ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
ОБЛАСТЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
[0001] Настоящее изобретение относится к способу производства соединения, способного ингибировать активность SHP2, и промежуточных продуктов, используемых в нем.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0002] Фосфатаза с Src-гомологичным доменом 2 (SHP2) представляет собой нерецепторную тирозинфосфатазу белка, кодируемую геном PTPN11, который участвует во многих клеточных функциях, включая пролиферацию, дифференциацию, поддержание клеточного цикла и миграцию. SHP2 участвует в сигнальном процессе через каскадные пути Ras-митоген-активированной протеинкиназы, JAK-STAT или фосфоинозитол-3-киназы-AKT.
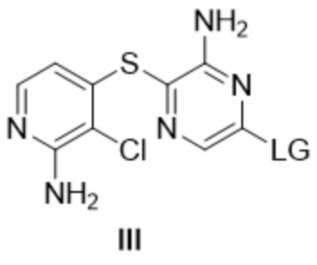
[0003] Соединение с названием (3S,4S)-8-(6-амино-5-((2-амино-3-хлорпиридин-4-ил)тио)пиразин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин, соответствующее формуле I:
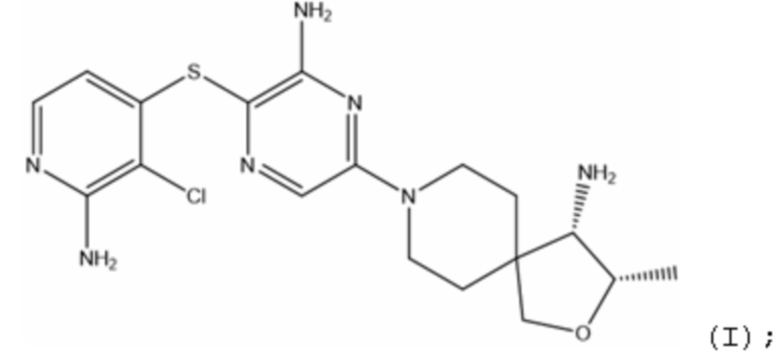
[0004] а также его фармацевтически приемлемые соли описаны в WO2015/107495 A1 в качестве ингибитора SHP2. Также описаны различные терапевтические и лечебные способы.
[0005] Фосфатаза с Src-гомологичным доменом 2 (SHP2) представляет собой нерецепторную тирозинфосфатазу белка, кодируемую геном PTPN11, который участвует во многих клеточных функциях, включая пролиферацию, дифференциацию, поддержание клеточного цикла и миграцию. SHP2 участвует в сигнальном процессе через каскадные пути Ras-митоген-активированной протеинкиназы, JAK-STAT или фосфоинозитол-3-киназы-AKT.
[0006] SHP2 имеет два N-концевых Src-гомологичных домена 2 (N-SH2 и C-SH2), каталитический домен (PTP) и C-концевой хвост. Два SH2 домена контролируют субклеточную локализацию и функциональную регуляцию SHP2. Молекула существует в неактивной, самоингибированной конформации, стабилизированной связывающей сетью, в которой участвуют остатки из обоих доменов N-SH2 и PTP. Стимулирование, например, цитокинов или факторов роста, приводит к обнажению каталитических сайтов, приводящему к ферментативной активации SHP2.
[0007] Мутации в гене PTPN11, а впоследствии и в SHP2, были обнаружены при некоторых заболеваниях человека, таких как синдром Нунан, синдром Leopard, ювенильные миеломоноцитарные лейкозы, нейробластома, меланома, острый миелоидный лейкоз и онкологические заболевания молочной железы, легкого и толстой кишки. SHP2, таким образом, представляет собой очень привлекательную цель для разработки новых способов терапевтического лечения различных заболеваний. Соединение, которое может быть получено согласно настоящему изобретению, направлено на удовлетворение потребности в малых молекулах, которые подавляют активность SHP2.
[0008] В WO/2015/107495 A1 описан способ производства соединения формулы I, который можно охарактеризовать следующей схемой синтеза 1:
Схема 1:

[0009] Последнее соединение, полученное на стадии g выше, подвергали затем реакции, как показано далее:
Схема 2:
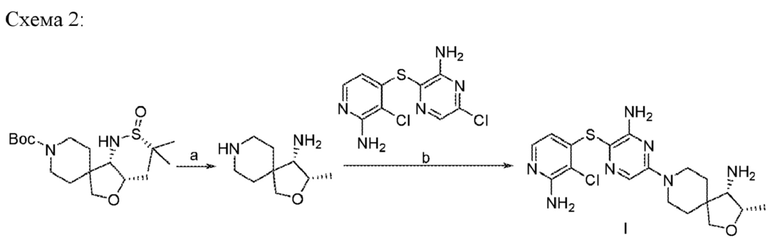
[0010] Таким образом получают соединение формулы I (последнее соединение на вышеуказанной схеме). Синтез требует как минимум 9 показанных стадий и подходит для синтеза в лабораторном масштабе.
[0011] Производство же является сложным и требует, например, разделения диастереомеров на стадии g в схеме синтеза, приведенной выше. Кроме того, многие из промежуточных продуктов не кристаллизуются, поэтому их приходится использовать, не рассчитывая на преимущества, которые обеспечивает более высокая чистота, получаемая при кристаллизации.
[0012] Кроме того, в способе используются хроматографические стадии.
[0013] Помимо этого, альдегидный исходный материал для реакции а на схеме 1 выше представляет собой соединение, известное из литературы, но недоступное в больших количествах (обычно в граммовых количествах, например, от Aldlab Chemicals), и характеризуется некоторой нестабильностью, поэтому предпочтительно его получать и немедленно использовать. Для крупномасштабного синтеза требуется, например, килограмм или более.
[0014] Кроме того, выход на стадии циклизации (стадия d на схеме 1) недостаточно высок, так как также присутствуют эдукт, тозилат желаемого продукта и дополнительные примеси, поэтому требуется разделение.
[0015] Кетоновый субстрат, полученный на стадии е на схеме 1), частично рацемизируется, даже если используется энантиомерный чистый альдегидный исходный материал, что приводит к образованию 4 диастереомеров на стадии f (которая фактически включает две стадии - восстановление и конденсацию), что приводит к двум основным дисатереомерам в соотношении 95:5, которые потребуют дальнейшего разделения.
[0016] Кроме того, в синтез вовлечено множество маслянистых промежуточных продуктов, поэтому они не идеальны для очистки, как показано на следующей схеме:
Схема 1A
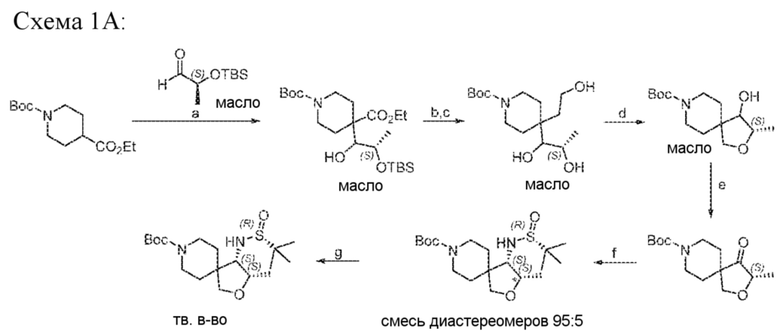
[0017] Следовательно, этот способ, хотя и технологичный особенно в лабораторном масштабе, не идеален для производства в больших масштабах.
[0018] Соединение, добавляемое в реакции b на схеме 2, получено согласно WO 2015/1107495 A1 как "промежуточное соединение 10", как указано ниже:
Схема 3:
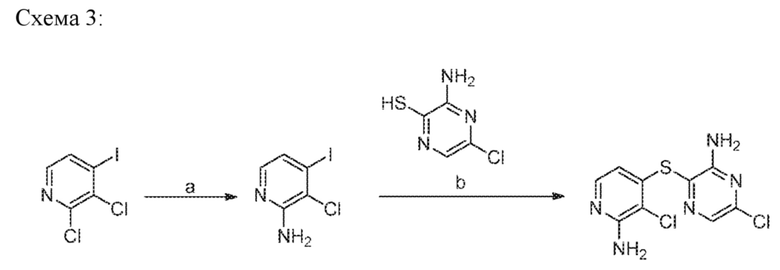
[0019] Хотя этот синтез и возможен, были бы желательны некоторые поправки, поскольку стадия аминирования «а» дает лишь умеренные выходы (например, примерно от 30 до 40%).
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0020] В одном аспекте настоящего изобретения предложен способ производства соединения формулы I, как упомянуто выше, или его фармацевтически приемлемой соли, сокристалла с кислотой, гидрата или другого сольвата.
[0021] В последующем аспекте настоящего изобретения предложен способ производства соединения формулы I, как указано выше, или его фармацевтически приемлемой соли, сокристалла с кислотой, гидрата или другого сольвата, причем указанный способ включает взаимодействие соединения формулы II с соединением формулы III согласно следующей схеме реакции:
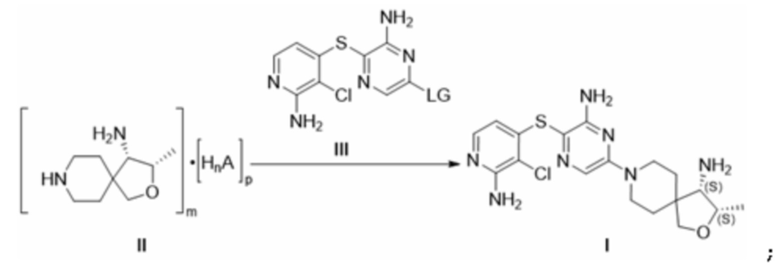
[0022] где LG представляет собой уходящую группу, A представляет собой анион протонной кислоты, а n и m являются целыми числами, которые предпочтительно принимают значения 1, 2 или 3 таким образом, что соль формулы II не имеет электрического заряда, предпочтительно m равно 1, n равно 1 и p равно 2; где соединение формулы II предпочтительно получают либо (i) снятием защиты, либо (ii) восстановлением соединения формулы IV:
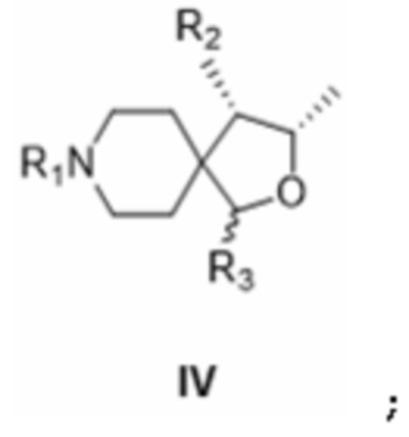
[0023] где в случае (i) R1 представляет собой защитную группу вторичной аминогруппы, и R2 представляет собой защищенную аминогруппу, и R3 представляет собой водород, или в случае (ii) R1 представляет собой защитную группу вторичной аминогруппы, R2 представляет собой аминогруппу, и R3 представляет собой гидроксил, и, если требуется (то есть, когда кислота еще не присутствует, например, из-за снятия защиты), взаимодействие полученного соединения формулы III:
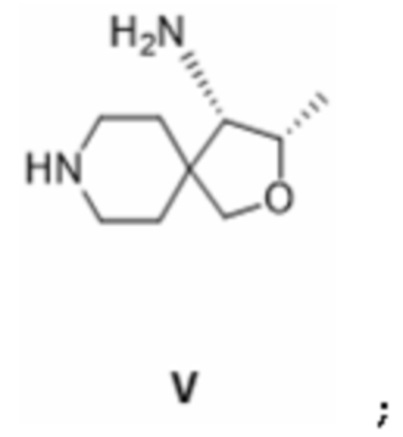
[0024] с кислотой формулы HnA с получением соединения формулы II.
[0025] В обоих случаях (i) и (ii), только что упомянутых, и в качестве предпочтительного второго аспекта изобретения, производство соединения формулы II на первой стадии, предпочтительно с последующими дальнейшими стадиями, определенными дополнительными вариантами осуществления изобретения, определенными ниже, включает взаимодействие соединения формулы V:
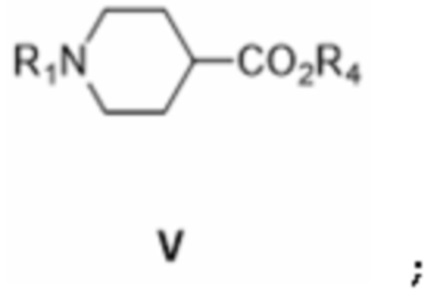
[0026] где R1 представляет собой защитную группу вторичной аминогруппы, а R4 представляет собой карбоксильную (-COOH) защитную группу, в присутствии сильного основания с L-лактидом формулы:
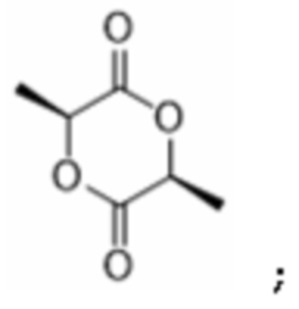
[0027] с получением соединения формулы VI:

