Область техники, к которой относится изобретение
Настоящее изобретение касается пиразольных соединений, замещенных гетероарилом, или их фармацевтически приемлемых солей, которые обладают SGLT1-ингибирующей активностью, содержащих их фармацевтических композиций и их применения в фармацевтике.
Уровень техники
SGLT1, т.е. натрий-зависимый котранспортер глюкозы 1, как известно, играет большую роль в абсорбции глюкозы и галактозы в тонком кишечнике. Сообщалось, что у пациентов с дефицитом SGLT1 наблюдался синдром малабсорбции глюкозы и галактозы. Кроме того, подтверждено, что экспрессия SGLT1 в тонком кишечнике повышается у пациентов с диабетом, и считается, что повышение абсорбции сахара у пациентов с диабетом вызвано высоким уровнем экспрессии SGLT1 в тонком кишечнике.
Исходя из этих данных, ожидается, что ингибитор SGLT1 нормализует уровень глюкозы в крови, блокируя абсорбцию глюкозы в тонком кишечнике. Поэтому ингибитор SGLT1 рассматривается как эффективное средство против диабета и осложнений диабета, связанных с гипергликемией. Он также рассматривается как эффективное средство для борьбы с ожирением, благодаря ингибированию поступления глюкозы в организм (Непатентные документы 1 и 2).
Воглибоз (непатентованное название) является лекарственным средством, одобренным для производства и продажи в рамках Japan Pharmaceutical Affairs Act, ст. 14 (Номер разрешения: 21600AMZ00368). Воглибоз снижает повышенный уровень глюкозы после еды путем подавления дисахаридазы, α-глюкозидазы, которая разлагает дисахариды, присутствующие в слизистой кишечника, на моносахариды, и подавления или замедления переваривания и абсорбции углеводов в желудочно-кишечном тракте. Известно, что такое фармакологическое действие эффективно против диабета 2-го типа с замедленным развитием при нарушенной толерантности к глюкозе.
Исходя из имеющихся данных, подавление абсорбции сахаров в тонком кишечнике с помощью ингибитора SGLT1 и снижение, благодаря этому, повышенного уровня сахара в крови после еды считается эффективным средством против диабета 2-го типа с замедленным развитием при нарушенной толерантности к глюкозе.
Подтверждена также экспрессия SGLT1 в кардиомиоцитах. Известно, что обычно в поступлении глюкозы в кардиомиоциты играют роль GLUT1 (переносчик глюкозы тип 1) и GLUT4 (переносчик глюкозы тип 4), а вклад SGLT1 - меньше. Однако экспрессия SGLT1 индуцируется в сердечной мышце у мышей, которым вводят мутированные гены PRKAG2 (гамма 2 субъединица АМФК (АМФ-активируемая протеинкиназа)), который является геном, ответственным за наследственную гипертрофическую кардиомиопатию (миокардоз с накоплением гликогена), или у мышей, проходящих лечение от ишемии миокарда, и сообщалось, что SGLT1 участвует в поглощении глюкозы кардиомиоцитами при этих патологиях. Считается, что глюкоза, усвоенная при участии SGLT1, избыточно накапливается или метаболизируется в кардиомиоцитах и дестабилизирует эти клетки. Для прошлых мышиных моделей сообщалось, что накопление гликогена в сердечной мышце действительно ингибируется при воздействии неселективного ингибитора SGLT, флоризина.
Исходя из имеющихся данных, считается, что ингибитор SGLT1 эффективен против гипертрофической кардиомиопатии и ишемической болезни сердца, благодаря ингибированию избыточного поглощения глюкозы кардиомиоцитами (Непатентные документы 3 и 4).
SGLT1 стабилизируется в раковых клетках рецепторами эпидермального фактора роста, т.е. белками на поверхности многих типов раковых клеток. Известно, что переносчики глюкозы, молочной кислоты, аминокислот и т.д. участвуют в поступлении питательных веществ в раковые клетки, и это особенно касается переноса глюкозы, т.к. SGLT1 и GLUT1 непрерывно поставляют глюкозу в раковые клетки. Когда глюкоза не поступает в течение длительного времени, клетки разрушаются вследствие аутофагии.
Исходя из имеющихся данных, считается, что ингибитор SGLT1 ингибирует поставку глюкозы в раковые клетки и оказывает противораковое действие (Непатентные документы 5 и 6).
Поскольку углеводы из пищи разлагаются до моносахаридов в желудочно-кишечном тракте и абсорбируются в верхних отделах желудочно-кишечного тракта, многие сахара никогда не попадают в нижние отделы желудочно-кишечного тракта. Однако при введении лекарственных средств, которые замедляют или ингибируют абсорбцию глюкозы, или при поглощении большого количества устойчивых полисахаридов непереваренные сахара попадают в нижние отделы желудочно-кишечного тракта и задерживаются в нижних отделах желудочно-кишечного тракта, вызывая осмотическую диарею.
Ингибитор SGLT1 ингибирует абсорбцию глюкозы и увеличивает количество моносахаридов в нижних отделах желудочно-кишечного тракта. Поэтому считается, что ингибитор SGLT1 будет эффективен против запора.
Непатентные документы
[Непатентный документ 1] Am J Physiol Gastrointest Liver Physiol. 2002; 282(2):G241-8
[Непатентный документ 2] Nature. 1991; 350(6316):354-6
[Непатентный документ 3] J Mol Cell Cardiol. 2010; 49(4):683-92
[Непатентный документ 4] Cardiovasc Res. 2009; 84(1):111-8
[Непатентный документ 5] Cancer Cell. 2008, 13: 385-93
[Непатентный документ 6] Pharmacol Ther. 2009, 121: 29-40
Краткое раскрытие сущности изобретения
В настоящем изобретении описаны пиразольные соединения, замещенные гетероарилом, которые обладают SGLT1-ингибирующей активностью и могут быть полезны в качестве лекарственного средства, или же их фармацевтически приемлемые соли; содержащие их фармацевтические композиции; и их применение в фармацевтике.
