Настоящее изобретение относится к гетерогенным катализаторам для прямого карбонилирования нитроароматических соединений в ароматические изоцианаты и к способу прямого карбонилирования нитроароматических соединений в ароматические изоцианаты.
Прямое карбонилирование нитроароматических соединений до соответствующих ароматических изоцианатов с гомогенными катализаторами описано в литературе. PdCl2(пиридин)2 и Fe(циклопентадиенил)2 в качестве сокатализатора достигали селективности по толуилендиизоцианату (TDI) от 9 до 67% при конверсии динитротолуола (DNT) от 82 до 100%, как описано в (DE19635723A1. Основными проблемами, которые предотвращают коммерческое использование, являются низкие число оборотов, сложное отделение катализатора, резкие условия реакции (Т = 250°С, р = 200-300 бар и.д.), образование побочных продуктов и полимеризация TDI.
В данной области техники известен катализатор, используемый для карбонилирования 2,4-динитротолуола, содержащий смесь комплекса палладия с изохинолином и Fe2Mo7O24, как описано в DE 2165355. 2,4-Толуилендиизоцианат получают с максимальным выходом 70% при 100% конверсия исходного соединения 2,4-динитротолуола. Когда вместо изохинолина используют пиридин, выход составляет 21-76% при конверсии исходного соединения 83-100%, как описано в патенте FR 2,120,110. Также известны катализаторы карбонилирования ароматических нитросоединений, содержащие Pd(пиридин)2Cl2 и MoO3 или Cr2O3 / Al2O3, как раскрыто в US3,823,174, и US3,828,089, соответственно. Другим гомогенным-гетерогенным катализатором для синтеза ароматических моноизоцианатов, в частности фенилизоцианата, является PdCl2 / V2O5, как раскрыто в US 3,523,964. В противоположность настоящему изобретению системы, описанные в вышеупомянутых документах, не являются действительно гетерогенными и соответствуют гибридной системе, содержащей гомогенные и гетерогенные компоненты. Недостатком является то, что хлорид палладия присутствует в жидкой фазе, что требует сложной системы для его отделения и регенерации.
В литературе сообщается только о нескольких гетерогенных катализаторах карбонилирования DNT до TDI. В US 4,207,212 сообщается, что PdO/MoO3/ZnO является высокоактивным и селективным катализатором для карбонилирования DNT. Все примеры этого патента были выполнены в присутствии пиридина в качестве добавки. Этот факт приводит к предположению, что образование пиридиновых комплексов необходимо для достижения карбонилирования нитроаренов с использованием этих катализаторов.
Помимо прямого превращения нитроароматических соединений в изоцианаты также известно и непрямое превращение с нитрозоароматическим соединением в качестве отделяемого промежуточного соединения. Превращение нитроаренов в нитрозоарены, а также превращение нитрозоаренов в ароматические изоцианаты в присутствии монооксида углерода описывается в литературе как две отдельные реакции. Это также имеет место, если исходный нитроарен имеет более одной нитрогруппы. Поскольку настоящее изобретение обеспечивает прямой синтез изоцианатов, а также непрямой синтез изоцианатов с нитрозоароматическими соединениями в качестве подходящих промежуточных соединений. Соответствующая литература цитируется ниже.
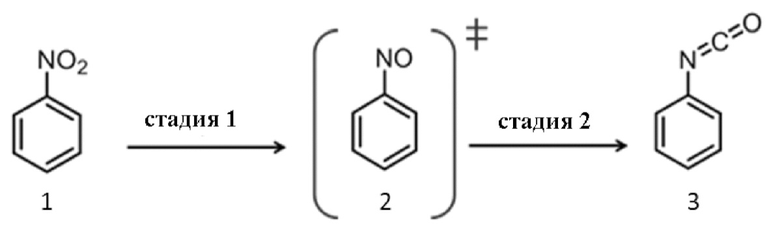
Схема 1
Получение нитрозоаренов согласно стадии 1 (схема 1), которое также можно рассматривать как селективное восстановление нитробензола до нитрозобензола, возможно с использованием Mn-содержащих катализаторов. DE 1810828 раскрывает каталитические системы общей формулы MxMnyOz, где M представляет собой Co, Fe, Pb или Ag, в качестве катализатора селективного восстановления нитробензола до нитрозобензола. Оксидное соединение, содержащее Mn и Pb при соотношении 70/30, дает выход 4,53% нитрозобензола в час реакции.
Превращение нитрозобензола в изоцианат согласно стадии 2 (схема 1) с той же Mn-содержащей системой, не сообщается. Карбонилирование нитрозоаренов до ароматических изоцианатов согласно реакции 2 (схема 1) можно проводить с гетерогенным катализатором, содержащим один или более из Pd, Rh и Ir, нанесенных на Al2O3, как сообщается в US 3,979,427.
GB 1 315 813 A описывает гетерогенно катализированное карбонилирование нитрозо- и нитроароматических соединений до изоцианатов в присутствии физических смесей MxMnyOz, где M представляет собой Fe, Ag или Pb, с металлами платиновой группы, выбранными из Pd, Ru и Rh, нанесенных на носители. такие как углерод или пемза. Нитробензол карбонилируется до фенилизоцианата в присутствии физической смеси PbxMnyOz и 5% Rh на углероде. Указанный выход изоцианата составляет 4,5% через 2 часа при 190°C.
Задачей настоящего изобретения является обеспечение гетерогенных катализаторов, обладающих высокой активностью и селективностью для гетерогенно катализируемого процесса, позволяющего синтезировать изоцианаты посредством прямого карбонилирования. Прямое карбонилирование в контексте настоящего изобретения следует понимать как проведение стадий реакции 1 и 2 однореакторным образом без выделения промежуточных соединений. Однако промежуточные продукты, такие как нитрозосоединения или частично карбонилированные нитроароматические соединения, могут быть получены в результате неполной реакции.
Задачей настоящего изобретения является обеспечение способа карбонилирования нитроароматических соединений до соответствующего изоцианата, показывающего значительное улучшение активности и селективности.
Композиция мультиметаллического материала
Задача настоящего изобретения решается с помощью катализатора прямого карбоксилирования нитроароматического соединения до соответствующего ароматического изоцианата и способа получения ароматического изоцианата путем прямого карбоксилирования нитроароматического соединения в присутствии катализатора.
Способ согласно настоящему изобретению осуществляют в виде гетерогенно катализируемого процесса
В таком гетерогенном катализируемом процессе катализатор и реагент(ы)/продукт(ы) находятся в разных фазах, которые находятся в контакте друг с другом. Реагент(ы)/продукт(ы) находятся в жидкой фазе и газовой фазе, тогда как катализатор будет в твердой фазе. Реакция будет происходить на границе раздела фаз между жидкой фазой, газовой фазой и твердой фазой.
Способ согласно настоящему изобретению проводят в присутствии катализатора. Катализатор содержит мультиметаллический материал, содержащий одну или более бинарных интерметаллических фаз общей формулы AxBy, где
A представляет собой один или более металлический элемент, выбранный из Ni, Ru, Rh, Pd, Ir, Pt и Ag,
B представляет собой один или более металлический элемент, выбранный из Sn, Sb, Pb, Zn, Ga, In, и As,
x в AxBy находится в интервале от 0,1 - 10, предпочтительно от 0,2 до 5, более предпочтительно от 0,5 до 2,
y в AxBy находится в интервале от 0,1 - 10, предпочтительно от 0,2 до 5, более предпочтительно от 0,5 до 2.
Более предпочтительно мультиметаллические материалы содержат одну или более из бинарных интерметаллических фаз общей формулы AxBy, где
A представляет собой один или более металлический элемент, выбранный из Ni, Rh, Pd, Ir и Pt,
B представляет собой один или более металлический элемент, выбранный из Sn, Sb, Pb, Ga и In,
x в AxBy находится в интервале от 0,1 - 10, предпочтительно от 0,2 до 5, более предпочтительно от 0,5 до 2,
y в AxBy находится в интервале от 0,1 - 10, предпочтительно от 0,2 до 5, более предпочтительно от 0,5 до 2.
Даже более предпочтительно мультиметаллические материалы содержат одну или более бинарных интерметаллических фаз общей формулы AxBy, где
A представляет собой Rh,
B представляет собой один или более элементов из Pb, Sn или Sb,
x в AxBy находится в интервале от 0,1 - 10, предпочтительно от 0,2 до 5, более предпочтительно от 0,5 до 2,
y в AxBy находится в интервале от 0,1 - 10, предпочтительно от 0,2 до 5, более предпочтительно от 0,5 до 2.
Задача настоящего изобретения также решается путем обеспечения непрерывного гетерогенного процесса с использованием жидкого и газообразного поступающего материала вместе с мультиметаллическим материалом.
В общем, мультиметаллический материал может содержать или состоять из одной или более бинарных интерметаллических фаз в виде общей формулы AxBy, как уточнено ранее. Кроме того, мультиметаллический материал определяется как материал, содержащий по меньшей мере два различных металла в макроскопически гомогенной фазе. В общем, мультиметаллические материалы содержит по меньшей мере 85 мас.%, предпочтительно по меньшей мере 90 мас.% и более предпочтительно 95 мас.% одной или более интерметаллических фаз общей формулы AxBy. Мультиметаллический материал может содержать один или более других компонентов C, где компонент C может состоять из или содержать A и/или B, не являющиеся частью интерметаллического соединения AxBy. Компонент C может также содержать или состоять из одного или более металлических или неметаллических элементов. Предпочтительно компонент C содержит O, N, C, H, Li, Na, K, Rb, Cs, Mg, Ca, Sr, Ba, Ti, Mn, Fe, Co, Ni, Zn, Ga. В более предпочтительном варианте выполнения настоящего изобретения компонент C содержит O, N, C, H, Mg, Ca, Mn, Fe, Co, Ni, Zn, Ga.
