Генетическая конструкция для экспрессии рекомбинантных дезоминаз на основе АРОВЕС1 для направленной модификации цитозиновых оснований митохондриальной ДНК.
Изобретение относится к медицине, терапии наследственных заболеваний и молекулярной биологии. Может быть использовано для редактирования точечных патогенных мутаций митохондриальной ДНК (мтДНК), ассоциированных с различными наследственными митохондриальными патологиями. Представляет линейку генетических конструкций для наработки рекомбинантных белков на основе цитидиновой дезаминазы APOBEC1. Данные белки нарабатываются в цитоплазме клетки и за счет наличия в их структуре митохондриальных детерминант импорта способны транспортироваться внутрь митохондрий, чтобы производить направленную специфическую модификацию цитозиновых оснований в адениновые в целевой последовательности митохондриальной ДНК (мтДНК). Специфичность белков к целевой последовательности обеспечивается наличием в их составе специально сконструированных TALE-доменов, которые с высокой степенью селективности связываются с ДНК-мишенями.
СК - синаптонемный комплекс,
мтДНК - митохондриальная ДНК,
п.н. - пара нуклеотидов,
ПЦР - полимеразная цепная реакция,
ПЦР-ПДРФ - полимеразная цепная реакция-полиморфизм длин рестрикционных фрагментов,
хнРНК - химерная направляющая РНК,
Cas - CRISPR-associated, ассоциированные с CRISPR,
CTCF - CCCTC-binding factor (CCCTC-связывающий фактор),
CRISPR - clustered regularly interspaced short palindromic repeats, кластеризованные регулярно расположенные короткие палиндромные повторы,
DFNB1A - несиндромальная аутосомно-рецессивная потеря слуха, 1A тип,
DFNA3 - несиндромальная аутосомно-доминантная потеря слуха,
EGFP - зеленый флуоресцентный белок,
GJB2 (gap junction protein, beta-2) - ген, кодирующий белок коннексин 26,
GRC - germline restricted chromosome (хромосомы, ограниченные клетками зародышевой линии),
gRNA - направляющая РНК,
HDR - homology directed repair (гомологичная рекомбинация),
HSR - homogeneously staining region (гомогенно окрашиваемые районы хромосом),
kb - kilobase (тысяча пар нуклеотидов),
Mb - megabase (миллион пар нуклеотидов),
MMEJ - microhomology-mediated end joining (соединение концов опосредованное микрогомологией),
NHEJ - non-homologous end joining (негомологичное соединение концов),
Sleeping Beauty - система внесения трансгенов «спящая красавица».
Известен «Способ лечения атрофии зрительного нерва различной этиологии» (патент РФ № 2375019). Он обеспечивает улучшение или стойкую стабилизацию зрительных функций на небольшом временном промежутке, но неприменим для долговременной коррекции зрительных показателей, так как не направлен на коррекцию наследственного материала митохондрий. Данный способ связан с введением аутологичных стволовых клеток в район диска зрительного нерва, и данные клетки лишь опосредованно оказывают благотворные влияние на функции пораженных клеток зрительного нерва.
Известно изобретение «Митохондриальные целевые антиоксиданты» (Mitochondrially targeted antioxidants) (патент США № US6331532 1998 г.), которое является технологией импорта функциональных молекул в митохондрии, с использованием липофильных агентов с иммобилизованными молекулами антиоксидантов, но не дает возможности осуществлять редактирование патогенных мутаций в митохондриальной ДНК.
Известно изобретение «Система доставки нуклеиновых кислот в митохондрии» (Mitochondrial nucleic acid delivery systems) (патент Канады № CA2678572 2008 г.). Недостатком данной технологии является то, что она не подразумевает доставку в митохондрии белков, поэтому она не может быть использована для импорта рекомбинантных белков на основе дезаминазы APOBEC1 в митохондрии.
Известно изобретение «Генетическая конструкция на основе системы редактирования генома CRISPR/Cas9, кодирующая нуклеазу Cas9, специфически импортируемую в митохондрии клеток человека» (патент РФ № 2634395, 2015 год). Технология позволяет направлять в митохондрии клеток человека нуклеазу SpCas9, и при совместном использовании с соответствующей направляющей РНК может обеспечивать сдвиг уровня гетероплазмии по определенным патогенным мутациям мтДНК. Недостаток данной системы состоит в том, что для ее корректного функционирования необходим импорт молекул направляющей РНК внутрь митохондрий, но к настоящему времени не существует эффективных технологий осуществления, доказанных или подтвержденных объективно методами контроля. Другим недостатком этой системы является неспособность редактировать точечные мутации в мтДНК в состоянии гомоплазмии, так как в этом случае она должна приводить к элиминации подавляющего числа копий мтДНК. Даже в состоянии гетероплазмии, система вносит двухцепочечные разрывы в мтДНК, что губительно сказывается на функционировании клеток при непрерывном использовании.
Существует похожее изобретение «Генетическая конструкция pMitoAsCpf1, кодирующая нуклеазу AsCpf1 с детерминантной импорта в митохондрии клеток человека» (патент РФ № 2662994, 2016 год). Технология позволяет доставлять в митохондрии клеток модифицированную нуклеазу AsCpf1 для специфической элиминации мутантной мтДНК. Как и в случае с системами для элиминации дефектных копий мтДНК на основе белка SpCas9, корректность функционирования системы связана с взаимодействием с молекулами направляющей РНК, для которых нет доказанного высокоэффективного способа импорта внутрь митохондрий. Более того, система на основе AsCpf1, даже в случае корректного функционирования, приводит к появлению двуцепочечных разрывов и последующему снижению общего числа копий мтДНК, что может негативно сказаться на метаболизме и жизнеспособности клеток. Также, в случае применения систем на основе SpCas9 или AsCpf1 велика вероятность неспецифического разрезания молекул мтДНК дикого типа, что еще больше ухудшит показатели жизнеспособности и функций клеток.
