УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
[0001] ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0002] Настоящее изобретение относится к соединениям, которые ингибируют SHP2 и могут использоваться для лечения гиперпролиферативных и неопластических заболеваний. Настоящее изобретение также относится к способам лечения рака или гиперпролиферативных заболеваний с помощью соединений настоящего изобретения.
[0003] ОПИСАНИЕ СУЩЕСТВУЮЩЕГО УРОВНЯ ТЕХНИКИ
[0004] SHP2 представляет собой протеинтирозинфосфатазу (PTP), содержащую Src-гомологичные домены 2 (SH2), кодируемые геном PTPN11. SHP2 вносит вклад во множество клеточных функций, включая пролиферацию, дифференциацию, поддержание клеточного цикла и миграцию. SHP2 необходима для полной активации пути Ras/ERK1/2, ключевого сигнального каскада в онкобиологии ниже широкого ряда рецепторных тирозинкиназ и других сигнальных трансдукторов. Было также показано, что SHP2 способствует передаче сигналов PI3K/AKT, JAK/STAT, JNK и NF-kB, которые также связаны с различными видами рака человека. SHP2 представляет собой онкобелок. См., Frankson, Rochelle, et al. «Therapeutic Targeting of Oncogenic Tyrosine Phosphatases». Cancer Research. Vol. 77, No. 21 (2017): pp. 5701-5705. Fedele, Carmine, et al. «SHP2 Inhibition Prevents Adaptive Resistance to MEK inhibitors in Multiple Cancer Models». Cancer Discovery. Vol. 8, No. 10 (2018): pp. 1237-49. Nichols, Robert J., et al. «Efficacy of SHP2 phosphatase inhibition in cancers with nucleotide-cycling oncogenic RAS, RAS-GTP dependent oncogenic BRAF and NF1 loss». bioRxiv 188730; doi: https://doi.org/10.1101/188730.
[0005] Следовательно, низкомолекулярные ингибиторы SHP2 были бы полезны для лечения широкого спектра раковых заболеваний, таких как, например, меланома, ювенильные миеломоноцитарные лейкозы, нейробластома, положительный по филадельфийской хромосоме хронический миелолейкоз, положительные по филадельфийской хромосоме острые лимфобластные лейкозы, острые миелоидные лейкозы, миелопролиферативные новообразования (такие как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз), рак молочной железы, рак легких, рак печени, колоректальный рак, рак пищевода, рак желудка, плоскоклеточная карцинома головы и шеи, глиобластома, анапластическая крупноклеточная лимфома, карцинома щитовидной железы, шпицоидные новообразования, а также нейрофиброматоз и синдром Нунан.
[0006] Ингибиторы SHP2 известны, см., например, WO 2015/107493; WO 2015/107494; WO 2015/107495; WO 2016/203404; WO 2016/203405; WO 2016/203406; WO 2017/210134; WO 2017/211303; WO 2017/216706; WO 2018/013597; WO 2018/057884; WO 2018/081091; WO 2018/136264; WO 2018/136265; WO 2018/172984 и WO 2019/051469. Однако хорошо известно, что существуют трудности при разработке соединения для получения одобренного лекарственного средства. DiMasi, Joseph A. «Success rates for new drugs entering clinical testing in the United States». Clinical Pharmacology & Therapeutics. Vol. 58, no. 1 (1995): pp. 1-14. Scannell, JW, Bosley J. «When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis». PloS ONE 11(2) (2016): e0147215. doi: 10.1371/journal.pone.0147215.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0007] Существует постоянная потребность в новых и оригинальных терапевтических агентах, которые могут использоваться для лечения рака и гиперпролиферативных состояний. Разработка и создание новых фармацевтических соединений жизненно необходима.
[0008] В частности, в одном из аспектов предложены соединения формулы I:
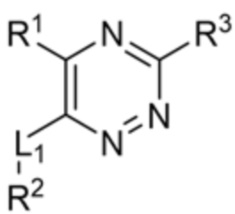
I
или их стереоизомер, таутомер или фармацевтически приемлемая соль, где L1, R1, R2 и R3 являются такими, как определено в настоящем описании.
[0009] В другом аспекте предложен способ лечения гиперпролиферативного нарушения путем введения терапевтически эффективного количества соединения формулы I или его стереоизомера, таутомера или фармацевтически приемлемой соли нуждающемуся в этом пациенту. Соединение может быть введено отдельно или совместно с по меньшей мере одним другим антигиперпролиферативным или химиотерапевтическим соединением.
[0010] В другом аспекте предлагается способ ингибирования активности протеинтирозинфосфатазы SHP2 в клетке, включающий обработку клетки соединением в соответствии с формулой I, или его стереоизомером, таутомером или фармацевтически приемлемой солью, в количестве, эффективном для ослабления или устранения активности киназы SHP2.
[0011] В еще одном аспекте предложены способы лечения или профилактики заболевания или нарушения, модулируемого SHP2, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I, или его стереоизомера, таутомера или фармацевтически приемлемой соли. Примеры таких заболеваний и нарушений включают, без ограничения, гиперпролиферативные нарушения, такие как рак.
[0012] В еще одном аспекте предложены способы лечения или профилактики рака, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I, или его стереоизомера, таутомера или фармацевтически приемлемой соли, по отдельности или в сочетании с одним или более дополнительными соединениями, обладающими противораковыми свойствами.
[0013] В другом аспекте предлагается способ лечения гиперпролиферативного заболевания у млекопитающего, включающий введение терапевтически эффективного количества соединения формулы I или его стереоизомера, таутомера или фармацевтически приемлемой соли млекопитающему.
[0014] В другом аспекте предложено использование соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли в производстве лекарственного средства для лечения гиперпролиферативного заболевания.
[0015] В другом аспекте предложено соединение формулы I, его стереоизомер, таутомер или фармацевтически приемлемая соль для использования в лечении гиперпролиферативных заболеваний.
[0016] В другом аспекте предложена фармацевтическая композиция, содержащая соединение формулы I, его стереоизомер, таутомер или фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или эксципиент.
[0017] В другом аспекте предложены промежуточные соединения для получения соединений формулы I. Некоторые соединения формул могут использоваться в качестве промежуточных соединений для других соединений формул.
[0018] Другой аспект включает в себя способы получения, способы разделения и способы очистки описанных здесь соединений.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0019] Ниже подробно описаны некоторые варианты осуществления, примеры которых проиллюстрированы в прилагаемых структурах и формулах. Хотя будут описаны перечисленные варианты осуществления, следует понимать, что они не предназначены для ограничения изобретения этими вариантами осуществления. Напротив, изобретение предназначено для охвата всех альтернатив, модификаций и эквивалентов, которые могут быть включены в объем настоящего изобретения, определенный формулой изобретения. Специалисту в данной области будут известны многие способы и материалы, сходные или эквивалентные описанным в настоящем изобретении, которые могут использоваться в практике применения настоящего изобретения. Настоящее изобретение никоим образом не ограничивается описанными способами и материалами. В случае, когда один или более из включенных литературных источников и аналогичных материалов отличается от данной заявки или противоречит ей, включая, без ограничения, определенные термины, применение терминов, описанные методики или тому подобное, варианты в данной заявке будут иметь преимущество.
[0020] ОПРЕДЕЛЕНИЯ
[0021] Единственное число, используемое в данном описании, относится к единственному или множественному числу, например, «соединение» относится к одному или более соединений или к по меньшей мере одному соединению. В связи с этим выражения «один», «один или более» и «по меньшей мере один» могут использоваться в настоящем описании взаимозаменяемо.
[0022] Фраза «как определено в настоящем документе» относится к наиболее широкому определению для каждой группы, как это предусмотрено в разделе «Подробное описание изобретения» или в наиболее широком пункте формулы изобретения. Во всех остальных вариантах осуществления, приведенных ниже, заместители, которые могут присутствовать в каждом варианте осуществления и которые специально не определены, сохраняют наиболее широкое значение из раздела «Подробное описание изобретения».
[0023] Как используется в данном описании, независимо от того, находится ли термин в переходной фразе или в теле пункта формулы изобретения, термины «содержит (содержат)» и «содержащий» следует толковать как имеющие открытое значение. То есть термины должны быть интерпретированы как синонимы фраз «имеющий по меньшей мере» или «включающий по меньшей мере». При использовании в контексте способа, термин «содержащий» означает, что способ включает по меньшей мере перечисленные стадии, но может включать и дополнительные стадии. При использовании в контексте соединения или композиции, термин «содержащий» означает, что соединение или композиция включает по меньшей мере перечисленные признаки или компоненты, но также может включать в себя и дополнительные признаки или компоненты. В дополнение к этому слова «включать», «включающий» и «включает» при использовании в данном описании и в следующих пунктах формулы изобретения предназначены для определения наличия заявленных признаков, целых чисел, компонентов или стадий, но они не исключают присутствия или добавления одного или более других признаков, целых чисел, компонентов, стадий или их групп.
[0024] Термин «независимо» употребляется в настоящем документе для обозначения того, что радикал применяется в любом отдельно взятом случае независимо от наличия или отсутствия радикала, имеющего такое же или другое определение в пределах одного соединения. Таким образом, в соединении, в котором R" встречается дважды и определяется как «независимо углерод или азот», оба радикала R" могут быть углеродом, оба R" могут быть азотом, или один R" может быть углеродом, а другой - азотом.
[0025] Когда любой радикал (например, R1, R4a, Ar, X1 или Het) встречается более одного раза в любом фрагменте или формуле, изображающей или описывающей соединения, используемые или заявленные в настоящем изобретении, их определение в каждом случае является независимым от их значения в каждом другом случае. К тому же, комбинации заместителей и/или радикалов возможны только в случае, если такие комбинации дают стабильные соединения.
[0026] Термин «необязательный» или «необязательно», используемый в данном описании, описывает последующие события или обстоятельства, которые могут, но не обязаны случиться, и что описание включает случаи, при которых события или обстоятельства случаются, и случаи, когда их нет. Например, «необязательно замещенный» означает, что необязательно замещенный фрагмент может включать водород или заместитель.
[0027] Используемый в настоящем документе термин «примерно» означает «приблизительно», «порядка», «ориентировочно» или «около». Когда термин «примерно» используется вместе с числовым диапазоном, он изменяет этот диапазон расширением верхней и нижней границ указанных числовых значений. В общем, термин «примерно» используется в настоящем изобретении для изменения числового значения выше и ниже указанного значения на отклонение в 20%.
[0028] Указание в данном описании числового диапазона для переменной предназначено для того, чтобы показать, что изобретение можно осуществлять на практике с переменной, равной любому из значений в пределах этого диапазона. Так, переменная, которая по определению является дискретной, может быть равна любому целому числу в этом числовом диапазоне, включая конечные значения диапазона. Аналогичным образом, переменная, которая по определению является непрерывной, может быть равна любому вещественному значению в этом числовом диапазоне, включая конечные значения диапазона. Например, переменная, которая описана как обладающая значениями, равными от 0 до 2, может быть равна 0, 1 или 2 в случае, если она по определению является дискретной, и может быть равна 0,0, 0,1, 0,01, 0,001 или любому другому вещественному значению, если она по определению является непрерывной.
[0029] Соединения формулы I проявляют таутомеризм. Таутомерные соединения могут существовать в виде двух или более взаимопревращающихся форм. Прототропные таутомеры образуются в результате миграции ковалентно связанного атома водорода между двумя атомами. Таутомеры обычно существуют в равновесии, и попытки изолировать отдельные таутомеры обычно приводят к получению смеси, химические и физические свойства которой соответствуют смеси соединений. Положение равновесия зависит от химических свойств внутри молекулы. Например, у многих алифатических альдегидов и кетонов, таких как ацетальдегид, преобладает кето-форма, тогда как у фенолов преобладает енольная форма. Обычные прототропные таутомеры включают кето/енольные (-C(=O)-CH2- ↔ -C(-OH)=CH-), амид/имидокислотные (-C(=O)-NH- ↔ -C(-OH)=N-) и амидиновые (-C(=NR)-NH- ↔ -C(-NHR)=N-) таутомеры. Последние два особенно распространены в гетероарильных и гетероциклических кольцах, и настоящее изобретение охватывает все таутомерные формы данных соединений.
[0030] Специалисту будет понятно, что некоторые из соединений формулы I могут содержать один или более хиральных центров и поэтому могут существовать в двух или более стереоизомерных формах. Рацематы этих изомеров, отдельные изомеры и смеси, обогащенные одним энантиомером, а также диастереоизомеры, в которых содержатся два хиральных центра, и смеси, частично обогащенные конкретными диастереоизомерами, входят в объем настоящего изобретения. Настоящее изобретение включает все отдельные стереоизомеры (например, энантиомеры), рацемические смеси или частично разделенные смеси соединений формулы I и, в случае необходимости, их отдельные таутомерные формы.
[0031] Соединения формулы I могут содержать основной центр, и подходящие кислотно-аддитивные соли образуются из кислот, которые формируют нетоксичные соли. Примеры солей неорганических кислот включают гидрохлорид, гидробромид, гидройодид, хлорид, бромид, йодид, сульфат, бисульфат, нитрат, фосфат и гидрофосфат. Примеры солей органических кислот включают ацетат, фумарат, памоат, аспартат, безилат, карбонат, бикарбонат, камзилат, D- и L-лактат, D- и L-тартрат, эзилат, мезилат, малонат, оротат, глюцептат, метилсульфат, стеарат, глюкуронат, 2-напсилат, тозилат, гибензат, никотинат, изетионат, малат, малеат, цитрат, глюконат, сукцинат, сахарат, бензоат, эзилат и памоат. В качестве обзора подходящих солей см. Berge, Stephen M., et al. «Pharmaceutical salts». J. Pharm. Sci. Vol. 66, No. 1 (1977): 1-19, и Paulekuhn, G. Steffen, et al. «Trends in Active Pharmaceutical Ingredient Salt Selection based on Analysis of the Orange Book Database». J. Med. Chem. Vol. 50, No. 26 (2007): 6665-6672.
[0032] Технические и научные термины, используемые в настоящем описании, имеют значение, обычно понимаемое специалистом в данной области, к которой относится настоящее изобретение, если не указано иное. В настоящем описании приведены ссылки на различные методологии и материалы, известные специалистам в данной области. Стандартные справочники, в которых изложены общие положения фармакологии, включают Hardman, Joel Griffith, et al. Goodman & Gilman's The Pharmacological Basis of Therapeutics. New York: McGraw-Hill Professional, 2001. Исходные материалы и реагенты, которые используют при получении таких соединений, либо являются доступными от коммерческих поставщиков, таких как Sigma-Aldrich (St. Louis, MO), либо могут быть получены способами, известными специалистам в данной области, в соответствии с процедурами, изложенными в ссылках. Материалы, реагенты и тому подобное, на которые сделана ссылка в нижеследующем описании и примерах, могут быть получены из коммерческих источников, если не указано иное. Общие процедуры синтеза были описаны в учебных пособиях, таких как Louis F. Fieser, Mary Fieser, Reagents for Organic Synthesis. v. 1-23, New York: Wiley 1967-2006 ed. (также доступно на веб-сайте Wiley InterScience®); LaRock, Richard C., Comprehensive Organic Transformations: A Guide to Functional Group Preparations. New York: Wiley-VCH, 1999; B. Trost, I. Fleming, eds. Comprehensive Organic Synthesis. v. 1-9, Oxford: Pergamon 1991; A. R. Katritzky, C. W. Rees, eds. Comprehensive Heterocyclic Chemistry. Oxford: Pergamon 1984; A. R. Katritzky, C. W. Rees, eds. Comprehensive Heterocyclic Chemistry II. Oxford: Pergamon 1996; и Paquette, Leo A., ed. Organic Reactions. v. 1-40, New York: Wiley & Sons 1991; и будут известны специалистам в данной области.
[0033] Термин «алкил» включает в себя линейные или разветвленные радикалы атомов углерода. Некоторые алкильные фрагменты обозначаются сокращениями, например, метил («Ме»), этил («Et»), пропил («Pr») и бутил («Bu»), и дополнительные сокращения используются для обозначения конкретных изомеров соединений, например, 1-пропил или н-пропил («н-Pr»), 2-пропил или изопропил («и-Pr»), 1-бутил или н-бутил («н-Bu»), 2-метил-1-пропил или изобутил («и-Bu»), 1-метилпропил или втор-бутил («втор-Bu»), 1,1-диметилэтил или трет-бутил («т-Bu») и тому подобное. Сокращения иногда применяются в сочетании с аббревиатурами, обозначающими элементы и химические структуры, например, метанол («MeOH») или этанол («EtOH»). В некоторых вариантах осуществления, алкил представляет собой C1-10 алкил. В некоторых вариантах осуществления, алкил представляет собой С1-6 алкил.
[0034] Дополнительные сокращения, используемые во всей заявке, могут включать, например, бензил («Bn»), фенил («Ph»), ацетат («Ac») и мезилат («Ms»).
[0035] Термин «BOC» или «boc» или «Boc» означает трет-бутилоксикарбонильную защитную группу.
[0036] Термины алкенил и алкинил также включают линейные или разветвленные радикалы атомов углерода.
[0037] Термин «алкокси», используемый в данном описании, означает алкильный заместитель, присоединенный через атом кислорода. Неограничивающие примеры включают метокси, этокси, пропокси, бутокси, пентокси, и гексилокси.
[0038] Термин «бициклический», используемый в данном описании, означает бициклическую, одновалентную углеводородную группу, содержащую от шести до десяти атомов углерода, в которых два кольца конденсированы, спиро-конденсированы или образуют мостиковую структуру. При использовании для модификации гетероцикла или гетероарила бициклическая группа может содержать от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы.
[0039] Термин «циклоалкил», используемый в настоящем документе, означает циклическую одновалентную углеводородную группу формулы -CnH(2n-1), содержащую по меньшей мере три атома углерода. Неограничивающие примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
[0040] Термин «арил», используемый в данном описании, означает фенил или нафталинил.
[0041] Термины «гетероцикл» и «гетероциклический» означают 4-7 членные насыщенные или частично ненасыщенные кольца, содержащие один, два или три гетероатома, выбранные из группы, состоящей из O, N, S, S(=O) и S(=O)2. В некоторых случаях эти термины могут быть специально дополнительно ограничены, например, «5-6 членное гетероциклическое соединение», включающее только пяти- и шестичленные кольца.
[0042] Термин «гетероарил» означает 5-6-членные ароматические кольца, содержащие один, два, три или четыре гетероатома, выбранные из группы, состоящей из O, N и S. В некоторых случаях эти термины могут быть специально дополнительно ограничены, например, «5-6 членный гетероарил», где гетероарил содержит один или два гетероатома азота. Как известно специалисту в данной области, гетероарильные кольца имеют менее ароматический характер по сравнению с их аналогами, содержащими все атомы углерода. Таким образом, для целей изобретения, гетероарильная группа должна обладать ароматическим характером лишь в некоторой степени. Такая гетероарильная группа может быть присоединена через атом углерода кольца или, где позволяет валентность, через атом азота кольца.
[0043] Термин «галоген», используемый в данном описании, относится к фтору, хлору, брому или йоду.
[0044] Связь, направленная в кольцевую систему (в отличие от связи, соединенной с конкретной вершиной), указывает на то, что связь может быть присоединена к любому подходящему атому кольца. Волнистая линия ( ) в пределах связи указывает на место присоединения.
) в пределах связи указывает на место присоединения.
[0045] Термины «лечить» или «лечение» относятся к терапевтическим, профилактическим, паллиативным или превентивным мерам. Благоприятные или желаемые клинические результаты включают, без ограничения, облегчение симптомов, минимизацию степени заболевания, стабилизированное (т.е. не ухудшающееся) состояние заболевания, задержку или замедление прогрессирования заболевания, улучшение или облегчение болезненного состояния и ремиссию (частичную или полную), независимо от того может ли это быть обнаружено или нет. «Лечение» также может означать увеличение продолжительности жизни по сравнению с ожидаемой продолжительностью жизни при отсутствии лечения. Нуждающиеся в лечении включают тех, у кого уже есть данное состояние или нарушение, а также тех, кто имеет предрасположенность к развитию данного состояния или нарушения, или тех, у кого данное состояние или нарушение следует предупреждать.
[0046] Термины «терапевтически эффективное количество» или «эффективное количество» означают количество соединения, описанного в данном изобретении, которое, при введении млекопитающему, нуждающемуся в таком лечении, является достаточным для (i) лечения или профилактики конкретного заболевания, состояния или нарушения, (ii) смягчения, улучшения или устранения одного или более симптомов конкретного заболевания, состояния или расстройства, или (iii) предупреждения или задержки развития одного или более симптомов конкретного заболевания, состояния или нарушения, описанного в данном изобретении. Количество соединения, которое будет соответствовать такому количеству, будет варьировать в зависимости от таких факторов, как конкретное соединение, состояние и тяжесть заболевания, параметры (например, масса тела) млекопитающего, нуждающегося в лечении, но в любом случае, может быть обычным образом определено специалистом в данной области.
[0047] Термины «рак» и «раковый» относятся к или описывают физиологическое состояние у млекопитающих, которое как правило характеризуется аномальным или нерегулируемым ростом клеток. «Опухоль» содержит одну или более раковых клеток. Примеры рака включают, без ограничения, карциному, лимфому, бластому, саркому и лейкоз или лимфоидные злокачественные образования. Более конкретные примеры такого рака включают плоскоклеточный рак (например, эпителиальный плоскоклеточный рак), рак легкого, включая мелкоклеточный рак легкого (SCLC), немелкоклеточный рак легкого (NSCLC), аденокарциному легкого и плоскоклеточную карциному легкого, рак брюшной полости, печеночноклеточный рак, рак желудка, включая рак желудочно-кишечного тракта, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак молочной железы, рак толстой кишки, рак прямой кишки, колоректальный рак, эндометриальную карциному или карциному матки, карциному слюнных желез, рак почек, рак простаты, рак вульвы, рак щитовидной железы, гепатокарциному, анальную карциному, карциному пениса, рак кожи, включая меланому, а также рак головы и шеи.
[0048] Фраза «фармацевтически приемлемый» указывает на то, что данное вещество или композиция совместимы с точки зрения химии и/или токсикологии с другими ингредиентами, входящими в состав препарата, и/или с млекопитающим, которого лечат с их помощью.
[0049] Фраза «фармацевтически приемлемая соль», используемая в данном описании, относится к фармацевтически приемлемым органическим или неорганическим солям описанного здесь соединения.
[0050] Соединения, описанные в данном документе, также включают и другие соли таких соединений, которые не обязательно являются фармацевтически приемлемыми солями и которые могут быть полезны в качестве промежуточных соединений для получения и/или очистки соединений, описанных в данном документе, и/или для разделения энантиомеров соединений, описанных в данном документе.
[0051] Термин «млекопитающее» означает теплокровное животное, имеющее или находящееся в группе риска возникновения описанного здесь заболевания, и включает, без ограничения, морских свинок, собак, кошек, крыс, мышей, хомяков и приматов, включая человека.
[0052] ИНГИБИТОРЫ SHP2
[0053] В настоящем документе предложены соединения и их фармацевтические композиции, которые потенциально пригодны для лечения заболеваний, состояний и/или нарушений, модулируемых SHP2.
[0054] Данное изобретение относится к новому классу триазиновых соединений. Изобретение также относится к получению этих соединений и промежуточных соединений, используемых в получении, к композициям, содержащим соединения, и к применению соединений, включая лечение гиперпролиферативных и неопластических заболеваний, таких как рак.
[0055] В одном варианте осуществления предложены соединения формулы I:
I
или их стереоизомер, таутомер или фармацевтически приемлемая соль, где:
L1 выбран из прямой связи, S, CH2, O, NH и Se;
R1 выбран из водорода и метила;
R2 выбран из (а) фенила, (b) 5-6-членного гетероарила, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, при этом один гетероатом азота может быть замещен кислородом с образованием оксида, (c) 8-10-членного бициклического циклоалкила, (d) 10-членного бициклического арила, (e) 9-10-членного бициклического гетероцикла, где гетероцикл содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, и (f) 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где фенил, гетероарил, бициклический циклоалкил, бициклический арил, бициклический гетероцикл и бициклический гетероарил необязательно замещены одной или более группами, выбранными из группы, состоящей из галогена, циано, оксо, C1-C3 алкила, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, C3-C6 циклоалкила, C1-C3 алкокси, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, NHRa, и 3-6 членного гетероцикла, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы;
R3 выбран из группы, состоящей из:
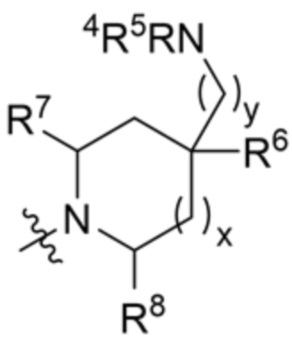 и
и 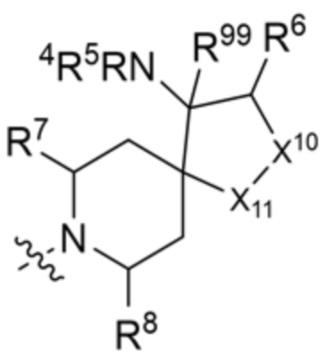 ;
;
X10 представляет собой CR9 или О;
X11 представляет собой CH2 или O, при этом только один из X10 и X11 может быть O;
R4 и R5 независимо выбраны из водорода и C1-C3 алкила;
R6 выбран из группы, состоящей из водорода, OH и C1-C3 алкила, необязательно замещенного OH-группой, или
R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил или 5-6-членный гетероарил, где гетероарил содержит 1 или 2 гетероатома, выбранных из азота, кислорода и серы, при этом арил и гетероарил необязательно замещены 1 или 2 группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила и C1-C3 алкокси;
R7 и R8 представляют собой водород, или
R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо;
R99 представляет собой водород или дейтерий;
x равно 1 или 2;
y равно 0 или 1; и
Ra представляет собой водород или C1-C4 алкил, необязательно замещенный 1-3 группами, выбранными из ОН, метокси, галогена и циано.
[0056] В определенном варианте осуществления предложены соединения формулы I, или их стереоизомер, таутомер или фармацевтически приемлемая соль, где:
L1 выбран из прямой связи, S, CH2, O, NH и Se;
R1 выбран из водорода и метила;
R2 выбран из (а) фенила, необязательно замещенного одной или двумя галогеновыми группами; (b) 5-6-членного гетероарила, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, C1-C3 алкила, необязательно замещенного 1-3 галогеновыми группами, C3-C6 циклоалкила, C1-C3 алкокси, NHRa и 3-6-членного гетероцикла, необязательно замещенного ОН-группой, при этом гетероцикл содержит один или два гетероатома, выбранных из азота и кислорода; (c) 8-10-членного бициклического частично ненасыщенного циклоалкила; (d) 9-10-членного бициклического частично ненасыщенного гетероцикла, где гетероцикл содержит от одного до трех гетероатомов, выбранных из азота, кислорода и серы, где гетероцикл необязательно замещен 1-3 группами, выбранными из галогена и оксо; (е) 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где бициклический гетероарил необязательно замещен 1-3 группами, выбранными из галогена и C1-C3 алкила;
R3 выбран из группы, состоящей из:
и ;
X10 представляет собой CR9 или О;
X11 представляет собой CH2 или O, при этом только один из X10 и X11 может быть O;
R4 и R5 независимо выбраны из водорода и C1-C3 алкила;
R6 выбран из группы, состоящей из водорода, OH и C1-C3 алкила, необязательно замещенного OH-группой, или
R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил или 5-6-членный гетероарил, где гетероарил содержит 1 или 2 гетероатома, выбранных из азота, кислорода и серы, при этом арил и гетероарил необязательно замещены 1 или 2 группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила и C1-C3 алкокси;
R7 и R8 представляют собой водород, или
R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо;
R99 представляет собой водород или дейтерий;
x равно 1 или 2;
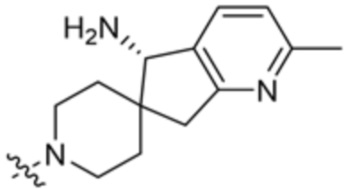
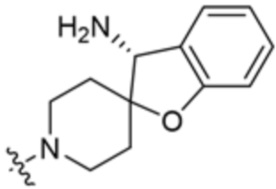
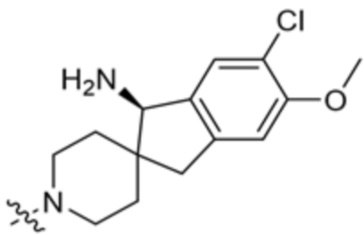
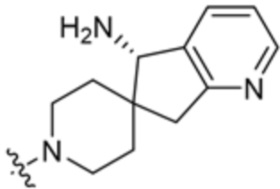
y равно 0 или 1; и
Ra представляет собой водород или C1-C4 алкил, необязательно замещенный 1-3 группами, выбранными из ОН и циано.
[0057] В определенном варианте осуществления предложены соединения формулы I, или их стереоизомер, таутомер или фармацевтически приемлемая соль, где:
L1 выбран из прямой связи, S, CH2, O, и NH;
R1 представляет собой водород;
R2 выбран из (а) фенила, необязательно замещенного одной или двумя галогеновыми группами; (b) 5-6-членного гетероарила, где гетероарил содержит один гетероатом азота, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, C1-C3 алкила, необязательно замещенного 1-3 галогеновыми группами, C3циклоалкила, C1-C3 алкокси, NHRa и 6-членного гетероцикла, необязательно замещенного ОН-группой, при этом гетероцикл содержит один или два гетероатома, выбранных из азота и кислорода; (c) 8-10-членного бициклического частично ненасыщенного циклоалкила; (d) 9-членного бициклического частично ненасыщенного гетероцикла, где гетероцикл содержит два или три гетероатома азота, где гетероцикл необязательно замещен 1-3 группами, выбранными из галогена и оксо; и (е) 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где бициклический гетероарил необязательно замещен одной группой, выбранной из галогена и C1-C3 алкила;
R3 выбран из группы, состоящей из:
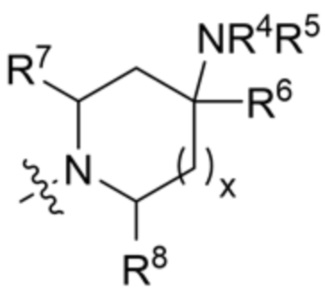 и ;
и ;
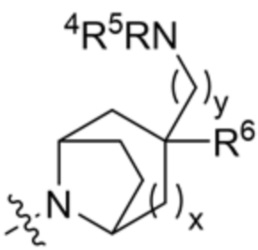
X10 представляет собой CR9 или О;
X11 представляет собой CH2 или O, при этом только один из X10 и X11 может быть O;
R4 и R5 независимо выбраны из водорода и метила;
R6 представляет собой метил, или
R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил или 5-6-членный гетероарил, где гетероарил содержит 1 или 2 гетероатома, выбранных из азота и серы, при этом арил и гетероарил необязательно замещены 1 или 2 группами, выбранными из группы, состоящей из галогена, метила, метокси и циано;
R7 и R8 представляют собой водород, или
R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо;
R99 представляет собой водород или дейтерий;
x равно 1 или 2; и
Ra представляет собой водород или C1-C4 алкил, необязательно замещенный одной группой, выбранной из ОН и циано.
[0058] В определенном варианте осуществления предложены соединения формулы I, или их стереоизомер, таутомер или фармацевтически приемлемая соль, где:
L1 выбран из прямой связи, S, CH2, O или NH;
R1 выбран из водорода и метила;
R2 выбран из фенила, 5-6 -членного гетероарила, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, 10-членного бициклического арила, и 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы,
где фенил, гетероарил, бициклический арил и бициклический гетероарил необязательно замещены одной или более группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила, необязательно замещенного галогеном, циано или ОН, -O(C1-C3 алкила), необязательно замещенного галогеном, циано или OH, NHRa, и 3-6 членного гетероцикла, необязательно замещенного галогеном, циано или ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы;
R3 выбран из группы, состоящей из:
и 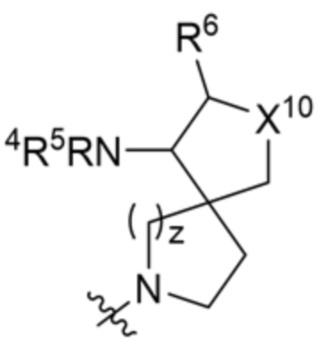 ;
;
X10 представляет собой CR9 или О;
R4 и R5 независимо выбраны из водорода и метила;
R6 выбран из группы, состоящей из водорода, метила, OH и CH2OH;
R7 и R8 представляют собой водород, или
R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо;
R9 представляет собой водород, или
R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил;
Ra представляет собой водород или C1-C3 алкил, необязательно замещенный ОН, метокси, галогеном или циано;
x равно 1 или 2;
y равно 0 или 1; и
z равно 1 или 2.
[0059] В некоторых вариантах осуществления:
L1 выбран из прямой связи или S;
R1 выбран из водорода и метила;
R2 выбран из фенила, необязательно замещенного галогеном, 5-6-членного гетероарила, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, метила и NH2, и 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где бициклический гетероарил необязательно замещен галогеном;
R3 выбран из группы, состоящей из:
 ,
,  ,
, 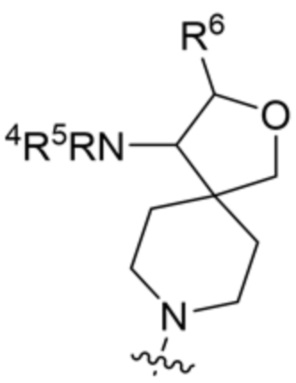 и
и 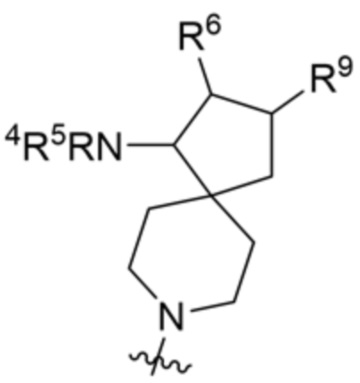 ;
;
R4 и R5 представляют собой водород;
R6 выбран из водорода и метила, или
R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил;
R7 и R8 представляют собой водород, или
R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо;
Rа представляет собой водород.
[0060] В другом варианте осуществления предложены соединения формулы I или их стереоизомер или фармацевтически приемлемая соль.
[0061] В другом варианте осуществления предложены соединения формулы I или их таутомер или фармацевтически приемлемая соль.
[0062] В другом варианте осуществления предложены соединения формулы I или их стереоизомер или таутомер.
[0063] В другом варианте осуществления предложены соединения формулы I или их стереоизомер.
[0064] В другом варианте осуществления предложены соединения формулы I или их таутомер.
[0065] В другом варианте осуществления предложены соединения формулы I или их фармацевтически приемлемая соль.
[0066] В некоторых вариантах осуществления L1 выбран из прямой связи, S, CH2, O, NH и Se. В некоторых вариантах осуществления L1 выбран из прямой связи и S. В некоторых вариантах осуществления L1 выбран из S, CH2, O или NH. В некоторых вариантах осуществления L1 представляет собой прямую связь. В некоторых вариантах осуществления L1 представляет собой S. В некоторых вариантах осуществления L1 представляет собой селен (Se).
[0067] В некоторых вариантах осуществления L1 выбран из прямой связи, S, CH2, O или NH. В некоторых вариантах осуществления L1 выбран из прямой связи и S. В некоторых вариантах осуществления L1 выбран из S, CH2, O или NH. В некоторых вариантах осуществления L1 представляет собой прямую связь. В некоторых вариантах осуществления L1 представляет собой S.
[0068] В некоторых вариантах осуществления R1 выбран из водорода и метила. В некоторых вариантах осуществления R1 представляет собой водород. В некоторых вариантах осуществления R1 представляет собой метил. В предпочтительном варианте осуществления R1 представляет собой водород.
[0069] В некоторых вариантах осуществления R2 выбран из (а) фенила, (b) 5-6-членного гетероарила, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, при этом один гетероатом азота может быть замещен кислородом с образованием оксида, (c) 8-10-членного бициклического циклоалкила, (d) 10-членного бициклического арила, (e) 9-10-членного бициклического гетероцикла, где гетероцикл содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, и (f) 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где фенил, гетероарил, бициклический циклоалкил, бициклический арил, бициклический гетероцикл и бициклический гетероарил необязательно замещены одной или более группами, выбранными из группы, состоящей из галогена, циано, оксо, C1-C3 алкила, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, C3-C6 циклоалкила, C1-C3 алкокси, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, NHRa, и 3-6 членного гетероцикла, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы. Группа R2 может быть замещена одной или более группами, что означает от одного до четырех заместителей, насколько позволяет валентность. В некоторых вариантах осуществления R2 выбран из (а) фенила, необязательно замещенного одной или двумя галогеновыми группами; (b) 5-6-членного гетероарила, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, C1-C3 алкила, необязательно замещенного галогеном, C3-C6-циклоалкила, C1-C3 алкокси, NHRa и 3-6-членного гетероцикла, необязательно замещенного ОН-группой, при этом гетероцикл содержит один или два гетероатома, выбранных из азота и кислорода; (c) 8-10-членного бициклического частично ненасыщенного циклоалкила; (d) 9-10-членного бициклического частично ненасыщенного гетероцикла, где гетероцикл содержит от одного до трех гетероатомов, выбранных из азота, кислорода и серы, где гетероцикл необязательно замещен 1-3 группами, выбранными из галогена и оксо; (е) 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где бициклический гетероарил необязательно замещен 1-3 группами, выбранными из галогена и C1-C3 алкила. В некоторых вариантах осуществления R2 выбран из (а) фенила, необязательно замещенного одной или двумя галогеновыми группами; (b) 5-6-членного гетероарила, где гетероарил содержит один гетероатом азота, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, C1-C3 алкила, необязательно замещенного 1-3 галогеновыми группами, C3циклоалкила, C1-C3 алкокси, NHRa и 6-членного гетероцикла, необязательно замещенного ОН-группой, при этом гетероцикл содержит один или два гетероатома, выбранных из азота и кислорода; (c) 8-10-членного бициклического частично ненасыщенного циклоалкила; (d) 9-членного бициклического частично ненасыщенного гетероцикла, где гетероцикл содержит два или три гетероатома азота, где гетероцикл необязательно замещен 1-3 группами, выбранными из галогена и оксо; и (е) 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где бициклический гетероарил необязательно замещен одной группой, выбранной из галогена и C1-C3 алкила. В некоторых вариантах осуществления R2 выбран из группы, состоящей из фенила, 2-хлорфенила, 3-хлорфенила, 2,3-дихлорфенила,2-амино-3-хлорпиридин-4-ила, 2,3-дихлорпиридин-4-ила, 3-хлор-2-метилпиридин-4-ила, 3-хлор-2-(4-гидроксипиперидин-1-ил)пиридин-4-ила, 3-хлор-2-(метиламино)пиридин-4-ила, 2,3-диметилпиридин-4-ила, 2-амино-3-циклопропилпиридин-4-ила, 2-амино-5-хлорпиридин-4-ила, 3-хлор-2-метоксипиридин-4-ила, 3-хлор-2-(трифторметил)пиридин-4-ила, 6-амино-2-(трифторметил)пиридин-3-ила, 2-амино-3-бромпиридин-4-ила, 6-аминопиридин-3-ила, 2-амино-3-фторпиридин-4-ила, 3-хлор-2-((2-цианo-2-метилпропил)амино)пиридин-4-ила, 2-амино-3-хлор-1-оксидопиридин-4-ила, 3-хлор-2-((2-гидроксиэтил)амино)пиридин-4-ила, бицикло[4.2.0]октa-1(6),2,4-триен-2-ила, 2,3-дигидро-1H-пирроло[3,2-c]пиридин-1-ила, 3,3-дифтор-2-оксо-2,3-дигидро-1H-пирроло[2,3-b]пиридин-4-ила, 2,3-дигидро-1H-пирроло[2,3-b]пиридин-4-ила, 1H-пирроло[2,3-b]пиридин-4-ила, 5-хлор-1H-пирроло[2,3-b]пиридин-4-ила, 1-метил-1H-пирроло[2,3-b]пиридин-4-ила, 1H-пирроло[2,3-b]пиридин-3-ила, имидазо[1,2-a]пиридин-3-ила, 1H-пиразоло[3,4-b]пиридин-3-ила, 1H-индазол-3-ила, 1H-пирроло[2,3-b]пиридин-5-ила, имидазо[1,2-a]пиримидин-3-ила, изохинолин-5-ила и 1H-индол-3-ила.
[0070] В некоторых вариантах осуществления R2 выбран из фенила, 5-6-членного гетероарила, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, 10-членного бициклического арила, и 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где фенил, гетероарил, бициклический арил и бициклический гетероарил необязательно замещены одной или более группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила, необязательно замещенного галогеном, циано или ОН, -O(C1-C3 алкила), необязательно замещенного галогеном, циано или OH, NHRa, и 3-6 членного гетероцикла, необязательно замещенного галогеном, циано или ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы. В некоторых вариантах осуществления R2 выбран из фенила, необязательно замещенного галогеном, 5-6-членного гетероарила, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, метила и NH2, и 9-10-членного бициклического гетероарила, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где бициклический гетероарил необязательно замещен галогеном. В некоторых вариантах осуществления R2 выбран из группы, состоящей из 2,3-дихлорфенила, 2-амино-3-хлорпиридин-4-ила, 2,3-дихлорпиридин-4-ила, 3-хлор-2-метилпиридин-4-ила, 1H-пирроло[2,3-b]пиридин-4-ила и 5-хлор-1H-пирроло[2,3-b]пиридин-4-ила.
[0071] В некоторых вариантах осуществления R2 представляет собой фенил, необязательно замещенный одной или более группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, C1-C3 алкокси, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, NHRa, и 3-6 членного гетероцикла, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы. В некоторых вариантах осуществления R2 представляет собой фенил, необязательно замещенный галогеном. В некоторых вариантах осуществления R2 представляет собой фенил, необязательно замещенный одной или двумя галогеновыми группами. В некоторых вариантах осуществления R2 выбран из группы, состоящей из фенила, 2-хлорфенила, 3-хлорфенила и 2,3-дихлорфенила.
[0072] В некоторых вариантах осуществления R2 представляет собой фенил, необязательно замещенный одной или более группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила, необязательно замещенного галогеном, циано или ОН, -O(C1-C3 алкила), необязательно замещенного галогеном, циано или OH, NHRa, и 3-6 членного гетероцикла, необязательно замещенного галогеном, циано или ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы. В некоторых вариантах осуществления R2 представляет собой фенил, необязательно замещенный галогеном. В некоторых вариантах осуществления R2 представляет собой 2,3-дихлорфенил.
[0073] В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, при этом один гетероатом азота может быть замещен кислородом с образованием оксида, где гетероарил может быть необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, C3-C6 циклоалкила, C1-C3 алкокси, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, NHRa, и 3-6 членного гетероцикла, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы. В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, при этом один гетероатом азота может быть замещен кислородом с образованием оксида, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, C1-C3 алкила, необязательно замещенного 1-3 галогеновыми группами, C3-C6 циклоалкила, C1-C3 алкокси, NHRa и 3-6-членного гетероцикла, необязательно замещенного ОН-группой, при этом гетероцикл содержит один или два гетероатома, выбранных из азота и кислорода. В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, C1-C3 алкила, необязательно замещенного 1-3 галогеновыми группами, C3-C6 циклоалкила, C1-C3 алкокси, NHRa и 3-6-членного гетероцикла, необязательно замещенного ОН-группой, при этом гетероцикл содержит один или два гетероатома, выбранных из азота и кислорода. В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, при этом один гетероатом азота может быть замещен кислородом с образованием оксида, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, метила, трифторметила, циклопропила, метокси, NH2, NHCH2C(CH3)2CN, NHCH2CH2OH и 4-гидроксипиперидин-1-ила. В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит один гетероатом азота, где гетероатом азота может быть замещен кислородом с образованием оксида, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, C1-C3 алкила, необязательно замещенного 1-3 галогеновыми группами, C3-C6 циклоалкила, C1-C3 алкокси, NHRa и 3-6-членного гетероцикла, необязательно замещенного ОН-группой, при этом гетероцикл содержит один или два гетероатома, выбранных из азота и кислорода. В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит один гетероатом азота, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, C1-C3 алкила, необязательно замещенного 1-3 галогеновыми группами, C3-C6 циклоалкила, C1-C3 алкокси, NHRa и 3-6-членного гетероцикла, необязательно замещенного ОН-группой, при этом гетероцикл содержит один или два гетероатома, выбранных из азота и кислорода. В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит один гетероатом азота, где гетероатом азота может быть замещен кислородом с образованием оксида, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, метила, трифторметила, циклопропила, метокси, NH2, NHCH2C(CH3)2CN, NHCH2CH2OH и 4-гидроксипиперидин-1-ила. В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит один гетероатом азота, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, метила, трифторметила, циклопропила, метокси, NH2, NHCH2C(CH3)2CN, NHCH2CH2OH и 4-гидроксипиперидин-1-ила. В некоторых вариантах осуществления R2 представляет собой пиридин, где гетероатом азота может быть замещен кислородом с образованием оксида, где пиридин необязательно замещен одним или двумя заместителями, выбранными из галогена, метила, трифторметила, циклопропила, метокси, NH2, NHCH2C(CH3)2CN, NHCH2CH2OH и 4-гидроксипиперидин-1-ила. В некоторых вариантах осуществления R2 выбран из группы, состоящей из2-амино-3-хлорпиридин-4-ила,2,3-дихлорпиридин-4-ила, 3-хлор-2-метилпиридин-4-ила, 3-хлор-2-(4-гидроксипиперидин-1-ил)пиридин-4-ила, 3-хлор-2-(метиламино)пиридин-4-ила, 2,3-диметилпиридин-4-ила, 2-амино-3-циклопропилпиридин-4-ила, 2-амино-5-хлорпиридин-4-ила, 3-хлор-2-метоксипиридин-4-ила, 3-хлор-2-(трифторметил)пиридин-4-ила, 6-амино-2-(трифторметил)пиридин-3-ила, 2-амино-3-бромпиридин-4-ила, 6-аминопиридин-3-ила, 2-амино-3-фторпиридин-4-ила, 3-хлор-2-((2-цианo-2-метилпропил)амино)пиридин-4-ила, 2-амино-3-хлор-1-оксидопиридин-4-ила и 3-хлор-2-((2-гидроксиэтил)амино)пиридин-4-ила.
[0074] В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, необязательно замещенный одной или более группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила, необязательно замещенного галогеном, циано или ОН, -O(C1-C3 алкила), необязательно замещенного галогеном, циано или OH, NHRa, и 3-6 членного гетероцикла, необязательно замещенного галогеном, циано или ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы. В некоторых вариантах осуществления R2 представляет собой 5-6-членный гетероарил, где гетероарил содержит от одного до четырех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где гетероарил необязательно замещен одним или двумя заместителями, выбранными из галогена, метила и NH2. В некоторых вариантах осуществления R2 выбран из группы, состоящей из 2-амино-3-хлорпиридин-4-ила,2,3-дихлорпиридин-4-ила и 3-хлор-2-метилпиридин-4-ила.
[0075] В некоторых вариантах осуществления R2 представляет собой 8-10-членный бициклический циклоалкил. В некоторых вариантах осуществления R2 представляет собой 8-10-членный бициклический частично ненасыщенный циклоалкил. В некоторых вариантах осуществления R2 представляет собой бицикло[4.2.0]октa-1(6),2,4-триен-2-ил.
[0076] В некоторых вариантах осуществления R2 представляет собой 9-10-членный бициклический гетероцикл, где гетероцикл содержит от одного до трех гетероатомов, выбранных из азота, кислорода и серы, где гетероцикл необязательно замещен одной или более группами, выбранными из галогена и оксо. В некоторых вариантах осуществления R2 представляет собой9-10-членный бициклический частично ненасыщенный гетероцикл, где гетероцикл содержит от одного до трех гетероатомов, выбранных из азота, кислорода и серы, где гетероцикл необязательно замещен 1-3 группами, выбранными из галогена и оксо. В некоторых вариантах осуществления R2 представляет собой 9-членный бициклический частично ненасыщенный гетероцикл, где гетероцикл содержит два или три гетероатома азота, где гетероцикл необязательно замещен 1-3 группами, выбранными из галогена и оксо. В некоторых вариантах осуществления R2 представляет собой 2,3-дигидро-1H-пирроло[3,2-c]пиридин-1-ил, 3,3-дифтор-2-оксо-2,3-дигидро-1H-пирроло[2,3-b]пиридин-4-ил и 2,3-дигидро-1H-пирроло[2,3-b]пиридин-4-ил.
[0077] В некоторых вариантах осуществления R2 представляет собой 9-10-членный бициклический гетероарил, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где бициклический гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, C1-C3 алкокси, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, NHRa, и 3-6 членного гетероцикла, необязательно замещенного 1-3 группами, выбранными из галогена, циано и ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы. В некоторых вариантах осуществления R2 представляет собой 9-10-членный бициклический гетероарил, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где бициклический гетероарил необязательно замещен 1-3 группами, выбранными из галогена и C1-C3 алкила. В некоторых вариантах осуществления R2 представляет собой 9-10-членный бициклический гетероарил, где бициклический гетероарил содержит от одного до трех гетероатомов азота, где бициклический гетероарил необязательно замещен одной группой, выбранной из галогена и метила. В некоторых вариантах осуществления R2 выбран из группы, состоящей из 1H-пирроло[2,3-b]пиридин-4-ила, 5-хлор-1H-пирроло[2,3-b]пиридин-4-ила, 1-метил-1H-пирроло[2,3-b]пиридин-4-ила, 1H-пирроло[2,3-b]пиридин-3-ила, имидазо[1,2-a]пиридин-3-ила, 1H-пиразоло[3,4-b]пиридин-3-ила, 1H-индазол-3-ила, 1H-пирроло[2,3-b]пиридин-5-ила, имидазо[1,2-a]пиримидин-3-ила, изохинолин-5-ила и 1H-индол-3-ила.
[0078] В некоторых вариантах осуществления R2 представляет собой 9-10-членный бициклический гетероарил, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, необязательно замещенный одной или более группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила, необязательно замещенного галогеном, циано или ОН, -O(C1-C3 алкила), необязательно замещенного галогеном, циано или OH, NHRa, и 3-6 членного гетероцикла, необязательно замещенного галогеном, циано или ОН, где гетероцикл содержит один или два гетероатома, выбранных из азота, кислорода и серы. В некоторых вариантах осуществления R2 представляет собой 9-10-членный бициклический гетероарил, где бициклический гетероарил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где бициклический гетероарил необязательно замещен галогеном. В некоторых вариантах осуществления R2 выбран из группы, состоящей из 1H-пирроло[2,3-b]пиридин-4-ила и 5-хлор-1H-пирроло[2,3-b]пиридин-4-ила.