[0028] где R1 такой, как определено для соединения формулы IV, и R5 представляет собой незамещенный или замещенный алкил, незамещенный или замещенный циклоалкил или незамещенный или замещенный арил, или, альтернативно, с получением соединения формулы VI*:
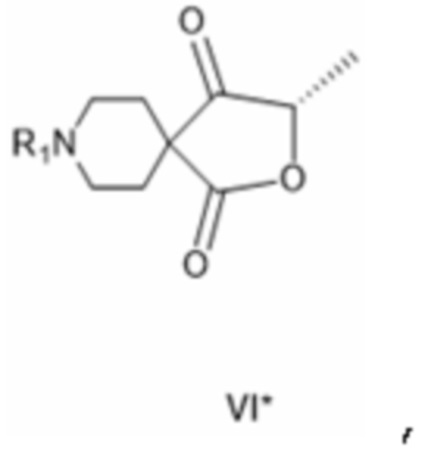
[0029] где R1 такой, как определено для соединения формулы IV.
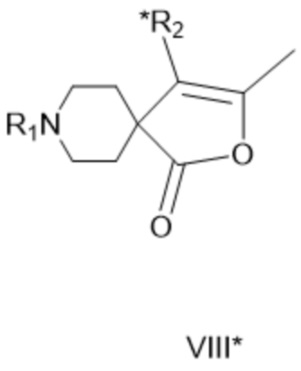
[0030] Каждый из этих двух вариантов реакции как таковой также представляет собой вариант осуществления настоящего изобретения.
[0031] В качестве дополнительного варианта осуществления изобретения или, предпочтительно, на следующей стадии соединение формулы VI, как только что описано, подвергают циклизации с гидроксиламином или его солью или, альтернативно, соединение формулы VI* циклизуют с гидроксиламином или его солью с получением производного гидроксиламина формулы VII, соответственно:
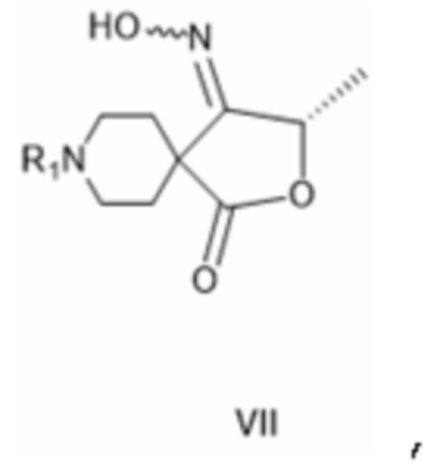
[0032] где R1 такой, как определено для соединения формулы IV.
[0033] В качестве дополнительного варианта осуществления изобретения или, предпочтительно, на следующей стадии соединение формулы VII либо (a-i) гидрируют с получением аминосоединения формулы VIII:
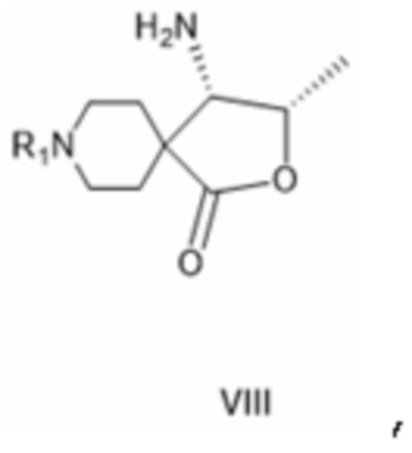
[0034] где R1 такой, как определено для соединения формулы IV, либо (a-ii) ацилируют в восстанавливающих условиях с получением соединения формулы VIII*:
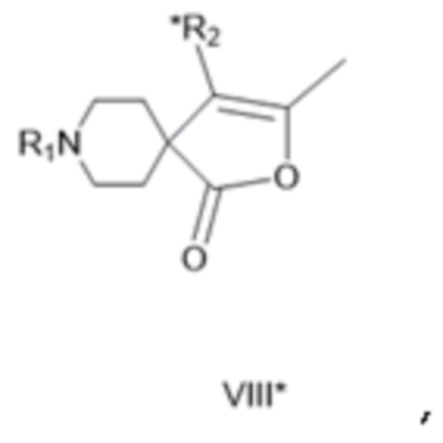
[0035] где R1 такой, как определено для соединения формулы IV, а *R2 представляет собой ацилированную аминогруппу (= защищенную ацилом аминогруппу).
[0036] В другом предпочтительном варианте осуществления изобретения или, предпочтительно, на следующей стадии после только что описанной реакции (a-i) соединение формулы VIII либо (b-i) восстанавливают с получением соединения формулы IX:
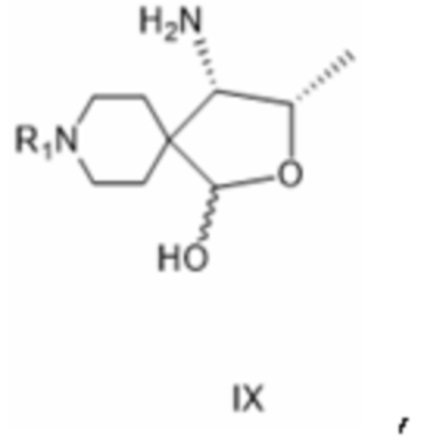
[0037] где R1 такой, как определено для соединения формулы IV, указанное соединение представляет собой соединение формулы IV, где R1 представляет собой защитную группу вторичной аминогруппы, R2 представляет собой аминогруппу, и R3 представляет собой гидроксил; где стадию восстановления (ii), упомянутую выше для соединения, соответствующего формуле IV, подпадающего под определение соединения формулы IX, предпочтительно, в качестве отдельного варианта осуществления изобретения или, более предпочтительно, на следующей стадии проводят с использованием триалкилсилана, чтобы получить после последующего добавления кислоты формулы HnA, как определено выше, соединение формулы II, как описано выше;
[0038] или (c-i), в качестве отдельного варианта осуществления изобретения или, предпочтительно, на следующей стадии взаимодействует с соединением, вставляющим защитную группу аминогруппы, с получением соединения формулы X:
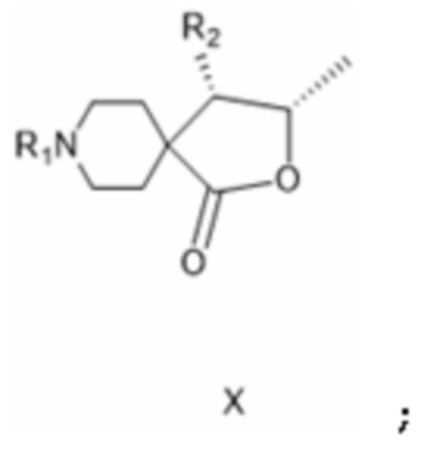
[0039] где R1 такой, как определено для соединения формулы IV, а R2 представляет собой защищенную аминогруппу, причем соединение формулы X в качестве отдельного варианта осуществления изобретения или, предпочтительно, на следующей стадии восстанавливают до соединения формулы XI:
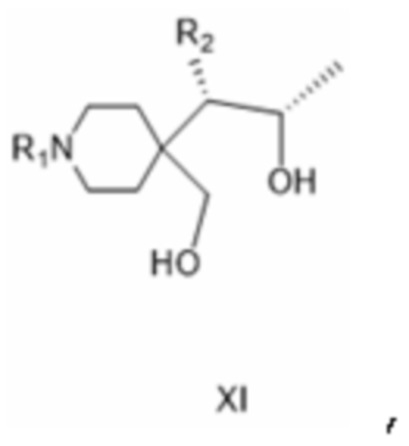
[0040] где R1 такой, как определено для соединения формулы IV, а R2 представляет собой защищенную аминогруппу; причем соединение формулы XI в качестве отдельного варианта осуществления изобретения или, предпочтительно, на следующей стадии реагирует по гидрокси-группе гидроксиметильной группы (непосредственно связанной с кольцом) с реагентом, образующим уходящую группу формулы LG*-X, в которой LG* представляет собой электрофильный радикал, способный вместе с гидрокси-группой, с которой он связан, образовывать уходящую группу LG2, а X представляет собой галоген, с образованием соединения формулы XII:
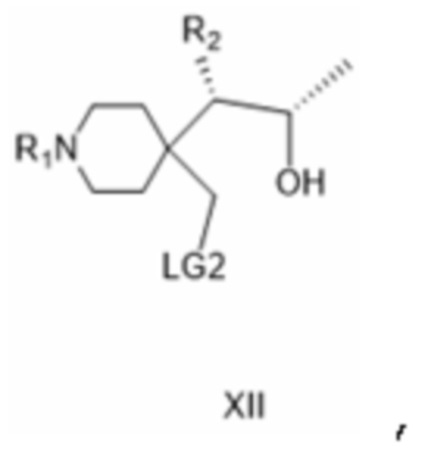
[0041] где R1 такой, как определено для соединения формулы IV, R2 представляет собой защищенную аминогруппу, а LG2 представляет собой уходящую группу;
[0042] причем соединение формулы XII затем в качестве отдельного варианта осуществления изобретения или, предпочтительно, на следующей стадии, подвергают циклизации в основных условиях с получением соединения формулы XIII:
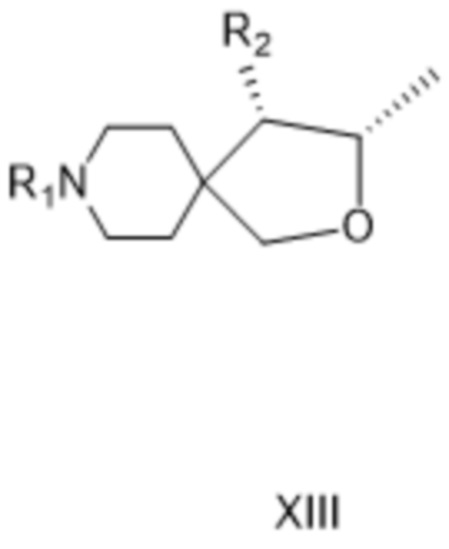
[0043] где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой защищенную аминогруппу, которая представляет собой соединение формулы IV, где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой защищенную аминогруппу, и R3 представляет собой водород, где стадия снятия защиты (i), упомянутая для соединения, соответствующего формуле IV выше соединения формулы XIII, предпочтительно, в качестве отдельного варианта осуществления изобретения или, более предпочтительно, на следующей стадии проводят с использованием кислоты HnA, как определено для c соединения формулы II, с получением соединения формулы II, как описано выше.
[0044] В другом предпочтительном варианте осуществления изобретения или, предпочтительно, на следующей стадии после реакции (a-ii), описанной выше, соединение формулы VIII* представляет собой (b-ii), в качестве отдельного варианта осуществления изобретения или, предпочтительно, на следующей стадии гидрируют в присутствии хирального катализатора гидрирования с получением соединения формулы X*:
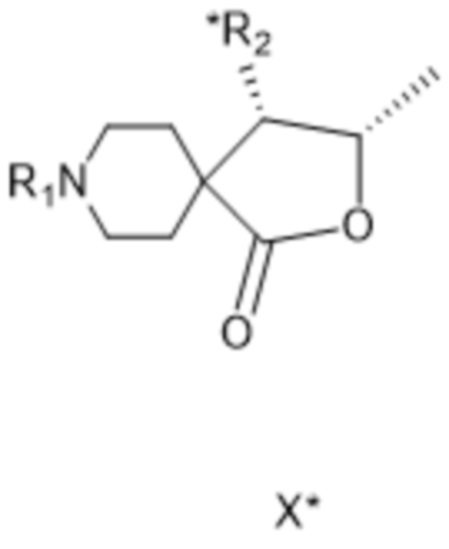
[0045] где R1 такой, как определено для соединения формулы IV, а *R2 представляет собой ацилированную аминогруппу, причем соединение формулы X* в качестве отдельного варианта осуществления изобретения или, предпочтительно, на следующей стадии восстанавливают до соединения формулы XI*:
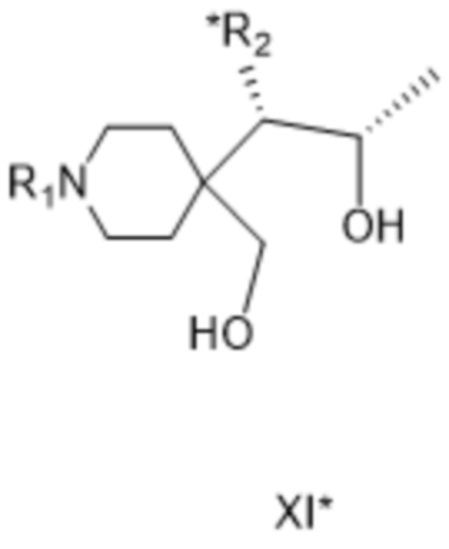
[0046] где R1 такой, как определено для соединения формулы IV, и *R2 представляет собой ацилированную аминогруппу;
[0047] где соединение формулы XI* в качестве отдельного варианта осуществления изобретения или, предпочтительно, на следующей стадии реагирует по гидрокси-группе гидроксиметильной группы (которая непосредственно связанна с кольцом в формуле XI*) с реагентом, образующим уходящую группу формулы LG*-X, в которой LG* представляет собой электрофильный радикал, способный вместе с гидрокси-группой, с которой он связан, образовывать уходящую группу LG2, а X представляет собой галоген, с образованием соединения формулы XII*:
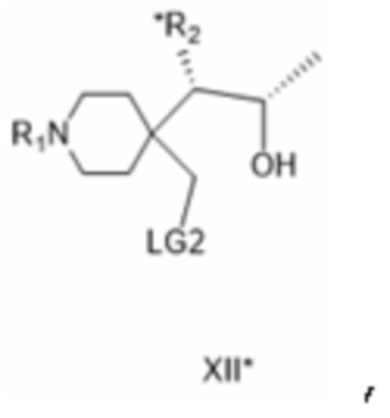
[0048] где R1 такой, как определено для соединения формулы IV, R2 представляет собой защищенную аминогруппу, а LG2 представляет собой уходящую группу;
[0049] причем соединение формулы XII затем в качестве отдельного варианта осуществления изобретения или, предпочтительно, на следующей стадии, подвергают циклизации в основных условиях с получением соединения формулы XIII*:

[0050] в которой R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу, которая соответствует соединению формулы IV, где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой ацилированную (= защищенную ацилом) аминогруппу, и R3 представляет собой водород, где стадия снятия защиты (i), упомянутая для соединения, соответствующего формуле IV выше (где в данном случае снятие защиты означает деацилирование) в соединении формулы XIII*, предпочтительно, в качестве отдельного варианта осуществления изобретения или, более предпочтительно, на следующей стадии проводят с использованием кислоты HnA, как определено для соединения формулы II, с получением соединения формулы II, как описано выше.
[0051] Каждое из следующих новых промежуточных соединений также представляет варианты осуществления изобретения:
[0052] (Соль) соединения формулы II:
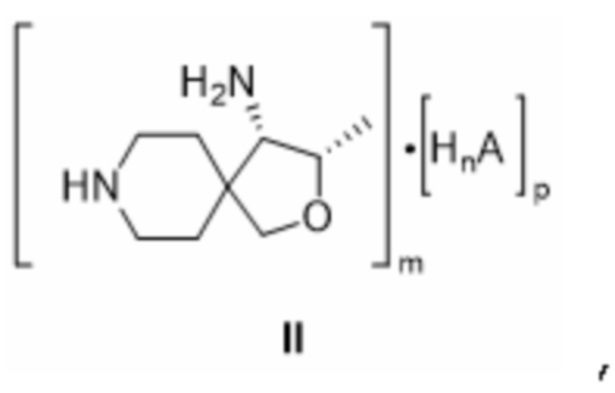
[0053] где A представляет собой анион протонной кислоты, особенно Cl, а n, m и p предпочтительно являются целыми числами, которые принимают значения 1, 2 или 3 так, чтобы соль формулы II не имела электрического заряда, где особенно n и m равны 1, а p равно 2.
[0054] Соединение формулы VI:
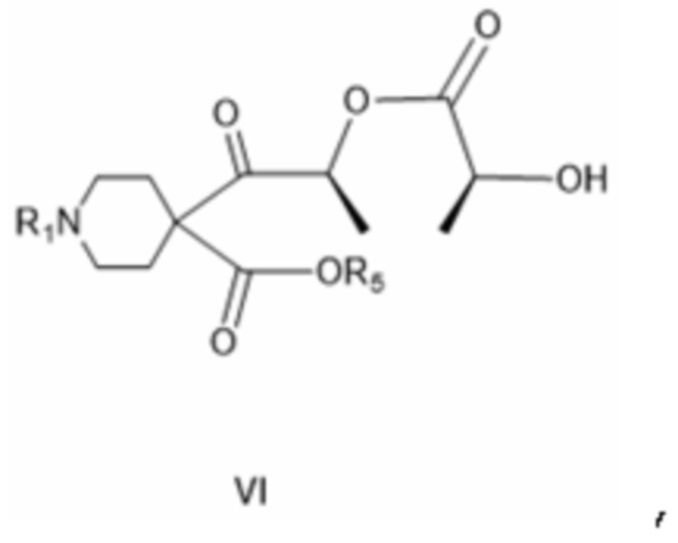
[0055] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутилоксикарбонил, а R5 представляет собой незамещенный или замещенный алкил, незамещенный или замещенный циклоалкил или незамещенный или замещенный арил, особенно - этил.
[0056] Соединение формулы VI*:
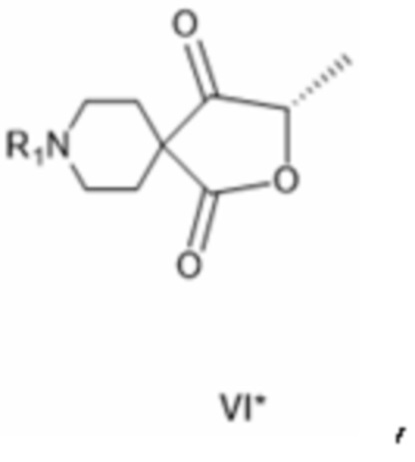
[0057] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил.
[0058] Соединение формулы VII:
[0059] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил.
[0060] Соединение формулы VIII:
[0061] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил.
[0062] Соединение формулы IX:
[0063] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил.
[0064] Соединение формулы VIII*:
[0065] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил, а *R2 представляет собой ацилированную аминогруппу, особенно - ацетиламиногруппу.
[0066] Соединение формулы X*:
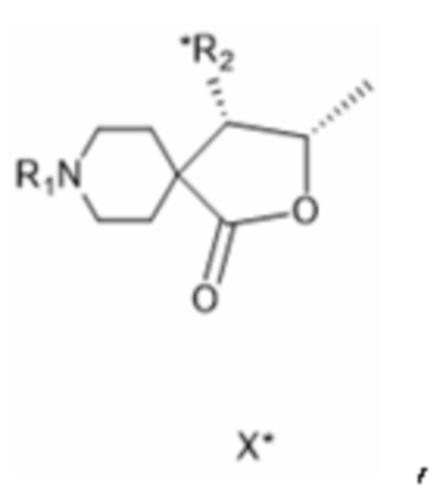
[0067] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил, а *R2 представляет собой ацилированную аминогруппу, особенно - ацетиламиногруппу.
[0068] Соединение формулы XI*:
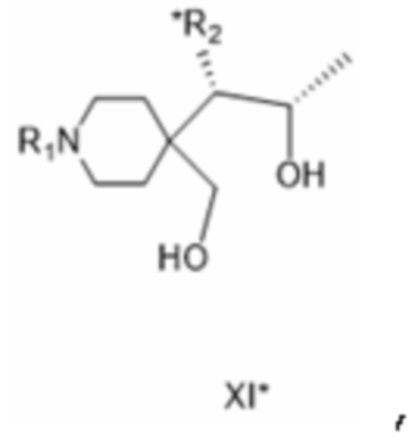
[0069] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил, а *R2 представляет собой ацилированную аминогруппу, особенно - ацетиламиногруппу.
[0070] Соединение формулы XII*:
[0071] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил, *R2 представляет собой ацилированную аминогруппу, особенно - ацетиламиногруппу, а LG2 представляет собой уходящую группу, особенно - толуолсульфонилокси-группу.
[0072] Соединение формулы XIII*:
[0073] в котором R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил, а *R2 представляет собой ацилированную аминогруппу, особенно - ацетиламиногруппу.
[0074] Соединение формулы X:
[0075] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил, а R2 представляет собой защищенную аминогруппу, особенно - трет-бутоксикарбониламиногруппу.
[0076] Соединение формулы XI:
[0077] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил, а R2 представляет собой защищенную аминогруппу, особенно - трет-бутоксикарбониламиногруппу.
[0078] Соединение формулы XII:
[0079] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил, R2 представляет собой защищенную аминогруппу, особенно - трет-бутоксикарбониламиногруппу, а LG2 представляет собой уходящую группу, особенно - толуолсульфонилокси-группу.
[0080] Соединение формулы XIII:

