ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способам инактивации вирусов с липидной оболочкой с использованием экологически приемлемых моющих средств и к способам получения биофармацевтического лекарственного средства с использованием экологически приемлемых моющих средств. Настоящее изобретение также относится к экологически приемлемым моющим средствам.
УРОВЕНЬ ТЕХНИКИ
Применение биофармацевтических лекарственных средств продолжалось с усилением их важности в качестве способа лечения многих заболеваний, нарушений или патологических состояний, которые влияют на здоровье индивидуума. Биофармацевтические лекарственные средства обычно получают путем очистки биологической жидкости или путем рекомбинантной выработки в клетках хозяина, таких как линии клеток млекопитающих. Однако в таких биофармацевтических производственных процессах вирусное загрязнение приводит к значительным затруднениям. Вирусное загрязнение может попасть в биофармацевтический производственный процесс с помощью очищаемых биологических жидкостей или вследствие использования продуктов, взятых у животных. В отличие от бактериального загрязнения, вирусное загрязнение трудно обнаружить. Однако, если вирусное загрязнение остается незамеченным и контагиозный вирус включается в состав биофармацевтического лекарственного средства, возникает значительная опасность для здоровья пациентов. Поэтому инактивация вирусов является первостепенной задачей в биофармацевтическом производстве.
Во многих биофармацевтических производственных процессах для инактивации вирусов используют моющие средства. Часто эти моющие средства объединяют с растворителями при так называемой обработке растворителем/моющим средством (S/D). Моющее средство Triton X-100 в течение многих лет использовали для S/D обработки коммерческих продуктов.
Однако последние экологические исследования показали, что Triton X-100 и продукты его разложения в водных организмах могут вести себя, как разрушители эндокринной системы, что усилило опасения, связанные с воздействием на окружающую среду (см. документ "ECHA Support, в котором описано установление того, что этоксилированный 4-(1,1,3,3-тетраметилбутил)фенол относится к веществам, вызывающим большие опасения, поскольку его разложение с образованием вещества, (4-(1,1,3,3-тетраметилбутил)фенола), вызывающего большие опасения, поскольку оно обладает способностью разрушать эндокринную систему, оно вызывает возможное сильное влияние на окружающую среду, что приводит к степени опасений, эквивалентной опасениям по отношению к CMRs и PBTs/vPvBs", принятый 12 декабря 2012 г.). Вследствие этого необходимы альтернативные экологически приемлемые моющие средства для инактивации вирусов.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает описанные выше потребности и решает указанные выше задачи в данной области техники с помощью вариантов осуществления, описанных ниже:
Токсическая активность Triton-X100 обусловлена фенольным фрагментом, который способен повреждать некоторые эндокринные рецепторы морских организмов. Поэтому на основании in silico прогнозов активности при разрушении эндокринной системы, ни для одного из моющих средств на основе не содержащего фенола простого эфира полиоксиэтилена, предлагаемых в настоящем изобретении, не обнаружено никаких свидетельств о том, что они активны, как разрушители эндокринной системы.
Авторы настоящего изобретения неожиданно установили, что экологически приемлемые моющие средства на основе не содержащего фенола простого эфира полиоксиэтилена, такие как Triton X-100 reduced, Triton N-101 reduced и Brij C10, эффективно инактивируют вирусы с липидной оболочкой при S/D обработке. Авторы настоящего изобретения также синтезировали экологически приемлемое моющее средство на основе не содержащего фенола простого эфира полиоксиэтилена, которое эффективно инактивирует вирусы с липидной оболочкой при S/D обработке и обработке одним моющим средством. Таким образом, авторы настоящего изобретения установили, что экологически приемлемые моющие средства на основе не содержащего фенола простого эфира полиоксиэтилена можно использовать в способах инактивации вирусов с липидной оболочкой, предлагаемых в настоящем изобретении.
Соответственно, настоящее изобретение относится к экологически приемлемым моющим средствам на основе не содержащего фенола простого эфира полиоксиэтилена, а также к улучшенным средствам инактивации вирусов с липидной оболочкой и предпочтительные варианты осуществления описаны ниже:
1. Соединение следующей общей формулы (VIII):
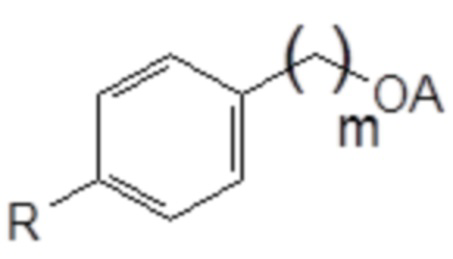
Формула (VIII)
в которой
R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи,
m является целым числом, равным от 1 до 4, и
A означает полиоксиэтиленовый остаток.
2. Соединение по параграфу 1, в котором A означает полиоксиэтиленовый остаток, содержащий от 4 до 16 оксиэтиленовых звеньев, предпочтительно 9 или 10 оксиэтиленовых звеньев.
3. Соединение по параграфу 1 или 2, в котором m равно 1.
4. Соединение по любому из параграфов 1-3, в котором R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 6 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи.
5. Соединение по любому из параграфов 1-4, в котором R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 6 атомов углерода и от 2 до 4 метильных групп в качестве заместителей указанной линейной цепи.
6. Соединение по любому из параграфов 1-5, в котором R означает углеводородную группу, включающую линейную цепь, содержащую 4 атома углерода и 4 метильные группы в качестве заместителей указанной линейной цепи.
7. Соединение по любому из параграфов 1-6, в котором R означает 2,4,4-триметилпент-2-ильную группу.
8. Соединение по параграфу 1, где соединением формулы (VIII) является следующее соединение:
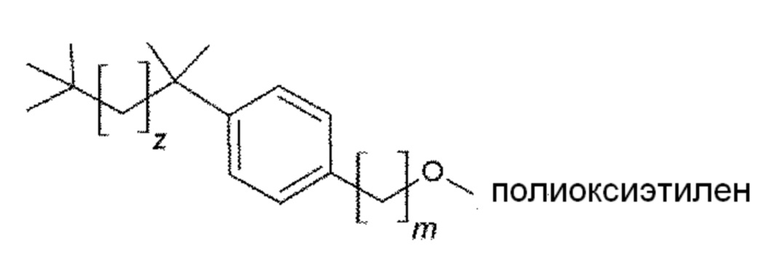
в котором m и z являются целыми числами, которые независимо выбраны из следующих групп:
m=от 1 до 4
z=от 1 до 5.
9. Соединение по параграфу 8, в котором m равно 1.
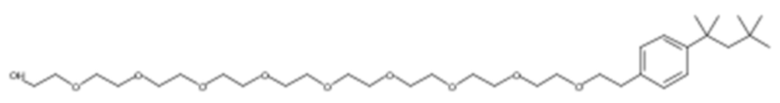
10. Соединение по параграфу 1, где соединением формулы (VIII) является следующее соединение:
в котором n является целым числом, равным от 4 до 16, предпочтительно в котором n равно 9 или 10.
11. Соединение по любому из параграфов 1-10 при условии, что исключен 29-[4-(1,1,3,3-тетраметилбутил)фенил]-3,6,9,12,15,18,21,24,27-нонаоксанонакозан-1-ол следующей структурной формулы:
12. Способ инактивации вирусов, обладающих липидной оболочкой, способ включает следующие стадии:
a) добавление моющего средства к жидкости для приготовления смеси указанного моющего средства и указанной жидкости; и
b) инкубирование указанной смеси для инактивации указанных вирусов;
где указанным моющим средством является простой эфир полиоксиэтилена и где указанным моющим средством является нефенольное моющее средство.
13. Способ по параграфу 12, в котором указанное моющее средство является экологически приемлемым.
14. Способ по параграфу 12 или 13, в котором указанным моющим средством является неионогенное моющее средство.
15. Способ по любому из параграфов 12-14, в котором указанным моющим средством является соединение по любому из параграфов 1-11.
16. Способ по любому из параграфов 12-14, в котором указанным моющим средством является алкиловый эфир полиоксиэтилена.
17. Способ по параграфу 16, в котором указанным алкиловым эфиром полиоксиэтилена является циклоалкиловый эфир полиоксиэтилена.
18. Способ по параграфу 17, в котором циклоалкильный фрагмент указанного циклоалкилового эфира полиоксиэтилена представляет собой алкилзамещенный циклоалкильный фрагмент.
19. Способ по параграфу 18, в котором указанный алкилзамещенный циклоалкильный фрагмент представляет собой разветвленный алкилзамещенный циклоалкильный фрагмент.
20. Способ по любому из параграфов 17-19, в котором указанный циклоалкиловый эфир полиоксиэтилена представляет собой циклогексиловый эфир полиоксиэтилена.
21. Способ по любому из параграфов 17-20, в котором указанный циклоалкиловый эфир полиоксиэтилена не представляет собой гетероциклический циклоалкиловый эфир полиоксиэтилена.
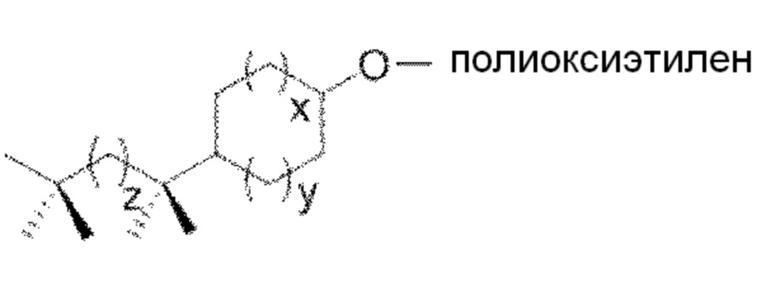
22. Способ по любому из параграфов 12-14 и 16-21, в котором указанное моющее средство обладает следующей структурой формулы (I):
(Формула (I));
где x, y и z являются целыми числами, которые независимо выбраны из следующих групп:
x=от 0 до 5
y=от 0 до 5
z=от 0 до 20
23. Способ по параграфу 22, в котором указанное моющее средство обладает следующей структурой формулы (II):
(Формула (II)).
24. Способ по параграфу 23, в котором указанное моющее средство обладает следующей структурой формулы (III):
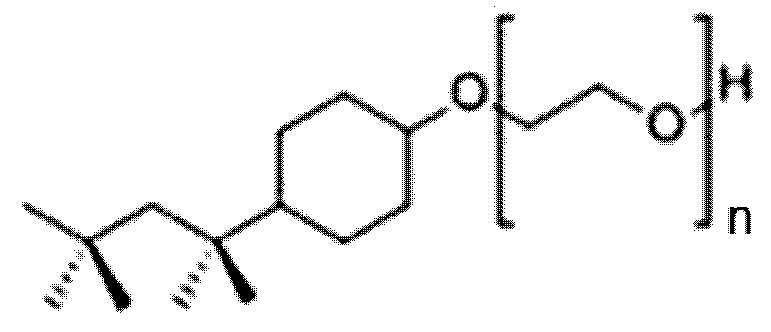
(Формула (III));
в котором n является целым числом, равным от 4 до 16.
25. Способ по параграфу 24, в котором n равно 9 или 10.
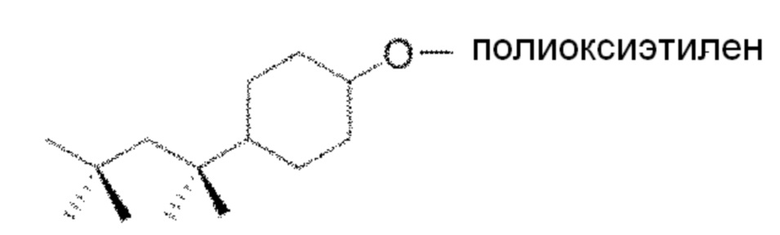
26. Способ по параграфу 22, в котором указанное моющее средство обладает следующей структурой формулы (IV):
(Формула (IV)).
27. Способ по параграфу 26, в котором указанное моющее средство обладает следующей структурой формулы (V):
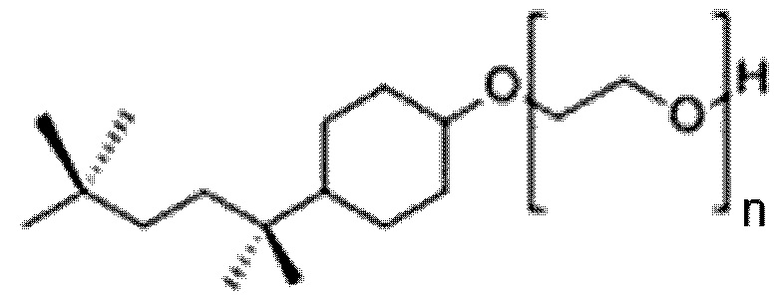
(Формула (V));
в котором n является целым числом, равным от 4 до 16.
28. Способ по параграфу 27, в котором n равно 9 или 10.
29. Способ по любому из параграфов 12-14 и 16, в котором указанным моющим средством является алкиловый эфир линейного полиоксиэтилена.
30. Способ по параграфу 29, в котором указанным моющим средством является линейный гексадециловый эфир полиоксиэтилена.
31. Способ по параграфу 30, в котором указанное моющее средство обладает следующей структурой формулы (VI):

(Формула (VI));
в которой x равно 15.
32. Способ по параграфу 31, в котором указанное моющее средство обладает следующей структурой формулы (VII):
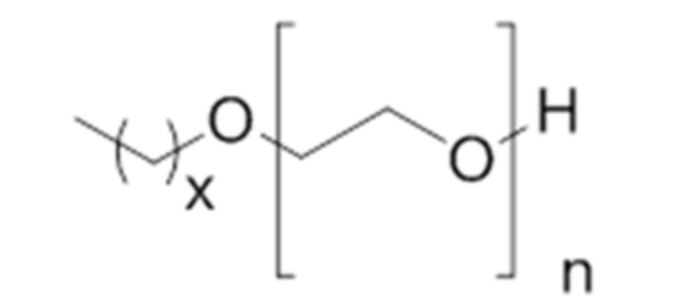
(Формула (VII));
в которой x равно 15 и в которой n является целым числом, равным от 5 до 15.
33. Способ по параграфу 32, в котором n равно 10.
34. Способ по любому из параграфов 12-33, в котором указанное моющее средство применимо для инактивации указанных вирусов.
35. Способ по любому из параграфов 12-34, в котором на стадии a) органический растворитель не добавляют к указанной жидкости.
36. Способ по любому из параграфов 12-34, в котором стадия a) дополнительно включает добавление растворителя к указанной жидкости и в котором на стадии a) смесь растворитель/моющее средство для инактивации указанных вирусов готовят путем добавления указанного моющего средства и указанного растворителя к указанной жидкости.
37. Способ по параграфу 36, в котором указанным растворителем является органический растворитель.
38. Способ по параграфу 36 или 37, в котором указанным растворителем является три-н-бутилфосфат.
39. Способ по любому из параграфов 12-38, в котором на стадии a) кроме указанного моющего средства не добавляют дополнительное моющее средство.
40. Способ по любому из параграфов 12-38, где в способе кроме указанного моющего средства не добавляют дополнительное моющее средство.
41. Способ по любому из параграфов 12-38, в котором стадия a) дополнительно включает добавление дополнительного моющего средства к указанной жидкости.
42. Способ по параграфу 41, в котором указанным дополнительным моющим средством является полисорбат 80.
43. Способ по любому из параграфов 12-42, в котором указанная жидкость включает биологический лечебный продукт.
44. Способ по любому из параграфов 12-43, в котором указанная жидкость включает биофармацевтическое лекарственное средство.
45. Способ по параграфу 44, в котором указанное биофармацевтическое лекарственное средство не является вирусной вакциной.
46. Способ по любому из параграфов 44 или 45, в котором указанным биофармацевтическим лекарственным средством является фактор крови, иммуноглобулин, такой как моноклональные антитела, замещающий фермент, вакцина, генотерапевтический вектор, фактор роста или рецептор фактора роста.
47. Способ по любому из параграфов 44-46, в котором указанным биофармацевтическим лекарственным средством является терапевтический белок.
48. Способ по любому из параграфов 44-47, в котором указанным биофармацевтическим лекарственным средством является фактор крови и в котором указанным фактором крови является фактор I (фибриноген), фактор II (протромбин), тканевый фактор, фактор V, фактор VII или VIIa, фактор VIII, фактор IX, фактор X, фактор XI, фактор XII, фактор XIII, фактор фон Виллебранда (VWF), прекалликреин, кининоген с большой молекулярной массой (HMWK), фибронектин, антитромбин III, кофактор II гепарина, белок C, белок S, белок Z, плазминоген, альфа-2-антиплазмин, тканевый активатор плазминогена (tPA), урокиназа, ингибитор-1 активатора плазминогена (PAI1) или ингибитор-2 активатора плазминогена (PAI2).
49. Способ по любому из параграфов 44-48, в котором указанным биофармацевтическим лекарственным средством является фактор VIII, предпочтительно рекомбинантный фактор VIII человека.
50. Способ по любому из параграфов 44-47, в котором указанным биофармацевтическим лекарственным средством является иммуноглобулин и в котором указанным иммуноглобулином является иммуноглобулин из плазмы человека или моноклональные антитела.
51. Способ по любому из параграфов 12-50, где до стадии a) или между стадией a) и стадией b) указанный способ дополнительно включает стадию фильтрования указанной жидкости или смеси через объемный фильтр.
52. Способ по любому из параграфов 12-51, в котором на стадии b) указанную смесь инкубируют в течение не менее 1 ч.
53. Способ по любому из параграфов 12-52, в котором на стадии b) указанную смесь инкубируют при температуре, равной от 0°C до 10°C, или в котором указанную смесь инкубируют при температуре, равной от 16°C до 25°C.
54. Способ по любому из параграфов 44-53, после стадии b) дополнительно включающий стадию
c) очистки указанного биофармацевтического лекарственного средства.
55. Способ по параграфу 54, в котором указанная очистка включает отделение указанного биофармацевтического лекарственного средства от указанного моющего средства.
56. Способ по параграфу 54 или 55, в котором указанная очистка включает отделение указанного биофармацевтического лекарственного средства от указанного дополнительного моющего средства.
57. Способ по любому из параграфов 54-56, в котором указанная очистка указанного биофармацевтического лекарственного средства включает очистку указанного биофармацевтического лекарственного средства с помощью по меньшей мере одной хроматографической очистки.
58. Способ по любому из параграфов 54-57, в котором указанная по меньшей мере одна хроматографическая очистка проводится с помощью анионообменной хроматографии и/или с помощью катионообменной хроматографии.
59. Способ получения биофармацевтического лекарственного средства, указанный способ включает способ по любому из параграфов 44-58, в котором указанное биофармацевтическое лекарственное средство является таким, как определено в любом из указанных параграфов 44-58.
60. Способ по параграфу 59, дополнительно включающий после указанного способа по любому из параграфов 44-58 стадию получения фармацевтического состава, включающего указанное биофармацевтическое лекарственное средство.
61. Применение моющего средства по любому из параграфов 12-34 в способе инактивации вирусов, обладающих липидной оболочкой.
62. Применение по параграфу 61, где при указанном применении кроме указанного моющего средства не используют дополнительное моющее средство.
63. Применение по параграфу 61 или 62, где при указанном применении не используют органический растворитель.
64. Применение по параграфу 61, в котором указанный способ указанной инактивации указанных вирусов представляет собой способ с использованием обработки растворителем/моющим средством, указанная обработка растворителем/моющим средством включает применение указанного моющего средства по любому из параграфов 12-34.
65. Применение по любому из параграфов 61-64, в котором указанная инактивация вирусов представляет собой инактивацию вирусов в жидкости, включающей биофармацевтическое лекарственное средство по любому из параграфов 44-50.
66. Композиция, включающая моющее средство по любому из параграфов 12-34.
67. Моющее средство по любому из параграфов 12-34.
68. Композиция по параграфу 66, дополнительно включающая биофармацевтическое лекарственное средство по любому из параграфов 44-50.
69. Композиция по параграфу 66 или 68, где композиция не включает какой-либо органический растворитель.
70. Композиция по параграфу 66 или 68, дополнительно включающая органический растворитель по любому из параграфов 37 и 38.
71. Композиция по любому из параграфов 66 или 68-70, где композиция не включает какое-либо дополнительное моющее средство кроме указанного моющего средства.
72. Композиция по любому из параграфов 66 или 68-70, дополнительно включающая дополнительное моющее средство по любому из параграфов 41 и 42.
73. Набор для инактивации вирусов, включающий моющее средство по параграфу 67 или композицию по любому из предыдущих параграфов и дополнительно включающий хроматографическую смолу для хроматографической очистки по любому из параграфов 57-58.
74. Набор по параграфу 73, дополнительно включающий объемный фильтр.
75. Способ синтеза соединения следующей общей формулы (VIII)
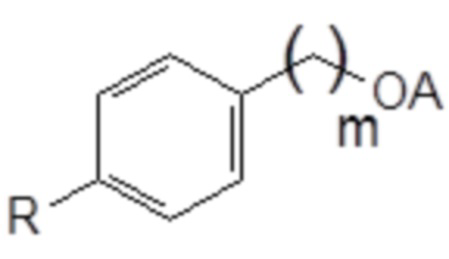
Формула (VIII)
в которой
R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи,
m является целым числом, равным от 1 до 4, и
A означает полиоксиэтиленовый остаток,
где способ включает стадии
A) превращение фенола следующей общей формулы (IX), в которой R является таким, как определено выше
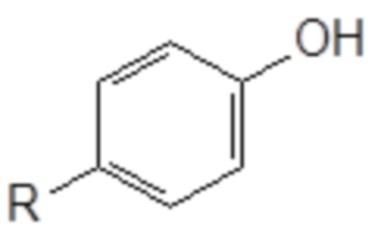
Формула (IX)
в спирт следующей общей формулы (X), в которой R и m являются такими, как определено выше
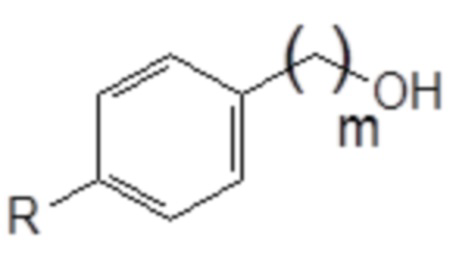
Формула (X)
и
(B) превращение спирта общей формулы (X) в простой эфир полиоксиэтилена общей формулы (VIII), определенной выше.
76. Способ синтеза соединения следующей общей формулы (VIII)
Формула (VIII)
в которой
R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи,
m является целым числом, равным от 1 до 4, и
A означает полиоксиэтиленовый остаток,
где способ включает стадии
(1) введение в реакцию толуола с получением замещенного толуола следующей общей формулы (XI), в которой R является таким, как определено выше
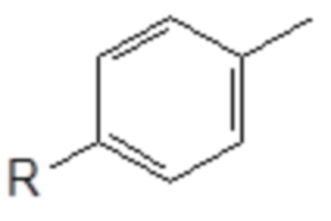
Формула (XI)
(2) превращение замещенного толуола общей формулы (XI) в соединение следующей общей формулы (XII), в которой R и m являются такими, как определено выше, и X выбран из группы, включающей гидроксигруппу, атом брома, атом йода и атом хлора
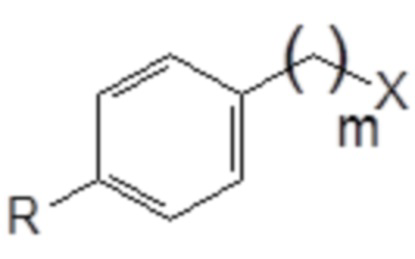
Формула (XII)
и
(3) превращение соединения общей формулы (XII) в простой эфир полиоксиэтилена общей формулы (VIII), определенной выше.
77. Способ синтеза соединения следующей общей формулы (VIIIa)
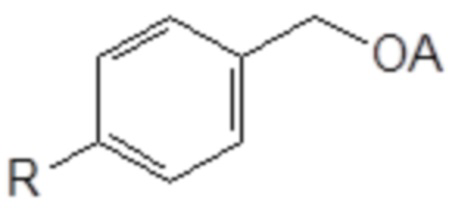
Формула (VIIIa)
в которой
R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи, и
A означает полиоксиэтиленовый остаток,
где способ включает стадии
(I) превращение бензилового спирта в соединение следующей общей формулы (XIII), в которой R является таким, как определено выше
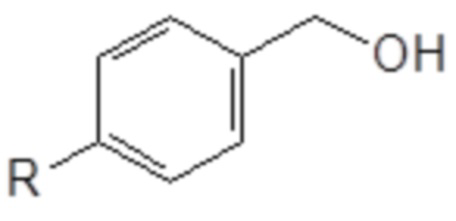
Формула (XIII)
и
(II) превращение соединения общей формулы (XIII) в простой эфир полиоксиэтилена общей формулы (VIIIa), определенной выше.
78. Способ по любому из параграфов 75-77, в котором A означает полиоксиэтиленовый остаток, содержащий от 4 до 16 оксиэтиленовых звеньев.
79. Способ по параграфу 78, в котором A означает полиоксиэтиленовый остаток, содержащий от 8 до 10 оксиэтиленовых звеньев.
80. Способ по параграфу 78, в котором A означает полиоксиэтиленовый остаток, содержащий 9 или 10 оксиэтиленовых звеньев.
81. Способ по любому из параграфов 75, 76 и 78-80, в котором m равно 1.
82. Способ по любому из параграфов 75-81, в котором R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 6 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи.
83. Способ по любому из параграфов 75-82, в котором R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 6 атомов углерода и от 2 до 4 метильных групп в качестве заместителей указанной линейной цепи.
84. Способ по любому из параграфов 75-83, в котором R означает углеводородную группу, включающую линейную цепь, содержащую 4 атома углерода и 4 метильные группы в качестве заместителей указанной линейной цепи.
85. Способ по любому из параграфов 75-84, в котором R означает 2,4,4-триметилпент-2-ильную группу.
86. Способ по любому из параграфов 75-77, в котором соединением формулы (VIII) является следующее соединение:
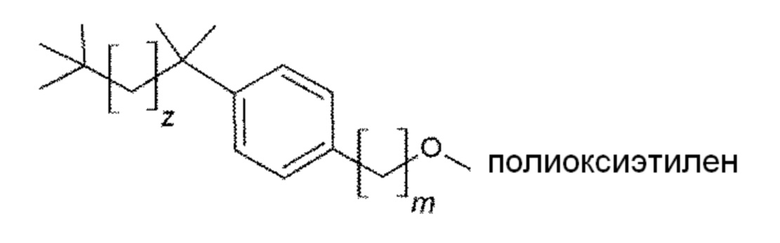
в котором m и z являются целыми числами, которые независимо выбраны из следующих групп:
m=от 1 до 4
z=от 1 до 5.
87. Способ по параграфу 86, в котором m равно 1.
88. Способ по любому из параграфов 75-77, в котором соединением формулы (VIII) является следующее соединение:
в котором n является целым числом, равным от 4 до 16, предпочтительно в котором n равно 9 или 10.
89. Способ по любому из параграфов 76 и 78-88, в котором превращение на стадии (2) представляет собой радикальную реакцию с использованием AIBN (азобис(изобутиронитрил)) в качестве радикального инициатора.
90. Способ по любому из параграфов 76 и 78-89, в котором X означает атом брома.
91. Способ по любому из параграфов 76 и 78-90, в котором при превращении на стадии (2) используется N-бромсукцинимид (NBS) в качестве реагента.
92. Способ по любому из параграфов 76 и 78-91, в котором при превращении на стадии (3) используется TBME (метил-трет-бутиловый эфир) в качестве растворителя.
93. Способ по любому из параграфов 76 и 78-92, в котором превращение на стадии (3) протекает в течение не менее 2 ч, предпочтительно при температуре окружающей среды.
94. Способ по любому из параграфов 76 и 78-93, в котором превращение на стадии (3) протекает в течение не более 5 ч, предпочтительно при температуре окружающей среды.
95. Способ по любому из параграфов 76 и 78-94, в котором превращение на стадии (3) протекает в течение 3 ч, предпочтительно при температуре окружающей среды.
96. Способ по любому из параграфов 75-95, где способ проводят в масштабе, который дает не менее 100 г, не менее 1 кг, не менее 10 кг, не менее 100 кг или не менее 1000 кг указанного соединения указанной формулы (VIII).
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1: Эффективность инактивации вирусов при низких концентрациях Triton X-100 reduced или Triton N-101 reduced при S/D обработке содержащей IVIG жидкости при 17°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,04% - 0,06% Triton X-100 reduced или Triton N-101 reduced, 0,01% - 0,02% полисорбата 80, 0,01% - 0,02% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с HIV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием Triton X-100 reduced ("TX-100 red.") или Triton N-101 reduced ("TN-101 red.") сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100").
Фиг. 2: Эффективность инактивации вирусов при низких концентрациях Triton X-100 reduced или Triton N-101 reduced при S/D обработке содержащей IVIG жидкости при 17°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,04% - 0,06% Triton X-100 reduced или Triton N-101 reduced, 0,01% - 0,02% полисорбата 80, 0,01% - 0,02% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с PRV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием Triton X-100 reduced ("TX-100 red.") или Triton N-101 reduced ("TN-101 red.") сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100").
Фиг. 3: Эффективность инактивации вирусов при низких концентрациях Brij C10 при S/D обработке содержащей IVIG жидкости при 17°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,04% - 0,06% Brij C10, 0,01% - 0,02% полисорбата 80, 0,01% - 0,02% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с HIV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием Brij C10 сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100").
Фиг. 4: Эффективность инактивации вирусов при низких концентрациях Brij C10 при S/D обработке содержащей IVIG жидкости при 17°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,04% - 0,06% Brij C10, 0,01% - 0,02% полисорбата 80, 0,01% - 0,02% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с PRV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием Brij C10 сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100").
Фиг. 5: Эффективность инактивации вирусов при низких концентрациях Brij C10 при S/D обработке содержащей IVIG жидкости при 17°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,04% - 0,06% Brij C10, 0,01% - 0,02% полисорбата 80, 0,01% - 0,02% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с BVDV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием Brij C10 сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100").
Фиг. 6: Эффективность инактивации вирусов при низких концентрациях Brij C10 при S/D обработке содержащей сывороточный альбумин человека (HSA) жидкости при 1°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,08% - 0,1% Brij C10, 0,02% - 0,03% полисорбата 80, 0,02% - 0,03% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с X-MuLV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием Brij C10 сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100").
Фиг. 7: Эффективность инактивации вирусов при низких концентрациях Brij C10 при S/D обработке содержащей HSA жидкости при 1°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,08% - 0,1% Brij C10, 0,02% - 0,03% полисорбата 80, 0,02% - 0,03% TnBP (сопоставление с имеющимися данными для Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с BVDV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием Brij C10 сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100").