[0079] В некоторых вариантах осуществления Ra представляет собой водород или C1-C4 алкил, необязательно замещенный 1-3 группами, выбранными из ОН, метокси, галогена и циано. В некоторых вариантах осуществления Ra представляет собой водород или C1-C4 алкил, необязательно замещенный 1-3 группами, выбранными из ОН и циано. В некоторых вариантах осуществления Ra представляет собой водород, метил, 2-циано-2-метилпропил или 2-гидроксиэтил. В некоторых вариантах осуществления Ra представляет собой водород или метил.
[0080] В некоторых вариантах осуществления Ra представляет собой водород или C1-C3 алкил, необязательно замещенный ОН, метокси, галогеном или циано. В некоторых вариантах осуществления Ra представляет собой водород или C1-C3 алкил, необязательно замещенный ОН. В некоторых вариантах осуществления Ra представляет собой водород.
[0081] В некоторых вариантах осуществления R3 выбран из группы, состоящей из:
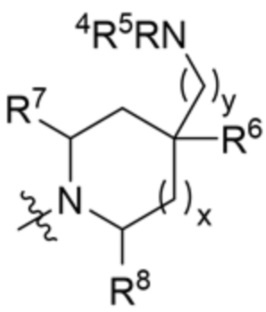 и
и 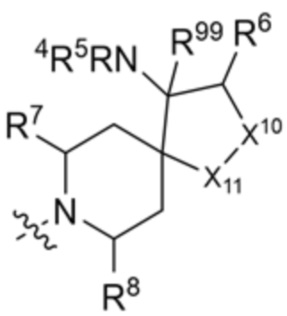 ;
;
где X10 представляет собой CR9 или О, X11 представляет собой CH2 или O, при этом только один из X10 и X11 может быть O; R4 и R5 независимо выбраны из водорода и C1-C3 алкила; R6 выбран из группы, состоящей из водорода, OH и C1-C3 алкила, необязательно замещенного OH-группой, или R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил или 5-6-членный гетероарил, где гетероарил содержит 1 или 2 гетероатома, выбранных из азота, кислорода и серы, при этом арил и гетероарил необязательно замещены 1 или 2 группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила и C1-C3 алкокси; R7 и R8 представляют собой водород, или R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо; R99 представляет собой водород или дейтерий; x равно 1 или 2; и y равно 0 или 1.
[0082] В некоторых вариантах осуществления R3 выбран из группы, состоящей из:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 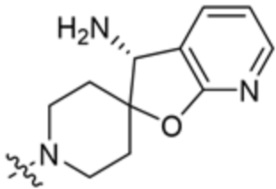 ,
, 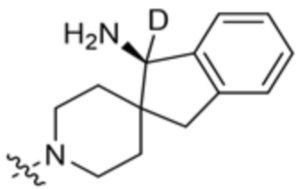 ,
, 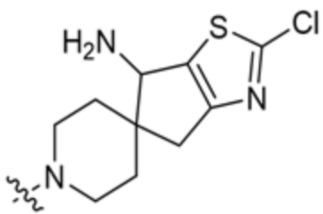 ,
, 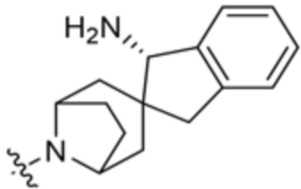 ,
,  и
и 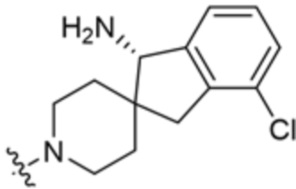 .
.
[0083] В некоторых вариантах осуществления R3 выбран из группы, состоящей из:
и ;
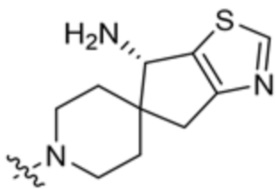
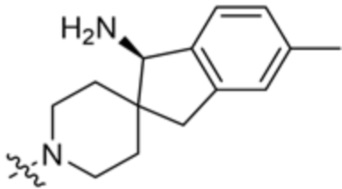
где X10 представляет собой CR9 или О; R4 и R5 независимо выбраны из водорода и метила; R6 выбран из группы, состоящей из водорода, метила, OH и CH2OH; R7 представляет собой водород, или R6 и R7 вместе с атомами, к которым они присоединены, образуют 6-членный арил; R7 и R8 представляют собой водород, или R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо; x равно 1 или 2; y равно 0 или 1; и z равно 1 или 2. В некоторых вариантах осуществления R3 выбран из группы, состоящей из (3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ила, (1R,3s,5S)-3-амино-8-азабицикло[3,2.1]октан-8-ила, 1-амино-1,3-дигидроспиро[инден-2,4’-пиперидин]-1’-ила и 4-амино-4-метилазепан-1-ила.
[0084] В некоторых вариантах осуществления x равно 1 или 2. В некоторых вариантах осуществления x равно 1. В некоторых вариантах осуществления x равно 2.
[0085] В некоторых вариантах осуществления y равно 0 или 1. В некоторых вариантах осуществления y равно 0. В некоторых вариантах осуществления y равно 1.
[0086] В некоторых вариантах осуществления z равно 1 или 2. В некоторых вариантах осуществления z равно 1. В некоторых вариантах осуществления z равно 2.
[0087] В некоторых вариантах осуществления x равно 1, и y равно 0, так что R3 имеет структуру:
.
[0088] В некоторых вариантах осуществления x равно 1, y равно 0, и R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо, так что R3 имеет структуру:
 .
.
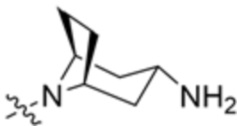
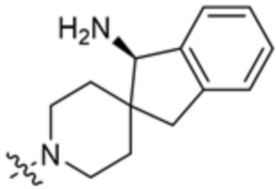
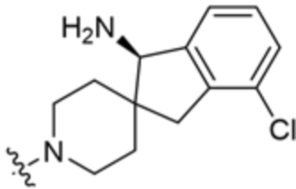
В некоторых вариантах осуществления R3 представляет собой(1R,3s,5S)-3-амино-8-азабицикло[3.2.1]октан-8-ил.
[0089] В некоторых вариантах осуществления x равно 1, y равно 0, и R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо, так что R3 имеет структуру:
 .
.
[0090] В некоторых вариантах осуществления x равно 2, и y равно 0, так что R3 имеет структуру:
.
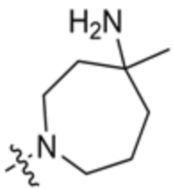
В некоторых вариантах осуществления R3 представляет собой 4-амино-4-метилазепан-1-ил.
[0091] В некоторых вариантах осуществления Х10 представляет собой О, и Х11 представляет собой СН2, так что R3 имеет структуру:
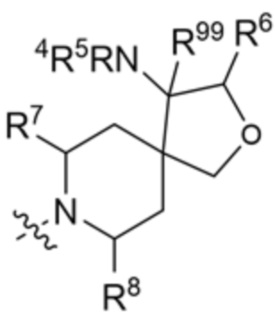 .
.
[0092] В некоторых вариантах осуществления z равно 2, и X10 представляет собой O, так что R3 имеет структуру:
.
В некоторых вариантах осуществления R3 представляет собой(3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил.
[0093] В некоторых вариантах осуществления Х10 представляет собой CR9, и Х11 представляет собой О, так что R3 имеет структуру:
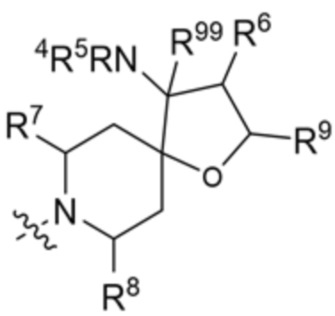 .
.
В этих вариантах осуществления R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил или 5-6-членный гетероарил, где гетероарил содержит 1 или 2 гетероатома, выбранных из азота, кислорода и серы, при этом арил и гетероарил необязательно замещены 1 или 2 группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила и C1-C3 алкокси.
[0094] В некоторых вариантах осуществления Х10 представляет собой CR9, и Х11 представляет собой CH2, так что R3 имеет структуру:
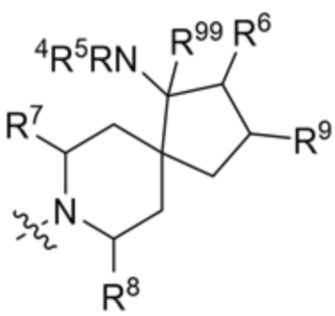 .
.
В этих вариантах осуществления R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил или 5-6-членный гетероарил, где гетероарил содержит 1 или 2 гетероатома, выбранных из азота, кислорода и серы, при этом арил и гетероарил необязательно замещены 1 или 2 группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила и C1-C3 алкокси.
[0095] В некоторых вариантах осуществления z равно 2, и X10 представляет собой CR9, так что R3 имеет структуру:
.
[0096] В некоторых вариантах осуществления z равно 2, X10 представляет собой CR9, и R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил, так что R3 имеет структуру:
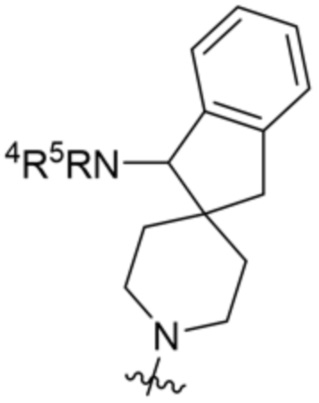 .
.
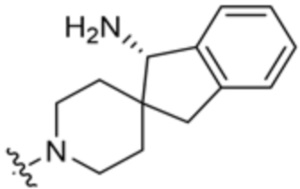
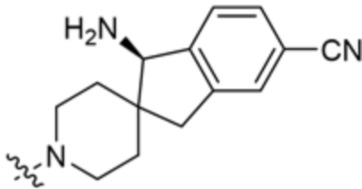
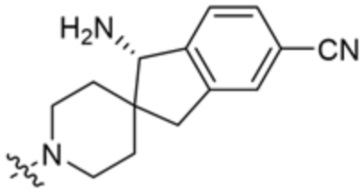
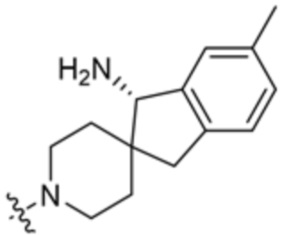
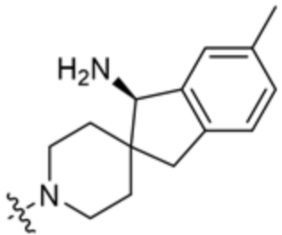
В некоторых вариантах осуществления R3 представляет собой 1-амино-1,3-дигидроспиро[инден-2,4’-пиперидин]-1’-ил.
[0097] В некоторых вариантах осуществления R3 выбран из группы, состоящей из:
, , и
[0098] В некоторых вариантах осуществления R4 и R5 независимо выбраны из водорода и метила. В некоторых вариантах осуществления R4 и R5 представляют собой водород.
[0099] В некоторых вариантах осуществления R6 выбран из группы, состоящей из водорода, метила, OH и CH2OH. В некоторых вариантах осуществления R6 выбран из водорода и метила. В некоторых вариантах осуществления R6 представляет собой метил. В некоторых вариантах осуществления R6 представляет собой водород.
[00100] В некоторых вариантах осуществления R6 выбран из группы, состоящей из водорода, метила, OH и CH2OH.
[00101] В некоторых вариантах осуществления X10 представляет собой CR9 или О. В некоторых вариантах осуществления X10 представляет собой CR9. В некоторых вариантах осуществления X10 представляет собой O.
[00102] В некоторых вариантах осуществления R6 выбран из группы, состоящей из водорода, метила, OH и CH2OH; и R9 представляет собой водород.
[00103] В некоторых вариантах осуществления X10 представляет собой CR9, иR6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил или 5-6-членный гетероарил, где гетероарил содержит 1 или 2 гетероатома, выбранных из азота, кислорода и серы, при этом арил и гетероарил необязательно замещены 1 или 2 группами, выбранными из группы, состоящей из галогена, циано, C1-C3 алкила и C1-C3 алкокси. В некоторых вариантах осуществления X10 представляет собой CR9, и R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил или 5-6-членный гетероарил с 1 или 2 гетероатомами, выбранными из азота и серы, при этом арил и гетероарил необязательно замещены 1 или 2 группами, выбранными из галогена, метила, метокси и циано. В некоторых вариантах осуществления X10 представляет собой CR9, и R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил или 5-6-членный гетероарил с 1 или 2 гетероатомами, выбранными из азота и серы, при этом арил необязательно замещен 1 или 2 группами, выбранными из галогена, метила, метокси и циано, и гетероарил необязательно замещен галогеном, метилом или метокси.
[00104] В некоторых вариантах осуществления X10 представляет собой CR9, и R6 и R9 вместе с атомами, к которым они присоединены, образуют 6-членный арил, так что R3 имеет структуру:
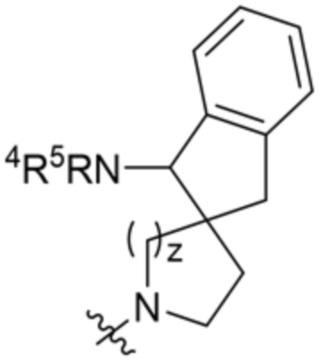 .
.
[00105] В некоторых вариантах осуществления R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо, так что R3 имеет структуру:
 или .
или .
[00106] В некоторых вариантах осуществления R7 и R8 представляют собой водород, или R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо. В некоторых вариантах осуществления R7 и R8 представляют собой водород. В некоторых вариантах осуществления R7 и R8 вместе с атомами, к которым они присоединены, образуют этильный мостик, так что R3 представляет собой азабициклическое кольцо, так что R3 имеет структуру:
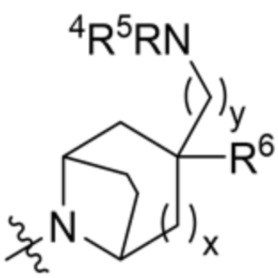 .
.
[00107] В некоторых вариантах осуществления R99 представляет собой водород или дейтерий. В предпочтительном варианте осуществления R99 представляет собой водород. В некоторых вариантах осуществления R99 представляет собой дейтерий.
[00108] В некоторых вариантах осуществления предложено соединение примеров 1-84. В некоторых вариантах осуществления предлагается соединение примеров 1, 2, 4-9, 11-14, 16-20, 22-27, 29-55, 58-71, 73, 74, 76, 77 и 79-84.В некоторых вариантах осуществления предлагается соединение примеров 1, 2, 5-8, 14, 17-19, 22-27, 29-35, 37-41, 44-48, 50-54, 58, 61, 62, 64-71, 73, 76, 77, 79, 81, 83 и 84. В некоторых вариантах осуществления предлагается соединение примеров 7, 17, 18, 30, 31, 32, 37, 38, 40, 45, 67, 69, 81 и 84. В некоторых вариантах осуществления предлагается соединение примеров 1, 5-8, 14, 16-20, 22-27, 29-39, 41, 44-55, 57-59, 61, 62, 64-67, 69-71, 73-77, 81, 83 и 84. В некоторых вариантах осуществления предлагается соединение примеров 1, 6-8, 14, 16-18, 22-25, 27, 30, 32, 34-39, 41, 44, 45, 47, 48, 50-55, 58, 61, 62, 65-67, 70, 73, 74, 76, 77, 81, 83 и 84. В некоторых вариантах осуществления предлагается соединение примеров 6, 17, 18, 22-25, 27, 30, 32, 34-36, 39, 41, 44, 45, 48, 50, 52-54, 58, 61, 62, 66, 70, 73, 74, 76, 81, 83 и 84.
[00109] В некоторых вариантах осуществления предлагается соединение примеров 1-14. В некоторых вариантах осуществления предлагается соединение примеров 1, 2, 4, 5, 6, 7, 8, 9, 11, 12, 13 и 14.
[00110] В некоторых вариантах осуществления предложено соединение формулы I. В некоторых вариантах осуществления изобретения предложено соединение формулы I при условии, что соединение не является примером 3, 10, 15, 21, 28, 56, 57, 72, 75 или 78. В некоторых вариантах осуществления изобретения предложено соединение формулы I при условии, что соединение не является примером 3, 4, 9-13, 15, 16, 20, 21, 28, 36, 42, 43, 49, 55-57, 60, 63, 72, 74, 75, 78, 80 или 82. В некоторых вариантах осуществления изобретения предложено соединение формулы I при условии, что соединение не является примером 1-6, 8-16, 19-29, 33-36, 39, 41-44, 46-66, 68, 70-80, 82 или 83. В некоторых вариантах осуществления изобретения предложено соединение формулы I при условии, что соединение не является примером 2-4, 9-13, 15, 21, 28, 40, 42, 43, 56, 60, 63, 68, 72, 78-80 или 82. В некоторых вариантах осуществления изобретения предложено соединение формулы I при условии, что соединение не является примером 2-5, 9-13, 15, 19-21, 26, 28, 29, 31, 33, 40, 42, 43, 46, 49, 56, 57, 59, 60, 63, 64, 68, 69, 71, 72, 75, 78-80 или 82. В некоторых вариантах осуществления изобретения предложено соединение формулы I при условии, что соединение не является примером 1-5, 7-16, 19-21, 26, 28, 29, 31, 33, 37, 38, 40, 42, 43, 46, 47, 49, 51, 55-57, 59, 60, 63-65, 67-69, 71, 72, 75, 77-80 или 82.
[00111] В некоторых вариантах осуществления предложено соединение формулы I. В некоторых вариантах осуществления изобретения предложено соединение формулы I при условии, что соединение не является примером 2, 3, 11, 12 или 13. В некоторых вариантах осуществления изобретения предложено соединение формулы I при условии, что соединение не является примером 2, 3, 4, 9, 10, 11, 12 или 13.
[00112] Соединение каждого примера или его фармацевтически приемлемая соль могут быть заявлены отдельно или сгруппированы вместе в любой комбинации с любым числом всех без исключения описанных здесь вариантов осуществления.
[00113] Следует иметь в виду, что некоторые соединения, описанные в данном документе, могут содержать асимметричные или хиральные центры и, следовательно, существовать в различных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений, описанных в данном документе, включая, без ограничения, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, включая рацемические смеси, образуют часть настоящих соединений.
[00114] В структурах, показанных в данном документе, где стереохимия какого-либо конкретного хирального атома не указана, все стереоизомеры рассматриваются и включаются в качестве описанных здесь соединений. В случае, когда стереохимия показана «жирным клином» или пунктирной линией, представляющими конкретную конфигурацию, в этом случае стереоизомер таким образом указан и определен.
[00115] Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченные соединения формулы I, в которых один или более атомов замещены атомами, имеющими тот же самый атомный номер, но атомную массу или массовое число, отличающиеся от атомной массы или массового числа, которое преобладает в природе.
[00116] Примеры изотопов, подходящих для включения в соединения данного изобретения, включают изотопы водорода, такие как 2Н и 3Н, углерода, такие как 11С, 13С и 14С, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32Р, и серы, такие как 35S.
[00117] Некоторые изотопно-меченные соединения формулы I, например, соединения, которые включают радиоактивный изотоп, можно использовать при исследованиях распределения лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы трития, т.е. 3Н, и углерод-14, т.е. 14C, особенно подходят для этой цели из-за простоты их включения и легкости способов обнаружения.
[00118] Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2Н, может дать определенные терапевтические преимущества в результате большей метаболической стабильности, например, увеличенного периода полураспада in vivo или снижение требуемых дозировок.
[00119] Изотопно-меченные соединения формулы I обычно могут быть получены традиционными методами, известными специалистам в данной области, или способами, аналогичными описанным в прилагаемых примерах и способах получения с использованием подходящего изотопно-меченного реагента вместо применявшегося ранее немеченного реагента.
[00120] Фармацевтически приемлемые сольваты в соответствии с изобретением включают сольваты, в которых растворитель кристаллизации может быть изотопно-замещенным, например, D2O, d6-ацетон (или (CD3)2CO), d6-ДМСО (или (CD3)2SO).
[00121] Также следует понимать, что некоторые соединения формулы I могут быть использованы в качестве промежуточных соединений для получения дополнительных соединений формулы I.
[00122] Кроме того, следует понимать, что описанные здесь соединения могут существовать в несольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное, и предполагается, что данные соединения охватывают как сольватированные, так и несольватированные формы.
[00123] Каждый из вариантов осуществления соединений настоящего изобретения, описанных здесь, может быть объединен с одним или более другими вариантами осуществления соединений настоящего изобретения, описанными здесь, не противоречащими варианту (вариантам) осуществления, с которым он (они) объединен (объединены).
[00124] СИНТЕЗ СОЕДИНЕНИЙ
[00125] Соединения, описанные в данном изобретении, могут быть синтезированы с использованием методов синтеза, которые включают процессы, аналогичные тем, которые хорошо известны в области химии, в частности, в свете приведенного в данном изобретении описания. Как правило, исходные материалы доступны из коммерческих источников, таких как Sigma-Aldrich (St. Louis, MO), Alfa Aesar (Ward Hill, MA), или TCI (Portland, OR), или их легко получить, используя способы, хорошо известные специалистам, (например, получить способами, в целом описанными в Louis F. Fieser, Mary Fieser, Reagents for Organic Synthesis. v. 1-23, New York: Wiley 1967-2006 ed. (также доступны через веб-сайт Wiley InterScience®), или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая приложения (также доступны через онлайн-базу данных Бейльштейна)).
[00126] В иллюстративных целях схемы 1 и 2 показывают общий способ получения соединений, описанных в данном документе, а также ключевые промежуточные соединения. Более подробное описание отдельных стадий реакции см. в разделе «Примеры» ниже. Специалистам в данной области будет ясно, что и другие пути синтеза могут использоваться для синтеза соединений. Хотя конкретные исходные материалы и реагенты представлены на схемах и обсуждаются ниже, их можно легко заменять другими исходными материалами и реагентами для получения различных производных и/или условий реакции. Кроме того, многие соединения, полученные способами, описанными ниже, можно далее модифицировать в свете данного описания с использованием общепринятых химических реакций, хорошо известных специалистам в данной области.
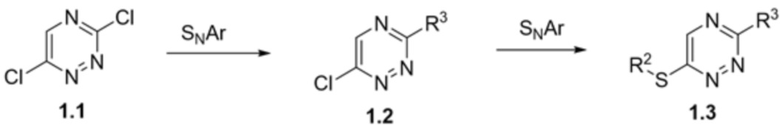
Схема 1
[00127] На схеме 1 представлена общая схема синтеза соединения 1.3. 3,6-Дихлор-1,2,4-триазин 1.1 может быть подвергнут реакции SNAr для получения триазина 1.2, где R3 является таким, как определено в данном документе. Триазин 1.2 может быть подвергнут реакции SNAr для получения триазина 1.3, где R3 является таким, как определено в данном документе.
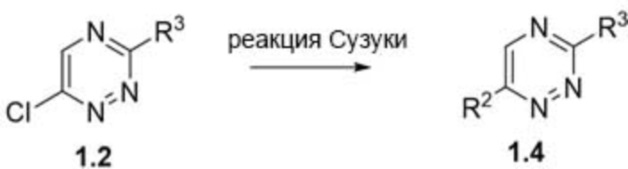
Схема 2
[00128] На схеме 1 представлена общая схема синтеза соединения 1.4. Триазин 1.2 может быть подвергнут реакции Сузуки для получения триазина 1.4, где R2 и R3 являются такими, как определено в данном документе.
[00129] СПОСОБЫ РАЗДЕЛЕНИЯ
[00130] Может быть желательно отделять продукты реакции друг от друга и/или от исходных материалов. Желаемые продукты каждой стадии или серии стадий разделяют и/или очищают (в дальнейшем, в настоящем документе, разделяют) до желаемой степени гомогенности способами, известными в данной области. Как правило, подобное разделение включает в себя многофазное экстрагирование, кристаллизацию из раствора или смеси растворов, дистилляцию, сублимацию или хроматографию. Хроматография может включать любое количество способов, включая, например: хроматографию с обращенной фазой и нормальной фазой; с исключением по размеру; ионообменную хроматографию; способы и оборудование для жидкостной хроматографии высокого, среднего и низкого давления; аналитическую хроматографию небольших количеств; с псевдодвижущимся слоем («SMB») и препаративную тонкослойную или толстослойную хроматографию, а также методики тонкослойной хроматографии небольших количеств и флэш-хроматографии. Специалист в данной области будет применять методики, наиболее подходящие для достижения требуемого разделения.
[00131] Диастереомерные смеси можно разделять на их индивидуальные диастереомеры на основании их физико-химических различий способами, хорошо известными специалистам в данной области, такими как хроматография и/или фракционная кристаллизация. Энантиомеры можно разделять путем преобразования энантиомерной смеси в диастереомерную смесь с помощью реакции с подходящим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорид кислоты Мошера), разделения диастереомеров и преобразования (например, гидролиза) индивидуальных диастереоизомеров в соответствующие чистые энантиомеры. Энантиомеры также могут быть разделены с помощью колонки хиральной ВЭЖХ.
[00132] Отдельный стереоизомер, например, энантиомер, по существу свободный от своего стереоизомера, может быть получен путем разделения рацемической смеси с использованием такого способа, как образование диастереомеров при использовании оптически активных разделяющих агентов (Eliel, E. and Wilen, S. Stereochemistry of Organic Compounds. New York: John Wiley & Sons, Inc., 1994; Lochmuller, C. H., et al. “Chromatographic resolution of enantiomers: Selective review.” J. Chromatogr. Vol.113, No. 3 (1975): pp. 283-302). Рацемические смеси хиральных соединений, описанные в данном документе, можно разделить и выделить любым подходящим способом, включая: (1) образование ионных, диастереомерных солей с хиральными соединениями и разделение фракционной кристаллизацией или другими способами, (2) образование диастереомерных соединений с хиральными дериватизирующими реагентами, разделение диастереомеров и превращение в чистые стереоизомеры, и (3) разделение по существу чистых или обогащенных стереоизомеров напрямую в хиральных условиях. См.: Wainer, Irving W., ed. Drug Stereochemistry: Analytical Methods and Pharmacology. New York: Marcel Dekker, Inc., 1993.
[00133] По способу (1) диастереомерные соли можно получить реакцией энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, α-метил-β-фенилэтиламин (амфетамин) и тому подобное, с асимметрическими соединениями, имеющими кислотную функциональную группу, такими как карбоновая кислота и сульфоновая кислота. Диастереомерные соли могут быть разделены при помощи фракционной кристаллизации или ионной хроматографии. Для разделения оптических изомеров аминосоединений добавление хиральной карбоновой или сульфоновой кислот, таких как камфорсульфоновая кислота, винная кислота, миндальная кислота или молочная кислота, может приводить к образованию диастереомерных солей.
[00134] В качестве альтернативы, по способу (2), подвергаемый разделению субстрат вводят в реакцию с одним энантиомером хирального соединения с целью образования пары диастереомеров (Eliel, E. and Wilen, S. Stereochemistry of Organic Compounds. New York: John Wiley & Sons, Inc., 1994, p. 322). Диастереомерные соединения могут быть образованы путем взаимодействия асимметричных соединений с энантиомерно чистыми хиральными дериватизирующими реагентами, такими как ментиловые дериваты, с последующим разделением диастереомеров и гидролизом с получением чистого или обогащенного энантиомера. Способ определения оптической чистоты включает получение хиральных сложных эфиров, таких как ментиловый сложный эфир, например, (-)ментилхлорформиат в присутствии основания, или сложный эфир Мошера, α-метокси-α-(трифторметил)фенилацетат (Jacob III, Peyton. «Resolution of (±)-5-Bromonornicotine. Synthesis of (R)- and (S)-Nornicotine of High Enantiomeric Purity» J. Org. Chem. Vol. 47, No. 21 (1982): pp. 4165-4167), из рацемической смеси и анализ 1H-ЯМР спектра на присутствие двух атропизомерных энантиомеров или диастереомеров. Стабильные диастереомеры атропизомерных соединений можно разделить и выделить с использованием хроматографии с нормальной или обращенной фазой, следуя способам разделения атропизомерных нафтилизохинолинов (WO 96/15111).
[00135] По способу (3) рацемическую смесь двух энантиомеров можно разделить хроматографией с применением хиральной неподвижной фазы (Lough, W.J., ed. Chiral Liquid Chromatography. New York: Chapman and Hall, 1989; Okamoto, Yoshio, et al. «Optical resolution of dihydropyridine enantiomers by high-performance liquid chromatography using phenylcarbamates of polysaccharides as a chiral stationary phase». J. of Chromatogr. Vol. 513 (1990): pp. 375-378). Обогащенные или очищенные энантиомеры можно различать способами, используемыми для различения других хиральных молекул с асимметричными атомами углерода, такими как оптическое вращение и круговой дихроизм.
[00136] БИОЛОГИЧЕСКАЯ ОЦЕНКА
[00137] Соединения изобретения представляют собой ингибиторы SHP2. В частности, они проявляют аффинность к SHP2.
[00138] Определение активности соединения формулы I в отношении активности SHP2 возможно с помощью ряда прямых и косвенных способов определения. Некоторые приводимые в качестве примера соединения, описанные в настоящем документе, анализировали на предмет их ингибирования SHP2 (Биологический пример 1). Анализ на основе клеток (Биологический пример 2) использовали для определения влияния ингибиторов SHP2 на нисходящую передачу сигналов путем оценки фосфорилирования ERK1/2.
[00139] ВВЕДЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
[00140] Соединения, описанные в настоящем документе, могут вводиться любым удобным способом, подходящим для состояния, подлежащего лечению. Соединения по изобретению вводят любым подходящим путем в форме фармацевтической композиции, адаптированной к такому пути, и в дозе, эффективной для предполагаемого лечения. Подходящие пути включают пероральный, парентеральный (в том числе подкожный, внутримышечный, внутривенный, внутриартериальный, интрадермальный, интратекальный и эпидуральный), трансдермальный, ректальный, назальный, местный (в том числе трансбуккальный и сублингвальный), вагинальный, интраперитонеальный, внутрилегочный и интраназальный пути.
[00141] Соединения можно вводить в любой подходящей форме введения, например, в форме таблеток, порошков, капсул, растворов, дисперсий, суспензий, сиропов, спреев, суппозиториев, гелей, эмульсий, пластырей и т.д. Такие композиции могут содержать компоненты, обычные в фармацевтических препаратах, например, разбавители, носители, модификаторы рН, подсластители, наполнители и другие активные агенты. Если желательно парентеральное введение, композиции должны быть стерильны и находиться в форме раствора или суспензии, пригодной для инъекции или инфузии. Подходящие устройства для парентерального введения включают игольные (включая микроигольные) инъекторы, безыгольные инъекторы и инфузионное оборудование.
[00142] Как правило, соединение по изобретению вводится в количестве, эффективном для лечения состояния, как описано в настоящем документе. Соединения по изобретению можно вводить как соединение per se или, в качестве альтернативы, в виде фармацевтически приемлемой соли. Для целей введения и дозирования, соединение per se или его фармацевтически приемлемая соль будут просто называться соединениями по изобретению.
[00143] Типичную композицию готовят путем смешивания соединения по настоящему изобретению и носителя, разбавителя или эксципиента. Подходящие носители, разбавители и эксципиенты хорошо известны специалистам в данной области и подробно описаны, например, в Ansel, Howard C., et al., Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004; Gennaro, Alfonso R., et al. Remington: The Science and Practice of Pharmacy. Philadelphia: Lippincott, Williams & Wilkins, 2000; и Rowe, Raymond C. Handbook of Pharmaceutical Excipients. Chicago, Pharmaceutical Press, 2005. Композиции также могут включать один или более буферов, стабилизирующих агентов, поверхностно-активных веществ, увлажняющих агентов, смазывающих агентов, эмульгаторов, суспендирующих агентов, консервантов, антиоксидантов, матирующих агентов, скользящих веществ, технологических добавок, красителей, подсластителей, ароматизирующих агентов, вкусовых агентов, разбавителей и других известных добавок для получения хорошего внешнего вида лекарственного средства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или чтобы способствовать изготовлению фармацевтического продукта (т.е. лекарственного средства).
[00144] Один вариант осуществления включает фармацевтическую композицию, содержащую соединение формулы I, или его стереоизомер, таутомер или фармацевтически приемлемую соль. Еще один вариант осуществления предлагает фармацевтическую композицию, содержащую соединение формулы I, или его стереоизомер, таутомер или фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
[00145] В другом варианте осуществления изобретение предлагает фармацевтическую композицию, содержащую соединение формулы I или его фармацевтически приемлемую соль, как определено в любом из описанных здесь вариантов осуществления, в смеси с по меньшей мере одним фармацевтически приемлемым эксципиентом.
[00146] В другом варианте осуществления изобретение включает фармацевтические композиции. Такие фармацевтические композиции содержат соединение по изобретению, представленное фармацевтически приемлемым носителем. Другие фармакологически активные вещества также могут присутствовать. Используемый в настоящем документе термин «фармацевтически приемлемый носитель» включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства, средства для поддержания изотоничности и средства, задерживающие поглощение, и тому подобное, которые являются физиологически совместимыми. Примеры фармацевтически приемлемых носителей включают одно или более из воды, физиологического раствора, фосфатно-солевого буферного раствора, декстрозы, глицерина, этанола и тому подобного, а также их комбинации, и могут включать изотонические агенты, например, сахара, хлорид натрия или многоатомные спирты, такие как маннит или сорбит в композиции. Фармацевтически приемлемые вещества, такие как увлажняющие вещества или незначительные количества вспомогательных веществ, таких как увлажняющие или эмульгирующие вещества, консерванты или буферы, которые увеличивают срок годности или эффективность антитела или части антитела.
[00147] Композиции данного изобретения могут быть в различных формах. Они включают, например, жидкие, полутвердые и твердые лекарственные формы, такие как жидкие растворы (например, растворы для инъекций и инфузий), дисперсии или суспензии, таблетки, пилюли, порошки, липосомы и суппозитории. Форма зависит от предполагаемого способа введения и терапевтического применения.
[00148] Типичные композиции находятся в форме растворов для инъекций или инфузий, таких как композиции, аналогичные тем, которые используются для пассивной иммунизации человека антителами в целом. Одним из способов введения является парентеральный (например, внутривенный, подкожный, интраперитонеальный, внутримышечный). В другом варианте осуществления антитело вводят посредством внутривенной инфузии или инъекции. В еще одном варианте осуществления антитело вводят путем внутримышечной или подкожной инъекции.
[00149] Пероральное введение твердой дозированной формы может быть, например, представлено в виде дискретных единиц, таких как твердые или мягкие капсулы, пилюли, крахмальные капсулы, пастилки или таблетки, каждая из которых содержит заранее определенное количество по меньшей мере одного соединения по изобретению. В другом варианте осуществления пероральное введение может осуществляться в виде порошка или гранул. В другом варианте осуществления пероральная дозированная форма является сублингвальной, как, например, пастилка. В таких твердых лекарственных формах соединения формулы I обычно объединяют с одним или несколькими вспомогательными веществами. Такие капсулы или таблетки могут содержать лекарственную форму с контролируемым высвобождением. В случае капсул, таблеток и пилюль, лекарственные формы также могут содержать буферные вещества или могут быть получены с энтеросолюбильными покрытиями.
[00150] В другом варианте осуществления пероральное введение может осуществляться в жидкой дозированной форме. Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно используемые в данной области (например, воду). Такие композиции также могут содержать вспомогательные вещества, такие как увлажняющие, эмульгирующие, суспендирующие, вкусовые (например, подсластители) и/или ароматизирующие агенты.
[00151] В другом варианте осуществления изобретение включает парентеральную дозированную форму. «Парентеральное введение» включает, например, подкожные инъекции, внутривенные инъекции, интраперитонеальные инъекции, внутримышечные инъекции, интрастернальные инъекции и инфузию. Препараты для инъекций (т.е. вводимые посредством инъекций стерильные водные или масляные суспензии), можно приготовить известным способом, используя подходящие диспергирующие, увлажняющие и/или суспендирующие вещества.
[00152] В другом варианте осуществления изобретение включает дозированную форму для местного применения. «Местное применение» включает, например, трансдермальное введение, например, посредством трансдермальных пластырей или устройств для ионтофореза, внутриглазное введение или интраназальное или ингаляционное введение. Композиции для местного применения также включают, например, гели для местного применения, спреи, мази и кремы. Лекарственная форма для местного применения может включать соединение, которое усиливает абсорбцию или проникновение активного ингредиента через кожу или другие области воздействия. Когда соединения данного изобретения вводят с помощью трансдермального устройства, введение будет осуществляться с использованием пластыря в виде резервуара и пористой мембраны или в виде различных твердых матриц. Типичные лекарственные формы для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, кожные пластыри, капсулы, имплантаты, губки, волокна, бандажи и микроэмульсии. Также можно использовать липосомы. Типичные носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены усилители проникновения, см., например, Finnin, Barrie C. and Timothy M. Morgan. «Transdermal penetration enhancers: Applications, limitations, and potential». J. Pharm. Sci. Vol. 88, No. 10 (1999): pp. 955-958.
[00153] Лекарственные формы, подходящие для местного введения в глаза, включают, например, глазные капли, при этом соединение настоящего изобретения растворено или суспендировано в подходящем носителе. Типичная лекарственная форма, подходящая для глазного или ушного введения, может быть в виде капель микронизированной суспензии или раствора в изотоническом стерильном физиологическом растворе с отрегулированным рН. Другие лекарственные формы, подходящие для глазного и ушного введения, включают мази, биоразлагаемые (т.е. абсорбируемые гелевые губки, коллаген) и небиоразлагаемые (т.е. силикон) имплантаты, капсулы, линзы и системы микрочастиц или везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например, геллановая камедь, может быть включен вместе с консервантом, таким как бензалконийхлорид. Такие лекарственные формы также могут доставляться ионтофорезом.
[00154] Для интраназального введения или введения посредством ингаляции соединения данного изобретения удобным образом доставляют в форме раствора или суспензии из контейнера-пульверизатора, который сжимает или накачивает пациент, или в форме аэрозольного спрея из находящегося под давлением контейнера или небулайзера, с использованием подходящего пропеллента. Препараты, подходящие для интраназального введения, обычно вводят в форме сухого порошка (или отдельно, или в виде смеси, например в сухой смеси с лактозой, или в виде частиц из смеси компонентов, например, смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора сухого порошка или в виде аэрозольного спрея из контейнера под давлением, насоса, спрея, распылителя (предпочтительно распылителя с использованием электродинамики для получения мелкодисперсного тумана), или небулайзера, с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Для интраназального применения порошок может содержать биоадгезивный агент, например, хитозан или циклодекстрин.
[00155] В другом варианте осуществления изобретение включает дозированную форму для ректального введения. Такая дозированная форма для ректального введения может быть в виде, например, суппозитория. Масло какао является традиционной основой суппозиториев, но в соответствующих случаях можно использовать различные альтернативы.
[00156] Также могут использоваться и другие материалы носителя и способы введения, известные в области фармацевтики. Фармацевтические композиции по изобретению могут быть получены с помощью любой из хорошо известных фармацевтических методик, таких как эффективные способы получения и введения. Приведенные выше соображения относительно эффективных способов получения и введения хорошо известны в данной области и описаны в стандартных учебниках.
[00157] Режим дозирования соединений по изобретению и/или композиций, содержащих указанные соединения, основан на ряде факторов, включающих тип, возраст, вес, пол и медицинское состояние пациента; тяжесть состояния; способ введения; и активность конкретного применяемого соединения. Таким образом, режим дозирования может варьировать в широких пределах. В одном варианте осуществления общая суточная доза соединения по изобретению обычно составляет от примерно 0,01 мг/кг до примерно 100 мг/кг (т.е. мг соединения по изобретению на кг массы тела) для лечения указанных в настоящем описании состояний. В другом варианте осуществления общая суточная доза соединения по изобретению составляет от примерно 0,1 мг/кг до примерно 50 мг/кг, и в другом варианте осуществления от примерно 0,5 мг/кг до примерно 30 мг/кг. Для взрослого весом 70 кг общая суточная доза может составлять 0,1 мг - 2 г; 1 мг - 500 мг и т.д. Нередко введение соединений по изобретению повторяют несколько раз в день (обычно не более 4 раз). Для увеличения общей суточной дозы, если требуется, можно использовать многократные суточные дозы.
[00158] Для перорального введения композиции могут быть представлены в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 75,0, 100, 125, 150, 175, 200, 250 и 500 мг активного ингредиента, для симптоматического регулирования дозирования для пациента. Лекарственное средство обычно содержит от примерно 0,01 мг до примерно 500 мг активного ингредиента или, в другом варианте осуществления, от примерно 1 мг до примерно 100 мг активного ингредиента. Для внутривенного введения дозы могут находиться в диапазоне от примерно 0,01 до примерно 10 мг/кг/мин при постоянной скорости инфузии.
[00159] Подходящие субъекты в соответствии с изобретением включают в себя субъекты млекопитающих. Млекопитающие согласно изобретению включают псовых, кошачьих, бычьих, козьих, лошадиных, овечьих, свиней, грызунов, зайцеобразных, приматов и тому подобное, и включают внутриутробных млекопитающих. В одном варианте осуществления подходящими субъектами являются люди. Субъекты-люди могут быть любого пола и на любой стадии развития.
[00160] СПОСОБЫ ЛЕЧЕНИЯ СОЕДИНЕНИЯМИ ИЗОБРЕТЕНИЯ
[00161] Соединения настоящего изобретения могут применяться при лечении широкого спектра заболеваний, нарушений или состояний, включая рак. Другие состояния, которые можно лечить соединениями настоящего изобретения, включают гиперпролиферативные заболевания, воспалительные нарушения или боль.
[00162] Также предложены способы лечения или профилактики заболевания или состояния путем введения одного или более соединений, описанных в данном документе, или его стереоизомера, таутомера или фармацевтически приемлемой соли. В одном варианте осуществления предлагается способ лечения гиперпролиферативного заболевания у млекопитающего, включающий введение терапевтически эффективного количества соединения формулы I или его стереоизомера, таутомера или фармацевтически приемлемой соли млекопитающему.
[00163] Другой вариант осуществления предусматривает способ лечения или профилактики рака у млекопитающего, нуждающегося в таком лечении, где способ включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли.
[00164] Изобретение также относится к фармацевтической композиции, содержащей соединение формулы I, или его фармацевтически приемлемую соль, как определено в любом из описанных здесь вариантов осуществления, для применения в лечении рака.
[00165] Другой вариант осуществления предусматривает способ лечения или профилактики боли у млекопитающего, нуждающегося в таком лечении, где способ включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли.
[00166] Другой вариант осуществления предусматривает способ лечения или профилактики воспалительного нарушения у млекопитающего, нуждающегося в таком лечении, где способ включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли.
[00167] Другой вариант осуществления предусматривает способ ингибирования активности протеинтирозинфосфатазы SHP2 в клетке, включающий обработку клетки соединением в соответствии с формулой I, или его стереоизомером, таутомером или фармацевтически приемлемой солью.
[00168] Другой вариант осуществления предусматривает способ ингибирования активности протеинтирозинфосфатазы SHP2 в клетке, включающий обработку клетки соединением в соответствии с формулой I, или его стереоизомером, таутомером или фармацевтически приемлемой солью, в количестве, эффективном для ослабления или устранения активности киназы SHP2.
[00169] Другой вариант осуществления предусматривает способ ингибирования активности протеинтирозинфосфатазы SHP2 у нуждающегося в этом пациента, включающий стадию введения указанному пациенту соединения в соответствии с формулой I, или его стереоизомера, таутомера или фармацевтически приемлемой соли.
[00170] В другом варианте осуществления изобретение предлагает способ ингибирования активности протеинтирозинфосфатазы SHP2 для лечения рака.
[00171] Другой вариант осуществления предусматривает способ лечения или уменьшения тяжести гиперпролиферативного нарушения у нуждающегося в этом пациента, включающий введение указанному пациенту соединения в соответствии с формулой I, или его стереоизомера, таутомера или фармацевтически приемлемой соли.
[00172] Другой вариант осуществления предусматривает способ лечения или уменьшения тяжести гиперпролиферативного нарушения у нуждающегося в этом пациента, включающий совместное введение указанному пациенту соединения в соответствии с формулой I, или его стереоизомера, таутомера или фармацевтически приемлемой соли, с по меньшей мере одним другим химиотерапевтическим агентом, используемым для лечения или облегчения гиперпролиферативного нарушения.
[00173] Другой вариант осуществления предусматривает способ лечения или уменьшения тяжести боли у нуждающегося в этом пациента, включающий введение указанному пациенту соединения в соответствии с формулой I, или его стереоизомера, таутомера или фармацевтически приемлемой соли.
[00174] Другой вариант осуществления предусматривает способ лечения или уменьшения тяжести воспалительного нарушения у нуждающегося в этом пациента, включающий введение указанному пациенту соединения в соответствии с формулой I, или его стереоизомера, таутомера или фармацевтически приемлемой соли.
[00175] В другом варианте осуществления предлагается способ лечения или профилактики заболевания или нарушения, модулируемого SHP2, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I, или его стереоизомера, таутомера или фармацевтически приемлемой соли. Примеры таких заболеваний и нарушений включают, без ограничения, гиперпролиферативные заболевания, такие как рак, и боль или воспалительные заболевания.
[00176] Другой вариант осуществления предусматривает использование соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли в производстве лекарственного средства для лечения гиперпролиферативного заболевания. Другой вариант осуществления предусматривает использование соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли в производстве лекарственного средства для лечения рака.
[00177] Другой вариант осуществления предусматривает использование соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли в производстве лекарственного средства для лечения боли.
[00178] Другой вариант осуществления предусматривает использование соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли в производстве лекарственного средства для лечения воспалительного заболевания.
[00179] Другой вариант осуществления предусматривает применение соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли для использования в лечении гиперпролиферативных заболеваний. Другой вариант осуществления предусматривает применение соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли для использования в лечении рака.
[00180] Другой вариант осуществления предусматривает применение соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли для использования в лечении боли.
[00181] Другой вариант осуществления предусматривает применение соединения формулы I, его стереоизомера, таутомера или фармацевтически приемлемой соли для использования в лечении воспалительных заболеваний.
[00182] Изобретение также включает в себя следующие варианты осуществления:
соединение формулы I или его фармацевтически приемлемая соль, как определено в любом из описанных здесь вариантов осуществления, для применения в качестве лекарственного средства;
соединение формулы I или его фармацевтически приемлемая соль, как определено в любом из описанных здесь вариантов осуществления, для применения в лечении рака;
способ лечения заболевания, для которого показан ингибитор SHP2, у субъекта, нуждающегося в таком лечении, включающий введение субъекту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, как определено в любом из описанных здесь вариантов осуществления;
применение соединения формулы I или его фармацевтически приемлемой соли, как определено в любом из описанных здесь вариантов осуществления, для производства лекарственного средства для лечения заболевания или состояния, для которых показан ингибитор SHP2;
соединение формулы I или его фармацевтически приемлемая соль, как определено в любом из описанных здесь вариантов осуществления, для применения при лечении заболевания или состояния, для которых показан ингибитор SHP2; или
фармацевтическая композиция для лечения заболевания или состояния, для которого показан ингибитор SHP2, содержащая соединение формулы I или его фармацевтически приемлемую соль, как определено в любом из описанных здесь вариантов осуществления.
[00183] В некоторых вариантах осуществления гиперпролиферативное заболевание представляет собой рак. В некоторых вариантах осуществления рак может быть выбран из следующего: меланома, ювенильные миеломоноцитарные лейкозы, нейробластома, положительный по филадельфийской хромосоме хронический миелолейкоз, положительные по филадельфийской хромосоме острые лимфобластные лейкозы, острые миелоидные лейкозы, миелопролиферативные новообразования (такие как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз), рак молочной железы, рак легких, рак печени, колоректальный рак, рак пищевода, рак желудка, плоскоклеточная карцинома головы и шеи, глиобластома, анапластическая крупноклеточная лимфома, карцинома щитовидной железы и шпицоидные новообразования. В некоторых вариантах осуществления рак представляет собой меланому. В некоторых вариантах осуществления рак представляет собой ювенильные миеломоноцитарные лейкозы. В некоторых вариантах осуществления рак представляет собой нейробластому. В некоторых вариантах осуществления рак представляет собой положительный по филадельфийской хромосоме хронический миелолейкоз. В некоторых вариантах осуществления рак представляет собой положительные по филадельфийской хромосоме острые лимфобластные лейкозы. В некоторых вариантах осуществления рак представляет собой острые миелоидные лейкозы. В некоторых вариантах осуществления рак представляет собой миелопролиферативные новообразования, такие как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз. В некоторых вариантах осуществления рак выбран из группы, состоящей из истинной полицитемии, эссенциальной тромбоцитемии и первичного миелофиброза. В некоторых вариантах осуществления рак представляет собой истинную полицитемию. В некоторых вариантах осуществления рак представляет собой эссенциальную тромбоцитемию. В некоторых вариантах осуществления рак представляет собой первичный миелофиброз. В некоторых вариантах осуществления рак представляет собой рак молочной железы. В некоторых вариантах осуществления рак представляет собой рак легкого. В некоторых вариантах осуществления рак представляет собой рак печени. В некоторых вариантах осуществления рак представляет собой колоректальный рак. В некоторых вариантах осуществления рак представляет собой рак пищевода. В некоторых вариантах осуществления рак представляет собой рак желудка. В некоторых вариантах осуществления рак представляет собой плоскоклеточную карциному головы и шеи. В некоторых вариантах осуществления рак представляет собой глиобластому. В некоторых вариантах осуществления рак представляет собой анапластическую крупноклеточную лимфому. В некоторых вариантах осуществления рак представляет собой карциному щитовидной железы. В некоторых вариантах осуществления рак представляет собой шпицоидные новообразования. В некоторых вариантах осуществления рак выбран из группы, состоящей из NSCLC, рака толстой кишки, рака пищевода, рака прямой кишки, ювенильного миеломоноцитарного лейкоза («JMML»), рака молочной железы, меланомы и рака поджелудочной железы.
[00184] В некоторых вариантах осуществления заболевание или нарушение может быть выбрано из нейрофиброматоза и синдрома Нунан. В некоторых вариантах осуществления заболевание или нарушение представляет собой нейрофиброматоз. В некоторых вариантах осуществления заболевание или нарушение представляет собой синдром Нунан.
[00185] В некоторых вариантах осуществления заболевание или нарушение представляет собой шванноматоз.
[00186] В некоторых вариантах осуществления заболевание или нарушение включает клетку, содержащую мутацию, кодирующую KRASG12C вариант. См. WO 2019/051084.
[00187] В некоторых вариантах осуществления гиперпролиферативное заболевание представляет собой заболевание или нарушение, содержащее клетку с мутацией, кодирующей вариант потери функции NF1 («NF1LOF»). В некоторых вариантах осуществления мутация NF1 представляет собой мутацию с потерей функции. В некоторых вариантах осуществления заболевание или нарушение представляет собой опухоль, содержащую клетки с мутацией с потерей функции NF1. В некоторых вариантах осуществления опухоль представляет собой NSCLC или меланому. В некоторых вариантах осуществления заболевание выбрано из нейрофиброматоза I типа, нейрофиброматоза II типа, шванноматоза и синдрома Уотсона.
