ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области синтеза лекарственных средств и в частности относится к ингибитору производного четырехчленного кольца, способу его получения и его применению.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Рецептор дофамина D3 является членом семейства рецепторов, сопряженных с G-белком, и является подтипом рецептора дофамина. Он принадлежит к D2-подобным ингибирующим рецепторам наряду с рецепторами дофамина D2 и D4. При связывании с DA (дофамин) он снижает уровень цАМФ (циклический аденозинмонофосфат) путем ингибирования G-белка. Рецепторы D3 в основном распределены в мезолимбической системе, особенно в прилежащем ядре, обонятельном бугорке и островках Калеха, которые не связаны с двигательной функцией. Высокоактивные модуляторы рецептора D3 могут обладать хорошей антишизофренической активностью. Рецептор D3 тесно связан с настроением, мыслительным процессом, характером, зависимостью и т.д. и может улучшать негативные симптомы у пациентов с шизофренией. Рецептор D3 может играть регуляторную роль в мыслительном процессе, регулируя высвобождение ацетилхолина и регулируя рецептор глутамата. Частичный агонизм рецептора D3 может улучшить мыслительный процесс.
Рецептор 5-гидрокситриптамина 2A (5-HT2A) является членом семейства рецепторов, сопряженных с G-белком, и является основным возбуждающим подтипом рецептора для рецептора 5-HT. Они распределены по центру и периферии и тесно связаны с характером, эмоциями, обучением, памятью и т.д. Высокоактивные ингибиторы рецептора 5-HT2A оказывают значительный антишизофренический эффект и могут уменьшать побочные эффекты экстрапирамидных симптомов.
Шизофрения представляет собой психическое заболевание с самой высокой распространенностью, с медленным течением заболевания, склонное к повторным приступам, усугублению или обострению, что приводит к серьезной нагрузке и неблагоприятным последствиям для пациентов и их семей. Психопаты могут испытывать положительные симптомы, такие как бред, галлюцинации и нарушения мышления, языка и поведения, отрицательные симптомы, такие как отсутствие эмоций и мимического выражения, плохая речь и отсутствие удовольствия, и другие симптомы, такие как когнитивное расстройство. Несмотря на то, что исследование, разработка и клиническое применение лекарственных средств против шизофрении значительно развились в последние несколько десятилетий, при лечении положительных симптомов как традиционные антипсихотики (первое поколение) (галоперидол, дроперидол, тиоридазин и т.д.), так и атипичные антипсихотики (второе поколение) (клозапин, рисперидон, оланзапин, арипипразол и т.д.), эффективны, в то время как они неэффективны при улучшении отрицательных симптомов и когнитивного расстройства. Поэтому существует острая необходимость в разработке лекарственных средств против шизофрении, которые могут улучшить не только положительные симптомы, но также и отрицательные симптомы и когнитивное расстройство. Высокоактивные модуляторы рецептора дофамина D3 могут улучшить отрицательные симптомы, положительные симптомы и когнитивное расстройство у пациентов с шизофренией без побочных эффектов антипсихотиков первого и второго поколения, таких как экстрапирамидные симптомы и набор веса.
Антагонисты или частичные агонисты рецептора D3 обладают хорошей эффективностью для улучшения положительных симптомов, отрицательных симптомов и когнитивного расстройства при шизофрении. В международных патентных заявках WO2007093540, WO2009013212A2, WO2010031735A1 и WO2012117001A1 сообщается о соединениях, являющихся двойным модулятором рецептора D3 и 5HT2A , но большая часть активностей связывания Ki соединений с рецептором D3 и 5HT2A составляет более 10 нМ. В заявке на патент WO2014086098A1, поданной Jiangsu Hengyi Pharmaceutical Co., LTD, сообщается о селективных ингибиторах D3, но не сообщается об исследовании активности связывания с 5HT2A. Карипразин, антагонист D3, разработанный Gedeon Richter Plc., был получен в 2015 году и подан на международную патентную заявку WO2005012266A1. Карипразин обладает мощной агонистической активностью в отношении рецепторов D3, и его применение в лечении отрицательных симптомах при шизофрении имеет значительные преимущества по сравнению с существующими лекарственными средствами. Однако карипразин обладает слабой ингибирующей активностью в отношении рецептора 5-HT2A, что приводит к тяжелым побочным эффектам экстрапирамидных симптомов (ЭПС). Поэтому существует острая необходимость в разработке высокоактивных модуляторов рецептора D3 с оптимизированной активностью связывания 5HT2A для уменьшения побочных эффектов экстрапирамидных симптомов и улучшения эффектов отрицательных симптомов и когнитивного улучшения при шизофрении.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Все содержимое патентной заявки PCT/CN2020/073153 включено в настоящее изобретение посредством ссылки.
Целью настоящего изобретения является получение соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли, где структура указанного соединения формулы (IX-A) является следующей:
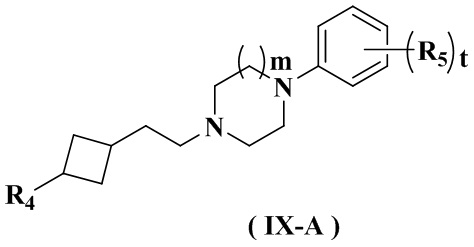
где:
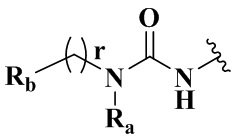
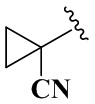
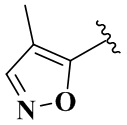
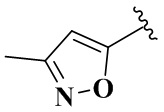
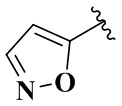
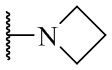
R4 выбран из группы, состоящей из 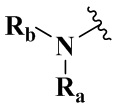 ,
, 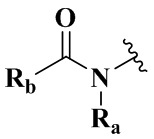 ,
, 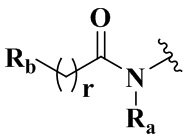 ,
,  и
и 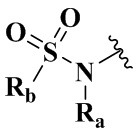 ;
;
Ra выбран из группы, состоящей из водорода или C1-6 алкила;
Rb выбран из группы, состоящей из водорода, амино, C1-6 алкила, C1-6 алкокси, C3-8 циклоалкила, 3-8-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из O, N, S и 5-10-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, причем каждый указанный амино, C1-6 алкил, C3-8 циклоалкил, 3-8-членный гетероциклил и 5-10-членный гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, C1-6 алкила, C1-6 алкокси или C3-8 циклоалкила;
R5 выбран из водорода, C1-6 алкила или галогена;
предпочтительно выбран из группы, состоящей из водорода, галогена, C1-3 алкила;
и более предпочтительно выбран из группы, состоящей из водорода и хлора;
или любые соседние два R5 связаны с образованием 5-6-членного гетероарила, содержащего 1 гетероатом, выбранный из N, S или O; и более предпочтительно тиенила;
r равен 0 или 1;
m равен 0 или 1; и
t равен 2.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли, когда 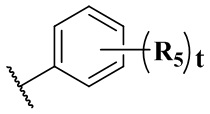 является
является  и m равно 1, то R4 не является
и m равно 1, то R4 не является 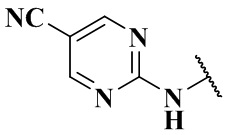 ,
, 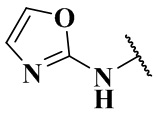 ,
, 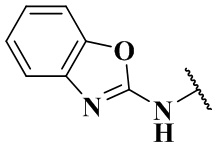 , -NHC(O)C2H5, -NHC(O)N(CH3)2, -NHC(O)NHCH3, -NHC(O)NC2H5CH3, -NHC(O)NHC2H5,
, -NHC(O)C2H5, -NHC(O)N(CH3)2, -NHC(O)NHCH3, -NHC(O)NC2H5CH3, -NHC(O)NHC2H5, 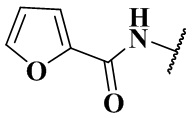 ,
, 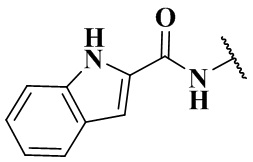 ,
, 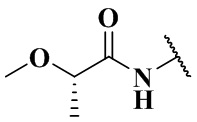 ,
, 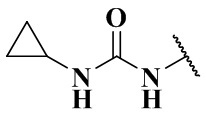 ,
, 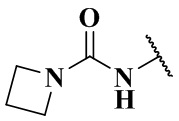 ,
,  ,
,  или
или  .
.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения, его стереоизомера или его фармацевтически приемлемой соли, в котором указанное соединение представлено формулой (X) или формулой (X-A):
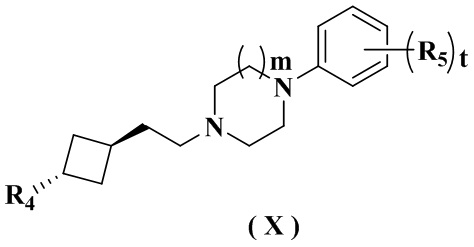
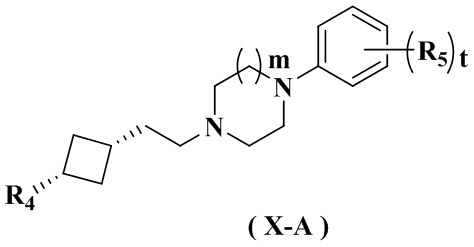
где:
R4 и m определены в формуле (IX-A).
В еще одном предпочтительном варианте осуществления настоящего изобретения Ra выбран из группы, состоящей из водорода или C1-3 алкила;
и более предпочтительно Ra выбран из группы, состоящей из водорода и метила;
Rb выбран из группы, состоящей из амино, C1-3 алкила, C1-3 алкокси, C3-6 циклоалкила, 3-6-членного гетероциклила и 5-10-членного гетероарила, причем каждый указанный амино, C1-3 алкил, C1-3 алкокси, C3-6 циклоалкил, 3-6-членный гетероциклил и 5-10-членный гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, C1-3 алкила и C1-3 алкокси;
и более предпочтительно Rb выбран из группы, состоящей из амино, метила, этила, метокси, циклопропила, азетидинила, пиридила, фуранила, пиримидинила, оксазолила, тиазолила, изоксазолила, индолила, хинолила и бензоксазолила, причем каждый указанный амино, метил, этил, метокси, циклопропил, азетидинил, пиридил, фуранил, пиримидинил, оксазолил, тиазолил, изоксазолил, индолил, хинолил и бензоксазолил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из фтора, циано, гидрокси, метила и метокси; и
r равен 0 или 1.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения, его стереоизомера или его фармацевтически приемлемой соли, в котором формула (IX-A) представлена формулой (XI):
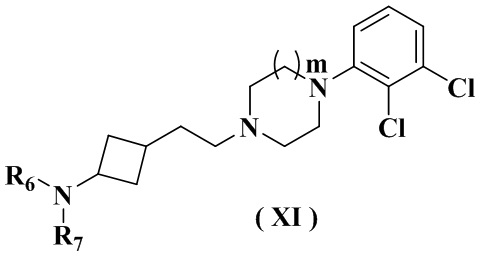
где:
R6 выбран из группы, состоящей из водорода или C1-6 алкила;
R7 выбран из группы, состоящей из -C(O)(CH2)n2Ree, -C(O)NReeRff, -C(O)NRff(CH2)n2NRee и -S(O)m2Ree;
Ree выбран из группы, состоящей из водорода, амино, C1-6 алкила, C1-6 алкокси, C3-8 циклоалкила, 3-8-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, и 5-10-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, причем каждый указанный амино, C1-6 алкил, C3-8 циклоалкил, 3-8-членный гетероциклил, и 5-10-членный гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из водорода, галогена, гидрокси, циано, C1-6 алкила и C1-6 алкокси;
Rff выбран из группы, состоящей из водорода, C1-6 алкила;
и более предпочтительно Ree выбран из группы, состоящей из:
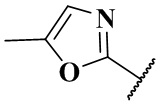
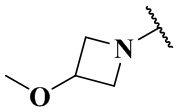
(CH3)2N-, CH3NH-, CH3-, CH3CH2-, CH3CH2NH-, CH3CH2NCH3-, (CH3)2COH-, (CH3)2COHCH2-, CH3OCH2-,  ,
,  ,
, 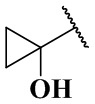 ,
, 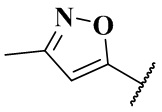 ,
,  ,
,  ,
, 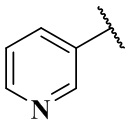 ,
, 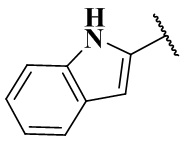 ,
, 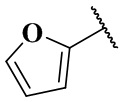 ,
, 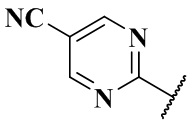 ,
, 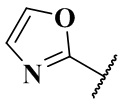 ,
, 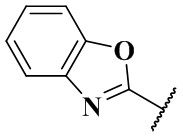 ,
, 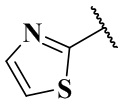 и
и 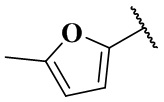 ;
;
Rff выбран из группы, состоящей из водорода, C1-3 алкила;
n2 выбран из группы, состоящей из 0 или 1;
m2 равен 2; и
m выбран из группы, состоящей из 0 или 1.
В еще одном предпочтительном варианте осуществления настоящего изобретения Ree выбран из группы, состоящей из водорода и следующих заместителей:
 ,
,  ,
,  ,
,  ,
, 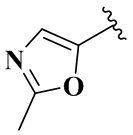 ,
, 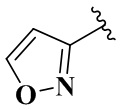 ,
, 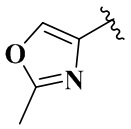 и
и 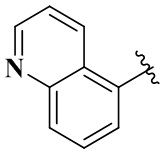 ;
;
Rff выбран из группы, состоящей из водорода и метила.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения, его стереоизомера или его фармацевтически приемлемой соли, в котором формула (XI) представлена формулой (XI-A) или формулой (XI-B):
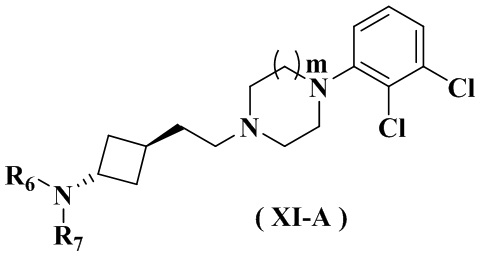
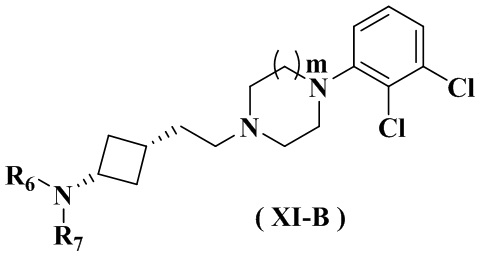 .
.
В предпочтительном варианте осуществления настоящего изобретения R6 выбран из группы, состоящей из водорода;
R7 выбран из группы, состоящей из -C(O)(CH2)n2Ree, -C(O)NRff(CH2)n2Ree или -S(O)m2Ree;
Ree выбран из группы, состоящей из C1-3 алкила, C3-6 циклоалкила, 4-членного гетероциклила, содержащего 1 гетероатом азота, и 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов кислорода, азота, серы, необязательно замещенной одним или более заместителями, выбранными из группы, состоящей из гидрокси, циано, C1-6 алкила и C1-6 алкокси;
Rff выбран из группы, состоящей из водорода и метила;
и более предпочтительно Ree выбран из группы, состоящей из:
(CH3)2N-, CH3NH-, CH3-, CH3CH2-, CH3CH2NH-, CH3CH2NCH3-, (CH3)2C(OH)-, (CH3)2C(OH)CH2-, CH3OCH2-, , , , , , , , , , , ,  , , , , , , и ;
, , , , , , и ;
Rff выбран из группы, состоящей из метила.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения, его стереоизомера или его фармацевтически приемлемой соли, характеризующихся тем, что  выбран из группы, состоящей из:
выбран из группы, состоящей из: 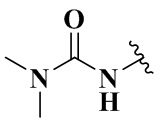 ,
, 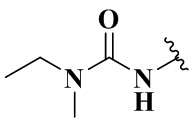 ,
, 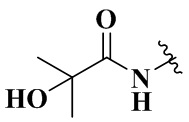 ,
, 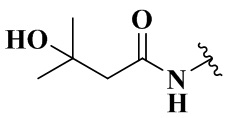 ,
, 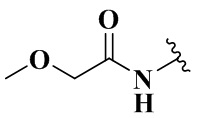 ,
, 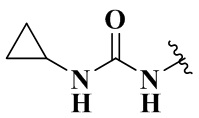 ,
, 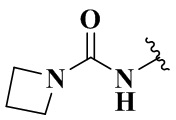 ,
, 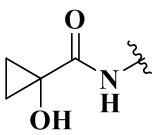 ,
, 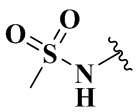 ,
, 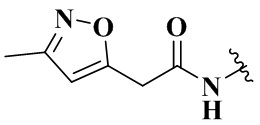 ,
, 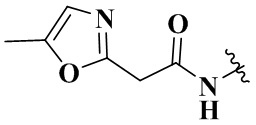 ,
, 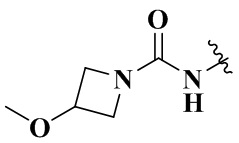 ,
, 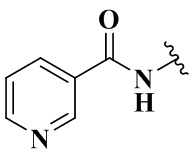 ,
, 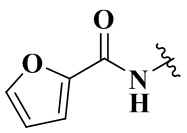 ,
, 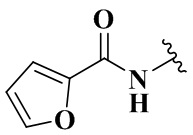 ,
, 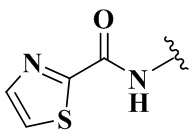 ,
, 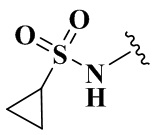 ,
, 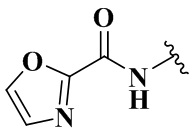 и
и 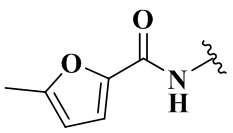 .
.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения, его стереоизомера или его фармацевтически приемлемой соли, в котором формула (XI) представлена формулой (XII):
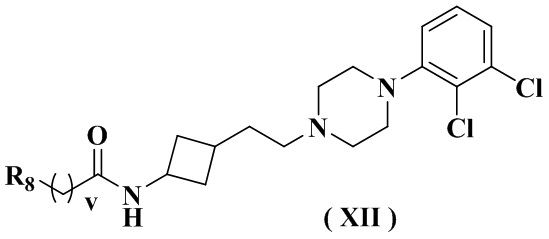
где:
R8 выбран из группы, состоящей из амино, C1-3 алкила, C3-6 циклоалкила, 3-6-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, и 5-10-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, причем каждый указанный амино, C1-3 алкил, C3-6 циклоалкил, 3-6-членный гетероциклил и 5-10-членный гетероарил необязательно дополнительно замещен одним заместителем, выбранным из группы, состоящей из водорода, гидрокси, циано, C1-3 алкила или C1-3 алкокси;
и дополнительно выбран из группы, состоящей из:
HOC(CH3)2-, , , , , , , , , , , , и ; и
v равен 0 или 1.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения, его стереоизомера или его фармацевтически приемлемой соли, в котором формула (XI) представлена формулой (XII):
где:
R8 выбран из группы, состоящей из амино, C1-3 алкила, C3-6 циклоалкила, 3-6-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, и 5-10-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, причем каждый указанный амино, C1-3 алкил, C3-6 циклоалкил, 3-6-членный гетероциклил и 5-10-членный гетероарил необязательно дополнительно замещен одним заместителем, выбранным из группы, состоящей из гидрокси, циано, C1-3 алкила или C1-3 алкокси;
и дополнительно выбран из группы, состоящей из:
HOC(CH3)2-, , ,, , , , , , , , , и ; и
v равен 0 или 1.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения формулы (XII), его стереоизомера или его фармацевтически приемлемой соли, в котором, когда v равен 0, то R8 не является -C2H5, -N(CH3)2, -NHCH3, -NC2H5CH3, -NHC2H5, 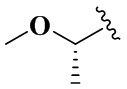 ,
, 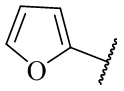 ,
, 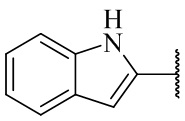 ,
, 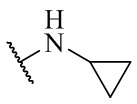 ,
,  или
или 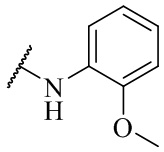 ;
;
когда v равен 1, то R8 не является фенилом.
В настоящем изобретении также предлагается предпочтительный вариант осуществления для указанного соединения, его стереоизомера или его фармацевтически приемлемой соли, в котором формула (XII) представлена формулой (XII-A) или формулой (XII-B):
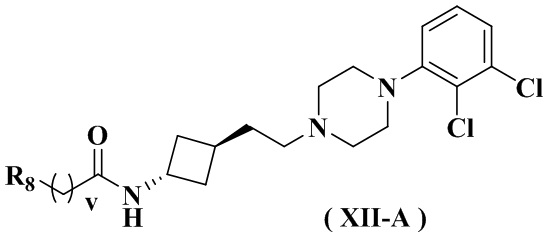 или
или 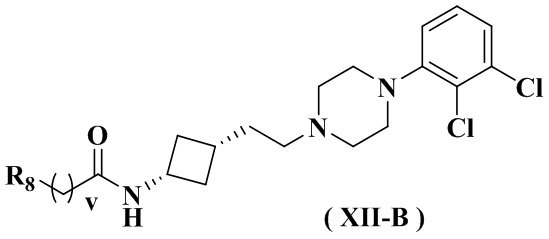 .
.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли, в котором:
R4 представляет собой 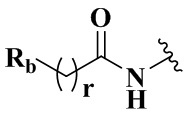 ;
;
Rb выбран из группы, состоящей из C3-6 циклоалкила и 5-10-членного гетероарила, содержащего от 1 до 2 атомов азота, кислорода, серы, необязательно дополнительно замещенных одним или более заместителями, выбранными из группы, состоящей из гидрокси, C1-3 алкила или C1-3 алкокси;
R5 выбран из группы, состоящей из водорода, галогена и C1-3 алкила;
m равен 1;
t равен 2.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли, причем когда r равен 0 и Rb является 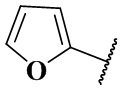 , то Rb замещен по меньшей мере одним заместителем;
, то Rb замещен по меньшей мере одним заместителем;
когда r равен 0 и Rb является 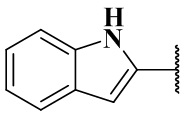 , то Rb замещен по меньшей мере одним заместителем.
, то Rb замещен по меньшей мере одним заместителем.
В настоящем изобретении также предлагается предпочтительный вариант осуществления указанного соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли, в котором:
Rb выбран из группы, состоящей из C3-6 циклоалкила, 5-6-членного гетероарила, содержащего азот или кислород, и 9-10-членного конденсированного гетероарила, содержащего азот, необязательно дополнительно замещенных одним заместителем, выбранными из группы, состоящей из C1-3 алкила или C1-3 алкокси;
R5 выбран из группы, состоящей из водорода, галогена, метила и этила.
В настоящем изобретении также предлагается более предпочтительный вариант осуществления указанного соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли, в котором:
Rb выбран из группы, состоящей из циклопропила, пиридила, фуранила, тиазолила, оксазолила, изоксазолила и хинолила, необязательно дополнительно замещенных одним заместителем, выбранным из группы, состоящей из C1-3 алкила.
Указанное соединение по настоящему изобретению не только обладает мощной агонистической активностью в отношении рецептора D3, но также обладает значительно лучшей ингибирующей активностью в отношении 5-HT2A, чем Карипразин, что приводит к хорошей клинической эффективности при лечении отрицательных симптомов при шизофрении и значительному снижению риска побочных эффектов ЭПС.
Настоящее изобретение также относится к способу получения указанного соединения формулы (XII), его стереоизомера или его фармацевтически приемлемой соли, включающему следующую стадию:
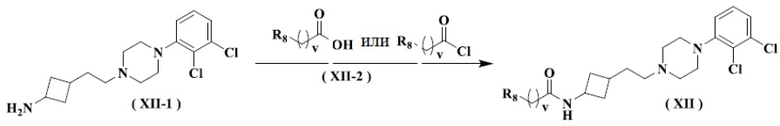
осуществление взаимодействия соединения формулы (XII-1) с ацилхлоридом или карбоновой кислотой формулы (XII-2) с получением соединения формулы (XII), его стереоизомера или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к соединению формулы (XII-1), его стереоизомеру или его фармацевтически приемлемой соли,
 .
.
Настоящее изобретение также относится к способу получения указанного соединения формулы (XII-1), его стереоизомера или его фармацевтически приемлемой соли, включающему следующую стадию:
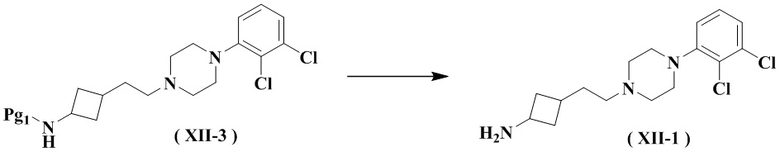
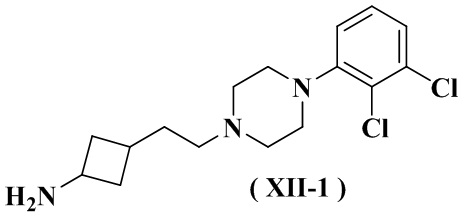
снятие защиты с соединения формулы (XII-3) с получением соединения формулы (XII-1), его стереоизомера или его фармацевтически приемлемой соли;
где:
Pg1 представляет собой аминозащитную группу, выбранную из группы, состоящей из аллилоксикарбонила (Alloc), трифторацетила, 2,4-диметоксибензила, нитробензолсульфонила, тритила, флуоренметоксикарбонила (FMOC), п-толуолсульфонила (Tos), формиата, ацетила, бензилоксикарбонила (Cbz), трет-бутоксикарбонила (Boc), бензила (Bn) и п-метоксифенила (PMP), и предпочтительно трет-бутоксикарбонила (Boc).
Настоящее изобретение также относится к способу получения промежуточного соединения формулы (XII-3), его стереоизомера или его фармацевтически приемлемой соли, включающему следующую стадию:
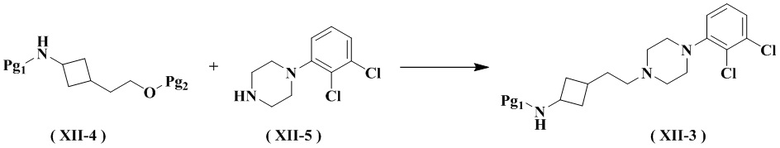
осуществление взаимодействия соединения формулы (XII-4) с соединением формулы (XII-5) с получением соединения формулы (XII-3), его стереоизомера или его фармацевтически приемлемой соли;
где:
Pg2 представляет собой гидроксизащитную группу, выбранную из группы, состоящей из метила (-CH3), трет-бутила (-C(CH3)3), трифенила (-CPh3), метилтиометилового эфира (MTM), 2-метоксиэтоксиметилового эфира (MEM), метоксиметилового эфира (MOM), п-метоксибензилового эфира (PMB), пивалоила (Piv), группы бензилового эфира (-CH2Ph), метоксиметила (-CH2OCH3), триметилсилила (-Si(CH3)3), тетрагидрофуранила (-THP), трет-бутилдисилила (-SiMe2(t-Bu)), ацетила (-Ac), бензоила (-COPh) и п-толуолсульфонила (-SO2PhMe) и предпочтительно п-толуолсульфонила.
Настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективную дозу указанного соединения с общей формулой, конкретных соединений, их стереоизомеров или их фармацевтически приемлемых солей, как описано выше, и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
Настоящее изобретение также относится к применению указанного соединения с общей формулой, конкретных соединений, их стереоизомеров или их фармацевтически приемлемых солей, как описано выше, или указанной фармацевтической композиции, как описано выше, для получения лекарственного средства, модулирующего рецептор, сопряженный с G-белком, в частности лекарственного средства, модулирующего рецептор дофамина D3, и лекарственного средства, модулирующего рецептор 5-HT2A.
Настоящее изобретение также относится к способу лечения воспалительного заболевания с помощью соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли или его фармацевтической композиции.
Настоящее изобретение также относится к способу лечения и/или предупреждения заболевания центральной нервной системы и/или психического заболевания или расстройства, включающему стадию введения пациенту терапевтически эффективной дозы соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли или его фармацевтической композиции.
Настоящее изобретение также относится к способу лечения состояния заболевания с помощью применения указанного соединения или фармацевтической композиции согласно настоящему изобретению, причем состояние заболевания включает, но не ограничивается этим, состояние, связанное с модулятором рецептора дофамина и модулятором рецептора 5-HT2A.
Настоящее изобретение также относится к способу лечения заболевания нервной системы и/или психического заболевания у млекопитающего, включающему стадию введения млекопитающему терапевтически эффективного количества указанного соединения или его фармацевтически приемлемой соли, сложного эфира, пролекарства, сольвата, гидрата или производного согласно настоящему изобретению.
В некоторых вариантах осуществления указанный способ включает лечение таких состояний, как рак, заболевание костей, воспалительное заболевание, иммунное заболевание, неврологическое заболевание, метаболическое заболевание, респираторное заболевание и сердечное заболевание.
В некоторых вариантах осуществления указанный способ включает лечение и/или предупреждение заболевания центральной нервной системы и/или психического заболевания или расстройства, выбранного из группы, состоящей из шизофрении, депрессии, расстройства сна, расстройства настроения, расстройства шизофренического спектра, спастического расстройства, расстройства памяти и/или когнитивного расстройства, двигательного расстройства, расстройства личности, расстройства аутистического спектра, боли, травматического повреждения головного мозга, сосудистого заболевания, расстройства, связанного со злоупотреблением психоактивными веществами, и/или абстинентного синдрома, тиннита, депрессии, аутизма, старческой деменции, болезни Альцгеймера, судорог, невралгии, симптомов большого депрессивного расстройства, связанных с отменой лекарственных средств, и мании.
Способ лечения, предложенный в настоящем документе, включает стадию введения субъекту терапевтически эффективного количества соединения по настоящему изобретению. В варианте осуществления настоящего изобретения предлагается способ лечения заболевания нервной системы и/или психического заболевания у млекопитающего. Указанный способ включает стадию введения млекопитающему терапевтически эффективного количества соединения или его фармацевтически приемлемой соли, сложного эфира, пролекарства, сольвата, гидрата или производного согласно настоящему изобретению.
ОПРЕДЕЛЕНИЯ
Если не указано иное, термины, применяемые в описании и формуле изобретения, имеют приведенные ниже значения.
Термин “алкил” относится к насыщенной алифатической углеводородной группе, которая представляет собой группу с прямой или разветвленной цепью, содержащую 1-20 атомов углерода, предпочтительно алкил содержит 1-8 атомов углерода, более предпочтительно алкил содержит 1-6 атомов углерода, и наиболее предпочтительно алкил содержит 1-3 атома углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их различные разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, содержащий 1-6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкильная группа может быть замещенной или незамещенной. При замещении замещающая(ие) группа(ы) может быть замещена в любом доступном месте соединения. Замещающая(ие) группа(ы) предпочтительно представляет собой одну или несколько групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила. Алкил по настоящему изобретению предпочтительно выбран из группы, состоящей из метила, этила, изопропила, трет-бутила, галогеналкила, дейтерированного алкила, алкоксизамещенного алкила и гидроксизамещенного алкила.
Термин “алкилен” относится к алкилу, в котором атом водорода дополнительно замещен, например, “метилен” относится к -CH2-, “этилен” относится к -(CH2)2-, “пропилен” относится к -(CH2)3-, “бутилен” относится к -(CH2)4- и т.п. Термин “алкенил” относится к алкилу, как определено выше, который состоит по меньшей мере из двух атомов углерода и по меньшей мере одной двойной связи углерод-углерод, например, этенил, 1-пропенил, 2-пропенил, 1-, 2- или 3-бутенила и т.п. Алкенильная группа может быть замещенной или незамещенной. При замещении замещающая(ие) группа(ы) предпочтительно представляет собой одну или несколько групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклилтио.