Чтобы избежать проблем с элиминацией копий мтДНК, исследователи разработали технологии, сочетающие связывание с целевой последовательностью мтДНК с помощью ДНК-связывающих доменов Cas9 c доменами, модифицирующими первичную последовательность ДНК без внесения двуцепочечных разрывов. Одним из примеров подобных технологий является изобретение «Генетические конструкции, кодирующие направляемые в митохондрии клеток человека нуклеазы Cas9-BE4-Gam и Cas9-ABE 7.10» (патент РФ № 2709739, 2018 год). В данной системе, рекомбинантные белки Cas9-BE4-Gam и Cas9-ABE 7.10, нарабатываемые в цитоплазме клеток с обозначенных в патенте конструкций, транспортируются в митохондрии за счет специально встроенных детерминант импорта. Затем предполагается взаимодействие рекомбинантных нуклеаз со специфическими целевыми последовательностями мтДНК, за счет их связывания с ДНК распознающим доменом белка SpCas9, действующего, в свою очередь, за счет транспортируемых в митохондрии молекул направляющей РНК. Недостатком, как и у других систем, зависящих от транспорта РНК внутрь митохондрий, является крайне низкая эффективность данного процесса.
Существует изобретение «Модификации структуры направляющей нуклеазу SpCas9 молекулы РНК для обеспечения импорта в митохондрии клеток человека» (патент РФ № 2717977, 2018 год). Данная технология призвана способствовать доставке молекул направляющей РНК (нРНК) для таргетирования нуклеазы SpCas9 в митохондрии клеток за счет внесения в структуру данных нРНК субдоменов транспортной РНК лизина HD и HF, что, в свою очередь, приводит к их взаимодействию с человеческой митохондриальной тРНК-лизил синтазой (pre-KARS2) и белком ENO2, которые импортируются в митохондрии совместно с направляющей РНК. В митохондриях направляющая РНК должна связываться с нуклеазой SpCas9, формируя функциональный комплекс, вносящий двухцепочечные разрывы в мтДНК, которые приводят к ее элиминации ферментами системы репликации митохондрий. Однако в мировой и отечественной литературе практически отсутствуют подтверждения эффективности подобных систем. Напротив, зачастую авторы отмечают неэффективность или невозможность импорта направляющих РНК. Кроме того, кроме данной патентной заявки, авторам не известны модификации направляющих нуклеазу SpCas9 молекул РНК, для которых достоверно продемонстрирована их способность функционально взаимодействовать с нуклеазой и при этом эффективно импортироваться в митохондрии клеток человека.
Из данных отечественной и зарубежной литературы, патентов и патентных заявок авторам не известно использование генетических конструкций на базе дезаминазы APOBEC1 и ДНК-связывающих TALE-доменов для редактирования точечных патогенных мутаций мтДНК. Описанные в литературе системы для изменения уровня гетероплазмии мтДНК на основе TALE-доменов (mitoTALENs) содержат в себе также и нуклеазные домены для внесения направленных двуцепочечных разрывов в месте мутации, и последующей элиминации мутантных копий мтДНК. Механизм действия подобных систем в целом напоминает таковой для ранее описанных нами систем на основе программируемых нуклеаз SpCas9 и AsCpf1, когда нежелательным побочным эффектом является избыточная элиминация копий мтДНК. Однако нацеливание на искомые последовательности мтДНК с помощью белковых TALE доменов - это заметное преимущество mitoTALEN технологии по сравнению с CRISPR-ассоциированными системами, так как они не требуют импорта молекул направляющей РНК внутрь митохондрий. В представляемой нами системе мы также устраняем и второй недостаток - чрезмерное снижение количества копий мтДНК вследствие внесения двуцепочечных разрывов. Наша система направлена на внесение точечных модификаций конкретных оснований мтДНК, не приводящих к запуску процессов элиминации поврежденных копий. Мы представляем 6 плазмидных конструкций (pNtGGS1_APOBEC1_TALE, pNt(GGS)2_APOBEC1_TALE, pNt(GGS)3_APOBEC1_TALE, pNtPAP_APOBEC1_TALE, pNt(PA)3_APOBEC1_TALE, и pNt(PA)4P_APOBEC1_TALE), кодирующих рекомбинантные дезаминазы на основе APOBEC1 и TALE доменов. Рекомбинантные дезаминазы отличаются композицией полипептидных линкеров, соединяющих дезаминазную (APOBEC1) и ДНК-связывающую (TALE) части. Кодирующие их векторные конструкции, соответственно, отличаются между собой последовательностью участков, кодирующих полипептидные линкеры. Новизну представляют нуклеотидные последовательности 6 разработанных нами векторных конструкций.
Задачей изобретения является исправление точечных патогенных мутаций митохондриальной ДНК, которые ассоциированы с наследственными митохондриальными патологиями.
Техническим результатом изобретения является обеспечение системы редактирования точечных мутаций в мтДНК и способа доставки рекомбинантных дезаминаз в митохондрии клеток человека. Дополнительным техническим результатом является исправление точечных патогенных мутаций митохондриальной ДНК.
Поставленная задача решается тем, что генетические конструкции заявляемого изобретения, после попадания в цитоплазму и ядро клеток, обеспечивает экспрессию кодируемых ими молекул рекомбинантных дезаминаз (NtGGS1_APOBEC1_TALE, Nt(GGS)2_APOBEC1_TALE, Nt(GGS)3_APOBEC1_TALE, NtPAP_APOBEC1_TALE, Nt(PA)3_APOBEC1_TALE, и Nt(PA)4P_APOBEC1_TALE; обобщенное название всей группы - Nt_APOBEC1_TALE), которые, используя собственный аппарат клетки, транспортируются в митохондрии, где обеспечивают исправление точечных мутаций в мтДНК путем распознавания специфического участка последовательности посредством взаимодействия своего TALE домена со специфическими целевыми районами мтДНК, находящимися поблизости (5-15 п.н. от соответствующей мутации) и последующим дезаминированием цитозинового основания в месте мутации. Далее, в ходе репликации молекулы мтДНК, напротив дезаминированного основания используется комплементарный ему нуклеотид - тимин, являющийся «исправленным вариантом» мутации.