[0081] где R1 представляет собой защитную группу вторичной аминогруппы, особенно - трет-бутоксикарбонил, а R2 представляет собой защищенную аминогруппу, особенно - трет-бутоксикарбониламиногруппу.
[0082] Упомянутые соединения могут присутствовать в свободной форме или в виде их солей, в которых присутствуют солеобразующие группы (такие как имино- или аминогруппы), особенно солей присоединения кислоты, таких как соли с неорганической кислотой, такой как галогеноводородная кислота, например HCl, серной кислотой или фосфорной кислотой, и/или с органической кислотой, такой как сульфокислота, такой как метил- или этилсульфокислота или толуолсульфокислота, фосфоновая кислота или карбоновая кислота, например алкановая кислота, такая как уксусная кислота или лимонная кислота, в качестве нескольких примеров.
Описание предпочтительных вариантов осуществления
[0083] Следующие ниже определения обозначают более общие признаки предпочтительным, более конкретным образом, и можно заменить один, несколько или все более общие признаки в вариантах изобретения=вариантах осуществления более конкретным определением, которое определяет более конкретные варианты осуществления изобретения.
[0084] Условия для описанных выше реакций выбирают особенно следующим образом:
[0085] Реакция соединения II с соединением формулы III, где LG представляет собой уходящую группу, предпочтительно галоген, особенно хлор или бром, предпочтительно происходит в присутствии слабого основания, такого как карбонат щелочного металла или гидрокарбонат металла, в апротонном растворителе, таком как N, N-диалкиламид алкановой кислоты, например диметилацетамид или диметилформамид, предпочтительно при повышенных температурах, например от 30°C до температуры кипения реакционных смесей, например, от 50 до 100°C.
[0086] Снятие защиты (i) с соединения формулы IV, где R1 представляет собой защитную группу вторичной аминогруппы, и R2 представляет собой защищенную аминогруппу, и R3 представляет собой водород, с получением соединения формулы II предпочтительно проводят в присутствие сильной кислоты HnA, такой как трифторуксусная кислота, трифторметансульфокислота, или предпочтительно - неорганической кислоты, например серной кислоты, фосфорной кислоты или особенно галогеноводородной кислоты, особенно хлористоводородной кислоты, в растворителе, например спирте или смеси спиртов (особенно, если R2 представляет собой бензилоксикарбонил или особенно алкоксикарбонил, такой как трет-бутоксикарбонил), или в присутствии воды (особенно, если R2 представляет собой ацил, особенно низший алканоил, например, ацетил) при предпочтительных температурах от 10°C до температуры кипения растворителя, например от 20°C до (особенно, когда R2 представляет собой ацил) 115°C.
[0087] Альтернативное восстановление (ii) соединения формулы IV, где R1 представляет собой защитную группу вторичной аминогруппы, R2 представляет собой аминогруппу, а R3 представляет собой гидроксил, предпочтительно проводят, используя триалкилсилан, особенно триэтилсилан, в присутствии сильной неорганической или, предпочтительно, (сильной) органической кислоты, особенно трифторметансульфоновой кислоты, в подходящем апротонном растворителе, таком как простой эфир или особенно ацетонитрил, с последующим добавлением кислоты HnA, получая (соль или сокристалл) соединения формулы II.
[0088] Реакцию соединения формулы V с L-лактидом с образованием соединения формулы VI или VI* предпочтительно проводить в присутствии сильного основания, особенно комплексов алкилов и щелочных металлов, таких как н-бутиллитий, и азотного основания, особенно диизопропиламина или диэтиламина, в растворителе, таком как ациклический или особенно циклический простой эфир, предпочтительно тетрагидрофуран, предпочтительно при низких температурах, например от -80 до -5°C. Если реакцию проводят при температуре, близкой к -80°C, то результатом является соединение формулы VI, если реакцию проводят при повышении температуры до температуры, близкой к -5°C, то результатом является соединение формулы VI*.
[0089] Циклизацию соединения формулы VI с гидроксиламином или его солью или реакцию соединения формулы VI* с гидроксиламином или его солью с получением соединения формулы VII, соответственно, предпочтительно проводить с солью присоединения кислоты гидроксиламина, например с его солью галогеноводородной кислоты, такой как его гидрохлоридная соль, в присутствии слабого основания, например алканоата щелочного металла, такого как ацетат натрия, в полярном органическом растворителе, например, спирте, таком как алканол, например метанол или этанол, при предпочтительных температурах от 0 до 80°C, например от 10 до 50°C.
[0090] Гидрирование (a-i) соединения гидроксиламина формулы VII до соответствующего амина формулы VIII предпочтительно проводить как гетерогенное гидрирование в присутствии катализатора гидрирования, например платины, палладия, родия или рутения или других высокоактивных катализаторов, которые работают при более низких температурах (например, от 0 до 40°C) и более низких давлениях (например, 1 бар) H2, или катализаторов из неблагородных металлов, особенно на основе никеля (например, никеля Ренея и никеля Урушибары) при повышенных температурах и более высоком давлении Н2, например, в пределах от 5 до 50 бар, например от 10 до 20 бар. Реакцию проводят в полярном растворителе, особенно в спирте, например в алканоле, таком как этанол или, особенно, метанол.
[0091] Ацилирование (a-ii) гидроксильного соединения формулы VII в восстанавливающих условиях до соединения формулы VIII* предпочтительно проводить в присутствии ацилирующего реагента, особенно ангидрида карбоновой кислоты, такого как ангидрид алкановой кислоты, особенно уксусного ангидрида, в присутствии неблагородного металла, такого как цинк (например, амальгама цинка) или, особенно, железа, и кислоты, либо неорганической кислоты, такой как галогеноводородная кислота, например хлористоводородная, серная кислота, либо органической кислоты, такой как карбоновая кислота, соответствующая ангидриду, особенно алкановая кислота, особенно уксусная кислота, в качестве восстановителя в инертном органическом растворителе, таком как углеводород или ароматическое соединение, например толуол или фенилендиметилен, предпочтительно при повышенных температурах от 25°C до точки кипения реакционной смеси, например от 40 до 80°C.
[0092] Ацил в контексте настоящего изобретения относится к фрагменту органической кислоты, где в ацильном остатке карбоксильная (-COOH) группа связана с углеродом (например, как в ацетил=H3CCOO-), а не (как, например, в трет-бутоксикарбониле) с кислородом.
[0093] Восстановление (b-i) соединения формулы VIII до соединения формулы IX предпочтительно проводить с помощью комплексного гидрида, восстанавливающего оксо-группу в формуле VIII до гидрокси-группы в формуле IX, такого как гидрид диизобутилалюминия, в апротонном растворителе, таком как простой эфир или, особенно, циклический простой эфир, такой как тетрагидрофуран, предпочтительно при низких температурах от -100 до -20°C, например от -80 до -70°C.
[0094] Если затем соединение формулы IX, как соединение, соответствующее соответствующему соединению формулы IV, восстанавливают до соединения формулы II, то восстановление предпочтительно проводить, используя триалкилсилан, особенно триэтилсилан, в кислоте, особенно сильной органической сульфокислоте, такой как трифторметансульфокислота, в апротонном растворителе, таком как углеводород, сложный эфир или, особенно, нитрил, такой как ацетонитрил, предпочтительно при повышенных температурах от 30°C до температуры кипения реакционной смеси, например от 50 до 95°C. Последующую реакцию с кислотой HnA предпочтительно проводить в протонном, предположительно водном растворителе, таком как изопропиловый спирт.
[0095] Реакцию (c-i) соединения формулы VIII с реагентом, вставляющим аминогруппу, особенно диалканоилдикарбонатом, особенно ди-трет-бутилдикарбонатом (= Boc-ангидридом), предпочтительно проводить в присутствии третичного амина, такого как триалкиламин, особенно диизопропилэтиламин, в апротонном растворителе, особенно в галогенированном углеводороде, таком как дихлорметан, при предпочтительных температурах вот 0 до 50°C, например от 20 до 30°C, с получением соединения формулы X.
[0096] Восстановление соединения формулы X до соединения формулы XI предпочтительно проводить в присутствии комплексного гидрида, способного восстанавливать лактонную группу в формуле X до открытого кольца в формуле XI с двумя гидроксильными группами, такого как борогидрид лития, в апротонном растворителе, таком как линейный или предпочтительно циклический простой эфир, например тетрагидрофуран, предпочтительно при температуре от 0 до 50°C, например от 20 до 30°C.
[0097] Реакцию соединения формулы XI, при которой вводят уходящую группу формулы LG2, с образующим уходящую группу реагентом LG*-X, где X представляет собой галоген, особенно хлор, LG* представляет собой электрофильный радикал, способный вместе с гидрокси-группой, с которой он связан, образовывать уходящую группу LG2, особенно сульфонилгалогенид, предпочтительно толуолсольфонилхлорид, с образованием соединения формулы XII, предпочтительно проводить в присутствии основания, такого как гидроксид щелочного металла, например гидроксида натрия, в водном органическом растворителе, таком как водный галогенированный углеводород, например дихлорметан, при предпочтительных температурах от 0 до 50°C, например от 20 до 30°C.
[0098] Циклизацию соединения формулы XII до соединения формулы XIII в основных условиях в присутствии катализатора межфазного переноса, например галогенида тетраалкиламмония, такого как тетра-н-бутиламмонийбромид, в присутствии основания, особенно гидроксида щелочного металла, такого как гидроксид натрия, в водном органическом растворителе, таком как водный галогенированный углеводород, например дихлорметан, при предпочтительных температурах от 0 до 50°C, например от 20 до 30°C.
[0099] Снятие защиты с соединения формулы XIII предпочтительно проводить с помощью кислоты HnA, которая является частью соли полученной формулы II, в полярном растворителе, таком как спирт, например алканол, такой как этанол или, особенно, метанол, в присутствии аминного основания, например изопропиламина, при предпочтительных температурах от 0 до 50°C, например при 20-30°C.
[00100] Гидрирование соединения формулы VIII* до соединения формулы X* в присутствии хирального катализатора гидрирования (обычно полученного из предшественника катализатора, например, на основе рутения(I), такого как бис(норборнадиен)родия(I) тетрафторборат и хиральный лиганд), например, как определено ниже, предпочтительно осуществлять водородом при повышенном давлении, например в пределах от 3 до 50 бар, например от 20 до 40 бар, в полярном растворителе, особенно и 2,2,2-трифторэтаноле, при температурах предпочтительно от 30 до 80°C, например от 40 до 60°C. Такое гидрирование обычно проводят водородом в присутствии катализатора - переходного металла, предпочтительно в присутствии катализатора - переходного металла, содержащего металлоорганический комплекс и хиральный лиганд. Восстановление можно проводить в условиях гетеро- или гомогенной гидрогенизации, предпочтительно в условиях гомогенной гидрогенизации. Переходный металл выбирают из группы 9 или 10 периодической таблицы элементов. Поэтому катализатор на основе переходного металла включает, например, кобальт (Co), родий (Rh), иридий (Ir), никель (Ni), палладий (Pd) и/или платину (Pt).
[00101] Среди хиральных катализаторов подходящими являются все те, которые позволяют гидрирование двойной связи в соединении формулы VIII* с получением конфигурации по предшествующей двойной связи, показанной в формуле X*. Кроме того, предпочтительно, чтобы хиральный лиганд содержал хиральный ферроцен.
[00102] Предпочтительный хиральный ферроцен соответствует формуле:
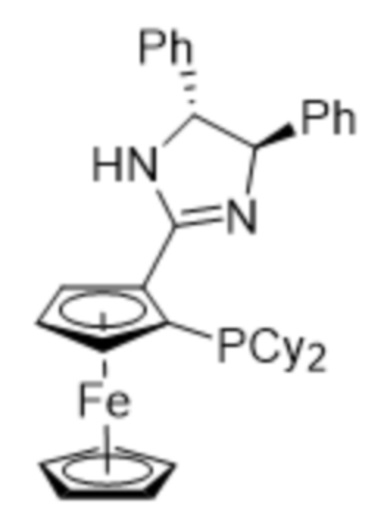
[00103] но возможны и другие, например, соответствующие любой из следующих формул:
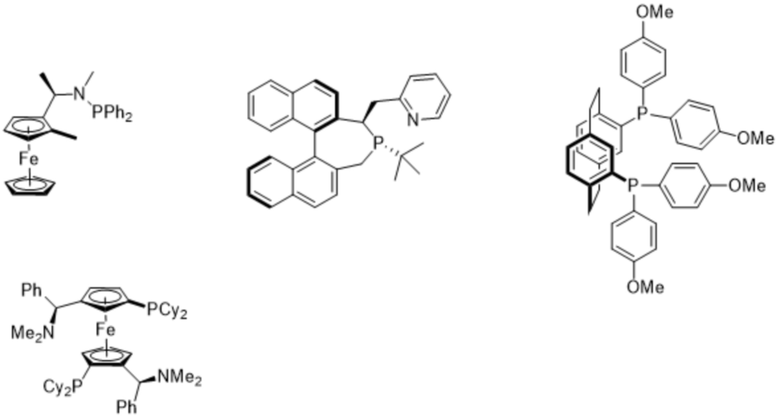
[00104] Также возможны смеси двух или более таких лигандов, особенно тех, которые определены в приведенных выше формулах.
[00105] Обычно активный катализатор получают путем смешивания 0,9-1,2, предпочтительно - от 1,0 до 1,1, более предпочтительно - от 1,0 до 1,05 моля хирального лиганда с 1,0 моля атомов переходного металла, содержащихся в катализаторе - переходном металле. Например, если используют димерный катализатор на основе переходного металла, предпочтительно взаимодействие двух молей хирального лиганда с одним молем катализатора - переходного металла с образованием «активного катализатора».
[00106] Хиральный лиганд обычно добавляют к реакционной смеси в растворе, приготовленном в том же растворителе, который использовался для реакции.
[00107] Восстановление соединения формулы X* до соединения формулы XI* при раскрытии цикла предпочтительно проводить в присутствии комплексного гидрида, способного восстанавливать лактонную группу в формуле X до открытого кольца в формуле XI с двумя гидроксильными группами, такого как борогидрид лития, в апротонном растворителе, таком как линейный или предпочтительно циклический простой эфир, например тетрагидрофуран, предпочтительно при температуре от 0 до 50°C, например от 20 до 30°C.
[00108] Защитные группы аминогруппы предпочтительно представляют собой группы, которые можно отщеплять в не слишком жестких кислотных условиях, например, в присутствии галогеноводородной кислоты, такой как HCl, или, если соединение формулы II является продуктом прямой реакции, то кислоты формулы HnA, как определено для соединения формулы II, особенно в которой n равно 1 и A представляет собой галогенид-анион, особенно хлорид-анион. Например, 9-флуоренилметоксикарбонил, аллилоксикарбонил или, особенно, трет-бутоксикарбонил.
[00109] Реакцию соединения формулы XI*, при которой вводят уходящую группу формулы LG2, с образующим уходящую группу реагентом LG*-X, где X представляет собой галоген, особенно хлор, LG* представляет собой электрофильный радикал, способный вместе с гидрокси-группой, с которой он (должен быть) связан, образовывать уходящую группу LG2, особенно сульфонилгалогенид, предпочтительно толуолсольфонилхлорид, с образованием соединения формулы XII*, предпочтительно проводить в присутствии основания, такого как гидроксид щелочного металла, например гидроксида натрия, в водном органическом растворителе, таком как водный галогенированный углеводород, например дихлорметан, при предпочтительных температурах от 0 до 50°C, например от 20 до 30°C.
[00110] Циклизацию соединения формулы XII* до соединения формулы XIII* предпочтительно проводить в основных условиях в присутствии катализатора межфазного переноса, например галогенида тетраалкиламмония, такого как тетра-н-бутиламмонийбромид, в присутствии основания, особенно гидроксида щелочного металла, такого как гидроксид натрия, в водном органическом растворителе, таком как водный галогенированный углеводород, например дихлорметан, при предпочтительных температурах от 0 до 50°C, например от 20 до 30°C.
[00111] Снятие защиты с соединения формулы XIII* предпочтительно проводить с помощью кислоты HnA, которая является частью соли полученной формулы II, в полярном растворителе, таком как спирт, например алканол, такой как этанол или, особенно, метанол, особенно при повышенных температурах от 50 до 120°C, например при 100-115°C.
[00112] Соединение формулы III в дополнительном одном варианте осуществления изобретения или в качестве части общего синтеза соединения формулы I согласно изобретению согласно одному варианту осуществления предпочтительно получают галогенированием соединения формулы XVIII:

[00113] где LG представляет собой уходящую группу, особенно галоген, такой как хлор, галогенирующим реагентом с образованием соединения формулы XIX.

