ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым конъюгатам лекарственного средства, лекарственным линкерным соединениям, к способам их получения, фармацевтическим композициям, содержащим указанные конъюгаты лекарственного средства, и к их использованию в качестве противоопухолевых средств.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Международные публикации №№ WO-A-2007/144423 и WO-A-2009/080761 раскрывают новые дигидропиран-2-оновые и тетрагидропиран-2-оновые производные, которые демонстрируют очень перспективную противоопухолевую активность. PM060184, раскрытый в WO-A-2007/144423, в настоящее время находится в Фазе I клинических испытаний для профилактики и лечения солидных опухолей.
Лечение рака заметно продвинулось в последние годы с развитием фармацевтических средств, которые таргетируют и убивают раковые клетки более эффективно. Исследователи использовали преимущество клеточно-поверхностных рецепторов и антигенов, селективно экспрессируемых клетками-мишенями, такими как раковые клетки, для разработки фармацевтических средств, основанных на антителах, которые связываются, на примере опухолей, с опухоль-специфическими или опухоль-ассоциированными антигенами. Для того чтобы этого добиться, осуществили химическое связывание цитотоксических молекул, таких как химиотерапевтические лекарственные средства, бактерии и растительные токсины и радионуклиды, с моноклональными антителами, которые связываются с опухоль-специфическими или опухоль-ассоциированными антигенами клеточной поверхности (см., например, международные заявки на патент WO-A-2004/010957, WO-A-2006/060533 и WO-A-2007/024536). Такие соединения обычно называют “конъюгатами” лекарственных средств, токсинов и радионуклидов. Гибель опухолевой клетки происходит при связывании конъюгата лекарственного средства с опухолевой клеткой и высвобождении или/и активации цитотоксической активности компонента лекарственного средства. Селективность, обеспечиваемая конъюгатами лекарственного средства, минимизирует токсичность для нормальных клеток, тем самым повышая переносимость лекарственного средства пациентом. Три примера конъюгатов антитела с лекарственным средством данного типа, которые получили разрешение для продажи на рынке, представляют собой: Гемтузумаб озогамицин для острого миелобластного лейкоза, Брентуксимаб ведотин для рецидивирующей и рефрактерной лимфомы Ходжкина и анапластической крупноклеточной лимфомы, и адо-Трастузумаб эмтансин для рака молочной железы, в частности HER2+.
Эффективность лекарственных средств для химиотерапии рака, как правило, опирается на различия в скорости роста, биохимических путях и физиологических особенностях между опухолевыми и нормальными тканями. Следовательно, большинство стандартных химиотерапевтических средств являются относительно неспецифическими и проявляют дозоограничивающую токсичность, которая способствуют субоптимальным терапевтическим эффектам. Один подход к избирательному таргетированию злокачественных клеток, а не здоровых тканей, является использование специфических моноклональных антител (mAbs), которые распознают опухоль-ассоциированные антигены, экспрессируемые на поверхности опухолевых клеток [Meyer, D.L. & Senter, P.D. (2003) Recent advances in antibody drug conjugates for cancer therapy. Annu. Rep. Med. Chem., 38, 229-237; Chari, R.V. (2008) Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc. Chem. Res. 41, 98−107]. mAbs и производные в настоящее время являются самым быстрорастущим классом терапевтических молекул. Более чем 30 иммуноглобулинов G-типа (IgG) и родственные средства были одобрены за последние 25 лет в основном для раковых и воспалительных заболеваний. В онкологии, mAbs часто объединяют с цитотоксическими лекарственными средствами для повышения их терапевтической эффективности. Альтернативно, небольшие антинеопластические молекулы могут быть химически конъюгированы с mAbs, использоваться как в качестве носителей (увеличенный период полураспада), так и в качестве таргетирующих средств (селективность). Значительные усилия были направлены на использование моноклональных антител (mAbs) для целевой доставки лекарственного средства, благодаря их высоким селективностям в отношении опухоль-ассоциированных антигенов, благоприятным фармакокинетическим свойствам и их относительно низкой собственной токсичности. Конъюгаты mAb-лекарственное средство (ADCs) получают путем ковалентного связывания противораковых лекарственных средств с mAbs, обычно через условно стабильную линкерную систему. При связывании с антигенами клеточной поверхности, mAbs, используемые для большинства ADCs, активно транспортируются в лизосомы или другие внутриклеточные компартменты, где ферменты, низкий уровень рН или восстановители облегчают высвобождение лекарственного средства.
Антигены должны обладать высокой селективностью в отношении опухолевой клетки для ограничения токсичности и побочных эффектов. Множество опухоль-ассоциированных антигенов были изучены в пред-клинических моделях и в клинических испытаниях, включая антигены, сверхэкспрессируемые в В-клетках (например, CD20, CD22, CD40, CD79), T-клетках (CD25, CD30), раковых клетках (HER2, EGFR, EpCAM, EphB2, PSMA), эндотелиальных (эндоглин) или стромальных клетках (фибробластный активированный белок), для примера [Teicher BA. Antibody-drug conjugate targets. Curr Cancer Drug Targets 9(8):982-1004, 2009]. Главным и важным свойством для ADC мишеней является их способность к интернализации; это может быть неотъемлемой чертой антигена как такового или может быть вызвано связыванием антитела с его антигеном. Действительно, интернализация ADC является критической для снижения токсичности, связанной с внеклеточной доставкой полезной нагрузки лекарственного средства.
Что касается конъюгированных малых молекул, и в отличие от огромного разнообразия предполагаемых антиген-мишеней, ограниченное количество семейств цитотоксических лекарственных средств, используемых в качестве полезных нагрузок в ADCs, в настоящее время активно исследуются в клинических испытаниях: калихимицин (Pfizer), дуокармицины (Synthon), пирролобензодиазепины (Spirogen), иринотекан (Immunomedics), майтанзиноиды (DM1 и DM4; ImmunoGen + Genentech/Roche, Sanofi-Aventis, Biogen Idec, Centocor/Johnson & Johnson, Millennium/Takeda) и ауристатины (MMAE и MMAF; Seattle Genetics + Genentech/Roche, MedImmune/AstraZeneca, Bayer-Schering, Celldex, Progenics, Genmab). Калихимицин, дуокармицины и пирролобензодиазепины представляют собой средства, связывающиеся с малой бороздкой ДНК, иринотекан представляет собой ингибитор Топоизомеразы I, а майтанзиноиды и ауристатины представляют собой деполимеризующие тубулин средства. Одной из их общих характерных черт является их высокая эффективность свободного лекарственного средства (от 10-9 до 10-11 M) по сравнению, например, с доксорубицином (10-7 M), используемым в ADCs первого поколения. Другим ключевым элементом успеха является четкое знание “допустимого” положения для присоединения линкера, что способствует высвобождению активных метаболитов, аналогичных традиционным пролекарствам.
Интересно, что представитель трех этих содержащих цитотоксическое средство ADCs достиг поздней стадии клинических испытаний. Трастузумаб эмтансин (T-DM1), трастузумаб, связанный с майтанзиноидным полусинтетическим лекарственным средством посредством стабильного линкера (одобрен управлением по контролю за продуктами и лекарствами от 22 февраля 2013 года для прогрессирующего HER2 положительного рака молочной железы); Инотузумаб озогамицин (CMC-544), гуманизированное анти-CD22 mAb (G5/44, IgG4), конъюгированное с калихимицином с использованием кислотолабильного линкера (ацетилфенокси-бутановый) (B-клеточная неходжкинская лимфома); Брентуксимаб ведотин, гуманизированное анти-CD30 mAb, связанное с монометилауристатином E (MMAE) через малеимидкапроил-валил-цитруллинил-пара-аминобензилкарбаматный линкер (одобрен управлением по контролю за продуктами и лекарствами от 19 августа 2011 года для анапластической крупно-клеточной лимфомы и лимфомы Ходжкина).
Линкеры представляют собой ключевой компонент ADC структур. Были исследованы несколько классов линкеров второго поколения, включая кислотно-лабильные гидразоновые линкеры (лизосомы) (например, гемтузумаб и инотузумаб озогамицин); линкеры на дисульфидной основе (восстановительная внутриклеточная среда); нерасщепляемые тиоэфирные линкеры (катаболическая деградация в лизосомах) (например, трастузумаб эмтансин); пептидные линкеры (например, цитрулин-валин) (лизосомальные протеазы подобные катепсину-В) (например, брентуксимаб ведотин): см., например, WO-A-2004/010957, WO-A-2006/060533 и WO-A-2007/024536. Также была описана очистка конъюгатов антитела с лекарственным средством при помощи эксклюзионной хроматографии (ЭХ) [см., например, Liu et al., Proc. Natl. Acad. Set (USA), 93: 8618-8623 (1996), и Chari et al., Cancer Research, 52: 127-131 (1992)].
Трастузумаб (Герцептин) представляет собой моноклональное антитело, которое перекрестно реагирует с HER2/neu рецептором. В основном, его применяют для лечения некоторых видов рака молочной железы. HER рецепторы представляют собой белки, которые встроены в клеточную мембрану и сообщают молекулярные сигналы снаружи клетки (молекулы, называемые EGFs) внутрь клетки, и включают и выключают гены. HER белки стимулируют клеточную пролиферацию. В некоторых видах раковых заболеваний, в особенности в некоторых видах рака молочной железы, HER2 сверхэкспрессируется и заставляет раковые клетки размножаться бесконтрольно.
Ген HER2 амплифицируется в 20-30% ранних стадий рака молочной железы, что заставляет его сверхэкспрессировать рецепторы эпидермального фактора роста (EGF) в клеточной мембране. В некоторых видах раковых заболеваний, HER2 может посылать сигналы без появления факторов роста и связывания с рецептором, что делает его эффект в клетке существенным; однако трастузумаб не эффективен в этом случае.
HER2 путь стимулирует клеточный рост и деление, когда он нормально функционирует; однако, когда он сверхэкспрессируется, клеточный рост ускоряется сверх его нормальных пределов. В некоторых видах раковых заболеваний этот путь используется для стимулирования быстрого клеточного роста и пролиферации и, следовательно, образованию опухолей. В раковых клетках HER2 белок может экспрессироваться до 100 раз больше, чем в нормальных клетках (2 миллиона по сравнению с 20000 на клетку). Это сверхэкспрессия приводит к сильной и постоянной пролиферативной сигнальной активности и, следовательно, образованию опухолей. Сверхэкспрессия HER2 также вызывает дезактивацию контрольных точек, допуская даже большее увеличение пролиферации.
Сущность изобретения
Существует необходимость в новых активных конъюгатах лекарственного средства, которые не основаны на семействах цитотоксических лекарственных средств, используемых в качестве полезной нагрузки на сегодняшний день. Настоящее изобретение удовлетворяет эту потребность. Оно также обеспечивает новые лекарственные линкерные соединения для использования в получении конъюгатов лекарственного средства по настоящему изобретению, способы получения новых конъюгатов лекарственного средства по настоящему изобретению, фармацевтические композиции, содержащие указанные конъюгаты лекарственного средства, и их применение в качестве противоопухолевых средств, а также набор, содержащий конъюгат лекарственного средства по настоящему изобретению, для использования при лечении рака.
В первом аспекте настоящего изобретения представлен конъюгат лекарственного средства, содержащий группу лекарственного средства, ковалентно связанную с остальной частью конъюгата лекарственного средства, при этом такое соединение имеет формулу [D-(X)b-(AA)w-(L)-]n-Ab, где
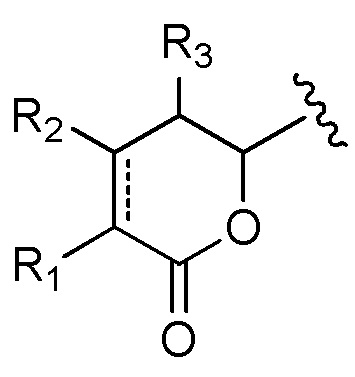
D означает группу, представляющую собой лекарственное средство, имеющее следующую формулу (I), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где
 (I)
(I)
A выбран из
 и
и  ;
;
каждый из R1, R2 и R3 независимо выбран из водорода, ORa, OCORa, OCOORa, NRaRb, NRaCORb, NRaC(=NRa)NRaRb, замещенного или незамещенного C1-C12 алкила, замещенного или незамещенного C2-C12 алкенила и замещенного или незамещенного C2-C12 алкинила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R3’ выбран из водорода, CORa, COORa, CONRaRb, S(O)Ra, SO2Ra, P(O)(Ra)Rb, замещенного или незамещенного C1-C12 алкила, замещенного или незамещенного C2-C12 алкенила и замещенного или незамещенного C2-C12 алкинила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из группы, состоящей из водорода, замещенного или незамещенного C1-C12 алкила, замещенного или незамещенного C2-C12 алкенила и замещенного или незамещенного C2-C12 алкинила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R11 выбран из группы, состоящей из водорода, CORa, COORa, замещенного или незамещенного C1-C12 алкила, замещенного или незамещенного C2-C12 алкенила и замещенного или незамещенного C2-C12 алкинила, или R11 и R12 вместе с соответствующим атомом N и атомом С, с которыми они связаны, могут образовывать 5-14-членную замещенную или незамещенную ненасыщенную или насыщенную гетероциклическую группу, содержащую одно или несколько колец и необязательно содержащую один или несколько дополнительных гетероатомов, выбранных из атомов кислорода, азота и серы, в указанном кольце(кольцах) в дополнение к атому азота группы NR11, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R13 и R14 независимо выбран из группы, состоящей из водорода, CORa, COORa, замещенного или незамещенного C1-C12 алкила, замещенного или незамещенного C2-C12 алкенила, замещенного или незамещенного C2-C12 алкинила и замещенного или незамещенного C4-C12 алкенинила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из Ra и Rb независимо выбран из группы, состоящей из водорода, замещенного или незамещенного C1-C12 алкила, замещенного или незамещенного C2-C12 алкенила, замещенного или незамещенного C2-C12 алкинила, замещенных или незамещенных арильных групп, содержащих от 6 до 18 атомов углерода в одном или нескольких кольцах, и 5-14-членных замещенных или незамещенных ненасыщенных или насыщенных гетероциклических групп, содержащих одно или несколько колец, где необязательные заместители представляют собой один или несколько заместителей Rx;
заместители Rx выбраны из группы, включающей C1-C12 алкильные группы, которые могут быть необязательно замещены по меньшей мере одной группой Ry, C2-C12 алкенильные группы, которые могут быть необязательно замещены по меньшей мере одной группой Ry, C2-C12 алкинильные группы, которые могут быть необязательно замещены по меньшей мере одной группой Ry, атомы галогена, оксо группы, тио группы, циано группы, нитро группы, ORy, OCORy, OCOORy, CORy, COORy, OCONRyRz, CONRyRz, S(O)Ry, SO2Ry, P(O)(Ry)ORz, NRyRz, NRyCORz, NRyC(=O)NRyRz, NRyC(=NRy)NRyRz, арильные группы, содержащие от 6 до 18 атомов углерода в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями, которые могут быть одинаковыми или отличными друг от друга, выбранными из группы, включающей Ry, ORy, OCORy, OCOORy, NRyRz, NRyCORz и NRyC(=NRy)NRyRz, аралкильные группы, содержащие алкильную группу, содержащую от 1 до 12 атомов углерода, замещенную необязательно замещенной арильной группой, как определено выше, аралкилокси группы, содержащие алкокси группу, содержащую от 1 до 12 атомов углерода, замещенную необязательно замещенной арильной группой, как определено выше, и 5-14-членную насыщенную или ненасыщенную гетероциклическую группу, содержащую одно или несколько колец и содержащую по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная гетероциклическая группа необязательно замещена одним или несколькими заместителями Ry, и где более чем один необязательный заместитель присутствует на любой данной группе, такие необязательные заместители Ry могут быть одинаковыми или отличными друг от друга;
каждый Ry и Rz независимо выбран из группы, включающей водород, C1-C12 алкильные группы, C1-12 алкильные группы, которые замещены по меньшей мере одним атомом галогена, аралкильные группы, включающие C1-12 алкильную группу, которая замещена арильной группой, содержащей от 6 до 18 атомов углерода в одном или нескольких кольцах, и гетероциклоалкильные группы, включающие C1-C12 алкильную группу, которая замещена 5-14-членной ненасыщенной или насыщенной гетероциклической группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах);
каждая пунктирная линия представляет собой необязательную дополнительную связь;
каждая волнистая линия указывает точку ковалентного присоединения группы A к остальной части группы лекарственного средства;
X представляет собой расширяющую группу;
каждый AA независимо представляет собой аминокислотное звено;
L представляет собой линкерную группу;
w представляет собой целое число, имеющее значение от 0 до 12;
b представляет собой целое число, имеющее значение 0 или 1;
Ab представляет собой компонент, содержащий по меньшей мере один антиген-связывающий сайт; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-] к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 1 до 20.
Как объясняется и проиллюстрировано более подробно ниже, конъюгаты лекарственных средств формулы [D-(X)b-(AA)w-(L)-]n-Ab по настоящему изобретению представляют собой прорыв в решении описанных выше проблем, требующих дополнительных конъюгатов лекарственных средств в дополнении к тем, которые основаны на трех основных семействах цитотоксических лекарственных средств, которые использовались в качестве полезных нагрузок до настоящего времени, которые демонстрируют превосходную противоопухолевую активность.
В предпочтительном варианте воплощения первого аспекта настоящего изобретения представлен конъюгат лекарственного средства по пункту 1 формулы изобретения, или его фармацевтически приемлемая соль, сложный эфир, сольват, таутомер или стереоизомер, где D означает группу, представляющую собой лекарственное средство, выбранную из формул (Ia) и (Ib):

где волнистые линии формулы (Ia) и (Ib) указывают точку ковалентного присоединения к (X)b, если это имеет место, или к (AA)w, если это имеет место, или к линкерной группе L;
A выбран из
 и
и 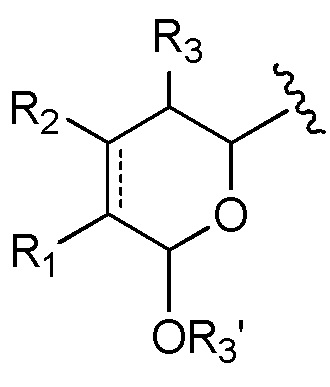 ,
,
где волнистые линии группы A указывают место ковалентного присоединения к остальной части группы лекарственного средства;
каждый из R1, R2, R3, R3’, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, Ra, Rb, Rx, Ry и Rz имеет значение, определенное в первом аспекте настоящего изобретения;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, включающей водород, замещенный или незамещенный C1-C12 алкил, замещенный или незамещенный C2-C12 алкенил и замещенный или незамещенный C2-C12 алкинил, где необязательные заместители представляют собой один или несколько заместителей Rx;
R18 выбран из группы, включающей водород, C1-12 алкильные группы, которые необязательно могут быть замещены по меньшей мере одной группой Rx, арильные группы, содержащие от 6 до 18 атомов углерода в одном или нескольких ароматических кольцах, при этом указанные арильные группы необязательно замещены одним или несколькими заместителями Rx, и 5-14-членные замещенные или незамещенные ненасыщенные или насыщенные гетероциклические группы, содержащие одно или несколько колец, где необязательные заместители представляют собой один или несколько заместителей Rx;
R27 выбран из водорода, замещенного или незамещенного C1-C12 алкила и галогена;
и каждая пунктирная линия представляет собой необязательную дополнительную связь, но, когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26, либо R27 отсутствуют.
Во втором аспекте настоящего изобретения представлено соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H, где
L1 представляет собой линкер, выбранный из группы формул, включающей:
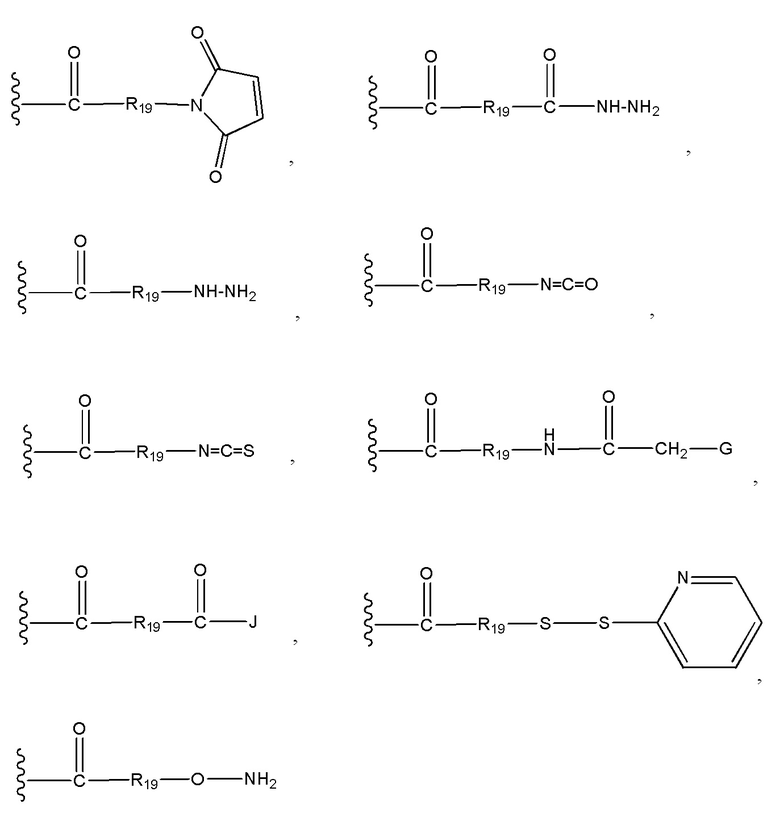
каждая из волнистых линий указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
G выбран из галогена, -O-мезила и -O-тозила;
J выбран из галогена, гидрокси, -N-сукцинимидокси, -O-(4-нитрофенил), -O-пентафторфенила, -O-тетрафторфенила и -O-C(O)-OR20;
R19 выбран из -C1-C12 алкилена-, -C3-C8 карбоцикло, -O-(C1-C12 алкилен), -C6-C18 арилена в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C1-C12 алкилен-C6-C18 арилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, - C6-C18 арилен-C1-C12 алкилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C1-C12 алкилен-(C3-C8 карбоцикло)-, -(C3-C8 карбоцикло)-C1-C12 алкилена-, -C5-C14 гетероцикло-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -C1-C12 алкилен-(C5-C14 гетероцикло)-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -(C5-C14 гетероцикло)-C1-C12 алкилена-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -(OCH2CH2)r- и -CH2-(OCH2CH2)r-, где каждый из указанных выше алкиленовых заместителей, либо отдельно, либо присоединенный к другому фрагменту углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx;
R20 представляет собой С1-C12 алкил или арильную группу, содержащую от 6 до 18 атомов углерода в одном или нескольких ароматических кольцах, при этом указанные арильные группы необязательно замещены одним или несколькими заместителями Rx;
r представляет собой целое число, имеющее значение от 1 до 10; и
каждый из D, X, AA и w имеет значение, определенное в первом аспекте настоящего изобретения.
В третьем аспекте настоящего изобретения представлен конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения, для использования в качестве лекарственного препарата.
В четвертом аспекте настоящего изобретения представлен конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения для использования в лечении рака, и более предпочтительно рака, выбранного из рака легких, колоректального рака, рака молочной железы, рака поджелудочной железы, рака почки, лейкоза, множественной миеломы, лимфомы и рака яичника. Наиболее предпочтительно рак выбран из колоректального рака, рака молочной железы, лейкоза, лимфомы и рака яичника.
В пятом аспекте настоящего изобретения представлена фармацевтическая композиция, содержащая конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения и фармацевтически приемлемый носитель.
В шестом аспекте настоящего изобретения представлен способ профилактики или лечения рака, включающий введение эффективного количества конъюгата лекарственного средства в соответствии с первым аспектом настоящего изобретения нуждающемуся в этом пациенту. Предпочтительно рак выбран из рака легких, колоректального рака, рака молочной железы, рака поджелудочной железы, рака почки, лейкоза, множественной миеломы, лимфомы и рака яичника. Наиболее предпочтительно рак выбран из колоректального рака, рака молочной железы, лейкоза, лимфомы и рака яичника.
В седьмом аспекте настоящего изобретения представлено применение конъюгата лекарственного средства в соответствии с первым аспектом настоящего изобретения для получения лекарственного препарата для лечения рака, и более предпочтительно рака, выбранного из рака легких, колоректального рака, рака молочной железы, рака поджелудочной железы, рака почки, лейкоза, множественной миеломы, лимфомы и рака яичника. Наиболее предпочтительно рак выбран из колоректального рака, рака молочной железы, лейкоза, лимфомы и рака яичника.
В восьмом аспекте настоящего изобретения представлен набор, содержащий терапевтически эффективное количество конъюгата лекарственного средства в соответствии с первым аспектом настоящего изобретения и фармацевтически приемлемый носитель. Набор предназначен для использования при лечении рака, и более предпочтительно рака, выбранного из рака легких, колоректального рака, рака молочной железы, рака поджелудочной железы, рака почки, лейкоза, множественной миеломы, лимфомы и рака яичника. Наиболее предпочтительно рак выбран из колоректального рака, рака молочной железы, лейкоза, лимфомы и рака яичника.
В девятом аспекте настоящего изобретения представлен способ получения конъюгата лекарственного средства в соответствии с первым аспектом настоящего изобретения, включающий конъюгирование компонента Ab, включающего по меньшей мере один антиген-связывающий сайт, и лекарственного средства D формулы (I), (Ia) или (Ib), где Ab и D имеют значение, определенное в первом аспекте настоящего изобретения.
Подробное описание предпочтительных вариантов воплощения
В соединениях по настоящему изобретению, алкильные группы в определениях заместителей R1, R2, R3, R3’, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R17’, R18, R18’, R20, R24, R24’, R25, R26, R27, Ra, Rb, Rx, Ry и Rz могут представлять собой алкильную группу с прямой или разветвленной цепью, содержащую от 1 до 12 атомов углерода, и они предпочтительно представляют собой алкильную группу, содержащую от 1 до 6 атомов углерода, более предпочтительно метильную группу, этильную группу или изо-пропильную группу, и наиболее предпочтительно метильную группу. В определениях заместителей M и Q, они могут представлять собой алкильную группу с прямой или разветвленной цепью, содержащую от 1 до 6 атомов углерода.
В соединениях по настоящему изобретению, алкенильные группы в определениях заместителей R1, R2, R3, R3’, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R17’, R18, R18’, R24, R24’, R25, R26, Ra, Rb и Rx являются разветвленными или неразветвленными, и могут содержать одну или несколько двойных связей и от 2 до 12 атомов углерода. Предпочтительно они содержат от 2 до 6 атомов углерода, и более предпочтительно они представляют собой разветвленные или неразветвленные алкенильные группы, содержащие 2, 3 или 4 атомов углерода.
В соединениях по настоящему изобретению, алкинильные группы в определениях заместителей R1, R2, R3, R3’, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R17’, R18’, R24, R24’, R25, R26, Ra, Rb и Rx являются разветвленными или неразветвленными, и могут содержать одну или несколько тройных связей и от 2 до 12 атомов углерода. Предпочтительно они содержат от 2 до 6 атомов углерода, и более предпочтительно они представляют собой разветвленные или неразветвленные алкинильные группы, содержащие 2, 3 или 4 атомов углерода.
В соединениях по настоящему изобретению, алкенинильные группы в определениях заместителей R13 и R14 являются разветвленными или неразветвленными и могут содержать одну или несколько двойных связей и одну или несколько тройных связей. Предпочтительно они содержат от 4 до 12 атомов углерода, и более предпочтительно они представляют собой разветвленные или неразветвленные алкинильные группы, содержащие от 6 до 10 атомов углерода.
В соединениях по настоящему изобретению галогеновые заместители в определениях заместителей R27, Rx, Ry и Rz включают F, Cl, Br и I, предпочтительно Cl.
В соединениях по настоящему изобретению 5-14-членные насыщенные или ненасыщенные гетероциклические группы в определениях заместителей Rx, Ra, Rb, R18, и гетероциклические группы, которые могут быть образованы группами R11 и R12 вместе с атомом азота и атомом углерода, к которым они присоединены, представляют собой гетероциклические группы, содержащие одно или несколько колец, содержащие по меньшей мере один атом кислорода, азота или серы, в указанном кольце(кольцах). Гетероциклические группы представляют собой группы, которые могут быть гетероароматическими группами или гетероалициклическими группами, последние из которых могут быть частично ненасыщенными, и как ароматическими, так и алициклическими гетероциклическиими группами, содержащими от 1 до 3 отдельных или конденсированных колец. Предпочтительно гетероароматические и гетероалициклические группы содержат от 5 до 10 кольцевых атомов. Подходящие гетероароматические группы в соединениях по настоящему изобретению содержат один, два или три гетероатома, выбранных из атомов N, O или S и включают, например, хинолин, включая 8-хинолин, изохинолин, кумаринил, включая 8-кумаринил, пиридил, пиразинил, пиразолил, пиримидинил, фурил, пирролил, тиенил, тиазолил, изотиазолил, триазолил, тетразолил, изоксазолил, оксазолил, имидaзолил, индолил, изоиндолил, индазолил, индолизинил, фталазинил, птеридинил, пуринил, оксадиазолил, тиадиазолил, фуразанил, пиридазинил, триазинил, циннолинил, бензимидaзолил, бензофуранил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридил. Подходящие гетероалициклические группы в соединениях по настоящему изобретению содержат один, два или три гетероатома, выбранных из атомов N, O или S, и включают, например, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидротиопиранил, пиперидил, морфолинил, тиоморфолинил, тиоксанил, пиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 1,2,3,6-тетрагидропиридил, 2-пирролинил, 3-пирролинил, индолинил, 2H-пиранил, 4H-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолинил, имидазолидинил, 3-азабицикло[3,1,0]гексил, 3-азабицикло[4,1,0]гептил, 3H-индолил и хинолизинил.
В соединениях по настоящему изобретению, арильные группы в определениях заместителей R18, R20, Ra, Rb, и Rx, представляют собой кольцевые соединения с одним или несколькими кольцами, которые содержат отдельные и/или конденсированные арильные группы и содержат от 6 до 18 кольцевых атомов и являются необязательно замещенными. Типичные арильные группы содержат от 1 до 3 отдельных или конденсированных колец. Предпочтительно арильные группы содержат от 6 до 12 углеродных кольцевых атомов. Особенно предпочтительные арильные группы включают замещенный или незамещенный фенил, замещенный или незамещенный нафтил, замещенный или незамещенный бифенил, замещенный или незамещенный фенантрил и замещенный или незамещенный антрил, и наиболее предпочтителен замещенный или незамещенный фенил, где заместители имеют значение, указанное выше, в зависимости от того, является ли арильная группа одним из заместителей R20, R28, Ra и Rb, или она представляет собой заместитель Rx.
В соединениях по настоящему изобретению, аралкильные группы в определениях заместителей Rx, Ry и Rz включают алкильную группу, которая определена и примеры которой представлены выше, которая замещена одной или несколькими арильными группами, которые определены и примеры которых представлены выше. Предпочтительные примеры включают необязательно замещенный бензил, необязательно замещенный фенилэтил и необязательно замещенный нафтилметил.
В соединениях по настоящему изобретению, аралкилокси группы в определениях заместителей Rx включают алкокси группу, содержащую от 1 до 12 атомов углерода, которая замещена одной или несколькими арильными группами, которые определены и примеры которых представлены выше. Предпочтительно алкокси группа содержит от 1 до 6 атомов углерода, и арильная группа содержит от 6 до около 12 углеродных кольцевых атомов, и наиболее предпочтительно аралкилокси группа представляет собой необязательно замещенный бензилокси, необязательно замещенный фенилэтокси и необязательно замещенный нафтилметокси.
В соединениях по настоящему изобретению, гетероциклоалкильные группы в определениях заместителей Ry и Rz включают алкильную группу, которая определена и примеры которой представлены выше, которая замещена одной или несколькими гетероциклильными группами, которые определены и примеры которых представлены выше. Предпочтительно гетероциклоалкильные группы включают алкильную группу, содержащую от 1 до 6 атомов углерода, которая замещена гетероциклильной группой, содержащей от 5 до 10 кольцевых атомов в 1 или 2 кольцах, и может быть ароматической, частично насыщенной или полностью насыщенной. Более предпочтительно гетероциклоалкильные группы включают метильную или этильную группу, которая замещена гетероциклильной группой, выбранной из группы, включающей пирролидинил, имидазолидинил, пиперидинил, пиперазинил, морфолинил, тетрагидрофуранил, тиоанил, оксанил, тианил, 8-хинолин, изохинолин, пиридил, пиразинил, пиразолил, пиримидинил, фурил, пирролил, тиенил, тиазолил, изотиазолил, триазолил, тетразолил, изоксазолил, оксазолил и бензимидазол.
В соединениях по настоящему изобретению, алкиленовые группы в определении R19 представляют собой алкиленовые группы с прямой или разветвленной цепью, содержащие от 1 до 12 атомов углерода, и алкиленовые группы в определениях заместителей M и X представляют собой алкиленовые группы с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода. Предпочтительно алкиленовые группы в определении R19 представляют собой алкиленовые группы с прямой или разветвленной цепью, содержащие от 1 до 8 атомов углерода, более предпочтительно алкиленовые группы с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода. Для заместителя M предпочтительными являются алкиленовые группы с прямой или разветвленной цепью, содержащие от 1 до 3 атомов углерода. В определении X, алкиленовые группы в определении X предпочтительно представляют собой алкиленовые группы с прямой или разветвленной цепью, содержащие от 2 до 4 атомов углерода.
В соединениях по настоящему изобретению, карбоцикло группы в определениях заместителей R19 и M представляют собой циклоалкильные группы, содержащие от 3 до 8 атомов углерода, которые содержат две ковалентные связи в любом положении на циклоалкильном кольце, связывая указанную циклоалкильную группу с остальной частью конъюгата лекарственного средства. Предпочтительно карбоцикло группы в определениях заместителей R19 и M представляют собой циклоалкильные группы, содержащие от 3 до 7 атомов углерода, и более предпочтительно карбоцикло группы, содержащие от 5 до 7 атомов углерода.
В соединениях по настоящему изобретению, ариленовые группы в определении R19 представляют собой арильные группы, содержащие от 6 до 18 атомов углерода в одном или нескольких кольцах, которые содержат две ковалентные связи в любом положении на ароматической кольцевой системе, связывая указанные ариленовые группы с остальной частью конъюгата лекарственного средства. Предпочтительно ариленовые группы в определении R19 представляют собой арильные группы, содержащие от 6 до 12 атомов углерода в одном или нескольких кольцах, которые содержат две ковалентные связи в любом положении на ароматической кольцевой системе, и наиболее предпочтительно они представляют собой фениленовые группы.
В соединениях по настоящему изобретению, гетероцикло группы в определении R19 представляют собой гетероциклильные группы, содержащие от 1 до 3 отдельных или конденсированных колец, содержащих от 5 до 14 кольцевых атомов и содержащих по меньшей мере один атом кислорода, азота и/или серы, в указанном кольце(кольцах), где присутствуют две ковалентные связи в любом положении на кольцевой системе указанных гетероциклических групп. Гетероциклические группы представляют собой группы, которые могут быть гетероароматическими группами или гетероалициклическими группами (последние могут быть частично ненасыщенными). Предпочтительно гетероцикло группы в определении R19 представляют собой гетероциклильные группы, содержащие от 1 до 3 отдельных или конденсированных колец, содержащих от 5 до 12 кольцевых атомов и содержащих по меньшей мере один атом кислорода, азота и/или серы, в указанном кольце(кольцах), где присутствуют две ковалентные связи в любом положении на кольцевой системе указанных гетероциклических групп.
В случае присутствия более чем одного необязательного заместителя Rx на заместителе, каждый заместитель Rx может иметь одинаковое или отличное друг от друга значение.
Предпочтительные конъюгаты лекарственного средства в соответствии с первым аспектом настоящего изобретения включают:
конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения, где L представляет собой линкерную группу, выбранную из группы, включающей:
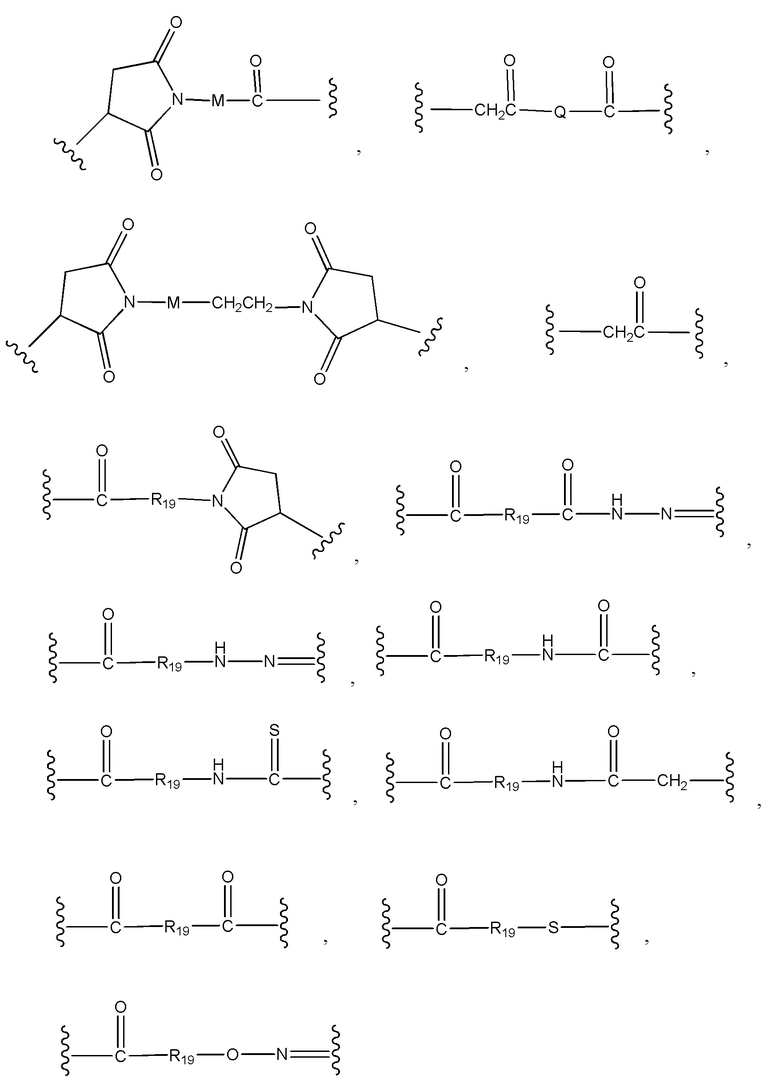
где
волнистые линии указывают точку ковалентных присоединений к Ab (волнистая линия справа) и (AA)w, если это имеет место, или (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева);
R19 выбран из -C1-C12 алкилена-, -C3-C8 карбоцикло, -O-(C1-C12 алкилена), -C6-C18 арилена в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C1-C12 алкилен-C6-C18 арилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, - C6-C18 арилен-C1-C12 алкилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C1-C12 алкилен-(C3-C8 карбоцикло)-, -(C3-C8 карбоцикло)-C1-C12 алкилена-, -C5-C14 гетероцикло-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -C1-C12 алкилен-(C5-C14 гетероцикло)-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -(C5-C14 гетероцикло)-C1-C12 алкилена-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -(OCH2CH2)r-, и - CH2-(OCH2CH2)r -, где каждый из указанных выше алкиленовых заместителей, или в отдельности или присоединенный к другому фрагменту углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx;
M выбран из группы, включающей -C1-C6 алкилен-, -C1-C6 алкилен-(C3-C8 карбоцикло)-, -(CH2CH2O)s-, -C1-C6 алкилен-(C3-C8 карбоцикло)-CON(H или C1-6алкил)-C1-C6 алкилен-, фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx, фенилен-C1-C6 алкилен-, где фениленовая группа может быть необязательно замещена одним или несколькими заместителями Rx, и -C1-C6 алкилен-CON(H или C1-6алкил)C1-C6 алкилен-;
Q выбран из группы, включающей -N(H или C1-6алкил)фенилен- и -N(H или C1-6алкил)-(CH2)s;
r представляет собой целое число, имеющее значение от 1 до 10; и
s представляет собой целое число, имеющее значение от 1 до 10.
Конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения, где L выбран из группы, включающей: 
где
волнистые линии указывают точку ковалентных присоединений к Ab (волнистая линия справа) и (AA)w, если это имеет место, или (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева);
R19 выбран из -C1-C12 алкилена-, -O-(C1-C12 алкилена), -C6-C12 арилена в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C1-C12 алкилен-C6-C12арилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, - C6-C12 арилен-C1-C12алкилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C5-C12гетероцикло-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -C1-C12 алкилен-(C5-C12 гетероцикло)-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -(C5-C12 гетероцикло)-C1-C12 алкилена-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -(OCH2CH2)r-, и - CH2-(OCH2CH2)r-, где каждый из указанных выше алкиленовых заместителей, или в отдельности или присоединенный к другому фрагменту углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx; и
M выбран из группы, включающей -C1-C6 алкилен-, -C1-C6 алкилен-(C3-C8 карбоцикло)- и фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx; и
r представляет собой целое число, имеющее значение от 1 до 6.
Конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения, выбранный из формул (IV) и (V):
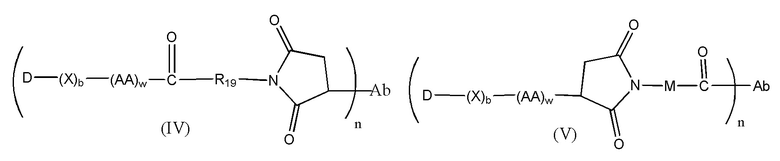
где
X представляет собой расширяющую группу, как определено в первом аспекте настоящего изобретения;
каждый AA независимо представляет собой аминокислотное звено, как определено в первом аспекте настоящего изобретения;
w представляет собой целое число, имеющее значение от 0 до 12;
b представляет собой целое число, имеющее значение 0 или 1;
D означает группу, представляющую собой лекарственное средство;
Ab представляет собой компонент, включающий по меньшей мере один антиген-связывающий сайт;
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формуле (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 1 до 20;
R19 выбран из -C1-C8 алкилена-, -O-(C1-C8 алкилена), -C1-C8 алкилен-C6-C12арилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, - C6-C12арилен-C1-C8алкилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, где каждый из указанных выше алкиленовых заместителей, или в отдельности или присоединенный к другому фрагменту углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx; и
M выбран из группы, включающей -C1-C3 алкилен- и -C1-C3 алкилен-(C5-C7 карбоцикло)-.
Конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения, выбранный из формул (IV) и (V):
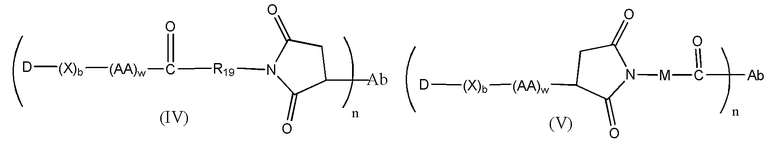
где
X представляет собой расширяющую группу;
каждый AA независимо представляет собой аминокислотное звено;
w представляет собой целое число, имеющее значение от 0 до 12;
b представляет собой целое число, имеющее значение 0 или 1;
D означает группу, представляющую собой лекарственное средство;
Ab представляет собой компонент, включающий по меньшей мере один антиген-связывающий сайт;
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 1 до 20;
R19 выбран из -C1-C6 алкилена-, фенилен-C1-C6 алкилена-, где фениленовая группа может быть необязательно замещена одним или несколькими заместителями Rx выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы и циано группы, где каждый из указанных выше алкиленовых заместителей, в отдельности или присоединенный к другой группе в углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, арильные группы, содержащие от 6 до 12 атомов углерода, атомы галогена, нитро группы и циано группы, и предпочтительно R19 представляет собой -C1-C6 алкиленовую группу; и
M представляет собой -C1-C3 алкилен-(C5-C7 карбоцикло)-.
Предпочтительно, когда в определении конъюгата лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab, L имеет значение, определенное выше в предпочтительных определениях для указанной группы, и (AA)w представляет собой формулу (II):
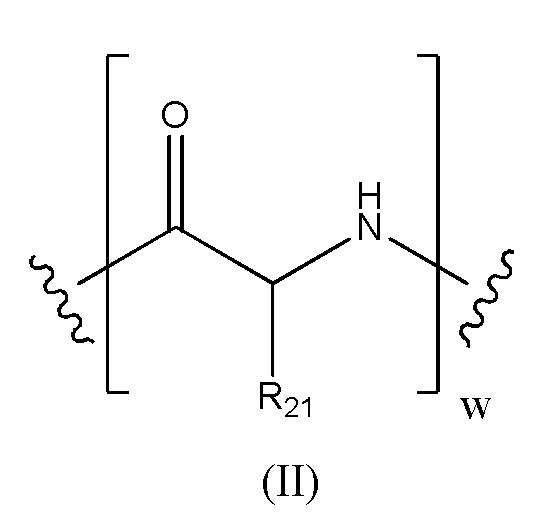
где волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа); и
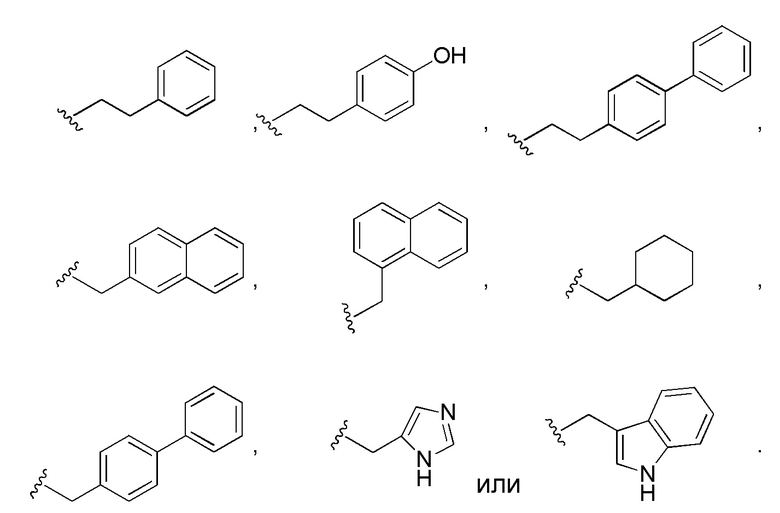
R21 в каждом случае выбран из группы, включающей водород, метил, изопропил, изобутил, втор-бутил, бензил, пара-гидроксибензил, -CH2OH, -CH(OH)CH3, -CH2CH2SCH3, -CH2CONH2, -CH2COOH, -CH2CH2CONH2, -CH2CH2COOH, (CH2)3NHC(=NH)NH2, -(CH2)3NH2, -(CH2)3NHCOCH3, -(CH2)3NHCHO, -(CH2)4NHC(=NH)NH2, -(CH2)4NH2, -(CH2)4NHCOCH3, -(CH2)4NHCHO, -(CH2)3NHCONH2, -(CH2)4NHCONH2, -CH2CH2CH(OH)CH2NH2, 2-пиридилметил-, 3-пиридилметил-, 4-пиридилметил-, фенил, циклогексил,
 ;
;
и w представляет собой целое число, имеющее значение от 0 до 12.
Конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения, где L имеет значение, определенное выше в предпочтительных определениях для указанной группы, и (AA)w представляет собой формулу (II) где
R21 в каждом случае выбран из группы, включающей водород, метил, изопропил, втор-бутил, бензил, индолилметил, -(CH2)3NHCONH2, -(CH2)4NH2, -(CH2)3NHC(=NH)NH2 и -(CH2)4NHC(=NH)NH2; и
w представляет собой целое число, имеющее значение от 0 до 6.
Конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения, где L имеет значение, определенное выше в предпочтительных определениях для указанной группы, где w имеет значение 0 или 2,и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):

где
волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
R22 выбран из метила, бензила, изопропила, втор-бутила и индолилметила; и
R23 выбран из метила, -(CH2)4NH2, -(CH2)3NHCONH2 и -(CH2)3NHC(=NH)NH2.
Далее, предпочтительно, когда в определении конъюгата лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab, L и AA имеют значение, определенное выше в предпочтительных определениях для указанных групп, и X представляет собой расширяющую группу, выбранную из группы, включающей:
-CONH-(C1-C6 алкилен)NH-;
-COO-CH2-(фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx)-NH-;
-CONH-(C1-C6 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx)-NH-;
-CH2-(фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx)-NH-;
-COCH2NH-COCH2-NH-;
-COCH2NH-;
-CONH-(C1-C6 алкилен)S-;
-CONH-(C1-C6 алкилен)NHCO(C1-C6 алкилен)S-;
-(C1-C6 алкилен)NHCO(C1-C6 алкилен)S-;
-(C1-C6 алкилен)S-;
-(C1-C6 алкилен)NH-; и
-(C1-C6 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx)-NH-.
Конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения, где L и AA имеют значение, определенное выше в предпочтительных определениях для указанных групп, и X представляет собой расширяющую группу, выбранную из группы, включающей:
-CONH-(C2-C4 алкилен)NH-;
-COO-CH2-фенилен-NH-, где указанная фениленовая группа может быть необязательно замещена одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы и циано группы;
-CONH-(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы и циано группы)-NH-;
-COCH2NH-COCH2-NH-;
-CONH-(C2-C4 алкилен)S-;
-CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-;
-(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-;
-(C2-C4 алкилен)S-;
-(C2-C4 алкилен)NH-; и
-(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы и циано группы)-NH-.
Конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения, где L и AA имеют значение, определенное выше в предпочтительных определениях для указанных групп, и X представляет собой расширяющую группу, выбранную из группы, включающей:
-CONH(CH2)3NHCOOCH2-фенилен-NH-;
-CONH(CH2)3NH-;
-CONH(CH2)3-S-;
-CONH(CH2)3NHCO(CH2)2S-;
-(CH2)3NHCO(CH2)2S-;
-(CH2)3S-;
-(CH2)3NH-; и
-(CH2)3NHCOOCH2-фенилен-NH-.
Предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где R2 и R3 каждый, независимо, выбран из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx, и более предпочтительно каждый из R2 и R3 представляет собой водород.
Другой предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где R1 выбран из водорода, ORa и OCORa, где Ra выбран из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx, и более предпочтительно R1 представляет собой водород или метокси.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где R3’ выбран из водорода, CORa и замещенного или незамещенного C1-C6 алкила, где Ra представляет собой замещенный или незамещенный C1-C6 алкил, где необязательные заместители представляют собой один или несколько заместителей Rx, и более предпочтительно R3’ представляет собой водород.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода и замещенного и незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx, и более предпочтительно каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода, замещенного и незамещенного метила, замещенного и незамещенного изопропила и замещенного и незамещенного трет-бутила, где необязательные заместители представляют собой один или несколько заместителей Rx.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где каждый из R5, R7, R8, R9 и R10 представляет собой водород.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где каждый из R4 и R6 представляет собой метил.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-A в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где R12 представляет собой изопропил, трет-бутил или бензил.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, AA и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где каждый из R11 и R13 независимо выбран из водорода и замещенного и незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx, и более предпочтительно каждый из R11 и R13 представляет собой водород.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, включающей водород, замещенный или незамещенный C1-C6 алкил, замещенный или незамещенный C2-C6 алкенил и замещенный или незамещенный C2-C6 алкинил, где необязательные заместители представляют собой один или несколько заместителей Rx, более предпочтительно каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители выбраны из группы, включающей алкокси группы, содержащие от 1 до 6 атомов углерода, гидроксильные группы, оксо группы, атомы галогена, OCORy, OCOORy, CORy, COORy, OCONRyRz, CONRyRz, NRyRz, и NRyCORz, где каждый из Ry и Rz выбран из атомов водорода и алкильных групп, содержащих от 1 до 6 атомов углерода, еще более предпочтительно каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо представляет собой водород или C1-C6 алкильную группу, и наиболее предпочтительно каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 представляет собой водород или метил.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где R18 выбран из водорода, С1-6 алкильной группы, которая может быть необязательно замещенной одним или несколькими заместителями Rx, арильной группы, содержащей от 6 до 12 атомов углерода в одном или нескольких ароматических кольцах, при этом указанные арильные группы необязательно замещены одним или несколькими заместителями Rx и 5-10-членной ненасыщенной или насыщенной гетероциклической группой, содержащей одно или несколько колец, при этом указанная гетероциклическая группа необязательно замещена одним или несколькими заместителями Rx, где заместители Rx выбраны из группы, включающей алкокси группы, содержащие от 1 до 6 атомов углерода, гидроксильные группы, атомы галогена, алкиламино группы, содержащие от 1 до 6 атомов углерода и диалкиламино группы, содержащие от 1 до 6 атомов углерода, более предпочтительно R18 выбран из водорода, С1-6 алкильной группы, которая может быть необязательно замещена по меньшей мере одной группой Rx и фенильной группой, которая может быть необязательно замещена по меньшей мере одной группой Rx, и наиболее предпочтительно R18 представляет собой водород или фенильную группу, особенно водород.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-A в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где R27 выбран из атома водорода, атома галогена или замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx, и более предпочтительно R27 выбран из атома водорода и атома хлора.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где каждая пара атомов углерода, соединенная одной или несколькими пунктирными линиями, связана посредством двойных связей.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где
R1 выбран из водорода, ORa и OCORa, где Ra выбран из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R2 и R3 каждый, независимо, выбран из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R3’ выбран из водорода, CORa и замещенного или незамещенного C1-C6 алкила, где Ra представляет собой замещенный или незамещенный C1-C6 алкил, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода и замещенного и незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R11 и R13 независимо выбраны из водорода и замещенного и незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, включающей:
водород и замещенные или незамещенные C1-C6 алкильные группы, где необязательные заместители выбраны из группы, включающей алкокси группы, содержащие от 1 до 6 атомов углерода, гидроксильные группы, оксо группы, атомы галогена, OCORy, OCOORy, CORy, COORy, OCONRyRz, CONRyRz, NRyRz и NRyCORz, где каждый из Ry и Rz выбран из атомов водорода и алкильных групп, содержащих от 1 до 6 атомов углерода;
R18 выбран из водорода, С1-6 алкильной группы, которая может быть необязательно замещена по меньшей мере одной группой Rx, арильной группы, содержащей от 6 до 12 атомов углерода в одном или нескольких ароматических кольцах, при этом указанные арильные группы необязательно замещены одним или несколькими заместителями Rx и 5-10-членной ненасыщенной или насыщенной гетероциклической группой, содержащей одно или несколько колец, при этом указанная гетероциклическая группа необязательно замещена одним или несколькими заместителями Rx, где заместители Rx выбраны из группы, включающей алкокси группы, содержащие от 1 до 6 атомов углерода, гидроксильные группы, атомы галогена, алкиламино группы, содержащие от 1 до 6 атомов углерода и диалкиламино группы, содержащие от 1 до 6 атомов углерода;
R27 выбран из водорода, галогена и замещенного и незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx; и
каждая пунктирная линия представляет собой необязательную дополнительную связь, но, когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26 либо R27 отсутствуют.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где
R1 представляет собой водород или метокси;
каждый из R2 и R3 представляет собой водород;
R3’ представляет собой водород;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода, замещенного или незамещенного метила, замещенного или незамещенного изопропила и замещенного или незамещенного трет-бутила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R11 и R13 представляет собой водород;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, включающей:
водород и замещенные или незамещенные C1-C6 алкильные группы, где необязательные заместители выбраны из группы, включающей алкокси группы, содержащие от 1 до 6 атомов углерода, гидроксильные группы, оксо группы, атомы галогена, OCORy, OCOORy, CORy, COORy, OCONRyRz, CONRyRz, NRyRz и NRyCORz, где каждый из Ry и Rz выбран из атомов водорода и алкильных групп, содержащих от 1 до 6 атомов углерода.
R18 выбран из водорода, С1-6 алкильной группы, которая может быть необязательно замещена по меньшей мере одной группой Rx, и фенильной группы, которая может быть необязательно замещена по меньшей мере одной группой Rx;
R27 представляет собой атом водорода или атома хлора; и
каждая пунктирная линия представляет собой необязательную дополнительную связь, но, когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26 либо R27 отсутствуют.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где
R1 представляет собой водород или метокси;
каждый из R2 и R3 представляет собой водород;
R3’ представляет собой водород;
каждый из R5, R7, R8, R9 и R10 представляет собой водород;
каждый из R4 и R6 представляет собой метил;
каждый из R11 и R13 представляет собой водород;
R12 представляет собой изопропил, трет-бутил или бензил;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из водорода и C1-C6 алкильной группы, предпочтительно водорода и метила;
R18 выбран из водорода и фенила, предпочтительно водорода;
R27 представляет собой атом водорода или атома хлора; и
каждая пара атомов углерода, соединенная одной или несколькими пунктирными линиями, связана через двойные связи.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, выбранное из:
 ,
,
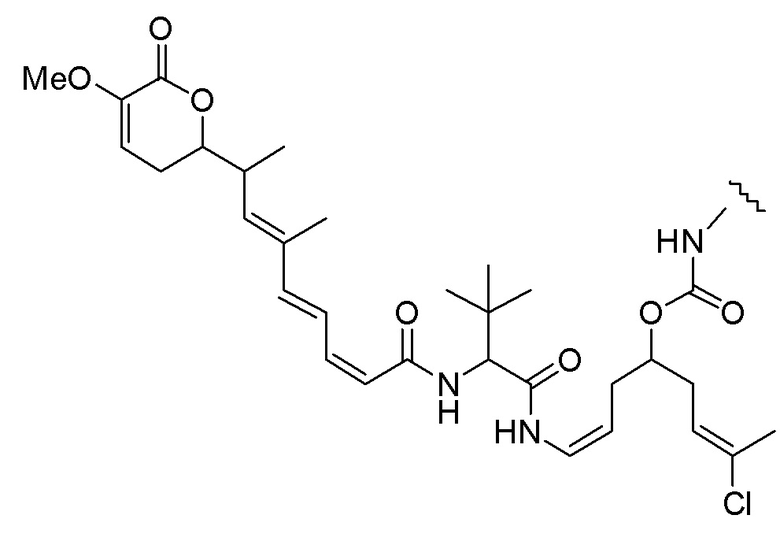 ,
,
 ,
,
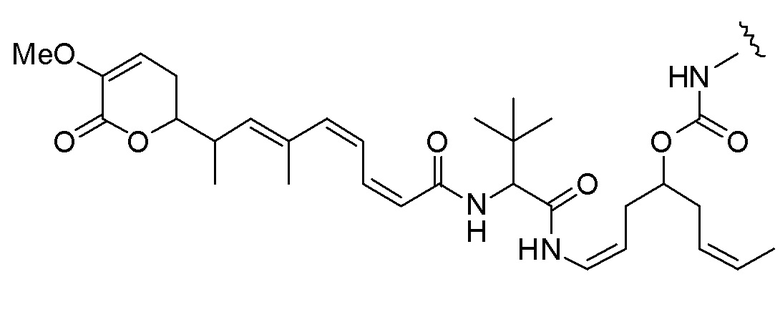 ,
,
 ,
,
 ,
,
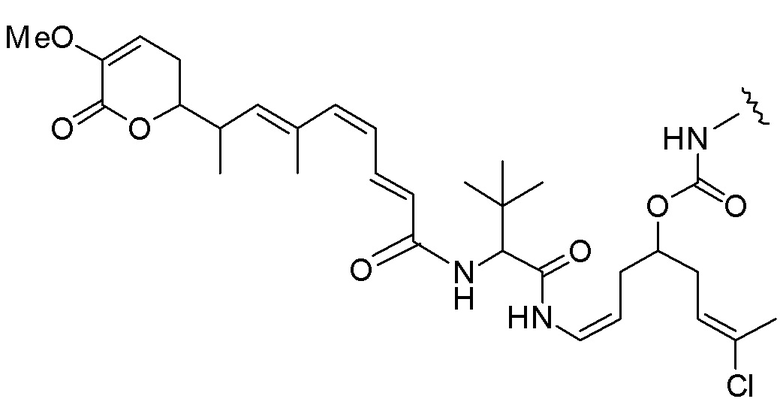 ,
,
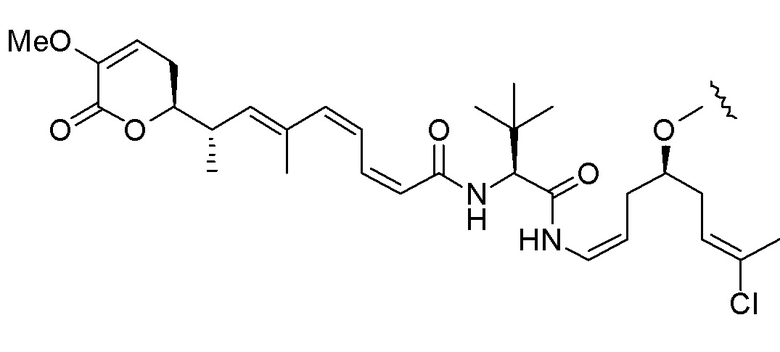 ,
,
 ,
,
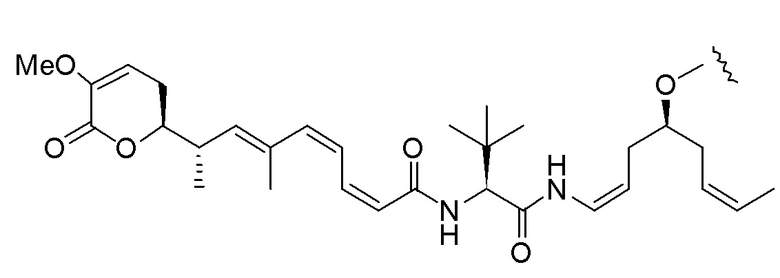 ,
,
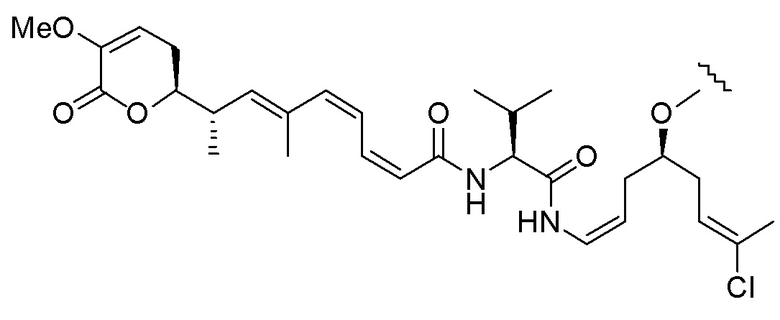 ,
,
 ,
,
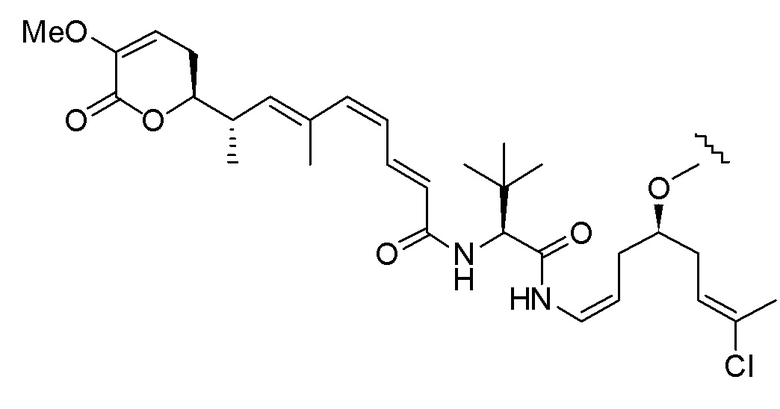 ,
,
 ,
,
 ,
,
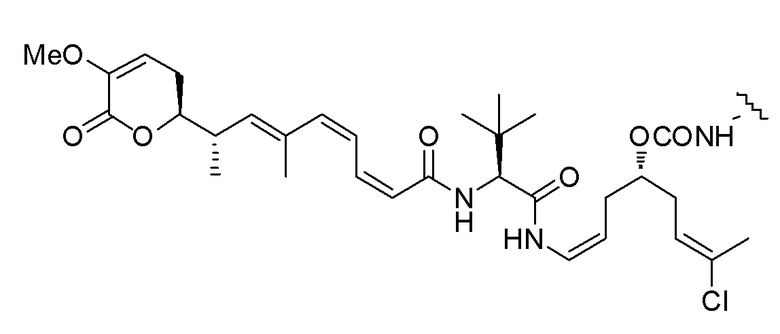 ,
,
 ,
,
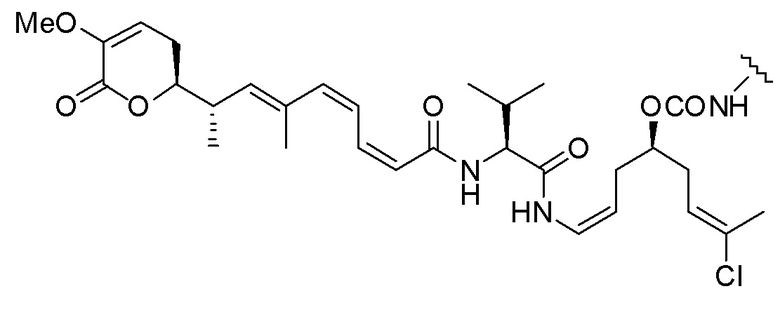 ,
,
 ’
’
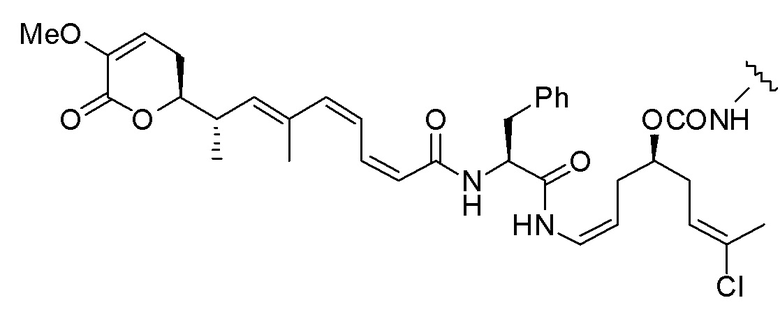 ,
,
 ,
,
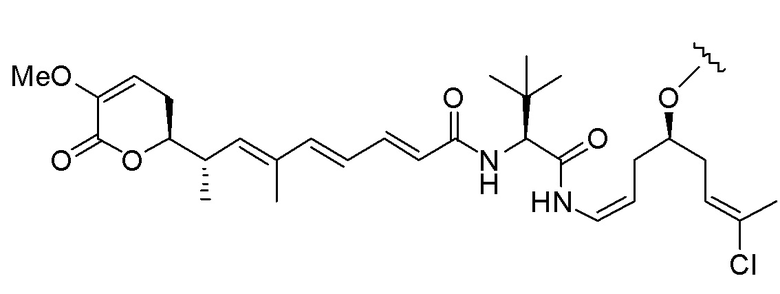 ,
,
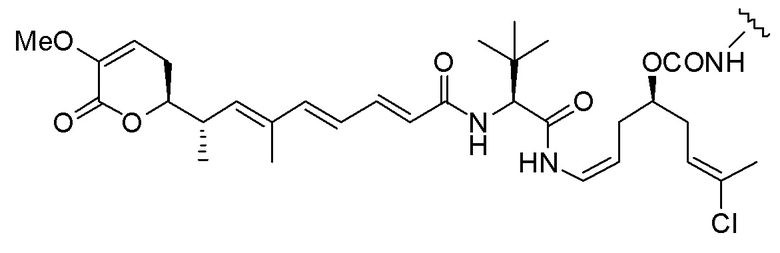 ,
,
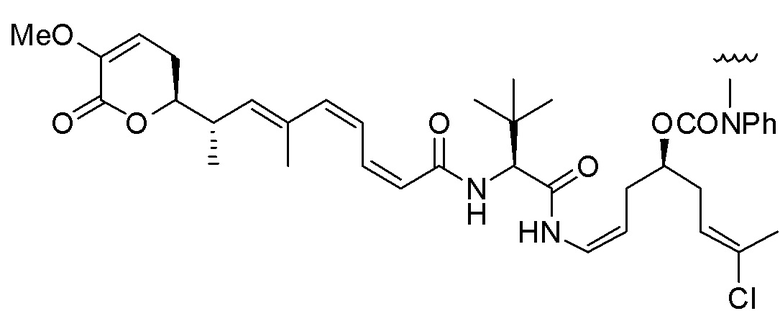 ,
, и
и ,
,
где волнистые линии указывают точку ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, выбранное из:

и
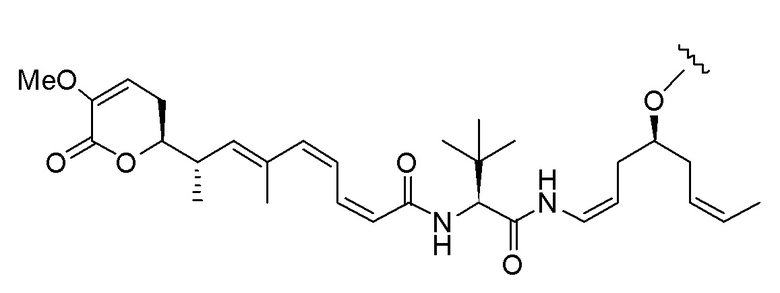
где волнистые линии указывают точку ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w и X имеют значение, определенное выше, и где D представляет собой соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, выбранное из:
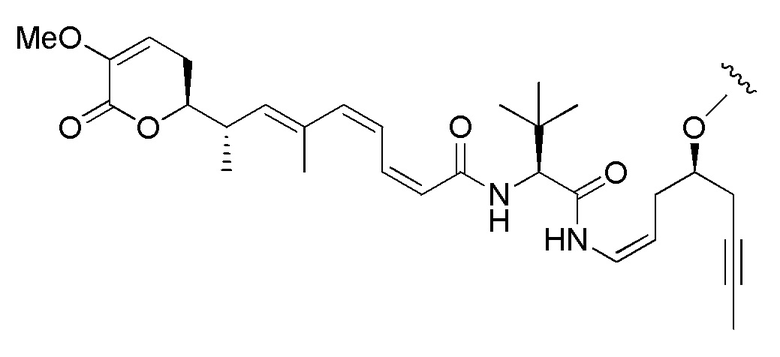 и
и
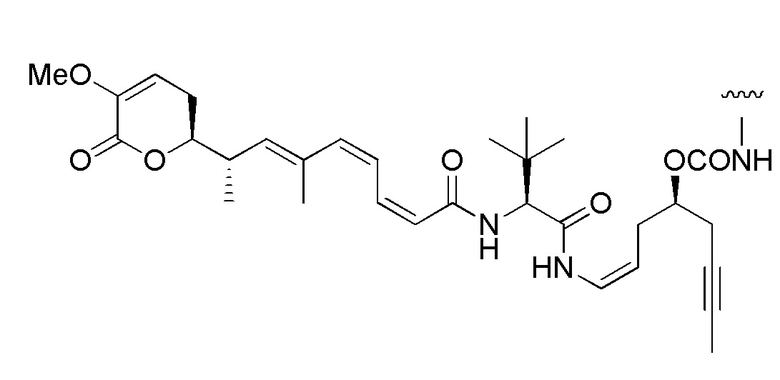 ,
,
где волнистые линии указывают точку ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w, X и D имеют значение, определенное выше, и где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой антиген-связывающий пептид.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w, X и D имеют значение, определенное выше, и компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой антитело, однодоменное антитело или его антиген-связывающий фрагмент.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w, X и D имеют значение, определенное выше, и компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт представляет собой моноклональное, поликлональное антитело или биспецифическое антитело, и где антитело или его антиген-связывающий фрагмент выделены из любых видов, предпочтительно человека, мыши или кролика.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w, X и D имеют значение, определенное выше, и компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой антитело или его антиген-связывающий фрагмент, который выбран из группы, включающей человеческое антитело, антиген-связывающий фрагмент человеческого антитела, гуманизированное антитело, антиген-связывающий фрагмент гуманизированного антитела, химерное антитело, антиген-связывающий фрагмент химерного антитела, гликозилированное антитело и гликозилированный антиген-связывающий фрагмент.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-A в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w, X и D имеют значение, определенное выше, и компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой антитело или его антиген-связывающий фрагмент, где антитело или его антиген-связывающий фрагмент представляет собой антиген-связывающий фрагмент, выбранный из группы, включающей Fab фрагмент, Fab’ фрагмент, F(ab’)2 фрагмент и Fv фрагмент.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w, X и D имеют значение, определенное выше, и компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой антитело или его антиген-связывающий фрагмент, где антитело или его антиген-связывающий фрагмент представляет собой моноклональное антитело, которое иммуноспецифически связывается с антигенами раковых клеток, вирусными антигенами, антигенами клеток, которые продуцируют аутоиммунные антитела, связанные с аутоиммунным заболеванием, микробными антигенами, и предпочтительно моноклональное антитело, которое иммуноспецифически связывается с антигенами раковых клеток.
Еще один предпочтительный конъюгат лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения представляет собой конъюгат, где L, (AA)w, X и D имеют значение, определенное выше, и компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой антитело, выбранное из группы, включающей Абциксимаб, Алемтузумаб, Базиликсимаб, Бевацизумаб, Цетуксимаб, Даклизумаб, Глембатумумаб, Гемтузумаб, Ибритутомаб, Инотузумаб, Лабетузумаб, Лорвотиузумаб, Милатузумаб, Нимотузумаб, Омализумаб, Паливизумаб, Панитумумаб, Пинатумумаб, Ритуксимаб, Ворсетузумаб, Трастузумаб, анти-CD4 антитело, анти-CD5 антитело и анти-CD13 антитело или его иммунологически активную часть, где предпочтительное антитело выбрано из Абциксимаба, Алемтузумаба, Базиликсимаба, Бевацизумаба, Цетуксимаба, Даклизумаба, Глембатумумаба, Гемтузумаба, Ибритутомаба, Инотузумаба, Лабетузумаба, Лорвотиузумаба, Милатузумаба, Нимотузумаба, Омализумаба, Паливизумаба, Панитумумаба, Пинатумумаба, Ритуксимаба, Ворсетузумаба, Трастузумаба и анти-CD4 антитела или его иммунологически активной части, и еще более предпочтительным является Абциксимаб, Алемтузумаб, Базиликсимаб, Бевацизумаб, Цетуксимаб, Даклизумаб, Гемтузумаб, Ибритутомаб, Нимотузумаб, Омализумаб, Паливизумаб, Панитумумаб, Ритуксимаб и Трастузумаб или его иммунологически активная часть. Из вышеперечисленного, особенно предпочтительными являются Трастузумаб, Ритуксимаб, анти-CD4 антитело, анти-CD5 антитело и анти-CD13 антитело или его иммунологически активная часть; или антитело выбрано из Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части, в особенности, Трастузумаба или его иммунологически активной части; или антитело выбрано из анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, в особенности, анти-CD13 антитела или его иммунологически активной части.
Особенно предпочтительные конъюгаты лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab в соответствии с первым аспектом настоящего изобретения включают следующие:
(a)конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения, где
L выбран из группы, включающей:

и
где
волнистые линии указывают точку ковалентных присоединений к Ab (волнистая линия справа) и (AA)w, если это имеет место, или (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева);
R19 выбран из -C1-C12 алкилена-, -O-(C1-C12 алкилена), -C6-C12 арилена в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C1-C12алкилен-C6-C12арилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, - C6-C12 арилен-C1-C12 алкилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C5-C12 гетероцикло-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -C1-C12 алкилен-(C5-C12 гетероцикло)-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -(C5-C12 гетероцикло)-C1-C12 алкилена-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -(OCH2CH2)r- и -CH2-(OCH2CH2)r-, где каждый из указанных выше алкиленовых заместителей, или в отдельности или присоединенный к другому фрагменту углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx;
M выбран из группы, включающей -C1-C6 алкилен-, -C1-C6 алкилен-(C3-C8 карбоцикло)- и фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx;
r представляет собой целое число, имеющее значение от 1 до 6;
(AA)w представляет собой формулу (II):
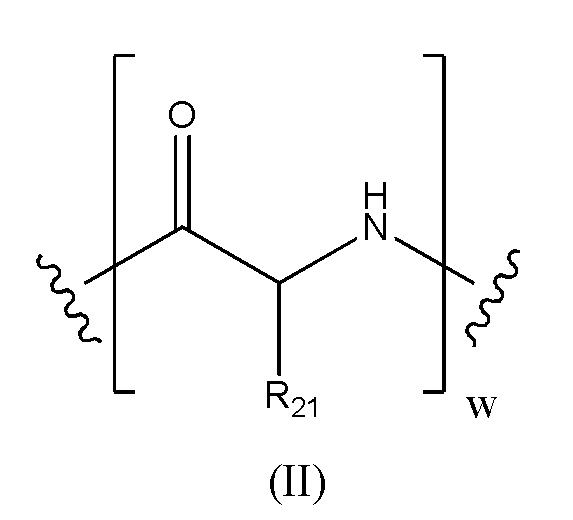
где волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
R21 в каждом случае выбран из группы, включающей водород, метил, изопропил, изобутил, втор-бутил, бензил, пара-гидроксибензил, -CH2OH, -CH(OH)CH3, -CH2CH2SCH3, -CH2CONH2, -CH2COOH, -CH2CH2CONH2, -CH2CH2COOH, -(CH2)3NHC(=NH)NH2, -(CH2)3NH2, -(CH2)3NHCOCH3, -(CH2)3NHCHO, -(CH2)4NHC(=NH)NH2, -(CH2)4NH2, -(CH2)4NHCOCH3, -(CH2)4NHCHO, -(CH2)3NHCONH2, -(CH2)4NHCONH2, -CH2CH2CH(OH)CH2NH2, 2-пиридилметил-, 3-пиридилметил-, 4-пиридилметил-, фенил, циклогексил,
w представляет собой целое число, имеющее значение от 0 до 12;
где X представляет собой расширяющую группу, выбранную из -CONH-(C1-C6 алкилен)NH-, -COO-CH2-(фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx)-NH-, -CONH-(C1-C6 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx)-NH-, -CH2-(фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx)-NH-, -COCH2NH-COCH2-NH-, -COCH2-NH-, -CONH-(C1-C6 алкилен)S-, -CONH-(C1-C6 алкилен)NHCO(C1-C6 алкилен)S-, -(C1-C6 алкилен)NHCO(C1-C6 алкилен)S-, -(C1-C6 алкилен)S-, -(C1-C6 алкилен)NH- и -(C1-C6 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где
R1 выбран из водорода, ORa и OCORa, где Ra выбран из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R2 и R3 каждый, независимо, выбран из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R3’ выбран из водорода, CORa и замещенного или незамещенного C1-C6 алкила, где Ra представляет собой замещенный или незамещенный C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода и замещенного и незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R11 и R13 независимо выбраны из водорода и замещенного и незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, включающей:
водород и замещенный или незамещенный C1-C6 алкильные группы, где необязательные заместители выбраны из группы, включающей алкокси группы, содержащие от 1 до 6 атомов углерода, гидроксильные группы, оксо группы, атомы галогена, OCORy, OCOORy, CORy, COORy, OCONRyRz, CONRyRz, NRyRz и NRyCORz, где каждый из Ry и Rz выбран из атомов водорода и алкильных групп, содержащих от 1 до 6 атомов углерода;
R18 выбран из водорода, С1-6 алкильной группы, которая может быть необязательно замещена по меньшей мере одной группой Rx, арильной группой, содержащей от 6 до 12 атомов углерода в одном или нескольких ароматических кольцах, при этом указанные арильные группы необязательно замещены одним или несколькими заместителями Rx и 5-10-членной ненасыщенной или насыщенной гетероциклической группой, содержащей одно или несколько колец, при этом указанная гетероциклическая группа необязательно замещена одним или несколькими заместителями Rx, где заместители Rx выбраны из группы, включающей алкокси группы, содержащие от 1 до 6 атомов углерода, гидроксильные группы, атомы галогена, алкиламино группы, содержащие от 1 до 6 атомов углерода и диалкиламино группы, содержащие от 1 до 6 атомов углерода;
R27 выбран из водорода, галогена и замещенного и незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждая пунктирная линия представляет собой необязательную дополнительную связь, но, когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26 либо R27 отсутствуют;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой антитело или его антиген-связывающий фрагмент, и выбран из группы, включающей человеческое антитело, антиген-связывающий фрагмент человеческого антитела, гуманизированное антитело, антиген-связывающий фрагмент гуманизированного антитела, химерное антитело, антиген-связывающий фрагмент химерного антитела, гликозилированное антитело и гликозилированный антиген-связывающий фрагмент; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-] к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 1 до 12;
(b)конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения, выбранный из формул (IV) и (V):

где
R19 выбран из -C1-C8 алкилена-, -O-(C1-C8 алкилена), -C1-C8 алкилен-C6-C12 арилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, и -C6-C12 арилен-C1-C8 алкилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, где каждый из указанных выше алкиленовых заместителей, или в отдельности или присоединенный к другому фрагменту углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx;
M выбран из группы, включающей -C1-C3 алкилен- и -C1-C3 алкилен-(C5-C7 карбоцикло)-;
(AA)w представляет собой формулу (II)
 ,
,
где
волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
R21 в каждом случае выбран из группы, включающей водород, метил, изопропил, втор-бутил, бензил, индолилметил, -(CH2)3NHCONH2, -(CH2)4NH2, -(CH2)3NHC(=NH)NH2 и -(CH2)4NHC(=NH)NH2;
w представляет собой целое число от 0 до 6;
X представляет собой расширяющую группу, выбранную из группы, включающей -CONH-(C2-C4 алкилен)NH-, -COO-CH2-фенилен-NH-, где указанная фениленовая группа может быть необязательно замещена одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы, -CONH-(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-, -COCH2NH-COCH2-NH-, -CONH-(C2-C4 алкилен)S-,-CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где
R1 представляет собой водород или метокси;
каждый из R2 и R3 представляет собой водород;
R3’ представляет собой водород;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода, замещенного и незамещенного метила, замещенного и незамещенного изопропила и замещенного и незамещенного трет-бутила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R11 и R13 представляет собой водород;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, включающей:
водород и замещенные или незамещенные C1-C6 алкильные группы, где необязательные заместители выбраны из группы, включающей алкокси группы, содержащие от 1 до 6 атомов углерода, гидроксильные группы, оксо группы, атомы галогена, OCORy, OCOORy, CORy, COORy, OCONRyRz, CONRyRz, NRyRz, NRyCORz, где каждый из Ry и Rz выбран из атомов водорода и алкильных групп, содержащих от 1 до 6 атомов углерода.
R18 выбран из водорода, С1-6 алкильной группы, которая может быть необязательно замещена по меньшей мере одной группой Rx, и фенильной группы, которая необязательно замещена одним или несколькими заместителями RX;
R27 представляет собой атом водорода или атома хлора;
каждая пунктирная линия представляет собой необязательную дополнительную связь, но, когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26 либо R27 отсутствуют;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой антитело или его антиген-связывающий фрагмент, где антитело или антиген-связывающий фрагмент представляет собой моноклональное антитело, которое иммуноспецифически связывается с антигенами раковых клеток, вирусными антигенами, антигенами клеток, которые продуцируют аутоиммунные антитела, связанные с аутоиммунным заболеванием, микробными антигенами, и предпочтительно моноклональное антитело, которое иммуноспецифически связывается с антигенами раковых клеток; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 8;
(c)конъюгат лекарственного средства в соответствии с первым аспектом, выбранный из формул (IV) и (V):

где
R19 выбран из -C1-C6 алкилена-, -фенилен-C1-C6 алкилена-, где фениленовая группа может быть необязательно замещена одним или несколькими заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы и циано группы, где каждый из указанных выше алкиленовых заместителей, в отдельности или присоединенный к другой группе в углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, арильные группы, содержащие от 6 до 12 атомов углерода, атомы галогена, нитро группы и циано группы, и предпочтительно R19 представляет собой С1-C6 алкиленовую группу;
M представляет собой -C1-C3 алкилен-(C5-C7 карбоцикло)-;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
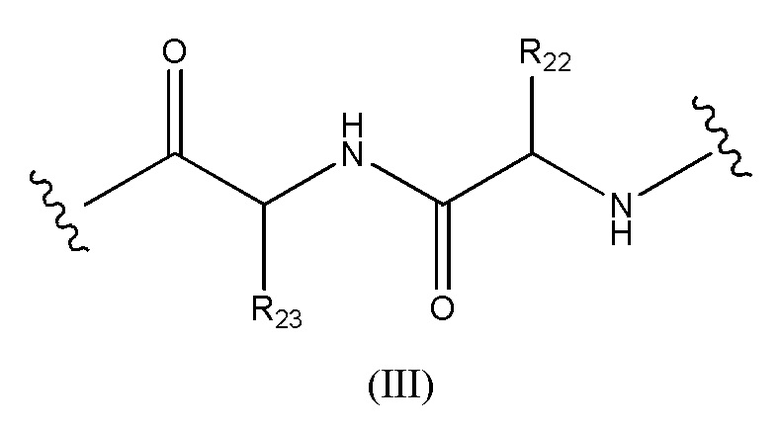
где волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
R22 выбран из метила, бензила, изопропила, втор-бутила и индолилметила;
R23 выбран из метила, -(CH2)4NH2, -(CH2)3NHCONH2 и -(CH2)3NHC(=NH)NH2;
X представляет собой расширяющую группу, выбранную из группы, включающей -CONH-(C2-C4 алкилен)NH-, -COO-CH2-фенилен-NH-, где указанная фениленовая группа может быть необязательно замещена одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы, -CONH-(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-, -COCH2NH-COCH2-NH-,-CONH-(C2-C4 алкилен)S-, -CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где
R1 представляет собой водород или метокси;
каждый из R2 и R3 представляет собой водород;
R3’ представляет собой водород;
каждый из R5, R7, R8, R9 и R10 представляет собой водород;
каждый из R4 и R6 представляет собой метил;
каждый из R11 и R13 представляет собой водород;
R12 представляет собой изопропил, трет-бутил или бензил;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из водорода и C1-C6 алкильной группы, предпочтительно из водорода и метила;
R18 выбран из водорода и фенила, предпочтительно из водорода;
R27 представляет собой атом водорода или атома хлора;
каждая пара атомов углерода, соединенная одной или несколькими пунктирными линиями, связана через двойные связи;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой моноклональное антитело, выбраное из группы, включающей Абциксимаб, Алемтузумаб, Базиликсимаб, Бевацизумаб, Цетуксимаб, Даклизумаб, Глембатумумаб, Гемтузумаб, Ибритутомаб, Инотузумаб, Лабетузумаб, Лорвотиузумаб, Милатузумаб, Нимотузумаб, Омализумаб, Паливизумаб, Панитумумаб, Пинатумумаб, Ритуксимаб, Ворсетузумаб, Трастузумаб, анти-CD4 антитело, анти-CD5 антитело и анти-CD13 антитело или его иммунологически активную часть, предпочтительно моноклональное антитело выбрано из группы, включающей Абциксимаб, Алемтузумаб, Базиликсимаб, Бевацизумаб, Цетуксимаб, Даклизумаб, Глембатумумаб, Гемтузумаб, Ибритутомаб, Инотузумаб, Лабетузумаб, Лорвотиузумаб, Милатузумаб, Нимотузумаб, Омализумаб, Паливизумаб, Панитумумаб, Пинатумумаб, Ритуксимаб, Ворсетузумаб, Трастузумаб и анти-CD4 антитело или его иммунологически активную часть, и еще более предпочтительным является Абциксимаб, Алемтузумаб, Базиликсимаб, Бевацизумаб, Цетуксимаб, Даклизумаб, Гемтузумаб, Ибритутомаб, Нимотузумаб, Омализумаб, Паливизумаб, Панитумумаб, Ритуксимаб и Трастузумаб или его иммунологически активная часть. Из вышеперечисленного, особенно предпочтительными являются антитела, выбранные из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части; более предпочтительное антитело выбрано из Трастузумаба, анти-CD13 антитела, анти-CD4 антитела, анти-CD5 антитела или его иммунологически активной части; или антитело выбрано из Трастузумаба и Ритуксимаба и анти-CD4 антитела или его иммунологически активной части, предпочтительно из Трастузумаба или его иммунологически активной части; или антитело выбрано из анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, особенно из анти-CD13 антитела или его иммунологически активной части; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5;
(d)конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения, выбранный из формул (IV) и (V):

где
R19 представляет собой -C3-C6 алкилен-;
M представляет собой -C1-C3 алкилен-(C5-C7 карбоцикло)-;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
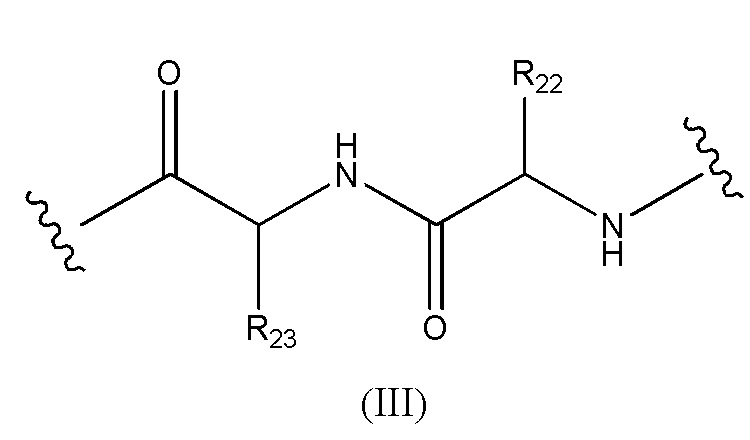 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, где волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
X представляет собой расширяющую группу, выбранную из группы, включающей -CONH-(C2-C4 алкилен)NH-, -COO-CH2-фенилен-NH-, где указанная фениленовая группа может быть необязательно замещена одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы, -CONH-(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-, -COCH2NH-COCH2-NH-, -CONH-(C2-C4 алкилен)S-, -CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, выбранную из следующей группы:
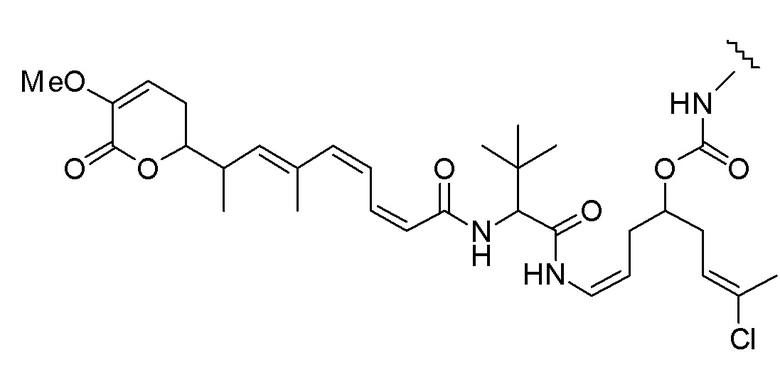 ,
,
 ,
,
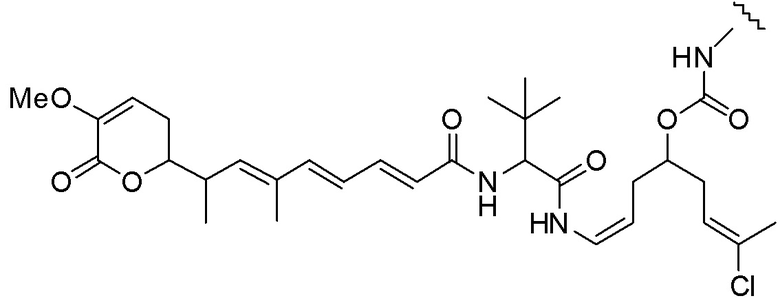 ,
,
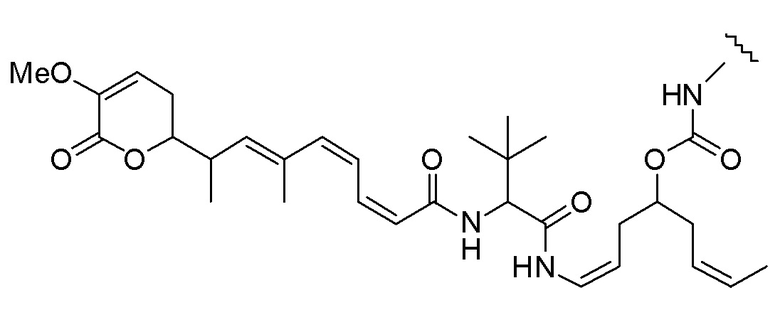 ,
,
 ,
,
 ,
,
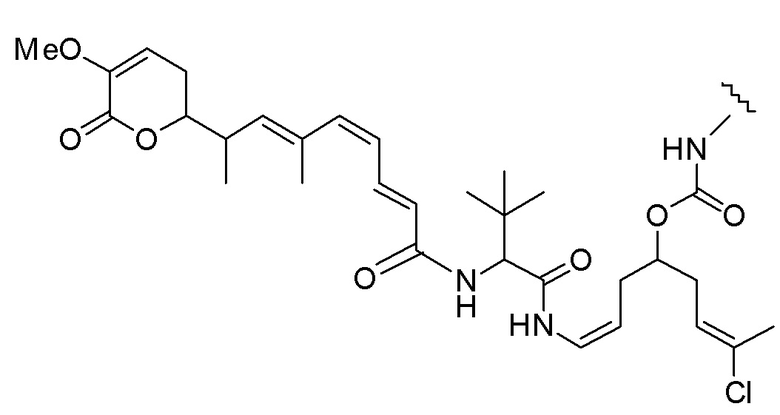 ,
,
 ,
,
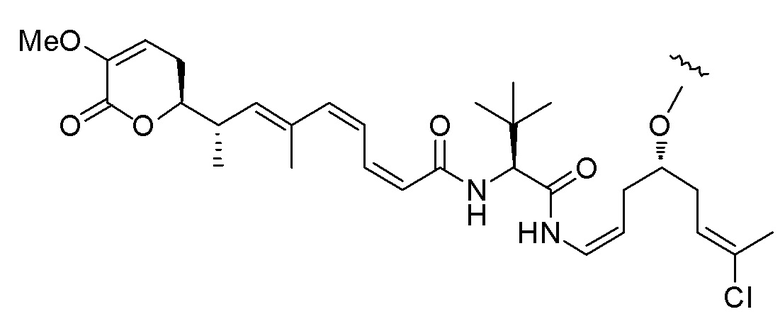 ,
,
 ,
,
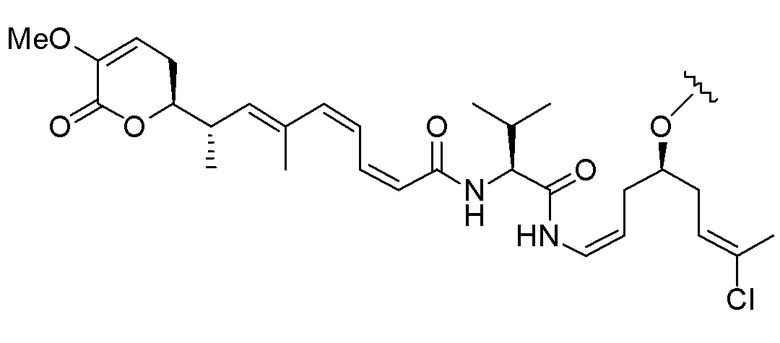 ,
,
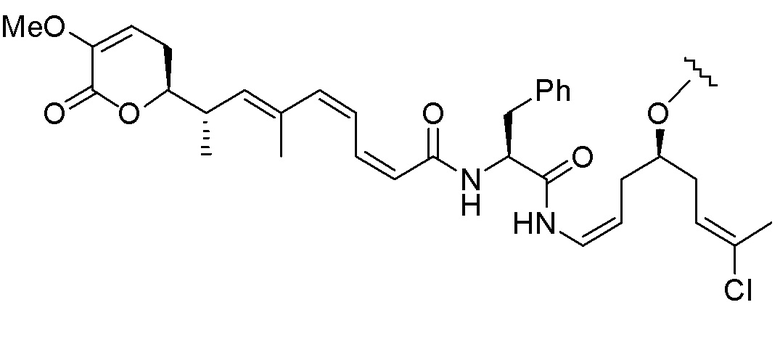 ,
,
 ,
,
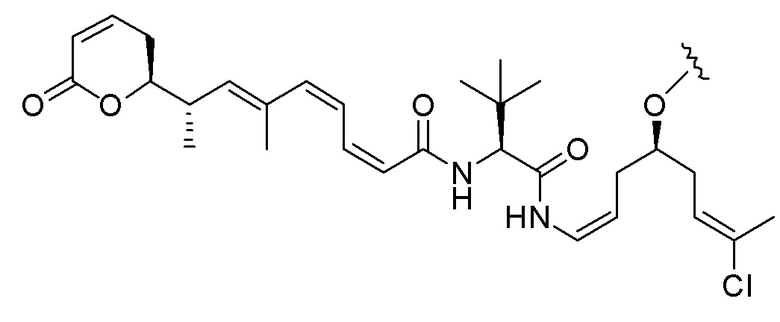 ,
,
 ,
,
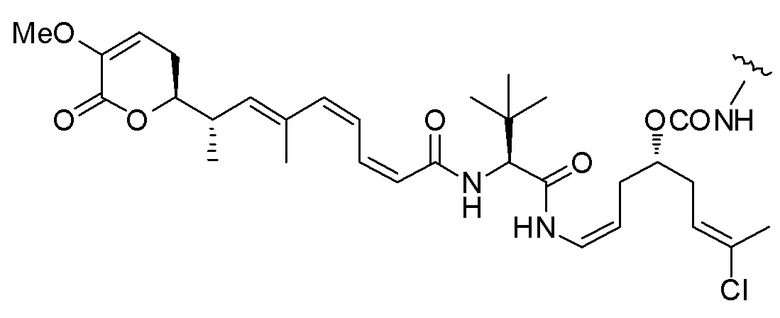 ,
,
 ,
,
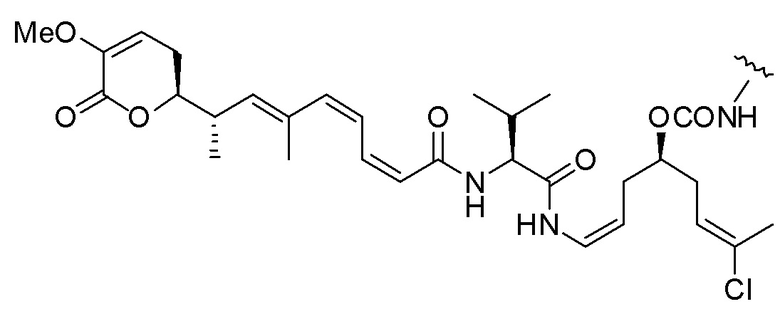 ,
,
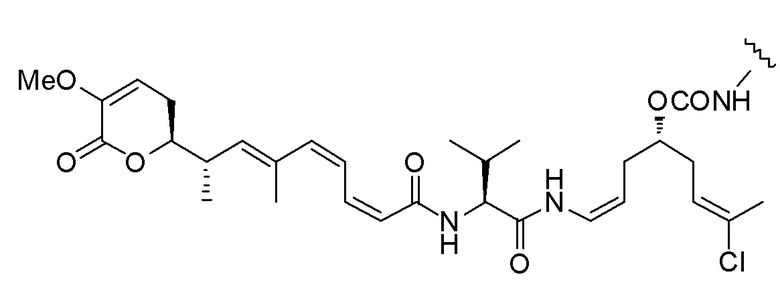 ,
,
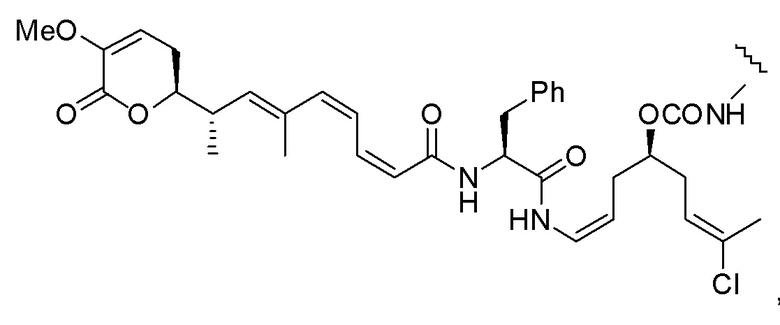 ,
,
 ,
,
 ,
,
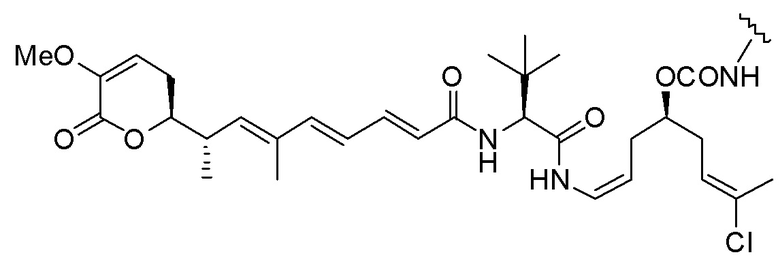 ,
,
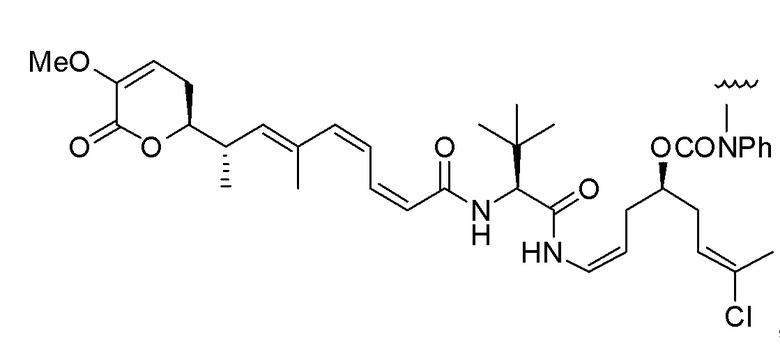 ,
,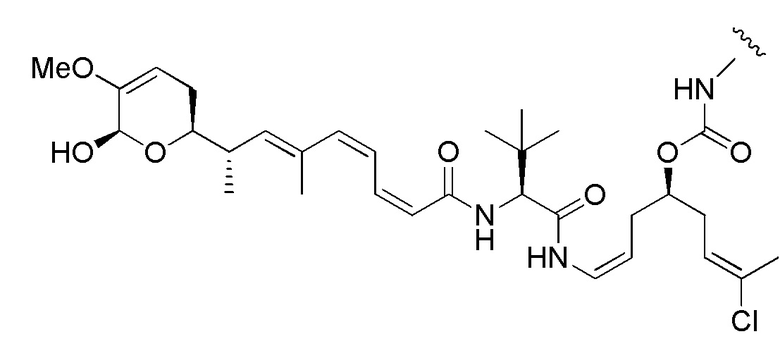 и
и
где волнистые линии указывают точку ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части и более предпочтительно он выбран из Трастузумаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, или он выбран из Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части, предпочтительно Трастузумаба или его иммунологически активной части; или он выбран из анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, в особенности, анти-CD13 антитела или его иммунологически активной части; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5;
(e)конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения, выбранный из формул (IV) и (V):
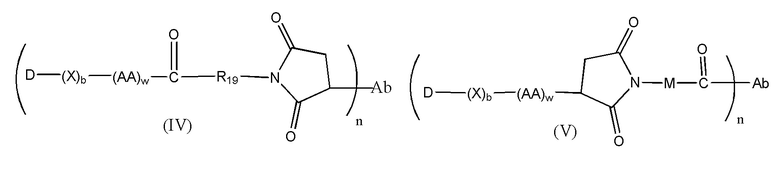
где
R19 представляет собой -C3-C6 алкилен-;
M представляет собой -C1-C3 алкилен-(C5-C7 карбоцикло)-;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
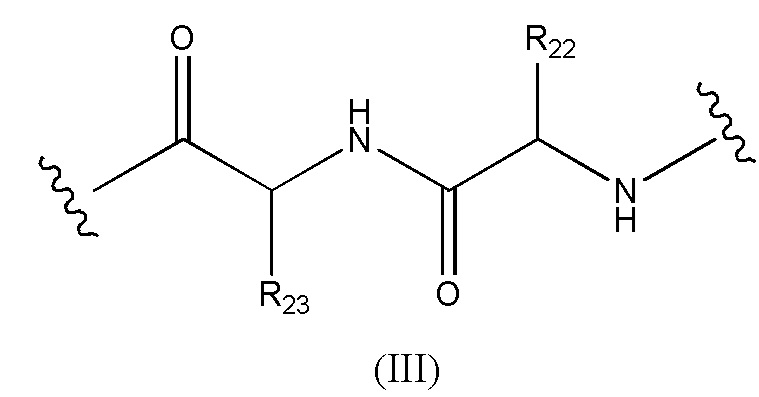 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, и волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
X представляет собой расширяющую группу, выбранную из группы, включающей -CONH-(C2-C4 алкилен)NH-, -COO-CH2-фенилен-NH-, где указанная фениленовая группа может быть необязательно замещена одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы, -CONH-(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-, -COCH2NH-COCH2-NH-, -CONH-(C2-C4 алкилен)S-, -CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, выбранную из:
 и
и
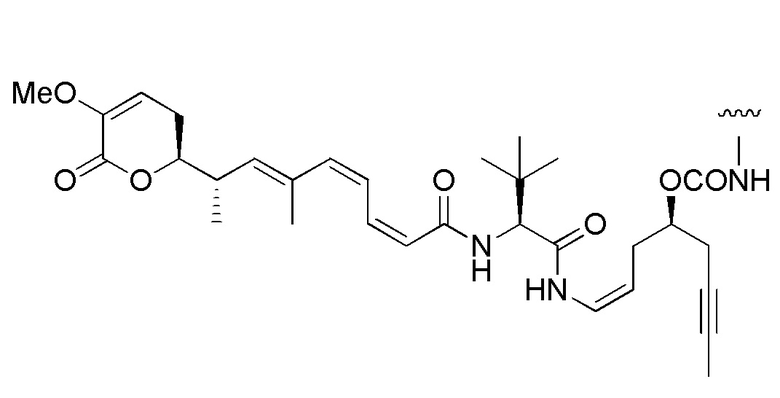 ,
,
где волнистые линии указывают точку ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части; более предпочтительно он выбран из Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части; и наиболее предпочтительно он представляет собой Трастузумаб или его иммунологически активную часть; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5;
(f)конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения формулы (IV):

где
R19 представляет собой C5 алкилен-;
b имеет значение 1;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
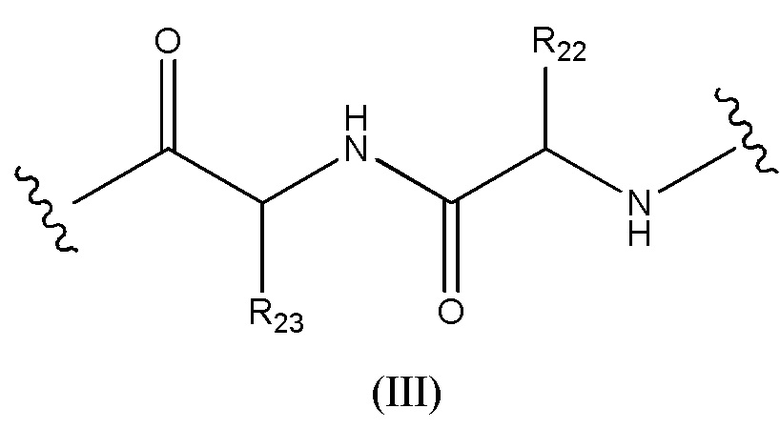 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, и волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа); и
X представляет собой расширяющую группу, выбранную из -(CH2)3NHCOOCH2-фенилен-NH- и -(CH2)3NH-;
или формулы (V)
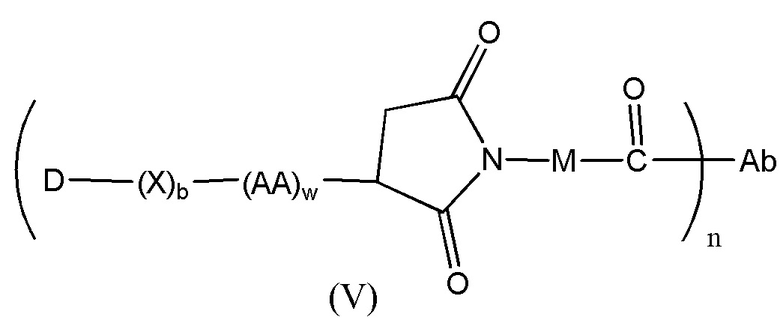 ,
,
где M представляет собой -метил-циклогексилен-;
b имеет значение 1;
w имеет значение 0; и
X представляет собой расширяющую группу, выбранную из -(CH2)3S- и -(CH2)3NHCO(CH2)2S-;
D означает группу, представляющую собой лекарственное средство формулы (Ia), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, выбранную из:
 и
и
 ,
,
где волнистые линии указывают точку ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой Трастузумаб, Ритуксимаб, анти-CD4 антитело, анти-CD5 антитело и анти-CD13 антитело или его иммунологически активную часть; более предпочтительно он выбран из Трастузумаба, анти-CD13 антитела, анти-CD4 антитела и анти-CD5 антитела или его иммунологически активной части; или он выбран из Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части, и наиболее предпочтительно он представляет собой Трастузумаб или его иммунологически активную часть; или он выбран из анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, и наиболее предпочтительно он представляет собой анти-CD13 антитело или его иммунологически активную часть; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (V) или (VI), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5, и предпочтительно 4;
(g)конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения формулы (IV):
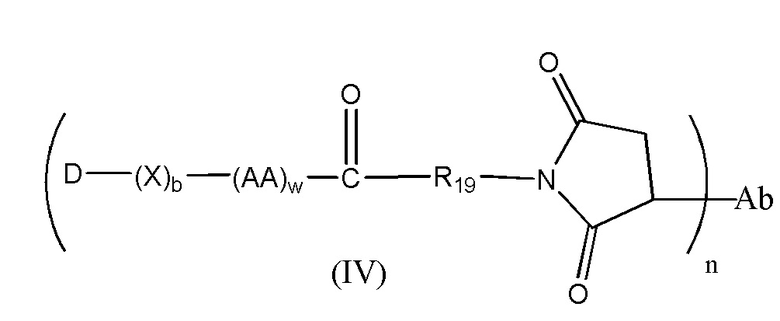 ,
,
где R19 представляет собой -C5 алкилен-;
b имеет значение 1;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, и волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа); и
X представляет собой расширяющую группу, выбранную из -(CH2)3NHCOOCH2-фенилен-NH- и -(CH2)3NH-;
или группу формулы (V)
 ,
,
где M представляет собой -метил-циклогексилен-;
b имеет значение 1;
w имеет значение 0; и
X представляет собой расширяющую группу, выбранную из -(CH2)3S- и -(CH2)3NHCO(CH2)2S-;
D означает группу, представляющую собой лекарственное средство формулы (Ia), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, выбранную из:
 и
и
 ,
,
где волнистые линии указывают точку ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбранный из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части; и более предпочтительно он выбран из Трастузумаба, анти-CD13 антитела, анти-CD4 антитела и анти-CD5 антитела или его иммунологически активной части; или он выбран из Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части; и наиболее предпочтительно он представляет собой Трастузумаб или его иммунологически активную часть; или он выбран из анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, и наиболее предпочтительно он представляет собой анти-CD13 антитело или его иммунологически активную часть; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (V) или (VI), к фрагменту Ab, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5, и предпочтительно 4;
h) конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения формулы (IV):

где
R19 представляет собой -C5 алкилен-;
b имеет значение 1;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, и волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа); и
X представляет собой расширяющую группу, выбранную из -(CH2)3NHCOOCH2-фенилен-NH- и -(CH2)3NH-;
или группу формулы (V)
 ,
,
где M представляет собой -метил-циклогексилен-;
b имеет значение 1;
w имеет значение 0; и
X представляет собой расширяющую группу, выбранную из -(CH2)3S- и -(CH2)3NHCO(CH2)2S-;
D означает группу, представляющую собой лекарственное средство формулы
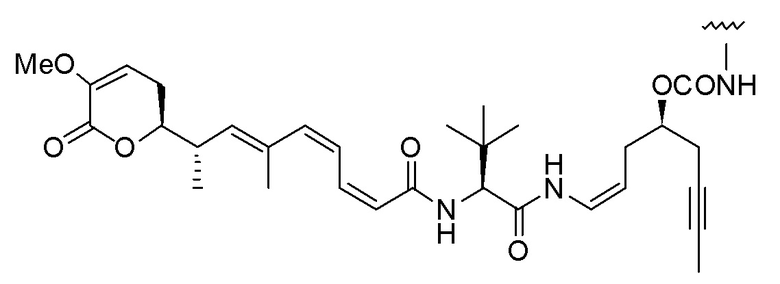
или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где волнистые линии указывают точку ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части или он выбран из Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части, и наиболее предпочтительно он представляет собой Трастузумаб или его иммунологически активную часть; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (V) или (VI), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5, и предпочтительно 4;
i) конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения формулы (IV):
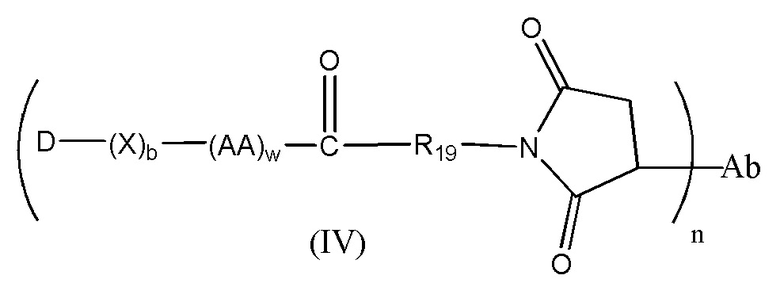
где
R19 представляет собой C5 алкилен-;
b имеет значение 1;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
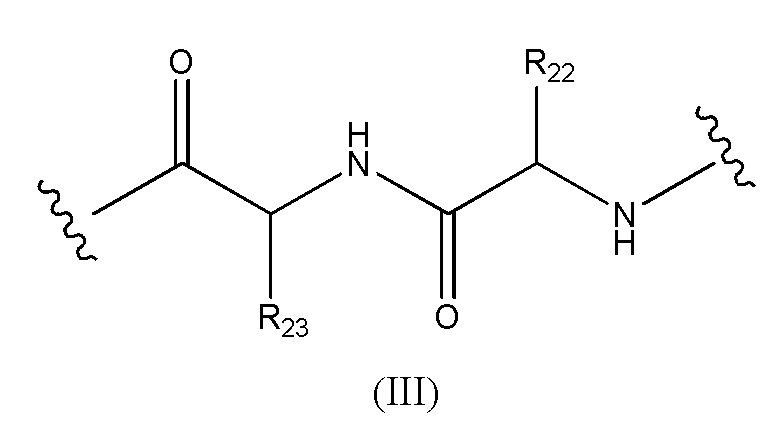 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, и волнистые линии указывают точку ковалентных присоединений к (X)b, если это имеет место, или группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа); и
X представляет собой расширяющую группу, выбранную из -(CH2)3NHCOOCH2-фенилен-NH- и -(CH2)3NH-;
или группу формулы (V)
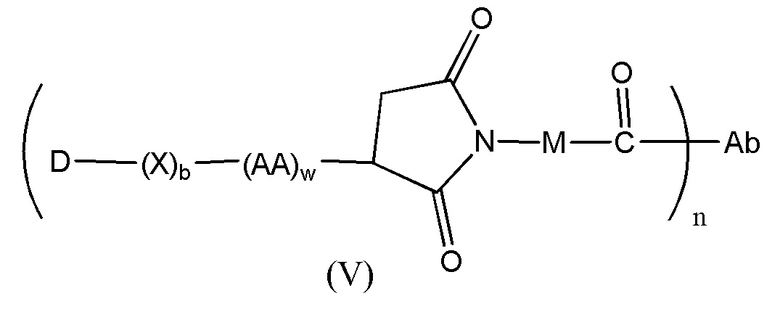 ,
,
где M представляет собой -метил-циклогексилен-;
b имеет значение 1;
w имеет значение 0; и
X представляет собой расширяющую группу, выбранную из -(CH2)3S- и -(CH2)3NHCO(CH2)2S-;
D означает группу, представляющую собой лекарственное средство формулы
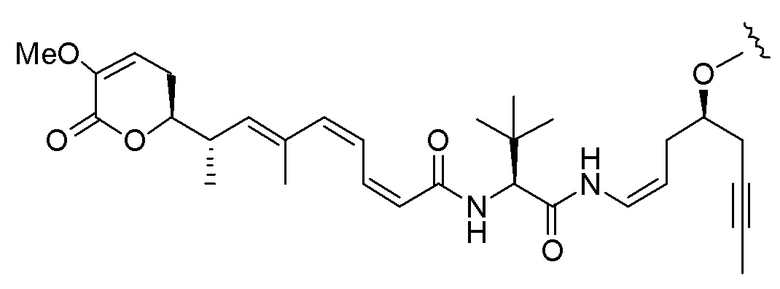
или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер,
где волнистые линии указывают точку ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части или он выбран из Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части, и наиболее предпочтительно он представляет собой Трастузумаб или его иммунологически активную часть; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5, и предпочтительно 4;
(j) конъюгат антитела с лекарственным средством в соответствии с первым аспектом настоящего изобретения, выбранный из группы, включающей:
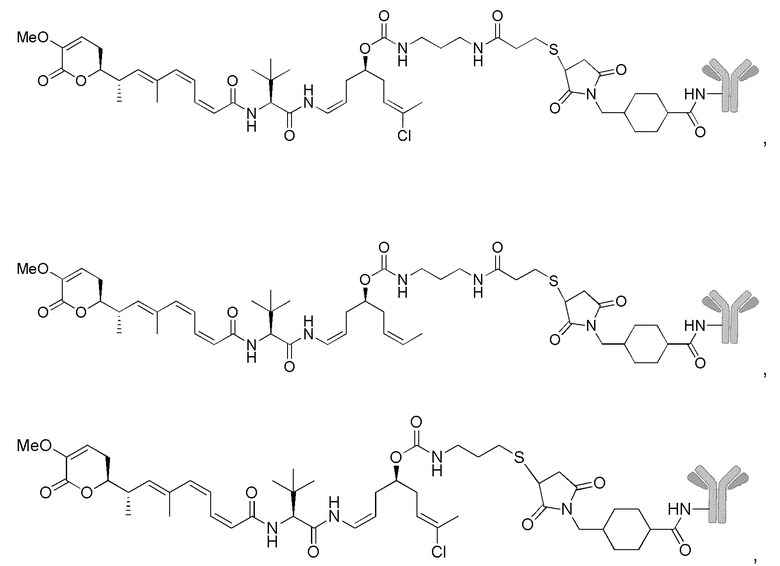

где каждый из  и
и  выбран из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, предпочтительно Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части, и наиболее предпочтительно Трастузумаба или его иммунологически активной части; или, альтернативно, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, и наиболее предпочтительно анти-CD13 антитела или его иммунологически активной части. Более предпочтительно конъюгат антитела с лекарственным средством выбран из группы, включающей:
выбран из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, предпочтительно Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части, и наиболее предпочтительно Трастузумаба или его иммунологически активной части; или, альтернативно, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, и наиболее предпочтительно анти-CD13 антитела или его иммунологически активной части. Более предпочтительно конъюгат антитела с лекарственным средством выбран из группы, включающей:
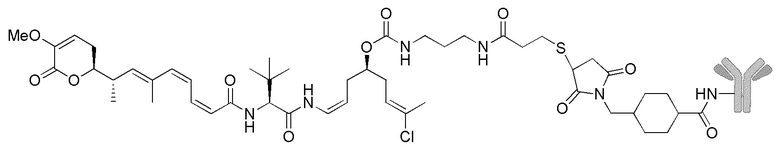
где выбран из Трастузумаба и анти-CD13 антитела или его иммунологически активной части, предпочтительно Трастузумаба или его иммунологически активной части; или предпочтительно анти-CD13 антитела или его иммунологически активной части,

где представляет собой Трастузумаб или его иммунологически активную часть,
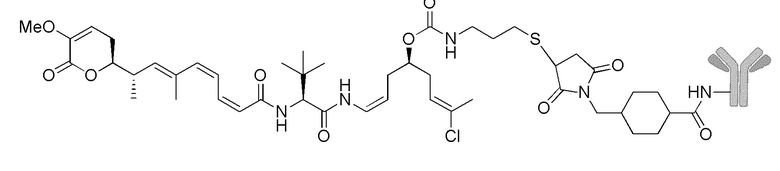
где представляет собой Трастузумаб или его иммунологически активную часть,
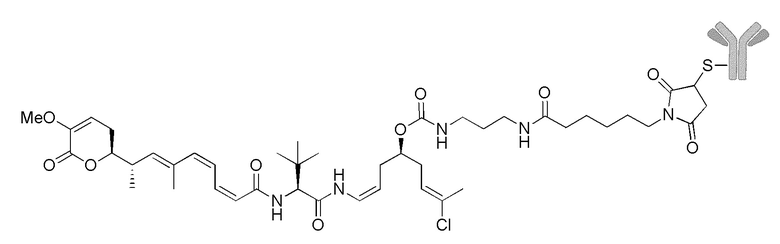
где выбран из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, предпочтительно Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части, и наиболее предпочтительно Трастузумаба или его иммунологически активной части; или, альтернативно, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части и наиболее предпочтительно анти-CD13 антитела или его иммунологически активной части,
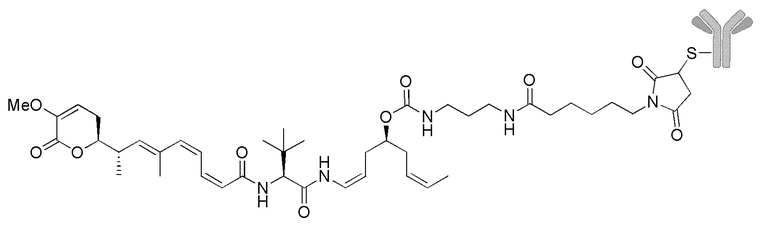
где представляет собой Трастузумаб или его иммунологически активную часть,
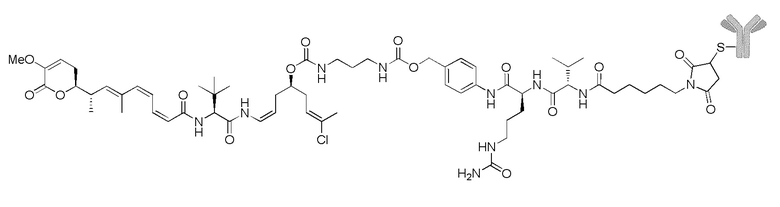
где выбран из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, предпочтительно Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части и наиболее предпочтительно Трастузумаба или его иммунологически активной части; или, альтернативно, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части и наиболее предпочтительно анти-CD13 антитела или его иммунологически активной части,

где представляет собой Трастузумаб или его иммунологически активную часть и
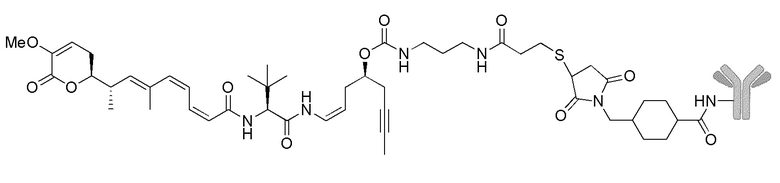
где представляет собой Трастузумаб или его иммунологически активную часть.
Особенно предпочтительно конъюгаты антитела с лекарственным средством в соответствии с настоящим изобретением должны быть в выделенной или очищенной форме.
Предпочтительные соединения формулы D-X-(AA)w-(L1)b в соответствии со вторым аспектом настоящего изобретения включают:
соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H в соответствии со вторым аспектом настоящего изобретения, где
L1 представляет собой линкер формулы:
 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 выбран из -C1-C12 алкилена-, -O-(C1-C12 алкилена), -C6-C12 арилена в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C1-C12 алкилен-C6-C12 арилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, - C6-C12 арилен-C1-C12 алкилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, -C5-C12 гетероцикло-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота и/или серы, в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -C1-C12 алкилен-(C5-C12 гетероцикло)-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx,-(C5-C12 гетероцикло)-C1-C12 алкилена-, где указанная гетероцикло группа может быть насыщенной или ненасыщенной группой, содержащей одно или несколько колец и содержащей по меньшей мере один атом кислорода, азота или серы в указанном кольце(кольцах), при этом указанная группа необязательно замещена одним или несколькими заместителями Rx, -(OCH2CH2)r- и -CH2-(OCH2CH2)r-, где каждый из указанных выше алкиленовых заместителей, или в отдельности или присоединенный к другому фрагменту углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx;
r представляет собой целое число, имеющее значение от 1 до 6; и
каждый из D, X, AA и w имеют значения, определенные в первом аспекте настоящего изобретения.
Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H в соответствии со вторым аспектом настоящего изобретения, где
L1 представляет собой линкер формулы:
 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 выбран из -C1-C8 алкилена-, -O-(C1-C8 алкилена), -C1-C8 алкилен-C6-C12 арилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, - C6-C12 арилен-C1-C8 алкилена-, где ариленовая группа присутствует в одном или нескольких кольцах, которые могут быть необязательно замещены одним или несколькими заместителями Rx, где каждый из указанных выше алкиленовых заместителей, или в отдельности или присоединенный к другому фрагменту углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx;
(AA)w представляет собой формулу (II):
 ,
,
где волнистые линии указывают точку ковалентных присоединений к X (волнистая линия слева) и к L1 или к атому водорода (волнистая линия справа);
где R21 в каждом случае выбран из группы, включающей водород, метил, изопропил, втор-бутил, бензил, индолилметил, -(CH2)3NHCONH2, -(CH2)4NH2, -(CH2)3NHC(=NH)NH2 и -(CH2)4NHC-(=NH)NH2, и w представляет собой целое число от 0 до 6;
X представляет собой расширяющую группу, выбранную из группы, включающей -CONH-(C2-C4 алкилен)NH-, -COO-CH2-фенилен-NH, где указанная фениленовая группа может быть необязательно замещена одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы, -CONH-(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-, -COCH2NH-COCH2-NH-, -CONH-(C2-C4 алкилен)S-, -CONH-(C2-C4 алкилен)-NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-; и
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, где
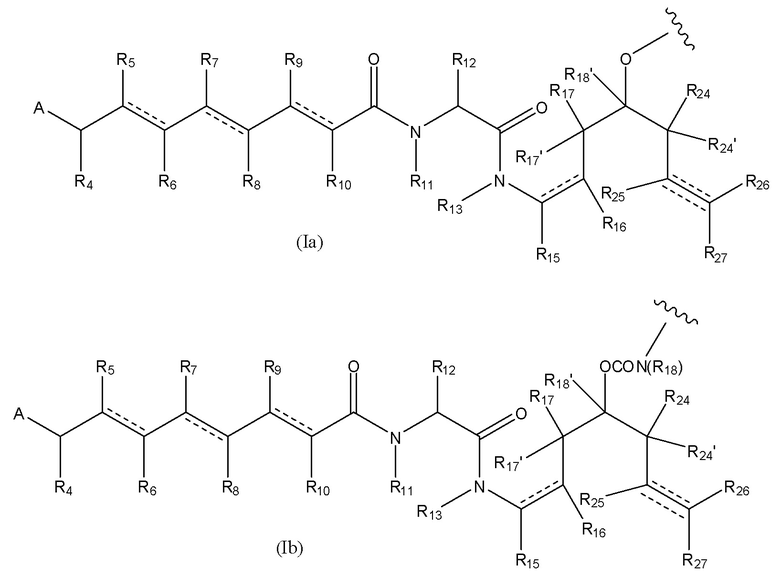 ,
,
где волнистые линии формул (Ia) и (Ib) указывают точку ковалентного присоединения к X;
A выбран из
 и
и  ,
,
где волнистые линии группы A указывают точку ковалентного присоединения к остальной части группы лекарственного средства;
R1 выбран из водорода, ORa и OCORa, где Ra выбран из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R2 и R3 каждый, независимо, выбран из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
R3’ выбран из водорода, CORa и замещенного или незамещенного C1-C6 алкила, где Ra представляет собой замещенный или незамещенный C1-C6 алкил, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода и замещенного и незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R11 и R13 независимо выбран из водорода и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, включающей:
водород и замещенные или незамещенные C1-C6 алкильные группы, где необязательные заместители выбраны из группы, включающей алкокси группы, содержащие от 1 до 6 атомов углерода, гидроксильные группы, оксо группы, атомы галогена, OCORy, OCOORy, CORy, COORy, OCONRyRz, CONRyRz, NRyRz, NRyCORz, где каждый из Ry и Rz выбран из атомов водорода и алкильных групп, содержащих от 1 до 6 атомов углерода.
R18 выбран из водорода, С1-6 алкильной группы, которая может быть необязательно замещена по меньшей мере одной группой Rx, и фенильной группы, которая необязательно замещена одним или несколькими заместителями RX;
R27 выбран из водорода, галогена и замещенного или незамещенного C1-C6 алкила, где необязательные заместители представляют собой один или несколько заместителей Rx;
и каждая пунктирная линия представляет собой необязательную дополнительную связь, но, когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26, либо R27 отсутствуют.
Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H в соответствии со вторым аспектом настоящего изобретения, где
L1 представляет собой группу формулы:
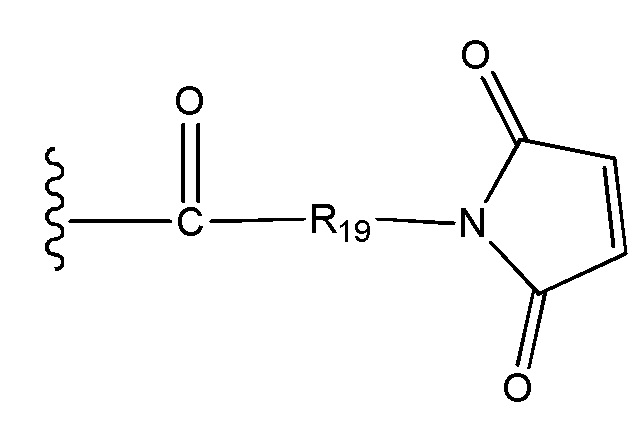 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 выбран из -C1-C6 алкилена-, фенилен-C1-C6 алкилена-, где фениленовая группа может быть необязательно замещена одним или несколькими заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы и циано группы, где каждый из указанных выше алкиленовых заместителей, в отдельности или присоединенный к другой группе в углеродной цепи, может быть необязательно замещен одним или несколькими заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, арильные группы, содержащие от 6 до 12 атомов углерода, атомы галогена, нитро группы и циано группы, и предпочтительно R19 представляет собой С1-C6 алкиленовую группу;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
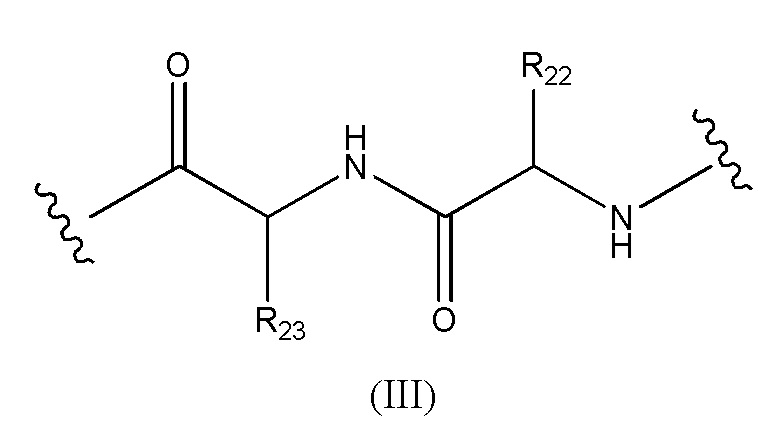 ,
,
где волнистые линии указывают точку ковалентных присоединений к X (волнистая линия слева) и к L1 или к атому водорода (волнистая линия справа);
R22 выбран из метила, бензила, изопропила, втор-бутила и индолилметила;
R23 выбран из метила, -(CH2)4NH2, -(CH2)3NHCONH2 и -(CH2)3NHC(=NH)NH2;
X представляет собой расширяющую группу, выбранную из -CONH-(C2-C4 алкилен)NH-,-CONH(C2-C4 алкилен)NHCOO-CH2-(фенилен, который может быть необязательно замещен одним или несколькими заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH, -CONH-(C2-C4 алкилен)S-,-CONH-(C2-C4 алкилен)NHCO-(C1-C3 алкилен)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-; и
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер:
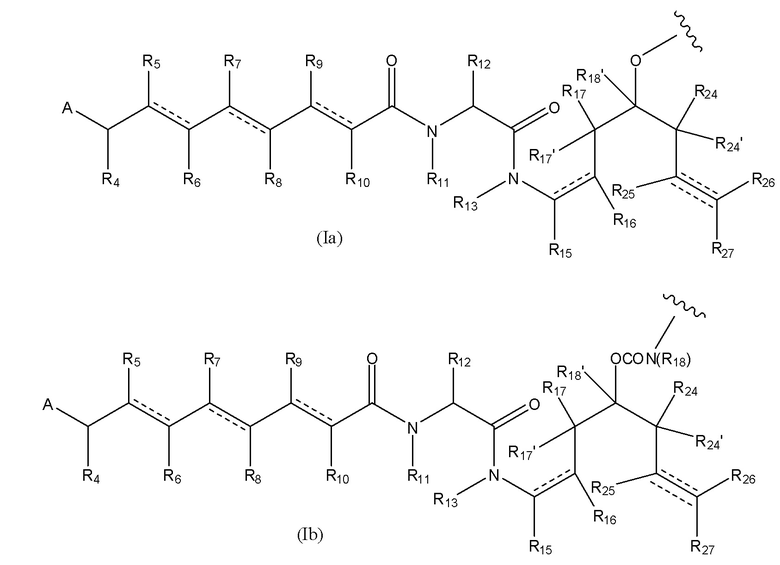 ,
,
где волнистые линии формулы (Ia) или формулы (Ib) указывают точку ковалентного присоединения к X;
A выбран из
 и
и 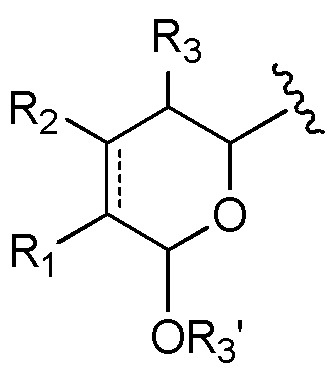 ,
,
где волнистые линии группы A указывают точку ковалентного присоединения к остальной части группы лекарственного средства;
R1 представляет собой водород или метокси;
каждый из R2 и R3 представляет собой водород;
R3’ представляет собой водород;
каждый из R5, R7, R8, R9 и R10 представляет собой водород;
каждый из R4 и R6 представляет собой метил;
R12 представляет собой изопропил, трет-бутил или бензил;
каждый из R11 и R13 представляет собой водород;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из водорода и C1-C6 алкильной группы, предпочтительно водорода и метила;
R18 выбран из водорода и фенила, и предпочтительно водорода;
R27 представляет собой водород или галоген;
и каждая пунктирная линия представляет собой необязательную дополнительную связь, но, когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26, либо R27 отсутствуют.
Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H в соответствии со вторым аспектом настоящего изобретения, где
L1 представляет собой линкер формулы:
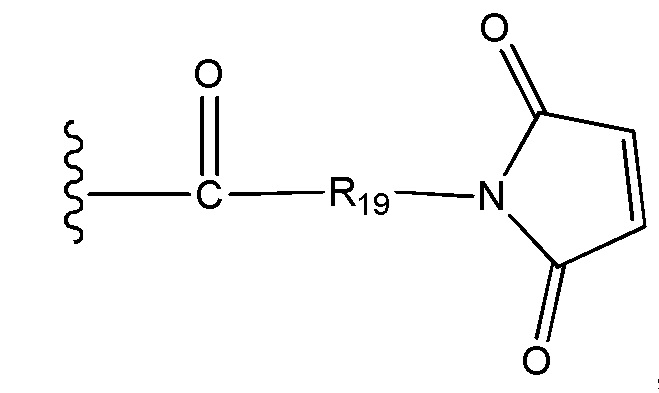 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 представляет собой -C3-C6 алкилен-;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
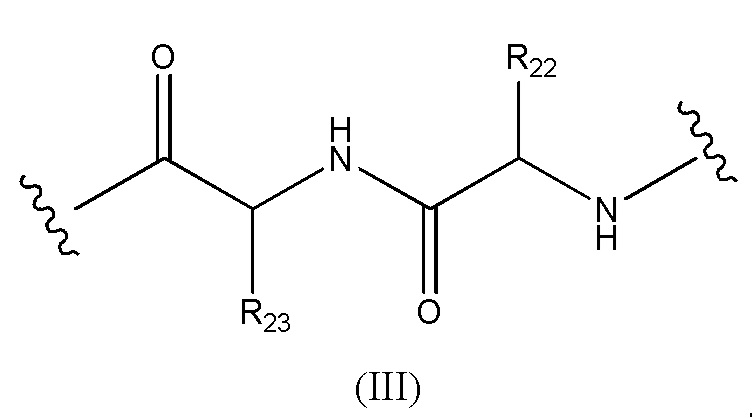 ,
,
R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, где волнистые линии указывают точку ковалентных присоединений к X (волнистая линия слева) и к L1 или к атому водорода (волнистая линия справа);
X представляет собой расширяющую группу, выбранную из группы, включающей -CONH-(C2-C4 алкилен)NH-, -COO-CH2-фенилен-NH-, где указанная фениленовая группа может быть необязательно замещена одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы, -CONH-(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-, -CONH-(C2-C4 алкилен)S-, -CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилен)NH-COO-CH2-(фенилен, который может быть необязательно замещен одним-четырьмя заместителями Rx, выбранными из группы, включающей алкильные группы, содержащие от 1 до 6 атомов углерода, алкокси группы, содержащие от 1 до 6 атомов углерода, атомы галогена, нитро группы или циано группы)-NH-; и
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, выбранные из следующей группы:
 ,
,
 ,
,
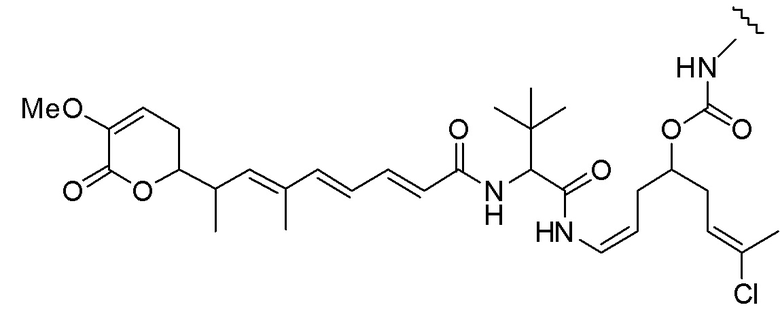 ,
,
 ,
,
 ,
,
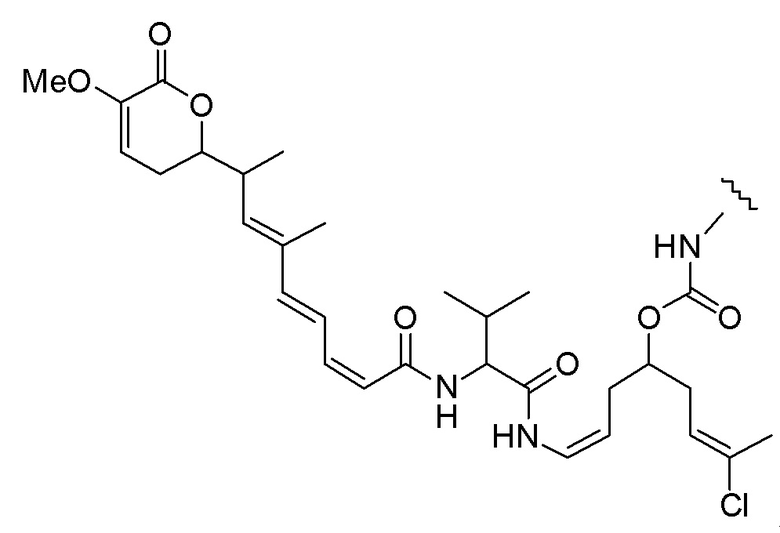 ,
,
 ,
,
 ,
,
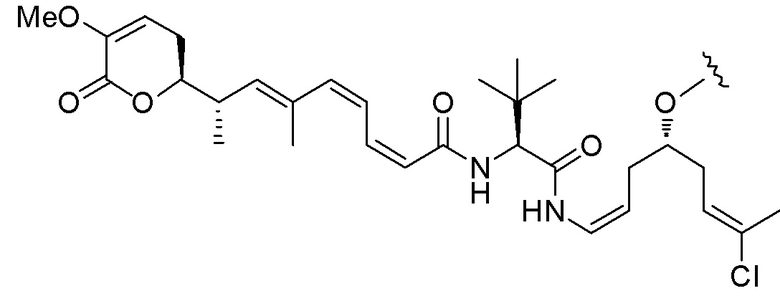 ,
,
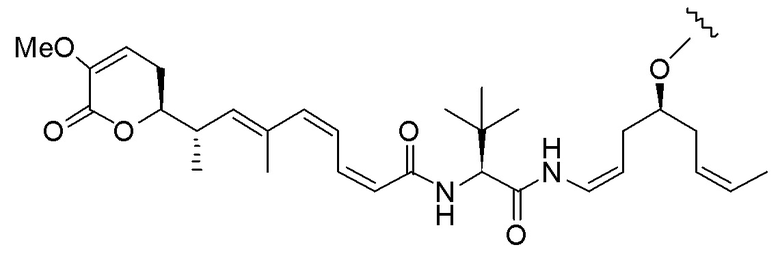 ,
,
 ,
,
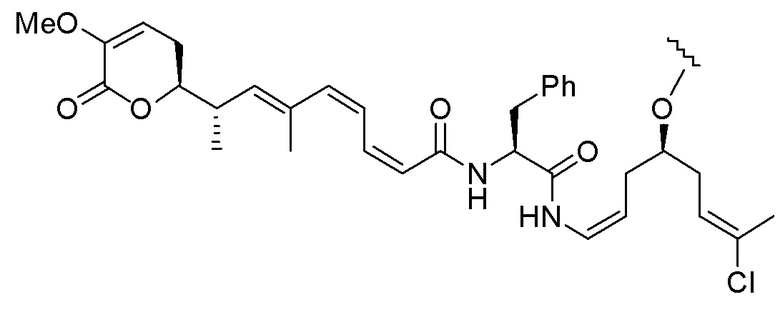 ,
,
 ,
,
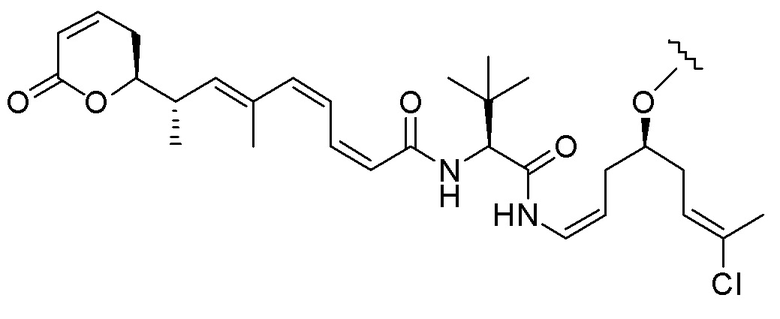 ,
,
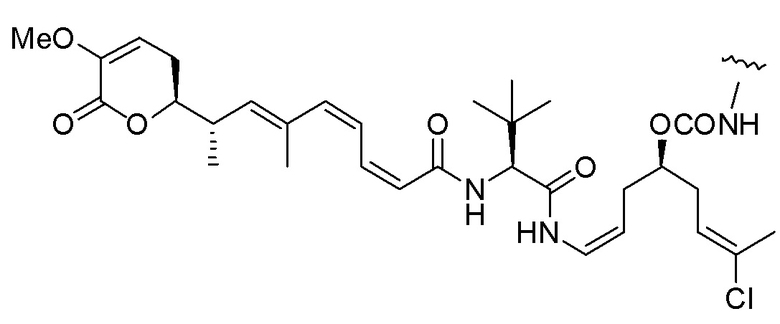 ,
,
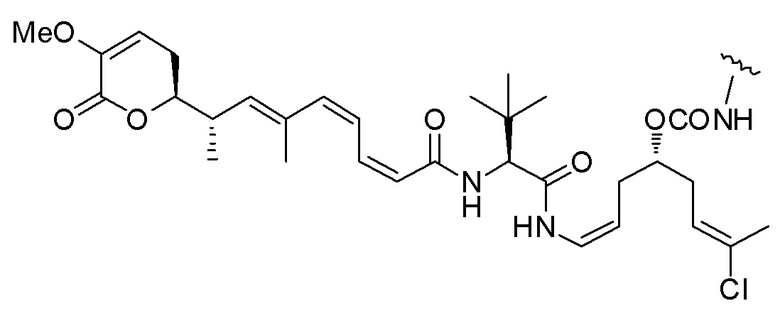 ,
,
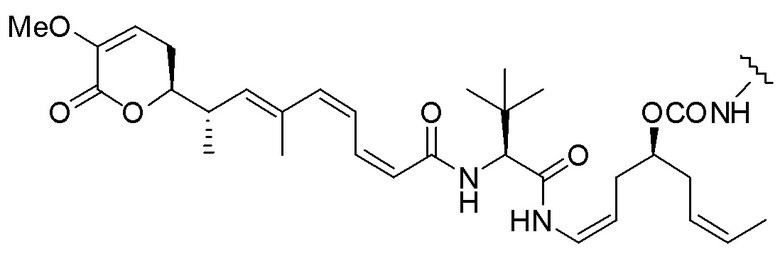 ,
,
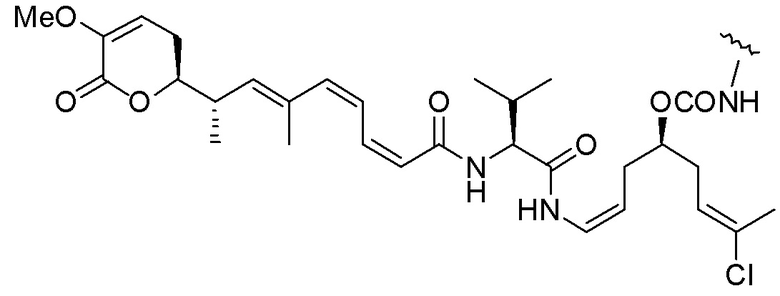 ,
,
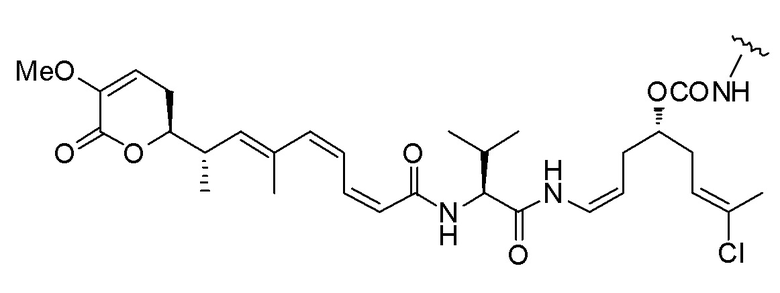 ,
,
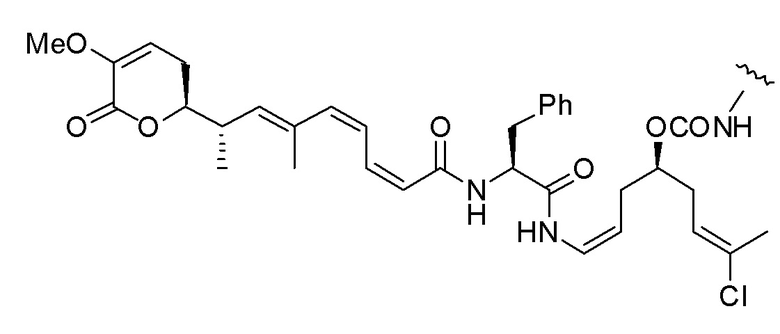 ,
,
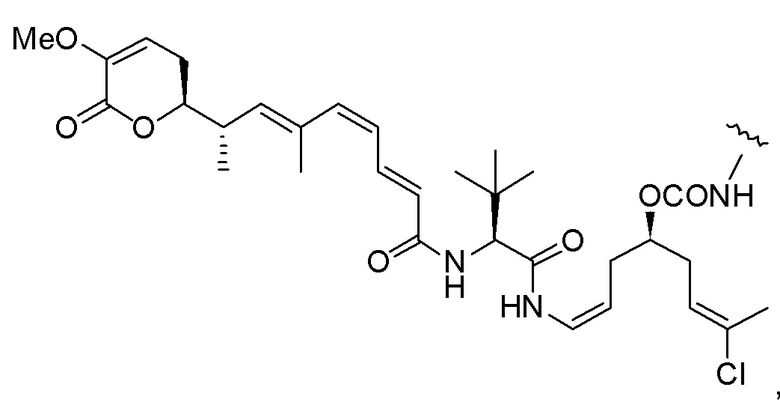
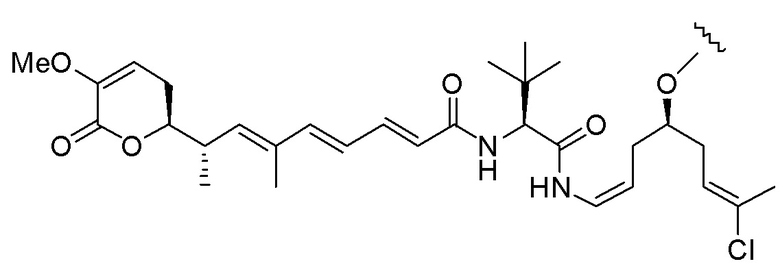 ,
,
 ,
,
 ,
,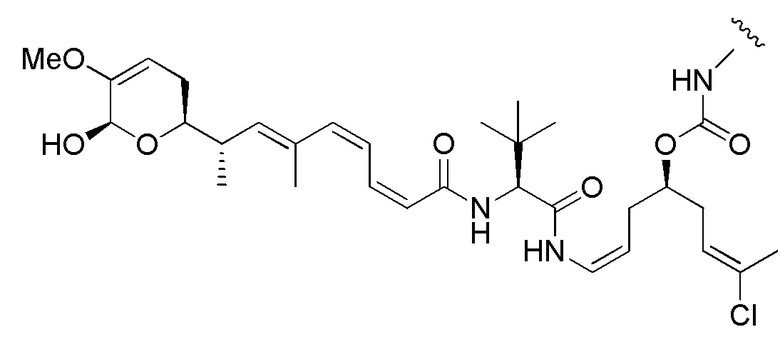 ,
, ,
,
 и
и  ,
,
где волнистая линия указывает точку ковалентного присоединения к X.
Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H в соответствии со вторым аспектом настоящего изобретения, где
L1 представляет собой группу формулы:
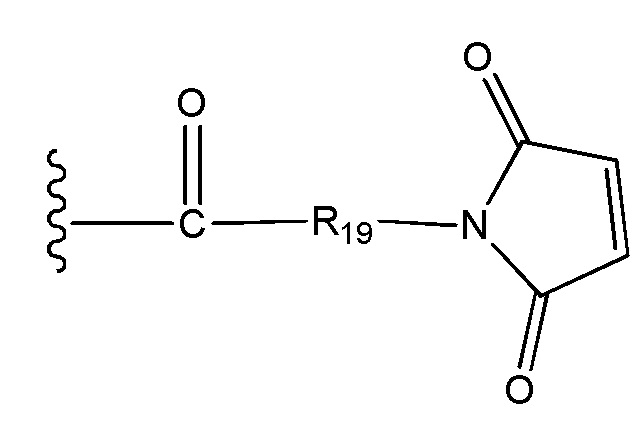 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 представляет собой -C5 алкилен-;
w имеет значение 0 или 2, и, когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
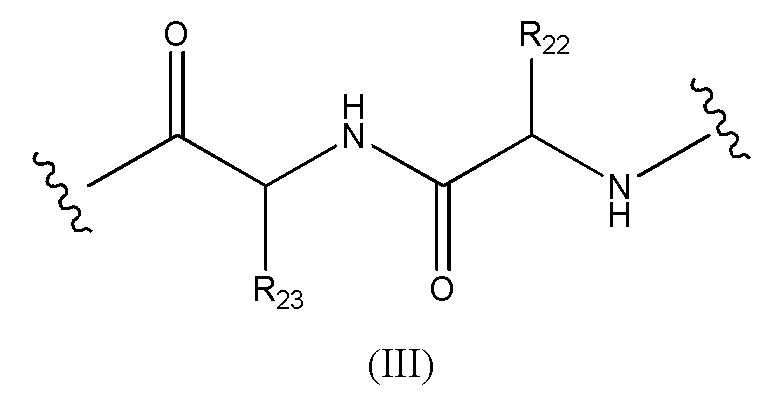 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, где волнистые линии указывают точку ковалентных присоединений к X (волнистая линия слева) и к L1 или к атому водорода (волнистая линия справа);
X представляет собой расширяющую группу, выбранную из -CONH(CH2)3NHCOOCH2-фенилен-NH-, и -CONH(CH2)3NH-, -CONH(CH2)3-S- и -CONH(CH2)3NHCO(CH2)2S-; и
D означает группу, представляющую собой лекарственное средство формулы (Ia), или его фармацевтически приемлемую соль, сложный эфир, сольват, таутомер или стереоизомер, выбранную из:
 и
и