[00188] В некоторых вариантах осуществления заболевание или нарушение связано с мутацией пути RAS в клетке субъекта, которое делает клетку по меньшей мере частично зависимой от активации сигнального пути через SHP2. В некоторых вариантах осуществления мутация пути RAS представляет собой мутацию RAS, выбранную из мутации KRAS, мутации NRAS, мутации SOS, мутации BRAF класса III, мутации MEKl класса I, мутации MEKl класса II и мутации F1. В некоторых вариантах осуществления мутация KRAS выбрана из мутации KRASG12A, мутации KRASG12C, мутации KRASG12D, мутации KRASG12F, мутации KRASG12l, мутации KRASG12L, мутации KRASG12R, мутации KRASG12S, мутации KRASG12V и мутации KRASG12Y. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12A. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12C. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12D. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12F. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12l. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12L. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12R. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12S. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12V. В некоторых вариантах осуществления мутация KRAS представляет собой мутацию KRASG12Y. В некоторых вариантах осуществления мутация BRAF класса III выбрана из одной или более следующих аминокислотных замен в BRAF человека: D287H; P367R; V459L; G466V; G466E; G466A; S467L; G469E; N581S; N581I; D594N; D594G; D594A; D594H; F595L; G596D; G596R и A762E. В некоторых вариантах осуществления мутация MEKl класса I выбрана из одной или более следующих аминокислотных замен в MEKl человека: D67N; P124L; P124S и L177V. В некоторых вариантах осуществления мутация MEKl класса II выбрана из одной или более следующих аминокислотных замен в MEKl человека: AE51-Q58; AF53-Q58; E203K; L177M; C121S; F53L; K57E; Q56P и K57N.
[00189] КОМБИНИРОВАННАЯ ТЕРАПИЯ
[00190] Соединения, описанные в данном документе, и их стереоизомеры, таутомеры и фармацевтически приемлемые соли могут быть использованы отдельно или в комбинации с другими терапевтическими агентами для лечения. Соединения, описанные в данном документе, могут быть использованы в комбинации с одним или более дополнительными лекарственными средствами, например, антигиперпролиферативным (или противораковым) агентом, который работает посредством воздействия на другой целевой белок. Второе соединение фармацевтического комбинированного препарата или режима дозирования предпочтительно обладает комплементарными активностями по отношению к соединению, описанному в данном документе, так что они не оказывают негативного влияния друг на друга. Такие молекулы в подходящем случае присутствуют в комбинации в количествах, которые являются эффективными для предполагаемой цели. Соединения могут вводиться вместе в единой фармацевтической композиции или по отдельности, и при раздельном введении это может происходить одновременно или последовательно в любом порядке. Такое последовательное введение может быть близким по времени или удаленным по времени.
[00191] Выражения «сопутствующее введение», «совместное введение», «одновременное введение» и «вводится одновременно» означают, что соединения вводятся в комбинации.
[00192] В другом варианте осуществления изобретение относится к способам лечения, которые включают введение соединений настоящего изобретения в комбинации с одним или более другими фармацевтическими агентами, где один или более других фармацевтических агентов может быть выбран из рассматриваемых в данном описании агентов.
[00193] В некоторых вариантах осуществления соединение формулы I вводится в комбинации с ингибитором пути RAS. В некоторых вариантах осуществления ингибитор пути RAS представляет собой ингибитор MEK или ингибитор ERK. В некоторых вариантах осуществления ингибитор пути Ras выбирают из одного или более из траметиниба, биниметиниба, селуметиниба, кобиметиниба, LErafAON (NeoPharm), ISIS 5132; вемурафениба, пимасертиба, TAK733, R04987655 (CH4987655); CI-1040; PD-0325901; CH5126766; MAP855; AZD6244; рефаметиниба (RDEA 119/BAY 86-9766); GDC-0973/XL581; AZD8330 (ARRY-424704/ARRY-704); R05126766; ARS-853; LY3214996; BVD523; GSK1 120212; уликсертиниба и абемациклиба, включая фармацевтически приемлемые соли конкретно названных соединений и фармацевтически приемлемые сольваты указанных конкретно названных соединений и солей. В некоторых вариантах осуществления ингибитор пути Ras выбирают из одного или более из траметиниба, биниметиниба, селуметиниба и кобиметиниба, включая фармацевтически приемлемые соли конкретно названных соединений и фармацевтически приемлемые сольваты указанных конкретно названных соединений и солей. В некоторых вариантах осуществления ингибитор пути Ras представляет собой биниметиниб, включая фармацевтически приемлемые соли и фармацевтически приемлемые сольваты биниметиниба.
[00194] В некоторых вариантах осуществления ингибитор пути RAS представляет собой ингибитор B-Raf. В некоторых вариантах осуществления ингибитор B-Raf выбирают из одного или более из энкорафениба, вемурафениба и дабрафениба, включая фармацевтически приемлемые соли конкретно названных соединений и фармацевтически приемлемые сольваты указанных конкретно названных соединений и солей. В некоторых вариантах осуществления ингибитор пути RAS представляет собой ингибитор B-Raf. В некоторых вариантах осуществления ингибитор B-Raf представляет собой энкорафениб, включая фармацевтически приемлемые соли и фармацевтически приемлемые сольваты энкорафениба.
[00195] В некоторых вариантах осуществления соединение формулы I вводится в комбинации с ингибитором MEK и ингибитором B-Raf. В некоторых вариантах осуществления ингибитор MEK выбран из траметиниба, биниметиниба, селуметиниба и кобиметиниба, и ингибитор B-Raf выбран из энкорафениба, вемурафениба и дабрафениба, включая фармацевтически приемлемые соли конкретно названных соединений и фармацевтически приемлемые сольваты указанных конкретно названных соединений и солей. В некоторых вариантах осуществления ингибитор MEK представляет собой биниметиниб, и ингибитор B-Raf представляет собой энкорафениб, включая фармацевтически приемлемые соли конкретно названных соединений и фармацевтически приемлемые сольваты указанных конкретно названных соединений и солей.
[00196] В некоторых вариантах осуществления соединение формулы I вводится в комбинации с ингибитором пути RAS. В некоторых вариантах осуществления ингибитор пути RAS представляет собой ингибитор KRAS. В некоторых вариантах осуществления ингибитор KRAS выбран из группы, состоящей из BI 1701963 и BBP-454. В некоторых вариантах осуществления ингибитор пути RAS представляет собой ингибитор KRAS G12C. В некоторых вариантах осуществления ингибитор KRAS G12C выбран из группы, состоящей из MRTX849, AMG 510 и ARS1620.
[00197] В некоторых вариантах осуществления соединение формулы I вводится в комбинации с ингибитором ALK. В некоторых вариантах осуществления ингибитор ALK выбран из группы, состоящей из лорлатиниба, кризотиниба, церитиниба, алектиниба и бригатиниба, включая фармацевтически приемлемые соли конкретно названных соединений и фармацевтически приемлемые сольваты указанных конкретно названных соединений и солей. В некоторых вариантах осуществления, ингибитор ALK представляет собой лорлатиниб, включая фармацевтически приемлемые соли и фармацевтически приемлемые сольваты лорлатиниба.
[00198] Эти агенты и соединения по изобретению можно комбинировать с фармацевтически приемлемыми носителями, такими как физиологический раствор, раствор Рингера, раствор декстрозы и тому подобное. Конкретный режим дозирования, т.е. доза, время и кратность, будет зависеть от конкретного индивидуума и его истории болезни.
[00199] Приемлемые носители, эксципиенты или стабилизаторы являются нетоксичными для реципиентов в используемых дозах и концентрациях, и могут включать буферы, такие как фосфатный, цитратный и другие буферы органических кислот; соли, такие как хлорид натрия; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензилхлорид аммония; гексаметонийхлорид; бензалконийхлорид, бензэтонийхлорид; феноловый, бутиловый или бензиловый спирт; алкилпарабены, такие как метилпарабен или пропилпарабен; катехин; резорцин; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее чем из приблизительно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как ЭДТА; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы металлов (например, Zn-белковые комплексы); и/или неионогенные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (PEG).
[00200] Липосомы, содержащие эти агенты и/или соединения по изобретению, получают способами, известными в данной области, например, описанными в патентах US №№ 4485045 и 4544545. Липосомы с увеличенным временем циркуляции описаны в патенте US № 5013556. Особенно пригодные липосомы можно получить методом обращенно-фазового выпаривания со смесью липидов, содержащей фосфатидилхолин, холестерин и PEG-дериватизированный фосфатидилэтаноламин (PEG-PE). Липосомы экструдируют через фильтры с определенным размером пор для получения липосом желаемого диаметра.
[00201] Эти вещества и/или соединения данного изобретения также могут быть заключены в микрокапсулы, приготовленные, например, способами коацервации или полимеризации на границе фаз, например, микрокапсулы из гидроксиметилцеллюлозы или желатиновые микрокапсулы и микрокапсулы из поли(метилметакрилата), соответственно, в коллоидные системы доставки лекарственных веществ (например, липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие способы описаны в Remington: The Science and Practice of Pharmacy, выше.
[00202] Можно использовать препараты с замедленным высвобождением. Подходящие примеры препаратов с замедленным высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие антитело/соединение по изобретению, где матрицы находятся в форме профилированных изделий, например, пленок или микрокапсул. Примеры матриц для замедленного высвобождения включают сложные полиэфиры, гидрогели (например, поли-(2-гидроксиэтилметакрилат) или поливиниловый спирт)), полилактиды (патент US 3773919), сополимеры L-глутаминовой кислоты и 7-этил-L-глутамата, недеградируемый этиленвинилацетат, деградируемые сополимеры молочной кислоты и гликолевой кислоты, такие как используются в LUPRON DEPOT™ (инъецируемые микросферы, состоящие из сополимера молочной кислоты и гликолевой кислоты и ацетата леупролида), ацетат-изобутират сахарозы и поли-D-(-)-3-гидроксимасляную кислоту.
[00203] Лекарственные формы, используемые для внутривенного введения, должны быть стерильными. Это легко осуществляется, например, фильтрованием через мембраны для стерилизующего фильтрования. Соединения данного изобретения обычно помещают в емкость, имеющую стерильное входное отверстие, например, пакет для внутривенного раствора или флакон, имеющий пробку, прокалываемую иглой для подкожной инъекции.
[00204] Подходящие эмульсии могут быть получены с использованием коммерчески доступных жировых эмульсий, таких как Intralipid™, Liposyn™, Infonutrol™, Lipofundin™ и Lipiphysan™. Активный ингредиент может быть либо растворен в предварительно смешанной эмульсионной композиции, либо, в качестве альтернативы, его можно растворить в масле (например, соевом масле, сафлоровом масле, хлопковом масле, кунжутном масле, кукурузном масле или миндальном масле) и образованную эмульсию затем смешать с фосфолипидом (например, яичными фосфолипидами, соевыми фосфолипидами или соевым лецитином) и водой. Следует иметь в виду, что могут быть добавлены и другие ингредиенты, например, глицерин или глюкоза, для регулирования тоничности эмульсии. Подходящие эмульсии обычно содержат до 20% масла, например, 5-20%. Эмульсия жира может содержать капельки жира, имеющие размер 0,1-1,0 мкм, в частности 0,1-0,5 мкм, и иметь рН в диапазоне 5,5-8,0.
[00205] Эмульсионные композиции могут быть композициями, полученными путем смешивания соединения по изобретению с Intralipid™ или его компонентами (соевое масло, яичные фосфолипиды, глицерин и вода).
[00206] Композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях или их смесях, а также порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые эксципиенты, указанные выше. В некоторых вариантах осуществления композиции вводят пероральным или назальным респираторным путем для местного или системного действия. Композиции в предпочтительно стерильных фармацевтически приемлемых растворителях можно распылять при помощи газов. Распыляемые растворы могут вдыхаться непосредственно из распыляющего устройства, или распыляющее устройство может быть присоединено к надеваемой на лицо маске, палатке или аппарату с перемежающимся положительным давлением. Композиции в виде растворов, суспензий или порошков можно вводить предпочтительно перорально или назально из устройств, которые доставляют готовую лекарственную форму подходящим образом.
[00207] Другой аспект изобретения относится к наборам, содержащим соединение по изобретению или фармацевтические композиции, содержащие соединение по изобретению. Набор может включать, в дополнение к соединению по изобретению или его фармацевтической композиции, диагностические или терапевтические агенты. Набор может также включать инструкции по применению в диагностическом или терапевтическом способе. В некоторых вариантах осуществления набор включает соединение или его фармацевтическую композицию и диагностическое средство. В других вариантах осуществления набор включает соединение или его фармацевтическую композицию, и один или более терапевтических агентов, таких как ингибитор пути Ras.
[00208] В еще одном варианте осуществления изобретение включает наборы, которые подходят для использования при осуществлении способов лечения, описанных в настоящей заявке. В одном варианте осуществления набор содержит первую лекарственную форму, содержащую одно или более соединений по изобретению в количествах, достаточных для осуществления способов изобретения. В другом варианте осуществления набор включает одно или более соединений по изобретению в количествах, достаточных для осуществления способов изобретения, а также контейнер для дозирования.
ПРИМЕРЫ
[00209] Для иллюстративных целей включены следующие примеры. Однако, следует понимать, что эти примеры не ограничивают изобретение, и предназначены только для того, чтобы предложить способ практического осуществления изобретения. Специалисты в данной области поймут, что описанные химические реакции могут быть легко адаптированы для получения целого ряда других соединений, описанных в данном документе, и альтернативные способы получения соединений находятся в пределах объема данного изобретения. Например, синтез не приведенных в примерах соединений может быть успешно проведен в результате модификаций, очевидных специалистам в данной области, например, путем надлежащей защиты мешающих реакции групп, путем использования других подходящих реагентов, известных в области техники, отличных от описанных реагентов, и/или путем проведения общепринятых модификаций условий реакции. В качестве альтернативы, другие реакции, описанные в настоящем документе или известные в данной области техники, будут признаны пригодными для получения других соединений, описанных в настоящем документе.
[00210] В описанных ниже примерах, если не указано иное, все температуры приведены в градусах Цельсия. Реагенты были приобретены у коммерческих поставщиков, таких как Sigma-Aldrich, Alfa Aesar или TCI, и использовались без дополнительной очистки, если не указано иное.
[00211] Описанные ниже реакции обычно проводили под избыточным давлением азота или аргона, или с применением осушающей трубки (если не указано иное) в безводных растворителях, и реакционные колбы в типичном случае снабжались резиновыми септами для введения субстратов и реагентов при помощи шприца. Стеклянную посуду сушили в сушильном шкафу и/или нагреванием.
[00212] Колоночную хроматографию проводили на системе Biotage (производитель: Dyax Corporation), имеющей колонку с силикагелем или картридж на основе диоксида кремния SepPak (Waters) (если не указано иное). 1H ЯМР спектры регистрировали на приборе Varian, работающем при 400 МГц. 1H ЯМР спектры получали для растворов CDCl3, CD3OD, D2O, (CD3)2SO, (CD3)2CO, C6D6, CD3CN (приведены в м.д.) с использованием тетраметилсилана (0,00 м.д.) или остаточного растворителя (CDCl3: 7,26 м.д.; CD3OD: 3,31 м.д.; D2O: 4,79 м.д.; (CD3)2SO: 2,50 м.д.; (CD3)2CO: 2,05 м.д.; C6D6: 7,16 м.д.; CD3CN: 1,94 м.д.) в качестве контрольного стандарта. При указании мультиплетности пиков использовались следующие сокращения: с (синглет), д (дублет), т (триплет), кв (квартет), м (мультиплет), ушир. (уширенный), дд (дублет дублетов), дт (дублет триплетов). Константы взаимодействия, когда они приводятся, указываются в герцах (Гц).
Биологический пример 1
Ферментный анализ SHP2
[00213] Кинетический анализ интенсивности флуоресценции конфигурировали для полноразмерного SHP2, с контролем количества 6,8-дифтор-7-гидрокси-4-метилкумарина («DiFMU»), образующегося при гидролизе 6,8-дифтор-4-метилумбеллиферилфосфата («DiFMUP») с помощью SHP2. Смеси для анализа состояли из 25 мМ K+HEPES, pH 7,4, 0,01% Triton X-100, 1 мМ DTT, 50 мМ KCl, 100 мкг/мл бычьего γ-глобулина, 50 мкМ DiFMUP, 1 мкМ SHP2-активирующего пептида (LN(pY)IDLDLV(dPEG8)LST(pY)ASINFQK-амида), 1 нМ полноразмерного SHP2 (His6-меченого SHP2(2-527), рекомбинантно экспрессированного в E. coli и очищенного в местной лаборатории), и 2% диметилсульфоксида («ДМСО») (из соединения). Соединения обычно разбавляли в ДМСО по 10-точечному дозовому диапазону, созданному с использованием 3-кратного протокола последовательного разбавления при верхней дозе 20 мкМ. Анализ проводили в 384-луночных полистирольных малообъемных необработанных черных микротитрационных планшетах (Costar 4511) в конечном объеме 20 мкл. Малообъемные контрольные лунки были без фермента. Анализы инициировали добавлением смеси SHP2 и активирующего пептида, и после 15 с перемешивания на орбитальном шейкере считывали в кинетическом режиме в течение 15 мин (30 с/цикл) при температуре окружающей среды на микропланшетном ридере PerkinElmer EnVision (λEx=355 нм, λEm=460 нм). Начальные скорости (наклоны касательных при t=0) оценивали по экспоненциальным приближениям к слегка нелинейным кривым прохождения процесса, и затем преобразовывали в процент контроля («POC») с использованием следующего уравнения:
где:  Средние неингибированные контроли;
Средние неингибированные контроли;
 Средний фон.
Средний фон.
Четырехпараметрическую логистическую модель использовали для соответствия данным POC для каждого соединения. Исходя из этого соответствия, оценивали IC50 и определяли как концентрацию соединения, при которой кривая пересекает 50 POC.
[00214] В таблице 1 приведены репрезентативные данные для примеров, описанных в настоящем документе. Значение IC50, указанное в таблице 1, может быть получено в результате одного анализа или как среднее значение из нескольких анализов. Примеры 1-14 тестировали в указанном выше анализе и обнаружили, что IC50 составляет менее 10 мкМ. Примеры 1, 2, 4, 5, 6, 7, 8, 9, 11, 12, 13 и 14 тестировали в указанном выше анализе и обнаружили, что IC50 составляет менее 500 мкМ. Примеры 1-14 тестировали в указанном выше анализе и обнаружили, что IC50 составляет 1 мкМ или менее.
[00215] В таблице 1 приведены примеры, протестированные в приведенном выше анализе:
IC50 (нМ)
Биологический пример 2
Клеточный анализ фосфо-p44/42 MAPK (Erk1/2) (Thr202/Tyr204)
[00216] Ингибирование фосфорилирования ERK1/2 (Thr202/Tyr204) определяли с помощью следующего клеточного анализа, который включает инкубацию клеток с соединением в течение 1 ч и количественную оценку сигнала pERK анализом In-Cell Western на фиксированных клетках и нормализацию сигнала GAPDH. Клетки KYSE520 получали из DSMZ и выращивали в RPMI с добавлением 10% фетальной бычьей сыворотки, пенициллина/стрептомицина, 2 мМ дипептида L-аланил-L-глутамина в 0,85% NaCl (Glutamax™), незаменимых аминокислот и пирувата натрия. Клетки высевали в 96-луночные планшеты при плотности 30000 клеток/лунку и давали возможность прикрепиться в течение ночи при 37°C и 5% CO2. Клетки обрабатывали соединениями, приготовленными в 10-точечной серии разбавлений, с последовательными разбавлениями 1:3 (диапазон: 20 мкМ - 1 нМ), с конечной концентрацией ДМСО 0,5%. После 1 ч инкубации клетки фиксировали в 3,7% формальдегиде в физиологическом растворе, забуференном фосфатом Дульбекко («dPBS»), при комнатной температуре в течение 20 мин. Затем клетки промывали dPBS и пермеабилизировали в 100% MeOH при комнатной температуре в течение 10 мин. После пермеабилизации клетки промывали в dPBS и инкубировали в блокирующем буфере LI-COR (LI-COR Biosciences, Cat#927-40000) в течение 1 ч или дольше. Затем планшеты инкубировали с антителом, специфичным для МЕК-зависимых сайтов фосфорилирования ERK1/2, треонина 202 и тирозина 204 (Cell Signaling Technologies; Cat # 9101), после SHP2 в пути МАР-киназной сигнальной трансдукции, а также GAPDH (Millipore; Cat# MAB374). Антитело pErk1/2 (Thr202/Tyr204) разбавляли в блокирующем буфере LI-COR, содержащем 0,05% полисорбата-20 (Tween-20), в соотношении 1:250; GAPDH разбавляли при 1:2500. Планшеты инкубировали в течение ночи при 4°C. После промывки PBS/0,05% Tween-20, клетки инкубировали флуоресцентно-меченными вторичными антителами (Anti-rabbit-Alexa Flour680, Invitrogen Cat#A21109; Anti-mouse-IRDye800CW, Li-cor Biosciences Cat#926-32210, оба в разбавлении 1:1000) в течение 1 ч. Затем клетки промывали, как указано выше, и анализировали на флуоресценцию на длинах волн 680 нм и 800 нм с использованием системы инфракрасной флуоресцентной визуализации Aerius (LI-COR Biosciences, модель 9250). Сигнал фосфорилированного Erk1/2 (Thr202/Tyr204) нормализовали по сигналу GAPDH для каждой лунки. Значения IC50 рассчитывали из нормализованных значений с использованием 4-параметрической модели соответствия в программном обеспечении BioAssay. В таблице 2 приведены репрезентативные данные для примеров, описанных в настоящем документе. Значение IC50, указанное в таблице 2, может быть получено в результате одного анализа или как среднее значение из нескольких анализов.
[00217] В таблице 2 приведены избранные примеры, протестированные в приведенном выше анализе:
IC50 (нМ)
Промежуточный пример А
2-амино-3-хлорпиридин-4-тиол
[00218] Стадия A: Сложный 2-этилгексиловый эфир 3-меркаптопропионовой кислоты (2,3 мл, 22 ммоль) и основание Хунига (6,9 мл, 39 ммоль) добавляли в смесь 3-хлор-4-иодпиридин-2-амина (5,0 г, 20 ммоль), Pd(OAc)2 (0,22 г, 0,98 ммоль) и ксантфоса (1,1 г, 2,0 ммоль) в диоксане (65 мл, 20 ммоль) в атмосфере газообразного Ar. Реакционную смесь нагревали до 100°С в атмосфере аргона в течение 18 ч. Реакционную смесь разбавляли этилацетатом (EtOAc) и фильтровали через диатомовый диоксид кремния (Celite®). Фильтрат концентрировали с получением метил-3-((2-амино-3-хлорпиридин-4-ил)тио)пропанoата (4,3 г, 17 ммоль, выход 88%).
[00219] Стадия B: NaOEt (7,1 мл, 19 ммоль) добавляли к метил-3-((2-амино-3-хлорпиридин-4-ил)тио)пропанoату (4,3 г, 17 ммоль) в тетрагидрофуране («THF») (87 мл, 17 ммоль) и перемешивали в атмосфере N2 в течение 1 ч при комнатной температуре. Добавляли дихлорметан («DCM») (20 мл) и данную смесь перемешивали в течение 5 мин. Реакционную смесь концентрировали, и твердое вещество титровали DCM, фильтровали и сушили. Твердые вещества растворяли в воде (суспензия), и добавляли 1 н. HCl для доведения рН до примерно 6. Твердые вещества отфильтровывали и промывали водой с получением 2-амино-3-хлорпиридин-4-тиола (1,4 г, 9,0 ммоль, выход 52%). 1H ЯМР (400 МГц, (CD3)2SO) δ 11,4 (ушир., 1H), 7,06 (д, 1H, J= 6,8 Гц), 6,70 (ушир., 2H), 6,65 (д, 1H, J=7,0 Гц); m/z (esi/APCI) M+1=161,0.
Промежуточный пример B
3-хлор-2-(метиламино)пиридин-4-тиол
[00220] Стадия A: 3-Хлор-2-фтор-4-йодпиридин (8,0 г, 31,1 ммоль) и метанамин (42,7 мл, 85,5 ммоль) помещали в ДМСО (20 мл) и нагревали до 70°С в течение 30 мин. Добавляли воду, и твердые вещества отфильтровывали с получением неочищенного продукта. Материал очищали силикагелем (0-5% MeOH в DCM с 2% NH4OH) с получением 3-хлор-4-йод-N-метилпиридин-2-амина (6,34 г, 23,6 ммоль, выход 76%).
[00221] Стадия B: 3-Хлор-4-йод-N-метилпиридин-2-амин (1 г, 3,7 ммоль) разбавляли диоксаном (5 мл), с последующим добавлением PdOAc2 (41,8 мг, 0,19 ммоль), ксантфоса (215,5 мг, 0,37 ммоль) и метил-3-меркаптопропанoата (453,8 мкл, 4,10 ммоль). Реакционную смесь помещали в атмосферу азота и добавляли DIEA (1301 мкл, 7,50 ммоль). Реакционную смесь нагревали до 100°С и перемешивали в течение 2,3 ч. Реакционной смеси давали возможность остыть, разбавляли этилацетатом и фильтровали через Celite®, промывая этилацетатом. Фильтрат концентрировали с получением метил-3-((3-хлор-2-(метиламино)пиридин-4-ил)тио)пропанoата (905 мг, 3,471 ммоль, выход 93%).
[00222] Метил-3-((3-хлор-2-(метиламино)пиридин-4-ил)тио)пропанoат (905 мг, 3,47 ммоль) разбавляли THF (15 мл), после чего добавляли этоксид натрия (1425 мкл, 3,82 ммоль). Реакционную смесь помещали в атмосферу азота и перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрировали и растирали с DCM. Материал разбавляли водой и рН доводили до примерно 6. Смесь отфильтровывали, промывали водой и сушили под вакуумом с получением 3-хлор-2-(метиламино)пиридин-4-тиола (400 мг, 2,29 ммоль, выход 66,0%). m/z (esi/APCI) M+1=175,0.
Промежуточный пример C
5-хлор-1H-пирроло[2,3-b]пиридин-4-тиол
[00223] Стадия A: Сложный 2-этилгексиловый эфир 3-меркаптопропионовой кислоты (0,12 мл, 1,1 ммоль) и основание Хунига (0,34 мл, 1,9 ммоль) добавляли в смесь 5-хлор-4-йод-1H-пирроло[2,3-b]пиридина (0,270 г, 0,97 ммоль), Pd(OAc)2 (0,011 г, 0,048 ммоль) и ксантфоса (0,056 г, 0,097 ммоль) в диоксане (3,2 мл, 0,97 ммоль) в атмосфере газообразного Ar. Реакционную смесь нагревали до 100°С в атмосфере аргона в течение 18 ч. Реакционную смесь разбавляли EtOAc и фильтровали через диатомовый диоксид кремния (Celite®). Фильтрат концентрировали и затем очищали на силикагеле, элюируя 10-100% EtOAc/гексаны, с получением метил-3-((5-хлор-1H-пирроло[2,3-b]пиридин-4-ил)тио)пропанoата (0,225 г, 0,83 ммоль, выход 85%).
[00224] Стадия B: NaOEt (0,34 мл, 0,91 ммоль) добавляли в метил-3-((5-хлор-1H-пирроло[2,3-b]пиридин-4-ил)тио)пропанoат (0,225 г, 0,83 ммоль) в тетрагидрофуране («THF») (4,146 мл, 0,83 ммоль) и перемешивали в течение 2 ч при комнатной температуре. Добавляли DCM (20 мл), и данную смесь перемешивали в течение 5 мин. Реакционную смесь концентрировали и затем очищали на силикагеле, элюируя 0-20% MeOH/DCM (2% NH4OH), с получением 5-хлор-1H-пирроло[2,3-b]пиридин-4-тиола (0,080 г, 0,43 ммоль, выход 52%). m/z (esi/APCI) M+1=185,0.
Промежуточный пример D
(R)-N-((S)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид
[00225] Стадия A: Трет-бутил-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (25 г, 83 ммоль) и (R)-2-метилпропан-2-сульфинамид (30 г, 249 ммоль) и Ti(OEt)4 (122 мл, 580 ммоль) нагревали до 90°С в течение 18 ч. Добавляли EtOAc, после чего воду. Твердые вещества отфильтровывали, и слои разделяли. Органический слой сушили над MgSO4, фильтровали и концентрировали, и полученный остаток очищали на силикагеле (0-40% EtOAc в гексанах), с получением трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (16 г, 39 ммоль, выход 47%).
[00226] Стадия B: Трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (6,5 г, 16 ммоль) помещали в THF (10 мл) и охлаждали до -78°С. Добавляли борогидрид лития (14 мл, 28 ммоль) и реакционную смесь оставляли медленно нагреваться до комнатной температуры и перемешивали в течение 18 ч. Добавляли воду и смесь экстрагировали DCM (3×25 мл). Экстракты объединяли и концентрировали, и полученный остаток очищали на силикагеле (0-5% MeOH в DCM с 2% NH4OH) с получением трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (3,2 г, 8 ммоль, выход 50%).
[00227] Стадия C: Трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (3,1 г, 7,6 ммоль) помещали в DCM (50 мл) и охлаждали до 0°С. Добавляли трифторуксусную кислоту («TFA») (7 мл), и реакционную смесь перемешивали в течение 2,5 ч. Реакционную смесь концентрировали и образующийся в результате остаток растворяли в насыщенном бикарбонате. Смесь экстрагировали DCM. Экстракты сушили, фильтровали и концентрировали с получением (R)-N-((S)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (1,8 г, 5,9 ммоль, выход 78%).
Промежуточный пример E
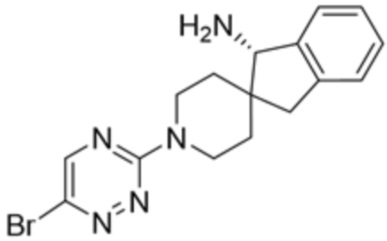
(S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00228] Дигидрохлорид (S)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (1,00 г, 3,63 ммоль) суспендировали в диоксане (15 мл), и к смеси добавляли триэтиламин (1,84 г, 18,2 ммоль). Смесь перемешивали при комнатной температуре в течение 20 мин, затем добавляли 3,6-дибром-1,2,4-триазин (0,868 г, 3,63 ммоль). Реакционную смесь нагревали до 50°С и перемешивали в течение 2 ч. Через 2 ч реакционную смесь фильтровали, и фильтрат выпаривали с получением остатка. Остаток очищали, используя колоночную хроматографию с 40 г силикагеля (смесь MeOH/DCM 2-20%) с получением (S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (1,19 г, 3,30 ммоль, выход 90,9%) в виде твердого вещества. m/z (esi/APCI) M+1 =400,2.
Промежуточный пример F
Трет-бутил-(R)-5-(((R)-трет-бутилсульфинил)амино)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат
[00229] Стадия A: 2-Хлорпиридин (458 мл, 4,84 ммоль) растворяли в сухом THF (15 мл), и раствор охлаждали до -70°C на бане со смесью сухой лед/IPA. Диизопропиламид лития («LDA») (2,75 мл, 5,50 ммоль) добавляли по каплям в смесь, и реакционную смесь нагревали до -60°С и перемешивали при этой температуре в течение 1,5 ч. Трет-бутил-4-формил-4-метилпиперидин-1-карбоксилат (1 г, 4,40 ммоль) в THF (3 мл) добавляли к смеси и перемешивали при -60°С в течение 1 ч. Реакционную смесь гасили водой и распределяли между EtOAc и водой. Органический слой отделяли, высушивали и концентрировали. Полученный остаток очищали, используя колонку с 40 г силикагеля (EtOAc/гексаны 10-80%), с получением трет-бутил-4-((2-хлорпиридин-3-ил)(гидрокси)метил)-4-метилпиперидин-1-карбоксилата (1,06 г, 3,11 ммоль, выход 71%).
[00230] Стадия B: Трет-бутил-4-((2-хлорпиридин-3-ил)(гидрокси)метил)-4-метилпиперидин-1-карбоксилат (1,06 г, 3,11 ммоль) растворяли в DCM (8 мл), и в раствор добавляли периодинан Десса-Мартина («DMP») (2,64 г, 6,22 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч, и ее гасили 10% раствором бисульфита натрия. Органический слой отделяли, промывали насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, сушили и выпаривали. Полученный остаток очищали, используя флэш-хроматографию на силикагеле (EtOAc/гексаны 10-100%), с получением трет-бутил-4-(2-хлорникотинoил)-4-метилпиперидин-1-карбоксилат (0,31 г, 0,92 ммоль, выход 29%) в виде масла.
[00231] Стадия C: Трет-бутил-4-(2-хлорникотинoил)-4-метилпиперидин-1-карбоксилат (8,13 г, 24,0 ммоль) растворяли в мезитилене (70 мл) в трубке высокого давления. Трициклогексилфосфоний тетрафторборат (0,884 г, 2,40 ммоль), диацетоксипaллaдий (0,269 г, 1,20 ммоль), пивалевую кислоту (0,735 г, 7,20 ммоль) и карбонат цезия (15,6 г, 48,0 ммоль) добавляли в реакционную смесь. Газообразный азот барботировали в течение 5 мин в реакционной смеси, затем трубку герметизировали и нагревали при 140°C в течение 72 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc (50 мл). Смесь фильтровали через слой Celite® и промывали EtOAc несколько раз. Фильтрат выпаривали под вакуумом с получением остатка. Остаток очищали, используя колонку с 330 г диоксида кремния (EtOAc/гексан 20-80%), с получением трет-бутил-5-оксо-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (2,23 г, 7,37 ммоль, выход 31%) в виде твердого вещества. m/z (esi/APCI) M+1 =303,1.
[00232] Стадия D: Трет-бутил-5-оксо-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат (2,23 г, 7,37 ммоль) суспендировали в тетраэтоксититане (6,98 мл, 51,62 ммоль), и в смесь добавляли (R)-2-метилпропан-2-сульфинамид (2,68 г, 22,12 ммоль). Реакционную смесь нагревали до 90°С и перемешивали в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли EtOAc (250 мл), а затем насыщенный солевой раствор (200 мл). Смесь энергично перемешивали в течение 10 мин и фильтровали для удаления осадка. Слой EtOAc отделяли и дважды промывали насыщенным солевым раствором, сушили и выпаривали. Образующийся в результате остаток очищали, используя колонку со 120 г силикагеля (EtOAc/гексаны 10-100%). Сбор второго элюируемого пика давал трет-бутил-(R, Z)-5-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат (2,8 г, 6,90 ммоль, выход 94%) в виде твердого вещества. m/z (esi/APCI) M+1 =406,2.
[00233] Стадия E: Трет-бутил-(R, Z)-5-((трет-бутилсульфинил)имино)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат (0,15 г, 0,37 ммоль) растворяли в THF (2 мл) во флаконе. Раствор охлаждали до -78°C, и в одну порцию добавляли LiBH4 (0,28 мл, 0,55 ммоль) 2 М в THF. Реакцию выдерживали при -78°C в течение 1 ч. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 18 ч. Реакцию гасили насыщенным NH4Cl с последующей экстракцией EtOAc (3 × 10 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили и выпаривали. Полученный остаток очищали с использованием колоночной хроматографии на колонке с 24 г диоксида кремния (EtOAc/гексаны 10-80%), с получением трет-бутил-(R)-5-(((R)-трет-бутилсульфинил)амино)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (30 мг, 0,27 ммоль, выход 24%) в виде твердого вещества в качестве второстепенного продукта, и другого диастереомера в качестве главного продукта - трет-бутил-(S)-5-(((R)-трет-бутилсульфинил)амино)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (51 мг, 0,27 ммоль, выход 35%). m/z (esi/APCI) M+1 =408,2.
Промежуточный пример G
2,3-диметилпиридин-4-тиол
[00234] 2,3-Диметилпиридин-4-тиол получали в соответствии с промежуточным примером A, заменив 3-хлор-4-йодпиридин-2-амин на 4-бром-2,3-диметилпиридин на стадии A. m/z (esi/APCI) M+1=140,1.
Промежуточный пример H
2-амино-5-хлорпиридин-4-тиол
[00235] 2-амино-5-хлорпиридин-4-тиол получали в соответствии с промежуточным примером A, заменив 3-хлор-4-йодпиридин-2-амин на 4-бром-5-хлорпиридин-2-амин на стадии A. m/z (esi/APCI) M+1=161,1.
Промежуточный пример I
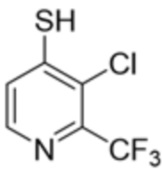
3-хлор-2-(трифторметил)пиридин-4-тиол
[00236] 3-хлор-2-(трифторметил)пиридин-4-тиол получали в соответствии с промежуточным примером A, заменив 3-хлор-4-йодпиридин-2-амин на 3-хлор-4-йод-2-(трифторметил)пиридин на стадии A. m/z (esi/APCI) M+1=214,0.
Промежуточный пример J
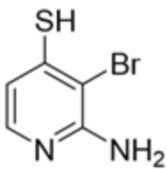
2-амино-3-бромпиридин-4-тиол
[00237] 2-амино-3-бромпиридин-4-тиол получали в соответствии с промежуточным примером A, заменив 3-хлор-4-йодпиридин-2-амин на 3-бром-4-йодпиридин-2-амин на стадии A. m/z (esi/APCI) M+1=205,0.
Промежуточный пример K
метил-3-((2-амино-3-фторпиридин-4-ил)тио)пропанoат
[00238] 2-Амино-3-фтор-4-йодпиридин (1,1 г, 4,4 ммоль) растворяли в 1,4-диоксане (10 мл, 4,4 ммоль). 4,5-Бис(дифенилфосфино)-9,9-диметилксантен (0,26 г, 0,45 ммоль) и метил-3-((2-амино-3-фторпиридин-4-ил)тио)пропанoат (0,95 г, 4,1 ммоль) добавляли в реакционный раствор. Реакционную смесь помещали в атмосферу азота. Добавляли N, N-диизопропилэтиламин (1,5 мл, 8,9 ммоль) и полученный раствор перемешивали при 100°С в течение ночи. Неочищенный материал загружали на колонку с 80 г силикагеля и очищали в градиенте 0-100% EtOAc:гексан, с получением метил-3-((2-амино-3-фторпиридин-4-ил)тио)пропанoата (0,95 г, выход 92%). m/z (esi/APCI) M+1=231,1.
Промежуточный пример L
метил-3-((6-амино-2-(трифторметил)пиридин-3-ил)тио)пропанoат
[00239] Метил-3-((6-амино-2-(трифторметил)пиридин-3-ил)тио)пропанoат получали в соответствии с промежуточным примером K, заменив 2-амино-3-фтор-4-йодпиридин на 5-бром-6-трифторметилпиридин-2-иламин на стадии A. m/z (esi/APCI) M+1=281.
Промежуточный пример M
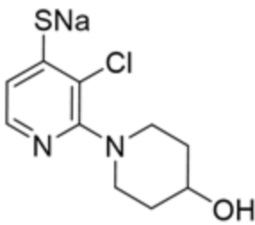
3-хлор-2-(4-гидроксипиперидин-1-ил)пиридин-4-тиолат натрия
[00240] Стадия A: Раствор 3-хлор-2-фтор-4-йодпиридина (2,0 г, 7,77 ммоль) и 4-гидроксипиперидина (1,18 г, 11,7 ммоль) в ДМСО (5 мл) нагревали до 75°С в течение 1 ч. Раствор охлаждали до комнатной температуры, разбавляли водой (10 мл) и распределяли с EtOAc/простым метил-трет-бутиловым эфиром («MTBE») (2/1). Твердое вещество, образованное между слоями, отфильтровывали и сушили под вакуумом с получением 1-(3-хлор-4-йодпиридин-2-ил)пиперидин-4-ола (1,08 г, 70%). m/z (esi) M+1=339,0.
[00241] Стадия B: В 20 мл флаконе 1-(3-хлор-4-йодпиридин-2-ил)пиперидин-4-ол (0,50 г, 1,48 ммоль) растворяли в диоксане (5 мл), и затем флакон барботировали азотом. Добавляли сложный метиловый эфир 3-меркаптопропионовой кислоты (0,195 г, 1,62 ммоль), после чего основание Хунига (0,514 мл, 2,95 ммоль). Через 5 мин добавляли Pd(OAc)2 (0,017 г, 0,074 ммоль), и затем ксантфос (0,086 г, 0,15 ммоль). Флакон закрывали крышкой, обрабатывали ультразвуком и нагревали до 105°C в течение 2,5 ч. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и фильтровали через слой Celite®. Слой промывали EtOAc, и раствор выпаривали досуха, получая в виде плотного твердого вещества, которое переносили на следующую стадию без дальнейшей очистки. m/z (esi) M+1=331,0.
[00242] Стадия C: Метил-3-((3-хлор-2-(4-гидроксипиперидин-1-ил)пиридин-4-ил)тио)пропанoат (0,58 г, 1,75 ммоль) растворяли в безводном диоксане (9 мл) и добавляли этанолат натрия (21% масс. в EtOH, 2,4 мл, 2,1 ммоль) при комнатной температуре в атмосфере азота. Смесь перемешивали в течение 3 ч, и растворитель выпаривали досуха. Полученное твердое вещество дважды растирали с MTBE (5 мл). Неочищенный материал сушили под вакуумом с получением неочищенного 3-хлор-2-(4-гидроксипиперидин-1-ил)пиридин-4-тиолата натрия (563 мг), который использовали непосредственно без дальнейшей очистки. m/z (esi) M+1=245,0-247,0.
Промежуточный пример N
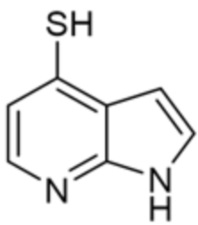
1H-пирроло[2,3-b]пиридин-4-тиол
[00243] Стадия A: Сложный 2-этилгексиловый эфир 3-меркаптопропионовой кислоты (0,35 мл, 3,27 ммоль) и основание Хунига (1,035 мл, 5,94 ммоль) добавляли к смеси 4-йод-1H-пирроло[2,3-b]пиридина (0,73 г, 2,971 ммоль), Pd(OAc)2 (0,033 г, 0,15 ммоль) и ксантфоса (0,17 г, 0,30 ммоль) в 1,4-диоксане (14,85 мл, 2,97 ммоль) в атмосфере газообразного Ar. Реакционную смесь нагревали до 100°С в атмосфере аргона в течение 3 ч. Реакционную смесь охлаждали, разбавляли EtOAc (100 мл) и затем фильтровали через диатомовый диоксид кремния (Celite®). Фильтрат концентрировали, и полученный неочищенный продукт очищали флэш-хроматографией, элюируя с градиентом 10-100% EtOAc в гексанах, с получением метил-3-((1H-пирроло[2,3-b]пиридин-4-ил)тио)пропанoата (0,69 г, 2,92 ммоль, выход 98%) в виде твердого вещества. m/z (esi/APCI) M+1=237,1.
[00244] Стадия B: NaOEt (1,14 мл, 3,07 ммоль) добавляли к метил-3-((1H-пирроло[2,3-b]пиридин-4-ил)тио)пропанoату (0,69 г, 2,92 ммоль) в THF (14,6 мл, 2,92 ммоль) и перемешивали в течение 1 ч при комнатной температуре. Добавляли DCM (20 мл), и данную смесь перемешивали в течение 5 мин. Реакционную смесь концентрировали, и твердое вещество растирали с DCM (25 мл), фильтровали и сушили на воздухе. Неочищенный продукт очищали флэш-хроматографией, элюируя с градиентом 0-20% МеОН в DCM с 2% NH4OH, с получением натриевой соли 1H-пирроло[2,3-b]пиридин-4-тиола (0,19 г, 1,07 ммоль, выход 37%) в виде твердого вещества. m/z (esi/APCI) M+1=151,1.
Промежуточный пример O
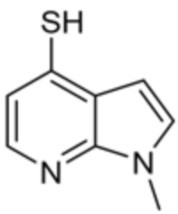
1-метил-1H-пирроло[2,3-b]пиридин-4-тиол
[00245] 1-Метил-1H-пирроло[2,3-b]пиридин-4-тиол получали в соответствии с промежуточным примером N, заменив 4-бром-1-метил-1H-пирроло[2,3-b]пиридин на 4-йод-1H-пирроло[2,3-b]пиридин на стадии A. m/z (esi/APCI) M+1=165,1.
Промежуточный пример P
Трет-бутил-(S)-(1'-(6-меркапто-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)карбамат
[00246] Стадия A: Раствор дигидрохлорида (S)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (11,3 г, 41,0 ммоль) в диметилацетамиде («DMA») (205 мл, 41,0 ммоль) продували аргоном в течение 15 мин перед добавлением N, N-диизопропилэтиламина («DIEA») (28,7 мл, 164 ммоль). Эту смесь нагревали до 60°C в атмосфере азота, и раствор 3,6-дибром-1,2,4-триазина (10,0 г, 41,9 ммоль) в DMA (10 мл) медленно добавляли в течение 45 мин с помощью капельной воронки. Смесь охлаждали, выливали в воду (1 л) и перемешивали в течение 30 мин. Полученные твердые вещества собирали фильтрованием и промывали водой (2 × 250 мл). Этот материал очищали флэш-хроматографией, элюируя с градиентом 1-10% MeOH в DCM с модификатором 1% NH4OH, с получением (S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (9,0 г, 25,0 ммоль, выход 61%). m/z (esi/APCI) M+1 =361,2.
[00247] Стадия B: Смесь (S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (3,0 г, 8,33 ммоль), ди-трет-бутилдикарбоната (5,45 г, 25,0 ммоль) и N, N-диметилпиридин-4-амина (0,051 г, 0,42 ммоль) в дихлорэтане («DCE») (41,6 мл, 8,33 ммоль) перемешивали при комнатной температуре в течение 2 ч. Смесь добавляли в воду (250 мл) и DCM (100 мл). Двухфазную смесь разделяли, и водную фазу экстрагировали DCM (2 × 150 мл). Органические слои объединяли, промывали насыщенным солевым раствором (150 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Материал очищали с помощью флэш-хроматографии, элюируя 10-100% EtOAc в гексанах, с получением трет-бутил-(S)-(1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)карбамата (0,75 г, 1,63 ммоль, выход 20%) в виде твердого вещества. m/z (esi/APCI) M, M+2 =460,1, 462,1.
[00248] Стадия C: Сложный 2-этилгексиловый эфир 3-меркаптопропионовой кислоты (0,10 мл, 0,91 ммоль) и основание Хунига (0,29 мл, 1,65 ммоль) добавляли к смеси трет-бутил-(S)-(1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)карбамата (0,38 г, 0,83 ммоль), Pd(OAc)2 (0,0093 г, 0,041 ммоль) и ксантфоса (0,048 г, 0,083 ммоль) в диоксане (8,25 мл, 0,83 ммоль) в атмосфере газообразного Ar. Данную реакционную смесь нагревали до 100°С в течение 1 ч. Смесь охлаждали, разбавляли EtOAc (25 мл) и затем фильтровали через диатомовый диоксид кремния (Celite®). Фильтрат концентрировали, и полученный неочищенный продукт очищали флэш-хроматографией, элюируя с градиентом 10-100% EtOAc в гексанах, с получением метил-(S)-3-((3-(1-((трет-бутоксикарбонил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)пропанoата (0,41 г, 0,83 ммоль, выход 100%) в виде твердого вещества. m/z (esi/APCI) M+1 =500,2.
[00249] Стадия D: NaOEt (0,62 мл, 1,65 ммоль) добавляли к раствору метил-(S)-3-((3-(1-((трет-бутоксикарбонил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)пропанoата (0,41 г, 0,83 ммоль) в THF (8,25 мл, 0,83 ммоль). Данную смесь перемешивали в течение 30 мин при комнатной температуре. Реакционную смесь концентрировали под вакуумом и очищали флэш-хроматографией, элюируя с градиентом 0-20% МеОН в EtOAc, с получением трет-бутил-(S)-(1'-(6-меркапто-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)карбамата (0,19 г, 0,45 ммоль, выход 54%) в виде твердого вещества. m/z (esi/APCI) M+1 =414,2.
Промежуточный пример Q
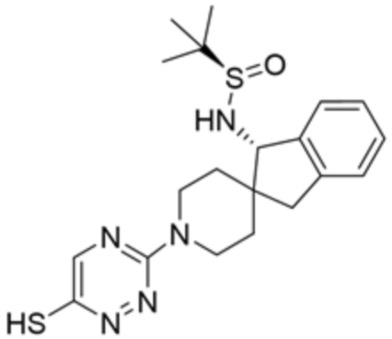
(R)-N-((S)-1'-(6-меркапто-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид
[00250] Стадия A: Смесь (R)-N-((S)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (6,41 г, 20,9 ммоль) и 3,6-дибром-1,2,4-триазина (5,00 г, 20,9 ммоль) в 1,4-диоксане (52,3 мл, 20,9 ммоль) продували аргоном в течение 15 мин. DIEA (4,39 мл, 25,1 ммоль) добавляли в смесь, и затем смесь нагревали до 60°C в течение 1 ч. Смесь охлаждали до комнатной температуры, выливали в воду (1 л) и перемешивали в течение 30 мин. Твердые вещества собирали фильтрованием и сушили, с получением (R)-N-((S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (9,5 г, 20,5 ммоль, выход 98%). m/z (esi/APCI) M, M+2 =464,1, 466,1.
[00251] Стадия B: Сложный 2-этилгексиловый эфир 3-меркаптопропионовой кислоты (0,51 мл, 4,74 ммоль) и основание Хунига (1,50 мл, 8,61 ммоль) добавляли к смеси (R)-N-((S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (2,0 г, 4,31 ммоль), Pd(OAc)2 (0,048 г, 0,22 ммоль) и ксантфоса (0,25 г, 0,43 ммоль) в 1,4-диоксане (43,06 мл, 4,30 ммоль) в атмосфере газообразного аргона. Данную реакционную смесь нагревали до 100°С в течение 1 ч. Смесь охлаждали, разбавляли EtOAc (25 мл) и затем фильтровали через диатомовый диоксид кремния (Celite®). Фильтрат концентрировали, и полученный неочищенный продукт очищали флэш-хроматографией, элюируя с градиентом 10-100% EtOAc в гексанах, с получением метил-3-((3-((S)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)пропанoата (0,88 г, 1,74 ммоль, выход 40%) в виде твердого вещества. m/z (esi/APCI) M+1=504,2.
[00252] Стадия C: NaOEt (0,96 мл, 2,58 ммоль) добавляли к раствору метил-3-((3-((S)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)пропанoата (0,65 г, 1,29 ммоль) в THF (12,9 мл, 1,29 ммоль). Данную смесь перемешивали в течение 30 мин при комнатной температуре. Смесь разбавляли EtOAc и фильтровали через диатомовый диоксид кремния (Celite®). Фильтрат концентрировали, и полученный неочищенный продукт очищали флэш-хроматографией, элюируя с градиентом 0-20% МеОН в EtOAc, с получением (R)-N-((S)-1'-(6-меркапто-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (0,34 г, 0,81 ммоль, выход 63%) в виде твердого вещества. m/z (esi/APCI) M+1=418,2.
