Изобретение относится к области медицины, фармакологии и химико-фармацевтической промышленности, а именно, к новому усовершенствованному способу получения (1R,2S,5S)-N-[(1S)-1-циано-2-[(3S)-2-оксопирролидин-3-ил]этил]-3-[(2S)-3,3-диметил-2-[(2,2,2-трифторацетил)амино]бутаноил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоксамида (соединение формулы (I)), а также способу получения промежуточного продукта, предназначенного для применения в способе получения соединения формулы (I).
Предпосылки создания изобретения.
Одна из наиболее актуальных проблем современной медицины - это борьба с вирусными инфекциями. По данным Всемирной организации здравоохранения (ВОЗ), новые инфекционные заболевания возникают с угрожающей скоростью, так с 1970-х годов было обнаружено около 40 новых инфекционных заболеваний. Одними из самых опасных среди новых возникающих вирусов являются РНК-вирусы, яркими примерами которых за последние 20 лет являются: коронавирус тяжелого острого респираторного синдрома (SARS-CoV) в 2002-2003 гг., грипп (свиной грипп) A/H1N1 в 2009 г., коронавирус ближневосточного респираторного синдрома (MERS) в 2012 году, эпидемия болезни, вызванной вирусом Эбола в 2013-2015 годах, пандемия вируса Зика в 2016-2017 годах, а теперь и пандемия COVID-19. В дополнение к этим недавно выявленным возбудителям такие ранее признанные вирусные инфекции, включая пандемический грипп, желтую лихорадку, корь и другие, продолжают представлять угрозу в связи с возникновением новых эпидемий (David A. Schwartz. Prioritizing the Continuing Global Challenges to Emerging and Reemerging Viral Infections // Front.Virol.- 2021).
Филогенетический анализ показал, что некоторые одноцепочечные (+)РНК-вирусы могут быть отнесены к пикорнавирусоподобному суперкластеру, который включает вирусы, принадлежащие к семействам Picornaviridae, Caliciviridae и Coronaviridae. Общей чертой вирусов пикорнавирусоподобного суперкластера является то, что они используют 3C или 3C-подобную протеазу (3Cpro или 3CLpro соответственно) в своем метаболизме. Консервативные сайты 3Cpro или 3CLpro могут служить привлекательными мишенями для разработки противовирусных препаратов широкого спектра действия для вирусов в пикорнавирусоподобном суперкластере. (Yunjeong Kim et al. Broad-Spectrum Antivirals against 3C or 3C-Like Proteases of Picornaviruses, Noroviruses, and Coronaviruses // J Virol.- 2012.- 86(21), p. 11754-11762).
Вирус SARS-CoV2 - это тяжёлая острая респираторная инфекция. Коронавирусная пандемия стартовала в мировом масштабе в декабре 2019 года. Согласно статистике, ежедневно регистрируются сотни тысяч заболевших и тысячи летальных исходов. По данным на 11 октября 2021 года количество заболевших насчитывает 251 млн. человек, а летальные исходы пересекли рубеж в 5 млн. человек. С начала всемирной пандемии был нанесен тяжелый урон как глобальному человеческому здоровью, так и общемировой экономике. Пандемия послужила вызовом для современной медицины, сформировав острую необходимость в разработке эффективных стратегий профилактики и лечения вирусных заболеваний. Отсутствие специальной терапии против нового вируса и его высокая изменчивость требует создания новых лекарственных средств. В дополнение к существующим средствам терапии необходимы специфические препараты, оказывающие непосредственное действие на вирус, приводящие к облегчению симптомов заболевания, ускоренному разрешению заболевания, блокированию передачи инфекции и уменьшению риска развития клинических осложнений.
Одним из перспективных путей блокирования вируса SARS-CoV2 является ингибирование протеазы SARS-CoV2, также называемой 3C-подобной протеазой (3CL или 3CLpro). Ингибирование протеазы 3CL приводит к предотвращению репликации вируса. (1R,2S,5S)-N-[(1S)-1-циано-2-[(3S)-2-оксопирролидин-3-ил]этил]-3-[(2S)-3,3-диметил-2-[(2,2,2-трифторацетил)амино]бутаноил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоксамид, который характеризуется Формулой (I), подавляет в протеазе активность остатка цистеина на стадии протеолиза. 3CLpro отвечает за процесс расщепления липопротеинов вируса, содержащих сам 3CLpro, PLpro (папаинподобную протеазу) и еще 14 прочих белков. Молекула показала высокую противовирусную активность в доклинических исследованиях на модели адаптированных к мышам SARS-CoV-2. Кроме того из уровня техники известно, что (1R,2S,5S)-N-[(1S)-1-циано-2-[(3S)-2-оксопирролидин-3-ил]этил]-3-[(2S)-3,3-диметил-2-[(2,2,2-трифторацетил)амино]бутаноил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоксамид может проявлять способность ингибирования широкого спектра 3C-подобной протеазы не только SARS-CoV-2, что подчеркивает возможность использования для других вирусов человека.

Соединение формулы (I)
При разработке лекарственных средств важно, чтобы вещество, которое является активным агентом, было доступным и соответствовало требованиям качества. Наиболее оптимальный способ увеличения доступности и контроля качества активного агента - это разработка уникального синтетического протокола. Синтез активного агента позволяет регулировать такие свойства субстанции, как форму активного агента, например, кристаллическую, аморфную или их соотношения; химическую чистоту, в частности, количество родственных примесей или реагентов, используемых в ходе химических превращений, стереоизомерную чистоту активного агента.
Химическая чистота имеет большое значение в производстве субстанций. Известно, что степень чистоты субстанций потенциально влияет на их безопасность и эффективность, как в медицине, так и в ветеринарии. Повышение химической чистоты субстанции обеспечивает улучшение ее исходных биологических свойств и снижение возможных побочных действий, связанных с наличием примесей.
Разработка новых подходов к синтезу активного агента открывает возможности улучшения профиля физико-химических и технологических характеристик фармацевтического продукта. Это расширяет спектр активных агентов, которые специалист по разработке препаратов имеет в своем распоряжении при разработке нового лекарственного средства с повышенным профилем безопасности. Также устойчивое развитие современных подходов к синтезу активных агентов позволяет внедрять в производства экологические, ресурсоэффективные, атомэкономные инновационные технологии, которые способствуют экономическому развитию и минимизации ущерба окружающей среде.
Таким образом, в настоящее время имеется постоянная потребность в разработке новых, улучшенных способов синтеза противовирусных агентов.
В связи с указом Президента РФ от 7 мая 2018 г. № 204 "О национальных целях и стратегических задачах развития Российской Федерации на период до 2024 года" в рамках программы по импортозамещению «Фарма-2030» авторами настоящего изобретения были приложены усилия по созданию инновационного способа получения противовирусного соединения (1R,2S,5S)-N-[(1S)-1-циано-2-[(3S)-2-оксопирролидин-3-ил]этил]-3-[(2S)-3,3-диметил-2-[(2,2,2-трифторацетил)амино]бутаноил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоксамида (соединение формулы (I)).
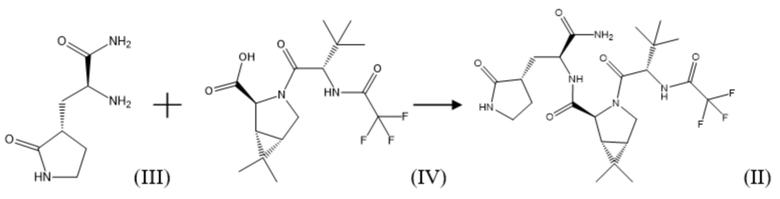
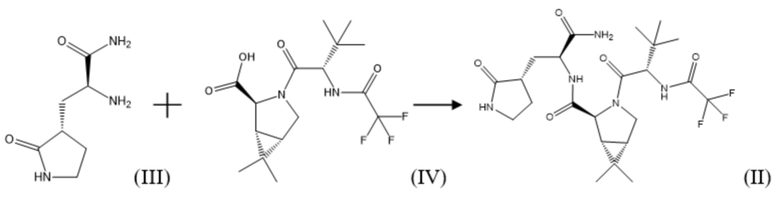
Важным промежуточным продуктом в синтезе соединения формулы (I) по настоящему изобретению является (1R,2S,5S)-N-{(2S)-1-амино-1-оксо-3-[(3S)-2-оксопирролидин-3-ил]пропан-2-ил]-6,6-диметил-3-[3-метил-N-(трифторацетил)-L-валил]-3-азабицикло[3.1.0]гексан-2-карбоксамид соединение формулы (II):
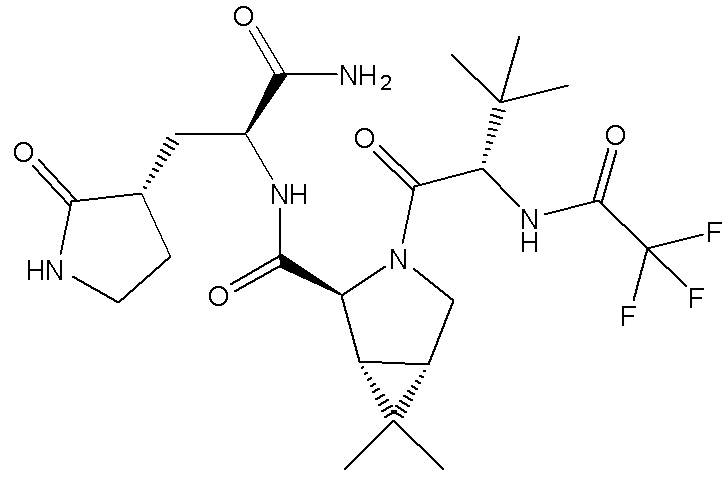
Соединение формулы (II)
В уровне техники (Owen D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19 // Science. - 2021. - Т. 374. - №. 6575. - С. 1586-1593 и WO2021250648) раскрыты способы получения сольватированной метилтретбутил эфирной формы соединения формулы (I) с последующим преобразованием в свободную форму под действием гептана с изопропилацетатом. При этом предшествующая стадия представляет собой получение промежуточного продукта соединения формулы (II) посредством взаимодействия (1R,2S,5S)-6,6-диметил-3-[3-метил-N-(трифторацетил)-L-валил]-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты с 3-[(3S)-2-оксопирролидин-3-ил]-L-аланинамидом в виде гидрохлорида в присутствии 2-гидроксипиридин-1-оксида в бутан-2-оне с дальнейшим добавлением 1-этил-3-(3-диметиламинопропил)карбодиимида.
