ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому производному 2-арилтиазола или его соли, способу для их получения и содержащей их фармацевтической композиции. В частности, настоящее изобретение относится к новому производному 2-арилтиазола или его фармацевтически приемлемой соли, имеющей определенную карбоксамидную группу, например, замещенную аминоалкилкарбоксамидную группу, азотсодержащую гетероцикл-алкилкарбоксамидную группу или азотсодержащую гетероцикл-карбоксамидную группу, способу для их получения и содержащей их фармацевтической композиции. Производное 2-арилтиазола или его фармацевтически приемлемая соль согласно настоящему изобретению обладает превосходной индуцирующей аутофагию активностью.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Аутофагия, которая также называется аутофагоцитозом, представляет собой естественный регулируемый механизм клетки, который разбирает ненужные или не функционирующие компоненты. Она обеспечивает упорядоченный распад и переработку клеточных компонентов. При осуществлении процесса аутофагии невосстановимые цитоплазматические компоненты изолируются от остальной части клетки внутри двухмембранной везикулы, известной как аутофагосома. Далее аутофагосома соединяется с доступной лизосомой, и в результате содержимое везикулы расщепляется и перерабатывается. Как правило, описывают три формы аутофагии: макроаутофагия, микроаутофагия и шаперон-опосредованная аутофагия (CMA). При заболевании аутофагия рассматривалась как адаптивный ответ на стресс, способствующий выживанию клетки; но в других случаях, по-видимому, способствует гибели клеток и патологическим проявлениям. В крайнем случае голода расщепление клеточных компонентов способствует выживанию клеток за счет поддержания клеточного уровня энергии.
В то же время, если аутофагия уменьшается, различные заболевания могут наступать вследствие накопления неправильно свернутых белков и т.п. Например, сообщалось, что индукция аутофагии может лечить нейродегенеративные заболевания, такие как болезнь Хантингтона (HD), болезнь Паркинсона (PD), болезнь Альцгеймера (AD), прионная болезнь, рассеянный склероз и боковой амиотрофический склероз (болезнь Лу Герига) (например, корейский патент № 10-1731908). И кроме того, сообщалось, что индукцией аутофагии можно лечить болезни печени, такие как фиброз печени, цирроз печени, гепатит и жировая болезнь печени (например, выложенный для всеобщего ознакомления корейский документ № 10-2017-0022790). Кроме того, сообщалось, что индукцией аутофагии можно лечить метаболические заболевания, такие как диабет, гиперлипидемия, ожирение и воспаление (например, выложенный для всеобщего ознакомления корейский документ № 10-2018-0007307). Более того, сообщалось, что индукция аутофагии может ингибировать чрезмерные иммунные ответы, связанные с сепсисом (например, выложенный для всеобщего ознакомления корейский документ № 10-2012-0131401).
Следовательно, предполагается, что вещество, индуцирующее аутофагию, может быть успешно применено для профилактики или лечения различных заболеваний, связанных с аутофагией, таких как нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и т.д.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
Авторы настоящего изобретения обнаружили, что новое производное 2-арилтиазола или его фармацевтически приемлемая соль, имеющая определенную карбоксамидную группу, например, замещенную аминоалкилкарбоксамидную группу, азотсодержащую гетероцикл-алкилкарбоксамидную группу или азотсодержащую гетероцикл-карбоксамидную группу, обладает превосходной индуцирующей аутофагию активностью и, следовательно, может эффективно применяться для профилактики или лечения различных заболеваний, связанных с аутофагией.
Таким образом, настоящее изобретение обеспечивает указанное производное 2-арилтиазола или его фармацевтически приемлемую соль, способ для их получения, содержащую их фармацевтическую композицию и их применение.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ
Согласно одному аспекту настоящего изобретения предлагается новое производное 2-арилтиазола или его фармацевтически приемлемая соль.
Согласно другому аспекту настоящего изобретения предлагается способ для приготовления указанного производного 2-арилтиазола или его фармацевтически приемлемой соли.
Согласно еще одному аспекту настоящего изобретения предлагается фармацевтическая композиция, содержащая указанное производное 2-арилтиазола или его фармацевтически приемлемую соль в качестве активного ингредиента.
Согласно еще одному аспекту настоящего изобретения предлагается способ для лечения заболевания, связанного с аутофагией, включающий введение указанного производного 2-арилтиазола или его фармацевтически приемлемой соли.
Согласно еще одному аспекту настоящего изобретения предлагается использование указанного производного 2-арилтиазола или его фармацевтически приемлемой соли для производства лекарственного препарата для профилактики или лечения заболевания, связанного с аутофагией.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
Соединение согласно настоящему изобретению, то есть новое производное 2-арилтиазола или его фармацевтически приемлемая соль, имеющая определенную карбоксамидную группу, например, замещенную аминоалкилкарбоксамидную группу, азотсодержащую гетероцикл-алкилкарбоксамидную группу или азотсодержащую гетероцикл-карбоксамидную группу, обладает превосходной индуцирующей аутофагию активностью. Следовательно, соединение или его фармацевтически приемлемая соль согласно настоящему изобретению может эффективно применяться для профилактики или лечения различных заболеваний, связанных с аутофагией, включая нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и т.д.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
ФИГ. 1 показывает результаты полученные в результате анализа аутофагических потоков соединений согласно настоящему изобретению в клетках N2a. ФИГ. 1a показывает результаты, полученные в результате получения изображения клеток в целом с помощью мозаичного метода, где стрелки указывают на аутофагию. ФИГ. 1b показывает результаты получения изображения под электронным микроскопом клеток N2a, обработанных соединением согласно примеру 29, где черные стрелки показывают аутофагосомы, а белые стрелки показвают аутолизосомы.
ФИГ. 2 показывает обобщенные экспериментальные способы согласно тестовому примеру 4 для того, чтобы оценить активности, улучшающие функцию печени при пероральном введении на модели поражения печени.
ФИГ. 3 показывает обобщенные экспериментальные способы согласно тестовому примеру 5 для того, чтобы оценить активности, улучшающие функцию печени при пероральном введении на модели поражения печени.
ФИГ. 4 показывает результаты, полученный окрашиванием гематоксилином и эозином (H&E) и трихромного окрашивания по Массону на образцах ткани, полученных в тестовом примере 5. Масштабный отрезок: левая черная линия 100 мкм, правая черная линия 100 мкм.
ЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
В контексте настоящего документа термин «алкил» означает линейный или разветвленный алифатический углеводородный радикал. Например, C1-C6-алкил означает линейный или разветвленный алифатический углеводород, имеющий от 1 до 6 атомов углерода, такой как метил, этил, пропил, н-бутил, н-пентил, н-гексил, изопропил, изобутил, втор-бутил, трет-бутил, неопентил и изопентил.
Термин «гидрокси» означает радикал -ОН. Термин «алкокси» означает радикал, образованный замещением атома водорода гидроксигруппы алкилом. Например, C1-C6-алкокси включает метокси, этокси, пропокси, н-бутокси, н-пентилокси, изопропокси, изобутокси, втор-бутокси, трет-бутокси, неопентилокси и изопентилокси.
Термин «амино» означает радикал -NH2. Термин «алкиламино» означает аминогруппу, замещенную моно- или диалкилом. Например, C1-C6-алкиламино включает аминогруппу, замещенную моно- или ди-C1-6-алкилом.
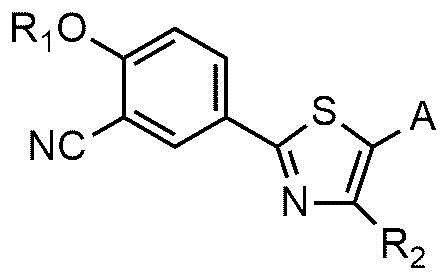
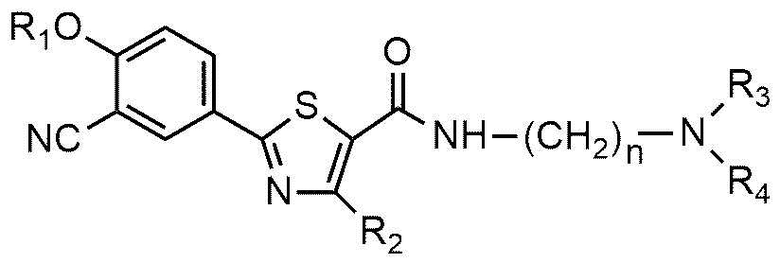
Настоящее изобретение обеспечивает соединение или его соль, обладающие превосходной индуцирующей аутофагию активностью, то есть соединение формулы 1 или его фармацевтически приемлемую соль:
<формула 1>
 ,
,
где
R1 представляет собой водород, C1~C4-алкильную группу или C1~C4-алкильную группу, замещенную моно- или ди-C1~C5-алкиламино,
R2 представляет собой C1~C4-алкильную группу,
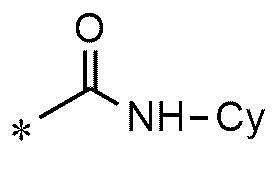
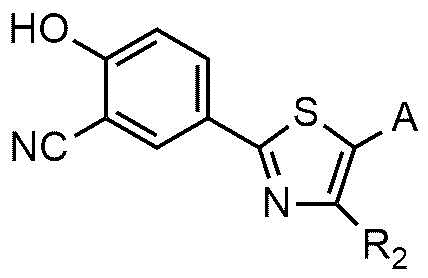
A представляет собой группу, выбранную из группы, состоящей из следующих групп формул от 1a до 1d:
<формула 1a>
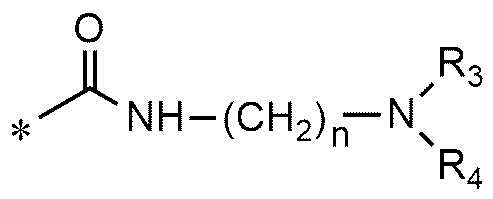
<формула 1b>
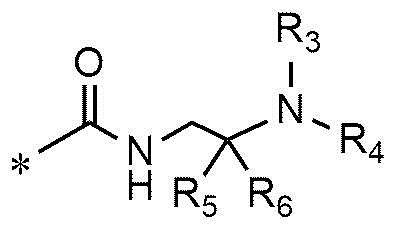
<формула 1c>
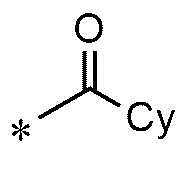
<формула 1d>
 ,
,
в формуле от 1a до 1d,
* означает место, присоединенное к соединению формулы 1,
n равно 1, 2 или 3,
R3 и R4, независимо друг от друга, представляют собой водород или C1~C4-алкил; или R3 и R4 соединены друг с другом, с атомом азота, к которому они присоединены, с образованием пиперидина, морфолина или пирролидина, где пиперидин, морфолин или пирролидин необязательно замещен C1~C4-алкилом,
R5 и R6 представляют собой C1~C4-алкил; или R5 и R6 образуют кольцо с образованием циклопентана, циклогексана или тетрагидропирана, и
Cy представляет собой азотсодержащую гетероциклическую группу, выбранную из группы, состоящей из гомопиперазинила, пиперидинила, морфолинила и пирролидинила, где азотсодержащая гетероциклическая группа необязательно замещена C1~C4-алкилом или бензилом.
В соединении или его фармацевтически приемлемой соли, согласно настоящему изобретению, R1 предпочтительно может представлять собой водород, изобутильную группу, диэтиламиноэтильную группу, и R2 предпочтительно может представлять собой метильную группу.
В соединении или его фармацевтически приемлемой соли согласно настоящему изобретению A представляет собой группу, выбранную из группы, состоящей из групп формул от 1a до 1d. В одном варианте осуществления настоящего изобретения A может представлять собой группу формулы 1a или группу формулы 1b. Соединения согласно настоящему изобретению, в которых A представляет собой группу формулы 1a или группу формулы 1b, имеют следующие структуры формул 11a и 11b.
<формула 11a>
<формула 11b>
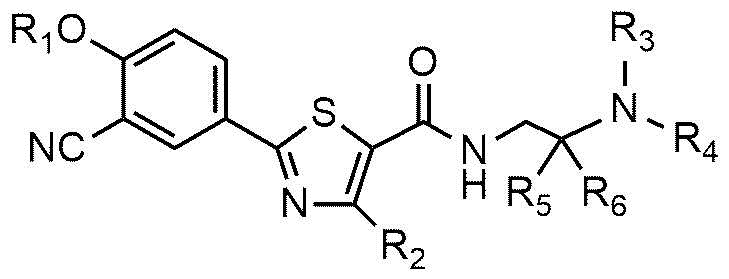
В соединениях формулы 11a и 11b R1, R2, R3, R4, R5 и R6 являются такими же, как определены в вышеизложенном.
В соединениях формулы 11a и 11b R3 и R4 предпочтительно могут быть соединены друг с другом, с атомом азота, к которому они присоединены, с образованием пиперидина, морфолина или пирролидина. Более предпочтительно, пиперидин, морфолин или пирролидин могут быть замещены C1~C4-алкилом.
Более предпочтительно, соединение или его фармацевтически приемлемая соль согласно настоящему изобретению могут представлять собой одно или несколько, выбранных из группы, состоящей из:
N-(2-(диэтиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диизопропиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(диметиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(3-(диэтиламино)пропил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(4-метилгомопиперазино)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(4-(1-метил)пиперидинил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(4-морфолино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(1-пирролидино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(1-пиперидино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-((1-(диметиламино)циклопентил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-((1-(диметиламино)циклогексил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(3-(1-бензил)пирролидинил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(2-(1-метил)пирролидино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-((S)-2-(1-этил)пирролидинометил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(4-морфолинамино)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(1-пиперидинамино)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(диизопропиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(диэтиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(диметиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(4-морфолино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-((S)-2-(1-этил)пирролидинометил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(диизопропиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(диэтиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(диметиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(4-морфолино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-((S)-2-(1-этил)пирролидинометил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид; и
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорид.
В частности, предпочтительно соединение или его фармацевтически приемлемая соль согласно настоящему изобретению могут представлять собой одно или несколько, выбранных из группы, состоящей из:
N-(2-(диизопропиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(диизопропиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-(2-(4-морфолино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорид;
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид; и
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорид.
Соединение формулы 1 может быть в форме фармацевтически приемлемой соли, например, в форме соли присоединения кислоты. Соль присоединения кислоты может представлять собой соль, полученную из неорганической или органической кислоты, такую как гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат, гидросульфат, фосфат, ацетат, лактат, цитрат, тартрат, сукцинат, малеат, малонат, оксалат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, памоат и т.п., но не ограниваясь этим. Соль присоединения кислоты может быть приготовлена реакцией соединения формулы 1 с неорганической кислотой или органической кислотой в обычном растворителе, например, воде, спирте, тетрагидрофуране, ацетоне или их смеси.
Соединение формулы 1 или его фармацевтически приемлемая соль согласно настоящему изобретению может быть в безводной форме, в форме гидрата или в форме сольвата. Кроме того, соединение формулы 1 или его фармацевтически приемлемая соль согласно настоящему изобретению может быть в аморфной или кристаллической формах. Указанные аморфные или кристаллические формы также могут быть в форме гидрата или в форме сольвата. Гидрат или сольват может содержать воду или органический растворитель в стехиометрическом или нестехиометрическом количестве по отношению к соединению формулы 1.
Соединение формулы 1 или его фармацевтически приемлемая соль может иметь заместитель(и), содержащие асимметричный углерод, и, следовательно, может быть в форме рацемической смеси (RS) или в формах оптических изомеров, таких как (R) или (S) изомер. Следовательно, если не указано иное, соединение формулы 1 или его фармацевтически приемлемая соль включает как рацемическую смесь (RS), так и оптические изомеры, такие как (R) или (S) изомер.
