/7 (% клеток), наблюдаемая для каждой обработки.
Фиг.17 На ксенотрансплантаты HCT-116 с мутантным KRAS воздействовали ингибитором MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-оном (СОЕДИНЕНИЕ A), ингибитором MEK траметинибом (СОЕДИНЕНИЕ B) и ингибитором BCL-2/-XL ABT-263 (СОЕДИНЕНИЕ C) или их комбинациями. В частности, на ксенотрансплантаты воздействовали носителем (G1), ABT-263 (G2, 100 мг/кг, каждые сутки), СОЕДИНЕНИЕМ A (G3, 100 мг/кг, три раза в неделю), траметинибом (G4, 0,3 мг/кг, каждые сутки), комбинацией СОЕДИНЕНИЯ A и траметиниба (G5), или комбинацией всех трех средств (G6). На 9 сутки к G3-G5 добавляли ABT-263. Показано среднее процентное изменение объема опухоли относительно первоначального объема опухоли. Планки погрешности соответствуют SEM.
Фиг.18 Поточные диаграммы, демонстрирующие процентное изменение объема опухоли (относительно исходного объема) для индивидуальных опухолей в группах G3-G6 (как описано в примере 10 и на фиг.17) после введения в течение 9 суток (A), и введения в течение 19 суток (10 суток после последовательного добавления ABT-263)(B).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЕ
В одном аспекте настоящее изобретение относится к фармацевтической комбинации, содержащей
(a) ингибитор MDM2, выбранный из (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она или его фармацевтически приемлемой соли, и (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она или его фармацевтически приемлемой соли; и (b)
(i) ингибитор MEK, выбранный из группы, состоящей из траметиниба, (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, (S)-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида, PD0325901, PD-184352, RDEA119, XL518, AS-701255, AS-701173, AS703026, RDEA436, E6201, RO4987655, RG7167 и RG7420 или их фармацевтически приемлемой соли; и/или
(ii) ингибитор Bcl2, выбранный из группы, состоящей из ABT-737, ABT-263 (навитоклакс) и ABT-199 или их фармацевтически приемлемой соли.
Было определено, что эту комбинацию можно использовать для эффективного лечения рака. В частности, было определено, что эту комбинацию можно использовать для эффективного лечения рака вследствие синергичного эффекта в отношении ингибирования клеточной пролиферации и/или индукции апоптоза. Таким образом, комбинации по настоящему изобретению, в частности, тройная и последующие комбинации, могут сдвинуть "цитостатический ответ" в сторону "цитотоксического" ответа, таким образом, достигая регрессии рака.
Форму единственного числа в контексте описания изобретения (особенно в контексте приведенной ниже формулы изобретения) следует истолковывать как охватывающую как единственное число, так и множественное число, если в настоящем описании нет иных указаний или если контекст явно не противоречит этому. Когда форму множественного числа используют для соединений, пациентов, раков и т.п., ее также следует понимать как означающую одно соединение, пациента и т.п.
Термин "синергический эффект", как используют в рамках изобретения, относится к действию двух или трех лекарственных средств, таких как, например, соединение формулы (I), например, соединение A, и по меньшей мере одно соединение-ингибитор MEK по настоящему изобретению, например, соединение A и по меньшей мере одно соединение-ингибитор BCL2 по настоящему изобретению, вызывающему эффект, например, замедление прогрессирования заболевания, в частности, рака или его симптомов, который превышает простую сумму эффектов каждого лекарственного средства, вводимого отдельно. Синергический эффект может быть вычислен, например, с использованием подходящих способов, таких как сигмоидальное уравнение для вычисления Emax (Holford, N. H. G. and Scheiner, L. B., Clin. Pharmacokinet. 6: 429-453 (1981)), уравнение аддитивности Леве (Loewe, S. and Muischnek, H., Arch. Exp. Pathol Pharmacol. 114: 313-326 (1926)) и уравнение медианного эффекта (Chou, T. C. and Talalay, P., Adv. Enzyme Regul. 22: 27-55 (1984)). Каждое уравнение, упомянутое выше, может быть применено к экспериментальным данным для получения соответствующего графика для облегчения оценки эффектов комбинации лекарственных средств. Соответствующими графиками, связанными с уравнениями, упоминаемыми выше, являются кривая концентрация-эффект, изоболограмма и кривая показателя аддитивности, соответственно.
В частности, было продемонстрировано, что комбинированное ингибирование MDM2 и MEK в линии рака ободочной и прямой кишки с TP53 дикого типа обеспечивает улучшенный (пример 1, фиг.1 и 2, таблица 2) и более длительный ответ (пример 1, фиг.5, таблица 3) по сравнению с каждым отдельным средством. Также комбинированное ингибирование MDM2 и Bcl2 в линии рака ободочной и прямой кишки с TP53 дикого типа продемонстрировало более мощную индукцию апоптоза по сравнению с отдельными средствами (пример 5, фиг.16). Более того, тройная комбинация ингибитора MDM2, ингибитора MEK и ингибитора Bcl2 вызвала синергичное ингибирование относительно пар лекарственных средств в 2/5 протестированных моделей на клетках рака ободочной и прямой кишки с TP53 дикого типа (пример 2, таблица 5), и в четырех из этих клеточных линий тройная комбинация продемонстрировала более мощный апоптоз по сравнению с парными комбинациями (пример 2, фиг.10). Таким образом, комбинации по настоящему изобретению обеспечивают возможность эффективной терапии, способной улучшать ответы по сравнению с каждым из отдельных средств, и может приводить к более длительным ответам в клинике.
Термины "ингибитор MDM2" или "ингибитор HDM2" или "ингибитор Mdm2", как используют в рамках изобретения, относятся к любому соединению, ингибирующему ассоциацию для взаимодействия HDM2/p53 (Mdm2/p53). HDM2 (человеческий гомолог double minute 2 мыши) является отрицательным регулятором p53. Ингибиторы Mdm2 являются пригодными в фармацевтических композициях для применения у человека или в ветеринарии, когда показано ингибирование ассоциации Mdm2/p53, например, для лечения опухолей и/или роста раковых клеток. В частности, ингибиторы Mdm2 являются пригодными для лечения рака у человека, поскольку прогрессирование этих раков может быть по меньшей мере частично зависимым от превышения функции "сдерживания" p53, например, сверхэкспрессии Mdm2.
Согласно настоящему изобретению, ингибитор Mdm2 представляет собой соединение, выбранное из группы, состоящей из
(6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она или его фармацевтически приемлемой соли, и
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она или его фармацевтически приемлемой соли.
Ингибитор MDM2 может представлять собой (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-он или его фармацевтически приемлемую соль. Ингибитор Mdm2 (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-он принадлежит к новому классу соединений имидазопирролидинонов и демонстрирует мощное ингибирование взаимодействия MDM2/p53 (этот термин включает, в частности, взаимодействие Hdm2/p53). В частности, это соединение выступает в качестве ингибитора взаимодействия MDM2 с p53 посредством связывания с MDM2. Ингибитор MDM2 (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-он, который является наиболее предпочтительным ингибитором Mdm2i согласно настоящему изобретению, представляет собой соединение формулы I, и описан в примере 102 WO2013/111105, которая включена в настоящее описание в качестве ссылки в полном объеме:
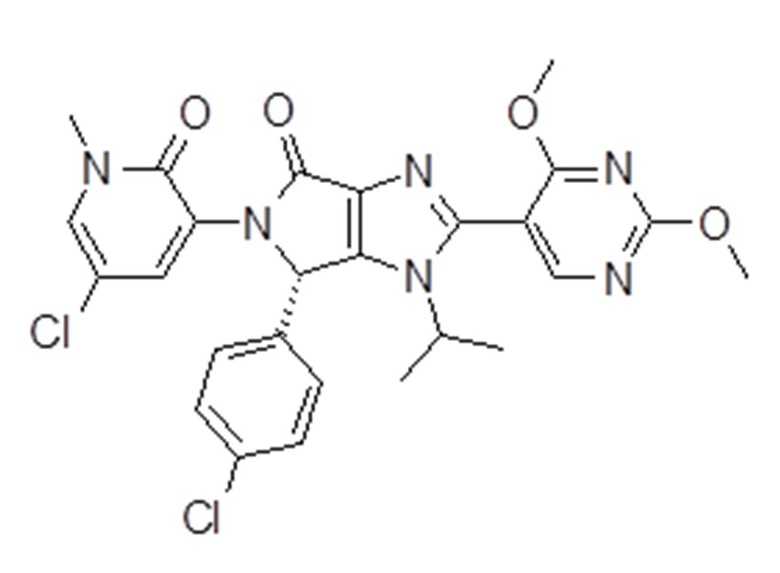 (I).
(I).
Кристаллические формы (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она описаны в качестве EX6, EX7 и EX8 в WO2013/111105. Изобретение охватывает сокристалл янтарной кислоты и соединения (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она. Соединение также может быть в форме сольвата этанола.
Ингибитор MDM2 также может представлять собой (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-он или его фармацевтически приемлемую соль. Ингибитор Mdm2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-он представляет собой соединение формулы II, и он описан в примере 106 WO2011/076786, которая включена в настоящее описание в качестве ссылки в полном объеме:
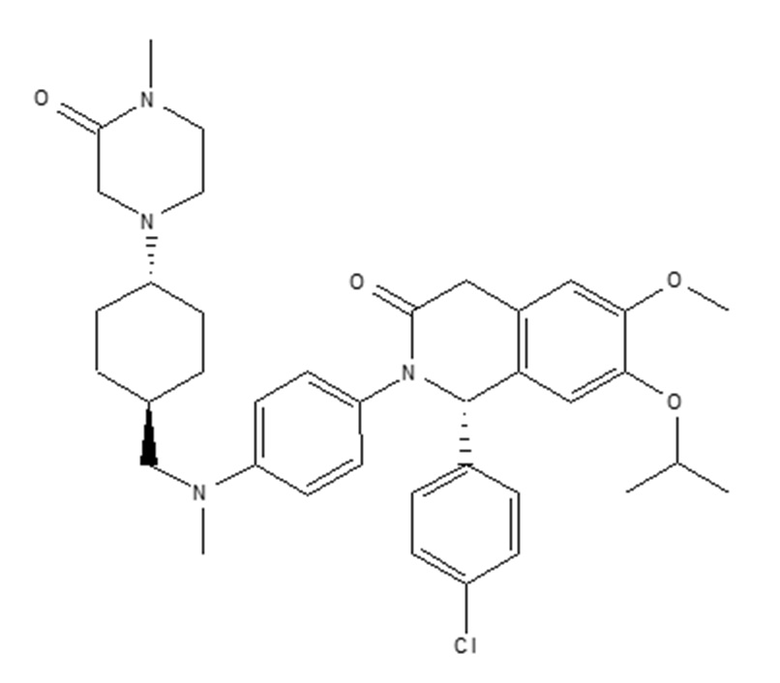 (II).
(II).
В одном варианте осуществления фармацевтически приемлемой солью (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она является бисульфат. Кристаллическая форма бисульфата (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она описана в WO2012/066095.
Термин "ингибитор MEK" определяют в настоящем описании как соединение, которое нацелено, снижает или ингибирует киназную активность MAP-киназы MEK. Мишень ингибитора MEK включает, но не ограничивается ими, ERK. Непрямая мишень ингибитора MEK включает, но не ограничивается ими, циклин D1.
Фармацевтические комбинации по настоящему изобретению могут включать по меньшей мере одно соединение ингибитора MEK, выбранное из группы, состоящей из траметиниба, (2-гидроксиэтокси)-амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, (S)-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида, PD0325901, PD-184352, RDEA119, XL518, AS-701255, AS-701173, AS703026, RDEA436, E6201, RO4987655, RG7167 и RG7420, или их фармацевтически приемлемой соли.
Предпочтительно ингибитор MEK представляет собой траметениб (N-(3-{3-циклопропил-5-[(2-фтор-4-йодфенил)амино]-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидропиридо[4,3-d]пиримидин-1(2H)-ил}фенил)ацетамид, также обозначаемый как JPT-74057 или GSK1120212). Траметиниб (GSK1120212) описан в публикации PCT № WO05/121142, которая включена в настоящее описание в качестве ссылки в полном объеме. Соединение было одобрено как Mekinist®.
Согласно настоящему изобретению, другим подходящим ингибитором MEK для комбинации по настоящему изобретению является соединение (2-гидроксиэтокси)-амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты формулы (III)
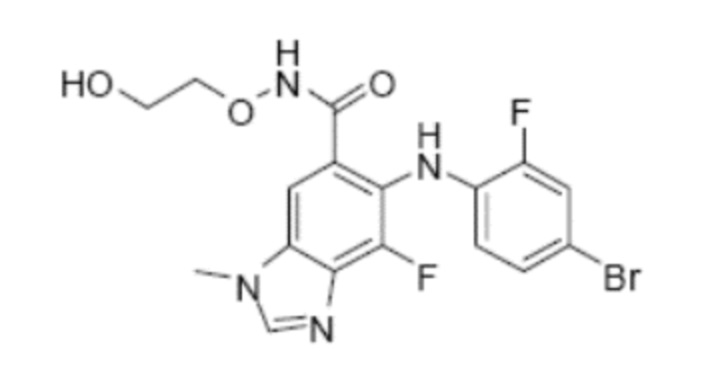 (III).
(III).
Соединение ингибитора MEK (2-гидроксиэтокси)-амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты описано в заявке PCT № WO 03/077914, и способы его получения описаны, например, в примере 18 указанной заявки.
Дополнительным пригодным ингибитором MEK для комбинации по настоящему изобретению является соединение (S)-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид формулы (IV)
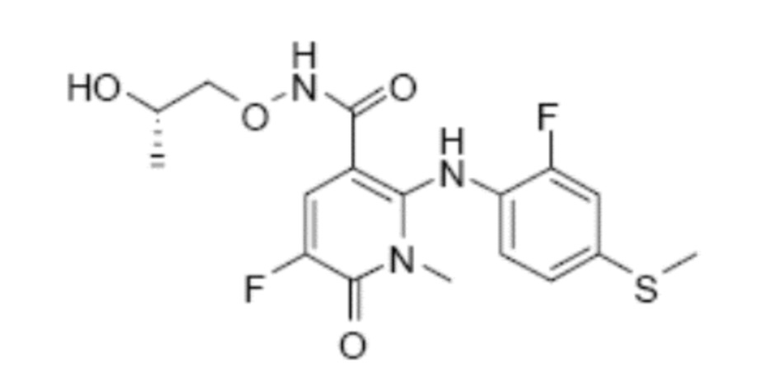 (IV).
(IV).
Соединение ингибитора MEK (S)-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид описано в примере 25-BB заявки PCT № WO2007/044084, и там же описаны способы его получения.
Особенно предпочтительной солью (2-гидроксиэтокси)-амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты является гидрохлорид или сульфат. Дополнительные фармацевтически приемлемые соли (2-гидроксиэтокси)-амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты и (S)-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида, пригодные в рамках настоящего изобретения, включают соли, описанные в заявке PCT № WO 03/077914 и заявке PCT № WO2007/044084, обе из которых включены в настоящее описание в качестве ссылок в полном объеме.
Дополнительные ингибиторы MEK, которые можно использовать в комбинации по настоящему изобретению, включают, но не ограничиваются ими, PD0325901 (Pfizer)(см. публикацию PCT № WO02/06213), PD-184352 (Pfizer), RDEA119 (Ardea Biosciences), XL518 (Exelexis), AS-701255 (Merck Serono), AS-701173 (Merck Serono), AS703026 (Merck Serono), RDEA436 (Ardea Biosciences, E6201 (Eisai)(cм. Goto et al, Journal of Pharmacology and Experimental Therapeutics, 3331(2): 485-495 (2009)), RO4987655 (Hoffmann-La Roche), RG7167 и/или RG7420.
Термины "ингибитор Bcl2" или "ингибитор BCL2" или "ингибитор BCL-2" или "ингибитор Bcl-2" определяют в настоящем описании как соединение, которое нацелено, снижает уровень или ингибирует семейство антиапоптотических белков B-клеточной лимфомы-2 (Bcl-2) (Bcl-2, Bcl-XL, Bcl-w, Mcl-1, Bfl1/A-1 и/или Bcl-B).
В одном варианте осуществления фармацевтическая комбинация по настоящему изобретению включает по меньшей мере одно соединение ингибитора Bcl2, выбранное из группы, состоящей из ABT-737, ABT-263 (навитоклакс) и ABT-199.
Особенно предпочтительным ингибитором Bcl2 по настоящему изобретению является навитоклакс (ABT-263) или его фармацевтически приемлемая соль. Навитоклакс представляет собой селективный высокоаффинный низкомолекулярный ингибитор Bcl-2 и родственного ингибитора апоптоза Bcl-xL (Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al.ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res, 2008;68:3421-8).
Согласно настоящему изобретению фармацевтическая комбинация может включать ингибитор MDM2 и ингибитор MEK; или она может включать ингибитор MDM2 и ингибитор Bcl2. Согласно настоящему изобретению, фармацевтическая комбинация, содержащая ингибитор MDM2 и ингибитор MEK или ингибитор MDM2 и Bcl2, кроме того, может преимущественно содержать дополнительный ингибитор, который еще более повышает противоопухолевую активность комбинации. Таким образом, тройная комбинация ингибитора MDM2, ингибитора MEK и ингибитора Bcl2 вызывала синергичное ингибирование относительно пар лекарственных средств в 2/5 моделях на клетках рака ободочной и прямой кишки с TP53 дикого типа (пример 2, таблица 5), и в четырех из этих клеточных линий комбинация продемонстрировала более мощный апоптоз по сравнению с парными комбинациями (пример 2, фиг.10).
Аналогично, фармацевтические комбинации по настоящему изобретению, содержащие (a) ингибитор MDM2 и (b)(i) ингибитор MEK, и/или (ii) ингибитор Bcl2 могут дополнительно преимущественно включать ингибитор EGFR.
Термин "ингибитор EGFR" определяют в настоящем описании как соединение, которое нацелено, снижает или ингибирует активность рецепторных тирозинкиназ семейства эпидермального фактора роста (EGFR, ErbB2, ErbB3, ErbB4 в качестве гомо- или гетеродимеров) или связывается с EGF или родственными EGF лигандами.
Соединение ингибитора EGFR, используемое в комбинации по настоящему изобретению, выбрано из группы, состоящей из эрлотиниба, гефитиниба, лапатиниба, канертиниба, пелитиниба, нератиниба, (R,E)-N-(7-хлор-1-(1-(4-(диметиламино)бут-2-еноил)азепан-3-ил)-1H-бензо[d]имидазол-2-ил)-2-метилизоникотинамида, панитумумаба, матузумаба, пертузумаба, нимотузумаба, залутумумаба, икотиниба, афатиниба и цетуксимаба, и их фармацевтически приемлемой солей.
Предпочтительно ингибитор EGFR представляет собой эрлотиниб или его фармацевтически приемлемую соль.
В одном варианте осуществления фармацевтическая комбинация, содержащая ингибитор MDM2 и ингибитор MEK, может дополнительно преимущественно включать ингибитор EGFR. Неожиданно было обнаружено, что эта тройная комбинация продемонстрировала более мощный апоптоз по сравнению с парными комбинациями (пример 3, фиг.12).
В предпочтительном варианте осуществления фармацевтическая комбинация содержит ингибитор MDM2, выбранный из (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она или его фармацевтически приемлемой соли, и (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она или его фармацевтически приемлемой соли; ингибитор MEK траметиниб или его фармацевтически приемлемую соль, и ингибитор EGFR эрлотиниб или его фармацевтически приемлемую соль.
Согласно настоящему изобретению, фармацевтические комбинации по настоящему изобретению, содержащие (a) ингибитор MDM2 и (b)(i) ингибитор MEK, и/или (ii) ингибитор Bcl2, могут дополнительно преимущественно содержать ингибитор PI3K.
Термин "ингибитор фосфатидилинозитол 3-киназы" или "ингибитор PI3K" определяют в настоящем описании как соединение, которое нацелено, снижает уровень или ингибирует PI3-киназу. Было показано, что активность PI3-киназы возрастает в ответ на ряд гормональных и обусловленных факторами роста стимулов, включая инсулин, тромбоцитарный фактор роста, инсулиноподобный фактор роста, эпидермальный фактор роста, колониестимулирующий фактор и фактор роста гепатоцитов, и она вовлечена в процессы, связанные с ростом и трансформацией клеток.
Ингибиторы фосфатидилинозитол-3-киназы (PI3K), пригодные в рамках настоящего изобретения, выбраны из группы, состоящей из 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)-фенил]-пропионитрила или его фармацевтически приемлемой соли, 5-(2,6-диморфолин-4-ил-пиримидин-4-ил)-4-трифторметилпиридин-2-иламина или его фармацевтически приемлемой соли; и 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амида) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты или его фармацевтически приемлемой соли.
В WO2006/122806 описаны производные имидазохинолина, которые, как было описано, ингибируют активность PI3K. Соединение 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидро-имидазо[4,5-c]хинолин-1-ил)-фенил]-пропионитрил имеет химическую структуру формулы (V)
 (V).
(V).
Соединение, его пригодность в качестве ингибитора PI3K и синтез 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидро-имидазо[4,5-c]хинолин-1-ил)-фенил]-пропионитрила и его монотозилата описаны в WO2006/122806, которая включена в настоящее описание в качестве ссылки в полном объеме, например, в примере 7 и примере 152-3, соответственно. Соединение 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидро-имидазо[4,5-c]хинолин-1-ил)-фенил]-пропионитрил может присутствовать в форме свободного основания или любой его фармацевтически приемлемой соли. Предпочтительно, 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидро-имидазо[4,5-c]хинолин-1-ил)-фенил]-пропионитрил имеет форму его монотозилата.
В WO07/084786 описаны конкретные производные пиримидина, которые, как было обнаружено, ингибируют активность PI3K. Соединение 5-(2,6-ди-морфолин-4-ил-пиримидин-4-ил)-4-трифторметил-пиридин-2-иламин имеет химическую структуру формулы (VI)
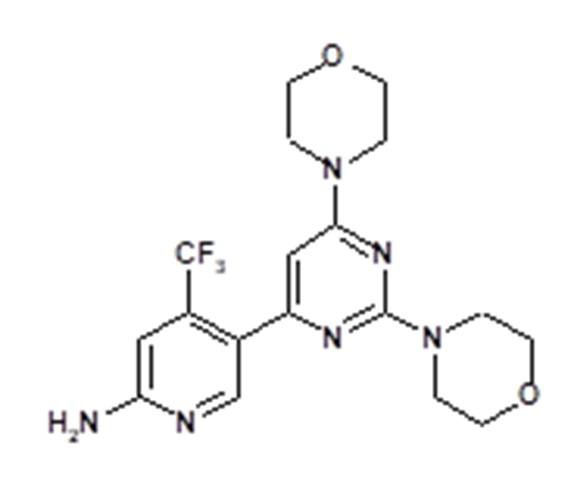 (VI).
(VI).