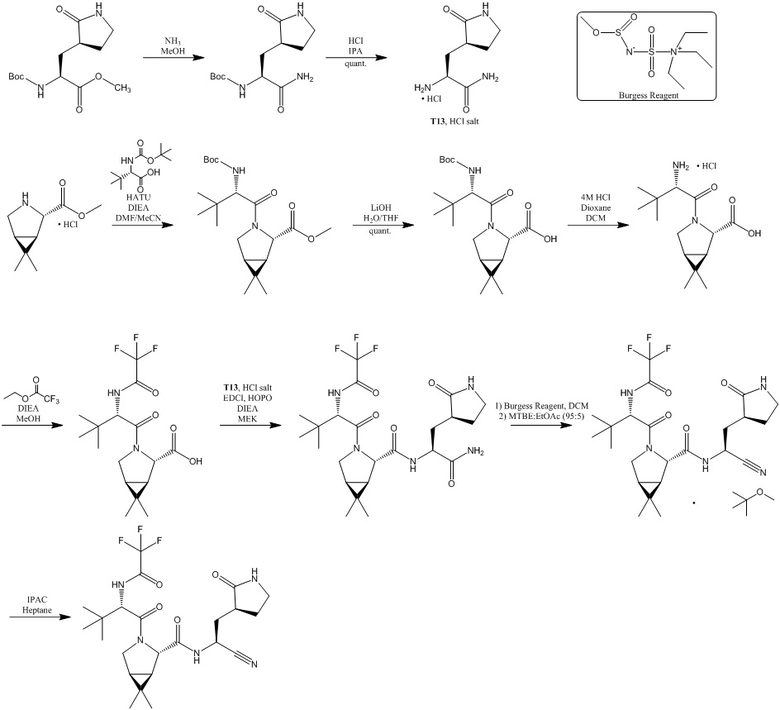 , где Burgess Reagent- реактив Берджесса;
, где Burgess Reagent- реактив Берджесса;
IPA- изопропанол;
HATU- О-(7-азабензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфат;
DIEA- N,N-диизопропилэтиламин;
DMF- диметилформамид;
THF- тетрагидрофуран;
Dioxane- диоксан;
DCM- дихлорметан;
EDCl- 1-этил-3-(3-диметиламинопропил)карбодиимид;
HOPO- 2-гидроксипиридин-N-оксид;
MEK- метилэтилкетон;
MTBE-метилтретбутиловый эфир;
IPAC- изопропилацетат;
Heptane- гептан.
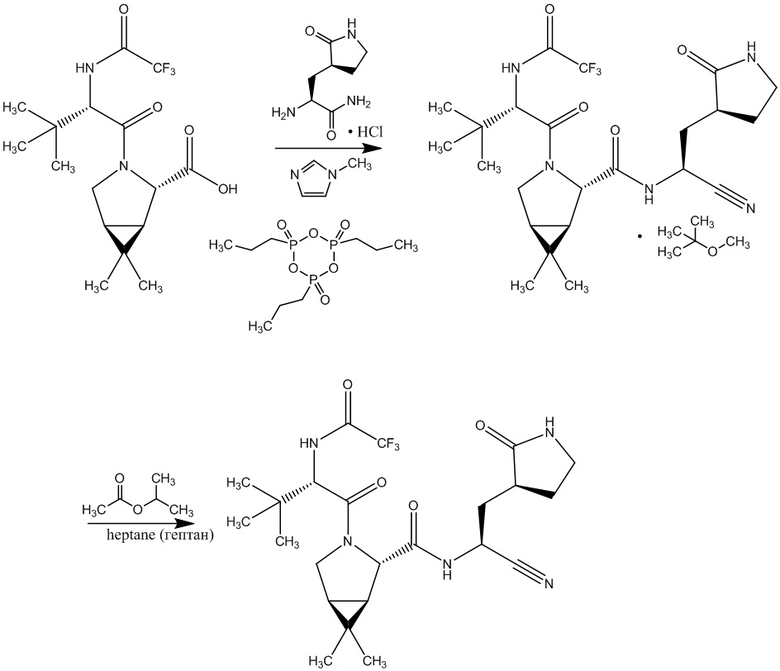
Недостатком указанного синтеза соединения формулы (I) является, в том числе, применение дорогостоящих токсичных реактивов, например, 1-этил-3-(3-диметиламинопропил)карбодиимида, который применяют только для синтеза в лабораторных условиях, что не позволяет масштабировать синтез в промышленное производство. При этом такие реактивы в процессе синтеза образуют побочные продукты: использование 1-этил-3-(3-диметиламинопропил)карбодиимид сопровождается образованием следующих побочных продуктов (продуктов присоединения карбодиимидной группы к амину и продуктов внутреннего ацилирования (ацилмочевин):
.
Также в заявке WO2021250648 описан синтез соединения формулы (I), представляющий собой взаимодействие метил (1R,2S,5S)-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоксилат гидрохлоридной соли с О-(7-азабензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфатом и N-(трет-бутоксикарбонил)-3 - метил-L-валином в присутствии N,N- диметилформамида с последующим прибавлением к полученному продукту гидроксида лития в присутствии смеси тетрагидрофурана и метанола, с последующим смешиванием полученного продукта с (2S)-2-амино-3-[(3S)-2-оксопирролидин-3-ил] пропаннитрил метансульфонатной солью в ацетонитриле при последовательном добавлении О-(7-азабензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфата, раствора 4-метилморфолина в ацетонитриле. Последняя стадия синтеза включает добавление метансульфокислоты к продукту, полученному на предыдущей стадии, в 1,1,1,3,3,3-гексафторпропан-2-оле, с дальнейшим концентрированием в смеси растворителей ацетонитрил/этилацетат и этилацетат/гептан с последующим растворением в дихлорметане и обработкой 4-метилмофолином и трифторуксусным ангидридом.
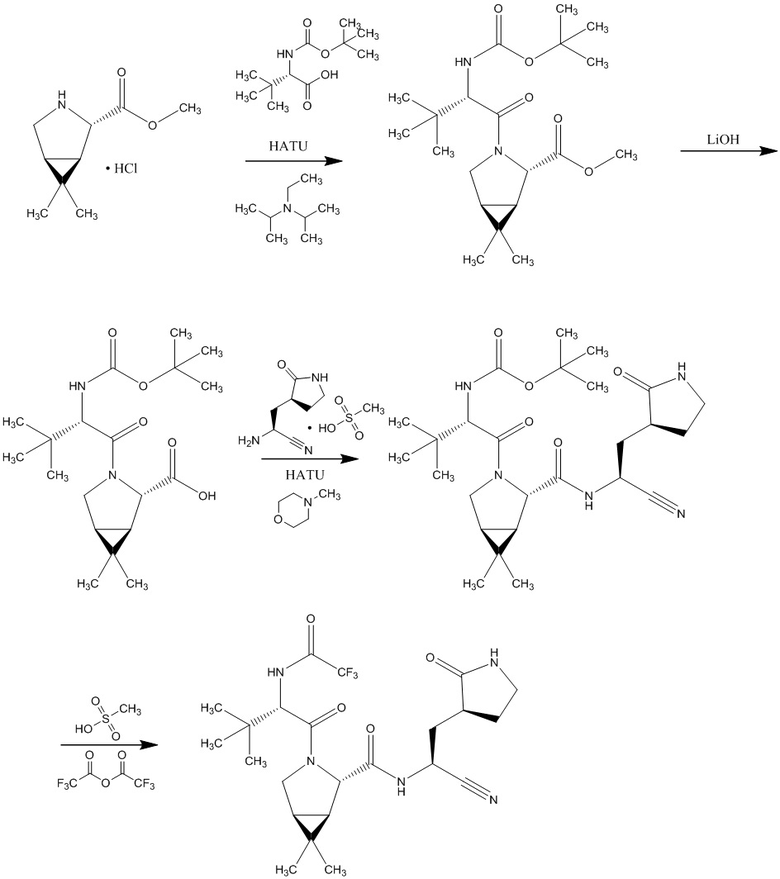
, где HATU- О-(7-азабензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфат.