Настоящее изобретение также включает в рамках своего объема способ для приготовления соединения формулы 1 или его фармацевтически приемлемой соли. Например, соединение формулы 1 или его фармацевтически приемлемую соль можно приготовить ацилированием соединения формулы 2 азотсодержащим соединением с приготовлением соединения формулы 1 или ацилированием соединения формулы 3 азотсодержащим соединением с приготовлением соединения формулы 4; и алкилированием соединения формулы 4 с использованием R1X формулы 5 с приготовлением соединения формулы 1, как показано на следующей схеме реакции 1:
<Схема реакции 1>
ацилирование
ацилирование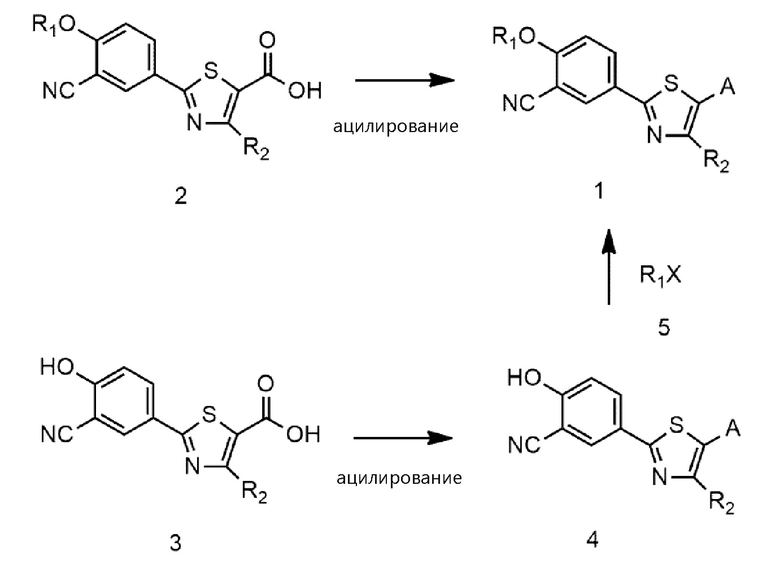
На схеме реакции 1 R1, R2, и A являются такими же, как определено в вышеизложенном, и X представляет собой галоген.
В одном варианте осуществления настоящее изобретение обеспечивает способ приготовления соединения формулы 1 или его фармацевтически приемлемой соли, причем способ включает ацилирование соединения формулы 2 азотсодержащим соединением, выбранным из группы, состоящей из NH2-(CH2)n-NR3R4, NH2-CH2-CR5R6-NR3R4, Cy и NH2-Cy (где n, R3, R4, R5 и R6 являются такими же, как определено в вышеизложенном; Cy представляет собой азотсодержащий гетероцикл, выбранный из группы, состоящей из гомопиперазина, пиперидина, морфолина и пирролидина, где азотсодержащий гетероцикл необязательно замещен C1~C4-алкилом или бензилом; и NH2-Cy представляет собой азотсодержащий гетероциклический амин, выбранный из группы, состоящей из гомопиперазиниламина, пиперидиниламина, морфолиниламина и пирролидиниламина, где азотсодержащий гетероциклический амин необязательно замещен C1~C4-алкилом или бензилом):
<формула 1>
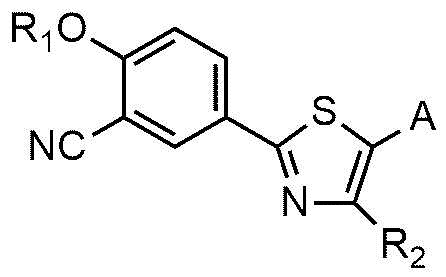
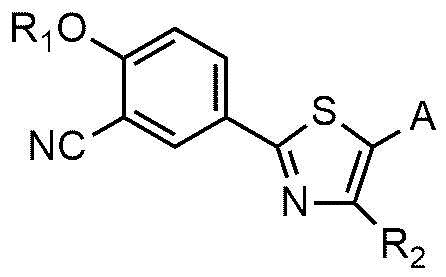
<формула 2>
где R1, R2 и A являются такими же, как определено в вышеизложенном.
В другом варианте осуществления настоящее изобретение обеспечивает способ для приготовления соединения формулы 1 или его фармацевтически приемлемой соли, причем способ включает ацилирование соединения формулы 3 азотсодержащим соединением, выбранным из группы, состоящей из: NH2-(CH2)n-NR3R4, NH2-CH2-CR5R6-NR3R4, Cy и NH2-Cy (где n, R3, R4, R5 и R6 являются такими же, как определено в вышеизложенном; Cy представляет собой азотсодержащий гетероцикл, выбранный из группы, состоящей из гомопиперазина, пиперидина, морфолина и пирролидина, где азотсодержащий гетероцикл необязательно замещен C1~C4-алкилом или бензилом; и NH2-Cy представляет собой азотсодержащий гетероциклический амин, выбранный из группы, состоящей из гомопиперазиниламина, пиперидиниламина, морфолиниламина и пирролидиниламина, где азотсодержащий гетероциклический амин необязательно замещен C1~C4-алкилом или бензилом) с приготовлением соединения формулы 4 и алкилирование соединения формулы 4 соединением формулы 5 с приготовлением соединения формулы 1:
<формула 1>
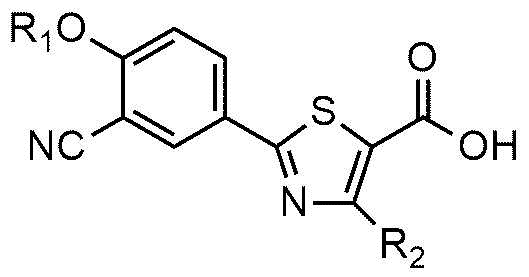
<формула 3>
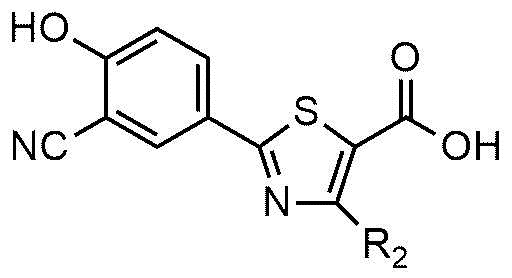
<формула 4>
<формула 5>
R1X,
где R1, R2 и A являются такими же, как определено в вышеизложенном, и X представляет собой галоген.
Соединение формулы 2, соединение формулы 3 и азотсодержащее соединение (т.е, NH2-(CH2)n-NR3R4, NH2-CH2-CR5R6-NR3R4, Cy или NH2-Cy) являются известными и, таким образом, доступны на рынке. Ацилирование может быть осуществлено с помощью ацилирующего агента, такого как 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), дициклогексилкарбодиимид (DCC), 1,1'-карбонилдиимидазол (CDI), N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (EEDQ). Ацилирование также может быть осуществлено реакцией соединения формулы 2 или формулы 3 с тионилхлоридом, оксалилхлоридом, хлоридом фосфора и т.п. с приготовлением ацилхлорида с последующей реакцией азотсодержащего соединения с ним. Ацилирование может быть осуществлено в обычном органическом растворителе, например, дихлорметане, диметилформамиде и т.д.
Алкилирование можно осуществлять в присутствии основания щелочного металла, такого как гидроксид натрия или карбонат натрия. Реакция между соединением формулы 4 и R1X формулы 5 может быть осуществлена при молярном соотношении, находящемся в диапазоне от 1 : 1 до 1 : 5, предпочтительно, при молярном соотношении примерно 1 : 1. Алкилирование можно осуществлять в воде, C1~C4-спирте, ацетоне, тетрагидрофуране, толуоле или их смеси.
Производное 2-арилтиазола согласно настоящему изобретению, то есть соединение формулы 1 или его фармацевтически приемлемая соль, обладает превосходной индуцирующей аутофагию активностью. Следовательно, соединение формулы 1 или его фармацевтически приемлемая соль согласно настоящему изобретению может эффективно применяться для профилактики или лечения различных заболеваний, связанных с аутофагией, включая нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и т.д.
Следовательно, настоящее изобретение включает фармацевтическую композицию для индуцирования аутофагии, содержащую терапевтически эффективное количество соединения формулы 1 или его фармацевтически приемлемой соли в качестве активного ингредиента.
Заболевания, связанные с аутофагией, включают без ограничения различные заболевания, которые можно предотвратить, купировать или лечить посредством индукции аутофагии. Например, фармацевтическая композиция согласно настоящему изобретению может представлять собой фармацевтическую композицию для профилактики или лечения нейродегенеративных заболеваний, выбранных из группы, состоящей из болезни Хантингтона, болезни Паркинсона, болезни Альцгеймера, прионной болезни, рассеянного склероза и болезни Лу Герига; заболеваний печени, выбранных из группы, состоящей из фиброза печени, цирроза печени, гепатита и жировой болезни печени; метаболических заболеваний, выбранных из группы, состоящей из диабета, гиперлипидемии, ожирения и воспаления; или сепсиса.
Фармацевтическая композиция согласно настоящему изобретению может включать фармацевтически приемлемый носитель, такой как разбавители, разрыхлители, подсластители, смазывающие вещества или вкусоароматические агенты. Фармацевтическая композиция может быть составлена для лекарственной формы перорального применения, такой как таблетки, капсулы, порошки, гранулы, суспензии, эмульсии или сиропы; или лекарственной формы парентерального применения, такой как наружные растворы, наружные суспензии, наружные эмульсии, гели (например, мази), лекарственные формы для ингаляции, спреи, инъекционные лекарственные формы, в соответствии с традиционными способами. Лекарственная форма может представлять собой различные формы, например лекарственные формы для однократного введения или для многократного введения.
Фармацевтическая композиция согласно настоящему изобретению может содержать, например, разбавитель (например, лактозу, кукурузный крахмал и т. д.); смазывающее вещество (например, стеарат магния); эмульгирующий агент; суспендирующий агент; стабилизатор; и/или изотонический агент. При необходимости композиция дополнительно содержит подсластители и/или вкусоароматические агенты.
Композицию согласно настоящему изобретению можно вводить перорально или парентерально, включая ингалируемый, внутривенный, интраперитонеальный, подкожный, интрацеребровентрикулярный, ректальный и местный пути введения. Таким образом, композиция согласно настоящему изобретению может быть составлена в виде различных форм, таких как таблетки, капсулы, водные растворы или суспензии. В случае таблеток для перорального введения обычно используют такие носители, как лактоза, кукурузный крахмал, и смазывающие агенты, например, стеарат магния. В случае капсул для перорального введения в качестве разбавителя можно использовать лактозу и/или сухой кукурузный крахмал. Если для перорального введения требуется водная суспензия, активный ингредиент можно объединять с эмульгирующими и/или суспендирующими агентами. При необходимости можно использовать некоторые подсластители и/или вкусоароматические агенты. Для внутримышечного, интраперитонеального, подкожного и внутривенного введения обычно готовят стерильные растворы активного ингредиента, и pH растворов должно быть надлежащим образом отрегулировано и создавать буферную систему. Для внутривенного введения суммарная концентрация растворенных веществ должна контролироваться, чтобы сделать препарат изотоничным. Композиция согласно настоящему изобретению может быть в форме водного раствора, содержащего фармацевтически приемлемые носители, например, физиологического раствора с уровнем pH 7,4. Указанные растворы можно вводить во внутримышечный кровоток пациента путем локальной болюсной инъекции.
Производное 2-арилтиазола согласно настоящему изобретению, то есть соединение формулы 1 или его фармацевтически приемлемая соль, может быть введено больному пациенту в терапевтически эффективном количестве, находящемся в диапазоне примерно от 0,0001 мг/кг до примерно 100 мг/кг в сутки, предпочтительно от примерно 0,001 мг/кг до примерно 100 мг/кг в сутки. Введение может быть осуществлено посредством перорального или парентерального пути введения один или несколько раз в сутки. Безусловно, доза может быть изменена в зависимости от возраста пациента, состояния здоровья, веса, чувствительности, степени заболевания, пути введения, продолжительности введения и т.п. В зависимости от способа введения фармацевтическая композиция согласно настоящему изобретению может содержать соединение формулы 1 или его фармацевтически приемлемую соль в количестве, находящемся в диапазоне от 0,001 до 99% по массе, предпочтительно от 0,01 до 60% по массе.
Настоящее изобретение также включает в рамках своего объема способ для индуцирования аутофагии у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли. Например, настоящее изобретение включает способ для профилактики или лечения нейродегенеративных заболеваний, выбранных из группы, состоящей из болезни Хантингтона, болезни Паркинсона, болезни Альцгеймера, прионной болезни, рассеянного склероза и болезни Лу Герига; заболеваний печени, выбранных из группы, состоящей из фиброза печени, цирроза печени, гепатита и жировой болезни печени; метаболических заболеваний, выбранных из группы, состоящей из диабета, гиперлипидемии, ожирения и воспаления; или сепсиса.
Настоящее изобретение также включает в пределах своего объема применение соединения формулы 1 или его фармацевтически приемлемой соли для производства лекарственного препарата для индуцирования аутофагии у млекопитающего, нуждающегося в этом. Например, настоящее изобретение вклюяает применение соединения формулы 1 или его фармацевтически приемлемой соли для производства лекарственного препарата для профилактики или лечения нейродегенеративных заболеваний, выбранных из группы, состоящей из болезни Хантингтона, болезни Паркинсона, болезни Альцгеймера, прионной болезни, рассеянного склероза и болезни Лу Герига; заболеваний печени, выбранных из группы, состоящей из фиброза печени, цирроза печени, гепатита и жировой болезни печени; метаболических заболеваний, выбранных из группы, сосостоящей из диабета, гиперлипидемии, ожирения и воспаления; или сепсиса.
Приведенные ниже примеры и тестовые примеры предоставлены исключительно в иллюстративных целях и не предназначены для ограничения объема изобретения.
Анализ соединений, полученных в приведенных ниже примерах, осуществляли, как изложено ниже: анализ спектра ядерного магнитного резонанса (ЯМР) осуществляли с использованием спектрометра Bruker 400 МГц и его химические сдвиги выражены в миллионных долях (м.д.). Колоночную хроматографию осуществляли на силикагеле (Merck, 70-230 меш). Если не указано иное, все исходные вещества были коммерчески приобретены и использовались без дополнительной очистки. Все реакции и хроматографические фракции анализировали с помощью тонкослойной хроматографии (ТСХ) на пластине силикагеля 250 нм и визуализировали с помощью ультрафиолетового излучения или окрашивания йодом (I2). Продукты реакции и промежуточные соединения очищали методом флэш-хроматографии или обращенно-фазовой высокоэффективной жидкостной хроматографией (ВЭЖХ).