Принцип функционирования предлагаемых систем базируется на пересечении особенностей работы систем mitoTALE и белка APOBEC1. TALE (transcription activator-like effectors) уникальны с точки зрения простоты и гибкости механизма их нацеливания на ДНК-мишени. Их специфичность определяется центральным доменом тандемных повторов, каждый из которых состоит из 33-35 аминокислотных остатков (а.о.), за которыми следует усеченный повтор из 20 а.о. Пара аминокислот в позициях 12 и 13 в структуре каждого повтора называется ‘repeat-variable di-residue’ (RVD). Именно эта пара определяет специфичность к ДНК-мишени, когда один RVD соответствует одному нуклеотиду в ДНК, и каждый из четырех наиболее часто встречающихся RVD преимущественно соотносится с одним из четырех оснований ДНК. Подобная прямолинейная зависимость последовательностей ДНК и TALE способствует возможности предсказывания сайтов связывания для TALE в ДНК, а также конструирования TALE доменов с учетом имеющейся таргетной последовательности ДНК.
APOBEC1 (rAPOBEC1, rat Apolipoprotein B-editing complex catalytic subunit 1) изначально был открыт как один из каталитических компонентов белкового комплекса, в пищеварительном тракте редактирующего РНК и дезаминирующего цитозин в положении 6666 в урацил в РНК аполипротеина B с последующим образованием преждевременного стоп кодона и обрыва рамки считывания. В то время как РНК является естественным субстратом APOBEC1, позднее в экспериментах in vitro на экстрактах трансформированных E.coli и эукариотических моделях, была неоднократно показана способность данного белка дезаминировать цитозин в одноцепочечных молекулах ДНК. Во время репликации мтДНК образуются одноцепочечные участки, которые являются подходящими субстратами для APOBEC1. При этом каталитическая активность APOBEC1 может зависеть от локальных особенностей модифицируемой нуклеотидной последовательности.
Активность домена APOBEC1 в составе рекомбинантного белка позволяет исправить комплементарную пару оснований C:G на Т:А. После доставки в митохондрии клеток, рекомбинантная дезаминаза за счет своего TALE домена специфически связывается с целевым участком мтДНК в районе исправляемой мутации. С помощью APOBEC1 происходит дезаминирование цитозина, который становится комплементарен аденину вместо гуанина. В ходе репликации мтДНК напротив аденина митохондриальная ДНК-полимераза интегрирует тимин.
Генетические конструкции, кодирующие рекомбинантные дезаминазы Nt_APOBEC1_TALE с детерминантой импорта в митохондрии клеток представляют собой плазмидные векторы, разработанные на базе ранее созданного нами вектора pMitoCas9 (патент РФ № 2634395, 2015 год). Плазмидные вектора обеспечивают возможность трансформации компетентных клеток E.coli с последующей наработкой большого количества копий, позволяют отбирать трансформированные колонии на селективной среде, содержащей антибиотик. Они обеспечивают экспрессию рекомбинантных дезаминаз Nt_APOBEC1_TALE в клетках млекопитающих и человека, а также доставку продуктов трансляции в митохондрии. Выбор мутации, однако ограничен несколькими параметрами таргетируемой последовательности ДНК. Преимущественный контекст для наиболее эффективного редактирования молекулы цитозина в молекулу тимина для рекомбинантных деаминаз на основе крысиного APOBEC1 - это дуплет тимин-цитозин TC, однако с более низкой эффективностью фермент способен дезаминировать цитозин и в мотивах CC. Искусственно сконструированные TALE домены в составе различных рекомбинантных белков обычно нацелены на ДНКовые мотивы длиной от 10 до 30 п.н., и эффективность связывания увеличивается, если подобному мотиву с 5’-конца предшествует тимин. Тем не менее, репертуар потенциально таргетируемых нашей системой сайтов мтДНК весьма широк, и впоследствии он может быть расширен методами генной инженерии.
Генетическая конструкция NtGGS1_APOBEC1_TALE содержит ориджин репликации, ген устойчивости к ампициллину, промотор цитомегаловируса, сигнал митохондриальной локализации гена COX8A, репортерный пептид 3xFLAG, последовательность, кодирующую дезаминазу APOBEC1, последовательность, кодирующую пептидную линкерную часть (в данном случае, глицин-глицин-серин, GGS), последовательность, кодирующую TALE домен (N- и С-концевые мотивы, 20 полных TALE повторов и один усеченный), а также последовательность, кодирующую сигнал полиаденилирования бычьего гормона роста (Bovine growth hormone polyadenylation signal). Последовательности шести различных конструкций в нашей линейке отличаются только мотивами, кодирующими линкерные участки - G-G-S (глицин-глицин-серин), (G-G-S)x2 (глицин-глицин-серин x2), (G-G-S)x3 (глицин-глицин-серин x3), P-A-P (пролин-аланин-пролин), (P-A)x3 (пролин-аланин x3), (P-A)x4P ((пролин-аланин x4 пролин).
Описание способа получения генетической конструкции заявляемого изобретения.
Карта генетической конструкции pNtGGS1_APOBEC1_TALE была построена при помощи программного обеспечения SnapGene. Для сборки был использован каркас разработанной ранее генетической конструкции pMitoCas9, а также плазмида BE4-Gam (Addgene #100806). Сначала с плазмиды pMitoCas9 мы в две стадии амплифицировали фрагмент, кодирующий Cox8 и 3xFLAG мотивы, которым предшествует фрагмент Козака перед сайтом инициации трансляции. В исходной последовательности имелся сайт разрезания ферментом рестрикции BamHI. Мы инактивировали его, с помощью 2-стадийной ПЦР внеся мутацию G>T в положении 1 данного сайта. На втором шаге, мы амплифицировали фрагмент, кодирующий дезаминазу APOBEC1 с плазмиды BE4-Gam (Addgene #100806) добавив на 3’-конце последовательности, кодирующие пептидный линкер GGS и дополнительный сайт рестрикции BamHI для последующего внесения последовательности, кодирующей соответствующий ДНК-связывающий TALE-домен. Также с помощью ПЦР с BE4-Gam нарабатывался третий фрагмент для сборки конструкции без TALE домена, включающий следующие элементы: ориджин репликации, ген устойчивости к ампициллину, промотор цитомегаловируса, а также последовательность, кодирующую сигнал полиаденилирования бычьего гормона роста (Bovine growth hormone polyadenylation signal). Далее, путем бесшовного клонирования по методу Гибсона данные три фрагмента соединяются в единую генетическую конструкцию, в которую впоследствии вставляется фрагмент, кодирующий TALE домен. Этот вектор назовем pNtGGS1.