В статье (Zhao Y. et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332 //Protein & cell. - 2021. - С. 1-5) описан синтез соединения формулы (I). Финальная стадия синтеза предполагает взаимодействие двух интермедиатов под действием O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и диизопропилэтиламина.
Также авторы изобретения хотели бы отметить, что при взаимодействии промежуточного продукта в указанном синтезе с гидроксидом натрия происходит отщепление трифторацетила, что противоречит указанному в синтезе (Albert Isidro-Llobet et al. Amino Acid-Protecting Groups // Chem. Rev.- 2009.-109, 6.- p. 2455-2504).
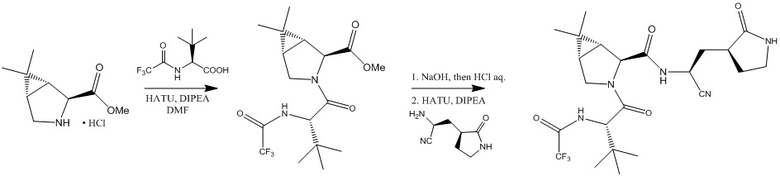
, где HATU- О-(7-азабензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфат; DIPEA- N,N-диизопропилэтиламин;
DMF- диметилформамид.
При этом основными недостатками указанных в уровне техники синтезов соединения формулы (I) являются применение дорогостоящих токсичных реактивов, в том числе, 2- гидроксипиридин-1-оксида, O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, 1-этил-3-(3-диметиламинопропил)карбодиимида, которые применяют только для синтеза в лабораторных условиях, что не позволяет масштабировать синтез в промышленное производство. При этом такие реактивы в процессе синтеза образуют побочные продукты: использование O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата сопровождается образованием побочного продукта тетраметилмочевины, обладающего 1 классом опасности по тератогенности и не поддающемуся дальнейшей переработке, что крайне негативно сказывается на окружающей среде. Помимо перечисленного описанные синтезы имеют низкий общий выход целевого продукта по всем стадиям синтеза, что является нецелесообразным для промышленного производства.
Задачей настоящего изобретения является разработка нового улучшенного, экологически безвредного, безопасного способа получения соединения формулы (I) с высоким выходом продукта, в котором используются легко доступные реактивы, при помощи которых можно воспроизвести синтез в промышленном масштабе. Также задачей настоящего изобретения является обеспечение нового, усовершенствованного синтеза промежуточных продуктов в синтезе соединения формулы (I).
В результате исследований авторами настоящего изобретения был разработан новый улучшенный по сравнению с известными из уровня техники способами способ получения соединения формулы (I), включающий новую стадию получения промежуточного продукта соединения формулы (II), пригодный для промышленного производства. При этом, авторами изобретения было неожиданно обнаружено, что способ получения соединения формулы (I) по настоящему изобретению позволяет получать целевой продукт (соединение формулы (I)) с общим выходом, превышающим выход в описанных в уровне техники способах получения соединения формулы (I). Также авторы изобретения в ходе исследований смогли разработать новый способ получения промежуточного продукта (соединения формулы (II)), в результате которого получили с высоким выходом стереоизомерно чистое вещество (соединение формулы (II)) без образования нежелательных побочных продуктов.
Таким образом, техническими результатами настоящего изобретения являются:
- высокий выход и химическая чистота (в том числе стереоизомерная) целевого продукта (соединения формулы (I)) и промежуточного продукта (соединения формулы (II));
- адаптирование способа получения соединения формулы (I) под промышленное производство;
- уменьшение экономических затрат на осуществление синтеза;
- обеспечение экологически безопасного производства;
- уменьшение образования нежелательных побочных продуктов в ходе синтеза.
Ниже приведены определения терминов, которые используются в описании настоящего изобретения.
Если не указано иное, все технические и специальные термины, использованные в данном контексте, имеют общепринятое в данной области техники значение.
Термин «содержащий», «содержит», «включающий» в контексте настоящего изобретения означает, что указанные фармацевтические составы, продукты, композиции и лекарственные средства включают перечисленные компоненты, но не исключают включение других компонентов, а также указанные способы получения соединений включают перечисленные этапы синтеза, но не исключают включение других.
Термин «эффективное количество» в контексте настоящего изобретения означает количество вещества (а также комбинации веществ), которое при ведении субъекту для лечения или предотвращения заболевания, или по меньшей мере одного из клинических симптомов заболевания или нарушения является достаточным для воздействия такого лечения и/или профилактики на заболевание, нарушение или симптом. «Эффективное количество» определяется в зависимости от заболевания, нарушения и/или симптомов заболевания или нарушения, тяжести заболевания, нарушения и/или симптомов заболевания или нарушения, клинической картины и общего состояния здоровья пациента, а также от возраста и/или веса субъекта, которому необходимо такое лечение (профилактика).
Используемый в тексте термины «примерно», «около» обозначает приблизительно плюс или минус десять процентов от указанной величины.
Термин «примеси» в контексте настоящего изобретения представляет собой любой компонент в продукте синтеза, который не является целевым веществом. Примеси в продукте синтеза включают: неорганические примеси, органические примеси (в том числе остаточные растворители, используемые в синтезе соединений). В перечисленные группы также входят родственные и технологические примеси. Источником примесей могут являться используемые в синтезе исходных веществ реактивы, побочные продукты, продукты разложения или полимеризации, реагенты, лиганды, структурные или пространственные изомеры, катализаторы, технологическое оборудование, остатки фильтрующих материалов, недостаточно очищенное исходное сырье, использующиеся для синтеза активного агента.
Термин «химическая чистота» в контексте настоящего изобретения характеризует содержание (в %) целевого вещества в образце по отношению к общему количеству образца. Химически чистым по настоящему изобретению является вещество, химическая чистота которого составляет 97,0% или более, более предпочтительно, 98,0% или более, и особенно предпочтительно, 99,0% или более.
Термин «стереоизомеры» в контексте настоящего изобретения, определяет все возможные изомеры, которые могут иметь соединения по настоящему изобретению, образованные из идентичных наборов атомов, соединенных одинаковой последовательностью связей, но имеющих разное пространственное расположение атомов или групп атомов относительно связей, при этом изомерные соединения не являются взаимозаменяемыми. Если не указано иное, химическое обозначение соединения включает в себя смесь всех возможных стереохимически изомерных форм, которые может иметь указанное соединение. Указанная смесь может содержать все диастереомеры и/или энантиомеры молекулярной структуры указанного соединения. Предполагается, что за исключением случая, когда указано иное, все стереоизомерные формы соединений, применяемых в настоящем изобретении, как в чистой (индивидуальной) форме, так и в смеси друг с другом, включены в объем настоящего изобретения.
Термин «диастереомеры» относится к стереоизомерам, в структуре которых содержатся два и более асимметричных атомов углерода, а именно атомов углерода, связанных с четырьмя различными заместителями, при условии, что стереоизомеры не являются зеркальным отражением друг друга. Однако, если конфигурация различается по меньшей мере у одного стереоцентра, то такие стереизомеры являются диастереомерами.
Термин «энантиомеры» относится к стереоизомерам, в структуре которых содержатся один или более асимметричных атомов углерода, а именно атомов углерода, связанных с четырьмя различными заместителями, при условии, что пара данных стереоизомеров являются зеркальным отражением друг друга, не совмещаемым в пространстве. Если два стереоизомера имеют противоположные конфигурации всех соответствующих стереоцентров, то они являются энантиомерами.
Термин «стереоизомерно чистый» в контексте настоящего изобретения определяется как относительный избыток одного стереоизомера в образце над другим. Стереоизомерно чистым по настоящему изобретению является вещество (образец), имеющее стереоизомерный избыток по меньшей мере 80% вплоть до стереоизомерного избытка 100% (т.е. 100% одного стереоизомера и никакого другого), более предпочтительно, вещество, имеющее стереоизомерный избыток от 90% до 100%, более предпочтительно, имеющее стереоизомерный избыток от 95% до 100%, более предпочтительно, имеющее стереоизомерный избыток от 97% до 100%. Термины «энантиомерно чистый» и «диастереомерно чистый» следует понимать таким же образом, но ссылаясь на энантиомерный избыток и диастереомерный избыток рассматриваемой смеси соответственно.
