Область изобретения
Настоящее изобретение относится к новому способу получения альфа-гидроксисоединений, полезных в качестве промежуточных соединений, как например гидроксианалоги аминокислот или полимерные предшественники.
Уровень техники
При кормлении животных существует большая потребность в кормовых добавках. Группой кормовых добавок являются аминокислоты и гидроксианалоги аминокислот. В частности, существует потребность в кормовых добавках на биологической основе, изготовленных из возобновляемого экологичного сырья. Аминокислоты лейцин (Leu), изолейцин (Ile), валин (Val), фенилаланин (Phe), гистидин (His), метионин (Met), цистеин (Cys), глутаминовая кислота (Glu), триптофан (Trp) и тирозин (Tyr) интересны как добавки в корма для животных. Но также могут использоваться соответствующие аналоги аминокислот. Чтобы их можно было использовать в качестве кормовых добавок, необходимы недорогие способы производства аминокислот или альфа-гидроксианалогов. Известные способы включают ферментацию и различные способы химического синтеза.
Как лизин, так и метионин могут быть упомянуты в качестве примеров аминокислот, которые успешно используются в качестве добавок в питание животных, как в природном виде, так и в виде гидроксианалогов. Их получают как ферментацией, так и химическим синтезом.
В WO 2017/118871 раскрывается способ ферментативного получения L-метионина и его производных из сахаров.
В WO 2016/174231 раскрывается способ получения альфа-гидроксианалога метионина и его производных посредством контакта одного или более Сахаров с металлосиликатной композицией в присутствии соединения, содержащего серу, и растворителя. Были получены выходы более 30%.
На сегодняшний день наиболее успешными и экономичными способами получения аминокислот для промышленного использования, по-видимому, является ферментация биосырья с использованием генетически модифицированных микроорганизмов. До сих пор не было обнаружено, что химический способ эффективен для получения аминокислот, отличных от метионина.
Таким образом, все еще существует потребность в экологичных, экономичных способах получения аминокислот и их гидроксианалогов на основе биологических веществ из возобновляемого экологичного сырья, которые являются экономичными, гибкими и подходящими для крупномасштабного промышленного производства.
Сущность изобретения
Авторы настоящего изобретения неожиданно обнаружили, что ряд важных аминокислот может быть получен в форме их гидроксианалога путем хемокаталитического объединения гликоальдегида (GA, первое соединение) и определенных химических соединений (второе соединение) простым и устойчивым способом, который является гибким и подходит для производства в промышленных масштабах.
Согласно первому аспекту настоящее изобретение относится к способу получения альфа-гидроксисоединения в качестве продукта реакции формулы I:
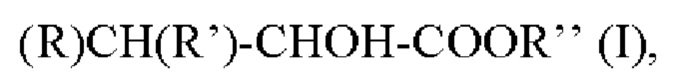
в которой
R представляет собой -Н или -СН3;
R' представляет собой -СН3, -СН2СН3, - СН(СН3)2, -С6Н5, -CH2SCH3, -C8H6N или -C3H3N2; и
Rʺ представляет собой -Н, -СН3, -СН2СН3, -СН(СН3)2, -СН2СН2СН3, -СН2СН2СН2СН3, -СН2СН(СН3)2, или -С(СН3)3;
причем способ включает стадии
a) обеспечения первого соединения формулы II:

b) обеспечения второго соединения формулы III:

где
R и R' имеют значения, как определено выше; и
с) реакции первого соединения со вторым соединением в присутствии катализатора на основе кислоты Льюиса с обеспечением альфа-гидроксисоединения в качестве продукта реакции.
Преимущества этого способа заключаются в том, что он подходит для масштабирования и, таким образом, подходит для крупномасштабного производства альфа-гидроксианалогов аминокислот с помощью способа, который является гибким и эффективным и позволяет использовать исходные материалы на основе биологических веществ, получаемых из возобновляемого экологичного сырья.
Стадию с) можно проводить в реакционной зоне, как, например, в жидкой среде реактора (т.е. реакционной смеси). После реакции первого и второго соединений (реактантов) реакционная смесь будет содержать любые непрореагировавшие реактанты и любые образующиеся альфа-гидроксисоединение в качестве продукта реакции. Предполагается, что реакция будет проходить в реакторе, содержащем катализатор на основе кислоты Льюиса. Система для получения альфа-гидроксисоединения в качестве продукта реакции в соответствии с настоящим изобретением является довольно гибкой, поскольку один и тот же катализатор может использоваться для производства различных альфа-гидроксисоединений в качестве продукта реакции, необязательно в однореакторном процессе с использованием двух или более разных соединения формулы (III). Кроме того, первое соединение является одним и тем же, независимо от того, какое альфа-гидроксисоединение в качестве продукта реакции желательно получить.
Согласно второму аспекту настоящее изобретение относится к соединению формулы I:
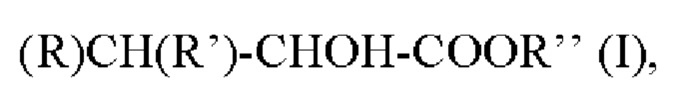
в котором
R представляет собой -Н или -СН3;
R' представляет собой -СН3, -СН2СН3, - СН(СН3)2, -С6Н5, -CH2SCH3, -C8H6N или -C3H3N2; и
Rʺ представляет собой -H, -СН3, -СН2СН3, -СН(СН3)2, -СН2СН2СН3, -СН2СН2СН2СН3, -СН2СН(СН3)2 или -С(СН3)3
Согласно третьему аспекту настоящее изобретение относится к композиции корма для животных, содержащей одно или более соединений формулы I и компонент корма для животных.
Согласно четвертому аспекту настоящее изобретение относится к применению одного или более соединений формулы I для получения композиции корма для животных.
Согласно четвертому аспекту настоящее изобретение относится к применению одного или более соединений формулы I для получения полимера.
Подробное описание изобретения
Определения
Если не указано иное, термин «активный металл» относится к атому металла в каталитически активной форме.
Соединение 1, как означает, относится к гликоальдегиду и может также упоминаться как первому соединению. Оно может находиться в мономерной, димерной или олигомерной форме. Соединение 2, как означает, относится к соединению альдегида или кетона формулы (III), и оно также может упоминаться как второе соединение. Оно может находиться в мономерной, димерной или олигомерной форме. Альтернативно, соединения 1 и 2 могут упоминаться как реактанты или субстраты. Альфа-гидроксисоединение в качестве продукта реакции может упоминаться просто как «продукт реакции». Если более чем одно соединение формулы (III) добавляют в одну и ту же реакционную смесь или реакционную зону, также будет получено более одного альфа-гидроксисоединения в качестве продукта реакции (формула (I)). Оно может упоминаться в единственном числе, даже если образуется несколько продуктов реакции.
Если не указано иное, концентрации, указанные в процентах, следует понимать как мас.% (т.е. масс х из расчета на общую массу раствора, умноженная на 100%). Если не указано иное, когда речь идет о концентрациях соединений, которые могут димеризоваться в растворе, указанные концентрации относятся к концентрации мономерных эквивалентов, например, для первого и второго соединения, а также для альфа-гидроксисоединения в качестве продукта реакции.
Термин «извлечение» предназначен для обозначения либо сбора альфа-гидроксисоединения в качестве продукта реакции, либо направления реакционной смеси, содержащей альфа-гидроксисоединение в качестве продукта реакции, на последующую стадию, такую как узел очистки.
Термин «выход» в контексте настоящего изобретения предназначен для обозначения молярного процента углерода первого соединения (гликоальдегид), которое извлекается в желаемое образованное альфа-гидроксисоединение в качестве продукта реакции. Соответственно, если 100 ммольреактантагликоальдегида (первого соединения) превратилось в 50 ммоль альфа-гидроксисоединения в качестве продукта реакции, то половина атомов углерода исходного гликоальдегида будет извлечена в альфа-гидроксисоединение в качестве продукта реакции, и таким образом, выход составит 50%; в случае образования MVG, 100 ммольгликоальдегида, превращенного в 50 ммоль MVG, будет соответствовать выходу 100%, поскольку для образования одной молекулы MVG необходимы две молекулы гликоальдегида.
Термин «превращение» в контексте настоящего изобретения предназначен для обозначения молярной доли гликоальдегида (первого соединения), которая прореагировала в ходе стадии с) с образованием либо желаемого альфа-гидроксисоединения в качестве продукта реакции, либо других соединений.
Термин «селективно», как означает, относится к молярной доле альфа-гидроксисоединения в качестве продукта реакции, образованной из расчета на превращенный гликоальдегид.
В контексте настоящего изобретения «реакционная зона» означает область вокруг катализатора, где первое и второе соединения вступают в контакт с катализатором на основе кислоты Льюиса, и два соединения вступают в реакцию. Согласно некоторым вариантам осуществления реакционная зона может быть ограничена стенками химического реактора. В реакторе непрерывного действия реакционная зона может быть ограничена стенками реактора и входом и выходом. Реакционная зона альтернативно может быть определена границей раздела между реакционной смесью, содержащейся внутри реактора, и окружающей средой.
