Изобретение относится к аналитической химии, а именно к анализу объектов природного и техногенного происхождения методом инверсионной вольтамперометрии для определения ионов осмия, и может быть использовано для его определения в присутствии растворенного кислорода в объектах природного и техногенного происхождения.
Изучено электрохимическое поведение осмия в присутствии пероксида водорода на разных фоновых электролитах: кислотные растворы, нейтральные и буферные растворы. Показана возможность определения осмия полярографическим методом с содержанием до 1·10-7 М. [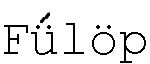 K. and Csányi L.J. On the polarographic study of the system hydrogen peroxide-osmium tetroxide // Acta Chimica Hungaricae, 1963. Т.38, №3, р.193-202]. Недостатком данного метода является то, что для исследуемых растворов осмия наблюдается смещение потенциала максимума в область положительных потенциалов и максимум осмия раздваивается. Одновременно нарушается пропорциональная зависимость между величиной тока и концентрацией осмия в растворе. Для устранения этих недостатков авторы предлагают использовать буферные растворы с pH ~ 9-10 с добавкой тиосульфата для стабилизации растворов, содержащих осмий.
K. and Csányi L.J. On the polarographic study of the system hydrogen peroxide-osmium tetroxide // Acta Chimica Hungaricae, 1963. Т.38, №3, р.193-202]. Недостатком данного метода является то, что для исследуемых растворов осмия наблюдается смещение потенциала максимума в область положительных потенциалов и максимум осмия раздваивается. Одновременно нарушается пропорциональная зависимость между величиной тока и концентрацией осмия в растворе. Для устранения этих недостатков авторы предлагают использовать буферные растворы с pH ~ 9-10 с добавкой тиосульфата для стабилизации растворов, содержащих осмий.
Известна методика определения осмия на платиновом или графитовом вращающемся электроде. Электродом сравнения служил нас. кал. электрод, время электролиза 10 минут при потенциале электролиза 0,0 В (для графитового электрода). Интервал определяемых содержаний составляет (1,2-4,0)·10-5 г-ион/л (25 мл - 2-7,6 мкг/мл). В случае разбавленных растворов используют платиновый электрод, время электролиза 30 минут при потенциале минус 0,3 В. Интервал определяемых содержаний составляет (2,0-8,0)·10-6 г-ион/л [Усатенко Ю.Н., Швыдка Л.Ф., Толубара А.И. и Тулюпа Ф.М. Пленочная полярография с концентрированием осмия на твердых электродах. // Известия ВУЗов. Серия: Химия и химическая технология. - 1969, т.12, №12, с.1654-1657]. Недостатком является проблема изготовления платинового электрода для других модификаций применяемых приборов. Низкая чувствительность даже в применении увеличенного времени электролиза (от 10 до 30 минут). Наблюдается уменьшение анодного тока при введении в раствор ацетат-, хлорид-иона и лимонной кислоты. Также влияние оказывает рутений при более, трехкратном избытке.
Разработана методика с использованием смесового фона 0,1 М KCl+xM KOH (рН 9-10). Используется графитовый или платиновый микродисковый электрод. Потенциал электролиза составляет минус 0,7 В и время накопления - 6 минут. Минимально определяемая концентрация составляет 1·10-7 М [Бардин М.Б., Горчаренко В.П. и Болоцкая Ф.З. Определение малых количеств осмия методом пленочной вольтамперометрии. // Журнал аналитической химии. - 1970, т.25, вып.11, с.2160-2166]. Недостатком является строгое выдерживание pH раствора (10, так как при рН 4-8 чувствительность падает, а при pH>12 вольтамперные кривые искажаются и максимум раздваивается, что приводит к нарушению пропорциональности между концентрацией осмия в растворе и величиной максимального тока. Для этого авторы рекомендуют использовать буферные растворы с pH 9-10 с добавками тиосульфатов, оксалатов или тартратов, что делает фоновый раствор многосоставным. Также указывается на мешающее влияние рутения, приводящего к искажению вольтамперных кривых.
