Настоящее изобретение относится к применению сульфидной композиции для электрокаталитического расщепления воды.
Предшествующий уровень техники изобретения
Водород в основном получают из природного газа путем паровой конверсии метана. Другими ископаемыми источниками являются другие углеводороды и уголь (см., например, John A. Turner, Sustainable Hydrogen Production, Science 2004, Vol 305, 972-974). Экологичными исходными материалами для получения водорода являются биомасса или вода. С водой в качестве исходного материала осуществляют различные производственные процессы, например, электролиз, термолиз и фотоэлектролиз. Существует два разных типа электролизеров при умеренной температуре: электролизеры со щелочной и протонообменной мембраной (РЕМ (от англ. - proton exchange membrane)) (см., например, Jamie D. Holladay, An overview of hydrogen production technologies, Catalysis Today 2009, 139, 244 - 260).
В коммерческих электролизерах РЕМ платину используются в качестве электрокатализатора для реакции выделения водорода (HER (от англ. - hydrogen evolution reaction)) (см., например, Peter С.K. Vesborg, Recent Development in Hydrogen Evolution Reaction Catalysts and Their Practical Implementation, The Journal of Physical Chemistry Letters 2015, 6, 951-957).
Например, в европейском патентном документе ЕР3222752 А1 раскрыт способ образования водорода и кислорода из воды, включающий в себя обеспечение сборки мембранного электрода, содержащей анодный катализатор электролизера, способный катализировать реакцию выделения кислорода при потенциалах, превышающих +1,23 В относительно обратимого водородного электрода, при этом анодный катализатор электролизера содержит наноструктурированные нитевидные кристаллы, имеющие на себе несколько чередующихся слоев, содержащих соответственно в любом порядке Pt и Ir, сборка мембранного электрода, кроме того, содержит катод; обеспечение контакта воды с анодным катализатором электролизера и обеспечение электрического потенциала с достаточным током через сборку мембранного электрода для конверсии по меньшей мере части воды в водород и кислород на катоде и аноде, соответственно.
Уже были предприняты попытки обеспечения для HER не содержащих платину электрокатализаторов. В частности, сульфиды металлов, карбиды металлов, селениды металлов, нитриды металлов и фосфиды металлов демонстрируют представляющие интерес свойства для этой реакции (см., например, Xiaoxin Zou, Noble metal-free hydrogen evolution catalysts for water splitting, Chem. Soc. Rev. 2015, 44, 5148-5180).
В публикации международной заявки WO 2017/062736 А1 раскрыты материалы на основе структурированного дисульфида молибдена для электрокаталитических применений.
В публикации международной заявки WO2018/098451 А1 раскрыты катализаторы, включающие в себя пленки халькогенидов переходных металлов, для реакции выделения водорода.
В патентном документе США US2015/0259810 А1 заявлено устройство, содержащее катализатор реакции выделения водорода. Катализатор включает в себя по меньшей мере один компонент, выбранный из группы, состоящей из фосфидов переходных металлов, сульфидов переходных металлов первого ряда и арсенидов переходных металлов в виде наночастиц и, в частности, СоР.
В патентном документе Китая CN105132941 А заявлен композитный материал для электрокатализа выделения водорода на основе диселенида молибдена/углеродной сажи и способ его получения.
В публикации международной заявки WO2015/021019 А1 заявлен катализатор для обеспечения реакции выделения водорода, при этом катализатор включает в себя нитрид металла, характеризующийся формулой M'xM''yNz, где М' выбран из группы, состоящей из Ag, Al, Са, Со, Cr, Cu, Fe, Ga, In, Li, Mg, Mn, Na, Ni, Sc, Ti, V, Y, Zn и их смесей; где М'' выбран из группы, состоящей из Hf, Mo, Nb, Re, Ru, Та, W, Zr и их смесей; где х представляет собой число от 0 до 1; где у представляет собой число от 1 до 2; где z представляет собой число более 1,8 и менее 2,2; и где нитрид металла содержит гексагональную решетку с четырехслойной последовательностью упаковки, которая включает в себя две формульных единицы смешанной плотноупакованной структуры с чередующимися слоями металлов М'' в тригональной призматической координации, а также металлов М' или М' и М'' в октаэдрической координации.
В европейском патентном документе ЕР2377971 А1 раскрыты электрокатализаторы для восстановления протонов с образованием Н2, состоящие из аморфных пленок сульфидов переходных металлов или твердых веществ, обладающих активностью при всех значениях рН. Кроме того, упоминаются MoS2 и WS2 в качестве возможных сульфидов переходных металлов.
Железо-никелевый пентландит оказался представляющим интерес составом для HER (см. Konkena et al., "Pentlandite rocks as sustainable and stable efficient electrocatalysts for hydrogen generation", Nature Communication 2016, 7, Article number 12269).
У Klein et al. ((таблица A2) "Chemical composition of Pentlandite of ODP holes 209-1268A, 209-1270D, 209-1271 В and 209-1274A"; 2010), Schrocke et al. ("Mineralogie: Ein Lehrbuch Auf Systematischer Grundlage", page 137, 1981), в Mineral Data Publishing ("Pentlandite", 2001) и у Rajamani et al. ("Crystal Chemistry of Natural Pentlandites", Canadian Mineralogist, Vol. 12, page 178-187, 1973) раскрыты встречающиеся в природе пентландиты.
У Knop et al. ("Chalcogenides of the Transition Elements", Can. J. Chem., Vol. 39, 1961) раскрыты синтетически полученные FeCoNiS-пентландиты.
Цель и краткое раскрытие изобретения
Композиции для электрокаталитического расщепления воды, раскрытые в известном уровне техники, не удовлетворяют всем требованиям каталитических характеристик, устойчивости к соединениям серы, стабильности каталитической активности и/или стартстопных свойств. Таким образом, цель настоящего изобретения заключается в том, чтобы предложить новое применение композиции для HER, характеризующейся более высокой плотностью тока, лучшей устойчивостью к соединениям серы и лучшими стартстопными свойствами по сравнению с материалами известного уровня техники.
Согласно одному аспекту настоящее изобретение относится к применению композиции формулы
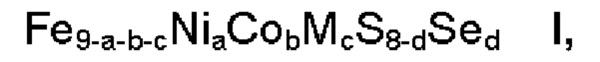
где
М обозначает один или несколько элементов, характеризующихся в ионном состоянии эффективным ионным радиусом в диапазоне 70-92 пм,
а представляет собой число в диапазоне 2,5≤а≤3,5, более предпочтительно 2,7≤а≤3,3,
b представляет собой число в диапазоне 1,5≤b≤5,0, более предпочтительно 1,5≤b≤4,0, наиболее предпочтительно 2,5≤b≤3,5,
с представляет собой число в диапазоне 0,0≤с≤2,0, более предпочтительно 0,0≤с≤1,0,
d представляет собой число в диапазоне 0,0≤d≤4,0, более предпочтительно 0,0≤d≤1,0,
где сумма a, b и с находится в диапазоне 5≤а+b+с≤8,
и где ≥90 масс. % композиции находится в пентландитовой фазе, для электрокаталитического расщепления воды, предпочтительно для реакции выделения водорода.
Краткое описание графических материалов
На фиг. 1 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, полученные в диапазоне сканирования от 0 до -0,65 V по сравнению с RHE (от англ. - reversible hydrogen electrode - обратимый водородный электрод) в 0,5 М серной кислоте при комнатной температуре.
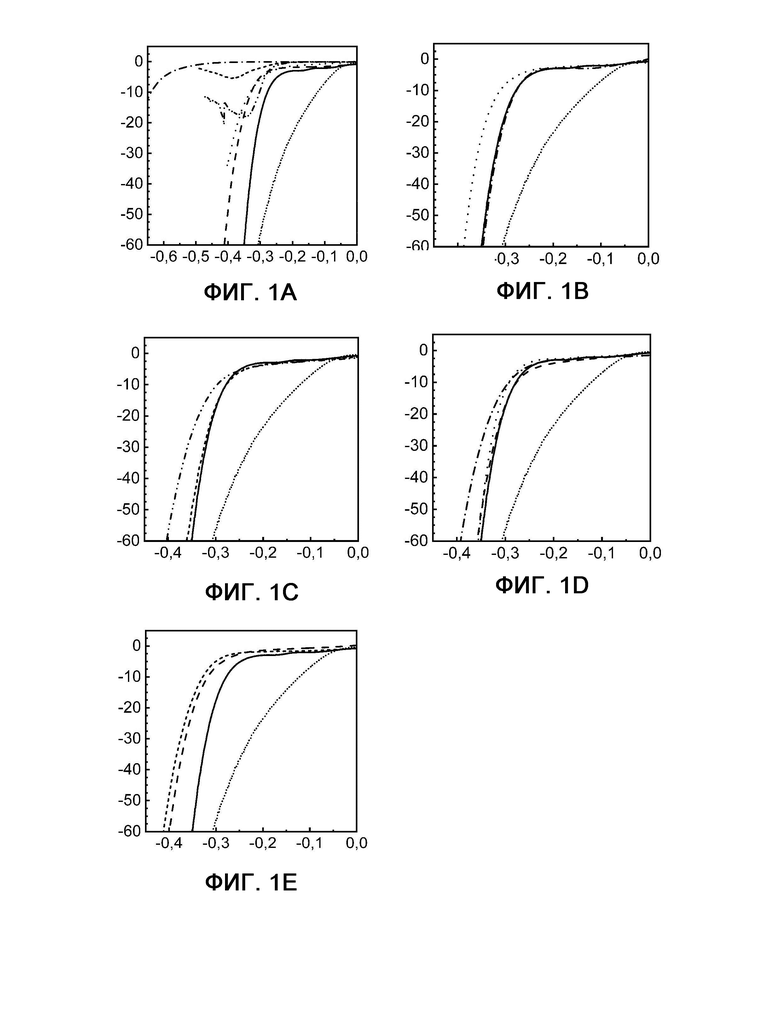
На фиг. 2 показаны результаты измерений кулонометрии с управляемым потенциалом при -0,5 В по сравнению с RHE при 20°С и 60°С для электродов на основе Pt (сравнительный пример 6) и Fe3Co3Ni3S8 (пример 1) в течение 20 часов.
На фиг. 3 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой при 20°С (А) и 60°С (В) до и после 20-часового электролиза для платины (сравнительный пример 6) и Fe3Co3Ni3S8 (пример 1).
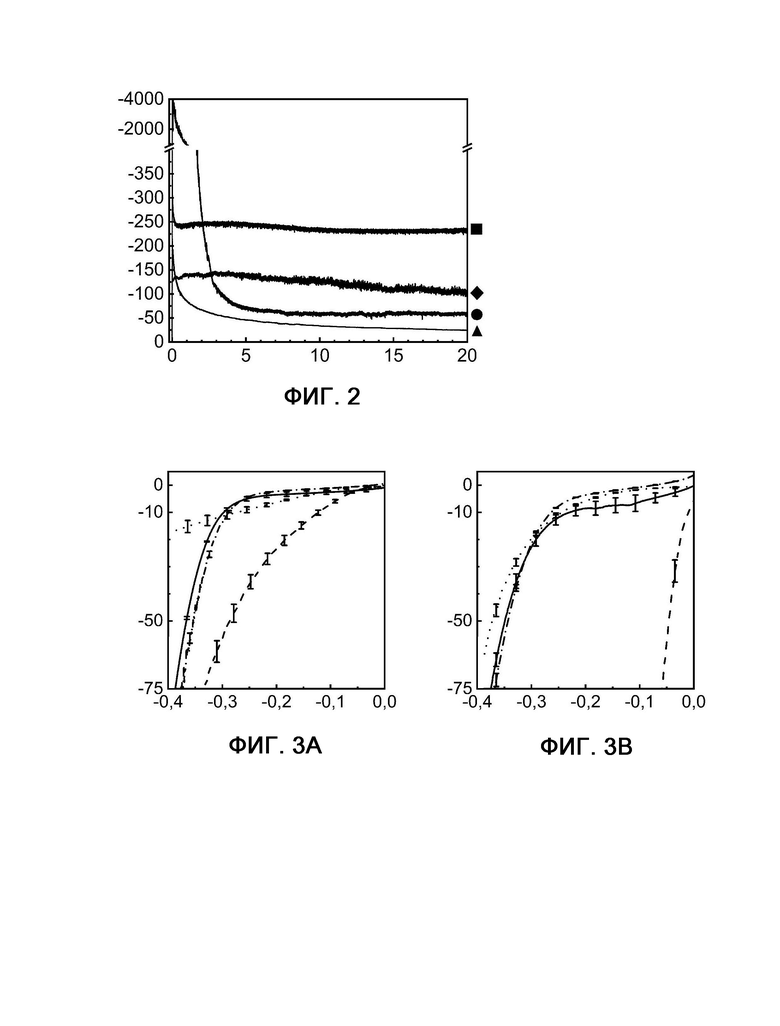
На фиг. 4 показана кулонометрия с управляемым потенциалом в стартстопном режиме, измеряемая при 60°С и -304 мВ по сравнению с RHE на протяжении 72 часов для Fe3Co3Ni3S8 (пример 1) и платины (сравнительный пример 6).