Также, отдельно, по методу Golden Gate клонирования собирается последовательность, кодирующая TALE-домен. Далее, она вырезается из несущего ее вектора по BamHI рестрикционным сайтам и по этим же сайтам встраивается в вектор pNtGGS1. Финальный вектор назовем pNtGGS1_TALE.
Чтобы получить базовые конструкции (без встроенного TALE домена) со встройками, кодирующими отличные от GGS линкерные участки, производилась ПЦР амплификация участка, содержащего фрагмент, кодирующий APOBEC1, с праймерами, к последовательности одного из которых добавлена последовательность, кодирующая один из линкеров - (GGS)2, (GGS)3, PAP, (PA)3, (PA)4P. Также на конце этого праймера после мотива соответствующего линкеру добавлен рестрикционный сайт BamHI. Наработанные таким образом фрагменты вставляются в исходную плазмиду pNt(GGS)1 путем рестрикции и лигирования по сайту BamHI. Полученная в результате плазмида, в зависимости от композиции линкерного участка закодированного ее белка называется pNt(GGS)2, pNt(GGS)3, pNtPAP, pNt(PA)3, pNt(PA)4P. Как и в случае плазмиды pNtGGS1_TALE, чтобы из базовых плазмид получить плазмиды со встройкой участка, кодирующего TALE, проводят рестрикцию несущего TALE вектора и базовой плазмиды с помощью фермента BamHI. Затем фрагмент, кодирующий TALE, лигируется в разрезанную по BamHI базовую плазмиду. Корректность сборки полученных векторов проверяется с помощью ПЦР и секвенирования по методу Сэнгера. Амплификацию фрагментов ДНК с конструкций проводили при помощи полимеразы Q5 на амплификаторе T100 Thermo Cycler (Bio-Rad, США). Олигонуклеотидные праймеры синтезировали с помощью фосфорамидитного метода на AMS-2000, очищали методом обращено-фазовой хроматографии на OPS-1000.
Для приготовления экстрактов (лизатов) тотальных белков клетки на 6-луночных культуральных планшетах промывали 1 мл PBS, затем добавляли 500 мкл Trypsin-EDTA (Trypsin-EDTA in HBSS (1x) with Phenol Red, Capricorn, cat # TRY-4B), инкубировали 5 мин при температуре 37°С в CO2-инкубаторе и переносили в 15 мл центрифужные пробирки. Клетки осаждали центрифугированием 5 мин при 500 об/мин. Для получения тотальных клеточных экстрактов лизировали в 4-х объемах (по отношению к плотному осадку клеток) RIPA буфера, содержащего 150 mM NaCl, 50 mM Tris-HCl pH 8.0, Nonidet P-40 1%, дезоксихолат Na (sodium deoxycholate) 0.5%, додецил сульфат Na (sodium dodecyl sulfate) 0.1%, коктейль ингибиторов протеаз и фосфатаз (Thermo Scientific, США) в разведении 1:100. Затем инкубировали лизаты 15 минут при 4°С. Экстракты подвергали процедуре соникации (интенсивность 35%, 3 раза по 3 цикла по 10 секунд) на ультразвуковом дезинтеграторе Sonopuls, затем центрифугировали 15 мин при 14000 g при 4°С и супернатанты использовали для последующих анализов.
Концентрацию тотального белка определяли с помощью набора реактивов BCA Protein Assay в соответствии с протоколом производителя. Для получения рабочего раствора смешивали реагент A c реагентом B в соотношении 50:1. К 2 мкл экстракта добавляли 100 мкл рабочего раствора и инкубировали полученный раствор 30 мин при 37°С. Оптическую плотность измеряли на спектрофотометре NanoPhotometer P360 (Implen). Концентрацию белка рассчитывали по калибровочной кривой, построенной по результатам измерений оптических плотностей образцов бычьего сывороточного альбумина с известной концентрацией.
Далее проводили электрофорез белков в денатурирующем полиакриламидном геле с додецилсульфатом натрия (SDS-PAGE) и иммуноблоттинг. Электрофорез проводили в 4-12% градиентном полиакриламидном геле Bolt 4-12% Bis-Tris Plus (Invitrogen, Thermo Fisher Scientific, cat # NW04120BOX). Объем образца брали из расчета 30 мкг общего белка на дорожку. Образцы смешивали с Laemmli буфером для нанесения образцов (Bolt LDS Sample Buffer (4x), Novex, Life Technologies, cat # B0007) и реагентом восстановителем (Bolt Sample Reducing Agent, Novex, Life Technologies, cat # B0009) в соотношении 10 : 4 : 1 (образец : буфер : реагент-восстановитель), инкубировали в термостате 10 мин при 70°С и наносили на полиакриламидный гель. Электрофоретическое