Фиг. 8: Эффективность инактивации вирусов при низких концентрациях Brij C10 при S/D обработке содержащей HSA жидкости при 19°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,08% - 0,1% Brij C10, 0,02% - 0,03% полисорбата 80, 0,02% - 0,03% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с X-MuLV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием Brij C10 сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100").
Фиг. 9: Эффективность инактивации вирусов при низких концентрациях полиэтоксилата 4-трет-октилбензилового спирта при S/D обработке содержащей IVIG жидкости при 17°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,04% - 0,06% полиэтоксилата 4-трет-октилбензилового спирта, 0,01% - 0,02% полисорбата 80, 0,01% - 0,02% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с PRV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием полиэтоксилата 4-трет-октилбензилового спирта сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100"). Следует отметить, что для (A) оба моющие средства приводят к идентичной кинетике инактивации. Зачерненными символами указаны значения с остаточной инфекционностью, зачерненные символы соответствуют коэффициентам уменьшения содержания ниже предела обнаружения.
Фиг. 10: Эффективность инактивации вирусов при низких концентрациях полиэтоксилата 4-трет-октилбензилового спирта при S/D обработке содержащей сывороточный альбумин человека содержащей (HSA) жидкости при 1°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,08% - 0,1% полиэтоксилата 4-трет-октилбензилового спирта, 0,02% - 0,03% полисорбата 80, 0,02% - 0,03% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с X-MuLV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием полиэтоксилата 4-трет-октилбензилового спирта сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100"). Зачерненными символами указаны значения с остаточной инфекционностью, зачерненные символы соответствуют коэффициентам уменьшения содержания ниже предела обнаружения.
Фиг. 11: Эффективность инактивации вирусов при низких концентрациях полиэтоксилата 4-трет-октилбензилового спирта при S/D обработке содержащей HSA жидкости при 19°C ± 1°C. Использовали трехкомпонентную смесь и получали конечные концентрации 0,08% - 0,1% полиэтоксилата 4-трет-октилбензилового спирта, 0,02% - 0,03% полисорбата 80, 0,02% - 0,03% TnBP (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает коэффициент уменьшения содержания вируса (RF) для двух экспериментов с X-MuLV (A и B соответственно). Инактивацию вирусов при S/D обработке с использованием полиэтоксилата 4-трет-октилбензилового спирта сопоставляли с инактивацией вирусов при S/D обработке с использованием Triton X-100 ("TX-100"). Зачерненными символами указаны значения с остаточной инфекционностью, зачерненные символы соответствуют коэффициентам уменьшения содержания ниже предела обнаружения.
Фиг. 12: (A) Эффективность инактивации вирусов при низких концентрациях полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced или Brij C10 при обработке моющим средством - буфером, содержащим HSA при 14°C ± 1°C. Проводили обработку одним моющим средством и получали конечные концентрации 0,09% - 0,11% полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced или Brij C10 (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает средний коэффициент уменьшения содержания вируса (RF) для двух экспериментов с BVDV. Инактивацию вирусов при обработке одним моющим средством с использованием полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced ("TX-100 red.") или Brij C10 сопоставляли с инактивацией вирусов при обработке одним моющим средством с использованием Triton X-100 ("TX-100"). Следует отметить, что полиэтоксилат 4-трет-октилбензилового спирта и Triton X-100 характеризуются такой же кинетикой инактивации, как Triton X-100. Зачерненными символами указаны значения с остаточной инфекционностью, зачерненные символы соответствуют коэффициентам уменьшения содержания ниже предела обнаружения. (B) Эффективность инактивации вирусов при низких концентрациях полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced при обработке моющим средством - буфером, содержащим HSA при 14°C ± 1°C. Проводили обработку одним моющим средством и получали конечные концентрации 0,02% - 0,04% полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает средний коэффициент уменьшения содержания вируса (RF) для двух экспериментов с BVDV. Инактивацию вирусов при обработке одним моющим средством с использованием полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced ("TX-100 red.") сопоставляли с инактивацией вирусов при обработке одним моющим средством с использованием Triton X-100 ("TX-100"). Зачерненными символами указаны значения с остаточной инфекционностью, зачерненные символы соответствуют коэффициентам уменьшения содержания ниже предела обнаружения.
Фиг. 13: (A) Эффективность инактивации вирусов при низких концентрациях полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced при обработке моющим средством содержащей IVIG жидкости при 17°C ± 1°C. Проводили обработку одним моющим средством и получали конечные концентрации 0,09% - 0,11% полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает средний коэффициент уменьшения содержания вируса (RF) для двух экспериментов с BVDV. Инактивацию вирусов при обработке одним моющим средством с использованием полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced ("TX-100 red.") сопоставляли с инактивацией вирусов при обработке одним моющим средством с использованием Triton X-100 ("TX-100"). Зачерненными символами указаны значения с остаточной инфекционностью, зачерненные символы соответствуют коэффициентам уменьшения содержания ниже предела обнаружения. (B) Эффективность инактивации вирусов при низких концентрациях полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced при обработке моющим средством содержащей IVIG жидкости при 17°C ± 1°C. Проводили обработку одним моющим средством и получали конечные концентрации 0,02% - 0,04% полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает средний коэффициент уменьшения содержания вируса (RF) для двух экспериментов с BVDV. Инактивацию вирусов при обработке одним моющим средством с использованием полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced ("TX-100 red.") сопоставляли с инактивацией вирусов при обработке одним моющим средством с использованием Triton X-100 ("TX-100"). Зачерненными символами указаны значения с остаточной инфекционностью, зачерненные символы соответствуют коэффициентам уменьшения содержания ниже предела обнаружения.
Фиг. 14: Эффективность инактивации вирусов при низких концентрациях полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced при обработке моющим средством содержащей фактор VIII жидкости при 23°C ± 1°C. Проводили обработку одним моющим средством и получали конечные концентрации 0,09% - 0,11% полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced (непосредственное сравнение с такой же концентрацией Triton X-100). На зависимость инактивации вирусов от времени указывает средний коэффициент уменьшения содержания вируса (RF) для двух экспериментов с BVDV. Инактивацию вирусов при обработке одним моющим средством с использованием полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced ("TX-100 red.") сопоставляли с инактивацией вирусов при обработке одним моющим средством с использованием Triton X-100 ("TX-100"). Зачерненными символами указаны значения с остаточной инфекционностью, зачерненные символы соответствуют коэффициентам уменьшения содержания ниже предела обнаружения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если ниже не приведено другое определение, термины, использующиеся в настоящем изобретении, обладают своими обычными значениями, известными специалисту в данной области техники.
Все публикации, патенты и заявки на патенты, цитированные в настоящем изобретении, во всей их полноте включены в настоящее изобретение в качестве ссылки для всех объектов.
Определения
Термины "вирус, обладающий липидной оболочкой", "вирус с липидной оболочкой" и "вирус с оболочкой" в настоящем изобретении используются взаимозаменяемым образом и обладают значениями, известными специалисту в данной области техники. Например, вирусами с липидной оболочкой могут быть Herpesviridae, такие как вирус псевдобешенства (PRV), вирус простого герпеса, вирус ветряной оспы, цитомегаловирус или вирус Эпштейна-Барра; Hepadnaviridae, такие как вирус гепатита B; Togaviridae, такие как вирус Синдбис, вирус краснухи или альфавирус; Arenaviridae, такие как вирус лимфоцитарного хориоменингита; Flaviviridae, такие как вирус Западного Нила, вирус диареи крупного рогатого скота (BVDV), вирус денге, вирус гепатита C или вирус желтой лихорадки; Orthomyxoviridae, такие как вирус гриппа A, вирус гриппа B, вирус гриппа C, изавирус или вирус Тогото; Paramyxoviridae, такие как вирус Сендай, вирус кори, вирус свинки, респираторно-синцитиальный вирус, вирус чумы или вирус чумки собачьих; Bunyaviridae, такие как вирус калифорнийского энцефалита или хантавирус; Rhabdoviridae, такие как вирус везикулярного стоматита или вирус бешенства; Filoviridae, такие как вирус Эбола или вирус, вызывающий "марбургскую болезнь"; Coronaviridae, такие как коронавирус или коронавирус тяжелого острого респираторного синдрома (SARS); Bornaviridae, такие как вирус болезни Борна; или Arteriviridae, такие как артеривирус или вирус артериита лошадей; Retroviridae, такие как вирус иммунодефицита человека (HIV), Т-лимфотропный вирус человека 1 (HTLV-1) или ксенотропный вирус лейкемии мышей (X-MuLV); Poxviridae, такие как вирус вакцины или ортопоксвирус натуральной оспы (вирус натуральной оспы).
Термин "инактивация вирусов, обладающих липидной оболочкой" при использовании в настоящем изобретении означает нарушение способности вируса с липидной оболочкой инфицировать клетки. Как должен понимать специалист в данной области техники, способность вируса с липидной оболочкой инфицировать клетки, т. е. инфекционность вируса с липидной оболочкой, обычно оценивают путем определения количества контагиозных вирусных частиц в жидкости. Таким образом, термин "инактивация вирусов, обладающих липидной оболочкой" или "инактивация вируса с липидной оболочкой" при использовании в настоящем изобретении означает уменьшение количества контагиозных вирусных частиц в растворе.
В настоящем изобретении термин "Log10 коэффициента уменьшения" или "LRV" используются взаимозаменяемым образом с терминами "коэффициент уменьшения содержания вируса", "коэффициент уменьшения", "RF" или "R". В одном варианте осуществления "Log10 коэффициента уменьшения" или "LRV" можно использовать в качестве меры уменьшения количества контагиозных вирусных частиц в жидкости. При использовании в настоящем изобретении "Log10 коэффициента уменьшения" или "LRV" определяется, как логарифм (с основанием 10) отношения количества контагиозных вирусных частиц до инактивации вирусов к количеству контагиозных вирусных частиц после инактивации вирусов. Значение LRV специфично для данного типа вируса. Для специалиста в данной области техники очевидно, что любое значение Log10 коэффициента уменьшения (LRV), превышающее 0, благоприятно для улучшения безопасности способов и процессов, таких как способы и процессы биофармацевтического производства. Значение Log10 коэффициента уменьшения (LRV), которое обеспечивается способами, предлагаемыми в настоящем изобретении, определяют так, как известно специалисту в данной области техники. Например, LRV можно определить путем установления количества контагиозных вирусных частиц в жидкости до и после обработки жидкости в соответствии со способом инактивации вирусов, предлагаемым в настоящем изобретении.
Специалисту в данной области техники известны многочисленные методики определения содержания контагиозных вирусных частиц в жидкости. Например и без наложения ограничений, концентрацию контагиозных вирусных частиц в жидкости предпочтительно можно определить с помощью анализа бляшкообразования или с помощью анализа TCID50, более предпочтительно с помощью анализа TCID50. "Анализ TCID50" при использовании в настоящем изобретении означает анализ инфекционной дозы культуры ткани. Анализ TCID50 представляет исследование при конечном разведении, при котором значение TCID50 представляет собой концентрацию вирусов, необходимую для инициирования гибели клеток или патологических изменений в 50% инокулированной культуры клеток.
Термин "поверхностно-активное вещество" при использовании в настоящем изобретении означает соединение, которое снижает поверхностное натяжение между двумя жидкостями или между жидкостью и твердым веществом. Поверхностно-активные вещества могут действовать, как моющие средства, смачивающие агенты, эмульгаторы, вспенивающие агенты и диспергирующие средства.
В соответствии с настоящим изобретением такие термины, как "добавление к", "добавить к" или "добавляли к" в связи с первым указанным и вторым указанным растворителем, моющим средством и/или жидкостью включают ситуацию, когда первый указанный растворитель, моющее средство и/или жидкость добавляют ко второму указанному растворителю, моющему средству и/или жидкости. Однако эти термины также включают ситуацию, когда второй указанный растворитель, моющее средство и/или жидкость добавляют к первому указанному растворителю, моющему средству и/или жидкости. Таким образом, такие термины, как "добавление к", "добавить к" или "добавляли к" специально не указывают, следует ли добавлять первый указанный растворитель, моющее средство и/или жидкость ко второму указанному растворителю, моющему средству и/или жидкости, или наоборот.
Как известно специалисту в данной области техники, термин "моющее средство" используют в соответствии с его обычным значением, известным в данной области техники, и он включает, в частности, поверхностно-активные вещества, которые могут проходить через липидные мембраны. Например, моющие средства Triton X-100 и дезоксихолат предложены для солюбилизации амфипатических мембранных белков путем связывания с гидрофобными сегментами белков (см. Simons et al., 1973). Моющие средства в соответствии с их электрическим зарядом разделяют на три обширные группы. Анионогенные моющие средства содержат анионогенную, т. е. отрицательно заряженную гидрофильную группу. Типичными анионогенными моющими средствами являются тетрадецилтриметиламмонийбромид, додецилтриметиламмонийбромид, лауретсульфат натрия, додецилсульфат натрия (SDS), цетримид и тетрагексилтриметиламмонийбромид. Катионогенные моющие средства содержат катионогенную, т. е. положительно заряженную гидрофильную группу. Типичными катионогенными моющими средствами являются бензалконийхлорид, цетилтриметиламмонийбромид (CTAB), цетилпиридинийхлорид (CPC) и бензэтонийхлорид (BZT). Термин "неионогенные моющие средства" при использовании в настоящем изобретении означает моющие средства, не обладающие ни положительным, ни отрицательным зарядом. Типичными неионогенными моющими средствами являются сорбитановые эфиры (сорбитанмонолаурат, сорбитанмонопальмитат, сорбитанмоностеарат, сорбитантристеарат, сорбитанмоноолеат, сорбитантриолеат), полисорбаты (полиоксиэтилен-(20)-сорбитанмонолаурат (полисорбат-20), полиоксиэтилен-(20)-сорбитанмонопальмитат, полиоксиэтилен-(20)-сорбитанмоностеарат, полиоксиэтилен-(20)-сорбитантристеарат, полиоксиэтилен-(20)-сорбитантриолеат, полиоксиэтилен-(20)-сорбитанмоноолеат (Tween 80/полисорбат 80)), полоксамеры (полоксамер 407, полоксамер 188) и кремофор. Моющие средства, предлагаемые в настоящем изобретении, являются такими, как описано в предпочтительных вариантах осуществления, и включают без наложения ограничений, Triton N-101 reduced, Triton X-100 reduced и Brij C10.
Термин "нефенольное" при использовании в настоящем изобретении используется взаимозаменяемым образом с термином "не содержащее фенол". Нефенольное моющее средство при использовании в настоящем изобретении означает моющее средство, которое не содержит фенольные функциональные группы. Термин "ароматическое" обладает значением, известным специалисту в данной области техники. Моющее средство, которое не является ароматическим, при использовании в настоящем изобретении означает моющее средство, которое не содержит какие-либо ароматические кольца.
Термин "экологически приемлемое" при использовании в настоящем изобретении обладает значением, известным специалисту в данной области техники. В предпочтительном варианте осуществления настоящего изобретения применительно к моющим средствам термин "экологически приемлемое" показывает, что моющее средство не ведет себя, как разрушитель эндокринной системы. Разрушители эндокринной системы являются эндогенными веществами, которые изменяют функцию (функции) эндокринной системы и поэтому приводят к вредным последствием для состояния здоровья непораженного организма или его потомства, или (суб)популяций. Специалисту в данной области техники известны разные методики выявления разрушителей эндокринной системы. Дополнительная информация о разрушителях эндокринной системы и их исследований приведена, например, в документе ECHA Support, в котором описано установление того, что этоксилированный 4-(1,1,3,3-тетраметилбутил)фенол относится к веществам, вызывающим большие опасения, поскольку его разложение с образованием вещества, (4-(1,1,3,3-тетраметилбутил)фенола), вызывающего большие опасения, поскольку оно обладает способностью разрушать эндокринную систему, оно вызывает возможное сильное влияние на окружающую среду, что приводит к степени опасений, эквивалентной опасениям по отношению к CMRs и PBTs/vPvBs", принятом 12 декабря 2012 г.), который во всей его полноте включен в настоящее изобретение в качестве ссылки для всех объектов; или в брошюре "Global Assessment of the Sate-of-the-Science of Endocrine Disruptors" (WHO/PCS/EDC/02,2), которая во всей ее полноте включена в настоящее изобретение в качестве ссылки для всех объектов, опубликованной the International Programme on Chemical Safety of the World Health Organisation.
Термин "растворитель" при использовании в настоящем изобретении обладает значением, известным специалисту в данной области техники. В предпочтительном варианте осуществления настоящего изобретения органические растворители используют в способах, предлагаемых в настоящем изобретении. Особенно подходящие органические растворители создают среду, стимулирующую взаимодействие между моющим средством и липопротеиновой оболочкой вируса с липидной оболочкой. Органический растворитель, который стимулирует такое взаимодействие, предпочтительно используют в способах, предлагаемых в настоящем изобретении, включая, без наложения ограничений, простой эфир, спирт, алкилфосфат, такой как диалкилфосфат или триалкилфосфат, или любую их комбинацию.
Простые эфирные растворители, применимые в способах, раскрытых в настоящем изобретении, включают описывающиеся формулой R1-O-R2, в которой R1 и R2 независимо означают C1-C18 алкил или C1-C18 алкенил, который может содержать атом кислорода или атом серы, предпочтительно C1-C18 алкил или C1-C18 алкенил. Неограничивающие примеры простых эфиров включают диметиловый эфир, диэтиловый эфир, этилпропиловый эфир, метилбутиловый эфир, метилизопропиловый эфир и метилизобутиловый эфир. Спиртовые растворители, применимые в способе, раскрытом в настоящем изобретении, включают содержащие C1-C8 алкильные группы или C1-C8 алкенильные группы. Неограничивающие примеры спиртов включают метанол, этанол, пропанол, изопропанол, н-бутанол, изобутанол, н-пентанол и изопентанолы. Алкилфосфатные растворители, применимые в способе, раскрытом в настоящем изобретении, включают содержащие C1-C18 алкильные группы или C1-C18 алкенильные группы, каждая из которых может содержать атом кислорода или атом серы. Неограничивающие примеры алкилфосфатов включают диалкилфосфаты, такие как ди-(н-бутил)фосфат, ди-(трет-бутил)фосфат, ди-(н-гексил)фосфат, ди-(2-этилгексил)фосфат, ди-(н-децил)фосфат, или этилди(н-бутил)фосфат; и триалкилфосфаты, такие как три-(н-бутил)фосфат, три-(трет-бутил)фосфат, три-(н-гексил)фосфат, три-(2-этилгексил)фосфат или три-(н-децил)фосфат.
Термин "биологический медицинский продукт", использующийся в настоящем изобретении, известен в данной области техники и означает продукт, активное вещество которого является биологическим веществом, например, биологическим веществом, которое вырабатывается клетками млекопитающего или микроорганизмов. При использовании в настоящем изобретении биологический медицинский продукт, использующийся в способах, предлагаемых в настоящем изобретении, не ограничивается конечным изготовленным продуктом, предпочтительно также включает промежуточные продукты на любой стадии процесса получения.
Термин "биофармацевтическое лекарственное средство" при использовании в настоящем изобретении обладает значением, известным специалисту в данной области техники. Биофармацевтические лекарственные средства включают рекомбинантные биофармацевтические лекарственные средства и биофармацевтические лекарственные средства из других источников, такие как биофармацевтические лекарственные средства, полученные из плазмы человека.
При использовании в настоящем изобретении термин "объемный фильтр" обладает значением, известным в данной области техники. В частности, такой фильтр (например, объемный фильтр с градиентом плотности) обеспечивает фильтрование в глубине фильтрующего материала. Обычным классом таких фильтров являются те, которые содержат матрицу из случайным образом распределенных волокон, связанных (или иным образом зафиксированных) с образованием комплекса, запутанного лабиринта проточных каналов. Разделение частиц в таких фильтрах обычно обусловлено захватом или адсорбцией в матрице из волокон. Чаще всего использующимися средами для объемного фильтра для биологической обработки бульонов клеточных культур и другого сырья являются целлюлозные волокна, фильтрующее средство, такое как DE, и положительно заряженная связующая смола. Среды объемного фильтра, в отличие от абсолютных фильтров, удерживают частицы в пористых средах, обеспечивающих удерживание частиц, более крупных или менее крупных, чем размер пор. Считается, что удерживание частиц включает исключение по размерам и адсорбцию посредством гидрофобных, ионных и других взаимодействий.
Термин "очистка биофармацевтического лекарственного средства" при использовании в настоящем изобретении обладает значением, известным специалисту в данной области техники, и означает отделение биофармацевтического лекарственного средства от других веществ, которые могут содержаться в смеси, предлагаемой в настоящем изобретении. В предпочтительном варианте осуществления настоящего изобретения термин "очистка биофармацевтического лекарственного средства" означает отделение биофармацевтического лекарственного средства от моющего средства, предлагаемого в настоящем изобретении.
Термин "хроматография" используется в соответствии с его значением, известным в данной области техники. Он включает любую хроматографическую методику, с помощью которой представляющее интерес анализируемое вещество (например, молекула-мишень, такая как биофармацевтическое лекарственное средство) отделяется от других молекул, содержащихся в смеси. Обычно представляющее интерес анализируемое вещество отделяется от других молекул вследствие различий скоростей, с которыми отдельные молекулы смеси перемещаются через неподвижную среду под влиянием подвижной фазы или вследствие процессов связывания и элюирования.
Термины "хроматографическая смола" и "хроматографические среды" в настоящем изобретении используются взаимозаменяемым образом и означают любой тип фазы (например, твердую фазу), которая отделяет представляющее интерес анализируемое вещество (например, молекулу-мишень, такую как биофармацевтическое лекарственное средство) от других молекул, содержащихся в смеси. Обычно представляющее интерес анализируемое вещество отделяется от других молекул вследствие различий скоростей, с которыми отдельные молекулы смеси перемещаются через неподвижную среду под влиянием подвижной фазы или вследствие процессов связывания и элюирования. Примеры хроматографических сред разных типов включают, например, катионообменные смолы, катионообменные мембраны, аффинные смолы, анионообменные смолы, анионообменные мембраны, смолы с гидрофобным взаимодействием и ионообменные монолиты.
Термин "фармацевтический состав" при использовании в настоящем изобретении обладает значением, известным специалисту в данной области техники и означает любой состав, который пригоден для введения пациенту. Фармацевтические составы можно получить по методикам, известным в данной области техники. Например, для любого биофармацевтического лекарственного средства, которое содержится в составе, специалист в данной области техники способен выбрать и добавить предпочтительные дополнительные ингредиенты, включая буферы, стабилизаторы, поверхностно-активные вещества, антиоксиданты, хелатные агенты и/или консерванты и т. п.
Термин "смесь растворитель/моющее средство" при использовании в настоящем изобретении обладает значением, известным специалисту в данной области техники. В предпочтительном варианте осуществления смесь растворитель/моющее средство, которую используют в соответствии с настоящим изобретением, содержит по меньшей мере один растворитель, не являющийся водой, и по меньшей мере одно моющее средство. Растворитель, использующийся в соответствии с настоящим изобретением, предпочтительно представляет собой органический растворитель и наиболее предпочтительно три-н-бутилфосфат. На количество разных растворителей и/или моющих средств, содержащихся в смеси, не налагаются особые ограничения. Например, смесь растворитель/моющее средство может состоять из три-н-бутилфосфата, полисорбата 80 и моющего средства на основе простого эфира полиоксиэтилена, предлагаемого в настоящем изобретении.
Следует понимать, что термин "от" при использовании для указания числового диапазона в настоящем изобретении включает указанные нижние и верхние граничные значения соответствующего диапазона. Например, если указано, что температура равна от 0°C до 10°C, это включает температуры, равные от 0°C до 10°C. Аналогичным образом, если указано, что переменная x является целым числом, равным, например, от 4 до 16, это включает целые числа, равные от 4 до 16.
Следует понимать, что термин "1 ч" при использовании в настоящем изобретении не ограничивается точно 60 минутами. При использовании в настоящем изобретении термин "1 ч" означает 60 мин ± 5 мин, предпочтительно 60 мин ± 2 мин.
Варианты осуществления
Способ инактивации вирусов, обладающих липидной оболочкой, предлагаемый в настоящем изобретении, включает стадию добавления моющего средства к жидкости для приготовления смеси указанного моющего средства и указанной жидкости и стадию инкубирования указанной смеси для инактивации указанных вирусов. Как указано выше, термин "инактивация вирусов, обладающих липидной оболочкой" при использовании в настоящем изобретении означает уменьшение концентрации контагиозных вирусных частиц в растворе. В способе инактивации вирусов, обладающих липидной оболочкой, предлагаемом в настоящем изобретении, предпочтительно, если способ обеспечивает по меньшей мере 1 Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса, или по меньшей мере 2 Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса, или по меньшей мере 3 Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса, или по меньшей мере 4 Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса, или по меньшей мере 5 Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса, или по меньшей мере 6 Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса, или по меньшей мере 7 Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса, или по меньшей мере 8 Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса, наиболее предпочтительно по меньшей мере 4 Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса. Разумеется, для специалиста в данной области техники очевидно, что любой Log10 коэффициента уменьшения (LRV) по меньшей мере для одного вируса является благоприятным, поскольку он приводит к увеличению безопасности, например, биофармацевтического производственного процесса. LRVs, указанные в настоящем изобретении, представляют собой LRVs для вируса с оболочкой.
Следует понимать, что, хотя способы, предлагаемые в настоящем изобретении, обычно предназначенные для инактивации вирусов с липидной оболочкой, моющие средства, предлагаемые в настоящем изобретении, также могут инактивировать вирусы без оболочки, например, если такие вирусы без оболочки приобретают липидную оболочку на некоторой стадии во время их цикла репликации. Таким образом, термин "инактивация вирусов, обладающих липидной оболочкой" не исключает возможности того, что в способах, предлагаемых в настоящем изобретении, в дополнение к вирусам, обладающим липидной оболочкой, также можно инактивировать вирусы без оболочки.
В способе инактивации вирусов, обладающих липидной оболочкой, предлагаемом в настоящем изобретении, моющее средство добавляют к жидкости для приготовления смеси указанного моющего средства и указанной жидкости, и указанную смесь инкубируют для инактивации указанных вирусов. Следует понимать, что жидкость, указанная в способе, предлагаемом в настоящем изобретении, может быть жидкостью любого типа или смесью нескольких жидкостей, включая раствор, суспензию, или смесь нескольких суспензий и/или растворов. Например, и без наложения ограничений указанная жидкость, предлагаемая в настоящем изобретении, может представлять собой кровь или может содержать кровь или фракцию крови, может представлять собой плазму или может содержать плазму или фракцию плазмы, может представлять собой сыворотку или может содержать сыворотку или фракцию сыворотки, может представлять собой клеточную культуральную среду или может содержать клеточную культуральную среду, может представлять собой буфер или может содержать буфер. Жидкость также может представлять собой промежуточный продукт процесса, например, промежуточный продукт процесса получения биофармацевтического лекарственного средства.
Жидкость, предлагаемая в настоящем изобретении, может содержать вирус, обладающий липидной оболочкой, или можно предполагать, что она содержит вирус, обладающий липидной оболочкой (например, в случае, если она представляет собой кровь или содержит кровь или фракцию крови, плазму или содержит плазму или фракцию плазмы, сыворотку или содержит сыворотку или фракцию сыворотки, или если она содержит биофармацевтическое лекарственное средство, выработанное в клеточной культуре). В предпочтительном варианте осуществления настоящего изобретения в соответствии со всеми остальными вариантами осуществления настоящего изобретения указанная жидкость, предлагаемая в настоящем изобретении, содержит вирус, обладающий липидной оболочкой. На природу указанного вируса, обладающего липидной оболочкой, в указанной жидкости, предлагаемой в настоящем изобретении, не налагаются особые ограничения. Например, вирус может происходить из крови человека, которую можно использовать для приготовления жидкости, предлагаемой в настоящем изобретении, или из плазмы человека, которую можно использовать для приготовления жидкости, предлагаемой в настоящем изобретении, или из сыворотки человека, которую можно использовать для приготовления жидкости, предлагаемой в настоящем изобретении, или из клеточной культуральной среды, которую можно использовать для приготовления жидкости, предлагаемой в настоящем изобретении. В частности, если вирус происходит из клеточной культуральной среды, которую используют для приготовления жидкости, предлагаемой в настоящем изобретении, вирус может происходить из образованных из животного компонентов указанной клеточной культуральной среды, такой как бычий сывороточный альбумин.
В способе инактивации вирусов, обладающих липидной оболочкой, предлагаемом в настоящем изобретении, моющее средство добавляют к жидкости для приготовления смеси указанного моющего средства и указанной жидкости. Указанное моющее средство является простым эфиром полиоксиэтилена.
Простые эфиры полиоксиэтилена известны специалисту в данной области техники и обладают следующей структурой формулы A:
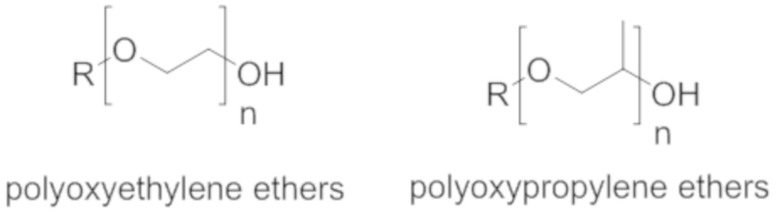
(Формула A);
в котором n больше или равно 1.
Как понятно специалисту в данной области техники, простые эфиры полиоксипропилена могут обладать характеристиками, очень сходными с характеристиками простых эфиров полиоксиэтилена, предлагаемых в настоящем изобретении. Таким образом, в соответствии со всеми остальными вариантами осуществления настоящего изобретения например, в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении, простые эфиры полиоксиэтилена можно заменить простыми эфирами полиоксипропилена.
Простые эфиры полиоксипропилена также известны специалисту в данной области техники и обладают следующей структурой формулы B:
(Формула B);
в котором n больше или равно 1.