На фиг. 5 показана кулонометрия с управляемым потенциалом в присутствии и в отсутствие катализаторного яда H2S, измеряемая при 20°С и -350 мВ по сравнению с RHE на протяжении 20 часов.
На фиг. 6 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
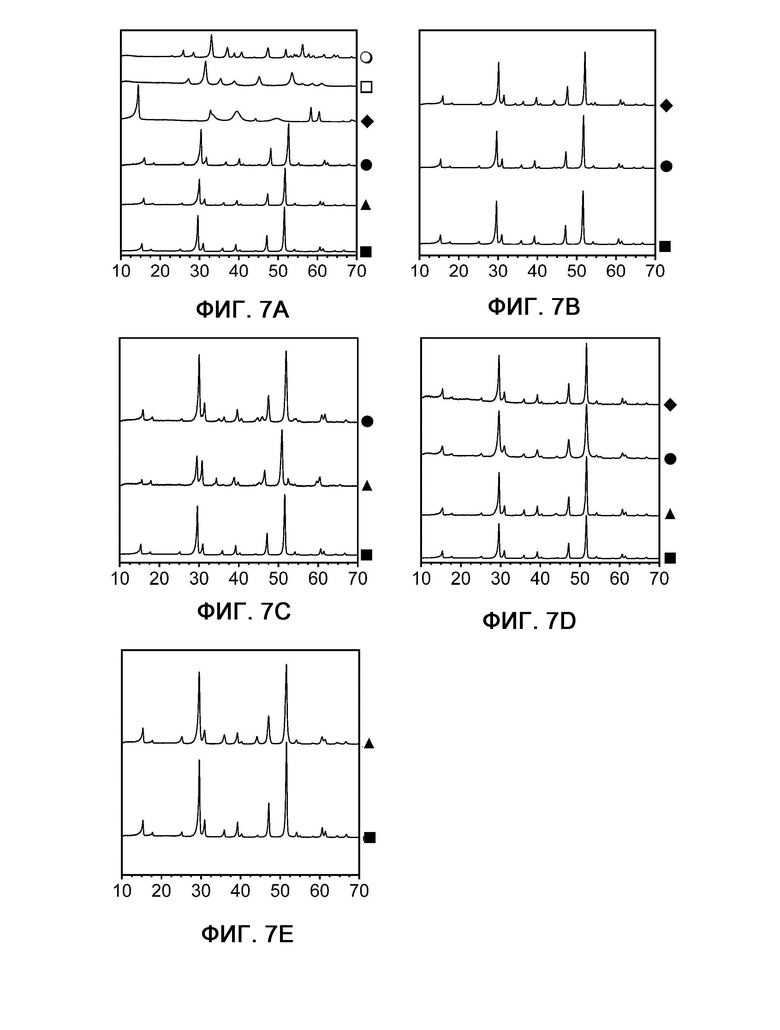
На фиг. 7 показана порошковая рентгенограмма (паттерны дифракции, (излучение Cu-Kα)) композиций, используемых в соответствии с настоящим изобретением и сравнительными примерами.
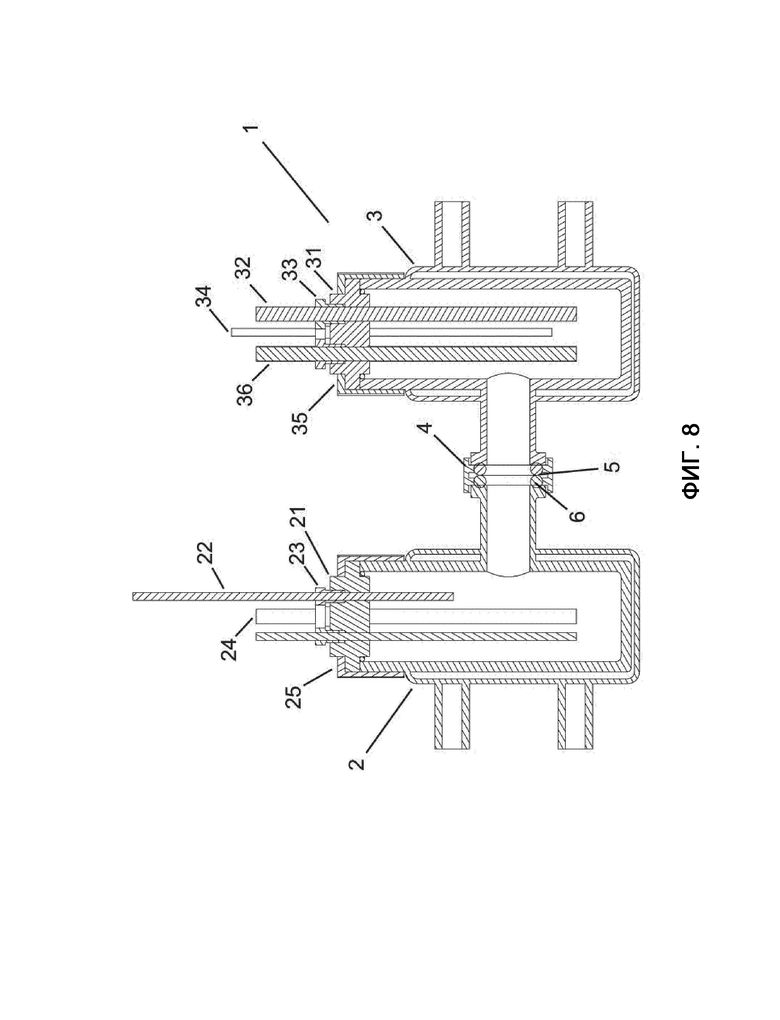
На фиг. 8 показано поперечное сечение электрохимической ячейки, используемой для испытаний, проводимых с композициями, используемыми в соответствии с настоящим изобретением и сравнительными примерами.
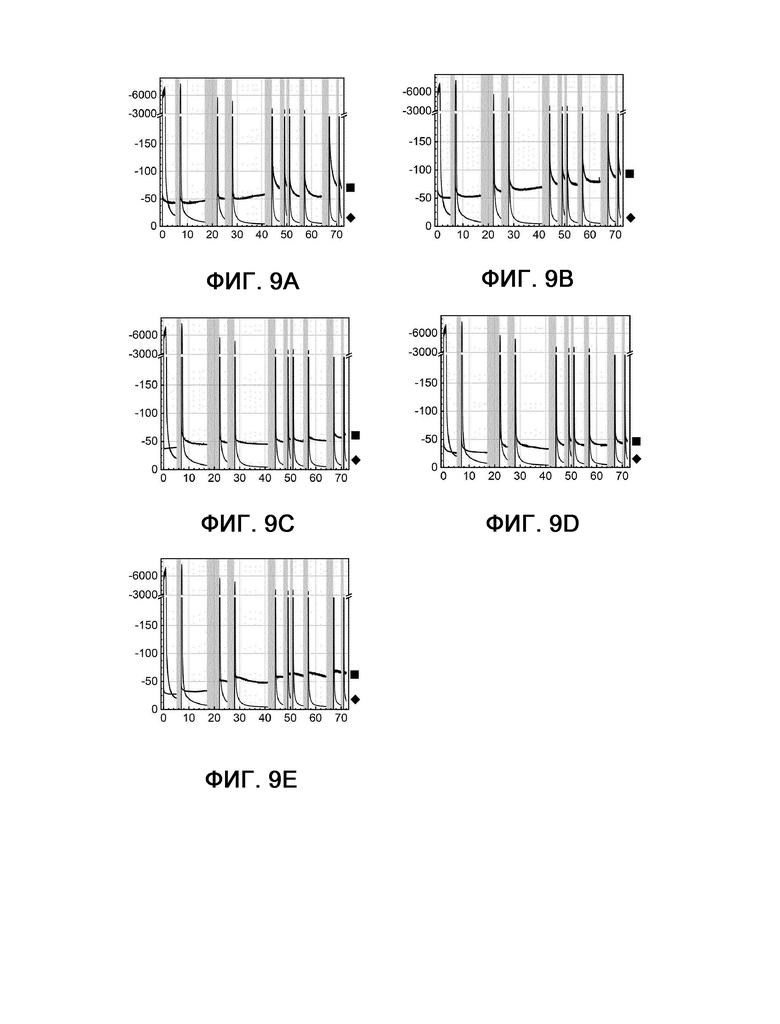
На фиг. 9 показана кулонометрия с управляемым потенциалом в стартстопном режиме, измеряемая при 60°С и -350 мВ по сравнению с RHE на протяжении 72 часов.
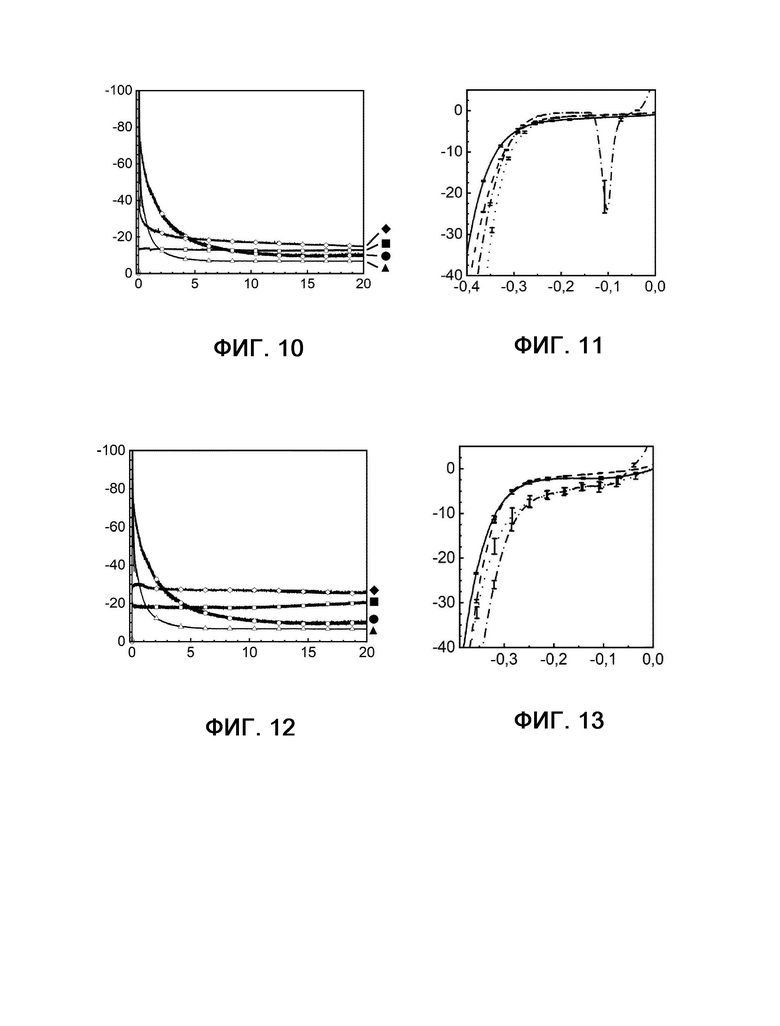
На фиг. 10 показана кулонометрия с управляемым потенциалом в присутствии и в отсутствие катализаторного яда H2S, измеряемая при 20°С и -350 мВ по сравнению с RHE на протяжении 20 часов.
На фиг. 11 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
На фиг. 12 показана кулонометрия с управляемым потенциалом в присутствии и в отсутствие катализаторного яда H2S, измеряемая при 20°С и -350 мВ по сравнению с RHE на протяжении 20 часов.
На фиг. 13 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
На фиг. 14 показана кулонометрия с управляемым потенциалом в присутствии и в отсутствие катализаторного яда H2S, измеряемая при 20°С и -350 мВ по сравнению с RHE на протяжении 20 часов.
На фиг. 15 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
На фиг. 16 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
На фиг. 17 показана кулонометрия с управляемым потенциалом в стартстопном режиме, измеряемая при 60°С и -350 мВ по сравнению с RHE на протяжении 72 часов.
На фиг. 18 показана кулонометрия с управляемым потенциалом в присутствии и в отсутствие катализаторного яда H2S, измеряемая при 20°С и -350 мВ по сравнению с RHE на протяжении 20 часов.