Пример 1: Получение N-(2-(диэтиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
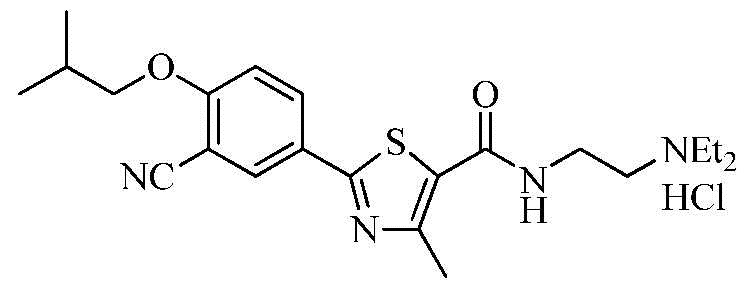
Смесь 2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоновой кислоты (9,49 г), дихлорметана (100 мл) и диметилформамида (0,25 мл) перемешивали в течение 10 минут. Тионилхлорид (4,28 г) добавили к смеси, которую затем нагревали с обратным холодильником при перемешивании в течение 3 часов. Реакционную смесь охладили до комнатной температуры и затем сконцентрировали при пониженном давлении. Добавили дихлорметан (90 мл) к полученному концентрату, который затем охладили до 0~10°C. Добавили K2CO3 (5,30 г) к реакционной смеси, которую затем перемешивали в течение 20 минут. К смеси добавили 2-(диэтиламино)этиламин (3,49 г). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, промыли очищенной водой (100 мл), высушили над безводным сульфатом натрия и затем сконцентрировали под вакуумом с получением N-(2-(диэтиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида. Полученное соединение растворили в смешанном растворителе из метанола (70 мл) и дихлорметана (7 мл), и затем туда добавили раствор 1N соляной кислоты в эфире (25 мл). Смесь нагрели до 40°C и затем сконцентрировали при пониженном давлении. К полученному концентрату добавили ацетон (70 мл). Смесь перемешивали в течение 1 часа и затем фильтровали. Полученное твердое вещество промыли ацетоном (10 мл) и затем высушили при 50~55°C с получением 6,50 г озаглавленного соединения. (выход: 66,3%)
ТСХ коэффициент удерживания (Rf) = 0,24 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,26 (д, 1H, J = 2,4 Гц), 8,22 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,33 (д, 1H, J = 8,8 Гц), 4,03 (д, 2H, J = 6,4 Гц), 3,78 (т, 2H, J = 6,4 Гц), 3,42 (т, 2H, J = 6,4 Гц), 3,40 ~ 3,32 (м, 4H), 2,74 (с, 3H), 2,25 ~ 2,15 (м, 1H), 1,40 (т, 6H, J = 7,2 Гц), 1,12 (д, 6H, J = 6,8 Гц).
Пример 2: Получение N-(2-(диизопропиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
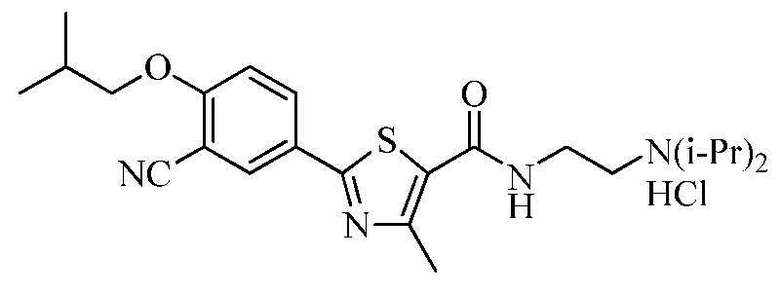
Озаглавленное соединение получили в соответствии с теми же способами, что и в примере 1, за исключением использования 2-(диизопропиламино)этиламина вместо 2-(диэтиламино)этиламина (выход: 55,5%)
ТСХ Rf = 0,29 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,23 (д, 1H, J = 2,4 Гц), 8,20 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,30 (д, 1H, J = 8,8 Гц), 4,01 (д, 2H, J = 6,4 Гц), 3,90 ~ 3,82 (м, 2H), 3,75 (т, 2H, J = 6,8 Гц), 3,38 (т, 2H, J = 6,8 Гц), 2,72 (с, 3H), 2,25 ~ 2,15 (м, 1H), 1,46 (д, 12H, J = 6,8 Гц), 1,12 (д, 6H, J = 6,8 Гц).
Пример 3: Получение N-(2-(диметиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
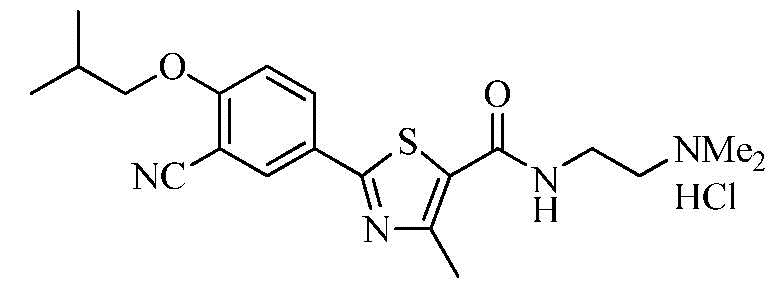
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 2-(диметиламино)этиламина вместо 2-(диэтиламино)этиламина. (выход: 90,6%)
ТСХ Rf = 0,34 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,26 (д, 1H, J = 2,4 Гц), 8,22 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,34 (д, 1H, J = 9,2 Гц), 4,03 (д, 2H, J = 6,4 Гц), 3,77 (т, 2H, J = 6,0 Гц), 3,42 (т, 2H, J = 6,0 Гц), 3,02 (с, 6H), 2,74 (с, 3H), 2,25 ~ 2,15 (м, 1H), 1,12 (д, 6H, J = 6,8 Гц).
Пример 4: Получение N-(3-(диэтиламино)пропил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
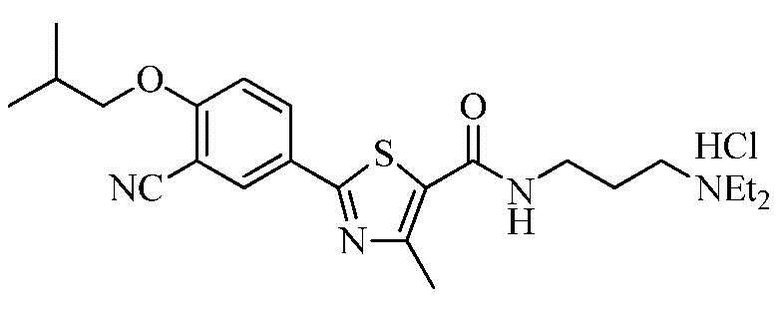
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 3-(диэтиламино)пропиламина вместо 2-(диэтиламино)этиламина. (выход: 44,4%)
ТСХ Rf = 0,17 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,27 (д, 1H, J = 2,4 Гц), 8,22 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,35 (д, 1H, J = 8,8 Гц), 4,03 (д, 2H, J = 6,4 Гц), 3,51 (т, 2H, J = 6,4 Гц), 3,34 ~ 3,22 (м, 6H), 2,73 (с, 3H), 2,25 ~ 2,15 (м, 1H), 2,15 ~ 2,05 (м, 2H), 1,37 (т, 6H, J = 7,2 Гц), 1,12 (д, 6H, J = 6,4 Гц).
Пример 5: Получение N-(4-метилгомопиперазино)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
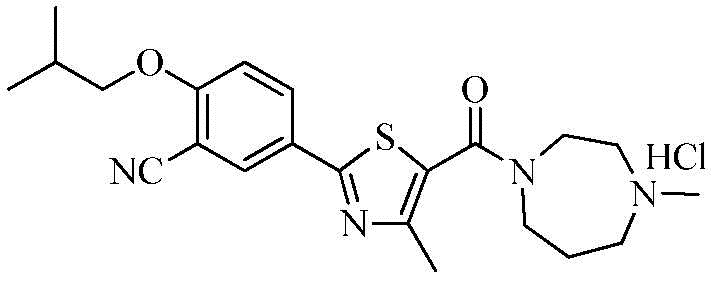
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования N-метилгомопиперазина вместо 2-(диэтиламино)этиламина. (выход: 45,3%)
ТСХ Rf = 0,39 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,23 (д, 1H, J = 2,4 Гц), 8,20 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,33 (д, 1H, J = 8,8 Гц), 4,32 ~ 4,18 (м, 1H), 4,02 (д, 2H, J = 6,4 Гц), 3,90 ~ 3,60 (м, 5H), 3,45 ~ 3,35 (м, 2H), 3,00 (с, 3H), 2,52 (с, 3H), 2,35 ~ 2,25 (м, 2H), 2,25 ~ 2,15 (м, 1H), 1,12 (д, 6H, J = 6,8 Гц).
Пример 6: Получение N-(4-(1-метил)пиперидинил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
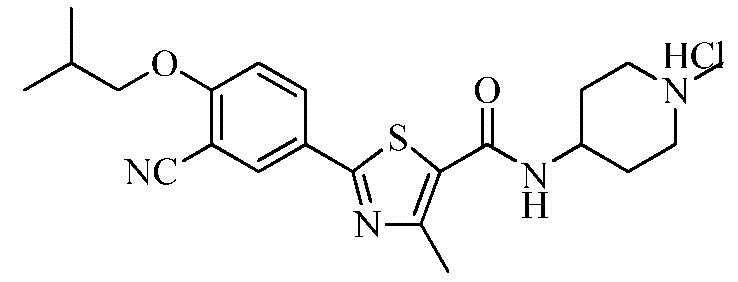
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 4-амино-1-метилпиперидина вместо 2-(диэтиламино)этиламина. (выход: 62,4%)
ТСХ Rf = 0,29 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,24 (д, 1H, J = 2,4 Гц), 8,20 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,33 (д, 1H, J = 8,8 Гц), 4,20 ~ 4,10 (м, 1H), 4,02 (д, 2H, J = 6,4 Гц), 3,65 ~ 3,55 (м, 2H), 3,25 ~ 3,15 (м, 2H), 2,97 (с, 3H), 2,68 (с, 3H), 2,35 ~ 2,25 (м, 2H), 2,25 ~ 2,15 (м, 1H), 2,05 ~ 1,90 (м, 2H), 1,12 (д, 6H, J = 6,8 Гц).
Пример 7: Получение N-(2-(4-морфолино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
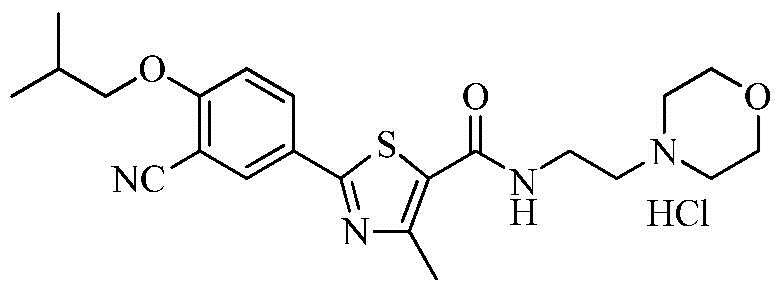
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 4-(2-аминоэтил)морфолина вместо 2-(диэтиламино)этиламина. (выход: 64,9%)
ТСХ Rf = 0,61 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,26 (д, 1H, J = 2,4 Гц), 8,22 (дд, 1H, J = 2,4 Гц, 9,2 Гц), 7,34 (д, 1H, J = 9,2 Гц), 4,12 (дд, 2H, J = 3,2, 12,8 Гц), 4,03 (д, 2H, J = 6,4 Гц), 3,92 ~ 3,85 (м, 2H), 3,85 ~ 3,78 (м, 2H), 3,73 (д, 2H, J = 12,8 Гц), 3,44 (т, 2H, J = 6,0 Гц), 3,30 ~ 3,20 (м, 2H), 2,74 (с, 3H), 2,25 ~ 2,15 (м, 1H), 1,12 (д, 6H, J = 6,8 Гц).
Пример 8: Получение N-(2-(1-пирролидино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
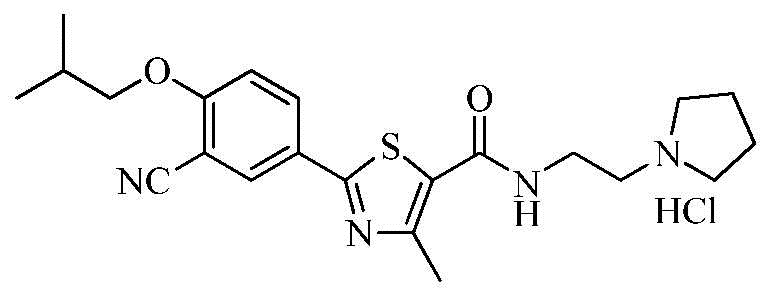
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 1-(2-аминоэтил)пирролидина вместо 2-(диэтиламино)этиламина. (выход: 59,5%)
ТСХ Rf = 0,14 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,26 (д, 1H, J = 2,4 Гц), 8,22 (дд, 1H, J = 2,4 Гц, 9,2 Гц), 7,35 (д, 1H, J = 8,8 Гц), 4,03 (д, 2H, J = 6,4 Гц), 3,88 ~ 3,80 (м, 2H), 3,77 (т, 2H, J = 6,0 Гц), 3,48 (т, 2H, J = 6,0 Гц), 3,25 ~ 3,15 (м, 2H), 2,75 (с, 3H), 2,25 ~ 2,15 (м, 3H), 2,15 ~ 2,05 (м, 2H), 1,12 (д, 6H, J = 6,8 Гц).
Пример 9: Получение N-(2-(1-пиперидино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
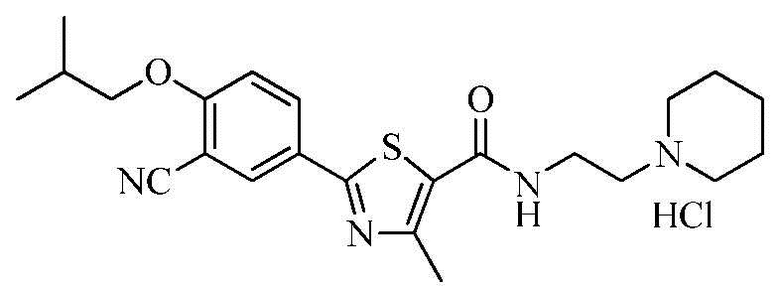
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 1-(2-аминоэтил)пиперидина вместо 2-(диэтиламино)этиламина. (выход: 72,7%)
ТСХ Rf = 0,33 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,25 (д, 1H, J = 2,4 Гц), 8,21 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,33 (д, 1H, J = 8,8 Гц), 4,03 (д, 2H, J = 6,4 Гц), 3,77 (т, 2H, J = 6,0 Гц), 3,73 (д, 2H, J = 11,6 Гц), 3,37 (т, 2H, J = 6,0 Гц), 3,10 ~ 3,00 (м, 2H), 2,73 (с, 3H), 2,25 ~ 2,15 (м, 1H), 2,05 ~ 1,95 (м, 2H), 1,93 ~ 1,83 (м, 3H), 1,62 ~ 1,53 (м, 1H), 1,12 (д, 6H, J = 6,8 Гц).
Пример 10: Получение N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
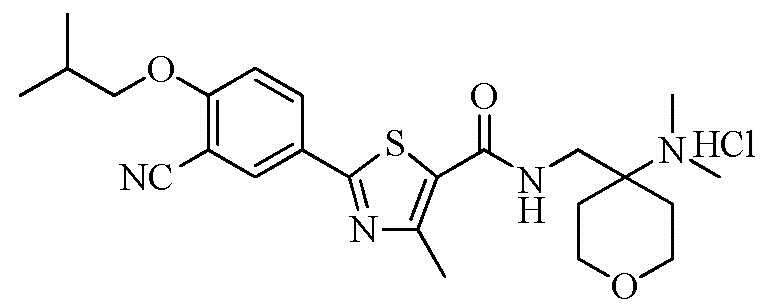
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 4-(аминометил)-N,N-диметилтетрагидро-2H-пиран-4-амина вместо 2-(диэтиламино)этиламина. (выход: 67,9%)
ТСХ Rf = 0,44 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, гексадейтеродиметилсульфоксид (ДМСО-D6)) δ 10,31 (т, 1H, J = 4,8 Гц), 8,67 (т, 1H, J = 6,4 Гц), 8,26 (д, 1H, J = 2,4 Гц), 8,20 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,40 (д, 1H, J = 8,8 Гц), 4,02 (д, 2H, J = 6,4 Гц), 3,95 ~ 3,85 (м, 2H), 3,83 (д, 2H, J = 6,4 Гц), 3,65 ~ 3,55 (м, 2H), 2,80 (с, 3H), 2,79 (с, 3H), 2,64 (с, 3H), 2,15 ~ 2,05 (м, 1H), 1,95 ~ 1,85 (м, 4H), 1,03 (д, 6H, J = 6,4 Гц).