[00114] в которой LG представляет собой уходящую группу, в частности, как только что определено, а Hal представляет собой галоген, особенно хлор.
[00115] Реакцию предпочтительно проводят с галогенсукцинимидом, таким как бромсукцинимид, так что предпочтительно Hal представляет собой бром. Реакцию проводят в одном или нескольких апротонных растворителях, таких как дихлорметан, ацетонитрил, тетрагидрофуран, N, N-диметилацетамид и т.п., предпочтительно в интервале температур 20°C ~ 100°C.
[00116] Соединение формулы XIX затем или сначала может быть замещено меркаптановым соединением формулы XX:
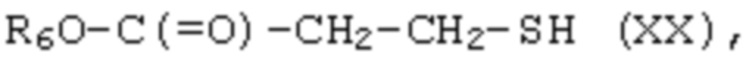
[00117] где R6 представляет собой незамещенный или замещенный алкил или незамещенный или замещенный арил, особенно C1-C6алкил, такой как этил, с получением соединения формулы XXI:

[00118] где LG представляет собой уходящую группу, а R6 представляет собой незамещенный или замещенный алкил или незамещенный или замещенный арил, особенно как только что определено.
[00119] Реакцию предпочтительно проводят в присутствии комплекса благородного металла, содержащего благородный металл, особенно палладий, и лиганд, такой как Xantphos, в присутствии третичного амина, такого как диизопропилэтиламин, в апротонном растворителе, например сложном эфире, предпочтительно сложном циклическом эфире, таком как диоксан, предпочтительно при повышенных температурах, например от 30°C до температуры кипения реакционной смеси.
[00120] В следующем самостоятельном или комбинированном варианте осуществления соединение формулы XXI затем обрабатывают алкоксилатом, особенно метоксилатом или этоксилатом щелочного металла, особенно лития, калия или особенно предпочтительно - натрия, с получением соединения формулы XXII:

[00121] где Mt представляет собой щелочной металл, особенно натрий. Эта реакция предпочтительно протекает в растворителе, таком как смесь спирта, например метанола или этанола (особенно спирта, соответствующего алкоксилату так, чтобы алкоксигруппа была идентична органическому остатку в спирте), и простого эфира, например циклического простого эфира, такого как тетрагидрофуран, предпочтительно при температуре от 0 до 50°C:
[00122] Соединение формулы XXII затем реагирует с соединением формулы XXIII:

[00123] с получением соединения формулы III:
[00124] где LG представляет собой уходящую группу, особенно такую, как определено выше для соединения формулы III.
[00125] Реакцию предпочтительно проводить в присутствии комплекса благородного металла, особенно полученного из Pd2(dbba)2, в присутствии лиганда, такого как Xantphos, и третичного азотного основания, такого как диизопропиламин, в апротонном растворителе, таком как простой эфир, например циклический простой эфир, особенно диоксан, предпочтительно при повышенных температурах, например, от 30°C до точки кипения реакционной смеси.
[00126] Соединение формулы XXIII можно предпочтительно получать взаимодействием соединения формулы XXIV:
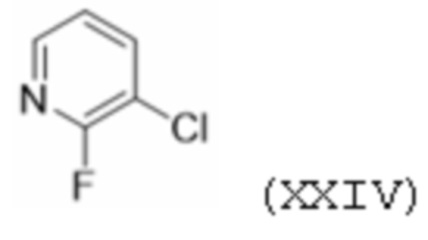
[00127] с йодом в присутствии сильного основания.
[00128] Эту реакцию предпочтительно проводить в присутствии сильного основания, особенно комплексов алкила с щелочным металлом, таких как н-бутиллитий, и азотного основания, особенно диизопропиламина или диэтиламина, в растворителе, таком как ациклический или, особенно, циклический простой эфир, предпочтительно в тетрагирофуране, предпочтительно при низких температурах, например, от -80 до -5°C.
[00129] Это приводит к соединению формулы XXV:

[00130] которое затем обрабатывают аммиаком с получением соединения формулы XXIII.
[00131] Эту реакцию затем предпочтительно проводить в присутствии газообразного аммиака и инертного полярного растворителя, такого как DMSO, особенно при повышенных температурах, предпочтительно от 30°C до температуры кипения реакционной смеси, например, при температуре от 85 до 95°C.
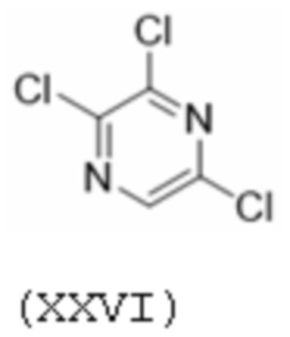
[00132] В качестве альтернативы синтезу из соединения формулы XVIII соединение формулы XIX, где Hal представляет собой хлор, и LG такой, как определено выше, также можно предпочтительно получать обработкой соединения формулы XXVI:
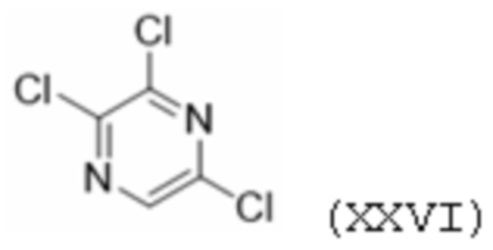
[00133] аммиаком с получением соединения формулы XIX, где Hal представляет собой хлор (условия реакции предпочтительно такие же, как только что описанные для реакции соединения формулы XXV), а затем путем дальнейших реакций, получая последовательно соединения формулы XXI, XXII и XXIII выше с получением соединения формулы III, как описано выше.
[00134] В следующем и особенно предпочтительном варианте осуществления только что описанное соединение формулы XXVI взаимодействует с аммиаком (предпочтительно в водной среде и при температурах от 0 до 80°C) с получением соединения формулы XIX, где Hal представляет собой галоген, предпочтительно хлор, которое затем подвергают реакции с (предпочтительно безводным) сульфидом щелочного металла формулы Mt2S, где Mt представляет собой щелочной металл, особенно натрий, и затем - с галогенидом четвертичного аммония формулы (alk)4NZ, в котором каждый alk независимо от других представляет собой алкил, особенно н-алкил, такой как C1-C6-алкил, и Z представляет собой галоген, особенно хлор или, более предпочтительно - бром, с получением соединения формулы XXVII:
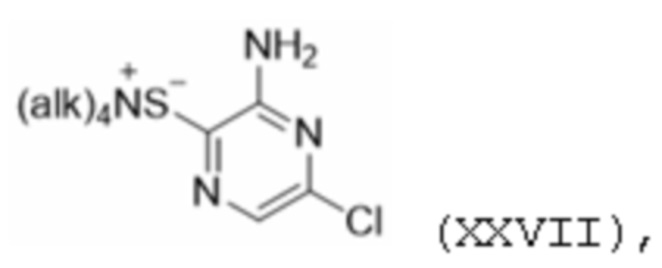
[00135] где alk не зависит от других алкилов, особенно н-алкилов, таких как C1-C6-алкил, которое затем можно подвергать реакции с соединением формулы XXIII (которое предпочтительно получают, как описано выше), предпочтительно в присутствии комплекса йодида меди (I), такого как CuI/фенантролин, в подходящем растворителе, например, в воде или спирте или их смеси, предпочтительно в воде и/или метаноле, этаноле или особенно изопропаноле, предпочтительно при температурах от -20 до 80°C, например, от 0 до 40°C, с получением соединения формулы III.
[00136] В другом варианте осуществления представлен способ производства соединения формулы I или его фармацевтически приемлемой соли, сокристалла с кислотой, гидрата или другого сольвата, причем указанный способ включает взаимодействие соединения формулы II с соединением формулы III согласно следующей схеме реакции:
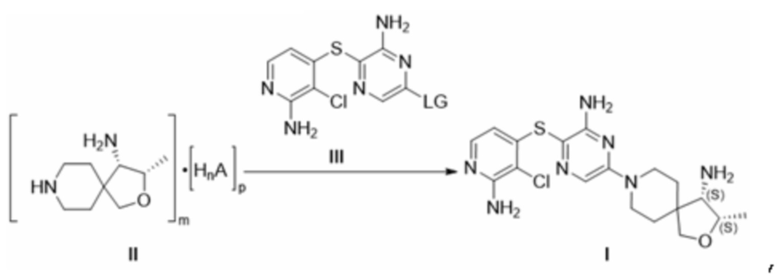
[00137] где LG представляет собой уходящую группу, A представляет собой анион протонной кислоты, а n, m и p независимо принимают значения 1, 2 или 3 так, чтобы соль формулы II не имела электрического заряда.
[00138] В последующем варианте осуществления предложен способ, где соединение формулы II получают либо (i) снятием защиты, либо (ii) восстановлением соединения формулы IV,
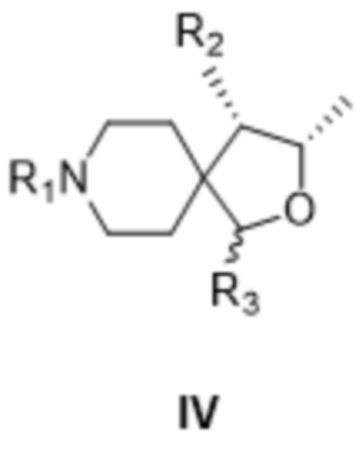
[00139] где в случае (i) R1 представляет собой защитную группу вторичной аминогруппы, и R2 представляет собой защищенную аминогруппу, и R3 представляет собой водород, или в случае (ii) R1 представляет собой защитную группу вторичной аминогруппы, R2 представляет собой аминогруппу, и R3 представляет собой гидроксил, и, если требуется, взаимодействием полученного соединения формулы IVa:
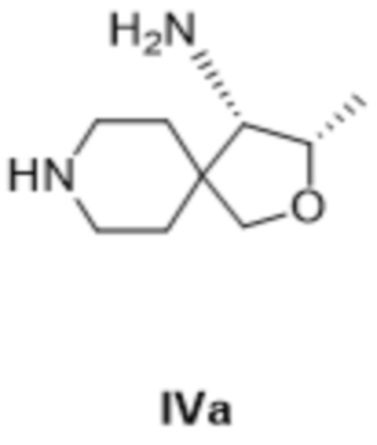
[00140] с кислотой формулы HnA с получением соединения формулы II.
[00141] В последующем варианте осуществления предложен способ производства соединения формулы II, включающий взаимодействие соединения формулы V:
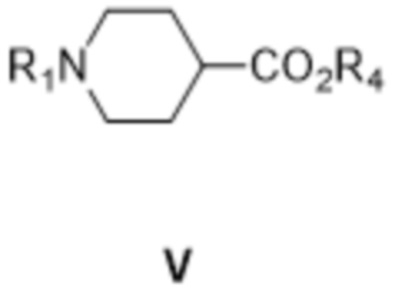
[00142] где R1 представляет собой защитную группу вторичной аминогруппы, а R4 представляет собой карбоксильную (-COOH) защитную группу, в присутствии сильного основания с L-лактидом формулы:
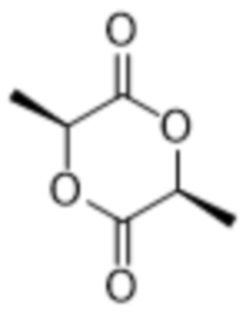
[00143] с получением соединения формулы VI:
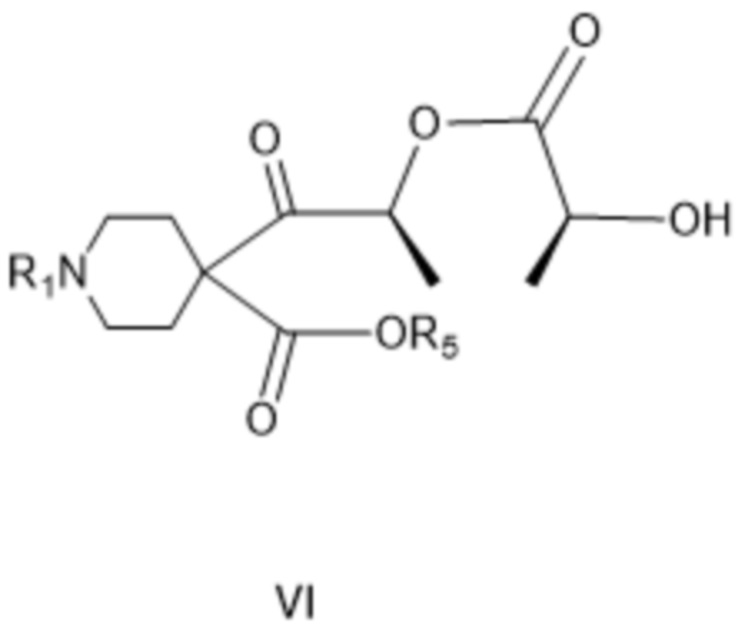
[00144] где R1 представляет собой защитную группу вторичной аминогруппы, а R5 представляет собой незамещенный или замещенный алкил, незамещенный или замещенный циклоалкил или незамещенный или замещенный арил; или, альтернативно, с получением соединения формулы VI*:

[00145] где R1 представляет собой защитную группу вторичной аминогруппы.
[00146] В следующем варианте осуществления способ дополнительно включает циклизацию соединения формулы VI:
[00147] где R1 - защитная группа вторичной аминогруппы, а R5 представляет собой незамещенный или замещенный алкил, незамещенный или замещенный циклоалкил или незамещенный или замещенный арил, с гидроксиламином или его солью; или, альтернативно, включает циклизацию соединения формулы VI*:
[00148] где R1 представляет собой защитную группу вторичной аминогруппы, с получением соединения формулы VII:
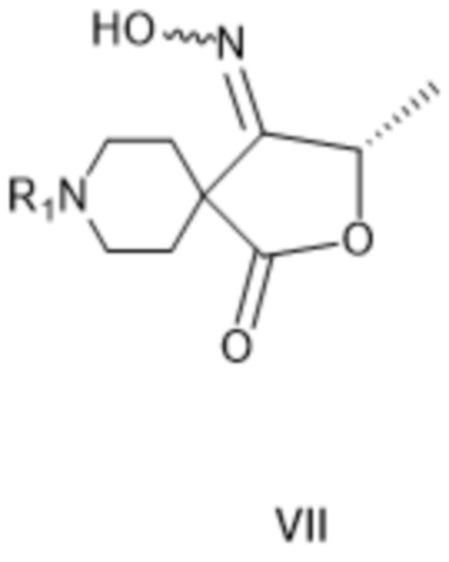
[00149] где R1 - защитная группа вторичной аминогруппы.
[00150] В другом варианте осуществления способ дополнительно включает либо: (a-i) гидрирование соединения формулы VII:
[00151] где R1 - защитная группа вторичной аминогруппы, с получением аминосоединения формулы VIII:
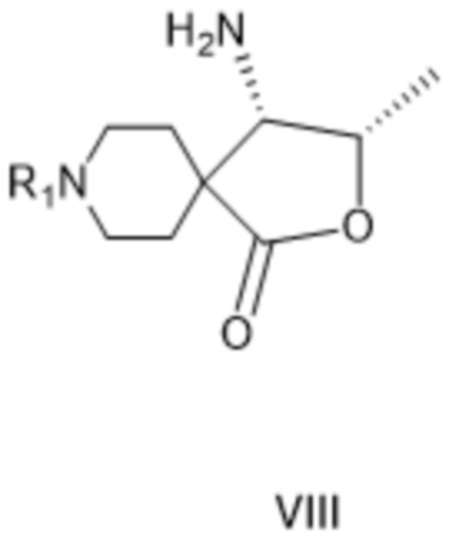
[00152] где R1 представляет собой защитную группу вторичной аминогруппы; либо (a-ii) ацилирование указанного соединения формулы VII в восстанавливающих условиях с получением соединения формулы VIII*:
[00153] где R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу.
[00154] В следующем варианте осуществления способ дополнительно включает восстановление соединения формулы VII:
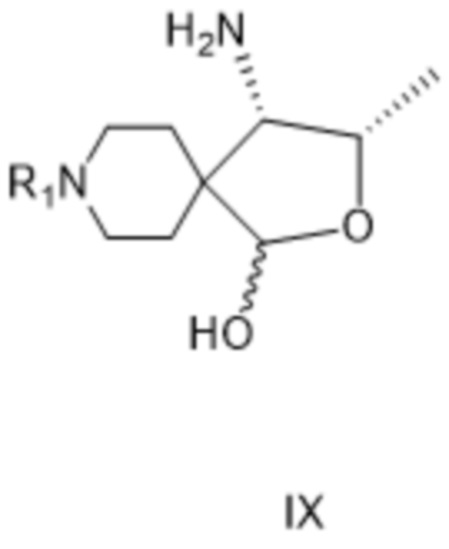
[00155] где R1 представляет собой защитную группу вторичной аминогруппы; указанное соединение представляет собой соединение формулы IV по п. 2, где R1 представляет собой защитную группу вторичной аминогруппы, R2 представляет собой аминогруппу, а R3 представляет собой гидроксил; с получением соединения формулы IX:
[00156] где R1 представляет собой защитную группу вторичной аминогруппы, указанное соединение представляет собой соединение формулы IV, где R1 представляет собой защитную группу вторичной аминогруппы, R2 представляет собой аминогруппу, а R3 представляет собой гидроксил; затем с использованием стадии восстановления (ii) по п. 2 для соединения соответствующей формулы IV, подпадающего под формулу IX, с использованием триалкилсилана получают соединение формулы II, как определено в п. 1, или соединение формулы V:

[00157] которое затем превращают в соединение формулы II обработкой кислотой формулы HnA, где A представляет собой кислотный анион, а n является целым числом.
[00158] В другом варианте осуществления способ включает взаимодействие аминосоединения формулы VIII:
[00159] где R1 представляет собой защитную группу вторичной аминогруппы; с защитной группой аминогруппы с получением соединения формулы Х:
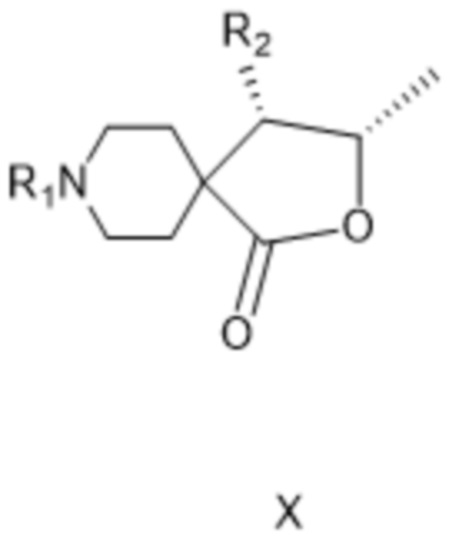
[00160] где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой защищенную аминогруппу.
[00161] В следующем варианте осуществления способ дополнительно включает восстановление соединения формулы Х:
[00162] где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой защищенную аминогруппу, с получением соединения формулы XI:

[00163] где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой защищенную аминогруппу; взаимодействием указанного соединения формулы XI по гидрокси-группе гидроксиметильной группы с реагентом, образующим уходящую группу формулы LG*-X, в которой LG* представляет собой электрофильный радикал, способный вместе с гидрокси-группой, с которой он связан, образовывать уходящую группу LG2, а X представляет собой галоген, получают соединение формулы XII:
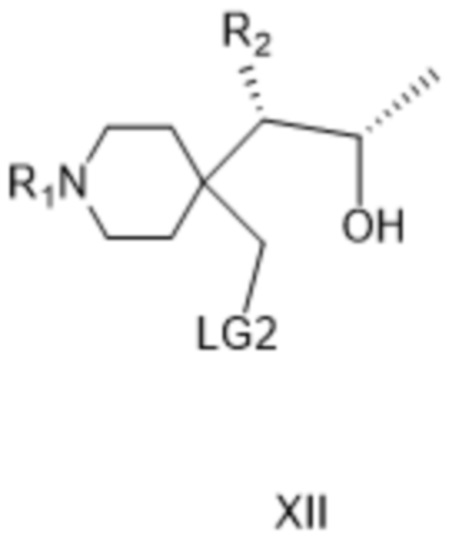
[00164] где R1 представляет собой защитную группу вторичной аминогруппы, R2 представляет собой защищенную аминогруппу, а LG2 представляет собой уходящую группу; указанное соединение формулы XII затем циклизуют в основных условиях с получением соединения формулы XIII:
[00165] где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой защищенную аминогруппу; где стадию (i) снятия защиты по п. 2 в соединении формулы IV соединения формулы XIII проводят обработкой кислотой формулы HnA.
[00166] В следующем варианте осуществления способ дополнительно включает гидрирование соединения формулы VIII*:
[00167] где R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу; в присутствии хирального катализатора гидрирования с получением соединения формулы X*:
[00168] где R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу; указанное соединение формулы X* затем восстанавливают до соединения формулы XI*:
[00169] где R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу; взаимодействием указанного соединения формулы XI* по гидрокси-группе гидроксиметильной группы с реагентом, образующим уходящую группу формулы LG*-X, в которой LG* представляет собой электрофильный радикал, способный образовывать вместе с гидрокси-группой, с которой он связан, уходящую группу LG2, а X представляет собой галоген, получают соединение формулы XII*:
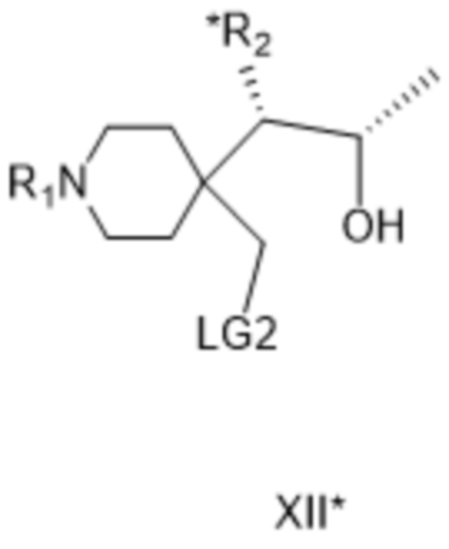
[00170] где R1 представляет собой защитную группу вторичной аминогруппы, R2 представляет собой защищенную аминогруппу, а LG2 представляет собой уходящую группу; указанное соединение формулы XII* циклизуют в основных условиях с получением соединения формулы XIII*:

[00171] где R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу; где стадию снятия защиты/деацилирования (i) для соединения, соответствующего формуле IV в п. 2 соединения формулы XIII* проводят с использованием кислоты HnA.
[00172] Еще одним вариантом осуществления является производство соединения формулы III:
[00173] где LG представляет собой уходящую группу, включающее получение сначала соединения формулы XIX:
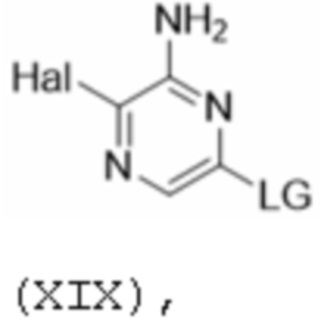
[00174] где LG представляет собой хлор, и Hal представляет собой хлор, обработкой соединения формулы XXVI:
[00175] аммиаком с получением соединения формулы XIX; затем взаимодействие соединения формулы XIX с сульфидом щелочного металла формулы Mt2S, где Mt представляет собой щелочной металл, и затем с галогенидом четвертичного аммония формулы (alk)4NZ, в котором каждый alk независимо от других - алкил, а Z - галоген, с получением соединения формулы XXVII:
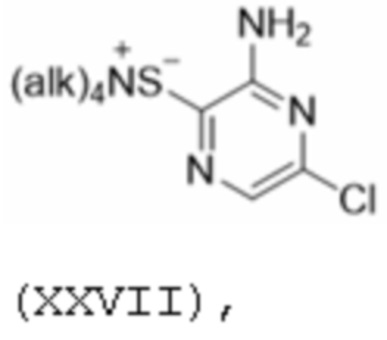
[00176] где каждый alk независимо представляет собой алкил, которое затем реагирует с соединением формулы XXIII:

[00177] с получением соединения формулы III.
[00178] В другом варианте осуществления способа щелочной металл Mt представляет собой натрий.
[00179] В другом варианте осуществления - соединение, выбранное из группы, состоящей из:
[00180] (i) соли соединения формулы II:

[00181] где A представляет собой анион протонной кислоты, а значения n, m и p выбраны из 1, 2 или 3 так, чтобы соль формулы II не имела электрического заряда; (ii) соединения формулы VI:
[00182] где R1 представляет собой защитную группу вторичной аминогруппы, а R5 представляет собой незамещенный или замещенный алкил, незамещенный или замещенный циклоалкил или незамещенный или замещенный арил;
[00183] (iii) соединения формулы VI*:
[00184] где R1 представляет собой защитную группу вторичной аминогруппы; (iv) соединения формулы VII:
[00185] где R1 представляет собой защитную группу вторичной аминогруппы; (v) соединения формулы VIII:
[00186] где R1 представляет собой защитную группу вторичной аминогруппы; (vi) соединения формулы IX:
[00187] где R1 представляет собой защитную группу вторичной аминогруппы; (vii) соединения формулы VIII*:
[00188] где R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу; (viii) соединения формулы X*:
[00189] где R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу; (ix) соединения формулы XI*:
[00190] где R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу; (х) соединения формулы XII*:
[00191] где R1 представляет собой защитную группу вторичной аминогруппы, *R2 представляет собой ацилированную аминогруппу, а LG2 представляет собой уходящую группу; (xi) соединения формулы XIII*:
[00192] где R1 представляет собой защитную группу вторичной аминогруппы, а *R2 представляет собой ацилированную аминогруппу; (xii) соединения формулы X:
[00193] где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой защищенную аминогруппу; (xiii) соединения формулы XI:
[00194] где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой защищенную аминогруппу; (xiv) соединения формулы XII:
[00195] где R1 представляет собой защитную группу вторичной аминогруппы, R2 представляет собой защищенную аминогруппу, а LG2 представляет собой уходящую группу; (xv) соединения формулы XIII:
[00196] где R1 представляет собой защитную группу вторичной аминогруппы, а R2 представляет собой защищенную аминогруппу; или их соли.
[00197] В другом варианте осуществления A представляет собой Cl.
[00198] В другом варианте осуществления R1 представляет собой трет-бутоксикарбонил.
Примеры
[00199] Следующие ниже примеры приведены в качестве иллюстрации изобретения без ограничения объема, определенного здесь иным образом. Используемые сокращения: Ас (ацетат); AcOH (уксусная кислота); Ac2O (уксусный ангидрид); Boc (трет-бутоксикарбонил); Boc2O (ди-трет-бутилдикарбонат); соляной раствор (насыщенный раствор хлорида натрия при к.т.); n-Bu4NBr (тетра-(н-бутил)аммония бромид); n-BuLi (н-бутиллитий); расчтн. (рассчитано); DCM (дихлорметан); DIBAL-Н (диизобутилалюминия гидрид); DIPEA (ди(изопропил)этиламин); DMAc (диметилацетамид); DMSO (диметилсульфоксид); DMSO-d6 (пердейтерированный диметилсульфоксид); эк. или экв. (эквиваленты); Et (этил); EtOAc (этилацетат); МСВР (масс-спектроскопия высокого разрешения); ч (час(-ы)); IPA (изопропиламин); IT (внутренняя температура (реакционной смеси)); LOQ (предел количественной оценки); МКЦ (микрокристаллическая целлюлоза); Me (метил); МеОН (метанол); MTBE (метил-трет-бутиловый эфир); MW (молекулярная масса); ЯМР (ядерный магнитный резонанс); iPrOH (изопропанпол); iPr2NH (диизопропиламин); к.т. (комнатная температура (примерно от 20 до 25°C)); TBAB (тетра-(н-бутил)аммония бромид); Tf-OH (трифликовая кислота); THF (тетрагидрофуран); TsCl (тозилхлорид); трифликовая кислота (трифторметансульфокислота); и Xantphos (4,5-бис(дифенилфосфино)-9,9-диметилксантен).
[00200] Экспериментальные процедуры: три основные процедуры (соответствующие примеру 1=путь B; примеру 2=путь C; и примеру 3=путь D) описаны в следующих обобщенных схемах реакций:
Пример 1
Путь В

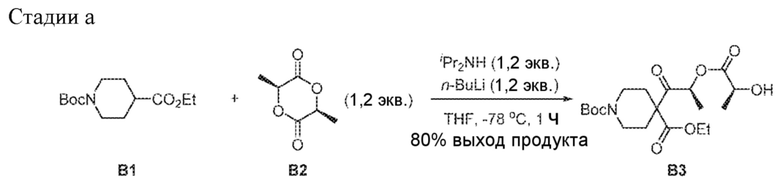
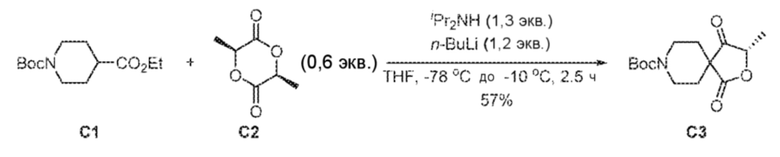
[00201] В трехгорлую круглодонную колбу A объемом 500 мл в атмосфере азота загружали диизопропиламин (9,44 г, 93,3 ммоль, 1,2 экв.) и THF (200 мл). Раствор охлаждали до IT = -20°C, по каплям в течение 30 мин добавляли 2,4 М n-BuLi в гексанах (38,9 мл, 1,2 экв.). Реакционную смесь перемешивали при -20°C в течение 30 мин, а затем охлаждали до -70°C. Раствор 1-(трет-бутил)4-этилпиперидин-1,4-дикарбоксилата (Jinan Welt Chem. Co., Ltd., Цзинань, Китай) (20,0 г, 77,7 ммоль, 1,0 экв.) в THF (20 мл) добавляли по каплям в течение 30 мин, поддерживая IT = (-70°C)-(-60°C). Реакционную смесь перемешивали при -70°C в течение 30 мин и получали бледно-желтый раствор.
[00202] В трехгорлую круглодонную колбу В объемом 500 мл в атмосфере азота загружали L-лактид (13,4 г, 93,3 ммоль, 1,2 экв.) и THF (120 мл). Раствор охлаждали до IT = -70°C. Раствор из колбы A медленно переносили в колбу B через канюлю в течение 30 мин, поддерживая IT = (-70°C)-(-60°C). Реакционную смесь перемешивали при -70°C в течение 30 мин. Реакционный раствор переносили в колбу C, содержащую 3% HCl (300 мл) через канюлю в течение 30 мин, поддерживая IT=0°C - 5°C. Смесь экстрагировали, используя EtOAc (400 мл ×2) и промывали 20%-ным соляным раствором (200 мл). Органический слой отделяли, сушили над безводным Na2SO4 и отфильтровывали. Фильтрат концентрировали досуха, получая B3 в виде бледно-желтого масла (36,0 г, 71 мас.%, выход продукта 81%), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ=5,42 (q, J=6,8 Гц, 1H), 4,36-4,27 (m, 1H), 4,27-4,16 (m, 2H), 3,71-3,49 (m, 2H), 3,39-3,24 (m, 2H), 2,81 (br d, J=5,0 Гц, 1H), 2,24-1,81 (m, 4H), 1,44 (s, 15H), 1,27 (t, J=7,1 Гц, 3H).
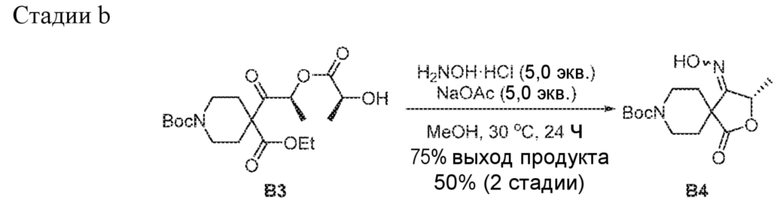
[00203] В круглодонную колбу на 250 мл вносили указанное выше желтое масло (15,0 г, 71 мас.%, 26,5 ммоль), гидрохлорид гидроксиламина (9,3 г, 132,6 ммоль, 5,0 экв.), ацетат натрия (10,9 г, 132,6 ммоль, 5,0 экв.) и метанол (150 мл). Смесь перемешивали в течение 24 ч при 20-25°C. Полученную суспензию фильтровали через МКЦ и осадок на фильтре промывали, используя МеОН (20 мл ×2). Фильтрат концентрировали до ок. 60 мл, затем по каплям добавляли воду (60 мл) в течение 15 минут, в осадок выпадало белое твердое вещество. Суспензию перемешивали в течение ночи и отфильтровывали. Осадок на фильтре промывали смесью МеОН (5 мл) и воды (25 мл) и сушили в вакууме, получая B4 в виде белого твердого вещества (4,9 г, 59%). 1H ЯМР (400 МГц, DMSO-d6) δ=11,45 (s, 1H), 5,33 (q, J=6,6 Гц, 1H), 3,73-3,58 (m, 2H), 3,56-3,43 (m, 1H), 3,43-3,35 (m, 1H), 1,87-1,65 (m, 4H), 1,52 (d, J=6,7 Гц, 3H), 1,41 (s, 9H).
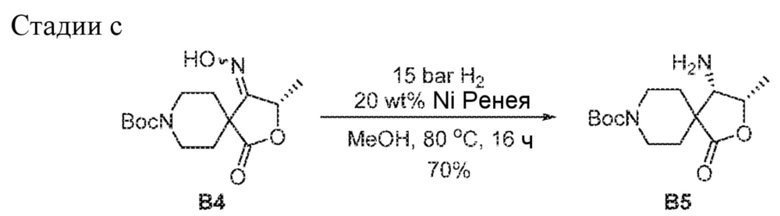
[00204] В реактор объемом 1 л с лопастной мешалкой в атмосфере азота вносили никель Ренея (5 г) и МеОН (250 мл), затем трет-бутил(S)-4-(гидроксиимино)-3-метил-1-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат B4 (25,0 г, 83,80 ммоль). Реактор трижды продували азотом, а затем трижды водородом. Смесь перемешивали в течение 16 ч под давлением водорода 20 бар при IT=80°C. Реакционную смесь фильтровали через микрокристаллическую целлюлозу и осадок на фильтре промывали, используя МеОН (10 мл). Фильтрат концентрировали досуха, получая белое твердое вещество (23,0 г). К твердому веществу добавляли EtOAc (220 мл), полученную суспензию нагревали до кипения с обратным холодильником (JT=100°C) и порциями добавляли н-гептан (550 мл). Полученный прозрачный раствор охлаждали до к.т. в течение 2 ч и оставляли на ночь, получая B5 в виде бесцветного кристаллического продукта (16,7 г, цис/транс > 99/1, 70%). 1H ЯМР (400 МГц, CDCl3) δ=4,75-4,64 (m, 1H), 3,89-3,80 (m, 1H), 3,68-3,58 (m, 1H), 3,48-3,33 (m, 3H), 1,92-1,61 (m, 4H), 1,46 (s, 9H), 1,40 (d, J=6,5 Гц, 3H).
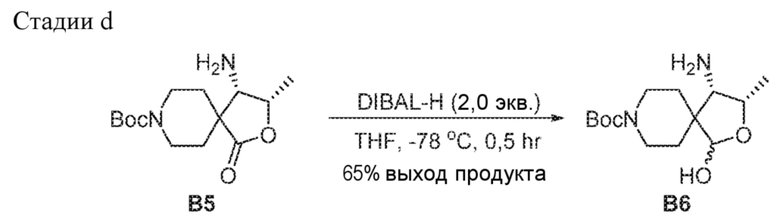
[00205] В трехгорлую круглодонную колбу на 500 мл в атмосфере азота загружали трет-бутил-(3S,4S)-4-амино-3-метил-1-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат B5 (6,0 г, 21,1 ммоль) и THF (200 мл). Раствор охлаждали до IT = -78°C, по каплям добавляли 1,0 M DIBAL (42,2 мл, 42,2 ммоль, 2,0 экв.) в течение 30 мин. Реакционную смесь перемешивали при -78°C в течение 30 мин. Реакцию гасили, осторожно добавляя насыщенный водный раствор Na, K-тартрата (150 мл), поддерживая IT = (-78°C)-(-60°C). Смесь интенсивно перемешивали при 20-25°C до получения двух прозрачных фаз (ок. 1,5 часа) и экстрагировали, используя EtOAc (200 мл ×2). Объединенные органические экстракты промывали 20 мас.% соляного раствора (200 мл), сушили над Na2SO4, отфильтровывали и концентрировали, получая B6 в виде вязкого масла (6,1 г, 64 мас.%, выход продукта 65%), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ=5,06 (s, 1H), 4,39-4,29 (m, 1H), 3,68-3,57 (m, 2H), 3,35-3,24 (m, 2H), 3,18 (d, J=4,4 Гц, 1H), 1,98-1,85 (m, 1H), 1,75-1,54 (m, 3H), 1,46 (s, 9H), 1,35 (d, J=6,6 Гц, 3H).
Стадия е
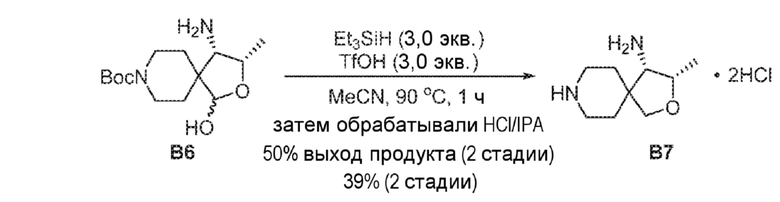
[00206] В круглодонную колбу на 100 мл вносили 6,0 г вышеуказанного вязкого масла и ацетонитрил (150 мл). Колбу охлаждали на ледяной бане и затем последовательно добавляли триэтилсилан (7,4 г, 63,3 ммоль) и трифликовую кислоту (9,5 г, 63,3 ммоль). Затем реакционную смесь перемешивали в течение 1 часа на масляной бане при 90°C. Затем реакционную смесь охлаждали до 20-25°C, выливали в делительную воронку и промывали н-гептаном (100 мл ×2). Слой ацетонитрила отделяли и концентрировали досуха, получая бесцветное масло, которое разбавляли с помощью EtOAc (150 мл). По каплям при перемешивании добавляли 6 н. HCl в изопропаноле (30 мл), в осадок выпадало белое твердое вещество. Добавляли МТВЕ (150 мл) и белую суспензию перемешивали в течение 2 ч и отфильтровывали. Осадок на фильтре промывали, используя EtOAc (50 мл ×2), получая белое твердое вещество, которое растворяли в MeOH (6,0 мл), по каплям добавляли EtOAc (18 мл) при перемешивании. Полученную белую суспензию отфильтровывали и промывали, используя EtOAc (10 мл ×2), получая B7 в виде белого твердого вещества (2,5 г, 81 мас.%, 39% за две стадии). 1H ЯМР (400 МГц, DMSO-d6) δ=9,37 (br s, 1H), 9,25 (br s, 1H), 8,42 (br s, 3H), 4,26-4,17 (m, 1H), 3,72 (ABq, J=9,1 Гц, 2H), 3,50-3,41 (m, 1H), 3,28-3,18 (m, 1H), 3,18-3,09 (m, 1H), 2,99-2,74 (m, 2H), 2,07-1,63 (m, 4H), 1,22 (d, J=6,5 Гц, 3H).
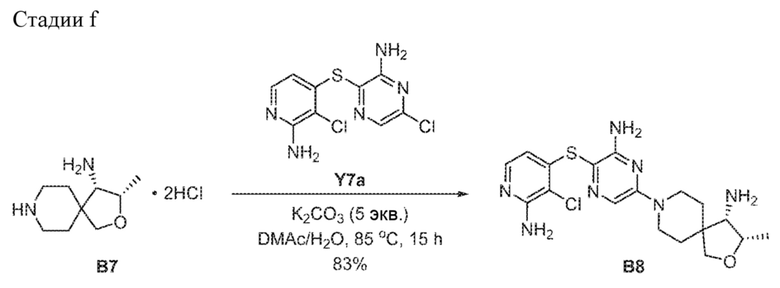
[00207] В сосуд Шленка на 10 мл вносили 3-((2-амино-3-хлорпиридин-4-ил)тио)-6-хлорпиразин-2-амин Y7a (0,1 г, 0,347 ммоль), (3S,4S)-3-метил-2-окса-8-азаспиро[4,5]декан-4-амина дигидрохлорид B7 (0,1 г, 0,416 ммоль, 1,2 экв.), DMAc (0,6 мл) и 36%-ный водн. K2CO3 (0,66 г, 1,735 ммоль, 5,0 экв.). Смесь перемешивали в течение 16 часов на масляной бане при 100°C и охлаждали до 20-25°C. Добавляли 20%-ный соляной раствор (10 мл) и смесь экстрагировали с помощью EtOAc (20 мл ×2). Объединенные экстракты промывали 20%-ным соляным раствором (10 мл ×4), сушили над безводным Na2SO4 и отфильтровывали. Фильтрат концентрировали досуха, получая B8 в виде твердого вещества желтого цвета (121 мг, 83%). 1H ЯМР (400 МГц, DMSO-d6) δ=7,64 (d, J=6,2 Гц, 1H), 7,62 (s, 1H), 6,26 (s, 2H), 6,13 (s, 2H), 5,74 (d, J=5,3 Гц, 1H), 4,12-4,02 (m, 1H), 3,90-3,78 (m, 2H), 3,67 (d, J=8,4 Гц, 1H), 3,49 (d, J=8,4 Гц, 1H), 3,33 (s, 2H), 2,91 (d, J=5,1 Гц, 1H), 1,78-1,68 (m, 1H), 1,67-1,57 (m, 1H), 1,56-1,41 (m, 2H), 1,08 (d, J=6,5 Гц, 3H).
Пример 2
Путь С
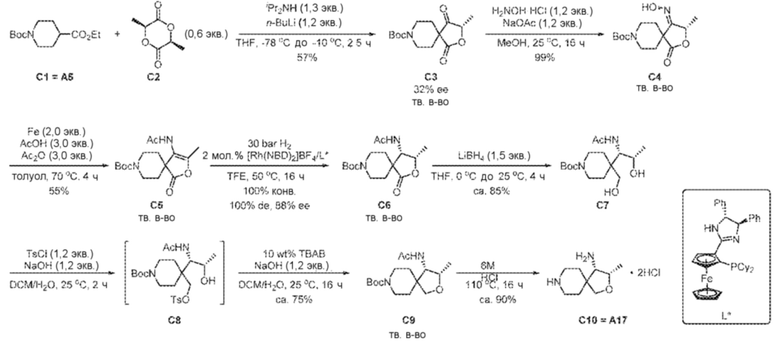
Стадия а:
[00208] В трехгорлую круглодонную колбу A объемом 1 л в атмосфере азота загружали диизопропиламин (10,2 г, 100,8 ммоль) и THF (200 мл). Раствор охлаждали до IT = -20°C, по каплям в течение 30 мин добавляли 2,5 M n-BuLi в гексанах (37,3 мл, 93,3 ммоль). Реакционную смесь перемешивали при -20°C в течение 30 мин, а затем охлаждали до -70°C. По каплям в течение 30 минут добавляли раствор 1-(трет-бутил)4-этилпиперидин-1,4-дикарбоксилата (20,0 г, 77,7 ммоль) в THF (30 мл), поддерживая IT = (-70°C)-(-60°С). Реакционную смесь перемешивали при -70°C в течение 1 ч и получали бледно-желтый раствор. Раствор L-лактида (13,4 г, 93,3 ммоль, 1,2 экв.) в THF (50 мл) добавляли по каплям в течение 30 мин, поддерживая IT = (-70°C)-(-60°С). Реакционную смесь перемешивали при -70°C в течение 1 ч.
[00209] В трехгорлую круглодонную колбу B объемом 500 мл в атмосфере азота загружали диизопропиламин (9,2 г, 90,9 ммоль) и THF (180 мл). Раствор охлаждали до IT = -20°С, по каплям в течение 30 мин добавляли 2,5 M n-BuLi в гексанах (33,6 мл, 84,0 ммоль). Реакционную смесь перемешивали при -20°С в течение 30 мин, а затем охлаждали до -70°С. Раствор 1-(трет-бутил)-4-этилпиперидин-1,4-дикарбоксилата (18,0 г, 70,0 ммоль) в THF (27 мл) добавляли по каплям в течение 30 мин, поддерживая IT = (-70°C)-(-60°С). Реакционную смесь перемешивали при -70°С в течение 1 ч.
[00210] Раствор из колбы B медленно переносили в колбу A через канюлю в течение 30 минут, поддерживая IT = (-70°C)-(-60°С). Реакционную смесь перемешивали при -70°С в течение 1 ч. Затем реакционную смесь постепенно нагревали до -10°С в течение 1 ч и перемешивали при -10°С в течение 30 мин. Реакционный раствор переносили в колбу C, содержащую 3% HCl (500 мл) через канюлю в течение 30 мин при поддержании IT=0°С-5°С. Смесь экстрагировали EtOAc (300 мл ×2) и промывали 20%-ным соляным раствором (200 мл). Органический слой отделяли, сушили над безводным Na2SO4 и отфильтровывали. Фильтрат концентрировали досуха, получая бесцветное масло, которое постепенно затвердевало при стоянии в течение ночи. Твердое вещество перекристаллизовывали из н-гептана/EtOAc, получая C3 в виде белого твердого вещества (24,0 г, 57%). 1H ЯМР (400 МГц, CDCl3) δ=4,83 (q, J=7,0 Гц, 1H), 3,78-3,62 (m, 4H), 1,90-1,72 (m, 4H), 1,53 (d, J=7,1 Гц, 3H), 1,47 (s, 9H).
Стадия b