Термин “циклоалкил” относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной замещающей группе, имеющей 3-20 атомов углерода, предпочтительно 3-12 атомов углерода, более предпочтительно 3-8 атомов углерода и наиболее предпочтительно 3-6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.п. Полициклический циклоалкил включает циклоалкил, имеющий спирокольцо, конденсированное кольцо или мостиковое кольцо. Циклоалкил предпочтительно представляет собой циклопропил, циклобутил, циклогексил, циклопентил и циклогептил и более предпочтительно циклопропил, циклобутил и циклогексил.
Термин “спироциклоалкил” относится к 5-20-членной полициклической группе с отдельными кольцами, соединенными через один общий атом углерода (называемый спироатомом), причем кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженную π-электронную систему. Спироциклоалкил предпочтительно представляет собой 6-14-членный спироциклоалкил и более предпочтительно 7-10-членный спироциклоалкил. В зависимости от количества спироатомов, общих между кольцами, спироциклоалкил можно разделить на моноспироциклоалкил, диспироциклоалкил или полиспироциклоалкил, и спироциклоалкил предпочтительно представляет собой моноспироциклоалкил или диспироциклоалкил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспироциклоалкил. Неограничивающие примеры спироциклоалкила включают:
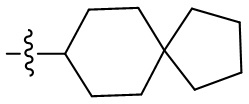
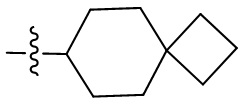
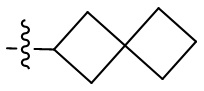
 ;
;
а также включают спироциклоалкил, в котором циклоалкил и гетероциклил соединены одним спироатомом, неограничивающие примеры которого включают:
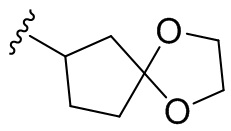
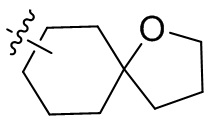
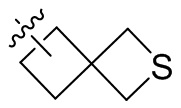
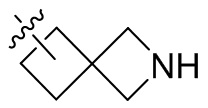 и
и  .
.
Термин “конденсированный циклоалкил” относится к 5-20-членной полностью углеродной полициклической группе, в которой каждое кольцо в системе соединяет пару соседних атомов углерода с другим кольцом, причем одно или несколько колец могут содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженную π-электронную систему. Конденсированный циклоалкил предпочтительно представляет собой 6-14-членный конденсированный циклоалкил и более предпочтительно 7-10-членный конденсированный циклоалкил. По количеству членных колец, конденсированный циклоалкил может быть разделен на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, и конденсированный циклоалкил предпочтительно представляет собой бициклический или трициклический конденсированный циклоалкил и более предпочтительно 4-членный/4-членный, 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:
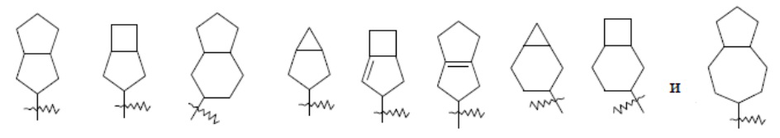 .
.
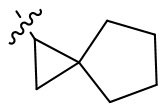
Термин “мостиковый циклоалкил” относится к 5-20-членной полностью углеродной полициклической группе, в которой каждые два кольца в системе имеют два общих несоединенных атома углерода, причем кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью сопряженную π-электронную систему. Мостиковый циклоалкил предпочтительно представляет собой 6-14-членный мостиковый циклоалкил и более предпочтительно 7-10-членный мостиковый циклоалкил. По количеству членных колец мостиковый циклоалкил может быть разделен на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, и мостиковый циклоалкил предпочтительно представляет собой бициклический, трициклический или тетрациклический мостиковый циклоалкил и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостикового циклоалкила включают:
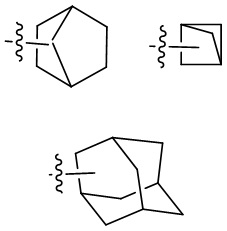
 .
.
Циклоалкильное кольцо может быть конденсировано с кольцом арила, гетероарила или гетероциклила, причем кольцо, связанное с исходной структурой, представляет собой циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и т.п. Циклоалкил может быть необязательно замещенным или незамещенным. При замещении замещающая(ие) группа(ы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин “гетероциклил” относится к 3-20-членной насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, в которой один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода, бора, фосфора, S(O)m (где m представляет собой целое число от 0 до 2) и P(O)n (где n представляет собой целое число от 0 до 2), но исключая -O-O-, -O-S- или -S-S- в кольце, причем остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно гетероциклил имеет 3-12 кольцевых атомов, где 1-4 атома представляют собой гетероатомы; более предпочтительно 3-8 кольцевых атомов; и наиболее предпочтительно 3-8 кольцевых атомов. Неограничивающие примеры моноциклического гетероциклила включают оксациклобутил, оксациклобутил, пирролидинил, группу оксазолидин-2-он, азепинил, имидазолидинил, тетрагидрофуранил, тетрагидротиенил, дигидроимидазолил, дигидрофуранил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил и т.п., предпочтительно оксациклобутил, тетрагидрофуранил, пирролидинил, пиразолидинил, пиперазинил, группу оксазолидин-2-он, морфолинил, пиперазинил и азепинил, и более предпочтительно оксациклобутил, пирролидинил, пиперидинил, пиперазинил, азепинил и группу оксазолидин-2-он. Полициклический гетероциклил включает гетероциклил, имеющий спирокольцо, конденсированное кольцо или мостиковое кольцо. Гетероциклил, имеющий спирокольцо, конденсированное кольцо или мостиковое кольцо, необязательно связан одной связью с другой группой или дополнительно связан с другим циклоалкилом, гетероциклилом, арилом и гетероарилом через любые два или более атомов в кольце.
Термин “спирогетероциклил” относится к 3-20-членной полициклической гетероциклильной группе с отдельными кольцами, соединенными одним общим атомом (называемым спироатом), в которой один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода, бора, фосфора, S (O)m (где m представляет собой целое число от 0 до 2) и P(O)n (где n представляет собой целое число от 0 до 2), причем остальные кольцевые атомы представляют собой атомы углерода, и кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженную π-электронную систему. Спирогетероциклил предпочтительно представляет собой 6-14-членный спирогетероциклил и более предпочтительно 7-10-членный спирогетероциклил. По количеству спироатомов, распределенных между кольцами, спирогетероциклил может быть разделен на моноспирогетероциклил, диспирогетероциклил или полиспирогетероциклил, и спирогетероциклил предпочтительно представляет собой моноспирогетероциклил или диспирогетероциклил и более предпочтительно 3-членный/5-членный, 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспирогетероциклил. Неограничивающие примеры спирогетероциклила включают:
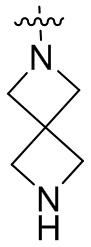
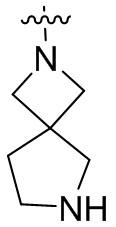
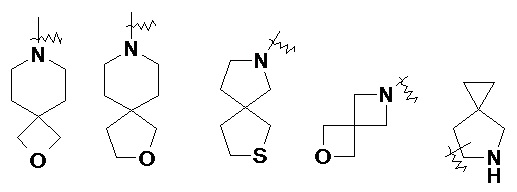 и т.п.
и т.п.
Термин “конденсированный гетероциклил” относится к 5-20-членной полициклической гетероциклильной группе, в которой каждое кольцо в системе соединено с другим кольцом парой соседних атомов, причем одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеют полностью сопряженную π-электронную систему, и один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое число от 0 до 2), причем оставшиеся кольцевые атомы являются атомами углерода. Конденсированный гетероциклил предпочтительно представляет собой 6-14-членный конденсированный гетероциклил и более предпочтительно 7-10-членный конденсированный гетероциклил. По количеству членных колец конденсированный гетероциклил может быть разделен на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил и предпочтительно бициклический или трициклический конденсированный гетероциклил и более предпочтительно 3-членный/5-членный, 4-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:
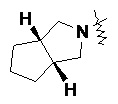
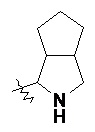
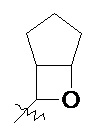
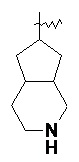
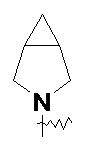
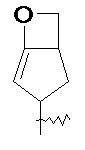
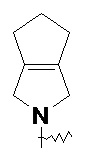
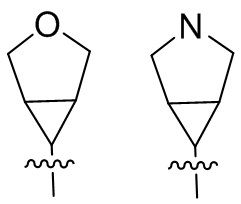
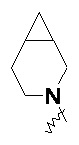
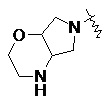
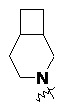
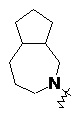
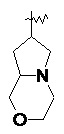
 и т.п.
и т.п.
Термин “мостиковый гетероциклил” относится к 5-14-членной полициклической гетероциклильной группе, в которой каждые два кольца в системе имеют два общих несвязанных атома, причем кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью сопряженную π-электронную систему, и один или более кольцевых атома представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое число от 0 до 2), причем остальные кольцевые атомы представляют собой атомы углерода. Мостиковый гетероциклил предпочтительно представляет собой 6-14-членный мостиковый гетероциклил и более предпочтительно 7-10-членный мостиковый гетероциклил. По количеству членных колец мостиковый гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, и мостиковый гетероциклил предпочтительно представляет собой бициклический, трициклический или тетрациклический мостиковый гетероциклил, и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостикового гетероциклила включают:
 .
.
Гетероциклильное кольцо может быть конденсировано с кольцом арила, гетероарила или циклоалкила, причем кольцо, связанное с исходной структурой, представляет собой гетероциклил. Его неограничивающие примеры включают:
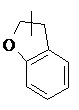
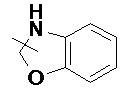
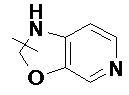
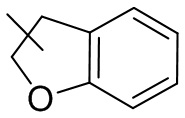
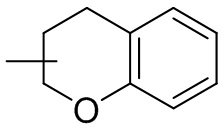
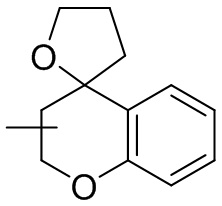
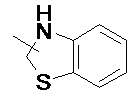 и т.п.
и т.п.
Гетероциклил может быть необязательно замещенным или незамещенным. При замещении замещающая(ие) группа(ы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин “арил” относится к 6-14-членному полностью углеродному моноциклическому или полициклическому конденсированному кольцу (т. е. каждое кольцо в системе имеет общую пару атомов углерода с другим кольцом в системе), имеющему сопряженную π-электронную систему, предпочтительно 6-10-членному арилу, например, фенилу и нафтилу. Арил более предпочтительно представляет собой фенил. Арильное кольцо может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, причем кольцо, связанное с исходной структурой, представляет собой арильное кольцо. Его неограничивающие примеры включают:
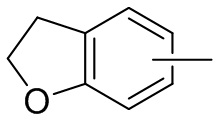
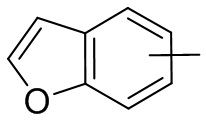
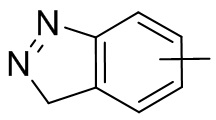
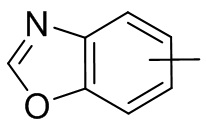
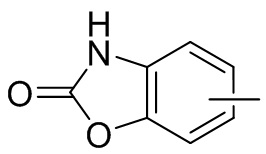
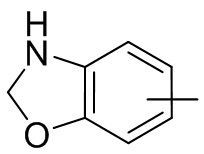
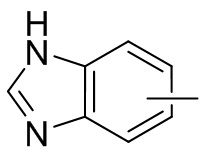
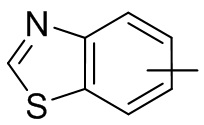
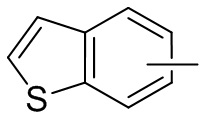
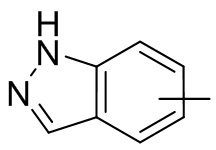
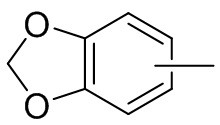
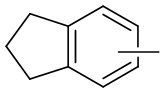
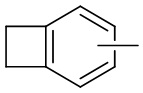
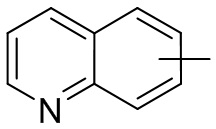
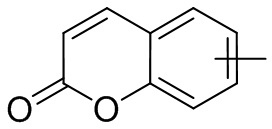
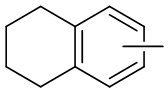
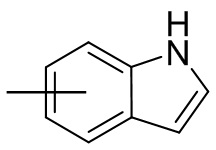
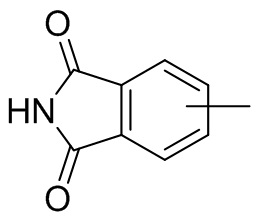
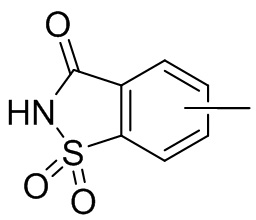 и т.п.
и т.п.
Арил может быть замещенным или незамещенным. При замещении замещающая(ие) группа(ы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
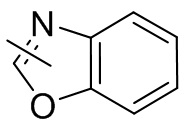
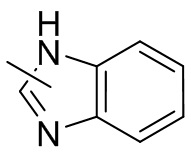
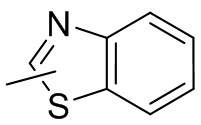
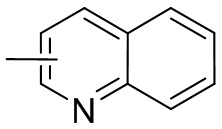
Термин “гетероарил” относится к 5-14-членной гетероароматической системе, имеющей 1-4 гетероатома, выбранных из группы, состоящей из кислорода, серы и азота. Гетероарил предпочтительно представляет собой 5-12-членный гетероарил, более предпочтительно 5-10-членный гетероарил и еще предпочтительно 5-6-членный гетероарил, в котором гетероатом представляет собой 1-2 гетероатома, выбранных из группы, состоящей из атома кислорода, серы и азота, например, имидазолил, фуранил, тиенил, тиазолил, пиразолил, оксазолил, пирролил, триазолил, тетразолил, пиридил, пиримидинил, тиадиазолил, изоксазолил, оксадиазолил, пиразинил и т.п., предпочтительно пиридил, тиазолил, оксазолил, изоксазолил, оксадиазолил, тетразолил, триазолил, тиенил, имидазолил, пиразолил, пиримидинил и тиазолил и более предпочтительно пиридил, тиазолил, оксазолил, изоксазолил, фуранил и пиримидинил. Гетероарильное кольцо может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, причем кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо. Его неограничивающие примеры включают:
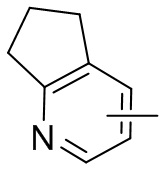
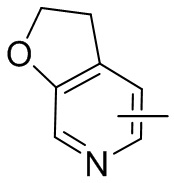
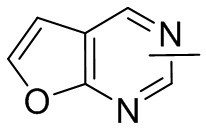
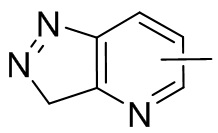
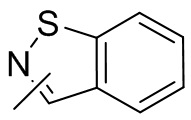
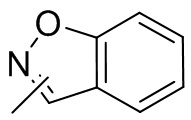
 и
и 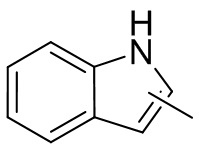 .
.
Гетероарил может быть необязательно замещенным или незамещенным. При замещении замещающая(ие) группа(ы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин “алкокси” относится к группе -O-(алкил) или -O-(незамещенный циклоалкил), в которой алкил является таким, как определено выше. Неограничивающие примеры алкокси включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси и циклогексилокси. Алкокси может быть необязательно замещенным или незамещенным. При замещении замещающая(ие) группа(ы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин “галогеналкил” относится к алкильной группе, замещенной одним или более галогенами, в которой алкил является таким, как определено выше.
Термин “галогеналкокси” относится к алкокси-группе, замещенной одним или более галогенами, в которой алкокси является таким, как определено выше.
Термин “гидроксиалкил” относится к алкильной группе, замещенной гидрокси-группой (гидрокси-группами), в которой алкил является таким, как определено выше.
Термин “алкенил” относится к цепочечному алкенилу, также известному как алкеновая группа. Алкенил может быть дополнительно замещен другой родственной группой, например, алкилом, алкенилом, алкинилом, алкокси, алкилтио, алкиламино, галогеном, тиолом, гидрокси, нитро, циано, циклоалкилом, гетероциклилом, арилом, гетероарилом, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси или алкоксикарбонилом.
Термин “алкинил” относится к (CH≡C-). Алкенил может быть дополнительно замещен другой родственной группой, например, алкилом, алкенилом, алкинилом, алкокси, алкилтио, алкиламино, галогеном, тиолом, гидрокси, нитро, циано, циклоалкилом, гетероциклилом, арилом, гетероарилом, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси или алкоксикарбонилом.
“Гидрокси” относится к группе -OH.
“Галоген” относится к фтору, хлору, брому или йоду.
“Амино” относится к группе -NH2.
“Циано” относится к группе -CN.
“Нитро” относится к группе -NO2.
“Карбокси” относится к группе -C(O)OH.
“THF” относится к тетрагидрофурану.
“EtOAc” относится к этилацетату.
“MeOH” относится к метанолу.
“DMF” относится к N,N-диметилформамиду.
“DIPEA” относится к диизопропилэтиламину.
“TFA” относится к трифторуксусной кислоте.
“MeCN” относится к ацетонитрилу.
“DMA” относится к N,N-диметилацетамиду.
“Et2O” относится к диэтиловому эфиру.
“DCE” относится к 1,2-дихлорэтану.
“DIPEA” относится к N,N-диизопропилэтиламину.
“NBS” относится к N-бромсукцинимиду.
“NIS” относится к N-йодсукцинимиду.
“Cbz-Cl” относится к бензилхлорформиату.
“Pd2(dba)3” относится к трис(дибензилиденацетон)дипалладию.
“Dppf” относится к 1,1’-бис(дифенилфосфино)ферроцену.
“HATU” относится к O-(7-Азабензотриазол-1-ил)-N,N,N',N'- тетраметилуроний гексафторфосфату.
“KHMDS” относится к гексаметилдисилазиду калия.
“LiHMDS” относится к бис(триметилсилил)амиду лития.
“MeLi” относится к метиллитию.
“n-BuLi” относится к н-бутиллитию.
“NaBH(OAc)3” относится к триацетоксиборгидриду натрия.
Различные выражения, такие как “X выбран из группы, состоящей из A, B или C”, “X выбран из группы, состоящей из A, B и C”, “X представляет собой A, B или C”, “X является A, B и C” и т.п., имеют одно и то же значение, т. е. X может быть любым одним или более из A, B и C.
Атом водорода по настоящему изобретению может быть замещен его изотопом - дейтерием. Любой атом водорода в соединениях из примеров настоящего изобретения также может быть замещен атомом дейтерия.
Термин “необязательный” или “необязательно” означает, что описанное далее событие или обстоятельство может, но не обязательно должно, иметь место, и такое описание включает ситуацию, в которой событие или обстоятельство имеет место или не имеет место. Например, “гетероциклил, необязательно замещенный алкилом” означает, что алкильная группа может, но не обязательно должна, присутствовать, и такое описание включает ситуацию, когда гетероциклил замещен алкилом, и когда гетероциклил не замещен алкилом.
Термин “замещенный” относится к одному или более атомам водорода в группе, предпочтительно до 5 атомов, и более предпочтительно от 1 до 3 атомов водорода, независимо замещенным соответствующим числом заместителей. Разумеется, заместители существуют только в возможных для них химических положениях. Специалист в данной области техники способен определить с помощью экспериментов или теоретических знаний, не прилагая чрезмерных усилий, возможно или невозможно провести замещение. Например, комбинация амино- или гидрокси-групп, имеющих свободные атомы водорода, и атомов углерода, имеющих ненасыщенные связи (такие как олефиновые), может быть нестабильной.
Термин “фармацевтическая композиция” относится к смеси одного или более соединений по настоящему изобретению или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами и другими компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Целью фармацевтической композиции является облегчение введения соединения в организм, т.е. способствование абсорбции активного ингредиента для проявления биологической активности.
Как правило, соединение формулы (IX-A) или его фармацевтически приемлемую соль вводят в терапевтически эффективном количестве любым приемлемым способом введения агента с аналогичным применением. Терапевтически эффективное количество соединения по настоящему изобретению может составлять от около 0,01 до около 500 мг на кг массы тела в сутки, которое можно вводить в виде однократной дозы или многократных доз. Подходящий уровень дозы может составлять от около 0,1 до около 250 мг/кг в сутки, от около 0,5 до около 100 мг/кг в сутки. Подходящий уровень дозы может составлять от около 0,01 до около 250 мг/кг в сутки, от около 0,05 до около 100 мг/кг в сутки или от около 0,1 до около 50 мг/кг в сутки. В этом диапазоне доза может составлять от около 0,05 до около 0,5, от около 0,5 до около 5 или от около 5 до около 50 мг/кг в сутки. При пероральном введении композиция может быть представлена в виде таблетки, содержащей от около 1,0 до около 1000 мг активного ингредиента, в частности около 0,1, 0,5, 1, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 6, 7,5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900 и 1000 мг активного ингредиента, и предпочтительно 0,1, 0,2, 0,25, 0,5, 1, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 6, 7,5, 10, 15 и 20 мг активного ингредиента. Фактическое количество соединения по настоящему изобретению, т. е. активного ингредиента, зависит от многих факторов, таких как тяжесть заболевания, подлежащего лечению, возраст и относительное состояние здоровья пациента, эффективность используемого соединения, путь и форма введения и т.п.
Термин “фармацевтически приемлемая соль” относится к соли соединения по настоящему изобретению, которая является безопасной и эффективной для млекопитающих и обладает желаемой биологической активностью.
ПОДРОБНОЕ ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение далее будет описано со ссылкой на следующие примеры, которые не следует рассматривать как на ограничивающие объем настоящего изобретения.
ПРИМЕРЫ
Структуры соединений по настоящему изобретению идентифицировали с помощью спектроскопии ядерного магнитного резонанса (ЯМР) и/или Жидкостный хромато-масс-спектрометрии (ЖХ-МС). Сдвиги ЯМР (δ) приведены в частях на миллион (млн-1). Спектры ЯМР получали с помощью прибора Bruker AVANCE-400. Растворители для ЯМР представляли собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный метанол (CD3OD) и дейтерированный хлороформ (CDCl3), а внутренним стандартом был тетраметилсилан (ТМС).
Жидкостную хромато-масс-спектрометрию (ЖХ-МС) проводили на масс-спектрометре Agilent 1200 Infinity Series. Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили на жидкостном хроматографе высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire C18 150 × 4,6 мм) и на жидкостном хроматографе высокого давления Waters 2695-2996 (хроматографическая колонка Gimini C18 150 × 4,6 мм).
Силикагелевые пластины Yantai Huanghai HSGF254 или Qingdao GF254 применяли в качестве пластины для тонкослойной хроматографии на силикагеле (ТСХ). Размер силикагелевой пластины, применяемой в ТСХ, составлял 0,15-0,2 мм, а размер силикагелевой пластины, применяемой при очистке продукта, составлял 0,4-0,5 мм. Силикагель Yantai Huanghai размером 200-300 меш обычно применяли в качестве носителя для колоночной хроматографии.
Исходные материалы, применяемые в примерах по настоящему изобретению, известны и коммерчески доступны, или могут быть синтезированы путем заимствования или в соответствии со способами, известными в данной области техники.
Если не указано иное, все реакции по настоящему изобретению проводили при непрерывном перемешивании на магнитном устройстве в атмосфере сухого азота или аргона, причем растворитель сухой, а температура реакции измерялась в градусах Цельсия.
Пример 1
1-Бензил-3-(транс-4-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)пропил)циклогексил)мочевина
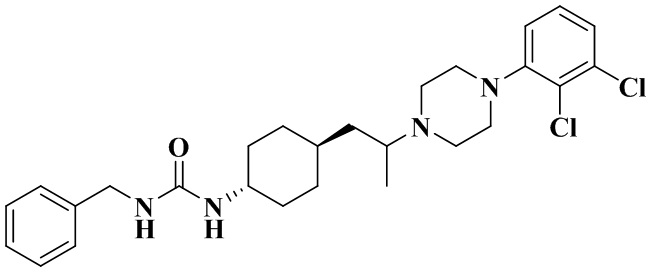
Стадия 1: 1-Бензил-3-(транс-4-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)пропил)циклогексил)мочевина
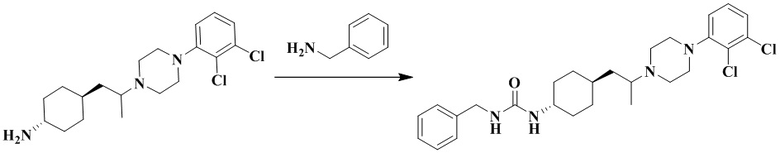
Транс-4-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)пропил)циклогексан-1-амин (60 мг, 0,162 ммоль) и триэтиламин (50 мг, 0,49 ммоль) добавляли к DCM (дихлорметан) (3 мл). Добавляли CDI (N'N-карбонилдиимидазол) (29 мг, 0,178 ммоль) и перемешивали реакционный раствор при комнатной температуре в течение 1 ч. Добавляли бензиламин (36 мг, 0,324 ммоль) и перемешивали реакционный раствор при комнатной температуре в течение 16 ч. К реакционному раствору добавляли воду (20 мл), затем смесь экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, промывали насыщенным солевым рассолом (30 мл), высушивали над безводным сульфатом натрия и отфильтровывали. Фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной хроматографии с получением 1-бензил-3-(транс-4-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)пропил)циклогексил)мочевины в виде белого твердого вещества (10 мг, выход: 12%).
1H ЯМР (400 МГц, CDCl3) δ 7,36 - 7,27 (m, 5H), 7,18 - 7,04 (m, 2H), 6,97 (d, J = 6,9 Гц, 1H), 4,58 (s, 1H), 4,37 (d, J = 5,7 Гц, 2H), 4,17 (d, J = 7,5 Гц, 1H), 3,48 (s, 1H), 3,11-2,79 (m, 8H), 2,00 (s, 2H), 1,79-1,64 (m, 4H), 1,33 - 1,00 (m, 9H).
MS m/z (масса/заряд) (ESI) (масс-спектрометрия с ионизацией распылением в электрическом поле): 503,2 [M+H]+.
Пример 2
3-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевина
Стадия 1: Трет-бутил (3-оксоциклобутил)карбамат
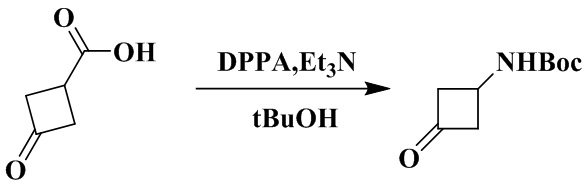
3-оксоциклобутан-1-карбоновую кислоту (1,5 г, 13,2 ммоль), триэтиламин (2,0 мл, 14,5 ммоль) и толуол (30 мл) последовательно добавляли в 100 мл грушевидную колбу. Дифенилфосфорилазид (4,0 г, 14,5 ммоль) медленно добавляли при минус 5-0 °C. Реакционный раствор перемешивали при 0 °C в течение 16 ч. Реакционный раствор промывали насыщенным водным раствором бикарбоната натрия (30 мл × 1) и насыщенным водным раствором хлорида натрия (30 мл × 1) при 0 °C, а органическую фазу сушили над безводным сульфатом натрия. К органической фазе добавляли трет-бутанол (7,5 мл, 74,8 ммоль), и реакционный раствор нагревали до 100 °С и перемешивали в течение 16 ч. Реакционный раствор концентрировали досуха с помощью роторного испарения с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат: 5/1) с получением трет-бутил (3-оксоциклобутил)карбамата (500 мг, выход: 20,5%).
1H ЯМР (400 МГц, CDCl3) δ 4,86 (s, 1H), 4,27 (s, 1H), 3,50 - 3,33 (m, 2H), 3,11 - 2,97 (m, 2H), 1,46 (s, 9H).
Стадия 2: Метил-2-(3-((трет-бутоксикарбонил)амино)циклобутилиден)ацетат
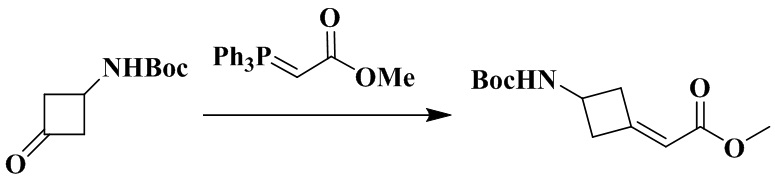