При использовании в настоящем изобретении "простой эфир полиоксиэтилена", предлагаемый в настоящем изобретении, предпочтительно представляет собой простой эфир полиоксиэтилена, соответствующий его обычному значению в данной области техники. Альтернативно, простой эфир полиоксиэтилена, предлагаемый в настоящем изобретении, также может представлять собой простой эфир, где часть, предпочтительно большая часть от полного количества молекул простого полиоксиэфира являются молекулами простого эфира полиоксиэтилена, но другая часть, предпочтительно меньшая часть от полного количества молекул простого полиоксиэфира являются смешанными полимерами, включающими звенья оксиэтилена и оксипропилена и/или полимерами, включающими звенья оксипропилена. В этом случае термин "большая часть от полного количества молекул простого полиоксиэфира являются молекулами простого эфира полиоксиэтилена" означает, что не менее 50% от полного количества молекул простого полиоксиэфира являются молекулами простого эфира полиоксиэтилена. Предпочтительно, если не менее 60% от полного количества молекул простого полиоксиэфира являются молекулами простого эфира полиоксиэтилена. Более предпочтительно, если не менее 70% от полного количества молекул простого полиоксиэфира являются молекулами простого эфира полиоксиэтилена. Еще более предпочтительно, если не менее 80% от полного количества молекул простого полиоксиэфира являются молекулами простого эфира полиоксиэтилена. Еще более предпочтительно, если не менее 90% от полного количества молекул простого полиоксиэфира являются молекулами простого эфира полиоксиэтилена. Наиболее предпочтительно, если не менее 95% от полного количества молекул простого полиоксиэфира являются молекулами простого эфира полиоксиэтилена. Альтернативно, простой эфир полиоксиэтилена, предлагаемый в настоящем изобретении, также может быть смешанным простым полиоксиэфиром, включающим большую часть (например, не менее 60%, предпочтительно не менее 70%, более предпочтительно не менее 80%, еще более предпочтительно не менее 90% и наиболее предпочтительно не менее 95%) оксиэтиленовых звеньев меньшую часть оксипропиленовых звеньев.
Моющие средства для применения в способах, предлагаемых в настоящем изобретении, являются нефенольными. В другом варианте осуществления в соответствии со всеми остальными вариантами осуществления настоящего изобретения моющие средства для применения в способах, предлагаемых в настоящем изобретении, не являются ароматическими.
Моющие средства на основе простого эфира полиоксиэтилена, предлагаемые в настоящем изобретении, можно синтезировать с помощью реакции этоксилирования. Этоксилирование является промышленной технологией, проводимой со спиртами (альтернативно можно использовать амины) для получения этоксилатов спиртов (простых эфиров полиоксиэтилена). Реакцию проводят путем пропускания этиленоксида через спирт при высокой температуре (например, около 180°C) и при высоком давлении (например, при давлении 1-2 бар), с основанием (таким как гидроксид калия, KOH) в качестве катализатора. Реакция является сильно экзотермической.

Реакция приводит к образованию продукта с широкой полидисперсностью длин повторяющихся звеньев (параметр n в приведенном выше уравнении является средней длиной полимера).
В лабораторном масштабе моющие средства на основе простого эфира полиоксиэтилена можно получить путем проводимого сначала формирования подходящей отщепляющейся группы (такой как Cl, Br, I, OMs или OTf) из гидроксигруппы спирта и последующей реакции этого субстрата с монодепротонированным полиэтиленгликолем. Альтернативно, подходящую отщепляющуюся группу (такую как Cl, Br, I, OMs или OTf) можно ввести в цепь полиэтиленгликоля, которую на следующей стадии вводят в реакцию с депротонированным спиртом.
Типичные методики синтеза простых эфиров полиоксиэтилена подробно описаны, например, в патенте US № 1970578 и Di Serio et al. (2005), которые во всей их полноте включены в настоящее изобретение в качестве ссылки для всех объектов.
Хотя TX-100, Triton N-101 reduced, Triton X-100 reduced и Brij C10 являются алкиловыми эфирами полиоксиэтилена (= реакцию этоксилирования проводят с газообразным этиленоксидом), аналогичную реакцию также можно провести в промышленном масштабе с использованием пропиленоксида (=пропоксилирование). Единственным отличием является замещение метильной группы в цепи PEG:
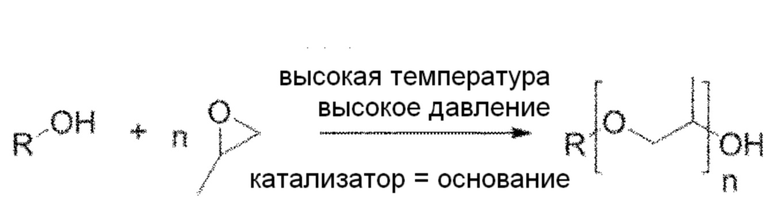
Этоксилирование и пропоксилирование также можно объединить в той же процедуре и получить смешанный продукт, такой как смешанный полимер простого полиоксиэфира, включающий оксиэтиленовые и оксипропиленовые звенья.
Токсическая активность Triton-X100 обусловлена фенольным фрагментом, который способен повреждать некоторые эндокринные рецепторы морских организмов. После вставки метиленовой группы между ароматическим кольцом и цепью PEG фенольная группа Triton-X100 в новой структуре больше не содержится. Кроме того, после разрыва цепи PEG в результате попадания в окружающую среду выделившийся бензиловый спирт легко окисляется в соответствующую бензойную кислоту. Этот метаболит обладает совершенно иной полярностью и геометрической структурой, чем производное фенола, который предотвратит любое ингибирование эндокринных рецепторов.
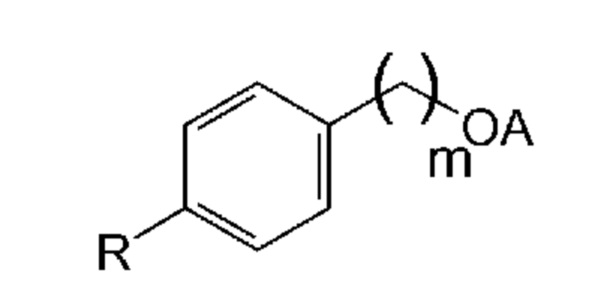
На основании указанных выше соображений авторы настоящего изобретения синтезировали не содержащие фенол простые эфиры полиоксиэтилена и исследовали их противовирусную активность (см. примеры 4-10). Таким образом, настоящее изобретение также относится к не содержащим фенол простым эфирам полиоксиэтилена следующей общей формулы (VIII):
Формула (VIII)
В формуле (VIII) R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи, m является целым числом, равным от 1 до 4, и A означает полиоксиэтиленовый остаток, необязательно при условии, что исключен 29-[4-(1,1,3,3-тетраметилбутил)фенил]-3,6,9,12,15,18,21,24,27-нонаоксанонакозан-1-ол следующей структурной формулы.
В формуле (VIII) R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12, предпочтительно от 2 до 8, более предпочтительно от 2 до 6 и наиболее предпочтительно 4 атомов углерода и одну или большее количество, предпочтительно от 2 до 6 и наиболее предпочтительно 4 метильные группы в качестве заместителей указанной линейной цепи.
Предпочтительно, если R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12, предпочтительно от 2 до 8, более предпочтительно от 2 до 6 и наиболее предпочтительно 4 атома углерода и 2 метильные группы в качестве заместителей указанной линейной цепи; углеводородную группу, включающую линейную цепь, содержащую от 2 до 12, предпочтительно от 2 до 8, более предпочтительно от 2 до 6 и наиболее предпочтительно 4 атома углерода и 4 метильные группы в качестве заместителей указанной линейной цепи; или углеводородную группу, включающую линейную цепь, содержащую от 2 до 12, предпочтительно от 2 до 8, более предпочтительно от 2 до 6 и наиболее предпочтительно 4 атома углерода и 6 метильных групп в качестве заместителей указанной линейной цепи.
Наиболее предпочтительно, если R означает 2,4,4-триметилпент-2-ильную группу.
В формуле (VIII) m является целым числом, равным от 1 до 4, предпочтительно целым числом, равным от 1 до 2 и наиболее предпочтительно целым числом, равным 1.
В формуле (VIII) A означает полиоксиэтиленовый остаток, предпочтительно полиоксиэтиленовый остаток, содержащий 2-20 оксиэтиленовых звеньев, более предпочтительно полиоксиэтиленовый остаток, содержащий от 4 до 16 оксиэтиленовых звеньев, еще более предпочтительно полиоксиэтиленовый остаток, содержащий от 8 до 12 оксиэтиленовых звеньев и наиболее предпочтительно полиоксиэтиленовый остаток, содержащий 9 или 10 оксиэтиленовых звеньев
Предпочтительным вариантом осуществления соединения общей формулы (VIII) является соединение, в котором R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 6 атомов углерода и от 2 до 4 метильных групп в качестве заместителей указанной линейной цепи; m является целым числом, равным от 1 до 2; и A означает полиоксиэтиленовый остаток, содержащий от 8 до 12 оксиэтиленовых звеньев.
Другим предпочтительным вариантом осуществления соединения общей формулы (VIII) является соединение, в котором R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 6 атомов углерода и от 2 до 4 метильных групп в качестве заместителей указанной линейной цепи; m является целым числом, равным 1; и A означает полиоксиэтиленовый остаток, содержащий от 8 до 12 оксиэтиленовых звеньев.
Конкретным предпочтительным вариантом осуществления соединения общей формулы (VIII) является следующее соединение:
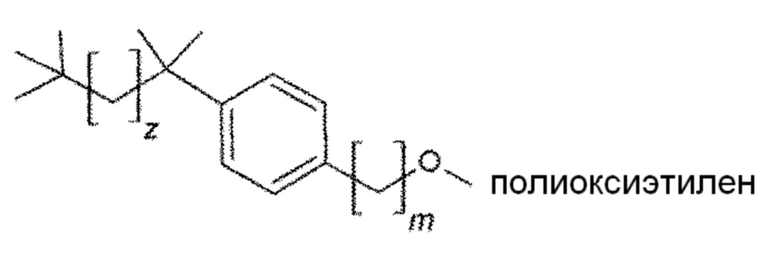
в котором m и z являются целыми числами, которые независимо выбраны из следующих групп:
m=от 1 до 4, предпочтительно в котором m равно 1;
z=от 1 до 5.
Другим конкретным предпочтительным вариантом осуществления соединения общей формулы (VIII) является следующее соединение:
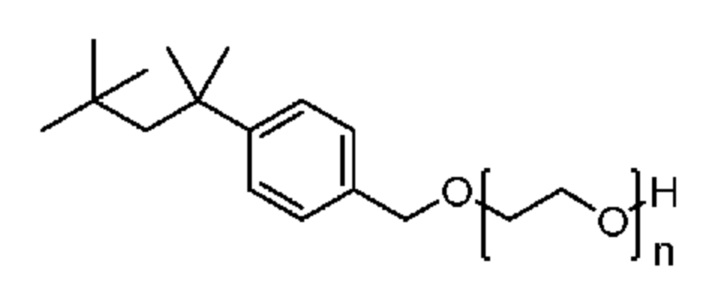
полиэтоксилат 4-трет-октилбензилового спирта
в котором n является целым числом, равным от 4 до 16, предпочтительно в котором n равно 9 или 10.
Соединение формулы (VIII) можно синтезировать по общеизвестным методикам синтеза, таким описанные в Vogel‘s Textbook of Practical Organic Chemistry (5th Edition, 1989, A.I. Vogel, A.R. Tatchell, B.S. Furnis, A.J. Hannaford, P.W.G. Smith). Например, полиэтоксилат 4-трет-октилбензилового спирта можно получить путем превращения фенола, содержащего заместитель, соответствующий группе R общей формулы (VIII), в соответствующую бензойную кислоту или гомобензойную кислоту, восстановления карбоксигруппы в гидроксигруппу и введения в реакцию спирта с образованием простого эфира полиэтиленгликоля (также известного, как простой эфир полиоксиэтилена; POE эфир). В настоящем изобретении этиленоксид или подходящий полиэтиленгликоль могут служить основой для введения функциональной группы простого эфира полиэтиленгликоля (схема 1; типичные примеры экспериментальных методик, которые эффективны для проведения отдельных превращений, приведены, например, в Bioorganic & Medicinal Chemistry, 16(9), 4883-4907, 2008; Journal of Medicinal Chemistry, 48(10), 3586-3604, 2005; Journal of Physical Chemistry B, 107(31), 7896-7902; 2003; Journal of Nanoparticle Research, 15(11), 2025/1-2025/12, 12 pp., 2013; и PCT Int. Appl., 2005016240, 24 Feb 2005.).
Схема 1
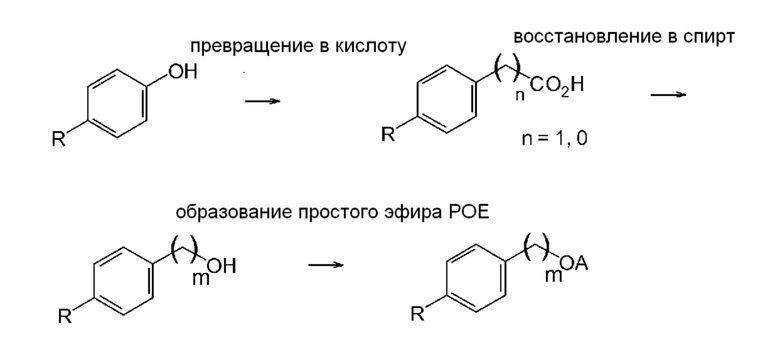
Альтернативно, соединение общей формулы (VIII) можно получить путем алкилирования толуола с последующей функционализацией бензильной метильной группы и с последующей реакцией с образованием простого эфира полиэтиленгликоля (схема 2; типичные примеры экспериментальных методик, которые эффективны для проведения отдельных превращений, приведены, например, в Russian Journal of Applied Chemistry, 82(6), 1029-1032, 2009; Journal of Organic Chemistry, 79(1), 223-229, 2014; и Chemistry - A European Journal, 23(60), 15133-15142, 2017)). Также рассматривается прямое алкилирование бензилового спирта дает подходящий промежуточный продукт для введения функциональной группы простого эфира полиэтиленгликоля (схема 3; типичные примеры экспериментальных методик для соответствующих превращений приведены, например, в Russian Journal of Organic Chemistry, 51(11), 1545-1550, 2015).
Схема 2
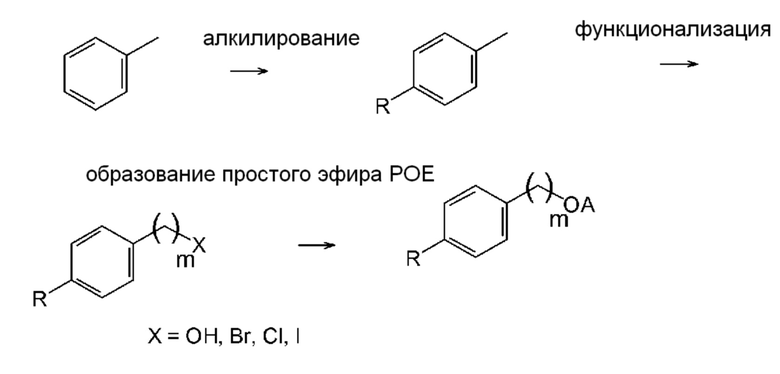
Схема 3
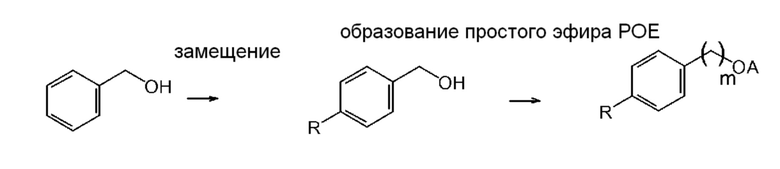
Указанные выше простые эфиры полиоксиэтилена со структурой формулы (VIII) можно использовать в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении.
Как показано выше и как понятно специалисту в данной области техники, синтез простых эфиров полиоксиэтилена обычно дает продукты с широкой полидисперсностью длин повторяющихся звеньев полиоксиэтилена. Следовательно, если приведено количество повторяющихся звеньев полиоксиэтилена для простых эфиров полиоксиэтилена, предлагаемых в настоящем изобретении, это количество указывает на среднюю длину повторяющихся звеньев полиоксиэтилена. Средняя длина повторяющихся звеньев полиоксиэтилена указывает на среднее количество повторяющихся звеньев полиоксиэтилена для всех молекул простого эфира полиоксиэтилена в образце. Например, в одном варианте осуществления способа инактивации вирусов, обладающих липидной оболочкой, предлагаемого в настоящем изобретении, моющее средство добавляют к жидкости для приготовления смеси указанного моющего средства и указанной жидкости, где указанным моющим средством может быть простой эфир полиоксиэтилена, который обладает следующей структурой формулы (III):
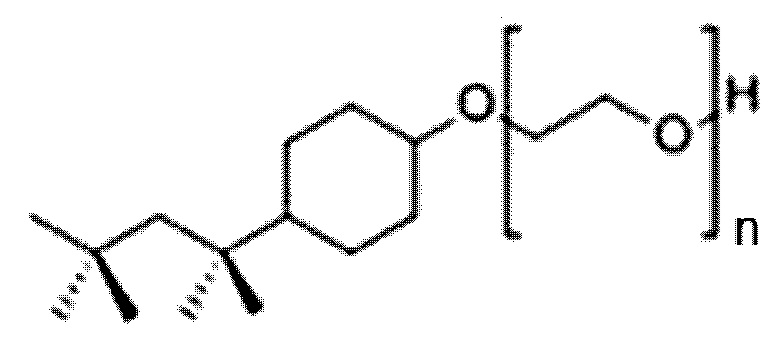
(Формула (III));
в котором n равно 10.
В этом варианте осуществления способа инактивации вирусов, обладающих липидной оболочкой, предлагаемого в настоящем изобретении, n=10 в приведенной выше формуле (III) означает среднее количество повторяющихся звеньев полиоксиэтилена для всех молекул простого эфира полиоксиэтилена, которые добавляют к жидкости, предлагаемой в настоящем изобретении.
Моющие средства на основе простого эфира полиоксиэтилена, использующегося в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении, обладают средним количеством повторяющихся звеньев полиоксиэтилена, равным от 2 до 100, от 2 до 50, от 2 до 20, или от 4 до 16, или 9 или 10. Предпочтительно, если среднее количество повторяющихся звеньев полиоксиэтилена равно от 4 до 16, более предпочтительно 9 или 10 и наиболее предпочтительно 10.
Как понятно специалисту в данной области техники, в моющих средствах на основе простых эфиров полиоксиэтилена/полиоксипропилена, предлагаемых в настоящем изобретении, метильная группа может быть присоединена к концевой гидроксигруппе фрагмента полиоксиэтилен/полиоксипропилен (т.е. концевая гидроксигруппа может быть блокирована). Такое блокирование концевой гидроксигруппы может облегчить синтез. Оно является особенно подходящим для соединений, не полученного путем этоксилирования или пропоксилирования, таких как соединения, полученных в соответствии с приведенной выше схемой 1 или схемой 2. Структуры с блокированным метильной группой фрагментом полиоксиэтилен/полиоксипропилен обычно называются производными mPEG, которые проиллюстрированы следующими типичными структурами:
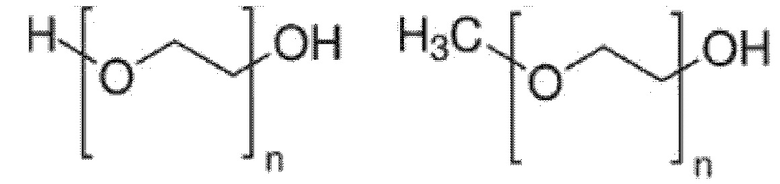
Поли(этиленгликоль) (PEG) Метоксиполиэтиленгликоль (mPEG)
Как понятно специалисту в данной области техники, и синтез простых эфиров полиоксипропилена обычно дает продукты с широкой полидисперсностью длин полиоксипропиленовых повторяющихся звеньев. Следовательно, если приведено количество повторяющихся звеньев полиоксипропилена для простых эфиров полиоксипропилена, предлагаемых в настоящем изобретении, это количество указывает на среднюю длину повторяющихся звеньев полиоксипропилена. Средняя длина повторяющихся звеньев полиоксипропилена указывает на среднее количество повторяющихся звеньев полиоксипропилена для всех молекул простого эфира полиоксипропилена в образце.
Моющие средства на основе простого эфира полиоксипропилена, использующегося в соответствии с настоящим изобретением, обладают средним количеством повторяющихся звеньев полиоксипропилена, равным от 2 до 100, от 2 до 50, от 2 до 20, или от 5 до 15, или 9 или 10. Предпочтительно, если среднее количество повторяющихся звеньев полиоксипропилена равно от 5 до 15, более предпочтительно 9 или 10 и наиболее предпочтительно 10.
В одном варианте осуществления настоящего изобретения простой эфир полиоксиэтилена, который используют в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении, обладает следующей структурой формулы (II):
(Формула (II))
В предпочтительном варианте осуществления настоящего изобретения соединение, описывающееся приведенной выше формулой (II), представляет собой имеющийся в продаже Triton X-100 reduced (CAS No. 92046-34-9).
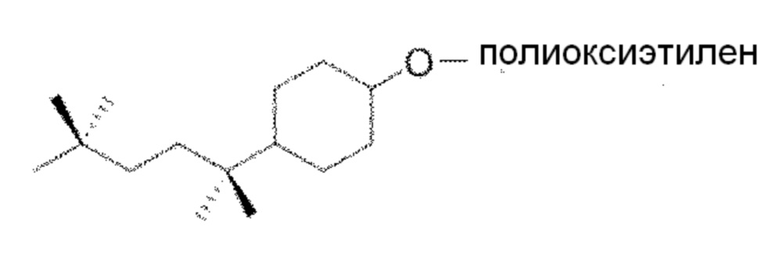
В одном варианте осуществления настоящего изобретения простой эфир полиоксиэтилена, который используют в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении, обладает следующей структурой формулы (IV):
(Формула (IV))
В предпочтительном варианте осуществления настоящего изобретения соединение, описывающееся приведенной выше формулой (IV), представляет собой имеющийся в продаже Triton N-101 reduced (CAS No. 123359-41-1).
В одном варианте осуществления настоящего изобретения простой эфир полиоксиэтилена, который используют в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении, обладает следующей структурой формулы (VI):
(Формула (VI));
в которой x равно 15.
В предпочтительном варианте осуществления настоящего изобретения соединение, описывающееся приведенной выше формулой (VI), представляет собой имеющийся в продаже Brij C10 (CAS No. 9004-95-9).
В соответствии со всеми остальными вариантами осуществления настоящего изобретения в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении, простые эфиры полиоксиэтилена также можно заменить простыми эфирами монооксиэтилена. В предпочтительном варианте осуществления настоящего изобретения указанные простые эфиры монооксиэтилена обладают следующей структурой формулы C:
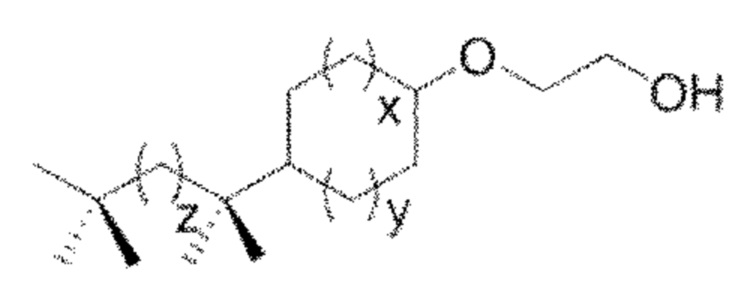
(Формула C);
где x, y и z являются целыми числами, которые независимо выбраны из следующих групп:
x=от 0 до 5
y=от 0 до 5
z=от 0 до 20
Предпочтительно, если указанные простые эфиры монооксиэтилена обладают следующей структурой формулы D:
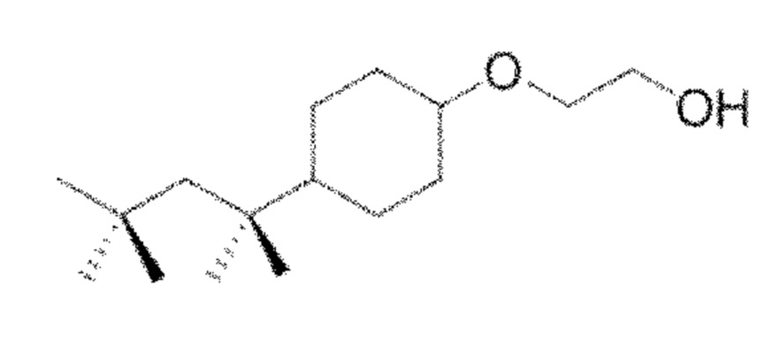
(Формула D);
или следующей структурой формулы E:
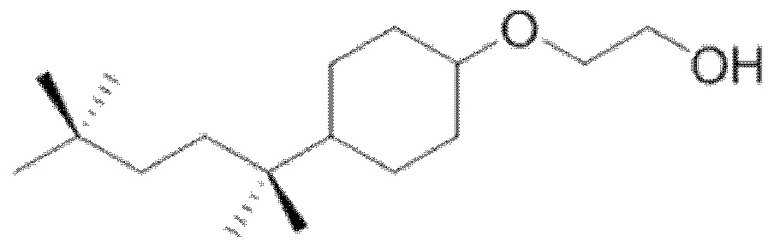
(Формула E).
В соответствии со всеми остальными вариантами осуществления настоящего изобретения настоящее изобретение также относится к простым эфирам монооксиэтилена, соответствующим простым эфирам полиоксиэтилена, предлагаемым в настоящем изобретении. В предпочтительном варианте осуществления настоящего изобретения указанные простые эфиры монооксиэтилена обладают следующей структурой формулы F:
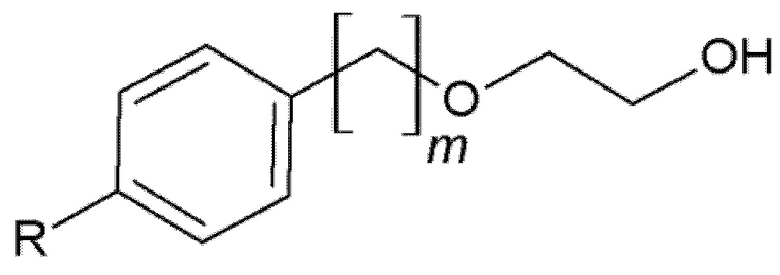
(Формула F);
в котором R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи; m является целым числом, равным от 1 до 4, и A означает полиоксиэтиленовый остаток.
В другом предпочтительном варианте осуществления простым эфиром монооксиэтилена формулы F является следующее соединение:
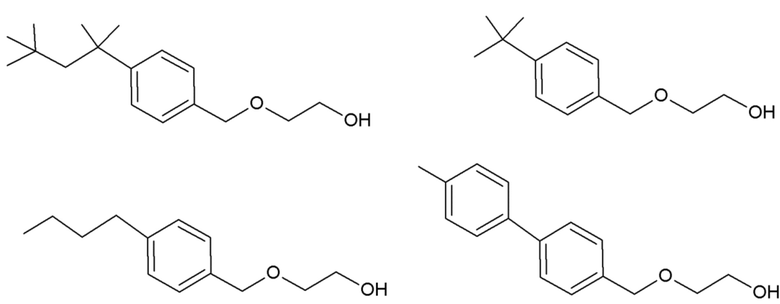
Указанные выше простые эфиры монооксиэтилена со структурами формулы F можно использовать в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении.
В способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении, моющее средство добавляют к жидкости для приготовления смеси указанного моющего средства и указанной жидкости. В одном варианте осуществления настоящего изобретения моющие средства на основе простого эфира полиоксиэтилена добавляют к жидкости и получали конечную концентрацию, равную примерно от 0,03% (мас./мас.) до 10% (мас./мас.), предпочтительно примерно от 0,05% (мас./мас.) до 10% (мас./мас.), более предпочтительно от 0,1% (мас./мас.) до 10% (мас./мас.), еще более предпочтительно примерно от 0,5% (мас./мас.) до 5% (мас./мас.), наиболее предпочтительно примерно от 0,5% (мас./мас.) до 2% (мас./мас.) моющего средства на основе простого эфира полиоксиэтилена в жидкости.
В предпочтительном варианте осуществления способов инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении, простые эфиры полиоксиэтилена, которые используют в способах, являются подходящими для инактивации указанного вируса, обладающего липидной оболочкой. Инактивация при использовании в настоящем изобретении означает нарушение способности вируса с липидной оболочкой инфицировать клетки. Как понятно специалисту в данной области техники, способность вируса с липидной оболочкой инфицировать клетки, т. е. инфекционность вируса с липидной оболочкой, обычно оценивают путем определения количества контагиозных вирусных частиц в растворе. Типичные методики определения количества контагиозных вирусных частиц в растворе описаны в настоящем изобретении.
В одном варианте осуществления в способе инактивации вирусов, обладающих липидной оболочкой, предлагаемом в настоящем изобретении, стадию добавления моющего средства и растворителя к жидкости проводят так, что получают смесь растворитель/моющее средство для инактивации указанных вирусов. Предпочтительно, если указанным растворителем является органический растворитель и еще более предпочтительно, если указанным растворителем является три-н-бутилфосфат. Следует понимать, что концентрацию моющего средства и тип и концентрацию растворителя надлежащим образом может выбрать специалист в данной области техники с учетом, например, потенциальных вирусов, содержащихся в жидкости, желательного LRV, характеристик биофармацевтического лекарственного средства, которое может содержаться в жидкости, и характеристик способа получения биофармацевтического лекарственного средства, которое может содержаться в жидкости (например, при какой температуре будет проводиться инактивация). Обычно конечные концентрации органического растворителя и одного моющего средства во время инкубации в соответствии с настоящим изобретением равны от примерно 0,01% (мас./мас.) до примерно 5% (мас./мас.) органического растворителя и от примерно 0,05% (мас./мас.) до примерно 10% (мас./мас.) моющего средства, предпочтительно от примерно 0,1% (мас./мас.) до примерно 5% (мас./мас.) органического растворителя и от примерно 0,1% (мас./мас.) до примерно 10% (мас./мас.) моющего средства, более предпочтительно от примерно 0,1% (мас./мас.) до примерно 1% (мас./мас.) органического растворителя и от примерно 0,5% (мас./мас.) до примерно 5% (мас./мас.) моющего средства, наиболее предпочтительно от примерно 0,1% (мас./мас.) до примерно 0,5% (мас./мас.) органического растворителя и от примерно 0,5% (мас./мас.) до примерно 2% (мас./мас.) моющего средства.