[00211] В круглодонную колбу на 500 мл добавляли трет-бутил-(S)-3-метил-1,4-диоксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат C3 (26,5 г, 93,5 ммоль, 1,0 экв), гидрохлорид гидроксиламина (7,8 г, 112,2 ммоль, 1,2 экв.), ацетат натрия (9,2 г, 112,2 ммоль, 1,2 экв.) и метанол (200 мл). Смесь перемешивали в течение ночи при 20-25°С. Реакционную смесь концентрировали досуха, полученное твердое вещество разбавляли, используя EtOAc (300 мл), и промывали водой (200 мл) и 20%-ным соляным раствором (200 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали досуха, получая C4 в виде белого твердого вещества (27,9 г, 99%, частично рацемизированный). 1H ЯМР (400 МГц, DMSO-d6) δ=11,45 (s, 1H), 5,33 (q, J=6,6 Гц, 1H), 3,73-3,58 (m, 2H), 3,56-3,43 (m, 1H), 3,43-3,35 (m, 1H), 1,87-1,65 (m, 4H), 1,52 (d, J=6,7 Гц, 3H), 1,41 (s, 9H).
Стадия с
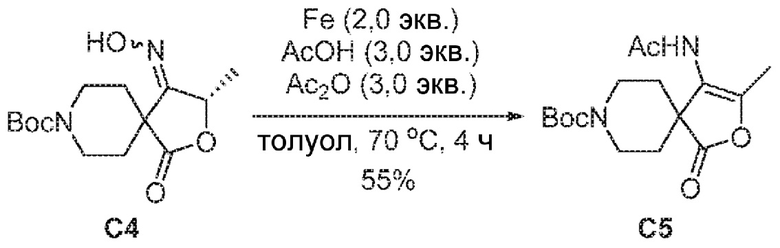
[00212] В круглодонную колбу на 500 мл в атмосфере азота загружали трет-бутил-4-(гидроксиимино)-3-метил-1-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат C4 (27,9 г, 93,5 моль), толуол (150 мл), уксусный ангидрид (29,1 г, 280,6 ммоль), уксусную кислоту (16,8 г, 280,6 ммоль) и железо (10,4 г, 187,0 ммоль). Смесь интенсивно перемешивали в течение 4 ч на масляной бане при 70°C и охлаждали до к.т. Суспензию фильтровали через микрокристаллическую целлюлозу для удаления твердого остатка, который затем промывали, используя EtOAc (150 мл ×2). Объединенные фильтраты охлаждали на ледяной бане и промывали 5%-ным NaHCO3 (300 мл) и 20%-ным соляным раствором (300 мл). Органический слой отделяли, сушили над Na2SO4 и отфильтровывали. Фильтрат упаривали досуха. Остаток очищали колоночной хроматографией (силикагель, EtOAc/н-гептан=от 1/1 до 3/1, об./об.) и дополнительно очищали перекристаллизацией из EtOAc/н-гептана, получая C5 в виде белых игольчатых кристаллов (16,7 г , 55%). 1H ЯМР (400 МГц, CDCl3) δ=7,43 (s, 1H), 4,10-3,78 (m, 2H), 3,55-3,38 (m, 2H), 2,10 (s, 3H), 1,94 (s, 3H), 1,76-1,58 (m, 4H), 1,45 (s, 9H).
Стадия d

[00213] Во флакон в атмосфере азота вносили [Rh(NBD)2]BF4 (2,0 мг, 0,005 ммоль), лиганд L* (получен от Johnson Matthey & Brandenberger AG, Цюрих, Швейцария) (3,3 мг, 0,005 ммоль) и DCM (1 мл). Полученный раствор перемешивали в течение 30 минут перед удалением растворителя, получая твердое вещество желтого цвета. Во флакон в атмосфере азота вносили трет-бутил-4-ацетамидо-3-метил-1-оксо-2-окса-8-азаспиро[4.5]дец-3-ен-8-карбоксилат C5 (86 мг, 0,27 ммоль) и 2,2,2-трифторэтанол (TFE) (2,7 мл). Флакон помещали в реактор гидрирования. Реактор трижды продували азотом, а затем трижды водородом. Смесь перемешивали в течение 16 ч под давлением водорода 30 бар при IT=50°C. Реакционную смесь охлаждали до 20-25°C, отфильтровывали через короткую подушку из диоксида кремния и концентрировали досуха, получая C6 в виде белого твердого вещества (86 мг, 100%). 1H ЯМР (400 МГц, DMSO-d6) δ=8,33 (br d, J=10,3 Гц, 1H), 4,94-4,84 (m, 1H), 4,71-4,56 (m, 1H), 3,78-3,65 (m, 2H), 3,22-3,02 (m, 1H), 2,87-2,69 (m, 1H), 1,89 (s, 3H), 1,64-1,50 (m, 4H), 1,40 (s, 9H), 1,19 (d, J=6,7 Гц, 3H).
Стадия е
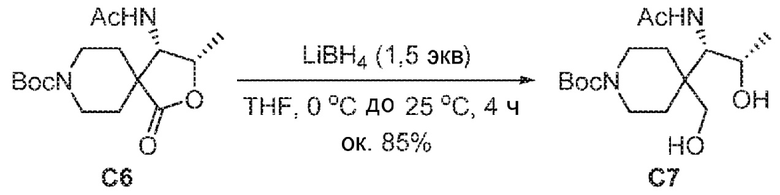
[00214] В колбу Шленка на 10 мл в атмосфере азота вносили трет-бутил-(3S,4S)-4-ацетамидо-3-метил-1-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат C6 (300 мг, 0,919 ммоль) и THF (3,0 мл). Колбу охлаждали на ледяной бане. 2,0 M LiBH4 в THF (0,7 мл) добавляли по каплям и реакционную смесь перемешивали в течение 4 ч при 20-25 °C. Реакционную смесь охлаждали на ледяной бане и гасили, добавляя по каплям 5%-ный NaHCO3 (1,0 мл). Смесь разделяли, и водный слой экстрагировали, используя EtOAc (10 мл ×3). Объединенные экстракты промывали 20%-ным соляным раствором (20 мл). Органический слой отделяли, сушили над Na2SO4 и отфильтровывали. Фильтрат упаривали досуха. Остаток очищали колоночной хроматографией (силикагель, EtOAc/н-гептан=от 1/1 до 1/3, об./об.), получая C7 в виде бесцветного вязкого масла (258 мг, 85%). 1H ЯМР (400 МГц, DMSO-d6) δ=7,48 (br d, J=10,1 Гц, 1H), 5,23 (br s, 1H), 5,15 (br s, 1H), 4,09-4,04 (m, 1H), 3,92-3,82 (m, 1H), 3,75 (d, J=10,1 Гц, 1H), 3,56 (d, J=5,1 Гц, 1H), 3,54-3,44 (m, 4H), 1,98 (s, 3H), 1,68-1,57 (m, 2H), 1,52-1,46 (m, 2H), 1,44 (s, 9H), 1,00 (d, J=6,2 Гц, 3H).
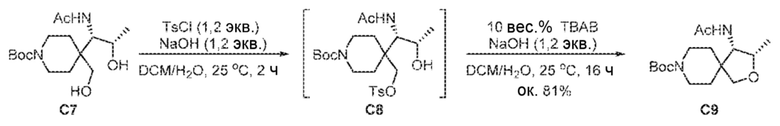
[00215] В сосуд Шленка на 25 мл в атмосфере азота вносили NaOH (94 мг, 2,35 ммоль) и воду (5,0 мл). Пробирку охлаждали на ледяной бане и по каплям добавляли раствор трет-бутил-4-((1S,2S)-1-ацетамидо-2-гидроксипропил)-4-(гидроксиметил)пиперидин-1-карбоксилата C7 (650 мг, 1,97 ммоль) и TsCl (450 мг, 2,36 ммоль) в DCM (5,0 мл). Затем смесь перемешивали в течение 16 ч при 20-25°C. Добавляли n-Bu4NBr (65 мг, 0,202 ммоль), а затем NaOH (94 мг, 2,35 ммоль) в воде (2,0 мл). Затем смесь перемешивали в течение 16 ч при 20-25°C. Органический слой отделяли, промывали 20%-ным соляным раствором (5 мл), сушили над Na2SO4 и отфильтровывали. Фильтрат упаривали досуха, получая C9 в виде белого твердого вещества (500 мг, 81%). 1H ЯМР (400 МГц, DMSO-d6) δ=7,82 (br d, J=10,0 Гц, 1H), 4,18-4,06 (m, 2H), 3,65-3,56 (m, 1H), 3,55 (ABq, J=8,7 Гц, 2H), 3,32-3,11 (m, 3H), 1,89 (s, 3H), 1,57-1,40 (m, 4H), 1,38 (s, 9H), 1,01 (d, J=6,1 Гц, 3H).
Стадия f