где волнистая линия указывает точку ковалентного присоединения к X.
соединение формулы D-X-(AA)w-L1, выбранное из:
 ,
,
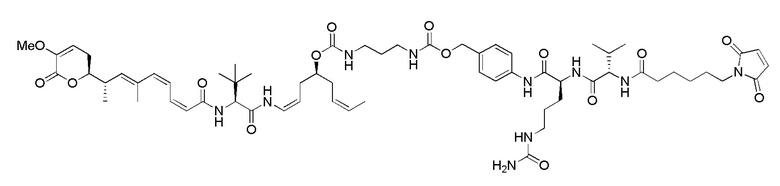
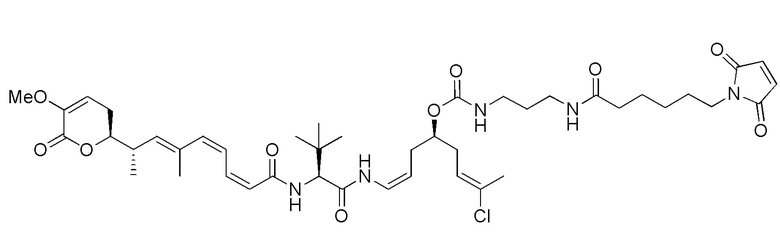 ,
,
 и
и
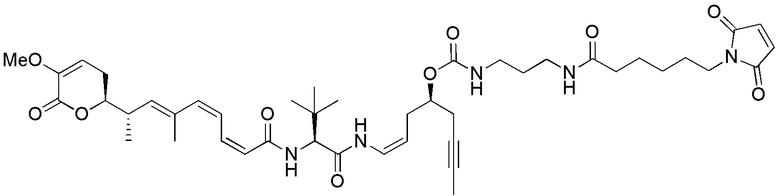 .
.
Соединение формулы D-X-(AA)w-H, выбранное из:

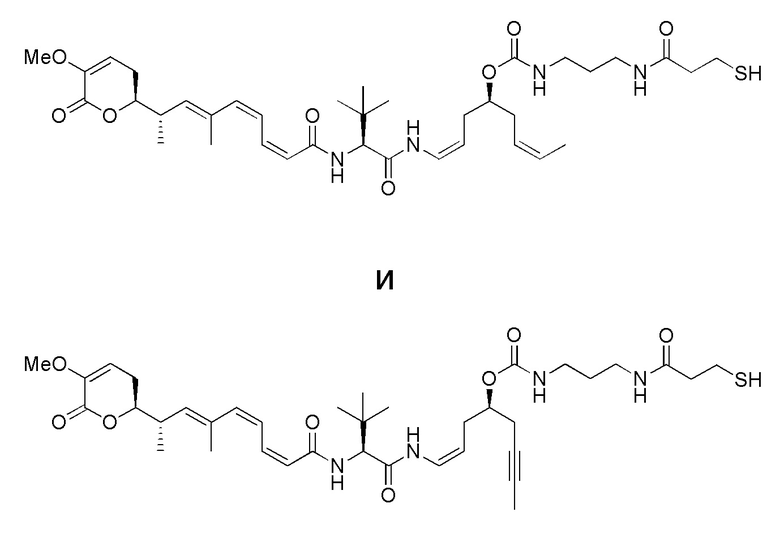
Термин “фармацевтически приемлемые соли, сложные эфиры, сольваты, таутомеры или стереоизомеры” в конъюгатах лекарственного средства по настоящему изобретению относится к любой фармацевтически приемлемой соли, сложному эфиру, сольвату, гидрату или стереоизомерной форме или любому другому соединению, которое, при введении пациенту способно обеспечивать соединение, описанное в настоящей заявке, либо непосредственно либо опосредованно. Однако следует иметь в виду, что фармацевтически неприемлемые соли также входят в объем изобретения, поскольку они могут быть полезны в получении фармацевтически приемлемых солей. Получение солей, пролекарств и производных можно осуществить при помощи способов, известных в данной области.
Например, фармацевтически приемлемые соли соединений, представленные в настоящей заявке, синтезируют из исходного соединения, которое содержит основную или кислотную группу, обычными химическими способами. Как правило, такие соли, например, получают путем взаимодействия форм свободной кислоты или основания этих соединений со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе или в смеси двух растворителей. Как правило, предпочтительны неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Примеры кислотно-аддитивных солей включают аддитивные соли минеральных кислот, такие как, например, гидрохлорид, гидробромид, гидройодид, сульфат, нитрат, фосфат, и аддитивные соли органической кислоты, такие как, например, ацетат, трифторацетат, малеат, фумарат, цитрат, оксалат, сукцинат, тартрат, малат, манделат, метансульфонат и пара-толуолсульфонат. Примеры основно-аддитивных солей включают неорганические соли, такие как, например, соли натрия, калия, кальция и аммония, и соли органических оснований, таких как, например, этилендиамин, этаноламин, N,N-диалкиленэтаноламин, триэтаноламин, и соли основных аминокислот.
Конъюгаты лекарственного средства по настоящему изобретению могут быть в кристаллической форме, либо в виде свободных соединений либо в виде сольватов (например, гидратов), и предполагается, что обе формы входят в объем настоящего изобретения. Способы сольватирования широко известны в данной области.
Любое соединение, которое представляет собой пролекарство конъюгированного лекарственного средства по настоящему изобретению, находится в рамках объема и сущности изобретения. Термин “пролекарство” используется в самом широком смысле и охватывает те производные, которые преобразуются in vivo в соединения по настоящему изобретению. Такие производные будут очевидны специалистам в данной области и включают, например, соединения, в которых свободная гидрокси группа преобразована в сложноэфирное производное. Многие подходящие пролекарства хорошо известны специалистам в данной области, и их можно найти, например, в Burger “Medicinal Chemistry and Drug Discovery 6th ed. (Donald J. Abraham ed., 2001, Wiley) и “Design and Applications of Prodrugs” (H. Bundgaard ed., 1985, Harwood Academic Publishers), содержание которых включено в настоящую заявку посредством ссылки.
В отношении соединений по настоящему изобретению, фармакологически приемлемые сложные эфиры не особо ограничены, и могут быть выбраны обычным специалистом в данной области. В случае указанных сложных эфиров предпочтительно, чтобы такие сложные эфиры могли расщепляться под воздействием биологического процесса, такого как гидролиз, in vivo. Группа, образующая указанные сложные эфиры (группа, показанная как R, когда сложные эфиры выражены как -COOR), может быть, например, С1-C4 алкокси C1-C4 алкильной группой, такой как метоксиэтил, 1-этоксиэтил, 1-метил-1-метоксиэтил, 1-(изопропокси)этил, 2-метоксиэтил, 2-этоксиэтил, 1,1-диметил-1-метоксиметил, этоксиметил, пропоксиметил, изопропоксиметил, бутоксиметил или трет-бутоксиметил; С1-C4 алкоксилированной C1-C4 алкокси C1-C4 алкильной группой, такой как 2-метоксиэтоксиметил; С6-C10 арилокси C1-C4 алкильной группой, такой как феноксиметил; галогенированной C1-C4 алкокси C1-C4 алкильной группой, такой как 2,2,2-трихлорэтоксиметил или бис(2-хлорэтокси)метил; С1-C4 алкоксикарбонил C1-C4 алкильной группой, такой как метоксикарбонилметил; циано C1-C4 алкильной группой, такой как цианометил или 2-цианоэтил; С1-C4 алкилтиометильной группой, такой как метилтиометил или этилтиометил; С6-C10 арилтиометильной группой, такой как фенилтиометил или нафтилтиометил; С1-C4 алкилсульфонил C1-C4 низшей алкильной группой, которая может быть необязательно замещенной атомом(атомами) галогена, такой как 2-метансульфонилэтил или 2-трифторметансульфонилэтил; С6-C10 арилсульфонил C1-C4 алкильной группой, такой как 2-бензолсульфонилэтил или 2-толуолсульфонилэтил; С1-C7 алифатической ацилокси C1-C4 алкильной группой, такой как формилоксиметил, ацетоксиметил, пропионилоксиметил, бутирилоксиметил, пивалоилоксиметил, валерилоксиметил, изовалерилоксиметил, гексаноилоксиметил, 1-формилоксиэтил, 1-ацетоксиэтил, 1-пропионилоксиэтил, 1-бутирилоксиэтил, 1-пивалоилоксиэтил, 1-валерилоксиэтил, 1-изовалерилоксиэтил, 1-гексаноилоксиэтил, 2-формилоксиэтил, 2-ацетоксиэтил, 2-пропионилоксиэтил, 2-бутирилоксиэтил, 2-пивалоилоксиэтил, 2-валерилоксиэтил, 2-изовалерилоксиэтил, 2-гексаноилоксиэтил, 1-формилоксипропил, 1-ацетоксипропил, 1-пропионилоксипропил, 1-бутирилоксипропил, 1-пивалоилоксипропил, 1-валерилоксипропил, 1-изовалерилоксипропил, 1-гексаноилоксипропил, 1-ацетоксибутил, 1-пропионилоксибутил, 1-бутирилоксибутил, 1-пивалоилоксибутил, 1-ацетоксипентил, 1-пропионилоксипентил, 1-бутирилоксипентил, 1-пивалоилоксипентил или 1-пивалоилоксигексил; С5-C6 циклоалкилкарбонилокси C1-C4 алкильной группой, такой как циклопентилкарбонилоксиметил, циклогексилкарбонилоксиметил, 1-циклопентилкарбонилоксиэтил, 1-циклогексилкарбонилоксиэтил, 1-циклопентилкарбонилоксипропил, 1-циклогексилкарбонилоксипропил, 1-циклопентилкарбонилоксибутил или 1-циклогексилкарбонилоксибутил; С6-C10 арилкарбонилокси C1-C4 алкильной группой, такой как бензоилоксиметил; С1-C6 алкоксикарбонилокси C1-C4 алкильной группой, такой как метоксикарбонилоксиметил, 1-(метоксикарбонилокси)этил, 1-(метоксикарбонилокси)пропил, 1-(метоксикарбонилокси)бутил, 1-(метоксикарбонилокси)пентил, 1-(метоксикарбонилокси)гексил, этоксикарбонилоксиметил, 1-(этоксикарбонилокси)этил, 1-(этоксикарбонилокси)пропил, 1-(этоксикарбонилокси)бутил, 1-(этоксикарбонилокси)пентил, 1-(этоксикарбонилокси)гексил, пропоксикарбонилоксиметил, 1-(пропоксикарбонилокси)этил, 1-(пропоксикарбонилокси)пропил, 1-(пропоксикарбонилокси)бутил, изопропоксикарбонилоксиметил, 1-(изопропоксикарбонилокси)этил, 1-(изопропоксикарбонилокси)бутил, бутоксикарбонилоксиметил, 1-(бутоксикарбонилокси)этил, 1-(бутоксикарбонилокси)пропил, 1-(бутоксикарбонилокси)бутил, изобутоксикарбонилоксиметил, 1-(изобутоксикарбонилокси)этил, 1-(изобутоксикарбонилокси)пропил, 1-(изобутоксикарбонилокси)бутил, трет-бутоксикарбонилоксиметил, 1-(трет-бутоксикарбонилокси)этил, пентилоксикарбонилоксиметил, 1-(пентилоксикарбонилокси)этил, 1-(пентилоксикарбонилокси)пропил, гексилоксикарбонилоксиметил, 1-(гексилоксикарбонилокси)этил или 1-(гексилоксикарбонилокси)пропил; С5-C6 циклоалкилоксикарбонилокси C1-C4 алкильной группой, такой как циклопентилоксикарбонилоксиметил, 1-(циклопентилоксикарбонилокси)этил, 1-(циклопентилоксикарбонилокси)пропил, 1-(циклопентилоксикарбонилокси)бутил, циклогексилоксикарбонилоксиметил, 1-(циклогексилоксикарбонилокси)этил, 1-(циклогексилоксикарбонилокси)пропил или 1-(циклогексилоксикарбонилокси)бутил; [5-(C1-C4 алкил)-2-оксо-1,3-диоксолен-4-ил]метильной группой, такой как (5-метил-2-оксо-1,3-диоксолен-4-ил)метил, (5-этил-2-оксо-1,3-диоксолен-4-ил)метил, (5-пропил-2-оксо-1,3-диоксолен-4-ил)метил, (5-изопропил-2-оксо-1,3-диоксолен-4-ил)метил или (5-бутил-2-оксо-1,3-диоксолен-4-ил)метил; [5-(фенил, который может быть необязательно замещен С1-C4 алкилом, C1-C4 алкокси или атомом(атомами) галогена)-2-оксо-1,3-диоксолен-4-ил]метильной группой, такой как (5-фенил-2-оксо-1,3-диоксолен-4-ил)метил, [5-(4-метилфенил)-2-оксо-1,3-диоксолен-4-ил]метил, [5-(4-метоксифенил)-2-оксо-1,3-диоксолен-4-ил]метил, [5-(4-фторфенил)-2-оксо-1,3-диоксолен-4-ил]метил или [5-(4-хлорфенил)-2-оксо-1,3-диоксолен-4-ил]метил; или фталидильной группой, которая может быть необязательно замещенной С1-C4 алкильной или C1-C4 алкокси группой(группами), такой как фталидил, диметилфталидил или диметоксифталидил, и предпочтительно представляет собой пивалоилоксиметильную группу, фталидильную группу или (5-метил-2-оксо-1,3-диоксолен-4-ил)метильную группу, и более предпочтительно (5-метил-2-оксо-1,3-диоксолен-4-ил)метильную группу.
Любое соединение, указанное в настоящей заявке, предназначено для представления такого конкретного соединения, а также некоторых вариантов или форм. В частности, соединения указанные в настоящей заявке, могут иметь асимметричные центры и поэтому могут существовать в различных энантиомерных формах. Все оптические изомеры и стереоизомеры соединений, указанных в настоящей заявке, и их смеси, рассматриваются в рамках настоящего изобретения. Таким образом, любое данное соединение, указанное в настоящей заявке, предназначено для представления любой из форм, выбранных из рацемата, одной или нескольких энантиомерных форм, одной или нескольких диастереомерных форм, одной или нескольких атропизомерных форм и их смесей. В частности, конъюгаты лекарственного средства формулы [D-(X)b-(AA)w-(L)]n-Ab и соединения формулы D-X-(AA)w-L1 или D-X-(AA)w-H могут включать энантиомеры в зависимости от их асимметрии или диастереоизомеры. Стереоизомерия вокруг двойной связи также возможна, поэтому в некоторых случаях молекула может существовать в виде (E)-изомера или (Z)-изомера. Если молекула содержит несколько двойных связей, каждая двойная связь будет обладать своей собственной стереоизомерией, которая может быть такой же или отличной от стереоизомерии других двойных связей в молекуле. Отдельные изомеры и смеси изомеров входят в объем настоящего изобретения.
Кроме того, соединения, указанные в настоящей заявке, могут существовать в виде геометрических изомеров (то есть цис- и транс-изомеры), в виде таутомеров или в виде атропизомеров. В частности, термин таутомер относится к одному из двух или нескольких структурных изомеров соединения, которые существуют в равновесии и легко преобразуются из одной изомерной формы в другую. Распространенные таутомерные пары представляют собой амин-имин, амид-имид, кето-енол, лактам-лактим и другие. Помимо этого, любое соединение, указанное в настоящей заявке, предназначено для представления гидратов, сольватов и полиморфов и их смесей, когда такие формы существуют в данной среде. Кроме того, соединения, указанные в настоящей заявке, могут существовать в изотопно-меченных формах. Все геометрические изомеры, таутомеры, атропизомеры, гидраты, сольваты, полиморфы и изотопно-меченные формы соединений, указанные в настоящей заявке, и их смеси включены в объем настоящего изобретения.
В соединениях по настоящему изобретению, Ab представляет собой компонент, включающий по меньшей мере один антиген-связывающий сайт. В альтернативном варианте воплощения, Ab может представлять собой любой подходящий агент, который способен к связыванию с клеткой-мишенью, предпочтительно с клеткой животного, и более предпочтительно с клеткой человека. Примеры таких агентов включают лимфокины, гормоны, факторы роста и молекулы-переносчики биогенных веществ (например, трансферрин).
Когда Ab представляет собой компонент, включающий по меньшей мере один антиген-связывающий сайт, такой компонент предпочтительно представляет собой антиген-связывающий пептид или полипептид. В предпочтительном варианте воплощения, такой компонент представляет собой антитело или его антиген-связывающий фрагмент.
Термин ‘антитело’ в конъюгатах лекарственного средства по настоящему изобретению относится к любому иммуноглобулину, предпочтительно полноразмерному иммуноглобулину. Предпочтительно термин охватывает моноклональные антитела, поликлональное антитела, мультиспецифические антитела, такие как биспецифические антитела, и фрагменты антител при условии, что они проявляют желаемую биологическую активность. Антитела могут происходить из любых видов, но предпочтительно имеют происхождение от грызунов, например крыс или мышей, или кролика и человека. Альтернативно, антитела, предпочтительно моноклональные антитела, могут быть гуманизированными, химерными или фрагментами таких антител. Термин ‘химерные антитела’ также может включать "приматизированные" антитела, включающие различные последовательности антиген-связывающих доменой, происходящие из отличного от человека примата (например, мартышки, обезьяны и других), и последовательности константной области человека. Иммуноглобулины также могут представлять собой любой тип (например, IgG, IgE, IgM, IgD, и IgA), класс (например, IgGl, IgG2, IgG3, IgG4, IgAl и IgA2) или подкласс молекулы иммуноглобулина.
Термин ‘моноклональное антитело’ относится к практически гомогенной популяции молекул антител (т.е. индивидуальные антитела, составляющие популяцию, являются идентичными за исключением возможных встречающихся в природе мутаций, которые могут присутствовать в незначительных количествах), продуцируемой одним клоном B-клеток, часто гибридомой. Важно, что каждое моноклональное антитело обладает одинаковой антигенной специфичностью - то есть оно направлено против одной детерминанты на антигене.
Получение моноклональных антител можно осуществить при помощи способов, известных в данной области. Однако, в качестве примера, моноклональные антитела могут быть получены методом гибридом (Kohler et al. (1975) Nature 256:495), методом B-клеточной гибридомы человека (Kozbor et al., 1983, Immunology Today 4: 72) или методом EBV-гибридомы (Cole et al., 1985, Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc., pp. 77-96). Альтернативно, моноклональное антитело может быть получено с использованием методов рекомбинантной ДНК (см., US 4816567) или выделено из фаговых библиотек антител с использованием методов, описанных в Clackson et al. (1991) Nature, 352:624-628; Marks et al. (1991) J. MoI. Biol., 222:581-597.
Поликлональное антитела представляют собой антитела, направленные против различных детерминант (эпитопов). Эта гетерогенная популяция антитела может быть получена из сыворотки крови вакцинированных животных с использованием различных процедур, хорошо известных в данной области.
Термин ‘биспецифическое антитело’ относится к искусственному антителу, состоящему из двух разных моноклональных антител. Они могут быть предназначены либо для связывания с двумя соседними эпитопами на одном антигене, тем самым увеличивая как авидность, так и специфичность, или связывания с двумя разными антигенами для множества различных изменений, но особенно для рекрутинга цитотоксических T- и природных киллерных клеток (NK) или переориентирования токсинов, радионуклидов или цитотоксических лекарственных средств для лечения рака (Holliger & Hudson, Nature Biotechnology, 2005, 9, 23). Биспецифическое антитело может иметь гибрид тяжелой цепи иммуноглобулина с первой специфичностью связывания в одном ответвлении, и гибрид пары тяжелой цепи-легкой цепи иммуноглобулина (обеспечивающую вторую специфичность связывания) в другом ответвлении. Эта асимметричная структура облегчает отделение желаемого биспецифического соединения от нежелательных комбинаций цепей иммуноглобулина, поскольку наличие легких цепей иммуноглобулина только в одной половине биспецифической молекулы обеспечивает легкий путь отделения (WO 94/04690; Suresh et al., Methods in Enzymology, 1986, 121:210; Rodrigues et al., 1993, J. of Immunology 151:6954-6961; Carter et al., 1992, Bio/Technology 10:163-167; Carter et al., 1995, J. of Hematotherapy 4:463-470; Merchant et al., 1998, Nature Biotechnology 16:677-681).
Способы получения гибрида или биспецифических антител известны в данной области. В одном способе, биспецифические антитела могут быть получены путем слияния двух гибридом в одну ‘квадрому’ путем химического сшивания или генетического слияния двух разных модулей Fab или scFv (Holliger & Hudson, Nature Biotechnology, 2005, 9, 23).
Термин ‘химерное’ антитело относится к антителу, в котором различные части происходят из различных видов животных. Например, химерное антитело может иметь вариабельную область мышиного происхождения и константную область человеческого происхождения. С другой стороны, a ‘гуманизированное антитело’ преимущественно имеет человеческое происхождение, даже если оно содержит части, происходящие не от человека. Конкретнее, гуманизированные антитела представляют собой человеческие иммуноглобулины (реципиентное антитело), в которых остатки из гипервариабельной области реципиента заменены остатками из гипервариабельных областей нечеловеческих видов (донорское антитело), таких как мышь, крыса, кролик или отличный от человека примат, обладающими желаемой специфичностью, аффинностью и способностью. В некоторых случаях, остатки каркасной области (FR) человеческого иммуноглобулина заменены соответствующими остатками отличных от человека видов. Кроме того, гуманизированные антитела могут включать остатки, которые не обнаружены в реципиентном антителе или в донорском антителе. Эти модификации осуществляют для дальнейшего улучшения функционирования антитела. В общем, гуманизированное антитело будет включать по существу все из по меньшей мере одного, и обычно двух, вариабельных доменов, в которых все или по существу все из гипервариабельных петель соответствуют тем, которые присутствуют в не-человеческом иммуноглобулине, и все или по существу все из FRs представляют собой такие, которые имеют последовательность иммуноглобулина человека. Гуманизированное антитело необязательно также будет включать, по меньшей мере, часть константной области иммуноглобулина (Fc), типично, иммуноглобулина человека.
Рекомбинантные антитела, такие как химерные и гуманизированные моноклональные антитела, могут быть получены при помощи методов рекомбинантной ДНК, известных в данной области. Полностью человеческие антитела могут быть получены с использованием трансгенных мышей, которые не могут экспрессировать гены тяжелой и легкой цепи эндогенного иммуноглобулина, но которые могут экспрессировать гены тяжелой и легкой цепи человека. Трансгенных мышей иммунизировали обычным способом выбранным антигеном. Моноклональные антитела, направленные против антигена, могут быть получены с использованием обычной гибридомной технологии. Трансгены иммуноглобулина человека, накопленные у трансгенных мышей, подвергаются перегруппировке в процессе B-клеточной дифференциации, и затем происходит переключение класса и соматическая мутация. Таким образом, с использованием такого метода можно получить терапевтически полезные IgG, IgA, IgM и IgE антитела. Для обзора этой технологии для получения антител человека см. Lonberg and Huszar (1995, Int. Rev. Immunol. 13:65-93). Подробное обсуждение этой технологии для получения человеческих антител и моноклональных человеческих антител и протоколы для получения таких антител см., например, в Патентах США №: 5625126; 5633425; 5569825; 5661016; 5545806; каждый из которых включен в настоящую заявку посредством ссылки во всей полноте. Другие человеческие антитела могут быть получены коммерчески, например, от компании Abgenix, Inc. (Freemont, CA) и Genpharm (San Jose, CA).
Термин ‘антиген-связывающий фрагмент’ в конъюгатах лекарственного средства по настоящему изобретению относится к части полноразмерного антитела, где такие антиген-связывающие фрагменты антител сохраняют антиген-связывающую функцию соответствующего полноразмерного антитела. Антиген-связывающий фрагмент может включать часть вариабельной области антитела, при этом такая часть содержит по меньшей мере одну, две, предпочтительно три CDR, выбранных из CDR1, CDR2 и CDR3. Антиген-связывающий фрагмент также может включать часть легкой и тяжелой цепи иммуноглобулина. Примеры фрагментов антител включают Fab, Fab', F(ab')2, scFv, di-scFv и BiTE (би-специфические задействующие T-клетки), Fv фрагменты, включая нанотела, диатела, диатело-Fc гибриды, триатела и тетратела; мини-тела; линейные антитела; фрагменты, полученные с использованием библиотеки Fab экспрессии, анти-идиотипических (анти-Id) антител, CDR (определяющая комплементарность область) и эпитоп-связывающих фрагментов любого из указанных выше, которые иммуноспецифически связываются с целевым антигеном, таким как антигены раковой клетки, вирусные антигены или микробные антигены, молекул одноцепочечных или одно-доменных антител, включая антитела, содержащие только тяжелую цепь, например, VHH домены верблюда и V-NAR акулы; и мультиспецифических антител, образованных из фрагментов антитела. Для сравнения, полноразмерное антитело, называемое ‘антитело’, представляет собой антитело, включающее VL и VH домены, а также полные константные области легких и тяжелых цепей.
Антитело также может иметь одну или несколько эффекторных функций, которые относятся к биологическим активностям, обусловленным FC областями (Fc область нативной последовательности или Fc область варианта аминокислотной последовательности, сконструированные в соответствии со способами, известными из уровня техники, для изменения связывания с рецептором) антитела. Примеры эффекторных функций антитела включают CIq связывание; комплемент-зависимую цитотоксичность; связывание Fc рецептора; антитело-зависимую клеточно-опосредованную цитотоксичность (ADCC); фагоцитоз; отрицательную негативную модуляцию рецепторов клеточной поверхности (например, B-клеточный рецептор; BCR) и т.п.
Антитело также может представлять собой функционально активный фрагмент, производное или аналог антитела, которое иммуноспецифически связывается с целевым антигеном, таким как антиген раковой клетки, вирусный антиген или микробный антиген, или другие антитела, связывающиеся с опухолевыми клетками. В этой связи, “функционально активный” означает, что фрагмент, производное или аналог способен индуцировать анти-идиотипические антитела, которые распознают тот же антиген, что распознается антителом, из которого получен фрагмент, производное или аналог. Более конкретно, в примере осуществления настоящего изобретения антигенность идиотипа молекулы иммуноглобулина может быть усилена путем делеции каркасной и CDR последовательностей, которые являются C-концевыми относительно CDR последовательности, которая специфически распознает антиген. Для определения, какие CDR последовательности связываются с антигеном, синтетические пептиды, содержащие CDR последовательности, могут быть использованы в анализах связывания с антигеном с использованием любого метода осуществления анализа связывания, известного в данной области (например, BIA анализ ядра) (см., например, Kabat et al., 1991, Sequences of Proteins of Immunological Interest, Fifth Edition, National Institute of Health, Bethesda, Md; Kabat E et al., 1980, J. of Immunology 125(3):961-969).
Термин ‘антитело’ также может включать гибридный белок антитела, или его функционально активный фрагмент, например, где слияние антитела происходит через ковалентную связь (например, пептидную связь), либо на N-конце либо C-конце, с аминокислотной последовательностью другого белка (или ее частью, такой как, по меньшей мере часть белка из 10, 20 или 50 аминокислот), который не является антителом. Антитело или его фрагмент могут быть ковалентно связаны с другим белком на N-конце константного домена.
Кроме того, антитело или антиген-связывающие фрагменты по настоящему изобретению могут включать аналоги и производные антител или их антиген-связывающие фрагменты, модифицированные, например, путем ковалентного присоединения любого типа молекулы, при условии, что такое ковалентное присоединение дает возможность антителу сохранить его антиген-связывающую иммуноспецифичность. Примеры модификаций включают гликозилирование, ацетилирование, пэгилирование, фосфорилирование, амидирование, дериватизацию известными защитными/блокирующими группами, протеолитическое расщепление, связывание с клеточной единицей антитела или другим белком, и т.п. Любую из многочисленных химических модификаций можно осуществлять известными способами, включая, но не ограничиваясь этим, специфическое химическое расщепление, ацетилирование, формилирование, метаболический синтез в присутствии туникамицина, и другие. Помимо этого, аналог или производное могут содержать одну или несколько не встречающихся в природе аминокислот.
Антитела или антиген-связывающие фрагменты по настоящему изобретению также могут иметь модификации (например, замещения, делеции или добавки) в Fc домене антитела. Более конкретно, модификации могут быть в Fc-шарнирной области и приводить к повышению связывания в отношении FcRn рецептора (WO 97/34631).
В одном варианте воплощения настоящего изобретения, антитело в конъюгате лекарственного средства по настоящему изобретению может представлять собой любое антитело или его антиген-связывающий фрагмент, предпочтительно моноклональное антитело, которое является полезным для лечения заболевания, предпочтительно рака. Рак может представлять собой рак молочной железы, колоректальный рак, рак эндометрия, рак почки, лейкемии, рак легких, множественную миелому, солидные опухоли, такие как лимфомы (например, болезнь Ходжкина и неходжкинская лимфома), саркому и карциномы, меланому, мезотелиому, остеосаркому, рак яичников и рак почки. В предпочтительном варианте воплощения рак представляет собой рак легких, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак почки, лейкоз, множественную миелому, лимфому и рак яичников. В более предпочтительном варианте воплощения рак представляет собой колоректальный рак, рак молочной железы, лейкоз, лимфому и рак яичников.
Антитела, которые могут быть полезными для лечения рака, включают, но не ограничиваются этим, антитела против следующих антигенов: CA125 (овариальный), CA15-3 (карциномы), CA19-9 (карциномы), L6 (карциномы), Lewis Y (карциномы), Lewis X (карциномы), альфа фетобелок (карциномы), CA 242 (колоректальный), плацентарная щелочная фосфатаза (карциномы), специфичный антиген простаты (простата), простатическая кислая фосфатаза (простата), эпидермальный фактор роста (карциномы) например, рецепторный белок 2 EGF (рак молочной железы), MAGE-I (карциномы), MAGE-2 (карциномы), MAGE-3 (карциномы), MAGE-4 (карциномы), анти-трансферриновый рецептор (карциномы), p97 (меланома), MUCl-KLH (рак молочной железы), CEA (колоректальный), gplOO (меланома), MARTl (меланома), PSA (простата), IL-2 рецептор (T-клеточный лейкоз и лимфомы), CD20 (неходжкинская лимфома), CD52 (лейкоз), CD33 (лейкоз),CD22 (лимфома), хорионический гонадотропин человека (карцинома), CD38 (множественная миелома), CD40 (лимфома), муцин (карциномы), P21 (карциномы), MPG (меланома) и Neu онкогенный продукт (карциномы). Некоторые специфические полезные антитела включают, но не ограничиваются этим, BR96 mAb (Trail, P. A., et al. Science (1993) 261, 212-215), BR64 (Trail, PA, et al. Cancer Research (1997) 57, 100-105), mAbs против CD40 антигена, такие как S2C6 mAb (Francisco, J. A., et al. Cancer Res. (2000) 60:3225-3231), mAbs против CD70 антигена, такие как 1F6 mAb, и mAbs против CD30 антигена, такие как AClO (Bowen, M. A., et al. (1993) J. Immunol., 151:5896-5906; Wahl et al., 2002 Cancer Res. 62(13):3736-42). Многие другие интернализированные антитела, которые связываются с опухоль-ассоциированными антигенами, могут быть использованы и были описаны в литературе (Franke, A. E., et al. Cancer Biother Radiopharm. (2000) 15:459-76; Murray, J. L., (2000) Semin Oncol, 27:64-70; Breitling, F., and Dubel, S., Recombinant Антитела, John Wiley, and Sons, New York, 1998).
Другие опухоль-ассоциированные антигены включают, но не ограничиваются этим, BMPR1B, E16, STEAP1, STEAP2, 0772P. MPF, Napi3b, Sema5b, PSCA hlg, ETBR, MSG783, TrpM4, CRIPTO, CD21, CD79b, FcRH2, HER2, NCA, MDP, IL20Rα, Brevican, EphB2R, ASLG659, PSCA, GEDA, BAFF-R, CD79A, CXCR5, HLA-DOB, P2X5, CD72, LY64, FCRH1, IRTA2 и TENB2.
В альтернативном варианте воплощения, антитело в конъюгате лекарственного средства по настоящему изобретению может представлять собой антитело или его антиген-связывающий фрагмент, предпочтительно моноклональное антитело, которое иммуноспецифически связывается с вирусным антигеном, микробным антигеном или антигеном клетки, которая продуцирует аутоиммунные антитела, связанные с аутоиммунным заболеванием.
Вирусный антиген может включать, но не ограничивается этим, любой вирусный пептид, полипептид или белок, такой как HIV gpl20, HIV nef, RSV F гликопротеин, нейраминидаза вируса гриппа, гемагглютинин вируса гриппа, HTLV tax, гликопротеин вируса простого герпеса (например, Gb, Gc, Gd, и Ge) и поверхностный антиген гепатита В), который может вызывать иммунный ответ.
Микробный антиген может включать, но не ограничивается этим, любой микробный пептид, полипептид, белок, сахарид, полисахарид или липидную молекулу (например, бактериальный, грибковый, патогенный протозойный или дрожжевой полипептид, включая, например, LPS и капсулярный полисахарид), который может вызывать иммунный ответ.
В другом варианте воплощения настоящего изобретения, антитело может представлять собой любое антитело, известное для лечения или профилактики вирусной или микробной инфекции - то есть, инфекционного заболевания. Примеры таких антител включают, но не ограничиваются этим, PRO542 (Progenies), которое представляет собой СD4 слитое антитело, полезное для лечения HIV инфекции; OsTAVIR (Protein Design Labs, Inc., CA), которое представляет собой человеческое антитело, полезное для лечения вируса гепатита В; PROTOVIR. (Protein Design Labs, Inc., CA), которое представляет собой гуманизированное IgG1 антитело, полезное для лечения цитомегаловируса (CMV); и анти-LPS антитела.
Другие антитела, полезные для лечения инфекционных заболеваний, включают, но не ограничиваются этим, антитела против антигенов из патогенных штаммов бактерий (Streptococcus pyogenes, Streptococcus pneumoniae, Neisseria gonorrheae, Neisseria meningitidis, Corynebacterium diphtheriae, Clostridium botulinum, Clostridium perfringens, Clostridium tetani, Hemophilus influenzae, Klebsiella pneumoniae, Klebsiella ozaenas, Klebsiella rhinoscleromotis, Staphylococcus aureus, Vibrio colerae, Escherichia coli, Pseudomonas aeruginosa, Campylobacter (Vibrio) fetus, Aeromonas hydrophila, Bacillus cereus, Edwardsiella tarda, Yersinia enterocolitica, Yersinia pestis, Yersinia pseudotuberculosis, Shigella dysenteriae, Shigella flexneri, Shigella sonnei, Salmonella typhimurium, Treponema pallidum, Treponema pertenue, Treponema carateneum, Borrelia vincentii, Borrelia burgdorferi, Leptospira icterohernorrhagiae, Mycobacterium tuberculosis, Pneumocystis carinii, Francisella tularensis, Brucella abortus, Brucella suis, Brucella melitensis, Mycoplasma spp., Rickettsia prowazeki, Rickettsia tsutsugumushi, Chlamydia spp.); патогенные грибы (Coccidioides immitis, Aspergillus fumigatus, Candida albicans, Blastomyces dermatitidis, Cryptococcus neoformans, Histoplasma capsulatum); простейшие (Entomoeba histolytica, Toxoplasma gondii, Trichomonas tenas, Trichomonas hominis, Trichomonas vaginalis, Tryoanosoma gambiense, Trypanosoma rhodesiense, Trypanosoma cruzi, Leishmania donovani, Leishmania tropica, Leishmania braziliensis, Pneumocystis pneumonia, Plasmodium vivax, Plasmodium falciparum, Plasmodium malaria); или гельминты (Enterobius vermicularis, Trichuris trichiura, Ascaris lumbricoides, Trichinella spiralis, Strongyloides stercoralis, Schistosoma japonicum, Schistosoma mansoni, Schistosoma haematobium и нематоды).
Другие антитела, полезные для лечения вирусных заболеваний, включают, но не ограничиваются этим, антитела против антигенов патогенных вирусов, включая в качестве примеров и без ограничения: поксвирус, вирус герпеса, вирус простого герпеса 1, вирус простого герпеса 2, аденовирус, паповавирус, энтеровирус, пикорнавирус, парвовирус, реовирус, ретровирус, вирусы гриппа, вирусы парагриппа, паротит, корь, респираторно-синцитиальный вирус, краснуху, арбовирус, рабдовирус, аренавирус, вирус гепатита A, вирус гепатита В, вирус гепатита C, вирус гепатита E, вирус "ни А, ни В"-гепатита, риновирус, коронавирус, ротовирус и Вирус Иммунодефицита Человека.
В альтернативном варианте воплощения, антитело конъюгата лекарственного средства по настоящему изобретению также может представлять собой любое антитело, известное для лечения или профилактики аутоиммунных расстройств, таких как, но не ограничиваясь этим, Th2 лимфоцит-зависимые расстройства (например, атопический дерматит, атопическая астма, риноконъюнктивит, аллергический ринит, синдром Оменна, системный склероз и реакция "трансплантат против хозяина"); Th1 лимфоцит-зависимые расстройства (например, ревматоидный артрит, рассеянный склероз, псориаз, синдром Шегрена, тиреоидит Хашимото, болезнь Грейвса, первичный билиарный цирроз, гранулематоз Вегенера и туберкулез); активированный B лимфоцит-зависимые расстройства (например, системная красная волчанка, синдром Гудпасчера, ревматоидный артрит и диабет I типа); и активный хронический гепатит, болезнь Аддисона, аллергический альвеолит, аллергические реакции, аллергический ринит, синдром Альпорта, анафилаксия, анкилозирующий спондилит, анти-фосфолипидный синдром, артрит, аскаридоз, аспергиллез, атопические аллергии, атопический дерматит, атопический ринит, болезнь Бехчета, легочная аллергия птицеводов, бронхиальная астма, синдром Каплана, кардиомиопатия, глютенчувствительная целиакия, болезнь Чагаса, хронический гломерулонефрит, синдром Когана, болезнь холодовых агглютининов, инфекционная врожденная краснуха, CREST-синдром, болезнь Крона, криоглобулинемия, синдром Кушинга, дерматомиозит, дискоидная волчанка, синдром Дресслера, синдром Итона-Ламберта, эховирусная инфекция, энцефаломиелит, эндокринная офтальмопатия, вирусная инфекция Эпштейна-Барра, Equine Heaves, эритематоз, синдром Эвана, синдром Фелти, фибромиалгия, циклит Фукса, атрофия желудка, желудочно-кишечные аллергические реакции, крупноклеточный артериит, гломерулонефрит, синдром Гудпасчера, реакция "трансплантат против хозяина", болезнь Грейва, болезнь Гийена-Барре, тиреоидит Хашимото, гемолитическая анемия, пурпура Шенлейна-Геноха, идиопатическая атрофия надпочечников, идиопатический легочный фибрит, IgA нефропатия, воспалительные заболевания кишечника, ннсулин-зависимый сахарный диабет, ювенильный артрит, ювенильный сахарный диабет (тип I), синдром Ламберта-Итона, ламинит, плоский лишай, люпоидный гепатит, волчанка, лимфопения, синдром Меньера, смешанное заболевание соединительной ткани, рассеянный склероз, тяжелая миастения, пернициозная анемия, полигландулярные синдромы, пресенильная деменция, первичная агаммаглобулинемия, первичный билиарный цирроз, псориаз, псориатический артрит, синдром Рейно, невынашивание беременности, синдром Рейтера, острая ревматическая лихорадка, ревматоидный артрит, синдром Сэмптера, шистосомоз, синдром Шмидта, склеродермия, синдром Шульмана, синдром Шегрена, синдром скованного человека, симпатическая офтальмия, системная красная волчанка, синдром Такаясу, височный артериит, тиреоидит, тромбоцитопения, тиреотоксикоз, токсический эпидермальный некролиз, инсулиновая резистентность типа B, диабет I типа, язвенный колит, увеит, витилиго, макроглобулинемия Вальденстрема и гранулематоз Вегенера.
Антитела, иммуноспецифичные для антигена клетки, которая отвечает за продукцию аутоиммунных антител, могут быть получены любым способом, известным обычному специалисту в данной области, таким как, например, химический синтез или методы рекомбинантной экспрессии. Примеры аутоиммунных антител включают, но не ограничиваются этим, антинуклеарное антитело; антитела к двуспиральной нативной ДНК; антитела к односпиральной нативной ДНК, анти-кардиолипиновое антитело IgM, IgG; анти-фосфолипидное антитело IgM, IgG; анти SM антитело; анти-митохондриальное Антитело; тироидное антитело; микросомальное антитело; тироглобулиновое антитело; анти SCL-70; Анти-Jo; Анти-U1RNP; Анти-La/SSB; Анти SSA; Анти SSB; Анти-перитальноклеточное антитело; анти-гистоны; анти-RNP; C-ANCA; пара-ANCA; анти-центромер; анти-фибрилларин и анти-GBM антитело.
В другом варианте воплощения настоящего изобретения, антитело конъюгата лекарственного средства по настоящему изобретению может представлять собой антитело, которое связывается и с рецептором или с рецепторным комплексом, экспрессированным на активированном лимфоците, например, антитело, связанное с аутоиммунным заболеванием. Рецептор или рецепторный комплекс могут включать члена суперсемейства иммуноглобулиновых генов, члена суперсемейства TNF рецепторов, интегрин, интерлейкин, цитокиновый рецептор, хемокиновый рецептор, белок главного комплекса гистосовместимости, лектин или контрольный белок комплемента. Неограничивающие примеры подходящих членов суперсемейства иммуноглобулина представляют собой CD2, CD3, CD4, CD5, CD8, CD13, CD22, CD28, CD79, CD90, CD152/CTLA-4, PD-I и ICOS. Неограничивающие примеры подходящих членов суперсемейства TNF рецепторов представляют собой CD27, CD40, CD95/Fas, CD134/OX40, CD137/4-1BB, TNF-Rl, TNFR-2, RANK, TACI, BCMA, остеопротегерин, Apo2/TRAEL-Rl, TRAIL-R2, TRAIL-R3, TRABL-R4 и APO-3. Неограничивающие примеры подходящих интегринов представляют собой CDl Ia, CDlIb, CDlIc, CD18, CD29, CD41, CD49a, CD49b, CD49c, CD49d, CD49e, CD49f, CD103 и CD104. Неограничивающие примеры подходящих лектинов представляют собой лектин C-типа, S- типа и I-типа.
Антитело, которое связывается с молекулярную мишенью или представляющим интерес антигеном, например, ErbB2 антигеном, представляет собой антитело, способное связываться с таким антигеном с достаточной аффинностью, такой, что антитело является полезным для таргетирования клетки, экспрессирующей такой антиген. Когда антитело представляют собой антитело, которое связывается с ErbB2, оно обычно предпочтительно связывается с ErbB2 в противоположность другим ErbB рецепторам, и может представлять собой антитело, которые существенным образом не вступает в перекрестные реакции с другими белками, такими как EGFR, ErbB 3 или ErbB4. В таких вариантах воплощения настоящего изобретения, степень связывания антитела с этими отличными от ErbB2 белками (например, связывание клеточной поверхности с эндогенным рецептором) будет меньше 10%, как определено анализом сортировки клеток с возбужденной флуоресценцией (FACS) или анализом радиоиммунопреципитации (RIA). Иногда, анти-ErbB2 антитело существенным образом не вступает в перекрестные реакции с белком крыс neu, например, как описано в Schecter et al., Nature 312:513 (1984) и Drebin et al., Nature 312:545-548 (1984).
В другом варианте воплощения настоящего изобретения, антитело конъюгата лекарственного средства или мишень по настоящему изобретению могут быть выбраны из антитела или мишени в представленной ниже таблице. Такие антитела являются иммуноспецифическими для антигена-мишени и могут быть получены коммерчески или получены любым способом, известным в данной области, таким как, например, рекомбинантные методы экспрессии.
Терапевтические моноклональные антитела
В дополнение к описанному выше, антитело конъюгата лекарственное средство-антитело по настоящему изобретению может представлять собой Vitaxin, который представляет собой гуманизированное антитело для лечения саркомы; Smart IDlO, который представляет собой гуманизированное анти-HLA-DR антитело для лечения неходжкинской лимфомы; Oncolym, который представляет собой меченое радиоактивным изотопом мышиное анти-HLA-DrlO антитело для лечения неходжкинской лимфомы; и Allomune, который представляет собой гуманизированное анти-CD2 mAb для лечения болезни Ходжкина или неходжкинской лимфомы.
Антитело конъюгата лекарственного средства по настоящему изобретению также может представлять собой любой фрагмент антитела, известный для лечения любого заболевания, предпочтительно рака. И в этом случае, такие фрагменты антитела являются иммуноспецифическими для антигена-мишени и могут быть получены коммерчески или получены любым методом, известным в данной области, таким как, например, рекомбинантные методы экспрессии. Примеры таких доступных антител включают любое из представленных в таблице ниже.
Фрагменты терапевтического моноклонального антитела
(абциксимаб)
(акритумомаб)
(ранибизумаб;
Rhu-Fab)
гуманизированное
гуманизированное
гуманизированное
CD64 (γFcR1)
(scFv)/гуманизированное
стрептавидином мышиное
антиген панкарциномы
человеческое
человеческое
Иммунолипосома
(VH-VL)2
человеческое
(VH-VL)2
человека
L19-γIFN
фибронектина
(VL-VH)2
человеческое
(scFv-CH3)2
мышиное- человеческое химерное
(минитело)
мышиное- человеческое химерное
(минитело)
(ScFv)2-Fc
мышиное- человеческое химерное
(минитело)
(VL-VH-VH-VL)
мышиное
(VL-VH-VH-VL)
неизвестного происхождения
(VL-VH-VH-VL)
неизвестного происхождения
(VH-VL- VH -VL) (мышиное)
верблюжье
(Holliger & Hudson, Nature Biotechnology, 2005, 9, 23).
В предпочтительных вариантах воплощения настоящего изобретения, антитело в конъюгатах лекарственного средства по настоящему изобретению может связываться с рецептором, кодируемым ErbB геном. Антитело может специфически связываться с ErbB рецептором, выбранным из EGFR, HER2, HER3 и HER4. Предпочтительно антитело в конъюгате лекарственного средства может специфически связываться с внеклеточным доменом HER2 рецептора и ингибировать рост опухолевых клеток, которые сверхэкспрессируют HER2 рецептор. Антитело конъюгата лекарственного средства может представлять собой моноклональное антитело, например, мышиное моноклональное антитело, химерное антитело или гуманизированное антитело. Предпочтительно гуманизированное антитело может представлять собой huMAb4D5-1, huMAb4D5-2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 или huMAb4D5-8 (Трастузумаб), особенно предпочтительно Трастузумаб. Антитело также может представлять собой фрагмент антитела, например, Fab фрагмент.
Другие предпочтительные антитела включают:
(i) анти-CD4 антитела. Антитело конъюгата лекарственного средства может представлять собой моноклональное антитело, например, мышиное моноклональное антитело, химерное антитело или гуманизированное антитело;
(ii)анти-CD5 антитела. Антитело конъюгата лекарственного средства может представлять собой моноклональное антитело, например, мышиное моноклональное антитело, химерное антитело, или гуманизированное антитело;
(iii)анти-CD13 антитела. Антитело конъюгата лекарственного средства может представлять собой моноклональное антитело, например, мышиное моноклональное антитело, химерное антитело, или гуманизированное антитело; и
(iv)анти-CD20 антитела. Антитело конъюгата лекарственного средства может представлять собой моноклональное антитело, например, мышиное моноклональное антитело, химерное антитело или гуманизированное антитело. Предпочтительно гуманизированное антитело представляет собой Ритуксимаб или фрагмент его антитела, например, Fab фрагмент.
Способы получения конъюгатов антитела с лекарственным средством
Конъюгаты антитела с лекарственным средством по настоящему изобретению могут быть получены в соответствии со способами, которые хорошо известны в данной области. Способы для конъюгирования компонентов, включающих по меньшей мере один антиген-связывающий сайт, таких как антитела, с множеством различных лекарственных средств с использованием различных способов были описаны и проиллюстрированы ранее, например, в WO-A-2004/010957, WO-A-2006/060533 и WO-A-2007/024536, содержание которых включено в настоящую заявку посредством ссылки. Эти способы включают использование линкерной группы, которая дериватизирует лекарственное средство, токсин или радионуклид таким образом, чтобы его затем можно было присоединить к компоненту, такому как антитело. Присоединение к компоненту, такому как антитело, обычно осуществляют одним из трех путей: через свободные тиольные группы в цистеинах после частичного восстановления дисульфидных групп в антителе; через свободные амино группы в лизинах в антителе; и через свободные гидроксильные группы в серинах и/или треонинах в антителе. Способ присоединения варьируется в зависимости от участка присоединения на компоненте, таком как антитело. Очистка конъюгатов антитела с лекарственным средством при помощи эксклюзионной хроматографии (SEC) также была описана [см., например, Liu et al., Proc. Natl. Acad. Set (USA), 93: 8618-8623 (1996), и Chari et al., Cancer Research, 52: 127-131 (1992)].
Как отмечалось ранее, полезные нагрузки лекарственного средства в конъюгатах лекарственного средства по настоящему изобретению представляют собой дигидропиран-2-оновые и тетрагидропиран-2-оновые производные, которые были раскрыты или которые входят в объем Международных публикаций №№ WO-A-2007/144423 и WO-A-2009/080761, содержание которых включено в настоящую заявку посредством ссылки. Эти соединения синтезируют в соответствии со способами, описанными и представленными в качестве примеров в этих международных заявках.
Как отмечалось ранее, в девятом аспекте настоящего изобретения представлен способ получения конъюгата лекарственного средства в соответствии с первым аспектом настоящего изобретения, включающий конъюгирование компонента Ab, включающего по меньшей мере один антиген-связывающий сайт, и лекарственного средства D формулы (I), (Ia) или (Ib), где Ab и D имеют значение, определенное в первом аспекте настоящего изобретения.
Один пример способа получения конъюгата лекарственного средства по настоящему изобретению включает получение конъюгатов антитела с лекарственным средством формулы (G) или (H) по настоящему изобретению, как показано ниже:

при этом указанный способ включает следующие стадии:
(i) взаимодействие лекарственного средства (D) формулы (Ia)-H:

где заместители в определениях (Ia) имеют значение, определенное выше, с соединением формулы X2-C(O)-X1, где X1 и X2 представляют собой отщепляемые группы, с получением соединения формулы (B):

и точка присоединения группы -(С=О)Х1 представляет собой свободную гидроксильную группу, присоединенную к тому же атому углерода, что и R18’;
(ii) взаимодействие соединения формулы (B), полученного на стадии (i), с диамином формулы H2N-(CH2)1-6NH2 с получением соединения формулы (C):

(iii) либо взаимодействие соединения формулы (C), полученного на стадии (ii), с соединением формулы (D’):

(D’)
с получением соединения формулы (F):

(F)
либо взаимодействие соединения (C), полученного на стадии (ii), с N-гидроксисукцинимидным эфиром 6-малеимидoгексановой кислоты с получением соединения формулы (E):

(iv) частичное восстановление одной или нескольких дисульфидных связей в антителе, которое должно быть конъюгировано, с получением восстановленного антитела Ab-SH, содержащего свободные тиольные группы:
 и
и
(v) взаимодействие частично восстановленного антитела Ab-SH, содержащего свободные тиольные группы, с соединением формулы (E) или (F), полученным на стадии (iv), с получением целевого конъюгата антитела с лекарственным средством формулы (G) или (H), соответственно:
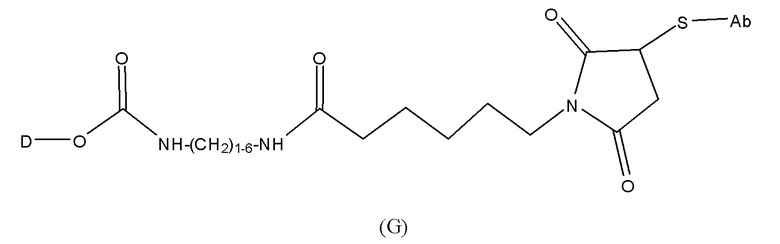

(H)
Предпочтительно соединение формулы X2-C(O)-X1, подвергаемое взаимодействию с лекарственным средством (A) на стадии (i), представляет собой 1,1’карбонилдиимидазол.
Предпочтительно диамин на стадии (ii) имеет формулу NH2-(CH2)2-4-NH2, и более предпочтительно он представляет собой пропилен-1,3-диамин.
В одном предпочтительном варианте воплощения данного способа, промежуточное соединение (C) подвергают взаимодействию с соединением формулы (D’), где R23 представляет собой -(CH2)3-NH-CO-NH2, и R22 представляет собой изопропил.
В другом предпочтительном варианте воплощения этого способа, антитело выбрано из Трастузумаба, Ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части, или оно выбрано из Трастузумаба, Ритуксимаба и анти-CD4 антитела или его иммунологически активной части и наиболее предпочтительно оно представляет собой Трастузумаб или его иммунологически активную часть; или оно выбрано из анти-CD5 антитела и анти-CD13 антитела, или его иммунологически активной части и наиболее предпочтительно оно представляет собой анти-CD13 антитело или его иммунологически активную часть. Кроме того, частичное восстановление этого моноклонального антитела осуществляют с использованием трис[2-карбоксиэтил]фосфин гидрохлорида (TCEP).
Другой пример способа получения конъюгата антитела с лекарственным средством по настоящему изобретению, включает получение конъюгатов антитела с лекарственным средством формулы (O) или (P), как показано ниже:

при этом указанный способ включает следующие стадии:
(i) либо:
взаимодействие лекарственного средства (D) формулы (Ia)-H:
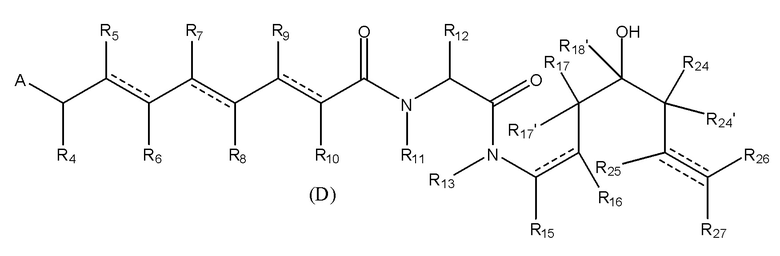
где заместители в определениях (Ia)-H имеют значение, определенное выше, с соединением формулы X2-C(O)-X1, где X1 и X2 представляют собой отщепляемые группы, с получением соединения формулы (B):
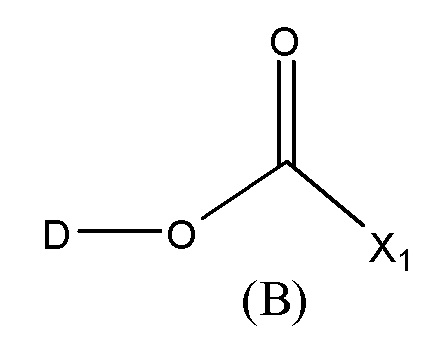
и точка присоединения группы X1(CO) представляет собой свободную гидроксильную группу, присоединенную к тому же атому углерода, как R18’, или
(b) взаимодействие указанного лекарственного средства (A) формулы (Ia)-H, определенного выше, с 4-нитро-фенилхлорформиатом с получением соединения формулы (J):
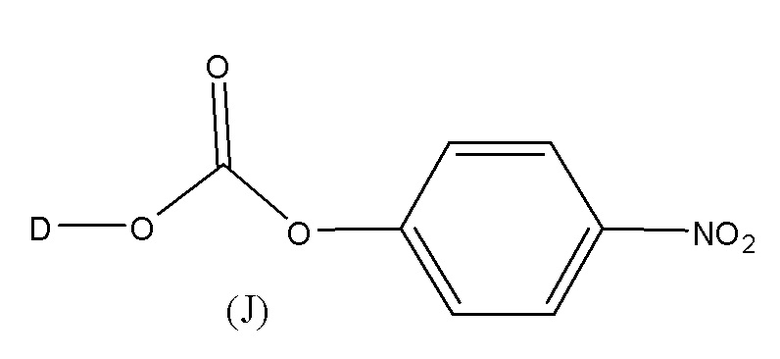
и точка присоединения группы (4-нитрофенил)-O-CO- представляет собой такую же, как для группы X1(CO) на стадии (a) выше;
(ii) либо:
(c) взаимодействие соединения формулы (B), полученного на стадии (i), с диамином формулы H2N-(CH2)1-6NH2 с получением соединения формулы (C):

и затем взаимодействие полученного соединения формулы (C) с соединением формулы Me-S-S-(CH2)1-3-CO2H с получением соединения формулы (K)

или
(d) взаимодействие соединения (J), полученного на стадии (i), с аминоалкилтио соединением формулы H2N-(CH2)1-3SH с получением соединения формулы (L):

(iii) взаимодействие соединения формулы (K) или (L), полученного на стадии (ii), с дитиотреитолом в дисульфид восстанавливающих условиях с получением соединений формулы (M) и (N), соответственно:

(iv) взаимодействие антитела, которое должно быть конъюгировано, с сукцининимидил-4-(N-малеимидoметил)циклогексан-1-карбоксилатом для дериватизирования указанного антитела на одной или нескольких лизиновых группах сукцининимидил-4-(N-малеимидoметил)циклогексан-1-карбонильной группой:
(v) взаимодействие дериватизированного антитела, полученного на стадии (iv), либо с соединением формулы (M), либо с соединением формулы (N), полученным на стадии (iii), с получением целевого конъюгата антитела с лекарственным средством формулы (O) или (P):
Что касается описанного выше способа, соединение формулы X2-C(O)-X1 предпочтительно представляет собой 1,1’-карбонилдиимидазол. Подобным образом, диаминовое соединение формулы (B) предпочтительно представляет собой NH2-(CH2)2-4-NH2, и более предпочтительно пропилен-1,3-диамин.
В одном предпочтительном варианте воплощения настоящего изобретения, соединение, подвергаемое взаимодействию с соединением формулы (C) для получения Соединения формулы (K), представляет собой 3-(метилдисульфанил)пропановую кислоту.
В другом предпочтительном варианте воплощения настоящего изобретения, аминоалкилтио соединение, подвергаемое взаимодействию взаимодействию с соединением формулы (J) для получения соединения формулы (L), представляет собой 3-аминопропан-1-тиол.
Где присоединение к линкерной группе лекарственного средства осуществляется через свободные тиольные группы в цистеинах после частичного восстановления дисульфидных групп в компоненте, включающем по меньшей мере один антиген-связывающий сайт, таком как моноклональное антитело, частичное восстановление обычно осуществляют сначала разбавлением до подходящей концентрации и буферизацией раствора перед частичным восстановлением дисульфидных связей путем добавления подходящего восстановителя, такого как трис[2-карбоксиэтил]фосфингидрохлорид (TCEP) или дитиотреитол (DTT). При выборе подходящих соотношений компонента, который подлежит восстановлению, такого как моноклональное антитело, и восстановителя, условий реакции и времени восстановления, можно получить нужное отношение свободного тиола к компоненту, например, четыре свободных тиольных группы на моноклональное антитело.
Частично восстановленный компонент, такой как частично восстановленное моноклональное антитело, содержащее свободные тиольные группы, полученное, как описано выше, затем подвергают взаимодействию с лекарственное средство-линкер соединениями по настоящему изобретению (где группа L в таком соединении представляет собой малеимидную группу, которая является свободной для взаимодействия с тиольными группами). Полученные конъюгаты антитела с лекарственным средством очищают с использованием любых подходящих способов, известных в данной области, например, при помощи эксклюзионной хроматографии (SEC) [см., например, Liu et al., Proc. Natl. Acad. Set (USA), 93: 8618-8623 (1996), и Chari et al., Cancer Research, 52: 127-131 (1992)].
В одном предпочтительном варианте воплощения настоящего изобретения моноклональное антитело представляет собой Трастузумаб или анти-CD13 антитело или его иммунологически активную часть; предпочтительно Трастузумаб или его иммунологически активную часть, или предпочтительно анти-CD13 антитело или его иммунологически активную часть.
В альтернативном варианте воплощения настоящего изобретения, лизины в компоненте, включающем по меньшей мере один антиген-связывающий сайт, таком как моноклональное антитело, сначала можно подвергнуть взаимодействию с сукцинимидил-4-(N-малеимидoметил)циклогексан-1-карбоксилатом. Свободная аминовая группа на антителе может взаимодействовать с N-гидроксисукцинимидным сложным эфиром с получением малеимид-активированного антитела:

Малеимид-активированное антитело затем подвергают взаимодействию с соединением формулы D-X-(AA)w-Н, содержащим реакционноспособную тиольную группу.
Два конкретных примера способов получения конъюгатов антитела с лекарственным средством формулы [D-(X)b-(AA)w-(L)-]n-Ab по настоящему изобретению путем конъюгирования через свободные тиольные группы в цистеинах после частичного восстановления дисульфидных групп в антителе и через свободные амино группы в лизинах в антителе с последующей активацией с малеимидной группой, показаны на Фиг. 1 и 2.
Композиции, содержащие конъюгат антитела с лекарственным средством по настоящему изобретению, и их применения
В пятом аспекте настоящего изобретения представлена фармацевтическая композиция, включающая конъюгат лекарственного средства в соответствии с настоящим изобретением и фармацевтически приемлемый носитель. Примеры формы введения конъюгата лекарственного средства, имеющего общую формулу [D-(X)b-(AA)w-(L)-]n-Ab, по настоящему изобретению включают, без ограничения, пероральную, местную, парентеральную, сублингвальную, ректальную, вагинальную, глазную и интраназальную. Парентеральное введение включает подкожные инъекции, внутривенные, внутримышечные, интрастернальные инъекции или инфузии. Предпочтительно композиции вводят парентерально. Фармацевтические композиции по настоящему изобретению могут быть сформулированы таким образом, чтобы конъюгат лекарственного средства по настоящему изобретению был биодоступным при введении композиции животному, предпочтительно человеку. Композиции могут принимать форму одной или нескольких единиц дозирования, при этом, например, таблетка может представлять собой одну единицу дозирования, и контейнер для конъюгата антитела с лекарственным средством по настоящему изобретению в форме аэрозоля может содержать множество единиц дозирования.
Фармацевтически приемлемый носитель или наполнитель может представлять собой вещество в виде мелких частиц, чтобы получать композиции, например, в форме таблеток или порошка. Носитель(носители) могут быть жидкими, при этом композиции представляют собой, например, пероральный сироп или жидкость для инъекций. Кроме того, носитель(носители) могут быть газообразными, чтобы можно было получить аэрозольную композицию, полезную, например, для введения путем ингаляции. Термин "носитель" относится к разбавителю, адъюванту или эксципиенту, с которым вводят конъюгат антитела с лекарственным средством по настоящему изобретению. Такие фармацевтические носители могут представлять собой жидкости, такие как вода и масла, включая масла нефтяного, животного, растительного или синтетического происхождения, такие как арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.п. Носители могут представлять собой физиологический раствор, аравийскую камедь, желатин, крахмальную пасту, тальк, кератин, коллоидный диоксид кремния, мочевину и т.п. Кроме того, можно использовать вспомогательные вещества, стабилизаторы, загустители, смазывающие вещества и красители. В одном варианте воплощения настоящего изобретения, при введении животному, конъюгаты антитела с лекарственным средством по настоящему изобретению или композиции и фармацевтически приемлемые носители являются стерильными. Вода является предпочтительным носителем, когда конъюгаты антитела с лекарственным средством по настоящему изобретению вводят внутривенно. Физиологические растворы и водные растворы декстрозы и глицерин также можно использовать в качестве жидких носителей, особенно для растворов для инъекций. Подходящие фармацевтические носители также включают эксципиенты, такие как крахмал, глюкоза, лактоза, сахароза, желатин, солод, рис, мука, мел, силикагель, стеарат натрия, глицеринмоностеарат, тальк, хлорид натрия, сухое снятое молоко, глицерин, пропиленгликоль, вода, этанол и т.п. Композиции по настоящему изобретению, если желательно, также могут содержать незначительные количества смачивающих веществ или эмульгаторов, или буферных веществ для регулирования pH.
Когда она предназначена для перорального введения, композиция предпочтительно представлена в твердой или жидкой форме, где полутвердые, полужидкие, суспендированные и гелевые формы включены формы, рассматриваемые в настоящей заявке либо как твердые, либо как жидкие.
Как твердая композиция для перорального введения, композиция может быть сформулирована в виде порошка, гранул, прессованных таблеток, пилюль, капсул, жевательных резинок, пастилок или подобной формы. Такая твердая композиция типично содержит один или несколько инертных разбавителей. Кроме того, может присутствовать одно или несколько из следующих веществ: связующие, такие как карбоксиметилцеллюлоза, этилцеллюлоза, микрокристаллическая целлюлоза или желатин; эксципиенты, такие как крахмал, лактоза или декстрины, разрыхлители, такие как альгиновая кислота, альгинат натрия, кукурузный крахмал и т.п.; смазывающие вещества, такие как стеарат магния; вещества, обеспечивающие скольжение, такие как коллоидный диоксид кремния; подсластители, такие как сахароза или сахарин; отдушка, такая как перечная мята, метилсалицилат или апельсиновая отдушка; и краситель.
Когда композиция представлена в форме капсулы (например, желатиновой капсулы), она содержит, в дополнение к веществам указанного выше типа, жидкий носитель, такой как полиэтиленгликоль, циклодекстрин или жирное масло.
Композиция может быть в форме жидкости, например, эликсир, сироп, раствор, эмульсия или суспензия. Жидкость может быть полезна для перорального введения или для доставки путем инъекции. Когда она предназначена для перорального введения, композиция может включать один или несколько из подсластителей, консервантов, красителей/окрашивающих веществ и отдушек. Композиция для введения путем инъекции также может включать одно или несколько из веществ, таких как поверхностно-активное вещество, консервант, смачивающее вещество, диспергирующее вещество, суспендирующее вещество, буфер, стабилизатор и изотонический агент.
Предпочтительный путь введения представляет собой парентеральное введение, включая, но не ограничиваясь этим, внутрикожное, внутримышечное, интраперитонеальное, внутривенное, подкожное, интраназальное, эпидуральное, интраназальное, интрацеребральное, интравентрикулярное, интратекальное, интравагинальное или чрескожное. Предпочтительный путь введения определяет лечащий врач, и это будет зависеть, частью, от зоны медицинского состояния (такой как зона рака). В более предпочтительном варианте воплощения, конъюгаты антитела с лекарственным средством по настоящему изобретению вводят внутривенно.
Жидкие композиции по настоящему изобретению, независимо от того находятся ли они в форме растворов, суспензий или в другой подобной форме, также могут включать одно или несколько из следующих веществ: стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно физиологический солевой раствор, раствор Рингера, изотонический раствор хлорида натрия, нелетучие масла, такие как синтетические моно- или диглицериды, полиэтиленгликоли, глицерин или другие растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабен; и агенты регулирования тоничности, такие как хлорид натрия или декстроза. Парентеральная композиция может быть заключена в ампулу, одноразовый шприц или многодозовый флакон из стекла, пластика или другого материала. Физиологический раствор является предпочтительным адъювантом.
Количество конъюгата лекарственного средства по настоящему изобретению, которое является эффективным для лечения определенного расстройства или состояния, будет зависеть от природы расстройства или состояния, и его можно определить стандартными клиническими методами. Кроме того, in vitro или in vivo анализы можно использовать для определения оптимальных диапазонов доз. Точная доза для использования в композициях также будет зависеть от пути введения и тяжести заболевания или расстройства, и ее следует определять в соответствии с мнением лечащего врача и обстоятельств каждого пациента.
Композиции включают эффективное количество конъюгата лекарственного средства по настоящему изобретению, так, чтобы можно было получить подходящую дозу. Подходящая доза соединений будет варьироваться в соответствии с конкретной композицией, способом применения и конкретным участком ее применения, хозяином и заболеванием, подлежащим лечению, например, рак, и, в случае такого заболевания, конкретным типом опухоли. Другие факторы, такие как возраст, масса тела, пол, режим питания, время введения, скорость экскреции, состояние хозяина, комбинации лекарственных средств, чувствительности реакций и тяжесть заболевания, следует принимать во внимание. Введение можно осуществлять непрерывно или периодически в пределах максимальной переносимой дозы.
Типично, это количество составляет по меньшей мере около 0,01% конъюгата лекарственного средства по настоящему изобретению в расчете на массу композиции. Когда композиция предназначена для перорального введения, это количество может варьироваться в пределах от около 0,1% до около 80% в расчете на массу композиции. Предпочтительные пероральные композиции могут включать от около 4% до около 50% конъюгата лекарственного средства по настоящему изобретению в расчете на массу композиции.
Предпочтительные композиции по настоящему изобретению получают таким образом, чтобы парентеральная единица дозирования содержала от около 0,01% до около 2% в расчете на массу конъюгата лекарственного средства по настоящему изобретению.
Для внутривенного введения, композиция может включать типично от около 0,1 мг/кг до около 250 мг/кг массы тела животного, предпочтительно от около 0,1 мг/кг до около 20 мг/кг массы тела животного, и более предпочтительно от около 1 мг/кг до около 10 мг/кг массы тела животного.
Конъюгат лекарственного средства по настоящему изобретению или композиции можно вводить любым удобным путем, например, путем инфузии или болюсной инъекции, путем абсорбции через эпителиальные или кожно-слизистые выстилки.
В конкретных вариантах воплощения настоящего изобретения, может быть желательным введение одного или нескольких конъюгатов лекарственного средства по настоящему изобретению или композиции локально в область, нуждающуюся в лечении. В одном варианте воплощения настоящего изобретения, введение можно осуществлять путем прямой инъекции в область (или формирующую область) рака, опухоли или опухолевой или пред-опухолевой ткани. В другом варианте воплощения настоящего изобретения, введение можно осуществлять путем прямой инъекции в область (или формирующую область) проявления аутоиммунного заболевания.
Также можно использовать внутрилегочное введение, например, с использованием ингалятора или небулайзера и композиции с распыляющим агентом, или через перфузию во фторуглеродном или синтетическом пульмональном поверхностно-активном веществе. В конкретных вариантах воплощения, конъюгат антитела с лекарственным средством по настоящему изобретению или композиции могут быть сформулированы в виде суппозитория, с использованием традиционных связующих и носителей, таких как триглицериды.
Композиции по настоящему изобретению могут принимать форму растворов, суспензий, эмульсии, таблеток, пилюль, гранул, капсул, капсул, содержащих жидкости, порошков, композиций замедленного высвобождения, суппозиториев, эмульсий, аэрозолей, спреев, суспензий или любую другую форму, подходящую для применения. Другие примеры подходящих фармацевтических носителей описаны в "Remington's Pharmaceutical Sciences" by E. W. Martin.
Фармацевтические композиции можно получить с использованием методов, хорошо известных в фармацевтике. Например, композицию, предназначенную для введения путем инъекции, можно получить путем объединения конъюгата лекарственного средства по настоящему изобретению с водой для получения раствора. Можно добавить поверхностно-активное вещество, способствующее образованию гомогенного раствора или суспензии.
Было обнаружено, что конъюгаты лекарственного средства и композиции по настоящему изобретению являются особенно эффективными для лечения рака.
Таким образом, как описано выше, шестой аспект настоящего изобретения обеспечивает способ лечения пациента, нуждающегося в этом, особенно человека, страдающего раком, который включает введение страдающему заболеванием субъекту терапевтически эффективного количества конъюгата лекарственного средства или композиции по настоящему изобретению. Четвертый аспект настоящего изобретения обеспечивает конъюгат лекарственного средства в соответствии с первым аспектом настоящего изобретения для применения в лечении рака, и более предпочтительно рака, выбранного из рака легких, колоректального рака, рака молочной железы, рака поджелудочной железы, рака почки, лейкоза, множественной миеломы и лимфомы.
Конъюгаты лекарственных средств и композиции по настоящему изобретению являются полезными для ингибирования размножения опухолевой клетки или раковой клетки, или для лечения рака у животного. Конъюгаты лекарственного средства и композиции по настоящему изобретению можно использовать соответственно в различных условиях для лечения рака животных. Конъюгаты по настоящему изобретению, включающие Лекарственное средство-Линкер-Компонент, содержащий по меньшей мере один антиген-связывающий сайт, можно использовать для доставки Лекарственного средства или Лекарственной группы к опухолевой клетке или раковой клетке. Не будучи связанным теорией, в одном варианте воплощения настоящего изобретения Компонент, содержащий по меньшей мере один антиген-связывающий сайт, конъюгата лекарственного средства по настоящему изобретению связывается с или вступает в ассоциацию с антигеном, ассоциациированным с раковой клеткой или опухолевой клеткой, и конъюгат лекарственного средства по настоящему изобретению может захватываться внутрь опухолевой клетки или раковой клетки через рецептор-опосредованный эндоцитоз. Антиген может быть связан с опухолевой клеткой или раковой клеткой или может представлять собой белок внеклеточного матрикса, ассоциированный с опухолевой клеткой или раковой клеткой. Попадая внутрь клетки, одна или несколько специфических последовательностей в Линкерной группе гидролитически расщепляются одной или несколькими ассоциациированными с опухолевой клеткой или раковой клеткой протеазами или гидролазами, приводя к высвобождению Лекарственного средства или Соединения Лекарственное средство-Линкер. Высвобожденное Лекарственное средство или Лекарственное средство-Линкер Соединение затем является свободным для миграции в клетке и индукции цитотоксических активностей. В альтернативном варианте воплощения, Лекарственное средство или группа Лекарственного средства отщепляется от конъюгата лекарственного средства по настоящему изобретению вне опухолевой клетки или раковой клетки, и Лекарственное средство или Соединение Лекарственное средство-Линкер затем проникает в клетку.
В одном варианте воплощения настоящего изобретения, Компонент, включающий по меньшей мере один антиген-связывающий сайт, связывается с опухолевой клеткой или раковой клеткой. В другом варианте воплощения настоящего изобретения, Компонент, включающий по меньшей мере один антиген-связывающий сайт, связывается с антигеном опухолевой клетки или раковой клетки, который находится на поверхности опухолевой клетки или раковой клетки. Еще в одном варианте воплощения настоящего изобретения, Компонент, включающий по меньшей мере один антиген-связывающий сайт, связывается с антигеном опухолевой клетки или раковой клетки, который представляет собой белок внеклеточного матрикса, ассоциированный с опухолевой клетки или раковой клетки.
Специфичность Компонента, включающего по меньшей мере один антиген-связывающий сайт, в отношении определенной опухолевой клетки или раковой клетки может быть важной для определения тех типов опухолей или рака, которые наиболее эффективно лечатся. Например, конъюгаты лекарственного средства по настоящему изобретению содержащие группу Трастузумаба (Heparin®), могут быть полезными для лечения антиген-положительных карцином, включая лейкозы, рак легких, рак толстой кишки, лимфомы (например, болезнь Ходжкина, неходжкинская лимфома), солидные опухоли, такие как саркома и карциномы, множественную миелому, рак почки и меланому. Рак предпочтительно может представлять собой рак легких, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак почки, лейкоз, множественную миелому, лимфому или рак яичника. Например, конъюгаты лекарственного средства по настоящему изобретению содержащие группу Ритуксимаба, могут быть полезными для лечения CD20-экспрессирующих опухолей, таких как гематологические типы рака, включая лейкозы и лимфомы. Например, конъюгаты лекарственного средства по настоящему изобретению содержащие группу анти-CD4 антитела, могут быть полезными для лечения CD4-экспрессирующих опухолей, таких как гематологические типы рака, включая лимфомы. Например, конъюгаты лекарственного средства по настоящему изобретению содержащие группу анти-CD5 антитела, могут быть полезными для лечения CD5-экспрессирующих опухолей, таких как гематологические типы рака, включая лейкозы и лимфомы, например, конъюгаты лекарственного средства по настоящему изобретению содержащие группу анти-CD13 антитела, могут быть полезными для лечения CD13-экспрессирующих опухолей, таких как гематологические типы рака, включая лейкозы и лимфомы.
Другие конкретные типы рака, которые можно лечить при помощи конъюгатов лекарственного средства по настоящему изобретению, включают, но не ограничиваются этим: гематологические типы рака, включая все формы лейкоза; лимфомы, такие как болезнь Ходжкина, неходжкинская лимфома, и множественная миелома.
В частности, конъюгаты лекарственного средства и композиции по настоящему изобретению демонстрируют превосходную активность для лечения рака, такого как рак легкого, колоректальный рака, рак молочной железы, рак поджелудочной железы, рак почки, лейкоз, множественная миелома, лимфома и рак яичника.
Конъюгаты лекарственного средства и композиции по настоящему изобретению обеспечивают конъюгация-специфическое таргетирование опухоли или рака, таким образом, снижая общую токсичность этих конъюгатов. Линкерные группы стабилизируют конъюгаты лекарственное средство-антитело в крови, при этом расщепляются опухоль-специфическими протеазами и гидролазами внутри клетки, высвобождая Лекарственное средство.
Конъюгаты лекарственных средств и композиции по настоящему изобретению можно вводить животному, которое также подвергалось хирургической операции для лечения рака. В одном варианте воплощения настоящего изобретения, дополнительный способ лечения представляет собой лучевую терапию.
В специальном варианте осуществления настоящего изобретения, конъюгат лекарственного средства или композицию по настоящему изобретению вводят одновременно с химиотерапевтическим средством или с лучевой терапией. В другом специальном варианте осуществления настоящего изобретения, химиотерапевтическое средство или лучевую терапию вводят до или после введения конъюгата лекарственного средства или композиции по настоящему изобретению, предпочтительно по меньшей мере с промежутком времени один час, пять часов, 12 часов, один день, одну неделю, один месяц, более предпочтительно несколько месяцев (например, до трех месяцев), до или после введения конъюгата антитела с лекарственным средством или композиции по настоящему изобретению.
Химиотерапевтическое средство можно вводить в течение ряда сеансов лечения, можно вводить любое одно или комбинацию химиотерапевтических средств, известных в данной области.
Что касается облучения, можно использовать любой протокол лучевой терапии, в зависимости от типа рака, подлежащего лечению. Например, но не в качестве ограничения, можно применять рентгеновское облучение; в частности, можно использовать высокомощное мегавольтное облучение (облучение мощностью больше чем 1 МэВ) для глубоких опухолей, и электроннолучевое и ортовольтажное рентгеновское излучение можно использовать для рака кожи. Также можно вводить испускающие гамма-лучи радиоизотопы, такие как радиоактивные изотопы радия, кобальта и других элементов.
В восьмом аспекте настоящего изобретения представлен набор, включающий терапевтически эффективное количество конъюгата лекарственного средства в соответствии с первым аспектом настоящего изобретения и фармацевтически приемлемый носитель.
В одном варианте воплощения настоящего изобретения, набор в соответствии с этим аспектом предназначен для использования в лечении рака, и более предпочтительно рака, выбранного из рака легких, колоректального рака, рака молочной железы, рака поджелудочной железы, рака почки, лейкоза, множественной миеломы, лимфомы и рака яичника.
Краткое описание чертежей
Изобретение схематически проиллюстрировано, посредством примера, на прилагаемых чертежах, где
Фиг. 1 представляет собой схематическую иллюстрацию одного способа в соответствии с настоящим изобретением, где конъюгация с антителом осуществляется через свободные тиольные группы;
Фиг. 2 представляет собой схематическую иллюстрацию другого способа в соответствии с настоящим изобретением, где конъюгация с антителом осуществляется через свободные лизиновые группы;
Фиг. 3 представляет репрезентативные кривые доза-ответ для ADC1 против различных раковых клеточных линий;
Фиг. 4 представляет гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC1 при 10 или 1 мкг/мл;
Фиг. 5 представляет репрезентативные кривые доза-ответ для ADC2 против различных раковых клеточных линий;
Фиг. 6 представляет гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC2 при 10 или 1 мкг/мл;
Фиг. 7 представляет репрезентативные кривые доза-ответ для ADC3 против различных раковых клеточных линий;
Фиг. 8 представляет гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC3 при 10 или 1 мкг/мл;
Фиг. 9 представляет репрезентативные кривые доза-ответ для ADC4 против различных раковых клеточных линий;
Фиг. 10 представляет гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC4 при 10 или 1 мкг/мл;
Фиг. 11 представляет репрезентативные кривые доза-ответ для ADC5 против различных раковых клеточных линий;
Фиг. 12 представляет гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC5 при 10 или 1 мкг/мл;
Фиг. 13 представляет репрезентативные кривые доза-ответ для ADC6 против различных раковых клеточных линий;
Фиг. 14 представляет гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC6 при 10 или 1 мкг/мл;
Фиг. 15 представляет репрезентативные кривые доза-ответ для ADC7 против различных раковых клеточных линий;
Фиг. 16 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC7 при 50 или 1 мкг/мл;
Фиг. 17 представляет репрезентативные кривые доза-ответ для ADC8 против различных раковых клеточных линий;
Фиг. 18 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC8 при 50 или 10 мкг/мл;
Фиг. 19 представляет репрезентативные кривые доза-ответ для ADC9 против различных раковых клеточных линий;
Фиг. 20 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC9 при 50 или 0,1 мкг/мл;
Фиг. 21 представляет репрезентативные кривые доза-ответ для ADC10 против различных раковых клеточных линий;
Фиг. 22 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC10 при 50 или 1 мкг/мл;
Фиг. 23 представляет репрезентативные кривые доза-ответ для ADC11 против различных раковых клеточных линий;
Фиг. 24 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC11 при 50 или 1 мкг/мл;
Фиг. 25 представляет репрезентативные кривые доза-ответ для ADC12 против различных раковых клеточных линий;
Фиг. 26 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC12 при 1 или 0,1 мкг/мл;
Фиг. 27 представляет репрезентативные кривые доза-ответ для ADC13 против различных раковых клеточных линий;
Фиг. 28 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC13 при 1 или 0,1 мкг/мл;
Фиг. 29 представляет репрезентативные кривые доза-ответ для ADC14 против различных раковых клеточных линий;
Фиг. 30 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC14 при 1 мкг/мл;
Фиг. 31 представляет репрезентативные кривые доза-ответ для ADC16 против различных раковых клеточных линий;
Фиг. 32 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC16 при 1 или 0,1 мкг/мл;
Фиг. 33 представляет репрезентативные кривые доза-ответ для ADC17 против различных раковых клеточных линий;
Фиг. 34 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC17 при 1 и 0,1 мкг/мл;
Фиг. 35 представляет репрезентативные кривые доза-ответ для ADC14 против двух Raji клеточных клонов;
Фиг. 36 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC14 при 10 мкг/мл;
Фиг. 37 представляет репрезентативные кривые доза-ответ для ADC15 против двух Raji клеточных клонов; и
Фиг. 38 представляет гистограммы, показывающие процент клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC15 при 10 мкг/мл;
Примеры
Настоящее изобретение далее проиллюстрировано при помощи следующих, неограничивающих примеров. В примерах использованы следующие аббревиатуры:
CDI - 1,1’-карбонилдиимидазол
DIPEA - диизопропилэтиламин
Hex - гексан
EtOAc - этилацетат
DCM - дихлорметан
NMP - N-метил-2-пирролидон
DMF - диметилформамид
EDC - N-(3-диметиламинопропил)-N’-этилкарбодиимид гидрохлорид
EDTA -этилендиаминтетрауксусная кислота
MeOH - метанол
DTT - дитиотреитол
Py - пиридин
THF - тетрагидрофуран
TCEP - Трис[2-карбоксиэтил]фосфингидрохлорид
MC - 6-малеимидoкапроил
Fmoc - 9-флуоренилметоксикарбонил
Cit - цитруллин
Val -валин
DMSO - диметилсульфоксид
Trt - трифенилметил
HOBt - 1-гидроксибензотриазол
DIPCDI, N,N’-диизопропилкарбодиимид
TFA - трифторуксусная кислота
PABOH - 4-аминобензиловый спирт
bis-PNP, бис(4-нитрофенил) карбонат
NAC - N-Ацетилцистеин
SEC - эксклюзионная хроматография
ВЭЖХ - высокоэффективная жидкостная хроматография
ADC - конъюгат антитела с лекарственным средством
ATCC - Американская коллекция типовых культур
DMEM - Модифицированная по способу Дульбекко среда Игла
RPMI - питательная среда Rosmell Park Memorial Institute
ITS - дополняющая среда инсулин-трансферрин-натрийселенит
FCS - Фетальная телячья сыворотка
SRB - сульфородамин В
PBS - фосфатно-солевой буферный раствор
DR - доза-ответ
UV - ультрафиолет
SMCC - Сукцинимидил-4-(N-малеимидoметил)циклогексан-1-карбоксилат
LAR - отношение линкера к антителу
Пример получения
Получение Соединения 9: MC-Val-Cit-PABC-PNP
Схема реакции