Трет-бутил (3-оксоциклобутил)карбамат (450 мг, 2,43 ммоль) и толуол (20 мл) последовательно добавляли в 50 мл грушевидную колбу с последующим медленным добавлением метил(трифенилфосфоранилиден)ацетата (1,22 г, 3,64 ммоль). Реакционный раствор нагревали до рефлюкса в атмосфере азота в течение 16 ч, охлаждали и концентрировали досуха с помощью роторного испарения с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат: 6/1) с получением метил-2-(3-((трет-бутоксикарбонил)амино)циклобутилиден)ацетата (450 мг, выход: 76,8%).
1H ЯМР (400 МГц, CDCl3) δ 5,76 - 5,66 (m, 1H), 4,80 (br, 1H), 4,24 (s, 1H), 3,69 (s, 3H), 3,63 - 3,49 (m, 1H), 3,27 - 3,10 (m, 1H), 3,00 - 2,86 (m, 1H), 2,82 - 2,64 (m, 1H), 1,45 (s, 9H).
Стадия 3: Метил-2-(3-((трет-бутоксикарбонил)амино)циклобутил)ацетат
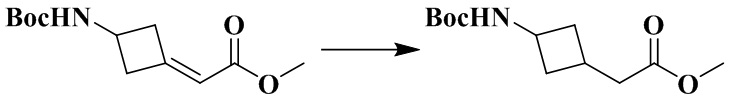
Метил-2-(3-((трет-бутоксикарбонил)амино)циклобутилиден)ацетат (450 мг, 1,9 ммоль) и метанол (10 мл) последовательно добавляли в 50 мл грушевидную колбу. Pd/C (палладиевая чернь) (45 мг, содержащая 10% палладия и 50% воды) медленно добавляли в атмосфере азота. Реакционный раствор перемешивали в атмосфере водорода (1 атм. (101,325 кПа)) в течение 5 ч, фильтровали и концентрировали досуха с помощью роторного испарения для удаления растворителя и получения неочищенного продукта метил-2-(3-((трет-бутоксикарбонил)амино)циклобутил)ацетата (450 мг), который применяли непосредственно на следующей стадии.
MS m/z (ESI): 244,2 [M+H]+.
Стадия 4: Трет-бутил (3-(2-гидроксиэтил)циклобутил)карбамат
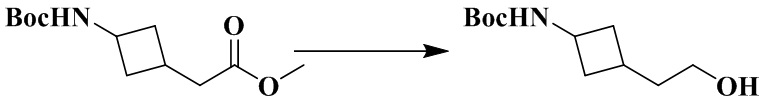
Метил-2-(3-((трет-бутоксикарбонил)амино)циклобутилиден)ацетат (450 мг, 1,9 ммоль) и безводный тетрагидрофуран (10 мл) последовательно добавляли в 50 мл грушевидную колбу. Тетрагидридоалюминат лития (210 мг, 5,6 ммоль) медленно добавляли при 0 °С в атмосфере азота. Реакционный раствор перемешивали при 0 °C в течение 2 ч и гасили реакцию насыщенным водным раствором бикарбоната натрия. Реакционный раствор непосредственно сушили над безводным сульфатом натрия и перемешивали в течение 15 мин. Органическую фазу фильтровали и концентрировали досуха с помощью роторного испарения с получением неочищенного продукта трет-бутил (3-(2-гидроксиэтил)циклобутил)карбамата (450 мг), который использовали непосредственно на следующей стадии.
MS m/z (ESI): 216,2 [M+H]+.
Стадия 5: 2-(3-((Трет-бутоксикарбонил)амино)циклобутил)этил-4-метилбензолсульфонат
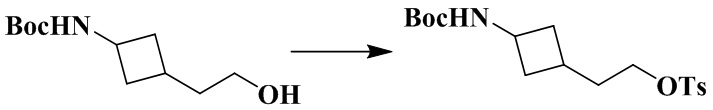
Трет-бутил (3-(2-гидроксиэтил)циклобутил)карбамат (450 мг, 2,1 ммоль), триэтиламин (634 мг, 6,3 ммоль) и дихлорметан (10 мл) последовательно добавляли в 50 мл грушевидную колбу с последующим медленным добавлением тозилхлорида (438 мг, 2,3 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение ночи с последующим добавлением дихлорметана (20 мл) и промывали водой (30 мл × 1). Органическую фазу сушили и концентрировали досуха с помощью роторного испарения с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат: 5/1) с получением 2-(3-((трет-бутоксикарбонил)амино)циклобутил)этил-4-метилбензолсульфоната (710 мг, выход: 84%).
MS m/z (ESI): 370,2 [M+H]+.
Стадия 6: Трет-бутил (3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)карбамат
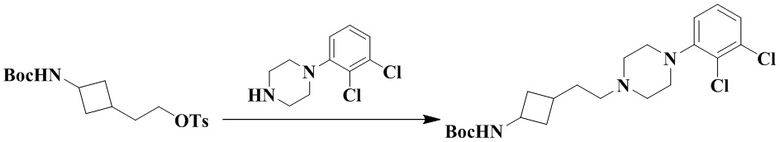
2-(3-((Трет-бутоксикарбонил)амино)циклобутил)этил-4-метилбензолсульфонат (350 мг, 0,95 ммоль), карбонат калия (392 мг, 2,84 ммоль) и ацетонитрил (10 мл) добавляли последовательно в 50 мл грушевидную колбу с последующим медленным добавлением 1-(2,3-дихлорфенил)пиперазина (219 мг, 0,95 ммоль). Реакционный раствор нагревали с рефлюксом в течение ночи. Реакционный раствор охлаждали с последующим добавлением дихлорметана (20 мл) и промывали водой (30 мл × 3). Органическую фазу сушили и концентрировали досуха с помощью роторного испарения с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (дихлорметан/метанол: 50/1) с получением трет-бутил (3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)карбамата (310 мг, выход: 76%).
MS m/z (ESI): 428,2 [M+H]+.
Стадия 7: 3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин гидрохлорид
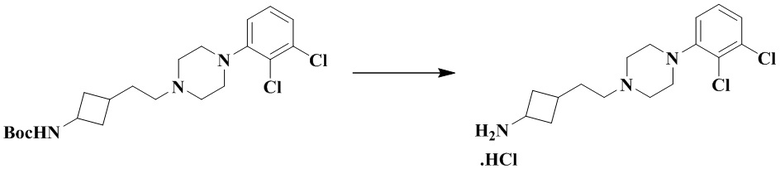
Трет-бутил (3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)карбамат (310 мг, 0,72 ммоль) и этилацетат (2 мл) последовательно добавляли в 25 мл грушевидную колбу с последующим добавлением соляной кислоты в этилацетат (10 мл, 4 М) при 0 °C. Реакционный раствор перемешивали при комнатной температуре в течение 1 ч и концентрировали досуха с помощью роторного испарения для удаления растворителя и получения неочищенного продукта 3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин гидрохлорида (310 мг), который использовали непосредственно на следующей стадии.
MS m/z (ESI): 328,1 [M+H]+.
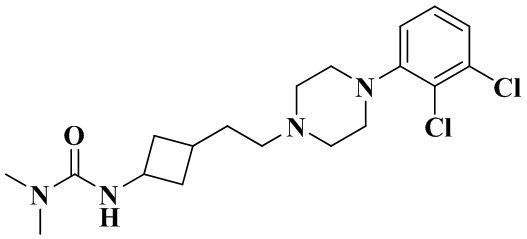
Стадия 8: 3-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевина
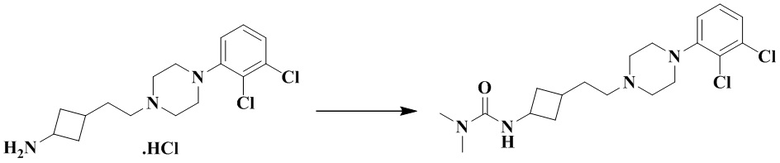
3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин гидрохлорид (50 мг, 0,11 ммоль), триэтиламин (69 мг, 0,69 ммоль) и дихлорметан (2 мл) последовательно добавляли в 10 мл реакционную колбу с последующим добавлением при перемешивании хлорида диметилкарбамоила (18,4 мг, 0,17 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 12 ч и концентрировали досуха с помощью роторного испарения для удаления растворителя и получения неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением 3-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевины (11 мг, выход: 24%).
1H ЯМР (400 МГц, CDCl3) δ 7,23 - 7,10 (m, 2H), 7,08 - 6,91 (m, 1H), 4,61 - 3,93 (m, 2H), 3,56 - 3,02 (m, 4H), 3,03 - 2,64 (m, 8H), 2,65 - 2,31 (m, 3H), 2,31 - 1,21 (m, 7H).
MS m/z (ESI): 399,2 [M+H]+.
Пример 2A
3-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевина
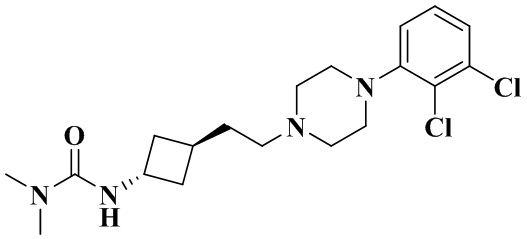
Стадия 1: Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин (промежуточное соединение 2-1) и цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин (промежуточное соединение 2-2)
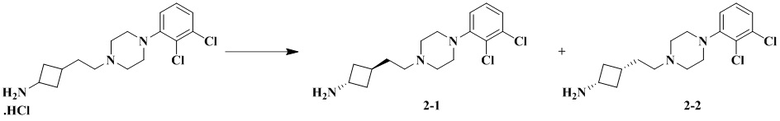
3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин гидрохлорид растворяли с получением транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амина (2-1) и цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амина (2-2).
Условия получения хиральных соединений:
Промежуточное соединение 2-1: tR = 1,285 мин.
1H ЯМР (400 МГц, Хлороформ-d) δ 7,18 - 7,12 (m, 2H), 6,99 - 6,93 (m, 1H), 3,63 - 3,53 (m, 1H), 3,16 - 3,02 (m, 4H), 2,74 - 2,54 (m, 4H), 2,39 - 2,30 (m, 2H), 2,26 - 2,13 (m, 1H), 2,06 - 1,99 (m, 2H), 1,99 - 1,93 (m, 2H), 1,91 - 1,84 (m, 2H), 1,72 - 1,64 (m, 2H).
MS m/z (ESI): 328,1 [M+H]+.
Промежуточное соединение 2-2: tR = 0,882 мин.
1H ЯМР (400 МГц, Хлороформ-d) δ 7,18 - 7,11 (m, 2H), 7,00 - 6,93 (m, 1H), 3,33 - 3,22 (m, 1H), 3,13 - 3,00 (m, 4H), 2,71 - 2,56 (m, 4H), 2,51 - 2,43 (m, 2H), 2,37 - 2,30 (m, 2H), 2,07 - 1,97 (m, 2H), 1,89 - 1,75 (m, 1H), 1,67 - 1,58 (m, 2H), 1,39 - 1,28 (m, 2H).
MS m/z (ESI): 328,1 [M+H]+.
Стадия 2: 3-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевина
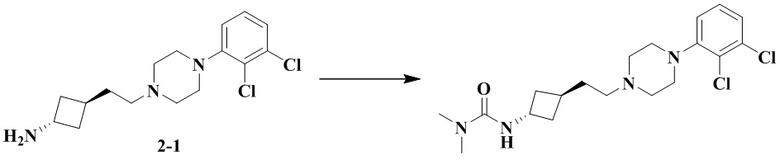
В соответствии с условиями реакции на стадии 8 примера 2 в качестве исходного материала применяли промежуточное соединение 2-1, соответственно получали 3-(транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевину.
1H ЯМР (400 МГц, Хлороформ-d) δ 7,23 - 7,07 (m, 2H), 7,07 - 6,91 (m, 1H), 4,49 (d, J = 7,1 Гц, 1H), 4,44 - 4,28 (m, 1H), 3,53 - 3,03 (m, 5H), 2,90 (s, 6H), 2,82 - 2,61 (m, 3H), 2,51 - 2,35 (m, 2H), 2,27 - 2,10 (m, 3H), 2,08 - 1,95 (m, 2H), 1,88 - 1,72 (m, 2H).
MS m/z (ESI): 399,1 [M+H]+.
Пример 2B
3-(Цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевина
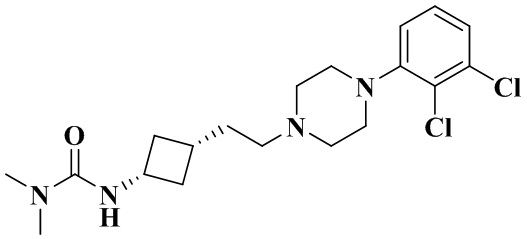
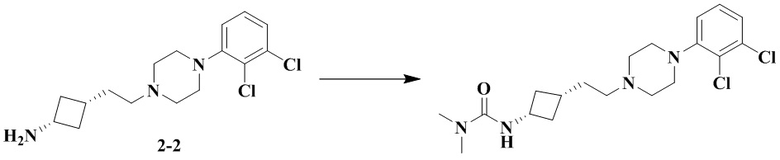
В соответствии с условиями реакции на стадии 8 примера 2 в качестве исходного материала применяли промежуточное соединение 2-2, соответственно получали 3-(цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевину.
1H ЯМР (400 МГц, Хлороформ-d) δ 7,20 - 7,12 (m, 2H), 6,97 (dd, J = 6,7, 2,8 Гц, 1H), 4,41 (d, J = 7,5 Гц, 1H), 4,21 - 4,08 (m, 1H), 3,21 - 3,04 (m, 4H), 2,89 (s, 6H), 2,81 - 2,59 (m, 4H), 2,53 (dd, J = 9,6, 7,0 Гц, 2H), 2,45 - 2,32 (m, 2H), 1,99 - 1,88 (m, 1H), 1,70 - 1,65 (m, 2H), 1,47 - 1,39 (m, 2H).
MS m/z (ESI): 399,1 [M+H]+.
Пример 3
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)пропионамид
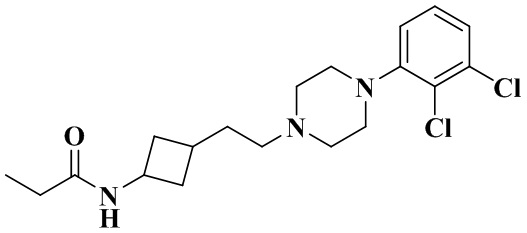
Стадия 1: N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)пропионамид
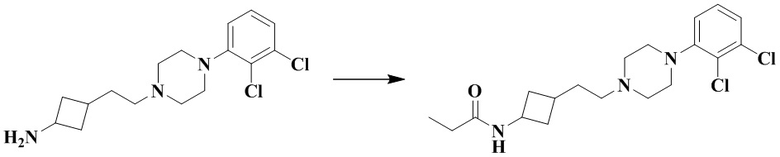
3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин гидрохлорид (50 мг, 0,11 ммоль), диизопропилэтиламин (88 мг, 0,69 ммоль) и дихлорметан (10 мл) последовательно добавляли в 10 мл реакционную колбу с последующим добавлением при перемешивании пропионилхлорида (12,7 мг, 0,14 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 12 ч и промывали водой. Органическую фазу сушили и концентрировали досуха с помощью роторного испарения для удаления растворителя и получения неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)пропионамида (18 мг, выход: 41%).
MS m/z (ESI): 384,2 [M+H]+.
Пример 4
1-Циклопропил-3-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)мочевина
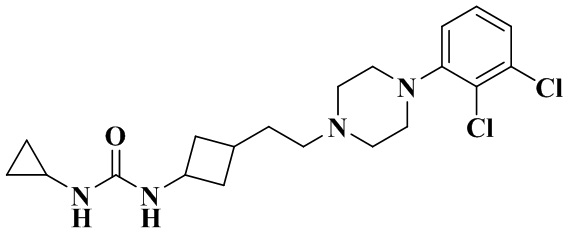
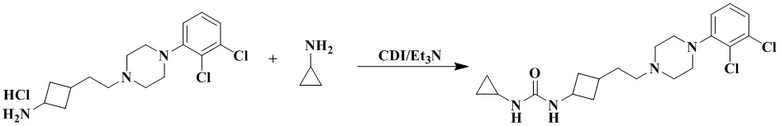
3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин гидрохлорид (33 мг, 0,09 ммоль), триэтиламин (46 мг, 0,45 ммоль) и N'N-карбонилдиимидазол (22 мг, 0,16 ммоль) растворяли в дихлорметане (2 мл). Реакционный раствор перемешивали при комнатной температуре в течение 2 ч до полного растворения исходных материалов. Добавляли циклопропиламин (10 мг, 0,18 ммоль) и перемешивали реакционный раствор при 35 °C в течение 48 ч. Реакционный раствор концентрировали досуха с помощью роторного испарения и полученный неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением 1-циклопропил-3-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)мочевины (12 мг, выход: 32,2%).
1H ЯМР (400 МГц, CDCl3) δ 7,20 - 7,12 (m, 2H), 6,97 (dd, J = 7,0, 2,4 Гц, 1H), 5,08 (dd, J = 28,8, 7,3 Гц, 1H), 4,64 (s, 1H), 4,43 - 4,09 (m, 1H), 3,14 (s, 4H), 2,73 (s, 4H), 2,56 (ddd, J = 16,2, 7,4, 2,8 Гц, 2H), 2,43 (s, 3H), 2,05 (dddd, J = 33,4, 24,1, 16,7, 8,5 Гц, 4H), 1,83 - 1,68 (m, 2H), 1,48 (dt, J = 9,6, 6,0 Гц, 2H), 0,76 (q, J = 6,3 Гц, 2H), 0,61 - 0,53 (m, 2H).
MS m/z (ESI): 411,2 [M+H]+.
Пример 5
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1H-индол-2-карбоксамид
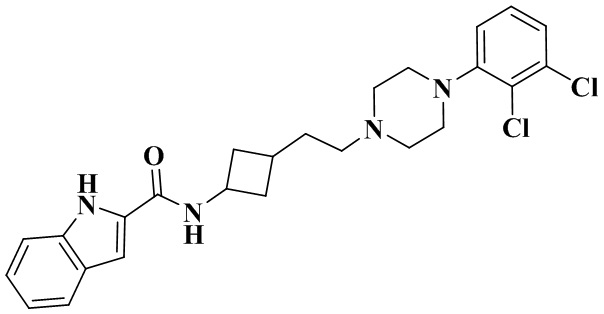
3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин (50 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (3 мл) с последующим добавлением 1H-индол-2-карбоновой кислоты (30 мг, 0,18 ммоль), HATU (86 мг, 0,23 ммоль) и диизопропилэтиламина (58 мг, 0,45 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение ночи и концентрировали досуха с помощью роторного испарения. Полученный неочищенный продукт очищали с помощью высокоэффективной Жидкостный хроматографии с получением N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1H-индол-2-карбоксамида.
MS m/z (ESI): 471,2 [M+H]+.
Пример 6
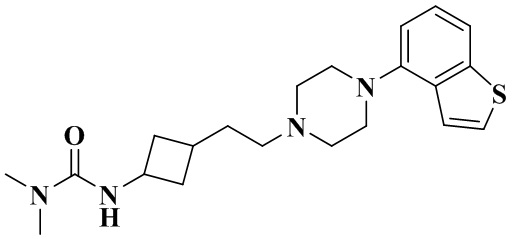
3-(3-(2-(4-(Бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевина
Стадия 1: Трет-бутил (3-(2-(4-(бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутил)карбамат
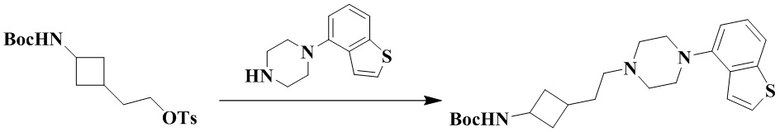
2-(3-((Трет-бутоксикарбонил)амино)циклобутил)этил-4-метилбензолсульфонат (200 мг, 0,54 ммоль), карбонат калия (224 мг, 1,62 ммоль) и ацетонитрил (10 мл) добавляли последовательно в 50 мл грушевидную колбу с последующим медленным добавлением 1-(бензо[b]тиофен-4-ил)пиперазина (118 мг, 0,54 ммоль). Реакционный раствор нагревали с рефлюксом в течение ночи. Реакционный раствор охлаждали с последующим добавлением дихлорметана (20 мл) и промывали водой (30 мл × 3). Органическую фазу сушили и концентрировали досуха с помощью роторного испарения с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (дихлорметан/метанол: 50/1) с получением трет-бутил (3-(2-(4-(бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутил)карбамата (120 мг, выход: 53%).
MS m/z (ESI): 416,2 [M+H]+.
Стадия 2: 3-(2-(4-(Бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутан-1-амин гидрохлорид
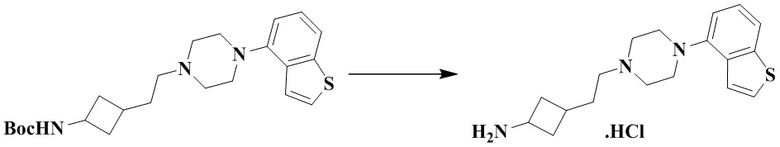
Трет-бутил (3-(2-(4-(бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутил)карбамат (120 мг, 0,29 ммоль) и этилацетат (1 мл) последовательно добавляли в 25 мл грушевидную колбу с последующим добавлением соляной кислоты в этилацетат (6 мл, 4М) при 0 °С. Реакционный раствор перемешивали при комнатной температуре в течение 1 ч и концентрировали досуха с помощью роторного испарения для удаления растворителя и получения неочищенного гидрохлорида (110 мг), который использовали непосредственно на следующей стадии.
MS m/z (ESI): 316,1 [M+H]+.
Стадия 3: 3-(3-(2-(4-(Бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевина
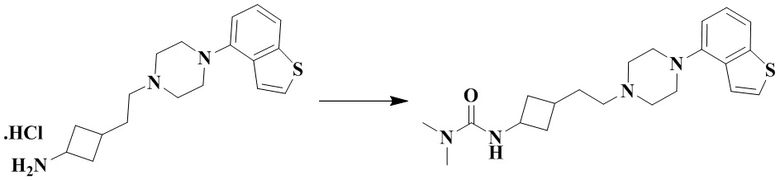
3-(2-(4-(Бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутан-1-амин гидрохлорид (50 мг, 0,12 ммоль), триэтиламин (71 мг, 0,70 ммоль) и дихлорметан (2 мл) добавляли последовательно в 10 мл реакционную колбу с последующим добавлением при перемешивании диметилкарбамоилхлорида (19 мг, 0,18 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 12 ч и концентрировали досуха с помощью роторного испарения для удаления растворителя и получения неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением 3-(3-(2-(4-(бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутил)-1,1-диметилмочевины (17 мг, выход: 37%).
MS m/z (ESI): 387,2 [M+H]+.
Пример 7
N-(3-(2-(4-(2,3-дихлорфенил)-1,4-диазепан-1-ил)этил)циклобутил)фуран-2-карбоксамид
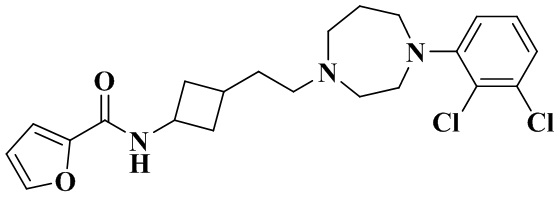
Способ получения такой же, как в примере 2.
MS m/z (ESI): 436,2 [M+H]+.
Пример 8
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-5-метилфуран-2-карбоксамид
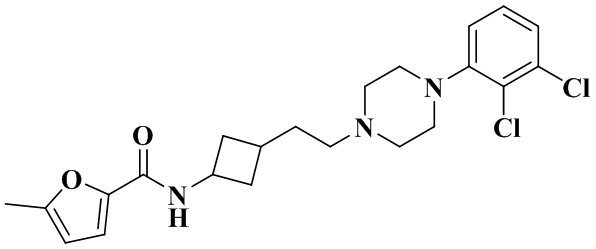
В соответствии со стадией 8 примера 2 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-5-метилфуран-2-карбоксамид (23 мг, белое твердое вещество, выход: 28,3%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,21 - 7,12 (m, 2H), 7,01 - 6,96 (m, 2H), 6,39 (dd, J = 34,1, 8,0 Гц, 1H), 6,11 - 6,06 (m, 1H), 4,52 (dq, J = 84,5, 8,0 Гц, 1H), 3,23 - 3,05 (m, 4H), 2,76 (s, 4H), 2,59 (td, J = 7,4, 6,8, 2,2 Гц, 1H), 2,48 - 2,46 (m, 1H), 2,35 (s, 3H), 2,24 - 2,13 (m, 2H), 2,04 - 1,97 (m, 1H), 1,84 - 1,75 (m, 2H), 1,62 (qd, J = 9,1, 2,8 Гц, 2H).
MS m/z (ESI): 436,1 [M+H]+.
Пример 9
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-метоксиацетамид
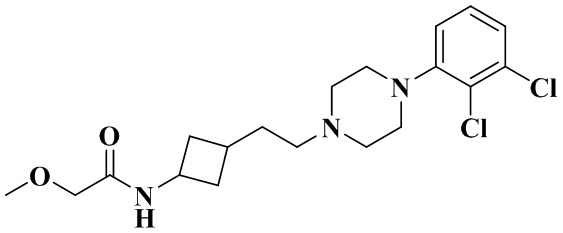
В соответствии со стадией 8 примера 2 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-метоксиацетамид (29 мг, белое твердое вещество, выход: 33%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,19 - 7,12 (m, 2H), 6,97 (dd, J = 7,0, 2,5 Гц, 1H), 6,63 (dd, J = 42,2, 8,2 Гц, 1H), 4,42 (dq, J = 87,5, 7,9 Гц, 1H), 3,86 (d, J = 4,7 Гц, 2H), 3,42 (d, J = 2,5 Гц, 3H), 3,22 - 3,06 (m, 4H), 2,81 - 2,61 (m, 4H), 2,58 - 2,52 (m, 1H), 2,45 - 2,32 (m, 2H), 2,21 - 2,03 (m, 2H), 2,02 - 1,91 (m, 1H), 1,79 - 1,73 (m, 1H), 1,70 - 1,67 (m, 1H), 1,57 - 1,49 (m, 1H).
MS m/z (ESI): 400,1 [M+H]+.
Пример 10
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)никотинамид
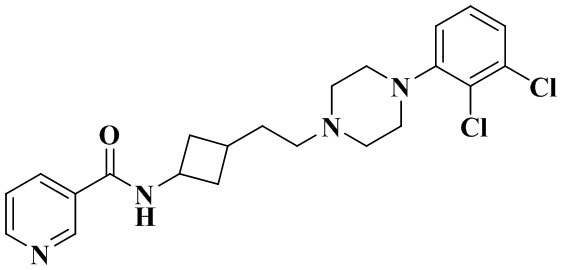
В соответствии с примером 2 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)никотинамид (25 мг, белое твердое вещество, выход: 29%).
Соединение по примеру 10 также может быть получено способом синтеза по примеру 5.
1H ЯМР (400 МГц, Хлороформ-d) δ 8,97 (d, J = 2,2 Гц, 1H), 8,72 (dd, J = 4,8, 1,8 Гц, 1H), 8,12 (dq, J = 8,0, 2,0 Гц, 1H), 7,44 - 7,34 (m, 1H), 7,19 - 7,12 (m, 2H), 6,97 (dt, J = 7,0, 2,7 Гц, 1H), 6,41 (dd, J = 14,4, 7,5 Гц, 1H), 4,85 - 4,34 (m, 1H), 3,12 (t, J = 5,0 Гц, 4H), 2,78 - 2,66 (m, 4H), 2,46 - 2,39 (m, 2H), 2,27 - 2,16 (m, 2H), 2,13 - 2,02 (m, 1H), 1,87 - 1,79 (m, 1H), 1,79 - 1,57 (m, 3H).
MS m/z (ESI): 433,1 [M+H]+.
Пример 11
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-гидрокси-2-метилпропанамид
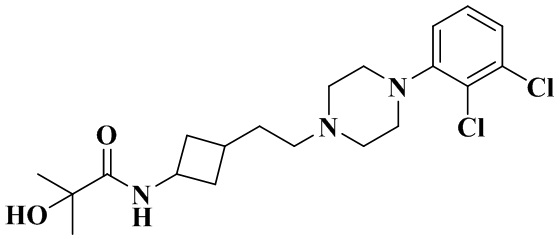
В соответствии с примером 2 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-гидрокси-2-метилпропанамид (32 мг, белое твердое вещество, выход: 30%).
Соединение по примеру 11 также может быть получено способом синтеза по примеру 5.
MS m/z (ESI): 414,1 [M+H]+.
Пример 12
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-3-метоксиазетидин-1-карбоксамид
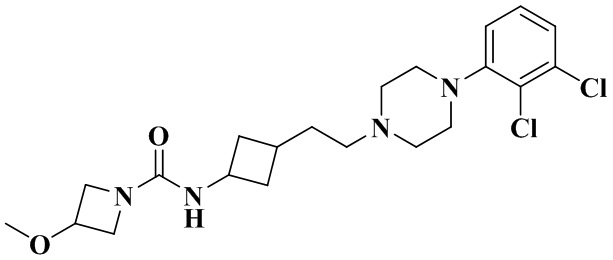
В соответствии с примером 4 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-3-метоксиазетидин-1-карбоксамид (22 мг, белое твердое вещество, выход: 23%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,18 - 7,12 (m, 2H), 6,96 (dd, J = 7,1, 2,6 Гц, 1H), 4,41 - 4,22 (m, 1H), 4,21 - 4,15 (m, 2H), 4,11 - 4,06 (m, 2H), 3,85 - 3,78 (m, 2H), 3,29 (s, 3H), 3,16 - 3,08 (m, 4H), 2,69 (s, 4H), 2,55 - 2,49 (m, 1H), 2,42 - 2,35 (m, 2H), 2,11 (ddd, J = 11,5, 7,3, 2,9 Гц, 1H), 2,04 - 1,85 (m, 2H), 1,71 (dq, J = 32,8, 7,8 Гц, 2H), 1,44 (td, J = 9,2, 2,9 Гц, 1H).
MS m/z (ESI): 441,1 [M+H]+.
Пример 13
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1-гидроксициклопропан-1-карбоксамид
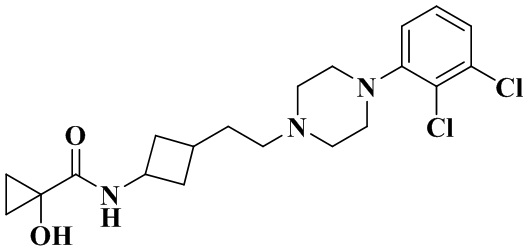
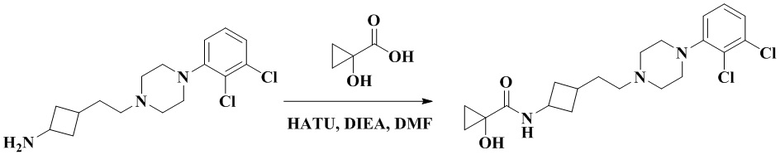
3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин (50 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (3 мл) с последующим добавлением 1-гидроксипропан-1-карбоновой кислоты (18 мг, 0,18 ммоль), HATU (86 мг, 0,23 ммоль) и диизопропилэтиламина (58 мг, 0,45 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение ночи и концентрировали досуха с помощью роторного испарения. Полученный неочищенный продукт очищали с помощью высокоэффективной Жидкостный хроматографии с получением N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1-гидроксициклопропан-1-карбоксамида (13 мг, белое твердое вещество, выход: 21%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,20 - 7,12 (m, 2H), 7,07 (dd, J = 28,3, 8,0 Гц, 1H), 6,96 (dd, J = 7,1, 2,5 Гц, 1H), 4,38 (dq, J = 87,6, 7,9 Гц, 1H), 3,20 - 3,05 (m, 4H), 2,72 (s, 4H), 2,56 (dd, J = 8,9, 2,9 Гц, 1H), 2,41 (dd, J = 9,5, 6,3 Гц, 2H), 2,25 (d, J = 8,4 Гц, 1H), 2,19 - 2,06 (m, 2H), 1,99 (dd, J = 14,2, 6,4 Гц, 1H), 1,82 - 1,69 (m, 2H), 1,55 (dd, J = 9,1, 2,9 Гц, 1H), 1,35 - 1,30 (m, 2H), 1,01 (q, J = 4,6 Гц, 2H).
MS m/z (ESI): 412,1 [M+H]+.
Пример 13A
N-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1-гидроксициклопропан-1-карбоксамид
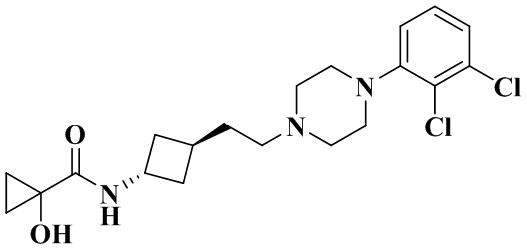
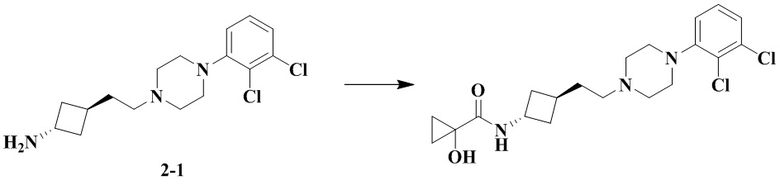
В соответствии с условиями реакции в примере 13 в качестве исходного материала применяли промежуточное соединение 2-1, соответственно получали N-(транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1-гидроксициклопропан-1-карбоксамид.
1H ЯМР (400 МГц, Хлороформ-d) δ 7,20 - 7,13 (m, 2H), 7,07 (d, J = 8,0 Гц, 1H), 6,97 (dd, J = 7,0, 2,6 Гц, 1H), 4,56 - 4,44 (m, 1H), 3,22 - 3,07 (m, 4H), 2,86 - 2,66 (m, 4H), 2,50 - 2,41 (мm, 2H), 2,32 - 2,24 (m, 1H), 2,20 - 2,05 (m, 5H), 1,84 - 1,76 (m, 2H), 1,38 - 1,32 (m, 2H), 1,06 - 1,00 (m, 2H).
MS m/z (ESI): 412,1 [M+H]+.
Пример 13B
N-(Цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1-гидроксициклопропан-1-карбоксамид
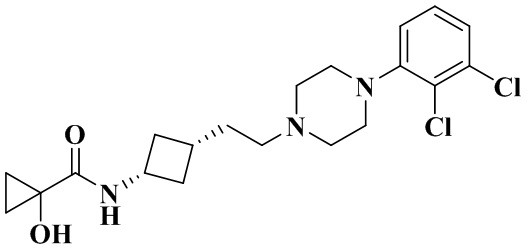
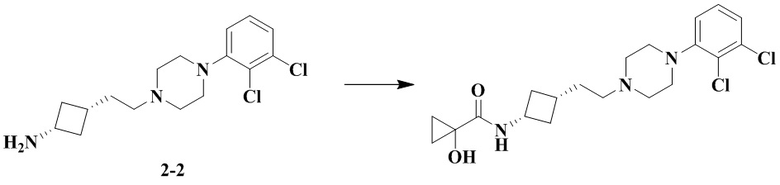
В соответствии с условиями реакции в примере 13 в качестве исходного материала применяли промежуточное соединение 2-2, соответственно получали N-(цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1-гидроксициклопропан-1-карбоксамид (13В).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,24 - 7,15 (m, 2H), 7,09 (d, J = 7,8 Гц, 1H), 7,02 - 6,98 (m, 1H), 4,36 - 4,25 (m, 1H), 3,33 (s, 4H), 3,18 - 2,95 (m, 3H), 2,73 - 2,65 (m, 2H), 2,63 - 2,54 (m, 2H), 2,05 - 1,89 (m, 4H), 1,71 - 1,59 (m, 3H), 1,36 - 1,30 (m, 2H), 1,06 - 1,00 (m, 2H).
MS m/z (ESI): 412,1 [M+H]+.
Пример 14
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-тиазол-2-карбоксамид
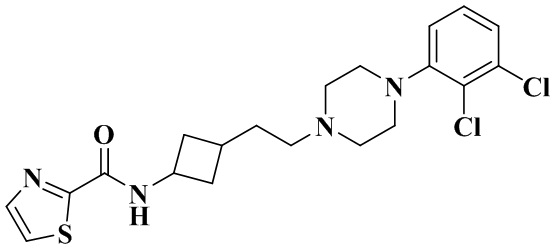
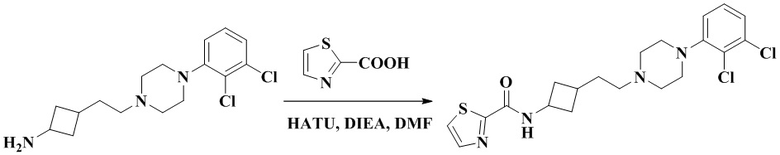
3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин (50 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (3 мл) с последующим добавлением тиазол-2-карбоновой кислоты (23 мг, 0,18 ммоль), HATU (86 мг, 0,23 ммоль) и диизопропилэтиламина (58 мг, 0,45 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение ночи и концентрировали досуха с помощью роторного испарения. Полученный неочищенный продукт очищали с помощью высокоэффективной Жидкостный хроматографии с получением N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)тиазол-2-карбоксамида (21 мг, белое твердое вещество, выход: 32%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,86 (dd, J = 3,1, 1,5 Гц, 1H), 7,57 (d, J = 3,1 Гц, 1H), 7,40 (dd, J = 42,4, 8,2 Гц, 1H), 7,20 - 7,12 (m, 2H), 7,01 - 6,93 (m, 1H), 4,73 - 4,28 (m, 1H), 3,18 - 3,03 (m, 4H), 2,78 - 2,54 (m, 6H), 2,44 - 2,35 (m, 2H), 2,24 - 2,19 (m, 1H), 2,10 - 1,98 (m, 1H), 1,81 - 1,75 (m, 1H), 1,71 - 1,65 (m, 2H).
MS m/z (ESI): 439,1 [M+H]+.
Пример 14A
N-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)тиазол-2-карбоксамид
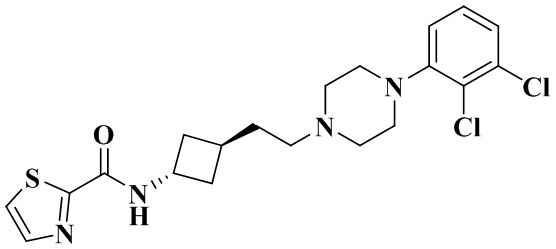
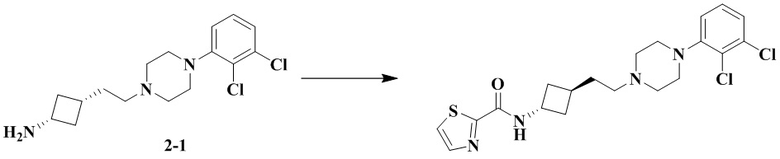
В соответствии с условиями реакции в примере 14 в качестве исходного вещества применяли промежуточное соединение 2-1, соответственно получали N-(транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)тиазол-2-карбоксамид (21 мг, белое твердое вещество, выход: 32%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,86 (d, J = 3,1 Гц, 1H), 7,57 (d, J = 3,1 Гц, 1H), 7,45 (d, J = 8,0 Гц, 1H), 7,21 - 7,13 (m, 2H), 7,00 - 6,95 (m, 1H), 4,74 - 4,59 (m, 1H), 3,18 - 3,02 (m, 4H), 2,79 - 2,58 (m, 4H), 2,47 - 2,38 (m, 2H), 2,35 - 2,27 (m, 1H), 2,25 - 2,18 (m, 4H), 1,87 - 1,77 (m, 2H).
MS m/z (ESI): 439,1 [M+H]+.
Пример 15
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-3-гидрокси-3-метилбутанамид
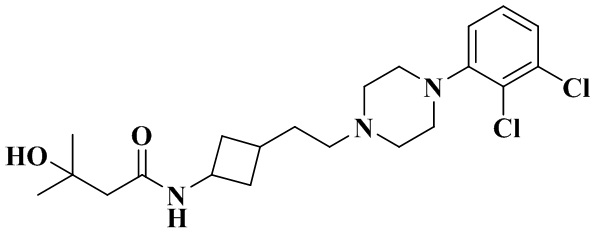
В соответствии с примером 2 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-3-гидрокси-3-метилбутинамид (21 мг, белое твердое вещество, выход: 20%).
Соединение по примеру 15 также может быть получено способом синтеза по примеру 5.
1H ЯМР (400 МГц, Хлороформ-d) δ 7,21 - 7,13 (m, 2H), 6,96 (dd, J = 7,0, 2,7 Гц, 1H), 6,08 (dd, J = 21,6, 7,5 Гц, 1H), 4,51 - 4,19 (m, 2H), 3,10 (d, J = 6,2 Гц, 4H), 2,76 - 2,62 (m, 4H), 2,56 (ddd, J = 8,9, 5,9, 2,7 Гц, 1H), 2,42 - 2,36 (m, 2H), 2,29 (d, J = 6,4 Гц, 2H), 2,05 - 1,96 (m, 3H), 1,72 (dq, J = 28,0, 7,6 Гц, 2H), 1,51 (td, J = 9,1, 2,8 Гц, 1H), 1,27 (d, J = 2,2 Гц, 6H).
MS m/z (ESI): 428,1 [M+H]+.
Пример 16
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-(5-метилоксазол-2-ил)ацетамид
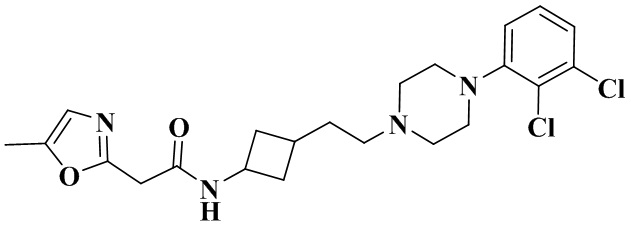
В соответствии с примером 2 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-(5-метилоксазол-2-ил)ацетамид (15 мг, белое твердое вещество, выход: 16%).
Соединение по примеру 16 также может быть получено способом синтеза по примеру 5.
MS m/z (ESI): 451,1 [M+H]+.
Пример 17
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-(3-метилоксазол-5-ил)ацетамид
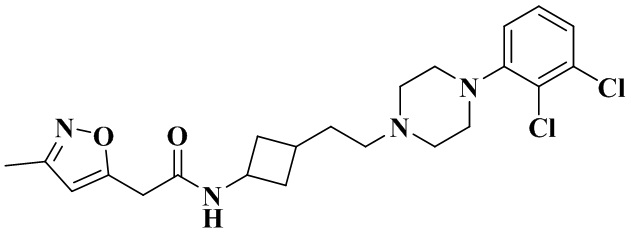
В соответствии с примером 2 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-(3-метилоксазол-5-ил)ацетамид (26 мг, белое твердое вещество, выход: 28%).
Соединение по примеру 17 также может быть получено способом синтеза по примеру 5.
MS m/z (ESI): 451,1 [M+H]+.
Пример 18
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)циклопропансульфонамид
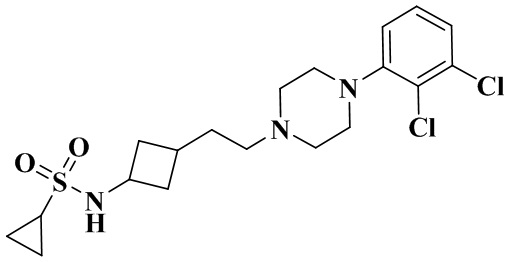
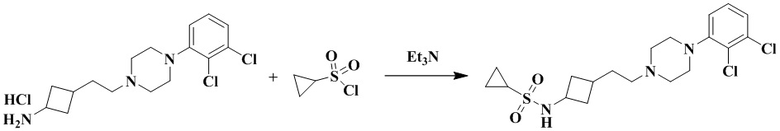
3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин гидрохлорид (40 мг, 0,11 ммоль), триэтиламин (44 мг, 0,44 ммоль) и циклопропансульфонилхлорид (31 мг, 0,22 ммоль) растворяли в дихлорметане (2 мл). Реакционный раствор перемешивали при комнатной температуре в течение 12 ч и концентрировали досуха с помощью роторного испарения для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)циклопропансульфонамида (15 мг, выход: 31,6%).
1H ЯМР (400 МГц, CDCl3) δ 7,20 - 7,10 (m, 2H), 7,00 - 6,92 (m, 1H), 4,75 - 4,60 (m, 1H), 4,14 - 3,73 (m, 1H), 3,09 (s, 4H), 2,67 (s, 4H), 2,62 - 2,49 (m, 2H), 2,42 - 2,29 (m, 3H), 2,25 - 1,89 (m, 5H), 1,79 - 1,54 (m, 4H), 1,16 (d, J = 4,8 Гц, 2H), 0,99 (q, J = 6,8 Гц, 2H).
MS m/z (ESI): 432,0 [M+H]+.
Пример 19
3-(3-(2-(4-(Бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутил)-1-этил-1-метилмочевина
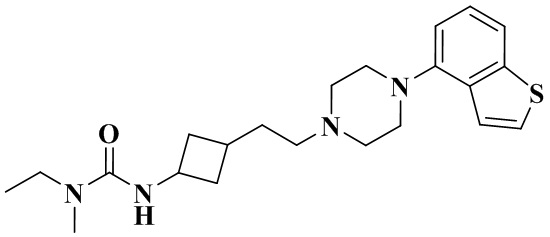
В соответствии с примером 6 в качестве исходного материала применяли 1-(бензо[b]тиофен-4-ил)пиперазин, соответственно получали 3-(3-(2-(4-(бензо[b]тиофен-4-ил)пиперазин-1-ил)этил)циклобутил)-1-этил-1-метилмочевину.
1H ЯМР (400 МГц, Хлороформ-d) δ 7,55 (d, J = 8,0 Гц, 1H), 7,40 (d, J = 3,7 Гц, 2H), 7,31 - 7,26 (m, 1H), 6,90 (d, J = 7,6 Гц, 1H), 4,54 - 4,09 (m, 2H), 3,41 - 3,15 (m, 6H), 2,85 (d, J = 4,0 Гц, 3H), 2,82 - 2,63 (m, 4H), 2,59 - 2,51 (m, 1H), 2,48 - 2,35 (m, 2H), 2,28 - 2,07 (m, 1H), 2,07 - 1,87 (m, 2H), 1,85 - 1,62 (m, 2H), 1,53 - 1,40 (m, 1H), 1,22 - 1,02 (m, 3H).
MS m/z (ESI): 401,2 [M+H]+.
Пример 20
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-оксазол-2-карбоксамид