Соединение, его соли, его применимость в качестве ингибитора PI3K и синтез соединения 5-(2,6-диморфолин-4-ил-пиримидин-4-ил)-4-трифторметил-пиридин-2-иламина описаны в WO 2007/084786, которая включена в настоящее описание в качестве ссылки в полном объеме, например, в примере 10. Соединение 5-(2,6-диморфолин-4-ил-пиримидин-4-ил)-4-трифторметил-пиридин-2-иламин может присутствовать в форме свободного основания или любой его фармацевтически приемлемой соли. Предпочтительно, 5-(2,6-диморфолин-4-илпиримидин-4-ил)-4-трифторметилпиридин-2-иламин имеет форму соли хлористоводородной кислоты.
В WO2010/029082 описаны конкретные производные 2-карбоксамидциклоаминомочевины, которые, как было обнаружено, являются высокоселективными в отношении альфа-изоформы PI3K и могут быть добавлены к комбинациям по настоящему изобретению. Соединение 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты имеет химическую структуру формулы (VII)
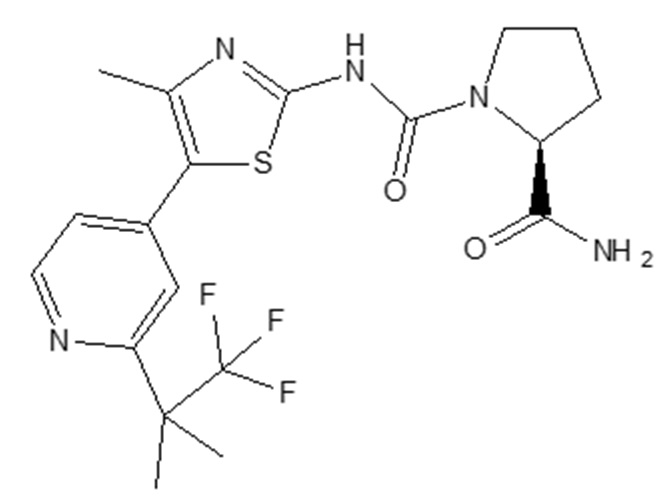 (VII).
(VII).
Соединение, его соли, его применимость в качестве селективного ингибитора альфа-изоформы PI3K и синтез соединения 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амида) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты описаны в WO2010/029082, которая включена в настоящее описание в качестве ссылки в полном объеме, например, в примере 15. Соединение 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты может присутствовать в форме свободного основания или любой его фармацевтически приемлемой соли. Предпочтительно, 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты имеет форму его свободного основания.
Предпочтительно, соединение ингибитора PI3K, используемое в комбинации по настоящему изобретению, представляет собой 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты или любую его фармацевтически приемлемую соль.
В одном варианте осуществления фармацевтическая комбинация, содержащая ингибитор MDM2 и ингибитор Bcl2, может дополнительно преимущественно включать ингибитор PI3K. Неожиданно, было обнаружено, что эта тройная комбинация обладает синергичным ингибированием (относительно пар лекарственных средств в 2/5 исследованных моделях на клетках (пример 4, таблица 9) и продемонстрировала более мощный апоптоз по сравнению с парными комбинациями (пример 4, фиг.14).
В предпочтительном варианте осуществления фармацевтическая комбинация содержит ингибитор MDM2, выбранный из (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она или его фармацевтически приемлемой соли, и (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она или его фармацевтически приемлемой соли; ингибитора Bcl2 навитоклакс или его фармацевтически приемлемой соли и ингибитора PI3K 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты или любой их фармацевтически приемлемой соли.
Более того, согласно настоящему изобретению, фармацевтические комбинации по настоящему изобретению, содержащие (a) ингибитор MDM2 и (b) (i) ингибитор MEK, и/или (ii) ингибитор Bcl2, могут дополнительно преимущественно включать ингибитор BRAF.
Более того, фармацевтическая комбинация по настоящему изобретению преимущественно может включать (a) ингибитор MDM2, (b) ингибитор MEK, (c) ингибитор Bcl2, и (d) ингибитор BRAF.
Термин "ингибитор BRAF" определяют в настоящем описании как соединение, которое нацелено, снижает или ингибирует активность серин/треониновой протеинкиназы B-Raf.
Фармацевтическая комбинация по любому из предшествующих п.п., где ингибитор BRAF выбран из группы, состоящей из RAF265, дабрафениба, (S)-метил-1-(4-(3-(5-хлор-2-фтор-3-(метилсульфонамидо)фенил)-1-изопропил-1H-пиразол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамата, метил N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамата и вемурафениба или их фармацевтически приемлемой соли.
Согласно настоящему изобретению, ингибитором BRAF предпочтительно является дабрафениб или его фармацевтически приемлемая соль. В одном варианте осуществления ингибитором BRAF, добавленным к комбинации, является RAF265.
Комбинация по настоящему изобретению, в частности, комбинация ингибитора MDM2 и ингибитора MEK (такого как траметиниб), кроме того, может включать ингибитор CDK4/6. "Ингибитор циклин-зависимой киназы 4/6 (CDK4/6)", как определяют в настоящем описании, относится к низкомолекулярному соединению, которое взаимодействует с комплексом циклин-CDK, блокируя киназную активность. Циклин-зависимые киназы (CDK) представляют собой большое семейство протеинкиназ, которые регулируют инициацию, прогрессирование и завершение клеточного цикла млекопитающих. Предпочтительно, ингибитор CDK4/6 представляет собой 7-циклопентил-N,N-диметил-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)-7H-пирроло[2,3-d]пиримидин-6-карбоксамид или его фармацевтически приемлемую соль.
Термин "фармацевтически приемлемые соли" относится к солям, которые сохраняют биологическую эффективность и свойства соединения и которые, как правило, не являются биологически или иным образом нежелательными. Соединение может быть способно образовывать кислотно-аддитивные соли вследствие присутствия аминогруппы.
Если нет иных указаний или если текст явно не указывает на иное, указание на лекарственные средства, пригодные в фармацевтической комбинации по настоящему изобретению, включает как свободное основание соединений, так и все фармацевтически приемлемые соли соединений.
Термин "комбинация" или "фармацевтическая комбинация" определяют в настоящем описании как либо фиксированную комбинацию в одной единичной дозированной форме, либо нефиксированную комбинацию, либо набор для комбинированного введения, где лекарственные средства можно вводить вместе, независимо в одно и то же время или по отдельности с временными интервалами, которые предпочтительно позволяют партнерам этой комбинации демонстрировать кооперативный, например, синергичный эффект. Таким образом, отдельные соединения фармацевтической комбинации по настоящему изобретению можно вводить одновременно или последовательно.
Более того, фармацевтическая комбинация по настоящему изобретению может быть в форме фиксированной комбинации или в форме нефиксированной комбинации.
Термин "фиксированная комбинация" означает, что лекарственные средства, например, отдельные соединения комбинации, имеют формы отдельных единиц или дозированных форм.
Термин "нефиксированная комбинация" означает, что лекарственные средства, например, отдельные соединения, комбинации вводят пациенту в качестве отдельных единиц или дозированных форм либо одновременно, либо последовательно, без конкретных временных пределов, где предпочтительно такое введение обеспечивает терапевтически эффективные уровни двух лекарственных средств в организме индивидуума, например, млекопитающего или человека, нуждающегося в этом.
Фармацевтические комбинации, кроме того, могут содержать по меньшей мере один фармацевтически приемлемый носитель. Таким образом, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтическую комбинацию по настоящему изобретению и по меньшей мере один фармацевтически приемлемый носитель.
Как используют в рамках изобретения, термин "носитель" или "фармацевтически приемлемый носитель" включает любые и все растворители, дисперсионные среды, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные средства, противогрибковые средства), обеспечивающие изотоничность средства, средства, замедляющие всасывание, соли, консерванты, стабилизаторы лекарственных средств, связующие вещества, эксципиенты, разрыхлители, смазывающие вещества, подсластители, вкусовые добавки, красители и т.п., и их комбинации, как будет известно специалистам в данной области (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). За исключением случаев, когда какой-либо общепринятый носитель является несовместимым с активным ингредиентом, предусматривается его применение в терапевтических или фармацевтических композициях.
Выражение "фармацевтически приемлемый" используют в настоящем описании для обозначения соединений, материалов, композиций и/или дозированных форм, которые по мнению медицинского специалиста пригодны для применения в контакте с человеком и животными без чрезмерной токсичности, раздражения, аллергического ответа или другой проблемы или осложнения, в соответствии с приемлемым соотношением польза/риск.
Как правило, термин "фармацевтическая композиция" определяют в настоящем описании как смесь или раствор, содержащие по меньшей мере одно лекарственное средство, подлежащее введению индивидууму, например, млекопитающему или человеку. Фармацевтические комбинации по настоящему изобретению могут быть составлены в виде подходящей фармацевтической композиции для энтерального или парентерального введения, как например, в виде единичных дозированных форм, таких как покрытые сахаром таблетки, таблетки, капсулы или суппозитории, или ампулы. Если нет иных указаний, их получают способом, по существу известным, например, посредством различных общепринятых процессов смешения, растирания, прямого прессования, гранулирования, покрытия сахаром, растворения, лиофилизации, или способов изготовления, хорошо известных специалистам в данной области. Будет понятно, что единичное содержание партнера комбинации, содержащееся в индивидуальной дозе каждой дозированной форме, само по себе не должно составлять эффективное количество, поскольку необходимое эффективное количество может быть достигнуто путем введения множества дозированных единиц. Фармацевтическая композиция может содержать от приблизительно 0,1% до приблизительно 99,9%, предпочтительно от приблизительно 1% до приблизительно 60%, лекарственного средства(средств). Специалист в данной области может выбрать один или несколько из вышеупомянутых носителей, исходя из конкретных желаемых свойств дозированной формы, посредством стандартного экспериментирования и без излишних затруднений. Количество каждого используемого носителя может варьироваться в диапазонах, общепринятых в данной области. В следующих источниках литературы описаны способы и эксципиенты, используемые для составления дозированных форм. См. The Handbook of Pharmaceutical Excipients, 4th edition, Rowe et al., Eds., American Pharmaceuticals Association (2003); и Remington: the Science and Practice of Pharmacy, 20th edition, Gennaro, Ed., Lippincott Williams & Wilkins (2003). Эти необязательные дополнительные общепринятые носители могут быть включены в пероральную дозированную форму либо путем включения одного или нескольких общепринятых носителей в исходную смесь до или в процессе гранулирования, либо путем комбинирования одного или нескольких общепринятых носителей с гранулами, содержащими комбинацию средств или индивидуальные средства комбинации средств в пероральной дозированной форме. В последнем из вариантов осуществления комбинированную смесь можно далее перемешивать, например, с использованием смесителя V-типа, и затем прессовать или формовать в таблетку, например, монолитную таблетку, инкапсулировать в капсулу или помещать в саше. Очевидно, фармацевтические комбинации по настоящему изобретению можно использовать для производства лекарственного средства.
Настоящее изобретение относится к таким фармацевтическим комбинациям или фармацевтическим композициям, которые являются особенно пригодными в качестве лекарственного средства.
В частности, комбинации или композиции по настоящему изобретению можно использовать для лечения рака.
Также настоящее изобретение относится к применению фармацевтических комбинаций или фармацевтических композиций по настоящему изобретению для получения лекарственного средства для лечения рака и к способу лечения рака у индивидуума, нуждающегося в этом, включающему введение индивидууму терапевтически эффективного количества фармацевтической комбинации по настоящему изобретению или фармацевтической композиции по настоящему изобретению.
Термин "лечение", как используют в рамках изобретения, включает лечение, смягчающее, уменьшающее или ослабляющее по меньшей мере один симптом у индивидуума, увеличивающее выживаемость без прогрессирования, общую выживаемость, продлевающее длительность ответа или замедляющее прогрессирование заболевания. Например, лечение может представлять собой уменьшение одного или нескольких симптомов нарушения или полное устранение нарушения, такого как рак. В пределах значения настоящего изобретения термин "лечение" также обозначает остановку, отсрочивание возникновения (т.е. период до клинического проявления заболевания) и/или снижение риска развития или ухудшения заболевания у пациента, например, млекопитающего, в частности, пациентом является человек. Термин "лечение", как используют в рамках изобретения, включает ингибирование роста опухоли, включающее прямое ингибирование первичного роста опухоли и/или системное ингибирование метастатических раковых клеток.
"Субъект", "индивидуум" или "пациент" используются в настоящем описании взаимозаменяемо, и они относятся к позвоночному, предпочтительно млекопитающему, более предпочтительно человеку. Млекопитающие включают, но не ограничиваются ими, мышей, обезьян, человека, сельскохозяйственных животных, спортивных животных и домашних питомцев.
Термин "терапевтически эффективное количество" соединения (например, химического соединения или биологического средства) по настоящему изобретению относится к количеству соединения по настоящему изобретению, которое будет индуцировать биологический или медицинский ответ у индивидуума, например, снижение или ингибирование активности фермента или белка, или смягчать симптомы, смягчать состояние, замедлять или отсрочивать прогрессирование заболевания, или предупреждать заболевание и т.д. В одном варианте осуществления терапевтически эффективное количество in vivo может варьироваться в диапазоне приблизительно 0,1-500 мг/кг или приблизительно 1-100 мг/кг в зависимости от пути введения.
Оптимальная дозировка каждого партнера комбинации для лечения рака может быть определена эмпирически для каждого индивидуума с использованием известных способов и зависит от множества факторов, включая, но не ограничиваясь ими, степень прогрессирования заболевания; возраст, массу тела, общее состояние здоровья, пол и рацион индивидуума; время и путь введения; и другие лекарственные средства, которые принимает индивидуум. Оптимальные дозировки могут быть установлены с использованием стандартного тестирования и процедур, которые хорошо известны в данной области. Количество каждого партнера комбинации, которое можно комбинировать с материалами носителей для получения единичной дозированной формы, варьируется в зависимости от подвергаемого лечению индивидуума и конкретного пути введения. В некоторых вариантах осуществления единичные дозированные формы, содержащие комбинацию средств, как описано в настоящем описании, содержат каждое средство комбинации, которое обычно вводят, когда средства вводят по отдельности.
Частота дозирования может варьироваться в зависимости от используемого соединения и конкретного состояния, подлежащего лечению или предупреждению. Как правило, применение минимальной дозировки, которая является достаточной для обеспечения эффективной терапии, является предпочтительным. Пациентов, как правило, можно подвергать мониторингу в отношении терапевтической эффективности с использованием способов анализа, пригодных для подвергаемого лечению или предупреждению состояния, которые хорошо известны специалистам в данной области.
Терапевтическое количество или доза (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она может находиться в диапазоне от 100 до 1500 мг раз в три недели, в частности, от 100 до 800 мг раз в три недели или от до 50 до 600 мг каждые сутки при введении перорально. Терапевтическое количество или доза (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она может составлять 400 мг, более предпочтительно, она составляет 300 мг для ежедневного введения в течение первых 21 суток каждого курса из 28 суток. Альтернативно общее терапевтическое количество или общая доза (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она составляет 560 мг на курс (40 мг qd 2 недели приема/2 недели покоя, или 80 мг qd 1 неделя приема/3 недели покоя). Внутривенные дозы потребуется снизить соответствующим образом.
Терапевтическое количество или доза (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она составляет от 500 до 2000 мг, в частности, от 500 до 1200 мг, при введении перорально. В предпочтительном варианте осуществления терапевтическое количество или доза (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она составляет 500 мг, более предпочтительно 800 мг. Внутривенные дозы потребуется снизить соответствующим образом.
Рекомендованная доза ингибитора MEK траметиниба составляет 2 мг в сутки. Контроль неблагоприятных реакций может потребовать снижения дозы вплоть до 1 мг в сутки.
Соединение ингибитора MEK (2-гидроксиэтокси)-амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты можно вводить подходящему индивидууму каждые сутки одной или разделенными дозами в эффективной дозировке в диапазоне от приблизительно 0,001 до приблизительно 100 мг на кг массы тела в сутки, предпочтительно от приблизительно 1 до приблизительно 35 мг/кг/сутки, одной или разделенными дозами. Для человека массой 70 кг предпочтительный диапазон дозировок будет составлять приблизительно 0,05-7 г/сутки, предпочтительно от приблизительно 0,05 до приблизительно 2,5 г/сутки.
Соединение ингибитора MEK (S)-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид можно вводить ежедневно подходящему индивидууму одной или разделенными дозами в эффективной дозировке в диапазоне от приблизительно 0,001 до приблизительно 100 мг на кг массы тела в сутки, предпочтительно от приблизительно 1 мг/кг/сутки до приблизительно 35 мг/кг/сутки одной или разделенными дозами. Для человека массой 70 кг предпочтительный диапазон дозировок будет составлять приблизительно от 0,07 до 2,45 г/сутки, предпочтительно от приблизительно 0,05 до приблизительно 1,0 г/сутки.
Эффективная доза ингибитора Bcl-2 навитоклакса может находиться в диапазоне от приблизительно 100 мг до приблизительно 500 мг в сутки. Доза может быть снижена или может быть использована начальная доза в течение 7 суток, составляющая 150 мг. После начальной дозы можно вводить дозу 325 мг или вплоть до 425 мг каждые сутки.
Рекомендованная доза ингибитора EGFR эрлотиниба составляет 100 мг или 150 мг в сутки.
Соединение ингибитора PI3K 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амида) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты обычно вводят перорально в дозе, находящейся в диапазоне приблизительно от 30 мг до 450 мг в сутки, например от 100 до 400 мг в сутки для взрослого человека. Суточную дозу можно вводить по схеме qd или bid. 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты можно вводить подходящему индивидууму каждые сутки одной или разделенными дозами в эффективной дозировке, находящейся в диапазоне от приблизительно 0,05 до приблизительно 50 мг на кг массы тела в сутки, предпочтительно приблизительно 0,1-25 мг/кг/сутки, более предпочтительно приблизительно 0,5-10 мг/кг/сутки, одной или разделенными дозами. Для человека массой 70 кг предпочтительный диапазон дозировок будет составлять приблизительно 35-700 мг в сутки. Более предпочтительно, диапазон дозировок составляет приблизительно 35-400 мг в сутки.
Соединение ингибитора PI3K 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)-фенил]-пропионитрила обычно вводят перорально в дозе, находящейся в диапазоне приблизительно от 100 мг до 1200 мг, или приблизительно от 200 мг до 1000 мг, или приблизительно от 300 мг до 800 мг, или приблизительно от 400 мг до 600 мг в сутки для взрослого человека. Суточную дозу можно вводить по схеме qd или bid.
Соединение ингибитора PI3K 5-(2,6-диморфолин-4-ил-пиримидин-4-ил)-4-трифторметил-пиридин-2-иламина обычно вводят перорально в дозе, находящейся в диапазоне приблизительно от 30 мг до 300 мг, или приблизительно от 60 мг до 120 мг, или приблизительно 100 мг в сутки для взрослого человека. Суточную дозу можно вводить по схеме qd или bid.
Рекомендованная доза ингибитора BRAF дабрафениба составляет 150 мг перорально два раза в сутки в качестве единственного средства или в комбинации с траметинибом 2 мг перорально один раз в сутки.
Понятно, что каждое лекарственное средство может быть удобно вводить, например, в одной индивидуальной дозированной единице или разделенным на множество дозированных единиц. Кроме того, понятно, что каждое лекарственное средство может быть удобно вводить в дозах, предназначенных для одного раза в сутки, или в дозах, предназначенных для вплоть до четырех раз в сутки.
Термин "рак" используют в настоящем описании для обозначения широкого спектра опухолей, в частности, солидных опухолей. Примеры таких опухолей включают, но не ограничиваются ими, доброкачественную или злокачественную опухоль легкого (включая мелкоклеточный рак легкого и немелкоклеточный рак легкого), бронха, предстательной железы, молочной железы (включая спорадические раки молочной железы и страдающих от болезни Каудена), поджелудочной железы, желудочно-кишечного тракта, толстой кишки, прямой кишки, карциному толстой кишки, рак ободочной и прямой кишки, щитовидной железы, печени, желчных путей, внутрипеченочных желчных протоков, печеночно-клеточный рак, надпочечников, желудка, гастральную, глиому, глиобластому, эндометрия, почки, почечной лоханки, мочевого пузыря, матки, шейки матки, влагалища, яичника, множественную миелому, пищевода, головы и шеи, головного мозга, полости рта и глотки, гортани, тонкого кишечника, меланому, ворсинчатую аденому толстого кишечника, саркому, новообразование, новообразование эпителиального характера, карциному молочной железы, базально-клеточную карциному, плоскоклеточную карциному, актинический кератоз, истинную полицитемию, эссенциальную тромбоцитемию, лейкоз (включая острый миелогенный лейкоз, хронический миелогенный лейкоз, лимфоцитарный лейкоз и миелоидный лейкоз), лимфому (включая неходжкинскую лимфому и лимфому Ходжкина), миелофиброз с миелоидной метаплазией, болезнь Вальденстрема и аденокарциному Баррета.
Предпочтительно рак представляет собой рак ободочной и прямой кишки, меланому, липосаркому, глиобластому, нейробластому, лимфому или лейкоз. В предпочтительном варианте осуществления рак представляет собой рак ободочной и прямой кишки. Термин "колоректальный рак", как используют в рамках изобретения, относится к раку ободочной и прямой кишки, также известному как рак ободочной кишки, рак прямой кишки или рак толстого кишечника. В одном варианте осуществления в рамках настоящего изобретения предусматривается метастазирующий рак ободочной и прямой кишки.
Ожидается, что комбинация достигнет лучших эффектов в раках с функциональным p53 или p53 дикого типа. Ген TP53 является одним из наиболее частых мутантных генов в злокачественных опухолях человека. Таким образом, опухолевый супрессор p53 функционально нарушается посредством мутации или делеции практически в 50% раков человека. В остальных раках человека p53 сохраняет статус дикого типа, однако его функция ингибируется первичным клеточным ингибитором мышиного двойного малого 2 (Mdm2, MDM2; HDM2 (человеческий гомолог мышиного двойного малого 2)). Mdm2 является отрицательным регулятором опухолевого супрессора p53. Белок Mdm2 функцинирует как в качестве убиквитинлигазы E3, которая приводит к протеасомной деградации p53, так и в качестве ингибитора активации транскрипции посредством p53. Часто Mdm2 обнаруживается амплифицированным в опухолях p53 дикого типа. Поскольку взаимодействие между Mdm2 и p53 является основным механизмом ингибирования функции p53 в раках, которые сохраняют p53 дикого типа, комбинация по настоящему изобретению, содержащая ингибитор MDM2, является особенно пригодной для лечения раков с функциональным p53 или p53 дикого типа.
Кроме того, ожидается, что эффективность комбинации будет повышенной в раке, который характеризуется одной или несколькими из мутации KRAS, и/или мутации BRAF, и/или мутации MEK1, и/или мутации PIK3CA и/или сверхэкспрессии PIK3CA.
