ОБЛАСТЬ ТЕХНИКИ
Настоящее раскрытие относится к соединениям со спиро- и ароматическими кольцами. В частности, изобретение относится к соединениям со спиро- и ароматическими кольцами, которые можно применять в качестве ингибитора SHP2, их фармацевтически приемлемым солям или их энантиомерам, диастереоизомерам, таутомерам, сольватам, изотопнозамещенным производным, пролекарствам или метаболитам. Кроме того, раскрытие также относится к способам получения соединений, фармацевтическим композициям, содержащим соединения, и применению соединений в получении лекарственных средств для профилактики или лечения заболеваний или состояний, связанных с аномальной активностью SHP2.
УРОВЕНЬ ТЕХНИКИ
Белок тирозинфосфатаза SHP2 играет важную роль в передаче клеточных сигналов и является мишенью для лечения социально значимых заболеваний, таких как диабет, аутоиммунные заболевания и рак. SHP2 мутирован или высоко экспрессируется при различных заболеваниях, таких как синдром Нунана, синдром леопарда, ювенильный миеломоноцитарный лейкоз, нейробластома, меланома, острый миелоидный лейкоз, рак молочной железы, рак пищевода, рак легких, рак толстой кишки, рак головы, нейробластома, плоскоклеточная карцинома головы и шеи, рак желудка, анапластическая крупноклеточная лимфома и глиобластома и т.д. Молекулярно-биологические исследования показывают, что SHP2 участвует во множестве сигнальных путей опухолевых клеток, таких как MAPK, JAK/STAT и PI3K/Akt и т.д. В то же время SHP2 также отвечает за передачу сигнала иммуносупрессивного пути PD1-PDL1. Следовательно, ингибирование активности SHP2 может реверсировать иммуносупрессию в микроокружении опухоли.
SHP2 состоит из двух N-концевых доменов Src-Homolgy-2 (N-SH2 и C-SH2) и каталитического домена протеинтирозинфосфатазы (PTP). В состоянии самоингибирования N-SH2 объединяется с PTP с образованием кольцевой структуры, которая препятствует связыванию PTP с субстратом, таким образом ингибируя каталитическую активность фермента; когда тирозин вышележащего рецепторного белка фосфорилируется и связывается с N-SH2, каталитический домен PTP высвобождается для проявления активности фосфатазы.
В настоящее время разработка ингибиторов SHP2 в основном сосредоточена на аллостерических ингибиторах в некаталитической области, таких как соединения, описанные в WO2015107493A1, WO2016203404A1, WO2016203406A1, WO2017216706A1, WO2017211303A1, CN201710062495, WO2018136265A1, WO2018057884 и т.д. Исследование этого года показывает, что SHP2 как новая лекарственная мишень привлекает все больше и больше внимания. Следовательно, в данной области техники существует острая необходимость в разработке ингибиторов SHP2 с новыми структурами, хорошей биологической активностью и высокой лекарственной способностью.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Одним из объектов раскрытия является предоставление соединения формулы I или его фармацевтически приемлемой соли, фармацевтической композиции, содержащей соединение или его фармацевтически приемлемую соль, и применение соединения или фармацевтической композиции в профилактике или лечении заболеваний или состояний, связанных с аномальной активностью SHP2.
Первый аспект раскрытия предоставляет соединение формулы I:
 ,
,
или его фармацевтически приемлемую соль, или его энантиомер, диастереоизомер, таутомер, сольват, изотопозамещенное производное, полиморф, пролекарство или метаболит, где:
X1 и X2 независимо выбраны из связи, O, CRaRb или NRc;
X3 выбран из связи, CRaRb, NRc, S или O;
X4 выбран из N или CRc; и Ra, Rb и Rc независимо выбраны из H, галогена, замещенного или незамещенного C1-6 алкила или замещенного или незамещенного C1-6 алкоксила;
R1, R2, R3, R4 и R7 независимо выбраны из H, -OH, галогена, замещенного или незамещенного амино, замещенного или незамещенного C1-6 алкила или замещенного или незамещенного C1-6 алкоксила; и R1, R2, R3, R4 и R7 не могут представлять собой -OH или -NH2 одновременно;
кольцо A выбрано из замещенного или незамещенного C4-8 циклического гидрокарбила, замещенного или незамещенного 4-8-членного гетероциклила, замещенного или незамещенного C5-10 арила или замещенного или незамещенного 5-10-членного гетероарила, где гетероциклил или гетероарил содержит 1-3 гетероатома, выбранных из следующих атомов: N, O, S или P;
кольцо C выбрано из замещенного или незамещенного C4-8 циклического гидрокарбила, замещенного или незамещенного 5-6-членного моноциклического гетероциклила, замещенного или незамещенного 8-10-членного бициклического гетероциклила, замещенного или незамещенного C5-10 моноциклического или бициклического арила, замещенного или незамещенного 5-6-членного моноциклического гетероарила или замещенного или незамещенного 8-10-членного бициклического гетероарила, где гетероциклил или гетероарил содержит 1-4 гетероатома, выбранных из следующих атомов: N, O, S или P;
R5 и R6 независимо выбраны из H, -OH, галогена, циано, замещенного или незамещенного амино, замещенного или незамещенного C1-6 алкила или замещенного или незамещенного C1-6 алкоксила;
n представляет собой любое целое число от 0 до 3; и
"замещенный" относится к одному или нескольким атомам водорода в группе, замещенным заместителем, выбранным из следующих заместителей: галоген, -OH, -NO2, -NH2, -NH(незамещенный или галогенированный C1-6 алкил), -N(незамещенный или галогенированный C1-6 алкил)2, -CN, незамещенный или галогенированный C1-8 алкил, незамещенный или галогенированный C1-8 алкоксил, незамещенный или галогенированный C1-8 алкоксил-C1-8 алкил, незамещенный или галогенированный C3-8 циклоалкил-C1-8 алкил, незамещенный или галогенированный C1-6 алкилкарбонил, незамещенный или галогенированный C1-6 алкоксилкарбонил, группа гидроксамовой кислоты, незамещенный или галогенированный C1-6 алкилтиол, -S(O)2N(незамещенный или галогенированный C1-6 алкил)2, -S(O)2 незамещенный или галогенированный C1-6 алкил, -N(незамещенный или галогенированный C1-6 алкил)S(O)2N(незамещенный или галогенированный C1-6 алкил)2, -S(O)N(незамещенный или галогенированный C1-6 алкил)2, -S(O)(незамещенный или галогенированный C1-6 алкил), -N(незамещенный или галогенированный C1-6 алкил)S(O)N(незамещенный или галогенированный C1-6 алкил)2, -N(незамещенный или галогенированный C1-6 алкил)S(O)(незамещенный или галогенированный C1-6 алкил), незамещенный или галогенированный C5-10 арил, незамещенный или галогенированный 5-10-членный гетероарил, незамещенный или галогенированный C4-8 циклический гидрокарбил или незамещенный или галогенированный 4-8-членный гетероциклил, где гетероциклил и гетероарил содержат 1-4 гетероатома, выбранных из следующих атомов: N, O или S.
В качестве предпочтительного варианта осуществления один из X1 и X2 представляет собой CH2, а другой представляет собой связь.
В качестве предпочтительного варианта осуществления X3 представляет собой S.
В качестве предпочтительного варианта осуществления X4 выбран из N или CH.
В качестве предпочтительного варианта осуществления R1, R2, R3, R4 и R7 независимо выбраны из H, -OH, -F, -Cl, -Br, -NH2, -NHC1-3 алкила, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси или изопропокси; C1-3 алкила, замещенного галогеном, -NH2, -OH, C1-3 алкилом или C1-3 алкоксилом; или C1-3 алкоксила, замещенного галогеном, -NH2, -OH, C1-3 алкилом или C1-3 алкоксилом.
В качестве предпочтительного варианта осуществления R5 и R6 независимо выбраны из H, -OH, -F, -Cl, -Br, -CN, -NH2, -NHC1-3 алкила, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси или изопропокси; C1-3 алкила, замещенного галогеном, -NH2, -OH, C1-3 алкилом или C1-3 алкоксилом; или C1-3 алкоксила, замещенного галогеном, -NH2, -OH, C1-3 алкилом или C1-3 алкоксилом.
В качестве предпочтительного варианта осуществления заместитель выбран из -F, -Cl, -Br, -OH, -NO2, -NH2, -NH(C1-6 алкил), -N(C1-6 алкил)2, -CN, C1-6 алкила, C1-4 алкоксила, C1-4 алкоксил-C1-6 алкила, C3-8 циклоалкил-C1-8 алкила, C1-6 алкилкарбонила, C1-6 алкоксилкарбонила, C1-6 алкилтиола, -S(O)2N(C1-6 алкил)2, -S(O)2 C1-6 алкила, -N(C1-6 алкил)S(O)2N(C1-6 алкил)2, -S(O)N(C1-6 алкил)2, -S(O)(C1-6 алкил), -N(C1-6 алкил)S(O)N(C1-6 алкил)2, -N(C1-6 алкил)S(O)(C1-6 алкил), замещенного или незамещенного C5-10 арила, замещенного или незамещенного 5-10-членного гетероарила, замещенного или незамещенного C4-8 циклического гидрокарбила или замещенного или незамещенного 4-8-членного гетероциклила, где гетероциклил и гетероарил содержат 1-4 гетероатома, выбранных из следующих атомов: N, O или S.
В качестве предпочтительного варианта осуществления заместитель выбран из -F, -Cl, -Br, -OH, -NO2, -NH2, -NH(C1-3 алкил), -N(C1-3 алкил)2, -CN, C1-3 алкила, C1-3 алкоксила, C1-3 алкилкарбонила, циклопропила, циклобутила, циклопентила, циклопентенила, циклогексила, циклогексенила, циклогексадиенила, циклогептила, циклооктила, пирролидинила, морфолинила, пиперазинила, гомопиперазинила, пиперидинила, тиоморфолинила, фенила, нафтила, антрацила, фенантрила, флуоренила, тиофенила, имидазолила, пиразолила, тиазолила, оксазолила, оксадиазолила, изоксазолила, пиридила, пиримидинила, пиразинила, пиридазинила, бензимидазолила, бензопиразолила, индолила, фуранила, пирролила, триазолила, тетразолила, триазинила, индолизинила, изоиндолила, индазолила, изоиндазолила, пуринила, хинолинила или изохинолинила.
В качестве предпочтительного варианта осуществления заместитель выбран из -F, -Cl, -Br, -OH, -NO2, -NH2, -NH(C1-3 алкил), -N(C1-3 алкил)2, -CN, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси, изопропокси или фенила.
В качестве предпочтительного варианта осуществления кольцо C выбрано из любой из следующих групп:
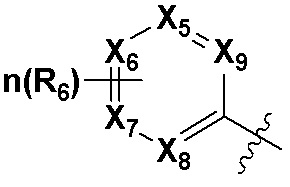 ,
,  или
или 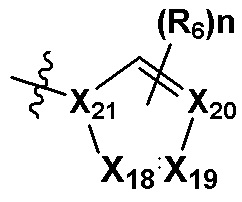 ;
;
где:
X5, X6, X7, X8 и X9 независимо выбраны из N или CRd; и по большей мере 3 из X5, X6, X7, X8 и X9 одновременно представляют собой N;
X10, X11, X12, X13, X14, X15, X16 и X17 независимо выбраны из N или CRd; и по большей мере 5 из X10, X11, X12, X13, X14, X15, X16 и X17 одновременно представляют собой N;
X18, X19, X20 и X21 независимо выбраны из N или CRd, и по большей мере 3 из X18, X19, X20 и X21 одновременно представляют собой N;
R6 и R8 независимо выбраны из H, -NH2, -CN, -OH, -NO2, галогена, незамещенного или галогенированного C1-6 алкила или незамещенного или галогенированного C1-6 алкоксила; и
Rd выбран из H, галогена, незамещенного или галогенированного C1-6 алкила или незамещенного или галогенированного C1-6 алкоксила.
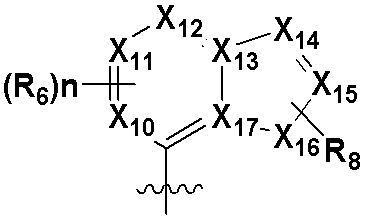
В качестве предпочтительного варианта осуществления кольцо C выбрано из любой из следующих групп:
 или
или 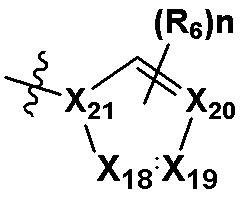 ;
;
в которых:
0, 1 или 2 из X5, X6, X7, X8 и X9 представляют собой N, остальные представляют собой CRd;
0, 1 или 2 из X18, X19, X20 и X21 представляют собой N, остальные представляют собой CRd;
R6 выбран из H, -NH2, -CN, -OH, -NO2, -F, -Cl, -Br, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси, изопропокси, фторированного или бромированного C1-3 алкила, фторированного или бромированного C1-3 алкоксила; и
Rd выбран из H, -F, -Cl, -Br, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси, изопропокси, фторированного или бромированного C1-3 алкила или фторированного или бромированного C1-3 алкоксила.
В качестве предпочтительного варианта осуществления кольцо C выбрано из любой из следующих групп:
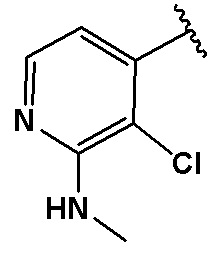 ,
, 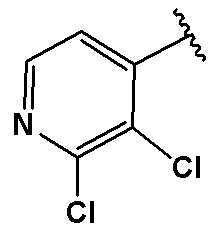 или
или  .
.
В качестве предпочтительного варианта осуществления кольцо A выбрано из замещенного или незамещенного C4-6 циклического гидрокарбила, замещенного или незамещенного 4-6-членного гетероциклила, замещенного или незамещенного C5-6 арила или замещенного или незамещенного 5-6-членного гетероарила, где гетероциклил или гетероарил содержит 1-3 атома N.
В качестве предпочтительного варианта осуществления кольцо A выбрано из любой из следующих групп:
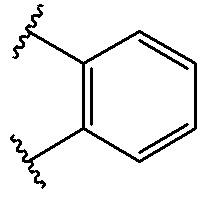 ,
, 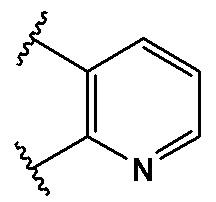 ,
, 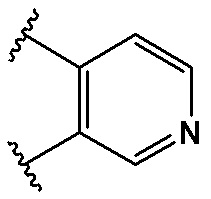 ,
, 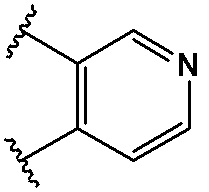 ,
, 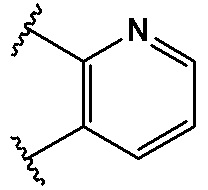 ,
,
 ,
, 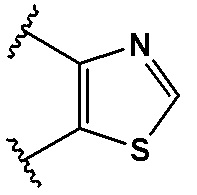 ,
,  ,
,  или
или  .
.
В качестве предпочтительного варианта осуществления кольцо A выбрано из любой из следующих групп:
 ,
,  ,
,  ,
,  ,
,  или
или  .
.
В качестве другого предпочтительного варианта осуществления соединение имеет структуру, выбранную из:
 ,
,  ,
, 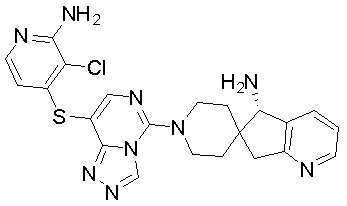 ,
, 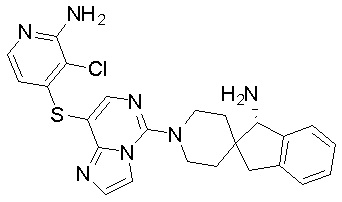 ,
,  ,
, 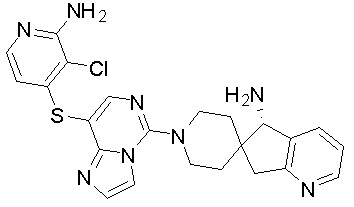 ,
, 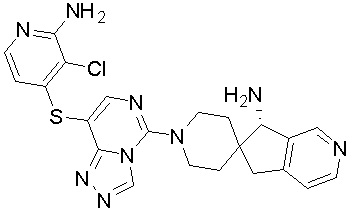 ,
, 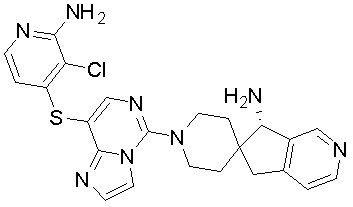 ,
, 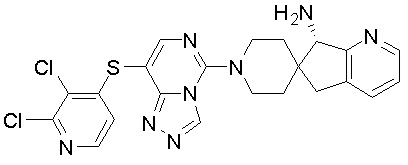 ,
, 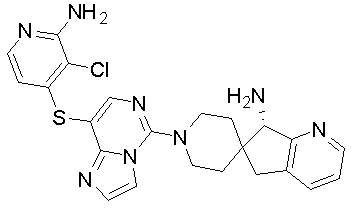 ,
,  ,
, 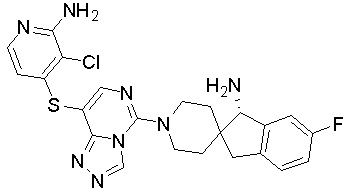 ,
,  ,
, 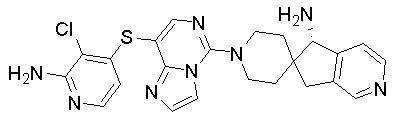 ,
,  ,
, 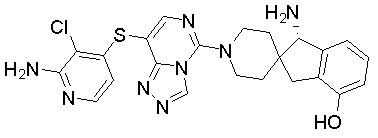 ,
, 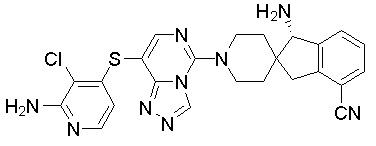 ,
, 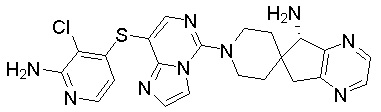 ,
,  ,
, 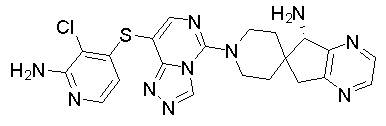 ,
, 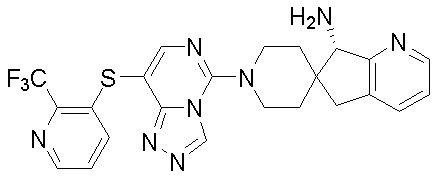 ,
,  или
или  .
.
В качестве предпочтительного варианта осуществления изотопное замещение изотопно-замещенного производного соединения в настоящем документе относится к атому, включающему, но не ограничиваясь ими, водород, углерод, азот, кислород, фтор, фосфор, хлор или иод; и предпочтительно представляет собой 2H, 3H, 11C, 13C, 14C, 15N, 17O, 18O, 18F, 31P, 32P, 35S, 36Cl или 125I.
Второй аспект раскрытия предоставляет способ получения соединения формулы I согласно раскрытию, где способ включает:
(i) проведение реакции нуклеофильного замещения между формулой Ib и формулой Ic с получением формулы Id;
(ii) проведение реакции замещения между формулой Id и формулой Ie с получением формулы If; и
(iii) удаление защитной группы в формуле If посредством кислоты с получением соединения формулы I:
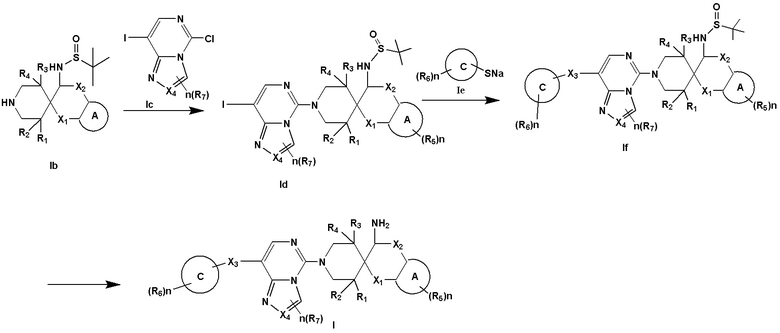
Третий аспект раскрытия предоставляет применение соединения формулы I согласно раскрытию в способе:
(a) получения лекарственного средства для профилактики или лечения заболеваний или состояний, связанных с аномальной активностью SHP2;
(b) получения лекарственного средства для профилактики или лечения SHP2-опосредованных заболеваний или состояний;
(c) получения лекарственного средства-ингибитора для ингибирования активности SHP2;
(d) нетерапевтического ингибирования активности SHP2 in vitro;
(e) нетерапевтического ингибирования пролиферации опухолевых клеток in vitro; или
(f) лечения заболеваний или состояний, связанных с аномальной SHP2.
В качестве предпочтительного варианта осуществления заболевание представляет собой рак, предпочтительно представляет собой синдром Нунана, синдром леопарда, ювенильный миеломоноцитарный лейкоз, нейробластому, меланому, острый миелоидный лейкоз, рак молочной железы, рак пищевода, рак легких, рак толстой кишки, рак головы, нейробластому, плоскоклеточную карциному головы и шеи, рак желудка, анапластическую крупноклеточную лимфому или глиобластому.
Четвертый аспект раскрытия предоставляет фармацевтическую композицию, содержащую:
(i) эффективное количество соединения формулы I или его фармацевтически приемлемой соли, энантиомера, диастереоизомера, таутомера, сольвата, изотопозамещенного производного, полиморфа, пролекарства или метаболита; и
(ii) фармацевтически приемлемый носитель.
Пятый аспект раскрытия предоставляет способ ингибирования активности SHP2, где способ включает следующую стадию: введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы I согласно раскрытию или его фармацевтически приемлемой соли, или введение субъекту, нуждающемуся в этом, эффективного количества фармацевтической композиции по настоящему изобретению.
Следует понимать, что вышеупомянутые технические характеристики раскрытия и технические характеристики, конкретно описанные ниже (такие как варианты осуществления), могут быть объединены в рамках объема раскрытия с образованием новых или предпочтительных технических решений. Для простоты они не будут описываться в настоящем документе по отдельности.
ПОДРОБНОЕ ОПИСАНИЕ РАСКРЫТИЯ
После долгих и интенсивных исследований изобретатели получили новые соединения-аллостерические ингибиторы формулы I, которые могут достигать цели ингибирования активности SHP2 путем связывания с некаталитической областью SHP2 и "блокировки" состояния самоингибирования со слабой активностью SHP2. Соединения по настоящему описанию проявляют очень хорошую биологическую активность и лекарственные свойства и имеют очень хорошие перспективы разработки лекарственных средств; они оказывают ингибирующее действие на SHP2 при очень низких концентрациях (до ≤100 нМ/л), и их ингибирующая активность довольно высока, поэтому их можно использовать для лечения заболеваний или состояний, связанных с SHP2, таких как опухоли. На основании вышеизложенного открытия изобретатели выполнили изобретение.
Термины
Если не указано иное, все технические и научные термины, используемые в настоящем документе, имеют те же значения, которые обычно понимаются специалистами в данной области, к которой принадлежит объект формулы изобретения. Если не указано иное, все патенты, патентные заявки и раскрытые материалы, цитируемые в настоящем документе, включены в настоящее раскрытие посредством ссылки во всей своей полноте.
Следует понимать, что приведенное выше краткое описание и последующее подробное описание являются иллюстративными и предназначены только для интерпретации, без какого-либо ограничения объектом раскрытия. В настоящем раскрытии, если не указано иное, множественное число также включено, когда используется единственное число. Следует отметить, что, если иное явно не указано в тексте, форма единственного числа, используемая в описании и формуле изобретения, включает форму множественного числа указанного объекта. Следует также отметить, что, если не указано иное, термин "или" используется для представления "и/или". Кроме того, термин "включать" и его грамматические варианты, такие как "содержать", "содержать в своем составе" и "иметь", не являются ограничительными и могут быть открытыми, полузакрытыми и закрытыми. Другими словами, термин "включать" и его грамматические варианты также включают значение "в основном состоять из" или "состоит из".
Определения стандартных химических терминов можно найти в справочных материалах (включая Carey и Sundberg “ADVANCED ORGANIC CHEMISTRY 4TH ED.” Тома A (2000) и B (2001), Plenum Press, New York). Если не указано иное, используются обычные методы в рамках данной области техники, например, масс-спектроскопия, ЯМР, спектроскопия в ИК и УФ/видимой области, а также фармакологические методы. Если не указано иное, используемые в настоящем описании термины, относящиеся к аналитической химии, органической синтетической химии, медицине и фармацевтической химии, известны в данной области техники. Стандартные методы можно использовать в химическом синтезе, химическом анализе, получении лекарственных средств, приготовлении и доставке, а также в лечении пациентов. Например, реакции и очистку можно проводить в соответствии с инструкциями производителя по использованию набора или способом, хорошо известным в данной области техники, или в соответствии с описанием раскрытия. Как правило, вышеупомянутые методы и способы могут быть реализованы в соответствии с обычными способами, хорошо известными в данной области техники, согласно описаниям во множестве общих и более конкретных публикаций, цитируемых и обсуждаемых в настоящем описании. В настоящем описании группы и заместители могут быть выбраны специалистом в данной области для обеспечения стабильных структурных частей и соединений.
Когда заместитель описывается обычной формулой, записанной слева направо, заместитель также включает химически эквивалентный заместитель, полученный, когда структурная формула записывается справа налево. Например, -CH2O- равно -OCH2-.
Заголовки разделов используются в настоящем документе только с целью систематизации статьи и не должны толковаться как ограничение объектом. Вся цитируемая в настоящем документе литература или части литературы, включая, помимо прочего, патенты, патентные заявки, статьи, книги, руководства по эксплуатации и документы, включены в настоящее раскрытие посредством ссылки во всей своей полноте.
Некоторым химическим группам, определенным в настоящем описании, предшествуют упрощенные символы, обозначающие общее количество атомов углерода в группе. Например, C1-C6 алкил относится к алкилу, как определено ниже, содержащему всего от 1 до 6 атомов углерода. Общее количество атомов углерода в упрощенных символах не включает атомы углерода, которые могут присутствовать в заместителях группы.
В дополнение к вышеизложенному, при использовании в описании и формуле изобретения, если специально не указано иное, следующие термины имеют следующие значения.
В раскрытии термин "галоген" относится к фтору, хлору, брому или иоду.
"Гидроксил" относится к группе -OH.
"Гидроксилалкил" относится к алкилу, как определено ниже, замещенному гидроксилом (-OH).
"Карбонил" относится к группе -C(=O)-.
"Нитрил" относится к -NO2.
"Циано" относится к -CN.
"Амино" относится к -NH2.
"Замещенный амино" относится к амино, замещенному одним или двумя из алкила, алкилкарбонила, арилалкила, гетероарилалкила, как определено ниже, например, замещенный амино может представлять собой моноалкиламино, диалкиламино, алкилациламино, арилалкиламино, гетероарилалкиламино.
"Карбоксил" относится к -COOH.
В раскрытии в качестве группы или части других групп (например, используемый в таких группах, как галогенированный (такой как фторированный, хлорированный, бромированный или иодированный) алкил) термин "алкил" относится к группе с полностью насыщенной прямой или разветвленной углеводородной цепью, состоящей только из атомов углерода и атомов водорода, например, содержащей от 1 до 12 (предпочтительно от 1 до 8, более предпочтительно от 1 до 6) атомов углерода и соединенной с остальной частью молекулы одинарной связью. Например, "алкил" включает, но не ограничивается ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-амил, 2-метилбутил, 2,2-диметилпропил, н-гексил, гептил, 2-метилгексил, 3-метилгексил, октил, нонил и децил и т.д. В случае настоящего описания термин "алкил" относится к алкильной группе, содержащей от 1 до 8 атомов углерода.
В раскрытии в качестве группы или части других групп термин "алкенил" относится к группе с прямой или разветвленной углеводородной цепью, состоящей только из атомов углерода и атомов водорода, например, содержащей от 2 до 20 (предпочтительно от 2 до 10, более предпочтительно от 2 до 6) атомов углерода, содержащей по меньшей мере одну двойную связь и связанную с остальной частью молекулы одинарной связью. Примеры включают, но не ограничиваются ими, винил, пропенил, аллил, бут-1-енил, бут-2-енил, пент-1-енил, пент-1,4-диенил и т.д.
В раскрытии в качестве группы или части других групп термин "циклический гидрокарбил" относится к стабильному неароматическому моноциклическому или полициклическому гидрокарбилу (такому как алкил, алкенил или алкинил), состоящему только из атомов углерода и атомов водорода, который может содержать конденсированную кольцевую систему, мостиковую кольцевую систему или спирокольцевую систему, содержит от 3 до 15 атомов углерода, предпочтительно содержит от 3 до 10 атомов углерода, более предпочтительно содержит от 3 до 8 атомов углерода, например, содержит 3, 4, 5, 6, 7 или 8 атомов углерода, и который является насыщенным или ненасыщенным и может быть связан с остальной частью молекулы через любой подходящий атом углерода одинарной связью. Если иное специально не указано в описании, атомы углерода в циклическом гидрокарбиле могут быть необязательно окислены. Варианты осуществления циклического гидрокарбила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклооктил, 1H-инденил, 2,3-дигидроинденил, 1,2,3,4-тетрагидро-нафтил, 5,6,7,8-тетрагидро-нафтил, 8,9-дигидро-7H-бензоциклогептен-6-ил, 6,7,8,9-тетрагидро-5H-бензоциклогептенил, 5,6,7,8,9,10-гексагидро-бензоциклооктенил, флуоренил, бицикло[2.2.1]гептил, 7,7-диметил-бицикло[2.2.1]гептил, бицикло[2.2.1]гептенил, бицикло[2.2.2]октил, бицикло[3.1.1]гептил, бицикло[3.2.1]октил, бицикло[2.2.2]октенил, бицикло[3.2.1]октенил, адамантил, октагидро-4,7-метилен-1H-инденил и октагидро-2,5-метилен-дициклопентадиенил и т.д.