В соответствии со стадией 1 примера 3 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)оксазол-2-карбоксамид (белое твердое вещество, выход: 26%).
Соединение по примеру 20 также может быть получено следующим способом:

3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутан-1-амин (50 мг, 0,15 ммоль) растворяли при комнатной температуре в N,N-диметилформамиде (3 мл) с последующим добавлением оксазол-2-карбоновой кислоты (20 мг, 0,18 ммоль), HATU (86 мг, 0,23 ммоль) и диизопропилэтиламина (58 мг, 0,45 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение ночи и концентрировали досуха с помощью роторного испарения. Полученный неочищенный продукт очищали с помощью высокоэффективной Жидкостный хроматографии с получением N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)оксазол-2-карбоксамида (13 мг, белое твердое вещество, выход: 21%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,79 (d, J = 1,9 Гц, 1H), 7,24 - 7,14 (m, 4H), 6,98 (m, 2,0 Гц, 1H), 4,68 - 4,59 (m, 0,3H), 4,48 - 4,38 (m, 0,7H), 3,26 - 3,15 (m, 4H), 2,98 - 2,81 (m, 4H), 2,64 - 2,56 (m, 2H), 2,22 (t, J = 7,0 Гц, 2H), 2,07 - 1,99 (m, 1H), 1,89 - 1,80 (m, 2H), 1,75 - 1,67 (m, 2H).
MS m/z (ESI): 423,1 [M+H]+.
Пример 20A и Пример 20B
N-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)оксазол-2-карбоксамид (20А)
N-(Цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)оксазол-2-карбоксамид (20B)
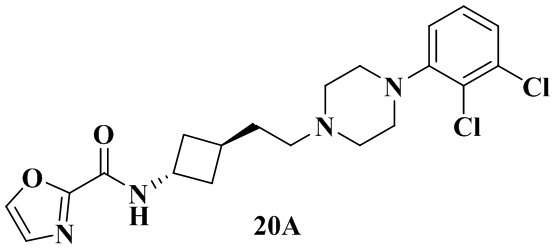
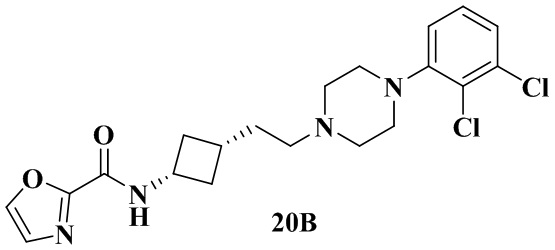
Соединение из примера 20 растворяли с получением N-(транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)оксазол-2-карбоксамида (20A) и N-(цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)оксазол-2-карбоксамида (20B), массовое соотношение 20A к 20B составляло около 1:2.
Условия получения хиральных соединений:
Пример 20A: tR = 2,473 мин.
1H ЯМР (400 МГц, Хлороформ-d) δ 7,79 (s, 1H), 7,24 - 7,20 (m, 2H), 7,17 - 7,11 (m, 2H), 6,97 (dd, J = 6,4, 3,1 Гц, 1H), 4,69 - 4,58 (m, 1H), 3,16 - 3,02 (m, 4H), 2,76 - 2,58 (m, 4H), 2,41 - 2,36 (m, 2H), 2,36 - 2,28 (m, 1H), 2,24 - 2,17 (m, 4H), 1,82 - 1,73 (m, 2H).
MS m/z (ESI): 423,1 [M+H]+.
Пример 20B: tR = 1,782 мин.
1H ЯМР (400 МГц, Хлороформ-d) δ 7,79 (s, 1H), 7,22 (s, 1H), 7,19 - 7,10 (m, 3H), 6,97 (dd, J = 7,0, 2,5 Гц, 1H), 4,49 - 4,37 (m, 1H), 3,28 - 3,03 (m, 4H), 2,84 - 2,67 (m, 4H), 2,67 - 2,54 (m, 2H), 2,53 - 2,35 (m, 2H), 2,15 - 2,02 (m, 1H), 1,75 - 1,63 (m, 4H).
MS m/z (ESI): 423,1 [M+H]+.
Пример 21
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-4-метилизоксазол-5-карбоксамид