(a) Получение Соединения 10: MC-Val-Cit-OH
Соединение 10
Cl-TrtCl-смолу (20 г, 1,49 ммоль/г) (Iris Biotech, Ref.: BR-1065, 2-хлортритилхлоридную смолу (200-400 меш, 1% DVB, 1,0-1,6 ммоль/г), CAS 42074-68-0) помещали на фильтровальную пластину. К смоле добавляли 100 мл DCM и смесь перемешивали в течение 1 часа. Растворитель отгоняли при помощи фильтрации в вакууме. Добавляли раствор Fmoc-Cit-OH (11,83 г, 29,78 ммоль) и DIPEA (17,15 мл, 98,45 ммоль) в DCM (80 мл) и смесь перемешивали в течение 10 минут. После этого добавляли DIPEA (34,82 ммоль, 199,98 ммоль) и смесь перемешивали в течение 1 часа. Реакцию останавливали путем добавления MeOH (30 мл) после перемешивания в течение 15 минут. Полученную в результате Fmoc-Cit-O-TrtCl-смолу подвергали следующим промывкам/обработкам: DCM (5×50 мл × 0,5 мин.), DMF (5×50 мл × 0,5 мин.), пиперидин:DMF (1:4, 1×1 мин., 2×10 мин.), DMF (5×50 мл × 0,5 мин.), DCM (5×50 мл × 0,5 мин.), конечная пиперидиновая промывка привела к получению NH2-Cit-O-TrtCl-смолы. Загрузка была рассчитана: 1,15 ммоль/г.
Полученную выше NH2-Cit-O-TrtCl-смолу промывали при помощи DMF (5×50 мл × 0,5 мин.) и добавляли раствор Fmoc-Val-OH (31,22 г, 91,98 ммоль), HOBt (11,23 г, 91,98 ммоль) в DMF (100 мл) к NH2-Cit-O-TrtCl-смоле, перемешивали и добавляли DIPCDI (14,24 мл, 91,98 ммоль) и смесь перемешивали в течение 1,5 часов. Реакцию останавливали путем промывания при помощи DMF (5×50 мл × 0,5 мин.). Полученную таким образом Fmoc-Val-Cit-O-TrtCl-смолу обрабатывали смесью пиперидин:DMF (1:4, 1×1 мин., 2 × 10 мин.) и промывали при помощи DMF (5×50 мл × 0,5 мин.), конечная пиперидиновая промывка привела к получению NH2-Val-Cit-O-TrtCl-смолы.
Раствор 6-малеимидoкапроновой кислоты (MC-OH) (9,7 г, 45,92 ммоль), HOBt (6,21 г, 45,92 ммоль) в DMF (100 мл) добавляли к полученной выше NH2-Val-Cit-O-TrtCl-смоле, перемешивали и добавляли DIPCDI (7,12 мл, 45,92 ммоль) и смесь перемешивали в течение 1,5 часов. Реакцию останавливали путем промывки при помощи DMF (5×50 мл × 0,5 мин.) и DCM (5×50 мл × 0,5 мин.).
Пептид отщепляли от смолы путем обработки смесью TFA:DCM (1:99, 5×100 мл). Смолу промывали при помощи DCM (7×50 мл × 0,5 мин.). Объединенные фильтраты упаривали досуха при пониженном давлении и полученное твердое вещество растирали в порошок с Et2O и фильтровали с получением Соединения 10 (7,60 г, 71%) в виде белого твердого вещества.
1H ЯМР (500 МГц, DMSO-d6): δ 12,47 (с, 1H), 8,13 (д, J=7,3 Гц, 1H), 7,74 (д, J=9,0 Гц, 1H), 6,99 (с, 2H), 5,93 (с, 1H), 5,35 (с, 2H), 4,20 (дд, J=9,0, 6,8 Гц, 1H), 4,15-4,07 (м, 1H), 3,36 (т, J=7,0 Гц, 2H), 3,00-2,88 (м, 2H), 2,21-2,12 (м, 1H), 2,11-2,03 (м, 1H), 1,98-1,86 (м, 1H), 1,74-1,62 (м, 1H), 1,61-1,50 (м, 1H), 1,50-1,31 (м, 6H), 1,21-1,11 (м, 2H), 0,84 (д, J=6,8 Гц, 3H), 0,80 (д, J=6,8 Гц, 3H).
ESI-MS m/z: Рассчитано для C21H33N5O7: 467,2. Найдено: 468,3 (M+H)+.
(b) Получение Соединения 11: MC-Val-Cit-PABOH
Соединение 11
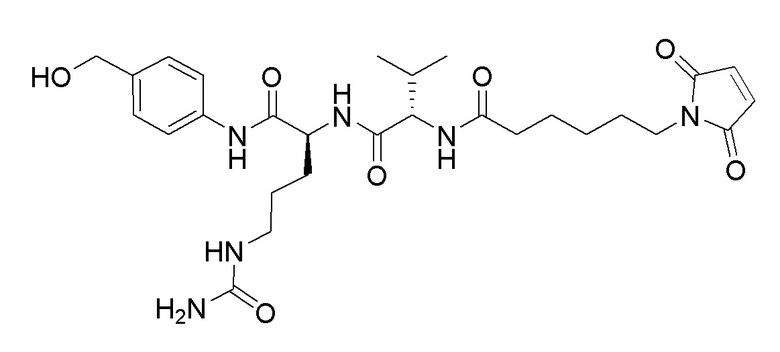
К раствору Соединения 10 (1,6 г, 3,42 ммоль) и 4-аминобензилового спирта (PABOH) (0,84 г, 6,84 ммоль) в DCM (60 мл) добавляли раствор HOBt (0,92 г, 6,84 ммоль) в DMF (5 мл). Добавляли DIPCDI (1,05 мл, 6,84 ммоль), реакционную смесь перемешивали в течение 2 часов при 23°C, добавляли Et2O (150 мл) и полученное твердое вещество фильтровали в фильтровальной пластине под вакуумом с получением Соединения 11 (1,31 г, 67%).
1H ЯМР (500 МГц, DMSO-d6): δ 9,88 (с, 1H), 8,03 (д, J=7,6 Гц, 1H), 7,77 (дд, J=12,2, 8,5 Гц, 1H), 7,53 (д, J=8,2 Гц, 2H), 7,21 (д, J=8,2 Гц, 2H), 6,99 (с, 3H), 6,01-5,92 (м, 1H), 5,39 (с, 2H), 5,07 (с, 1H), 4,41 (с, 2H), 4,39-4,31 (м, 1H), 4,23-4,12 (м, 1H), 3,36 (т, J=7,0 Гц, 2H), 3,06-2,97 (м, 1H), 2,96-2,90 (м, 1H), 2,22-2,03 (м, 2H), 2,01-1,88 (м, 1H), 1,76-1,62 (м, 1H), 1,63-1,28 (м, 6H), 1,25-1,11 (м, 2H), 0,84 (д, J=6,9 Гц, 3H), 0,81 (д, J=6,8 Гц, 3H).
ESI-MS m/z: Рассчитано для C28H40N6O7: 572,3. Найдено: 573,3 (M+H)+.
(c) Получение Соединения 9: MC-Val-Cit-PAB-PNP
Соединение 9
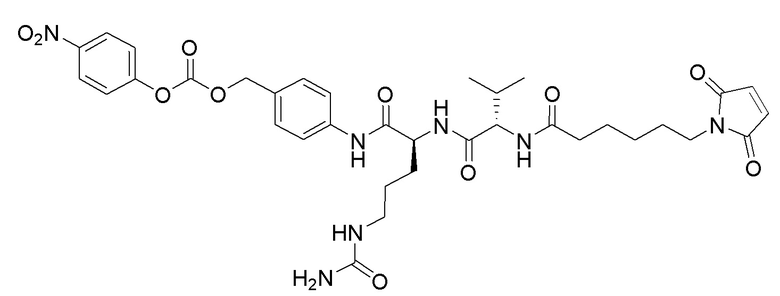
К раствору Соединения 11 (500 мг, 0,87 ммоль) и бис(4-нитрофенил)карбоната (bis-PNP ) (2,64 г, 8,72 ммоль) в смеси DCM:DMF (8:2, 25 мл) добавляли DIPEA (0,45 мл, 2,61 ммоль). Реакционную смесь перемешивали в течение 20 часов при 23°C и вносили в колонку с силикагелем (DCM:CH3OH, от 50:1 до 10:1) с получением чистого целевого Соединения 9 (364 мг, 57%).
Rf=0,40 (CH2Cl2:CH3OH, 9:1).
1H ЯМР (400 МГц, CDCl3/CD3OD): δ 9,45 (с, 1H), 8,23 (д, J=8,3 Гц, 2H), 7,59 (д, J=8,5 Гц, 2H), 7,35 (д, J=8,3 Гц, 2H), 7,34 (д, J=8,5 Гц, 2H), 6,65 (с, 2H), 5,20 (с, 2H), 4,56 (дт, J=10,5, 5,4 Гц, 1H), 4,15 (д, J=7,2 Гц, 1H), 3,46 (дд, J=8,0, 6,4 Гц, 2H), 3,16-2,89 (м, 2H), 2,21 (дд, J=8,3, 6,6 Гц, 2H), 2,06-1,97 (м, 1H), 1,90-1,83 (м, 1H), 1,73-1,46 (м, 7H), 1,34-1,20 (м, 2H), 0,91 (д, J=6,7 Гц, 3H), 0,90 (д, J=6,7 Гц, 3H).
13C ЯМР (125 МГц, CDCl3/CD3OD) δ 174,4, 172,4, 171,1, 170,6, 160,5, 155,5, 152,5, 145,3, 138,7, 134,1, 129,9, 129,5, 125,2, 121,8, 120,0, 70,6, 59,0, 53,2, 37,5, 35,8, 30,6, 29,6, 29,3, 28,1, 26,2, 26,2, 25,1, 19,1, 18,1.
ESI-MS m/z: Рассчитано для C35H43N7O11: 737,3. Найдено: 738,3 (M+H)+.
Пример 1
Получение Соединения 1
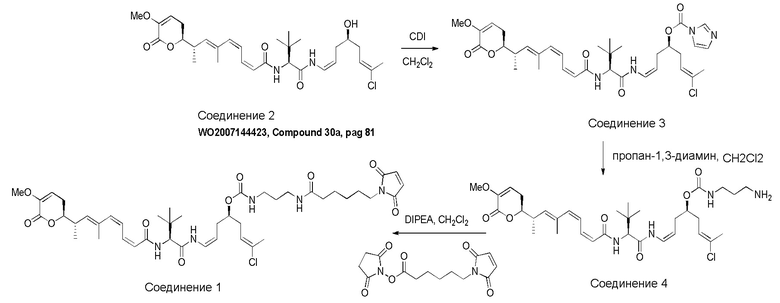
(a) Получение Соединения 3
К раствору Соединения 2 (Соединение 30a, получено, как описано в WO 2007144423, содержание которого включено в настоящую заявку посредством ссылки) (1,014 г, 1,8 ммоль) в DCM (45 мл) добавляли 1,1’-карбонилдиимидазол (876 мг, 5,4 ммоль). После перемешивания при 23°C в течение ночи реакционную смесь концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc, от 99:1 до 85:15) с получением чистого Соединения 3 (1,176 г, 86%).
1H ЯМР (500 МГц, CDCl3): δ 8,39 (д, J=9,0 Гц, 1H), 8,12 (шир.с, 1H), 7,40 (шир.с, 1H), 7,30 (т, J=11,5 Гц, 1H), 7,08 (шир.с, 1H), 6,91 (т, J=12,0 Гц, 1H), 6,86 (т, J=10,0 Гц, 1H), 6,22 (д, J=9,0 Гц, 1H), 6,18 (д, J=11,0 Гц, 1H), 5,67 (д, J=12,0 Гц, 1H), 5,63-5,61 (м, 2H), 5,28 (д, J=11,5 Гц, 1H), 4,94-4,91 (м, 1H), 4,81-4,76 (м, 1H), 4,42 (д, J=9,0 Гц, 1H), 4,23-4,19 (м, 1H), 3,66 (с, 3H), 2,87-2,82 (м, 1H), 2,58-2,46 (м, 3H), 2,42-2,35 (м, 3H), 2,08 (с, 3H), 1,83 (с, 3H), 1,16 (д, J=6,6 Гц, 3H), 1,06 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 168,5, 166,4, 161,5, 148,7, 145,2, 140,4, 137,6, 137,0, 134,4, 133,9, 133,0, 130,9, 124,2, 123,9, 121,0, 120,5, 117,03, 110,0, 108,1, 104,1, 81,7, 77,8, 60,4, 55,4, 37,2, 34,5, 26,6, 26,3, 21,0, 17,1, 16,6.
ESI-MS m/z: Рассчитано для C34H45ClN4O7: 656,30. Найдено: 657,3 (M+H)+.
(b) Получение Соединения 4
К раствору Соединения 3 (1,160 мг, 1,78 ммоль) в DCM (45 мл), полученного, как описано на стадии (a) выше, добавляли пропан 1,3-диамин (0,19 мл, 2,22 ммоль). Реакционную смесь перемешивали при 23°C в течение ночи и затем концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiNH2, DCM:CH3OH, от 100:0 до 97:3) с получением чистого Соединения 4 (800 мг, 68%).
1H ЯМР (300 МГц, CDCl3): δ 8,90 (д, J=11,7 Гц, 1H), 7,34-7,26 (м, 1H), 6,99-6,74 (м, 2H), 6,50 (д, J=9,0 Гц, 1H), 6,15 (д, J=12,9 Гц, 1H), 5,83 (т, J=11,5 Гц, 1H), 5,70 (д, J=11,5 Гц, 1H), 5,68-5,57 (м, 2H), 5,27 (д, J=9,4 Гц, 1H), 4,80 (кв., J=8,3 Гц, 1H), 4,52-4,44 (м, 2H), 4,24-4,17 (м, 1H), 3,66 (с, 3H), 3,39-3,17 (м, 2H), 2,93-2,82 (м, 1H), 2,78 (т, J=6,5 Гц, 2H), 2,50-2,34 (м, 2H), 2,34-2,24 (м, 2H), 2,19-1,99 (м, 2H), 2,06 (с, 3H), 1,84 (с, 3H), 1,72-1,50 (м, 2H), 1,16 (д, J=6,6 Гц, 3H), 1,04 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 168,4, 166,1, 161,5, 156,7, 145,2, 139,9, 137,1, 134,0, 133,9, 131,8, 124,3, 124,2, 122,5, 120,9, 108,1, 105,5, 81,8, 74,3, 60,6, 55,4, 39,81, 39,30 37,2, 34,7, 33,1, 31,5, 29,6, 26,7, 26,2, 21,0, 17,1, 16,6.
ESI-MS m/z: Рассчитано для C34H51ClN4O7: 662,30. Найдено: 663,3 (M+H)+.
(c) Получение Соединения 1
К раствору Соединения 4 (52 мг, 0,078 ммоль), полученного, как описано на стадии (b) выше, и N-гидроксисукцинимидного эфира 6-малеимидoгексановой кислоты (27,1 мг, 0,088 ммоль) в DCM (2 мл) добавляли DIPEA (15 мкл, 0,086 ммоль). Реакционную смесь перемешивали при 23°C в течение ночи и концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого целевого Соединения 1 (29,7 мг, 44%).
1H ЯМР (500 МГц, CDCl3): δ 8,86 (д, J=10,8 Гц, 1H), 7,30 (т, J=11,6 Гц, 1H), 6,90 (тд, J=11,5, 1,2 Гц, 1H), 6,78 (т, J=9,7 Гц, 1H), 6,68 (шир.с, 2H), 6,63 (д, J=9,4 Гц, 1H), 6,51 (т, J=6,4 Гц, 1H), 6,16 (д, J=11,8 Гц, 1H), 5,76 (т, J=6,4 Гц, 1H), 5,72 (д, J=11,6 Гц, 1H), 5,65 (дд, J=6,4, 2,9 Гц, 1H), 5,62-5,57 (м, 1H), 5,29 (д, J=11,1 Гц, 1H), 4,81 (кв., J=8,3 Гц, 1H), 4,52-4,48 (м, 2H), 4,24 (ддд, J=11,4, 7,3, 4,3 Гц, 1H), 3,66 (с, 3H), 3,50 (т, J=7,3 Гц, 2H), 3,33-3,10 (м, 4H), 2,93-2,81 (м, 1H), 2,45-2,31 (м, 5H), 2,17 (т, J=7,6 Гц, 2H), 2,10-1,98 (м, 1H), 2,07 (с, 3H), 1,84 (с, 3H), 1,72-1,54 (м, 8H), 1,16 (д, J=6,6 Гц, 3H), 1,05 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 173,8, 170,8, 168,3, 166,5, 161,6, 157,1, 145,1, 140,4, 137,5, 134,2, 134,1, 134,0, 131,9, 124,2, 124,0, 122,5, 120,6, 108,3, 106,0, 81,8, 74,5, 60,6, 55,4, 37,7, 37,6, 37,2, 36,3, 36,1, 34,7, 33,4, 31,0, 29,8, 28,3, 26,7, 26,3, 26,2, 25,6, 21,0, 17,2, 16,6.
ESI-MS m/z: Рассчитано для C44H62ClN5O10: 855,42. Найдено: 856,5 (M+H)+.
Пример 2
Получение Соединения 5

(a) Получение Соединения 7
К раствору Соединения 6 (Соединение 30b, получено, как описано в WO 2007144423, содержание которого включено в настоящую заявку посредством ссылки) (750 мг, 1,42 ммоль) в DCM (35,5 мл) добавляли 1,1’-карбонилдиимидазол (691 мг, 4,26 ммоль). После перемешивания при 23°C в течение ночи реакционную смесь концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого Соединения 7 (717 мг, 81%).
1H ЯМР (300 МГц, CDCl3): δ 8,24 (д, J=11,0 Гц, 1H), 8,09 (с, 1H), 7,40 (с, 1H), 7,36-7,23 (м, 1H), 7,05 (с, 1H), 6,95-6,83 (м, 2H), 6,34 (д, J=9,2 Гц, 1H), 6,14 (д, J=11,8 Гц, 1H), 5,74-5,57 (м, 3H), 5,43-5,34 (м, 1H), 5,28 (д, J=10,2 Гц, 1H), 4,98-4,88 (м, 1H), 4,78 (кв., J=7,8 Гц, 1H), 4,47 (д, J=9,2 Гц, 1H), 4,25-4,16 (м, 1H), 3,64 (с, 3H), 2,92-2,76 (м, 1H), 2,59-2,37 (м, 6H), 1,83 (с, 3H), 1,64 (д, J=6,7 Гц, 3H), 1,14 (д, J=6,7 Гц, 3H), 1,03 (с, 9H).
(b) Получение Соединения 8
К раствору Соединения 7 (1,68 г, 2,7 ммоль), полученного, как описано на стадии (a) выше, в DCM (80 мл) добавляли пропан 1,3-диамин (0,27 мл, 3,24 ммоль). Реакционную смесь перемешивали при 23°C в течение ночи и концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, DCM:CH3OH, от 100:0 до 97:3) с получением Соединения 8 (854 мг, 50%).
1H ЯМР (300 МГц, CDCl3): δ 8,90 (д, J=11,7 Гц, 1H), 7,39-7,18 (м, 1H), 6,92-6,84 (м, 2H), 6,50 (д, J=9,0 Гц, 1H), 6,15 (д, J=12,9 Гц, 1H), 5,75-5,67 (м, 2H), 5,66-5,54 (м, 2H), 5,46-5,33 (м, 1H), 5,26 (д, J=10,3 Гц, 1H), 4,83 (кв., J=8,3 Гц, 1H), 4,50-4,48 (м, 2H), 4,30-4,04 (м, 1H), 3,66 (с, 3H), 3,39-3,17 (м, 2H), 2,93-2,82 (м, 1H), 2,78 (т, J=6,5 Гц, 2H), 2,50-2,34 (м, 3H), 2,34-2,24 (м, 2H), 2,19-1,99 (м, 1H), 1,83 (с, 3H), 1,72-1,50 (м, 2H), 1,62 (д, J=6,7 Гц, 3H), 1,16 (д, J=6,6 Гц, 3H), 1,04 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 168,3, 166,2, 161,6, 157,1, 145,1, 139,9, 137,1, 134,0, 133,9, 126,9, 124,9, 124,2, 123,9, 120,9, 108,2, 106,3, 81,8, 75,0, 60,6, 55,4, 39,6, 37,2, 34,7, 32,8, 31,5, 31,1, 29,6, 26,7, 26,2, 17,1, 16,6, 12,9.
ESI-MS m/z: Рассчитано для C34H52N4O7: 628,4. Найдено: 629,5 (M+H)+.
(c) Получение Соединения 5
К раствору Соединения 8 (150 мг, 0,24 ммоль), полученного, как описано на стадии (b) выше, в DCM (8 мл) при 23°C добавляли N-гидроксисукцинимидный эфир 6-малеимидoгексановой кислоты (88,3 мг, 0,28 ммоль). Реакционную смесь перемешивали при 23°C в течение 18 часов и концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого целевого Соединения 5 (75 мг, 38%).
1H ЯМР (500 МГц, CDCl3): δ 8,87 (д, J=10,7 Гц, 1H), 7,32-7,22 (м, 1H), 6,89 (т, J=11,6 Гц, 1H), 6,78 (т, J=9,7 Гц, 1H), 6,68 (с, 2H), 6,61 (д, J=9,4 Гц, 1H), 6,54 (т, J=6,0 Гц, 1H), 6,15 (д, J=11,6 Гц, 1H), 5,77-5,51 (м, 2H), 5,64 (дд, J=6,4, 3,0 Гц, 1H), 5,60-5,55 (м, 1H), 5,38 (ддд, J=13,0, 8,8, 6,6 Гц, 1H), 5,28 (д, J=10,0 Гц, 1H), 4,83 (кв., J=8,3 Гц, 1H), 4,59-4,44 (м, 2H), 4,23 (ддд, J=11,5, 7,2, 4,4 Гц, 1H), 3,65 (с, 3H), 3,49 (т, J=7,2 Гц, 2H), 3,29-3,12 (м, 4H), 2,87-2,81 (м, 1H), 2,48-2,32 (м, 5H), 2,18-2,09 (м, 3H), 1,88-1,82 (м, 1H), 1,83 (с, 3H), 1,67-1,55 (м, 7H),1,62 (д, J=6,8 Гц, 3H), 1,15 (д, J=6,7 Гц, 3H), 1,04 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 173,6, 170,8, 168,2, 166,4, 161,6, 157,4, 145,2, 140,2, 137,4, 134,2, 134,1, 134,0, 127,0, 124,9, 123,9, 120,7, 108,3, 106,5, 81,8, 75,3, 60,6, 55,4, 37,7, 37,6, 37,2, 36,3, 36,0, 34,7, 31,8, 31,6, 31,1, 29,9, 28,3, 26,7, 26,4, 26,2, 25,2, 22,6, 17,2, 16,6.
ESI-MS m/z: Рассчитано для C44H63N5O10: 821,5. Найдено: 822,4 (M+H)+.
Пример 3
Получение Соединения 12
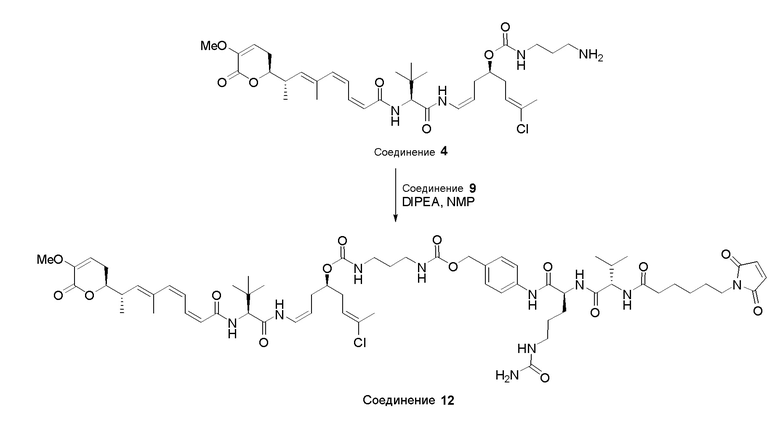
(a) Получение Соединения 12
DIPEA (25 мкл, 0,14 ммоль) добавляли к раствору Соединения 9 (94,5 мг, 0,13 ммоль), полученного, как показано в примере получения выше, и Соединения 4 (85 мг, 0,13 ммоль), полученного, как описано в Пример 1(b) выше, в NMP (6,5 мл) при 23°C. Через 9 часов реакционную смесь разбавляли при помощи H2O и экстрагировали при помощи EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, DCM:CH3OH, от 100:0 до 90:10). В конце, очистку целевого Соединения 12 (35,7 мг, 22%) осуществляли при помощи полупрепаративной ВЭЖХ (Symmetry C18, 7 мкм, 19×150 мм, градиент H2O+CH3CN, скорость потока 15 мл/мин, УФ детекция).
1H ЯМР (500 МГц, CDCl3/CD3OD): δ 7,49 (д, J=8,1 Гц, 2H), 7,22 (д, J=8,1 Гц, 2H), 7,19 (т, J=11,8 Гц, 1H), 6,96 (дд, J=23,5, 8,9 Гц, 1H), 6,84 (т, J=11,5 Гц, 1H), 6,70-6,64 (м, 1H), 6,64 (с, 2H), 6,10 (д, J=11,6 Гц, 1H), 5,93 (т, J=6,2 Гц, 1H), 5,82 (т, J=6,2 Гц, 1H), 5,69 (д, J=11,4 Гц, 1H), 5,61 (дд, J=6,3, 3,1 Гц, 1H), 5,54 (т, J=7,8 Гц, 1H), 5,22 (д, J=9,7 Гц, 1H), 4,96 (с, 2H), 4,75 (кв., J=8,1 Гц, 1H), 4,55-4,49 (м, 2H), 4,43-4,36 (м, 1H), 4,23-4,10 (м, 2H), 3,59 (с, 3H), 3,44 (т, J=7,2 Гц, 2H), 3,18-3,04 (м, 8H), 2,82-2,72 (м, 1H), 2,49-2,34 (м, 3H), 2,27 (т, J=7,1 Гц, 2H), 2,18 (т, J=7,2 Гц, 2H), 2,16-2,06 (м, 1H), 2,01-1,95 (м, 1H), 2,00 (с, 3H), 1,87-1,79 (м, 1H), 1,78 (с, 3H), 1,73-1,40 (м, 11H), 1,35-1,20 (м, 2H), 1,09 (д, J=10,0 Гц, 3H), 0,96 (с, 9H), 0,87 (дд, J=6,8, 4,3 Гц, 6H).
13C ЯМР (125 МГц, CDCl3): δ 174,1, 172,2, 171,0, 170,3, 168,8, 166,8, 162,1, 160,2, 157,0, 156,9, 144,9, 140,2, 137,7, 137,5, 134,0, 132,4, 131,7, 128,7, 124,0, 123,4, 122,4, 120,4, 119,8, 111,5, 108,6, 107,0, 81,9, 73,8, 68,6, 66,2, 60,3, 58,8, 55,4, 53,0, 37,6, 37,5, 37,1, 35,9, 34,6, 33,2, 30,6, 29,9, 29,2, 28,0, 26,5, 26,2, 26,0, 25,0, 22,6, 20,8, 19,1, 18,2, 17,04, 16,4.
ESI-MS m/z: Рассчитано для C63H89ClN10O15: 1260,6. Найдено: 1261,6 (M+H)+.
Пример 4
Получение Соединения 13
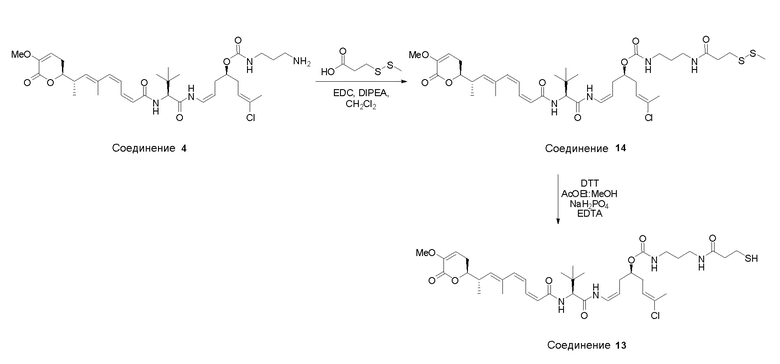
(a) Получение Соединения 14
К раствору Соединения 4 (110 мг, 0,17 ммоль), полученного, как описано в Примере 1(b) выше, и 3-(метилдисульфанил)пропановой кислоты (34 мг, 0,22 ммоль) в DCM (5 мл) добавляли N-(3-диметиламинопропил)-N′-этилкарбодиимид гидрохлорид (EDC) (47,8 мг, 0,22 ммоль) и N,N′-диизопропилэтиламин (3,8 мкл, 0,22 ммоль). Реакционную смесь перемешивали при 23°C в течение 6 часов, разбавляли при помощи H2O и экстрагировали при помощи DCM. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого Соединения 14 (123 мг, 93%).
1H ЯМР (500 МГц, CDCl3): δ 8,88 (д, J=10,8 Гц, 1H), 7,29-7,24 (м, 1H), 6,90 (т, J=11,5 Гц, 1H), 6,82 (т, J=9,1 Гц, 1H), 6,63 (т, J=6,1 Гц, 1H), 6,49 (д, J=9,4 Гц, 1H), 6,16 (дд, J=11,5, 1,5 Гц, 1H), 5,70 (д, J=11,5 Гц, 1H), 5,68-5,51 (м, 3H), 5,29 (д, J=9,7 Гц, 1H), 4,81 (кв., J=8,2 Гц, 1H), 4,52 (д, J=9,5 Гц, 1H), 4,52-4,43 (м, 1H), 4,24 (ддд, J=11,5, 7,3, 4,3 Гц, 1H), 3,66 (с, 3H), 3,37-3,21 (м, 3H), 3,21-3,12 (м, 1H), 2,97 (т, J=7,2 Гц, 2H), 2,90-2,81 (м, 1H), 2,60 (т, J=7,2 Гц, 2H), 2,49-2,35 (м, 3H), 2,39 (с, 3H), 2,33 (т, J=7,0 Гц, 2H), 2,14-2,07 (м, 1H), 2,07 (с, 3H), 1,84 (с, 3H), 1,73-1,64 (м, 2H), 1,16 (д, J=6,7 Гц, 3H), 1,05 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 171,6, 168,2, 166,4, 161,6, 157,2, 145,2, 140,3, 137,4, 134,2, 134,0, 131,9, 124,4, 124,1, 122,4, 120,7, 108,3, 105,6, 81,8, 74,8, 60,6, 60,4, 55,5, 37,8, 37,2, 36,2, 35,6, 34,7, 33,1, 31,0, 29,8, 26,7, 26,2, 23,0, 21,0, 17,2, 16,6.
(b) Получение Соединения 13
Раствор Соединения 14 (100 мг, 0,125 ммоль), полученного, как описано на стадии (a) выше, в смеси EtOAc (4,3 мл) и CH3OH (4,3 мл) обрабатывали раствором дитиотреитола (154,8 мг, 1,0 ммоль) в 0,05 M калийфосфатном буфере (4,3 мл) при °C pH 7,5, содержащем 2 мМ этилендиаминтетрауксусную кислоту (EDTA). Смесь перемешивали при 23°C в течение 4 часов. Реакционную смесь обрабатывали 0,2 M раствором калийфосфатного буфера (13 мл) при pH 6,0, содержащего 2 мМ EDTA, и затем экстрагировали при помощи EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Полученное неочищенное вещество очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого целевого Соединения 13 (35 мг, 37%).
1H ЯМР (300 МГц, CDCl3): δ 8,91 (д, J=10,8 Гц, 1H), 7,27-7,24 (м, 1H), 6,91 (т, J=11,5 Гц, 1H), 6,82 (т, J=9,7 Гц, 1H), 6,67 (т, J=6,1 Гц, 1H), 6,49 (д, J=9,4 Гц, 1H), 6,16 (д, J=11,6 Гц, 1H), 5,71 (д, J=11,6 Гц, 1H), 5,66-5,57 (м, 3H), 5,29 (д, J=9,9 Гц, 1H), 4,84 (кв., J=8,3 Гц, 1H), 4,51 (д, J=9,5 Гц, 1H), 4,50-4,45 (м, 1H), 4,24-4,20 (м, 1H), 3,66 (с, 3H), 3,36-3,12 (м, 4H), 2,90-2,71 (м, 3H), 2,64-2,24 (м, 7H), 2,14-2,04 (м, 1H), 2,06 (с, 3H), 1,83 (с, 3H), 1,73-1,68 (м, 2H), 1,15 (д, J=6,7 Гц, 3H), 1,05 (с, 9H).
ESI-MS m/z: Рассчитано для C37H55ClN4O8S: 750,3. Найдено: 773,2 (M+Na)+.
Пример 5
Получение Соединения 15
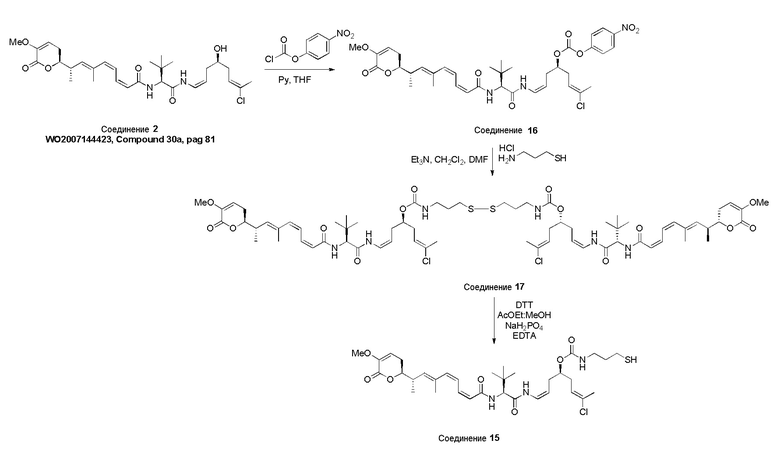
(a) Получение Соединения 16
К раствору Соединения 2 (Соединение 30a, получено, как описано в WO 2007144423, содержание которого включено в настоящую заявку посредством ссылки) (300 мг, 0,53 ммоль) в DCM (5 мл) добавляли пиридин (85 мкл, 1,06 ммоль) и 4-нитрофенилхлорформиат (214,7 мг, 1,06 ммоль) при 0°C. Реакционную смесь перемешивали при 23°C в течение 1,5 часов, разбавляли при помощи 10% раствора лимонной кислоты и экстрагировали при помощи DCM. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого Соединения 16 (307 мг, 80%).
1H ЯМР (500 МГц, CDCl3): δ 8,29 (д, J=9,2 Гц, 2H), 8,08 (д, J=10,9 Гц, 1H), 7,44 (д, J=9,1 Гц, 2H), 7,27-7,22 (м, 1H), 6,92-6,83 (м, 2H), 6,20 (д, J=9,2 Гц, 1H), 6,17 (дд, J=11,6, 1,4 Гц, 1H), 5,67-5,58 (м, 3H), 5,29 (д, J=10,0 Гц, 1H), 4,84 (кв., J=8,2 Гц, 1H), 4,77-4,72 (м, 1H), 4,41 (д, J=9,3 Гц, 1H), 4,22 (ддд, J=11,5, 7,5, 4,4 Гц, 1H), 3,67 (с, 3H), 2,89-2,82 (м, 1H), 2,54-2,33 (м, 6H), 2,10 (д, J=1,2 Гц, 3H), 1,85 (д, J=1,3 Гц, 3H), 1,17 (д, J=6,7 Гц, 3H), 1,02 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 168,4, 166,1, 161,5, 155,3, 152,5, 145,5, 145,2, 140,4, 137,6, 134,3, 134,0, 133,2, 125,3, 124,4, 124,1, 121,8, 121,2, 120,4, 108,1, 104,6, 81,8, 79,1, 60,4, 55,5, 37,3, 34,7, 32,7, 30,1, 26,6, 26,3, 21,1, 17,2, 16,6.
(b) Получение Соединения 17
К раствору Соединения 16 (156,3 мг, 0,21 ммоль) в DCM (2,5 мл) добавляли суспензию 3-аминопропан-1-тиолгидрохлорида (44,8 мг, 0,26 ммоль) в DCM (2,5 мл), триэтиламин (58 мкл, 0,34 ммоль) и DMF (0,1 мл) при 23°C. Реакционную смесь перемешивали при 23°C в течение 3 часов, разбавляли при помощи H2O и экстрагировали при помощи DCM. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого Соединения 17 (80 мг, 95%).
1H ЯМР (300 МГц, CDCl3): δ 8,68 (д, J=10,6 Гц, 1H), 7,28 (т, J=11,6 Гц, 1H), 6,89 (т, J=11,5 Гц, 1H), 6,76 (т, J=9,6 Гц, 1H), 6,65 (д, J=9,1 Гц, 1H), 6,13 (д, J=11,7 Гц, 1H), 5,87-5,51 (м, 4H), 5,28 (д, J=5,0 Гц, 1H), 4,77 (кв., J=8,2 Гц, 1H), 4,61-4,39 (м, 2H), 4,29-4,00 (м, 1H), 3,65 (с, 3H), 3,31-3,18 (м, 2H), 2,98-2,77 (м, 1H), 2,68 (т, J=7,4 Гц, 2H), 2,55-2,22 (м, 6H), 2,04 (с, 3H), 2,00-1,80 (м, 2H), 1,83 (с, 3H), 1,15 (д, J=6,6 Гц, 3H), 1,05 (с, 9H).
ESI-MS m/z: Рассчитано для C68H98Cl2N6O14S2: 1356,6. Найдено: 1357,3 (M+H)+.
(c) Получение Соединения 15
Раствор Соединения 17 (59,4 мг, 0,044 ммоль) в смеси EtOAc (1,5 мл) и CH3OH (1,5 мл) обрабатывали раствором дитиотреитола (0,35 мл, 0,35 ммоль) в 0,05 M калийфосфатном буфере (1,5 мл) при pH 7,5, содержащем 2 мМ этилендиаминтетрауксусной кислоты (EDTA). Смесь перемешивали при 23°C в течение 4 часов. Реакционную смесь обрабатывали раствором 0,2 M калийфосфатного буфера при pH 6,0, содержащим 2 мМ раствор EDTA, и экстрагировали при помощи EtOAc (× 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого целевого Соединения 15 (31 мг, 59%).
1H ЯМР (500 МГц, CDCl3): δ 8,66 (д, J=10,7 Гц, 1H), 7,29 (т, J=11,2 Гц, 1H), 6,91 (т, J=11,5 Гц, 1H), 6,83 (т, J=9,7 Гц, 1H), 6,38 (д, J=9,4 Гц, 1H), 6,17 (д, J=11,8 Гц, 1H), 5,70 (д, J=11,4 Гц, 1H), 5,65-5,51 (м, 2H), 5,34 (т, J=6,3 Гц, 1H), 5,29 (д, J=10,0 Гц, 1H), 4,82 (кв., J=8,3 Гц, 1H), 4,56-4,48 (м, 1H), 4,45 (д, J=9,3 Гц, 1H), 4,22 (ддд, J=11,4, 7,5, 4,3 Гц, 1H), 3,67 (с, 3H), 3,31 (кв., J=6,4 Гц, 2H), 2,88-2,83 (м, 1H), 2,55 (кв., J=7,7 Гц, 2H), 2,47-2,30 (м, 5H), 2,12-2,07 (м, 1H), 2,08 (с, 3H), 1,88-1,76 (м, 5H), 1,17 (д, J=6,6 Гц, 3H), 1,06 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 168,2, 166,2, 161,5, 156,7, 145,2, 140,2, 137,3, 134,2, 134,0, 132,0, 124,4, 124,2, 122,3, 120,8, 108,1, 105,5, 81,8, 74,5, 60,6, 55,4, 39,6, 37,3, 34,6, 33,9, 33,3, 30,8, 26,7, 26,3, 21,8, 21,1, 17,2, 16,7.
ESI-MS m/z: Рассчитано для C34H50ClN3O7S: 679,3. Найдено: 702,4 (M+Na)+.
Пример 6
Получение Соединения 18
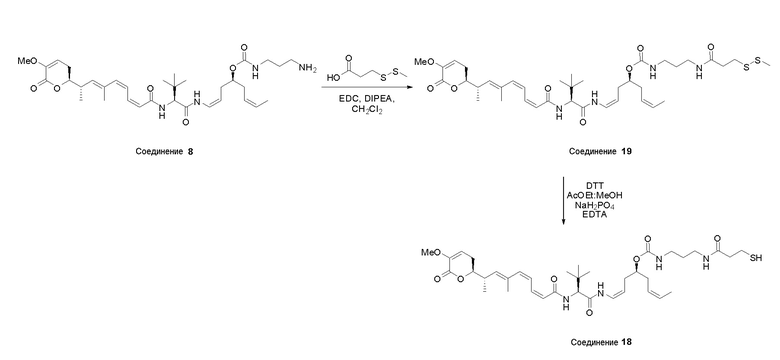
(a) Получение Соединения 19
К раствору Соединения 8 (280 мг, 0,45 ммоль), полученного, как описано в Примере 2(b) выше, и 3-(метилдисульфанил)пропановой кислоты (88 мг, 0,58 ммоль) в DCM (7,5 мл) добавляли N-(3-диметиламинопропил)-N′-этилкарбодиимидгидрохлорид (EDC) (126 мг, 0,58 ммоль) и N,N′-диизопропилэтиламин (0,1 мл, 0,58 ммоль). Реакционную смесь перемешивали при 23°C в течение 3 часов, разбавляли при помощи H2O и экстрагировали при помощи DCM. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого Соединения 19 (240 мг, 71%).
1H ЯМР (500 МГц, CDCl3): δ 8,91 (д, J=10,8 Гц, 1H), 7,34-7,20 (м, 1H), 6,89 (т, J=11,4 Гц, 1H), 6,83-6,72 (м, 2H), 6,51 (д, J=9,5 Гц, 1H), 6,16 (д, J=11,3Гц, 1H), 5,70 (д, J=11,5 Гц, 1H), 5,64 (дд, J=6,1, 3,3 Гц, 1H), 5,61-5,55 (м, 2H), 5,47-5,33 (м, 1H), 5,28 (д, J=9,3 Гц, 1H), 4,84 (кв., J=8,3 Гц, 1H), 4,51 (д, J=9,6 Гц, 1H), 4,52-4,47 (м, 1H), 4,27-4,19 (м, 1H), 3,66 (с, 3H), 3,37-3,21 (м, 3H), 3,21-3,12 (м, 1H), 2,96 (т, J=7,2 Гц, 2H), 2,90-2,81 (м, 1H), 2,60 (т, J=7,2 Гц, 2H), 2,43-2,35 (м, 5H), 2,39 (с, 3H), 2,16-2,04 (м, 1H), 1,84 (с, 3H), 1,70-1,61 (м, 5H), 1,16 (д, J=6,7 Гц, 3H), 1,05 (с, 9H).
13C ЯМР (125 МГц, CDCl3) δ 171,5, 168,1, 166,3, 157,4, 145,2, 140,3, 137,4, 134,2, 134,0, 127,0, 124,9, 124,0, 120,8, 108,3, 106,3, 81,8, 75,5, 60,6, 55,4, 53,4, 37,8, 37,2, 36,2, 35,9, 34,7, 33,1, 31,8, 31,2, 29,8, 26,7, 26,2, 23,0, 17,2, 16,6, 13,0.
ESI-MS m/z: Рассчитано для C38H58N4O8S2: 763,4. Найдено: 762,4 (M+H)+.
(b) Получение Соединения 18
Раствор Соединения 19 (240 мг, 0,31 ммоль), полученного, как описано на стадии (a) выше, в смеси EtOAc (15 мл) и CH3OH (22 мл) обрабатывали раствором дитиотреитола (0,79 мл 1,0 M раствор, 0,79 ммоль) в 0,05 M калийфосфатном буфере (17,4 мл) при pH 7,5, содержащем 2 мМ этилендиаминтетрауксусной кислоты (EDTA). Смесь перемешивали при 23°C в течение 4 часов. Реакционную смесь обрабатывали раствором 0,2 M калийфосфатного буфера (21 мл) при pH 6,0, содержащим 2 мМ EDTA, и экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Полученное неочищенное вещество очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого целевого Соединения 18 (105 мг, 47%)
1H ЯМР (300 МГц, CDCl3): δ 8,93 (д, J=10,6 Гц, 1H), 7,30-7,22 (м, 1H), 6,90 (т, J=11,6 Гц, 1H) 6,86-6,72 (м, 2H), 6,48 (д, J=9,4 Гц, 1H), 6,16 (д, J=11,6 Гц, 1H), 5,70 (д, J=11,4 Гц, 1H), 5,65-5,55 (м, 3H), 5,48-5,34 (м, 1H), 5,29 (д, J=9,9 Гц, 1H), 4,84 (кв., J=9,4, 8,7 Гц, 1H), 4,52 (д, J=9,5 Гц, 1H), 4,57-4,44 (м, 1H), 4,32-4,17 (м, 1H), 3,66 (с, 3H), 3,39-3,20 (м, 3H), 3,22-3,09 (м, 1H), 2,90-2,75 (м, 3H), 2,50 (т, J=6,9, 2H), 2,45-2,28 (м, 5H), 2,16-2,08 (м, 1H), 1,83 (с, 3H), 1,72-1,65 (m 2H), 1,64 (д, J=6,6 Гц, 3H) 1,16 (д, J=6,7 Гц, 3H), 1,05 (с, 9H).
ESI-MS m/z: Рассчитано для C37H56N4O8S: 716,4. Найдено: 717,3 (M+H)+.
Пример 7
Получение Соединения 25

