Область техники
Настоящее изобретение относится к конъюгату антитела с лекарственным средством, содержащему противоопухолевое лекарственное средство, конъюгированное с анти-HER2 антителом через связывающую структурную группу, причем указанный конъюгат используют в качестве противоопухолевого лекарственного средства.
Уровень техники
Ожидается, что конъюгат антитела с лекарственным средством (КАЛС), содержащий лекарственное средство с цитотоксичностью, конъюгированное с антителом, антиген которого экспрессируется на поверхность раковых клеток, и которое также связывается с антигеном, способным к клеточной интернализации, и поэтому может доставлять лекарственное средство селективно в раковые клетки, вызывает аккумулирование лекарственного средства в раковых клетках и убивает раковые клетки (см. непатентные документы 1-3). В качестве КАЛС, Милотарг (зарегистрированный товарный знак; Гемтузумаб озогамицин) в котором калихеамицин конъюгирован с анти-CD33 антителом, одобрен в качестве терапевтического агента для острого миелоидного лейкоза. Далее, Адцетрис (зарегистрированный товарный знак; Брентуксимаб ведотин), в котором ауристатин E конъюгирован с анти-CD30 антителом, недавно был одобрен в качестве агента для Ходжкинской лимфомы и анапластической крупноклеточной лимфомы (см. не патентный документ 4). Лекарственные средства, содержащиеся в КАЛС, которые были апробированы по настоящее время, нацелены на ДНК или тубулин.
Что касается противоопухолевого агента, известны производные камптотецина, низкомолекулярные соединения, которые ингибируют топоизомеразу I для оказания противоопухолевого действия. Среди них, противоопухолевое соединение, представленное формулой ниже
Формула 1
(экзатекан, химическое наименование: (1S,9S)-1-амино-9-этил-5-фтор-2,3-дигидро-9-гидрокси-4-метил-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-10,13(9H,15H)дион) является водорастворимым производным камптотецина (патентные документы 1 и 2). В отличие от иринотекана, применяемого в настоящее время в клинических условиях, это соединение не требует активации ферментом для оказания противоопухолевого действия. Кроме того, его наблюдаемое ингибирующее действие на топоизомеразу I выше, чем у SN-38, который является основным фармацевтически активным веществом иринотекана и топотекана, также применяемого в клинических условиях, и его более высокая in vitro активность, разрушающая клетки, подтверждена в отношении различных раковых клеток. В частности, было подтверждено, что оно обладает действием против раковых клеток, которые резистентны к SN-38 или подобным, благодаря экспрессии P-гликопротеина. Далее, в мышиной модели подкожно трансплантированной человеческой опухоли было подтверждено, что оно обладает мощным противоопухолевым действием, прошло клинические исследования, но еще не выведено на рынок (см. непатентные документы 5-10). Остается неясным, может ли экзатекан эффективно действовать в качестве КАЛС.
DE-310 представляет собой комплекс, в котором экзатекан конъюгирован с биоразлагаемым карбоксиметилдекстрановым полиспиртовым полимером через GGFG пептидный спейсер (патентный документ 3). Превращение экзатекана в полимерное пролекарство позволяет поддерживать свойство высокого содержания в крови, а также свойство сильной нацеленности на опухолевые области пассивно повышается благодаря повышенной проницаемости новых кровеносных сосудов в опухолях и свойству удерживания в опухолевых тканях. В DE-310 благодаря расщеплению пептидного спейсера ферментом, экзатекан и экзатекан с глицином, связанный с аминогруппой, непрерывно выделяется в качестве основного активного вещества, и в результате улучшается фармакокинетика. Было обнаружено, что DE-310 обладает большей эффективностью, чем экзатекан, вводимый отдельно, даже если общая доза экзатекана, содержащаяся в D310, ниже, чем при введении экзатекана отдельно, согласно различным моделям оценки опухолей в не клинических исследованиях. Для DE-310 проводили клинические исследования, которые также подтвердили эффективность, включая отчет, в котором предполагается, что основное активное вещество аккумулируется в опухолях сильнее, чем в нормальных тканях. Однако имеется также отчет, показывающий, что аккумуляция DE-310 и основного активного вещества в опухолях не сильно отличается от аккумуляции в нормальных тканях у человека, таким образом, у человека пассивный таргетинг не наблюдается (см. Непатентные документы 11-14). В результате, DE-310 также не коммерциализирован, и остается непонятным, может ли экзатекан эффективно действовать в качестве лекарственного средства, направленного на такие цели.
В качестве соединения, родственного к DE-310, также известен комплекс, в котором структурная группа, представленная -NH-(CH2)4-C(=O)-, вставлена между -GGFG-спейсером и экзатеканом с получением -GGFG-NH-(CH2)4-C(=O)-, применяемого в качестве спейсерной структуры (патентный документ 4). Однако противоопухолевое действие указанного комплекса вообще не известно.
HER2 является одним из продуктов типового онкогена рецептора фактора роста, идентифицированного как онкоген, связанный с рецептором эпидермального фактора роста 2, и является трансмембранным рецепторным белком, имеющим молекулярную массу 185 кДа, и имеющим тирозинкиназный домен (непатентный документ 15). Последовательность ДНК и последовательность аминокислот HER2 описаны в публичной базе данных, и могут быть указаны здесь, например, как образец № M11730 (GenBank), NP_004439.2 (NCBI) или подобный.
HER2 (neu, ErbB-2) является одним из членов семейства РЭФР РЭФР (рецептора эпидермального фактора роста) и активируется аутофосфорилированием на внутриклеточных тирозиновых остатках через образование его гомодимера или образование его гетеродимера с другим РЭФР рецептором HER1 (РЭФР, ErbB-1), HER3 (ErbB-3) или HER4 (ErbB-4) (непатентные документы 16-18), тем самым играя важную роль в росте, дифференциации и выживании клеток в нормальных клетках и раковых клетках (непатентные документы 19 и 20). HER2 чрезмерно экспрессируется при различных типах рака, таких как рак молочной железы, рак желудка и рак яичников (непатентные документы 21-26), и был описан как отрицательный фактор прогноза для рака молочной железы (непатентные документы 27-28).
Трастузумаб представляет собой гуманизированное антитело мыши анти-HER2 антитело 4D5 (непатентный документ 29 и патентный документ 5), названное как рекомбинантное гуманизированное анти-HER2 моноклональное антитело (huMAb4D5-8, rhuMAb HER2, Herceptin(R)) (патентный документ 6). Трастузумаб специфически связывается с внеклеточным доменом IV HER2 и вызывает антитело-зависимую клеточную цитотоксичность (КАЛСС) или оказывает противораковое действие через ингибирование трансдукции сигнала из HER2 (непатентные документы 30 и 31). Трастузумаб является высокоэффективным для опухолей, чрезмерно экспрессирующих HER2 (непатентный документ 32), и, как таковой, был выпущен в 1999 в США и в 2001 в Японии в виде терапевтического агента для пациентов с метастатическим раком молочной железы, чрезмерно экспрессирующим HER2.
Хотя терапевтическое действие трастузумаба на рак молочной железы было должным образом доказано (непатентный документ 33), по неподтвержденным данным, около 15% пациентов с раком молочной железы, чрезмерно экспрессирующим HER2, которые получали широкий спектр обычных противораковых терапий, реагируют на трастузумаб. Около 85% пациентов из этой выборки не давали, или давали незначительную реакцию на лечение трастузумабом.
Таким образом, необходимость в терапевтическом агенте, нацеленном на заболевания, связанные с экспрессией HER2, была определена для пациентов, пораженных опухолями, чрезмерно экспрессирующими HER2, дающих отсутствие или слабую реакцию на трастузумаб, или связанными с HER2 расстройствами. T-DM1 (трастузумаб эмтансин, Kadcyla (R); непатентный документ 34), содержащий противоопухолевое лекарственное средство, конъюгированное с трастузумабом через связующую структуру, пертузумаб (Perjeta(R); непатентный документ 35 и патентный документ 7) были разработаны для поражения внеклеточного домена II HER2 и ингибируют образование гетеродимера. Однако их реактивность, сила действия и приемлемые показания до сих пор не являются достаточными, и существует неудовлетворенная потребность в поражении HER2.
Список источников
Патентные документы
Патентный документ 1: выложенный патент Японии № 5-59061
Патентный документ 2: выложенный патент Японии № 8-337584
Патентный документ 3: международная публикация № WO 1997/46260
Патентный документ 4: международная публикация № WO 2000/25825
Патентный документ 5: патент США № 5677171
Патентный документ 6: патент США № 5821337
Патентный документ 7: международная публикация № WO 01/00244
Непатентные документы
Непатентный документ 1: Ducry, L., et al., Bioconjugate Chem. (2010) 21, 5-13.
Непатентный документ 2: Alley, S. C., et al., Current Opinion in Chemical Biology (2010) 14, 529-537.
Непатентный документ 3: Damle N.K. Expert Opin. Biol. Ther. (2004) 4, 1445-1452.
Непатентный документ 4: Senter P. D., et al., Nature Biotechnology (2012) 30, 631-637.
Непатентный документ 5: Kumazawa, E., Tohgo, A., Exp. Opin. Invest. Drugs (1998) 7, 625-632.
Непатентный документ 6: Mitsui, I., et al., Jpn J. Cancer Res. (1995) 86, 776-782.
Непатентный документ 7: Takiguchi, S., et al., Jpn J. Cancer Res. (1997) 88, 760-769.
Непатентный документ 8: Joto, N. et al. Int J Cancer (1997) 72, 680-686.
Непатентный документ 9: Kumazawa, E. et al., Cancer Chemother. Pharmacol. (1998) 42, 210-220.
Непатентный документ 10: De Jager, R., et al., Ann N Y Acad Sci (2000) 922, 260-273.
Непатентный документ 11: Inoue, K. et al., Polymer Drugs in the Clinical Stage, Edited by Maeda et al. (2003) 145-153.
Непатентный документ 12: Kumazawa, E. et al., Cancer Sci (2004) 95, 168-175.
Непатентный документ 13: Soepenberg, O. et al., Clinical Cancer Research, (2005) 11, 703-711.
Непатентный документ 14: Wente M. N. et al., Investigational New Drugs (2005) 23, 339-347.
Непатентный документ 15: Coussens L, et al., Science. 1985;230(4730):1132-1139.
Непатентный документ 16: Graus-Porta G, et al., EMBO J. 1997;16;1647-1655.
Непатентный документ 17: Karnagaran D, et al., EMBO J. 1996;15:254-264.
Непатентный документ 18: Sliwkowski MX, et al., J. Biol. Chem. 1994;269:14661-14665.
Непатентный документ 19: Di Fore PP, et al., Science. 1987;237:178-182.
Непатентный документ 20: Hudziak RM, et al., Proc Natl Acad Sci U S A. 1987;84:7159-7163.
Непатентный документ 21: Hardwick R, et al., Eur. J Surg Oncol. 1997 (23):30-35.
Непатентный документ 22: Korkaya H, et al., Oncogene. 2008;27(47):6120-6130.
Непатентный документ 23: Yano T, et al., Oncol Rep. 2006;15(1):65-71.
Непатентный документ 24: Slamon DJ, et al., Science. 1987;235:177-182.
Непатентный документ 25: Gravalos C, et al., Ann Oncol 19: 1523-1529, 2008.
Непатентный документ 26: Fukushige S et al., Mol Cell Biol 6: 955-958, 1986.
Непатентный документ 27: Slamon DJ, et al. Science. 1989;244:707-712.
Непатентный документ 28: Kaptain S et al., Diagn Mol Pathol 10:139-152, 2001.
Непатентный документ 29: Fendly. et al., Cancer Research 1990(50):1550-1558.
Непатентный документ 30: Sliwkowski MX, et al., Semin Oncol. 1999;26(4,Suppl 12):60-70.
Непатентный документ 31: Hudis CA, et al., N Engl J Med. 357: 39-51, 2007.
Непатентный документ 32: Vogel CL, et al., J Clin Oncol. 2002;20(3):719-726.
Непатентный документ 33: Baselga et al., J. Clin. Oncol. 14:737-744 (1996).
Непатентный документ 34: Howard A. et al., J Clin Oncol 2011;29:398-405.
Непатентный документ 35: Adams CW, et al., Cancer Immunol Immunother. 2006;6:717-727.
Сущность изобретения
Техническая задача
Что касается лечения опухолей антителами, недостаточное противоопухолевое действие может наблюдаться, даже если антитело распознает антиген для связывания с опухолевыми клетками, и существуют случаи, в которых необходимо более эффективное противоопухолевое антитело. Далее, многие противоопухолевые низкомолекулярные соединения имеют проблемы с безопасностью, такие как побочные эффекты и токсичность, даже если соединения обладают превосходным противоопухолевым эффектом. Все еще сохраняется потребность в достижении превосходного терапевтического эффекта дальнейшим улучшением безопасности. Таким образом, объектом настоящего изобретения является получение противоопухолевого лекарственного средства, обладающего превосходным противоопухолевым действием и безопасностью.
Решение задачи
Авторы настоящего изобретения считают, что анти-HER2 антитело представляет собой антитело, которое способно поражать опухолевые клетки, то есть, обладающее свойством распознавания опухолевых клеток, свойством связываться с опухолевыми клетками, свойством поглощаться опухолевыми клетками, цитотоксическим действием против опухолевых клеток, активностью, разрушающей опухолевые клетки, или подобными; таким образом, когда противоопухолевое соединение экзатекан превращают в конъюгат антитела с лекарственным средством, через линкерную структурную группу, через конъюгирование с этим антителом, противоопухолевое соединение может быть более надежно доставлено в опухолевые клетки для специфического оказания противоопухолевого действия соединения в опухолевых клетках, и таким образом, противоопухолевое действие может быть надежно оказано, а также может ожидаться улучшение действия, разрушающего клетки, анти-HER2 антитела, и доза противоопухолевого соединения может быть снижена по сравнению с введением соединения отдельно, и таким образом, влияние противоопухолевого соединения на нормальные клетки может быть снижено настолько, чтобы достичь высокой степени безопасности.
В связи с этим, авторы настоящего изобретения создали связующую группу определенной структуры, и им удалось получить конъюгат антитела с лекарственным средством, в котором анти-HER2 антитело и экзатекан конъюгированы друг с другом через связующую группу, и они подтвердили превосходное противоопухолевое действие, демонстрируемое конъюгатом, тем самым завершив настоящее изобретение.
Более конкретно, настоящее изобретение относится к следующему.
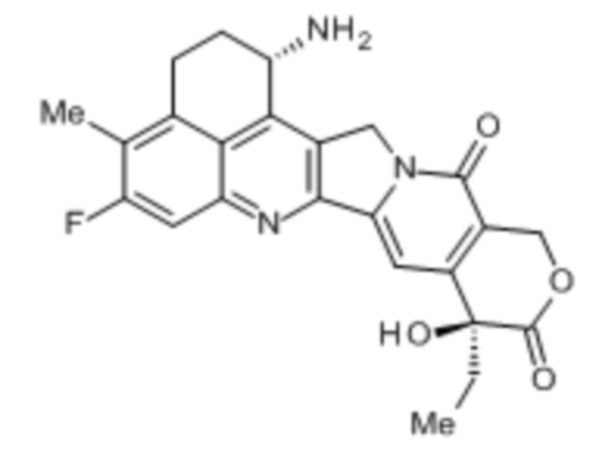
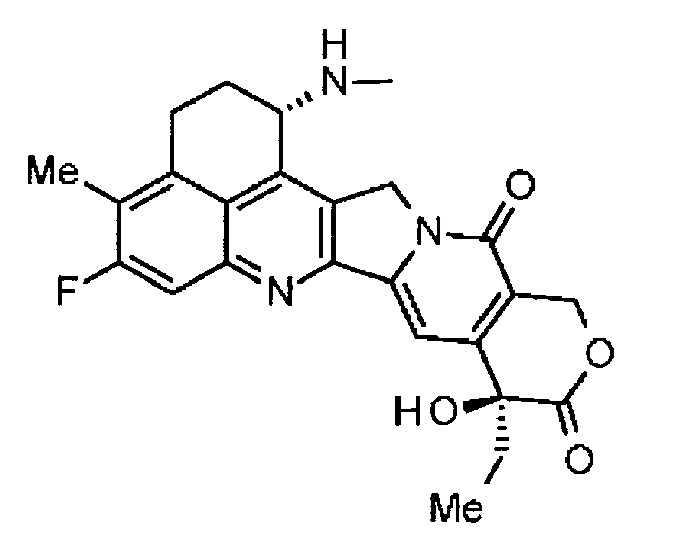
[1] Конъюгат антитела с лекарственным средством, где противоопухолевое соединение представлено следующей формулой:
Формула 2
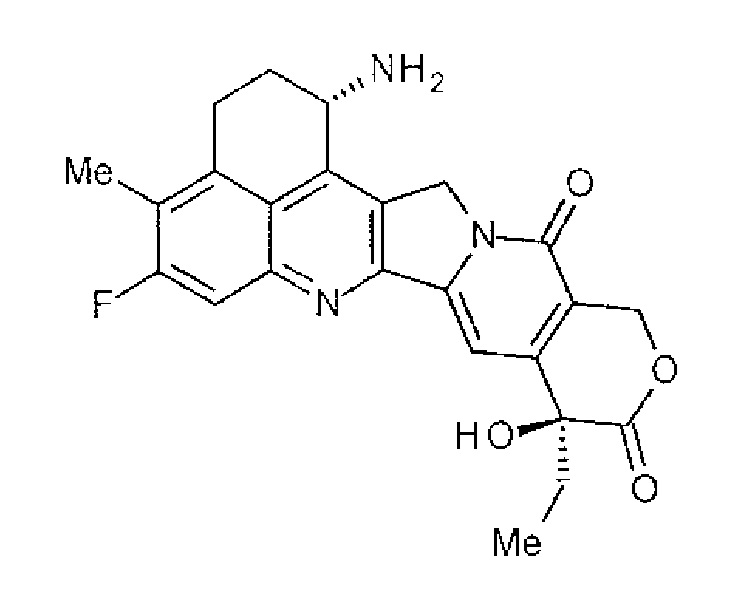
конъюгировано с анти-HER2 антителом через связующую группу, имеющую структуру, представленную следующей формулой:
-L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-
через тиоэфирную связь, которая образуется на дисульфидной связи, присутствующей в шарнирной части анти-HER2 антитела.
Здесь анти-HER2 антитело соединено с концевой группой L1,
противоопухолевое соединение соединено с карбонильной группой -(CH2)n2-C(=O)- части с атомом азота аминогруппы в положении 1 в качестве связующего положения,
где
n1 является целым числом от 0 до 6,
n2 является целым числом от 0 до 5,
L1 является -(сукцинимид-3-ил-N)-(CH2)n3-C(=O)-,
где n3 является целым числом от 2 до 8,
L2 является -NH-(CH2CH2-O)n4-CH2CH2-C(=O)- или одинарной связью,
где n4 является целым числом от 1 до 6,
LP является пептидным остатком, состоящим из от 2 до 7 аминокислот,
La является -O- или одинарной связью, и
-(Сукцинимид-3-ил-N)- имеет структуру, представленную следующей формулой:
Формула 3
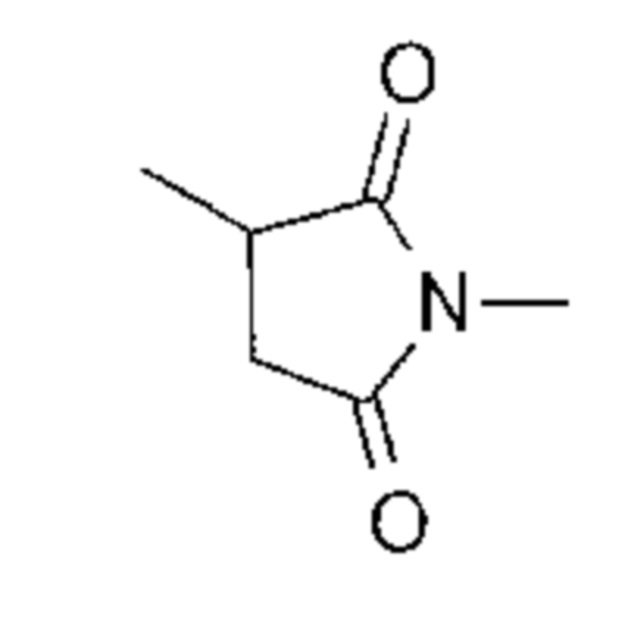 ,
,
которая соединена с анти-HER2 антителом в положении 3 и связанную с метиленовой группой в связующей структуре, содержащей эту структуру на атоме азота в положении 1.
Настоящее изобретение также относится к следующему.
[2] Конъюгат антитела с лекарственным средством по пункту [1], где пептидным остатком LP является пептидный остаток, содержащий аминокислоту, выбранную из фенилаланина, глицина, валина, лизина, цитруллина, серина, глутаминовой кислоты и аспарагиновой кислоты.
[3] Конъюгат антитела с лекарственным средством по пунктам [1] или [2], где LP является пептидный остаток, выбранный из следующей группы:
-GGF-,
-DGGF-,
-(D-)D-GGF-,
-EGGF-,
-GGFG-,
-SGGF-,
-KGGF-,
-DGGFG-,
-GGFGG-,
-DDGGFG-,
-KDGGFG-, и
-GGFGGGF-;
где "(D-)D" является D-аспарагиновой кислотой.
[4] Конъюгат антитела с лекарственным средством по пунктам [1] или [2], где LP является пептидный остаток, состоящий из 4 аминокислот.
[5] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [4], где LP является тетрапептидный остаток -GGFG-.
[6] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [5], где n3 является целым числом от 2 до 5, и L2 является одинарной связью.
[7] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [5], где n3 является целым числом от 2 до 5, L2 является -NH-(CH2CH2-O)n4-CH2CH2-C(=O)- и n4 равно 2 или 4.
[8] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [7], где -NH-(CH2)n1-La-(CH2)n2-C(=O)- является подструктурой, имеющей длину цепи от 4 до 7 атомов.
[9] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [7], где -NH-(CH2)n1-La-(CH2)n2-C(=O)- является подструктурой, имеющей длину цепи от 5 до 6 атомов.
[10] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [9], где -NH-(CH2)n1-La-(CH2)n2-C(=O)- является
-NH-CH2CH2-C(=O)-,
-NH-CH2CH2CH2-C(=O)-,
-NH-CH2CH2CH2CH2-C(=O)-,
-NH-CH2CH2CH2CH2CH2-C(=O)-,
-NH-CH2-O-CH2-C(=O)-,
-NH-CH2CH2-O-CH2-C(=O)- или
-NH-CH2CH2-O-C(=O)-.
[11] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [9], где -NH-(CH2)n1-La-(CH2)n2-C(=O)- является
-NH-CH2CH2CH2-C(=O)-,
-NH-CH2-O-CH2-C(=O)- или
-NH-CH2CH2-O-CH2-C(=O)-.
[12] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [9], где структурой лекарственное средство-связующая группа, в которой лекарственное средство соединено с -L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-, является структура лекарственное средство-связующая группа, выбранное из следующей группы:
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
где -(сукцинимид-3-ил-N)- имеет структуру, представленную следующей формулой:
Формула 4
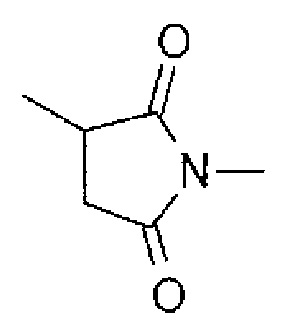 ,
,
которая связана с анти-HER2 антителом в положении 3, и связана с метиленовой группой в связующей структуре, содержащей эту структуру на атоме азота в положении 1,
-(NH-DX) является группой, представленной следующей формулой:
Формула 5
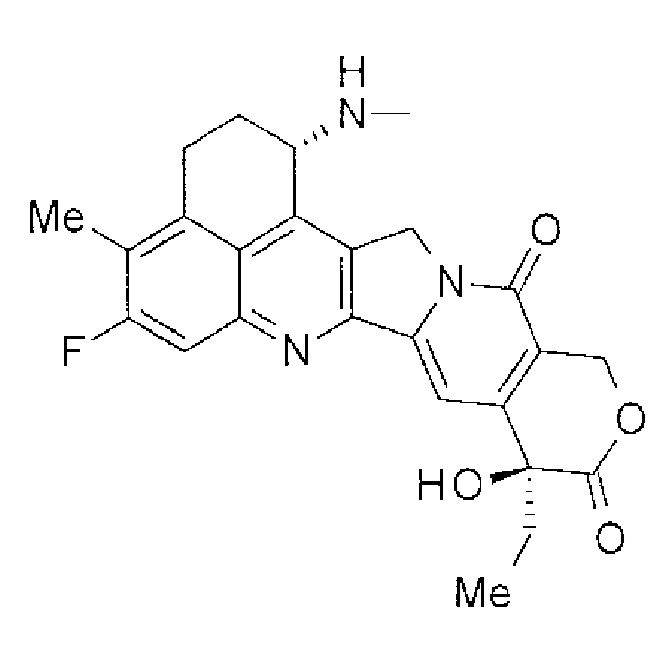 ,
,
где атом азота аминогруппы в положении 1 является связующим положением, и
-GGFG- является тетрапептидным остатком -Gly-Gly-Phe-Gly-.
[13] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [9], где структурой лекарственное средство-связующая группа, содержащей лекарственное средство, присоединенное к -L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-, является структура лекарственное средство-связующая группа, выбранная из следующей группы:
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
Здесь, -(сукцинимид-3-ил-N)-, -(NH-DX) и -GGFG- такие, как определены выше.
[14] Конъюгат антитела с лекарственным средством, где противоопухолевое соединение, представленное следующей формулой:
Формула 6
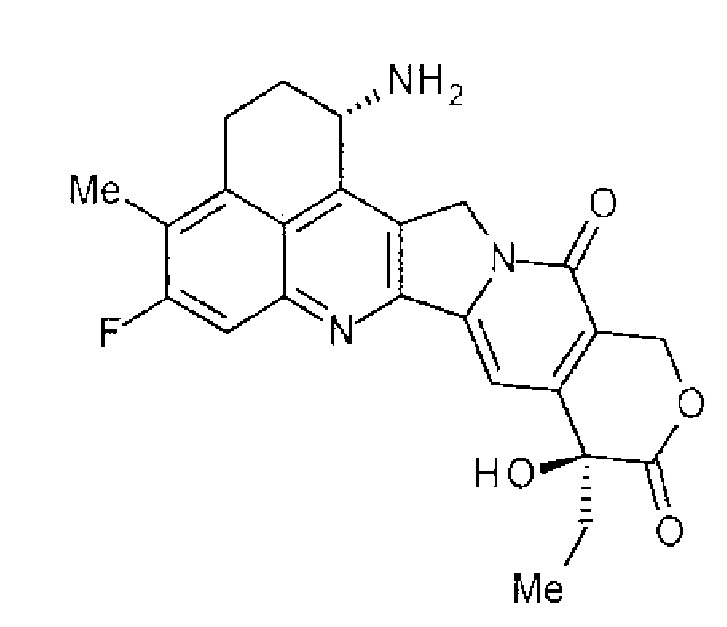 ,
,
конъюгировано с анти-HER2 антителом через связующую группу, имеющую структуру, представленную следующей формулой:
-L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-
через тиоэфирную связь, которая образуется на дисульфидной группе, присутствующей в шарнирной части анти-HER2 антитела,
где
анти-HER2 антитело соединено с концевой группой L1, где противоопухолевое соединение соединено с карбонильной группой -(CH2)n2-C(=O)- части,
где
n1 является целым числом от 0 до 6,
n2 является целым числом от 0 до 5,
L1 является -(сукцинимид-3-ил-N)-(CH2)n3-C(=O)-,
где n3 является целым числом от 2 до 8,
L2 является -NH-(CH2CH2-O)n4-CH2CH2-C(=O)- или одинарной связью,
где n4 является целым числом от 1 до 6,
LP является тетрапептидным остатком -GGFG-,
La является -O- или одинарной связью, и
-(сукцинимид-3-ил-N)- имеет структуру, представленную следующей формулой:
Формула 7
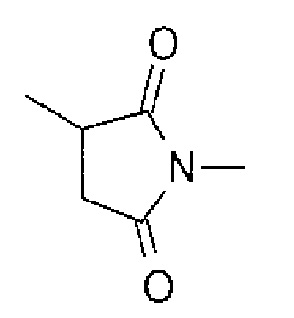 ,
,
которая соединена с анти-HER2 антителом в положении 3, и соединена с метиленовой группой в связующей структуре , содержащей эту структуру на атоме азота в положении 1.
[15] Конъюгат антитела с лекарственным средством по пункту [14], где
n1 равно 3, n2 равно 0, n3 равно 2, L2 является -NH-(CH2CH2-O)n4-CH2CH2-C(=O)-, n4 является 2 и La является одинарной связью,
n1 равно 1, n2 равно 1, n3 равно 5, L2 является одинарной связью и La является -O-, или
n1 равно 2, n2 равно 1, n3 равно 5, L2 является одинарной связью и La является -O-.
[16] Конъюгат антитела с лекарственным средством по пункту [14] или [15], где n3 равно 2 или 5 и L2 является одинарной связью.
[17] Конъюгат антитела с лекарственным средством по пункту [14] или [15], где n3 равно 2 или 5, L2 является -NH-(CH2CH2-O)n4-CH2CH2-C(=O)- и n4 равно 2 или 4.
[18] Конъюгат антитела с лекарственным средством по любому из пунктов от [14] до [17], где -NH-(CH2)n1-La-(CH2)n2-C(=O)- является
-NH-CH2CH2CH2-C(=O)-,
-NH-CH2-O-CH2-C(=O)-, или
-NH-CH2CH2-O-CH2-C(=O)-.
[19] Конъюгат антитела с лекарственным средством по любому из пунктов от [14] до [18], где структурой лекарственное средство-связующая группа, связанной с -L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-, является одна из структур лекарственное средство-связующая группа, выбранных из группы, включающей следующие:
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
где -(сукцинимид-3-ил-N)- имеет структуру, представленную формулой:
Формула 8
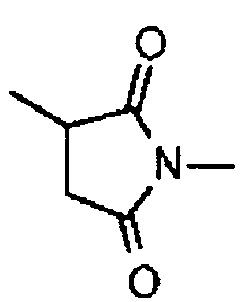 ,
,
которая соединена с анти-HER2 антителом в положении 3 и соединена с метиленовой группой в связующей структуре, содержащей эту структуру на атоме азота в положении 1,
-(NH-DX) является группой, представленной следующей формулой:
Формула 9
 ,
,
где атом азота аминогруппы в положении 1 является связующим положением, и
-GGFG- является тетрапептидным остатком -Gly-Gly-Phe-Gly-.
[20] Конъюгат антитела с лекарственным средством по любому из пунктов от [14] до [18], где структурой лекарственное средство-связующая группа, содержащей лекарственное средство, соединенное с -L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-, является структура лекарственное средство-связующая группа, выбранная из следующей группы:
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX), и
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
Здесь, -(сукцинимид-3-ил-N)-, -(NH-DX) и -GGFG- такие, как определены выше.
[21] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [20], где среднее количество единиц в выбранной структуре лекарственное средство-связующая группа, конъюгированной с молекулой антитела, составляет от 1 до 10.
[22] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [20], где среднее количество единиц в выбранной структуре лекарственное средство-связующая группа, конъюгированной с молекулой антитела, составляет от 2 до 8.
[23] Конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [20], где среднее количество единиц в выбранной структуре лекарственное средство-связующая группа, конъюгированной с молекулой антитела, составляет от 3 до 8.
[24] Лекарственное средство, содержащее конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [23], его соль или его гидрат.
[25] Противоопухолевое лекарственное средство и/или противораковое лекарственное средство, содержащее конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [23], его соль или его гидрат.
[26] Противоопухолевое лекарственное средство и/или противораковое лекарственное средство по пункту [25], которое применяют при раке легкого, уротелиальном раке, раке ободочной и прямой кишки, раке предстательной железы, раке яичников, раке поджелудочной железы, раке молочной железы, раке мочевого пузыря, раке желудка, гастроинтестинальной стромальной опухоли, раке шейки матки, раке пищевода, плоскоклеточном раке, перитонеальном раке, раке печени, печеночно-клеточном раке, раке толстой кишки, раке прямой кишки, раке ободочной и прямой кишки, раке эндометрия, раке матки, раке слюнной железы, раке почки, раке вульвы, раке щитовидной железы, раке полового члена, лейкемии, злокачественной лимфоме, плазмоцитоме, миеломе или саркоме.
[27] Фармацевтическая композиция, содержащая конъюгат антитела с лекарственным средством по любому из пунктов от [1] до [23], его соль или его гидрат в качестве активного компонента, и фармацевтически приемлемый наполнитель.
[28] Фармацевтическая композиция по пункту [27], которая предназначена для использования при раке легкого, уротелиальном раке, раке ободочной и прямой кишки, раке предстательной железы, раке яичников, раке поджелудочной железы, раке молочной железы, раке мочевого пузыря, раке желудка, гастроинтестинальной стромальной опухоли, раке шейки матки, раке пищевода, плоскоклеточном раке, перитонеальном раке, раке печени, печеночно-клеточном раке, раке толстой кишки, раке прямой кишки, раке ободочной и прямой кишки, раке эндометрия, раке матки, раке слюнной железы, раке почки, раке вульвы, раке щитовидной железы, раке полового члена, лейкемии, злокачественной лимфоме, плазмоцитоме, миеломе или саркоме.
[29] Способ лечения опухоли и/или рака, включающий введение конъюгата антитела с лекарственным средством по любому из пунктов от [1] до [23], его соли или его гидрата.
[30] Способ получения конъюгата антитела с лекарственным средством, включающий взаимодействие соединения, представленного следующей формулой:
(малеимид-N-ил)-(CH2)n3-C(=O)-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-(NH-DX)
с анти-HER2 антителом или его реакционноспособным производным и конъюгирование группы лекарственное средство-связующая группа с антителом способом получения тиоэфирной связи на дисульфидной связи, присутствующей в шарнирной части антитела.
В формуле, n3 является целым числом от 2 до 8,
L2 является -NH-(CH2CH2-O)n4-CH2CH2-C(=O)- или одинарной связью,
где n4 является целым числом от 1 до 6,
LP является пептидным остатком, состоящим из от 2 до 7 аминокислот, выбранных из фенилаланина, глицина, валина, лизина, цитруллина, серина, глутаминовой кислоты и аспарагиновой кислоты,
n1 является целым числом от 0 до 6,
n2 является целым числом от 0 до 5,
La является -O- или одинарной связью,
(малеимид-N-ил)- является группой, представленной следующей формулой:
Формула 10
 ,
,
где атом азота является связующим положением, и
-(NH-DX) является группой, представленной следующей формулой:
Формула 11
 ,
,
где атом азота аминогруппы в положении 1 является связующим положением.
[31] Способ получения по пункту [30], где способом конъюгирования группы лекарственное средство-связующая группа с анти-HER2 антителом является способ восстановления антитела для превращения антитела в реакционноспособное производное.
[32] Способ получения по пункту [30] или [31], где среднее количество единиц выбранной одной структуры лекарственное средство-связующая группа, конъюгированной с молекулой антитела составляет от 1 до 10.
[33] Способ получения по пункту [30] или [31], где среднее количество единиц выбранной одной структуры лекарственное средство-связующая группа, конъюгированной с молекулой антитела составляет от 2 до 8.
[34] Способ получения по пункту [30] или [31], где среднее количество единиц выбранной одной структуры лекарственное средство-связующая группа, конъюгированной с молекулой антитела составляет от 3 до 8.
[35] Конъюгат антитела с лекарственным средством, полученный способом получения по любому пункту от [30] до [34].
[36] Конъюгат антитела с лекарственным средством, полученный образованием тиоэфирной связи на сульфидном месте связывания в шарнирной части антитела, где анти-HER2 антитело обрабатывают в восстанавливающих условиях с последующим взаимодействием с соединением, выбранным из группы, показанной ниже:
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX), и
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX).
В перечисленном выше, (малеимид-N-ил)- является группой, представленной формулой:
Формула 12
 ,
,
где атом азота является положением связывания, и
-(NH-DX) является группой, представленной следующей формулой:
Формула 13
 ,
,
где атом азота аминогруппы в положении 1 является положением связывания, и
-GGFG- является тетрапептидным остатком -Gly-Gly-Phe-Gly-.
[37] Конъюгат антитела с лекарственным средством, полученный образованием тиоэфирной связи на сульфидном месте связывания в шарнирной части антитела, где анти-HER2 антитело обрабатывают в восстанавливающих условиях с последующим взаимодействием с соединением, выбранным из группы, показанной ниже:
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX), и
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX).
Здесь, (малеимид-N-ил)-, -(NH-DX) и -GGFG- такие, как определены выше.
[38] Конъюгат антитела с лекарственным средством по пунктам [36] или [37], где среднее количество единиц выбранной одной структуры лекарственное средство-связующая группа, конъюгированной с молекулой антитела составляет от 1 до 10.
[39] Конъюгат антитела с лекарственным средством по пунктам [36] или [37], где среднее количество единиц выбранной одной структуры лекарственное средство-связующая группа, конъюгированной с молекулой антитела составляет от 2 до 8.
[40] Конъюгат антитела с лекарственным средством по пункту [36] или [37], где среднее количество единиц выбранной одной структуры лекарственное средство-связующая группа, конъюгированной с молекулой антитела составляет от 3 до 8.
Преимущества настоящего изобретения
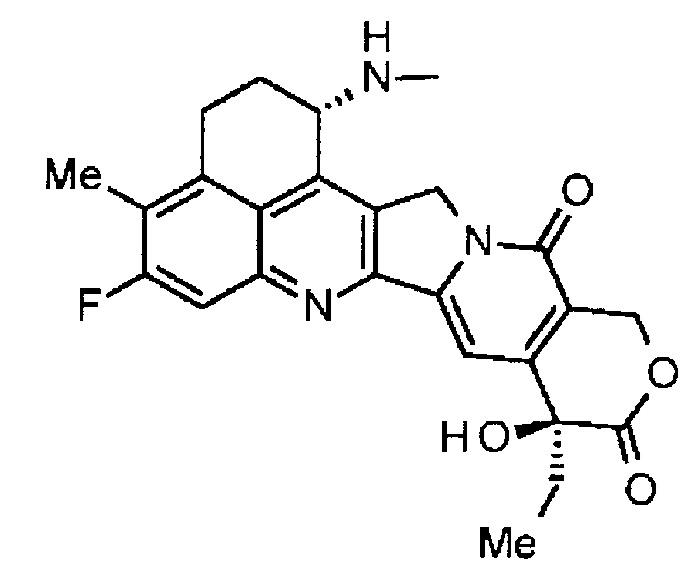
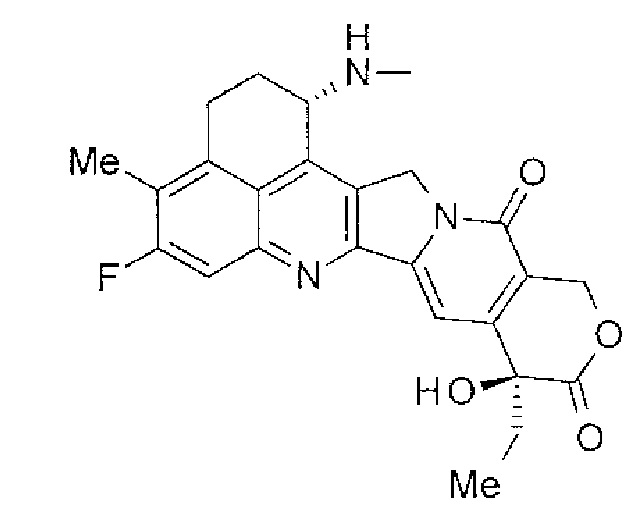
При использовании анти-HER2 конъюгата антитела с лекарственным средством, содержащего противоопухолевое соединение экзатекан, конъюгированное через связующую группу с определенной структурой, может быть достигнуто превосходное противоопухолевое действие.
Краткое описание фигур
[Фигура 1] На фигуре 1 показана последовательность аминокислоты тяжелой цепи гуманизированного анти-HER2 моноклонального антитела (SEQ ID NO: 1).
[Фигура 2] На фигуре 2 показана последовательность аминокислоты легкой цепи гуманизированного анти-HER2 моноклонального антитела (SEQ ID NO: 2).
[Фигура 3] На фигуре 3 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (27) или трастузумабом на безтимусных мышей с трансплантированной подкожно колонией клеток рака молочной железы человека KPL-4. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 4] На фигуре 4 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (8), (28) или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток рака желудка человека NCI-N87. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 5] На фигуре 5 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (8), (29), (30), трастузумабом или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток рака молочной железы человека JIMT-1. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 6] На фигуре 6 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (31), трастузумабом или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток рака поджелудочной железы человека Capan-1. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 7] На фигуре 7 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (50) на безтимусных мышей с трансплантированной подкожно колонией клеток рака желудка человека NCI-N87. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 8] На фигуре 8 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (50), трастузумабом или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток рака молочной железы человека ST225. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 9] На фигуре 9 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (50), трастузумабом или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток рака молочной железы человека ST910. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 10] На фигуре 10 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (50), трастузумабом или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток рака прямой и ободочной кишки человека CTG-0401. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 11] На фигуре 11 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (50), трастузумабом или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток не мелкоклеточного рака легких человека CTG-0860. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 12] На фигуре 12 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (50), трастузумабом или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток рака желчных протоков человека CTG-0927. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 13] На фигуре 13 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (50), трастузумабом или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток рака пищевода человека CTG-0137. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
[Фигура 14] На фигуре 14 представлена диаграмма, показывающая противоопухолевое действие конъюгата антитела с лекарственным средством (50), трастузумабом или трастузумабом эмтанзином на безтимусных мышей с трансплантированной подкожно колонией клеток рака яичников человека SK-OV-3. На фигуре, на абсциссе указаны дни после инокуляции опухоли, и на ординате указан объем опухоли.
Описание вариантов
Далее предпочтительные варианты осуществления настоящего изобретения описаны со ссылкой на фигуры. Описанные ниже варианты даны только для иллюстрации одного примера типового варианта настоящего изобретения и не ограничивают объем настоящего изобретения.
Анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением представляет собой противоопухолевое лекарственное средство, в котором анти-HER2 антитело конъюгировано с противоопухолевым соединением через связующую структурную группу, оно более подробно описано ниже.
[Антитело]
Анти-HER2 антитело, применяемое в анти-HER2 конъюгате антитела с лекарственным средством в соответствии с данным изобретением, может быть получено из любых видов, и предпочтительные варианты видов могут включать человека, крыс, мышей и кроликов. В случае, когда антитело получено из вида, отличного от человека, но, предпочтительно, химеризовано или гуманизировано с применением хорошо известной методики. Антителом в соответствии с данным изобретением может быть поликлональное антитело или моноклональное антитело и, предпочтительно, является мноклональным антителом.
Анти-HER2 антителом является антитело, которое способно поражать опухолевые клетки, то есть, обладает свойством распознавания опухолевых клеток, свойством связывания с опухолевой клеткой, свойством интернализироваться в опухолевой клетке, активностью, разрушающей опухолевые клетки или подобными, и может быть конъюгировано с лекарственным средством, обладающим противоопухолевым действием, через связующую группу с получением конъюгата антитела с лекарственным средством.
Связывающая активность антитела против опухолевых клеток может быть подтверждена с применением проточной цитометрии. Интернализация антитела в опухолевые клетки может быть подтверждена с применением (1) анализа визуализации антитела, введенного в клетки, под флуоресцентным микроскопом с применением связывания вторичного антитела (флуоресцентно меченного) с терапевтическим антителом (Cell Death and Differentiation (2008) 15, 751-761), (2) анализа измерения интенсивности флуоресценции, введенной в клетки, с применением связывания вторичного антитела ((флуоресцентно меченного) с терапевтическим антителом (Molecular Biology of the Cell, Vol. 15, 5268-5282, December 2004) или (3) анализа Mab-ZAP с применением связывания иммунотоксина с терапевтическим антителом, где токсин выделяется при введении в клетки для ингибирования роста клетки (Bio Techniques 28: 162-165, January 2000). В качестве иммунотоксина может применяться рекомбинантный комплекс белка каталитического домена токсина дифтерии и белка G.
Противоопухолевое действие антитела может быть подтверждено in vitro определением ингибирующего действия против роста клеток. Например, колонию раковых клеток, чрезмерно экспрессирующую целевой белок для антитела, культивируют, и антитело добавляют в различных концентрациях в культуральную систему для определения ингибирующего действия против формирования очагов, формирования колонии и роста сфероида. Противоопухолевая активность может быть подтверждена in vivo, например, введением антитела безтимусным мышам с трансплантированной колонией опухолевых клеток, сильно экспрессирующей целевой белок, и определением изменений в раковых клетках.
Так как соединение, конъюгированное в конъюгат антитела с лекарственным средством, обладает противоопухолевым действием, предпочтительно, но не существенно, чтобы само антитело также обладало противоопухолевым действием. Для целей специфического и селективного оказания цитотоксического действия противоопухолевого соединения в отношении раковых клеток, важно и предпочтительно, чтобы антитело обладало свойством интернализации для миграции в опухолевые клетки.
Анти-HER2 антитело может быть получено методикой, известной в данной области техники. Например, антитело в соответствии с данным изобретением может быть получено способом, обычно применяемым в данной области техники, который включает иммунизацию животных антигенным полипептидом и сбор, и очистку антител, полученных in vivo. Происхождение антигена не ограничено человеком, и животные могут быть иммунизированы антигеном, полученным от не человекообразного животного, такого как мышь, крыса и подобные. В этом случае, перекрестная реакционная способность антител, связывающихся с полученным гетерологичным антигеном с человеческими антигенами, может быть тестирована для выбора антитела, применимого для человеческих заболеваний.
Альтернативно, производящие антитело клетки, которые производят антитела против антигена, сливают с клетками миеломы способом, известным в данной области техники (например, Kohler and Milstein, Nature (1975) 256, p. 495-497; и Kennet, R. ed., Monoclonal Antibodies, p. 365-367, Plenum Press, N.Y. (1980)) для установления гибридом, из которых, в свою очередь, могут быть получены моноклональные антитела.
Антиген может быть получен генной инженерией клеток-хозяев с получением гена, кодирующего антигенный белок. Более конкретно, векторы, которые позволяют экспрессию гена антигена, получают и переносят в клетки-хозяева так, чтобы экспрессировать ген. Экспрессированный таким образом антиген может быть очищен. Антитело также может быть получено способом иммунизации животных описанными выше полученными генной инженерией экспрессирующими антиген клетками или колонией клеток, экспрессирующих антиген.
Анти-HER2 антитела, которые могут применяться в соответствии с данным изобретением, особенно не ограничены и, предпочтительно, включают такие, которые имеют описанные ниже свойства.
(1) Анти-HER2 антитело, имеющее следующие свойства:
(a) специфическое связывание с HER2, и
(b) обладание активностью интернализации в HER2-экспрессирующих клетках через связывание с HER2.
(2) Антитело, такое как описано в (1) выше, где антитело связывается с внеклеточным доменом HER2.
(3) Антитело, такое как описано в (1) или (2) выше, где антителом является моноклональное антитело.
(4) Антитело, такое как описано в любом из (1)-(3) выше, где антитело обладает антитело-зависимой клеточной цитотоксичностью (АЗКЦ) и/или комплемент-зависимой цитотоксичностью (КЗЦ)
(5) Антитело, такое как описано в любом из (1)-(4) выше, где антителом является мышиное моноклональное антитело, химерное моноклональное антитело или гуманизированное моноклональное антитело.
(6) Антитело, такое как описано в любом из (1)-(5) выше, где антителом является гуманизированное моноклональное антитело, содержащее тяжелую цепь, состоящую из последовательности аминокислот, представленной SEQ ID NO: 1, и легкую цепь, состоящую из последовательности аминокислот, представленной SEQ ID NO: 2.
(7) Антитело, такое как описано в любом из (1)-(6) выше, где антител не содержит лизиновый остаток на карбоксильном окончании тяжелой цепи.
(8) Антитело, такое как описано в (7) выше, где антитело содержит тяжелую цепь, состоящую из последовательности аминокислот, состоящей из остатков аминокислот от 1 до 449 из SEQ ID NO: 1, и легкую цепь, состоящую из последовательности аминокислот, состоящей из остатков аминокислот от 1 до 214 из SEQ ID NO: 2.
(9) Антитело, полученное способом получения антитела, такого как любое из (1)-(8) выше, где способ включает стадии: культивирования клеток-хозяев, трансформированных вектором экспрессии, содержащим полинуклеотид, кодирующий антитело; и сбора целевых антител из культур, полученных на предыдущей стадии.
Далее описано анти-HER2 антитело, применяемое в соответствии с данным изобретением.
Термины "рак" и "опухоль" в данном описании применяют взаимозаменяемо.
Термин "ген" в данном описании включает не только ДНК, но также ее мРНК, кДНК и кРНК.
Термин "полинуклеотид" в данном описании имеет то же значение, что и нуклеиновая кислота, и также включает ДНК, РНК, пробы, олигонуклеотиды и праймеры.
Термины "полипептид", "белок" и "белок" в данном описании применяют взаимозаменяемо.
Термин "клетка" в данном описании также включает клетки животных и культивированные клетки.
Термин "HER2" в данном описании имеет то же значение, что и HER2 белок.
Примеры анти-HER2 антител в данном описании могут включать, но не ограничены ими, пертузумаб (публикация международного патента № WO 01/00245) и трастузумаб (патент США № 5821337). Трастузумаб является предпочтительным. Однако анти-HER2 антитело в соответствии с данным изобретением не ограничено в той мере, в какой оно является анти-HER2 антителом, специфически связывающимся с HER2, и более предпочтительно, обладающим способностью интернализироваться в экспрессирующих HER2 клетках через связывание с HER2.
Термин "трастузумаб" в данном описании также называют HERCEPTIN(R), huMAb4D5-8 или rhuMAb4D5-8, и он является гуманизированным антителом, содержащим тяжелую цепь, состоящую из последовательности аминокислот, состоящей из остатков аминокислот от 1 до 449 из SEQ ID NO: 1 (фигура 1), и легкую цепь, состоящую из последовательности аминокислот, состоящей из остатков аминокислот от 1 до 214 из SEQ ID NO: 2 (фигура 2).
Термин "специфическое связывание" в данном описании означает связывание, которое не является неспецифической адсорбцией. Примеры критериев для определения того, является ли связывание специфическим или нет, могут включать константу диссоциаций (далее обозначенную как "КД"). Значение КД антитела для HER2 белка предпочтительно составляет 1×10-5 M или менее, 5×10-6 M или менее, 2×10-6 M или менее или 1×10-6 M или менее, более предпочтительно, 5×10-7 M или менее, 2×10-7 M или менее или 1×10-7 M или менее, еще более предпочтительно, 5×10-8 M или менее, 2×10-8 M или менее или 1×10-8 M или менее, и наиболее предпочтительно, 5×10-9 M или менее, 2×10-9 M или менее или 1×10-9 M или менее. Связывание между HER2 белком и антителом может быть измерено способом, известным в данной области техники, таким как резонанс поверхностных плазмонов, ELISA или РИА.
Термин "ОКО" в данном описании относится к определяющей комплементарность области (ОКО). Известно, что каждая тяжелая и легкая цепь молекулы антитела имеет три определяющих комплементарность области (ОКО). ОКО также называют гипервариабельным доменом, и она присутствует в вариабельной области каждой тяжелой и легкой цепи антитела. Она представляет собой сайт, который обладает необычно высокой вариабельностью в своей первичной структуре, и имеется три отдельных ОКО в первичной структуре каждой тяжелой и легкой полипептидной цепи. В данном описании, что касается ОКО антитела, ОКО тяжелой цепи представлены ОКОТ1, ОКОТ2 и ОКОТ3 из амино-концевой стороны последовательности аминокислот тяжелой цепи, и ОКО легкой цепи представлены ОКОЛ1, ОКОЛ2 и ОКОЛ3 из амино-концевой стороны последовательности аминокислот легкой цепи. Эти сайты расположены рядом друг с другом в третичной структуре и определяют специфичность антигена, с которым связывается антитело.
Фраза "гибридизацию проводят в жестких условиях" в данном описании относится к процессу, в котором гибридизацию проводят в условиях, при которых идентификация может быть достигнута проведением гибридизации при 68°C в коммерчески доступном растворе для гибридизации ExpressHyb Hybridization Solution (производства Clontech, Inc.), или проведением гибридизации при 68°C в присутствии от 0,7 до 1,0 M NaCl с применением фильтра, содержащего обездвиженную ДНК, с последующим промыванием при 68°C с применением от 0,1 до 2 x SSC раствора (1 x SSC раствор состоит из 150 мМ NaCl и 15 мМ цитата натрия) или в эквивалентных условиях.