В соответствии с примером 5 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-4-метилизоксазол-5-карбоксамид.
1H ЯМР (400 МГц, Хлороформ-d) δ 8,16 (s, 1H), 7,23 - 7,09 (m, 2H), 7,05 - 6,91 (m, 1H), 6,75 - 6,51 (m, 1H), 4,70 - 4,33 (m, 1H), 3,41 - 3,00 (m, 4H), 2,90 - 2,54 (m, 4H), 2,54 - 2,40 (m, 2H), 2,34 (s, 3H), 2,26 - 2,02 (m, 3H), 1,91 - 1,58 (m, 4H).
MS m/z (ESI): 437,1 [M+H]+.
Пример 21A
N-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-4-метилизоксазол-5-карбоксамид
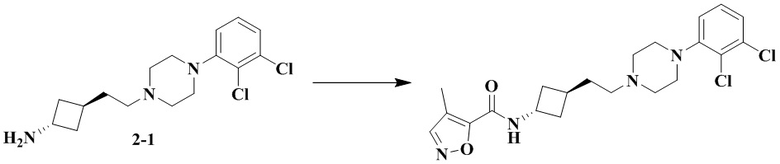
В соответствии с условиями реакции в примере 5 в качестве исходного материала применяли промежуточное соединение 2-1, соответственно получали N-(транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-4-метилизоксазол-5-карбоксамид (21А) (21 мг, белое твердое вещество, выход: 25%).
1H ЯМР (400 МГц, Хлороформ-d) δ 8,17 (s, 1H), 7,22 - 7,09 (m, 2H), 7,02 - 6,90 (m, 1H), 6,71 (d, J = 7,5 Гц, 1H), 4,69 - 4,54 (m, 1H), 3,30 - 3,00 (m, 4H), 2,86 - 2,58 (m, 4H), 2,53 - 2,39 (m, 2H), 2,34 (s, 3H), 2,33 - 2,27 (m, 1H), 2,26 - 2,12 (m, 4H), 1,91 - 1,72 (m, 2H).
MS m/z (ESI): 437,1 [M+H]+.
Пример 21B
N-(Цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-4-метилизоксазол-5-карбоксамид

