Настоящее изобретение относится к олигонуклеотиду, который представляет собой эффективный ингибитор микроРНК miR-132, и к его применению в медицине, в особенности, для профилактики или лечения нарушений сердечной деятельности и/или фиброзных нарушений.
Сердечная недостаточность представляет собой одну из основных патологий в мире, ведущих к смертности. Инфаркт миокарда (ИМ) – это наиболее важная причина сердечной недостаточности, поскольку ИМ вызывает последующее прогрессирующее ремоделирование сердца, что приводит к сердечной недостаточности с плохим прогнозом. Применяемые в настоящее время терапевтические фармакологические варианты лечения сердечной недостаточности включают ангиотензин-модулирующие средства, β-блокаторы, диуретики, антагонисты альдостерона, комбинированный с блокатором рецепторов ангиотензина-II ингибитор неприлизина, вазодилататоры или инотропные средства. Хотя для всех этих средств ряд клинических исследований показали значительное снижение смертности, вызванной сердечной недостаточностью, 5-летняя смертность остается на недопустимом уровне, равном почти 50%. Таким образом, существует большая потребность в разработке новых и более эффективных терапевтических подходов при сердечной недостаточности.
Патологический гипертрофический рост кардиомиоцитов может привести к развитию ремоделирования сердца, сердечной недостаточности и внезапной остановке сердца. Гипертрофический рост кардиомиоцитов представляет собой ответ на повышенную нагрузку на стенку сердца, вызванную перегрузкой сердечного объема и/или давления. Первоначально, гипертрофия сердца представляет собой компенсаторный механизм, направленный на снижение напряжения на стенке и увеличение объёмной скорости кровотока в сердце. Однако длительная гипертрофия сердца прогрессирует до сократительной дисфункции, сердечной декомпенсации и, наконец, сердечной недостаточности (Hill and Olson, 2008; Barry and Townsend, 2010). Переход от физиологической к патологической гипертрофии может происходить в зависимости от многих факторов, включая потерю миоцитов в результате апоптоза или некроза, изменения в аутофагии, дефекты сократительной реакции, нарушение регуляции гомеостаза кальция, десенсибилизацию адренергических рецепторов или фиброз сердца (Hill and Olson, 2008; Barry and Townsend, 2010). Гипертрофическую передачу сигналов в значительной степени опосредует сигнальный путь инсулина (DeBosch and Muslin, 2008; Barry and Townsend, 2010). Как инсулин, так и инсулиноподобный фактор роста-1 (IGF-1) активируют про-гипертрофические пути в кардиомиоцитах через рецептор IGF-1, который активирует фосфоинозитин-3-киназу (PI3K) (McMullen et al., 2004). Активность PI3K приводит к активации серин/треонинкиназы Akt посредством ее фосфорилирования, и активные Akt фосфорилируют антигипертрофические факторы транскрипции FoxO, что приводит к их дестабилизации и предотвращению ядерной локализации (Datta et al., 1999; Skurk et al., 2005; Ronnebaum and Patterson, 2010). Напротив, ацетилирование факторов FoxO сиртуином-1 (Sirt-1) приводит к их стабилизации и ядерной транслокации (Frescas et al., 2005). Стабилизированные факторы транскрипции FoxO локализуются в ядре для регуляции экспрессии антигипертрофических генов. Антигипертрофические функции белков FoxO в значительной степени опосредованы подавлением про-гипертрофического сигнального пути кальциневрина посредством экспрессии антигипертрофических генов-мишеней факторов FoxO, таких как атрогин-1 (Ni et al., 2006; Ronnebaum and Patterson, 2010; Glas, 2010). Кроме того, транскрипционные факторы FoxO также вызывают апоптоз и регулируют аутофагию в кардиомиоцитах (Ronnebaum and Patterson, 2010).
Было показано, что микроРНК играют ключевую роль в неблагоприятном ремоделировании сердца. В WO 2013/034653 описано, что miR-132 и/или miR-212 могут вызывать гипертрофию сердца и, таким образом, представляют собой потенциальные терапевтические мишени для лечения сердечной недостаточности.
В WO 2016/042561 описан способ лечения связанного с липидами нарушения путем введения субъекту терапевтически эффективного количества полинуклеотидного агента, который в значительной степени комплементарен нуклеотидной последовательности человеческой miR-132.
Авторы настоящего изобретения идентифицировали новый аналог олигонуклеотида, который представляет собой эффективный ингибитор экспрессии miR-132 в кардиомиоцитах. Аналог олигонуклеотида, в дальнейшем обозначаемый как CDR132L, представляет собой миксмер, состоящий из структурных блоков ДНК и LNA, имеющих межнуклеозидные фосфоротиоатные связи. Он не обладает значительной токсичностью в клеточной линии печени человека и изолированных кардиомиоцитах новорожденных крыс. Кроме того, было показано, что CDR132L демонстрирует превосходные эффекты по сравнению с другими аналогами олигонуклеотидов, имеющими ту же нуклеотидную последовательность, но другое распределение структурных блоков LNA.
Эффективность CDR132L была протестирована на нескольких моделях на животных. В модели гипертрофии сердца у трансгенных мышей CDR132L приводил к обратному ремоделированию сердца, связанному со сниженной экспрессией miR-132. В мышиной модели сердечной недостаточности после ИМ было обнаружено, что обработка CDR132L снижает дисфункцию левого желудочка после ИМ и загружает независимые параметры систолической сократительной функции. Кроме того, введение CDR132L приводило к улучшению сердечной функции и уменьшению экспрессии miR-132, передачи сигналов сердечного стресса и гипертрофии после ИМ. Было обнаружено, что на модели свиней с сердечной недостаточностью после ИМ лечение с помощью CDR132L предотвращает дезадаптивное ремоделирование и улучшает функцию левого желудочка. Кроме того, CDR132L нормализует тканевую экспрессию маркеров патологической сердечной недостаточности, таких как BNP, ANP, и обеспечивает сдвиг в изоформах тяжелой цепи миозина (соотношение MYH7/6).
В дополнительном исследовании авторы настоящего изобретения идентифицировали антифибротические терапевтические эффекты для олигонуклеотидного аналога CDR132L на мышиной модели in vivo инфаркта миокарда и на моделях in vitro фиброза печени и фиброза легких.
Таким образом, олигонуклеотид CDR132L полезен в качестве активного агента в медицине, в особенности, в профилактике или лечении нарушений сердечной деятельности и/или фиброзных нарушений.
Соответственно, первый аспект настоящего изобретения обеспечивает олигонуклеотидный аналог, включающий последовательность формулы I:
5’-ATGGCTGTAGACTGTT-3’
в которой A, T, G и C представляют собой дезоксирибонуклеотидные структурные блоки и
в которой, по меньшей мере, один структурный блок G или T представляет собой мостиковый нуклеотидный структурный блок и/или морфолино-нуклеотидный структурный блок.
В конкретном воплощении, олигонуклеотид включает или имеет последовательность формулы II:
5’-A+TG+GC+TG+TA+GACTG+T+T-3’
в которой A, T, G и C представляют собой дезоксирибонуклеотидные структурные блоки и
в которой +G и +T представляют собой мостиковые нуклеотидные структурные блоки и/или морфолино-нуклеотидные структурные блоки, в особенности, в которой +G и +T представляют собой структурные блоки LNA.
Олигонуклеотидный аналог формулы I или формулы II может включать, по меньшей мере, одну модифицированную межнуклеозидную связь, например, межнуклеозидную связь, которая стабилизирована против расщепления нуклеазой, например, фосфоротиоатную или фосфородиамидатную связь. В конкретных воплощениях все межнуклеозидные связи представляют собой модифицированные связи, в особенности, фосфоротиоатные связи.
В более конкретном воплощении, изобретение относится к олигонуклеотиду CDR132L, как описан в настоящем документе.
Олигонуклеотид CDR132L имеет последовательность формулы III:
5’-dA*+T*dG*+G*dC*+T*dG*+T*dA*+G*dA*dC*dT*dG*+T*+T-3’
в которой dA представляет собой 2’-дезоксиаденозин, dG представляет собой 2’-дезоксигуанозин, dC представляет собой 2’-дезоксицитидин и Т представляет собой тимидин,
в которой +T представляет собой структурный блок LNA-T и +G представляет собой структурный блок LNA-G и в которой * представляет собой фосфоротиоатную связь.