Известна методика определения осмия методом ИВ с использованием графитового электрода, ацетатного буферного раствора с pH 4,6. Методика разработана для медно-никелевых сульфидных руд. Нижняя граница определяемых содержаний составила 5·10-6%. [Колпакова Н.А., Швец Л.А. Определение осмия методом инверсионной вольтамперометрии в минеральном сырье. // Журнал аналитической химии, - 1983, т.38, вып.8, с.1858-1862]. Недостатком является то, что для стабилизации растворов используют тиосульфат и определение осмия возможно при не более чем 25-кратном количестве рутения. В отгонке используется HClO4, которая при разложении образует HCl, что приводит к образованию летучих хлоридов (AsCl3, SbCl5, BiCl3 и др.), мешающих определению осмия и искажающих вид вольтамперных кривых.
Разработана методика с использованием в качестве индикаторного электрода графитового, а, электрода сравнения использовался каломельный. Фоном служила муравьиная кислота концентрацией 0,01 М. Нижняя граница определяемых содержаний осмия составила 2·10-8 М [Швец Л.А., Колпакова Н.А. Определение осмия методом инверсионной вольтамперометрии в технологических продуктах. // Журнал аналитической химии, - 1983, т.38, вып.8, с.1858-1862]. Недостатком является то, что проводят продувку анализируемого раствора азотом. При этом происходит улетучивание OsO4 и занижение результатов анализа. Вследствие этого выбирают соответствующее время накопления в зависимости от содержания осмия в технологическом продукте от 1 до 10 минут.
Разработан гибридный вариант метода инверсионной вольтамперометрии - кинетический метод инверсионной вольтамперометрии (Кин-ИВ). Метод реализован на примере определения осмия. Установлено, что при электроокислении Os+OsO2, предварительно осажденного на поверхность графитового электрода, в кислых электролитах и в присутствии пероксида водорода на анодных вольтамперных кривых наблюдаются «обратные пики». Показано, что определение осмия методом Кин-ИВ можно проводить в сложных матрицах без его отделения. Нижняя граница определяемых содержаний осмия на фоне 0,1 М серной кислоты при концентрации пероксида водорода 30% равна 8·10-10 моль/л [Колпакова Н.А., Каминская О.В., Яговкина Е.В. «Определение осмия в минеральном сырье методом кинетической инверсионной вольтамперометрии» // Заводская лаборатория. Диагностика материалов. - 1997, т.64, №4, с.9-12]. Основным недостатком является мешающее влияние серебра и 105-кратные избытки неблагородных компонентов. Без отделения осмия дистилляцией мешают как все благородные металлы, так и некоторые компоненты пробы (Fe, Se, Те, As и др.).
Для инверсионно-вольтамперометрического определения осмия в природных и промышленных объектах была разработана методика [Колпакова Н.А., Кропоткина С.В., Сухомлинова О.В. Авторское свидетельство №1746285, 07.07.1992 г. Бюл. №25 // Инверсионно-вольтамперометрический способ определения осмия в природных и промышленных объектах] (прототип). Определение осмия проводят по следующей методике. Для определения осмия в качестве рабочего электрода был использован графитовый электрод, а электродом сравнения служил каломельный. Фоновым электролитом была 0,01 М муравьиная кислота. Потенциал электролиза составлял 0,0 В при времени электролиза 1 минута. Потенциал обратного максимума регистрировали в кислой среде и в присутствии 10-3 - 30% перекиси водорода от 0,4 до 0,5 В при скорости развертки 50 мВ/с. Недостатком данного метода является то, что ток обратного максимума сильно зависит от концентрации перекиси водорода. Низкая чувствительность определения осмия в бедных продуктах с содержанием 10-7-10-6%. Очень сильное влияние оказывает серебро, искажающее вид вольтамперной кривой.
В работе была поставлена задача снизить предел и нижнюю границу определяемых содержаний осмия (VIII) по пику электрокаталитического разложения пероксида водорода, полученному после электроконцентрирования OsO2 на сажевом электроде (СЭ) методом ИВ.
