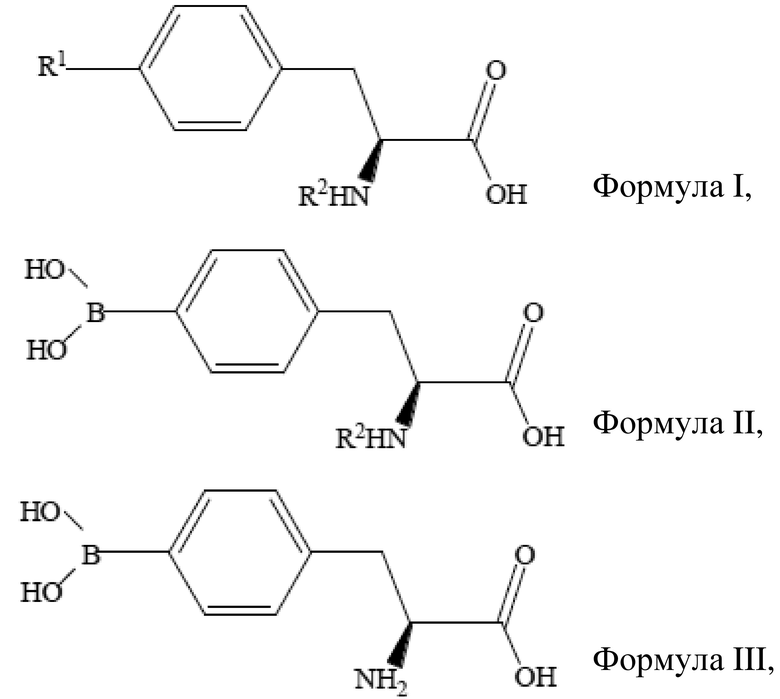
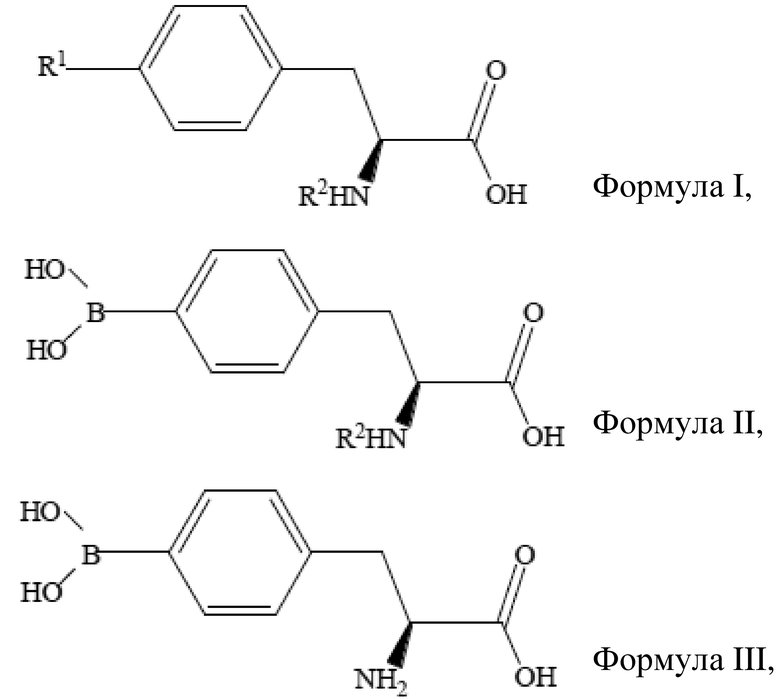
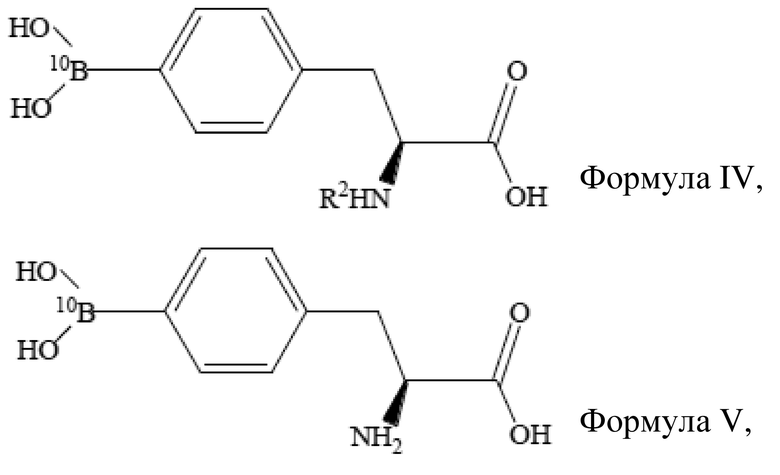
Область техники, к которой относится изобретение
[01] Настоящее изобретение относится к способу получения борсодержащего лекарственного средства для бор-нейтронозахватной терапии и, более конкретно, относится к способу получения L-BPA.
Уровень техники
[02] С развитием ядерной физики такие виды радиотерапии, как, например, с применением кобальта-60, линейных ускорителей, пучка электронов, стали одними из основных способов лечения рака. Однако традиционные способы фотонной или электронной терапии ограничены физическими свойствами излучения - хотя лучи и уничтожают опухолевые клетки, они также повреждают большое количество нормальных тканей на своем пути; кроме того, вследствие различной чувствительности опухолевых клеток к излучению, стандартные способы радиотерапии часто являются неэффективными при лечении злокачественных опухолей с высокой устойчивостью к радиации (например, мультиформной глиобластомы и меланомы).
[03] С целью уменьшить радиационное повреждение нормальных тканей вокруг опухоли в радиотерапии применяют средства направленной терапии из химиотерапии; при этом, в случае опухолевых клеток с высокой устойчивостью к радиации, в настоящее время активно разрабатываются источники излучения с высокой относительной биологической эффективностью (RBE) для видов терапии, таких как протонная терапия, терапия тяжелыми частицами, нейтронозахватная терапия и т.д. Один из них, нейтронозахватная терапия сочетает в себе оба вышеуказанные способа; например, бор-нейтронозахватная терапия заключается в специфичном накоплении борсодержащих лекарственных средств в опухолевых клетках в сочетании с точной настройкой нейтронного пучка, что обеспечивает более эффективное лечение рака по сравнению с традиционными способами радиотерапии.
[04] Бор-нейтронозахватная терапия (BNCT) основана на применении лекарственных средств, содержащих бор (10B), который характеризуется большим сечением захвата тепловых нейтронов, и в результате захвата нейтронов 10B(n,α)7Li и реакции расщепления ядра образуются две тяжелые заряженные частицы 4He и 7Li. Две заряженные частицы обладают средней энергией, которая составляет приблизительно 2,33 МэВ, и характеризуются линейной передачей энергии (LET), и коротким пробегом излучения; для α-частиц линейная передача энергии и пробег излучения составляют соответственно 150 кэВ/мкм и 8 мкм, и для тяжелых частиц 7Li - соответственно 175 кэВ/мкм и 5 мкм, при этом общий пробег излучения обеих частиц приблизительно равняется размеру клетки; таким образом радиационное повреждение по отношению к живым организмам может быть ограничено на клеточном уровне; когда борсодержащие лекарственные средства селективно накапливаются в опухолевых клетках, с применением подходящего источника излучения нейтронов можно уничтожать опухолевые клетки локально без значительного повреждения нормальных тканей.
[05] Эффективность бор-нейтронозахватной терапии зависит от концентрации борсодержащих лекарственных средств в опухолевых клетках и от количества тепловых нейтронов, поэтому ее относят к бинарной терапии рака; таким образом, кроме разработки источников нейтронного излучения, разработка борсодержащих лекарственных средств играет большую роль в изучении бор-нейтронозахватной терапии.
[06] Известно, что 4-(10B)бороно-L-фенилаланин (L-10BPA) является важным борсодержащим лекарственным средством, которое применяют в бор-нейтронозахватной терапии (BNCT) при лечении рака.
[07] Вследствие этого были разработаны различные способы синтеза L-BPA. Как показано ниже на формуле (A), способы синтеза L-BPA, известные из уровня техники, предусматривают два способа: образование связи (a) или образование связи (b).
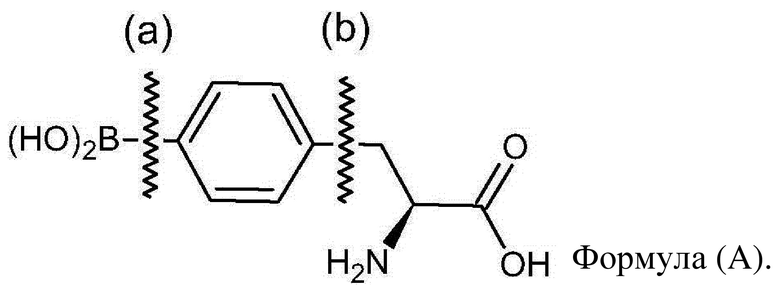
[08] Один из них, способ синтеза L-BPA с образованием связи (a), представляет собой введение заместителя, содержащего бороно, в кольцо фенилаланина, при этом L-BPA образуется вследствие образования связи углерод-бор в пара-положении относительно амида.
[09] В J. Org. Chem. 1998, 63, 8019 приведен способ проведения катализируемой палладием реакции перекрестного сочетания (S)-4-йодфенилаланина (например, (S)-N-трет-бутоксикарбонил-4-йодфенилаланина) с защитной группой при аминогруппе и соединения дибора (например, бис(пинаколато)дибора) с получением (S)-N-трет-бутоксикарбонил-4-пинаколатоборонофенилаланина с защитной группой (S)-4-боратфенилаланина при аминогруппе с последующим получением L-BPA путем удаления защитной группы с аминогруппы и заместителя с бора.
[10] Однако, поскольку бис(пинаколато)дибор с высоким содержанием 10B не является коммерчески доступным соединением, в данном способе также необходимо предварительное получение борирующего средства, в результате чего повышается сложность и длительность способа, при этом невозможно получить высокий выход L-BPA. Кроме того, карбоксильную группу (S)-4-йодфенилаланина с защитной группой при аминогруппе необходимо защитить заместителем с образованием бензиловой сложноэфирной группы, чтобы повысить выход способа до 88%; однако, в данном случае для получения L-BPA требуется дополнительная стадия снятия защиты с карбоксильной группы, что, по сути, повышает сложность способа получения L-BPA.
[11] Таким образом, в приведенном в литературе способе не только необходимо предварительное получение борирующего средства, но также необходимо увеличение длительности и добавление стадий применения и удаления защитной группы с карбоксильной группы, что негативно влияет на основной промышленный способ синтеза L-BPA.
[12] В другом аспекте способ синтеза L-BPA с образованием связи (a), представляет собой проведение реакции сочетания аминокислоты с борсодержащим бензильным фрагментом или борсодержащим бензальдегидным фрагментом с образованием L-BPA. В Biosci. Biotech. Biochem. 1996, 60, 683 приведен энантиоселективный способ синтеза L-BPA путем проведения реакции сочетания циклических эфиров борной кислоты с хиральными производными L-валина с получением L-BPA. Однако, в данном способе необходимо сначала из 4-боронобензилбромида получить циклический эфир борной кислоты, который затем вводят в реакцию сочетания с хиральным производным L-валина, при этом на второй стадии синтеза легко вызвать нежелательную рацемизацию аминокислоты, в результате, чтобы получить L-BPA с определенной оптической чистотой, в способе требуется стадия ферментативного расщепления, что еще больше снижает выход.