Пример 11: Получение N-((1-(диметиламино)циклопентил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
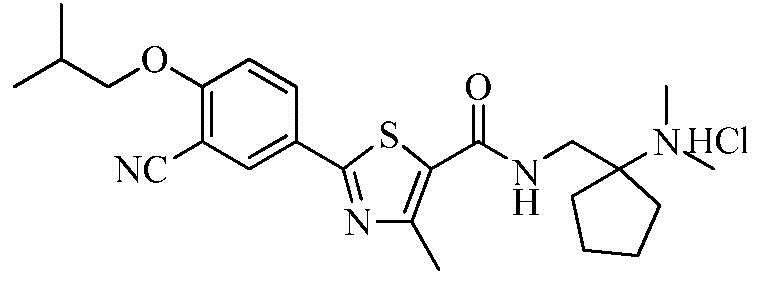
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 1-(аминометил)-N,N-диметилциклопентиламина вместо 2-(диэтиламино)этиламина. (выход: 72,9%)
ТСХ Rf = 0,34 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 10,73 (т, 1H, J = 4,8 Гц), 8,73 (т, 1H, J = 6,4 Гц), 8,26 (д, 1H, J = 2,4 Гц), 8,20 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,40 (д, 1H, J = 8,8 Гц), 4,02 (д, 2H, J = 6,4 Гц), 3,61 (д, 2H, J = 6,4 Гц), 2,81 (с, 3H), 2,80 (с, 3H), 2,64 (с, 3H), 2,15 ~ 2,05 (м, 1H), 2,00 ~ 1,90 (м, 4H), 1,80 ~ 1,70 (м, 4H), 1,02 (д, 6H, J = 6,8 Гц).
Пример 12: Получение N-((1-(диметиламино)циклогексил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
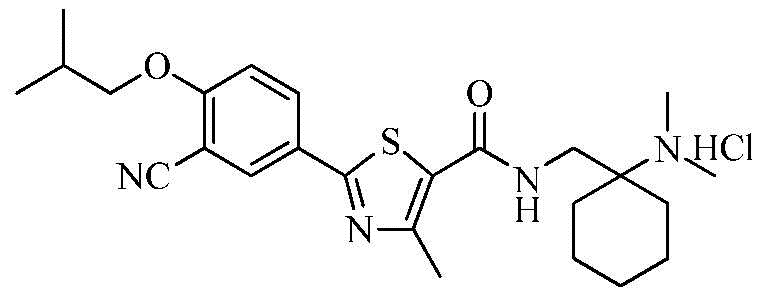
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 1-(аминометил)-N,N-диметилциклогексиламина вместо 2-(диэтиламино)этиламина. (выход: 58,3%)
ТСХ Rf = 0,31 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 9,90 (с, 1H), 8,56 (т, 1H, J = 6,0 Гц), 8,26 (д, 1H, J = 2,4 Гц), 8,20 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,40 (д, 1H, J = 8,8 Гц), 4,02 (д, 2H, J = 6,4 Гц), 3,74 (д, 2H, J = 6,4 Гц), 2,81 (с, 3H), 2,79 (с, 3H), 2,64 (с, 3H), 2,15 ~ 2,05 (м, 1H), 1,95 ~ 1,88 (м, 2H), 1,75 ~ 1,52 (м, 7H), 1,22 ~ 1,13 (м, 1H), 1,02 (д, 6H, J = 6,8 Гц).
Пример 13: Получение N-(3-(1-бензил)пирролидинил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
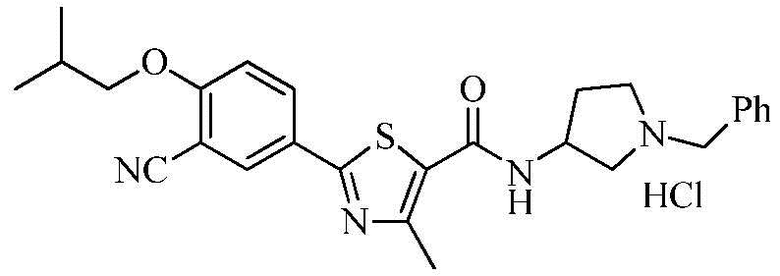
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 1-бензил-3-аминопирролидина вместо 2-(диэтиламино)этиламина. (выход: 43,4%)
ТСХ Rf = 0,41 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 11,20 (с, 1H), 8,73 (дд, 1H, J = 6,4, 26 Гц), 8,25 ~ 8,15 (м, 2H), 7,65 ~7,60 (м, 2H), 7,50 ~ 7,40 (м, 4H), 4,73 ~ 4,45 (м, 1H), 4,42 (дд, 2H, J = 6,0, 14 Гц), 4,02 (дд, 2H, J = 2,8, 6,4 Гц), 3,60 ~ 3,40 (м, 2H), 3,40 ~ 3,30 (м, 1H), 3,22 ~ 3,10 (м, 1H), 2,62 (д, 3H, J = 4,0 Гц), 2,15 ~ 2,05 (м, 1H), 1,02 (дд, 6H, J = 1,6, 6,8 Гц).
Пример 14: Получение N-(2-(2-(1-метил)пирролидино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
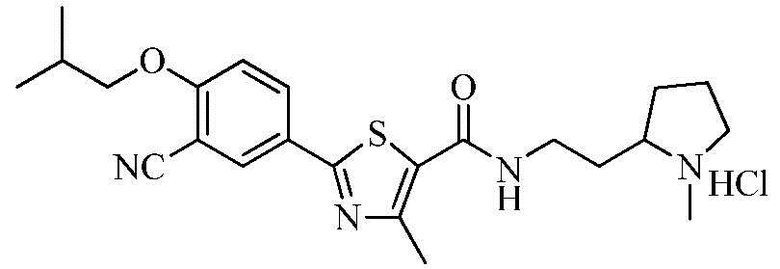
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 2-(2-аминоэтил)-1-метил-пирролидина вместо 2-(диэтиламино)этиламина. (выход: 85,3%)
ТСХ Rf = 0,14 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, D2O) δ 7,58 (дд, 1H, J = 2,4, 8,8 Гц), 7,51 (д, 1H, J = 2,4 Гц), 6,88 (д, 1H, J = 8,8 Гц), 3,72 (д, 2H, J = 6,8 Гц), 3,68 ~ 3,60 (м, 1H), 3,37 (т, 2H, J = 6,8 Гц), 3,32 ~ 3,25 (м, 1H), 3,27 (с, 2H), 3,15 ~ 3,05 (м, 1H), 2,87 (с, 3H), 2,35 ~ 2,25 (м, 1H), 2,25 ~ 2,15 (м, 1H), 2,15 ~ 1,92 (м, 3H), 1,85 ~ 1,70 (м, 2H), 0,92 (д, 6H, J = 6,8 Гц).
Пример 15: Получение N-((S)-2-(1-этил)пирролидинометил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
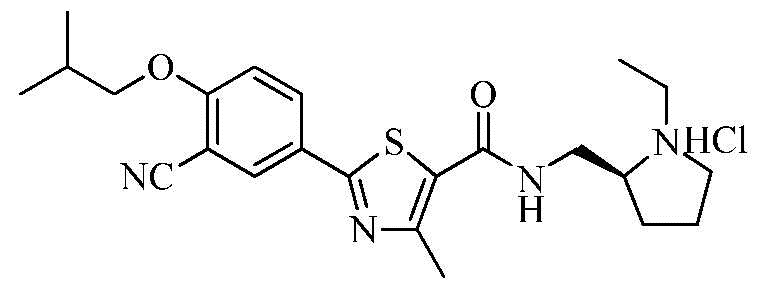
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования (S)-2-(аминометил)-1-этил-пирролидина вместо 2-(диэтиламино)этиламина. (выход: 63,6%)
ТСХ Rf = 0,24 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 10,39 (с, 1H), 8,74 (т, 1H, J = 5,6 Гц), 8,25 (д, 1H, J = 2,0 Гц), 8,19 (дд, 1H, J = 2,0 Гц, 9,2 Гц), 7,40 (д, 1H, J = 9,2 Гц), 4,01 (д, 2H, J = 6,4 Гц), 3,75 ~ 3,52 (м, 4H), 3,45 ~ 3,35 (м, 1H), 3,13 ~ 3,03 (м, 2H), 2,65 (с, 3H), 2,20 ~ 1,80 (м, 5H), 1,29 (т, 3H, J = 7,2 Гц), 1,02 (д, 6H, J = 6,8 Гц).
Пример 16: Получение N-(4-морфолинамино)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
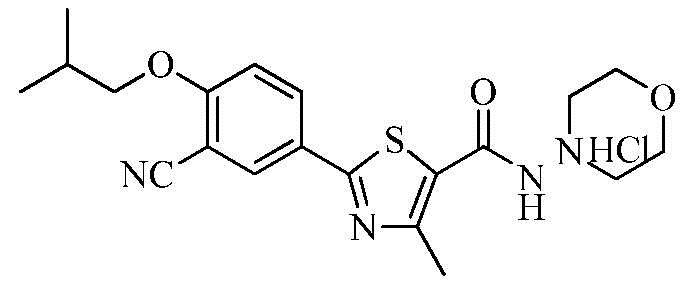
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 4-аминоморфолина вместо 2-(диэтиламино)этиламина. (выход: 59,9%)
ТСХ Rf = 0,69 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 9,10 (с, 1H), 8,35 (с, 1H), 8,30 ~ 8,20 (м, 1H), 7,35 ~ 7,40 (м, 1H), 4,01 (д, 2H, J = 6,4 Гц), 3,85 ~ 3,65 (м, 4H), 2,95 ~ 2,70 (м, 4H), 2,67 (с, 3H), 2,15 ~ 2,05 (м, 1H), 1,29 (т, 3H, J = 7,2 Гц), 1,03 (д, 6H, J = 6,8 Гц).
Пример 17: Получение N-(1-пиперидинамино)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
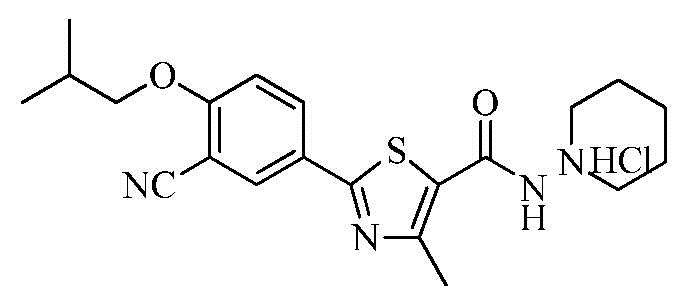
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 1, за исключением использования 1-аминопиперидина вместо 2-(диэтиламино)этиламина. (выход: 45,8%)
ТСХ Rf = 0,39 в 10% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 9,01 (с, 1H), 8,28 (д, 1H, J = 2,4 Гц), 8,23 (дд, 1H, J = 2,0, 8,8 Гц), 7,38 (д, 1H, J = 8,8 Гц), 4,01 (д, 2H, J = 6,4 Гц), 3,25 ~ 3,20 (м, 1H), 3,05 ~ 2,95 (м, 2H), 2,72 (с, 3H), 2,70 ~ 2,65 (м, 1H), 2,15 ~ 2,05 (м, 1H), 1,82 ~ 1,65 (м, 6H), 1,03 (д, 6H, J = 6,4 Гц)
Пример 18: Получение N-(2-(диизопропиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
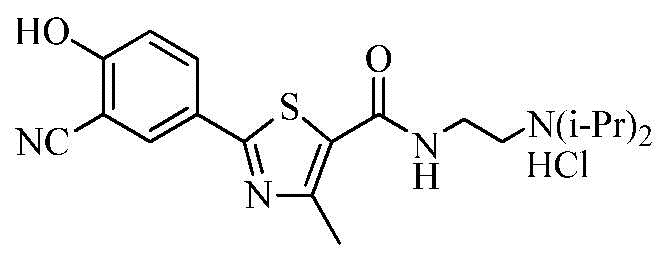
Смесь 2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоновой кислоты (3,00 г), гидроксибензотриазола (1,71 г), дициклогексилкарбодиимида (2,62 г) и диметилформамида (30 мл) перемешивали в течение 10 минут. N-метилморфолин (1,28 г) и 2-(диизопропиламино)этиламин (1,83 г) добавили к смеси, которую нагревали до 50°C и затем перемешивали в течение 3 часов. Реакционную смесь охладили до 0~5°C и затем фильтровали для отделения полученного твердого вещества. Фильтрат сконцентрировали при пониженном давлении. Полученный маслообразный концентрат растворили в 0,5N растворе соляной кислоты (60 мл) и очищенной воде (60 мл) и затем промыли этилацетатом (60 мл x 2 раза). Водный слой доводили до pH 7~8 гидроксидом натрия (1,2 г) и экстрагировали дихлорметаном (60 мл x 2 раза). Объединенный экстракт высушили над безводным сульфатом натрия и сконцентрировали под вакуумом с получением N-(2-(диизопропиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида. Соединение растворили в метаноле (10 мл) и затем туда добавили раствор 1N соляной кислоты в эфире (20 мл). Смесь нагрели до 40°C и затем туда добавили ацетон (30 мл). Смесь перемешивали в течение 1 часа и затем фильтровали. Полученное твердое вещество промыли ацетоном (10 мл) и затем высушили при 50~55°C с получением 4,17 г озаглавленного соединения. (выход: 85,5%)
ТСХ Rf = 0,20 в 20% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,18 (д, 1H, J = 2,4 Гц), 8,09 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,11 (д, 1H, J = 8,8 Гц), 3,90 ~ 3,82 (м, 2H), 3,74 (т, 2H, J = 6,8 Гц), 3,38 (т, 2H, J = 6,8 Гц), 2,73 (с, 3H), 1,46 (д, 12H, J = 6,8 Гц).
Пример 19: Получение N-(2-(диэтиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
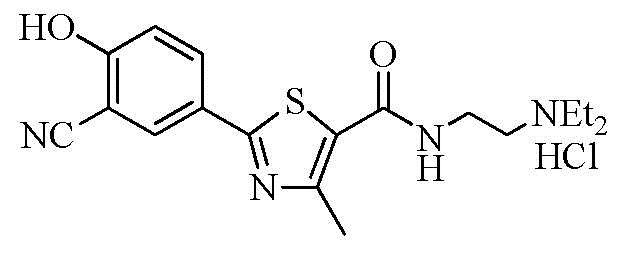
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 18, за исключением использования 2-(диэтиламино)этиламина вместо 2-(диизопропиламино)этиламина. (выход: 40,0%)
ТСХ Rf = 0,39 в 40% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 11,99 (с, 1H), 10,40 (с, 1H), 8,61 (т, 1H, J = 6,0 Гц), 8,13 (д, 1H, J = 2,4 Гц), 7,95 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,23 (д, 1H, J = 8,8 Гц), 3,64 (q, 2H, J = 6,4 Гц), 3,22 (q, 2H, J = 6,4 Гц), 3,20 ~ 3,12 (м, 4H), 2,64 (с, 3H), 1,25 (т, 6H, J = 7,2 Гц).