На фиг. 19 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
На фиг. 20 показана кулонометрия с управляемым потенциалом в присутствии и в отсутствие катализаторного яда H2S, измеряемая при 20°С и -350 мВ по сравнению с RHE на протяжении 20 часов.
На фиг. 21 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
На фиг. 22 показана кулонометрия с управляемым потенциалом в присутствии и в отсутствие катализаторного яда H2S, измеряемая при 20°С и -350 мВ по сравнению с RHE на протяжении 20 часов.
На фиг. 23 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
На фиг. 24 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
На фиг. 25 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
Подробное раскрытие изобретения
Неожиданно обнаружили, что благодаря введению Со в Fe,Ni-пентландит композиции, используемые в соответствии с настоящим изобретением, демонстрируют более низкое перенапряжение для HER по сравнению с известным из уровня техники пентландитом или другими не содержащими металлы платиновой группы (МПГ) материалами. Также выяснили, что в композицию могут быть добавлены дополнительные металлы, имеющие эффективный ионный радиус, сравнимый с радиусами Fe2+, Ni2+ или Со2+. Кроме того, неожиданно обнаружили, что, хотя Se2- имеет другой ионный радиус по сравнению с S2- (198 пм против 184 пм), можно заменить до 50% S на Se без потери целостности молекулярной структуры пентландитовой фазы, электрокаталитической активности или стабильности.
Эффективный ионный радиус ионов металлов может быть определен согласно способу, раскрытому в Hollemann, Wieberg, Lehrbuch der Anorganischen Chemie, Verlag: De Gruyter; 102th edition (2007).
Композиции, используемые в соответствии с настоящим изобретением, демонстрируют более высокие плотности тока, чем материалы на основе платины, после нескольких часов электролиза и не теряют значительной активности в ходе получения водорода. Кроме того, электроды, полученные из композиций, используемых в соответствии с настоящим изобретением, имеют существенно улучшенные стартстопные характеристики по сравнению с платиновыми электродами. Наконец, композиции, используемые в соответствии с настоящим изобретением, устойчивы к отравлению серой.
Кристаллическая фаза немодифицированного пентландита раскрывается, например, в A. Pearson, М. Buerger, Am. Mineral. 1956, 41, 804-805. В композиции, используемой в соответствии с настоящим изобретением, ≥90 масс. % композиции находится в пентландитовой фазе.
Согласно одному варианту осуществления настоящего изобретения М не присутствует в композиции формулы I.
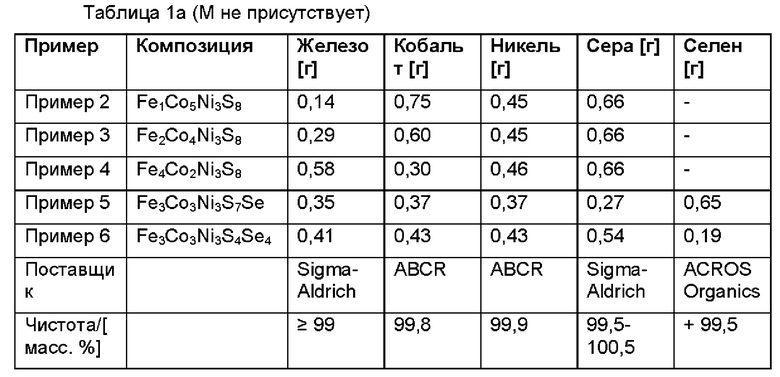
Согласно данному варианту осуществления композиция может быть выбрана из группы, состоящей из Fe3Ni3Co3S8, Fe1Co5Ni3S8, Fe2Co4Ni3S8, Fe4Co2Ni3S8, Fe3Ni3Co3S4Se4, Fe3Ni3Co3S7Se и их смесей.
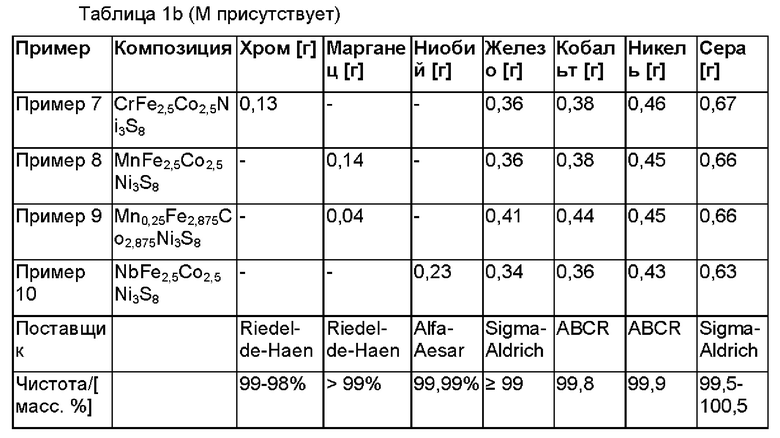
Согласно другому варианту осуществления настоящего изобретения М присутствует в композиции формулы I.
М предпочтительно характеризуется эффективным ионным радиусом, сравнимым с таковым у Fe2+, Ni2+ или Со2+.
В частности, М может быть выбран из группы, состоящей из Nb, Cu, Mn или Cr и их смесей, более предпочтительно из Mn, Cr и их смесей, наиболее предпочтительно из Cr.
В частности, композиция может быть выбрана из группы, состоящей из CrFe2,5Co2,5Ni3S8, MnFe2,5Co2,5Ni3S8, Mn0,25Fe2,875Со2,875Ni3S8,
NbFe2,5Co2,5Ni3S8 и их смесей.
Композиция, используемая в соответствии с настоящим изобретением, предпочтительно характеризуется показателем перенапряжения для реакции выделения водорода (HER) 328 мВ и меньше, предпочтительно 300 мВ и меньше, при плотности тока -10 мА/см2, в частности, в соответствии с условиями испытания, подробно изложенными ниже.
Согласно следующему аспекту представлен электрод, содержащий композицию, используемую в соответствии с настоящим изобретением.
Электрод в соответствии с настоящим изобретением характеризуется показателем перенапряжения для реакции выделения водорода (HER) 328 мВ и меньше, предпочтительно 300 мВ и меньше, при плотности тока -10 мА/см2, в частности, в соответствии с условиями испытания, подробно изложенными ниже.
Согласно следующему аспекту настоящее изобретение относится к различным процессам получения композиции, используемой в соответствии с настоящим изобретением. Первый процесс (далее - процесс №1) характеризуется тем, что композиции синтезируют термическим синтезом при отсутствии кислорода в вакууме. Второй процесс (процесс №2) характеризуется тем, что композиции синтезируют термическим синтезом при отсутствии кислорода в инертном газе. Для не содержащих селена композиций, используемых в соответствии с настоящим изобретением, подходят два дополнительных процесса. В одном из этих процессов в атмосфере инертного газа композиции синтезируют с помощью способа совместного осаждения и стадии дополнительной обработки (процесс №3). Другой процесс (процесс №4) характеризуется тем, что композиции синтезируют с помощью способа золь-гель и стадии дополнительной обработки.
Термический синтез при отсутствии кислорода для получения композиций, используемых в соответствии с настоящим изобретением, представляет собой способ, аналогичный пути термического синтеза, раскрытого в Piontek et al. ACS Catalysis 2018, 8, 987-996.
Более конкретно, термический синтез при отсутствии кислорода в вакууме (процесс №1) включает в себя следующие стадии:
а) получения гомогенной смеси порошка Fe, порошка Ni, порошка Со, порошка S и необязательно порошка Se и/или порошка М путем смешивания или измельчения; при этом средний размер частиц конечной смеси предпочтительно ниже 100 мкм;
b) переноса смеси, полученной на стадии а), в закрытый сосуд и откачивания воздуха до состояния вакуума в диапазоне от 30 мбар до 10-15 мбар, предпочтительно от 1 мбар до 10-7 мбар;
c) нагревания закрытого сосуда при скорости 0,1-20 K минута-1 в диапазоне температур 600-1100°С и охлаждения в течение 1-120 часов, предпочтительно 700-1100°С, в течение 1-10 часов при скорости 2-5 K минут-1, более предпочтительно с двумя температурами охлаждения - первой температурой 600-800°С в течении 3-10 часов и второй температурой 900-1100°С в течение 10-20 часов - при скорости 2-5 K минута-1, наиболее предпочтительно с двумя температурами охлаждения - первой температурой 700°С в течение 3 часов и второй температурой 1000°С в течение 10 часов.
Более конкретно, термический синтез при отсутствии кислорода в инертном газе (процесс №2) включает в себя следующие стадии:
a) получения гомогенной смеси порошка Fe, порошка Ni, порошка Со, порошка S и необязательно порошка Se и/или порошка М путем смешивания или измельчения; при этом средний размер частиц конечной смеси предпочтительно ниже 100 мкм;
b) переноса смеси, полученной на стадии а), в закрытый сосуд и замены воздуха инертным газом; при этом инертный газ может представлять собой азот, диоксид углерода, неон, аргон, криптон, ксенон или их смесь, предпочтительно азот, диоксид углерода и аргон, более предпочтительно азот и аргон, наиболее предпочтительно азот;
c) нагревания закрытого сосуда при скорости 0,1-20 K минута-1 в диапазоне температур 600-1100°С и охлаждения в течение 1-120 часов, предпочтительно 700-1100°С, в течение 1-10 часов при скорости 2-5 K минут-1, более предпочтительно с двумя температурами охлаждения - первой температурой 600-800°С в течении 3-10 часов и второй температурой 900-1100°С в течение 10-20 часов - при скорости 2-5 K минута-1, наиболее предпочтительно с двумя температурами охлаждения - первой температурой 700°С в течение 3 часов и второй температурой 1000°С в течение 10 часов.
Более конкретно, способ совместного осаждения со стадией дополнительной обработки (процесс №3) включает в себя следующие стадии:
а) получения раствора соли железа, соли никеля, соли кобальта и необязательно одной или нескольких солей М, определяемого в соединении формулы I, и необязательно неорганической кислоты в воде;
b) добавления одного или нескольких источников сульфида в воде, необязательно с неорганическим основанием, на протяжении от 1 до 120 минут, предпочтительно от 10 до 60 минут;
c) фильтрации и промывания осадка водой;
d) сушки в диапазоне температур 60-120°С в течение 1-120 часов, предпочтительно 80-100°С, в течение 3-15 часов, или необязательно сушки замораживанием;
e) нагревания осадка в диапазоне температур 200-500°С при скорости 0,1-20 K минута-1 в течение 1-120 часов, предпочтительно 250-400°С, в течение 2-10 часов при скорости 2-5 K минута-1, в атмосфере водорода и сероводорода;
f) и необязательно нагревания образца в течение 0,5-20 часов, предпочтительно 2-5 часов, в атмосфере водорода, необязательно разбавленного инертным газом, например, N2 или Ar.
В данном процессе подходящая соль железа, никеля, кобальта и М включает в себя, например, нитраты, оксиды, гидроксиды, карбонаты, сульфаты, ацетаты, галогениды и их смеси, предпочтительно нитраты, сульфаты, ацетаты и их смеси, наиболее предпочтительно нитраты.
В данном процессе неорганическая кислота на стадии а) включает в себя соответствующие неорганические кислоты, например, азотную кислоту, серную кислоту, хлористоводородную кислоту и их смеси, предпочтительно азотную кислоту, серную кислоту и их смеси, наиболее предпочтительно азотную кислоту.
В данном процессе подходящий источник сульфида включает в себя соли сульфида, например, сульфид натрия, гидросульфид натрия, сульфид аммония, гидросульфид аммония, сульфид калия, гидросульфид калия и сероводород, предпочтительно сульфид натрия, сероводород и сульфид аммония, более предпочтительно сульфид натрия и сульфид аммония, наиболее предпочтительно сульфид натрия.
В данном процессе неорганическое основание на стадии b) включает в себя подходящее неорганическое основание, например, гидроксид натрия, оксид натрия, аммиак, оксид калия, гидроксид калия, предпочтительно гидроксид натрия, оксид натрия и аммиак, более предпочтительно гидроксид натрия и аммиак, наиболее предпочтительно гидроксид натрия.
Смесь водорода и сероводорода может характеризоваться отношением от 95:5 до 0:100, предпочтительно от 90:10 до 50:50, более предпочтительно от 90:10 до 80:20, например, 85:15. Такая смесь может быть разбавлена инертным газом, например, аргоном, азотом.