[13] Таким образом, приведенный в литературе способ все же содержит стадии предварительного получения борирующего средства и последующего ферментативного расщепления, в результате чего повышается сложность и длительность способа, при этом невозможно получить высокий выход L-BPA.
[14] Кроме того, в настоящее время, как известно, (10B)борсодержащий L-10BPA (4-(10B)бороно-L-фенилаланин) представляет собой главное средство, которое накапливают в опухолевых клетках, после чего с помощью пучка тепловых нейтронов облучают бор, накопленный в опухолевой клетке, в результате захвата образуются частицы с высокой энергией, уничтожающие опухолевую клетку, и таким образом достигается главная цель терапии рака. Таким образом, (10B)бор способствует применению L-10BPA в бор-нейтронозахватной терапии в качестве средства для терапии рака.
[15] Однако, существующий в природе бор содержит приблизительно 19,9% (10B)бора и приблизительно 80,1% (11B)бора. Таким образом, многие исследователи все еще активно разрабатывают новые способы, пригодные для синтеза L-BPA, в частности, пригодные для синтеза L-BPA с высоким содержанием (10B)бора.
[16] В J. Org. Chem. 1998, 63, 8019 приведен и другой способ синтеза (10B)борирующего средства, но, поскольку данный способ содержит большое количество стадий, то при получении значительно снижается содержание (10B)бора в веществах с высоким содержанием (10B)бора. Соответственно, приведенный в литературе способ не является пригодным для синтеза L-BPA с высоким содержанием (10B)бора.
[17] Также в Biosci. Biotech. Biochem. 1996, 60, 683 приведен способ без проведения стадии ферментативного расщепления, но в нем не удалось получить L-BPA с определенной оптической чистотой; и способ получения (10B)борирующего средства также содержит большое количество стадий, что приводит к превращениям веществ с высоким содержанием (10B)бора при получении. Соответственно, приведенный в литературе способ также не является пригодным для синтеза L-BPA с высоким содержанием (10B)бора.
[18] Кроме того, в Bull. Chem. Soc. Jpn. 2000, 73, 231 приведен способ проведения катализируемой палладием реакции сочетания 4-йод-L-фенилаланина и 4,4,5,5-тетраметил-1,3,2-диоксаборолана (тривиальное название: пинаколборан). Однако, в литературе не указано, как применить данный способ для получения L-BPA с высоким содержанием (10B)бора, и 4,4,5,5-тетраметил-1,3,2-диокса(10B)боролан не является коммерчески доступным соединением, поэтому приведенный в литературе способ также не является пригодным для синтеза L-BPA с высоким содержанием (10B)бора.
[19] Кроме того, в Synlett. 1996, 167 приведен способ проведения реакции сочетания йодфенилбората и цинковых производных L-серина, при этом в способе необходимо включение стадий получения йодфенилбората и цинкового производного L-серина, что приводит к меньшему выходу полученного L-BPA. Кроме того, выбранный в данном способе трийод(10B)бор с высоким содержанием (10B)бора и 1,3-дифенилпропан-1,3-диол не являются коммерчески доступными соединениями, поэтому приведенный в литературе способ не является пригодным для синтеза L-BPA с высоким содержанием (10B)бора.
Краткое описание изобретения
[20] В связи с тем, что способ, известный из уровня техники, обладает такими недостатками, как длительность, сложность стадий и необходимость получать заранее борирующее средство, в одном аспекте настоящего изобретения предусматривается способ получения L-BPA (4-бороно-L-фенилаланина), для которого не требуются сложные стадии очистки, и который является безопасным для окружающей среды, что снижает длительность и стоимость способа и увеличивает его эффективность. Соответственно, L-BPA, полученный с помощью способа получения по настоящему изобретению, также обладает преимуществами высокой химической чистоты и высокой оптической чистоты.
[21] В другом аспекте настоящего изобретения предусматривается способ получения L-10BPA (4-(10B)бороно-L-фенилаланина), для которого не требуются сложные стадии очистки, и который является безопасным для окружающей среды, что снижает длительность и стоимость способа и увеличивает его эффективность. В одном варианте осуществления способ получения по настоящему изобретению может способствовать получению L-10BPA с высокой химической чистотой, высокой оптической чистотой и высокой изотопной чистотой.
[22] В еще одном аспекте настоящего изобретения предусматривается подходящий способ получения L-BPA и L-10BPA, для которого не требуются сложные стадии очистки, и который обладает такими преимуществами как снижение длительности и стоимости, повышение эффективности, безопасность для окружающей среды и удобство.
[23] С целью осуществления каждого из указанных выше аспектов, в одном варианте осуществления способ получения L-BPA по настоящему изобретению включает стадии:
проведения реакции (S)-4-галогенфенилаланина формулы I с защитной группой при аминогруппе, борирующего средства, реактива Гриньяра и бис-(2-метиламиноэтилового) эфира с получением реакционной смеси, где реакционная смесь содержит (S)-4-бороно-L-фенилаланин формулы II с защитной группой при аминогруппе, при этом и в формуле I, и в формуле II R2 представляет собой защитную группу;
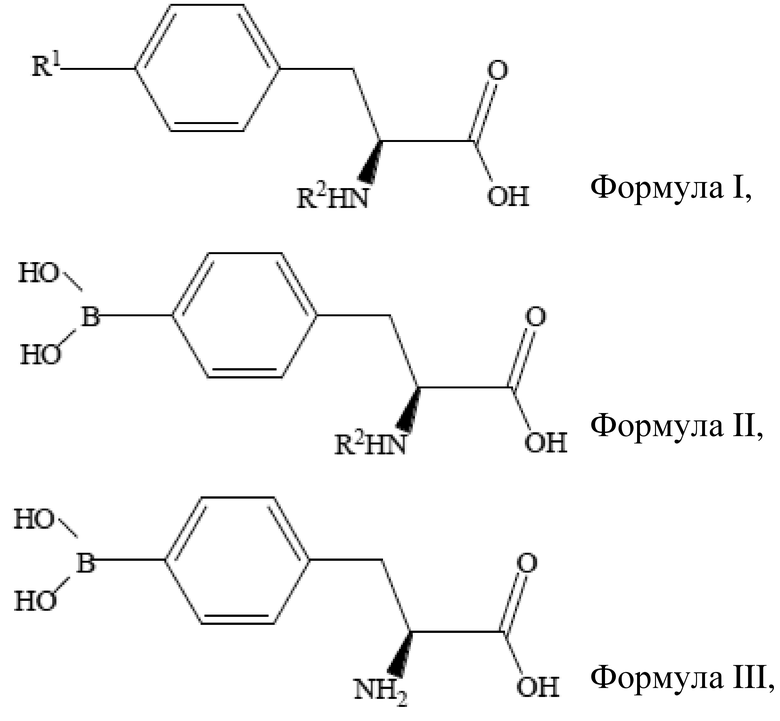
разделения реакционной смеси с получением (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе;
удаления защитной группы с аминогруппы (S)-4-бороно-L-фенилаланина с получением L-BPA, который имеет структуру, представленную формулой III выше.
[24] В другом варианте осуществления способ получения L-BPA по настоящему изобретению включает стадии:
проведения реакции (S)-4-галогенфенилаланина формулы I с защитной группой при аминогруппе, борирующего средства и реактива Гриньяра с получением реакционной смеси, где реакционная смесь содержит (S)-4-бороно-L-фенилаланин формулы II с защитной группой при аминогруппе, при этом и в формуле I, и в формуле II R2 представляет собой защитную группу;
разделения реакционной смеси с получением (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе;
удаления защитной группы с аминогруппы (S)-4-бороно-L-фенилаланина с получением L-BPA, который имеет структуру, представленную формулой III.
[25] В соответствии с настоящим изобретением, термин «борирующее средство» означает любой реагент, способный, после стадии проведения реакции (S)-4-галогенфенилаланина с защитной группой при аминогруппе с борирующим средством и реактивом Гриньяра (с добавлением бис-(2-метиламиноэтилового) эфира или без него), заменять заместитель R1 (т.е. R1, изображенный на формуле I) (S)-4-галогенфенилаланина с защитной группой при аминогруппе на борсодержащий заместитель (такой как без ограничения бороно).
[26] В соответствии с настоящим изобретением, бор в борирующем средстве может представлять собой любую форму бора, такую как (11B)бор, (10B)бор или их комбинация (например, существующий в природе бор: (10B)бор составляет приблизительно 19,9% от всего бора). специалисту в данной области техники будет понятно, что содержание (10B)бора также может быть другим, например, без ограничения содержание (10B)бора ≥ 95% в одном варианте осуществления настоящего изобретения.
[27] Предпочтительно, борирующее средство включает триалкилборат, однако специалисту в данной области техники будет понятно, что приведенные в данном документе виды борирующих средств не подразумеваются как ограничивающие. Триалкилборат включает: трибутилборат, триэтилборат, триметилборат, триизопропилборат, трипропилборат, три-трет-бутилборат или любой подходящий триалкилборат. Предпочтительно, борирующее средство представляет собой трибутилборат.
[28] Указанный выше способ получения обладает следующими преимуществами.
(1) При проведении реакции (S)-4-галогенфенилаланина с защитной группой при аминогруппе, борирующего средства и реактива Гриньяра (с добавлением бис-(2-метиламиноэтилового) эфира или без него) можно получить (S)-4-бороно-L-фенилаланин с защитной группой при аминогруппе без необходимости предварительной защиты карбоксильных групп (S)-4-галогенфенилаланина с защитной группой при аминогруппе. Соответственно, в данном способе получения нет необходимости в стадии удаления защитной группы с карбоксильной группы, что значительно сокращает длительность способа.
(2) Борирующее средство применяют непосредственно в реакции, в результате упрощается предварительное получение борирующего средства.
(3) Вследствие простоты способа получения L-BPA обладает преимуществами высокой химической чистоты, высокой оптической чистоты и высокого общего выхода. Таким образом, обеспечивается способ получения, который характеризуется малой продолжительностью, низкой стоимостью и высокой эффективностью;
(4) Исходным материалом для получения борирующего средства является борная кислота, ее преимуществами являются доступность и низкая цена, соответственно, преимуществами полученного борирующего средства также являются доступность и низкая цена.
[29] Получение неочищенного (S)-4-галогенфенилаланина также включает стадию
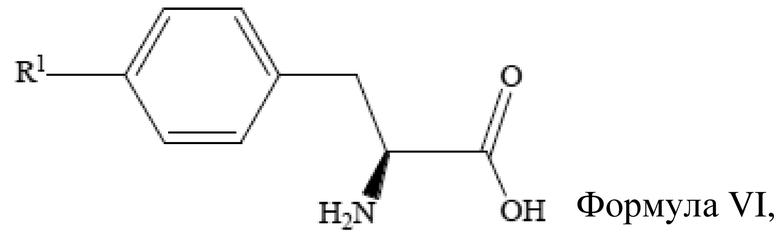
введения защитной группы для аминогруппы (S)-4-галогенфенилаланина, который имеет структуру, представленную формулой VI выше, с получением (S)-4-галогенфенилаланина с защитной группой при аминогруппе.