В другом варианте осуществления в способе инактивации вирусов, обладающих липидной оболочкой, предлагаемом в настоящем изобретении, стадию добавления моющего средства (и необязательно также растворителя) к жидкости проводят таким образом, что к жидкости добавляют дополнительное моющее средство. Предпочтительно, если указанным дополнительным моющим средством является полиоксиэтилен-(80)-сорбитанмоноолеат (также известный, как, например, полисорбат 80 или TWEEN 80). Предпочтительно, если указанным растворителем является органический растворитель и еще более предпочтительно, если указанным растворителем является три-н-бутилфосфат. Следует понимать, что концентрацию моющего средства, предлагаемого в настоящем изобретении, а также тип и дополнительного моющего средства, а также тип и концентрацию растворителя надлежащим образом может выбрать специалист в данной области техники с учетом, например, потенциальных вирусов, содержащихся в жидкости, желательного LRV, характеристик биофармацевтического лекарственного средства, которое может содержаться в жидкости, и характеристик способа получения биофармацевтического лекарственного средства, которое может содержаться в жидкости (например, при какой температуре будет проводиться инактивация). Обычно конечная концентрация органического растворителя равна от примерно 0,01% (мас./мас.) до примерно 5% (мас./мас.), конечная концентрация моющего средства, предлагаемого в настоящем изобретении, равна от примерно 0,05% (мас./мас.) до примерно 10% (мас./мас.) и конечная концентрация дополнительного моющего средства равна от примерно 0,01% (мас./мас.) до примерно 5% (мас./мас.). Предпочтительно, если конечная концентрация органического растворителя равна от примерно 0,1% (мас./мас.) до примерно 5% (мас./мас.), конечная концентрация моющего средства, предлагаемого в настоящем изобретении, равна от примерно 0,1% (мас./мас.) до примерно 10% (мас./мас.) и конечная концентрация дополнительного моющего средства равна от примерно 0,1% (мас./мас.) до примерно 5% (мас./мас.). Более предпочтительно, если конечная концентрация органического растворителя равна от примерно 0,1% (мас./мас.) до примерно 1% (мас./мас.), конечная концентрация моющего средства, предлагаемого в настоящем изобретении, равна от примерно 0,5% (мас./мас.) до примерно 5% (мас./мас.) и конечная концентрация дополнительного моющего средства равна от примерно 0,1% (мас./мас.) до примерно 1% (мас./мас.). Наиболее предпочтительно, если конечная концентрация органического растворителя равна от примерно 0,1% (мас./мас.) до примерно 0,5% (мас./мас.), конечная концентрация моющего средства, предлагаемого в настоящем изобретении, равна от примерно 0,5% (мас./мас.) до примерно 2% (мас./мас.) и конечная концентрация дополнительного моющего средства равна от примерно 0,1% (мас./мас.) до примерно 0,5% (мас./мас.).
В другом варианте осуществления настоящего изобретения используют только одно моющее средство. Например, в одном варианте осуществления способа, предлагаемого в настоящем изобретении, на стадии a) не добавляют дополнительное моющее средство кроме моющего средства, предлагаемого в настоящем изобретении. В другом варианте осуществления способа, предлагаемого в настоящем изобретении, в способе не добавляют дополнительное моющее средство кроме моющего средства, предлагаемого в настоящем изобретении. В другом варианте осуществления настоящего изобретения при применении моющего средства, предлагаемого в настоящем изобретении, в способе инактивации вирусов, обладающих липидной оболочкой, не используют дополнительное моющее средство кроме моющего средства, предлагаемого в настоящем изобретении. Одним преимуществом этих вариантов осуществления является то, что одно моющее средство легче можно удалить на последующих стадиях (способа). Например, одно моющее средство легче можно удалить, чем три компонента, использующиеся при стандартной обработке растворителем/моющим средством (S/D), которое обычно включает два моющих средства и один растворитель, предпочтительно органический растворитель. Таким образом, в другом варианте осуществления настоящего изобретения композиция, содержащая моющее средство, предлагаемое в настоящем изобретении, не содержит какое-либо дополнительное моющее средство кроме указанного моющего средства.
В другом варианте осуществления настоящего изобретения не используют органический растворитель. Например, в одном варианте осуществления способа, предлагаемого в настоящем изобретении, на стадии a) не добавляют органический растворитель. В другом варианте осуществления настоящего изобретения при применении моющего средства, предлагаемого в настоящем изобретении, в способе инактивации вирусов, обладающих липидной оболочкой, не используют органический растворитель. В другом варианте осуществления настоящего изобретения композиция, включающая моющее средство, предлагаемое в настоящем изобретении, не содержит какой-либо органический растворитель.
Как отмечено выше, способ инактивации вирусов, обладающих липидной оболочкой, предлагаемый в настоящем изобретении, является особенно подходящим для биофармацевтических производственных процессов, в которых для гарантии безопасности для пациента должно быть обеспечено отсутствие активного (т. е. контагиозного) вируса из конечного продукта. Таким образом, в одном варианте осуществления в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении, моющее средство добавляют к жидкости, которая содержит биологический медицинский продукт или биофармацевтическое лекарственное средство, предпочтительно биофармацевтическое лекарственное средство. В предпочтительном варианте осуществления настоящего изобретения указанное биофармацевтическое лекарственное средство не является вирусной вакциной. На биофармацевтические лекарственные средства, предлагаемые в настоящем изобретении, не налагаются особые ограничения. Они включают рекомбинантные биофармацевтические лекарственные средства и биофармацевтические лекарственные средства из других источников, такие как биофармацевтические лекарственные средства, полученные из плазмы человека. Биофармацевтические лекарственные средства, предлагаемые в настоящем изобретении, включают без наложения ограничений факторы крови, иммуноглобулины, замещающие ферменты, вакцины, генотерапевтические векторы, фактор роста и их рецепторы. В предпочтительном варианте осуществления биофармацевтическое лекарственное средство представляет собой терапевтический белок. Предпочтительные факторы крови включают фактор I (фибриноген), фактор II (протромбин), тканевый фактор, фактор V, фактор VII и фактор VIIa, фактор VIII, фактор IX, фактор X, фактор XI, фактор XII, фактор XIII, фактор фон Виллебранда (VWF), прекалликреин, кининоген с большой молекулярной массой (HMWK), фибронектин, антитромбин III, кофактор II гепарина, белок C, белок S, белок Z, плазминоген, альфа-2-антиплазмин, тканевый активатор плазминогена (tPA), урокиназа, ингибитор-1 активатора плазминогена (PAI1), и ингибитор-2 активатора плазминогена (PAI2). Фактор VIII является особенно предпочтительным фактором крови и рекомбинантный фактор VIII является еще более предпочтительным. Факторы крови, которые можно использовать в соответствии с настоящим изобретением, включают функциональные варианты полипептидов и полинуклеотиды, которые кодируют факторы крови или кодируют такие функциональные варианты полипептидов. Предпочтительные иммуноглобулины включают иммуноглобулины из плазмы человека, моноклональные антитела и рекомбинантные антитела. Биофармацевтические лекарственные средства, предлагаемые в настоящем изобретении, предпочтительно представляют собой соответствующие белки человека или рекомбинантные белки человека, или их функциональные варианты.
Как отмечено выше, биофармацевтическое лекарственное средство, предлагаемое в настоящем изобретении, также может представлять собой генотерапевтический вектор, включая вирусный генотерапевтический вектор. Как понятно специалисту в данной области техники, способы инактивации вирусов, обладающих липидной оболочкой, предлагаемые в настоящем изобретении, обычно не инактивируют вирусы без оболочки. Таким образом, в предпочтительном варианте осуществления, в котором биофармацевтическое лекарственное средство, предлагаемое в настоящем изобретении, представляет собой вирусный генотерапевтический вектор, такой вирусный генотерапевтический вектор основан на вирусе без оболочки. В предпочтительном варианте осуществления такой вирусный генотерапевтический вектор основан на аденоассоциированном вирусе (AAV).
В соответствии со всеми остальными вариантами осуществления настоящего изобретения способ инактивации вирусов, обладающих липидной оболочкой, предлагаемый в настоящем изобретении, может включать после стадии инкубирование указанной смеси для инактивации указанных вирусов стадию очистки указанного биофармацевтического лекарственного средства. Предпочтительно, если указанная очистка включает отделение указанного биофармацевтического лекарственного средства от указанного моющего средства (средств). Специалисту в данной области техники известны разные методики отделения биофармацевтического лекарственного средства от моющего средства (средств). Такие методики может выбрать специалист в данной области техники с учетом характеристик биофармацевтического лекарственного средства, источника, из которого оно получено (например, рекомбинантно или из других источников, например, из плазмы человека), и желательного биофармацевтического применения (например, вводят ли его подкожно или внутривенно и т. п.). Например, биофармацевтическое лекарственное средство можно отделить от моющего средства (средств) с помощью хроматографии, такой как анионообменная хроматография или катионообменная хроматография. В одном варианте осуществления указанная очистка от указанного моющего средства (средств) включает более, чем одну хроматографическую очистку.
В соответствии со всеми остальными вариантами осуществления настоящего изобретения способ инактивации вирусов, обладающих липидной оболочкой, предлагаемый в настоящем изобретении, может включать стадию фильтрования указанной смеси, предпочтительно через объемный фильтр. Эту стадию фильтрования можно провести до стадии добавления моющего средства к жидкости для приготовления смеси указанного моющего средства и указанной жидкости. Альтернативно, эту стадию фильтрования можно провести между стадией добавления моющего средства к жидкости для приготовления смеси указанного моющего средства и указанной жидкости и стадией инкубирования указанной смеси для инактивации указанных вирусов.
В другом варианте осуществления в соответствии со всеми остальными вариантами осуществления настоящего изобретения в способах инактивации вирусов, обладающих липидной оболочкой, стадию инкубирования указанной смеси для инактивации указанных вирусов можно провести таким образом, что указанную смесь инкубируют в течение не менее 10 мин, в течение не менее 30 мин, в течение не менее 1 ч, в течение не менее 2 ч, в течение не менее 4 ч, в течение не менее 12 ч или в течение не менее 24 ч.
В другом варианте осуществления в соответствии со всеми остальными вариантами осуществления настоящего изобретения на стадии инкубирования указанной смеси для инактивации указанных вирусов указанную смесь инкубируют при низкой температуре, такой как температура, равная от 0°C до 15°C, от 0°C до 10°C, от 0°C до 8°C, от 0°C до 6°C, от 0°C до 4°C или от 0°C до 2°C, предпочтительно от 0°C до 10°C. В альтернативном варианте осуществления в соответствии со всеми остальными вариантами осуществления настоящего изобретения на стадии инкубирования указанной смеси для инактивации указанных вирусов указанную смесь инкубируют при комнатной температуре или температуре, близкой к комнатной температуре, такой как температура, равная от 16°C до 25°C, от 18°C до 24°C или от 20°C до 23°C.
Следует понимать, что на то, как проводят стадии способа, предлагаемого в настоящем изобретении, не налагаются особые ограничения. В частности, стадии способа можно провести периодическим образом. Альтернативно, стадии способа также можно провести полунепрерывным или непрерывным образом.
Как указано выше, способ инактивации вирусов, обладающих липидной оболочкой, предлагаемый в настоящем изобретении, является особенно подходящим для биофармацевтических производственных процессов. Таким образом, настоящее изобретение также относится к способу получения биофармацевтического лекарственного средства, указанный способ включает стадии способа инактивации вирусов, обладающих липидной оболочкой, предлагаемого в настоящем изобретении, и любыми его вариантами осуществления. Предпочтительно, если способ получения биофармацевтического лекарственного средства, предлагаемый в настоящем изобретении, включает стадию получения фармацевтического состава, включающего указанное биофармацевтическое лекарственное средство, которую проводят после стадий способа инактивации вирусов, обладающих липидной оболочкой, предлагаемого в настоящем изобретении. Такой фармацевтический состав можно получить в соответствии с известными стандартами для получения фармацевтического состава. Например, состав можно получить таким образом, чтобы его можно было соответствующим образом хранить и вводить, например, путем использования фармацевтически приемлемых компонентов, таких как носители, инертные наполнители или стабилизаторы. Такие фармацевтически приемлемые компоненты нетоксичны в количествах, использующихся при введении фармацевтического состава пациенту.
Авторы настоящего изобретения неожиданно установили, что моющие средства на основе простого эфира полиоксиэтилена, предлагаемые в настоящем изобретении, являются особенно подходящими для инактивации вирусов, обладающих липидной оболочкой. Таким образом, настоящее изобретение также относится к применению раскрытых моющих средств на основе простого эфира полиоксиэтилена, предлагаемых в настоящем изобретении, в любом способе инактивации вирусов, обладающих липидной оболочкой. Предпочтительно, если указанный способ указанной инактивации указанных вирусов представляет собой способ с использованием обработки растворителем/моющим средством, в котором указанная обработка растворителем/моющим средством включает применение указанного моющего средства, предлагаемого в настоящем изобретении. В другом варианте осуществления указанная инактивация вирусов представляет собой инактивацию вирусов в жидкости, включающей биофармацевтическое лекарственное средство.
Поскольку авторы настоящего изобретения неожиданно установили, что моющие средства на основе не содержащего фенола простого эфира полиоксиэтилена, предлагаемые в настоящем изобретении, являются особенно подходящими для инактивации вирусов, обладающих липидной оболочкой, настоящее изобретение также относится к моющим средствам на основе простого эфира полиоксиэтилена, предлагаемым в настоящем изобретении, и к композиции, включающей моющее средство на основе простого эфира полиоксиэтилена, предлагаемое в настоящем изобретении. В другом варианте осуществления композиция, включающая моющее средство на основе простого эфира полиоксиэтилена, предлагаемое в настоящем изобретении, дополнительно включает биофармацевтическое лекарственное средство и/или органический растворитель и/или дополнительное моющее средство.
Настоящее изобретение также относится к набору для инактивации вирусов, включающему моющее средство на основе простого эфира полиоксиэтилена, предлагаемое в настоящем изобретении, или композицию, включающую моющее средство на основе простого эфира полиоксиэтилена, предлагаемое в настоящем изобретении, и дополнительно включающему хроматографическую смолу для хроматографической очистки. В другом варианте осуществления указанный набор дополнительно включает объемный фильтр.
Настоящее изобретение относится к не содержащим фенол простым эфирам полиоксиэтилена. Как указано выше, эти простые эфиры полиоксиэтилена можно использовать в способах инактивации вирусов, обладающих липидной оболочкой, предлагаемых в настоящем изобретении. Однако эти не содержащие фенол простые эфиры полиоксиэтилена, а также все другие не содержащие фенол простые эфиры полиоксиэтилена, предлагаемые в настоящем изобретении, также можно использовать для разных других целей, например, для тех, в которых обычно используют Triton X-100. Например, не содержащие фенол простые эфиры полиоксиэтилена, предлагаемые в настоящем изобретении, можно использовать в лаборатории. В лаборатории их можно использовать, например, для лизиса клеток с целью экстрагирования белка или органелл, или для придания проницаемости мембранам живых клеток; для придания проницаемости мембранам нефиксированных (или слабофиксированных) эукариотных клеток; для солюбилизации мембранных белков в их нативном состоянии совместно с цвиттерионными моющими средствами, такими как CHAPS; в качестве части литического буфера (обычно в 5% растворе в щелочном литическом буфере) при экстракции DNA; для снижения поверхностного натяжения водных растворов во время иммунного окрашивания (обычно при концентрации, равной 0,1-0,5% в буфере TBS или PBS); для ограничения размножения колоний Aspergillus nidulans в микробиологии; для извлечения клеток из взятых у животных тканей; или для удаления SDS из SDS-PAGE гелей до ренатурации белков в геле. В другом варианте осуществления не содержащие фенол простые эфиры полиоксиэтилена, предлагаемые в настоящем изобретении, можно использовать в электронной промышленности, например, в качестве смачивающего агента для пластин с целью улучшения и ускорения некоторых процедур и операций. В другом варианте осуществления не содержащие фенол простые эфиры полиоксиэтилена, предлагаемые в настоящем изобретении, можно использовать в медицине, например, их можно использовать в качестве заменителя спермицида ноноксинол 9 или в качестве фармацевтического инертного наполнителя, или в качестве ингредиента в вакцине от гриппа (Fluzone). В других вариантах осуществления не содержащие фенол простые эфиры полиоксиэтилена, предлагаемые в настоящем изобретении, можно использовать в разных типах очищающих систем в диапазоне от сильно действующих промышленных продуктов до мягких моющих средств; их можно использовать в качестве ингредиента в самодельных жидкостях для очистки виниловых пластинок вместе с дистиллированной водой и изопропиловым спиртом; их можно использовать при очистке алмаз дисков; их можно использовать в составах для полимеризации эмульсий; их можно использовать для покрышек; их можно использовать в моющих и очищающих средствах; их можно использовать в промышленности в качестве стандартного химиката для получения полимеров и клеев; их можно использовать в бытовых или промышленных очищающих устройствах, в красках или покрытиях, в целлюлозно-бумажной промышленности, в месторождениях нефти, в текстильной промышленности, в агрохимикатах, в жидкостях для обработки металлов; их можно использовать для диспергирования углеродных материалов в мягких композитных материалах; или их можно использовать при нанесении металлических покрытий.
Ниже настоящее изобретение проиллюстрировано примерами, но оно не ограничивается только ими.
ПРИМЕРЫ
Как указано выше, последние экологические исследования усилили опасения, связанные с воздействием на окружающую среду в случае применения Triton X-100 в биофармацевтических производственных процессах. На основании исследований структуры авторы настоящего изобретения выявили возможные моющие средства в качестве подходящих альтернатив для Triton X-100, которые не связаны с неблагоприятным воздействием на окружающую среду. В частности, авторы настоящего изобретения неожиданно установили, что простые эфиры полиоксиэтилена являются подходящими альтернативами для Triton X-100 для инактивации вирусов с липидной оболочкой путем обработки растворителем/моющим средством (S/D) в биофармацевтическом производстве. В описанных ниже экспериментах применимость типичных возможных моющих средств для S/D обработки исследовали с использованием содержащих внутривенный иммуноглобулин (IVIG) и сывороточный альбумин человека в содержащих (HSA) жидкостях. Кроме того, в описанных ниже экспериментах синтезировали полиэтоксилат 4-трет-октилбензилового спирта и для разных объектов исследовали применимость полиэтоксилата 4-трет-октилбензилового спирта и других простых эфиров полиоксиэтилена для S/D обработки, а также обработки одним моющим средством.
Пример 1: Инактивация HIV и PRV с использованием Triton X-100 reduced или Triton N-101 reduced в жидкостях, содержащих внутривенный иммуноглобулин
Применимость Triton X-100 reduced или Triton N-101 reduced для S/D обработки для инактивации вирусов с липидной оболочкой, вируса иммунодефицита человека (HIV) и вирус псевдобешенства (PRV) в жидкостях, содержащих внутривенный иммуноглобулин (IVIG) исследовали и сопоставляли с Triton X-100. Для этого вирус добавляли к жидкостям, содержащим IVIG. Содержащие вирус жидкости, включающие IVIG, затем инкубировали с S/D смесями, обладающими низкими концентрациями Triton X-100 reduced, Triton N-101 reduced или Triton X-100, в течение разных периодов времени и определяли оставшуюся инфекционность вирусов. Triton X-100 reduced, Triton N-101 reduced и Triton X-100 использовали при низких концентрациях для исследования кинетики инактивации вирусов, т. е. эффективности инактивации вирусов (выраженной с помощью RF) в зависимости от времени. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, моющие средства, предлагаемые в настоящем изобретении, включая Triton X-100 reduced или Triton N-101 reduced, можно использовать при значительно более высоких концентрациях, что увеличит скорость инактивации вирусов и также предположительно увеличит обеспечиваемый LRV.
Материалы
Жидкость, содержащая IVIG
Жидкость, содержащую IVIG (содержащую IVIG жидкость) замораживали твердым диоксидом углерода и хранили при ≤ -60°C для использования в течение не более одного года после даты сбора.
Вирусы
Вирус псевдобешенства (PRV; семейство Herpesviridae; с оболочкой; dsDNA; Ø=120-200 нм) использовали в качестве модели для больших DNA-вирусов с оболочкой.
Вирус иммунодефицита человека (HIV; семейство Retroviridae; с оболочкой; ssRNA; Ø=80-100 нм) использовали в качестве соответствующего вируса-мишени и модели для других RNA-вирусов с липидной оболочкой (рибонуклеиновая кислота), таких как HIV-2.
HIV-1 IIIB
Таблица 1: Исходные растворы вирусов, использованные в экспериментах.
Исходные растворы вирусов исследовали до использования. Это исследование включало определение титра вируса посредством не менее 10 независимых титрований и спецификации приемлемости диапазона титра вируса для применения в качестве положительного контроля, определение содержания белка в исходном растворе вирусов, исследование с помощью PCR идентичности вирусов и загрязнения другими вирусами и микоплазмой и исследования агрегации вирусов в фильтрах, не пропускающих крупные вирусные агрегаты. Использовали только исходные растворы вирусов, удовлетворяющие требованиям исследований с помощью PCR идентичности/загрязнения при отсутствии значительной агрегации (т. е. разность титров инфекционности исходного раствора вирусов и фильтрованного исходного раствора была меньше 1,0 log).
Смеси реагентов Triton X-100 reduced или Triton N-101 reduced
Компоненты S/D полисорбат 80 (PS80, Crillet 4 HP, Tween 80) и три-н-бутилфосфат (TnBP) объединяли с соответствующим моющим средством (т. е. Triton X-100, Triton X-100 reduced или Triton N-101 reduced) в следующем отношении (см. таблицы 2-4):
Таблица 2: Количество компонентов для смеси Triton X-100 реагент S/D
Таблица 3: Количество компонентов для смеси Triton X-100 reduced реагент S/D
Таблица 4: Количество компонентов для смеси Triton N-101 reduced реагент S/D
Каждую смесь перемешивали в течение не менее 15 мин. Смеси реагента S/D хранили при комнатной температуре для использования в течение не более 1 года. До использования смесь соответствующего реагента S/D повторно перемешивали в течение не менее 15 мин для обеспечения гомогенности.
Методики
Способность к инактивации вирусов и эффективность S/D обработки исследовали при условиях, неблагоприятных для инактивации вирусов, т. е. при коротких периодах инкубации и относительно низких температурах. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, можно использовать более продолжительные периоды инкубации и более высокие температуры, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV. Кроме того, как уже отмечено выше, низкие концентрации компонентов S/D использовали. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, можно использовать более высокие концентрации компонентов S/D, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV.
Поскольку концентрация белка не оказывает значительное влияние на инактивацию вирусов во время обработки S/D (см. также Dichtelmüller et al., 2009), стабильность содержания белка не исследовали.
Содержащую IVIG жидкость размораживали и все остальные стадии проводили в камере класса II биологической безопасности. Содержащую IVIG жидкость инкубировали при перемешивании при температуре, равной +17°C ± 1°C с использованием для инкубации сосуда с двойными стенками, соединенного с криостатом. Содержащую IVIG жидкость затем фильтровали через объемный фильтр с отверстиями размером 0,2 мкм с эффективной площадью фильтра, равной 25 см2 (Zeta Plus® VR06, Cuno/3M или эквивалентный), соединенный с изготовленным из нержавеющей стали держателем фильтра Sartorius (SM16249) с использованием сжатого азота при целевом давлении, равном 0,9 бар (предельное значение: 0,5 бар - 1,5 бар). Для кондиционирования до фильтрования жидкости на материал фильтра предварительно наносили покрытие с помощью 55 л/м2 суспензии Hyflo Supercel (5,0 г ± 0,05 г Hyflo Supercel на 1 л; проводимость устанавливали равной 3,5 мСм/см (установленный диапазон: 2,5-6,0 мСм/см) с использованием 3 M NaCl) (при давлении ≤ 0,5 бар). Во время фильтрования держатель фильтра охлаждали и фильтрат собирали при заданной температуре, равной ≤ 18°C с использованием сосуда с двойными стенками, соединенного с криостатом. Сосуд для фильтрата охлаждали. В случае забивания фильтра для продолжения фильтрования оставшейся жидкости использовали свежие предварительно кондиционированные фильтры. После фильтрования измеряли температуру (целевое значение: ≤ 18°C) и объем.
После измерения объема содержащей IVIG жидкости при перемешивании с помощью холодного (от +2°C до +8°C) буфера для разведения (раствор NaCl с целевой проводимостью, равной 3,5 мСм/см (диапазон: 2,5 мСм/см - 6,0 мСм/см)) для нее устанавливали рассчитанную целевую оптическую плотность, равную 28,9 AU280-320/см (диапазон: 14,5-72,3 AU280-320/см). С учетом последующего добавления вируса в отношении 1:31 это дает рассчитанную целевую оптическую плотность, равную 28 AU280-320/см (диапазон: 14-70 AU280-320/см) для инкубации с реагентами S/D после фильтрования.
Температуру фильтрованной содержащей IVIG жидкости с отрегулированным содержанием белка при перемешивании повторно устанавливали равной +17°C ± 1°C с использованием сосуда с двойными стенками, соединенного с криостатом. Этот диапазон температуры при перемешивании поддерживали до завершения инкубации фильтрованной содержащей IVIG жидкости с реагентами S/D и непрерывно регистрировали. К заданному объему фильтрованной содержащей IVIG жидкости, перенесенному в сосуд с винтовой крышкой, для которого определяли массу тары, добавляли вирус при отношении 1:31, например, к 30 мл содержащей IVIG жидкости добавляли 1 мл исходного раствора вируса. Содержащую IVIG жидкость с добавкой дополнительно инкубировали при непрерывном перемешивании при 17°C ± 1°C. Не позже чем через 1-2 мин после добавления отбирали образцы для титрования вируса (0,5 мл контроля добавления, SC, и 2 мл контроля удерживания, HC).
После размораживания контроль удерживания (HC) выдерживали при такой же температуре, т. е. при +17°C ± 1°C, как обрабатываемый материал с добавкой после добавления реагентов S/D, т. е. его хранили при таком же цикле охлаждения, как сосуд с содержащей IVIG жидкостью, до конца S/D обработки. Температуру охлаждающей жидкости определяли до введения образца контроля удерживания и повторно незадолго до удаления контроля удерживания для титрования после S/D обработки.
Массу содержащей IVIG жидкости с добавкой определяли для расчета количества добавляемой смеси реагента S/D. Температуру взвешенного материала, при необходимости при перемешивании, повторно устанавливали равной +17°C ± 1°C. Смесь соответствующего реагента S/D добавляли к содержащей IVIG жидкости и получали конечную концентрацию, равную 0,05% соответствующего моющего средства на основе простого эфира полиоксиэтилена. Смесь реагента S/D при перемешивании добавляли в течение 1 мин с помощью шприца и фактическое количество добавленной смеси реагента S/D определяли путем повторного взвешивания шприца. Содержащую IVIG жидкость с добавкой дополнительно инкубировали с реагентами S/D при непрерывном перемешивании при +17°C ± 1°C в течение 59 ± 1 мин. Во время инкубации образцы по 1 мл для титрования вируса отбирали через 1-2 мин, 10 ± 1 мин, 30 ± 1 мин и 59 ± 1 мин. Для предупреждения дополнительной инактивации вируса реагентами S/D после отбора образца, образцы сразу разводили в отношении 1:20 холодной (от +2°C до +8°C) клеточной культуральной средой (т. е. 1 объем образца и 19 объемов клеточной культуральной среды).
Для титрования образцов серийные 0,5 log разведения образцов готовили в подходящей культуральной среде для тканей и 100 мкл каждого разведения добавляли в каждую из 8 лунок планшета для микротитрования, засеянных клетками соответствующей индикаторной линии. Клетки инкубировали в течение 7 дней при 36,0°C (заданное значение) и затем цитопатический эффект определяли путем визуального осмотра под микроскопом. Медианные инфекционные дозы культуры ткани (TCID50) рассчитывали по распределению Пуассона и представляли в виде log10[TCID50/мл].
Расчет степени удаления вируса проводили с помощью следующей формулы:
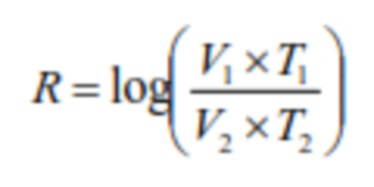
где,
R=коэффициент уменьшения содержания вируса
V1=объем исходного материала [мл]
T1=концентрация вируса в исходном материале [TCID50/мл]
V2=объем материала после инактивации вирусов [мл]
T2=концентрация вируса после инактивации вирусов [TCID50/мл]
Объемы и титры каждого образца с добавкой до и после обработки использовали для расчета R. Если вирус не обнаруживался, то для расчета за предел обнаружения принимали титр вируса.
Результаты
Когда S/D обработку содержащей IVIG жидкости проводили с использованием смеси Triton X-100 reduced, PS80 и TnBP или смеси Triton N-101 reduced, PS80 и TnBP, HIV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным не менее 4, не более, чем за 10 мин (фиг. 1A). Аналогичные результаты получали в повторном эксперименте (фиг. 1B).
В дополнительных экспериментах, когда S/D обработку содержащей IVIG жидкости проводили с использованием смеси Triton X-100 reduced, PS80 и TnBP или смеси Triton N-101 reduced, PS80 и TnBP, PRV инактивировался с RF, равным примерно 4, не более, чем за 10 мин (фиг. 2A). Аналогичные результаты получали в повторном эксперименте (фиг. 2B).
Эти эксперименты показали, что замена Triton X-100 на Triton X-100 reduced или Triton N-101 reduced для S/D обработки жидкостей, содержащих биофармацевтическое лекарственное средство, приводит к эффективной инактивации вирусов с липидной оболочкой, даже при низких концентрациях моющих средств.
Пример 2: Инактивация HIV, PRV и BVDV с использованием Brij C10 в жидкостях, содержащих внутривенный иммуноглобулин
Материалы и методики
Эти эксперименты проводили, как описано выше в примере 1. Однако S/D обработку смесью S/D, содержащей Brij C10, исследовали и сопоставляли со смесью S/D, содержащей Triton X-100. Композицию смеси S/D, содержащей Triton X-100, получали, как описано выше в примере 1. Композицию смеси S/D, содержащей Brij C10, получали следующим образом.
Компоненты S/D полисорбат 80 (PS80, Crillet 4 HP, Tween 80) и три-н-бутилфосфат (TnBP) объединяли с моющим средством Brij C10 в следующих отношениях (см. таблицу 5):
Таблица 5: Количество компонентов для смеси Brij C10 реагент S/D
Смесь перемешивали в течение не менее 15 мин. Смесь реагента S/D хранили при комнатной температуре для использования в течение не более 1 года. До использования смесь реагента S/D повторно перемешивали в течение не менее 15 мин для обеспечения гомогенности.