Интерметаллическая фаза или интерметаллическое соединение в соответствии с настоящим изобретением представляет собой соединение, изготовленное по меньшей мере из двух разных металлов в упорядоченной или частично упорядоченной структуре с определенной стехиометрией. Структура может быть подобной или отличной от структуры чистых составляющих металлов. Примерами интерметаллических соединений являются упорядоченные, частично упорядоченные и эвтектические сплавы, фазы Лавеса, фазы Цинтля, фазы Гейслера, фазы Юм-Розери и другие интерметаллические фазы, известные специалистам в данной области техники. Также включены соединения, содержащие элементы, относящиеся к группе полуметаллов, такие как селениды, теллуриды, арсениды, антимониды, силизиды, германиды и бориды.
Примерами интерметаллических фаз согласно настоящему изобретению являются RhPb, RhPb2, Rh4Pb5, Rh2Sn, RhSn, RhSn2, RhSn4, Rh2Sb, RhSb, RhSb2, RhSb3, RhGa, Rh10Ga17, Rh3Ga5, Rh2Ga9, Rh4Ga21, Rh3Ga16, RhGa3, RhIn, RhIn3, Rh5Ge3, Rh2Ge, RhGe, Rh17Ge22, RhGe4, IrPb, IrSn, Ir5Sn7, IrSn2, Ir3Sn7, IrSn4, IrSn, Ir5Sn7, IrSn2, Ir3Sn7, IrSn4, Pd3Pb, Pd13Pb9, Pd5Pb3, PdPb, Pd3Sn, Pd20Sn13, Pd2Sn, PdSn, Pd5Sn7, PdSn2, PdSn3, PdSn4, Pd3Sb, Pd20Sb7, Pd5Sb2, Pd8Sb3, Pd2Sb, PdSb, PdSb2, Pd2Ga, Pd5Ga2, Pd5Ga3, PdGa, PdGa5, Pd7Ga3, Ru2Sn3, RuSn2, Ru3Sn7, RuSb, RuSb2, RuSb3, NiPb, Ni3Sn4, Ni3Sn2, Ni3Sn, NiSn, Ni5Sb2, Ni3Sb, NiSb2 и NiSb3, , где RhPb, RhPb2, RhSb, Rh2Sb, RhSb2 и Rh2Sn являются предпочтительными.
Присутствие интерметаллических фаз в мультиметаллическом материале можно обнаружить стандартными методами для определения характеристик твердых веществ, такими как, например, электронная микроскопия, ЯМР в твердом состоянии или порошковая дифракция рентгеновских лучей (PXRD), где предпочтителен PXRD-анализ.
В общем, форма, в которой обеспечивается мультиметаллический материал согласно настоящему изобретению, не ограничена.
Мультиметаллический материал может использоваться в виде отдельного соединения или в смеси с другими соединениями, где предпочтительным является нанесение на носитель способами, включающими импрегнирование в неглубоком слое, импрегнирование распылением, пропитку по влагоемкости, импрегнирование расплавом и другие способы импрегнирования, известные специалистам в данной области техники. Описание, как разместить мультиметаллический материал на носители, приведено ниже.
Материал-носитель в соответствии с настоящим изобретением может быть кристаллическим или аморфным оксидным материалом. Он включает как бинарные, так и полинарные оксиды. Примерами подходящих бинарных оксидов являются: Al2O3, CaO, CeO2, Ce2O3, Fe2O3, La2O3, MgO, MnO2, Mn2O3, SiO2, TiO2, Ti2O3, ZrO2 и ZnO. Они в частности включают нестехиометрические оксиды или оксиды со смешанной валентностью, где некислородный элемент присутствует в более чем одном состоянии окисления, например: CeO2-x, WOx, Fe0,95O Mn3O4, Fe3O4, Ti4O7 и другие нестехиометрические оксиды, известные специалистам в данной области техники. Также включены полинарные оксиды, такие как, например, MgAl2O4, LaAlO3, CaTiO3, CeZrO4 H2Al14Ca12O34. Также включены физические смеси бинарных, полинарных и нестехиометрических оксидов.
В группу оксидных носителей также включены цеолитные носители, как указано ниже. Она включает носители, содержащие один или более цеолитов, микропористых молекулярных сит, алюмосиликатов и алюмофосфатов, а также смеси цеолитов с бинарными, полинарными и/или нестехиометрическими оксидами. В общем, возможно, что тип цеолитной каркасной структуры является одним из следующих ABW, ACO, AEI, AEL, AEN, AET, AFG, AFI, AFN, AFO, AFR, AFS, AFT, AFV, AFX, AFY, AHT, ANA, APC, APD, AST, ASV, ATN, ATO, ATS, ATT, ATV, AVL, AWO, AWW, BCT, BEA, BEC, BIK, BOF, BOG, BOZ, BPH, BRE, BSV, CAN, CAS, CDO, CFI, CGF, CGS, CHA, -CHI, -CLO, CON, CSV, CZP, DAC, DDR, DFO, DFT, DOH, DON, EAB, EDI, EEI, EMT, EON, EPI, ERI, ESV, ETR, EUO, *-EWT, EZT, FAR, FAU, FER, FRA, GIS, GIU, GME, GON, GOO, HEU, IFO, IFR, -IFU, IFW, IFY, IHW, IMF, IRN, IRR, -IRY, ISV, ITE, ITG, ITH, *-ITN, ITR, ITT, -ITV, ITW, IWR, IWS, IWV, IWW, JBW, JNT, JOZ, JRY, JSN, JSR, JST, JSW, KFI, LAU, LEV, LIO, -LIT, LOS, LOV, LTA, LTF, LTJ, LTL, LTN, MAR, MAZ, MEI, MEL, MEP, MER, MFI, MFS, MON, MOR, MOZ, *MRE, MSE, MSO, MTF, MTN, MTT, MTW, MVY, MWF, MWW, NAB, NAT, NES, NON, NPO, NPT, NSI, OBW, OFF, OKO, OSI, OSO, OWE, -PAR, PAU, PCR, PHI, PON, POS, PSI, PUN, RHO, -RON, RRO, RSN, RTE, RTH, RUT, RWR, RWY, SAF, SAO, SAS, SAT, SAV, SBE, SBN, SBS, SBT, SEW, SFE, SFF, SFG, SFH, SFN, SFO, SFS, *SFV, SFW, SGT, SIV, SOD, SOF, SOS, SSF, *-SSO, SSY, STF, STI, *STO, STT, STW, -SVR, SVV, SZR, TER, THO, TOL, TON, TSC, TUN, UEI, UFI, UOS, UOV, UOZ, USI, UTL, UWY, VET, VFI, VNI, VSV, WEI, -WEN, YUG, ZON, или смешанный тип двух или более из них. Более предпочтительно, цеолитный материл содержит, более предпочтительно представляет собой, один или более из цеолитных материалов, имеющих тип каркасной структуры MFI, MOR, BEA, и FAU.
Дополнительными примерами носителей являются углерод или углеродоподобные материалы, такие как активированный уголь, графит или графен. Также включены модифицированные материалы на основе углерода, такие как интеркалированные соединения, и карбиды, такие как W-C, B-C, Si-C. Также включены нитриды, бориды, силициды, фосфиды, антимониды, арсениды, сульфиды, селениды и теллуриды.
Также включены сплавы, сплавы со структурой твёрдого раствора, сплавы со структурой частичного раствора и интерметаллические соединения, а также соединения, называемые соединениями металлов в контексте данного изобретения. Также в группу носителей включены бинарные и полинарные оксидные носители, содержащие один или более элементов из основных групп (исключая благородные газы и галогениды), переходные элементы и/или лантаноиды в комбинации с кислородом и их соответствующие модификации.
Материал-носитель может быть обеспечен в виде порошка, дисперсии, коллоида, гранулята, фасонных изделий, таких как кольца, сферы, экструдаты, шарики и другие формованные изделия, известные специалистам в данной области техники.
Предпочтительными материалами-носителями являются углерод, бинарные и полинарные оксиды и смеси бинарных и полинарных оксидов.
Синтез мультиметаллического материала
Катализатор согласно настоящему изобретению может быть получен способом, включающим стадии в порядке (i) - (iv):
(i) Обеспечение предшественника металла предпочтительно в форме раствора;
(ii) Нанесение предшественника металла на материал-носитель, необязательно после сушки;
(iii) Восстановительная обработка композитного материала;
(iv) Термическая обработка композитного материала.
(i) Эта стадия включает получение предшественника металла путем растворения или разбавления металлсодержащего компонента, такого как соли металлов, коллоидные металлы или органические соединения металлов, в подходящем растворителе, таком как вода, спирты, полиолы, кислоты, основания и другие растворители, известные специалистам в данной области техники. Этот раствор может быть получен либо в виде раствора, содержащего один металл, A или B, либо в виде раствора множества металлов, содержащего любую концентрацию A и B. В конкретном варианте выполнения настоящего изобретения получают дополнительный раствор, содержащий промоторный компонент C. В очень конкретном варианте выполнения настоящего изобретения помоторный компонент может быть частью раствора из одного металла, содержащего А или В, или частью раствора из множества металлов, содержащего А и В.