Дополнительный аспект настоящего изобретения относится к фармацевтической композиции, включающей в качестве активного агента олигонуклеотидный аналог, включающий последовательность формул I, II или III и фармацевтически приемлемый носитель.
Еще один аспект настоящего изобретения относится к медицинскому применению аналога олигонуклеотида, включающего последовательность формулы I, II или III. В определенных воплощениях медицинское применение относится к применению для лечения или профилактики нарушения, связанного с, сопровождаемого и/или вызванного патологической экспрессией miR-132. В определенных воплощениях медицинское применение относится к лечению или профилактике сердечных нарушений, в особенности, нарушений, связанных с гипертрофией сердца. В определенных воплощениях медицинское применение относится к лечению фиброзных нарушений, например, нарушений, связанных с, сопровождаемых и/или вызванных патологическим фиброзом, в особенности, сердечных фиброзных нарушений, легочных фиброзных нарушений или печеночных фиброзных нарушений.
Олигонуклеотид формулы I, II или III может состоять из структурных блоков дезоксирибонуклеотида ДНК и структурных блоков с мостиковыми нуклеотидами и/или морфолино-нуклеотидных структурных блоков (DNA). Термин «мостиковый нуклеотид» относится к модифицированному рибонуклеотиду, в котором фрагмент рибозы включает двух- или трехатомный мостик, соединяющий 2'- и 4'-атом углерода. Например, мостик может включать структуру 2'-O-CH2-4', 2'-O-CH2-CH2-4', 2'-O-CH(CH3)-4' или соответствующую структуру, в которой O заменен на S или NH. В конкретном воплощении, по меньшей мере, один мостиковый нуклеотидный структурный блок представляет собой структурный блок с запертой нуклеиновой кислотой (LNA), имеющий мостик 2'-O-CH2-4'. Морфолино-нуклеотидный структурный блок относится к модифицированному нуклеотиду, в котором рибоза или дезоксирибоза заменены морфолино-фрагментом.
Олигонуклеотид формулы I, II или III имеет длину, по меньшей мере, 16 структурных блоков, например, длину от 16 до 20 структурных блоков. В конкретном воплощении олигонуклеотид формулы I, II или III имеет длину 16 структурных блоков.
В некоторых воплощениях, олигонуклеотид формулы I, II или III включает от 5 до 10, например, от 6 до 8, в особенности, 7 мостиковых нуклеотидных структурных блоков, например, структурные блоки LNA и/или морфолино-нуклеотидные структурные блоки.
В некоторых воплощениях, олигонуклеотид формулы I, II или III представляет собой «голый» олигонуклеотид. В некоторых воплощениях олигонуклеотид может быть конъюгирован, по меньшей мере, с одним гетерологичным фрагментом, например, с фрагментом, который не способствует связыванию олигонуклеотида с miR-132. Гетерологичный фрагмент может быть фрагментом, который улучшает нацеливание и/или клеточное поглощение, например, липидный фрагмент, такой как холестерин или жирная кислота, сахаридный или аминосахаридный фрагмент, такой как N-галактозамин-содержащий фрагмент, пептидный или полипептидный фрагмент или нуклеозидный или нуклеотидный фрагмент, такой как аптамер. Гетерологичный фрагмент может быть конъюгирован с 5'- и/или 3'-концом олигонуклеотидного аналога посредством ковалентной связи или спейсера.
Олигонуклеотид по настоящему изобретению подходит для применения в области медицины, включая медицину человека и ветеринарию. В конкретных воплощениях, соединение полезно для предотвращения или лечения нарушения, связанного с патологической экспрессией, сопровождаемой и/или вызванной, например, сверхэкспрессией miR-132. Было обнаружено, что введение соединения значительно снижает экспрессию miR-132 in vitro и in vivo.
В некоторых воплощениях, соединение может быть введено пациентам, демонстрирующим сверхэкспрессию miR-132 по сравнению со здоровыми субъектами. В некоторых воплощениях соединение может быть введено пациентам, не демонстрирующим сверхэкспрессию miR-132 по сравнению со здоровыми субъектами, но все еще нуждающимся в снижении уровня miR-132.
Термин «профилактика» в контексте настоящего изобретения относится к введению соединения пациенту, о котором известно, что он имеет повышенный риск развития определенного нарушения. Термин «лечение» в контексте настоящего изобретения относится к введению соединения пациенту, у которого уже развились признаки и/или симптомы определенного нарушения. Термин «пациент» относится к субъекту, нуждающемуся во введении соединения по изобретению в области медицины человека или ветеринарии. В конкретных воплощениях пациент представляет собой пациента-человека.
В конкретных воплощениях, соединение по настоящему изобретению полезно для профилактики или лечения сердечных нарушений, в особенности, сердечнососудистых нарушений, связанных с гипертрофией. Например, соединение полезно для профилактики или лечения сократительной дисфункции, декомпенсация сердечной деятельности, сердечной недостаточности или для профилактики или лечения ремоделирования сердца после инфаркта миокарда, миокардита, нарушений деятельности клапанов сердца, таких как аортальный стеноз или недостаточность митрального клапана, генетические сердечные нарушения с гипертрофией сердца, например, гипертрофическая необструктивная и обструктивная кардиомиопатия или болезнь Фабри.
Соединение полезно для введения пациентам, которых выбирают из
(i) пациентов с повышенным риском развития сердечной недостаточности,
(ii) пациентов, страдающие (застойной) сердечной недостаточностью, например, пациентов с повышенным риском прогрессирования сердечной недостаточности,
(iii) пациентов после инфаркта миокарда и/или
(iv) пациентов с врожденными пороками сердца, связанными с гипертрофией сердца, такими как стеноз легочной вены, дефекты предсердной или желудочковой перегородки.
В конкретных воплощениях, соединение по настоящему изобретению полезно для профилактики или лечения фиброзных нарушений, в особенности, нарушений, связанных с, сопровождаемых и/или вызванных патологическим фиброзом.
Патологический фиброз представляет собой образование избыточной волокнистой соединительной ткани в органе или ткани, в особенности, связанное с патологическим состоянием, сопровождаемое и/или вызванное патологическим состоянием. Патологический фиброз может возникать во многих различных органах и тканях организма, как правило, в результате воспаления или повреждения.
В определенном воплощении, фиброз представляет собой фиброз сердца, например, состояние, включающее патологический фиброз в сердце. Типичные типы фиброза сердца включают фиброз предсердий, фиброз эндомиокарда или фиброз, возникший в результате предшествующего инфаркта миокарда.
В дополнительном воплощении, фиброз представляет собой легочный фиброз, например, состояние, включающее патологический фиброз в легких. Типичные типы легочного фиброза включают фиброзные нарушения, вызванные профессиональными или экологическими факторами, например, воздействием токсинов и загрязняющих веществ, таких как кремнеземная пыль, асбестовые волокна, металлическая пыль, угольная пыль, зерновая пыль, птичий и животный помет. Другие типы легочных фиброзных нарушений вызываются лучевой терапией и/или лечением лекарственными средствами, такими как химиотерапевтические средства, кардиотропные средства, антибиотики или противовоспалительные средства. Другие типы легочных фиброзных нарушений вызываются такими нарушениями, как идиопатический легочный фиброз, дерматит, полимиозит, смешанное заболевание соединительной ткани, аутоиммунное заболевание, такое как ревматоидный артрит, склеродермия, синдром Шегрена или системная красная волчанка, саркоидоз, пневмония, вирусная инфекция или гастроэзофагеальная рефлюксная болезнь (ГЭРБ).
В дополнительном воплощении, фиброз может представлять собой фиброз печени, например, условия, включающие патологический фиброз в печени. Обычные типы фиброза печени вызываются вирусной инфекцией, например, вирусами гепатита B и/или C, наследственными нарушениями обмена веществ, аутоиммунным гепатитом, обструкцией желчных путей, перегрузкой железом, неалкогольной жировой болезнью печени, включая неалкогольную жировую болезнь печени (НЖБП) и неалкогольный стеатогепатит (NASH), и алкогольную болезнь печени.