В раскрытии в качестве группы или части других групп термин "гетероциклил" относится к стабильной 3-20-членной неароматической циклической группе, состоящей из от 2 до 14 атомов углерода (например, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 атомов углерода) и от 1 до 6 гетероатомов, выбранных из азота, фосфора, кислорода или серы. Если иное специально не указано в описании, гетероциклил может представлять собой моноциклическую кольцевую систему, бициклическую кольцевую систему, трициклическую кольцевую систему или кольцевую систему с большим количеством колец и может содержать конденсированную кольцевую систему, мостиковую кольцевую систему или спиро-кольцевую систему; атомы азота, фосфора или серы в гетероциклиле необязательно могут быть окислены; атомы азота в гетероциклиле необязательно могут быть кватернизованы; и гетероциклил может быть частично или полностью насыщенным. Гетероциклил может быть связан с остальной частью молекулы через атом углерода или гетероатом посредством одинарной связи. В конденсированном кольце, содержащем гетероциклил, одно или несколько колец могут представлять собой арил или гетероарил, как определено ниже, при условии, что точкой соединения между группой и остальной частью молекулы является неароматический кольцевой атом. Для целей настоящего раскрытия гетероциклил предпочтительно представляет собой стабильную 4-11-членную неароматическую моноциклическую, бициклическую, мостиковую или спиро-кольцевую группу, содержащую от 1 до 3 гетероатомов, выбранных из азота, кислорода или серы, более предпочтительно стабильную 4-8-членную неароматическую моноциклическую, бициклическую, мостиковую или спиро-кольцевую группу, содержащую от 1 до 3 гетероатомов, выбранных из азота, кислорода или серы. Примеры гетероциклилов включают, но не ограничиваются ими: пирролидинил, морфолинил, пиперазинил, гомопиперазинил, пиперидинил, тиоморфолинил, 2,7-диаза-спиро[3.5]нонан-7-ил, 2-окса-6-аза-спиро[3.3]гептан-6-ил, 2,5-диаза-бицикло[2.2.1]гептан-2-ил, азациклобутанил, пиранил, тетрагидропиранил, тиопиранил, тетрагидрофуранил, оксазинил, диоксоланил, тетрагидроизохинолинил, декагидроизолидизохинолинил, имидазолинил, имидазолидинил, хинолизинил, тиазолидинил, изотиазолидинил, изоксазолидинил, дигидроиндолил, октагидроиндолил, октагидроизоиндолил, пирролидинил, пиразолидинил, фталимидо и т.д.
В раскрытии в качестве группы или части других групп термин "арил" относится к группе конъюгированной углеводородной кольцевой системы, содержащей от 6 до 18 атомов углерода (предпочтительно, содержащей от 6 до 10 атомов углерода, например, 6, 7, 8, 9 или 10 атомов углерода). Для целей настоящего раскрытия арил может представлять собой моноциклическую кольцевую систему, бициклическую кольцевую систему, трициклическую кольцевую систему или кольцевую систему с большим количеством колец и может быть конденсирован с циклическим гидрокарбилом или гетероциклилом, как определено выше, при условии, что арил и остальная часть молекулы связаны через атом ароматического кольца одинарной связью. Примеры арилов включают, но не ограничиваются ими, фенил, нафтил, антрацил, фенантренил, флуоренил, 2,3-дигидро-1H-изоиндолил, 2-бензоксазолинон, 2H-1,4-бензоксазин-3(4H)-он-7-ил и т.д.
В раскрытии термин "арилалкил" относится к алкилу, как определено выше, который замещен арилом, как определено выше.
В раскрытии в качестве группы или части других групп термин "гетероарил" относится к 5-16-членной группе сопряженной кольцевой системы, содержащей от 1 до 15 атомов углерода (предпочтительно, содержащей от 1 до 10 атомов углерода, например, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода) и от 1 до 6 гетероатомов, выбранных из азота, кислорода или серы. Если иное специально не указано в описании, гетероарил может представлять собой моноциклическую кольцевую систему, бициклическую кольцевую систему, трициклическую кольцевую систему или кольцевую систему с большим количеством колец, и может быть конденсирован с циклоалкилом или гетероциклилом при условии, что гетероарил и остальная часть молекулы связаны через атом ароматического кольца одинарной связью. Атомы азота, углерода или серы в гетероариле необязательно могут быть окислены; атомы азота в гетероариле могут быть необязательно кватернизованы. Для целей настоящего описания гетероарил предпочтительно представляет собой стабильную 5-12-членную ароматическую группу, содержащую от 1 до 5 гетероатомов, выбранных из азота, кислорода или серы, и более предпочтительно представляет собой стабильную 5-10-членную ароматическую группу, содержащую от 1 до 4 гетероатомов, выбранных из азота, кислорода или серы, или 5-6-членную ароматическую группу, содержащую от 1 до 3 гетероатомов, выбранных из азота, кислорода или серы. Примеры гетероарилов включают, но не ограничиваются ими, тиофенил, имидазолил, пиразолил, тиазолил, оксазолил, оксадиазолил, изоксазолил, пиридил, пиримидинил, пиразинил, пиридазинил, бензимидазолил, бензопиразолил, индолил, фуранил, пирролил, триазолил, тетразолил, триазинил, индолизинил, изоиндолил, индазолил, изоиндазолил, пуринил, хинолинил, изохинолинил, диазанафталенил, нафтиридинил, хиноксалинил, птеридинил, карбазолил, карболинил, фенантридинил, фенантролинил, акридинил, феназинил, изотиазолил, бензотиазолил, бензотиофенил, оксатриазолил, циннолинил, хиназолил, фенилтио, индолизинил, фенантролинил, изоксазолил, феноксазинил, фенотиазинил, 4,5,6,7-тетрагидробензо[b]тиофенил, нафтопиридинил, [1,2,4]триазоло[4,3-b]пиридазин, [1,2,4]триазоло[4,3-a]пиразин, [1,2,4]триазоло[4,3-c]пиримидин, [1,2,4]триазоло[4,3-a]пиридин, имидазо[1,2-a]пиридин, имидазо[1,2-b]пиридазин, имидазо[1,2-a]пиразин и т.д.
В раскрытии термин "гетероарилалкил" относится к алкилу, как определено выше, который замещен гетероарилом, как определено выше.
В раскрытии "необязательный" или "необязательно" означает, что описанное далее событие или условие может или не может произойти, и такое описание включает в себя как наступление, так и ненаступление события или условия. Например, "необязательно замещенный арил" означает арил, замещенный или незамещенный, и такое описание включает как замещенный арил, так и незамещенный арил. "Необязательный" заместитель, используемый в формуле изобретения и описании раскрытия, выбран из алкила, алкенила, алкинила, галогена, галогеналкила, галогеналкенила, галогеналкинила, циано, нитро, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного циклического гидрокарбила или необязательно замещенного гетероциклила.
"SHP2" относится к "фосфатазе Src Homolgy-2", также именуемой SH-PTP2, SH-PT3, Syp, PTP1D, PTP2C, SAP-2 или PTPN11.
В контексте настоящего раскрытия термины "часть", "структурная часть", "химическая часть", "группа", "химическая группа" относятся к конкретной части или функциональной группе в молекуле. Химическая часть обычно считается химическим структурным элементом, встроенным в молекулу или присоединенным к ней.
"Стереизомер" относится к соединению, состоящему из одинаковых атомов, связанных одинаковыми связями, но имеющему разные трехмерные структуры. Настоящее раскрытие будет охватывать различные стереоизомеры и их смеси.
Если соединение согласно настоящему раскрытию содержит олефиновую двойную связь, если не указано иное, предполагается, что соединение согласно настоящему раскрытию включает E- и Z-геометрические изомеры.
"Таутомер" относится к изомеру, образованному переносом протона от одного атома молекулы к другому атому той же молекулы. Все таутомерные формы соединения согласно настоящему раскрытия также будут включены в объем настоящего раскрытия.
Соединение согласно раскрытию или его фармацевтически приемлемая соль может содержать один или несколько хиральных атомов углерода и, таким образом, может давать энантиомеры, диастереомеры и другие стереоизомерные формы. Каждый хиральный атом углерода может быть определен как (R)- или (S)- на основе стереохимии. Раскрытие предназначено для включения всех возможных изомеров, а также их рацематов и их оптически чистых форм. Соединение согласно раскрытию может быть получено с использованием рацемата, диастереомера или энантиомера в качестве исходного вещества или промежуточного соединения. Оптически активные изомеры могут быть получены с использованием хиральных синтонов или хиральных реагентов или разделены обычными методами, такими как кристаллизация и хиральная хроматография.
Традиционные методы получения/разделения отдельных изомеров включают хиральный синтез из подходящих оптически чистых предшественников или разделение рацематов (или рацематов солей или производных) с использованием, например, хиральной высокоэффективной жидкостной хроматографии, например, см. Gerald Gübitz и Martin G. Schmid (Eds.), Chiral Separations, Methods and Protocols, Methods in Molecular Biology, Vol. 243, 2004; A.M. Stalcup, Chiral Separations, Annu. Rev. Anal. Chem. 3:341-63, 2010; Fumiss с соавт. (под ред.), VOGEL’S ENCYCLOPEDIA OF PRACTICAL ORGANIC CHEMISTRY 5.sup.TH ED., Longman Scientific and Technical Ltd., Essex, 1991, 809-816; Heller, Acc. Chem. Res. 1990, 23, 128.
Раскрытие также включает все подходящие изотопные варианты соединений согласно настоящему раскрытию или их фармацевтически приемлемых солей. Изотопные варианты соединений согласно настоящему раскрытию или их фармацевтически приемлемых солей определяются как те, в которых по меньшей мере один атом заменен атомом, имеющим тот же атомный номер, но атомную массу, отличную от атомной массы, часто встречающейся в природе. Изотопы, которые могут быть включены в соединения согласно настоящему раскрытию и их фармацевтически приемлемые соли, включают, но не ограничиваются ими, H, C, N и O, например, 2H, 3H, 11C, 13C, 14C, 15N, 17O, 18O, 35S, 18F, 36Cl и 125I. Подходящие изотопные варианты соединений или их фармацевтически приемлемых солей согласно настоящему раскрытию могут быть получены обычными методами с использованием соответствующих изотопных вариантов подходящих реагентов.
В раскрытии термин "фармацевтически приемлемая соль" включает фармацевтически приемлемые кислотно-аддитивные соли и фармацевтически приемлемые основно-аддитивные соли.
"Фармацевтически приемлемая кислотно-аддитивная соль" относится к соли, образованной с неорганической кислотой или органической кислотой, которая может сохранять биологическую эффективность свободного основания без других побочных эффектов. Соли неорганических кислот включают, но не ограничиваются ими, гидрохлорид, гидробромид, сульфат, нитрат, фосфат и т.д.; соли органических кислот включают, но не ограничиваются ими, формиат, ацетат, 2,2-дихлорацетат, трифторацетат, пропионат, гексаноат, каприлат, капрат, ундециленат, гликолят, глюконат, лактат, себацинат, адипат, глутарат, малонат, оксалат, малеат, сукцинат, фумарат, тартрат, цитрат, пальмитат, стеарат, олеат, циннамат, лаурат, малат, глутамат, пироглутамат, аспартат, бензоат, метансульфонат, бензолсульфонат, п-толуолсульфонат, альгинат, аскорбат, салицилат, 4-аминосалицилат, нафталиндисульфонат. Эти соли могут быть получены способами, известными в данной области техники.
"Фармацевтически приемлемые основно-аддитивные соли" относятся к соли, образованной с неорганическим основанием или органическим основанием, которая может сохранять биологическую эффективность свободной кислоты без других побочных эффектов. Соли, полученные из неорганических оснований, включают, но не ограничиваются ими, натриевую соль, калиевую соль, литиевую соль, аммониевую соль, кальциевую соль, магниевую соль, железную соль, цинковую соль, медную соль, марганцевую соль, алюминиевую соль и т.д. Предпочтительными неорганическими солями являются аммониевая соль, натриевая соль, калиевая соль, кальциевая соль и магниевая соль. Соли, полученные из органических оснований, включают, но не ограничиваются ими, следующие соли: соли первичных аминов, вторичных аминов и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, такие как соли аммиака, изопропиламина, триметиламина, диэтиламина, триэтиламина, трипропиламина, этаноламина, диэтаноламина, триэтаноламина, диметилэтаноламина, 2-диметиламиноэтанола, 2-диэтиламиноэтанола, дициклогексиламина, лизина, аргинина, гистидина, кофеина, прокаина, холина, бетаина, этилендиамина, глюкозамина, метилглюкозамина, теобромина, пурина, пиперазина, пиперидина, N-этилпиперидина, полиаминовой смолы и т.д. Предпочтительные органические основания включают изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин. Эти соли могут быть получены способами, известными в данной области техники.
В раскрытии "фармацевтическая композиция" относится к составу соединения согласно настоящему раскрытию и среде, общепринятой в данной области техники для доставки биологически активного соединения млекопитающему (например, человеку). Среда включает фармацевтически приемлемый носитель. Назначение фармацевтической композиции состоит в том, чтобы способствовать введению в живые организмы, что облегчает абсорбцию активных ингредиентов и, таким образом, проявляет биологическую активность.
Термин "фармацевтически приемлемый" в контексте настоящего раскрытия относится к веществу (такому как носитель или разбавитель), которое не влияет на биологическую активность или свойства соединения согласно настоящему раскрытию и является относительно нетоксичным, то есть веществу, которое можно вводить индивидууму, не вызывая побочных биологических реакций или нежелательного взаимодействия с любыми компонентами, содержащимися в композиции.
В раскрытии "фармацевтически приемлемый эксципиент" включает, но не ограничивается ими, любой адъювант, носитель, эксципиент, глидант, подсластитель, разбавитель, консервант, краситель/пигмент, корригирующее вещество, поверхностно-активное вещество, смачивающий агент, диспергирующее вещество, суспендирующий агент, стабилизатор, изотонический агент, растворитель или эмульгатор, одобренный соответствующим государственным регулирующим органом как приемлемый для применения людьми или домашними животными.
"Опухоль" в раскрытии включает, но не ограничивается ими, синдром Нунана, синдром леопарда, ювенильный миеломоноцитарный лейкоз, нейробластому, саркому, меланому, суставную хондрому, холангиому, лейкемию, рак молочной железы, стромальную опухоль желудочно-кишечного тракта, гистиоцитарную лимфому, немелкоклеточный рак легких, мелкоклеточный рак легких, рак пищевода, рак поджелудочной железы, плоскоклеточный рак легких, аденокарцинома легких, рак молочной железы, рак простаты, рак печени, рак кожи, рак эпителиальных клеток, рак шейки матки, рак яичников, рак кишечника, рак носоглотки, рак мозга, рак кости, рак почки, рак ротовой полости/рак головы, нейробластому, плоскоклеточный рак головы и шеи, анапластическую крупноклеточную лимфому или глиобластому и другие заболевания.
Термины "профилактический", "предотвращать" и "профилактика" в контексте настоящего раскрытия включают уменьшение вероятности возникновения или ухудшения заболевания или состояния у пациента.
Термин "лечение" и другие подобные синонимы в контексте настоящего раскрытия включают следующие значения:
(i) профилактика возникновения заболевания или состояния у млекопитающих, особенно когда такие млекопитающие восприимчивы к заболеванию или состоянию, но у них не диагностировано заболевание или состояние;
(ii) подавление заболевания или состояния, то есть подавление развития заболевания или состояния;
(iii) облегчение заболевания или состояния, то есть уменьшение степени заболевания или состояния; или
(iv) облегчение симптомов, вызванных заболеванием или состоянием.
Термин "эффективное количество", "терапевтически эффективное количество" или "фармацевтически эффективное количество" в контексте настоящего раскрытия относится к количеству по меньшей мере одного лекарственного средства или соединения, достаточному для облегчения одного или нескольких симптомов заболевания или заболевания, подвергающегося лечению, до определенной степени после введения. Результатом может быть ослабление и/или ремиссия признаков, симптомов или причин или любые другие желаемые изменения в биологической системе. Например, "эффективное количество" для лечения представляет собой количество композиции, содержащей соединение, раскрытое в настоящем документе, необходимое для обеспечения клинически значимого эффекта облегчения заболевания. Для определения эффективного количества, подходящего для каждого отдельного случая, можно использовать такие методы, как испытания с повышением дозы.
Термины "прием", "применение", "введение" и т.п. относятся к способу доставки соединения или композиции в желаемое место для биологического действия. Эти способы включают, но не ограничиваются ими, пероральный путь, трансдуоденальный путь, парентеральную инъекцию (включая внутривенную, подкожную, внутрибрюшинную, внутримышечную, внутриартериальную инъекцию или инфузию), местное применение и трансректальное введение. Специалист в данной области техники знаком с методами введения, которые можно использовать для соединений и способов, описанных в настоящем документе, например, описанными в Goodman и Gilman, The Pharmacological Basis of Therapeutics, current ed.; Pergamon; и Remington’s, Pharmaceutical Sciences (current edition), Mack Publishing Co., Easton, Pa. В предпочтительном варианте осуществления обсуждаемые в настоящем документе соединения и композиции вводят перорально.
Термины "комбинация лекарственных средств", "совместное введение лекарственных средств", "комбинированное лекарственное средство", "применение других методов лечения", "введение других терапевтических агентов" и тому подобное, используемые в настоящем документе, относятся к медицинскому лечению, полученному путем смешивания или объединения более чем одного активного ингредиента, и включают фиксированные и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" относится к одновременному введению по меньшей мере одного соединения, описанного в настоящем документе, и по меньшей мере одного синергического лекарственного средства пациенту в форме единого целого или одной дозированной формы. Термин "нефиксированная комбинация" относится к одновременному введению, совместному введению или последовательному введению с различными интервалами по меньшей мере одного соединения, описанного в настоящем документе, и по меньшей мере одного синергетического состава пациенту в форме отдельных единиц. Это также относится к коктейльной терапии, такой как введение трех или более активных ингредиентов.
Специалист в данной области техники также должен понимать, что в способе, описанном ниже, функциональная группа промежуточного соединения может нуждаться в защите соответствующей защитной группой. Такие функциональные группы включают гидроксил, амино, сульфидрил и карбоновую кислоту. Подходящие защитные группы для гидроксила включают триалкилсилил или диарилалкилсилил (например, трет-бутилдиметилсилил, трет-бутилдифенилсилил или триметилсилил), тетрагидропиранил, бензил и т.д. Подходящие защитные группы для амино, амидино и гуанидино включают трет-бутоксикарбонил, бензилоксикарбонил и т.д. Подходящие защитные группы для сульфидрила включают -C(O)-R” (где R” представляет собой алкил, арил или аралкил), п-метоксибензил, тритил, и т.д. Подходящие карбокси-защитные группы включают сложные алкиловые, ариловые или аралкиловые эфиры.
Защитные группы могут быть введены и удалены в соответствии со стандартными методами, известными специалистам в данной области техники, и как описано в настоящем документе. Использование защитных групп подробно описано в Greene, T. W. и P. G. M. Wuts, Protective Groups in Organi Synthesis, (1999), 4-е изд., Wiley. Защитные группы также могут представлять собой полимерные смолы.
Получение соединения формулы I
Соединение формулы I, предоставленное настоящим описанием, может быть получено следующим способом: проведение реакции нуклеофильного замещения между формулой Ib и формулой Ic с получением формулы Id; проведение реакции замещения между формулой Id и формулой Ie с получением формулы If; и удаление защитной группы в формуле If при помощи кислоты с получением соединения Формулы I:
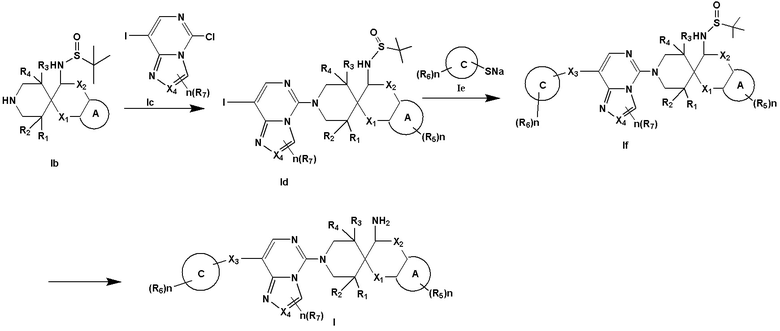
В формуле определение каждой группы такое, как описано выше.
Фармакология и применение
Фосфатаза Src Homology-2 (SHP2) представляет собой протеинтирозинфосфатазу, кодируемую геном PTPN11, которая промотирует различные функции клетки, включая пролиферацию, дифференциацию, поддержание клеточного цикла и миграцию. SHP2 участвует в передаче сигналов через Ras-митоген-активируемую протеинкиназу, JAK-STAT или путь фосфоинозитид-3-киназа-AKT. SHP2 опосредует активацию рецепторных тирозинкиназ, таких как ErbB1 и ErbB2, и Erk1 и Erk2 MAP-киназ c-Met.
SHP2 имеет два N-концевых домена Src Homolgy-2 (N-SH2 и C-SH2), каталитический домен (PTP) и C-концевой "хвост". Два домена SH2 контролируют субклеточную локализацию и функциональную регуляцию SHP2. Молекула существует в неактивной конформации, подавляя свою собственную активность через сеть связывания, включающую остатки из доменов N-SH2 и PTP. В ответ на стимуляцию фактора роста SHP2 связывается со специфическими сайтами фосфорилирования тирозина, такими как Gab1 и Gab2, на докинг-белках через SH2-домен SHP2. Это вызывает конформационные изменения, приводящие к активации SHP2.
Мутации в PTPN11 были идентифицированы при различных заболеваниях человека, таких как синдром Нунана, синдром леопарда, ювенильный миеломоноцитарный лейкоз, нейробластома, меланома, острый миелоидный лейкоз и раковые заболеания молочной железы, легких и толстой кишки. SHP2 является важной молекулой нисходящей передачи сигнала для различных рецепторных тирозинкиназ, включая рецепторы тромбоцитарного фактора роста (PDGF-R), фактора роста фибробластов (FGF-R) и эпидермального фактора роста (EGF-R). SHP2 также является важной молекулой нисходящей передачи сигнала, которая активирует путь митоген-активированной протеинкиназы (MAP), что может привести к клеточной трансформации (необходимое условие для развития рака). Нокдаун SHP2 значительно подавляет рост клеток линий клеток рака легких, имеющих мутации SHP2 или транслокацию EML4/ALK, а также EGFR-амплифицированного рака молочной железы и пищевода. SHP2 также представляет собой регулирующий последующие звенья сигнальных каскадов ген активации онкогенов при раке желудка, анапластической крупноклеточной лимфоме и глиобластоме.
Синдром Нунана (NS) и синдром леопарда (LS) - Мутация PTPNll вызывает LS (синдром множественных пигментированных невусов, аномальная проводимость ЭКГ, слишком большое расстояние между глазами, стеноз легочного клапана, аномальные гениталии, задержка роста, нейросенсорная потеря слуха) и NS (включая врожденные пороки сердца, черепно-лицевую деформацию и низкий рост). Эти два расстройства являются частью семейства аутосомно-доминантного синдрома, вызванного герминальными мутациями в компонентах пути митоген-активируемой протеинкиназы RAS/RAF/MEK/ERK (необходимого для нормального роста и дифференцировки клеток). Аномальная регуляция этого пути имеет далеко идущие последствия, особенно на развитие сердца, приводя к множеству аномалий, включая вальвулосептальные дефекты и/или гипертрофическую кардиомиопатию (ГКМП). Было определено, что нарушение сигнального пути MAPK является важным для этих расстройств, и некоторые гены-кандидаты, которые следуют этому пути, были идентифицированы у людей, включая мутации в KRAS, NRAS, SOS1, RAF1, BRAF, MEK1, MEK2, SH0C2 и CBL. Наиболее часто мутирующим геном при NS и LS является PTPN11. Герминальные мутации PTPN11 (SHP2) были обнаружены примерно в 50% случаев NS и почти у всех пациентов с LS, имеющих определенные характеристики NS. Для NS наиболее частыми мутациями являются Y62D и Y63C в белке. Эти две мутации влияют на конформацию некаталитической активности SHP2, но не мешают связыванию фосфатазы и ее фосфорилированного сигнального лиганда.
Ювенильный миеломоноцитарный лейкоз (JMML) - Соматические мутации в PTPN11 (SHP2) встречаются примерно у 35% пациентов с JMML (детским миелодиспластическим заболеванием (MPD)). Эти мутации с приобретением функции обычно представляют собой точечные мутации в домене N-SH2 или в домене фосфатазы, которые предотвращают самоингибирование между каталитическим доменом и доменом N-SH2, становятся причиной активности SHP2.
Острый миелоидный лейкоз - Мутации PTPNll были выявлены примерно в 10% случаев острых лейкозов у детей, таких как миелодиспластический синдром (МДС), примерно в 7% случаев острого лимфобластного лейкоза В-клеток (В-ОЛЛ) и примерно в 4% случаев острого миелоидного лейкоза (ОМЛ).
Мутации при NS и лейкозе вызывают изменения аминокислот на границе раздела, образованного доменами N-SH2 и PTP в самоингибирующейся конформации SHP2, нарушают ингибирующие внутримолекулярные взаимодействия и приводят к гиперактивности каталитического домена.
SHP2 действует как положительный регулятор в передаче сигналов рецепторной тирозинкиназы (RTK). Раковые образования, содержащие изменения RTK (EGFRamp, Her2amp, FGFRamp, Metamp, транслоцированная/активированная RTK, а именно ALK, BCR/ABL), включают рак пищевода, рак молочной железы, рак легких, рак толстой кишки, рак желудка, глиому и рак головы и шеи.
Рак пищевода (или эзофагеальный рак) представляет собой злокачественное заболевание пищевода. Существует множество подтипов рака пищевода, в основном плоскоклеточный рак (˂50%) и аденокарцинома. Уровень экспрессии RTK выше при аденокарциноме пищевода и плоскоклеточной карциноме. Следовательно, ингибитор SHP2 согласно раскрытию может быть использован для инновационных стратегий лечения.
Рак молочной железы является важным типом рака и основной причиной смерти женщин, когда у пациентов развивается устойчивость к существующим лекарственным средствам. Существует четыре основных подтипа рака молочной железы, включая просветной A, просветной B, Her21ik и тройной отрицательный/базальный. Тройной отрицательный рак молочной железы (TNBC) представляет собой инвазивный рак молочной железы, для которого отсутствуют специфические целевые методы лечения. Рецептор эпидермального фактора роста I (EGFR) был продемонстрирован в качестве многообещающей мишени при TNBC. Ингибирование HER2 и EGFR через SHP2 может быть многообещающим методом лечения рака молочной железы.
Рак легких - НМРЛ в настоящее время является важной причиной смертности от рака. На его долю приходится около 85% случаев рака легких (в основном аденокарцинома и плоскоклеточный рак). Хотя цитотоксическая химиотерапия по-прежнему является важной частью лечения, типы таргетной терапии, основанные на генетических изменениях (таких как EGFR и ALK) в опухолях, с большей вероятностью пользуются преимуществом за счет таргетной терапии.
Рак толстой кишки - Известно, что от 30% до 50% колоректальных опухолей имеют мутировавшие (аномальные) KRAS, а мутации BRAF встречаются в 10-15% случаев колоректального рака. Для подгруппы пациентов, у которых было показано, что колоректальные опухоли сверхэкспрессируют EGFR, эти пациенты демонстрируют благоприятный клинический ответ на терапию против EGFR.
Рак желудка является одним из самых популярных видов рака. Аномальная экспрессия тирозинкиназы (что отражается в аномальном фосфорилировании тирозина в клетках рака желудка) известна в данной области техники. Три рецепторные тирозинкиназы, а именно c-met (рецептор HGF), FGF рецептор 2 и erbB2/neu, часто ампилифицированы при раке желудка. Следовательно, разрушение различных сигнальных путей может способствовать прогрессированию различных типов рака желудка.
Нейробластома представляет собой педиатрическую опухоль развивающейся симпатической нервной системы, на которую приходится около 8% всех онкологических заболеваний у детей. Было высказано предположение, что геномные изменения гена киназы анапластической лимфомы (ALK) способствуют патогенезу нейробластомы.
Плоскоклеточная карцинома головы и шеи (SCCHN). Высокие уровни экспрессии EGFR связаны с плохим прогнозом и устойчивостью к лучевой терапии при различных формах рака, чаще всего при плоскоклеточной карциноме головы и шеи (SCCHN). Блокирование сигнала EGFR приводит к ингибированию стимуляции рецептора и снижению клеточной пролиферации, инвазии и метастазирования. Следовательно, EGFR являются лучшей мишенью новой противоопухолевой терапии при SCCHN.
Настоящее раскрытие относится к соединениям, способным ингибировать активность SHP2. В раскрытии также предложен способ получения соединения согласно раскрытию и фармацевтический препарат, содержащий это соединение. Другой аспект раскрытия относится к способу лечения SHP2-опосредованного заболевания или состояния, который включает стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I согласно раскрытию.
В некоторых вариантах осуществления раскрытие относится к способу, описанному выше, в котором SHP2-опосредованное заболевание или состояние выбрано из следующих видов рака, но не ограничивается ими: JMML, AML, MDS, B-ALL, нейробластома, рак пищевода, рак молочной железы, рак легких, рак толстой кишки, рак желудка, рак головы и шеи.
Соединение согласно раскрытию также можно использовать для лечения других заболеваний или состояний, связанных с аномальной активностью SHP2. Следовательно, в качестве предпочтительного варианта осуществления изобретение относится к способу лечения заболевания или состояния, выбранного из: NS, LS, JMML, AML, MDS, B-ALL, нейробластомы, рака пищевода, рака молочной железы, рака легких, рака толстой кишки, рака желудка, рака головы и шеи.
Ингибиторы SHP2 согласно настоящему раскрытию можно комбинировать с другим фармакологически активным соединением или с двумя или более другими фармакологически активными соединениями, особенно при лечении рака. Например, соединение формулы (I) согласно настоящему раскрытию или его фармацевтически приемлемая соль можно вводить одновременно, последовательно или отдельно в комбинации с одним или несколькими веществами, выбранными из: химиотерапевтических агентов, таких как ингибиторы митоза, такие как таксан, алкалоиды барвинка, паклитаксел, доцетаксел, винкристин, винбластин, винорелбин или винфлунин, и других противораковых агентов, таких как цисплатин, 5-фторурацил или 5-фтор-2-4(1H,3H)-пиримидиндион (5FU), флутамид или гемциатбин.
Некоторые комбинации могут обеспечить значительные преимущества в терапии, включая синергетическую активность.
В некоторых вариантах осуществления раскрытие относится к описанному выше способу, в котором соединение вводят парентерально.
В некоторых вариантах осуществления раскрытие относится к описанному выше способу, в котором соединение вводят внутримышечно, внутривенно, подкожно, перорально, ингаляционно, интратекально, местно или интраназально.
В некоторых вариантах осуществления раскрытие относится к описанному выше способу, в котором соединение вводят системно.
В некоторых вариантах осуществления раскрытие относится к описанному выше способу, в котором пациент представляет собой млекопитающее.