(ii) Раствор(ы) металла, полученный на стадии (i), наносят на материал-носитель с использованием стандартных методик, таких как импрегнирование в неглубоком слое, импрегнирование распылением, пропитка по влагоемкости, импрегнирование расплавом и другие способы импрегнирования, известные специалистам в данной области техники. Импрегнирование может быть проведено за одну стадию с использованием растворов одного или множества металлов или смесей растворов одного или множества металлов. Импрегнирование может быть проведено за несколько стадий с использованием растворов одного или множества металлов или смесей растворов одного или множества металлов в нескольких стадиях. Настоящее изобретение также охватывает способы осаждения, где носитель получают in situ из растворов металлов или на отдельной стадии. Эта стадия также включает одну или более стадий сушки (iia).
Продуктом стадии (ii) или соответственно стадии (iia) является композитный материал.
(iii) Восстановительная обработка включает воздействие на композитный материал, полученный на стадии (ii) или, соответственно, на стадии (iia), восстанавливающим агентом или восстановление композитного материала путем термического восстановления. Этот восстанавливающий агент может быть обеспечен в твердой, жидкой или газообразной форме. Стадия восстановления может быть выполнена с или без выполнения стадии (iia) перед ней. Восстанавливающие агенты в соответствии с настоящим изобретением представляют собой газы, такие как, например, H2, СО и газообразные углеводороды, такие как CH4, C2H4, и другие восстановительные газы, известные специалисту в данной области техники, жидкие восстанавливающие агенты, такие как спирты, углеводороды и амины, такие как, например, полиолы и гидразин, а также восстанавливающие агенты, обеспеченные в твердой форме, такой как, например, порошок металла.
(iv) Термическая обработка восстановленного композитного материала проводится путем нагревания восстановленного композитного материала, взятого со стадии (iii), до желаемой температуры в химически инертных условиях, где присутствующая газовая смесь не содержит никаких реакционноспособных компонентов, которые могут подвергаться химической реакции с композитным материалом. В частности, смесь не должна содержать окисляющих агентов, таких как, например, кислород, вода, NOx, галогениды или тому подобное. Нагревание может быть выполнено любым способом, подходящим для нагревания твердых или влажных твердых веществ, таким как нагревание в муфельных печах, микроволновых печах, вращающихся печах, трубчатых печах, псевдоожиженном слое и других нагревательных устройствах, известных специалисту в данной области техники.
В конкретном варианте выполнения настоящего изобретения, стадии (iii) и (iv) могут быть объединены в одну стадию путем термической обработки композитного материала в присутствии восстанавливающего агента или при температуре, при которой термическое восстановление происходит.
Способ синтеза изоцианатов из нитроароматических соединений и монооксида углерода
Задача настоящего изобретения также решается способом получения ароматического изоцианата посредством прямого карбонилирования нитроароматического соединения реакцией нитроароматического соединения с монооксидом углерода в присутствии катализатора, отличающимся тем, что катализатор содержит мультиметаллический материал, как уточнено ранее, содержащий одну или более из бинарных интерметаллических фаз общей формулы AxBy, с или без компонента C.
Способ может проводиться периодически или непрерывно.
Настоящее изобретение обеспечивает новые каталитические материалы, способные катализировать стадии реакции 1 и 2 всей реакции. Каталитический материал представляет собой не физическую смесь двух отдельных катализаторов, каждый из которых способен катализировать только одну из двух последовательных стадий реакции, как описано в GB 1315813A, а катализатор, который катализирует обе стадии реакции 1 и 2.
В документе GB 1 315 813 описано гетерогенно катализируемое карбонилирование нитрозо- и нитроароматических соединений до изоцианатов. Однако, в отличие от настоящего изобретения, применяются физические смеси одного катализатора общей формулы MxMnyOz, где М представляет собой Fe, Ag или Pb, со вторым катализатором, содержащим металлы платиновой группы, выбранные из Pd, Ru и Rh, на носителе, таком как углерод или пемза. Указанный выход изоцианата составляет 4,5% через 2 часа при 190 °С. Согласно настоящему изобретению, один мультиметаллический материал, содержащий одну или более интерметаллических фаз AxBy, обеспечивает требуемый изоцианат со значительно более высокой селективностью при более высоких конверсиях (смотрите Таблицу 4). Полагают, что присутствие одной или более интерметаллических фаз является причиной значительно более высоких выходов.
Кроме того, настоящее изобретение обеспечивает способ синтеза изоцианатов из нитроароматических соединений и монооксида углерода, включающий следующие стадии:
a) обеспечение смеси реагентов M1, содержащей нитроароматические соединения и по меньшей мере один дополнительный компонент D, где D содержит подходящий растворитель;
b1) Обеспечение смеси реагентов M2, содержащей смесь реагентов M1 и монооксид углерода или смесь монооксида углерода и инертного газа G, и/или
b2) обеспечение реакционной смеси R1, содержащей смесь реагентов M1 катализатор карбонилирования, содержащий мультиметаллический материал, который более подробно описан выше;
c) контактирование смеси реагентов M2 с катализатором карбонилирования, содержащим, предпочтительно состоящим из мультиметаллического материалп I), который более подробно описан выше; и/или
d) контактирование смеси реагентов R1 с монооксидом углерода или смесью монооксида углерода и инертного газа G;
e) получение реакционной смеси, содержащей изоцианаты.
Вышеуказанные стадии могут быть выполнены с использованием либо стадии b1), либо стадии b2), либо обеих.
Предпочтительно концентрация нитроароматическиго соединения в смеси M1 находится в интервале от в интервале от 0,01мас.% до 60 мас.%, более предпочтительно в интервале от 0,1 мас.% до 50 мас.%, более предпочтительно в интервале от 1 мас.% до 40 мас.%.
Предпочтительно концентрация компонента D в смеси M1 находится в интервале от 40 мас.% до 99,99 мас.%, более предпочтительно в интервале от 50мас.% до 99,9 мас.%, и более предпочтительно в интервале от 60 мас.% до 99 мас.%.
Подходящие нитроароматические соединения (или нитроароматическое соединение), подлежащие реакции согласно настоящему изобретению, представляют собой простые или полиароматические соединения с одной или более нитрогруппами, такие как нитробензол, динитробензол, нитротолуол, динитротолуол, тринитротолуол, нитронафталин, нитроантрацен, нитродифенил, бис(нитрофенил)метан и другие простые и полиароматические соединения, имеющие одну или более нитрогрупп. Нитроароматические соединения также могут содержать другие функциональные группы. В контексте данного изобретения функциональные группы представляют собой заместители, связанные с ароматическим кольцом. Функциональные группы могут содержать один или более гетероатомов, выбранных из группы, состоящей из H, B, C, N, P, O, S, F, Cl, Br и I.
Примерами функциональных групп являются гидроксильные группы, галогены, алифатические боковые цепи, карбонильные группы, изоцианатные группы, нитрозогруппы, карбоксильные группы и аминогруппы.
Также включены нитроорганические соединения, содержащие одну или более нитрогрупп, связанных с алифатической цепью или боковой цепью или кольцом, такие как 1,6-динитрогексен или нитроциклогексен, нитроциклопентен, нитрометан, нитрооктан и бис(нитроциклогексил)-метан.
Подходящим источником нитроароматических соединений является любой источник, содержащий по меньшей мере частично нитроароматические соединения. Источником может быть нитроароматическое соединение, свеже обеспечиваемое в поток реагента М1. Кроме того, нитроароматические соединения могут представлять собой непрореагировавшие нитроароматические соединения, которые после отделения от потока продукта рециркулируют после одной или более стадий переработки. Нитроароматическое соединение также может представлять собой соединение, которое содержит по меньшей мере одну нитро- и/или по меньшей мере одну нитрозогруппу, которое рециркулируется после его частичного превращения с монооксидом углерода. Комбинация свежеобеспеченного нитроароматического соединения и рециркулированного нитроароматического соединения также может быть использована. Также возможно применение нитроароматических аддуктов или предшественников, таких как, например, нитрозоароматические соединения.
Подходящим источником монооксида углерода является также любой источник, содержащий по меньшей мере частично монооксид углерода. Источником может быть только монооксид, свеже обеспечиваемый в поток реагента М1. Кроме того, монооксид углерода может представлять собой непрореагировавший монооксид углерода, который после отделения от потока продукта рециркулируется после одной или более стадий переработки. Комбинация свеже обеспеченного монооксида углерода и рециркулированного монооксида углерода также может быть использована. Также возможно применение аддуктов или предшественников монооксида углерода, таких как, например, муравьиная кислота.
В дополнение к нитроароматическим соединениям и необязательно монооксиду углерода поток реагентов M1 может содержать один или более компонентов D, содержащих растворители S, добавки X и инертные газы G.
Подходящими растворителями S являются апротонные органические растворители, такие как арены и замещенные арены, такие как хлорбензол, дихлорбензол, бензол, толуол, 1,2-дифенилбензол, 1,2-диметилнафталин, гексадецилбензол, Solvesso 150 ND и Solvesso 200 ND.
Другими подходящими апротонными растворителями являются (цикло)алканы и замещенные (цикло)алканы, такие как н-алканы, циклоалканы, хлороформ, дихлорметан, дифенилметан, дибензил.
Другими подходящими растворителями являются простые эфиры с открытой цепью и циклические простые эфиры, такие как диоктиловый простой эфир или ТГФ.
Предпочтительны растворители с температурой кипения в диапазоне от 50°C до 300°C, более предпочтительно от 100°C до 275°C, и более предпочтительно от 125° до 255°C.
Растворитель также может представлять собой изоцианат, соответствующий соответствующему нитроароматическому соединению.