Термин "реакционная смесь", как означает, относится к смеси, присутствующей в реакционной зоне, включающей, например, любое непрореагировавшее первое и второе соединения (реактанты) и образованное соединение альфа-гидроксикислоты (альфа-гидроксисоединение в качестве продукта реакции) и любые присутствующие побочные продукты или растворители или разбавители. Согласно варианту осуществления, стадия с) происходит в такой реакционной смеси. Реакционная смесь может также называться "текучая среда реактора". При извлечении потока продукта из реакционной зоны все соединения, присутствующие в реакционной смеси, будут присутствовать в некоторой степени.
Термин «непрерывный процесс», как означает, относится к процессу, осуществляемому в непрерывных условиях или в условиях установившегося состояния. Соответственно, не будет существенных колебаний концентрации. В непрерывном процессе первые и/или вторые соединения (реактанты) непрерывно подают в реакционную зону, а продукт реакции непрерывно извлекают из реакционной зоны. В этом контексте «непрерывная подача» и «непрерывное извлечение» включает многократную подачу небольших порций реактантов в реакционную зону и многократное извлечение небольших порций композиции продукта альфа-гидроксикислоты из реакционной зоны. Кроме того, реактанты можно подавать в реакционную зону в нескольких положениях, и продукт можно извлекать из нескольких положений (как, например, в реакторе с псевдоожиженным слоем или реакторе с уплотненным слоем, необязательно с рециркуляцией избыточного второго соединения в поток поступающего материала или во вход реактора)
Если иное не указано, радикалы R, R' и Rʺ имеют следующие значения:
R представляет собой -Н или -СН3;
R' представляет собой -СН3, -СН2СН3, - СН(СН3)2, -С6Н5, -CH2SCH3, -C8H6N или -C3H3N2; и
Rʺ представляет собой -Н, -СН3, -СН2СН3, -СН(СН3)2, -СН2СН2СН3, -СН2СН2СН2СН3, -СН2СН(СН3)2, или -С(СН3)3
Существует потребность в кормовых добавках для животных на биологической основе, которые можно получить с помощью экономичных процессов, подходящих для крупномасштабного производства. Такой способ обеспечивается Способом согласно настоящему изобретению. Авторы настоящего изобретения обнаружили, что катализаторы на основе кислоты Льюиса обладают превосходной каталитической активностью при облегчении реакции между гликоальдегидом (первое соединение) и одним или более конкретными альдегидами или кетонами (второе соединение) в соответствии со схемой реакции:
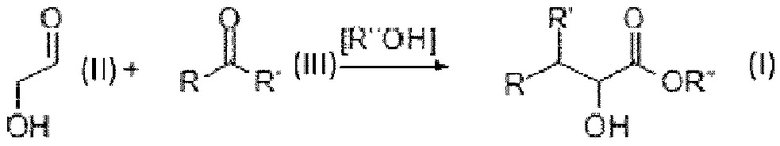
Возникающие в результате одно или более альфа-гидроксисоединений в качестве продуктов реакции представляют собой альфа-гидроксисоединения. Альфа-гидроксисоединение имеет структуру, которая напоминает основную цепь аминокислот, за исключением альфа-гидроксигруппы, которая должна быть замещена аминогруппой, чтобы превратиться в соответствующую аминокислоту. Соответственно, радикалы R и R' могут быть выбраны так, чтобы соответствовать боковым группам аминокислот. В зависимости от окружающей среды в реакционной зоне альфа-гидроксисоединение в качестве продукта реакции может быть в кислотной форме (в этом случае Rʺ представляет собой Н) или в форме сложного эфира (в этом случае Rʺ представляет собой алкильную группу).
Авторы настоящего изобретения неожиданно обнаружили, что катализаторы на основе кислоты Льюиса катализируют реакцию конденсации между гликоальдегидом и кетонами или альдегидами структур, представленных в настоящем документе. Мало того, что катализаторы на основе кислоты Льюиса катализируют вышеуказанную реакцию, они также отдают предпочтение изомерам, важным для получения альфа-гидроксианалогов аминокислот, и выходы являются довольно высокими. Авторы настоящего изобретения неожиданно обнаружили, что для гидроксианалога метионина преимущество Способа согласно настоящему изобретению состоит в том, что метилмеркаптогруппа будет расположена исключительно на углероде номер 4 от карбоксильной группы, если исходить из соединений, соответствующих первому и второму соединениям, в присутствии катализатора на основе кислоты Льюиса.
При контакте первого соединения со вторым соединением в присутствии катализатора на основе кислоты Льюиса, эти два соединения вступают в реакцию, и, неожиданно, предпочтение отдается альфа-гидроксисоединению в качестве продукта реакции. Не ограничиваясь теорией полагают, что первое соединение (которым является гликоальдегид) действует как более реакционноспособное вещество, способствуя в первую очередь образованию сложного альфа-гидрокси эфира. Приведенная ниже реакционная схема иллюстрирует предполагаемый механизм реакции гликоальдегида, реагирующего со вторым соединением.
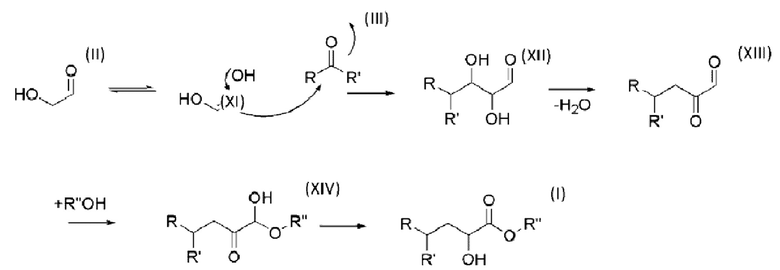
Альфа-гидроксисоединение в качестве продукта реакции представляет собой карбоновую кислоту или сложный эфир с гидроксигруппой в положении углерода C2, относительно группы карбоновой кислоты/сложного эфира. Природа радикала R" будет зависеть от окружения первого и второго соединений на стадии с). В водном растворе предпочтительна кислотная форма (Rʺ представляет собой -Н). В растворителе, содержащем спирт, предпочтителен соответствующий сложный эфир. Соответственно, метанол будет способствовать образованию сложного метилового эфира (Rʺ представляет собой - СН3), этанол будет способствовать образованию сложного этилового эфира (Rʺ представляет собой -С2Н5) и т.д. Согласно варианту осуществления настоящего изобретения альфа-гидроксисоединение в качестве продукта реакции формулы I представляет собой альфа-гидроксианалог аминокислоты. Согласно варианту осуществления настоящего изобретения альфа-гидроксисоединение в качестве продукта реакции выбрано из группы, состоящей из: метил 2-гидрокси-3-фенилпропаноата (IV), метил 2-гидрокси-4-метилпентаноата (V), метил 2-гидрокси-3-(1Н-индол-3-ил)-пропаноата (VI), метил 2-гидрокси-3-метилбутаноата (VII), метил 2-гидрокси-4-метилсульфанилбутаноата (VIII), метил 2-гидрокси-3-метилпентаноата (IX) и метил 2-гидрокси-3-(1Н-имидазол-4-ил)-пропаноата (X). Соединения могут быть представлены структурами:
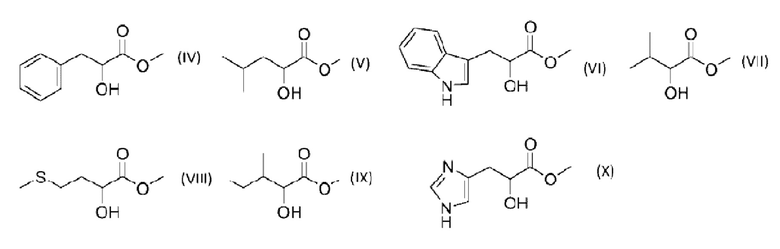
В Способе согласно настоящему изобретению первое соединение представляет собой гликоальдегид. Согласно варианту осуществления настоящего изобретения концентрация первого соединения в реакционной смеси находится в интервале от 0.1 до 30 мас. %, как например от 0.1 до 20 мас. % или от 1 до 5 мас. %.
В Способе согласно настоящему изобретению второе соединение представляет собой кетон или альдегид, несущий заместитель (R'), который соответствует боковой цепи аминокислоты. Если R представляет собой СН3, второе соединение представляет собой кетон. Если R представляет собой Н, второе соединение представляет собой альдегид. Второе соединение может быть обеспечено в растворителе. Согласно варианту осуществления может применяться смесь растворителей. В этом случае более одного альфа-гидроксисоединения в качестве продукта реакции могут быть совместно получены. Согласно варианту осуществления настоящего изобретения концентрация вторых соединений в реакционной смеси находится в интервале от 0.9 до 60 мас. %, как например от 0.9 до 40 или от 7 до 15 мас. %. Согласно варианту осуществления настоящего изобретения объединенная концентрация первого и второго соединений в реакционной смеси находится в интервале от 1 до 50 мас. %, как например от 5 до 20 или от 8 до 15 мас. %.