В ходе интенсивных исследований авторы настоящего изобретения обнаружили специфические пиразольные соединения, замещенные гетероарилом, и таким образом было сделано настоящее изобретение.
В одном варианте осуществления описано соединение, имеющее формулу [X]:
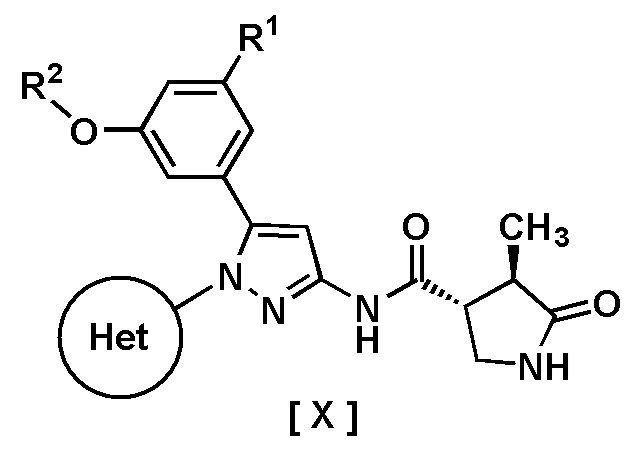
где R1 представляет собой атом водорода или галоген;
R2 представляет собой C1-6 алкил or галоген-C1-6 алкил;
Кольцо Het представляет собой:
(1) пиридил, замещенный R3; или
(2) пиразинил, пиримидинил или пиридазинил, необязательно замещенный R4;
R3 представляет собой циано-группу, галоген или галоген-C1-3 алкил;
R4 представляет собой галоген, гидрокси-группу, C1-3 алкил, галоген-C1-3 алкил, C1-3 алкокси-группу или -N(R5)(R6); и
R5 и R6 каждый независимо представляет собой атом водорода или C1-3 алкил,
или его фармацевтически приемлемая соль и его применение в фармацевтике.
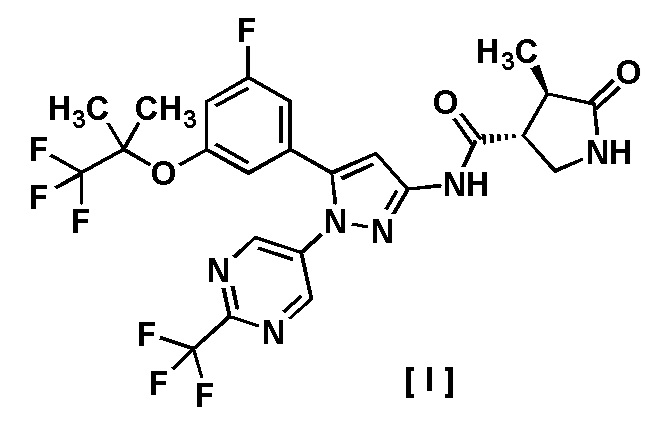
В другом варианте осуществления описано соединение, имеющее формулу [I], или его фармацевтически приемлемая соль и его применение в фармацевтике.
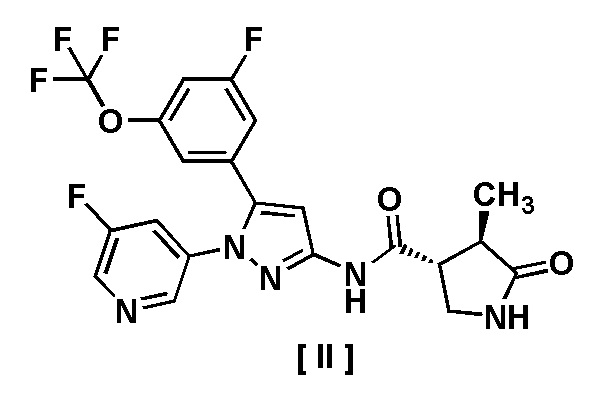
В другом варианте осуществления описано соединение, имеющее формулу [II], или его фармацевтически приемлемая соль и его применение в фармацевтике.
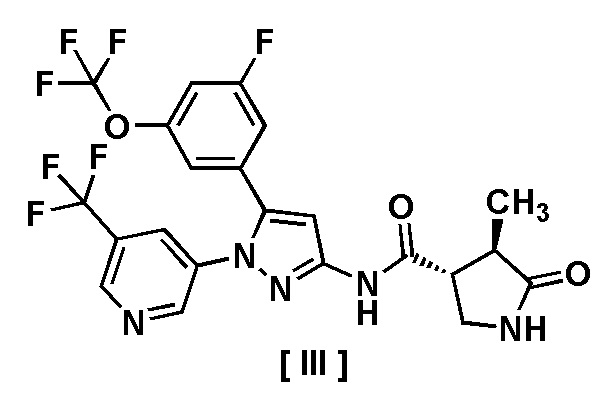
В другом варианте осуществления описано соединение, имеющее формулу [III], или его фармацевтически приемлемая соль и его применение в фармацевтике.