Промежуточный пример R
3,3-дифтор-4-йод-1,3-дигидро-2H-пирроло[2,3-b]пиридин-2-он
[00253] 35% водный раствор перекиси водорода (2,52 мл, 85,22 ммоль) добавляли к раствору 2-амино-4-йодпиридина (2,50 г, 11,36 ммоль), этилбромдифторацетата (3,64 мл, 28,41 ммоль) и бис(циклопентадиенил)железа (0,22 г, 1,14 ммоль) в ДМСО (21,85 мл, 11,36 ммоль) при -5°С, одновременно перемешивая. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 22 ч. Смесь выливали в воду (100 мл), и органические соединения экстрагировали из водной фазы EtOAc (3×50 мл). После объединения органические слои промывали водой (50 мл) и насыщенным солевым раствором (3×50 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали флэш-хроматографией, используя градиент 10-100% EtOAc в гексанах, с получением 3,3-дифтор-4-йод-1,3-дигидро-2H-пирроло[2,3-b]пиридин-2-она (0,93 г, 3,13 ммоль, выход 27%). m/z (esi/APCI) M+1=296,9.
Промежуточный пример S
дигидрохлорид (R)-3H-спиро[фуро[2,3-b]пиридин-2,4'-пиперидин]-3-амина
[00254] Стадия A: 3-(1,3-Дитиан-2-ил)-2-фторпиридин (1 г, 4,64 ммоль) в THF (4,64 мл) добавляли к перемешиваемому раствору LDA (4,06 мл, 8,13 ммоль) в THF (4,64 мл) при -78°С. Реакционную смесь нагревали до 0°С и перемешивали в течение 20 мин. Реакционную смесь снова охлаждали до -78°C с последующим добавлением по каплям 1-boc-4-пиперидона (0,97 г, 4,88 ммоль) в THF (4,64 мл). Реакционную смесь перемешивали в течение 1 ч, давали нагреться до 0°C и перемешивали в течение 6 ч. Смесь выливали в насыщенный NH4Cl. Продукт трижды экстрагировали этилацетатом, объединяли органические слои, фильтровали через бумагу 1PS, выпаривали под вакуумом, концентрировали, очищали хроматографией на силикагеле (0-100% EA/гексан), с получением трет-бутил-диспиро[пиперидин-4,2'-фуро[2,3-b]пиридин-3',2''-[1,3]дитиан]-1-карбоксилата (993 мг, 2,52 ммоль, выход 54%). ЖХМС (ММ-ES+APCI, положит.): m/z 395,1 (M+H).
[00255] Стадия B: Пербромид пиридиния (1,2 г, 3,78 ммоль) и бромид тетрабутиламмония (81,1 мг, 0,25 ммоль) добавляли к перемешиваемому раствору трет-бутилдиспиро[пиперидин-4,2'-фуро[2,3-b]пиридина-3',2"-[1,3]дитиан]-1-карбоксилата (993 мг, 2,52 ммоль) и пиридина (0,2 мл, 2,52 ммоль) в смеси DCM-H2O 5:1 (16,8 мл). После перемешивания при комнатной температуре в течение 24 ч реакционную смесь выливали в воду и DCM. Слой DCM промывали водой, насыщенным солевым раствором, фильтровали через бумагу 1PS, концентрировали и очищали с помощью хроматографии на силикагеле (0-100% этилацетат/гексаны) с получением трет-бутил-3-оксо-3H-спиро[фуро[2,3-b]пиридин-2,4'-пиперидин]-1'-карбоксилата (735 мг, 1,93 ммоль, выход 77%). ЖХМС (ММ-ES+APCI, положит.): m/z 205,1 (M+H - Boc).
[00256] Стадия C: (R)-(+)-2-Метил-2-пропансульфинамид (608,8 мг, 5,02 ммоль) и тетраэтоксититан (2,5 мл, 11,72 ммоль) добавляли в раствор трет-бутил-3-оксо-3H-спиро[фуро[2,3-b]пиридин-2,4'-пиперидин]-1'-карбоксилата (637 мг, 1,67 ммоль) в THF (8,4 мл), и реакционную смесь перемешивали в течение ночи при 90°С. Реакционную смесь разбавляли этилацетатом и промывали водой. Слои разделяли. Водный слой экстрагировали этилацетатом. Объединенные органические вещества промывали водой, насыщенным солевым раствором, фильтровали через бумагу 1PS и концентрировали под вакуумом. Материал хроматографировали с использованием 0-50% EtOAc/гексанов в качестве элюента, с получением трет-бутил-(R, E)-3-((трет-бутилсульфинил)имино)-3H-спиро[фуро[2,3-b]пиридин-2,4'-пиперидин]-1'-карбоксилата (426 мг, 1,04 ммоль, выход 62%). ЖХМС (ММ-ES+APCI, положит.): m/z 408,2 (M+H).
[00257] Стадия D: Трет-бутил-(R, E)-3-((трет-бутилсульфинил)имино)-3H-спиро[фуро[2,3-b]пиридин-2,4'-пиперидин]-1'-карбоксилат (426 мг, 1,05 ммоль) растворяли в THF (2,6 мл), и раствор охлаждали до -78°С. 1 М раствор BH3·THF (2,2 мл, 2,20 ммоль) добавляли в раствор с помощью шприца в течение 30 мин. Реакционную смесь перемешивали при -78°C в течение 1 ч и нагревали до комнатной температуры при перемешивании в течение 3 дней. Реакцию гасили насыщенным NH4Cl и экстрагировали EtOAc. Объединенные органические слои промывали водой, насыщенным солевым раствором, сушили, выпаривали под вакуумом и очищали хроматографией на силикагеле (0-20% MeOH/DCM) с получением трет-бутил-(R)-3-(((R)-трет-бутилсульфинил)амино)-3H-спиро[фуро[2,3-b]пиридин-2,4'-пиперидин]-1'-карбоксилата (180 мг, 0,44 ммоль, выход 42%). ЖХМС (ММ-ES+APCI, положит.): m/z 410,2 (M+H).
[00258] Стадия E: Смесь трет-бутил-(R)-3-(((S)-трет-бутилсульфинил)амино)-3H-спиро[фуро[2,3-b]пиридин-2,4'-пиперидин]-1'-карбоксилата (80 мг, 0,20 ммоль) в DCM (1 мл) продували N2 и обрабатывали HCl в диоксане (0,25 мл, 0,98 ммоль) с помощью шприца при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 2 ч. Смесь выпаривали под вакуумом с получением дигидрохлорида (R)-3H-спиро[фуро[2,3-b]пиридин-2,4'-пиперидин]-3-амина (44 мг, 0,13 ммоль, выход 65%).
Промежуточный пример T
дигидрохлорид (1'S)-1',3'-дигидро-8-азаспиро[бицикло[3.2.1]октaн-3,2'-инден]-1'-амина
[00259] Стадия A: Диизопропиламид лития, 2M раствор в THF/н-гептане (2,78 мл, 5,57 ммоль) добавляли по каплям к раствору при -70°С 8-(трет-бутил)-3-метил-(1R,5S)-8-азабицикло[3.2.1]октaн-3,8-дикарбоксилата (1 г, 3,71 ммоль) в THF (9,28 мл). После перемешивания при -70°С в течение 90 мин, медленно добавляли бензилбромид (0,57 мл, 4,83 ммоль). Реакционную смесь перемешивали при -70°С в течение 3 ч. Реакционную смесь осторожно гасили насыщенным водным NH4Cl. Водный слой отделяли, экстрагировали этилацетатом, объединенные экстракты фильтровали через бумагу 1PS, концентрировали и очищали с помощью хроматографии на силикагеле (0-50% этилацетат/гексаны) с получением 8-(трет-бутил)-3-метил-(1R,3r,5S)-3-бензил-8-азабицикло[3.2.1]октaн-3,8-дикарбоксилата (1,1 г, 3,06 ммоль, выход 82%). ЖХМС (ММ-ES+APCI, положит.): m/z 260,1 (M+H -Boc).
[00260] Стадия B: Смесь 8-(трет-бутил)-3-метил (1R,3r,5S)-3-бензил-8-азабицикло[3.2.1]октaн-3,8-дикарбоксилата (1 г, 2,78 ммоль) в полифосфорной кислоте (5,54 мл, 139 ммоль) перемешивали при 120°С в течение 5 дней. Смесь разбавляли водой, рН доводили до 10 с помощью NaOH. Добавляли Boc-ангидрид (0,97 мл, 4,17 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Смесь экстрагировали этилацетатом, объединенные органические вещества фильтровали через бумагу 1PS, концентрировали и очищали с помощью хроматографии на силикагеле (0-50% этилацетат/гексаны) с получением трет-бутил-1'-оксо-1',3'-дигидро-8-азаспиро[бицикло[3.2.1]октaн-3,2'-инден]-8-карбоксилата (650 мг, 1,39 ммоль, выход 50%). ЖХМС (ММ-ES+APCI, положит.): m/z 228,1 (M+H-Boc).
[00261] Стадия C: (R)-(+)-2-Метил-2-пропансульфинамид (721,8 мг, 5,95 ммоль) и тетраэтоксититан (2,9 мл, 13,90 ммоль) добавляли к раствору трет-бутил-1'-оксо-1',3'-дигидро-8-азаспиро[бицикло[3.2.1]октaн-3,2'-инден]-8-карбоксилата (0,65 г, 1,98 ммоль) в THF (9,9 мл), и реакционную смесь перемешивали в течение ночи при 90°С. Реакционную смесь разбавляли этилацетатом и промывали водой, и слои разделяли. Водный слой экстрагировали этилацетатом. Объединенные органические вещества промывали водой, насыщенным солевым раствором, фильтровали через бумагу 1PS и концентрировали под вакуумом. Материал хроматографировали (0-50% EtOAc/гексаны) с получением трет-бутил-(Z)-1'-(((R)-трет-бутилсульфинил)имино)-1',3'-дигидро-8-азаспиро[бицикло[3.2.1]октaн-3,2'-инден]-8-карбоксилата (307 мг, 0,71 ммоль, выход 36%). ЖХМС (ММ-ES+APCI, положит.): m/z 431,2 (M+H).
[00262] Стадия D: Борогидрид натрия (108 мг, 2,85 ммоль) добавляли к раствору трет-бутил-(Z)-1'-(((R)-трет-бутилсульфинил)имино)-1',3'-дигидро-8-азаспиро[бицикло[3.2.1]октaн-3,2'-инден]-8-карбоксилата (307 мг, 0,71 ммоль) в THF (0,79 мл), охлажденному до -70°С, и реакционную смесь перемешивали в течение 18 ч, одновременно нагревая до комнатной температуры. Реакционную смесь переливали в воду и экстрагировали этилацетатом. Органические вещества промывали насыщенным солевым раствором, фильтровали через бумагу 1PS и концентрировали под вакуумом с получением трет-бутил-(1'S)-1'-(((R)-трет-бутилсульфинил)амино)-1',3'-дигидро-8-азаспиро[бицикло[3.2.1]октaн-3,2'-инден]-8-карбоксилата (274 мг, 0,63 ммоль, выход 89%). ЖХМС (ММ-ES+APCI, положит.): m/z 333,2 (M+H-Boc).
[00263] Стадия E: Смесь трет-бутил-(1'S)-1'-(((R)-трет-бутилсульфинил)амино)-1',3'-дигидро-8-азаспиро[бицикло[3.2.1]октaн-3,2'-инден]-8-карбоксилата (274 мг, 0,63 ммоль) в DCM (3,2 мл) продували N2 и обрабатывали 4 н. HCl в диоксане (0,79 мл, 3,17 ммоль) с помощью шприца при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 90 мин. Смесь выпаривали под вакуумом, остаток встряхивали в MTBE, твердые вещества собирали фильтрованием и сушили с получением дигидрохлорида (1'S)-1',3'-дигидро-8-азаспиро[бицикло[3.2.1]октaн-3,2'-инден]-1'-амина (144 мг, 0,48 ммоль, выход 76%). ЖХМС (ММ-ES+APCI, положит.): m/z 229,2.
Промежуточный пример U
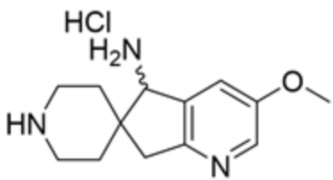
гидрохлорид 3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина
[00264] Стадия A: MeI (1,65 мл, 26,58 ммоль) добавляли К перемешиваемому раствору 5-бром-6-хлорпиридин-3-ола (5,0 г, 24,16 ммоль) и K2CO3 (5,0 г, 36,25 ммоль) в ACN (30 мл), и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь экстрагировали EtOAc. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (0-10% EtOAc/гексан) с получением 3-бром-2-хлор-5-метоксипиридина (3,48 г, выход 65%) в виде твердого вещества. m/z (esi) M+1=223,9.
[00265] Стадия B: Бромид изопропилмагния (19,76 мл, 1,3 M в THF, 25,68 ммоль) добавляли по каплям к перемешиваемому раствору трет-бутил-4-формил-4-метилпиперидин-1-карбоксилата (3,89 г, 17,12 ммоль) в THF (55 мл) при 0°С и перемешивали при комнатной температуре в течение 2 ч. 3-Бром-2-хлор-5-метоксипиридин (5,68 г, 25,68 ммоль) в THF (10 мл) добавляли по каплям при 0°С и перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь гасили насыщенным раствором NH4Cl и экстрагировали EtOAc. Органическую часть высушивали над Na2SO4, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (15-20% EtOAc/гексан) с получением трет-бутил-4-((2-хлор-5-метоксипиридин-3-ил)(гидрокси)метил)-4-метилпиперидин-1-карбоксилата (2,157 г, выход 83%) в виде клейкого твердого вещества. m/z (esi) M+1=371,4.
[00266] Стадия C: Периодинан Десса-Мартина (1,89 г, 4,45 ммоль) добавляли к перемешиваемому раствору трет-бутил-4-((2-хлор-5-метоксипиридин-3-ил)(гидрокси)метил)-4-метилпиперидин-1-карбоксилата (1,1 г, 2,97 ммоль) в DCM (16 мл) при 0°С и перемешивали при комнатной температуре в течение 3 ч в атмосфере азота. Реакционную смесь гасили насыщенным раствором тиосульфата натрия и экстрагировали DCM. Органическую фазу промывали 1 н. раствором NaOH, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (15-20% EtOAc/гексан) с получением трет-бутил-4-(2-хлор-5-метоксиникотинoил)-4-метилпиперидин-1-карбоксилата (860 мг, выход 78%) в виде масла. m/z (esi) M+1=369,1.
[00267] Стадия D: Трициклогексилфосфонийтетрафторборат (145 мг, 0,39 ммоль), ацетат пaллaдия(II) (44 мг, 0,19 ммоль), пивалевую кислоту (121 мг, 1,18 ммоль), карбонат цезия (1,54 г, 4,72 ммоль) и трет-бутил-4-(2-хлор-5-метоксиникотинoил)-4-метилпиперидин-1-карбоксилат (1,45 г, 3,94 ммоль) в мезитилене (20 мл) добавляли в высушенную пламенем герметичную пробирку в атмосфере аргона. Смесь дегазировали аргоном в течение 10 мин и нагревали при 140°C в течение 48 ч. Реакционную смесь охлаждали до комнатной температуры, фильтровали через слой Celite® и промывали EtOAc. Органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (0-10% EtOAc/гексан) с получением трет-бутил-3-метокси-5-оксо-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (0,84 г, выход 64%) в виде твердого вещества. m/z (esi) M+1=333,2.
[00268] Стадия E: (R)-(+)-2-Метил-2-пропансульфинамид (629 мг, 5,19 ммоль) добавляли к перемешиваемому раствору трет-бутил-3-метокси-5-оксо-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (575 мг, 1,73 ммоль) в этоксиде титана(IV) (2,2 мл, 10,38 ммоль), и реакционную смесь перемешивали при 110°С в течение 4 ч. Реакционной смеси давали возможность остыть до комнатной температуры, и реакционную смесь разбавляли EtOAc и водой. Полученную смесь энергично перемешивали в течение 15 мин при комнатной температуре и затем фильтровали через слой Celite®. Фильтрат экстрагировали EtOAc, промывали насыщенным солевым раствором, сушили над Na2SO4 и фильтровали. Органическую фазу концентрировали и полученный остаток очищали колоночной хроматографией на силикагеле (40-50% EtOAc/гексан) с получением трет-бутил-(R, Z)-5-((трет-бутилсульфинил)имино)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата в виде полутвердого вещества (0,64 г, выход 84%). m/z (esi) M+1=437,2.
[00269] Стадия F: Борогидрид натрия (256 мг, 6,77 ммоль) добавляли к перемешиваемому раствору трет-бутил-(R, Z)-5-((трет-бутилсульфинил)имино)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (590 мг, 1,35 ммоль) в MeOH (10 мл) при 0°С, и реакционную смесь перемешивали в течение 3 ч. Реакционной смеси давали нагреться до комнатной температуры. Насыщенный водный раствор NH4Cl медленно добавляли для гашения реакции. МеОН выпаривали, и реакционную смесь экстрагировали EtOAc. Органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (0-4% MeOH/DCM) с получением трет-бутил-5-(((R)-трет-бутилсульфинил)амино)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (0,58 г, выход 97%) в виде твердого вещества. m/z (esi) M+1=437,9.
[00270] Стадия G: Диоксан-HCl (4M; 12 мл) добавляли к перемешиваемому раствору трет-бутил-5-(((R)-трет-бутилсульфинил)амино)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (600 мг, 1,37 ммоль) в MeOH (12 мл) при 0°С, и реакционную смесь перемешивали в течение 2 ч. Реакционную смесь концентрировали и неочищенный материал растирали с простым диэтиловым эфиром с получением гидрохлорида 3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (418 мг, выход 99%) в виде твердого вещества. Неочищенный продукт использовали непосредственно без дальнейшей очистки. m/z (esi) M+1=234,3.
Промежуточный пример V
Трет-бутил-(R)-1-(((S)-трет-бутилсульфинил)амино)-6-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00271] Стадия A: NaH (60% в минеральном масле) (1,45 г, 36,14 ммоль) добавляли к перемешиваемому раствору 6-хлор-2,3-дигидро-1H-инден-1-она (2,0 г, 12,05 ммоль) в диметилформамиде («DMF») (40 мл) при 0°С, и смесь перемешивали в течение 30 мин при 0-5 °С. Гидрохлорид N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амина (3,06 г, 13,25 ммоль) добавляли порциями, и смесь перемешивали при 60°С в течение 16 ч. Реакцию гасили насыщенным солевым раствором и экстрагировали EtOAc. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью силикагеля (EtOAc/гексан) для получения 1'-бензил-6-хлороспиро[инден-2,4'-пиперидин]-1(3Н)-она (1,55 г, выход 40%) в виде масла. m/z (esi) M+1: 325,9.
[00272] Стадия B: 1-Хлорэтилхлорформиат (1,55 мл, 14,34 ммоль) добавляли к перемешиваемому раствору 1'-бензил-6-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (1,55 г, 4,78 ммоль) в DCE (24 мл), и реакционную смесь нагревали с обратным холодильником в течение 1 ч. Реакционную смесь концентрировали и добавляли MeOH (24 мл), и нагревали с обратным холодильником в течение 1 ч. MeOH выпаривали досуха с получением 6-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (1,13 г, неочищенный материал) в виде твердого вещества. m/z (esi) M+1: 236,2.
[00273] Стадия C: Триэтиламин (2,66 мл, 19,16 ммоль) добавляли к перемешиваемому раствору 6-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (1,13 г, 4,79 ммоль) в DCM (20 мл), после чего добавляли boc-ангидрид (1,65 мл, 7,19 ммоль) при 0°С, и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь выпаривали досуха и очищали колоночной хроматографией на силикагеле (15-20% EtOAc/гексан) с получением трет-бутил-6-хлор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,3 г, 81%, 2-стадийный выход) в виде твердого вещества. m/z (esi) M+1: 336,3.
[00274] Стадия D: Раствор трет-бутил-6-хлор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (800 мг, 2,38 ммоль), этоксида титана(IV) (1,99 мл, 9,5 ммоль) и (R)-(+)-2-метилпропан-2-сульфинамида (577,5 мг, 4,76 ммоль) в THF (15 мл) перемешивали при 90°С в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры и гасили водой. Соединение экстрагировали EtOAc. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали с помощью колоночной хроматографии на силикагеле (20-25% EtOAc/гексан) с получением трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-6-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (861 мг, выход 94%) в виде твердого вещества. m/z (esi) M+1: 438,8.
Промежуточный пример W
Трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-5-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00275] Стадия A: NaH (60% дисперсия в минеральном масле, 1,97 г, 82,19 ммоль) добавляли порциями к перемешиваемому раствору 5-метил-2,3-дигидро-1H-инден-1-она (4,0 г, 27,39 ммоль) в DMF (80 мл) при 0°С. Смесь перемешивали при 0°С в течение 30 мин. Гидрохлорид N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амина (8,09 г, 30,13 ммоль) добавляли порциями в реакционную смесь, и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли насыщенным солевым раствором и экстрагировали EtOAc. Органические части объединяли и промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (25% EtOAc/гексан) с получением 1'-бензил-5-метилспиро[инден-2,4'-пиперидин]-1(3H)-она (4,0 г, выход 48%) в виде клейкого твердого вещества. m/z (esi) M+1=305,6.
[00276] Стадия B: Хлорэтилхлорформиат (6,97 г, 49,11 ммоль) добавляли к перемешиваемому раствору 1'-бензил-5-метилспиро[инден-2,4'-пиперидин]-1(3H)-она (5,0 г, 16,37 ммоль) в DCE (100 мл) при 0°С и перемешивали в течение 10 мин. Реакционную смесь перемешивали при 80°С в течение 1 ч. Реакционную смесь концентрировали досуха, и добавляли MeOH (100 мл) и перемешивали при 75°C в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении с получением 5-метилспиро[инден-2,4'-пиперидин]-1(3H)-она (3,5 г, неочищенное вещество) в виде жидкости, которую использовали для следующей стадии без дальнейшей очистки. m/z (esi) M+1=215,9.
[00277] Стадия C: Триэтиламин (9,07 мл, 65,11 ммоль) добавляли к перемешиваемому раствору 5-метилспиро[инден-2,4'-пиперидин]-1(3H)-она (3,5 г, 16,27 ммоль) в DCM (35 мл) при 0°С. Boc-ангидрид (5,61 мл, 24,42 ммоль) добавляли при 0°С, и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (10% EtOAc/гексан) с получением трет-бутил-5-метил-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (520 мг, выход 68%, 2 стадии) в виде твердого вещества. m/z (esi) M+1=316,2.
[00278] Стадия D: Трет-бутил-5-метил-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (1,9 мг, 6,03 ммоль) и (R)-(+)-2-метилпропан-2-сульфинамид (2,19 г, 18,09 ммоль) добавляли в теплый (100°С) этоксид титана (IV) (4,12 г, 18,09 ммоль) и перемешивали при 100°С в течение 19 ч. Реакционную смесь выливали в EtOAc и насыщенный солевой раствор и перемешивали в течение 15 мин. Твердые вещества отфильтровывали, и жидкую часть отделяли. Органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (1% MeOH/DCM) с получением трет-бутил-(R, E)-1-((трет-бутил-сульфинил)имино)-5-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,25 г, выход 49%) в виде твердого вещества. m/z (esi) M+1=418,9.
[00279] Стадия E: Борогидрид натрия (470 мг, 12,42 ммоль) добавляли к раствору при 0°C трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-5-метил-1,3-дигидро-спиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,3 г, 3,10 ммоль) в MeOH (30 мл), и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь гасили ледяной водой и экстрагировали EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (30% EtOAc/гексан) с получением трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-5-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (310 мг, выход 24%) (m/z (esi) M-1=419,3).
Промежуточный пример Х
Трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-5-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00280] Трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-5-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат получали в соответствии с промежуточным примером W, собирая второй пик на стадии E. (m/z (esi) M-1=419,3).
Промежуточный пример Y
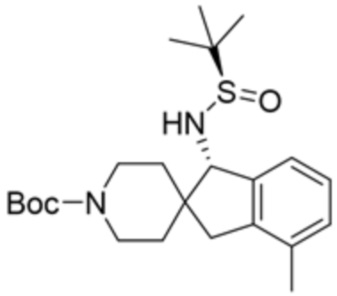
Трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-4-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00281] Стадия A: NaH (60%, 0,98 г, 15,06 ммоль) добавляли к перемешиваемому раствору 4-метил-2,3-дигидро-1H-инден-1-она (2 г, 13,69 ммоль) в DMF (25 мл) при 0°С, и реакционную смесь перемешивали в течение 30 мин при 0°С. Гидрохлоридную соль N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амина добавляли и перемешивали в течение 18 ч при комнатной температуре. Реакцию гасили водным насыщенным раствором NH4Cl и экстрагировали этилацетатом. Объединенный органический слой промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью колонки Combi-Flash (элюировали 25% этилацетатом в гексане) с получением 1'-бензил-4-метилспиро[инден-2,4'-пиперидин]-1(3H)-она (1,5 г, 39%) в виде твердого вещества. m/z (esi) M+1=305,8.
[00282] Стадия B: 1-Хлорэтилхлорформиат (1,59 мл, 14,73 ммоль) добавляли к перемешиваемому раствору 1'-бензил-4-метилспиро[инден-2,4'-пиперидин]-1(3H)-она (1,5 г, 4,91 ммоль) в DCE (10 мл) и нагревали с обратным холодильником в течение 1 ч. Летучие компоненты концентрировали при пониженном давлении с получением неочищенного масла. Добавляли метанол и нагревали с обратным холодильником в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении с получением 4-метилспиро[инден-2,4'-пиперидин]-1(3Н)-она в качестве неочищенного продукта, который использовали на следующей стадии без дальнейшей очистки. m/z (esi) M+1=215,7.
[00283] Стадия C: Триэтиламин («TEA») (3,23 мл, 23,22 ммоль) добавляли к перемешиваемому раствору 4-метилспиро[инден-2,4'-пиперидин]-1(3H)-она (1 г, неочищенный) в DCM (15 мл), после чего добавляли boc-ангидрид (2,13 мл, 9,28 ммоль). Реакционную смесь перемешивали в течение 18 ч при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении для получения неочищенного продукта, который очищали с помощью колонки Combi-Flash (элюировали 10-15% этилацетатом в гексане) с получением трет-бутил-4-метил-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,5 г, 66%, 2 стадии) в виде жидкости.
[00284] Стадия D: Ti(OEt)4 (8,77 мл, 41,84 ммоль) добавляли к трет-бутил-4-метил-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилату (1 г, 3,17 ммоль) и нагревали до 90°С. Добавляли (R)-2-метилпропан-2-сульфинамид (1,1 г, 9,51 ммоль) и нагревание продолжали при 90°С в течение 24 ч. Реакционную смесь выливали в этилацетат (50 мл), и добавляли водный насыщенный солевой раствор (50 мл). Осажденное твердое вещество отфильтровывали и фильтрат промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали для получения неочищенного продукта, который очищали с помощью колонки Combi-Flash (элюировали 20% этилацетатом в гексане), с получением трет-бутил-(R, Z)-1-((трет-бутилсульфинил)имино)-4-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (800 мг, 60%) в виде твердого вещества. m/z (esi) M+1=418,8.
[00285] Стадия E: Борогидрид натрия (600 мг, 15,76 ммоль) добавляли к перемешиваемому раствору трет-бутил-(R, Z)-1-((трет-бутилсульфинил)имино)-4-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,1 г, 2,62 ммоль) в метаноле (60 мл) при комнатной температуре и перемешивали в течение 4 ч. Реакцию гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом. Объединенную органическую часть промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью колонки Combi-Flash (элюировали 25% этилацетатом в гексане) с получением трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-4-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (440 мг, 40%) (m/z (esi) M+1= 421,4) в виде твердого вещества.
Промежуточный пример Z
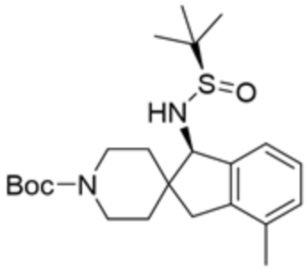
трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-4-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00286] Трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-4-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат получали в соответствии с промежуточным примером Y, собирая второй пик. m/z (esi) M+1=421,4.
Промежуточный пример АB
трет-бутил-(S)-5-(((R)-трет-бутилсульфинил)амино)-2-метил-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат
[00287] Стадия A: Смесь циклопентaн-1,3-диона (20,0 г, 204,08 ммоль), бут-3-ен-2-она (26,78 мл, 306,12 ммоль), молекулярных сит 4Å (100 г) и NH4OAc (31,42 г, 408,16 ммоль) в толуоле (800 мл) перемешивали при нагревании с обратным холодильником в течение 24 ч. Реакционную смесь фильтровали через слой Celite® и концентрировали, и полученный остаток очищали колоночной хроматографией на силикагеле (50% EtOAc/гексан) с получением 2-метил-6,7-дигидро-5H-циклопентa[b]пиридин-5-она (6,0 г, выход 20%) в виде жидкости. m/z (esi) M+1=148,3.
[00288] Стадия B: NaH (60% масс. в парафине) (4,0 г, 102,04 ммоль) добавляли к перемешиваемому раствору 2-метил-6,7-дигидро-5H-циклопентa[b]пиридин-5-она (5,0 г, 34,01 ммоль) в DMF (60 мл) при 0°С и перемешивали в течение 30 мин при комнатной температуре. Гидрохлорид N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амина (7,31 г, 27,21 ммоль) добавляли порциями при 0°С и перемешивали в течение 16 ч при комнатной температуре. Реакционную смесь гасили ледяной водой и экстрагировали этилацетатом. Органическую часть высушивали (Na2SO4), фильтровали и концентрировали, и неочищенный материал очищали с помощью силикагеля (0-15% MeOH в DCM) с получением 1'-бензил-2-метилспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5(7H)-она (1,2 г, выход 11%) в виде жидкости. m/z (esi) M+1=306,9.
[00289] Стадия C: Формиат аммония (247,14 мг, 3,92 ммоль) и Pd/C (200 мг) добавляли к перемешиваемому раствору 1'-бензил-2-метилспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5(7H)-она (400,0 мг, 1,31 ммоль) в этаноле (15 мл), и реакционную смесь продували аргоном в течение 10 мин. Реакционную смесь нагревали с обратным холодильником при 80°С в течение 16 ч и концентрировали с получением 2-метилспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5(7H)-она, который использовали для следующей стадии без дальнейшей очистки. m/z (esi) M+1=217,2.
[00290] Стадия D: Триэтиламин (0,72 мл, 5,18 ммоль) добавляли к перемешиваемому раствору 2-метилспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5(7H)-она (280,0 мг, 1,29 ммоль) в DCM (10 мл) при 0°С, с последующим добавлением Boc-ангидрида (0,45 мл, 1,94 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали и очищали колоночной хроматографией на силикагеле (30% EtOAc/гексан) с получением трет-бутил-2-метил-5-оксо-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (200 мг, выход 48%, 2 стадии) в виде твердого вещества. m/z (esi) M+1=317,2.
[00291] Стадия E: Трет-бутил-2-метил-5-оксо-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат (400 мг, 1,26 ммоль) и (R)-2-метилпропан-2-сульфинамид (460,21 мг, 3,80 ммоль) добавляли к этоксиду титана (IV) (866,20 мг, 3,79 ммоль) при 90°С и перемешивали при 90°С в течение 5 ч. Реакционную смесь выливали в этилацетат и насыщенный солевой раствор. После перемешивания в течение 15 мин осажденное твердое вещество отфильтровывали, и жидкую часть отделяли. Органический слой промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с получением трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-5-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (410 мг, выход 77%) в виде смолообразной жидкости. m/z (esi) M+1=420,2.
[00292] Стадия F: NaBH4 (185 мг, 4,89 ммоль) добавляли к раствору трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-5-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (410 мг, 0,98 ммоль) в MeOH (10 мл) при 0°С и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь гасили ледяной водой и экстрагировали EtOAc. Объединенный органический слой сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (1% MeOH-DCM) и затем препаративной ВЭЖХ (Chiralpak IG (21,0×250 мм), 5µ н-гексан/EtOH/изопропиламин 80/20/0,1, 21,0 мл/мин, 20 мин, 276 нм, MeOH), с получением трет-бутил-(S)-5-(((R)-трет-бутилсульфинил)амино)-2-метил-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (40 мг, выход 10%). m/z (esi) M+1=422,4.
Промежуточный пример АC
трет-бутил-(R)-5-(((R)-трет-бутилсульфинил)амино)-2-метил-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат
[00293] Трет-бутил-(R)-5-(((R)-трет-бутилсульфинил)амино)-2-метил-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат получали в соответствии с промежуточным примером АВ, собирая второй пик на стадии F. m/z (esi) M+1=422,5.
Промежуточный пример АD
3-хлор-2-((2-гидроксиэтил)амино)пиридин-4-тиолат натрия
[00294] Стадия A: 2-Аминоэтан-1-ол (0,47 мл, 7,78 ммоль) добавляли к перемешиваемому раствору 3-хлор-2-фтор-4-йодпиридина (1,0 г, 3,89 ммоль) в ДМСО (5 мл) и перемешивали при 70°С в течение 16 ч. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органическую часть сушили (Na2SO4) и концентрировали, и полученный остаток очищали колоночной хроматографией на силикагеле (40% EtOAc-гексан) с получением 2-((3-хлор-4-йодпиридин-2-ил)амино)этан-1-ола (820 мг, выход 70%) в виде твердого вещества. m/z (esi) M+1=298,8.
[00295] Стадия B: DIEA (0,6 мл, 3,35 ммоль) добавляли к перемешиваемому раствору 2-((3-хлор-4-йодпиридин-2-ил)амино)этан-1-ола (500 мг, 1,67 ммоль) и метил-3-меркаптопропанoата (0,2 мл, 1,84 ммоль) в диоксане (5 мл) и дегазировали аргоном в течение 10 мин. Добавляли ксантфос (48 мг, 0,08 ммоль) и Pd(OAC)2 (23 мг, 0,10 ммоль) и дегазировали в течение еще 10 мин. Реакционную смесь перемешивали в предварительно нагретой масляной бане в герметичной пробирке при температуре 100°C в течение 4 ч. Реакционную смесь фильтровали через слой Celite® и промывали этилацетатом. Растворитель выпаривали, и неочищенный материал очищали колоночной хроматографией на силикагеле (60% EtOAc/гексан) с получением метил-3-((3-хлор-2-((2-гидроксиэтил)амино)пиридин-4-ил)тио)пропанoата (460 мг, выход 94%) в виде твердого вещества. m/z (esi) M+1=290,9.
[00296] Стадия C: NaOEt (21% масс. в EtOH; 1,5 мл, 2,06 ммоль) добавляли к перемешиваемому раствору метил-3-((3-хлор-2-((2-гидроксиэтил)амино)пиридин-4-ил)тио)пропанoата (500 мг, 1,72 ммоль) в THF (10 мл) при 0°С и перемешивали в течение 30 мин при 0°С. Реакционную смесь концентрировали и неочищенный материал растирали с DCM. Твердый осадок отфильтровывали с получением 3-хлор-2-((2-гидроксиэтил)амино)пиридин-4-тиолата натрия (350 мг, выход 90%) в виде твердого вещества. m/z (esi) M+1=205,1.
Промежуточный пример АE
трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-6-хлор-5-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00297] Стадия A: AlCl3 (10,3 г, 77,14 ммоль) добавляли порциями к раствору 1-хлор-2-метоксибензола (10 г, 70,13 ммоль) и 3-хлорпропаноилхлорида (7,4 мл, 77,14 ммоль) в DCM (80 мл) при 0°С. Через 30 мин в реакционную смесь медленно вливали серную кислоту (240 мл). DCM удаляли на роторном испарителе при пониженном давлении, и вязкий остаток перемешивали при 100°С в течение 2 ч. После охлаждения до 20°C вязкую реакционную смесь осторожно выливали в лед (500 мл) и оставляли стоять в течение ночи. Смесь отфильтровывали, и осадок неочищенного продукта промывали водой (100 мл). Неочищенный продукт растворяли в 5% метаноле в DCM (200 мл), разбавляли водой (100 мл), экстрагировали 5% MeOH в DCM (100 мл), и промывали тщательно насыщенным солевым раствором (100 мл). Органическую часть высушивали над Na2SO4, концентрировали под вакуумом и очищали хроматографией на силикагеле (100-200) методом флэш-хроматографии (элюент: 20% этилацетат в гексане) с получением 6-хлор-5-метокси-2,3-дигидро-1H-инден-1-она (4,5 г, 32%). m/z (esi) M+1=197,0.
[00298] Стадия B: NaH (2,7 г, 68,65 ммоль) добавляли порциями к раствору 6-хлор-5-метокси-2,3-дигидро-1H-инден-1-она (4,5 г, 22,88 ммоль) в DMF (40 мл) при температуре от 0°С до 5°С, и реакционную смесь перемешивали в течение 30 мин при этой же температуре. Бензилбис(2-хлорэтил)амин (6,76 г, 25,17 ммоль) затем добавляли порциями в раствор, и смесь перемешивали в течение 18 ч при комнатной температуре. Реакционную смесь экстрагировали этилацетатом (2 × 250 мл) и тщательно промывали холодной водой (400 мл). Объединенную органическую часть высушивали над безводным Na2SO4, концентрировали и очищали флэш-хроматографией на силикагеле (элюент: 40% этилацетат в гексане) с получением 1'-бензил-6-хлор-5-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-она (2,2 г, 27%). m/z (esi) M+1=356,0.
[00299] Стадия C: 1-Хлорэтилхлорформиат (2,72 мл, 24,78 ммоль) добавляли к перемешиваемому раствору 1'-бензил-6-хлор-5-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-она (2,2 г, 6,19 ммоль) в DCE (40 мл) и нагревали с обратным холодильником в течение 1 ч при 80°С. Затем DCE выпаривали, и в реакционную смесь добавляли метанол (40 мл). Реакционную смесь нагревали с обратным холодильником в течение 1 ч при 65°С. Растворитель выпаривали при пониженном давлении с получением 6-хлор-5-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-она в виде неочищенного продукта, который использовали непосредственно на следующей стадии без дальнейшей очистки (количество неочищенного продукта: 1,7 г). m/z (esi) M+1=266,3.
[00300] Стадия D: TEA (1,78 мл, 12,83 ммоль) добавляли к перемешиваемому раствору 6-хлор-5-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-она (1,7 г, 6,41 ммоль) в DCM (20 мл), пока pH реакционной смеси не становился основным. Boc-ангидрид (2,21 мл, 9,62 ммоль) добавляли в реакционную смесь и перемешивали в течение 16 ч. Реакционную смесь разбавляли водой (30 мл) и экстрагировали DCM (2 × 30 мл). Органическую часть высушивали над безводным Na2SO4, концентрировали и очищали флэш-хроматографией на силикагеле (элюент: 40% этилацетат в гексане) с получением трет-бутил-6-хлор-5-метокси-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,3 г, 55%). m/z (esi) M+1=366,0.
[00301] Стадия E: Этоксид титана (9,85 мл, 47,01 ммоль) добавляли к трет-бутил-6-хлор-5-метокси-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилату (1,3 г, 3,56 ммоль) и нагревали при 90°С в течение 5 мин, после чего добавляли (R)-2-метилпропан-2-сульфинамид (1,3 г, 10,68 ммоль). Реакционную смесь нагревали при 90°С в течение 18 ч. Реакционную смесь гасили водой (30 мл) и разбавляли этилацетатом (90 мл). Твердый остаток отфильтровывали, и органическую часть отделяли. Экстрагированную объединенную органическую часть промывали насыщенным солевым раствором (50 мл), сушили над безводным Na2SO4 и концентрировали в вакууме для получения неочищенной массы, которую очищали флэш-хроматографией на силикагеле (элюент: 40% этилацетата в гексане) с получением трет-бутил-(1Z)-6-хлор-5-метокси-1-{[(S)-2-метилпропан-2-сульфинил]имино}-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата в виде твердого вещества (800 мг, 48%). m/z (esi) M+1=469,3.
[00302] Стадия F: NaBH4 (388 мг, 10,28 ммоль) добавляли порциями к раствору трет-бутил-(1Z)-6-хлор-5-метокси-1-{[(S)-2-метилпропан-2-сульфинил]имино}-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (800 мг, 1,7 ммоль) в MeOH (9 мл) и THF (18 мл) при 0°С и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь гасили насыщенным раствором NH4Cl (20 мл) и экстрагировали этилацетатом (2 × 20 мл). Органическую часть высушивали над безводным Na2SO4, концентрировали и подвергали очистке препаративной ВЭЖХ. Сбор первого элюируемого пика давал трет-бутил-(1S)-6-хлор-5-метокси-1-{[(R)-2-метилпропан-2-сульфинил]амино}-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (60,3 мг, 7,51%) (m/z (esi) M+1= 471,5).
Промежуточный пример АF
трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-6-хлор-5-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00303] Трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-6-хлор-5-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (37,8 мг, 4,7%) получали в соответствии с промежуточным примером AE, собирая второй элюируемый пик на стадии F. (m/z (esi) M+1=471,5).
Промежуточный пример АG
трет-бутил-(S)-4-(((R)-трет-бутилсульфинил)амино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилат
[00304] Стадия A: н-BuLi (2M в гексане) (7,5 мл, 14,37 ммоль) добавляли по каплям к перемешиваемому раствору диизопропиламина («DIPA») (2,19 мл, 15,56 ммоль) в THF (10 мл) при -78°С и перемешивали при -78°С в течение 1 ч. 1-(Трет-бутил)-4-этилпиперидин-1,4-дикарбоксилат (3,09 г, 11,97 ммоль) в THF (15 мл) добавляли по каплям при -78°С и перемешивали при этой температуре в течение 1 ч. 2-хлор-5-(хлорметил)тиазол (2,0 г, 11,97 ммоль) в THF (10 мл) добавляли по каплям при -78°С и перемешивали в течение еще 1 ч при -78°С. Реакционную смесь гасили насыщенным раствором NH4Cl (30 мл) и экстрагировали EtOAc. Органическую часть высушивали над безводным Na2SO4, фильтровали и концентрировали, и неочищенный продукт очищали хроматографией на силикагеле (0-30% EtOAc/гексан) с получением 1-(трет-бутил) 4-этил-4-((2-хлортиазол-5-ил)метил)пиперидин-1,4-дикарбоксилата (1,0 г, выход 21%) в виде жидкости. m/z (esi) M+1=389,4.
[00305] Стадия B: н-BuLi (19,62 мл, 2M в гексане) добавляли по каплям к перемешиваемому раствору DIPA (5,78 мл, 41,23 ммоль) в THF (75 мл) при -78°С и перемешивали в течение 1 ч. Раствор 1-(трет-бутил)-4-этил-4-((2-хлортиазол-5-ил)метил)пиперидин-1,4-дикарбоксилата (10 г, 25,77 ммоль) в THF (75 мл) добавляли по каплям в реакционную смесь и перемешивали при -78°С в течение 1 ч. Реакционную смесь гасили насыщенным солевым раствором (30 мл), и органический слой экстрагировали EtOAc. Органический слой выпаривали при пониженном давлении, получая неочищенный продукт, который очищали колоночной хроматографией на силикагеле (0-40% EtOAc/гексан), с получением трет-бутил-2-хлор-4-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (3,3 г, выход 37%) в виде твердого вещества. m/z (esi) M+1=343,3.
[00306] Стадия C: Этоксид титана (IV) (4,32 мл, 20,46 ммоль) добавляли к перемешиваемому раствору трет-бутил-2-хлор-4-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (1,4 г, 4,09 ммоль) и (R)-(+)-2-метилпропан-2-сульфинамида (1,48 г, 12,28 ммоль) и перемешивали при 100°С в течение 5 ч. Реакционную смесь разбавляли водой (30 мл) и экстрагировали EtOAc. Органическую часть сушили над безводным Na2SO4, концентрировали с получением трет-бутил-(R, Z)-4-((трет-бутилсульфинил)имино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (1,1 г, выход 60%) в виде смолообразной жидкости. m/z (esi) M+1=445,8.
[00307] Стадия D: Борогидрид натрия (0,14 г, 3,70 ммоль) добавляли к перемешиваемому раствору трет-бутил-(R, Z)-4-((трет-бутилсульфинил)имино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (1,1 г, 2,47 ммоль) в THF (10 мл) при -50°С и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили льдом и концентрировали до сухого состояния. Неочищенное вещество разбавляли EtOAc и промывали водой и насыщенным солевым раствором. Органическую часть высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (0-1% MeOH/DCM). Сбор первого элюируемого пика давал трет-бутил-(S)-4-(((R)-трет-бутилсульфинил)амино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилат (400 мг, выход 36%) (m/z (esi) M+1=448,4).
Промежуточный пример АH
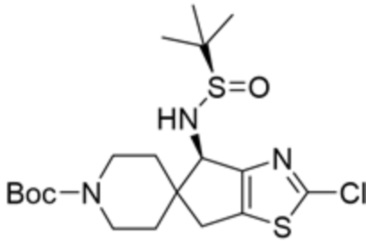
трет-бутил-(R)-4-(((R)-трет-бутилсульфинил)амино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилат
[00308] Трет-бутил-(R)-4-(((R)-трет-бутилсульфинил)амино)-2-хлор-4,6- дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилат (30 мг, выход 3%) получали в соответствии с промежуточным примером AG, собирая второй элюируемый пик на стадии D (m/z (esi) M+1=448,3).
Промежуточный пример АI
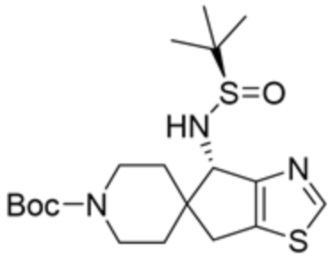
трет-бутил-(S)-4-(((R)-трет-бутилсульфинил)амино)-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилат
[00309] TEA (0,31 мл, 2,23 ммоль) и Pd/C (500 мг) добавляли к перемешиваемому раствору трет-бутил-(S)-4-(((R)-трет-бутилсульфинил)амино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (500 мг, 1,11 ммоль) в EtOH (20 мл) и перемешивали в течение 16 ч в атмосфере H2 (100 фунт/кв. дюйм (0,69 МПа)). Реакционную смесь фильтровали через слой Celite® и промывали MeOH (50 мл). Растворитель выпаривали при пониженном давлении, и неочищенный материал очищали колоночной хроматографией на силикагеле (0-4% MeOH-DCM) с получением трет-бутил-(S)-4-(((R)-трет-бутилсульфинил)амино)-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (220 мг, выход 47%) в виде твердого вещества. m/z (esi) M+1=419,4.
Промежуточный пример AJ
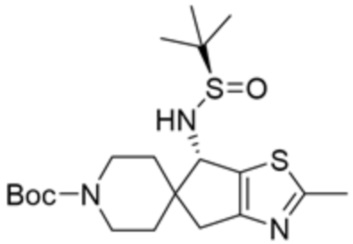
трет-бутил-(S)-6-метил-(((R)-трет бутилсульфинил]амино)-2-метил-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилат
[00310] Стадия A: Борогидрид натрия (6,91 г, 182,63 ммоль) добавляли порциями к раствору этил-2-хлортиазол-4-карбоксилата (10 г, 52,18 ммоль) в этаноле (150 мл) при комнатной температуре. Реакционной смеси давали перемешиваться при 50°С в течение 2 ч. Реакционную смесь гасили водным раствором NH4Cl, и EtOH удаляли при пониженном давлении. Остаток разбавляли водой (200 мл) и экстрагировали EtOAc. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении для получения неочищенной массы, которую очищали колоночной хроматографией на силикагеле (5-20% EtOAc/гексан), с получением (2-хлортиазол-4-ил)метанола (5,28 г, выход 68%) в виде жидкости. m/z (esi) M+1=150,1.
[00311] Стадия B: Et3N (36,5 мл, 267,38 ммоль) добавляли к раствору (2-хлортиазол-4-ил)метанола (10 г, 66,85 ммоль) в DCM (500 мл) и охлаждали до 0°С в атмосфере Ar. Метансульфонилхлорид (7,8 мл, 100,27 ммоль) медленно добавляли в реакционную смесь в течение 10 минут. Реакционную смесь оставляли перемешиваться при той же температуре в течение 30 мин. Реакцию гасили раствором NaHCO3, и органическую часть отделяли. Органическую часть промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением (2-хлортиазол-4-ил)метилметансульфоната (13,7 г, 90%, неочищенный), который использовали непосредственно на следующей стадии без дальнейшей очистки. m/z (esi) M+1=228,0.
[00312] Стадия C: Раствор DIPA (4 мл, 28,55 ммоль) в THF (12 мл) охлаждали до -78°С в атмосфере Ar. К нему медленно добавляли н-BuLi (2М в гексане) (13,2 мл). Реакционной смеси давали перемешиваться в течение 30 мин при -78°С и затем в течение 30 мин при комнатной температуре. Раствор LDA снова охлаждали до -78°C, и к нему медленно добавляли 1-трет-бутил-4-этилпиперидин-1,4-дикарбоксилат (6,215 г, 24,16 ммоль) в THF (6 мл). Реакционную смесь перемешивали в течение 1 ч при -78°С. Затем добавляли раствор (2-хлортиазол-4-ил)метилметансульфоната (5 г, 21,96 ммоль) в THF (12 мл), и реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь гасили насыщенным солевым раствором (100 мл) и экстрагировали EtOAc. Объединенные органические части сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле (5-15% EtOAc/гексан) с получением 1-(трет-бутил)-4-этил-4-((2-хлортиазол-4-ил)метил)пиперидин-1,4-дикарбоксилата (2,56 г, выход 30%) в виде жидкости. m/z (esi) M+1=388,9.
[00313] Стадия D: Раствор DIPA (4,7 мл, 33,43 ммоль) в THF (30 мл) охлаждали до -78°C в атмосфере Ar с последующим добавлением н-BuLi (2М в гексане) (16,1 мл) в течение 25 мин. Реакционной смеси давали перемешиваться в течение 30 мин при -78°С и затем при комнатной температуре в течение 10 мин. Раствор LDA медленно добавляли к 1-(трет-бутил)-4-этил-4-((2-хлортиазол-4-ил)метил)пиперидин-1,4-дикарбоксилату (5,0 г, 12,86 ммоль) в THF (40 мл) при -78°С в течение 30 мин. Реакционную смесь гасили водным раствором NH4Cl и экстрагировали EtOAc. Объединенные органические части сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (5-10% EtOAc/гексан) с получением трет-бутил-2-хлор-6-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (1,95 г, выход 44%) в виде твердого вещества. m/z (esi) M+1=343,4.
[00314] Стадия E: К раствору трет-бутил-2-хлор-6-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (500 мг, 1,46 ммоль) в диоксане (10 мл) добавляли K2CO3 (403 мг, 2,92 ммоль) и дегазировали Ar в течение 5 мин, после чего добавляли Pd(dppf)Cl2⋅DCM (357 мг, 0,44 ммоль) и триметилбороксин (0,8 мл, 5,83 ммоль). Реакционную смесь герметизировали и нагревали при 100°C в течение 2 ч. Реакционную смесь фильтровали через спеченную воронку и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (10-30% EtOAc/гексан) с получением трет-бутил-2-метил-6-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (330 мг, выход 70%) в виде твердого вещества. m/z (esi) M+1=323,1.