Первое и второе соединения могут присутствовать в различных формах, таких как мономер, димер, ацеталь или олигомер, в зависимости от их физического состояния и химической среды (как например, растворителя). Все формы первого и второго соединений охватываются настоящим изобретением. Согласно варианту осуществления настоящего изобретения гликоальдегид обеспечивается в виде димерагликоальдегида, диэтилацеталягликоальдегида или диметилацеталягликоальдегида. Когда первое и второе соединение обеспечены в растворе, они могут быть предоставлены, например, в форме ацеталя в растворе, а затем в реакционной зоне они гидролизуется с получением альдегида или кетона, соответствующего первому и/или второму соединению. Соответственно, первое и/или второе соединение может, например, быть обеспечено в растворителях, или они могут быть барботированы через текучую среду реактора.
Также альфа-гидроксисоединение в качестве продукта реакции может присутствовать в различных формах, таких как мономер, димер, ацеталь или олигомер, в зависимости от его физического состояния и химической среды (как например, растворителя). Все вышеупомянутые формы альфа-гидроксисоединения в качестве продукта реакции охватываются настоящим изобретением. Альфа-гидроксисоединение в качестве продукта реакции может быть извлечено в растворителе.
Согласно варианту осуществления настоящего изобретения обеспечивают два или более вторых соединений, и на стадии с) два или более альфа-гидроксисоединения в качестве продукта реакции соответственно получают. Согласно варианту осуществления настоящего изобретения общий выход одного или более альфа-гидроксисоединения в качестве продукта реакции формулы I находится в интервале 10-99%, как например 15-99%.
Согласно варианту осуществления настоящего изобретения второе соединение обеспечивают при стехиометрическом избытке относительно первого соединения. Преимущество избытка второго соединения состоит в том, что самоконденсация гликоальдегида в ходе реакции снижается. Соответственно, молярное соотношение между первым и вторым соединением предпочтительно находится в интервале от 0.01 до 1, как например от 0.01 до 0.8 или от 0.01 до 0.5.
Согласно варианту осуществления настоящего изобретения второе соединение представляет собой кетон (в соединении формулы I радикал R=СН3). Согласно предпочтительному варианту осуществления, молярное соотношение между кетоном и первым соединением находится в интервале от 1 до 50, как например от 5 до 30 или от 8 до 30.
Согласно варианту осуществления настоящего изобретения второе соединение представляет собой альдегид (в соединении формулы I радикал R=Н). Согласно предпочтительному варианту осуществления молярное соотношение между альдегидом и первым соединением находится в интервале от 1 до 50, как например от 5 до 30 или от 8 до 30.
Катализатор на основе кислоты Льюиса (или материал на основе кислоты Льюиса) действует как акцептор электронной пары для увеличения реакционной способности субстрата. Это может быть металлосиликатный материал, в этом случае это гетерогенный катализатор. Однако гомогенные катализаторы на основе кислоты Льюиса, такие как соли металлов, также могут быть подходящими в настоящем изобретении.
Металлосиликатный материал (также известный как металлосиликаты, металлосиликатная композиция или металлосиликатный катализатор) относится к одному или более твердым материалам, содержащим оксид кремния и активный металл (необязательно в форме компонента оксида металла), где активный металл и/или компоненты оксида металла встраиваются в (например, прививаются на) поверхность структуры оксида кремния (т.е. структура оксида кремния содержит связи МО-Si). Структура оксида кремния также известна как силикат, а структура оксида кремния, включающая активный металл, соответственно известна как металлосиликат. Металлосиликатные материалы могут быть кристаллическими или некристаллическими. Некристаллические металлосиликатные материалы включают упорядоченные мезопористые аморфные формы и другие мезопористые аморфные формы. Кристаллический микропористый материал включает цеолитные материалы и материалы цеотипа. Согласно варианту осуществления настоящего изобретения катализатор на основе кислоты Льюиса имеет цеолитную каркасную структуру, которая выбрана из группы, состоящей из BEA, MFI, FAU, MOR, FER и MWW. Согласно другому варианту осуществления катализатор на основе кислоты Льюиса имеет мезопористую структуру МСМ-41 и SBA-15.
Цеолитные материалы представляют собой кристаллические алюмосиликаты с микропористой кристаллической структурой, согласно Corma et al., Chem. Rev. 1995, 95 рр 559-614. Атомы алюминия в цеолитном материале могут быть частично или полностью замещены активным металлом (см., например, WO/2015/067654); эти материалы относятся к классу материалов цеотипа. Для целей настоящего изобретения материалы цеотипа включают цеолитные материалы, а металлосиликат замещен активным металлом, придающим материалу кислотность Льюиса. Катализаторы на основе кислоты Льюиса действуют как акцептор электронной пары, увеличивая реакционную способность субстрата. В контексте настоящего изобретения катализаторы на основе кислоты Льюиса катализируют реакцию альдольной конденсации между соединением 1 (гликоальдегид) и выбранным соединением 2 с получением целевого альфа-гидроксианалога аминокислоты. Согласно варианту осуществления настоящего изобретения катализатор на основе кислоты Льюиса содержит активный металл, выбранный из одного или более из группы, состоящей из Al, Sn, Ti, Pb, Zr, Zn, V, Nb, Та, Ge и Hf, предпочтительно из Sn, Zr, Ge и Hf, наиболее предпочтительно он представляет собой Sn.
Согласно варианту осуществления настоящего изобретения катализатор на основе кислоты Льюиса выбран из группы, состоящей из Sn-BEA, Sn-MCM-41 и растворимой соли олова. Растворимая соль олова может быть выбрана из группы, состоящей из хлорида олова (SnC14 и SnC12), фторида олова (SnF4 и SnF2), бромида олова (SnBr4 и SnBr2), иодида олова (SnI4 и SnI2), ацетилацетоната олова (SnC10H14O4), пирофосфата олова (Sn2P2O7), ацетата олова (Sn(CH3CO2)4 и Sn(CH3CO2)2), оксалата олова (Sn(C2O4)2 и SnC2O4), трифлата олова (Sn(CF3S03)2 и Sn(CF3S03)4). Соответствующие соли, например, Al, Ti, Pb, Zr, Zn, V, Nb, Та, Ge и Hf также подходят для использования в качестве катализаторов на основе кислоты Льюиса в настоящем изобретении.
Согласно варианту осуществления настоящего изобретения на стадии с) не присутствуют другие альдегиды или кетоны, кроме первого и второго соединений
Согласно варианту осуществления настоящего изобретения стадию с) проводят при температуре в интервале от 30 до 220°С, как например от 60 до 180°С. Согласно варианту осуществления настоящего изобретения на стадии с) первое и второе соединения реагируют в течение периода времени в интервале от 10 секунд до 48 часов. Необходимое время будет зависеть от различных факторов, таких как соотношение и концентрации первого и второго соединений, а также от количества добавляемого катализатора по отношению к двум реагентам, а также от выбранной температуры.
Способ, описанный в настоящем документе, может проводиться в реакторе, содержащем реакционный сосуд, один или более входов для реактантов и один или более выходов для продукта. Способ может осуществляться как периодический или непрерывный процесс.
Согласно варианту осуществления настоящего изобретения способ, раскрытый в настоящем документе, проводят как периодический процесс. Согласно варианту осуществления раскрытую в настоящей документе систему обеспечивают для проведения периодического процесса, как описано в настоящем документе, причем указанная система включает реактор периодического действия или реактор периодического действия с подпиткой.
Согласно варианту осуществления настоящего изобретения способ, раскрытый в настоящем документе, работает как непрерывный процесс, и исходный материал подают в реакционную зону со скоростью 0,01-400 г (гликоальдегид)/(г (катализатор)/час) (среднечасовая скорость подачи сырья, WHSV). Согласно варианту осуществления, раскрытому в настоящем документе, обеспечивается система для непрерывного выполнения способа согласно настоящему изобретению, причем указанная система содержит реактор с неподвижным слоем (реактор с поршневым потоком, PFR) или реактор с постоянным перемешиванием (CSTR).
Согласно варианту осуществления стадию с) проводят в присутствии растворителя. Соответственно, первое и/или второе соединение обеспечивают в форме исходного сырья, содержащего соединение 1 или соединение 2 и растворитель. Согласно варианту осуществления настоящего изобретения растворитель представляет собой полярный или слабополярный растворитель. Согласно варианту осуществления настоящего изобретения растворитель имеет диэлектрическую проницаемость выше 15. Примерными растворителями являются DMSO, диметилформамид, уксусная кислота, ацетонитрил, метанол, этанол, пропанол, изопропанол, н-бутанол, трет-бутанол, ацетон, бензальдегид, бутанон, изобутиральдегид, 1Н-имидазол-4-карбальдегид, 1Н-индол-3-карбальдегид и метилсульфанилацетальдегид, вода или их смеси. Согласно варианту осуществления растворитель выбран из группы, состоящей из воды, метанола и этанола или их смеси. Согласно варианту осуществления второе соединение и растворитель представляет собой одно и то же. В этом случае используется второе соединение с избытком по меньшей мере 1:2, как например 1: 5 или 1:10 (первое соединение: второе соединение). Преимущество использования полярных или слабополярных растворителей заключается в том, что растворимость первого соединения высока, что приводит к выходам альфа-гидроксисоединения в качестве продукта реакции в количестве, превышающем 10%. Предпочтительно выход альфа-гидрокси аналога аминокислоты превышает 10%, 20%, 30%, 40%, 50%, 60% или даже достигает 70%. Согласно варианту осуществления настоящего изобретения выход альфа-гидроксисоединения в качестве продукта реакции составляет от 10 до 99%, например, от 10 до 70% или от 30 до 60%.