В некоторых вариантах осуществления раскрытие относится к описанному выше способу, в котором пациент представляет собой примата.
В некоторых вариантах осуществления раскрытие относится к описанному выше способу, в котором пациент представляет собой человека.
В некоторых вариантах осуществления раскрытие относится к способу лечения SHP2-опосредованного заболевания или состояния, где способ включает следующую стадию: введение пациенту, который в этом нуждается, комбинации терапевтически эффективного количества химиотерапевтического агента и терапевтически эффективного количества соединения формулы I согласно раскрытию.
Основные преимущества раскрытия включают:
1. Раскрытие предоставляет соединение формулы I.
2. Раскрытие предоставляет ингибитор SHP2 новой структуры, его получение и применение, причем ингибитор обладает высокой ингибирующей активностью в отношении SHP2.
3. Раскрытие предоставляет фармацевтическую композицию для лечения заболеваний или состояний, связанных с SHP2.
Раскрытие дополнительно описывается ниже в сочетании с конкретными примерами. Следует понимать, что эти примеры используются только для иллюстрации настоящего раскрытия, а не для ограничения объема настоящего раскрытия. Экспериментальные способы без конкретных условий в следующих примерах обычно следуют обычным условиям или условиям, рекомендованным производителем. Если не указано иное, проценты и части рассчитываются по массе.
Исходные вещества, используемые в следующих примерах, могут быть приобретены у химических дистрибьюторов, таких как Aldrich, TCI, Alfa Aesar, Bide, Energy и т.д., или могут быть синтезированы известными способами.
Значения английских аббревиатур, используемых в следующих примерах, описаны в следующей таблице.
В следующих примерах ледяная баня относится к температуре от -5°C до 0°C, комнатная температура относится к температуре от 10°C до 30°C, а температура кипения с обратным холодильником обычно относится к температуре кипения растворителя при нормальном давлении. Ночная реакция обычно длится 8-15 часов. В следующих примерах все операции без определенной рабочей температуры выполняют при комнатной температуре.
В следующих примерах разделение и очистку промежуточных соединений и конечных продуктов выполняют с помощью разделения на хроматографической колонке с нормальной фазой или с обращенной фазой или другими подходящими методами. В колонках для флэш-хроматографии с нормальной фазой в качестве подвижных фаз используют этилацетат и н-гексан или метанол и метиленхлорид. В препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) с обращенной фазой используют колонку C18 и УФ при 214 нм и 254 нм с подвижными фазами A (вода и 0,1% муравьиной кислоты) и B (ацетонитрил) или подвижными фазами A (вода и 0,1% бикарбоната аммония) и B (ацетонитрил).
В примерах: прибор ЖХМС: насос Agilent 1260, УФ-детектор: Agilent 1260 DAD масс-спектрометр API 3000;
Хроматографическая колонка: Waters Sunfire C18, 4,6 × 50 мм, 5 мкм;
Подвижные фазы: A-H2O (0,1% HCOOH); B-ацетонитрил
Прибор ЯМР: Bruker Ascend 400M (1H ЯМР: 400 МГц; 13C ЯМР: 100 МГц).
Синтез промежуточного соединения A1: (R)-N-((S)-1,3-дигидроспиро[инден-2,4’-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида
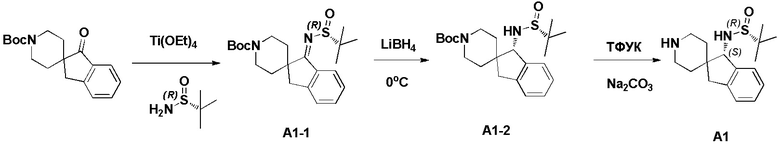
Стадия 1: Сложный трет-бутиловый эфир 1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (1,51 г, 5 ммоль), тетраэтилтитанат (6,84 г, 30 ммоль) и (R)-(+)-трет-бутилсульфинамид (2,41 г, 20 ммоль) последовательно добавляли в сухую одногорлую колбу объемом 100 мл, и смесь перемешивали при нагревании и кипячении с обратным холодильником в течение 15 часов. После охлаждения реакционной системы до комнатной температуры к реакционному остатку добавляли насыщенный рассол (60 мл), после чего полученную смесь перемешивали в течение 15 минут и затем фильтровали через диатомит. Водную смесь экстрагировали этилацетатом (3 × 80 мл). Органическую фазу сушили над Na2SO4 и фильтровали, и летучие вещества удаляли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент от 0 до 30% этилацетата:петролейный эфир) с получением сложного трет-бутилового эфира (R, Z)-1-((трет-бутилсульфинил)имино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A1-1, 1,61 г, выход: 80%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,34 (д, J=7,2 Гц, 1H), 7,48-7,39 (м, 1H), 7,35-7,28 (м, 2H), 4,21-3,92 (м, 2H), 3,00 (с, 2H), 2,88 (т, J=11,9 Гц, 2H), 2,00-1,80 (м, 2H), 1,48-1,30 (м, 11H), 1,24 (д, J=13,1 Гц, 9H); ЖХМС: м/з 405,1 [M+H]+
Стадия 2: Сложный трет-бутиловый эфир (R, Z)-1-((трет-бутилсульфинил)имино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'- карбоновой кислоты (A1-1, 0,802 г, 2 ммоль) и ТГФ (10 мл) последовательно добавляли в сухую одногорлую колбу объемом 100 мл, смесь охлаждали до 0°C, и затем добавляли боргидрид лития (66 мг, 3 ммоль). Полученную смесь затем перемешивали в течение 1 часа. Медленно добавляли метанол, чтобы погасить избыток боргидрида. Реакционный раствор фильтровали и концентрировали, и летучие вещества удаляли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент от 0 до 50% этилацетата:петролейный эфир) с получением сложного трет-бутилового эфира (S)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A1-2, 0,63 г, выход: 78%) в виде светло-желтого твердого вещества.
1H ЯМР (400 МГц, ДМСО) δ 7,30-7,15 (м, 4H), 5,64 (д, J=10,5 Гц, 1H), 4,38 (д, J=10,5 Гц, 1H), 3,86 (с, 2H), 3,05 (д, J=15,8 Гц, 1H), 2,87 (с, 2H), 2,62 (д, J=15,8 Гц, 1H), 1,89 (с, 1H), 1,61-1,35 (м, 12H), 1,27-1,10 (м, 11H); ЖХМС: м/з 407,1 [M+H]+
Стадия 3: Сложный трет-бутиловый эфир (S)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A1-2, 0,406 г, 1 ммоль), дихлорметан (5 мл) и ТФУК (1 мл) последовательно добавляли в сухую одногорлую колбу объемом 50 мл, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор Na2CO3 до тех пор, пока pH не достигал 7, и водную смесь экстрагировали с помощью ДХМ (3 × 30 мл). Объединенную органическую фазу промывали рассолом, сушили над Na2SO4 и фильтровали, и летучие вещества удаляли при пониженном давлении. Полученный остаток охлаждали с получением (R)-N-((S)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (A1, 0,183 г, выход 70%) в виде бесцветного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,26-7,11 (м, 5H), 4,45 (д, J=10,1 Гц, 1H), 3,77 (с, 1H), 3,18 (с, 2H), 3,04 (д, J=15,9 Гц, 3H), 2,67 (д, J=15,8 Гц, 1H), 2,20 (тд, J=12,7, 3,5 Гц, 1H), 1,82 (т, J=11,1 Гц, 1H), 1,61 (д, J=12,9 Гц, 1H), 1,34-1,11 (м, 10H);
ЖХМС: м/з 307,1 [M+H]+
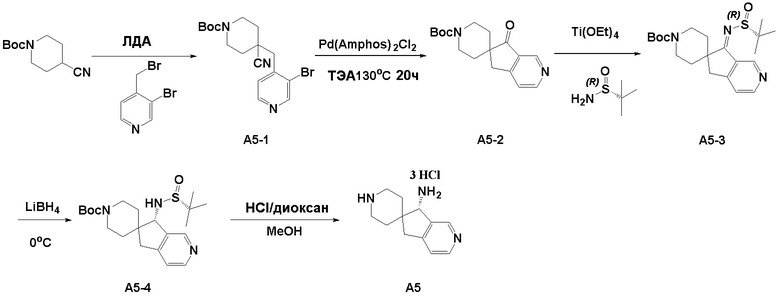
Синтез промежуточного соединения A2: R-N-((S)-5,7-дигидроспиро[циклопента[b]пиридин-6,4’-пиперидин]-5-ил)-2-метилпропан-2-сульфинамида

Стадия 1: Сложный трет-бутиловый эфир 4-цианопиперидин-1-карбоновой кислоты (1,05 г, 5 ммоль) и ТГФ (20 мл) последовательно добавляли в сухую колбу объемом 100 мл. В атмосфере азота смесь охлаждали до -78°C, а затем к реакционной смеси медленно добавляли 2M ЛДА (3,3 мл, 6,5 ммоль). Реакционной смеси давали прореагировать в течение 1 часа, а затем к ней добавляли 3-бром-2-(бромметил)пиридин (1,24 г, 5 ммоль), а затем реакционной смеси давали возможность продолжить реагировать в течение 2 часов. После реакции добавляли насыщенный раствор хлорида аммония (15 мл) для гашения реакции, полученную смесь экстрагировали этилацетатом (3 × 30 мл), органические слои объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия и фильтровали, и остаток, полученный концентрированием при пониженном давлении, очищали хроматографией на силикагеле (градиент от 0 до 30% этилацетата/петролейный эфир) с получением сложного трет-бутилового эфира 4-((3-бромпиридин-2-ил)метил)-4-цианопиперидин-1'-карбоновой кислоты (A2-1, 1,40 г, выход: 75%) в виде белого твердого вещества.
Стадия 2: В атмосфере азота сложный трет-бутиловый эфир 4-((3-бромпиридин-2-ил)метил)-4-цианопиперидин-1'-карбоновой кислоты (A2-1, 379 мг, 1 ммоль), триметиламин (404 мг , 4 ммоль), бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладий (II) Pd(AmPhos)2Cl2 (71 мг, 0,1 ммоль) и ДМА:H2O=10:1 (6 мл) последовательно добавляли в сухую одногорлую колбу объемом 25 мл, а затем смесь перемешивали при 130°C в течение 18 часов. После завершения реакции полученный остаток фильтровали, и остаток, полученный путем концентрирования при пониженном давлении, очищали хроматографией на силикагеле (градиент от 0 до 50% этилацетата:петролейный эфир) с получением сложного трет-бутилового эфира 5-оксо-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A2-2, 180 мг, выход: 60%) в виде желтого твердого вещества. ЖХМС: м/з 303,1 [M+H]+
Стадия 3: Сложный трет-бутиловый эфир 5-оксо-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A2-2, 0,302 г, 1 ммоль), тетраэтилтитанат (1,37 г, 6 ммоль) и (R)-(+)-трет-бутилсульфинамид (0,480 г, 4 ммоль) последовательно добавляли в сухую одногорлую колбу объемом 100 мл, и смесь перемешивали при нагревании и кипячении с обратным холодильником в течение 15 часов. После охлаждения реакционной системы до комнатной температуры к реакционному остатку добавляли насыщенный рассол (15 мл), после чего полученную смесь перемешивали в течение 15 минут и затем фильтровали через диатомит. Водную смесь экстрагировали этилацетатом (3 × 300 мл). Органическую фазу сушили над Na2SO4 и фильтровали, и летучие вещества удаляли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент от 0 до 50% этилацетата:петролейный эфир) с получением сложного трет-бутилового эфира (R, Z)-5-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A2-3, 0,333 г, выход: 82%) в виде желтого твердого вещества. ЖХМС: м/з 406,1 [M+H]+
Стадия 4: Сложный трет-бутиловый эфир (R, Z)-5-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A2-3, 0,20 г, 0,491 ммоль) и ТГФ (50 мл) последовательно добавляли в сухую одногорлую колбу объемом 100 мл, смесь охлаждали до 0°C, а затем добавляли боргидрид лития (0,018 г, 0,737 ммоль). Полученная смесь продолжала реагировать при перемешивании в течение 1 часа. Медленно добавляли метанол, чтобы погасить избыток боргидрида. Реакционный раствор фильтровали и концентрировали, и летучие вещества удаляли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент от 0 до 80% этилацетата:петролейный эфир) с получением сложного трет-бутилового эфира (S)-5-((R)-трет-бутилсульфонамидо)-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A2-4, 0,130 г, выход: 65%) в виде белого твердого вещества. ЖХМС: м/з 408,1 [M+H]+
Стадия 5: Сложный трет-бутиловый эфир (S)-5-((R)-трет-бутилсульфонамидо)-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A2-4, 0,100 г, 0,245 ммоль), дихлорметан (5 мл) и ТФУК (1 мл) последовательно добавляли в сухую одногорлую колбу объемом 50 мл, и полученную смесь подвергали взаимодействию при перемешивании при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор Na2CO3 до тех пор, пока pH не достигал 7, и водную смесь экстрагировали с помощью ДХМ (3 × 30 мл). Объединенную органическую фазу промывали рассолом, сушили над Na2SO4 и фильтровали, и летучие вещества удаляли при пониженном давлении. Полученный остаток охлаждали с получением R-N-((S)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамида (A2, 0,056 г, выход 75%) в виде бесцветного маслянистого вещества. ЖХМС: м/з 308,1 [M+H]+
Синтез промежуточного соединения A3: (S)-1,3-дигидроспиро[инден-2,4’-пиперидин]-1-амина
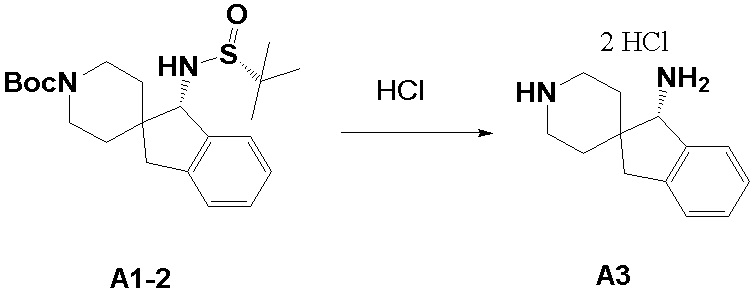
Стадия: Метиловый спирт (1 мл), дихлорметан (1 мл) и A1-2 (540 мг, 1,33 ммоль, 1,0 экв.) добавляли в сухую круглодонную колбу объемом 100 мл. По каплям при комнатной температуре добавляли HCl/1,4-диоксан (3,3 мл, 4M), выпадало белое твердое вещество, и реакционную систему нагревали до 50°C и перемешивали в течение 2 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении с получением (S)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (A3, 373 мг, выход: 97,1%, HCl-соль) в виде белого твердого вещества. ЖХМС: м/з 203,1 [M+H]+
Синтез промежуточного соединения A4: (S)-5,7-дигидроспиро [циклопента[b]пиридин-6,4’-пиперидин]-5-амина
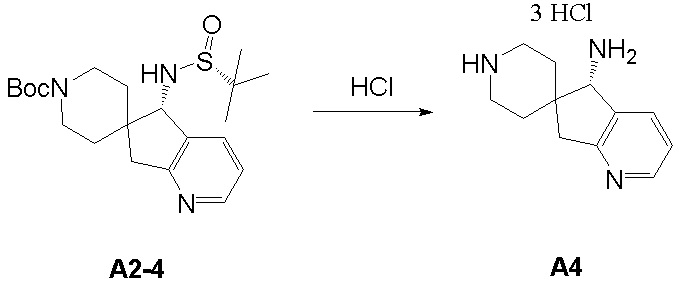
Промежуточное соединение (S)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин (A4) синтезировали в соответствии с протоколом синтеза промежуточного соединения A3, используя промежуточное соединение A2-4 вместо промежуточного соединения A1-2. ЖХМС: м/з 204,1 [M+H]+
Синтез промежуточного соединения A5: (S)-5,7-дигидроспиро[циклопента[c]пиридин-6,4’-пиперидин]-7-амина
A5 синтезировали в соответствии с протоколом синтеза промежуточных соединений A2 и A3. ЖХМС: м/з 204,1 [M+H]+
Синтез промежуточного соединения A6: (S)-5,7-дигидроспиро[циклопента[b]пиридин-6,4’-пиперидин]-7-амина
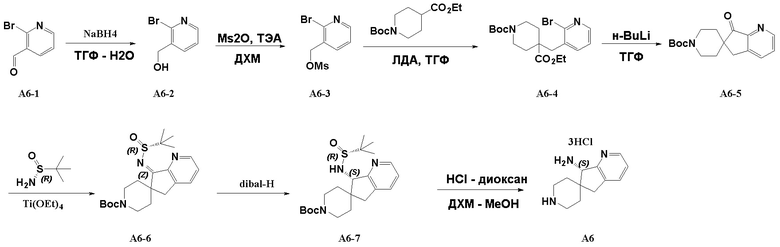
Стадия 1: A6-1 (11,1 г, 60 ммоль) и NaBH4 (2,51 г, 66 ммоль) последовательно добавляли к 300 мл ТГФ и 60 мл H2O в колбе объемом 1 л, и смесь подвергали реакции при 20°C в течение 2 часов. После подтверждения детектированием пятен на пластине того, что исходные вещества полностью прореагировали, реакционную смесь гасили насыщенным раствором NH4Cl, разбавляли водой и экстрагировали этилацетатом. Объединенную органическую фазу промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 2-бромпиридин-3-илметанола (A6-2, 11,2 г, выход: 100%) в виде белого твердого вещества.
Стадия 2: A6-2 (6,73 г, 36 ммоль) и Ms2O (6,96 г, г, 40 ммоль) последовательно добавляли к 120 мл дихлорметана в сухой одногорлой колбе объемом 250 мл, затем смесь охлаждали до 0°C, затем медленно добавляли ТЭА (5,45 г, 54 ммоль), а затем смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. После завершения реакции реакционный раствор промывали водой. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент от 0 до 10% этилацетата/дихлорметан) с получением сложного (2-бромпиридин-3-ил)метилового эфира метилсульфоновой кислоты (A6-3, 8,02 г, выход: 84%) в виде бесцветного маслянистого вещества.
Стадия 3: Сложный трет-бутиловый эфир 4-этоксикарбонилпиперидин-1-карбоновой кислоты (9,3 г, 36,2 ммоль) и ТГФ (133 мл) последовательно добавляли в сухую трехгорлую колбу объемом 500 мл. В атмосфере азота смесь охлаждали до -70°C, а затем к реакционной смеси медленно добавляли 2M ЛДА (21,1 мл, 42,3 ммоль). Реакционной смеси давали возможность прореагировать в течение 1 часа, затем к ней добавляли сложный (2-бромпиридин-3-ил)метиловый эфир метилсульфоновой кислоты (A6-3, 8,0 г, 30,2 ммоль), растворенный в 65 мл ТГФ, затем реакционной смеси давали возможность продолжать реагировать в течение 0,5 часа, а затем реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 1 часа. После завершения реакции реакционную смесь гасили насыщенным рассолом и экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент от 8 до 12% этилацетата/дихлорметан) с получением сложного трет-бутилового эфира 4-этоксикарбонил-4-(2-бром-3-пиридил)метил-пиперидин-1-карбоновой кислоты (A6-4, 10,5 г, выход: 81%) в виде бесцветного маслянистого вещества.
Стадия 4: Сложный трет-бутиловый эфир 4-этоксикарбонил-4-(2-бром-3-пиридил)метил-пиперидин-1-карбоновой кислоты (A6-4, 7,85 г, 18,4 ммоль) и ТГФ (120 мл) последовательно добавляли в сухую трехгорлую колбу объемом 250 мл. В атмосфере азота смесь охлаждали до -70°C, а затем к реакционной смеси медленно добавляли 2,5М н-бутиллитий (11 мл, 27,6 ммоль). Реакционной смеси давали возможность прореагировать в течение 1,5 часов. После завершения реакции к ней добавляли насыщенный водный раствор хлорида аммония для гашения реакции, а затем добавляли насыщенный рассол для разбавления и разделения жидкости. Водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент от 0 до 60% этилацетата/петролейный эфир) с получением сложного трет-бутилового эфира 7-оксо-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A6-5, 1,4 г, выход: 25%) в виде светло-коричневого твердого вещества.
Стадия 5. Промежуточное соединение сложный трет-бутиловый эфир (R, Z)-7-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A6-6, 1,46 г, выход: 68%) синтезировали в соответствии с протоколом синтеза промежуточного соединения A2-3, используя промежуточное соединение A6-5 вместо промежуточного соединения A2-2.
Стадия 6: Сложный трет-бутиловый эфир (R, Z)-7-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A6-6, 530 мг, 1,31 ммоль) и ТГФ (10 мл) последовательно добавляли в сухую трехгорлую колбу объемом 50 мл. В атмосфере азота смесь охлаждали до -70°C, а затем к реакционной смеси медленно добавляли 1,5М диизобутилалюминийгидрид в толуоле (1,3 мл, 1,95 ммоль). Реакционной смеси давали прореагировать в течение 0,5 часа, а затем смесь медленно нагревали до комнатной температуры, гасили насыщенным водным раствором тартрата калия-натрия и перемешивали в течение 0,5 часа. Реакционную смесь экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент от 0 до 5% метилового спирта/дихлорметан) с получением сложного трет-бутилового эфира (S)-7-((R)-трет-бутилсульфонамидо)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A6-7, 466 мг, выход: 87%) в виде светло-желтого вспененного твердого вещества.
Стадия 7: Сложный трет-бутиловый эфир (S)-7-((R)-трет-бутилсульфонамидо)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A6-7, 312 мг, 0,766 ммоль), MeOH (5 мл) и 4M раствор HCl/1,4-диоксана (3,83 мл, 15,3 ммоль) последовательно добавляли в сухую одногорлую колбу объемом 25 мл, а затем смесь нагревали до 50°C и подвергали реакции в течение 8 часов с получением белой суспензии. Суспензию концентрировали при пониженном давлении с получением HCl-соли (S)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амина (A6, 232 мг, выход: 97%) в виде белого твердого вещества. ЖХМС: м/з 204,1 [M+H]+
Синтез промежуточного соединения A7: (S)-6-фтор-1,3-дигироспиро[инден-2,4'-пиперидин]-1-амина
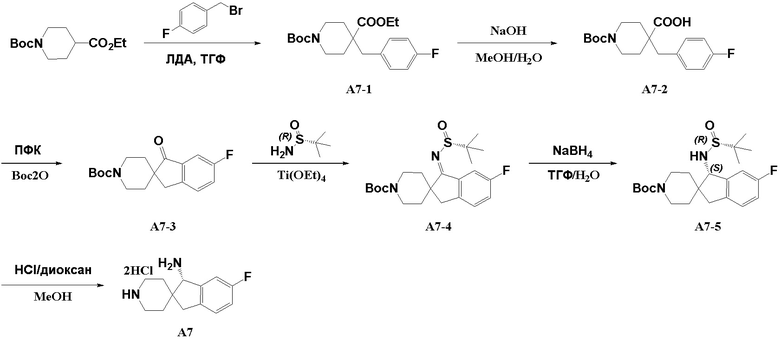
Стадия 1: Промежуточное соединение сложный трет-бутиловый эфир 4-этоксикарбонил-4-(4-фторбензил)пиридин-1-карбоновой кислоты A7-1 синтезировали в соответствии с протоколом синтеза промежуточного соединения A2 с использованием исходного вещества 4-фторбензилбромида вместо исходного вещества 3-бром-2(бромметил)пиридина.
Стадия 2: Сложный трет-бутиловый эфир 4-этоксикарбонил-4-(4-фторбензил)пиридин-1-карбоновой кислоты (A7-1, 3,40 г, 9,30 ммоль) и гидроксид натрия (1,86 г, 46,5 ммоль) последовательно добавляли к 20 мл метилового спирта и 20 мл воды в одногорлой колбе объемом 100 мл. Реакционный раствор подвергали реакции при 70°C в течение 17 часов. Реакционную смесь охлаждали до комнатной температуры, а затем концентрировали при пониженном давлении, чтобы избавиться от летучих веществ. Полученный остаток разбавляли водой (50 мл), и добавляли разбавленную соляную кислоту для доведения pH до 3, а затем смесь экстрагировали 3 раза этилацетатом (80 мл). Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1-трет-бутоксикарбонил-4-(4-фторбензил)-пиридин-4-карбоновой кислоты (A7-2, 3,0 г, сырой продукт) в виде желтого твердого вещества.
Стадия 3: 1-Трет-бутоксикарбонил-4-(4-фторбензил)-пиридин-4-карбоновую кислоту (A7-2, 2,0 г, 5,93 ммоль) и ПФК (15 мл) последовательно добавляли в сухую одногорлую колбу объемом 50 мл. Реакционный раствор подвергали реакции при 120°C в течение 2 часов. Реакционный раствор выливали в смесь лед-вода (50 мл), пока он был еще горячим, и добавляли твердый NaOH, чтобы довести pH до 10. Затем к полученной смеси добавляли Boc2O (1,94 г, 8,90 ммоль), и реакционный раствор перемешивали при 20°C в течение 1 часа. Реакционный раствор экстрагировали 3 раза этилацетатом (80 мл). Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (петролейный эфир/этилацетат=5:1) с получением сложного трет-бутилового эфира 6-фтор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A7-3, 1,20 г, выход: 63,5%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,46-7,38 (м, 2H), 7,37-7,30 (м, 1H), 4,20-4,06 (м, 2H), 3,07-2,95 (м, 4H), 1,96-1,85 (м, 2H), 1,48 (с, 9H), 1,44-1,35 (м, 2H).
Стадия 4. Промежуточное соединение (R, Z)-1-((трет-бутилсульфинил)имино)-6-фтор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-формиат A7-4 синтезировали в соответствии с протоколом синтеза промежуточного соединения A2, используя промежуточное соединение A7-3 вместо промежуточного соединения A2-2.
Стадия 5: (R, Z)-1-((трет-бутилсульфинил)имино)-6-фтор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-формиат (A7-4, 1,59 г, 3,76 ммоль), тетрагидрофуран/воду (98:2, 32 мл) и боргидрид натрия (427 мг, 11,3 ммоль) последовательно добавляли в круглодонную колбу при -50°C, и реакционный раствор нагревали до 20°C за 3 часа при перемешивании. Полнота реакции подтверждена пятнами на пластине ТСХ. Реакционный раствор разбавляли водой (30 мл) и 3 раза экстрагировали этилацетатом (30 мл). Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (петролейный эфир/этилацетат=2:1) с получением сложного трет-бутилового эфира (S)-1-(((R)-трет-бутилсульфинил)амино)-6-фтор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A7-5, 850 мг, выход: 53%) в виде белого твердого вещества.