[00315] Стадия F: Этоксид титана (IV) (2,1 мл, 10,24 ммоль) добавляли к трет-бутил-2-метил-6-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилату (250 мг, 0,78 ммоль) и нагревали при 100°С в течение 5 мин, после чего добавляли (R)-2-метилпропан-2-сульфинамид (282 мг, 2,33 ммоль). Смесь нагревали при 100°С в течение 16 ч. Реакцию гасили насыщенным солевым раствором (50 мл) и экстрагировали EtOAc. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (10-40% EtOAc/гексан) с получением трет-бутил-(R, Z)-6-((трет-бутилсульфинил)имино)-2-метил-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (255 мг, выход 77%) в виде твердого вещества. m/z (esi) M+1=425,9.
[00316] Стадия G: Борогидрат натрия (251 мг, 6,63 ммоль) добавляли порциями к раствору трет-бутил-(R, Z)-6-((трет-бутилсульфинил)имино)-2-метил-4,6-дигидроспиро[циклопентa[d]тиазол-5,4’-пиперидин]-1’-карбоксилата (470 мг, 1,10 ммоль) в THF (10 мл) и метанола (5 мл) при 0°С. Реакционную смесь перемешивали в течение 30 мин при той же температуре. Реакционную смесь гасили водным раствором NH4Cl и экстрагировали EtOAc. Объединенные органические части сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали обращенно-фазовой препаративной ВЭЖХ (5-95% ACN:вода) с получением трет-бутил-(S)-6-метил-(((R)-трет-бутилсульфинил]амино)-2-метил-4,6-дигидроспиро[циклопентa[d]тиазол-5,4’-пиперидин]-1’-карбоксилата (120 мг, выход 25%). m/z (esi) M+1=428,2.
Промежуточный пример АK
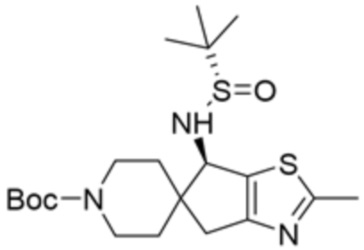
трет-бутил-(R)-6-(((S)-трет-бутилсульфинил)амино)-2-метил-4,6-дигидроспиро[циклопентa[d]тиазол-5,4’-пиперидин]-1’-карбоксилат
[00317] Стадия A: K2CO3 (403 мг, 2,92 ммоль) добавляли к раствору трет-бутил-2-хлор-6-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (500 мг, 1,46 ммоль) в диоксане (10 мл) и дегазировали Ar в течение 5 мин, после чего добавляли Pd(dppf)Cl2⋅CH2Cl2 (357 мг, 0,44 ммоль) и триметилбороксин (0,8 мл, 5,83 ммоль). Реакционную смесь герметизировали и нагревали при 100°C в течение 2 ч. Реакционную смесь фильтровали через спеченную воронку и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (10-30% EtOAc/гексан) с получением трет-бутил-2-метил-6-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (330 мг, выход 70%) в виде твердого вещества. m/z (esi) M+1=323,1.
[00318] Стадия B: Этоксид титана (IV) (5,84 мл, 25,8 ммоль) добавляли к трет-бутил-2-метил-6-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилату (630 мг, 1,95 ммоль) и нагревали при 100°С в течение 5 мин, после чего добавляли (S)-2-метилпропан-2-сульфинамид (711 мг, 5,86 ммоль). Смесь нагревали при 100°С в течение 6 ч. Реакцию гасили насыщенным солевым раствором. Твердое вещество фильтровали через спеченную воронку и экстрагировали EtOAc. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (10-40% EtOAc/гексан) с получением трет-бутил-(S, Z)-6-((трет-бутилсульфинил)имино)-2-метил-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (450 мг, выход 54%) в виде твердого вещества. m/z (esi) M+1=425,9.
[00319] Стадия C: Борогидрат натрия (240 мг, 6,35 ммоль) добавляли к раствору трет-бутил-(S, Z)-6-((трет-бутилсульфинил)имино)-2-метил-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (450 мг, 1,05 ммоль) в THF (9 мл) и метаноле (4,5 мл) при 0°С порциями. Реакционную смесь перемешивали в течение 1 ч при 0°С, гасили водным раствором NH4Cl и экстрагировали EtOAc. Объединенные органические части сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали обращенно-фазовой препаративной ВЭЖХ (5-95% ACN:вода, 20 мМ бикарбоната аммония, 16 мл/мин) с получением трет-бутил-(R)-6-(((S)-трет-бутилсульфинил)амино)-2-метил-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (46 мг, выход 10%). m/z (esi) M+1=428,2.
Промежуточный пример АL
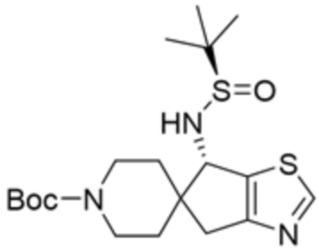
трет-бутил-(S)-6-(((R)-трет-бутилсульфинил)амино)-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилат
[00320] Стадия A: Этоксид титана (IV) (13,1 мл, 57,76 ммоль) добавляли к трет-бутил-2-хлор-6-оксо-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилату (1,5 г, 4,38 ммоль) и нагревали при 100°С в течение 5 мин, после чего добавляли (R)-2-метилпропан-2-сульфинамид (1,59 г, 13,13 ммоль). Смесь нагревали при 100°С в течение 16 ч. Реакцию гасили насыщенным солевым раствором (500 мл). Твердое вещество фильтровали через спеченную воронку и промывали EtOAc. Органическую часть отделяли и водную часть экстрагировали EtOAc. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (5-15% EtOAc/гексан) с получением трет-бутил-(R,Z)-6-((трет-бутилсульфинил)имино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (1050 мг, выход 54%) в виде твердого вещества. m/z (esi) M+1=446,1.
[00321] Стадия B: Борогидрид натрия (267 мг, 7,05 ммоль) добавляли порциями к раствору трет-бутил-(R,Z)-6-((трет-бутилсульфинил)имино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (500 мг, 1,18 ммоль) в THF (10 мл) и метанола (5 мл) в течение 1 ч при 0°С и перемешивали в течение еще 1 ч при 0°С. Реакционную смесь гасили водным раствором NH4Cl и экстрагировали EtOAc. Объединенную органическую часть сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали обращенно-фазовой препаративной ВЭЖХ (5-95% ACN:вода, 10 мМ бикарбоната аммония, 16 мл/мин) с получением трет-бутил-(S)-6-(((R)-трет-бутилсульфинил)амино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (115 мг, выход 22%) в виде твердого вещества. (m/z (esi) M-1=446,2).
[00322] Стадия C: Триэтиламин (0,2 мл, 1,43 ммоль) добавляли к раствору трет-бутил-(S)-6-(((R)-трет-бутилсульфинил)амино)-2-хлор-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (320 мг, 0,71 ммоль) в метаноле (40 мл) и дегазировали Ar в течение 10 мин, после чего добавляли Pd-C (10% загрузка, 50% масс.) (640 мг), при дегазации Ar. Через 5 мин реакционную смесь помещали в атмосферу водорода, под действием давления баллона. Через 18 ч реакционную смесь фильтровали через слой Celite® и концентрировали при пониженном давлении. Образующийся в результате остаток очищали колоночной хроматографией, используя аминосиликагель (10-30% EtOAc/гексан) с получением трет-бутил-(S)-6-(((R)-трет-бутилсульфинил)амино)-4,6-дигидроспиро[циклопентa[d]тиазол-5,4'-пиперидин]-1'-карбоксилата (185 мг, выход 63%) в виде твердого вещества. m/z (esi) M+1=414,4.
Промежуточный пример АM
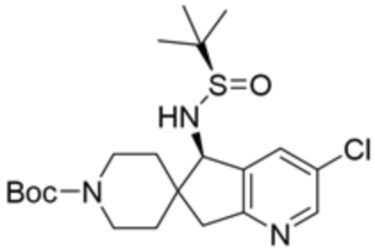
трет-бутил-(R)-5-(((R)-трет-бутилсульфинил)амино)-3-хлор-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат
[00323] Стадия A: H2SO4 (5,76 мл, 105,7 ммоль) добавляли к перемешиваемому раствору 3-бром-5-хлорпиколиновой кислоты (5 г, 21,1 ммоль) в MeOH (50 мл) при 0°С и смесь перемешивали при 90°С в течение 4 ч. Реакционную смесь охлаждали до 25°C и MeOH выпаривали досуха. Остаток нейтрализовали 2 н. раствором NaHCO3 и экстрагировали EtOAc. Объединенные слои промывали насыщенным солевым раствором и концентрировали при пониженном давлении с получением метил-3-бром-5-хлорпиколината (4,8 г, выход 91%) в виде твердого вещества. m/z (esi) M+1=252,0.
[00324] Стадия B: Борогидрид натрия (4,37 г, 115,7 ммоль) добавляли небольшими порциями к охлажденному (0°С) раствору метил-3-бром-5-хлорпиколината (4,8 г, 19,3 ммоль) в метаноле (70 мл) в течение приблизительно 30 мин. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Затем реакционную смесь разбавляли насыщенным солевым раствором, и MeOH выпаривали при пониженном давлении. Остаток экстрагировали EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и выпаривали с получением (3-бром-5-хлорпиридин-2-ил)метанола (3,72 г, выход 88%) в виде полутвердого вещества. m/z (esi) M+1=224,0.
[00325] Стадия C: MsCl (1,42 мл, 18,4 ммоль) добавляли по каплям к раствору (при -15°C) (3-бром-5-хлорпиридин-2-ил)метанола (3,72 г, 16,7 ммоль) и триэтиламина (4,66 мл, 33,4 ммоль) в DCM (50 мл). Полученную смесь оставляли перемешиваться при той же температуре в течение 2 ч. Реакционную смесь гасили водой и разбавляли EtOAc, и водный слой отделяли. Органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (15-20% EtOAc/гексан) с получением (3-бром-5-хлорпиридин-2-ил)метилметансульфоната (3,8 г, выход 76%) в виде полутвердого вещества. m/z (esi) M+1=302,0.
[00326] Стадия D: н-BuLi (1,72 M раствор в THF/гексане, 6,1 мл, 10,5 ммоль) добавляли по каплям к раствору (-70°C) DIPA (1,69 мл, 11,8 ммоль) в THF (10 мл). Полученной смеси давали нагреться до -40°C в течение 1 ч. Раствор 1-(трет-бутил)-4-этилпиперидин-1,4-дикарбоксилата (1,8 г, 6,9 ммоль) в THF (20 мл) добавляли к LDA при -70°С и перемешивали при -70°С в течение 1 ч. Затем по каплям добавляли раствор (3-бром-5-хлорпиридин-2-ил)метилметансульфоната (2,52 г, 8,4 ммоль) в THF (10 мл), и смесь перемешивали при -70°С в течение 1 ч. Реакцию гасили водой. Водный слой отделяли и экстрагировали EtOAc. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (30-40% EtOAc/гексан) с получением 1-(трет-бутил)-4-этил-4-((3-бром-5-хлорпиридин-2-ил)метил)пиперидин-1,4-дикарбоксилата (2,35 г, выход 73%) в виде смолы. m/z (esi) M+1=462,9.
[00327] Стадия E: NaOH (1,95 мг, 48,8 ммоль) добавляли к перемешиваемому раствору 1-(трет-бутил)-4-этил-4-((3-бром-5-хлорпиридин-2-ил)метил)пиперидин-1,4-дикарбоксилата (4,5 г, 9,7 ммоль) в MeOH (50 мл) и воде (10 мл) при 25°С и перемешивали при 65°С в течение 16 ч. Смесь концентрировали досуха, и неочищенный продукт растворяли водой и промывали Et2O. Водный слой отделяли и поддерживали при рН 7-6 с помощью 2 н. HCl. Соединение экстрагировали EtOAc. Органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 4-((3-бром-5-хлорпиридин-2-ил)метил)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоты (4,1 г, выход 97%) в виде твердого вещества. m/z (esi) M+1=435,3.
[00328] Стадия F: NaH (60% дисперсия в минеральном масле, 456 мг, 11,4 ммоль) добавляли порциями в раствор (с температурой -15°C) 4-((3-бром-5-хлорпиридин-2-ил)метил)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоты (4,1 г, 9,5 ммоль) в THF (23 мл) в атмосфере N2. После перемешивания в течение 15 мин при данной температуре смесь охлаждали до -60°C. н-BuLi (1,72 M раствор в гексане, 7,66 мл, 13,2 ммоль) добавляли по каплям к смеси, перемешивали в течение 30 мин, и затем температуру повышали до -20°C в течение 30 мин. Реакционную смесь гасили водой и экстрагировали EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный материал очищали колоночной хроматографией на силикагеле (30% EtOAc/гексан) с получением трет-бутил-3-хлор-5-оксо-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (1,1 г, выход 34%) в виде полутвердого вещества. m/z (esi) M+1=337,3.
[00329] Стадия G: Этоксид титана (IV) (4,11 мл, 19,6 ммоль) добавляли к перемешиваемому раствору трет-бутил-3-хлор-5-оксо-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (1,1 г, 3,3 ммоль) и перемешивали в течение 5 мин при 95°С. Добавляли (R)-(+)-2-метилпропан-2-сульфинамид (1,18 г, 9,8 ммоль) и перемешивали при 95°С в течение 5 ч. Смеси давали остыть при комнатной температуре и разбавляли EtOAc и водой. Полученную смесь энергично перемешивали в течение 15 мин при комнатной температуре и затем фильтровали через слой Celite®. Фильтрат экстрагировали EtOAc и промывали насыщенным солевым раствором. Органическую фазу высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный материал очищали колоночной хроматографией на силикагеле (30% EtOAc/гексан) с получением трет-бутил-(R, Z)-5-((трет-бутилсульфинил)имино)-3-хлор-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (911 мг, выход 63%) в виде полутвердого вещества. m/z (esi) M+1=440,2.
[00330] Стадия H: Борогидрид натрия (391 мг, 10,3 ммоль) добавляли к перемешиваемому раствору трет-бутил-(R, Z)-5-((трет-бутилсульфинил)имино)-3-хлор-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (910 мг, 2,1 ммоль) в MeOH (28,0 мл) при 0°С и перемешивали в течение 3 ч. Реакционную смесь оставляли нагреваться до комнатной температуры, насыщенный водный раствор NH4Cl медленно добавляли для гашения избытка борогидрида натрия, и MeOH выпаривали. Остаток разбавляли EtOAc и водой. Органические слои разделяли. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали с помощью нормально-фазовой препаративной ВЭЖХ (Chiralpak IG (21,0×250 мм), 5μ, гексан/этанол: 85/15) с получением трет-бутил-(R)-5-(((R)-трет-бутилсульфинил)амино)-3-хлор-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилата (580 мг, выход 50%) (m/z (esi) M+1=442,1) в виде твердого вещества.
Промежуточный пример AN
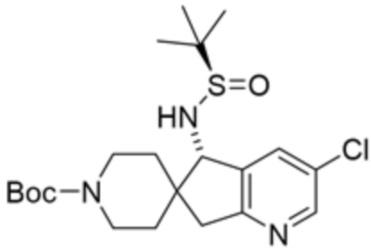
трет-бутил-(S)-5-(((R)-трет-бутилсульфинил)амино)-3-хлор-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат
[00331] Трет-бутил-(S)-5-(((R)-трет-бутилсульфинил)амино)-3-хлор-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат получали в соответствии с промежуточным примером AM, выделяя материал на стадии G, и очищали с помощью нормально-фазовой препаративной ВЭЖХ (Chiralpak IG (21,0×250 мм), 5μ, гексан/этанол: 85/15) с получением чистого материала в виде клейкого твердого вещества (117 мг, выход 11%, m/z (esi) M+1=442,2).
Промежуточный пример АO
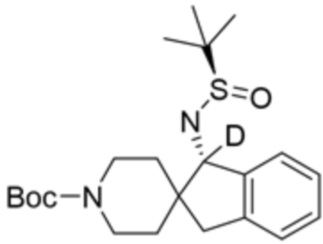
трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4’-пиперидин]-1’-карбоксилат-1-d
[00332] Стадия A: NaH (60% в минеральном масле) (2,72 г, 68,08 ммоль) добавляли к перемешиваемому раствору 2,3-дигидро-1H-инден-1-она (3,0 г, 22,69 ммоль) в DMF (60 мл) при 0°С. Реакционную смесь перемешивали в течение 30 мин при 5°С. Гидрохлорид N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амина (5,76 г, 24,96 ммоль) добавляли порциями, и смесь перемешивали при 60°С в течение 16 ч. Реакцию гасили насыщенным солевым раствором, экстрагировали EtOAc. Органические слои объединяли и промывали избытком воды, затем насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (35-40% EtOAc/гексан) с получением 1'-бензилспиро[инден-2,4'-пиперидин]-1(3H)-она (2,7 г, выход 41%) в виде масла. m/z (esi) M+1=292,5.
[00333] Стадия B: 1-Хлорэтилхлорформиат (3,07 мл, 28,44 ммоль) добавляли к перемешиваемому раствору 1'-бензилспиро[инден-2,4'-пиперидин]-1(3H)-она (2,76 г, 9,48 ммоль) в DCE (45 мл) и нагревали с обратным холодильником в течение 1 ч. DCE выпаривали досуха и добавляли метанол (45 мл). Реакционную смесь нагревали с обратным холодильником еще в течение 1 ч. Реакционную смесь выпаривали досуха с получением спиро[инден-2,4'-пиперидин]-1(3H)-она (1,91 г, неочищенный продукт) в виде полутвердого вещества. m/z (esi) M+1=202,3.
[00334] Стадия C: Триэтиламин (5,28 мл, 37,95 ммоль) добавляли к перемешиваемому раствору спиро[инден-2,4'-пиперидин]-1(3H)-она (1,91 г, 9,49 ммоль) в DCM (45 мл), после чего добавляли boc-ангидрид (3,27 мл, 14,23 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали и полученный остаток очищали колоночной хроматографией на силикагеле (15-17% EtOAc/гексан) с получением трет-бутил-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,47 г, 2-стадийный выход 52%) в виде твердого вещества. m/z (esi) M+1=ЖХМС: 302,1.
[00335] Стадия D: Этоксид титана (IV) (2,29 мл, 10,93 ммоль) и (R)-(+)-2-метил-2-пропансульфинамид (662,40 мг, 5,46 ммоль) добавляли к перемешиваемому раствору трет-бутил-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (823 мг, 2,73 ммоль) в THF (5 мл), и реакционную смесь нагревали при 90°С в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, и концентрировали с получением неочищенного продукта. Неочищенный продукт смешивали с другой партией или трет-бутил-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилатом (200 мг) и очищали колоночной хроматографией на силикагеле (20-25% EtOAc/гексан), с получением трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-6-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (545 мг, выход 42%). m/z (esi) M+1=405,0.
[00336] Стадия E: NaBD4 (46,62 мг, 1,11 ммоль) добавляли к перемешиваемому раствору трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-6-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (300 мг, 0,74 ммоль) в THF (12 мл) при -50°С, и температуре позволяли подняться до комнатной температуры в течение 16 ч. Реакцию гасили насыщенным раствором NH4Cl и экстрагировали EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью СФХ [Chiralpak IG (250×21 мм) 5μ, 25 г/мин. Подвижная фаза: 60% CO2+40% метанол. ABPR: 100 бар (10 МПа). Сбор первого элюируемого пика давал трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат-1-d (152 мг, выход 51%). m/z (esi) M+1=408,4.
Промежуточный пример АP
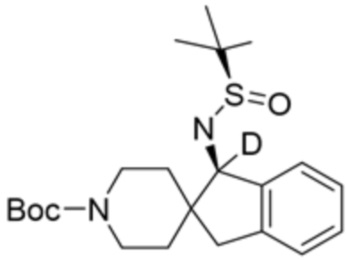
трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат-1-d
[00337] Трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат-1-d получали в соответствии с промежуточным примером AО, собирая второй элюируемый пик на стадии D (34 мг, выход 11%) m/z (esi) M+1=408,4.
Промежуточный пример АQ
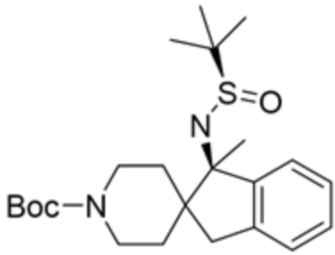
Трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-1-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00338] MeMgBr (3M в простом эфире) (0,4 мл, 1,23 ммоль) добавляли к перемешиваемому раствору трет-бутил-(R, Z)-1-((трет-бутилсульфинил)имино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (100 мг, 0,247 ммоль) в THF (0,5 мл) при 0°С и перемешивали при этой температуре в течение 3 ч. Реакцию гасили насыщенным раствором NH4Cl и экстрагировали EtOAc. Органические слои объединяли, промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт смешивали с другой партией трет-бутил-(R, Z)-1-((трет-бутилсульфинил)имино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (120 мг) и очищали с помощью нормально-фазовой препаративной ВЭЖХ (Chiralpak IG (21,0×250 мм), 5μ, гексан/EtOH/iPrNH2 80/20/0,1, 1,0 мл/мин)). Сбор второго элюируемого пика давал клейкое твердое вещество (76 мг, выход 33%). m/z (esi) M+1=421,4.
Промежуточный пример AR
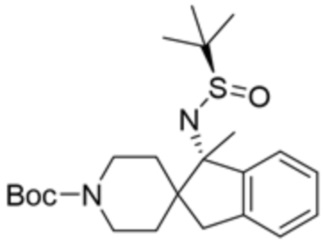
трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-1-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00339] Трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-1-метил-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат получали в соответствии с промежуточным примером AQ, собирая первый элюируемый пик. m/z (esi) M+1=421,4.
Промежуточный пример AS
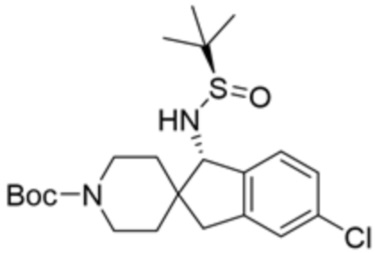
трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00340] Стадия A: NaH (60% дисперсия в минеральном масле, 500 мг, 12,05 ммоль) добавляли к перемешиваемому раствору 5-хлор-2,3-дигидро-1H-инден-1-она (1 г, 6,02 ммоль) в DMF (10 мл) при 0°С и перемешивали в течение 30 мин. Гидрохлорид N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амина (1,5 г, 6,63 ммоль) добавляли в реакционную смесь при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили водой и экстрагировали EtOAc. Органический слой концентрировали досуха, и неочищенный материал очищали колоночной хроматографией на силикагеле (30% EtOAc/гексан) с получением 1'-бензил-5-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (300 мг, выход 15%) в виде твердого вещества. m/z (esi) M+1=325,8.
[00341] Стадия B: Хлорэтилхлорформиат (270 мг, 1,84 ммоль) добавляли к перемешиваемому раствору 1'-бензил-5-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (300 мг, 0,92 ммоль) в DCE (10 мл) при 0°С и перемешивали в течение 10 мин. Реакционную смесь перемешивали при 80°С в течение 1 ч. Реакционную смесь концентрировали досуха. Неочищенный продукт растворяли в MeOH (10 мл) и снова перемешивали при температуре 75°C в течение еще 1 ч. Реакционную смесь концентрировали при пониженном давлении с получением 5-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она в виде клейкого твердого вещества (250 мг), которое непосредственно использовали на следующей стадии без дополнительной очистки. m/z (esi) M+1=236,1.
[00342] Стадия C: Триэтиламин (0,3 мл, 2,13 ммоль) добавляли к перемешиваемому раствору 5-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (250 мг, 1,06 ммоль) в DCM (10 мл) при 0°С. Boc-ангидрид (0,5 мл, 2,13 ммоль) добавляли в раствор при 0°С, и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (10% EtOAc/гексан) с получением трет-бутил-5-хлор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (130 мг, выход 30%, 2 стадии) в виде твердого вещества. m/z (esi) M+1=335,3.
[00343] Стадия D: Трет-бутил-5-хлор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (700 мг, 2,09 ммоль) и (R)-(+)-2-метилпропан-2-сульфинамид (760,0 мг, 6,26 ммоль) добавляли в теплый (100°С) этоксид титана (IV) (1,43 г, 6,26 ммоль) и перемешивали при 100°С в течение 16 ч. Реакционную смесь выливали в EtOAc и насыщенный солевой раствор. Смесь перемешивали в течение 15 мин, и осажденное твердое вещество отфильтровывали. Жидкую часть отделяли. Органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (1% MeOH/DCM) с получением трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (600 мг, выход 41%) в виде твердого вещества. m/z (esi) M+1=439,2.
[00344] Стадия E: Борогидрид натрия (217 мг, 5,70 ммоль) добавляли в охлажденный льдом раствор трет-бутил-(R, E)-1-((трет-бутилсульфинил)имино)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (500 мг, 1,14 ммоль) в MeOH (15 мл) и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь гасили ледяной водой и экстрагировали EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (30% EtOAc/гексан) с получением трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (195 мг, выход 39%) (m/z (esi) M-1=439,2) в виде твердого вещества.
Промежуточный пример AT
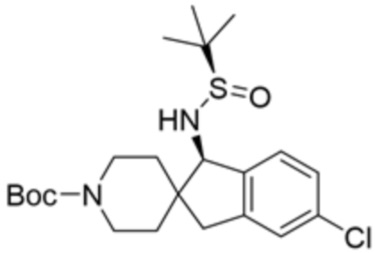
трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00345] Трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат получали в соответствии с промежуточным примером AS, собирая второй элюируемый пик на стадии Е (255 мг, выход 23%) (m/z (esi) M+1=441,4).
Промежуточный пример AU
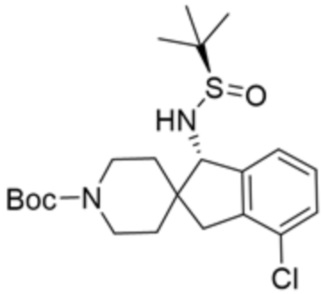
трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00346] Стадия A: NaH (60% дисперсия в минеральном масле, 2,16 г, 90,0 ммоль) добавляли порциями к перемешиваемому раствору 4-хлор-2,3-дигидро-1H-инден-1-она (5,0 г, 30,0 ммоль) в DMF (10 мл) в условиях охлаждения льдом и перемешивали в течение 30 мин. Гидрохлорид N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амина добавляли медленно в реакционную смесь и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили водой, и органический слой экстрагировали EtOAc. Объединенные органические слои промывали холодной водой и насыщенным солевым раствором, сушили над безводным Na2SO4 и фильтровали. Органический слой выпаривали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле (10% EtOAc/гексан) с получением 1'-бензил-4-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (2,9 г, выход 30%) в виде смолистого материала. m/z (esi) M+1=326,0.
[00347] Стадия B: Хлорэтилхлорформиат (2,78 мл, 25,84 ммоль) добавляли к перемешиваемому раствору 1'-бензил-4-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (2,8 г, 8,61 ммоль) в DCE (10 мл), и реакционную смесь нагревали с обратным холодильником в течение 2 ч. Растворитель выпаривали досуха и реакционную смесь растворяли в MeOH (5 мл) и далее нагревали при 80°С в течение 1 ч. Реакционную смесь выпаривали досуха с получением неочищенной реакционной смеси гидрохлорида 4-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (3,2 г, неочищенный продукт), которую использовали на следующей стадии без дополнительной очистки. m/z (esi) M+1= 235,7.
[00348] Стадия C: Неочищенный гидрохлорид 4-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (3,2 г, 11,76 ммоль) растворяли в DCM (5 мл) в условиях охлаждения льдом, и по каплям добавляли триэтиламин (6,68 мл, 47,83 ммоль). Boc-ангидрид (2,63 мл, 11,47 ммоль) добавляли к реакционной смеси и продолжали перемешивать в течение 16 ч при комнатной температуре. Воду добавляли к реакционной смеси и ее экстрагировали DCM. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и выпаривали при пониженном давлении для получения неочищенного продукта, который очищали колоночной хроматографией на силикагеле (5% EtOAc/гексан), с получением трет-бутил-4-хлор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,6 г, 50% выход, 2 стадии) в виде твердого вещества. m/z (esi) M+1= 336,2.
[00349] Стадия D: R-(+)-2-Метилпропан-2-сульфинамид (705 мг, 5,82 ммоль) добавляли к раствору трет-бутил-4-хлор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,5 г, 4,47 ммоль) в теплый (100°С) этоксид титана (IV) (4,0 мл,19,08 ммоль), и полученную смесь нагревали при 100°С в течение 16 ч. Реакционную смесь перемешивали с EtOAc (15 мл) и H2O (15 мл) в течение 20 мин и фильтровали через слой Celite®. Органический слой отделяли, и водную часть экстрагировали EtOAc. Объединенные органические слои выпаривали при пониженном давлении, и неочищенную реакционную смесь очищали колоночной хроматографией на силикагеле (20% EtOAc/гексан) с получением трет-бутил-(S, E)-1-((трет-бутилсульфинил)имино)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (870 мг, выход 44%) в виде твердого вещества. m/z (esi) M+1=438,8.
[00350] Стадия E: Борогидрид натрия (368 мг, 9,7 ммоль) добавляли порциями к перемешиваемому раствору трет-бутил-(S, E)-1-((трет-бутилсульфинил)имино)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (850 мг, 1,94 ммоль) в MeOH (5 мл) при 0°С и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь выпаривали досуха, и к реакционной смеси добавляли воду. Смесь экстрагировали EtOAc. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хиральной препаративной ВЭЖХ (Chiralpak IC 4,6×250 мм, 5μ, DCM/EtOH/iPrNH2 50/50/0,1, 1,0 мл/мин) с получением трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата, (231 мг, выход 27%; (m/z (esi) M+1=441,2) в виде клейкого твердого вещества.
Промежуточный пример AV
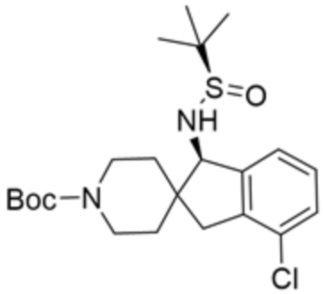
трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат
[00351] Трет-бутил-(R)-1-(((R)-трет-бутилсульфинил)амино)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат получали в соответствии с промежуточным примером AU, собирая второй элюируемый пик на стадии Е в виде клейкого твердого вещества (295 мг, выход 34%) (m/z (esi) M+1=441,2).
Промежуточный пример AW
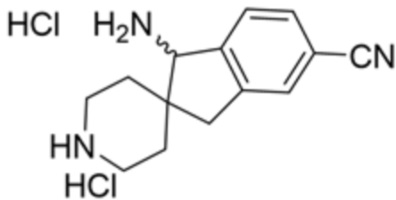
дигидрохлорид 1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-5-карбонитрила
[00352] Стадия A: NaH (60% масс. в парафине) (3,61 г, 90,36 ммоль) добавляли к перемешиваемому раствору 5-хлор-2,3-дигидро-1H-инден-1-она (5,0 г, 30,12 ммоль) в DMF (50 мл) при 0°С и перемешивали при комнатной температуре в течение 30 мин. N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амин (8,89 г, 33,13 ммоль) добавляли порциями при 0°С и перемешивали при комнатной температуре в течение еще 5 ч. Реакционную смесь гасили ледяной водой и экстрагировали этилацетатом. Органическую часть высушивали (Na2SO4), фильтровали, концентрировали и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (25% EtOAc-гексан) с получением 1'-бензил-5-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (2 г, выход 20%) в виде жидкости. m/z (esi) M+1=325,9.
[00353] Стадия B: 1-Хлорэтилхлорформиат (3,49 г, 24,61 ммоль) добавляли к перемешиваемому раствору 1'-бензил-5-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (2,0 г, 6,15 ммоль) в DCE (20 мл) при 0°С и перемешивали в течение 10 мин. Реакционную смесь перемешивали при 80°С в течение 16 ч. Реакционную смесь концентрировали и неочищенный материал растворяли в MeOH (20 мл) и перемешивали при 80°C в течение 1 ч. Реакционную смесь концентрировали с получением гидрохлорида 5-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (1,45 г, неочищенный продукт) в виде смолистой жидкости, который использовали на следующей стадии без дополнительной очистки. m/z (esi) M+1=236,1.
[00354] Стадия C: Триэтиламин (3,43 мл, 24,68 ммоль) и boc-ангидрид (2,12 мл, 9,25 ммоль) добавляли к перемешиваемому раствору гидрохлорида 5-хлорспиро[инден-2,4'-пиперидин]-1(3H)-она (1,45 г, 6,17 ммоль) в DCM (15 мл) при 0°С, и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь концентрировали и очищали колоночной хроматографией на силикагеле (30% EtOAc-гексан) с получением трет-бутил-5-хлор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (400 мг, выход 19%, 2 стадии) в виде твердого вещества. m/z (esi) M+1=336,3.
[00355] Стадия D: Zn(CN)2 (982,0 мг, 8,35 ммоль) и цинковый порошок (55,0 мг, 0,83 ммоль) добавляли к перемешиваемому раствору трет-бутил-5-хлор-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,4 г, 4,14 ммоль) в DMF (10 мл) и перемешивали в течение 10 мин. Реакционную смесь дегазировали аргоном, затем добавляли TrixiePhos (383,0 мг, 0,41 ммоль), после чего Pd(OAc)2 (232,0 мг, 0,41 ммоль), и реакционную смесь перемешивали при 120°C в течение 16 ч. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органическую часть высушивали (Na2SO4), фильтровали и концентрировали, и неочищенный материал очищали с помощью колоночной хроматографии на силикагеле (30% EtOAc-гексан) с получением трет-бутил-5-цианo-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (250 мг, выход 18%) в виде клейкого твердого вещества. m/z (esi) M+1=326,3.
[00356] Стадия E: (R)-2-Метилпропан-2-сульфинамид (204,5 мг, 1,68 ммоль) добавляли к перемешиваемому раствору трет-бутил-5-цианo-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (500 мг, 1,53 ммоль) в этоксиде титана (IV) (1,62 мл, 7,66 ммоль) и перемешивали при 90°С в течение 1 ч. Реакционную смесь выливали в EtOAc и насыщенный солевой раствор и перемешивали в течение 15 мин. Твердый осадок отфильтровывали. Органический слой промывали насыщенным солевым раствором, сушили (Na2SO4) и концентрировали с получением трет-бутил-1-(((R)-трет-бутилсульфинил)амино)-5-цианo-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (600 мг, неочищенный) в виде смолистой жидкости, который использовали на следующей стадии без дополнительной очистки. m/z (esi) M+1=430,3.
[00357] Стадия F: NaBH4 (105,8 мг, 2,79 ммоль) добавляли к перемешиваемому раствору трет-бутил-1-(((R)-трет-бутилсульфинил)амино)-5-цианo-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (600,0 мг, 1,39 ммоль) в MeOH (10 мл) при -10°С и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали, разбавляли этилацетатом и промывали водой. Органический слой сушили (Na2SO4), фильтровали, концентрировали и очищали колоночной хроматографией на силикагеле (40% EtOAc-гексан) с получением трет-бутил-1-(((R)-трет-бутилсульфинил)амино)-5-цианo-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (430 мг, выход 71%) в виде твердого вещества. m/z (esi) M+1=432,1.
[00358] Стадия G: 4M HCl в диоксане (3 мл) добавляли к перемешиваемому раствору трет-бутил-1-(((R)-трет-бутилсульфинил)амино)-5-цианo-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (430,0 мг, 0,99 ммоль) в MeOH (3 мл) при 0°С и перемешивали при 0°С в течение 2 ч. Реакционную смесь концентрировали и неочищенный материал растирали с простым диэтиловым эфиром с получением дигидрохлорида 3-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-6-карбонитрила (250 мг, выход 84%) в виде твердого вещества. m/z (esi) M+1=228,4.
Промежуточный пример AX
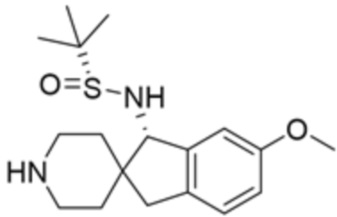
(R)-N-((S)-5-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-3-ил)-2-метилпропан-2-сульфинамид-2,2,2-трифторацетат
[00359] Стадия A: Гидрид натрия, 60% дисперсия в минеральном масле (0,74 г, 18 ммоль) добавляли порциями к раствору 6-метокси-1-инданона (1г, 6,2 ммоль) в DMF (6,9 мл, 6,2 ммоль) в атмосфере аргона. Смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амин (1,6 г, 6,8 ммоль), и смесь продолжали перемешивать в течение ночи. Реакционную смесь выливали в воду и распределяли с EtOAc. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (0-10% DCM:MeOH (2% NH4OH)) с получением 1'-бензил-6-метоксиспиро[инден-2,4'-пиперидин]-1(3H)-она (1,6 г, 5 ммоль, выход 81%). m/z (esi) M+1=322,2.
[00360] Стадия B: Раствор 1'-бензил-6-метоксиспиро[инден-2,4'-пиперидин]-1(3H)-она (1,6 г, 5 ммоль) и ди-трет-бутилдикарбоната (1,2 г, 5,5 ммоль) в EtOH (25 мл, 5 ммоль) и THF (25 мл, 5 ммоль) продували N2 в течение 5 мин. Пaллaдий (тип Degussa, 10% масс., 50% H2O) (1,3 г, 1,2 ммоль) добавляли в данный раствор и сразу же закрывали крышкой и продували N2 в течение дополнительных 5 мин. Затем раствор перемешивали под давлением 1 атм (0,1 МПа) H2. Смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь разбавляли MeOH и фильтровали через упакованный Celite®. Фильтрат концентрировали под вакуумом с получением неочищенного трет-бутил-6-метокси-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,12 г, 3,38 ммоль, выход 68%). m/z (esi) M+1=232,2.
[00361] Стадия C: (R)-(+)-2-Метил-2-пропансульфинамид (1,2 г, 10,14 ммоль) и тетраэтоксититан (5,4 г, 23,66 ммоль) добавляли к раствору трет-бутил-6-метокси-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (1,12 г, 3,34 ммоль) в THF (16,9 мл, 3,34 ммоль), и реакцию перемешивали в течение ночи при 90°С. Добавляли EtOAc, после чего воду. Твердые вещества отфильтровывали, и слои разделяли. Органический слой высушивали, фильтровали и концентрировали. Полученный остаток очищали с помощью нормально-фазовой хроматографии (0-100% гексаны:EtOAc) с получением трет-бутил-(R, Z)-1-((трет-бутилсульфинил)имино)-6-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (0,79 г, 1,83 ммоль, выход 54%). m/z (esi) M+1=435,2.
[00362] Стадия D: Трет-бутил-(R, Z)-1-((трет-бутилсульфинил)имино)-6-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (0,79 г, 1,83 ммоль) помещали в THF (15 мл) и охлаждали до 0°С. Добавляли NaBH4 (0,1 г, 2,74 ммоль), и реакционную смесь оставляли медленно нагреваться до комнатной температуры и перемешивали в течение 18 ч. Добавляли воду и смесь экстрагировали DCM (3×25 мл). Экстракты объединяли и концентрировали. Полученный остаток очищали на силикагеле (0-5% MeOH в DCM с 2% NH4OH). Первый элюируемый пик собирали с получением трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-6-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (0,343 г, 0,79 ммоль, выход 43%). m/z (esi) M+1=437,3.
[00363] Стадия E: TFA (303 мкл, 3,93) добавляли к раствору трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-6-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилата (343 мг, 0,79 ммоль) в DCM (1,57 мл, 0,79 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали под вакуумом и использовали далее неочищенный (R)-N-((S)-5-метокси-1,3-дигидроспиро[инден-2,4'-пиперидин]-3-ил)-2-метилпропан-2-сульфинамид-2,2,2-трифторацетат (354 мг, 0,79 ммоль, выход 100%). m/z (esi) M+1=337,2.
Пример 1
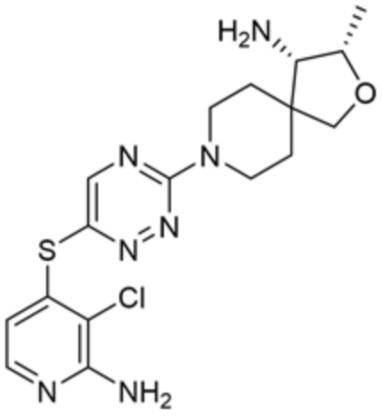
(3 S ,4 S )-8-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
[00364] Стадия A: Дигидрохлорид (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (320 мг, 1,3 ммоль) разбавляли диоксаном (6 мл), после чего добавляли DIEA (815 мкл, 4,67 ммоль) и 3,6-дихлор-1,2,4-триазин (200 мг, 1,33 ммоль). Реакционную смесь нагревали до 50°С и перемешивали в течение 3 ч. Реакционной смеси давали возможность остыть и разбавляли DCM/IPA и 10% карбонатом натрия. Слои разделяли и водный слой дважды экстрагировали DCM/IPA. Органические фазы объединяли, сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 1-10% метанол/DCM (1% NH4OH) с получением (3S,4S)-8-(6-хлор-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (290 мг, 1,02 ммоль, выход 76,6%).
[00365] (3S,4S)-8-(6-хлор-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (30 мг, 0,11 ммоль) и 2-амино-3-хлорпиридин-4-тиол (20 мг, 0,13 ммоль) разбавляли диоксаном, после чего добавляли DIEA (55 мкл, 0,32 ммоль). Реакционную смесь помещали в атмосферу азота и нагревали до 100°С. После перемешивания в течение 4 ч реакционную смесь разбавляли DCM/IPA и 10% карбонатом натрия. Материал экстрагировали еще два раза DCM/IPA. Органические фазы объединяли, сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 10% метанол/DCM (1% NH4OH). Материал снова очищали на C-18 силикагеле, элюируя 5-95% ACN/вода (0,1% TFA). Чистые фракции разбавляли DCM/IPA и насыщенным бикарбонатом натрия. Слои разделяли и органические слои сушили над MgSO4, фильтровали и концентрировали с получением (3S,4S)-8-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (6 мг, 0,015 ммоль, выход 14%). 1H ЯМР (400 МГц, CDCl3) δ 8,2 (с, 1H), 7,75 (д, 1H, J=5,5 Гц), 6,12 (д, 1H, 5,5 Гц) 4,9 (ушир., 2H), 4,18-4,40 (м, 3H), 3,84 (д, 1H, J=8,6 Гц), 3,6-3,8 (м, 3H), 3,02 (д, 1H, J=4,7 Гц), 1,4-1,95 (м, 6H); m/z (esi/APCI) M+1=408,2.
Пример 2
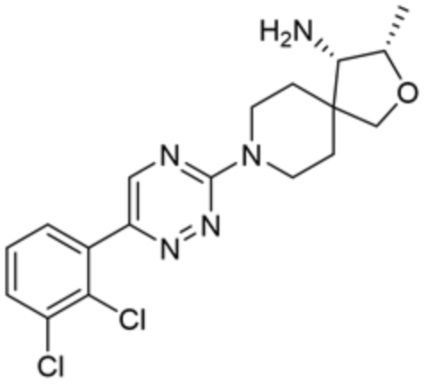
(3 S ,4 S )-8-(6-(2,3-дихлорфенил)-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
[00366] Стадия A: Дигидрохлорид (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (320 мг, 1,3 ммоль) разбавляли диоксаном (6 мл), после чего добавляли DIEA (815 мкл, 4,67 ммоль) и 3,6-дихлор-1,2,4-триазин (200 мг, 1,3 ммоль). Реакционную смесь нагревали до 50°С и перемешивали в течение 3 ч. Реакционной смеси давали возможность остыть и разбавляли DCM/IPA и 10% карбонатом натрия. Слои разделяли и водный слой дважды экстрагировали DCM/IPA. Органические фазы объединяли, сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 1-10% метанол/DCM (1% NH4OH) с получением (3S,4S)-8-(6-хлор-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (290 мг, 1,0 ммоль, выход 76,6%).
[00367] Стадия B: (3S,4S)-8-(6-хлор-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (30 мг, 0,11 ммоль), (2,3-дихлорфенил)бороновую кислоту (30 мг, 0,16 ммоль) и тетракис (3,7 мг, 0,0032 ммоль) разбавляли диоксаном (1,0 мл), после чего добавляли Na2CO3 (132 мкл, 0,26 ммоль). Реакционную смесь продували аргоном, герметизировали и нагревали до 95°C в течение 4 ч. Реакционной смеси давали возможность остыть, разбавляли этилацетатом и водой. Слои разделяли и этилацетат сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 10% метанол/DCM (1% NH4OH) с получением (3S,4S)-8-(6-(2,3-дихлорфенил)-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (10 мг, 0,025 ммоль, выход 24%). 1H ЯМР (400 МГц, CDCl3) δ 8,51 (с, 1H), 7,61 (дд, 1H, J=7,83, 1,57 Гц), 7,54 (дд, 1H, J=7,83, 1,57 Гц), 7,33 (т, 1H), 4,18-4,40 (м, 3H), 3,84 (д, 1H, J=9,0 Гц), 3,55-3,75 (м, 3H), 3,05 (д, 1H, J=4,7 Гц), 1,4-1,95 (м, 6H); m/z (esi/APCI) M+1=394,1.
Пример 3
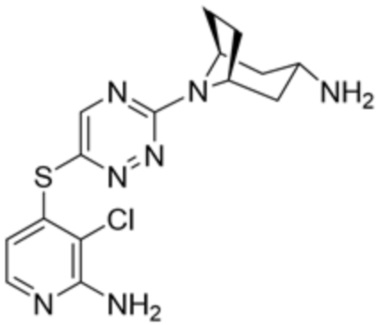
(1R,3 s ,5S)-8-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-8-азабицикло[3.2.1]октан-3-амин
[00368] Стадия A: Трет-бутил-((1R,3s,5S)-8-азабицикло[3.2.1]октан-3-ил)карбамат (226 мг, 1,00 ммоль) разбавляли диоксаном (5 мл), после чего добавляли DIEA (611 мкл, 3,50 ммоль) и 3,6-дихлор-1,2,4-триазин (150 мг, 1,00 ммоль). Реакционную смесь нагревали до 50°С и перемешивали в течение 3 ч. Реакционной смеси давали возможность остыть, разбавляли DCM/IPA и 10% карбонатом натрия. Слои разделяли и водный слой дважды экстрагировали DCM/IPA. Органические фазы объединяли, сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 10-50% этилацетат/гексаны с получением трет-бутил ((1R,3s,5S)-8-(6-хлор-1,2,4-триазин-3-ил)-8-азабицикло[3.2.1]октан-3-ил)карбамата (235 мг, 0,692 ммоль, выход 69,1%).
[00369] Стадия B: Трет-бутил-((1R,3s,5S)-8-(6-хлор-1,2,4-триазин-3-ил)-8-азабицикло[3.2.1]октан-3-ил)карбамат (30 мг, 0,088 ммоль) и 2-амино-3-хлорпиридин-4-тиол (14 мг, 0,088 ммоль) разбавляли диоксаном, после чего добавляли DIEA (46 мкл, 0,26 ммоль). Реакционную смесь помещали в атмосферу азота и нагревали до 90°С. После перемешивания в течение 4 ч реакционную смесь разбавляли DCM/IPA и 10% карбонатом натрия. Материал экстрагировали еще два раза DCM/IPA. Органические фазы объединяли, сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 1-10% метанол/DCM (1% NH4OH) с получением трет-бутил-((1R,3s,5S)-8-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-8-азабицикло[3.2.1]октан-3-ил)карбамата (25 мг, 0,054 ммоль, выход 61%).
[00370] Стадия C: Трет-бутил-((1R,3s,5S)-8-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-8-азабицикло[3.2.1]октан-3-ил)карбамат (25 мг, 0,054 ммоль) разбавляли DCM (1 мл), после чего добавляли TFA (1 мл). После перемешивания в течение 2 ч реакционную смесь концентрировали. Материал очищали на C-18 силикагеле, элюируя смесью 5-95% ACN/вода (0,1% TFA). Чистые фракции разбавляли DCM/IPA и насыщенным бикарбонатом натрия. Слои разделяли и органические слои сушили над MgSO4, фильтровали и концентрировали. Материал снова очищали на силикагеле, элюируя смесью 10% метанол/DCM (1% NH4OH) с получением (1R,3s,5S)-8-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-8-азабицикло[3.2.1]октан-3-амина (3 мг, 0,0082 ммоль, выход 15%). 1H ЯМР (400 МГц, CDCl3) δ 8,2 (с, 1H), 7,76 (д, 1H, J=5,48 Гц), 6,17 (д, 1H, J=5,48 Гц), 5,10 (ушир., 1H), 4,92 (ушир., 2H), 4,74 (ушир., 1H), 3,37 (м, 1H), 1,2-2,2 (м, 10H); m/z (esi/APCI) M+1=364,1.
Пример 4
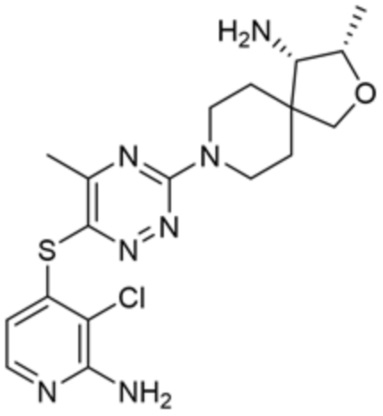
(3 S ,4 S )-8-(6-((2-амино-3-хлорпиридин-4-ил)тио)-5-метил-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
[00371] Стадия A: Дигидрохлорид (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (175 мг, 0,720 ммоль) разбавляли диоксаном (4 мл), после чего добавляли DIEA (377 мкл, 2,16 ммоль) и 3,6-дихлор-5-метил-1,2,4-триазин (118 мг, 0,720 ммоль). Реакционную смесь продували азотом, герметизировали и нагревали до 120°C. После перемешивания в течение 12 ч реакционной смеси давали возможность остыть, разбавляли DCM (25% IPA) и 10% карбонатом натрия. Слои разделяли и водный слой еще дважды экстрагировали DCM/IPA. Органические фазы объединяли, сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 1-10% метанол/DCM с получением (3S,4S)-8-(6-хлор-5-метил-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (125 мг, 0,420 ммоль, выход 58,3%).