[00216] В запаянную пробирку на 10 мл добавляли трет-бутил-(3S,4S)-4-ацетамидо-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилат C9 (25 мг, 0,077 ммоль) и 6 н. водн. HCl (1,0 мл). Реакционную смесь перемешивали в течение 16 ч на масляной бане при 110°C. Затем реакционную смесь охлаждали до 20-25°C и концентрировали досуха, получая C10 в виде белого твердого вещества (17,0 мг, 90%). 1H ЯМР (400 МГц, DMSO-d6) δ=9,37 (br s, 1H), 9,25 (br s, 1H), 8,42 (br s, 3H), 4,26-4,17 (m, 1H), 3,72 (ABq, J=9,1 Гц, 2H), 3,50-3,41 (m, 1H), 3,28-3,18 (m, 1H), 3,18-3,09 (m, 1H), 2,99-2,74 (m, 2H), 2,07-1,63 (m, 4H), 1,22 (d, J=6,5 Гц, 3H).
Стадия g
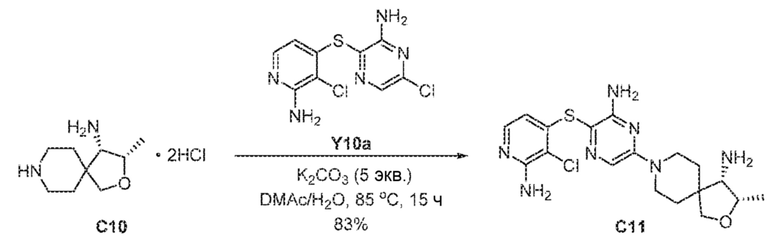
[00217] В сосуд Шленка на 10 мл вносили 3-((2-амино-3-хлорпиридин-4-ил)тио)-6-хлорпиразин-2-амин Y10a (0,1 г, 0,347 ммоль), (3S,4S)-3-метил-2-окса-8-азаспиро[4,5]декан-4-амина дигидрохлорид C10 (0,1 г, 0,416 ммоль, 1,2 экв.), DMAc (0,6 мл) и 36%-ный водн. K2CO3 (0,66 г, 1,735 ммоль, 5,0 экв.). Смесь перемешивали в течение 16 часов на масляной бане при 100°C и охлаждали до 20-25°C. Добавляли 20%-ный соляной раствор (10 мл) и смесь экстрагировали с помощью EtOAc (20 мл ×2). Объединенные экстракты промывали 20%-ным соляным раствором (10 мл ×4), сушили над безводным Na2SO4 и отфильтровывали. Фильтрат концентрировали досуха, получая C11 в виде твердого вещества желтого цвета (121 мг, 83%). 1H ЯМР (400 МГц, DMSO-d6) δ=7,64 (d, J=6,2 Гц, 1H), 7,62 (s, 1H), 6,26 (s, 2H), 6,13 (s, 2H), 5,74 (d, J=5,3 Гц, 1H), 4,12-4,02 (m, 1H), 3,90-3,78 (m, 2H), 3,67 (d, J=8,4 Гц, 1H), 3,49 (d, J=8,4 Гц, 1H), 3,33 (s, 2H), 2,91 (d, J=5,1 Гц, 1H), 1,78-1,68 (m, 1H), 1,67-1,57 (m, 1H), 1,56-1,41 (m, 2H), 1,08 (d, J=6,5 Гц, 3H).
Пример 3
Путь D
Стадия а:

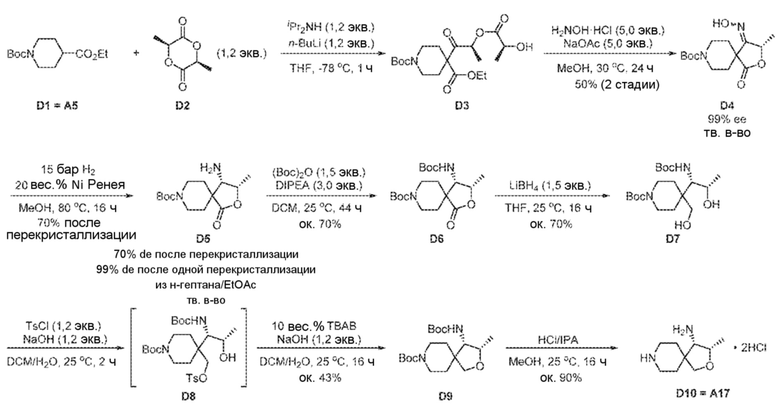
[00218] В трехгорлую круглодонную колбу A объемом 500 мл в атмосфере азота загружали диизопропиламин (9,44 г, 93,3 ммоль, 1,2 экв.) и THF (200 мл). Раствор охлаждали до IT = -20°C, по каплям в течение 30 мин добавляли 2,4 М n-BuLi в гексанах (38,9 мл, 1,2 экв.). Реакционную смесь перемешивали при -20°C в течение 30 мин, а затем охлаждали до -70°C. Раствор 1-(трет-бутил)4-этилпиперидин-1,4-дикарбоксилата (D1) (20,0 г, 77,7 ммоль, 1,0 экв.) в THF (20 мл) добавляли по каплям в течение 30 мин, поддерживая IT = (-70°C)-(-60°C). Реакционную смесь перемешивали при -70°C в течение 30 мин и получали бледно-желтый раствор.
[00219] В трехгорлую круглодонную колбу В объемом 500 мл в атмосфере азота загружали L-лактид (13,4 г, 93,3 ммоль, 1,2 экв.) и THF (120 мл). Раствор охлаждали до IT = -70°C. Раствор из колбы A медленно переносили в колбу B через канюлю в течение 30 мин, поддерживая IT = (-70°C)-(-60°C). Реакционную смесь перемешивали при -70°C в течение 30 мин. Реакционный раствор переносили в колбу C, содержащую 3% HCl (300 мл) через канюлю в течение 30 мин, поддерживая IT=0°C - 5°C. Смесь экстрагировали, используя EtOAc (400 мл ×2), и промывали 20%-ным соляным раствором (200 мл). Органический слой отделяли, сушили над безводным Na2SO4 и отфильтровывали. Фильтрат концентрировали досуха, получая D3 в виде бледно-желтого масла (36,0 г, 71 мас.%, выход продукта 81%), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ=5,42 (q, J=6,8 Гц, 1H), 4,36-4,27 (m, 1H), 4,27-4,16 (m, 2H), 3,71-3,49 (m, 2H), 3,39-3,24 (m, 2H), 2,81 (br d, J=5,0 Гц, 1H), 2,24-1,81 (m, 4H), 1,44 (s, 15H), 1,27 (t, J=7,1 Гц, 3H).
Стадия b

[00220] В круглодонную колбу на 250 мл вносили указанное выше желтое масло (15,0 г, 71 мас.%, 26,5 ммоль), гидрохлорид гидроксиламина (9,3 г, 132,6 ммоль, 5,0 экв.), ацетат натрия (10,9 г, 132,6 ммоль, 5,0 экв.) и метанол (150 мл). Смесь перемешивали в течение 24 ч при 20-25°C. Полученную суспензию фильтровали через МКЦ и осадок на фильтре промывали, используя МеОН (20 мл ×2). Фильтрат концентрировали до ок. 60 мл, затем по каплям добавляли воду (60 мл) в течение 15 минут, в осадок выпадало белое твердое вещество. Суспензию перемешивали в течение ночи и отфильтровывали. Осадок на фильтре промывали смесью МеОН (5 мл) и воды (25 мл) и сушили в вакууме, получая D4 в виде белого твердого вещества (4,9 г, 59%). 1H ЯМР (400 МГц, DMSO-d6) δ=11,45 (s, 1H), 5,33 (q, J=6,6 Гц, 1H), 3,73-3,58 (m, 2H), 3,56-3,43 (m, 1H), 3,43-3,35 (m, 1H), 1,87-1,65 (m, 4H), 1,52 (d, J=6,7 Гц, 3H), 1,41 (s, 9H).
Стадия с

[00221] В реактор объемом 1 л с лопастной мешалкой в атмосфере азота вносили никель Ренея (5 г) и МеОН (250 мл), затем трет-бутил(S)-4-(гидроксиимино)-3-метил-1-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат D4 (25,0 г, 83,80 ммоль). Реактор трижды продували азотом, а затем трижды водородом. Смесь перемешивали в течение 16 ч под давлением водорода 20 бар при IT=80°C. Реакционную смесь фильтровали через микрокристаллическую целлюлозу и осадок на фильтре промывали, используя МеОН (10 мл). Фильтрат концентрировали досуха, получая белое твердое вещество (23,0 г). К твердому веществу добавляли EtOAc (220 мл), полученную суспензию нагревали до кипения с обратным холодильником (IT=100°C) и порциями добавляли н-гептан (550 мл). Полученный прозрачный раствор охлаждали до к.т. в течение 2 ч и оставляли на ночь, чтобы получить D5 в виде бесцветных кристаллов (16,7 г, цис/транс > 99/1, 70%). 1H ЯМР (400 МГц, CDCl3) δ=4,75-4,64 (m, 1H), 3,89-3,80 (m, 1H), 3,68-3,58 (m, 1H), 3,48-3,33 (m, 3H), 1,92-1,61 (m, 4H), 1,46 (s, 9H), 1,40 (d, J=6,5 Гц, 3H).
Стадия d

[00222] В сосуд Шленка на 10 мл загружали трет-бутил-(3S,4S)-4-амино-3-метил-1-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат D5 (100 мг, 0,352 ммоль) и DCM (5,0 мл). Сосуд охлаждали на ледяной бане. По каплям добавляли диизопропиламин (182 мг, 1,41 ммоль), а затем Boc2O (230 мг, 1,05 ммоль). Затем реакционную смесь перемешивали в течение 44 ч при 20-25°C. Органический слой отделяли, промывали 20%-ным соляным раствором (5 мл), сушили над Na2SO4 и отфильтровывали. Фильтрат упаривали досуха, получая D6 в виде бесцветного масла (95 мг, 70%), которое постепенно затвердевало при стоянии. МСВР масса/заряд расчтн. для C19H33N2O6 [M+H]+ 385,2333, найдено 385,2334.
Стадия е
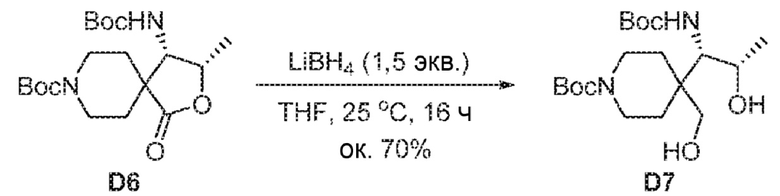
[00223] В колбу Шленка на 10 мл в атмосфере азота загружали трет-бутил-(3S,4S)-4-((трет-бутоксикарбонил)амино)-3-метил-1-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат D6 (126 мг, 0,335 ммоль) и THF (3,0 мл). Колбу охлаждали на ледяной бане. Добавляли по каплям 2,0 M LiBH4 в THF (0,25 мл) и реакционную смесь перемешивали в течение 16 ч при 20-25°C. Реакцию охлаждали на ледяной бане и гасили, добавляя по каплям 5%-ный NaHCO3 (1,0 мл). Смесь разделяли, и водный слой экстрагировали, используя EtOAc (10 мл ×3). Объединенные экстракты промывали 20%-ным соляным раствором (20 мл). Органический слой отделяли, сушили над Na2SO4 и отфильтровывали. Фильтрат упаривали досуха, получая D7 в виде вязкого бесцветного масла (91 мг, 70%). МСВР масса/заряд расчтн. для C19H37N2O6 [M+H]+ 389,2646, найдено 389,2628.
Стадия f
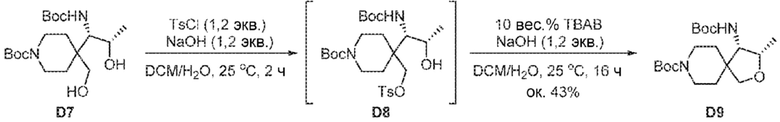
[00224] В сосуд Шленка на 25 мл в атмосфере азота вносили NaOH (14 мг, 0,34 ммоль) и воду (2,0 мл). Сосуд охлаждали на ледяной бане и загружали раствор трет-бутил-4-(1S,2S)-1-((трет-бутоксикарбонил)амино)-2-(гидроксиметил)пиперидин-1-карбоксилата D7 (110 мг, 0,283 ммоль) и TsCl (65 мг, 0,34 ммоль) в DCM (2,0 мл). Смесь перемешивали в течение 16 ч при 20-25°C. Вносили n-Bu4NBr (9,1 мг, 0,028 ммоль), а затем - NaOH (14 мг, 0,34 ммоль) в воде (1,0 мл). Затем смесь перемешивали в течение 16 ч при 20-25°C. Органический слой отделяли, промывали 20%-ным соляным раствором (2 мл), сушили над Na2SO4 и отфильтровывали. Фильтрат упаривали досуха, получая D9 в виде бесцветного масла (45 мг, 43%). МСВР масса/заряд расчтн. для C19H35N2O5 [M+H]+ 371,2540, найдено 371,2533.
Стадия g

[00225] В сосуд Шленка на 10 мл загружали трет-бутил-(3S,4S)-4-((трет-бутоксикарбонил)амино)-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилат D9 (100 мг, 0,27 ммоль), 6 н. HCl в изопропаноле (1,0 мл) и метанол (3,0 мл). Реакционную смесь перемешивали в течение 16 ч при 20-25°C и концентрировали досуха, получая D10 в виде белого твердого вещества (59 мг, 90%). 1H ЯМР (400 МГц, DMSO-d6) δ=9,37 (br s, 1H), 9,25 (br s, 1H), 8,42 (br s, 3H), 4,26-4,17 (m, 1H), 3,72 (ABq, J=9,1 Гц, 2H), 3,50-3,41 (m, 1H), 3,28-3,18 (m, 1H), 3,18-3,09 (m, 1H), 2,99-2,74 (m, 2H), 2,07-1,63 (m, 4H), 1,22 (d, J=6,5 Гц, 3H).
Стадия h
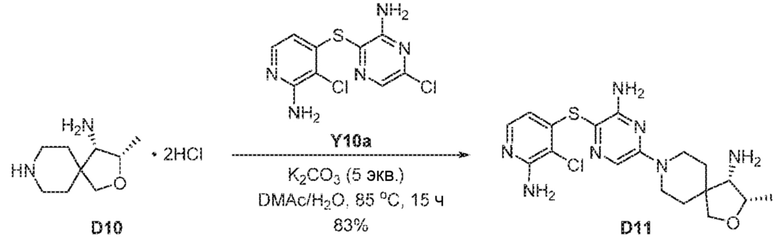
[00226] В сосуд Шленка на 10 мл вносили 3-((2-амино-3-хлорпиридин-4-ил)тио)-6-хлорпиразин-2-амин Y10a (0,1 г, 0,347 ммоль), (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина дигидрохлорид D10 (0,1 г, 0,416 ммоль, 1,2 экв.), DMAc (0,6 мл) и 36%-ный водн. K2CO3 (0,66 г, 1,735 ммоль, 5,0 экв.). Смесь перемешивали в течение 16 часов на масляной бане при 100°C и охлаждали до 20-25°C. Добавляли 20%-ный соляной раствор (10 мл) и смесь экстрагировали с помощью EtOAc (20 мл ×2). Объединенные экстракты промывали 20%-ным соляным раствором (10 мл ×4), сушили над безводным Na2SO4 и отфильтровывали. Фильтрат концентрировали досуха, получая D11 в виде твердого вещества желтого цвета (121 мг, 83%). 1H ЯМР (400 МГц, DMSO-d6) δ=7,64 (d, J=6,2 Гц, 1H), 7,62 (s, 1H), 6,26 (s, 2H), 6,13 (s, 2H), 5,74 (d, J=5,3 Гц, 1H), 4,12-4,02 (m, 1H), 3,90-3,78 (m, 2H), 3,67 (d, J=8,4 Гц, 1H), 3,49 (d, J=8,4 Гц, 1H), 3,33 (s, 2H), 2,91 (d, J=5,1 Гц, 1H), 1,78-1,68 (m, 1H), 1,67-1,57 (m, 1H), 1,56-1,41 (m, 2H), 1,08 (d, J=6,5 Гц, 3H).
Пример 4
(3-((2-амино-3-хлорпиридин-4-ил)тио)-6-хлорпиразин-2-амин)
[00227] Указанное выше соединение Y7a=Y10a (3-((2-амино-3-хлорпиридин-4-ил)тио)-6-хлорпиразин-2-амин) получали следующим образом:

Стадия а
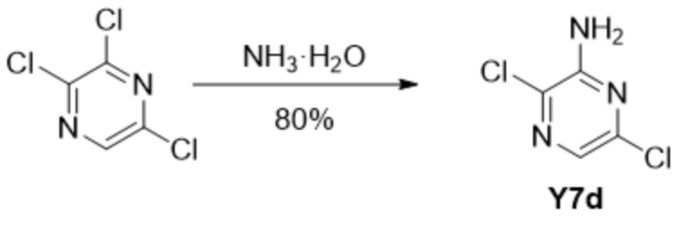
[00228] 2,3,5-трихлорпиразин (70,50 г, 384,36 ммоль, 1 экв.) и раствор аммиака (25 мас.%, 364,00 г, 400 мл, 2,68 моль, 6,14 экв.) загружали в герметичный реактор объемом 1 л. Смесь нагревали до 80°C и перемешивали в течение 24 ч, и реакция была завершена. Реакционную смесь охлаждали до 30°C и фильтровали, получая коричневый осадок на фильтре. Коричневый осадок на фильтре растворяли в ацетоне (50 мл) и фильтровали. К фильтрату добавляли петролейный эфир (300 мл). Суспензию перемешивали в течение 4 часов и отфильтровывали, получая сырой продукт. Неочищенный продукт суспендировали в смешанном растворителе из петролейного эфира и ацетона (10/1, 200 мл) и отфильтровывали, получая продукт Y7d (51,00 г, 307,91 ммоль, выход 80%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ=7,63 (s, 1H).Преимущество этого (также обобщенного) метода заключается в том, что для получения Y7d не требуется колоночная хроматография.
Стадия b