1. HER2
HER2 является одним из онкогенных продуктов типового онкогена рецептора фактора роста, идентифицированного в качестве онкогена, связанного с рецептором 2 эпидермального фактора роста человека, и является белком трансмембранного рецептора, имеющего молекулярную массу 185 кДа и имеющего домен тирозинкиназы. HER2 является членом семейства РЭФР, состоящего из HER1 (РЭФР, ErbB-1), HER2 (neu, ErbB-2), HER3 (ErbB-3) и HER4 (ErbB-4), и как известно, он аутофосфорилирован на внутриклеточных тирозиновых остатках путем образования гомодимера или гетеродимера с другим рецептором РЭФР HER1, HER3 или HER4, и сам он активируется именно таким образом, тем самым играя важную роль в росте, дифференциации и выживании нормальных клеток и опухолевых клеток.
Что касается белка HER2, применяемого в соответствии с данным изобретением, белок HER2 может быть непосредственно очищен из экспрессирующих HER2 клеток человека или не человекообразного млекопитающего (такого как крыса или мышь) и использован, или фракция мембран клеток описанных выше клеток может быть получена и использована. Кроме того, HER2 может быть получен его in vitro синтезом или его получением в клетках-хозяевах с применением генной инженерии. В генной инженерии, конкретно, после того, как кДНК HER2 интегрируют в вектор, способный экспрессировать кДНК HER2, белок HER2 может быть получен синтезом в растворе, содержащем фермент, субстрат и энергетическое вещество, требуемое для транскрипции и трансляции, или экспрессированием HER2 в другой прокариотной или эукариотной трансформированной клетке-хозяине. Альтернативно, описанные выше генно-инженерные HER2-экспрессирующие клетки или колония клеток, экспрессирующих HER2, могут применяться в качестве белка HER2.
Последовательность ДНК и последовательность аминокислот HER2 описаны в общественной базе данных, и могут быть обозначены как, например, образец № M11730 (GenBank), NP_004439.2 (NCBI) или подобные.
Далее белок, который состоит из последовательности аминокислот, где одна или несколько аминокислот замещены, удаляют и/или добавляют в любую из описанных выше последовательностей аминокислот HER2, и он также обладает биологическим действием, эквивалентным действию белка, также включенного в HER2.
Человеческий белок HER2 состоит из сигнальной последовательности, состоящей из N-концевых 22 аминокислотных остатков, внеклеточного домена, состоящего из 630 аминокислотных остатков, трансмембранного домена, состоящего из 23 аминокислотных остатков и внутриклеточного домена, состоящего из 580 аминокислотных остатков.
2. Получение анти-HER2 антитела
Антитело против HER2 в соответствии с данным изобретением может быть получено, например, способом, обычно проводимым в данной области техники, который включает иммунизацию животных HER2 или произвольным полипептидом, выбранным из последовательности аминокислот HER2, и сбор и очистку антител, полученных in vivo. Биологические виды HER2, применяемого в качестве антигена, не ограничены только человеком, и животное, которое может быть иммунизировано HER2, выбирают из животных, отличных от человека, таких как мыши или крысы, или крысы p185neu. В этом случае, исследование перекрестной реакционной способности между антителом, связывающимся с полученным гетерологическим HER2, и человеческим HER2 позволяет выбрать антитело, применимое при заболеваниях человека.
Далее, моноклональное антитело может быть получено из гибридомы, образованной сливанием производящих антитело клеток, которые производят антитело против HER2, с клетками миеломы с применением известного способа (например, Kohler and Milstein, Nature, (1975) 256, pp. 495-497; Kennet, R. ed., Monoclonal Antibodies, pp. 365-367, Plenum Press, N.Y. (1980)).
HER2, применяемый в качестве антигена, может быть получен экспрессией гена HER2 в клетках-хозяевах с применением генной инженерии.
Более конкретно, получают вектор, способный экспрессировать ген HER2, и полученный вектор трансфицируют в клетки-хозяева для экспрессии гена, и затем экспрессированный HER2 очищают.
Альтернативно, описанные выше генно-инженерные клетки, экспрессирующие HER2, или колония клеток, экспрессирующих HER2, могут применяться в качестве белка HER2. Анти-HER2 антитело может быть получено методикой, известной в данной области техники. Далее способ получения антитела против HER2 описан более подробно.
(1) Получение антигена
Примеры антигена, применяемого для получения анти-HER2 антитела, включают HER2, или полипептид, состоящий из частичной последовательности аминокислот, содержащей, по крайней мере, 6 последовательных аминокислот HER2, или производное, полученное добавлением к нему данной последовательности аминокислот или носителя.
HER2 может быть очищен непосредственно из опухолевых тканей человека или опухолевых клеток, и использован. Далее, HER2 может быть получен синтезом in vitro или получением в клетках-хозяевах с применением генной инженерии.
Что касается генной инженерии, более конкретно, после того, как кДНК HER2 интегрируют в вектор, способный экспрессировать кДНК HER2, HER2 может быть получен синтезом в растворе, содержащем фермент, субстрат и энергетическое вещество, требуемое для транскрипции и трансляции, или экспрессированием HER2 в других прокаритоных и эукариотных трансформированных клетках-хозяевах.
Кроме того, антиген также может быть получен в виде секреторного белка через экспрессию слитого белка, полученного замыканием внеклеточного домена HER2, который является белком мембраны, с постоянной областью антитела в подходящей системе хозяин-вектор.
кДНК HER2 может быть получена, например, так называемым способом ПЦР, в котором реакцию полимеразной цепи проводят с применением библиотеки кДНК, экспрессирующей кДНК HER2 в виде матрицы, и праймеров, которые специфически амплифицируют кДНК HER2 (PCR; Saiki, R. K., et al., Science, (1988) 239, pp. 487-489).
In vitro синтез полипептида может быть представлен, но не ограничен им, Rapid Translation System (RTS) производства Roche Diagnostics, Inc.
Примеры прокариотных клеток-хозяев включают Escherichia coli и Bacillus subtilis. Для трансформации клеток-хозяев в целевом гене, клетки-хозяева трансформируют плазмидным вектором, содержащим репликон, т.е., ориджин репликацию, полученную из видов, совместимых с хозяином, и регуляторную последовательность. Далее, вектор предпочтительно имеет последовательность, способную налагать фенотипическую селективность на трансформированную клетку.
Примеры эукариотных клеток-хозяев включают клетки позвоночных, клетки насекомых и клетки дрожжей. В качестве клеток позвоночных часто применяют обезьяньи клетки COS (Gluzman, Y., Cell, (1981) 23, pp. 175-182, ATCC CRL-1650; ATCC: American Type Culture Collection), мышиные фибробласты NIH3T3 (ATCC No. CRL-1658) и штаммы с дефицитом дигидрофолатредуктазы (Urlaub, G. and Chasin, L. A., Proc. Natl. Acad. Sci. USA (1980) 77, pp. 4126-4220) клеток яичников китайского хомячка (клетки CHO; ATCC: CCL-61); и подобные, однако, клетки ими не ограничены.
Полученный таким образом трансформант может быть культивирован с применением способа, обычно применяемого в данной области техники, и через культивирование трансформанта получают целевой полипептид, внутриклеточно или внеклеточно.
Подходящая среда, применяемая для культивирования, может быть выбрана из различных широко применяемых культуральных сред, в зависимости от применяемых клеток-хозяев. Если применяется Escherichia coli, например, может применяться среда LB с добавлением антибиотика, такого как ампициллин или IPMG, при необходимости.
Рекомбинантный белок, полученный внутриклеточно или внеклеточно, с применением описанного культивирования трансформанта, может быть отделен и очищен любым из множества известных способов отделения с применением физического или химического свойства белка.
Конкретные примеры способов включают обработку обычным осадителем белка, ультрафильтрацией, различные типы жидкостной хроматографии, такие как хроматография с молекулярными ситами (гель-фильтрация), адсорбционная хроматография, ионообменная хроматография и аффинная хроматография, диализ, и их сочетание.
Далее, присоединением маркера из шести гистидиновых остатков к экспрессируемому рекомбинантному белку, бело может быть эффективно очищен с применением никелевой аффинной колонки. Альтернативно, присоединением IgG Fc области к экспрессируемому рекомбинантному белку, белок может быть эффективно очищен с применением колонки с белком A.
При объединении описанных выше способов большое количество целевого полипептида может быть легко получено с высоким выходом и высокой степенью чистоты.
писанный выше трансформант сам по себе может применяться в качестве антигена. Колония клеток, экспрессирующих HER2, также может применяться в качестве антигена. Примеры таких колоний клеток могут включать колонии клеток рака молочной железы человека SK-BR-3, BT-474, KPL-4 и JIMT-1, колонии клеток рака желудка человека NCI-N87 и колонии клеток рака яичников человека SK-OV-3. Колония клеток в соответствии с данным изобретением не ограничена этими колониями клеток, при условии, что они экспрессируют HER2.
(2) Получение анти-HER2 моноклонального антитела
Примеры антитела, специфически связывающегося с HER2, включают моноклональное антитело, специфически связывающееся с HER2, и способ получения такого антитела описан ниже.
Получение моноклонального антитела обычно требует следующих рабочих стадий:
(a) очистка биополимера, применяемого в качестве антигена, или получение экспрессирующих антиген клеток;
(b) получение производящих антитело клеток иммунизацией животного инъекцией антигена, сбор крови, исследование ее титра антител для определения момента удаления селезенки;
(c) получением клеток миеломы (далее названных "миелома");
(d) сливанием производящих антитело клеток с миеломой;
(e) скрининг группы гибридом, производящих желаемое антитело;
(f) деление гибридом на отдельные клоны клеток (клонирование);
(g) необязательно, культивирование гибридомы или выведение животного с имплантированной гибридомой для получения большого количества моноклональных антител;
(h) исследование полученных моноклональных антител для определения биологической активности и специфичности связывания, или анализ их свойств в виде меченого реагента; и подобные.
Далее способ получения моноклональных антител описан более подробно по указанным выше стадиям, однако, данный способ не является ограничивающим, и, например, могут применяться производящие антитело клетки, отличные от клеток селезенки и миеломы.
(a) Очистка антигена
В качестве антигена может применяться HER2, полученный способом, описанным выше, или его частичный пептид.
Далее, мембранная фракция, полученная из рекомбинантных клеток, экспрессирующих HER2, или сами рекомбинантные клетки, экспрессирующие HER2, а также частичный пептид белка в соответствии с данным изобретением, химически синтезированный способом, известным специалистам в данной области техники, также могут применяться в качестве антигена.
Более того, колония клеток, экспрессирующих HER2, также может применяться в качестве антигена.
(b) Получение клеток, производящих антитело
Антиген, полученный на стадии (a), смешивают с адъювантом, таким как полный или неполный адъювант Фрейнда, или вспомогательным агентом, таким как сульфат калия-алюминия, и полученную смесь применяют в качестве иммуногена для иммунизации экспериментального животного. Другой способ включает иммунизацию экспериментального животного экспрессирующими антиген клетками в качестве иммуногена. В качестве экспериментального животного может применяться любое животное, применяемое в известном способе получения гибридомы, без ограничений. Более конкретно, например, может применяться мышь, крыса, коза, овца, крупный рогатый скот, лошадь или подобные. Однако, с точки зрения доступности клеток миеломы, сливаемых с экстрагированными производящими антитело клетками, предпочтительно применяют мышь или крысу в качестве иммунизируемого животного.
Далее, вид применяемых мышей или крыс особенно не ограничен, и в случае мышей, например, применяют такие виды, как A, AKR, BALB/c, BDP, BA, CE, C3H, 57BL, C57BL, C57L, DBA, FL, HTH, HT1, LP, NZB, NZW, RF, R III, SJL, SWR, WB и 129 и подобные, и в случае крыс, применяют такие виды, как, например, Wistar, Low, Lewis, Sprague, Dawley, ACI, BN, Fischer и подобные.
Эти мыши и крысы коммерчески доступны от заводчиков/дистрибьюторов экспериментальных животных, например, CLEA Japan, Inc. и Charles River Laboratories Japan, Inc.
В качестве иммунизируемых животных, в точки зрения совместимости слияния с клетками миеломы, описанными выше, особенно предпочтительны, в случае мышей, вид BALB/c, и в случае крыс, виды Wistar и Low.
Далее, с точки зрения антигенной гомологии между человеком и мышами, также предпочтительно применять мышей, имеющих пониженную биологическую функцию для удаления ауто-антител, то есть, мышей с аутоиммунным заболеванием.
Возраст таких мышей или крыс в момент иммунизации предпочтительно составляет от 5 до 12 недель, более предпочтительно, от 6 до 8 недель.
Для иммунизации животного HER2 или его рекомбинанта, например, может применяться известный способ, описанный подробно в, например, Weir, D. M., Handbook of Experimental Immunology Vol. I. II. III., Blackwell Scientific Publications, Oxford (1987); Kabat, E. A. и Mayer, M. M., Experimental Immunochemistry, Charles C Thomas Publisher Springfield, Illinois (1964) или подобные.
Среди этих способов иммунизации, предпочтительным конкретным способом в данном изобретении является, например, следующий.
То есть, сначала, фракцию мембранного белка, являющегося антигеном, или клетки, экспрессирующие антиген, внутрикожно или внутрибрюшинно вводят животному. Однако, сочетание обоих способов введения предпочтительно для повышения эффективности иммунизации, и внутрикожное введение проводят в первой половине, и внутрибрюшинное введение проводят в последней половине или только в виде последней дозы, эффективность иммунизации может быть особенно повышена.
Режим введения антигена варьируется в зависимости от типа иммунизируемого животного, индивидуальных особенностей или подобных. Однако, в общем, режим введения, при котором частота введения антигена составляет от 3 до 6 раз, и интервал введения составляет от 2 до 6 недель, является предпочтительным, и режим введения, при котором частота введения антигена составляет от 3 до 4 раз и интервал введения составляет от 2 до 4 недель, является особенно предпочтительным.
Далее, доза антигена варьируется в зависимости от типа животного, индивидуальных особенностей и подобных, однако, обычно доза составляет от 0,05 до 5 мг, предпочтительно, от 0,1 до 0,5 мг.
Бустер-иммунизацию проводят в течение от 1 до 6 недель, предпочтительно, от 1 до 4 недель, более предпочтительно, от 1 до 3 недель после введения антигена как описано выше. Если иммуногеном являются клетки, применяют от 1×106 до 1×107 клеток.
Доза антигена во время проведения бустер-иммунизации варьируется в зависимости от типа или размера животного и подобных факторов, однако, для, например, мыши, доза обычно составляет от 0,05 до 5 мг, предпочтительно, от 0,1 до 0,5 мг, более предпочтительно, от около 0,1 до 0,2 мг. Если иммуногеном являются клетки, применяют от 1×106 до 1×107 клеток.
Клетки селезенки или лимфоциты, включающие производящие антитело клетки, асептически берут у иммунизированного животного через период времени от 1 до 10 дней, предпочтительно от 2 до 5 дней, более предпочтительно, от 2 до 3 дней от момента бустерной иммунизации. В этот момент измеряют титр антител, и если животное, имеющее достаточно повышенный титр антител, используют в качестве источника производящих антитело клеток, последующая процедура может проводиться более эффективно.
Примеры способа измерения титра антител, применяемого в соответствии с данным изобретением, включают способ РИА и способ ELISA, но они не являются ограничивающими для способа. Например, если применяется способ ELISA, измерение титра антител в данном изобретении может проводиться согласно описанным ниже методикам.
Во-первых, очищенный или частично очищенный антиген адсорбируют на поверхность твердой фазы, такой как 96-луночный планшет для ELISA, и поверхность твердой фазы, содержащую адсорбированный в нее антиген, покрывают белком, не родственным антигену, таким как альбумин бычье сыворотки (АБС). После промывания поверхности, поверхность подвергают контакту с серийно разведенным образцом (например, сывороткой мыши) в качестве первичного антитела для того, чтобы антитело в образце связалось с антигеном.
Затем, в качестве вторичного антитела, добавляют антитело, меченное ферментом против мышиного антитела, и оставляют связываться с мышиным антителом. После промывания, добавляют субстрат для фермента и измеряют изменение абсорбции, которое возникает в результате проявления цвета, вызванного разложением субстрата или подобным, и титр антител рассчитывают на основе этих измерений.
Отделение производящих антитело клеток от клеток селезенки или лимфоцитов иммунизированного животного может проводиться известным способом (например, Kohler et al., Nature (1975), 256, p. 495; Kohler et al., Eur. J. Immunol. (1977), 6, p. 511; Milstein et al., Nature (1977), 266, p. 550; Walsh, Nature (1977), 266, p. 495). Например, в случае клеток селезенки, может применяться общий способ, в котором производящие антитело клетки отделяют гомогенизацией селезенки с получением клеток фильтрацией через сито из нержавеющей стали, и суспендированием клеток в минимальной эссенциальной среде Игла (МЭС).
(c) Получение клеток миеломы (далее обозначенных как "миелома")
Клетки миеломы, применяемые для слияния клеток, особенно не ограничены, и подходящие клетки могут быть выбраны из известных колоний клеток. Однако, с точки зрения удобства выбора гибридомы из слитых клеток, предпочтительно применять штамм с дефицитом ГГФРТ (гипоксантин-гуанинфосфорибозил трансферазы), методика выбора которых известна.
Более конкретно, примеры штамма с дефицитом ГГФРТ включают X63-Ag8(X63), NS1-ANS/1(NS1), P3X63-Ag8.U1(P3U1), X63-Ag8.653(X63.653), SP2/0-Ag14(SP2/0), MPC11-45.6TG1,7(45.6TG), FO, S149/5XXO и BU.1 полученные у мышей; 210.RSY3.Ag.1.2.3(Y3) полученные у крыс; и U266AR(SKO-007), GM1500⋅GTG-A12(GM1500), UC729-6, LICR-LOW-HMy2(HMy2) и 8226AR/NIP4-1(NP41), полученные у человека. Эти штаммы с дефицитом ГГФРТ доступны, например, из ATCC или подобных.
Эти штаммы клеток субкультивируют в подходящей среде, такой как 8-азагуаниновая среда [среда, полученная добавлением 8-азагуанина к среде RPMI 1640 с добавлением глутамина, 2-меркаптоэтанола, гентамицина и фетальной телячьей сыворотки (далее обозначенной как "ФТС")], среда Дульбекко, модифицированная по способу Исков (далее обозначенная как "СДМСИ") или среда Игла, модифицированная по способу Дульбекко (далее обозначен как "СИМСД"). В этом случае, за 3-4 дня до проведения слияния клеток, клетки субкультивируют в нормальной среде (например, среде ASF104 (производства Ajinomoto Co., Ltd.), содержащей 10% ФТС) для того, чтобы обеспечить не менее 2×107 клеток в день слияния клеток.
(d) Слияние клеток
Слияние производящих антитело клеток и клеток миеломы может быть соответствующим образом проведено известным способом (Weir, D. M. Handbook of Experimental Immunology Vol. I. II. III., Blackwell Scientific Publications, Oxford (1987); Kabat, E. A. and Mayer, M. M., Experimental Immunochemistry, Charles C Thomas Publisher, Springfield, Illinois (1964), etc.), в таких условиях, чтобы степень выживания клеток чрезмерно не снижалась.
Как таковой может применяться способ, например, химический способ, в котором производящие антитело клетки и клетки миеломы смешивают в растворе, содержащем полимер, такой как полиэтиленгликоль, в высокой концентрации, физический способ с применением электрической стимуляции или подобные. Среди этих способов конкретный пример химического способа описан ниже.
То есть, в случае, когда полиэтиленгликоль применяют в растворе, содержащем полимер в высокой концентрации, производящие антитело клетки и клетки миеломы смешивают в растворе полиэтиленгликоля, имеющего молекулярную массу от 1500 до 6000, более предпочтительно, от 2000 до 4000, при температуре от 30 до 40°C, предпочтительно, от 35 до 38°C, в течение от 1 до 10 минут, предпочтительно, от 5 до 8 минут.
(e) Выбор группы гибридом
Способ выбора гибридом, полученных описанным выше слиянием клеток, особенно не ограничен. Обычно применяют способ выбора ГАТ (гипоксантин, аминоптерин, тимидин) (Kohler et al., Nature (1975), 256, p. 495; Milstein et al., Nature (1977), 266, p. 550).
Этот способ является эффективным, если гибридомы получают с применением клеток миеломы штамма с дефицитом ГГФРТ, которые не могут выживать в присутствии аминоптерина. То есть, при культивировании не слитых клеток и гибридом в среде ГАТ только гибридомы, резистентные к аминоптерину, селективно выживают и пролиферируют.
(f) Деление на отдельные клоны клеток (клонирование)
В качестве способа клонирования гибридом может применяться известный способ, такой как способ метилцеллюлозы, способ мягкой агарозы или метод серийных разведений (см., например, Barbara, B. M. and Stanley, M. S.: Selected Methods in Cellular Immunology, W. H. Freeman and Company, San Francisco (1980)). Среди этих способов особенно предпочтителен способ трехмерного культивирования, такой как способ метилцеллюлозы. Например, группу гибридом, полученных слиянием клеток, суспендируют в метицеллюлозной среде, такой как ClonaCell-HY Selection Medium D (производства StemCell Technologies, Inc., #03804) и культивируют. Затем полученные колонии гибридом собирают с получением моноклональных гибридом. Собранные соответствующие колонии гибридом культивируют, и гибридомы, у которых подтвержден стабильный титр антител в полученной надосадочной жидкости культуры гидридом, выбирают в качестве штамма гибридом, производящих HER2 моноклональное антитело.
(g) Получение моноклонального антитела культивированием гибридомы
Культивированием выбранной таким образом гибридомы может быть эффективно получено моноклональное антитело. Однако до культивирования предпочтительно проводить скрининг гибридомы, которая производит целевые моноклональные антитела.
Для такого скрининга может применяться известный способ.
Измерение титра антител в данном изобретении может проводиться, например, способом ELISA, подробно описанным в параграфе (b), описанном выше.
Гибридому, полученную описанным выше способом, можно хранить в замороженном состоянии в жидком азоте или в холодильнике при температуре -80°C или ниже.
После завершения клонирования среду меняют со среды HT на нормальную среду, и гибридомы культивируют.
Массовое культивирование проводят культивированием вращением с применением большого культурального сосуда, или спин-культивированием. Из надосадочной жидкости, полученной массовым культивированием, моноклональные антитела, которые специфически связываются с белком в соответствии с данным изобретением, могут быть получены очисткой с применением способа, известного специалистам в данной области техники, такого как гель-фильтрация.
Далее, гибридомы впрыскивают в брюшную полость мыши того же вида, у которого брали гибридому (например, описанным выше BALB/c) или мышам Nu/Nu для пролиферации гибридомы, с получением асцита, содержащего большое количество моноклональных антител в соответствии с данным изобретением.
В случае введения гибридомы в брюшную полость, если за 3-7 дней до впрыскивания вводить минеральное масло, такое как 2,6,10,14-тетраметилпентадекан (пристан), можно получить большее количество асцитов.
Например, иммунодепрессант предварительно вводят в брюшную полость мыши того же вида, у которого были получены гибридомы, для инактивации T клеток. Через 20 дней от 106 до 107 клонированных клеток гибридомы суспендируют в не содержащей сыворотку среде (0,5 мл), и суспензию вводят в брюшную полость мыши. В общем, когда живот раздувается и заполняется асцитами, асциты собирают у мышей. Таким способом моноклональные антитела могут быть получены в концентрации, которая в около 100 раз выше, чем концентрация в культуральном растворе.
Моноклональные антитела, полученные описанным выше способом, могут быть очищены способом, описанным в, например, Weir, D. M.: Handbook of Experimental Immunology Vol. I, II, III, Blackwell Scientific Publications, Oxford (1978).
Полученные таким образом моноклональные антитела обладают высокой антигенной специфичностью к HER2. Примеры моноклональных антител в соответствии с данным изобретением могут включать, но особенно не ограничены ими, мышиные моноклональные антитела 4D5 (ATCC CRL 10463).
(h) Исследование моноклональных антител
Изотип и подкласс полученных таким образом моноклональных антител может быть определен следующим образом.
Сначала, примеры способов идентификации включают способ Оухтерлони, способ ELISA и способ РИА.
Способ Оухтерлони является простым, но если концентрация моноклональных антител является низкой, требуется операция конденсации.
С другой стороны, если применяется способ ELISA или способ РИА, прямое взаимодействие культуральной надосадочной жидкости с твердой фазой в которой адсорбирован антиген, и применение антител, соответствующих различным типам изотипов и подклассов иммуноглобулина в качестве вторичных антител, позволяет идентифицировать изотип и подкласс моноклональных антител.
В дополнение, в качестве еще более простого способа, может применяться коммерчески доступный набор для идентификации (например, Mouse Typer Kit производства Bio-Rad Laboratories, Inc.) или подобные.
Далее, количественное определение белка может проводиться способом Фолина-Лоури и способа расчета, основанного на абсорбции при 280 нм (1,4 (OD 280) = иммуноглобулин 1 мг/мл).
Далее, даже если моноклональные антитела отдельно и независимо получают проведением снова стадий (a)-(h) в (2), возможно получить антитело, обладающее цитотоксическим действием, эквивалентным действию HER2 антитела, полученного на стадии (g). Один из примеров такого антитела включает антитело, которое связывается с тем же эпитопом, как и HER2 антитело, полученное на стадии (g). Если свежее полученные моноклональные антитела связываются с частичным пептидом или частичной третичной структурой, с которыми связывается анти-HER2 антитело, можно сделать вывод, что моноклональные антитела связываются с тем же эпитопом, как и анти-HER2 антитела. Далее, подтверждение того, что моноклональные антитела соревнуются с анти-HER2 антителами за связывание с HER2 (то есть, моноклональное антитело ингибирует связывание между анти-HER2 антителом и HER2), позволяет определить, что моноклональное антитело связывается с тем же эпитопом, что и анти-HER2 антитело, даже если определенная последовательность или структура эпитопа не определена. Если подтверждено, что моноклональное антитело связывается с тем же эпитопом, что и анти-HER2 антитело, то имеется значительная уверенность в том, что моноклональное антитело обладает антиген-связывающим сродством или биологическим действием, эквивалентным анти-HER2 антителу.
(3) Другие антитела
Антитело в соответствии с данным изобретением включает не только описанное выше of моноклональное антитело против HER2, но также рекомбинантное антитело, полученное искусственной модификацией в целях снижения гетерологичной антигенности к человеку, такое как химерное антитело, гуманизированное антитело и человеческое антитело. Эти антитела могут быть получены известными способами.
В качестве химерного антитела может быть указано антитело, в котором переменные и постоянные области получают у различных видов, например, химерное антитело, в котором полученная у мыши или крысы переменная область антитела соединена с полученной у человека постоянной областью антитела (см. Proc. Natl. Acad. Sci. USA, 81, 6851-6855, (1984)). Примеры химерного антитела в соответствии с данным изобретением могут включать, но не ограничены ими, химерное антитело 4D5, содержащее постоянную область с тяжелой цепью человеческого IgG1 или IgG2.
В качестве гуманизированного антитела может быть указано антитело, полученное интеграцией только определяющей комплементарность области (ОКО) в полученное у человека антитело (см. Nature (1986) 321, pp. 522-525), и антитело, полученное трансплантацией части аминокислотных остатков каркасного участка, а также последовательности ОКО в человеческое антитело способом ОКО-трансплантации (WO 90/07861), и антитело, гуманизированное с применением стратегии мутагенеза конверсии генов (патент США № 5821337).
Термин "несколько" в данном описании включает от 1 до 10, от 1 до 9, от 1 до 8, от 1 до 7, от 1 до 6, от 1 до 5, от 1 до 4, от 1 до 3 или 1 или 2.
В качестве аминокислотного замещения в соответствии с данным изобретением предпочтительно консервативное аминокислотное замещение. Консервативным аминокислотным замещением является замещение, имеющее место в группе аминокислот, относящихся к боковым цепям аминокислот. Предпочтительными аминокислотными группами являются следующие: кислотная группа (аспартиновая кислота и глутаминовая кислота); основная группа (лизин, аргинин и гистидин); не полярная группа (аланин, валин, лейцин, изолейцин, пролин, фенилаланин, метионин и типтофан); и не измененное полярное семейство (глицин, аспарагин, глутамин, цистеин, серин, треонин и тирозин). Более предпочтительно, аминокислотными группами являются следующие: алифатическая гидроксигруппа (серин и треонин); амидсодержащая группа (аспарагин и глутамин); алифатическая группа (аланин, валин, лейцин и изолейцин); и ароматическая группа (фенилаланин, триптофан и тирозин). Такое аминокислотное замещение, предпочтительно, проводят в интервале, который не ухудшает свойства вещества, имеющего оригинальную последовательность аминокислот.
Объединение последовательности, имеющей высокую гомологию с описанной выше последовательностью аминокислот с тяжелой цепью, с последовательностью, имеющей высокую гомологию с описанной выше последовательностью аминокислот с легкой цепью, позволяет выбрать антитело, обладающее биологическим действием, эквивалентным действию каждого из писанных выше антител. Такой гомологией обычно является гомология 80% или более, предпочтительно, гомология 90% или более, более предпочтительно, гомология 95% или более, наиболее предпочтительно, гомология 99% или более. Далее, объединение последовательности аминокислот, в которой от одного до нескольких остатков аминокислот замещены, удалены или добавлены в тяжелую цепь или легкую цепь последовательности аминокислот, также позволяет выбрать антитело, обладающее биологическим действием, эквивалентным действию каждого из описанных выше антител. Термин "гомология" в данном описании применяют в значении "идентичность".
Гомология между двумя последовательностями аминокислот может быть определена с применением параметров по умолчанию алгоритма Бласта, версия 2,2,2 (Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaeffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25: 3389-3402). Алгоритм Бласта может применяться также через интернет, оценкой на сайте www.ncbi.nlm.nih.gov/blast.
Далее, антитело в соответствии с данным изобретением включает человеческое антитело, которое связывается с HER2. Анти-HER2 человеческое антитело относится к человеческому антителу, имеющему только последовательность антитела, полученную из человеческой хромосомы. Анти-HER2 человеческое антитело может быть получено способом с применением мыши, производящей человеческое антитело, имеющей фрагмент человеческой хромосомы, включающий гены с тяжелой и легкой цепью человеческого антитела (см. Tomizuka, K. et al., Nature Genetics (1997) 16, pp. 133-143; Kuroiwa, Y. et al., Nucl. Acids Res. (1998) 26, pp. 3447-3448; Yoshida, H. et al., Animal Cell Technology: Basic and Applied Aspects vol. 10, pp. 69-73 (Kitagawa, Y., Matuda, T. and Iijima, S. eds.), Kluwer Academic Publishers, 1999; Tomizuka, K. et al., Proc. Natl. Acad. Sci. USA (2000) 97, pp. 722-727, etc.).
Такая мышь, производящая человеческое антитело, может быть выведена специально следующим образом. Генетически модифицированное животное, у которого локусы генов эндогенного иммуноглобулина с тяжелой и легкой цепью были разорваны, и вместо этого, локусы генов человеческого иммуноглобулина с тяжелой и легкой цепью были введены через вектор искусственной хромосомы дрожжей (ИХД) или подобные, может быть выведено при спаривании животного с выключенными генами и трансгенного животного.
Далее, согласно методу рекомбинантной ДНК, с применением кДНК, кодирующей каждую из таких тяжелых и легких цепей человеческого антитела, и, предпочтительно, вектора, содержащего такие кДНК, трансформируют эукариотные клетки, и культивируют клетки трансформанта, которые производят рекомбинантное человеческое моноклональное антитело, где антитело также может быть получено из культуральной надосадочной жидкости.
Здесь, в качестве хозяина, могут применяться, например, эукариотные клетки, предпочтительно, клетки млекопитающих, такие как клетки CHO, лимфоциты или клетки миеломы.
Далее, также известен способ получения производных из фагового отображения человеческих антител, выбранных из библиотеки человеческих антител (см. Wormstone, I. M. et al., Investigative Ophthalmology & Visual Science. (2002) 43 (7), pp. 2301-2308; Carmen, S. et al., Briefings in Functional Genomics and Proteomics (2002), 1 (2), pp. 189-203; Siriwardena, D. et al., Ophthalmology (2002) 109 (3), pp. 427-431, etc.).
Например, может применяться способ фагового отображения, в котором переменную область человеческого антитела экспрессируют на поверхность фага в виде антитела с одной цепью (scFv) и выбирают фаг, который связывается с антигеном (Nature Biotechnology (2005), 23, (9), pp. 1105-1116).
Анализ гена фага, выбранного на основе связывания с антигеном, позволяет определить последовательность ДНК, кодирующую переменную область человеческого антитела, которое связывается с антигеном.
Если определена последовательность ДНК scFv, которое связывается с антигеном, человеческое антитело может быть получено приготовлением вектора экспрессии, содержащего последовательность, и введением вектора в подходящего хозяина для его экспрессии (WO 92/01047, WO 92/20791, WO 93/06213, WO 93/11236, WO 93/19172, WO 95/01438, WO 95/15388; Annu. Rev. Immunol. (1994) 12, pp. 433-455, Nature Biotechnology (2005) 23 (9), pp. 1105-1116).
В качестве одного из примеров других показателей, применяющихся для сравнения свойств антител, может быть указана стабильность антител. Дифференциальная сканирующая калориметрия (ДСК) является одним из средств, способных быстро и точно измерить среднюю температуру термической денатурации (Tm), которая применяется в качестве подходящего показателя относительной конформационной стабильности белков. Измерение значений Tm с помощью ДСК и сравнение значений позволяет сравнить разность в тепловой стабильности. Известно, что стабильность антител при хранении демонстрирует определенную корреляцию с тепловой стабильностью антител (Lori Burton, et. al., Pharmaceutical Development and Technology (2007) 12, pp. 265-273), и предпочтительные антитела могут быть выбраны с применением тепловой стабильности в качестве показателя. Примеры других показателей выбора антител включают следующие характеристики: выход в подходящей клетке-хозяине является высоким; и способность к агрегации вводном растворе является низкой. Например, антитело, которое демонстрирует наивысший выход, не всегда демонстрирует наивысшую термостабильность, и поэтому необходимо выбирать антитело, наиболее подходящее для введения человеку с помощью комплексной оценки на основе описанных выше критериев.
В настоящее изобретение также включен модифицированный вариант антитела. Модифицированный вариант относится к варианту, полученному химической или биологической модификацией антитела в соответствии с данным изобретением. Примеры химически модифицированного варианта включают варианты, химически модифицированные присоединением химической группы к скелету аминокислоты, варианты, химически модифицированные N-связанной или O-связанной углеводородной цепью, и т.д. Примеры биологически модифицированного варианта включают варианты, полученные пост-трансляционной модификацией (такой как N-связанное или O-связанное гликозилирование, N- или C-концевую обработку, деамидирование, изомеризацию аспарагиновой кислоты или окисление метионина), и варианты, в которых метиониновый остаток добавлен был добавлен к N окончанию через экспрессирование в прокариотной клетке-хозяине. Далее, антитело, меченное так, чтобы сделать возможным определение или выделение антитела или антигена в соответствии с данным изобретением, например, меченое ферментом антитело, флуоресцентно меченое антитело и аффинно меченое антитело, также включены в термин модифицированный вариант. Такой модифицированный вариант антитела в соответствии с данным изобретением применяют для улучшение стабильности и удержания в крови антитела, снижения его антигенности, определения или выделения антитела или антигена, и так далее.
Далее, регулирование модификации гликана, который связан с антителом в соответствии с данным изобретением (гликозилирование, дефукозилирование и т.д.) позволяет улучшить зависимое от антитела клеточное цитотоксичное действие. Методика регулирования модификации гликана антител описана в WO 99/54342, WO 00/61739, WO 02/31140 и т.д. Однако методики ими не ограничены. В понятие антитела в соответствии с данным изобретением также включено антител, в котором отрегулирована модификация гликана.
В случае получения антитела выделением гена антитела с последующим введение гена в подходящего хозяина, может применяться сочетание подходящего хозяина и подходящего вектора экспрессии. Конкретные примеры гена антитела включают сочетание гена, кодирующего последовательность тяжелой цепи антитела, описанного в данном описании, и гена, кодирующего его последовательность легкой цепи. Если клетка-хозяин трансформирована, возможно вставить ген последовательности тяжелой цепи и ген последовательности легкой цепи в один и тот же вектор экспрессии, а также в различные векторы экспрессии, по отдельности.
В случае, когда эукариотные клетки применяют в качестве хозяина, могут применяться животные клетки, растительные клетки и эукариотные микроорганизмы. В качестве животных клеток могут применяться клетки млекопитающих, например, обезьяньи клетки COS (Gluzman, Y., Cell, (1981) 23, pp. 175-182, ATCC CRL-1650), мышиные фибробласты NIH3T3 (ATCC No. CRL-1658) и штаммы с дефицитом дигидрофолатредуктазы (Urlaub, G. and Chasin, L. A., Proc. Natl. Acad. Sci. USA (1980) 77, pp. 4126-4220) клеток яичников китайского хомяка (клетки CHO; ATCC: CCL-61).
В случае применения прокариотных клеток могут быть упомянуты, например, Escherichia coli и Bacillus subtilis.
Введение желаемого гена антитела в эти клетки через трансформацию и культивирование полученных трансформированных клеток in vitro позволяет получать антитело. В описанном выше способе культивирования, выход иногда может варьироваться в зависимости от последовательности антитела, и, поэтому, возможно выбрать антитело, которое можно легко получить в виде фармацевтического препарата, учитывая выход в качестве критерия отбора среди антител, имеющих эквивалентное связующее действие. Поэтому в понятие антитела в соответствии с данным изобретением также включено антитело, полученное способом получения антитела, характеризуемым добавлением стадии культивирования трансформированной клетки-хозяина и стадии сбора желаемого антитела из культивированного продукта, полученного на стадии культивирования.
Известно, что лизиновый остаток на карбоксильном конце тяжелой цепи антитела, полученного в культивированной клетке млекопитающего, удален (Journal of Chromatography A, 705: 129-134 (1995)), и также известно, что два аминокислотных остатка (глицин и лизин) на карбоксильном конце тяжелой цепи антитела, полученного в культивированной клетке млекопитающего, удалены, и пролиновый остаток, только что расположенный на карбоксильном конце, амидирован (Analytical Biochemistry, 360: 75-83 (2007)). Однако такое удаление и модификация последовательности тяжелой цепи не влияет на антиген-связывающее сродство и эффекторную функцию (активацию комплемента, зависимую от антитела клеточную цитотоксичность и т.д.) антитела. Поэтому в антитело в соответствии с данным изобретением также включено антитело, подвергнутое такой модификации и функциональный фрагмент антитела, и также включены варианты с удалением, в которых одна или две аминокислоты были удалены на карбоксильной конце тяжелой цепи, вариант, полученный амидированием варианта с удалением (например, тяжелой цепи, в которой пролиновый остаток карбоксильного конца был амидирован), и подобные. Тип варианта с удалением на карбоксильном конце тяжелой цепи антитела в соответствии с данным изобретением не ограничен указанными выше вариантами, пока сохраняется антиген-связывающее сродство и эффекторная функция. Две тяжелых цепи, составляющие антитело в соответствии с данным изобретением, могут быть одного типа, выбранные из группы, включающей первичную тяжелую цепь и указанный выше вариант с удалением, или могут быть двух типов в сочетании, выбранных из них. На соотношение количества каждого варианта с удалением может влиять тип культивированных клеток млекопитающих, которые производят антитело в соответствии с данным изобретением, и условия культивирования, однако, возможен случай, когда один аминокислотный остаток на карбоксильном конце был удален в обеих из двух тяжелых цепей, являющихся основными компонентами в антителе в соответствии с данным изобретением.
В качестве изотипа антитела в соответствии с данным изобретением могут быть указаны, например, IgG (IgG1, IgG2, IgG3, IgG4), и, предпочтительно, IgG1 или IgG2.
В качестве биологической активности антитела обычно указывают связывание с антигеном, интернализацию в клетках, экспрессируюших антиген, через связывание с антигеном, нейтрализацию активности антигена, улучшение активности антигена, антитело-зависимую клеточную цитотоксичность (АЗКЦ), комплемент-зависимую цитотоксичность (КЗЦ) и антитело-зависимый медиированный клеткой фагоцитоз (АЗМКФ). Биологической активностью антитела в соответствии с данным изобретением является связывание с HER2, и, предпочтительно, интернализация в HER2-экспрессирующих клеток через связывание с HER2. Далее, антитело в соответствии с данным изобретением может иметь АЗКЦ, КЗЦ и/или АЗМКФ в дополнение с интернализации в клетках.
Полученное антитело может быть очищено до гомогенности. Разделение и очистка антитела может проводиться с применением обычного способа разделения и очистки белков. Например, антитело может быть выделено и очищено подходящим выбором и объединением хроматографии на колонке, фильтрации на фильтре, ультрафильтрации, осаждения соли, диализа, препаративного электрофореза в полиакриламидном геле, электрофорез с изоэлектрическим фокусированием и подобных (Strategies for Protein Purification and Characterization: A Laboratory Course Manual, Daniel R. Marshak et al. eds., Cold Spring Harbor Laboratory Press (1996); Antibodies: A Laboratory Manual. Ed Harlow and David Lane, Cold Spring Harbor Laboratory (1988)), но способ ими не ограничен.
Примеры такой хроматографии включают аффинную хроматографию, ионообменную хроматографию, гидрофобную хроматографию, гельпроникающую хроматографию, хроматографию с обращенной фазой и адсорбционную хроматографию.
Такая хроматография может быть проведена с применением жидкостной хроматографии, такой как ВЭЖХ или ЖЭХБ.
Колонку, применяемую в аффинной хроматографии, выбирают из колонки Protein A и колонки Protein G. Например, колонку, применяемую в качестве колонки Protein A, выбирают из Hyper D, POROS, Sepharose FF (Pharmacia Corporation) и подобных.
Далее, при применении носителя, содержащего обездвиженный в нем антиген, антитело может быть очищено с применением свойства связывания антитела с антигеном.
[Противоопухолевое соединение]
Далее подробно описано противоопухолевое соединение, конъюгированное с анти-HER2 конъюгатом антитела с лекарственным средством в соответствии с данным изобретением. Противоопухолевое соединение, применяемое в соответствии с данным изобретением, особенно не ограничено при условии, что оно является соединением, обладающим противоопухолевым действием, и имеет группу заместителя или подструктуру, позволяющую соединение со связующей структурой. Когда часть или вся связующая группа отщепляется в опухолевых клетках, группа противоопухолевого соединения высвобождается для оказания противоопухолевого действия противоопухолевого соединения. Так как связующая группа отщепляется в положении соединения с лекарственным средством, противоопухолевое соединение выделяется в не модифицированной структуре для оказания противоопухолевого действия.
В качестве противоопухолевого соединения, применяемого в соответствии с данным изобретением, предпочтительно применяют экзатекан (((1S,9S)-1-амино-9-этил-5-фтор-2,3-дигидро-9-гидрокси-4-метил-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-10,13(9H,15H)дион; показанный на формуле ниже), одно из производных камптотецина.
[Формула 14]
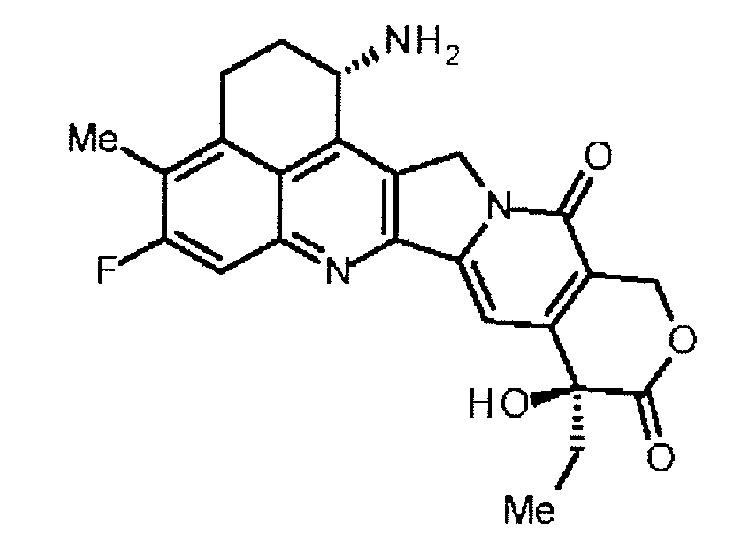
Хотя он обладает превосходным противоопухолевым действием, экзатекан не был выведен на рынок в качестве противоопухолевого лекарственного средства. Соединение может быть легко получено известным способом, и аминогруппа в положении 1 может, предпочтительно, применяться в качестве связующего положения со связующей структурой. Далее, хотя экзатекан также может выделяться в опухолевых клетках, в то время как часть связующей группы все еще присоединена к нему, он является превосходным соединением, обладающим превосходным противоопухолевым действием даже в такой структуре.
Так как экзатекан имеет структуру камптотецина, известно, что равновесие сдвигается к структуре с замкнутым лактоновым кольцом (замкнутым кольцом) в водной кислой среде (например, pH 3 или около того), но оно сдвигается к структуре с открытым лактоновым кольцом (открытым кольцом) в водной щелочной среде (например, pH 10 или около того). Конъюгат лекарственного средства, введенный в остаток экзатекана, соответствующий замкнутой кольцевой структуре и открытой кольцевой структуре , также будет обладать таким же противоопухолевым действием, и необходимо отметить, что любые такие структуры включены в объем настоящего изобретения.
Другие примеры противоопухолевого соединения могут включать доксорубицин, даунорубицин, митомицин C, блеомицин, циклоцитидин, винкристин, винбластин, метотрексат, противоопухолевый агент на основе платины (цисплатин или его производные), таксол или его производные, и другие камптотецины или их производные (противоопухолевый агент, описанный в выложенном патенте Японии № 6-87746).
Что касается конъюгата антитела с лекарственным средством, количество конъюгированных молекул лекарственного средства на молекулу антитела является ключевым фактором, влияющим на эффективность и безопасность. Получение конъюгата антитела с лекарственным средством проводят при определении условий реакции, включая такие количества сырья и реагентов, применяемых для реакции, чтобы иметь постоянное число конъюгированных молекул лекарственного средства. Смесь, содержащую различные количества конъюгированных молекул лекарственного средства, обычно получают в отличие от химической реакции низкомолекулярного соединения. Количество лекарственных средств, конъюгированных в молекуле антитела, выражается или определяется средним значением, то есть, средним количеством конъюгированных молекул лекарственного средства. Если специально не указано иное, в принципе, количество конъюгированных молекул лекарственного средства является средним значением, за исключением случая, в котором рассматривается конъюгат антитела с лекарственным средством, имеющий определенное количество конъюгированных молекул лекарственного средства, который включен в смесь конъюгата антитела с лекарственным средством, имеющую различные количества конъюгированных молекул лекарственного средства.
Количество молекул экзатекана, конъюгированных с молекулой антитела, регулируется, и в среднем, в качестве конъюгированных молекул лекарственного средства на молекулу антитела, подсоединяют от около 1 до 10 молекул экзатекана. Предпочтительно, от 2 до 8, и более предпочтительно, от 3 до 8. В то же время, специалист в данной области техники может разработать реакцию для конъюгирования требуемого количества молекул лекарственного средства с молекулой антитела на основе описания, представленного в примерах в соответствии с данным изобретением, и может получить конъюгат антитела с лекарственным средством, конъюгированный с контролируемым количеством молекул экзатекана.
[Связующая структура]
Что касается анти-HER2 конъюгата антитела с лекарственным средством в соответствии с данным изобретением, далее объясняется связующая структура для конъюгирования противоопухолевого соединения с анти-HER2 антителом. Связующая группа имеет структуру следующей формулы:
-L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-.
Антитело соединено с концевым L1 (концом, противоположным соединению с L2), и противоопухолевое соединение соединено с карбонильной группой -La-(CH2)n2-C(=O)- части.
n1 является целым числом от 0 до 6 и, предпочтительно целым числом от 1 до 5, и более предпочтительно, от 1 до 3.
1. L1
L1 представлен следующей структурой:
-(Сукцинимид-3-ил-N)-(CH2)n3-C(=O)-.
В формулах выше, n3 является целым числом от 2 до 8, "-(Сукцинимид-3-ил-N)-" имеет структуру, представленную следующей формулой:
[Формула 15]
Положение 3 указанной выше частичной структуры является связующим положением для анти-HER2 антитела. Связь с антителом в положении 3 характеризуется связыванием с тиоэфирным образованием. Атом азота в положении 1 структурной группы соединен с атомом углерода метилена, который присутствует в связующей группе, включенной в структуру. Более конкретно, -(Сукцинимид-3-ил-N)-(CH2)n3-C(=O)-L2- является структурой, представленной следующей формулой (далее "антитело-S-", полученное из антитела).
[Формула 16]
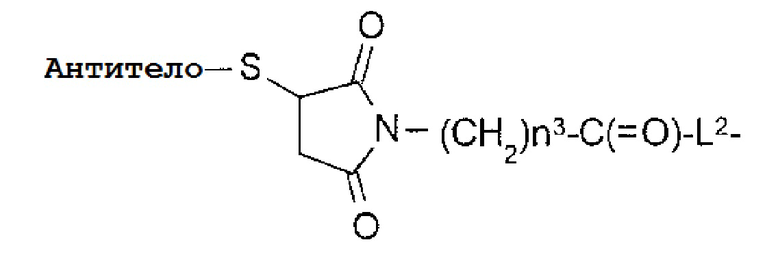 .
.
В формуле, n3 является целым числом от 2 до 8, и, предпочтительно, от 2 до 5.
Конкретные примеры L1 могут включать
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2-C(=O)- и
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-.
2. L2
L2 представлен следующей структурой:
-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-,
L2 может отсутствовать, и в таком случае, L2 является одинарной связью. n4 является целым числом от 1 до 6 и, предпочтительно, от 2 до 4. L2 соединен с L1 на его концевой аминогруппе, и соединен с LP на его карбонильной группе на другом конце.
Конкретные примеры L2 могут включать
-NH-CH2CH2-O-CH2CH2-C(=O)-,
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-,
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-,
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-,
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-,
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-.
3. LP
LP является пептидным остатком, состоящим из 2-7 аминокислот. Более конкретно, он состоит из олигопептидного остатка, в котором от 2 до 7 аминокислот связаны пептидным связыванием. LP соединен с L2 на его N конце и соединен с аминогруппой -NH-(CH2)n1-La-(CH2)n2-C(=O)- части связующей группы на его C конце. Здесь термин "пептидный остаток" или "олигопептидный остаток" является группой, полученной из пептида, состоящего из двух или более аминокислотных остатков и относится к двухвалентной группе, N конец и C конец которой являются связующими положениями.
Аминокислоты, составляющие LP в связующей группе, особенно не ограничены, однако, их примеры включают L- или D-аминокислоту, предпочтительно, L-аминокислоту. И она может быть аминокислотой, имеющей структуру, такую как β-аланин, ε-аминокапроновая кислота или γ-аминомасляная кислота в добавление к α-аминокислоте, также она может быть не натуральной аминокислотой, такой как N-метилированная аминокислота.
Последовательность аминокислот LP особенно не ограничена, но примеры составляющих его аминокислот включают фенилаланин (Phe; F), тирозин (Tyr; Y), лейцин (Leu; L), глицин (Gly; G), аланин (Ala; A), валин (Val; V), лизин (Lys; K), цитруллин (Cit), серин (Ser; S), глутаминовую кислоту (Glu; E) и аспарагиновую кислоту (Asp; D). Среди них предпочтительные примеры включают фенилаланин, глицин, валин, лизин, цитруллин, серин, глутаминовую кислоту и аспарагиновую кислоту. Из этих аминокислот может быть построен LP, имеющий последовательность аминокислот, необязательно выбранных с или без наложения. В зависимости от типа аминокислот, может контролироваться модель высвобождения лекарственного средства. Количество аминокислот может составлять от 2 до 7.
Конкретные примеры LP могут включать
-GGF-,
-DGGF-,
-(D-)D-GGF-,
-EGGF-,
-GGFG-,
-SGGF-,
-KGGF-,
-DGGFG-,
-GGFGG-,
-DDGGFG-,
-КДGGFG-, и
-GGFGGGF-.
В формулах выше, "(D-)D" является D-аспарагиновой кислотой.
Особенно предпочтительные примеры LP для конъюгата антитела с лекарственным средством в соответствии с данным изобретением могут включать тетрапептидный остаток -GGFG-.
4. La-(CH2)n2-C(=O)-
La в La-(CH2)n2-C(=O)- является структурой -O- или одинарной связью. n2 является целым числом от 0 до 5, предпочтительно, от 0 до 3, и более предпочтительно, 0 или 1.