Пример 20: Получение N-(2-(диметиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
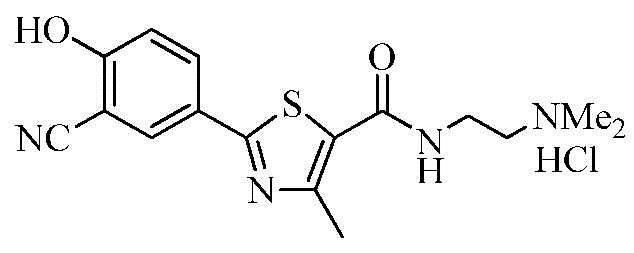
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 18, за исключением использования 2-(диметиламино)этиламина вместо 2-(диизопропиламино)этиламина. (выход: 83,7%)
ТСХ Rf = 0,18 в 40% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 11,97 (с, 1H), 10,19 (с, 1H), 8,52 (т, 1H, J = 6,0 Гц), 8,13 (д, 1H, J = 2,4 Гц), 8,05 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,22 (д, 1H, J = 8,8 Гц), 3,62 (q, 2H, J = 6,0 Гц), 3,25 (q, 2H, J = 6,0 Гц), 2,83 (с, 3H), 2,82 (с, 3H), 2,64 (с, 3H).
Пример 21: Получение N-(2-(4-морфолино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
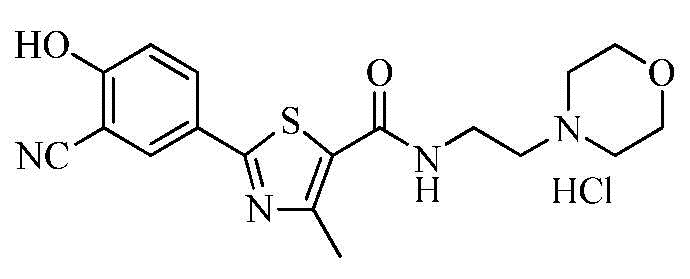
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 18, за исключением использования 4-(2-аминоэтил)морфолина вместо 2-(диизопропиламино)этиламина. (выход: 73,2%)
ТСХ Rf = 0,67 в 20% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 12,03 (с, 1H), 11,24 (с, 1H), 8,64 (т, 1H, J = 5,6 Гц), 8,13 (д, 1H, J = 2,4 Гц), 8,05 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,24 (д, 1H, J = 8,8 Гц), 4,00 ~ 3,80 (м, 4H), 3,68 (q, 2H, J = 6,0 Гц), 3,58 ~ 3,48 (м, 2H), 3,45 ~ 3,35 (м, 2H), 3,25 ~ 3,15 (м, 2H), 2,64 (с, 3H).
Пример 22: Получение N-((S)-2-(1-этил)пирролидинометил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
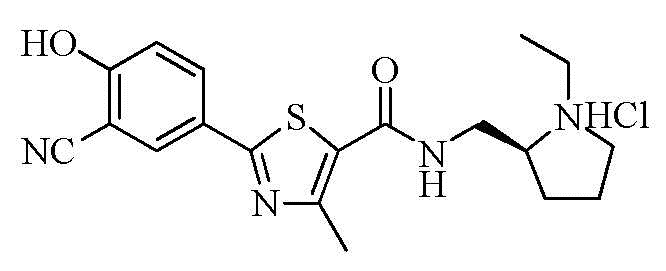
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 18, за исключением использования (S)-2-(аминометил)-1-этил-пирролидина вместо 2-(диизопропиламино)этиламина. (выход: 23,5%)
ТСХ Rf = 0,47 в 40% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 11,96 (с, 1H), 10,43 (с, 1H), 8,71 (т, 1H, J = 5,6 Гц), 8,14 (д, 1H, J = 2,4 Гц), 8,06 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,21 (д, 1H, J = 8,8 Гц), 3,75 ~ 3,52 (м, 4H), 3,45 ~ 3,35 (м, 1H), 3,13 ~ 3,03 (м, 2H), 2,64 (с, 3H), 2,20 ~ 2,10 (м, 1H), 2,05 ~ 1,80 (м, 3H), 1,29 (т, 3H, J = 7,2 Гц).
Пример 23: Получение N-(2-(диизопропиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида
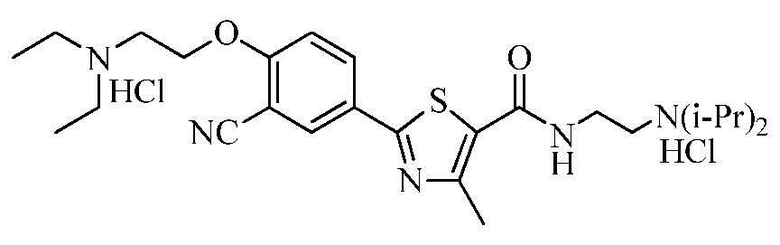
N-(2-(диизопропиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорид (3,17 г), полученный в примере 18, добавили к раствору гидроксида натрия (1,26 г), растворенного в очищенной воде (8 мл) и тетрагидрофуране (35 мл). К смеси добавили гидрохлорид 2-диэтиламиноэтилхлорида (1,30 г). Смесь перемешивали примерно при 70°C в течение 4 часов. Реакционную смесь охладили до комнатной температуры и затем водный слой отделили методом расслоения. Органический слой сконцентрировали при пониженном давлении, растворили в дихлорметане (50 мл) и затем водный слой отделили методом расслоения. Органический слой высушили над безводным сульфатом натрия, сконцентрировали под вакуумом и затем кристаллизовали диизопропиловым эфиром (50 мл). Полученный кристалл отфильтровали и затем высушили при 50~55°C с получением N-(2-(диизопропиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида. Соединение растворили в метаноле (5 мл) и затем туда добавили раствор 1N соляной кислоты в эфире (15 мл). Смесь нагрели до 40°C и затем туда добавили ацетон (30 мл). Смесь перемешивали в течение 1 часа и затем фильтровали. Полученное твердое вещество промыли ацетоном (10 мл) и затем сушили при 50~55°C с получением 2,50 г озаглавленного соединения. (выход: 60,0%)
ТСХ Rf = 0,33 в 20% MeOH в хлороформе
1H ЯМР (400 МГц, MeOH-d4) δ 8,34 (д, 1H, J = 2,4 Гц), 8,30 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,43 (д, 1H, J = 8,8 Гц), 4,65 (т, 2H, J = 4,8 Гц), 3,90 ~ 3,80 (м, 2H), 3,80 ~ 3,70 (м, 4H), 3,52 ~ 3,43 (м, 4H), 3,38 (т, 2H, J = 6,8 Гц), 2,73 (с, 3H), 1,50 ~ 1,46 (м, 18H)
1H ЯМР (400 МГц, ДМСО-D6) δ 10,90 (с, 1H), 10,14 (с, 1H), 8,77 (т, 1H, J = 5,6 Гц), 8,30 (д, 1H, J = 2,4 Гц), 8,24 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,44 (д, 1H, J = 9,2 Гц), 4,67 (т, 2H, J = 4,4 Гц), 3,70 ~ 3,60 (м, 6H), 3,30 ~ 3,15 (м, 6H), 2,68 (с, 3H), 1,40 ~ 1,40 (м, 18H).
Пример 24: Получение N-(2-(диэтиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида
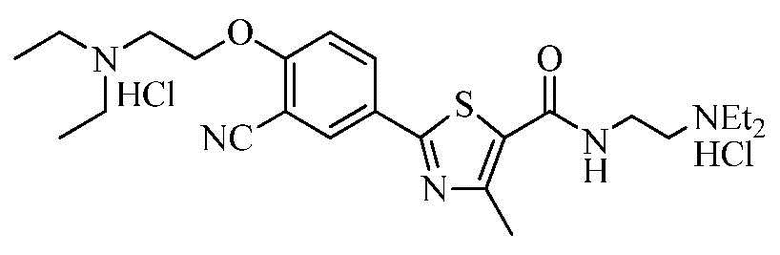
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 23, за исключением использования N-(2-(диэтиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида, полученного в примере 19. (выход: 46,3%)
ТСХ Rf = 0,30 в 20% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 10,54 (с, 1H), 10,31 (с, 1H), 8,65 (т, 1H, J = 5,2 Гц), 8,30 (д, 1H, J = 2,4 Гц), 8,25 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,44 (д, 1H, J = 8,8 Гц), 4,64 (с, 2H), 3,70 ~ 3,60 (м, 4H), 3,40 ~ 3,15 (м, 10H), 1,35 ~ 1,20 (м, 12H).
Пример 25: Получение N-(2-(диметиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида
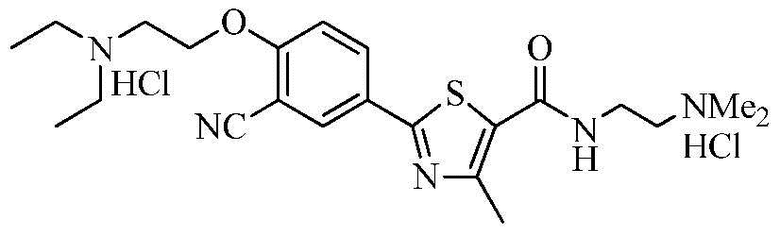
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 23, за исключением использования N-(2-(диметиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида, полученного в примере 20. (выход: 55,5%)
ТСХ Rf = 0,25 в 20% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 10,80 (с, 1H), 10,45 (с, 1H), 8,89 (т, 1H, J = 5,2 Гц), 8,30 (д, 1H, J = 2,4 Гц), 8,25 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,44 (д, 1H, J = 8,8 Гц), 4,66 (т, 2H, J = 4,8 Гц), 3,65 ~ 3,55 (м, 4H), 3,30 ~ 3,20 (м, 6H), 2,83 (с, 3H), 2,82 (с, 3H), 2,66 (с, 3H), 1,35 ~ 1,28 (м, 6H).
Пример 26: Получение N-(2-(4-морфолино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида
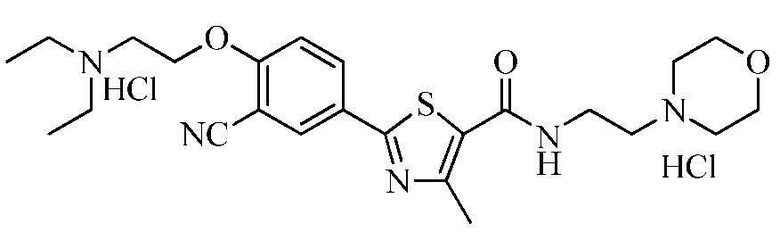
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 23, за исключением использования N-(2-(4-морфолино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида, полученного в примере 21 (выход: 52,1%)
ТСХ Rf = 0,40 в 20% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 11,24 (с, 1H), 10,86 (с, 1H), 8,70 (т, 1H, J = 5,6 Гц), 8,30 (д, 1H, J = 2,4 Гц), 8,24 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,44 (д, 1H, J = 8,8 Гц), 4,66 (т, 2H, J = 4,8 Гц), 4,05 ~ 3,95 (м, 2H), 3,90 ~ 3,80 (м, 4H), 3,75 ~ 3,65 (м, 2H), 3,65 ~ 3,60 (м, 2H), 3,60 ~ 3,50 (м, 2H), 3,45 ~ 3,25 (м, 6H), 3,20 ~ 3,10 (м, 2H), 2,66 (с, 3H), 1,30 (т, 6H, J = 7,2Гц).
Пример 27: Получение N-((S)-2-(1-этил)пирролидинометил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида
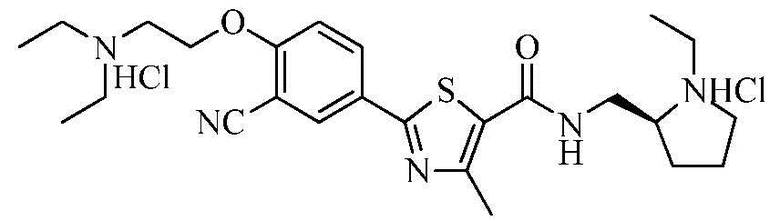
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 23, за исключением использования N-((S)-2-(1-этил)пирролидинометил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида, полученного в примере 22. (выход: 28,8%)
ТСХ Rf = 0,08 в 20% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 10,70 (с, 1H), 10,66 (с, 1H), 8,80 (т, 1H, J = 5,6 Гц), 8,31 (д, 1H, J = 2,4 Гц), 8,25 (дд, 1H, J = 2,4 Гц, 8,8 Гц), 7,45 (д, 1H, J = 8,8 Гц), 4,65 (т, 2H, J = 4,4 Гц), 3,80 ~ 3,50 (м, 6H), 3,45 ~ 3,35 (м, 5H), 3,15 ~ 3,05 (м, 2H), 2,67 (с, 3H), 2,20 ~ 2,10 (м, 1H), 2,05 ~ 1,80 (м, 3H), 1,35 ~ 1,25 (м, 9H).
Пример 28: Получение N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида
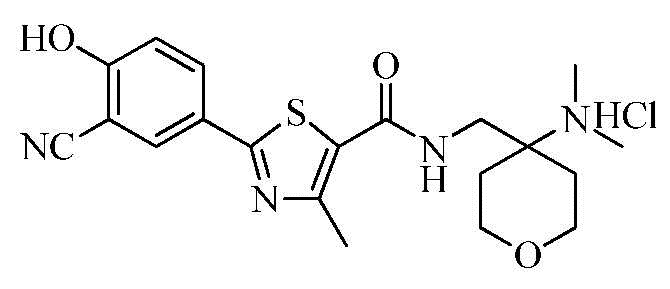
Смесь 2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоновой кислоты (5,00 г), гидроксибензотриазола (2,86 г), дициклогексилкарбодиимида (4,36 г) и диметилформамида (45 мл) перемешивали в течение 10 минут. N-метилморфолин (2,14 г) и 4-(аминометил)-N,N-диметилтетрагидро-2H-пиран-4-амин (3,50 г) добавили к смеси, которую нагревали до 50°C и затем перемешивали в течение 4 часов. Реакционную смесь охладили до 0~5°C и затем фильтровали для отделения полученного твердого вещества. Фильтрат сконцентрировали при пониженном давлении с получением N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида. Соединение растворили в 2N растворе соляной кислоты (25 мл) и очищенной воде (100 мл), промыли этилацетатом (100 мл) и затем фильтровали. Полученный фильтрат перемешивали в течение 1 часа и затем фильтровали. Полученное твердое вещество промыли холодной очищенной водой (10 мл) и затем туда добавили ацетон (50 мл). Смесь перемешивали в течение 30 минут и затем фильтровали. Полученное твердое вещество промыли ацетоном (20 мл) и затем сушили при 50~55°C с получением 7,10 г озаглавленного соединения. (выход: 84,6%)
ТСХ Rf = 0,48 в 20% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 11,98 (с, 1H), 10,48 (с, 1H), 8,67 (т, 1H, J = 6,4 Гц), 8,15 (д, 1H, J = 2,4 Гц), 8,06 (дд, 1H, J = 6,4 Гц, 8,8 Гц), 7,21 (д, 1H, J = 8,8 Гц), 3,92 ~ 3,85 (м, 2H), 3,83 (д, 2H, J = 6,4 Гц), 3,68 ~ 3,58 (м, 2H), 2,79 (с, 6H), 2,63 (с, 3H), 2,00 ~ 1,88 (м, 4H).