Подходящие инертные газы G включают газы, такие как азот, гелий, неон, аргон или диоксид углерода, из которых предпочтительными являются азот, аргон и диоксид углерода.
Карбонилирование в общем проводят при температуре в интервале от 50 до 250 °C, предпочтительно в интервале от 80 до 190 °C, и более предпочтительно в интервале от 100 до 170 °C.
Общее давление в ходе реакции находится в интервале от 1 до 200 бар, предпочтительно от 10 до 150 бар и более предпочтительно в интервале от 15 до 100 бар.
Парциальное давление монооксида углерода находится в интервале от 1 до 150 бар, предпочтительно в интервале от 1 до 120 бар и более предпочтительно в интервале от 1 до 100 бар.
В общем контактирование реакционной смеси M1 с катализатором, содержащим предпочтительно состоящим из мультиметаллического материала, и с монооксидом углерода может проводиться непрерывным или периодическим образом.
Предпочтительно настоящее изобретение проводят в реакторах периодического действия, каскаде реакторов периодического действия, реакторе полупериодического действия или непрерывных реакторах. Подходящими реакторами являются реакторы с мешалкой, петлевые реакторы, реакторы с петлей вентури, петлевые реакторы с реверсированным потоком, реакторы с пульсирующим потоком, трубчатые реакторы, суспензионные реакторы, реакторы со слоем носителя, реакторы с орошаемым слоем, реакторы с движущимся катализатором, реакторы с корпусом ротора, другие типы реакторов, известные специалистам в данной области техники, и комбинации различных типов реакторов.
В одной установке реакция включает следующие стадии реакции:
a) обеспечение смеси реагентов M1, содержащей нитроароматические соединения и по меньшей мере один дополнительный компонент D, где D содержит подходящий растворитель;
b) обеспечение реакционной смеси R1, содержащей смесь реагентов M1 и катализатор карбонилирования, содержащий мультиметаллический материал, который описан более подробно выше;
c) контактирование реакционной смеси R1 с монооксидом углерода или смесью монооксида углерода и инертного газа G;
d) получение реакционной смеси, содержащей изоцианаты.
В альтернативной установке реакция включает следующие стадии реакции:
a) Обеспечение смеси реагентов M1, содержащей нитроароматические соединения и по меньшей мере один дополнительный компонент D, где D содержит подходящий растворитель;
b) Обеспечение смеси реагентов M2, содержащей смесь реагентов M1 и монооксид углерода или смесь монооксида углерода и инертного газа G, с получением смеси реагентов M2.
c) Контактирование смеси реагентов M2 с ктализатором карбонилирования, содержащим, предпочтительно состоящим из мультиметаллического материала I), который более подробно описан выше,
d) Получение реакционной смеси, содержащей изоцианаты.
В общем реакционная смесь R1 содержит катализатор карбонилирования, содержащий мультиметаллический материал. Концентрация катализатора карбонилирования находится в интервале от 0.1 до 10 мас.%, предпочтительно в интервале от 0.1 до 7.5 мас.%, и более предпочтительно в интервале от 0.1 до 5 мас.%.
В общем, реакционная смесь R1 контактирует с монооксидом углерода в течение от 0.5 до 24 ч, предпочтительно от 2 до 20 ч, и более предпочтительно от 4 до 12 ч.
В общем, в реакционной смеси M2 парциальное давление монооксида углерода находится в интервале от 1 до 150 бар, предпочтительно в интервале от 1 до 120 бар и более предпочтительно в интервале от 1 до 100 бар.
Предпочтительные варианты выполнения настоящего изобретения
Настоящее изобретение дополнительно проиллюстрировано следующими вариантами выполнения настоящего изобретения и комбинациями вариантов выполнения настоящего изобретения, как указано ниже.
В общем, настоящее изобретение обеспечивает способ получения ароматического изоцианата посредством прямого карбонилирования нитроароматического соединения, катализированного мультиметаллическим материалом, содержащим одну или более из бинарных интерметаллических фаз общей формулы AxBy, где:
A представляет собой один или более элемент, выбранный из группы, состоящей из Ni, Ru, Rh, Pd, Ir, Pt и Ag;
B представляет собой один или более элемент, выбранный из группы, состоящей из Sn, Sb, Pb, Zn, Ga, In, Ge и As;
x в AxBy находится в интервале от 0,1 - 10, предпочтительно от 0,2 до 5, и более предпочтительно от 0,5 до 2;
y в AxBy находится в интервале от 0,1 - 10, предпочтительно от 0,2 до 5, и более предпочтительно от 0,5 до 2.
Предпочтительный катализатор содержит одну или более из бинарных интерметаллических фаз общей формулы AxBy, где
A представляет собой один или более элемент, выбранный из группы, состоящей из Ni, Rh, Pd, Ir и Pt;
B представляет собой один или более элемент, выбранный из группы, состоящей из Sn, Sb, Pb, Ga и In.
Более предпочтительные катализаторы содержат одну или более бинарных интерметаллических фаз общей формулы AxBy, где
A представляет собой Rh;
B представляет собой один или более элементов, выбранных из группы, состоящей из Pb, Sn и Sb.
Предпочтительно, мультиметаллический материал составляет до по меньшей мере 85 мас.%, более предпочтительно до по меньшей мере 90 мас.% и даже более предпочтительно до по меньшей мере 95 мас.% одной или более из интерметаллических фаз AxBy.
В одном варианте выполнения настоящего изобретения, мультиметаллический материал содержит один или более компонентов C, где компонент C состоит из или содержит A и/или B, не являющиеся частью интерметаллического соединения AxBy.
В другом варианте выполнения настоящего изобретения, мультиметаллический материал содержит один или более компонентов C, где компонент C содержит или состоит из одного или более элементов, выбранных из группы, состоящей из O, N, C, H, Li, Na, K, Rb, Cs, Mg, Ca, Sr и Ba, Ti, Mn, Fe, Co, Ni, Zn, Ga, предпочтительно один или более элементов из группы, состоящей из O, N, C, H, Mg, Ca, Mn, Fe, Co, Ni, Zn и Ga.
Предпочтительно, мультиметаллический материал наносят на материал-носитель, в общем кристаллический или аморфный материал-носитель. В первом предпочтительном варианте выполнения настоящего изобретения, материал-носитель содержит углерод, графит, графен или интеркалированное соединение. Во втором предпочтительном варианте выполнения настоящего изобретения, материал-носитель содержит карбид, нитрид, борид, силицид, фосфид, антимонид, арсенид, сульфид, селенид или теллурид. В третьем предпочтительном варианте выполнения настоящего изобретения, материал-носитель содержит один или более из бинарных и полинарных оксидов, таких как MgO, CaO, ZnO, CeO2, SiO2, Al2O3. TiO2, ZrO2, Mn2O3, Fe2O3, Fe3O4, MgAl2O4, LaAlO3, CaTiO3, CeZrO4 H2Al14Ca12O34 и других бинарных и полинарных оксидов, известных специалистам в данной области техники в их соответствующих модификациях. В четвертом предпочтительном варианте выполнения настоящего изобретения, материал-носитель содержит, предпочтительно состоит из одного или более цеолитных материалов, где цеолитный материал предпочтительно имеет каркасную структуру типа ZSM, MFI, MOR, BEA или FAU.
Материал-носитель может быть обеспечен в форме, содержащей порошки, дисперсии, коллоиды, грануляты, формованные тела, такие как кольца, сферы, экструдаты или шарики.
Мультиметаллический материал предпочтительно содержит одну или более интерметаллических кристаллических фаз, выбранных из RhPb, RhPb2, Rh4Pb5, Rh2Sn, RhSn, RhSn2, RhSn4, Rh2Sb, RhSb, RhSb2, RhSb3, RhGa, Rh10Ga17, Rh3Ga5, Rh2Ga9, Rh4Ga21, Rh3Ga16, RhGa3, RhIn, RhIn3, Rh5Ge3, Rh2Ge, RhGe, Rh17Ge22, RhGe4, IrPb, IrSn, Ir5Sn7, IrSn2, Ir3Sn7, IrSn4, IrSn, Ir5Sn7, IrSn2, Ir3Sn7, IrSn4, Pd3Pb, Pd13Pb9, Pd5Pb3, PdPb, Pd3Sn, Pd20Sn13, Pd2Sn, PdSn, Pd5Sn7, PdSn2, PdSn3, PdSn4, Pd3Sb, Pd20Sb7, Pd5Sb2, Pd8Sb3, Pd2Sb, PdSb, PdSb2, Pd2Ga, Pd5Ga2, Pd5Ga3, PdGa, PdGa5, Pd7Ga3, Ru2Sn3, RuSn2, Ru3Sn7, RuSb, RuSb2, RuSb3, NiPb, Ni3Sn4, Ni3Sn2, Ni3Sn, NiSn, Ni5Sb2, Ni3Sb, NiSb2 и NiSb3, В частности мультиметаллические материалы содержит одну или более интерметаллических кристаллических фаз, выбранных из RhPb, RhPb2, RhSb, Rh2Sb, RhSb2 и Rh2Sn.
Мультиметаллический материал согласно любому из предшествующих вариантов выполнения настоящего изобретения получают способом, включающим стадии (i) - (iv):
(i) Обеспечение предшественника металла предпочтительно в форме раствора;
(ii) Нанесение предшественника металла на материал-носитель;
(iia) необязательно стадия сушки;
(iii) Восстановительная обработка композитного материала;
(iv) Термическая обработка композитного материала.