Примеры La-(CH2)n2-C(=O)- могут включать следующие структуры:
-O-CH2-C(=O)-,
-O-CH2CH2-C(=O)-,
-O-CH2CH2CH2-C(=O)-,
-O-CH2CH2CH2CH2-C(=O)-,
-O-CH2CH2CH2CH2CH2-C(=O)-,
-CH2-C(=O)-,
-CH2CH2-C(=O)-,
-CH2CH2CH2-C(=O)-,
-CH2CH2CH2CH2-C(=O)-,
-CH2CH2CH2CH2CH2-C(=O)-,
-O-C(=O)-.
Среди них,
-O-CH2-C(=O)-,
-O-CH2CH2-C(=O)-,
-O-C(=O)-,
или вариант, в котором La является одинарной связью и n2 равно 0 являются предпочтительными.
Конкретные примеры структуры, представленной -NH-(CH2)n1-La-(CH2)n2-C(=O)- связующей группы могут включать
-NH-CH2-C(=O)-,
-NH-CH2CH2-C(=O)-,
-NH-CH2-O-CH2-C(=O)-,
-NH-CH2CH2-O-CH2-C(=O)-,
-NH-CH2CH2CH2-C(=O)-,
-NH-CH2CH2CH2CH2-C(=O)-,
-NH-CH2CH2CH2CH2CH2-C(=O)-,
-NH-CH2-O-C(=O)-,
-NH-CH2CH2-O-C(=O)-,
-NH-CH2CH2CH2-O-C(=O)-,
-NH-CH2CH2CH2CH2-O-C(=O)-.
Среди них,
-NH-CH2CH2CH2-C(=O)-,
-NH-CH2-O-CH2-C(=O)-,
-NH-CH2CH2-O-CH2-C(=O)-
являются более предпочтительными.
В связующей группе длина цепи -NH-(CH2)n1-La-(CH2)n2-C(=O)-, предпочтительно, является длиной цепи от 4 до 7 атомов, и более предпочтительно, длиной цепи в 5 или 6 атомов.
Что касается анти-HER2 конъюгата антитела с лекарственным средством в соответствии с данным изобретением, когда его переносят внутрь опухолевых клеток, можно предположить, что связующая группа отщепляется, и производное лекарственного средства, имеющее структуру, представленную NH2-(CH2)n1-La-(CH2)n2-C(=O)-(NH-DX), выделяется для оказания противоопухолевого действия. Примеры противоопухолевого производного, обладающего противоопухолевым действием при выделении из конъюгата антитела с лекарственным средством в соответствии с данным изобретением, включают противоопухолевое производное, имеющее структурную группу, в которой конец структуры, представленной -NH-(CH2)n1-La-(CH2)n2-C(=O)- связующей группой, имеет аминогруппу, и особенно предпочтительные группы включают следующие.
NH2-CH2CH2-C(=O)-(NH-DX),
NH2-CH2CH2CH2-C(=O)-(NH-DX),
NH2-CH2-O-CH2-C(=O)-(NH-DX),
NH2-CHCH2-O-CH2-C(=O)-(NH-DX).
В то же время, в случае NH2-CH2-O-CH2-C(=O)-(NH-DX), было подтверждено, что, так как аминальная структура в молекуле является нестабильной, она снова претерпевает саморазложение с выделением следующей группы
HO-CH2-C(=O)-(NH-DX).
Эти соединения также могут, предпочтительно, применяться в качестве промежуточного соединения при производстве конъюгата антитела с лекарственным средством в соответствии с данным изобретением.
Для конъюгата антитела с лекарственным средством в соответствии с данным изобретением, в котором в качестве лекарственного средства применяют экзатекан, предпочтительно, чтобы структура лекарственное средство-связующая группа [-L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-(NH-DX)], имеющая следующую структуру, была соединена с антителом. Среднее число конъюгатов в указанной структуре лекарственное средство-связующая группа на молекулу антитела может составлять от 1 до 10. Предпочтительно, оно составляет от 2 до 8, и более предпочтительно, от 3 до 8.
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
Среди них более предпочтительными являются следующие.
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
Особенно предпочтительными являются следующие.
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
Что касается связующей структуры для конъюгирования анти-HER2 антитела и лекарственного средства в конъюгат антитела с лекарственным средством в соответствии с данным изобретением, предпочтительная связующая группа может быть сконструирована соединением предпочтительных структур, показанных для каждой части связующей группы, описанной выше. Что касается связующей структуры, предпочтительно применяют группы со следующей структурой. При этом левый конец этих структур является связующим положением с антителом, и правый конец является связующим положением с лекарственным средством.
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-.
Среди них более предпочтительными являются следующие.
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-.
Особенно предпочтительными являются следующие.
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-,
-(Сукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-.
[Способ получения]
Далее представлено подробное описание типового способа получения конъюгата антитела с лекарственным средством в соответствии с данным изобретением или производное соединения для его получения. При этом, соединения ниже описаны с тем номером соединения, который показан в каждой реакционной формуле. Более конкретно, они обозначены как "соединение формулы (1)", "соединение (1)" или подобное. Соединения с номерами, отличными от указанного выше, описаны так же.
1. Способ получения 1
Конъюгат антитела с лекарственным средством, представленный формулой (1), в которой антитело соединено со структурой лекарственное средство-связующая группа через тиоэфир, может быть получено следующим способом, например.
[Формула 17]
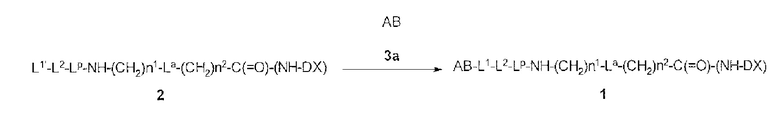
[В формуле, AB является антителом, имеющим сульфгидрильную группу, и L1' является L1 связующей структурой, в которой окончанием связующей группы является малеимидильная группа (формула показана ниже)
[Формула 18]
(в формуле, атом азота является связующим положением), и, в частности, является группой, в которой -(сукцинимид-3-ил-N)- часть в -(сукцинимид-3-ил-N)-(CH2)n3-C(=O)- в L1 является малеимидильной группой. Далее, -(NH-DX) является структурой, представленной следующей формулой:
[Формула 19]
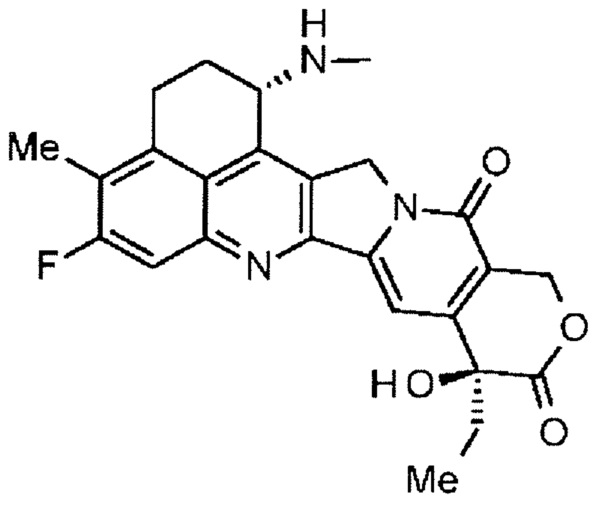
и является группой, которую получают удалением одного атома водорода аминогруппы в положении 1 экзатекана.]
Далее, соединение формулы (1) в указанной выше реакции может быть истолковано как структура, в которой одна структурная часть, соответствующая концу от лекарственного средства к связующей группе, соединена с одним антителом. Однако, данное описание дано только для удобства, и существует множество случаев, в которых множество структурных частей соединены с одной молекулой антитела. То же самое применимо к объяснению способа получения, описанного ниже.
Конъюгат антитела с лекарственным средством (1) может быть получен взаимодействием соединения (2), которое получают описанным ниже способом, с антителом (3a), имеющим сульфгидрильную группу.
Антитело (3a), имеющее сульфгидрильную группу, может быть получено способом, хорошо известным в данной области техники (Hermanson, G.T, Bioconjugate Techniques, pp. 56-136, pp. 456-493, Academic Press (1996)). Примеры включают: реагент Трота подвергают взаимодействию с аминогруппой антитела; N-сукцинимидил S-ацетилтиоалканоаты подвергают взаимодействию с аминогруппой антитела с последующим взаимодействием с гидроксиламином; после взаимодействия с N-сукцинимидил 3-(пиридилдитио)пропионатом, антитело подвергают взаимодействию с восстанавливающим агентом; антитело подвергают взаимодействию с восстанавливающим агентом, таким как дитиотретиол, 2-меркаптоэтанол и гидрохлорид трис(2-карбоксиэтил)фосфина (ТКЭФ) с восстановлением дисульфидной связи в шарнирной части антитела с получением сульфгидрильной группы, но данный пример не является ограничивающим.
Более конкретно, применение от 0,3 до 3 молярных эквивалентов ТКЭФ в качестве восстанавливающего агента на дисульфид в шарнирной части в антителе и взаимодействие с антителом в буферном растворе, содержащем хелатирующий агент, позволяет получить антитело с частично или полностью восстановленным дисульфидом в шарнирной части в антителе. Примеры хелатирующего агента включают этилендиаминтетрауксусную кислоту (ЭДТК) и диэтилентриаминпентауксусную кислоту (ДТПК). Он может применяться в концентрации от 1 мМ до 20 мМ. Примеры буферного раствора, который может применяться, включают раствор фосфата натрия, бората натрия или ацетата натрия. Более конкретно, взаимодействие антитела с ТКЭФ при температуре от 4°C до 37°C в течение от 1 до 4 часов позволяет получить антитело (3a), имеющее частично или полностью восстановленную сульфгидрильную группу.
При этом проведение реакции добавления сульфгидрильной группы к группе лекарственное средство-связующая группа позволяет конъюгировать группу лекарственное средство-связующая группа через тиоэфирную связь.
Применение от 2 до 20 молярных эквивалентов соединения (2) на антитело (3a), имеющее сульфгидрильную группу, позволяет получить конъюгат антитела с лекарственным средством (1), в котором от 2 до 8 молекул лекарственного средства конъюгированы с молекулой антитела. Более конкретно, для реакции достаточно добавить раствор, содержащий соединение (2), растворенное в нем, к буферному раствору, содержащему антитело (3a), имеющее сульфгидрильную группу. Здесь примеры буферного раствора, который может применяться, включают раствор ацетата натрия, фосфата натрия и бората натрия. pH реакции составляет от 5 до 9, и более предпочтительно, реакцию проводят при pH около 7. Примеры растворителя для растворения соединения (2) включают органический растворитель, такой как диметилсульфоксид (ДМСО), диметилформамид (ДМФ), диметилацетамид (ДМА) и N-метил-2-пиридон (NMP).
Для реакции достаточно добавлять раствор органического растворителя, содержащий соединение (2), в количестве от 1 до 20% об./об. к буферному раствору, содержащему антитело (3a), имеющее сульфгидрильную группу. Температура реакции составляет от 0 до 37°C, более предпочтительно, от 10 до 25°C, и время реакции составляет от 0,5 до 2 часов. Реакция может быть остановлена деактивированием реакционной способности непрореагировавшего соединения (2) агентом, содержащим тиол. Примеры реагента, содержащего тиол, включают цистеин и N-ацетил-L-цистеин (NAC). Более конкретно, от 1 до 2 молярных эквивалентов NAC добавляют к применяемому соединению (2),и инкубирование при комнатной температуре в течение от 10 до 30 минут позволяет остановить реакцию.
Полученный конъюгат антитела с лекарственным средством (1) может, после концентрации, замены буфера, очистки и измерения концентрации антитела и среднего количества конъюгированных молекул лекарственного средства обычными методами, описанными ниже, быть подвергнут идентификации конъюгата антитела с лекарственным средством (1).
Общая методика A: Концентрация водного раствора антитела или конъюгата антитела с лекарственным средством
В контейнер Amicon Ultra (50,000 MWCO, Millipore Co.) добавляют раствор антитела или конъюгата антитела с лекарственным средством, и раствор антитела или конъюгата антитела с лекарственным средством концентрируют центрифугированием (центрифуга в течение от 5 до 20 минут при от 2000 G до 3800 G) с применением центрифуги (Allegra X-15R, Beckman Coulter, Inc.).
Общая методика B: Измерение концентрации антитела
С применением УФ датчика (Nanodrop 1000, Thermo Fisher Scientific Inc.), измерение концентрации антитела проводят способом, указанным производителем. В это время используют 280 нм коэффициент абсорбции, различный для каждого антитела (от 1,3 млмг-1см-1 до 1,8 млмг-1см-1).
Общая методика C-1: Замена буфера для антитела
Колонку NAP-25 (№ по каталогу 17-0852-02, GE Healthcare Japan Corporation) с применением носителя Sephadex G-25 уравновешивают фосфатным буфером (10 мМ, pH 6.0; в спецификации он обозначен как ФРФБ6,0/ЭДТК), содержащим хлорид натрия (137 мМ) и этилендиаминтетрауксусную кислоту (ЭДТК, 5 мМ) с применением способа, указанного производителем. Водный раствор антитела применяют в количестве 2,5 мл для одной колонки NAP-25, и затем собирают фракцию (3,5 мл), элюированную с 3,5 мл ФРФБ6,0/ЭДТК. Полученную фракцию концентрируют с применением общей методики A. После измерения концентрации антитела с применением общей методики B, концентрацию антитела доводят до 10 мг/мл с применением ФРФБ6,0/ЭДТК.
Общая методика C-2: Замена буфера для антитела
Колонку NAP-25 (№ по каталогу 17-0852-02, GE Healthcare Japan Corporation) с применением носителя Sephadex G-25 уравновешивают фосфатным буфером (50 мМ, pH 6.5; в спецификации он обозначен как ФРФБ6,5/ЭДТК), содержащим хлорид натрия (50 мМ) и ЭДТК (2 мМ) с применением способа, указанного производителем. Водный раствор антитела применяют в количестве 2,5 мл для одной колонки NAP-25, и затем собирают фракцию (3,5 мл), элюированную с 3,5 мл ФРФБ6,5/ЭДТК. Полученную фракцию концентрируют с применением общей методики A. После измерения концентрации антитела с применением общей методики B, концентрацию антитела доводят до 20 мг/мл с применением ФРФБ6,5/ЭДТК.
Общая методика D: Очистка конъюгата антитела с лекарственным средством
Колонку NAP-25 уравновешивают любым буфером, выбранным из коммерчески доступного фосфатного буфера (ФРФБ7,4, № по каталогу 10010-023, Invitrogen), буфера на основе фосфата натрия (10 мМ, pH 6.0; обозначен как ФРФБ6,0), содержащего хлорид натрия (137 мМ), и ацетатного буфера, содержащего сорбит (5%) (10 мМ, pH 5.5; обозначен как ABS в описании). Водный раствор конъюгата антитела с лекарственным средством помещают в количестве от около 1,5 мл в колонку NAP-25 и затем элюируют буфером в количестве, определенном производителем для сбора фракции антитела. Собранную фракцию снова помещают в колонку NAP-25 и, повторяя от 2 до 3 раз методику очистки гель-фильтрацией для элюирования буфером, получают конъюгат антитела с лекарственным средством, не содержащий не конъюгированную связующую группу лекарственного средства и низкомолекулярное соединение (гидрохлорид трис(2-карбоксиэтил)фосфина (ТКЭФ), N-ацетил-L-цистеин (NAC) и диметилсульфоксид).
Общая методика E: Измерение концентрации антитела в конъюгате антитела с лекарственным средством и среднего количества конъюгированных молекул лекарственного средства на молекулу антитела (1).
Концентрация конъюгированного лекарственного средства в конъюгате антитела с лекарственным средством может быть рассчитана измерением УФ абсорбции водного раствора конъюгата антитела с лекарственным средством при двух длинах волн 280 нм и 370 нм, с последующим проведением указанных ниже расчетов.
Так как общая абсорбция при любой длине волны равна сумме абсорбции каждого абсорбирующего свет химического вещества, которое присутствует в системе (аддитивность абсорбции), когда молярные коэффициенты абсорбции антитела и лекарственного средства остаются одинаковыми до и после конъюгирования антитела и лекарственного средства, концентрацию антитела и концентрацию лекарственного средства в конъюгате антитела с лекарственным средством выражают следующими уравнениями.
A280=AD.280+AA.280=εD.280CD+εA.280CA Уравнение (I)
A370=AD.370+AA.370=εD.370CD+εA.370CA Уравнение (II)
В уравнениях выше, A280 является абсорбцией водного раствора конъюгата антитела с лекарственным средством при 280 нм, A370 является абсорбцией водного раствора конъюгата антитела с лекарственным средством при 370 нм, AA.280 является абсорбцией антитела при 280 нм, AA.370 является абсорбцией антитела при 370 нм, AD.280 является абсорбцией предшественника конъюгата при 280 нм, AD.370 является абсорбцией предшественника конъюгата при 370 нм, εA.280 является молярным коэффициентом абсорбции антитела при 280 нм, εA.370 является молярным коэффициентом абсорбции антитела при 370 нм, εD.280 является молярным коэффициентом абсорбции предшественника конъюгата при 280 нм, εD.370 является молярным коэффициентом абсорбции предшественника конъюгата при 370 нм, CA является концентрацией антитела в конъюгате антитела с лекарственным средством, и CD является концентрацией антитела в конъюгате антитела с лекарственным средством.
Для εA.280, εA.370, εD.280 и εD.370 в уравнениях выше применяют полученные заранее значения (расчетные значения, основанные на расчете или измерении, полученном УФ измерением соединений). Например, εA.280 может быть получена из последовательности аминокислот антитела с применением известного способа расчета (Protein Science, 1995, vol. 4, 2411-2423). εA.370 обычно равно нулю. В примерах, в качестве молярного коэффициента абсорбции трастузумаба используют εA.280=215400 (расчетное значения на основе расчета) и εA.370=0. εD.280 и εD.370 могут быть получены на основе закона Ламберта-Бера (абсорбция=молярная концентрация×молярный коэффициент абсорбции×длина клеточного пути) измерением абсорбции раствора, в котором растворен применяемый предшественник конъюгата в определенной молярной концентрации. В качестве молярного коэффициента абсорбции связующей группы лекарственного средства в примерах применяют εD.280=5000 (измеренное среднее значение) и εD.370=19000 (измеренное среднее значение), если не указано иначе. Измерением A280 и A370 водного раствора конъюгата антитела с лекарственным средством и решением системы уравнений (I) и (II) с применением значений получают CA и CD. Далее, делением CD на CA получают среднее количество конъюгированных молекул лекарственного средства на молекулу антитела.
Общая методика F: Измерение (2) среднего количества конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством.
Среднее количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством также может быть определено высокоэффективной жидкостной хроматографией (ВЭЖХ) с применением следующего способа в дополнение к указанной выше общей методике E.
[F-1. Получение образца для ВЭЖХ (восстановление конъюгата антитела с лекарственным средством)]
Раствор конъюгата антитела с лекарственным средством (около 1 мг/мл, 60 мкл) смешивают с водным раствором дитиотреитола (ДТТ) (100 мМ, 15 мкл). Образец, в котором дисульфидная связь между L цепью и H цепью конъюгата антитела с лекарственным средством была расщеплена инкубированием смеси в течение 30 минут при 37°C, применяют для ВЭЖХ.
[F-2. ВЭЖХ]
ВЭЖХ проводят при следующих условиях измерения:
Система ВЭЖХ: система ВЭЖХ Agilent 1290 (Agilent Technologies, Inc.)
Датчик: спектрометр ультрафиолетового поглощения (длина волны измерения: 280 нм)
Колонка: PLRP-S (2,1×50 мм, 8 мкм, 1000 ангстрем; Agilent Technologies, Inc., P/N PL1912-1802)
Температура колонки: 80°C
Подвижная фаза A: водный раствор, содержащий 0,04% трифторуксусной кислоты (ТФК)
Подвижная фаза B: раствор ацетонитрила, содержащий 0,04% ТФК
Градиент: 29%-36% (0-12,5 мин), 36%-42% (12,5-15 мин), 42%-29% (15-15,1 мин) и 29%-29% (15,1-25 мин)
Объем впрыска образца: 15 мкл
[F-3. Анализ данных]
[F-3-1] По сравнению с цепями не конъюгированного антитела L (L0) и H (H0), конъюгированные с лекарственным средством цепи L (цепь L, соединенная с одной молекулой лекарственного средства: L1) и H (цепь H, соединенная с одной молекулой лекарственного средства: H1, цепь H, соединенная с двумя молекулами лекарственного средства: H2, цепь H, соединенная с тремя молекулами лекарственного средства: H3) обладают более высокой гидрофобностью пропорционально количеству конъюгированных молекул лекарственного средства и, следовательно, имеют большее время удержания. Эти цепи, поэтому, элюируются в порядке L0 и L1 или H0, H1, H2 и H3. Пики определения могут быть присвоены любой из L0, L1, H0, H1, H2 и H3 сравнением времени удержания с L0 и H0.
[F-3-2] Так как связующая группа лекарственного средства имеет УФ абсорбцию, значения площади пика корректируют в зависимости от количества конъюгированных молекул связующей группы лекарственного средства в соответствии со следующим выражением с применением молярных коэффициентов абсорбции цепи L или H и связующей группы лекарственного средства.
[Выражение 1]

[Выражение 2]

Здесь, в качестве молярного коэффициента абсорбции (280 нм) цепи L или H каждого антитела может применяться значение, рассчитанное из последовательности аминокислот цепи L или H каждого антитела известным способом расчета (Protein Science, 1995, vol. 4, 2411-2423). Для трастузумаба, молярный коэффициент абсорбции 26150 и молярный коэффициент абсорбции 81290 применяют в качестве расчетных значений для цепей L и H, соответственно, согласно их последовательности аминокислот. В качестве молярного коэффициента абсорбции (280 нм) связующей группы лекарственного средства используют измеренный молярный коэффициент абсорбции (280 нм) соединения, в котором малеимидная группа превращена в тиоэфир сукцинимида взаимодействием каждой связующей группы лекарственного средства с меркаптоэтанолом или N-ацетилцистеином.
[F-3-3] Долю площади пика (%) каждой цепи рассчитывают для всех скорректированных значений площадей пика согласно следующему выражению.
[Выражение 3]
[F-3-4] Среднее количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством рассчитывают согласно следующему выражению.
Среднее количество конъюгированных молекул лекарственного средства = (L0 доля площади пика×0 + L0 доля площади пика×1 + H0 доля площади пика×0 + H1 доля площади пика×1 + H2 доля площади пика×2 + H3 доля площади пика×3)/100×2
Далее описано технологическое промежуточное соединение для получения, применяемое в способе получения 1. Соединение, представленное формулой (2) в способе получения 1, является соединением, представленным следующей формулой:
(Малеимид-N-ил)-(CH2)n3-C(=O)-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-(NH-DX).
В формуле
n3 является целым числом от 2 до 8,
L2 является -NH-(CH2CH2-O)n4-CH2CH2-C(=O)- или одинарной связью,
где n4 является целым числом от 1 до 6,
LP является пептидным остатком, состоящим из от 2 до 7 аминокислот, выбранных из фенилаланина, глицина, валина, лизина, цитруллина, серина, глутаминовой кислоты и аспарагиновой кислоты.
n1 является целым числом от 0 до 6,
n2 является целым числом от 0 до 5,
La является -O- или одинарной связью,
(малеимид-N-ил)- является малеимидильной группой (2,5-диоксо-2,5-дигидро-1H-пиррол-1-ильной группой), представленной следующей формулой:
[Формула 20]
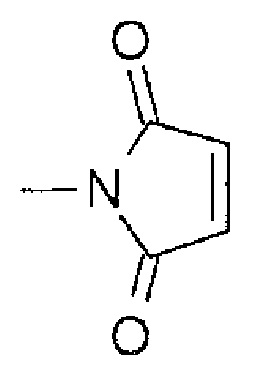 ,
,
где атом азота является связующим положением, и
-(NH-DX) является группой, представленной следующей формулой:
[Формула 21]
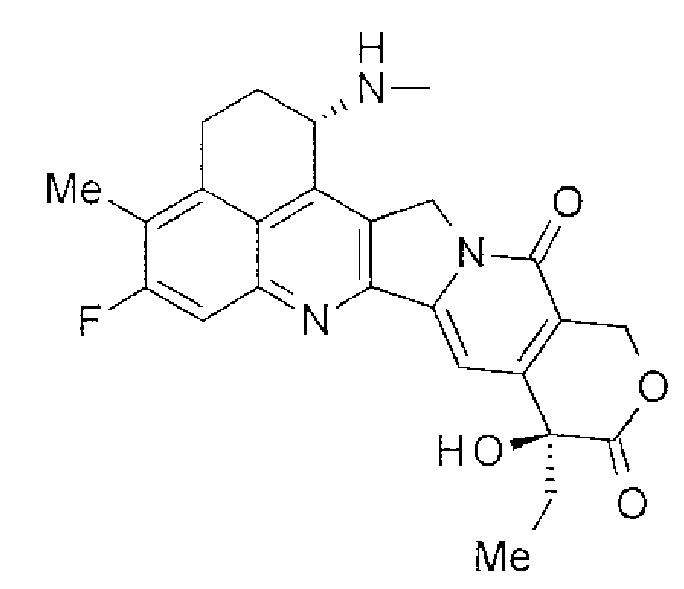 ,
,
где атом азота аминогруппы в положении 1 является связующим положением.
Если L2 является одинарной связью или -NH-(CH2CH2-O)n4-CH2CH2-C(=O)-, предпочтительно, чтобы в технологическом промежуточном соединении n4 было равно целому числу от 2 до 4.
Что касается пептидного остатка LP, соединение, имеющее пептидный остаток, состоящий из аминокислоты, выбранной из фенилаланина, глицина, валина, лизина, цитруллина, серина, глутаминовой кислоты и аспарагиновой кислоты, является предпочтительным в качестве технологического промежуточного средства. Среди этих пептидных остатков, соединение, в котором LP является пептидным остатком, состоящим из 4 аминокислот, является предпочтительным технологическим промежуточным соединением. Более конкретно, соединение, в котором LP является тетрапептидным остатком -GGFG-, является предпочтительным в качестве технологического промежуточного соединения.
Далее, что касается -NH-(CH2)n1-La-(CH2)n2-, соединение, содержащее -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2CH2CH2CH2-, -NH-CH2CH2CH2CH2CH2-, -NH-CH2-O-CH2- или -NH-CH2CH2-O-CH2-, является предпочтительным технологическим промежуточным соединением. Соединение, содержащее -NH-CH2CH2CH2-, -NH-CH2-O-CH2- или -NH-CH2CH2-O-CH2, является более предпочтительным.
Соединение, представленное формулой (2), в которой n3 является целым числом от 2 до 5, L2 является одинарной связью, и -NH-(CH2)n1-La-(CH2)n2- является -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2CH2CH2CH2-, -NH-CH2CH2CH2CH2CH2-, -NH-CH2-O-CH2- или -NH-CH2CH2-O-CH2-, является предпочтительным технологическим промежуточным соединением. Соединение, в котором -NH-(CH2)n1-La-(CH2)n2- является -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2-O-CH2- или -NH-CH2CH2-O-CH2-, является более предпочтительным. Соединение, в котором n3 является целым числом 2 или 5, особенно предпочтительно.
Соединение, представленное формулой (2), в котором n3 является целым числом от 2 до 5, L2 является -NH-(CH2CH2-O)n4-CH2CH2-C(=O)-, n4 является целым числом от 2 до 4, и -NH-(CH2)n1-La-(CH2)n2- является -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2CH2CH2CH2-, -NH-CH2CH2CH2CH2CH2-, -NH-CH2-O-CH2- или -NH-CH2CH2-O-CH2-, является предпочтительным технологическим промежуточным соединением. Соединение, в котором n4 является целым числом 2 или 4, является более предпочтительным. Соединение, в котором -NH-(CH2)n1-La-(CH2)n2- является -NH-CH2CH2CH2-, -NH-CH2-O-CH2- или -NH-CH2CH2-O-CH2-, является еще более предпочтительным.
Предпочтительные примеры промежуточного соединения, применяемого для получения соединения в соответствии с данным изобретением, могут включать следующие.
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
Анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением может быть получен взаимодействием соединения лекарственное средство-связующая группа, выбранного из указанных выше технологических промежуточных соединений, с анти-HER2 антителом или его реакционноспособным производным с получением тиоэфирной связи на месте дисульфидной связи, присутствующей в шарнирной части анти-HER2 антитела. В этом случае, предпочтительно применяют реакционноспособное производное анти-HER2 антитела и особенно предпочтительно реакционноспособное производное, полученное восстановлением анти-HER2 антитела.
Далее представлены соединения, более предпочтительные в качестве технологического промежуточного соединения.
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2-CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
Среди указанных выше промежуточных соединений соединение, представленное следующей формулой:
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX) или
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
являются более предпочтительными соединениями.
Для того чтобы обеспечить количество конъюгата, множество конъюгатов, полученных в одинаковых условиях получения для того, чтобы получить эквивалентное количество лекарственных средств (например, около ±1), могут быть смешаны для получения новых партий. В этом случае, среднее количество лекарственных средств попадает в интервалы средних количества лекарственных средств в конъюгатах до смешивания.
2. Способ получения 2
Соединение, представленное формулой (2) в качестве промежуточного соединения, применяемого в описанном выше способе получения, и его фармакологически приемлемая соль могут быть получены следующим способом, например.
[Формула 22]
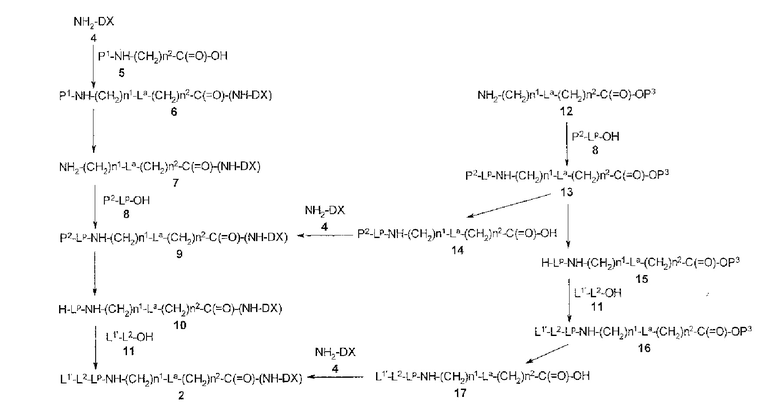
В формуле, L1' является концевой малеимидильной группой, и P1, P2 и P3 каждый является защитной группой.
Соединение (6) может быть получено превращением карбоновой кислоты (5) в активный сложный эфир, смешанный ангидрид кислоты, галоидангидрид или подобные, и взаимодействием его с NH2-DX (4) или его фармакологически приемлемой солью. NH2-DX (4) означает экзатекан (химическое наименование: (1S,9S)-1-амино-9-этил-5-фтор-2,3-дигидро-9-гидрокси-4-метил-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-10,13(9H,15H)-дион).
Реагенты и условия реакции, обычно применяемые для пептидного синтеза, могут применяться для реакции. Существуют различные виды активного сложного эфира. Например, он может быть получен взаимодействием фенолов, таких как п-нитрофенол, N-гидроксибензотриазол, N-гидроксисукцинимид или подобные, с карбоновой кислотой (5) с применением конденсирующего агента, такого как N,N'-дициклогексилкарбодиимид или гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимид. Далее, активный сложный эфир также может быть получен взаимодействием карбоновой кислоты (5) с трифторацетатом пентафторфенила или подобными; взаимодействием карбоновой кислоты (5) с гексафторфосфитом 1-бензотриазолил окситрипирролидинофосфония; реакцией карбоновой кислоты (5) с диэтил цианофосфонатом (способ всаливания); реакцией карбоновой кислоты (5) с трифенилфосфином и 2,2'-дипиридилдисульфидом (способ Мукаямы); реакцией карбоновой кислоты (5) с производным триазина, таким как хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMTMM); или подобными. Далее, реакция также может быть проведена, например, способом галоидангидрида, в котором карбоновую кислоту (5) обрабатывают галоидангидридом, таким как тионилхлорид и оксалилхлорид в присутствии основания.
Взаимодействие активного сложного эфира, смешанного ангидрида кислоты или галоидангидрида карбоновой кислоты (5), полученного как описано выше с соединением (4) в присутствии подходящего основания в инертном растворителе при температуре от -78°C до 150°C, дает соединение (6). ("Инертный растворитель" означает растворитель, который не ингибирует реакцию, для которой применяется растворитель.)
Типовые примеры основания, применяемого на каждой стадии, описанной выше, включают карбонат, алкоксид, гидроксид или гидрид щелочного металла или щелочноземельного металла, такой как карбонат натрия, карбонат калия, этоксид натрия, бутоксид калия, гидроксид натрия, гидроксид калия, гидрид натрия и гидрид калия; металлорганическое основание, представленное алкиллитием, такое как н-бутиллитий, или диалкиламинолитием, такое как диизопропиламид лития; металлорганическое основание биссилиламина, такое как бис(триметилсилил)амид лития; и органическое основание, включающее третичный амин или азотсодержащее гетероциклическое соединение, такое как пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, N-метилморфолин, диизопропилэтиламин и диазабицикло[5,4,0]ундец-7-ен (ДБУ).
Примеры инертного растворителя, который применяют для взаимодействия в соответствии с данным изобретением, включают галогенированный углеводородный растворитель, такой как дихлорметан, хлороформ и четыреххлористый углерод; простой эфирный растворитель, такой как тетрагидрофуран, 1,2-диметоксиэтан и диоксан; ароматический углеводородный растворитель, такой как бензол и толуол; и амидный растворитель, такой как N,N-диметилформамид, N,N-диметилацетамид и N-метилпирролидин-2-он. Кроме них, сульфоксидный растворитель, такой как диметилсульфоксид и сульфолан; и кетоновый растворитель, такой как ацетон и метилэтилкетон и спиртовой растворитель, такой как метанол и этанол, могут применяться в некоторых случаях. Далее, может применяться их смешанный растворитель.
В качестве защитной группы может применяться защитная группа P1 для концевой аминогруппы соединения (6), защитная группа для аминогруппы, которая обычно применяется для пептидного синтеза, например, трет-бутоксилоксикарбонильная группа, 9-флуоренилметилоксикарбонильная группа или бензилоксикарбонильная группа. Примеры других защитных групп для аминогруппы включают алканоильную группу, такую как ацетильная группа; алкоксикарбонильную группу, такую как метоксикарбонильная группа и этоксикарбонильная группа; арилметоксикарбонильную группу, такую как параметоксибензилоксикарбонильня группа и пара (или орто)нитробензилоксикарбонильная группа; арилметильную группу, такую как бензильная группа и трифенилметильная группа; ароильную группу, такую как бензоильная группа; и арилсульфонильную группу, такую как 2,4-динитробензолсульфонильная группа и ортонитробензолсульфонильная группа. Защитная группа P1 может быть выбрана в зависимости от, например, свойств соединения, имеющего защищаемую аминогруппу.
Снятие защитной группы P1 для концевой аминогруппы полученного соединения (6) дает соединение (7). Для снятия защиты реагенты и условия выбирают в зависимости от защитной группы.
Соединение (9) может быть получено превращением пептидной карбоновой кислоты (8), имеющей конец N, защищенный P2, в активный сложный эфир, смешанный кислотный ангидрид или подобные, и его взаимодействием с полученным соединением (7). Условия реакции, реагенты, основание и инертный растворитель, применяемые для получения пептидной связи между пептидной карбоновой кислотой (8) и соединением (7) могут быть подходящим образом выбраны из тех, которые описаны для синтеза соединения (6). Защитная группа P2 может быть подходящим образом выбрана из тех, которые описаны для защитной группы соединения (6), и выбор может быть основан на, например, свойствах соединения, имеющего защищаемую аминогруппу. Как это обычно применяют для пептидного синтеза, последовательное повторение реакции и снятия защиты аминокислоты или пептида, составляющего пептидную карбоновую кислоту (8) для удлинения, также может быть получено соединение (9).
Снятие защитной группы P2 аминогруппы полученного соединения (9) дает соединение (10). Для снятия защиты реагенты и условия выбирают в зависимости от защитной группы.
Возможно получить соединение (2) превращением карбоновой кислоты (11) в активный сложный эфир, смешанный кислотный ангидрид, галогенид кислоты и подобные и взаимодействие его с полученным соединением (10). Условия реакции, реагенты, основание и инертный растворитель, применяемые для получения пептидной связи между карбоновой кислотой (11) и соединением (10), могут быть подходящим образом выбраны из тех, которые описаны для синтеза соединения (6).
Соединение (9) также может быть получено следующим способом, например.
Соединение (13) может быть получено превращением пептидной карбоновой кислоты (8), имеющее концевой N, защищенный P2, в активный сложный эфир, смешанный кислотный ангидрид и подобные, и взаимодействием его в присутствии основания с соединением амина (12), имеющим карбоксигруппу, защищенную P3. Условия реакции, реагенты, основание и инертный растворитель, применяемые для получения пептидной связи между пептидной карбоновой кислотой (8) и соединением (12), могут быть подходящим образом выбраны из тех, которые описаны для синтеза соединения (6). Защитная группа P2 для аминогруппы соединения (13) особенно не ограничена, если она является типовой защитной группой.
Более конкретно, примеры защитной группы для гидроксильной группы могут включать алкоксиметильную группу, такую как метоксиметильная группа; арилметильную группу, такую как бензильная группа, 4-метоксибензильная группа и трифенилметильная группа; алканоильную группу, такую как ацетильная группа; ароильную группу, такую как бензоильная группа; и силильную группу, такую как трет-бутилдифенилсилильная группа. Карбоксигруппа может быть защищена, например, в виде сложного эфира алкильной группой, такой как метильная группа, этильная группа и трет-бутильная группа, аллильной группой или арилметильной группой, такой как бензильная группа. Примеры защитной группы для аминогруппы могут включать: алкилоксикарбонильную группу, такую как трет-бутилоксикарбонильная группа, метоксикарбонильная группа и этоксикарбонильная группа; аллилоксикарбонильную группу или арилметоксикарбонильную группу, такую как 9-флуоренилметилоксикарбонильная группа, бензилоксикарбонильная группа, параметоксибензилоксикарбонильная группа и пара- (или орто)нитробензилоксикарбонильная группа; алканоильную группу, такую как ацетильная группа; арилметильную группу, такую как бензильная группа и трифенилметильная группа; ароильную группу, такую как бензоильная группа; и арилсульфонильную группу, такую как 2,4-динитробензолсульфонильная группа или тртонитробензолсульфонильная группа.
В качестве защитной группы P3 для карбоксигруппы может применяться защитная группа, обычно применяемая в качестве защитной группы для карбоксигруппы в органическом синтезе, в частности, пептидном синтезе. Более конкретно, она может быть подходящим образом выбрана из защитных групп, описанных выше, например, сложных эфиров с алкильной группой, такой как метильная группа, этильная группа или трет-бутильная группа, аллильные сложные эфиры и бензильные сложные эфиры.
В таких случаях, защитная группа для аминогруппы и защитная группа для карбоксигруппы могут быть, предпочтительно, удалены различными способами или в различных условиях. Например, типовой пример включает сочетание, в котором P2 является трет-бутилоксикарбонильной группой и P3 является бензильной группой. Защитные группы могут быть выбраны из указанных выше групп в зависимости от, например, свойств соединений, имеющих защищаемую аминогруппу и карбоксигруппу. Для удаления защитных групп реагенты и условия могут быть выбраны в зависимости от защитной группы.
Снятие защитной группы P3 с карбоксигруппы полученного соединения (13) дает соединение (14). Для снятия защиты реагенты и условия выбирают в зависимости от защитной группы.
Соединение (9) может быть получено превращением полученного соединения (14) в активный сложный эфир, смешанный ангидрид кислоты, галоидангидрид или подобные и взаимодействием с соединением (4) в присутствии основания. Для реакции также могут применяться реагенты и условия, которые обычно применяют для пептидного синтеза, и условия реакции, реагенты, основание и инертный растворитель для реакции могут быть подходящим образом выбраны из тех, которые описаны для синтеза соединения (6).
Соединение (2) также может быть получено следующим способом, например.
Снятие защитной группы P2 с аминогруппы соединения (13) дает соединение (15). Для снятия защиты реагенты и условия выбирают в зависимости от защитной группы.
Соединение (16) может быть получено превращением производного карбоновой кислоты (11) в активный сложный эфир, смешанный ангидрид кислоты, галогенид кислоты или подобные, и взаимодействие его в присутствии основания с полученным соединением (15). Условия реакции, реагенты, основание и инертный растворитель, применяемый для получения амидной связи между пептидной карбоновой кислотой (11) и соединением (15), могут быть подходящим образом выбраны из тех, которые описаны для синтеза соединения (6).
Снятие защиты с карбоксигруппы полученного соединения (16) дает соединение (17). Снятие защиты может проводиться так же, как для снятия защиты на карбоксигруппе для получения соединения (14).
Соединение (2) может быть получено превращением соединения (17) в активный сложный эфир, смешанный ангидрид кислоты, галогенид кислоты или подобные, и взаимодействие его с соединением (4) в присутствии основания. Для реакции также могут применяться реагенты и условия реакции, которые обычно применяют для пептидного синтеза, и условия реакции, реагенты, основание и инертный растворитель, применяемые для реакции, могут быть подходящим образом выбраны из тех, которые описаны для синтеза соединения (6).
3. Способ получения 3
Соединение, представленное формулой (2) промежуточного соединения также может быть получено следующим способом.
[Формула 23]
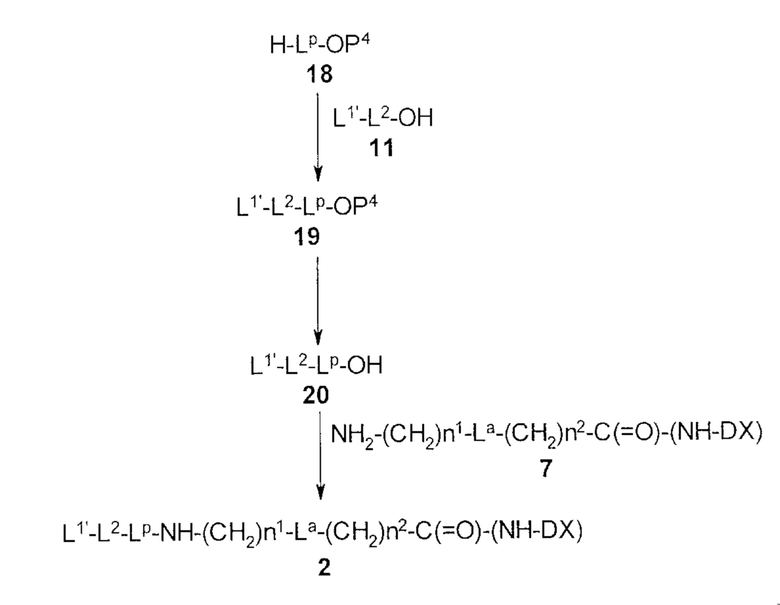
В формуле L1' соответствует L1, имеющему структуру, в которой концевая группа превращена в малеимидильную группу и P4 является защитной группой.
Соединение (19) может быть получено превращением соединения (11) в активный сложный эфир, смешанный ангидрид кислоты или подобные и взаимодействием его в присутствии основания с пептидной карбоновой кислотой (18), имеющей конец C, защищенный P4. Условия реакции, реагенты, основание и инертный растворитель, применяемые для образования пептидной связи между пептидной карбоновой кислотой (18) и соединением (11), могут быть подходящим образом выбраны из тех, которые описаны для синтеза соединения (6). Защитная группа P4 для карбоксигруппы соединения (18) может быть подходящим образом выбрана из защитных групп, описанных выше.
Снятие защитной группы карбоксигруппы полученного соединения (19) дает соединение (20). Снятие защиты может проводиться так же, как снятие защиты карбоксигруппы при получении соединения (14).
Соединение (2) может быть получено превращением полученного соединения (20) в активный сложный эфир, смешанный ангидрид кислоты или подобные и взаимодействием его с соединением (7). Для реакции также могут применяться реагенты и условия, которые обычно применяют для пептидного синтеза, и условия реакции, реагенты, основание и инертный растворитель, применяемые для реакции могут быть подходящим образом выбраны из тех, которые описаны для синтеза соединения (6).
4. Способ получения 4
Ниже подробно описан способ получения соединения (10b), в котором n1=1, La=O в технологическом промежуточном соединении (10), описанном в способе получения 2. Соединение, представленное формулой (10b), его соль или сольват могут быть получены следующим способом, например.
[Формула 24]
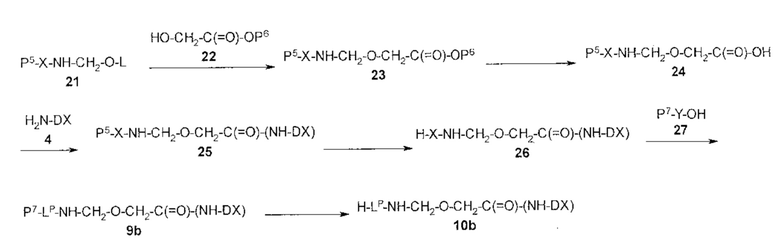
В формуле LP такой, как описан выше, L является ацильной группой, которая является алканоильной группой, такой как ацетильная группа, или ароильной группой, такой как бензоильная группа, или атомом водорода, X и Y каждый является олигопептидом, состоящим из от 1 до 3 аминокислот, P5 и P7 каждый является защитной группой для аминогруппы и P6 является защитной группой для карбоксигруппы.
Соединение, представленное формулой (21), может быть получено с применением способа, описанного в выложенном патенте Японии № 2002-60351 или в литературе (J. Org. Chem., Vol. 51, page 3196, 1986), и проведением удаления защитных групп или модификации функциональных групп, при необходимости. Более того, оно также может быть получено обработкой аминокислоты с защищенной концевой аминогруппой или амида кислоты олигопептида с защищенной аминогруппой альдегидом или кетоном.
Взаимодействие соединения (21) с соединением (22), имеющим гидроксильную группу, при температуре от низкотемпературного охлаждения до комнатной температуры в инертном растворителе в присутствии кислоты или основания, дает соединение (23).
В данном способе примеры кислоты, которая может применяться, включают неорганическую кислоту, такую как фтористоводородная кислота, хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и борная кислота; органическую кислоту, такую как уксусная кислота, лимонная кислота, паратолуолсульфоновая кислота и метансульфоновая кислота; и кислоту Льюиса, такую как тетрафторборат, хлорид цинка, хлорид олова, хлорид алюминия и хлорид железа. Среди них сульфоновая кислота, в частности, паратолуолсульфоновая кислота, является предпочтительной. В качестве основания может быть подходящим образом выбрано и использовано любое основание из указанных выше. Предпочтительные примеры включают алкоксид щелочного металла, такой как трет-бутоксид калия, гидроксид щелочного металла, такой как гидроксид натрия и гидроксид калия; гидрид щелочного металла или щелочноземельного металла, такой как гидрид натрия и гидрид калия; металлорганическое основание, представленное диалкиламинолитием, таким как диизопропиламид лития; и металлорганическое основание биссилиламина, такое как бис(триметилсилил)амид лития.
Примеры растворителя, применяемого для реакции, включают простой эфирный растворитель, такой как тетрагидрофуран и 1,4-диоксан; и ароматический углеводородный растворитель, такой как бензол и толуол. Эти растворители могут быть приготовлены в виде смеси с водой.
Далее, защитная группа для аминогруппы, представленная P5, особенно не ограничена при условии, что она является группой, обычно применяемой для защиты аминогруппы. Типовые примеры включают защитные группы для аминогруппы, которые описаны в способе получения 2. Однако, в данной реакции, могут быть варианты, в которых защитная группа для аминогруппы, представленная P5, отщеплена. В таких случаях защитная группа может быть введена снова подходящей реакцией с подходящим реагентом для защиты аминогруппы, при необходимости.
Соединение (24) может быть получено удалением защитной группы P6 соединения (23). Здесь типовые примеры защитной группы для карбоксигруппы, представленной P6, описаны в способе получения 2, и они могут быть подходящим образом выбраны из них. В соединении (23) желательно, в таком случае, чтобы защитная группа P5 для аминогруппы и защитная группа P6 для карбоксигруппы являлись защитными группами, которые могут быть удалены различными способами или в различных условиях. Например, типовой пример включает сочетание, в котором P5 является 9-флуоренилметилоксикарбонильной группой и P6 является бензильной группой. Защитные группы могут быть выбраны в зависимости от, например, свойств соединения, имеющего защищаемую аминогруппу и карбоксигруппу. Для удаления защитных групп, реагенты и условия выбирают в зависимости от защитной группы.
Соединение (26) может быть получено превращением карбоновой кислоты (24) в активный сложный эфир, смешанный ангидрид кислоты, галоидангидрид или подобное, и взаимодействие его с соединением (4) или его фармакологически приемлемой солью с получением соединения (25) с последующим удалением защитной группы P5 полученного соединения (25). Для реакции между соединением (4) и карбоновой кислотой (24) и реакции удаления защитной группы P6 могут применяться те же реагенты и условия реакции, которые применяют для способа получения 2.
Соединение (10b) может быть получено взаимодействием соединения (26) с аминокислотой с защищенной концевой аминогруппой или олигопептидом (27) с защищенной аминогруппой с получением соединения (9b) и удалением защитной группы P7 полученного соединения (9b). Защитная группа для аминогруппы, представленной P7, особенно не ограничена при условии, что ее обычно применяют для защиты аминогруппы. Типовые примеры включают защитные группы для аминогруппы, которые описаны в способе получения 2. Для удаления защитной группы регенты и условия выбирают в зависимости от защитной группы. Для реакции между соединением (26) и соединением (27), могут применяться реагенты и условия реакции, которые обычно применяют для пептидного синтеза. Соединение (10b), полученное указанным выше способом, может быть превращено в соединение (1) в соответствии с данным изобретением описанным выше способом.
5. Способ получения 5
Далее подробно описан способ получения соединения (2), в котором n1=1, n2=1, La=O в технологическом промежуточном соединении (2), описанном в способе получения 2. Соединение, представленное формулой (2), его соль или сольват может быть получено следующим способом, например.
[Формула 25]
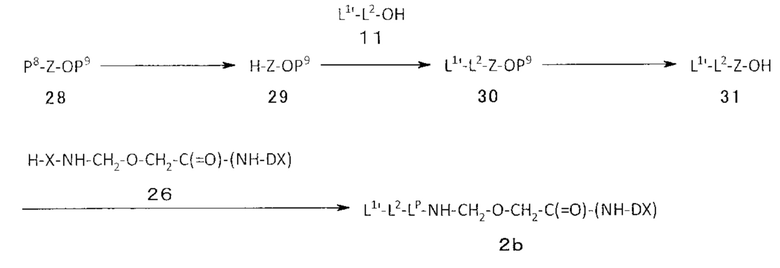
В формуле, L1', L2, LP такие, как определены выше, Z является олигопептидом, состоящим из от 1 до 3 аминокислот, P8 является защитной группой для аминогруппы и P9 является защитной группой для карбоксигруппы.
Соединение (30) может быть получено удалением защитной группы P8 аминокислоты или олигопептида (28) с защищенной концевой аминогруппой и карбоксигруппой с получением соединения (29) и взаимодействием полученного амина (29) с соединением (11). Защитная группа для аминогруппы, представленная P8, особенно не ограничена при условии, что она является группой, обычно применяемой для защиты аминогруппы. Типовые примеры включают защитные группы для аминогруппы, которые описаны в способе получения 2. Далее, для удаления защитной группы P8, реагенты и условия могут быть выбраны в зависимости от защитной группы. Для реакции между соединением (29) и карбоновой кислотой (11) могут применяться реагенты и условия реакции, описанные в способе получения 2.
Технологическое промежуточное соединение (2b) может быть получено удалением защитной группы P9 соединения (30) с получением соединения (31) и взаимодействием полученной карбоновой кислоты (31) с соединением (26). Типовые примеры защитной группы для карбоксигруппы, представленной P8, описаны в способе получения 2. Для снятия защиты могут применяться те же реагенты и условия реакции, которые описаны в способе получения 2. Для реакции между соединением (26) и карбоновой кислотой (31) также могут применяться реагенты и условия, которые обычно применяют для пептидного синтеза. Соединение (2b), полученное описанным выше способом, может быть превращено в соединение (1) в соответствии с данным изобретением описанным выше способом.
6. Способ получения 6
Ниже подробно описан способ получения соединения (17b), в котором n1=1, n2=1, La=O в технологическом промежуточном соединении (17), описанном в способе получения 2. Соединение, представленное формулой (17b), его соль или сольват также может быть получено следующим способом, например.