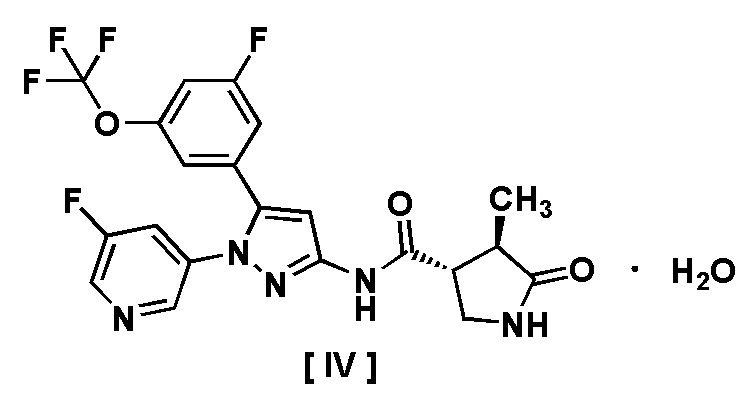
В другом варианте осуществления описано соединение, имеющее формулу [IV], или его фармацевтически приемлемая соль и его применение в фармацевтике.
Описание вариантов осуществления
Настоящее изобретение включает проиллюстрированные ниже варианты осуществления.
Пункт 1. Соединение, имеющее формулу [X]:
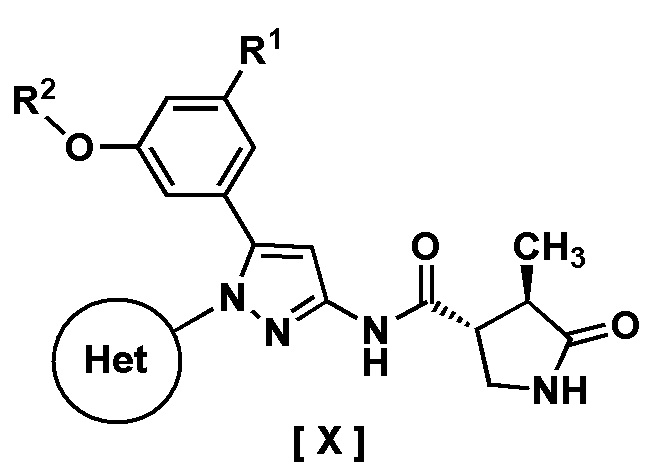
где R1 представляет собой атом водорода или галоген;
R2 представляет собой C1-6 алкил или галоген-C1-6 алкил;
Кольцо Het представляет собой:
(1) пиридил, замещенный R3; или
(2) пиразинил, пиримидинил или пиридазинил, необязательно замещенный R4;
R3 представляет собой циано-группу, галоген или галоген-C1-3 алкил;
R4 представляет собой галоген, гидрокси-группу, C1-3 алкил, галоген-C1-3 алкил, C1-3 алкокси-группу или -N(R5)(R6); и
R5 и R6 каждый независимо представляют собой атом водорода или C1-3 алкил,
или его фармацевтически приемлемая соль.
Пункт 2. Соединение или его фармацевтически приемлемая соль по пункту 1, где R1 представляет собой галоген.
Пункт 3. Соединение или его фармацевтически приемлемая соль по любому из пунктов 1 или 2, где R2 представляет собой галоген-C1-6 алкил.
Пункт 4. Соединение или его фармацевтически приемлемая соль по любому из пунктов 1 - 3, где кольцо Het представляет собой пиридил, замещенный R3.
Пункт 5. Соединение или его фармацевтически приемлемая соль по любому из пунктов 1 - 3, где кольцо Het представляет собой пиразинил, пиримидинил или пиридазинил, необязательно замещенный R4.
Пункт 6. Соединение, имеющее формулу [I]:
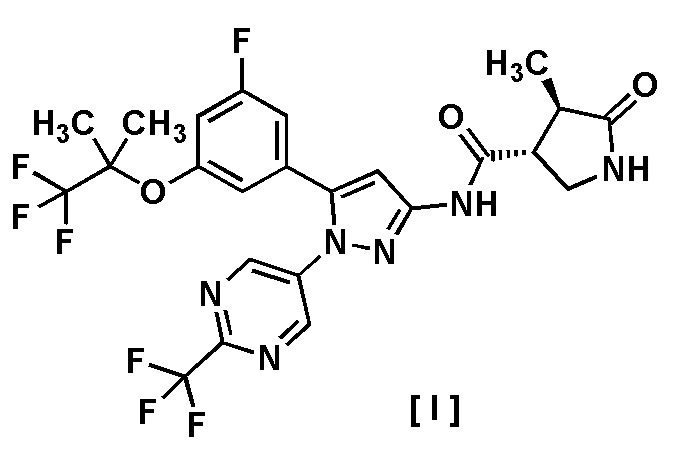
или его фармацевтически приемлемая соль.
Пункт 7. Соединение, имеющее формулу [II]:
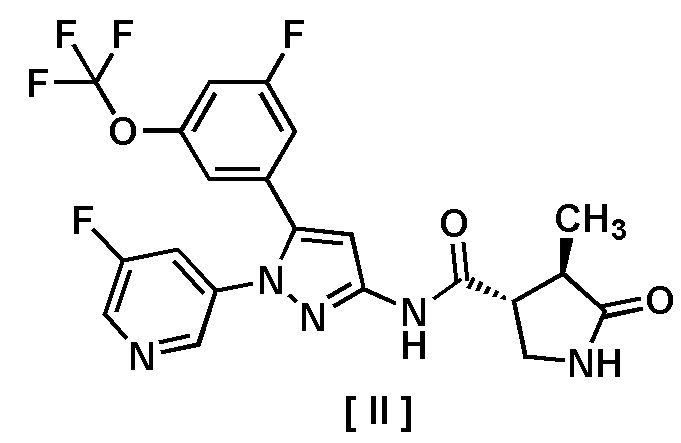
или его фармацевтически приемлемая соль.