[00372] Стадия B: (3S,4S)-8-(6-хлор-5-метил-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (23 мг, 0,077 ммоль) и 2-амино-3-хлорпиридин-4-тиол (15 мг, 0,093 ммоль) разбавляли диоксаном, после чего добавляли DIEA (40 мкл, 0,23 ммоль). Реакционную смесь помещали в атмосферу азота и нагревали до 100°С. После перемешивания в течение 4 ч реакционную смесь разбавляли DCM/IPA и 10% карбонатом натрия. Слои разделяли и органические слои сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 10% метанол/DCM (1% NH4OH) с получением (3S,4S)-8-(6-((2-амино-3-хлорпиридин-4-ил)тио)-5-метил-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (2,5 мг, 0,0059 ммоль, выход 7,7%). 1H ЯМР (400 МГц, CDCl3) δ 7,34 (дд, 1H, J=7,8, 1,6 Гц), 7,09 (т, 1H, J=7,8 Гц), 7,02 (дд, 1H, J=7,8, 1,6 Гц), 4,1-4,3 (м, 3H), 3,82 (д, 1H, J=8,6 Гц), 3,70 (д, 1H, J=8,6 Гц), 3,6 (м, 2H), 3,04 (д, 1H, J=4,7 Гц), 2,22 (с, 3H), 1,55-1,91 (м, 6H), 1,2 (д, 3H, J=6,3 Гц); m/z (esi/APCI) M+1=422,2.
Пример 5
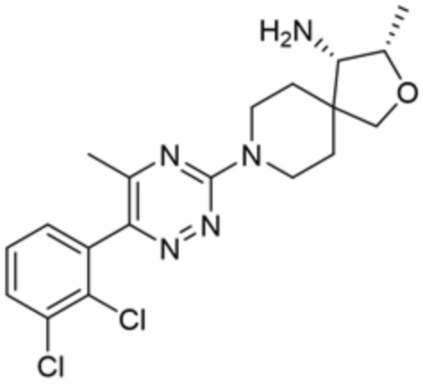
(3 S ,4S)-8-(6-(2,3-дихлорфенил)-5-метил-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
[00373] Стадия A: Дигидрохлорид (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (175 мг, 0,720 ммоль) разбавляли диоксаном (4 мл), после чего добавляли DIEA (377 мкл, 2,16 ммоль) и 3,6-дихлор-5-метил-1,2,4-триазин (118 мг, 0,720 ммоль). Реакционную смесь продували азотом, герметизировали и нагревали до 120°C. После перемешивания в течение 12 ч реакционной смеси давали возможность остыть и разбавляли DCM (25% IPA) и 10% карбонатом натрия. Слои разделяли и водный слой еще дважды экстрагировали DCM/IPA. Органические фазы объединяли, сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 1-10% метанол/DCM с получением (3S,4S)-8-(6-хлор-5-метил-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (125 мг, 0,420 ммоль, выход 58,3%).
[00374] Стадия B: (3S,4S)-8-(6-Хлор-5-метил-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (100 мг, 0,336 ммоль), (2,3-дихлорфенил)бороновую кислоту (96,1 мг, 0,504 ммоль) и тетракис (11,6 мг, 0,0101 ммоль) разбавляли диоксаном (1,5 мл), после чего добавляли Na2CO3 (420 мкл, 0,840 ммоль). Реакционную смесь продували аргоном, герметизировали и нагревали до 130°C в течение 4 ч. Реакционной смеси давали возможность остыть и разбавляли этилацетатом и водой. Слои разделяли и этилацетат сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 10% метанол/DCM (1% NH4OH) с получением (3S,4S)-8-(6-(2,3-дихлорфенил)-5-метил-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (32 мг, 0,0784 ммоль, выход 23,3%). 1H ЯМР (400 МГц, CDCl3) δ 7,56 (м, 1H), 7,31 (м, 2H), 4,33 (м, 2H), 4,21 (м, 1H), 3,86 (д, 1H, J=8,9 Гц), 3,73 (д, 1H, J=8,9 Гц), 3,5-3,7 (м, 2H), 3,04 (д, 1H, J=4,7 Гц), 2,22 (с, 3H), 1,55-1,91 (м, 6H), 1,25 (д, 3H, J=6,65 Гц); m/z (esi/APCI) M+1=408,1.
Пример 6
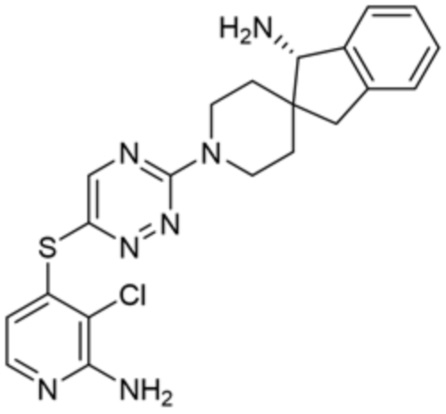
(S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00375] Стадия A: (R)-N-((S)-1,3-Дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (163 мг, 0,53 ммоль) разбавляли диоксаном (2 мл), после чего добавляли DIEA (326 мкл, 1,9 ммоль). После перемешивания в течение 5 мин добавляли 3,6-дихлор-1,2,4-триазин (80 мг, 0,53 ммоль). Реакционную смесь нагревали до 50°С и перемешивали в течение 3 ч. Реакционной смеси давали возможность остыть и разбавляли DCM/IPA и 10% карбонатом натрия. Слои разделяли и водный слой дважды экстрагировали DCM/IPA. Органические фазы объединяли, сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 1-10% метанол/DCM (1% NH4OH) с получением (R)-N-((S)-1'-(6-хлор-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (25 мг, 0,060 ммоль, выход 11%).
[00376] Стадия B: (R)-N-((S)-1'-(6-Хлор-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (55 мг, 0,13 ммоль) и 2-амино-3-хлорпиридин-4-тиолат натрия (48 мг, 0,26 ммоль) разбавляли NMP (437 мкл, 0,13 ммоль), после чего добавляли DIEA (46 мкл, 0,26 ммоль). Реакционную смесь продували аргоном, герметизировали и нагревали до 110°C. После перемешивания в течение 12 ч реакционной смеси давали возможность остыть, разбавляли этилацетатом и промывали водой и насыщенным солевым раствором. Этилацетат сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя 100% этилацетатом, с получением (R)-N-((S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (55 мг, 0,10 ммоль, выход 77%).
[00377] Стадия C: (R)-N-((S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (20 мг, 0,037 ммоль) разбавляли диоксаном (1 мл), после чего добавляли HCl (92 мкл, 0,37 ммоль). После перемешивания в течение 30 мин реакционную смесь разбавляли DCM и насыщенным водным бикарбонатом натрия. После перемешивания смеси в течение 10 мин слои разделяли и DCM сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 20% метанол/этилацетат, с получением (S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (15 мг, 0,034 ммоль, выход 93%). 1H ЯМР (400 МГц, CDCl3) δ 8,2 (с, 1H), 7,76 (д, 1H, J=5,2 Гц), 7,2-7,35 (м, 4H), 6,15 (д, 1H, J=5,2 Гц), 4,92 (ушир., 2H), 4,78 (ушир., 1H), 4,01 (с, 1H), 3,37 (м, 2H), 3,14 (д, 1H, J=15,6 Гц), 2,78 (д, 1H, J=15,6), 1,3-1,91 (м, 7H); m/z (esi/APCI) M+1=440,1.
[00378] Следующие соединения в таблице 3 были получены в соответствии с вышеуказанными процедурами, с использованием соответствующих исходных материалов и промежуточных соединений.
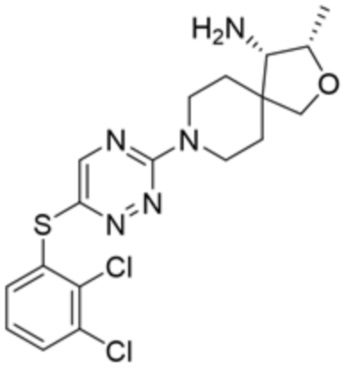
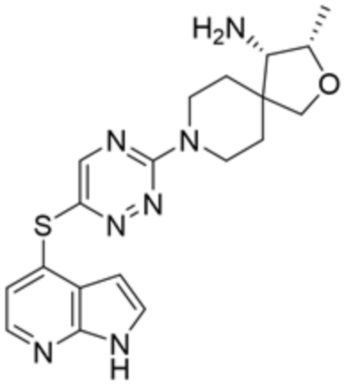
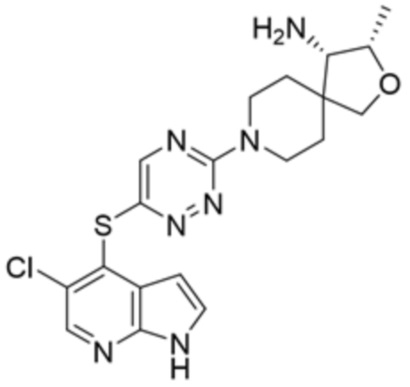
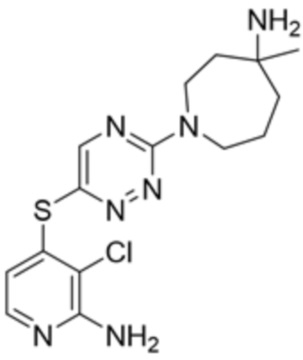
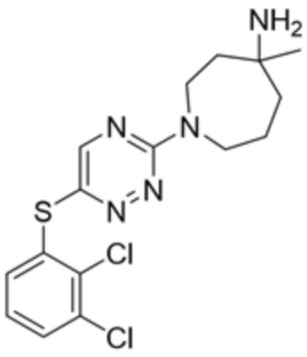
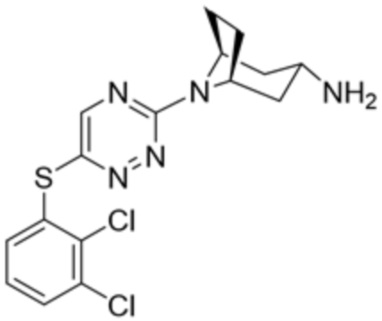
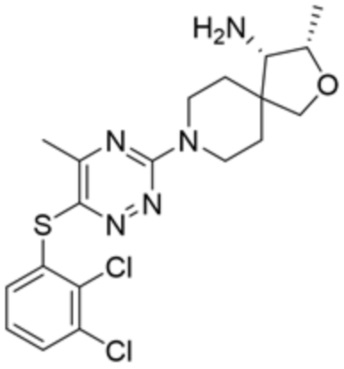
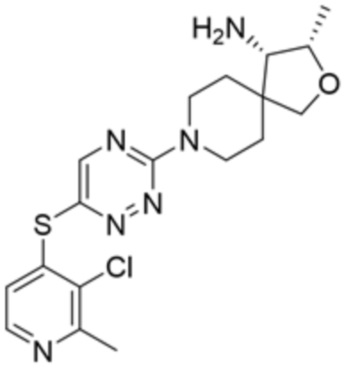
Пример 15
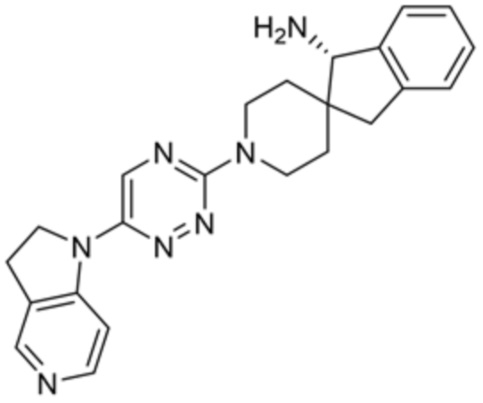
(S)-1'-(6-(2,3-дигидро-1H-пирроло[3,2-c]пиридин-1-ил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00379] В небольшом флаконе с тефлоновой крышкой Pd2(dba)3 (13 мг, 0,014 ммоль), ксантфос (16 мг, 0,028 ммоль), карбонат цезия (113 мг, 0,35 ммоль), 2,3-дигидро-1H-пирроло[3,2-c]пиридин (22 мг, 0,18 ммоль) и (S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин (50 мг, 0,14 ммоль) смешивали вместе с диоксаном (3 мл). Флакон вакуумировали и вновь заполняли Ar трижды. Затем флакон нагревали до 90°C в течение 18 ч. Реакционную смесь разбавляли DCM (5 мл), и смесь фильтровали через слой Celite®. Фильтрат выпаривали и остаток очищали, используя колонку с 12 г силикагеля (смесь 2-20% MeOH/DCM), с получением (S)-1'-(6-(2,3-дигидро-1H-пирроло[3,2-c]пиридин-1-ил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (9 мг, 0,023 ммоль, выход 16%) в виде твердого вещества. m/z (esi/APCI) M+1=360,0; 1H ЯМР (400 МГц, CDCl3) δ 8,32 (с, 2 H), 8,17 (с, 1 H), 7,64 (с, 1 H), 7,33 (д, J=5,5 Гц, 1 H), 7,23 (с, 3 H), 4,62-4,49 (м, 2 H), 4,18 (т, J=8,7 Гц, 2 H), 4,00 (с, 1 H), 3,39-3,24 (м, 4 H), 3,13 (д, J=15,6 Гц, 1 H), 2,76 (д, J=15,6 Гц, 1 H), 1,87 (тд, J=12,5, 4,3 Гц, 1 H), 1,81-1,71 (м, 1 H), 1,48-1,33 (м, 3 H).
Пример 16
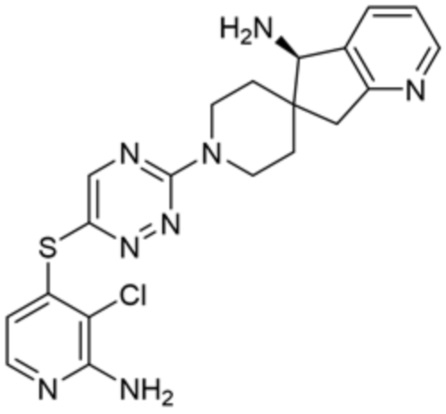
тригидрохлорид (R)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина
[00380] Стадия A: Трет-бутил-(R)-5-(((R)-трет-бутилсульфинил)амино)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат (0,193 г, 0,474 ммоль) растворяли в DCM (3 мл), и затем добавляли 2,2,2-трифторуксусную кислоту (0,270 г, 2,37 ммоль). После перемешивания в течение 1 ч реакционную смесь выпаривали, и оставшуюся соль TFA использовали на следующем этапе без дополнительной очистки.
[00381] Стадия B: (R)-N-((R)-5,7-Дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (0,07 г, 0,23 ммоль) в виде соли TFA растворяли в диоксане (4 мл), и в раствор добавляли TEA (0,16 мл, 1,1 ммоль). Добавляли 3,6-дихлор-1,2,4-триазин (0,032 г, 0,22 ммоль) и смесь нагревали до 50°С в течение 3 ч. 2-Амино-3-хлорпиридин-4-тиолат натрия (0,050 г, 0,27 ммоль) добавляли в реакционную смесь, и смесь нагревали до 90°С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры, гасили водой (10 мл) и экстрагировали EtOAc (3 × 15 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили и выпаривали с получением остатка. Остаток очищали, используя колонку с 24 г силикагеля (смесь 2-20% MeOH/DCM), получая (R)-N-((R)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (0,081 г, 0,15 ммоль, выход 65%) в виде твердого вещества. m/z (esi/APCI) M+1 =545,2.
[00382] Стадия C: (R)-N-((R)-1'-(6-((2-Амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (0,081 г, 0,15 ммоль) растворяли в DCM (2 мл), и к смеси добавляли HCl в диоксане (4M) (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем к смеси добавляли простой эфир (5 мл). Продукт отфильтровывали и промывали эфиром трижды, с получением тригидрохлорида (R)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (0,057 г, 0,13 ммоль, выход 87%). m/z (esi/APCI) M+1 =441,1; 1H ЯМР (400 МГц, (CD3)2SO)) δ 8,73 (с, 1H), 8,57-8,52 (м, 2 H), 8,08 (д, J=7,4 Гц, 1 H), 7,75 (д, J=6,3 Гц, 1 H), 7,44-7,38 (м, 1 H), 6,20 (д, J=6,2 Гц, 1 H), 4,50 (с, 1 H), 3,15 (д, J=17,1 Гц, 2 H), 1,86 (д, J=11,8 Гц, 2 H), 1,62 (д, J=12,5 Гц, 2 H), 1,07 (т, J=7,0 Гц, 1 H).
Пример 17
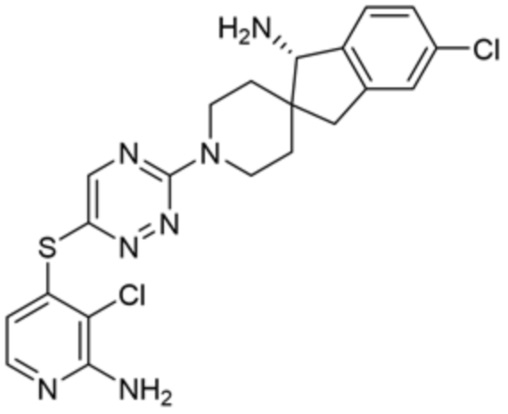
(S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00383] Трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (0,100 г, 0,227 ммоль) растворяли в DCM (3 мл), и 4M HCl в диоксане (1 мл) добавляли в раствор. Смесь перемешивали в течение 1 ч при комнатной температуре и добавляли Et2O (10 мл). Образовавшееся твердое вещество отфильтровывали и высушивали. Дигидрохлорид (S)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (30 мг, 0,097 ммоль) суспендировали в диоксане (3 мл), и к смеси добавляли триэтиламин (29 мг, 0,29 ммоль). Смесь перемешивали при комнатной температуре в течение 30 мин, затем добавляли 3,6-дибром-1,2,4-триазин (23 мг, 0,097 ммоль). Реакционную смесь нагревали до 50°С и перемешивали в течение 1 ч. Добавляли 2-амино-3-хлорпиридин-4-тиолат натрия (18 мг, 0,097 ммоль) и реакционную смесь нагревали до 90°С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры, гасили водой (10 мл), и смесь экстрагировали смесью DCM/IPA (3 × 10 мл). Объединенные органические слои промывали насыщенным солевым раствором (1×10 мл), сушили и выпаривали с получением остатка. Остаток очищали, используя колонку с 12 г силикагеля (смесь 2-20% MeOH/DCM), получая (S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин (15 мг, 0,032 ммоль, выход 33%). m/z (esi/APCI) M+1=475,2. 1H ЯМР (400 МГц, CDCl3) δ 8,20 (с, 1 H), 7,76 (с, 1 H), 7,22 (с, 2 H), 6,13 (с, 1 H), 4,91 (с, 2 H), 4,74 (с, 2 H), 3,97 (с, 1 H), 3,35 (д, J=12,1 Гц, 2 H), 3,12 (д, J=16,2 Гц, 1 H), 2,75 (д, J=16,1 Гц, 1 H), 1,87 (д, J=12,4 Гц, 1 H), 1,76 (д, J=11,5 Гц, 1 H), 1,67 (д, J=13,0 Гц, 1 H), 1,39 (д, J=12,5 Гц, 2 H).
Пример 18
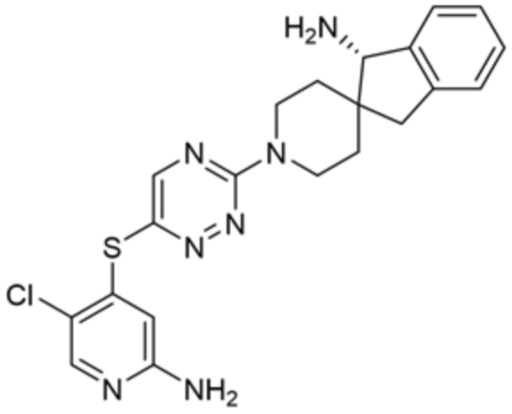
(S)-1'-(6-((2-амино-5-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00384] Стадия A: (R)-N-((S)-1'-(6-Хлор-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (.10 г, 0,24 ммоль) и 2-амино-5-хлорпиридин-4-тиолат натрия (0,088 г, 0,48 ммоль) растворяли в N, N-диметилацетамиде (1,2 мл, 0,24 ммоль). Добавляли триэтиламин (0,13 мл, 0,96 ммоль), и полученный раствор перемешивали при 100°С в течение 96 ч. Неочищенный материал загружали на колонку с 40 г силикагеля и выделяли в градиенте 0-10% MeOH:DCM+NH4OH с получением (R)-N-((S)-1'-(6-((2-амино-5-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (0,066 г, 0,12 ммоль, 48%). m/z (esi/APCI) M+1=544,2.
[00385] Стадия B: (R)-N-((S)-1'-(6-((2-амино-5-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (0,028 г, 0,051 ммоль) растворяли в дихлорметане (0,51 мл, 0,051 ммоль). Добавляли раствор соляной кислоты (4,0 M в 1,4-диоксане) (0,10 мл, 0,404 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 30 мин. Неочищенную реакционную смесь фильтровали под вакуумом с получением (S)-1'-(6-((2-амино-5-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (0,015 г, 0,034 ммоль, выход 68%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,52 (с, 1H), 7,86 (с, 1H), 7,31 (с, 1H), 7,19 (д, 2H, J= 8,2 Гц), 6,15 (с, 2H), 5,86 (с, 1H), 5,76 (с, 1H), 4,64 (ушир., 3H), 3,88 (с, 1H), 3,14 (д, 1H, J=15,7 Гц), 2,68 (д, 1H, J=14,9), 1,76 (м, 4H), 1,20 (м, 3H); m/z (esi/APCI) M+1=440,2.
Пример 19
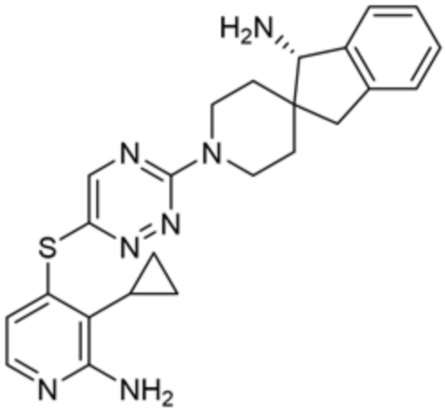
(S)-1'-(6-((2-амино-3-циклопропилпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00386] Стадия A: 4-Бром-3-циклопропилпиридин-2-амин (0,013 г, 0,061 ммоль), трис(дибензилиденацетон)дипалладий (0) (0,0044 г, 0,0048 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (0,0035 г, 0,0061 ммоль) и трет-бутил-(S)-(1'-(6-меркапто-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)карбамат (0,025 г, 0,061 ммоль) растворяли в 1,4-диоксане (0,30 мл, 0,061 ммоль). Добавляли N-этил-N-изопропилпропaн-2-амин (0,022 мл, 0,12 ммоль), и полученный раствор перемешивали при 100°С в течение 4 ч. Неочищенный материал загружали на 60 г колонку С-18 и выделяли в градиенте 5-95% MeCN:H2O+0,1% TFA. Фракции конденсировали, ресуспендировали в DCM, промывали насыщенным NaHCO3 и сушили над Na2SO4. Раствор фильтровали и конденсировали с получением (R)-N-((S)-1'-(6-((2-амино-3-циклопропилпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (0,0089 г, 0,016 ммоль, 27%) в виде твердого вещества. m/z (esi/APCI) M+1=550,2.
[00387] Стадия B: (R)-N-((S)-1'-(6-((2-амино-3-циклопропилпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (0,0080 г, 0,015 ммоль) растворяли в дихлорметане (0,10 мл, 0,015 ммоль). Добавляли раствор соляной кислоты (4,0 н. в диоксане) (0,018 мл, 0,073 ммоль), и раствор перемешивали при комнатной температуре в течение 30 мин. Неочищенную реакционную смесь фильтровали под вакуумом. Осадок растворяли в MeOH и конденсировали, затем ресуспендировали в DCM. Раствор промывали насыщенным Na2HCO3, а затем водой. Органические вещества сушили над Na2SO4 и конденсировали с получением (S)-1'-(6-((2-амино-3-циклопропилпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (0,0054 г, 0,011 ммоль, выход 76%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,46 (с, 1H), 7,60 (с, 1H), 7,31 (д, 1H, J= 6,3 Гц), 7,18 (м, 2H), 5,81 (с, 1H), 5,75 (д, 2H, J=2,2 Гц), 4,57 (ушир., 2H), 3,87 (с, 1H), 3,87 (м, 3H), 3,13 (м, 1H), 2,67 (м, 1H), 1,71 (м, 5H), 1,20 (м, 2H), 0,85 (м, 2H), 0,58 (м, 2H); m/z (esi/APCI) M+1=446,2.
Пример 20
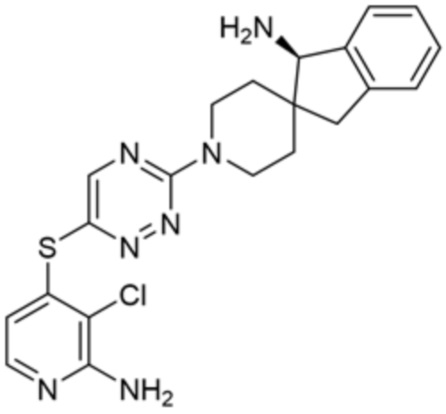
(R)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00388] Стадия A: Трет-бутил-(1R)-1-(3,3-диметил-1-оксидо-1,2-тиазиридин-2-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (1,2 г, 3,2 ммоль) растворяли в дихлорметане (8,0 мл, 3,2 ммоль). Добавляли раствор соляной кислоты (4,0 M в 1,4-диоксане, 7,9 мл, 32 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 2 ч. Неочищенную реакционную смесь фильтровали, и осадок промывали простым эфиром, с получением (R)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин (0,82 г, 3,0 ммоль, 94%) в виде твердого вещества. m/z (esi/APCI) M+1=203,2.
[00389] Стадия B: (R)-1,3-Дигидроспиро[инден-2,4'-пиперидин]-1-амин (0,494 г, 2,44 ммоль) и 3,6-дибром-1,2,4-триазин (.58 г, 2,4 ммоль) растворяли в 1,4-диоксане (9,8 мл, 2,4 ммоль). Добавляли триэтиламин (1,0 мл, 7,3 ммоль), и полученный раствор перемешивали при 50°С в течение 1,5 ч. Реакционную смесь разбавляли раствором 3:1 DCM:IPA (25 мл) и промывали 2M NaCO3 (15 мл). Органические соединения отделяли от водного слоя. Органические слои объединяли и промывали водой и насыщенным NaHCO3. Неочищенный материал сушили над Na2SO4 и конденсировали с получением (R)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина. m/z (esi/APCI) M+1=362,2.
[00390] Стадия C: (R)-1'-(6-Бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин (0,88 г, 2,4 ммоль) растворяли в N, N-диметилацетамиде (9,8 мл, 2,4 ммоль). Добавляли 2-амино-3-хлорпиридин-4-тиолат натрия (0,450 г, 2,4 ммоль), а затем гидрохлорид триэтиламина (0,34 г, 2,5 ммоль). Полученный раствор перемешивали при 80°С в течение 12 ч. Неочищенную реакционную смесь разбавляли 3:1 IPA:DCM. Органические вещества промывали 2М Na2CO3 и насыщенным NaHCO3. Органические вещества объединяли, сушили над Na2SO4 и конденсировали. Неочищенный материал загружали на колонку с 80 г силикагеля и элюировали 0-10% MeOH:EtOAc с получением (R)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (0,14 г, 0,33 ммоль, выход 13,5%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,48 (с, 1H), 7,70 (с, 1H), 7,31 (д, 1H, J=6,2 Гц), 7,18 (м, 2H), 6,42 (с, 1H), 5,93 (д, 1H, J=5,3 Гц), 4,54 (ушир., 2H), 3,88 (с, 1H), 3,39 (м, 1H), 3,13 (д, 1H, J=15,6 Гц), 2,67 (д, 1H, J=15,6), 1,85 (м, 5H), 1,59 (д, 1H, J=13,2 Гц), 1,20 (м, 3H); m/z (esi/APCI) M+1=440,1.
Пример 21
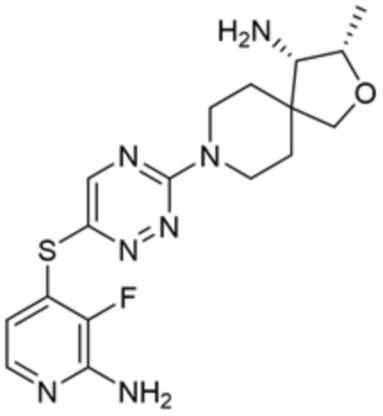
(3 S ,4S)-8-(6-((2-амино-3-фторпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
[00391] (3S,4S)-8-(6-Хлор-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4,5]декан-4-амин (0,026 г, 0,090 ммоль) и метил-3-((2-амино-3-фторпиридин-4-ил)тио)пропанoат (0,023 г, 0,099 ммоль) растворяли в N, N-диметилацетамиде (0,50 мл, 0,10 ммоль). Добавляли трет-бутоксид калия (0,090 мл, 0,090 ммоль) и полученный раствор перемешивали в течение ночи при 80°C. Неочищенную реакционную смесь загружали на колонку с 40 г силикагеля и выделяли в градиенте 0-8% MeOH:DCM+NH4OH с получением (3S,4S)-8-(6-((2-амино-3-фторпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (0,0092 г, 0,024 ммоль, выход 26,0%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,23 (с, 1H), 7,92 (м, 1H), 7,57 (м, 1H), 6,67 (с, 2H), 5,73 (с, 2H), 3,99 (м, 1H), 3,64 (д, 3H, J= 8,4 Гц), 2,88 (д, 2H, J=5,0 Гц), 1,96 (с, 2H), 1,54 (м, 4H), 1,15 (м, 1H), 1,05 (д, 2H, J= 6,2 Гц); m/z (esi/APCI) M+1=393,2.
Пример 22
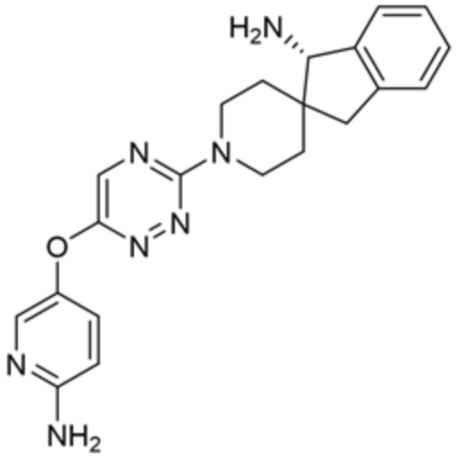
(S)-1'-(6-((6-аминопиридин-3-ил)oкси)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00392] Стадия A: (S)-N-((S)-1'-(6-Бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (0,10 г, 0,22 ммоль), 2-амино-5-гидроксипиридин HCl (0,033 г, 0,23 ммоль) и карбонат цезия (0,22 г, 0,66 ммоль) растворяли в (метилсульфинил)метане (2,0 мл, 0,22 ммоль). Полученный раствор перемешивали при 100°С в течение ночи. Неочищенный материал загружали на колонку с 40 г силикагеля и выделяли изократическим способом в 10% MeOH:EtOAc+NH4OH, с получением (R)-N-((S)-1'-(6-((6-аминопиридин-3-ил)oкси)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (0,015 г, 0,030 ммоль, выход 14%). m/z (esi/APCI) M+1=494,2.
[00393] Стадия B: (R)-N-((S)-1'-(6-((6-аминопиридин-3-ил)oкси)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (0,015 г, 0,030 ммоль) растворяли в дихлорметане (0,10 мл, 0,030 ммоль). Добавляли соляную кислоту (4,0 н. в диоксане, 0,076 мл, 0,30 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Неочищенную реакционную смесь фильтровали под вакуумом с получением осадка. Осадок промывали MeOH в отдельную колбу для фильтрования. Неочищенный материал загружали на колонку с 4 г силикагеля и выделяли с использованием изократического градиента 10% MeOH:EtOAc+NH4OH, с получением (S)-1'-(6-((6-аминопиридин-3-ил)oкси)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (0,0050 г, 0,013 ммоль, выход 42%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,37 (с, 1H), 7,80 (м, 1H), 7,30 (м, 2H), 7,17 (м, 2H), 6,46 (д, 1H, J= 8,8 Гц), 5,87 (с, 2H), 4,33 (м, 1H), 3,87 (м, 1H), 3,20 (м, 1H), 3,08 (д, 2H, J=16,0 Гц), 2,65 (м, 1H), 1,96 (с, 2H), 1,73 (м, 1H), 1,62 (м, 1H), 1,47 (м, 1H), 1,21 (с, 1H), 1,15 (м, 2H); m/z (esi/APCI) M+1=390,2.
Пример 23
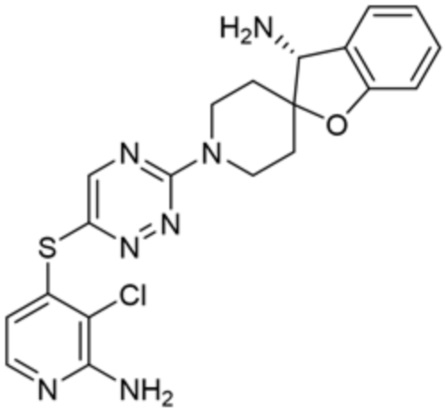
(R)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3H-спиро[бензофуран-2,4'-пиперидин]-3-амин
[00394] Стадия A: (R)-N-((R)-1'-(6-Хлор-1,2,4-триазин-3-ил)-3H-спиро[бензофуран-2,4'-пиперидин]-3-ил)-2-метилпропан-2-сульфинамид (0,037 г, 0,088 ммоль) и 2-амино-3-хлорпиридин-4-тиолат натрия (0,034 г, 0,18 ммоль) разбавляли в N, N-диметилацетамиде (0,44 мл, 0,088 ммоль). Добавляли триэтиламин (0,049 мл, 0,35 ммоль), и полученный раствор перемешивали в течение ночи при 100°С. Неочищенный материал загружали на колонку с 40 г силикагеля и выделяли в градиенте 0-10% MeOH:EtOAc с получением (R)-N-((R)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3H-спиро[бензофуран-2,4'-пиперидин]-3-ил)-2-метилпропан-2-сульфинамида. m/z (esi/APCI) M+1=546,1.
[00395] Стадия B: (R)-N-((R)-1'-(6-((2-Амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3H-спиро[бензофуран-2,4'-пиперидин]-3-ил)-2-метилпропан-2-сульфинамид (0,039 г, 0,071 ммоль) разбавляли в 1,4-диоксане (0,71 мл, 0,071 ммоль). Добавляли соляную кислоту (4,0 M в 1,4-диоксане, 0,14 мл, 0,57 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 30 мин. Реакцию гасили насыщенным Na2SO4 (2,0 мл) до достижения рН=9 раствора и экстрагировали EtOAc. Неочищенную реакционную смесь загружали на колонку с 24 г силикагеля и выделяли в градиенте 0-10% MeOH:EtOAc+NH4OH с получением (R)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3H-спиро[бензофуран-2,4'-пиперидин]-3-амина (0,0077 г, 0,017 ммоль, выход 24%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 8,22 (с, 1H), 7,71 (д, 1H, J=4,4 Гц), 7,34 (д, 1H, J= 7,3 Гц), 7,23 (м, 1H), 6,94 (м, 1H), 6,84 (д, 1H, J=8,0 Гц), 6,14 (д, 1H, J=5,4 Гц), 4,80 (ушир., 2H), 4,14 (с, 1H), 3,01 (с, 1H), 2,49 (с, 1H), 2,00 (м, 4H), 1,81 (м, 1H), 1,26 (м, 3H); m/z (esi/APCI) M+1=442,1.
Пример 24
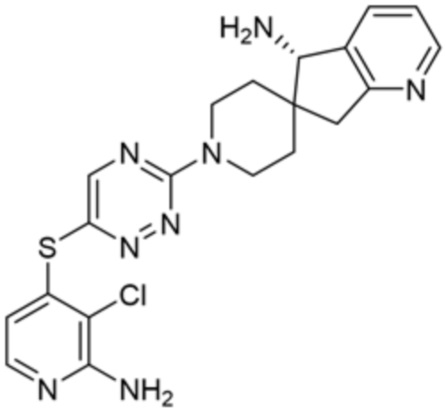
(S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амин
[00396] Стадия A: Трет-бутил-(S)-5-(((R)-трет-бутилсульфинил)амино)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-1'-карбоксилат (0,20 г, 0,49 ммоль) растворяли в дихлорметане (2,5 мл, 0,49 ммоль). Добавляли трифторуксусную кислоту (0,38 мл, 4,9 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали с получением (R)-N-((S)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамида (0,15 г, 0,49 ммоль, выход 99%) в виде стекловидного твердого вещества. m/z (esi/APCI) M+1=308,2.
[00397] Стадия B: (R)-N-((S)-5,7-Дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (0,15 г, 0,49 ммоль) и 3,6-дихлор-1,2,4-триазин (0,084 г, 0,56 ммоль) растворяли в 1,4-диоксане (1,5 мл, 0,375 ммоль). Добавляли триэтиламин (0,21 мл, 1,47 ммоль) и полученный раствор перемешивали при 100°С в течение ночи. Неочищенный материал загружали на колонку с 40 г силикагеля и выделяли в градиенте 50-100% EtOAc/гексан с получением (R)-N-((S)-1'-(6-хлор-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамида (0,099 г, 0,23 ммоль, выход 47%) в виде твердого вещества. m/z (esi/APCI) M+1=421,2.
[00398] Стадия C: (R)-N-((S)-1'-(6-хлор-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (0,099 г, 0,23 ммоль) и 2-амино-3-хлорпиридин-4-тиолат натрия (0,10 г, 0,56 ммоль) разбавляли в N, N-диметилацетамиде (1,2 мл, 0,23 ммоль). Добавляли триэтиламин (0,13 мл, 0,94 ммоль), и полученный раствор перемешивали при 100°С в течение 12 ч. Неочищенную реакционную смесь загружали на колонку с 40 г силикагеля и выделяли в градиенте 0-10% MeOH:EtOAc с получением (R)-N-((S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамида (0,027 г, 0,050 ммоль, выход 21%) в виде твердого вещества. m/z (esi/APCI) M+1=545,1.
[00399] Стадия D: (R)-N-((S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид (0,027 г, 0,05 ммоль) разбавляли в 1,4-диоксане (0,50 мл, 0,05 ммоль). Добавляли раствор соляной кислоты (4,0 M в 1,4-диоксане, 0,099 мл, 0,40 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 15 мин. Реакцию гасили насыщенным Na2HCO3 и целевой продукт экстрагировали EtOAc. Органические вещества сушили над Na2SO4, конденсировали и загружали на колонку с 24 г силикагеля и выделяли в градиенте 0-20% MeOH:EtOAc+NH4OH с получением (S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (0,0073 г, 0,017 ммоль, выход 33%). 1H ЯМР (400 МГц, (CD3)2SO) δ 8,45 (с, 1H), 8,20 (с, 1H), 7,77 (д, 1H, J=5,2 Гц), 7,65 (д, 1H, J=7,5 Гц), 7,15 (м, 1H), 6,15 (д, 1H, J=5,3 Гц), 4,91 (с, 3H), 4,79 (ушир., 2H), 4,06 (с, 1H), 3,35 (м, 2H), 3,26 (д, 1H, J=16,3), 2,93 (д, 1H, J= 16,3 Гц), 1,86 (м, 2H), 1,70 (м, 1H), 1,43 (д, 1H, J= 13,2 Гц), 1,25 (с, 1H); m/z (esi/APCI) M+1=441,2.
Пример 25
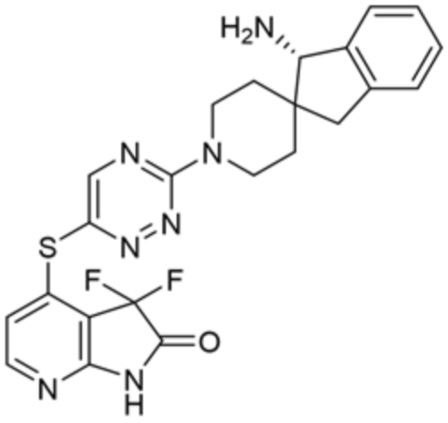
(S)-4-((3-(1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)-3,3-дифтор-1,3-дигидро-2H-пирроло[2,3-b]пиридин-2-он
[00400] Стадия A: DIEA (0,030 мл, 0,17 ммоль) добавляли к смеси 3,3-дифтор-4-йод-1,3-дигидро-2H-пирроло[2,3-b]пиридин-2-она (0,028 г, 0,093 ммоль), трет-бутил-(S)-(1'-(6-меркапто-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)карбамата (0,035 г, 0,085 ммоль), трис(дибензилиденацетон)дипалладия (0) (0,0039 г, 0,0042 ммоль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантена (0,0049 г, 0,0085 ммоль) в 1,4-диоксане (0,85 мл, 0,085 ммоль) при комнатной температуре при перемешивании. Смесь дегазировали аргоном в течение 5 мин, после чего нагревали до 100°С в течение 1 ч. Реакционную смесь концентрировали под вакуумом, и затем очищали с помощью флэш-хроматографии, элюируя в градиенте 0-20% MeOH в EtOAc с 2% NH4OH, получая трет-бутил-(S)-(1'-(6-((3,3-дифтор-2-оксо-2,3-дигидро-1H-пирроло[2,3-b]пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)карбамат (предположительно количественный выход). m/z (esi/APCI) M+1=582,2.
[00401] Стадия B: Трет-бутил-(S)-(1'-(6-((3,3-дифтор-2-оксо-2,3-дигидро-1H-пирроло[2,3-b]пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)карбамат суспендировали в 5 мл DCM и подвергали воздействию TFA (5 мл) при перемешивании при комнатной температуре в течение 15 мин. Смесь концентрировали под вакуумом и ресуспендировали в 25 мл смеси 3:1 DCM:IPA. Добавляли насыщенный NaHCO3 (25 мл) и оставляли перемешиваться в течение 5 мин. Слои разделяли и затем экстрагировали дополнительные органические вещества из водного слоя с помощью DCM:IPA (2×15 мл). Органические слои объединяли и промывали насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением (S)-4-((3-(1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)-3,3-дифтор-1,3-дигидро-2H-пирроло[2,3-b]пиридин-2-она (0,023 г, 0,048 ммоль, выход 57%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,54 (с, 1 H), 8,09 (д, J=5,7 Гц, 1 H), 7,33 (м, 1 H), 7,24-7,16 (м, 4 H), 6,56 (д, J=5,9 Гц, 1 H), 4,55 (ушир., 2 H), 3,94 (с, 1 H), 3,41 (ушир., 2 H), 3,14 (д, J=15,5 Гц, 1 H), 2,72 (д, J=15,85 Гц, 1 H), 1,86-1,66 (м, 2 H), 1,60 (д, J=13,5 Гц, 1 H), 1,24 (м, 2 H). m/z (esi/APCI) M+1=482,1.
Пример 26
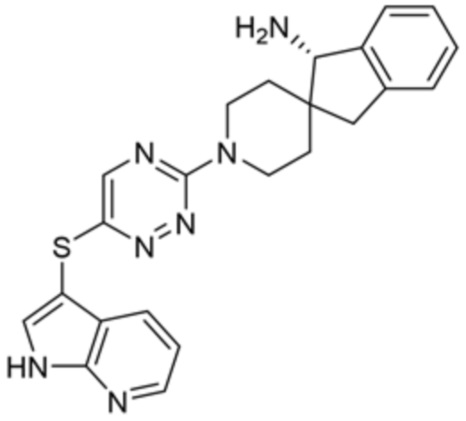
(S)-1'-(6-((1H-пирроло[2,3-b]пиридин-3-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00402] Смесь 3-йод-1H-пирроло[2,3-b]пиридина (0,015 г, 0,061 ммоль), трет-бутил-(S)-(1'-(6-меркапто-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)карбамата (0,025 г, 0,061 ммоль), карбоната калия (0,015 г, 0,091 ммоль) и йодида меди (I) (0,0031 мл, 0,091 ммоль) в DMF (0,61 мл, 0,061 ммоль) нагревали до 75°С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и затем гасили EtOAc (25 мл) и водой (25 мл). Эту двухфазную смесь фильтровали через бумагу GF/F, и затем слои разделяли. Органическую фазу промывали насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и затем концентрировали под вакуумом. Полученный остаток ресуспендировали в DCM (5 мл) и подвергали воздействию TFA (5 мл) при перемешивании при комнатной температуре в течение 15 мин. Смесь концентрировали под вакуумом и ресуспендировали в DCM:IPA (3:1) (25 мл). Добавляли насыщенный NaHCO3 (25 мл) и перемешивали при комнатной температуре в течение 5 мин. Двухфазную смесь разделяли, и оставшиеся органические вещества из водного слоя экстрагировали DCM:IPA (2×15 мл). Полученные органические слои объединяли и промывали насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Неочищенный материал очищали флэш-хроматографией, элюируя с градиентом 0-20% МеОН в EtOAc с 2% NH4OH, с получением (S)-1'-(6-((1H-пирроло[2,3-b]пиридин-3-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (0,011 г, 0,025 ммоль, выход 41%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 12,28 (с, 1 H), 8,30 (дд, J=4,7, 1,6 Гц, 1 H), 8,18 (ушир., 2 H), 8,10 (с, 1 H), 7,94 (д, J=2,7 Гц, 1 H), 7,90 (дд, J=8,2, 1,6 Гц, 1 H), 7,46 (д, J=7,2 Гц, 1 H), 7,36-7,24 (м, 4 H), 7,15 (дд, J=7,8, 4,7 Гц, 1 H), 4,45-4,31 (м, 3 H), 3,24 (т, J=12,9 Гц, 2 H), 3,15 (д, J=16,0 Гц, 1 H), 2,97 (д, J=16,2 Гц, 1 H), 1,71-1,56 (м, 2 H), 1,47 (д, J=13,3 Гц, 2 H). m/z (esi/APCI) M+1=430,2.
Пример 27
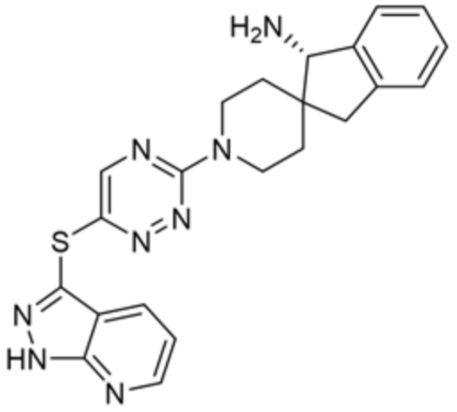
(S)-1'-(6-((1H-пиразоло[3,4-b]пиридин-3-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00403] Смесь 1-boc-3-йод-1H-пиразоло[3,4-b]пиридина (0,027 г, 0,079 ммоль), (R)-N-((S)-1'-(6-меркапто-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (0,030 г, 0,072 ммоль), карбоната калия (0,015 г, 0,11 ммоль) и йодида меди(I) (0,0037 мл, 0,11 ммоль) в DMF (0,72 мл, 0,072 ммоль) нагревали до 100°С в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и гасили EtOAc (25 мл) и водой (25 мл). Эту двухфазную смесь фильтровали через бумагу GF/F, и слои разделяли. Органическую фазу промывали насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток ресуспендировали в 1,4-диоксане (5 мл) и подвергали воздействию 4 н. HCl в диоксане (5 мл) при перемешивании при комнатной температуре в течение 15 мин. Смесь концентрировали под вакуумом и ресуспендировали в DCM:IPA (3:1) (25 мл). Добавляли насыщенный NaHCO3 (25 мл) и данную смесь перемешивали при комнатной температуре в течение 5 мин. Двухфазную смесь разделяли, и оставшиеся органические вещества из водного слоя экстрагировали DCM:IPA (2×15 мл). Полученные органические слои объединяли и промывали насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Неочищенный материал очищали с помощью препаративной ВЭЖХ, элюируя с градиентом 5-95% ACN в воде с модификатором 0,1% TFA. Фракции продукта были переведены в свободное основание с использованием насыщенного NaHCO3 (25 мл). Образующуюся в результате двухфазную смесь разделяли, и оставшиеся органические вещества из водного слоя экстрагировали DCM:IPA (2×15 мл). Полученные органические слои объединяли и промывали насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением (S)-1'-(6-((1H-пиразоло[3,4-b]пиридин-3-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (0,011 г, 0,025 ммоль, выход 41%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,59 (дд, J=4,5, 1,6 Гц, 1 H), 8,36 (с, 1 H), 8,12 (дд, J=8,0, 1,6 Гц, 1 H), 7,30 (д, J=6,3 Гц, 1 H), 7,27 (дд, J=8,2, 4,5 Гц, 1 H), 7,21-7,14 (м, 3 H), 4,42 (т, J=14,3 Гц, 2 H), 3,87 (с, 1 H), 3,24 (м, 2 H), 3,08 (д, J=15,8 Гц, 1 H), 2,65 (д, J=15,8 Гц, 1 H), 1,73 (тд, J=12,3, 4,5 Гц, 1 H), 1,62 (тд, J=12,7, 4,3 Гц, 1 H), 1,51 (д, J=13,3 Гц, 1 H), 1,13 (д, J=13,7 Гц, 1 H). m/z (esi/APCI) M+1=431,2.
Пример 28
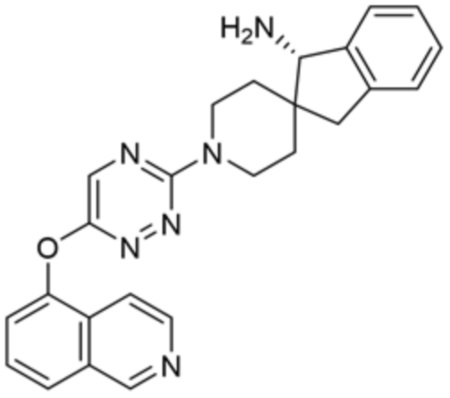
(S)-1'-(6-(изохинолин-5-илoкси)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00404] Стадия A: Раствор 5-гидроксиизохинолина (0,031 г, 0,22 ммоль), (R)-N-((S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (0,050 г, 0,11 ммоль) и карбоната цезия (0,11 г, 0,32 ммоль) в ДМСО (1,1 мл, 0,11 ммоль) нагревали до 100°С в течение 72 ч. Реакционную смесь охлаждали и добавляли в воду (25 мл) и EtOAc (25 мл). Две фазы разделяли, и водную фазу экстрагировали EtOAc (2 × 25 мл). Органические слои объединяли, промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученное ресуспендировали в DCM (2 мл) и очищали с помощью флэш-хроматографии, элюируя с градиентом 20-100% EtOAc в гексанах, с получением (R)-N-((S)-1'-(6-(изохинолин-5-илoкси)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (предположительно количественный выход). m/z (esi/APCI) M+1 =529,2.