[Формула 26]
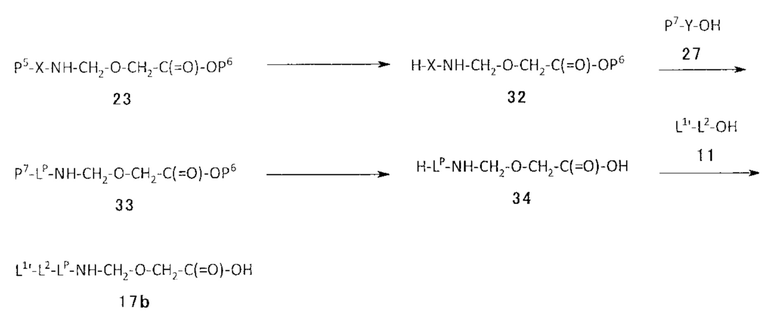
В формуле L1', L2, LP, X, Y, P5, P6 и P7 такие, как описаны выше.
Соединение (33) может быть получено снятием защитной группы P5 для аминогруппы соединения (23), имеющего защищенную концевую аминогруппу и карбоксигруппу, с получением соединения (32) и взаимодействием полученного производного амина (32) с олигопептидом (27), имеющим защищенную концевую аминогруппу или защищенную аминогруппу. Защитная группа для аминогруппы, представленная P5, особенно не ограничена при условии, что она является группой, обычно применяемой для защиты аминогруппы. Типовые примеры защитных групп для аминогруппы описаны в способе получения 2. Далее, для удаления защитной группы P5, реагенты и условия могут быть выбраны в зависимости от защитной группы. Здесь, типовые примеры защитной группы для карбоксигруппы, представленной P6, и защитной группы для аминогруппы, представленной P7, включают защитные группы для карбоксигруппы и аминогруппы, которые описаны способе получения 2. Желательно, чтобы в соединении (33), защитная группа P6 для карбоксигруппы и защитная группа P7 для аминогруппы являлись защитными группами, которые могут быть удалены одним способом или в одних и тех же условиях. Например, типовой пример включает сочетание, в котором P6 является группой бензилового эфира и P7 является бензилоксикарбонильной группой.
Соединение (34) может быть получено удалением защитной группы P6 для карбоксигруппы соединения (33) и защитной группы P7 для аминогруппы соединения (33). Соединение (37) также может быть получено последовательным удалением защитной группы P6 для карбоксигруппы и защитной группы P7 для аминогруппы, и затем соединение (34) может быть получено простым одновременным удалением защитных групп P6 и P7, которые могут быть удалены одним способом или в одних и тех же условиях.
Соединение (17b) может быть получено взаимодействием полученного соединения (34) с соединением (11). Для взаимодействия между соединение (34) и соединением (11) могут применяться те же реагенты и условия реакции, которые описаны в способе получения 2.
Анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением, когда его оставляют на воздухе или перекристаллизовывают или очищают, может абсорбировать влагу или содержать адсорбированную воду или превращаться в гидрат, и такое соединение или соли, содержащие воду, также включены в объем настоящего изобретения.
Соединения, меченные различными радиоактивными или не радиоактивными изотопами, также включены в объем настоящего изобретения. Один или более атомов, составляющих конъюгат антитела с лекарственным средством в соответствии с данным изобретением, может содержать атомный изотоп в не природном соотношении. Примеры атомных изотопов включают дейтерий (2H), тритий (3H), йод-125 (125I) и углерод-14 (14C). Кроме того, соединение в соответствии с данным изобретением может быть радиоактивно меченым радиоактивным изотопом, таким как тритий (3H), йод-125 (125I), углерод-14 (14C), медь-64 (64Cu), цирконий-89 (89Zr), йод-124 (124I), фтор-18 (18F), индий-111 (111I), углерод-11 (11C) и йод-131 (131I). Соединение, меченное радиоактивным изотопом, применяют в качестве терапевтического или профилактического агента, реагента для исследований, такого как химический реактив, и агента для диагностики, такого агент для диагностической визуализации in vivo. Независимо от радиоактивности, любой изотопный вариант конъюгата антитела с лекарственным средством в соответствии с данным изобретением включен в объем настоящего изобретения.
[Лекарственные средства]
Анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением обладает цитотоксичным действием против раковых клеток и, следовательно, может применяться в качестве лекарственного средства, в частности, в качестве терапевтического агента и/или профилактического агента для рака.
То есть, анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением может селективно применяться в качестве лекарственного средства для химиотерапии, которая является основным способом лечения рака, и в результате, может задерживать развитие раковых клеток, ингибировать их рост и далее убивать раковые клетки. Это освобождает пациентов, страдающих раковыми заболеваниями, от симптомов, вызываемых раком, или позволяет достичь улучшения в качестве жизни пациентов, страдающих раковыми заболеваниями, и достичь терапевтического эффекта через увеличение длительности жизни пациентов, страдающих раковыми заболеваниями. Даже если анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением не полностью убивает раковые клетки, он позволяет улучшить качество жизни пациентов, страдающих раковыми заболеваниями путем увеличения длительности жизни через ингибирование или контроль роста раковых клеток.
В такой лекарственной терапии он может применяться в качестве лекарственного средства отдельно и, кроме того, он может применяться в качестве лекарственного средства в сочетании с дополнительной терапией во вспомогательной терапии, и может быть объединен с хирургическим вмешательством, радиотерапией, гормональной терапией или подобными. Кроме того, он также может применяться в качестве лекарственного средства для лекарственной терапии в неоадъювантной терапии.
Кроме терапевтического применения, описанного выше, также может ожидаться действие, направленное на подавление роста мелких клеток метастатического рака и дальнейшее их умерщвление. В частности, если экспрессия HER2 подтверждена в первичных раковых клетках, ингибирование раковых метастазов или профилактическое действие может ожидаться при введении анти-HER2 конъюгата антитела с лекарственным средством в соответствии с данным изобретением. Например, может ожидаться эффект ингибирования и смерти раковых клеток в жидкости тела в курсе метастазов или эффект, например, ингибирования и смерти мелких раковых клеток сразу же после имплантации в любые ткани. Следовательно, ингибирование раковых метастазов или профилактическое действие может ожидаться, в частности, после хирургического удаления рака.
Анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением могу оказывать терапевтическое действие через введение в виде системной терапии пациентам и, дополнительно, местное введение в раковые ткани.
Примеры типов рака, к которым примени анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением, могут включать рак легкого, рак уротелия, рак прямой и ободочной кишки, рак предстательной железы, рак яичников, рак поджелудочной железы, рак молочной железы, рак мочевого пузыря, рак желудка, желудочно-кишечную стромальную опухоль, рак шейки матки, рак пищевода, плоскоклеточный рак, перитонеальный рак, рак печени, печеночно-клеточный рак, рак толстой кишки, рак прямой кишки, рак ободочной и прямой кишки, рак эндометрия, рак матки, рак слюнной железы, рак почки, рак вульвы, рак щитовидной железы или рак полового члена. Объектом лечения анти-HER2 конъюгатом антитела с лекарственным средством в соответствии с данным изобретением является раковая клетка, экспрессирующая, в раковой клетке в качестве объекта лечения, белок HER2, который антитело, содержащееся в конъюгате антитела с лекарственным средством, может распознать. Термин "рак, экспрессирующий белок HER2" в данном описании относится к раку, содержащему клетки, имеющие белок HER2 на своей поверхности. Белок HER2 чрезмерно экспрессируется в различных опухолях человека и может быть оценен с применением способа, обычно применяемого в данной области техники, такого как способ иммуногистохимического окрашивания (ИГХ) для оценки чрезмерной экспрессии белка HER2, или способ флуоресцентной гибридизации in situ (FISH) для оценки амплификации гена HER2.
Далее, анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением демонстрирует противоопухолевое действие посредством распознавания, через его анти-HER2 антитело, белка HER2, экспрессированного на поверхности раковых клеток, и интернализирования в раковых клетках. Таким образом, лечение пациента анти-HER2 конъюгатом антитела с лекарственным средством в соответствии с данным изобретением не ограничено "раком, экспрессирующим белок HER2" и также может быть применимо к, например, лейкозу, злокачественной лимфоме, плазмацитоме, миеломе или саркоме.
Анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением, предпочтительно, вводят млекопитающим, но более предпочтительно, вводят человеку.
Вещества, применяемые в фармацевтической композиции, содержащей анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением, могут быть подходящим образом выбраны, использованы из композиционных добавок или подобных, которые обычно применяют в данной области техники, с учетом дозы и концентрации при введении.
Анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением может вводиться в виде фармацевтической композиции, содержащей, по крайней мере, один фармацевтически приемлемый ингредиент. Например, фармацевтическая композиция, описанная выше, обычно содержит, по крайней мере, один фармацевтический носитель (например, стерилизованную жидкость). В данном описании жидкость включает, например, воду и масло (нефтяное масло и масло животного происхождения, растительного происхождения или синтетического происхождения). Маслом может быть, например, арахисовое масло, соевое масло, минеральное масло или конопляное масло. Вода является более часто применяемым носителем при внутривенном введении описанной выше фармацевтической композиции. Физиологический раствор, водный раствор декстрозы и водный раствор глицерина также могут применяться в качестве жидкого носителя, в частности, в качестве раствора для инъекций. Подходящий фармацевтический носитель может быть выбран из носителей, известных в данной области техники. При желании, описанная выше композиция может также содержать незначительные количества увлажняющего агента, эмульгирующего агента или pH буферного агента. Примеры подходящего фармацевтического носителя описаны в "Remington's Pharmaceutical Sciences" by E. W. Martin. Композиции соответствуют способу введения.
Известны различные системы доставки, и они могут применяться для введения анти-HER2 конъюгата антитела с лекарственным средством в соответствии с данным изобретением. Примеры способа введения могут включать внутрикожное, внутримышечное, внутрибрюшинное, внутривенное и подкожное введение, но не ограничены ими. Введение может проводиться инъекцией или болюсной инъекцией, например. В особенно предпочтительном варианте, введение конъюгата антитела с лекарственным средством проводят инъекцией. Парентеральное введения является предпочтительным способом введения.
Согласно типовому варианту, фармацевтическую композицию прописывают, в качестве фармацевтической композиции, подходящей для внутривенного введения человеку, в соответствии с обычными методами. Композиция для внутривенного введения обычно представляет собой раствор в стерильном и изотоническом буферном растворе. при необходимости, лекарственное средство может содержать солюбилизирующий агент и местные анестетики для облегчения боли в месте инъекции (например, лигнокаин). В общем, указанный выше ингредиент представлен индивидуально виде любого из лиофилизированного порошка или безводного концентрата, содержащегося в контейнере, который получают герметичным закупориванием в ампуле или саше определенного количества активного ингредиента, или в виде смеси в стандартной лекарственной форме. Если лекарственное средство должно вводиться инъекцией, оно может вводиться из сосуда для инъекций, содержащего воду или физиологический раствор стерильной фармацевтической степени чистоты. Если лекарственное средство вводят инъекцией, ампула со стерильной водой или физиологическим раствором для инъекций может быть предоставлена так, чтобы указанные выше ингредиенты смешивали друг с другом перед введением.
Фармацевтической композицией в соответствии с данным изобретением может быть фармацевтическая композиция, содержащая только анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением, или фармацевтическая композиция, содержащая анти-HER2 конъюгат антитела с лекарственным средством и, по крайней мере, один агент для лечения рака, отличный от конъюгата. Анти-HER2 конъюгат антитела с лекарственным средством в соответствии с данным изобретением может вводиться с другими агентами для лечения рака. Следовательно, противораковое действие может быть усилено. Другие противораковые агенты, применяемые для этой цели, могут вводиться пациенту одновременно с, отдельно от или последовательно с конъюгатом антитела с лекарственным средством, и могут вводиться при изменении интервала введения для каждого из них. Примеры агентов для лечения рака включают 5-FU, пертузумаб, паклитаксел, карбоплатин, цисплатин, гемцитабин, капецитабин, иринотекан (CPT-11), паклитаксел, доцетаксел, пеметрексед, сорафениб, винбластин, винорелбин, эверолимус, танеспимицин, бевацизумаб, оксалиплатин, лапатиниб, адо-трастузумаб эмтанзин(T-DM1) или лекарственные средства, описанные в международной публикации № WO 2003/038043, аналоги LH-RH (лейпрорелин, госерелин или подобные), эстрамустин фосфат, антагонисты эстрогена (тамоксифен, ралоксифен или подобные) и ингибиторы ароматазы (анастрозол, летрозол, экземестан или подобные), но не ограничены ими, до тех пор, пока они являются лекарственными средствами, обладающими противоопухолевым действием.
Фармацевтическая композиция может быть составлена в лиофизилированную композицию или жидкую композицию в виде состава, имеющего желаемую композицию и требуемую чистоту. Если она составлена в виде лиофилизированной композиции, она может представлять собой композицию, содержащую подходящие композиционные добавки, которые применяются в данной области техники. Что касается жидкой композиции, она может быть составлена виде жидкой композиции, содержащей различные композиционные добавки, которые применяются в данной области техники.
Композиция и концентрация фармацевтической композиции может варьироваться в зависимости от способа введения. Однако анти-HER2 конъюгат антитела с лекарственным средством, содержащийся в фармацевтической композиции в соответствии с данным изобретением, может оказывать фармацевтическое действие даже в небольших дозах, если конъюгат антитела с лекарственным средством имеет высшее сродство с антигеном, то есть, высшее сродство (= низшее значение Kd) относительно константы диссоциации (то есть значения Kd) для антигена. Таким образом, для определения дозы конъюгата антитела с лекарственным средством, доза может быть определена с учетом ситуации, относящейся к сродству между конъюгатом антитела с лекарственным средством и антигеном. Если конъюгат антитела с лекарственным средством в соответствии с данным изобретением вводят человеку, например, доза от около 0,001 до 100 мг/кг может быть введена один раз или несколько раз с интервалом от 1 до 180 дней.
в одно время с интервалом от 1 до 180 дней
Примеры
Настоящее изобретение отдельно описано в примерах, представленных ниже. Однако настоящее изобретение ими не ограничено. Далее, они не должны быть истолкованы в ограничивающей форме. Далее, если конкретно не указано иначе, реагент, растворитель и исходный материал, описанные в описании, могут быть легко получены от коммерческих поставщиков.
Ссылочный пример 1. Получение трастузумаба
Четырнадцать флаконов с 440 мг/флакон Герцептина (Genentech, Inc.) растворяют в 2 л катионообменного хроматографического буфера A (25 мМ цитратный буфер, 30 мМ NaCl, pH 5,0) и фильтруют через 0,2 мкл фильтр (Millipore Corp.: Stericup 0,22 мкм, мембрана GVPVDF). Образцы помещают в катионообменную хроматографическую колонку (SP Sepharose HP 240 мл, колонка XK50) с последующим элюированием с линейным градиентом концентрации NaCl от 30 мМ до 500 мМ с применением катионообменного хроматографического буфера B (25 мМ цитратный буфер, 500 мМ NaCl, pH 5,0) для отделения фракций IgG мономера. Образцы мономера, имеющие более высокую чистоту свыше 98% по данным анализа гель-фильтрационной хроматографии, объединяют и концентрируют с UF30K (Millipore Corp.: фильтр PELLICON XL, BIOMAX 30K, PXB030A50), и буфер меняют на буфер CBS (10 мМ цитрата/140 мМ NaCl, pH 6,0). Образцы в замененном буфере CBS фильтруют через 0,2 мкм фильтр (Sartorius AG: Minisart-Plus 0,2 мкм, 17823K).
Ссылочный пример 2. Получение трастузумаба эмтанзина T-DM1
SMCC превращение антитела: с применением общей методики C-2 (ФРФБ6,5/ЭДТК применяют в качестве буферного раствора), общей методики A и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,37 млмг-1см-1), описанных в способе получения 1, проводят замену буфера на ФРФБ6,5/ЭДТК для трастузумаба, полученного в ссылочном примере 1 с получением раствора, содержащего трастузумаб (160,0 мг), растворенный в ФРФБ6,5/ЭДТК (7,60 мл) в 15 мл полипропиленовой пробирке. Затем ДМСО раствор SMCC (1,84 мг) (0,40 мл; что соответствует около 5,1 эквивалента на молекулу антитела) добавляют при комнатной температуре. Реакционную смесь корректируют до концентрации антитела 20 мг/мл, и реакцию проводят при комнатной температуре с применением трубчатого ротатора (MTR-103, производства AS ONE Corporation) в течение 2 часов. Этот реакционный раствор очищают согласно общей методике D-2 (ФРФБ6,5/ЭДТК применяют в качестве буферного раствора) с получением 12 мл раствора, содержащего 154,9 мг SMCC-дериватизированного антитела.
Конъюгирование антитела и связующей группы лекарственного средства: Добавляют ФРФБ6,5/ЭДТК (2,56 мл) и N2-деацетил-N2-(3-меркапто-1-оксопропил)майтанзин (4,67 мг; DM1, Journal of Medicinal Chemistry, 2006, Vol. 49, No. 14, p. 4392) раствор ДМА (диметилцетамида) (0,93 мл; что соответствует около 5,8 эквивалента на молекулу SMCC-дериватизированного антитела) к полученному выше раствору в 50 мл полипропиленовой пробирке при комнатной температуре, реакционный раствор доводят до концентрации антитела 10 мг/мл, и реакцию проводят при комнатной температуре с применением трубчатого ротатора в течение 16,5 часов.
Методика очистки: Полученный выше раствор подвергают очистке с применением общей методики D-1 с применением раствор буфера на основе фосфата натрия (10 мМ, pH 6,5), содержащего хлорид натрия (137 мМ) с получением 35 мл раствора, содержащего целевое соединение ссылочного примера.
Физико-химические характеристики: С применением общей методики E с применением УФ абсорбции при двух длинах волн 252 нм и 280 нм, получают следующие характеристики.
Концентрация антитела: 4,14 мг/мл, выход антитела: 144,9 мг (91%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,0.
Пример 1. Промежуточное соединение (1)
[Формула 27]
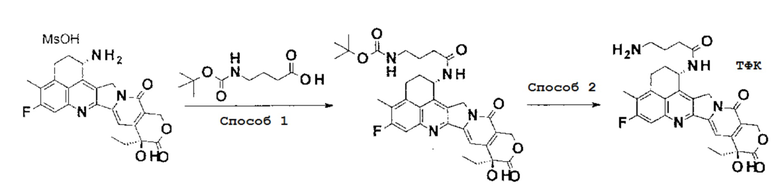
Способ 1: трет-Бутил (4-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-4-оксобутил)карбамат
4-(трет-Бутоксикарбониламино)бутановую кислоту (0,237 г, 1,13 ммоль) растворяют в дихлорметане (10 мл), добавляют N-гидроксисукцинимид (0,130 г, 1,13 ммоль) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,216 г, 1,13 ммоль) и перемешивают в течение 1 часа. Реакционный раствор добавляют по каплям в раствор N,N-диметилформамида (10 мл) с добавлением соли метансульфоновой кислоты экзатекана (0,500 г, 0,94 ммоль) и триэтиламина (0,157 мл, 1,13 ммоль), и перемешивают при комнатной температуре в течение 1 дня. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=8:2 (об./об.)] с получением указанного в заголовке соединения (0,595 г, количественный).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,2 Гц), 1,31 (9H, с), 1,58 (1H, т, J=7,2 Гц), 1,66 (2H, т, J=7,2 Гц), 1,82-1,89 (2H, м), 2,12-2,21 (3H, м), 2,39 (3H, с), 2,92 (2H, т, J=6,5 Гц), 3,17 (2H, с), 5,16 (1H, д, J=18,8 Гц), 5,24 (1H, д, J=18,8 Гц), 5,42 (2H, с), 5,59-5,55 (1H, м), 6,53 (1H, с), 6,78 (1H, т, J=6,3 Гц), 7,30 (1H, с), 7,79 (1H, д, J=11,0 Гц), 8,40 (1H, д, J=8,6 Гц).
МС (ХИАД) m/z: 621 (M+H)+
Способ 2: 4-Амино-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]бутанамид
Соединение (0,388 г, 0,61 ммоль), полученное в способе 1 выше, растворяют в дихлорметане (9 мл). Добавляют трифторуксусную кислоту (9 мл) и перемешивают в течение 4 часов. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформа:метанол:вода=7:3:1 (об./об./об.)] с получением трифторацетата указанного в заголовке соединения (0,343 г, количественный).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,2 Гц), 1,79-1,92 (4H, м), 2,10-2,17 (2H, м), 2,27 (2H, т, J=7,0 Гц), 2,40 (3H, с), 2,80-2,86 (2H, м), 3,15-3,20 (2H, м), 5,15 (1H, д, J=18,8 Гц), 5,26 (1H, д, J=18,8 Гц), 5,42 (2H, с), 5,54-5,61 (1H, м), 6,55 (1H, с), 7,32 (1H, с), 7,72 (3H, шс), 7,82 (1H, д, J=11,0 Гц), 8,54 (1H, д, J=8,6 Гц).
МС (ХИАД) m/z: 521 (M+H)+
Пример 2. Конъюгат антитела с лекарственным средством (2)
[Формула 28]
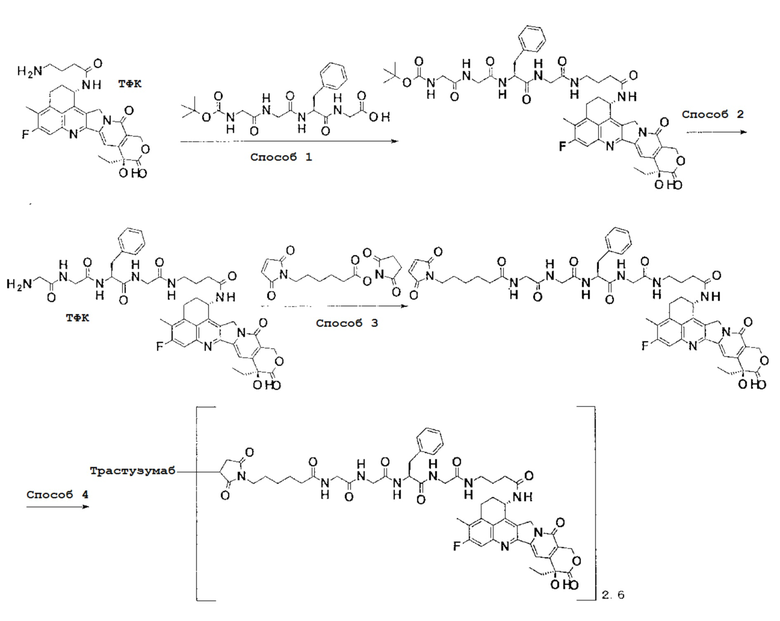
Способ 1: N-(трет-Бутоксикарбонил)глицилглицил-L-фенилаланил-N-(4-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-4-оксобутил)глицинамид
N-(трет-Бутоксикарбонил)глицилглицил-L-фенилаланилглицин (0,081 г, 0,19 ммоль) растворяют в дихлорметане (3 мл), добавляют N-гидроксисукцинимид (0,021 г, 0,19 ммоль) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,036 г, 0,19 ммоль) и затем перемешивают в течение 3,5 часов. Реакционный раствор добавляют по каплям к раствору N,N-диметилформамида (1,5 мл) с добавлением соединения (0,080 г, 0,15 ммоль), полученного в способе 2 примера 1, и перемешивают при комнатной температуре в течение 4 часов. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=8:2 (об./об.)] с получением указанного в заголовке соединения (0,106 г, 73%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,4 Гц), 1,36 (9H, с), 1,71 (2H, м), 1,86 (2H, т, J=7,8 Гц), 2,15-2,19 (4H, м), 2,40 (3H, с), 2,77 (1H, дд, J=12,7, 8,8 Гц), 3,02 (1H, дд, J=14,1, 4,7 Гц), 3,08-3,11 (2H, м), 3,16-3,19 (2H, м), 3,54 (2H, д, J=5,9 Гц), 3,57-3,77 (4H, м), 4,46-4,48 (1H, м), 5,16 (1H, д, J=19,2 Гц), 5,25 (1H, д, J=18,8 Гц), 5,42 (2H, с), 5,55-5,60 (1H, м), 6,53 (1H, с), 7,00 (1H, т, J=6,3 Гц), 7,17-7,26 (5H, м), 7,31 (1H, с), 7,71 (1H, т, J=5,7 Гц), 7,80 (1H, д, J=11,0 Гц), 7,92 (1H, т, J=5,7 Гц), 8,15 (1H, д, J=8,2 Гц), 8,27 (1H, т, J=5,5 Гц), 8,46 (1H, д, J=8,2 Гц).
МС (ХИАД) m/z: 939 (M+H)+
Способ 2: Глицилглицил-L-фенилаланил-N-(4-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-4-оксобутил)глицинамид
Соединение (1,97 г, 2,10 ммоль), полученное в способе 1 выше, растворяют в дихлорметане (7 мл). После добавления трифторуксусной кислоты (7 мл) раствор перемешивают в течение 1 часа. Растворитель удаляют при пониженном давлении, и к остаткам добавляют толуол для азеотропной дистилляции. Полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением трифторацетата указанного в заголовке соединения (1,97 г, 99%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,4 Гц), 1,71-1,73 (2H, м), 1,82-1,90 (2H, м), 2,12-2,20 (4H, м), 2,40 (3H, с), 2,75 (1H, дд, J=13,7, 9,4 Гц), 3,03-3,09 (3H, м), 3,18-3,19 (2H, м), 3,58-3,60 (2H, м), 3,64 (1H, д, J=5,9 Гц), 3,69 (1H, д, J=5,9 Гц), 3,72 (1H, д, J=5,5 Гц), 3,87 (1H, дд, J=16,8, 5,9 Гц), 4,50-4,56 (1H, м), 5,16 (1H, д, J=19,2 Гц), 5,25 (1H, д, J=18,8 Гц), 5,42 (2H, с), 5,55-5,60 (1H, м), 7,17-7,27 (5H, м), 7,32 (1H, с), 7,78-7,81 (2H, м), 7,95-7,97 (3H, м), 8,33-8,35 (2H, м), 8,48-8,51 (2H, м).
МС (ХИАД) m/z: 839 (M+H)+
Способ 3: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-(4-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-4-оксобутил)глицинамид
К раствору в N,N-диметилформамиде (1,2 мл) соединения (337 мг, 0,353 ммоль), полученного в способе 2 выше, добавляют триэтиламин (44,3 мл, 0,318 ммоль) и гексаноат N-сукцинимидил 6-малеимида (119,7 мг, 0,388 ммоль) и перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=5:1 (об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (278,0 мг, 76%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,3 Гц), 1,12-1,22 (2H, м), 1,40-1,51 (4H, м), 1,66-1,76 (2H, м), 1,80-1,91 (2H, м), 2,05-2,21 (6H, м), 2,39 (3H, с), 2,79 (1H, дд, J=14,0, 9,8 Гц), 2,98-3,21 (5H, м), 3,55-3,77 (8H, м), 4,41-4,48 (1H, м), 5,15 (1H, д, J=18,9 Гц), 5,24 (1H, д, J=18,9 Гц), 5,40 (1H, д, J=17,1 Гц), 5,44 (1H, д, J=17,1 Гц), 5,54-5,60 (1H, м), 6,53 (1H, с), 6,99 (2H, с), 7,20-7,27 (5H, м), 7,30 (1H, с), 7,70 (1H, т, J=5,5 Гц), 7,80 (1H, д, J=11,0 Гц), 8,03 (1H, т, J=5,8 Гц), 8,08 (1H, т, J=5,5 Гц), 8,14 (1H, д, J=7,9 Гц), 8,25 (1H, т, J=6,1 Гц), 8,46 (1H, д, J=8,5 Гц).
МС (ХИАД) m/z: 1032 (M+H)+
Способ 4: Конъюгат антитела с лекарственным средством (2)
Восстановление антитела: трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл с ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (применяют коэффициент абсорбции при 280 нм, 1,37 млмг-1см-1), описанной в способе получения 1. Раствор (3,0 мл) помещают в 15 мл полипропиленовую пробирку и загружают водным раствором 10 мМ гидрохлорида трис(2-карбоксиэтил)фосфина (ТКЭФ, Tokyo Chemical Industry Co., Ltd.) (0,0934 мл; 4,6 эквивалента на молекулу антитела) и водным раствором 1M вторичного кислого фосфата калия (Nacalai Tesque, Inc.; 0,150 мл). После подтверждения того, что раствор имеет pH 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После инкубирования раствора при температуре выше 22°C в течение 10 минут, туда добавляют раствор ДМСО (0,187 мл; 9,2 эквивалента на молекулу антитела), содержащий 10 мМ соединения, полученного в способе 3, и инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 22°C в течение 40 минут. Затем туда добавляют водный раствор (0,0374 мл; 18,4 эквивалента на молекулу антитела) N-ацетилцистеина (NAC, Sigma-Aldrich Co. LLC) и инкубируют при 22°C для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D-1 (ФРФБ6,0 применяют в качестве буферного раствора), описанной в способе получения 1 с получением 6 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством. Затем раствор концентрируют с применением общей методики A.
Физико-химические характеристики: с помощью общей методики E, описанной в способе получения 1, получают следующие характеристики.
Концентрация антитела: 3,21 мг/мл, выход антитела: 22,5 мг (75%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 2,6.
Пример 3 Конъюгат антитела с лекарственным средством (3)
[Формула 29]
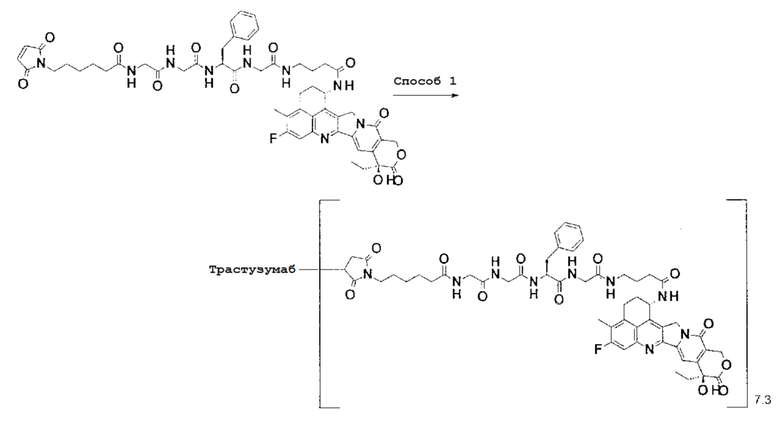
Способ 1: Конъюгат антитела с лекарственным средством (3)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1 получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,487 млмг-1см-1). Раствор (1,25 мл) помещают в 1,5 мл полипропиленовую пробирку и загружают водным раствором 10 мМ ТКЭФ (0,039 мл; 4,6 эквивалента на молекулу антитела) и водным раствором 1M вторичного кислого фосфата калия (0,0625 мл). После подтверждения того, что раствор имеет pH 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После добавления ДМСО (0,072 мл) и раствора ДМСО, содержащего 10 мМ соединения из способа 3 примера 2 (0,078 мл; 9,2 эквивалента на молекулу антитела) к полученному выше раствору при комнатной температуре, смесь перемешивают с применением трубчатого ротатора для конъюгирования связующей группы лекарственного средства с антителом при комнатной температуре в течение 40 минут. Затем добавляют водный раствор (0,0155 мл; 18,4 эквивалента на молекулу антитела) 100 мМ NAC и перемешивают при комнатной температуре для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D (ABS применяют в качестве буферного раствора), описанной в способе получения 1 с получением 6 мл раствора, содержащего целевое соединение. Раствор затем концентрируют по общей методике A. После этого с применением общей методики E получают следующие характеристики.
Концентрация антитела: 9,85 мг/мл, выход антитела: 6,9 мг (55%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 7,3.
Пример 4. Конъюгат антитела с лекарственным средством (4)
[Формула 30]
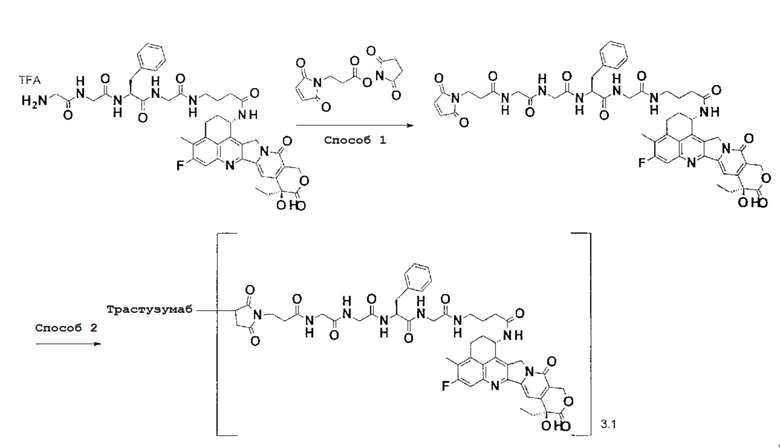
Способ 1: N-[3-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)пропаноил]глицилглицил-L-фенилаланил-N-(4-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-4-оксобутил)глицинамид
Соединение (80 мг, 0,084 ммоль) из примера 1 подвергают взаимодействию по методике способа 3 из примера 2 с применением пропиоата N-сукцинимидил 3-малеимида (24,6 мг, 0,0924 ммоль) вместо гексаноата N-сукцинимидил 6-малеимида с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (60,0 мг, 73%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,89 (3H, т, J=7,3 Гц), 1,70-1,78 (2H, м), 1,81-1,94 (2H, м), 2,12-2,23 (4H, м), 2,42 (3H, с), 2,81 (1H, дд, J=13,7, 9,8 Гц), 3,01-3,15 (3H, м), 3,16-3,23 (2H, м), 3,30-3,35 (1H, м), 3,58-3,71 (6H, м), 3,71-3,79 (1H, м), 4,44-4,51 (1H, м), 5,19 (1H, д, J=19,0 Гц), 5,27 (1H, д, J=19,0 Гц), 5,43 (1H, д, J=17,6 Гц), 5,47 (1H, д, J=17,6 Гц), 5,57-5,63 (1H, м), 6,56 (1H, с), 7,02 (2H, с), 7,17-7,22 (1H, м), 7,22-7,30 (5H, м), 7,34 (1H, с), 7,73 (1H, т, J=5,6 Гц), 7,83 (1H, д, J=10,7 Гц), 8,08 (1H, т, J=5,6 Гц), 8,15 (1H, д, J=7,8 Гц), 8,30 (2H, дт, J=18,7, 5,7 Гц), 8,49 (1H, д, J=8,8 Гц).
МС (ХИАД) m/z: 990 (M+H)+
Способ 2: Конъюгат антитела с лекарственным средством (4)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и Общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (1 мл) помещают в 1,5 мл полипропиленовую пробирку и загружают водным раствором 10 мМ ТКЭФ (0,0155 мл; 2,3 эквивалента на молекулу антитела) и водным раствором 1M вторичного кислого фосфата калия (0,050 мл). После подтверждения того, что раствор имеет pH of 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После добавления ДМСО (0,072 мл) и раствора ДМСО, содержащего 10 мМ соединения из способа 3 примера 2 (0,031 мл; 4,6 эквивалента на молекулу антитела) к полученному выше раствору при комнатной температуре, смесь перемешивают с применением трубчатого ротатора для конъюгирования связующей группы лекарственного средства с антителом при комнатной температуре в течение 40 минут. Затем добавляют водный раствор (0,0078 мл; 9,2 эквивалента на молекулу антитела) 100 мМ NAC и перемешивают при комнатной температуре для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D (ABS применяют в качестве буферного раствора) с получением 6 мл раствора, содержащего целевое соединение. С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 1,32 мг/мл, выход антитела: 7,9 мг (79%), и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,1.
Пример 5. Конъюгат антитела с лекарственным средством (5)
[Формула 31]
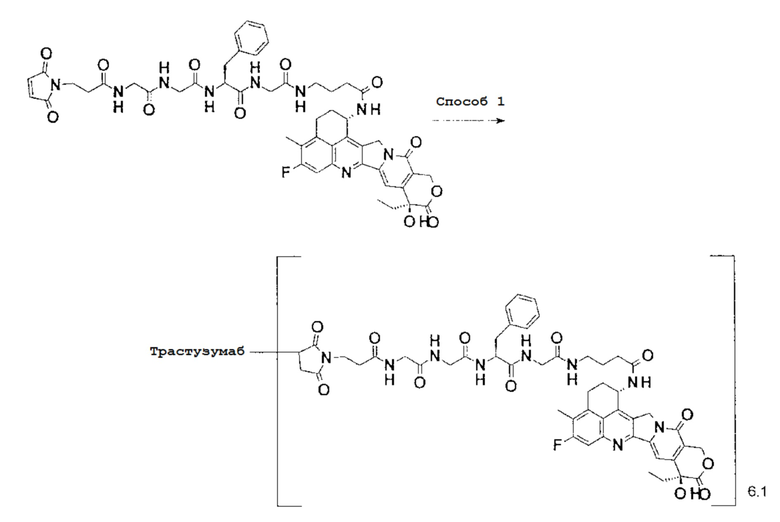
Способ 1: Конъюгат антитела с лекарственным средством (5)
Количество водного раствора 10 мМ ТКЭФ корректируют таким образом, чтобы молярное отношение ТКЭФ к антителу при восстановлении антитела составляло 4,6. И количество добавляемого 10 мМ раствора связующей группы лекарственного средства корректируют так, чтобы молярное отношение соединения из способа 1 примера 4 к антителу при конъюгировании связующей группы лекарственного средства составляло 9,2. Затем количество добавленного водного раствора 100 мМ NAC корректируют так, чтобы молярное отношение NAC к антителу в конце реакции составляло 18,4. По методикам способа 2 примера 4, получают 6 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством, и получают следующие характеристики.
Концентрация антитела: 1,23 мг/мл, выход антитела: 7,4 мг (74%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,1.
Пример 6: Конъюгат антитела с лекарственным средством (6)
[Формула 32]
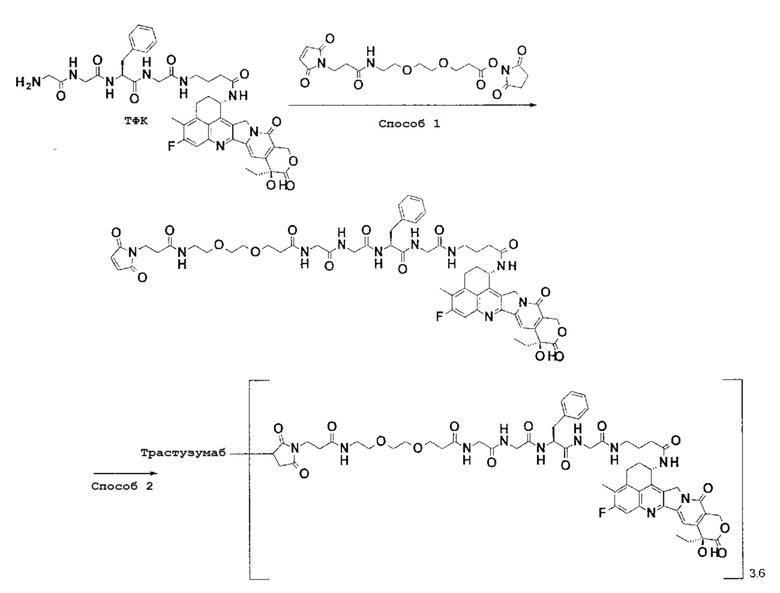
Способ 1: N-{3-[2-(2-{[3-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)пропаноил]амино}этокси)этокси]пропаноил}глицилглицил-L-фенилаланил-N-(4-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-4-оксобутил)глицинамид
Соединение (100 мг, 0,119 ммоль), полученное в способе 2 примера 2 подвергают взаимодействию по методике способа 3 из примера 2 с применением диизопропилэтиламина (20,8 мкл, 0,119 ммоль) вместо триэтиламина и 3-(2-(2-(3-малеинимидпропанамид)этокси)этокси)пропаноата N-сукцинимидила (50,7 мг, 0,119 ммоль) вместо гексаноата N-сукцинимидил 6-малеимида с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (66,5 мг, 48%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,85 (3H, т, J=7,4 Гц), 1,65-1,74 (2H, м), 1,77-1,90 (2H, м), 2,07-2,19 (4H, м), 2,30 (2H, т, J=7,2 Гц), 2,33-2,36 (2H, м), 2,38 (3H, с), 2,76 (1H, дд, J=13,7, 9,8 Гц), 2,96-3,18 (9H, м), 3,42-3,44 (4H, м), 3,53-3,76 (10H, м), 4,43 (1H, td, J=8,6, 4,7 Гц), 5,14 (1H, д, J=18,8 Гц), 5,23 (1H, д, J=18,8 Гц), 5,38 (1H, д, J=17,2 Гц), 5,42 (1H, д, J=17,2 Гц), 5,52-5,58 (1H, м), 6,52 (1H, с), 6,98 (2H, с), 7,12-7,17 (1H, м), 7,18-7,25 (4H, м), 7,29 (1H, с), 7,69 (1H, т, J=5,5 Гц), 7,78 (1H, д, J=11,3 Гц), 7,98-8,03 (2H, м), 8,11 (1H, д, J=7,8 Гц), 8,16 (1H, т, J=5,7 Гц), 8,23 (1H, т, J=5,9 Гц), 8,44 (1H, д, J=9,0 Гц).
МС (ХИАД) m/z: 1149 (M+H)+
Способ 2: Конъюгат антитела с лекарственным средством (6)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (1,25 мл) помещают в 1,5 мл полипропиленовую пробирку и загружают водным раствором 10 мМ ТКЭФ (0,019 мл; 2,3 эквивалента на молекулу антитела) и водным раствором 1M вторичного кислого фосфата калия (0,0625 мл). После подтверждения того, что раствор имеет pH of 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После добавления ДМСО (Sigma-Aldrich Co. LLC; 0,109 мл) и раствора ДМСО, содержащего 10 мМ соединения из способа 1 (0,039 мл; 4,6 эквивалента на молекулу антитела) к полученному выше раствору при комнатной температуре, смесь перемешивают с применением трубчатого ротатора для конъюгирования связующей группы лекарственного средства с антителом при комнатной температуре в течение 40 минут. Затем добавляют водный раствор (0,008 мл) 100 мМ NAC и перемешивают при комнатной температуре для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D (ABS применяют в качестве буферного раствора), описанной в способе получения 1 с получением 6 мл раствора, содержащего целевое соединение.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 1,76 мг/мл, выход антитела: 10,6 мг (85%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,6.
Пример 7. Конъюгат антитела с лекарственным средством (7)
[Формула 33]
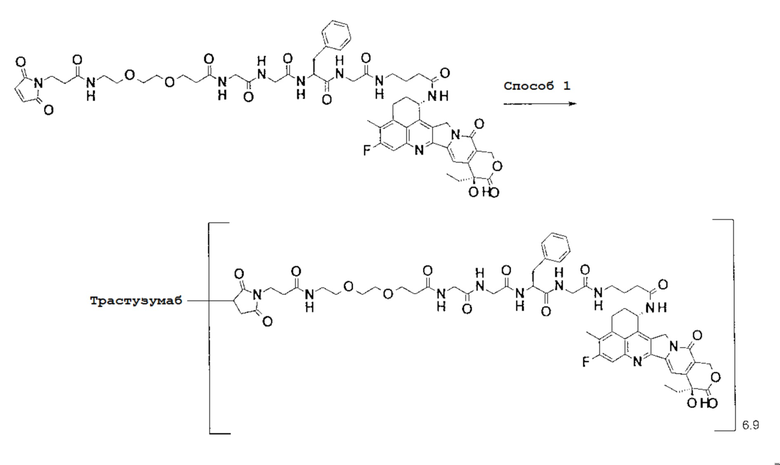
Способ 1: Конъюгат антитела с лекарственным средством (7)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (1,25 мл) помещают в 1,5 мл полипропиленовую пробирку и загружают водным раствором 10 мМ ТКЭФ (0,039 мл; 4,6 эквивалента на молекулу антитела) и водным раствором 1M вторичного кислого фосфата калия (0,0625 мл). После подтверждения того, что раствор имеет pH of 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После добавления ДМСО (0,072 мл) и раствора ДМСО, содержащего 10 мМ соединения из способа 1 примера 6 (0,078 мл; 9,2 эквивалента на молекулу антитела) к полученному выше раствору при комнатной температуре, смесь перемешивают с применением трубчатого ротатора для конъюгирования связующей группы лекарственного средства с антителом при комнатной температуре в течение 40 минут. Затем добавляют водный раствор (0,0155 мл) 100 мМ NAC и перемешивают при комнатной температуре для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D (ABS применяют в качестве буферного раствора) с получением 6 мл раствора, содержащего целевое соединение.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 1,93 мг/мл, выход антитела: 11,6 мг (93%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,9.
Пример 8. Конъюгат антитела с лекарственным средством (8)
[Формула 34]
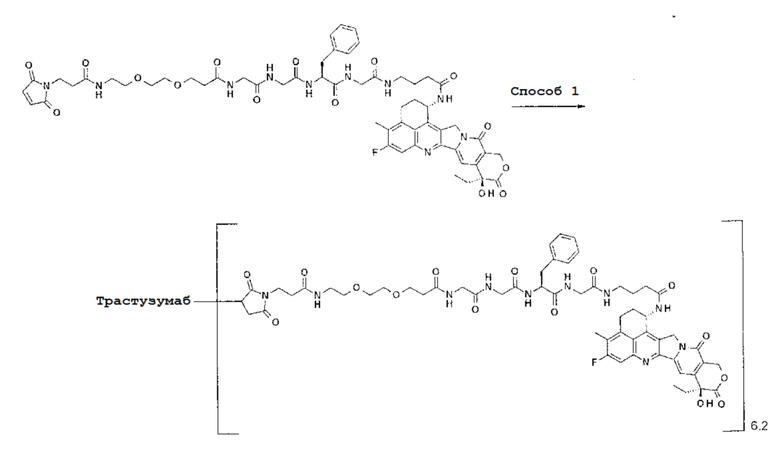
Способ 1: Конъюгат антитела с лекарственным средством (8)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (1,25 мл) помещают в 1,5 мл полипропиленовую пробирку и загружают водным раствором 10 мМ ТКЭФ (0,039 мл; 4,6 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,0625 мл). После подтверждения того, что раствор имеет pH of 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После добавления ДМСО (0,072 мл) и раствора ДМСО, содержащего 10 мМ соединения из способа 1 примера 6 (0,078 мл; 9,2 эквивалента на молекулу антитела) к полученному выше раствору при комнатной температуре, смесь перемешивают с применением трубчатого ротатора для конъюгирования связующей группы лекарственного средства с антителом при комнатной температуре в течение 40 минут. Затем добавляют водный раствор (0,0155 мл) 100 мМ NAC и перемешивают при комнатной температуре для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D-1 (ABS применяют в качестве буферного раствора) с получением 5,7 мл раствора, содержащего целевое соединение.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 1,50 мг/мл, выход антитела: 8,55 мг (86%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,2.
Пример 9. Конъюгат антитела с лекарственным средством (9)
[Формула 35]
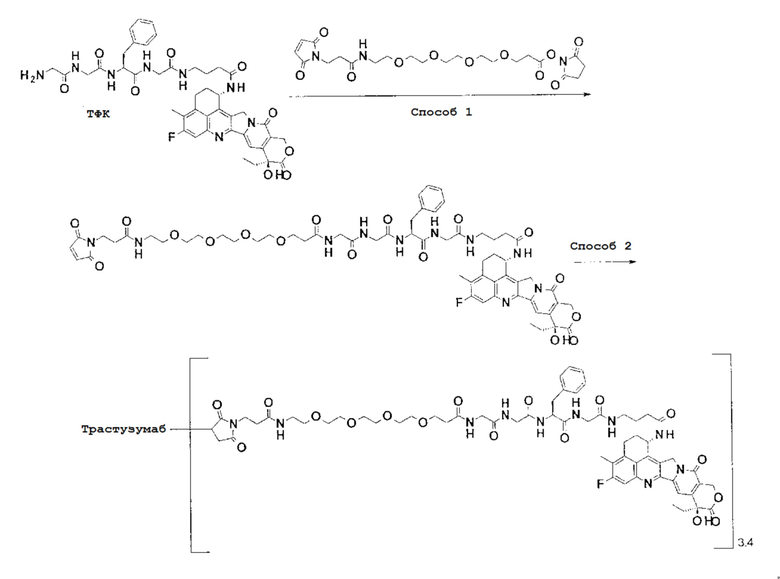
Способ 1: N-[19-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраоксо-16-азанонадекан-1-оил]глицилглицил-L-фенилаланил-N-(4-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-4-оксобутил)глицинамид
Соединение (90 мг, 0,107 ммоль), полученное в способе 2 примера 2 подвергают взаимодействию по методике способа 3 примера 2 с применением диизопропилэтиламина (18,7 мкл, 0,107 ммоль) вместо триэтиламина и 1-малеинимид-3-оксо-7,10,13,16-тетраокса-4-азанонадекан-19-оата N-сукцинимидила (55,1 мг, 0,107 ммоль) вместо гексаноата N-сукцинимидила 6-малеимида с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (50 мг, 37%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,85 (3H, т, J=7,2 Гц), 1,64-1,74 (2H, м), 1,77-1,90 (2H, м), 2,06-2,19 (4H, м), 2,27-2,32 (2H, м), 2,33-2,37 (2H, м), 2,38 (3H, с), 2,72-2,80 (3H, м), 2,96-3,19 (6H, м), 3,39-3,48 (10H, м), 3,52-3,75 (10H, м), 4,39-4,48 (1H, м), 5,14 (1H, д, J=18,8 Гц), 5,23 (1H, д, J=18,8 Гц), 5,38 (1H, д, J=17,0 Гц), 5,42 (1H, д, J=17,0 Гц), 5,52-5,58 (1H, м), 6,52 (1H, с), 6,98 (1H, с), 7,13-7,24 (5H, м), 7,29 (1H, с), 7,69 (1H, т, J=5,5 Гц), 7,78 (1H, д, J=10,9 Гц), 7,98-8,03 (2H, м), 8,10 (1H, д, J=7,8 Гц), 8,16 (1H, т, J=5,7 Гц), 8,23 (1H, т, J=5,7 Гц), 8,44 (1H, д, J=8,6 Гц).
МС (ХИАД) m/z: 1237 (M+H)+
Способ 2: Конъюгат антитела с лекарственным средством (9)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 2 примера 6.
Концентрация антитела: 1,75 мг/мл, выход антитела: 10,5 мг (84%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,4.
Пример 10. Конъюгат антитела с лекарственным средством (10)
[Формула 36]
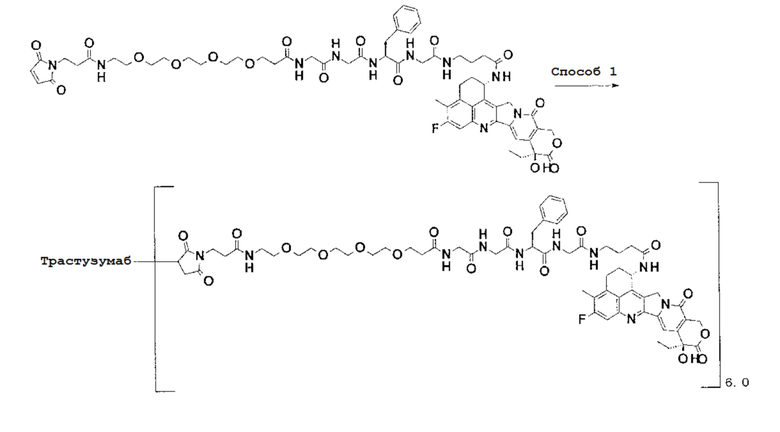
Способ 1: Конъюгат антитела с лекарственным средством (10)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1 примера 9, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 примера 7.
Концентрация антитела: 1,79 мг/мл, выход антитела: 10,7 мг (86%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,0.