В еще других воплощении, фиброз также может представлять собой сосудистый фиброз, например, артериальную жёсткость, кожный фиброз, например, келоидное образование или нефрогенный системный фиброз, артрофиброз, некоторые формы спаечного капсулита, фиброз мягких тканей, такой как медиастинальный фиброз или ретроперитонеальный фиброз, или фиброз костного мозга, такой как миелофиброз.
В конкретных воплощениях, изобретение охватывает определение количества и/или активности определенных физиологических параметров у субъекта, подлежащего лечению, до, во время и/или после введения соединения по изобретению. Эта сопутствующая диагностическая процедура может помочь в медицинском применении, как описано выше. Например, диагностическая процедура может обеспечить помощь в оценке риска, стратификации пациента, мониторинге курса лечения и/или контроле после лечения.
В конкретных воплощениях, изобретение охватывает определение количества и/или активности miR-132 у субъекта, подлежащего лечению, до, во время и/или после введения соединений по изобретению. В других воплощениях, изобретение включает определение количества и/или активности сердечных маркеров, таких как BNP, ANP или изоформы тяжелых цепей миозина, например, соотношения MYH7/6, и/или уровней FoxO3 и/или SERCA2. В еще других воплощениях, изобретение охватывает определение количества и/или активности фиброзных маркеров, таких как коллаген, например, отложение коллагена и/или экспрессия генов-маркеров фиброза, таких как коллаген 1A1, коллаген 1A2, коллаген 3A1, и/или матриксной металлопептидазы, такой как матриксная металлопептидаза 2.
Определение вышеуказанных параметров может быть выполнено в образцах жидкости организма, таких как кровь, плазма или сыворотка, или в образцах ткани в соответствии с известными способами на уровне нуклеиновой кислоты и/или белка и может обеспечить полезную диагностическую информацию, например, о ходе и/или успешности лечения.
Соединение по изобретению можно вводить в виде фармацевтической композиции, включающей фармакологически приемлемый носитель. Введение может быть осуществлено известными способами, в которых соединение вводят в желаемую целевую клетку или целевой орган субъекта, подлежащего лечению.
Соединение можно вводить как таковое или в виде конъюгата с гетерологичным фрагментом, как описано выше.
Для фармацевтических применений композиция может находиться в форме раствора, например, раствора для инъекций, эмульсии, ингаляции, суспензии или тому подобного.
Композицию можно вводить любым подходящим способом, например, парентерально, в особенности, путем инъекции, такой как подкожная, внутримышечная, внутривенная или внутриартериальная инъекция, или инфузии, путем перорального или ингаляционного приема и/или путем дермального применения. Носитель может представлять собой любой подходящий фармацевтический носитель. Предпочтительно применяют носитель, который способен повысить эффективность проникновения молекул олигонуклеотидов в клетки-мишени. Липосомы, например, катионные липосомы, или предварительно разработанные экзосомы представляют собой подходящие примеры таких носителей.
Соединение вводят в фармацевтически эффективной дозировке в зависимости от пути введения и типа или серьезности заболевания.
Соединение можно вводить в виде монотерапии или в сочетании с другим лекарственным средством, в особенности, с лекарственным средством, подходящим для профилактики или лечения сердечных или фиброзных нарушений, как описано выше.
Ангиотензинмодулирующие средства, β-блокаторы, диуретики, антагонисты альдостерона, вазодилататоры, ионотропные средства, статины или ингибиторы неприлизина или их комбинации, например, комбинация ингибитора неприлизина, например, сакубитрила, с блокатором рецепторов ангиотензина II, например, валсартаном, представляют собой примеры дополнительных лекарственных средств, подходящих для профилактики или лечения сердечных нарушений.
Лекарственные средства, предназначенные для профилактики или лечения фиброза сердца, такие как ингибиторы ACE, например, лизиноприл, блокаторы рецепторов ангиотензина II, например, кандесартан, лозартан или олмесартан, антагонисты альдостерона, например, ингибиторы спиронолактона и/или TGF β, например, пирфенидон или транлиласт, лекарственные средства, предназначенные для профилактики или лечения легочного фиброза, такие как антифиброзные средства, например нинтеданиб или пирфенидон, противовоспалительные средства, например кортикостероиды, азатиоприн, циклофосфамид и микофенолатемофетил, противорефлюксные средства, например, ингибиторы протонного насоса или Н2-блокаторы и/или средства против кашля и лекарственные средства для профилактики и/или лечения фиброза печени, такие как ингибиторы ACE, например, беназеприл, лизиноприл или рамиприл, противовирусные препараты или PPAR α-агонисты, представляют собой примеры дополнительных лекарственных средств, подходящих для предотвращения или лечения фиброзных нарушений.
Кроме того, изобретение относится к применению соединения по изобретению, как описано выше, для изготовления лекарственного средства для профилактики или лечения сердечного нарушения.
Кроме того, изобретение относится к применению соединения по изобретению, как описано выше, для изготовления лекарственного средства для профилактики или лечения фиброзного нарушения.
Кроме того, изобретение относится к способу профилактики или лечения сердечного нарушения, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества, по меньшей мере, одного соединения как описано в настоящем документе.
Кроме того, изобретение относится к способу профилактики или лечения фиброзного нарушения, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества, по меньшей мере, одного соединения, как описано в настоящем документе.
Дополнительно, настоящее изобретение будет описано более подробно с помощью следующих чертежей и примеров.
Пример 1. Подавление экспрессии miR-132 в кардиомиоцитах
Был проведен количественный анализ in vitro активности, ингибирующей miRNA, для многих соединений, представляющих собой структурные аналоги, полученные из библиотеки анти-miR-132. Сайленсинг экспрессии miRNA определяли количественно с помощью анализов TaqMan® и количественной ПЦР в реальном времени.
Исследование проводили на изолированных кардиомиоцитах крысы после гипертрофической стимуляции путем обработки фенилэфрином/изопротеренолом в концентрациях 10 мкМ. Клетки инкубировали в течение 48-ми часов в стандартной среде для культивирования клеток. Тестируемые соединения вводили индивидуально в концентрации 100 нМ. Испытания проводили в трех повторностях.
Соединение CDR132L, миксмер LNA-ДНК, имеющий фосфоротиоатный остов, было идентифицировано как наиболее активное соединение из библиотеки структурных аналогов anti-miR-132.
Структура CDR132L выглядит следующим образом:
5’-dA*+T*dG*+G*dC*+T*dG*+T*dA*+G*dA*dC*dT*dG*+T*+T-3’
в которой dA представляет собой 2’-дезоксиаденозин, dG представляет собой 2’-дезоксигуанозин, dC представляет собой 2’-дезоксицитидин и Т представляет собой тимидин,
в которой +T представляет собой структурный блок LNA-T и +G представляет собой структурный блок LNA-G и в которой * представляет собой фосфоротиоатную связь.