В соответствии с условиями реакции в примере 5 в качестве исходного материала применяли промежуточное соединение 2-2, соответственно получали N-(цис-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-4-метилизоксазол-5-карбоксамид (21В).
MS m/z (ESI): 437,1 [M+H]+.
Пример 22
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-3-метилизоксазол-5-карбоксамид
В соответствии с примером 5 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-3-метилизоксазол-5-карбоксамид (21 мг, белое твердое вещество, выход: 32%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,21 - 7,11 (m, 2H), 7,02 - 6,93 (m, 1H), 6,77 - 6,57 (m, 2H), 4,69 - 4,33 (m, 1H), 3,30 - 3,01 (m, 4H), 2,89 - 2,56 (m, 5H), 2,49 - 2,38 (m, 2H), 2,36 (s, 3H), 2,27 - 2,15 (m, 1H), 2,10 - 1,99 (m, 1H), 1,87 - 1,57 (m, 4H).
MS m/z (ESI): 437,0 [M+H]+.
Пример 23
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)изоксазол-5-карбоксамид

В соответствии с примером 5 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)изоксазол-5-карбоксамид (14 мг, белое твердое вещество, выход: 22%).
1H ЯМР (400 МГц, Хлороформ-d) δ 8,46 (d, J = 1,6 Гц, 1H), 7,21 - 7,11 (m, 2H), 7,06 - 6,87 (m, 2H), 6,84 - 6,77 (m, 1H), 4,70 - 4,34 (m, 1H), 3,24 - 3,01 (m, 4H), 2,76 - 2,57 (m, 5H), 2,46 - 2,35 (m, 2H), 2,34 - 2,14 (m, 2H), 2,11 - 1,99 (m, 1H), 1,83 - 1,59 (m, 3H).
MS m/z (ESI): 423,0 [M+H]+.
Пример 23A
N-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)изоксазол-5-карбоксамид


В соответствии с условиями реакции в примере 5 в качестве исходного материала применяли промежуточное соединение 2-1, соответственно получали N-(транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)изоксазол-5-карбоксамид (23A) (белое твердое вещество).
1H ЯМР (400 МГц, Хлороформ-d) δ 8,34 (d, J = 1,8 Гц, 1H), 7,22 - 7,11 (m, 2H), 6,98 (dd, J = 6,7, 2,9 Гц, 1H), 6,91 (d, J = 1,9 Гц, 1H), 6,78 (d, J = 7,6 Гц, 1H), 4,70 - 4,57 (m, 1H), 3,28 - 3,02 (m, 4H), 2,86 - 2,56 (m, 4H), 2,50 - 2,39 (m, 2H), 2,39 - 2,30 (m, 1H), 2,28 - 2,14 (m, 4H), 1,88 - 1,76 (m, 2H).
MS m/z (ESI): 423,2 [M+H]+.
Пример 24
N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-метилоксазол-5-карбоксамид

В соответствии с примером 5 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-метилоксазол-5-карбоксамид.
1H ЯМР (400 МГц, CDCl3) δ 7,55 (s, 1H), 7,16 (dd, J = 7,2, 4,3 Гц, 2H), 6,96 (dd, J = 6,6, 2,8 Гц, 1H), 6,28 (d, J = 7,7 Гц, 1H), 4,47 - 4,32 (m, 1H), 3,09 (s, 4H), 2,68 - 2,58 (m, 7H), 2,41 - 2,33 (m, 2H), 2,18 (td, J = 20,3, 12,2 Гц, 2H), 2,02 (dd, J = 15,7, 8,3 Гц, 1H), 1,78 (dd, J = 15,3, 7,6 Гц, 1H), 1,71 - 1,55 (m, 3H).
MS m/z (ESI): 437,1 [M+H]+.
Пример 25
N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)изоксазол-3-карбоксамид
В соответствии с примером 5 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)изоксазол-3-карбоксамид.
1H ЯМР (400 МГц, CDCl3) δ 8,33 (d, J = 1,7 Гц, 1H), 7,19 - 7,14 (m, 2H), 7,02 - 6,93 (m, 1H), 6,91 (d, J = 1,6 Гц, 1H), 4,48-4,64 (m, 1H), 3,29-3,11 (m, 4H), 2,79 - 2,60 (m, 4H), 2,45 - 2,39 (m, 2H), 2,26-2,22 (m, 2H), 2,05-2,01 (m, 1H), 1,76-1,60 (m, 4H).
MS m/z (ESI): 423,1 [M+H]+.
Пример 25A
N-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)изоксазол-3-карбоксамид

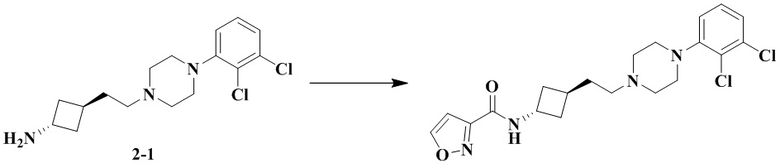
В соответствии с условиями реакции в примере 5 в качестве исходного материала применяли промежуточное соединение 2-1, соответственно получали N-(транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)изоксазол-3-карбоксамид.
1H ЯМР (400 МГц, CDCl3) δ 8,46 (d, J = 1,4 Гц, 1H), 7,22-7,17 (m, 2H), 7,00 (d, J = 6,8 Гц, 2H), 6,81 (d, J = 1,4 Гц, 1H), 4,64 (dd, J = 15,1, 7,5 Гц, 1H), 3,29 (s, 4H), 2,79-2,77 (m, 4H), 2,36 (s, 2H), 2,24 (d, J = 7,0 Гц, 2H), 2,01 (s, 1H), 1,60 (s, 4H).
MS m/z (ESI): 423,1 [M+H]+.
Пример 26
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-метилоксазол-4-карбоксамид
В соответствии с примером 5 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-метилоксазол-4-карбоксамид (белое твердое вещество).
1H ЯМР (400 МГц, Хлороформ-d) δ 8,06 (d, J = 1,6 Гц, 1H), 7,17 - 7,13 (m, 2H), 7,05 - 6,90 (m, 2H), 4,66 - 4,36 (m, 1H), 3,19 - 3,05 (m, 4H), 2,74 - 2,63 (m, 3H), 2,64 - 2,53 (m, 2H), 2,50 - 2,46 (m, 3H), 2,42 - 2,34 (m, 2H), 2,21 - 2,13 (m, 1H), 2,08 - 1,94 (m, 1H), 1,68 - 1,60 (m, 4H).
MS m/z (ESI): 437,1 [M+H]+.
Пример 26A
N-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-метилизоксазол-4-карбоксамид


В соответствии с условиями реакции в примере 5 в качестве исходного материала применяли промежуточное соединение 1-1, соответственно получали N-(транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-2-метилоксазол-4-карбоксамид.
1H ЯМР (400 МГц, Хлороформ-d) δ 8,06 (s, 1H), 7,20 - 7,11 (m, 2H), 7,05 - 6,92 (m, 2H), 4,67 - 4,54 (m, 1H), 3,19 - 3,06 (m, 4H), 2,76 - 2,63 (m, 4H), 2,48 (s, 3H), 2,44 - 2,41 (m, 2H), 2,21 - 2,11 (m, 5H), 1,84 - 1,75 (m, 2H).
MS m/z (ESI): 437,1 [M+H]+.
Пример 27
N-(3-(2-(4-(2,3-Дихлорфенил)пиперазин-1-ил)этил)циклобутил)хинолин-5-карбоксамид

В соответствии со стадией 1 примера 5 получали N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)хинолин-5-карбоксамид (35 мг, белое твердое вещество).
1H ЯМР (400 МГц, Хлороформ-d) δ 8,96 (dd, J = 4,2, 1,7 Гц, 1H), 8,76 (d, J = 8,7 Гц, 1H), 8,25 - 8,16 (m, 1H), 7,75 - 7,67 (m, 2H), 7,51 - 7,45 (m, 1H), 7,16 (dd, J = 7,0, 2,0 Гц, 2H), 7,03 - 6,95 (m, 1H), 6,29 - 6,15 (m, 1H), 4,84 - 4,52 (m, 1H), 3,23 - 3,05 (m, 4H), 2,80 - 2,67 (m, 4H), 2,52 - 2,41 (m, 2H), 2,35 - 2,04 (m, 3H), 1,70 - 1,57 (m, 4H).
MS m/z (ESI): 483,1 [M+H]+.
Пример 27A
N-(Транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)хинолин-5-карбоксамид


В соответствии с условиями реакции в примере 5 в качестве исходного материала применяли промежуточное соединение 1-1, соответственно получали N-(транс-3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)хинолин-5-карбоксамид.
1H ЯМР (400 МГц, Хлороформ-d) δ 8,96 (dd, J = 4,3, 1,7 Гц, 1H), 8,76 (d, J = 8,6 Гц, 1H), 8,25 - 8,14 (m, 1H), 7,69 (d, J = 5,0 Гц, 2H), 7,47 (dd, J = 8,6, 4,2 Гц, 1H), 7,19 - 7,14 (m, 2H), 6,98 (dd, J = 7,2, 2,4 Гц, 1H), 6,29 (d, J = 7,5 Гц, 1H), 4,84 - 4,71 (m, 1H), 3,22 - 3,14 (m, 4H), 2,94 - 2,88 (m, 1H), 2,86 - 2,74 (m, 4H), 2,56 - 2,50 (m, 2H), 2,32 - 2,26 (m, 2H), 2,25 - 2,18 (m, 2H), 1,93 - 1,84 (m, 2H).
MS m/z (ESI): 483,1 [M+H]+.
Пример 28
1-Циклопропил-3-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1-метилмочевина
В соответствии со стадией 1 примера 1 получали 1-циклопропил-3-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)-1-метилмочевину (43 мг, белое твердое вещество, выход: 33%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,22 - 7,11 (m, 2H), 7,03 - 6,92 (m, 1H), 5,32 (dd, J = 36,5, 7,6 Гц, 1H), 4,45 - 4,10 (m, 1H), 3,25 - 3,02 (m, 4H), 2,88 (d, J = 1,7 Гц, 3H), 2,82 - 2,57 (m, 4H), 2,57 - 2,51 (m, 1H), 2,47 - 2,32 (m, 3H), 2,24 - 2,10 (m, 1H), 2,08 - 1,90 (m, 1H), 2,06 - 1,75 (m, 2H), 1,50 - 1,39 (m, 1H), 0,88 - 0,78 (m, 2H), 0,75 - 0,67 (m, 2H).
MS m/z (ESI): 425,1 [M+H]+.
Пример 29
1-Циано-N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)циклопропан-1-карбоксамид
В соответствии со стадией 1 примера 5 получали 1-циано-N-(3-(2-(4-(2,3-дихлорфенил)пиперазин-1-ил)этил)циклобутил)циклопропан-1-карбоксамид (31 мг, белое твердое вещество).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,19 - 7,12 (m, 2H), 7,00 - 6,94 (m, 1H), 6,56 - 6,36 (m, 1H), 4,54 - 4,17 (m, 1H), 3,21 - 3,02 (m, 4H), 2,79 - 2,60 (m, 4H), 2,57 - 2,53 (m, 1H), 2,43 - 2,36 (m, 2H), 2,18 - 2,12 (m, 1H), 2,06 - 1,95 (m, 1H), 1,68 - 1,65 (m, 3H), 1,63 - 1,56 (m, 3H), 1,51 - 1,45 (m, 2H).
MS m/z (ESI): 421,1 [M+H]+.
Пример 30
(R)-N-(3-(2-(4-(2,3-дихлорфенил)-3-метилпиперазин-1-ил)этил)циклобутил)-2-гидрокси-2-метилпропанамид

Стадия 1: Трет-бутил (R)-4-(2,3-дихлорфенил)-3-метилпиперазин-1-карбоксилат
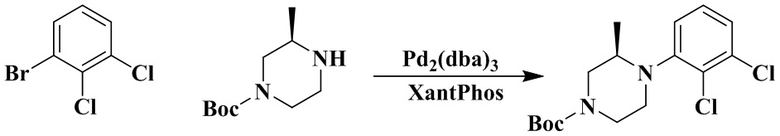
В соответствии со стадией 1 примера 2 в качестве исходных материалов применяли 1-бром-2,3-дихлорбензол и трет-бутил (R)-3-метилпиперазин-1-карбоксилат, соответственно получали трет-бутил (R)-4-(2,3-дихлорфенил)-3-метилпиперазин-1-карбоксилат (600 мг, желтое твердое вещество, выход: 32,6%).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,26 - 7,21 (m, 1H), 7,21 - 7,11 (m, 1H), 7,11 - 6,94 (m, 1H), 3,99 - 3,00 (m, 7H), 1,49 (s, 9H), 0,91 (d, J = 6,3 Гц, 3H).
MS m/z (ESI): 345,1 [M+H]+.
Стадия 2: (R)-1-(2,3-Дихлорфенил)-2-метилпиперазин