Пример 11. Промежуточное соединение (11)
[Формула 37]
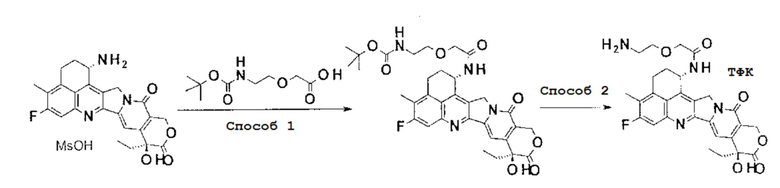
Способ 1: трет-Бутил [2-(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)этил]карбамат
Соль метансульфоновой кислоты экзатекана (3,10 г, 5,47 моль) подвергают взаимодействию по методике способа 1 примера 1 с применением {2-[(трет-бутоксикарбонил)амино]этокси}уксусной кислоты (J. Med. Chem., 1992, vol. 35, pp. 292; 1,55 г, 6,01 ммоль) вместо 4-(трет-бутоксикарбониламино)бутановой кислоты с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (2,56 г, 73%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,3 Гц), 1,26 (9H, с), 1,81-1,91 (2H, м), 2,13-2,22 (2H, м), 2,40 (3H, с), 3,08-3,26 (4H, м), 3,43-3,53 (2H, м), 4,00 (1H, д, J=15,1 Гц), 4,05 (1H, д, J=15,1 Гц), 5,14 (1H, д, J=18,7 Гц), 5,22 (1H, д, J=18,7 Гц), 5,40 (1H, д, J=16,6 Гц), 5,44 (1H, д, J=16,6 Гц), 5,59-5,66 (1H, м), 6,53 (1H, с), 6,86 (1H, т, J=5,4 Гц), 7,31 (1H, с), 7,79 (1H, д, J=10,9 Гц), 8,49 (1H, д, J=9,1 Гц).
МС (ХИАД) m/z: 637 (M+H)+
Способ 2: 2-(2-Аминоэтокси)-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]ацетамид
Соединение (1,50 г, 2,36 моль), полученное в способе 1 выше, подвергают взаимодействию по методике способа 2 примера 1 с получением соли трифторуксусной кислоты указанного в заголовке соединения в виде бледно-желтого твердого вещества (1,50 г, количественный).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,5 Гц), 1,81-1,92 (2H, м), 2,15-2,23 (2H, м), 2,41 (3H, с), 3,05 (2H, т, J=5,1 Гц), 3,15-3,23 (2H, м), 3,71 (2H, т, J=5,1 Гц), 4,10 (2H, с), 5,19 (1H, д, J=18,7 Гц), 5,24 (1H, д, J=18,7 Гц), 5,43 (2H, с), 5,58-5,66 (1H, м), 6,55 (1H, с), 7,33 (1H, с), 7,73-7,84 (4H, м), 8,55 (1H, д, J=9,1 Гц).
МС (ХИАД) m/z: 537 (M+H)+
Пример 12. Конъюгат антитела с лекарственным средством (12)
[Формула 38]
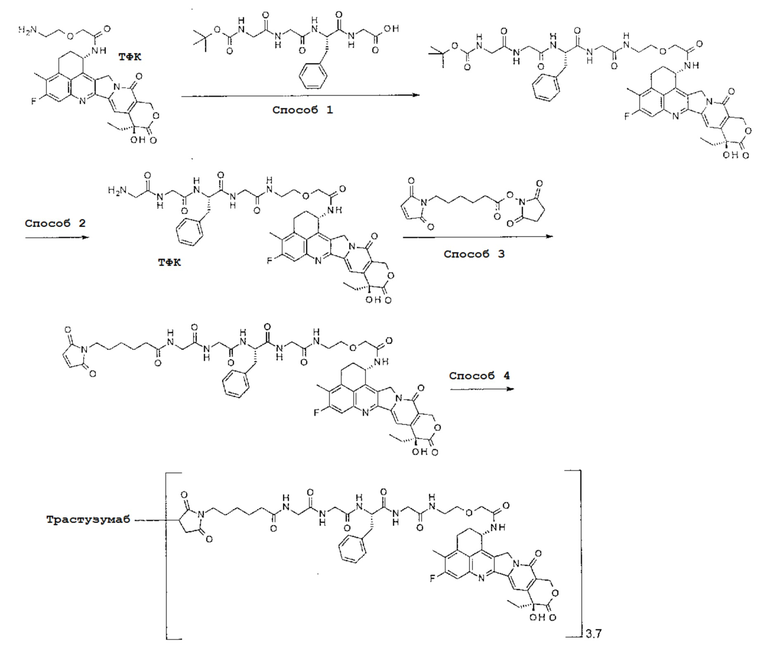
Способ 1: N-(трет-Бутоксикарбонил)глицилглицил-L-фенилаланил-N-[2-(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)этил]глицинамид
Соединение (554 мг, 0,85 ммоль) способа 2 примера 11 подвергают взаимодействию по методике способа 1 примера 2 с получением указанного в заголовке соединения (775 мг, 95%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,85 (3H, т, J=7,3 Гц), 1,36 (9H, с), 1,78-1,89 (2H, м), 2,13-2,22 (2H, м), 2,39 (3H, с), 2,71 (1H, дд, J=13,4, 9,8 Гц), 2,95 (1H, дд, J=13,4, 4,3 Гц), 3,09-3,23 (1H, м), 3,23-3,32 (2H, м), 3,40-3,62 (8H, м), 3,73 (1H, дд, J=16,5, 5,5 Гц), 4,03 (2H, с), 4,39-4,47 (1H, м), 5,17 (1H, д, J=18,9 Гц), 5,25 (1H, д, J=18,9 Гц), 5,41 (1H, д, J=16,8 Гц), 5,45 (1H, д, J=16,8 Гц), 5,57-5,64 (1H, м), 6,54 (1H, с), 6,99 (1H, т, J=5,8 Гц), 7,13-7,26 (5H, м), 7,31 (1H, с), 7,76-7,82 (2H, м), 7,90 (1H, т, J=5,2 Гц), 8,13 (1H, д, J=7,9 Гц), 8,27 (1H, т, J=5,8 Гц), 8,49 (1H, д, J=8,5 Гц).
МС (ХИАД) m/z: 955 (M+H)+
Способ 2: Глицилглицил-L-фенилаланил-N-[2-(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)этил]глицинамид
Соединение (630 мг, 0,659 ммоль), полученное в способе 1 выше подвергают взаимодействию по методике способа 2 примера 2 с получением соли трифторуксусной кислоты указанного в заголовке соединения (588 мг, 92%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,3 Гц), 1,79-1,90 (2H, м), 2,13-2,22 (2H, м), 2,39 (3H, с), 2,71 (1H, дд, J=13,4, 10,1 Гц), 2,99 (1H, дд, J=13,4, 4,3 Гц), 3,09-3,23 (1H, м), 3,24-3,32 (3H, м), 3,41-3,71 (7H, м), 3,86 (1H, дд, J=16,8, 5,8 Гц), 4,04 (2H, с), 4,52 (1H, тд, J=9,0, 4,1 Гц), 5,17 (1H, д, J=18,9 Гц), 5,25 (1H, д, J=18,9 Гц), 5,41 (1H, д, J=16,5 Гц), 5,45 (1H, д, J=16,5 Гц), 5,56-5,65 (1H, м), 6,55 (1H, с), 7,13-7,26 (5H, м), 7,32 (1H, с), 7,80 (1H, д, J=11,0 Гц), 7,87-8,01 (4H, м), 8,29-8,36 (2H, м), 8,46-8,55 (2H, м).
МС (ХИАД) m/z: 855 (M+H)+
Способ 3: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-[2-(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)этил]глицинамид
Соединение (240 мг, 0,247 ммоль), полученное в способе 2 выше подвергают взаимодействию по методике способа 3 примера 2 с получением указанного в заголовке соединения (162 мг, 62%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,6 Гц), 1,13-1,22 (2H, м), 1,40-1,51 (4H, м), 1,78-1,90 (2H, м), 2,09 (2H, т, J=7,6 Гц), 2,14-2,21 (2H, м), 2,39 (3H, с), 2,74 (1H, дд, J=13,6, 9,7 Гц), 2,96 (1H, дд, J=13,6, 4,5 Гц), 3,08-3,24 (1H, м), 3,24-3,30 (1H, м), 3,33-3,40 (4H, м), 3,47-3,68 (7H, м), 3,72 (1H, дд, J=16,6, 5,7 Гц), 4,03 (2H, с), 4,42 (1H, тд, J=8,6, 4,2 Гц), 5,17 (1H, д, J=18,7 Гц), 5,25 (1H, д, J=18,7 Гц), 5,40 (1H, д, J=17,2 Гц), 5,44 (1H, д, J=17,2 Гц), 5,57-5,64 (1H, м), 6,52 (1H, с), 6,99 (2H, с), 7,13-7,25 (5H, м), 7,31 (1H, с), 7,74-7,81 (2H, м), 7,99 (1H, т, J=5,7 Гц), 8,03-8,11 (2H, м), 8,22 (1H, т, J=5,7 Гц), 8,47 (1H, д, J=9,1 Гц).
МС (ХИАД) m/z: 1048 (M+H)+
Способ 4: Конъюгат антитела с лекарственным средством (12)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 3, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 2 примера 6. Раствор затем концентрируют по общей методике A. Затем, с применением общей методики E, получают следующие характеристики.
Концентрация антитела: 10,77 мг/мл, выход антитела: 7,5 мг (60%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,7.
Пример 13. Конъюгат антитела с лекарственным средством (13)
[Формула 39]
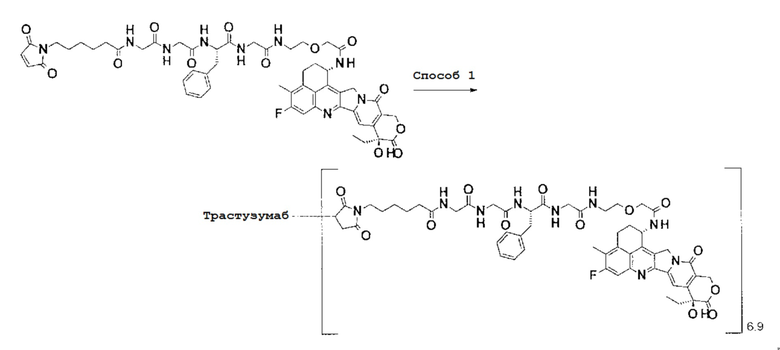
Способ 1: Конъюгат антитела с лекарственным средством (13)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 3 примера 12, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 примера 7. Раствор затем концентрируют по общей методике A. Затем, с применением общей методики E, получают следующие характеристики.
Концентрация антитела: 10,69 мг/мл, выход антитела: 7,5 мг (60%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,9.
Пример 14. Промежуточное соединение (14)
[Формула 40]
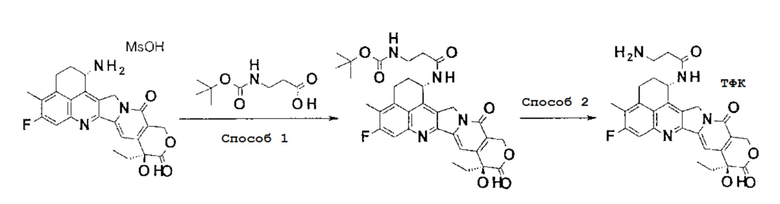
Способ 1: трет-Бутил (3-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-3-oxoпропил)карбамат
Соль метансульфоновой кислоты экзатекана (500 мг, 0,941 ммоль) подвергают взаимодействию по методике способа 1 из примера 1 с применением N-(трет-бутоксикарбонил)-β-аланина вместо 4-(трет-бутоксикарбониламино)бутановой кислоты с получением указанного в заголовке соединения в виде желто-коричневого твердого вещества (616 мг, количественный).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,2 Гц), 1,29 (9H, с), 1,86 (2H, дт, J=15,1, 7,3 Гц), 2,04-2,22 (2H, м), 2,31 (2H, т, J=6,8 Гц), 2,40 (3H, с), 3,10-3,26 (4H, м), 5,15 (1H, д, J=18,8 Гц), 5,26 (1H, д, J=19,2 Гц), 5,42 (2H, дд, J=18,8, 16,4 Гц), 5,57 (1H, дт, J=8,5, 4,2 Гц), 6,53 (1H, с), 6,78 (1H, т, J=5,5 Гц), 7,30 (1H, с), 7,80 (1H, д, J=11,0 Гц), 8,46 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 607 (M+H)+
Способ 2: N-[(1S,9S)-9-Этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-β-аланинамид
Соединение, полученное в способе 1 выше подвергают взаимодействию по методике способа 2 из примера 1 с получением соли трифторуксусной кислоты указанного в заголовке соединения в виде желтого твердого вещества (499 мг, 86%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,2 Гц), 1,86 (2H, дквин, J=14,6, 7,2, 7,2, 7,2, 7,2 Гц), 2,06-2,27 (1H, м), 2,41 (3H, с), 2,46-2,57 (2H, м), 3,08 (2H, т, J=6,8 Гц), 3,14-3,24 (2H, м), 5,22 (1H, д, J=18,8 Гц), 5,29 (1H, д, J=18,8 Гц), 5,43 (2H, с), 5,58 (1H, дт, J=8,5, 4,5 Гц), 6,55 (1H, с), 7,32 (1H, с), 7,74 (3H, шс), 7,82 (1H, д, J=11,0 Гц), 8,67 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 507 (M+H)+
Пример 15 Конъюгат антитела с лекарственным средством (15)
[Формула 41]
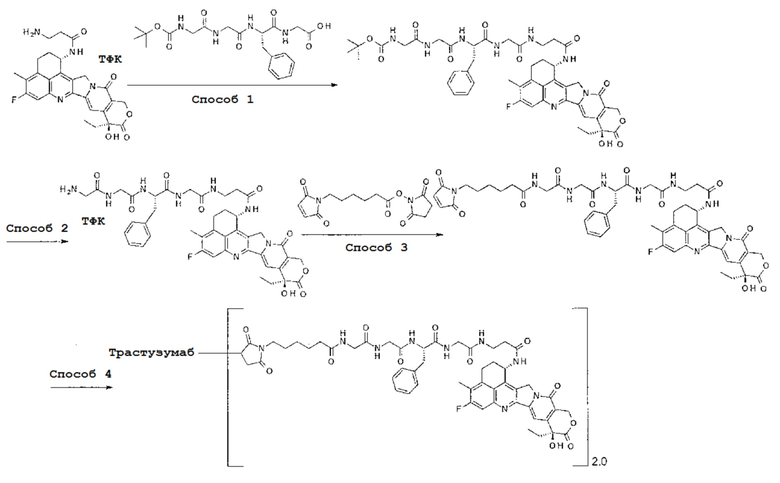
Способ 1: N-(трет-Бутоксикарбонил)глицилглицил-L-фенилаланилглицил-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-β-аланинамид
Соединение (484 мг, 0,780 ммоль), полученное в способе 2 из примера 14 подвергают взаимодействию по методике способа 1 из примера 2 с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (626 мг, 87%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,4 Гц), 1,27-1,42 (9H, м), 1,77-1,93 (2H, м), 2,06-2,22 (2H, м), 2,36 (2H, т, J=7,2 Гц), 2,40 (3H, д, J=1,6 Гц), 2,44-2,54 (2H, м), 2,76 (1H, дд, J=14,5, 10,2 Гц), 3,02 (1H, дд, J=13,9, 4,5 Гц), 3,12-3,22 (2H, м), 3,52 (6H, д, J=6,3 Гц), 4,42-4,54 (1H, м), 5,19 (1H, д, J=19,2 Гц), 5,26 (1H, д, J=18,4 Гц), 5,42 (1H, дд, J=18,4, 16,4 Гц), 5,57 (1H, дт, J=8,7, 4,4 Гц), 6,53 (1H, с), 6,98 (1H, т, J=5,9 Гц), 7,14-7,28 (5H, м), 7,31 (1H, с), 7,77-7,84 (1H, м), 7,91 (1H, т, J=5,5 Гц), 8,16 (1H, д, J=7,8 Гц), 8,27 (1H, т, J=5,1 Гц), 8,52 (1H, д, J=9,0 Гц).
Способ 2: Трифторацетат глицилглицил-L-фенилаланилглицил-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-β-аланинамида
Соединение (624 мг, 0,675 ммоль), полученное в способе 1 выше, подвергают взаимодействию по методике способа 2 из примера 2 с получением указанного в заголовке соединения в виде желтого твердого вещества (626 мг, 92%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,4 Гц), 1,86 (2H, tt, J=14,5, 7,2 Гц), 2,07-2,22 (2H, м), 2,36 (2H, т, J=7,2 Гц), 2,40 (3H, с), 2,44-2,54 (2H, м), 2,75 (1H, дд, J=13,7, 9,8 Гц), 3,04 (1H, дд, J=13,7, 4,3 Гц), 3,12-3,22 (2H, м), 3,58 (2H, д, J=4,7 Гц), 3,69 (3H, тд, J=11,2, 5,7 Гц), 3,87 (1H, дд, J=17,0, 5,7 Гц), 4,54 (1H, m, J=17,8, 4,5 Гц), 5,19 (1H, д, J=19,2 Гц), 5,26 (1H, д, J=18,8 Гц), 5,43 (2H, с), 5,51-5,60 (1H, м), 6,55 (1H, с), 7,14-7,29 (5H, м), 7,32 (1H, с), 7,81 (1H, д, J=10,9 Гц), 7,88 (1H, т, J=5,7 Гц), 7,97 (3H, шс), 8,29-8,38 (2H, м), 8,50 (1H, т, J=5,7 Гц), 8,55 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 825 (M+H)+
Способ 3: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланилглицил-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-β-аланинамид
Соединение (60,0 мг, 0,0646 ммоль), полученное в способе 2 выше, подвергают взаимодействию по методике способа 3 из примера 2 с получением указанного в заголовке соединения в виде твердого вещества (14,0 мг, 21%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,2 Гц), 1,12-1,22 (2H, м), 1,39-1,51 (4H, м), 1,79-1,91 (2H, м), 2,02-2,20 (2H, м), 2,07 (2H, т, J=7,4 Гц), 2,30-2,42 (4H, м), 2,40 (3H, с), 2,78 (1H, дд, J=14,1, 9,4 Гц), 3,02 (1H, дд, J=14,7, 4,9 Гц), 3,12-3,21 (2H, м), 3,26-3,42 (2H, м), 3,50-3,80 (6H, м), 4,40-4,51 (1H, м), 5,19 (1H, д, J=19,6 Гц), 5,26 (1H, д, J=19,2 Гц), 5,42 (2H, шс), 5,51-5,62 (1H, м), 6,53 (1H, с), 6,99 (2H, с), 7,13-7,28 (5H, м), 7,31 (1H, с), 7,74-7,84 (2H, м), 8,01 (1H, т, J=5,3 Гц), 8,06 (1H, т, J=5,7 Гц), 8,14 (1H, д, J=8,2 Гц), 8,25 (1H, т, J=5,7 Гц), 8,53 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 1018 (M+H)+
Способ 4: Конъюгат антитела с лекарственным средством (15)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1 получают с концентрацией антитела 10 мг/мл с ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,37 млмг-1см-1). Раствор (1,0 мл) собирают в 2 мл пробирку и загружают водным раствором 10 мМ ТКЭФ (0,0155 мл; 2,3 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,050 мл). После подтверждения того, что раствор имеет pH 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: после инкубирования раствора при 22°C в течение 10 минут туда добавляют раствор ДМСО (0,0311 мл; 4,6 эквивалента на молекулу антитела), содержащий 10 мМ соединения, полученного в способе 3 и инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 22°C в течение 40 минут. Затем добавляют водный раствор (0,00622 мл; 9,2 эквивалента на молекулу антитела) 100 мМ NAC и инкубируют при 22°C для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D-1 (ФРФБ6,0 применяют в качестве буферного раствора) с получением 6 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 1,18 мг/мл, выход антитела: 7,08 мг (71%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 2,0.
Пример 16. Конъюгат антитела с лекарственным средством (16)
[Формула 42]
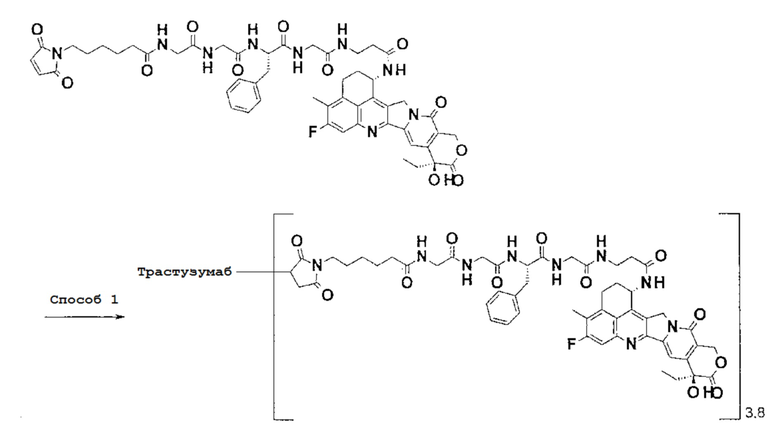
Способ 1: Конъюгат антитела с лекарственным средством (16)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл с ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,37 млмг-1см-1). Раствор (1,0 мл) собирают в 2 мл пробирку и загружают водным раствором 10 мМ ТКЭФ (0,0311 мл; 4,6 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,050 мл). После подтверждения того, что раствор имеет pH 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После инкубирования раствора при 22°C в течение 10 минут, туда добавляют раствор ДМСО (0,0622 мл; 9,2 эквивалента на молекулу антитела), содержащий 10 мМ соединения, полученного в способе 3 из примера 15, и инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 22°C в течение 40 минут. Затем добавляют водный раствор (0,0124 мл; 18,4 эквивалента на молекулу антитела) 100 мМ NAC и инкубируют при 22°C для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D-1 (ФРФБ6,0 применяют в качестве буферного раствора) с получением 6 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 1,03 мг/мл, выход антитела: 6,18 мг (62%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,8.
Пример 17. Конъюгат антитела с лекарственным средством (17)
[Формула 43]
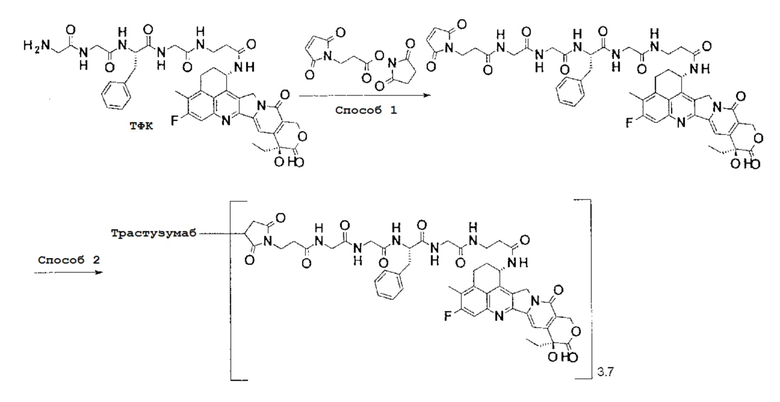
Способ 1: N-[3-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)пропаноил]глицилглицил-L-фенилаланилглицил-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-β-аланинамид
Соединение (60,0 мг, 0,0646 ммоль), полученное в способе 2 из примера 15, подвергают взаимодействию по методике способа 3 из примера 2 с применением пропионата N-сукцинимидил 3-малеимида вместо гексаноата N-сукцинимидил 6-малеимида с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (36,0 мг, 57%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,4 Гц), 1,85 (2H, дт, J=14,4, 7,5 Гц), 2,05-2,22 (2H, м), 2,40 (3H, с), 2,30-2,44 (5H, м), 2,73-2,84 (1H, м), 3,02 (1H, дд, J=13,9, 4,5 Гц), 3,17 (3H, д, J=5,1 Гц), 3,26-3,40 (2H, м), 3,41-3,81 (6H, м), 4,40-4,51 (1H, м), 5,19 (1H, д, J=19,2 Гц), 5,26 (1H, д, J=18,8 Гц), 5,42 (2H, шс), 5,52-5,61 (1H, м), 6,53 (1H, с), 6,99 (2H, с), 7,13-7,28 (5H, м), 7,31 (1H, с), 7,80 (2H, д, J=10,2 Гц), 8,03 (1H, т, J=5,5 Гц), 8,12 (1H, д, J=8,2 Гц), 8,20-8,31 (2H, м), 8,52 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 976 (M+H)+
Способ 2: Конъюгат антитела с лекарственным средством (17)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1 выше, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 2 из примера 6.
Концентрация антитела: 1,74 мг/мл, выход антитела: 10,4 мг (83%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,7.
Пример 18. Конъюгат антитела с лекарственным средством (18)
[Формула 44]
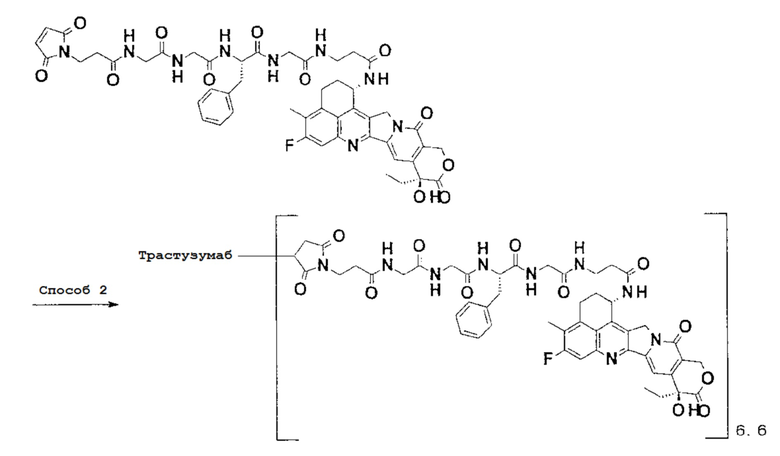
Способ 1: Конъюгат антитела с лекарственным средством (18)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1 из примера 17, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 из примера 7.
Концентрация антитела: 1,98 мг/мл, выход антитела: 11,9 мг (95%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,6.
Пример 19. Конъюгат антитела с лекарственным средством (19)
[Формула 45]
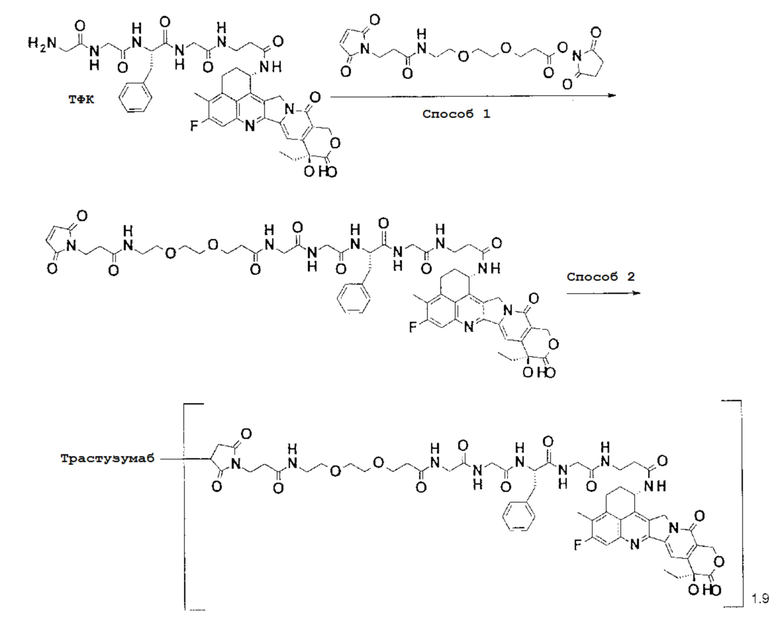
Способ 1: N-{3-[2-(2-{[3-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)пропаноил]амино})этокси]пропаноил}глицилглицил-L-фенилаланилглицил-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-β-аланинамид
Соединение (60,0 мг, 0,0646 ммоль), полученное в способе 2 из примера 15, подвергают взаимодействию по методике способа 3 из примера 2 с применением 3-(2-(2-(3-малеинимидпропанамид)этокси)этокси)пропаноата N-сукцинимидила вместо гексаноата N-сукцинимидил 6-малеимида с получением указанного в заголовке соединения в виде твердого вещества (23,0 мг, 31%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,4 Гц), 1,77-1,92 (2H, м), 2,07-2,21 (2H, м), 2,27-2,42 (6H, м), 2,40 (3H, с), 2,74-2,84 (1H, м), 2,97-3,06 (1H, м), 3,09-3,21 (4H, м), 3,25-3,39 (6H, м), 3,45 (4H, с), 3,50-3,80 (8H, м), 4,41-4,51 (1H, м), 5,19 (1H, д, J=18,4 Гц), 5,26 (1H, m, J=18,4 Гц), 5,42 (2H, шс), 5,51-5,61 (1H, м), 6,54 (1H, с), 7,00 (2H, с), 7,13-7,28 (5H, м), 7,31 (1H, с), 7,74-7,87 (2H, м), 7,93-8,07 (2H, м), 8,09-8,21 (2H, м), 8,26 (1H, шс), 8,54 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 1135 (M+H)+
Способ 2: Конъюгат антитела с лекарственным средством (19)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1 выше, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 2 из примера 6.
Концентрация антитела: 1,60 мг/мл, выход антитела: 9,6 мг (77%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 1,9.
Пример 20. Конъюгат антитела с лекарственным средством (20)
[Формула 46]
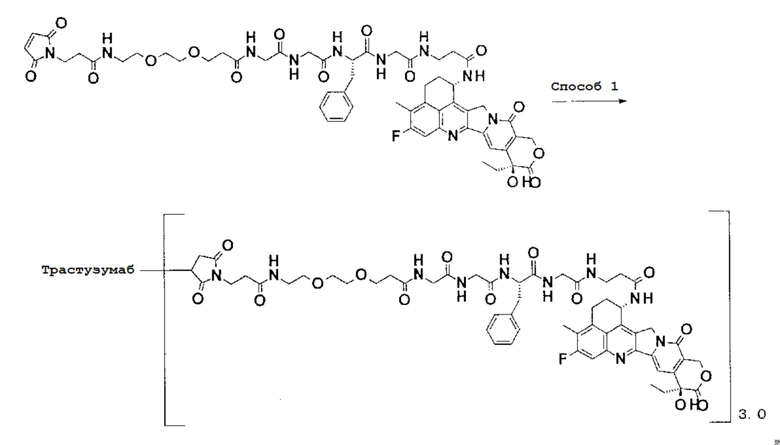
Способ 1: Конъюгат антитела с лекарственным средством (20)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1 из примера 19, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 из примера 7.
Концентрация антитела: 1,69 мг/мл, выход антитела: 10,1 мг (81%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,0.
Пример 21. Конъюгат антитела с лекарственным средством (21)
[Формула 47]
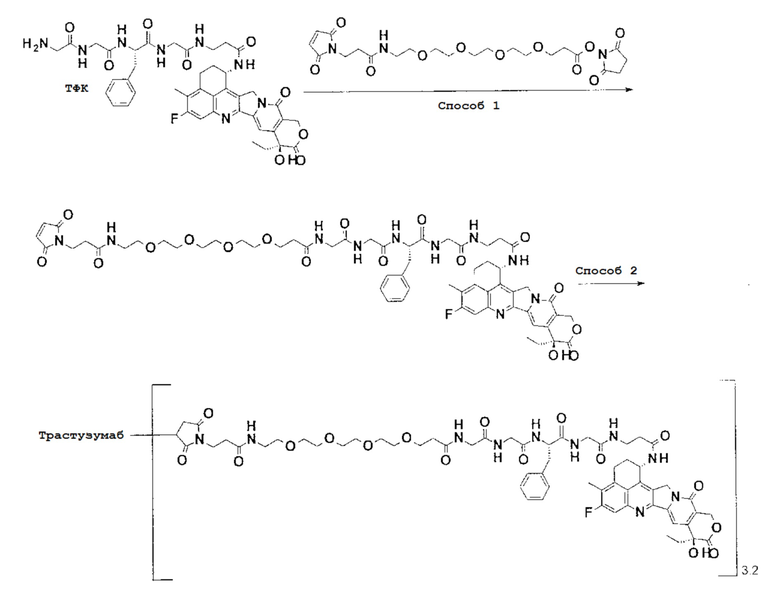
Способ 1: N-[19-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраокса-16-азанонадекан-1-оил]глицилглицил-L-фенилаланилглицил-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-β-аланинамид
Соединение (60,0 мг, 0,0646 ммоль), полученное в способе 2 из примера 15, подвергают взаимодействию по методике способа 3 из примера 2 с применением 1-малеинимид-3-оксо-7,10,13,16-тетраокса-4-азанонадеканоата N-сукцинимидил вместо гексаноата N-сукцинимидил 6-малеимида с получением указанного в заголовке соединения в виде твердого вещества (23,0 мг, 29%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,0 Гц), 1,85 (2H, tt, J=14,6, 7,1 Гц), 2,06-2,22 (2H, м), 2,40 (3H, с), 2,28-2,43 (6H, м), 2,78 (1H, дд, J=13,7, 9,4 Гц), 3,02 (1H, дд, J=14,1, 3,9 Гц), 3,09-3,22 (4H, м), 3,27-3,41 (4H, м), 3,47 (12H, д, J=8,6 Гц), 3,53-3,81 (10H, м), 4,41-4,51 (1H, м), 5,19 (1H, д, J=19,2 Гц), 5,26 (1H, д, J=18,8 Гц), 5,42 (2H, шс), 5,53-5,61 (1H, м), 6,54 (1H, с), 7,00 (2H, с), 7,12-7,29 (5H, м), 7,31 (1H, с), 7,74-7,85 (2H, м), 8,03 (2H, д, J=6,6 Гц), 8,11-8,21 (2H, м), 8,27 (1H, т, J=5,9 Гц), 8,54 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 1224 (M+H)+
Способ 2: Конъюгат антитела с лекарственным средством (21)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 2 из примера 6.
Концентрация антитела: 1,77 мг/мл, выход антитела: 10,6 мг (85%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,2.
Пример 22. Конъюгат антитела с лекарственным средством (22)
[Формула 48]
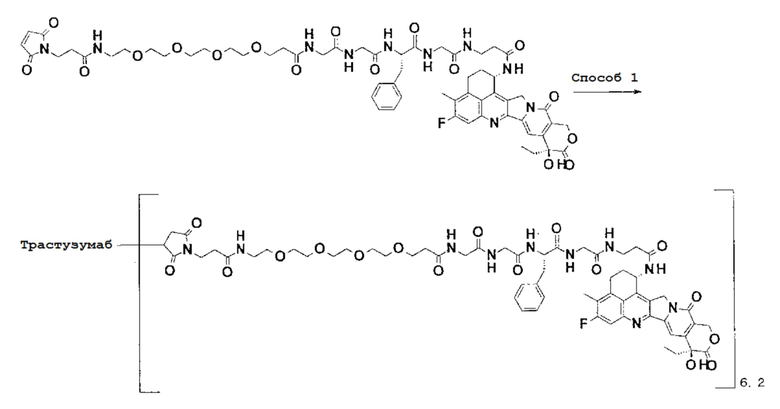
Способ 1: Конъюгат антитела с лекарственным средством (22)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1 из примера 21, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 из примера 7.
Концентрация антитела: 1,89 мг/мл, выход антитела: 11,3 мг (90%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,2.
Пример 23. Промежуточное соединение (23)
[Формула 49]
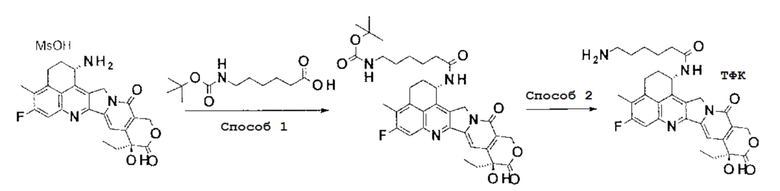
Способ 1: трет-Бутил (6-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-6-оксогексил)карбамат
Соль метансульфоновой кислоты экзатекана (0,500 г, 0,882 ммоль) подвергают взаимодействию по методике способа 1 из примера 1 с применением 6-(трет-бутоксикарбониламино)гексановой кислоты вместо 4-(трет-бутоксикарбониламино)бутановой кислоты с получением указанного в заголовке соединения (0,620 г, количественный).
1H-ЯМР (ДМСО-d6) δ: 0,83 (3H, т, J=7,8 Гц), 1,14-1,28 (2H, м), 1,31 (9H, с), 1,47-1,61 (2H, м), 1,75-1,89 (2H, м), 2,04-2,17 (4H, м), 2,35 (3H, с), 2,81-2,88 (2H, м), 3,09-3,16 (2H, м), 5,10 (1H, д, J=19,4 Гц), 5,16 (1H, д, J=19,4 Гц), 5,39 (2H, с), 5,48-5,55 (1H, м), 6,50 (1H, с), 6,73-6,78 (1H, м), 7,26 (1H, с), 7,74 (1H, д, J=10,9 Гц), 8,39 (1H, д, J=9,0 Гц).
Способ 2: 6-Амино-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]гексанамид
Соединение (0,397 г, 0,611 ммоль), полученное в способе 1 выше, подвергают взаимодействию по методике способа 2 из примера 1 с получением соли трифторуксусной кислоты указанного в заголовке соединения (0,342 г, 84%).
1H-ЯМР (ДМСО-d6) δ: 0,88 (3H, т, J=7,2 Гц), 1,31-1,41 (2H, м), 1,52-1,70 (4H, м), 1,80-1,94 (2H, м), 2,05-2,18 (2H, м), 2,21 (2H, т, J=7,4 Гц), 2,40 (3H, с), 2,81 (2H, т, J=7,4 Гц), 3,10-3,25 (2H, м), 3,33 (2H, шс), 5,18 (1H, д, J=19,8 Гц), 5,22 (1H, д, J=19,8 Гц), 5,41 (2H, д, J=16,6 Гц), 5,45 (2H, д, J=16,6 Гц), 5,53-5,60 (1H, м), 6,55 (1H, с), 7,32 (1H, с), 7,80 (1H, д, J=10,9 Гц), 8,49 (1H, д, J=9,2 Гц).
Пример 24. Конъюгат антитела с лекарственным средством (24)
[Формула 50]
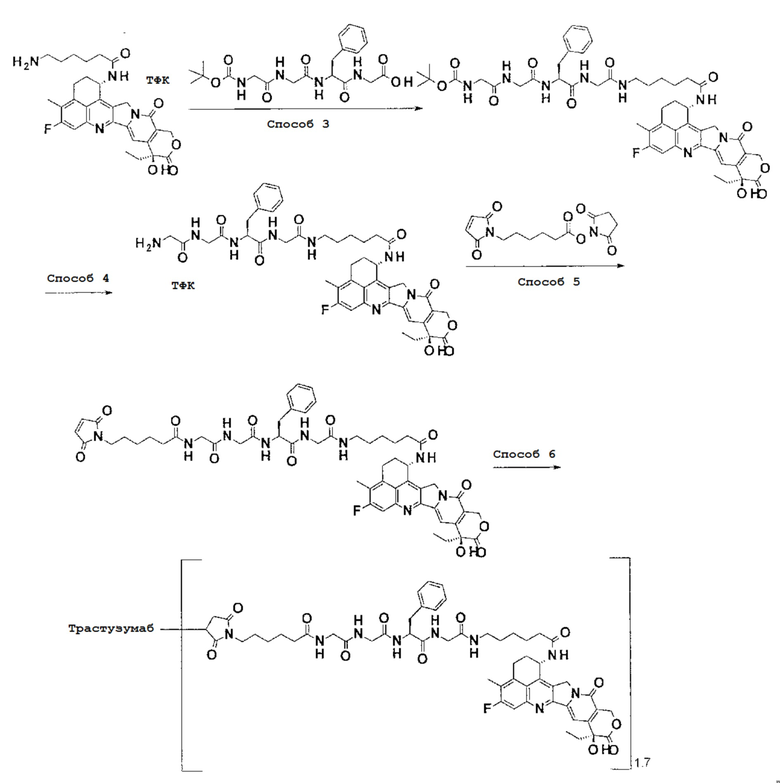
Способ 1: N-(трет-Бутоксикарбонил)глицилглицил-L-фенилаланил-N-(6-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-6-оксогексил)глицинамид
Соединение (0,170 г, 0,516 ммоль), полученное в способе 2 из примера 23, подвергают взаимодействию по методике способа 1 из примера 2 с получением указанного в заголовке соединения (0,225 г, 91%).
1H-ЯМР (ДМСО-d6) δ: 0,88 (3H, т, J=7,4 Гц), 1,43-1,70 (6H, м), 1,87 (2H, тд, J=15,0, 7,4 Гц), 2,10-2,22 (3H, м), 2,28-2,37 (1H, м), 2,42 (3H, с), 2,78-2,85 (1H, м), 3,01-3,10 (3H, м), 3,15-3,22 (2H, м), 3,54-3,61 (5H, м), 3,62-3,69 (1H, м), 4,44-4,53 (1H, м), 5,17 (1H, д, J=19,2 Гц), 5,25 (1H, д, J=19,2 Гц), 5,45 (2H, с), 5,54-5,61 (1H, м), 6,55 (1H, с), 7,02 (1H, т, J=6,1 Гц), 7,11-7,28 (5H, м), 7,33 (1H, с), 7,63-7,69 (1H, м), 7,82 (1H, д, J=11,0 Гц), 7,90-7,96 (1H, м), 8,17 (1H, д, J=7,8 Гц), 8,28 (1H, т, J=5,5 Гц), 8,46 (1H, д, J=9,0 Гц).
Способ 2: Глицилглицил-L-фенилаланил-N-(6-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-6-оксогексил)глицинамид
Соединение (0,105 г, 0,108 ммоль), полученное в способе 1 выше, подвергают взаимодействию по методике способа 2 из примера 2 с получением указанного в заголовке соединения (0,068 мг, 65%).
1H-ЯМР (ДМСО-d6) δ: 0,89 (3H, т, J=7,4 Гц), 1,15-1,67 (6H, м), 1,79-1,97 (2H, м), 2,08-2,24 (4H, м), 2,42 (3H, с), 2,76-2,82 (1H, м), 3,00-3,10 (5H, м), 3,19 (1H, с), 3,50-3,63 (2H, м), 3,64-3,76 (3H, м), 3,84-3,92 (1H, м), 4,51-4,59 (1H, м), 5,17 (1H, д, J=19,4 Гц), 5,24 (1H, д, J=19,4 Гц), 5,44 (2H, с), 5,53-5,61 (1H, м), 6,55 (1H, шс), 7,15-7,29 (5H, м), 7,33 (1H, с), 7,72-7,78 (1H, м), 7,82 (1H, д, J=11,0 Гц), 7,96-8,08 (2H, м), 8,30-8,38 (2H, м), 8,46-8,56 (2H, м).
Способ 3: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-(6-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-6-оксогексил)глицинамид
Соединение (58 мг, 0,060 ммоль), полученное в способе 2 выше, подвергают взаимодействию по методике способа 3 из примера 2 с получением указанного в заголовке соединения (39 мг, 62%).
1H-ЯМР (CD3OD) δ: 0,99 (3H, т, J=7,4 Гц), 1,27 (2H, тд, J=11,6, 6,1 Гц), 1,38-1,44 (2H, м), 1,50-1,63 (6H, м), 1,65-1,80 (2H, м), 1,89-1,98 (2H, м), 2,17-2,25 (3H, м), 2,26-2,36 (3H, м), 2,40 (3H, с), 2,95 (1H, дд, J=14,3, 9,2 Гц), 3,12 (1H, дд, J=13,7, 5,7 Гц), 3,15-3,25 (4H, м), 3,44 (2H, т, J=7,2 Гц), 3,65 (1H, д, J=17,2 Гц), 3,76 (1H, д, J=17,2 Гц), 3,79-3,86 (4H, м), 4,43 (1H, дд, J=8,9, 6,0 Гц), 5,10 (1H, д, J=18,9 Гц), 5,25 (1H, д, J=18,9 Гц), 5,35 (1H, д, J=16,6 Гц), 5,56 (1H, д, J=16,0 Гц), 5,60-5,64 (1H, м), 6,76 (2H, с), 7,12-7,24 (6H, м), 7,58 (1H, с), 7,60 (1H, д, J=10,9 Гц), 7,68 (1H, т, J=5,7 Гц).
МС (ИЭР) m/z: 1060 (M+H)+
Способ 4: Конъюгат антитела с лекарственным средством (24)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл с ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,37 млмг-1см-1). Раствор (9,0 мл) собирают в 50 мл пробирку и загружают водным раствором 10 мМ ТКЭФ (0,140 мл; 2,3 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,450 мл). После подтверждения того, что раствор имеет pH 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После инкубирования раствора при 22°C в течение 10 минут, добавляют раствор ДМСО (0,280 мл; 4,6 эквивалента на молекулу антитела), содержащий 10 мМ соединения из способа 3 и инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 22°C в течение 40 минут. Затем добавляют водный раствор (0,0559 мл; 9,2 эквивалента на молекулу антитела) 100 мМ NAC и инкубируют при 22°C для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D-1 (ФРФБ7,4 применяют в качестве буферного раствора) с получением раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 3,30 мг/мл, выход антитела: 53,5 мг (59%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 1,7.
Пример 25 Конъюгат антитела с лекарственным средством (25)
[Формула 51]
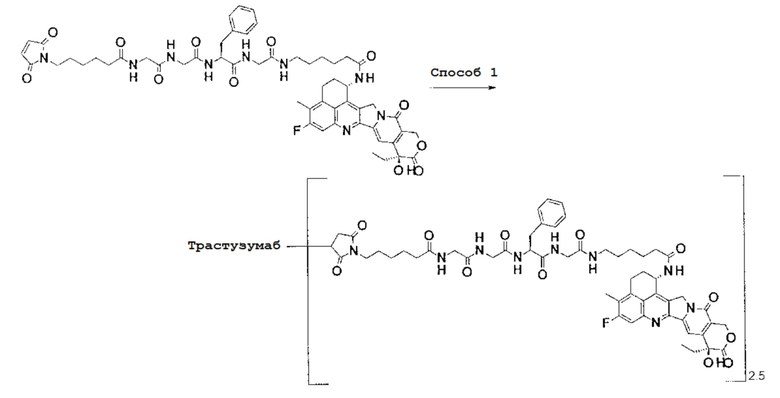
Способ 1: Конъюгат антитела с лекарственным средством (25)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл с ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,37 млмг-1см-1). Раствор (9,0 мл) собирают в 50 мл пробирку и загружают водным раствором 10 мМ ТКЭФ (0,280 мл; 4,6 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,450 мл). После подтверждения того, что раствор имеет pH 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После инкубирования раствора при 22°C в течение 10 минут, добавляют раствор ДМСО (0,559 мл; 9,2 эквивалента на молекулу антитела), содержащий 10 мМ соединения из способа 3 из примера 24, и инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 22°C в течение 40 минут. Затем добавляют водный раствор (0,112 мл; 18,4 эквивалента на молекулу антитела) 100 мМ NAC и инкубируют при 22°C для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D-1 (ФРФБ6,0 применяют в качестве буферного раствора) с получением раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 10,65 мг/мл, выход антитела: 55,1 мг (61%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 2,5.
Пример 26. Конъюгат антитела с лекарственным средством (26)
[Формула 52]
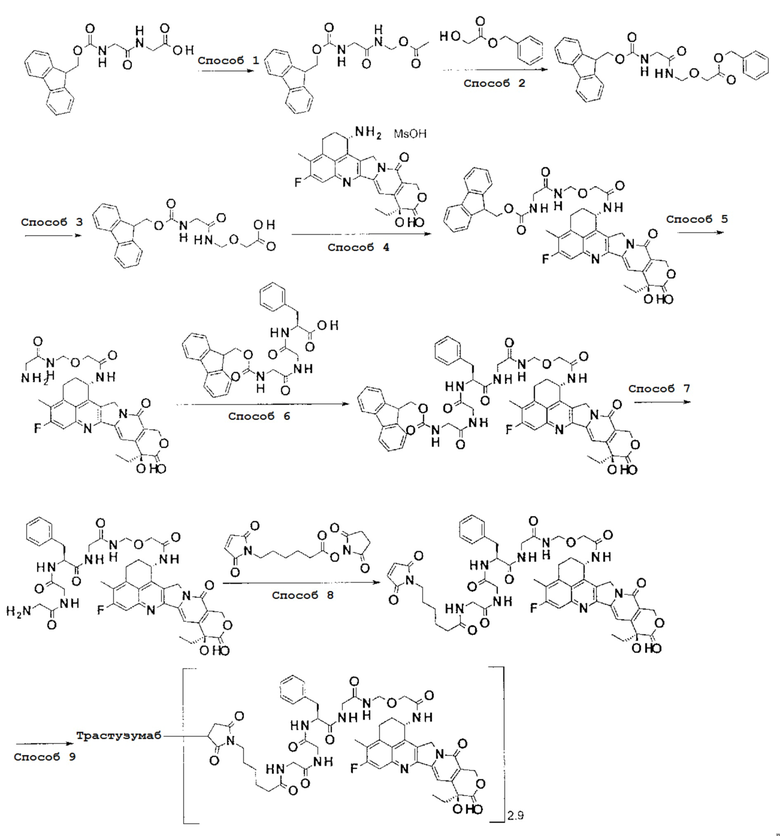
Способ 1: Ацетат ({N-[(9H-флуорен-9-илметокси)карбонил]глицил}амино)метила
К смеси, содержащей N-9-флуоренилметоксикарбонилглицилглицин (4,33 г, 12,2 ммоль), тетрагидрофуран (ТГФ; 120 мл) и толуол (40,0 мл), добавляют пиридин (1,16 мл, 14,7 ммоль) и тетраацетат свинца (6,84 г, 14,7 ммоль) и нагревают с обратным холодильником в течение 5 часов. Затем реакционный раствор охлаждают до комнатной температуры, нерастворимые вещества удаляют фильтрацией через Целит и концентрируют при пониженном давлении. Полученные остатки растворяют в этилацетате и промывают водой и насыщенным раствором соли и затем органический слой сушат над безводным сульфатом магния. Затем растворитель удаляют при пониженном давлении, полученные остатки очищают хроматографией на колонке с силикагелем [гексан:этилацетат=9:1 (об./об.)-этилацетат] с получением указанного в заголовке соединения в виде бесцветного твердого вещества (3,00 г, 67%).
1H-ЯМР (400 МГц, CDCl3) δ: 2,07 (3H, с), 3,90 (2H, д, J=5,1 Гц), 4,23 (1H, т, J=7,0 Гц), 4,46 (2H, д, J=6,6 Гц), 5,26 (2H, д, J=7,0 Гц), 5,32 (1H, шс), 6,96 (1H, шс), 7,32 (2H, т, J=7,3 Гц), 7,41 (2H, т, J=7,3 Гц), 7,59 (2H, д, J=7,3 Гц), 7,77 (2H, д, J=7,3 Гц).
Способ 2: Бензил [({N-[(9H-флуорен-9-илметокси)карбонил]глицил}амино)метокси]ацетат
К раствору в ТГФ (40,0 мл) соединения (3,68 г, 10,0 ммоль), полученного в способе 1 выше, и гликолята бензила (4,99 г, 30,0 ммоль) добавляют трет-бутоксид калия (2,24 г, 20,0 ммоль) при 0°C и перемешивают при комнатной температуре в течение 15 минут. В реакционный раствор загружают этилацетат и воду при 0°C и экстрагируют этилацетатом и хлороформом. Полученный органический слой сушат над сульфатом натрия и фильтруют. Растворитель удаляют при пониженном давлении. Полученные остатки растворяют в диоксане (40,0 мл) и воде (10,0 мл), загружают гидрокарбонатом натрия (1,01 г, 12,0 ммоль) и 9-флуоренилметилхлороформиатом (2,59 г, 10,0 ммоль) и перемешивают при комнатной температуре в течение 2 часов. В реакционный раствор загружают водой и экстрагируют этилацетатом. Полученный органический слой сушат над сульфатом натрия и фильтруют. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [гексан:этилацетат=100:0 (об./об.)-0:100] с получением указанного в заголовке соединения в виде бесцветного маслянистого вещества (1,88 г, 40%).
1H-ЯМР (400 МГц, CDCl3) δ: 3,84 (2H, д, J=5,5 Гц), 4,24 (3H, т, J=6,5 Гц), 4,49 (2H, д, J=6,7 Гц), 4,88 (2H, д, J=6,7 Гц), 5,15-5,27 (1H, м), 5,19 (2H, с), 6,74 (1H, шс), 7,31-7,39 (7H, м), 7,43 (2H, т, J=7,4 Гц), 7,61 (2H, д, J=7,4 Гц), 7,79 (2H, д, J=7,4 Гц).
Способ 3: [({N-[(9H-Флуорен-9-илметокси)карбонил]глицил}амино)метокси]уксусная кислота
Соединение (1,88 г, 3,96 ммоль), полученное в способе 2 выше, растворяют в этаноле (40,0 мл) и этилацетате (20,0 мл). После добавления катализатора на основе палладия на угле (376 мг), раствор перемешивают в атмосфере водорода при комнатной температуре в течение 2 часов. Нерастворимые вещества удаляют фильтрацией через Целит, и растворитель удаляют при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (1,52 г, количественный).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 3,62 (2H, д, J=6,3 Гц), 3,97 (2H, с), 4,18-4,32 (3H, м), 4,60 (2H, д, J=6,7 Гц), 7,29-7,46 (4H, м), 7,58 (1H, т, J=5,9 Гц), 7,72 (2H, д, J=7,4 Гц), 7,90 (2H, д, J=7,4 Гц), 8,71 (1H, т, J=6,5 Гц).