Стереоизомерно чистые формы соединений по настоящему изобретению можно получить посредством использования известных в данной области методик. Например, стереоизомеры по настоящему изобретению можно отделить друг от друга селективной перекристаллизацией их диастереомерных солей с оптически активными кислотами; стереоизомеры по настоящему изобретению можно разделить хроматографическими способами с применением хиральных неподвижных фаз. Чистые стереохимически изомерные формы соединений по настоящему изобретению можно также получить из соответствующих чистых стереохимически изомерных форм подходящих исходных веществ, при условии, что реакция протекает стереоселективно. Если требуется определенный стереоизомер, указанное соединение предпочтительно будет синтезировано стереоизбирательными способами получения. В этих способах преимущественно будут применяться стереоизомерно чистые исходные вещества.
Одним из основных методов для контроля стереоизомерной чистоты химических соединений по настоящему изобретению является метод высокоэффективной жидкостной хроматографии (ВЭЖХ), который основан на различном распределении веществ между двумя не смешивающимися фазами. Жидкость, являющаяся подвижной фазой, проходит через неподвижную фазу, помещенную в колонку с высоким гидравлическим сопротивлением. Жидкостная хроматография, в которой используются колонки с уменьшенным размером частиц (например, менее 2 мкм) называется сверхвысокоэффективной жидкостной хроматографией (СВЭЖХ). Жидкостная хроматография в зависимости от механизма разделения веществ может быть адсорбционной, распределительной, ионообменной, эксклюзионной, хиральной и др. в соответствии с характером основных проявляющихся межмолекулярных взаимодействий. Каждый из этих видов может быть высокоэффективным, если для проведения хроматографии используется режим хроматографирования с высоким давлением в колонке.
Еще одним способом разделения изомеров может выступать перекристаллизация. Перекристаллизация - это способ очистки соединений от различных примесей (например, родственных, технологических), основанный на различии растворимости веществ в различных растворителях или их смесях при различных температурах. Одним из вариантов осуществления перекристаллизации является добавление в раствор вещества антирастворителя (растворителя, в котором вещество имеет низкую растворимость), в результате чего растворяющая способность среды ухудшается и растворенное вещество переходит из раствора в твердую форму. Преимуществами метода перекристаллизации являются высокая степень очистки, однако в некоторых случаях недостатком может быть высокая потеря вещества.
В рамках настоящего изобретения, поставленная задача решается, а заявленный технический результат достигается посредством нового способа получения соединения формулы (II), который включает взаимодействие соединения формулы (III) или его соли, например, гидрохлоридом, с соединением формулы (IV) с использованием соединения формулы R1O-CO-Cl, где R1 представляет собой C1-C5 алкил; C1-C5 алкил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор; фенил; фенил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор, нитро группой или C1-C5 алкилом; бензил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор, нитро группой или C1-C5 алкилом, в присутствии основания, по следующей схеме синтеза:
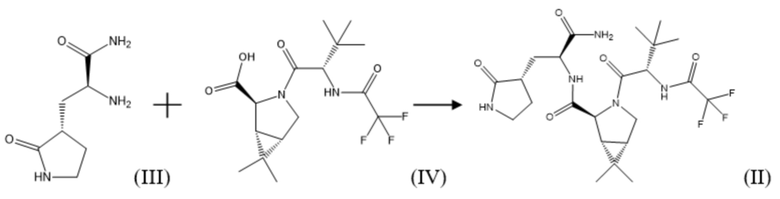
Одним из вариантов воплощения настоящего изобретения является способ получения соединения формулы (II), в котором взаимодействие соединения формулы (III) или его соли, например, гидрохлоридом, с соединением формулы (IV) проходит в органическом растворителе.
Более предпочтительно органический растворитель в указанной реакции представляет собой апротонный органический растворитель.
Более предпочтительно органический растворитель в указанной реакции выбран из группы, включающей, не ограничиваясь указанным, диметилформамид, диметилацетамид, N-метилпирролидон, тетрагидрофуран, дихлорметан, этилацетат или их смеси.
Более предпочтительно соединение формулы R1O-CO-Cl по настоящему изобретению выбрано из группы, включающей, не ограничиваясь указанным, метилхлорформиат, этилхлорформиат, пропилхлорформиат, бутилхлорформиат, изобутилхлорформиат или третбутилхлорформиат.
Более предпочтительно в качестве основания по настоящему изобретению используют органическое основание.
Более предпочтительно в качестве основания по настоящему изобретению используют, не ограничиваясь указанным, пиридин; пиридин, замещенный C1-C5 алкилом; хинолин или хинолин, замещенный C1-C5 алкилом.
Более предпочтительно органическое основание по настоящему изобретению представляет собой третичный амин.
Более предпочтительно третичный амин по настоящему изобретению представляет собой соединение формулы R2R3R4N, где R2, R3, R4 независимо друг от друга представляют собой C1-C5 алкил или, когда R2 с R3 вместе с атомом азота образуют С4-С8 циклоалкильную группу или С4-С8 гетероциклическую группу, где гетероатом представляет собой кислород, R4 представляет собой C1-C5 алкил.
Более предпочтительно третичный амин по настоящему изобретению представляет собой соединение формулы R2R3R4N, где R2, R3, R4 независимо друг от друга представляют собой C1-C5 алкил или, когда R2 с R3 вместе с атомом азота образуют С4-С8 циклоалкильную группу или С4-С8 гетероциклическую группу, где гетероатом представляет собой кислород, R4 представляет собой C1-C5 алкил, при этом R4 связан с атомом.
Более предпочтительно третичный амин по настоящему изобретению представляет собой соединение формулы R2R3R4N, где R2, R3, R4 независимо друг от друга выбраны из группы, включающей, метил, этил, пропил или изопропил.
Более предпочтительно третичный амин по настоящему изобретению представляет собой, не ограничиваясь указанным, N-метилформалин или триэтиламин.
Более предпочтительно реакцию получения соединения формулы (II) по настоящему изобретению проводят при температуре в интервале от -50°С до 50°С.
Более предпочтительно реакцию получения соединения формулы (II) по настоящему изобретению проводят при температуре в интервале от -40°С до 40°С.
Более предпочтительно реакцию получения соединения формулы (II) по настоящему изобретению проводят при температуре в интервале от -30°С до 30°С.
Более предпочтительно реакцию получения соединения формулы (II) по настоящему изобретению проводят при температуре в интервале от -20°С до 20°С.
Более предпочтительно реакцию получения соединения формулы (II) по настоящему изобретению проводят при температуре в интервале от -10°С до 10°С.
Более предпочтительно реакцию получения соединения формулы (II) по настоящему изобретению проводят при температуре в интервале от -5°С до 5°С.
Одним из вариантов воплощения настоящего изобретения является способ получения соединения формулы (II), в котором к раствору соединения формулы (III) или его соли, например, гидрохлорида, в присутствии основания, например, охарактеризованного выше, добавляют соединение формулы R1O-CO-Cl, где R1 представляет собой C1-C5 алкил, C1-C5 алкил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор; фенил; фенил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор, нитро группой или C1-C5 алкилом; бензил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор, нитро группой или C1-C5 алкилом, при температуре от -50°С до 10°С и выдерживают, далее к реакционной массе добавляют соединение формулы (IV) и основание при температуре от -50°С до 10°С, а затем выдерживают полученную реакционную массу при температуре от -10°С до 50°С до окончания реакции.
Одним из вариантов воплощения настоящего изобретения является способ получения соединения формулы (II), в котором к раствору соединения формулы (III) или его соли, например, гидрохлорида, в присутствии основания, например, охарактеризованного выше, добавляют соединения формулы R1O-CO-Cl, где R1 представляет собой C1-C5 алкил; C1-C5 алкил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор; фенил; фенил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор, нитро группой или C1-C5 алкилом; бензил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор, нитро группой, или C1-C5 алкилом, при температуре от -30°С до -15°С и выдерживают, далее к реакционной массе добавляют соединение формулы (IV) и основание при температуре от -25°С до -15°С, а затем выдерживают полученную реакционную массу при температуре от -5°С до 5°С до окончания реакции.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (II), в котором стадия взаимодействия соединения формулы (III) или его соли, например, гидрохлорида, с соединением формулы (IV) дополнительно включает следующие стадии: перекристаллизации продукта из органического растворителя и/или очистки продукта методом хроматографии.
Более предпочтительно стадия перекристаллизации полученного продукта включает в себя суспендирование осажденного продукта в органическом растворителе, нагрев получаемой смеси до получения однородного раствора и последующее охлаждение.
Более предпочтительно охлаждение осуществляется естественным путем при комнатной температуре.