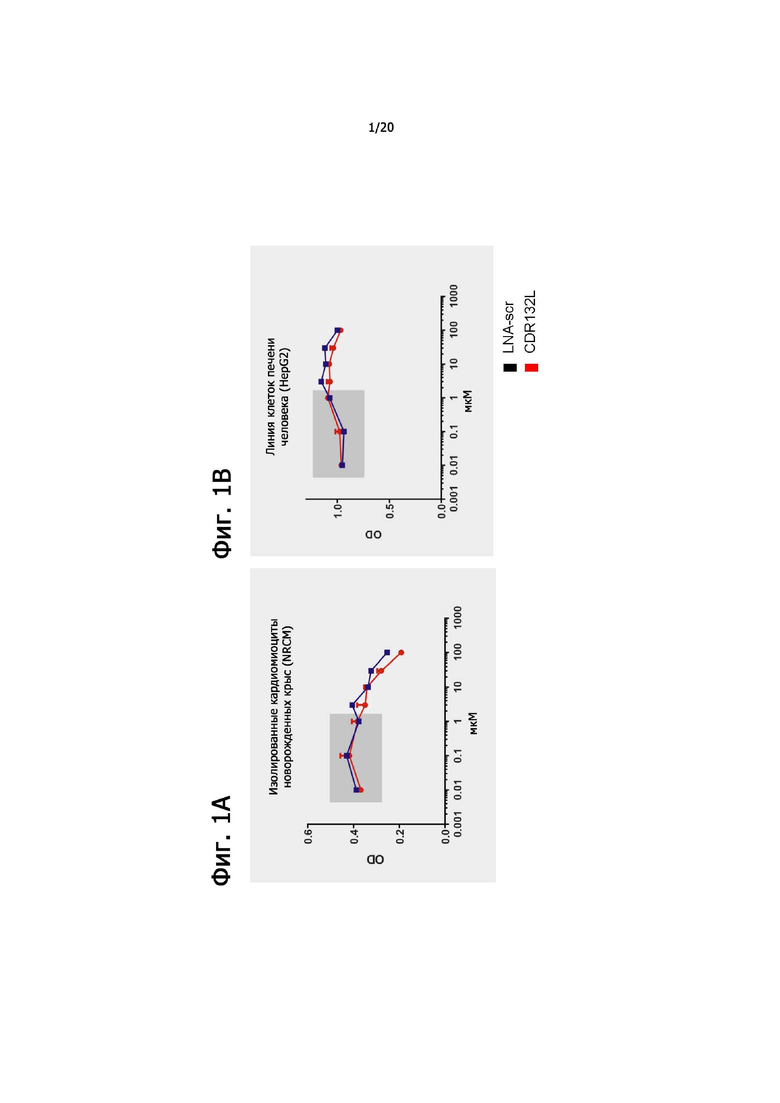
Пример 2. Получение токсикологического профиля
2.1. Цель исследования: Определение профиля токсичности CDR132L
2.2. Описание исследования:
Анализ цитотоксичности in vitro проводили на клетках печени человека (HepG2) и изолированных неонатальных кардиомиоцитах (NRCM). Для оценки цитотоксичности применяли колориметрический коммерческий анализ МТТ. После добавления отдельных соединений клетки инкубировали в среде DMEM в течение 48-ми часов. Эффект CDR132L (красный) сравнивали со рандомизированным LNA-олигонуклеотидом, примененным в качестве контроля (синий) в диапазоне доз 0,01-100 мкМ. Диапазон терапевтических доз отмечен серым цветом.
2.3. Результаты: В диапазоне терапевтических доз не было обнаружено значительной токсичности CDR132L в клетках HepG2 и NRCM (см. фиг. 1А и 1В).
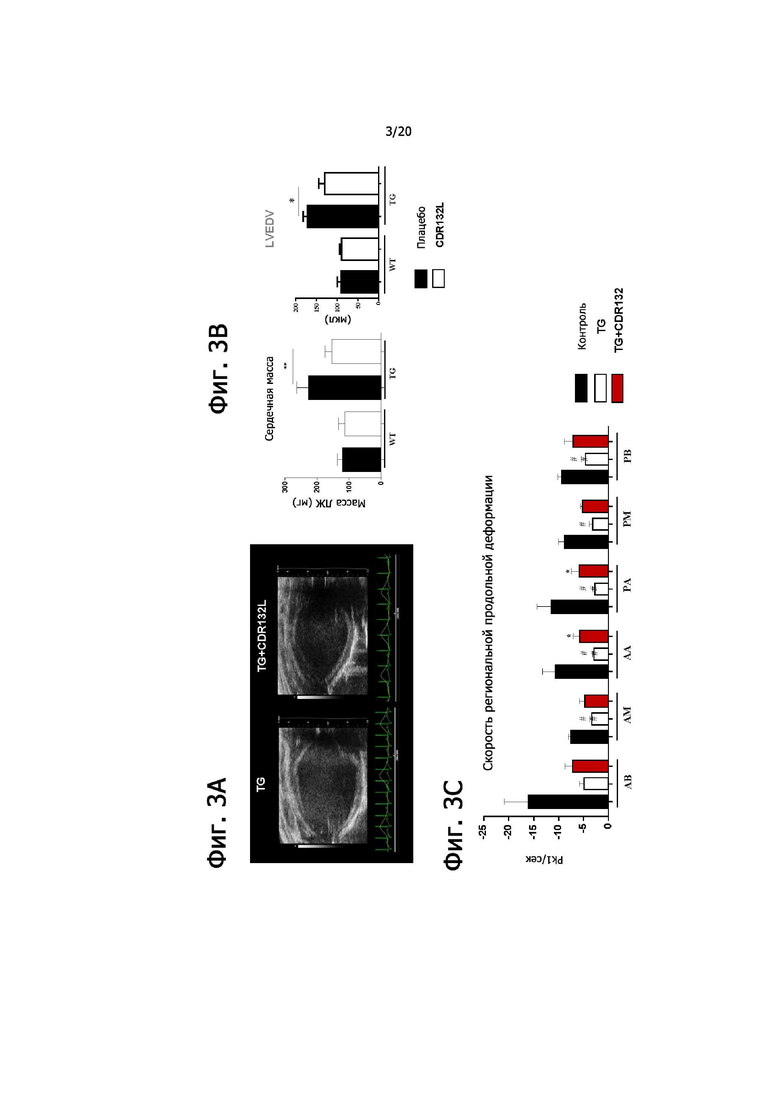
Пример 3. Обратное ремоделирование левого желудочка при сердечной недостаточности путем введения CDR132L на модели трансгенной мыши
3.1. Цель исследования: Анализ эффективности CDR132L в обращении сердечной недостаточности на мышиной модели сердечной недостаточности.
3.2. Описание исследования:
Модель гипертрофии сердца: трансгенные (TG) мыши со сверхэкспрессией miR-132 в сердце (Ucar et al. 2012).
Обработка: еженедельно 20 мг/кг внутрибрюшинно CDR132L или плацебо (см. фиг. 2А)
Группы: однопометник дикого типа (WT) + плацебо, WT + CDR132L, TG + плацебо, TG + CDR132L. n = 6/группа.
Уровни экспрессии miR-132 определяли с помощью qPCR. Статистический тест: непарный t-критерий (фиг. 2В).
**p<0,01, n = 6/группа.
3.3. Результаты:
Репрезентативные эхокардиографические изображения сердца (фиг. 3А).
CDR132L обращает гипертрофию, измеренную по массе сердца и конечному диастолическому объему (LVEDV) (фиг. 3B).
CDR132L улучшает локальную сократительную функцию в большинстве сегментов левого желудочка (AB, передний базальный; AM, передний средний; AA, передний верхушечный; PA задний верхушечный; PM, задний средний и PB, задний базальный) (фиг. 3C).
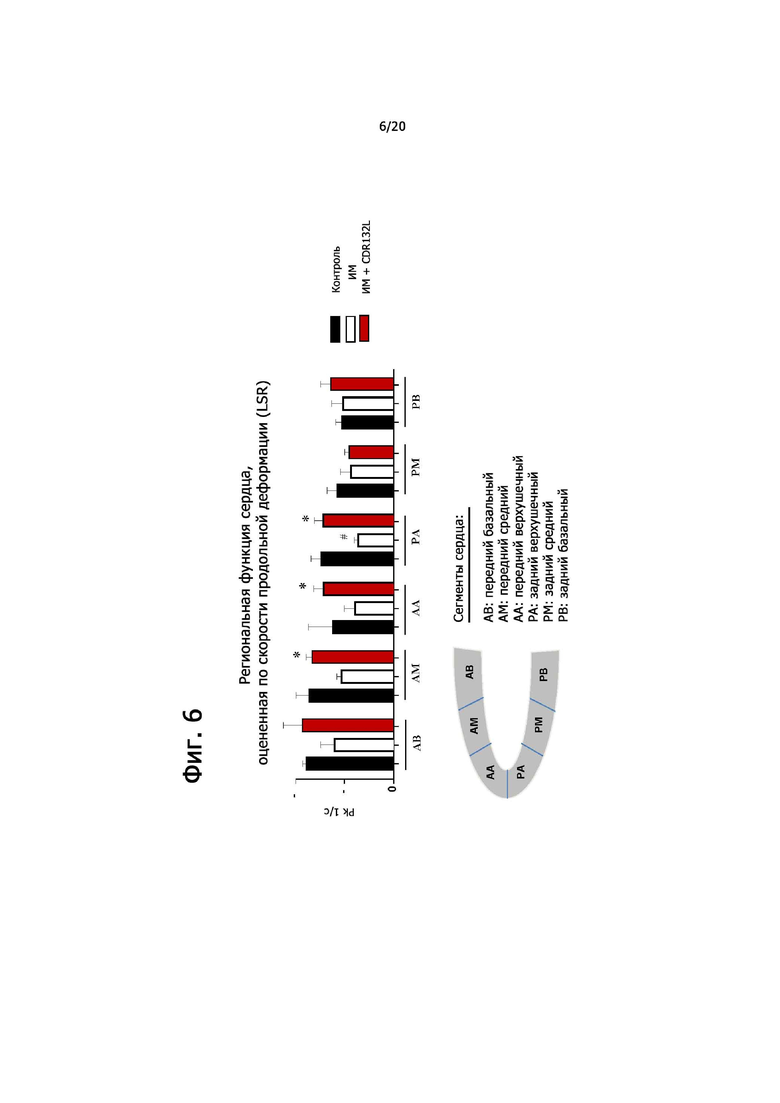
Пример 4. Введение CDR132L в мышиной модели сердечной недостаточности после ИМ
4.1. Цель исследования: Тест эффективности CDR132L на мышиной модели сердечной недостаточности после ИМ
4.2. Описание исследования
Модель инфаркта миокарда (ИМ) на мышах: постоянная перевязка коронарной артерии (LAD) у мышей C57BL/6N (Kolk et al., 2009).