Способ 4: 9H-Флуорен-9-илметил(2-{[(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)метил]амино}-2-оксоэтил)карбамат
При охлаждении на льду к раствору в N,N-диметилформамиде (10,0 мл) соли метансульфоновой кислоты экзатекана (0,283 г, 0,533 ммоль), N-гидроксисукцинимида (61,4 мг, 0,533 ммоль) и соединения (0,205 г, 0,533 ммоль), полученного в способе 3 выше, добавляют N,N-диизопропилэтиламин (92,9 мкл, 0,533 ммоль) и N,N'-дициклогексилкарбодиимид (0,143 г, 0,693 ммоль) и перемешивают при комнатной температуре в течение 3 дней. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением указанного в заголовке соединения в виде бледно-коричневого твердого вещества (0,352 г, 82%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,81 (3H, т, J=7,4 Гц), 1,73-1,87 (2H, м), 2,06-2,20 (2H, м), 2,34 (3H, с), 3,01-3,23 (2H, м), 3,58 (2H, д, J=6,7 Гц), 3,98 (2H, с), 4,13-4,25 (3H, м), 4,60 (2H, д, J=6,7 Гц), 5,09-5,22 (2H, м), 5,32-5,42 (2H, м), 5,50-5,59 (1H, м), 6,49 (1H, с), 7,24-7,30 (3H, м), 7,36 (2H, т, J=7,4 Гц), 7,53 (1H, т, J=6,3 Гц), 7,66 (2H, д, J=7,4 Гц), 7,75 (1H, д, J=11,0 Гц), 7,84 (2H, д, J=7,4 Гц), 8,47 (1H, д, J=8,6 Гц), 8,77 (1H, т, J=6,7 Гц).
МС (ИЭР) m/z: 802 (M+H)+
Способ 5: N-[(2-{[(1S,9S)-9-Этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)метил]глицинамид
К раствору в N,N-диметилформамиде (11,0 мл) соединения (0,881 г, 1,10 ммоль), полученного в способе 4 выше, добавляют пиперидин (1,1 мл) и перемешивают при комнатной температуре в течение 2 часов. Растворитель удаляют при пониженном давлении с получением смеси, содержащей указанное в заголовке соединение. Смесь применяют в следующей реакции без дальнейшей очистки.
Способ 6: N-[(9H-Флуорен-9-илметокси)карбонил]глицилглицил-L-фенилаланил-N-[(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)метил]глицинамид
При охлаждении на льду, к раствору в N,N-диметилформамиде (50,0 мл) смеси (0,439 ммоль), полученной в способе 5 выше, добавляют N-гидроксисукцинимид (0,101 г, 0,878 ммоль) и N-[(9H-флуорен-9-илметокси)карбонил]глицилглицил-L-фенилаланин (соединение, описанное в выложенном патенте Японии № 2002-60351; 0,440 г, 0,878 ммоль), N,N'-дициклогексилкарбодиимид (0,181 г, 0,878 ммоль) и перемешивают при комнатной температуре в течение 4 дней. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=9:1 (об./об.)] с получением указанного в заголовке соединения в виде бледно-оранжевого твердого вещества (0,269 г, 58%).
МС (ИЭР) m/z: 1063 (M+H)+
Способ 7: Глицилглицил-L-фенилаланил-N-[(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)метил]глицинамид
К раствору в N,N-диметилформамиде (4,00 мл) соединения (0,269 г, 0,253 ммоль), полученного в способе 6 выше, добавляют пиперидин (0,251 мл, 2,53 ммоль) и перемешивают при комнатной температуре в течение 2 часов. Растворитель удаляют при пониженном давлении с получением смеси, содержащей указанное в заголовке соединение. Смесь применяют в следующей реакции без дальнейшей очистки.
Способ 8: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-[(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)метил]глицинамид
К раствору в N,N-диметилформамиде (10,0 мл) соединения (0,253 ммоль), полученного в способе 7 выше, добавляют гексаноат N-сукцинимидил 6-малеимида (0,156 г, 0,506 ммоль) и перемешивают при комнатной температуре в течение 3 дней. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=9:1 (об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,100 г, 38%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,83 (3H, т, J=7,2 Гц), 1,09-1,21 (2H, м), 1,33-1,47 (4H, м), 1,75-1,90 (2H, м), 2,00-2,23 (4H, м), 2,36 (3H, с), 2,69-2,81 (1H, м), 2,94-3,03 (1H, м), 3,06-3,22 (2H, м), 3,23-3,74 (6H, м), 3,98 (2H, с), 4,39-4,50 (1H, м), 4,60 (2H, д, J=6,7 Гц), 5,17 (2H, с), 5,39 (2H, с), 5,53-5,61 (1H, м), 6,50 (1H, с), 6,96 (2H, с), 7,11-7,24 (5H, м), 7,28 (1H, с), 7,75 (1H, д, J=11,0 Гц), 7,97 (1H, т, J=5,7 Гц), 8,03 (1H, т, J=5,9 Гц), 8,09 (1H, д, J=7,8 Гц), 8,27 (1H, т, J=6,5 Гц), 8,48 (1H, д, J=9,0 Гц), 8,60 (1H, т, J=6,5 Гц).
МС (ИЭР) m/z: 1034 (M+H)+
Способ 9: Конъюгат антитела с лекарственным средством (26)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 8 выше, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 2 из примера 6.
Концентрация антитела: 1,61 мг/мл, выход антитела: 9,7 мг (77%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 2,9.
Пример 27. Конъюгат антитела с лекарственным средством (27)
[Формула 53]
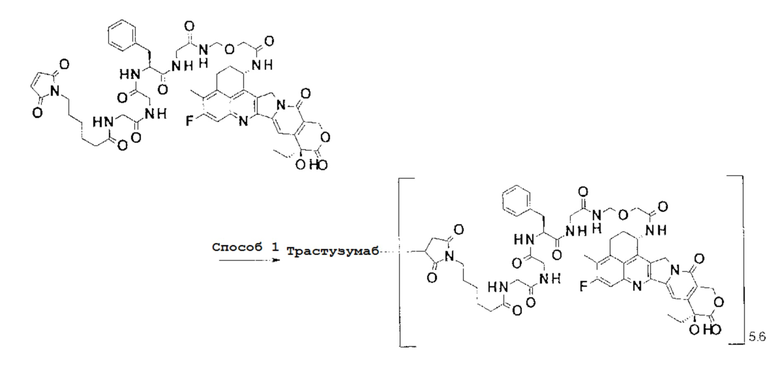
Способ 1: Конъюгат антитела с лекарственным средством (27)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 8 из примера 26, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 из примера 7.
Концентрация антитела: 1,58 мг/мл, выход антитела: 9,5 мг (76%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 5,6.
Пример 28. Конъюгат антитела с лекарственным средством (28)
[Формула 54]
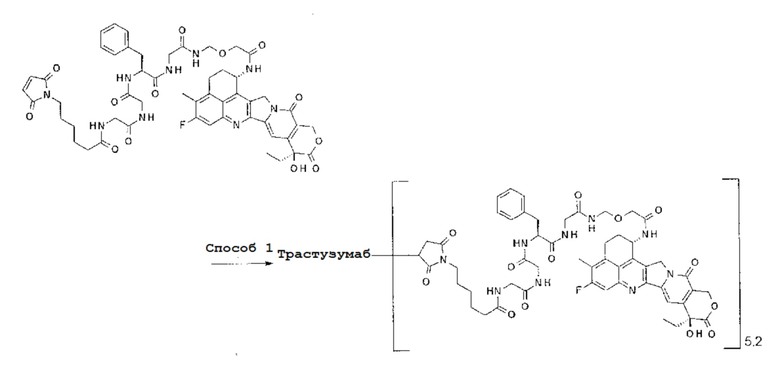
Способ 1: Конъюгат антитела с лекарственным средством (28)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (1,25 мл) помещают в две 1,5 мл полипропиленовых пробирки и загружают водным раствором 10 мМ ТКЭФ (0,039 мл; 4,6 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,0625 мл). После подтверждения того, что раствор имеет pH 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После добавления ДМСО (0,072 мл) и раствора ДМСО, содержащего 10 мМ соединения из способа 8 из примера 26 (0,078 мл; 9,2 эквивалента на молекулу антитела) к полученному выше раствору при комнатной температуре, его перемешивают с применением трубчатого ротатора для конъюгирования связующей группы лекарственного средства с антителом при комнатной температуре в течение 40 минут. Затем добавляют водный раствор 100 мМ NAC (0,0155 мл) и перемешивают при комнатной температуре для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D-1 (ABS применяют в качестве буферного раствора) с получением всего 11,7 мл раствора, содержащего целевое соединение.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 1,60 мг/мл, выход антитела: 18,7 мг (94%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 5,2.
Пример 29. Конъюгат антитела с лекарственным средством (29)
[Формула 55]
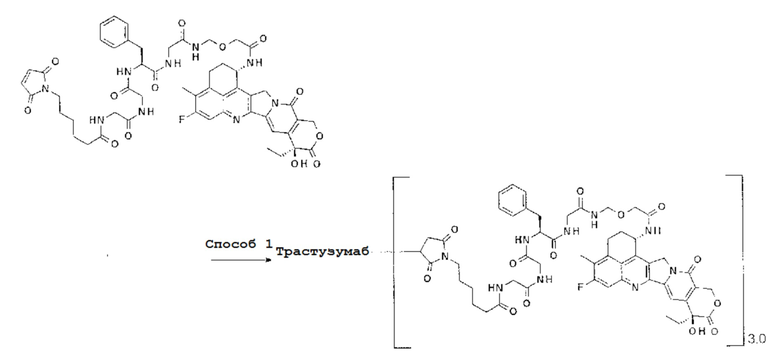
Способ 1: Конъюгат антитела с лекарственным средством (29)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (6 мл) помещают в полипропиленовую пробирку и загружают водным раствором 10 мМ ТКЭФ (0,108 мл; 2,5 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,091 мл). После подтверждения того, что раствор имеет pH 7,0±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После добавления ДМСО (0,146 мл) и раствора ДМСО, содержащего 10 мМ соединения из способа 8 из примера 26 (0,193 мл; 4,5 эквивалента на молекулу антитела) к полученному выше раствору при комнатной температуре, раствор инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 15°C в течение 1 часа. Затем добавляют водный раствор (0,029 мл) 100 мМ NAC и перемешивают при комнатной температуре для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D (ABS применяют в качестве буферного раствора) с получением 24 мл раствора, содержащего целевое соединение.
Физико-химические характеристики: С применением общих методик E и F (применяют εD,280=5178 (измеренное значение) и εD,370=20217 (измеренное значение)), получают следующие характеристики.
Концентрация антитела: 1,77 мг/мл, выход антитела: 42 мг (85%), среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела, измеренное с применением общей методики E: 3,0 и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела, измеренное с применением общей методики F: 3,4.
Пример 30. Конъюгат антитела с лекарственным средством (30)
[Формула 56]
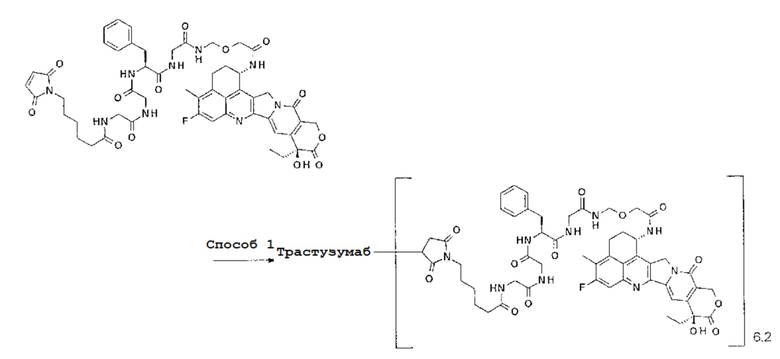
Способ 1: Конъюгат антитела с лекарственным средством (30)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (6 мл) помещают в полипропиленовую пробирку и загружают водным раствором 10 мМ ТКЭФ (0,215 мл; 5 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,094 мл). После подтверждения того, что раствор имеет pH 7,0±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После добавления раствора ДМСО, содержащего 10 мМ соединения из способа 8 из примера 26 (0,370 мл; 8,6 эквивалента на молекулу антитела), к полученному выше раствору при комнатной температуре, его инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 15°C в течение 1 часа. Затем добавляют водный раствор (0,056 мл) 100 мМ NAC и перемешивают при комнатной температуре для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D (ABS применяют в качестве буферного раствора) с получением 24 мл раствора, содержащего целевое соединение.
Физико-химические характеристики: С применением общих методики E и F (применяют εD,280=5178 (измеренное значение) и εD,370=20217 (измеренное значение)), получают следующие характеристики.
Концентрация антитела: 1,92 мг/мл, выход антитела: 46 мг (92%), среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела, измерено с применением общей методики E: 6,2 и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела измерено с применением общей методики F: 7,1.
Пример 31. Конъюгат антитела с лекарственным средством (31)
[Формула 57]
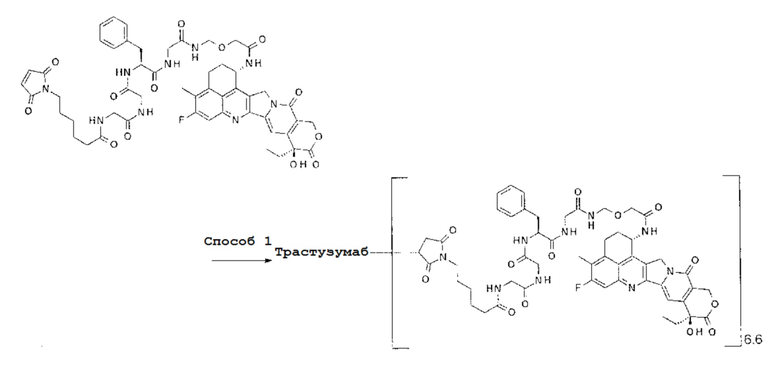
Способ 1: Конъюгат антитела с лекарственным средством (31)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (50,00 мл) помещают в полипропиленовый контейнер и загружают водным раствором 1M вторичного кислого фосфата калия (0,745 мл) при комнатной температуре при перемешивании, и затем водным раствором 10 мМ ТКЭФ (1,868 мл; 5,4 эквивалента на молекулу антитела). После подтверждения того, что раствор имеет pH 7,0±0,1, перемешивание заканчивают и дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После охлаждения полученного выше раствора до 15°C, туда постепенно по каплям добавляют раствор ДМСО, содержащий 10 мМ соединения из способа 8 из примера 26 (2,958 мл; 8,6 эквивалента на молекулу антитела) при перемешивании. Сохраняя температуру на уровне 15°C, реакционный раствор перемешивают в течение первых 30 минут и инкубируют без перемешивания для конъюгирования связующей группы лекарственного средства с антителом в течение следующего 1 часа. Затем добавляют водный раствор (0,444 мл) 100 мМ NAC при перемешивании и перемешивают при комнатной температуре для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Постепенным добавлением 20% водного раствора уксусной кислоты (около 0,25 мл) и ABS (50 мл) к полученному выше раствору при перемешивании pH раствора доводят до 5,5±0,1. Этот раствор подвергают микрофильтрации (Millipore Corp., фильтр Millex-HV, 0,45 мкм, мембрана ПВДФ) для удаления беловатого вещества. Этот раствор подвергают ультрафильтрационной очистке с применением аппарата для ультрафильтрации, состоящего из ультрафильтрационной мембраны (Merck Japan, Pellicon XL Cassette, Biomax 50 КДa), трубчатого насоса (Cole-Parmer International, MasterFlex Pump модель 77521-40, Pump Head модель 7518-00) и трубки (Cole-Parmer International, MasterFlex Tube L/S16). Более конкретно, добавляя по каплям ABS (всего 800 мл) в качестве буферного раствора для очистки реакционного раствора, ультрафильтрационную очистку проводят для удаления не конъюгированных связующих групп лекарственного средства и других низкомолекулярных реагентов, также заменяя буферный раствор на ABS и затем на концентрирующий раствор. Полученный очищенный раствор подвергают микрофильтрации (0,22 мкм (Millipore Corp., фильтр Millex-GV, мембрана ПВДФ) и 0,10 мкм (Millipore Corp., фильтр Millex-VV, мембрана ПВДФ)) с получением раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общих методик E и F (применяют εD,280=5178 (измеренное значение) и εD,370=20217 (измеренное значение)), получают следующие характеристики.
Концентрация антитела: 11,28 мг/мл, выход антитела: 451 мг (90%), среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела измерено с применением общей методики E: 6,6 и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела измерено с применением общей методики F: 7,7.
Пример 32 (Альтернативный способ синтеза соединения из способа 8 из примера 26)
[Формула 58]
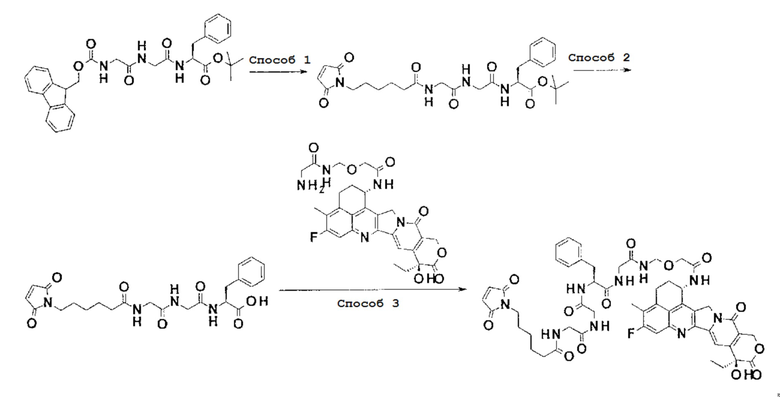
Способ 1: трет-Бутил N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланинат
При охлаждении на льду к раствору в ТГФ (12,0 мл) трет-бутил N-[(9H-флуорен-9-илметокси)карбонил]глицилглицил-L-фенилаланината (J. Pept. Res., 1999, vol. 53, pp. 393; 0,400 г, 0,717 ммоль) добавляют 1,8-диазабицикло[5,4,0]-7-ундецен (0,400 мл) и перемешивают при комнатной температуре в течение 4 дней, и затем добавляют гексаноат N-сукцинимидил 6-малеимида (0,221 г, 0,717 ммоль) и перемешивают в течение 3 часов. Реакционный раствор разбавляют этилацетатом и промывают водным раствором 10% лимонной кислоты, насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли и затем органический слой сушат над безводным сульфатом магния. Затем растворитель удаляют при пониженном давлении, полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=9:1 (об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,295 г, 78%).
1H-ЯМР (400 МГц, CDCl3) δ: 1,28-1,36 (2H, м), 1,41 (9H, с), 1,57-1,71 (4H, м), 2,23 (2H, т, J=7,6 Гц), 3,09 (2H, д, J=6,0 Гц), 3,51 (2H, т, J=7,6 Гц), 3,85-4,02 (4H, м), 4,69-4,78 (1H, м), 6,15 (1H, т, J=4,6 Гц), 6,33 (1H, д, J=7,3 Гц), 6,60 (1H, т, J=5,0 Гц), 6,68 (2H, с), 7,10-7,16 (2H, м), 7,22-7,31 (3H, м).
МС (ИЭР) m/z: 529 (M+H)+
Способ 2: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланин
К раствору в дихлорметане (8,00 мл) соединения (0,295 г, 0,558 ммоль), полученного в способе 1 выше, добавляют трифторуксусную кислоту (4,00 мл) и перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют при пониженном давлении с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,240 г, 91%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 1,15-1,23 (2H, м), 1,40-1,53 (4H, м), 2,10 (2H, т, J=7,6 Гц), 2,88 (1H, дд, J=13,7, 8,9 Гц), 3,04 (1H, дд, J=13,7, 5,0 Гц), 3,35-3,43 (2H, м), 3,58-3,77 (4H, м), 4,41 (1H, тд, J=7,8, 5,0 Гц), 7,00 (2H, с), 7,16-7,31 (5H, м), 8,00 (1H, т, J=5,7 Гц), 8,06 (1H, т, J=5,7 Гц), 8,13 (1H, д, J=7,8 Гц).
Способ 3: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-[(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)метил]глицинамид
Соединение (0,572 г, 1,21 ммоль), полученное в способе 2 выше, растворяют в дихлорметане (12,0 мл), загружают N-гидроксисукцинимид (0,152 г, 1,32 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,253 г, 1,32 ммоль) и перемешивают в течение 1 часа. Реакционный раствор добавляют к раствору в N,N-диметилформамиде (22,0 мл) смеси (1,10 ммоль), полученной в способе 5 из примера 26, и перемешивают при комнатной температуре в течение 3 часов. Реакционный раствор загружают водным раствором 10% лимонной кислоты и экстрагируют хлороформом. Полученный органический слой сушат над сульфатом натрия и фильтруют. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=8:2 (об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,351 г, 31%). Результаты измерения соединения такие же, как для соединения из способа 8 из примера 26.
Пример 33 (Альтернативный способ синтеза соединения из способа 8 из примера 26)
[Формула 59]
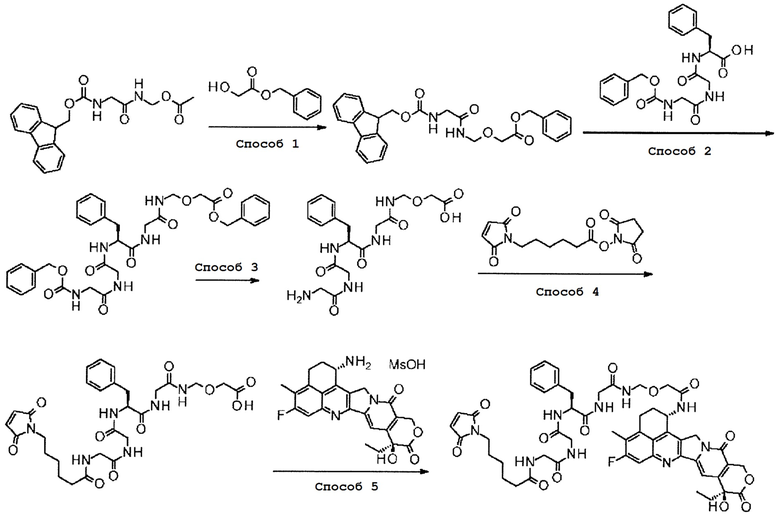
Способ 1: Бензил [({N-[(9H-флуорен-9-илметокси)карбонил]глицил}амино)метокси]ацетат
К раствору в ТГФ (200 мл) соединения (7,37 г, 20,0 ммоль), полученного в способе 1 из примера 26, добавляют бензилгликолят (6,65 г, 40,0 ммоль) и моногидрат п-толуолсульфоновой кислоты (0,381 г, 2,00 ммоль) при 0°C и перемешивают при комнатной температуре в течение 2 часов 30 минут. Реакционный раствор загружают насыщенным водным раствор гидрокарбоната натрия и экстрагируют этилацетатом. Полученный органический слой сушат над сульфатом натрия и фильтруют. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [гексан:этилацетат=100:0 (об./об.) - 0:100] с получением указанного в заголовке соединения в виде бесцветного твердого вещества (6,75 г, 71%). Результаты измерения соединения такие же, как для соединения из способа 2 из примера 26.
Способ 2: N-[(Бензилокси)карбонил]глицилглицил-L-фенилаланин-N-{[(2-(бензилокси)-2-оксоэтокси]метил}глицинамид
К раствору в N,N-диметилформамиде (140 мл) соединения (6,60 г, 13,9 ммоль), полученного в способе 1 выше, добавляют 1,8-диазабицикло[5,4,0]ундец-7-ен (2,22 г, 14,6 ммоль) при 0°C и перемешивают при комнатной температуре в течение 15 минут. Реакционный раствор загружают раствором в N,N-диметилформамиде (140 мл) N-[(бензилокси)карбонил]глицилглицил-L-фенилаланина (6,33 г, 15,3 ммоль), N-гидроксисукцинимида (1,92 г, 16,7 ммоль) и гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (3,20 г, 16,7 ммоль) заранее перемешивают при комнатной температуре в течение 1 часа и перемешивают при комнатной температуре в течение 4 часов. Реакционный раствор загружают 0,1N хлористоводородной кислотой и экстрагируют хлороформом. Полученный органический слой сушат над сульфатом натрия и фильтруют. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=8:2 (об./об.)] с получением указанного в заголовке соединения в виде бесцветного твердого вещества (7,10 г, 79%).
1H-ЯМР (ДМСО-d6) δ: 2,78 (1H, дд, J=13,9, 9,6 Гц), 3,05 (1H, дд, J=13,9, 4,5 Гц), 3,56-3,80 (6H, м), 4,15 (2H, с), 4,47-4,55 (1H, м), 4,63 (2H, д, J=6,6 Гц), 5,03 (2H, с), 5,15 (2H, с), 7,16-7,38 (15H, м), 7,52 (1H, т, J=5,9 Гц), 8,03 (1H, т, J=5,5 Гц), 8,17 (1H, д, J=8,2 Гц), 8,36 (1H, т, J=5,7 Гц), 8,61 (1H, т, J=6,6 Гц).
Способ 3: Глицилглицил-L-фенилаланил-N-[(карбоксиметокси)метил]глицинамид
К раствору в N,N-диметилформамиде (216 мл) соединения (7,00 г, 10,8 ммоль), полученного в способе 2 выше, добавляют катализатор на основе палладия на угле (7,00 г) и перемешивают в атмосфере водорода при комнатной температуре в течение 24 часов. Нерастворимые вещества удаляют фильтрацией через Целит, и растворитель удаляют при пониженном давлении. Полученные остатки растворяют в воде, нерастворимый материал удаляют фильтрацией через Целит, и растворитель удаляют при пониженном давлении. Эту методику повторяют дважды с получением указанного в заголовке соединения в виде бесцветного твердого вещества (3,77 г, 82%).
1H-ЯМР (ДМСО-d6) δ: 2,84 (1H, дд, J=13,7, 9,8 Гц), 3,08 (1H, дд, J=13,7, 4,7 Гц), 3,50-3,72 (4H, м), 3,77-3,86 (2H, м), 3,87 (2H, с), 4,52-4,43 (1H, м), 4,61 (2H, д, J=6,6 Гц), 7,12-7,30 (5H, м), 8,43 (1H, т, J=5,9 Гц), 8,54 (1H, д, J=7,8 Гц), 8,70 (1H, т, J=6,3 Гц), 8,79 (1H, т, J=5,5 Гц).
Способ 4: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-[(карбоксиметокси)метил]глицинамид
К раствору в N,N-диметилформамиде (85,0 мл) соединения (3,59 г, 8,48 ммоль), полученного в способе 3 выше, добавляют гексаноат N-сукцинимидил 6-малеимида (2,88 г, 9,33 ммоль) и триэтиламин (0,858 г, 8,48 ммоль) и перемешивают при комнатной температуре в течение 1 часа. Реакционный раствор загружают 0,1N хлористоводородной кислотой и экстрагируют хлороформом и смешанным растворителем хлороформа и метанола [хлороформ:метанол=4:1 (об./об.)]. Полученный органический слой сушат над сульфатом натрия и фильтруют. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением указанного в заголовке соединения в виде бесцветного твердого вещества (3,70 г, 71%).
1H-ЯМР (ДМСО-d6) δ: 1,13-1,24 (2H, м), 1,42-1,53 (4H, м), 2,11 (2H, т, J=7,4 Гц), 2,80 (1H, дд, J=13,7, 9,8 Гц), 3,06 (1H, дд, J=13,9, 4,5 Гц), 3,37 (2H, т, J=7,2 Гц), 3,56-3,78 (6H, м), 3,97 (2H, с), 4,46-4,53 (1H, м), 4,61 (2H, д, J=6,3 Гц), 7,00 (2H, с), 7,15-7,29 (5H, м), 8,03-8,20 (3H, м), 8,32 (1H, т, J=5,9 Гц), 8,60 (1H, т, J=6,7 Гц).
Способ 5: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-[(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)метил]глицинамид
К раствору в N,N-диметилформамиде (40,0 мл) соли метансульфоновой кислоты экзатекана (1,14 г, 2,00 ммоль), триэтиламина (0,202 г, 2,00 ммоль) добавляют соединение (1,48 г, 2,40 ммоль), полученное в способе 4 выше, и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (0,993 г, 3,00 ммоль), содержащее 16,4% воды при 0°C и перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=8:2 (об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (1,69 г, 82%). Спектральные данные соединения были такими же, как для соединения из способа 8 из примера 26.
[0273]
Пример 34. Промежуточное соединение (34)
[Формула 60]
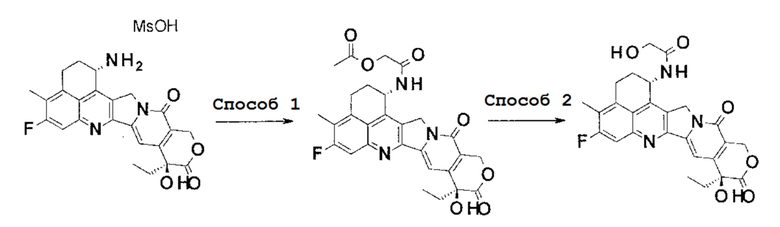
Способ 1: 2-{[(1S,9S)-9-Этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтилацетат
При охлаждении на льду, к суспензии в N,N-диметилформамиде (20,0 мл) соли метансульфоновой кислоты экзатекана (0,500 г, 0,941 ммоль) добавляют N,N-диизопропилэтиламин (0,492 мл, 2,82 ммоль) и хлорид ацетоксиацетила (0,121 мл, 1,13 ммоль) и перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,505 г, количественный).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,4 Гц), 1,81-1,92 (2H, м), 2,08 (3H, с), 2,08-2,22 (2H, м), 2,41 (3H, с), 3,14-3,21 (2H, м), 4,51 (2H, дд, J=19,4, 14,7 Гц), 5,22 (2H, дд, J=40,1, 19,0 Гц), 5,43 (2H, с), 5,56-5,61 (1H, м), 6,53 (1H, с), 7,31 (1H, с), 7,81 (1H, д, J=11,0 Гц), 8,67 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 536 (M+H)+
Способ 2: N-[(1S,9S)-9-Этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-2-гидроксиацетамид
К суспензии в метаноле (50,0 мл) соединения (0,504 г, 0,941 ммоль), полученного в способе 1 выше, добавляют ТГФ (20,0 мл) и водный раствор 1N гидроксида натрия (4,00 мл, 4,00 ммоль) и перемешивают при комнатной температуре в течение 1 часа. Реакцию останавливают добавлением 1N хлористоводородной кислоты (5,00 мл, 5,00 ммоль), и растворитель удаляют при пониженном давлении. Полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,412 г, 89%). Это соединение подтверждают в опухоли мыши, которая получает конъюгат антитела с лекарственным средством (45) или (46).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,3 Гц), 1,78-1,95 (2H, м), 2,09-2,28 (2H, м), 2,39 (3H, с), 3,07-3,27 (2H, м), 3,96 (2H, д, J=6,0 Гц), 5,11-5,26 (2H, м), 5,42 (2H, с), 5,46-5,54 (1H, м), 5,55-5,63 (1H, м), 6,52 (1H, с), 7,30 (1H, с), 7,78 (1H, д, J=10,9 Гц), 8,41 (1H, д, J=9,1 Гц). МС (ИЭР) m/z: 494 (M+H)+
Пример 35. (Альтернативный способ синтеза соединения из примера 34)
[Формула 61]

Способ 1: N-[(1S,9S)-9-Этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-2-гидроксиацетамид
Гликолевую кислоту (0,0201 г, 0,27 ммоль) растворяют в N,N-диметилформамиде (1,0 мл), загружают N-гидроксисукцинимид (0,0302 г, 0,27 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,0508 г, 0,27 ммоль) и перемешивают в течение 1 часа. Реакционный раствор добавляют к суспензии N,N-диметилформамида (1,0 мл), загружают соль метансульфоновой кислоты экзатекана (0,1 г, 0,176 ммоль) и триэтиламин (0,025 мл, 0,18 ммоль), и перемешивают при комнатной температуре в течение 24 часов. Растворитель удаляют при пониженном давлении, и полученные остатки очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=10:1 (об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,080 г, 92%). Спектральные данные соединения такие же, как для соединения, полученного в способе 2 из примера 34.
Пример 36. Конъюгат антитела с лекарственным средством (36)
[Формула 62]
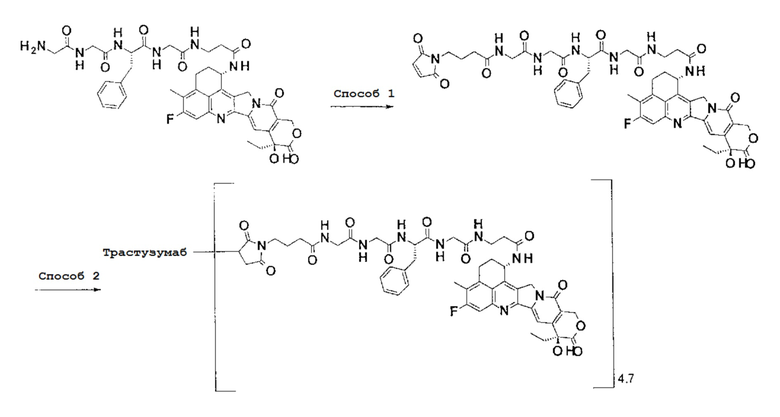
Способ 1: N-[4-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)бутаноил]глицилглицил-L-фенилаланилглицил-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]-β-аланинамид
Соединение (60,0 мг, 0,0646 ммоль), полученное в способе 2 из примера 15, подвергают взаимодействию по методике способа 3 из примера 2 с применением бутирата N-сукцинимидил 4-малеимида вместо гексаноата N-сукцинимидил 6-малеимида с получением указанного в заголовке соединения в виде беловатого твердого вещества (24,0 мг, 38%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,2 Гц), 1,68 (2H, квин, J=7,4 Гц), 1,78-1,92 (2H, м), 2,06-2,22 (2H, м), 2,10 (2H, т, J=7,8 Гц), 2,31-2,43 (2H, м), 2,40 (3H, с), 2,78 (1H, дд, J=13,7, 9,4 Гц), 3,01 (1H, дд, J=13,7, 4,7 Гц), 3,17 (4H, д, J=5,1 Гц), 3,29-3,40 (2H, м), 3,52-3,80 (6H, м), 4,40-4,51 (1H, м), 5,19 (1H, д, J=18,4 Гц), 5,26 (1H, д, J=18,8 Гц), 5,42 (2H, с), 5,52-5,61 (1H, м), 6,53 (1H, с), 6,99 (2H, с), 7,12-7,28 (5H, м), 7,31 (1H, с), 7,74-7,84 (2H, м), 8,02 (1H, т, J=5,9 Гц), 8,08-8,16 (2H, м), 8,25 (1H, т, J=5,9 Гц), 8,52 (1H, д, J=8,2 Гц).
МС (ИЭР) m/z: 990 (M+H)+
Способ 2: Конъюгат антитела с лекарственным средством (33)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1 выше, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 2 из примера 6.
Концентрация антитела: 1,75 мг/мл, выход антитела: 10,5 мг (84%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 4,7.
Пример 37. Конъюгат антитела с лекарственным средством (37)
[Формула 63]
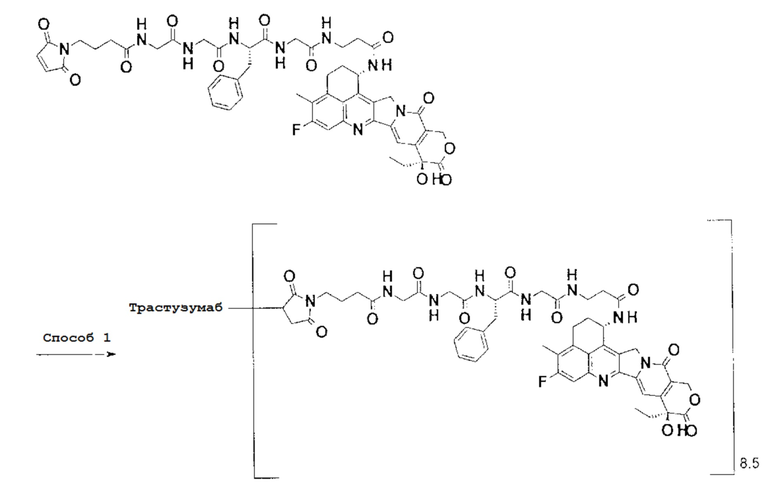
Способ 1: Конъюгат антитела с лекарственным средством (37)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1 из примера 36, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 из примера 7.
Концентрация антитела: 1,89 мг/мл, выход антитела: 11,3 мг (90%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 8,5.
Пример 38. Промежуточное соединение (38)
[Формула 64]
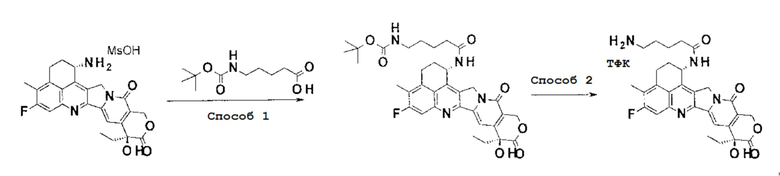
Способ 1: трет-Бутил (5-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-5-оксопентил)карбамат
Соль метансульфоновой кислоты экзатекана (500 мг, 0,941 ммоль) подвергают взаимодействию по методике способа 1 из примера 1 с применением 5-(трет-бутоксикарбониламино)валериановой кислоты вместо 4-(трет-бутоксикарбониламино)бутановой кислоты с получением указанного в заголовке соединения в виде желто-коричневого твердого вещества (571 мг, 96%). Соединение применяют в следующей реакции без дальнейшей очистки.
МС(ИЭР)m/z:635(M+H)+
Способ 2: 5-Амино-N-[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]пентанамид
Соединение (558 мг, 0,879 ммоль), полученное в способе 1 выше, подвергают взаимодействию по методике способа 2 из примера 1 с получением трифторацетата указанного в заголовке соединения в виде желтого твердого вещества (363 мг, 64%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,88 (3H, т, J=7,4 Гц), 1,52-1,71 (4H, м), 1,87 (2H, тт, J=14,4, 6,9 Гц), 2,07-2,18 (2H, м), 2,22 (2H, т, J=7,0 Гц), 2,40 (3H, с), 2,76-2,88 (2H, м), 3,13-3,22 (2H, м), 5,18 (1H, д, J=18,8 Гц), 5,24 (1H, д, J=18,8 Гц), 5,43 (2H, с), 5,53-5,61 (1H, м), 6,55 (1H, с), 7,33 (1H, с), 7,65 (3H, шс), 7,81 (1H, д, J=11,3 Гц), 8,49 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 535 (M+H)+
Пример 39. Конъюгат антитела с лекарственным средством (39)
[Формула 65]
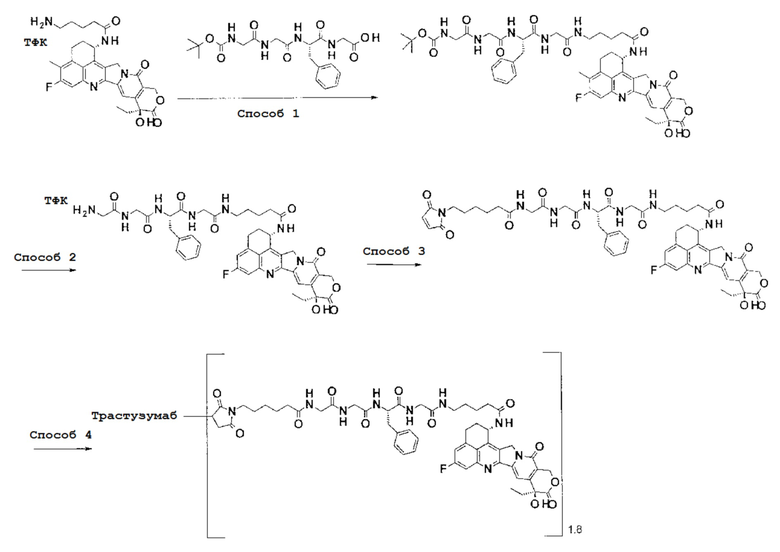
Способ 1: N-(трет-Бутоксикарбонил)глицилглицил-L-фенилаланил-N-(5-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-5-оксопентил)глицинамид
Соединение (348 мг, 0,537 ммоль), полученное в способе 2 из примера 38, подвергают взаимодействию по методике способа 1 из примера 2 с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (429 мг, 84%). Соединение применяют в следующей реакции без дальнейшей очистки.
Способ 2: Глицилглицил-L-фенилаланил-N-(5-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-5-оксопентил)глицинамид
Соединение (427 мг, 0,448 ммоль), полученное в способе 1 выше, подвергают взаимодействию по методике способа 2 из примера 2 с получением трифторацетат указанного в заголовке соединения в виде желтого твердого вещества (430 мг, 99%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,2 Гц), 1,38-1,49 (2H, м), 1,54-1,66 (2H, м), 1,86 (2H, тт, J=14,5, 7,0 Гц), 2,08-2,16 (2H, м), 2,19 (2H, т, J=7,2 Гц), 2,40 (3H, с), 2,76 (1H, дд, J=13,9, 10,0 Гц), 3,00-3,12 (3H, м), 3,14-3,21 (2H, м), 3,57 (2H, д, J=4,7 Гц), 3,60-3,75 (3H, м), 3,87 (1H, дд, J=16,8, 5,9 Гц), 4,55 (1H, тд, J=9,0, 4,7 Гц), 5,16 (1H, д, J=18,8 Гц), 5,23 (1H, д, J=18,4 Гц), 5,44 (2H, с), 5,53-5,60 (1H, м), 6,55 (1H, с), 7,14-7,29 (5H, м), 7,32 (1H, с), 7,74 (1H, т, J=5,5 Гц), 7,81 (1H, д, J=10,9 Гц), 7,96 (3H, шс), 8,30-8,37 (1H, м), 8,44-8,53 (2H, м).
МС (ИЭР) m/z: 853 (M+H)+
Способ 3: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-(5-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-5-оксопентил)глицинамид
Соединение (60,0 мг, 0,0621 ммоль), полученное в способе 2 выше, подвергают взаимодействию по методике способа 3 из примера 2 с получением указанного в заголовке соединения в виде твердого вещества (16,0 мг, 25%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,4 Гц), 1,13-1,21 (2H, м), 1,36-1,52 (6H, м), 1,53-1,65 (2H, м), 1,79-1,92 (2H, м), 2,05-2,15 (4H, м), 2,19 (2H, с), 2,40 (3H, с), 2,79 (1H, дд, J=13,7, 10,2 Гц), 2,98-3,10 (3H, м), 3,12-3,21 (2H, м), 3,29-3,37 (2H, м), 3,53-3,79 (6H, м), 4,41-4,50 (1H, м), 5,16 (1H, д, J=18,8 Гц), 5,23 (1H, д, J=18,8 Гц), 5,43 (2H, с), 5,52-5,60 (1H, м), 6,53 (1H, с), 6,99 (2H, с), 7,12-7,28 (5H, м), 7,31 (1H, с), 7,63 (1H, т, J=5,7 Гц), 7,80 (1H, д, J=10,6 Гц), 8,02 (1H, т, J=5,9 Гц), 8,08 (1H, т, J=5,7 Гц), 8,12 (1H, д, J=7,8 Гц), 8,24 (1H, т, J=5,7 Гц), 8,45 (1H, д, J=8,6 Гц).
МС (ИЭР) m/z: 1046 (M+H)+
Способ 4: Конъюгат антитела с лекарственным средством (39)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл с ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,37 млмг-1см-1). Раствор (1,0 мл) собирают в 2 мл пробирку и загружают водным раствором 10 мМ ТКЭФ (0,0155 мл; 2,3 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,050 мл). После подтверждения того, что раствор имеет pH 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После инкубирования раствора в течение 10 минут при 22°C, туда добавляют раствор ДМСО (0,0311 мл; 4,6 эквивалента на молекулу антитела), содержащий 10 мМ соединения, полученного в способе 3, и инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 22°C в течение 40 минут. Затем добавляют водный раствор (0,00622 мл; 9,2 эквивалента на молекулу антитела) 100 мМ NAC и инкубируют при 22°C для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D-1 (ФРФБ6,0 применяют в качестве буферного раствора) с получением 6 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 1,12 мг/мл, выход антитела: 6,72 мг (67%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 1,8.
Пример 40. Конъюгат антитела с лекарственным средством (40)
[Формула 66]
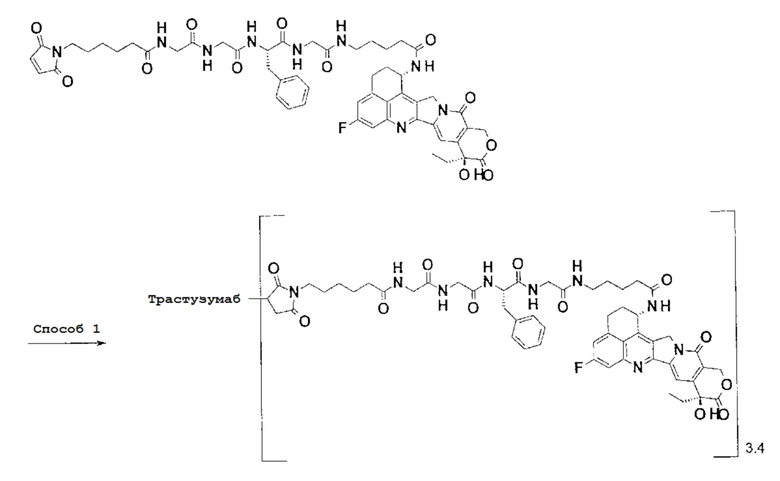
Способ 1: Конъюгат антитела с лекарственным средством (40)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл с ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,37 млмг-1см-1). Раствор (1,0 мл) собирают в 2 мл пробирку и загружают водным раствором 10 мМ ТКЭФ (0,0311 мл; 4,6 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,050 мл). После подтверждения того, что раствор имеет pH 7,4±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После инкубирования раствора при 22°C в течение 10 минут, туда добавляют раствор ДМСО (0,0622 мл; 9,2 эквивалента на молекулу антитела), содержащий 10 мМ соединения, полученного в способе 3 из примера 39, и инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 22°C в течение 40 минут. Затем добавляют водный раствор (0,0124 мл; 18,4 эквивалента на молекулу антитела) 100 мМ NAC и инкубируют при 22°C для остановки реакции связующей группы лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D-1 (ФРФБ6,0 применяют в качестве буферного раствора) с получением 6 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общей методики E, получают следующие характеристики.
Концентрация антитела: 0,98 мг/мл, выход антитела: 5,88 мг (59%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,4.
Пример 41. Конъюгат антитела с лекарственным средством (41)
[Формула 67]
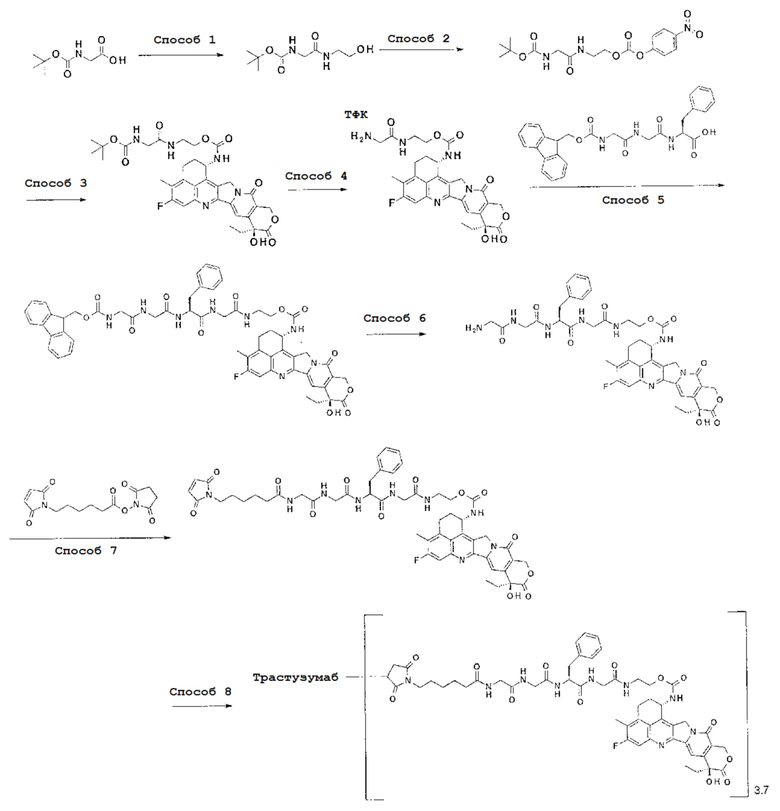
Способ 1: трет-Бутил {2-[(2-гидроксиэтил)амино]-2-оксоэтил}карбамат
N-(трет-Бутоксикарбонил)глицин (4,2 г, 24 ммоль) растворяют в диметилформамиде (40 мл). После добавления аминоэтанола (2,9 г, 48 ммоль) и 1-гидроксибензотриазола (3,7 г, 24 ммоль) и добавления гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (6,9 г, 36 ммоль), смесь перемешивают при комнатной температуре в течение 12 часов. Растворитель удаляют при пониженном давлении и в остаток загружают толуол для азеотропной дистилляции. Полученный остаток очищают хроматографией на колонке с силикагелем [этилацетат-этилацетат:метанол=10:1 (об./об.)] с получением указанного в заголовке соединения в виде бесцветного маслянистого вещества (3,8 г, 72%).
1H-ЯМР (400 МГц, CDCl3) δ: 1,44 (9H, с), 1,69 (1H, шс), 3,43 (2H, тд, J=5,9, 5,1 Гц), 3,71 (2H, т, J=5,1 Гц), 3,79 (2H, д, J=5,9 Гц), 5,22 (1H, шс), 6,62 (1H, шс).
Способ 2: 2-{[N-(трет-Бутоксикарбонил)глицил]амино}этил4-нитрофенилкарбонат
К раствору в ТГФ (23 мл) соединения (1,0 г, 4,59 ммоль), полученного в способе 1 выше, добавляют диизопропилэтиламин (0,80 мл, 4,59 ммоль) и карбонат бис(4-нитрофенила) (1,32 г, 6,88 ммоль) и перемешивают при комнатной температуре в течение 12 часов. Растворитель удаляют при пониженном давлении, и полученный остаток очищают хроматографией на колонке с силикагелем [гексан-гексан:этилацетат=1:3 (об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (1,13 г, 64%).
1H-ЯМР (400 МГц, CDCl3) δ: 1,44 (1H, с), 3,66 (2H, тд, J=5,1, 5,9 Гц), 3,81 (2H, д, J=5,9 Гц), 4,36 (2H, т, J=5,1 Гц), 5,07 (1H, с), 6,48-6,53 (1H, м), 7,38 (2H, дт, J=9,9, 2,7 Гц), 8,27 (2H, дт, J=9,9, 2,7 Гц).
Способ 3: 2-({[(трет-Бутоксикарбонил)амино]ацетил}амино)этил[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]карбамат
К соли метансульфоновой кислоты экзатекана (0,70 г, 1,2 ммоль) добавляют соединение (0,57 г, 1,5 ммоль), полученное в способе 2, и 1-гидроксибензотриазол (3,7 г, 24 ммоль), диметилформамид (23 мл) и диизопропилэтиламин (0,43 мл, 2,5 ммоль) и перемешивают при комнатной температуре в течение 12 часов. Растворитель удаляют при пониженном давлении, и в остаток загружают толуол для азеотропной дистилляции. Полученный остаток очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=10:1 (об./об.)] с получением указанного в заголовке соединения (0,86 г, количественный) в виде бледно-желтого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,4 Гц), 1,35 (9H, с), 1,78-1,94 (1H, м), 2,07-2,17 (1H, м), 2,17-2,27 (1H, м), 2,37 (3H, с), 3,05-3,16 (1H, м), 3,19-3,26 (1H, м), 3,34-3,39 (2H, м), 3,50-3,56 (2H, м), 4,00-4,07 (1H, м), 4,13-4,21 (1H, м), 5,15-5,34 (3H, м), 5,44 (2H, с), 6,54 (1H, с), 6,90-6,96 (1H, м), 7,32 (1H, с), 7,78 (1H, д, J=11,0 Гц), 7,93-8,07 (2H, м).