Поставленная задача решается, а заявленный технический результат достигается посредством нового способа получения соединения формулы (I), который включает дегидратацию соединения формулы (II), полученного по любому из способов по настоящему изобретению, с образованием соединения формулы (I) с использованием 2,4,6-трихлор-1,3,5-триазина в среде растворителя, по следующей схеме:
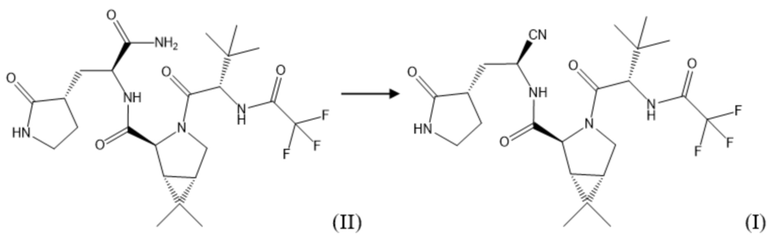
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), в котором в качестве растворителя используется N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), в котором дегидратацию соединения формулы (II) проводят при температуре в интервале от 0°С до 100°С.
Более предпочтительно дегидратацию соединения формулы (II) проводят при температуре в интервале от 15°С до 25°С.
Одним из вариантов воплощения настоящего изобретения является способ получения соединения формулы (I), в котором стадия дегидратации соединения формулы (II) дополнительно включает следующие стадии: a) выделение продукта и/или b) промывка выделенного продукта и/или; c) сушка продукта.
Одним из вариантов воплощения настоящего изобретения является способ получения соединения формулы (I), в котором стадия дегидратации соединения формулы (II) дополнительно включает следующие стадии: a) выделение продукта; b) промывка выделенного продукта; c) сушка продукта.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), в котором стадия дегидратации соединения формулы (II) дополнительно включает следующие стадии: a) выделение целевого продукта и/или b) промывка выделенного целевого продукта и/или; c) сушка целевого продукта.
Более предпочтительно дополнительную стадию выделения (a) продукта проводят методом осаждения или экстракции.
Более предпочтительно дополнительную стадию выделения продукта (a) проводят методом фильтрования.
Более предпочтительно дополнительную стадию выделения продукта (a) проводят методом осаждения с последующим фильтрованием.
Более предпочтительно дополнительную стадию промывки выделенного продукта (b) проводят при помощи воды, раствора лимонной кислоты или водного раствора гидрокарбоната натрия.
Более предпочтительно дополнительная стадия сушки (c) представляет собой стадию сушки при пониженном давлении.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), в котором стадия дегидратации соединения формулы (II) дополнительно включает следующие стадии: a) выделение продукта и/или b) промывка выделенного продукта и/или; c) сушка продукта, где стадия промывки выделенного продукта дополнительно включает стадию перекристаллизации продукта из органического растворителя.
Более предпочтительно стадия перекристаллизации полученного продукта включает в себя суспендирование осажденного продукта в органическом растворителе, нагрев получаемой смеси до получения однородного раствора и последующее охлаждение.
Более предпочтительно охлаждение осуществляется естественным путем при комнатной температуре.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I) (целевого продукта), в котором стадия дегидратации соединения формулы (II) дополнительно включает следующие стадии: a) выделение продукта и/или b) промывка выделенного продукта и/или; c) сушка продукта, где стадия промывки выделенного продукта дополнительно включает стадию перекристаллизации продукта посредством суспендирования осажденного продукта в органическом растворителе, нагрева получаемой смеси до получения однородного раствора и последующего охлаждения естественным путем при комнатной температуре.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), в котором стадия дегидратации соединения формулы (II) дополнительно включает следующие стадии: a) выделение продукта и/или b) промывка выделенного продукта и/или; c) сушка продукта, где стадия промывки продукта дополнительно включает стадию очистки продукта методом хроматографии.
Поставленная задача решается, а заявленный технический результат достигается за счет применения способа получения соединения формулы (I) по настоящему изобретению для периодического химического промышленного производства, полунепрерывного промышленного производства или непрерывного промышленного производства соединения формулы (I).
Одним из вариантов воплощения изобретения является применение способа получения соединения формулы (I), который включает дегидратацию соединения формулы (II) с образованием соединения формулы (I) с использованием 2,4,6-трихлор-1,3,5-триазина в среде растворителя, для периодического химического промышленного производства, полунепрерывного промышленного производства или непрерывного промышленного производства соединения формулы (I).
Поставленная задача решается, а заявленный технический результат достигается за счет применения способа получения соединения формулы (I) по настоящему изобретению для периодического химического промышленного производства, полунепрерывного промышленного производства или непрерывного промышленного производства продукта, содержащего соединение формулы (I).
Поставленная задача решается, а заявленный технический результат достигается за счет применения способа получения соединения формулы (I) по настоящему изобретению для получения продукта, содержащего соединение формулы (I).
Поставленная задача решается, а заявленный технический результат достигается посредством продукта, содержащего соединение формулы (I), полученное способом по настоящему изобретению.
Продукт в контексте настоящего изобретения может представлять собой субстанцию, содержащую соединение формулы (I), полученное любым способом по настоящему изобретению. Термин «субстанция» в контексте настоящего изобретения означает продукт синтеза, содержащий любое вещество или смесь веществ синтетического или иного (биологического, биохимического, минерального, растительного, животного, микробного и прочего) происхождения, предназначенный для получения лекарственных средств, который в процессе производства лекарственного средства (препарата) становится активным ингредиентом этого лекарственного средства и определяет его эффективность.
Одним из вариантов воплощения изобретения является фармацевтический продукт, содержащий соединение формулы (I), полученное способом по настоящему изобретению.
Одним из вариантов воплощения изобретения является фармацевтический продукт, содержащий соединение формулы (I), полученное способом по настоящему изобретению, отличающийся тем, что общее содержание примесей в его составе не превышает 1,5 мас. % от массы фармацевтического продукта.
Одним из вариантов воплощения изобретения является фармацевтический продукт, содержащий соединение формулы (I), полученное способом по настоящему изобретению, отличающийся тем, что общее содержание примесей в его составе менее 1,5 мас. %, но более 0,001 мас.% от массы фармацевтического продукта.
Одним из вариантов воплощения настоящего изобретения является фармацевтический продукт, содержащий соединение формулы (I), полученное способом по настоящему изобретению, для лечения и/или профилактики вирусных заболеваний, где вирус представляет собой, не ограничиваясь указанным, пикорнавирус, норовирус, вирус ящура, вирус гепатита А, поливирус, риновирус, энтеровирус, калицивирус или коронавирус.
Одним из вариантов воплощения настоящего изобретения является фармацевтический продукт, содержащий соединение формулы (I), полученное способом по настоящему изобретению, для лечения и/или профилактики вирусных заболеваний, где вирус представляет собой SARS-CoV, MERS-CoV или SARS-CoV-2.
Одним из вариантов воплощения настоящего изобретения является фармацевтический продукт, содержащий соединение формулы (I), полученное способом по настоящему изобретению, который проявляет профилактическую и/или терапевтическую противовирусную активность в отношении вируса SARS-CoV-2.
Одним из вариантов воплощения настоящего изобретения является противовирусный фармацевтический продукт, содержащий соединение формулы (I), полученное способом по настоящему изобретению.
Поставленная задача решается, а заявленный технический результат достигается посредством применения продукта, содержащего соединение формулы (I), полученное способом по настоящему изобретению, для лечения и/или профилактики вирусных заболеваний.
Поставленная задача решается, а заявленный технический результат достигается посредством применения фармацевтического продукта, содержащего соединение формулы (I), полученное способом по настоящему изобретению, для лечения и/или профилактики вирусных заболеваний.
Одним из вариантов воплощения настоящего изобретения является применения продукта, содержащего соединение формулы (I), полученное способом по настоящему изобретению, для лечения и/или профилактики вирусных заболеваний, выбранных из группы, не ограничиваясь указанным, пикорнавирус, норовирус, вирус ящура, вирус гепатита А, поливирус, риновирус, энтеровирус, калицивирус или коронавирус.
Поставленная задача решается, а заявленный технический результат достигается посредством состава, содержащего соединение формулы (I), полученное способом по настоящему изобретению.
Одним из вариантов воплощения изобретения является фармацевтический состав, содержащий соединение формулы (I), полученное способом по настоящему изобретению.
Одним из вариантов воплощения изобретения является фармацевтический состав, содержащий соединение формулы (I), полученное способом по настоящему изобретению, отличающийся тем, что общее содержание примесей в его составе не превышает 1,5 мас. % от массы фармацевтического состава.
Одним из вариантов воплощения изобретения является фармацевтический состав, содержащий соединение формулы (I), полученное способом по настоящему изобретению, отличающийся тем, что общее содержание примесей в его составе менее 1,5 мас. %, но более 0,001 мас.% от массы фармацевтического состава.
Одним из вариантов воплощения настоящего изобретения является фармацевтический состав для лечения и/или профилактики вирусных заболеваний, где вирус представляет собой, не ограничиваясь указанным, пикорнавирус, норовирус, вирус ящура, вирус гепатита А, поливирус, риновирус, энтеровирус, калицивирус или коронавирус.
Одним из вариантов воплощения настоящего изобретения является фармацевтический состав для лечения и/или профилактики вирусных заболеваний, где вирус представляет собой SARS-CoV, MERS-CoV или SARS-CoV-2.