[30] Введение защитной группы для аминогруппы (S)-4-галогенфенилаланина с получением (S)-4-галогенфенилаланина с защитной группой при аминогруппе также включает стадии
добавления в реакционный сосуд (S)-4-галогенфенилаланина, 1,4-диоксана, воды, гидроксида натрия и ди-трет-бутилбикарбоната с проведением реакции;
регулирования pH до ≤2, предпочтительно до pH = 2, с кристаллизацией (S)-4-галогенфенилаланина с защитной группой при аминогруппе;
выделения (S)-4-галогенфенилаланина с защитной группой при аминогруппе с помощью первого экстрагента.
[31] Получение неочищенной борной кислоты включает стадию проведение реакции борной кислоты, серной кислоты и н-бутанола в первом органическом растворителе с получением триалкилбората.
[32] Предпочтительно, в (S)-4-галогенфенилаланине формулы I с защитной группой при аминогруппе R1 представляет собой йодид или бромид; и предпочтительно, в (S)-4-галогенфенилаланине формулы I с защитной группой при аминогруппе R1 представляет собой йодид.
[33] Предпочтительно, в (S)-4-галогенфенилаланине формулы I с защитной группой при аминогруппе и в (S)-4-бороно-L-фенилаланине формулы II с защитной группой при аминогруппе защитная группа R2 выбрана из группы, которая включает: трет-бутоксикарбонил (t-Boc или Boc), тритил (Trt), 3,5-диметоксифенилизопропоксикарбонил (Ddz), 2-(4-бифенил)изопропоксикарбонил (Bpoc) и 2-нитрофенилсульфенил (Nps); и предпочтительно, в (S)-4-галогенфенилаланине формулы I с защитной группой при аминогруппе и в (S)-4-бороно-L-фенилаланине формулы II с защитной группой при аминогруппе защитная группа представляет собой трет-бутоксикарбонил.
[34] В соответствии с настоящим изобретением, термин «реактив Гриньяра» означает любой реагент, способный, после стадии проведения реакции (S)-4-галогенфенилаланина с защитной группой при аминогруппе с борирующим средством и реактивом Гриньяра (с добавлением бис-(2-метиламиноэтилового) эфира или без него), заменять заместитель R1 (т.е. R1, изображенный на формуле I) (S)-4-галогенфенилаланина с защитной группой при аминогруппе на борсодержащий заместитель (такой как без ограничения бороно). Предпочтительно, реактив Гриньяра представляет собой алкилхлорид магния, алкилбромид магния, арилхлорид магния или арилбромид магния. Более предпочтительно, реактив Гриньяра представляет собой трет-бутилхлорид магния, циклогексилхлорид магния или трет-пентилхлорид магния.
[35] В соответствии с настоящим изобретением, применение трет-бутилхлорида магния, циклогексилхлорида магния или трет-пентилхлорида магния в способе получения в качестве реактива Гриньяра обладает следующими преимуществами:
(1) реакция проходит при комнатной температуре (0-30°C, температуру также можно немного понижать, например, до -10°C, или температуру можно немного повышать, например, до 60°C) такие условия реакции легко обеспечить, и они являются экономически эффективными;
(2) его применяют непосредственно в реакции, в результате упрощается получение и повышается выход.
[36] В соответствии с настоящим изобретением, применение трет-бутоксикарбонила в качестве защитной группы обладает следующими преимуществами:
(1) (S)-N-трет-бутоксикарбонил-4-галогенфенилаланин представляет собой твердое вещество, что облегчает проведение способа;
(2) исходным материалом для получения (S)-N-трет-бутоксикарбонил-4-галогенфенилаланина является ди-трет-бутилдикарбонат (Boc2O), его преимуществами являются доступность и низкая цена, соответственно, преимуществами полученного (S)-N-трет-бутоксикарбонил-4-галогенфенилаланина также являются доступность и низкая цена; и
(3) после удаления защитной группы трет-бутоксикарбонила с (S)-N-трет-бутоксикарбонил-4-боронофенилаланина трет-бутоксикарбонил распадается на CO2 и трет-бутанол, которые представляют собой химические вещества с низкой токсичностью, соответственно, преимуществами получения L-BPA из (S)-N-трет-бутоксикарбонил-4-галогенфенилаланина являются безопасность и низкая токсичность.
[37] Предпочтительным является проведение реакции (S)-4-галогенфенилаланина с защитной группой при аминогруппе, борирующего средства, реактива Гриньяра и бис-(2-метиламиноэтилового) эфира при температуре 0°C-60°C с получением реакционной смеси.
[38] Таким образом, в соответствии с настоящим изобретением реакцию можно осуществлять при температуре выше 0°C, в результате исключается необходимость создания среды с низкой температурой, оборудование и работники не подвергаются неблагоприятным условиям реакции, что является экономически эффективным.
[39] Предпочтительно, разделение реакционной смеси с получением (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе включает стадии
добавления в реакционную смесь второго органического растворителя и раствора кислоты, регулирования pH до ≤5, предпочтительно до pH = 3, и экстрагирования органической фазы;
добавления в органическую фазу раствора основания, регулирования pH в диапазоне 7,1-14, предпочтительно, в pH диапазоне 12,1-12,6;
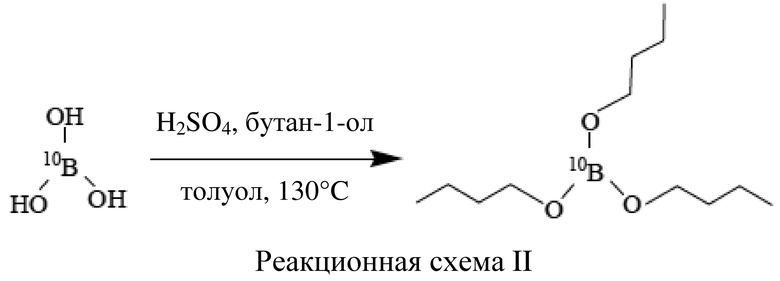
выделения (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе с помощью второго экстрагента.
[40] Таким образом, в соответствии с настоящим изобретением не требуются сложные стадии очистки, что означает простой способ получения и разделение реакционной смеси с получением (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе. Соответственно, в способе получения в соответствии с настоящим изобретением можно не производить в больших количествах отходы растворителей и силикагеля, что также является преимуществом в отношении безопасности для окружающей среды.
[41] Предпочтительным является удаление защитной группы с аминогруппы (S)-4-бороно-L-фенилаланина при температуре 30°C-60°C с получением 4-бороно-L-фенилаланина.
[42] Таким образом, в соответствии с настоящим изобретением не требуются сложные стадии очистки, стадию удаления защитной группы с (S)-4-бороно-L-фенилаланин с защитной группой при аминогруппе можно провести простым способом, что обеспечивает L-BPA с высокой химической чистотой и высокой оптической чистотой. Соответственно, в способе получения в соответствии с настоящим изобретением можно не производить в больших количествах отходы растворителей и силикагеля, что также является преимуществом в отношении безопасности для окружающей среды.
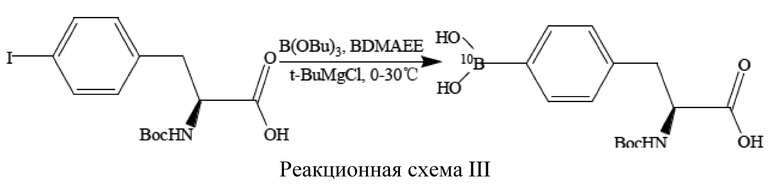
[43] Предпочтительно, указанное борирующее средство содержит ≥95% (10B)бора.
[44] Предпочтительно, (S)-4-бороно-L-фенилаланин с защитной группой при аминогруппе представляет собой (S)-4-(10B)бороно-L-фенилаланин с защитной группой при аминогруппе.
[45] Предпочтительно, (S)-4-бороно-L-фенилаланин с защитной группой при аминогруппе, который имеет структуру, представленную формулой II, представляет собой (S)-4-(10B)бороно-L-фенилаланин с защитной группой при аминогруппе.
[46] В соответствии с настоящим изобретением, борирующее средство предусматривает триалкил(10B)борат или любой другой реагент с ≥98% (10B)бора. Триалкил(10B)борат включает трибутил(10B)борат (10B(OBu)3), триметил(10B)борат (10B(OCH3)3) или любой другой указанный выше триалкилборат с 98% (10B)бора, однако специалисту в данной области техники будет понятно, что приведенные в данном документе виды борирующих средств не подразумеваются как ограничивающие. Предпочтительно, борирующее средство с ≥98% (10B)бора представляет собой коммерчески доступный реагент, такой как трибутил(10B)борат.
[47] Предпочтительно,
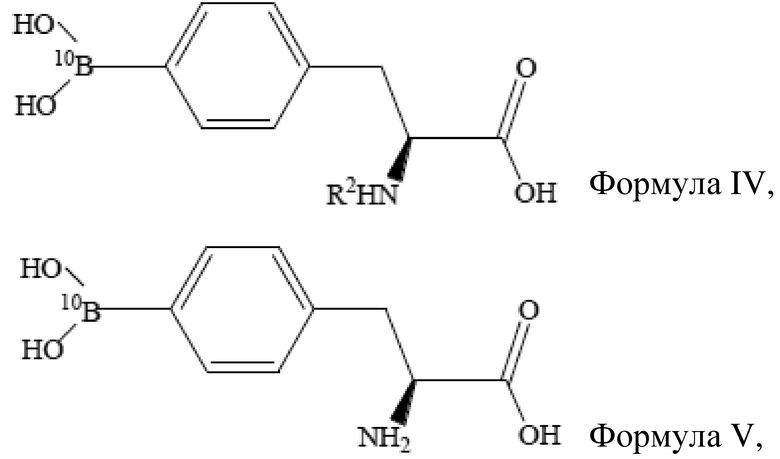
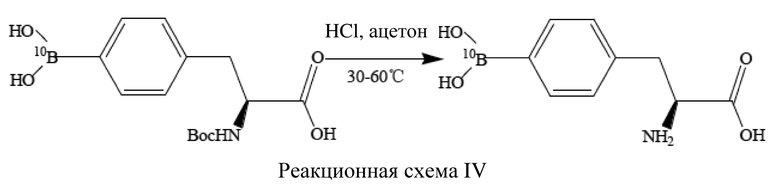
борирующее средство содержит ≥95% 10B, (S)-4-бороно-L-фенилаланин с защитной группой при аминогруппе представляет собой (S)-4-(10B)бороно-L-фенилаланин с защитной группой при аминогруппе, который имеет структуру, представленную формулой IV выше, L-BPA представляет собой L-10BPA, который имеет структуру, представленную формулой V выше.