(a) Получение Соединения 23
К раствору Соединения 22 (333 мг, 0,63 ммоль) (Соединение 71, получено, как описано в WO 2009080761, содержание которого включено в настоящую заявку посредством ссылки) в CH2Cl2 (12,5 мл) добавляли 1,1’-карбонилдиимидазол (308 мг, 1,90 ммоль). После перемешивания при 23°C в течение ночи реакционную смесь концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого Соединения 23 (344 мг, 88%).
1H ЯМР (300 МГц, CDCl3): δ 8,14 (с, 1H), 8,11 (д, J=10,8 Гц, 1H), 7,43 (с, 1H), 7,28 (т, J=11,6 Гц, 1H), 7,08 (дд, J=1,7, 0,9 Гц, 1H), 6,96-6,81 (м, 2H), 6,26 (д, J=9,3 Гц, 1H), 6,17 (д, J=11,6 Гц, 1H), 5,66 (дт, J=11,4, 1,4 Гц, 1H), 5,62 (дд, J=5,9, 3,5 Гц, 1H), 5,28 (д, J=10,0 Гц, 1H), 5,04-4,89 (м, 1H), 4,80 (кв., J=8,3 Гц, 1H), 4,40 (д, J=9,3 Гц, 1H), 4,21 (ддд, J=10,3, 7,5, 5,3 Гц, 1H), 3,65 (с, 3H), 2,91-2,78 (м, 1H), 2,68-2,49 (м, 3H), 2,45-2,31 (м, 2H), 1,89 (т, J=2,5 Гц, 3H), 1,84 (с, 3H), 1,15 (д, J=6,7 Гц, 3H), 1,05 (с, 9H).
ESI-MS m/z: Рассчитано для C34H44N4O7: 620,32. Найдено: 621,3 (M+H)+.
(b) Получение Соединения 24
К раствору Соединения 23 (0,130 г, 0,21 ммоль), полученного, как описано на стадии (a) выше, в CH2Cl2 (3,5 мл) добавляли пропан 1,3-диамин (0,022 мл, 0,26 ммоль). Реакционную смесь перемешивали при 23°C в течение 6 часов и затем концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, DCM:CH3OH, от 100:0 до 97:3) с получением Соединения 24 (120 мг, 91%).
1H ЯМР (300 МГц, CDCl3): δ 8,71 (д, J=10,7 Гц, 1H), 7,28 (т, J=11,6 Гц, 1H), 6,91-6,77 (м, 2H), 6,44 (д, J=9,4 Гц, 1H), 6,14 (д, J=11,6 Гц, 1H), 5,77-5,57 (м, 3H), 5,27 (д, J=10,0 Гц, 1H), 4,83 (кв., J=8,3 Гц, 1H), 4,57-4,53 (м, 1H), 4,46 (д, J=12,4 Гц, 1H), 4,31-4,11 (м, 1H), 3,65 (с, 3H), 3,31-3,24 (м, 2H), 2,93-2,67 (м, 3H), 2,56-2,24 (м, 6H), 2,16 (с, 3H), 1,83 (с, 3H), 1,63 (дд, J=9,6, 3,5 Гц, 2H), 1,15 (д, J=6,6 Гц, 3H), 1,04 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 168,3, 166,1, 161,6, 156,5, 145,2, 140,0, 137,2, 134,1, 134,0, 124,3, 124,2, 120,9, 108,1, 106,0, 81,8, 78,4, 74,5, 73,2, 60,6, 55,4, 39,8, 39,2, 37,3, 34,8, 33,0, 30,9, 30,2, 26,7, 26,3, 17,2, 16,7, 3,6.
ESI-MS m/z: Рассчитано для C34H50N4O7: 626,37. Найдено: 627,3 (M+H)+.
(c) Получение Соединения 25
К раствору Соединения 24 (40 мг, 0,064 ммоль), полученного, как описано на стадии (b) выше, в CH2Cl2 (2 мл) добавляли N-гидроксисукцинимидный эфир 6-малеимидoгексановой кислоты (21,6 мг, 0,07 ммоль). Реакционную смесь перемешивали при 23°C в течение ночи и затем концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого Соединения 25 (33,5 мг, 64%).
1H ЯМР (400 МГц, CDCl3): δ 8,67 (д, J=10,7 Гц, 1H), 7,29-7,23 (м, 1H), 6,90 (т, J=11,5 Гц, 1H), 6,80 (т, J=9,6 Гц, 1H), 6,68 (с, 2H), 6,46 (д, J=9,4 Гц, 1H), 6,42 (шир.с, 1H), 6,16 (д, J=11,6 Гц, 1H), 5,73-5,67 (м, 2H), 5,64 (дд, J=6,2, 3,1 Гц, 1H), 5,30 (д, J=9,6 Гц, 1H), 4,86 (кв., J=8,4 Гц, 1H), 4,63-4,54 (м, 1H), 4,44 (д, J=9,4 Гц, 1H), 4,30-4,18 (м, 1H), 3,66 (с, 3H), 3,50 (т, J=7,2 Гц, 2H), 3,34-3,10 (м, 3H), 2,85 (дт, J=9,9, 6,9 Гц, 1H), 2,54-2,37 (м, 4H), 2,36-2,28 (м, 1H), 2,16 (т, J=7,6 Гц, 2H), 1,84 (д, J=1,3 Гц, 3H), 1,83-1,81 (м, 3H), 1,70-1,51 (м, 8H), 1,34-1,23 (м, 2H), 1,16 (д, J=6,6 Гц, 3H), 1,05 (с, 9H).
13C ЯМР (100 МГц, CDCl3): δ 173,4, 170,8, 168,1, 166,4, 161,6, 157,0, 145,2, 140,3, 137,5, 134,3, 134,1, 133,9, 124,2, 120,7, 108,2, 106,2, 81,8, 78,4, 74,3, 73,5, 60,7, 55,4, 37,7, 37,6, 37,3, 36,4, 35,9, 34,6, 32,8, 30,3, 29,9, 28,3, 26,7, 26,4, 26,2, 25,4, 25,2, 24,4, 17,2, 16,6, 3,6.
ESI-MS m/z: Рассчитано для C44H61ClN5O10: 819,44. Найдено: 820,4 (M+H)+.
Пример 8
Получение Соединения 27
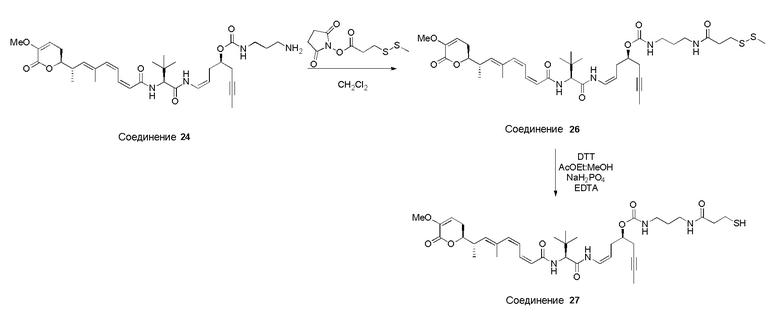
(a) Получение Соединения 26
К раствору Соединения 24 (70 мг, 0,11 ммоль), полученного, как описано в Примере 7(b) выше, в CH2Cl2 (2 мл) добавляли N-гидроксисукцинимидный эфир 3-(метилдисульфанил)пропановой кислоты (36,2 мг, 0,12 ммоль). Реакционную смесь перемешивали при 23°C в течение 16 часов и затем концентрировали под вакуумом. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого Соединения 26 (46,3 мг, 61%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 8,72 (д, J=10,8 Гц, 1H), 7,29-7,19 (м, 1H), 6,90 (т, J=11,3 Гц, 1H), 6,80 (т, J=9,7 Гц, 1H), 6,69 (т, J=6,1 Гц, 1H), 6,47 (д, J=9,4 Гц, 1H), 6,16 (д, J=11,0 Гц, 1H), 5,69 (д, J=11,5 Гц, 1H), 5,64 (дд, J=6,3, 3,1 Гц, 1H), 5,30 (д, J=0,5 Гц, 1H), 4,86 (кв., J=8,4 Гц, 1H), 4,60-4,54 (м, 1H), 4,46 (д, J=9,4 Гц, 1H), 4,23 (ддд, J=11,5, 7,4, 4,8 Гц, 1H), 3,66 (с, 3H), 3,33-3,22 (м, 3H), 3,19-3,14 (м, 1H), 2,96 (т, J=7,2 Гц, 2H), 2,66-2,54 (м, 1H), 2,59 (т, J=7,2 Гц, 2H), 2,48-2,42 (м, 5H), 2,40 (с, 3H), 2,38-2,28 (м, 1H) 1,83 (с, 3H), 1,82 (с, 3H), 1,73-1,64 (м, 2H), 1,16 (д, J=6,7 Гц, 3H), 1,05 (с, 9H).
13C ЯМР (100 МГц, CDCl3): δ 171,5, 168,1, 166,4, 165,2, 157,0, 145,1, 140,4, 137,5, 134,3, 134,0, 124,20, 124,0, 120,7, 108,2, 106,2, 81,8, 78,4, 74,2, 73,6, 60,7, 55,4, 37,8, 37,3, 35,9, 34,6, 33,1, 30,3, 29,8, 26,7, 26,2, 24,4, 23,0, 17,2, 16,6, 3,6.
(b) Получение Соединения 27
Раствор Соединения 26 (44,3 мг, 0,064 ммоль), полученного, как описано на стадии (a) выше, в смеси EtOAc (3,6 мл) и CH3OH (3,6 мл) обрабатывали раствором дитиотреитола (0,19 мл 1,0 M раствора, 0,19 ммоль) в растворе 0,05 M калийфосфатного буфера (3,6 мл) при pH 7,5, который содержал 2 мМ этилендиаминтетрауксусной кислоты (EDTA). Смесь перемешивали при 23°C в течение 4 часов. Реакционную смесь обрабатывали раствором 0,2 M калийфосфатного буфера (21 мл) при pH 6,0, содержащим 2 мМ EDTA, и затем экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc) с получением чистого целевого Соединения 27 (33,6 мг, 74%) в виде белого твердого вещества.
1H ЯМР (500 МГц, CDCl3): δ 8,73 (д, J=10,7 Гц, 1H), 7,27 (т, J=11,5 Гц, 1H), 6,91 (тд, J=11,5, 1,1 Гц, 1H), 6,94-6,86 (м, 1H), 6,85-6,77(м, 1H), 6,43 (д, J=9,4 Гц, 1H), 6,17 (дд, J=12,0, 1,6 Гц, 1H), 5,71 (д, J=11,4 Гц, 1H), 5,65 (дд, J=6,5, 2,9 Гц, 1H), 5,54 (т, J=6,3 Гц, 1H), 5,30 (д, J=10,5 Гц, 1H), 4,84 (кв., J=8,3 Гц, 1H), 4,53 (д, J=9,5 Гц, 1H), 4,48-4,44 (м, 1H), 4,24 (ддд, J=11,5, 7,3, 4,2 Гц, 1H), 3,67 (с, 3H), 3,336-3,22 (м, 3H), 3,21-3,10 (м, 1H), 2,89-2,84 (м, 1H), 2,80 (дт, J=8,2, 6,8 Гц, 2H), 2,49 (т, J=7,2 Гц, 2H), 2,45-2,36 (м, 5H), 2,31-2,28 (м, 1H), 1,84 (с, 3H), 1,83 (с, 3H), 1,72-1,69 (м, 2H), 1,16 (д, J=6,7 Гц, 3H), 1,05 (с, 9H).
ESI-MS m/z: Рассчитано для C37H54N4O8S: 714,37. Найдено: 737,3 (M+Na)+.
Пример 9
Получение конъюгата антитело-лекарственное средство ADC1 с Трастузумабом и Соединением 1
(a) Частичное восстановление Трастузумаба с получением частично восстановленного Трастузумаба (Соединение 20)
Трастузумаб (Трастузумаб закупали у компании Roche в виде белого лиофилизированного порошка для получения концентрированного раствора для инфузии) в виде раствора (9,52 мл, 200 мг, 1,38 мкмоль) разбавляли до концентрации 5 мг/мл с использованием 20 мМ гистидин/ацетатного буфера (pH 5,5, 30,5 мл) с последующим доведением pH с использованием фосфат/EDTA буфера (pH 8,4, 13 мл). Частичное восстановление дисульфидных связей в антителе осуществляли путем добавления 5,17 мМ трис[2-карбоксиэтил]фосфин гидрохлоридного (TCEP) раствора (689 мкл, 3,562 мкмоль, 2,6 экв.) Восстановительную реакцию оставляли для перемешивания в течение 90 минут при 20°C. Сразу после восстановления, осуществляли анализ Элмана с получением отношения свободного тиола к антителу (FTAR) 4,1, очень близко к значению 4,0, которое было запланированным.
(b) Получение ADC1
К раствору частично восстановленного Трастузумаба, Соединение 20 (24,98 мл, 93,0 мг, 0,64 мкмоль), полученного, как описано в Примере 7(a) выше, в DMSO добавляли (1,25 мл) с последующим добавлением свежеприготовленного раствора Соединения 1, полученного, как описано в Примере 1, (10 мМ в DMSO, 366 мкл, 3,66 мкмоль, 5,7 экв.). После добавления Соединения 1 раствор становился очень мутным, следовательно, дополнительно добавляли DMSO (1 мл). Реакционную смесь для конъюгации перемешивали в течение 30 минут при 20°C, и мутность исчезала в процессе реакции конъюгации. Избыток лекарственного средства гасили путем добавления N-ацетилцистеина (NAC) (10 мМ, 366 мкл, 3,66 мкмоль) с последующим перемешиванием раствора в течение 20 минут. После гашения реакции конъюгации смесь очищали при помощи Vivaspin центрифугирования и буфер заменяли на конечный PBS буфер. Конечный целевой продукт ADC1 концентрировали до конечной концентрации 8,56 мг/мл, как было определено при помощи УФ анализа, и получали 7,4 мл (63,3 мг, 0,43 мкмоль, 68,0%) ADC раствора. Осуществляли анализы методом эксклюзионной ВЭЖХ для определения чистоты продукта (61,4%).
ADC1 дополнительно очищали препаративной гель-фильтрационной хроматографией на системе очистки Äkta с использованием колонки HiLoad 16/600 superdex 200 из-за присутствия больших количеств агрегатов. После объединения конечную концентрацию (1,6 мг/мл) определяли при помощи УФ анализа и чистоту (90,9%) конечных лекарственных продуктов определяли методом эксклюзионной ВЭЖХ, с получением 8,65 мл (13,7 мг, 0,09 мкмоль, 14,7%) раствора ADC (ADC1).
Пример 10
Получение конъюгата антитело-лекарственное средство ADC2 с Трастузумабом и Соединением 5
(a) Частичное восстановление Трастузумаба с получением частично восстановленного Трастузумаба (Соединение 20)
Раствор Трастузумаба (14,29 мл, 300 мг, 2,06 мкмоль) разбавляли до концентрации 5 мг/мл с использованием 20 мМ гистидин/ацетатного буфера (pH, 5,5, 45,74 мл), с последующим доведением pH с использованием фосфат/EDTA буфера (pH 8,4, 14,4 мл). Частичное восстановление дисульфидных связей в антителе осуществляли путем добавления 5 мМ трис[2-карбоксиэтил]фосфин гидрохлоридного (TCEP) раствора (1,07 мл, 5,36 мкмоль, 2,6 экв.). Восстановительную реакцию оставляли для перемешивания в течение 90 минут при 20°C. Сразу после восстановления осуществляли анализ Элмана с получением отношения свободного тиола к антителу (FTAR) 4,1, очень близко к значению 4,0, которое было запланированным.
(b) Получение ADC2
К раствору частично восстановленного Трастузумаба, Соединение 20 (23,6 мл, 93,8 мг, 0,645 мкмоль), полученного, как описано в Примере 8(a) выше, добавляли DMSO (1,18 мл) с последующим добавлением свежеприготовленного раствора Соединения 5, полученного, как описано в Примере 2, (10 мМ в DMSO, 368 мкл, 3,68 мкмоль, 5,7 экв.). Лекарственное средство-линкер осторожно добавляли 10 порциями. После добавления шестой порции раствор становился слегка мутным, и мутность не исчезала в процессе реакции конъюгации и гашения. Реакционную смесь для конъюгации перемешивали в течение 30 минут при 20°C. Избыток лекарственного средства гасили путем добавления N-ацетилцистеина (NAC) (10 мМ, 368 мкл, 3,68 мкмоль) при перемешивании раствора в течение 46 минут. Раствор фильтровали через 0,2 мкм шприцевой фильтр. После гашения реакции конъюгации смесь концентрировали до 16,5 мг/мл при помощи Vivaspin центрифугирования и очищали на NAP-25 колонках. Осуществляли анализы методом эксклюзионной ВЭЖХ для определения чистоты продукта (36,1%).
ADC2 дополнительно очищали препаративной гель-фильтрационной хроматографией на системе очистки Äkta с использованием колонки HiLoad 16/600 superdex 200 из-за присутствия больших количеств агрегатов. После объединения конечную концентрацию (3,7 мг/мл) определяли при помощи УФ анализа и чистоту (78,3%) конечного целевого продукта ADC определяли методом эксклюзионной ВЭЖХ, с получением 5,3 мл (19,4 мг, 19,4%) раствора ADC (ADC2).
Пример 11
Получение конъюгата антитело-лекарственное средство ADC3 с Трастузумабом и Соединением 12
(a) Получение ADC3
К раствору частично восстановленного Трастузумаба, Соединение 20 (23,6 мл, 93,8 мг, 0,645 мкмоль), полученного, как описано в Примере 10(a) выше, добавляли DMSO (1,18 мл), с последующим добавлением свежеприготовленного раствора Соединения 12, полученного, как описано в Примере 3, (10 мМ в DMSO, 369 мкл, 3,69 мкмоль, 5,7 экв.). Лекарственное средство-линкер осторожно добавляли 10 порциями, несмотря на это, раствор начинал мутнеть после добавления третьей порции. Сильную мутность наблюдали при добавлении двух последних порций. Раствор не становился прозрачным вплоть до стадии фильтрования. Реакционную смесь для конъюгации перемешивали в течение 31 минуты при 20°C. Избыток лекарственного средства гасили путем добавления N-ацетилцистеина (NAC) (10 мМ, 369 мкл, 3,69 мкмоль) при перемешивании реакционной смеси в течение 50 минут. После гашения реакции конъюгации раствор фильтровали через 0,2 мкм шприцевой фильтр и концентрировали до 14,2 мг/мл при помощи Vivaspin центрифугирования. Затем смесь очищали на NAP-25 колонках. Осуществляли анализы методом эксклюзионной ВЭЖХ для определения чистоты продукта (34,2%).
ADC3 дополнительно очищали препаративной гель-фильтрационной хроматографией на системе очистки Äkta с использованием колонки HiLoad 16/600 superdex 200 из-за присутствия больших количеств агрегатов. После объединения конечную концентрацию (2,3 мг/мл) определяли при помощи УФ анализа и чистоту (78,6%) конечных лекарственных продуктов определяли методом эксклюзионной ВЭЖХ, с получением 7,3 мл (16,6 мг, 16,6%) раствора ADC (ADC3).
Пример 12
Получение конъюгатов антитело-лекарственное средство ADCs 4, 5 и 6 с Трастузумабом и Соединениями 13, 15 и 18, соответственно
(a) SMCC конъюгация с Трастузумабом (Соединение 21)
Буфер раствора Трастузумаба (262 мг, 1,8 мкмоль) заменяли фосфатным буфером (50 мМ фосфата, 2 мМ EDTA, pH 6,5) с использованием NAP-25 колонок. К объединенному раствору Трастузумаба в стеклянных реакторах (16-17 г/л) добавляли DMSO (5%). Конъюгацию линкера начинали путем добавления SMCC (20,0 мМ, 8,0 экв.) к раствору Трастузумаба. Реакционную смесь перемешивали при 18°C в течение 3 часов. Реакционную смесь затем очищали на NAP-25 колонках с получением соединения 21. Осуществляли обращенно-фазовый анализ Элмана с определением LAR 3,7.
(b) Конъюгация соединений 13, 15 и 18 с Трастузумабом-MCC: Получение ADC4, ADC5 и ADC6.
Для реакции конъюгации, на первой стадии раствор конъюгата антитела, Соединение 21, разбавляли фосфатным буфером (50 мМ фосфата, 2 мМ EDTA, pH 6,5) до концентрации 10 г/л. Затем DMSO (5%) добавляли к раствору Соединения 21. Реакции конъюгации с соединениями 13, 15 и 18 осуществляли путем медленного добавления лекарственного средства (10 мМ, 6,3-6,6 экв.) к раствору Соединения 21 и перемешивания в течение четырех часов при 18°C. После завершения реакции конъюгации, реакционные смеси фильтровали через 0,2 мкм фильтр и снова очищали на NAP-25 колонках с заменой буфера на 1 × PBS буфер. Осуществляли анализы методом эксклюзионной ВЭЖХ для определения чистоты продукта и концентрацию конечного продукта измеряли при помощи УФ анализа.
ADC из получения образца с соединением 15 выделяли с хорошей чистотой (74,5%) и выходом 56% (49 мг), и дополнительная очистка не требовалась. Конечную концентрацию (5,7 мг/мл) ADC5 раствора (87 мл) определяли при помощи УФ анализа.
Однако в двух получениях образцов с соединением 13 и 18, присутствовали низкомолекулярные соединения. Эти соединения имели очень схожее время удерживания с пиком продукта и трудно поддавались разделению на SEC колонке. Для удаления возможных остаточных количеств лекарственных средств, все еще присутствующих в растворе, растворы пропускали снова через NAP-25 колонки. Хроматограммы после первой и второй NAP-25 очистки были идентичными, поэтому соединения не образовывались из свободных лекарственных средств, все еще присутствующих в растворе, и должны быть более крупного происхождения. Образцы затем дополнительно очищали гель-фильтрационной хроматографией на эксклюзионной колонке.
После объединения, конечные концентрации (2,8 и 3,9 мг/мл) определяли при помощи УФ анализа и чистоту (62,3% и 50,9%) конечных лекарственных продуктов определяли методом эксклюзионной ВЭЖХ, с получением 6,7 мл (19,3 мг, 22,0%) ADC4 раствора и 6,8 мл (26,7 мг, 30,5%) ADC6 раствора, соответственно.
Пример 13
Получение конъюгатов антитело-лекарственное средство ADCs 7 и 8 с Трастузумабом и Соединениями 25 и 27
(a) Общие процедуры
Концентрацию антитела проверяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм с использованием коэффициента молярной экстинкции 2,18E5 M-1 см-1 и молекулярной массы 150 кДа. Буферы, используемые в этих способах, представляли собой либо буфер A (50 мМ фосфата натрия pH 6,5 с 2 мМ EDTA), либо буфер B (50 мМ фосфата натрия pH 8,0), либо фосфатно-солевой буферный раствор (“PBS”). Отношение лекарственного средства к антителу (“DAR”) was выводили из отношения линкера к антителу (“LAR”) в случае конъюгаци через Lys, или из отношения свободных Cys на моль антитела в случае Cys-ориентированной конъюгации, исходя из того, что реакция конъюгации лекарственного средства-линкера либо с малеимидным линкером, либо со свободным Cys является количественной. Оба определения были основаны на колориметрической реакции 5,5′-дитиобис(2-нитробензойной кислоты) (“DTNB”) со свободными тиольными группами с образованием окрашенного тионитробензоатного продукта присоединения. Для LAR определения, продукт присоединения был предварительно образован путем смешивания равных объемов 200 мкМ раствора DTNB в буфере B с 200 мкМ раствором N-ацетил-цистеина в таком же буфере. 75 мкл этой смеси затем смешивали с 75 мкл испытываемого образца и после 1-часовой инкубации поглощательную способность при 412 нм определяли спектрофотометрически и полученное значение сравнивали со значением, полученным из стандартной кривой, с использованием известных концентраций N-гидроксисукцинимидного эфира 4-(N-малеимидoметил)циклогексанкарбоновой кислоты (“SMCC”) с получением концентрации малеимидов в образце. Эту концентрацию затем соотносили с концентрацией антитела для расчета LAR. Подобным образом, свободные Cys определяли путем смешивания 50 мкл испытываемого образца с 150 мкл 133 мкМ DTNB в буфере B, отслеживая поглощательную способность при 412 нм и сравнивая полученное значение со значениями, полученными из стандартной кривой с использованием известных концентраций Cys: рассчитанную концентрацию свободных Cys в испытываемом образце затем соотносили с концентрацией антитела для расчета отношения.
(b) Получение конъюгатов антитело-лекарственное средство
Когда цитотоксическую полезную нагрузку конъюгировали с Cys остатками (как в случае соединения 25 для получения ADC7), антитело предварительно восстанавливали с использованием трис(2-карбоксиэтил)фосфин гидрохлорида (“TCEP”). Вкратце, 70 мкМ (10,5 мг/мл) раствора антитела в буфере B смешивали с подходящим количеством 5 мМ раствора TCEP в воде для поддержания восстановителя в 2,5-кратном избытке по отношению к антителу. Смесь инкубировали и перемешивали в течение 60 минут при 20°C и затем небольшую аликвоту полученного восстановленного антитела выделяли для расчета отношения свободных Cys к антителу, при этом оставшийся образец смешивали с подходящим объемом 10 мМ раствора Соединения 25 в DMSO для достижения 6-кратного избытка соединения по отношению к антителу: учитывая, что восстановленное антитело обычно представляет меньше чем 6 свободных Cys на молекулу белка, молярное отношение соединения к доступным свободным Cys никогда не бывает ниже 1. Добавляли DMSO, если необходимо, с поддержанием его концентрации при 5% (об./об.) и смесь инкубировали в течение 30 минут при 20°C. Затем добавляли N-ацетил-цистеин для гашения реакции с использованием подходящего объема 10 мМ раствора в воде для соответствия концентрации лекарственного средства-линкера. Полученный конъюгат в конце очищали от оставшихся реагентов гель-фильтрацией в Sephadex G-25 с использованием PD-10 колонок от GE Healthcare. Присутствие агрегатов проверяли при помощи аналитической эксклюзионной хроматографии с использованием Äkta FPLC системы, снабженной колонкой Superdex-100 10/300, изократным способом с использованием PBS при 1 мл/мин: если площадь пика, соответствующего агрегатам, превышала 10% от общей площади пиков, мономеры очищали с использованием такой же системы хроматографии с Superdex 200 16/600 препаративной колонкой, осуществляя такой способ, который описан выше. Конечную ADC концентрацию определяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм с использованием такого же коэффициента молярной экстинкции, как для исходного антитела: если ADC концентрация была ниже 2 мг/мл, осуществляли концентрирование с использованием Vivaspin устройств от GE Healthcare и снова определяли новую концентрацию, как указано выше.
Когда цитотоксическую полезную нагрузку конъюгировали с Lys остатками (как в случае соединения 27 для получения ADC8), антитело предварительно активировали при помощи SMCC. Вкратце, 70 мкМ (10,5 мг/мл) раствор антитела в буфере A смешивали с подходящим количеством 20 мМ раствор SMCC в DMSO с поддержанием активирующего реагента в 8-кратном избытке по отношению к антителу. Добавляли DMSO, если необходимо, для достижения конечной DMSO концентрации 5% (об./об.). Смесь инкубировали и перемешивали в течение 3 часов при 18°C и избыток SMCC затем удаляли гель-фильтрационной хроматографией на Sephadex G-25 с использованием PD-10 колонок от GE Healthcare. Небольшую аликвоту полученного активированного антитела выделяли для расчета LAR и оставшийся образец смешивали с подходящим объемом 10 мМ раствора Соединения 27 в DMSO с получением 8-кратного избытка соединения по отношению к антителу: учитывая, что LAR значение никогда не превышает 8, это гарантирует, что молярное отношение соединения к доступным участкам взаимодействия никогда не бывает ниже 1. Добавляли DMSO, если необходимо, с поддержанием его концентрации при 5% (об./об.). Смесь инкубировали в течение 4 часов при 18°C и полученный конъюгат очищали от оставшихся реагентов гель-фильтрацией в Sephadex G-25 с использованием PD-10 колонок от GE Healthcare. Присутствие агрегатов проверяли при помощи аналитической эксклюзионной хроматографии с использованием Äkta FPLC системы, снабженной колонкой Superdex-100 10/300, изократным способом с использованием PBS при 1 мл/мин: если площадь пика, соответствующего агрегатам, превышала 10% от общей площади пиков, мономеры очищали с использованием такой же системы хроматографии с Superdex 200 16/600 препаративной колонкой, осуществляя такой же способ, который описан выше. Конечную ADC концентрацию определяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм с использованием такого же коэффициента молярной экстинкции, как для исходного антитела: если ADC концентрация была ниже 2 мг/мл, осуществляли концентрирование с использованием Vivaspin устройств от GE Healthcare и снова определяли новую концентрацию, как указано выше.
Пример 14
Получение анти-CD4, анти-CD5 и анти-CD13 моноклональных антител
Анти-CD4, анти-CD5 и анти-CD13 моноклональные антитела получали, следуя хорошо известным процедурам, традиционно используемым в данной области. Вкратце, BALB/c мышей иммунизировали HPB-ALL клетками (для конечной продукции анти-CD4 антитела) или человеческими T-клетками, активированными смесью форбол 12-миристат 13-ацетата и коммерчески доступного анти-CD3 моноклонального антитела, как описано Cebrian et al. (1988, J. Exp. Med. 168:1621-1637) (для конечной продукции анти-CD5 антитела) или человеческими эндотелиальными клетками, выделенными из пуповины (для конечной продукции анти-CD13 антитела). Для этой цели, 1.5E7 соответствующих клеток вводили мышам путем инъекции интраперитонеально в дни −45 и −30 и внутривенно в день −3. В день 0 у этих животных удаляли селезенку и осуществляли слияние клеток селезенки с SP2 мышиными миеломными клетками при соотношении 4:1 в соответствии со стандартными процедурами с получением соответствующих гибридом и распределяли в 96-луночные планшеты для культур ткани (Costar Corp., Cambridge, MA). Через 2 недели супернатанты культуры гибридом собирали и их реактивность против клеточной линии, используемой на стадии иммунизации, испытывали методом проточной цитометрии. Положительные супернатанты анализировали методом иммунофлуоресценции, окрашивая соответствующие клетки, используемые в качестве антигенов. Гибридомы, показывающие специфическое окрашивание, картину иммунопреципитации и клеточную дистрибуцию, отбирали и клонировали и субклонировали методом серийных разведений.
После отбора клонов, клетки культивировали в RPMI-1640 среде, дополненной 10% (об./об.) фетальной телячьей сывороткой, 2 мМ глутамина, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина при 37°C в течение 3-4 дней, пока вреда не становилась бледно-желтой. В этот момент, две трети объема среды удаляли, центрифугировали при 1000×g в течение 10 минут для осаждения клеток и супернатант либо снова центрифугировали для дальнейшее очистки при 3000×g в течение 10 минут, либо фильтровали через мембраны с размером пор 22 мкм. Очищенный супернатант подвергали преципитации с использованием 55% насыщенного раствора сульфата аммония и полученный осадок ресуспендировали в 100 мМ Tris-HCl pH 7,8 (1 мл на 100 мл исходного очищенного супернатанта) и диализовали при 4°C в течение 16-24 часов против 5 л 100 мМ Tris-HCl pH 7,8 с 150 мМ NaCl, заменяя диализирующий раствор по меньшей мере три раза. Диализированное вещество в конце загружали на Protein A-Sepharose колонку и соответствующее моноклональное антитело элюировали при помощи 100 мМ цитрата натрия pH 3,0, или альтернативно при помощи 1M глицина pH 3,0. Те фракции, которые содержали антитело, нейтрализовали при помощи 2M Tris-HCl pH 9,0 и в завершение диализовали против PBS и хранили при -80°C до использования.
Пример 15
Получение конъюгатов антитело-лекарственное средство ADC 9, 10 и 11 с Анти-CD13 и Соединениями 1, 12 и 13
(a) Общие процедуры
Во всех способах, описанных в настоящей заявке, концентрацию антитела проверяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм, с использованием коэффициента молярной экстинкции 2,25E5 M-1 см-1 и молекулярной массы 150 кДа. Буферы, используемые в этих способах, представляли собой либо буфер A (50 мМ фосфата натрия pH 6,5 с 2 мМ EDTA), либо буфер B (50 мМ фосфата натрия pH 8,0), либо фосфатно-солевой буферный раствор (“PBS”). Отношение лекарственного средства к антителу (“DAR”) выводили из отношения линкера к антителу (“LAR”) в случае конъюгации через Lys, или из отношения свободных Cys на моль антитела в случае Cys-ориентированной конъюгации, исходя из того, что реакция конъюгации лекарственного средства-линкера либо с малеимидным линкером, либо со свободным Cys является количественной. Оба определения были основаны на колориметрической реакции 5,5′-дитиобис(2-нитробензойной кислоты) (“DTNB”) со свободными тиольными группами с образованием окрашенного тионитробензоатного продукта присоединения. Для LAR определения, продукт присоединения был предварительно образован путем смешивания равных объемов 200 мкМ раствора DTNB в буфере B с 200 мкМ раствором N-ацетил-цистеина в таком же буфере. 75 мкл этой смеси затем смешивали с 75 мкл испытываемого образца и после 1-часовой инкубации поглощательную способность при 412 нм определяли спектрофотометрически и полученное значение сравнивали со значением, полученным из стандартной кривой с использованием известных концентраций N-гидроксисукцинимидного эфира 4-(N-малеимидoметил)циклогексанкарбоновой кислоты (“SMCC”) с получением концентрации малеимидов в образце. Эту концентрацию затем соотносили с концентрацией антитела для расчета LAR. Подобным образом, свободные Cys определяли путем смешивания 50 мкл испытываемого образца с 150 мкл 133 мкМ DTNB в буфере B, отслеживая поглощательную способность при 412 нм и сравнивая полученное значение со значениями, полученными из стандартной кривой, с использованием известных концентраций Cys: рассчитанную концентрацию свободных Cys в испытываемом образце затем соотносили с концентрацией антитела для расчета отношения.
(b) Получение конъюгатов антитело-лекарственное средство
Когда цитотоксическую полезную нагрузку конъюгировали с Cys остатками (как в случае соединения 1 для получения ADC9 или Соединения 12 для получения ADC10), антитело предварительно восстанавливали с использованием Трис(2-карбоксиэтил)фосфин гидрохлорида (“TCEP”). Вкратце, 70 мкМ (10,5 мг/мл) раствор антитела в буфере B смешивали с подходящим количеством 5 мМ раствора TCEP в воде с поддержанием восстановителя в 2,5-кратном избытке по отношению к антителу. Смесь инкубировали и перемешивали в течение 60 минут при 20°C и затем небольшую аликвоту полученного восстановленного антитела выделяли для расчета отношения свободных Cys к антителу, при этом оставшийся образец смешивали с подходящим объемом 10 мМ раствора содержащего лекарственное средство линкера (Соединение 1 для ADC9 или Соединение 12 для ADC10) в DMSO для достижения 6-кратного избытка соединения по отношению к антителу: учитывая, что восстановленное антитело обычно представляет меньше чем 6 свободных Cys на молекулу белка, молярное отношение соединения к доступным свободным Cys никогда не бывает ниже 1. Добавляли DMSO, если необходимо, с поддержанием его концентрации при 5% (об./об.), и смесь инкубировали в течение 30 минут при 20°C. Затем добавляли N-ацетил-цистеин для гашения реакции, с использованием подходящего объема 10 мМ раствора в воде для соответствия концентрации лекарственного средства-линкера. Полученный конъюгат в конце очищали от оставшихся реагентов гель-фильтрацией в Sephadex G-25 с использованием PD-10 колонок от GE Healthcare. Присутствие агрегатов проверяли при помощи аналитической эксклюзионной хроматографии с использованием Äkta FPLC системы, снабженной колонкой Superdex-100 10/300, изократным способом с использованием PBS при 1 мл/мин: если площадь пика, соответствующего агрегатам, превышала 10% от общей площади пиков, мономеры очищали с использованием такой же системы хроматографии с Superdex 200 16/600 препаративной колонкой, осуществляя такой же способ, который описан выше. Конечную ADC концентрацию определяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм, с использованием такого же коэффициента молярной экстинкции, как для исходного антитела: если ADC концентрация была ниже 2 мг/мл, осуществляли концентрирование с использованием Vivaspin устройств от GE Healthcare и снова определяли новую концентрацию, как указано выше.
Когда цитотоксическую полезную нагрузку конъюгировали с Lys остатками (как в случае соединения 13 для получения ADC11), антитело предварительно активировали при помощи SMCC. Вкратце, 70 мкМ (10,5 мг/мл) раствор антитела в буфере A смешивали с подходящим количеством 20 мМ раствора SMCC в DMSO, с поддержанием активирующего реагента в 8-кратном избытке по отношению к антителу. Добавляли DMSO, если необходимо, для достижения конечной DMSO концентрации 5% (об./об.). Смесь инкубировали и перемешивали в течение 3 часов при 18°C и избыток SMCC затем удаляли гель-фильтрационной хроматографией на Sephadex G-25 с использованием PD-10 колонок от GE Healthcare. Небольшую аликвоту полученного активированного антитела выделяли для расчета LAR и оставшийся образец смешивали с подходящим объемом 10 мМ раствора Соединения 13 в DMSO с получением 8-кратного избытка соединения по отношению к антителу: учитывая, что LAR значение никогда не превышает 8, это гарантирует, что молярное отношение соединения к доступным участкам взаимодействия никогда не бывает ниже 1. Добавляли DMSO, если необходимо, с поддержанием его концентрации при 5% (об./об.). Смесь инкубировали в течение 4 часов при 18°C и полученный конъюгат очищали от оставшихся реагентов гель-фильтрацией в Sephadex G-25 с использованием PD-10 колонок от GE Healthcare. Присутствие агрегатов проверяли при помощи аналитической эксклюзионной хроматографии с использованием Äkta FPLC системы, снабженной колонкой Superdex-100 10/300, изократным способом с использованием PBS при 1 мл/мин: если площадь пика, соответствующего агрегатам, превышала 10% от общей площади пиков, мономеры очищали с использованием такой же системы хроматографии с Superdex 200 16/600 препаративной колонкой, осуществляя такой же способ, который описан выше. Конечную ADC концентрацию определяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм, с использованием такого же коэффициента молярной экстинкции, как для исходного антитела: если ADC концентрация была ниже 2 мг/мл, осуществляли концентрирование с использованием Vivaspin устройств от GE Healthcare и снова определяли новую концентрацию, как указано выше.
Пример 16
Получение конъюгатов антитело-лекарственное средство ADCs 12 и 13 с Ритуксимабом и Соединениями 1 и 12
(a) Общие процедуры
Концентрацию антитела проверяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм, с использованием коэффициента молярной экстинкции 2,08E5 M-1 см-1 и молекулярной массы 150 кДа. Буферы, используемые в этих способах, представляли собой либо буфер B (50 мМ фосфата натрия pH 8,0), либо фосфатно-солевой буферный раствор (“PBS”). Отношение лекарственного средства к антителу (“DAR”) выводили из отношения свободных Cys на моль антитела, исходя из того, что реакция конъюгации лекарственного средства-линкера со свободными Cys является количественной. Определение было основано на колориметрической реакции 5,5′-дитиобис(2-нитробензойной кислоты) (“DTNB”) со свободными тиольными группами с образованием окрашенного тионитробензоатного продукта присоединения. 50 мкл испытываемого образца смешивали с 150 мкл 133 мкМ DTNB в буфере B, затем измеряли поглощательную способность при 412 нм и полученное значение сравнивали со значениями, полученными из стандартной кривой, с использованием известных концентраций Cys. Рассчитанную концентрацию свободных Cys в испытываемом образце затем соотносили с концентрацией антитела для расчета отношения.
(b) Получение конъюгатов антитело-лекарственное средство
Перед конъюгацией с лекарственным средством-линкерами через Cys, антитело восстанавливали с использованием трис(2-карбоксиэтил)фосфин гидрохлорида (“TCEP”). Вкратце, 70 мкМ (10,5 мг/мл) раствор антитела в буфере B смешивали с подходящим количеством 5 мМ раствора TCEP в воде, с поддержанием восстановителя в 2,5-кратном избытке по отношению к антителу. Смесь инкубировали и перемешивали в течение 60 минут при 20°C и затем небольшую аликвоту полученного восстановленного антитела выделяли для расчета отношения свободных Cys к антителу, при этом оставшийся образец смешивали с подходящим объемом 10 мМ раствора лекарственного средства-линкера (Соединение 1 для ADC12 или Соединение 12 для ADC13) в DMSO для достижения 6-кратного избытка соединения по отношению к антителу: учитывая, что восстановленное антитело обычно представляет меньше чем 6 свободных Cys на молекулу белка, молярное отношение соединения к доступным свободным Cys никогда не бывает ниже 1. Добавляли DMSO, если необходимо, с поддержанием его концентрации при 5% (об./об.), и смесь инкубировали в течение 30 минут при 20°C. Затем добавляли N-ацетил-цистеин для гашения реакции, с использованием подходящего объема 10 мМ раствора в воде для соответствия концентрации лекарственного средства-линкер. Полученный конъюгат в конце очищали от оставшихся реагентов гель-фильтрацией в Sephadex G-25 с использованием PD-10 колонок от GE Healthcare. Присутствие агрегатов проверяли при помощи аналитической эксклюзионной хроматографии с использованием Äkta FPLC системы, снабженной колонкой Superdex-100 10/300, изократным способом с использованием PBS при 1 мл/мин: если площадь пика, соответствующего агрегатам, превышала 10% от общей площади пиков, мономеры очищали с использованием такой же системы хроматографии с Superdex 200 16/600 препаративной колонкой, осуществляя такой же способ, который описан выше. Конечную ADC концентрацию определяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм, с использованием такого же коэффициента молярной экстинкции, как для исходного антитела: если ADC концентрация была ниже 1 мг/мл, осуществляли концентрирование с использованием Vivaspin устройств от GE Healthcare и снова определяли новую концентрацию, как указано выше.
Пример 17
Получение конъюгатов антитело-лекарственное средство ADCs 14 и 15 с Анти-CD5 и Соединениями 1 и 12
(a) Общие процедуры
Концентрацию антитела проверяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм, с использованием коэффициента молярной экстинкции 2,25E5 M-1 см-1 и молекулярной массы 150 кДа. Буферы, используемые в этих способах, представляли собой либо буфер B (50 мМ фосфата натрия pH 8,0), либо фосфатно-солевой буферный раствор (“PBS”). Отношение лекарственного средства к антителу (“DAR”) выводили из отношения свободных Cys на моль антитела, исходя из того, что реакция конъюгации лекарственного средства-линкера со свободными Cys является количественной. Определение было основано на колориметрической реакции 5,5′-дитиобис(2-нитробензойной кислоты) (“DTNB”) со свободными тиольными группами с образованием окрашенного тионитробензоатного продукта присоединения. 50 мкл испытываемого образца смешивали с 150 мкл 33 мкМ DTNB в буфере B, поглощательную способность при 412 нм затем измеряли и полученное значение сравнивали со значениями, полученными из стандартной кривой, с использованием известных концентраций Cys. Рассчитанную концентрацию свободных Cys в испытываемом образце затем соотносили с концентрацией антитела для расчета отношения.
(b) Получение конъюгатов антитело-лекарственное средство
Перед конъюгацией с лекарственным средством-линкерами через Cys, антитело восстанавливали с использованием трис(2-карбоксиэтил)фосфин гидрохлорида (“TCEP”). Вкратце, 70 мкМ (10,5 мг/мл) раствор антитела в буфере B смешивали с подходящим количеством 5 мМ раствора TCEP в воде, с поддержанием восстановителя в 2,5-кратном избытке по отношению к антителу. Смесь инкубировали и перемешивали в течение 60 минут при 20°C и затем небольшую аликвоту полученного восстановленного антитела выделяли для расчета отношения свободных Cys к антителу, при этом оставшийся образец смешивали с подходящим объемом 10 мМ раствора лекарственного средства-линкер (Соединение 1 для ADC14 или Соединение 12 для ADC15) в DMSO для достижения 6-кратного избытка соединения по отношению к антителу: учитывая, что восстановленное антитело обычно представляет меньше чем 6 свободных Cys на молекулу белка, молярное отношение соединения к доступным свободным Cys никогда не бывает ниже 1. Добавляли DMSO, если необходимо, с поддержанием его концентрации при 5% (об./об.), и смесь инкубировали в течение 30 минут при 20°C. Затем добавляли N-ацетил-цистеин для гашения реакции, с использованием подходящего объема 10 мМ раствора в воде для соответствия концентрации лекарственного средства-линкер. Полученный конъюгат в конце очищали от оставшихся реагентов гель-фильтрацией в Sephadex G-25 с использованием PD-10 колонок от GE Healthcare. Присутствие агрегатов проверяли при помощи аналитической эксклюзионной хроматографии с использованием Äkta FPLC системы, снабженной колонкой Superdex-100 10/300, изократным способом с использованием PBS при 1 мл/мин: если площадь пика, соответствующего агрегатам, превышала 10% от общей площади пиков, мономеры очищали с использованием такой же системы хроматографии с Superdex 200 16/600 препаративной колонкой, осуществляя такой же способ, который описан выше. Конечную ADC концентрацию определяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм с использованием такого же коэффициента молярной экстинкции, как для исходного антитела: если ADC концентрация была ниже 1 мг/мл, осуществляли концентрирование с использованием Vivaspin устройств от GE Healthcare и снова определяли новую концентрацию, как указано выше.
Пример 18
Получение конъюгатов антитело-лекарственное средство ADCs 16 и 17 с Анти-CD4 и Соединениями 1 и 12
(a) Общие процедуры
Концентрацию антитела проверяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм, с использованием коэффициента молярной экстинкции 2,25E5 M-1 см-1 и молекулярной массы 150 кДа. Буферы, используемые в этих способах, представляли собой либо буфер B (50 мМ фосфата натрия pH 8,0), либо фосфатно-солевой буферный раствор (“PBS”). Отношение лекарственного средства к антителу (“DAR”) выводили из отношения свободных Cys на моль антитела, исходя из того, что реакция конъюгации лекарственного средства-линкера со свободными Cys является количественной. Определение было основано на колориметрической реакции 5,5′-дитиобис(2-нитробензойной кислоты) (“DTNB”) со свободными тиольными группами с образованием окрашенного тионитробензоатного продукта присоединения. 50 мкл испытываемого образца смешивали с 150 мкл 133 мкМ DTNB в буфере B, затем измеряли поглощательную способность при 412 нм и полученное значение сравнивали со значениями, полученными из стандартной кривой, с использованием известных концентраций Cys. Рассчитанную концентрацию свободных Cys в испытываемом образце затем соотносили с концентрацией антитела для расчета отношения.
(b) Получение конъюгатов антитело-лекарственное средство
Перед конъюгацией с лекарственным средством-линкерами через Cys, антитело восстанавливали с использованием трис(2-карбоксиэтил)фосфин гидрохлорида (“TCEP”). Вкратце, 70 мкМ (10,5 мг/мл) раствор антитела в буфере B смешивали с подходящим количеством 5 мМ раствора TCEP в воде, с поддержанием восстановителя в 2,5-кратном избытке по отношению к антителу. Смесь инкубировали и перемешивали в течение 60 минут при 20°C и затем небольшую аликвоту полученного восстановленного антитела выделяли для расчета отношения свободных Cys к антителу, при этом оставшийся образец смешивали с подходящим объемом 10 мМ раствора лекарственного средства-линкера (Соединение 1 для ADC16 или Соединение 12 для ADC17) в DMSO для достижения 6-кратного избытка соединения по отношению к антителу: учитывая, что восстановленное антитело обычно представляет меньше чем 6 свободных Cys на молекулу белка, молярное отношение соединения к доступным свободным Cys никогда не бывает ниже 1. Добавляли DMSO, если необходимо, с поддержанием его концентрации при 5% (об./об.), и смесь инкубировали в течение 30 минут при 20°C. Затем добавляли N-ацетил-цистеин для гашения реакции, с использованием подходящего объема 10 мМ раствора в воде для соответствия концентрации лекарственного средства-линкера. Полученный конъюгат в конце очищали от оставшихся реагентов гель-фильтрацией в Sephadex G-25 с использованием PD-10 колонок от GE Healthcare. Присутствие агрегатов проверяли при помощи аналитической эксклюзионной хроматографии с использованием Äkta FPLC системы, снабженной колонкой Superdex-100 10/300, изократным способом с использованием PBS при 1 мл/мин: если площадь пика, соответствующего агрегатам, превышала 10% от общей площади пиков, мономеры очищали с использованием такой же системы хроматографии с Superdex 200 16/600 препаративной колонкой, осуществляя такой же способ, который описан выше. Конечную ADC концентрацию определяли спектрофотометрически, осуществляя контроль его поглощательной способности при 280 нм, с использованием такого же коэффициента молярной экстинкции, как для исходного антитела: если ADC концентрация была ниже 1 мг/мл, осуществляли концентрирование с использованием Vivaspin устройств от GE Healthcare и снова определяли новую концентрацию, как указано выше.
Пример 19 Синтез соединения формулы D-X-(AA)w-L1
Получение Соединения 28
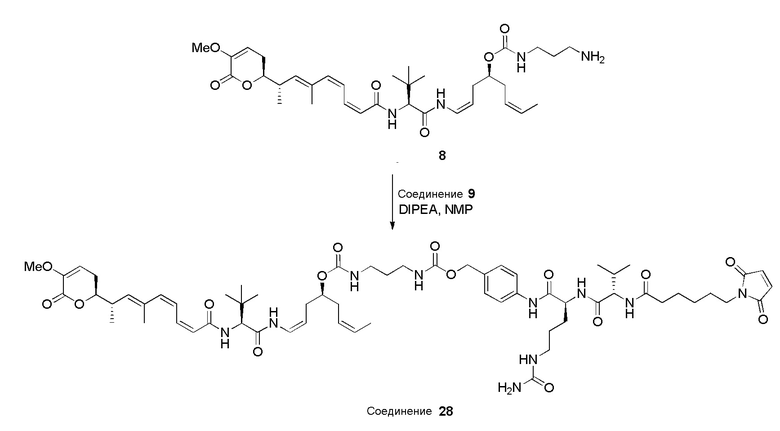
(a) Получение Соединения 28
DIPEA (10 мкл, 0,06 ммоль) добавляли к раствору Соединения 9 (13 мг, 0,02 ммоль), полученного, как показано в Примере получения выше, и Соединения 8 (20 мг, 0,02 ммоль), полученного, как описано в Примере 2 выше, в NMP (6,5 мл) при 23°C. Через 9 часов реакционную смесь разбавляли при помощи H2O и экстрагировали при помощи EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, DCM:CH3OH, от 100:0 до 90:10) с получением чистого целевого Соединения 28 (9 мг, 38%).
1H ЯМР (400 МГц, CDCl3/CD3OD): δ 9,40 (с, 1H), 8,92 (д, J=10,6 Гц, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,47 (д, J=8,0 Гц, 2H), 7,21 (д, J=7,9 Гц, 2H), 7,16 (т, J=11,9 Гц, 1H), 6,97 (т, J=9,6 Гц, 2H), 6,82 (т, J=11,4 Гц, 1H), 6,67-6,64 (м, 1H), 6,64 (с, 2H), 6,08 (д, J=11,7 Гц, 1H), 5,68 (д, J=11,4 Гц, 1H), 5,62-5,58 (м, 1H), 5,54-5,47 (м, 1H), 5,35-5,29 (м, 1H), 5,20 (д, J=9,9 Гц, 1H), 4,94 (с, 2H), 4,77 (кв., J=8,1 Гц, 1H), 4,54-4,45 (м, 2H), 4,37 (д, J=9,2 Гц, 1H), 4,20-4,12 (м, 1H), 4,08 (т, J=7,8 Гц, 1H), 3,58 (с, 3H), 3,42 (т, J=7,2 Гц, 2H), 3,18- 3,00 (м, 7H), 2,81-2,75 (м, 1H), 2,35-2,30 (м, 3H), 2,29-2,25 (м, 3H), 2,17 (т, J=7,2 Гц, 2H), 2,14-2,06 (м, 1H), 2,04-1,92 (м, 1H), 1,86-1,74 (м, 1H), 1,76 (с, 3H), 1,61-1,42 (м, 10H), 1,54 (д, J=6,3 Гц, 3H), 1,30-1,14 (м, 4H), 1,08 (д, J=6,4Гц, 3H), 0,94 (с, 9H), 0,86 (дд, J=6,8, 4,3 Гц, 6H).
13C ЯМР (100 МГц, CDCl3): δ 173,0, 172,1, 171,0, 170,2, 168,5, 167,1, 162,0, 161,3, 157,2, 157,0, 145,0, 140,2, 137,7, 137,5, 137,1, 134,0, 132,4, 128,8, 126,9, 124,9, 124,4, 124,3, 124,0, 120,5, 119,8, 108,6, 107,3. 81,9, 74,8, 66,1, 60,4, 58,8, 55,4, 37,6, 37,2, 36,0, 34,8, 31,9, 31,6, 30,9, 30,7, 30,0, 29,7, 29,3, 28,1, 26,5, 26,2, 26,1, 25,1, 19,2, 18,3, 17,1, 16,5, 12,9.
ESI-MS m/z: Рассчитано для C63H90N10O15: 1226,7. Найдено: 1267,4 (M+H)+.
Пример 20 Синтез соединения формулы D-X-(AA)w-H
Получение Соединения 36
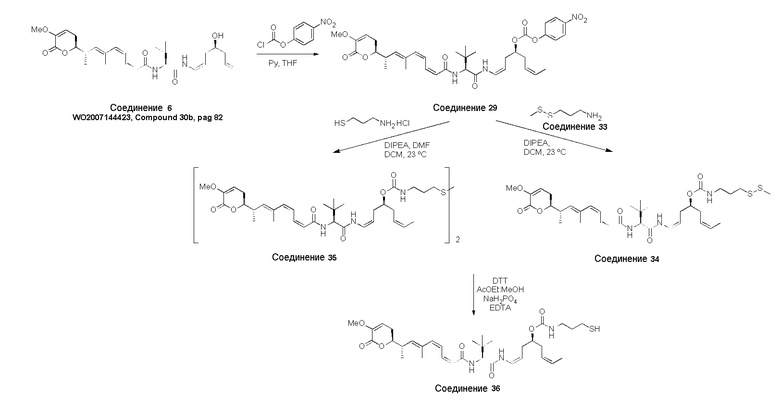
(a) Получение Соединения 29
К раствору Соединения 6 (1,01 г, 1,91 ммоль) (Соединение 30b, полученное, как описано в WO 2007144423, содержание которого включено в настоящую заявку посредством ссылки) в DCM (40 мл) добавляли пиридин (0,31 мл, 3,82 ммоль) и 4-нитрофенил хлорформиат (769,7 мг, 3,82 ммоль) при 0°C. Реакционную смесь перемешивали при 23°C в течение 1,5 часов, разбавляли лимонной кислотой 10% и экстрагировали при помощи DCM. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc смеси) с получением чистого Соединения 29 (783 мг, 59%).
1H ЯМР (400 МГц, CDCl3): δ 8,26 (д, J=9,2 Гц, 2H), 8,02 (д, J=10,9 Гц, 1H), 7,43 (д, J=9,2 Гц, 2H), 7,22 (т, J=9,2 Гц, 1H), 6,92-6,76 (м, 2H), 6,21 (д, J=9,3 Гц, 1H), 6,17-6,12 (м, 1H), 5,73-5,64 (м, 1H), 5,65-5,56 (м, 2H), 5,46-5,38 (м, 1H), 5,27 (д, J=9,9 Гц, 1H), 4,86 (кв., J=8,2 Гц, 1H), 4,76 (п, J=6,2 Гц, 1H), 4,40 (д, J=9,3 Гц, 1H), 4,21 (ддд, J=10,7, 7,6, 4,9 Гц, 1H), 3,66 (с, 3H), 2,89-2,79 (м, 1H), 2,59-2,29 (м, 6H), 1,83 (д, J=1,3 Гц, 3H), 1,61 (с, 3H), 1,16 (д, J=6,7 Гц, 3H), 1,00 (с, 9H).
(b) Получение Соединения 33

Получение Соединения 30
К раствору 3-бромпропиламин гидробромида (1,22 г, 2,98 ммоль) в CH2Cl2 (30 мл) добавляли 4-метокситрифенилметил хлорид (5,89 г, 19,1 ммоль) и DIPEA (6,3, мл, 36,38 ммоль). Реакционную смесь перемешивали при 23°C в течение ночи, разбавляли при помощи H2O и экстрагировали при помощи CH2Cl2. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc смесь) с получением чистого Соединения 30 (8,16 г, 100%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 7,50-7,42 (м, 4H), 7,39-7,33 (м, 2H), 7,29-7,22 (м, 4H), 7,22-7,15 (м, 2H), 6,81 (д, J=8,9 Гц, 2H), 3,78 (с, 3H), 3,56 (т, J=6,8 Гц, 2H), 2,26 (т, J=6,7 Гц, 2H), 2,07-1,96 (м, 2H).
Получение Соединения 31
К раствору Соединения 30 (1,49 г, 3,63 ммоль) в этаноле (36 мл) добавляли этилксантогенат калия (1,46 г, 9,08 ммоль). Реакционную смесь перемешивали при 23°C в течение ночи и осажденный бромид калия затем фильтровали из раствора. Фильтрат упаривали при пониженном давлении и затем твердый остаток растирали в порошок с гексаном. Образовавшееся твердое вещество удаляли фильтрованием и фильтрат упаривали и очищали флэш-хроматографией (SiO2, Hex/EtOAc смеси) с получением Соединения 31 (1,31 г, 80%).
1H ЯМР (400 МГц, CDCl3): δ 7,50-7,42 (м, 4H), 7,39-7,33 (м, 2H), 7,29-7,22 (м, 4H), 7,22-7,15 (м, 2H), 6,81 (д, J=8,9 Гц, 2H), 4,62 (кв., J=7,1 Гц, 2H), 3,78 (с, 3H), 3,32-3,20 (дд, J=7,7, 6,9 Гц, 2H), 2,23 (т, J=6,7 Гц, 2H), 1,91-1,80 (м, 2H), 1,41 (т, J=7,1 Гц, 3H).
Получение Соединения 32
К раствору Соединения 31 (3 г, 6,64 ммоль) в DCM (10 мл) добавляли 1-пропиламин (4,4 мл, 66,4 ммоль). Реакционную смесь перемешивали при 23°C в течение 10 минут и концентрировали в вакууме. Полученный остаток использовали на следующей стадии без дополнительной очистки. Его растворяли в безводном метаноле (50 мл) и охлаждали до 0°C. Добавляли метилметантиосульфонат (9,7 мл, 7,97 ммоль) и раствор перемешивали в течение 16 часов при 23°C. Растворитель удаляли в вакууме, остаточное масло растворяли в дихлорметане, промывали pH 7 буфером и насыщенным солевым раствором, сушили и упаривали. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc смеси) с получением Соединения 32 (1,6 г, 59%).
1H ЯМР (400 МГц, CDCl3): δ 7,52-7,44 (м, 4H), 7,38 (д, J=8,9 Гц, 2H), 7,32-7,22 (м, 4H), 7,19 (д, J=7,3 Гц, 2H), 6,82 (д, J=8,9 Гц, 2H), 3,79 (с, 3H), 2,90-2,72 (м, 2H), 2,39 (с, 3H), 2,24 (т, J=6,7 Гц, 2H), 1,88 (п, J=6,9 Гц, 2H).
Получение Соединения 33
К раствору Соединения 32(1,22 г, 2,98 ммоль) в CH2Cl2 (30 мл) добавляли дихлоруксусную кислоту (0,9 мл, 10,9 ммоль). Реакционную смесь перемешивали при 23°C в течение 20 минут и разбавляли водой. Органический слой экстрагировали и водную фазу подщелачивали при помощи KOH 10%. Затем смесь тщательно экстрагировали дихлорметаном (3×), и объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали с получением Соединения 33 (409 мг, 100%).
1H ЯМР (400 МГц, CDCl3): δ 2,83-2,75 (м, 2H), 2,41 (с, 3H), 1,88-1,81 (м, 2H).
(c) Получение Соединения 34
К раствору Соединения 29 (230,2 мг, 0,33 ммоль), полученного, как описано на стадии (a) выше, в CH2Cl2 (10 мл) добавляли Соединение 33 (50 мг, 0,36 ммоль) и DIPEA (0,06 мл, 0,36 ммоль). Реакционную смесь перемешивали при 23°C в течение 3 часов, разбавляли при помощи H2O и экстрагировали при помощи CH2Cl2. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc смеси) с получением чистого Соединения 34 (150 мг, 66%).
1H ЯМР (400 МГц, CDCl3): δ 8,68 (д, J=10,7 Гц, 1H), 7,31 (т, J=6,0 Гц, 1H), 6,90 (дд, J=12,7, 10,2 Гц, 1H), 6,81 (дд, J=10,9, 8,5 Гц, 1H), 6,38 (д, J=9,4 Гц, 1H), 6,16 (д, J=11,7 Гц, 1H), 5,69 (д, J=11,5 Гц, 1H), 5,65-5,52 (м, 3H), 5,41-5,37 (м, 1H), 5,29-5,56 (д, J=8,9 Гц, 1H), 4,83 (кв., J=8,3 Гц, 1H), 4,54-4,50 (м, 1H), 4,46 (д, J=9,4 Гц, 1H), 4,24-4,19 (м, 1H), 3,65 (с, 3H), 3,36-3,16 (м, 2H), 2,86-2,82 (м, 1H), 2,70 (т, J=7,2 Гц, 2H), 2,44-2,35 (м, 5H), 2,40 (с, 3H), 2,19-2,04 (м, 1H), 1,98-1,83 (м, 2H), 1,84 (с, 3H), 1,63 (д, J=6,6 Гц, 3H), 1,16 (д, J=6,6 Гц, 3H), 1,05 (с, 9H).
ESI-MS m/z: Рассчитано для C35H53N3O7S2: 691,33. Найдено: 692,4 (M+H)+.
(d) Получение Соединения 35
К раствору Соединения 29 (121,3 мг, 0,17 ммоль), полученного, как описано для стадии (a) выше, в DCM (2,5 мл) добавляли суспензию 3-аминопропан-1-тиол гидрохлорида (37,2 мг, 0,29 ммоль) в DCM (2,5 мл), DIPEA (59 мкл, 0,34 ммоль) и DMF (0,1 мл) при 23°C. Реакционную смесь перемешивали при 23°C в течение 7 часов, разбавляли при помощи H2O и экстрагировали при помощи EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc смеси) с получением чистого Соединения 35 (40,5 мг, 37%).
1H ЯМР (300 МГц, CDCl3): δ 8,63 (д, J=10,6 Гц, 1H), 7,34- 7,21 (м, 1H), 6,88 (т, J=11,4 Гц, 1H), 6,76 (т, J=9,6 Гц, 1H), 6,68 (д, J=9,3 Гц, 1H), 6,13 (д, J=11,6 Гц, 1H), 5,7-5,67 (м, 3H), 5,66-5,48 (м, 3H), 4,81 (кв., J=8,1 Гц, 1H), 4,66-4,50 (м, 1H), 4,46 (д, J=9,2 Гц, 1H), 4,25-4,18 (м, 1H), 3,64 (с, 3H), 3,27-3,34 (м, 1H), 2,88-2,77 (м, 1H), 2,66 (т, J=7,2 Гц, 2H), 2,42-2,30 (м, 5H), 2,22-2,06 (м, 2H), 1,89-1,74 (м, 5H), 1,62 (д, J=6,6 Гц, 3H), 1,15 (д, J=6,7 Гц, 3H), 1,05 (с, 9H).
ESI-MS m/z: Рассчитано для C68H100N6O14S2: 1288,67. Найдено: 1289,4(M+H)+.
(e) Получение Соединения 36
Раствор Соединения 35 (40,5 мг, 0,03 ммоль) в смеси EtOAc (1,5 мл) и CH3OH (1,5 мл) обрабатывали раствором дитиотреитола (0,36 мл, 0,36 ммоль) в 0,05 M калийфосфатном буфере (1,2 мл) при pH 7,5, содержащем 2 мМ этилендиаминтетрауксусной кислоты (EDTA). Смесь перемешивали при 23°C в течение 4 часов. Реакционную смесь обрабатывали раствором 0,2 M калийфосфатного буфера при pH 6,0, содержащего 2 мМ EDTA, и экстрагировали при помощи EtOAc (×3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали в системе для ВЭЖХ (SiO2, Hex:EtOAc смеси) с получением Соединения 36 (15 мг, 38%).
1H ЯМР (300 МГц, CDCl3): δ 8,70 (д, J=10,7 Гц, 1H), 7,35- 7,21 (м, 1H), 6,89 (т, J=11,4 Гц, 1H), 6,80 (т, J=9,8 Гц, 1H), 6,42 (д, J=9,4 Гц, 1H), 6,16 (д, J=11,3 Гц, 1H), 5,75-5,48 (м, 3H), 5,52-5,13 (м, 3H), 4,83 (кв., J=8,3 Гц, 1H), 4,65-4,38 (м, 2H), 4,32-4,16 (м, 1H), 3,65 (с, 3H), 3,29 (кв., J=6,6 Гц, 2H), 2,85 (дт, J=9,8, 6,9 Гц, 1H), 2,54 (кв., J=7,3 Гц, 2H), 2,48-2,26 (м, 5H), 2,19-2,01 (м, 1H), 1,89-1,74 (м, 5H), 1,63 (дд, J=6,8, 1,7 Гц, 3H), 1,16 (д, J=6,6 Гц, 3H), 1,05 (с, 9H).
ESI-MS m/z: Рассчитано для C34H51N3O7S: 645,85. Найдено: 668,4 (M+Na)+.
Пример 21. Альтернативный синтез Соединения 14

Получение Соединения 37
К раствору N-Fmoc-1,3-пропандиамин гидробромида (377 мг, 1 ммоль) в CH2Cl2 (15 мл) добавляли DIPEA (0,52 мл, 3 ммоль) и N-гидроксисукцинимидный эфир 6-малеимидогексановой кислоты (323,7 мг, 1,1 ммоль). Реакционную смесь перемешивали при 23°C в течение ночи и концентрировали в вакууме. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc смеси) с получением чистого Соединения 37 (430 мг, 100%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 7,76 (дд, J=7,6, 1,0 Гц, 2H), 7,64-7,55 (м, 2H), 7,45-7,37 (м, 2H), 7,31 (тд, J=7,5, 1,2 Гц, 2H), 6,16 (bs, 1H), 5,24 (bs, 1H), 4,42 (д, J=6,9 Гц, 2H), 4,21 (т, J=6,8 Гц, 1H), 3,27 (дкв., J=18,3, 6,3 Гц, 4H), 2,99 (т, J=7,0 Гц, 2H), 2,62 (т, J=7,0 Гц, 2H), 2,41 (с, 3H), 1,71-1,59 (м, 2H).
Получение Соединения 38
К раствору Соединения 37 (430 мг, 1 ммоль), полученного, как описано для стадии (a) выше, в CH2Cl2 (8 мл) добавляли диэтиламин (1,4 мл, 13,5 ммоль). Реакционную смесь перемешивали при 23°C в течение 6 часов и концентрировали в вакууме. Полученный остаток растирали в порошок с Et2O и фильтровали с получением Соединения 38 (148 мг, 71%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,29 (тд, J=6,5, 2,3 Гц, 2H), 3,01-2,86 (м, 2H), 2,78 (т, J=6,6 Гц, 2H), 2,57 (т, J=7,1 Гц, 2H), 2,37 (с, 3H), 1,69 (п, J=6,6 Гц, 2H).
Получение Соединения 14
К раствору Соединение 16 (60 мг, 0,08 ммоль), полученного, как описано в Примере 5(a) выше, в CH2Cl2 (2 мл) добавляли раствор Соединения 38 (58 мг, 0,28 ммоль), полученного, как описано для стадии (b) выше, и DIPEA (0,1 мл, 0,56 ммоль) в CH2Cl2 (2 мл). Реакционную смесь перемешивали при 23°C в течение 3 часов, разбавляли при помощи H2O и экстрагировали при помощи CH2Cl2. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc смеси) с получением чистого Соединения 14 (60,5 мг, 95%).
1H ЯМР (500 МГц, CDCl3): δ 8,88 (д, J=10,8 Гц, 1H), 7,29-7,24 (м, 1H), 6,90 (т, J=11,5 Гц, 1H), 6,82 (т J=9,1 Гц, 1H), 6,63 (т, J=6,1 Гц, 1H), 6,49 (д, J=9,4 Гц, 1H), 6,16 (дд, J=11,5, 1,5 Гц, 1H), 5,70 (д, J=11,5 Гц, 1H), 5,68-5,51 (м, 3H), 5,29 (д, J=9,7 Гц, 1H), 4,81 (кв., J=8,2 Гц, 1H), 4,52 (д, J=9,5 Гц, 1H), 4,52-4,43 (м, 1H), 4,24 (ддд, J=11,5, 7,3, 4,3 Гц, 1H), 3,66 (с, 3H), 3,37-3,21 (м, 3H), 3,21-3,12 (м, 1H), 2,97 (т, J=7,2 Гц, 2H), 2,90-2,81 (м, 1H), 2,60 (т, J=7,2 Гц, 2H), 2,49-2,35 (м, 3H), 2,39 (с, 3H), 2,33 (т, J=7,0 Гц, 2H), 2,14-2,07 (м, 1H), 2,07 (с, 3H), 1,84 (с, 3H), 1,73-1,64 (м, 2H), 1,16 (д, J=6,7 Гц, 3H), 1,05 (с, 9H).
13C ЯМР (125 МГц, CDCl3): δ 171,6, 168,2, 166,4, 161,6, 157,2, 145,2, 140,3, 137,4, 134,2, 134,0, 131,9, 124,4, 124,1, 122,4, 120,7, 108,3, 105,6, 81,8, 74,8, 60,6, 60,4, 55,5, 37,8, 37,2, 36,2, 35,6, 34,7, 33,1, 31,0, 29,8, 26,7, 26,2, 23,0, 21,0, 17,2, 16,6.
Пример 22. Альтернативный синтез Соединения 15
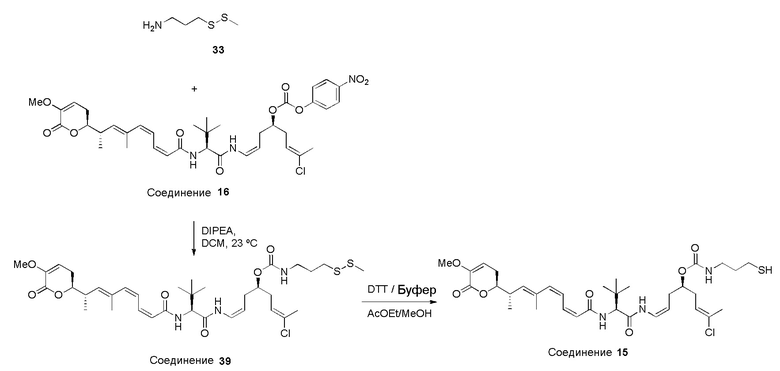
(a) Получение Соединения 39
К раствору Соединения 16 (178,7 мг, 0,25 ммоль), полученного, как описано в Примере 5(a) выше, в CH2Cl2 (2,5 мл) добавляли Соединение 33 (120 мг, 0,88 ммоль), полученное, как описано в Примере 20(b) выше, и DIPEA (0,05 мл, 0,25 ммоль). Реакционную смесь перемешивали при 23°C в течение 2 часов, разбавляли при помощи H2O и экстрагировали при помощи CH2Cl2. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc смеси) с получением чистого Соединения 39 (100 мг, 56%).
1H ЯМР (400 МГц, CDCl3): δ 8,65 (д, J=10,7 Гц, 1H), 7,28 (т, J=11,6 Гц, 1H), 6,90 (т, J=11,5 Гц, 1H), 6,82 (т, J=9,6 Гц, 1H), 6,37 (д, J=9,4 Гц, 1H), 6,17 (д, J=11,7 Гц, 1H), 5,69 (д, J=11,4 Гц, 1H), 5,64-5,57 (м, 2H), 5,34 (т, J=6,2 Гц, 1H), 5,29 (д, J=10,0 Гц, 1H), 4,81 (кв., J=8,3 Гц, 1H), 4,54-4,49 (м, 1H), 4,45 (д, J=9,4 Гц, 1H), 4,28-4,17 (м, 1H), 3,66 (с, 3H), 3,37-3,21 (м, 2H), 2,85 (дт, J=9,7, 6,9 Гц, 1H), 2,71 (т, J=7,2 Гц, 2H), 2,44-2,30 (м, 5H), 2,38 (с, 3H) 2,13-2,06 (м, 1H), 2,07 (с, 3H), 1,93 (п, J=6,9 Гц, 2H), 1,84 (с, 3H), 1,16 (д, J=6,6 Гц, 3H), 1,06 (с, 9H).
ESI-MS m/z: Рассчитано для C35H52ClN3O7S2: 725,9. Найдено: 748,3 (M+Na)+.
(b) Получение Соединения 15
Раствор Соединения 39 (90 мг, 0,12 ммоль), полученного, как описано в Примере 5(a) выше, в смеси EtOAc (6,7 мл) и CH3OH (6,7 мл) обрабатывали раствором дитиотреитола (0,36 мл, 0,36 ммоль) в 0,05 M калийфосфатном буфере (6,7 мл) при pH 7,5, содержащем 2 мМ этилендиаминтетрауксусной кислоты (EDTA). Смесь перемешивали при 23°C в течение 4 часов. Реакционную смесь обрабатывали раствором 0,2 M калийфосфатного буфера при pH 6,0, содержащего 2 мМ EDTA, и экстрагировали при помощи EtOAc (×3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали в системе для флэш-хроматографии (SiO2, Hex:EtOAc смеси) с получением чистого целевого Соединения 15 (40,2 мг, 46%).
1H ЯМР (500 МГц, CDCl3): δ 8,66 (д, J=10,7 Гц, 1H), 7,29 (т, J=11,2 Гц, 1H), 6,91 (т, J=11,5 Гц, 1H), 6,83 (т, J=9,7 Гц, 1H), 6,38 (д, J=9,4 Гц, 1H), 6,17 (д, J=11,8 Гц, 1H), 5,70 (д, J=11,4 Гц, 1H), 5,65-5,51 (м, 2H), 5,34 (т, J=6,3 Гц, 1H), 5,29 (д, J=10,0 Гц, 1H), 4,82 (кв., J=8,3 Гц, 1H), 4,56-4,48 (м, 1H), 4,45 (д, J=9,3 Гц, 1H), 4,22 (ддд, J=11,4, 7,5, 4,3 Гц, 1H), 3,67 (с, 3H), 3,31 (кв., J=6,4 Гц, 2H), 2,88-2,83 (м, 1H), 2,55 (кв., J=7,7 Гц, 2H), 2,47-2,30 (м, 5H), 2,12-2,07 (м, 1H), 2,08 (с, 3H), 1,88-1,76 (м, 5H), 1,17 (д, J=6,6 Гц, 3H), 1,06 (с, 9H).
13C ЯМР (125 МГц, CDCl3): d 168,2, 166,2, 161,5, 156,7, 145,2, 140,2, 137,3, 134,2, 134,0, 132,0, 124,4, 124,2, 122,3, 120,8, 108,1, 105,5, 81,8, 74,5, 60,6, 55,4, 39,6, 37,3, 34,6, 33,9, 33,3, 30,8, 26,7, 26,3, 21,8, 21,1, 17,2, 16,7.
ESI-MS m/z: Рассчитано для C34H50ClN3O7S: 679,3. Найдено: 702,4 (M+Na)+.
Примеры, демонстрирующие цитотоксичность конъюгатов антитело-лекарственное средство по настоящему изобретению
Биоанализы для определения противоопухолевой активности
Целью этого анализа было in vitro определение цитостатической (способность замедлять или останавливать рост опухолевых клеток) или цитотоксической (способность убивать опухолевые клетки) активности испытываемых образцов.
Клеточные линии и клеточная культура
Все опухолевые клеточные линии, используемые в этом испытании, были получены от American Type Cylture Collection (ATCC), если не указано иное: BT-474 (ATCC HTB-20, дуктальная карцинома молочной железы), SK-BR-3 (ATCC HТB-30, аденокарцинома молочной железы) и HCC-1954 (ATCC CRL-2338, дуктальная карцинома молочной железы), все HER2+; MDA-MB-231 (ATCC HTB-26, аденокарцинома молочной железы) и MCF-7 (ATCC HTB-22, аденокарцинома молочной железы, плевральный выпот), все HER2-; SK-OV-3 (ATCC HTB-77, аденокарцинома яичников), HER2+; NB-4 (Острый Промиелоцитарный Лейкоз, APL, CD13+, M. Lanotte et al. (1991) NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood 77, 1080-1086), (CD13+) и U937 (ATCC CRL-1593,2, Гистиоцитарная Лимфома, CD13+ и CD4+); Raji (ATCC CCL-86, Лимфома Беркитта) (CD13-, CD20+, CD5- и CD4-); RPMI-8226 (ATCC CRM-CCL-155, Множественная Миелома) (CD13-, CD20-, CD5- и CD4-); Karpas-299 (DSMZ ACC-31, Не-ходжкинская Лимфома) (CD20-, CD5+ и CD4+); MOLT-4 (ATCC CRL-1582, Острый Лимфобластный Лейкоз, CD5+). Кроме того, два Raji клеточных (ATCC CCL-86, Лимфома Беркитта) клона использовали в этом испытании Raji-клон#10 (высокая CD5 экспрессия) и Raji-клон18 (нулевая CD5 экспрессия), были получены от Dr. Juan M. Zapata (Instituto de Investigaciones Biomédicas “Alberto Sols”, CSIC-UAM, Madrid, Spain). Клетки поддерживали при 37°C, 5% CO2 и 95% влажности в модифицированной Дульбекко среде Игла (DMEM) (для MCF и MDA-MB-231 клеток), RPMI-1640 (для SK-BR-3, HCC-1954, NB-4, U937, Raji, RPMI-8226, Karpas-299, MOLT-4, Raji-клон#10 и Raji-клон#18 клеток), RPMI-1640+1% ITS (для BT-474 клеток) или McCOyS (для SK-OV-3 клеток), все среды были дополнены 10% фетальной телячьей сывороткой (FCS) и 100 единиц/мл пенициллина и стрептомицина.
Анализ цитотоксичности
Для адгезивных клеток: Колориметрический анализ с использованием сульфородамина B (SRB) адаптировали для количественного определения клеточного роста и цитотоксичности, как описано в V. Vichai и K. Kirtikara (2006) Sulforhodamine B colorimetric assay for cytotoxicity screening. Nature Protocols, 1, 1112-1116. Вкратце, клетки высевали в 96-луночные микротитровальные планшеты и давали выстояться в течение 24 часов в среде, не содержащей лекарственное средство, перед обработкой только носителем или указанными соединениями в течение 72 часов. Для количественного анализа клетки промывали два раза фосфатно-буферным солевым раствором (PBS), фиксировали в течение 15 минут в 1% растворе глутаральдегида, промывали два раза при помощи PBS, окрашивали в растворе 0,4% SRB-1% уксусной кислоты в течение 30 минут, промывали несколько раз 1% раствором уксусной кислоты и сушили на воздухе. SRB затем экстрагировали в 10 мМ trizma исходном растворе и оптическую плотность измеряли при 490 нм в спектрофотометре для микропланшетов. Клеточное выживание выражали как процент от контроля, выживания необработанных клеток.
Для клеток в суспензии: Стандартный метаболический анализ с использованием MTT (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолийбромид) адаптировали для количественного определения клеточного роста и цитотоксичности, как описано в T. Mosmann (1983) Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth., 65, 55-63. Вкратце, MTT раствор добавляли к клеточным культурам при конечной концентрации 0,5 мг/мл и инкубировали в течение 1-4 часов при 37°C вплоть до образования кристаллов формазана. Культуральную среду осторожно удаляли из клеточных культур и кристаллы формазана ресуспендировали в 100 мл DMSO. После перемешивания в течение обеспечения солюбилизации количество формазана (предположительно прямо пропорциональное количеству жизнеспособных клеток) измеряли путем регистрации изменений поглощающей способности при 570 нм с использованием спектрофотометра для считывания планшетов. Клеточное выживание выражали как процент от контроля, выживания необработанных клеток.
IC50 значение относится к концентрации соединения, вызывающей 50% клеточной гибели по сравнению с контрольным клеточным выживанием.
Пример биоактивности 1 - Цитотоксичность ADC1 и соответствующих реагентов против HER2 положительных и отрицательных клеток рака молочной железы
In vitro цитотоксичность ADC1 наряду с исходными цитотоксическими Соединениями 1 и 4 и Трастузумабом оценивали против различных клеточных линий рака молочной железы человека, сверхэкспрессирующих или не экспрессирующих HER2 рецептор, включая BT-474, HCC-1954 и SK-BR-3 (HER2 положительные клетки) и MDA-MB-231 и MCF-7 (HER отрицательные клетки). SK-OV-3, HER2+ клеточная линия рака яичника, также была включена в испытание в качестве модели, отличной от non-клеток ткани молочной железы. Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Трастузумаба
Прежде всего, in vitro цитотоксичность Трастузумаба испытывали против различных опухолевых клеточных линий. В полученных в трех повторностях DR кривых в диапазоне от 5,0E01 до 2,6E-05 мкг/мл (3,1E-07-8,0E-11 M), в двух независимых экспериментах, Трастузумаб был совершенно неактивным, не достигая значений IC50 в какой-либо из испытываемых клеточных линий, независимо от их HER2 статуса (см. Таблицу 3).
Обобщенные результаты in vitro цитотоксичности Трастузумаба
(мкг/мл)
(молярный)
Цитотоксичность Соединения 4
Цитотоксичность промежуточного Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 1E-01 до 2,6E-05 мкг/мл (1,5E-07 до 3,9E-11 M).
Цитотоксичность этого соединения, в двух независимых эксперименах, была относительно гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 8,9E-05 до 1,7E-03 мкг/мл (1,34E-10 до 2,6E-09 M), со средним IC50 значением для серии цельноклеточных анализов 5,69E-04 мкг/мл (8,57E-10 M). Кроме того, цитотоксичность Соединения 4 не зависела от HER2 статуса опухолевых клеточных линий (см. Таблицу 4).
Обобщенные результаты in vitro цитотоксичности Соединения 4
(мкг/мл)
(молярный)
Цитотоксичность Соединения 1
Активность исходного соединения 1 испытывали с использованием таких же условий, которые описаны выше, от 1E-01 до 2,6E-05 мкг/мл (1,1E-07 до 3,0E-11 M). Цитотоксичность этого соединения, в двух независимых экспериментах, также была относительно гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 8,9E-04 до 6,4E-03 мкг/мл (1,04E-09 до 7,47E-09 M), со средним IC50 значением для серии цельноклеточных анализов 3,41E-03 мкг/мл (3,98E-09 M). Малеимидный линкер по-видимому, несколько снижал цитотоксический эффект соединения. В этом случае также цитотоксичность Соединения 1 не зависела от HER2 статуса опухолевых клеточных линий (см. Таблицу 5).
Обобщенные результаты in vitro цитотоксичности Соединения 1
(мкг/мл)
Цитотоксичность ADC1
Цитотоксичность ADC1 испытывали против различных клеточных линий. Для обеспечения подходящего диапазона концентраций, конъюгат анализировали в шести различных, полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1, 0,1, 0,01 и 0,001 мкг/мл (эквивалентно 3,3E-07, 6,6E-08, 6,6E-09, 6,6E-10, 6,6E-11 и 6,6E-12 молярной концентрации), в двух независимых экспериментах.
Репрезентативная DR кривая представлена на Фиг. 3.
После корректировки различных DR кривых, получали среднее IC50 значение, рассчитанное для ADC1 против различных клеточных линий, которое представлено в Таблице 6 ниже. ADC1 показал цитотоксичность сопоставимую с цитотоксичностью исходного Соединения 1, используемого отдельно, и, что важно, четкую специфичность против HER2+ экспрессирующих клеток. Поэтому считают, что конъюгат действительно действовал через взаимодействие mAb с мембраносвязанным HER2 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью. Между HER2 положительными клеточными линиями были существенные различия в чувствительности к ADC1. Наиболее чувствительными клеточными линиями были HCC-1954 и SK-BR-3, демонстрирующие значения IC50 3,88E-02 и 2,45E-02 мкг/мл (эквивалентно 2,581E-10 и 1,63E-10 M), затем BT-474 клетки, которые показали значительно более высокое IC50 значение 7,4E-01 мкг/мл (эквивалентно 4,93E-09 M). Овариальная клеточная линия SK-OV-3 показала еще более высокое IC50 значение 7,0E+00 мкг/мл (эквивалентно 4,67E-08 M). Две HER отрицательные клетки показали одинаковую чувствительность порядка 2,0E+01 мкг/мл (эквивалентно примерно 1,0E-07 M) (см. Таблицу 6).
Обобщенные результаты in vitro цитотоксичности ADC1
(мкг/мл)
Таким образом, наиболее чувствительные HER2 положительные клетки были примерно в 300-800 раз более чувствительными, чем HER2 отрицательные клеточные линии, указывая на специфичность конъюгата против HER2 экспрессирующих клеток.
Для графического сравнения цитотоксичности mAb Трастузумаба, используемого отдельно, с цитотоксичностью конъюгата ADC1, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC при 10 или 1 мкг/мл, представлены на Фиг. 4. Как можно видеть из Фиг. 4, при равной концентрации 10 мкг/мл, mAb Трастузумаб, используемый отдельно, показал незначительную или вообще никакой цитотоксичности (<20% máx) против любой из испытываемых клеточных линий, независимо от их HER2 статуса. С другой стороны, ADC1 показал сильную цитотоксичность против HER2 экспрессирующих клеток, HCC-1954 и SK-BR-3, и в меньшей степени BT-474 и SK-OV-3, индуцируя среднее ингибирование клеточного выживания 88%, 82%, 52% и 47%, соответственно, по сравнению с контрольными клетками. При этой концентрации, ADC1 показал некоторую цитотоксичность против HER отрицательных клеток MCF-7 и MDA-MB-231, со средними с процентными значениями ингибирования клеточного выживания 38% и 32% соответственно. При концентрации 1 мкг/мл, ADC1 конъюгат показал довольно схожую цитотоксичность против HER2 положительных клеток с той, которую наблюдали при 10 мкг/мл, но в этом случае без заметных эффектов на HER2 отрицательные клетки (Фиг. 4).
Эти результаты наглядно демонстрируют поразительную цитотоксичность и специфичность ADC1 конъюгата против HER2 экспрессирующих опухолевых клеток человека in vitro.
Пример биоактивности 2 - Цитотоксичность ADC2 и соответствующих реагентов против HER2 положительных и отрицательных клеток рака молочной железы
In vitro цитотоксичность ADC2, Трастузумаб-Соединение 5 ADC, наряду с исходными цитотоксическими Соединениями 5 и 8 и mAb Трастузумабом оценивали против различных клеточных линий рака молочной железы человека, экспрессирующих или не экспрессирующих HER2 рецептор, включая HCC-1954 и SK-BR-3 (HER2 положительные клетки) и MDA-MB-231 и MCF-7 (HER отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 8
Цитотоксичность промежуточного Соединения 8 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E+00 до 2,6E-04 мкг/мл (1,6E-06 до 4,0E-10 M).
Цитотоксичность этого соединения, в двух независимых экспериментах, была относительно гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 3,40E-03 до 6,75E-03 мкг/мл (5,4E-09 до 1,0E-08 M), со средним IC50 значением для серии цельноклеточных анализов 5,53E-03 мкг/мл (эквивалентно 8,79E-09 M). Кроме того, цитотоксичность Соединения 8 не зависела от HER2 статуса опухолевых клеточных линий (Таблица 7).
Обобщенные данные in vitro цитотоксичности Соединения 8
Цитотоксичность Соединения 5
Активность Соединения 5, модифицированное Соединение 8, содержащее малеимидный линкер, испытывали в таких же условиях, которые описаны выше, от 01E+00 до 2,6E-04 мкг/мл (1,2E-06 до 3,1E-10 M).
Цитотоксичность этого соединения, в двух независимых экспериментах, также была относительно гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 1,9E-02 до 7,7E-02 мкг/мл (2,32E-08 до 9,41E-08 M), со средним IC50 значением для серии цельноклеточных анализов 4,33E-02 мкг/мл (5,26E-08 M). Присутствие малеимидного линкера в Соединении 5 слегка снижал цитотоксичность этого соединения по сравнению с Соединением 8. Также, цитотоксичность Соединения 5 по-видимому, не зависела от HER2 статуса опухолевых клеточных линий (Таблица 8).
Обобщенные данные in vitro цитотоксичности Соединения 5
Цитотоксичность ADC2
Цитотоксическую активность ADC2 испытывали против различных клеточных линий. Для обеспечения подходящего диапазона концентраций, конъюгат испытывали в пяти различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1, 0,1, и 0,01 мкг/мл, в двух независимых экспериментах. Репрезентативная DR кривая представлена на Фиг. 5. Полученные после корректировки всех разных DR кривых, средние IC50 значения, рассчитанные для ADC2 против различных клеточных линий, показаны в Таблице 9. Конъюгат ADC2 показал специфичность против HER2+ экспрессирующих клеток, HCC-1954 и SK-BR-3, в которых соединение продемонстрировало цитотоксичность, подобную цитотоксичности исходного соединения 5, со средними IC50 значениями 5,8E+00 и 2,2E-01 мкг/мл (эквивалентно 4,0E-08 и 1,5E-09 M) соответственно. Две HER- клеточные линии, MCF-7 и MDA-MB-231, были фактически невосприимчивыми к ADC2 в испытываемом диапазоне концентраций, не достигая IC50 значения (>5,0E+01 мкг/мл) (см. Фиг. 5 и Таблицу 9).
Поэтому считают, что конъюгат действительно действовал через взаимодействие mAb с мембраносвязанным HER2 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC2 (Трастузумаб-Соединение 5 ADC)
Для графического сравнения цитотоксичности Трастузумаба, используемого отдельно, с цитотоксичностью конъюгата ADC2, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий только одним Трастузумабом (10 мкг/мл) или ADC при 10 или 1 мкг/мл, представлены на Фиг. 6. При концентрации 10 мкг/мл, mAb Трастузумаб, используемый отдельно, не показал никакой цитотоксичности против любой из испытываемых клеточных линий, независимо от их HER2 статуса. С другой стороны, ADC2 конъюгат продемонстрировал значительную и специфическую цитотоксичность против HER2 экспрессирующих клеток HCC-1954 и SK-BR-3, индуцируя среднее ингибирование клеточного выживания 57% и 78%, соответственно, по сравнению с контрольными клетками.
При концентрации 1 мкг/мл, ADC2 конъюгат показал довольно схожую цитотоксичность против HER2 положительных клеток с той, которую наблюдали при 10 мкг/мл, в этом случае также без заметных эффектов на HER2 отрицательные клетки (Фиг. 6).
Эти результаты наглядно демонстрируют поразительную цитотоксичность и специфичность ADC2 конъюгата против HER2 экспрессирующих опухолевых клеток человека in vitro.
Пример биоактивности 3 - Цитотоксичность ADC3 и соответствующих реагентов против HER2 положительных и отрицательных клеток рака молочной железы
In vitro цитотоксичность ADC3, наряду с исходными цитотоксическими Соединениями 12 и 4 и mAb Трастузумабом оценивали против различных клеточных линий рака молочной железы человека, экспрессирующих или не экспрессирующих HER2 рецептор, включая HCC-1954 и SK-BR-3 (HER2 положительные клетки) и MDA-MB-231 и MCF-7 (HER отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 4
Цитотоксичность исходного Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-07 до 3,9E-12 M).
Цитотоксичность Соединения 4, в двух независимых экспериментах, была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в пикомолярном диапазоне, от 1,16E-04 до 2,80E-04 мкг/мл (1,75E-10 до 4,23E-10 M), со средним IC50 значением для серии цельноклеточных анализов 1,97E-04 мкг/мл (эквивалентно 2,96E-10 M). Кроме того, цитотоксичность Соединения 4 не зависела от HER2 статуса опухолевых клеточных линий (Таблица 10).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 12
Активность Соединения 12, модифицированное Соединение 4, содержащее расщепляемый пептидный линкер, испытывали в таких же экспериментальных условиях, которые описаны выше, в диапазоне концентраций от 01E+01 до 2,6E-03 мкг/мл (7,9E-06 до 2,0E-09 M).
Цитотоксичность Соединения 12, в двух независимых экспериментах, также была относительно гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 7,60E-03 до 3,05E-02 мкг/мл (6,02E-09 до 2,42E-08 M), со средним IC50 значением для серии цельноклеточных анализов 1,63E-02 мкг/мл (1,29E-08 M) (Таблица 11). Присутствие пептидного линкера в Соединении 12 имело отрицательный эффект на цитотоксичность соединения, по сравнению с Соединением 4. Цитотоксичность Соединения 12 была достаточно независимой от HER2 статуса опухолевых клеточных линий.
Обобщенные данные in vitro цитотоксичности Соединения 12
Цитотоксичность ADC 3
В завершение, цитотоксичность ADC3 испытывали против различных клеточных линий. Для обеспечения подходящего диапазона концентраций, конъюгат испытывали в шести различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1, 0,1, 0,01 и 0,001 мкг/мл (эквивалентно 3,33E-07, 6,64E-08, 6,64E-09, 6,64E-10, 6,64E-11 и 6,64E-12 молярной концентрации), в двух независимых экспериментах. Репрезентативная DR кривая представлена на Фиг. 7. Средние IC50 значения, рассчитанные для ADC3 против различных испытываемых клеточных линиях, показаны в Таблице 12.
Конъюгат ADC3 совершенно ясно показал существенную специфичность против HER2+ экспрессирующих клеток, в которых соединение продемонстрировало сильную цитотоксичность, подобную цитотоксичности исходного соединения 4 (примерно 1 log более активное, чем промежуточное Соединение 12, содержащее пептидный линкер). Обе HER2+ клеточные линии, HCC-1954 и SK-BR-3, показали сопоставимую чувствительность против ADC3, со средними IC50 значениями 8,83E-02 и 6,77 E-02 мкг/мл (эквивалентно 5,86E-10 и 4,49E-10 M), соответственно. Две HER отрицательные клеточные линии, MCF-7 и MDA-MB-231, показали существенно более низкую чувствительность против ADC3, со средними IC50 значениями 9,40E+00 и >5,0E+01 мкг/мл (эквивалентно примерно 6,24E-08 M и >3,32E-07 M), соответственно.
Фиг. 8 представляет график цитотоксичности ADC3 против HER2 положительных и отрицательных клеток рака молочной железы. Было обнаружено, что HER2+ клеточные линии (среднее значение IC50 7,80E-02 мкг/мл) были по меньшей мере в >120 раз более чувствительными к ADC3, чем HER2 отрицательные MCF-7 клетки (среднее значение IC50 9,40E+00 мкг/мл), и намного более чувствительными, чем MDA-MB-231 клетки, совершенно ясно демонстрируя специфичность ADC3 против HER2 экспрессирующих клеток (Фиг. 7 и Таблица 12). Поэтому считают, что конъюгат действительно действовал через взаимодействие mAb с мембраносвязанным HER2 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC3 (Трастузумаб-Соединение 12)
Для графического сравнения цитотоксичности mAb Трастузумаба, используемого отдельно, с цитотоксичностью конъюгата ADC3, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC3 при 10 или 1 мкг/мл, представлены на Фиг. 8, которая показывает цитотоксическую активность Трастузумаба vs ADC3 против различных клеточных линий рака молочной железы человека.
При одинаковой концентрации 10 мкг/мл, Трастузумаб, используемый отдельно, не показал никакой цитотоксичности против ни одной из испытываемых клеточных линий, независимо от их HER2 статуса. С другой стороны, ADC3 конъюгат показал сильную цитотоксичность против HER2 экспрессирующих клеток, HCC-1954 и SK-BR-3. В этих клеточных линиях, ADC3 продемонстрировал ингибирование клеточного выживания 83% и 84%, соответственно, по сравнению с контрольными клетками. При этой концентрации, ADC3 также оказывал некоторое действие на HER2 отрицательные клетки, MCF-7 и MDA-MB-231, обеспечивая небольшое ингибирование клеточного выживания 33% и 20%, соответственно. При концентрации 1 мкг/мл, ADC3 конъюгат показал такую же цитотоксичность против HER2 положительных клеток, которую наблюдали при 10 мкг/мл, но без заметных эффектов на HER2 отрицательные клетки (Фиг. 8). Эти результаты наглядно демонстрируют поразительную цитотоксичность и специфичность ADC3 против HER2 экспрессирующих опухолевых клеток человека in vitro.
Пример биоактивности 4 - Цитотоксичность ADC4 и соответствующих реагентов против HER2 положительных и отрицательных клеток рака молочной железы
In vitro цитотоксичность ADC4, наряду с исходными цитотоксическими Соединениями 13 и 4 и mAb Трастузумабом, оценивали против различных клеточных линий рака молочной железы человека, экспрессирующих или не экспрессирующих HER2 рецептор, включая HCC-1954 и SK-BR-3 (HER2 положительные клетки) и MDA-MB-231 и MCF-7 (HER2 отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 4
Цитотоксичность исходного Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 1E-01 до 2,6E-05 мкг/мл (1,5E-07 до 3,0E-11 M)
Цитотоксичность Соединения 4, в двух независимых экспериментах, была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 2,43E-04 до 4,45E-04 мкг/мл (3,6E-10 до 6,7E-10 M), со средним IC50 значением для серии цельноклеточных анализов 3,3E-04 мкг/мл (эквивалентно 4,98E-10 M). Таким образом, цитотоксичность Соединения 4 не зависела от HER2 статуса опухолевых клеточных линий (Таблица 13).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 13
Активность Соединения 13, модифицированное Соединение 4 с тиол-содержащей группой, испытывали в таких же условиях, которые описаны выше, от 1E-01 до 2,6E-05 мкг/мл (1,3E-07 до 2,0E-11 M)
Цитотоксическая активность Соединения 13, в двух независимых экспериментах, также была относительно гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 7,95E-04 до 2,63E-03 мкг/мл (1,0E-09 до 3,5E-09 M), со средним IC50 значением для серии цельноклеточных анализов 1,83E-03 мкг/мл (2,44E-09 M) (Таблица 14). Присутствие тиол-содержащей концевой группы в Соединении 13 слегка снижало (примерно в 5 раз) цитотоксическую активность соединения по сравнению с Соединением 4. Также, цитотоксичность Соединения 13 по-видимому, не зависела от HER2 статуса опухолевых клеточных линий (Таблица 14).
Обобщенные данные in vitro цитотоксичности Соединения 13
Цитотоксичность ADC4
Цитотоксичность ADC4 испытывали против различных клеточных линий. Для обеспечения подходящего диапазона концентраций, конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, в двух независимых экспериментах. Репрезентативная DR кривая представлена на Фиг. 9. После корректировки всех разных DR кривых, среднее IC50 значения, рассчитанные для ADC4 против различных испытываемых клеточных линиях, показаны в Таблице 15.
Конъюгат ADC4 показал специфичность против HER2+ экспрессирующих клеток, HCC-1954 и SK-BR-3, в которых соединение продемонстрировало сильную цитотоксичность, подобную цитотоксичности исходных Соединений 4 и 13, со средними IC50 значениями 1,17E-01 и 4,80E-02 мкг/мл, соответственно. Две HER отрицательные клеточные линии, MCF-7 и MDA-MB-231, показали значительно более низкую чувствительность против ADC4, со средними IC50 значениями 5,35E+00 и 6,50E+00 мкг/мл, соответственно. По-видимому, конъюгат преимущественно действовал через взаимодействие mAb с мембраносвязанным HER2 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC4
Для графического сравнения цитотоксичности mAb Трастузумаба, используемого отдельно, с цитотоксичностью конъюгата ADC4, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC4 при 10 или 1 мкг/мл, представлены на Фиг. 10. При концентрации 10 мкг/мл, mAb Трастузумаб, используемый отдельно, не показал никакой цитотоксичности против любой из испытываемых клеточных линий, независимо от их HER2 статуса. С другой стороны, ADC4 конъюгат продемонстрировал значительную и специфическую цитотоксичность против HER2 экспрессирующих клеток, HCC-1954 и SK-BR-3, индуцируя среднее ингибирование клеточного выживания 80% и 75%, соответственно, по сравнению с контрольными клетками. При этой концентрации, ADC4 также оказывал действие на HER2 отрицательные клетки, MCF-7 и MDA-MB-231, вызывая ингибирование клеточного выживания 53% и 40%, соответственно. При концентрации 1 мкг/мл, ADC4 конъюгат показал довольно схожую цитотоксичность против HER2 положительных клеток с той, которую наблюдали при 10 мкг/мл, но без заметных эффектов на HER2 отрицательные клетки (Фиг. 10). Эти результаты наглядно демонстрируют поразительную цитотоксичность и специфичность ADC4 конъюгата против HER2 экспрессирующих опухолевых клеток человека in vitro.
Пример биоактивности 5 - Цитотоксичность ADC5 и соответствующих реагентов против HER2 положительных и отрицательных клеток рака молочной железы.
In vitro цитотоксичность ADC5, наряду с исходными цитотоксическими Соединениями 15 и 28, оценивали против различных клеточных линий рака молочной железы человека, экспрессирующих или не экспрессирующих HER2 рецептор, включая HCC-1954 и SK-BR-3 (HER2 положительные клетки) и MDA-MB-231 и MCF-7 (HER отрицательные клетки).
Цитотоксичность Соединения 40