Одним из вариантов воплощения настоящего изобретения является фармацевтический состав, который проявляет профилактическую и/или терапевтическую противовирусную активность в отношении вируса SARS-CoV-2.
Поставленная задача решается, а заявленный технический результат достигается посредством применения фармацевтического состава, содержащего соединение формулы (I), полученное способом по настоящему изобретению, для лечения и/или профилактики вирусных заболеваний.
Одним из вариантов воплощения настоящего изобретения является применения состава, содержащего соединение формулы (I), полученное способом по настоящему изобретению, для лечения и/или профилактики вирусных заболеваний, выбранных из группы, не ограничиваясь указанным, пикорнавирус, норовирус, вирус ящура, вирус гепатита А, поливирус, риновирус, энтеровирус, калицивирус или коронавирус.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), в котором стереохимия реагентов, промежуточных продуктов и целевого продукта находится под контролем, что позволяет синтезировать конкретные стереоизомеры соединения формулы (I) и соединения формулы (II).
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), который позволяет получать целевой продукт с выходом, превышающим выход в описанных в уровне техники способах.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (II), который позволяет получать промежуточный продукт в синтезе соединения формулы (I) с выходом, превышающим выход в описанных в уровне техники способах.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (II), который позволяет получать продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (II) более 80%.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (II), который позволяет получать продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (II) более 85%.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (II), который позволяет получать продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (II) более 90%.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (II), который позволяет получать продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (II) более 95%.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (II), который позволяет получать продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (II) более 98%.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), который позволяет получать целевой продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (I) более 80%.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), который позволяет получать целевой продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (I) более 85%.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), который позволяет получать целевой продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (I) более 90%.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), который позволяет получать целевой продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (I) более 95%.
Одним из вариантов воплощения изобретения является способ получения соединения формулы (I), который позволяет получать целевой продукт с выходом, превышающим выход в описанных в уровне техники способах, и со стереоизомерным избытком соединения формулы (I) более 98%.
Стереоизомерная чистота соединений по настоящему изобретению может контролироваться перекристаллизацией промежуточных и целевого продуктов в синтезе. Для перекристаллизации могут быть использованы следующие растворители, их дейтеропроизводные или их смеси: спирты, такие как метанол, этанол, н-пропанол, изо-пропанол, н-бутанол, 2-бутанол, изо-бутанол, трет-бутанол, амиловый спирт и его изомеры, циклогексанол, бензиловый спирт, этиленгликоль, 1,2-пропандиол, 1,3-пропандиол, кетоны, такие как ацетон, бутанон, метилизобутилкетон, 2-гексанон, циклопентанон, циклогексонон, эфиры, такие как этилформиат, метилацетат, этилацетат, н-пропилацетат, изо-пропилацетат, н-бутилацетат, изо-бутилацетат, этиленгликоль диацетат, пропиленгликоль диацетат, глицерол триацетат, диэтиловый эфир, диизопропиловый эфир, дибутиловый эфир, тетрагидрофуран, метилтетрагидрофуран, диоксан, анизол, метилтретбутиловый эфир, этилтретбутиловый эфир, циклопентилметиловый эфир, диметоксиэтан, диэтоксиметан, диглим, лактоны, такие как бутиролактон, валеролактон, углеводороды, такие как н-пентан, н-гексан, н-гептан, н-октан, циклогексан, метилциклогексан, бензол, толуол, ксилолы, кумол, тетралин, лимонен, терпентин, галогенуглеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод, 1,2-дихлорэтан, 1,1-дихлорэтилен, цис-дихлорэтилен, транс-дихлорэтилен, 1,1,2-трихлорэтилен, 1,1,1-трихлорэтан, 1-хлорбутан, хлорбензол, дихлорбензол, апротонные полярные растворители, такие как ацетонитрил, пропионитрил, диметилформамид, диметилацетамид, N-метилпирролидон, диметилолэтиленмочевина, диметилолпропиленмочевина, диметилсульфоксид, сульфолан, нитрометан, бифункциональные растворители, такие как метоксиэтанол, этоксиэтанол, 1-метоксипропан-2-ол, фурфуриловый спирт, тетрагидрофурфуриловый спирт, этиллактат, протонные растворители, такие как вода, кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, трифторуксксная кислота, метансульфоновая кислота, трифторметансульфоновая кислота, ангидриды, такие как уксусный ангидрид, амиак, амины, такие как пиридин, тиэтиламин, а также сверхкритические жидкости, такие как диоксид углерода, вода, метан, этан, пропан, этилен, пропилен, метанол, этанол, ацетон, оксиды азота, оксид серы, и/или их смеси.
Контроль стереоизомерной чистоты соединений по настоящему изобретению может проводиться, в том числе, не ограничиваясь указанным, с помощью аналитической ВЭЖХ.
Подробное описание изобретения
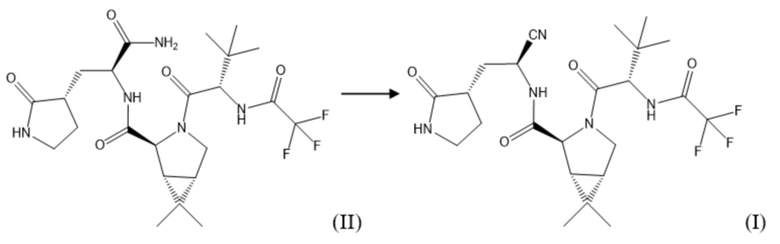
Предложенный способ получения (1R,2S,5S)-N-[(1S)-1-циано-2-[(3S)-2-оксопирролидин-3-ил]этил]-3-[(2S)-3,3-диметил-2-[(2,2,2-трифторацетил)амино]бутаноил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоксамида (соединения формулы (I)) предусматривает получение соединения формулы (I) по следующей схеме синтеза:
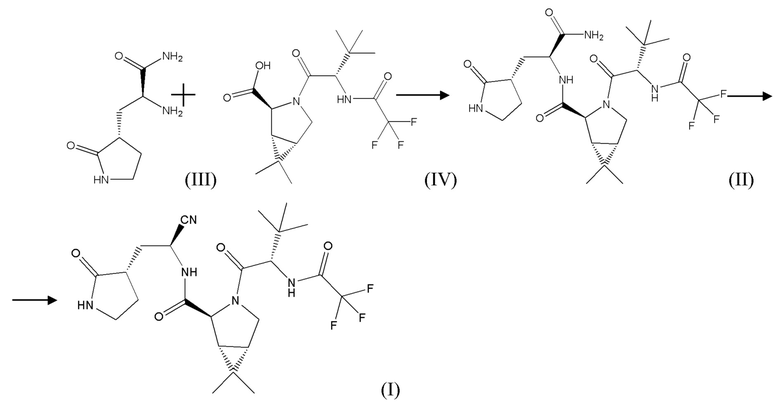
Соединение формулы (II) в рамках настоящего изобретения получают путем взаимодействия коммерчески доступных соединения формулы (III) или его соли, например, не ограничиваясь указанным, гидрохлорида, и соединения формулы (IV) (данные соединения можно также синтезировать известными в уровне техники способами) в подходящем растворителе, в том числе апротонном, выбранном из группы, не ограничиваясь указанным: диметилформамид, диметилацетамид, N-метилпирролидон, ТГФ, дихлорметан, этилацетат и т.д., а так же их смесей, в присутствии соединения формулы R1O-CO-Cl, где R1 представляет собой C1-C5 алкил, в частности, не ограничиваясь указанным, метил, этил, пропил, изопропил, бутил, изобутил и т.д, C1-C5 алкил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор; фенил; фенил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор, нитро группой, C1-C5 алкилом; бензил, замещенный галогеном, выбранным из группы, не ограничиваясь указанным, хлор, бром, фтор, нитро группой, C1-C5 алкилом, и основания, в том числе, не ограничиваясь указанным, пиридин, пиридин, замещенный C1-C5 алкилом, хинолин или хинолин, замещенный C1-C5 алкилом, третичный амин, в том числе, третичный амин формулы R2R3R4N, где R2, R3, R4 независимо друг от друга представляют собой C1-C5 алкил, в частности, метил, этил, пропил, изопропил и т.д, или, когда R2 с R3 вместе с атомом азота образуют С4-С8 циклоалкильную группу или С4-С8 гетероциклическую группу, где гетероатом представляет собой кислород, R4 представляет собой C1-C5 алкил, при этом R4 может быть связан с атомом, при температуре от -50оС до 50оС.
В рамках настоящего изобретения возможно как одностадийное, так и двухстадийное проведение указанного процесса. В случае одностадийного проведения процесса к раствору или суспензии, содержащей соединение формулы (III) или его соль, например, гидрохлорид, соединение формулы (IV) и указанное основание, например, третичный амин, охарактеризованный выше, в подходящем растворителе, например, апротонном, добавляют соединение формулы R1O-CO-Cl, охарактеризованное выше, при температуре от -50°С до 10°С, и затем выдерживают полученную реакционную массу при температуре от 10°С до 50°С до окончания реакции.