Поставленная задача достигается тем, что ионы осмия (VIII) электрохимически концентрируют на поверхности различных типов сажевых электродов в форме OsO2 с последующим разложением пероксида водорода на осадке и регистрацией анодных вольтамперных кривых с обратным пиком. Проводят накопление ионов осмия на сажевом электроде в перемешиваемом растворе в присутствии пероксида водорода в течение 60-120 секунд с последующей регистрацией анодных пиков электрокаталитического разложения пероксида водорода на осадке OsO2 при скорости развертки потенциала 80-120 мВ/с при потенциалах электролиза минус 0,2 В на фоновом электролите 0,05-0,1 М CH3COOH, концентрацию ионов осмия определяют по высоте анодного обратного пика каталитического разложения пероксида водорода на вольтамперной кривой в диапазоне потенциалов от плюс 0,1 до плюс 0,3 В относительно насыщенного хлоридсеребряного электрода методом добавок аттестованных смесей.
Новым в способе является то, что для получения полезного сигнала, зависящего от концентрации осмия (VIII), используется избыток пероксида водорода для полноты осуществления каталитической реакции ее разложения на осадке, полученном в ходе электроконцентрирования Os из раствора.
В предлагаемом способе установлена способность осадка осмия разлагать пероксид водорода на поверхности различных типов сажевых электродов. В качестве индикаторного применяли СЭ (в прототипе применяли графитовый электрод). Использование таких электродов обусловлено высокой химической и электрохимической устойчивостью сажи, широкой областью рабочих потенциалов, а также простотой механического обновления поверхности и требованиями техники безопасности. В анализе не используется ртуть, т.к. ртуть токсична. Нижняя граница определяемых содержаний по данному методу составила 1·10-4 мг/дм3 (в прототипе 1·10-3 мг/дм3).
Результаты определения осмия (VIII) на СЭ приведены в таблице 1 и таблице 2. Как видно из таблицы 1, максимальная погрешность измерений составляет порядка 15,8%, в стандартных образцах она составила ~40%. Расчет определяемых концентраций осмия проводится по методу «Введено-найдено».
Проводят накопление ионов осмия (VIII) на поверхность сажевого электрода в перемешиваемом растворе в присутствии пероксида водорода в течение 60-120 с при потенциале электролиза минус 0,2 В. Дальнейшее уменьшение потенциала электролиза вызывает уменьшение пика осмия (VIII), что снижает чувствительность определения. Измерения проводились на фоне 0,05-0,1 М CH3COOH с последующей регистрацией анодных пиков в накопительном режиме и съемки вольтамперограмм при скорости развертки 80-120 мВ/с. Концентрацию ионов осмия (VIII) определяют по высоте анодного обратного пика разложения пероксида водорода в диапазоне потенциалов от плюс 0,1 до плюс 0,3 В относительно насыщенного хлоридсеребряного электрода (нас.х.с.). На фиг.1 представлены вольтамперные кривые электрокаталитического разложения пероксида водорода на осадке OsO2, предварительно электроосажденного на поверхности сажевого электрода на фоне 0,1 М CH3COOH. Кривая 1 - фон 0,1 М CH3COOH+1 мл 30% H2O2, кривая 2 - COs(VIII) равно 0,002 мг/дм3, кривая 3 - COs(VIII) равно 0,004 мг/дм3.
Таким образом, установленные условия впервые позволили количественно определять содержание ионов осмия (VIII) на основе реакции каталитического разложения пероксида водорода с выделением кислорода на поверхности сажевого электрода.
Предлагаемый вольтамперометрический способ позволил существенно улучшить метрологические характеристики анализа осмия (VIII); повысить чувствительность определения (1·10-4 мг/дм3), что на порядок ниже по сравнению с прототипом.