В соответствии со стадией 2 примера 2 в качестве исходного материала применяли трет-бутил (R)-4-(2,3-дихлорфенил)-3-метилпиперазин-1-карбоксилат, соответственно получали (R)-1-(2,3-дихлорфенил)-2-метилпиперазин (420 мг, желтое твердое вещество, выход: 98,8%).
1H ЯМР (400 МГц, Метанол-d4) δ 7,36 - 7,29 (m, 1H), 7,27 - 7,16 (m, 2H), 3,60 - 3,44 (m, 1H), 3,42 - 3,27 (m, 2H), 3,21 - 3,13 (m, 2H), 3,02 - 2,81 (m, 2H), 0,88 (d, J = 6,3 Гц, 3H).
MS m/z (ESI): 245,1 [M+H]+.
Стадия 3: (R)-3-(2-(4-(2,3-Дихлорфенил)-3-метилпиперазин-1-ил)этил)циклобутан-1-амин
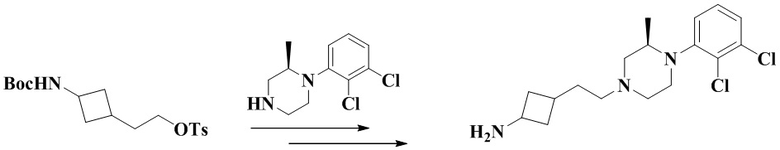
В соответствии со стадиями 6 и 7 примера 2 получали (R)-3-(2-(4-(2,3-дихлорфенил)-3-метилпиперазин-1-ил)этил)циклобутан-1-амин (280 мг).
MS m/z (ESI): 342,1 [M+H]+.
Стадия 4: (R)-N-(3-(2-(4-(2,3-Дихлорфенил)-3-метилпиперазин-1-ил)этил)циклобутил)-2-гидрокси-2-метилпропанамид
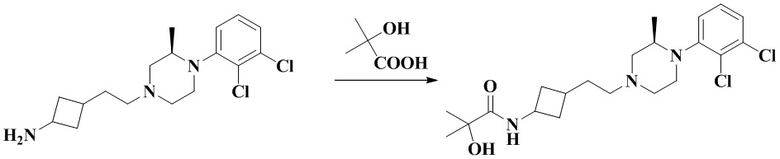
В соответствии с примером 5 получали (R)-N-(3-(2-(4-(2,3-дихлорфенил)-3-метилпиперазин-1-ил)этил)циклобутил)-2-гидрокси-2-метилпропанамид (18 мг).
1H ЯМР (400 МГц, Хлороформ-d) δ 7,25 - 7,20 (m, 1H), 7,16 (t, J = 7,9 Гц, 1H), 7,10 - 7,04 (m, 1H), 6,91 - 6,75 (m, 1H), 4,49 - 4,14 (m, 1H), 3,47 - 3,34 (m, 1H), 3,21 - 3,13 (m, 1H), 2,91 - 2,82 (m, 1H), 2,83 - 2,68 (m, 2H), 2,59 - 2,48 (m, 2H), 2,38 - 2,31 (m, 2H), 2,24 - 2,12 (m, 2H), 2,11 - 1,93 (m, 2H), 1,82 - 1,73 (m, 1H), 1,70 - 1,65 (m, 1H), 1,56 - 1,46 (m, 2H), 1,44 (d, J = 2,4 Гц, 6H), 0,90 (d, J = 6,2 Гц, 3H).
MS m/z (ESI): 428,1 [M+H]+.
Биологический анализ и оценка
Настоящее изобретение дополнительно описано ниже вместе со следующими тестовыми примерами, которые не предназначены для ограничения объема настоящего изобретения.
I. Анализ радиолиганд-рецепторного связывания
Тестовый пример 1. Определение способности соединений по настоящему изобретению связываться с рецептором дофамина D3
1. Цель эксперимента:
Целью данного тестового примера является определение аффинности указанных соединений к рецептору дофамина D3.
2.1 Приборы для эксперимента:
Вихревая мешалка (IKA, MS3 Basic)
Электронагревательный инкубатор постоянной температуры (Shanghai Yiheng Scientific Instruments Co., Ltd, DHP-9032)
Шейкер для микропланшетов (VWR, 12620-928)
Жидкостный сцинтилляционный счетчик TopCount (PerkinElmer, NTX)
Универсальный клеточный комбайн (PerkinElmer, UNIFILTER-96).
2.2 Реагенты и расходные материалы для эксперимента:
[3H]-метилспиперон (PerkinElmer, NET856250UC)
Мембрана рецептора дофамина человека D3 (PerkinElmer, ES-173-M400UA)
GR 103691 (4′-ацетил-N-[4-[4-(2-метоксифенил)-1-пиперазинил]бутил]-[1,1′бифенил]-4-карбоксамид) (Sigma, 162408-66-4)
Сцинтиллятор ULTIMA GOLD (PerkinElmer, 77-16061)
Глубоколуночные планшеты на 96 лунок объемом 1,1 мл с круглым дном (PerkinElmer, P-DW-11-C)
Планшет фильтрационный UNIFILTER-96 GF/B (PerkinElmer, 6005174)
Полиэтиленимин, разветвленный (Sigma, 408727)
Центрифужные пробирки (BD, 352096, 352070)
Загрузочная кювета для жидкости (JET BIOFIL, LTT001050)
Подложка для кюветы (JET BIOFIL, LTT001050)
Наконечники для пипеток (Axygen, T-300-R-S, T-200-Y-R-S, T-1000-B-R-S)
Хлорид магния (Sigma, 7786-30-3)
Трис-основание (Sigma, 77-86-1)
3. Экспериментальный способ:
0,5-5 мкл тестируемых соединений (0,005-100 нМ, всего по 10 концентраций) и 100 мкл буфера добавляли в 96-луночный аналитический планшет. В каждую лунку добавляли 0,5 мкл клеточной мембраны и 300 мкл буфера. К буферу добавляли [3H]-метилспиперон и инкубировали планшет при 27 °С в течение 30 мин. Фильтрационный планшет UNIFILTER-96 GF/B, предварительно инкубированный в течение 1 ч с 0,5% PEI (полиэтиленимин), дважды промывали буфером (1 мл/лунку). Суспензию клеточной мембраны добавляли к фильтрационному планшету UNIFILTER-96 GF/B, промывали 4 раза и инкубировали при 55 °С в течение 10 мин. В каждую лунку добавляли 40 мкл ULTIMA GOLD и проводили подсчет сцинтилляции в жидкости.
4. Способ обработки экспериментальных данных:
Значения CPM (число импульсов в минуту) были определены с помощью счетчика TopCount. Процент скорости ингибирования связывания [3H]-метилспиперона рассчитывали из значений экспериментальной группы с высоким контролем (контроль ДМСО) и экспериментальной группы с низким контролем (100 нМ положительного соединения) {% скорости ингибирования = (CPMобразец - CPMнизкий контроль) / (CPMвысокий контроль - CPMнизкий контроль) × 100}. После 3-кратного разбавления реакционной системы 10-ть концентраций указанного соединения составляли от 100 нМ до 0,005 нМ. Данные о проценте скорости ингибирования и о концентрации в десяти точках ввели в параметрическое нелинейное логистическое уравнение с помощью GraphPad Prism для расчета значений IC50 (концентрация полумаксимального ингибирования) указанного соединения.
5. Результаты эксперимента:
Связывающую активность соединений по настоящему изобретению с D3 определяли с помощью вышеуказанного анализа, и полученные значения IC50 приведены в таблице 1.
Таблица 1. IC50 связывающей активности соединений по настоящему изобретению с D3
IC50 (нM)
6. Вывод из эксперимента:
Соединения по настоящему изобретению обладают хорошей аффинностью к рецептору дофамина D3.
Тестовый пример 2. Определение способности соединений по настоящему изобретению связываться с рецептором 5-HT2A
1. Цель эксперимента:
Целью данного тестового примера является определение аффинности указанных соединений к рецептору 5-HT2A.
2.1 Приборы для эксперимента:
Вихревая мешалка (IKA, MS3 Basic)
Электронагревательный инкубатор постоянной температуры (Shanghai Yiheng Scientific Instruments Co., Ltd, DHP-9032)
Шейкер для микропланшетов (VWR, 12620-928)
Жидкостный сцинтилляционный счетчик TopCount (PerkinElmer, NTX)
Универсальный клеточный комбайн (PerkinElmer, UNIFILTER-96).
2.2 Реагенты и расходные материалы для эксперимента:
[3H]-Кетансерин (PerkinElmer, NET791)
Мембрана рецептора дофамина человека 5-HT2A (PerkinElmer)
GR 103691 (Sigma, 162408-66-4)
Сцинтиллятор ULTIMA GOLD (Perkin Elmer, 77-16061)
Глубоколуночные планшеты на 96 лунок объемом 1,1 мл с круглым дном (Perkin Elmer, P-DW-11-C)
Планшет фильтрационный UNIFILTER-96 GF/B (PerkinElmer, 6005174)
Полиэтиленимин, разветвленный (Sigma, 408727)
Центрифужные пробирки (BD, 352096, 352070)
Загрузочная кювета для жидкости (JET BIOFIL, LTT001050)
Подложка для кюветы (JET BIOFIL, LTT001050)
Наконечники для пипеток (Axygen, T-300-R-S, T-200-Y-R-S, T-1000-B-R-S)
Хлорид магния (Sigma, 7786-30-3)
Трис-основание (Sigma, 77-86-1)
3. Экспериментальный способ:
0,5-5 мкл указанных тестируемых соединений (0,005-100 нМ, всего по 10 концентраций) и 100 мкл буфера добавляли в 96-луночный аналитический планшет. В каждую лунку добавляли 0,5 мкл клеточной мембраны и 300 мкл буфера. К буферу добавляли [3H]-кетансерин и инкубировали планшет при 27 °С в течение 30 мин. Фильтрационный планшет UNIFILTER-96 GF/B, предварительно инкубированный в течение 1 ч с 0,5% PEI, дважды промывали буфером (1 мл/лунку). Суспензию клеточной мембраны добавляли к фильтрационному планшету UNIFILTER-96 GF/B, промывали 4 раза и инкубировали при 55 °С в течение 10 мин. В каждую лунку добавляли 40 мкл ULTIMA GOLD и проводили подсчет сцинтилляции в жидкости.
4. Способ обработки экспериментальных данных:
Значения CPM (число импульсов в минуту) были определены с помощью счетчика TopCount. Процент скорости ингибирования связывания [3H]-кетансерина рассчитывали из значений экспериментальной группы с высоким контролем (контроль ДМСО) и экспериментальной группы с низким контролем (100 нМ положительного соединения) {% скорости ингибирования = CPMобразец - CPMнизкий контроль) / (CPMвысокий контроль - CPMнизкий контроль) × 100}. После 3-кратного разбавления реакционной системы 10-ть концентраций указанного соединения составляли от 100 нМ до 0,005 нМ. Данные о проценте скорости ингибирования и о концентрации в десяти точках ввели в параметрическое нелинейное логистическое уравнение с помощью GraphPad Prism для расчета значений IC50 указанного соединения.
5. Результаты эксперимента:
Связывающую активность соединений по настоящему изобретению с 5-HT2A определяли с помощью вышеуказанного анализа, и полученные значения IC50 приведены в таблице 2.
Таблица 2. IC50 связывающей способности соединений по настоящему изобретению с 5-HT2A
IC50 (нM)
6. Вывод из эксперимента:
Приведенные выше данные показывают, что соединения по настоящему изобретению обладают хорошей аффинностью к 5-HT2A.
II. Анализ клеточной функции
Тестовый пример 1. Определение влияния соединений по настоящему изобретению на содержание цАМФ в клетках, стабильно экспрессирующих рецепторы D3
1. Цель эксперимента:
Определить активирующее влияние указанных соединений на рецептор D3.
2.1 Приборы для эксперимента:
384-луночный аналитический планшет (Perkin Elmer, 6007680)
96-луночный полипропиленновый планшет без РНКаз/ДНКаз с коническим дном (ThermoFisher, 249944)
Пипетка (Axygen)
Считыватель микропланшетов EnVision (Perkin Elmer).
2.2 Реагенты для эксперимента:
Фетальная бычья сыворотка (Gibco, 10999141)
Среда Хэма F-12K (Kaighn) (Hyclone, SH30526.01)
Пенициллин-стрептомицин, жидкий (Gibco, 15140122)
G418 (Invitrogen, 0131-027)
Форсколин (Selleck, S2449)
Стабилизатор BSA (бычий сывороточный альбумин) (Perkin Elmer, CR84-100)
Набор для определения цАМФ (Cisbio, 62AM4PEC)
IBMX (изобутилметилксантин) (Sigma, I5879)
HEPES (2-этансульфоновая кислота) (Gibco, 15630080).
3. Экспериментальный способ:
1. Получение буфера: 1 × HBSS (сбалансированный солевой раствор Хенкса) + 20 мМ HEPES + 0,1% BSA + 500 мкМ IBMX.
Полная среда: среда Хэма F12K + 10% фетальная бычья сыворотка + 1 × пенициллин-стрептомицин + 400 мкг/мл G418.
2. Клетки CHO-D3 культивировали в полной среде при 37 °C, 5%-ным CO2. После расщепления ферментом TrypLE клетки ресуспендировали в экспериментальном буфере и высевали в 384-луночный планшет для культивирования клеток с плотностью посева 8000 клеток на лунку.
3. Получали экспериментальный буфер (1 × HBSS, 0,1% BSA, 20 мМ HEPES и 500 мкМ IBMX). Соединение разбавляли буфером. В каждую лунку добавляли 2,5 мкл раствора соединения и инкубировали планшет при 37 °С в течение 10 мин. Форсколин восьмикратно разбавляли экспериментальным буфером до 8 мкМ (8 ×). Добавляли 2,5 мкл разбавленного 8 × форсколина и инкубировали планшет при 37 °С в течение 30 мин. цАМФ-d2 и Анти-цАМФ-Eu3+ размораживали и разбавляли в 20 раз лизирующим буфером. В экспериментальную лунку добавляли 10 мкл цАМФ-d2 с последующим добавлением 10 мкл Анти-цАМФ-Eu3+. Реакционный планшет центрифугировали при 200 g при комнатной температуре в течение 30 с и оставляли при 25 °С в течение 1 ч. Данные собирали с помощью Envision.
4. Способ обработки экспериментальных данных:
1) показатель Z’= 1 - 3 × (SD(стандартное отклонение)макс + SDмин) / (Среднеемакс - Среднеемин);
2) CVмакс (коэффициент вариации максимальный) = (SDмакс / Среднеемакс) × 100%;
3) CVмин (коэффициент вариации минимальный) = (SDмин / Среднеемин) × 100%;
4) S/B = Сигнал/Фон;
5) EC50 (полумаксимальная эффективная концентрация) соединения рассчитывали с помощью уравнения нелинейной аппроксимации GraphPad:
Y = Нижнее плато + (Верхнее плато - Нижнее плато) / (1 + 10 ^ ((LogEC50 - X) × HillSlope (угловой коэффициент)))
X: логарифмическое значение концентрации соединения; Y: % активации
5. Результаты эксперимента:
Таблица 3. Значения EC50 соединений на содержание цАМФ в клетках, стабильно экспрессирующих рецепторы D3
6. Вывод из эксперимента:
Из данных в таблице видно, что соединения из примеров настоящего изобретения демонстрируют хорошую агонистическую активность при анализе влияния содержания цАМФ в клетках, стабильно экспрессирующих рецепторы D3.
Тестовый пример 2. Определение влияния соединений по настоящему изобретению на подвижность иона кальция в клетках, стабильно экспрессирующих рецепторы 5-HT2A
1. Цель эксперимента:
Определить ингибирующее влияние указанных соединений на рецептор 5-HT2A.
2. Приборы и реагенты для эксперимента:
2.1 Приборы для эксперимента:
384-луночный аналитический планшет (Corning, 3712)
Пипетка (Axygen)
Система FLIPR (Molecular Devices)
2.2 Реагенты для эксперимента:
DMEM (Модифицированная по способу Дульбекко среда Игла) (Invitrogen, 11965)
Фетальная бычья сыворотка (Biowest, S1810-500)
Фетальная бычья сыворотка, диализированная (S-FBS-AU-065, Serana)
Пенициллин-стрептомицин (Biowest, L0022-100)
Гигромицин B (CABIOCHEM, 400052)
Matrigel (BD, 354230)
ДМСО (Sigma, D2650)
HBSS (Invitrogen, 14065)
HEPES (Invitrogen, 15630080)
Пробенецид (Sigma, P8761)
BSA (Renview, FA016)
Фермент TrypLE (ThermoFisher, 12604021).
3. Экспериментальный способ:
1) Получение буфера: 1 × HBSS, 20 мМ HEPES, 2,5 мМ пробенецида (пробенецид представлял собой 400 мМ исходный раствор в 1М NaOH), 0,1% BSA. Пробенецид и BSA добавляли свежими в день эксперимента. Экспериментальные буферы включают буфер-краситель и буфер для разбавления соединения.
2) Клеточная культуральная среда: среда Хэма F-12K + 10% фетальная бычья сыворотка + 600 мкг/мл гигромицина B + 1 × пенициллин-стрептомицин. Среда для посева: среда Хэма F-12K + 10% диализированная сыворотка. Аналитический буфер: 1 × HBSS + 20 мМ HEPES. Клеточная линия: стабильный пул Flp-In-CHO-5HT2A.
3) Клетки культивировали в полной среде при 37 °C, 5% CO2 до 70%-90% конфлюэнтности. Клетки расщепляли трипсином TrypLE, высевали в 384-луночный аналитический планшет с плотностью 1 × 104 клеток/лунку и инкубировали в течение 16-24 ч (по меньшей мере в течение ночи).
4) 20× компонент A размораживали до комнатной температуры, разбавляли до 2× рабочей концентрации (содержащей 5 мМ пробенецида) буфером для анализа и помещали при комнатной температуре для последующего применения.
5) Планшет для культивирования клеток извлекали и оставляли при комнатной температуре в течение 10 мин. FBS (фетальная бычья сыворотка) разбавляли до концентрации 0,03% с помощью Абрикосового и аналитического буфера, и в конце оставляли 20 мкл раствора в культуральном планшете на 3764 лунок. В каждую экспериментальную лунку добавляли 20 мкл 2× компонента A (содержащего 5 мМ пробенецида), центрифугировали при комнатной температуре при 200 g в течение 3-5 с и инкубировали при 37 °С в течение 2 ч.
6) Среду отбрасывали и добавляли 20 мкл красителя. Планшет инкубировали при 37 °С в темноте в течение 60 мин и определяли кальциевый сигнал.
7) Антагонист получали перед экспериментом: рабочий раствор тестируемого соединения (6×) был составлен с ДМСО. Планшет для культивирования клеток извлекали и оставляли при комнатной температуре в течение 10 мин. 6× тестируемое соединение добавляли к 384-луночному аналитическому планшету (10 мкл/лунку), который затем инкубировали при комнатной температуре в темноте в течение 35 мин. Планшет для анализа переносили в FLIPR. В каждую экспериментальную лунку добавляли 10 мкл разбавленного 5HT с последующим добавлением соединения агониста при 6× концентрации (5 мкл/лунку). Значения определяли с помощью системы FLIPR и сохраняли. Общий анализируемый объем составлял 30 мкл, включая 20 мкл/лунку буфера-красителя, 5 мкл/лунку тестируемого соединения при 5× концентрации и 5 мкл/лунку соединения агониста при 6× концентрации.
4. Способ обработки экспериментальных данных:
Значения кальциевого сигнала определяли с помощью FLIPR. Расчетным выходом для каждой временной точки выборки в эксперименте было отношение сигналов с длиной волны от 340/510 нм до 380/510 нм. Расчет максимум минус минимум был выведен из кривой сигнала отношения. Данные о проценте скорости ингибирования и о концентрации в десяти точках были введены в параметрическое нелинейное логистическое уравнение с помощью GraphPad Prism для расчета значений IC50 соединения.
5. Результаты эксперимента:
Таблица 4. Значения IC50 соединений на подвижность ионов кальция в клетках, стабильно экспрессирующих рецепторы 5-HT2A
6. Вывод из эксперимента:
Из данных, приведенных в таблице, видно, что соединения из примеров по настоящему изобретению демонстрируют хорошую ингибирующую активность при анализе подвижности ионов кальция в клетках, стабильно экспрессирующих рецепторы 5-HT2A.
III. Фармакокинетический анализ в линии мышей Balb/c
1. Цель исследования:
В качестве тестируемых животных использовали мышей линии Balb/c. Изучали фармакокинетическое поведение в организме мыши (плазме и ткани головного мозга) соединений по примерам настоящего изобретения, вводимых перорально в дозе 5 мг/кг.
2. Протокол эксперимента:
2.1 Тестируемые соединения:
Соединения из примеров настоящего изобретения, полученные заявителем.
2.2 Тестируемые животные:
Самцы мышей линии Balb/c (12 мышей в группе), приобретенные у Shanghai Jiesijie Laboratory Animal Co., LTD, с сертификатом №: SCXK (Шанхай) 2013-0006 N0.311620400001794.
2.3 Состав препарата:
Тестируемое соединение растворяли в 0,5% CMC-Na (карбоксиметилцеллюлоза-натрий) (1% Tween80 (полисорбат 80)) с помощью ультразвуковой обработки для получения прозрачного раствора или однородной суспензии.
2.4 Введение:
После ночного голодания самцам мышей линии Balb/c (12 мышей в группе) вводили перорально тестируемое соединение в дозе 5 мг/кг и объеме 10 мл/кг.
2.5 Отбор образцов:
0,2 мл крови отбирали из сердца мыши перед введением и через 1, 2, 4, 8 и 24 ч после введения, и мышей умерщвляли CO2. Образцы хранили в пробирках EDTA-K2 и центрифугировали в течение 6 мин при 4 °C при 6000 об/мин для отделения плазмы. Образцы плазмы хранили при минус 80 °C. Цельную ткань головного мозга извлекали, взвешивали, помещали в 2 мл центрифужную пробирку и хранили при минус 80 °C.
2.6 Работа с образцом:
1) 160 мкл ацетонитрила добавляли к 40 мкл образца плазмы для осаждения, а затем смесь центрифугировали в течение 5-20 мин при 3500 g.
2) 90 мкл ацетонитрила, содержащего внутренний стандарт (100 нг/мл), добавляли к 30 мкл плазмы и образца гомогената головного мозга для осаждения, а затем смесь центрифугировали в течение 8 минут при 13000 об/мин.
3) 70 мкл воды добавляли к 70 мкл обработанного супернатанта и перемешивали на вихревой мешалке в течение 10 мин. 20 мкл супернатанта отбирали для анализа концентрации тестируемого соединения с помощью ЖХ-МС/МС (жидкостная хроматография с тандемной масс-спектрометрией). Аналитический прибор для ЖХ-МС/МС: AB Sciex API 4000 Qtrap.
2.7 Анализ с помощью Жидкостный хроматографии:
Условие Жидкостный хроматографии: насос Shimadzu LC-20AD
Хроматографическая колонка: Agilent ZORBAX XDB-C18 (50 × 2,1 мм, 3,5 мкм); Подвижная фаза: Элюент A представлял собой 0,1% муравьиной кислоты в воде, а Элюент B представлял собой ацетонитрил
Скорость потока: 0,4 мл/мин
Время элюирования: 0-4,0 мин, элюент следующий:
. Результаты эксперимента и анализ:
Основные параметры фармакокинетики рассчитывали с помощью WinNonlin 6.1. Результаты фармакокинетического теста на мышах приведены в следующей таблице 5:
Таблица 5. Результаты фармакокинетического анализа на мышах
№
(нг/мл)
(нг/мл)
(нг/мл × ч)
(нг/мл × ч)
(ч)
(ч)
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Плазма
Головной мозг
Вывод из эксперимента:
Из результатов фармакокинетического анализа на мышах из таблицы видно, что соединения из примеров по настоящему изобретению демонстрировали хорошие фармакокинетические свойства, как экспозиция AUC (площадь под кривой), так и максимальная концентрация в плазме Cмакс были хорошими.
IV. Анализ стабильности в микросоме печени in vitro
1. Цель эксперимента:
Оценить метаболическую стабильность соединений по настоящему изобретению в микросоме печени in vitro.
2. Приборы для эксперимента:
2.1 Приборы:
2.2 Реагенты:
3. Экспериментальный способ:
3.1. Состав рабочего раствора соединения
Состав рабочего раствора соединения: 2 мкл исходного раствора соединения добавляли к 998 мкл фосфатного буфера, и конечная концентрация составляла 20 мкМ.
Состав рабочего раствора контрольного соединения (7-гидроксикумарин): Состав соответствовал составу соединения.
3.2. Состав рабочего раствора микросомы печени
78,1 мкл микросомы с концентрацией 20 мг/мл разбавляли 100 мМ фосфатным буфером до 2,5 мл и хорошо перемешивали, и конечная концентрация составляла 0,625 мг/мл.
3.3. Составление NADPH и UDPGA
33,3 мг NADPH и 25,8 мг UDPGA взвешивали соответственно с последующим добавлением 2 мл 100 мМ фосфатного буфера. Конечные концентрации составляли 20 мМ.
3.4. Состав каналообразующего реагента (аламетицина)
1 мг аламетицина взвешивали, к нему добавляли 200 мкл ДМСО для получения раствора с концентрацией 5 мг/мл. 10 мкл этого раствора добавляли к 990 мкл фосфатного буфера (pH 7,4), и конечная концентрация составляла 50 мкг/мл.
3.5. Состав останавливающего раствора для реакции
Останавливающий раствор: холодный ацетонитрил, содержащий 100 нг/мл гидрохлорида лабеталола и 400 нг/мл толбутамида в качестве внутренних стандартов, хранят в холодильнике при температуре 2-8 °C.
3.6. Способ инкубирования
400 мкл приготовленной микросомы печени, 25 мкл рабочего раствора соединения (10 мкМ) и 25 мкл аламетицина (50 мкг/мл) последовательно добавляли в 96-луночный планшет, который затем предварительно инкубировали при 37 °С в течение 10 мин. Для инициирования реакции добавляли 50 мкл состава NADPH/UDPGA, и инкубировали планшет при 37 °С. Общий объем реакционной системы составлял 500 мкл. Окончательное содержание компонентов выглядит следующим образом:
50 мкл образца отбирали в моменты времени 0 мин, 5 мин, 10 мин, 20 мин, 30 мин и 60 мин, соответственно, с последующим добавлением 200 мкл холодного останавливающего раствора, содержащего внутренние стандарты, для остановки реакции в образцах. Полученный образец центрифугировали при 4000 g в течение 10 мин, и супернатант собирали для анализа ЖХ-МС/МС.
4. Результаты эксперимента:
Таблица 6. Стабильность в микросоме печени in vitro
мин
(мкл/мин/мг белка)
(%, 120 мин.)
Примечание:
5. Вывод из эксперимента:
Приведенные выше данные показывают, что соединения из примеров настоящего изобретения умеренно метаболизируются в микросомах печени человека, крысы и собаки in vitro.
V. Фармакодинамическая модель эксперимента активного побега на крысах
1. Цель эксперимента:
Оценить антишизофренический эффект соединений с помощью фармакодинамической модели эксперимента активного побега на крысах.
2. Приборы и реагенты для эксперимента:
2.1 Приборы:
2.2 Реагенты:
2.3 Тестируемые соединения:
Соединения по примерам настоящего изобретения, полученные заявителем.
3. Тестируемые животные:
4. Состав носителя и соединений:
4.1 Носитель (0,5% CMC-Na + 1% Tween80)
Определенную массу (например, 1,0 г) CMC-Na взвешивали в стеклянной бутылке, добавляли определенный объем (например, 200 мл) очищенной воды и полученную смесь перемешивали для равномерного диспергирования. Добавляли 1% (об./об.) Tween 80 соответственно к объему раствора, и полученную смесь перемешивали в течение ночи для получения однородного прозрачного раствора, который хранили при 2-8 °C для последующего применения.
4.2 Состав соединений:
Взвешивали предписанное количество соединения с последующим добавлением предписанного объема 0,5% раствора CMC-Na + 1% Tween 80. Раствор соединения готовили перед введением, хранили при температуре 2-8 ° С и применяли в течение 4 дней.
Фактическое количество образца необходимо рассчитать во время приготовления и введения раствора соединения. Расчетное уравнение выглядит следующим образом: фактическое количество образца соединения = теоретическое количество навески образца × чистота / коэффициент соли.
5. Способ эксперимента:
После прибытия в экспериментальную лабораторию животных акклиматизировали в течение одной недели перед началом эксперимента.
5.1 Создание фармакодинамической модели:
5.1.1 Животное помещали в челночную камеру и адаптировали в течение 5 секунд с последующей 10-секундной световой и звуковой стимуляцией;
5.1.2 Если животное в течение 10-секундной световой и звуковой стимуляции сбегало на другую сторону, то электрошок не применялся, это записывалось как побеги, и единичная тренировка заканчивалась;
5.1.3 Если животное в течение 10-секундной световой и звуковой стимуляции не перемещалось на другую сторону, то применялся электрошок, сила тока составляла 0,6 мА, продолжительность - 10 секунд, если животное в течение 10 секунд электрошока сбегало на другую сторону, то электрошок прекращался, это записывалось как побеги, и единичная тренировка заканчивалась;
5.1.4 Если животное в течение 10 секунд электрошока не перемещалось на другую сторону, то электрошок прекращался, это записывалось как неудачные побеги, и единичная тренировка заканчивалась;
5.1.5 Каждое животное подвергалось тренировке 30 раз в день в общей сложности в течение 6 дней и возвращалось в клетку после тренировки.
5.2 Базовый тест и группировка
За день до скринингового теста соединения проводили базовый тест. Тест был таким же, как в пунктах 5.1.1-5.1.3, а количество базовых тестов составляло 20. Животных, чье число побегов достигало 16 (80%), группировали по количеству побегов, 10 животных на группу. Первой группе вводили носитель перорально, а другим группам вводили соответствующие тестируемые соединения в соответствии со схемой эксперимента.
5.3 Скрининговое тестирование соединения
Соединение вводили перорально (5 мл/кг) за один час до теста;
Тест был таким же, как в пунктах 5.1.1-5.1.4, а количество тестов составляло 20.
6. Способ обработки данных:
Следующие данные были собраны программным обеспечением для анализа данных:
Количество побегов животного;
Количество неудачных побегов животного;
Латентность побега животного;
Все данные измерений были выражены как среднее ± стандартная погрешность (среднее ± SEM) и проанализированы с помощью статистического программного обеспечения Graphpad 6. Разница считалась значимой, когда p было меньше 0,05.
7. Результаты эксперимента:
Таблица 7
(мг/кг)
8. Вывод из эксперимента:
Из приведенных выше данных видно, что соединения из примеров по настоящему изобретению демонстрируют хорошие эффекты в фармакодинамической модели эксперимента активного побега на крысах, что указывает на то, что они обладают антишизофреническим эффектом.
Настоящее изобретение относится к соединению формулы (IX-A), его стереоизомеру или его фармацевтически приемлемой соли, к способу получения данного соединения и к его применению:
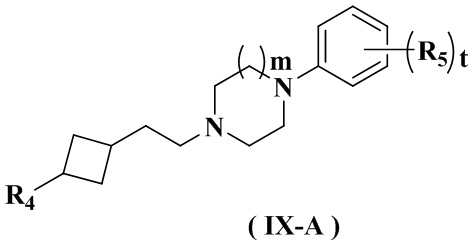 .
.
В формуле IX-A R4 выбран из группы, состоящей из  ,
, 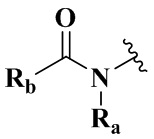 ,
,  ,
,  и
и  ; Ra выбран из группы, состоящей из водорода или C1-6 алкила; Rb выбран из группы, состоящей из водорода, амино, C1-6 алкила, C1-6 алкокси, C3-8 циклоалкила, 3-8-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, и 5-10-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, причем каждый указанный амино, C1-6 алкил, C3-8 циклоалкил, 3-8-членный гетероциклил и 5-10-членный гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, C1-6 алкила, C1-6 алкокси или C3-8 циклоалкила; R5 выбран из водорода, C1-6 алкила или галогена; или любые соседние два R5 связаны с образованием 5-6-членного гетероарила, содержащего 1 гетероатом, выбранный из N, S или O; r равен 0 или 1; m равен 0 или 1; t равен 2. Технический результат – получение новых соединений, которые могут быть использованы в качестве модуляторов рецептора, сопряженного с G-белком, а также в лечении или предупреждении заболеваний центральной нервной системы и/или психических заболеваний. 8 н. и 23 з.п. ф-лы, 7 табл., 31 пр.
; Ra выбран из группы, состоящей из водорода или C1-6 алкила; Rb выбран из группы, состоящей из водорода, амино, C1-6 алкила, C1-6 алкокси, C3-8 циклоалкила, 3-8-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, и 5-10-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, причем каждый указанный амино, C1-6 алкил, C3-8 циклоалкил, 3-8-членный гетероциклил и 5-10-членный гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, C1-6 алкила, C1-6 алкокси или C3-8 циклоалкила; R5 выбран из водорода, C1-6 алкила или галогена; или любые соседние два R5 связаны с образованием 5-6-членного гетероарила, содержащего 1 гетероатом, выбранный из N, S или O; r равен 0 или 1; m равен 0 или 1; t равен 2. Технический результат – получение новых соединений, которые могут быть использованы в качестве модуляторов рецептора, сопряженного с G-белком, а также в лечении или предупреждении заболеваний центральной нервной системы и/или психических заболеваний. 8 н. и 23 з.п. ф-лы, 7 табл., 31 пр.
1. Соединение формулы (IX-A), его стереоизомер или его фармацевтически приемлемая соль,
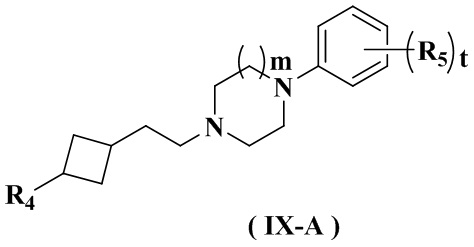
где:
R4 выбран из группы, состоящей из  ,
, 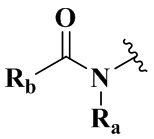 ,
, 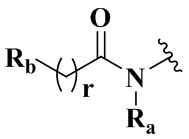 ,
,  и
и  ;
;
Ra выбран из группы, состоящей из водорода или C1-6 алкила;
Rb выбран из группы, состоящей из водорода, амино, C1-6 алкила, C1-6 алкокси, C3-8 циклоалкила, 3-8-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, и 5-10-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, причем каждый указанный амино, C1-6 алкил, C3-8 циклоалкил, 3-8-членный гетероциклил и 5-10-членный гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, C1-6 алкила, C1-6 алкокси или C3-8 циклоалкила;
R5 выбран из водорода, C1-6 алкила или галогена;
или любые соседние два R5 связаны с образованием 5-6-членного гетероарила, содержащего 1 гетероатом, выбранный из N, S или O;
r равен 0 или 1;
m равен 0 или 1; и
t равен 2;
когда  является
является 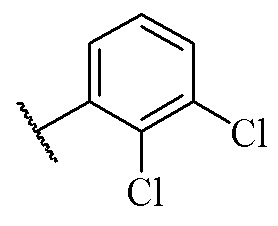 и m равно 1,
и m равно 1,
то R4 не является 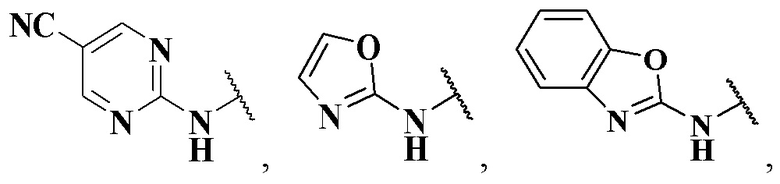 -NHC(O)C2H5, -NHC(O)N(CH3)2, -NHC(O)NHCH3, -NHC(O)N(C2H5)CH3, -NHC(O)NHC2H5,
-NHC(O)C2H5, -NHC(O)N(CH3)2, -NHC(O)NHCH3, -NHC(O)N(C2H5)CH3, -NHC(O)NHC2H5, 
 или
или 
2. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, характеризующиеся тем, что указанное соединение также представлено формулой (X) или формулой (X-A):
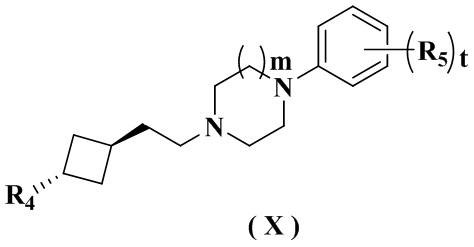
 .
.
3. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, характеризующиеся тем, что указанное соединение также представлено формулой (XI):
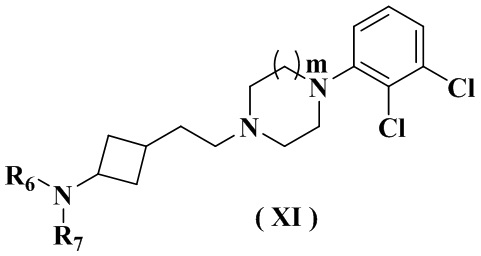
где:
R6 выбран из группы, состоящей из водорода или C1-6 алкила;
R7 выбран из группы, состоящей из -C(O)(CH2)n2Ree, -C(O)NReeRff, -C(O)NRff(CH2)n2Ree и -S(O)m2Ree;
Ree выбран из группы, состоящей из водорода, амино, C1-6 алкила, C1-6 алкокси, C3-8 циклоалкила, 3-8-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, и 5-10-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, причем каждый указанный амино, C1-6 алкил, C3-8 циклоалкил, 3-8-членный гетероциклил и 5-10-членный гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, C1-6 алкила и C1-6 алкокси;
Rff выбран из группы, состоящей из водорода, C1-6 алкила;
n2 выбран из группы, состоящей из 0 или 1;
m2 равен 2; и
m выбран из группы, состоящей из 0 или 1.
4. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, характеризующиеся тем, что Ree выбран из группы, состоящей из следующих заместителей:
(CH3)2N-, CH3NH-, CH3-, CH3CH2-, CH3CH2NH-, CH3CH2NCH3-, (CH3)2C(OH)-, (CH3)2C(OH)CH2-, CH3OCH2-, 