Пример 29: Получение N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида
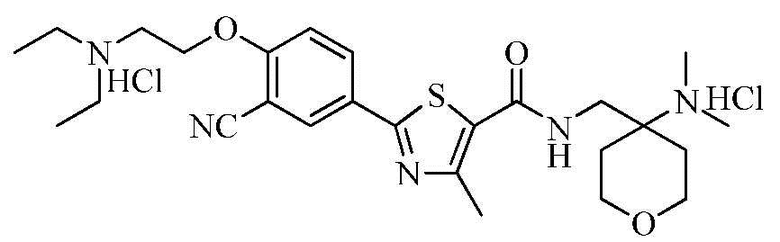
Озаглавленное соединение приготавливали в соответствии с теми же способами, что и в примере 23, за исключением использования N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида, полученного в примере 28. (выход: 74,0%)
ТСХ Rf = 0,38 в 20% MeOH в хлороформе
1H ЯМР (400 МГц, ДМСО-D6) δ 10,45 ~ 10,35 (м, 2H), 8,72 (т, 1H, J = 6,4 Гц), 8,33 (д, 1H, J = 2,4 Гц), 8,27 (дд, 1H, J = 6,4 Гц, 8,8 Гц), 7,45 (д, 1H, J = 8,8 Гц), 4,64 (т, 2H, J = 4,4 Гц), 3,98 ~ 3,85 (м, 2H), 3,84 (д, 2H, J = 6,4 Гц), 3,70 ~ 3,60 (м, 4H), 3,35 ~ 3,25 (м, 4H), 2,80 (д, 6H, J = 4,8Гц), 2,65 (с, 3H), 1,95 ~ 1,88 (м, 4H), 1,30 (т, 6H, J = 7,2 Гц).
Тестовый пример 1: Индуцирующая аутофагию активность
Индуцирующую аутофагию активность соединений согласно настоящему изобретению измеряли на клетках Hela (корейский банк клеточных линий), используя набор для обнаружения аутофагии (DAPGreen Autophagy detection (Dojindo, D676-10)) в соответствии с инструкциями производителя. В частности, клетки HeLa (8 X 104 клеток) вместе с минимальной эссенциальной средой Игла, модифицированной по способу Дульбекко (DMEM) FluroBrite (FluroBrite DMEM), содержащей 10% фетальной бычьей сыворотки (FBS), засевали в каждую лунку 96-луночного планшета и затем инкубировали на протяжении ночи при 37°C в CO2-инкубаторе, чтобы стабилизировать клетки. Раствор DAPGreen (каталог, D676-10, Dojindo) (0,1 ммоль в диметилсульфоксиде) разбавили до 1/1000 в среде (FluroBrite DMEM, содержащей 10% FBS) для приготовления среды, содержащей 0,1 мкмоль DAPGreen (среды, содержащей DAPGreen/FBS). После того, как была удалена надосадочная жидкость из каждой лунки, к которой были прикреплены клетки монослойной культуры, каждую лунку промыли FluroBrite DMEM. Среду, содержащую DAPGreen/FBS, полученную в вышеизложенном (100 мкл), добавили в каждую лунку, которую инкубировали в CO2-инкубаторе в течение 30 минут. Значения флуоресценции (длина волны возбуждения (EX): 450 нм /длина волны детекции (EM): 535 нм) измерили в начальный момент и в момент 30 минут, исчисляемые с момента обработки DAPGreen, соответственно. Значение их разности было принято за базовое значение поглощения клеток.
Подобно измерению базового значения поглощения клеток, надосадочную жидкость удалили из каждой лунки, к которой были прикреплены клетки монослойной культуры, и затем каждую лунку промывали FluroBrite DMEM. Соединения примеров, контроль (2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоновая кислота) и положительный контроль (рапамицин) растворили в диметилсульфоксиде, соответственно. Каждый раствор разбавили до концентраций 500 нмоль и 1000 нмоль в среде, содержащей DAPGreen//FBS (100 мкл) и затем обрабатывали с этим. Для каждой концентрации тестировали три лунки (n = 3). После инкубирования при 37°C в течение 4 часов измерили значения флуоресценции (EX: 450 нм / EM: 535 нм). После того, как базовое значение поглощения вычитали из значения флуоресценции каждой лунки, каждое полученное значение нормализовали на основании среднего значения поглощения в группе обработки диметилсульфоксидом.
Относительные значения флуоресценции в соответствии с обработками тестируемых веществ (1000 нм и 500 нм) и контроля (2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоновая кислота: 1000 нм и 500 нм) рассчитали на основании значений флуоресценции в соответствии с обработкой положительного контроля (рапамицин: 1000 нм и 500 нм). Их результаты показаны ниже в таблице 1. В таблице 1 относительное значение флуоресценции равно 1, если индуцирующая аутофагию активность тестируемого вещества, приготовленного в примере, равна таковой у положительного контроля. Относительное значение флуоресценции превышает 1, если тестируемое вещество, приготовленное в примере, показывает более высокую индуцирующую аутофагию активность, чем положительный контроль, в то время, как относительное значение флуоресценции не превышает 1, если тестируемое вещество, приготовленное в примере, показывает более низкую индуцирующую аутофагию активность, чем положительный контроль.
Таблица 1
Относительное значение флуоресценции (инкубация в течение 4 часов)
Из результатов в вышеизложенной таблице 1 можно ведить, что при инкубации в течение 4 часов после лечения тестируемыми веществами, соединения согласно настоящему изобретению показывали индуцирующую аутофагию активность, равную или выше, чем положительный контроль (т.е. рапамицин), и показывали в высшей степени превосходную индуцирующую аутофагию активность, по сравнению с 2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоновой кислотой, содержащей группу карбоновой кислоты. Следовательно, соединения согласно настоящему изобретению проявляют превосходную индуцирующую аутофагию активность, благодаря чему могут эффективно применяться для профилактики и лечения различных заболеваний, связанных с аутофагией, включая нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и т.д.
Тестовый пример 2: Анализ формирования аутофагической везикулы
Активацию лизосомального пути аутофагии (ALP) соединениями согласно настоящему изобретению проверяли путем измерения степени формирования аутофагической везикулы в клетках нейронов с помощью набора для обнаружения аутофагии CYTO-ID™ (ENZ-51031-K200, Enzo Life Sciences, Inc.). Клетки нейронов (т.е. первичные клетки кортикальных нейронов мыши) выделены из мыши с использованием способа ферментативного гидролизата. В частности, первичные клетки кортикальных нейронов мыши были выделены путем инкубирования тканей коры головного мозга, полученных от 16-дневных эмбрионов мышей C57BL6, при 37°C в течение 30 минут с 20 ед/мл папаина (Worthington Biochemical Corporation, LK003176) и 0,005% дезоксирибонуклеазой I (Worthington Biochemical Corporation, LK003170).
Первичные клетки кортикальных нейронов мыши (8 X 104 клеток) и среду (культуральную среду для нейрональных клеток, содержащую 2 ммоль L-глутамина (Gibco, 25030-081), добавку N2 (Gibco, 17502-048), добавку B27 (Gibco, 17504-044) и 50 мкг/мл пенициллина-стрептомицина (Gibco, 15140-122)) добавили в каждую лунку 96-луночного планшета и затем инкубировали на протяжении ночи при 37°C в CO2-инкубаторе. Соединением примера 10 (5 мкмоль), соединением примера 29 (5 мкмоль) и рапамицином (10 мкмоль), соответственно, воздействовали на клетки, которые инкубировали при 37°C в CO2-инкубаторе в течение 24 часов. После того, как каждую лунку промыли однократно аналитическим буферным раствором (Enzo Life Science, ENZ-51031-K200), воздействовали красителем для аутофагии Cyto-ID 0,2 мкл и Hoechst 33342 0,1 мкл на клетки, которые инкубировали при 37°С в CO2-инкубаторе в течение 30 минут. После того, как каждую лунку промыли однократно аналитическим буферным раствором (Enzo Life Science, ENZ-51031-K200), значения флуоресценции Cyto-ID (EX: 480 нм, EM: 530 нм) и Hoechst 33342 (EX: 340 нм, EM: 480 нм) соответственно, измерили флуорометром. Для каждой группы тестировали три лунки. После того, как была осуществлена нормализация посредством деления значения флуоресценции Cyto-ID каждой лунки на значение флуоресценции Hoechst 33342, каждую относительную интенсивность рассчитывали, положив относительную интенсивность необработанного контроля за 1. Соответствующие результаты показаны ниже в таблице 2.
Таблица 2
Значение флуоресценции Cyto-ID (относительная интенсивность)
Из результатов в вышеизложенной таблице 2 можно видеть, что соединения примеров 10 и 29 значительно увеличивают образование аутофагических везикул даже в концентрации 1/2 по сравнению с положительным контролем (т.е. рапамицином). Следовательно, соединения согласно настоящему изобретению проявляют превосходную индуцирующую аутофагию активность, благодаря чему могут эффективно применяться для профилактики и лечения различных заболеваний, связанных с аутофагией, включая нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и т.д.
Тестовый пример 3: Анализ аутофагического потока
Клетки N2a (ATCC, CCL-131, клеточная линия нейробластомы мыши) (3 X 104 клеток) и среду (DMEM (среду Игла, модифицированную по Дульбекко, Gibco,11995-065), дополненную 5% термоинактивированной фетальной бычьей сывороткой, FBS; Gibco, 16000-044) и 50 мкг/мл пенициллина-стрептомицина (Gibco, 15140-122) добавили в каждую лунку 6-луночного планшета и инкубировали на протяжении ночи при 37°C в CO2-инкубаторе. Соединением примера 29 (5 мкмоль) и рапамицином (10 мкмоль), соответственно, воздействовали и затем инкубировали при 37°C в CO2-инкубаторе в течение 24 часов. Три лунки в каждой группе являлись объектом для блока для электронного микроскопа, при этом осуществляли префиксацию, постфиксацию, обезвоживание, замещение и заливку в соответствии с методом заключения объекта в блок эпоксидной смолы для приготовления ультратонкого среза. После того, как приготовили ультратонкие срезы с толщиной 70 нм, осуществляли окрашивание тяжелыми металлами (уранилацетатом, цитратом свинца) на каждом срезе, который фотографировали с помощью метода получения мозаичного изображения для получения изображения клеток в целом. Результаты получения изображения под электронным микроскопом показаны на ФИГ. 1. Кроме того, результаты, полученные путем измерения количества аутофагий в целом (аутофагических везикул), аутофагосом и аутолизосом на клетку, в зависимости от экспериментальной группы, показаны в таблице 3. В Таблице 3 ниже контроль означает необработанный контроль.
Таблица 3
Количество аутофагий на клетку
Из результатов в вышеизложенной таблице 3 можно видеть, что соединение примера 29 значительно увеличивает образование аутофагических везикул, в частности, образование аутолизосом даже в концентрации 1/2 по сравнению с положительным контролем (т.е. рапамицином). Следовательно, соединения согласно настоящему изобретению проявляют превосходную индуцирующую аутофагию активность, благодаря чему могут эффективно применяться для профилактики и лечения различных заболеваний, связанных с аутофагией, включая нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и т.д.
Тестовый пример 4: Оценка активностей, улучшающих функцию печени при пероральном введении на модели поражения печени (1)
Соединения согласно настоящему изобретению вводили перорально самцам крыс линии Спраг-Доули (SD) с поражением печени, вызванным диметилнитрозамином (ДМН), в течение 3 недель, чтобы оценить активность, улучшающую функцию печени. В частности, семинедельных самцов крыс SD (Orient Bio, Корея) помещали в лабораторные условия при комнатной температуре на 7 дней. Наблюдали общие симптомы и затем для эксперимента использовали только здоровых животных. Крыс разделили на 4 группы (n = 5 для каждой группы), т.е. нормальную контрольную группу, группу, которой вводили только ДМН, и группы, которым вводили как соединение согласно настоящему изобретению (соединения примера 2 или 23), так и ДМН. ДМН растворяли в очищенной воде и затем вводили внутрибрюшинно в дозе 10 мг/кг в течение 3 дней в неделю подряд (3 раза в неделю в течение 4 недель). Пробы крови собирали в день 3 после завершения первой недели индуцирования поражения печени и затем измеряли значения сывороточной АЛТ (аланин-трансаминазы) и значения сывороточной АСТ (аспартат-трансаминазы), чтобы подтвердить поражение печени, соответственно. Соединения согласно настоящему изобретению вводили в течение 3 недель, а именно, со дня 4 после завершения индуцирования поражения печени первой недели до периода введения ДМН. Соединения согласно настоящему изобретению растворили в очищенной воде и затем вводили перорально с помощью перорального зонда в дозе 25 мг/кг один раз в день в течение 3 недель. Пробы крови собирали в день 0 (через 3 дня после завершения первой недели индуцирования поражения печени) и в день 7, 14 и 21 после введения тестируемых веществ. Собранную кровь впрыскивали в вакуумную пробирку, содержащую активатор свертывания, и затем выдерживали при комнатной температуре в течение примерно 20 минут, чтобы коагулировать каждый образец крови. После центрифугирования в течение 10 минут полученные сыворотки подвергали биохимическим анализам крови. Экспериментальные способы приведены на ФИГ. 2. Результаты, полученные путем измерений, подвергли групповому сравнению путем проведения параметрических множественных сравнений или непараметрических множественных сравнений с помощью системы программного обеспечения высшей производительности (SPSS). Если P < 0,05, это было признано статистически значимым.
Значения сывороточных АЛТ и АСТ, полученные при осуществлении биохимических анализов крови, как описано выше, показаны в таблицах 4 и 5 ниже.
Таблица 4
Таблица 5
Как можно видеть из результатов таблиц 4 и 5, значения сывороточных АЛТ и АСТ в группе, которой вводили ДМН, увеличились соответственно примерно в 3 раза и примерно в 2 раза через 3 недели. Тем не менее, в группах, которым вводили как соединение согласно настоящему изобретению, так и ДМН, значения АЛТ через 3 недели после введения составили 101,04 и 117,02 (то есть уменьшились на 35% и 25% по сравнению с таковыми у группы, которой вводили ДМН); и значения АСТ через 3 недели после введения составили 203,44 и 231,54 (то есть уменьшились на 32% и 23% по сравнению с таковыми у группы, которой вводили ДМН). Таким образом, можно подтвердить, что соединения согласно настоящему изобретению обладают превосходной ингибирующей активностью в отношении фиброза печени.