Стадия 6. Промежуточное соединение (S)-6-фтор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин A7 синтезировали в соответствии с протоколом синтеза промежуточного соединения A3, используя промежуточное соединение A7-5 вместо промежуточного соединения A1-2. ЖХМС: м/з 220,1 [M+H]+
Промежуточное соединение A8: (S)-5,7-дигидроспиро[циклопента[c]пиридин-6,4’-пиперидин]-7-амин
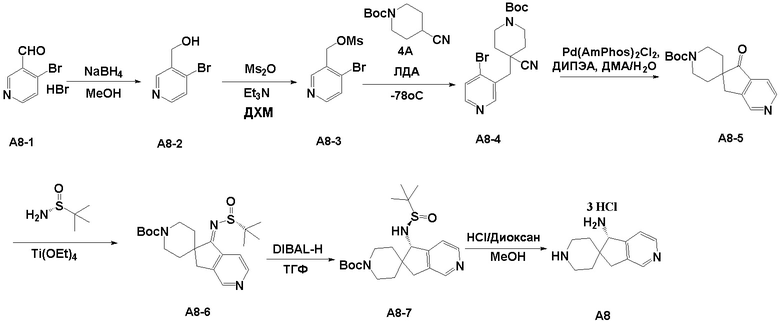
Стадия 1: гидробромидную соль 4-бромникотинового альдегида (A8-1, 2,5 г, 9,36 ммоль) растворяли в метиловом спирте (50 мл), порциями добавляли боргидрид натрия (0,72 г, 18,93 ммоль, 2,0 экв.) на ледяной бане, и смесь подвергали реакции при 0°C в течение 1 часа. После завершения реакции на ледяной бане добавляли насыщенный водный раствор хлорида аммония (50 мл), чтобы погасить реакцию, и реакционную смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия. Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением (4-бромпиридин-3-ил)метанола (A8-2, 1,7 г, выход: 100%) в виде белого твердого вещества. ЖХМС: м/з 190,3 [M+H]+
Стадия 2: (4-Бромпиридин-3-ил)метанол (A8-2, 1,7 г, 9,04 ммоль) растворяли в дихлорметане (100 мл) и добавляли триэтиламин (2,30 г, 22,7 ммоль) на ледяной бане. В атмосфере азота порциями добавляли ангидрид метансульфоновой кислоты (1,95 г, 11,2 ммоль), смесь подвергали реакции при 0°C в течение 2 часов, а затем реакцию завершали. Для гашения реакции на ледяной бане добавляли насыщенный хлорид натрия (50 мл), органическую фазу отделяли и промывали насыщенным водным раствором хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением сложного (4-бромпиридин-3-ил)метилового эфира метансульфоновой кислоты (A8-3, 1,7 г, выход: 70,7%) в виде красновато-коричневого твердого вещества. ЖХМС: м/з 266,2 [M+H]+
Стадия 3: Сложный трет-бутиловый эфир 4-цианопиперидин-1-карбоновой кислоты (1,61 г, 7,67 ммоль) растворяли в безводном тетрагидрофуране (60 мл). В бане с сухим льдом и ацетоном (-78°C) и в атмосфере азота медленно по каплям добавляли ЛДА (4,6 мл, 9,2 ммоль), смесь подвергали реакции в течение 0,5 часа при поддержании температуры, и выпадало много белого твердого вещества из реакционного раствора. Раствор (4-бромпиридин-3-ил)метилового эфира метансульфоновой кислоты (A8-3, 1,7 г, 6,39 ммоль) в безводном тетрагидрофуране (50 мл) медленно добавляли по каплям при -78°C, смесь продолжала реагировать в течение 2 часов, пока поддерживали температуру, и затем реакцию завершали. Для гашения реакции добавляли насыщенный хлорид аммония (100 мл), и смесь экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле (градиент от 0 до 50% дихлорметана/петролейный эфир) с получением сложного трет-бутилового эфира 4-((4-бромпиридин-3-ил)метил)-4-цианопиперидин-1-карбоновой кислоты (A8-4, 1,58 г, выход: 54%) в виде белого твердого вещества. ЖХМС: м/з 326,0 [M+H-56]+
Стадия 4: Сложный трет-бутиловый эфир 4-((4-бромпиридин-3-ил)метил)-4-цианопиперидин-1-карбоновой кислоты (A8-4, 1,5 г, 3,95 ммоль) растворяли в DMAc/H2O (100 мл/10 мл), добавляли ДИПЭА (1,34 г, 15,8 ммоль) и Pd(AmPhos)2Cl2 (142 мг, 0,20 ммоль), атмосферу в реакционной системе трижды заменяли азотом, смесь подвергали реакции при 130°C в течение 2 часов, и затем реакцию завершали. Добавляли воду (50 мл), и смесь экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира 5-оксо-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A8-5, 750 мг, выход: 62%) в виде твердого вещества. ЖХМС: м/з 303,3 [M+H]+
Стадия 5: Сложный трет-бутиловый эфир 5-оксо-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A8-5, 750 мг, 2,48 ммоль) растворяли в ТГФ (10 мл), и добавляли и (R)-(+)-трет-бутилсульфинамид (390 мг, 3,22 ммоль) и тетраэтилтитанат (10 мл). В атмосфере азота смесь нагревали до 90°C и кипятили с обратным холодильником в течение 18 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (50 мл), добавляли насыщенный рассол (10 мл), и белое твердое вещество выпадало в осадок. Смесь фильтровали, и осадок на фильтре промывали этилацетатом. Фильтрат промывали насыщенным рассолом, и органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира (R, Z)-5-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A8-6, 800 мг, выход: 80%) в виде белого твердого вещества. ЖХМС: м/з 406,2 [M+H]+
Стадия 6: Сложный трет-бутиловый эфир (R, Z)-5-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A8-6, 800 мг, 1,98 ммоль) растворяли в ТГФ (50 мл), и смесь охлаждали до -78°C в атмосфере азота. В реакционный раствор медленно по каплям добавляли 1,5М DIBAL-H (2 мл, 3 ммоль). После того, как добавление было завершено, смесь подвергали реакции при -78°C в течение 1 часа, а затем реакцию завершали. Для гашения реакции добавляли воду, добавляли насыщенный раствор тартрата калия-натрия (20 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органическую фазу один раз промывали насыщенным водным раствором хлорида натрия. Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира (S)-5-(((R)-трет-бутилсульфинил)амино)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A8-7, 680 мг, выход: 84%) в виде белого твердого вещества. ЖХМС: м/з 408,3 [M+H]+
Стадия 7: Сложный трет-бутиловый эфир (S)-5-(((R)-трет-бутилсульфинил)амино)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-1'-карбоновой кислоты (A8-7, 680 мг, 1,67 ммоль) растворяли в MeOH (50 мл), смесь охлаждали до 0°C, по каплям добавляли 4M раствор хлористоводородной кислоты в диоксане (10 мл, 40 ммоль), полученную смесь подвергали реакции при комнатной температуре в течение 3 часов, а затем реакцию завершали. Реакционный раствор концентрировали с получением гидрохлорида (S)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-7-амина (A8, 400 мг, выход: 99%) в виде белого твердого вещества. ЖХМС: м/з 204,1 [M+H]+
Промежуточное соединение A9: (S)-1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-4-ол
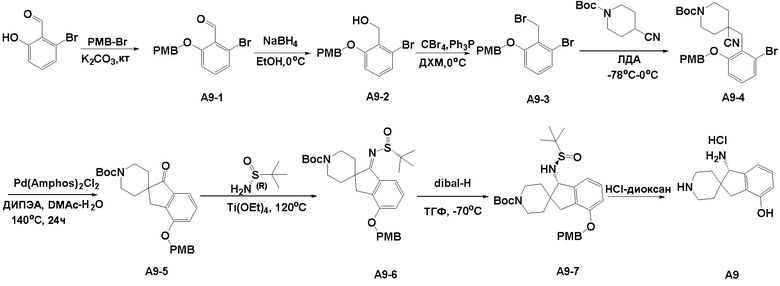
Стадия 1: 2-Бром-6-гидроксибензальдегид (5 г, 24,9 ммоль) растворяли в ДМФА (100 мл), добавляли безводный карбонат калия (6,88 г, 49,8 ммоль) и 4-метоксибензилбромид (5,26 г, 26,1 ммоль), смесь подвергали реакции при комнатной температуре в течение 18 часов в атмосфере азота, а затем реакцию завершали. Реакционный раствор выливали в ледяную воду и дважды экстрагировали этилацетатом (250 мл). Органические фазы объединяли и промывали насыщенным рассолом. Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением 2-бром-6-((4-метоксибензил)окси)бензальдегида (A9-1, 8 г, выход 100%) в виде светло-желтого твердого вещества. ЖХМС: м/з 343,0 [M+Na]+
Стадия 2: 2-Бром-6-((4-метоксибензил)окси)бензальдегид (A9-1, 8 г, 24,9 ммоль) растворяли в этаноле (100 мл), и смесь охлаждали до 0°C на ледяной бане. Боргидрид натрия (942 мг, 24,9 ммоль) осторожно добавляли несколькими порциями. Смесь подвергали реакции при 0°C в течение 0,5 часа, а затем реакцию завершали. Реакционный раствор выливали в ледяную воду и дважды экстрагировали этилацетатом (200 мл). Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением (2-бром-6-((4-метоксибензил)окси)фенил)метанола (A9-2, 7,3 г, выход: 90%) в виде твердого вещества. ЖХМС: м/з 345,0 [M+Na]+
Стадия 3: (2-Бром-6-((4-метоксибензил)окси)фенил)метанол (A9-2, 7,3 г, 22,6 ммоль) растворяли в дихлорметане (200 мл) и смесь охлаждали на ледяной бане до 0°C. Четырехбромистый углерод (11,2 г, 33,9 ммоль) и трифенилфосфин (8,88 г, 33,9 ммоль) добавляли в атмосфере азота. Смесь подвергали реакции при 0°C в течение 5 часов, а затем реакцию завершали. Реакционный раствор выливали в ледяную воду и дважды экстрагировали этилацетатом (200 мл). Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением 1-бром-2-(бромметил)-3-((4-метоксибензил)окси)бензола (A9-3, 4 г, выход: 46%) в виде твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,40 (т, J=4,4 Гц, 2H), 7,17-7,20 (м, 1H), 7,11 (т, J=8,2 Гц, 1H), 6,92-6,95 (м, 2H), 6,87-6,89 (м, 1H), 5,30 (с, 2H), 4,76 (с, 2H), 3,83 (с, 3H);
Стадия 4: Сложный трет-бутиловый эфир 4-цианопиперидин-1-карбоновой кислоты (2,6 г, 12,4 ммоль) растворяли в безводном ТГФ (60 мл), и смесь охлаждали до -78°C. Медленно по каплям добавляли 2М ЛДА (7,5 мл, 14,9 ммоль). Смесь подвергали реакции при -78°C в течение 0,5 часа, а затем по каплям добавляли раствор 1-бром-2-(бромметил)-3-((4-метоксибензил)окси)бензола (A9-3, 4 г, 10,4 ммоль) в безводном ТГФ (40 мл). Смесь подвергали реакции при -78°C в течение 2 часов, а затем ее медленно нагревали до 0°. Реакцию завершали. Реакционный раствор выливали в ледяную воду и дважды экстрагировали этилацетатом (200 мл). Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира 4-(2-бром-6-((4-метоксибензил)окси)бензил)-4-цианопиперидин-1-карбоновой кислоты (A9-4, 2,8 г , выход: 44%) в виде твердого вещества. ЖХМС: м/з 537,0 [M+Na]+
Стадия 5: Сложный трет-бутиловый эфир 4-(2-бром-6-((4-метоксибензил)окси)бензил)-4-цианопиперидин-1-карбоновой кислоты (A9-4, 2,6 г, 5,0 ммоль) растворяли в DMAc (100 мл), добавляли H2O (10 мл), ДИПЭА (3,225 г, 25,0 ммоль) и PdCl2(AmPhos)2 (354 мг, 0,5 ммоль). Смесь подвергали реакции при 130°C в атмосфере аргона в течение 18 часов, после чего реакцию завершали. После охлаждения до комнатной температуры реакционный раствор выливали в ледяную воду и дважды экстрагировали этилацетатом (300 мл). Органические фазы объединяли, промывали один раз водой и насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира 4-((4-метоксибензил)окси)-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты. (A9-5, 1,8 г, выход: 82%) в виде твердого вещества. ЖХМС: м/з 382,2 [M-56]+
Стадия 6: Сложный трет-бутиловый эфир 4-((4-метоксибензил)окси)-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A9-5, 1,8 г 4,12 ммоль) растворяли в ТГФ (20 мл), добавляли (R)-2-метилпропан-2-сульфинамид (748 мг, 6,18 ммоль) и тетраэтилтитанат (50 мл). Реакционную смесь нагревали до 100°C и кипятили с обратным холодильником в течение 18 часов в атмосфере азота. Реакцию завершали, и после охлаждения смеси до комнатной температуры добавляли этилацетат (200 мл) для разбавления, добавляли насыщенный рассол (50 мл), и белое твердое вещество выпадало в осадок. Смесь фильтровали, и осадок на фильтре промывали этилацетатом. Фильтрат промывали насыщенным рассолом, органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира (R, Z)-1-((трет-бутилсульфинил)имино)-4-((4-метоксибензил)окси)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A9-6, 1,7 г, выход: 76%) в виде белого твердого вещества. ЖХМС: м/з 541,3 [M+H]+
Стадия 7: Сложный трет-бутиловый эфир (R, Z)-1-((трет-бутилсульфинил)имино)-4-((4-метоксибензил)окси)-1,3-дигидроспиро [инден-2,4'-пиперидин]-1'-карбоновой кислоты (A9-6, 1,7 г, 3,15 ммоль) растворяли в ТГФ (100 мл), и смесь охлаждали до -78°C в атмосфере азота. К реакционному раствору медленно по каплям добавляли 1,5М DIBAL-H (3,15 мл, 4,7 ммоль). После завершения добавления реакционный раствор подвергали реакции при -78°C в течение 1 часа до завершения реакции. Для гашения реакции добавляли воду, добавляли насыщенный раствор тартрата калия-натрия (50 мл), смесь экстрагировали этилацетатом (200 мл × 2), и органическую фазу один раз промывали насыщенным водным раствором хлорида натрия. Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира (S)-1-(((R)-трет-бутилсульфинил)амино)-4-((4-метоксибензил)окси)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A9-7, 1,5 г, выход: 88%) в виде белого твердого вещества. ЖХМС: м/з 543,3 [M+H]+
Стадия 8: Сложный трет-бутиловый эфир (S)-1-(((R)-трет-бутилсульфинил)амино)-4-((4-метоксибензил)окси)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A9-7, 1,5 г, 2,77 ммоль) растворяли в MeOH (50 мл), смесь охлаждали до 0°C, и по каплям добавляли 4M раствор хлористоводородной кислоты в диоксане (10 мл, 40 ммоль). Смесь подвергали реакции при комнатной температуре в течение 3 часов, а затем реакцию завершали. Реакционный раствор концентрировали с получением (S)-1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-4-ола гидрохлорида (A9, 600 мг, выход: 85%) в виде белого твердого вещества. ЖХМС: м/з 219,2 [M+H]+
Промежуточное соединение A10: (S)-1-(((R)-трет-бутилсульфинил)амино)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]
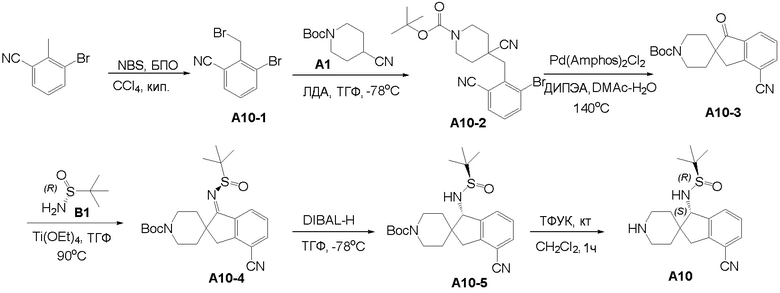
Стадия 1: 2-Метил-3-цианобромбензол (3,0 г, 15,3 ммоль, 1,0 экв.), N-бромсукцинимид (2,72 г, 15,3 ммоль, 1,0 экв.), пероксид дибензоила (371 мг, 1,53 ммоль, 0,1 экв.) и четыреххлористый углерод (40 мл) последовательно добавляли в круглодонную колбу объемом 100 мл, и смесь перемешивали при 80°C в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении с получением остатка. Остаток растворяли в этилацетате (200 мл), дважды промывали 2н водным раствором NaOH (50 мл) и один раз насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого продукта. Сырой продукт очищали хроматографией на силикагеле с получением 1-бром-2-(бромметил)-3-цианобензола (A10-1, 2,0 г, выход: 47,5%) в виде белого твердого вещества.
Стадия 2: Тетрагидрофуран (30 мл) и сложный трет-бутиловый эфир 4-цианопиперидин-1-карбоновой кислоты (1,84 г, 8,73 ммоль, 1,2 экв.) последовательно добавляли в круглодонную колбу объемом 100 мл, смесь охлаждали до -78°C, затем добавляли 2,0М ЛДА (5,1 мл, 10,2 ммоль, 1,4 экв.), и смесь перемешивали при -78°C в течение одного часа. Затем добавляли раствор 1-бром-2-(бромметил)-3-цианобензола (A10-1, 2,0 г, 7,27 ммоль, 1,0 экв.) в тетрагидрофуране (15 мл), и смесь перемешивали при -78°C в течение 0,5 часа. Затем низкотемпературную баню удаляли, смеси давали возможность вернуться к комнатной температуре и затем продолжали перемешивание в течение одного часа. После подтверждения завершения реакции исходного вещества с помощью ТСХ, для гашения добавляли насыщенный рассол (30 мл), а затем смесь экстрагировали этилацетатом (100 мл * 2). Объединенную органическую фазу промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого продукта. Сырой продукт очищали хроматографией на силикагеле с получением трет-бутил-4-(2-бром-6-цианобензил)-4-цианопиперидин-1-карбоксилата (A10-2, 1,5 г, выход: 51%) в виде светло-желтого маслянистого вещества. ЖХ-МС: м/з 404,1, 406,1 [M+H]+
Стадия 3. Трет-бутил-4-(2-бром-6-цианобензил)-4-цианопиперидин-1-карбоксилат (A10-2, 1,5 г, 3,71 ммоль, 1,0 экв.), Pd(AmPhos)2Cl2 (262 мг, 0,37 ммоль 0,1 экв.), диизопропилэтиламин (2,4 г, 18,5 ммоль, 5,0 экв.), N, N-диметилацетамид (30 мл) и воду (4 мл) последовательно добавляли в сухую круглодонную трехгорлую колбу объемом 100 мл. При перемешивании атмосферу в реакционной системе трижды заменяли азотом, а затем реакционную смесь нагревали до 140°C и подвергали реакции в течение 16 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом (100 мл) и фильтровали при пониженном давлении. Осадок на фильтре промывали этилацетатом (20 мл), и полученный фильтрат промывали насыщенным рассолом (30 мл) 3 раза, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого продукта. Сырой продукт очищали хроматографией на силикагеле с получением трет-бутил-4-циано-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-формиата (A10-3, 0,90 г, выход: 74,3%) в виде белого твердого вещества. ЖХ-МС: м/з 327,2 [M+H]+
Стадия 4: Трет-бутил-4-циано-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-формиат (A10-3, 900 мг, 2,76 ммоль), тетраэтоксилат титана (3,78 г, 16,6 ммоль), (R)-(+)-трет-бутилсульфинамид (401 мг, 3,31 ммоль) и тетрагидрофуран (20 мл) последовательно добавляли в сухую одногорлую колбу объемом 100 мл, и смесь перемешивали при нагревании и кипячении с обратным холодильником в течение 16 часов. После охлаждения до комнатной температуры к реакционному остатку добавляли насыщенный рассол (60 мл), а затем полученную смесь перемешивали в течение 15 минут и затем фильтровали через диатомит. Водную смесь экстрагировали этилацетатом (3 × 80 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент от 0 до 30% этилацетата:петролейный эфир) с получением сложного трет-бутилового эфира (R, Z)-1-((трет-бутилсульфинил)имид)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A10-4, 980 мг, выход: 82,7%) в виде белого твердого вещества. ЖХМС: м/з 430,2 [M+H]+
Стадия 5: Тетрагидрофуран (15 мл) и сложный трет-бутиловый эфир (R, Z)-1-((трет-бутилсульфинил)имид)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A10-4, 980 мг, 2,28 ммоль, 1,0 экв.) последовательно добавляли в сухую трехгорлую колбу объемом 50 мл. По каплям добавляли диизобутилалюминийгидрид (6,8 мл, 1,5М в толуоле, 10,3 ммоль, 4,5 экв.) при -78°C и в атмосфере азота, и смесь продолжали перемешивать в течение получаса. Затем смесь нагревали до 0°C и продолжали перемешивать в течение получаса. После завершения реакции реакционную смесь гасили тартратом калия-натрия (4 г, растворенных в 20 мл воды), перемешивали в течение получаса и экстрагировали этилацетатом (30 мл * 3). Полученную органическую фазу промывали насыщенным рассолом (30 мл), сушили безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (градиент 0-70% этилацетата:петролейный эфир) с получением сложного трет-бутилового эфира (S)-1-((R)-трет-бутилсульфинил)амино)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A10-5, 800 мг, выход: 81,3%) в виде желтого твердого вещества. ЖХ-МС: м/з 432,2 [M+H]+
Стадия 6. Сложный трет-бутиловый эфир (S)-1-((R)-трет-бутилсульфинил)амино)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоновой кислоты (A10-5, 800 мг, 1,85 ммоль), дихлорметан (15 мл) и трифторуксусную кислоту (5 мл) последовательно добавляли в сухую одногорлую колбу объемом 50 мл, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор Na2CO3 до pH=7, и водную смесь экстрагировали дихлорметаном (3 × 50 мл). Объединенную органическую фазу промывали рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (градиент 0-70% этилацетата:петролейный эфир) с получением (R)-N-((S)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропил-2-сульфинамида (A10, 490 мг, выход: 79,8%) в виде желтого твердого вещества. ЖХМС: м/з 332,2 [M+H]+
Промежуточное соединение A11: (S)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
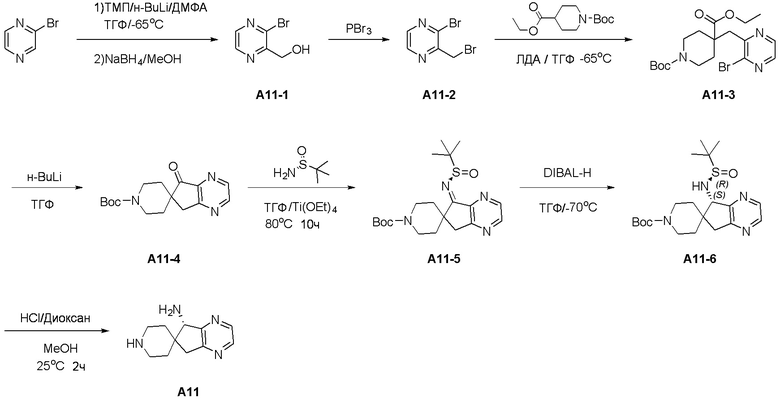
Стадия 1: 2,2,6,6-Тетраметилпиперидин (5,33 г, 37,8 ммоль) растворяли в 100 мл тетрагидрофурана, смесь охлаждали до -65°C в атмосфере аргона на бане с сухим льдом и ацетоном, по каплям добавляли н-бутиллитий (2,5M, 15,8 мл, 39,5 ммоль), затем температуру смеси поддерживали на уровне 0°C в течение 30 минут, а затем снова понижали до -65°C, по каплям добавляли смесь 2-бромпиразина (5,0 г, 31,4 ммоль) и 10 мл тетрагидрофурана, температуру поддерживали в течение 30 минут, по каплям добавляли смесь ДМФА (5,75 г, 78,6 ммоль) и 10 мл тетрагидрофурана, полученную смесь продолжали перемешивать в течение 2 часов, после чего реакцию завершали. Затем по каплям добавляли 75 мл метанола при -65°C, смесь перемешивали в течение 10 минут, медленно добавляли NaBH4 (2,38 г, 62,9 ммоль), температуру смеси медленно повышали до 0°C и выдерживали в течение 1 часа, и затем реакцию завершали. Реакционную жидкость выливали в 500 мл ледяного водного раствора NH4Cl и трижды экстрагировали этилацетатом. Органическую фазу один раз промывали насыщенным рассолом. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и пропускали через колонку с получением (3-бромпиридин-2-ил)метанола (A11-1, 4,33 г, выход: 72,9%) в виде желтого твердого вещества. ЖХМС: м/з 191 [M+H]+
Стадия 2: (3-Бромпиридин-2-ил)метанол (A11-1, 4,33 г, 22,9 ммоль) растворяли в 80 мл эфира, по каплям добавляли PBr3 (6,83 г, 25,2 ммоль) при 0°C в атмосфере азота, затем смесь нагревали до кипячения с обратным холодильником примерно при 40°C и подвергали реакции в течение 4 часов, после чего реакцию завершали. Реакционную жидкость выливали в ледяной водный раствор NaHCO3, дважды экстрагировали дихлорметаном и один раз промывали насыщенным рассолом. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и пропускали через колонку с получением 2-бром-3-(бромметил)пиразина (A11-2, 5,14 г, выход: 89,0%) в виде почти бесцветного прозрачного маслянистого вещества. ЖХМС: м/з 252,9 [M+H]+
Стадия 3: Этил N-Boc-4-пиперидин-формиат (6,81 г, 26,5 ммоль) растворяли в 160 мл безводного тетрагидрофурана, смесь охлаждали до -65°C на бане с сухим льдом и ацетоном в атмосфере аргона, медленно по каплям добавляли ЛДА (2,0M, 16,8 мл, 33,6 ммоль), и смесь подвергали реакции в течение 1 часа при поддержании температуры. По каплям добавляли раствор 2-бром-3-(бромметил)пиразина (A11-2, 5,14 г, 24,1 ммоль) в безводном тетрагидрофуране (30 мл) при -65°C, реакция продолжалась в течение 2 часов при поддержании температуры, и затем реакцию завершали. Реакционный раствор выливали в 300 мл ледяного водного раствора NH4Cl и трижды экстрагировали этилацетатом. Органическую фазу промывали насыщенным рассолом один раз. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и пропускали через колонку с получением 1-(трет-бутил)-4-этил-4-((3-бромпиразин-2-ил)метил) пиперидин-1,4-дикарбоксилата (A11-3, 5,81 г, выход: 56,4%). ЖХМС: м/з 328,1 [M+H-100]+
Стадия 4: 1-(Трет-бутил)-4-этил-4-((3-бромпиразин-2-ил)метил)пиперидин-1,4-дикарбоксилат (A11-3, 5,8 г, 13,5 ммоль) растворяли в 300 мл тетрагидрофурана, смесь охлаждали до -65°C на бане с сухим льдом и ацетоном в атмосфере аргона, по каплям добавляли н-бутиллитий (2,5М, 8,2 мл, 20,3 ммоль), смесь естественным образом нагревали до -10°C и выдерживали в течение 3 часов, после чего реакцию завершали. Реакционную жидкость выливали в 300 мл ледяного водного раствора NH4Cl и трижды экстрагировали этилацетатом. Органическую фазу один раз промывали насыщенным рассолом. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и пропускали через колонку с получением сложного трет-бутилового эфира 5-оксо-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-1'-карбоновой кислоты (A11-4, 2,23 г, выход: 54,4%) в виде желтого твердого вещества.
ЖХМС: м/з 304,2 [M+H]+ слабый
Стадия 5: Сложный трет-бутиловый эфир 5-оксо-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-1'-карбоновой кислоты (A11-4, 2,23 г, 7,35 ммоль) растворяли в 350 мл тетрагидрофурана, и добавляли (R)-2-метилпропан-2-сульфинамид (980 мг, 8,09 ммоль) и тетраэтил титанат (21,8 г, 95,6 ммоль). В атмосфере азота смесь нагревали до 80°C и кипятили с обратным холодильником в течение 10 часов. Реакцию завершали. Реакционную смесь охлаждали до комнатной температуры, выливали в 400 мл ледяной воды и 3 раза экстрагировали 300 мл этилацетата (водная фаза содержала большое количество белого хлопьевидного твердого вещества, а органическая фаза почти не содержала твердого вещества). Органическую фазу промывали один раз 200 мл воды и один раз 200 мл насыщенного рассола. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и пропускали через колонку с получением сложного трет-бутилового эфира (R, Z)-5-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-1'-карбоновой кислоты (A11-5, 2,38 г, выход: 79,6%) в виде светло-оранжевого вспененного твердого вещества. ЖХМС: м/з 407,1 [M+H]+
Стадия 6: Сложный трет-бутиловый эфир (R, Z)-5-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-1'-карбоновой кислоты (A11-5, 2,38 г, 5,85 ммоль) растворяли в 280 мл тетрагидрофурана, и смесь охлаждали до -65°C на бане с сухим льдом и ацетоном в атмосфере аргона. Медленно по каплям добавляли 1,5М DIBAL-H (5,07 мл, 7,61 ммоль), и температуру смеси продолжали поддерживать в течение 2 часов, после чего реакцию завершают. Реакционный раствор выливали в 300 мл насыщенного водного раствора тартрата калия-натрия, экстрагировали этилацетатом 3 раза. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали досуха с получением сырого продукта сложного трет-бутилового эфира (S)-5-((R)-трет-бутилсульфинил)амино)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-1'-карбоновой кислоты (А11-6, 2,48 г) в виде золотисто-желтого вспененного твердого вещества. ЖХМС: м/з 309,3 [M+H-100]+
Стадия 7: Сырой продукт сложный трет-бутиловый эфир (S)-5-((R)-трет-бутилсульфинил)амино)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-1'-карбоновой кислоты (A11-6, 2,48 г, 5,85 ммоль) растворяли в 120 мл метанола, смесь охлаждали в ледяной воде примерно до 0°C в атмосфере аргона, по каплям добавляли 4M раствор гидрохлорида в диоксане (18 мл, 72 ммоль), смесь подвергали реакции при комнатной температуре в течение 2 часов, а затем реакцию завершали. Реакционный раствор концентрировали досуха, добавляли безводный ацетонитрил для измельчения, и смесь фильтровали в атмосфере аргона с получением (S)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-5-амина гидрохлорида (A11, 1,77 г, выход: 99%) в виде темно-зеленого твердого порошка (очень легко впитывающего влагу и становящегося темно-зеленой масляной каплей), который сохраняли в атмосфере аргона. ЖХМС: м/з 205,3 [M+H]+
Промежуточное соединение A12: (S)-1-(((R)-трет-бутилсульфинил)амино)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]

Стадия 1: 2-Метил-3-метоксибромбензол (2,0 г, 9,95 ммоль, 1,0 экв.), N-бромсукцинимид (1,77 г, 9,95 ммоль, 1,0 экв.), пероксид дибензоила (241 мг, 0,995 ммоль, 0,1 экв.) и четыреххлористый углерод (40 мл) последовательно добавляли в круглодонную колбу объемом 100 мл, смесь перемешивали при 80°C в течение 16 часов, и реакционный раствор концентрировали при пониженном давлении с получением остатка. Остаток растворяли в этилацетате (200 мл), дважды промывали 2н водным раствором NaOH (50 мл × 2) и один раз насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого продукта. Сырой продукт очищали хроматографией на силикагеле с получением 1-бром-2-(бромметил)-3-метоксибензола (A12-1, 2,0 г, выход: 71,8%) в виде белого твердого вещества.
Стадия 2: Тетрагидрофуран (30 мл) и N-Boc-4-цианопиридин (1,8 г, 8,57 ммоль, 1,2 экв.) последовательно добавляли в круглодонную колбу объемом 100 мл, смесь охлаждали до -78°C, затем добавляли 2,0М ЛДА (5 мл, 10 ммоль, 1,4 экв.), и смесь перемешивали при -78°C в течение одного часа. Затем добавляли раствор 1-бром-2-(бромметил)-3-метоксибензола (A12-1, 2,0 г, 7,14 ммоль, 1,0 экв.) в тетрагидрофуране (15 мл), и смесь перемешивали при -78°C в течение 0,5 часа. Затем убирали низкотемпературную баню. Смеси давали возможность нагреться до комнатной температуры естественным путем и продолжали перемешивать в течение одного часа. После подтверждения с помощью ТСХ того, что исходные вещества полностью прореагировали, добавляли насыщенный рассол (30 мл) для гашения реакции, а затем смесь экстрагировали этилацетатом (100 мл * 2). Объединенную органическую фазу промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого продукта. Сырой продукт очищали хроматографией на силикагеле с получением N-Boc-4-(2-бром-6-метоксибензил)-4-цианопиридина (A12-2, 1,5 г, выход: 51,3%) в виде светло-желтого маслянистого вещества. ЖХМС: м/з 409,1, 411,1 [M+H]+
Стадия 3: N-Boc-4-(2-бром-6-метоксибензил)-4-цианопиридин (A12-2, 1,5 г, 3,66 ммоль, 1,0 экв.), Pd(AmPhos)2Cl2 (259 мг, 0,37 ммоль, 0,1 экв.), диизопропилэтиламин (2,37 г, 18,3 ммоль, 5,0 экв.), N, N-диметилацетамид (30 мл) и воду (4 мл) последовательно добавляли в сухую круглодонную колбу объемом 100 мл. При перемешивании атмосферу в реакционной системе трижды заменяли азотом, а затем реакционную смесь нагревали до 140°C и подвергали реакции в течение 16 часов. После завершения реакции реакционную жидкость охлаждают до комнатной температуры, разбавляют этилацетатом (100 мл) и фильтруют при пониженном давлении. Осадок на фильтре промывали этилацетатом (20 мл). Полученный фильтрат трижды промывали насыщенным рассолом (30 мл * 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого продукта. Сырой продукт очищали хроматографией на силикагеле, получая N-Boc-4-метокси-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин] (A12-3, 0,90 г, выход: 74,1%) в виде белого твердого вещества.