[00405] Стадия B: (R)-N-((S)-1'-(6-(Изохинолин-5-илoкси)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид суспендировали в 1,4-диоксане (5 мл) и добавляли 4 н. HCl в 1,4-диоксане (1 мл) при перемешивании при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали под вакуумом и затем ресуспендировали в DCM (15 мл). Неочищенный продукт переводили в свободное основание насыщенным NaHCO3 (15 мл) и оставляли перемешиваться в течение 10 мин при комнатной температуре. Образующуюся в результате двухфазную смесь разделяли, и оставшиеся органические вещества экстрагировали DCM (2×15 мл). Органические слои объединяли, промывали насыщенным солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Неочищенный материал очищали с помощью препаративной ВЭЖХ, элюируя с градиентом 5-95% ACN в воде с модификатором 0,1% TFA. Фракции продукта были переведены в свободное основание с использованием насыщенного NaHCO3 (25 мл), и затем добавляли DCM (25 мл). Образующуюся в результате двухфазную смесь разделяли, и оставшиеся органические вещества из водного слоя экстрагировали DCM (2×15 мл). Полученные органические слои объединяли и промывали насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением (S)-1'-(6-(изохинолин-5-илoкси)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (0,0013 г, 0,0031 ммоль, выход 3% за две стадии) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 9,22 (с, 1 H), 9,16 (с, 1 H), 8,45 (д, J=5,9 Гц, 1 H), 8,24 (д, J=5,9 Гц, 1 H), 8,12 (д, J=9,0 Гц, 1 H), 7,64 (д, J=8,8 Гц, 1 H), 7,39 (т, J=4,3 Гц, 1 H), 7,29-7,21 (м, 4 H), 4,69 (м, 2 H), 4,04 (с, 1 H), 3,46 (м, 2 H), 3,22 (д, J=15,6 Гц, 1 H), 2,91 (д, J=15,5 Гц, 1 H), 1,93-1,78 (м, 2 H), 1,66 (д, J=13,1 Гц, 1 H), 1,52 (д, J=13,9 Гц, 1 H). m/z (esi/APCI) M+1=425,2.
Пример 29
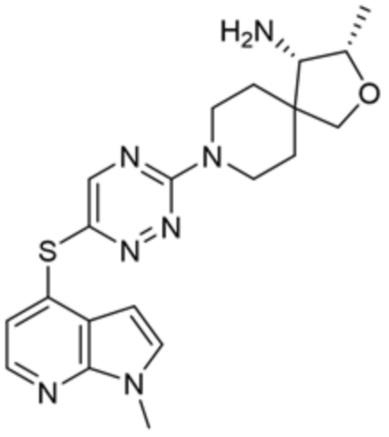
(3 S ,4S)-3-метил-8-(6-((1-метил-1H-пирроло[2,3-b]пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-2-окса-8-азаспиро[4.5]декан-4-амин
[00406] Стадия A: DIEA (0,77 мл, 4,32 ммоль) добавляли к раствору 3,6-дихлор-1,2,4-триазина (0,19 г, 1,23 ммоль) и дигидрохлорида (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (0,30 г, 1,23 ммоль) в 1,4-диоксане (6,2 мл, 1,23 ммоль) при комнатной температуре. Данную смесь нагревали до 50°С и перемешивали в течение 4 ч. Реакционной смеси давали возможность остыть и разбавляли 3:1 DCM:IPA (25 мл) и 1М карбонатом натрия (25 мл). Слои разделяли, и дополнительные органические вещества в водном слое экстрагировали DCM:IPA (3×15 мл). Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали флэш-хроматографией, элюируя с градиентом 0-15% MeOH в DCM с модификатором 1% NH4OH, с получением (3S,4S)-8-(6-хлор-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4,5]декан-4-амина (0,25 г, 0,88 ммоль, выход 71%) в виде твердого вещества. m/z (esi/APCI) M+1=284,1.
[00407] Стадия B: 1-Метил-1H-пирроло[2,3-b]пиридин-4-тиол, натриевую соль (0,035 г, 0,19 ммоль), (3S,4S)-8-(6-хлор-1,2,4-триазин-3-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (0,035 г, 0,12 ммоль) и DIEA (0,065 мл, 0,37 ммоль) помещали в DMA (1,23 мл, 0,12 ммоль), и смесь нагревали до 100°С в течение 16 ч. Смесь охлаждали и очищали флэш-хроматографией с градиентом 0-20% MeOH в DCM с 2% NH4OH. Фракции продукта концентрировали под вакуумом и далее очищали с помощью препаративной ВЭЖХ, элюируя с градиентом 5-95% ACN в воде с модификатором 0,1% TFA. Фракции продукта были переведены в свободное основание с использованием насыщенного NaHCO3 (25 мл), и затем добавляли 3:1 DCM:IPA (25 мл). Образующуюся в результате двухфазную смесь разделяли, и оставшиеся органические вещества из водного слоя экстрагировали DCM:IPA (2×15 мл). Полученные органические слои объединяли и промывали насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением (3S,4S)-3-метил-8-(6-((1-метил-1H-пирроло[2,3-b]пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-2-окса-8-азаспиро[4,5]декан-4-амина (0,013 г, 0,032 ммоль, выход 26%) в виде твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,42 (с, 1 H), 8,11 (д, J=5,1 Гц, 1 H), 7,55 (д, J=3,5 Гц, 1 H), 6,75 (д, J=5,1 Гц, 1 H), 6,35 (д, J=3,3 Гц, 1 H), 4,06 (м, 3 H), 3,80 (с, 3 H), 3,68 (д, J=8,4 Гц, 1 H), 3,60 (м, 2 H), 3,48 (д, J=8,4 Гц, 1 H), 2,92 (д, J=5,3 Гц, 1 H), 1,76 (м, 1 H), 1,66 (м, 1 H), 1,58-1,45 (м, 2 H), 1,07 (д, J=6,5 Гц, 3 H). m/z (esi/APCI) M+1=412,2.
Пример 30
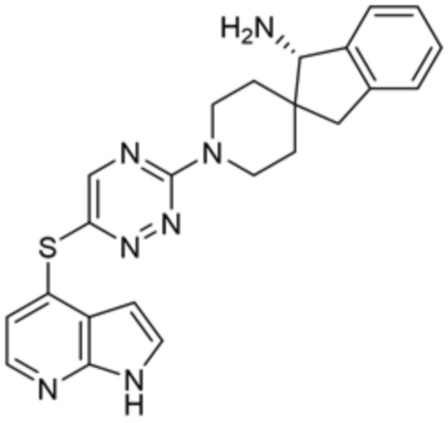
(S)-1'-(6-((1H-пирроло[2,3-b]пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00408] Стадия A: 1H-Пирроло[2,3-b]пиридин-4-тиол (0,043 г, 0,29 ммоль), основание Хунига (0,17 мл, 0,95 ммоль) и N-((S)-1'-(6-хлор-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (0,080 г, 0,19 ммоль) помещали в DMA (1,90 мл, 0,19 ммоль), и смесь нагревали до 70°С в течение 16 ч. Реакционную смесь охлаждали и очищали непосредственно флэш-хроматографией, элюируя с градиентом 0-20% MeOH в DCM с 2% NH4OH в качестве добавки, с получением (S)-N-((S)-1'-(6-((1H-пирроло[2,3-b]пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида. Предполагался количественный выход. m/z (esi/APCI) M+1=534,2.
[00409] Стадия B: (S)-N-((S)-1'-(6-((1H-пирроло[2,3-b]пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид суспендировали в 1,4-диоксане (5 мл), и 4 н. HCl в 1,4-диоксане (5 мл) добавляли при перемешивании при комнатной температуре в течение 30 мин. Смесь концентрировали под вакуумом и ресуспендировали в 3:1 DCM:IPA (25 мл). Неочищенный продукт переводили в свободное основание насыщенным NaHCO3 (15 мл) и оставляли перемешиваться в течение 10 мин при комнатной температуре. Образующуюся в результате двухфазную смесь разделяли, и оставшиеся органические вещества экстрагировали дополнительным количеством DCM:IPA (2×15 мл). Органические слои объединяли и промывали насыщенным солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Неочищенный материал очищали с помощью препаративной ВЭЖХ, элюируя с градиентом 5-95% ACN в воде с модификатором 0,1% TFA. Фракции продукта были переведены в свободное основание с использованием насыщенного NaHCO3 (25 мл), и добавляли DCM:IPA (25 мл). Образующуюся в результате двухфазную смесь разделяли, и оставшиеся органические вещества из водного слоя экстрагировали DCM:IPA (2×15 мл). Полученные органические слои объединяли и промывали насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением (S)-1'-(6-((1H-пирроло[2,3-b]пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (0,015 г, 0,035 ммоль, выход 19%) в виде твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 11,87 (с, 1 H), 8,45 (с, 1 H), 8,09 (д, J=5,1 Гц, 1 H), 7,51 (т, J=2,9 Гц, 1 H), 7,31 (д, J=6,1, 1 H), 7,22-7,14 (м, 3 H), 6,73 (д, J=8,7 Гц, 1 H), 6,36 (дд, J=3,3, 1,4 Гц, 1 H), 4,64-4,42 (ушир., 2 H), 3,87 (с, 1 H), 3,34 (м, 2 H), 3,12 (д, J=15,5 Гц, 1 H), 2,66 (д, J=15,8 Гц, 1 H), 1,80 (тд, J=12,5, 3,7 Гц, 1 H), 1,69 (тд, J=12,3, 4,1 Гц, 1 H), 1,58 (д, J=13,1 Гц, 1 H), 1,17 (д, J=13,1 Гц, 1 H). m/z (esi/APCI) M+1=430,2.
Пример 31
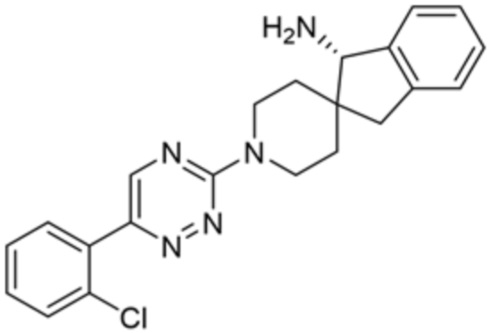
(S)-1'-(6-(2-хлорфенил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00410] Стадия A: N-((S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (50 мг, 0,11 ммоль), (2-хлорфенил)бороновую кислоту (20 мг, 0,13 ммоль) и тетракис (12 мг, 0,011 ммоль) растворяли в диоксане (1 мл), после чего добавляли Na2CO3 (135 мкл, 0,27 ммоль). Реакционную смесь продували аргоном, герметизировали и нагревали до 90°C. После перемешивания в течение 12 ч реакционной смеси давали возможность остыть и разбавляли этилацетатом и водой. Слои разделяли. Этилацетат сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на силикагеле, элюируя 20-70% этилацетатом/гексанами, с получением N-((S)-1'-(6-(2-хлорфенил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (20 мг, 0,040 ммоль, выход 37%). m/z (esi/APCI) M+1=496,2.
[00411] Стадия B: N-((S)-1'-(6-(2-хлорфенил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (20 мг, 0,040 ммоль) разбавляли DCM (500 мкл) и HCl (101 мкл 4M раствора, 0,4 ммоль). После перемешивания в течение 1 ч реакционную смесь концентрировали. Остаток разбавляли этилацетатом и насыщенным бикарбонатом натрия. Смесь перемешивали в течение 10 мин и помещали в разделительную воронку. Слои разделяли. Этилацетат сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 10% метанол/этилацетат, с получением (S)-1'-(6-(2-хлорфенил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (9,5 мг, 0,024 ммоль, выход 60%). 1H ЯМР (400 МГц, CDCl3) δ 8,55 (с, 1 H), 7,75 (дд, J=7,04, 2,3 Гц, 1 H), 7,48 (дд, J=7,82, 1,57 Гц, 1 H), 7,38 (м, 3 H), 7,23 (м, 3H), 4,74 (с, 2 H), 4,03 (с, 1H), 3,38 (м, 2H), 3,15 (д, J=15,65 Гц, 1H), 2,79 (д, J=15,65 Гц, 1 H), 1,84 (м, 4H), 1,66 (м, 1 H), 1,42 (м, 1 H). m/z (esi/APCI) M+1=392,1.
Пример 32
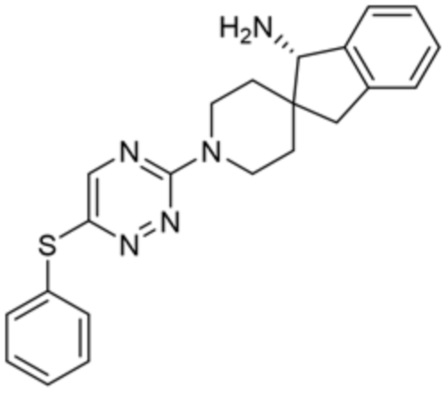
(S)-1'-(6-(фенилтио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00412] Стадия A: N-((S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (50 мг, 0,11 ммоль) разбавляли ДМСО (300 мкл), после чего добавляли бензолтиол (24 мг, 0,22 ммоль) и Cs2CO3 (88 мг, 0,27 ммоль). Реакционную смесь помещали в атмосферу азота и нагревали до 100°С. После перемешивания в течение 1 ч реакционной смеси давали возможность остыть, разбавляли этилацетатом и водой. Слои разделяли. Этилацетат сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 30% этилацетат/гексаны, с получением 2-метил-N-((S)-1'-(6-(фенилтио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил) пропан-2-сульфинамида (25 мг, 0,051 ммоль, выход 47%). m/z (esi/APCI) M+1=494,1.
[00413] Стадия B: 2-Метил-N-((S)-1'-(6-(фенилтио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил) пропан-2-сульфинамид (25 мг, 0,051 ммоль) разбавляли DCM (500 мкл) и HCl (127 мкл 4M раствора, 0,51 ммоль). После перемешивания в течение 1 ч реакционную смесь концентрировали. Остаток разбавляли этилацетатом и насыщенным бикарбонатом натрия. Смесь перемешивали в течение 10 мин и помещали в разделительную воронку. Слои разделяли. Этилацетат сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 10% метанол/этилацетат, с получением (S)-1'-(6-(фенилтио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (11 мг, 0,028 ммоль, выход 56%). 1H ЯМР (400 МГц, CDCl3) δ 8,05 (с, 1 H), 7,47 (м, 2H), 7,32 (м, 4H), 7,22 (м, 3 H), 4,62 (с, 2 H), 3,98 (с, 1H), 3,30 (м, 2H), 3,11 (д, J=15,65 Гц, 1H), 2,75 (д, J=15,65 Гц, 1 H), 1,35-1,85 (м, 6H), m/z (esi/APCI) M+1=390,2.
Пример 33
(S)-1'-(6-бензил-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00414] Стадия A: N-((S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (20 мг, 0,043 ммоль) и тетракис (5,0 мг, 0,0043 ммоль) разбавляли бромидом бензилцинка (II) (129 мкл, 0,065 ммоль). Реакционную смесь продували аргоном, герметизировали и нагревали до 70°C, и перемешивали в течение 12 ч. Реакционную смесь охлаждали, разбавляли этилацетатом и насыщенным бикарбонатом натрия. Слои разделяли и этилацетат сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 30% этилацетат/гексаны, с получением N-((S)-1'-(6-бензил-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамида (5 мг, 0,011 ммоль, выход 24%). m/z (esi/APCI) M+1=476,2.
[00415] Стадия B: N-((S)-1'-(6-бензил-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (8 мг, 0,017 ммоль) разбавляли DCM (100 мкл) и HCl (42 мкл, 0,17 ммоль). После перемешивания в течение 1 ч реакционную смесь концентрировали. Остаток разбавляли этилацетатом и насыщенным бикарбонатом натрия. Смесь перемешивали в течение 10 мин и помещали в разделительную воронку. Слои разделяли и этилацетат сушили над MgSO4, фильтровали и концентрировали. Материал очищали на силикагеле, элюируя смесью 10% метанол/этилацетат, с получением (S)-1'-(6-бензил-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (5 мг, 0,013 ммоль, выход 80%). 1H ЯМР (400 МГц, CDCl3) δ 7,97 (с, 1 H), 7,20-7,35 (м, 9H), 4,61 (с, 2 H), 4,15 (с, 2H), 3,98 (с, 1H), 3,28 (м, 2H), 3,11 (д, J=15,65 Гц, 1H), 2,75 (д, J=15,65 Гц, 1 H), 1,50-1,85 (м, 5H), 1,34 (м, 1H). m/z (esi/APCI) M+1=372,2.
Пример 34
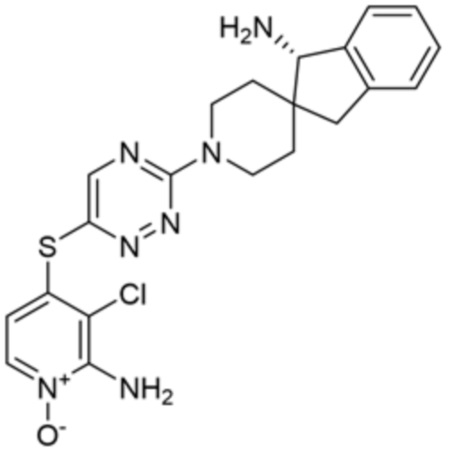
(S)-2-амино-4-((3-(1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)-3-хлорпиридин 1-оксид
[00416] (S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин (100 мг, 0,23 ммоль) помещали в MeOH (36,4 мл, 0,23 ммоль) и добавляли мета-хлорпероксибензойную кислоту («м-CPBA») (84,0 мг, 0,34 ммоль), и перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь концентрировали до примерно 10 мл, и добавляли насыщенный бикарбонат и экстрагировали DCM. Экстракты объединяли и концентрировали. Полученный остаток очищали обращенно-фазовой хроматографией (5-70% ACN:вода с 0,1% TFA). Материал растворяли в 10% MeOH в DCM. Добавляли насыщенный бикарбонат и слои разделяли. Органический слой высушивали, фильтровали и концентрировали, с получением (S)-2-амино-4-((3-(1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)-3-хлорпиридин 1-оксида (3,6 мг, 0,0079 ммоль, выход 3%). 1H ЯМР (400 МГц, (CD3)2SO) δ 8,47 (с, 1H), 7,99 (д, 1H, J=7,3 Гц), 7,31 (д, 1H, J=5,5 Гц), 7,26-7,11 (м, 5H), 6,21 (д, 1H, J=7,3 Гц), 4,54 (ушир., 2H), 4,09 (м, 2H), 3,87 (с, 1H), 3,12 (д, 1H, J=14,6 Гц), 2,68 (с, 1H), 1,86-1,54 (м, 3H), 1,19 (м, 1H); m/z (esi/APCI) M+1=456,1.
Пример 35
(S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00417] Стадия A: Трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат (100 мг, 0,227 ммоль) растворяли в CH2Cl2 (0,1 мл), и затем добавляли 4 н. HCl в диоксане (0,5 мл) и перемешивали при комнатной температуре в течение 30 мин. Твердые вещества дополнительно осаждали добавлением ET2O (3 мл) и фильтровали. Клейкое твердое вещество сушили в вакуумном сушильном шкафу и использовали без дополнительного анализа.
[00418] Стадия B: Дигидрохлорид (S)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (35 мг, 0,113 ммоль) суспендировали в диоксане (1 мл). 3,6-Дибром-1,2,4-триазин (27,0 мг, 0,113 ммоль) добавляли в суспензию, после чего добавляли триэтиламин (78,8 мкл, 0,565 ммоль). Реакционную смесь нагревали до 50°С в течение 30 мин. Реакционную смесь продували азотом. Добавляли 3-амино-2-хлорбензолтиолaт натрия (20,5 мг, 0,113 ммоль) и реакционную смесь нагревали до 90°С в течение 2 ч. Реакционную смесь концентрировали и очищали на силикагеле (0-10% MeOH в EtOAc с 1% NH4OH) с получением (S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-4-хлор-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (25,1 мг, 0,053 ммоль, выход 47%) в виде твердого вещества. 1H (400 МГц, CDCl3) δ 8,22 (с, 1H), 7,73 (д, J=5,5 Гц, 1H), 7,26-7,18 (м, 3H), 6,13 (д, J=5,5 Гц, 1H), 4,75 (ушир. с, 2H), 4,04 (с, 1H, 3,45-3,34 (м, 2H), 3,20 (д, J=16,2 Гц, 1H), 2,81 (д, J=16,2 Гц, 1H), 2,10-2,00 (м, 2H), 1,93-1,84 (м, 1H), 1,83-1,74 (м, 1H), 1,73-1,66 (м, 1H), 1,47-1,41 (м, 1H). m/z (esi/APCI) M+1=474,1.
Пример 36
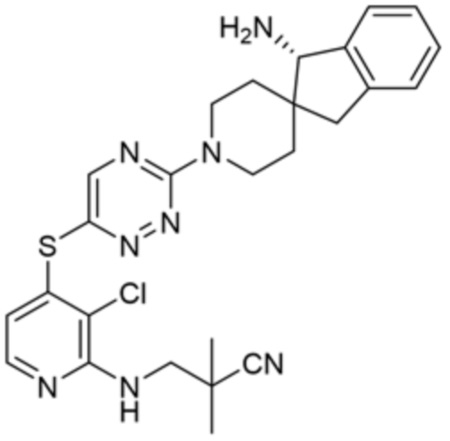
(S)-3-((4-((3-(1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)-3-хлорпиридин-2-ил)амино)-2,2-диметилпропаннитрил
[00419] Стадия A: 3-Амино-2,2-диметилпропаннитрил (115 мг, 1,16 ммоль) добавляли к перемешиваемому раствору 3-хлор-2-фтор-4-йодпиридина (250 мг, 0,97 ммоль) в ДМСО (2,5 мл) и перемешивали при 120°С в течение 16 ч. Реакционную смесь разбавляли водой и экстрагировали EtOAc. Органическую часть сушили, концентрировали, полученный остаток очищали колоночной хроматографией на силикагеле (30% EtOAc-гексан) с получением 3-((3-хлор-4-йодпиридин-2-ил)амино)-2,2-диметилпропаннитрила (250 мг, выход 77%) в виде твердого вещества. m/z (esi) M+1=335,7.
[00420] Стадия B: DIEA (0,26 мл, 1,49 ммоль) добавляли к перемешиваемому раствору 3-((3-хлор-4-йодпиридин-2-ил)амино)-2,2-диметилпропаннитрила (250 мг, 0,74 ммоль) и метил-3-меркаптопропанoата (0,09 мл, 0,82 ммоль) в диоксане (5 мл), и дегазировали аргоном в течение 10 мин. Добавляли ксантфос (22 мг, 0,03 ммоль) и Pd(OAC)2 (10 мг, 0,04 ммоль), и дегазировали в течение еще 10 мин. Реакционную смесь перемешивали в предварительно нагретой масляной бане при 100°C в течение 4 ч. Реакционную смесь фильтровали через слой Celite® и промывали EtOAc. Растворитель выпаривали, и неочищенный материал очищали колоночной хроматографией на силикагеле (50% EtOAc-гексан) с получением метил-3-((3-хлор-2-((2-цианo-2-метилпропил)амино)пиридин-4-ил)тио)пропанoата (230 мг, выход 94%) в виде твердого вещества. m/z (esi) M+1=327,9.
[00421] Стадия C: NaOEt (21% масс. в EtOH) (0,25 мл, 0,77 ммоль) добавляли к перемешиваемому раствору 3-((3-хлор-2-((2-цианo-2-метилпропил)амино)пиридин-4-ил)тио)пропанoата (210 мг, 0,64 ммоль) в THF (5 мл) при 0°С и перемешивали в течение 30 мин при 0°С. Реакционную смесь концентрировали при 25°С и добавляли простой диэтиловый эфир (10 мл). Полученное твердое вещество фильтровали с получением 3-хлор-2-((2-цианo-2-метилпропил)амино)пиридин-4-тиолата натрия (130 мг, выход 90%) в виде твердого вещества. m/z (esi) M+1=242,1.
[00422] Стадия D: 3-Хлор-2-((2-цианo-2-метилпропил)амино)пиридин-4-тиолат натрия (132,34 мг, 0,5 ммоль) добавляли к перемешиваемому раствору (S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (60,0 мг, 0,1 ммоль) в н-бутаноле (1,5 мл) и перемешивали при 120°С в течение 16 ч в герметично закрытой пробирке. Реакционную смесь концентрировали и неочищенный материал очищали обращенно-фазовой препаративной ВЭЖХ (30-95% ACN:вода (0,1% NH3)) с получением (S)-3-((4-((3-(1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-ил)-1,2,4-триазин-6-ил)тио)-3-хлорпиридин-2-ил)амино)-2,2-диметилпропаннитрила (8 мг, 0,017 ммоль, выход 9%) в виде твердого вещества. 1H ЯМР (400 МГц, Метанол-d4) δ 8,37 (с, 1H), 7,76 (д, J=5,5 Гц, 1H), 7,40-7,33 (м, 1H), 7,25-7,16 (м, 3H), 6,05 (д, J=5,5 Гц, 1H), 4,69 (с, 2H), 3,98 (с, 1H), 1,40-1,22 (м, 7H), 3,72 (с, 2H), 3,51-3,35 (м, 2H), 3,20 (д, J=15,7 Гц, 1H), 2,84 (д, J=15,8 Гц, 1H), 1,97-1,70 (м, 2H), 1,65 (д, J=13,3 Гц, 1H), 1,46 (д, J=13,4 Гц, 1H), 1,36 (с, 6H); m/z (esi) M+1=521,5.
Пример 37
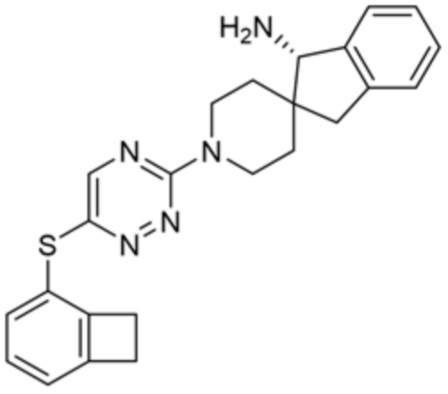
(S)-1'-(6-(бицикло[4.2.0]октa-1(6),2,4-триен-2-илтио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00423] Стадия A: т-BuLi (56 мл, 96 ммоль, 1,7 M в пентaне) добавляли по каплям к перемешиваемому раствору 1-фтор-2-(2-йодэтил)бензола (6,0 г, 24,0 ммоль) в пентaне (60 мл) и Et2O (20 мл) при -78°С в атмосфере аргона. Реакционную смесь перемешивали при -78°C в течение дополнительных 30 мин, и в нее добавляли безводный THF (12,0 мл). Охлаждающую баню убирали, и реакционную смесь перемешивали в течение дополнительных 15 мин. Добавляли I2 (7,31 г, 28,8 ммоль) путем растворения в Et2O (60 мл). Перед добавлением эфирный раствор I2 дегазировали в течение 5 мин аргоном. Реакционную смесь гасили NH4Cl и экстрагировали Et2O. Объединенный органический слой сушили (Na2SO4) и выпаривали с получением 2-йодбицикло[4.2.0]октa-1,3,5-триена (6,6 г, неочищенный), который использовали на следующей стадии. m/z (ГХ-МС) M=230.
[00424] Стадия B: DIEA (4,24 мл, 24,35 ммоль) добавляли к перемешиваемому раствору 2-йодбицикло[4.2.0]октa-1,3,5-триена (6,6 г, неочищенный) и метил-3-меркаптопропанoата (1,48 мл, 13,39 ммоль) в диоксане (25 мл), и дегазировали аргоном в течение 10 мин. Добавляли ксантфос (352 мг, 0,61 ммоль) и Pd(OAC)2 (164 мг, 0,73 ммоль), и дегазировали в течение еще 10 мин. Реакционную смесь перемешивали в предварительно нагретой масляной бане в герметичной пробирке при температуре 100°C в течение 4 ч. Реакционную смесь фильтровали через слой Celite® и промывали этилацетатом. Растворитель выпаривали, и неочищенный материал очищали колоночной хроматографией на силикагеле (5% MeOH/DCM) с получением метил-3-(бицикло[4.2.0]октa-1,3,5-триен-2-илтио)пропанoата (2,5 г, выход 46%, 2 стадии) в виде твердого вещества. m/z (esi) M+1=223.
[00425] Стадия C: NaOEt (1,7 мл, 5,40 ммоль, 21% масс. в EtOH) добавляли к перемешиваемому раствору метил-3-(бицикло[4.2.0]октa-1,3,5-триен-2-илтио)пропанoата (1 г, 4,50 ммоль) в THF (15 мл) при -78°С и перемешивали в течение 30 мин при той же температуре. Реакционную смесь концентрировали и неочищенное вещество растирали в эфире. Твердую фазу отфильтровывали с получением бицикло[4.2.0]октa-1,3,5-триен-2-тиолата натрия (520 мг, выход 73%) в виде твердого вещества. m/z (esi) M-Na=134,9.
[00426] Стадия D: Бицикло[4.2.0]октa-1,3,5-триен-2-тиолат натрия (88 мг, 0,56 ммоль) добавляли к перемешиваемому раствору (S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (100 мг, 0,28 ммоль) в бутаноле (5 мл) и перемешивали при 100°С в течение 4 ч. Реакционную смесь концентрировали и очищали с помощью обращенно-фазовой препаративной ВЭЖХ (50-90% ACN:0,1% NH4OH в H2O) с получением (S)-1'-(6-(бицикло[4.2.0]октa-1(6),2,4-триен-2-илтио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (30 мг, выход 26%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,36 (с, 1H), 7,30 (д, J=6,5 Гц, 1H), 7,23-7,09 (м, 5H), 7,00 (д, J=7,1 Гц, 1H), 4,49 (с, 2H), 3,86 (с, 1H), 3,40-3,34 (м, 1H), 3,29-3,21 (м, 1H), 3,16-3,03 (м, 3H), 2,82 (т, J=4,2 Гц, 2H), 2,66 (д, J=15,3 Гц, 1H), 2,16-1,83 (м, 2H), 1,77 (т, J=11,6 Гц, 1H), 1,66 (т, J=11,6 Гц, 1H), 1,55 (д, J=13,0 Гц, 1H), 1,14 (д, J=13,1 Гц, 1H). m/z (esi) M+1=416,2.
Пример 38
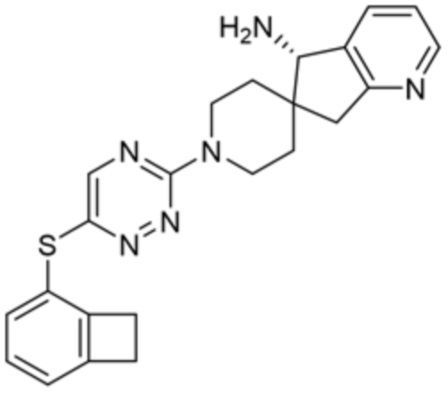
(S)-1'-(6-(бицикло[4.2.0]октa-1(6),2,4-триен-2-илтио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амин
[00427] Стадия A: Триэтиламин (0,30 мл, 2,12 ммоль) добавляли к перемешиваемому раствору дигидрохлоридной соли (S)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (117 мг, 0,42 ммоль) в 1,4-диоксане (2,8 мл) при комнатной температуре и перемешивали в течение 20 мин. Добавляли 3,6-дибром-1,2,4-триазин (100 мг, 0,42 ммоль), перемешивали в течение 2 ч при комнатной температуре и нагревали при 50°С в течение 1 ч. Растворитель выпаривали и очищали хроматографией на силикагеле (3% MeOH-DCM) с получением (S)-1'-(6-бром-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (75 мг, выход 49%) в виде твердого вещества. m/z (esi) M+1=360,8.
[00428] Стадия B: Бицикло[4.2.0]октa-1,3,5-триен-2-тиолaт натрия (63,2 мг, 0,4 ммоль) добавляли к перемешиваемому раствору (S)-1'-(6-бром-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (72 мг, 0,2 ммоль) в н-бутаноле (4 мл) и перемешивали при 100°С в течение 4 ч. Реакционную смесь выпаривали, и неочищенную реакционную смесь растворяли в ДМСО и очищали обращенно-фазовой препаративной ВЭЖХ (30-70% ACN:вода (0,1% NH3)) с получением (S)-1'-(6-(бицикло[4.2.0]октa-1(6),2,4-триен-2-илтио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (19,3 мг, выход 24%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,37 (с, 1H), 8,32 (д, J=5,0 Гц, 1H), 7,65 (д, J=7,5 Гц, 1H), 7,23-7,09 (м, 3H), 7,01 (д, J=7,1 Гц, 1H), 4,50 (с, 2H), 3,91 (с, 1H), 3,44-3,36 (м, 1H), 3,30-3,23 (м, 1H), 3,20-3,02 (м, 3H), 2,86-2,73 (м, 3H), 2,18-2,13 (м, 2H), 1,85-1,62 (м, 2H), 1,58 (д, J=13,3 Гц, 1H), 1,17 (д, J=13,6 Гц, 1H). m/z (esi) M+1=417,2.
Пример 39
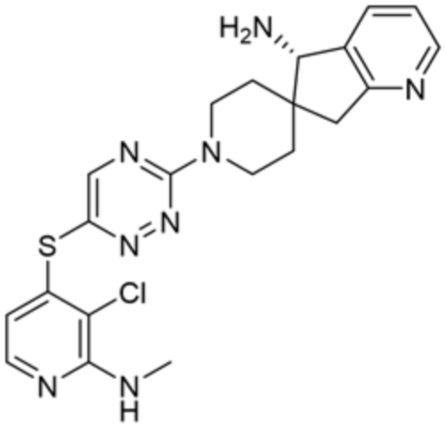
(S)-1'-(6-((3-хлор-2-(метиламино)пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амин
[00429] Стадия A: TEA (0,15 мл, 1,06 ммоль) добавляли к перемешиваемому раствору гидрохлорида (S)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (58,47 мг, 0,21 ммоль) в 1,4-диоксане (1,4 мл) при комнатной температуре и перемешивали в течение 20 мин. Добавляли 3,6-дибром-1,2,4-триазин (50 мг, 0,21 ммоль) и перемешивали в течение 2 ч при комнатной температуре, и затем нагревали при 50°С в течение 1 ч. Растворитель выпаривали и очищали колоночной хроматографией на силикагеле (7-14% MeOH/DCM) с получением (S)-1'-(6-бром-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (70 мг, выход 91%) в виде твердого вещества. m/z (esi) M+1=362,9.
[00430] Стадия B: 3-Хлор-2-(метиламино)пиридин-4-тиол (72,5 мг, 0,42 ммоль) добавляли к перемешиваемому раствору (S)-1'-(6-бром-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (50 мг, 0,14 ммоль) в н-BuOH (5 мл) и нагревали до 90°С в течение 16 ч. Реакционную смесь очищали обращенно-фазовой препаративной ВЭЖХ (20-50% ACN:вода (0,1% NH3)) с получением (S)-1'-(6-((3-хлор-2-(метиламино)пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (13 мг, выход 21%) в виде клейкого твердого вещества. 1H ЯМР (400 МГц, Метанол-d4) δ 8,38 (с, 2H), 7,85 (д, J=7,6 Гц, 1H), 7,74 (д, J=5,6 Гц, 1H), 7,30 (т, J=6,3 Гц, 1H), 5,98 (д, J=5,4 Гц, 1H), 4,79-4,67 (м, 2H), 4,15 (с, 1H), 3,47-3,42 (м, 2H), 3,02 (д, J=16,7 Гц, 1H), 2,94 (с, 3H), 1,98-1,76 (м, 2H), 1,69 (д, J=13,6 Гц, 1H), 1,52 (д, J=13,4 Гц, 1H). m/z (esi) M+1=455,4.
Пример 40
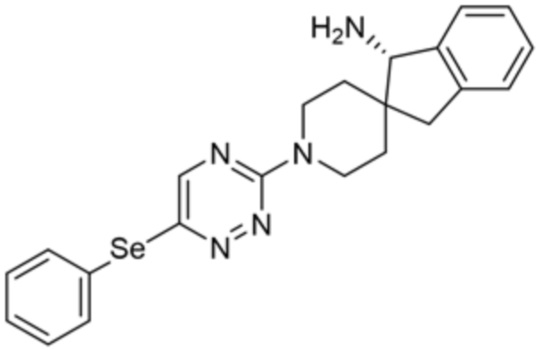
(S)-1'-(6-(фенилселанил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
[00431] Стадия A: NaBH4 (163 мг, 4,3 ммоль) добавляли к перемешиваемому раствору дифенилдиселена (161 мг, 0,5 ммоль) в PEG-400 (5 мл), и реакционную смесь нагревали при 70°С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли N-((S)-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид (200 мг, 0,43 ммоль). Реакционную смесь снова нагревали при 70°С в течение 16 ч в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, выливали в воду, экстрагировали этилацетатом (3×150 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле, используя 70% этилацетат в гексане, с получением 2-метил-N-((S)-1'-(6-(фенилселанил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)пропан-2-сульфинамида (100 мг, 43%) в виде твердого вещества. МС (m/z)=542,2 (M+H).
[00432] Стадия B: 4 н. HCl в диоксане (1 мл) добавляли к перемешиваемому раствору 2-метил-N-((S)-1'-(6-(фенилселанил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)пропан-2-сульфинамида (50 мг, 0,09 ммоль) в MeOH (1 мл), и реакционную смесь перемешивали в течение 30 мин при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, и неочищенный материал промывали простым диэтиловым эфиром, сушили при пониженном давлении и очищали препаративной ВЭЖХ (Xbridge C18 (50×19 мм, 5 μ), работающей при температуре окружающей среды и скорости потока 20 мл/мин. Подвижная фаза: А=20 мМ бикарбоната аммония в воде, B=ацетонитрил; Профиль градиента: Исходная композиция подвижной фазы 80% A и 20% B, затем до 20% A и 80% B за 12 мин, затем до 5% A и 95% B за 13 мин, выдерживали данный состав до 15 мин), с получением (S)-1'-(6-(фенилселанил)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амина (15 мг, 37%) в виде твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 8,36 (с, 1H), 7,52 (дд, J=3,1, 6,5 Гц, 2H), 7,39-7,26 (м, 4H), 7,23-7,10 (м, 3H), 4,46 (т, J=13,9 Гц, 2H), 3,84 (с, 1H), 3,42-3,33 (м, 1H), 3,24 (д, J=12,8 Гц, 1H), 3,10 (д, J=15,6 Гц, 1H), 2,64 (д, J=15,7 Гц, 1H), 1,82-1,70 (м, 2H), 1,69-1,58 (м, 2H), 1,54 (д, J=13,4 Гц, 1H), 1,13 (д, J=14,2 Гц, 1H); МС (m/z)=438,4 (M+H).
Пример 41
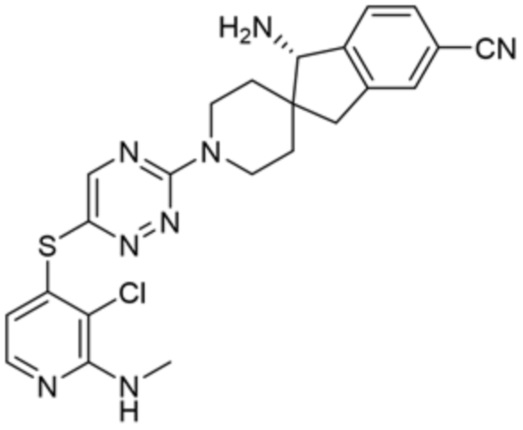
(S)-1-амино-1'-(6-((3-хлор-2-(метиламино)пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-5-карбонитрил
[00433] Стадия A: Триэтиламин (0,26 мл, 1,88 ммоль) добавляли к перемешиваемому раствору 3,6-дибром-1,2,4-триазина (180 мг, 0,75 ммоль) в диоксане (5 мл) при 0°С и перемешивали в течение 5 мин. Гидрохлорид 1-амино-1,3-дигидроспиро[инден-2,4'-пиперидин]-5-карбонитрила (224 мг, 0,75 ммоль) в диоксане (1 мл) добавляли по каплям при 0°С и перемешивали в течение 1 ч. Реакционную смесь разбавляли водой и экстрагировали EtOAc. Органическую часть сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле (10% MeOH/DCM) с получением (S)-1-амино-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-5-карбонитрила (200 мг, выход 69%) в виде твердого вещества. m/z (esi) M+1=385,2.
[00434] Стадия B: 3-Хлор-2-(метиламино)пиридин-4-тиолaт натрия (92 мг, 0,46 ммоль) добавляли к перемешиваемому раствору (S)-1-амино-1'-(6-бром-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-5-карбонитрила (90 мг, 0,23 ммоль) в н-бутаноле (5 мл) и перемешивали в микроволновой печи при 120°С в течение 1 ч. Реакционную смесь концентрировали и полученный остаток очищали обращенно-фазовой препаративной ВЭЖХ (40-75% ACN: вода (0,1% NH3)) с получением рацемического продукта, который подвергали хиральному разделению (Chiralpak OJ-H (250×20 мм) 5μ, Поток: 25 г/мин, Подвижная фаза: 60% CO2+40% (0,5% изопропиламин в метаноле), ABPR: 120 бар (12 МПа), температура: 35°C). Сбор первого элюируемого пика давал (S)-1-амино-1'-(6-((3-хлор-2-(метиламино)пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-5-карбонитрил (10 мг, выход 9%). 1H ЯМР (400 МГц, (CD3)2SO) δ 8,48 (с, 1H), 7,80 (д, J=5,3 Гц, 1H), 7,67 (д, J=6,9 Гц, 2H), 7,50 (д, J=7,7 Гц, 1H), 6,66 (д, J=4,8 Гц, 1H), 5,93 (д, J=5,3 Гц, 1H), 4,57 (с, 2H), 3,93 (с, 1H), 3,20 (д, J=15,9 Гц, 1H), 2,85 (д, J=4,5 Гц, 3H), 2,72 (д, J=16,0 Гц, 1H), 2,15-1,90 (м, 2H), 1,90-1,76 (м, 2H), 1,64 (т, J=14,3 Гц, 2H), 1,11 (д, J=13,8 Гц, 1H). m/z (esi) M+1=479,1.
Пример 42
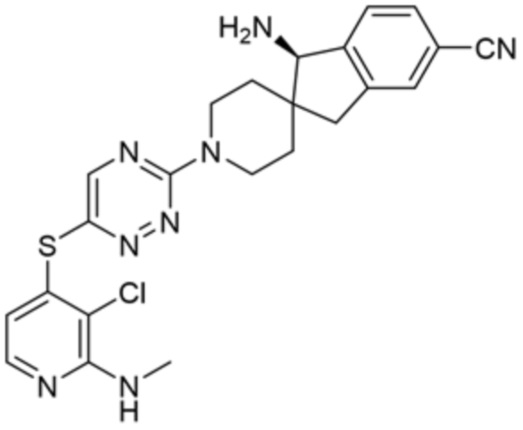
(R)-1-амино-1'-(6-((3-хлор-2-(метиламино)пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-5-карбонитрил
[00435] (R)-1-амино-1'-(6-((3-хлор-2-(метиламино)пиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-5-карбонитрил (10 мг, выход 9%) получали в соответствии с примером 41, собирая второй элюируемый пик на стадии В. 1H ЯМР (400 МГц, Метанол-d4) δ 8,38 (с, 1H), 7,76 (д, J=5,6 Гц, 1H), 7,61 (д, J=6,3 Гц, 2H), 7,55 (д, J=8,0 Гц, 1H), 6,00 (д, J=5,6 Гц, 1H), 3,49-3,34 (м, 3H), 3,31-3,17 (м, 2H), 2,97 (с, 3H), 2,88 (д, J=16,1 Гц, 1H), 2,02-1,88 (м, 1H), 1,86-1,73 (м, 1H), 1,69 (д, J=13,3 Гц, 1H), 1,38 (д, J=14,0 Гц, 1H), 1,30 (с, 2H), 0,91 (д, J=7,2 Гц, 1H), m/z (esi) M+1=479,1.
Пример 43
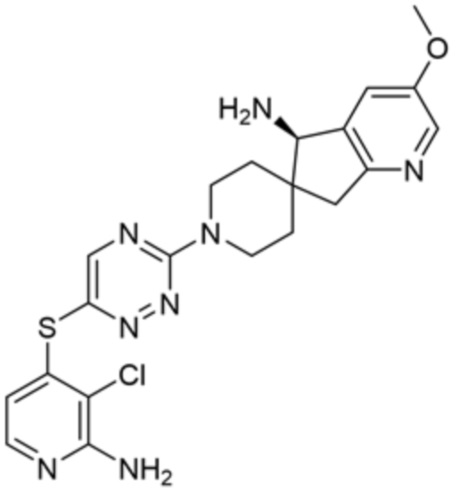
(R)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амин
[00436] Стадия A: Триэтиламин (0,74 мл, 5,0 ммоль) добавляли к перемешиваемому раствору гидрохлорида 3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (418 мг, 1,37 ммоль) в диоксане (10 мл) и перемешивали при комнатной температуре в течение 20 мин. Добавляли 3,6-дибром-1,2,4-триазин (249 мг, 1,05 ммоль) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь нагревали при 50°С в течение 1 ч. Растворитель концентрировали досуха и промывали Et2O, с получением 1'-(6-бром-1,2,4-триазин-3-ил)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (410 мг, неочищенный) в виде твердого вещества. Неочищенную массу использовали на следующей стадии без очистки. m/z (esi) M+1=391,1.
[00437] Стадия B: 2-Амино-3-хлорпиридин-4-тиолaт натрия (93 мг, 0,511 ммоль) добавляли к перемешиваемому раствору 1'-(6-бром-1,2,4-триазин-3-ил)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (100 мг, 0,256 ммоль) в н-бутаноле (2,0 мл) и перемешивали при 120°С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли 2-амино-3-хлорпиридин-4-тиолат натрия (93 мг, 0,51 ммоль), и перемешивали при 120°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и промывали Et2O. Неочищенный материал смешивали с другой порцией (S)-1'-(6-бром-1,2,4-триазин-3-ил)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амина (100 мг) и очищали хиральной ВЭЖХ (Chiralpak IG (21,0×250 мм), 5μ, DCM/EtOH/изопропиламин 40/60/0,1, 9,0 мл/мин). Сбор первого элюируемого пика давал (S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амин (12 мг, 5%, трехстадийный выход) в виде твердого вещества. 1H ЯМР (400 МГц, Метанол-d4) δ 8,42 (с, 1H), 8,24 (д, J=2,8 Гц, 1H), 7,66 (д, J=5,5 Гц, 1H), 7,53 (с, 1H), 6,04 (д, J=5,5 Гц, 1H), 4,77-4,66 (м, 2H), 4,46 (с, 1H), 3,91 (с, 3H), 3,50-3,39 (м, 2H), 3,28-3,12 (м, 2H), 1,95-1,60 (м, 4H).
Пример 44
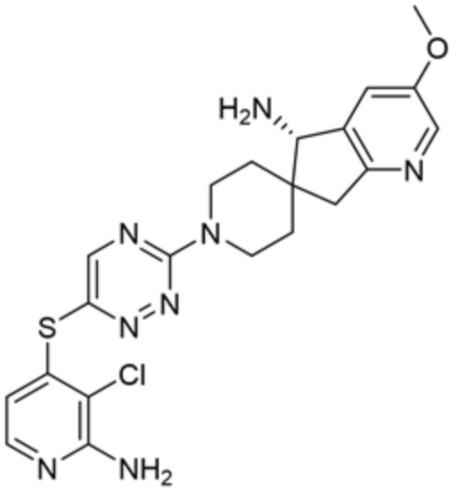
(S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амин
[00438] (S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-3-метокси-5,7-дигидроспиро[циклопентa[b]пиридин-6,4'-пиперидин]-5-амин (5 мг, 5%, двухстадийный выход) получали в соответствии с примером 43, собирая второй элюируемый пик в виде твердого вещества. 1H ЯМР (400 МГц, Метанол-d4) δ 8,40 (с, 1H), 8,16 (с, 1H), 7,66 (д, J=5,4 Гц, 1H), 7,50 (с, 1H), 6,04 (д, J=5,5 Гц, 1H), 4,84-4,64 (м, 2H), 4,30 (с, 1H), 3,89 (с, 3H), 3,44 (т, J=12,7 Гц, 2H), 3,26-3,17 (м, 1H), 3,06 (д, J=16,5 Гц, 1H), 1,94-1,75 (м, 2H), 1,76-1,58 (м, 2H); m/z (esi) M+1=471,1.
[00439] Следующие соединения в таблице 4 были получены в соответствии с вышеуказанными процедурами, с использованием соответствующих исходных материалов и промежуточных соединений.