Способ золь-гель со стадией дополнительной обработки (процесс №4) включает в себя следующие стадии:
a) получения раствора соли железа, соли никеля, соли кобальта и необязательно одной или нескольких солей М, определяемого в соединении формулы I, и одного или нескольких полимерных предшественников, а также необязательно одного или нескольких комплексообразующих средств в воде;
b) необязательно добавления неорганической кислоты в раствор, полученный на стадии а);
c) нагревания раствора при 90°С в течение 1-120 часов до образования вязкого геля;
d) прокаливания геля в диапазоне температур 300-700°С в течение 1-120 часов, предпочтительно 400-500°С в течение 2-20 часов, в воздухе;
e) нагревания смешанного оксида в диапазоне температур 200-500°С в течение 1-120 часов, предпочтительно 250-400°С, в течение 2-10 часов в атмосфере водорода и сероводорода;
f) и необязательно нагревания образца в течение 0,5-20 часов, предпочтительно 2-5 часов, в атмосфере водорода, необязательно разбавленного инертным газом, например, N2 или Ar.
В данном процессе комплексообразующее средство также может служить полимерным предшественником.
В данном процессе подходящий полимерный предшественник включает в себя многоосновные карбоновые кислоты, гидроксикарбоновые кислоты, многоатомные спирты и их смеси, предпочтительно многоатомные спирты, многоосновные карбоновые кислоты и их смеси, более предпочтительно многоосновные карбоновые кислоты, наиболее предпочтительно лимонную кислоту.
В данном процессе подходящее комплексообразующее средство включает в себя органические соединения, например, органические кислоты, кетоны, альдегиды, спирты, амины и их смеси, предпочтительно многоосновные карбоновые кислоты, более предпочтительно лимонную кислоту и щавелевую кислоту, а наиболее предпочтительно лимонную кислоту.
В данном процессе подходящая соль железа, никеля, кобальта и М включает в себя соли этих элементов, например, нитраты, оксиды, гидроксиды, карбонаты, сульфаты, ацетаты, галогениды и их смеси, предпочтительно нитраты, сульфаты, ацетаты и их смеси, наиболее предпочтительно нитраты.
В данном процессе неорганическая кислота на стадии а) включает в себя подходящие неорганические кислоты, например, азотную кислоту, серную кислоту, хлористоводородную кислоту и их смеси, предпочтительно азотную кислоту, серную кислоту и их смеси, наиболее предпочтительно азотную кислоту.
Примеры
Пример 1 (согласно процессу №1)
Fe3Co3Ni3S8
Порошок железа (0,43 г, Sigma-Aldrich, ≥99 масс. %), порошок кобальта (0,46 г, ABCR, 99,8 масс. %), порошок никеля (0,45 г, ABCR, 99,9 масс. %) и сера (0,66 г, Sigma-Aldrich, 99,5 масс. % - 100,5 масс. %) смешивали вместе в течение 10 минут до получения визуально гомогенной смеси элементов. Гомогенной смесью элементов наполняли кварцевую ампулу (диаметром 10 мм, общим объемом 15 мл), из которой откачивали воздух при давлении ниже 4×10-2 мбар в течение 16 часов, а затем запечатывали в вакууме. Образец помещали в печь и нагревали до 700°С при скорости нагревания 4,5 K минута-1. После выдерживания изотермы температур в течение 3 часов, чтобы сера прореагировала со смесью металлов без повреждения сосуда, температуру повышали до 1000°С (скорость нагревания 3,33 K минута-1) для усиления диффузии. Затем образец выдерживали при этой температуре в течение 10 часов. После этого обеспечивали охлаждение образца до комнатной температуры.
Пример 2-10
Следующие композиции синтезировали подобно процессу, раскрываемому в примере 1.
Пример 11 (согласно процессу №2)
Fe3Co3Ni3S8
Порошок железа (0,43 г, Sigma-Aldrich, ≥99 масс. %), порошок кобальта (0,46 г, ABCR, 99,8 масс. %), порошок никеля (0,45 г, ABCR, 99,9 масс. %) и серу (0,66 г, Sigma-Aldrich, 99,5 масс. % - 100,5 масс. %) смешивали вместе в течение 10 минут до получения визуально гомогенной смеси элементов. Гомогенной смесью элементов наполняли кварцевую ампулу (диаметром 10 мм, общим объемом 15 мл), из которой откачивали воздух при давлении ниже 4×10-2 мбар, заполняли аргоном, а затем запечатывали в статической атмосфере аргона. Образец помещали в печь и нагревали до 700°С при скорости нагревания 4,5 K минута-1. После выдерживания изотермы температур в течение 3 часов температуру повышали до 1000°С (скорость нагревания 3,33 K минута-1). Затем образец выдерживали при этой температуре в течение 10 часов. После этого обеспечивали охлаждение образца до комнатной температуры.
Пример 12 (согласно процессу №3)
Fe3Co3Ni3S8
Нитрат железа(III) (Fe(NO3)3⋅9H2O, 1,35 г), нитрат никеля(II) (Ni(NO3)2⋅6H2O, 0,97 г) и нитрат кобальта(II) (Co(NO3)2⋅6H2O, 0,97 г) растворяли в 300 мл воды. В отдельном контейнере растворяли сульфид натрия (Na2S⋅9H2O, 4,80 г) в 200 мл воды. Добавляли раствор сульфида к раствору нитрата за 30 минут при энергичном взбалтывании, что приводило к образованию черного осадка. Черный осадок отфильтровывали и промывали 200 мл воды. Затем осадок сушили замораживанием. Затем в потоке N2 черный порошок нагревали до 40°С (10 K минута-1) и выдерживали при этой температуре в течение 10 минут. После этого температуру повышали до 300°С (10 K минута-1) с применением потока водорода (85 об.%)/сероводорода (15 об.%). Через 4 часа при таких условиях к образцу подавали чистый водород в течение 4 часов подряд. Реакцию быстро останавливали путем подачи холодного (25°С) потока газообразного азота. На протяжении всего эксперимента поток газа регулировали до 40 мл минута-1 и держали постоянным.
Пример 13 (согласно процессу №3)
Fe3Co3Ni3S8
Нитрат железа(III) (Fe(NO3)3⋅9H2O, 1,35 г), нитрат никеля(II) (Ni(NO3)2⋅6H2O, 0,97 г) и нитрат кобальта(II) (Co(NO3)2⋅6H2O, 0,97 г) растворяли в 300 мл воды. В отдельном контейнере растворяли сульфид аммония ((NH4)2S (20 масс. % в H2O), 20 ммоль, 6,81 г) в 200 мл воды. Добавляли раствор сульфида к раствору нитрата за 30 минут при энергичном взбалтывании, что приводило к образованию черного осадка. Черный осадок отфильтровывали и промывали 200 мл воды. Затем осадок сушили замораживанием. Затем в потоке N2 черный порошок нагревали до 40°С (10 K минута-1) и выдерживали при этой температуре в течение 10 минут. После этого температуру повышали до 300°С (10 K минута-1) с применением потока водорода (85 об.%)/сероводорода (15 об.%). Через 4 часа при таких условиях к образцу подавали чистый водород в течение 4 часов подряд. Реакцию быстро останавливали путем подачи холодного (25°С) потока газообразного азота. На протяжении всего эксперимента поток газа регулировали до 40 мл минута-1 и держали постоянным.
Пример 14 (согласно процессу №4)
Fe3Co3Ni3S8
Нитрат железа(II) (Fe(NO3)3⋅9H2O, 2,02 г), нитрат кобальта(II) (Co(NO3)2⋅6H2O, 1,46 г) и нитрат никеля(II) (Ni(NO3)2⋅6H2O, 1,45 г) растворяли в 15 мл воды, содержащей 2,88 г лимонной кислоты. Раствор нагревали до 90°С в течение 2-6 часов, что приводило к образованию вязкого геля. Полученный гель прокаливали при 500°С в течение 18 часов и полученным черным твердым веществом наполняли кювету из кварцевого стекла, которую помещали в печь. Затем печь продували инертным газом (N2) в течение 10 минут при 40°С. Затем поток газа заменяли на смесь водорода (85 об.%) и сероводорода (15 об.%) и температуру повышали до 300°С при скорости нагревания 10 K минута-1. Через 15 минут продувания образца водородом (85 об.%) и сероводородом (15 об.%) при 300°С через аппарат продували чистый водород при 300°С в течение 4 часов. После этого печь выключали и образец быстро охлаждали в холодном (25°С) потоке N2. На протяжении всего эксперимента поток газа регулировали до 40 мл минута-1 и держали постоянным.
Сравнительный пример 1
Fe4,5Ni4,5S8 (Piontek et al. ACS Catalysis 2018, 8, 987-996)
Порошок металлического железа (1,75 г, Sigma-Aldrich, ≥99 масс. %), никель (1,75 г, ABCR, 99,9 масс. %) и серу (1,70 г, Sigma-Aldrich, 99,5 масс. % -100,5 масс. %) смешивали вместе в течение 10 минут до получения визуально гомогенной смеси элементов. Эту смесь помещали в 10-мм кварцевую трубку. После этого кварцевую трубку запечатывали в статическом вакууме и нагревали до 700°С со скоростью 5°С минута-1. После 3 часов отжига при 700°С температуру повышали до 1100°С за 30 минут. После 10 часов при 1100°С обеспечивали охлаждение смеси до комнатной температуры.
Сравнительный пример 2
Co9S8
Порошок кобальта (1,35 г, ABCR, 99,8 масс. %) и сера (0,66 г, Sigma-Aldrich, 99,5 масс. % - 100,5 масс. %) смешивали вместе в течение 10 минут до получения визуально гомогенной смеси элементов. Гомогенной смесью элементов наполняли кварцевую ампулу (диаметром 10 мм, общим объемом 15 мл), из которой откачивали воздух при давлении ниже 4 x 10-2 мбар в течение 16 часов, а затем запечатывали в вакууме. Образец помещали в печь и нагревали до 700°С при скорости нагревания 4,5 K минута-1. После выдерживания изотермы температур в течение 3 часов, чтобы сера прореагировала со смесью металлов без повреждения сосуда, температуру повышали до 1000°С (скорость нагревания 3,33 K минута-1) для усиления диффузии. Затем образец выдерживали при этой температуре в течение 10 часов. После этого обеспечивали охлаждение образца до комнатной температуры.
Получение электрода согласно примеру 1-10, а также согласно сравнительному примеру 1-2
В качестве контакта для электродов (диаметром 3 мм) использовали специально изготовленный тефлоновый кожух с латунным стержнем. Соответствующий материал, используемый для изготовления электрода (50 мг), измельчали до получения мелкоизмельченного порошкового материала. Измельченный порошок загружали в прессовальный инструмент (диаметром 3 мм) и прессовали измельченный порошок с максимальной силой веса 800 кг/см2. На латунный стержень в полости тефлонового кожуха наносили двухкомпонентный эпоксидный клей с серебром для соединения латунной подложки с материалом электрода. Затем гранулу вдавливали в тефлоновый кожух и удаляли любые загрязнения с тефлонового кожуха. Контакт между латунным стержнем и гранулой испытывали с помощью вольтметра, чтобы убедиться в надлежащей проводимости. После этого электрод выдерживали при 60°С в течение 12 часов для обеспечения отверждения двухкомпонентного клея. После охлаждения до комнатной температуры электрод полировали наждачной бумагой (зернистость 20 мкм, 14 мкм, 3 мкм и 1 мкм) для получения блестящей гладкой плоской поверхности внутри тефлонового корпуса. После очистки поверхности деионизированной водой и сушки в условиях окружающей среды электрод можно было использовать без дополнительной обработки.
Сравнительный пример 3
Нанолисты MoS2
Объемные кристаллы MoS2 синтезировали способом химического паропереноса. При обычном синтезе порошки элементарных частиц Мо и S смешивали в стехиометрических пропорциях (1:1) и помещали в кварцевую трубку. Из кварцевой трубки откачивали воздух до ~10-6 мбар и запечатывали. Запечатанную кварцевую трубку помещали в трубчатую печь при 800°С на 2 недели для обеспечения образования кристаллов. Кварцевую трубку охлаждали до комнатной температуры и открывали для сбора образовавшихся кристаллов.
Эти кристаллы MoS2 эксфолиировали путем диспергирования 5 мг/мл кристаллов в растворе поверхностно-активного вещества цетилтриметиламмония бромид ЦТАБ (2 мг/мл) в воде с последующей обработкой ультразвуком в течение 10 часов в ультразвуковой ванне мощностью 100 Вт. После обработки ультразвуком дисперсии подвергали дифференциальному центрифугированию для сужения распределения по размерам. В типичном способе дисперсии центрифугировали при 1000 оборотах в минуту в течение 1 часа. Супернатант отделяли и подвергали последовательному центрифугированию при 2000 и 4000 оборотах в минуту на протяжении периодов по 2 часа каждый. На этой стадии процесс останавливали (при 4000 оборотах в минуту). Осадок собирали и повторно диспергировали в воде при обработке ультразвуком. После обработки ультразвуком дисперсия оставалась стабильной в течение 3 месяцев без какой-либо флокуляции и использовалась для дальнейших исследований. Для получения электрода готовили водную суспензию с концентрацией 5 г MoS2 на литр с последующей обработкой ультразвуком в течение 30 минут. Объем 5 мл данной суспензии наносили каплями на полированный стеклоуглеродный электрод с геометрической площадью 0,126 см2 и сушили на воздухе при комнатной температуре. Модифицированные электроды подвергали непрерывному циклированию потенциала в окне потенциалов от -0,5 до 0,5 В относительно Ag/AgCl/3 М KCl до получения воспроизводимых диаграмм вольтамперометрии.