Соединение 40 было получено, как описано в WO2007144423 (Соединение 1 в этой патентной заявке), содержание которой включено в настоящую заявку посредством ссылки.
Цитотоксичность исходного Соединения 40 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 1E-02 до 2,6E-06 мкг/мл (1,65E-08 до 4,29E-12 M).
Цитотоксичность Соединения 40, в двух независимых экспериментах, была в высшей степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 4,90E-05 до 1,73E-04 мкг/мл (8,10E-11 до 2,84E-10 M), со средним IC50 значением для серии цельноклеточных анализов 1,06E-04 мкг/мл (эквивалентно 1,75E-10 M). Таким образом, цитотоксичность Соединения 40 не зависела от HER2 статуса опухолевых клеточных линий (Таблица 16).
Обобщенные данные in vitro цитотоксичности Соединения 40
Цитотоксичность Соединения 15
Активность Соединения 15, модифицированное Соединение 40, с тиол-содержащей группой испытывали в таких же условиях, которые описаны выше, от 1E-01 до 2,6E-05 мкг/мл (1,47E-07 до 3,82E-11 M).
Цитотоксичность Соединения 15, в двух независимых экспериментах, также была довольно гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 4,80E-04 до 1,49E-03 мкг/мл (7,06E-10 до 2,19E-09 M), со средним IC50 значением для серии цельноклеточных анализов 1,03E-03 мкг/мл (1,51E-09 M). Присутствие тиол-содержащей концевой группы в Соединении 15 слегка снижало (примерно в 8 раз) цитотоксическую активность соединения по сравнению с Соединением 40. Также, цитотоксичность Соединения 15 не зависела от HER2 статуса опухолевых клеточных линий. (Таблица 17)
Обобщенные данные in vitro цитотоксичности Соединения 15
Цитотоксичность ADC 5
Цитотоксичность ADC5 испытывали против различных клеточных линий. Для обеспечения подходящего диапазона концентраций, конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, в двух независимых экспериментах. Репрезентативная DR кривая (исходная концентрация 1 мкг/мл) представлена на Фиг. 11. После корректировки всех разных DR кривых, средние IC50 значения, рассчитанные для ADC5 против различных испытываемых клеточных линиях, показаны в Таблице 18.
Конъюгат ADC5 показал специфичность против HER2+ экспрессирующих клеток, HCC-1954 и SK-BR-3, в которых соединение продемонстрировало цитотоксическую активность, подобную активности исходных соединений Соединения 28 и Соединения 15, со средними IC50 значениями 1,13E-01 и 4,61E-02 мкг/мл, соответственно. Две HER отрицательные клеточные линии, MCF-7 и MDA-MB-231, показали a существенно более низкую чувствительность против ADC5, со средними IC50 значениями 1,23E+00 и 1,45E+00 мкг/мл, соответственно.
По-видимому, конъюгат ADC5 преимущественно действовал через взаимодействие mAb с мембраносвязанным HER2 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC5
Для графического сравнения цитотоксический активности mAb Трастузумаба, используемого отдельно, с цитотоксичностью конъюгата ADC5, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC5 при 10 мкг/мл или 1 мкг/мл, представлены на Фиг. 12. При концентрации 10 мкг/мл mAb Трастузумаб, используемый отдельно, не показал никакой цитотоксичнеской активности против любой из испытываемых клеточных линий, независимо от их HER2 статуса. С другой стороны, ADC5 конъюгат продемонстрировал значительную и специфическую цитотоксичность против HER2 экспрессирующих клеток, HCC-1954 и SK-BR-3, индуцируя среднее ингибирование клеточного выживания 82% и 72%, соответственно, по сравнению с контрольными клетками. При этой концентрации, ADC5 также оказывал действие на HER2 отрицательные клетки, MCF-7 и MDA-MB-231, вызывая ингибирование клеточного выживания 58% и 54%, соответственно. При концентрации 1 мкг/мл, ADC5 конъюгат показал цитотоксическую активность против HER2 положительных клеток примерно аналогичную той, которую наблюдали при 10 мкг/мл (77% и 75%, соответственно), но значительно более низкую активность против HER2 отрицательных клеток, с ингибированием клеточного выживания 24% и 15%, соответственно (Фиг. 12). Эти результаты продемонстрировали поразительную цитотоксическую активность и относительную специфичность ADC5 конъюгата против HER2 экспрессирующих опухолевых клеток человека in vitro.
Пример биоактивности 6 - Цитотоксичность ADC6 и соответствующих реагентов против HER2 положительных и отрицательных клеток рака молочной железы
In vitro цитотоксичность ADC6 наряду с исходными цитотоксическими Соединениями 18 и 8 оценивали против различных клеточных линий рака молочной железы человека, экспрессирующих или не экспрессирующих HER2 рецептор, включая HCC-1954 и SK-BR-3 (HER2 положительные клетки) и MDA-MB-231 и MCF-7 (HER2 отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для нескольких часов.
Цитотоксичность Соединения 8
Цитотоксичность исходного соединения 8 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 1E+00 до 2,6E-04 мкг/мл (1,59E-06 до 4,13E-10 M). Цитотоксическая активность этого соединения, в двух независимых экспериментах, была относительно гомогенной в различных испытываемых клеточных линиях (несколько более высокая активность против SK-BR-3 клеток), с IC50 значениями в низком наномолярном диапазоне, от 1,85E-03 до 9,50E-03 мкг/мл (2,94E-09 до 1,51 E-08 M), со средним IC50 значением для серии цельноклеточных анализов 5,45E-03 мкг/мл (эквивалентно 8,67E-09 M). Таким образом, цитотоксичность Соединения 8, по-видимому, не зависела от HER2 статуса опухолевых клеточных линий (Таблица 19).
Обобщенные данные in vitro цитотоксичности Соединения 8
Цитотоксичность Соединения 18
Активность Соединения 18, модифицированное Соединение 8 с тиол-содержащей группой, испытывали в таких же условиях, которые описаны выше, от 1E+00 до 2,6E-04 мкг/мл (1,39E-06 до 3,63E-10 M). Цитотоксичность этого соединения, в двух независимых экспериментах, также была довольно гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 4,40E-03 до 1,85E-02 мкг/мл (6,14E-09 до 2,58E-08 M), со средним IC50 значением для серии цельноклеточных анализов 1,06E-02 мкг/мл (1,48E-08 M). Присутствие тиол-содержащей концевой группы в Соединении 18 оказывало незначительный эффект на активность соединения, по сравнению с Соединением 8. Также, цитотоксичность Соединения 18, по- видимому, была в достаточной степени независимой от HER2 статуса опухолевой клеточной линии (Таблица 20).
Обобщенные данные in vitro цитотоксичности Соединения 18
Цитотоксичность ADC6
Цитотоксичность ADC6 испытывали против различных клеточных линий. Для обеспечения подходящего диапазона концентраций, конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, в двух независимых экспериментах. Репрезентативная кривая DR представлена на Фиг. 13. После корректировки всех разных DR кривых, средние IC50 значения, рассчитанные для ADC6 против различных испытываемых клеточных линиях показаны в Таблице 21.
Конъюгат ADC6 показал некоторую специфичность, хотя и ограниченную, в отношении HER2+ экспрессирующих клеток, HCC-1954 и SK-BR-3. В этих клеточных линиях, конъюгат был несколько менее цитотоксическим, чем исходные Соединения 8 и 18 отдельно (в 5,6 и 3,2 раз, соответственно), со средними IC50 значениями 1,04 E+01 и 3,80E+00 мкг/мл, соответственно. Две HER отрицательные клеточные линии, MCF-7 и MDA-MB-231, показали несколько более низкую чувствительность против ADC6 (в5 раз меньше), со средними IC50 значениями 3,50E+01 и 4,40E+01 мкг/мл, соответственно. По-видимому, конъюгат ADC6 оказывал некоторое предпочтительное действие на HER2 экспрессирующие клетки, действуя через взаимодействие mAb с мембраносвязанным HER2 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC6
Для графического сравнения цитотоксический активности mAb Трастузумаба, используемого отдельно, с цитотоксичностью конъюгата ADC6, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (10 мкг/мл) или ADC6 при 10 или 1 мкг/мл, представлены на Фиг. 14. При концентрации 10 мкг/мл, mAb Трастузумаб, используемый отдельно, не продемонстрировал никакой цитотоксической активности против любой из испытываемых клеточных линий, независимо от их HER2 статуса. С другой стороны, ADC6 конъюгат показал специфическую цитотоксичность против HER2 экспрессирующих клеток, HCC-1954 и SK-BR-3, индуцируя среднее ингибирование клеточного выживания 57% и 70%, соответственно, по сравнению с контрольными клетками. При этой концентрации, ADC6 также имел остаточный эффект на HER2 отрицательные клетки, MCF-7 и MDA-MB-231, вызывая ингибирование клеточного выживания 9% и 7%, соответственно. При концентрации 1 мкг/мл, ADC6 конъюгат все же обладал цитотоксической активностью против HER2 положительных клеток, хотя и меньше той, которую наблюдали при 10 мкг/мл (19% и 38%, соответственно). При этой концентрации, ADC6 был совершенно неактивным против HER2 отрицательных клеток (Фиг. 14). Эти результаты продемонстрировали преимущественную цитотоксическую активность ADC6 конъюгата против HER2 экспрессирующих опухолевых клеток человека in vitro.
Пример биоактивности 7 - Цитотоксичность ADC7 и соответствующих реагентов против HER2 положительных и отрицательных клеток рака молочной железы
In vitro цитотоксичность ADC7 наряду с исходными цитотоксическими Соединениями 24, 25 и 41 оценивали против различных клеточных линий рака молочной железы человека, экспрессирующих или не экспрессирующих HER2 рецептор, включая HCC-1954 и SK-BR-3 (HER2 положительные клетки) и MDA-MB-231 и MCF-7 (HER2 отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 41
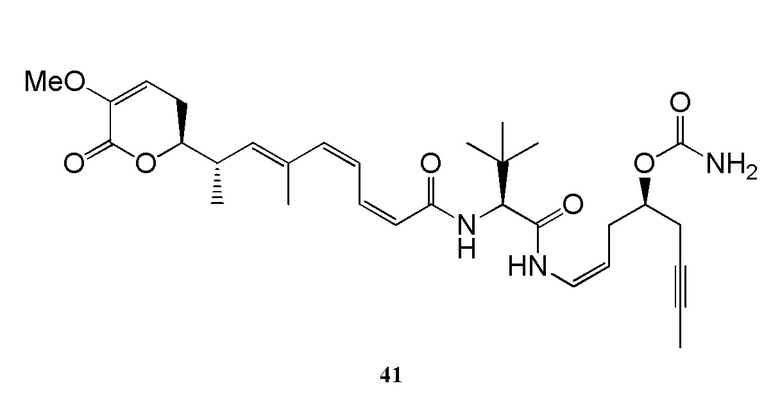
Соединение 41 было получено, как описано в WO 2009/080761 (Соединение 72 в этой патентной заявке), содержание которой включено в настоящую заявку посредством ссылки.
Цитотоксичность исходного Соединения 41 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 1E-02 до 2,6E-06 мкг/мл (1,8E-08 до 4,6E-12 M). Цитотоксическая активность этого соединения, в двух независимых экспериментах, была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 1,0E-04 до 2,6E-04 мкг/мл (1,8E-10 до 4,6E-10 M), со средним IC50 значением для серии цельноклеточных анализов 1,6E-04 мкг/мл (эквивалентно 2,9E-10 M). Таким образом, цитотоксичность Соединения 41 не зависела от HER2 статуса опухолевых клеточных линий (Таблица 22)
Обобщенные данные in vitro цитотоксичности Соединения 41
Цитотоксичность Соединения 24
Активность Соединения 24 испытывали в таких же условиях, которые описаны выше, от 1E+00 до 2,6E-04 мкг/мл (1,6E-06 до 4,1E-10 M). Цитотоксическая активность этого соединения, в двух независимых экспериментах, была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 9,0E-03 до 1,8E-02 мкг/мл (1,4E-08 до 2,8E-08 M), со средним IC50 значением для серии цельноклеточных анализов 1,5E-02 мкг/мл (2,4E-08 M). Присутствие 1,3-пропилендиаминовой группы в Соединении 24 существенно снижало (примерно 2 logs) цитотоксическую активность соединения по сравнению с Соединением 41. Также, цитотоксичность Соединения 24, по-видимому, не зависела от HER2 статуса опухолевых клеточных линий (Таблица 23).
Обобщенные данные in vitro цитотоксичности Соединения 24
Цитотоксичность Соединения 25
Активность Соединения 25, модифицированноу Соединение 24, содержащего MC линкер, испытывали в таких же условиях, которые описаны выше, от 1E-01 до 2,6E-05 мкг/мл (1,2E-07 до 3,2E-11 M). Цитотоксическая активность этого соединения, в двух независимых экспериментах, была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 2,5E-02 до 5,3E-02 мкг/мл (3,1E-08 до 6,5E-08 M), со средним IC50 значением для серии цельноклеточных анализов 4,1E-02 мкг/мл (4,9E-08 M). Присутствие MC линкера в Соединении 25 очень незначительно снижало цитотоксическую активность соединения по сравнению с Соединением 24, особенно в MDA-MB-231 клетках. Цитотоксичность Соединения 25, по-видимому, не зависела от HER2 статуса опухолевых клеточных линий (Таблица 24).
Обобщенные данные in vitro цитотоксичности Соединения 25
Цитотоксичность ADC7
Цитотоксичность ADC7 испытывали против различных клеточных линий. Для обеспечения подходящего диапазона концентраций, конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл в двух независимых экспериментах. Репрезентативная DR кривая (исходная концентрация 10 мкг/мл) представлена на Фиг. 15. После корректировки всех разных DR кривых, средние IC50 значения, рассчитанные для ADC7 против различных испытываемых клеточных линий показаны в Таблице 25.
Конъюгат ADC7 показал специфичность против HER2+ экспрессирующих клеток, HCC-1954 и SK-BR-3, в которых соединение продемонстрировало цитотоксическую активность очень схожую с активностью исходного соединения 29, со средними IC50 значениями 3,7E-01 и 8,9E-02 мкг/мл, соответственно. Две HER отрицательные клеточные линии, MCF-7 и MDA-MB-231, были фактически невосприимчивыми к ADC7. Конъюгат, по-видимому, действовал через взаимодействие mAb с мембраносвязанным HER2 рецептором на положительных опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC7
Для графического сравнения цитотоксический активности mAb Трастузумаба, используемого отдельно, с цитотоксичностью конъюгата ADC7, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC7 при 50 или 1 мкг/мл, представлены на Фиг. 16. При концентрации 50 мкг/мл, mAb Трастузумаб, используемый отдельно, не показал никакой значимой цитотоксической активности против любой из испытываемых клеточных линий, независимо от их HER2 статуса. С другой стороны, ADC7 конъюгат продемонстрировал значительную и специфическую цитотоксичность против HER2 экспрессирующих клеток, HCC-1954 и SK-BR-3, индуцируя среднее ингибирование клеточного выживания 77% и 76%, соответственно, по сравнению с контрольными клетками. При этой концентрации, ADC7 имел лишь остаточный эффект на HER2 отрицательные клетки, MCF-7 и MDA-MB-231, вызывая ингибирование клеточного выживания 13% и 15%, соответственно. Подобную активность и специфичность определяли при более низких концентрациях ADC7 (таких как 1 мкг/мл) в HCC-1954 и SK-BR-3 клетках, вызывающую ингибирование клеточного выживания 68% и 79%, соответственно (Фиг. 16). Взятые вместе, эти результаты наглядно демонстрируют поразительную цитотоксическую активность и специфичность ADC7 конъюгата против HER2 экспрессирующих опухолевых клеток человека in vitro.
Пример биоактивности 8 - Цитотоксичность ADC8 и соответствующих реагентов против HER2 положительных и отрицательных клеток рака молочной железы
In vitro цитотоксическую активность ADC8, наряду с исходными цитотоксическими Соединениями 24, 27 и 41, оценивали против различных клеточных линий рака молочной железы человека, экспрессирующих или не экспрессирующих HER2 рецептор, включая HCC-1954 и SK-BR-3 (HER2 положительные клетки) и MDA-MB-231 и MCF-7 (HER отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 41
Цитотоксический активность исходного Соединения 41 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,8E-08 до 4,6E-12 M). Цитотоксическая активность этого соединения, в двух независимых экспериментах, была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 7,25E-05 до 1,4E-04 мкг/мл (1,3E-10 до 2,4E-10 M), со средним IC50 значением для серии цельноклеточных анализов 1,1E-04 мкг/мл (эквивалентно 1,9E-10 M). Таким образом, цитотоксичность Соединения 41 не зависела от HER2 статуса опухолевых клеточных линий (Таблица 26).
Обобщенные данные in vitro цитотоксичности Соединения 41
Цитотоксичность Соединения 24
Активность Соединения 24 (испытывали в таких же условиях, которые описаны выше, от 01E+00 до 2,6E-04 мкг/мл (1,6E-06 до 4,1E-10 M). Цитотоксическая активность этого соединения, в двух независимых экспериментах, была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 9,0E-03 до 1,8E-02 мкг/мл (1,4E-08 до 2,9E-08 M), со средним IC50 значением для серии цельноклеточных анализов 1,5E-02 мкг/мл (2,4E-08 M). Присутствие 1,3-пропилендиаминовой группы в Соединении 24 существенно снижало (примерно 2 logs) цитотоксическую активность соединения по сравнению с Соединением 41. Цитотоксичность Соединения 24, по-видимому, не зависела от HER2 статуса опухолевых клеточных линий (Таблица 27).
Обобщенные данные in vitro цитотоксичности Соединения 24
Цитотоксичность Соединения 27
Активность Соединения 27 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E+00 до 2,6E-04 мкг/мл (1,4E-06 до 3,6E-10 M). Цитотоксическая активность Соединения 27, в двух независимых экспериментах, была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в микромолярном диапазоне, от 1,05E-02 до 3,9E-02 мкг/мл (1,5E-08 до 5,5E-08 M), со средним IC50 значением для серии цельноклеточных анализов 2,5E-02 мкг/мл (3,5E-08 M). Присутствие MPA линкера в Соединении 27 не имело никакого значительного эффекта на цитотоксическую активность соединения по сравнению с Соединением 24. Активность Соединения 27 не зависела от HER2 статуса опухолевых клеточных линий (Таблица 28).
Обобщенные данные in vitro цитотоксичности Соединения 27
Цитотоксичность ADC8
Цитотоксическую активность ADC8 испытывали против различных клеточных линий. Конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, в двух независимых экспериментах. Репрезентативная DR кривая (максимальная концентрация 10 мкг/мл) представлена на Фиг. 17. ADC8 показал некоторую специфичность против HER2+ экспрессирующих клеток, особенно в SK-BR-3, наиболее чувствительной клеточной линии. За исключением этих клеток, которые примерно в 4 раза более чувствительны, чем HER2 отрицательные клетки (IC50 2,3E-09M), ADC8 показал подобную цитотоксическую активность, HER2 независимую, как у исходного соединения 27, с IC50 значениями в наномолярном диапазоне (Таблица 29).
Обобщенные данные in vitro цитотоксичности ADC8
Для графического сравнения цитотоксический активности mAb Трастузумаба, используемого отдельно, с цитотоксичностью конъюгата ADC8, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC8 (50 или 10 мкг/мл), представлены на Фиг. 18. При 50 мкг/мл, mAb отдельно не показал никакой активности в любой из испытываемых клеточных линий, за исключением SK-BR-3, в которой он вызывал ингибирование клеточного выживания меньше чем 20%. ADC8, в свою очередь, не показал никакой значимой специфичности в отношении HER2+ клеточных линий, вызывая сильное ингибирование клеточного выживания более чем 60% во всех анализируемых клетках. При концентрации 10 мкг/мл, ADC8 показал некоторую, но незначительную, специфичность против HER2+ клеток, вызывая 78% ингибирование клеточного выживания в HCC-1954 и SK-BR-3 клетках, обе HER2 положительные, при этом он имел меньший эффект на HER2 отрицательные клетки, 59% и 41%, в MCF7 и MDA-MB-231, соответственно. HER2 положительные клетки были примерно в 1,5 - 2 раз более чувствительными к ADC8, чем HER2 отрицательные клетки.
Пример биоактивности 9 - Цитотоксичность ADC9 и соответствующих реагентов против CD13 положительных и отрицательных опухолевых клеток человека
In vitro цитотоксическую активность ADC9, наряду с исходными цитотоксическими Соединениями 1 и 4, оценивали против различных опухолевых клеточных линий человека, экспрессирующих или не экспрессирующих CD13 рецептор, включая NB4 и U937 (CD13 положительные клетки) и Raji и RPMI-8226 (CD13 отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность анти-CD13 мышиного моноклонального антитела
Прежде всего, in vitro цитотоксическую активность анти-CD13 мышиного mAb отдельно испытывали против различных опухолевых клеточных линий. В полученных в трех повторностях DR кривых в диапазоне от 5,0E+01 до 1,3E-02 мкг/мл (3,3E-07-8,7E-11 M), в двух независимых экспериментах, антитело было фактически неактивным, не достигая значений IC50 в какой-либо из испытываемых клеточных линий, независимо от их CD13 статуса (Таблица 30).
Обобщенные данные in vitro цитотоксической активности антиCD13 мышиного mAb
Цитотоксичность Соединения 4
Цитотоксическую активность Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 4, в двух независимых экспериментах, была в высшей степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 7,9E-05 до 2,65E-03 мкг/мл (1,2E-10 до 4,0E-09 M), со средним IC50 значением для серии цельноклеточных анализов 8,4E-04 мкг/мл (эквивалентно 1,2E-09 M). Таким образом, цитотоксичность Соединения 4 была достаточно независимой от CD13 статуса опухолевых клеточных линий (Таблица 31).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 1
Активность соединения 1 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-01 до 2,6E-05 мкг/мл (1,1E-07 до 3,0E-11 M). Цитотоксическая активность Соединения 1, в двух независимых экспериментах, была в некоторой степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 8,0E-04 до 6,3E-03 мкг/мл (9,4E-10 до 7,3E-09 M), со средним IC50 значением для серии цельноклеточных анализов 2,8E-03 мкг/мл (3,3E-09 M). Присутствие малеимидного линкера в Соединении 1 не изменяло существенным образом цитотоксическую активность соединения, по сравнению с Соединением 4. Кроме того, цитотоксичность соединения не зависела от CD13 статуса опухолевых клеточных линий (Таблица 32).
Обобщенные данные in vitro цитотоксичности Соединения 1
Цитотоксичность ADC9
Цитотоксическую активность ADC9 испытывали против различных клеточных линий. Конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, в двух независимых экспериментах. Репрезентативная DR кривая (максимальная концентрация 0,1 мкг/мл) представлена на Фиг. 19.
Конъюгат ADC9 показал существенную специфичность против CD13+ экспрессирующих клеток, в которых соединение продемонстрировало цитотоксическую активность, подобную или несколько выше, чем у исходных Соединений 4 и 1. Обе CD13+ клеточные линии, NB4 и U937, показали сопоставимую чувствительность против ADC9, со средними IC50 значениями 8,7E-03 и 2,4E-02 мкг/мл, соответственно. Две CD13 отрицательные клеточные линии, Raji и RPMI-8226, показали существенно более низкую чувствительность против ADC9, со средними IC50 значениями 1,6E+00 и 5,9E-01 мкг/мл, соответственно. В среднем, CD13+ клеточные линии (среднее значение IC50 1,66E-02 мкг/мл) были примерно в 65 раз более чувствительными к ADC9, чем CD13- клетки (среднее значение IC50 1,08E+00 мкг/мл). При сравнении активности ADC9 в NB4 клетках (наиболее чувствительные) vs Raji клеток (наименее чувствительные); была обнаружена разница примерно в 180 раз. Эти результаты совершенно ясно показали специфичность конъюгата против CD13 экспрессирующих клеток (Таблица 33). Поэтому считают, что ADC9, по меньшей мере частично, действовал через взаимодействие mAb с мембраносвязанным CD13 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC9
Для графического сравнения цитотоксической активности mAb, используемого отдельно, с цитотоксичностью конъюгата ADC9, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC при 50 или 0,1 мкг/мл, представлены на Фиг. 20. При равной концентрации 50 мкг/мл, анти-CD13 антитело отдельно не продемонстрировало никакой цитотоксической активности против любой из испытываемых клеточных линий, независимо от их CD13 статуса. С другой стороны, ADC9 конъюгат показал сильную цитотоксическую активность против всех клеточных линий, индуцируя ингибирование клеточного выживания более чем 80%. При концентрации 0,1 мкг/мл, конъюгат ADC9 показал цитотоксическую активность против CD13 положительных клеток, подобную той, которую наблюдали при 50 мкг/мл, но без какого-либо заметного эффекта на CD13 отрицательные клетки. Эти результаты дополнительно продемонстрировали поразительную цитотоксическую активность и специфичность ADC9 против CD13 экспрессирующих опухолевых клеток человека in vitro.
Пример биоактивности 10 - Цитотоксичность ADC10 и соответствующих реагентов против CD13 положительных и отрицательных опухолевых клеток человека
In vitro цитотоксическую активность ADC10, наряду с исходными цитотоксическими Соединениями 12 и 4, оценивали против различных опухолевых клеточных линий человека, экспрессирующих или не экспрессирующих CD13 рецептор, включая NB4 и U937 (CD13 положительные клетки) и Raji и RPMI-8226 (CD13 отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 4
Цитотоксическую активность Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 4, в двух независимых экспериментах, была в высшей степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком наномолярном диапазоне, от 7,9E-05 до 2,65E-03 мкг/мл (1,2E-10 до 4,0E-09 M), со средним IC50 значением для серии цельноклеточных анализов 8,4E-04 мкг/мл (эквивалентно 1,2E-09 M). Таким образом, цитотоксичность Соединения 4 была достаточно независимой от CD13 статуса опухолевых клеточных линий (Таблица 34).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 12
Активность соединения 12 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E+00 до 2,6E-04 мкг/мл (7,9E-07 до 2,0E-10 M). Цитотоксическая активность Соединения 12, в двух независимых экспериментах, была в некоторой степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 4,4E-03 до 4,8E-02 мкг/мл (3,5E-09 до 3,8E-08 M), со средним IC50 значением для серии цельноклеточных анализов 2,0E-02 мкг/мл (1,6E-08 M). Присутствие длинного линкера в Соединении 12 снижало (примерно 1 log) цитотоксическую активность соединения, по сравнению с Соединением 4. Кроме того, цитотоксичность соединения не зависела от CD13 статуса опухолевых клеточных линий (Таблица 35).
Обобщенные данные in vitro цитотоксичности Соединения 12
Цитотоксичность ADC10
Цитотоксическую активность ADC10 испытывали против различных клеточных линий. Конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, в двух независимых экспериментах. Репрезентативная DR кривая (максимальная концентрация 1 мкг/мл) представлена на Фиг. 21.
Конъюгат ADC10 показал существенную специфичность против CD13+ экспрессирующих клеток, в которых соединение продемонстрировало цитотоксическую активность, которая подобна или несколько выше активности исходных Соединений 4 и 12. Обе CD13+ клеточные линии, NB4 и U937, показали сопоставимую чувствительность против ADC10, со средними IC50 значениями 7,2E-03 и 9,8E-03 мкг/мл, соответственно. Две CD13 отрицательные клеточные линии, Raji и RPMI-8226, показали существенно более низкую чувствительность против ADC10, со средними IC50 значениями 1,0E+01 и 5,3E+00 мкг/мл, соответственно. В среднем, CD13+ клеточные линии (среднее значение IC50 8,50E-03 мкг/мл) были примерно в 900 раз более чувствительными к ADC10, чем CD13- клетки (среднее значение IC50 7,83E+00 мкг/мл). При сравнении активности ADC10 в NB4 клетках (наиболее чувствительные) vs Raji клеток (наименее чувствительные) была обнаружена разница примерно в 1440 раз. Эти результаты совершенно ясно показали специфичность ADC10 против CD13 экспрессирующих клеток (Таблица 36). Поэтому считают, что ADC10, по меньшей мере частично, действовал через взаимодействие mAb с мембраносвязанным CD13 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC10
Для графического сравнения цитотоксической активности mAb, используемого отдельно, с цитотоксичностью конъюгата ADC10, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC при 50 или 1 мкг/мл, представлены на Фиг. 22. При равной концентрации 50 мкг/мл, анти-CD13 антитело отдельно, не продемонстрировало никакой цитотоксической активности против любой из испытываемых клеточных линий, независимо от их CD13 статуса. С другой стороны, ADC10 конъюгат показал сильную цитотоксическую активность против всех клеточных линий, индуцируя ингибирование клеточного выживания более чем 80%, за исключением Raji клеток, в которых он вызывал более низкое, но все еще существенное, ингибирование примерно 70%. При концентрации 1 мкг/мл, ADC10 показал цитотоксическую активность против CD13 положительных клеток, подобную той, которую наблюдали при 50 мкг/мл, но без какого-либо заметного эффекта на CD13 отрицательные клетки. Эти результаты дополнительно продемонстрировали поразительную цитотоксическую активность и специфичность ADC10 против CD13 экспрессирующих опухолевых клеток человека in vitro.
Пример биоактивности 11 - Цитотоксичность ADC11 и соответствующих реагентов против CD13 положительных и отрицательных опухолевых клеток человека
In vitro цитотоксическую активность ADC11, наряду с исходными цитотоксическими Соединениями 13 и 40, оценивали против различных опухолевых клеточных линий человека, экспрессирующих или не экспрессирующих CD13 рецептор, включая NB4 и U937 (CD13 положительные клетки) и Raji и RPMI-8226 (CD13 отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 40
Цитотоксическую активность Соединения 40 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-03 до 2,6E-07 мкг/мл (1,7E-09 до 4,3E-13 M). Цитотоксическая активность Соединения 40, в двух независимых экспериментах, была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком пикомолярном диапазоне, от 3,1E-05 до 1,7E-04 мкг/мл (5,2E-11 до 2,8E-10 M), со средним IC50 значением для серии цельноклеточных анализов 8,6E-05 мкг/мл (эквивалентно 1,4E-10 M). Цитотоксичность Соединения 40 не зависела от уровней экспрессии CD13 на опухолевых клеточных линиях (Таблица 37).
Обобщенные данные in vitro цитотоксичности Соединения 40
Цитотоксичность Соединения 13
Активность соединения 13 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-01 до 2,6E-05 мкг/мл (1,3E-07 до 3,4E-11 M). Цитотоксическая активность Соединения 13, в двух независимых экспериментах, была в некоторой степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 1,7E-03 до 1,0E-02 мкг/мл (2,7E-09 до 1,4E-08 M), со средним IC50 значением для серии цельноклеточных анализов 4,3E-03 мкг/мл (5,7E-09 M). Присутствие тиол-содержащей концевой группы в Соединении 13 снижало (меньше чем 1 log) цитотоксическую активность соединения по сравнению с Соединением 40. Цитотоксичность соединения была достаточно независимой от CD13 статуса опухолевых клеточных линий (Таблица 38).
Обобщенные данные in vitro цитотоксичности Соединения 13
Цитотоксичность ADC11
Цитотоксическую активность ADC11 испытывали против различных клеточных линий. Конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, в двух независимых экспериментах. Репрезентативная DR кривая (максимальная концентрация 1 мкг/мл) представлена на Фиг. 23. ADC11 показал некоторую, но небольшую, специфичность против CD13 экспрессирующих клеток. Конъюгат имел примерно схожую цитотоксическую активность, за исключением Raji клеток, которые являются несколько менее чувствительными, во всех испытываемых клеточных линиях. Активность ADC11 была сопоставима с активностью исходного соединения 13, с IC50 значениями в низком наномолярном диапазоне (Таблица 39).
Обобщенные данные in vitro цитотоксичности ADC11
Для графического сравнения цитотоксической активности анти-CD13 mAb, используемого отдельно, с цитотоксичностью конъюгата ADC11, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC11 (50 или 1 мкг/мл), представлены на Фиг. 24. При концентрации 50 мкг/мл, анти-CD13 антитело отдельно, показало некоторую цитотоксическую активность против CD13+ клеточных линий, вызывая ингибирование клеточного выживания примерно 30%. В CD13- клетках, антитело было фактически неактивным. При такой же концентрации, ADC11 конъюгат показал сильную цитотоксическую активность против всех клеточных линий, с некоторой, но очень низкой специфичностью против CD13 экспрессирующих клеток, в которых он индуцировал снижение клеточного выживания около 100%. В CD13- клетках, ADC11 индуцировал более чем 80% снижение клеточного выживания. При концентрации 1 мкг/мл, ADC11 показал более высокую специфичность против CD13+ клеток, NB-4 и U937, в которых он индуцировал снижение клеточного выживания 99 и 85%, соответственно. В CD13- клетках, Raji и RPMI8226, конъюгат индуцировал снижение клеточного выживания 38 и 60%, соответственно.
Пример биоактивности 12 - Цитотоксичность ADC12 и соответствующих реагентов против CD20 положительных и отрицательных опухолевых клеток человека
In vitro цитотоксическую активность ADC12, наряду с исходными цитотоксическими Соединениями 1, 4 и 40, оценивали против различных раковых клеточных линий человека, экспрессирующих или не экспрессирующих CD20 антиген, включая Raji (CD20 положительные клетки); RPMI-8226 и Karpas-299 (CD20 отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Ритуксимаба
Прежде всего, in vitro цитотоксическую активность mAb Ритуксимаба, используемого отдельно, испытывали против различных раковых клеточных линий человека, экспрессирующих или не экспрессирующих CD20 антиген, включая Raji (CD20 положительные клетки); RPMI-8226 и Karpas-299 (CD20 отрицательные клетки). В полученных в трех повторностях DR кривых охватывающих от 5,0E+01 до 1,3E-02 мкг/мл (3,3E-07 до 8,7E-11 M), антитело было достаточно неактивным, не достигая значений IC50 в какой-либо из испытываемых клеточных линий, независимо от их CD20 статуса (Таблица 40).
Обобщенные данные in vitro цитотоксичности Ритуксимаба
Цитотоксичность Соединения 40
Цитотоксическую активность исходного соединения Соединение 40 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-03 до 2,6E-07 мкг/мл (1,7E-09 до 4,3E-13 M). Цитотоксическая активность Соединения 40 была в высшей степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком субнаномолярном диапазоне, от 4,6E-05 до 1,3E-04 мкг/мл (7,6E-11 до 2,1E-10 M), со средним IC50 значением для серии цельноклеточных анализов 9,6E-05 мкг/мл (эквивалентно 1,6E-10 M). Цитотоксичность Соединения 40 не зависела от CD20 статуса опухолевых клеточных линий (Таблица 41).
Обобщенные данные in vitro цитотоксичности Соединения 40
Цитотоксичность Соединения 4
Цитотоксическую активность Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 4 была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 5,7E-04 до 1,4E-03 мкг/мл (8,6E-10 до 2,1E-09 M), со средним IC50 значением для серии цельноклеточных анализов 9,7E-04 мкг/мл (1,5E-09 M). Присутствие амин-содержащей группы в Соединении 4 слегка снижало цитотоксическую активность соединения, по сравнению с Соединением 40. Цитотоксическая активность Соединения 4 также не зависела от CD20 статуса опухолевых клеточных линий (Таблица 42).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 1
Активность соединения 1 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-01 до 2,6E-05 мкг/мл (1,2E-07 до 3,0E-11 M). Цитотоксическая активность Соединения 1, в двух независимых экспериментах, была в высшей степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 1,6E-03 до 2,8E-03 мкг/мл (1,9E-09 до 3,3E-09 M), со средним IC50 значением для серии цельноклеточных анализов 2,1E-03 мкг/мл (2,5E-09 M). Присутствие малеимидного линкера в Соединении 1 существенно не изменяло цитотоксическую активность соединения по сравнению с Соединением 4. Кроме того, цитотоксичность соединения также не зависела от CD20 статуса опухолевых клеточных линий (Таблица 43).
Обобщенные данные in vitro цитотоксичности Соединения 1
Цитотоксичность ADC12
Цитотоксическую активность ADC12 испытывали против различных опухолевых клеточных линий. Конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, соответственно. Репрезентативная DR кривая (исходная концентрация 1 мкг/мл) представлена на Фиг. 25. Хотя он показал более высокую активность в CD20 положительных Raji клетках, ADC12 продемонстрировал примерно одинаковую цитотоксическую активность во всех испытываемых клеточных линиях. Raji клетки (CD20+) показали среднее значение IC50 9,5E-02 мкг/мл, тогда как RPMI-8226 и Karpas-299 (обе CD20-) показали 4,0E-01 и 4,1E-01 мкг/мл, соответственно (Таблица 44). Таким образом, CD20 положительные клетки были несколько более чувствительными (в 4 раза) к ADC12, чем CD20 отрицательные клетки.
Обобщенные данные in vitro цитотоксичности ADC12
Для графического сравнения цитотоксической активности mAb Ритуксимаба, используемого отдельно, с цитотоксичностью конъюгата ADC12, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC12 (1 и 0,1 мкг/мл), представлены на Фиг. 26. Ритуксимаб отдельно, при концентрации 50 мкг/мл, был фактически неактивным во всех испытываемых клеточных линиях, независимо от их CD20 статуса. С другой стороны, ADC12 конъюгат, при концентрации 1 мкг/мл, показал сильную цитотоксическую активность во всех испытываемых клеточных линиях, вызывая более чем 70% снижение клеточного выживания после 72 часов обработки. При более низкой концентрации, 0,1 мкг/мл, ADC12 конъюгат показал некоторую специфичность, вызывая снижение клеточного выживания около 60% в CD20 положительных клетках (Raji), при этом он был фактически неактивным в CD20 отрицательных клетках (RPMI-8226 и Karpas-299).
Пример биоактивности 13 - Цитотоксичность ADC13 и соответствующих реагентов против CD20 положительных и отрицательных опухолевых клеток человека
In vitro цитотоксическую активность ADC13, наряду с исходными цитотоксическими Соединения 1, 4 и 40, оценивали против различных раковых клеточных линий человека, экспрессирующих или не экспрессирующих CD20 антиген, включая Raji (CD20 положительные клетки); RPMI-8226 и Karpas-299 (CD20 отрицательные клетки). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 40
Цитотоксическую активность исходного соединения Соединения 40 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-03 до 2,6E-07 мкг/мл (1,7E-09 до 4,3E-13 M). Цитотоксическая активность Соединения 40 была в высшей степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком субнаномолярном диапазоне, от 8,6E-05 до 1,1E-04 мкг/мл (1,4E-10 до 1,9E-10 M), со средним IC50 значением для серии цельноклеточных анализов 9,6E-05 мкг/мл (эквивалентно 1,6E-10 M). Цитотоксичность Соединения 40 не зависела от CD20 статуса опухолевых клеточных линий (Таблица 45).
Обобщенные данные in vitro цитотоксичности Соединения 40
Цитотоксичность Соединения 4
Цитотоксическую активность Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 4 была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 5,7E-04 до 1,4E-03 мкг/мл (8,6E-10 до 2,1E-09 M), со средним IC50 значением для серии цельноклеточных анализов 9,7E-04 мкг/мл (1,5E-09 M). Присутствие амин-содержащей группы в Соединении 4 слегка снижало цитотоксическую активность соединения, по сравнению с Соединением 40. Цитотоксическая активность Соединения 4 также не зависела от CD20 статуса опухолевых клеточных линий (Таблица 46).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 12
Активность соединения 12 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E+00 до 2,6E-04 мкг/мл (7,9E-07 до 2,1E-10 M). Цитотоксическая активность Соединения 12, в двух независимых экспериментах, была в высшей степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 2,2E-02 до 6,7E-02 мкг/мл (1,8E-08 до 5,5E-08 M), со средним IC50 значением для серии цельноклеточных анализов 3,9E-02 мкг/мл (3,1E-08 M). Присутствие малеимид-содержащего линкера в Соединении 12 снижало цитотоксическую активность соединения, по сравнению с Соединением 4 и Соединением 40. Кроме того, цитотоксичность соединения также не зависела от CD20 статуса опухолевых клеточных линий (Таблица 47).
Обобщенные данные in vitro цитотоксичности Соединения 12
Цитотоксичность ADC13
Цитотоксическую активность ADC13 испытывали против различных опухолевых клеточных линий. Конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, соответственно. Репрезентативная DR кривая (исходная концентрация 1 мкг/мл) представлена на Фиг. 27. Хотя и более высокую в CD20 положительных клетках Raji, ADC13 показал практически одинаковую цитотоксическую активность, в наномолярном диапазоне, во всех испытываемых клеточных линиях. Raji клетки (CD20+) показали среднее IC50 значение 2,5E+01 мкг/мл, тогда как соответствующие значения для RPMI-8226 и Karpas-299 клеток (обе CD20-) были 1,1E+00 мкг/мл (Таблица 48). Таким образом, CD20 положительные клетки были несколько более чувствительными (примерно в 5 раз) к ADC13, чем CD20 отрицательные клетки.
Обобщенные данные in vitro цитотоксичности ADC13
Для графического сравнения цитотоксической активности mAb Ритуксимаба, используемого отдельно, с цитотоксичностью конъюгата ADC13, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий mAb отдельно (50 мкг/мл) или конъюгатом ADC13 (1 и 0,1 мкг/мл), представлены на Фиг. 28. Ритуксимаб, используемый отдельно, при концентрации 50 мкг/мл был фактически неактивным во всех испытываемых клеточных линиях, независимо от их CD20 статуса. С другой стороны, ADC13 показал, при обеих концентрациях 1 и 0,1 мкг/мл, цитотоксическую активность, с некоторой специфичностью в отношении CD20 экспрессирующих Raji клеток. При концентрации 1 мкг/мл, ADC13 вызывал, после 72 часов обработки, более чем 65% снижение клеточного выживания Raji клеток (CD20+), при этом индуцируя 35-45% в RPMI-8226 и Karpas-299 клетках (CD20-), соответственно. При 0,1 мкг/мл, ADC13 конъюгат показал более четкую специфичность, вызывая снижение клеточного выживания примерно 50% в CD20 положительных клетках, будучи при этом фактически неактивным в CD20 отрицательных клетках.
Пример биоактивности 14 - Цитотоксичность ADC14 и соответствующих реагентов против CD5 положительных и отрицательных опухолевых клеток человека
In vitro цитотоксическую активность ADC14, наряду с исходными цитотоксическими Соединениями 1, 4 и 40, оценивали против различных раковых клеточных линий человека, экспрессирующих или не экспрессирующих CD5 антиген, включая Karpas-299 и MOLT-4 (обе CD5+); Raji и RPMI-8226 (обе CD5-). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Анти-CD5 mAb
Прежде всего, in vitro цитотоксическую активность анти-CD5 мышиного mAb отдельно испытывали против различных раковых клеточных линий человека, экспрессирующих или не экспрессирующих CD5 антиген, включая Karpas-299 и MOLT-4 (обе CD5+); Raji и RPMI-8226 (обе CD5-). В полученных в трех повторностях DR кривых, охватывающих от 5,0E+01 до 1,3E-02 мкг/мл (3,3E-07 до 8,7E-11 M), в двух независимых экспериментах, антитело было фактически неактивным, не достигая значений IC50 в какой-либо из испытываемых клеточных линий, независимо от их CD5 статуса (Таблица 49).
Цитотоксичность Соединения 40
Цитотоксическую активность исходного Соединения 40 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-03 до 2,6E-07 мкг/мл (1,7E-09 до 4,3E-13 M). Цитотоксическая активность Соединения 40 была в высшей степени гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком субнаномолярном диапазоне, от 7,5E-05 до 3,6E-04 мкг/мл (1,2E-10 до 5,9E-10 M), со средним IC50 значением для серии цельноклеточных анализов 1,6E-04 мкг/мл (эквивалентно 2,6E-10 M). Цитотоксичность Соединения 40 не зависела от уровней экспрессии CD5 в опухолевых клеточных линиях (Таблица 50).
Обобщенные данные in vitro цитотоксичности Соединения 40
Цитотоксичность Соединения 4
Цитотоксическую активность Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 4 была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 6,3E-04 до 2,7E-03 мкг/мл (9,5E-10 до 4,1E-09 M), со средним IC50 значением для серии цельноклеточных анализов 1,3E-03 мкг/мл (1,9E-09 M). Присутствие амин-содержащей группы в Соединении 4 снижало цитотоксическую активность соединения (примерно 1 log), по сравнению с Соединением 40. Цитотоксическая активность Соединения 4 также не зависела от уровней экспрессии CD5 в опухолевых клеточных линиях (Таблица 51).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 1
Активность соединения 1 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-01 до 2,6E-05 мкг/мл (1,2E-07 до 3,0E-11 M). Цитотоксическая активность Соединения 1 была практически гомогенной в различных испытываемых клеточных линиях, за исключением Raji клеток, в которых соединение было менее активным, с IC50 значениями в наномолярном диапазоне, от 1,8E-03 до 1,1E-02 мкг/мл (2,2E-09 до 1,3E-09 M), со средним IC50 значением для серии цельноклеточных анализов 4,6E-03 мкг/мл (5,3E-09 M). Присутствие малеимид-содержащего линкера в Соединении 1 не изменяло цитотоксическую активность соединения по сравнению с Соединением 4. Цитотоксичность соединения также не зависела от уровней экспрессии CD5 в опухолевых клеточных линиях (Таблица 52).
Обобщенные данные in vitro цитотоксичности Соединения 1
Цитотоксичность ADC14
Цитотоксическую активность ADC14 испытывали против различных опухолевых клеточных линий. Конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, соответственно. Репрезентативная DR кривая (исходная концентрация 1 мкг/мл) представлен на Фиг. 29. ADC14 показал некоторую тенденцию селективности против CD5 положительных клеток, Karpas-299 и хотя средние IC50 значения, в среднем наномолярном диапазоне, были примерно одинаковыми во всех испытываемых клеточных линиях, независимо от их CD5 статуса (Таблица 53).
Обобщенные данные in vitro цитотоксичности ADC14
Для графического сравнения цитотоксической активности анти-CD5 mAb, используемого отдельно, с цитотоксичностью конъюгата ADC14, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC14 (1 и 0,1 мкг/мл), представлены на Фиг. 30. Анти-CD5 mAb, используемый отдельно, при концентрации 50 мкг/мл, был фактически неактивным во всех испытываемых клеточных линиях. ADC14, при концентрации 1 мкг/мл, показал специфическую цитотоксическую активность против CD5 положительных клеток, Karpas-299 и MOLT-4, индуцируя ингибирование клеточного выживания примерно 84% и 70%, соответственно, при этом был фактически неактивным в CD5 отрицательных клетках (Фиг. 30).
Пример биоактивности 15 - Цитотоксичность ADC16 и соответствующих реагентов против CD4 положительных и отрицательных опухолевых клеток человека
In vitro цитотоксическую активность ADC16, наряду с исходными цитотоксическими Соединениями 1, 4 и 40, оценивали против различных раковых клеточных линий человека, экспрессирующих или не экспрессирующих CD4 антиген, включая Karpas-299 и U937 (обе CD4+); Raji и RPMI-8226 (обе CD4-). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Анти-CD4 mAb
Прежде всего, in vitro цитотоксическую активность анти-CD4 мышиного mAb, используемого отдельно, испытывали против различных раковых клеточных линий человека, экспрессирующих или не экспрессирующих CD4 антиген, включая Karpas-299 и U937 (обе CD4+); Raji и RPMI-8226 (обе CD4-). В полученных в трех повторностях DR кривых, охватывающих от 5,0E+01 до 1,3E-02 мкг/мл (3,3E-07 до 8,7E-11 M), в двух независимых эксперименах, антитело было фактически неактивным, не достигая значений IC50 в какой-либо из испытываемых клеточных линий, независимо от их CD4 статуса (Таблица 54).
Обобщенные данные in vitro цитотоксичности Анти-CD4 mAb
Цитотоксичность Соединения 40
Цитотоксическую активность исходного Соединения 40 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-03 до 2,6E-07 мкг/мл (1,7E-09 до 4,3E-13 M). Цитотоксическая активность Соединения 40 была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком субнаномолярном диапазоне, от 7,9E-05 до 2,8E-04 мкг/мл (1,3E-10 до 4,7E-10 M), со средним IC50 значением для серии цельноклеточных анализов 1,5E-04 мкг/мл (эквивалентно 2,5E-10 M). Цитотоксичность Соединения 40 не зависела от уровней экспрессии CD4 в опухолевых клеточных линиях (Таблица 55).
Обобщенные данные in vitro цитотоксичности Соединения 40
Цитотоксичность Соединения 4
Цитотоксическую активность Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 4 также была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 6,1E-04 до 2,7E-03 мкг/мл (9,2E-10 до 4,1E-09 M), со средним IC50 значением для серии цельноклеточных анализов 1,2E-03 мкг/мл (1,8E-09 M). Присутствие амин-содержащей группы в Соединении 4 снижало цитотоксическую активность соединения по сравнению с Соединением 40. Цитотоксическая активность Соединения 4 также не зависела от уровней экспрессии CD4 в опухолевых клеточных линиях (Таблица 56).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 1
Активность соединения 1 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-01 до 2,6E-05 мкг/мл (1,2E-07 до 3,0E-11 M). Цитотоксическая активность Соединения 1 также была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 1,5E-03 до 7,3E-03 мкг/мл (1,7E-09 до 8,6E-09 M), со средним IC50 значением для серии цельноклеточных анализов 3,4E-03 мкг/мл (3,9E-09 M). Присутствие малеимид-содержащего линкера в Соединении 1 не изменяло цитотоксическую активность соединения по сравнению с Соединением 4. Цитотоксичность соединения также не зависела от уровней экспрессии CD4 в опухолевых клеточных линиях (Таблица 57).
Обобщенные данные in vitro цитотоксичности Соединения 1
Цитотоксичность ADC16
Цитотоксическую активность ADC16 испытывали против различных опухолевых клеточных линий. Конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, соответственно. Репрезентативная DR кривая (исходная концентрация 1 мкг/мл) представлен на Фиг. 31. ADC16 показал некоторую специфичность против CD4 положительных клеток, хотя средняя разница в чувствительности в отношении CD4 отрицательных клеток была относительно низкой, приблизительно в 7 раз (с максимальной разницей между наименее и наиболее чувствительной клеточной линией, Raji и Karpas-299, соответственно, примерно в 14 раз) (Таблица 58). Хотя демонстрируя небольшое терапевтическое окно, по-видимому, по меньшей мере часть наблюдаемой цитотоксичности ADC18 была опосредована взаимодействием mAb и CD4 гликопротеина в клеточной мембране опухолевых клеток.
Обобщенные данные in vitro цитотоксичности ADC16
Для графического сравнения цитотоксической активности анти-CD4 mAb, используемого отдельно, с цитотоксичностью конъюгата ADC16, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC16 (1 и 0,1 мкг/мл), представлены на Фиг. 32. Анти-CD4 mAb, используемый отдельно, при концентрации 50 мкг/мл, был фактически неактивным во всех испытываемых клеточных линиях. С другой стороны, ADC16, при концентрации 1 мкг/мл, показал сильную цитотоксическую активность во всех испытываемых клеточных линиях, независимо от их CD4 статуса, вызывая более чем 60% уменьшение (в пределах 60-90%) клеточного выживания после 72 часов обработки. Даже при концентрации 0,1 мкг/мл, ADC16 показал специфическую цитотоксическую активность против CD4 положительных клеток, Karpas-299 и U937, индуцируя ингибирование клеточного выживания примерно 80% и 70%, соответственно, будучи при этом неактивным против CD4 отрицательных клеток (Фиг. 32).
Пример биоактивности 16 - Цитотоксичность ADC17 и соответствующих реагентов против CD4 положительных и отрицательных опухолевых клеток человека
In vitro цитотоксическую активность ADC17, наряду с исходными цитотоксическими Соединения 12, 4 и 40, оценивали против различных раковых клеточных линий человека, экспрессирующих или не экспрессирующих CD4 антиген, включая Karpas-299 и U937 (обе CD4+); Raji и RPMI-8226 (обе CD4-). Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 40
Цитотоксическую активность исходного Соединения 40 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-03 до 2,6E-07 мкг/мл (1,7E-09 до 4,3E-13 M). Цитотоксическая активность Соединения 40 была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в низком субнаномолярном диапазоне, от 7,9E-05 до 2,8E-04 мкг/мл (1,3E-10 до 4,7E-10 M), со средним IC50 значением для серии цельноклеточных анализов 1,5E-04 мкг/мл (эквивалентно 2,5E-10 M). Цитотоксичность Соединения 40 не зависела от уровней экспрессии CD4 в опухолевых клеточных линиях (Таблица 59).
Обобщенные данные in vitro цитотоксичности Соединения 40
Цитотоксичность Соединения 4
Цитотоксическую активность Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 4 также была гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 6,1E-04 до 2,7E-03 мкг/мл (9,2E-10 до 4,1E-09 M), со средним IC50 значением для серии цельноклеточных анализов 1,2E-03 мкг/мл (1,8E-09 M). Присутствие амин-содержащей группы в Соединении 4 снижало цитотоксическую активность соединения по сравнению с Соединением 40. Цитотоксическая активность Соединения 4 также не зависела от уровней экспрессии CD4 в опухолевых клеточных линиях (Таблица 60).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 12
Активность Соединения 12 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E+00 до 2,6E-04 мкг/мл (7,9E-07 до 2,1E-10 M). Цитотоксическая активность Соединения 12, в двух независимых экспериментах, была относительно гомогенной в различных испытываемых клеточных линиях, с IC50 значениями в наномолярном диапазоне, от 7,3E-02 до 4,1E-01 мкг/мл (5,8E-08 до 3,2E-07 M), со средним IC50 значением для серии цельноклеточных анализов 1,7E-01 мкг/мл (1,3E-07 M). Присутствие длинного малеимид-содержащего линкера в Соединении 12 сильно снижало цитотоксическую активность соединения по сравнению с Соединением 40 (около 3 log) и Соединением 4 (около 2 log). Кроме того, цитотоксичность соединения также не зависела от уровней экспрессии CD4 опухолевых клеточных линий (Таблица 61).
Обобщенные данные in vitro цитотоксичности Соединения 12
Цитотоксичность ADC17
Цитотоксическую активность ADC17 испытывали против различных опухолевых клеточных линий. Конъюгат испытывали в четырех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 50, 10, 1 и 0,1 мкг/мл, соответственно. Репрезентативная DR кривая (исходная концентрация 1 мкг/мл) представлена на Фиг. 33. ADC17 показал специфичность против CD4 положительных клеток, со средней разницей в чувствительности в отношении CD4 отрицательных клеток примерно в 40 раз (в пределах от 11 до 64 раз) (Таблица 62). По-видимому основная часть наблюдаемой цитотоксической активности ADC17 была опосредована взаимодействием mAb и CD4 гликопротеина в клеточной мембране опухолевых клеток.
Обобщенные данные in vitro цитотоксичности ADC17
Для графического сравнения цитотоксической активности анти-CD4 mAb, используемого отдельно, с цитотоксичностью конъюгата ADC17, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC17 (1 и 0,1 мкг/мл), представлены на Фиг. 34. Анти-CD4 mAb отдельно, при концентрации 50 мкг/мл, был фактически неактивным во всех испытываемых клеточных линиях. С другой стороны, ADC17, при концентрации 1 мкг/мл, показал сильную цитотоксическую активность в трех из четырех испытываемых клеточных линиях (за исключением Raji клеток), вызывая более чем 70% уменьшение (в пределах от 72 до 95%) клеточного выживания после 72 часов обработки. Даже при концентрации 0,1 мкг/мл, ADC17 показал специфическую цитотоксическую активность против CD4 положительных клеток, Karpas-299 и U937, индуцируя ингибирование клеточного выживания примерно 70% и 75%, соответственно, будучи при этом совершенно неактивным против CD4 отрицательных клеток (Фиг. 34).
Пример биоактивности 17 - Цитотоксичность ADC14 и соответствующих реагентов против Raji клеточных клонов с высокой или нулевой CD5 экспрессией
In vitro цитотоксическую активность ADC14, наряду с исходными цитотоксическими Соединениями 1, 4 и 40, оценивали против Raji клеточных клонов, экспрессирующих или не экспрессирующих CD5 антиген. Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Анти-CD5 mAb
Прежде всего, in vitro цитотоксическую активность анти-CD5 мышиного mAb, используемого отдельно, испытывали против Raji клеточных клонов, экспрессирующих (С#10) или не экспрессирующих (С#18) CD5 антиген. В полученных в трех повторностях DR кривых, охватывающих от 5,0E+01 до 1,3E-02 мкг/мл (3,3E-07 до 8,7E-11 M), в двух независимых экспериментах, антитело было фактически неактивным, не достигая значений IC50 в какой-либо из испытываемых клеточных линий, независимо от их CD5 статуса (Таблица 63).
Обобщенные данные in vitro цитотоксичности Анти-CD5 mAb
Цитотоксичность Соединения 40
Цитотоксическую активность исходного Соединения 40 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-03 до 2,6E-07 мкг/мл (1,7E-09 до 4,3E-13 M). Цитотоксическая активность Соединения 40 была примерно одинаковой для CD5 экспрессирующих (клон#10) или не экспрессирующих (клон#18) Raji клеток, со средними IC50 значениями в субнаномолярном диапазоне, 4,95E-04 и 8,90E-04 мкг/мл (эквивалентно 8,17E-10 и 1,47E-0,9 M) соответственно. Хотя и несколько выше в CD5 положительных клетках, цитотоксичность Соединения 40, по-видимому, была достаточно независимой от уровней экспрессии CD5 в опухолевых клеточных линиях (Таблица 64).
Обобщенные данные in vitro цитотоксичности Соединения 40
Цитотоксичность Соединения 4
Цитотоксическую активность Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 4 была примерно одинаковой для CD5 экспрессирующих (клон#10) или не экспрессирующих (клон#18) Raji клеток, хотя в нулевых клетках соединение не достигало IC50 значений. В CD5 положительных клетках соединение показало среднее IC50 значение 9,9E-03 мкг/мл (эквивалентно 1,57E-08 M). Хотя и несколько выше в CD5 положительных клетках, цитотоксичность Соединения 4, по-видимому, была достаточно независимой от уровней экспрессии CD5 в опухолевых клеточных линиях (см. IC50 значения в Таблице 65 в качестве ссылки).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 1
Активность Соединения 1 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-01 до 2,6E-05 мкг/мл (1,2E-07 до 3,0E-11 M). Цитотоксическая активность Соединения 1 является разной в двух Raji клеточных клонах, соединение более активно в сверхэкспрессирующих CD5 клетках (клон#10), чем в CD5-нулевых клетках (клон#18),со средними IC50 значениями 2,9E-03 и 3,8E-02 мкг/мл (эквивалентно 3,4E-09 и 4,4E-08 M), соответственно (Таблица 66). В этом случае цитотоксичность Соединения 1, по-видимому, не была независимой от CD5 статуса опухолевых клеточных линий.
Обобщенные данные in vitro цитотоксичности Соединения 1
Цитотоксичность ADC14
Цитотоксическую активность ADC14 испытывали против двух Raji клонов. Конъюгат испытывали в трех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 10, 1 и 0,1 мкг/мл, соответственно. Репрезентативная DR кривая (исходная концентрация 10 мкг/мл) представлена на Фиг. 35. ADC14 показал специфичность против CD5 экспрессирующих клеток (клон#10), в которых соединение продемонстрировало цитотоксическую активность, такую же или даже более высокую, чем цитотоксичность исходных Соединений 1, 4 и 40. В CD5 экспрессирующих Raji клетках, конъюгат показал среднее IC50 значение 1,6E-01 мкг/мл. В CD5-нулевых клетках конъюгат был более чем в 50 раз менее активным, чем в CD5 положительных клетках, показывая среднее IC50 значение 9,0E-00 мкг/мл. Хотя и с некоторыми оговорками, из-за несколько разной чувствительности, наблюдаемой между двумя Raji клеточными клонами против некоторых исходных соединений, эти результаты показывают, что ADC14 обладал специфичностью против CD5 экспрессирующих клеток (Таблица 67). Поэтому считают, что ADC14 действовал, по меньшей мере частично, через взаимодействие mAb с мембраносвязанным CD5 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC14
Для графического сравнения цитотоксической активности анти-CD5 mAb, используемого отдельно, с цитотоксичностью конъюгата ADC14, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC14 (10 мкг/мл), представлены на Фиг. 36. Анти-CD5 mAb отдельно, при концентрации 50 мкг/мл, был неактивным против двух Raji клеточных клонов. Наоборот, ADC14, при концентрации 10 мкг/мл, показал сильную и несколько селективную цитотоксическую активность против CD5 положительных Raji клеток (клон#10), вызывая почти 90% снижение клеточного выживания после 72 часов обработки. В тех же условиях, ADC14 вызывал 30% снижение клеточного выживания CD5-нулевых клеток (клон#18) (Фиг.36).
Пример биоактивности 18 - Цитотоксичность ADC15 и соответствующих реагентов против Raji клеточных клонов с высокой или нулевой CD5 экспрессией
In vitro цитотоксическую активность ADC15, наряду с исходными цитотоксическими Соединениями 12, 4 и 40, оценивали против Raji клеточных клонов, экспрессирующих или не экспрессирующих CD5 антиген. Были получены стандартные кривые доза-ответ (DR) для 72 часов.
Цитотоксичность Соединения 40
Цитотоксическую активность исходного Соединения 40 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-03 до 2,6E-07 мкг/мл (1,7E-09 до 4,3E-13 M). Цитотоксическая активность Соединения 40 была примерно одинаковой для CD5 экспрессирующих (клон#10) или не экспрессирующих (клон#18) Raji клеток, со средними IC50 значениями в субнаномолярном диапазоне, 4,95E-04 и 8,90E-04 мкг/мл (эквивалентно 8,17E-10 и 1,47E-0,9 M), соответственно. Хотя и несколько выше в CD5 положительных клетках, цитотоксичность Соединения 40, по-видимому, была достаточно независимой от уровней экспрессии CD5 в опухолевых клеточных линиях (Таблица 68).
Обобщенные данные in vitro цитотоксичности Соединения 40
Цитотоксичность Соединения 4
Цитотоксическую активность Соединения 4 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 4 была примерно одинаковой для CD5 экспрессирующих (клон#10) или не экспрессирующих (клон#18) Raji клеток, хотя в нулевых клетках соединение не достигало IC50 значений. В CD5 положительных клетках соединение показало среднее IC50 значение 9,9E-03 мкг/мл (эквивалентно 1,57E-08 M). Хотя и несколько выше в CD5 положительных клетках, цитотоксичность Соединения 4, по-видимому, была достаточно независимой от уровней экспрессии CD5 в опухолевых клеточных линиях (см. IC50 значения в Таблице 69 в качестве ссылки).
Обобщенные данные in vitro цитотоксичности Соединения 4
Цитотоксичность Соединения 12
Активность Соединения 12 оценивали в DR кривых с использованием десяти серийных разведений (1/2,5 отношение) от 01E-02 до 2,6E-06 мкг/мл (1,5E-08 до 4,0E-12 M). Цитотоксическая активность Соединения 12 была примерно одинаковой для CD5 экспрессирующих (клон#10) или не экспрессирующих (клон#18) Raji клеток, хотя в нулевых клетках соединение не достигало IC50 значений. В CD5 положительных клетках соединение показало среднее IC50 значение 2,7E-01 мкг/мл (эквивалентно 2,15E-07 M). Хотя и несколько выше в CD5 положительных клетках, цитотоксичность Соединения 12, по-видимому, была достаточно независимой от уровней экспрессии CD5 в опухолевых клеточных линиях (см. IC50 значения в Таблице 70 в качестве ссылки).
Обобщенные данные in vitro цитотоксичности Соединения 12
Цитотоксичность ADC15
Цитотоксическую активность ADC15 испытывали против двух Raji клонов. Конъюгат испытывали в трех различных диапазонах концентраций, каждый в полученных в трех повторностях DR кривых (десять серийных разведений, 1/2,5 отношение), исходя из 10, 1 и 0,1 мкг/мл, соответственно. Репрезентативная DR кривая (исходная концентрация 10 мкг/мл) представлена на Фиг. 37. ADC15 показал значительную специфичность против CD5 сверхэкспрессирующих клеток (клон#10), в которых соединение продемонстрировало цитотоксическую активность, такую же, как у исходного Соединения 40, и даже более высокую, чем у Соединений 4 и 12. В CD5 экспрессирующих Raji клетках, конъюгат показал среднее IC50 значение 9,3E-01 мкг/мл. В CD5-нулевых клетках конъюгат был более чем в 10 раз менее активным, чем в CD5 положительных клетках, не достигая IC50 значения. Хотя и с некоторыми оговорками, из-за потенциальной чувствительности, наблюдаемой между двумя Raji клеточными клонами против некоторых исходных Соединений 4 и 12, эти результаты показывают, что ADC15 обладал специфичностью против CD5 экспрессирующих клеток (Таблица 71). Поэтому считают, что ADC15 действовал, по меньшей мере частично, через взаимодействие mAb с мембраносвязанным CD5 рецептором на опухолевых клетках, с последующей внутриклеточной доставкой цитотоксического лекарственного средства в ткань, которая является мишенью.
Обобщенные данные in vitro цитотоксичности ADC15
Для графического сравнения цитотоксической активности анти-CD5 mAb, используемого отдельно, с цитотоксичностью конъюгата ADC15, гистограммы, показывающие проценты клеточного выживания после обработки различных клеточных линий одним только mAb (50 мкг/мл) или ADC15 (10 мкг/мл), представлены на Фиг. 38. Анти-CD5 mAb отдельно, при концентрации 50 мкг/мл, был неактивным против двух Raji клеточных клонов, независимо от их CD5 статуса. Наоборот, ADC15, при концентрации 10 мкг/мл, показал сильную и селективную цитотоксическую активность против CD5 положительных Raji клеток (клон#10), вызывая 80% снижение клеточного выживания после 72 часов обработки. В тех же условиях, ADC15 был неактивным на CD5-нулевых клетках (клон#18) (Фиг.38).
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ ПРОИЗВОДНОГО АНТРАЦИКЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2523419C2 |
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, НАГРУЖЕННЫЙ БИНАРНЫМИ ТОКСИНАМИ, И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2795148C2 |
| СУЛЬФОНАМИДСОДЕРЖАЩИЕ СВЯЗЫВАЮЩИЕ СИСТЕМЫ ДЛЯ ЛЕКАРСТВЕННЫХ КОНЪЮГАТОВ | 2014 |
|
RU2729194C2 |
| ЦИТОТОКСИЧЕСКИЕ И АНТИМИТОТИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2723651C2 |
| ПИРРОЛОБЕНЗОДИАЗЕПИНОВЫЕ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2736725C1 |
| КОНЪЮГАТЫ АНТИ-CD22 АНТИТЕЛО-МАЙТАНСИН И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2744895C2 |
| ЦИТОТОКСИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2012 |
|
RU2631498C2 |
| СИСТЕМЫ ДОСТАВКИ ЛЕКАРСТВ НА ОСНОВЕ МАЙТАНЗИНОИДА | 2018 |
|
RU2783076C2 |
| ПЕПТИДОМИМЕТИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ КОНЪЮГАТЫ АНТИТЕЛ С ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ | 2014 |
|
RU2689388C1 |
| АНТИ-НЕR2 АНТИТЕЛО И ЕГО КОНЪЮГАТ | 2014 |
|
RU2656161C1 |
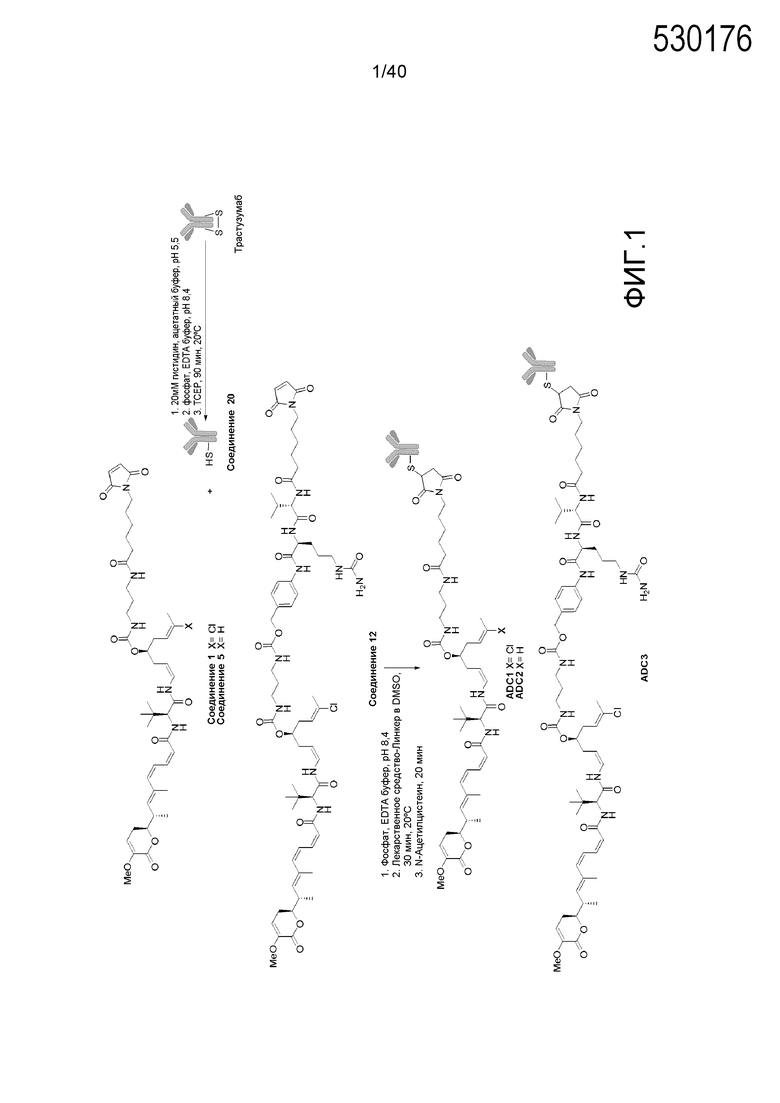