[48] В способе получения в соответствии с настоящим изобретением также не требуется сложное предварительное получение борирующего средства для успешного получения L-10BPA. Кроме того, вследствие малой длительности и простоты способа получения в соответствии с настоящим изобретением, L-10BPA, полученный с помощью данного способа, обладает преимуществами высокой химической чистоты, высокой оптической чистоты, высокой изотопной чистоты и высокого общего выхода, при этом способ, как было указано выше, обладает такими преимуществами как отсутствие необходимости в сложных стадиях очистки, снижение длительности и стоимости, повышение эффективности и безопасность для окружающей среды.
[49] В соответствии с настоящим изобретением, растворители для реакции включают без ограничения эфирный растворитель или любой другой пригодный органический растворитель. Эфирный растворитель в соответствии с настоящим изобретением включает тетрагидрофуран, 2-метилтетрагидрофуран, диэтиловый эфир или любой другой пригодный эфирный растворитель.
[50] В соответствии с настоящим изобретением, органический растворитель относится к органическому веществу, в котором реагенты могут по меньшей мере частично растворяться, и относится к органическим растворителям, таким как без ограничения: алканы, эфирные растворители или любой другой пригодный органический растворитель, такой как толуол, ацетон. Указанные алканы включают: гексаны, гептан, циклогексан, пентан или любой другой пригодный алкан, однако специалисту в данной области техники будет понятно, что приведенные в данном документе виды органических растворителей не подразумеваются как ограничивающие. Эфирные растворители включают: метил-трет-бутиловый эфир (MTBE), тетрагидрофуран (THF), диэтиловый эфир, диэтоксиметан, дибутиловый эфир, 2-метилтетрагидрофуран (2-MeTHF) или любой другой пригодный органический растворитель. Предпочтительно, первый органический растворитель представляет собой толуол, второй органический растворитель представляет собой метил-трет-бутиловый эфир, третий органический растворитель представляет собой ацетон.
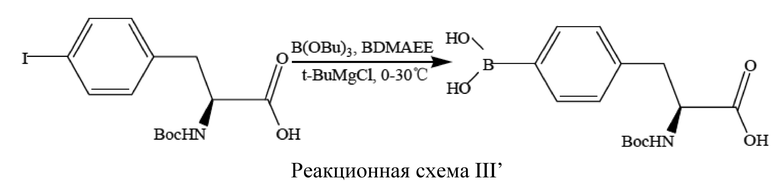
[51] В соответствии с настоящим изобретением, экстрагент относится к любому нерастворимому или частично растворимому в воде растворителю. Экстрагенты включают: изобутанол, толуол, н-бутанол, изопропилацетат, этилацетат или любой другой пригодный экстрагент, однако специалисту в данной области техники будет понятно, что приведенные в данном документе виды экстрагентов не подразумеваются как ограничивающие. предпочтительно, первый экстрагент представляет собой этилацетат, второй экстрагент представляет собой н-бутанол.
[52] В соответствии с настоящим изобретением, раствор кислоты включает раствор HCl или любой другой пригодный раствор кислоты, однако специалисту в данной области техники будет понятно, что приведенные в данном документе виды растворов кислот не подразумеваются как ограничивающие. Предпочтительно, раствор кислоты представляет собой 37% HCl.
[53] В соответствии с настоящим изобретением, раствор основания включает раствор NaOH или любой другой пригодный раствор основания, однако специалисту в данной области техники будет понятно, что приведенные в данном документе виды растворов оснований не подразумеваются как ограничивающие. Предпочтительно, раствор основания представляет собой водный раствор NaOH.
Подробное описание изобретения
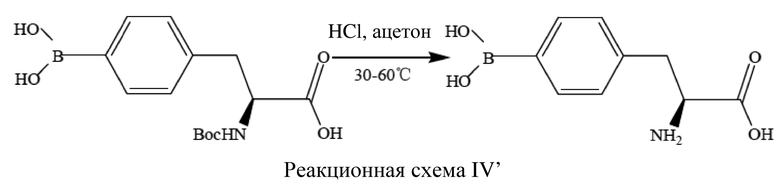
[54] Специалисту в данной области техники будет понятно, что способ получения L-BPA и способ получения L-10BPA практически не различаются, отличие состоит только в применении разных (10B)борсодержащих или (11B)борсодержащих борирующих средств в качестве исходного материала. Содержание бора в борирующем средстве, применяемом в качестве исходного материала для получения L-BPA, может представлять собой содержание бора, существующее в природе, т. е. приблизительно 19,9% (10B)бора и приблизительно 80,1% (11B)бора, также можно применять L-10BPA с ≥95% (10B)бора (т. е. L-BPA с высоким содержанием (10B)бора обычно называют L-10BPA). Соответственно, способ получения L-10BPA, раскрытый в варианте осуществления настоящего изобретения, также является применимым к способу получения L-BPA, например, в варианте осуществления 1 настоящего изобретения с помощью борирующего средства с ≥95% (10B)бора, бис-(2-метиламиноэтилового) эфира и трет-бутилхлорида магния в качестве реактива Гриньяра в качестве исходных материалов можно получать L-10BPA, другое содержание (10B)бора можно получить с помощью того же способа получения; в варианте осуществления 2 настоящего изобретения описана стадия получения (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина и (10B)борсодержащего борирующего средства, бис-(2-метиламиноэтилового) эфира и трет-бутилхлорида магния в качестве реактива Гриньяра в качестве исходных материалов, а также стадия удаления защитной группы (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина с получением L-BPA; в варианте осуществления 3 настоящего изобретения описана стадия получения (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина и (10B)борсодержащего борирующего средства, бис-(2-метиламиноэтилового) эфира и трет-бутилхлорида магния в качестве реактива Гриньяра в качестве исходных материалов; в варианте осуществления 4 настоящего изобретения описана стадия получения (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина и (10B)борсодержащего борирующего средства, бис-(2-метиламиноэтилового) эфира и циклогексилхлорида магния в качестве реактива Гриньяра в качестве исходных материалов; в варианте осуществления 5 настоящего изобретения описана стадия получения (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина и (10B)борсодержащего борирующего средства, бис-(2-метиламиноэтилового) эфира и трет-пентилхлорида магния в качестве реактива Гриньяра в качестве исходных материалов; в вариантах осуществления 6-7 настоящего изобретения описана стадия получения (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина и (10B)борсодержащего борирующего средства и трет-бутилхлорида магния в качестве реактива Гриньяра в качестве исходных материалов без добавления бис-(2-метиламиноэтилового) эфира.
[55] В вариантах осуществления 2-7 настоящего изобретения порядок стадий получения L-BPA в целом такой же, как и в варианте осуществления 1, за исключением того, что содержание (10B)бора отличается, поэтому далее описание данных стадий в данных вариантах осуществления повторяться не будет. В вариантах осуществления 3-7 настоящего изобретения стадия удаления защитной группы с аминогруппы при получении L-BPA в целом такая же, как и в варианте осуществления 2, поэтому далее описание данной стадии в данных вариантах осуществления повторяться не будет.
[56] В следующем варианте осуществления применение (S)-N-трет-бутоксикарбонил-4-йодфенилаланина в качестве предпочтительного варианта осуществления (S)-4-галогенфенилаланина с защитной группой при аминогруппе и применение (S)-N-трет-бутоксикарбонил-4-йодфенилаланина с получением L-BPA или L-10BPA, демонстрирует преодоление недостатков предшествующего уровня техники в настоящем изобретении. Специалисту в данной области техники после ознакомления с данным документом легко станут понятны преимущества и возможности настоящего изобретения, и что различные модификации и изменения настоящего изобретения для применения или осуществления на практике настоящего изобретения можно осуществлять без отклонения от его сути и объема, при этом подразумевается, что приведенные в данном документе варианты осуществления не предназначены для ограничения объема настоящего изобретения.
Вариант осуществления 1
[57] Перед получением (S)-N-трет-бутоксикарбонил-4-боронофенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина необходимо изложить способ получения (S)-N-трет-бутоксикарбонил-4-йодфенилаланина из (S)-4-йодфенилаланина в качестве исходного материала и способ получения трибутил(10B)бората из (10B)борной кислоты.
Стадия 1. Получение (S)-N-трет-бутоксикарбонил-4-йодфенилаланина из (S)-4-йодфенилаланина
[58] См. реакционную схему I ниже, где изображена реакция (S)-4-йодфенилаланина в растворе 1,4-диоксана и воды с гидроксидом натрия (NaOH) и ди-трет-бутилдикарбонатом (Boc2O) с получением (S)-N-трет-бутоксикарбонил-4-йодфенилаланина.
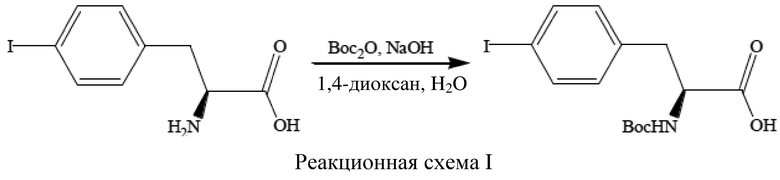
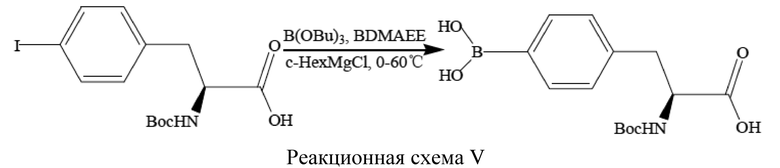
[59] В данном способе для проведения реакций использовали два реакционных сосуда.
[60] Способ получения включает следующие операции.
1. Реакцию проводили в трехгорлой колбе объемом 3 л.
2. Затем в реакционную систему добавляли (S)-4-йод-L-фенилаланин (200,00 г, 687,10 ммоль, 1,00 экв.).
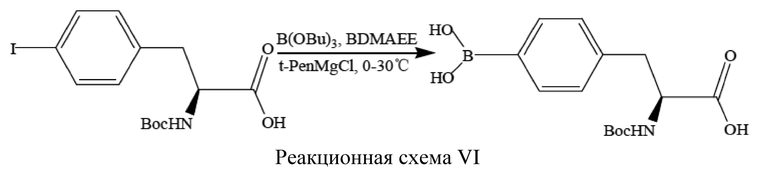
3. В реакционную систему добавляли 1,4-диоксан (1,00 л) и воду (1,00 л).
4. Затем в реакционную систему добавляли NaOH (68,71 г, 1,72 моль, 2,50 экв.); раствор медленно становился прозрачным, и температура повышалась до 19°C.
5. После охлаждения системы до 0-10°C в реакционную систему добавляли ди-трет-бутилдикарбонат (254,93 г, 1,17 моль, 268,35 мл, 1,70 экв.), после чего температура реакционной системы повышалась до 10-30°C, и систему перемешивали при комнатной температуре (примерно 30°C) в течение 8 часов.
6. С помощью высокоэффективной жидкостной хроматографии (HPLC) измеряли степень превращения исходного материала до завершения реакции.
7. Температуру системы контролировали ниже 40°C, концентрировали 1,4-диоксан в реакционном растворе.
8. Затем охлаждали реакционную систему до комнатной температуры (примерно 25°C), добавляли 100 мл воды и регулировали pH до 1,8-2 с помощью 2 М HCl.