Сравнительный пример 4
Нанолисты NiS2
Нанолисты NiS2 синтезировали одностадийным гидротермическим способом. В типичном способе смешивали 4 ммоль хлорида никеля гексагидрата (NiCl2-6H2O) и 4 ммоль Na2S2O3⋅5H2O в химическом стакане, содержащем 30 мл воды качества Milli-Q и взбалтывали в течение 1 часа. Смешанный раствор переносили в 60-мл покрытый тефлоном автоклав из нержавеющей стали и нагревали в течение 24 часов при 180°С. Осадок собирали центрифугированием и несколько раз промывали смесью этанола и воды (1:2), а затем сушили. Для получения электрода готовили водную суспензию с концентрацией 5 г MoS2 на литр с последующей обработкой ультразвуком в течение 30 минут. Объем 5 мл данной суспензии наносили каплями на полированный стеклоуглеродный электрод с геометрической площадью 0,126 см2 и сушили на воздухе при комнатной температуре. Модифицированные электроды подвергали непрерывному циклированию потенциала в окне потенциалов от -0,5 до 0,5 В относительно Ag/AgCl/3 М KCl до получения воспроизводимых диаграмм вольтамперометрии.
Сравнительный пример 5
Нанолисты FeS2
Нанолисты FeS2 синтезировали одностадийным гидротермическим способом. В типичном способе смешивали 4 ммоль хлорида трехвалентного железа тетрагидрата (FeCl2⋅4H2O) и 4 ммоль Na2S2O3⋅5H2O в химическом стакане, содержащем 30 мл воды качества Milli-Q и взбалтывали в течение 1 часа. Смешанный раствор переносили в 60-мл покрытый тефлоном автоклав из нержавеющей стали и нагревали в течение 24 часов при 180°С. Осадок собирали центрифугированием и несколько раз промывали смесью этанола и воды (1:2), а затем сушили. Для получения электрода готовили водную суспензию с концентрацией 5 г MoS2 на литр с последующей обработкой ультразвуком в течение 30 минут. Объем 5 мл данной суспензии наносили каплями на полированный стеклоуглеродный электрод с геометрической площадью 0,126 см2 и сушили на воздухе при комнатной температуре. Модифицированные электроды подвергали непрерывному циклированию потенциала в окне потенциалов от -0,5 до 0,5 В относительно Ag/AgCl/3 М KCl до получения воспроизводимых диаграмм вольтамперометрии.
Сравнительный пример 6
Pt
В качестве контакта для электродов (диаметром 3 мм) использовали специально изготовленный тефлоновый кожух с латунным стержнем. Измельчали 200 мг платиновой сетки с мелкими ячейками (99,99 мас. %) с получением мелкоизмельченного порошкового материала. Мелкоизмельченный порошок загружали в прессовальный инструмент (диаметром 3 мм) и прессовали порошок с максимальной силой веса 800 кг/см2. На латунный стержень в полости тефлонового кожуха наносили двухкомпонентный эпоксидный клей с серебром для соединения латунной подложки с материалом электрода. Затем гранулу вдавливали в тефлоновый кожух и удаляли любые загрязнения с тефлонового кожуха. Контакт между латунным стержнем и гранулой испытывали с помощью вольтметра, чтобы убедиться в надлежащей проводимости. После этого электрод выдерживали при 60°С в течение 12 часов для обеспечения отверждения двухкомпонентного клея. После охлаждения до комнатной температуры электрод полировали наждачной бумагой (зернистость 20 мкм, 14 мкм, 3 мкм и 1 мкм) для получения блестящей гладкой плоской поверхности внутри тефлонового корпуса. Сетку очищали на первой стадии концентрированной хлористоводородной кислотой, а затем деионизированной водой и сушили в условиях окружающей среды. Электрод можно было использовать без дополнительной обработки.
Условия для электрохимического испытания
Для электрохимического исследования материалов катализатора соответствующий катализатор прессовали в гранулы и вставляли в специально изготовленные электроды со следующими стадиями:
а) материал катализатора (50 мг) измельчали в мелкоизмельченный порошковый материал, загружали в специально созданный прессовальный инструмент (диаметром 3 мм), а затем прессовали с максимальной силой веса 800 кг/см2;
b) осуществляли контакт специально созданного тефлонового кожуха с латунным стержнем в качестве контакта для электродов (диаметром 3 мм); наносили эпоксидный клей с серебром (Polytec ЕС 151 L) на латунный стержень в полости тефлонового кожуха для соединения латунной подложки с материалом электрода;
c) гранулу вдавливали в тефлоновый кожух и удаляли какие-либо загрязнения с тефлонового кожуха;
d) контакт между латунным стержнем и гранулой испытывали с помощью вольтметра (VOLTCRAFT VC840), чтобы убедиться в надлежащей проводимости;
e) электрод выдерживали при 60°С в течение 12 часов для обеспечения отверждения двухкомпонентного клея;
f) электрод охлаждали до комнатной температуры и полировали наждачной бумагой (зернистость 20 мкм, 14 мкм, 3 мкм и 1 мкм, 3М Lapping Film Bogen 261Х/262Х) для получения блестящей гладкой плоской поверхности внутри тефлонового корпуса; после очистки поверхности деионизированной водой (Millipore Milli-Q Academic Water Purification System) и сушки в условиях окружающей среды электрод можно было использовать без дополнительной обработки.
Эксперименты проводили на стандартной трехэлектродной установке с использованием полученного электрода (Агеом.=0,071 см2) в качестве рабочего электрода, электрода из Ag/AgCl (насыщенный раствор KCl или 3 М KCl) в качестве эталонного электрода и проволоки из Pt (диаметр 1 мм) или сетки из Pt (Aгеом.=1,25 см2) в качестве противоэлектрода. Затем специально созданную газонепроницаемую ячейку Н-типа оснащали перемешивающим стержнем и наполняли электролитом, состоящим из 0,5 М H2SO4, для всех электрохимических экспериментов. Замену электролита во время электрохимического испытания электрода не производили. Все потенциалы относятся к ERHE (RHE=обратимый водородный электрод) согласно ERHE=EAg/AgCl+X+0,059 рН, где X=0,197 В (насыщенный раствор KCl), или X=0,210 В (3 М KCl), если не указано иное. В качестве потенциостата использовали прибор Gamry Reference 600+.
Измерение каталитических характеристик
Электрокаталитические эксперименты выполняли со следующими стадиями:
a) электролит (0,5 М H2SO4, 25 мл) добавляли в электрохимическую ячейку и электроды регулировали для обеспечения полного погружения электродов в раствор;
b) включили магнитную мешалку (IKA Topolino);
c) проводили эксперимент по циклической вольтамперометрии (CV) для получения быстрого обзора электрохимических процессов, которые можно было наблюдать;
d) эксперимент CV повторяли, чтобы обеспечить быструю электрохимическую очистку поверхности в диапазоне потенциалов от 0,2 до -0,2 В. Эксперименты проводили со скоростью сканирования 100 мВ/с (некаталитическая область потенциала) и количество циклов устанавливали на 20. Перед началом эксперимента значение компенсации iR для электрохимической установки определяли с использованием потенциостата GAMRY Reference 600 и установленной программы системы программного обеспечения. Диапазон потенциалов для экспериментов вольтамперометрии с линейной разверткой потенциала (LSV (от англ. - linear sweep voltametry) устанавливали от -0,2 до -0,8 В по сравнению с RHE, а скорость сканирования 5 мВ/с с учетом падения iR в эксперименте. Эксперименты с линейной разверткой повторяли по меньшей мере три раза для обеспечения воспроизводимости.
Испытание стабильности и газовый анализ
Для оценивания стабильности материалов согласно примерам электроды держали под постоянным потенциалом (эксперименты с СРС) на протяжении длительного периода времени (по меньшей мере 12 часов). Электроды оценивали в 0,5 М H2SO4 при постоянном потенциале -0,71 В по сравнению с RHE. Кроме того, одновременно отбирали образцы газа из свободного пространства. Для этого к электрохимической ячейке подключали газовый хроматограф (ГХ; система Agilent с системой впрыска JAS), оснащенный детектором теплопроводности. В петлевой дозатор ГХ непрерывно подавали газ из свободного пространство от электролизера. Аргон (10-20 мл минута-1) использовали в качестве газа-носителя для продувки реактора и петлевого дозатора ГХ. Таким образом, определяли количество газообразного водорода, образующегося во время электролиза, и рассчитывали эффективность по Фарадею, коррелируя измеренное количество, полученное из протекающего заряда во время экспериментов с СРС, с максимально теоретически возможным количеством.
Стартстопные характеристики
При работе с возобновляемыми источниками энергии возможность запускать и останавливать электрохимический процесс имеет первостепенное значение. Таким образом, исследовали стартстопные свойства пентландитовых электродов. Для этой цели раскрываемые выше длительные эксперименты (см. тестирование стабильности) с пониженным перенапряжением (-350 мВ по сравнению с RHE или -304 мВ по сравнению с RHE) выполняли с определенными перерывами (таблица 2). Затем измерения автоматически возобновляли при идентичных условиях. Измерения на пентландитовых электродах стандартно сравнивали с платиновым электродом того же диаметра.

Эксперименты по отравлению
Поскольку отравление малыми молекулами (например, H2S) делает многие катализаторы неприменимыми или требует тщательной очистки исходных материалов, авторы настоящего изобретения исследовали применимость пентландитов в качестве стабильных электрокатализаторов также в присутствии обычных катализаторных ядов. Таким образом, длительные эксперименты (см. приведенную выше экспериментальную процедуру «Испытание стабильности и газовый анализ») проводили в присутствии H2S. Для этого электролит постоянно продували H2S, а общее давление поддерживали на уровне 1,5 бар.
Характеристика
Композиции, используемые в соответствии с настоящим изобретением, частично характеризовали применительно к XRD (от англ. - X-ray diffraction -рентгеновская дифракция), DSC (от англ. - differential scanning calorimetry -дифференциальная сканирующая калориметрия), ICP-OES (от англ. - inductively coupled plasma optical emission spectrometry - оптическая эмиссионная спектрометрия с индуктивно связанной плазмой) и SEM-EDX (от англ. - scanning electron microscopy-electron dispersive X-ray spectroscopy - сканирующая электронная микроскопия с электронно-дисперсионной рентгеновской спектроскопией).
Сканирующий электронный микроскоп (SEM (от англ. - scanning electron microscope)) LEO (Zeiss) 1530 Gemini FESEM работал при напряжении 20 кВ (SEM) и 4,4 кВ (электронно-дисперсионная рентгеновская спектроскопия, EDX (от англ. - electron dispersive X-ray spectroscopy)).
Порошковую рентгеновскую дифракцию (PXRD (от англ. - powder X-ray diffraction)) выполняли с использованием дифрактометра от компании HUBER с излучением Мо-Kα (0,709 А) и Bruker Advance D8 с излучением Cu-Kα (0,154 А) при сканировании в диапазоне углов 3-50° с размером шага 0,037 с. Все позиции отражения преобразовывали из излучения Мо в излучение Cu по закону Брэгга.
Состав Fe и Ni для всех различных образцов определяли с помощью оптической эмиссионной спектрометрии с индуктивно связанной плазмой (ICP-OES) с Thermo Scientific iCAP 6500 Duo, оснащенным автоматическим пробоотборником СЕТАС ASX 520. Сбор данных осуществляли на iTEVA. Калибровочные кривые строили по дважды деионизированной воде (ddw (от англ. - double deionized water)) с 3 об.% азотной кислоты в диапазоне 5000-100 частей на миллиард (6 точек). Стандарты и образцы готовили свежими в ddw с 3 об.% азотной кислоты, не содержащей металлов. Показания регистрировали в безгазовом режиме. Образцы (1 мг) разбавляли 3 мл царской водки, не содержащей металлов, помещали в 55-мл сосуды из TFM (от англ. - modified polytetrafluoroethylene - модифицированный политетрафторэтилен) и разлагали в микроволновой печи СЕМ Mars Xpress (160°С, скорость изменения 15 минут, выдержка 15 минут). Затем разлагаемые смеси разбавляли до 10 мл, добавляя дважды деионизированную воду.