Согласно варианту осуществления настоящего изобретения молярное соотношение кремния и активного металла составляет от 10 до 1000, например, от 20 до 400, от 50 до 200 или от 75 до 125.
В соответствии с другим вариантом осуществления настоящего изобретения Sn-ВЕА получают способом прямого синтеза с использованием фтористого водорода или путем последующей обработки. Примеры способов прямого синтеза описаны в ЕР 1 010 667 B1. Пример способа последующей обработки для получения Sn-ВЕА проиллюстрирован в WO 2015/024875 А1.
Согласно варианту осуществления, как раскрыто в настоящем документе, первое соединение получают из возобновляемого, экологичного, биосырья. Гликоальдегид может, например, быть получен из этиленгликоля или сахара. Согласно варианту осуществления первое соединение, как раскрыто в настоящем документе, получают путем пиролиза сахара, такого как глюкоза или сахароза, как, например, описано в US 2004/0022912. Гликоальдегид согласно варианту осуществления может быть обеспечен в виде водного раствора, содержащего гликоальдегид в количестве 1-99 мас./мас. % ипирувальдегид в количестве 0.1-60 мас./мас. %, как, например, в количестве 0.1-40 мас./мас. %, как например в количестве 0.1-30 мас./мас. %. Согласно другому варианту осуществления, водный раствор дополнительно содержит ацетол в количестве 0.1-40 мас./мас. %, как, например, в количестве 0.1-20 мас./мас. %, как, например, в количестве 0.1-10 мас./мас. %. Согласно другому варианту осуществления водный раствор дополнительно содержит глиоксал в количестве 0.1-40 мас./мас. %, как, например, в количестве 0.1-20 мас./мас. %, как, например, в количестве 0.1-10 мас./мас. %. Согласно другому варианту осуществления, водный раствор дополнительно содержит формальдегид в количестве 0.1-60 мас./мас. %, как, например, в количестве 0.1-40 мас./мас. %, как, например, в количестве 0.1-20 мас./мас. %.
Согласно варианту осуществления настоящего изобретения способ включает дополнительную стадию d) извлечения одного или более альфа-гидроксисоединения в качестве продукта реакции. Подходящим образом одно или более альфа-гидроксисоединения в качестве продуктов реакции извлекают посредством дистилляции или экстракции.
Согласно варианту осуществления способ согласно настоящему изобретению содержит дополнительную стадию е) аминирования альфа-гидроксисоединения в качестве продукта реакции с получением соединения соответствующей аминокислоты. Это может быть удобно осуществлено ферментативным способом. Соответственно, стадии с) и е), описанные выше, можно проводить в одном и том же реакторе в ходе объединенного однореакторного процесса. Реактор может представлять собой реактор периодического действия, реактор периодического действия с подпиткой или хемостат.
Альфа-гидроксисоединения в качестве продукта реакции согласно настоящему изобретению, а также соответствующие аминированные альфа-гидроксисоединения в качестве продукта реакции согласно настоящему изобретению подходят в качестве кормовых добавок для животных. Подобным образом альфа-гидроксисоединения в качестве продукта реакции согласно настоящему изобретению, а также соответствующее аминированное альфа-гидроксисоединения в качестве продукта реакции подходят в качестве пищевых добавок для человека. Для обоих применений они могут быть смешаны с одним или более компонентами корма для животных или продуктов питания для людей, такими как материал-носитель, углевод, адъювант, агент против слеживания, антиоксидант или поверхностно-активное вещество, с получением композиции корма для животных или продукта питания для человека. Добавки или композиции могут быть приготовлены в виде раствора, суспензии, гранул, порошка и т.д., как известно в данной области техники.
Альфа-гидроксисоединения в качестве продукта реакции согласно настоящему изобретению, как также предполагается, подходят в качестве мономеров для получения полимеров. Они также могут быть объединены с другими мономерами, такими как молочная кислота, лактид, этиленгликоль или гликолевая кислота, для получения сополимера.
Примеры
В следующих примерах проиллюстрировано получение катализаторов и получение альфа-гидрокс аналогов аминокислот из гликоальдегида.
Пример 1: Получение металлосиликатных материалов с помощью методики постсинтеза:
Пример 1 А. Способ получения Sn-BEA посредством способа пост-обработки
Постсинтезированные цеолиты/материалы цеотипа Sn-BEA получали в соответствии с методикой, описанной в ChemSusChem 2015, 8, 613-617. Коммерческий цеолит ВЕА, имеющий каркас *ВЕА (СР7119, Zeolyst, Si/Al=12.5, NH4+-форма), сначала кальцинировали при 550°С в течение 6 часов с получением цеолита в его Н+-форме с последующим кислотным деалюминированием следующим образом: 10 г концентрированной азотной кислоты (HNO3, Sigma-Aldrich, ≥65%) добавляли на 1 г материала цеолита *ВЕА и суспензию нагревали до 80°С в течение 12-24 часов. Деалюминированное твердое вещество извлекали фильтрацией, тщательно промывали деионизированной водой и кальцинировали при 550°С в течение 6 часов со скоростью нагревания 2°С/мин. Затем олово вводили в образовавшиеся вакансии в каркасе цеолита путем пропитки по начальной влажности с использованием хлорида олова (II) в растворе в качестве источника олова. Раствор получали растворением 0,128 г хлорида олова (II) (Sigma-Aldrich, 98%) в 5,75 мл воды, и раствор добавляли к 5 г образца деалюминированного цеолита *ВЕА. После пропитки образец сушили в течение ночи при 110°С, а затем кальцинировали при 550°С в течение 6 часов.
Пример 1 В. Получение Zr-BEA посредством способа пост-обработки.
Проводили ту же самую процедуру как для 1A за исключением того, что вместо 0,128 г хлорида олова (II) применяли 0.121 г ZrOCl2⋅8H2O или ZrCl4; в качестве источника циркония.
Пример 1С. Получение Ti-BEA посредством способа пост-обработки
Проводили ту же самую процедуру как для 1A за исключением того, что вместо 0.128 г хлорида олова (II) применяли 0.154 г этоксида титана (IV) (Ti(OC2H5)4, Sigma-Aldrich) в качестве источника титана. Кроме того, источник титана растворяли в смеси 50:50 воды и пероксида водорода вместо чистой воды в ходе пропитки.
Пример 1D. Получение Zn-BEA посредством способа пост-обработки
Проводили ту же самую процедуру как для 1A за исключением того, что вместо 0.128 г хлорида Sn (II) применяли 0.091 г хлорида Zn (II) в качестве источника цинка.
Пример 1E. Получение Hf-BEA посредством способа пост-обработки
Проводили ту же самую процедуру как для 1A за исключением того, что вместо 0.128 г хлорида Sn (II) применяли 0.216 г Hf(IV) в качестве источника гафния.
Пример 1F. Получение Ge-BEA посредством способа пост-обработки
Проводили ту же самую процедуру как для 1A за исключением того, что вместо 0.128 г хлорида Sn (II) применяли 0.070 г оксида Ge (IV) в качестве источника германия.
Применение 2: Получение металлосиликатных материалов посредством методики прямого синтеза:
Пример 2А. Получение Sn-BEA посредством метода прямого синтеза
Цеолиты Sn-BEA, полученные прямым гидротермальным синтезом, были синтезированы способом, описанным в J. Mater. Chem А 2014, 2, 20252-20262. В этом получении 30,6 г тетраэтилортосиликата (TEOS, 98%, Aldrich) добавляли к 33,1 г гидроксида тетраэтиламмония (ТЕАОН, 35% раствор, Aldrich) при перемешивании. После получения одной фазы 0,336 г пентагидрата хлорида олова (IV)(SnC14⋅H2O, Sigma-Aldrich) растворяли в 2,0 мл H2O и медленно добавляли. После нескольких часов перемешивания (>5 часов) образовывался густой гель, который затем окончательно обрабатывали добавлением 3,1 г HF в 1,6 г деминерализованной H20. Образец гомогенизировали и переносили в покрытый тефлоном контейнер, помещали в автоклав из нержавеющей стали и статически нагревали при 140°С в течение 14 дней. Твердое вещество извлекали фильтрацией, тщательно промывали деминерализованной водой и сушили в течение ночи при 80°С на воздухе. Чтобы удалить органический темплат и окончательно обработать материал, его кальцинировали при 550°С в течение 6 часов, используя скорость нагрева 2°С/мин.
Пример 2В. Получение Zr-BEA посредством метода прямого синтеза
Проводили ту же самую процедуру как для 2А за исключением того, что вместо 0.336 г пентагидрата хлорида олова (IV) применяли 0.318 г ZrOCl2⋅8H2O или ZrCl4, в качестве источника циркония.
Пример 2С. Получение Ti-BEA посредством метода прямого синтеза
Проводили ту же самую процедуру как для 2A1D за исключением того, что вместо 0.336 г пентагидрата хлорида олова (IV) применяли 0.405 г этоксида титана (IV) (Ti(OC2H5)4, Sigma-Aldrich) в качестве источника титана. Кроме того, источник титана растворяли в смеси 50:50 воды и пероксида водорода вместо чистой воды во время пропитки.