Пункт 8. Соединение, имеющее формулу [III]:
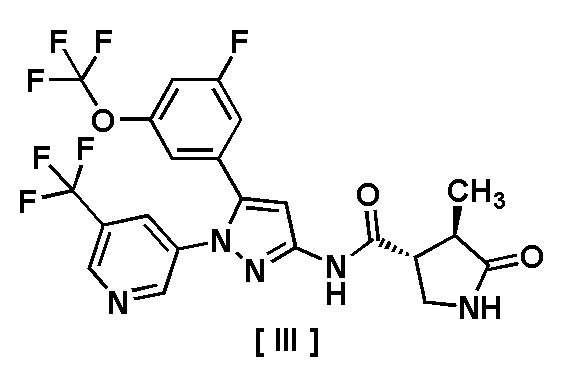
или его фармацевтически приемлемая соль.
Пункт 9. Соединение, имеющее формулу [IV]:
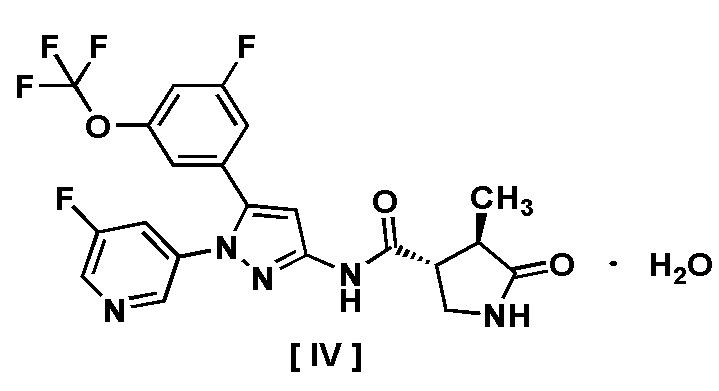
или его фармацевтически приемлемая соль.
Пункт 10. Фармацевтическая композиция, содержащая соединение или его фармацевтически приемлемую соль по любому из пунктов 1 - 9 и фармацевтически приемлемый носитель.
Пункт 11. Ингибитор SGLT1, содержащий соединение или его фармацевтически приемлемую соль по любому из пунктов 1 - 9.
Пункт 12. Терапевтическое или профилактическое средство против диабета, содержащее соединение или его фармацевтически приемлемую соль по любому из пунктов 1 - 9.
Пункт 13. Терапевтическое или профилактическое средство по пункту 12, где диабет представляет собой диабет 2-го типа.
Пункт 14. Способ ингибирования SGLT1, включающий введение млекопитающему терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пунктов 1 - 9.
Пункт 15. Способ лечения или предотвращения диабета, включающий введение млекопитающему терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пунктов 1 - 9.
Пункт 16. Способ по пункту 15, где диабет представляет собой диабет 2-го типа.
Пункт 17. Применение соединения или его фармацевтически приемлемой соли по любому из пунктов 1 - 9 для производства ингибитора SGLT1.
Пункт 18. Применение соединения или его фармацевтически приемлемой соли по любому из пунктов 1 - 9 для производства терапевтического или профилактического средства против диабета.
Пункт 19. Применение по пункту 18, где диабет представляет собой диабет 2-го типа.
Пункт 20. Соединение или его фармацевтически приемлемая соль по любому из пунктов 1 - 9 для ингибирования SGLT1.
Пункт 21. Соединение или его фармацевтически приемлемая соль по любому из пунктов 1 - 9 для применения в лечении или предотвращении диабета.
Пункт 22. Соединение или его фармацевтически приемлемая соль по пункту 21, где диабет представляет собой диабет 2-го типа.
Пункт 23. Коммерческая упаковка, содержащая композицию по пункту 10 и относящиеся к ней письменные указания на то, что данная композиция может или должна применяться для лечения или предотвращения диабета.
Пункт 24. Набор, содержащий композицию по пункту 10 и относящиеся к ней письменные указания на то, что данная композиция может или должна применяться для лечения и/или предотвращения диабета.
Изображенная ниже двойная волнистая линия:

в фрагменте структуры указывает место связывания данного фрагмента.
Термин “галоген” включает фтор, хлор, бром и иод.
Термин “C1-3 алкил” означает линейную или разветвленную насыщенную углеводородную группу, содержащую 1 - 3 атомов углерода. “C1-3 алкильная” группа включает метил, этил, н-пропил и изопропил.
Термин “C1-6 алкил” означает линейную или разветвленную насыщенную углеводородную группу, содержащую 1 - 6 атомов углерода. “C1-6 алкильная” группа включает, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил и н-гексил.
Термин “галоген-C1-3 алкил” означает “C1-3 алкильную” группу, замещенную 1 - 5 атомами галогена, независимо выбранными из группы, соответствующей термину “галоген”. “Галоген-C1-3 алкильная” группа включает, например, монофторметил, дифторметил, трифторметил, 2-фторэтил, 2-хлорэтил, 2-бромэтил, 1,1-дифторэтил, 2,2,2-трифторэтил, пентафторэтил, 3-фторпропил, 3-хлорпропил, 1,1-дифторпропил и 3,3,3-трифторпропил.
Термин “фтор-C1-3 алкил” означает “C1-3 алкильную” группу, замещенную 1 - 5 атомами фтора. “Фтор-C1-3 алкильная” группа включает, например, монофторметил, дифторметил, трифторметил, 2-фторэтил, 1,1-дифторэтил, 2,2,2-трифторэтил, пентафторэтил, 3-фторпропил, 1,1-дифторпропил и 3,3,3-трифторпропил.
Термин “галоген-C1-6 алкил” означает “C1-6 алкильную” группу, замещенную 1 - 5 атомами галогена, независимо выбранными из группы, соответствующей термину “галоген”. “Галоген-C1-6 алкильная” группа включает, например, монофторметил, дифторметил, трифторметил, 2-фторэтил, 2-хлорэтил, 2-бромэтил, 1,1-дифторэтил, 2,2,2-трифторэтил, пентафторэтил, 3-фторпропил, 3-хлорпропил, 1,1-дифторпропил, 3,3,3-трифторпропил, 4,4,4-трифторбутил, 5,5,5-трифторпентил и 6,6,6-трифторгексил.