Смеси соответствующего реагента S/D добавляли к содержащей IVIG жидкости и получали конечную концентрацию, равную 0,05% соответствующего моющего средства на основе простого эфира полиоксиэтилена.
Кроме того, также исследовали инактивацию вируса BVDV:
Таблица 6: Исходные растворы вирусов, использованные в экспериментах.
Результаты
Когда S/D обработку содержащей IVIG жидкости проводили с использованием смеси Brij C10, PS80 и TnBP, HIV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным более 4, не более, чем за 10 мин (фиг. 3A). Аналогичные результаты получали в повторном эксперименте (фиг. 3B).
В дополнительных экспериментах, когда S/D обработку содержащей IVIG жидкости проводили с использованием смеси Brij C10, PS80 и TnBP, PRV инактивировался с RF, равным примерно 4, не более, чем за 10 мин (фиг. 4A). Аналогичные результаты получали в повторном эксперименте (фиг. 4B).
В дополнительных экспериментах, когда S/D обработку содержащей IVIG жидкости проводили с использованием смеси Brij C10, PS80 и TnBP, BVDV инактивировался с RF, равным примерно 5, не более, чем за 10 мин (фиг. 5A). Аналогичные результаты получали в повторном эксперименте (фиг. 5B).
Эти эксперименты показали, что замена Triton X-100 на Brij C10 для S/D обработки жидкостей, содержащих биофармацевтическое лекарственное средство, приводит к эффективной инактивации вирусов с липидной оболочкой, даже при низких концентрациях моющего средства.
Пример 3: Инактивация X-MuLV и BVDV с использованием Brij C10 в жидкостях, содержащих сывороточный альбумин человека
Применимость Brij C10 для S/D обработки для инактивации вирусов с липидной оболочкой, ксенотропного вируса лейкемии мышей (X-MuLV) и вируса диареи крупного рогатого скота (BVDV) в жидкостях, содержащих сывороточный альбумин человека (HSA) в качестве модельного белка для биофармацевтического лекарственного средства исследовали и сопоставляли с Triton X-100. Для этого вирус добавляли к жидкостям, содержащим HSA. Содержащие вирус жидкости, включающие HSA, затем инкубировали с S/D смесями, обладающими низкими концентрациями Brij C10, в течение разных периодов времени и определяли оставшуюся инфекционность вирусов. Brij C10 использовали при низких концентрациях, чтобы было можно исследовать кинетику инактивации вируса, т. е. эффективности инактивации вирусов (выраженной с помощью RF) в зависимости от времени. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, Brij C10 можно использовать при значительно более высоких концентрациях, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый RF.
Материалы
Жидкость, содержащая HSA
Сывороточный альбумин человека в содержащей (HSA) жидкости использовали в качестве модели для жидкости, содержащей биофармацевтическое лекарственное средство.
Вирусы
Ксенотропный вирус лейкемии мышей (X-MuLV; семейства Retroviridae; вирус с оболочкой ssRNA; Ø=80-110 нм) использовали в качестве модели для эндогенных ретровирусных частиц и RNA-вирусов с оболочкой. Кроме того, использовали BVDV.
pNFS Th1
Таблица 7: Исходные растворы вирусов, использованные в экспериментах.
Исходные растворы вирусов исследовали до использования. Это исследование включало определение титра вируса посредством не менее 10 независимых титрований и спецификации приемлемости диапазона титра вируса для применения в качестве положительного контроля, определение содержания белка в исходном растворе вирусов, исследование с помощью PCR идентичности вирусов и загрязнения другими вирусами и микоплазмой и исследования агрегации вирусов в фильтрах, не пропускающих крупные вирусные агрегаты. Использовали только исходные растворы вирусов, удовлетворяющие требованиям исследований с помощью PCR идентичности/загрязнения при отсутствии значительной агрегации (т. е. разность титров инфекционности исходного раствора вирусов и фильтрованного исходного раствора была меньше 1,0 log).
Смесь реагента Brij
Компоненты S/D полисорбат 80 (PS80, Crillet 4 HP, Tween 80) и три-н-бутилфосфат (TnBP) объединяли с соответствующим моющим средством (т. е. Triton X-100 или Brij C10) в следующем отношении (см. таблицы 8-9):
Таблица 8: Количество компонентов для смеси Triton X-100 реагент S/D
Таблица 9: Количество компонентов для смеси Brij C10 реагент S/D
Каждую смесь перемешивали в течение не менее 15 мин. Смеси реагента S/D хранили при комнатной температуре для использования в течение не более 1 года. До использования смесь соответствующего реагента S/D повторно перемешивали в течение не менее 15 мин для обеспечения гомогенности.
Методики
Способность к инактивации вирусов и эффективность S/D обработки исследовали при условиях, неблагоприятных для инактивации вирусов, т. е. при коротких периодах инкубации и относительно низких температурах. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, можно использовать более продолжительные периоды инкубации и более высокие температуры, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV. Кроме того, как уже отмечено выше, низкие концентрации компонентов S/D использовали. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, можно использовать более высокие концентрации компонентов S/D, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV.
Поскольку концентрация белка не оказывает значительное влияние на инактивацию вирусов во время обработки S/D (см. также Dichtelmüller et al., 2009), стабильность содержания белка не исследовали.
Все остальные стадии проводили в камере класса II биологической безопасности. Исходный материал инкубировали при перемешивании при температуре, равной +1°C ± 1°C с использованием для инкубации сосуда с двойными стенками, соединенного с криостатом. Содержащую HSA жидкость затем фильтровали через обладающий отверстиями размером 0,2 мкм PVDF мембранный шприцевый фильтр (гидрофильный PVDF фильтр, Millipak 60 или эквивалентный). После фильтрования измеряли температуру и объем.
Температуру фильтрованной содержащей HSA жидкости при перемешивании повторно устанавливали равной +1°C ± 1°C с использованием сосуда с двойными стенками, соединенного с криостатом. Этот диапазон температуры при перемешивании поддерживали до завершения инкубации фильтрованной содержащей HSA жидкости с реагентами S/D и непрерывно регистрировали. К заданному объему фильтрованной содержащей HSA жидкости, перенесенному в сосуд с винтовой крышкой, для которого определяли массу тары, добавляли вирус при отношении 1:31, например, к 48 мл содержащей HSA жидкости добавляли 1,6 мл исходного раствора вируса. Содержащую HSA жидкость с добавкой дополнительно инкубировали при непрерывном перемешивании при 1°C ± 1°C. Не позже чем через 1-2 мин после добавления отбирали образцы для титрования вируса (0,5 мл контроля добавления, SC, и 2 мл контроля удерживания, HC).
Незадолго до удаления контроль удерживания для титрования (HC) выдерживали при такой же температуре, т. е. при +1°C ± 1°C, как содержащую HSA жидкость с добавкой после добавления реагентов, т. е. ее хранили при таком же цикле охлаждения, как сосуд с содержащей с содержащей HSA жидкостью, до конца S/D обработки. Температуру охлаждающей жидкости определяли до введения образца контроля удерживания и повторно незадолго до удаления контроля удерживания для титрования после S/D обработки.
Массу содержащей HSA жидкости с добавкой определяли для расчета количества добавляемой смеси реагента S/D. Температуру взвешенного материала, при необходимости при перемешивании, повторно устанавливали равной +1°C ± 1°C. Смесь соответствующего реагента S/D добавляли к содержащей HSA жидкости и получали конечную концентрацию, равную от 0,08% до 0,1% (мас./мас.) соответствующего моющего средства на основе простого эфира полиоксиэтилена. Смесь реагента S/D при перемешивании добавляли в течение 1 мин с помощью шприца и фактическое количество добавленной смеси реагента S/D определяли путем повторного взвешивания шприца. Содержащую HSA жидкость с добавкой дополнительно инкубировали с реагентами S/D при непрерывном перемешивании при +1°C ± 1°C в течение 59 ± 1 мин. Во время инкубации образцы по 1 мл для титрования вируса отбирали через 1-2 мин, 10 ± 1 мин, 30 ± 1 мин и 59 ± 1 мин. Для предупреждения дополнительной инактивации вируса реагентами S/D после отбора образца, образцы сразу разводили в отношении 1:20 холодной (от +2°C до +8°C) клеточной культуральной средой (т. е. 1 объем образца и 19 объемов клеточной культуральной среды).
Титрование образцов и расчет степени удаления вируса проводили, как описано выше в примере 1.
Результаты
Когда S/D обработку содержащей HSA жидкости проводили при 1°C ± 1°C с использованием смеси Brij C10, PS80 и TnBP, X-MuLV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным более 2, не более, чем за 60 мин (фиг. 6A). Аналогичные результаты получали в повторном эксперименте (фиг. 6B).
В дополнительных экспериментах, когда S/D обработку содержащей HSA жидкости проводили при 1°C ± 1°C с использованием смеси Brij C10, PS80 и TnBP, BVDV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным примерно 4, не более, чем за 10 мин (фиг. 7A). Аналогичные результаты получали в повторном эксперименте (фиг. 7B).
Дополнительные эксперименты проводили точно так, как описано в разделе Материалы и методики этого примера, только S/D обработку содержащей HSA жидкости проводили при 19°C ± 1°C вместо 1°C ± 1°C. Когда S/D обработку содержащей HSA жидкости проводили при 19°C ± 1°C с использованием смеси Brij C10, PS80 и TnBP (Brij C10), X-MuLV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным более 3, не более, чем за 10 мин (фиг. 8A). Аналогичные результаты получали в повторном эксперименте (фиг. 8B).
Эти эксперименты показали, что замена Triton X-100 на Brij C10 для S/D обработки жидкостей, содержащих биофармацевтическое лекарственное средство, приводит к эффективной инактивации вирусов с липидной оболочкой при комнатной температуре или температуре, близкой к комнатной температуре, и при температурах, равных лишь 1°C ± 1°C, даже при низких концентрациях моющих средств.
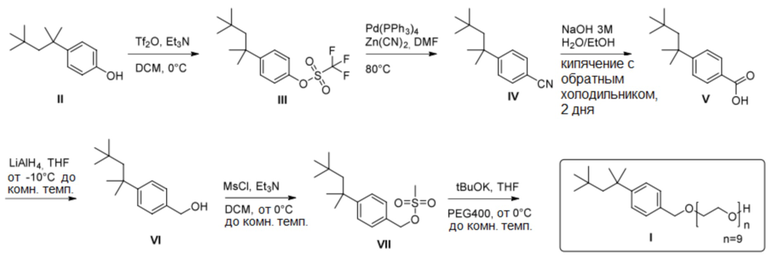
Пример 4: Синтез полиэтоксилата 4-трет-октилбензилового спирта I (методика 1)
Синтез промежуточного продукта III:
Фенол II (170,3 г, 800 ммоля) помещали в 3-горлую колбу объемом 2 л, снабженную внутренним термометром и стержнем для магнитной мешалки. В колбу добавляли безводный CH2Cl2 (1000 мл) и начинали перемешивание. После растворения исходного вещества раствор охлаждали до 0°C. После полного растворения за 10 мин добавляли NEt3 (225 мл, 1,6 моля). К реакционной смеси в течение 120 мин при 0°C добавляли раствор Tf2O (256 г, 907 ммоля) в CH2Cl2 (180 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли насыщенный водный раствор NaHCO3 (400 мл) и реакционную смесь экстрагировали. Органическую фазу несколько раз промывали водой (2×400 мл) и рассолом (500 мл). Затем органическую фазу концентрировали в вакууме. К остатку добавляли толуол (300 мл) и неочищенный продукт концентрировали и получали 314 г неочищенной черной жидкости. Этот остаток сверху помещали на слой SiO2 и элюировали петролейным эфиром/EtOAc (от 0 до 2%). Концентрирование чистых фракций давало 269,5 г (выход: 99,6%) прозрачного бесцветного масла. Rf=0,74 (петролейный эфир/EtOAc 5%).
Синтез промежуточного продукта IV:
Трифлат III (269 г, 795 ммоля) растворяли в безводном и дегазированном DMF (1,3 л) и последовательно добавляли Zn(CN)2 (95,3 г, 795 ммоля) и Pd(PPh3)4 (25 г, 21,5 ммоля). Реакционную смесь нагревали при 80°C в течение 3 ч, затем DMF удаляли в вакууме. К остатку добавляли толуол (300 мл) и неочищенный продукт концентрировали и получали 432 г черного остатка. Этот неочищенный продукт сверху помещали на слой SiO2 и элюировали петролейным эфиром/EtOAc (от 0 до 10%). Концентрирование чистых фракций давало 128,1 г (выход: 74,8%) прозрачного бесцветного масла. Rf=0,23 (петролейный эфир/EtOAc 2%).
Синтез промежуточного продукта V:
Нитрил IV (127,6 г, 592,5 ммоля) растворяли в MeOH (500 мл), добавляли водный раствор NaOH 4M (750 мл) и смесь нагревали до кипения и выдерживали при этой температуре (80°C) в течение ночи. К теплой смеси дополнительно добавляли водный раствор NaOH 10M (150 мл) и раствор дополнительно нагревали в течение 20 ч. После охлаждения до температуры окружающей среды содержимое сосуда для проведения реакции переносили в большой стакан и охлаждали в бане со льдом. Водный раствор HCl 4M (1,1 л) добавляли в течение 30 мин, в этот момент по данным индикаторной бумаги среда была кислой и осаждалось белое твердое вещество. Осадок отфильтровывали и промывали водой (500 мл). Влажный осадок на фильтре переносили в колбу объемом 2 л и сушили в вакууме в течение 3 дней и получали 127,5 г (выход: 91,9%) белого порошкообразного вещества. Rf=0,42 (петролейный эфир/EtOAc 2:1).
Синтез промежуточного продукта VI:
Карбоновую кислоту V (127 г, 542 ммоля) суспендировали в сухом THF (1,2 л) и охлаждали до -10°C. Раствор LiAlH4 в THF (1 M, 575 мл, 575 ммоля) добавляли в течение 60 мин, затем реакционную смесь медленно нагревали до температуры окружающей среды и дополнительно перемешивали в течение ночи. Содержимое сосуда для проведения реакции переносили в большой стакан и избыток гидрида осторожно разлагали льдом (15 г). Водный раствор HCl 3M (500 мл) добавляли в течение 20 мин, в этот момент по данным индикаторной бумаги среда была кислой. К неочищенной смеси добавляли EtOAc (300 мл) и 2 фазы энергично перемешивали. Водную фазу дважды подвергали обратной экстракции с помощью EtOAc (300 мл и 500 мл). Объединенные органические фазы последовательно промывали насыщенным водным раствором NaHCO3 (300 мл), водой (300 мл) и рассолом (500 мл). Затем органическую фазу концентрировали в вакууме. К остатку добавляли толуол (300 мл) и неочищенный продукт концентрировали и получали 123,3 г прозрачного желтоватого масла. Этот остаток сверху помещали на слой SiO2 и элюировали петролейным эфиром/EtOAc (от 0 до 15%). Концентрирование чистых фракций давало 85,3 г (выход: 71,4%) аморфного белого твердого вещества. Rf=0,37 (петролейный эфир/EtOAc 4:1).
Синтез промежуточного продукта VII:
Бензиловый спирт VI (84,8 г, 385 ммоля) растворяли в безводном CH2Cl2 (1 л) и охлаждали в бане лед/вода. Добавляли NEt3 (110 мл, 770 ммоля), затем за 60 мин медленно добавляли раствор MsCl (45 мл, 577 ммоля) в безводном CH2Cl2 (25 мл). Реакционную смесь медленно нагревали до температуры окружающей среды и дополнительно перемешивали в течение ночи. Добавляли насыщенный водный раствор NaHCO3 (420 мл) и реакционную смесь энергично перемешивали. Органическую фазу несколько раз промывали водой (2×500 мл) и рассолом (300 мл). Затем органическую фазу концентрировали в вакууме. К остатку добавляли толуол (200 мл) и CH2Cl2 (100 мл) и неочищенный продукт концентрировали и получали 90 г (выход: 78,4%) оранжевого полужидкого вещества. Rf=0,71 (петролейный эфир/EtOAc 4:1).
Синтез I:
PEG400 (360 г, 900 ммоля) растворяли в безводном THF (1 л) и при температуре окружающей среды порциями за 15 мин добавляли tBuOK (90 г, 802 ммоля) и смесь перемешивали 60 мин при температуре окружающей среды и охлаждали в бане со льдом. Одновременно мезилат VII (89,5 г, 300 ммоля) суспендировали в THF (300 мл) и мутный оранжевый раствор за 20 мин добавляли к охлажденному раствору депротонированного PEG400 раствору. Реакционную смесь медленно нагревали до температуры окружающей среды и дополнительно перемешивали в течение ночи. Добавляли лед (500 г), а также водный раствор HCl 1M (820 мл). THF удаляли в вакууме и добавляли EtOAc (1 л). Фазы перемешивали и органическую фазу последовательно промывали водой (2×500 мл). Каждую водную фазу подвергали обратной экстракции с помощью EtOAc (300 мл). Объединенные органические фазы промывали водой (500 мл) и концентрировали в вакууме. К остатку добавляли толуол (250 мл) и неочищенный продукт концентрировали и получали 125,2 г прозрачного желтоватого масла. Этот остаток сверху помещали на слой SiO2 и элюировали с помощью CH2Cl2/MeOH (от 0 до 8%). Концентрирование чистых фракций давало 107,5 г (выход: 59,5%) прозрачного светло-коричневого масла. Rf=0,37-0,22 (CH2Cl2/MeOH 20:1). MS (ESI): m/z =[M+H]+= 573,5, 617,5 (100%), 661,6; [M+Ac]- = 631,4, 675,4 (100%), 719,5. 1H ЯМР (600 МГц, CDCl3): δ=7,33 (д, J=8,3 Гц, 2H), 7,23 (д, J=8,3 Гц, 2H), 4,52 (с, 2H), 3,72-3,58 (м, 33H), 2,54 (уш с, 1H), 1,72 (с, 2H), 1,34 (с, 6H), 0,70 (с, 9H). 13C ЯМР (150 МГц, CDCl3): δ=149,7, 135,1, 127,4 (2C), 126,2 (2C), 73,2, 72,6, 70,7 (м), 70,5, 69,4, 61,9, 57,0, 38,6, 32,5, 31,9 (3C), 31,6 (2C).
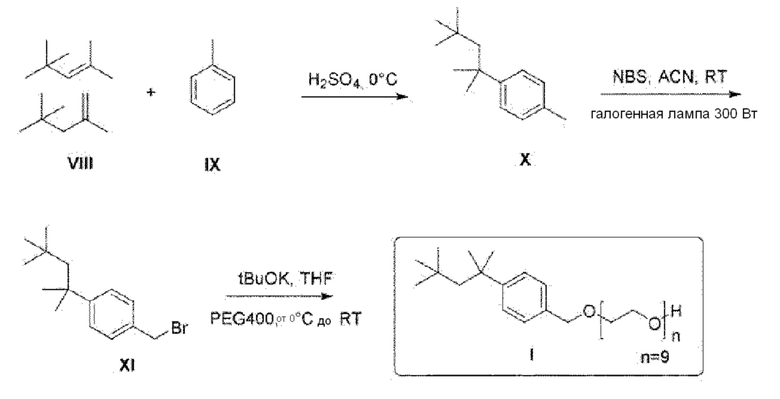
Пример 5a: Синтез полиэтоксилата 4-трет-октилбензилового спирта I (методика 2)
Толуол (35 мл, 328 ммоля) и концентрированную H2SO4 (10 мл) охлаждали до 0°C в круглодонной колбе. Смесь толуола (27 мл, 256 ммоля) и диизобутилена (в виде смеси 3:1 2,4,4-триметил-1-пентен+2,4,4-триметил-2-пентен, 10 мл, 64 ммоля) к реакционной смеси медленно добавляли в течение 2 ч. Реакционную смесь дополнительно перемешивали при 0°C в течение 2 ч. Добавляли воду (100 мл). После разделения фаз органическую фазу промывали насыщенным водным раствором NaHCO3 (100 мл), сушили над MgSO4 и концентрировали и получали 14 г прозрачного бесцветного масла. Масло помещали в круглодонную колбу объемом 100 мл и перегоняли в аппарате для молекулярной перегонки (1·10-1 мбар). Собирали фракцию (8,1 г, выход 61,9%), которая отгонялась при температуре от 62 до 70°C. Rf=0,65 (петролейный эфир 40-60 100%).
п-Замещенный толуол X (500 мг, 2,45 ммоля) растворяли в ацетонитриле (ACN) (5 мл). Добавляли N-бромсукцинимид (NBS) (460 мг, 2,57 ммоля) и после растворения круглодонную колбу Duran объемом 25 мл помещали на расстоянии 15 см от галогеновой лампы (300 Вт). После облучения в течение 60 мин (температура при проведении реакции=45°C) растворитель удаляли. Добавляли петролейный эфир 40-60 (25 мл) и осаждалось твердое вещество. Жидкую фазу промывали водой (2×20 мл), сушили над MgSO4 и концентрировали и получали 540 мг (выход: 77,9%) неочищенного желтоватого масла. Rf=0,43 (петролейный эфир 40-60 100%).
PEG400 (2,25 г, 5,61 ммоля) растворяли в безводном THF (5 мл) при температуре окружающей среды. Порциями в течение 1 мин добавляли tBuOK (420 мг, 3,74 ммоля) и смесь перемешивали 90 мин и затем охлаждали до 0°C в бане лед/вода. Одновременно промежуточный бензилбромид XI (530 мг, 1,87 ммоля) суспендировали в THF (2 мл) и раствор добавляли к охлажденному раствору депротонированного PEG400. Реакционную смесь медленно нагревали до температуры окружающей среды и дополнительно перемешивали в течение ночи. К реакционной смеси добавляли HCl (1M, 20 мл), а также EtOAc (50 мл) и воду (20 мл). Раствор переносили в делительную воронку и энергично экстрагировали. После разделения фаз органическую фазу последовательно промывали водой (5×15 мл) и в заключение сушили над MgSO4 и получали 0,8 г неочищенного маслообразного остатка. Этот остаток сверху помещали в колонку с SiO2 и элюировали с помощью CH2Cl2/MeOH (от 0 до 8%). Концентрирование чистых фракций давало 588 мг (выход: 52,2%) прозрачного желтоватого масла. Rf=0,37-0,22 (CH2Cl2/MeOH 20:1).
Пример 5b: Синтез полиэтоксилата 4-трет-октилбензилового спирта I (методика 2, варианты)
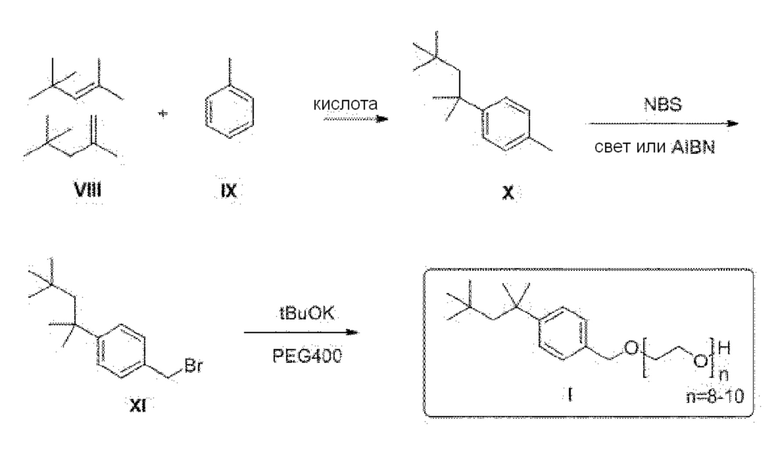
Стадия 1:
Эту стадию проводили следующим образом:
Толуол (750 мл, 7,04 моля) и концентрированную H2SO4 (20 мл, 0,375 моля) охлаждали до 0°C в круглодонной колбе. Смесь толуола (250 мл, 2,35 моля) и диизобутилена (в виде смеси 3:1 2,4,4-триметил-1-пентен+2,4,4-триметил-2-пентен, 200 мл, 1,28 моля) медленно добавляли к реакционной смеси в течение 90 мин. Реакционную смесь дополнительно перемешивали при 0°C и нагревали до температуры окружающей среды в течение ночи. Добавляли лед (400 г) и смесь переносили в делительную воронку. После разделения фаз органическую фазу последовательно промывали насыщенным водным раствором NaHCO3 (300 мл) и водой (2×250 мл). Органическую фазу концентрировали и получали 199,1 г прозрачного бесцветного масла. Масло помещали в круглодонную колбу объемом 500 мл и перегоняли в аппарате для молекулярной перегонки (20 мбар). Собирали фракции, которые отгонялись при температуре от 138°C до 161°C (134,1 г, выход 51,4%). Rf=0,65 (петролейный эфир 40-60 100%),1H ЯМР (600 МГц, CDCl3): δ=7,28 (д, J=8,2 Гц, 2H), 7,10 (д, J=8,2 Гц, 2H), 2,34 (с, 3H), 1,75 (с, 2H), 1,38 (с, 6H), 0,75 (с, 9H). 13C ЯМР (150 МГц, CDCl3): δ=147,3, 134,7, 128,6 (2C), 126,1 (2C), 57,1, 38,4, 32,5, 31,9 (3C), 31,7 (2C), 21,0.
Альтернативно, эту стадию проводили следующим образом:
Толуол (400 мл, 3,83 моля) и нонафтор-1-бутансульфоновую кислоту (4 мл, 24 ммоля) перемешивали при температуре окружающей среды в круглодонной колбе. Смесь толуол (200 мл, 1,92 моля) и диизобутилен (в виде смеси 3:1 2,4,4-триметил-1-пентен+2,4,4-триметил-2-пентен, 100 мл, 0,64 моля) медленно добавляли к реакционной смеси в течение 60 мин. Реакционную смесь дополнительно перемешивали при температуре окружающей среды в течение ночи. Добавляли насыщенный водный раствор NaHCO3 (200 мл) и смесь перемешивали в течение 20 мин. Смесь переносили в делительную воронку и водную фазу отбрасывали. Органическую фазу последовательно промывали водой (3×300 мл). Органическую фазу концентрировали и получали 132,5 г прозрачного бесцветного масла. Масло помещали в круглодонную колбу объемом 500 мл и перегоняли в аппарате для молекулярной перегонки (26-16 мбар). Собирали фракции, которые отгонялись при температуре от 115°C до 135°C (80,5 г, выход 62%). Rf=0,65 (петролейный эфир 40-60 100%). 1H ЯМР (600 МГц, CDCl3): δ=7,28 (д, J=8,2 Гц, 2H), 7,10 (д, J=8,2 Гц, 2H), 2,34 (с, 3H), 1,75 (с, 2H), 1,38 (с, 6H), 0,75 (с, 9H). 13C ЯМР (150 МГц, CDCl3): δ=147,3, 134,7, 128,6 (2C), 126,1 (2C), 57,1, 38,4, 32,5, 31,9 (3C), 31,7 (2C), 21,0.
Стадия 2:
Эту стадию проводили следующим образом:
п-Замещенный толуол X (63,6 г, 311 ммоля) растворяли в ацетонитриле (ACN) (650 мл). Добавляли N-бромсукцинимид (NBS) (58,2 г, 327 ммоля) и после растворения круглодонную колбу Duran объемом 25 мл помещали на расстоянии 5-25 см от галогеновой лампы (300 Вт) при перемешивании (350 об/мин). После облучения в течение 6 ч (температура при проведении реакции=до 46°C) растворитель удаляли в вакууме. Добавляли петролейный эфир 40-60 (450 мл) и осаждалось темное твердое вещество. Жидкую фазу концентрировали и получали темно-коричневый остаток (75 г). Этот остаток сверху помещали на слой SiO2 и элюировали петролейным эфиром 40-60 (100%). Концентрирование чистых фракций давало 43,8 г (выход: 49,7%) прозрачного оранжевого масла. Rf=0,43 (петролейный эфир 40-60 100%). 1H ЯМР (600 МГц, CDCl3): δ=7,34 (д, J=8,4 Гц, 2H), 7,30 (д, J=8,4 Гц, 2H), 4,50 (с, 2H), 1,74 (с, 2H), 1,36 (с, 6H), 0,72 (с, 9H). 13C ЯМР (150 МГц, CDCl3): δ=150,9, 134,8, 128,6 (2C), 126,7 (2C), 57,0, 38,7, 33,9, 32,5, 31,9 (3C), 31,6 (2C).
Альтернативно, эту стадию проводили следующим образом:
п-Замещенный толуол X (85,2 г, 417 ммоля) растворяли в трифтортолуоле (830 мл). Другие растворители, такие как сложные эфиры или алканы (EtOAc и гексан) также являются эффективными. Добавляли N-бромсукцинимид (NBS) (74,2 г, 417 ммоля), а также AIBN (азобис(изобутиронитрил)) (3,4 г, 21 ммоля) при перемешивании (450 об/мин). Смесь нагревали при 80°C в течение 5 ч. Растворитель удаляли в вакууме. Добавляли петролейный эфир 40-60 (350 мл) и осаждалось белое твердое вещество. Жидкую фазу концентрировали и получали прозрачный остаток (108 г). Этот остаток сверху помещали на слой SiO2 и элюировали петролейным эфиром 40-60 (100%). Концентрирование чистых фракций давало 64,1 г (выход: 54,3%) прозрачного почти бесцветного масла, которое кристаллизовали с получением бесцветных иголок, что указывало на высокую чистоту продукта. Rf=0,43 (петролейный эфир 40-60 100%). 1H ЯМР (600 МГц, CDCl3): δ=7,34 (д, J=8,4 Гц, 2H), 7,30 (д, J=8,4 Гц, 2H), 4,50 (с, 2H), 1,74 (с, 2H), 1,36 (с, 6H), 0,72 (с, 9H). 13C ЯМР (150 МГц, CDCl3): δ=150,9, 134,8, 128,6 (2C), 126,7 (2C), 57,0, 38,7, 33,9, 32,5, 31,9 (3C), 31,6 (2C). Предполагается, что использование AIBN (азобис(изобутиронитрил)) в качестве радикального инициатора способствует высокой чистоте продукта, который получен на этой стадии реакции.