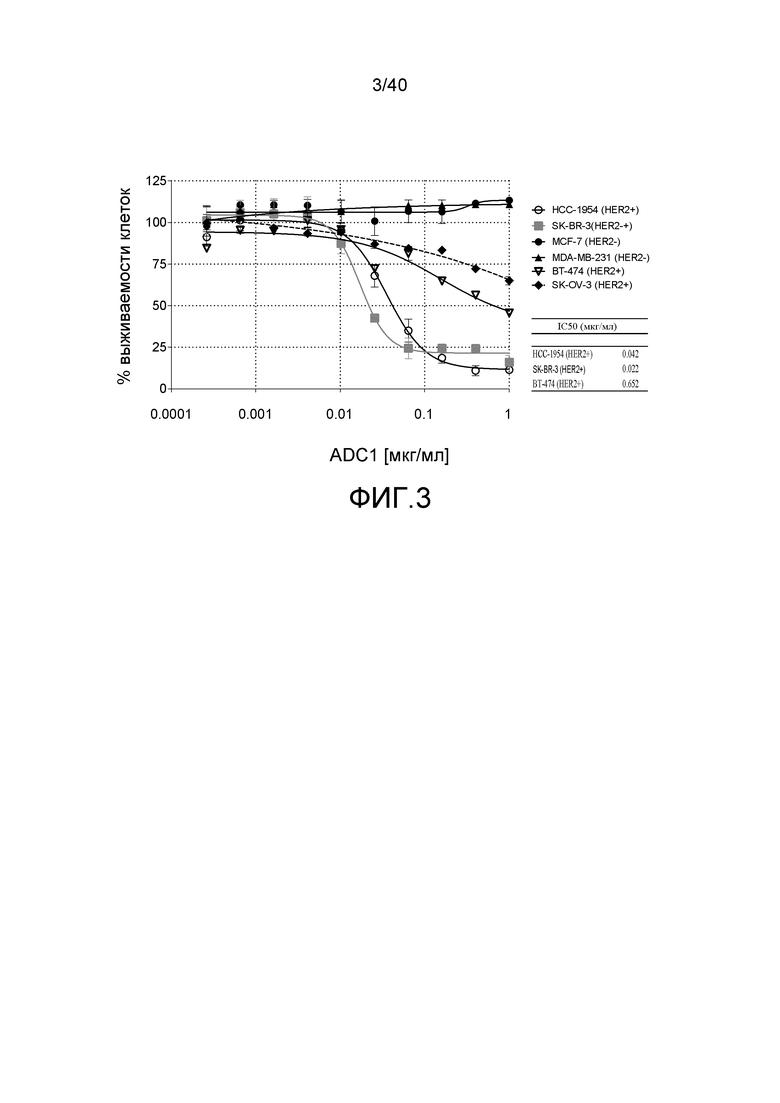

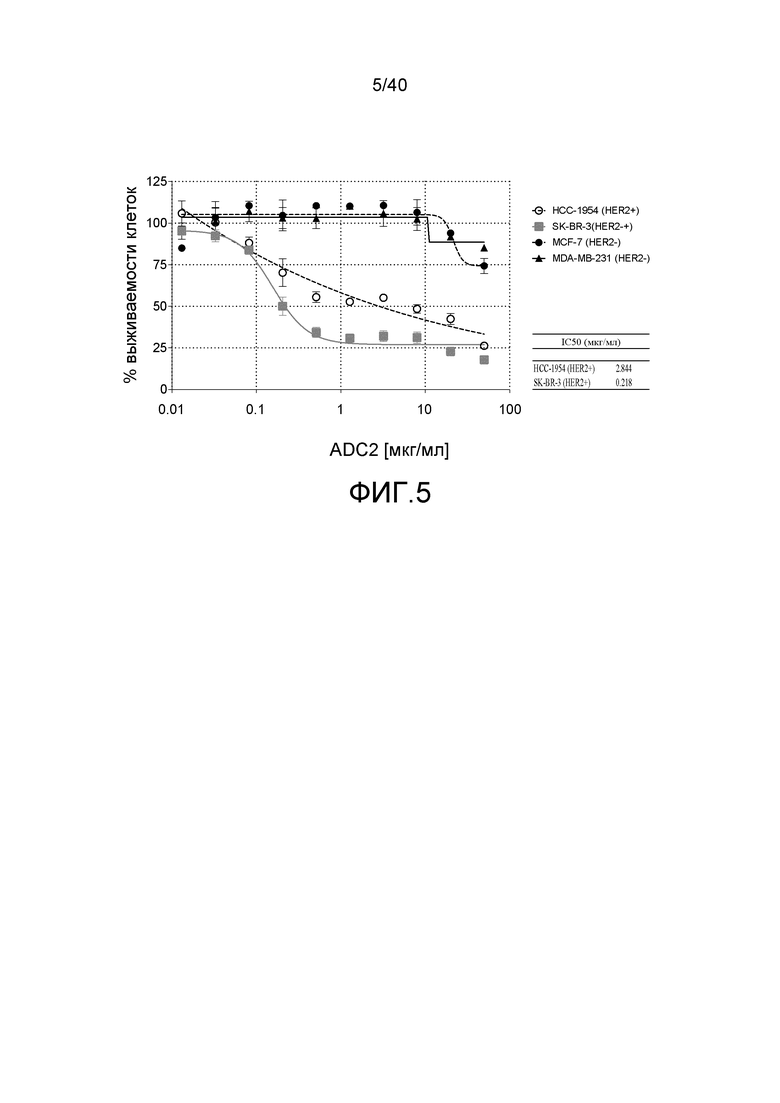


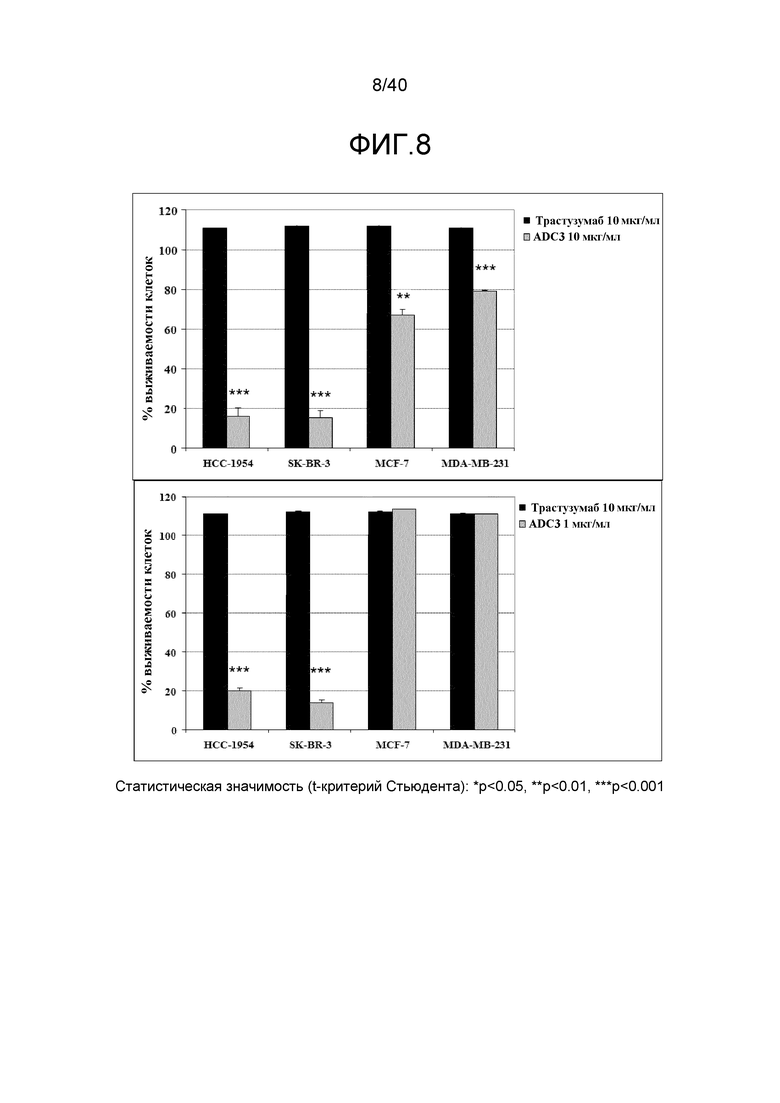
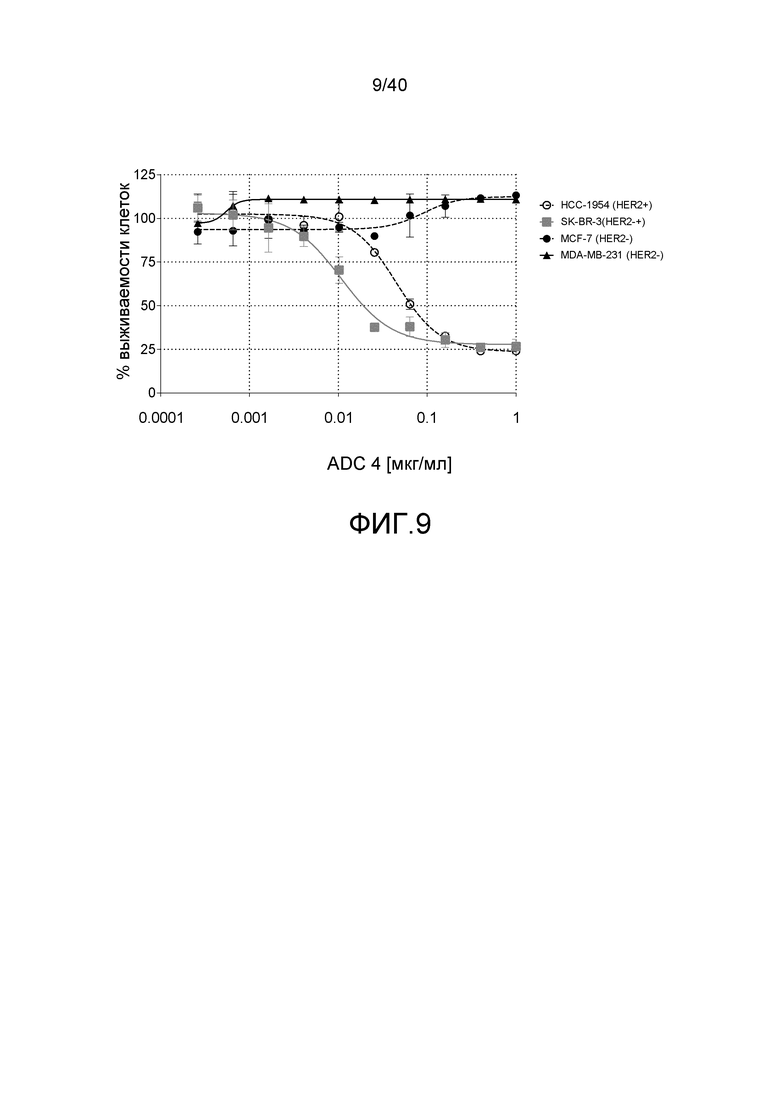
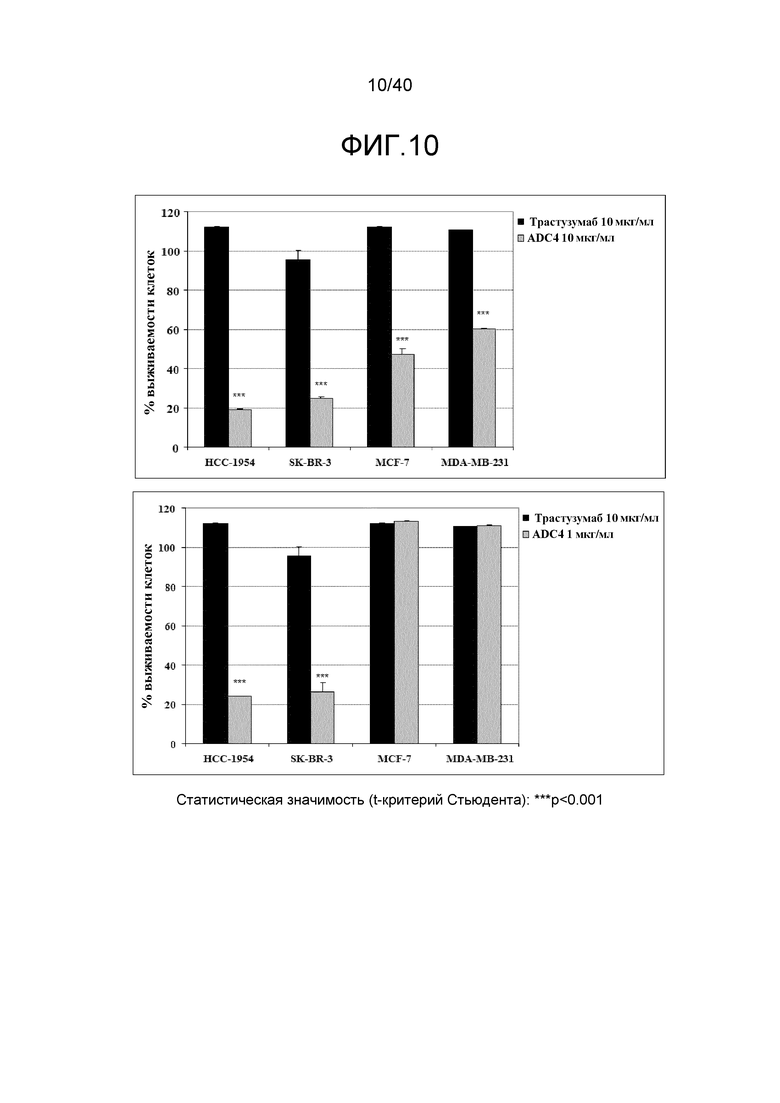
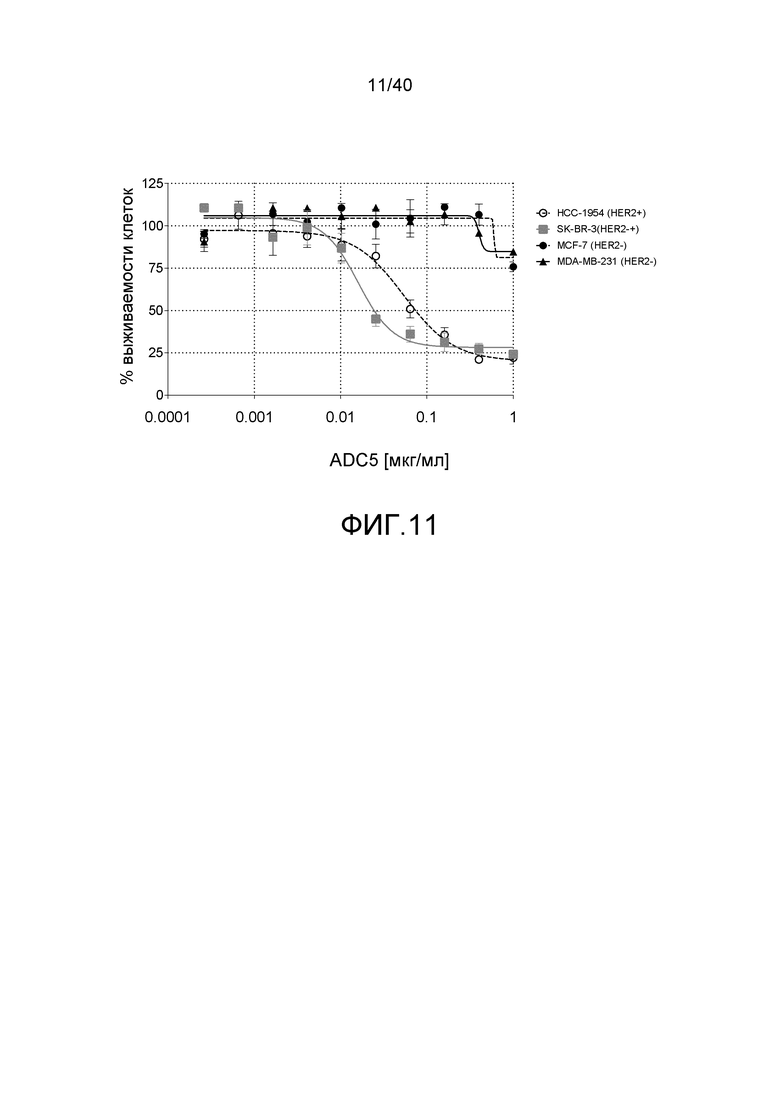

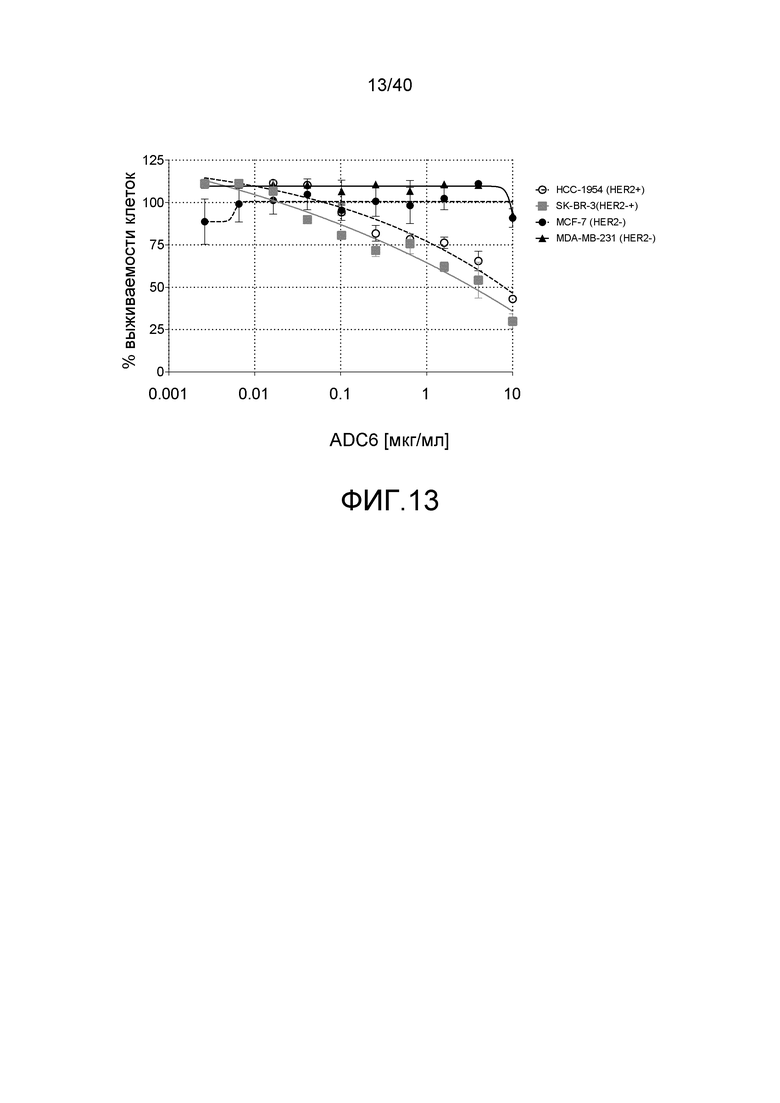
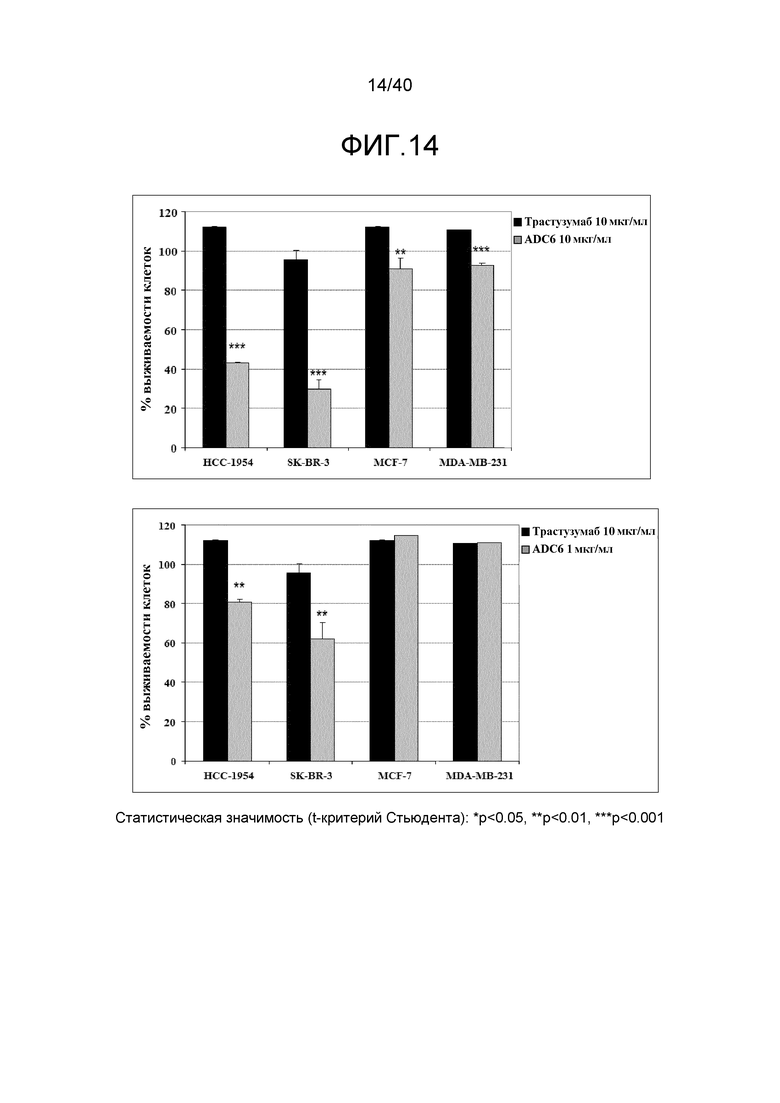

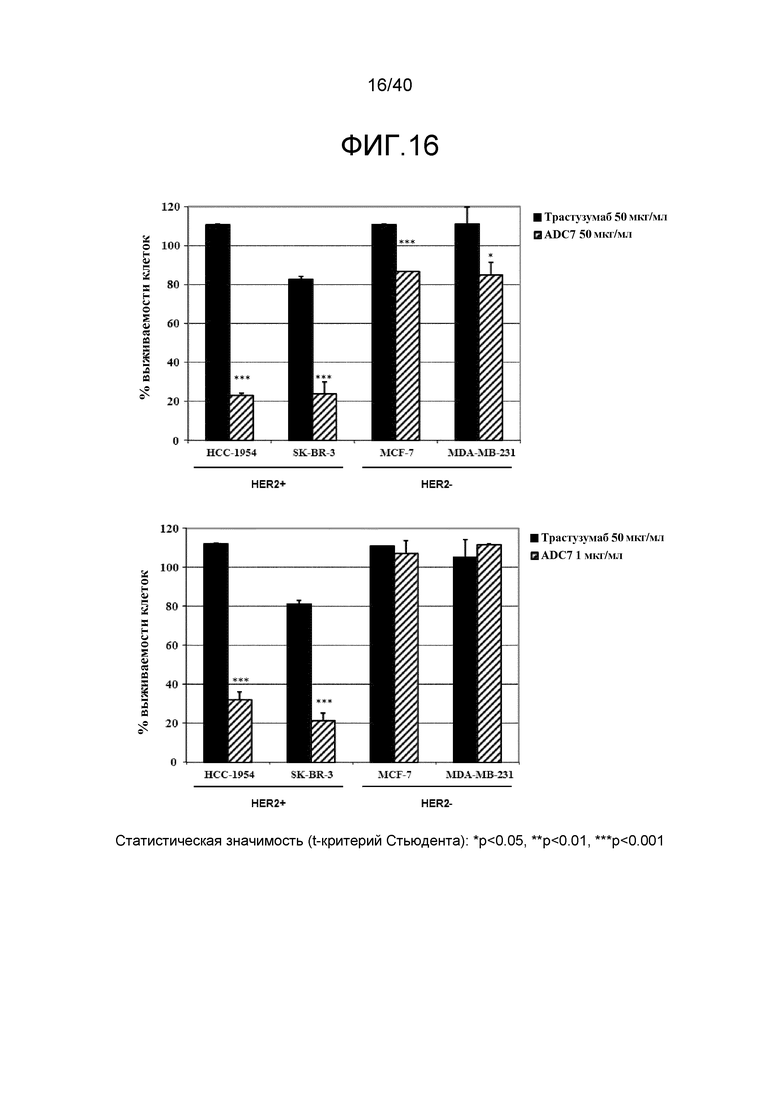
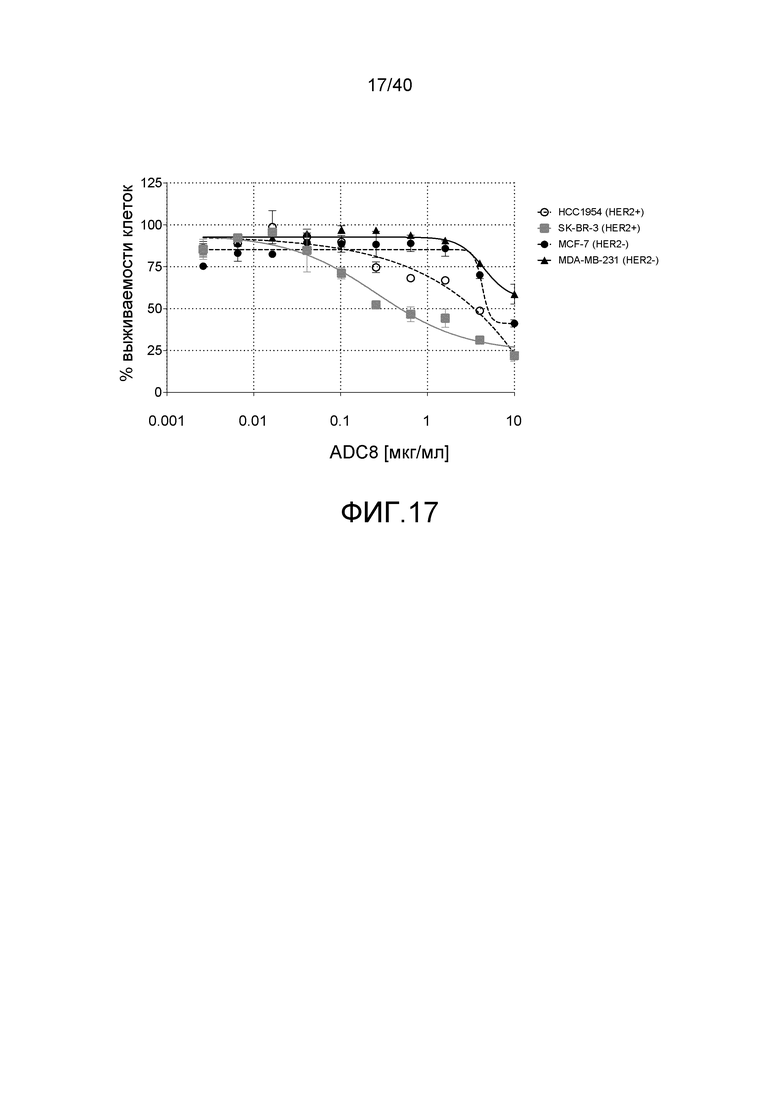
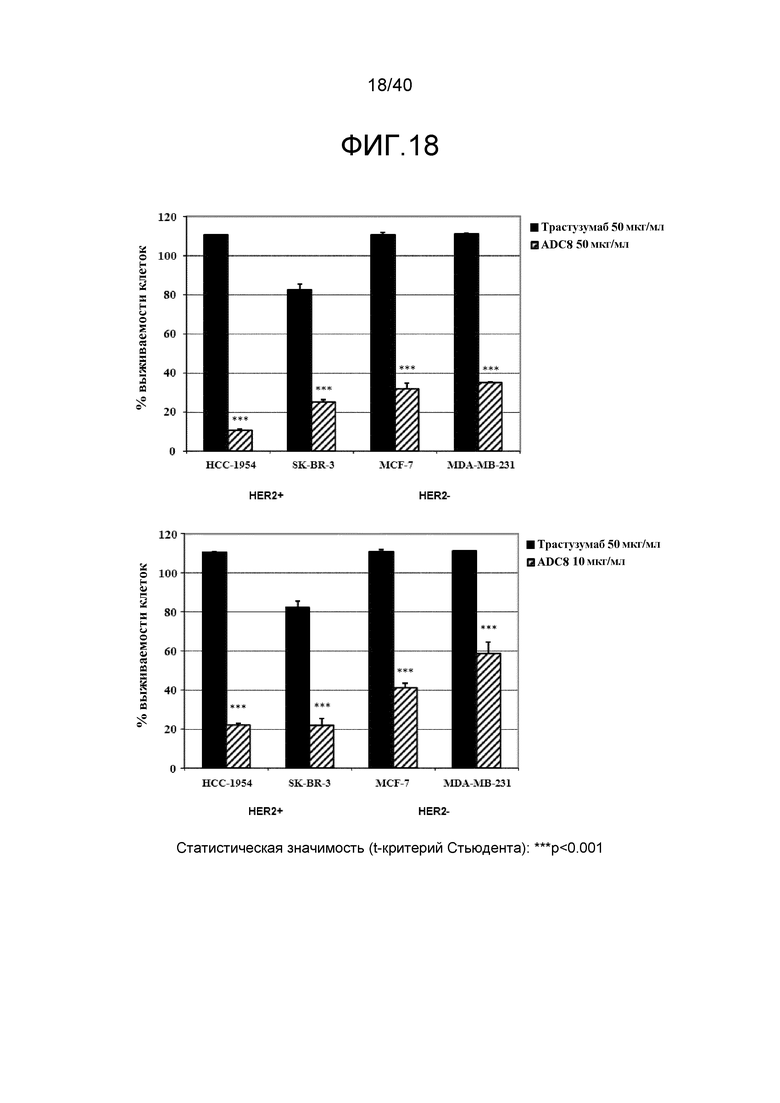
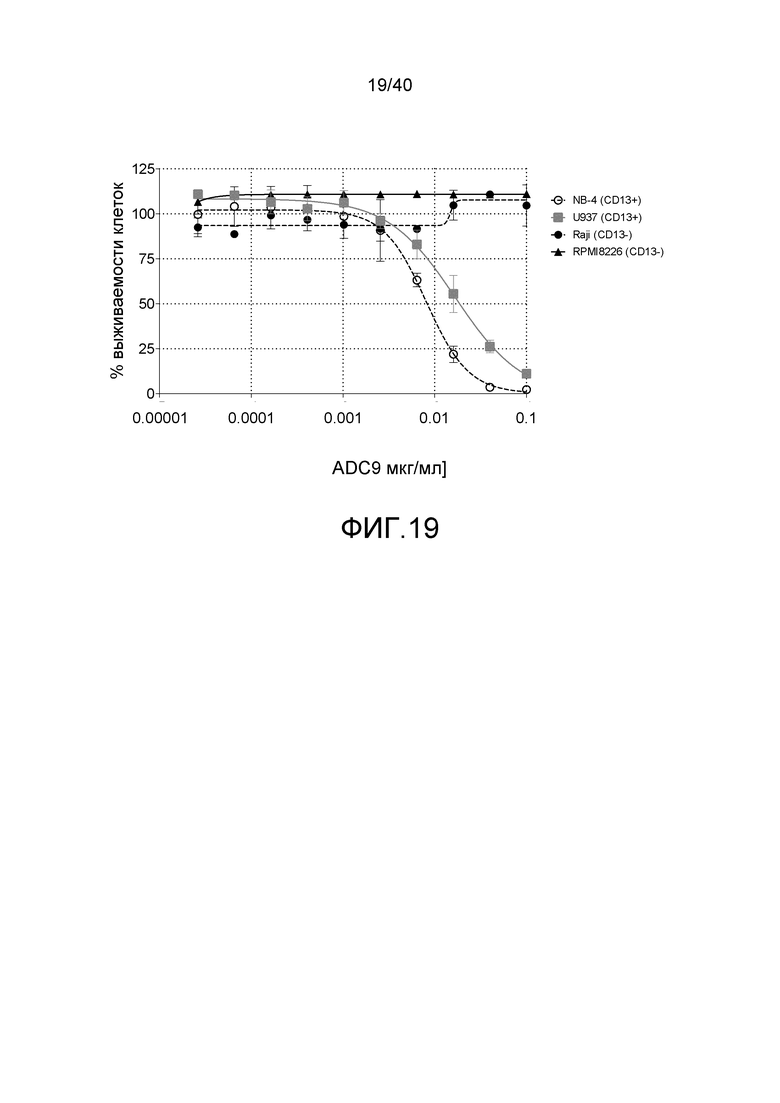
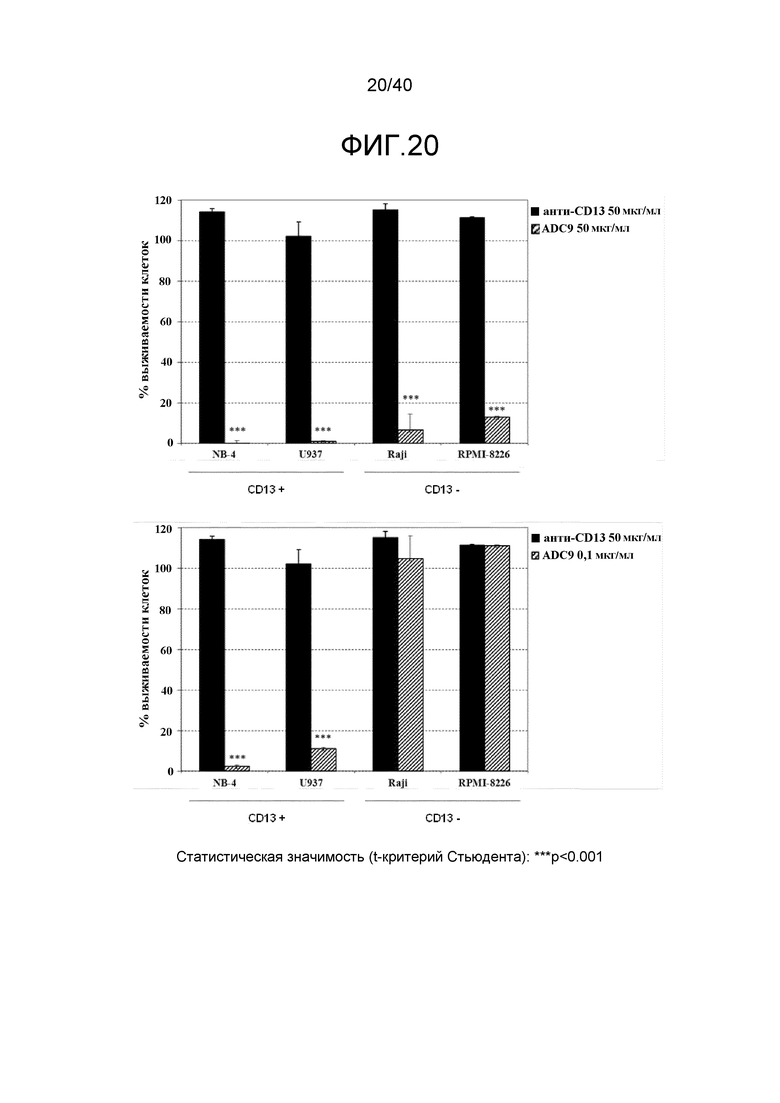

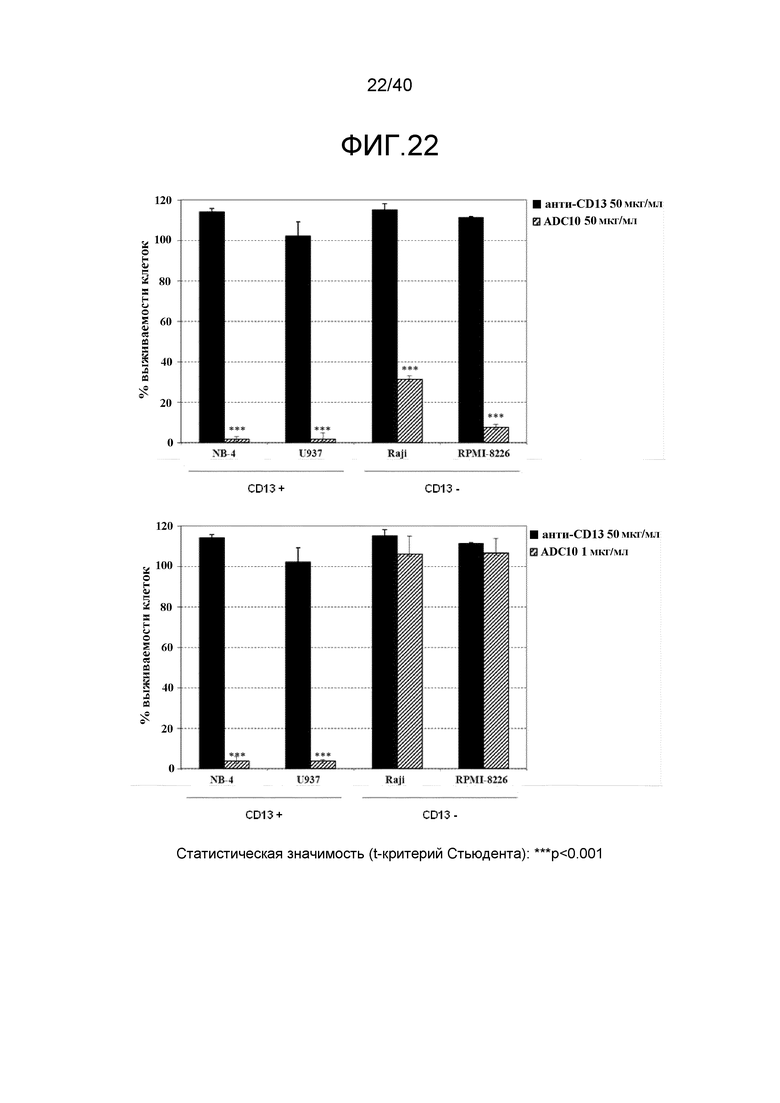
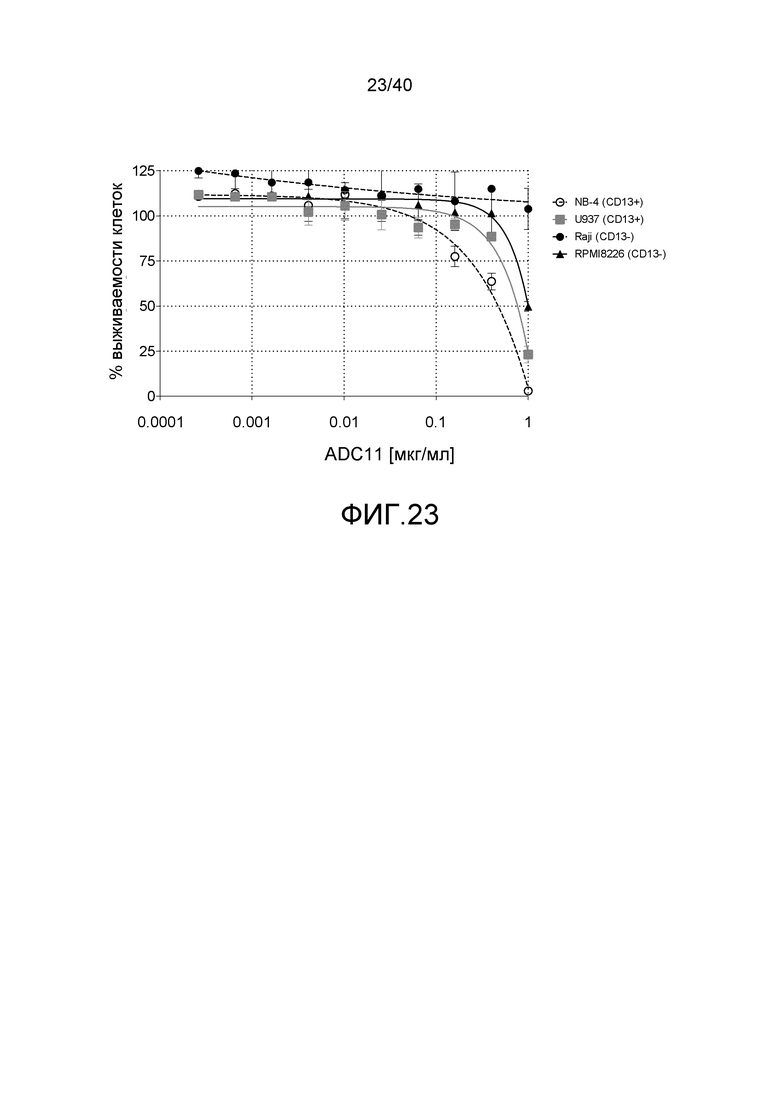
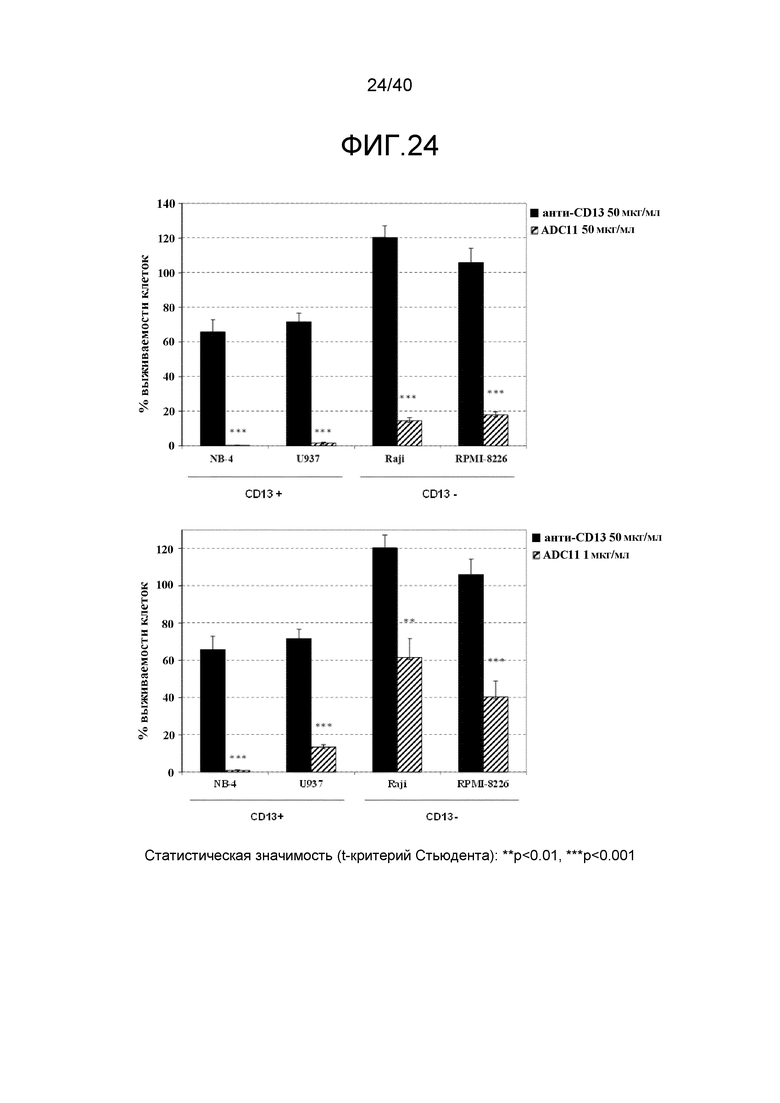
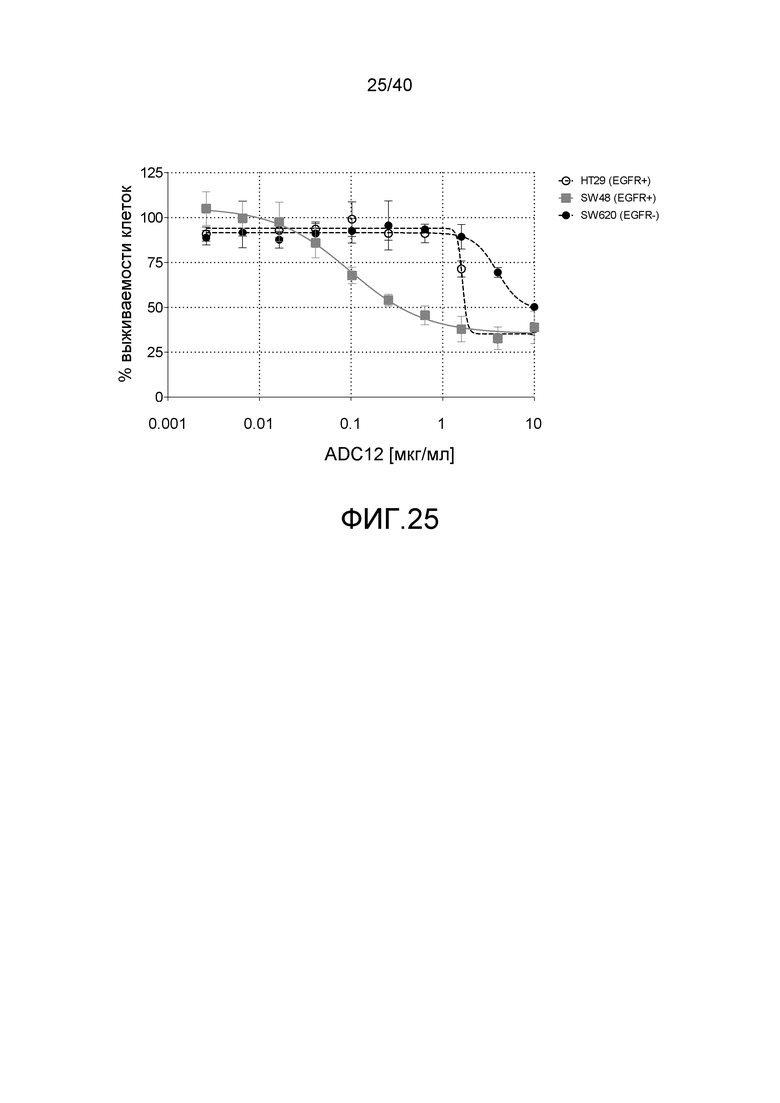
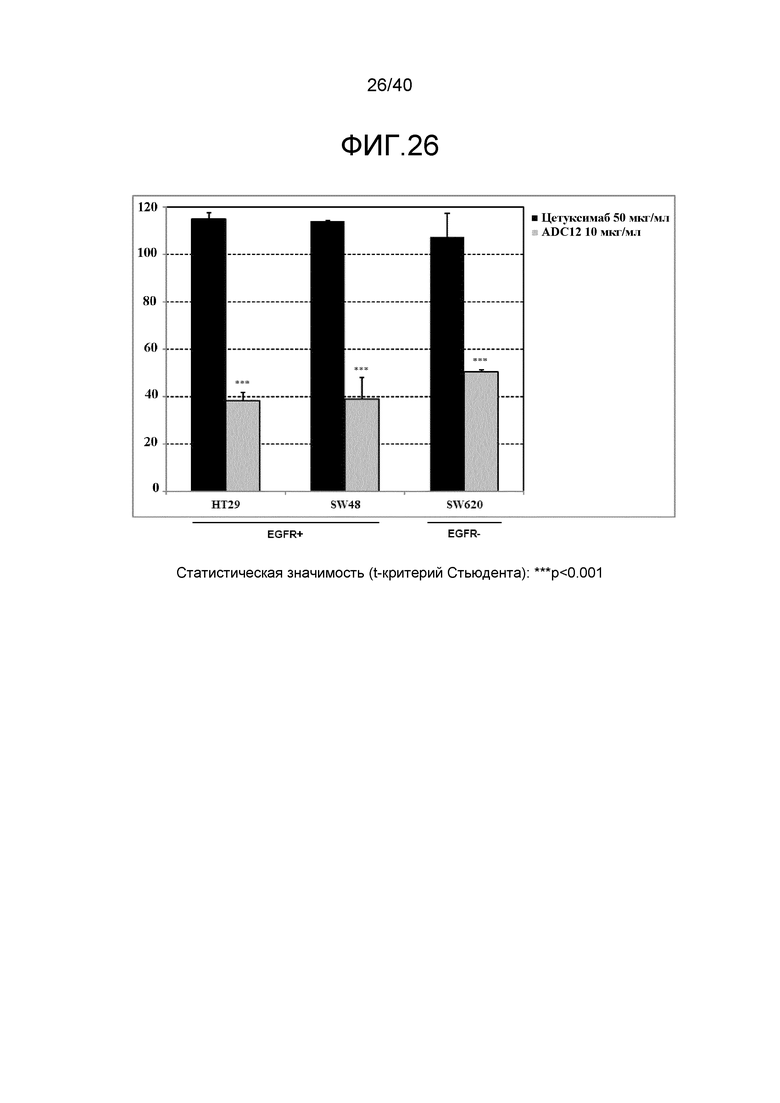

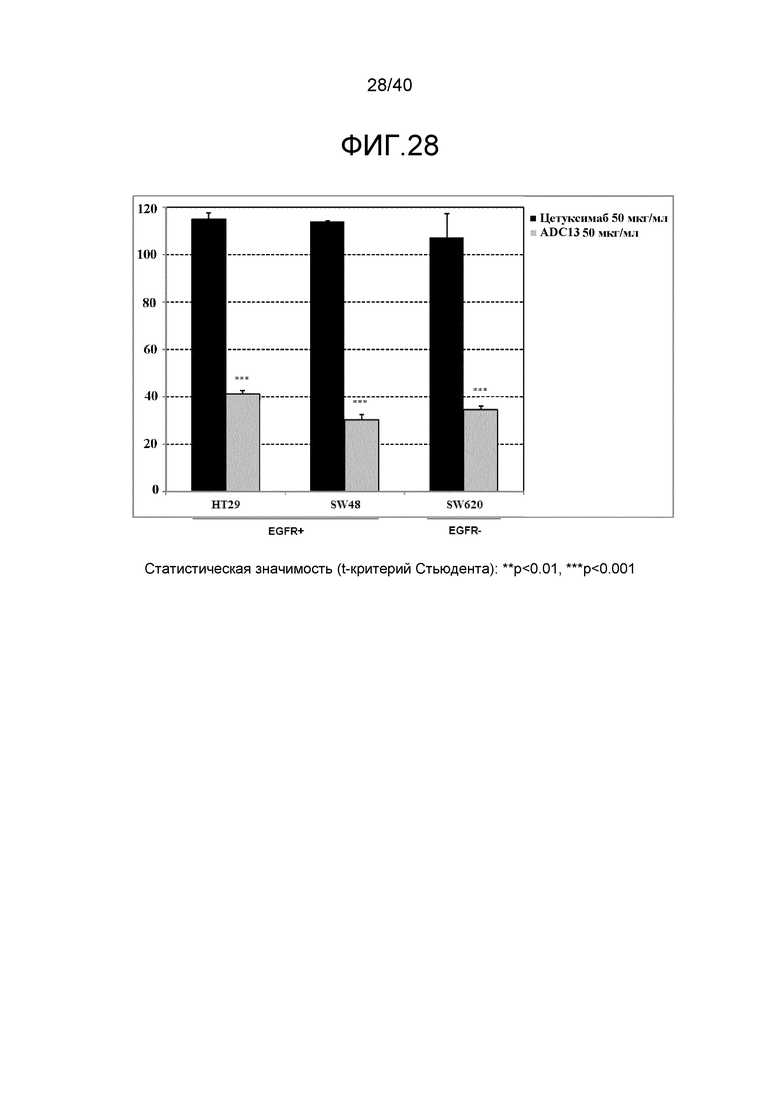
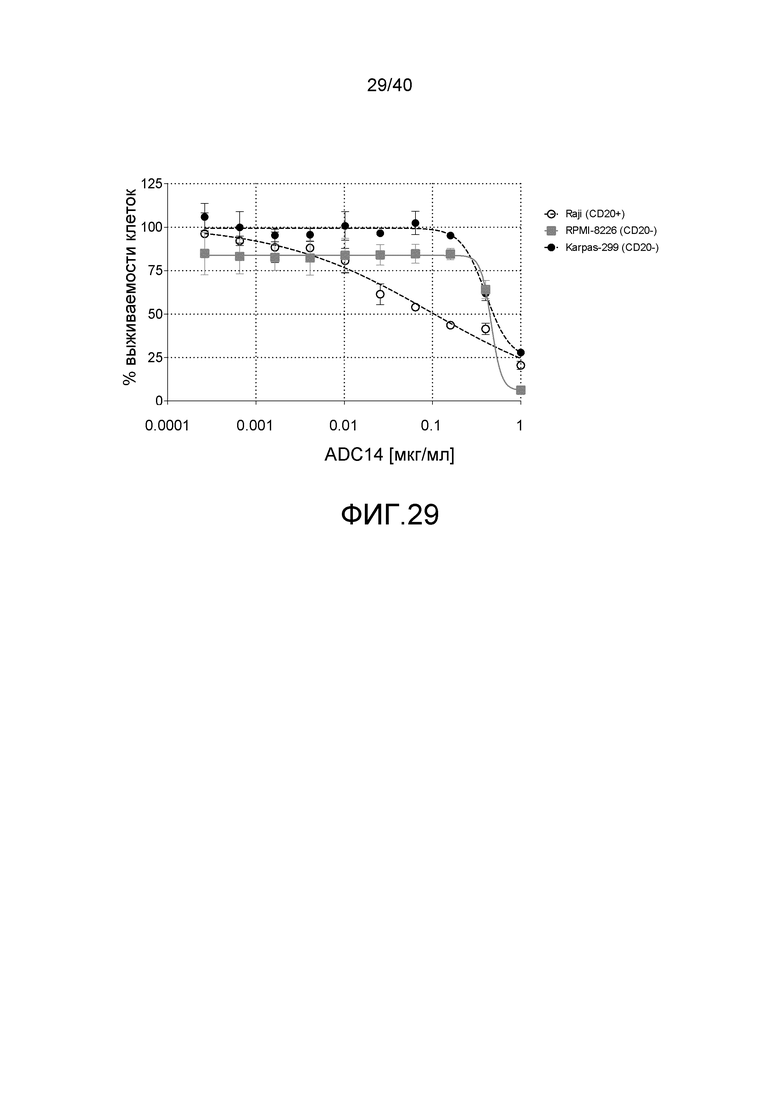
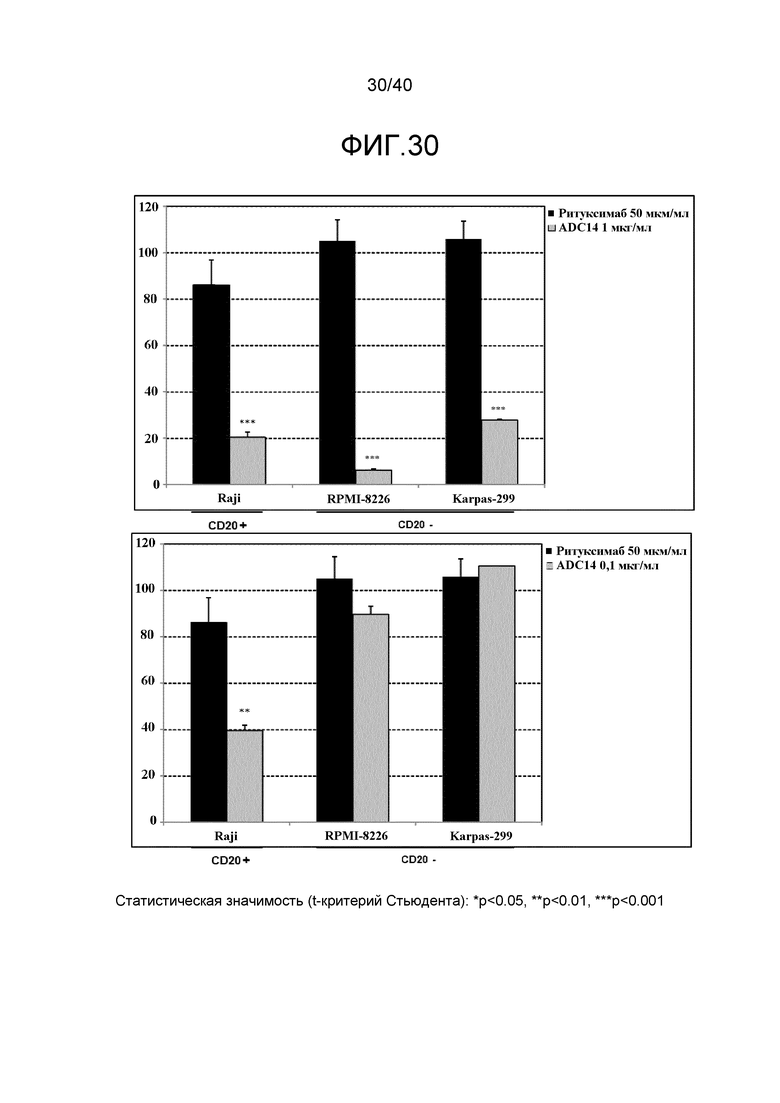
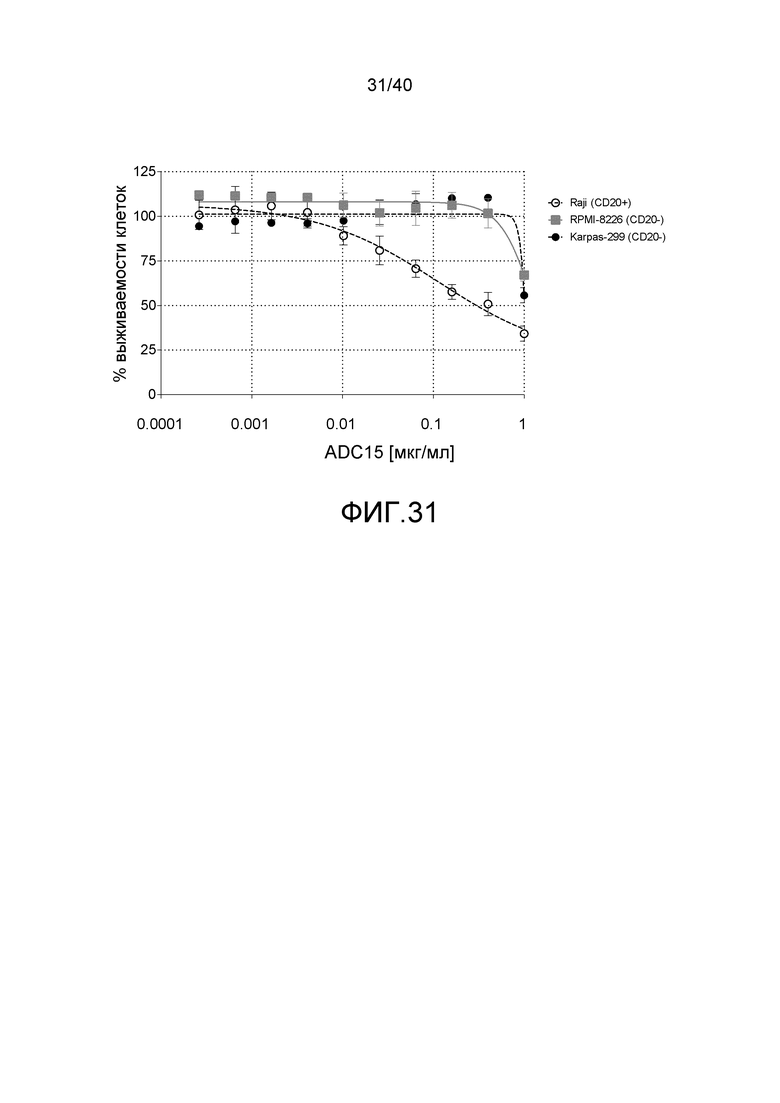
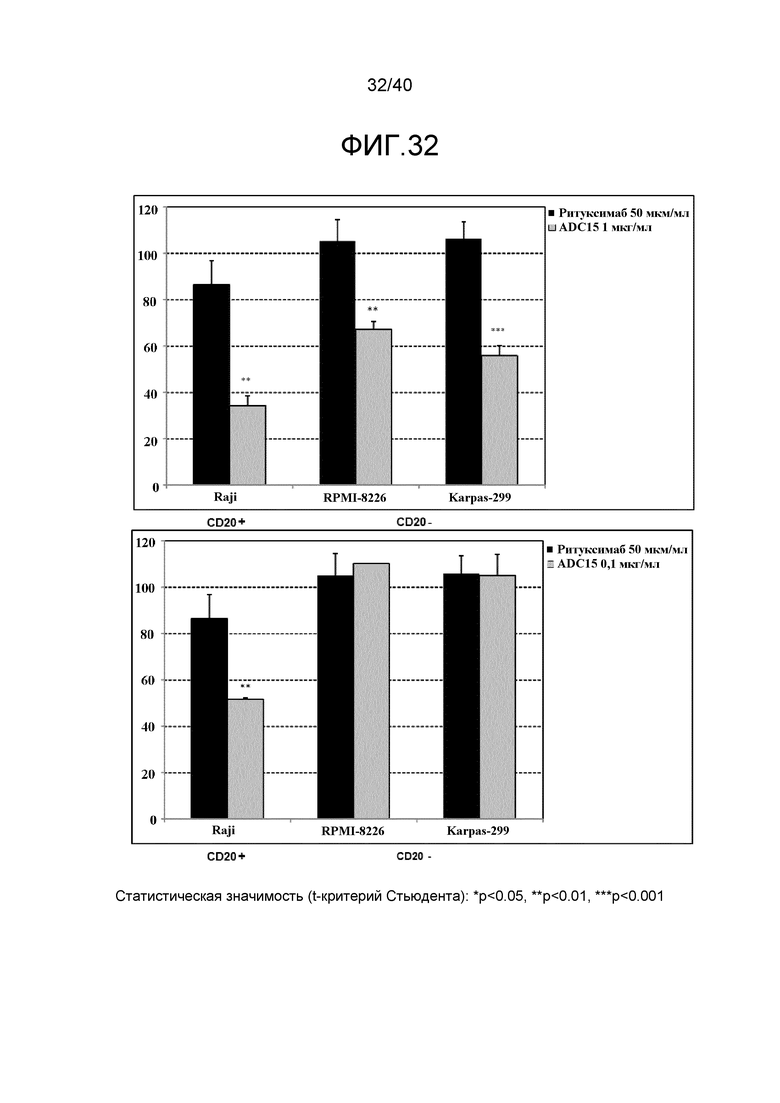

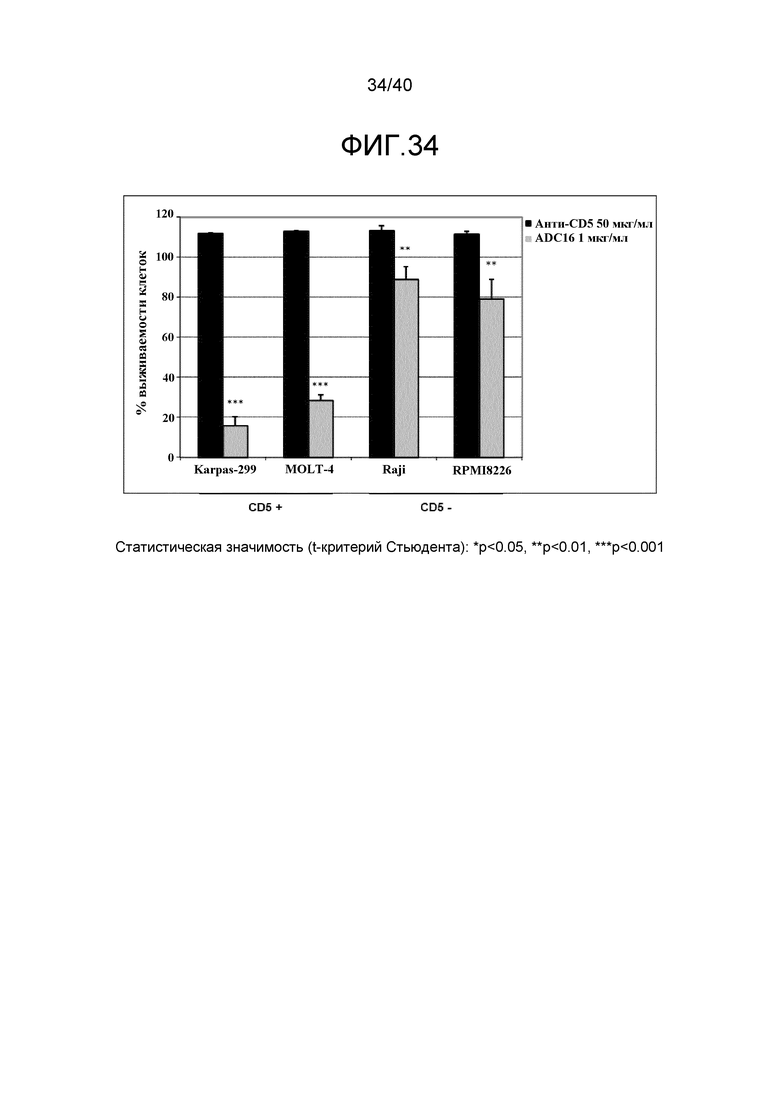
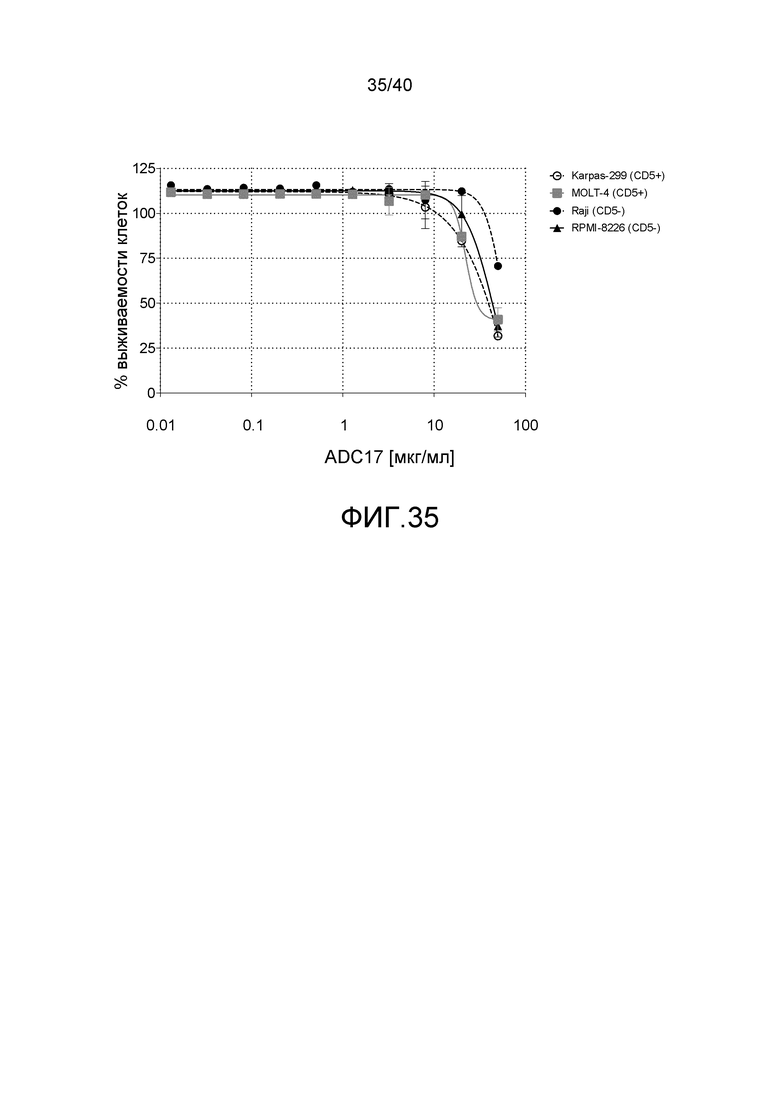
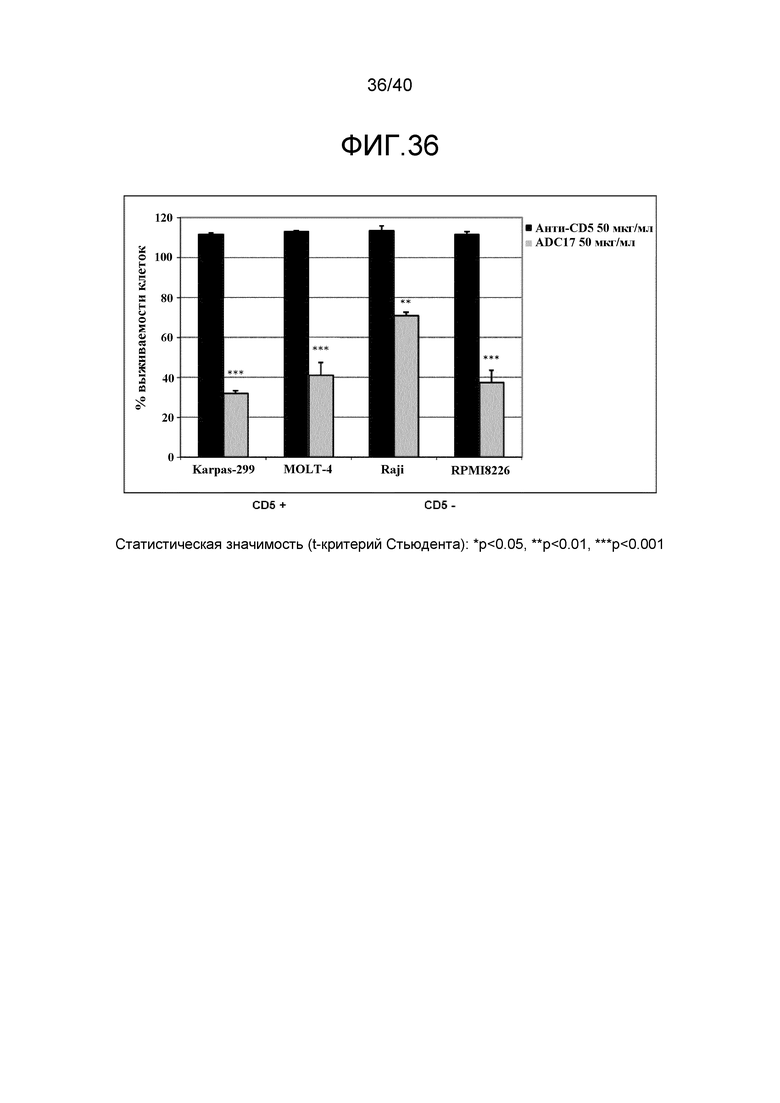
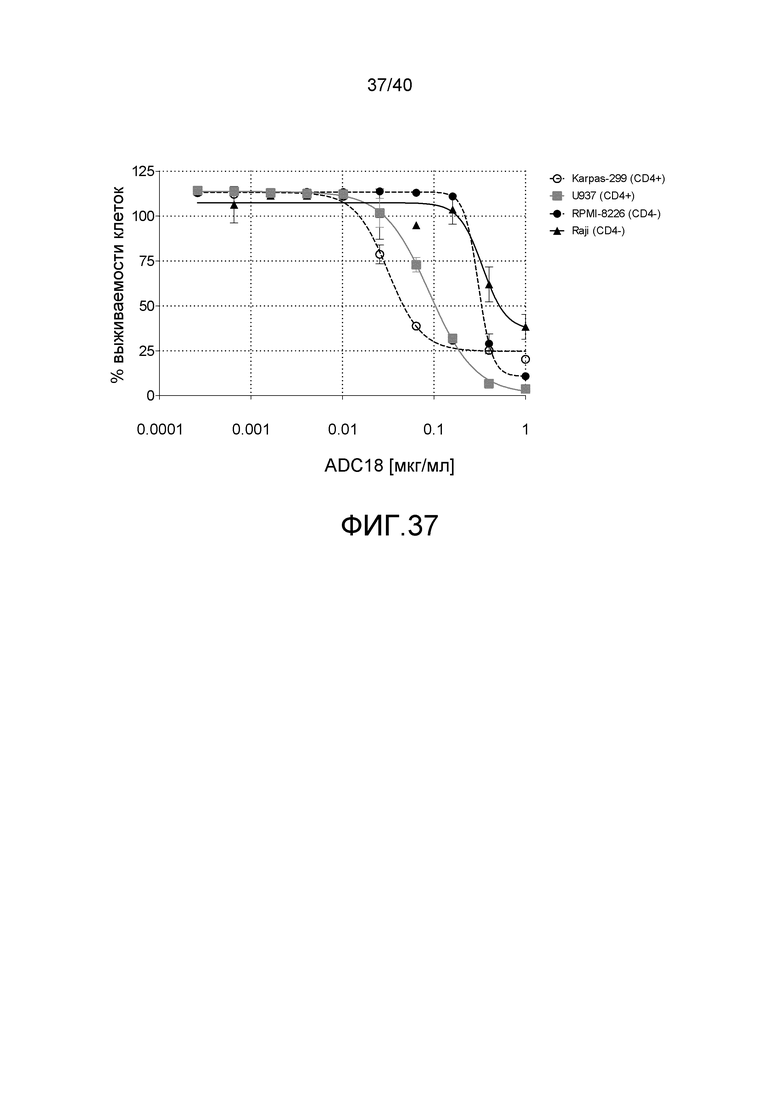
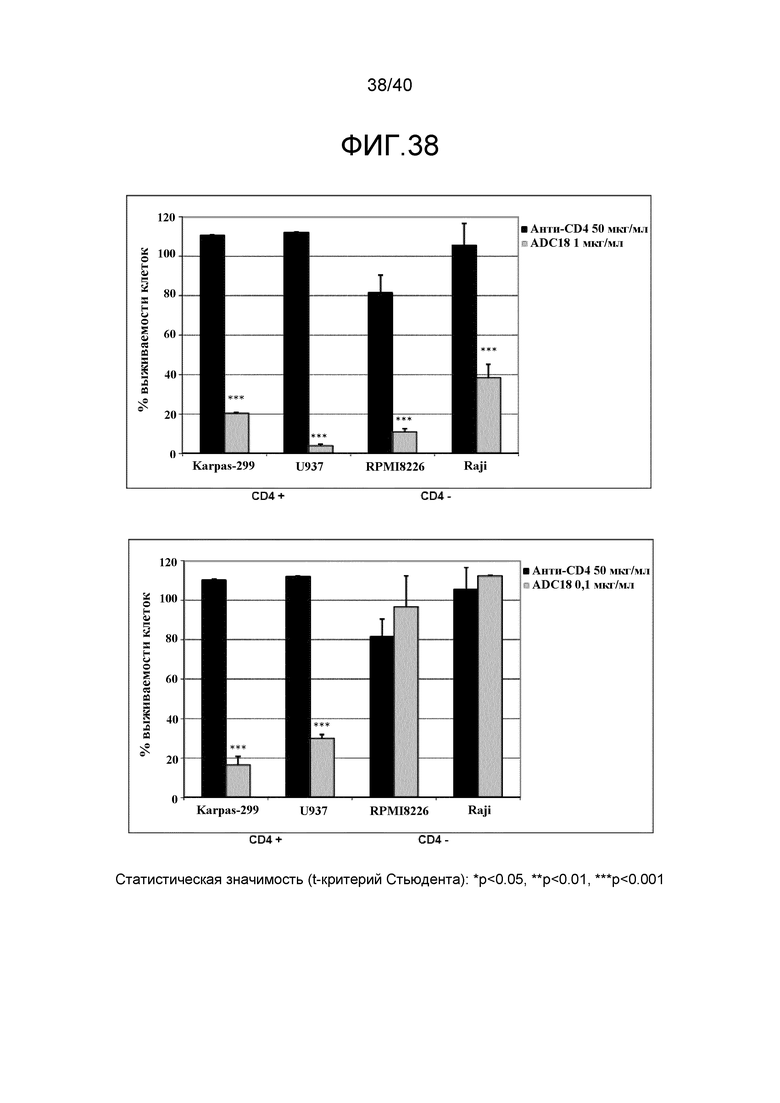