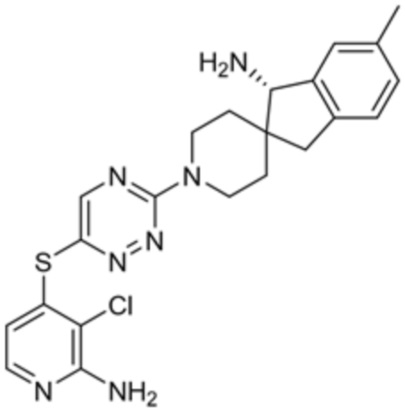
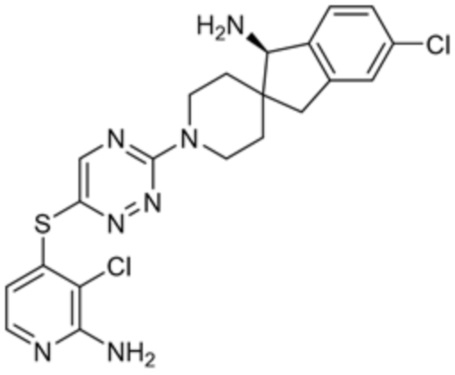
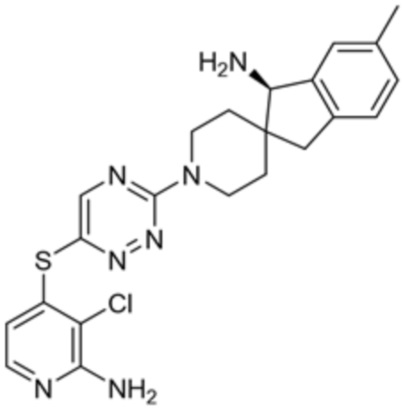
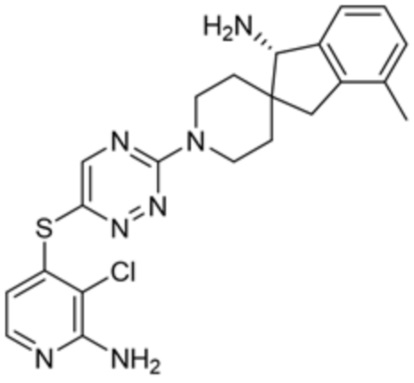
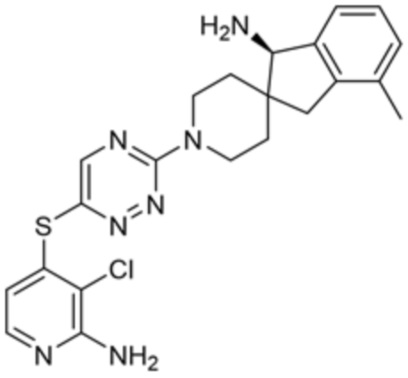
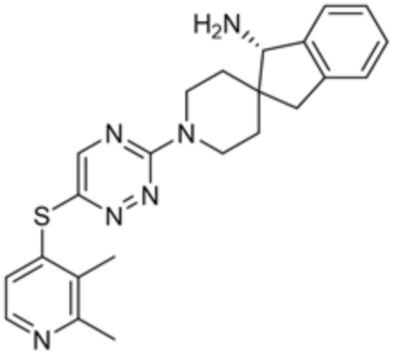
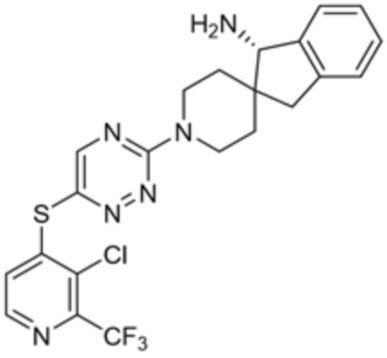
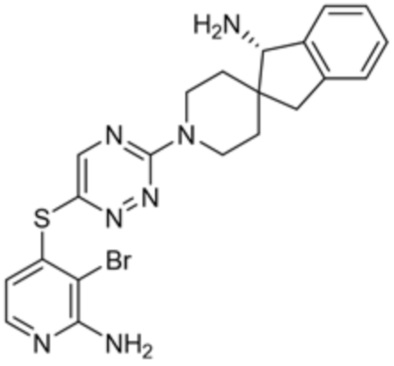
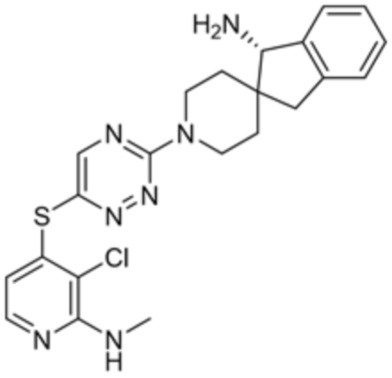
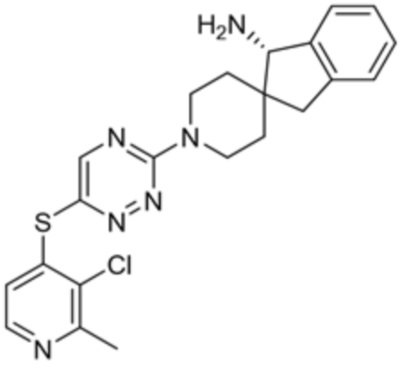
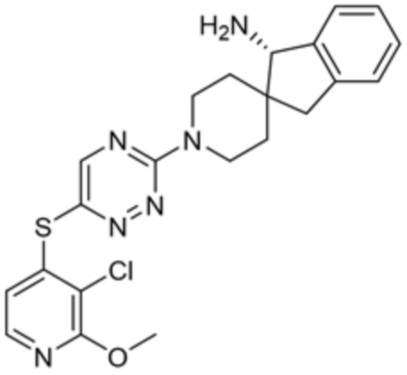
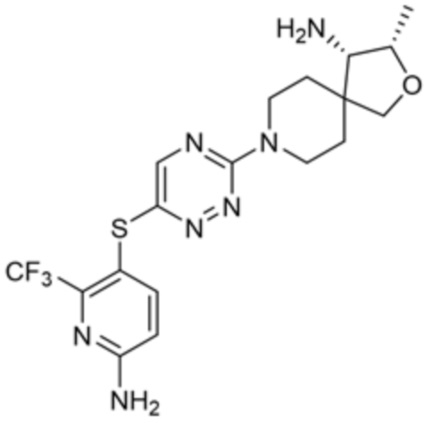
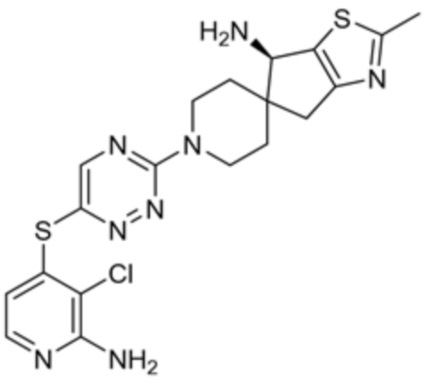
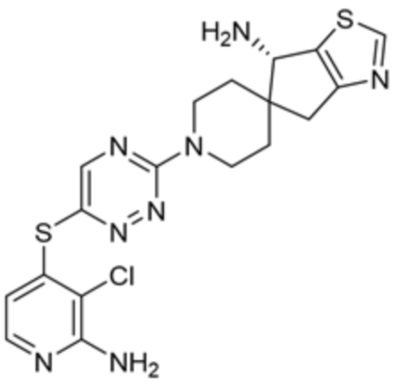
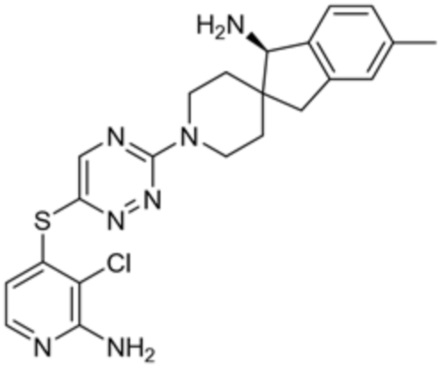
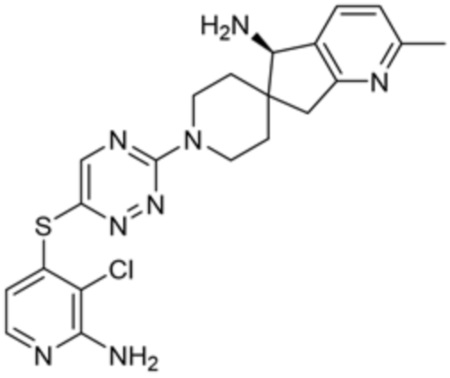
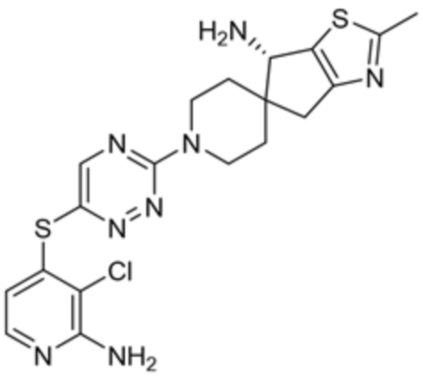
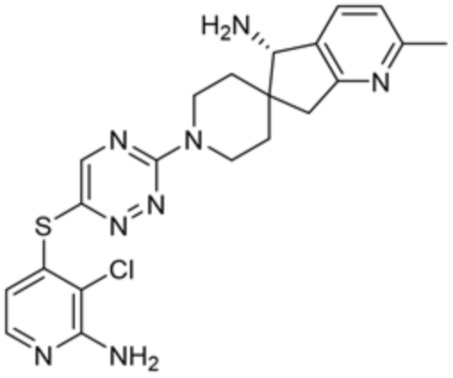
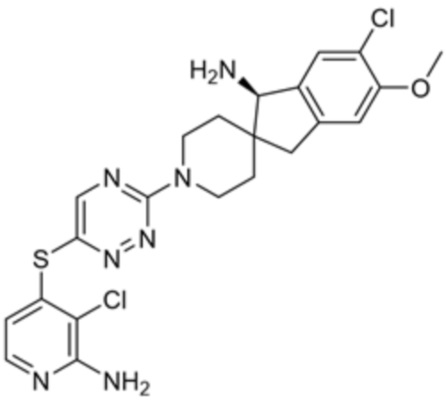
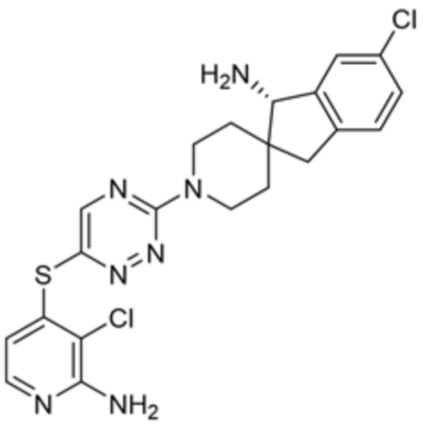
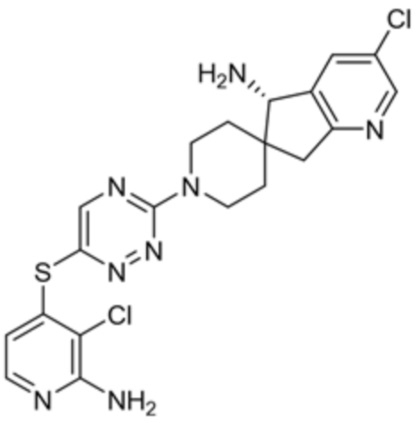
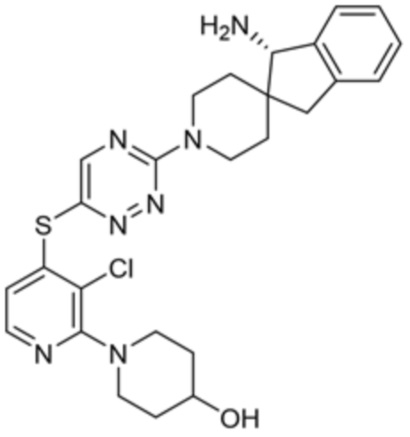
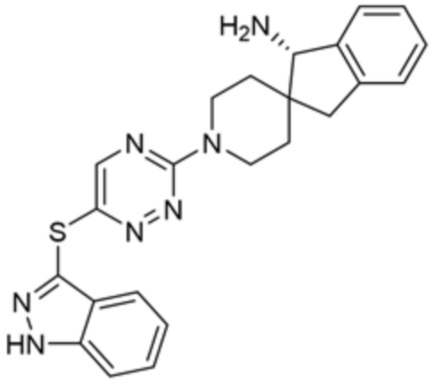
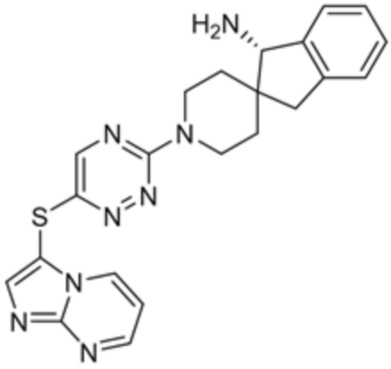
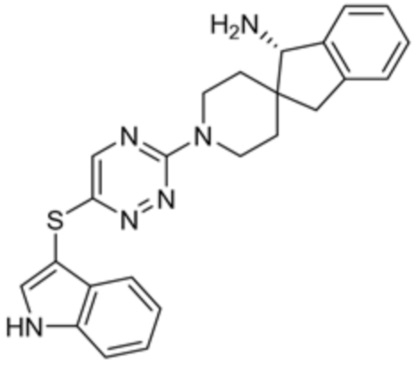
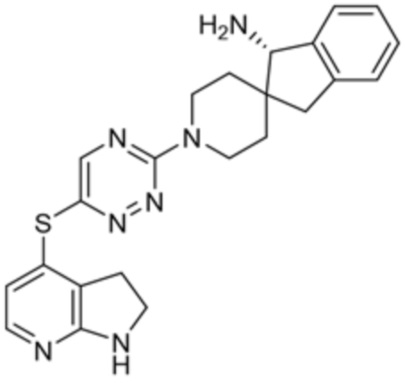
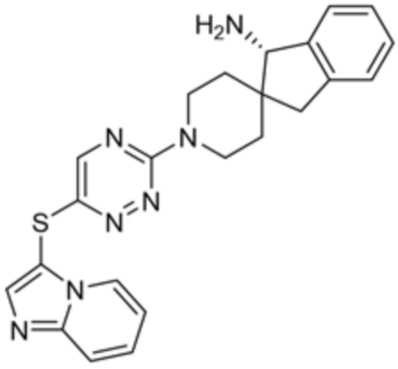
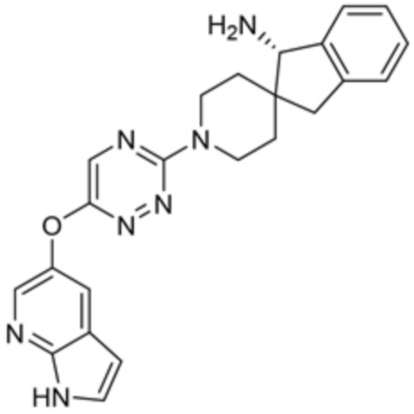
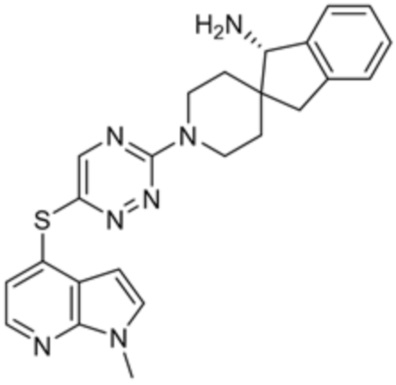
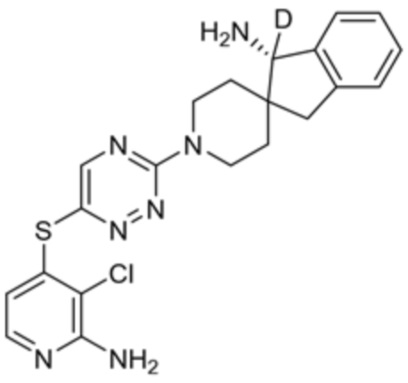
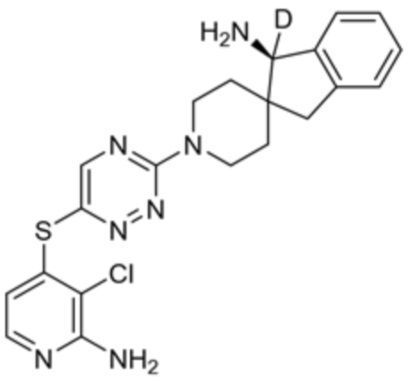
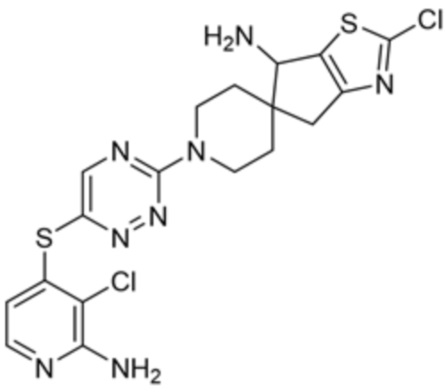
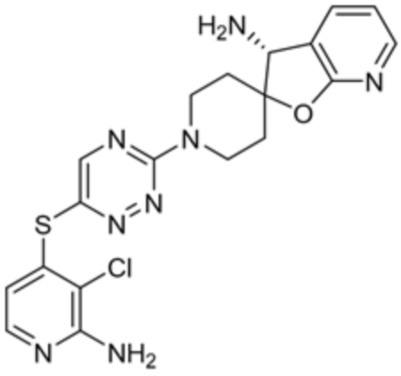
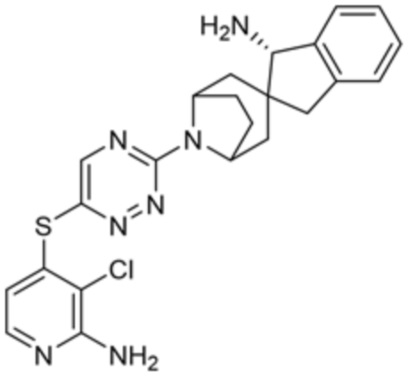
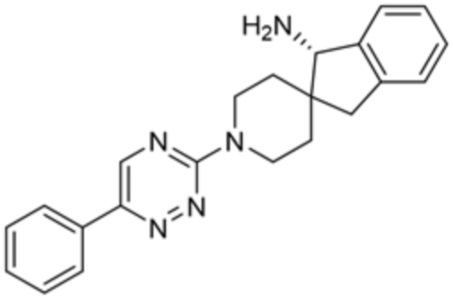
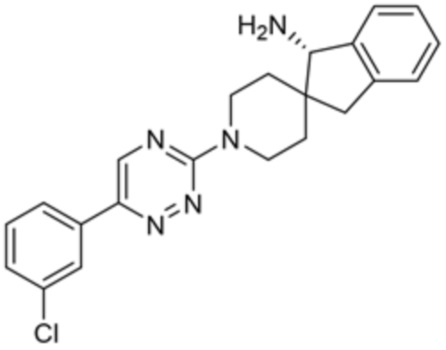
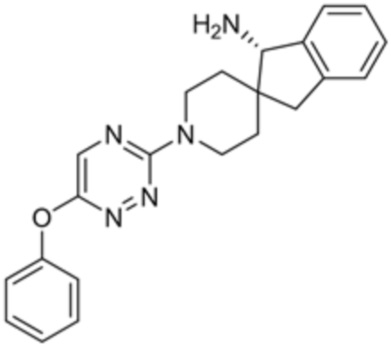
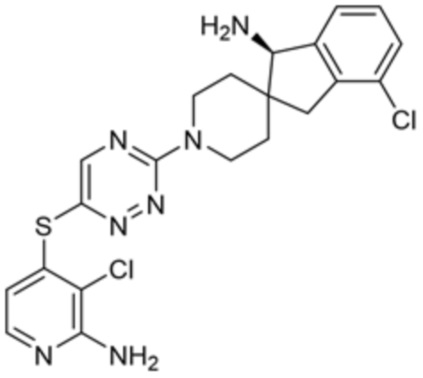
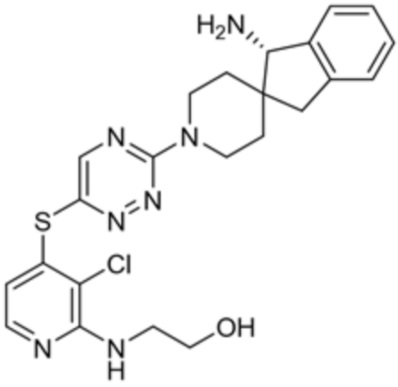
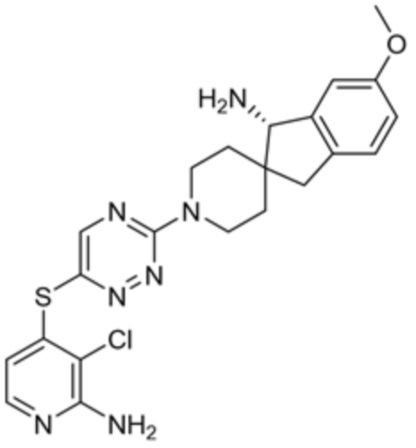
[00440] Следует понимать, что перечисленные варианты осуществления не предназначены для ограничения изобретения этими вариантами осуществления. Напротив, изобретение предназначено для охвата всех альтернатив, модификаций и эквивалентов, которые могут быть включены в объем настоящего изобретения, определенный формулой изобретения. Соответственно, вышеприведенное описание рассматривается только в качестве иллюстрации принципов изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ СО СПИРО- И АРОМАТИЧЕСКИМИ КОЛЬЦАМИ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2781100C1 |
| ГЕМИНАЛЬНО-ЗАМЕЩЕННЫЕ ЦИАНОЭТИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ JANUS КИНАЗ | 2014 |
|
RU2664533C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ SUV39H2 | 2016 |
|
RU2729187C1 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2019 |
|
RU2811601C1 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| АГОНИСТЫ РЕЦЕПТОРА GLP-1 И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2740135C1 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2015 |
|
RU2685230C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2666538C2 |
Группа изобретений относится к фармацевтической химии и включает производные триазина, приведенные в формуле изобретения, лекарственное средство, фармацевтическую композицию, способ ингибирования и способ лечения на их основе, а также их применение. Технический результат – производные триазина, обладающие ингибирующей активностью в отношении SHP2 и применяемые для лечения гиперпролиферативного заболевания, связанного с активностью SHP2. 11 н. и 6 з.п. ф-лы, 4 табл., 44 пр.
1. Соединение или его стереоизомер, таутомер или фармацевтически приемлемая соль, где соединение выбрано из:
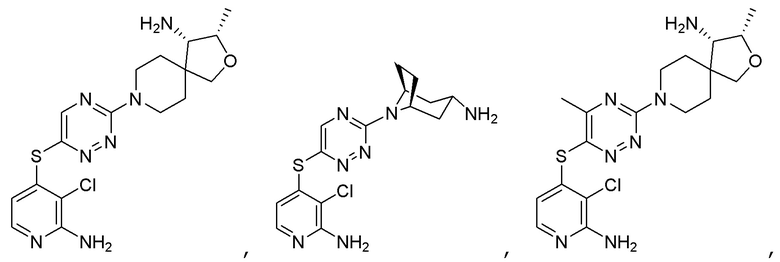
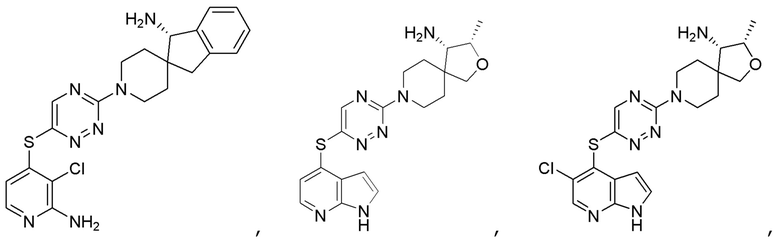
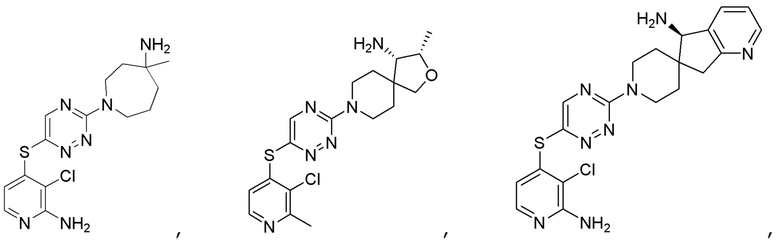
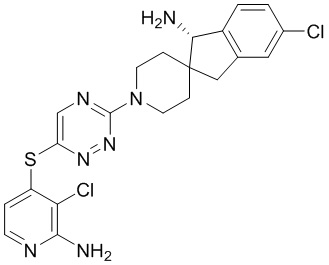 ,
, 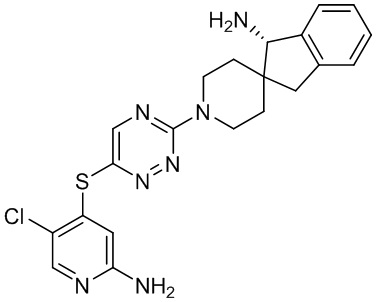 ,
, 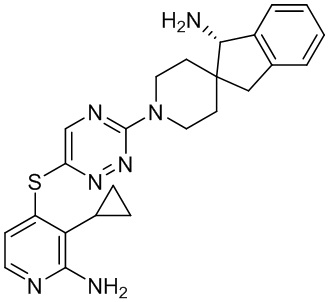 ,
, 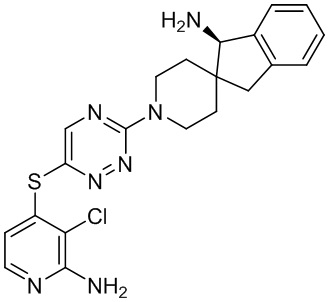 ,
,  ,
,  ,
,  ,
,  ,
, 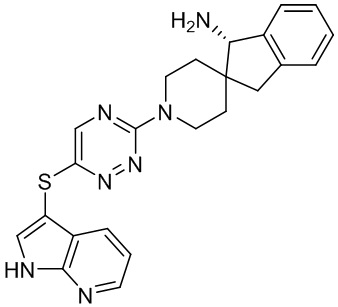 ,
, 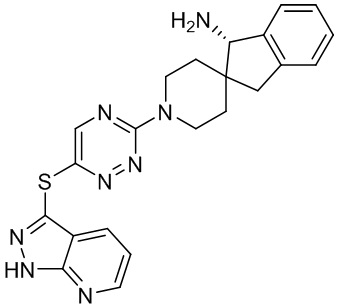 ,
, 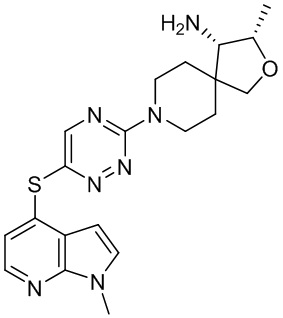 ,
, 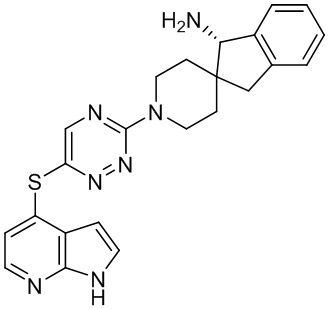 ,
,  ,
,  ,
,  ,
,  ,
, 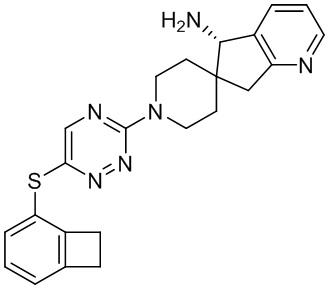 ,
, 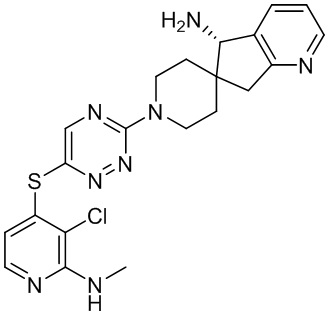 ,
, 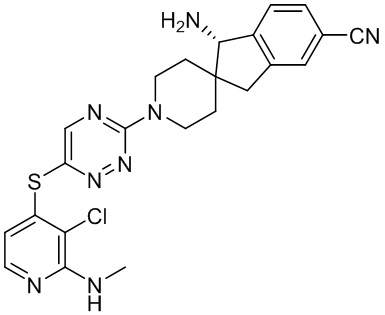 ,
, 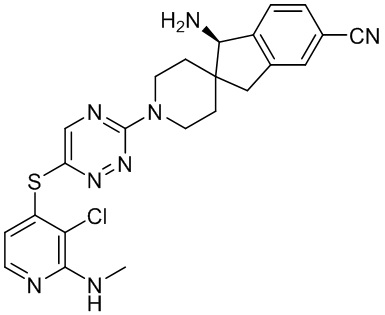 ,
, 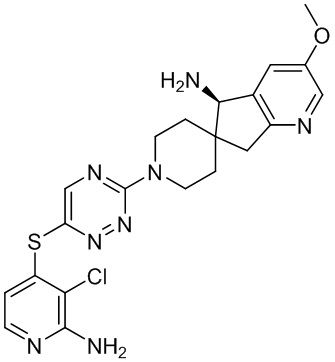 ,
, 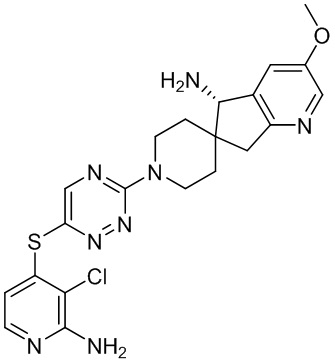 ,
, 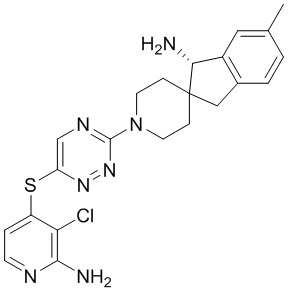 ,
, 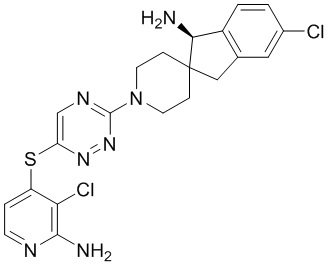 ,
, 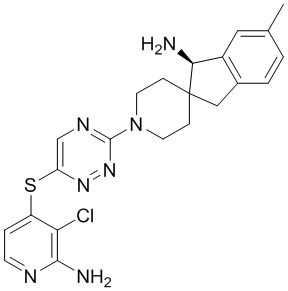 ,
, 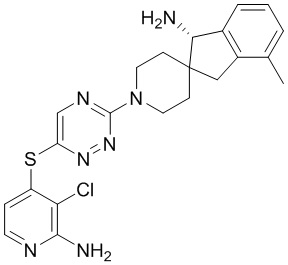 ,
, 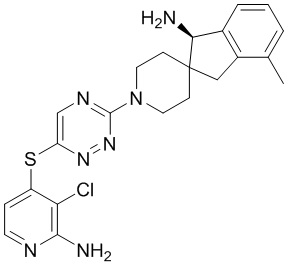 ,
, 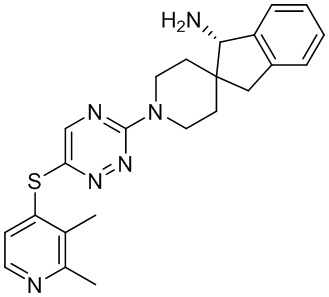 ,
, 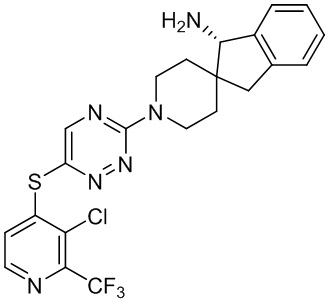 ,
, 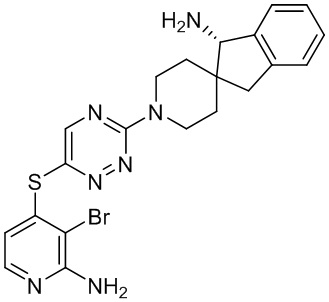 ,
, 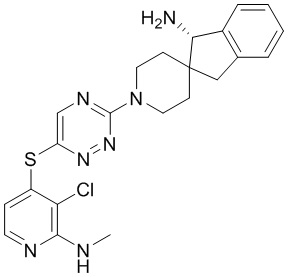 ,
, 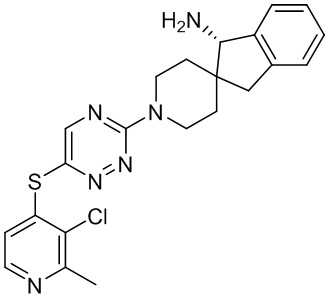 ,
,  ,
,  ,
,  ,
,  ,
, 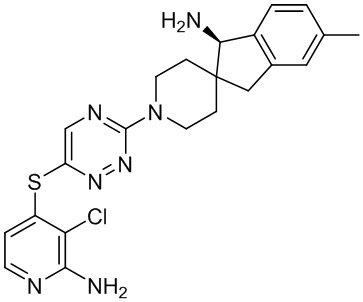 ,
, 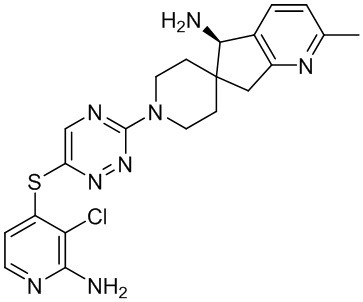 ,
, 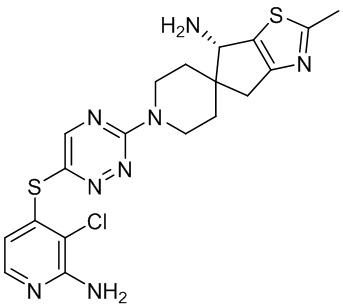 ,
, 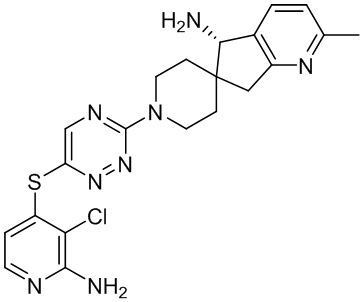 ,
, 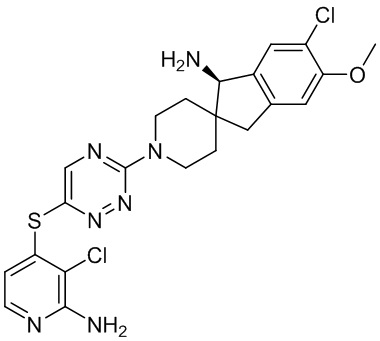 ,
, 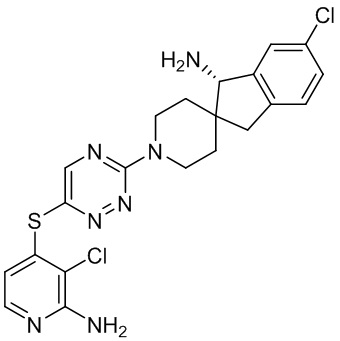 ,
, 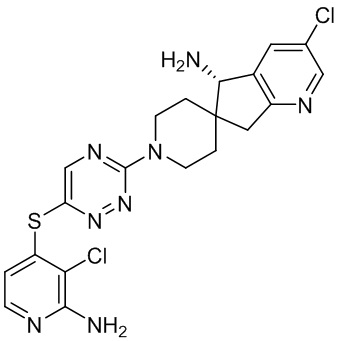 ,
, 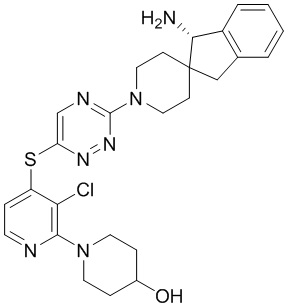 ,
, 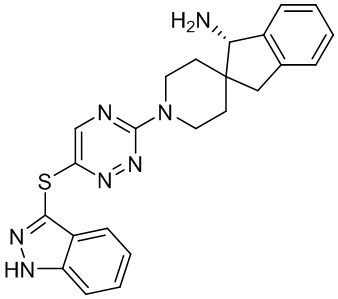 ,
, 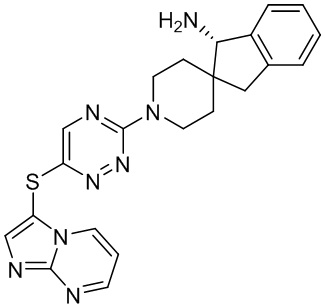 ,
, 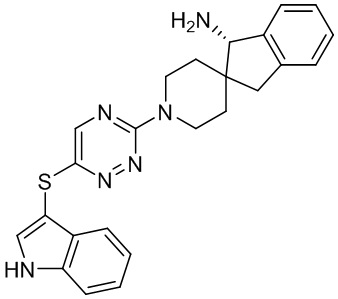 ,
, 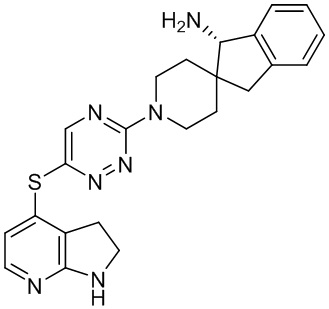 ,
, 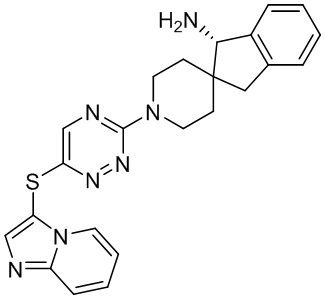 ,
, 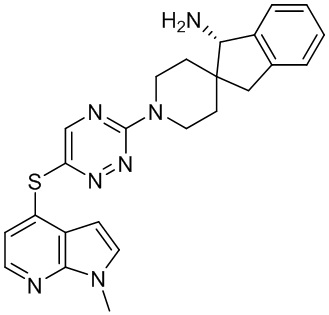 ,
, 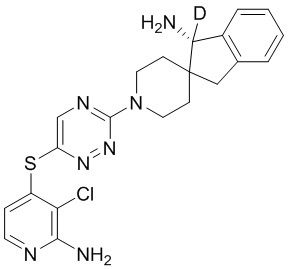 ,
, 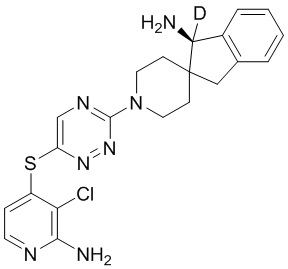 ,
, 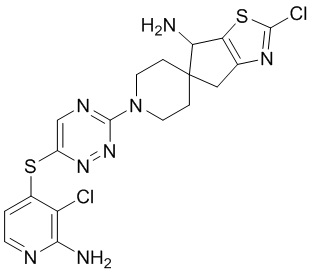 ,
, 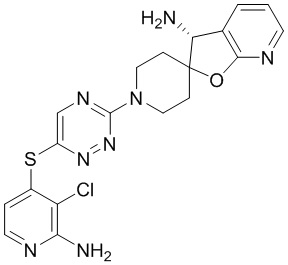 ,
, 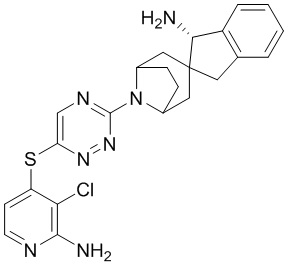 ,
, 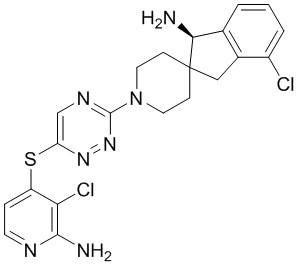 ,
, 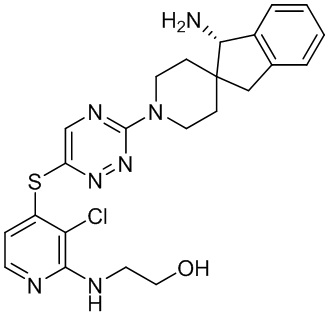 и
и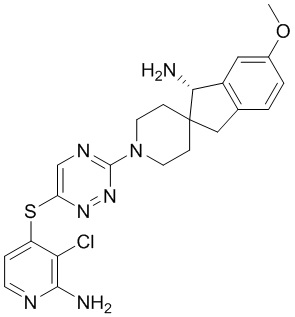 .
.
2. Соединение по п.1, или его таутомер или фармацевтически приемлемая соль, где соединение выбрано из:
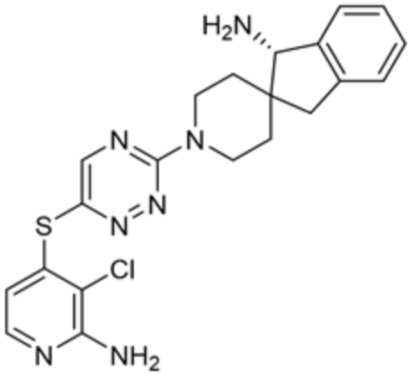 ,
, 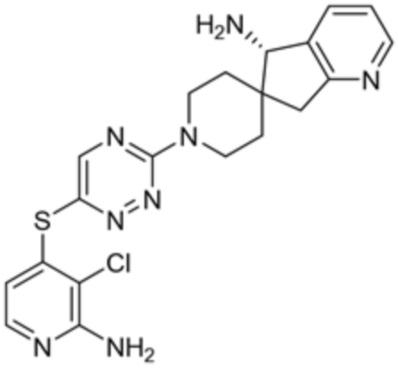 ,
, 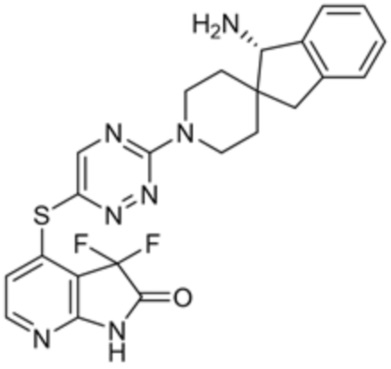 ,
, 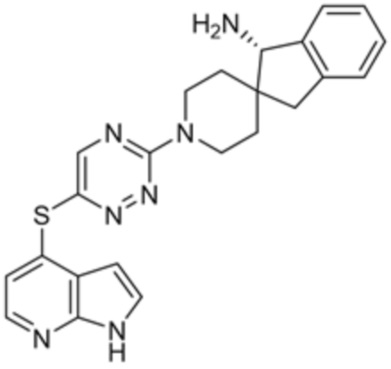 ,
, 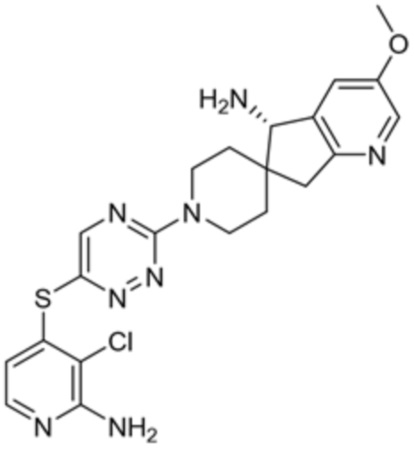 ,
, 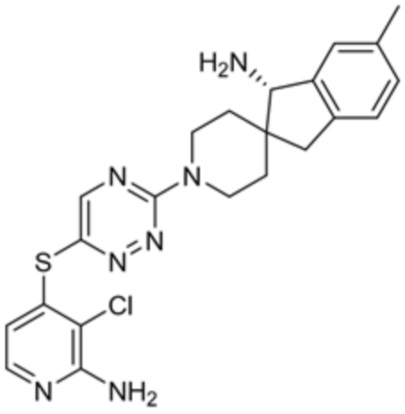 ,
, 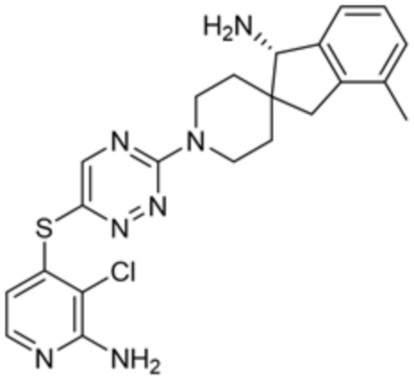 ,
, 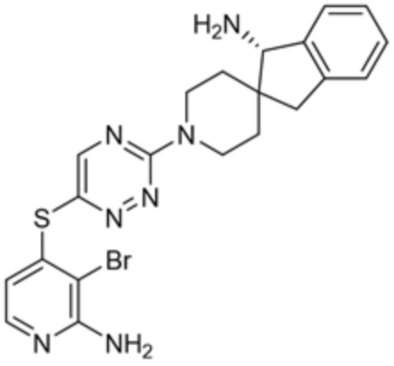 ,
, 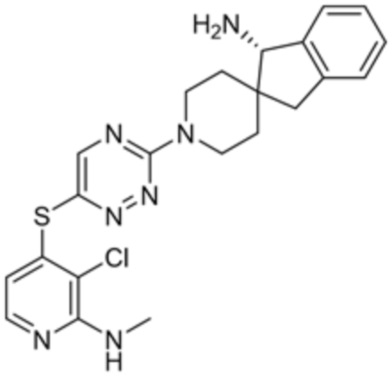 ,
, 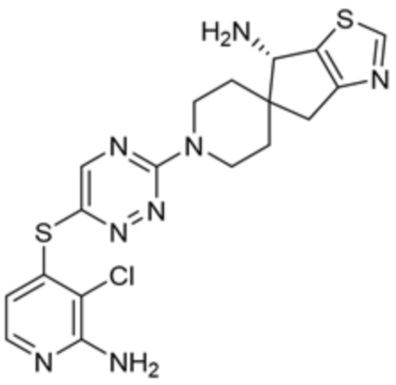 ,
, 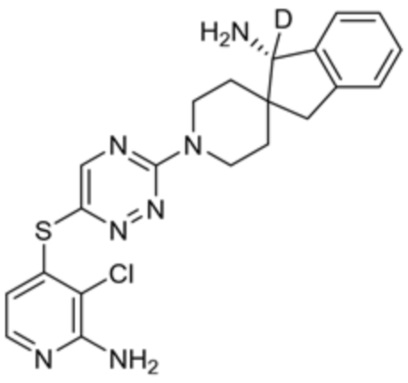 ,
, 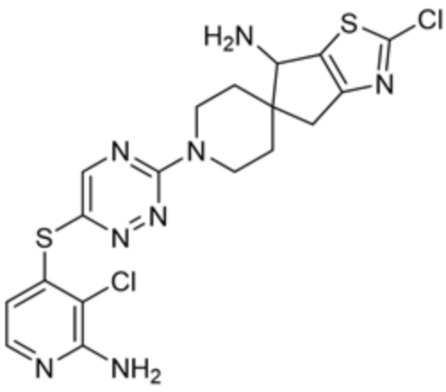 ,
, 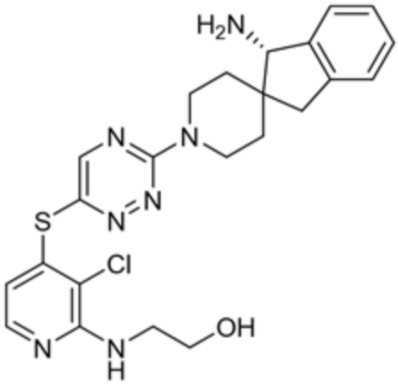 и
и 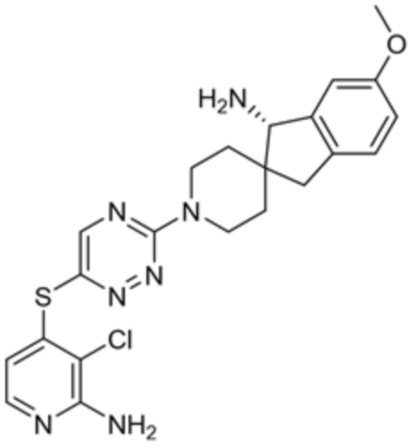 .
.
3. Соединение:
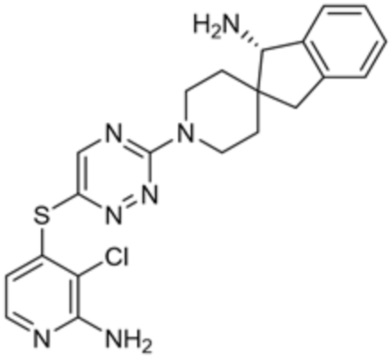
и его таутомеры и фармацевтически приемлемые соли.
4. Соединение:
и его таутомеры и фармацевтически приемлемые соли.
5. Соединение:
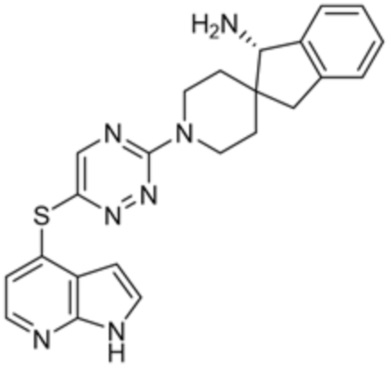
и его таутомеры и фармацевтически приемлемые соли.
6. Лекарственное средство, обладающее ингибирующей активностью в отношении SHP2, содержащее терапевтически эффективное количество соединения по любому из пп.1-5 или его стереоизомера, таутомера или фармацевтически приемлемой соли.
7. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении SHP2, содержащая терапевтически эффективное количество соединения по любому из пп.1-5 или его стереоизомера, таутомера или фармацевтически приемлемой соли, и по меньшей мере один фармацевтически приемлемый носитель, эксципиент или разбавитель.
8. Применение соединения по любому из пп.1-5, его стереоизомера, таутомера или фармацевтически приемлемой соли в производстве лекарственного средства для лечения гиперпролиферативного заболевания, связанного с активностью SHP2, где гиперпролиферативное заболевание выбрано из группы, состоящей из следующего: меланома, ювенильные миеломоноцитарные лейкозы, нейробластома, положительный по филадельфийской хромосоме хронический миелолейкоз, положительные по филадельфийской хромосоме острые лимфобластные лейкозы, острые миелоидные лейкозы, миелопролиферативные новообразования (такие как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз), рак молочной железы, рак легкого, рак печени, колоректальный рак, рак пищевода, рак желудка, плоскоклеточная карцинома головы и шеи, глиобластома, анапластическая крупноклеточная лимфома, карцинома щитовидной железы и шпицоидные новообразования.
9. Соединение по любому из пп.1-5 или его стереоизомер, таутомер или фармацевтически приемлемая соль для применения в качестве лекарственного средства, обладающего ингибирующей активностью в отношении SHP2.
10. Соединение по любому из пп.1-5 или его стереоизомер, таутомер, или фармацевтически приемлемая соль для применения в ингибировании активности протеинтирозинфосфатазы SHP2 в клетке.
11. Соединение по любому из пп.1-5 или его стереоизомер, таутомер, или фармацевтически приемлемая соль для применения в лечении рака, связанного с активностью SHP2.
12. Способ ингибирования активности протеинтирозинфосфатазы SHP2 в клетке, включающий обработку клетки соединением по пп.1-5 или его стереоизомером, таутомером или фармацевтически приемлемой солью.
13. Способ ингибирования активности протеинтирозинфосфатазы SHP2 у нуждающегося в этом пациента, включающий стадию введения пациенту терапевтически эффективного количества соединения в соответствии с пп.1-5 или его стереоизомера, таутомера или фармацевтически приемлемой соли.
14. Способ лечения или уменьшения тяжести гиперпролиферативного нарушения, связанного с активностью SHP2, у нуждающегося в этом пациента, включающий введение пациенту терапевтически эффективного количества соединения в соответствии с пп.1-5 или его стереоизомера, таутомера или фармацевтически приемлемой соли, где гиперпролиферативное заболевание выбрано из группы, состоящей из следующего: меланома, ювенильные миеломоноцитарные лейкозы, нейробластома, положительный по филадельфийской хромосоме хронический миелолейкоз, положительные по филадельфийской хромосоме острые лимфобластные лейкозы, острые миелоидные лейкозы, миелопролиферативные новообразования (такие как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз), рак молочной железы, рак легкого, рак печени, колоректальный рак, рак пищевода, рак желудка, плоскоклеточная карцинома головы и шеи, глиобластома, анапластическая крупноклеточная лимфома, карцинома щитовидной железы и шпицоидные новообразования.
15. Применение соединения по любому из пп.1-5 или его стереоизомера, таутомера или фармацевтически приемлемой соли в производстве лекарственного средства для лечения рака, связанного с активностью SHP2, где рак выбран из группы, состоящей из следующего: меланома, ювенильные миеломоноцитарные лейкозы, нейробластома, положительный по филадельфийской хромосоме хронический миелолейкоз, положительные по филадельфийской хромосоме острые лимфобластные лейкозы, острые миелоидные лейкозы, миелопролиферативные новообразования (такие как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз), рак молочной железы, рак легкого, рак печени, колоректальный рак, рак пищевода, рак желудка, плоскоклеточная карцинома головы и шеи, глиобластома, анапластическая крупноклеточная лимфома, карцинома щитовидной железы и шпицоидные новообразования.
16. Применение по п.15, где рак представляет собой рак легкого.
17. Применение по п.15, где рак представляет собой колоректальный рак.
| WO 2015107494 А1, 23.07.2015 | |||
| ЕА 201691442 А1, 30.12.2016 | |||
| Database REGISTRY [online], RN 2111623-79-9, 10.08.2017; 2111485-38-0, 10.08.2017; 2110890-17-8, 09.08.2017; 2110200-53-6, 09.08.2017; 2109948-31-2, 08.08.2017; 2107245-21-4, 02.08.2017; 2106311-53-7, 01.08.2017; 2105086-20-0, 30.07.2017; 2104243-38-9, 30.07.2017 | |||
| Retrieved from STN | |||
| WO |