9. Экстрагировали 3 раза этилацетатом (2 л).
10. Затем органические фазы объединяли и 2 раза промывали насыщенным солевым раствором (1 л).
11. Затем органическую фазу высушивали над Na2SO4 (200 г) с получением неочищенного светло-желтого твердого вещества.
12. Высушивание продолжали в сушильном шкафу (40-45°C) с получением (S)-N-трет-бутоксикарбонил-4-йод-L-фенилаланина в виде белого твердого вещества (250,00 г, 626,28 ммоль, анализ с помощью HPLC, выход 93,00%,чистота 98%).
[61] Результаты анализа (S)-N-трет-бутоксикарбонил-4-йод-L-фенилаланина, полученные с помощью 1H-ЯМР, представлены ниже.
[62] 1H-ЯМР: (400 МГц DMSO-d6)
δ7,49 (d, J = 7,8 Гц, 2H), 6,88 (d, J = 7,8 Гц, 2H), 5,80 (d, J = 5,9 Гц, 1H), 3,68 (d, J = 5,5 Гц, 1H), 3,00-2,90 (m, 1H), 2,87-2,75 (m, 1H), 1,35-1,15 (m, 9H).
Стадия 2. Получение трибутил(10B)бората из (10B)борной кислоты
[63] См. реакционную схему II ниже, где изображена реакция (10B)борной кислоты в растворе н-бутанола и толуола с серной кислотой (H2SO4) с получением трибутил(10B)бората (10B(OBu)3).
[64] Способ получения включает следующие операции.
1. Устанавливали реактор R1, который представлял собой трехгорлую колбу объемом 3 л, оборудованную сверху устройством для отвода воды.
2. Затем при комнатной температуре (примерно 25°C) добавляли (10B)борную кислоту (150,00 г, 2,46 моль, 1,00 экв.) в реакционную смесь R1.
3. Затем при комнатной температуре (примерно 25°C)добавляли н-бутанол (1,00 л) в реакционную смесь R1 и перемешивали, большая часть борной кислоты не растворилась.
4. Затем при комнатной температуре (примерно 25°C) добавляли толуол (1,00 л) в реакционную смесь R1 и перемешивали.
5. Затем при комнатной температуре (примерно 25°C) добавляли по каплям концентрированную H2SO4 (4,82 г, 49,16 ммоль, 2,62 мл, 0,02 экв.) в реакционную смесь, при этом значительная часть твердых веществ оставалась нерастворенной.
6. Затем реакционную систему нагревали до 130°C, перемешивали 3,5 часа с постоянным отводом воды, удаляли воду (приблизительно 140 г) из устройства для отвода воды, твердое вещество полностью растворялось, и раствор менял цвет с бесцветного на коричневый.
7. Завершение реакции определяли с помощью TLC (DCM:MeOH = 5:1, Rf = 0,43, краситель бромкрезоловый зеленый).
8. Выпаривали большую часть толуола при атмосферном давлении.
9. После выпаривания большей части толуола температуру системы понижали до 20-30°C, затем два реакционных раствора объединяли и меняли устройство для выпаривания.
10. На масляной бане с внешней температурой 108-110°C с помощью водяного насоса при пониженном давлении выпаривали н-бутанол при температуре 45°C, измеренной термометром Кельвина.
11. На масляной бане с внешней температурой 108-110°C с помощью масляного насоса при пониженном давлении выпаривали остаточный бутанол.
12. На масляной бане с внешней температурой 118-120°C с помощью масляного насоса при пониженном давлении начинали выделение продукта при температуре 55°C, измеренной термометром Кельвина.
13. Затем температуру повышали до 135-140°C и с помощью масляного насоса при пониженном давлении получали конечный продукт.
14. Полученный продукт представлял собой трибутил(10B)борат в виде бесцветной жидкости (830,00 г, 3,62 моль, выход 73,58%).
[65] Результаты анализа трибутил(10B)бората, полученные с помощью 1H-ЯМР, представлены ниже.
[66] 1H-ЯМР: (400 МГц CDCl3)
δ3,82-3,68 (m, 6H), 1,57-1,42 (m, 6H), 1,34 (qd, J = 7,4, 14,9 Гц, 6H), 0,95-0,80 (m, 9H).
Стадия 3. Получение (S)-N-трет-бутоксикарбонил-4-(10B)бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина
[67] См. реакционную схему III ниже, где изображена реакция (S)-N-трет-бутоксикарбонил-4-йодфенилаланина с трибутил(10B)боратом, трет-бутилхлоридом магния (t-BuMgCl) и бис-(2-метиламиноэтиловым) эфиром (BDMAEE) с получением (S)-N-трет-бутоксикарбонил-4-(10B)бороно-L-фенилаланина.
[68] В данном способе для проведения реакций использовали два реакционных сосуда.
[69] Способ получения включает следующие операции.
1. Реакцию проводили в трехгорлой колбе объемом 3 л.
2. Затем при комнатной температуре (примерно 22°C) вносили трибутил(10B)борат (187,60 г, 817,98 ммоль, 3,20 экв.) в реакционную систему.
3. Затем при комнатной температуре (примерно 22°C) добавляли одной порцией гидрид натрия (20,45 г, 511,24 ммоль, чистота 60%, 2,00 экв.) в реакционную систему, при этом реакционный раствор образовывал суспензию, которую перемешивали при комнатной температуре (примерно 22°C) в течение 5 минут.
4. Затем при комнатной температуре (примерно 22°C) добавляли бис-(2-метиламиноэтиловый) эфир (327,73 г, 2,04 моль, 8,00 экв.) в реакционную смесь.
5. Затем при комнатной температуре (примерно 22°C) добавляли N-трет-бутоксикарбонил-4-йод-L-фенилаланин (100,00 г, 255,62 ммоль, 1,00 экв.) в реакционную систему, значительная часть твердых веществ не растворялась.
6. Понижали температуру реакционной системы до 0-5°C, затем по каплям в течение приблизительно 1,5 минут добавляли трет-бутилхлорид магния (1,7 М, 1,20 л, 2,04 моль, 8,00 экв.) в реакционную смесь, контролировали температуру в диапазоне 0-10°C.
7. После завершения добавления температура реакционной системы повышалась до комнатной температуры (20-30°C), при которой систему перемешивали в течение 12 часов.
8. С помощью высокоэффективной жидкостной хроматографии (HPLC) определяли количество остаточного исходного материала, которое составляло приблизительно 9,00%.
9. Понижали температуру реакционной системы до -5-0°C и систему гасили путем добавления по каплям 500 мл воды.
10. Температуру системы понижали до 0-5°C, в реакционную систему добавляли метил-трет-бутиловый эфир (500 мл) и регулировали pH до 2,9-3,1 (с помощью pH-метра) с помощью 37% HCl (приблизительно 500 мл), регулирование pH сопровождалось выделением тепла, контролировали температуру системы в диапазоне 0-15°C.
11. Водную фазу отделяли и 1 раз экстрагировали метил-трет-бутиловым эфиром (500 мл); полученные органические фазы объединяли с получением приблизительно 1,1 л органической фазы.
12. В полученную органическую фазу медленно добавляли водный раствор NaOH (1 М, 400 мл), добавление сопровождалось выделением тепла, контролировали температуру системы в диапазоне 0-15°C.
13. После завершения добавления pH системы составлял примерно 10, регулировали pH в диапазоне 12,10-12,6 с помощью водного раствора NaOH (4 М) (определяли с помощью pH-метра).
14. Слои разделяли.
15. Затем водную фазу 1 отделяли и 1 раз экстрагировали н-бутанолом (500 мл) с получением водной фазы 2.
16. Затем объединяли водную фазу 2 из двух реакционных сосудов.
17. Регулировали pH водной фазы до 2,9-3,1 с помощью 37% HCl, перемешивали приблизительно 40 минут с образованием большого количества твердого осадка.
18. Фильтровали с промыванием дихлорметаном (50 мл) с получением белого твердого вещества.
19. Затем при 25°C выделенное твердое вещество суспендировали в дихлорметане (150 мл) и перемешивали 10 минут.
20. После фильтрования получали (S)-N-трет-бутоксикарбонил-4-(10B)бороно-L-фенилаланин в виде белого твердого вещества (75,00 г, 240,82 ммоль, анализ с помощью HPLC, выход 47,11%, чистота 99%).
[70] Результаты анализа (S)-N-трет-бутоксикарбонил-4-(10B)бороно-L-фенилаланина, полученные с помощью 1H-ЯМР, представлены ниже.
[71] 1H-ЯМР: (400 МГц DMSO-d6)
δ12,55 (br. s., 1H), 7,91 (s, 2H), 7,66 (d, J = 7,5 Гц, 2H), 7,17 (d, J = 7,5 Гц, 2H), 4,08-4,01 (m, 1H), 3,61-3,53 (m, 1H), 2,98 (dd, J = 4,2, 13,9 Гц, 1H), 2,79 (dd, J = 10,4, 13,5 Гц, 1H), 1,79-1,67 (m, 1H), 1,35-1,17 (m, 9H).
Стадия 4. Получение L-10BPA из (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина
[72] См. реакционную схему IV ниже, где изображена реакция удаления защитной группы с аминогруппы (S)-N-трет-бутоксикарбонил-4-(10B)бороно-L-фенилаланина с получением L-10BPA.
[73] Способ получения включает следующие операции.
1. Реакцию проводили в трехгорлой колбе объемом 1 л.
2. Затем при комнатной температуре (20-30°C) добавляли (S)-N-трет-бутоксикарбонил-4-(10B)бороно-L-фенилаланин (67,00 г, 217,31 ммоль, 1,00 экв.) в реакционную систему.
3. Затем при комнатной температуре (20-30°C) по отдельности, по каплям добавляли воду (23,75 мл) и ацетон (420,00 мл) в реакционный сосуд, перемешивали с (S)-N-трет-бутоксикарбонил-4-(10B)бороно-L-фенилаланином.
4. Затем при комнатной температуре (20-30°C) по каплям добавляли концентрированную HCl (23,93 г, 656,28 ммоль, 23,46 мл, 3,02 экв.) в реакционную систему, после завершения добавления температуру реакционной системы повышали до 55-60°C и перемешивали 4,5 часа.
5. С помощью HPLC обнаруживали завершение реакции исходного материала.
6. Контролировали температуру ниже 40°C, концентрировали ацетон в реакционной системе.
7. После концентрирования температуру системы понижали до ниже 15°C, регулировали pH системы до примерно 1,5 (измеряли с помощью pH-метра) с помощью раствора NaOH (4 М), после перемешивания в течение 40 минут дополнительно регулировали pH системы до 6,15-6,25 с помощью раствора NaOH (4 М) с образованием большого количества осадка в виде белого твердого вещества, фильтровали с промыванием ацетоном (200 мл) с получением белого твердого вещества.
8. Получали L-10BPA в виде белого твердого вещества (36,00 г, 171,17 ммоль, анализ с помощью HPLC, выход 78,77%, чистота 99%).
[74] Результаты анализа L-10BPA, полученные с помощью 1H-ЯМР, представлены ниже.