Материалы исследовали с помощью дифференциальной сканирующей калориметрии (DSC) с использованием NETZSCH STA 449 F3 Jupiter. Приблизительно 50 мг образца помещали в закрытый корундовый тигль и обрабатывали при температуре от комнатной до 1000°С и наоборот при 10 К минута-1 в непрерывном потоке газообразного N2.
Электрохимическая ячейка
Большинство частей ячейки изготовлены специально, если не указано иное, и поясняются далее со ссылкой на фигуру 8, на которой показано поперечное сечение ячейки. Специально изготовленная электрохимическая ячейка 1 состоит из двух стеклянных отсеков 2, 3. В правом отсеке 3 находятся рабочий электрод 32 и эталонный электрод (36). В левом отсеке 2 находится противоэлектрод 24. Ячейка 1 оснащена специально созданным держателем 4 алюминиевой мембраны, необязательно обернутым лентой из политетрофторэтилена ПТФЭ (обычной герметизирующей лентой), соединяющей части. Держатель 4 мембраны фиксирует мембрану 5 (Fumatech FUMASEP F 10100), отделяющую катод от анодного пространства, двумя резиновыми кольцами 6 (18×5 мм). Держатель 4 мембраны размещается между стеклянными фланцами отсеков 2, 3 ячейки, а алюминиевое удерживающее устройство (трехточечный фиксатор NeoLab KF16) фиксирует все на месте. Отсеки 2, 3 электрохимической ячейки 1 можно термически обрабатывать с помощью жидкостного рециркуляционного охладителя или нагревателя. Специально изготовленные вставки 21, 31 держателя электродов для отсеков ячейки выполнены из полимера полиарилэфирэфиркетон ПЭЭК и имеют просверленные отверстия для размещения электродов 24, 32, 36 и датчика температуры (необязательно) в электролите. Специально изготовленные винты 23, 33 из ПЭЭК и компрессионные кольца (не показаны) служат для фиксирования электродов 24, 32, 36 и необязательных датчиков температуры на месте. Для необязательных устройств 22, 34 продувки газом выполнены дополнительные просверленные отверстия. Вставки крышки фиксируются на месте крышками 25, 35 GL45 SCHOTT с нарезками (соединительные системы Duran).
Результаты Перенапряжение
В таблице 3 кратко характеризуются потенциалы, измеряемые с композициями, используемыми в соответствии с настоящим изобретением и сравнительными примерами. Отрицательные значения являются результатом того, что потенциал измеряли относительно обратимого водородного электрода.
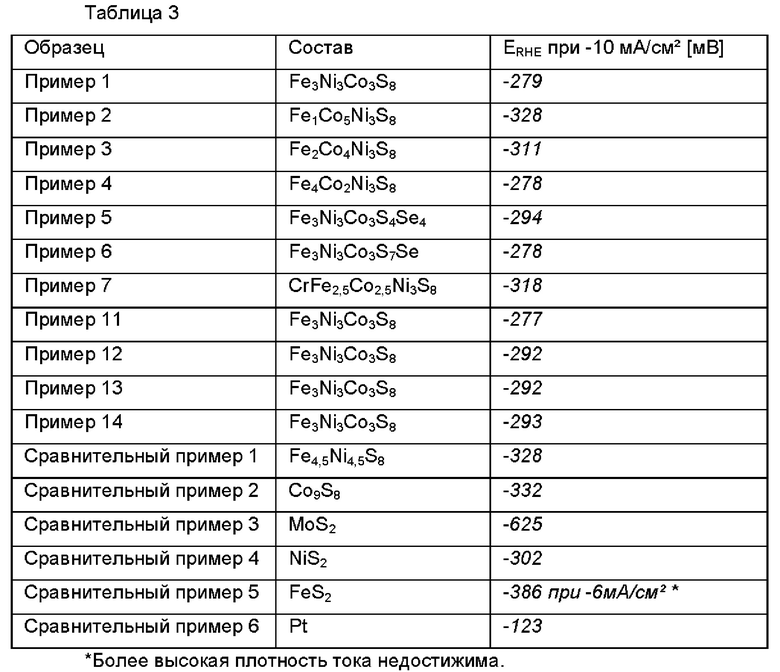
В таблице 3 показано, что композиции, используемые в соответствии с настоящим изобретением, демонстрируют более высокое перенапряжение по сравнению с катализаторами на основе платины, но, что удивительно, преимущественно они демонстрируют более низкое перенапряжение для HER по сравнению с другим известным из уровня техники катализатором (сравнительный пример 1).
На фигурах 1А - 1Е представлены диаграммы вольтамперометрии с линейной разверткой потенциала, полученные в диапазоне сканирования от 0 до -0,65 В по сравнению с RHE в 0,5 М серной кислоты при комнатной температуре. По оси X показан потенциал в В, а по оси Y показана плотность тока в мА см-2.
Фиг. 1А: пример 1 (сплошная линия), сравнительный пример 1 (штриховая линия), сравнительный пример 2 (пунктирная линия), сравнительный пример 3 (штрихпунктирная линия), сравнительный пример 4 (штрих-дважды пунктирная линия), сравнительный пример 5 (короткопунктирная линия) и сравнительный пример 6 (короткопунктирная линия).
Фиг. 1В: пример 1 (сплошная линия), пример 3 (пунктирная линия), пример 4 (штрихпунктирная линия) и сравнительный пример 6 (короткопунктирная линия).
Фиг. 1С: пример 1 (сплошная линия), пример 5 (штрих-дважды пунктирная линия), пример 6 (короткопунктирная линия) и сравнительный пример 6 (короткопунктирная линия).
Фиг. 1D: пример 1 (сплошная линия), пример 11 (штриховая линия), пример 12/13 (пунктирная линия), пример 14 (штрихпунктирная линия) и сравнительный пример 6 (короткопунктирная линия).
Фиг. 1Е: пример 1 (сплошная линия), пример 7 (штриховая линия), сравнительный пример 1 (короткопунктирная линия) и сравнительный пример 6 (короткопунктирная линия).
Плотности тока после нескольких часов электролиза
Композиции, используемые в соответствии с настоящим изобретением, демонстрируют более высокое перенапряжение для HER по сравнению с катализаторами на основе платины, но, что удивительно, они демонстрируют более высокие плотности тока, чем материалы на основе платины, после нескольких часов электролиза. При более высоких плотностях тока можно получать большее количество водорода с количественной эффективностью Фарадея. Следовательно, промышленное получение водорода с использованием этих композиций более эффективно и более дешевое, чем с коммерческими катализаторами.
На фигуре 2 показаны результаты измерений кулонометрии с управляемым потенциалом при -0,5 В по сравнению с RHE при 20°С и 60°С для электродов на основе Pt (сравнительный пример 6) и Fe3Co3Ni3S8 (пример 1) в течение 20 часов. Показаны кривые примера 1 при 20°С (шашка) и при 60°С (квадрат), а также сравнительного примера 6 при 20°С (треугольник) и 60°С (кружок).
По оси X показано время в часах, а по оси Y показана плотность тока в мА см-2.
Уровень активности при получении водорода
Известные из уровня техники материалы теряют значительную активность во время использования по сравнению с их свежим состоянием. Удивительно, что электроды, полученные из композиций, используемых в соответствии с настоящим изобретением, не теряют значительной активности во время получения водорода, как показано на фигуре 3.
На фигуре 3 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой при 20°С (А) и 60°С (В) до и после 20-часового электролиза для платины (сравнительный пример 6) и Fe3Co3Ni3S8 (пример 1).
Фигура 3А: пример 1 до (сплошная линия) и после (штрихпунктирная линия) и сравнительный пример 6 до (штриховая линия) и после (пунктирная линия) электролиза при 20°С.
Фигура 3 В: пример 1 до (сплошная линия) и после (штрихпунктирная линия) и сравнительный пример 6 до (штриховая линия) и после (пунктирная линия) электролиза при 60°С.
По оси X в каждом случае показан потенциал в В, а по оси Y показана плотность тока в мА см-2.
Стартстопная характеристика
Стартстопная характеристика - по сравнению со сравнительным примером 6
Удивительно то, что электроды, полученные из композиций, используемых в соответствии с настоящим изобретением, обладают значительно лучшими стартстопными характеристиками, чем платиновые электроды, о чем свидетельствуют фигуры 4 и 9. Тогда как плотность тока платины снижается до менее чем 20% от ее начальной плотности тока, композиции, используемые в соответствии с настоящим изобретением, могут сохранять значительно большее значение своих исходных плотностей тока. Благодаря этим улучшенным стартстопным характеристикам можно в полной мере использовать максимумы возобновляемых источников энергии (например, ветра или фотоэлектрической системы) для устойчивого получения водорода.
На фигуре 4 показана кулонометрия с управляемым потенциалом в стартстопном режиме, измеряемая при 60°С и -304 мВ по сравнению с RHE на протяжении 72 часов для Fe3Co3Ni3S8 (пример 1, квадрат) и платины (сравнительный пример 6, шашка).
По оси X показано время в часах, а по оси Y показана плотность тока в мА см-2.
На фигуре 9А также показана кулонометрия с управляемым потенциалом в стартстопном режиме для Fe3Co3Ni3S8 (пример 1, квадрат) и платины (сравнительный пример 6, шашка), однако измеренная при 60°С и -350 мВ по сравнению с RHE на протяжении 72 часов. Плотности тока в примере 1 и сравнительном примере 6 на фигуре 4 являются более низкими по сравнению с полученными плотностями тока с теми же катализаторами на фигуре 9А. Эти различия возникают из-за более низкого потенциала, приложенного при измерении на фигуре 4 (-304 мВ) по сравнению с фигурами 9А - 9Е (-350 мВ). Тем не менее, на обеих фигурах (фиг. 4 и 9А) показано, что стартстопная характеристика в примере 1 превосходит таковую в сравнительном примере 6.
На фигурах 9В - 9Е показана кулонометрия с управляемым потенциалом в стартстопном режиме, измеряемая при 60°С и -350 мВ по сравнению с RHE на протяжении 72 часов. При измерении с одним и тем же набором параметров результаты, показанные на фигурах 9А и 9В - 9Е, могут быть сравнимы друг с другом.
Фигура 9В: пример 2 (квадрат) и сравнительный пример 6 (шашка).
Фигура 9С: пример 4 (квадрат) и сравнительный пример 6 (шашка).
Фигура 9D: пример 6 (квадрат) и сравнительный пример 6 (шашка).
Фигура 9Е: пример 7 (квадрат) и сравнительный пример 6 (шашка).
По оси X показано время в часах, а по оси Y показана плотность тока в мА см-2.
Стартстопная характеристика - по сравнению со сравнительным примером 1
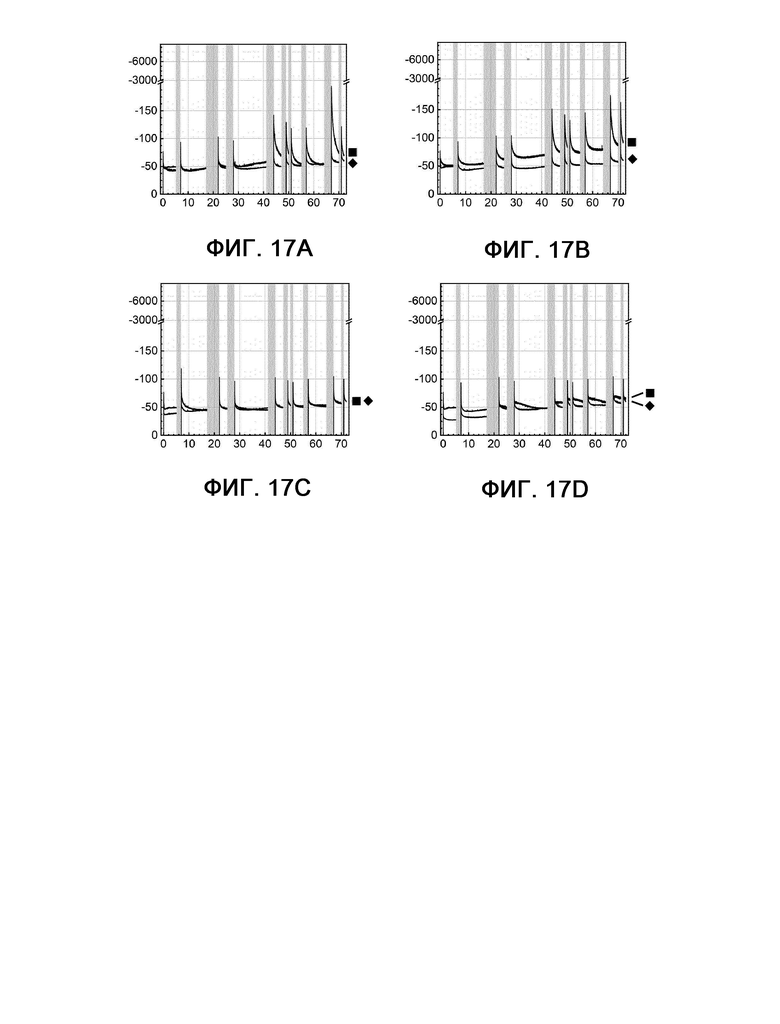
Удивительно то, что электроды, полученные из композиций, используемых в соответствии с настоящим изобретением, также обладают схожими или даже лучшими стартстопными характеристиками, чем электроды из других известных из уровня техники композиций, а именно Fe4,5Ni4,5S8 (сравнительный пример 1), как представлено на фигуре 17. Композиции, используемые в соответствии с настоящим изобретением, могут сохранять такое же или даже большее значение своих исходных плотностей тока по сравнению с композицией сравнительного примера 1, особенно после нескольких циклов старт/стоп. Благодаря этим улучшенным стартстопным характеристикам можно в полной мере использовать максимумы возобновляемых источников энергии (например, ветра или фотоэлектрической системы) для устойчивого получения водорода.