Термин “фтор-C1-6 алкил” означает “C1-6 алкильную” группу, замещенную 1 - 5 атомами фтора. “Фтор-C1-6 алкильная” группа включает, например, монофторметил, дифторметил, трифторметил, 2-фторэтил, 1,1-дифторэтил, 2,2,2-трифторэтил, пентафторэтил, 3-фторпропил, 1,1-дифторпропил, 3,3,3-трифторпропил, 4,4,4-трифторбутил, 5,5,5-трифторпентил и 6,6,6-трифторгексил.
Термин “C1-3 алкокси” означает группу, в которой “C1-3 алкильная” группа связана с атомом кислорода. “C1-3 алкокси” группа включает метокси, этокси, н-пропокси и изопропокси.
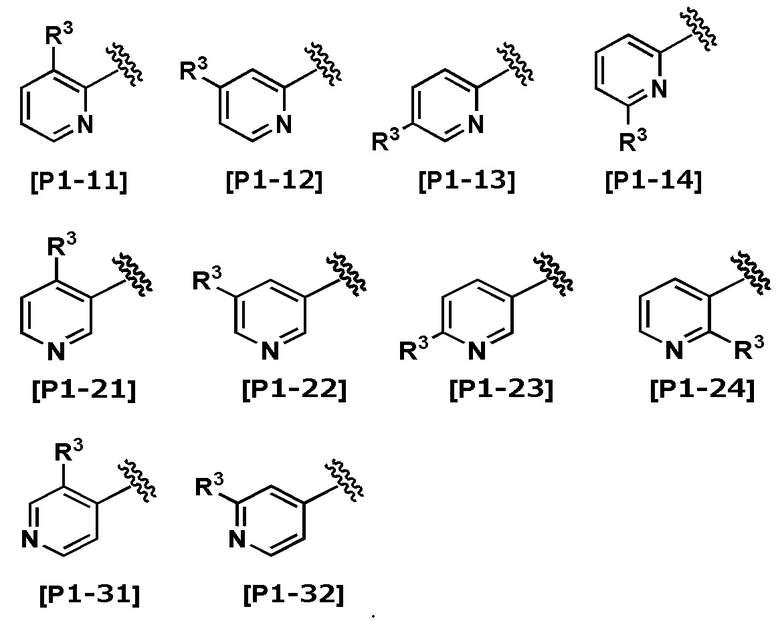
Термин “пиридил” означает любую из следующих групп:
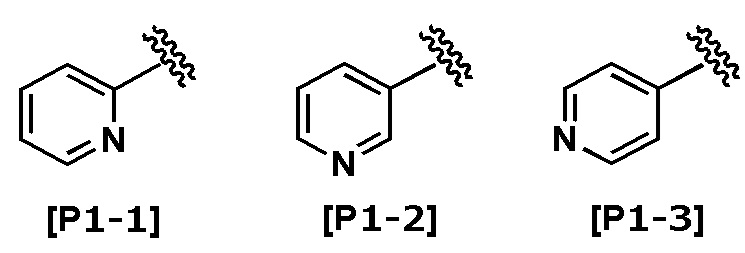 .
.
Термин “пиразинил” означает следующую группу:
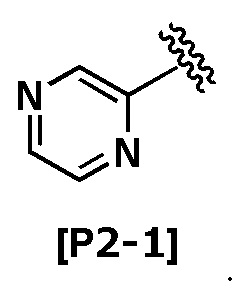
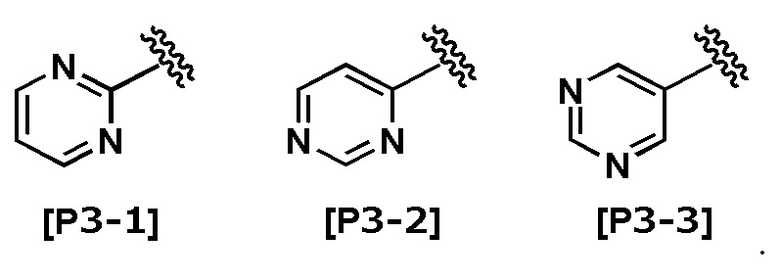
Термин “пиримидинил” означает любую из следующих групп:
 .
.
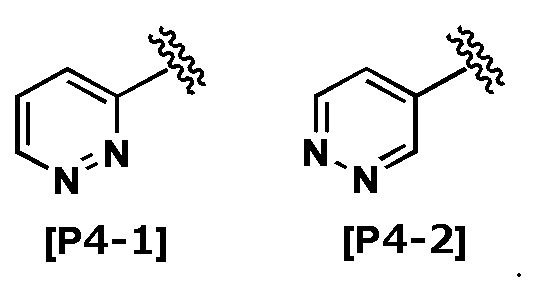
Термин “пиридазинил” означает любую из следующих групп:
 .
.
Термин “замещенный” включает любые химически приемлемые замещения. Например, выражение “пиридил, замещенный R3” означает любую из следующих групп:
Варианты каждого заместителя в соединении, имеющем формулу [X], проиллюстрированы ниже, но каждый заместитель в соединении, имеющем формулу [X], не ограничивается только указанными вариантами. Соединение, имеющее формулу [X], включает любую комбинацию двух или более вариантов осуществления, и элементы, необязательно выбранные из вариантов осуществления и элементов в каждом заместителе.
В одном варианте осуществления R1 представляет собой атом водорода или фтор. В другом варианте осуществления, R1 представляет собой фтор.