[75] 1H-ЯМР: (400 МГц D2O, CF3COOH)
δ7,44 (d, J = 7,9 Гц, 1H), 7,03 (d, J = 7,9 Гц, 1H), 4,06 (dd, J = 5,7, 7,5 Гц, 1H), 3,11-3,01 (m, 1H), 2,98-2,87 (m, 1H).
Вариант осуществления 2
Стадия 1. Получение (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина
[76] См. реакционную схему III’, где изображена реакция (S)-N-трет-бутоксикарбонил-4-йодфенилаланина с трибутилборатом, трет-бутилхлоридом магния (t-BuMgCl) и бис-(2-метиламиноэтиловым) эфиром (BDMAEE) с получением (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина.
[77] Способ получения включает следующие операции.
1. Устанавливали реактор, который представлял собой трехгорлую колбу объемом 100 мл.
2. Затем при температуре 20-30°C вносили трибутилборат (5,65 г, 24,54 ммоль, 3,20 экв.) в реакционный сосуд объемом 100 мл.
3. Затем при температуре 20-30°C добавляли гидрид натрия (613,50 мг, 15,34 ммоль, 2,00 экв.) в реакционный сосуд объемом 100 мл.
4. Затем при температуре 20-30°C добавляли бис-(2-метиламиноэтиловый) эфир (9,83 г, 61,35 моль, 8,00 экв.) в реакционный сосуд объемом 100 мл.
5. Затем при температуре 20-30°C добавляли (S)-N-трет-бутоксикарбонил-4-йодфенилаланин (3,00 г, 7,67 ммоль, 1,00 экв.) в реакционный сосуд объемом 100 мл.
6. Затем в атмосфере азота температуру реакционной системы понижали до 0°C, добавляли по каплям в течение приблизительно 30 минут трет-бутилхлорид магния (1,7 М в тетрагидрофуране, 36 мл, 8,00 экв.) в реакционную смесь, контролировали температуру в диапазоне 0°C-10°C.
7. Полученное перемешивали при 20-30°C в течение 16 часов.
8. С помощью HPLC обнаруживали, что реакция не завершилась, количество остаточного исходного материала составляло приблизительно 13%.
9. При температуре 0°C реакционную смесь гасили путем добавления по каплям 1,5 мл воды, после окончания гашения перемешивание продолжали в течение 10 минут.
10. Температуру понижали до 0°C, в реакционную смесь добавляли метил-трет-бутиловый эфир (15 мл) и регулировали pH до 3 (определяли с помощью pH-метра) с помощью 37% HCl (приблизительно 15 мл), регулирование pH сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
11. Водную фазу отделяли и 1 раз экстрагировали метил-трет-бутиловым эфиром (15 мл), две органические фазы объединяли.
12. Регулировали pH полученной органической фазы в диапазоне 12,10-12,6 путем добавления по каплям раствора NaOH (1 М, 17 мл), это сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
13. Водную фазу отделяли и 1 раз экстрагировали н-бутанолом (15 мл) с удалением большей части примесей.
14. Водную фазу отделяли и регулировали pH до 3 с помощью 37% HCl, перемешивали приблизительно 30 минут с образованием большого количества осадка в виде белого твердого вещества.
15. Фильтровали с промыванием дихлорметаном (15 мл) с получением белого твердого вещества.
16. Затем при 25°C выделенное твердое вещество суспендировали в 8 мл дихлорметана и перемешивали 10 минут.
17. После фильтрования получали (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланин в виде белого твердого вещества (1,80 г, 5,82 ммоль, анализ с помощью HPLC, выход 75,92%, чистота 100%).
[78] Результаты анализа (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина, полученные с помощью 1H-ЯМР, представлены ниже.
[79] 1H-ЯМР: (400 МГц DMSO-d6)
δ = 7,95 (s, 2H), 7,69 (d, J = 7,7 Гц, 2H), 7,19 (d, J = 7,7 Гц, 2H), 7,07 (d, J = 8,4 Гц, 1H), 4,14-4,04 (m, 1H), 3,00 (br dd, J = 4,4, 13,7 Гц, 1H), 2,82 (br dd, J = 10,5, 13,8 Гц, 1H), 1,32 (s, 9H).
Стадия 2. Получение L-BPA из (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина
[80] См. реакционную схему IV’ ниже, где изображена реакция удаления защитной группы с аминогруппы (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина с получением L-BPA.
[81] Способ получения включает следующие операции.
1. Устанавливали реактор, который представлял собой трехгорлую колбу объемом 100 мл.
2. Затем при температуре 20-30°C добавляли (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланин (1,80 г, 5,82 ммоль, 1,00 экв.) в трехгорлую колбу объемом 100 мл.
3. Затем при температуре 20-30°C по отдельности добавляли воду (0,63 мл) и ацетон (11,30 мл) в реакционный сосуд.
4. Затем при температуре 20-30°C по каплям добавляли HCl (17,46 ммоль, 1,46 мл, 3,00 экв.) в реакционную смесь, после завершения добавления температуру повышали до 60°C и перемешивали 4 часа.
5. С помощью HPLC обнаруживали завершение реакции.
6. При 40°C реакционный раствор концентрировали при пониженном давлении, большую часть ацетона выпаривали.
7. Температуру понижали до 0-15°C, регулировали pH до 1,5 с помощью раствора NaOH (4 М), появлялся твердый осадок.
8. Дополнительно регулировали pH до 6,2 с образованием большого количества осадка в виде белого твердого вещества, перемешивали 15 минут.
9. Фильтровали с промыванием ацетоном (6 мл) с получением белого твердого вещества, затем твердое вещество высушивали на роторном испарителе.
10. Получали L-BPA в виде твердого вещества белого цвета (0,85 г, 4,07 ммоль, анализ с помощью HPLC, выход 70,18%, чистота 98%).
[82] Результаты анализа L-BPA, полученные с помощью 1H-ЯМР, представлены ниже.
[83] 1H NMR: (400 МГц D2O)
δ = 7,62 (d, J = 7,5 Гц, 2H), 7,22 (d, J = 7,9 Гц, 2H), 3,86 (dd, J = 5,5, 7,5 Гц, 1H), 3,20-3,13 (m, 1H), 3,05-2,97 (m, 1H).
Вариант осуществления 3
Получение (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина
[84] В соответствии с реакционной схемой III’ выше, способ получения включает следующие операции.
1. Устанавливали реактор, который представлял собой трехгорлую колбу объемом 1 л.
2. Затем при 20-30°C вносили трибутилборат (56,48 г, 245,40 ммоль, 3,20 экв.) в реакционный сосуд объемом 1 л.
3. Затем при 20-30°C добавляли гидрид натрия (6,13 г, 153,37 ммоль, 2,00 экв.) в реакционный сосуд объемом 1 л.
4. Затем при 20-30°C добавляли бис-(2-метиламиноэтиловый) эфир (98,32 г, 613,50 моль, 8,00 экв.) в реакционный сосуд объемом 1 л.
5. Затем при 20-30°C добавляли (S)-N-трет-бутоксикарбонил-4-йодфенилаланин (30,00 г, 76,69 ммоль, 1,00 экв.) в реакционный сосуд объемом 1 л.
6. Затем в атмосфере азота температуру реакционной системы понижали до 0°C, добавляли по каплям в течение приблизительно 1 часа трет-бутилхлорид магния (1,7 М в тетрагидрофуране, 360 мл, 8,00 экв.) в реакционную смесь, контролировали температуру в диапазоне 0°C-10°C.
7. Полученное перемешивали при 20-30°C в течение 16 часов.
8. С помощью HPLC обнаруживали, что реакция не завершилась, количество остаточного исходного материала составляло приблизительно 8%.
9. При температуре 0°C реакционную смесь гасили путем добавления по каплям 15 мл воды, после окончания гашения перемешивание продолжали в течение 10 минут.
10. Температуру понижали до 0°C, в реакционную смесь добавляли метил-трет-бутиловый эфир (150 мл) и регулировали pH до 3 (определяли с помощью pH-метра) с помощью 37% HCl (приблизительно 160 мл), регулирование pH сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
11. Водную фазу отделяли и 1 раз экстрагировали метил-трет-бутиловым эфиром (150 мл), две органические фазы объединяли.
12. Регулировали pH полученной органической фазы в диапазоне 12,10-12,6 путем добавления по каплям раствора NaOH (1 М, 190 мл), это сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
13. Водную фазу отделяли и 1 раз экстрагировали н-бутанолом (150 мл) с удалением большей части примесей.
14. Водную фазу отделяли и регулировали pH до 3 с помощью 37% HCl, перемешивали 30 минут с образованием большого количества осадка в виде белого твердого вещества.
15. Фильтровали с промыванием дихлорметаном (150 мл) с получением белого твердого вещества.
16. Затем при 25°C выделенное твердое вещество суспендировали в 80 мл дихлорметана и перемешивали 10 минут.
17. После фильтрования получали (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланин в виде белого твердого вещества (15,00 г, 48,52 ммоль, анализ с помощью HPLC, выход 63,27%, чистота 100%).
[85] Результаты анализа (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина, полученные с помощью 1H-ЯМР, представлены ниже.
[86] 1H-ЯМР: (400 МГц DMSO-d6)
δ = 7,97 (s, 2H), 7,68 (d, J = 7,9 Гц, 2H), 7,23-7,16 (m, 2H), 7,08 (d, J = 8,4 Гц, 1H), 4,12-4,04 (m, 1H), 3,00 (dd, J = 4,4, 13,7 Гц, 1H), 2,81 (dd, J = 10,4, 13,7 Гц, 1H), 1,34-1,23 (m, 9H).
Вариант осуществления 4
Получение (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина
[87] См. реакционную схему V ниже, где изображена реакция (S)-N-трет-бутоксикарбонил-4-йодфенилаланина с трибутилборатом, циклогексилхлоридом магния (c-HexMgCl) и бис-(2-метиламиноэтиловым) эфиром (BDMAEE) с получением (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина.
[88] Способ получения включает следующие операции.
1. Устанавливали реактор, который представлял собой трехгорлую колбу объемом 100 мл.
2. Затем при 20-30°C вносили трибутилборат (2,82 г, 12,27 ммоль, 3,20 экв.) в реакционный сосуд объемом 100 мл.
3. Затем при 20-30°C добавляли бис-(2-метиламиноэтиловый) эфир (4,92 г, 30,67 моль, 8,00 экв.) в реакционный сосуд объемом 100 мл.
4. Затем при 20-30°C добавляли (S)-N-трет-бутоксикарбонил-4-йод-L-фенилаланин (1,50 г, 3,83 ммоль, 1,00 экв.) в реакционный сосуд объемом 100 мл.
5. Затем в атмосфере азота температуру реакционной системы понижали до 0°C, добавляли по каплям в течение приблизительно 20 минут циклогексилхлорид магния (2 М в диэтиловом эфире, 15 мл, 8,00 экв.) в реакционную смесь, контролировали температуру в диапазоне 0°C-10°C.
6. Полученное перемешивали при 20-30°C в течение 24 часов.
7. Температуру повышали до 60 ± 5°C.
8. Полученное перемешивали при 60 ± 5°C в течение 24 часов.