Группы: ИМ или симуляцию, обработанные CDR132L или плацебо
Обработка: 20 мг/кг внутрибрюшинно, на 7 и 14 день после ИМ
Конечная точка: функция ЛЖ на 28 день после ИМ. n = 6-7/группа. (фиг. 4).
4.3. Результаты:
Обработка CDR132L ослабляет дисфункцию левого желудочка после ИМ (фиг. 5A).
Не зависящие от нагрузки параметры систолической сократительной функции также были улучшены (фиг. 5B) (*p<0,05).
Обработка CDR132L также улучшает скорость продольной деформации (LSR) и, таким образом, обращает сократительную дисфункцию после ИМ в отдельных сегментах сердца в отдаленной области сердца. (*p<0,05) (фиг. 6).
Обработка CDR132L эффективно подавляет экспрессию miR-132 в тканях сердца, например, в отдаленной (неинфарктной) области и периинфарктной зоне (фиг. 7A).
На гистологическом уровне, CDR132L уменьшает размер кардиомиоцитов в отдаленной области сердца после ИМ (фиг. 7B).
На уровне ткани, CDR132L снижает экспрессию сигнала ANP сердечного стресса в сердце после ИМ (фиг. 7C).
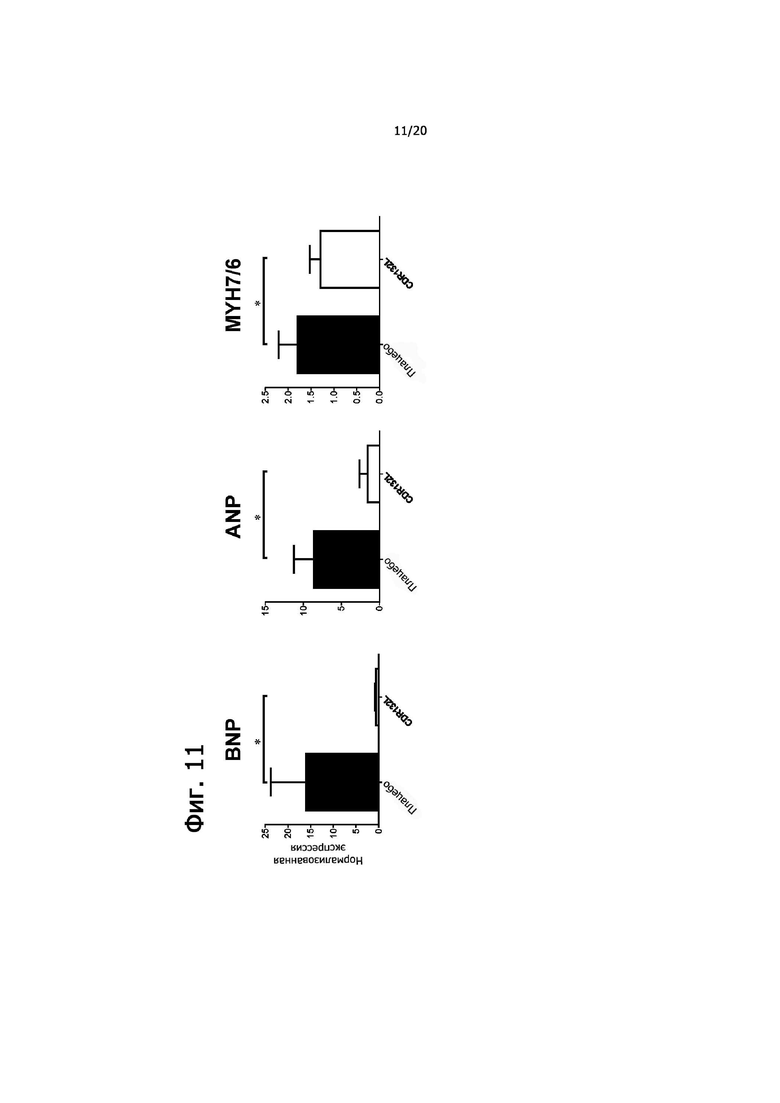
Пример 5. Тестирование CDR132L на модели сердечной недостаточности после ИМ у свиней
5.1. Цель исследования: Доказательство in vivo эффективности CDR132L в клинически значимой модели сердца после ИМ.
5.2. Условия исследования:
• Свинья модель инфаркта миокарда, вызванного 90-минутной ишемией (окклюзия LAD) и последующей реперфузией.
• Группы: плацебо или CDR132L, n = 6 на группу.
• Обработка: дважды, в день 3 и в день 28 после ИМ, 0,3 мг/кг внутрикоронарно и 0,5 мг/кг внутривенно, соответственно (фиг. 8).
• Конечная точка: 8 недель после ИМ. Основной результат: ремоделирование EF и ЛЖ.
5.3. Результаты:
• Обработка CDR132L предотвращает дезадаптивное ремоделирование и улучшает функцию, как было определено измерением конечного диастолического объема, конечного систолического объема, фракции выброса и функции левого желудочка. (фиг. 9A-D).
• Обработка CDR132L улучшает сегментарную сократимость в сегментах, соответствующих выжившему/внеинфарктному миокарду, МРТ сердца в конечной точке: n = 6/группа, красная область: p<0,05 (фиг. 9E).
• Обработка CDR132L предотвращает дезадаптивное ремоделирование и улучшает жизнеспособность ЛЖ в апикальной области, как определено с помощью NOGA, электро-анатомическое картирование в конечной точке: n = 6/группа, плацебо против CDR132L: p<0,05 (фиг. 10).
• CDR132L нормализует тканевую экспрессию маркеров патологической сердечной недостаточности в ANP и BNP и обеспечивает сдвиг в изоформах тяжелых цепей миозина, т.е. соотношение MYH7/6 (фиг. 11).
• На гистологическом уровне обработка CDR132L эффективно снижает гипертрофию кардиомиоцитов в отдаленных областях ЛЖ, репрезентативная микрофотография изображений области ЛЖ (WGA/DAPI, окрашивание 20x) (фиг. 12A) и графическое изображение (фиг. 12B), n = 6/группа, плацебо и CDR132L: p<0,05.
Пример 6. PD-профиль/связывание с мишенью для CDR132L у свиней
6.1. Условия исследования:
Обработка: 1x, день 0, внутрикоронарная перфузия 0,5 мг/кг или 5 мг/кг, n = 3 свиньи/группа, плацебо против CDR132L: p<0,05.
• анализ микроРНК ткани с помощью qPCR в конечной точке (24 часа после обработки).
6.2. Результаты:
• Однократная доза обработки CDR132L дозо-зависимым образом подавляет сердечные уровни miR-132 (фиг. 13).
Пример 7. Определение токсикологического профиля органов
7.1. Условия исследования:
• Обработка: дважды, в день 3 и в день 28 после ИМ, 0,3 мг/кг внутрикоронарно и 0,5 мг/кг внутривенно, соответственно.