В одном варианте осуществления, R2 представляет собой галоген-C1-6 алкил. В другом варианте осуществления R2 представляет собой фтор-C1-6 алкил.
В одном варианте осуществления Кольцо Het представляет собой:
(1) пиридил, замещенный R3; или
(2) пиразинил или пиримидинил, необязательно замещенный R4.
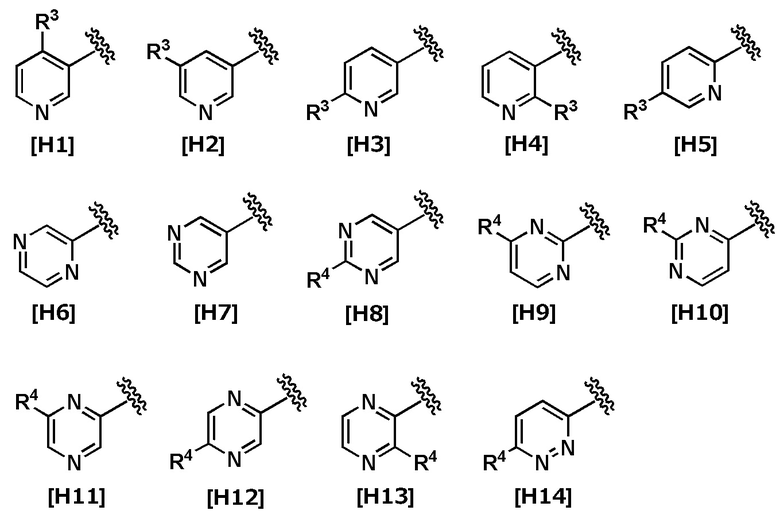
В другом варианте осуществления Кольцо Het выбрано из группы, состоящей из Формул [H1] - [H14].
В другом варианте осуществления Кольцо Het выбрано из группы, состоящей из Формул [H2], [H3], [H5], [H8] - [H12] и [H14].
В другом варианте осуществления Кольцо Het соответствует Формуле [H2] или [H8].
В одном варианте осуществления R3 представляет собой галоген или галоген-C1-3 алкил. В другом варианте осуществления R3 представляет собой фтор или фтор-C1-3 алкил.
В одном варианте осуществления R4 представляет собой галоген или галоген-C1-3 алкил. В другом варианте осуществления R4 представляет собой фтор-C1-3 алкил.
В одном варианте осуществления R5 и R6 каждый независимо представляют собой C1-3 алкил.
Термин “фармацевтически приемлемая соль” включает любые известные соли, не ассоциируются избыточной токсичностью. Такая фармацевтически приемлемая соль включает, в частности, соли с неорганическими кислотами, соли с органическими кислотами, соли с неорганическими основаниями и соли с органическими основаниями. Различные формы фармацевтически приемлемых солей хорошо известны в данной области техники и описаны, например, в следующих источниках:
(a) Berge et al., J. Pharm. Sci., 66, p1-19 (1977),
(b) Stahl et al., “Handbook of Pharmaceutical Salt: Properties, Selection, and Use” (Wiley-VCH, Weinheim, Германия, 2002),
(c) Paulekuhn et al., J. Med. Chem., 50, p6665-6672 (2007).
Соединение, имеющее формулу [X], можно вводить в реакцию с неорганической кислотой, органической кислотой, неорганическим основанием или органическим основанием согласно известным методикам, получая его соответствующую фармацевтически приемлемую соль.
Такая соль с неорганической кислотой включает соль с фтористоводородной кислотой, соляной кислотой, бромистоводородной кислотой, иодистоводородной кислотой, азотной кислотой, фосфорной кислотой и серной кислотой. Такая соль предпочтительно включает соль с соляной кислотой, азотной кислотой, серной кислотой, фосфорной кислотой и бромистоводородной кислотой.
Такая соль с органической кислотой включает соль с уксусной кислотой, адипиновой кислотой, альгиновой кислотой, 4-аминосалициловой кислотой, дегидрометиленлимонной кислотой, бензойной кислотой, бензолсульфокислотой, эдетатом кальция, камфорной кислотой, камфор-10-сульфокислотой, угольной кислотой, лимонной кислотой, эдетовой кислотой, этан-1,2-дисульфокислотой, додецилсерной кислотой, этансульфокислотой, фумаровой кислотой, глюкогептоновой кислотой, глюконовой кислотой, глюкуроновой кислотой, глюкогептоновой кислотой, гликоллиларсаниловой кислотой, гексилрезорциновой кислотой, гидроксинафтойной кислотой, 2-гидрокси-1-этансульфокислотой, молочной кислотой, лактобионовой кислотой, яблочной кислотой, малеиновой кислотой, миндальной кислотой, метансульфокислотой, метилсерной кислотой, метилазотной кислотой, метиленбис(салициловой кислотой), галактаровой кислотой, нафталин-2-сульфокислотой, 2-нафтоевой кислотой, 1,5-нафталиндисульфокислотой, олеиновой кислотой, щавелевой кислотой, памоевой кислотой, пантотеновой кислотой, пектиновой кислотой, пикриновой кислотой, пропионовой кислотой, полигалактуроновой кислотой, салициловой кислотой, стеариновой кислотой, янтарной кислотой, дубильной кислотой, винной кислотой, теоклиновой кислотой, тиоциановой кислотой, трифторуксусной кислотой, п-толуолсульфокислотой, ундекановой кислотой, аспартамовой кислотой и глутаминовой кислотой. Такая соль предпочтительно представляет собой соль с щавелевой кислотой, малеиновой кислотой, лимонной кислотой, фумаровой кислотой, молочной кислотой, яблочной кислотой, янтарной кислотой, винной кислотой, уксусной кислотой, трифторуксусной кислотой, бензойной кислотой, глюкуроновой кислотой, олеиновой кислотой, памоевой кислотой, метансульфокислотой, бензолсульфокислотой, п-толуолсульфокислотой и 2-гидрокси-1-этансульфокислотой.