Способ 4: 2-(Глициламино)этил[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]карбамат
Соединение (0,86 г, 2,1 ммоль), полученное в способе 3 выше, растворяют в дихлорметане (15 мл). После добавления трифторуксусной кислоты (15 мл) раствор перемешивают в течение 1 часа. Растворитель удаляют при пониженном давлении, и в остаток загружают толуол для азеотропной дистилляции. Полученный остаток очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,86 г, 99%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,2 Гц), 1,79-1,95 (2H, м), 2,06-2,18 (1H, м), 2,18-2,29 (1H, м), 2,38 (3H, с), 3,07-3,17 (1H, м), 3,20-3,29 (1H, м), 3,36-3,50 (2H, м), 3,51-3,62 (2H, м), 3,99-4,08 (1H, м), 4,22-4,31 (1H, м), 5,16-5,35 (3H, м), 5,42 (1H, д, J=18,8 Гц), 5,46 (1H, д, J=18,8 Гц), 6,56 (1H, с), 7,34 (1H, с), 7,65 (2H, шс), 7,79 (1H, д, J=10,6 Гц), 7,99-8,06 (1H, м), 8,51 (1H, т, J=5,5 Гц).
МС (ХИАД) m/z: 939 (M+H)+
Способ 5: N-[(9H-Флуорен-9-илметокси)карбонил]глицилглицил-L-фенилаланил-N-[2-({[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]карбамоил}окси)этил]глицинамид
N-[(9H-флуорен-9-илметокси)карбонил]глицилглицил-L-фенилаланин (выложенный патент Японии № 2002-60351; 0,21 г, 0,41 ммоль) растворяют в N,N-диметилформамиде (3 мл). После добавления N-гидроксисукцинимида (0,052 г, 0,45 ммоль) и гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (0,086 г, 0,45 ммоль), смесь перемешивают в течение 1 часа. Реакционный раствор добавляют по каплям к раствору N,N-диметилформамида (2 мл), загружают соединение (0,24 г, 0,35 ммоль), полученное в способе 4, и триэтиламин (0,078 мл, 0,45 ммоль) и перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют при пониженном давлении, и полученный остаток очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=8:2 (об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,24 г, 65%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,3 Гц), 1,79-1,90 (2H, м), 2,05-2,27 (2H, м), 2,36 (3H, с), 2,73-2,81 (1H, м), 2,98-3,12 (2H, м), 3,17-3,26 (1H, м), 3,35-3,42 (2H, м), 3,55-3,79 (6H, м), 4,00-4,10 (1H, м), 4,12-4,23 (2H, м), 4,23-4,29 (2H, м), 4,45-4,55 (1H, м), 5,13-5,33 (3H, м), 5,40 (1H, д, J=17,2 Гц), 5,44 (1H, д, J=17,2 Гц), 6,53 (1H, с), 7,11-7,26 (5H, м), 7,26-7,33 (3H, м), 7,38 (2H, т, J=7,6 Гц), 7,57 (1H, т, J=5,9 Гц), 7,68 (2H, д, J=7,4 Гц), 7,77 (1H, д, J=11,0 Гц), 7,85 (2H, д, J=9,0 Гц), 7,91-7,97 (1H, м), 7,98-8,05 (2H, м), 8,14 (1H, д, J=7,8 Гц), 8,31-8,26 (1H, м).
МС (ХИАД) m/z: 1063 (M+H)+
Способ 6: Глицилглицил-L-фенилаланил-N-[2-({[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]карбамоил}окси)этил]глицинамид
Соединение (0,24 г, 0,35 ммоль), полученное в способе 5 выше, подвергают взаимодействию по методике способа 7 из примера 26 с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,12 г, 65%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,4 Гц), 1,78-1,94 (2H, м), 2,06-2,27 (2H, м), 2,37 (3H, с), 2,72-2,81 (1H, м), 2,98-3,07 (1H, м), 3,12-3,17 (2H, м), 3,57-3,81 (6H, м), 4,00-4,21 (3H, м), 4,45-4,54 (1H, м), 5,15-5,35 (3H, м), 5,41 (1H, д, J=17,2 Гц), 5,45 (1H, д, J=17,2 Гц), 6,54 (1H, с), 7,11-7,26 (6H, м), 7,32 (1H, с), 7,78 (1H, д, J=11,0 Гц), 7,93-8,00 (1H, м), 8,03 (1H, д, J=9,4 Гц), 8,06-8,13 (1H, м), 8,21-8,27 (2H, м), 8,30-8,36 (1H, м).
МС (ХИАД) m/z: 841 (M+H)+
Способ 7: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-[2-({[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]карбамоил}окси)этил]глицинамид
Соединение (42,0 мг, 0,0499 ммоль), полученное в способе 6, подвергают взаимодействию по методике способа 3 из примера 2 с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (38,3 мг, 74%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,87 (3H, т, J=7,4 Гц), 1,12-1,23 (2H, м), 1,40-1,51 (4H, м), 1,80-1,95 (2H, м), 2,05-2,27 (4H, м), 2,38 (3H, с), 3,43-2,40 (8H, м), 3,53-3,78 (6H, м), 4,00-4,21 (2H, м), 4,44-4,55 (1H, м), 5,17-5,36 (3H, м), 5,43 (2H, с), 6,54 (1H, с), 6,99 (2H, с), 7,19 (5H, д, J=23,9 Гц), 7,33 (1H, с), 7,78 (1H, д, J=10,6 Гц), 7,91-8,16 (5H, м), 8,24-8,31 (1H, м).
МС (ИЭР) m/z: 1034 (M+H)+
Способ 8: Конъюгат антитела с лекарственным средством (41)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 7 выше, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 2 из примера 6.
Концентрация антитела: 1,54 мг/мл, выход антитела: 9,2 мг (74%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 3,7.
Пример 42. Конъюгат антитела с лекарственным средством (42)
[Формула 68]
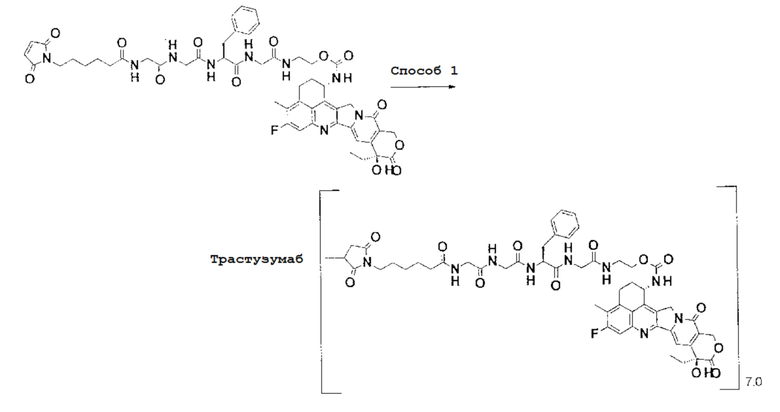
Способ 1: Конъюгат антитела с лекарственным средством (42)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 7 из примера 41, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 из примера 7.
Концентрация антитела: 1,47 мг/мл, выход антитела: 8,8 мг (71%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 7,0.
Пример 43. Конъюгат антитела с лекарственным средством (43)
[Формула 69]
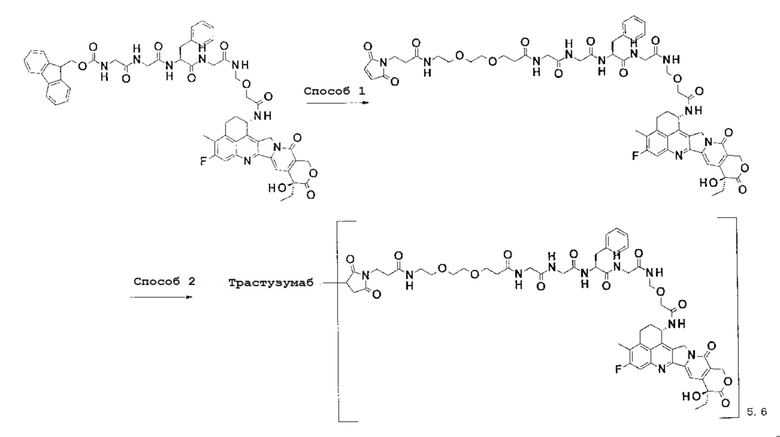
Способ 1: N-{3-[2-(2-{[3-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)пропаноил]амино}этокси)этокси]пропаноил}глицилглицил-L-фенилаланил-N-[(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)метил]глицинамид
Соединение (53,7 мг, 50,5 мкмоль), полученное в способе 6 из примера 26, растворяют в N,N-диметилформамиде (1,50 мл). После добавления 1,8-диазабицикло(5,4,0)-7-ундецена (7,5 мкл, 50,5 мкммоль), смесь перемешивают при комнатной температуре в течение 30 минут. Реакционный раствор п-толуолсульфонатом пиридиния (14,0 мг, 5,56 мкмоль), затем загружают 3-(2-(2-(3-малеинимидопропанамидо)этокси)этокси)пропаноат N-сукцинимидила (32,3 мг, 75,8 мкмоль) и перемешивают при комнатной температуре в течение 2,25 часов. Растворитель удаляют при пониженном давлении, и полученный остаток очищают хроматографией на колонке с силикагелем [[хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (27,1 мг, 47%).
1H-ЯМР (ДМСО-d6) δ: 0,87 (3H, т, J=7,0 Гц), 1,79-1,91 (2H, м), 2,18 (2H, т, J=15,1 Гц), 2,29-2,33 (4H, м), 2,39 (3H, с), 2,76 (1H, дд, J=13,9, 9,2 Гц), 3,02 (1H, дд, J=13,7, 3,9 Гц), 3,13-3,15 (2H, м), 3,44-3,46 (6H, м), 3,57-3,59 (6H, м), 3,69-3,75 (6H, м), 4,01 (2H, с), 4,46-4,48 (1H, м), 4,63 (2H, д, J=6,3 Гц), 5,21 (2H, с), 5,42 (2H, с), 5,60 (1H, дд, J=13,5, 5,7 Гц), 6,54 (1H, с), 7,00 (2H, с), 7,17-7,24 (6H, м), 7,31 (1H, с), 7,79 (1H, д, J=11,0 Гц), 8,00-8,02 (2H, м), 8,13 (1H, д, J=7,8 Гц), 8,17 (1H, т, J=6,3 Гц), 8,52 (1H, д, J=9,0 Гц), 8,65 (1H, т, J=6,5 Гц).
МС (ИЭР) m/z=1151 (M+H)+
Способ 2: Конъюгат антитела с лекарственным средством (43)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 1, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 из примера 7.
Концентрация антитела: 1,96 мг/мл, выход антитела: 17,6 мг (88%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 5,6.
Пример 44. Конъюгат антитела с лекарственным средством (44)
[Формула 70]
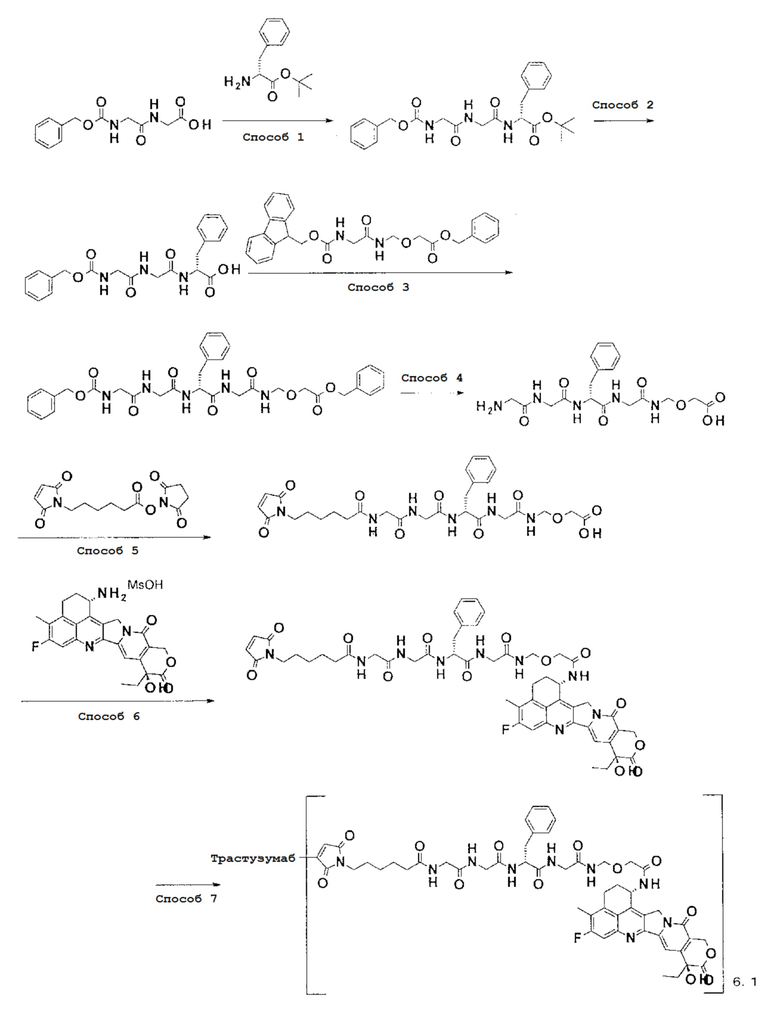
Способ 1: трет-Бутил N-[(бензилокси)карбонил]глицилглицил-D-фенилаланинат
N-[(Бензилокси)карбонил]глицилглицин (3,00 г, 11,3 ммоль) растворяют в N,N-диметилформамиде (20,0 мл). После добавления N-гидроксисукцинимида (1,43 г, 12,4 ммоль) и гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (2,37 г, 12,4 ммоль), смесь перемешивают в течение 1 часа. В раствор N,N-диметилформамида (10 мл) загружают трет-бутил D-фенилаланина (2,74 г, 12,38 ммоль) и по каплям добавляют триэтиламин (1,73 мл, 12,4 ммоль) к реакционному раствору и перемешивают при комнатной температуре в течение 2 hours. Реакционный раствор загружают дихлорметаном и промывают водой, 1N хлористоводородной кислотой и насыщенным водным раствором бикарбоната натрия, и затем органический слой сушат над безводным сульфатом натрия. Растворитель удаляют при пониженном давлении, и полученный остаток очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=9:1 (об./об.)] с получением указанного в заголовке соединения в виде бесцветного твердого вещества (4,21 г, 80%).
1H-ЯМР (CDCl3) δ: 1,41 (9H, с), 3,03-3,14 (2H, м), 3,86-3,97 (4H, м), 4,70-4,77 (1H, м), 5,13 (2H, с), 5,43 (1H, шс), 6,42 (1H, д, J=10,0 Гц), 6,64-6,71 (1H, м), 7,11-7,15 (2H, м), 7,20-7,31 (4H, м), 7,31-7,38 (4H, м).
МС (ХИАД) m/z: 470 (M+H)+
Способ 2: N-[(Бензилокси)карбонил]глицилглицил-D-фенилаланин
Соединение (4,21 г, 8,97 ммоль), полученное в способе 1, растворяют в этилацетате (20 мл). После добавления раствора в этилацетате (20,0 мл) 4N гидрохлорида, смесь выстаивают в течение ночи при комнатной температуре. Растворитель удаляют при пониженном давлении, затем добавляют толуол и растворитель удаляют при пониженном давлении. Полученный остаток очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением указанного в заголовке соединения в виде бесцветного твердого вещества (1,66 г, 45%).
1H-ЯМР (CDCl3) δ: 2,92-3,01 (1H, м), 3,10-3,18 (1H, м), 3,65-3,81 (3H, м), 3,88-3,98 (1H, м), 4,64-4,73 (1H, м), 5,06 (2H, с), 5,87 (1H, шс), 7,10-7,37 (13H, м).
МС (ХИАД) m/z: 412 (M+H) -
Способ 3: N-[(Бензилокси)карбонил]глицилглицил-D-фенилаланил-N-{[2-(бензилокси)-2-оксоэтокси]метил}глицинамид
К раствору в диоксане (25,0 мл) соединения (1,25 г, 2,63 ммоль), полученного в способе 1 из примера 32, добавляют пиперидин (5,00 мл) и N,N-диметилформамид (5,00 мл) и перемешивают при комнатной температуре в течение 30 минут. Растворитель удаляют при пониженном давлении и полученный остаток растворяют в N,N-диметилформамиде (20,0 мл). После добавления соединения (1,20 г, 2,90 ммоль) из способа 2 выше и хлорида 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (1,03 г, 3,16 ммоль), содержащего 16,4% воды, смесь перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор загружают хлороформом и промывают водой, и затем органический слой сушат над безводным сульфатом натрия. Растворитель удаляют при пониженном давлении и полученный остаток очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=9:1 (об./об.)] с получением указанного в заголовке соединения в виде бесцветного твердого вещества (270 мг, 16%).
1H-ЯМР (ДМСО-d6) δ: 2,78 (1H, дд, J=13,6, 10,0 Гц), 3,05 (1H, дд, J=13,9, 4,2 Гц), 3,56-3,79 (6H, м), 4,15 (2H, с), 4,47-4,54 (1H, м), 4,63 (2H, д, J=6,7 Гц), 5,03 (2H, с), 5,15 (2H, с), 7,14-7,39 (15H, м), 7,50 (1H, т, J=5,7 Гц), 8,02 (1H, т, J=5,4 Гц), 8,16 (1H, д, J=7,9 Гц), 8,34 (1H, т, J=6,0 Гц), 8,60 (1H, т, J=7,0 Гц).
МС (ХИАД) m/z: 648 (M+H)+
Способ 4: Глицилглицил-D-фенилаланил-N-[(карбоксиметокси)метил]глицинамид
Соединение (200 мг, 0,31 ммоль), полученное в способе 3 выше, растворяют в N,N-диметилформамиде (5,0 мл). После добавления катализатора 5% палладия на угле (0,12 г), смесь перемешивают в атмосфере водорода при комнатной температуре в течение 9 часов. Реакционный раствор фильтруют через Целит и остаток промывают смешанным растворителем воды и N,N-диметилформамида. Фильтрат и промывку объединяют, и растворитель удаляют при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (0,15 г, количественный).
1H-ЯМР (ДМСО-d6) δ: 2,85 (1H, дд, J=13,3, 9,7 Гц), 3,08 (1H, дд, J=13,9, 5,4 Гц), 3,43-3,52 (4H, м), 3,62-3,89 (7H, м), 4,36-4,44 (1H, м), 4,58-4,67 (2H, м), 7,12-7,29 (5H, м), 8,44 (1H, т, J=5,7 Гц), 8,67 (1H, д, J=7,3 Гц), 8,78 (1H, т, J=5,4 Гц), 8,91 (1H, шс).
МС (ХИАД) m/z: 424 (M+H)+
Способ 5: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-D-фенилаланил-N-[(карбоксиметокси)метил]глицинамид
Соединение (0,15 г, 0,35 ммоль), полученное в способе 4 выше, растворяют в N,N-диметилформамиде (10 мл). После добавления гексаноата N-сукцинимидил 6-малеимида (0,11 г, 0,35 ммоль), смесь перемешивают при комнатной температуре в течение 1 часа. Реакционный раствор загружают хлороформом и промывают водой, и затем органический слой сушат над безводным сульфатом натрия. Растворитель удаляют при пониженном давлении, и полученный остаток очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением указанного в заголовке соединения в виде бесцветного твердого вещества (41 мг, 26%).
1H-ЯМР (ДМСО-d6) δ: 1,13-1,24 (2H, м), 1,42-1,53 (4H, м), 2,12 (2H, т, J=7,3 Гц), 2,82 (1H, дд, J=13,9, 10,0 Гц), 3,09 (1H, дд, J=13,9, 4,8 Гц), 3,17 (2H, д, J=4,2 Гц), 3,47-3,89 (8H, м), 4,08-4,14 (1H, м), 4,41-4,49 (1H, м), 4,58-4,69 (2H, м), 7,00 (2H, с), 7,14-7,27 (5H, м), 8,31 (1H, т, J=6,0 Гц), 8,39 (1H, шс), 8,55 (2H, шс), 8,93 (1H, шс).
МС (ХИАД) m/z: 615 (M-H) -
Способ 6: N-[6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицилглицил-D-фенилаланил-N-[(2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтокси)метил]глицинамид
К раствору в N,N-диметилформамиде (10 мл) соли метансульфоновой кислоты экзатекана (22 мг, 0,388 ммоль) добавляют триэтиламин (5,42 мкл, 0,388 ммоль), соединение (29 мг, 0,466 ммоль), полученное в способе 5 выше, и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (19 мг, 0,686 ммоль), содержащий 16,4% воды при 0°C и перемешивают при комнатной температуре в течение 1 часа. Растворитель в реакционном растворе удаляют при пониженном давлении, и затем полученный остаток очищают хроматографией на колонке с силикагелем [хлороформ-разделенный органический слой хлороформ:метанол:вода=7:3:1 (об./об./об.)] с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (26 мг, 65%).
1H-ЯМР (ДМСО-d6) δ: 0,87 (3H, т, J=7,3 Гц), 1,12-1,22 (2H, м), 1,40-1,51 (4H, м), 1,79-1,92 (2H, м), 2,09 (2H, т, J=7,6 Гц), 2,13-2,23 (2H, м), 2,39 (3H, с), 2,78 (1H, дд, J=13,6, 9,4 Гц), 2,98-3,05 (1H, м), 3,13-3,23 (2H, м), 3,54-3,78 (8H, м), 4,02 (2H, с), 4,41-4,50 (1H, м), 4,61-4,66 (2H, м), 5,21 (2H, с), 5,42 (2H, с), 5,56-5,64 (1H, м), 6,53 (1H, с), 6,99 (2H, с), 7,14-7,27 (5H, м), 7,31 (1H, с), 7,79 (1H, д, J=10,9 Гц), 8,01 (1H, т, J=5,4 Гц), 8,07 (1H, т, J=5,7 Гц), 8,14 (1H, д, J=7,9 Гц), 8,31 (1H, т, J=5,7 Гц), 8,53 (1H, д, J=9,1 Гц), 8,63 (1H, т, J=6,3 Гц).
МС (ХИАД) m/z: 1034 (M+H)+
Способ 7: Конъюгат антитела с лекарственным средством (44)
С применением трастузумаба, полученного в ссылочном примере 1, и соединения, полученного в способе 6 выше, указанный в заголовке конъюгат антитела с лекарственным средством получают по методике способа 1 из примера 7.
Концентрация антитела: 1,87 мг/мл, выход антитела: 16,8 мг (84%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,1.
Пример 45. Промежуточное соединение (45)
[Формула 71]
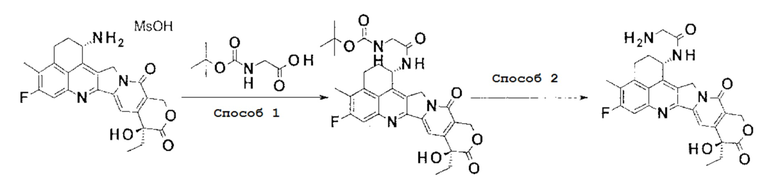
Способ 1: трет-Бутил (2-{[(1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]амино}-2-оксоэтил)карбамат
К раствору в дихлорметане (3,00 мл) N-(трет-бутоксикарбонил)глицина (0,395 г, 2,26 ммоль) добавляют N-гидроксисукцинимид (0,260 г, 2,26 ммоль) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,433 мг, 2,26 ммоль) и перемешивают при комнатной температуре в течение 1 часа. Этот раствор добавляют к раствору, содержащему соль метансульфоновой кислоты экзатекана (1,00 г, 1,88 ммоль), триэтиламин (0,315 мл, 2,26 ммоль) и N,N-диметилформамид (3,00 мл), и перемешивают при комнатной температуре в течение 16,5 часов. Реакционный раствор разбавляют хлороформом и промывают 10% раствором лимонной кислоты и затем органический слой сушат над безводным сульфатом натрия. Растворитель удаляют при пониженном давлении и полученный остаток очищают хроматографией на колонке с силикагелем [хлороформ-хлороформ:метанол=9:1 (об./об.)] с получением указанного в заголовке соединения в виде желтого твердого вещества (1,16 г, 99%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,86 (3H, т, J=7,2 Гц), 1,30 (9H, с), 1,81-1,89 (2H, м), 2,09-2,21 (2H, м), 2,38 (3H, с), 3,15-3,17 (2H, м), 3,55-3,56 (2H, м), 5,15 (1H, д, J=18,8 Гц), 5,23 (1H, д, J=19,2 Гц), 5,41 (2H, с), 5,55-5,56 (1H, м), 6,53 (1H, с), 6,95 (1H, т, J=5,5 Гц), 7,28 (1H, с), 7,77 (1H, д, J=11,0 Гц), 8,39 (1H, д, J=8,6 Гц).
МС (ХИАД) m/z: 593 (M+H)+
Способ 2: N-[(1S,9S)-9-Этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[de]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил]глицинамид
Соединение (0,513 г, 1,01 ммоль), полученное в способе 1 выше, подвергают взаимодействию по методике способа 2 из примера 1 с получением указанного в заголовке соединения в виде желтого твердого вещества (0,463 г, 93%).
1H-ЯМР (400 МГц, CD3OD) δ: 0,96 (3H, т, J=7,0 Гц), 1,89-1,91 (2H, м), 2,14-2,16 (1H, м), 2,30 (3H, с), 2,40-2,42 (1H, м), 3,15-3,21 (2H, м), 3,79-3,86 (2H, м), 4,63-4,67 (1H, м), 5,00-5,05 (1H, м), 5,23 (1H, д, J=16,0 Гц), 5,48 (1H, д, J=16,0 Гц), 5,62-5,64 (1H, м), 7,40-7,45 (2H, м).
МС (ХИАД) m/z: 493 (M+H)+
Пример 46. Конъюгат антитела с лекарственным средством (46)
[Формула 72]
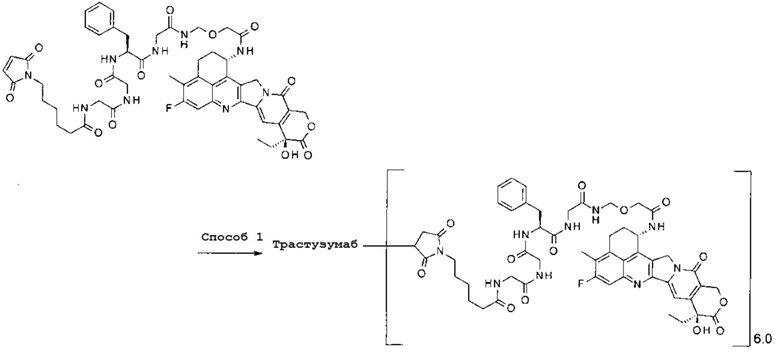
Способ 1: Конъюгат антитела с лекарственным средством (46)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (50 мл) помещают в 125 мл поликарбонатную колбу Эрленмейера, загружают водным раствором 1M вторичного кислого фосфата калия при комнатной температуре (0,750 мл) при перемешивании с применением магнитной мешалки, и затем загружают водным раствором 10 мМ ТКЭФ (1,857 мл; 5,4 эквивалента на молекулу антитела). После подтверждения того, что раствор имеет pH 7,0±0,1, перемешивание прекращают, и дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 1 часа.
Конъюгирование между антителом и связующей группой лекарственного средства: После охлаждения полученного выше раствора до 15°C постепенно по каплям добавляют раствор ДМСО, содержащий 10 мМ соединения из способа 8 из примера 26 (2,958 мл; 8,6 эквивалента на молекулу антитела) при перемешивании. При 15°C реакционный раствор перемешивают в течение первых 30 минут и инкубируют без перемешивания для конъюгирования связующей группы лекарственного средства с антителом в течение следующего 1 часа. Затем добавляют водный раствор (0,444 мл; 12,9 эквивалента на молекулу антитела) 100 мМ NAC при перемешивании и перемешивают при комнатной температуре для остановки реакционной способности непрореагировавших связующих групп лекарственного средства в течение еще 20 минут.
Очистка: Постепенное добавление 20% водного раствора уксусной кислоты (около 0,25 мл) и ABS (50 мл) к полученному выше раствору при перемешивании позволяет довести pH раствора до 5,5±0,1. Этот раствор подвергают микрофильтрации (Millipore Corp., фильтр Millex-HV, 0,45 мкм, мембрана ПВДФ) для удаления беловатого вещества. Этот раствор подвергают очистке ультрафильтрацией с применением аппарата для ультрафильтрации, состоящего из ультрафильтрационной мембраны (Merck Japan, Pellicon XL Cassette, Biomax 50 КДa), трубчатого насоса (Cole-Parmer International, MasterFlex Pump модель 77521-40, Pump Head модель 7518-00) и трубки (Cole-Parmer International, MasterFlex Tube L/S16). Более конкретно, добавляя по каплям ABS (всего 800 мл) в качестве буферного раствора для очистки реакционного раствора, ультрафильтрационную очистку проводят для удаления не конъюгированных связующих групп лекарственного средства и других низкомолекулярных реагентов, также заменяя буферный раствор на ABS и затем на концентрирующий раствор. Полученный очищенный раствор подвергают микрофильтрации (0,22 мкм (Millipore Corp., фильтр Millex-GV, мембрана ПВДФ) и 0,10 мкм (Millipore Corp., фильтр Millex-VV, мембрана ПВДФ)) с получением 42,5 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общих методик E и F (применяют εD,280=5178 (измеренное значение) и εD,370=20217 (измеренное значение)), получают следующие характеристики.
Концентрация антитела: 10,4 мг/мл, выход антитела: 442 мг (88,5%), среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела измерено с применением общей методики E: 6,0 и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела измерено с применением общей методики F: 7,5.
Пример 47. Конъюгат антитела с лекарственным средством (47)
[Формула 73]
Способ 1: Конъюгат антитела с лекарственным средством (47)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (15 мл) помещают в полипропиленовую пробирку и загружают водным раствором 10 мМ ТКЭФ (0,567 мл; 5,5 эквивалента на молекулу антитела) и водным раствором 1М вторичного кислого фосфата калия (0,225 мл). После подтверждения того, что раствор имеет pH 7,0±0,1, дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 2 часов.
Конъюгирование между антителом и связующей группой лекарственного средства: После добавления ДМСО (0,146 мл) и раствора ДМСО, содержащего 10 мМ соединения из способа 8 из примера 26 (0,928 мл; 9,0 эквивалента на молекулу антитела), к полученному выше раствору при комнатной температуре, его инкубируют для конъюгирования связующей группы лекарственного средства с антителом при 15°C в течение 30 минут. Затем добавляют водный раствор (0,133 мл; 12,9 эквивалента на молекулу антитела) 100 мМ NAC и перемешивают при комнатной температуре для превращения реакционной способности непрореагировавших связующих групп лекарственного средства в течение еще 20 минут.
Очистка: Полученный выше раствор очищают с применением общей методики D (ABS применяют в качестве буферного раствора) с получением 49 мл раствора, содержащего целевое соединение.
Физико-химические характеристики: С применением общей методики E (применяют εD,280=5178 и εD,370=20217), получают следующие характеристики.
Концентрация антитела: 2,91 мг/мл, выход антитела: 143 мг (95%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,2.
Пример 48. Конъюгат антитела с лекарственным средством (48)
[Формула 74]
Способ 1: Конъюгат антитела с лекарственным средством (48)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (280 мл) помещают в 1000 мл поликарбонатную колбу Эрленмейера, загружают водным раствором 1M вторичного кислого фосфата калия при комнатной температуре (4,200 мл) при перемешивании с применением магнитной мешалки и затем загружают водным раствором 10 мМ ТКЭФ (10,594 мл; 5,5 эквивалента на молекулу антитела). После подтверждения того, что раствор имеет pH 7,4±0,1, перемешивание останавливают и дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 2 часов.
Конъюгирование между антителом и связующей группой лекарственного средства: После охлаждения полученного выше раствора до 15°C постепенно по каплям добавляют раствор ДМСО, содержащий 10 мМ соединения из способа 8 из примера 26 (17,335 мл; 9,0 эквивалента на молекулу антитела) при перемешивании. Реакционный раствор перемешивают при 15°C для конъюгирования связующей группы лекарственного средства с антителом в течение 30 минут. Затем добавляют водный раствор (2,485 мл; 12,9 эквивалента на молекулу антитела) 100 мМ NAC при перемешивании и перемешивают при комнатной температуре для остановки реакционной способности непрореагировавших связующих групп лекарственного средства в течение еще 20 минут.
Очистка: Постепенное добавление 20% водного раствора уксусной кислоты (около 1,4 мл) и ABS (280 мл) к полученному выше раствору при перемешивании позволяет довести pH раствора до 5,5±0,1. Этот раствор подвергают микрофильтрации (0,45 мкм, мембрана ПВДФ) для удаления беловатого вещества, получая около 600 мл фильтрата. Этот раствор подвергают очистке ультрафильтрацией с применением аппарата для ультрафильтрации, состоящего из ультрафильтрационной мембраны (Merck Japan, Pellicon XL Cassette, Biomax 50 КДa), трубчатого насоса (Cole-Parmer International, MasterFlex Pump модель 77521-40, Pump Head модель 7518-00) и трубки (Cole-Parmer International, MasterFlex Tube L/S16). Более конкретно, добавляя по каплям ABS (всего 4800 мл) в качестве буферного раствора для очистки реакционного раствора, ультрафильтрационную очистку проводят для удаления не конъюгированных связующих групп лекарственного средства и других низкомолекулярных реагентов, также заменяя буферный раствор на ABS и затем на концентрирующий раствор. Полученный очищенный раствор подвергают микрофильтрации (дважды с 0,22 мкм и 0,10 мкм, мембрана ПВДФ) с получением 70 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общей методики E (применяют εD,280=5178 и εD,370=20217), получают следующие характеристики.
Концентрация антитела: 35,96 мг/мл, выход антитела: 2517 мг (90%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,2.
Пример 49. Конъюгат антитела с лекарственным средством (49)
[Формула 75]
Способ 1: Конъюгат антитела с лекарственным средством (49)
Восстановление антитела: Трастузумаб, полученный в ссылочном примере 1, получают с концентрацией антитела 10 мг/мл заменой среды на ФРФБ6,0/ЭДТК с применением общей методики C-1 и общей методики B (в качестве коэффициента абсорбции при 280 нм применяют 1,48 млмг-1см-1). Раствор (280 мл) помещают в 1000 мл поликарбонатную колбу Эрленмейера, загружают водным раствором 1M вторичного кислого фосфата калия при комнатной температуре (4200 мл) при перемешивании с применением магнитной мешалки и затем загружают водным раствором 10 мМ ТКЭФ (10,594 мл; 5,5 эквивалента на молекулу антитела). После подтверждения того, что раствор имеет pH 7,0±0,1, перемешивание останавливают и дисульфидную связь в шарнирной части антитела восстанавливают инкубированием при 37°C в течение 2 часов.
Конъюгирование между антителом и связующей группой лекарственного средства: После охлаждения полученного выше раствора до 15°C постепенно по каплям добавляют раствор ДМСО, содержащий 10 мМ соединения из способа 8 из примера 26 (17,335 мл; 9,0 эквивалента на молекулу антитела) при перемешивании. Реакционный раствор перемешивают при 15°C для конъюгирования связующей группы лекарственного средства с антителом в течение 30 минут. Затем добавляют водный раствор (2,485 мл; 12,9 эквивалента на молекулу антитела) 100 мМ NAC при перемешивании и перемешивают при комнатной температуре для остановки реакционной способности непрореагировавших связующих групп лекарственного средства в течение еще 20 минут.
Очистка: Постепенное добавление 20% водного раствора уксусной кислоты (около 1,4 мл) и ABS (280 мл) к полученному выше раствору при перемешивании позволяет довести pH раствора до 5,5±0,1. Этот раствор подвергают микрофильтрации (0,45 мкм, мембрана ПВДФ) для удаления беловатого вещества, получая около 600 мл фильтрата. Этот раствор подвергают очистке ультрафильтрацией с применением аппарата для ультрафильтрации, состоящего из ультрафильтрационной мембраны (Merck Japan, Pellicon XL Cassette, Biomax 50 КДa), трубчатого насоса (Cole-Parmer International, MasterFlex Pump модель 77521-40, Pump Head модель 7518-00) и трубки (Cole-Parmer International, MasterFlex Tube L/S16). Более конкретно, добавляя по каплям ABS (всего 4800 мл) в качестве буферного раствора для очистки реакционного раствора, ультрафильтрационную очистку проводят для удаления не конъюгированных связующих групп лекарственного средства и других низкомолекулярных реагентов, также заменяя буферный раствор на ABS и затем на концентрирующий раствор. Полученный очищенный раствор подвергают микрофильтрации (дважды с 0,22 мкм и 0,10 мкм, мембрана ПВДФ) с получением 130 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общей методики E (применяют εD,280=5178 и εD,370=20217), получают следующие характеристики.
Концентрация антитела: 21,00 мг/мл, выход антитела: 2730 мг (97,5%) и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела: 6,3.
Пример 50. Конъюгат антитела с лекарственным средством (50)
Способ 1: Конъюгат антитела с лекарственным средством (50)
Конъюгат антитела с лекарственными препаратами (47), (48) и (49), полученными в примерах 47, 48 и 49, смешивают (243 мл) и затем загружают ABS (39,75 мл) с получением 283 мл раствора, содержащего указанный в заголовке конъюгат антитела с лекарственным средством.
Физико-химические характеристики: С применением общих методик E и F (применяют εD,280=5178 и εD,370=20217), получают следующие характеристики.
Концентрация антитела: 20,0 мг/мл, выход антитела: 5655 мг, среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела измерено с применением общей методики E: 6,3 и среднее количество конъюгированных молекул лекарственного средства (n) на молекулу антитела измерено с применением общей методики F: 7,8.
Экспериментальный пример 1. Ингибирование роста клеток (1) конъюгатом антитела с лекарственным средством
Колонию клеток рака молочной железы человека KPL-4 HER2 антиген-положительных клеток (Dr. Junichi Kurebayashi, Kawasaki Medical School, British Journal of Cancer, (1999) 79 (5/6). 707-717) или колонию клеток рака молочной железы человека MCF7 антиген-отрицательных клеток (European Collection of Cell Cultures; ECACC) культивируют в RPMI1640 (GIBCO), содержащей 10% фетальной телячьей сыворотки (MOREGATE) (далее обозначена как среда). KPL-4 или MCF7 получают в концентрации 2,5×104 клеток/мл с применением среды, добавленной в концентрации 100 мкл/лунку в 96-луночный микропланшет для культивирования клеток, и культивируют в течение ночи.
На следующий день трастузумаб или конъюгат антитела с лекарственным средством, разведенный в 1000 нм, 200 нм, 40 нм, 8 нм, 1,6 нм, 0,32 нм и 0,064 нМ с применением среды, добавляют в концентрации 10 мкл/лунку в микропланшет. Среду добавляют в концентрации 10 мкл/лунку в не содержащие антитело лунки. Клетки культивируют под 5% CO2 при 37°C в течение от 5 до 7 дней. После культивирования микропланшет вынимают из инкубатора и выдерживают при комнатной температуре в течение 30 минут. В культуральный раствор загружают равное количество CellTiter-Glo Luminescent Cell Viability Assay (Promega) и перемешивают. После выдерживания микропланшета при комнатной температуре в течение 10 минут, интенсивность люминесценции каждой лунки измеряют с применением планшетного ридера (PerkinElmer). Значение IC50 рассчитывают согласно следующему уравнению:
IC50 (нМ)=antilog((50-d)×(LOG10(b)-LOG10(a))/(d-c)+LOG10(b))
a: Концентрация образца a
b: Концентрация образца b
c: Жизнеспособность клетки образца a
d: Жизнеспособность клетки образца b
Жизнеспособность клетки в каждой концентрации рассчитывают согласно следующему уравнению:
Жизнеспособность клетки (%)=a/b×100
a: Средняя интенсивность люминесценции в лунках с образцом (n=2)
b: Средняя интенсивность люминесценции в лунках без добавления антитела (n=10)
Конъюгат антитела с лекарственными препаратами (2), (3), (5), (7), (10), (12), (13), (16), (18), (40) и (42) демонстрирует ингибирование роста клеток IC50<0,1 (нМ) против клеток KPL-4.
Конъюгат антитела с лекарственными препаратами (4), (6), (9), (15), (17), (21), (22), (25), (36), (37), (39), (41) и (43) демонстрирует ингибирование роста клеток 0,1<IC50<1 (нМ) против клеток.
Конъюгат антитела с лекарственными препаратами (20), (24) и (27) демонстрирует ингибирование роста клеток 1<IC50<100 (нМ) против клеток. Ни один из конъюгатов антитела с лекарственными препаратами (19) или (26) не демонстрирует ингибирование роста клеток против клеток (>100 (нМ)).
С другой стороны, конъюгат антитела с лекарственными препаратами (5), (13) и (43) демонстрирует ингибирование роста клеток 1<IC50<100 (нМ) против клеток MCF7, в то время как ни один из конъюгатов антитела с лекарственными препаратами (2), (3), (4), (6), (7), (9), (10), (12), (15), (16), (17), (18), (25), (26), (27), (39), (40), (41), (42) и (44) не демонстрирует ингибирование роста клеток против клеток (>100 (нМ)).
Трастузумаб демонстрирует ингибирование роста клеток ни против клеток KPL-4, ни против клеток MCF7 (>100 (нМ)).
Экспериментальный пример 2. Противоопухолевый тест (1)
Мышь: 5-6-недельные самки безтимусных мышей (Charles River Laboratories Japan, Inc.) акклиматизируют в течение от 4 до 7 дней в условиях SPF перед применением в эксперименте. Мышей кормят стерилизованным твердым кормом (FR-2, Funabashi Farms Co., Ltd) и дают стерилизованную водопроводную воду (полученную добавлением от 5 до 15 ч./млн. раствора гипохлорита натрия).
Анализ и расчетное уравнение: во всех экспериментах, большие оси и малые оси опухоли измеряют дважды в неделю с применением штангенциркуля с электронным цифровым отсчетом (CD-15CX, Mitutoyo Corp.) и рассчитывают объем опухоли (мм3). Расчетное уравнение показано ниже.
Объем опухоли (мм3)=1/2×большие оси (мм)×[малые оси (мм)]2
Все конъюгаты антитела с лекарственными препаратами и антитела разводят физиологическим раствором (Otsuka Pharmaceutical Factory, Inc.) и применяют в объеме 10 мл/кг для внутривенного введения в хвостовую вену каждой мыши.
Клетки KPL-4 суспендируют в физиологическом растворе и 1,5×107 клеток подкожно трансплантируют на правую сторону тела каждой самки безтимусной мыши (День 0) и мышей произвольно делят на группы на 15 день. Конъюгат антитела с лекарственным средством (27) или анти-HER2 антитело трастузумаб (Ссылочный пример 1) для контрольной группы внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши в дни 15 и 22. Необработанную группу назначают контрольной группой.
Результаты показаны на фигуре 3. Введение трастузумаба ингибирует рост опухоли, в то время как введение конъюгата антитела с лекарственным средством (27) дает более значительное ингибирование роста опухоли. На фигуре, на абсциссе даны дни после инокуляции опухоли, и на ординате дан объем опухоли. Кроме того, мыши, получавшие трастузумаб или конъюгат антитела с лекарственным средством (27) не имели видимых признаков, таких как потеря веса, что позволяет предположить, что конъюгат антитела с лекарственным средством (27) в высокой степени безопасен. В экспериментальных примерах ниже, относящихся к противоопухолевому тесту, тест проводят по методике, применяемой в данном экспериментальном примере, если не указано иначе.
Экспериментальный пример 3. Противоопухолевый тест (2)
Колонию клеток рака желудка человека NCI-N87, купленную в ATCC (American Type Culture Collection), суспендируют в физиологическом растворе и 1×107 клеток подкожно трансплантируют на правую сторону тела каждой самки безтимусной мыши (день 0), и мышей произвольно делят на группы на 7 день. Конъюгат антитела с лекарственным средством (8) или (28) или трастузумаб эмтанзин (Ссылочный пример 2) внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 7 день. Необработанную группу назначают контрольной группой.
Результаты показаны на фигуре 4. Подтверждается, что конъюгат антитела с лекарственными препаратами (8) и (28) обладает сильным противоопухолевым действием при регрессии опухоли эквивалентной той, которая достигается с трастузумабом эмтанзином. В дополнение, введение конъюгата антитела с лекарственным средством (8) или (28) или трастузумаба эмтанзина не вызывает потерю веса у мышей.
Экспериментальный пример 4. Противоопухолевый тест (3)
Колонию клеток рака молочной железы человека JIMT-1, купленных у DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH), суспендируют в физиологическом растворе и 3×106 клеток подкожно трансплантируют на правую сторону тела каждой самки безтимусной мыши (день 0) и мышей произвольно делят на группы на 12 день. Конъюгат антитела с лекарственным средством (8), (29) или (30), трастузумаб или трастузумаб эмтанзин внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 12 и 19 дни. Группу, которой вводят физиологический раствор, назначают контрольной группой.
Результаты показаны на фигуре 5. Введение трастузумаба или трастузумаба эмтанзин не ингибирует роста опухоли JIMT-1. С другой стороны, введение конъюгата антитела с лекарственным средством (8), (29) или (30) ингибирует роста опухоли. Кроме того, введение конъюгата антитела с лекарственным средством (8), (29) или (30), трастузумаба или трастузумаба эмтанзина не вызывает потерю веса у мыши.
Экспериментальный пример 5. Ингибирование роста клеток (2) конъюгатом антитела с лекарственным средством
Колонию клеток немелкоклеточного рака легких человека Calu-3 (ATCC) культивируют в минимальной эссенциальной среде Игла (GIBCO), содержащей 10% фетальную бычью сыворотку (MOREGATE) (далее названа МЭС среда).
Колонию клеток рака желудка человека NCI-N87 (ATCC) или колонию клеток рака желудка человека MKN-45 (Health Science Research Resources Bank) культивируют в среде RPMI1640 (GIBCO), содержащей 10% фетальную бычью сыворотку (далее названа RPMI среда).
Колонию клеток рака молочной железы человека MDA-MB-453 (ATCC) или колонию клеток рака молочной железы человека MDA-MB-468 (ATCC) культивируют в среде Лейбовица L-15 (GIBCO), содержащей 10% фетальную бычью сыворотку (далее названа среда Лейбовица).
Среди этих 5 типов колоний клеток Calu-3, NCI-N87 и MDA-MB-453 являются HER2-положительными клетками и MKN-45 и MDA-MB-468 являются HER2-отрицательными клетками.
Calu-3, NCI-N87 или MKN-45 получают в концентрации 4×104 клеток/мл с применением МЭС среды или RPMI среды, добавленной в концентрации 25 мкл/лунку в 96-луночный микропланшет для культивирования клеток, загружают 65 мкл/лунку среды и культивируют в течение ночи под 5% CO2 при 37°C. Также, MDA-MB-453 или MDA-MB-468 получают в концентрации 4×104 клеток/мл с применением среды Лейбовица, добавленной в концентрации 25 мкл/лунку в 96-луночный микропланшет для культивирования клеток, загружают 65 мкл/лунку среды и культивируют в течение ночи при 37°C без установки концентрации CO2.
На следующий день тестируемое вещество, разведенное в 1000 нм, 200 нм, 40 нм, 8 нм, 1,6 нм, 0,32 нм и 0,064 нМ с применением RPMI среды или среды Лейбовица или RPMI среды или среды Лейбовица, добавляют в концентрации 10 мкл/лунку в микропланшет. Клетки культивируют под 5% CO2 при 37°C или при 37°C без установки концентрации CO2 в течение 6 дней.
Для Calu-3, NCI-N87 и MDA-MB-468, конъюгат антитела с лекарственным средством (46) добавляют в качестве тестируемого вещества. Для других клеток, конъюгат антитела с лекарственным средством (50) добавляют в качестве тестируемого вещества. После культивирования микропланшет вынимают из инкубатора и выдерживают при комнатной температуре в течение 30 минут. В культуральный раствор загружают в равном количестве CellTiter-Glo Luminescent Cell Viability Assay (Promega) и перемешивают с применением планшетного миксера для полного лизиса клеток. После выдерживания микропланшета при комнатной температуре в течение 10 минут, интенсивность люминесценции измеряют с помощью планшетного ридера.
Жизнеспособность клетки рассчитывают согласно следующего уравнения:
Жизнеспособность клетки (%)=a/b×100
a: Средняя интенсивность люминесценции лунок с добавлением тестируемого вещества
b: Средняя интенсивность люминесценции лунок с добавлением среды
Значение IC50 рассчитывают согласно следующему уравнению:
IC50 (нМ)=antilog((50-d)×(LOG10(b)-LOG10(a))/(d-c)+LOG10(b))
a: Концентрация a тестируемого вещества
b: Концентрация b тестируемого вещества
c: Жизнеспособность клетки с добавлением тестируемого вещества, имеющего концентрацию a
d: Жизнеспособность клетки с добавлением тестируемого вещества, имеющего концентрацию b
Концентрации a и b устанавливают отношение a>b переходящее 50% жизнеспособности клетки.
Конъюгат антитела с лекарственным средством (46) демонстрирует ингибирование роста клеток IC50<1 (нМ) против HER2-положительных клеток Calu-3 и NCI-N87. С другой стороны, конъюгат антитела с лекарственным средством (46) не демонстрирует ингибирование роста клеток против HER2-отрицательных клеток MDA-MB-468 (>100 (нМ)).
Конъюгат антитела с лекарственным средством (50) демонстрирует ингибирование роста клеток IC50<1 (нМ) против HER2-положительных клеток MDA-MB-453. С другой стороны, конъюгат антитела с лекарственным средством (50) не демонстрирует ингибирование роста клеток против HER2-отрицательных клеток MKN-45 (>100 (нМ)).
Экспериментальный пример 6. Противоопухолевый тест (4)
Колонию клеток рака поджелудочной железы Capan-1 (ATCC), слабо экспрессирующих HER2, суспендируют в физиологическом растворе и 4×107 клеток подкожно трансплантируют на правую сторону тела каждой самки безтимусной мыши для получения солидной опухоли Capan-1. Затем эту твердую опухоль поддерживают в несколько заходов трансплантацией самкам безтимусных мышей, применяемым в этом тесте. Часть солидной опухоли подкожно трансплантируют на правую сторону тела каждой самки безтимусной мыши (день 0) и мышей произвольно делят на группы на 20 день.
Конъюгат антитела с лекарственным средством (31), трастузумаб или трастузумаб эмтанзин внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 20 день. Группу, которой вводят физиологический раствор, назначают контрольной группой.
Результаты показаны на фигуре 6. Введение трастузумаба или трастузумаба эмтанзина не ингибирует рост опухоли Capan-1. Наоборот, введение конъюгата антитела с лекарственным средством (31) ингибирует рост опухоли, демонстрируя, что конъюгат антитела с лекарственным средством (31) является эффективным даже для опухоли с низкой экспрессией HER2. Конъюгат антитела с лекарственным средством (31) не ингибирует рост GCIY колонии клеток рака желудка опухоли не экспрессирующей HER2.
Что касается экспрессии HER2 в опухоли, на основе результатов измерений иммуногистохимического окрашивания, описанного в 3-м издании Руководства для тестирования HER2 (разработанного Japanese Pathology Board for Optimal Use of Trastuzumab, The Japanese Society of Pathology), классификацию проводят таким образом, что оценка 3+: высокая экспрессия, 2+: умеренная экспрессия и 1+: низкая экспрессия. Даже если оценка 0 в данном способе измерения, опухоль определяется как HER2-положительная с помощью других способов измерения, таких как измерение с применением проточного цитометра и классифицируется как опухоль с низкой экспрессией.
Экспериментальный пример 7. Противоопухолевый тест (5)
Колонию клеток рака желудка человека NCI-N87, купленную у ATCC, суспендируют в физиологическом растворе и 1×107 клеток подкожно трансплантируют на правую сторону тела каждой самки безтимусной мыши (день 0), и мышей произвольно делят на группы на 6 день. Конъюгат антитела с лекарственным средством (50) внутривенно вводят в каждой дозе 0,3, 1, 3 или 10 мг/кг в хвостовую вену каждой мыши на 6 день. Группу, которой вводят ацетатный буферный раствор, назначают контрольной группой.
Результаты показаны на фигуре 7. Конъюгат антитела с лекарственным средством (50) демонстрирует дозозависимое противоопухолевое действие. Кроме того, введение конъюгата антитела с лекарственным средством (50) не вызывает потерю веса мыши.
Экспериментальный пример 8. Противоопухолевый тест (6)
Этот тест проводят следующим способом.
Мышь: 6-12-недельные самки безтимусных мышей (Charles River Laboratories Japan, Inc.) подвергают эксперименту.
Анализ и расчетное уравнение: большие оси и малые оси опухоли измеряют дважды в неделю с применением штангенциркуля с электронным цифровым отсчетом и рассчитывают объем опухоли (мм3). Расчетное уравнение показано ниже.