На стадии (i) получают смесь, содержащую растворитель и один или более источников для A, B и C, где растворитель содержит одно или более из воды, спиртов, полиолов, кислот и оснований.
На стадии (ii) смесь, полученную в соответствии со стадией (i), приводят в контакт с материалом-носителем, используя метод, выбранный из импрегнирования в неглубоком слое, импрегнирования распылением, пропитка по влагоемкости и импрегнирования расплавом. Для удаления растворителя используется метод, выбранный из выпаривания, нагревания или сушки замораживанием. Также включены методы осаждения, где материал-носитель получают in situ из растворов металлов или на отдельной стадии. Этот метод также включает необязательную стадию сушки.
Стадия восстановительной обработки и стадии термической обработки (iii) и (iv) предпочтительно включают
(iii) Контактирование материала, полученного на стадии (ii), с одним или более восстанавливающими агентами, где восстанавливающий агент может быть обеспечен в твердой, жидкой или газообразной форме и содержать спирты, углеводороды, амины, полиолы, Zn-порошок, H2, CO, CH4 и C2H4;
(iv) Восстановление материала, полученного на стадии (iii), термическим восстановлением в химически инертных условиях.
Термическая обработка включает нагревание материала, полученного в химически инертных условиях, например, в инертных газах, таких как газы, такие как азот, аргон и гелий. Нагревание можно проводить в муфельных печах, микроволновых печах, вращающихся печах, трубчатых печах и псевдоожиженных слоях.
Мультиметаллический материал и катализатор, содержащий мультиметаллический материал согласно любому из предшествующих вариантов выполнения настоящего изобретения, применяются для прямого карбонилирования нитроароматических соединений до изоцианатов.
В общем, способ синтеза изоцианатов из нитроароматических соединений и монооксиди углерода включает стадии a) - d):
a) обеспечение смеси реагентов M1, содержащей нитроароматические соединения и по меньшей мере один дополнительный компонент D, где D содержит подходящий растворитель;
b1) Обеспечение смеси реагентов M2, содержащей смесь реагентов M1 и монооксид углерода или смесь монооксида углерода и инертного газа G, and/or
b2) обеспечение реакционной смеси R1, содержащей смесь реагентов M1 и катализатор карбонилирования, содержащий мультиметаллический материал, который более подробно описан ниже;
c) контактирование смеси реагентов M2 с катализатором карбонилирования, содержащим, предпочтительно состоящим из мультиметаллического материала I), который более подробно описан выше; и/или
d) контактирование смеси реагентов R1 с монооксидом углерода или смесью монооксида углерода и инертного газа G;
e) получение реакционной смеси, содержащей изоцианаты.
Концентрация нитроароматического соединения в смеси M1 находится в общем в интервале от 0,01 мас.% до 60 мас.%, более предпочтительно в интервале от 0.1 мас.% до 50 мас.%, и более предпочтительно в интервале от 0,1мас.% до 40 мас.%. Концентрация компонента D в смеси M1 находится в общем в интервале от 40 мас.% до 99 мас.%, более предпочтительно в интервале от 50 мас.% до 99 мас.%, и более предпочтительно в интервале от 60 мас.% до 99 мас.%.
Подходящие нитроароматические соединения включают простые или полиароматические соединения с одной или более нитрогруппами: нитробензол, динитробензол, нитротолуол, динитротолуол, тринитротолуол, нитронафталин, нитроантрацен, нитродифенил, бис(нитрофенил)метан и, кроме того, простые и полиароматические соединения, имеющие одну или более нитрогрупп. Нитроароматические соединения могут также содержать другие функциональные группы. В контексте настоящего изобретения функциональные группы представляют собой заместители, связанные с ароматическим кольцом. Функциональные группы могут содержать один или более гетероатомов, выбранных из группы, состоящей из Н, В, С, N, Р, О, S, F, Cl, Br и I. Примерами функциональных групп являются гидроксильные группы, галогены, алифатические боковые цепи, карбонильные группы, изоцианатные группы, нитрозогруппы, карбоксильные группы и аминогруппы.
Также включены нитроорганические соединения, содержащие одну или более нитрогрупп, связанных с алифатической цепью, боковой цепью или кольцом, такие как 1,6-динитрогексен или нитроциклогексен, нитроциклопентен, нитрометан, нитрооктан, бис-(нитроциклогексил) метан.
В предпочтительных вариантах выполнения настоящего изобретения нитроароматическое соединение обеспечивают в одном или более апротонных органических растворителях, выбранных из хлорбензола, дихлорбензола, бензола, толуола, ТГФ, диоктилового простого эфира, хлороформа, дихлорметана, н-алканов, циклоалканов, 1,2-дифенилбензола, 1-фенилнафталина, дибензила, 1,2-диметилнафталина, дифенилметана, гексадецилбензола, тетрадецилбензола, додецилбензола или Solvesso 150 ND и Solvesso 200 ND.
В общем, точка кипения одного или более апротонных органических растворителей находится в интервале от 50°C до 300°C, предпочтительно от 100°C до 275°C, более предпочтительно от 125° до 255°C.
В конкретном варианте выполнения настоящего изобретения растворителем может быть изоцианат, соответствующий соответствующем нитроароматическому соединению.
В общем, получение изоцианатов проводят при температуре в интервале от 50 до 250 °C, предпочтительно от 80 до 190 °C, более предпочтительно от 100 до 170 °C. В общем, получение изоцианатов проводят при общем давлении в интервале от 1 до 200 бар, предпочтительно от 10 до 150 бар и более предпочтительно от 15 до 100 бар. Парциальное давление монооксида углерода составляет в общем от 1 до 150 бар, предпочтительно от 1 до 120 бар и более предпочтительно от 1 до 100 бар.
В первом варианте выполнения настоящего изобретения, изоцианаты получают периодически, где для партии проводятся стадии:
a) обеспечение смеси реагентов M1, содержащей нитроароматические соединения и по меньшей мере один дополнительный компонент D, где D содержит подходящий растворитель;
b) обеспечение реакционной смеси R1, содержащей смесь реагентов M1 и катализатор карбонилирования, содержащий мультиметаллический материал, который описан выше;
c) контактирование реакционной смеси R1 с монооксидом углерода или смесью монооксида углерода и инертного газа G;
d) получение реакционной смеси, содержащей изоцианаты.
В общем, концентрация катализатора карбонилирования в реакционной смеси R1 находится в интервале от 0.1 до 10 мас.%, предпочтительно в интервале от 0.1 до 7.5 мас.%, более предпочтительно в интервале от 0.2 до 5 мас.%. Время реакции находится в общем в интервале от 0.5 до 24 ч, предпочтительно от 2 до 20 ч и более предпочтительно от 4 до 12 ч.
Во втором варианте выполнения настоящего изобретения, изоцианаты получают непрерывно в способе, включающем стадии:
a) обеспечение смеси реагентов M1, содержащей нитроароматические соединения и по меньшей мере один дополнительный компонент D, где D содержит подходящий растворитель;
b) обеспечение смеси реагентов M2, содержащей смесь реагентов M1 и монооксид углерода или смесь монооксида углерода или смесь монооксида углерода и инертного газа G, с получением смеси реагентов M2.
c) контактирование смеси реагентов M2 с катализатором карбонилирования, содержащим, предпочтительно состоящим из мультиметаллического материала I), который подробно описан выше
d) получение реакционной смеси, содержащей изоцианаты.
В общем, в реакционной смеси M2 парциальное давление монооксида углерода находится в интервале от 1 до 150 бар, предпочтительно в интервале от 1 до 120 бар и более предпочтительно в интервале от 1 до 100 бар.
Примеры
Фиг. 1 показывает каталитические результаты для примерных катализаторов H, I, J согласно Таблице 2.
Фиг. 2 показывает PXRD картину образца J α: Рефлексы RhPb2. β: Рефлекс графита.
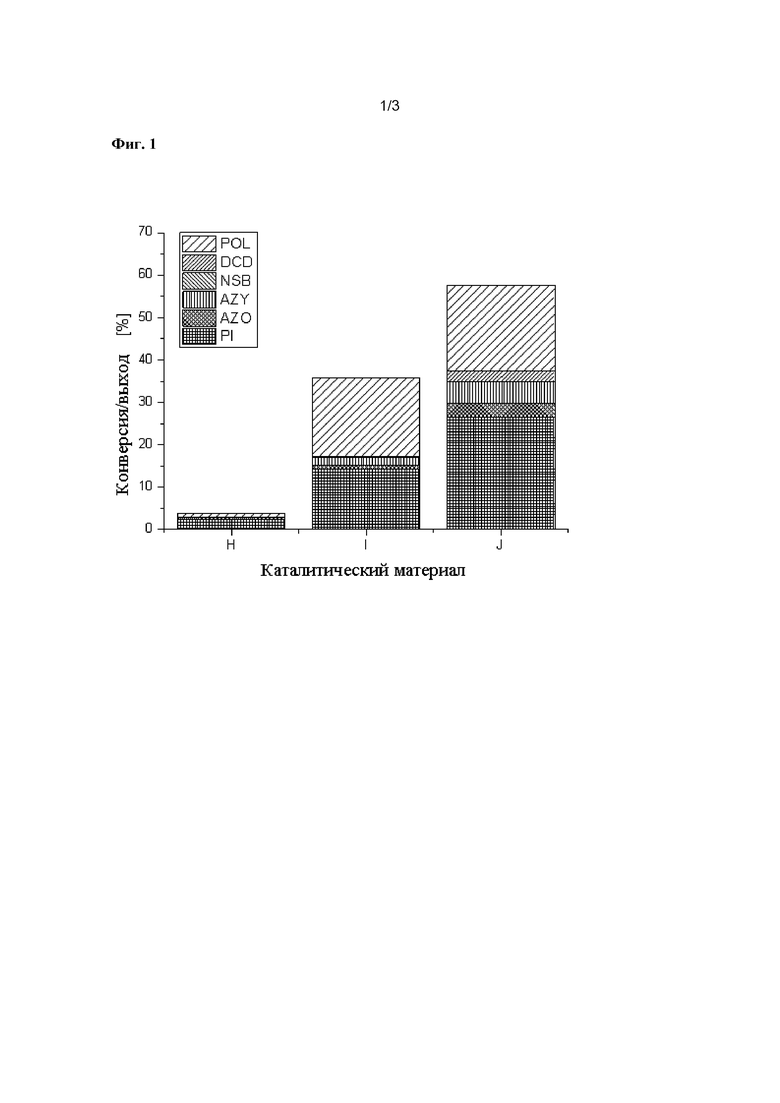
Фиг. 3 показывает каталитические результаты для примерных катализаторов K - O, согласно Таблице 3.