Пациенты с раком ободочной и прямой кишки, имеющие мутации KRAS или BRAF, которые вместе составляют 50%-60% от сообщенных случаев рака ободочной и прямой кишки (Fearon 2011), как правило, имеют плохой прогноз (Arrington, Heinrich et al. 2012, Safaee Ardekani, Jafarnejad et al. 2012). Комбинации по настоящему изобретению являются особенно пригодными для лечения рака, который имеет одну или несколько мутаций KRAS или одну или несколько мутаций BRAF.
Примеры мутаций BRAF включают, но не ограничиваются ими, V600E, R461I, I462S, G463E, G463V, G465A, G465E, G465V, G468A, G468E, N580S, E585K, D593V, F594L, G595R, L596V, T598I, V599D, V599E, V599K, V599R, V600K, A727V. Большинство из этих мутаций кластеризуются в двух областях: глицин-богатая P-петля N-доли, и сегмент активации и фланкирующие области. Мутация V600E была выявлена в различных раках и является результатом замены тимина аденином в нуклеотиде 1799. Это приводит к замене валина (V) глутаматом (E) в кодоне 600 (в настоящее время обозначаемой как V600E).
Мутация MEK1 может представлять собой, например, мутацию S72G MEK1.
Примеры мутации PIK3CA и/или сверхэкспрессии PIK3CA включают, но не ограничиваясь ими, амплификацию альфа-изоформы PI3K, соматическую мутацию PIK3CA, генеративные мутации или соматические мутации PTEN, мутации и транслокацию p85α, которые служат для активации комплекса p85-p110, или амплификацию или сверхэкспрессию бета-изоформы PI3K.
Фармацевтическая комбинация по настоящему изобретению является особенно пригодной для лечения рака, в частности, рака ободочной и прямой кишки, где рак является резистентным к лечению ингибитором EGFR, или приобретает резистентность к лечению ингибитором EGFR, или имеет высокий риск развития резистентности к лечению ингибитором EGFR, в частности, где ингибитор EGFR выбран из группы, состоящей из эрлотиниба, гефитиниба и афатиниба.
Фармацевтическая комбинация по настоящему изобретению также пригодна для лечения пациентов с плохим прогнозом, особенно таких пациентов с плохим прогнозом, которые имеют рак, в частности, рак ободочной и прямой кишки, который становится резистентным к лечению с использованием ингибитора EGFR, например, рак у таких пациентов, которые изначально отвечали на лечение ингибитором EGFR и у которых затем произошел рецидив. В следующем примере указанному пациенту не проводили лечение с использованием ингибитора FGFR. Этот рак может приобрести резистентность в ходе предшествующего лечения одним или несколькими ингибиторами EGFR. Например, направленная на EGFR терапия может включать лечение гефитинибом, эрлотинибом, лапатинибом, XL-647, HKI-272 (Нератиниб), BIBW2992 (Афатиниб), EKB-569 (Пелитиниб), AV-412, канертиниб, PF00299804, BMS 690514, HM781-36b, WZ4002, AP-26113, цетуксимабом, панитумумабом, матузумабом, трастузумабом, пертузумабом или их фармацевтически приемлемой солью. В частности, направленная на EGFR терапия может включать лечение гефитинибом, эрлотинибом и афатинибом. Механизмы приобретенной резистентности включают, но не ограничиваются ими, появление второй мутации непосредственно в гене EGFR, например, T790M, амплификацию EGFR и/или нарушение регуляции FGFR, мутацию FGFR, мутацию лиганда FGFR, амплификацию FGFR или амплификацию лиганда FGFR.
Следующие примеры иллюстрируют изобретение, описанное выше, однако они не предназначены для ограничения объема изобретения каким-либо образом. Другие модели исследования, по существу известные специалисту в данной области, также могут определить полезные эффекты заявленного изобретения.
Примеры
"СОЕДИНЕНИЕ A", "СОЕДИНЕНИЕ B" и т.п. обозначают в настоящем описании конкретные соединения. Обозначение соответствующих соединений может не быть одинаковым для всех примеров или комбинаций. Вместо этого, соединения в каждом примере обозначены заново.
Пример 1: Эффект комбинирования ингибитора MDM2 и ингибитора MEK на пролиферацию in vitro.
Это исследование было разработано для исследования эффекта на пролиферацию in vitro комбинирования ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) или ингибитора MDM2 (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она (СОЕДИНЕНИЕ B) с ингибитором MEK траметинибом (СОЕДИНЕНИЕ C) в клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа.
СПОСОБЫ
СОЕДИНЕНИЯ A, B и C растворяли в 100% DMSO (Sigma, каталожный номер D2650) в концентрации 20 мМ и хранили при -20°C до применения.
Клеточные линии рака ободочной и прямой кишки, использованные для этого исследования, были получены, культивировались и обрабатывались коммерческими поставщиками ATCC, ECACC, DSMZ и CellBank Australia (таблица 1). Все среды клеточных линий дополняли 10% FBS (HyClone, каталожный номер SH30071.03). Среды для LIM2405 дополнительно дополняли 0,6 мкг/мл инсулина (SIGMA, каталожный номер I9278), 1 мкг/мл гидрокортизона (SIGMA, каталожный номер H0135) и 10 мкМ 1-тиоглицерином (SIGMA, каталожный номер M6145).
Таблица 1. Информация о клеточных линиях
Клеточные линии культивировали в инкубаторе при 37°C и 5% CO2 и увеличивали в количестве во флаконах T-75. Во всех случаях клетки размораживали из замороженных исходных культур, увеличивали в количестве в течение ≥1 пассажа с использованием разведений 1:3, подсчитывали и оценивали в отношении жизнеспособности с использованием счетчика ViCell (Beckman-Coulter) перед посевом. Для разделения и увеличения в количестве клеточных линий клетки извлекали из флаконов с использованием 0,25% трипсина-EDTA (GIBCO, каталожный номер 25200). Было определено, что все клеточные линии были свободны от контаминации микоплазмой, как определяли с помощью методологии определения с использованием ПЦР, проведенной Idexx Radil (Columbia, MO, США), и были правильно идентифицированы посредством выявления панели SNP.
Для исследования эффекта комбинации СОЕДИНЕНИЯ A или СОЕДИНЕНИЯ B с СОЕДИНЕНИЕМ C на клеточную пролиферацию клетки высевали в черные 384-луночные микропланшеты с прозрачным дном (Matrix/Thermo Scientific, каталожный номер 4332) в 50 мкл среды на лунку с плотностью клеток от 500 до 1250 клеток/лунка (таблица 1), и им позволяли инкубироваться при 37 градусах, 5% CO2, в течение 24 ч. Через 24 ч один 384-луночный планшет на клеточную линию подготавливали для подсчета клеток посредством микроскопии (см. ниже) без проведения обработки (="исходный уровень"). Другие планшеты с клетками обрабатывали с использованием цифрового дозатора HP D300 (Tecan) для построения кривых доза-эффект из 6 точек с шагом разведения 2,5X.
СОЕДИНЕНИЕ A использовали в диапазоне конечных концентраций 51 нМ - 5 мкМ, СОЕДИНЕНИЕ B использовали в диапазоне конечных концентраций 10 нМ - 1 мкМ, и СОЕДИНЕНИЕ C использовали в диапазоне конечных концентраций 1 нМ-100 нМ.
Для комбинаций СОЕДИНЕНИЯ A или СОЕДИНЕНИЯ B с СОЕДИНЕНИЕМ C отдельные средства комбинировали в 6 дозах отдельных средств с получением матрицы доз 6×6=36 комбинированных обработок. Кроме того, отрицательные контроли (DMSO="носитель") и положительные контроли (стауроспорин=уничтожение клеток, серия разведений 1:2 из 7-точек для диапазона доз 16 нМ-1 мкМ) переносили в качестве контролей обработки. Клетки обрабатывали в течение от 72 ч до 96 ч в зависимости от их времени удвоения (таблица 1). В конце обработки клетки подготавливали для подсчета клеток посредством микроскопии. Клетки фиксировали и пермеабилизировали в течение 45 минут в 4% PFA (Electron Microscopy Sciences, каталожный номер 15714), 0,12% TX-100 (Electron Microscopy Sciences, каталожный номер 22140) в PBS (Boston Bioproducts, каталожный номер BM-220). После промывания клеток три раза PBS их ДНК окрашивали в течение 30 красителем Хехста 33342 (ThermoFisher, каталожный номер H3570) в конечной концентрации 4 мкг/мл. Клетки промывали три раза PBS, а затем планшеты запаивали с использованием PlateLoc (Agilent Technologies) алюминиевыми уплотнителями (Agilent Technologies, каталожный номер 06644-001) и хранили при 4°C до визуализации. Все клетки на лунку/обработку фотографировали в одном изображении посредством флуоресцентной микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективом 4X и фильтрами возбуждения/испускания DAPI.
Для тестирования эффектов комбинаций на индукцию апоптоза проводили анализ с каспазой 3/7 с использованием экспериментальных условий, сходных с анализом пролиферации, описанным выше, и тестируя только комбинацию СОЕДИНЕНИЯ A c СОЕДИНЕНИЕМ C. Соединения наносили на основные планшеты для лекарственных средств (Greiner, каталожный номер 788876) и подвергали 3-кратным серийным разведениям (7 шагов) при концентрации 2000X. Клетки обрабатывали посредством переноса 25 нл соединения 2000X из основных планшетов для лекарственных средств с использованием акустического жидкостного дозатора ATS (ECD Biosystems) к 50 мкл клеток, достигая конечной концентрации 1X.
СОЕДИНЕНИЕ A использовали в диапазоне конечных концентраций 13 нМ-10 мкМ, и СОЕДИНЕНИЕ C использовали в диапазоне конечных концентраций 0,4 нМ-0,3 мкМ. Кроме того, отрицательные контроли (DMSO="носитель") и положительные контроли (стауроспорин=уничтожение клеток, серия разведений 1:2 из 7-точек для диапазона доз 16 нМ-1 мкМ) переносили в качестве контролей лечения.
После добавления соединения к одной из трех реплик добавляли 50 нл 2 мМ реагента CellEvent Caspase-3/7 Green Detection Reagent (ThermoFisher, каталожный номер C10423) с использованием цифрового дозатора HP D300 (Tecan). Индукцию каспазы 3/7 количественно определяли в качестве показателя апоптоза, индуцированного обработкой. Клетки обрабатывали в течение от 72 ч до 96 ч в зависимости от их времени удвоения (таблица 1), и активацию каспазы 3/7 количественно определяли каждые 24 ч посредством микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективном 4X и фильтрами возбуждения/испускания FITC. В конце обработки клетки подготавливали для подсчета клеток посредством микроскопии и получения изображений, как описано выше для анализа клеточной пролиферации.
Изображения анализировали после адаптации ранее описанных способов (Horn, Sandmann et al. 2011) и с использованием пакета EBImage пакета Bioconductor в R (Pau, Fuchs et al. 2010). Объекты в обоих каналах, DAPI (для красителя Хехста/ДНК) и FITC (для каспазы 3/7), сегментировали по отдельности посредством адаптивного определения порогового значения и подсчета. Пороговое значение для положительных по каспазе 3/7 объектов определяли вручную на клеточную линию после сравнения отрицательных контролей (DMSO) и положительных контролей (стауроспорин). Посредством анализа 17 дополнительных признаков объектов/ядер в канале ДНК (признаки формы и интенсивности) идентифицировали дебрис/фрагментированные ядра. Для этого проводили сравнение вручную распределений дополнительных признаков между положительными контролями (стауроспорин) и отрицательными контролями (DMSO) для каждой клеточной линии. Признаки, которые могли отличаться между условиями (например, сдвиг распределения результата измерения признака при сравнении DMSO со стауроспорином) использовали для определения популяции "дебриса" против популяции "жизнеспособных" ядер. Результат подсчета дебриса вычитали из результата исходного подсчета ядер. Полученное количество ядер использовали в качестве показателя клеточной пролиферации ("подсчитанное количество клеток").
Эффект соединения на клеточную пролиферацию вычисляли из подсчитанных количеств обработанных клеток относительно подсчитанных количеств клеток, обработанных отрицательным контролем (DMSO), на фиг.1 и 2 обозначаемых как "нормализованное подсчитанное количество клеток" по оси y. Синергию комбинаций оценивали посредством анализа с использованием изоболограммы (Greco, Bravo et al. 1995)(фиг.3) и посредством вычисления показателей аддитивности (Chou, Talalay 1984)(таблица 2), который является тестом на синергию согласно модели Леве (Loewe 1928). Анализ CI проводили для 75% уровня изоэффекта (75% ингибирование при обработке отдельным средством по сравнению с комбинированной обработкой). "наилучший CI" (красные точки на фиг.3 и таблица 2) является наиболее низким показателем аддитивности, наблюдаемым для этой комбинации в конкретной клеточной линии.
Показатель аддитивности (CI) является показателем комбинированного эффекта, причем
CI < 1: синергия
CI=1: аддитивный эффект
CI > 1: антагонизм,
например, показатель аддитивности 0,5 указывает на то, что в комбинации только половина каждого отдельного средства требуется по сравнению с требуемыми дозами отдельных средств для достижения того же эффекта (в данном случае 75% ингибирование).
IC75 представляет собой концентрация соединения, которая приводит к 75% подсчитанных количеств клеток относительно DMSO. Вычисления IC75 (см. таблица 2) проводили посредством проведения 3-параметрической логистической регрессии данных.
Таблица 2. Величины IC75 отдельных средств для каждого соединения и наилучшие показатели аддитивности (наилучший CI) для комбинаций СОЕДИНЕНИЯ A и траметиниба, и СОЕДИНЕНИЯ B и траметиниба.
Эффект соединения на апоптоз определяли посредством вычисления процента клеток с активированной каспазой 3/7 на обработку и момента времени относительно исходного подсчитанного количества клеток (до вычитания дебриса) (ось y на фиг.4). Подсчитанные количества клеток в моменты времени, которые не были измерены экспериментально, получали посредством регрессионного анализа путем аппроксимации линейной модели для логарифмически преобразованных количеств клеток на 0 сутки и в конце обработки (предполагая экспоненциальный рост клеток).
Для анализа образования колоний (фиг.5) клетки высевали в 1 мл среды в 12-ячеечных планшетах для культивирования тканей (Costar, каталожный номер 3513): для COLO-678 6000 клеток/лунка, для SW48 5000 клеток/лунка, для GP2d 2000 клеток/лунка, для LoVo 2500 клеток/лунка, для LS-180 2500 клеток/лунка, для LIM2405 2500 клеток/лунка, для RKO 1000 клеток/лунка и для HCT-116 1000 клеток/лунка. Клетки выращивали в течение 72 ч перед добавлением соединений и обработку обновляли каждые 48 ч (в свежей среде) в течение вплоть до 14 суток с использованием цифрового дозатора HP D300 (Tecan). В конце обработки клетки промывали PBS один раз, фиксировали и окрашивали в течение 30 минут при комнатной температуре с использованием раствора, содержавшего 4% PFA (Electron Microscopy Sciences, каталожный номер 15714) и 2 мг/мл кристаллического фиолетового (EMD, каталожный номер 192-12), и промывали 3 раза водой. Планшеты сушили в течение ночи и сканировали с использованием устройства для визуализации Odyssee (Licor). Для количественного определения сигнала кристаллического фиолетового на фиг.5 использовали программное обеспечение ImageStudio (Licor). Для тестирования значимости см. таблицу 3.
Таблица 3. Значимость отличий результатов анализа образования колоний (фиг.4) комбинации СОЕДИНЕНИЯ A и траметиниба по сравнению с соответствующими дозами СОЕДИНЕНИЯ A или траметиниба отдельно (односторонний t-критерий). *p<0,05, **p<0,01, ***p<0,001.
Для анализа клеточного цикла (фиг.6) клетки высевали в 10 мл среды в 10-см обработанных чашках для культивирования тканей (Corning, каталожный номер 430167): для COLO-678 3,5 миллиона клеток, для SW48 2,75 миллиона клеток, для GP2d 2,5 миллиона клеток, для LoVo 2,5 миллиона клеток, для LS-180 2,5 миллиона клеток, для LIM2405 1,5 миллиона клеток, для RKO 1,5 миллиона клеток и для HCT-116 2 миллиона клеток. Обработку лекарственным средством проводили вручную через 24 ч. Образцы собирали через 24 ч после обработки посредством сбора супернатанта и сбора клеток трипсинизацией с использованием 0,25% трипсина-EDTA (GIBCO, каталожный номер 25200). Клетки осаждали, промывали один раз PBS и вновь осаждали, а затем фиксировали в 1 мл ледяного 70% этанола (добавленного капельно к осадку при встряхивании) в течение 30 минут при 4°C. Далее клетки осаждали при более высокой скорости (850×g) в течение 5 мин и осадок промывали два раза фосфатно-цитратным буфером (0,2 M NA2HPO4, 0,1 M лимонная кислота, доведенный до pH 7,8) и центрифугировали при той же скорости (в течение 5 минут). Наконец, осадок растворяли в 0,5 мл буфера для окрашивания PI/РНК-азой (BD Pharmingen, каталожный номер 550825). После инкубации в течение 15 минут при к.т. клеточный цикл анализировали на системе BD FACSCanto II (анализирующей 10000 событий на каждые условия). Данные анализировали с использованием программного обеспечения FlowJo.
Для вестерн-блоттинга (фиг.7) клетки высевали в 10 мл среды в 10-см обработанные чашки для культивирования тканей (Corning, каталожный номер 430167): для COLO-678 8 миллиона клеток, для SW48 4 миллиона клеток, для GP2d 3 миллиона клеток, для LoVo 4 миллиона клеток, для LS-180 4,5 миллиона клеток, для LIM2405 2 миллиона клеток, для RKO 3,5 миллиона клеток и для HCT-116 3,5 миллиона клеток. Клетки выращивали в течение 24 ч перед добавлением соединений. После обработки в течение 24 ч клетки собирали в ледяной PBS посредством соскребания, лизировали лизирующим буфером (Cell Signaling, каталожный номер 9803), содержащим ингибиторы фосфатазы (Roche, каталожный номер 04906837001) и ингибиторы протеаз (Roche, каталожный номер 04693116001), в течение 30 минут. Лизаты количественно определяли с использованием набора для анализа белка BCA (ThermoFisher, каталожный номер 23225), концентрацию нормализовывали, добавляли буфер для нанесения (ThermoFisher, NP0007), 25-50 мкг белка наносили на предварительно залитые 4-15% полиакриламидные градиентные гели (Biorad, каталожный номер 5671084), и разделяли на системе электрофореза (Biorad) в течение 30-35 мин при 300 В. Белок переносили на нитроцеллюлозные мембраны с использованием системы для переноса iBlot (Invitrogen) и набора для переноса iBlotGel (ThermoFisher, каталожный номер IB301001). Детекцию белков, показанных на фиг.5, проводили с использованием следующих первичных антител против: p53 (Santa Cruz Biotechnology, sc-126, 1:500), MDM2 (CalBiochem, #OP46, 1:500), ERK (Cell Signaling Technology, #4695, 1:1000), pERK (Cell Signaling Technology #4370, 1:1000), p21 (Cell Signaling Technology, #2947, 1:1000), p27 (Santa Cruz Biotechnology, sc-528, 1:500), CyclinD1 (Santa Cruz Biotechnology, sc-718, 1:500), BIM (Cell Signaling Technology, #2819, 1:500), cPARP (Cell Signaling Technology, #9541, 1:500), PUMA (Cell Signaling Technology, #4976, 1:500) и бета-актина (Ambion, AM4302, 1:10000). Использовали следующие вторичные антитела: антитело козы против антител колика с HRP (Biorad, 170-5046, 1:10000), антитело козы против антител мыши с HRP (Biorad, 170-5047, 1:10000) и антитело козы против антител мыши с IRDye® 800CW (Licor, 925-32210, 1:10000). Мембраны проявляли на пленке (Carestream, каталожный номер 178 8207) на проявочном устройстве (Kodak X-OMAT 2000A) или (для бета-актина) визуализировали на устройстве для визуализации Odyssee (Licor).
Для анализа кОТ-ПЦР (фиг.8) клетки высевали в 6-ячеечные обработанные планшеты для культивирования тканей (Corning, каталожный номер 3516): для COLO-678 0,75 миллиона клеток, для SW48 0,5 миллиона клеток, для GP2d 0,4 миллиона клеток, для LoVo 0,4 миллиона клеток, для LS-180 0,4 миллиона клеток, для LIM2405 0,25 миллиона клеток, для RKO 0,25 миллиона клеток и для HCT-116 0,3 миллиона клеток. Обработку лекарственным средством проводили вручную через 24 ч. Образцы собирали после обработки в течение 10 ч. Супернатант удаляли, клетки промывали один раз PBS, а затем использовали набор RNeasy Mini Kit (Qiagen, каталожный номер 74104) для экстракции РНК. РНК количественно определяли с использованием NanoDrop 1000 и 1 мкг тотальной РНК использовали для синтеза кДНК с использованием набора для синтеза кДНК iScript (Biorad, каталожный номер 170-8891). КОТ-ПЦР Taqman проводили в 384-луночных планшетах на ABI ViiA 7 (Applied Biosystems). На реакцию использовали 6 мкл a 2x основной смеси для ПЦР (ThermoFisher, каталожный номер 435 2042), 0,6 мкл набора праймеров/зондов для бета-актина (20X), 0,6 мкл набора праймеров/зондов для мишени (20X) и 4,8 мкл кДНК (разведение продукта синтеза кДНК 1:4). Проводили следующую программу: 2 минуты при 50°C, 10 мин при 95°C и 40 циклов с 10 секундами при 95°C и 1 минутой при 60°C. Использовали следующие наборы зондов/праймеров (анализ экспрессии генов ThermoFisher TaqMan): CDKN1A/p21 (Hs00355782_m1), BAX (Hs00180269_m1), BBC3/PUMA (Hs00248075_m1), BMF (Hs00372937_m1), NOXA1 (Hs00736699_m1).