Объем опухоли (мм3)=0,52×большие оси (мм)×[малые оси (мм)]2
Конъюгат антитела с лекарственным средством, трастузумаб и трастузумаб эмтанзин разводят ацетатным буферным раствором и применяют в объеме 10 мл/кг для внутривенного введения в хвостовую вену каждой мыши.
Опухоль (ST225; South Texas Accelerated Research Therapeutics (START)) иссекают у пациента с раком молочной железы и сохраняют в несколько заходов трансплантацией самкам безтимусных мышей, применяемым в этом тесте. Эта опухоль умеренно экспрессирует HER2 (получила оценку 2+ при иммуногистохимическом окрашивании).
Часть солидной опухоли подкожно трансплантируют на сторону тела каждой самки безтимусной мыши, и мышей произвольно делят на группы, когда объем опухоли достигает от 100 до 300 мм3. Дату разбивки на группы определяют как день 0. Конъюгат антитела с лекарственным средством (50), трастузумаб или трастузумаб эмтанзин внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 0 день. Группу, которой вводят ацетатный буферный раствор, назначают контрольной группой.
Результаты показаны на фигуре 8. Введение трастузумаба не ингибирует рост опухоли молочной железы ST225, умеренно экспрессирующей HER2. Наоборот, введение трастузумаба эмтанзина или конъюгата антитела с лекарственным средством (50) значительно ингибирует рост опухоли.
Экспериментальный пример 9. Противоопухолевый тест (7)
Опухоль (ST910; START) иссекают у пациента с раком молочной железы и сохраняют в несколько заходов трансплантацией самкам безтимусных мышей, применяемым в этом тесте. Эта опухоль слабо экспрессирует HER2 (получила оценку 1+ при иммуногистохимическом окрашивании).
Часть солидной опухоли подкожно трансплантируют на сторону тела каждой самки безтимусной мыши и мышей произвольно группируют, когда объем опухоли достигает от 100 до 300 мм3. Дату разделения на группы определяют как день 0. Конъюгат антитела с лекарственным средством (50), трастузумаб или трастузумаб эмтанзин внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 0 день. Группу, которой вводят ацетатный буферный раствор, назначают контрольной группой.
Результаты показаны на фигуре 9. Введение трастузумаба или трастузумаба эмтанзина не ингибирует рост клеток рака молочной железы ST910, слабо экспрессирующих HER2. Наоборот, введение конъюгата антитела с лекарственным средством (50) значительно ингибирует рост опухоли, демонстрируя, что конъюгат антитела с лекарственным средством (50) является эффективным для рака молочной железы, слабо экспрессирующего HER2. Этот экспериментальный пример 9 проводят по методике экспериментального примера 8.
Экспериментальный пример 10. Противоопухолевый тест (8)
Этот тест проводят по следующей методике. Экспериментальные примеры 11-13 также проводят по этой методике.
Мышь: 5-8-недельных самок безтимусных мышей (Harlan Laboratories Ltd.) используют в эксперименте.
Анализ и расчетное уравнение: большие оси и малые оси опухоли измеряют дважды в неделю с применением штангенциркуля с электронным цифровым отсчетом и рассчитывают объем опухоли (мм3). Расчетное уравнение показано ниже.
Объем опухоли (мм3)=0,52×большие оси (мм)×[малые оси (мм)]2
Конъюгат антитела с лекарственным средством, трастузумаб и трастузумаб эмтанзин разводят в растворе ацетатного буфера и применяют в объеме 10 мл/кг для внутривенного введения в хвостовую вену каждой мыши.
Опухоль (CTG-0401; Champions Oncology Inc.) иссекают у пациента с раком прямой и ободочной кишки и сохраняют в несколько заходов трансплантацией самкам безтимусных мышей, применяемым в этом тесте. Эта опухоль слабо и умеренно экспрессирует HER2 (получила оценку 1+ или 2+ при иммуногистохимическом окрашивании).
Часть солидной опухоли подкожно трансплантируют на левую сторону тела каждой самки безтимусной мыши и мышей произвольно группируют, когда объем опухоли достигает от 100 до 300 мм3. Дату разделения на группы определяют как день 0. Конъюгат антитела с лекарственным средством (50), трастузумаб или трастузумаб эмтанзин внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 0 день. Группу, которой вводят ацетатный буферный раствор, назначают контрольной группой.
Результаты показаны на фигуре 10. Введение трастузумаба или трастузумаба эмтанзина не ингибирует рост опухоли прямой и ободочной кишки CTG-0401, слабо или умеренно экспрессирующей HER2. Наоборот, введение конъюгата антитела с лекарственным средством (50) значительно ингибирует рост опухоли.
Экспериментальный пример 11 Противоопухолевый тест (9)
Опухоль (CTG-0860; Champions Oncology Inc.) иссекают у пациента с не мелкоклеточным раком легких и сохраняют в несколько заходов трансплантацией самкам безтимусных мышей, применяемым в этом тесте. Эта опухоль умеренно экспрессирует HER2 (получила оценку 2+ при иммуногистохимическом окрашивании).
Часть солидной опухоли подкожно трансплантируют на левую сторону тела каждой самки безтимусной мыши и мышей произвольно группируют, когда объем опухоли достигает от 100 до 300 мм3. Дату разделения на группы определяют как день 0. Конъюгат антитела с лекарственным средством (50), трастузумаб или трастузумаб эмтанзин внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 0 день. Группу, которой вводят ацетатный буферный раствор, назначают контрольной группой.
Результаты показаны на фигуре 11. Введение трастузумаба или трастузумаба эмтанзина не ингибирует рост немелкоклеточной опухоли легких CTG-0860, умеренно экспрессирующей HER2. Наоборот, введение конъюгата антитела с лекарственным средством (50) значительно ингибирует рост опухоли.
Экспериментальный пример 12. Противоопухолевый тест (10)
Опухоль (CTG-0927; Champions Oncology Inc.) иссекают у пациента с раком желчных протоков и сохраняют в несколько заходов трансплантацией самкам безтимусных мышей, применяемым в этом тесте. Эта опухоль сильно экспрессирует HER2 (получила оценку 3+ при иммуногистохимическом окрашивании).
Часть солидной опухоли подкожно трансплантируют на левую сторону тела каждой самки безтимусной мыши и мышей произвольно группируют, когда объем опухоли достигает от 100 до 300 мм3. Дату разделения на группы определяют как день 0. Конъюгат антитела с лекарственным средством (50), трастузумаб или трастузумаб эмтанзин внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 0 день. Группу, которой вводят ацетатный буферный раствор, назначают контрольной группой.
Результаты показаны на фигуре 12. Введение трастузумаба не ингибирует рост опухоли желчных протоков CTG-0927, сильно экспрессирующей HER2. Наоборот, введение трастузумаба эмтанзина ингибирует рост опухоли. Более того, введение конъюгата антитела с лекарственным средством (50) вызывает регрессию опухоли.
Экспериментальный пример 13. Противоопухолевый тест (11)
Опухоль (CTG-0137; Champions Oncology Inc.) иссекают у пациента с раком пищевода и сохраняют в несколько заходов трансплантацией самкам безтимусных мышей, применяемым в этом тесте. Эта опухоль сильно экспрессирует HER2 (получила оценку 3+ при иммуногистохимическом окрашивании).
Часть солидной опухоли подкожно трансплантируют на левую сторону тела каждой самки безтимусной мыши и мышей произвольно группируют, когда объем опухоли достигает от 100 до 300 мм3. Дату разделения на группы определяют как день 0. Конъюгат антитела с лекарственным средством (50), трастузумаб или трастузумаб эмтанзин внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 0 день. Группу, которой вводят ацетатный буферный раствор, назначают контрольной группой.
Результаты показаны на фигуре 13. Введение трастузумаба не ингибирует рост опухоли желчных протоков CTG-0137, сильно экспрессирующей HER2. Наоборот, введение трастузумаба эмтанзина или конъюгата антитела с лекарственным средством (50) значительно ингибирует рост опухоли.
Экспериментальный пример 14. Противоопухолевый тест (12)
Колонию клеток рака яичников человека SK-OV-3, сильно экспрессирующих HER2, купленную у ATCC, суспендируют в физиологическом растворе и 4×107 клеток подкожно трансплантируют на правую сторону тела каждой самки безтимусной мыши с получением солидной опухоли SK-OV-3. Затем эту твердую опухоль сохраняют в несколько заходов трансплантацией самкам безтимусных мышей и применяют в этом тесте.
Часть солидной опухоли подкожно трансплантируют на правую сторону тела каждой самки безтимусной мыши и мышей произвольно группируют, когда объем опухоли достигает от 100 до 300 мм3. Дату разделения на группы определяют как день 0. Конъюгат антитела с лекарственным средством (50), трастузумаб или трастузумаб эмтанзин внутривенно вводят в дозе 10 мг/кг в хвостовую вену каждой мыши на 0 день. Группу, которой вводят физиологический раствор, назначают контрольной группой.
Результаты показаны на фигуре 14. Ведение трастузумаба не ингибирует рост SK-OV-3 опухоли. Наоборот, введение трастузумаба эмтанзина или конъюгата антитела с лекарственным средством (50) значительно ингибирует рост опухоли.
Произвольный текст списка последовательностей
SEQ ID NO: 1 - Последовательность аминокислот тяжелой цепи гуманизированного анти-HER2 моноклонального антитела
SEQ ID NO: 2 - Последовательность аминокислот легкой цепи гуманизированного анти-HER2 моноклонального антитела
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Daiichi Sankyo Company, Limited
<120> КОНЪЮГАТ АНТИ-HER2 АНТИТЕЛА С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ
<130> PD20-9008WO
<150> JP2014-017777
<151> 2014-01-31
<150> JP2014-168944
<151> 2014-08-22
<150> JP2014-227886
<151> 2014-11-10
<160> 2
<170> PatentIn версия 3.5
<210> 1
<211> 450
<212> PRT
<213> Искусственная последовательность
<220>
<223> Последовательность аминокислот тяжелой цепи Трастузумаба
<400> 1
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Asn Ile Lys Asp Thr
20 25 30
Tyr Ile His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Arg Ile Tyr Pro Thr Asn Gly Tyr Thr Arg Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Ala Asp Thr Ser Lys Asn Thr Ala Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ser Arg Trp Gly Gly Asp Gly Phe Tyr Ala Met Asp Tyr Trp Gly Gln
100 105 110
Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val
115 120 125
Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala
130 135 140
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser
145 150 155 160
Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val
165 170 175
Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190
Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys
195 200 205
Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp
210 215 220
Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly
225 230 235 240
Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile
245 250 255
Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu
260 265 270
Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His
275 280 285
Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg
290 295 300
Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys
305 310 315 320
Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu
325 330 335
Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr
340 345 350
Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu
355 360 365
Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp
370 375 380
Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val
385 390 395 400
Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp
405 410 415
Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His
420 425 430
Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro
435 440 445
Gly Lys
450
<210> 2
<211> 214
<212> PRT
<213> Искусственная последовательность
<220>
<223> Последовательность аминокислот легкой цепи Трастузумаба
<400> 2
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Asp Val Asn Thr Ala
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Phe Leu Tyr Ser Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Arg Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln His Tyr Thr Thr Pro Pro
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИ-HER2 КОНЪЮГАТ АНТИТЕЛА С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ | 2015 |
|
RU2683780C2 |
| КОНЪЮГАТ АНТИ-HER3 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2015 |
|
RU2711640C2 |
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2013 |
|
RU2664465C2 |
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2013 |
|
RU2776489C2 |
| КОНЪЮГАТ АНТИ-TROP2 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2014 |
|
RU2705367C2 |
| КОНЪЮГАТ АНТИ-HER3 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2015 |
|
RU2802211C2 |
| КОНЪЮГАТ АНТИ-TROP2 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2021 |
|
RU2840864C2 |
| КОНЪЮГАТ АНТИ-TROP2 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2014 |
|
RU2743077C2 |
| КОНЪЮГАТЫ АНТИТЕЛА С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ | 2014 |
|
RU2669812C2 |
| КОНЪЮГАТ АНТИТЕЛО-ПРОИЗВОДНОЕ ПИРРОЛОБЕНЗОДИАЗЕПИНА | 2018 |
|
RU2820928C2 |


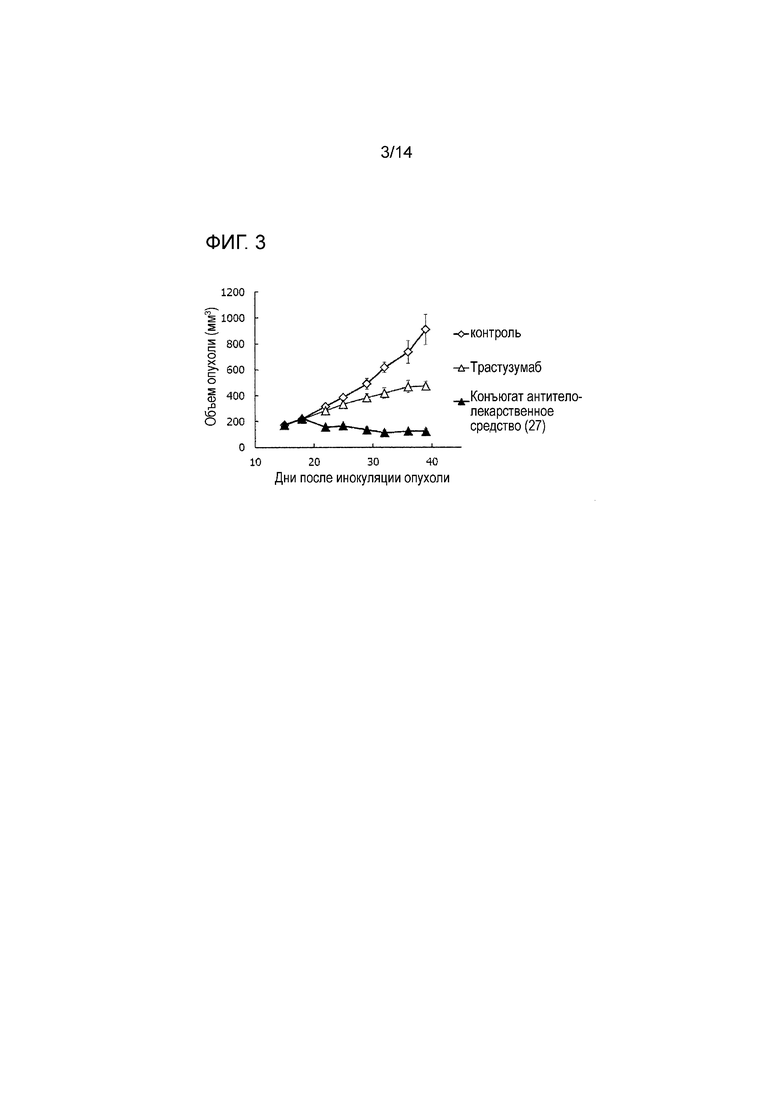
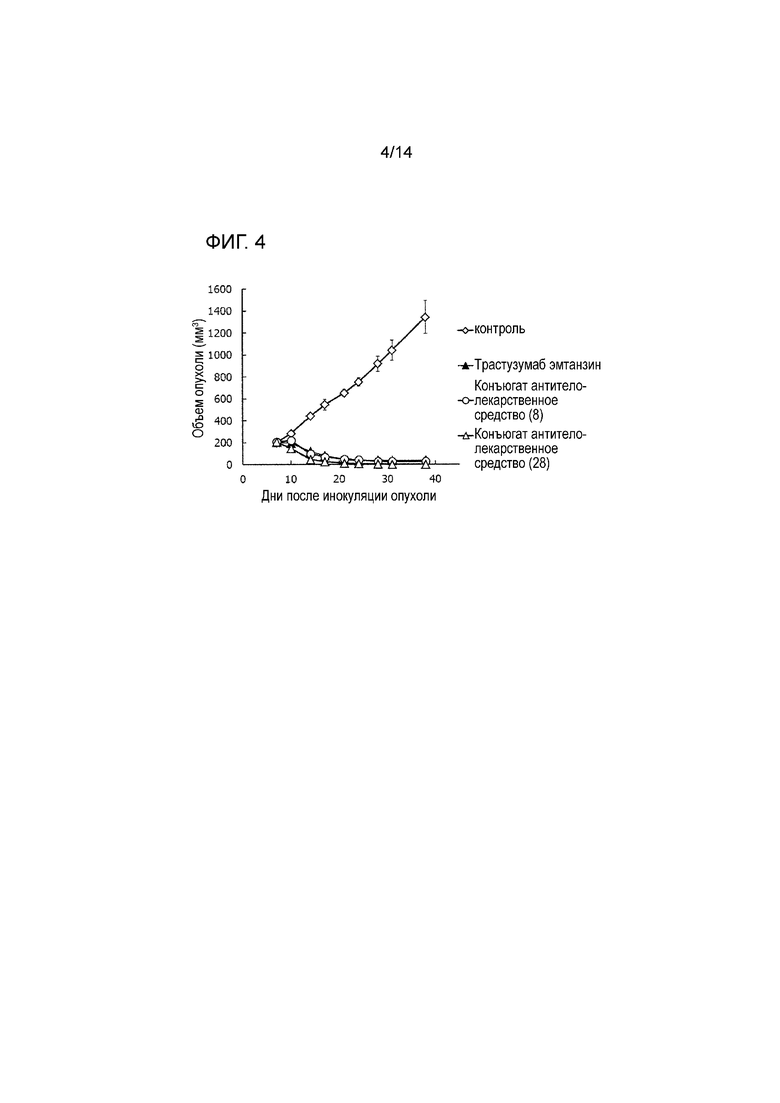
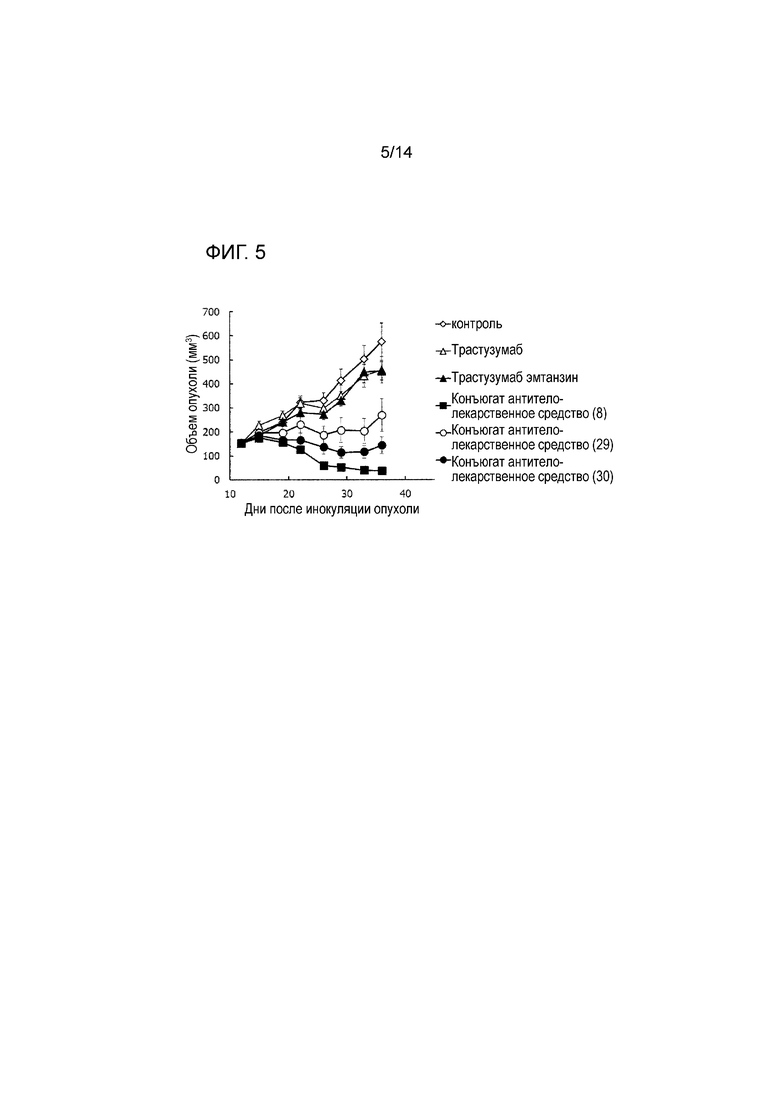
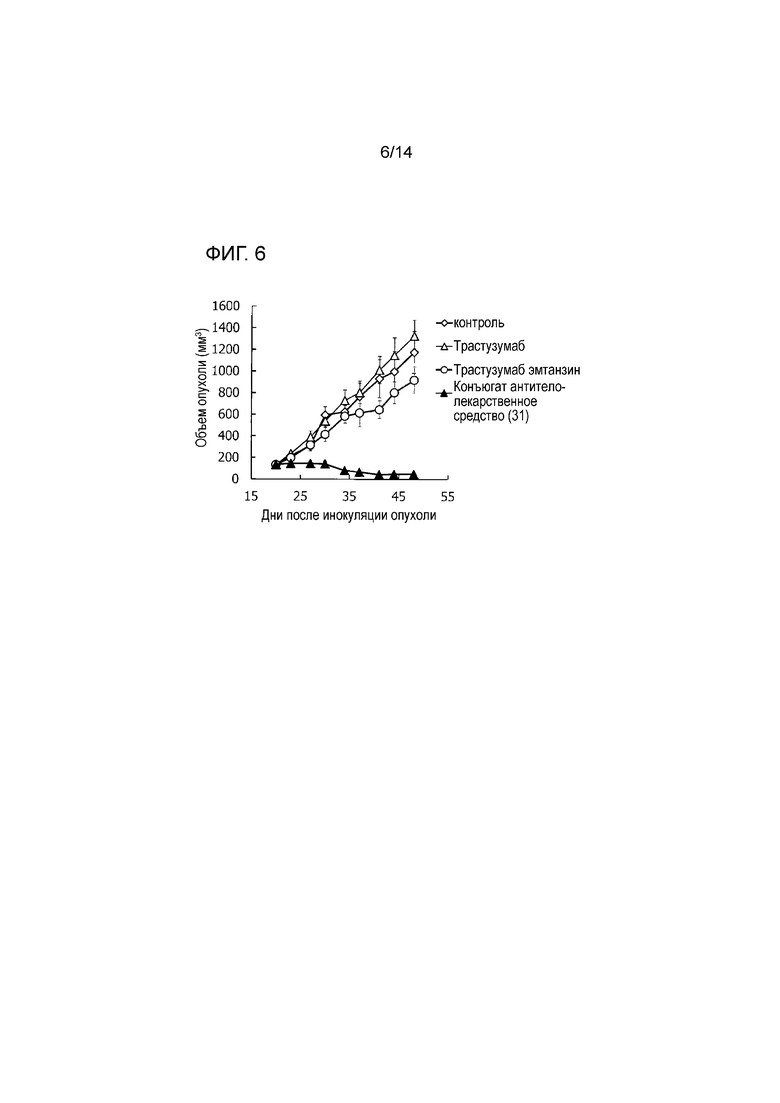
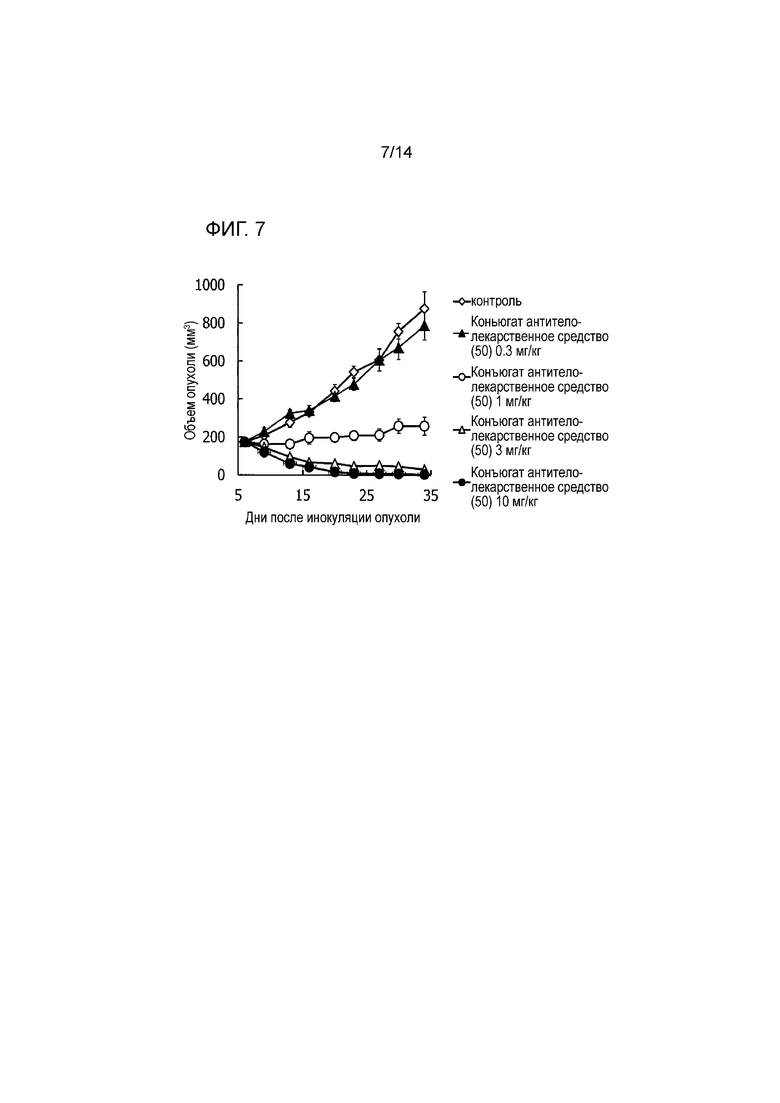
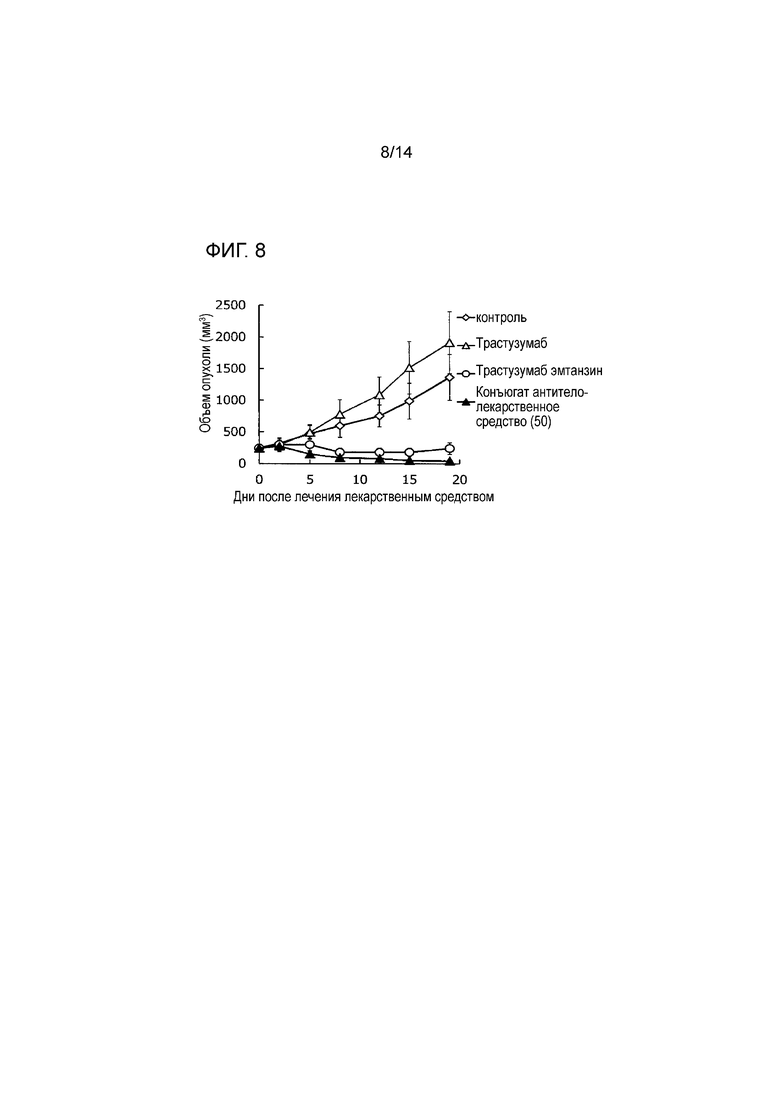
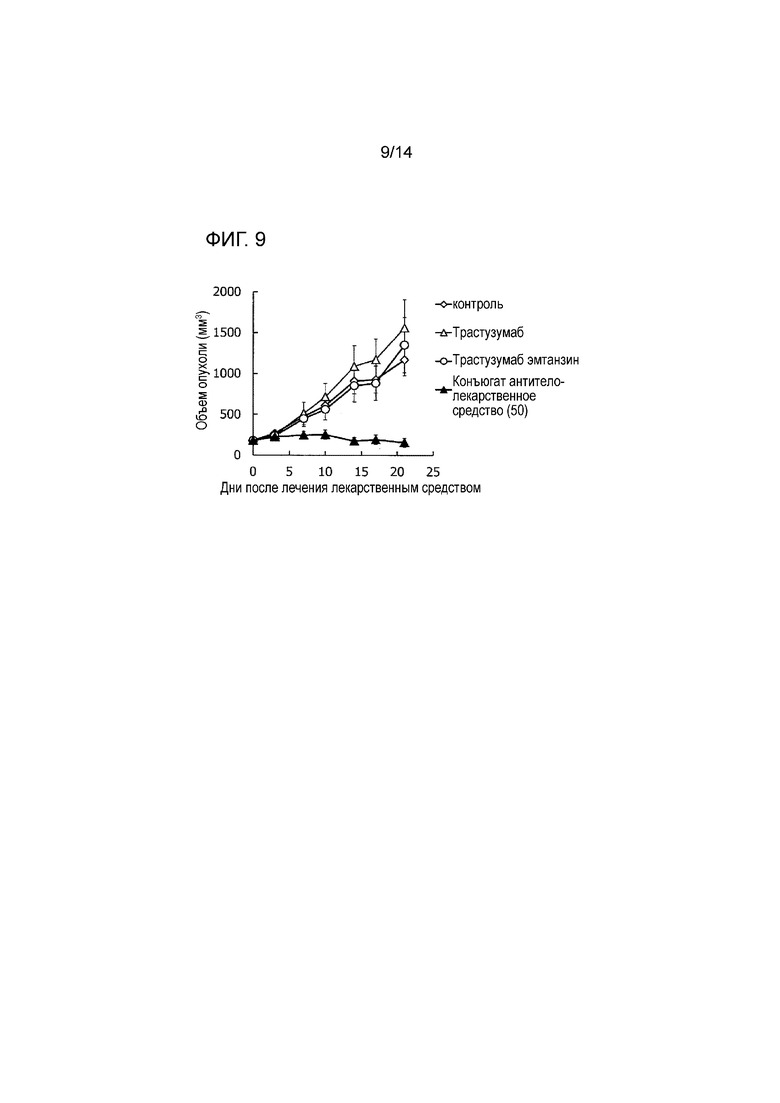
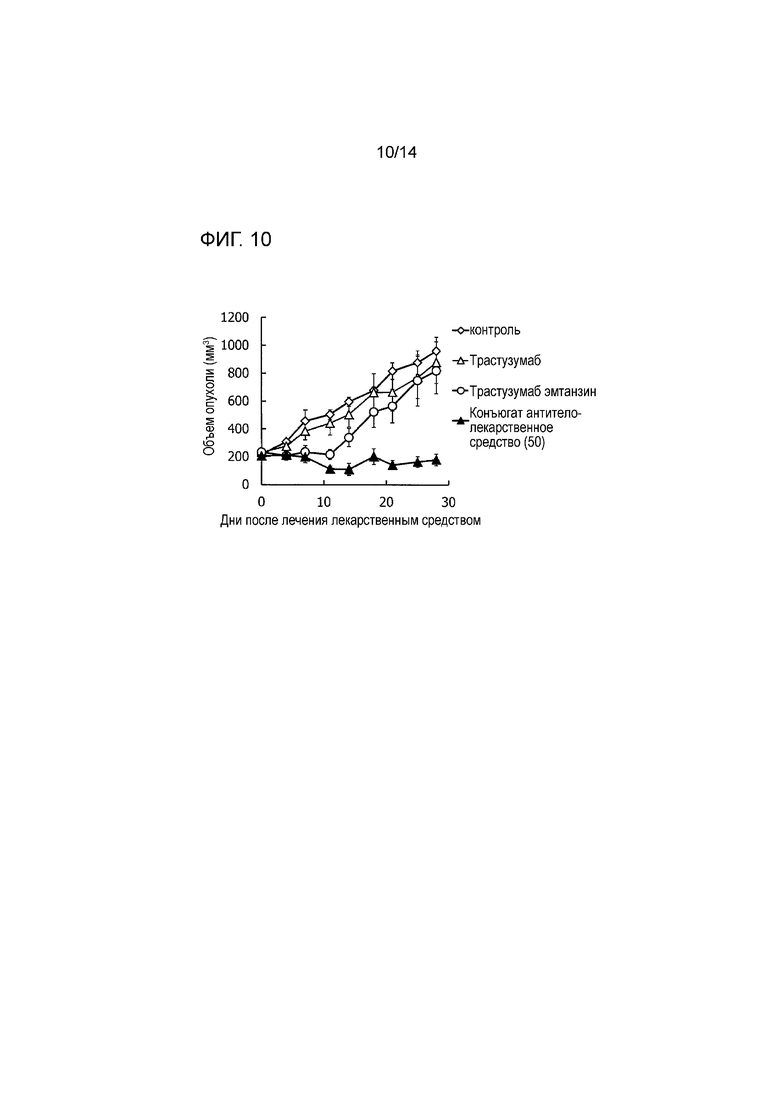
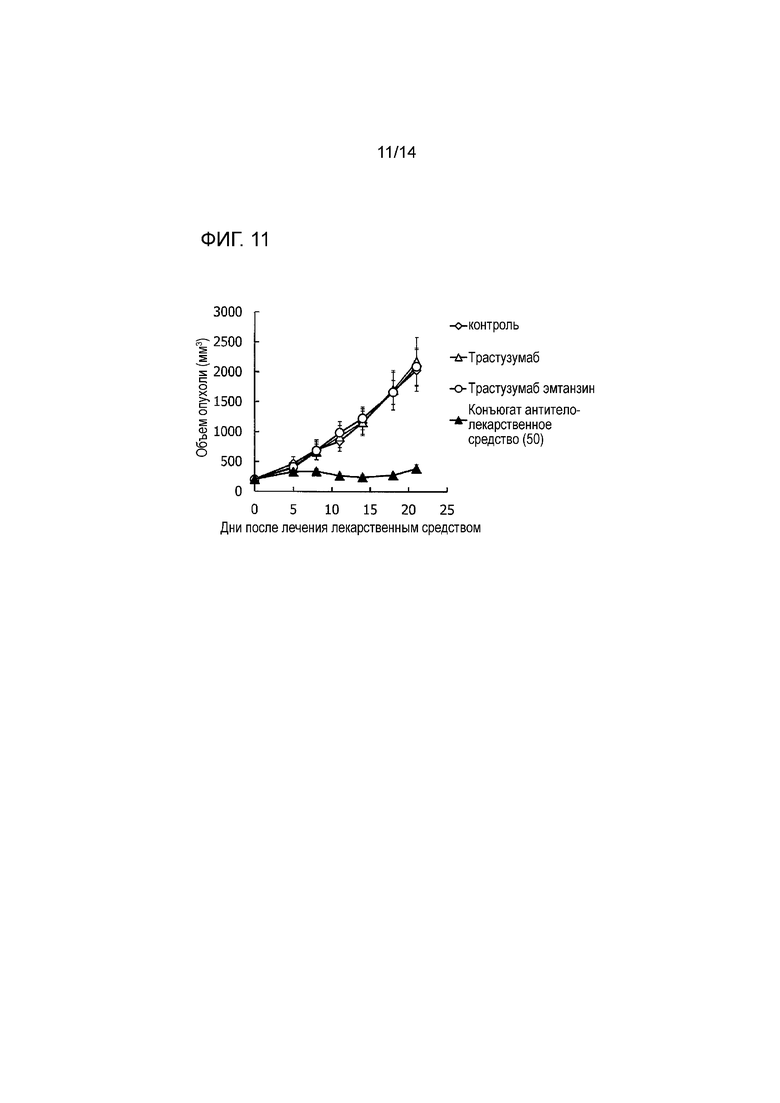
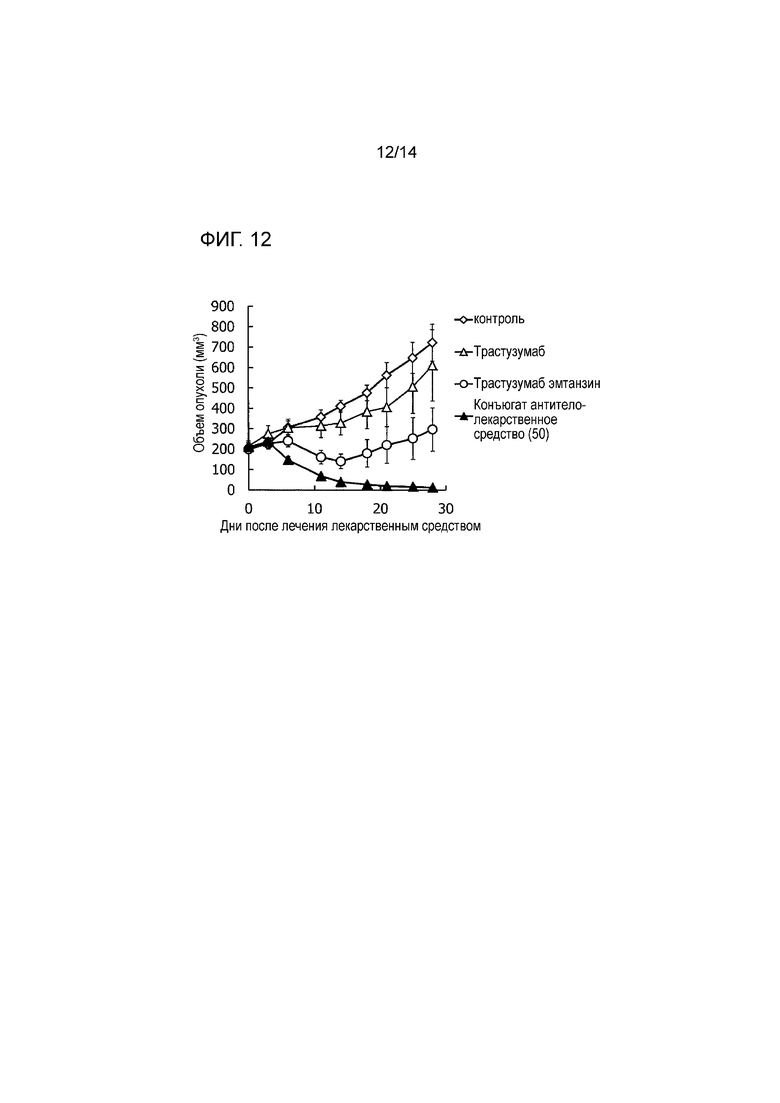
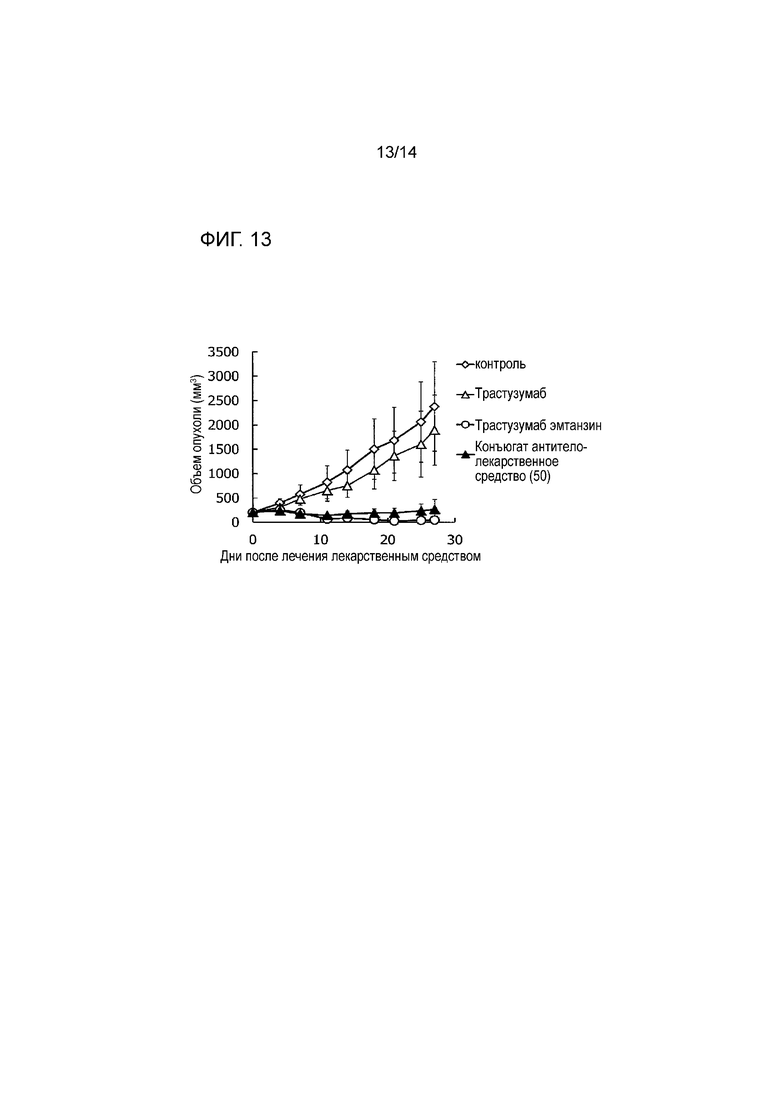

Изобретение относится к способу получения конъюгата антитела с лекарственным средством, представленного следующей формулой: (малеимид-N-ил)-(CH2)n3-C(=O)-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-(NH-DX)с анти-HER2 антителом или его реакционно-способным производным, и конъюгированию группы линкер-лекарственное средство с антителом способом с образованием тиоэфирной связи на участке дисульфидных связей, представленных в шарнирной части антитела, где n1 является целым числом от 0 до 6, n2 является целым числом от 0 до 5, n3 является целым числом от 2 до 8, L2 представляет собой -NH-(CH2CH2-O)n4-CH2CH2-C(=O)- или одинарную связь, где n4 является целым числом от 1 до 6, LP представляет собой тетрапептидный остаток -GGFG-, La представляет собой -O- или одинарную связь, и -(NH-DX) представляет собой группу, представленную следующей формулой:
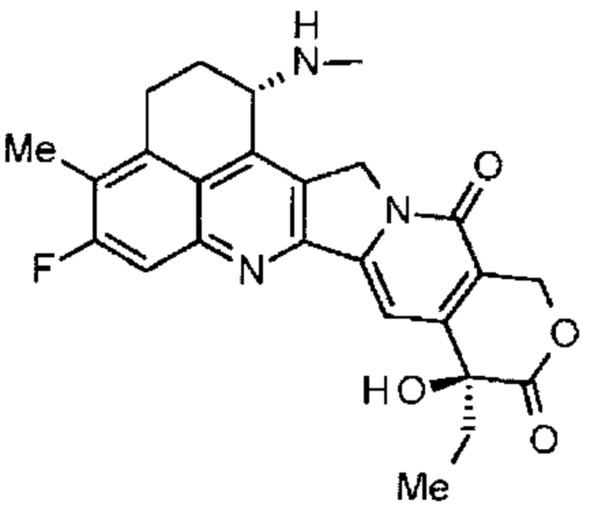
где атом азота аминогруппы в положении 1 находится в связующем положении. Конъюгат обладает превосходной противоопухолевой активностью. 2 н. и 17 з.п. ф-лы, 14 ил., 49 пр.
1. Способ получения конъюгата антитела с лекарственным средством, включающий взаимодействие соединения, представленного следующей формулой:
(малеимид-N-ил)-(CH2)n3-C(=O)-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-(NH-DX)
с анти-HER2 антителом или его реакционно-способным производным, и конъюгирование группы линкер-лекарственное средство с антителом способом с образованием тиоэфирной связи на участке дисульфидных связей, представленных в шарнирной части антитела,
где
n1 является целым числом от 0 до 6,
n2 является целым числом от 0 до 5,
n3 является целым числом от 2 до 8,
L2 представляет собой -NH-(CH2CH2-O)n4-CH2CH2-C(=O)- или одинарную связь,
где n4 является целым числом от 1 до 6,
LP представляет собой тетрапептидный остаток -GGFG-,
La представляет собой -O- или одинарную связь, и
(малеимид-N-ил)- представляет собой группу, представленную следующей формулой:
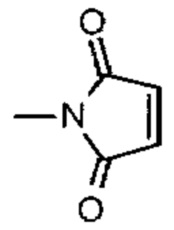
где атом азота находится в связующем положении, и
-(NH-DX) представляет собой группу, представленную следующей формулой:
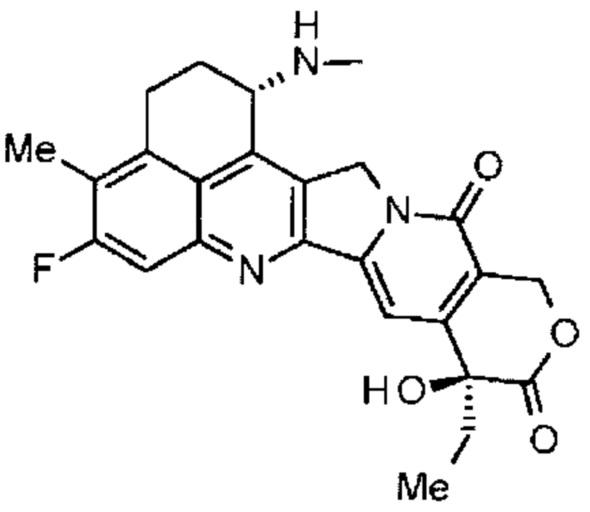
где атом азота аминогруппы в положении 1 находится в связующем положении.
2. Способ по п.1, где соединение является соединением, выбранным из следующей группы:
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX), и
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
3. Способ по п.2, где соединение является соединением, выбранным из следующей группы:
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX), и
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
4. Способ по п.3, где соединение представляет собой:
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX).
5. Способ по п.3, где структурная группа линкер-лекарственное средство представляет собой:
(малеимид-N-ил)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX).
6. Способ по п.3, где структурная группа линкер-лекарственное средство представляет собой:
(малеимид-N-ил)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
7. Способ по п.1, где конъюгат антитела с лекарственным средством содержит структуру линкер-лекарственное средство, представленную следующей формулой:
-(cукцинимид-3-ил-N)-(CH2)n3-C(=O)-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-(NH-DX),
которая конъюгирована с анти-HER2 антителом через тиоэфирную связь, образованную на участке дисульфидных связей, представленных в шарнирной части анти-HER2 антитела.
8. Способ по п.4, где конъюгат антитела с лекарственным средством содержит структуру линкер-лекарственное средство, представленную следующей формулой:
-(cукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
которая конъюгирована с анти-HER2 антителом через тиоэфирную связь, образованную на участке дисульфидных связей, представленных в шарнирной части анти-HER2 антитела.
9. Способ по п.5, где конъюгат антитела с лекарственным средством содержит структуру линкер-лекарственное средство, представленную следующей формулой:
-(cукцинимид-3-ил-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX),
которая конъюгирована с анти-HER2 антителом через тиоэфирную связь, образованную на участке дисульфидных связей, представленных в шарнирной части анти-HER2 антитела.
10. Способ по п.6, где конъюгат антитела с лекарственным средством содержит структуру линкер-лекарственное средство, представленную следующей формулой:
-(cукцинимид-3-ил-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
которая конъюгирована с анти-HER2 антителом через тиоэфирную связь, образованную на участке дисульфидных связей, представленных в шарнирной части анти-HER2 антитела.
11. Способ по любому из пп.1-10, где анти-HER2 антитело содержит тяжелую цепь, состоящую из аминокислотной последовательности, состоящей из остатков аминокислот 1-449 последовательности SEQ ID NO: 1, и легкую цепь, состоящую из аминокислотной последовательности, представленной как SEQ ID NO: 2.
12. Способ по любому из пп.1-10, где анти-HER2 антитело содержит тяжелую цепь, состоящую из аминокислотной последовательности, представленной как SEQ ID NO: 1, и легкую цепь, состоящую из аминокислотной последовательности, представленной как SEQ ID NO: 2.
13. Способ по любому из пп.1-10, где среднее количество структурных единиц выбранных конъюгированных структур линкер-лекарственное средство на одно антитело составляет от 2 до 8.
14. Способ по любому из пп.1-10, где среднее количество структурных единиц выбранных конъюгированных структур линкер-лекарственное средство на одно антитело составляет от 3 до 8.
15. Способ получения конъюгата антитела с лекарственным средством, представленного следующей формулой:
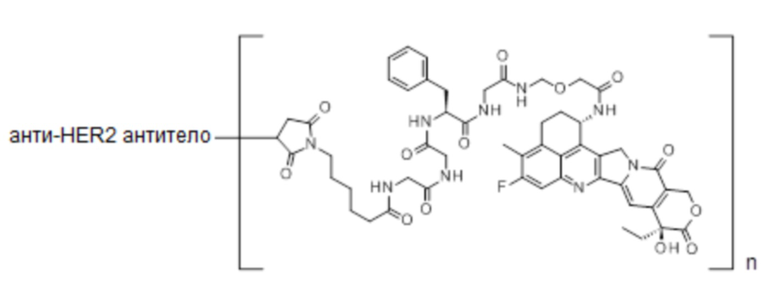
где n представляет среднее количество структурных единиц конъюгированных структур линкер-лекарственное средство на одно анти-HER2 антитело,
и где анти-HER2 антитело содержит:
тяжелую цепь, состоящую из аминокислотной последовательности, состоящей из остатков аминокислот 1-449 последовательности SEQ ID NO: 1, и аминокислотной последовательности легкой цепи, состоящую из остатков аминокислот 1-214 последовательности SEQ ID NO: 2, или
аминокислотную последовательность тяжелой цепи, состоящую из последовательности SEQ ID NO: 1, и легкую цепь, состоящую из аминокислотной последовательности SEQ ID NO: 2,
и где способ включает стадии:
обработки анти-HER2 антитела в восстанавливающих условиях, а затем
взаимодействия анти-HER2 антитела с соединением, представленным следующей формулой:
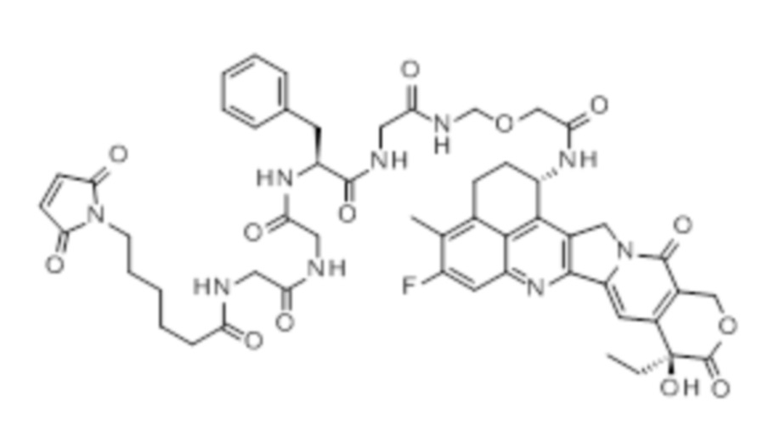
16. Способ по п.15, где аминокислотная последовательность тяжелой цепи анти-HER2 антитела состоит из остатков аминокислот 1-449 последовательности SEQ ID NO: 1, и аминокислотная последовательность легкой цепи анти-HER2 антитела состоит из остатков аминокислот 1-214 последовательности SEQ ID NO: 2.
17. Способ по п.15, где аминокислотная последовательность тяжелой цепи анти-HER2 антитела состоит из аминокислотной последовательности SEQ ID NO: 1, и аминокислотная последовательность легкой цепи анти-HER2 антитела состоит из аминокислотной последовательности SEQ ID NO: 2.
18. Способ по любому из пп.15-17, где значение n находится в диапазоне от 2 до 8.
19. Способ по любому из пп.15-17, где значение n находится в диапазоне от 3 до 8.
| PATRICK J | |||
| BURKE ET AL., "Design, Synthesis, and Biological Evaluation of Antibody-Drug Conjugates Comprised of Potent Camptothecin Analogues", BIOCONJUGATE CHEMISTRY, 2009, vol | |||
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| EP 916348 B1, 18.08.2004 | |||
| WO 2011011474 A1, 27.01.2011. | |||