Тестовый пример 5: Оценка активности, улучшающей функцию печени при пероральном введении на модели поражения печени (2)
Соединения согласно настоящему изобретению вводили перорально самцам крыс SD с поражением печени, вызванным диметилнитрозамином (ДМН), в течение 4 недель, чтобы оценить активность, улучшающую функцию печени. В частности, семинедельных самцов крыс SD (Orient Bio, Корея) помещали в лабораторные условия при комнатной температуре на 7 дней. Наблюдали общие симптомы и затем для эксперимента использовали только здоровых животных. Крыс разделили на 5 групп (n = 10 для каждой группы), т. е. нормальную контрольную группу, группу, которой вводили только ДМН, и группу, которой вводили как соединение согласно настоящему изобретению (соединение примера 23, 26 или 29), так и ДМН. ДМН растворяли в очищенной воде и затем вводили внутрибрюшинно в дозе 10 мг/кг в течение 3 дней подряд в неделю (3 раза в неделю в течение 4 недель). Пробы крови собирали после завершения четвертой недели индуцирования поражения печени и затем измерили значения сывороточной АЛТ (аланинтрансаминазы) и сывороточной АСT (аспартаттрансаминазы), чтобы подтвердить поражение печени. Соединения согласно настоящему изобретению вводили в течение 4 недель, со дня 1 после завершения четвертой недели индуцирования поражения печени. Соединения согласно настоящему изобретению растворили в очищенной воде и затем вводили перорально с помощью перорального зонда в дозе 25 мг/кг один раз в день в течение 4 недель. Пробы крови собирали в День 0 (через 1 день после завершения четвертой недели индуцирования поражения печени) и в день 7, 14, 21 и 28 после введения тестируемых веществ. Собранную кровь впрыскивали в вакуумную пробирку, содержащую активатор свертывания, и затем выдерживали при комнатной температуре в течение примерно 20 минут, чтобы коагулировать каждый образец крови. После центрифугирования в течение 10 минут полученные сыворотки подвергали биохимическим анализам крови. Кроме того, через 24 часа после последнего введения осуществили вскрытие для гепатэктомии и фиксацию для приготовления образцов ткани, соответственно. После приготовления предметных стекол с образцами тканей вслед за этим осуществляли окрашивание гематоксилином и эозином (H&E) для микроскопического исследования поражений на ткани печени и инфильтраций воспалительных клеток в тканях печени. Кроме того, на них также провели трихромное окрашивание по Массону для микроскопического исследования отложений коллагенового волокна в тканях печени. Экспериментальные способы приведены на ФИГ. 3. Результаты, полученные путем измерений, подвергли групповому сравнению путем проведения параметрических множественных сравнений или непараметрических множественных сравнений с помощью SPSS. Если P < 0,05, это было признано статистически значимым. Значения сывороточных АЛТ и АСТ, полученные при осуществлении биохимических анализов крови, как описано выше, показаны в таблицах 6 и 7.
Таблица 6
Таблица 7
Как можно видеть из результатов таблиц 6 и 7, значения сывороточных АЛТ и АСТ в группе, которой вводили ДМН, увеличились соответственно примерно в 4-5 раз и примерно в 3-4 раза через 4 недели. Тем не менее, в группах, которым вводили как соединение согласно настоящему изобретению, так и ДМН, значения АЛТ через 4 недели после введения составили 52,93 ~ 59,12 (то есть уменьшились на 9,9 ~ 19% по сравнению с таковыми у группы, которой вводили ДМН); и значения АСТ через 4 недели после введения составили 114,45 ~ 125,59 (то есть уменьшились на 21 ~ 28% по сравнению с таковыми у группы, которой вводили ДМН). В частности, группа, получавшая соединение примера 26, показала значение АСТ, эквивалентное таковому нормальной контрольной группы, на 4 неделе после введения. Таким образом, можно подтвердить, что соединения согласно настоящему изобретению обладают превосходным лечебным действием в отношении фиброза печени. Кроме того, результаты, полученные путем приготовления образцов ткани печени посредством вскрытия нормальной контрольной группы, группы, которой вводили ДМН (в течение 4 недель) и группы, которой вводили соединение согласно настоящему изобретению (соединение примера 23 или 26) и ДМН, и затем осуществления окрашивания гематоксилином и эозином (H&E) и трихромного окрашивания по Массону, как описано выше, показаны на ФИГ. 4. Исходя из результатов на ФИГ. 4 измерили степень поражения ткани печени и инфильтрацию воспалительных клеток в тканях печени. И кроме того, исходя из результатов на ФИГ. 4 измерили степень отложения коллагенового волокна в тканях печени. Результаты, соответственно, показаны в таблице 8 ниже.
Таблица 8
Как можно видеть из результатов H&E окрашивания (ФИГ. 4) и таблицы 8, поражение ткани печени и инфильтрация воспалительных клеток в тканях печени в группе, которой вводили ДМН, соответственно увеличилось примерно в 17 раз и примерно в 5 раз через 4 недели по сравнению с таковыми у нормальной контрольной группы. Тем не менее, в группе, которой вводили как соединение согласно настоящему изобретению (соединение примера 23 или 26), так и ДМН, поражения в ткани печени и инфильтрация воспалительных клеток в тканях печени составили 259,14 ~ 319,14 клеток/1000 клеток и 90,00 ~ 101,43 клеток/мм2, соответственно (т.е. улучшены примерно на 43 ~ 54% и 46 ~ 52%, по сравнению с таковыми у группы, которой вводили ДМН). И кроме того, как можно увидеть из результатов трихромного окрашивания по Массону (ФИГ. 4) и таблицы 8, отложение коллагенового волокна в тканях печени в группе, которой вводили ДМН, увеличились примерно в 12 раз через 4 недели по сравнению с таковым у нормальной контрольной группы. Тем не менее, в группе, которой вводили как соединение согласно настоящему изобретению (соединение примера 23 или 26), так и ДМН, отложение коллагенового волокна в тканях печени составило 10,75 ~ 12,44 %/мм2 (то есть уменьшилось примерно на 57 ~ 63%, по сравнению с таковым у группы, которой вводили ДМН). Таким образом, можно подтвердить, что соединения согласно настоящему изобретению (соединения примеров 23 и 26) обладают превосходной ингибирующей активностью в отношении фиброза печени.
Тестовый пример 6: Анализ ингибирующей активности в отношении апоптоза нейронов по действием бета-амилоида
Ингибирующие активности соединений согласно настоящему изобретению в отношении апоптоза нейронов под действием бета-амилоида, известного как основное цитотоксическое вещество при нейродегенеративных заболеваниях, проанализировали на клетках N2a (ATCC, CCL-131, клеточная линия нейробластомы мыши) и клетках нейронов (первичные клетки кортикальных нейронов мыши), выделенных также, как и в тестовом примере 2. Среды, используемые в тестовом примере 3 и тестовом примере 2 использовали в качестве среды для клеток N2a и в качестве среды для клеток нейронов, соответственно. В частности, клетки N2a (3 X 104 клеток) и клетки нейронов (8 X 104 клеток) вместе с соответствующей средой добавили в каждую лунку 96-луночного планшета и затем инкубировали на протяжении ночи при 37°C в CO2-инкубаторе. Клетки обрабатывали соединением примера 10 и соединением примера 29 в концентрациях 5 мкмоль и 500 нмоль, соответственно, по истечении 30 минут от этого обрабатывали 20 мкмоль бета-амилоида, а затем инкубировали при 37°С в CO2-инкубаторе в течение 72 часов. Клетки обрабатывали 10 мкл реагента CCK-8 (Enzo Life Science, ALX-850-039-KI02) и затем инкубировали при 37°С в CO2-инкубаторе в течение 2 часов. Три лунки тестировали для каждой группы и каждую абсорбцию измеряли при 450 нм. Значение абсорбции необработанной контрольной группы было принято за 100% жизнеспособность клеток. Жизнеспособность клеток каждой группы рассчитывали, исходя из соответствующих значений абсорбции. Результаты показаны в таблице 9 ниже. В таблице 9 контрольная группа обозначает группу, обработанную только 20 мкмоль бета-амилоида.
Таблица 9
Исходя из результатов таблицы 9, можно подтвердить, что как соединение примера 10, так и соединение примера 29 в значительной степени ингибируют цитотоксичность, вызванную обработкой бета-амилоидом. Поэтому соединение согласно настоящему изобретению можно эффективно применять для профилактики и лечения различных нейродегенеративных заболеваний, связанных с бета-амилоидом.
Тестовый пример 7: Анализ ингибирующей активности в отношении митохондриального повреждения под действием ротенона
Ингибирующую активность соединений согласно настоящему изобретению в отношении митохондриального повреждения под действием ротенона, известного как основное цитотоксическое вещество при нейродегенеративных заболеваниях, анализировали на клетках N2a (ATCC, CCL-131, клеточная линия нейробластомы мыши). В частности, клетки N2a (3 X 104 клеток) и среду (DMEM (среду Игла в модификации Дульбекко, Gibco,11995-065), дополненную 5% термоинактивированной фетальной бычьей сывороткой (FBS; Gibco, 16000-044), и 50 мкг/мл пенициллина-стрептомицина (Gibco, 15140-122) добавили в каждую лунку XF24-луночного культурального планшета и затем инкубировали на протяжении ночи при 37°С в CO2-инкубаторе. Клетки обрабатывали соединением примера 10 (5 мкмоль), по истечении 30 минут от этого обрабатывали 10 мкмоль ротенона и затем инкубировали при 37°С в CO2-инкубаторе в течение 24 часов. После обработки тест-набором XF Cell Mito Stress Test Kit (Seahorse Bioscience, 103015-100) в соответствии с инструкциями производителя измерили уровень митохондриального дыхания, используя анализатор XF24 Extracellular Flux Analyzer (Seahorse Bioscience). Для каждой группы тестировали четыре лунки, измеряли основное дыхание, выработку АТФ, максимальное дыхание и резервную дыхательную емкость. Уровни каждого митохондриального дыхания были нормализованы по значению количественного показателя белка. Основное дыхание необработанной контрольной группы было принято за 100%, и остальные значения показаны. Результаты показаны в таблице 10 ниже. В таблице 10 контрольная группа представляет собой группу, обработанную только 10 мкмоль ротенона.
Таблица 10
Исходя из результатов таблицы 10, можно подтвердить, что соединение примера 10 восстанавливает митохондриальное повреждение обработкой ротеноном на статистически значимом уровне. Поэтому соединение согласно настоящему изобретению можно эффективно применять для профилактики и лечения различных нейродегенеративных заболеваний, связанных с митохондриальным повреждением.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ КАТЕХОЛА ИЛИ ИХ СОЛЬ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2019 |
|
RU2795227C2 |
| НОВЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СОДЕРЖАЩИЙ ИХ | 2019 |
|
RU2793138C2 |
| Производное пирроло[3,2-b]пиридина, полезное в качестве модулятора мускаринового ацетилхолинового рецептора (mAChR)M1 (mAChR M1) | 2014 |
|
RU2688938C2 |
| ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННОГО 1,4-ДИГИДРОДИОКСИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА ТРАНСКРИПЦИИ 1 БЕЛКОВ ТЕПЛОВОГО ШОКА | 2014 |
|
RU2671979C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2016 |
|
RU2718915C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| СОЕДИНЕНИЯ ДИБЕНЗИЛАМИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ТЕРАПЕВТИЧЕСКИЙ ИЛИ ПРОФИЛАКТИЧЕСКИЙ АГЕНТ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ГИПЕРЛИПИДЕМИИ ИЛИ АРТЕРИОСКЛЕРОЗА | 2003 |
|
RU2293078C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ УСИЛЕНИЯ ДЕГРАДАЦИИ БЕЛКОВ-МИШЕНЕЙ И ДРУГИХ ПОЛИПЕПТИДОВ С ПОМОЩЬЮ Е3 УБИКВИТИН ЛИГАЗЫ | 2013 |
|
RU2666530C2 |
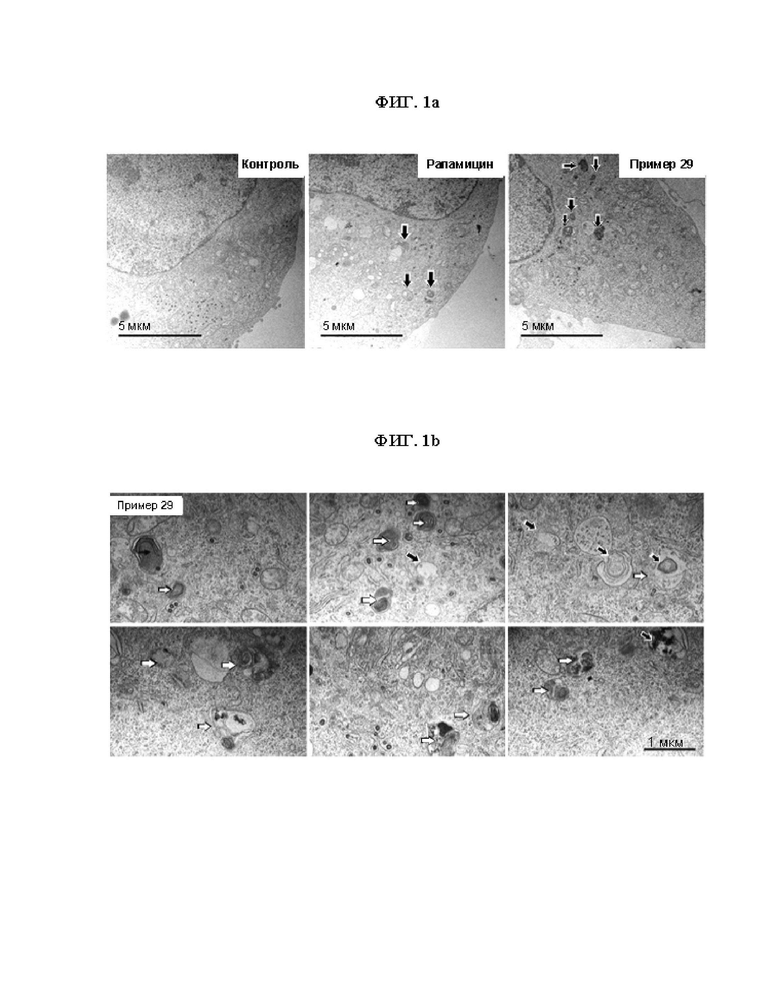
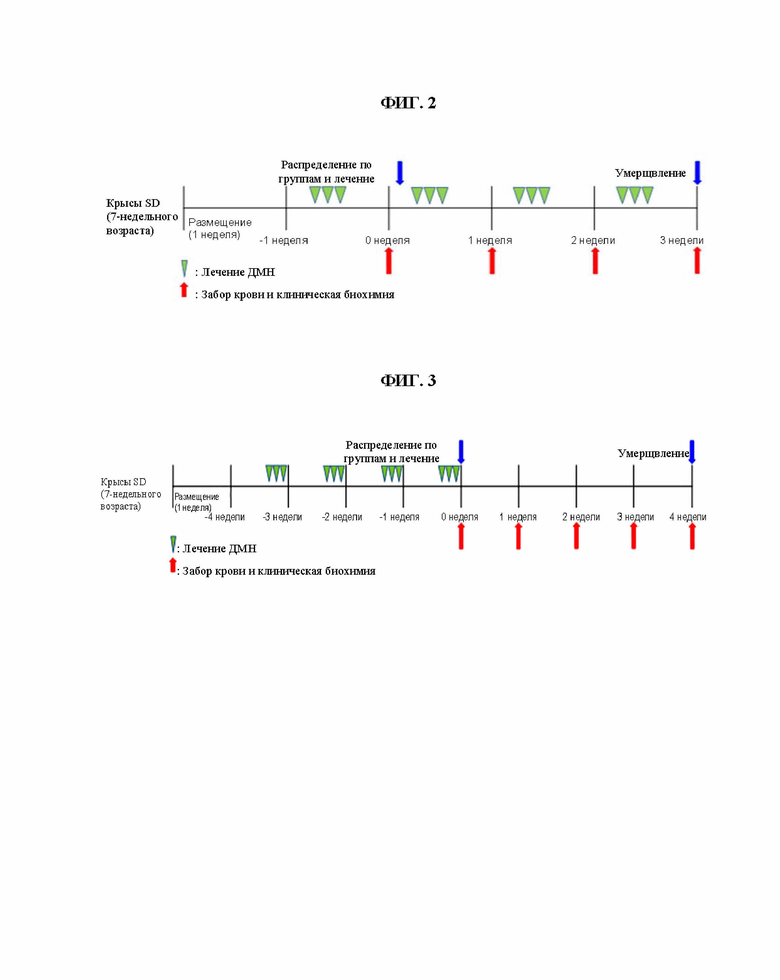
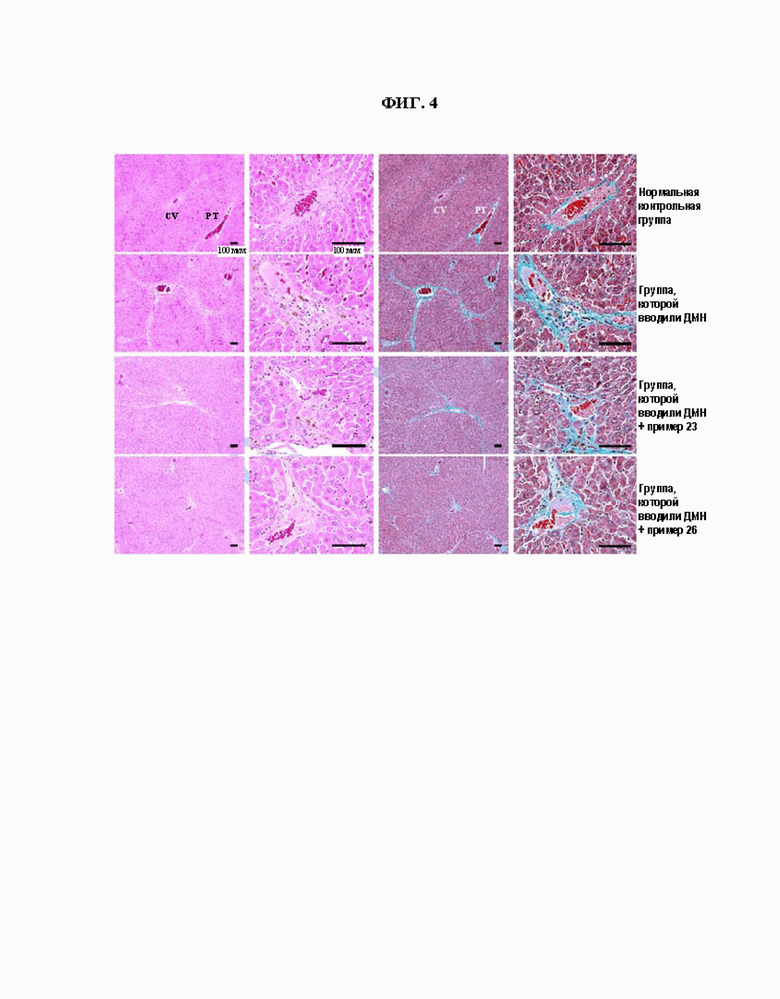
Изобретение относится к производному 2-арилтиазола и его солям, представленному общей формулой 1, где R1 представляет собой водород, C1~C4-алкильную группу или C1~C4-алкильную группу, замещенную моно- или ди-C1~C5-алкиламино; R2 представляет собой C1~C4-алкильную группу; A представляет собой группу, выбранную из группы, состоящей из следующих групп формул: 1a, 1b, 1c и 1d; в формулах 1a, 1b, 1c и 1d * означает место, присоединенное к соединению формулы 1; n равно 1, 2 или 3; R3 и R4, независимо друг от друга, представляют собой водород или C1~C4-алкил; или R3 и R4 соединены друг с другом, с атомом азота, к которому они присоединены, с образованием пиперидина, морфолина или пирролидина, где пиперидин, морфолин или пирролидин необязательно замещен C1~C4-алкилом; R5 и R6 представляют собой C1~C4-алкил; или R5 и R6 образуют кольцо с образованием циклопентана, циклогексана или тетрагидропирана; Cy представляет собой азотсодержащую гетероциклическую группу, выбранную из группы, состоящей из гомопиперазинила, пиперидинила, морфолинила и пирролидинила, где азотсодержащая гетероциклическая группа необязательно замещена C1~C4-алкилом или бензилом. Также изобретение относится к способу получения соединения формулы 1 и фармацевтической композиции, содержащей соединение формулы 1. Технический результат – производные 2-арилтиазола, применяемые для лечения заболеваний, связанных с аутофагией. 4 н. и 8 з.п. ф-лы, 10 табл., 36 пр., 5 ил.