В случае двухстадийного процесса к раствору соединения формулы (IV) и указанного основания, например, третичного амина, охарактеризованного выше, в подходящем растворителе, например, апротонном, добавляют соединение формулы R1O-CO-Cl, охарактеризованное выше, при температуре от -50°С до 10°С, и затем выдерживают полученную реакционную массу при температуре от -50°С до 10°С до окончания реакции образования смешанного ангидрида. Затем к реакционной массе добавляют соединение формулы (III) или его соль, например, гидрохлорид, и указанное основание, например, третичный амин, охарактеризованный выше, при температуре от -50°С до 10°С и затем выдерживают полученную реакционную массу при температуре от -10°С до 50°С до окончания реакции.
Так же в двухстадийном процессе возможно обратное прибавление, когда к раствору соединения формулы (IV) и соединения формулы R1O-CO-Cl, охарактеризованного выше, в подходящем растворителе, например, апротонном, добавляют указанное основание, например, третичный амин, при температуре от -50°С до 10°С, и затем выдерживают полученную реакционную массу при температуре от -50°С до 10°С до окончания реакции образования смешанного ангидрида. Затем к реакционной массе добавляют соединение формулы (III) или его соль, например, гидрохлорид, и указанное основание, например, третичный амин, охарактеризованный выше, при температуре от -50°С до 10° С и затем выдерживают полученную реакционную массу при температуре от 10°С до 50°С до окончания реакции. После окончания реакции реакционную массу упаривают или разбавляют водой, соединение формулы (II) экстрагируют подходящим органическим растворителем, не смешивающимся с водой.
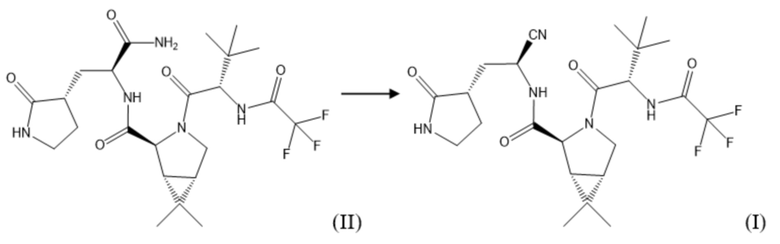
Соединение формулы (I) получают путем взаимодействия соединения формулы (II), полученного на предыдущей стадии, и 2,4,6-трихлор-1,3,5-триазина в подходящем растворителе, например, протонном, выбранном из группы, включающей, не ограничиваясь указанным: диметилформамид, диметилацетамид, N-метилпирролидон, при температуре от 0°С до 100°С в течение 1,5-3,0 часов. После окончания реакции реакционную массу упаривают или разбавляют водой, соединение формулы (I) экстрагируют подходящим органическим растворителем, не смешивающимся с водой.
Полученные экстракты на каждой стадии синтеза при необходимости промывают водой, растворами лимонной кислоты, гидрокарбоната натрия или раствором хлорида натрия, сушат водоотнимающими агентами, такими как, не ограничиваясь указанным, хлорид кальция, сульфат натрия, сульфат магния и тд., упаривают и остаток перекристаллизовывают из подходящего органического растворителя, либо очищают методом жидкостной хроматографии, или используют в следующей стадии без дополнительной очистки. При этом, перекристаллизация продукта может включать в себя суспендирование осажденного продукта в органическом растворителе, нагрев получаемой смеси до получения однородного раствора и последующее охлаждение (в том числе естественным путем при комнатной температуре). Промежуточные продукты синтеза, а также конечный продукт соединение формулы (I) по настоящему изобретению могут быть также выделены методом осаждения посредством фильтрации.
Далее приводятся примеры осуществления изобретения, которые иллюстрируют изобретение, но не охватывают все возможные варианты его осуществления и не ограничивают изобретение.
Специалисту в данной области понятно, что возможны и другие частные варианты осуществления изобретения.
Пример 1. Способ получения (1R,2S,5S)-N-[(1S)-1-циано-2-[(3S)-2-оксопирролидин-3-ил]этил]-3-[(2S)-3,3-диметил-2-[(2,2,2-трифторацетил)амино]бутаноил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоксамида (соединения формулы (I)).
36,40 г соединения формулы (IV), 20,80 г соединения формулы (III) растворяли в 250,00 мл диметилформамида и добавляли 22,20 г (2,20 эквивалента) N-метилморфолина. Реакционную массу перемешивали до растворения и затем по каплям добавляли 15,00 г (1,10 эквивалент) изобутилхлорформиата при температуре 0°С. После прибавления всего изобутилхлорформиата выдерживали полученную реакционную массу при температуре 25°С в течение 12-18 ч. После окончания реакции реакционную массу упаривали до постоянного веса под вакуумом на роторном испарителе при температуре бани 45°С и остаточном давлении 30 мбар, полученное соединение формулы (II) экстрагировали этилацетататом, экстракт промывали водой и раствором гидрокарбоната натрия, а затем упаривали и сушили. На выходе получали 94 % промежуточного продукта соединения формулы (II) со стереоизомерной чистотой более 97 %.
Далее 51,70 г полученного на предыдущей стадии соединения формулы (II) смешивали с 500,00 мл N-метилпирролидона и добавляли при перемешивании 9,22 г (1,50 эквивалента) 2,4,6-трихлор-1,3,5-триазина при температуре 25°С. После окончания реакции реакционную массу упаривали до постоянного веса под вакуумом на роторном испарителе при температуре бани 45°С и остаточном давлении 30 мбар, полученную смесь экстрагировали этилацетатом. Далее экстракт промывали раствором гидрокарбоната натрия, упаривали и перекристаллизовывали из гептана с последующей вакуумной сушкой при температуре 40°С и остаточном давлении 10 мбар. После сушки получали целевой продукт соединение формулы (I) в виде порошка с выходом 96%.
Идентификация соединения формулы (I) проводилась аналитическими методами: 1H ЯМР, 13C ЯМР и масс-спектральным анализом.
1H ЯМР (600 MГц, DMSO-d6) δ 9.45 (d, J = 8.4 Гц, 1H), 9.00 (d, J = 8.6 Гц, 1H), 7.69 (s, 1H), 4.97 (ddd, J = 10.9, 8.6, 5.1 Гц, 1H), 4.45 (d, J = 8.4 Гц, 1H), 4.12 (s, 1H), 3.90 (dd, J = 10.4, 5.5 Гц, 1H), 3.65 (d, J = 10.4 Гц, 1H), 3.13 (t, J = 9.1 Гц, 1H), 3.01 (td, J = 9.4, 7.1 Гц, 1H), 2.43 (tdd, J = 10.4, 8.4, 4.4 Гц, 1H), 2.25-2.00 (m, 2H), 1.75 - 1.65 (m, 2H), 1.53 (dd, J = 7.6, 5.4 Гц, 1H), 1.34 (d, J = 7.6 Гц, 1H), 1.05 (s, 3H), 0.97 (s, 9H), 0.83 (s, 3H).
13C ЯМР (151 MГц, DMSO-d6) δ 178.11, 171.20, 167.94, 158.15, 157.65, 157.19, 156.65, 120.05, 118.23, 116,85, 114.37, 60.54, 58.65, 48.04, 38.25, 37.19, 35.08, 34.65, 30.75, 27.80, 27.32, 26.70, 26.19, 19.30, 12.80.
Масс-спектр: m/z рассчитано для C23H33F3N5O4 [M+H]+ 500.2474, найдено 500.2471.
Следует также отметить, что в качестве реагентов для дегидратации амидной группы соединения формулы (II) и его структурных аналогов из уровня техники также известны Реактив Бёрджесса, трифторуксусный ангидрид, трихлороксид фосфора и 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6- триоксид. Авторами настоящего изобретения был проделан ряд экспериментов по изучению данной реакции при помощи различных реагентов. Было обнаружено, что выход целевого продукта соединения формулы (I) посредством дегидратации амидной группы соединения формулы (II) известными в уровне техники способами не превышал 50% в отличие от выхода соединения формулы (I), полученного способом по настоящему изобретению. Некоторые результаты экспериментов представлены в таблице 1.
Таблица 1. Изучение дегидратации амидной группы соединения формулы (II) при различных условиях реакции.
Пример 2. Получение продукта, содержащего (1R,2S,5S)-N-[(1S)-1-циано-2-[(3S)-2-оксопирролидин-3-ил]этил]-3-[(2S)-3,3-диметил-2-[(2,2,2- трифторацетил)амино]бутаноил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоксамид (соединение формулы (I)).