Для рентгеновской порошковой дифракции (XRPD) данные собирали на Bruker AXS D8 Advance. Для сбора данных использовалось излучение Cu Kα. Луч был сужен с использованием коллиматора для фокусировки линии (щель Соллера, 2,5°) и моторизованной щели с расходимостью. Использовались настройки генератора 40 кВ и 40 мА. Образцы осторожно измельчали в ступке с помощью пестика, а затем упаковывали в круглое крепление. Сбор данных из круглого крепления охватывал диапазон 2θ от 5° до 70° с использованием пошагового сканирования с размером шага 0,02° и временем измерения 0,2 с на шаг. Программное обеспечение DIFFRAC.EVA использовалось для всех этапов анализа данных. Фазы, присутствующие в каждом образце, были определены путем поиска и сопоставления данных, имеющихся в Международном центре дифракционных данных (ICDD, версия 2015 года).
Тестирование реактора периодического действия:
Скрининг в реакторе периодического действия проводился в серии одиночных экспериментов с использованием периодических автоклавов, изготовленных из хастеллоя C276. Общая экспериментальная методика для каждого эксперимента по скринингу была следующей:
На первой стадии готовили реакционную смесь путем растворения нитробензола в хлорбензоле. Концентрация нитробензола в реакционной смеси была установлена между 1 мас.% и 5 мас.%. Соответствующее количество катализатора помещали в пустой реактор и нагревали до 160 °С и 10-1 бар в течение по меньшей мере 12 часов. На второй стадии реакционную смесь загружали в реактор, не понижая температуру и не открывая реактор, с помощью специального загрузочного устройства. После загрузки реакционной смеси автоклав нагревали или охлаждали до желаемой температуры. На последней стадии в автоклаве повышали давление с помощью газа CO и азота до желаемого общего давления. Реакционную смесь перемешивали при 1000 оборотах в минуту в течение соответствующего времени.
Соответствующий спектр продуктов анализировали с помощью узла GC-MS (GC-MS от Agilent Technologies), оборудованного детекторами FID, MS и TCD. Общее превращение реакции рассчитывали как разницу в начальной и конечной концентрациях нитроароматического соединения, поделенную на исходную концентрацию нитроароматического соединения. Концентрацию соответствующих продуктов в реакционной смеси определяли с помощью GC-аналитики с использованием соответствующих факторов отклика. Выход определяли путем деления соответствующей концентрации продукта (в ммоль/кг) на исходную концентрацию нитроароматического соединения (в ммоль/кг) и умножения полученного значения на моль (моли) исходного нитроароматического соединения, необходимый для получения моля соответствующий продукт.
Разница между объединенным выходом всех продуктов и общей рассчитанной конверсией представлена термином «полимер». «Полимер» содержит образовавшиеся продукты, которые невозможно проанализировать с помощью применяемого GC-метода.
Сравнительные примеры A - C
Синтез оксидов согласно DE 1 810 828
Синтез Pb0.3Mn0.7Oz
Для получения образцов с соотношением Mn : Pb, равным 0.7 : 0.3, 0.1752 моль Mn в виде Mn(NO3)2x6H2O и 0.075 моль Pb в виде Pb(NO3)2 растворяли в 1 л деионизированной воды при перемешивании. После растворения нитратов добавляли деионизированную воду до 2,5 л. К раствору добавляли 1,04 моль активированного угля. Значение рН доводили до 10 путем добавления 8 мас.% раствора NaOH. Продукт выпадал в осадок, и суспензию перемешивали в течение 30 минут для состаривания.
Жидкую среду отделяли от твердых веществ путем декантации. К твердым веществам добавляли деионизированную воду и перемешивали в течение 15 минут. Процедуру повторяли до тех пор, пока значение рН не стало идентичным используемой деионизированной воде. Твердые вещества отделяли фильтрованием и сушили при 100 °С в течение ночи.
Синтез FexMnyOz
Для получения образцов MnFe с соотношением Mn : Fe, равным 0.8 : 0.2, описанная выше методика была применена, за исключением того, что источником Mn и Fe были не нитраты, а хлориды (0.2 моль Mn в виде MnCl2x4H2O и 0.05 моль Fe в виде FeCl3x6H2O).
1:1 физические смеси оксидов с 5 мас.% Rh или Pd, импрегнированном на активированном угле, получали согласно GB1315813 A и каталитически тестировали. Таблица 2 показывает результаты восстановления нитробензола и включения CO в нитрозобензол для смесей (A) 5 мас.% Pd на C и Pb0.3Mn0.7Ox, (B) 5 мас.% Rh на C и Pb0.3Mn0.7Ox и (C) 5 мас.% Rh на C и Fe0.2Mn0.8Ox. Условиями реакции были p = 100 бар и.д., T = 190oC и время реакции 6 ч. Физическая смесь (A) 5 мас.%Pd на C и PbxMnyOz вообще не дала фенилизоцианат, но наблюдалось образование нитрозобензола, азо- и азоксибензола. Однако другие две протестированные системы, (B) 5 мас.% Rh на C и PbxMnyOz и (C) 5 мас.% Rh на C и FexMnyOz, дали образование фенилизоцианата, азо- и азоксибензола, а также “полимера”.
Сравнительные примеры D - G
Получение сравнительных примеров D-G осуществляли путем получения растворов отдельных металлов, как описано на стадии (i), и импрегнирования растворов на носитель из активированного угля, как описано на стадии (ii). Методом пропитки, которому следовали, была пропитка по влагоемкости. Стадия сушки (iia) при 80 C была выполнена после импрегнирования. Количество металла, нанесенного на носитель, составило 5 мас.% от массы носителя. Соответствующие металлсодержащие компоненты и растворители могут быть взяты из Таблицы 1.
Таблица 1
Каталитические результаты для примеров A - G
Сравнительные примеры А - G были каталитически протестированы. В таблице 2 показаны выходы восстановления нитробензола (стадия 1) и включения СО в нитрозобензол с образованием фенилизоцианата (стадия 2). Для смесей (A) 5 мас.% Pd на C и Pb0.3Mn0.7Ox, (B) 5 мас.% Rh на C и Pb0.3Mn0.7Ox и (C) 5 мас.% Rh на C и Fe0.2Mn0.8Ox условиями реакции были: р = 100 бар и.д., Т = 190 °С и время реакции 6 часов. Физическая смесь (A) 5 мас.% Pd на C и PbxMnyOz вообще не давала фенилизоцианат, но наблюдалось образование нитрозобензола, азо- и азоксибензола. Однако две другие тестируемые системы, (B) 5 мас.% Rh на C и PbxMnyOz и (C) 5 мас.% Rh на C и FexMnyOz, привели к образованию фенилизоцианата, азо- и азоксибензола, а также «полимера» ,
Для металлических катализаторов на основе отдельных металлов D - G, условиями реакции были p = 100 бар и.д., T = 160°C и время реакции 4 ч. Никакие металлические катализаторы на основе отдельных металлов не дают фенилизоцианат.
Таблица 2: Результаты для сравнительных примеров. PI = Фенилизоцианат; AZO = Азобензол; AZY = Азоксибензол; NSB = Нитрозобензол; DCD = Дифенилкарбодиамид; POL = Полимер
[%]
[%]
[%]
[%]
[%]
[%]
Результаты сравнительных примеров B и C показывают, что обе стадии реакции происходили при однореакторном синтезе путем объединения функциональных возможностей двух катализаторов: оксида, ответственного за стадию 1 реакции, и основного металла, ответственного за стадию 2 реакции.
H - O
Получение примеров H-J согласно настоящему изобретению было выполнено путем получения двух отдельных растворов отдельных металлов, как описано на стадии (i). После этого из этих растворов получали смесь. Концентрация растворов отдельных металлов и соответствующий объем, использованный для получения смеси, показаны в таблице 3. Смесь импрегнировали на носитель из активированного угля, как описано на стадии (ii). Применяли метод импрегнирования, которым был метод пропитки по влагоемкости, и стадию сушки (iia) проводили при 80 °C после каждой стадии импрегнирования.
Количество металла А, нанесенного на носитель, должно было составлять 5 мас.% от массы носителя. Количество металла B рассчитывали согласно общей формуле. После стадии сушки композитные материалы из примеров H-J согласно настоящему изобретению получили комбинированную восстановительную и термическую обработку в течение 5 часов при 500 °C (стадии iii и iv) с использованием муфельной печи и N2 атмосферы. Соответствующие массы, концентрации и объемы носителя могут быть взяты из таблицы 3. Металлосодержащие компоненты и растворители могут быть взяты из таблицы 1.