• Серийный забор крови, конечная точка: 72 часа после обработки, n = 6 свиней/группа, плацебо против CDR132L: p<0,05.
7.2. Результаты:
• Обработка CDR132L in vivo не вызывает какой-либо токсичности в органах свиней (фиг. 14).
Пример 8. Уровни мРНК FoxO3 и SERCA2 в сердце на мышиной модели сердечной недостаточности
Уровни мРНК FoxO3 и Serca2 в сердце измеряли у контрольных мышей и мышей miR-132 TG, получавших внутрибрюшинную инъекцию либо контрольного рандомизированного олигонуклеотида, либо CDR132L, еженедельно, 4 раза. Все значения представляют собой среднее ± SEM. *Р < 0,05 (фиг. 15А и 15В)
Пример 9. Сравнение разных олигонуклеотидов
Цель этого исследования заключалась в оценке терапевтического эффекта нового ингибитора miR-132-3p CDR132L в соответствии с настоящим изобретением с двумя сравнительными олигонуклеотидами. Два сравнительных олигонуклеотида имели такую же последовательность олигонуклеотидов и фосфоротиоатный остов, что и CDR132L, но отличались по распределению структурных блоков LNA в молекуле. CDR2u1 содержал два структурных блока LNA на 5'- и 3'-конце, в то время как для CDR301 каждый нуклеотид нес структурный блок LNA.
Для проверки эффективности различные олигонуклеотиды вводили в кардиомиоциты новорожденных крыс (NRCM). Эффекты этой обработки контролировали с помощью количественной ПЦР в реальном времени (qRT-PCR) после изменений в экспрессии miR-132-3p и его известного целевого гена FoxO3 (Forkhead box O3).
Описание эксперимента и результаты показаны на фиг. 16: (A) Обзор экспериментальных условий. Кардиомиоциты новорожденных крыс высевали в день 0 и обрабатывали олигонуклеотидами CDR132L, CDR2u1 или CDR301 (100 нМ каждый) в день 1. В конечной точке клетки собирали для анализа экспрессии генов. (B) Уровни экспрессии miR-132-3p после обработки CDR132L, CDR2u1 или CDR301. (C) Уровни экспрессии целевого гена miR-132-3p Forkhead box O3 (FoxO3) после обработки CDR132L, CDR2u1 или CDR301. Данные представляют собой среднее ± SD. Значения P олигонуклеотидов в сравнении с плацебо определяли с помощью двустороннего критерия Стьюдента.
Обработка NRCMs CDR132L и CDR301 привела к значительному снижению уровней miR-132-3p на 96%, тогда как CDR2u1 снижал экспрессию miRNA на 30% (фиг. 16A-B). Кроме того, обработка CDR132L привела к значительному снижению целевого гена miR-132-3p Foxo3, чего не удалось достичь с помощью CDR2u1 и CDR301 (фиг. 16C).
Таким образом, наши данные демонстрируют превосходный ингибирующий эффект CDR132L по сравнению с CDR2u1 и CDR301, на что указывают значительно сниженные уровни экспрессии miR-132-3p и значительное подавление его целевого гена FoxO3.
Пример 10. Эффекты CDR132L при фиброзе сердца
Целью данного исследования было оценить антифибротические терапевтические эффекты CDR132L на модели фиброза in vivo.
Для доказательства in vivo антифибротической активности применяли мышиную модель инфаркта миокарда (ИМ) путем постоянного лигирования левой передней нисходящей коронарной артерии (LAD) у мышей C57BL/6N. Обработку CDR132L применяли на 7 и 14 день, плацебо (рандомизированный олиго-аналог CDR132L, 20 мг/кг) и CDR132L (20 мг/кг). Группы включали контрольную оперированную группу (ложнооперированных мышей) и LAD-лигированных мышей (ИМ, инфаркт миокарда), получавших либо плацебо, либо CDR132L: симуляция + плацебо, симуляция + CDR132L, МИ + плацебо, МИ + CDR132L. n = 6-7/группа. Описание эксперимента приведено на фиг. 17.
Оценка антифибротического эффекта CDR132L in vivo при сердечной недостаточности после ИМ показана на фиг. 18. Фиброз (показан как % отложения коллагена, выявленного путем окрашивания пикросириусом красным (PSR) и регистрацией экспрессии гена альфа-1-цепи коллагена типа III (Col3a1) относительно β-актина) был ослаблен после ИМ в результате обработки CDR132L. Группы включали контрольную группу (ложнооперированных мышей) и LAD-лигированных мышей (MI), получавших либо плацебо (черная колонка), либо CDR132L (белая колонка): симуляция + плацебо, симуляция + CDR132L, ИМ + плацебо, ИМ + CDR132L. Статистический тест (непарный t-критерий) проводили между мышами с ИМ, получавшими плацебо или CDR132L. **р<0,01, n = 6-7/группа.
Согласно гистологическим результатам, фиброз ослабевал после обработки CDR132L (фиг. 18A). Это было подтверждено на молекулярном уровне по пониженной экспрессии генов-маркеров фиброза, таких как альфа-1-цепь коллагена типа III (Col3a1) (фиг. 18B).
Пример 11. Эффекты CDR132L при легочном и печеночном фиброзе
Антифибротический эффект CDR132L протестировали in vitro для легочного и печеночного фиброза. Для этого первичные фибробласты человека, полученные из печени (первичные фибробласты печени человека, HPLF, «PeloBiotech») и легких (нормальные первичные фибробласты легких человека, NHLF, «Lonza») стимулировали про-фиброзными средствами и обрабатывали CDR132L. Терапевтические эффекты CDRL132L контролировали по следующим ключевым процессам в фиброзном пути, включая скорость пролиферации и изменения в экспрессии генов фиброзного маркера в конечной точке. Кроме того, оценивали экспрессию miR-132-3p для подтверждения эффективности обработки CDR132L.
Для определения пролиферации клеток применяли набор ELISA для клеточной пролиферации (иммуноферментный анализ) (предоставленный компанией «Roche»). Этот колориметрический анализ позволяет количественно оценить пролиферацию клеток на основе измерения BrdU (бромдезоксиуридина), включенного во вновь синтезированную ДНК пролиферирующих клеток. Количество включенного BrdU детектировали и количественно определяли. Значения абсорбции напрямую коррелируют с количеством синтезированной ДНК и, тем самым, с количеством пролиферирующих клеток в соответствующих микрокультурах. Экспрессию генов оценивали с помощью количественной ПЦР в реальном времени (qRT-PCR), измеряющей уровни экспрессии miR-132-3p и фиброзных маркеров, включая коллаген 1A1 (COL1A1), коллаген 1A2 (COL1A2) и матриксную металлопептидазу 2 (MMP2).