ЖХ-МС: м/з 332,2 [M+H]+
Стадия 4: N-Boc-4-метокси-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин] (A12-3, 900 мг, 2,72 ммоль), тетраэтоксититан (3,72 г, 16,3 ммоль), (R)-(+)-трет-бутилсульфинамид (395 мг, 3,26 ммоль) и тетрагидрофуран (20 мл) последовательно добавляли в сухую одногорлую колбу объемом 100 мл, и смесь перемешивали в течение 16 часов при нагревании и кипячении с обратным холодильником. После охлаждения реакционной смеси до комнатной температуры к реакционному остатку добавляли насыщенный рассол (60 мл), а затем полученную смесь перемешивали в течение 15 минут и фильтровали через диатомит. Водную смесь экстрагировали этилацетатом (3 × 80 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент 0-30% этилацетата:петролейный эфир), получая N-Boc-(R, Z)-1-((трет-бутилсульфинил)имид)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин] (A12-4, 800 мг, выход: 67,8%) в виде белого твердого вещества.
ЖХ-МС: м/з 435,2 [M+H]+
Стадия 5: Тетрагидрофуран (15 мл) и N-Boc-(R, Z)-1-((трет-бутилсульфинил)имид)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин] (A12-4, 800 мг, 1,84 ммоль, 1,0 экв.) последовательно добавляли в сухую трехгорлую колбу объемом 50 мл. По каплям добавляли диизобутилалюминийгидрид (5,52 мл, 1,5М в толуоле, 8,28 ммоль, 4,5 экв.) при -78°C в атмосфере азота, и смесь продолжали перемешивать в течение получаса. Затем смесь нагревали до 0°C и продолжали перемешивать в течение получаса. После завершения реакции реакционную смесь гасили тартратом калия-натрия (4 г, растворенных в 20 мл воды), перемешивали в течение получаса и экстрагировали этилацетатом (30 мл * 3). Полученную органическую фазу промывали насыщенным рассолом (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищают хроматографией на силикагеле (градиент 0-70% этилацетата:петролейный эфир) с получением трет-бутил (S)-1-(((R)-трет-бутилсульфинил)амино)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-формиата (A12-5, 720 мг, выход: 89,6%) в виде желтого твердого вещества. ЖХМС: м/з 437,2 [M+H]+
Стадия 6. Трет-бутил (S)-1-(((R)-трет-бутилсульфинил)амино)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-формиат (A12-5, 720 мг, 1,65 ммоль), дихлорметан (15 мл) и трифторуксусную кислоту (5 мл) последовательно добавляли в сухую одногорлую колбу объемом 50 мл, и полученную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли насыщенный водный раствор Na2CO3 до pH=7, и водную смесь экстрагировали дихлорметаном (3 × 50 мл). Объединенную органическую фазу промывали рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (градиент 0-70% этилацетата:петролейный эфир) с получением (R)-N-((S)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропил-2-сульфинамида (A12, 520 мг, выход: 93,7%) в виде желтого твердого вещества. ЖХМС: м/з 337,2 [M+H]+
Промежуточное соединение A13: (S)-2-хлор-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-4-амин

Стадия 1: 1-Трет-бутил 4-этилпиперидин-1,4-дикарбоксилат (10,7 г, 41,6 ммоль, 1,2 экв.) растворяли в ТГФ (50 мл), добавляли по каплям ЛДА (2M, 9,8 мл, 19,6 ммоль, 1,1 экв.) при -78°C, смесь перемешивали в течение 2 часов при этой температуре, и по каплям в систему добавляли 2-хлор-5-(хлорметил)тиазол (A13-1, 3 г, 34,7 ммоль, 1,0 экв.), растворенный в ТГФ (10 мл). Затем реакция продолжалась при этой температуре в течение 1,5 ч. Тестирование с помощью ТСХ показало, что небольшое количество исходных веществ не прореагировало. Реакционную смесь гасили водой, экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли и затем промывали насыщенным водным раствором хлорида натрия. Органическую фазу отделяли, затем сушили над безводным сульфатом натрия, фильтровали и концентрировали, смешивали с силикагелем и пропускали через колонку (ПЭ:ЭА:ДХМ=4:1:1) с получением 1-трет-бутил-4-этил 4-(2-хлортиазол-5-ил)метил)пиперидин-1,4-дикарбоксилата (A13-2, 2,4 г, выход: 35%) в виде желтого маслянистого вещества.
Стадия 2: 1-Трет-бутил-4-этил 4-(2-хлортиазол-5-ил)метил)пиперидин-1,4-дикарбоксилат (A13-2, 4 г, 10,3 ммоль) растворяли в ТГФ (100 мл), и ЛДА (2M, 8,5 мл, 16,6 ммоль) добавляли по каплям при -78°C. Смесь подвергали реакции в течение 1 ч, а затем реакцию завершали. Для гашения реакции добавляли насыщенный хлорид амина (100 мл), и смесь экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия. Фильтрат очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира 2-хлор-4-оксо-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-1'-карбоновой кислоты (A13-3, 1,9 г, выход: 54%) в виде желтого маслянистого вещества. ЖХМС: м/з 343 [M+H]+
Стадия 3: Сложный трет-бутиловый эфир 2-хлор-4-оксо-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-1'-карбоновой кислоты (A13-3, 710 мг, 2,07 ммоль), Ti(OEt)4 и (R)-2-метилпропан-2-сульфинамид (276 мг, 2,28 ммоль) добавляли в реакционную колбу, и смесь нагревали до 100°C в атмосфере азота в течение 5 часов. Реакцию завершали. Смесь охлаждали до комнатной температуры и разбавляли этилацетатом (50 мл), добавляли насыщенный рассол (15 мл), и белое твердое вещество выпадало в осадок. Смесь фильтровали. Осадок на фильтре промывали этилацетатом. Фильтрат промывали насыщенным рассолом. Органическую фазу отделяли, а затем сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира трет-бутил (E)-4-((трет-бутилсульфинил)имино)-2-хлор-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-1'-карбоновой кислоты (A13-4, 600 мг, выход: 68%) в виде светло-желтого твердого вещества. ЖХМС: м/з 426 [M+H]+
Стадия 4: Сложный трет-бутиловый эфир трет-бутил (E)-4-((трет-бутилсульфинил)имино)-2-хлор-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-1'-карбоновой кислоты (A13-4, 200 мг, 0,47 ммоль) растворяли в ТГФ (10 мл), и смесь охлаждали до -78°C в атмосфере азота. К реакционному раствору медленно по каплям добавляли 1,5М DIBAL-H (0,5 мл, 0,75 ммоль), и после того, как добавление DIBAL-H было завершено, смесь подвергали реакции при -78°C в течение 1 часа, а затем реакцию завершали. Для гашения реакции добавляли воду, а затем добавляли насыщенный раствор тартрата калия-натрия (20 мл). Смесь экстрагировали этилацетатом (50 мл × 2), и органическую фазу один раз промывали насыщенным водным раствором хлорида натрия. Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением сложного трет-бутилового эфира трет-бутил (S)-4-(((R)-трет-бутилсульфинил)амино)-2-хлор-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-1'-карбоновой кислоты A13-5 (155 мг, выход: 76%) в виде светло-желтого твердого вещества. ЖХМС: м/з 448 [M+H]+
Стадия 5: Сложный трет-бутиловый эфир трет-бутил (S)-4-(((R)-трет-бутилсульфинил)амино)-2-хлор-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-1'-карбоновой кислоты (A13-5, 155 мг, 0,35 ммоль) растворяли в 4М растворе гидрохлорида в диоксане (5 мл, 20 ммоль), и смесь подвергали реакции при комнатной температуре в течение 3 часов. Реакцию завершали. Реакционный раствор концентрировали с получением (S)-2-хлор-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-4-амина (A13, 145 мг, гидрохлорид) в виде светло-желтого масла. ЖХМС: м/з 244 [M+H]+
Синтез промежуточного соединения B2: 5-хлор-8-иод-[1,2,4]триазоло[4,3-c]пиримидина

Стадия 1: 2,4-Дихлор-5-иодпиримидин (1,1 г, 4 ммоль) и 20 мл безводного этанола добавляли в сухую колбу объемом 100 мл. К смеси медленно добавляли 80% смесь гидразингидрата (601 мг, 12 ммоль) при 0°C в атмосфере азота, и смесь продолжали перемешивать в течение 1 часа. После завершения реакции смесь фильтровали и промывали безводным этанолом с получением 2-хлор-4-гидразино-5-иодпиримидина (B2-1, 850 мг, выход: 78,7%).
1H ЯМР (400 МГц, CDCl3) δ 8,29 (с, 1H), 6,67 (с, 1H), 4,08 (с, 2H);
ЖХМС: м/з 271,1 [M+H]+
Стадия 2: 2-Хлор-4-гидразино-5-иодпиримидин (810 мг, 3 ммоль) и триметилортоформиат (10 мл) последовательно добавляли в высушенную колбу объемом 100 мл. В атмосфере азота смесь нагревали до 85°C и перемешивали в течение 5 часов. После завершения реакции полученный остаток выливали в насыщенный раствор NaCl (50 мл), экстрагировали этилацетатом (3 × 30 мл) и промывали насыщенным рассолом. Органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент 0-50% этилацетата/петролейный эфир) с получением 5-хлор-8-иод-[1,2,4]триазоло[4,3-c]пиримидина (B2, 420 мг, выход 50%) в виде светло-желтого твердого вещества.
ЖХМС: м/з 280,9 [M+H]+
Синтез промежуточного соединения B3: 5-хлор-8-иодимидазо[1,2-c]пиримидина

Стадия 1: 2,4-Дихлор-5-иодпиримидин (1,37 г, 5 ммоль), 2,2-диметоксиэтиламин (8,4 г, 10 ммоль) и безводный этанол (50 мл) последовательно добавляли в сухую колбу объемом 100 мл. Затем к реакционной смеси медленно по каплям добавляли триэтиламин (1,01 г, 10 ммоль) в атмосфере азота при 0°C, а затем смесь перемешивали при комнатной температуре в течение 10 часов. После завершения реакции реакционную смесь концентрировали в вакууме. К полученному концентрату добавляли 15 мл воды, смесь экстрагировали дихлорметаном (3 × 50 мл) и промывали насыщенным рассолом. Органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением 2-хлор-N-(2,2-диметоксиэтил)-5-иодпиримидин-4-амина (B3-1, 1,46 г, выход: 85%) в виде белого твердого вещества.
ЖХ-МС: м/з 344,2 [M+H]+
Стадия 2: 2-Хлор-N-(2,2-диметоксиэтил)-5-иодпиримидин-4-амин (B3-1, 1,03 г, 3 ммоль) и 10 мл концентрированной серной кислоты последовательно добавляли в высушенную колбу объемом 100 мл. В атмосфере азота смесь нагревали до 65°C и перемешивали в течение 2 часов. После завершения реакции реакционную жидкость охлаждали до комнатной температуры. Смесь медленно выливали в ледяную воду, затем pH смеси доводили до примерно 6-7 с помощью 4M раствора NaOH, а затем смесь фильтровали с получением 8-иодимидазо[1,2-c]пиримидин-5-ола (B3-2, 407 мг, выход: 52%) в виде грязно-белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,81 (с, 1H), 7,93 (д, J=1,4 Гц, 1H), 7,60 (с, 1H), 7,40 (д, J=1,4 Гц, 1H);
ЖХ-МС: м/з 262,2 [M+H]+
Стадия 3: 8-Иодимидазо[1,2-c]пиримидин-5-ол (B3-2, 522 мг, 2 ммоль) и оксихлорид фосфора (8 мл) последовательно добавляли в сухую одногорлую колбу объемом 50 мл. В атмосфере азота медленно по каплям добавляли N, N-диизопропилэтиламин (1 мл), а затем смесь нагревали до 120°C и перемешивали в течение 5 часов. После завершения реакции реакционную жидкость охлаждали до комнатной температуры и концентрировали в вакууме, затем гасили насыщенным раствором бикарбоната натрия, экстрагировали этилацетатом (3 × 40 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали хроматографией на силикагеле (градиент 0-30% этилацетата:петролейный эфир) с получением 5-хлор-8-иодимидазо[1,2-c]пиримидина (B3, 360 мг, выход: 55%) в виде светло-желтого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,24 (с, 1H), 8,20 (д, J=1,4 Гц, 1H), 7,81 (д, J=1,4 Гц, 1H);
ЖХ-МС: м/з 280,1 [M+H]+
Синтез промежуточного соединения C1: 2-амино-3-хлорпиридин-4-сульфида натрия
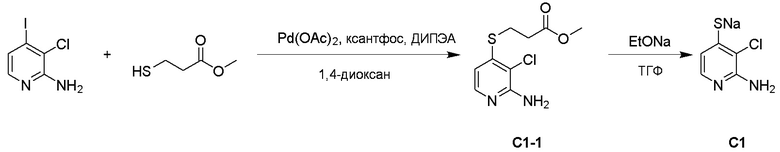
Стадия 1: 3-Хлор-4-иодпиридин-2-амин (2,5 г, 9,82 ммоль, 1,0 экв.), Ксантфос (341 мг, 0,59 ммоль, 0,06 экв.), ацетат палладия (110 мг, 0,49 ммоль, 0,05 экв.), ДИПЭА (3,25 мл, 19,6 ммоль, 2,0 экв.), метил 3-меркаптопропионат (1,19 мл, 10,8 ммоль, 1,1 экв.) и 1,4-диоксан (32,5 мл) последовательно добавляли в сухую трехгорлую круглодонную колбу объемом 100 мл. При перемешивании атмосферу в реакционной системе трижды заменяли азотом, затем смесь нагревали до 100°C и подвергали реакции в течение 3 часов. После завершения реакции реакционную жидкость охлаждали до комнатной температуры, разбавляли этилацетатом (50 мл) и фильтровали при пониженном давлении, и осадок на фильтре промывали этилацетатом (25 мл). Полученный фильтрат концентрировали в вакууме и полученный остаток очищали хроматографией на силикагеле (градиент 0-30% этилацетата:петролейный эфир) с получением метил 3-((2-амино-3-хлорпиридин-4-ил)тио)пропионата (C1-1, 2,0 г, выход: 78%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,89 (д, J=5,4 Гц, 1H), 6,53 (д, J=5,5 Гц, 1H), 4,87 (с, 2H), 3,74 (с, 3H), 3,24 (т, J=7,5 Гц, 2H), 2,75 (т, J=7,5 Гц, 2H).
Стадия 2: Соединение C1-1 (2 г, 8,11 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (28 мл) в сухой круглодонной трехгорлой колбе объемом 100 мл, и по каплям добавляли натрий в этаноле (2,9 г, 8,51 ммоль, 1,05 экв., 20% масс.) к реакционному раствору при комнатной температуре в атмосфере азота, а затем смесь перемешивали в течение одного часа. После завершения реакции смесь разбавляли дихлорметаном (60 мл) и обрабатывали ультразвуком в течение 5 минут, а затем фильтровали при пониженном давлении. Осадок на фильтре сушили в вакууме с получением 2-амино-3-хлорпиридин-4-сульфида натрия (C1, 1,4 г, выход: 89%) в виде желтого твердого вещества.
Синтез промежуточного соединения C2: 2,3-дихлорпиридин-4-сульфида натрия
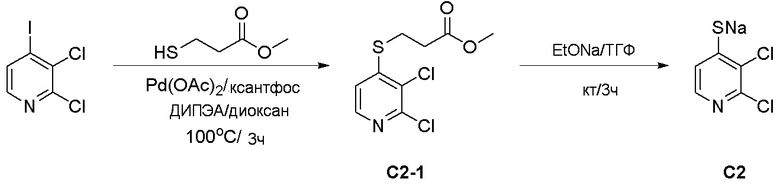
Стадия 1: 2,3-Дихлор-4-иодпиридин (1,0 г, 3,65 ммоль, 1,0 экв.), метил 3-меркаптопропионат (480 мг, 4,02 ммоль, 1,1 экв.) И N, N-диизопропилэтиламин (950 мг, 7,3 ммоль) растворяли в 1,4-диоксане (15 мл), атмосферу в реакционной системе трижды заменяли аргоном, и добавляли ацетат палладия (82 мг, 0,37 ммоль, 0,1 экв.) и Ксантфос (211 мг, 0,37 ммоль, 0,1 экв.) в атмосфере аргона. Затем смесь нагревали до 100°C и подвергали реакции в течение 3 часов, после чего реакцию завершали. Смесь экстрагировали этилацетатом (100 мл), фильтровали через диатомит и концентрировали. Остаток очищали высокоэффективной жидкостной хроматографией с получением метил 3-((2,3-дихлорпиридин-4-ил)тио)пропионата (C2-1, 550 мг, выход: 56,5%) в виде не совсем белого твердого вещества. ЖХМС: м/з 266,0 [M+H]+
Стадия 2: Метил 3-((2,3-дихлорпиридин-4-ил)тио)пропионат (C2-1, 100 мг, 0,37 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (10 мл) и этаноле (0,5 мл). Натрий в этаноле (27 мг, 0,39 ммоль, 1,05 экв.) добавляли в атмосфере азота, смесь подвергали реакции при комнатной температуре в течение 3 часов, реакцию не завершали, добавляли натрий в этаноле (27 мг, 0,39 ммоль, 1,05 экв.), смесь подвергали реакции при комнатной температуре в течение 3 часов, а затем реакцию завершали. Добавляли очищенную воду, и смесь лиофилизировали с получением сырого продукта 2,3-дихлорпиридин-4-сульфида натрия (C21, 50 мг, выход: 100%) в виде светло-желтого твердого вещества. ЖХМС: м/з 180,0 [M+H]+
Синтез промежуточного соединения C3: 2-(трифторметил)пиридин-3-сульфида натрия
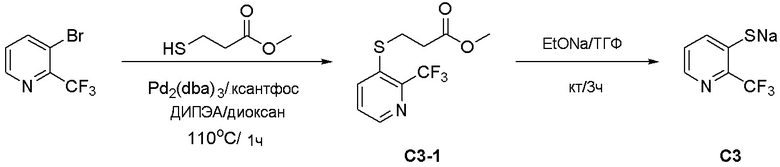
Стадия 1: 3-Бром-2-трифторметилпиридин (400 мг, 1,77 ммоль, 1,0 экв.), метил 3-меркаптопропионат (235 мг, 1,95 ммоль, 1,1 экв.) и N, N-диизопропилэтиламин (460 мг, 3,54 ммоль, 2,0 экв.) растворяли в 1,4-диоксане (15 мл), атмосферу в реакционной системе трижды заменяли аргоном, и Pd2(dba)3 (160 мг, 0,18 ммоль, 0,1 экв.) и Ксантфос (205 мг, 0,36 ммоль, 0,2 экв.) добавляли в атмосфере аргона. Затем смесь нагревали до 110°C и подвергали реакции в течение одного часа, после чего реакцию завершали. Смесь экстрагировали этилацетатом (100 мл), фильтровали через диатомит, концентрировали и пропускали через колонку (ПЭ:ЭА=8/1 ~ 6/1) с получением метил 3-((2-(трифторметил)пиридин-3-ил)тио)пропионата (C3-1, 450 мг, выход: 95,7%) в виде светло-желтого маслянистого вещества. ЖХМС: м/з 266,1 [M+H]+
Стадия 2: Метил 3-((2-(трифторметил)пиридин-3-ил)тио)пропионат (C3-1, 300 мг, 1,13 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (10 мл) и этаноле (0,5 мл). Натрий в этаноле (84 мг, 1,23 ммоль, 1,1 экв.) добавляли в атмосфере азота, смесь подвергали реакции при комнатной температуре в течение 3 часов, реакцию не завершали, добавляли натрий в этаноле (83 мг, 1,23 ммоль, 1,1 экв.), смесь подвергали реакции при комнатной температуре в течение 3 часов, а затем реакцию завершали. Тетрагидрофуран удаляли при пониженном давлении при комнатной температуре. Добавляли очищенную воду, и смесь лиофилизировали с получением сырого продукта 2-(трифторметил)пиридин-3-сульфида натрия (C3, 500 мг, выход: более 100%) в виде светло-желтого твердого вещества. ЖХМС: м/з 179,9 [M+H]+
Синтез промежуточного соединения C4: 2-(трифторметил)пиридин-4-сульфида натрия
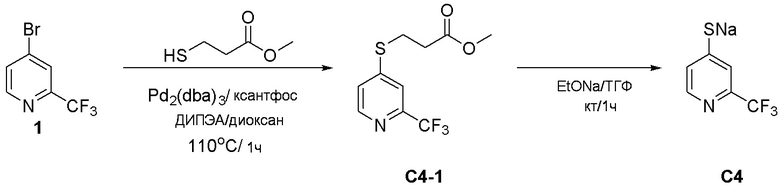
Стадия 1: 4-Бром-2-трифторметилпиридин (1,0 г, 4,4 ммоль, 1,0 экв.), метил 3-меркаптопропионат (760 мг, 6,3 ммоль, 1,4 экв.) и N, N-диизопропилэтиламин (2,17 г, 16,8 ммоль, 3,8 ммоль, 3,8 экв.) растворяли в 1,4-диоксане (25 мл), атмосферу в реакционной системе трижды заменяли аргоном, и добавляли Pd2(dba)3 (200 мг, 0,22 ммоль, 0,05 экв.) и Ксантфос (124 мг, 0,22 ммоль, 0,05 экв.) в атмосфере аргона. Затем смесь нагревали до 110°C и подвергали реакции в течение 1 часа, после чего реакцию завершали. Смесь экстрагировали этилацетатом (100 мл), фильтровали через диатомит, концентрировали и пропускали через колонку (ПЭ:ЭА=5/1) с получением метил 3-((2-(трифторметил)пиридин-4-ил)тио)пропионата (C4-1, 1,08 г, выход: 97%) в виде светло-желтого маслянистого вещества. ЖХМС: м/з 266,2 [M+H]+
Стадия 4: 3-((2-(Трифторметил)пиридин-4-ил)тио)пропионат (C4-1, 230 мг, 0,85 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (10 мл) и этаноле (0,5 мл). Натрий в этаноле (294 мг, 0,87 ммоль, 1,02 экв.) добавляли в атмосфере азота, смесь подвергали реакции при комнатной температуре в течение 1 часа, и реакцию завершали. Тетрагидрофуран удаляли при пониженном давлении при комнатной температуре. Добавляли очищенную воду, и смесь лиофилизировали с получением сырого продукта 2-(трифторметил)пиридин-4-сульфида натрия (C4, 150 мг) в виде светло-желтого твердого вещества. ЖХМС: м/з 180,0 [M+H]+
Синтез промежуточного соединения C5: 2-(трифторметил)пиридин-4-сульфида натрия
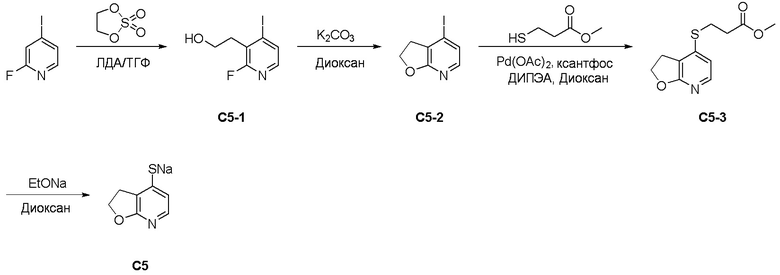
Стадия 1: 2-Фтор-4-иодпиридин (2,0 г, 8,97 ммоль) растворяли в ТГФ (30 мл). Атмосферу в реакционной системе трижды заменяли азотом, затем температуру понижали до -65°C, и по каплям добавляли ЛДА (2,0M в ТГФ, 5,4 мл, 10,80 ммоль). Реакционный раствор постепенно становился коричневым. После завершения добавления ЛДА температуру поддерживали в течение 1,5 часов. По каплям добавляли раствор 1,3,2-диоксотиофен-2,2-диоксида (1,45 г, 11,7 ммоль) в ТГФ (30 мл). Затем смесь нагревали до комнатной температуры естественным путем и перемешивали в течение ночи. Исчезновение исходного вещества подтверждали с помощью ТСХ (петролейный эфир/этилацетат=1/1). Реакционную смесь охлаждали до 0°C, и по каплям добавляли концентрированную соляную кислоту (4,48 мл, 40,2 ммоль). Затем смесь нагревали до комнатной температуры и перемешивали 3 часа. Реакционную жидкость выливали в насыщенный водный раствор NaHCO3 (50 мл), экстрагировали этилацетатом (3 × 30 мл), промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха, а затем очищали хроматографией на силикагеле с получением 2-(2-фтор-4-иодпиридин-3-ил)этан-1-ола (C5-1, 2,07 г, выход: 86,2%).
Стадия 2: Карбонат калия (4,28 г, 30,9 ммоль) добавляли к раствору 2-(2-фтор-4-иодпиридин-3-ил)этан-1-ола (C5-1, 2,07 г, 8,97 ммоль) в диоксане (60 мл). Смесь перемешивали при 115°C в течение 48 часов в атмосфере азота. ЖХМС и ТСХ (петролейный эфир/этилацетат=1/1) показали, что в реакционной системе были в основном исходные вещества и лишь небольшое количество продуктов. Добавляли карбонат цезия (7,56 г, 23,2 ммоль), и реакция продолжалась при 115°C в течение 6 часов. Было подтверждено с помощью ЖХМС и ТСХ (петролейный эфир/этилацетат=1/1), что исходные вещества почти полностью прореагировали. Реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, фильтровали с помощью диатомита и элюировали этилацетатом. Фильтрат промывали насыщенным рассолом (2 × 50 мл), сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха и затем очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир/дихлорметан=от 0/5/2 до 1/5/2) с получением 4-иод-2,3-дигидрофурано[2,3-b]пиридина (C5-2, 1,21 г, выход: 63,2%) в виде белого твердого вещества. ЖХМС: м/з 248,2 [M+H]+
Стадия 3: Метил 3-меркаптопропионат (437 мг, 3,64 ммоль), ДИПЭА (1,26 г, 9,71 ммоль), Ксантфос (70 мг, 0,12 ммоль) и Pd(OAC)2 (30 мг, 0,13 ммоль) добавляли к раствору 4-иод-2,3-дигидрофурано[2,3-b]пиридина (C5-2, 600 мг, 2,43 ммоль) в диоксане (10 мл). В атмосфере азота смесь нагревали до 100°C и подвергали реакции в течение 3 часов. ТСХ (петролейный эфир/этилацетат=1/1) и ЖХМС показали, что реакция завершилась. Реакционный раствор разбавляли этилацетатом и фильтровали. Фильтрат концентрировали досуха и очищали колоночной хроматографией на силикагеле с получением метил 3-((2,3-дигидрофурано[2,3-b]пиридин-4-ил)тио)пропионата (C5-3, 565 мг, выход: 97,2%). ЖХМС: м/з 240,3 [M+H]+
Стадия 4: Этанольный раствор этоксида натрия (20% (масс./масс.), 185 мг, 0,54 ммоль) добавляли к раствору метил 3-((2,3-дигидрофурано[2,3-b]пиридин-4-ил)тио)пропионата (C5-3, 145 мг, 0,61 ммоль) в ТГФ (5 мл) при 0°C. Смесь подвергали реакции при комнатной температуре в течение 2 часов, и ТСХ (этилацетат/петролейный эфир=1/1) и ЖХМС показали, что реакция завершилась. Без дополнительной обработки реакционную жидкость напрямую использовали для следующей реакции.
Синтез промежуточного соединения C6: 2-метиламино-3-хлорпиридин-4-сульфида натрия

Стадия 1: 2,3-Дихлор-4-иодпиридин (1,0 г, 3,65 ммоль) реагировал с раствором метиламин/этанол (27%, 25 мл) в закрытом резервуаре при 100°C в течение 12 часов. Мониторинг с помощью ТСХ показал, что реакция завершилась (петролейный эфир:этилацетат=5:1). Реакционный раствор концентрировали при пониженном давлении и очищали хроматографией на силикагеле (градиент 0-5% этилацетата:петролейный эфир) с получением 3-хлор-4-иод-N-метилпиридин-2-амина (C6-1, 400 мг, выход: 41%). ЖХМС: м/з 268,9 [M+H]+
Стадия 2: Ксантфос (72 мг, 0,15 ммоль), Pd(OAC)2 (34 мг, 0,15 ммоль), ДИПЭА (770 мг, 5,96 ммоль) и метил 3-меркаптопропионат (270 мг, 2,23 ммоль) добавляли к раствору 3-хлор-4-иод-N-метилпиридин-2-амина (C6-1, 400 мг, 1,49 ммоль) в безводном 1,4-диоксане (20 мл). Смесь подвергали реакции в течение 5 ч при 100°C. ТСХ (петролейный эфир:этилацетат=5/1) показала, что реакция завершилась. После охлаждения реакционной смеси до комнатной температуры добавляли воду (50 мл) и этилацетат (30 мл), и слои разделяли. Водную фазу экстрагировали этилацетатом (2 × 40 мл). Органическую фазу объединяли, промывали насыщенным рассолом (3 × 50 мл), сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали хроматографией на силикагеле (петролейный эфир:этилацетат=10:1) с получением метил 3-((3-хлор-2-(метиламино)пиридин-4-ил)тио)пропионата (C6-2, 250 мг, выход: 65%). ЖХМС: м/з 261,0 [M+H]+
Стадия 3. Метил 3-((3-хлор-2-(метиламино)пиридин-4-ил)тио)пропионат (C6-2, 114 мг, 0,44 ммоль) растворяли в диоксане (6 мл). В атмосфере аргона по каплям добавляли раствор EtONa (20% (масс./масс.), 150 мг, 0,44 ммоль) в этаноле, и смесь перемешивали при комнатной температуре в течение примерно 2,5 часов. ЖХМС показала, что реакция завершилась. Реакционный раствор непосредственно использовали для следующей реакции (выход: 100%). ЖХМС: м/з 174,8 [M+H-23]+
Синтез промежуточного соединения C7: 1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-4-меркаптана натрия
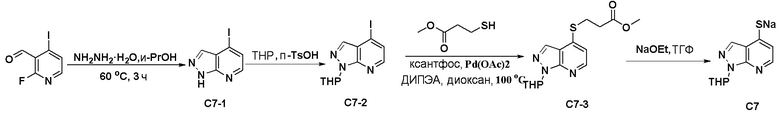
Стадия 1. Гидразингидрат (3032 мг, 60,56 ммоль) добавляли к раствору 2-фтор-3-формил-4-иодпиридина (1900 мг, 7,57 ммоль) в изопропаноле (30 мл), и смесь перемешивали при 60°C в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении для удаления части растворителя, затем выливали в воду и фильтровали. Осадок на фильтре промывали водой с получением 4-иод-1H-пиразоло[3,4-b]пиридина (C7-1, 1,8 г, выход: 97%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,19 (д, J=4,4 Гц, 1H), 7,98 (с, 1H), 7,68 (д, J=4,8 Гц, 1H).