Получение примеров К-О согласно настоящему изобретению выполняли путем приготовления растворов отдельных металлов, как описано на стадии (i), и последовательного импрегнирования растворов на оксидный носитель, как описано на стадии (ii). Методом пропитки, которому следовали, был метод пропитки по влагоемкости. Для примеров K, L и M сначала был импрегнирован раствор отдельного металла, содержащий металл A. Для примеров N и O сначала был импрегнирован раствор отдельного металла, содержащий металл B. В случае примера N и O потребовалось множество импрегнирований для каждого раствора (подробности смотрите в таблице 3). Стадия сушки (iia) выполнялась при 80 °С после каждой стадии импрегнирования. Количество металла А, нанесенного на носитель, должно было составлять 5 мас.% от массы подложки. Количество металла B рассчитывали по общей формуле. После заключительной стадии сушки композитный материал суспендировали в полиэтиленгликоле (полиоле) и подвергали восстановительной обработке, как описано на стадии (iii). Восстановление проводилось в течение 20 минут при 200 °C с использованием микроволновой печи мощностью 1000 Вт и атмосферы N2. Восстановленный композитный материал отделяли от полиола и подвергали термической обработке, как описано на стадии (iv), в течение 5 часов при 500 °C с использованием муфельной печи и N2 атмосферы.
Соответствующие массы, концентрации и объемы носителя могут быть взяты из таблицы 3. Металлосодержащие компоненты и растворители могут быть взяты из таблицы 1.
Таблица 3
1: Материал-носитель. *) удельная площадь поверхности: 100 м2
**) удельная площадь поверхности: 5 м2
***) Рутил
2: Количество материала-носителя [г].
3: Концентрация раствора, содержащего металл A [мол/л].
4: Общий объем раствора, содержащего металл A, применяемого для импрегнирования [мл].
5: Число стадий импрегнирования раствора, содержащего металл A.
6: Концентрация раствора, содержащего металл B [мол/л].
7: Общий объем раствора, содержащего металл B, применяемого для импрегнирования [мл].
8: Число стадий импрегнирования раствора, содержащего металл B.
9: Способ нагревания. a) Муфельная печь 500°C; 5ч; N2 атмосфера.
b) Микроволновая печь (1000W) 200°C; 20 минут; N2 атмосфера
Каталитические результаты примеров H - O
Таблица 4 и Фиг.1 показывает результаты восстановления нитробензола и включения CO в нитрозобензол. Условиями реакции были p = 100 бар и.д., T = 160°C и время реакции 4 ч.
Таблица 4: Результаты примеров H - O
[%]
[%]
[%]
[%]
[%]
Результаты показывают, что - в отличие от металлических катализаторов на основе отдельных металлов- мультиметаллические катализаторы дают фенилизоцианат в качестве продукта.
На Фиг. 2 показана PXRD-диаграмма образца J, доказывающая, что мультиметаллический катализатор состоит из интерметаллического соединения RhPb2 (α) на носителе из аморфного углерода. Однако некоторые следы графита (β), поступающие от углеродного носителя, также были идентифицированы. Кроме того, рефлексы графита появились как следствие стадии термической обработки.
Тестирование в непрерывном реакторе:
Скрининг в реакторе непрерывного действия был проведен в серии экспериментов с использованием реакторной системы с орошаемым слоем. Общая экспериментальная методика для каждого эксперимента скрининга была следующей:
На первом стадии получали реакционную смесь, растворяя нитробензол или динитротолуол в хлорбензоле. Концентрация соответствующего нитроароматического соединения в реакционной смеси составляла от 1 мас.% до 3 мас.%.
Используемый реактор представлял собой трубчатый реактор длиной 40 см с внутренним диаметром 0,4 см.
Внутри реактора был загружен 1 мл соответствующего катализатора, просеянного до размера фракции 125-160 мкм. SiO2 использовали в качестве инертного материала до и после слоя.
Реактор нагревали до 160°C в N2 атмосфере в течение по меньшей мере 12 часов для удаления остаточной воды. После этого температуру в реакторе доводили до желаемого значения.
На следующей стадии реакционную смесь смешивали с CO или смесью CO и N2 и подавали в реактор. Часовая объемная скорость жидкой среды (LHSV) была установлена между 1 ч-1 и 4 ч-1. Тогда как часовая объёмная скорость газа (GHSV) w была установлена между 500 и 3500 ч-1.
Полученную смесь продуктов собирали с течением времени и анализировали посредством GC.
Все экспериментальные параметры приведены в Таблице 6.
Соответствующий спектр продуктов анализировали с помощью узла GC-MS (GC-MS от Agilent Technologies), оборудованного детекторами FID, MS и TCD. Общее превращение каждой реакции рассчитывали как разницу между входной в реактор (исходной) концентрацией реактора и концентрацией нитроароматического соединения в смеси продуктов, поделенную на исходную концентрацию нитроароматического соединения. Концентрация соответствующих продуктов в смеси продуктов была определена с помощью GC-аналитики с использованием соответствующих факторов отклика. Выход определяли путем деления соответствующей концентрации продукта (в ммоль/кг) на концентрацию нитроароматического соединения (в ммоль/кг) и умножения полученного значения на моль (молии) исходного нитроароматического соединения, необходимый для образования моль соответствующего продукта.
Разница между объединенными выходами всех продуктов и общей рассчитанной конверсией нитроароматических соединений представлена термином «полимер». «Полимер» содержит образовавшиеся продукты, которые невозможно проанализировать с помощью примененного GC-метода.
Получение примеров H-BI согласно настоящему изобретению путем получения отдельных растворов отдельных металлов, как описано на стадии (i). После этого из этих растворов готовили смесь. Концентрация растворов отдельных металлов и соответствующий объем, используемые для получения смеси, показаны в таблице 5. Смесь импрегнировали на различные носители, как описано на стадии (ii). Применяли метод импрегнирования, который заключался в пропитке по влагоемкости, и стадию сушки (iia) проводили при 80 C после каждой стадии импрегнирования.
В некоторых примерах носитель получали из смеси двух оксидов. В этом случае оксиды были физически смешаны с использованием ручной мельницы. Полученную оксидную смесь кальцинировали при 500 °С перед импрегнированием.
Количество металла A, нанесенного на носитель, должно составлять от 1 до 5 мас.% от массы носителя. Количество металла B рассчитывали по общей формуле. После стадии сушки композитные материалы примеров H-BI согласно настоящему изобретению подвергались восстановительной и термической обработке в течение 5 часов при 500 °C (стадии iii и iv) с использованием муфельной печи и N2 атмосферы. Массы, концентрации и объемы соответствующих носителей можно взять из Таблицы 5. Металлсодержащие компоненты и растворители можно взять из Таблицы 1.
Таблица 5:
AxBy
1: Материал-носитель. *) удельная площадь поверхности: >5м2
**) Рутил
2: Количество материала-носителя I [г].
3: Количество материала-носителя II [г].
4: Концентрация раствора, содержащего металл A [мол/л].
5: Общий объем раствора, содержащего металл A, применяемого для импрегнирования [мл].
6: Число стадий импрегнирования раствора, содержащего металл A.
7: Концентрация раствора, содержащего металл B [мол/л].
8: Общий объем раствора, содержащего металл B, применяемого для импрегнирования [мл].
9: Число стадий импрегнирования раствора, содержащего металл B.
Таблица 6: Обзор экспериментальных параметров.
1a: Загрузочная смесь
NB) Нитробензол
2,4-DNT) 2,4 -Динитротолуол
2,4-DNT и 2,6-DNT) Смесь 20 мас.% 2,6-DNT и 80мас.% 2,4-DNT
2a: Поступающая концентрация [мас.%]
3a: Температура [°C]
4a: Общее давление [бар]
5a: Концентрация CO [об.%]
6a: Концентрация N2 [об.%]
7a: LHSV (Часовая объемная скорость жидкой среды) [ч-1]
8a: GHSV (Часовая объемная скорость газа) [ч-1]
Таблица 7: Результаты каталитических испытаний в установке с орошаемым слоем с нитробензолом: PI = Фенилизоцианат; AZO = Азобензол; AZY = Азоксибензол; NSB = Нитрозобензол; DCD = Дифенилкарбодиамид; POL = Полимер. X = Общее превращение.
Результаты показывают, что изоцинаты могут быть получены из нитроароматических соединений в непрерывном процессе.
Таблица 8: Результаты каталитических испытаний в установке с орошаемым слоем с DNT загрузочной смесью: TDI = 2,4-Толуолдиизоцианат; TNI = Толуолнитроизоцианаты, AZOC = Азосоединения; AZYC = Азоксисоединения; NSC = Нитрозосоединения; AC = Аминные соединения; POL = Полимер.
Результаты показывают, что нитроароматические соединения, содержащие множество нитрогрупп, могут быть непосредственно превращены в изоцианаты. Поскольку число структурных изомеров увеличивается с увеличением количества нитрогрупп, выходы представлены в виде выходов групп.
Как указано выше, промежуточные продукты, такие как нитрозосоединения или частично карбонилированные нитроароматические соединения, такие как толуолнитроизоцианаты (TNI), могут быть получены в результате неполной реакции, но все еще рассматриваются как успешный результат с точки зрения данного изобретения.