РЕЗУЛЬТАТЫ
В данном протоколе эффективность двух ингибиторов MDM2 (СОЕДИНЕНИЕ A и СОЕДИНЕНИЕ B) и ингибитора MEK траметиниба (СОЕДИНЕНИЕ C) оценивали индивидуально и в комбинации всего в 8 клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа. Пять из линий имели мутантный KRAS (GP2d, LS-180, HCT-116, LoVo, COLO-678), две линии имели мутантный BRAF (RKO, LIM2405), и одна линия имела мутантную MEK1 (SW48). Четыре из этих линий также были мутантными по PIK3CA (GP2d, RKO, LS-180, HCT-116) (таблица 1). СОЕДИНЕНИЕ A в качестве единственного средства ингибировало рост всех клеточных линий с величинами IC75 от субмикромолярных до микромолярных, СОЕДИНЕНИЕ B ингибировало их рост с субмикромолярными величинами IC75 (за исключением COLO-678) и СОЕДИНЕНИЕ C ингибировало их рост с наномолярными величинами IC75 (за исключением COLO-678, RKO и GP2d) (фиг.1 и 2, таблица 2). Комбинированная обработка вызывала синергичное ингибирование (согласно модели Леве) во всех протестированных клеточных моделях, на что указывали показатели аддитивности (CI) (фиг.3 и таблица 2). Следующие механистические исследования были сфокусированы на комбинации СОЕДИНЕНИЯ A с траметинибом. Комбинация продемонстрировала слабую индукцию апоптоза (оцененную посредством измерения индукции каспазы 3/7) (фиг.4). Комбинация препятствовала росту клонов в анализах образования колонов значительно лучше, чем каждое средство отдельно, также была продемонстрирована длительная эффективность комбинации (фиг.5 и таблица 3). FACS-анализ проводили после обработки в течение 24 ч, и он показал, что ингибирование MDM2 истощало клетки в S-фазе и останавливало их в G1- и/или G2-фазах клеточного цикла (фиг.6). Ответы на ингибирование MEK были более зависимыми от клеточной линии, но в большинстве моделей оно приводило к увеличению популяций G1. Комбинация в основном продемонстрировала истощение в S-фазе и в 5/8 моделей также увеличивала субпопуляции G1, что указывает на клеточному смерть. Для идентификации факторов, которые играли роль в остановке клеточного цикла и клеточной смерти после обработки комбинацией авторы настоящего изобретения провели вестерн-анализ (через 24 ч после обработки) и кПЦР (через 10 ч после обработки) в отношении регуляторов клеточного цикла и апоптоза (фиг.7 и 8). Результаты вестерн-анализа и кПЦР указывали на то, что остановка в G1 при ингибировании MDM2 была следствием индукции p21 (CDKN1A). Белок p27 (CDKN1B) индуцировался посредством ингибирования MEK в некоторых из протестированных моделей (см. вестерн-блот на фиг.7), что потенциально могло усилить остановку клеточного цикла в комбинации. Ингибирование MDM2 транскрипционно индуцировало экспрессию проапоптотических факторов PUMA и BAX (фиг.7), и индукция PUMA также была подтверждена посредством вестерн-блоттинга (фиг.8). Ингибирование MEK продемонстрировало повышенные уровни проапоптотического белка BIM (фиг.7), и транскрипционно индуцированную экспрессию проапоптотических факторов BMF и NOXA1 (фиг.8). В совокупности, индукция этих генов и белков в комбинации лекарственных средств может объяснить увеличенное расщепление PARP (cPARP), наблюдаемое для комбинации во всех моделях кроме COLO-678 (фиг.7). cPARP является признаком индукции апоптоза.
ЗАКЛЮЧЕНИЯ
В заключение, данные позволили предположить, что комбинированное ингибирование MDM2 и MEK могло регулировать комплементарные наборы белков остановки клеточного цикла (индукция p21 и p27) для индукции остановки клеточного цикла в G1 и/или G2, и проапоптотические белки (например, индукция BAX, BIM и PUMA) для индукции клеточной смерти посредством апоптоза. Комбинирование ингибирование MDM2 и MEK в клетках рака ободочной и прямой кишки с TP53 дикого типа может обеспечивать эффективный терапевтический режим, способный улучшать ответы по сравнению с каждым средством отдельно и приводить к более длительным ответам в клинике.
Пример 2: Эффект in vitro на пролиферацию комбинирования ингибитора MDM2 и ингибитора MEK с ингибитором Bcl2.
Данное исследование было предназначено для изучения эффекта in vitro на пролиферацию комбинирования ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) и ингибитора MEK траметиниба (СОЕДИНЕНИЕ B) с ингибитором BCL-2/-XL навитоклаксом (ABT-263) (СОЕДИНЕНИЕ C) в клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа.
СПОСОБЫ
СОЕДИНЕНИЯ A, B и C растворяли в 100% DMSO (Sigma, каталожный номер D2650) в концентрации 20 мМ и хранили при -20°C до применения. Соединения размещали в основных планшетах для лекарственных средств (Greiner, каталожный номер 788876) и подвергали 3-кратным серийным разведениям (7 шагов) в концентрации 2000X.
Клеточные линии рака ободочной и прямой кишки, использованные для этого исследования, были получены, культивировались и обрабатывались коммерческими поставщиками ATCC и ECACC (таблица 4). Все среды клеточных линий дополняли 10% FBS (HyClone, каталожный номер SH30071.03).
Таблица 4. Информация о клеточных линиях
Клеточные линии культивировали в инкубаторе при 37°C и 5% CO2 и увеличивали в количестве во флаконах T-75. Во всех случаях клетки размораживали из замороженных исходных культур, увеличивали в количестве в течение ≥1 пассажа с использованием разведений 1:3, подсчитывали и оценивали в отношении жизнеспособности с использованием счетчика ViCell (Beckman-Coulter) перед посевом. Для разделения и увеличения в количестве клеточных линий клетки извлекали из флаконов с использованием 0,25% трипсина-EDTA (GIBCO, каталожный номер 25200). Было определено, что все клеточные линии были свободны от контаминации микоплазмой, как определяли с помощью методологии определения с использованием ПЦР, проведенной Idexx Radil (Columbia, MO, США), и были правильно идентифицированы посредством выявления панели SNP.
Для исследования эффекта комбинации СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B и СОЕДИНЕНИЯ C на клеточную пролиферацию клетки высевали в черные 384-луночные микропланшеты с прозрачным дном (Matrix/Thermo Scientific, каталожный номер 4332) в 50 мкл среды на лунку с плотностью клеток от 500 до 1250 клеток/лунка (таблица 4) и им позволяли инкубироваться при 37 градусах, 5% CO2, в течение 24 ч. Через 24 ч один 384-луночный планшет на клеточную линию подготавливали для подсчета клеток посредством микроскопии (см. ниже) без проведения обработки (="исходный уровень"). Другие планшеты с клетками обрабатывали посредством переноса 25 нл 2000X соединения из основных планшетов с лекарственными средствами с использованием акустического жидкостного дозатора ATS (ECD Biosystems), достигая конечной концентрации 1X. СОЕДИНЕНИЕ A использовали в диапазоне конечных концентраций 13 нМ-10 мкМ, СОЕДИНЕНИЕ B использовали в диапазоне конечных концентраций 13 нМ-10 мкМ, и СОЕДИНЕНИЕ C использовали в диапазоне конечных концентраций 0,4 нМ-0,3 мкМ (7 шагов с разведением 1:3). Для оценки эффекта тройной комбинации все СОЕДИНЕНИЯ (A, B, C) отдельно, все три попарных комбинации (A+B, A+C, B+C) и тройную комбинацию (A+B+C) исследовали в одном эксперименте. Парные комбинации и тройную комбинацию тестировали в фиксированном соотношении 1:1 (для пар лекарственных средств) и 1:1:1 (для трех лекарственных средств) в каждом разведении с получением 7 комбинированных условий на обработку. Кроме того, отрицательные контроли (DMSO="носитель") и положительные контроли (стауроспорин=уничтожение клеток, серия разведений 1:2 из 7-точек для диапазона доз 16 нМ-1 мкМ) переносили в качестве контролей обработки, и соединения без эффективности в протестированных клеточных линиях использовали в комбинациях с СОЕДИНЕНИЕМ A и СОЕДИНЕНИЕМ B в качестве контролей в виде комбинаций (комбинации, которые не превышают эффективности более эффективного единственного средства="не взаимодействующие" комбинации). После добавления соединения к одной из трех реплик добавляли 50 нл 2 мМ реагента CellEvent Caspase-3/7 Green Detection Reagent (ThermoFisher, каталожный номер C10423) с использованием цифрового дозатора HP D300 (Tecan). Индукцию каспазы 3/7 количественно определяли в качестве показателя апоптоза, индуцированного обработкой. Клетки обрабатывали в течение от 72 ч до 96 ч в зависимости от их времени удвоения (таблица 4), и активацию каспазы 3/7 количественно определяли каждые 24 ч посредством микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективном 4X и фильтрами возбуждения/испускания FITC. В конце обработки клетки подготавливали для подсчета клеток посредством микроскопии. Клетки фиксировали и пермеабилизировали в течение 45 минут в 4% PFA (Electron Microscopy Sciences, каталожный номер 15714), 0,12% TX-100 (Electron Microscopy Sciences, каталожный номер 22140) в PBS (Boston Bioproducts, каталожный номер BM-220). После промывания клеток три раза PBS их ДНК окрашивали в течение 30 красителем Хехста 33342 (ThermoFisher, каталожный номер H3570) в конечной концентрации 4 мкг/мл. Клетки промывали три раза PBS, а затем планшеты запаивали с использованием PlateLoc (Agilent Technologies) алюминиевыми уплотнителями (Agilent Technologies, каталожный номер 06644-001) и хранили при 4°C до визуализации. Все клетки на лунку/обработку фотографировали в одном изображении посредством флуоресцентной микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективом 4X и фильтрами возбуждения/испускания DAPI.
Изображения анализировали после адаптации ранее описанных способов (Horn, Sandmann et al. 2011) и с использованием пакета EBImage пакета Bioconductor в R (Pau, Fuchs et al. 2010). Объекты в обоих каналах, DAPI (для красителя Хехста/ДНК) и FITC (для каспазы 3/7), сегментировали по отдельности посредством адаптивного определения порогового значения и подсчета. Пороговое значение для положительных по каспазе 3/7 объектов определяли вручную на клеточную линию после сравнения отрицательных контролей (DMSO) и положительных контролей (стауроспорин). Посредством анализа 17 дополнительных признаков объектов/ядер в канале ДНК (признаки формы и интенсивности) идентифицировали дебрис/фрагментированные ядра. Для этого проводили сравнение вручную распределений дополнительных признаков между положительными контролями (стауроспорин) и отрицательными контролями (DMSO) для каждой клеточной линии. Признаки, которые могли отличаться между условиями (например, сдвиг распределения результата измерения признака при сравнении DMSO со стауроспорином) использовали для определения популяции "дебриса" против популяции "жизнеспособных" ядер. Результат подсчета дебриса вычитали из результата исходного подсчета ядер. Полученное количество ядер использовали в качестве показателя клеточной пролиферации ("подсчитанное количество клеток").
Эффект соединения на клеточную пролиферацию вычисляли из подсчитанных количеств обработанных клеток относительно подсчитанных количеств клеток, обработанных отрицательным контролем (DMSO), на фиг.9 обозначаемых как "нормализованное подсчитанное количество клеток" (="xnorm") по оси y. Синергические комбинации идентифицировали с использованием модели наилучшего средства отдельно (HSA) в качестве нуль-гипотезы (Berenbaum 1989). Избыток относительно модели HSA прогнозирует функциональную взаимосвязь между ингибируемыми мишенями (Lehar, Zimmermann et al. 2007, Lehar, Krueger et al. 2009). Входными данными в модели были величины ингибирования на дозу лекарственного средства:
I=1 - xnorm
I: ингибирование
xnorm: нормализованное подсчитанное количество клеток (среднее значение для трех реплик)
В каждой точке дозирования комбинированной обработки вычисляли разность между ингибированием посредством комбинации и ингибированием наиболее мощного из двух отдельных средств (= остатки модели). Аналогично, для оценки синергии тройных комбинаций в каждой точке дозирования вычисляли разность между ингибированием тройной комбинации лекарственных средств и ингибированием наиболее мощной пары лекарственных средств. Для определения эффектов комбинации c высоким ингибированием остатки взвешивали с наблюдаемым ингибированием в той же точке дозы. Общий показатель комбинирования C для комбинации лекарственных средств представляет собой сумму взвешенных остатков для всех концентраций:
C=ΣConc (Idata * (Idata - Imodel))
Idata: измеренное ингибирование
Imodel: ингибирование согласно нуль-гипотезе HSA
Робастные z-показатели (zC) для комбинаций вычисляли в качестве соотношений показателей комбинирования C для обработок и срединное абсолютное отклонение (mad) от не взаимодействующих комбинаций:
zC=C/mad(Czero)
Czero: показатели комбинирования для не взаимодействующих комбинаций
zC является индикатором силы комбинирования, причем:
zC ≥ 3: синергия
3 > zC ≥ 2: слабая синергия
zC < 2: нет синергии
IC50 представляет собой концентрацию соединения, которая приводит к 50% подсчитанному количеству клеток относительно DMSO. Вычисления IC50 (см. таблицу 5) проводили с использованием пакета DRC в R (Ritz and Streibig 2005) и аппроксимации четерехпараметрической логарифмически-логистической функции к данным.
Эффект соединения на апоптоз определяли посредством вычисления процента клеток с активированной каспазой 3/7 на обработку и моментом времени относительно исходных подсчитанных количеств клеток (до вычитания дебриса) (ось y на фиг.10). Подсчитанные количества клеток в моменты времени, которые не были измерены экспериментально, получали посредством регрессионного анализа путем аппроксимации линейной модели для логарифмически преобразованных подсчитанных количеств клеток на 0 сутки и конца обработки (предполагая экспоненциальный рост клеток).
Таблица 5. Величины IC50 отдельных средств для каждого соединения и результаты измерения z-показателя синергии для комбинации СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B и СОЕДИНЕНИЯ C.
РЕЗУЛЬТАТЫ
В данном протоколе эффективность ингибитора MDM2 (СОЕДИНЕНИЕ A), ингибитора MEK (траметиниб, СОЕДИНЕНИЕ B) и ингибитора BCL-2/-XL (ABT-263, СОЕДИНЕНИЕ C) оценивали индивидуально и в комбинации всего в 5 клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа. Четыре из линий имели мутантный KRAS (GP2d, LS-180, HCT-116, LoVo), одна линия имела мутантный BRAF (RKO) (таблица 4). СОЕДИНЕНИЕ A в качестве единственного средства ингибировало рост клеточных линий с величинами IC50 от субмикромолярных до микромолярных. СОЕДИНЕНИЕ B в качестве единственного средства ингибировало рост всех кроме одной клеточных линий (GP2d) с наномолярными величинами IC50, в то время как СОЕДИНЕНИЕ C не имело эффективности в качестве единственного средства (фиг.9 и таблица 5). Тройная комбинация (A+B+C) обеспечивала синергичное ингибирование (согласно модели HSA) относительно пар лекарственных средств в 2/5 протестированных клеточных моделей (таблица 5). В четырех из линий (GP2d, HCT-116, RKO, LoVo) тройная комбинация продемонстрировала более мощный апоптоз (оцениваемый посредством измерения индукции каспазы 3/7) по сравнению с парными комбинациями (фиг.10).
ЗАКЛЮЧЕНИЯ
В совокупности, комбинированное ингибирование MDM2, MEK и BCL-2/-XL в клетках CRC с TP53 дикого типа может обеспечить эффективный терапевтический режим, способный улучшать ответы по сравнению с каждым из единственных средств и приводить к более длительным ответам в клинике.
Кроме того, ингибитор PI3K 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты добавляли к комбинации (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) и ингибитора MEK траметиниба (СОЕДИНЕНИЕ B) с ингибитором BCL-2/-XL навитоклаксом (ABT-263) (СОЕДИНЕНИЕ C) с получением четырехкомпонентной комбинации и вместе исследовали в 1 клеточной линии с мутантным KRAS, и было обнаружено, что они являются слабо синергичными (LS-180, z-показатель комбинирования 2,63) и сильно индуцирующими апоптоз (максимум 61%).
Пример 3: Эффект in vitro на пролиферацию комбинирования ингибитора MDM2 и ингибитора MEK с ингибитором EGFR.
Данное исследование было предназначено для изучения эффекта in vitro на пролиферацию комбинирования ингибитора (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) и ингибитора MEK траметиниба (СОЕДИНЕНИЕ B) с ингибитором EGFR эрлотинибом (СОЕДИНЕНИЕ C) в клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа.
СПОСОБЫ
СОЕДИНЕНИЯ A, B и C растворяли в 100% DMSO (Sigma, каталожный номер D2650) в концентрации 20 мМ и хранили при -20°C до применения. Соединения размещали в основных планшетах для лекарственных средств (Greiner, каталожный номер 788876) и подвергали 3-кратным серийным разведениям (7 шагов) в концентрации 2000X.
Клеточные линии рака ободочной и прямой кишки, использованные для этого исследования, были получены, культивировались и обрабатывались коммерческими поставщиками ATCC и ECACC (таблица 6). Все среды клеточных линий дополняли 10% FBS (HyClone, каталожный номер SH30071.03).
Таблица 6. Информация о клеточных линиях
Клеточные линии культивировали в инкубаторе при 37°C и 5% CO2 и увеличивали в количестве во флаконах T-75. Во всех случаях клетки размораживали из замороженных исходных культур, увеличивали в количестве в течение ≥1 пассажа с использованием разведений 1:3, подсчитывали и оценивали в отношении жизнеспособности с использованием счетчика ViCell (Beckman-Coulter) перед посевом. Для разделения и увеличения в количестве клеточных линий клетки извлекали из флаконов с использованием 0,25% трипсина-EDTA (GIBCO, каталожный номер 25200). Было определено, что все клеточные линии были свободны от контаминации микоплазмой, как определяли с помощью методологии определения с использованием ПЦР, проведенной Idexx Radil (Columbia, MO, США), и были правильно идентифицированы посредством выявления панели SNP.
Для исследования эффекта комбинации СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B и СОЕДИНЕНИЯ C на клеточную пролиферацию клетки высевали в черные 384-луночные микропланшеты с прозрачным дном (Matrix/Thermo Scientific, каталожный номер 4332) в 50 мкл среды на лунку с плотностью клеток от 500 до 1250 клеток/лунка (таблица 6) и им позволяли инкубироваться при 37 градусах, 5% CO2, в течение 24 ч. Через 24 ч один 384-луночный планшет на клеточную линию подготавливали для подсчета клеток посредством микроскопии (см. ниже) без проведения обработки (="исходный уровень"). Другие планшеты с клетками обрабатывали посредством переноса 25 нл 2000X соединения из основных планшетов с лекарственными средствами с использованием акустического жидкостного дозатора ATS (ECD Biosystems), достигая конечной концентрации 1X. СОЕДИНЕНИЕ A использовали в диапазоне конечных концентраций 13 нМ-10 мкМ, СОЕДИНЕНИЕ B использовали в диапазоне конечных концентраций 13 нМ-10 мкМ, и СОЕДИНЕНИЕ C использовали в диапазоне конечных концентраций 13 нМ-10 мкМ (7 шагов с разведением 1:3). Для оценки эффекта тройной комбинации все СОЕДИНЕНИЯ (A, B, C) отдельно, все три попарных комбинации (A+B, A+C, B+C) и тройную комбинацию (A+B+C) исследовали в одном эксперименте. Парные комбинации и тройную комбинацию тестировали в фиксированном соотношении 1:1 (для пар лекарственных средств) и 1:1:1 (для трех лекарственных средств) в каждом разведении с получением 7 комбинированных условий на обработку. Кроме того, отрицательные контроли (DMSO="носитель") и положительные контроли (стауроспорин=уничтожение клеток, серия разведений 1:2 из 7-точек для диапазона доз 16 нМ-1 мкМ) переносили в качестве контролей обработки, и соединения без эффективности в протестированных клеточных линиях использовали в комбинациях с СОЕДИНЕНИЕМ A и СОЕДИНЕНИЕМ B в качестве контролей в виде комбинаций (комбинации, которые не превышают эффективности более эффективного единственного средства="не взаимодействующие" комбинации). После добавления соединения к одной из трех реплик добавляли 50 нл 2 мМ реагента CellEvent Caspase-3/7 Green Detection Reagent (ThermoFisher, каталожный номер C10423) с использованием цифрового дозатора HP D300 (Tecan). Индукцию каспазы 3/7 количественно определяли в качестве показателя апоптоза, индуцированного обработкой. Клетки обрабатывали в течение от 72 ч до 96 ч в зависимости от их времени удвоения (таблица 4), и активацию каспазы 3/7 количественно определяли каждые 24 ч посредством микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективном 4X и фильтрами возбуждения/испускания FITC. В конце обработки клетки подготавливали для подсчета клеток посредством микроскопии. Клетки фиксировали и пермеабилизировали в течение 45 минут в 4% PFA (Electron Microscopy Sciences, каталожный номер 15714), 0,12% TX-100 (Electron Microscopy Sciences, каталожный номер 22140) в PBS (Boston Bioproducts, каталожный номер BM-220). После промывания клеток три раза PBS их ДНК окрашивали в течение 30 красителем Хехста 33342 (ThermoFisher, каталожный номер H3570) в конечной концентрации 4 мкг/мл. Клетки промывали три раза PBS, а затем планшеты запаивали с использованием PlateLoc (Agilent Technologies) алюминиевыми уплотнителями (Agilent Technologies, каталожный номер 06644-001) и хранили при 4°C до визуализации. Все клетки на лунку/обработку фотографировали в одном изображении посредством флуоресцентной микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективом 4X и фильтрами возбуждения/испускания DAPI.
Изображения анализировали после адаптации ранее описанных способов (Horn, Sandmann et al. 2011) и с использованием пакета EBImage пакета Bioconductor в R (Pau, Fuchs et al. 2010). Объекты в обоих каналах, DAPI (для красителя Хехста/ДНК) и FITC (для каспазы 3/7), сегментировали по отдельности посредством адаптивного определения порогового значения и подсчета. Пороговое значение для положительных по каспазе 3/7 объектов определяли вручную на клеточную линию после сравнения отрицательных контролей (DMSO) и положительных контролей (стауроспорин). Посредством анализа 17 дополнительных признаков объектов/ядер в канале ДНК (признаки формы и интенсивности) идентифицировали дебрис/фрагментированные ядра. Для этого проводили сравнение вручную распределений дополнительных признаков между положительными контролями (стауроспорин) и отрицательными контролями (DMSO) для каждой клеточной линии. Признаки, которые могли отличаться между условиями (например, сдвиг распределения результата измерения признака при сравнении DMSO со стауроспорином) использовали для определения популяции "дебриса" против популяции "жизнеспособных" ядер. Результат подсчета дебриса вычитали из результата исходного подсчета ядер. Полученное количество ядер использовали в качестве показателя клеточной пролиферации ("подсчитанное количество клеток").
Эффект соединения на клеточную пролиферацию вычисляли из подсчитанных количеств обработанных клеток относительно подсчитанных количеств клеток, обработанных отрицательным контролем (DMSO), на фиг.9 обозначаемых как "нормализованное подсчитанное количество клеток" (="xnorm") по оси y. Синергические комбинации идентифицировали с использованием модели наилучшего средства отдельно (HSA) в качестве нуль-гипотезы (Berenbaum 1989). Избыток относительно модели HSA прогнозирует функциональную взаимосвязь между ингибируемыми мишенями (Lehar, Zimmermann et al. 2007, Lehar, Krueger et al. 2009). Входными данными в модели были величины ингибирования на дозу лекарственного средства:
I=1 - xnorm
I: ингибирование
xnorm: нормализованное подсчитанное количество клеток (среднее значение для трех реплик)
В каждой точке дозирования комбинированной обработки вычисляли разность между ингибированием посредством комбинации и ингибированием наиболее мощного из двух отдельных средств (= остатки модели). Аналогично, для оценки синергии тройных комбинаций в каждой точке дозирования вычисляли разность между ингибированием тройной комбинации лекарственных средств и ингибированием наиболее мощной пары лекарственных средств. Для определения эффектов комбинации c высоким ингибированием остатки взвешивали с наблюдаемым ингибированием в той же точке дозы. Общий показатель комбинирования C для комбинации лекарственных средств представляет собой сумму взвешенных остатков для всех концентраций:
C=ΣConc (Idata * (Idata - Imodel))
Idata: измеренное ингибирование
Imodel: ингибирование согласно нуль-гипотезе HSA
Робастные z-показатели (zC) для комбинаций вычисляли в качестве соотношений показателей комбинирования C для обработок и срединное абсолютное отклонение (mad) от не взаимодействующих комбинаций:
zC=C/mad(Czero)
Czero: показатели комбинирования для не взаимодействующих комбинаций
zC является индикатором силы комбинирования, причем:
zC ≥ 3: синергия
3 > zC ≥ 2: слабая синергия
zC < 2: нет синергии
IC50 представляет собой концентрацию соединения, которая приводит к 50% подсчитанному количеству клеток относительно DMSO. Вычисления IC50 (см. таблицу 7) проводили с использованием пакета DRC в R (Ritz and Streibig 2005) и аппроксимации четерехпараметрической логарифмически-логистической функции к данным.