Стадия 2: п-Толуолсульфоновую кислоту (28 мг, 0,16 ммоль) и THP (206 мг, 2,45 ммоль) последовательно добавляли к раствору 4-иод-1H-пиразоло[3,4-b]пиридина (C7-1, 400 мг, 1,63 ммоль) в тетрагидрофуране (10 мл). Смесь перемешивали при 60°C в течение 16 часов. Реакционный раствор разбавляли этилацетатом (40 мл), промывали насыщенным рассолом (2 × 40 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент 0-50% этилацетата:петролейный эфир) с получением 4-иод-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-B] пиридина (C7-2, 390 мг, выход: 73%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,17 (д, J=4,8 Гц, 1H), 7,93 (с, 1H), 7,58 (д, J=5,2 Гц, 1H), 6,10 (дд, J=10,4, 2,4 Гц, 1H), 4,14-4,10 (м, 1H), 3,82 (тд, J=11,6, 2,8 Гц, 1H), 2,69-2,59 (м, 1H), 2,17-2,13 (м, 1H), 2,00-1,96 (м, 1H), 1,83-1,74 (м, 2H), 1,64-1,62 (м, 1H).
Стадия 3: 4-Иод-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-B]пиридин (C7-2, 390 мг, 1,18 ммоль), метил 3-меркаптопропионат (157 мг, 1,30 ммоль) и N, N-диизопропилэтиламин (306 мг, 2,37 ммоль) растворяли в 1,4-диоксане (10 мл), атмосферу в реакционной системе трижды заменяли аргоном, и Pd(OAc)2 (27 мг, 0,12 ммоль) и Ксантфос (137 мг, 0,24 ммоль) добавляли в атмосфере аргона. После завершения добавления смесь нагревали до 110°C и подвергали реакции в течение 2 часов, после чего реакцию завершали. Смесь экстрагировали этилацетатом (30 мл), промывали насыщенным рассолом (2 × 30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент 0-50% этилацетата:петролейный эфир) с получением метил 3-((1H-пиразоло[3,4-b]пиридин-4-ил)тио)пропионата (C7-3, 280 мг, выход: 74%) в виде желтовато-коричневого маслянистого вещества. ЖХМС: м/з 322,3 [M+H]+
1H ЯМР (400 МГц, CDCl3): δ 8,40 (д, J=5,2 Гц, 1H), 8,08 (с, 1H), 6,95 (д, J=5,2 Гц, 1H), 6,10 (дд, J=10,8, 2,4 Гц, 1H), 4,14 ~ 4,10 (м, 1H), 3,83 (тд, J=11,2 Гц, 1H), 3,73 (с, 3H), 3,40 (т, J=7,2 Гц, 2H), 2,79 (т, J=7,2 Гц, 2H), 2,68-2,58 (м, 1H), 2,16-2,13 (м, 1H), 2,00-1,55 (м, 2H), 1,63-1,60 (м, 1H).
Стадия 4: Метил 3-((1H-пиразоло[3,4-b]пиридин-4-ил)тио)пропионат (C7-3, 280 мг, 0,87 ммоль) растворяли в 1,4-диоксане (7 мл) в трехгорлой колбе с трубчатым холодильником. В атмосфере азота смесь охлаждали до 0°C, и добавляли этанольный раствор этоксида натрия в (20% (масс./масс.), 266 мг, 0,78 ммоль). Смесь перемешивали 2 ч при комнатной температуре. ТСХ (дихлорметан/метанол=20/1) показала, что реакция завершилась, и реакционную жидкость, содержащую 1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-4-меркаптан натрия (C7) непосредственно использовали на следующей стадии.
Пример 1: Синтез соединения 1
(S)-1'-((2,3-дигидрофуран[2,3-b]пиридин-4-ил)тио)имидазоло[1,2-c]пиримидин-5-ил)-5,7-дигидроспироциклопента[b]пиридин-6,4'-пиперидин]-5-амина
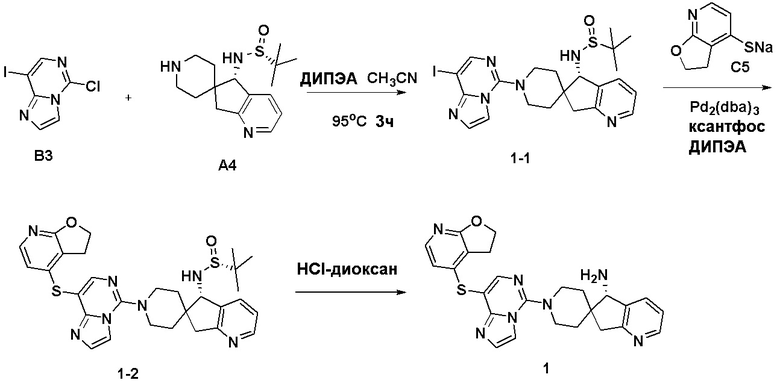
Стадия 1: В атмосфере азота 5-хлор-8-иодимидазо[1,2-c]пиримидин (B3, 80 мг, 0,285 ммоль), ((R)-N-((S)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (A4 105 мг, 0,342 ммоль), ДИПЭА (55 мг, 0,428 ммоль) и CH3CN (5 мл) последовательно добавляли в сухую одногорлую колбу объемом 25 мл, а затем смесь перемешивали при 95°C в течение 3 часов. После завершения реакции полученный реакционный раствор фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент 0-10% метанола/этилацетат) с получением соединения (1-1, 101 мг, выход: 65%). ЖХМС: м/з 551,1 [M+H]+
Стадия 2: 2,3-Дигидрофуран[2,3-b]пиридин-4-тиоформиат натрия (107 мг, 0,61 ммоль) добавляли к диоксану (15 мл) для разбавления, а затем добавляли (S)-N-((S)-1'-(8-иодимидазоло[1,2-C]пиримидин-5-ил)-5,7-дигидроспиро[циклопентан[B]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (1-1, 200 мг, 0,36 ммоль), ДИПЭА (141 мг, 1,09 ммоль), Ксантфос (63 мг, 0,11 ммоль) и Pd2(dba)3 (50 мг, 0,06 ммоль). В атмосфере азота смесь нагревали до 100°C и перемешивали в течение 3 часов. ТСХ (петролейный эфир/этилацетат=1/1) и ЖХМС показали, что реакция завершилась. Реакционный раствор разбавляли этилацетатом и фильтровали. Остаток на фильтре элюировали этилацетатом. Фильтрат концентрировали досуха и очищали колоночной хроматографией на силикагеле с получением (S)-N-((S)-1'-(8-((2,3-дигидрофуран[2,3-b]пиридин-4-ил)тио)имидазоло[1,2-c]пиримидин-5-ил)-5,7-дигидроспироциклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфаниламида (1-2, 217 мг, выход: 98,9%). ЖХМС: м/з 576,6 [M+H]+
Стадия 3: В атмосфере азота HCl/диоксан (4M, 0,2 мл, 0,80 ммоль) медленно добавляли к раствору (S)-N-((S)-1'-(8-((2,3-дигидрофуран[2,3-b]пиридин-4-ил)тио)имидазоло[1,2-c]пиримидин-5-ил)-5,7-дигидроспироциклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфаниламида (50 мг, 0,29 ммоль) в дихлорметане (5 мл) при 0°C, смесь перемешивали при комнатной температуре в течение 2 часов, и ТСХ (дихлорметан/метанол=8/1) и ЖХМС показали, что реакция завершилась. Реакционный раствор охлаждали до 0°C, медленно добавляли раствор аммиака и метанола, чтобы довести pH до примерно 10, а затем смесь концентрировали при пониженном давлении и очищали препаративной ВЭЖХ с получением формиата (S)-1'-(8-((2,3-дигидрофуран[2,3-b]пиридин-4-ил)тио)имидазоло[1,2-c]пиримидин-5-ил)-5,7-дигидроспироциклопентадиен[b]пиридин-6,4'-пиперидин]-5-амина (соединение 1, 8,86 мг, выход: 20,0%). ЖХМС: м/з 472,5 [M+H]+
1H ЯМР (400 МГц, MeOD) δ 8,47 (д, J=4,0 Гц, 2H), 8,09 (с, 1H), 7,92 (д, J=7,6 Гц, 1H), 7,85 (д, J=1,6 Гц, 1H), 7,59 (д, J=1,6 Гц, 1H), 7,55 (д, J=6,0 Гц, 1H), 7,37 ~ 7,34 (м, 1H), 6,21 (д, J=5,6 Гц, 1H), 4,72 ~ 4,67 (м, 2H), 4,38 (с, 1H), 4,06 ~ 4,03 (м, 2H), 3,48 ~ 3,42 (м, 2H), 3,28 (с, 2H), 3,14 (д, J=16,8 Гц, 2H) , 2,11 ~ 2,078 (м, 2H), 1,76 ~ 1,70 (м, 2H).
Пример 2: Синтез соединения 2
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина
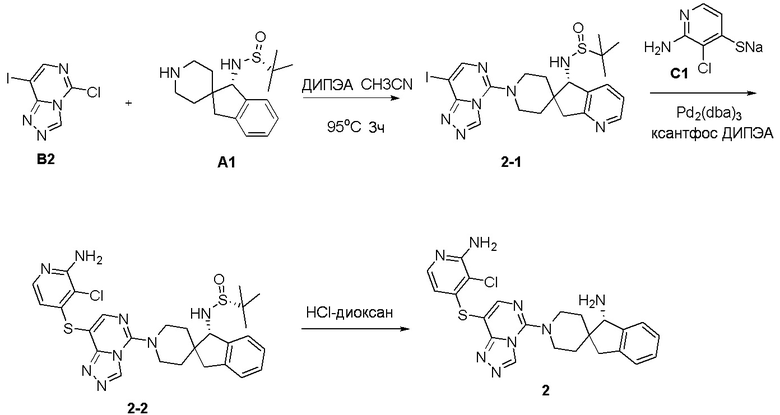
Стадия 1: В атмосфере азота 5-хлор-8-иод-[1,2,4]триазоло[4,3-c]пиримидин (B2, 80 мг, 0,285 ммоль), ((R)-N-((S)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (A1, 105 мг, 0,342 ммоль), ДИПЭА (55 мг, 0,428 ммоль) и CH3CN (5 мл) последовательно добавляли в сухую одногорлую колбу объемом 25 мл, а затем смесь перемешивали в течение 3 часов при 95°C. После завершения реакции полученный остаток фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент 0-10% метанола/этилацетат) с получением (R)-N-((S)-1'-(8-иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (2-1, 101 мг, выход: 65%) в виде желтого твердого вещества, ЖХМС: м/з 551,1 [M+H]+
Стадия 2: В атмосфере азота (R)-N-((S)-1'-(8-иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (2-1, 100 мг, 0,18 ммоль), 2-амино-3-хлорпиридин-4-меркаптан (49 мг, 0,27 ммоль), Pd2(dba)3 (16 мг, 0,018 ммоль), ксантфос (21 мг, 0,036 ммоль), ДИПЭА (58 мг, 0,45 ммоль) и раствор 1,4-диоксана (10 мл) последовательно добавляли в сосуд для реакции при микроволновом излучении объемом 5 мл, и смесь нагревали в микроволновом излучении до 100°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент 0-10% метанола/этилацетат) с получением (R)-N-((R)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (2-2, 65 мг, выход: 63%). ЖХ-МС: м/з 584,2 [M+H]+
Стадия 3: В атмосфере азота (R)-N-((R)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (2-2, 60 мг, 0,10 ммоль) и метанол (0,6 мл) последовательно добавляли в одногорлую колбу объемом 50 мл, по каплям добавляли раствор хлористоводородной кислоты в 1,4-диоксане (0,06 мл, 4M) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией с получением (S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (соединение 2, 20 мг, выход: 42%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,38 (с, 1H), 7,98 (с, 1H), 7,58 (д, J=5,4 Гц, 1H), 7,36 (д, J=4,8 Гц, 1H), 7,22 (дт, J=8,0, 4,0 Гц, 3H), 6,33 (с, 2H), 5,96 (д, J=5,6 Гц, 1H), 4,22-4,07 (м, 2H), 4,00 (с, 1H), 3,49 (дд, J=22,5, 11,2 Гц, 2H), 3,12 (д, J=15,6 Гц, 1H), 2,75 (д, J=15,6 Гц, 1H), 2,03-1,86 (м, 2H), 1,63 (д, J=13,6 Гц, 1H), 1,39-1,29 (м, 1H);
ЖХ-МС: м/з 479,1 [M+H]+
Пример 3: Синтез соединения 3
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амина

Соединение 3 синтезировали в три стадии в соответствии со способом синтеза соединения 2, используя промежуточное соединение A2 вместо промежуточного соединения A1.
1H ЯМР (400 МГц, ДМСО-d6)) δ 9,40 (с, 1H), 8,35 (д, J=4,3 Гц, 1H), 7,99 (с, 1H), 7,71 (д, J=7,4 Гц, 1H), 7,58 (д, J=5,4 Гц, 1H), 7,21 (дд, J=7,4, 5,0 Гц, 1H), 6,36 (с, 2H), 5,96 (д, J=5,4 Гц, 1H), 4,23-4,08 (м, 2H), 4,02 (с, 1H), 3,51 (дд, J=23,3, 11,6 Гц, 2H), 3,16 (д, J=16,3 Гц, 1H), 2,84 (д, J=16,3 Гц, 1H), 2,04-1,91 (м, 2H), 1,65 (д, J=13,4 Гц, 1H), 1,32 (д, J=14,8 Гц, 1H);
ЖХ-МС: м/з 480,1 [M+H]+
Пример 4: Синтез соединения 4
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазоло[1,2-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина
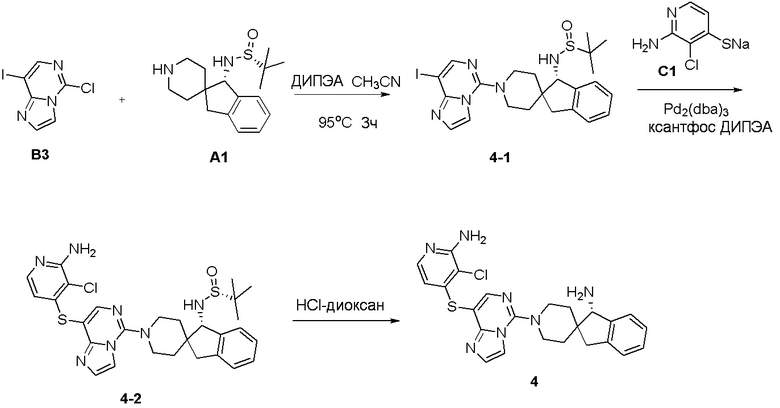
Соединение 4 (формиат, белое твердое вещество) синтезировали в три стадии в соответствии со способом синтеза соединения 2, используя промежуточное соединение B3 вместо промежуточного соединения B2.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,03 (с, 1H), 7,83 (с, 1H), 7,64-7,49 (м, 2H), 7,43 (с, 1H), 7,30-7,17 (м, 3H), 6,33 (с, 2H), 5,79 (д, J=5,2 Гц, 1H), 4,12 (с, 1H), 3,95 (д, J=12,8 Гц, 2H), 3,33 (дд, J=20,0, 10,8 Гц, 2H), 3,14 (д, J=15,6 Гц, 1H), 2,82 (д, J=15,6 Гц, 1H), 1,98 (д, J=9,6 Гц, 2H), 1,61 (д, J=12,8 Гц, 1H), 1,42 (д, J=12,8 Гц, 1H), 1,23 (с, 2H);
ЖХ-МС: м/з 478,1 [M+H]+
Пример 5: Синтез соединения 5
(S)-1'-(8-((3-хлор-2-(метиламино)пиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амина

Стадия 1: 2-Метиламино-3-хлорпиридин-4-сульфид натрия (C6, 86 мг) растворяли в 10 мл раствора диоксана, и добавляли (R)-N-((S)-1'-(8-иодимидазоло[1,2-C]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (161 мг, 0,29 ммоль), Pd2(dba)3 (86 мг, 0,094 ммоль), ксантфос (108 мг, 0,19 ммоль) и ДИПЭА (400 мг, 3,12 ммоль). Смесь подвергали реакции при 100°C в течение 5 ч в атмосфере аргона. ЖХМС-тестирование показало, что реакция завершилась. После охлаждения смеси до комнатной температуры добавляли дихлорметан (30 мл), и смесь фильтровали. К маточному раствору добавляли воду (40 мл), и слои разделяли. Водную фазу экстрагировали дихлорметаном (2 × 30 мл). Органические фазы объединяли, промывали насыщенным рассолом (3 × 40 мл), сушили над безводным сульфатом натрия, фильтровали и очищали хроматографией на силикагеле (дихлорметан:метанол=15:1) с получением (R)-N-((s)-1'-(8-((3-хлор-2-(метиламино)пиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамида (5-1, 135 мг, выход: 73%) в виде светло-желтого твердого вещества. ЖХМС: м/з 596,9 [M+H]+
Стадия 2: (R)-N-((s)-1'-(8-((3-хлор-2-(метиламино)пиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (135 мг, 0,23 ммоль) растворяли в метаноле (20 мл), медленно по каплям добавляли HCl/диоксан (4M, 1,2 мл) в атмосфере азота, и смесь подвергали реакции при комнатной температуре в течение 40 минут. ЖХМС-тестирование показало, что реакция завершилась. Реакционный раствор выливали в ледяной насыщенный раствор бикарбоната натрия. Смешанный раствор лиофилизировали, и добавляли дихлорметан/метанол (10:1, 20 мл). Смесь фильтровали. Фильтрат концентрировали досуха, а затем очищали препаративной ВЭЖХ с получением белого твердого вещества (соединение 5, 33 мг, выход: 30%). ЖХМС: м/з 493,0 [M+H]+
1H ЯМР (400 МГц, ДМСО-d6) δ 8,33 (д, J=4,4 Гц, 1H), 8,03 (с, 1H), 7,84 (д, J=1,2 Гц, 1H), 769 (д, J=7.6 Гц, 1H), 7,65 (д, J=5,2 Гц, 1H), 7,57 (д, J=1,2 Гц, 1H), 7,20 (дд, J=7,2, 5,2 Гц, 1H), 6,60 (кв, J=4,4 Гц, 1H), 5,81 (д, J=5,2 Гц, 1H), 3,98-3,89 (м, 3H), 3,38-3,22 (м, 2H), 3,15-3,11 (д, J=16,0 Гц, 1H), 2,85 (д, J=4,8 Гц, 3H), 2,81 (д, J=16,4 Гц, 1H), 2,50-2,39 (м, 1H), 2,05-1,90 (м, 3H), 1,67 (д, J=13,2 Гц, 1H), 1,28 ~ 1,25 (д, J=13,2 Гц, 1H).
Пример 6: Синтез соединения 6
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амина
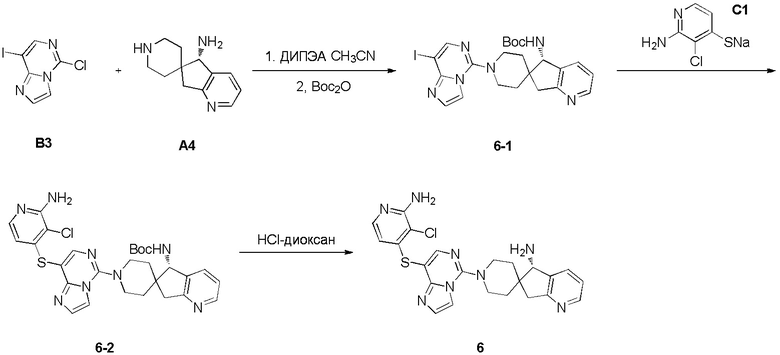
Стадия 1: B3 (1,37 г, 4,9 ммоль), A4 (1,35 г, 4,9 ммоль) и ДИПЭА (4,86 мл, 29,41 ммоль) последовательно добавляли к 3 мл ацетонитрила в одногорлой колбе объемом 25 мл, а затем смесь перемешивали в течение 2 часа при 80°C. После завершения реакции смесь охлаждали до комнатной температуры, затем добавляли Boc2O (1,6 г, 7,35 ммоль, 1,5 экв.), и имесь нагревали до 50°C и подвергали реакции до завершения реакции. Реакционный раствор концентрировали при пониженном давлении с получением остатка. Остаток очищали хроматографией на силикагеле (градиент 0-100% этилацетата/петролейный эфир) с получением 6-1 (1,7 г, выход: 63,4%) в виде желтого твердого вещества. ЖХ-МС: м/з=547,0 [M+H]+.
Стадия 2: В атмосфере азота 6-1 (1,7 г, 3,11 ммоль), 2-амино-3-хлорпиридин-4-меркаптан натрия (596 мг, 3,27 ммоль), Pd2(dba)3 (285 мг, 0,311 ммоль), ксантфос (360 мг, 0,622 ммоль), ДИПЭА (804 мг, 6,22 ммоль) и раствор 1,4-диоксана (30 мл) последовательно добавляли в сосуд для реакции при микроволновом излучении объемом 5 мл, смесь нагревали в микроволновом излучении до 100°C и перемешивали в течение 3 часов в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали хроматографией на силикагеле (градиент 0-10% этилацетата/метанол) с получением (S)-трет-бутил (1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазоло[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)карбоната (6-2, 1,2 г, 66,7%).
Стадия 3: В атмосфере азота ТФУК (5 мл) медленно добавляли к раствору (S)-трет-бутил (1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазоло[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)карбоната (6-2, 1,2 г, 2,07 ммоль) в дихлорметане (5 мл) при 0°C, смесь перемешивали при комнатной температуре в течение 1 часа, и ТСХ и ЖХМС показали, что реакция завершилась. Реакционный раствор концентрировали при пониженном давлении, затем добавляли смешанный раствор дихлорметана/метанола для растворения, и pH смеси доводили до нейтрального значения с помощью NaHCO3. Смесь очищали, пропуская через колонку с силикагелем, с получением (S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амина (соединение 6, 400 мг, выход: 40,0%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,35 (д, J=4,0 Гц, 1H), 8,03 (с, 1H), 7,83 (д, J=1,2 Гц, 1H), 7,72 (д, J=7,6 Гц, 1H), 7,56 (дд, J=10,4, 3,4 Гц, 2H), 7,20 (дд, J=7,6, 5,2 Гц, 1H), 6,33 (с, 2H), 5,80 (д, J=5,4 Гц, 1H), 4,02 (с, 1H), 3,95 (дд, J=11,6, 7,6 Гц, 2H), 3,31 (д, J=13,6 Гц, 2H), 3,15 (д, J=16,4 Гц, 1H), 2,83 (д, J=16,4 Гц, 1H), 2,00 (тт, J=12,4, 6,4 Гц, 2H), 1,64 (д, J=13,2 Гц, 1H), 1,48 (дд, J=13,6, 6,4 Гц, 1H), 1,34-1,29 (м, 2H);
ЖХМС: м/з 479,0 [M+H]+
Пример 7: Синтез соединения 7
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-7-амина
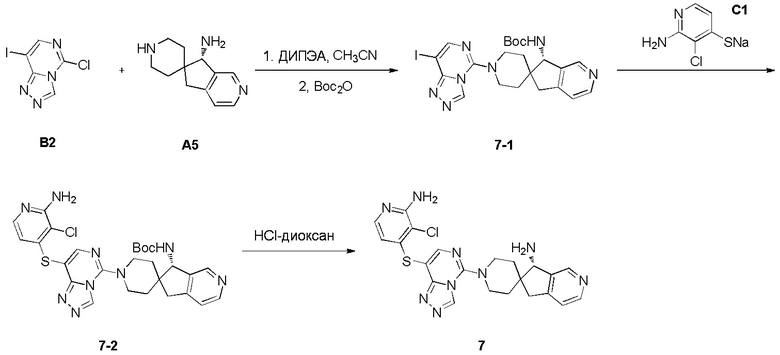
Соединение 7 синтезировали в три стадии в соответствии со способом синтеза соединения 6, используя промежуточное соединение B2 вместо промежуточного соединения 6 и промежуточное соединение A5 вместо промежуточного соединения A4.
1H ЯМР (400 МГц, ДМСО) δ 8,51 (с, 2H), 8,38 (д, J=4,8 Гц, 1H), 8,22 (с, 1H), 7,57 (д, J=5,6 Гц, 1H), 7,28 (д, J=4,8 Гц, 1H), 6,36 (с, 2H), 5,88 (д, J=5,6 Гц, 1H), 4,92 (т, J=13,6 Гц, 2H), 4,03 (с, 1H), 3,61 (дд, J=26,8, 12,0 Гц, 2H), 3,16 (д, J=16,8 Гц, 1H), 2,78 (с, 1H), 1,92 (тд, J=11,2, 4,0 Гц, 2H), 1,63 (д, J=13,2 Гц, 1H), 1,33-1,24 (м, 1H).
ЖХМС: м/з 480,1 [M+H]+
Пример 8: Синтез соединения 8
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-7-амина
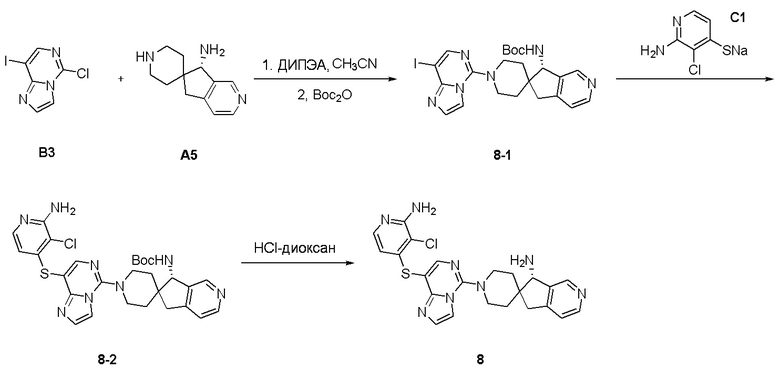
Соединение 8 синтезировали в соответствии с методом синтеза соединения 6 с использованием промежуточного соединения A5 вместо промежуточного соединения A4.
1H ЯМР (400 МГц, ДМСО) δ 8,53 (с, 1H), 8,39 (д, J=4,8 Гц, 1H), 8,04 (с, 1H), 7,84 (д, J=1,2 Гц, 1H), 7,58 (д, J=1,2 Гц, 1H), 7,56 (д, J=5,6 Гц, 1H), 7,28 (д, J=4,8 Гц, 1H), 6,34 (с, 2H), 5,80 (д, J=5,6 Гц, 1H), 4,07 (с, 1H), 3,93 (д, J=4,0 Гц, 2H), 3,37 (дд, J=19,6, 8,0 Гц, 2H), 3,13 (д, J=16,4 Гц, 1H), 2,76 (д, J=16,4 Гц, 1H), 1,98 (д, J=11,6 Гц, 2H), 1,62 (д, J=13,6 Гц, 1H), 1,30 (с, 1H).
ЖХМС: м/з 479,1 [M+H]+
Пример 9: Синтез соединения 9
(S)-1'-(8-(2,3-дихлорпиридин-4-ил)тио-[1,2,4]-триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амина
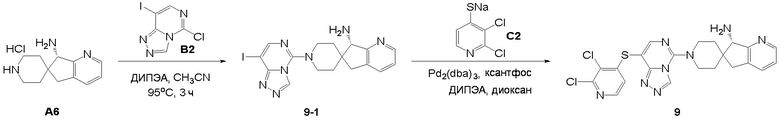
Стадия 1: (S)-5,7-Дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амина гидрохлорид (A6, 200 мг, 1,28 ммоль) растворяли в ацетонитриле (20 мл), добавляли ДИПЭА (1,6 мл, 9,68 ммоль) и 5-хлор-8-иод-[1,2,4]-триазоло[4,3-c]пиримидин (B2, 180 мг, 0,64 ммоль), смесь нагревали до 90°C и кипятили с обратным холодильником в течение 3 часов в атмосфере азота, и реакцию завершали. Реакционную жидкость охлаждали до комнатной температуры, выливали в насыщенный водный раствор бикарбоната натрия и дважды экстрагировали дихлорметаном (80 мл). Органические фазы объединяли и промывали насыщенным рассолом. Органическую фазу отделяли, а затем сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле (ДХМ:MeOH=20/1) с получением (S)-1'-(8-иодо[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амина (9-1, 260 мг, выход 90%) в виде светло-желтого твердого вещества. ЖХМС: м/з 448,2 [M+H]+
Стадия 2: (S)-1'-(8-иод[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амин (9-1, 55 мг, 0,12 ммоль) растворяли в 1,4-диоксане (3 мл), и добавляли ДИПЭА (35 мг, 0,27 ммоль) и 2,3-дихлорпиридин-4-меркаптан натрия (C2, 100 мг, сырой продукт). Ксантфос (30 мг, 0,05 ммоль) и Pd2(dba)3 (17 мг, 0,02 ммоль) добавляли в атмосфере азота. После того как атмосферу в реакционной системе трижды заменяли азотом, смесь подвергали реакции при 100°C в течение 3 часов, и реакцию завершали. Реакционный раствор концентрировали и очищали высокоэффективной жидкостной хроматографией с получением (S)-1'-(8-(2,3-дихлорпиридин-4-ил)тио-[1,2,4]-триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амина (соединение 9, 27 мг, соль ТФУК, выход: 40%) в виде светло-желтого твердого вещества. ЖХМС: м/з 499,1 [M+H]+
1H ЯМР (400 МГц, MeOD) δ 9,35 (с, 1H), 8,56 (д, J=4,8 Гц, 1H), 8,14 (с, 1H), 7,95 (д, J=5,2 Гц, 1H), 7,84 (д, J=7,6 Гц, 1H), 7,44-7,41 (м, 1H), 6,80 (д, J=5,2 Гц, 1H), 4,53 (с, 1H), 4,36-4,32 (м, 2H), 3,69-3,60 (м, 2H), 3,40 (д, J=16,4 Гц, 1H), 3,19 (д, J=16,4 Гц, 1H), 2,26-2,19 (м, 1H),2,05-1,93 (м, 2H), 1,73-1,70 (м, 1H).