разделение белков проводили при постоянном напряжении, составляющем 250 В на гель, в камере для вертикального электрофореза белков (Minigel Tank, Invitrogen, A25977) в однократном буфере для электрофореза белков, приготовленном из двадцатикратного Bolt MES SDS Running Buffer (20x). Белки, разделенные электрофорезом, переносили с геля на 0.45 um мембрану PVDF (Thermo Fisher Scientific) с помощью влажного электропереноса в камере MiniBlot Module (Invitrogen, cat # B1000) в течение 1 часа при постоянном напряжении, составляющем 25 В на гель в однократном буфере для переноса, приготовленном из двадцатикратного Bolt Transfer Buffer (20x) (Novex, Life Technologies, cat # BT00061), с добавлением 10% метанола. Для блокировки сайтов неспецифического связывания антител мембрану инкубировали 1 ч в блокирующем буфере на основе 5% бычьего сывороточного альбумина в TBST (150 мМ NaCl, 10 мМ Трис-HCl (pH 8.0), 0.05% Tween 20) Starting Block T20 (TBS) Blocking Buffer (Thermo Scientific, cat # 37543). TBST готовили из первичных компонентов, либо разводили из двадцатикратного 20x TBS Tween-20 (Thermo Scientific, cat # 28360). Антитела для гибридизации разводили в блокирующем растворе в соответствии с рекомендациями производителей. В качестве первичных антител использовали антитела против эпитопа FLAG - Monoclonal Anti-FLAG M2 Antibody (Sigma-Aldrich, cat # F3165-2MG) в разведении 1:2000. В качестве вторичных антител использовали видоспецифичные антитела, конъюгированные с пероксидазой хрена (HRP) в разведении 1:5000 (Rabbit anti-Mouse HRP-conjugated antibodies, Invitrogen, cat # 31450). HRP на мембране визуализировали с помощью реактива SuperSignal West PicoPLUS Chemiluminescent Substrate (Thermo Scientific, cat # 34580). Для этого компоненты набора смешивали в соотношении 1:1, из расчета 0.1 мл раствора на 1 см2 мембраны, наносили на мембрану и инкубировали в течение 5 мин. Изображения препаратов, активированных с помощью хемилюминесценции, получали на приборе iBright FL1500. В дальнейшем необходимо было оценить функциональность полученных белков путем их проверки в экспериментах по in vitro дезаминированию цитозиновых оснований в искусственно синтезированных ДНК-субстратах, меченых флуорохромом Cy3. 6 вариантов слитого белка на основе 6 выше обозначенных генетических конструкций были синтезированы с использованием протокола по транскрипции и трансляции в искусственной in vitro системе TNT Quick Coupled Transcription/Translation System (cat. # L1170, Promega, США) в соответствии с инструкциями производителя. Вкратце, реакция проводится в 1 стадию, в условиях чистоты от нуклеаз и стерильности. В реакционную смесь добавляют 40 мкл TNT Quick Master Mix, включающую в себя все необходимые ферменты и буферные компоненты, необходимые для реакции транскрипции и трансляции. Добавляют 1 мкл метионина (стоковая концентрация 1мМ), 2 мкл плазмидной ДНК (в концентрации 0.5 мкг/мкл) и 7 мкл деионизованной дистиллированной свободной от примесей нуклеаз воды. Полученную реакционную смесь инкубировали 90 мин при 30°С. Результативность реакции синтеза рекомбинантных белков проверяли с использованием системы Transcend™ в соответствии с инструкциями производителя.
Помимо рекомбинантных белков, необходимо было также получить ДНК-субстраты, на основе коммерчески синтезированных одноцепочечных ДНК олигонуклеотидов, часть из которых содержали на 5’ или 3’ конце флуоресцентную метку Cyanine3 -Cy3.
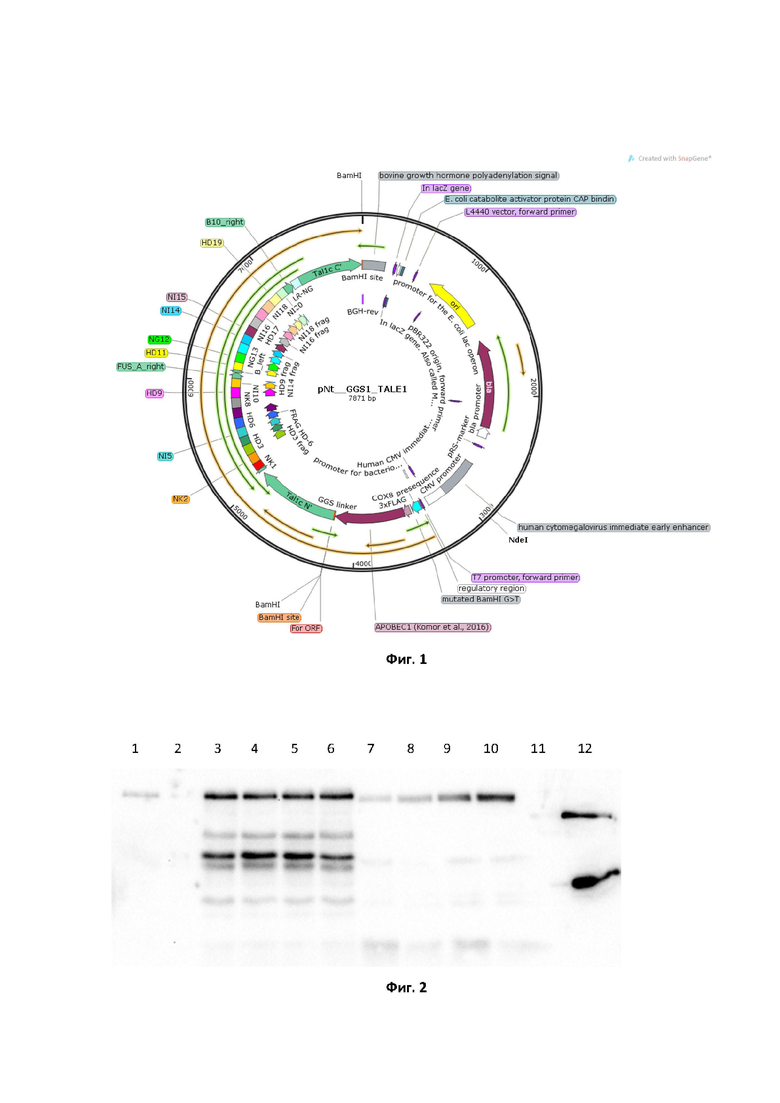
На фиг. 1 представлена карта генетической конструкции pNt(GGS)1_TALE, кодирующей рекомбинантную дезаминазу Nt(GGS)1_TALE, импортируемую в митохондрии, где: Ori - ориджин репликации, bla - ген устойчивости к антибиотику ампицилину, CMV promoter - промотор цитомегаловируса, Tal1c N’ - N-концевой мотив TAL-домена, TAL1c C’ - C-концевой мотив TAL-домена. Оценка эффективности наработки дезаминаз Nt(GGS)1_TALE - Nt(PA)4P_TALE в клетках HEK293T производилась методом вестерн блоттинга с использованием антител против эпитопа FLAG, который присутствует в составе разработанных нами рекомбинантных дезаминаз.