Такая соль с неорганическим основанием включает соль лития, натрия, калия, магния, кальция, бария, алюминия, цинка, висмута и аммония. Такая соль предпочтительно включает соль натрия, калия, кальция, магния и цинка.
Такая соль с органическим основанием включает соль с ареколином, бетаином, холином, клемизолом, этилендиамином, N-метилглюкамином, N-бензилфенетиламином, трис(гидроксиметил)метиламином, аргинином и лизином. Такая соль предпочтительно включает соль с трис(гидроксиметил)метиламином, N-метилглюкамином и лизином.
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут существовать в форме сольвата. Термин “сольват” означает соединение, в котором молекула растворителя скоординирована с соединением, имеющим формулу [X], или его фармацевтически приемлемой солью, и включает в себя гидрат. Сольват предпочтительно представляет собой фармацевтически приемлемый сольват; и включает, например, гидрат, этанолят и диметилсульфоксидный сольват. Такой сольват в частности включает полугидрат, моногидрат, дигидрат и моноэтанолят соединения, имеющего формулу [X], [I], [II] или [III]; и моногидрат натриевой соли соединения, имеющего формулу [X], [I], [II] или [III]; и 2/3 этанолят его дигидрохлоридной соли. Эти сольваты можно получить любым из известных способов.
Например, соединение, имеющее формулу [II], может существовать в форме моногидрата, как показано на изображенной ниже Формуле [IV]:
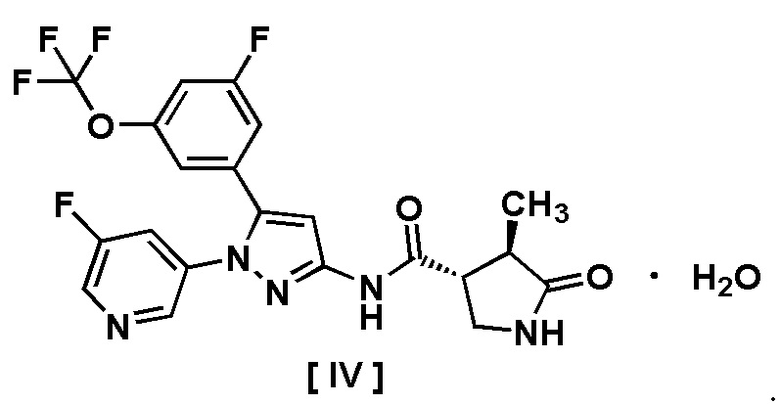 .
.
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут существовать в таутомерной форме. Так, соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут существовать в каждой таутомерной форме или в форме смеси таутомеров.
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут содержать двойную связь углерод-углерод. Так, соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут существовать в E или Z форме, или в форме смеси E- и Z-изомеров.
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут иметь стереоизомеры, идентифицируемые как цис/транс изомеры. Так, соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут существовать в цис- или транс-форме или в форме смеси цис- и транс-изомеров.
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут содержать один или больше асимметричных атомов углерода. Так, соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут существовать в форме индивидуального энантиомера или индивидуального диастереомера, или в форме смеси энантиомеров или диастереомеров.
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут существовать в атропоизомерных формах. Так, соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут существовать в виде отдельной атропоизомерной формы или в форме смеси атропоизомеров.
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут одновременно содержать несколько структурных признаков из описанных выше видов изомеров. Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль могут содержать описанные выше изомеры в любых соотношениях.
Формулы, химические структуры или названия соединений в настоящем документе, приведенные без указания определенной стереохимии, включают любые возможные изомеры из числа перечисленных выше, если иное не указано особо.
Смесь диастереомеров можно разделить на отдельные диастереомеры общеизвестными методами, такими как хроматография и кристаллизация. Каждый диастереомер может быть также получен из стереохимически индивидуального исходного вещества или методами синтеза с применением стереоселективных реакций.
Смесь энантиомеров можно разделить на отдельные индивидуальные энантиомеры методами, хорошо известными в данной области техники. Например, смесь энантиомеров можно ввести в реакцию с практически чистым энантиомером, известным как хиральное вспомогательное вещество, с образованием смеси диастереомеров, после чего полученную смесь диастереомеров разделяют известными методами, такими как дробная кристаллизация и хроматография, получая определенный диастереомер с увеличенным соотношением изомеров или практически чистый индивидуальный диастереомер. Затем выделенный диастереомер можно превратить в целевой энантиомер путем удаления хирального вспомогательного вещества по реакции расщепления.
Смесь энантиомеров можно также напрямую разделить на отдельные энантиомеры хроматографическими методами с применением хиральной неподвижной фазы, хорошо известной в данной области. В альтернативном варианте каждый из энантиомеров можно также получить из по существу чистого оптически активного исходного вещества или методами стереоселективного синтеза, т.е. с применением асимметричной индукции к прохиральному интермедиату с помощью хирального вспомогательного вещества или асимметричного катализатора.
Абсолютную конфигурацию можно определить методом рентгеноструктурного анализа в случае кристаллических продуктов или интермедиатов. Для определения конфигурации можно также использовать кристаллические продукты или интермедиаты, дериватизованные реагентом, имеющим известную конфигурацию и асимметричный центр.
Соединение, имеющее формулу [X], может быть помечено изотопом, таким как 2H, 3H, 14C и 35S.
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль предпочтительно представляют собой по существу очищенное соединение, имеющее формулу [X], или его фармацевтически приемлемую соль, а более предпочтительно - соединение, имеющее формулу [X], или его фармацевтически приемлемую соль, имеющие чистоту 80% или выше.