Отмеренное количество полученного в примере 1 порошка соединения формулы (I) при перемешивании помещали в реактор, снабженный мешалкой, рубашкой, обратным холодильником, термометром и манометром, в атмосфере азота, где предварительно было загружено отмеренное количество изопропилового спирта. По окончанию загрузки полученную суспензию перемешивали при температуре 40°С до полного растворения соединения формулы (I). Далее полученный раствор фильтровали на нутч-фильтре. Фильтрат подавали в кристаллизатор, снабженный рубашкой, термометром, мешалкой, обратным холодильником. При непрерывном перемешивании в кристаллизатор в атмосфере азота подавали отмеренное количество гептана. Перемешивание продолжали при комнатной температуре. Суспензию охлаждали до температуры 20°С и перемешивали при установленной температуре в течение 7,0-10,0 часов для полного осаждения соединения формулы (I), после чего полученную суспензию передавали на стадию центрифугирования. Полученный осадок соединения формулы (I) промывали водой с последующей сушкой в конусной вакуумной сушилке при температуре 40°С и остаточном давлении 7 мбар в течение 5 часов. Далее полученный порошок загружали в бункер мельницы для измельчения. В результате получали продукт в виде порошка, содержащий соединение (I), с химической чистотой более 99 %, а его стереоизомерной чистотой 100 %.
Группа изобретений относится к области медицины, фармакологии и химико-фармацевтической промышленности и включает способ получения (1R,2S,5S)-N-[(1S)-1-циано-2-[(3S)-2-оксопирролидин-3-ил]этил]-3-[(2S)-3,3-диметил-2-[(2,2,2-трифторацетил)амино]бутаноил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоксамида (соединение формулы (I)) и его применение для периодического химического промышленного производства, полунепрерывного промышленного производства или непрерывного промышленного производства, а также способ получения промежуточного соединения формулы (IV), предназначенного для применения в способе получения соединения формулы (I). Способ получения соединения формулы (I) заключается в дегидратации соединения формулы (II) с использованием 2,4,6-трихлор-1,3,5-триазина в среде растворителя. При этом способ получения промежуточного соединения формулы (II) включает взаимодействие соединения формулы (III) или его соли с соединением формулы (IV) с использованием соединения формулы R1O-CO-Cl, где R1 представляет собой C1-C5 алкил; C1-C5 алкил, замещенный галогеном; фенил; фенил, замещенный галогеном, нитро группой или C1-C5 алкилом; бензил, замещенный галогеном, нитро группой или C1-C5 алкилом, в присутствии основания. Технический результат: усовершенствованный способ получения, приводящий к повышению выхода и химической чистоты соединения формулы (I). 3 н. и 33 з.п. ф-лы, 1 табл., 2 пр.
1. Способ получения соединения формулы (II), который включает взаимодействие соединения формулы (III) или его соли с соединением формулы (IV) с использованием соединения формулы R1O-CO-Cl, где R1 представляет собой C1-C5 алкил; C1-C5 алкил, замещенный галогеном; фенил; фенил, замещенный галогеном, нитро группой или C1-C5 алкилом; бензил, замещенный галогеном, нитро группой или C1-C5 алкилом, в присутствии основания:
2. Способ по п. 1, в котором реакция проходит в органическом растворителе.
3. Способ по п. 2, в котором растворитель представляет собой апротонный органический растворитель.
4. Способ по п. 2, в котором органический растворитель выбран из группы, включающей диметилформамид, диметилацетамид, N-метилпирролидон, тетрагидрофуран, дихлорметан, этилацетат или их смеси.
5. Способ по п. 1, в котором соединение формулы R1O-CO-Cl выбрано из группы, включающей метилхлорформиат, этилхлорформиат, пропилхлорформиат, бутилхлорформиат, изобутилхлорформиат или третбутилхлорформиат.
6. Способ по п. 1, в котором в качестве основания используют органическое основание.
7. Способ по п. 6, в котором в качестве основания используют пиридин; пиридин, замещенный C1-C5 алкилом; хинолин или хинолин, замещенный C1-C5 алкилом.
8. Способ по п. 6, в котором органическое основание представляет собой третичный амин.
9. Способ по п. 8, в котором третичный амин представляет собой соединение формулы R2R3R4N, где R2, R3, R4 независимо друг от друга представляют собой C1-C5 алкил или когда R2 с R3 вместе с атомом азота образуют С4-С8 циклоалкильную группу или С4-С8 гетероциклическую группу, где гетероатом представляет собой кислород, R4 представляет собой C1-C5 алкил.
10. Способ по п. 9, в котором третичный амин представляет собой соединение формулы R2R3R4N, где R2, R3, R4 независимо друг от друга выбраны из группы, включающей метил, этил, пропил или изопропил.
11. Способ по п. 9, в котором третичный амин представляет собой N-метилформалин или триэтиламин.
12. Способ по п. 1, в котором реакцию проводят при температуре в интервале от -50 до 50°С.
13. Способ по п. 12, в котором реакцию проводят при температуре в интервале от -40 до 40°С.
14. Способ по п. 13, в котором реакцию проводят при температуре в интервале от -30 до 30°С.
15. Способ по п. 14, в котором реакцию проводят при температуре в интервале от -20 до 20°С.
16. Способ по п. 15, в котором реакцию проводят при температуре в интервале от -10 до 10°С.
17. Способ по п. 16, в котором реакцию проводят при температуре в интервале от -5 до 5°С.
18. Способ по п. 1, в котором к раствору соединения формулы (III) или его соли в присутствии основания добавляют соединение формулы R1O-CO-Cl, где R1 представляет собой C1-C5 алкил, C1-C5 алкил, замещенный галогеном; фенил; фенил, замещенный галогеном, нитро группой или C1-C5 алкилом; бензил, замещенный галогеном, нитро группой или C1-C5 алкилом, при температуре от -50 до 10°С и выдерживают, далее к реакционной массе добавляют соединение формулы (IV) и основание при температуре от -50 до 10°С, а затем выдерживают полученную реакционную массу при температуре от -10 до 50°С до окончания реакции.
19. Способ по п. 18, в котором к раствору соединения формулы (III) или его соли в присутствии основания добавляют соединения формулы R1O-CO-Cl, где R1 представляет собой C1-C5 алкил; C1-C5 алкил, замещенный галогеном; фенил; фенил, замещенный галогеном, нитро группой или C1-C5 алкилом; бензил, замещенный галогеном, нитро группой, или C1-C5 алкилом, при температуре от -30 до -15°С и выдерживают, далее к реакционной массе добавляют соединение формулы (IV) и основание при температуре от -25 до -15°С, а затем выдерживают полученную реакционную массу при температуре от -5 до 5°С до окончания реакции..
20. Способ по пп. 1-19, который дополнительно включает стадии:
a) перекристаллизации продукта из органического растворителя и/или
b) очистки продукта методом хроматографии.
21. Способ по п. 20, где стадия перекристаллизации полученного продукта включает в себя суспендирование осажденного продукта в органическом растворителе, нагрев получаемой смеси до получения однородного раствора и последующее охлаждение.
22. Способ по п. 21, в котором охлаждение выполняют естественным путем при комнатной температуре.
23. Способ получения соединения формулы (I), который включает дегидратацию соединения формулы (II), полученного способом по любому из пп. 1-22, с использованием 2,4,6-трихлор-1,3,5-триазина в среде растворителя:
24. Способ по п. 23, в котором в качестве растворителя используют N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон.
25. Способ по п. 23, в котором дегидратацию соединения формулы (II) проводят при температуре в интервале от 0 до 100°С.
26. Способ по п. 25, в котором дегидратацию соединения формулы (II) проводят при температуре в интервале от 15 до 25°С.
27. Способ по пп. 23-26, в котором стадия дегидратации соединения формулы (II) дополнительно включает следующие стадии:
a) выделение продукта и/или
b) промывка выделенного продукта и/или;
c) сушка продукта.
28. Способ по п. 27, в котором стадию a) проводят методом осаждения или экстракции.
29. Способ по п. 27, в котором стадию a) проводят методом фильтрования.
30. Способ по п. 27, в котором стадия b) дополнительно включает стадию перекристаллизации продукта из органического растворителя.
31. Способ по п. 27, в котором стадию b) проводят при помощи воды, раствора лимонной кислоты или водного раствора гидрокарбоната натрия.
32. Способ по п. 30, где стадия перекристаллизации полученного продукта включает в себя суспендирование осажденного продукта в органическом растворителе, нагрев получаемой смеси до получения однородного раствора и последующее охлаждение.
33. Способ по п. 32, в котором охлаждение выполняют естественным путем при комнатной температуре.
34. Способ по п. 27, в котором стадия b) дополнительно включает стадию очистки продукта методом хроматографии.
35. Способ по п. 27, в котором стадия c) представляет собой стадию сушки при пониженном давлении.
36. Применение способа по пп. 23-35 для периодического химического промышленного производства, полунепрерывного промышленного производства или непрерывного промышленного производства соединения формулы (I).
| WO 2021250648 A1, 16.12.2021 | |||
| Мелентьева Г.А., Антонова Л.А., Фармацевтическая химия | |||
| Москва: "МЕДИЦИНА", 1985, 480 с. | |||
| CN 114213275 A, 22.03.2022 | |||
| Owen D | |||
| R | |||
| et al., An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19 | |||
| Science, 24.12.2021, vol.374, no.6575, pp.1586-1593 | |||
| RU 2013153533 A, 10.06.2015. |