Таблица 9: Результаты каталитических испытаний в установке с орошаемым слоем с DNT загрузочной смесью: TDI = 2,4+2.6-Толуолдиизоцианат; TNI = (изомеры толуолнитроизоцианата, Побочные продукты = Азосоединения, Азоксисоединения, Нитрозосоединения, Аминные соединения, Полимер.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ АЛИЦИКЛИЧЕСКИЙ СПИРТ | 2011 |
|
RU2564416C2 |
| НОВЫЙ АЛИЦИКЛИЧЕСКИЙ СПИРТ | 2011 |
|
RU2564417C2 |
| ЧЕТЫРЕХХОДОВЫЕ КАТАЛИЗАТОРЫ КОНВЕРСИИ ДЛЯ СИСТЕМ ОБРАБОТКИ ВЫБРОСОВ БЕНЗИНОВОГО ДВИГАТЕЛЯ | 2017 |
|
RU2759005C2 |
| СПОСОБ КАРБОНИЛИРОВАНИЯ, ПРЕДНАЗНАЧЕННЫЙ ДЛЯ ПРОИЗВОДСТВА УКСУСНОЙ КИСЛОТЫ С ПРИМЕНЕНИЕМ МЕТАЛЛИЧЕСКИХ КАТАЛИЗАТОРОВ С КЛЕШНЕОБРАЗНЫМИ ЛИГАНДАМИ | 2008 |
|
RU2459800C2 |
| АВТОМОБИЛЬНЫЕ КАТАЛИТИЧЕСКИЕ КОМПОЗИТЫ, ИМЕЮЩИЕ СЛОЙ С ДВУМЯ МЕТАЛЛАМИ | 2014 |
|
RU2658002C2 |
| Способ получения уксусной кислоты | 1989 |
|
SU1715203A3 |
| УДАЛЕНИЕ ПРИМЕСЕЙ УГЛЕВОДОРОДОВ ИЗ ПОЛУПРОДУКТОВ ПРОИЗВОДСТВА УКСУСНОЙ КИСЛОТЫ | 2010 |
|
RU2523910C2 |
| КАТАЛИЗАТОР КАРБОНИЛИРОВАНИЯ И СПОСОБ | 2014 |
|
RU2696266C2 |
| СПОСОБ ПОЛУЧЕНИЯ УКСУСНОЙ КИСЛОТЫ БЕЗВОДНЫМ КАРБОНИЛИРОВАНИЕМ | 1999 |
|
RU2197470C2 |
| СПОСОБ КАРБОНИЛИРОВАНИЯ С ИСПОЛЬЗОВАНИЕМ КАТАЛИЗАТОРОВ С МЕТАЛЛТРИДЕНТАТНЫМИ ЛИГАНДАМИ | 2004 |
|
RU2348607C2 |

Настоящее изобретение относится к гетерогенным катализаторам, способу их получения и к способу прямого карбонилирования нитроароматических соединений в ароматические изоцианаты. Способ получения ароматического изоцианата посредством прямого карбонилирования нитроароматического соединения посредством реакции нитроароматического соединения с монооксидом углерода в присутствии катализатора, который содержит мультиметаллический материал, содержащий одну или более бинарных интерметаллических фаз общей формулы AxBy, где A представляет собой один или более элементов, выбранных из Rh и Pd, B представляет собой один или более элементов, выбранных из Sn, Sb, Pb, Ga и In, x находится в интервале от 0,1 - 10, y находится в интервале от 0,1 - 10. Изобретение также относится к катализатору, содержащему мультиметаллический материал, который содержит одну или более бинарных интерметаллических фаз общей формулы AxBy, указанных выше. Способ получения катализатора включает стадии (i) и (iv): (i) обеспечение одного или более предшественников для элементов A и B и необязательно компонента C в форме раствора; (ii) нанесение предшественников металлов на материал-носитель; (iii) восстановительная обработка композитного материала; (iv) термическая обработка композитного материала. Технический результат изобретения заключается в обеспечении способа карбонилирования нитроароматических соединений до соответствующего изоцианата, показывающего значительное улучшение активности и селективности. 3 н. и 16 з.п. ф-лы, 9 табл., 3 ил., 15 пр.
1. Способ получения ароматического изоцианата посредством прямого карбонилирования нитроароматического соединения посредством реакции нитроароматического соединения с монооксидом углерода в присутствии катализатора, отличающийся тем, что катализатор содержит мультиметаллический материал, содержащий одну или более бинарных интерметаллических фаз общей формулы AxBy,
где A представляет собой один или более элементов, выбранных из Rh и Pd,
B представляет собой один или более элемент, выбранный из Sn, Sb, Pb, Ga и In,
x находится в интервале от 0,1 - 10,
y находится в интервале от 0,1 - 10.
2. Способ по п. 1, отличающийся тем, что
A представляет собой Rh,
B представляет собой один или более элементов, выбранных из Pb, Sn и Sb.
3. Способ по п. 1, отличающийся тем, что мультиметаллический материал содержит один или более компонентов C, где компонент C содержит или состоит из А или В, не являющихся частью интерметаллической фазы AxBy.
4. Способ по п. 1, отличающийся тем, что мультиметаллический материал содержит один или более компонентов C, где компонент C содержит или состоит из одного или более элементов, выбранных из O, N, C, H, Mg, Ca, Mn, Fe, Co, Ni, Zn, Ga.
5. Способ по п. 1, отличающийся тем, что мультиметаллический материал расположен на материале-носителе.
6. Способ по п. 1, отличающийся тем, что нитроароматическое соединение выбрано из нитробензола, динитробензола, нитротолуола, динитротолуола, тринитротолуола, нитронафталина, нитроантрацена, нитродифенила, бис(нитрофенил)метана и других моно- и полиароматических соединений, имеющих одну или более нитрогрупп.
7. Способ по любому из пп. 1-6, отличающийся тем, что его проводят периодически.
8. Способ по любому из пп. 1-6, отличающийся тем, что его проводят непрерывно.
9. Катализатор для прямого карбонилирования нитроароматического соединения до соответствующего ароматического изоцианата, содержащий мультиметаллический материал, содержащий одну или более бинарных интерметаллических фаз общей формулы AxBy,
где A представляет собой один или более элементов, выбранных из Rh и Pd,
B представляет собой один или более элементов, выбранных из Sn, Sb, Pb, Ga и In,
x находится в интервале от 0,1 – 10,
y находится в интервале от 0,1 - 10.
10. Катализатор по п. 9, отличающийся тем, что
A представляет собой Rh,
B представляет собой один или более элемент, выбранный из Pb, Sn и Sb.
11. Катализатор по п. 9, отличающийся тем, что мультиметаллический материал содержит один или более компонентов C, где компонент C содержит или состоит из A или B, не являющихся частью интерметаллического соединения AxBy.
12. Катализатор по п. 9, отличающийся тем, что мультиметаллический материал содержит один или более компонентов C, где компонент C содержит или состоит из одного или более элементов, выбранных из O, N, C, H, Mg, Ca, Mn, Fe, Co, Ni, Zn и Ga.
13. Катализатор по п. 9, отличающийся тем, что мультиметаллический материал содержит одну или более бинарных интерметаллических кристаллических фаз, выбранных из RhPb, RhPb2, Rh4Pb5, Rh2Sn, RhSn, RhSn2, RhSn4, Rh2Sb, RhSb, RhSb2, RhSb3, RhGa, Rh10Ga17, Rh3Ga5, Rh2Ga9, Rh4Ga21, Rh3Ga16, RhGa3, RhIn, RhIn3, Pd3Pb, Pd13Pb9, Pd5Pb3, PdPb, Pd3Sn, Pd20Sn13, Pd2Sn, PdSn, Pd5Sn7, PdSn2, PdSn3, PdSn4, Pd3Sb, Pd20Sb7, Pd5Sb2, Pd8Sb3, Pd2Sb, PdSb, PdSb2, Pd2Ga, Pd5Ga2, Pd5Ga3, PdGa, PdGa5 и Pd7Ga3.
14. Катализатор по п. 13, отличающийся тем, что мультиметаллический материал содержит одну или более бинарных интерметаллических кристаллических фаз, выбранных из RhPb, RhPb2, RhSb, Rh2Sb, RhSb2 и Rh2Sn.
15. Катализатор по любому из пп. 9-14, отличающийся тем, что мультиметаллический материал расположен на материале-носителе.
16. Способ получения катализатора как определено в п. 15, включающий стадии (i) и (iv):
(i) обеспечение одного или более предшественников для элементов A и B и необязательно компонента C в форме раствора;
(ii) нанесение предшественников металлов на материал-носитель;
(iii) восстановительная обработка композитного материала;
(iv) термическая обработка композитного материала.
17. Способ по п. 16, где раствор содержит один или более растворителей, выбранных из воды, спиртов, полиолов, кислот и оснований.
18. Способ по п. 16, включающий стадии:
(iii) контакт материала-носителя, содержащего предшественники металлов, с одним или более восстанавливающими агентами в твердой, жидкой или газообразной форме, выбранными из спиртов, углеводородов, аминов, полиолов, Zn-порошка, H2, CO, CH4 и C2H4, и
(iv) восстановление материала-носителя, содержащего предшественники металлов, путем термического восстановления в химически инертных условиях.
19. Способ по любому из пп. 16-18, где стадию (iii) и (iv) проводят за одну стадию.
| US 3523966 A1, 11.08.1970 | |||
| DE 19635723 A1, 05.03.1998 | |||
| US 3523964 A1, 11.08.1970 | |||
| JP 2000256764 A, 19.09.2000 | |||
| 0 |
|
SU368743A1 | |
| Способ получения метилизоцианатов | 1987 |
|
SU1586510A3 |