9. С помощью HPLC обнаруживали, что реакция не завершилась, количество остаточного исходного материала составляло приблизительно 83%.
10. При температуре 0°C реакционную смесь гасили путем добавления по каплям 0,75 мл воды, после окончания гашения перемешивание продолжали в течение 10 минут.
11. Температуру понижали до 0°C, в реакционную смесь добавляли метил-трет-бутиловый эфир (7,5 мл) и регулировали pH до 3 (определяли с помощью pH-метра) с помощью 37% HCl (приблизительно 7,5 мл), регулирование pH сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
12. Водную фазу отделяли и 1 раз экстрагировали метил-трет-бутиловым эфиром (7,5 мл), две органические фазы объединяли.
13. Регулировали pH полученной органической фазы в диапазоне 12,10-12,6 путем добавления по каплям раствора NaOH (1 М, 8,5 мл), это сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
14. Водную фазу отделяли и 1 раз экстрагировали н-бутанолом (7,5 мл) с удалением большей части примесей.
15. Водную фазу отделяли и регулировали pH до 3 с помощью 37% HCl, перемешивали приблизительно 30 минут с образованием осадка в виде белого твердого вещества.
16. Фильтровали с промыванием дихлорметаном (7,5 мл) с получением белого твердого вещества.
17. Затем при 25°C выделенное твердое вещество суспендировали в 4 мл дихлорметана и перемешивали 10 минут.
18. После фильтрования получали (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланин в виде белого твердого вещества (0,2 г, анализ с помощью HPLC, выход 14,83%, чистота 99%).
[89] Результаты анализа (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина, полученные с помощью 1H-ЯМР, представлены ниже.
[90] 1H-ЯМР: (400 МГц DMSO-d6)
δ = 7,96 (s, 2H), 7,67 (d, J = 7,9 Гц, 2H), 7,21 (d, J = 7,7 Гц, 2H), 7,09 (d, J = 8,4 Гц, 1H), 4,12-4,04 (m, 1H), 3,00 (br dd, J = 4,4, 13,7 Гц, 1H), 2,83 (dd, J = 10,4, 13,7 Гц, 1H), 1,33-1,21 (m, 9H).
Вариант осуществления 5
Получение (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина
[91] См. реакционную схему VI ниже, где изображена реакция (S)-N-трет-бутоксикарбонил-4-йодфенилаланина с трибутилборатом, трет-пентилхлоридом магния (t-PenMgCl) и бис-(2-метиламиноэтиловым) эфиром (BDMAEE) с получением (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина.
[92] Способ получения включает следующие операции.
1. Устанавливали реактор, который представлял собой трехгорлую колбу объемом 100 мл.
2. Затем при 20-30°C вносили трибутилборат (2,82 г, 12,27 ммоль, 3,20 экв.) в реакционный сосуд объемом 100 мл.
3. Затем при 20-30°C добавляли бис-(2-метиламиноэтиловый) эфир (4,92 г, 30,67 моль, 8,00 экв.) в реакционный сосуд объемом 100 мл.
4. Затем при 20-30°C порциями добавляли (S)-N-трет-бутоксикарбонил-4-йод-L-фенилаланин (1,50 г, 3,83 ммоль, 1,00 экв.) в реакционный сосуд объемом 100 мл.
5. Затем в атмосфере азота температуру реакционной системы понижали до 0°C, добавляли по каплям в течение приблизительно 20 минут трет-пентилхлорид магния (1,0 М в 2-MeTHF, 31 мл, 8,00 экв.) в реакционную смесь, контролировали температуру в диапазоне 0°C-10°C.
6. Полученное перемешивали при 20-30°C в течение 24 часов.
7. Температуру повышали до 60 ± 5°C.
8. Полученное перемешивали при 60 ± 5°C в течение 24 часов.
9. С помощью HPLC обнаруживали, что реакция не завершилась, количество остаточного исходного материала составляло приблизительно 47%.
10. При температуре 0°C реакционную смесь гасили путем добавления по каплям 0,75 мл воды, после окончания гашения перемешивание продолжали в течение 10 минут.
11. Температуру понижали до 0°C, в реакционную смесь добавляли метил-трет-бутиловый эфир (7,5 мл) и регулировали pH до 3 (определяли с помощью pH-метра) с помощью 37% HCl (приблизительно 7,5 мл), регулирование pH сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
12. Водную фазу отделяли и 1 раз экстрагировали метил-трет-бутиловым эфиром (7,5 мл), две органические фазы объединяли.
13. Регулировали pH полученной органической фазы в диапазоне 12,10-12,6 путем добавления по каплям раствора NaOH (1 М, 8,5 мл), это сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
14. Водную фазу отделяли и 1 раз экстрагировали н-бутанолом (7,5 мл) с удалением большей части примесей.
15. Водную фазу отделяли и регулировали pH до 3 с помощью 37% HCl, перемешивали приблизительно 30 минут с образованием осадка в виде белого твердого вещества.
16. Фильтровали с промыванием дихлорметаном (7,5 мл) с получением белого твердого вещества.
17. Затем при 25°C выделенное твердое вещество суспендировали в 4 мл дихлорметана и перемешивали 10 минут.
18. После фильтрования получали (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланин в виде белого твердого вещества (0,5 г, анализ с помощью HPLC, выход 46,25%, чистота 98%).
[93] Результаты анализа (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина, полученные с помощью 1H-ЯМР, представлены ниже.
[94] 1H-ЯМР: (400 МГц DMSO-d6)
δ = 7,98 (s, 2H), 7,70 (d, J = 7,9 Гц, 2H), 7,23 (d, J = 7,7 Гц, 2H), 7,10 (d, J = 8,4 Гц, 1H), 4,13-4,04 (m, 1H), 3,00 (br dd, J = 4,4, 13,7 Гц, 1H), 2,84 (dd, J = 10,4, 13,7 Гц, 1H), 1,33-1,24 (m, 9H).
Вариант осуществления 6
Получение (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина из (S)-N-трет-бутоксикарбонил-4-йодфенилаланина
[95] См. реакционную схему VII ниже, где изображена реакция (S)-N-трет-бутоксикарбонил-4-йодфенилаланина с трибутилборатом и трет-бутилхлоридом магния (t-BuMgCl) с получением (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланина.
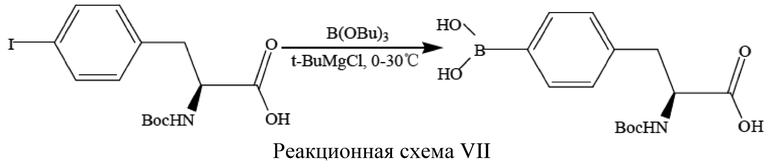
[96] Способ получения включает следующие операции.
1. Устанавливали реактор, который представлял собой трехгорлую колбу объемом 250 мл.
2. Затем при 20-30°C вносили трибутилборат (17,65 г, 76,68 ммоль, 3,00 экв.) в реакционный сосуд объемом 250 мл.
3. Затем при 20-30°C добавляли гидрид натрия (1,02 г, 25,56 ммоль, 1,00 экв.) в реакционный сосуд объемом 250 мл.
4. Затем при 20-30°C добавляли (S)-N-трет-бутоксикарбонил-4-йод-L-фенилаланин (10,00 г, 25,56 ммоль, 1,00 экв.) в реакционный сосуд объемом 250 мл.
5. Затем в атмосфере азота температуру реакционной системы понижали до 0°C, добавляли медленно, по каплям в течение приблизительно 30 минут трет-бутилхлорид магния (1,7 М в THF, 120 мл, 8,00 экв.) в реакционную смесь, контролировали температуру в диапазоне 0°C-10°C.
6. Полученное перемешивали при 20-30°C в течение 20 часов.
7. С помощью HPLC обнаруживали, что реакция почти полностью завершилась, количество остаточного исходного материала составляло приблизительно 0,7%.
8. При температуре 0°C реакционную смесь гасили путем добавления по каплям 5 мл воды, после окончания гашения перемешивание продолжали в течение 10 минут.
9. Температуру понижали до 0°C, в реакционную смесь добавляли метил-трет-бутиловый эфир (50 мл) и регулировали pH до 3 (определяли с помощью pH-метра) с помощью 37% HCl (приблизительно 50 мл), регулирование pH сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
10. Водную фазу отделяли и 1 раз экстрагировали метил-трет-бутиловым эфиром (50 мл), две органические фазы объединяли.
11. Регулировали pH полученной органической фазы в диапазоне 12,10-12,6 путем добавления по каплям раствора NaOH (1 М, 55 мл), это сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
12. Водную фазу отделяли и 1 раз экстрагировали н-бутанолом (50 мл) с удалением большей части примесей.
13. Водную фазу отделяли и регулировали pH до 3 с помощью 37% HCl, перемешивали приблизительно 30 минут с образованием осадка в виде белого твердого вещества.
14. Фильтровали с промыванием дихлорметаном (50 мл) с получением белого твердого вещества.
15. Затем при 25°C выделенное твердое вещество суспендировали в 25 мл дихлорметана и перемешивали 10 минут.
16. После фильтрования получали (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланин в виде белого твердого вещества (6,8 г, анализ с помощью HPLC, выход 83,15%, чистота 98%).
Вариант осуществления 7
[97] В соответствии с реакционной схемой VII выше, способ получения включает следующие операции.
1. Устанавливали реактор, который представлял собой трехгорлую колбу объемом 250 мл.
2. Затем при 20-30°C добавляли трибутилборат (8,82 г, 38,34 ммоль, 3,00 экв.) в реакционный сосуд объемом 250 мл.
3. Затем при 20-30°C добавляли гидрид натрия (511,25 мг, 12,78 ммоль, 1,00 экв.) в реакционный сосуд объемом 250 мл.
4. Затем при 20-30°C добавляли (S)-N-трет-бутоксикарбонил-4-йод-L-фенилаланин (5,00 г, 12,78 ммоль, 1,00 экв.) в реакционный сосуд объемом 250 мл.
5. Затем в атмосфере азота температуру реакционной системы понижали до 0°C, добавляли по каплям в течение приблизительно 30 минут трет-бутилхлорид магния (1,7 М в THF, 60 мл, 8,00 экв.) в реакционную смесь, контролировали температуру в диапазоне 0°C-10°C.
6. Полученное перемешивали при 22-30°C в течение 20 часов.
7. С помощью HPLC обнаруживали завершение реакции исходного материала.
8. При температуре 0°C реакционную смесь гасили путем добавления по каплям 2,5 мл воды, после окончания гашения перемешивание продолжали в течение 10 минут.
9. Температуру понижали до 0°C, в реакционную смесь добавляли метил-трет-бутиловый эфир (25 мл) и регулировали pH до 3 (определяли с помощью pH-метра) с помощью 37% HCl (приблизительно 25 мл), регулирование pH сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
10. Водную фазу отделяли и 1 раз экстрагировали метил-трет-бутиловым эфиром (25 мл), две органические фазы объединяли.
11. Регулировали pH полученной органической фазы в диапазоне 12,10-12,6 путем добавления по каплям раствора NaOH (1 М, 30 мл), это сопровождалось выделением тепла, контролировали температуру в диапазоне 0°C-15°C.