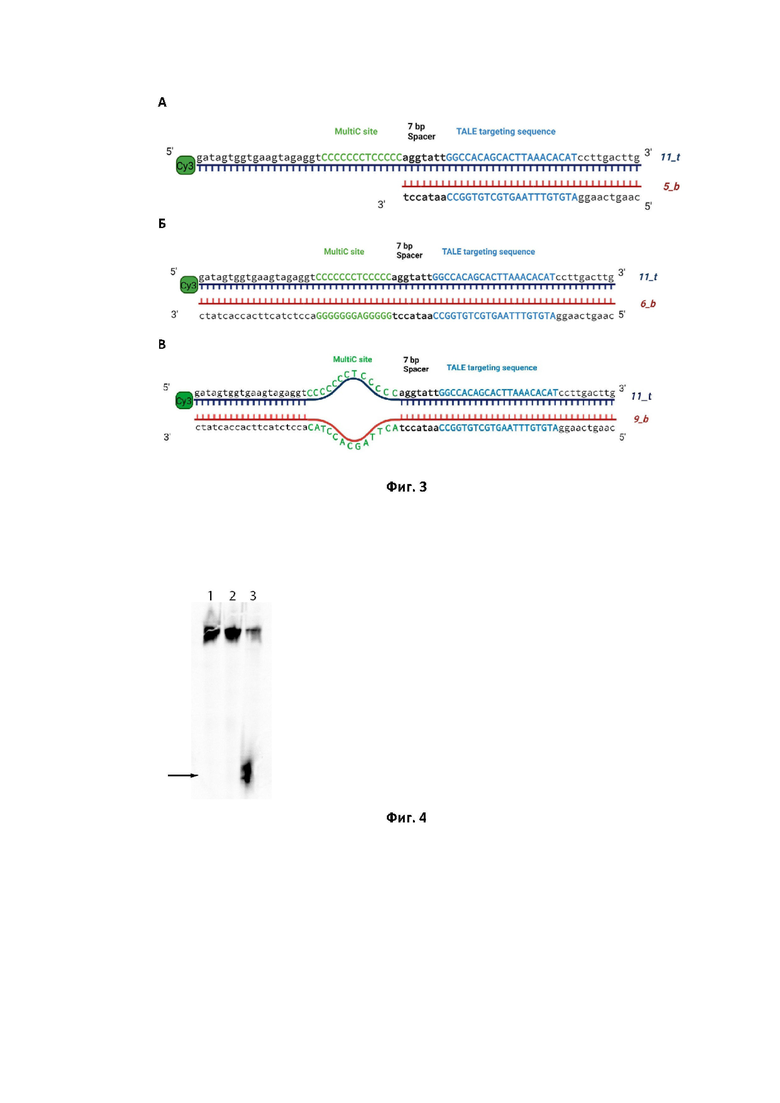
На фиг. 2 представлена фотография мембраны вестерн блота, на которой проведена детекция рекомбинантных дезаминаз на основе APOBEC1 с помощью антител против эпитопа FLAG (входящего в структуру дезаминаз), где 1 - положительный контроль (белок MitoCas9, также содержащий эпитоп FLAG), 2 - отрицательный контроль (лизат клеток при проведении трасфекции без добавления плазмиды в трансфекционную смесь), 3 - лизат после трансфекции с помощью NtPAP_TALE1, 4 - лизат после трансфекции с помощью Nt(GGS)3_TALE1, 5 - лизат после трансфекции с помощью Nt(GGS)2_TALE1, 6 - лизат после трансфекции с помощью Nt(GGS)1_TALE1, 7 - лизат после трансфекции с помощью CtPAP_TALE1, 8 - лизат после трансфекции с помощью Ct(GGS)3_TALE1, 9 - лизат после трансфекции с помощью Ct(GGS)2_TALE1, 10 - лизат после трансфекции с помощью Ct(GGS)1_TALE1, 11 - пустая дорожка, 12 - маркер молекулярного веса iBight (30 и 80 КДа).
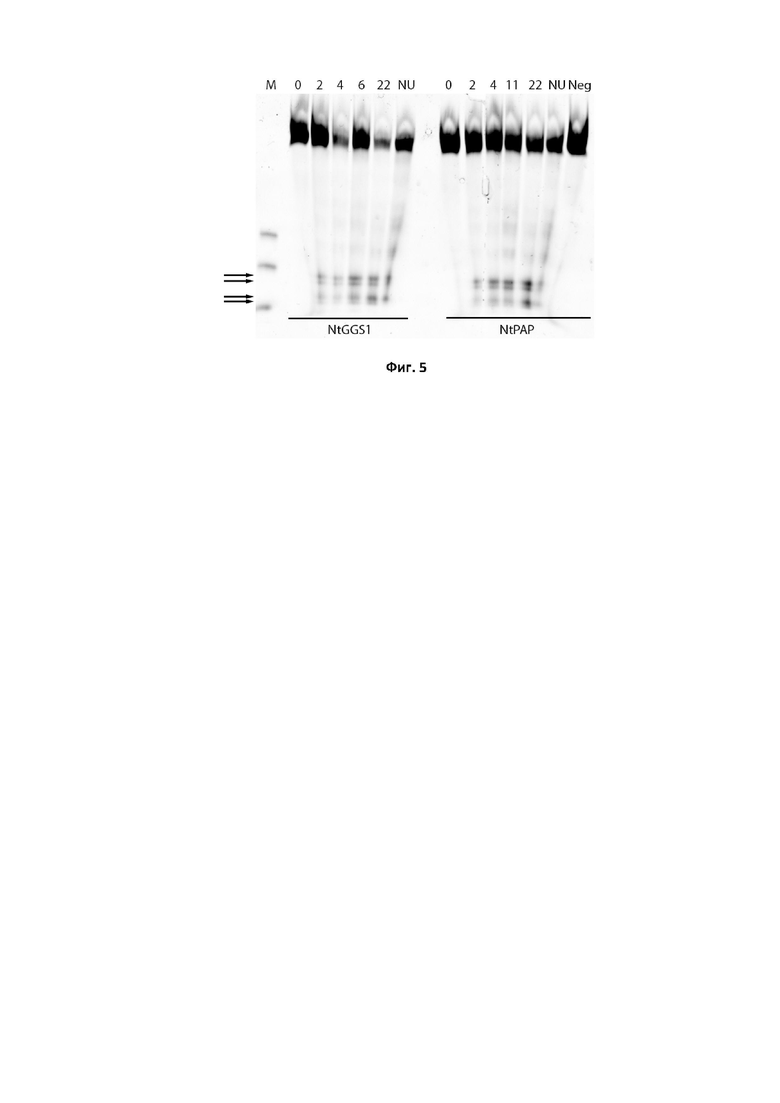
На фиг. 3 отражена структура трех основных типов ДНК-субстратов, где А - 3' Спейсер 7bp, Б - 3' Двуцепочечный 7bp, В - 3' Петля 7bp.