 и
и 
Rff выбран из группы, состоящей из водорода, C1-3 алкила.
5. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, характеризующиеся тем, что Ree выбран из группы, состоящей из водорода и следующих заместителей:
 ,
,  ,
,  ,
, 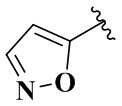 ,
, 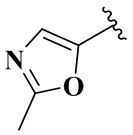 ,
,  ,
,  и
и  ;
;
Rff выбран из группы, состоящей из водорода и метила.
6. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, характеризующиеся тем, что:
R6 выбран из группы, состоящей из водорода;
R7 выбран из группы, состоящей из -C(O)(CH2)n2Ree, -C(O)NRff(CH2)n2Ree или -S(O)m2Ree;
Ree выбран из группы, состоящей из C1-3 алкила, C3-6 циклоалкила, 4-членного гетероциклила, содержащего 1 гетероатом азота, и 5-10-членного гетероарила, содержащего от 1 до 2 гетероатомов кислорода, азота, серы, необязательно дополнительно замещенной одним или более заместителями, выбранными из группы, состоящей из гидрокси, циано, C1-6 алкила и C1-6 алкокси;
Rff выбран из группы, состоящей из водорода и метила.
7. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 6, характеризующиеся тем, что Ree выбран из группы, состоящей из:
CH3-, CH3CH2-, (CH3)2C(OH)-, (CH3)2C(OH)CH2-, CH3OCH2-, 
 , , , , , , и ;
, , , , , , и ;
Rff выбран из группы, состоящей из метила.
8. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, характеризующиеся тем, что указанное соединение также представлено формулой (XI-A) или формулой (XI-B):

 .
.
9. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3 или 8, характеризующиеся тем, что  выбран из группы, состоящей из:
выбран из группы, состоящей из:  ,
, 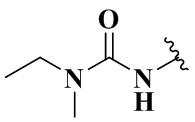 ,
, 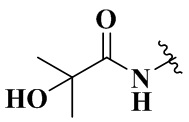 ,
,  ,
,  ,
, 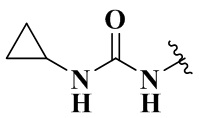 ,
, 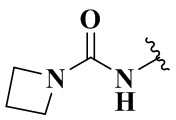 ,
,  ,
, 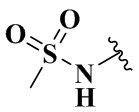 ,
,  ,
,  ,
,  ,
, 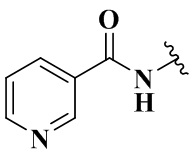 ,
, 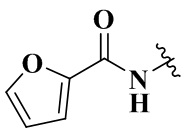 ,
, 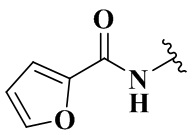 ,
, 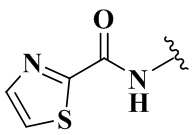 ,
,  ,
,  и
и  .
.
10. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3 или 8, характеризующиеся тем, что выбран из группы, состоящей из:  ,
, 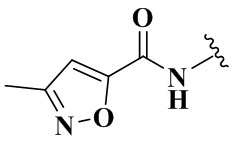 ,
,  ,
, 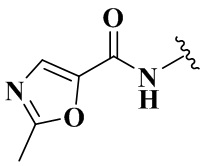 ,
,  ,
,  ,
,  ,
,  и
и  .
.
11. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, характеризующиеся тем, что указанное соединение также представлено формулой (XII):

где:
R8 выбран из группы, состоящей из амино, C1-3 алкила, C3-6 циклоалкила, 3-6-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, и 5-10-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из O, N, S, причем указанный амино, C1-3 алкил, C3-6 циклоалкил, 3-6-членный гетероциклил и 5-10-членный гетероарил необязательно дополнительно замещен одним заместителем, выбранным из группы, состоящей из гидрокси, циано, C1-3 алкила или C1-3 алкокси;
v равен 0 или 1;
когда v равен 0, то R8 не является -C2H5, -N(CH3)2, -NHCH3, -NC2H5CH3, -NHC2H5, 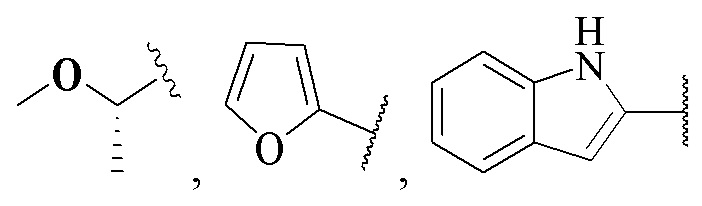 или
или  .
.
12. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 11, характеризующееся тем, что R8 дополнительно выбран из группы, состоящей из:
HOC(CH3)2-, 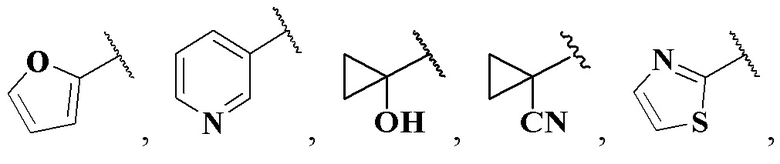 , , , , , и .
, , , , , и .
13. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 11, характеризующиеся тем, что указанное соединение с формулой (XII) также представлено формулой (XII-A) или формулой (XII-B):
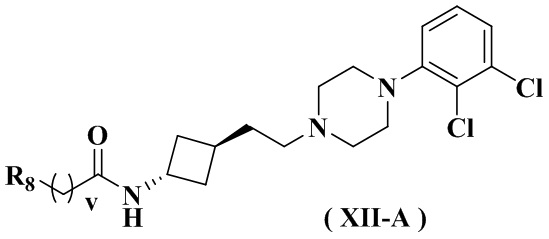 или
или  .
.
14. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, характеризующиеся тем, что:
R4 представляет собой 
Rb выбран из группы, состоящей из C3-6 циклоалкила и 5-10-членного гетероарила, содержащего от 1 до 2 атомов азота, кислорода, серы, необязательно дополнительно замещенной одним или более заместителями, выбранными из группы, состоящей из гидрокси, C1-3 алкила или C1-3 алкокси;
R5 выбран из группы, состоящей из водорода, галогена и C1-3 алкила;
m равен 1;
t равен 2;
когда r равен 0 и Rb является  , то Rb замещен по меньшей мере одним заместителем;
, то Rb замещен по меньшей мере одним заместителем;
когда r равен 0 и Rb является  , то Rb замещен по меньшей мере одним заместителем.
, то Rb замещен по меньшей мере одним заместителем.
15. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 14, характеризующиеся тем, что Rb выбран из группы, состоящей из C3-6 циклоалкила, 5-6-членного гетероарила, содержащего азот или кислород, и 9-10-членного конденсированного гетероарила, содержащего азот, необязательно дополнительно замещенной одним заместителем, выбранным из группы, состоящей из C1-3 алкила или C1-3 алкокси.
16. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 14, характеризующиеся тем, что Rb выбран из группы, состоящей из циклопропила, пиридила, фуранила, тиазолила, оксазолила, изоксазолила и хинолила, необязательно дополнительно замещенных одним заместителем, выбранным из группы, состоящей из C1-3 алкила.
17. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1-16, характеризующиеся тем, что конкретная структура указанного соединения является следующей:
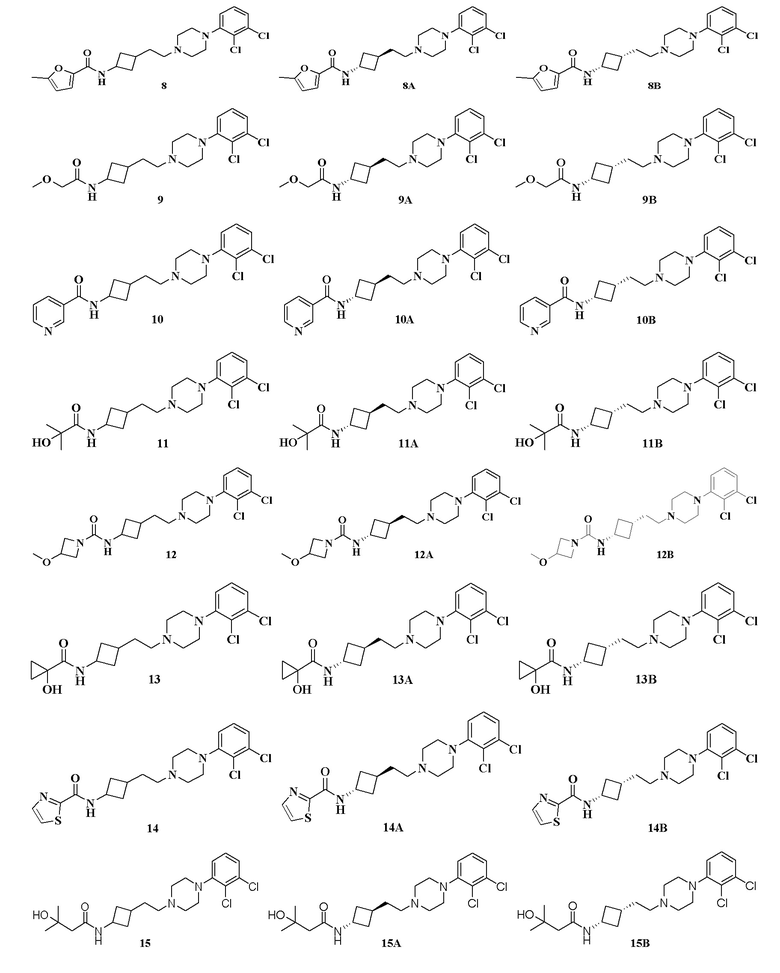
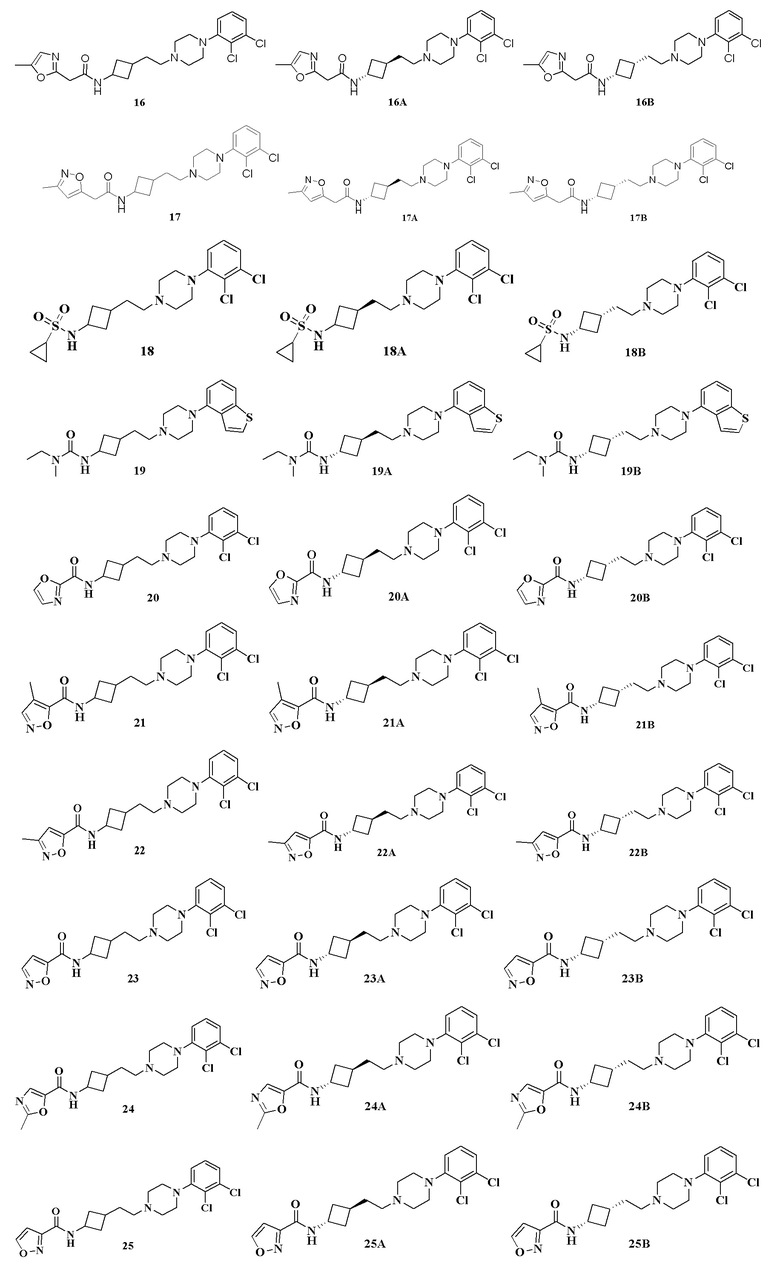


18. Соединение, его стереоизомер или его фармацевтически приемлемая соль, характеризующиеся тем, что конкретная структура указанного соединения является следующей:
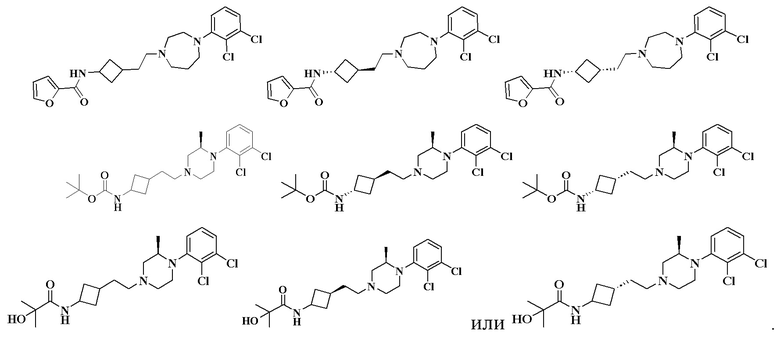
19. Способ получения соединения формулы (XII), его стереоизомера или его фармацевтически приемлемой соли по п. 11, характеризующийся тем, что он включает следующую стадию:
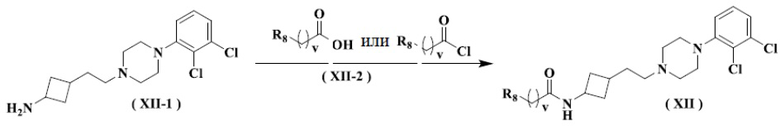
осуществление взаимодействия соединения формулы (XII-1) с ацилхлоридом или карбоновой кислотой формулы (XII-2) с получением соединения формулы (XII), его стереоизомера или его фармацевтически приемлемой соли.
20. Соединение формулы (XII-1), его стереоизомер или его соль, представляющее собой промежуточное соединение для получения соединения формулы (XII) и его стереоизомера или фармацевтически приемлемой соли, как определено по п. 11,

21. Способ получения соединения формулы (XII-1), его стереоизомера или его соли по п. 20, характеризующийся тем, что он включает следующую стадию:
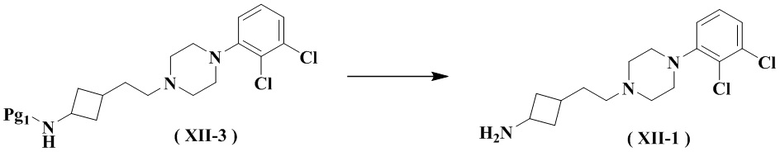
снятие защиты с соединения формулы (XII-3) с получением соединения формулы (XII-1), его стереоизомера или его фармацевтически приемлемой соли;
где:
Pg1 представляет собой аминозащитную группу, выбранную из группы, состоящей из аллилоксикарбонила, трифторацетила, 2,4-диметоксибензила, нитробензолсульфонила, тритила, флуоренметоксикарбонила, п-толуолсульфонила, формиата, ацетила, бензилоксикарбонила, трет-бутоксикарбонила, бензила и п-метоксифенила;
необязательно, осуществление взаимодействия соединения формулы (XII-4) с соединением формулы (XII-5) происходит с получением соединения формулы (XII-3), его стереоизомера или его фармацевтически приемлемой соли;
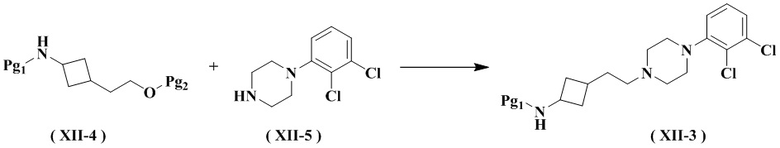
Pg2 представляет собой гидроксизащитную группу, выбранную из группы, состоящей из метила, трет-бутила, трифенила, метилтиометилового эфира, 2-метоксиэтоксиметилового эфира, метоксиметилового эфира, п-метоксибензилового эфира, пивалоила, группы бензилового эфира, метоксиметила, триметилсилила, тетрагидрофуранила, трет-бутилдизилила, ацетила, бензоила и п-толуолсульфонила.
22. Способ по п. 21, характеризующийся тем, что
Pg1 представляет собой трет-бутоксикарбонил;
Pg2 представляет собой п-толуолсульфонил.
23. Фармацевтическая композиция для модулирования рецептора, связанного с G-белком, выбранного из рецептора дофамина D3 и рецептора 5-HT2A, содержащая терапевтически эффективную дозу соединения, его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-18 и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
24. Фармацевтическая композиция по п. 23, где терапевтически эффективная доза составляет 0,1-500 мг.
25. Фармацевтическая композиция по п. 23, где терапевтически эффективная доза составляет 0,1-100 мг.
26. Фармацевтическая композиция по п. 23, где терапевтически эффективная доза составляет 0,1-50 мг.
27. Фармацевтическая композиция по п. 23, где терапевтически эффективная доза составляет 0,1-20 мг.
28. Фармацевтическая композиция по п. 23, где терапевтически эффективная доза составляет 0,5-10 мг.
29. Фармацевтическая композиция по п. 23, где терапевтически эффективная доза составляет 0,5-5 мг.
30. Применение соединения, его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-18 или фармацевтической композиции по п. 23 для получения лекарственного средства, модулирующего рецептор, связанный с G-белком, выбранный из рецептора дофамина D3 и рецептора 5-HT2A.
31. Применение соединения, его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-18 или фармацевтической композиции по п. 23 для получения лекарственного средства для лечения психического заболевания или расстройства, выбранного из шизофрении, расстройства шизофренического спектра, расстройства аутистического спектра, депрессии, аутизма, болезни Альцгеймера, судорог и мании.
| CN 105339357 A, 17.02.2016 | |||
| CN 104854103 A, 19.08.2015 | |||
| T.R | |||
| Belliotti et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Bioorganic & Medicinal Chemistry Letters, 1997, 7(18), 2403-2408 | |||
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2194048C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА/ПИПЕРАЗИНА | 2008 |
|
RU2478628C2 |