Примеры конкретного выполнения
Пример 1. Измерения были проведены на искусственных смесях (фиг.1). 10 мл фонового электролита (0,1 М CH3COOH) помещают в кварцевый стаканчик и добавляют 1 мл 30% H2O3. Не прекращая перемешивания, проводят электролиз раствора, при Еэ=-0,2 В и при τэ=60 сек, снимают вольтамперную кривую электрокаталитического разложения пероксида водорода при скорости развертки 100 мВ/с. Отсутствие пиков на вольтамперной кривой в интервале от плюс 0,1 до плюс 0,3 В свидетельствует о чистоте фона. Затем добавляют аттестованный раствор осмия (VIII) 0,02 мл из 0,1 мг/дм3 и проводят электрохимическое концентрирование осадка при аналогичных условиях. На вольтамперной кривой в интервале от плюс 0,1 до плюс 0,3 В появляется пик электрокаталитического разложения пероксида водорода. Вносили добавку стандартного раствора осмия (VIII) 0,02 мл из 0,1 мг/дм3 и снова регистрировали аналитический сигнал при потенциале накопления -0,2 В. По разнице токов пиков каталитического разложения пероксида водорода вычисляли концентрацию осмия в растворе. Пик тока каталитического разложения пероксида водорода на осадке OsO2 предварительно электроосажденного на поверхности сажевого электрода регистрировали в диапазоне потенциалов от плюс 0,1 до плюс 0,3 В (отн. нас.х.с.э.).
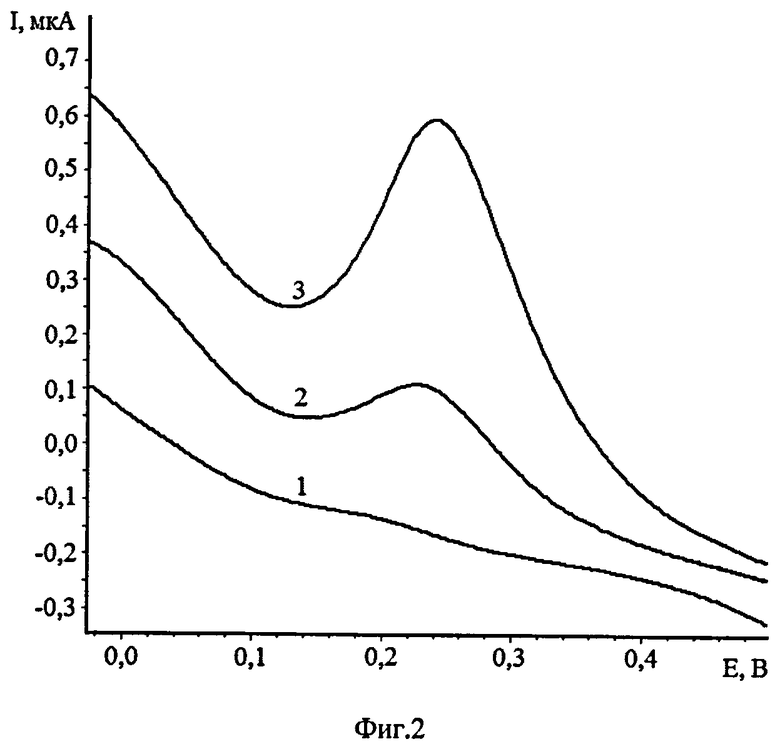
Пример 2. Измерения осмия были проведены после его выделения из пробы (фиг.2). 1 г пробы смешивают с пятикратным избытком пероксида натрия. Ставят в муфельную печь и сплавляют при температуре от 550 до 700°C в течение 30 мин. После остывания тигля пробу выщелачивают водой и количественно переносят в перегонную колбу. Добавляют концентрированную серную кислоту до полной нейтрализации щелочного раствора. Далее добавляют от 3 до 5 мл 30% пероксида водорода. Колбу помещают на предварительно разогретую до 115°C плитку и проводят отгонку осмия в виде OsO4. Во время отгонки периодически добавляют по 1 мл 30% пероксида водорода. Время отгонки составляет 30 минут. После отгонки из приемников собирают полученный раствор.