Эффект соединения на апоптоз определяли посредством вычисления процента клеток с активированной каспазой 3/7 на обработку и моментом времени относительно исходных подсчитанных количеств клеток (до вычитания дебриса) (ось y на фиг.12). Подсчитанные количества клеток в моменты времени, которые не были измерены экспериментально, получали посредством регрессионного анализа путем аппроксимации линейной модели для логарифмически преобразованных подсчитанных количеств клеток на 0 сутки и конца обработки (предполагая экспоненциальный рост клеток).
Таблица 7. Величины IC50 отдельных средств для каждого соединения и результаты измерения z-показателя синергии для комбинации СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B и СОЕДИНЕНИЯ C.
РЕЗУЛЬТАТЫ
В данном протоколе эффективность ингибитора MDM2 (СОЕДИНЕНИЕ A), ингибитора MEK (траметиниб, СОЕДИНЕНИЕ B) и ингибитора EGFR (эрлотиниб, СОЕДИНЕНИЕ C) оценивали индивидуально и в комбинации всего в 5 клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа. Четыре из линий имели мутантный KRAS (GP2d, LS-180, HCT-116, LoVo), одна линия имела мутантный BRAF (RKO) (таблица 6). СОЕДИНЕНИЕ A в качестве единственного средства ингибировало рост клеточных линий с величинами IC50 от субмикромолярных до микромолярных. СОЕДИНЕНИЕ B в качестве единственного средства ингибировало рост всех кроме одной клеточных линий (GP2d) с наномолярными величинами IC50, в то время как СОЕДИНЕНИЕ C не имело эффективности в качестве единственного средства в 4/5 линий и микромолярную IC50 у LoVo с мутантным KRAS (фиг.11 и таблица 7). Тройная комбинация (A+B+C) обеспечивала синергичное ингибирование (согласно модели HSA) относительно пар лекарственных средств в модели с мутантом KRAS LoVo (таблица 7). В этой клеточной линии тройная комбинация продемонстрировала более сильный апоптоз (оцениваемый посредством измерения индукции каспазы 3/7) по сравнению с парными комбинациями (фиг.12).
ЗАКЛЮЧЕНИЯ
Комбинированное ингибирование MDM2, MEK и EGFR в клетках CRC с TP53 дикого типа может обеспечить эффективный терапевтический режим, способный улучшать ответы по сравнению с каждым из единственных средств и приводить к более длительным ответам в клинике.
Пример 4: Эффект in vitro на пролиферацию комбинирования ингибитора PI3K и ингибитора MDM2 с ингибитором Bcl2.
Данное исследование было предназначено для изучения эффекта in vitro на пролиферацию комбинирования ингибитора PIK3CA 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амида) 2-амида (S)-пирролидин-1,2-дикарбоновой кислоты (СОЕДИНЕНИЕ A) и ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ B) с ингибитором BCL-2/-XL навитоклаксом (ABT-263) (СОЕДИНЕНИЕ C) в клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа.
СПОСОБЫ
СОЕДИНЕНИЯ A, B и C растворяли в 100% DMSO (Sigma, каталожный номер D2650) в концентрации 20 мМ и хранили при -20°C до применения. Соединения размещали в основных планшетах для лекарственных средств (Greiner, каталожный номер 788876) и подвергали 3-кратным серийным разведениям (7 шагов) в концентрации 2000X.
Клеточные линии рака ободочной и прямой кишки, использованные для этого исследования, были получены, культивировались и обрабатывались коммерческими поставщиками ATCC и ECACC (таблица 8). Все среды клеточных линий дополняли 10% FBS (HyClone, каталожный номер SH30071.03).
Таблица 8. Информация о клеточных линиях
Клеточные линии культивировали в инкубаторе при 37°C и 5% CO2 и увеличивали в количестве во флаконах T-75. Во всех случаях клетки размораживали из замороженных исходных культур, увеличивали в количестве в течение ≥1 пассажа с использованием разведений 1:3, подсчитывали и оценивали в отношении жизнеспособности с использованием счетчика ViCell (Beckman-Coulter) перед посевом. Для разделения и увеличения в количестве клеточных линий клетки извлекали из флаконов с использованием 0,25% трипсина-EDTA (GIBCO, каталожный номер 25200). Было определено, что все клеточные линии были свободны от контаминации микоплазмой, как определяли с помощью методологии определения с использованием ПЦР, проведенной Idexx Radil (Columbia, MO, США), и были правильно идентифицированы посредством выявления панели SNP.
Для исследования эффекта комбинации СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B и СОЕДИНЕНИЯ C на клеточную пролиферацию клетки высевали в черные 384-луночные микропланшеты с прозрачным дном (Matrix/Thermo Scientific, каталожный номер 4332) в 50 мкл среды на лунку с плотностью клеток от 500 до 1250 клеток/лунка (таблица 4) и им позволяли инкубироваться при 37 градусах, 5% CO2, в течение 24 ч. Через 24 ч один 384-луночный планшет на клеточную линию подготавливали для подсчета клеток посредством микроскопии (см. ниже) без проведения обработки (="исходный уровень"). Другие планшеты с клетками обрабатывали посредством переноса 25 нл 2000X соединения из основных планшетов с лекарственными средствами с использованием акустического жидкостного дозатора ATS (ECD Biosystems), достигая конечной концентрации 1X. СОЕДИНЕНИЕ A использовали в диапазоне конечных концентраций 13 нМ-10 мкМ, СОЕДИНЕНИЕ B использовали в диапазоне конечных концентраций 13 нМ-10 мкМ, и СОЕДИНЕНИЕ C использовали в диапазоне конечных концентраций 13 нМ-10 мкМ (7 шагов с разведением 1:3). Для оценки эффекта тройной комбинации все СОЕДИНЕНИЯ (A, B, C) отдельно, все три попарных комбинации (A+B, A+C, B+C) и тройную комбинацию (A+B+C) исследовали в одном эксперименте. Парные комбинации и тройную комбинацию тестировали в фиксированном соотношении 1:1 (для пар лекарственных средств) и 1:1:1 (для трех лекарственных средств) в каждом разведении с получением 7 комбинированных условий на обработку. Кроме того, отрицательные контроли (DMSO="носитель") и положительные контроли (стауроспорин=уничтожение клеток, серия разведений 1:2 из 7-точек для диапазона доз 16 нМ-1 мкМ) переносили в качестве контролей обработки, и соединения без эффективности в протестированных клеточных линиях использовали в комбинациях с СОЕДИНЕНИЕМ A и СОЕДИНЕНИЕМ B в качестве контролей в виде комбинаций (комбинации, которые не превышают эффективности более эффективного единственного средства="не взаимодействующие" комбинации). После добавления соединения к одной из трех реплик добавляли 50 нл 2 мМ реагента CellEvent Caspase-3/7 Green Detection Reagent (ThermoFisher, каталожный номер C10423) с использованием цифрового дозатора HP D300 (Tecan). Индукцию каспазы 3/7 количественно определяли в качестве показателя апоптоза, индуцированного обработкой. Клетки обрабатывали в течение от 72 ч до 96 ч в зависимости от их времени удвоения (таблица 4), и активацию каспазы 3/7 количественно определяли каждые 24 ч посредством микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективном 4X и фильтрами возбуждения/испускания FITC. В конце обработки клетки подготавливали для подсчета клеток посредством микроскопии. Клетки фиксировали и пермеабилизировали в течение 45 минут в 4% PFA (Electron Microscopy Sciences, каталожный номер 15714), 0,12% TX-100 (Electron Microscopy Sciences, каталожный номер 22140) в PBS (Boston Bioproducts, каталожный номер BM-220). После промывания клеток три раза PBS их ДНК окрашивали в течение 30 красителем Хехста 33342 (ThermoFisher, каталожный номер H3570) в конечной концентрации 4 мкг/мл. Клетки промывали три раза PBS, а затем планшеты запаивали с использованием PlateLoc (Agilent Technologies) алюминиевыми уплотнителями (Agilent Technologies, каталожный номер 06644-001) и хранили при 4°C до визуализации. Все клетки на лунку/обработку фотографировали в одном изображении посредством флуоресцентной микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективом 4X и фильтрами возбуждения/испускания DAPI.
Изображения анализировали после адаптации ранее описанных способов (Horn, Sandmann et al. 2011) и с использованием пакета EBImage пакета Bioconductor в R (Pau, Fuchs et al. 2010). Объекты в обоих каналах, DAPI (для красителя Хехста/ДНК) и FITC (для каспазы 3/7), сегментировали по отдельности посредством адаптивного определения порогового значения и подсчета. Пороговое значение для положительных по каспазе 3/7 объектов определяли вручную на клеточную линию после сравнения отрицательных контролей (DMSO) и положительных контролей (стауроспорин). Посредством анализа 17 дополнительных признаков объектов/ядер в канале ДНК (признаки формы и интенсивности) идентифицировали дебрис/фрагментированные ядра. Для этого проводили сравнение вручную распределений дополнительных признаков между положительными контролями (стауроспорин) и отрицательными контролями (DMSO) для каждой клеточной линии. Признаки, которые могли отличаться между условиями (например, сдвиг распределения результата измерения признака при сравнении DMSO со стауроспорином) использовали для определения популяции "дебриса" против популяции "жизнеспособных" ядер. Результат подсчета дебриса вычитали из результата исходного подсчета ядер. Полученное количество ядер использовали в качестве показателя клеточной пролиферации ("подсчитанное количество клеток").
Эффект соединения на клеточную пролиферацию вычисляли из подсчитанных количеств обработанных клеток относительно подсчитанных количеств клеток, обработанных отрицательным контролем (DMSO), на фиг.9 обозначаемых как "нормализованное подсчитанное количество клеток" (="xnorm") по оси y. Синергические комбинации идентифицировали с использованием модели наилучшего средства отдельно (HSA) в качестве нуль-гипотезы (Berenbaum 1989). Избыток относительно модели HSA прогнозирует функциональную взаимосвязь между ингибируемыми мишенями (Lehar, Zimmermann et al. 2007, Lehar, Krueger et al. 2009). Входными данными в модели были величины ингибирования на дозу лекарственного средства:
I=1 - xnorm
I: ингибирование
xnorm: нормализованное подсчитанное количество клеток (среднее значение для трех реплик)
В каждой точке дозирования комбинированной обработки вычисляли разность между ингибированием посредством комбинации и ингибированием наиболее мощного из двух отдельных средств (= остатки модели). Аналогично, для оценки синергии тройных комбинаций в каждой точке дозирования вычисляли разность между ингибированием тройной комбинации лекарственных средств и ингибированием наиболее мощной пары лекарственных средств. Для определения эффектов комбинации c высоким ингибированием остатки взвешивали с наблюдаемым ингибированием в той же точке дозы. Общий показатель комбинирования C для комбинации лекарственных средств представляет собой сумму взвешенных остатков для всех концентраций:
C=ΣConc (Idata * (Idata - Imodel))
Idata: измеренное ингибирование
Imodel: ингибирование согласно нуль-гипотезе HSA
Робастные z-показатели (zC) для комбинаций вычисляли в качестве соотношений показателей комбинирования C для обработок и срединное абсолютное отклонение (mad) от не взаимодействующих комбинаций:
zC=C/mad(Czero)
Czero: показатели комбинирования для не взаимодействующих комбинаций
zC является индикатором силы комбинирования, причем:
zC ≥ 3: синергия
3 > zC ≥ 2: слабая синергия
zC < 2: нет синергии
IC50 представляет собой концентрацию соединения, которая приводит к 50% подсчитанному количеству клеток относительно DMSO. Вычисления IC50 (см. таблицу 9) проводили с использованием пакета DRC в R (Ritz and Streibig 2005) и аппроксимации четерехпараметрической логарифмически-логистической функции к данным.
Эффект соединения на апоптоз определяли посредством вычисления процента клеток с активированной каспазой 3/7 на обработку и моментом времени относительно исходных подсчитанных количеств клеток (до вычитания дебриса) (ось y на фиг.14). Подсчитанные количества клеток в моменты времени, которые не были измерены экспериментально, получали посредством регрессионного анализа путем аппроксимации линейной модели для логарифмически преобразованных подсчитанных количеств клеток на 0 сутки и конца обработки (предполагая экспоненциальный рост клеток).
Таблица 9. Величины IC50 отдельных средств для каждого соединения и результаты измерения z-показателя синергии для комбинации СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B и СОЕДИНЕНИЯ C.
РЕЗУЛЬТАТЫ
В данном протоколе эффективность ингибитора PIK3CA (BYL719, СОЕДИНЕНИЕ A), ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ B) и ингибитора BCL-2/-XL (ABT-263, СОЕДИНЕНИЕ C) оценивали индивидуально и в комбинации всего в 5 клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа. Четыре из линий имели мутантный KRAS (GP2d, LS-180, HCT-116, LoVo), одна линия имела мутантный BRAF (RKO). СОЕДИНЕНИЕ A в качестве единственного средства ингибировало рост 2 клеточных линий с микромолярными величинами IC50 и было активным только в наиболее высокой дозе (10 мкМ) в 3 других линиях (фиг.13 и таблица 9). СОЕДИНЕНИЕ B в качестве единственного средства ингибировало рост клеточных линий с величинами IC50 от субмикромоляных до микромолярных, в то время как СОЕДИНЕНИЕ C не имело эффективности в качестве единственного средства (фиг.13 и таблица 9). Тройная комбинация (A+B+C) обеспечивала синергичное ингибирование (согласно модели HSA) относительно пар лекарственных средств в 2/5 протестированных клеточных моделей (таблица 9). В четырех из линий (HCT-116, LoVo, RKO, LS-180) тройная комбинация продемонстрировала более мощный апоптоз (оцениваемый посредстовм измерения индукции каспазы 3/7) по сравнению с парными комбинациями (фиг.14).
ЗАКЛЮЧЕНИЯ
В совокупности, комбинированное ингибирование PIK3CA, MDM2 и BCL-2/-XL в клетках CRC с TP53 дикого типа может обеспечить эффективный терапевтический режим, способный улучшать ответы по сравнению с каждым из единственных средств и приводить к более длительным ответам в клинике.
Пример 5: Эффект in vitro на пролиферацию комбинирования ингибитора MDM2 и ингибитора Bcl2
Данное исследование было предназначено для изучения эффекта in vitro на пролиферацию комбинирования ингибитора MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она (СОЕДИНЕНИЕ A) и ингибитора BCL-2/-XL навитоклакса (ABT-263) (СОЕДИНЕНИЕ B) в клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа.
СПОСОБЫ
СОЕДИНЕНИЯ A и B растворяли в 100% DMSO (Sigma, каталожный номер D2650) в концентрации 20 мМ и хранили при -20°C до применения. Соединения размещали в основных планшетах для лекарственных средств (Greiner, каталожный номер 788876) и подвергали 3-кратным серийным разведениям (7 шагов) в концентрации 2000X.
Клеточные линии рака ободочной и прямой кишки, использованные для этого исследования, были получены, культивировались и обрабатывались коммерческими поставщиками ATCC и ECACC (таблица 10). Все среды клеточных линий дополняли 10% FBS (HyClone, каталожный номер SH30071.03).
Таблица 10. Информация о клеточных линиях
Клеточные линии культивировали в инкубаторе при 37°C и 5% CO2 и увеличивали в количестве во флаконах T-75. Во всех случаях клетки размораживали из замороженных исходных культур, увеличивали в количестве в течение ≥1 пассажа с использованием разведений 1:3, подсчитывали и оценивали в отношении жизнеспособности с использованием счетчика ViCell (Beckman-Coulter) перед посевом. Для разделения и увеличения в количестве клеточных линий клетки извлекали из флаконов с использованием 0,25% трипсина-EDTA (GIBCO, каталожный номер 25200). Было определено, что все клеточные линии были свободны от контаминации микоплазмой, как определяли с помощью методологии определения с использованием ПЦР, проведенной Idexx Radil (Columbia, MO, США), и были правильно идентифицированы посредством выявления панели SNP.
Для исследования эффекта комбинации СОЕДИНЕНИЯ A и СОЕДИНЕНИЯ B на клеточную пролиферацию клетки высевали в черные 384-луночные микропланшеты с прозрачным дном (Matrix/Thermo Scientific, каталожный номер 4332) в 50 мкл среды на лунку с плотностью клеток от 500 до 1250 клеток/лунка (таблица 4) и им позволяли инкубироваться при 37 градусах, 5% CO2, в течение 24 ч. Через 24 ч один 384-луночный планшет на клеточную линию подготавливали для подсчета клеток посредством микроскопии (см. ниже) без проведения обработки (="исходный уровень"). Другие планшеты с клетками обрабатывали посредством переноса 25 нл 2000X соединения из основных планшетов с лекарственными средствами с использованием акустического жидкостного дозатора ATS (ECD Biosystems), достигая конечной концентрации 1X. СОЕДИНЕНИЕ A использовали в диапазоне конечных концентраций 13 нМ-10 мкМ и СОЕДИНЕНИЕ B использовали в диапазоне конечных концентраций 13 нМ-10 мкМ (7 шагов с разведением 1:3). Для комбинации СОЕДИНЕНИЯ A с СОЕДИНЕНИЕМ B отдельные средства комбинировали в фиксированном соотношении 1:1 при каждом разведении с получением 7 комбинированных обработок. Кроме того, отрицательные контроли (DMSO="носитель") и положительные контроли (стауроспорин=уничтожение клеток, серия разведений 1:2 из 7-точек для диапазона доз 16 нМ-1 мкМ) переносили в качестве контролей обработки, и соединения без эффективности в протестированных клеточных линиях использовали в комбинациях с СОЕДИНЕНИЕМ A и СОЕДИНЕНИЕМ B в качестве контролей в виде комбинаций (комбинации, которые не превышают эффективности более эффективного единственного средства="не взаимодействующие" комбинации). После добавления соединения к одной из трех реплик добавляли 50 нл 2 мМ реагента CellEvent Caspase-3/7 Green Detection Reagent (ThermoFisher, каталожный номер C10423) с использованием цифрового дозатора HP D300 (Tecan). Индукцию каспазы 3/7 количественно определяли в качестве показателя апоптоза, индуцированного обработкой. Клетки обрабатывали в течение от 72 ч до 96 ч в зависимости от их времени удвоения (таблица 4), и активацию каспазы 3/7 количественно определяли каждые 24 ч посредством микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективном 4X и фильтрами возбуждения/испускания FITC. В конце обработки клетки подготавливали для подсчета клеток посредством микроскопии. Клетки фиксировали и пермеабилизировали в течение 45 минут в 4% PFA (Electron Microscopy Sciences, каталожный номер 15714), 0,12% TX-100 (Electron Microscopy Sciences, каталожный номер 22140) в PBS (Boston Bioproducts, каталожный номер BM-220). После промывания клеток три раза PBS их ДНК окрашивали в течение 30 красителем Хехста 33342 (ThermoFisher, каталожный номер H3570) в конечной концентрации 4 мкг/мл. Клетки промывали три раза PBS, а затем планшеты запаивали с использованием PlateLoc (Agilent Technologies) алюминиевыми уплотнителями (Agilent Technologies, каталожный номер 06644-001) и хранили при 4°C до визуализации. Все клетки на лунку/обработку фотографировали в одном изображении посредством флуоресцентной микроскопии с использованием InCell Analyzer 2000 (GE Healthcare), оборудованного объективом 4X и фильтрами возбуждения/испускания DAPI.
Изображения анализировали после адаптации ранее описанных способов (Horn, Sandmann et al. 2011) и с использованием пакета EBImage пакета Bioconductor в R (Pau, Fuchs et al. 2010). Объекты в обоих каналах, DAPI (для красителя Хехста/ДНК) и FITC (для каспазы 3/7), сегментировали по отдельности посредством адаптивного определения порогового значения и подсчета. Пороговое значение для положительных по каспазе 3/7 объектов определяли вручную на клеточную линию после сравнения отрицательных контролей (DMSO) и положительных контролей (стауроспорин). Посредством анализа 17 дополнительных признаков объектов/ядер в канале ДНК (признаки формы и интенсивности) идентифицировали дебрис/фрагментированные ядра. Для этого проводили сравнение вручную распределений дополнительных признаков между положительными контролями (стауроспорин) и отрицательными контролями (DMSO) для каждой клеточной линии. Признаки, которые могли отличаться между условиями (например, сдвиг распределения результата измерения признака при сравнении DMSO со стауроспорином) использовали для определения популяции "дебриса" против популяции "жизнеспособных" ядер. Результат подсчета дебриса вычитали из результата исходного подсчета ядер. Полученное количество ядер использовали в качестве показателя клеточной пролиферации ("подсчитанное количество клеток").
Эффект соединения на клеточную пролиферацию вычисляли из подсчитанных количеств обработанных клеток относительно подсчитанных количеств клеток, обработанных отрицательным контролем (DMSO), на фиг.9 обозначаемых как "нормализованное подсчитанное количество клеток" (="xnorm") по оси y. Синергические комбинации идентифицировали с использованием модели наилучшего средства отдельно (HSA) в качестве нуль-гипотезы (Berenbaum 1989). Избыток относительно модели HSA прогнозирует функциональную взаимосвязь между ингибируемыми мишенями (Lehar, Zimmermann et al. 2007, Lehar, Krueger et al. 2009). Входными данными в модели были величины ингибирования на дозу лекарственного средства:
I=1 - xnorm
I: ингибирование
xnorm: нормализованное подсчитанное количество клеток (среднее значение для трех реплик)
В каждой точке дозирования комбинированной обработки вычисляли разность между ингибированием посредством комбинации и ингибированием наиболее мощного из двух отдельных средств (= остатки модели). Аналогично, для оценки синергии тройных комбинаций в каждой точке дозирования вычисляли разность между ингибированием тройной комбинации лекарственных средств и ингибированием наиболее мощной пары лекарственных средств. Для определения эффектов комбинации c высоким ингибированием остатки взвешивали с наблюдаемым ингибированием в той же точке дозы. Общий показатель комбинирования C для комбинации лекарственных средств представляет собой сумму взвешенных остатков для всех концентраций:
C=ΣConc (Idata * (Idata - Imodel))
Idata: измеренное ингибирование
Imodel: ингибирование согласно нуль-гипотезе HSA
Робастные z-показатели (zC) для комбинаций вычисляли в качестве соотношений показателей комбинирования C для обработок и срединное абсолютное отклонение (mad) от не взаимодействующих комбинаций:
zC=C/mad(Czero)
Czero: показатели комбинирования для не взаимодействующих комбинаций
zC является индикатором силы комбинирования, причем:
zC ≥ 3: синергия
3 > zC ≥ 2: слабая синергия
zC < 2: нет синергии
IC50 представляет собой концентрацию соединения, которая приводит к 50% подсчитанному количеству клеток относительно DMSO. Вычисления IC50 (см. таблицу 11) проводили с использованием пакета DRC в R (Ritz and Streibig 2005) и аппроксимации четерехпараметрической логарифмически-логистической функции к данным.