Стадия 3:
Эту стадию проводили следующим образом:
PEG400 (184,1 г, 460 ммоля) растворяли в безводном THF (550 мл) при температуре окружающей среды. В течение 15 мин порциями добавляли tBuOK (27,2 г, 245 ммоля) и смесь перемешивали в течение 90 мин. Одновременно промежуточный бензилбромид XI (43,5 г, 153 ммоля) суспендировали в THF (300 мл) и раствор добавляли к охлажденному раствору депротонированного PEG400. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды. К реакционной смеси добавляли HCl (1M, 270 мл). Летучие вещества удаляли и добавляли EtOAc (600 мл). Раствор переносили в делительную воронку и энергично экстрагировали. После разделения фаз водную фазу дополнительно экстрагировали с помощью EtOAc (300 мл). Объединенные органические фазы последовательно промывали смесью вода/рассол (1:1, 3×300 мл) и в заключение концентрировали и получали 90,4 г неочищенного оранжевого маслообразного остатка. Этот остаток сверху помещали в колонку с SiO2 и элюировали с помощью CH2Cl2/MeOH (от 0 до 8%). Концентрирование чистых фракций давало 82,2 г (выход: 89,0%) прозрачного светло-коричневого масла. Rf=0,37-0,22 (CH2Cl2/MeOH 20:1). MS (ESI): m/z =[M+H]+= 573,5, 617,5 (100%), 661,6; [M+Ac]- = 631,4, 675,4 (100%), 719,5. 1H ЯМР (600 МГц, CDCl3): δ=7,33 (д, J=8,3 Гц, 2H), 7,23 (д, J=8,3 Гц, 2H), 4,52 (с, 2H), 3,72-3,58 (м, 33H), 2,54 (уш с, 1H), 1,72 (с, 2H), 1,34 (с, 6H), 0,70 (с, 9H). 13C ЯМР (150 МГц, CDCl3): δ=149,7, 135,1, 127,4 (2C), 126,2 (2C), 73,2, 72,6, 70,7 (м), 70,5, 69,4, 61,9, 57,0, 38,6, 32,5, 31,9 (3C), 31,6 (2C).
Альтернативно, эту стадию проводили следующим образом:
PEG400 (475 г, 1,19 моля) растворяли в TBME (метил-трет-бутиловый эфир) (1,0 л) при температуре окружающей среды. В течение 20 мин порциями добавляли tBuOK (43,2 г, 385 ммоля) и смесь перемешивали 90 мин. Одновременно промежуточный бензилбромид XI (84 г, 297 ммоля) суспендировали в TBME (250 мл) и раствор добавляли к раствору депротонированного PEG400 при температуре окружающей среды. Реакционную смесь перемешивали 3 ч при температуре окружающей среды. К реакционной смеси добавляли лед (200 г) и HCl (1M, 400 мл). Раствор переносили в делительную воронку и EtOAc (1 л) и добавляли воду (500 мл) и энергично экстрагировали. После разделения фаз органическую фазу последовательно промывали смесью вода/рассол (1:1, 3×500 мл) и в заключение концентрировали и получали 165 г неочищенного оранжевого маслообразного остатка. Этот остаток сверху помещали в колонку с SiO2 и элюировали с помощью CH2Cl2/MeOH (от 0 до 10%). Концентрирование чистых фракций давало 151,7 г (выход: 84,9%) прозрачного светло-коричневого масла. Rf=0,37-0,22 (CH2Cl2/MeOH 20:1). MS (ESI): m/z =[M+H]+= 573,5, 617,5 (100%), 661,6; [M+Ac]- = 631,4, 675,4 (100%), 719,5. 1H ЯМР (600 МГц, CDCl3): δ=7,33 (д, J=8,3 Гц, 2H), 7,23 (д, J=8,3 Гц, 2H), 4,52 (с, 2H), 3,72-3,58 (м, 33H), 2,54 (уш с, 1H), 1,72 (с, 2H), 1,34 (с, 6H), 0,70 (с, 9H). 13C ЯМР (150 МГц, CDCl3): δ=149,7, 135,1, 127,4 (2C), 126,2 (2C), 73,2, 72,6, 70,7 (м), 70,5, 69,4, 61,9, 57,0, 38,6, 32,5, 31,9 (3C), 31,6 (2C). Предполагается, что использование TBME (метил-трет-бутиловый эфир) в качестве растворителя, средняя продолжительность реакции, равная 3 ч (которая предположительно сводит к минимуму образование побочных продуктов реакции), получение в более крупном масштабе и чистота исходного вещества (т. е. промежуточного бензилбромида XI) все способствуют высокому выходу, наблюдающемуся на этой стадии реакции.
Пример 6: Инактивация PRV с использованием полиэтоксилата 4-трет-октилбензилового спирта в жидкостях, содержащих внутривенный иммуноглобулин
Материалы и методики
Эти эксперименты проводили, как описано выше в примере 1. Однако S/D обработку смеси S/D, содержащей полиэтоксилат 4-трет-октилбензилового спирта, полученной в примере 5a исследовали и сопоставляли со смесью S/D, содержащей Triton X-100. Композицию смеси S/D, содержащей Triton X-100, получали, как описано выше в примере 1. Композицию смеси S/D, содержащей полиэтоксилат 4-трет-октилбензилового спирта, получали следующим образом.
Компоненты S/D полисорбат 80 (PS80, Crillet 4 HP, Tween 80) и три-н-бутилфосфат (TnBP) объединяли с моющим средством полиэтоксилатом 4-трет-октилбензилового спирта, полученным в примере 5a, в следующих отношениях (см. таблицу 10):
Таблица 10: Количество компонентов смеси полиэтоксилата 4-трет-октилбензилового спирта (аббревиатура "4-TOBAPE") со смесью реагента S/D
Смесь перемешивали в течение не менее 15 мин. Смесь реагента S/D хранили при комнатной температуре для использования в течение не более 1 года. До использования смесь реагента S/D повторно перемешивали в течение не менее 15 мин для обеспечения гомогенности.
Смеси соответствующего реагента S/D добавляли к содержащей IVIG жидкости и получали конечную концентрацию, равную 0,05% мас./мас., соответствующего моющего средства на основе простого эфира полиоксиэтилена.
Исследовали только инактивацию вируса PRV.
Результаты
Когда S/D обработку содержащей IVIG жидкости проводили с использованием смеси полиэтоксилата 4-трет-октилбензилового спирта, PS80 и TnBP, PRV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным примерно 4, не более, чем за 1-2 мин (фиг. 9A). Аналогичные результаты получали в повторном эксперименте (фиг. 9B).
Эти эксперименты показали, что замена Triton X-100 полиэтоксилатом 4-трет-октилбензилового спирта для S/D обработки жидкостей, содержащих биофармацевтическое лекарственное средство, приводит к эффективной инактивации вирусов с липидной оболочкой, даже при низких концентрациях моющего средства.
Пример 7: Инактивация X-MuLV с использованием полиэтоксилата 4-трет-октилбензилового спирта в жидкостях, содержащих сывороточный альбумин человека
Материалы и методики
Эксперименты проводили, как описано выше в примере 3. Однако S/D обработку смеси S/D, содержащей полиэтоксилат 4-трет-октилбензилового спирта исследовали и сопоставляли со смесью S/D, содержащей Triton X-100. Композицию смеси S/D, содержащей Triton X-100, получали, как описано выше в примере 3. Композицию смеси S/D, содержащей полиэтоксилат 4-трет-октилбензилового спирта, получали следующим образом.
Компоненты S/D полисорбат 80 (PS80, Crillet 4 HP, Tween 80) и три-н-бутилфосфат (TnBP) объединяли с моющим средством полиэтоксилатом 4-трет-октилбензилового спирта в следующих отношениях (см. таблицу 11):
Таблица 11: Количество компонентов смеси полиэтоксилата 4-трет-октилбензилового спирта (аббревиатура "4-TOBAPE") со смесью реагента S/D
Смесь перемешивали в течение не менее 15 мин. Смесь реагента S/D хранили при комнатной температуре для использования в течение не более 1 года. До использования смесь реагента S/D повторно перемешивали в течение не менее 15 мин для обеспечения гомогенности.
Смеси соответствующего реагента S/D добавляли к содержащей HSA жидкости и получали конечную концентрацию, равную 0,1% соответствующего моющего средства на основе простого эфира полиоксиэтилена.
Исследовали только инактивацию вируса X-MuLV.
Результаты
Когда S/D обработку содержащей HSA жидкости проводили при 1°C ± 1°C с использованием смеси полиэтоксилата 4-трет-октилбензилового спирта, PS80 и TnBP, X-MuLV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным более 2, не более, чем за 10 мин (фиг. 10A). Аналогичные результаты получали в повторном эксперименте (фиг. 10B).
Дополнительные эксперименты проводили точно так, как описано в разделе Материалы и методики этого примера, только S/D обработку содержащей HSA жидкости проводили при 19°C ± 1°C вместо 1°C ± 1°C. Когда S/D обработку содержащей HSA жидкости проводили при 19°C ± 1°C с использованием смеси полиэтоксилата 4-трет-октилбензилового спирта, PS80 и TnBP, X-MuLV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным более 2, не более, чем за 10 мин (фиг. 11A), и с RF, равным примерно 4, не более, чем за 30 мин. Аналогичные результаты получали в повторном эксперименте (фиг. 11B).
Эти эксперименты показали, что замена Triton X-100 полиэтоксилатом 4-трет-октилбензилового спирта для S/D обработки жидкостей, содержащих биофармацевтическое лекарственное средство, приводит к эффективной инактивации вирусов с липидной оболочкой при комнатной температуре или температуре, близкой к комнатной температуре, и при температурах, равных лишь 1°C ± 1°C, даже при низких концентрациях моющих средств.
Пример 8: Инактивация BVDV с использованием полиэтоксилата 4-трет-октилбензилового спирта, Triton X-100 reduced или Brij C10 в буфере, содержащем HSA
Применимость полиэтоксилата 4-трет-октилбензилового спирта, Triton X-100 reduced или Brij C10 для обработки одним моющим средством для инактивации вируса с липидной оболочкой, вируса диареи крупного рогатого скота (BVDV) в буфере, содержащем сывороточный альбумин человека (HSA) в качестве модельного белка, для терапевтических антител исследовали и сопоставляли с Triton X-100. Для этого соответствующие моющего средства в низких концентрациях добавляли к буферу, содержащему HSA, и затем к смеси добавляли вирус. После инкубации в течение разных периодов времени определяли оставшуюся инфекционность вирусов. Соответствующие моющие средства использовали при низких концентрациях, чтобы было можно исследовать кинетику инактивации вирусов, т. е. эффективности инактивации вирусов (выраженной с помощью RF) в зависимости от времени. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, моющие средства, предлагаемые в настоящем изобретении, можно использовать при значительно более высоких концентрациях, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый RF.
Материалы
Буфер, содержащий HSA
Буфер, содержащий сывороточный альбумин человека (HSA), использовали в качестве модели для терапевтических антител.
Вирусы
Таблица 12: Исходные растворы, использованные в экспериментах.
Исходные растворы вирусов исследовали до использования. Это исследование включало определение титра вируса посредством не менее 10 независимых титрований и спецификации приемлемости диапазона титра вируса для применения в качестве положительного контроля, определение содержания белка в исходном растворе вирусов, исследование с помощью PCR идентичности вирусов и загрязнения другими вирусами и микоплазмой и исследования агрегации вирусов в фильтрах, не пропускающих крупные вирусные агрегаты. Использовали только исходные растворы вирусов, удовлетворяющие требованиям исследований с помощью PCR идентичности/загрязнения при отсутствии значительной агрегации (т. е. разность титров инфекционности исходного раствора вирусов и фильтрованного исходного раствора была меньше 1,0 log).
Моющие средства
До использования соответствующее моющее средство или его разведение 1:10 (т. е. 1 г ± 2% соответствующего моющего средства с добавлением 9 г ± 2% Aqua dest.) перемешивали в течение не менее 15 мин для обеспечения гомогенности.
Методики
Способность к инактивации вирусов и эффективность обработки одним моющим средством исследовали при условиях, неблагоприятных для инактивации вирусов, т. е. при коротких периодах инкубации и относительно низких температурах. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, можно использовать более продолжительные периоды инкубации и более высокие температуры, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV. Кроме того, как уже отмечено выше, использовали низкие концентрации соответствующего моющего средства. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, более высокие концентрации соответствующего моющее средство можно использовать, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV.
Поскольку концентрация белка не оказывает значительное влияние на инактивацию вирусов (см. также Dichtelmüller et al., 2009), стабильность содержания белка не исследовали.
Все остальные стадии проводили в камере класса II биологической безопасности. Исходный материал инкубировали при перемешивании при температуре, равной +14°C ± 1°C, с использованием для инкубации сосуда с двойными стенками, соединенного с криостатом.
Буфер, содержащий HSA, переносили в сосуд с винтовой крышкой, для которого определяли массу тары. Массу буфера, содержащего HSA (в закрытом сосуде)определяли для расчета количества добавляемого одного моющего средства. Необходимое количество моющего средства ([мг]) или соответствующее количество его разведения 1:10 добавляли в пересчете на 1 г буфера, содержащего HSA, и получали следующие конечные концентрации соответствующего моющего средства: 0,1% ± 0,01% (мас./мас.) или 0,03% ± 0,01% (мас./мас.). Моющее средство при перемешивании добавляли в течение 1 мин с помощью шприца и фактическое добавленное количество определяли путем повторного взвешивания шприца. Буфер, содержащий HSA, дополнительно перемешивали после завершения добавления моющего средства в течение не менее 10 мин. Буфер, содержащий HSA, смешанный с моющим средством, фильтровали через мембранный шприцевый фильтр с отверстиями размером 0,2 мкм Supor (или эквивалентный) и фильтрат собирали при заданной температуре, равной 14°C ± 1°C, с использованием сосуда с двойными стенками, соединенного с криостатом. Сосуд для фильтрата охлаждали. В случае забивания фильтра для продолжения фильтрования буфера, содержащего HSA, использовали свежие фильтры. После фильтрования измеряли температуру (целевое значение: 14°C ± 1°C) и объем.
Температуру фильтрованного буфера, содержащего HSA с соответствующим моющим средством при перемешивании устанавливали равной 14°C ± 1°C с использованием сосуда с двойными стенками, соединенного с криостатом. Этот диапазон температуры при перемешивании поддерживали до завершения инкубации буфера, содержащего HSA, с моющим средством и непрерывно регистрировали. К заданному объему буфера, содержащего HSA с моющим средством, добавляли вирус в отношении 1:31, например, к 48 мл буфера, содержащего HSA, добавляли 1,6 мл исходного раствора вируса. Буфер с добавкой, содержащий HSA, дополнительно инкубировали при непрерывном перемешивании при 14°C ± 1°C в течение 59 ± 1 мин. Во время инкубации образцы для титрования вируса отбирали через 1-2 мин, 5 ± 1 мин, 29 ± 1 мин и 59 ± 1 мин. Для предупреждения дополнительной инактивации вируса моющим средством после отбора образца, образцы сразу разводили в отношении 1:20 соответствующей холодной (от +2°C до +8°C) клеточной культуральной средой (т. е. 1 объем образца и 19 объемов клеточной культуральной среды).
Контроли добавления и удерживания проводили в тот же день: Буфер, содержащий HSA, фильтровали, как указано выше, но без предварительного добавления моющего средства. Затем температуру фильтрата при перемешивании устанавливали равной 14°C ± 1°C, как указано выше. К буферу, содержащему HSA, добавляли вирус в отношении 1:31 и дополнительно инкубировали при 14°C ± 1°C в течение 59 ± 1 мин, как указано выше. Не позже чем через 1-2 мин после добавления отбирали образцы для титрования вируса (контроль добавления). После инкубации в течение 59 ± 1 мин отбирали образец для титрования вируса (т. е. образец контроля удерживания) (HC).
Титрование образцов и расчет степени удаления вируса проводили, как описано выше в примере 1.
Результаты
Если обработку одним моющим средством буфера, содержащего HSA, проводили при 14°C ± 1°C с использованием 0,1% ± 0,01% Triton X-100, Brij C10, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced, BVDV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным примерно 5, не более, чем за 5 мин (фиг. 12A). Если обработку одним моющим средством буфера, содержащего HSA, проводили при 14°C ± 1°C с использованием 0,03% ± 0,01% Triton X-100, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced, BVDV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным более 3, не более, чем за 5 мин, и равным более 4, не более, чем за 29 мин (фиг. 12B).
Эти эксперименты показали, что обработка одним моющим средством жидкостей, содержащих биофармацевтическое лекарственное средство, с использованием Triton X-100, Brij C10, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced приводит к эффективной инактивации вирусов с липидной оболочкой, даже при низких концентрациях моющих средств.
Пример 9: Инактивация BVDV с использованием полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced в содержащей IVIG жидкости
Применимость полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced для обработки одним моющим средством для инактивации вирусов с липидной оболочкой, вируса диареи крупного рогатого скота (BVDV) в жидкостях, содержащих внутривенный иммуноглобулин (IVIG) исследовали и сопоставляли с Triton X-100. Для этого вирус добавляли к жидкостям, содержащим IVIG. Содержащие вирус жидкости, включающие IVIG, после этого инкубировали при низких концентрациях Triton X-100 reduced, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100, в течение разных периодов времени и определяли оставшуюся инфекционность вирусов. Triton X-100 reduced, полиэтоксилат 4-трет-октилбензилового спирта и Triton X-100 использовали при низких концентрациях для исследования кинетики инактивации вирусов, т. е. эффективности инактивации вирусов (выраженной с помощью RF) в зависимости от времени. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, моющие средства, предлагаемые в настоящем изобретении, включая Triton X-100 reduced или полиэтоксилат 4-трет-октилбензилового спирта можно использовать при значительно более высоких концентрациях, что увеличит скорость инактивации вирусов и также предположительно увеличит обеспечиваемый LRV.
Материалы
Жидкость, содержащая IVIG
Жидкость, содержащую IVIG (содержащую IVIG жидкость) замораживали твердым диоксидом углерода и хранили при ≤ -60°C для использования в течение не более одного года после даты сбора.
Вирусы
Таблица 13: Исходные растворы вирусов, использованные в экспериментах.
Исходные растворы вирусов исследовали до использования. Это исследование включало определение титра вируса посредством не менее 10 независимых титрований и спецификации приемлемости диапазона титра вируса для применения в качестве положительного контроля, определение содержания белка в исходном растворе вирусов, исследование с помощью PCR идентичности вирусов и загрязнения другими вирусами и микоплазмой и исследования агрегации вирусов в фильтрах, не пропускающих крупные вирусные агрегаты. Использовали только исходные растворы вирусов, удовлетворяющие требованиям исследований с помощью PCR идентичности/загрязнения при отсутствии значительной агрегации (т.е. разность титров инфекционности исходного раствора вирусов и фильтрованного исходного раствора была меньше 1,0 log).
Моющие средства
До использования соответствующее моющее средство или его разведение 1:10 (т.е. 1 г ± 2% соответствующего моющего средства с добавлением 9 г ± 2% Aqua dest.) перемешивали в течение не менее 15 мин для обеспечения гомогенности.
Методики
Способность к инактивации вирусов и эффективность обработки одним моющим средством исследовали при условиях, неблагоприятных для инактивации вирусов, т.е. при коротких периодах инкубации и относительно низких температурах. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, можно использовать более продолжительные периоды инкубации и более высокие температуры, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV. Кроме того, как уже отмечено выше, использовали низкие концентрации соответствующего моющее средство. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, более высокие концентрации соответствующего моющие средства можно использовать, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV.
Поскольку концентрация белка не оказывает значительное влияние на инактивацию вирусов (см. также Dichtelmüller et al., 2009), стабильность содержания белка не исследовали.
Содержащую IVIG жидкость размораживали и все остальные стадии проводили в камере класса II биологической безопасности. Содержащую IVIG жидкость инкубировали при перемешивании при температуре, равной +17°C ± 1°C с использованием для инкубации сосуда с двойными стенками, соединенного с криостатом. Содержащую IVIG жидкость затем фильтровали через объемный фильтр с отверстиями размером 0,2 мкм с эффективной площадью фильтра, равной 25 cм2 (Cuno VR06 или эквивалентный), соединенный с изготовленным из нержавеющей стали держателем фильтра Sartorius (SM16249) с использованием сжатого азота при целевом давлении, равном 0,9 бар (предельное значение: 0,5 бар - 1,5 бар). Для кондиционирования на материал фильтра предварительно наносили покрытие с помощью 55 л/м2 суспензии Hyflo Supercel (5,0 г ± 0,05 г Hyflo Supercel на 1 л; проводимость устанавливали равной 3,5 мСм/см (установленный диапазон: 2,5-6,0 мСм/см) с использованием 3 M NaCl) (при давлении ≤ 0,5 бар) до фильтрования жидкости. Во время фильтрования держатель фильтра охлаждали и фильтрат собирали при заданной температуре, равной +17°C ± 1°C с использованием сосуда с двойными стенками, соединенного с криостатом. Сосуд для фильтрата охлаждали. В случае забивания фильтра для продолжения фильтрования оставшейся жидкости использовали свежие предварительно кондиционированные фильтры. После фильтрования измеряли объем.
После измерения объема содержащей IVIG жидкости при перемешивании с помощью холодного (от +2°C до +8°C) буфера для разведения (раствор NaCl с целевой проводимостью, равной 3,5 мСм/см (диапазон: 2,5 мСм/см - 6,0 мСм/см)) для нее устанавливали рассчитанную целевую оптическую плотность, равную 28,9 AU280-320/см (диапазон: 14,5-72,3 AU280-320/см). С учетом последующего добавления вируса в отношении 1:31 это дает рассчитанную целевую оптическую плотность, равную 28 AU280-320/см (диапазон: 14-70 AU280-320/см), для инкубации с моющим средством после фильтрования.
Температуру фильтрованной содержащей IVIG жидкости с отрегулированным содержанием белка при перемешивании повторно устанавливали равной +17°C ± 1°C с использованием сосуда с двойными стенками, соединенного с криостатом. Этот диапазон температуры при перемешивании поддерживали до завершения инкубации фильтрованной содержащей IVIG жидкости с моющим средством и непрерывно регистрировали. К заданному объему фильтрованной содержащей IVIG жидкости, перенесенному в сосуд с винтовой крышкой, для которого определяли массу тары, добавляли вирус при отношении 1:31, например, к 30 мл содержащей IVIG жидкости добавляли 1 мл исходного раствора вируса. Содержащую IVIG жидкость с добавкой дополнительно инкубировали при непрерывном перемешивании при 17°C ± 1°C. Не позже чем через 1-2 мин после добавления отбирали образцы для титрования вируса (контроль добавления, SC, и контроль удерживания, HC).
После размораживания контроль удерживания (HC) выдерживали при такой же температуре, т.е. при +17°C ± 1°C, как содержащую IVIG жидкость с добавкой после добавления моющего средства, т.е. его хранили при таком же цикле охлаждения, как сосуд с содержащей IVIG жидкостью, до завершения обработки моющим средством. Температуру охлаждающей жидкости определяли до введения образца контроля удерживания и повторно незадолго до удаления контроля удерживания для титрования после обработки моющим средством.
Массу содержащей IVIG жидкости с добавкой определяли для расчета количества добавляемого моющего средства. Температуру взвешенного материала, при необходимости при перемешивании, повторно устанавливали равной +17°C ± 1°C. Необходимое количество моющего средства ([мг]) или соответствующее количество его разведения 1:10 добавляли в пересчете на 1 г содержащей IVIG жидкости и получали следующие конечные концентрации соответствующего моющего средства: 0,1% ± 0,01% (мас./мас.) или 0,03% ± 0,01% (мас./мас.). Моющее средство при перемешивании добавляли в течение 1 мин с помощью шприца и фактическое количество добавленного моющего средства определяли путем повторного взвешивания шприца. Содержащую IVIG жидкость с добавкой дополнительно инкубировали с соответствующим моющим средством при непрерывном перемешивании при +17°C ± 1°C в течение 59 ± 1 мин. Во время инкубации образцы по 1 мл для титрования вируса отбирали через 1-2 мин, 10 ± 1 мин, 30 ± 1 мин и 59 ± 1 мин. Для предупреждения дополнительной инактивации вируса реагентами S/D после отбора образца, образцы сразу разводили в отношении 1:20 холодной (от +2°C до +8°C) клеточной культуральной средой (т.е. 1 объем образца и 19 объемов клеточной культуральной среды).
Титрование образцов и расчет степени удаления вируса проводили, как описано выше в примере 1.
Результаты
Если обработку одним моющим средством содержащей IVIG жидкости проводили при 17°C ± 1°C с использованием 0,1% ± 0,01% Triton X-100, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced, BVDV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным примерно 5, не более, чем за 10 мин (фиг. 13A). Если обработку одним моющим средством содержащей IVIG жидкости проводили при 17°C ± 1°C с использованием 0,03% ± 0,01% Triton X-100, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced, BVDV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным более 3, не более чем за 10 мин, и равным более 4, не более чем за 30 мин (фиг. 13B).
Эти эксперименты подтвердили, что обработка одним моющим средством жидкостей, содержащих биофармацевтическое лекарственное средство, с использованием Triton X-100, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced приводит к эффективной инактивации вирусов с липидной оболочкой, даже при низких концентрациях моющих средств.
Пример 10: Инактивация BVDV с использованием полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced в содержащей FVIII жидкости
Применимость полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced для обработки одним моющим средством для инактивации вирусов с липидной оболочкой, вируса диареи крупного рогатого скота (BVDV) в жидкостях, содержащих полученный из плазмы фактор VIII (pdFVIII) исследовали и сопоставляли с Triton X-100. Для этого вирус добавляли к жидкостям, содержащим pdFVIII. Затем содержащие вирус жидкости, включающие pdFVIII инкубировали при низких концентрациях Triton X-100 reduced, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100, в течение разных периодов времени и определяли оставшуюся инфекционность вирусов. Triton X-100 reduced, полиэтоксилат 4-трет-октилбензилового спирта и Triton X-100 использовали при низких концентрациях для исследования кинетики инактивации вирусов, т.е. эффективности инактивации вирусов (выраженной с помощью RF) в зависимости от времени. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, моющие средства, предлагаемые в настоящем изобретении, включая Triton X-100 reduced или полиэтоксилат 4-трет-октилбензилового спирта можно использовать при значительно более высоких концентрациях, что увеличит скорость инактивации вирусов и также предположительно увеличит обеспечиваемый LRV.
Материалы
Жидкость, содержащая pdFVIII
Жидкость, содержащую pdFVIII (содержащую pdFVIII жидкость) замораживали твердым диоксидом углерода и хранили при ≤ -60°C для использования в течение не более одного года после даты сбора.
Вирусы
Таблица 14: Исходные растворы, использованные в экспериментах.
Исходные растворы вирусов исследовали до использования. Это исследование включало определение титра вируса посредством не менее 10 независимых титрований и спецификации приемлемости диапазона титра вируса для применения в качестве положительного контроля, определение содержания белка в исходном растворе вирусов, исследование с помощью PCR идентичности вирусов и загрязнения другими вирусами и микоплазмой и исследования агрегации вирусов в фильтрах, не пропускающих крупные вирусные агрегаты. Использовали только исходные растворы вирусов, удовлетворяющие требованиям исследований с помощью PCR идентичности/загрязнения при отсутствии значительной агрегации (т.е. разность титров инфекционности исходного раствора вирусов и фильтрованного исходного раствора была меньше 1,0 log).
Моющие средства
До использования соответствующее моющее средство или его разведение 1:10 (т.е. 1 г ± 2% соответствующего моющего средства с добавлением 9 г ± 2% Aqua dest.) перемешивали в течение не менее 15 мин для обеспечения гомогенности.
Методики
Способность к инактивации вирусов и эффективность обработки одним моющим средством исследовали при условиях, неблагоприятных для инактивации вирусов, т.е. при коротких периодах инкубации. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, можно использовать более продолжительные периоды инкубации, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV. Кроме того, как уже отмечено выше, низкие концентрации соответствующего моющие средства использовали. Как должно быть понятно специалисту в данной области техники, в коммерческих производственных процессах, таких как биофармацевтическое производство, более высокие концентрации соответствующих моющих средств можно использовать, что увеличит скорость инактивации вирусов и также может увеличить обеспечиваемый LRV.
Поскольку концентрация белка не оказывает значительное влияние на инактивацию вирусов (см. также Dichtelmüller et al., 2009), стабильность содержания белка не исследовали.
Содержащую pdFVIII жидкость размораживали и все остальные стадии проводили в камере класса II биологической безопасности. Для удаления возможных крупных агрегатов содержащую pdFVIII жидкость фильтровали через мембранный фильтр с отверстиями размером 0,45 мкм (например, Sartorius SartoScale Sartobran или эквивалентный). Содержащую pdFVIII жидкость переносили в сосуд с винтовой крышкой, для которого определяли массу тары и при перемешивании устанавливали целевую температуру, равную +23°C ± 1°C с использованием сосуда с двойными стенками, соединенного с криостатом. К заданному объему содержащей pdFVIII жидкости добавляли вирус в отношении 1:31, например, к 48 мл содержащей pdFVIII жидкости добавляли 1,6 мл исходного раствора вируса. Затем отбирали образец каждого вируса для титрования (контроль добавления, SC) и для контроля удерживания (HC).
Контроль удерживания выдерживали при такой же температуре, т.е. при +23°C ± 1°C, как содержащую pdFVIII жидкость с добавкой после добавления моющего средства, т.е. его хранили при таком же цикле охлаждения, как сосуд с содержащей pdFVIII жидкостью, до завершения обработки моющим средством. Температуру охлаждающей жидкости определяли до введения образца контроля удерживания и повторно незадолго до удаления контроля удерживания для титрования после обработки моющим средством.
Массу содержащей pdFVIII жидкости с добавкой определяли для расчета количества добавляемого моющего средства. Температуру взвешенного материала при перемешивании устанавливали равной +23°C ± 1°C с использованием сосуда с двойными стенками, соединенного с криостатом. Этот диапазон температуры при перемешивании поддерживали до завершения инкубации содержащей pdFVIII жидкости с добавкой и с моющим средством и непрерывно регистрировали. Необходимое количество моющего средства ([мг]) или соответствующее количество его разведения 1:10 добавляли в пересчете на 1 г содержащей pdFVIII жидкости и получали конечную концентрацию моющего средства, равную 0,1% ± 0,01% (мас./мас.). Моющее средство при перемешивании добавляли в течение 1 мин с помощью шприца и фактическое количество добавленного моющего средства определяли путем повторного взвешивания шприца. После завершения добавления моющего средства содержащую pdFVIII жидкость с добавкой дополнительно инкубировали при непрерывном перемешивании при +23°C ± 1°C в течение 59 ± 1 мин. Во время инкубации образцы по 1 мл для титрования вируса отбирали через 1-2 мин, 5 ± 1 мин, 30 ± 1 мин и 59 ± 1 мин. Для предупреждения дополнительной инактивации вируса моющим средством после отбора образца, образцы сразу разводили в отношении 1:20 соответствующей холодной (от +2 до +8°C) клеточной культуральной средой (т.е. 1 объем образца и 19 объемов клеточной культуральной среды).