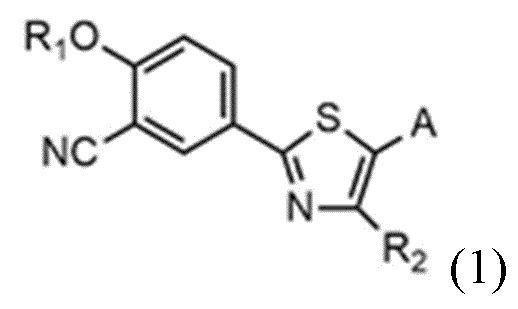
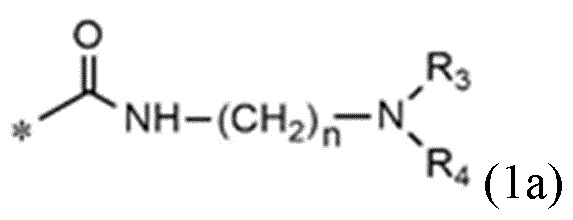
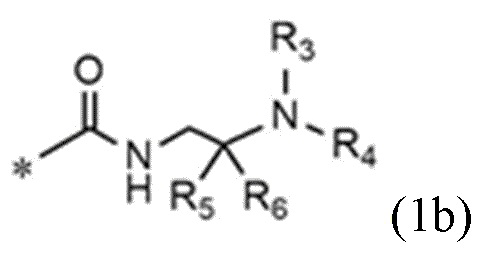
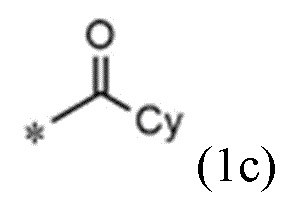
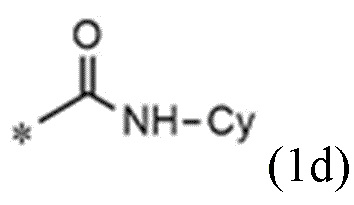
1. Соединение формулы 1 или его фармацевтически приемлемая соль:
<формула 1>
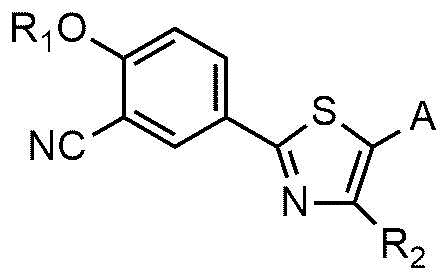 ,
,
где R1 представляет собой водород; C1~C4-алкильную группу или C1~C4-алкильную группу, замещенную моно- или ди-C1~C5-алкиламино,
R2 представляет собой C1~C4-алкильную группу,
A представляет собой группу, выбранную из группы, состоящей из следующих групп формул от 1a до 1d:
<формула 1a>
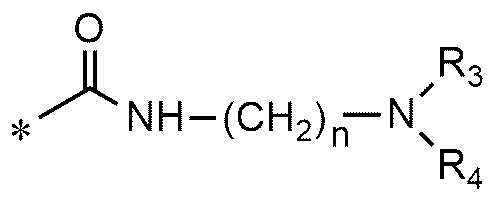
<формула 1b>
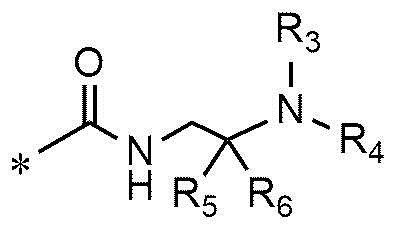
<формула 1c>
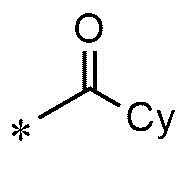
<формула 1d>
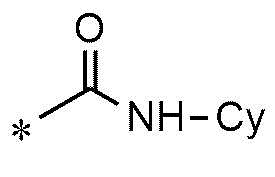 ,
,
в формуле от 1a до 1d
* означает место, присоединенное к соединению формулы 1,
n равно 1, 2 или 3,
R3 и R4, независимо друг от друга, представляют собой водород или C1~C4-алкил; или R3 и R4 соединены друг с другом, с атомом азота, к которому они присоединены, с образованием пиперидина, морфолина или пирролидина, где пиперидин, морфолин или пирролидин необязательно замещен C1~C4-алкилом,
R5 и R6 представляют собой C1~C4-алкил; или R5 и R6 образуют кольцо с образованием циклопентана, циклогексана или тетрагидропирана, и
Cy представляет собой азотсодержащую гетероциклическую группу, выбранную из группы, состоящей из гомопиперазинила, пиперидинила, морфолинила и пирролидинила, где азотсодержащая гетероциклическая группа необязательно замещена C1~C4-алкилом или бензилом.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой водород; изобутильную группу; или диэтиламиноэтильную группу.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где R2 представляет собой метильную группу.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где A представляет собой группу формулы 1a или группу формулы 1b.
5. Соединение или его фармацевтически приемлемая соль по п. 4, где R3 и R4 соединены друг с другом, с атомом азота, к которому они присоединены, с образованием пиперидина, морфолина или пирролидина.
6. Соединение или его фармацевтически приемлемая соль по п. 5, где пиперидин, морфолин или пирролидин замещен C1~C4-алкилом.
7. Соединение или его фармацевтически приемлемая соль по п.1, которое выбрано из группы, состоящей из:
N-(2-(диэтиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диизопропиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диметиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(3-(диэтиламино)пропил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(4-метилгомопиперазино)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(4-(1-метил)пиперидинил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(4-морфолино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(1-пирролидино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(1-пиперидино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-((1-(диметиламино)циклопентил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-((1-(диметиламино)циклогексил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(3-(1-бензил)пирролидинил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(2-(1-метил)пирролидино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-((S)-2-(1-этил)пирролидинометил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(4-морфолинамино)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(1-пиперидинамино)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диизопропиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диэтиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диметиламино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(4-морфолино)этил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-((S)-2-(1-этил)пирролидинометил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диизопропиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диэтиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диметиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(4-морфолино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-((S)-2-(1-этил)пирролидинометил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида; и
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида.
8. Соединение или его фармацевтически приемлемая соль по п.1, которое выбрано из группы, состоящей из:
N-(2-(диизопропиламино)этил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-изобутоксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(диизопропиламино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-(2-(4-морфолино)этил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида;
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(3-циано-4-гидроксифенил)-4-метилтиазол-5-карбоксамида гидрохлорида; и
N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-2-(4-(2-диэтиламино)этокси-3-цианофенил)-4-метилтиазол-5-карбоксамида гидрохлорида.
9. Способ для приготовления соединения формулы 1 или его фармацевтически приемлемой соли, причем способ включает ацилирование соединения формулы 2 азотсодержащим соединением, выбранным из группы, состоящей из NH2-(CH2)n-NR3R4, NH2-CH2-CR5R6-NR3R4, Cy и NH2-Cy (где n, R3, R4, R5 и R6 являются такими же, как определено в п. 1; Cy представляет собой азотсодержащий гетероцикл, выбранный из группы, состоящей из гомопиперазина, пиперидина, морфолина и пирролидина, где азотсодержащий гетероцикл необязательно замещен C1~C4-алкилом или бензилом; и NH2-Cy представляет собой азотсодержащий гетероциклический амин, выбранный из группы, состоящей из гомопиперазиниламина, пиперидиниламина, морфолиниламина и пирролидиниламина, где азотсодержащий гетероциклический амин необязательно замещен C1~C4-алкилом или бензилом):
<формула 1>
<формула 2>
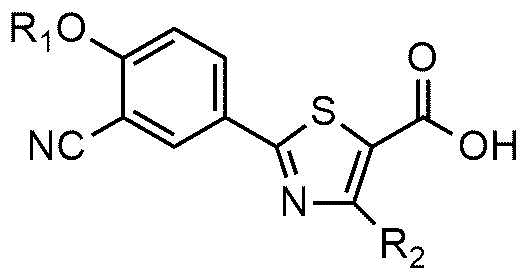 ,
,
где R1, R2 и A являются такими же, как определено в п. 1.
10. Способ для приготовления соединения формулы 1 или его фармацевтически приемлемой соли, причем способ включает
ацилирование соединения формулы 3 азотсодержащим соединением, выбранным из группы, состоящей из NH2-(CH2)n-NR3R4, NH2-CH2-CR5R6-NR3R4, Cy и NH2-Cy (где n, R3, R4, R5 и R6 являются такими же, как определено в п. 1; Cy представляет собой азотсодержащий гетероцикл, выбранный из группы, состоящей из гомопиперазина, пиперидина, морфолина и пирролидина, где азотсодержащий гетероцикл необязательно замещен C1~C4-алкилом или бензилом; и NH2-Cy представляет собой азотсодержащий гетероциклический амин, выбранный из группы, состоящей из гомопиперазиниламина, пиперидиниламина, морфолиниламина и пирролидиниламина, где азотсодержащий гетероциклический амин необязательно замещен C1~C4-алкилом или бензилом), с приготовлением соединения формулы 4; и
алкилирование соединения формулы 4 соединением формулы 5 с приготовлением соединения формулы 1:
<формула 1>
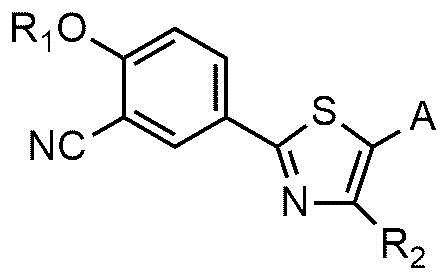
<формула 3>
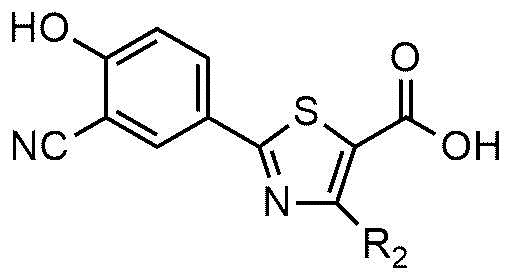
<формула 4>
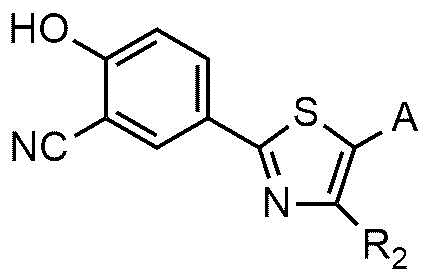
<формула 5>
R1X,
где R1, R2 и A являются такими же, как определено в п. 1, и X представляет собой галоген.
11. Фармацевтическая композиция для индуцирования аутофагии, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1-8 в качестве активного ингредиента.
12. Фармацевтическая композиция по п. 11 для профилактики или лечения нейродегенеративных заболеваний, выбранных из группы, состоящей из болезни Хантингтона, болезни Паркинсона, болезни Альцгеймера, прионной болезни, рассеянного склероза и болезни Лу Герига; заболеваний печени, выбранных из группы, состоящей из фиброза печени, цирроза печени, гепатита и жировой болезни печени; метаболических заболеваний, выбранных из группы, состоящей из диабета, гиперлипидемии, ожирения и воспаления; или сепсиса.
| US 7728009 B1, 01.06.2010 | |||
| US 20160075669 A1, 17.03.2016 | |||
| CN 110066258 A, 30.07.2019 | |||
| WO 2006103045 A1, 05.10.2006 | |||
| WO 2012066561 A1, 24.05.2012 | |||
| KR 1020180007307 A, 22.01.2018 | |||
| KR 1020170022790 A, 02.03.2017 | |||
| SEKHAR, K.C | |||
| et al | |||
| Augmenting the Xanthine Oxidase Inhibitory Activity of Febuxostat by its Structural Modification | |||
| Letters in drug |