Эффект соединения на апоптоз определяли посредством вычисления процента клеток с активированной каспазой 3/7 на обработку и моментом времени относительно исходных подсчитанных количеств клеток (до вычитания дебриса) (ось y на фиг.16). Подсчитанные количества клеток в моменты времени, которые не были измерены экспериментально, получали посредством регрессионного анализа путем аппроксимации линейной модели для логарифмически преобразованных подсчитанных количеств клеток на 0 сутки и конца обработки (предполагая экспоненциальный рост клеток).
Таблица 11. Величины IC50 отдельных средств для каждого соединения и результаты измерения z-показателя синергии для комбинации СОЕДИНЕНИЯ A и СОЕДИНЕНИЯ B.
РЕЗУЛЬТАТЫ
В данном протоколе эффективность ингибитора MDM2 (СОЕДИНЕНИЕ A) и ингибитора BCL-2/-XL (СОЕДИНЕНИЕ B) оценивали индивидуально и в комбинации всего в 5 клеточных линиях рака ободочной и прямой кишки с TP53 дикого типа. Четыре из линий имели мутантный KRAS (GP2d, LS-180, HCT-116, LoVo), одна линия имела мутантный BRAF (RKO), и четыре из линий также имели мутантный PIK3CA (GP2d, RKO, LS-180, HCT-116) (Таблица 10). СОЕДИНЕНИЕ A в качестве единственного средства ингибировало рост клеточных линий с величинами IC50 от субмикромолярных до микромолярных (фиг.15 и таблица 11). СОЕДИНЕНИЕ B не имело эффективности в качестве единственного средства (фиг.15 и таблица 11). Комбинированная обработка обеспечивала синергичное ингибирование (согласно модели HSA) в 3/5 клеточных моделей, и слабое синергичное ингибирование еще в 1 модели (таблица 11). Комбинация также продемонстрировала сильную индукцию апоптоза (оцениваемый посредством измерения индукции каспазы 3/7) по сравнению с отдельными средствами (фиг.16), причем наиболее сильную индукцию наблюдали в GP2d, HCT-116 и LoVo.
ЗАКЛЮЧЕНИЯ
Комбинированное ингибирование MDM2 и BCL-2/-XL в клетках рака ободочной и прямой кишки с TP53 дикого типа, мутантных по KRAS и BRAF, может обеспечить эффективный терапевтический режим, способный улучшать ответы по сравнению с каждым из единственных средств и приводить к более длительным ответам в клинике.
Пример 6: Эффект in vitro на пролиферацию комбинирования ингибитора MDM2, и ингибитора MEK, и ингибитора Bcl2
Аналогично, как описано в предшествующих примерах, тройную комбинацию (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она, траметиниба и RAF265 тестировали в 4 моделях с мутантным KRAS (HCT-116, GP2d, LoVo и LS-180). Комбинация оказывало синергичное действие в 2/4 моделей (HCT-116, GP2d, с z-показателями комбинирования 9,2 и 5,5, соответственно), и слабое синергичное действие в 1/4 моделей (LoVo, с z-показателем комбинирования 2,8). В модели LS-180 синергии не наблюдали (z-показатель комбинирования 0,2). Кроме того, тройная комбинация продемонстрировала более сильную индукцию апоптоза по сравнению с парами лекарственных средств (оцениваемая путем измерения активации каспазы 3/7) в моделях Gp2D и LoVo (в GP2d максимум 67% апоптоз, в LoVo 83%). Наблюдаемые уровни апоптоза, вызванные тройной комбинацией в HCT-116 и LS-180, составляли 49% и 15%, соответственно.
Пример 7: Эффект in vitro на пролиферацию комбинирования ингибитора MDM2, и ингибитора MEK, и ингибитора BRAF
Аналогично, как описано в предшествующих примерах, тройную комбинацию (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она, траметиниба и дабрафениба тестировали в 1 модели с мутантным BRAF (RKO). Авторы настоящего изобретения обнаружили, что тройная комбинация является синергичной (z-показатель комбинирования 3,5), и продемонстрировала более мощную (но в целом слабую, максимум 11%) индукцию апоптоза по сравнению с парами лекарственных средств (оцениваемую посредством измерения активации каспазы 3/7).
Пример 8: Эффект in vitro на пролиферацию комбинирования ингибитора MDM2, и ингибитора BRAF, и ингибитора Bcl2, необязательно вместе с ингибитором PI3K или ингибитором cMET
Аналогично, как описано в предшествующих примерах, тройную комбинацию (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она, RAF265 и навитоклакса (ABT-263) тестировали в 4 моделях с мутантным KRAS (HCT-116, GP2d, LoVo и LS-180). Комбинация оказывало синергичное действие в 1/4 моделей (GP2d, z-показатель комбинирования 5) и продемонстрировала более мощную индукцию апоптоза по сравнению с парами лекарственных средств (оцениваемую посредством измерения активации каспазы 3/7) во всех 4 исследованных моделях (в GP2d и LoVo, максимум 100% апоптоз, в HCT-116 64%, и в LS-180 32%). Не наблюдали синергии в HCT-116, LS-180 и LoVo (z-показатели комбинирования 1,3, 1,2 и 0,5, соответственно).
В той же категории тройную комбинацию (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она, дабрафениба и навитоклакса (ABT-263) тестировали в 1 модели с мутантным BRAF (RKO). Авторы настоящего изобретения обнаружили, что тройная комбинация является синергичной (z-показатель комбинирования 3) и демонстрирует мощную индукцию апоптоза (максимум 100%), в то время как все пары продемонстрировали только слабую индукцию апоптоза или отсутствие индукции апоптоза (оцениваемые путем измерения активации каспазы 3/7).
Пример 9: Эффект in vitro на пролиферацию комбинирования ингибитора MDM2, и ингибитора MEK, и ингибитора CD4/6 или паклитаксела
Добавление ингибитора CD4/6 (в частности, 7-циклопентил-N,N-диметил-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)-7H-пирроло[2,3-d]пиримидин-6-карбоксамида) к комбинации (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она и траметиниба приводило к синергичному эффекту в 2/5 протестированных моделей (HCT-116, LoVo; z-показатели 5,1, 3,3, соответственно) и имело слабо синергичное действие в 2/5 моделей (LS-180, RKO; z-показатели 2, 2,8, соответственно). Не наблюдали синергии в GP2d (z-показатель 1,7). Не наблюдали индукции апоптоза.
Добавление паклитаксела (стандарт лечения) к комбинации приводило к синергичному эффекту в 1/5 протестированных моделей (GP2d, z-показатель 4,3), и слабо синергичному эффекту в 3/5 протестированных моделей (HCT-116, LoVo, LS-180; z-показатели 2,4, 2 и 2,4, соответственно). Не наблюдали синергии в RKO (z-показатель 1,1). Более того, комбинация мощно индуцировала апоптоз (GP2d: 100%, HCT-116: 59%, LoVo: 61%, LS-180: 20% и RKO 65%).
Пример 10: Эффект in vivo на пролиферацию комбинирования ингибитора MDM2 с ингибитором MEK и ингибитором BCL-2/-XL в модели рака ободочной и прямой кишки с TP53 дикого типа
Получение имеющих опухоль мышей и лечение
Все эксперименты на животных проводили в строгом соответствии со швейцарским законодательством в отношении защиты животных. Самок мышей Crl:NU(NCr)-Foxn1nu приобретали от Charles River Laboratories International Inc (Germany) и держали в условиях с контролем патогенов. Подкожные опухоли HCT-116 (мутантный KRAS, мутантный PIK3CA, p53 дикого типа) индуцировали путем концентрирования 3 миллионов клеток в 100 мкл PBS и инъецирования их в правый бок мышей nude. Мышей случайным образом распределяли на группы и лечение начинали, когда размер опухолей достигал 50-250 мм3. Каждая группа включала 8 мышей. Мониторинг размеров опухолей проводили три раза в неделю и объемы опухолей вычисляли по следующей формуле: (мм3)=длина×ширина2 × 0,5.
Ингибитор MDM2 (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-он (СОЕДИНЕНИЕ A), ингибитор MEK траметиниб (СОЕДИНЕНИЕ B) и ингибитор BCL-2/-XL ABT-263 (СОЕДИНЕНИЕ C) в порошковой форме хранили при +4°C. СОЕДИНЕНИЕ A растворяли в 0,5% гидроксипропилметилцеллюлозе, СОЕДИНЕНИЕ B растворяли в 1% карбоксиметилцеллюлозе, содержавшей 0,5% Tween-80% в дистиллированной воде (pH7,6-8,0), и СОЕДИНЕНИЕ C растворяли в преконцентрате микроэмульсии 5. Все лекарственные средства дозировали перорально с использованием 5-10 мл/кг. СОЕДИНЕНИЕ A вводили три раза в неделю (3qw) в дозе 100 мг/кг. СОЕДИНЕНИЕ B и СОЕДИНЕНИЕ C вводили каждые сутки (q24h) в дозе 0,3 и 100 мг/кг, соответственно. Схему дозирования комбинации и дозировки были такими же, как и для отдельных реагентов.
Исследовали шесть групп лечения (G1-G6):
- G1: носитель (DMSO)
- G2: СОЕДИНЕНИЕ C
- G3: СОЕДИНЕНИЕ A => после лечения в течение 9 суток добавляли СОЕДИНЕНИЕ C
- G4: СОЕДИНЕНИЕ B => после лечения в течение 9 суток добавляли СОЕДИНЕНИЕ C
- G5: СОЕДИНЕНИЕ A+СОЕДИНЕНИЕ B => после лечения в течение 9 суток добавляли СОЕДИНЕНИЕ C
- G6: СОЕДИНЕНИЕ A+СОЕДИНЕНИЕ B+СОЕДИНЕНИЕ C
Статистический анализ
Для каждой опухоли в каждый момент времени размер нормализовывали к размеру до начала лечения с получением "% изменения объема опухоли" (фиг.17-18, ось y). Для фиг.17 в каждый момент времени вычисляли средний размер опухолей на группу и ошибку размера с использованием стандартной ошибки среднего (SEM). Для фиг.18 значения p вычисляли с использованием одностороннего t-критерия.
Результаты
В модели с ксенотрансплантатом клеток HCT-116 комбинированное лечение СОЕДИНЕНИЕМ A и траметинибом (G5) было значительно лучшим (стабильное заболевание) по сравнению с обработкой каждым средством отдельно (G3-G4, демонстрирующие прогрессирующее заболевание), и тройная комбинация СОЕДИНЕНИЯ A, траметиниба и ABT-263 (G6) привела к выраженной регрессии опухоли и имела значимо лучший ответ по сравнению c G5 (фиг.17 и фиг.18A). На фиг.17 обобщенно продемонстрированы кривые выживаемости и на фиг.18 представлены каскадные диаграммы на 9 сутки и 19 сутки после начала лечения. Последовательное добавление ABT-263 через 9 суток к единственному средству СОЕДИНЕНИЮ A не имела дополнительной пользы, хотя она останавливала прогрессирование опухоли при добавлении к единственному средству траметинибу. ABT-263 приводило к выраженной регрессии опухолей при добавлении к комбинации СОЕДИНЕНИЯ A и траметиниба, и на 19 сутки ответы сопутствующих и последовательных лечений были неотличимы (фиг.18B).
ССЫЛКИ
Arrington, A. K., E. L. Heinrich, W. Lee, M. Duldulao, S. Patel, J. Sanchez, J. Garcia-Aguilar and J. Kim (2012). "Prognostic and predictive roles of KRAS mutation in colorectal cancer." Int J Mol Sci 13(10): 12153-12168.
Berenbaum, M. C. (1989). "What is synergy?" Pharmacol Rev 41(2): 93-141.
Bozic, I., J. G. Reiter, B. Allen, T. Antal, K. Chatterjee, P. Shah, Y. S. Moon, A. Yaqubie, N. Kelly, D. T. Le, E. J. Lipson, P. B. Chapman, L. A. Diaz, Jr., B. Vogelstein and M. A. Nowak (2013). "Evolutionary dynamics of cancer in response to targeted combination therapy." Elife 2: e00747.
Brana, I. and L. L. Siu (2012). "Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment." BMC Med 10: 161.
Cancer Genome Atlas, N. (2012). "Comprehensive molecular characterization of human colon and rectal cancer." Nature 487(7407): 330-337.
Chandarlapaty, S. (2012). "Negative feedback and adaptive resistance to the targeted therapy of cancer." Cancer Discov 2(4): 311-319.
Chapman, P. B., D. B. Solit and N. Rosen (2014). "Combination of RAF and MEK inhibition for the treatment of BRAF-mutated melanoma: feedback is not encouraged." Cancer Cell 26(5): 603-604.
Chatterjee, M. S., J. E. Purvis, L. F. Brass and S. L. Diamond (2010). "Pairwise agonist scanning predicts cellular signaling responses to combinatorial stimuli." Nat Biotechnol 28(7): 727-732.
Chou, T. C. and P. Talalay (1981). "Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors." Eur J Biochem 115(1): 207-216.
DeVita, V. T., Jr. (1975). "Single agent versus combination chemotherapy." CA Cancer J Clin 25(3): 152-158.
Doebele, R. C., A. B. Pilling, D. L. Aisner, T. G. Kutateladze, A. T. Le, A. J. Weickhardt, K. L. Kondo, D. J. Linderman, L. E. Heasley, W. A. Franklin, M. Varella-Garcia and D. R. Camidge (2012). "Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer." Clin Cancer Res 18(5): 1472-1482.
Druker, B. J. (2008). "Translation of the Philadelphia chromosome into therapy for CML." Blood 112(13): 4808-4817.
Duncan, J. S., M. C. Whittle, K. Nakamura, A. N. Abell, A. A. Midland, J. S. Zawistowski, N. L. Johnson, D. A. Granger, N. V. Jordan, D. B. Darr, J. Usary, P. F. Kuan, D. M. Smalley, B. Major, X. He, K. A. Hoadley, B. Zhou, N. E. Sharpless, C. M. Perou, W. Y. Kim, S. M. Gomez, X. Chen, J. Jin, S. V. Frye, H. S. Earp, L. M. Graves and G. L. Johnson (2012). "Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer." Cell 149(2): 307-321.
Faber, A. C., E. M. Coffee, C. Costa, A. Dastur, H. Ebi, A. N. Hata, A. T. Yeo, E. J. Edelman, Y. Song, A. T. Tam, J. L. Boisvert, R. J. Milano, J. Roper, D. P. Kodack, R. K. Jain, R. B. Corcoran, M. N. Rivera, S. Ramaswamy, K. E. Hung, C. H. Benes and J. A. Engelman (2014). "mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1." Cancer Discov 4(1): 42-52.
Fearon, E. R. (2011). "Molecular genetics of colorectal cancer." Annu Rev Pathol 6: 479-507.
Horn, T., T. Sandmann, B. Fischer, E. Axelsson, W. Huber and M. Boutros (2011). "Mapping of signaling networks through synthetic genetic interaction analysis by RNAi." Nat Methods 8(4): 341-346.
Ji, Z., C. N. Njauw, M. Taylor, V. Neel, K. T. Flaherty and H. Tsao (2012). "p53 rescue through HDM2 antagonism suppresses melanoma growth and potentiates MEK inhibition." J Invest Dermatol 132(2): 356-364.
Katayama, R., A. T. Shaw, T. M. Khan, M. Mino-Kenudson, B. J. Solomon, B. Halmos, N. A. Jessop, J. C. Wain, A. T. Yeo, C. Benes, L. Drew, J. C. Saeh, K. Crosby, L. V. Sequist, A. J. Iafrate and J. A. Engelman (2012). "Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers." Sci Transl Med 4(120): 120ra117.
Lehar, J., A. Krueger, G. Zimmermann and A. Borisy (2008). "High-order combination effects and biological robustness." Mol Syst Biol 4: 215.
Lito, P., N. Rosen and D. B. Solit (2013). "Tumor adaptation and resistance to RAF inhibitors." Nat Med 19(11): 1401-1409.
Pau, G., F. Fuchs, O. Sklyar, M. Boutros and W. Huber (2010). "EBImage--an R package for image processing with applications to cellular phenotypes." Bioinformatics 26(7): 979-981.
Porter, K., A. Babiker, K. Bhaskaran, J. Darbyshire, P. Pezzotti, K. Porter, A. S. Walker and C. Collaboration (2003). "Determinants of survival following HIV-1 seroconversion after the introduction of HAART." Lancet 362(9392): 1267-1274.
Robert, C., B. Karaszewska, J. Schachter, P. Rutkowski, A. Mackiewicz, D. Stroiakovski, M. Lichinitser, R. Dummer, F. Grange, L. Mortier, V. Chiarion-Sileni, K. Drucis, I. Krajsova, A. Hauschild, P. Lorigan, P. Wolter, G. V. Long, K. Flaherty, P. Nathan, A. Ribas, A. M. Martin, P. Sun, W. Crist, J. Legos, S. D. Rubin, S. M. Little and D. Schadendorf (2015). "Improved overall survival in melanoma with combined dabrafenib and trametinib." N Engl J Med 372(1): 30-39.
Safaee Ardekani, G., S. M. Jafarnejad, L. Tan, A. Saeedi and G. Li (2012). "The prognostic value of BRAF mutation in colorectal cancer and melanoma: a systematic review and meta-analysis." PLoS One 7(10): e47054.
Shoemaker, R. H. (2006). "The NCI60 human tumour cell line anticancer drug screen." Nat Rev Cancer 6(10): 813-823.
Solit, D. B. and N. Rosen (2014). "Towards a unified model of RAF inhibitor resistance." Cancer Discov 4(1): 27-30.
Sullivan, R. J. and K. T. Flaherty (2013). "Resistance to BRAF-targeted therapy in melanoma." Eur J Cancer 49(6): 1297-1304.