Пример 2D. Получение Sn-MFI посредством метода прямого синтеза
Цеолиты/цеотипы MFI получали в соответствии с методикой, описанной в MicroporousMater. 1997, 12, 331-340. Для получения Sn-MFI (Si/Sn=100) 0,257 г пентагидрата хлорида олова (IV) (SnC14⋅5H2O, Aldrich, 98%) растворяли в 5 г деминерализованной воды и добавляли к 15,6 г тетраэтилортосиликата (TEOS, 98%, Aldrich) и перемешивали в течение 30 мин. К этому раствору затем добавляли 13,4 г гидроксида тетрапропиламмония (ТРАОН, 40%, AppliChem) в 13,4 г деминерализованной воды и перемешивали в течение 1 часа. После этого добавляли еще 60 г деминерализованной воды, и раствор перемешивали в течение еще 20 часов, после чего раствор добавляли в автоклав с тефлоновым покрытием и синтезировали при 160°С в течение 2 дней в статических условиях. Твердое вещество извлекали центрифугированием, тщательно промывали деминерализованной водой и сушили в течение ночи при 80°С на воздухе. Чтобы удалить органический темплат и окончательно обработать материал, его кальцинировали при 550°С в течение 6 часов, используя скорость нагрева 2°С/мин.
Пример 2Е. Способ получения TS-1 (Ti-MFI) гидротермальным способом
Проводили ту же самую процедуру как для 2D за исключением того, что вместо 0.257 г пентагидрата хлорида олова (IV) применяли 0.167 г этоксида титана (IV) (Ti(OC2H5)4, Sigma-Aldrich) в качестве источника титана. Кроме того, во время пропитки источник титана растворяли в смеси воды и пероксида водорода 50:50 вместо чистой воды.
Пример 2F. Получение Sn-MCM-41 посредством гидротермального способа
Упорядоченный мезопористыйстанносиликат Sn-MCM-41 получали согласно способу, описанному в GreenChem. 2011, 13, 1175-1181. 26,4 г силиката тетраэтиламмония (TMAS, Aldrich, 15-20 мас.% в воде, ≥99,99%) медленно добавляли к 13,0 г гексадецилтриметиламмония бромида (CTABr, Sigma,>99,0%), растворенного в 38,0 г воды. Затем эту смесь перемешивали в течение 1 ч с последующим добавлением 0,239 г пентагидрата хлорида олова (IV) (SnC14⋅5H2O, 98%, Aldrich) и 0,537 г соляной кислоты (HC1, Sigma-Aldrich, мин. 37%) в 2,1 г воды. Этот раствор перемешивали в течение 1,5 часов, затем добавляли 12,2 г тетраэтилортосиликата (TEOS, 98%, Aldrich) и перемешивали еще 3 часа. Полученную смесь переносили в покрытый тефлоном контейнер, помещенный в автоклав из нержавеющей стали, и нагревали до 140°С в течение 15 часов. Твердое вещество выделяли фильтрацией, тщательно промывали деминерализованной водой и сушили в течение ночи при 80°С на воздухе. Чтобы удалить органический темплат и окончательно обработать материал, его кальцинировали при 550°С в течение 6 часов, используя скорость нагрева 2°С/мин.
Пример 3: Получение гидроксианалога валина из GA и ацетона
Пример 3А. Для получения гидроксианалога валина 10 г раствора ацетона/GA, состоящего из 0,1 г GA, 5 г ацетона и безводного метанола, предварительно смешивали и добавляли в сосуд высокого давления из нержавеющей стали (40 см, Swagelock) вместе с 0,50 г пост-синтезированного Sn-BEA (Si/Sn=125). Этот реактор периодического действия затем герметично закрывали и помещали на предварительно нагретую масляную баню при 160°С при перемешивании при 700 оборотах в минуту и оставляли реагировать в течение 20 часов. По окончании Эксперимента сосуды быстро охлаждали в холодной воде. Затем реактор открывали, реакционную смесь извлекали фильтрованием, продукты идентифицировали и количественно определяли с помощью GC-MS (Agilent 6890 с колонкой Zebron ZB-5MS (Phenomenex), оснащенной масс-селективным детектором Agilent 5973) и GC-FID (Agilent 7890 с колонкой Zebron ZB-5 (Phenomenex), оснащенной пламенно-ионизационным детектором). Чистый стандарт гидроксианалога валина (Enamine, 95%), диметилацеталягликоальдегида (SigmaAldrich, 98%) и гликоальдегида (>99%) использовали для количественного определения выхода продукта и непревращенного субстрата.
Пример 3 В. Следовали условиям реакции из Примера 3А, за исключением изменения молярного соотношения ацетона/GA. В растворе 1-5 г ацетона добавляли к 0.1 г гликоальдегида, и метанол добавляли до общей массы раствора 10 г. Это дало молярное соотношение ацетона/GA от 10 до 55, показывая оптимальное значение/плато при молярном соотношении ацетона/GA от 35 до 55.
Таблица 1
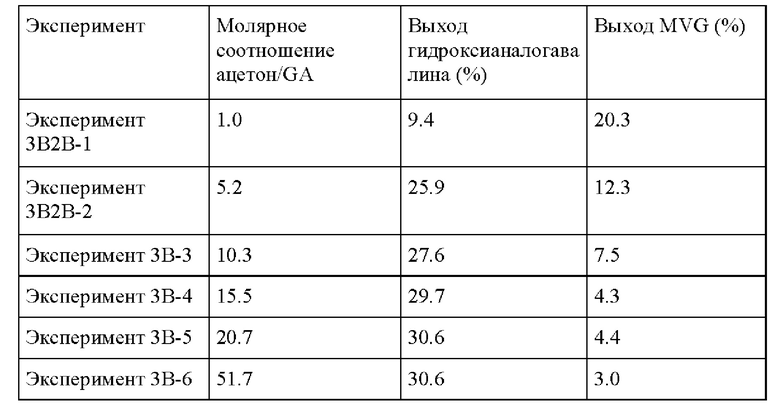
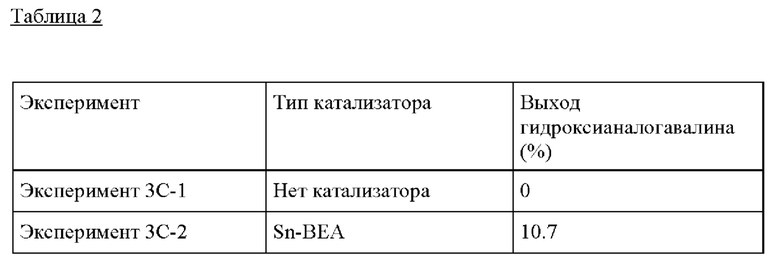
Пример 3С. Следовали условиям реакции из Примера 3А за исключением того, что загрузки катализатора и субстрата доводили до 0,1 г катализатора и до 20 г ацетона (0,4 г GA, 1,6 г ацетона, МеОН), и образование гидроксианалога валина в присутствии Sn-BEA, Ti-BEA и TS-1, соответственно, тестировали, также в отсутствие катализатора. Оловосодержащий катализатор Sn-BEA показал самую высокую активность в отношении образования желаемого продукта реакции в выбранных условиях по сравнению с титансодержащими катализаторами (Ti-BEA и TS-1).

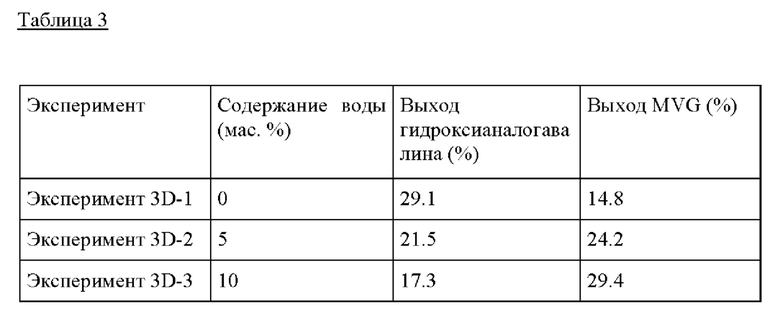
Пример 3D. Следовали условиям реакции из Примера 3А, за исключением того, воду добавляли к раствору ацетона/GA, применяемому в Эксперименте, применяя 0.2 г GA, 0.8 г ацетона, 0-1 г воды, МеОН до общей массы раствора 10 г. Понятно, что для образования гидроксианалога валина предпочтительно использовать <5 мас.% воды.
Пример 3Е. Следовали условиям реакции из Примера 3А, изменяя температуру от 140°С до 180°С, и изменяя применяемою композицию ацетона/GA до 0.2 г GA, 0.8 г ацетона, МеОН до получения 10 г раствора. Здесь для образования гидроксианалога валина желательны более низкие температуры, предпочтительно <160°С.