[00229] В круглодонную колбу на 200 мл загружали Na2S (10,816 г, 44 мас.%, содержит кристаллическую воду, 60,978 ммоль) и толуол (100 мл). Смесь кипятили с обратным холодильником и воду удаляли, используя ловушку Дина-Старка (отгоняли примерно 5~6 мл воды). После охлаждения смесь концентрировали досуха.
[00230] В вышеуказанную круглодонную колбу вносили Y7d (5,000 г, 30,489 ммоль) и 2-метилбутан-2-ол (50 мл), реакционную смесь кипятили с обратным холодильником и перемешивали в течение 36 ч. После охлаждения до 25°C смесь фильтровали. Растворитель фильтрата заменяли н-гептаном (5 об., 3 раза, из расчета на Y7d) и, наконец, концентрировали до 1 об. остатка. К остатку добавляли THF (25 мл) при 25°C и перемешивали. Суспензию отфильтровывали и промывали THF/н-гептаном (5 мл/5 мл), получая коричневое твердое вещество (6,200 г).
[00231] В другую круглодонную колбу на 200 мл загружали вышеуказанное коричневое твердое вещество (6,200 г), 10% соляной раствор (25 мл), Me-THF (30 мл) и n-Bu4NBr (9,829 г, 30,489 ммоль). Смесь перемешивали 0,5 ч при комнатной температуре и фазы разделяли. Органическую фазу промывали 20%-ным соляным раствором (25 мл), заменяли растворитель на изо-пропанол (5 об., 3 раза, на основе Y7d) с получением раствора Y7c в изо-пропаноле (27,000 г, чистота 99,2% на основании площади ВЭЖХ, выход продукта 58,08%). 1H ЯМР (400 МГц, DMSO-d6) δ=6,88 (s, 1H), 2,97-2,92 (m, 14H), 1,38-1,31 (m, 14H), 1,13-1,04 (m, 14H), 0,73-0,69 (t, 21H). Преимущество использования n-Bu4NBr (или другого соответствующего трет-алкиламиногалогенида) заключается в более легкой переработке и очистке.
Стадия с

[00232] В круглодонную колбу на 25 мл загружали Y7c (4,7 г, 23,27 мас.%, раствор в IPA, 2,723 ммоль, 1,0 экв.), Y7b (1,052 г, 4,085 ммоль, 1,5 экв.), 1,10-фенантролин (0,05 г, 0,272 ммоль) и воду (8 мл). Смесь трижды продували газообразным азотом и вносили CuI (0,026 г, 0,136 ммоль) в атмосфере азота. Смесь нагревали до 65°C и перемешивали в течение 3 ч, и реакция была завершена. Реакционную смесь охлаждали до комнатной температуры и отфильтровывали, а осадок на фильтре промывали водой (4 мл*3). Осадок на фильтре суспендировали в МТВЕ (6 мл) в течение 30 минут и отфильтровывали. Осадок на фильтре промывали, используя МТВЕ (6 мл) и сушили, получая Y7a=Z17a, который является соединением формулы Y10a, упомянутым на стадии g) примера 2 и стадии h) примера 3 (565 мг, выход 72%). Реакцию можно проводить с использованием меди вместо палладия для катализа реакции сочетания Y7b с Y7c.
Пример 5
(3-((2-амино-3-хлорпиридин-4-ил)тио)-6-хлорпиразин-2-амин)
[00233] Альтернативно соединение формулы Z17a=Y10a получают в соответствии со следующей схемой реакции:


[00234] В трехгорлую круглую колбу загружали Y7d (200 мг, 1,22 ммоль, 1 экв.), диоксан (4 мл). Раствор вакуумировали и трижды продували газообразным азотом. Добавляли Xantphos (14 мг, 0,024 ммоль, 0,02 экв.), PdCl2 (dppf) (8,9 мг, 0,012 ммоль, 0,1 экв.) и DIPEA (0,32 г, 2,44 ммоль, 2,0 экв.) в атмосфере азота. Раствор нагревали при 85°C в течение ночи. Реакционную смесь охлаждали и упаривали. Остаток очищали колоночной хроматографией (элюент/этилацетат/гептан=1/1), получая Z17d (259 мг, 0,99 ммоль, 81%). 1H ЯМР (400 МГц, CDCl3) δ=7,83 (s, 1H), 4,88 (bs, 2H), 3,73 (s, 3H), 3,47 (t, J=9,2 Гц, 2H), 2,79 (t, J=9,2 Гц, 2H).

[00235] К раствору Z17d (8,0 кг, содержание 95%, 30,68 моль) в THF (70 л) добавляли EtONa (приготовленный из 776 г Na и 13,6 л EtOH) при комнатной температуре и смесь перемешивали при температуре окружающей среды в течение 1 часа. Затем смесь концентрировали до получения влажного твердого вещества желтого цвета на роторном испарителе и остаток суспендировали в DCM (40 л). Смесь перемешивали в атмосфере N2 в течение 16 ч. Твердое вещество собирали вакуумной фильтрацией и осадок промывали DCM (около 15 л) до тех пор, пока фильтрат не стал бесцветным (PSC-2). Затем твердое вещество сушили в вакууме, получая Z17c (6,93 кг, qnmr 72%, выход 88%). 1H ЯМР (400 МГц, D2O) δ=7,37 (s, 1H).

[00236] К смеси Z17c (6,95 кг, содержание 72%, 27,23 моль) в 1,4-диоксане (72 л) добавляли Xantphos (233 г, 411 ммоль, 0,015 экв.), Pd2(dba)3 (186 г, 206 ммоль, 0,0075 экв.), Z17b (7,13 кг, 28,02 моль) и DIPEA (7,02 кг, 54,46 моль). Систему вакуумировали и трижды продували газообразным азотом. Смесь перемешивали при 65°C в течение 16 ч в атмосфере N2. Смесь охлаждали до к.т. и добавляли воду (50 л), отфильтровывали. Осадок на фильтре промывали, используя EA (25л). Фильтрат экстрагировали, используя ЕА (4 × 20 л). Органическую фазу концентрировали в вакууме, получая сырой продукт, который объединяли с осадком на фильтре. Затем к неочищенному продукту добавляли DCM (60 л) и перемешивали при 25-30°C в течение 18 ч, а затем отфильтровывали. Осадок на фильтре суспендировали в CH2Cl2 (30 л) в течение 4 ч и отфильтровывали. Осадок на фильтре суспендировали в CH2Cl2 (30 л) в течение 16 ч и отфильтровывали. Затем осадок на фильтре сушили в вакууме, получая Z17a (9,1 кг, 84%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6)δ=7,89 (s, 1H), 7,7 (d, J=7,6 Гц, 1H), 7,18 (bs, 2H), 6,40 (bs, 2H), 5,97 (d, J=7,6 Гц, 1H)
Пример 6
(3-((2-амино-3-хлорпиридин-4-ил)тио)-6-хлорпиразин-2-амин)
[00237] Альтернативно соединение формулы Z17a=Y10a получают в соответствии со следующей схемой реакции:

Более детально синтез соединения Z17a проводили следующим образом:
Стадия а

[00238] В атмосфере азота, n-BuLi (2,5 М, 7,6 л) по каплям добавляли к раствору 3-хлор-2-фторпиридина (2 кг) в THF (15 л) при -78°C. Затем полученную смесь перемешивали в течение 1 ч. Затем по каплям добавляли раствор I2 (4,82 кг) в THF (6 л). По завершении добавления реакционную смесь перемешивали в течение 30 мин, а затем гасили насыщ. Na2SO3 (10 л) и подогревали до 20-25°C. Фазы разделяли. Водную фазу экстрагировали с помощью EA (2 × 10 л). Объединенную органическую фазу промывали насыщенным Na2SO3 (2×8 л), соляным раствором (8 л) и сушили над Na2SO4. Органическую фазу концентрировали под вакуумом. Остаток суспендировали в МеОН (4 л), фильтровали и сушили, получая 3-хлор-2-фтор-4-йодпиридин 1c (2,2 кг, выход 68%).
Стадия b
[00239] Через раствор соединения 1c (8 кг) в DMSO (48 л) пропускали NH3 (газ) при 80°C в течение ночи. ТСХ показала, что реакция завершилась. Реакционную смесь охлаждали до к.т. Реакционную смесь вносили в воду (140 л). Твердое вещество собирали и промывали водой (25 л), сушили с получением Z17b (6,91 кг, выход 87%). 1H ЯМР (400 МГц, CDCl3) δ=7,61 (d, J=6,8 Гц, 1H), 7,14 (s, J=6,8 Гц, 1H), 5,09 (bs, 2H).
Стадия с

[00240] Раствор 2-амино-6-хлорпиразина 1a (1 кг, 7,69 моль) в DCM (15 л) кипятили с обратным холодильником, и в который загружали NBS (417 г) порциями в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры. Реакционную смесь промывали водой (3 л) и соляным раствором (3 л). Органическую фазу упаривали и остаток очищали колоночной хроматографией, получая продукт Z17f (3-бром-6-хлорпиразин-2-амин) (180 г, выход 11%).
Стадия d

[00241] В раствор 3-бром-6-хлорпиразин-2-амина Z17f (6,0 кг, 28,78 моль) в 1,4-диоксане (40 л) вносили Pd(OAc)2 (64,56 г, 287,6 ммоль), Xantphos (333 г, 575,6 ммоль) и DIPEA (7,44 кг, 57,56 моль) при комнатной температуре в атмосфере азота. Еще через 30 минут продувки азотом вносили метил-3-меркаптопропаноат (3,81 кг, 31,70 моль), что приводило к потемнению смеси оранжевого цвета. Смесь нагревали до 90°C. ВЭЖХ показала полную конверсию исходного материала. Смеси давали остыть примерно до комнатной температуры, затем разбавляли, используя EtOAc (40 л). После выдержки в течение 30 мин при перемешивании всю смесь отфильтровывали и твердое вещество промывали, используя EtOAc (3 × 15 л). Объединенный оранжевый фильтрат концентрировали досуха и твердый остаток суспендировали в DCM (45 л). Смесь нагревали до 35-40°C и перемешивали в течение 1 ч до полного растворения твердых веществ. Затем по каплям добавляли н-гептан (45 л). По окончании добавления смесь охлаждали до 15-20°C при перемешивании в течение 1 ч. Твердое вещество собирали вакуумной фильтрацией и твердое вещество промывали холодным 1:1 DCM/гептаном (25 л), затем гептаном (25 л) (PSC-2). Твердое вещество сушили в течение выходных с получением Z17d (5,32 кг, выход 75%). 1H ЯМР (400 МГц, CDCl3) δ=7,83 (s, 1H), 4,88 (bs, 2H), 3,73 (s, 3H), 3,47 (t, J=9,2 Гц, 2H), 2,79 (t, J=9,2 Гц, 2H).
Стадия е

[00242] К раствору Z17d (8,0 кг, содержание 95%, 30,68 моль) в THF (70 л) добавляли EtONa (приготовленный из 776 г Na и 13,6 л EtOH) при комнатной температуре и смесь перемешивали при температуре окружающей среды в течение 1 часа. Затем смесь концентрировали до получения влажного твердого вещества желтого цвета на роторном испарителе и остаток суспендировали в DCM (40 л). Смесь перемешивали в атмосфере N2 в течение 16 ч. Твердое вещество собирали вакуумной фильтрацией и осадок промывали DCM (около 15 л) до тех пор, пока фильтрат не стал бесцветным (PSC-2). Затем твердое вещество сушили в вакууме, получая Z17c (6,93 кг, qnmr 72%, выход 88%). 1H ЯМР (400 МГц, D2O) δ=7,37 (s, 1H).
Стадия f
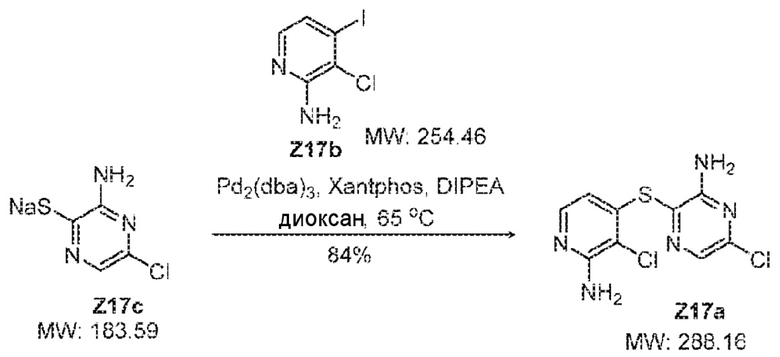
[00243] К смеси Z17c (6,95 кг, содержание 72%, 27,23 моль) в 1,4-диоксане (72 л) добавляли Xantphos (233 г, 411 ммоль, 0,015 экв.), Pd2(dba)3 (186 г, 206 ммоль, 0,0075 экв.), Z17b (7,13 кг, 28,02 моль) и DIPEA (7,02 кг, 54,46 моль). Систему вакуумировали и трижды продували газообразным азотом. Смесь перемешивали при 65°C в течение 16 ч в атмосфере N2. Смесь охлаждали до к.т. и добавляли воду (50 л), отфильтровывали. Осадок на фильтре промывали, используя EA (25 л). Фильтрат экстрагировали, используя ЕА (4 × 20 л). Органическую фазу концентрировали в вакууме, получая сырой продукт, который объединяли с осадком на фильтре. Затем к неочищенному продукту добавляли DCM (60 л) и перемешивали при 25-30°C в течение 18 ч, а затем отфильтровывали. Осадок на фильтре суспендировали в CH2Cl2 (30 л) в течение 4 ч и отфильтровывали. Осадок на фильтре суспендировали в CH2Cl2 (30 л) в течение 16 ч и отфильтровывали. Затем осадок на фильтре сушили в вакууме, получая Z17a (3-((2-амино-3-хлорпиридин-4-ил)тио)-6-хлорпиразин-2-амин; 9,1 кг, 84%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ=7,89 (s, 1H), 7,7 (d, J=7,6 Гц, 1H), 7,18 (bs, 2H), 6,40 (bs, 2H), 5,97 (d, J=7,6 Гц, 1H).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ L-BPA | 2016 |
|
RU2688676C1 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ | 2015 |
|
RU2701206C2 |
| НОВЫЕ АМИНОГЛИКОЗИДНЫЕ АНТИБИОТИКИ | 2007 |
|
RU2458931C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| ТИОФЕНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2709473C1 |
| СИНТЕЗ ЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ ПИРАЗОЛИН КАРБОКСАМИДИНА | 2011 |
|
RU2550694C2 |
| ПРОИЗВОДНЫЕ [4-(1-АМИНОЭТИЛ)ЦИКЛОГЕКСИЛ]МЕТИЛАМИНА И [6-(1-АМИНОЭТИЛ)ТЕТРАГИДРОПИРАН-3-ИЛ]МЕТИЛАМИНА | 2009 |
|
RU2515906C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-2-ФЕНИЛАЛКАНОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2009 |
|
RU2486174C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
Изобретение относится к способу производства соединения формулы I, причем указанный способ включает взаимодействие соединения формулы II с соединением формулы III согласно следующей схеме реакции:
 , где LG представляет собой Cl, A представляет собой анион протонной кислоты, и протонная кислота представляет собой HCl, m равно 1, n равно 1, и p равно 2 так, чтобы соль формулы II не имела электрического заряда, где реакцию проводят при температуре от 30°C до 100°C. Технический результат: разработан новый способ получения соединения формулы I, способного ингибировать активность SHP2, с высоким выходом, пригодный для производства в больших масштабах. 1 з.п. ф-лы, 6 пр.
, где LG представляет собой Cl, A представляет собой анион протонной кислоты, и протонная кислота представляет собой HCl, m равно 1, n равно 1, и p равно 2 так, чтобы соль формулы II не имела электрического заряда, где реакцию проводят при температуре от 30°C до 100°C. Технический результат: разработан новый способ получения соединения формулы I, способного ингибировать активность SHP2, с высоким выходом, пригодный для производства в больших масштабах. 1 з.п. ф-лы, 6 пр.
1. Способ производства соединения формулы I, причем указанный способ включает взаимодействие соединения формулы II с соединением формулы III согласно следующей схеме реакции:
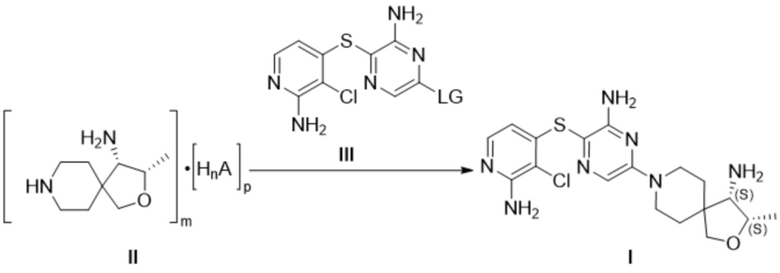 ,
,
где LG представляет собой Cl, A представляет собой анион протонной кислоты, и протонная кислота представляет собой HCl, m равно 1, n равно 1, и p равно 2 так, чтобы соль формулы II не имела электрического заряда, где реакцию проводят при температуре от 30°C до 100°C.
2. Способ согласно п. 1, где реакцию проводят при температуре от 50°C до 100°C.
| WO2018172984A1, 27.09.2018 | |||
| WO2017211303A1, 14.12.2017 | |||
| WO2015107494A1, 23.07.2015 | |||
| WO2016203404A1, 22.12.2016 | |||
| WO2016203406A1, 22.12.2016 | |||
| WO2018013597A1, 18.01.2018 | |||
| EA201691442A1, 30.12.2016. |