Титрование образцов и расчет степени удаления вируса проводили, как описано выше в примере 1.
Результаты
Если обработку одним моющим средством содержащей pdFVIII жидкости проводили при 23°C ± 1°C с использованием 0,1% ± 0,01% Triton X-100, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced, BVDV инактивировался с коэффициентом уменьшения содержания вируса (RF), равным примерно 5, не более, чем за 5 мин (фиг. 14).
Эти эксперименты подтвердили, что обработка одним моющим средством жидкостей, содержащих биофармацевтическое лекарственное средство, с использованием Triton X-100, полиэтоксилата 4-трет-октилбензилового спирта или Triton X-100 reduced приводит к эффективной инактивации вирусов с липидной оболочкой, даже при низких концентрациях моющих средств.
Пример 11: Токсикологические исследования
По данным in silico ни для одного из моющих средств, предлагаемых в настоящем изобретении, не обнаружено никаких свидетельств о том, что они активны, как разрушители эндокринной системы.
Промышленное применение:
Методики, способы и продукты, предлагаемые в настоящем изобретении, используются в промышленности, например, для экологически приемлемой инактивации вирусов с липидной оболочкой в промышленных процессах. Например, настоящее изобретение можно использовать в промышленном производстве биофармацевтических лекарственных средств. Таким образом, настоящее изобретение является промышленно применимым.
ЛИТЕРАТУРА
Dichtelmüller et al. (2009): Robustness of solvent/detergent treatment of plasma derivatives: a data collection from Plasma Protein Therapeutics Association member companies. Transfusion 49(9): 1931-1943.
Di Serio et al. (2005): Comparison of Different Reactor Types Used in the Manufacture of Ethoxylated, Propoxylated Products. Ind. Eng. Chem. Res. 44(25): 9482-9489.
ECHA Support document for for identification of 4-(1,1,3,3-tetramethylbutyl)phenol, ethoxylated as substances of very high concern because, due to their degradation to a substance of very high concern (4-(1,1,3,3-tetramethylbutyl)phenol) with endocrine disrupting properties, they cause probable serious effects to the environment which give rise to an equivalent level of concern to those of CMRs and PBTs/vPvBs, adopted on 12 December 2012
Brochure "Global Assessment of the Sate-of-the-Science of Endocrine Disruptors" (WHO/PCS/EDC/02.2), published by the International Programme on Chemical Safety of the World Health Organisation
Simons et al. (1973): Solubilization of the membrane proteins from Semliki Forest virus with Triton X100. J Mol Biol. 80(1):119-133.
US patent no. 1,970,578
Vogel‘s Textbook of Practical Organic Chemistry ((5th Edition, 1989, A.I. Vogel, A.R. Tatchell, B.S. Furnis, A.J. Hannaford, P.W.G. Smith)
Bioorganic & Medicinal Chemistry, 16(9), 4883-4907; 2008: Effects of modifications of the linker in a series of phenylpropanoic acid derivatives: Synthesis, evaluation as PPARα/γ dual agonists, and X-ray crystallographic studies; Casimiro-Garcia, Agustin; Bigge, Christopher F.; Davis, Jo Ann; Padalino, Teresa; Pulaski, James; Ohren, Jeffrey F.; McConnell, Patrick; Kane, Christopher D.; Royer, Lori J.; Stevens, Kimberly A.; Auerbach, Bruce J.; Collard, Wendy T.; McGregor, Christine; Fakhoury, Stephen A.; Schaum, Robert P.; Zhou, Hairong.
DOI:10.1016/j.bmc.2008.03.
Chemistry - A European Journal, 23(60), 15133-15142; 2017: Solid Phase Stepwise Synthesis of Polyethylene Glycols; Khanal, Ashok; Fang, Shiyue.DOI:10.1002/chem.201703004
Journal of Medicinal Chemistry, 48(10), 3586-3604; 2005: Synthesis and Structure-Activity Relationships of Novel Selective Factor Xa Inhibitors with a Tetrahydroisoquinoline Ring; Ueno, Hiroshi; Yokota, Katsuyuki; Hoshi, Jun-Ichi; Yasue, Katsutaka; Hayashi, Mikio; Hase, Yasunori; Uchida, Itsuo; Aisaka, Kazuo; Katoh, Susumu; Cho, Hidetsura. DOI:10.1021/jm058160e
Journal of Nanoparticle Research, 15(11), 2025/1-2025/12, 12 pp.; 2013: Magnetic nanoparticles conjugated to chiral imidazolidinone as recoverable catalyst; Mondini, Sara; Puglisi, Alessandra; Benaglia, Maurizio; Ramella, Daniela; Drago, Carmelo; Ferretti, Anna M.; Ponti, Alessandro. DOI:10.1007/s11051-013-2025-3
Journal of Organic Chemistry, 79(1), 223-229; 2014: A Scalable Procedure for Light-Induced Benzylic Brominations in Continuous Flow; Cantillo, David; de Frutos, Oscar; Rincon, Juan A.; Mateos, Carlos; Kappe, C. Oliver. DOI:10.1021/jo402409k
Journal of Physical Chemistry B, 107(31), 7896-7902; 2003: Anellated hemicyanine dyes with large symmetrical solvatochromism of absorption and fluorescence; Huebener, Gerd; Lambacher, Armin; Fromherz, Peter. DOI:10.1021/jp0345809
PCT Int. Appl., 2005016240, 24 Feb 2005: Preparation of aryl carbamate oligomers for hydrolyzable prodrugs and prodrugs comprising same; Ekwuribe, Nnochiri N.; Odenbaugh, Amy L. WO 2004-US15004, May 6, 2004
Russian Journal of Applied Chemistry, 82(6), 1029-1032; 2009: Relative activity of alkenyl-gem-dichlorocyclopropanes in the reactions of hydrogenation and alkylation; Brusentsova, E. A.; Zlotskii, S. S.; Kutepov, B. I.; Khazipova, A. N. DOI:10.1134/S1070427209060196
Russian Journal of Organic Chemistry, 51(11), 1545-1550; 2015: Alkylation of aromatic compounds with 1-bromoadamantane in the presence of metal complex catalysts; Khusnutdinov, R. I.; Shchadneva, N. A.; Khisamova, L. F.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БЕЗОПАСНЫХ В ВИРУСНОМ ОТНОШЕНИИ БИОЛОГИЧЕСКИХ ТЕКУЧИХ СРЕД | 2005 |
|
RU2411959C2 |
| СПОСОБ ОЧИСТКИ РАСТВОРА АЛЬФА-1-АНТИТРИПСИНА | 2004 |
|
RU2370500C2 |
| СПОСОБЫ ИНАКТИВАЦИИ ВИРУСОВ С ПРИМЕНЕНИЕМ N-МЕТИЛГЛЮКАМИДА И ЕГО ПРОИЗВОДНЫХ | 2018 |
|
RU2783667C2 |
| СПОСОБ ИНКУБАЦИИ ЖИДКОСТЕЙ И ВИРУСНОЙ ИНАКТИВАЦИИ | 2018 |
|
RU2779191C2 |
| ПРЕПАРАТ ИММУНОГЛОБУЛИНА IgG И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2337109C2 |
| СПОСОБЫ ИНАКТИВАЦИИ ВИРУСА С ИСПОЛЬЗОВАНИЕМ ЭКОЛОГИЧНЫХ ДЕТЕРГЕНТОВ | 2014 |
|
RU2707951C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЛИОФИЛИЗИРОВАННОГО ПРЕПАРАТА АКТИВИРОВАННОГО ПРОТРОМБИНОВОГО КОМПЛЕКСА, ОБЛАДАЮЩЕГО ФАКТОР VIII-ШУНТИРУЮЩЕЙ АКТИВНОСТЬЮ | 2017 |
|
RU2648517C1 |
| ПОЛУЧЕНИЕ ИММУНОГЛОБУЛИНОВ С ПОНИЖЕННЫМ СОДЕРЖАНИЕМ ТРОМБОГЕННЫХ АГЕНТОВ И ИММУНОГЛОБУЛИНОВАЯ КОМПОЗИЦИЯ | 2012 |
|
RU2627162C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВИРУСБЕЗОПАСНОГО ПОЛНОГО ПРОТРОМБИНОВОГО КОМПЛЕКСА | 2014 |
|
RU2559576C1 |
| СПОСОБЫ ИНАКТИВАЦИИ ВИРУСА ДЛЯ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АНТИТЕЛ | 2019 |
|
RU2807176C2 |
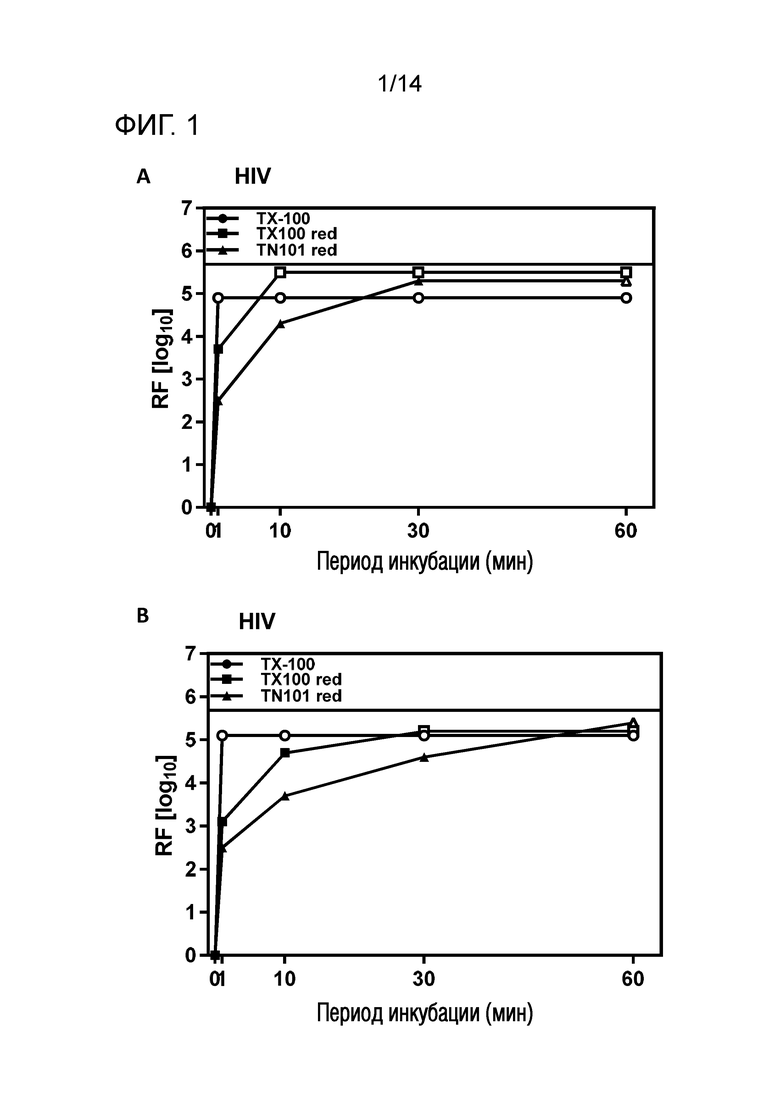
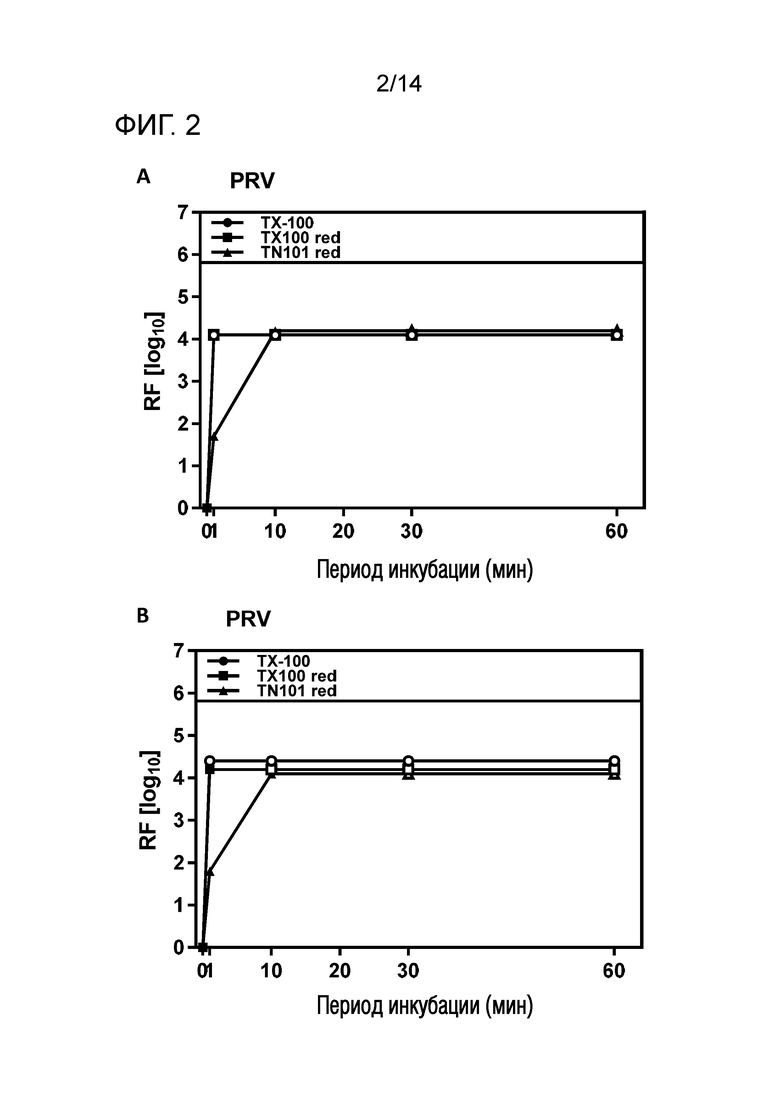
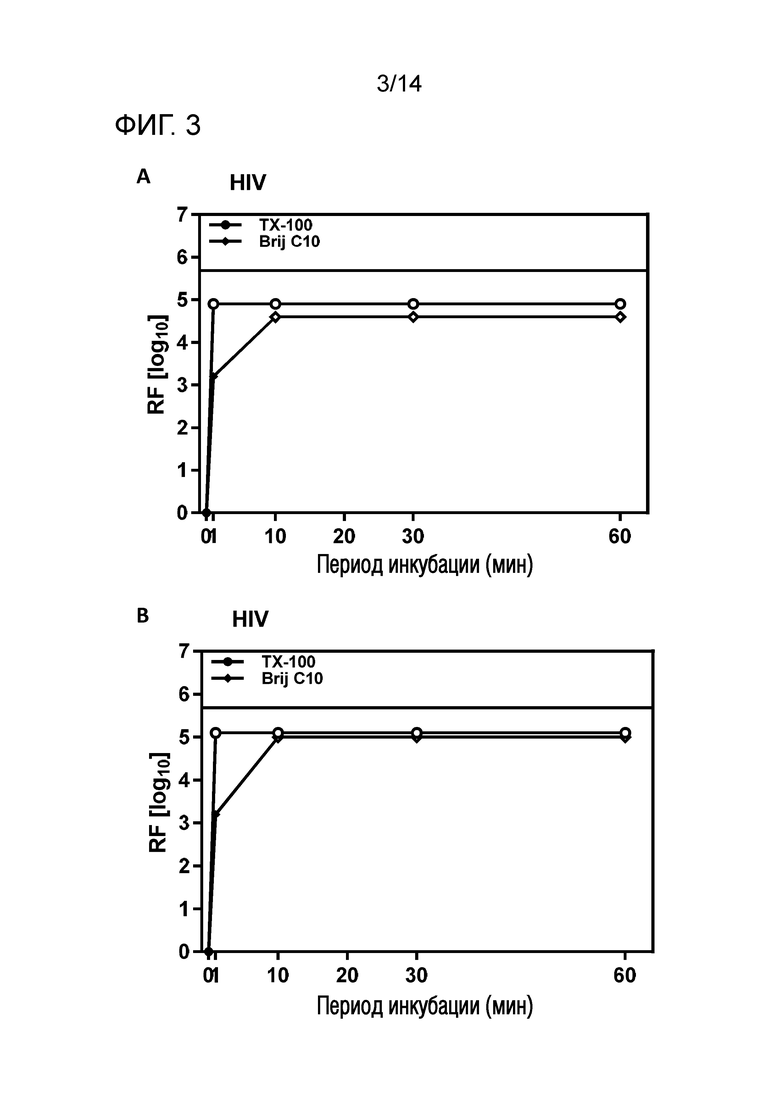
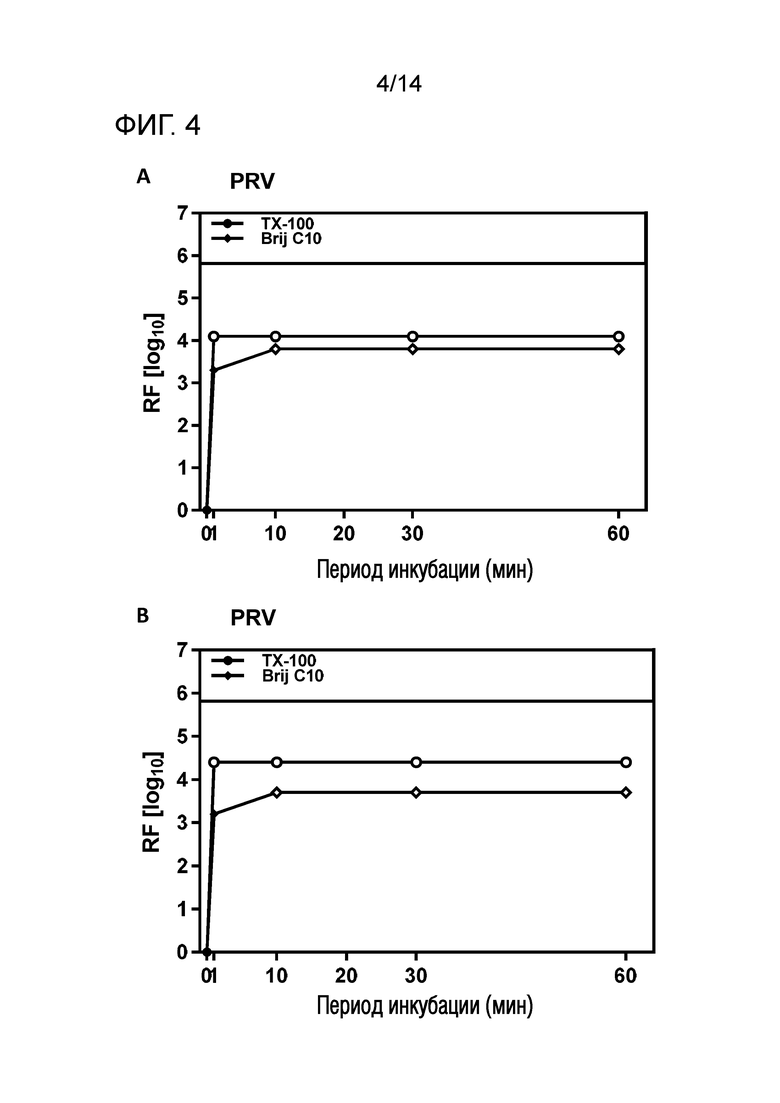
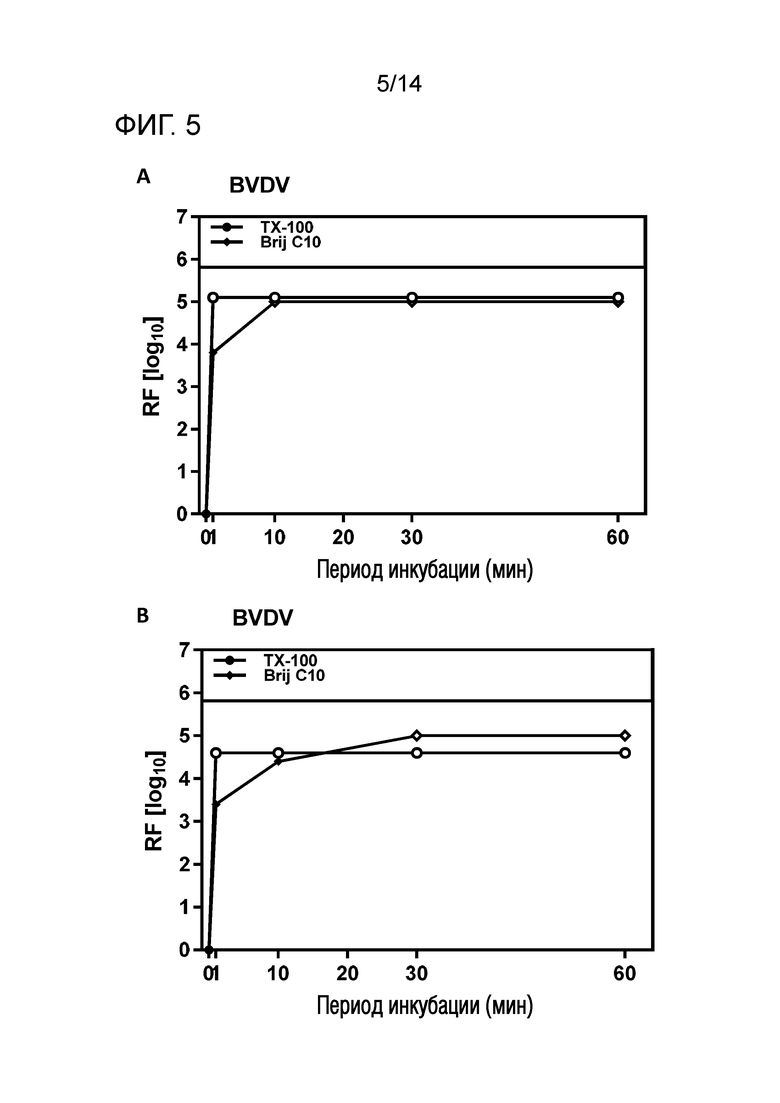
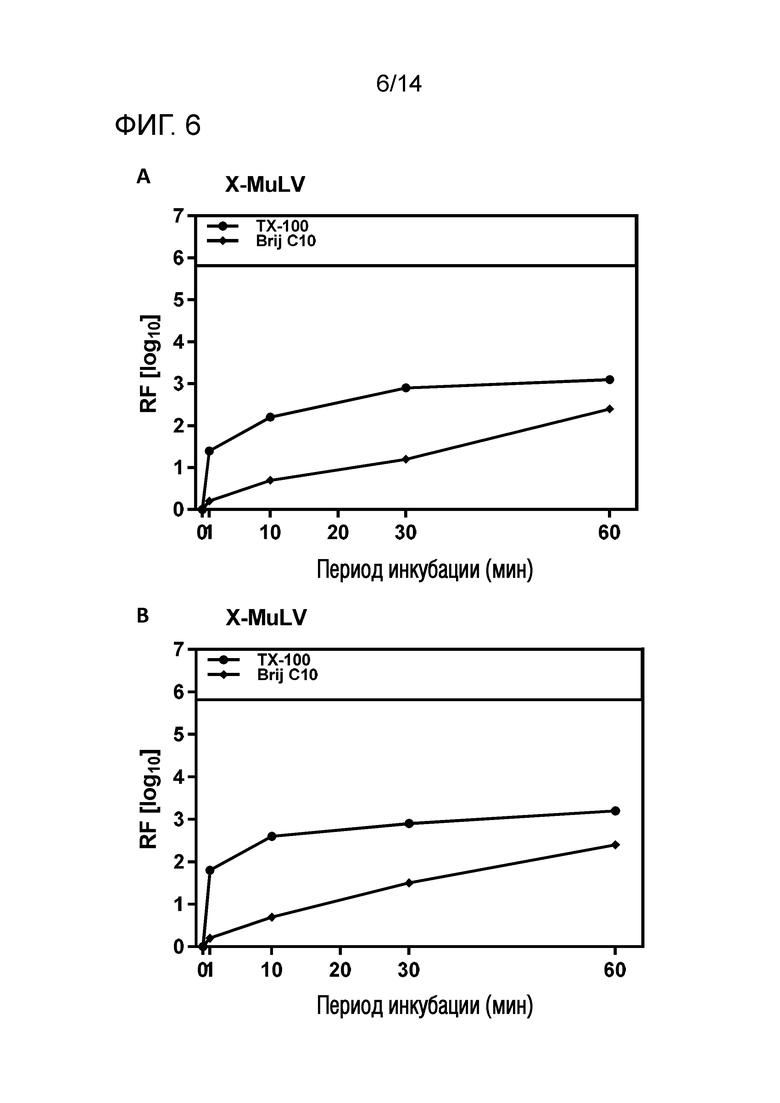
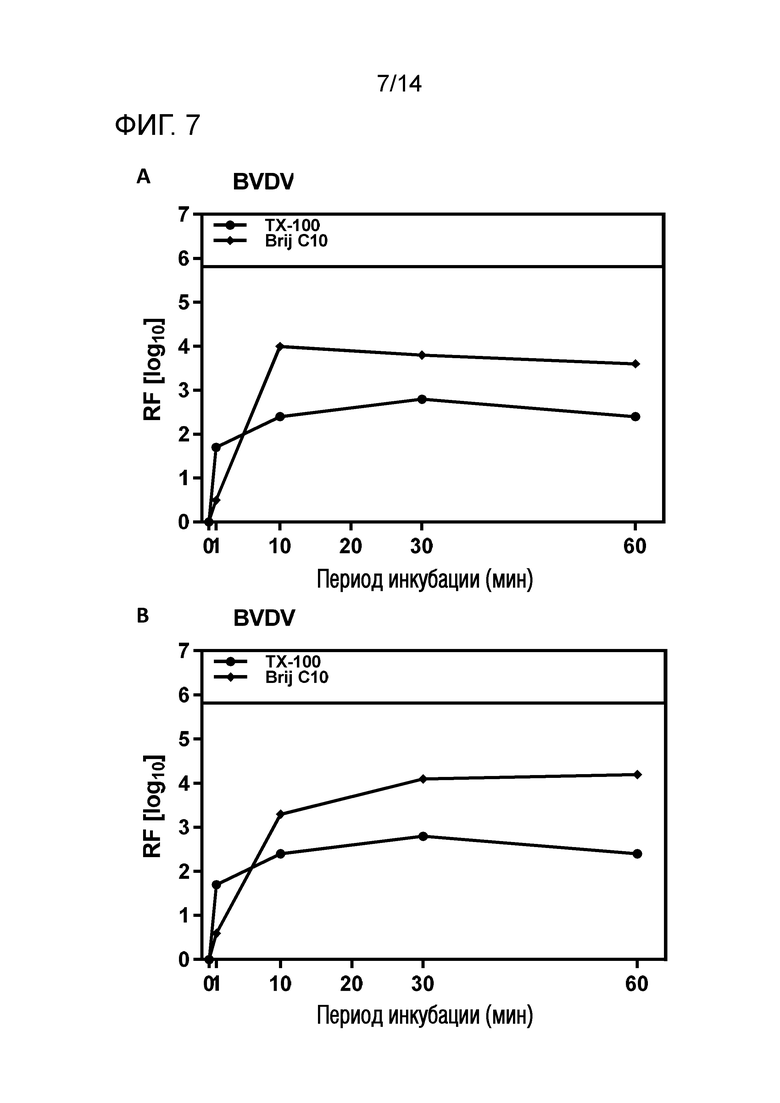
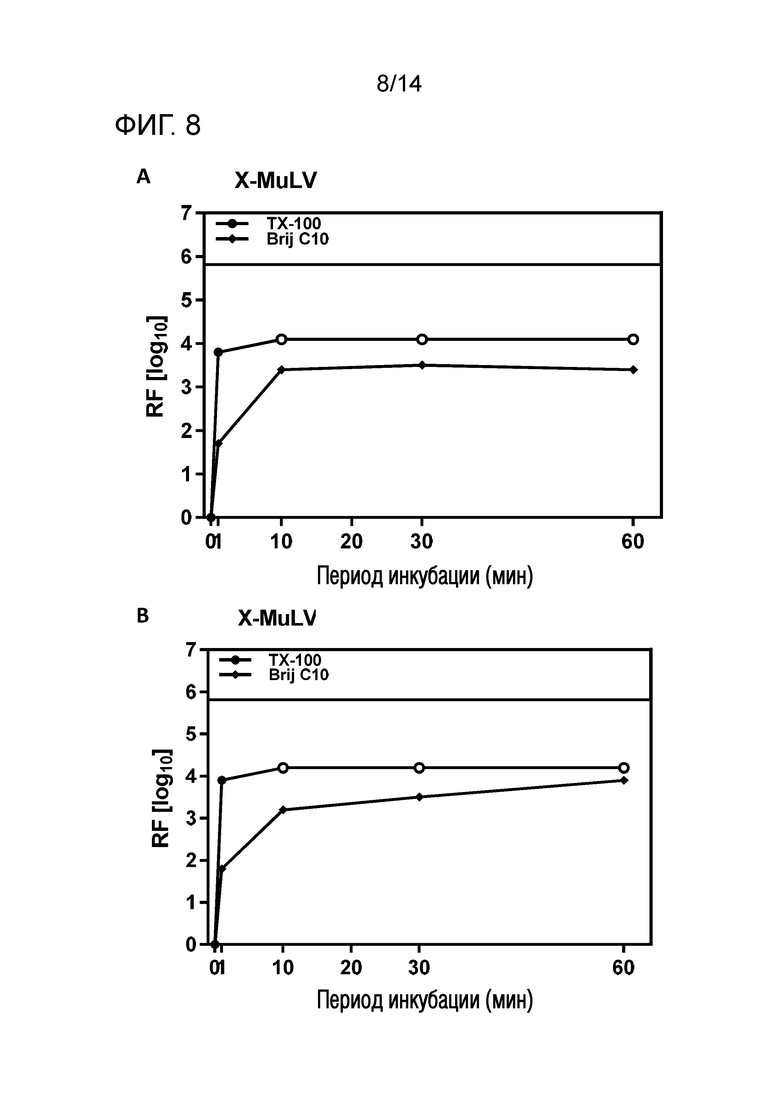
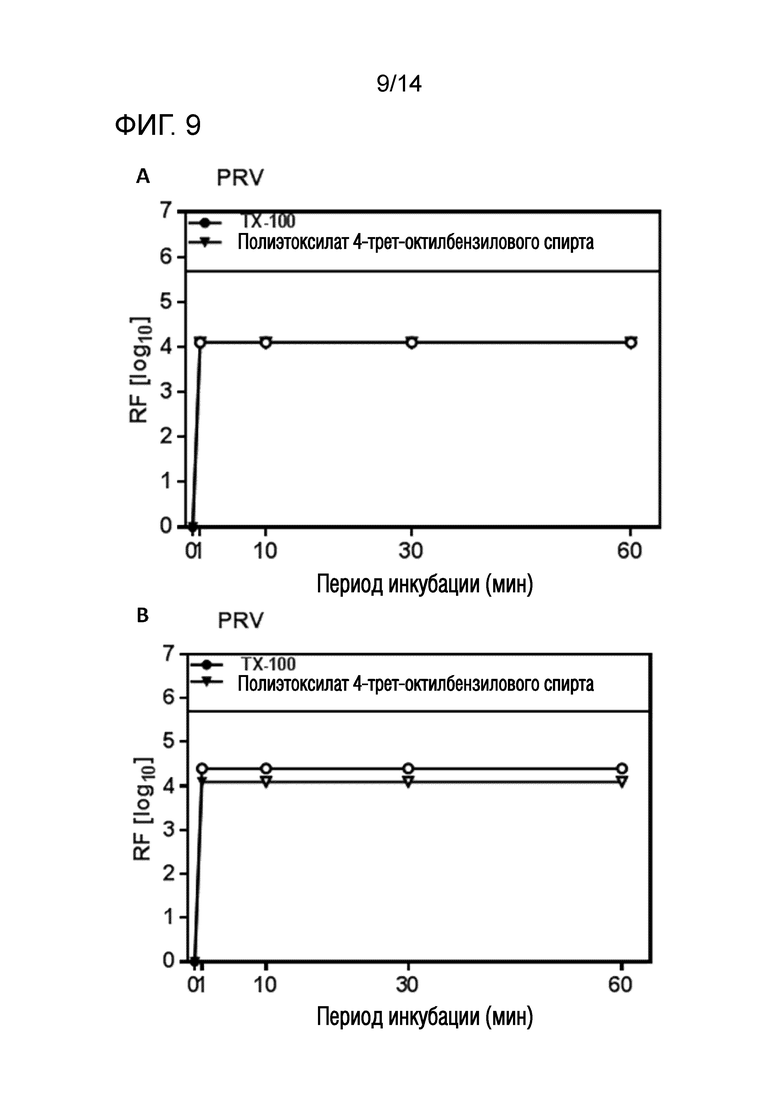
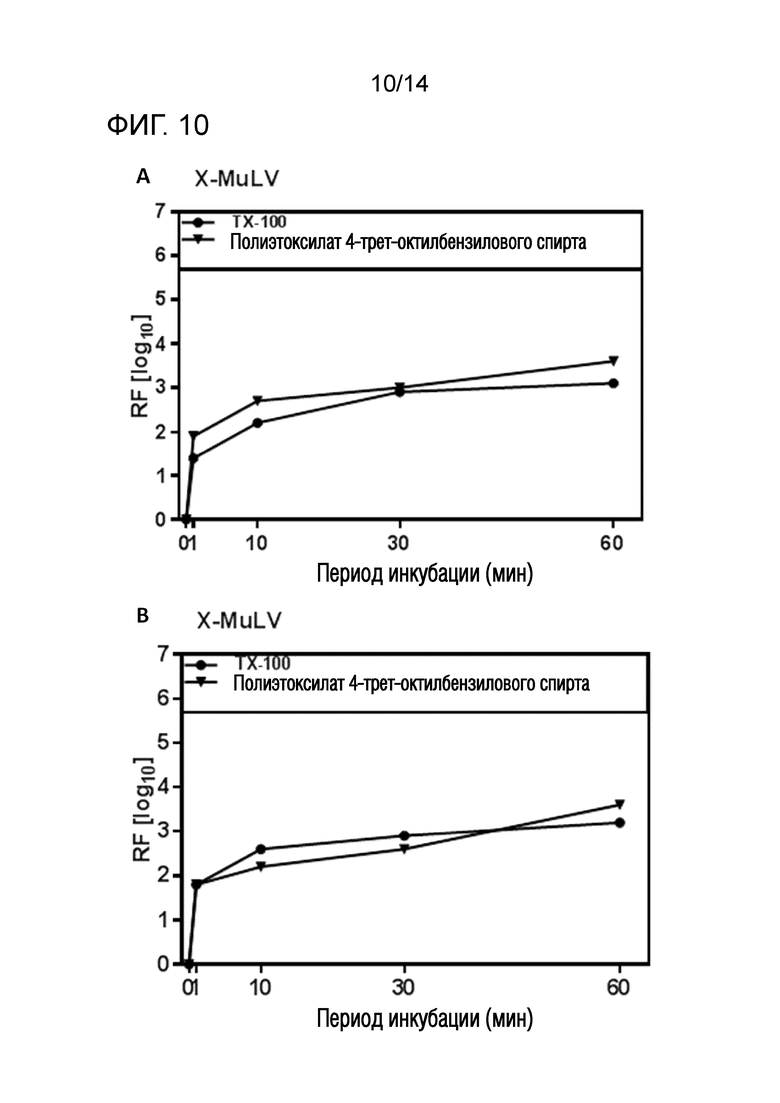
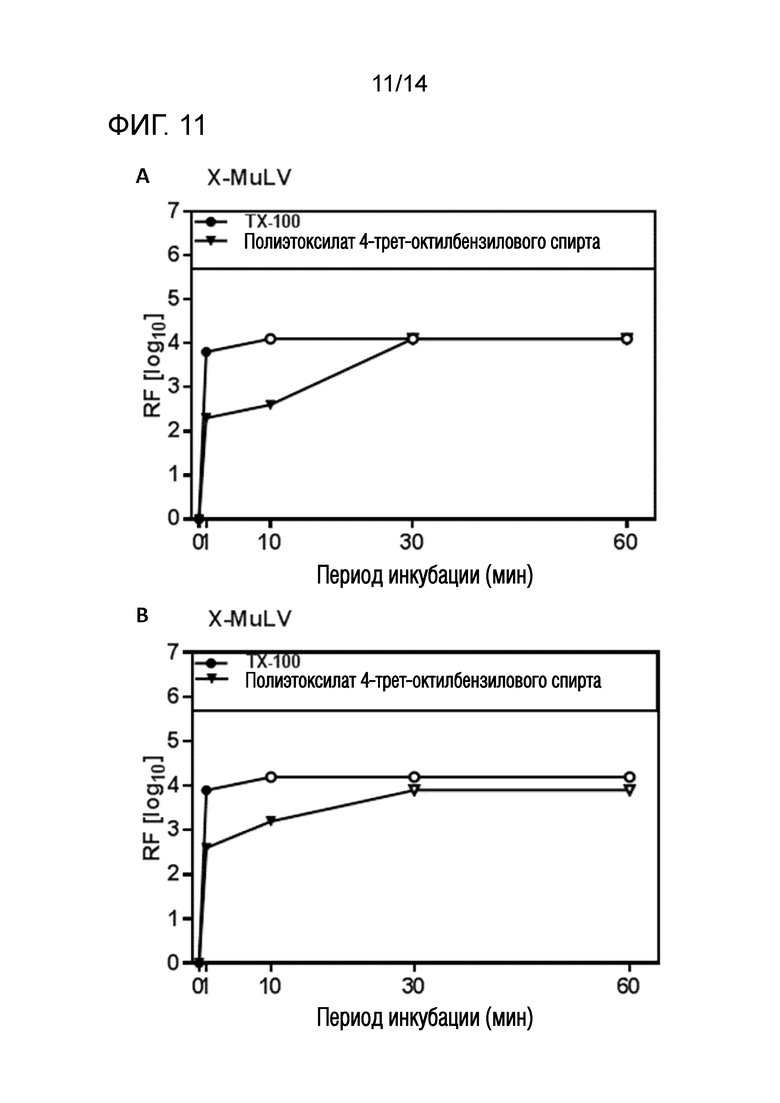
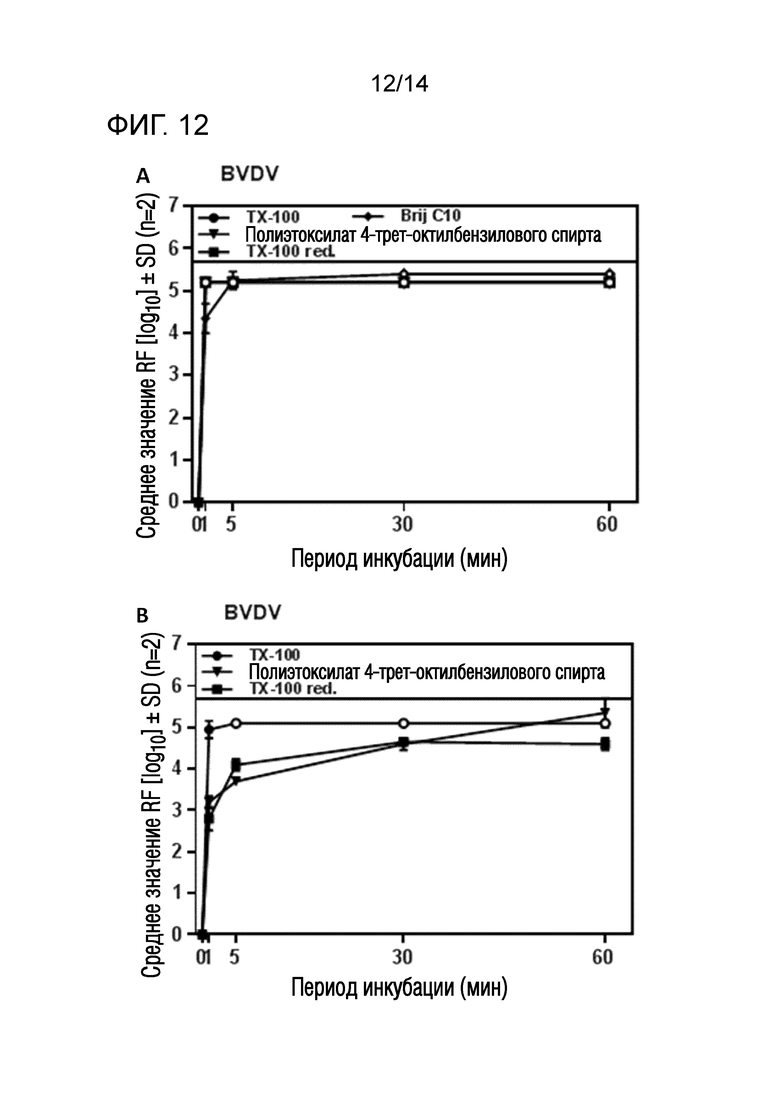
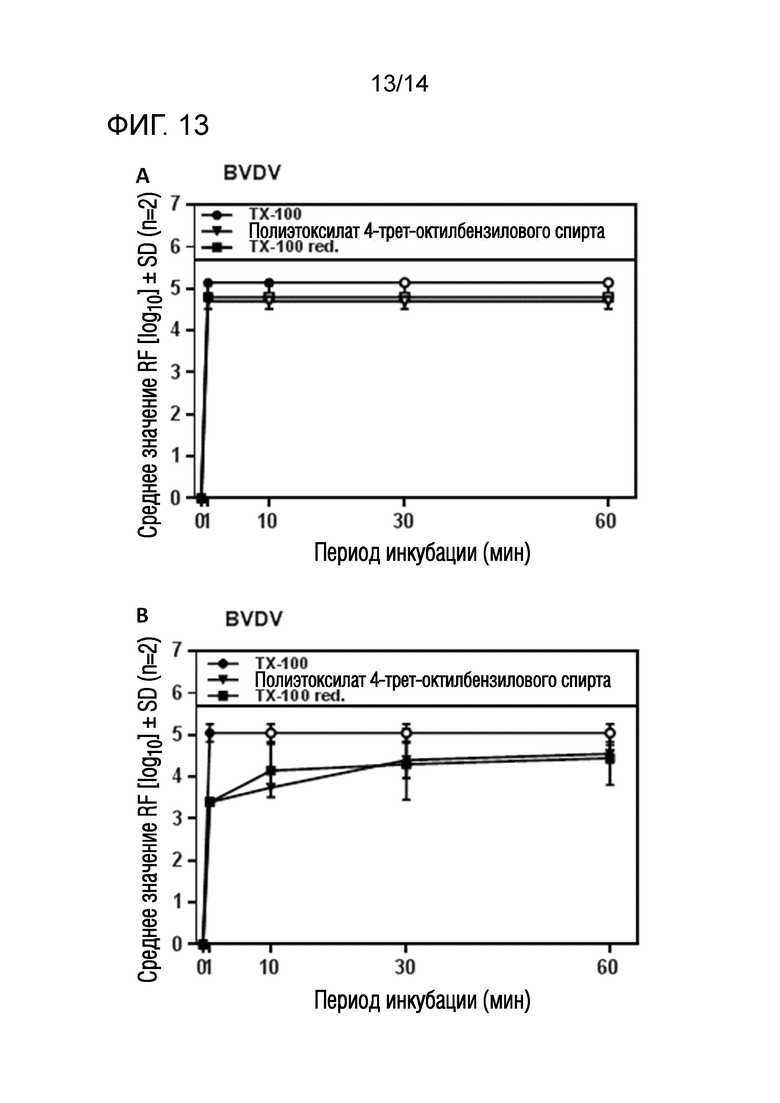
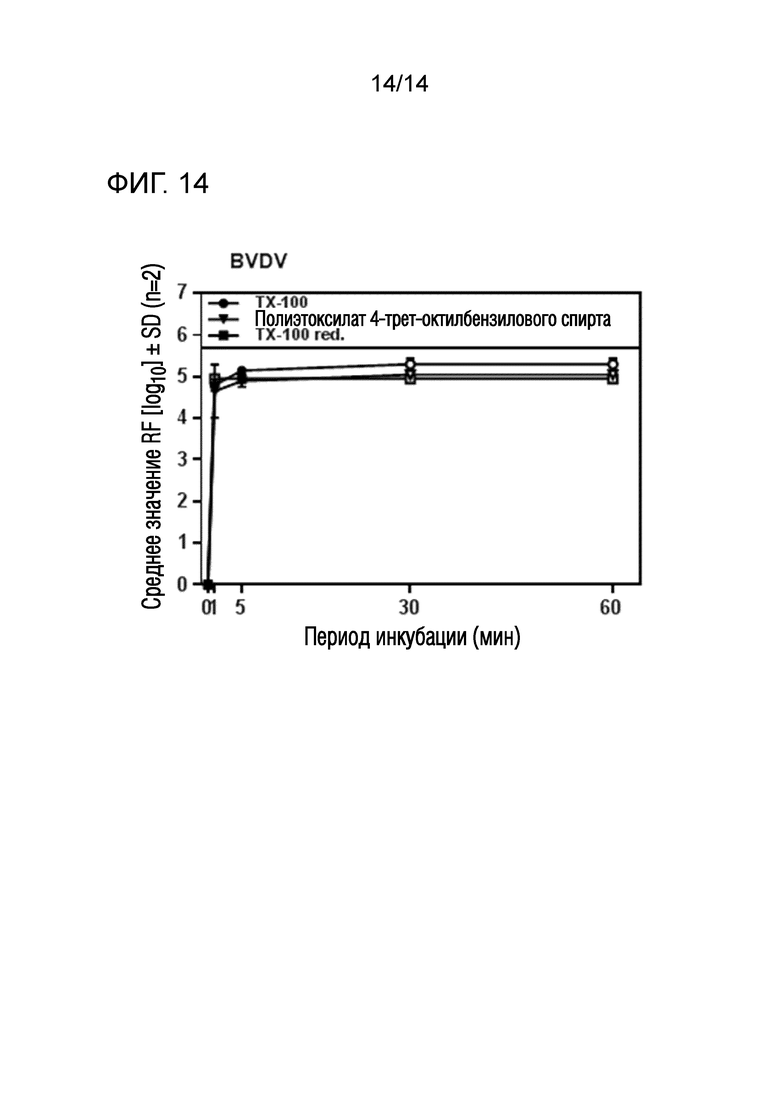
Группа изобретений относится к экологически приемлемым моющим средствам, а именно к моющим средствам, предназначенным для инактивации вирусов с липидной оболочкой.
В качестве активного соединения используется соединение со структурной формулой:
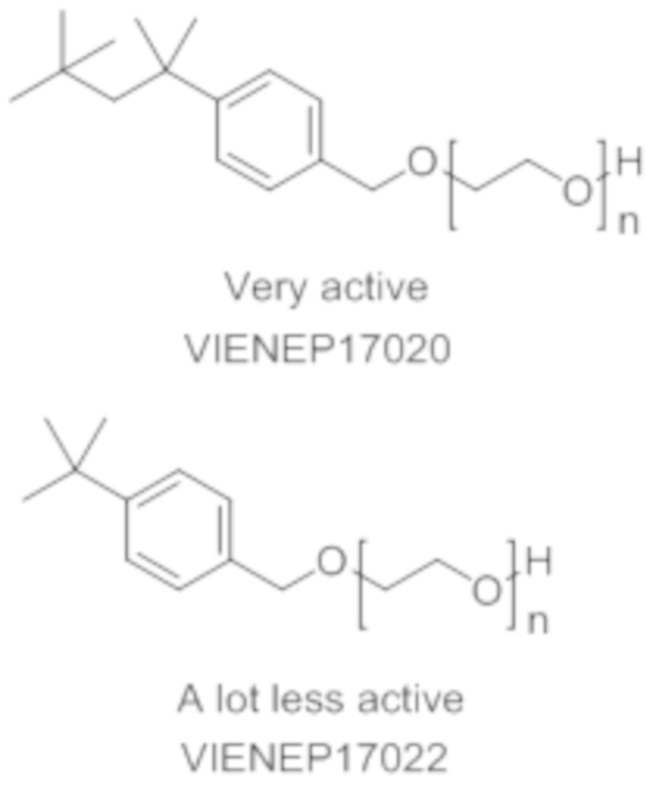
в котором n является целым числом между 4 и 16. Использование предложенного в изобретении подхода позволяет получить моющее средство, наносящее меньший вред здоровью живых организмов. 8 н. и 15 з.п. ф-лы, 14 ил., 14 табл., 11 пр.
1. Соединение, которым является следующее соединение:
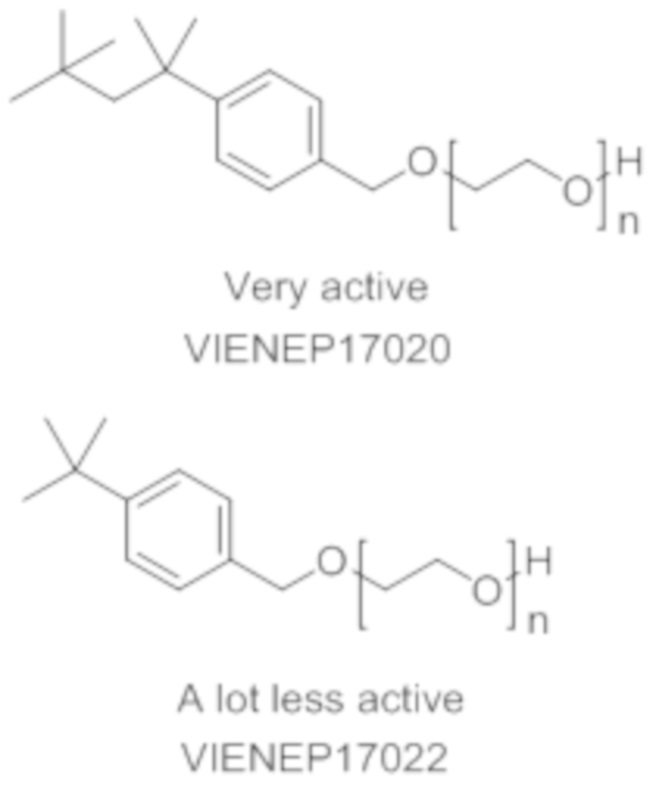
в котором n является целым числом, между 4 и 16.
2. Соединение по п. 1, где n равно 9 или 10.
3. Способ инактивации вирусов, обладающих липидной оболочкой, способ включает следующие стадии:
a) добавление моющего средства к жидкости для приготовления смеси указанного моющего средства и указанной жидкости; и
b) инкубирование указанной смеси для инактивации указанных вирусов;
где указанным моющим средством является соединение по любому из пп. 1-2.
4. Способ по п. 3, в котором указанное моющее средство является экологически приемлемым.