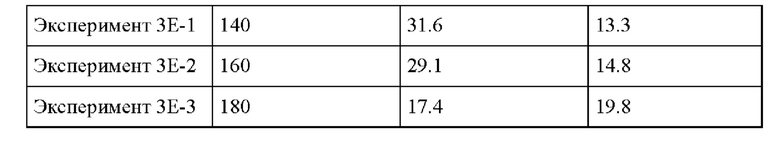
Пример 3F. Следовали условиям реакции из Примера 3А, за исключением изменения количества катализатора Sn-BEA, применяемого в Эксперименте, от 0.1 г до 1 г, и изменения состава раствора ацетона/GA. Реакционная смесь, применяемая в Эксперименте, состояла из 0.4 г GA, 1.6 г ацетона и МеОН до массы раствора 20 г раствора. В этих условиях реакции избыток катализатора предпочтителен для получения гидроксианалога валина.
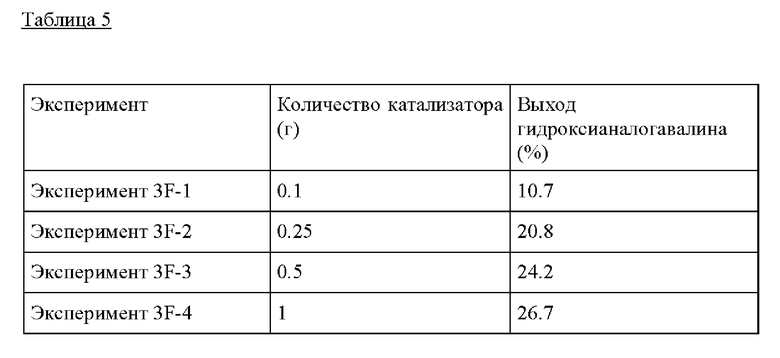
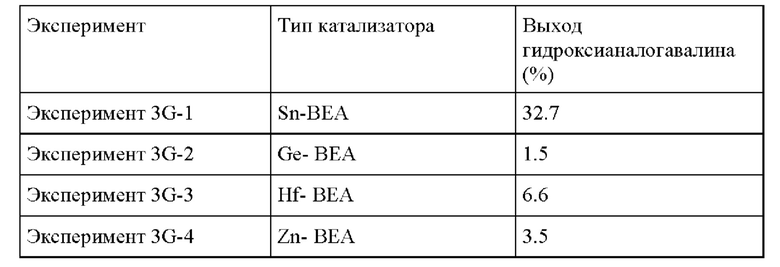
Пример 3G. Следовали условиям реакции из Примера 3А, за исключением изменения загрузки катализатора и субстрата до 0.5 г катализатора и 10 г раствора ацетона (0.1 г GA, 2 г ацетона, МеОН), тестируя образование гидроксианалога валина в присутствии Sn-BEA, Ge-BEA, Hf-BEA и Zn-BEA. Оловосодержащий катализатор Sn-BEA показал наивысшую активность в отношении образования продукта в выбранных условиях по сравнению с остальными катализаторами на основе кислоты Льюиса, но, что важно, все материалы были способны производить гидроксианалог валина.
Таблица 6
Пример 4: Получение гидроксианалога фенилаланина из GA и бензальдегида
Пример 4А. Для получения гидроксианалога фенилаланина 10 г раствора бензальдегида/GA, состоящего из 0,1 г GA, 5 г бензальдегида и безводного метанола, предварительно смешивали и добавляли в сосуд высокого давления из нержавеющей стали (40 см3, Swagelock) вместе с 0,50 г пост-синтезированного Sn-BEA (Si/Sn=125). Этот реактор периодического действия затем герметично закрывали и помещали на предварительно нагретую масляную баню при 160°С при перемешивании при 700 оборотах в минуту и оставляли реагировать в течение 20 часов. По окончании Эксперимента сосуды быстро охлаждали в холодной воде. Затем реактор открывали, реакционную смесь извлекали фильтрованием, продукты идентифицировали и количественно определяли с помощью GC-MS (Agilent 6890 с колонкой Zebron ZB-5MS (Phenomenex), оснащенной масс-селективным детектором Agilent 5973) и GC-FID (Agilent 7890 с колонкой Zebron ZB-5 (Phenomenex), оснащенной пламенно-ионизационным детектором). Чистый стандарт гидроксианалога фенилаланина (ArkPharm, 97%), диметилацеталягликоальдегида (SigmaAldrich, 98%) и гликоальдегида (>99%) использовали для количественного определения выхода продукта и непревращенного субстрата.
Пример 4 В. Следовали условиям реакции из Примера 4А, изменяя состав раствора бензальдегида/GA. В растворе 1-5 г бензальдегида добавляли к 0.1 г гликоальдегида, и метанол добавляли до общей массы раствора 10 г. Это давало молярные соотношения бензальдегида/GA от 5 до 30, показывая самый высокий выход при молярном соотношении бензальдегида/GA, равном 5.

Пример 4С. Следовали условиям реакции из Примера 4А с тем изменением, что добавляли воду к раствору бензальдегида/GA. В Эксперименте раствор бензальдегида/GA изменяли до следующего состава; 0.2 г GA, 0.8 г бензальдегида, 0-1 г воды и МеОН до общей массы раствора 10 г. Самый высокий выход гидроксианалога фенилаланина обнаруживали при содержании воды 5 мас.%.
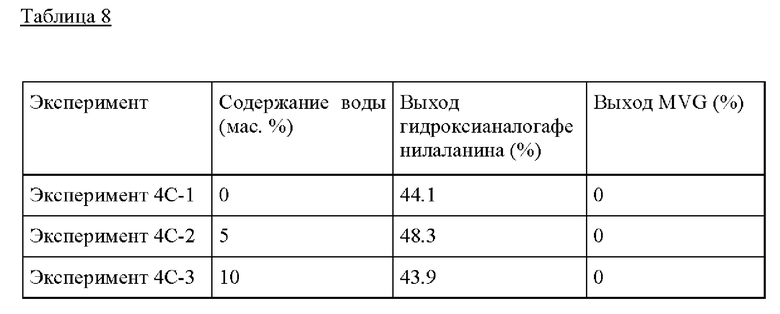
Пример 4D. Следовали условиям реакции из Примера 4А, изменяя температуру от 140°С до 180°С. Композиция бензальдегида/GA, применяемая в этом Эксперименте, представляла собой 0.1 г GA, 0.95 г бензальдегида и МеОН до получения 10 г раствора. Здесь для образования гидроксианалога фенилаланина желательны более высокие температуры, предпочтительно >160°С.
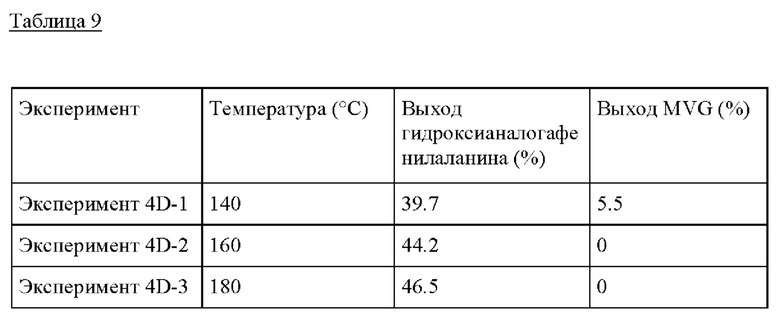
Пример 5
Получение гидроксианалога изолейцина из GA и бутанона
Пример 5А. Для получения гидроксианалога изолейцина 10 г раствора бутанона/GA, состоящего из 0,1 г GA, 1 г бутанона и безводного метанола, предварительно смешивали и добавляли в сосуд высокого давления из нержавеющей стали (40 см3, Swagelock) вместе с 0,50 г пост-синтезированного Sn-BEA (Si/Sn=125). Этот реактор периодического действия затем герметично закрывали и помещали на предварительно нагретую масляную баню при 160°С при перемешивании при 700 оборотах в минуту и оставляли реагировать в течение 20 часов. По окончании Эксперимента сосуды быстро охлаждали в холодной воде. Затем реактор открывали, реакционную смесь извлекали фильтрованием, продукты идентифицировали и количественно определяли с помощью GC-MS (Agilent 6890 с колонкой Zebron ZB-5MS (Phenomenex), оснащенной масс-селективным детектором Agilent 5973) и GC-FID (Agilent 7890 с колонкой Zebron ZB-5 (Phenomenex), оснащенной пламенно-ионизационным детектором). Чистый стандарт гидроксианалога изолейцина (Enamine, 95%), бутанона (SigmaAldrich), диметилацеталягликоальдегида (SigmaAldrich, 98%) и гликоальдегида (>99%) использовали для количественного определения выхода продукта и непревращенного субстрата.
Пример 5 В. Следовали условиям реакции из Примера 5А, изменяя состав раствора бутанона/GA. В растворе 0.1-3 г бутанона добавляли к 0.1 г гликоальдегида, и метанол добавляли до общей массы раствора 10 г. Это обеспечивало молярные соотношения бутанона/GA от 0.8 до 25, показывая самый высокий выход при молярном соотношении бутанона/GA, равном 25.