Пример 10: Синтез соединения 10
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амина
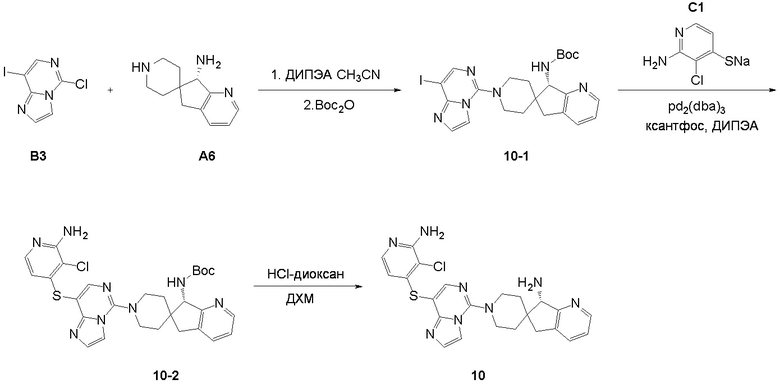
Соединение 10 синтезировали в три стадии в соответствии со способом синтеза соединения 6 с использованием промежуточного соединения A6 вместо промежуточного соединения A4.
1H ЯМР (400 МГц, ДМСО) δ 8,38 (д, J=4,3 Гц, 1H), 8,24 (с, 1H), 8,03 (с, 1H), 7,84 (д, J=1,4 Гц, 1H), 7,65 (д, J=7,4 Гц, 1H), 7,56 (дд, J=9,1, 3,4 Гц, 2H), 7,29-7,13 (м, 1H), 6,33 (с, 2H), 5,80 (д, J=5,4 Гц, 1H), 4,01 (с, 1H), 3,95-3,86 (м, 2H), 3,38 (дд, J=23,6, 11,2 Гц, 3H), 3,11 (д, J=16,0 Гц, 1H), 2,75 (д, J=16,0 Гц, 1H), 2,05-1,91 (м, 2H), 1,69 (д, J=13,6 Гц, 1H), 1,31 (д, J=14,0 Гц, 2H).
ЖХ-МС: м/з=478,0 [M+H+].
Пример 11: Синтез соединения 11
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амина
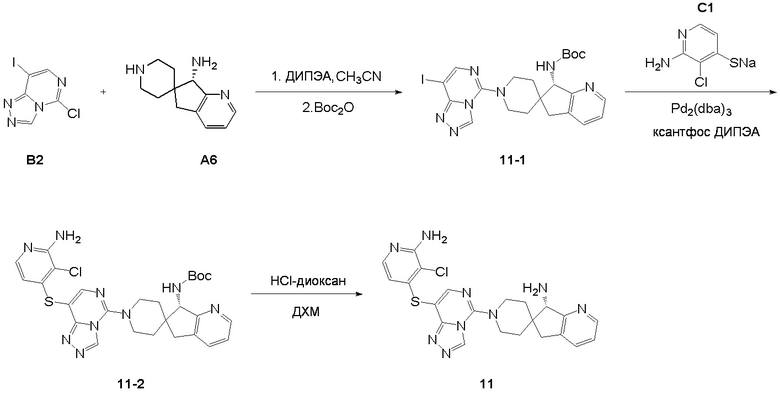
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амин (11, формиат, белое твердое вещество) синтезировали в три стадии согласно протоколу синтеза соединения 6, используя промежуточное соединение B2 вместо промежуточного B3 и промежуточный A6 вместо промежуточного A4.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,40 (с, 1H), 8,48 (д, J=4,5 Гц, 1H), 8,00 (с, 1H), 7,76 (д, J=7,3 Гц, 1H), 7,58 (д, J=5,4 Гц, 1H), 7,33 (дд, J=7,5, 5,0 Гц, 1H), 6,35 (с, 2H), 5,94 (д, J=5,4 Гц, 1H), 4,31 (с, 1H), 4,26-4,09 (м, 2H), 3,53 (дд, J=27,1, 12,0 Гц, 2H), 3,24 (д, J=16,4 Гц, 1H), 2,92 (д, J=16,3 Гц, 1H), 2,09 (с, 1H), 1,92 (с, 1H), 1,76 (д, J=13,2 Гц, 1H), 1,42 (д, J=13,2 Гц, 1H).
ЖХМС: м/з=479,0 [M+H+].
Пример 12: Синтез соединения 12
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-6-фтор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина
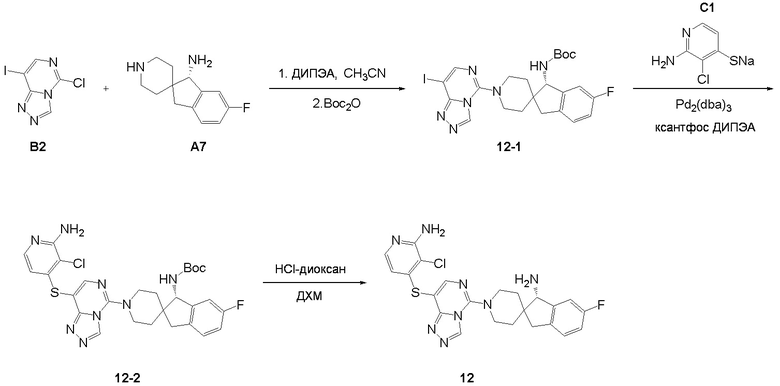
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-6-фтор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин (12, формиат, белое твердое вещество) синтезировали в три стадии согласно протоколу синтеза соединения 6, используя промежуточное соединение B2 вместо промежуточного соединения B3 и промежуточное соединение A7 вместо промежуточного соединения A4.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,38 (с, 1H), 8,22 (с, 1H), 7,98 (с, 1H), 7,57 (д, J=5,4 Гц, 1H), 7,31-7,20 (м, 1H), 7,16 (д, J=7,5 Гц, 1H), 7,01 (т, J=8,7 Гц, 1H), 6,34 (с, 2H), 5,95 (д, J=5,4 Гц, 1H), 4,14 (т, J=12,0 Гц, 2H), 4,00 (с, 1H), 3,47 (дд, J=25,4, 12,0 Гц, 3H), 3,10 (д, J=15,5 Гц, 1H), 2,71 (д, J=15,5 Гц, 1H), 2,08-1,85 (м, 2H), 1,64 (д, J=13,3 Гц, 1H), 1,30 (д, J=13,5 Гц, 1H). ЖХМС: м/з=495,0 [M+H+].
Пример 13: Синтез соединения 13
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-6-фтор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина
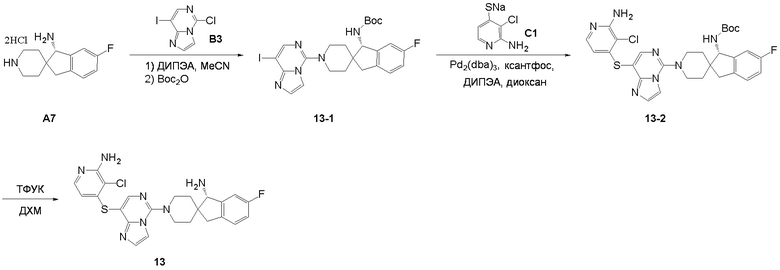
Сложный трет-бутиловый эфир (S)-(1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5-фтор-1,3-дигидроспиро[инден-2,4'-пиперидин]-3-ил)карбоновой кислоты (13-2) синтезировали в две стадии согласно протоколу синтеза соединения 6, используя промежуточное соединение A7 вместо промежуточного соединения A4.
Стадия 3: Сложный трет-бутиловый эфир (S)-(1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5-фтор-1,3-дигидроспиро[инден-2,4'-пиперидин]-3-ил)карбоновой кислоты (13-2, 60 мг, 0,101 ммоль), дихлорметан (1 мл) и трифторуксусную кислоту (0,2 мл) последовательно добавляли в сухую одногорлую колбу. Реакционный раствор перемешивали при 20°C в течение 1 часа. Реакционную жидкость концентрировали при пониженном давлении. pH полученной остаточной реакционной жидкости доводили до 8 насыщенным водным раствором бикарбоната натрия. Смесь трижды экстрагировали смешанным растворителем ДХМ/MeOH (10:1) (10 мл), и объединенную органическую фазу концентрировали при пониженном давлении. Полученный остаток очищали с помощью ВЭЖХ с получением (S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-6-фтор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (13, 32 мг, формиат, выход: 58,2%) в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,15 (с, 1H), 8,03 (с, 1H), 7,83 (д, J=1,2 Гц, 1H), 7,58 (д, J=1,6 Гц, 1H), 7,55 (д, J=5,6 Гц, 1H), 7,32-7,26 (м, 1H), 7,23-7,18 (м, 1H), 7,12-7,02 (м, 1H), 6,32 (с, 2H), 5,80 (д, J=5,2 Гц, 1H), 4,14 (с, 1H), 4,00-3,90 (м, 2H), 3,40-3,30 (м, 2H), 3,15-3,05 (м, 1H), 2,85-2,75 (м, 1H), 2,05-1,85 (м, 2H), 1,70-1,55 (м, 1H), 1,45-1,36 (м, 1H); ЖХ-МС: м/з 496,0 [M+H]+
Пример 14: Синтез соединения 14
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-5-амина
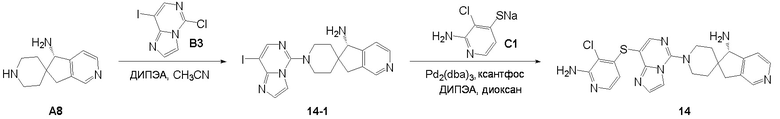
Стадия 1: (S)-5,7-Дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-5-амина гидрохлорид (A8, 200 мг, 0,83 ммоль) растворяли в ацетонитриле (20 мл), добавляли ДИПЭА (1,07 г, 8,3 ммоль) и 5-хлор-8-иодимидазо[1,2-c]пиримидин (B3, 208 мг, 0,75 ммоль), смесь нагревали до 90°C и кипятили с обратным холодильником в течение 5 часов в атмосфере азота, и реакцию завершали. Реакционную жидкость охлаждали до комнатной температуры, выливали в насыщенный водный раствор бикарбоната натрия и дважды экстрагировали дихлорметаном (50 мл). Органические фазы объединяли и затем промывали насыщенным рассолом. Органическую фазу отделяли, а затем сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением 1-(8-иодимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-4-амина (14-1, 250 мг, выход: 74%) в виде светло-желтого твердого вещества. ЖХМС: м/з 447,1 [M+H]+
Стадия 2: 1'-(8-Иодимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-4-амин (14-1, 50 мг, 0,11 ммоль) растворяли в 1,4-диоксане (10 мл), добавляли ДИПЭА (36 мг, 0,28 ммоль) и 2-амино-3-хлорпиридин-4-меркаптан натрия (C1, 31 мг, 0,17 ммоль). Ксантфос (13 мг, 0,02 ммоль) и Pd2(dba)3 (10 мг, 0,01 ммоль) добавляли в атмосфере азота. После того как атмосферу в реакционной системе трижды заменяли азотом, смесь подвергали реакции при 100°C в течение 3 часов, и реакцию завершали. Реакционную жидкость концентрировали, а затем очищали с помощью высокоэффективной жидкостной хроматографии с получением (S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-5-амина (14, 12 мг, выход: 22,4%) в виде светло-желтого твердого вещества.
ЖХМС: м/з 479,2 [M+H]+
1H ЯМР (400 МГц, MeOD) δ 8,64 (с, 1H), 8,56 (д, J=4,4 Гц, 1H), 8,09 (с, 1H), 7,62-7,60 (м, 2H), 7,60-7,50 (м, 1H), 5,95-5,94 (м, 1H), 4,64 (с, 1H), 4,22-4,07 (м, 2H), 3,60 (с, 2H),3,6-3,49 (м, 2H), 2,20-2,10 (м, 2H), 2,10-2,00 (м, 1H), 1,85-1,63(м, 2H).
Пример 15: Синтез соединения 15
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-5-амина
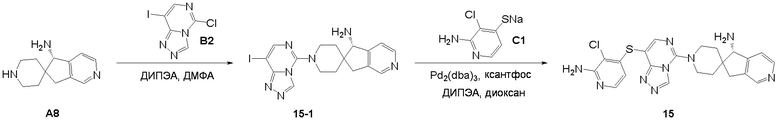
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[c]пиридин-6,4'-пиперидин]-5-амин (15) синтезировали в две стадии в соответствии с протоколом синтеза соединения 14, используя промежуточное соединение B2 вместо промежуточного соединения B3.
ЖХМС: м/з 480,0 [M+H]+
1H ЯМР (400 МГц, MeOD) δ 9,31 (с, 1H), 8,43-8,46 (м, 2H), 8,02 (с, 1H), 7,49-7,54 (м, 2H), 6,05 (д, J=5,6 МГц, 1H), 4,17-4,29 (м, 3H), 3,48-3,60 (м, 3H), 2,96 (д, J=16,0 МГц, 1H), 1,98-2,15 (м, 2H), 1,76-1,79 (м, 1H), 1,22-1,25 (м, 1H).
Пример 16: Синтез соединения 16
(S)-1-амино-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-4-ола
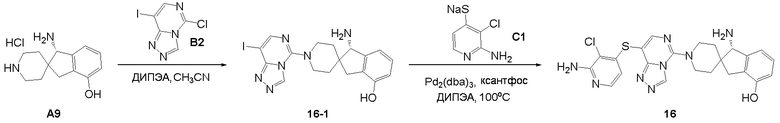
Стадия 1: (S)-1-Амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-4-ола гидрохлорид (A9, 200 мг, 0,79 ммоль) растворяли в ацетонитриле (40 мл), добавляли ДИПЭА (1,07 г, 8,3 ммоль) и 5-хлор-8-иод-[1,2,4]триазоло[4,3-c]пиримидин (B2, 208 мг, 0,75 ммоль), смесь подвергали реакции при комнатной температуре в атмосфере азота в течение 18 часов, и реакцию завершали. Реакционную жидкость охлаждали до комнатной температуры, выливали в насыщенный водный раствор бикарбоната натрия и дважды экстрагировали дихлорметаном (100 мл). Органические фазы объединяли, а затем промывали насыщенным рассолом. Органическую фазу отделяли, а затем сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением (S)-1-амино-1'-(8-иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-4-ола (16-1, 220 мг, выход: 63%) в виде светло-желтого твердого вещества. ЖХМС: м/з 463,0 [M+H]+
Стадия 2: (S)-1-Амино-1'-(8-иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-4-ол (16-1, 200 мг, 0,43 ммоль) растворяли в 1,4-диоксане (30 мл), и добавляли ДИПЭА (111 мг, 0,86 ммоль) и 2-амино-3-хлорпиридин-4-меркаптан натрия (C1, 118 мг, 0,65 ммоль). В атмосфере азота добавляли ксантфос (52,0 мг, 0,08 ммоль) и Pd2(dba)3 (41 мг, 0,04 ммоль). После того как атмосферу в реакционной системе трижды заменяли азотом, смесь подвергали реакции при 100°C в течение 3 часов, и реакцию завершали. Реакционную жидкость концентрировали, а затем очищали с помощью высокоэффективной жидкостной хроматографии с получением (S)-1-амино-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-4-ола (16, 100 мг, выход: 47%) в виде белого твердого вещества. ЖХМС: м/з 495,3 [M+H]+
1H ЯМР (400 МГц, MeOD) δ 9,32 (с, 1H), 8,01 (с, 1H), 7,53 (д, J=5,6 МГц, 1H), 7,04 (т, J=7,6 МГц, 1H), 6,84 (д, J=5,6 МГц, 1H), 6,64 (д, J=8,0 МГц, 1H), 6,06 (д, J=5,6 МГц, 1H), 4,20-4,24 (м, 2H), 3,95 (с, 1H), 3,54-3,62 (м, 2H), 3,13(д, J=15,6 МГц, 1H), 2,74 (д, J=15,6 МГц, 1H), 1,93-2,06 (м, 2H), 1,70 (д, J=12,8 МГц, 1H), 1,55 (д, J=12,8 МГц, 1H).
Пример 17: Синтез соединения 17
Соединение (S)-1-амино-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-4-нитрила
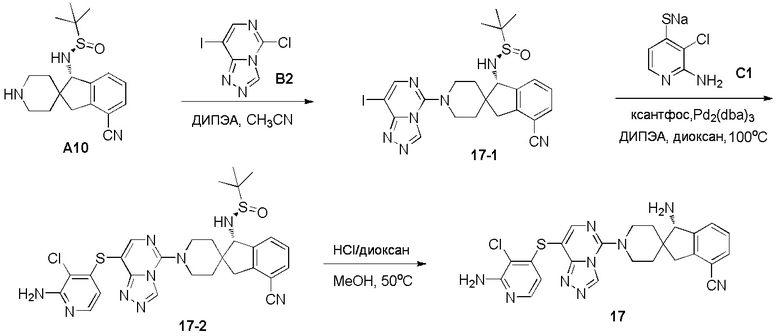
Стадия 1: (S)-1-(((R)-трет-бутилсульфинил)амино)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин] (A10, 490 мг, 1,48 ммоль) растворяли в ацетонитриле (20 мл), добавляли ДИПЭА (1,91 г, 14,8 ммоль) и 5-хлор-8-иод-[1,2,4]триазоло[4,3-c]пиримидин (B2, 415 мг, 1,48 ммоль), смесь подвергали реакции при 85°C в атмосфере азота в течение 3 часов, и реакцию завершали. Остаток, полученный концентрированием при пониженном давлении, очищали хроматографией на силикагеле (градиент 0-10% метанола/этилацетат) с получением (R)-N-((S)-1'-(8-иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-трет-бутилсульфонамида (17-1, 360 мг, выход: 42,3%) в виде желтого твердого вещества. ЖХМС: м/з 576,1 [M+H]+
Стадия 2: (R)-N-((S)-1'-(8-иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-трет-бутилсульфонамид (17-1, 360 мг, 0,63 ммоль) растворяли в 1,4-диоксане (10 мл), и добавляли ДИПЭА (202 мг 1,56 ммоль) и 2-амино-3-хлорпиридин-4-меркаптан натрия (C1, 171 мг, 0,94 ммоль). Ксантфос (73 мг, 0,126 ммоль) и Pd2(dba)3 (57,7 мг, 0,063 ммоль) добавляли в атмосфере азота. После того как атмосферу в реакционной системе трижды заменяли азотом, смесь подвергали реакции при 100°C в течение 3 часов, и реакцию завершали. Реакционный раствор концентрировали и затем очищали хроматографией на силикагеле с получением (R)-N-((S)-1'-((8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-трет-бутилсульфонамида (17-2, 130 мг, 34,2%) в виде белого твердого вещества ЖХ-МС: м/з 608,2 [M+H]+
Стадия 3: (R)-N-((S)-1'-((8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-циано-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-трет-бутилсульфонамид (17-2, 130 мг, 0,21 ммоль) и метанол (6 мл) последовательно добавляли в одногорлую колбу объемом 50 мл в атмосфере азота, и по каплям добавляли раствор хлористоводородной кислоты в 1,4-диоксане (2 мл, 4M) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали высокоэффективной жидкостной хроматографией с получением (S)-1-амино-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-4-нитрила (17, 10 мг, выход: 9,3%).
1H ЯМР (400 МГц, CDCl3) δ 9,32 (с, 1H), 8,02(с, 1H), 7,71 (д, J=7,2 Гц, 1H), 7,59 (д, J=7,6 Гц, 1H), 7,53 (д, J=5,6 Гц, 1H), 7,43 (д, J=7,6 Гц, 1H), 6,06 (д, J=5,6 Гц, 1H), 4,23 (д, J=10,8 Гц, 2H), 4,12 (с, 1H), 3,60 (м, 2H), 3,37 (м, 1H), 3,03 (д, J=16,4 Гц, 1H), 2,06 (м, 2H), 1,74 (т, 2H), 1,57 (д, J=19,2 Гц, 2H), 1,52 (с, 2H).
ЖХМС: м/з 504,1 [M+H]+
Пример 18: Синтез соединения 18
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-5-амина

Стадия 1: (S)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амина гидрохлорид (A11, 1,50 г, 4,95 ммоль) растворяли в 150 мл ацетонитрила, добавляли ДИПЭА (5,68 г, 44,0 ммоль) и 5-хлор-8-иодимидазо[1,2-c]пиримидин (B3, 1,28 г, 4,59 ммоль), смесь нагревали до 95°C и кипятили с обратным холодильником в течение 3 часов в атмосфере азота, и реакцию завершали. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали досуха, смешивали с силикагелем и пропускали через колонку с получением (S)-1'-(8-иодимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-5-амина (18-1, 2,05 г, выход: 92,7%) в виде бежевого порошка. ЖХМС: м/з 448,0 [M+H]+
Стадия 2: (S)-1'-(8-иодимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-5-амин (18-1, 2,05 г, 4,59 ммоль) растворяли в 120 мл 1,4-диоксана в стеклянной герметичной трубке объемом 350 мл, последовательно добавляли ДИПЭА (1,78 г, 13,8 ммоль), 2-амино-3-хлорпиридин-4-меркаптан натрия (C1, 1,26 г, 6,9 ммоль), ксантфос (797 мг, 1,38 ммоль) и Pd2(dba)3 (630 мг, 0,69 ммоль), реакционную смесь продували аргоном в течение 30 секунд и нагревали до 100°C в атмосфере аргона и выдерживали в течение 3 часов до завершения реакции. Смесь охлаждали до комнатной температуры, разбавляли дихлорметаном и фильтровали с отсасыванием. Фильтрат сушили центрифугированием и пропускали через колонку. Полученную чистую фракцию сушили центрифугированием, смешивали с дихлорметаном/н-гексаном (1/1) и измельчали в течение ночи, а затем фильтровали и сушили с получением (S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-5-амина (соединение 18, 440 мг, выход 19,9%). ЖХМС: м/з 480,1 [M+H]+
1H ЯМР (400 МГц, MeOD): δ 8,44-8,39 (м, 2H), 8,05 (с, 1H), 7,85 (д, J=1,6 Гц, 1H), 7,56 (д, J=1,6 Гц, 1H), 7,49 (д, J=5,6 Гц, 1H), 5,89 (д, J=5,6 Гц, 1H), 4,14 (с, 1H), 4,06-4,02 (м, 2H), 3,49-3,40 (м, 2H), 3,33 (с, 1H), 3,01 (д, J=16,8 Гц, 1H), 2,24-2,10 (м, 2H), 1,82 (д, J=13,2 Гц, 1H), 1,50 (д, J=13,6 Гц, 1H).
Пример 19: Синтез соединения 19
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина
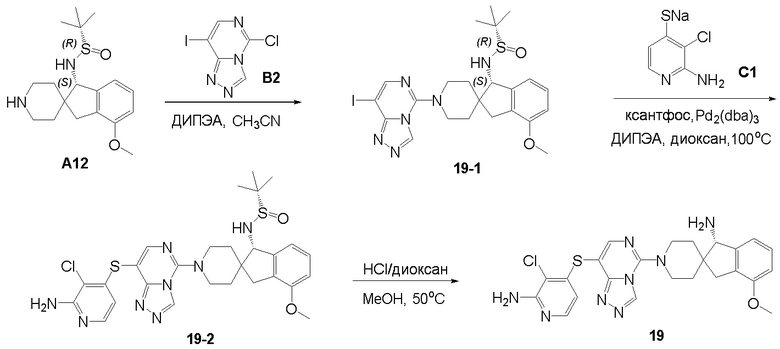
Стадия 1: (S)-1-(((R)-трет-бутилсульфинил)амино)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин] (A12, 520 мг, 1,55 ммоль) растворяли в ацетонитриле (20 мл), добавляли ДИПЭА (2,0 г, 15,5 ммоль) и 5-хлор-8-иод-[1,2,4]триазоло[4,3-c]пиримидин (B2, 435 мг, 1,55 ммоль), смесь подвергали реакции при 85°C в течение 3 часов в атмосфере азота, и реакцию завершали. Остаток, полученный концентрированием при пониженном давлении, очищали хроматографией на силикагеле (градиент 0-10% метанола/этилацетат) с получением (R)-N-((S)-1'-(8-иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-трет-бутилсульфонамида (19-1, 420 мг, выход: 46,8%) в виде желтого твердого вещества.
ЖХМС: м/з 581,1 [M+H]+
Стадия 2: (R)-N-((S)-1'-(8-иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-трет-бутилсульфонамид (19-1, 420 мг, 0,72 ммоль) растворяли в 1,4-диоксане (10 мл), и добавляли ДИПЭА (234 мг 1,81 ммоль) и 2-амино-3-хлорпиридин-4-меркаптан натрия (C1, 198 мг, 1,09 ммоль). Ксантфос (83 мг, 0,144 ммоль) и Pd2(dba)3 (66 мг, 0,072 ммоль) добавляли в атмосфере азота. После того как атмосферу в реакционной системе трижды заменяли азотом, смесь подвергали реакции при 100°C в течение 3 часов, и реакцию завершали. Реакционный раствор концентрировали и затем очищали хроматографией на силикагеле с получением (R)-N-((S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-трет-бутилсульфонамида (19-2, 310 мг, выход: 69,9%) в виде белого твердого вещества.
ЖХМС: м/з 613,2 [M+H]+
Стадия 3: (R)-N-((S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-трет-бутилсульфонамид (19-2, 310 мг, 0,51 ммоль) и метанол (6 мл) добавляли последовательно в одногорлую колбу объемом 50 мл в атмосфере азота, по каплям добавляли раствор хлористоводородной кислоты в 1,4-диоксане (2 мл, 4М) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией с получением (S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (соединение 19, 114 мг, выход: 44,3%).
1H ЯМР (400 МГц, CDCl3) δ 8,80 (с, 1H), 7,95(с, 1H), 7,65 (д, J=5,6 Гц, 1H), 7,26 (т, 2H), 6,96 (д, J=7,6 Гц, 1H), 6,77 (д, J=8,4 Гц, 1H), 6,02 (д, J=5,6 Гц, 1H), 4,89 (с, 2H), 4,13-4,04 (м, 4H), 3,86 (с, 3H), 3,53-3,48 (м, 2H), 3,11 (д, J=16 Гц, 1H), 2,70 (д, J=16 Гц, 1H), 2,08-2,01 (м, 3H), 1,76 (д, J=14 Гц, 1H), 1,53 (д, J=14 Гц, 2H).
ЖХМС: м/з 509,2 [M+H]+
Пример 20: Синтез соединения 20
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-5-амина
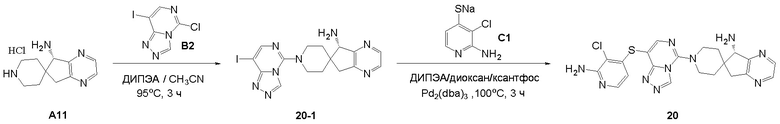
Стадия 1: (S)-5,7-Дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амина гидрохлорид (A11, 89 мг, 0,29 ммоль) растворяли в 20 мл ацетонитрила, добавляли ДИПЭА (212 мг, 1,64 ммоль) и 5-хлор-8-иод-[1,2,4]триазоло[4,3-c]пиримидин (B2, 77 мг, 0,275 ммоль), смесь нагревали до 95°C и кипятили с обратным холодильником в течение 3 часов в атмосфере азота, и реакцию завершали. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали досуха, смешивали с силикагелем и пропускали через колонку с получением (S)-1'-(8-иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-5-амина (20-1, 65 мг, выход: 52,7%). ЖХМС: м/з 449,2 [M+H]+
Стадия 2: (S)-1'-(8-Иод-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-5-амин (20-1, 65 мг, 0,145 ммоль) растворяли в 5 мл 1,4-диоксана в трубке для реакции при микроволновом излучении объемом 5 мл, и последовательно добавляли ДИПЭА (56 мг, 0,434 ммоль), 2-амино-3-хлорпиридин-4-сульфид натрия (C1, 40 мг, 0,219 ммоль), ксантфос (34 мг, 0,059 ммоль) и Pd2(dba)3 (27 мг, 0,029 ммоль), через реакционную смесь барботировали аргон в течение 30 секунд, после чего смесь нагревали до 100°C в атмосфере аргона и выдерживали в течение 3 часов до завершения реакции. Смесь охлаждали до комнатной температуры, разбавляли дихлорметаном и фильтровали с отсасыванием. Фильтрат сушили центрифугированием и очищали с помощью препаративной пластинки с силикагелем. Полученную чистую фракцию отправляли на подготовку с получением чистого продукта (S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиразин-6,4'-пиперидин]-5-амина (соединение 20, 19,4 мг, выход: 27,8%). ЖХМС: м/з 481,2 [M+H]+
1H ЯМР (400 МГц, MeOD) δ 9,35 (с, 1H), 8,59-8,57 (м, 2H), 8,11 (с, 1H), 7,53 (д, J=6,8 Гц, 1H), 6,37 (д, J=6,4 Гц, 1H), 4,65 (с, 1H), 4,37-4,35 (м, 2H), 3,69-3,62 (м, 2H), 3,47 (д, J=17,2 Гц, 1H), 3,26 (с, 1H), 2,31-2,23 (м, 1H), 2,07-1,97 (м, 2H), 1,68 (д, J=11,6 Гц, 1H).
Пример 21: Синтез соединения 21
(S)-1'-(8-((2-(трифторметил)пиридин-3-ил)тио)-[1,2,4]-триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амина
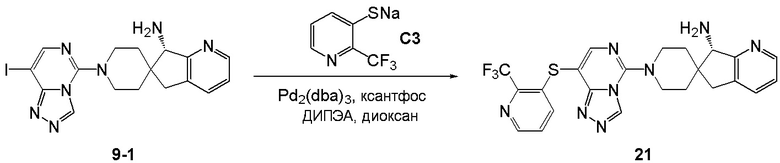
(S)-1'-(8-Иод[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амин (9-1, 100 мг, 0,22 ммоль) растворяли в 1,4-диоксане (12 мл), и добавляли ДИПЭА (300 мг, 2,32 ммоль) и 2-(трифторметил)пиридин-3-меркаптан натрия (C3, 200 мг, сырой продукт). Ксантфос (100 мг, 0,17 ммоль) и Pd2(dba)3 (100 мг, 0,11 ммоль) добавляли в атмосфере азота. После того как атмосферу в реакционной системе трижды заменяли азотом, смесь подвергали реакции при 100°C в течение 3 часов, и реакцию завершали. Реакционный раствор пропускали через колонку (ЭА:MeOH=5/1, 0,5% гидроксида аммония) с получением (S)-1'-(8-((2-(трифторметил)пиридин-3-ил)тио)-[1,2,4]-триазоло[4,3-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-7-амина (соединение 21, 35 мг, выход: 31%) в виде белого твердого вещества. ЖХМС: м/з 499,1 [M+H]+
1H ЯМР (400 МГц, MeOD) δ 9,32 (с, 1H), 8,41-8,37 (м, 2H), 8,03 (с, 1H), 7,72 (д, J=7,6 Гц, 1H), 7,60 (д, J=8,4 Гц, 1H), 7,38-7,35 (м, 1H), 7,28-7,25 (м, 1H), 4,18-4,14 (м, 2H), 4,04 (с, 1H), 3,62-3,59 (м, 2H), 3,23 (д, J=16,4 Гц, 1H), 2,90 (д, J=16,4 Гц, 1H), 2,11-2,06 (м, 2H), 1,79-1,75 (м, 1H), 1,56-1,53 (м, 1H).