5. Способ по любому из пп. 3-4, в котором стадия a) дополнительно включает добавление растворителя к указанной жидкости и в котором на стадии a) смесь растворитель/моющее средство для инактивации указанных вирусов готовят путем добавления указанного моющего средства и указанного растворителя к указанной жидкости.
6. Способ по п. 5, в котором указанным растворителем является органический растворитель
и в котором указанным растворителем необязательно является три-н-бутилфосфат.
7. Способ по любому из пп. 3-6, в котором указанная жидкость включает биологический лечебный продукт и/или биофармацевтическое лекарственное средство.
8. Способ по п. 7, в котором указанным биофармацевтическим лекарственным средством является:
(a) фактор крови, иммуноглобулин, такой как моноклональное антитело, замещающий фермент, вакцина, генотерапевтический вектор, фактор роста или рецептор фактора роста;
(b) терапевтический белок;
(c) фактор крови и в котором указанным фактором крови является фактор I (фибриноген), фактор II (протромбин), тканевый фактор, фактор V, фактор VII или VIIa, фактор VIII, фактор IX, фактор X, фактор XI, фактор XII, фактор XIII, фактор фон Виллебранда (VWF), прекалликреин, кининоген с большой молекулярной массой (HMWK), фибронектин, антитромбин III, кофактор II гепарина, белок C, белок S, белок Z, плазминоген, альфа-2-антиплазмин, тканевый активатор плазминогена (tPA), урокиназа, ингибитор-1 активатора плазминогена (PAI1) или ингибитор-2 активатора плазминогена (PAI2); и/или
(d) фактор VIII, предпочтительно рекомбинантный фактор VIII человека.
9. Способ по любому из пп. 3-8, где (a) до стадии a) или между стадией a) и стадией b) указанный способ дополнительно включает стадию фильтрования указанной жидкости или смеси через объемный фильтр;
(b) на стадии b) указанную смесь инкубируют в течение не менее 1 ч; и/или
(c) на стадии b) указанную смесь инкубируют при температуре, равной от 0°C до 10°C, или в котором указанную смесь инкубируют при температуре, равной от 16°C до 25°C.
10. Способ по любому из пп. 7-9, после стадии b) дополнительно включающий стадию
c) очистки указанного биофармацевтического лекарственного средства;
и в котором:
(i) указанная очистка включает отделение указанного биофармацевтического лекарственного средства от указанного моющего средства;
(ii) указанная очистка включает отделение указанного биофармацевтического лекарственного средства от указанного дополнительного моющего средства; и/или
(iii) указанная очистка указанного биофармацевтического лекарственного средства включает очистку указанного биофармацевтического лекарственного средства посредством по меньшей мере одной хроматографической очистки и указанная по меньшей мере одна хроматографическая очистка проводится необязательно с помощью анионообменной хроматографии и/или с помощью катионообменной хроматографии.
11. Способ получения биофармацевтического лекарственного средства, указанный способ включает способ по любому из пп. 7-10, в котором указанное биофармацевтическое лекарственное средство является таким, как определено в любом из указанных пп. 7-10.
12. Способ по п. 11, дополнительно включающий после указанного способа по любому из пп. 7-10 стадию получения фармацевтического состава, включающего указанное биофармацевтическое лекарственное средство.
13. Применение моющего средства в способе инактивации вирусов, обладающих липидной оболочкой, где моющим средством является соединение по любому из пп. 1, 2.
14. Применение по п. 13, в котором указанный способ указанной инактивации указанных вирусов представляет собой способ с использованием обработки растворителем/моющим средством, указанная обработка растворителем/моющим средством включает применение указанного моющего средства, которым является соединение по любому из пп. 1, 2.
15. Применение по любому из пп. 13-14, в котором указанная инактивация вирусов представляет собой инактивацию вирусов в жидкости, включающей биофармацевтическое лекарственное средство по любому из пп. 7-8.
16. Композиция для инактивации вирусов, включающая моющее средство, в которой моющим средством является соединение по любому из пп. 1, 2.
17. Композиция по п. 16, дополнительно включающая органический растворитель по п. 6.
18. Способ синтеза соединения следующей общей формулы (VIII)
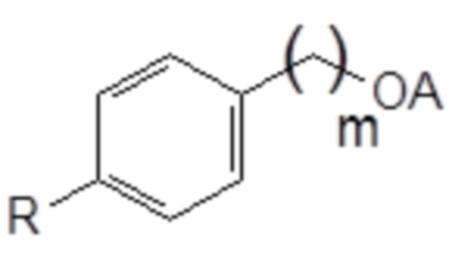
Формула (VIII)
в которой
R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи,
m равно 1, и
A означает полиоксиэтиленовый остаток,
где способ включает стадии
A) превращение фенола следующей общей формулы (IX), в которой R является таким, как определено выше
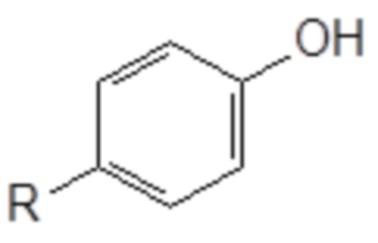
Формула (IX)
в спирт следующей общей формулы (X), в которой R и m являются такими, как определено выше
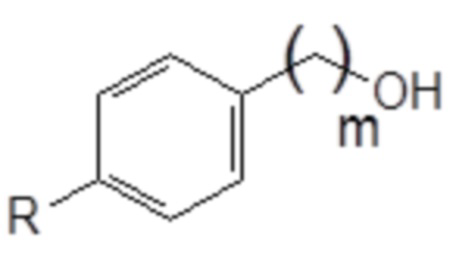
Формула (X)
и
(B) превращение спирта общей формулы (X) в простой эфир полиоксиэтилена общей формулы (VIII), определенной выше;
где соединением формулы (VIII) является следующее соединение:
в котором n является целым числом между 4 и 16.
19. Способ синтеза соединения следующей общей формулы (VIII)
Формула (VIII)
в которой
R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи,
m равно 1, и
A означает полиоксиэтиленовый остаток,
где способ включает стадии
(1) введение в реакцию толуола с получением замещенного толуола следующей общей формулы (XI), в которой R является таким, как определено выше
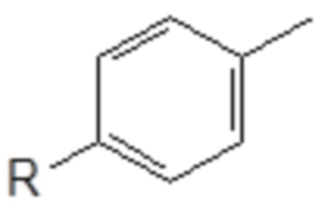
Формула (XI)
(2) превращение замещенного толуола общей формулы (XI) в соединение следующей общей формулы (XII), в которой R и m являются такими, как определено выше, и X выбран из группы, включающей гидроксигруппу, атом брома, атом йода и атом хлора
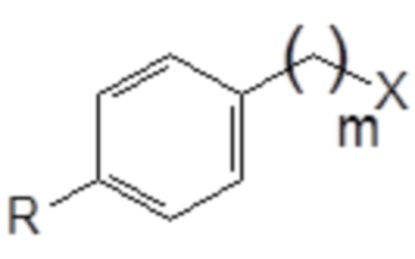
Формула (XII)
и
(3) превращение соединения общей формулы (XII) в простой эфир полиоксиэтилена общей формулы (VIII), определенной выше;
где соединением формулы (VIII) является следующее соединение:
в котором n является целым числом, между 4 и 16.
20. Способ синтеза соединения следующей общей формулы (VIIIa)
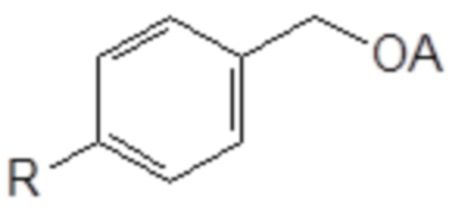
Формула (VIIIa)
в которой
R означает углеводородную группу, включающую линейную цепь, содержащую от 2 до 12 атомов углерода и одну или большее количество метильных групп в качестве заместителей указанной линейной цепи, и
A означает полиоксиэтиленовый остаток,
где способ включает стадии
(I) превращение бензилового спирта в соединение следующей общей формулы (XIII), в которой R является таким, как определено выше
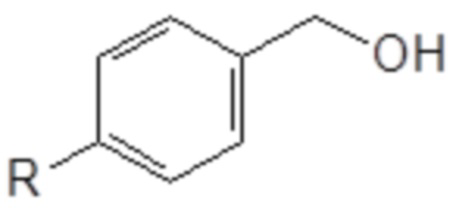
Формула (XIII)
и
(II) превращение соединения общей формулы (XIII) в простой эфир полиоксиэтилена общей формулы (VIIIa), определенной выше;
где соединением формулы (VIIIa) является следующее соединение:
в котором n является целым числом между 4 и 16.
21. Способ по любому из пп. 18-20, в котором n равно 9 или 10.
22. Способ по любому из пп. 19 и 21, в котором: (i) превращение на стадии (2) представляет собой радикальную реакцию с использованием AIBN (азобис(изобутиронитрил)) в качестве радикального инициатора; и/или
(ii) X означает атом брома.
23. Способ по любому из пп. 19 и 21-22, в котором
(a) при превращении на стадии (2) используется N-бромсукцинимид (NBS) в качестве реагента;
(b) при превращении на стадии (3) используется TBME (метил-трет-бутиловый эфир) в качестве растворителя;
(c) превращение на стадии (3) протекает в течение не менее 2 ч, предпочтительно при температуре окружающей среды;
(d) превращение на стадии (3) протекает в течение не более 5 ч, предпочтительно при температуре окружающей среды; и/или
(e) превращение на стадии (3) протекает в течение 3 ч, предпочтительно при температуре окружающей среды.
| YAJUAN LI et al | |||
| Microcalorimetric Study on Micellization of Nonionic Surfactants with a Benzene Ring or Adamantane in Their Hydrophobic Chains, Journal of Physical Chemistry Part B: Condenced Matter, Materials, Surfaces, Interfaces & Biophysical, US, (20050801), vol | |||
| Шкив для канатной передачи | 1920 |
|
SU109A1 |
| Способ сопряжения брусьев в срубах | 1921 |
|
SU33A1 |
| US 2016186094 A1, 30.06.2016 | |||
| US 2015094240 A1, 02.04.2015 | |||
| LYNN | |||