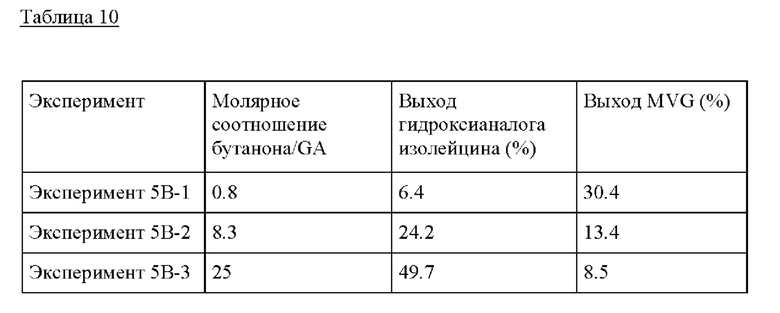
Пример 5С. Следовали условиям реакции из Примера 5А, изменяя температуру от 140°С до 180°С. Композиция бутанона/GA, применяемая в этом Эксперименте, представляла собой 0.1 GA, 1 г бутанона и МеОН до 10 г раствора. Как и в случае другого кетона в Примере 3, более низкая температура, предпочтительно <160°С, желательна для образования гидроксианалога изолейцина.
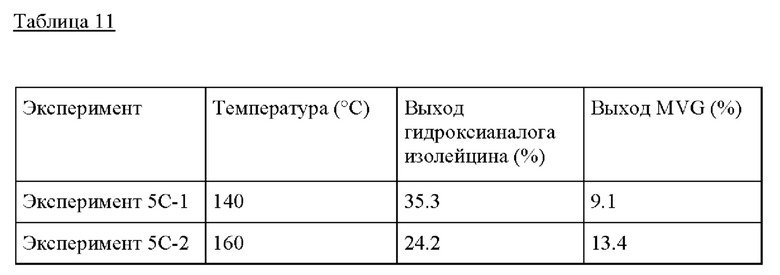

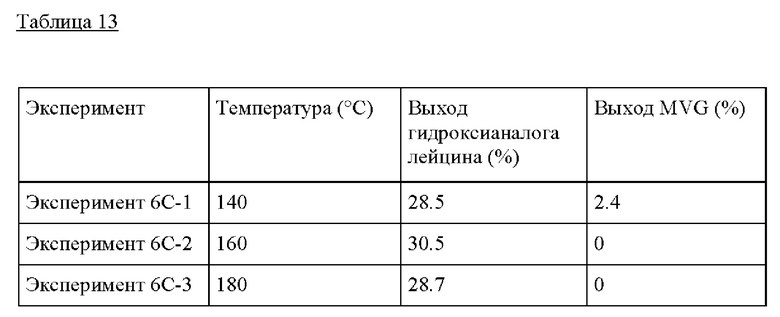
Пример 6
Получение гидроксианалога лейцина их GA и изобутиральдегида
Пример 6А. Для получения гидроксианалога лейцина 10 г раствора изобутиральдегида/GA, состоящего из 0,1 г GA, 1 г изобутиральдегида и безводного метанола, предварительно смешивали и добавляли в сосуд высокого давления из нержавеющей стали (40 см3, Swagelock) вместе с 0,50 г пост-синтезированного Sn-BEA (Si/Sn=125). Этот реактор периодического действия затем герметично закрывали и помещали на предварительно нагретую масляную баню при 160°С при перемешивании при 700 оборотах в минуту и оставляли реагировать в течение 20 часов. По окончании Эксперимента сосуды быстро охлаждали в холодной воде. Затем реактор открывали, реакционную смесь извлекали фильтрованием, продукты идентифицировали и количественно определяли с помощью GC-MS (Agilent 6890 с колонкой Zebron ZB-5MS (Phenomenex), оснащенной масс-селективным детектором Agilent 5973) и GC-FID (Agilent 7890 с колонкой Zebron ZB-5 (Phenomenex), оснащенной пламенно-ионизационным детектором). Чистый стандарт гидроксианалога лейцина (Enamine, 95%), изобутиральдегида (TCI), диметилацеталягликоальдегида (SigmaAldrich, 98%) и гликоальдегида (>99%) использовали для количественного определения выхода продукта и непревращенного субстрата.
Пример 6 В. Следовали условиям реакции из Примера 6А, изменяя состав раствора изобутиральдегида/GA. В растворе 0.1-3 г изобутиральдегида добавляли к 0.1 г гликоальдегида, и метанол добавляли до общей массы раствора 10 г. Это обеспечило молярные соотношения изобутиральдегида/GA от 0.8 до 25, показывая самый высокий выход при молярном соотношении изобутиральдегида/GA, равном 25.
Таблица 12
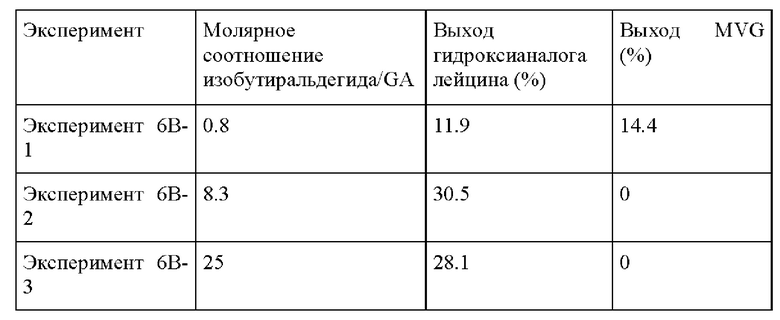
Пример 6С. Следовали условиям реакции из Примера 6А, изменяя температуру от 140°С до 180°С. Композиция изобутиральдегида/GA, применяемая в этом Эксперименте, представляла собой 0.1 г GA, 1 г изобутиральдегида и МеОН до 10 г раствора. Как и в случае другого альдегида в Примере 4, температура не оказывает большого влияния на образование гидроксианалога лейцина.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ МОЛОЧНОЙ КИСЛОТЫ ИЗ САХАРОВ | 2015 |
|
RU2710014C2 |
| ГЕТЕРОГЕННЫЕ КАТАЛИЗАТОРЫ ДЛЯ ПРЯМОГО КАРБОНИЛИРОВАНИЯ НИТРОАРОМАТИЧЕСКИХ СОЕДИНЕНИЙ В ИЗОЦИАНАТЫ | 2018 |
|
RU2777912C2 |
| КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ КАУЧУКОВЫЕ СМЕСИ | 2002 |
|
RU2285697C2 |
| КОНВЕРСИЯ С1-3 ОКСИГЕНАТНЫХ СОЕДИНЕНИЙ ДО С4-ОКСИГЕНАТНЫХ СОЕДИНЕНИЙ, ОПОСРЕДОВАННАЯ КРИСТАЛЛИЧЕСКИМ МИКРОПОРИСТЫМ МАТЕРИАЛОМ | 2015 |
|
RU2710598C2 |
| СПОСОБ ПЕРЕРАБОТКИ ОРГАНИЧЕСКОГО СЫРЬЯ И КАТАЛИЗАТОР ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1999 |
|
RU2142931C1 |
| ЗАМЕЩЕННЫЕ 2-МЕТИЛТИОЭТИЛОМ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ДОБАВОК К КОРМАМ | 2008 |
|
RU2516833C2 |
| КРАСИТЕЛИ И СМЕСИ ДЛЯ ПРИДАНИЯ ОТТЕНКА ВО ВРЕМЯ СТИРКИ | 2011 |
|
RU2545461C2 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ОЛИГОМЕРИЗАЦИИ ОЛЕФИНОВ, СПОСОБ ЕЁ ПРИГОТОВЛЕНИЯ И СПОСОБ ОЛИГОМЕРИЗАЦИИ | 2001 |
|
RU2212936C2 |
| СПОСОБ НУКЛЕАЦИИ ПОЛИОЛЕФИНОВОЙ КОМПОЗИЦИИ АЦЕТАЛЬНЫМИ СОЕДИНЕНИЯМИ | 2005 |
|
RU2348637C2 |
| СПОСОБ РАЗДЕЛЕНИЯ СМЕСИ ОПТИЧЕСКИХ ИЗОМЕРОВ ПИНОКЕМБРИНА, В ЧАСТНОСТИ РАЦЕМАТА ПИНОКЕМБРИНА | 2008 |
|
RU2480460C2 |
Настоящее изобретение относится к способу получения альфа-гидроксисоединения формулы (I) в качестве продукта реакции  в которой R представляет собой -Н или -СН3; R' представляет собой -СН3, -СН2СН3, -СН(СН3)2, -С6Н5, -CH2SCH3, -C8H6N или -C3H3N2; и R'' представляет собой -Н, -СН3, -СН2СН3, -СН(СН3)2, -СН2СН2СН3, -СН2СН2СН2СН3, -СН2СН(СН3)2 или -С(СН3)3. Данный способ включает следующие стадии: a) обеспечение первого соединения формулы II:
в которой R представляет собой -Н или -СН3; R' представляет собой -СН3, -СН2СН3, -СН(СН3)2, -С6Н5, -CH2SCH3, -C8H6N или -C3H3N2; и R'' представляет собой -Н, -СН3, -СН2СН3, -СН(СН3)2, -СН2СН2СН3, -СН2СН2СН2СН3, -СН2СН(СН3)2 или -С(СН3)3. Данный способ включает следующие стадии: a) обеспечение первого соединения формулы II: 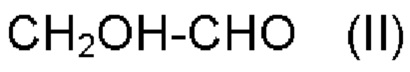 b) обеспечение второго соединения формулы III:
b) обеспечение второго соединения формулы III:  где R и R' имеют значения, как определено выше; c) реакцию первого соединения со вторым соединением и R''OH, где R'' имеет значения, как определено выше, в присутствии катализатора на основе кислоты Льюиса. Также изобретение относится к способу получения аминокислоты, включающему стадию аминирования альфа-гидроксисоединения формулы I, полученного с использованием стадий а)-с). Технический результат - получение аминокислот и их гидроксианалогов с использованием способов, которые являются гибкими и подходящими для крупномасштабного промышленного производства. 2 н. и 34 з.п. ф-лы, 13 табл., 6 пр.