Turner, N. C., J. Ro, F. Andre, S. Loi, S. Verma, H. Iwata, N. Harbeck, S. Loibl, C. Huang Bartlett, K. Zhang, C. Giorgetti, S. Randolph, M. Koehler and M. Cristofanilli (2015). "Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer." N Engl J Med.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНЕРГИЧЕСКИЕ КОМПОЗИЦИИ ИНГИБИТОРОВ PI3K И МЕК | 2012 |
|
RU2607944C2 |
| ПРЕРЫВИСТОЕ ВВЕДЕНИЕ ИНГИБИТОРА MDM2 | 2015 |
|
RU2695228C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ | 2018 |
|
RU2815400C2 |
| КОМПОЗИЦИИ НА ОСНОВЕ ИНГИБИТОРА SHP2 И СПОСОБЫ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2805355C2 |
| СОЕДИНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2014 |
|
RU2708247C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ НА ОСНОВЕ ИНГИБИТОРОВ PRMT5 | 2020 |
|
RU2830439C1 |
| СИНЕРГИСТИЧЕСКИЕ КОМБИНАЦИИ АУРИСТАНА | 2015 |
|
RU2717570C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ | 2014 |
|
RU2684106C2 |
| ТЕРАПЕВТИЧЕСКИЕ КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР RAF И ИНГИБИТОР ERK | 2017 |
|
RU2774612C2 |
| Комбинация ингибитора EGFR и ингибитора MEK для применения в лечении рака, вызванного мутировавшим NRAS | 2015 |
|
RU2683276C2 |
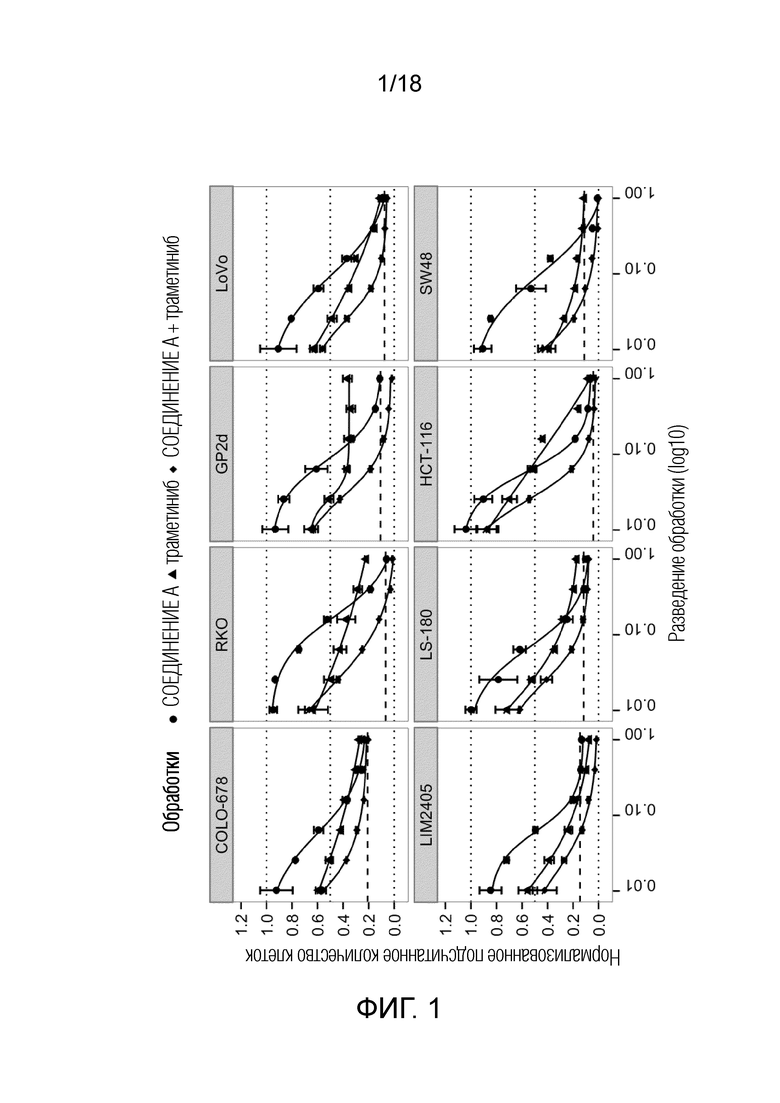
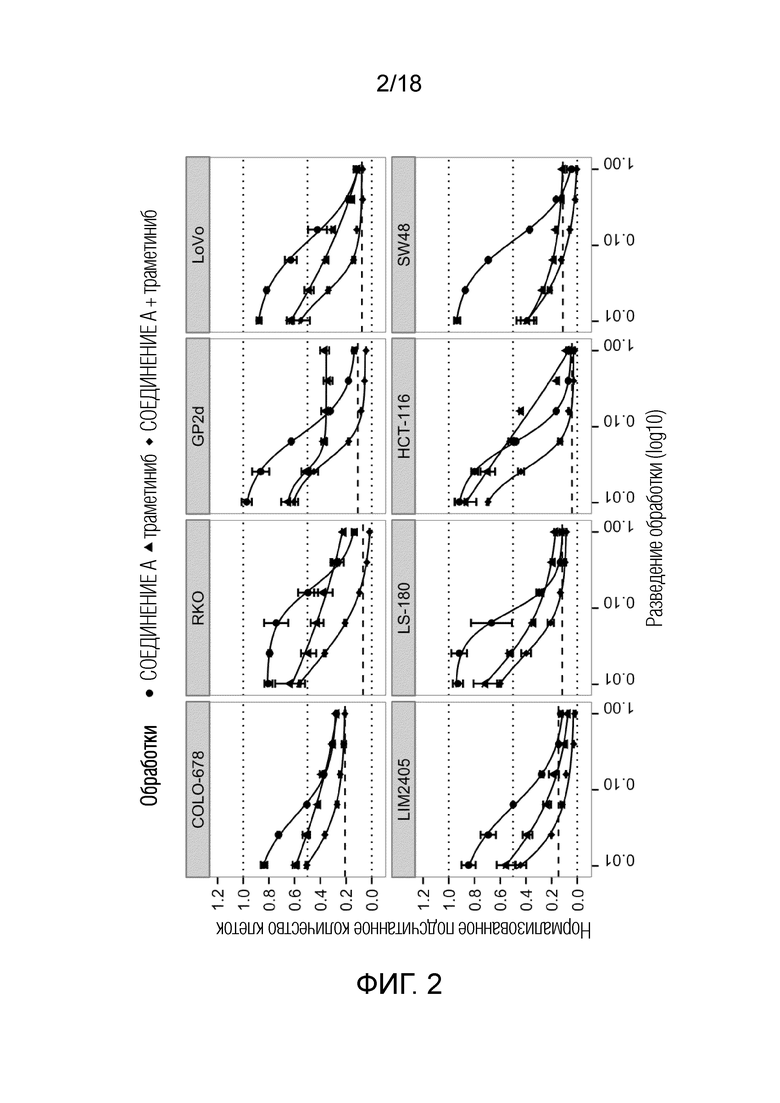
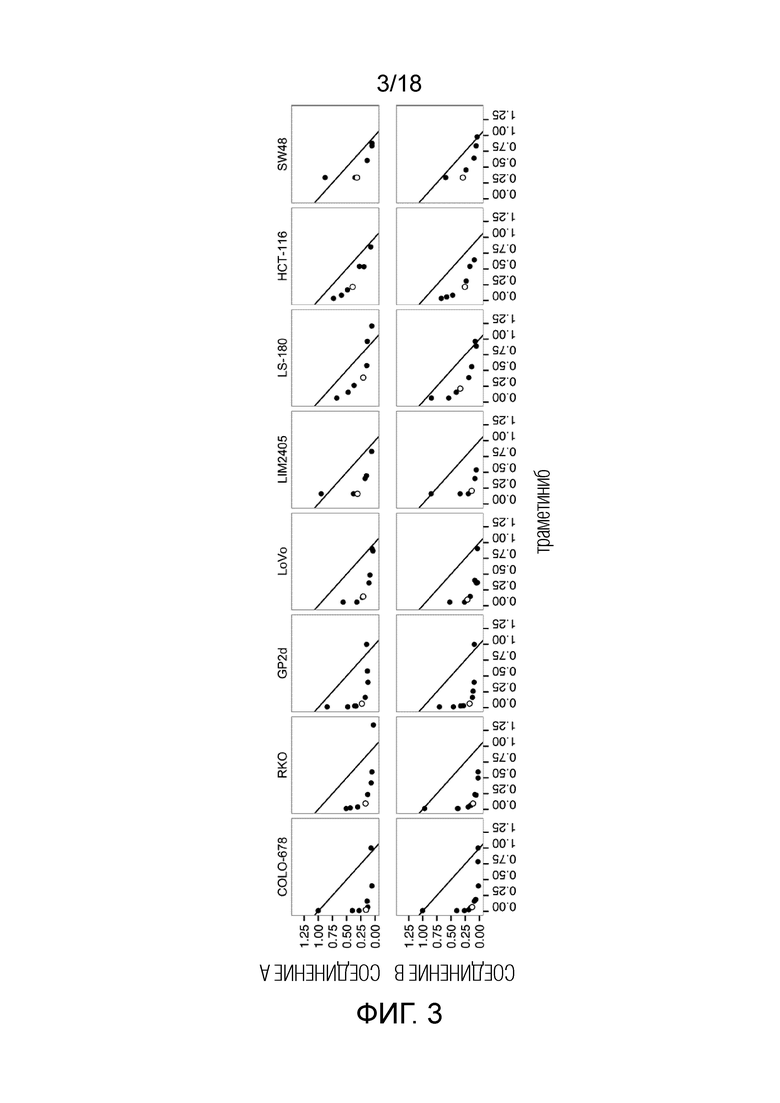
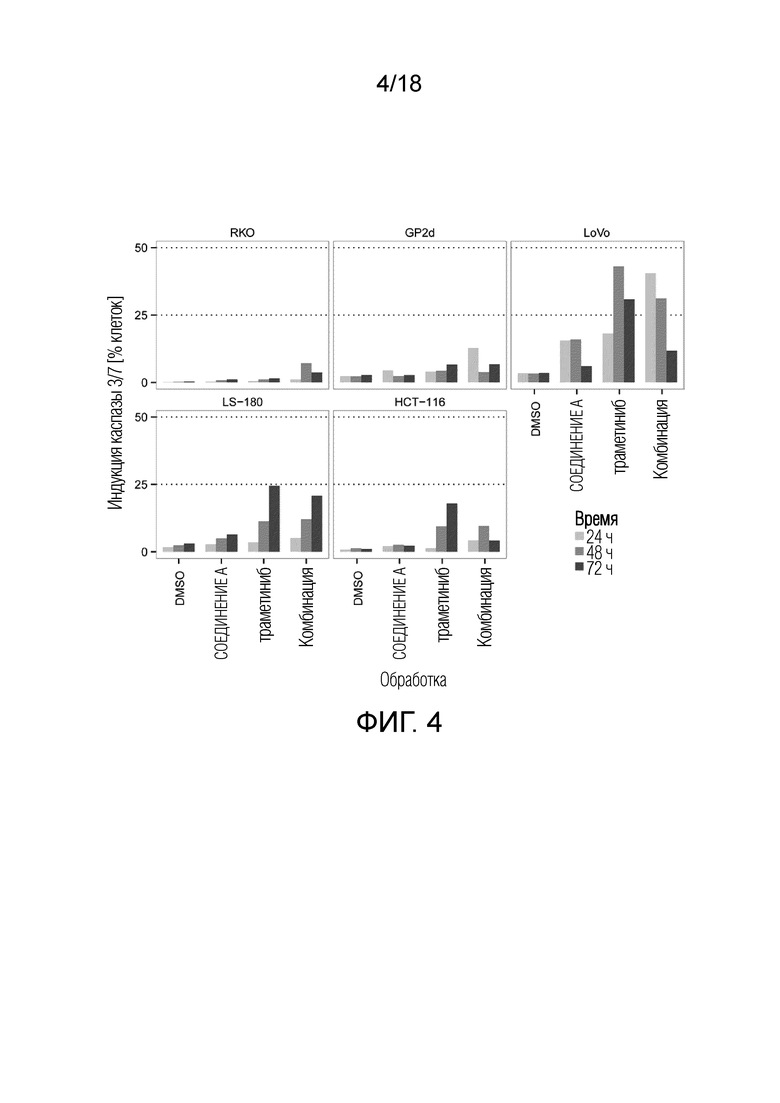
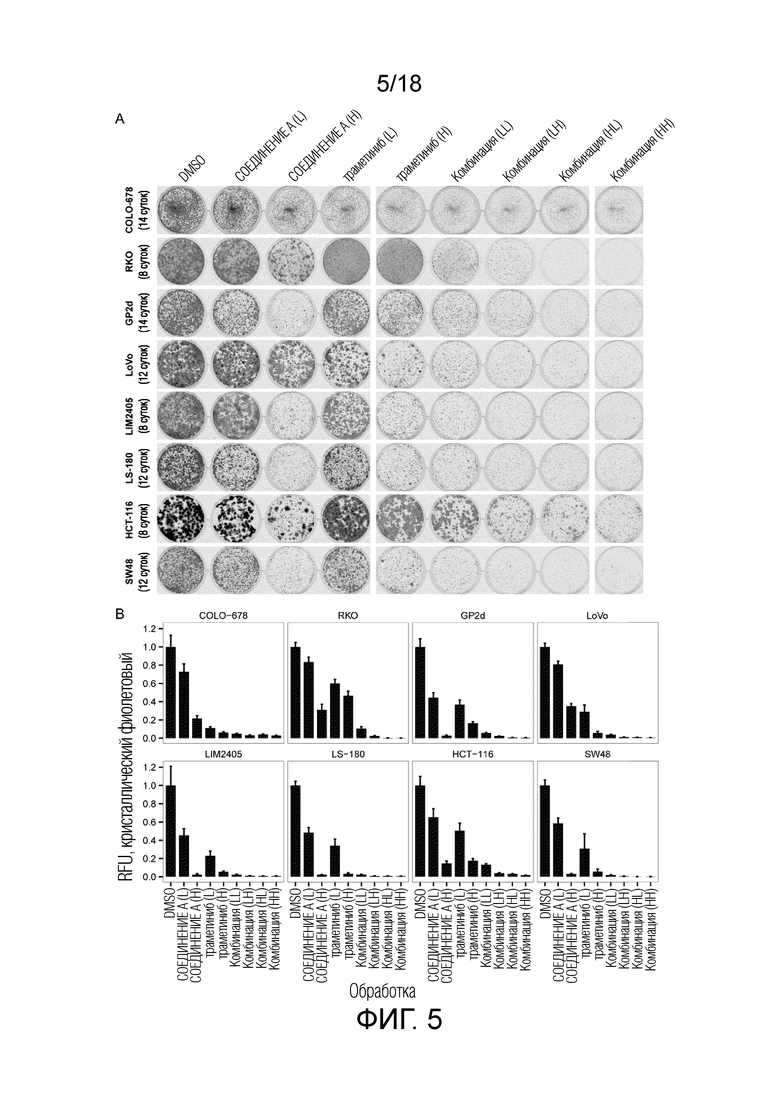
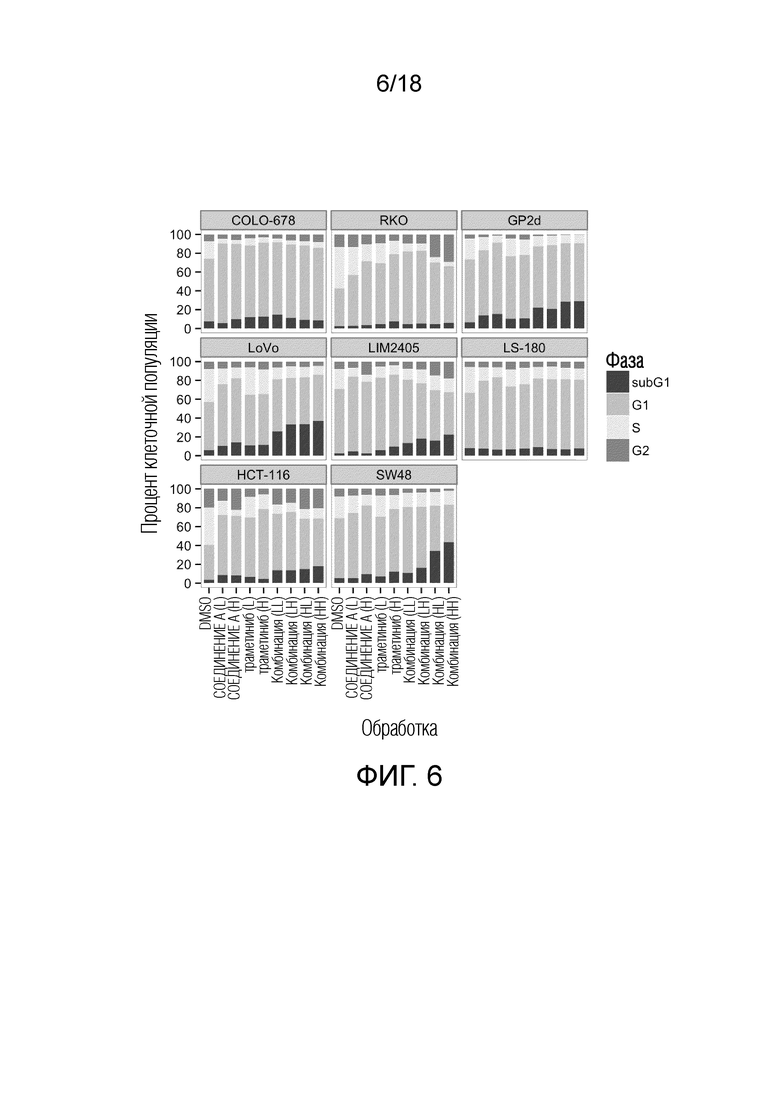

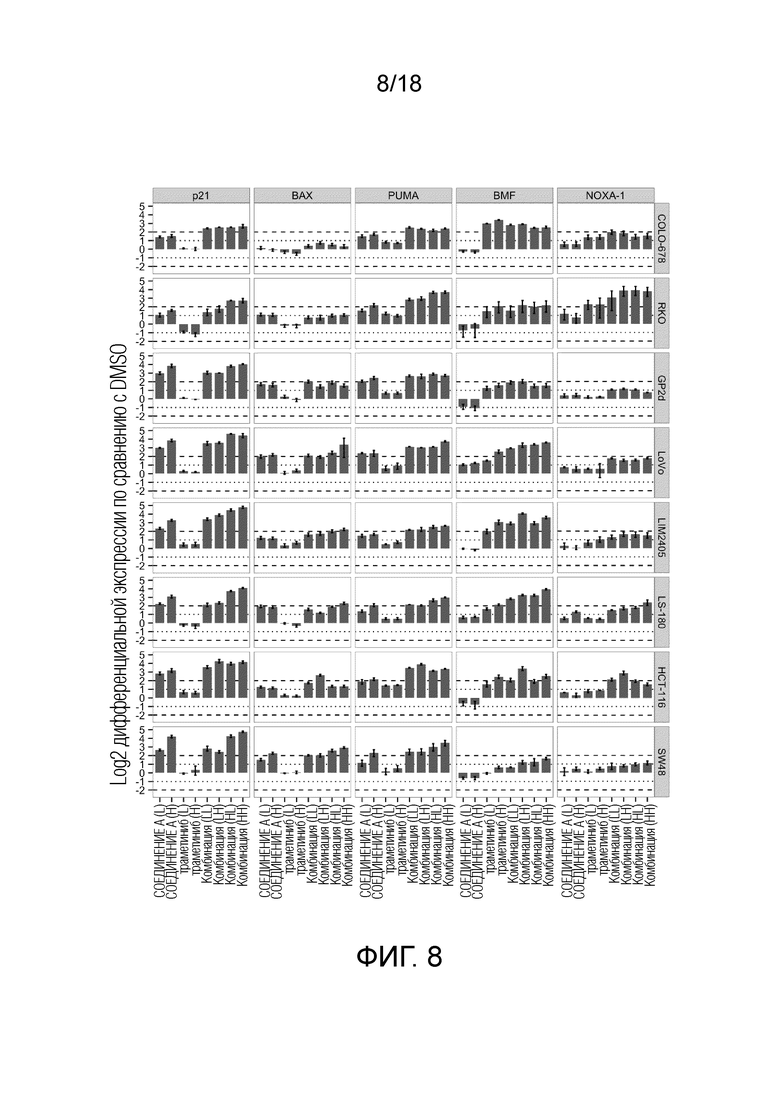
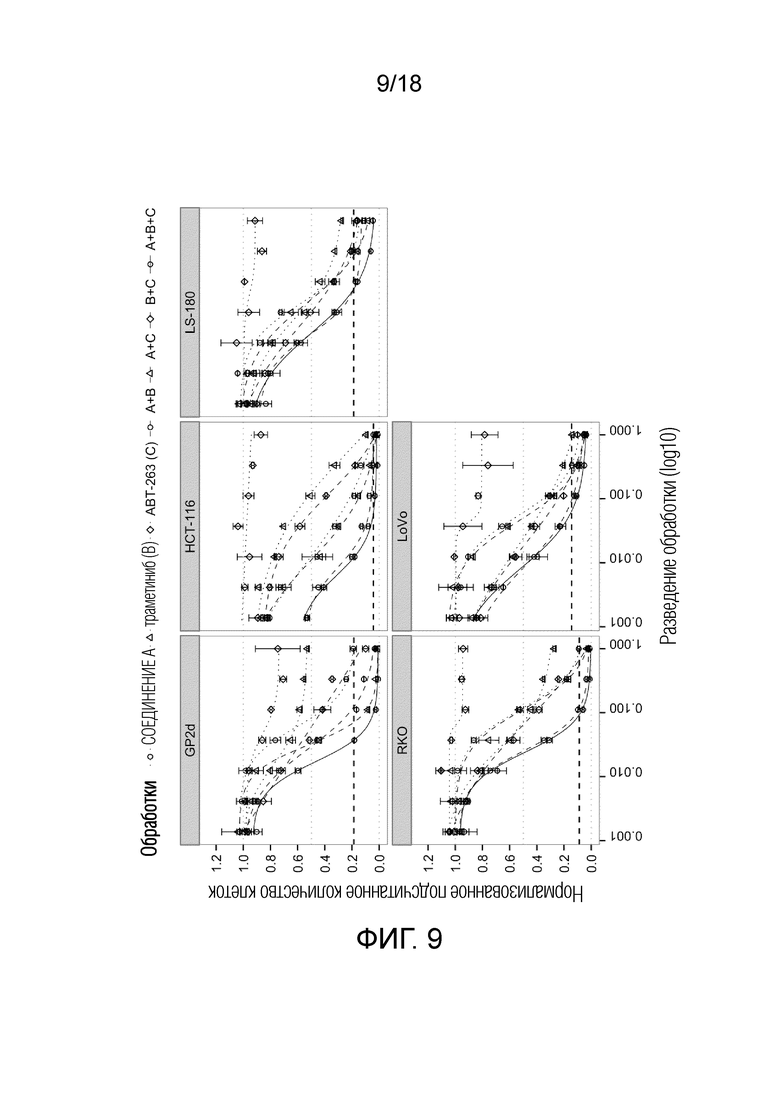
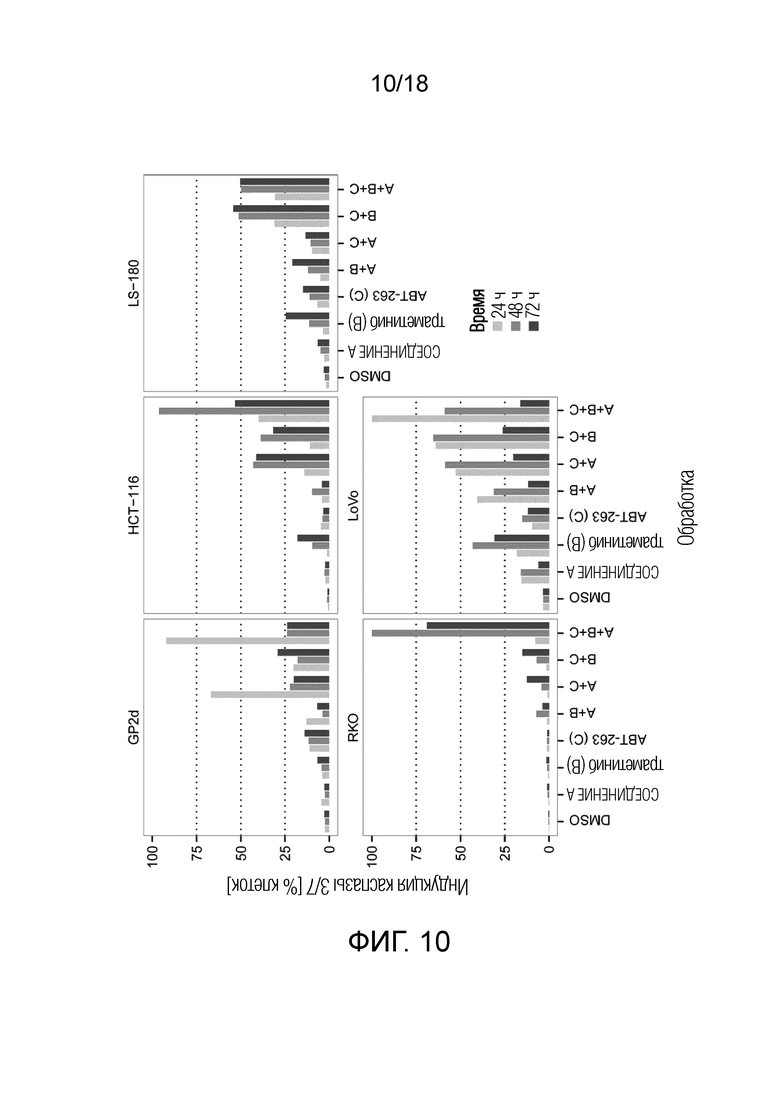
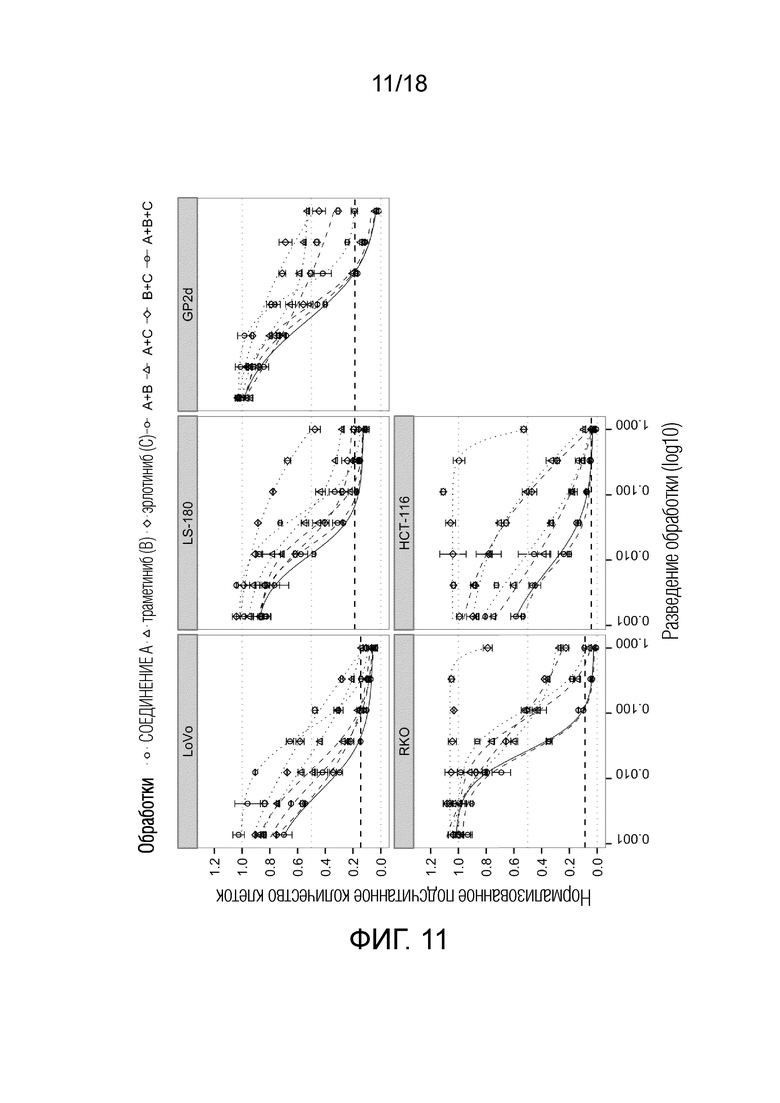
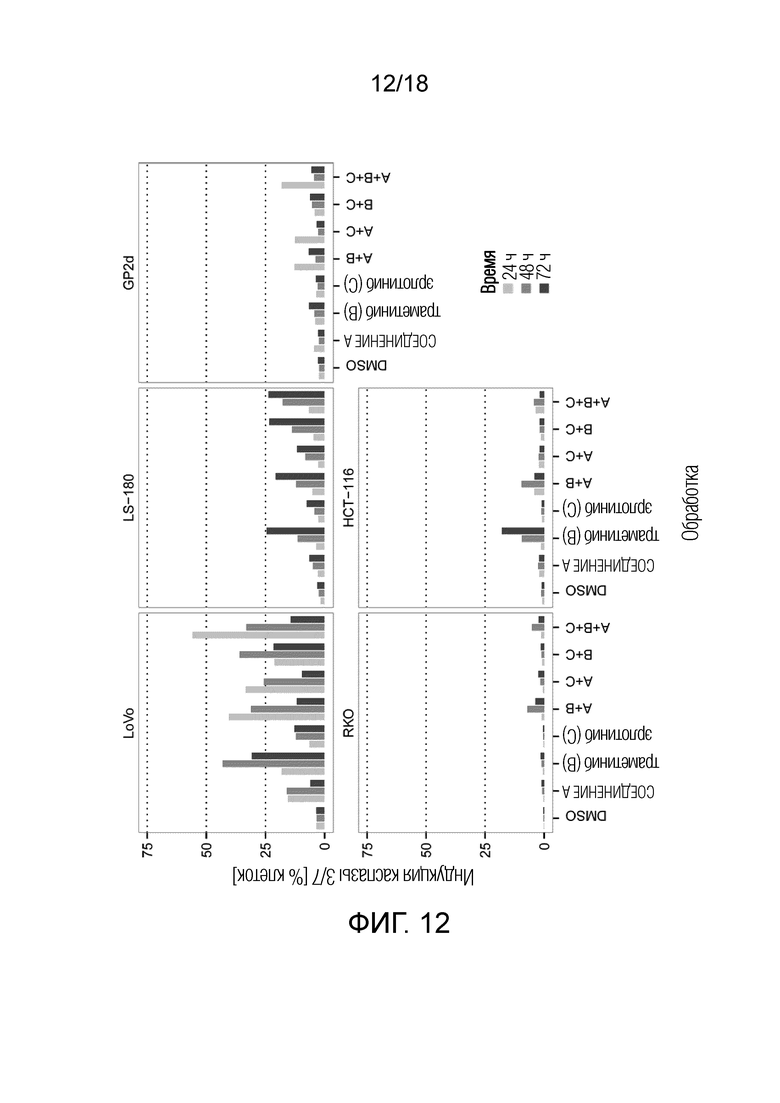
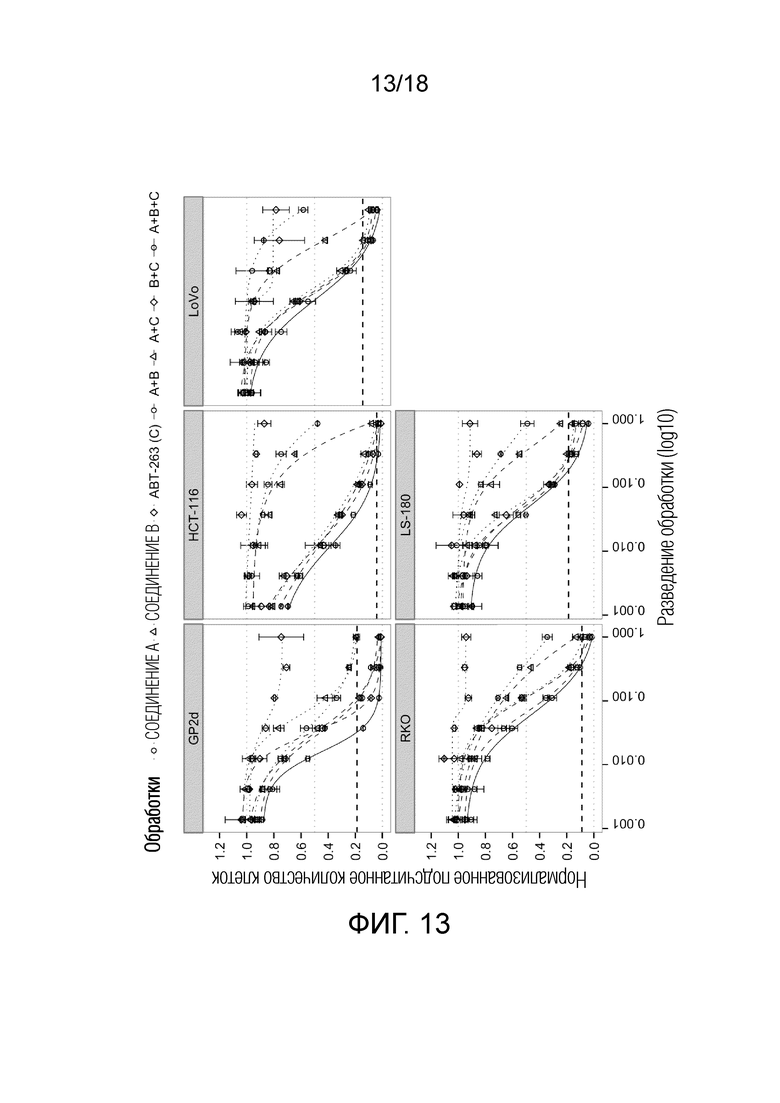
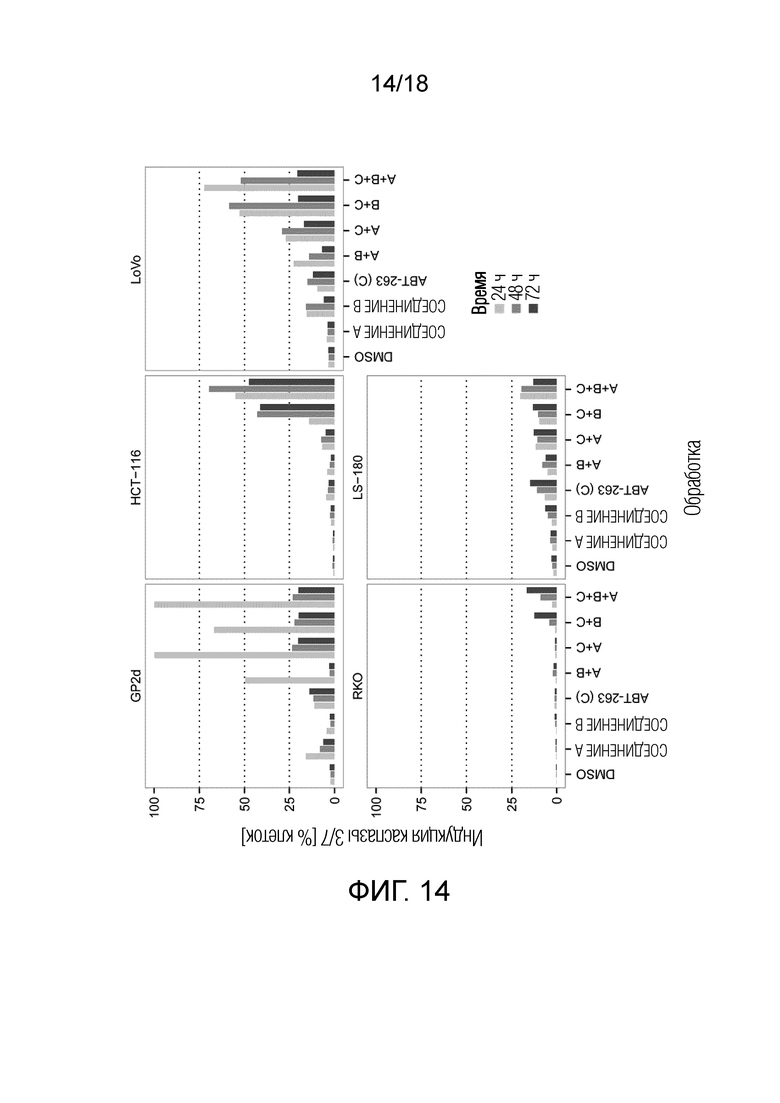
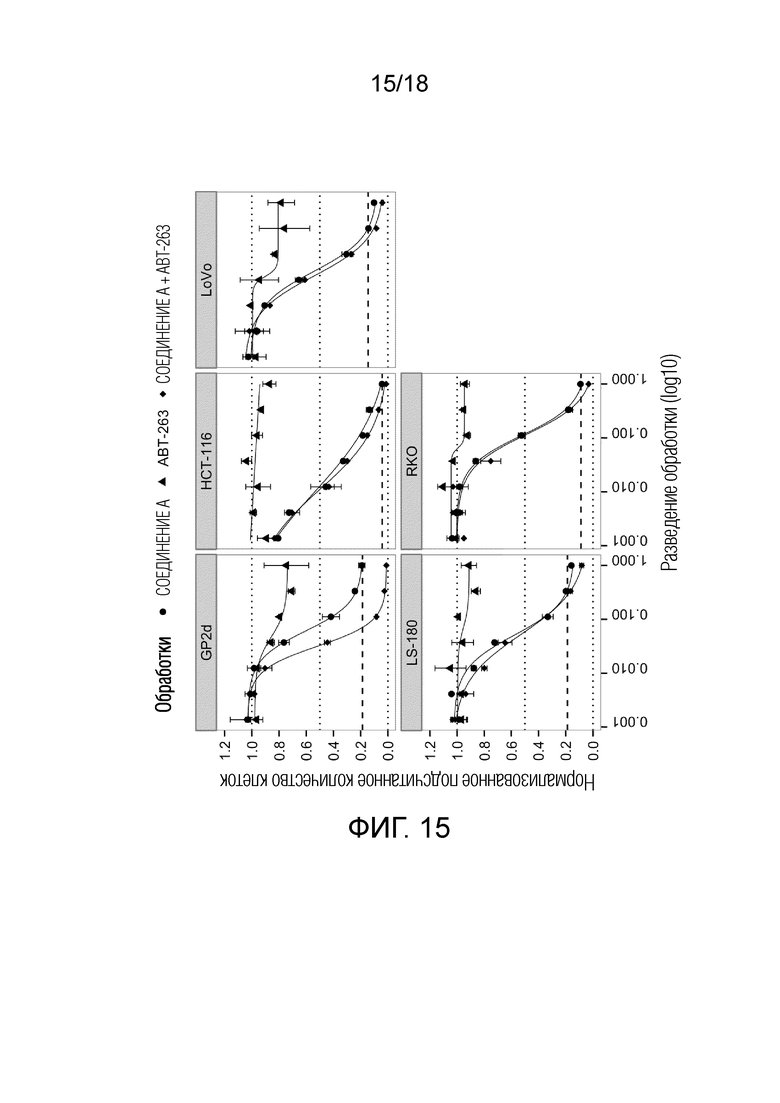
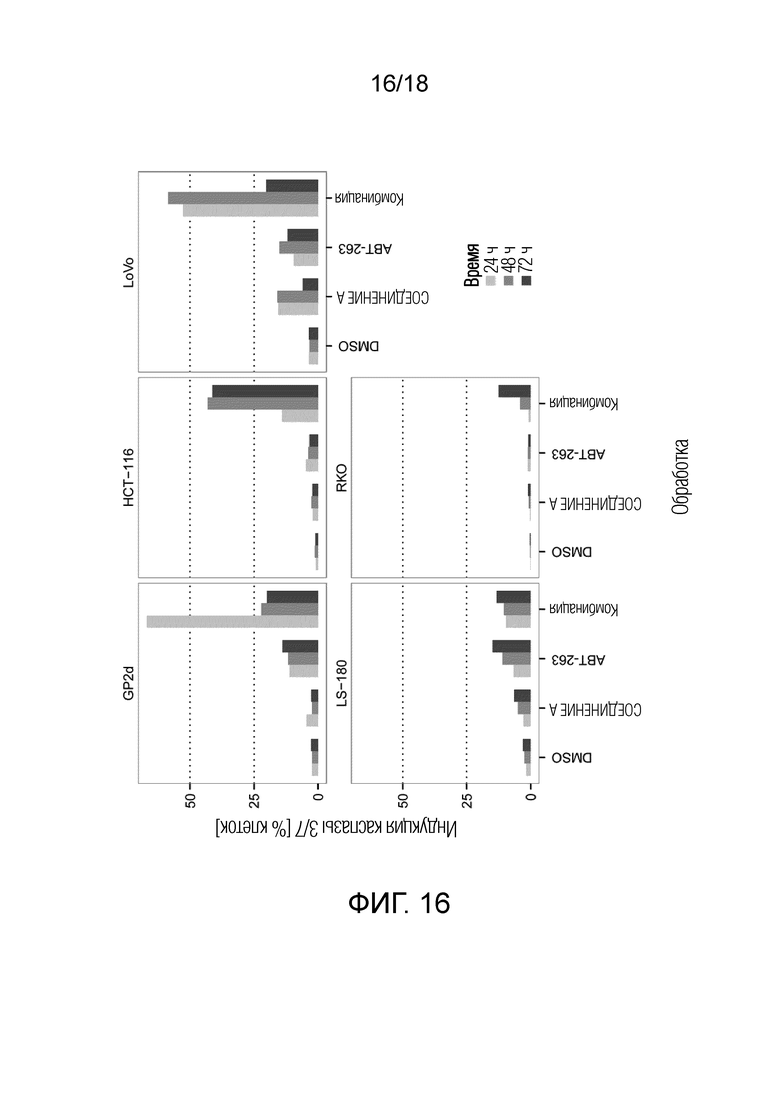
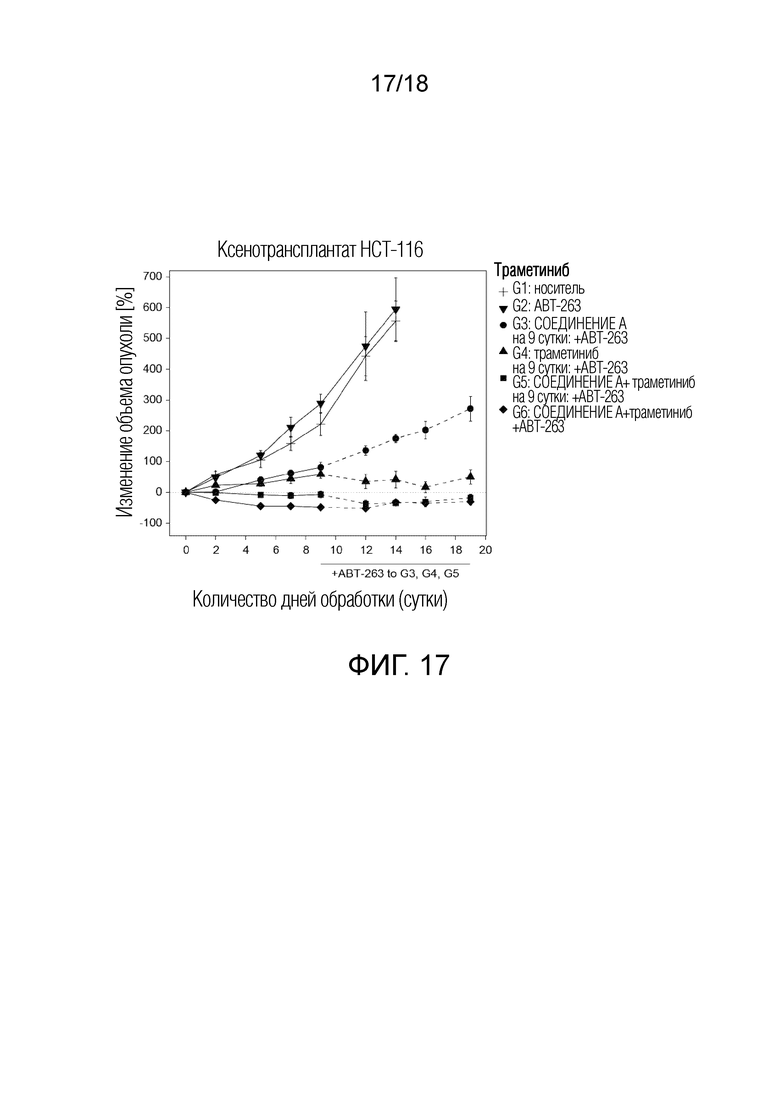
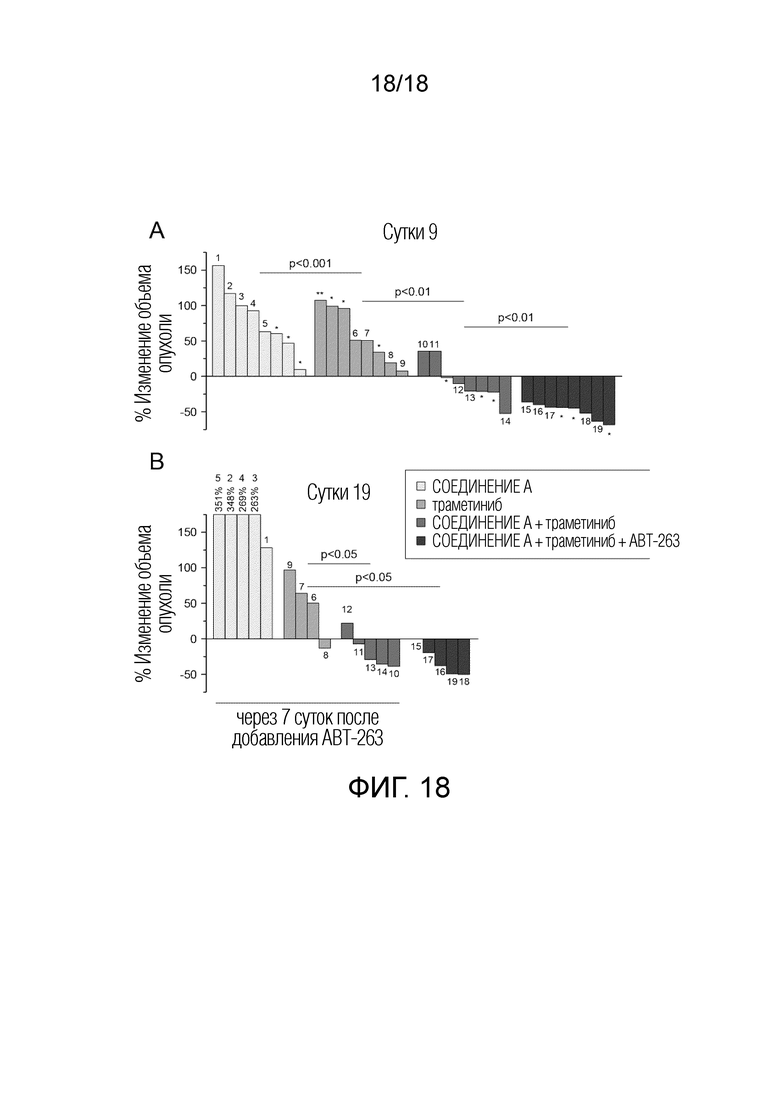
Группа изобретений относится к области медицины и фармацевтики. 1 объект представляет собой фармацевтическую комбинацию для лечения рака, содержащую ингибитор MDM2 (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-он или его фармацевтически приемлемую соль и ингибитор Bcl2 ABT-199 или его фармацевтически приемлемую соль. 2 объект - фармацевтическую композицию для лечения рака. 3 и 5 объекты - применение фармацевтической комбинации или фармацевтической композиции для лечения и способ лечения острого миелогенного лейкоза. 