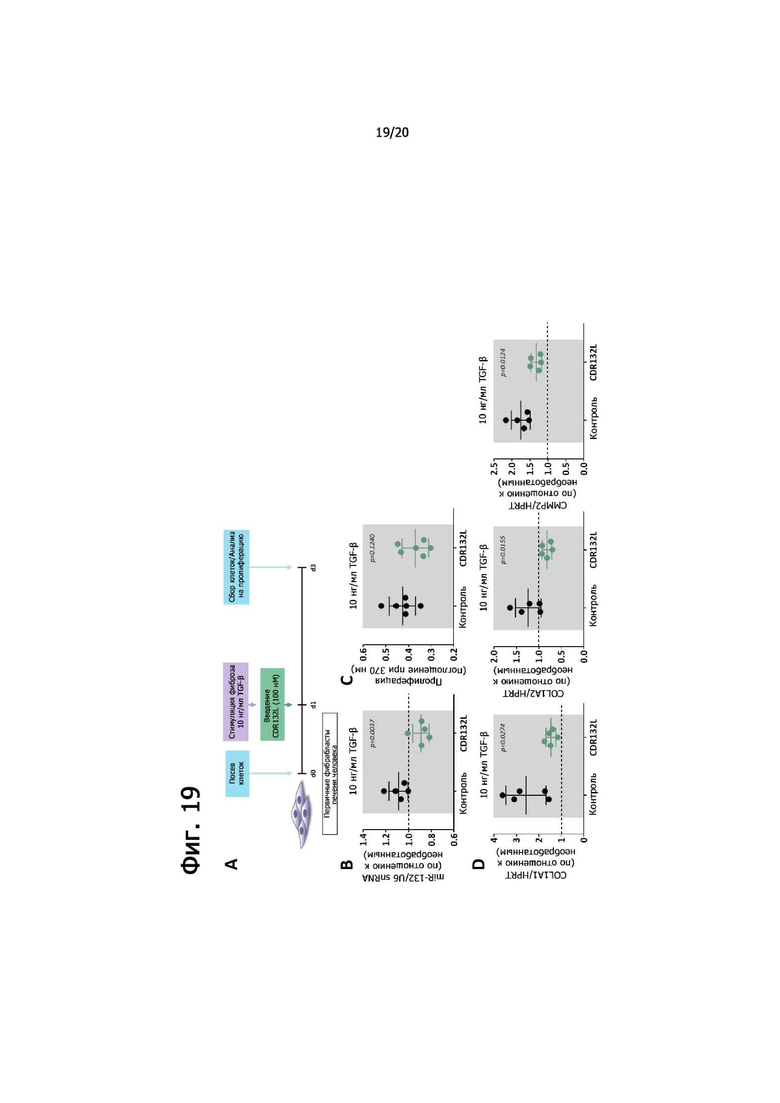
Фиг. 19 относится к модели фиброза печени in vitro: (A) Обзор экспериментальных условий. Первичные фибробласты печени человека (предоставленные компанией «PeloBiotech») высевали в день 0 и обрабатывали фиброзным стимулом (10 нг/мл TGF-β в нормальной питательной среде (полная среда для фибробластов с добавлением 10% FBS)) и CDR132L (100 нМ) в день 1. В конечной точке оценивали пролиферацию клеток и экспрессию генов фиброзных маркеров. (B) Уровни экспрессии miR-132-3p после обработки CDR132L. (C) Пролиферация, оцененная путем мониторинга включения BrdU (бромдезоксиуридина) во время синтеза ДНК. Реагент BrdU был добавлен в среду за 20 ч до конечной точки. (D) Уровни экспрессии генов-маркеров фиброза (коллаген 1A1 (COL1A1), коллаген 1A2 (COL1A2) и матриксная металлопептидаза 2 (MMP2)). В (B) и (D) пунктирная линия указывает уровень экспрессии нестимулированных контрольных клеток. Данные представляют собой среднее ± SD. Значения P для CDR132L в сравнении с контролем определяли с помощью двустороннего t-критерий Стьюдента.
Стимуляция HPLF трансформирующим фактором роста бета (TGF-β) (фиг. 19A) приводила к небольшой индукции miR-132-3p, которая значительно снижалась при обработке CDR132L (фиг. 19B). Кроме того, это соединение уменьшало пролиферацию фибробластов (фиг. 19C) и экспрессию генов фиброза, включая COL1A1, COL1A2 и MMP2 (фиг. 19D).
Фиг. 20 относится к модели фиброза легких in vitro: (A) Обзор экспериментальных условий. Нормальные фибробласты легких человека (предоставленные компанией «Lonza») высевали в день 0 и обрабатывали фиброзным стимулом (высокий FBS (5%) в среде для роста фибробластов и CDR132L (100 нМ) в день 1. В конечной точке оценивали пролиферацию клеток и экспрессию генов. (B) Уровни экспрессии miR-132-3p после обработки CDR132L. (C) Пролиферация, оцененная путем мониторинга включения BrdU (бромдезоксиуридина) во время синтеза ДНК. Реагент BrdU добавляли в среду за 20 ч до конечной точки. В (B) пунктирная линия показывает уровень экспрессии нестимулированных контрольных клеток. Данные представляют собой среднее значение ± SD. Значения P CDR132L в сравнении с контролем определяли с помощью двустороннего t-критерий Стьюдента.
В клетках NHLF после стимуляции фиброза высоким FBS (5%-ный FBS по сравнению с 2%-ным FBS в нормальных условиях роста) (фиг. 20A) не наблюдали повышения miR-132-3p (фиг. 20B). Тем не менее, обработка CDR132L привела к значительному снижению целевой микроРНК (фиг. 20B) и пролиферации фибробластов (фиг. 20C).
Таким образом, наши данные демонстрируют значительный антифиброзный эффект олигонуклеотидного аналога CDR132L в фибробластах печени или легких, а также в сердечной ткани. Мы предполагаем, что этот эффект может быть основан на антипролиферативной способности препарата и/или на его влиянии на экспрессию белка внеклеточного матрикса.
Список литературы
1. Barry, S.P.; Townsend, P.A. (2010). What causes a broken heart-Molecular insights into heart failure. Int Rev Cell Mol Biol 284, 113-179.
2. Datta, S.R.; Brunet, A.; Greenberg, M.E. (1999). Cellular survival: a play in three Akts. Genes Dev. 13, 2905-2927.
3. DeBosch, B.J.; Muslin, A.J. (2008). Insulin signaling pathways and cardiac growth. J Mol Cell Cardiol. 44, 855-864.
4. Frescas, D.; Valenti, L.; Accili, D. (2005). Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt1-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 280, 20589-20595.
5. Glas, D.J. (2010). PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr Top Microbiol Immunol. 346, 267-278.
6. Gottlieb, R.A.; Gustafsson, A.B. (2011). Mitochondrial turnover in the heart. Biochim Biophys Acta. 1813, 1295-1301.
7. Kolk, M.V.; Meyberg, D.; DenseT.; Tang-Quam, K. R.; Robbins R.C.; Reichenspurner, H.; Schrepfer. S (2009), J. Vis Exp. 32, pii: 1438. doi: 103791/1438
8. McMullen, J.R.; Shioi, T.; Huang, W.Y.; Zhang, L.; Tarnavski, O.; Bisping, E.; Schinke, M.; Kong, S.; Sherwood, M.C.; Brown, J. et al. (2004). The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase (p110alpha) pathway. J Biol Chem. 279, 4782-4793.
9. Ni, Y.G.; Berenji, K.; Wang, N.; Oh, M.; Sachan, N.; Dey, A.; Cheng, J.; Lu, G.; Morris, D.J.; Castrillon, D.H. et al. (2006). Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation. 114, 1159-1168.
10. Ronnebaum, S.M.; Patterson, C. (2010). The foxO family in cardiac function and dysfunction. Annu Rev Physiol. 72, 81-94.
11. Skurk, C.; Izumiya, Y.; Maatz, H.; Razeghi, P.; Shiojima, I.; Sandri, M.; Sato, K.; Zeng, L.; Schiekofer, S.; Pimentel, D. et al. (2005). The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 280, 20814-23.
12. Ucar, A. et al. (2012), Nat. Commun. 3: 1078. doi: 10.1038/ncomms2009.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕЧЕНИЕ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ У ЧЕЛОВЕКА | 2020 |
|
RU2811365C2 |
| ДВОЙНОЕ НАЦЕЛИВАНИЕ НП MIR-208 И MIR 499 В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ СЕРДЦА | 2010 |
|
RU2515926C2 |
| РЕГУЛЯЦИЯ МЕТАБОЛИЗМА С ПОМОЩЬЮ MIR-378 | 2011 |
|
RU2585491C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ КАРДИОМИОПАТИИ, ПЕРЕНЕСЕННОГО ИНФАРКТА МИОКАРДА И ХРОНИЧЕСКОЙ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ | 2018 |
|
RU2795320C2 |
| КОНЪЮГАТЫ ОЛИГОНУКЛЕОТИДОВ | 2013 |
|
RU2653438C2 |
| Рекомбинантная плазмидная ДНК, кодирующая микроРНК hsa-miR-145-3p, и ее применение в качестве положительного контроля при ПЦР-РВ для диагностики сердечно-сосудистых заболеваний и выявления риска их развития | 2025 |
|
RU2837873C1 |
| ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ ИНГИБИТОРОВ АКТИВАЦИИ ИЛИ СТИМУЛЯЦИИ Т-КЛЕТОК | 2016 |
|
RU2779308C2 |
| КОНЪЮГАТЫ УГЛЕВОДА И LNA-ОЛИГОНУКЛЕОТИДА | 2014 |
|
RU2649367C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНКАПСУЛИРОВАННЫЙ АНТАГОМИР | 2014 |
|
RU2668794C2 |
| Способ лечения келоидов или гипертрофических рубцов с использованием антисмысловых соединений, направленно действующих на фактор роста соединительной ткани(CTGF) | 2012 |
|
RU2608655C2 |















Изобретение относится к области биотехнологии. Описана группа изобретений, включающая олигонуклеотид-ингибитор микроРНК miR-132, ингибитор микроРНК miR-132, фармацевтическую композицию для ингибирования miR-132, применение олигонуклеотида, ингибитора miR-132 или фармацевтической композиции в профилактике или лечении нарушения, связанного с, сопровождаемого и/или вызванного патологической экспрессией miR-132, в профилактике или лечении нарушений сердечной деятельности, в профилактике или лечении сократительной дисфункции, декомпенсации сердечной деятельности или сердечной недостаточности, в профилактике или лечении нарушений сердечной деятельности, в лечении или профилактике нарушений сердечной деятельности в виде монотерапии или в комбинации с дополнительным лекарственным средством, в лечении или профилактике фиброзных нарушений, в лечении или профилактике сердечных фиброзных нарушений, при профилактике или лечении фиброзных нарушений в качестве монотерапии или в комбинации с дополнительным лекарственным средством. Изобретение расширяет арсенал средств, ингибирующих микроРНК miR-132. 11 н. и 6 з.п. ф-лы, 32 ил., 11 пр.