где R и R' имеют значения, как определено выше; c) реакцию первого соединения со вторым соединением и R''OH, где R'' имеет значения, как определено выше, в присутствии катализатора на основе кислоты Льюиса. Также изобретение относится к способу получения аминокислоты, включающему стадию аминирования альфа-гидроксисоединения формулы I, полученного с использованием стадий а)-с). Технический результат - получение аминокислот и их гидроксианалогов с использованием способов, которые являются гибкими и подходящими для крупномасштабного промышленного производства. 2 н. и 34 з.п. ф-лы, 13 табл., 6 пр.
1. Способ получения альфа-гидроксисоединения в качестве продукта реакции формулы I:

в которой
R представляет собой -Н или -СН3;
R' представляет собой -СН3, -СН2СН3, -СН(СН3)2, -С6Н5, -CH2SCH3, -C8H6N или -C3H3N2; и
R'' представляет собой -Н, -СН3, -СН2СН3, -СН(СН3)2, -СН2СН2СН3, -СН2СН2СН2СН3, -СН2СН(СН3)2 или -С(СН3)3, включающий стадии
a) обеспечения первого соединения формулы II:
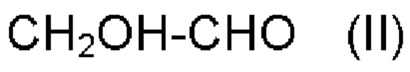
b) обеспечения второго соединения формулы III:

где
R и R' имеют значения, как определено выше;
c) реакции первого соединения со вторым соединением и R''OH, где R'' имеет значения, как определено выше, в присутствии катализатора на основе кислоты Льюиса с обеспечением альфа-гидроксисоединения в качестве продукта реакции.
2. Способ по п. 1, в котором альфа-гидроксисоединение в качестве продукта реакции представляет собой аналог аминокислоты.
3. Способ по п. 1, в котором R представляет собой СН3.
4. Способ по п. 1, в котором R представляет собой Н.
5. Способ по п. 1, в котором R" представляет собой -Н, -СН3, -СН2СН3 или -СН(СН3)2.
6. Способ по п. 1, в котором катализатор на основе кислоты Льюиса имеет каркасную структуру, которая выбрана из группы, состоящей из BEA, MFI, FAU, MOR, FER, MWW, МСМ-41 и SBA-15.
7. Способ по п. 1, в котором катализатор на основе кислоты Льюиса содержит один или более активных металлов, выбранных из группы, состоящей из Sn, Ti, Pb, Zr, Ge и Hf.
8. Способ по п. 1, в котором катализатор на основе кислоты Льюиса представляет собой Sn-BEA.
9. Способ по п. 1, в котором катализатор на основе кислоты Льюиса представляет собой Sn-MCM-41.
10. Способ по п. 1, в котором катализатор на основе кислоты Льюиса представляет собой растворимую соль олова.
11. Способ по п. 1, в котором на стадии с) первое и второе соединения подвергают реакции при температуре в интервале от 30 до 220°С, в частности в интервале от 60 до 180°С.
12. Способ по п. 1, в котором стадию с) проводят в растворителе.
13. Способ по п. 12, в котором растворитель выбран из группы, состоящей из воды, метанола, этанола, пропанола, изопропанола, н-бутанола, трет-бутанола, ацетона, бензальдегида, бутанона, изобутиральдегида, 1Н-имидазол-4-карбальдегида, 1Н-индол-3-карбальдегида и метилсульфанилацетальдегида или их смесей.
14. Способ по п. 1, в котором на стадии с) образуется метилвинилгликолят в качестве побочного продукта, и молярное соотношение альфа-гидроксисоединения в качестве продукта реакции и метилвинилгликолята находится в интервале от 1:1 до 100:1, в частности в интервале от 2:1 до 100:1.
15. Способ по п. 1, включающий последующую стадию d) извлечения альфа-гидроксисоединения в качестве продукта реакции.
16. Способ по п. 15, в котором альфа-гидроксисоединение в качестве продукта реакции извлекают посредством дистилляции и/или экстракции.
17. Способ по п. 15, в котором на стадии d) альфа-гидроксисоединение в качестве продукта реакции отделяют от метилвинилгликолята посредством дистилляции и/или экстракции.
18. Способ по любому из пп. 1-17, в котором никакие другие альдегиды или кетоны не присутствуют на стадии с) помимо первого и второго соединений.
19. Способ получения аминокислоты из альфа-гидроксисоединения формулы I:

в которой
R представляет собой -Н или -СН3;
R' представляет собой -СН3, -СН2СН3, -СН(СН3)2, -С6Н5, -CH2SCH3, -C8H6N или -C3H3N2; и
R'' представляет собой -Н, -СН3, -СН2СН3, -СН(СН3)2, -СН2СН2СН3, -СН2СН2СН2СН3, -СН2СН(СН3)2 или -С(СН3)3, включающий стадии
a) обеспечения первого соединения формулы II:
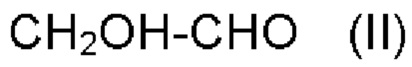
b) обеспечения второго соединения формулы III:
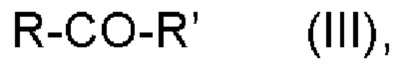
где
R и R' имеют значения, как определено выше;
с) реакции первого соединения со вторым соединением и R''OH, где R'' имеет значения, как определено выше, в присутствии катализатора на основе кислоты Льюиса с обеспечением альфа-гидроксисоединения,
е) аминирования альфа-гидроксисоединения в соответствующую аминокислоту.
20. Способ по п. 19, в котором альфа-гидроксисоединение представляет собой аналог аминокислоты.
21. Способ по п. 19, в котором R представляет собой СН3.
22. Способ по п. 19, в котором R представляет собой Н.
23. Способ по п. 19, в котором R" представляет собой -Н, -СН3, -СН2СН3 или -СН(СН3)2.
24. Способ по п. 19, в котором катализатор на основе кислоты Льюиса имеет каркасную структуру, которая выбрана из группы, состоящей из BEA, MFI, FAU, MOR, FER, MWW, МСМ-41 и SBA-15.
25. Способ по п. 19, в котором катализатор на основе кислоты Льюиса содержит один или более активных металлов, выбранных из группы, состоящей из Sn, Ti, Pb, Zr, Ge и Hf.
26. Способ по п. 19, в котором катализатор на основе кислоты Льюиса представляет собой Sn-BEA.
27. Способ по п. 19, в котором катализатор на основе кислоты Льюиса представляет собой Sn-MCM-41.
28. Способ по п. 19, в котором катализатор на основе кислоты Льюиса представляет собой растворимую соль олова.
29. Способ по п. 19, в котором на стадии с) первое и второе соединения подвергают реакции при температуре в интервале от 30 до 220°С, в частности в интервале от 60 до 180°С.
30. Способ по п. 19, в котором стадию с) проводят в растворителе.
31. Способ по п. 30, в котором растворитель выбран из группы, состоящей из воды, метанола, этанола, пропанола, изопропанола, н-бутанола, трет-бутанола, ацетона, бензальдегида, бутанона, изобутиральдегида, 1Н-имидазол-4-карбальдегида, 1Н-индол-3-карбальдегида и метилсульфанилацетальдегида или их смесей.
32. Способ по п. 19, в котором на стадии с) образуется метилвинилгликолят в качестве побочного продукта, и молярное соотношение альфа-гидроксисоединения и метилвинилгликолята находится в интервале от 1:1 до 100:1, в частности в интервале от 2:1 до 100:1.
33. Способ по п. 19, включающий последующую стадию d) извлечения альфа-гидроксисоединения.
34. Способ по п. 33, в котором альфа-гидроксисоединение извлекают посредством дистилляции и/или экстракции.
35. Способ по п. 33, в котором на стадии d) альфа-гидроксисоединение отделяют от метилвинилгликолята посредством дистилляции и/или экстракции.
36. Способ по любому из пп. 19-35, в котором никакие другие альдегиды или кетоны не присутствуют на стадии с) помимо первого и второго соединений.
| Andrew J | |||
| Humphrey et al | |||
| Synthesis of Enantiomerically Pure alfa-Hydroxyaldehydes from the Corresponding alfa-Hydroxycarboxylic Acids: Novel Substrates for Escherichia coli Transketolase | |||
| J | |||
| CHEM | |||
| SOC., CHEM | |||
| COMMUN., 1995, (24), 2475-2476 | |||
| Daniel Bur et al | |||
| An evaluation of the substrate specificity and asymmetric synthesis potential of the |