10 мл фонового электролита (0,1 М CH3COOH) помещают в кварцевый стаканчик и добавляют 1 мл 30% H2O2. Не прекращая перемешивания, проводят электролиз раствора, при Еэ=-0,2 В и при τэ=60 сек, снимают вольтамперную кривую электрокаталитического разложения пероксида водорода при скорости развертки 100 мВ/с. Отсутствие пиков на вольтамперной кривой в интервале от плюс 0,1 до плюс 0,3 В свидетельствует о чистоте фона. Затем добавляют 1 мл раствора, полученного после перегонки, и проводят электрохимическое концентрирование осадка при аналогичных условиях. На вольтамперной кривой в интервале от плюс 0,1 до плюс 0,3 В появляется пик электрокаталитического разложения пероксида водорода. Если на вольтамперной кривой не появляется пик, то вносят очередную аликвоту исследуемого раствора. Далее вносят добавку стандартного раствора осмия (VIII) 0,02 мл из 1 мг/дм3 и снова регистрируют аналитический сигнал при потенциале электроконцентрирования - 0,2 В. По разнице токов пиков каталитического разложения пероксида водорода вычисляли концентрацию осмия в пробе. Пик тока каталитического разложения пероксида водорода на осадке OsO2, предварительно электроосажденного на поверхности сажевого электрода, регистрировали в диапазоне потенциалов от плюс 0,1 до плюс 0,3 В (отн. нас.х.с.э.).
Таким образом, впервые установлена способность количественного анализа осмия по пикам каталитического электроокисления пероксида водорода на предварительно электросконцентрированном осадке в виде OsO2 на поверхности сажевого электрода на фоне 0,1 М CH3COOH. Предложенный способ прост, не используется ртуть из-за ее токсического действия. Способ может быть применен в любой химической лаборатории, имеющей компьютеризированные анализаторы типа СТА, ТА или полярограф.
Предложенный способ может быть использован для определения осмия в объектах природного и техногенного происхождения.
Способ определения осмия инверсионно - вольтамперометрическим методом в природном и техногенном сырье

Изобретение может быть использовано в различных отраслях народного хозяйства для определения содержания в растворах различных концентраций ионов осмия. Способ определения осмия инверсионно-вольтамперометрическим методом в природном и техногенном сырье заключается в том, что осмий (VIII) переводят в растворе в виде летучего тетраоксида осмия и проводят каталитическое, вольтамперометрическое определение, при этом проводят накопление ионов осмия на сажевом электроде в перемешиваемом растворе в присутствии пероксида водорода в течение 60-120 секунд с последующей регистрацией анодных пиков электрокаталитического разложения пероксида водорода на осадке OsO2 при скорости развертки потенциала 80-120 мВ/с при потенциалах электролиза минус 0,2 В на фоновом электролите 0,05-0,1 М СН3СООН, концентрацию ионов осмия определяют по высоте анодного обратного пика каталитического разложения пероксида водорода на вольтамперной кривой в диапазоне потенциалов от плюс 0,1 до плюс 0,3 В относительно насыщенного хлоридсеребряного электрода методом добавок аттестованных смесей. Изобретение обеспечивает возможность снизить предел и нижнюю границу определяемых содержаний осмия (VIII) по пику электрокаталитического разложения пероксида водорода, полученному после электроконцентрирования OsO2 на сажевом электроде (СЭ) методом инверсионной вольтамперометрии. 2 пр., 2 табл., 2 ил.
Способ определения осмия инверсионно-вольтамперометрическим методом в природном и техногенном сырье, заключающийся в том, что осмий (VIII) переводят в растворе в виде летучего тетраоксида осмия и проводят каталитическое, вольтамперометрическое определение, отличающийся тем, что проводят накопление ионов осмия на сажевом электроде в перемешиваемом растворе в присутствии пероксида водорода в течение 60-120 с с последующей регистрацией анодных пиков электрокаталитического разложения пероксида водорода на осадке OsO2 при скорости развертки потенциала 80-120 мВ/с при потенциалах электролиза минус 0,2 В на фоновом электролите 0,05-0,1 М СН3СООН, концентрацию ионов осмия определяют по высоте анодного обратного пика каталитического разложения пероксида водорода на вольтамперной кривой в диапазоне потенциалов от плюс 0,1 до плюс 0,3 В относительно насыщенного хлоридсеребряного электрода методом добавок аттестованных смесей.
| Инверсионно-вольтамперометрический способ определения осмия в природных и промышленных объектах | 1990 |
|
SU1746285A1 |
| Способ определения осмия | 1986 |
|
SU1460691A1 |
| Способ определения осмия | 1982 |
|
SU1054758A1 |