На фигурах 17А - 17D показана кулонометрия с управляемым потенциалом в стартстопном режиме, измеряемая при 60°С и -350 мВ по сравнению с RHE на протяжении 72 часов.
Фигура 17А: пример 1 (квадрат) и сравнительный пример 1 (шашка).
Фигура 17В: пример 2 (квадрат) и сравнительный пример 1 (шашка).
Фигура 17С: пример 4 (квадрат) и сравнительный пример 1 (шашка).
Фигура 17D: пример 7 (квадрат) и сравнительный пример 1 (шашка).
По оси X показано время в часах, а по оси Y показана плотность тока в мА см-2.
Стабильность в отношении отравления серой
Стабильность в отношении отравления серой - по сравнению со сравнительным примером 6
Следующим преимуществом композиций, используемых в соответствии с настоящим изобретением, является стабильность в отношении отравления серой (фигуры 5, 6 и 10-16). Тогда как платина очень чувствительна к соединениям серы и теряет активность, композиции, используемые в соответствии с настоящим изобретением, демонстрируют почти такую же или даже более высокую (например, фиг. 10 и 12) активность при обработке соединениями серы. Следующий эффект соединений серы заключается в том, что отравление платиновых электродов является необратимым эффектом, в то время как на композиции, используемые в соответствии с настоящим изобретением, соединения серы оказывают обратимое действие. Следовательно, необходима очень чистая вода в качестве исходных материала для электролизеров РЕМ с платиной в качестве электрокатализатора для HER, в то время как с композициями, используемыми в соответствии с настоящим изобретением, можно использовать различные источники воды.
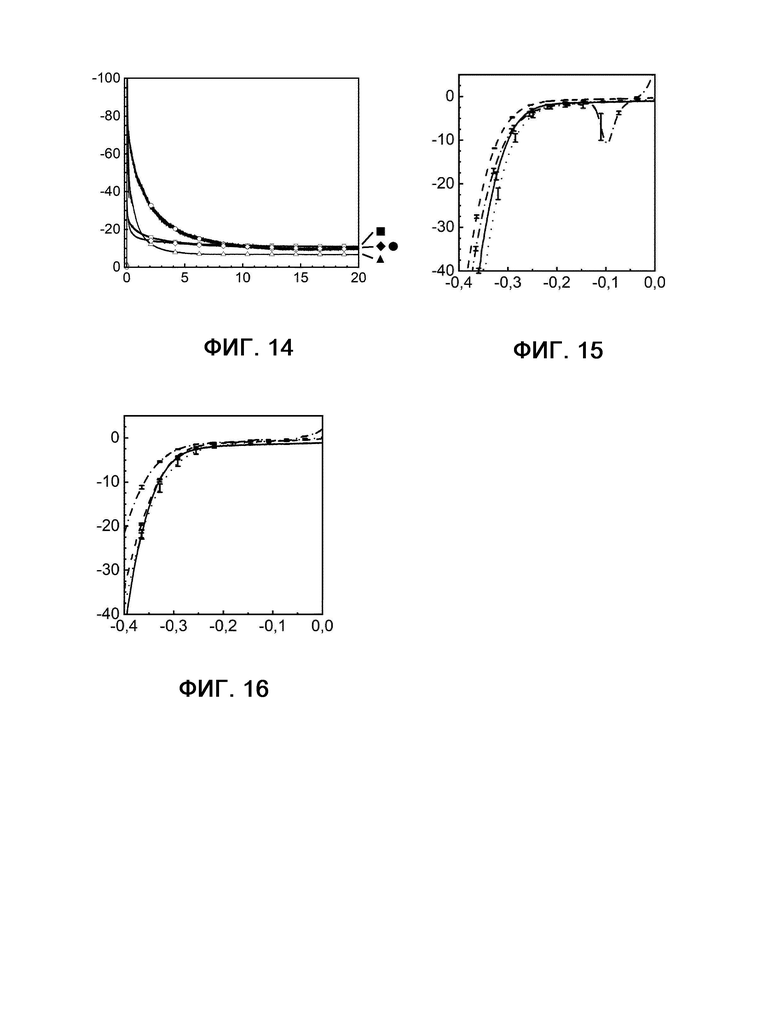
На фигурах 5, 10, 12 и 14 показана кулонометрия с управляемым потенциалом в присутствии и в отсутствие катализаторного яда H2S, измеряемая при 20°С и -350 мВ по сравнению с RHE на протяжении 20 часов.
Фигура 5: кривые примера 1 без присутствия (квадрат) и в присутствии (шашка) H2S, а также сравнительного примера 6 без присутствия (кружок) и в присутствии (треугольник) H2S.
Фигура 10: кривые примера 2 без присутствия (квадрат) и в присутствии (шашка) H2S, а также сравнительного примера 6 без присутствия (кружок) и в присутствии (треугольник) H2S.
Фигура 12: кривые примера 4 без присутствия (квадрат) и в присутствии (шашка) H2S, а также сравнительного примера 6 без присутствия (кружок) и в присутствии (треугольник) H2S.
Фигура 14: кривые примера 6 без присутствия (квадрат) и в присутствии (шашка) H2S, а также сравнительного примера 6 без присутствия (кружок) и в присутствии (треугольник) H2S.
По оси X показано время в часах, а по оси Y показана плотность тока в мА см-2.
На фигурах 6, 11, 13, 15 и 16 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
Фигура 6А: кривые примера 1 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 1 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
Фигура 6В: кривые сравнительного примера 6 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми сравнительного примера 6 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
Фигура 11: кривые примера 2 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 2 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
Фигура 13: кривые примера 4 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 4 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
Фигура 15: кривые примера 6 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 6 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
Фигура 16: кривые примера 7 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 7 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
По оси X в каждом случае показан потенциал в В, а по оси Y показана плотность тока в мА см-2.
Стабильность в отношении отравления серой - по сравнению со сравнительным примером 1
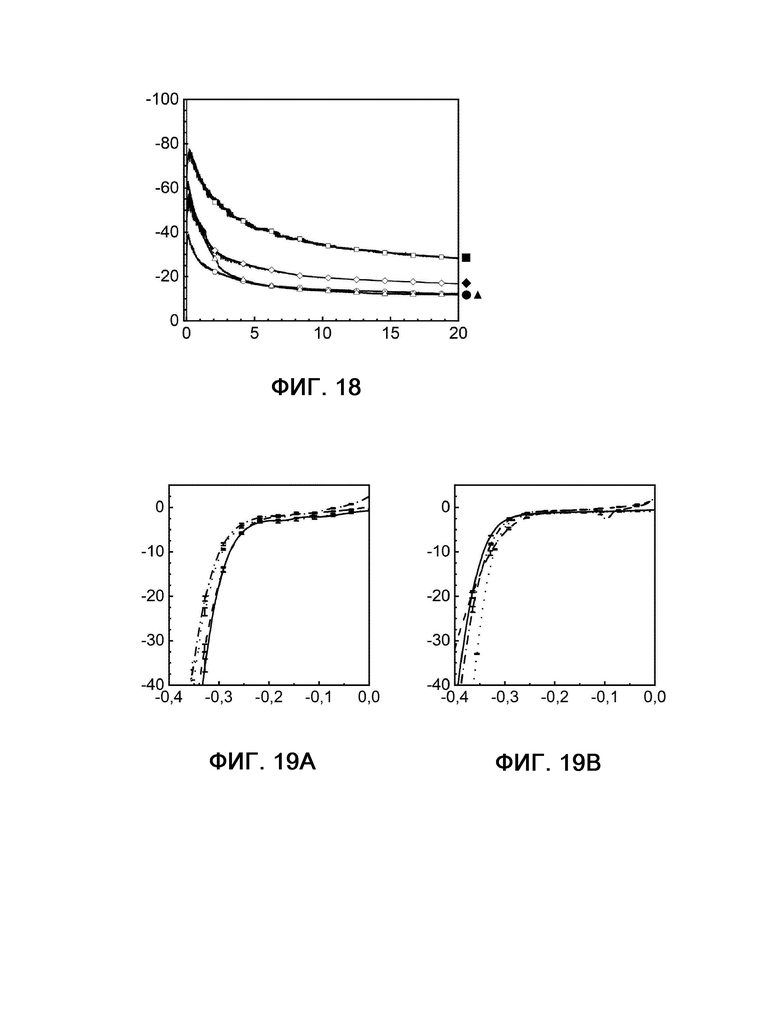
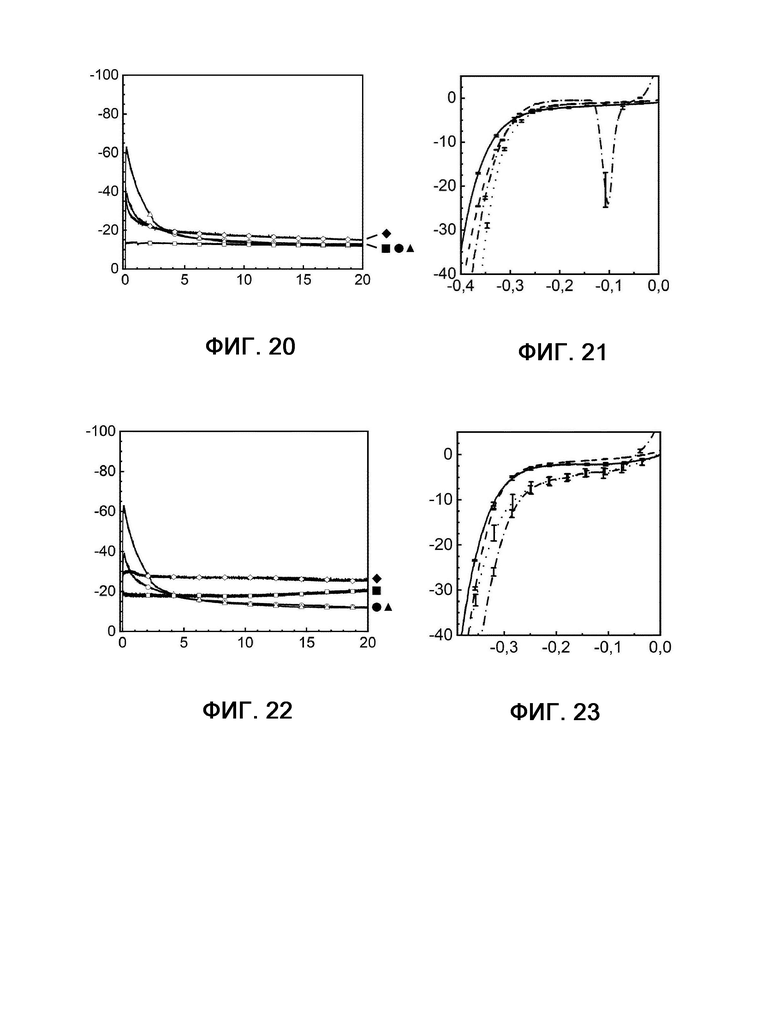
Стабильность в отношении отравления серой композиций, используемых в соответствии с настоящим изобретением, далее сравнивают с другим известным из уровня техники электродной композицией, а именно Fe45Ni4,5S8 (сравнительный пример 1) (фигуры 18-25). Композиции, используемые в соответствии с настоящим изобретением, демонстрируют сопоставимую или даже лучшую активность при обработке соединениями серы по сравнению со сравнительным примером 1.