На фиг. 4 показаны результаты денатурирующего электрофореза с продуктами in vitro расщепления меченых нуклеотидов ферментом USER после предшествующего дезаминирования сконструированными нами рекомбинантными дезаминазами, где слева направо цифры соответствуют образцам: 1 - 11_t, 2 - 11_t, 3 - 11_tU, а стрелкой отмечен целевой продукт ферментативного разрезания.
На фиг. 5 представлены результаты in vitro расщепления ДНК субстрата 3’ Спейсер 7bp после дезаминирования при 30°C вариантами слитых белков NtGGS1 (слева) и NtPAP (справа), где слева направо: М - Ммв, 0 - без дезаминирования, 2 - дезаминирование 2 ч, 4 - дезаминирование 4 ч, 6 - дезаминирование 6 ч, 22 - дезаминирование 22 ч, NU - дезаминирование 22 ч без последующей реакции расщепления ферментом USER, Neg - образец отрицательного контроля, без белка дезаминазы при инкубации в течение 22 ч, и стрелками отмечены продукты специфического ферментативного расщепления. В анализ был включен контрольный образец (NU), где после дезаминирования в течение 22 часов, расщепление с ферментом USER не производилось (контроль на неспецифическую деградацию субстрата).
Нуклеотидная последовательность использованных нуклеотидов отражена в Таблице 1.
Комбинации одноцепочечных олигонуклеотидов, использованных для получения ДНК-сусубстратов для реакций дезаминирования in vitro приведены в Таблице 2.
Структура «верхней» цепи ДНК-субстрата включала в себя три основных элемента - сайт распознавания для ДНК-связывающего домена слитого белка, мультицитозиновый тракт для направленной модификации рекомбинантной дезаминазой, а также спейсер (участок, разделяющий сайт связывания и мультицитозиновый тракт) длиной 7 или 21 нуклеотид. Варьировалось относительное расположение первых двух элементов (ДНК-связывающий домен с 3’-конца и мультицитозиновый тракт с 5’-конца, или наоборот), а также длина разделяющего их спейсера (7 или 21 нуклеотид). Применялись (1) полностью комплементарные двуцепочечные ДНК-субстраты - 11t_6b, 20t_15b, 29t_24b, 37t_32b; (2) частично комплементарные (спейсер и ДНК-связывающий домен) - 11t_5b, 20t_14b, 29t_23b, 37t_31b; а также ДНК-субстраты, имеющие неспаренный (некомплементарный) мотив (петлю или пузырек) мультицитозинового тракта - 11t_9b, 20t_18b, 29t_27b, 37t_35b (Таблица 2). Протокол эксперимента по дезаминированию флуоресцентно меченных ДНК-субстратов in vitro включает в себя несколько этапов: (1) приготовление Cy-3 коньюгированных двуцепочечных ДНК (дцДНК) субстратов, (2) собственно in vitro реакция дезаминирования дцДНК субстратов с помощью приготовленных in vitro рекомбинантных дезаминаз (см. выше), (3) оценка и анализ результатов реакций дезаминирования с помощью разделения продуктов реакции в 12.5% денатурирующем полиакриламидном геле на основе мочевины и Трис-борат-ЭДТА электрофорезного буфера (12.5% TBE Urea PAGE).
Подробнее о первом этапе протокола. Сначала смешивали 2 олигонуклеотида, входящих в состав конкретного дцДНК-субстрата - меченый и немеченый (5 мкл 100 мМ раствора каждого олигонуклеотида), и добавляли 0.25 мкл NEBuffer 2. Грели полученную смесь при 95°С в течение 5 мин, затем медленно охлаждали до 45°С с инкрементом 0.1°С. После охлаждения смесь разбавляли в 17.8 мкл трисового буфера (10 мМ Tris-Cl pH 8.5).
Подробнее о втором этапе протокола. Берут 3 мкл экстракта с наработанным рекомбинантным белком после реакции in vitro транскрипции и трансляции и добавляют 1 мкл итогового продукта реакции получения дцДНК субстратов. Инкубируют полученную смесь в течение ночи при 30°С. Чтобы отделить дцДНК от молекул рекомбинантного белка добавляют 100 мкл буфера PB (Qiagen, Германия) и 25 мкл изопропанола. Далее очищают дцДНК на колонках QIAprep spin column (Qiagen, Германия) в соответствии с рекомендациями производителя, элюируя с колонки с помощью 20 мкл буфера CutSmart. К полученному продукту добавляют 1 мкл (1 ед. активности) фермента USER для ферментативного расщепления (фрагментации) ДНК цепей по тем сайтам, где в результате дезаминирования цитозин был заменен на урацил. Инкубируют 1 час при 37°С. Полученную в результате реакционную смесь можно непосредственно наносить на дорожки денатурирующего полиакриламидного геля с мочевиной, предварительно проинкубировав 10 мин при 70°С.