12. Водную фазу отделяли и 1 раз экстрагировали н-бутанолом (25 мл) с удалением большей части примесей.
13. Водную фазу отделяли и регулировали pH до 3 с помощью 37% HCl, перемешивали приблизительно 30 минут с образованием осадка в виде белого твердого вещества.
14. Фильтровали с промыванием дихлорметаном (25 мл) с получением белого твердого вещества.
15. Затем при 25°C выделенное твердое вещество суспендировали в 15 мл дихлорметана и перемешивали 10 минут.
16. После фильтрования получали (S)-N-трет-бутоксикарбонил-4-бороно-L-фенилаланин в виде белого твердого вещества (3,4 г, анализ с помощью HPLC, выход 85,26%, чистота 98%).
[98] Бис-(2-метиламиноэтиловый) эфир представляет собой Mg-комплексообразующее средство, которое уменьшает возникновение в реакции побочных реакций. В вариантах осуществления 6 и 7 реакцию можно успешно проводить без добавления бис-(2-метиламиноэтилового) эфира, в соответствии с анализом, в варианте осуществления 6 выделялось приблизительно 17% примесей йода, и в варианте осуществления 7 выделялось приблизительно 28% примесей йода. Таким образом, с другой стороны, добавление бис-(2-метиламиноэтилового) эфира в реакцию может предотвратить выделение йода.
[99] В соответствии с анализом BPA или 10BPA, полученных в вариантах осуществления выше, с помощью хиральной HPLC, соотношение L-энантиомера и D-энантиомера составляло 100:0.
[100] Описанное в настоящем изобретении применение L-BPA в качестве борсодержащего средства для нейтронозахватной терапии не ограничивается сведениями, приведенными в вариантах осуществления. Описанные выше варианты осуществления приведены исключительно для иллюстративных целей, и объем настоящего изобретения будет ограничен только прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ F-БФА И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2019 |
|
RU2776178C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ФТОР-4-БОРОНО-L-ФЕНИЛАЛАНИНА И ВЕЩЕСТВА-ПРЕДШЕСТВЕННИКА 2-ФТОР-4-БОРОНО-L-ФЕНИЛАЛАНИНА | 2014 |
|
RU2668166C1 |
| ХИРАЛЬНОЕ 4-БОРОНОФЕНИЛАЛАНИНОВОЕ (ВРА) ПРОИЗВОДНОЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ F-МЕЧЕННОГО ВРА С ИСПОЛЬЗОВАНИЕМ УКАЗАННОГО ПРОИЗВОДНОГО | 2013 |
|
RU2660433C2 |
| ПРОИЗВОДНОЕ ПАРА-БОРФЕНИЛАЛАНИНА И КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОВОЕ, И НАБОР ДЛЯ ПОЛУЧЕНИЯ УПОМЯНУТЫХ ПРОИЗВОДНОГО И КОМПОЗИЦИИ | 2019 |
|
RU2797343C2 |
| ИНЪЕКЦИОННЫЙ РАСТВОР, СОДЕРЖАЩИЙ П-БОРОНОФЕНИЛАЛАНИН | 2020 |
|
RU2832338C1 |
| ПРОИЗВОДСТВО СОЕДИНЕНИЙ И КОМПОЗИЦИЙ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ SHP2 | 2019 |
|
RU2797951C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ИНГИБИРОВАНИЯ ВИРУСНОЙ ПОЛИМЕРАЗЫ | 2013 |
|
RU2654482C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОАЛКИЛЗАМЕЩЕННОГО ГЛУТАРАМИДА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2108322C1 |
| НОВЫЙ ЭФФЕКТИВНЫЙ АМИНОГЛИКОЗИДНЫЙ АНТИБИОТИК ПРОТИВ БАКТЕРИЙ С МНОЖЕСТВЕННОЙ ЛЕКАРСТВЕННОЙ РЕЗИСТЕНТНОСТЬЮ | 2016 |
|
RU2751634C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА ДИПЕПТИДИЛПЕПТИДАЗЫ-IV И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2010 |
|
RU2499792C2 |
Изобретение относится к способу получения L-BPA. Способ включает следующие стадии: проведение реакции (S)-4-галогенфенилаланина формулы I с защитной группой при аминогруппе, борирующего средства, реактива Гриньяра и бис-(2-метиламиноэтилового) эфира с получением реакционной смеси, где реакционная смесь содержит (S)-4-бороно-L-фенилаланин формулы II с защитной группой при аминогруппе, при этом R1 представляет собой галоген, и в формуле I, и в формуле II R2 представляет собой защитную группу, и при этом реактив Гриньяра представляет собой трет-бутилхлорид магния;
разделение реакционной смеси с получением (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе; и удаление защитной группы с аминогруппы (S)-4-бороно-L-фенилаланина с получением L-BPA, который имеет структуру, представленную формулой III выше. Также предложен вариант способа получения L-BPA. Изобретение позволяет упростить способ и обеспечить получение L-BPA с высокой химической чистотой и высокой оптической чистотой. 2 н. и 9 з.п. ф-лы, 7 пр.
1. Способ получения L-BPA, который включает следующие стадии:
проведение реакции (S)-4-галогенфенилаланина формулы I с защитной группой при аминогруппе, борирующего средства, реактива Гриньяра и бис-(2-метиламиноэтилового) эфира с получением реакционной смеси, где реакционная смесь содержит (S)-4-бороно-L-фенилаланин формулы II с защитной группой при аминогруппе, при этом R1 представляет собой галоген, и в формуле I, и в формуле II R2 представляет собой защитную группу, и при этом реактив Гриньяра представляет собой трет-бутилхлорид магния;
разделение реакционной смеси с получением (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе;
удаление защитной группы с аминогруппы (S)-4-бороно-L-фенилаланина с получением L-BPA, который имеет структуру, представленную формулой III выше.
2. Способ получения L-BPA по п. 1, где проведение реакции (S)-4-галогенфенилаланина с защитной группой при аминогруппе, борирующего средства, реактива Гриньяра и бис-(2-метиламиноэтилового) эфира с получением реакционной смеси предусматривает
проведение реакции (S)-4-галогенфенилаланина с защитной группой при аминогруппе, борирующего средства, реактива Гриньяра и бис-(2-метиламиноэтилового) эфира при температуре 0°C-60°C с получением реакционной смеси.
3. Способ получения L-BPA по п. 1, где удаление защитной группы с аминогруппы (S)-4-бороно-L-фенилаланина с получением 4-бороно-L-фенилаланина предусматривает
удаление защитной группы с аминогруппы (S)-4-бороно-L-фенилаланина при температуре 30°C-60°C с получением 4-бороно-L-фенилаланина.
4. Способ получения L-BPA по п. 1, где
борирующее средство содержит ≥ 95% 10B, (S)-4-бороно-L-фенилаланин с защитной группой при аминогруппе представляет собой (S)-4-(10B)бороно-L-фенилаланин с защитной группой при аминогруппе, который имеет структуру, представленную формулой IV выше, L-BPA представляет собой L-10BPA, который имеет структуру, представленную формулой V выше.
5. Способ получения L-BPA по п. 1, где способ получения L-BPA включает стадию
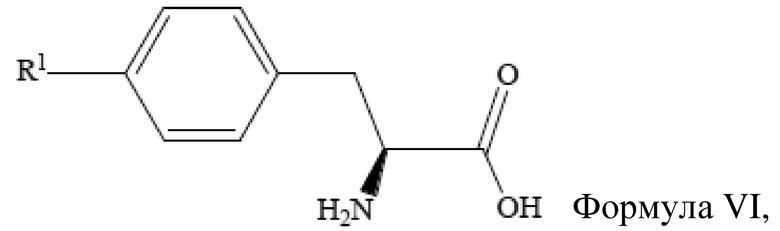
введения защитной группы для аминогруппы (S)-4-галогенфенилаланина, который имеет структуру, представленную формулой VI выше, с получением (S)-4-галогенфенилаланина с защитной группой при аминогруппе.
6. Способ получения L-BPA по п. 5, где введение защитной группы для аминогруппы (S)-4-галогенфенилаланина с получением (S)-4-галогенфенилаланина с защитной группой при аминогруппе включает
добавление в реакционный сосуд (S)-4-галогенфенилаланина, 1,4-диоксана, воды, гидроксида натрия и ди-трет-бутилбикарбоната с проведением реакции;
регулирование pH до ≤ 2 с кристаллизацией (S)-4-галогенфенилаланина с защитной группой при аминогруппе;
выделение (S)-4-галогенфенилаланина с защитной группой при аминогруппе с помощью первого экстрагента.
7. Способ получения L-BPA по п. 1, где способ получения L-BPA включает стадию
проведения реакции борной кислоты, серной кислоты и н-бутанола в первом органическом растворителе с получением триалкилбората.
8. Способ получения L-BPA по п. 1, где выделение (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе из реакционной смеси включает
добавление в реакционную смесь второго органического растворителя и раствора кислоты, регулирование pH до ≤ 5 и экстрагирование органической фазы;
добавление в органическую фазу раствора основания, регулирование pH в диапазоне 7,1-14;
выделение (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе с помощью второго экстрагента.
9. Способ получения L-BPA по п. 1, где удаление защитной группы с аминогруппы (S)-4-бороно-L-фенилаланина с получением L-BPA включает
добавление в реакционный сосуд (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе, воды, раствора кислоты и третьего органического растворителя;
регулирование pH до 6,15-6,25 с получением L-BPA.
10. Способ получения L-BPA, который включает следующие стадии:
проведение реакции (S)-4-галогенфенилаланина формулы I с защитной группой при аминогруппе, борирующего средства и реактива Гриньяра с получением реакционной смеси, где реакционная смесь содержит (S)-4-бороно-L-фенилаланин формулы II с защитной группой при аминогруппе, при этом R1 представляет собой галоген, и в формуле I, и в формуле II R2 представляет собой защитную группу, и при этом реактив Гриньяра представляет собой трет-бутилхлорид магния;
разделение реакционной смеси с получением (S)-4-бороно-L-фенилаланина с защитной группой при аминогруппе;
удаление защитной группы с аминогруппы (S)-4-бороно-L-фенилаланина с получением L-BPA, который имеет структуру, представленную формулой III выше.
11. Способ получения L-BPA по п. 10, где проведение реакции (S)-4-галогенфенилаланина с защитной группой при аминогруппе, борирующего средства и реактива Гриньяра с получением реакционной смеси предусматривает
проведение реакции (S)-4-галогенфенилаланина с защитной группой при аминогруппе, борирующего средства и реактива Гриньяра при температуре 0°C-30°C с получением реакционной смеси.
| CN 104447823 A, 25.03.2015 | |||
| CN 104447822 A, 25.03.2015 | |||
| СОЕДИНЕНИЯ 7-ФЕНИЛПИРАЗОЛОПИРИДИНА | 2003 |
|
RU2327699C2 |
| WANG X.-J | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Lett., 2006, v | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Держатель для поленьев при винтовом колуне | 1920 |
|
SU305A1 |
| SIVAEV I.B | |||
| et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ очищения сернокислого глинозема от железа | 1920 |
|
SU47A1 |