1. Олигонуклеотид-ингибитор микроРНК miR-132, где олигонуклеотид включает последовательность формулы II:
5’-A+TG+GC+TG+TA+GACTG+T+T-3’,
в которой A, T, G и C представляют собой дезоксирибонуклеотидные структурные блоки и
в которой +G и +T представляют собой мостиковые нуклеотидные структурные блоки и/или морфолиновые структурные блоки, в особенности, в которой +G и +T представляют собой структурные блоки LNA.
2. Олигонуклеотид по п. 1, в котором, по меньшей мере, один мостиковый нуклеотидный структурный блок представляет собой структурный блок запертого нуклеотида (LNA).
3. Олигонуклеотид по п. 1 или 2, имеющий в длину 16 структурных блоков.
4. Олигонуклеотид по любому из пп. 1-3, включающий, по меньшей мере, одну модифицированную межнуклеозидную связь, в особенности, по меньшей мере, одну фосфоротиоатную или фосфородиамидатную межнуклеозидную связь.
5. Олигонуклеотид по любому из пп. 1-4, в котором все межнуклеозидные связи представляют собой фосфоротиоатные связи.
6. Олигонуклеотид по любому из пп. 1-5, включающий последовательность формулы III:
5’-dA*+T*dG*+G*dC*+T*dG*+T*dA*+G*dA*dC*dT*dG*+T*+T-3’,
в которой dA представляет собой 2’-дезоксиаденозин, dG представляет собой 2’-дезоксигуанозин, dC представляет собой 2’-дезоксицитидин и Т представляет собой тимидин,
в которой +T представляет собой структурный блок запертого нуклеотида LNA-T и +G представляет собой структурный блок запертого нуклеотида LNA-G и в которой * представляет собой фосфоротиоатную связь.
7. Ингибитор микроРНК miR-132, содержащий олигонуклеотид, включающий последовательность формулы II:
5’-A+TG+GC+TG+TA+GACTG+T+T-3’,
в которой A, T, G и C представляют собой дезоксирибонуклеотидные структурные блоки и
в которой +G и +T представляют собой мостиковые нуклеотидные структурные блоки и/или морфолиновые структурные блоки, в особенности, в которой +G и +T представляют собой структурные блоки LNA,
или включающий последовательность формулы III:
5’-dA*+T*dG*+G*dC*+T*dG*+T*dA*+G*dA*dC*dT*dG*+T*+T-3’,
в которой dA представляет собой 2’-дезоксиаденозин, dG представляет собой 2’-дезоксигуанозин, dC представляет собой 2’-дезоксицитидин и Т представляет собой тимидин,
в которой +T представляет собой структурный блок запертого нуклеотида LNA-T и +G представляет собой структурный блок запертого нуклеотида LNA-G и в которой * представляет собой фосфоротиоатную связь,
где указанный олигонуклеотид конъюгирован с гетерологичным фрагментом.
8. Фармацевтическая композиция для ингибирования miR-132, включающая олигонуклеотид по любому из пп. 1-6 или ингибитор miR-132 по п. 7 и фармацевтически приемлемый носитель.
9. Применение олигонуклеотида по любому из пп. 1-6, ингибитора miR-132 по п. 7 или фармацевтической композиции по п. 8 в профилактике или лечении нарушения, связанного с, сопровождаемого и/или вызванного патологической экспрессией miR-132.
10. Применение олигонуклеотида по любому из пп. 1-6, ингибитора miR-132 по п. 7 или фармацевтической композиции по п. 8 в профилактике или лечении нарушений сердечной деятельности, в особенности, нарушений, связанных с гипертрофией сердца.
11. Применение олигонуклеотида по любому из пп. 1-6 или ингибитора miR-132 по п. 7 или фармацевтической композиции по п. 8 в профилактике или лечении сократительной дисфункции, декомпенсации сердечной деятельности или сердечной недостаточности.
12. Применение олигонуклеотида по любому из пп. 1-6, ингибитора miR-132 по п. 7 или фармацевтической композиции по п. 8 в профилактике или лечении нарушений сердечной деятельности, при которых соединение вводят пациентам, выбранным из:
(i) пациентов с повышенным риском развития сердечной недостаточности,
(ii) пациентов, страдающих сердечной недостаточностью и/или застойной сердечной недостаточностью, например, пациенты с повышенным риском прогрессирования сердечной недостаточности;
(iii) пациентов после инфаркта миокарда и/или
(iv) пациентов с врожденными пороками сердца, связанными с гипертрофией сердца, такими как стеноз легочной вены, дефекты предсердной или желудочковой перегородки.
13. Применение олигонуклеотида по любому из пп. 1-6, ингибитора miR-132 по п. 7 или фармацевтической композиции по п. 8 в лечении или профилактике нарушений сердечной деятельности в виде монотерапии или в комбинации с дополнительным лекарственным средством, в особенности, выбранным из ангиотензин-модулирующих средств, β-блокаторов, диуретиков, антагонистов альдостерона, вазодилататоров, ионотропных средств или их комбинаций.
14. Применение олигонуклеотида по любому из пп. 1-6, ингибитора miR-132 по п. 7 или фармацевтической композиции по п. 8 в лечении или профилактике фиброзных нарушений.
15. Применение олигонуклеотида по любому из пп. 1-6, ингибитора miR-132 по п. 7 или фармацевтической композиции по п. 8 в лечении или профилактике сердечных фиброзных нарушений, в особенности, фиброза предсердий, эндомиокардиального фиброза или фиброза, вызванного предыдущим инфарктом миокарда, легочных фиброзных нарушений, в особенности, легочного фиброза, вызванного профессиональными факторами или факторами окружающей среды, легочного фиброза, вызванного лучевой терапией и/или приемом лекарственных средств, или идиопатический легочный фиброз, или фиброзные нарушений печени, в особенности, алкогольная болезнь печени или неалкогольная жировая болезнь печени (НЖБП).
16. Применение олигонуклеотида по любому из пп. 1-6, ингибитора miR-132 по п. 7 или фармацевтической композиции по п. 8 при профилактике или лечении фиброзных нарушений в качестве монотерапии или в комбинации с дополнительным лекарственным средством.
17. Применение по любому из пп. 9-16, включающее этап определения количества и/или активности (i) miR-132;(ii), по меньшей мере, одного сердечного маркера и/или (iii), по меньшей мере, одного фиброзного маркера у подлежащего лечению пациента до, во время и/или после введения олигонуклеотида, ингибитора miR-132 или фармацевтической композиции.
| WO 2010105096 A2, 16.09.2010 | |||
| WO 2013034653 A1, 14.03.2013 | |||
| WO 2016042561 А2, 24.03.2016 | |||
| АНТИТЕЛА К РОСТОВОМУ ФАКТОРУ СОЕДИНИТЕЛЬНОЙ ТКАНИ | 2004 |
|
RU2330861C2 |