На фигурах 18, 20 и 22 показана кулонометрия с управляемым потенциалом в присутствии и в отсутствие катализаторного яда H2S, измеряемая при 20°С и -350 мВ по сравнению с RHE на протяжении 20 часов.
Фигура 18: кривые примера 1 без присутствия (квадрат) и в присутствии (шашка) H2S, а также сравнительного примера 1 без присутствия (кружок) и в присутствии (треугольник) H2S.
Фигура 20: кривые примера 2 без присутствия (квадрат) и в присутствии (шашка) H2S, а также сравнительного примера 1 без присутствия (кружок) и в присутствии (треугольник) H2S.
Фигура 22: кривые примера 4 без присутствия (квадрат) и в присутствии (шашка) H2S, а также сравнительного примера 1 без присутствия (кружок) и в присутствии (треугольник) H2S.
По оси X показано время в часах, а по оси Y показана плотность тока в мА см-2.
На фигурах 19, 21, 23, 24 и 25 показаны диаграммы вольтамперометрии с линейной разверткой потенциала, выполняемой в присутствии и в отсутствие катализаторного яда H2S с измерением при 20°С и -350 мВ по сравнению с RHE до и после 20-часового электролиза.
Фигура 19А: кривые примера 1 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 1 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
Фигура 19 В: кривые сравнительного примера 1 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми сравнительного примера 1 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
Фигура 21: кривые примера 2 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 2 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
Фигура 23: кривые примера 4 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 4 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
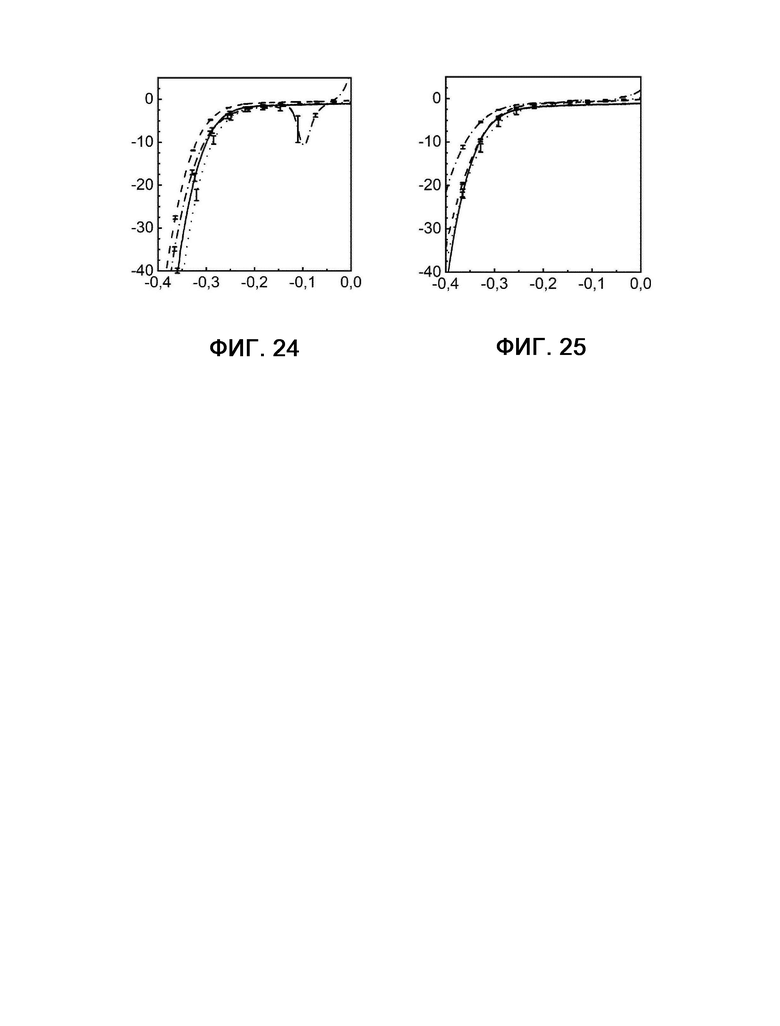
Фигура 24: кривые примера 6 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 6 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
Фигура 25: кривые примера 7 в отсутствие H2S до (сплошная линия) и после (штриховая линия) электролиза рядом с кривыми примера 7 в присутствии H2S до (пунктирная линия) и после (штрихпунктирная линия) электролиза.
По оси X в каждом случае показан потенциал в В, а по оси Y показана плотность тока в мА см-2.
Порошковая рентгенограмма (паттерны дифракции)
На фигуре 7 показана порошковая рентгенограмма (паттерны дифракции, (излучение Cu-Kα)) композиций, используемых в соответствии с настоящим изобретением и сравнительными примерами.
Фиг. 7А: паттерны примера 1 (квадрат), сравнительного примера 1 (треугольник), сравнительного примера 2 (кружок), сравнительного примера 3 (шашка), сравнительного примера 4 (незаштрихованный квадрат) и сравнительного примера 5 (незаштрихованный кружок).
Фигура 7В: пример 1 (квадрат), пример 3 (кружок) и пример 4 (шашка).
Фигура 7С: пример 1 (квадрат), пример 5 (треугольник) и пример 6 (кружок).
Фигура 7D: пример 1 (квадрат), пример 11 (треугольник), примеры 12/13 (кружок) и пример 14 (шашка).
Фигура 7Е: пример 1 (квадрат) и пример 7 (треугольник).
По осям X в каждом случае показаны углы 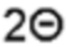 в°, а по осям Y - интенсивность в безразмерных величинах.
в°, а по осям Y - интенсивность в безразмерных величинах.
Из фиг. 7А видно, что композиции, используемые в соответствии с настоящим изобретением (например, в примере 1, квадрат), демонстрирую паттерн XRD, очень похожий на таковой немодифицированного пентландита (сравнительный пример 1, треугольник).
Удивительно, что композиции, используемые в соответствии с настоящим изобретением, работают в широкой области рН от 0 до 14 при приложении повышенных потенциалов, тогда как другие сульфидные композиции с железом, кобальтом или никелем не стабильны в такой широкой области рН. Пентландит как кристаллическая фаза, по-видимому, является причиной такой высокой стабильности.
Композиции, используемые в соответствии с настоящим изобретением, применимы сами по себе или в комбинации с электропроводящим материалом подложки (например, графеном) в качестве электрокатализатора для реакции выделения водорода в электролизере РЕМ.
Композиция, используемая в соответствии с настоящим изобретением, также может быть применима для получения водорода в электролизе в щелочной воде или с электролизерами высокого давления.
Настоящее изобретение относится к применению композиции формулы I
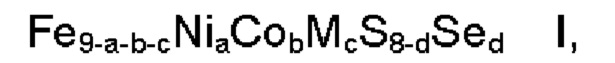
где М обозначает один или несколько элементов, характеризующихся в ионном состоянии эффективным ионным радиусом в диапазоне 70-92 пм, а представляет собой число в диапазоне 2,5≤а≤3,5, b представляет собой число в диапазоне 1,5≤b≤5,0, с представляет собой число в диапазоне 0,0≤с≤2,0, d представляет собой число в диапазоне 0,0≤d≤4,0, где сумма a, b и с находится в диапазоне 5≤а+b+с≤8, и где ≥90 масс. % композиции находится в пентландитовой фазе, для электрокаталитического расщепления воды. Также изобретение относится к электроду. Использование предлагаемого изобретения позволяет использовать более высокую плотность тока, также композиция обладает лучшей устойчивостью к соединениям серы и лучшими стартстопными свойствами по сравнению с материалами известного уровня техники. 2 н. и 7 з.п. ф-лы, 25 ил., 3 табл., 14 пр.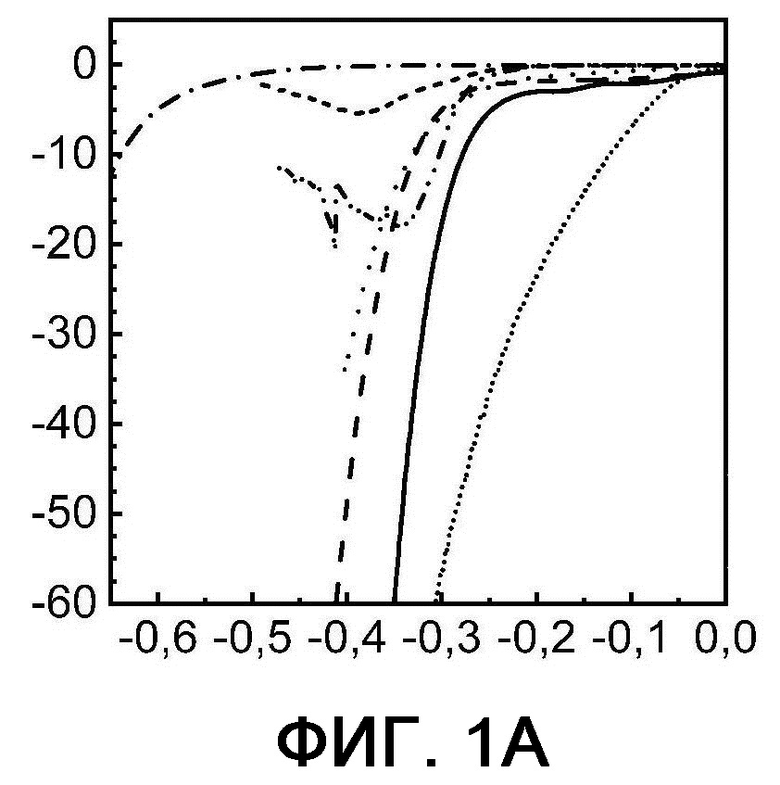
1. Применение композиции формулы
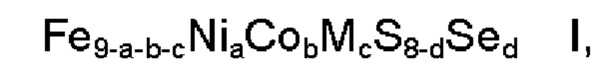
где
М обозначает один или более элементов, характеризующихся в ионном состоянии эффективным ионным радиусом в диапазоне от 70 до 92 пм,
а представляет собой число в диапазоне 2,5≤а≤3,5, более предпочтительно 2,7≤а≤3,3,
b представляет собой число в диапазоне 1,5≤b≤5,0, более предпочтительно 1,5≤b≤4,0, наиболее предпочтительно 2,5≤b≤3,5,
с представляет собой число в диапазоне 0,0≤с≤2,0, более предпочтительно 0,0≤с≤1,0,
d представляет собой число в диапазоне 0,0≤d≤4,0, более предпочтительно 0,0≤d≤1,0,
где сумма a, b и с находится в диапазоне 5≤а+b+с≤8,
и где ≥90 масс. % композиции находится в пентландитовой фазе, для электрокаталитического расщепления воды, предпочтительно для реакции выделения водорода.
2. Применение по п. 1, где М не присутствует.
3. Применение по п. 2, где композиция выбрана из группы, состоящей из
Fe3Ni3Co3S8,
Fe1Co5Ni3S8,
Fe2Co4Ni3S8,
Fe4Co2Ni3S8,
Fe3Ni3Co3S4Se4,
Fe3Ni3Co3S7Se
и их смесей.
4. Применение по п. 1, где М присутствует.
5. Применение по п. 4, где М выбран из группы, состоящей из Nb, Cu, Mn или Cr и их смесей, более предпочтительно из Mn, Cr и их смесей, наиболее предпочтительно из Cr.
6. Применение по п. 5, где композиция выбрана из группы, состоящей из
CrFe2,5Co2,5Ni3S8,
MnFe2,5Co2,5Ni3S8,
Mn0,25Fe2,875Co2,875Ni3S8,
NbFe2,5Co2,5Ni3S8
и их смесей.
7. Применение по любому из предыдущих пунктов, отличающееся тем, что композиция характеризуется перенапряжением для реакции выделения водорода (HER) 328 мВ и меньше, предпочтительно 300 мВ и меньше, при плотности тока -10 мА/см2.
8. Электрод, содержащий композицию формулы I, как определено в любом из предыдущих пунктов.
9. Электрод по п. 8, отличающийся тем, что характеризуется перенапряжением для реакции выделения водорода (HER) 328 мВ и меньше при плотности тока -10 мА/см2.
| KLEIN ET AL., CHEMICAL COMPOSITION OF PENTLANDITE OF ODP HOLES, PAGES 3-7, 01.01.2010 | |||
| HELMUT SCHROCKE ET AL., MINERALOGIE: EIN LEHRBUCH AUF SYSTEMATISCHER GRUNDLAGE, PAGE 137, 01.01.1981 | |||
| ANONYMOUS: "PENTLANTIDE", MINERAL DATA PUBLISHING, 01.01.2001, PAGE 307, RETRIEVED FROM THE INTERNET: |