Денатурирующий электрофорез нуклеиновых кислот представляет из себя стандартную процедуру и проводится как, например, описано в протоколе. Гель содержит 48 грамм мочевины на 100 мл. После заливки геля, он инкубируется полчаса при комнатной температуре. После этого достают гребенку, промывают карманы для нанесения образцов с помощью 1x TBE буфера. Чтобы гель был готов к разделению образцов его необходимо прогреть до температуры 45-55°С и параллельно убрать остатки не растворившейся мочевины. Для этого проводят т.н. «предразгон» в течение 30 мин при суммарной мощности электрического тока 15-25 Ватт. В тот же самый момент подготавливают образцы - их смешивают 1:1 с буфером для нанесения, содержащим 5 мМ Трис, 0.5 мМ ЭДТА, 12.5% глицерола, 0.02% бромфенолового синего, 0.02% ксилен цианола, 80% ДМСО (диметилсульфоксида). После смешивания образцы нагревают при 70°С в течение 10 мин и наносят на дорожки геля. Разделение образцов производится в геле в течение 1 - 1.5 часов при температуре 45-55°С и суммарной мощности электрического тока 15-25 Ватт. После этого гель извлекается, промывается 3 раза 1x TBE буфере содержащем 5% этанола и 5% метанола. Далее его фотографируют и анализируют результат на флуоресцентном имеджере iBright FL1500.
В качестве контрольной реакции для проверки корректности работы фермента USER было проведено разрезание этим ферментом субстрата 11_tU, аналогичного по композиции субстрату 11_t, только несущего в своем составе урацил (последовательность: 5`/Cy3/gatagtggtgaagtagaggtCCCCCCCUCCCCCaggtattGGCCACAGCACTTAAACACATccttgacttg-3`). При тестировании белков с помощью протокола дезаминирования in vitro все 6 вариантов рекомбинантных слитых белков испытывали в реакциях с каждым из выше обозначенных вариантов ДНК-субстратов индивидуально (Таблица 2). Не были испытаны только полностью 5’ двуцепочечные субстраты, сводные результаты данных экспериментов приведены в таблице 3.
Помимо тестов нескольких дцДНК субстратов с 6 вариантами рекомбинантной дезаминазы, мы также оценили временную динамику реакций дезаминирования на примере конструкции NtGGS1 и NtPAP и различным временным интервалам инкубирования с дезаминазой. На фиг. 5 видно, что с ростом времени, затраченного на реакцию дезаминирования, возрастает относительная интенсивность бэндов, соответствующих продуктам расщепления.
Группа изобретений относится к молекулярной биологии. Предложены генетическая конструкция для экспрессии рекомбинантных дезоминаз на основе АРОВЕС1 и способ ее получения. Генетическая конструкция pNtGGS1_TALE представлена на фиг. 1 и обеспечивает экспрессию рекомбинантных дезоминаз на основе АРОВЕС1 для направленной модификации цитозиновых оснований митохондриальной ДНК. Изобретения могут быть использованы для редактирования точечных патогенных мутаций митохондриальной ДНК (мтДНК), ассоциированных с различными наследственными митохондриальными патологиями. 2 н.п. ф-лы, 5 ил., 3 табл.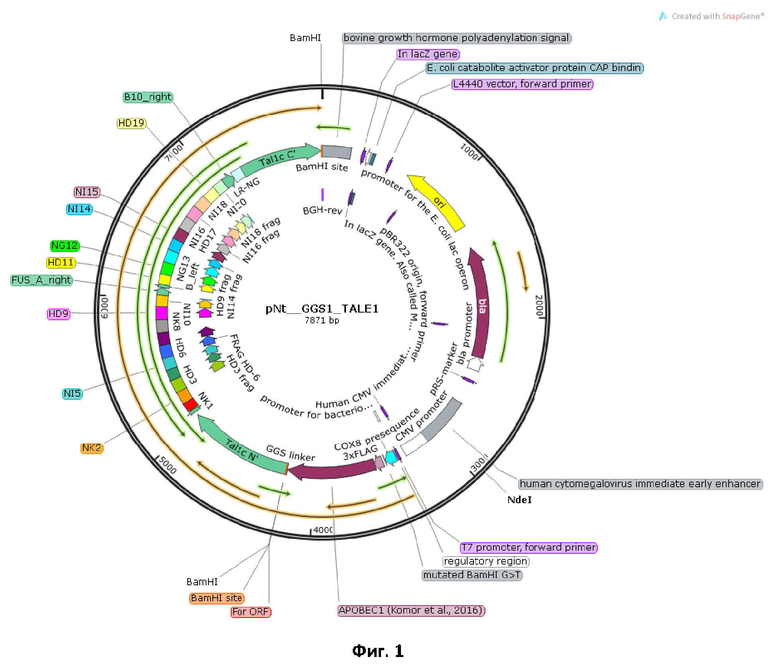
1. Генетическая конструкция pNtGGS1_TALE для обеспечения экспрессии рекомбинантной дезаминазы на основе АРОВЕС1, импортируемой в митохондрии, которая содержит, как показано на фиг.1, ориджин репликации, ген устойчивости к ампициллину, промотор цитомегаловируса, сигнал митохондриальной локализации гена COX8, репортерный пептид 3×FLAG, последовательность, кодирующую дезаминазу APOBEC1, последовательность, кодирующую пептидную линкерную часть глицин-глицин-серин, последовательность, кодирующую TALE домен, последовательность, кодирующую сигнал полиаденилирования бычьего гормона роста.
2. Способ получения генетической конструкции рNtGGS1_TALE по п.1, характеризующийся тем, что с плазмиды pMitoCas9 амплифицируют фрагмент, кодирующий COX8 и 3×FLAG мотивы, амплифицируют фрагмент, кодирующий дезаминазу APOBEC1 с плазмиды BE4-Gam, с добавлением последовательностей, кодирующих пептидный линкер GGS и дополнительный сайт рестрикции BamHI, с помощью ПЦР с BE4-Gam получают третий фрагмент для сборки конструкции без TALE домена, включающий следующие элементы: ориджин репликации, ген устойчивости к ампициллину, промотор цитомегаловируса, последовательность, кодирующую сигнал полиаденилирования бычьего гормона роста, путем бесшовного клонирования по методу Гибсона три фрагмента соединяют в единую генетическую конструкцию, в которую вставляют фрагмент, кодирующий TALE домен, методом рестрикции и лигирования и получают вектор pNtGGS1_TALE.