Пример 22: Синтез соединения 22
(S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-2-хлор-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-4-амина
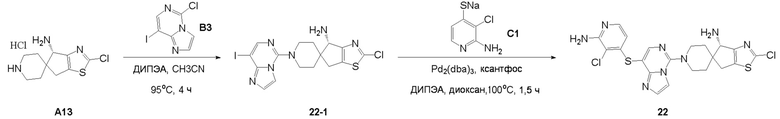
Стадия 1: (S)-2-Хлор-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-4-амина гидрохлорид (A13, 155 мг, 0,35 ммоль) растворяли в ацетонитриле (10 мл), и добавляли DIPEA (450 мг, 3,5 ммоль) и 5-хлор-8-иодимидазо[1,2-c]пиримидин (B3, 108 мг, 0,39 ммоль), смесь нагревали до 100°C и кипятили с обратным холодильником в течение 5 часов в атмосфере. азота, и реакцию завершали. Реакционный раствор охлаждали до комнатной температуры, выливали в насыщенный водный раствор бикарбоната натрия и экстрагировали дихлорметаном (50 мл × 2). Органические фазы объединяли, а затем промывали насыщенным рассолом. Органическую фазу отделяли, а затем сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с получением (S)-2-хлор-1'-(8-иодимидазо[1,2-c]пиримидин-5-ил)-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-4-амина (22-1, 72 мг, выход: 43%) в виде светло-желтого твердого вещества. ЖХМС: м/з 487 [M+H]+
Стадия 2: (S)-2-Хлор-1'-(8-иодимидазо[1,2-c]пиримидин-5-ил)-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-4-амин (22-1, 50 мг, 0,1 ммоль) растворяли в 1,4-диоксане (5 мл), и добавляли ДИПЭА (30 мг, 0,25 ммоль) и 2-амино-3-хлорпиридин-4-меркаптан натрия (C1 , 30 мг, 0,15 ммоль). Ксантфос (13 мг, 0,02 ммоль) и Pd2(dba)3 (20 мг, 0,02 ммоль) добавляли в атмосфере азота. После того как атмосферу в реакционной системе трижды заменяли азотом, смесь подвергали реакции при 100°C в течение 3 часов. Реакционный раствор концентрировали, а затем очищали с помощью высокоэффективной жидкостной хроматографии с получением (S)-1'-(8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-2-хлор-4,6-дигидроспиро[циклопента[d]тиазол-5,4'-пиперидин]-4-амина (соединение 22, 3 мг, выход: 5%) в виде светло-желтого твердого вещества. ЖХМС: м/з 519 [M+H]+
1H ЯМР (400 МГц, MeOD) δ 8,06 (с, 1H),7,85 (с, 1H), 7,57 (с, 1H), 7,50-7,49 (д, 1H), 5,89-5,87 (д, 1H), 4,15 (с, 1H), ,4,07-3,99 (дд, 2H), 3,53-3,39 (м, 4H), 3,11-3,09 (д, 2H), 2,21-2,04 (м, 4H)
Пример 23: Синтез соединения 23

Стадия 1. Диоксан (20 мл) добавляли к 1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-4-меркаптану натрия (C7, 224 мг, 0,8 ммоль) в реакционной колбе, а затем добавляли (S)-N-((S)-1'-(8-иодимидазоло[1,2-C]пиримидин-5-ил)-5,7-дигидроспиро[циклопентан[B]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (1-1, 287 мг, 0,52 ммоль), ДИПЭА (304 мг, 2,35 ммоль), ксантфос (91 мг, 0,16 ммоль) и Pd2(dba)3 (72 мг, 0,08 ммоль). Смесь нагревали до 100°C и перемешивали в течение 3 ч в атмосфере азота, и ТСХ (дихлорметан/метанол=20/1) и ЖХМС показали, что реакция завершилась. Реакционный раствор разбавляли этилацетатом и фильтровали. Остаток на фильтре элюировали этилацетатом. Фильтрат концентрировали досуха и очищали хроматографией на силикагеле с получением (R)-2-метил-N-((5S)-1'-((8-((1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопентадиен[b]пиридин-6,4'-пиперидин]-5-ил)пропан-2-сульфинамида (23-1, 352 мг, выход: 97,2%).
ЖХМС: м/з 658,4 [M+H]+
Стадия 2. В атмосфере азота HCl/диоксан (4M, 1,3 мл, 5,20 ммоль) медленно добавляли к раствору (R)-2-метил-N-((5S)-1'-((8-((1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопентадиен[b]пиридин-6,4'-пиперидин]-5-ил)пропан-2-сульфинамида (23-1, 176 мг, 0,26 ммоль) в дихлорметане (10 мл) при 0°C, смесь перемешивали при комнатной температуре в течение 2 ч, и ТСХ (дихлорметан/метанол=8/1) и ЖХМС показали, что реакция завершилась. Реакционный раствор охлаждали до 0°C, медленно добавляли раствор аммиака в метаноле, чтобы довести pH до примерно 10, а затем смесь концентрировали при пониженном давлении и очищали препаративной ТСХ с получением (S)-1'-(8-(((1H-пиразоло[3,4-b]пиридин-4-ил]тио]имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопентадиен[b]пиридин-6,4'-пиперидин]-5-амина (23, 73,01 мг, выход: 58,1%). ЖХМС: м/з 470,5 [M+H]+
1H ЯМР (400 МГц, MeOD): δ 8,42 (д, J=4,0 Гц, 1H), 8,16 ~ 8,14 (м, 2H), 8,10 (с, 1H), 7,91 ~ 7,87 (м, 2H), 7,56 (д, J=1,2 Гц, 1H), 7,34 ~ 7,31 (м, 1H), 4,26 (с, 1H), 4,11 ~ 4,06 (м, 1H), 3,49 ~ 3,43 (м, 2H), 3,28 (с, 1H), 3,07 (д, J=16,8 Гц, 1H), 2,15 ~ 2,07 (м, 2H), 1,76 (д, J=12,8 Гц, 2H), 1,64 (д, J=13,6 Гц, 2H).
Синтез контрольного соединения: Во время применения раскрытия наиболее близким контрольным соединением к описанию является пример 25 из WO2018136265. В соответствии с синтетическим путем и стадиями процесса примера 25 из WO2018136265 получали контрольное соединение аа.
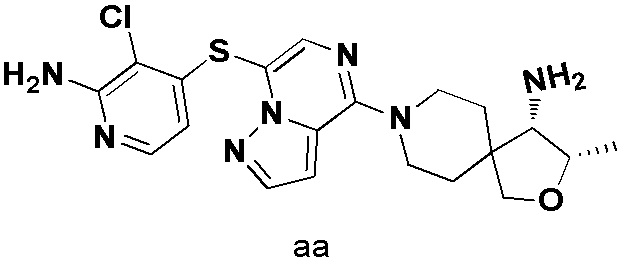
Биологическая функция соединения согласно раскрытию подтверждена испытаниями на ферментативную активность и на клеточном уровне. Например, в испытании на ингибирование активности фермента SHP2 соединение согласно раскрытию может достигать сильной ингибирующей активности (IC50 может достигать 1 нМ). На клеточном уровне соединение согласно настоящему раскрытию также показало очень хорошую активность в отношении ингибирования пролиферации раковых клеток, и ингибирующая активность в отношении пролиферации клеточной линии MV4-11 может достигать 1 нМ. По сравнению с SHP099 (6-(4-амино-4-метилпиперидин-1-ил)-3-(2,3-дихлорфенил)пиразин-2-амином) или контрольным соединением аа, соединение согласно раскрытию показало превосходную активность как на ферментативном уровне, так и на клеточном уровне.
Пример испытания 1: Метод испытания активности фермента SHP2
Метод определения активности фермента SHP2:
Порошок соединения растворяли в ДМСО для получения маточного раствора. Во время эксперимента исходный раствор соединения разбавляли ДМСО 3-кратным градиентом, и для каждого соединения устанавливали 10 различных тестовых концентраций. 1 мкл соединений в каждой точке концентрации помещали в лунку планшета для обнаружения (Corning, Costa 3915), и каждую концентрацию тестировали в двух повторностях. 6,8-Дифтор-4-метил-7-гидроксикумарин фосфат (DiFMUP) использовали в качестве субстрата, и он может быть гидролизован при катализе SHP2 E76A с образованием 6,8-дифтор-4-метил-7-гидроксикумарина (DiFMU). Для определения ферментативной активности SHP2 значение флуоресценции при 455 нм регистрировали многофункциональным ридером PE Enspire с использованием длины волны возбуждения 358 нм.
Буфер SHP2 для реакции состоял из 60 ммоль/л Hepes, PH7.2, 75 ммоль/л NaCl, 75 ммоль/л KCl, 1 ммоль/л ЭДТА, 5 ммоль/л ДТТ.
Система скрининга состояла из буфера SHP2, фермента белка SHP2 E76A, субстрата DiFMUP и тестируемых соединений.
Метод определения IC50:
В 96-луночном планшете для скрининга 50 нг белка SHP2 E76A подвергали взаимодействию с тестируемым соединением в буфере SHP2 в течение 20 минут, а затем инкубировали с 10 мкМ DiFMUP при комнатной температуре в течение 20 минут. Интенсивность света при 455 нм считывали многофункциональным ридером PE Enspire с возбуждающим светом 358 нм. Степень ингибирования ферментативной активности образца рассчитывали как отношение измеренного значения флуоресценции группы, обработанной соединением, к измеренному значению флуоресценции для контрольной лунки с ДМСО. Значения IC50 соединений рассчитывали с использованием программного обеспечения Graphpad's Prism путем нелинейного согласования скорости ингибирования с концентрацией ингибитора. Кривую активности фермента как функцию концентрации соединения подбирали по уравнению Y=Нижняя точка+(Верхняя точка-Нижняя точка)/(1+10˄((LogIC50-X)*Угловой коэффициент Хилла)).
Рассчитывали значение IC50 каждого соединения. В таблице 1 показаны значения IC50 некоторых соединений согласно раскрытию.
Таблица 1
Пример испытания 2: эксперимент по ингибированию пролиферации клеток MV4-11
Количество живых клеток в культуре определяли путем количественного определения внутриклеточного АТФ с использованием набора для люминесцентного анализа жизнеспособности клеток CellTiter-Glo®.
На первом этапе клетки MV4-11 инокулировали в 96-луночные планшеты с плотностью 2500 клеток на лунку и объемом 100 мкл на лунку. Планшеты помещали в инкубатор на ночь при 37°C с 5% углекислого газа.
На втором этапе клетки обрабатывали соединениями. Тестируемые соединения разбавляли в 3 раза и устанавливали 8 градиентов концентрации. В каждую лунку добавляли определенный объем ДМСО или тестируемого соединения, и каждую концентрацию тестировали в 2 повторностях, и конечная концентрация ДМСО составляла 0,5%. Планшеты помещали в инкубатор на 72 часа при 37°C с 5% углекислого газа.
На третьем этапе жизнеспособность клеток контрольных групп и групп обработки определяли с использованием набора для люминесцентного анализа жизнеспособности клеток CellTiter-Glo® (Promega, G7570). В каждую лунку добавляли 50 мкл CellTiter-Glo. Культуру хорошо перемешивали и инкубировали при комнатной температуре в течение 10 минут. Сигналы считывали с помощью EnSpire (Perkin Elmer). Процент ингибирования (%) рассчитывали по следующей формуле:
Процент ингибирования (%) = (1-значение сигнала группы обработки соединением/значение сигнала группы обработки ДМСО)*100. Результаты показаны в таблице 2.
Таблица 2
Пример испытания 3: Эксперимент по определению фармакокинетики соединения
Определяли фармакокинетику соединения согласно раскрытию. Следующий метод использовали для определения фармакокинетических параметров соединения согласно раскрытию.
В исследовании использовали здоровых взрослых мышей-самцов. Каждой группе внутрижелудочно вводили разовую дозу 5-100 мг/кг. Воздержание от пищи длилось от 10 часов до введения до 4 часов после введения. Образцы крови собирали в разные моменты времени после введения, и содержание соединения в плазме определяли с помощью ЖХ-МС/МС. Связь между концентрацией в плазме и временем анализировали с помощью профессионального программного обеспечения (winnonlin) и рассчитывали фармакокинетические параметры соединений. Согласно таблице 3, соединения согласно раскрытию обладают превосходными фармакокинетическими свойствами.
Таблица 3
AUC:мкМ*час
Все публикации, упомянутые в настоящем документе, цитируются посредством ссылки в настоящем раскрытии в той же степени, как если бы каждая публикация отдельно цитировалась посредством ссылки. Предпочтительные варианты осуществления настоящего раскрытия подробно описаны выше, но раскрытие не ограничивается конкретными деталями в вышеупомянутых вариантах осуществления. В рамках технической концепции настоящего раскрытия различные простые модификации могут быть внесены в технические решения раскрытия, и все модификации попадают в объем настоящего раскрытия.
Кроме того, следует отметить, что каждый конкретный технический признак, описанный в вышеупомянутых конкретных вариантах осуществления, может быть скомбинирован любым подходящим способом без противоречий. Чтобы избежать ненужного дублирования, настоящее раскрытие отдельно не описывает различные возможные комбинации. Кроме того, различные варианты осуществления настоящего раскрытия также могут быть объединены любым способом, и комбинации также должны рассматриваться как раскрытые в раскрытии без отступления от существа раскрытия.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ПИРИМИДИНА И ПЯТИЧЛЕННОГО АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2775229C1 |
| ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНФОСФАТАЗЫ | 2020 |
|
RU2799449C2 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2687093C1 |
| ПРОИЗВОДНЫЕ 2-АМИНОПИРАЗИНА В КАЧЕСТВЕ ИНГИБИТОРОВ CSF-1R КИНАЗЫ | 2013 |
|
RU2642777C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2829484C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
| СПИРОИНДОЛИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ РЕСПИРАТОРНО-СИНЦИТИАЛЬНОГО ВИРУСА (RSV) | 2015 |
|
RU2734248C2 |
Изобретение относится к новому соединению формулы I, или к его фармацевтически приемлемой соли, или его энантиомеру или таутомеру, где X1 и X2 независимо выбраны из связи или CRaRb; X3 представляет собой S; X4 выбран из N или CRc и Ra, Rb и Rc независимо выбраны из H, незамещенного C1-6 алкила или незамещенного C1-6 алкоксила; R1, R2, R3, R4 и R7 независимо выбраны из H, незамещенного C1-6 алкила или незамещенного C1-6 алкоксила; кольцо A выбрано из замещенного или незамещенного C5-6 арила или замещенного или незамещенного 5-6-членного гетероарила, где гетероарил содержит 1 или 2 гетероатома, выбранных из следующих атомов: N или S; кольцо C выбрано из замещенного или незамещенного 9-членного бициклического гетероциклила, замещенного или незамещенного C5-6 моноциклического арила, замещенного или незамещенного 5-6-членного моноциклического гетероарила или замещенного или незамещенного 9-членного бициклического гетероарила, где гетероциклил или гетероарил содержит 1-4 гетероатома, выбранных из следующих атомов: N, O или S; R5 и R6 независимо выбраны из H, -OH, галогена, циано, замещенного или незамещенного амино, замещенного или незамещенного C1-6 алкила или незамещенного C1-6 алкоксила; n представляет собой любое целое число от 0 до 3; и где термин "замещенный" относится к одному или нескольким атомам водорода в группе, замещенным заместителем, выбранным из: галогена, -OH, -NH2, -NH(незамещенный C1-6 алкил), -CN, галогенированного C1-8 алкила, незамещенного C1-8 алкоксила. Также предоставлен способ получения соединения формулы I. Соединение формулы I обладает более высокой ингибирующей активностью против SHP2 и, таким образом, может применяться для профилактики или лечения заболевания, связанного с SHP2. 5 н. и 16 з.п. ф-лы, 4 табл., 23 пр.
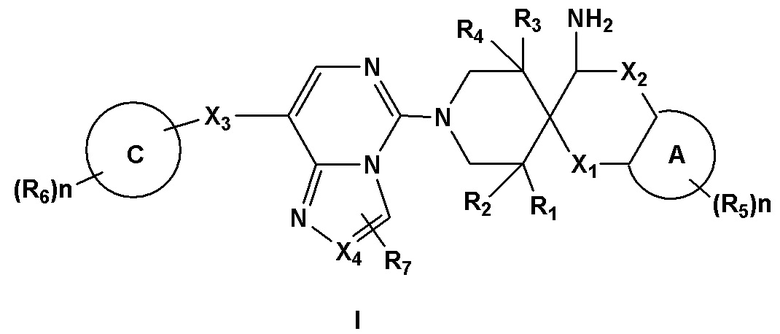
1. Соединение формулы I:
 ,
,
или его фармацевтически приемлемая соль, или его энантиомер или таутомер, где:
X1 и X2 независимо выбраны из связи или CRaRb;
X3 представляет собой S;
X4 выбран из N или CRc; и Ra, Rb и Rc независимо выбраны из H, незамещенного C1-6 алкила или незамещенного C1-6 алкоксила;
R1, R2, R3, R4 и R7 независимо выбраны из H, незамещенного C1-6 алкила или незамещенного C1-6 алкоксила;
кольцо A выбрано из замещенного или незамещенного C5-6 арила или замещенного или незамещенного 5-6-членного гетероарила, где гетероарил содержит 1 или 2 гетероатома, выбранных из следующих атомов: N или S;
кольцо C выбрано из замещенного или незамещенного 9-членного бициклического гетероциклила, замещенного или незамещенного C5-6 моноциклического арила, замещенного или незамещенного 5-6-членного моноциклического гетероарила или замещенного или незамещенного 9-членного бициклического гетероарила, где гетероциклил или гетероарил содержит 1-4 гетероатома, выбранных из следующих атомов: N, O или S;
R5 и R6 независимо выбраны из H, -OH, галогена, циано, замещенного или незамещенного амино, замещенного или незамещенного C1-6 алкила или незамещенного C1-6 алкоксила;
n представляет собой любое целое число от 0 до 3; и
где термин "замещенный" относится к одному или нескольким атомам водорода в группе, замещенным заместителем, выбранным из: галогена, -OH, -NH2, -NH(незамещенный C1-6 алкил), -CN, галогенированного C1-8 алкила, незамещенного C1-8 алкоксила.
2. Соединение формулы I по п.1, в котором один из X1 и X2 представляет собой CH2, а другой представляет собой связь.
3. Соединение формулы I по п.1, в котором X3 представляет собой S.
4. Соединение формулы I по п.1, в котором X4 выбран из N или CH.
5. Соединение формулы I по п.1, в котором R1, R2, R3, R4 и R7 независимо выбраны из H, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси или изопропокси.
6. Соединение формулы I по п.1, в котором R5 и R6 независимо выбраны из H, -OH, -F, -Cl, -Br, -CN, -NH2, -NHC1-3 алкила, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси или изопропокси.
7. Соединение формулы I по п.1, в котором заместитель выбран из -F, -Cl, -Br, -OH, -NH2, -NH(C1-6 алкил), -CN, C1-6 алкила, C1-4 алкоксила.
8. Соединение формулы I по п.7, в котором заместитель выбран из -F, -Cl, -Br, -OH, -NH2, -NH(C1-3 алкил), -CN, C1-3 алкила, C1-3 алкоксила.
9. Соединение формулы I по п.8, в котором заместитель выбран из -F, -Cl, -Br, -OH, -NH2, -NH(C1-3 алкил), -CN, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси или изопропокси.
10. Соединение формулы I по п.9, в котором кольцо C выбрано из любой из следующих групп:
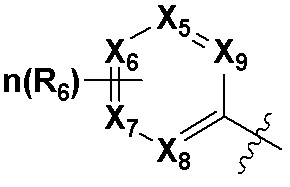 ,
, 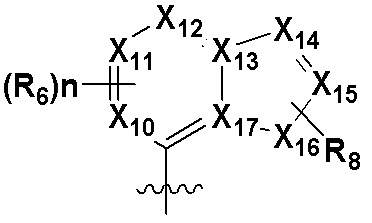 или
или  ;
;
где:
X5, X6, X7, X8 и X9 независимо выбраны из N или CRd; и по большей мере 3 из X5, X6, X7, X8 и X9 одновременно представляют собой N;
X10, X11, X12, X13, X14, X15, X16 и X17 независимо выбраны из N или CRd; и по большей мере 5 из X10, X11, X12, X13, X14, X15, X16 и X17 одновременно представляют собой N;
X18, X19, X20 и X21 независимо выбраны из N или CRd, и по большей мере 3 из X18, X19, X20 и X21 одновременно представляют собой N;
R6 и R8 независимо выбраны из H, -NH2, -CN, -OH, галогена, незамещенного или галогенированного C1-6 алкила или незамещенного C1-6 алкоксила; и
Rd выбран из H, галогена, незамещенного или галогенированного C1-6 алкила или незамещенного или галогенированного C1-6 алкоксила.
11. Соединение формулы I по п.10, в котором кольцо C выбрано из любой из следующих групп:
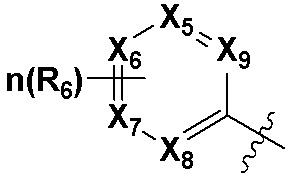 или
или 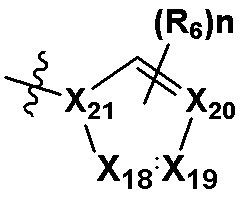 ;
;
где:
0, 1 или 2 из X5, X6, X7, X8 и X9 представляют собой N, остальные представляют собой CRd;
0, 1 или 2 из X18, X19, X20 и X21 представляют собой N, остальные представляют собой CRd;
R6 выбран из H, -NH2, -CN, -OH, -F, -Cl, -Br, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси, изопропокси, фторированного или бромированного C1-3 алкила; и
Rd выбран из H, -F, -Cl, -Br, метила, этила, пропила, изопропила, бутила, метокси, этокси, пропокси, изопропокси, фторированного или бромированного C1-3 алкила или фторированного или бромированного C1-3 алкоксила.
12. Соединение формулы I по п.11, в котором кольцо C выбрано из любой из следующих групп:
 ,
,  или
или  .
.
13. Соединение формулы I по п.1, в котором кольцо A выбрано из замещенного или незамещенного C5-6 арила или замещенного или незамещенного 5-6-членного гетероарила, где гетероарил содержит 1 или 2 атома N.
14. Соединение формулы I по п.13, в котором кольцо A выбрано из любой из следующих групп:
 ,
,  ,
,  ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,  или
или  .
.
15. Соединение формулы I по п.14, в котором кольцо A выбрано из любой из следующих групп:
 ,
,  ,
,  ,
,  ,
,  или
или  .
.
16. Соединение формулы I по п.1, где соединение имеет структуру, выбранную из:
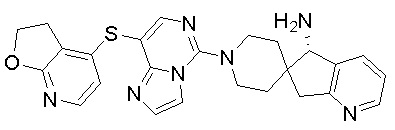 ,
, 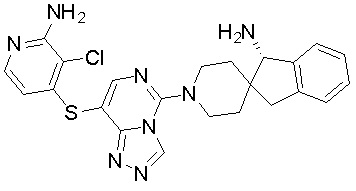 ,
, 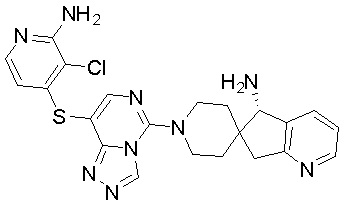 ,
, 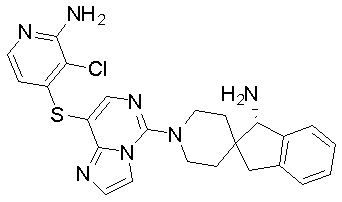 ,
, 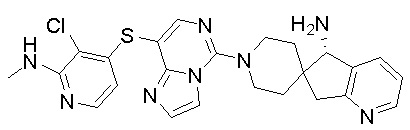 ,
, 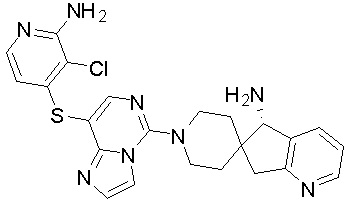 ,
, 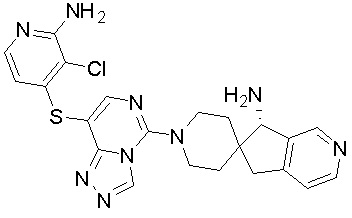 ,
, 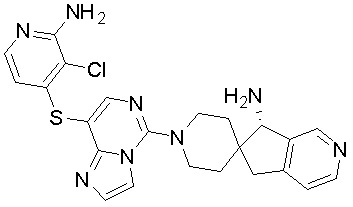 ,
, 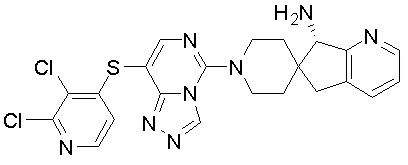 ,
, 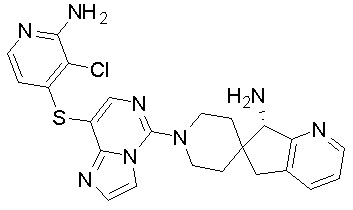 ,
, 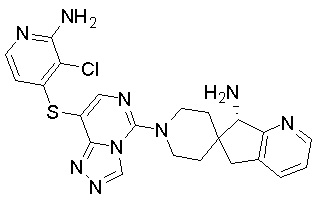 ,
, 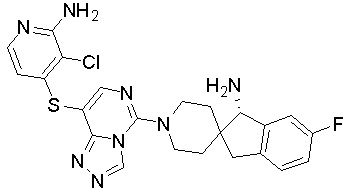 ,
, 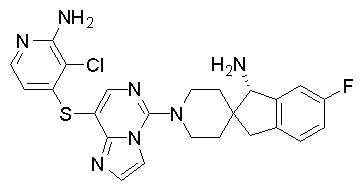 ,
, 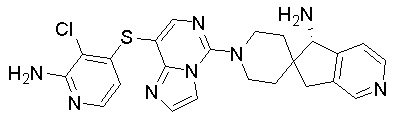 ,
, 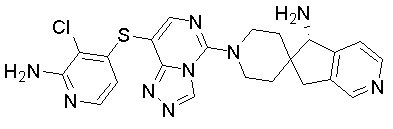 ,
, 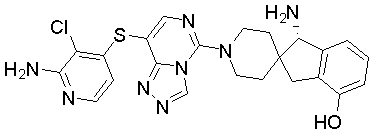 ,
,  ,
, 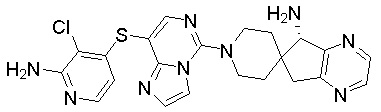 ,
,  ,
, 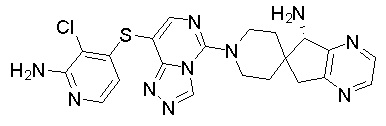 ,
,  ,
, 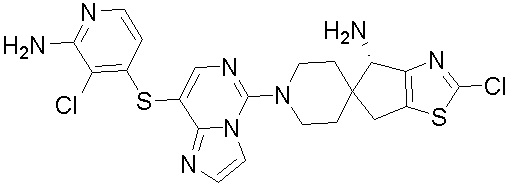 или
или 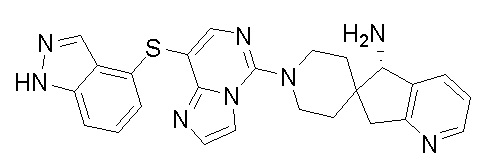 .
.
17. Способ получения соединения формулы I по любому из пп.1-16, включающий следующе стадии:
(i) проводят реакцию нуклеофильного замещения между соединением формулы Ib и соединением формулы Ic с получением соединения формулы Id;
(ii) проводят реакцию замещения между соединением формулы Id и соединением формулы Ie с получением соединения формулы If; и
(iii) удаляют защитную группу в соединении формулы If посредством кислоты с получением соединения формулы I:
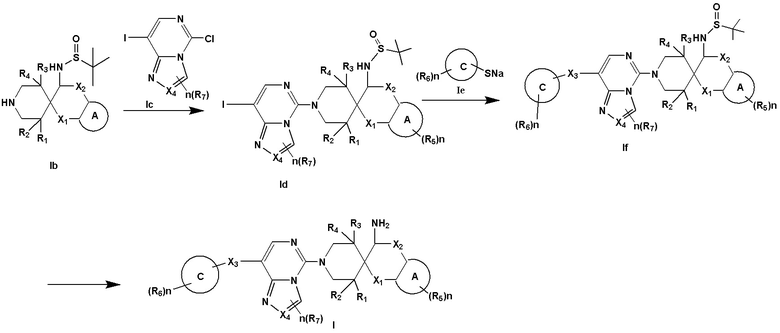
18. Применение соединения формулы I по любому из пп.1-16 в способе:
(a) получения лекарственного средства для профилактики или лечения заболеваний или состояний, связанных с аномальной активностью SHP2;
(b) получения лекарственного средства для профилактики или лечения SHP2-опосредованных заболеваний или состояний;
(c) получения лекарственного средства-ингибитора для ингибирования активности SHP2;
(d) нетерапевтического ингибирования активности SHP2 in vitro;
(e) нетерапевтического ингибирования пролиферации опухолевых клеток in vitro; или
(f) лечения заболеваний или состояний, связанных с аномальной активностью SHP2.
19. Применение по п.18, где заболевание представляет собой рак, включающий, но не ограничиваясь ими, синдром Нунана, синдром леопарда, ювенильный миеломоноцитарный лейкоз, нейробластому, меланому, острый миелоидный лейкоз, рак молочной железы, рак пищевода, рак легких, рак толстой кишки, рак головы, нейробластому, плоскоклеточную карциному головы и шеи, рак желудка, анапластическую крупноклеточную лимфому или глиобластому.
20. Фармацевтическая композиция для ингибирования активности SHP2, содержащая:
(i) эффективное количество соединения формулы I по любому из пп.1-16, или его фармацевтически приемлемой соли, или его энантиомера или таутомера; и
(ii) фармацевтически приемлемый носитель.
21. Способ ингибирования активности SHP2, включающий: введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы I по любому из пп.1-16 или его фармацевтически приемлемой соли или введение субъекту, нуждающемуся в этом, эффективного количества фармацевтической композиции по п.20.
| WO2018172984 А1, 27.09.2018 | |||
| EA 201691442 A1, 30.12.2016 | |||
| CN 105899491 A, 24.08.2016. |