Изобретение относится к конъюгатам лекарственного средства формулы [D-(X)b-(AA)w-(L)-]n-Ab, где D означает группу, представляющую собой лекарственное средство формулы (Ia) или (Ib), или его фармацевтически приемлемую соль или стереоизомер, где волнистые линии формулы (Ia) и (Ib) указывают место ковалентного присоединения к (X)b, если это имеет место, или (AA)w, если это имеет место, или к линкерной группе L; каждый из R1, R2 и R3 независимо выбран из водорода и ORa; каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из группы, состоящей из водорода и незамещенного C1-C12 алкила; R11 представляет собой водород; R13 представляет собой водород; каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из водорода и незамещенного C1-C12 алкила; R18 представляет собой водород; R27 выбран из водорода, незамещенного C1-C12 алкила и галогена; каждый Ra представляет собой незамещенный C1-C12 алкил; и каждая пунктирная линия представляет собой необязательную дополнительную связь, но когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26, либо R27 отсутствуют; L представляет собой линкерную группу, (AA)w представляет собой аминокислотное звено, X представляет собой расширяющую группу, Ab представляет собой компонент, содержащий по меньшей мере один антиген-связывающий сайт, n представляет собой отношение группы [D-(X)b-(AA)w-(L)-] к компоненту Ab, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 1 до 20. Конъюгаты предназначены для лечения рака. 3 н. и 33 з.п. ф-лы, 40 ил., 71 табл., 22 пр.
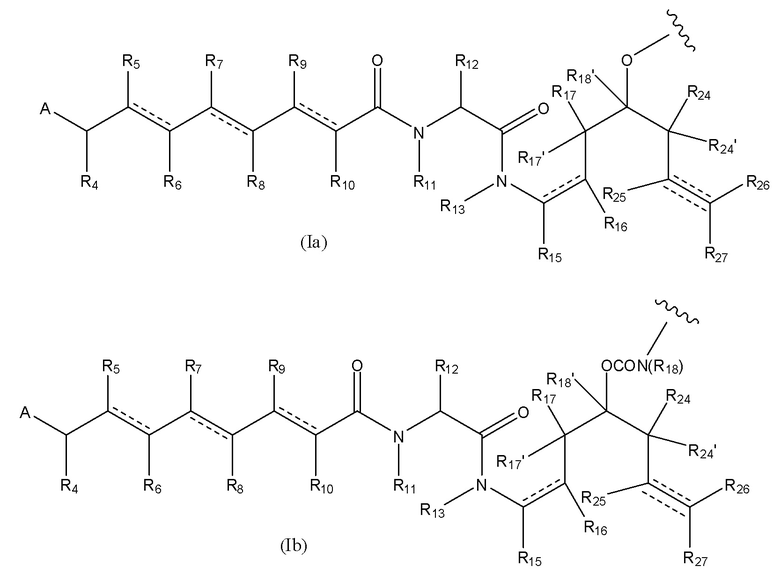
А: 
1. Конъюгат лекарственного средства, включающий группу лекарственного средства, ковалентно присоединенную к остальной части конъюгата лекарственного средства, который представляет собой соединение, имеющее формулу [D-(X)b-(AA)w-(L)-]n-Ab, где D означает группу, представляющую собой лекарственное средство, имеющее формулу, выбранную из (Ia) и (Ib), или его фармацевтически приемлемую соль, или его стереоизомер,
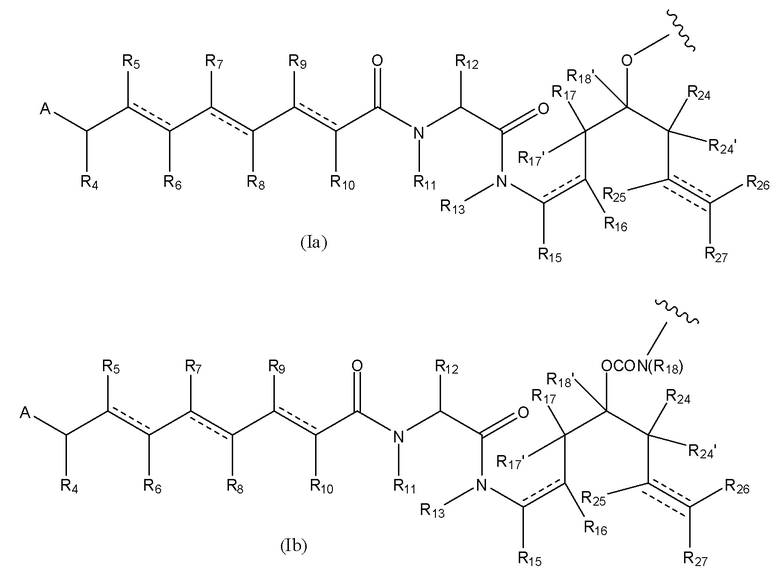
где волнистые линии формулы (Ia) и (Ib) указывают место ковалентного присоединения к (X)b, если это имеет место, или к (AA)w, если это имеет место, или к линкерной группе L;
A представляет собой
 ,
,
где волнистая линия группы A указывает место ковалентного присоединения к остальной части группы лекарственного средства;
каждый из R1, R2 и R3 независимо выбран из водорода и ORa;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из группы, состоящей из водорода и незамещенного C1-C12 алкила;
R11 представляет собой водород;
R13 представляет собой водород;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из водорода и незамещенного C1-C12 алкила;
R18 представляет собой водород;
R27 выбран из водорода, незамещенного C1-C12 алкила и галогена;
каждый Ra представляет собой незамещенный C1-C12 алкил;
и каждая пунктирная линия представляет собой необязательную дополнительную связь, но когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26, либо R27 отсутствуют;
L представляет собой линкерную группу, выбранную из группы, состоящей из:

где
волнистые линии указывают место ковалентных присоединений к Ab (волнистая линия справа) и (AA)w, если это имеет место, или к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева);
R19 выбран из -C1-C12 алкилена-;
M представляет собой -C1-C6 алкилен-(C3-C8 карбоцикло)-;
(AA)w представляет собой группу формулы (II):
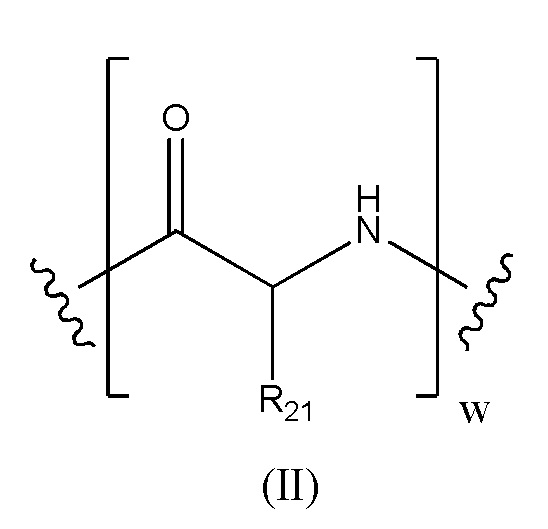
где волнистые линии указывают место ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
R21 в каждом случае выбран из группы, состоящей из изопропила и -(CH2)3NHCONH2;
и w представляет собой целое число, имеющее значение от 0 до 2;
X представляет собой расширяющую группу, выбранную из группы, состоящей из:
-CONH-(C1-C6 алкилена)NH-;
-CONH-(C1-C6 алкилена)NH-COO-CH2-(фенилен)-NH-;
-CONH-(C1-C6 алкилена)S-;
-CONH-(C1-C6 алкилен)NHCO(C1-C6 алкилена)S-;
-(C1-C6 алкилен)NHCO(C1-C6 алкилена)S-;
-(C1-C6 алкилена)S-;
-(C1-C6 алкилена)NH-; и
-(C1-C6 алкилен)NH-COO-CH2-(фенилен)-NH-;
b представляет собой целое число от 0 или 1;
Ab представляет собой компонент, содержащий по меньшей мере один антиген-связывающий сайт, выбранный из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела, анти-CD13 антитела или их иммунологически активной части; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-] к компоненту Ab, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 1 до 20.
2. Конъюгат лекарственного средства по п. 1, где
L представляет собой линкерную группу, выбранную из группы, состоящей из:
и
где
волнистые линии указывают место ковалентных присоединений к Ab (волнистая линия справа) и (AA)w, если это имеет место, или к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева);
R19 выбран из -C1-C12 алкилена-;
M представляет собой -C1-C6 алкилен-(C3-C8 карбоцикло)-;
(AA)w представляет собой группу формулы (II):
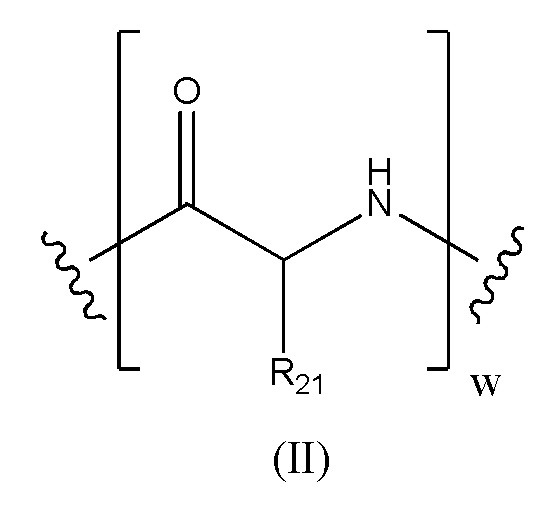
где волнистые линии указывают место ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
R21 в каждом случае выбран из группы, состоящей из изопропила и -(CH2)3NHCONH2;
w представляет собой целое число, имеющее значение от 0 до 2;
где X представляет собой расширяющую группу, выбранную из -CONH-(C1-C6 алкилен)NH-, -CONH-(C1-C6 алкилен)NH-COO-CH2-(фенилен)-NH-, -CONH-(C1-C6 алкилен)S-, -CONH-(C1-C6 алкилен)NHCO(C1-C6 алкилен)S-, -(C1-C6 алкилен)NHCO(C1-C6 алкилен)S-, -(C1-C6 алкилен)S-, -(C1-C6 алкилен)NH- и -(C1-C6 алкилен)NH-COO-CH2-(фенилен)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, или его стереоизомер, где
R1 выбран из водорода и ORa, где Ra представляет собой незамещенный C1-C6 алкил;
R2 и R3 представляют собой водород;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода и незамещенного C1-C6 алкила;
R11 и R13 представляют собой водород;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из:
водорода и незамещенного C1-C6 алкила;
R18 представляет собой водород;
R27 выбран из водорода, галогена и незамещенного C1-C6 алкила;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-] к компоненту Ab, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 1 до 12.
3. Конъюгат лекарственного средства по п. 1, выбранный из формул (IV) и (V):

где
R19 выбран из -C1-C8 алкилена-;
M представляет собой -C1-C3 алкилен-(C5-C7 карбоцикло)-;
(AA)w представляет собой формулу (II):
 ,
,
где волнистые линии указывают место ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
R21 в каждом случае выбран из группы, состоящей из изопропила и -(CH2)3NHCONH2;
w представляет собой целое число, имеющее значение от 0 до 2;
X представляет собой расширяющую группу, выбранную из группы, состоящей из -CONH-(C2-C4 алкилен)NH-, -CONH-(C2-C4 алкилен)NH-COO-CH2-(фенилен)-NH-, -CONH-(C2-C4 алкилен)S-, -CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилен)NH-COO-CH2-(фенилен)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, или его стереоизомер, где
R1 представляет собой водород или метокси;
каждый из R2 и R3 представляет собой водород;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода, незамещенного метила, незамещенного изопропила и незамещенного трет-бутила;
каждый из R11 и R13 представляет собой водород;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из:
водорода и незамещенных C1-C6 алкильных групп;
R18 представляет собой водород;
R27 представляет собой атом водорода или атом хлора;
каждая пунктирная линия представляет собой необязательную дополнительную связь, но когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26, либо R27 отсутствуют;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 8.
4. Конъюгат лекарственного средства по п. 1, выбранный из формул (IV) и (V):

где
R19 выбран из -C1-C6 алкилена-;
M представляет собой -C1-C3 алкилен-(C5-C7 карбоцикло)-;
w имеет значение 0 или 2, и когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
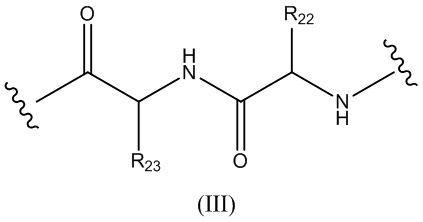
где волнистые линии указывают место ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
R22 представляет собой изопропил;
R23 представляет собой -(CH2)3NHCONH2;
X представляет собой расширяющую группу, выбранную из группы, состоящей из -CONH-(C2-C4 алкилен)NH-, -CONH-(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-, -CONH-(C2-C4 алкилена)S-, -CONH-(C2-C4 алкилена)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилена)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилена)S-, -(C2-C4 алкилена)NH- и -(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, или его стереоизомер, где
R1 представляет собой водород или метокси;
каждый из R2 и R3 представляет собой водород;
каждый из R5, R7, R8, R9 и R10 представляет собой водород;
каждый из R4 и R6 представляет собой метил;
каждый из R11 и R13 представляет собой водород;
R12 представляет собой изопропил или трет-бутил;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из водорода и незамещенной C1-C6 алкильной группы, предпочтительно водорода и метила;
R18 представляет собой водород;
R27 представляет собой атом водорода или атом хлора;
каждая пара атомов углерода, соединенных одной или несколькими пунктирными линиями, связана через двойные связи;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой моноклональное антитело, выбранное из группы, состоящей из трастузумаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части, и предпочтительно антитело, выбранное из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или его иммунологически активной части; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту Ab, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5.
5. Конъюгат лекарственного средства по п. 4, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба и анти-CD4 антитела или его иммунологически активной части.
6. Конъюгат лекарственного средства по п. 4, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой трастузумаб или его иммунологически активную часть.
7. Конъюгат лекарственного средства по п. 1, выбранный из формул (IV) и (V):
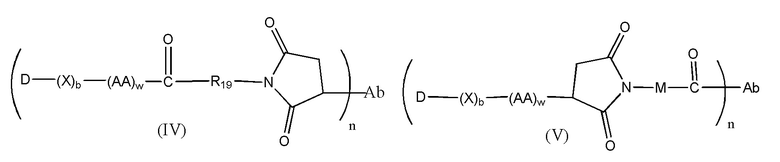
где
R19 представляет собой -C3-C6 алкилен-;
M представляет собой -C1-C3 алкилен-(C5-C7 карбоцикло)-;
w имеет значение 0 или 2, и когда w имеет значение 2, тогда (AA)w представляет собой группу формулы (III):
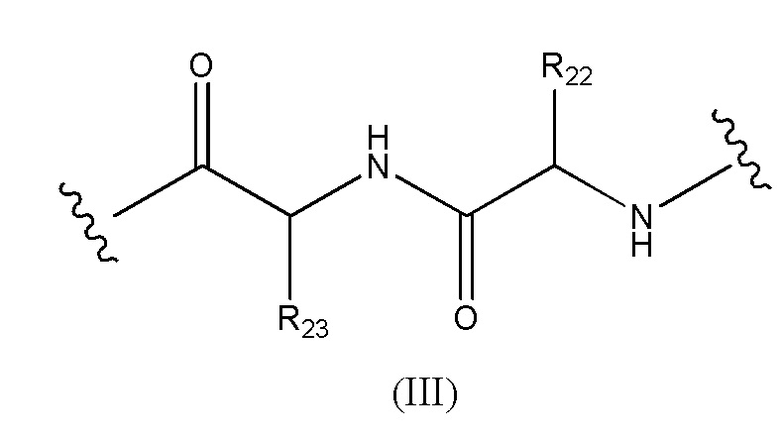 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, где волнистые линии указывают место ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
X представляет собой расширяющую группу, выбранную из группы, состоящей из -CONH-(C2-C4 алкилена)NH-, -CONH-(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-, -CONH-(C2-C4 алкилена)S-, -CONH-(C2-C4 алкилена)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилена)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилена)S-, -(C2-C4 алкилена)NH- и -(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, или его стереоизомер, выбранную из следующей группы:
 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
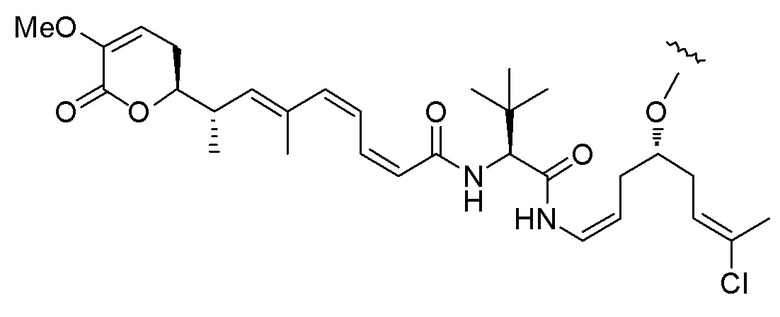 ,
,
 ,
,
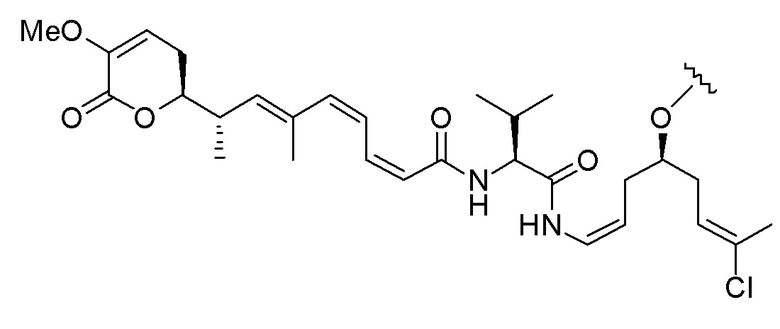 ,
,
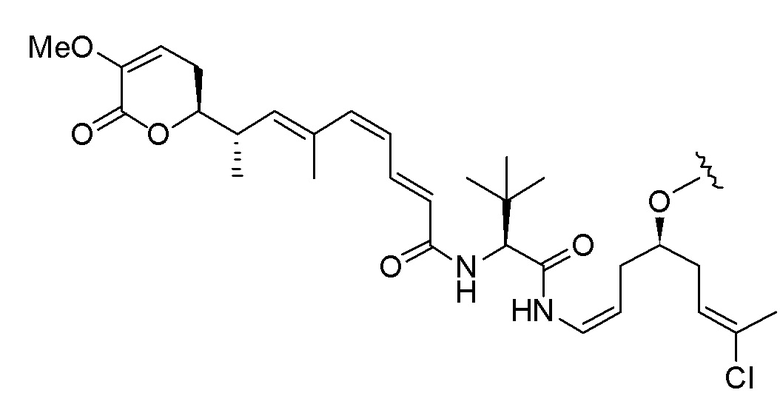 ,
,
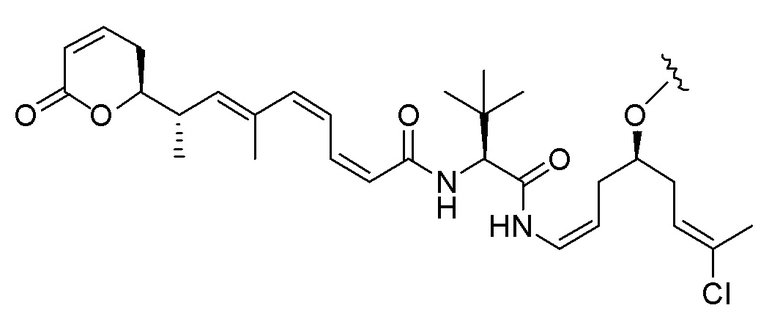 ,
,
 ,
,
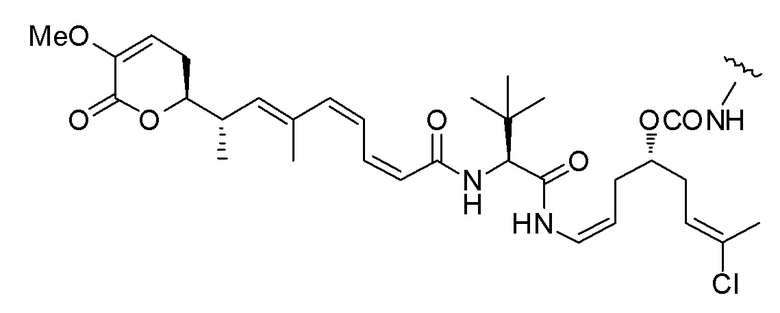 ,
,
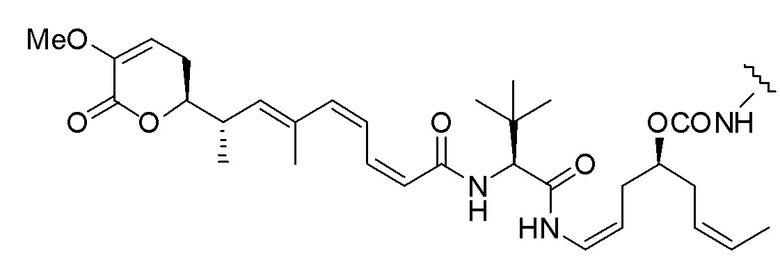 ,
,
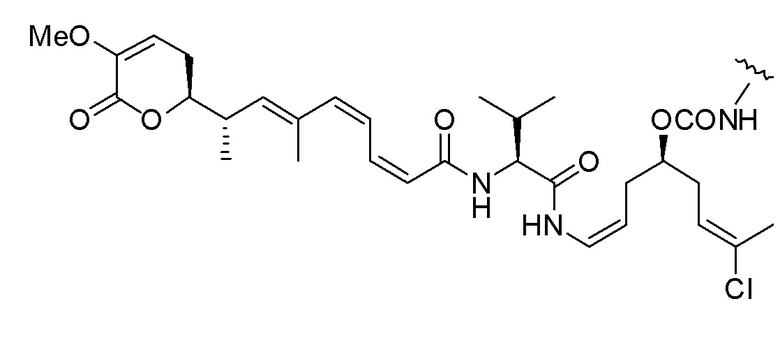 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
 и
и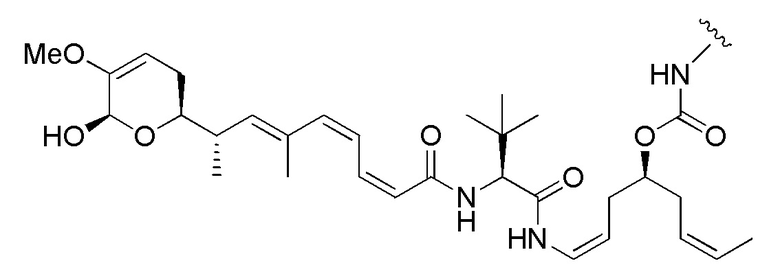
где волнистые линии указывают место ковалентного присоединения к (X)b, если это имеет место, или к (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту Ab, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5.
8. Конъюгат лекарственного средства по п. 7, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба и анти-CD4 антитела или их иммунологически активной части.
9. Конъюгат лекарственного средства по п. 7, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой трастузумаб или его иммунологически активную часть.
10. Конъюгат лекарственного средства по п. 1, выбранный из формул (IV) и (V):
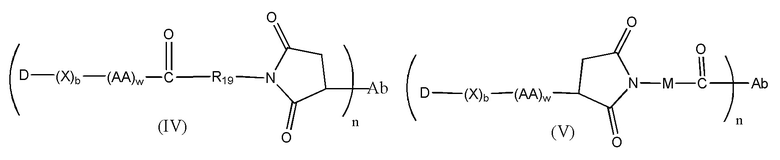
где
R19 представляет собой -C3-C6 алкилен-;
M представляет собой -C1-C3 алкилен-(C5-C7 карбоцикло)-;
w имеет значение 0 или 2, и когда w имеет значение 2, тогда (AA)w представляет собой группу формулы (III):
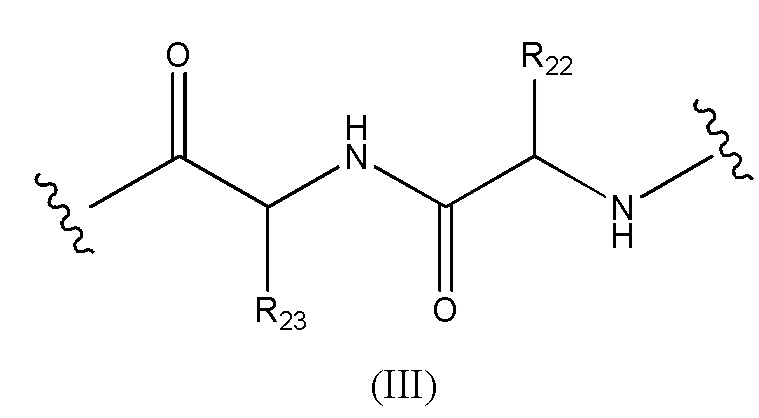 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, и волнистые линии указывают место ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа);
X представляет собой расширяющую группу, выбранную из группы, состоящей из -CONH-(C2-C4 алкилена)NH-, -CONH-(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-, -CONH-(C2-C4 алкилена)S-, -CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, или его стереоизомер, выбранную из:
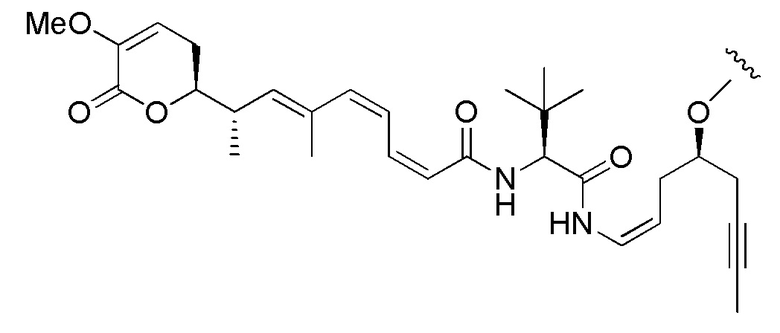 и
и
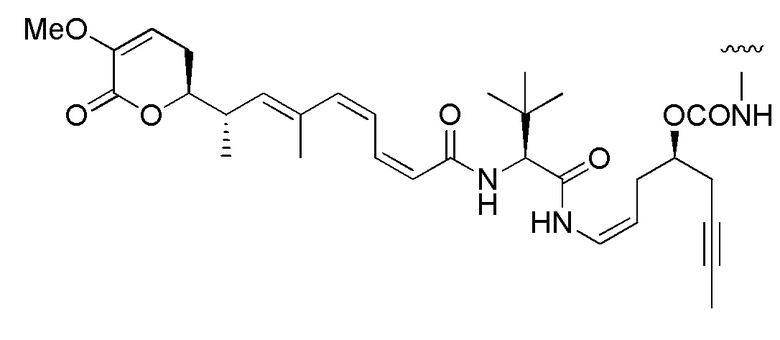 ,
,
где волнистые линии указывают место ковалентного присоединения к (X)b, если это имеет место, или к (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой трастузумаб, ритуксимаб, анти-CD4 антитело, анти-CD5 антитело и анти-CD13 антитело или их иммунологически активную часть; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5.
11. Конъюгат лекарственного средства по п. 10, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба, анти-CD4 антитела или их иммунологически активной части.
12. Конъюгат лекарственного средства по п. 10, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой трастузумаб или его иммунологически активную часть.
13. Конъюгат лекарственного средства по п. 1 формулы (IV):
 ,
,
где
R19 представляет собой -C5 алкилен-;
b имеет значение 1;
w имеет значение 0 или 2, и когда w имеет значение 2, тогда (AA)w представляет собой группу формулы (III):
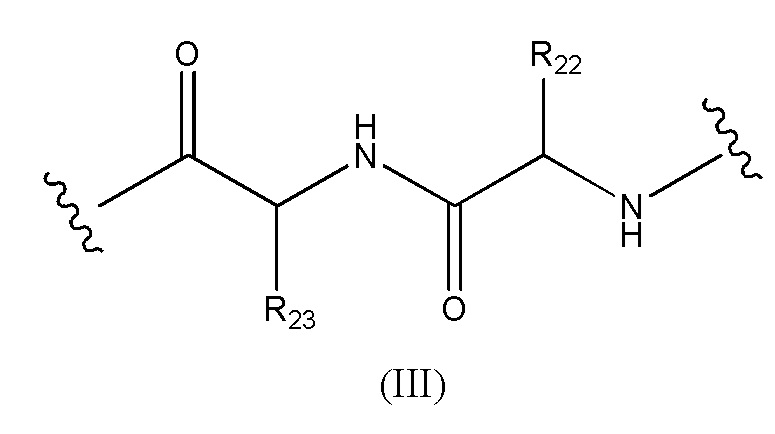 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, и волнистые линии указывают место ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа); и
X представляет собой расширяющую группу, выбранную из -CONH(CH2)3NHCOOCH2-фенилен-NH- и -CONH(CH2)3NH- или группы, имеющей формулу (V)
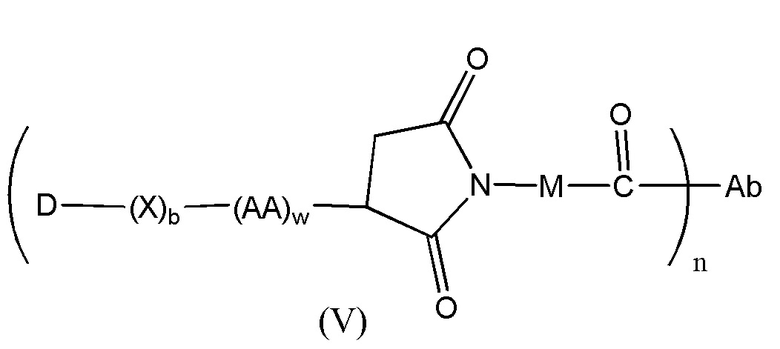 ,
,
где M представляет собой -метил-циклогексилен-;
b имеет значение 1;
w имеет значение 0; и
X представляет собой расширяющую группу, выбранную из -CONH(CH2)3-S- и -CONH(CH2)3NHCO(CH2)2S-;
D означает группу, представляющую собой лекарственное средство формулы (Ia) или его фармацевтически приемлемую соль, или его стереоизомер, выбранную из:
 и
и
 ,
,
где волнистые линии указывают место ковалентного присоединения к (X)b, если это имеет место, или к (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5 и предпочтительно 4.
14. Конъюгат лекарственного средства по п. 13, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба и анти-CD4 антитела или их иммунологически активной части.
15. Конъюгат лекарственного средства по п. 13, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой трастузумаб или его иммунологически активную часть.
16. Конъюгат лекарственного средства по п. 1, формулы (IV):
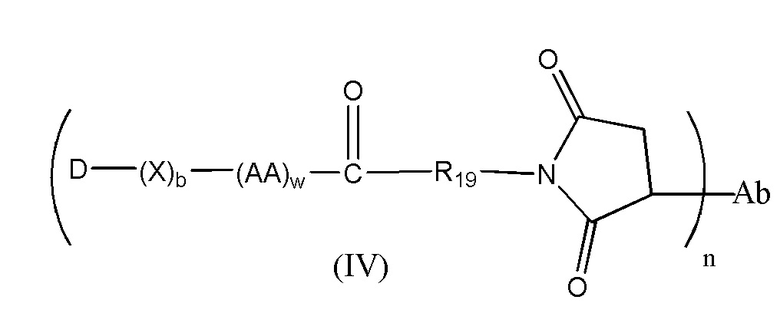 ,
,
где R19 представляет собой -C5 алкилен-;
b имеет значение 1;
w имеет значение 0 или 2, и когда w имеет значение 2, тогда (AA)w представляет собой формулу (III):
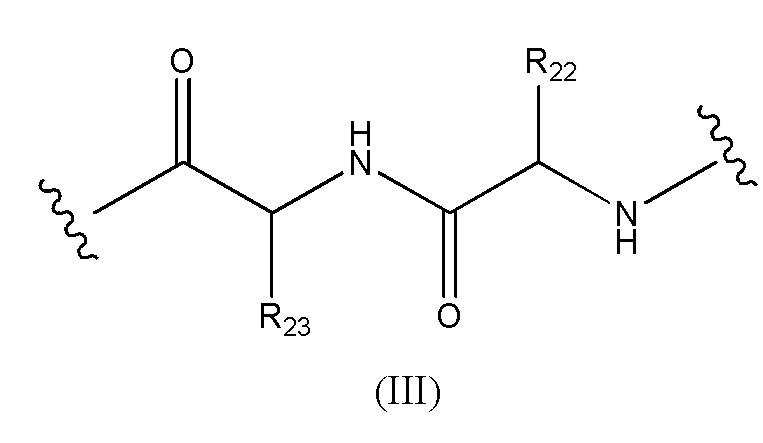 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, и волнистые линии указывают место ковалентных присоединений к (X)b, если это имеет место, или к группе лекарственного средства (волнистая линия слева) и к линкеру (волнистая линия справа); и
X представляет собой расширяющую группу, выбранную из -(CH2)3NHCOOCH2-фенилен-NH- и -(CH2)3NH- или группы, имеющей формулу (V)
 ,
,
где M представляет собой -метил-циклогексилен-;
b имеет значение 1;
w имеет значение 0; и
X представляет собой расширяющую группу, выбранную из -(CH2)3S- и -(CH2)3NHCO(CH2)2S-;
D означает группу, представляющую собой лекарственное средство формулы (Ib) или его фармацевтически приемлемую соль, или его стереоизомер, выбранную из:
 и
и
 ,
,
где волнистые линии указывают место ковалентного присоединения к (X)b, если это имеет место, или к (AA)w, если это имеет место, или к линкерной группе L;
компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части; и
n представляет собой отношение группы [D-(X)b-(AA)w-(L)-], где L имеет значение, определенное в формулах (IV) или (V), к компоненту, содержащему по меньшей мере один антиген-связывающий сайт, и находится в диапазоне от 3 до 5 и предпочтительно 4.
17. Конъюгат лекарственного средства по п. 16, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба и анти-CD4 антитела или их иммунологически активной части.
18. Конъюгат лекарственного средства по п. 16, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой трастузумаб или его иммунологически активную часть.
19. Конъюгат антитела с лекарственным средством по п. 13, выбранный из группы, включающей:
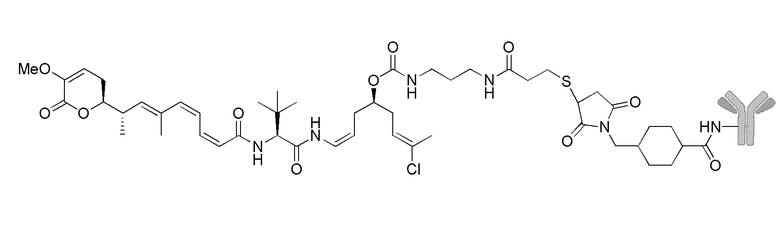 ,
,
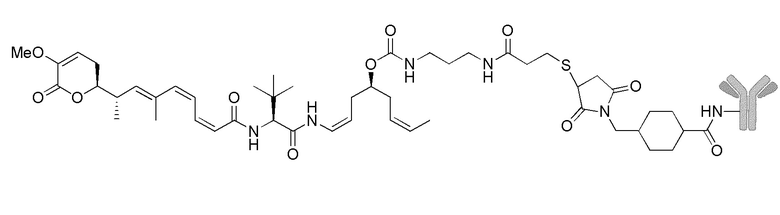 ,
,
 ,
,
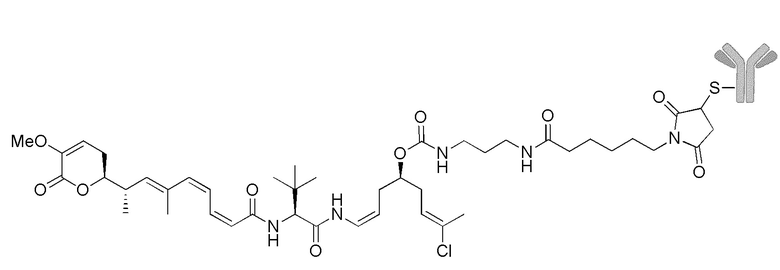 ,
,
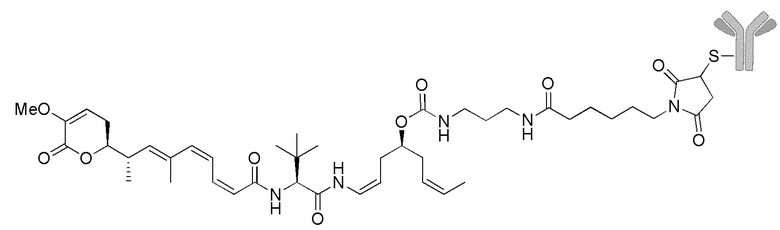 , и
, и
 ,
,
где каждый из  и
и  выбран из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части.
выбран из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части.
20. Конъюгат лекарственного средства по п. 10, выбранный из группы, состоящей из:
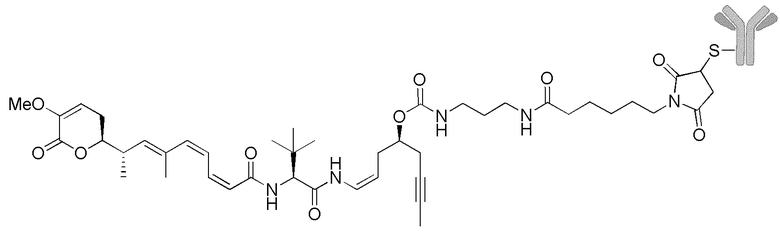 и
и
 ,
,
где каждый из  и
и  выбран из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части.
выбран из трастузумаба, ритуксимаба, анти-CD4 антитела, анти-CD5 антитела и анти-CD13 антитела или их иммунологически активной части.
21. Конъюгат лекарственного средства по п. 19 или 20, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, выбран из трастузумаба, ритуксимаба и анти-CD4 антитела или их иммунологически активной части.
22. Конъюгат лекарственного средства по п. 19 или 20, где компонент Ab, содержащий по меньшей мере один антиген-связывающий сайт, представляет собой трастузумаб или его иммунологически активную часть.
23. Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H, где
L1 представляет собой линкер формулы:
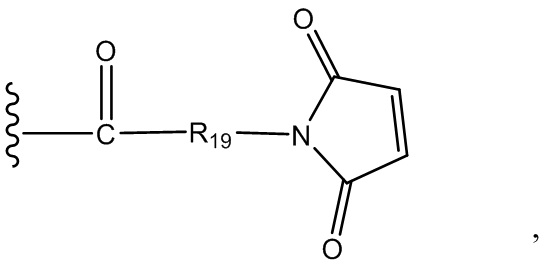
где волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 выбран из -C1-C12 алкилена-;
D означает группу, представляющую собой лекарственное средство, имеющую формулу, выбранную из (Ib) и (Ib) или его фармацевтически приемлемую соль, или его стереоизомер,
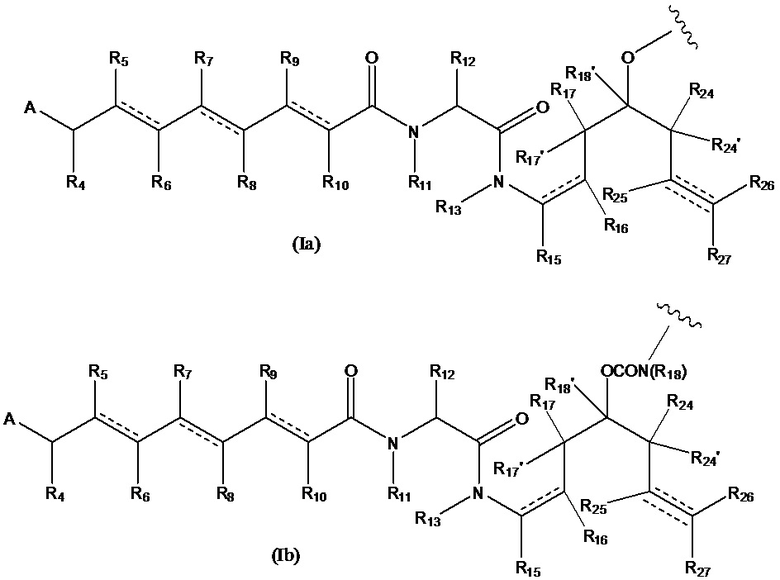
где волнистые линии (Ia) и (Ib) указывают место ковалентного присоединения к Х;
А представляет собой:
 ,
,
где волнистая линия группы A указывает место ковалентного присоединения к остальной части группы лекарственного средства;
каждый из R1, R2 и R3 независимо выбран из водорода и ORa;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из группы, состоящей из водорода и незамещенного C1-C12 алкила;
R11 представляет собой водород;
R13 представляет собой водород;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из водорода и незамещенного C1-C12 алкила,
R18 представляет собой водород;
R27 выбран из водорода, незамещенного C1-C12 алкила и галогена;
каждый Ra представляет собой незамещенный C1-C12 алкил;
и каждая пунктирная линия представляет собой необязательную дополнительную связь, но когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26, либо R27 отсутствуют;
X представляет собой расширяющую группу, выбранную из группы, состоящей из:
-CONH-(C1-C6 алкилена)NH-;
–CONH-(C1-C6 алкилена)NH-COO-CH2-( фенилен)-NH-;
–CONH-(C1-C6 алкилена)S-;
–CONH-(C1-C6 алкилен)NHCO(C1-C6 алкилена)S-;
-(C1-C6 алкилен)NHCO(C1-C6 алкилена)S-;
-(C1-C6 алкилена)S-;
-(C1-C6 алкилена)NH-; и
-(C1-C6 алкилена)NH-COO-CH2-(фенилен)-NH-;
(AA)w представляет собой формулу (II):

где волнистые линии указывают место ковалентных присоединений к Х (волнистая линия справа) и к L1 или атому водорода (волнистая линия справа);
R21 в каждом случае выбран из группы, состоящей из изопропила и -(CH2)3NHCONH2; и
w представляет собой целое число, имеющее значение от 0 до 2.
24. Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H по п. 23, где
L1 представляет собой линкер формулы:
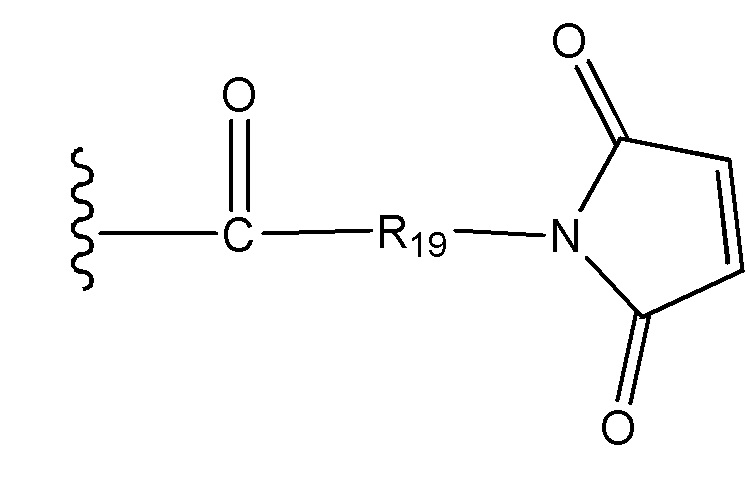 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 выбран из -C1-C8 алкилена-; и
каждый из D, X, AA и w имеет значение, определенное в п.23.
25. Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H по п. 23, где
L1 представляет собой линкер формулы:
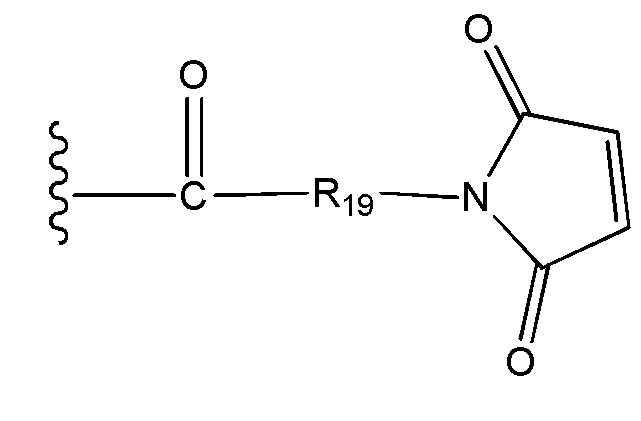 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 выбран из -C1-C8 алкилена-;
(AA)w представляет собой формулу (II):
 ,
,
где волнистые линии указывают место ковалентных присоединений к X (волнистая линия слева) и к L1 или к атому водорода (волнистая линия справа);
где R21 в каждом случае выбран из группы, состоящей из изопропила и -(CH2)3NHCONH2, и w представляет собой целое число, имеющее значение от 0 до 2;
X представляет собой расширяющую группу, выбранную из группы, состоящей из -CONH-(C2-C4 алкилена)NH-, -CONH-(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-, -CONH-(C2-C4 алкилена)S-, -CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилена)S-, -(C2-C4 алкилена)NH- и -(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-; и
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, или его стереоизомер:
 ,
,
где волнистые линии в формулах (Ia) и (Ib) указывают место ковалентного присоединения к X;
A выбран из
 ,
,
где волнистая линия группы A указывает место ковалентного присоединения к остальной части группы лекарственного средства;
R1 выбран из водорода и ORa, где Ra представляет собой незамещенный C1-C6 алкил;
R2 и R3 представляет собой водород;
каждый из R4, R5, R6, R7, R8, R9, R10 и R12 независимо выбран из водорода и незамещенного C1-C6 алкила;
R11 и R13 представляют собой водород;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из:
водорода и незамещенного C1-C6 алкила;
R18 представляет собой водород;
R27 выбран из водорода, галогена и незамещенного C1-C6 алкила;
и каждая пунктирная линия представляет собой необязательную дополнительную связь, но когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26, либо R27 отсутствуют.
26. Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H по п. 23, где
L1 представляет собой группу формулы:
 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 выбран из -C1-C6 алкилена-;
w имеет значение 0 или 2, и когда w имеет значение 2, тогда (AA)w представляет собой группу формулы (III):
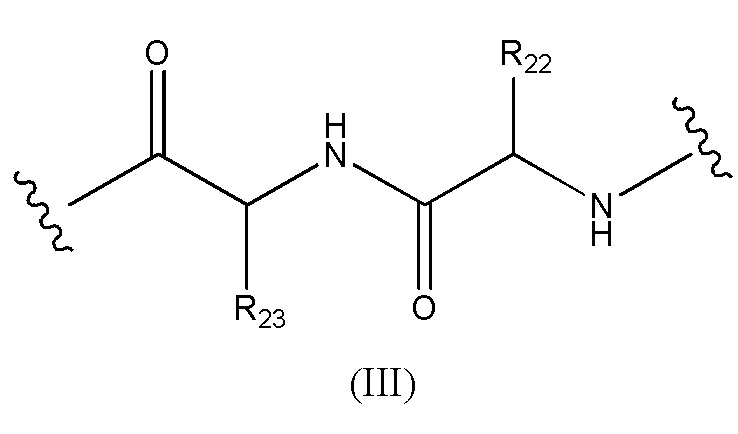 ,
,
где волнистые линии указывают место ковалентных присоединений к X (волнистая линия слева) и к L1 или к атому водорода (волнистая линия справа);
R22 представляет собой изопропил;
R23 представляет собой-(CH2)3NHCONH2;
X представляет собой расширяющую группу, выбранную из -CONH-(C2-C4 алкилен)NH-, -CONH(C2-C4 алкилен)NHCOO-CH2-(фенилен)-NH, -CONH-(C2-C4 алкилен)S-, -CONH-(C2-C4 алкилен)NHCO-(C1-C3 алкилен)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилен)S-, -(C2-C4 алкилен)S-, -(C2-C4 алкилен)NH- и -(C2-C4 алкилен)NH- COO-CH2-(фенилен)-NH-; и
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, или его стереоизомер:
 ,
,
где волнистые линии в формулах (Ia) и (Ib) указывают место ковалентного присоединения к X;
A представляет собой
 ,
,
где волнистая линия группы A указывает место ковалентного присоединения к остальной части группы лекарственного средства;
R1 представляет собой водород или метокси;
каждый из R2 и R3 представляет собой водород;
каждый из R5, R7, R8, R9 и R10 представляет собой водород;
каждый из R4 и R6 представляет собой метил;
R12 представляет собой изопропил или трет-бутил;
каждый из R11 и R13 представляет собой водород;
каждый из R15, R16, R17, R17’, R18’, R24, R24’, R25 и R26 независимо выбран из группы, состоящей из водорода и C1-C6 алкильной группы, предпочтительно водорода и метила;
R18 представляет собой водород;
R27 представляет собой водород или галоген;
и каждая пунктирная линия представляет собой необязательную дополнительную связь, но когда существует тройная связь между атомом С, к которому присоединен R25, и атомом С, к которому присоединены R26 и R27, тогда R25 и либо R26, либо R27 отсутствуют.
27. Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H по п. 23, где
L1 представляет собой линкер формулы:
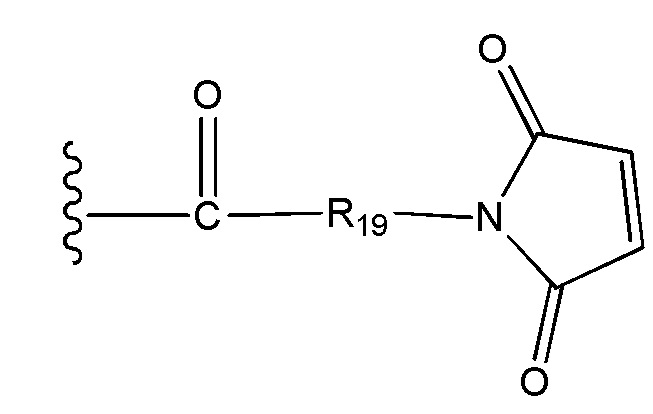 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 представляет собой -C3-C6 алкилен-;
w имеет значение 0 или 2, и когда w имеет значение 2, тогда (AA)w представляет собой группу формулы (III):
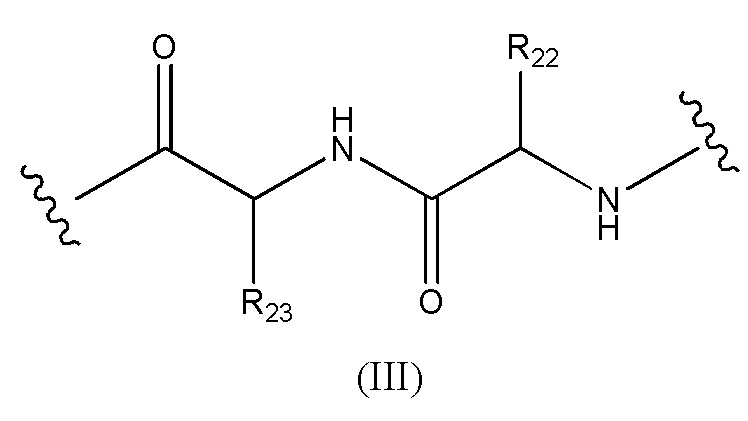 ,
,
R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, где волнистые линии указывают место ковалентных присоединений к X (волнистая линия слева) и к L1 или к атому водорода (волнистая линия справа);
X представляет собой расширяющую группу, выбранную из группы, состоящей из -CONH-(C2-C4 алкилена)NH-, -CONH-(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-, -CONH-(C2-C4 алкилена)S-, -CONH-(C2-C4 алкилен)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилен)NHCO(C1-C3 алкилена)S-, -(C2-C4 алкилена)S-, -(C2-C4 алкилена)NH- и -(C2-C4 алкилена)NH-COO-CH2-(фенилен)-NH-; и
D означает группу, представляющую собой лекарственное средство формулы (Ia) или формулы (Ib), или его фармацевтически приемлемую соль, или его стереоизомер, выбранную из следующей группы:
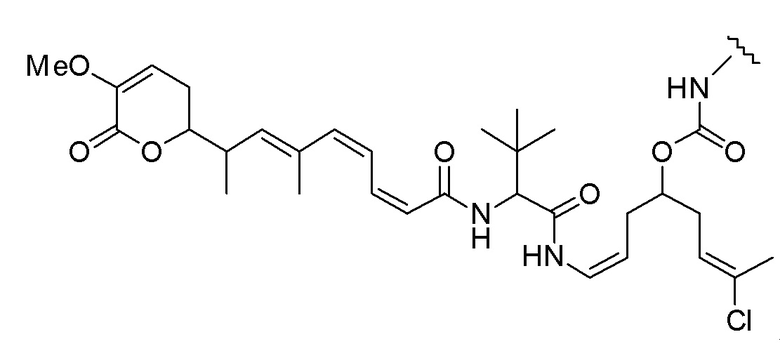 ,
,
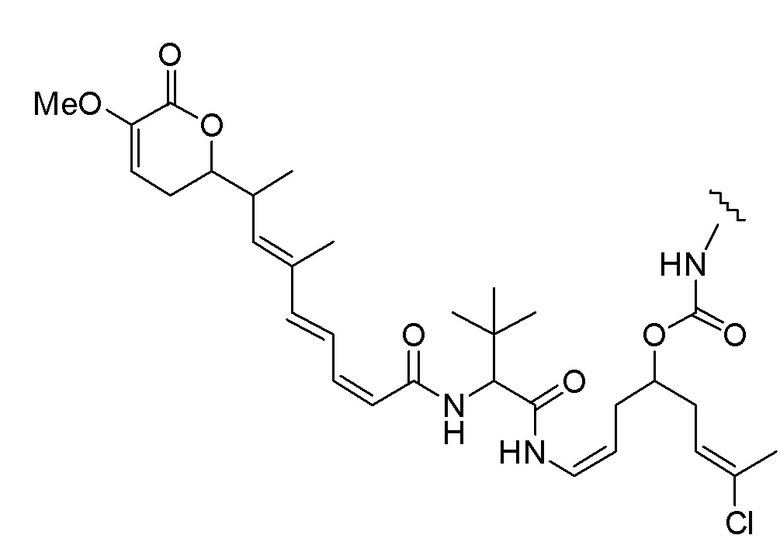 ,
,
 ,
,
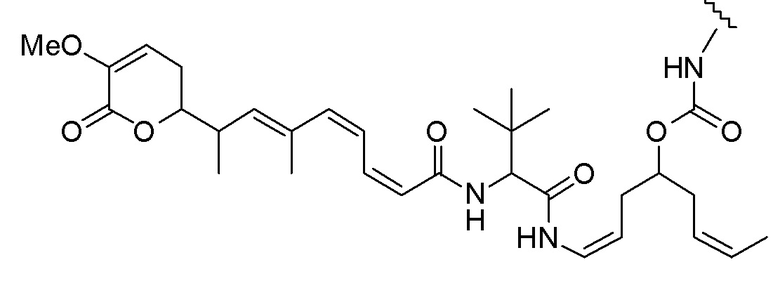 ,
,
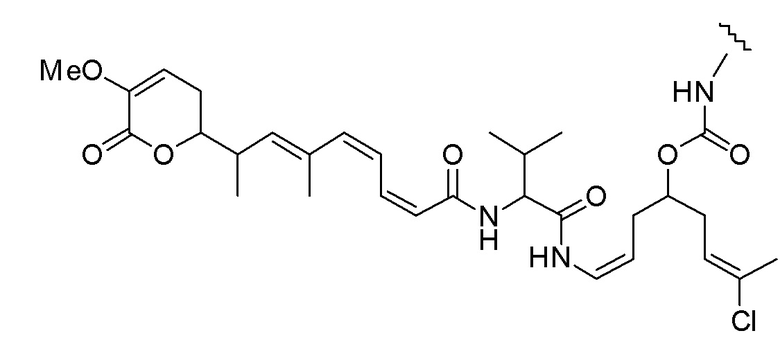 ,
,
 ,
,
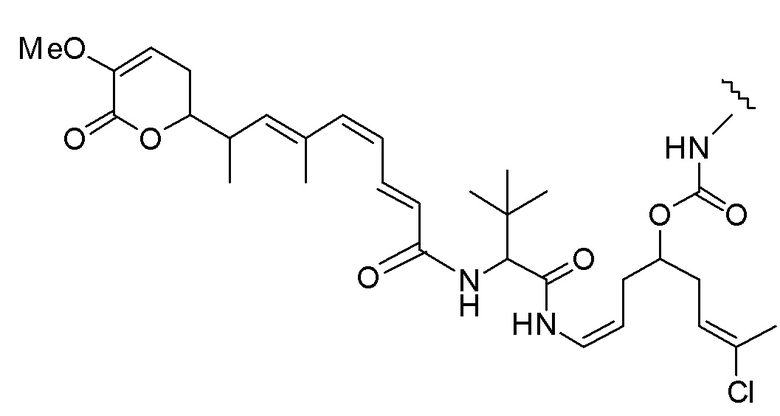 ,
,
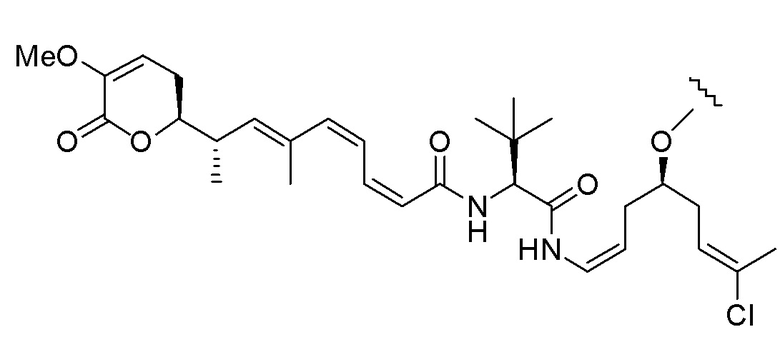 ,
,
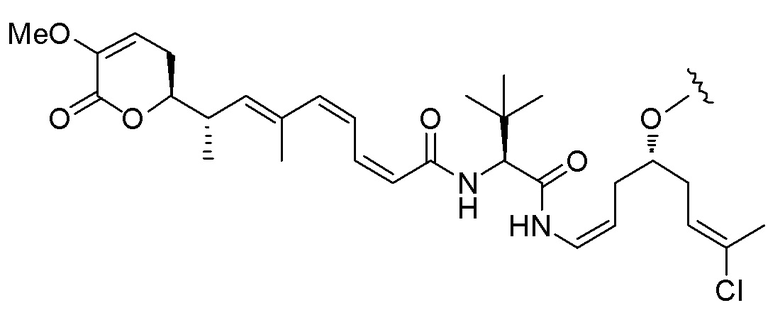 ,
,
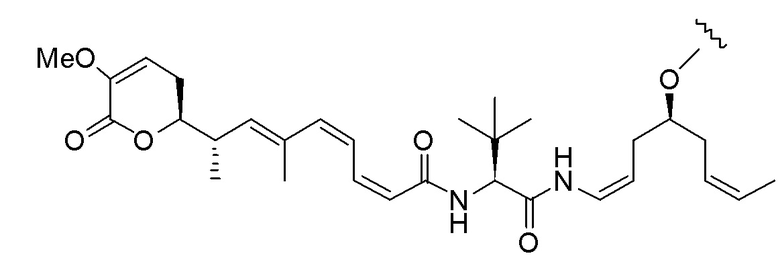 ,
,
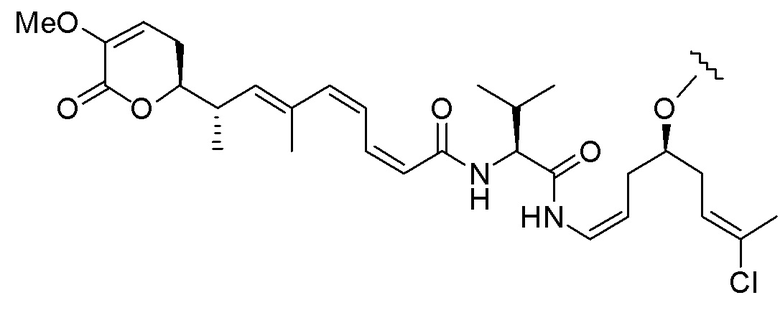 ,
,
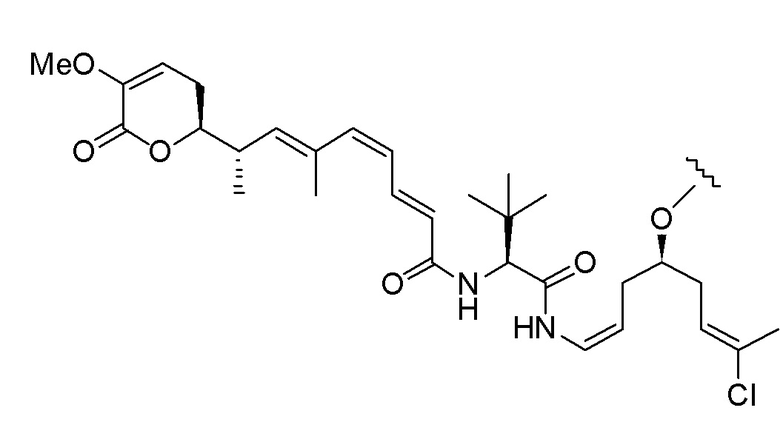 ,
,
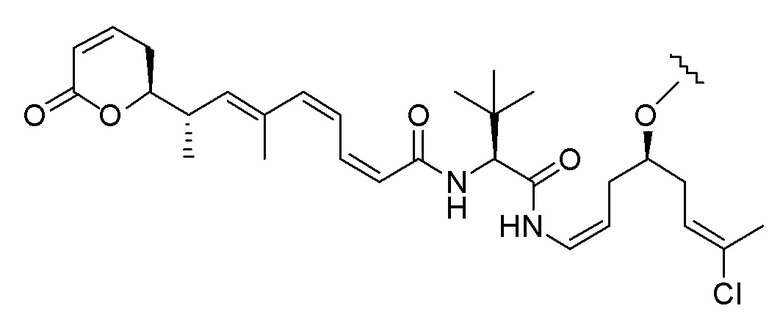 ,
,
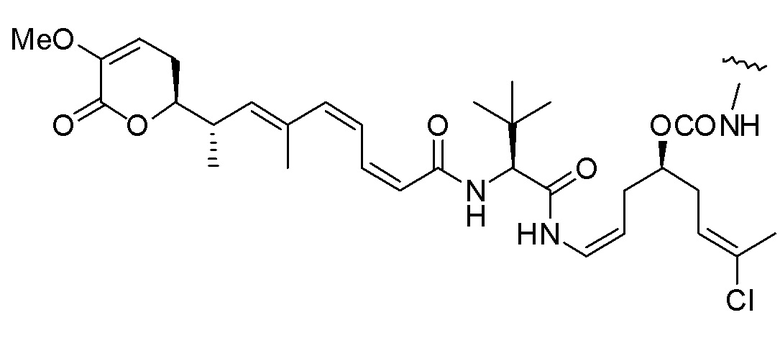 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
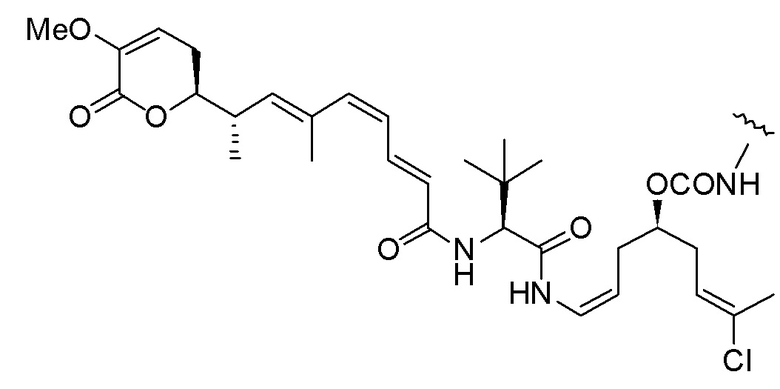 ,
,
 ,
,
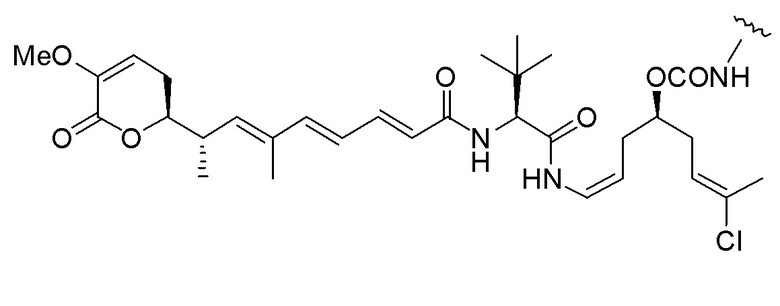 ,
,
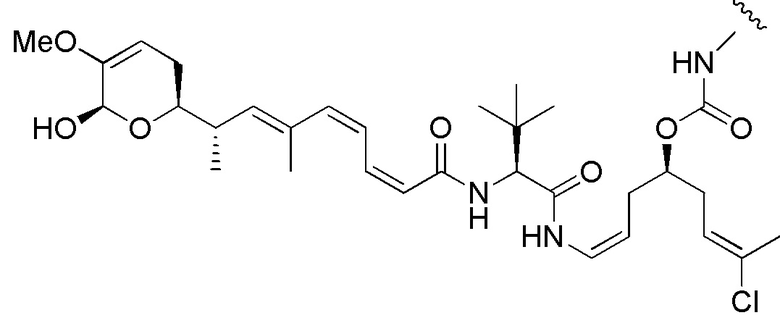 ,
, ,
,
 и
и  ,
,
где волнистая линия указывает точку ковалентного присоединения к X.
28. Соединение формулы D-X-(AA)w-L1 или формулы D-X-(AA)w-H по п. 23, где
L1 представляет собой группу формулы:
 ,
,
где
волнистая линия указывает точку ковалентного присоединения к (AA)w, если это имеет место, или к X;
R19 представляет собой -C5 алкилен-;
w имеет значение 0 или 2, и когда w имеет значение 2, тогда (AA)w представляет собой группу формулы (III):
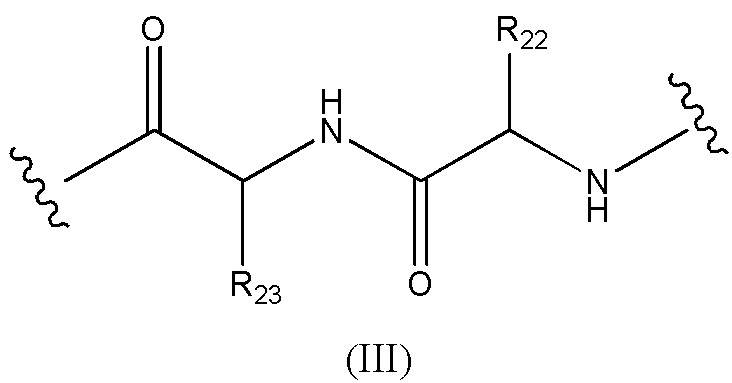 ,
,
где R22 представляет собой изопропил, R23 представляет собой -(CH2)3NHCONH2, где волнистые линии указывают место ковалентных присоединений к X (волнистая линия слева) и к L1 или к атому водорода (волнистая линия справа);
X представляет собой расширяющую группу, выбранную из -CONH(CH2)3NHCOOCH2-фенилен-NH-, -CONH(CH2)3NH-, -CONH(CH2)3-S- и -CONH(CH2)3NHCO(CH2)2S-; и
D означает группу, представляющую собой лекарственное средство формулы (Ia) или его фармацевтически приемлемую соль, или его стереоизомер, выбранную из:
 и
и
 ,
,
где волнистая линия указывает точку ковалентного присоединения к X.
29. Соединение формулы D-X-(AA)w-L1 по любому одному из пп. 23-28, выбранное из:
 ,
,
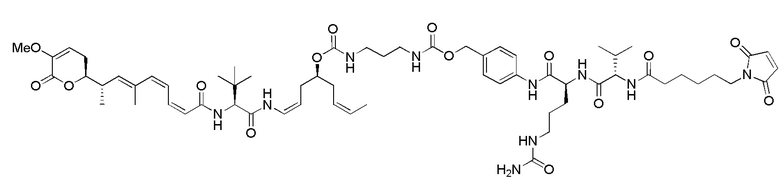 ,
,
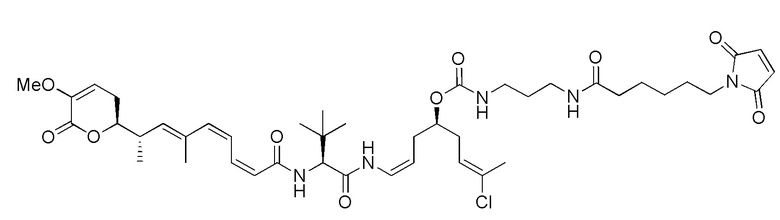 ,
,
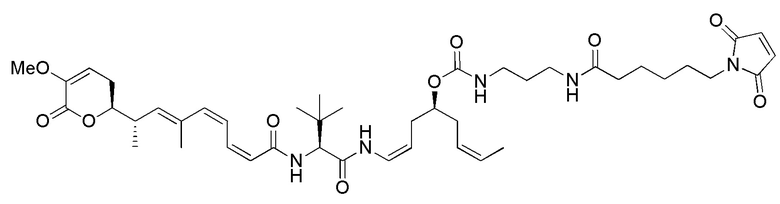 , и
, и
 .
.
30. Соединение формулы D-X-(AA)w-H по любому одному из пп. 23-28, выбранное из:
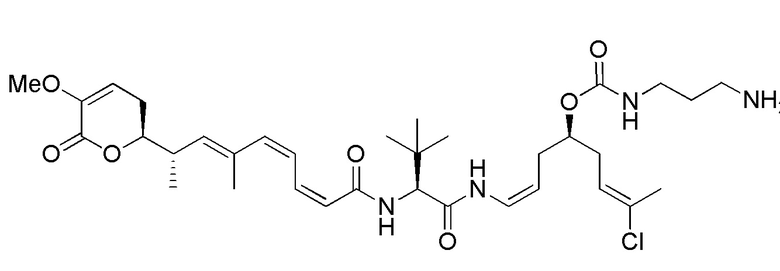 ,
,
 ,
,
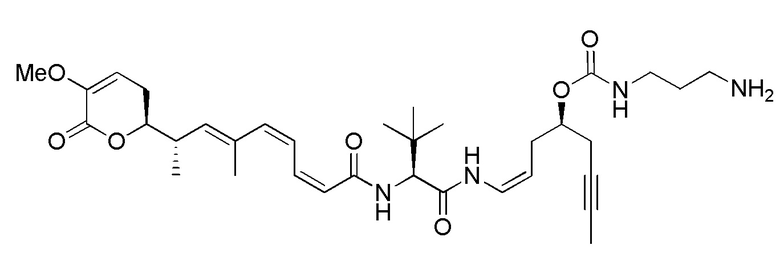 ,
,
 ,
,
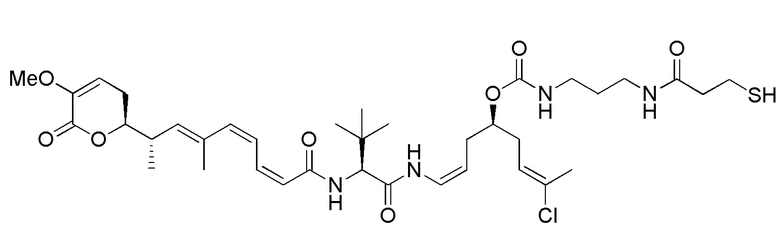 ,
,
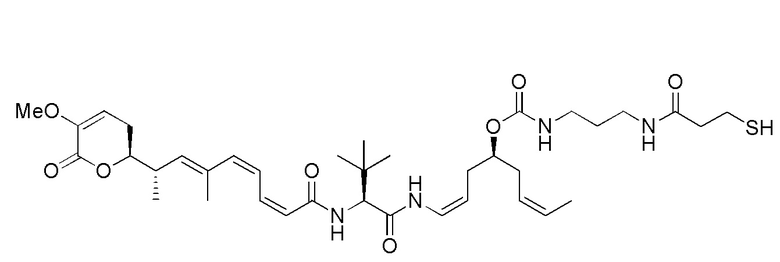 и
и
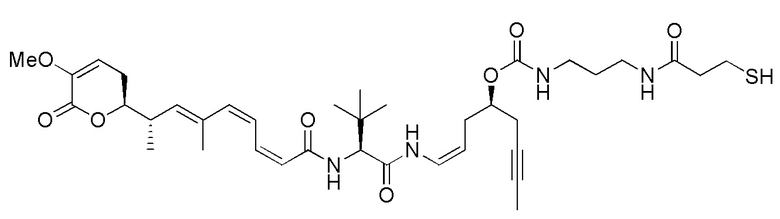 .
.
31. Конъюгат лекарственного средства по любому одному из пп. 1-22 для применения в качестве лекарственного средства для лечения рака.
32. Конъюгат лекарственного средства по п. 31 для лечения рака, выбранного из рака легких, колоректального рака, рака молочной железы, рака поджелудочной железы, рака почки, лейкоза, множественной миеломы, лимфомы и рака яичников.
33. Фармацевтическая композиция для лечения рака, включающая эффективное количество конъюгата лекарственного средства по любому одному из пп. 1-22 и фармацевтически приемлемый носитель.
| RU 2009101150 A, 27.07.2010 | |||
| WO 2009080761 A1, 02.07.2009 | |||
| EP 1864682 A1, 12.12.2007. |