4 объект - применение фармацевтической комбинации для получения лекарственного средства для лечения острого миелогенного лейкоза. Технический результат заключается в повышенной эффективности противоопухолевой терапии комбинацией (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она и ABT-199 по сравнению с их применением в отдельности. 5 н. и 3 з.п. ф-лы, 11 табл., 10 пр., 18 ил.
1. Фармацевтическая комбинация для лечения рака, содержащая
(a) ингибитор MDM2 (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-(пропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-он или его фармацевтически приемлемую соль; и
(b) ингибитор Bcl2 ABT-199 или его фармацевтически приемлемую соль.
2. Фармацевтическая комбинация по п. 1 для одновременного или последовательного применения.
3. Фармацевтическая комбинация по п. 1 или 2 в форме фиксированной комбинации.
4. Фармацевтическая комбинация по п. 1 или 2 в форме нефиксированной комбинации.
5. Фармацевтическая композиция для лечения рака, содержащая фармацевтическую комбинацию по любому из пп. 1-4 и по меньшей мере один фармацевтически приемлемый носитель.
6. Применение фармацевтической комбинации по любому из пп. 1-4 или фармацевтической композиции по п. 5 для лечения острого миелогенного лейкоза.
7. Применение фармацевтической комбинации по любому из пп. 1-4 для получения лекарственного средства для лечения острого миелогенного лейкоза.
8. Способ лечения острого миелогенного лейкоза у индивидуума, нуждающегося в этом, включающий введение индивидууму терапевтически эффективного количества фармацевтической комбинации по любому из пп. 1-4 или фармацевтической композиции по п. 5.
| WO 2015084804 A1, 11.06.2015 | |||
| WO 2015070224 A2, 14.05.2015 | |||
| WO 2015095834 A2, 25.06.2015 | |||
| WO 2012175520 A1, 27.12.2012 | |||
| WO 2012178038 A1, 27.12.2012 | |||
| US 2012115896 A1, 10.05.2012 | |||
| МАШКОВСКИЙ М.Д | |||
| Лекарственные средства, Москва, Новая волна, 2001, в 2-х томах, том 1, стр.11 | |||
| БЕЛИКОВ В.Г., Фармацевтическая химия, М., Высшая школа, 1993, с | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |