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль имеют SGLT1-ингибирующую активность, и поэтому могут применяться для лечения и/или предотвращения различных заболеваний или состояний, для которых ожидается улучшение при регулировке активности SGLT1, например диабета (например, диабет 1-го типа и диабет 2-го типа), ожирения, осложнений диабета (например, ретинопатия, нефропатия и нейропатия, которые вместе известны как микроангиопатия; и цереброваскулярная болезнь, ишемическая болезнь сердца и облитерирующий артериосклероз нижней конечности, которые вместе известны как макроангиопатия), гипертрофической кардиомиопатии, ишемической болезни сердца, рака и запора.
Термин “ингибирование SGLT1” означает, что функция SGLT1 подавляется с исчезновением или снижением ее активности; и, например, он означает, что функция SGLT1 подавляется по результатам описанного далее Примера Теста 1. Термин “ингибирование SGLT1” предпочтительно означает “ингибирование человеческого SGLT1”. Ингибирование функции или исчезновение или снижение активности предпочтительно осуществляется при клиническом показании у людей.
Термин “ингибитор SGLT1” означает любое вещество, которое ингибирует SGLT1, и включает низкомолекулярные соединения, нуклеиновые кислоты, полипептиды, белки, антитела и вакцины. Термин “ингибитор SGLT1” предпочтительно означает “ингибитор человеческого SGLT1”.
Термин “лечение” включает облегчение состояния, предотвращение ухудшения, поддержание ремиссии, предотвращение обострения и предотвращение рецидива.
Термин “предотвращение” или “профилактика” включает замедление появления состояний. Например, выражение “предотвращение диабета” включает замедление появления диабета 1-го типа и/или диабета 2-го типа при нарушении толерантности к глюкозе.
Фармацевтическую композицию по настоящему изобретению можно получить из терапевтически эффективного количества соединения, имеющего формулу [X], или его фармацевтически приемлемой соли и по меньшей мере одного или более фармацевтически приемлемых носителей, необязательно с последующим перемешиванием, согласно известным методикам из области медицинских препаратов. Количество соединения, имеющего формулу [X], или его фармацевтически приемлемой соли, содержащееся в фармацевтической композиции, варьируется в зависимости от таких факторов, как вид дозированной формы и размер дозировки, и находится, например, в диапазоне от 0,1 до 100 мас.% от общего количества композиции.
Дозированная форма препарата соединения, имеющего формулу [X], или его фармацевтически приемлемой соли включает пероральные препараты, такие как таблетки, капсулы, гранулы, порошки, литые таблетки, сиропы, эмульсии и суспензии; и парентеральные препараты, такие как препараты для наружного применения, суппозитории, инъекции, глазные капли, назальные препараты и легочные препараты.
Термин “фармацевтически приемлемый носитель” включает различные органические или неорганические вещества, которые обычно применяются в качестве компонента препарата. Такие вещества включают, например, вспомогательные вещества, разрыхлители, связующие вещества, улучшители текучести, и лубриканты для твердых препаратов; растворители, солюбилизаторы, суспендирующие агенты, агенты для поддержания тоничности, буферные вещества, и успокаивающие вещества для жидких препаратов; а также основы, эмульгаторы, увлажняющие агенты, стабилизаторы, диспергаторы, пластификаторы, регуляторы рН, стимуляторы абсорбции, загеливатели, антисептические агенты, наполнители, солюбилизаторы, и суспендирующие агенты для полутвердых препаратов. При необходимости можно дополнительно добавлять добавки, такие как консерванты, антиоксиданты, красители и подсластители.
Такое “вспомогательное вещество” включает, например, лактозу, белый мягкий сахар, D-маннит, D-сорбит, кукурузный крахмал, декстрин, микрокристаллическую целлюлозу, кристаллическую целлюлозу, кармеллозу, кармеллозу кальция, натрия карбоксиметилкрахмал, низкозамещенную гидроксипропилцеллюлозу и аравийскую камедь.
Такой “разрыхлитель” включает, например, кармеллозу, кальция кармеллозу, натрия кармеллозу, натрия карбоксиметилкрахмал, натрия кроскармеллозу, кросповидон, низкозамещенную гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и кристаллическую целлюлозу.
Такое “связующее вещество” включает, например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, повидон, кристаллическую целлюлозу, белый мягкий сахар, декстрин, крахмал, желатин, натрия кармеллозу и аравийскую камедь.
Такой “улучшитель текучести” включает, например, легкий безводный силикагель и стеарат магния.
Такой “лубрикант” включает, например, стеарат магния, стеарат кальция и тальк.
Такой “растворитель” включает, например, очищенную воду, этанол, пропиленгликоль, макрогол, сезамовое масло, кукурузное масло и оливковое масло.
Такой “солюбилизатор” включает, например, пропиленгликоль, D-маннит, бензилбензоат, этанол, триэтаноламин, карбонат натрия и цитрат натрия.
Такое “суспендирующий агент” включает, например, бензалкония хлорид, кармеллозу, гидроксипропилцеллюлозу, пропиленгликоль, повидон, метилцеллюлозу и моностеарат глицерина.
Такой “агент для поддержания тоничности” включает, например, глюкозу, D-сорбит, натрия хлорид и D-маннит.
Такое “буферное вещество” включает, например, динатрия гидрофосфат, натрия ацетат, карбонат натрия и цитрат натрия.
Такое “успокаивающее вещество” включает, например, бензиловый спирт.
Такая “основа” включает, например, воду, масла животного или растительного происхождения, такие как оливковое масло, кукурузное масло, арахисовое масло, сезамовое масло и касторовое масло, низшие спирты, такие как этанол, пропанол, пропиленгликоль, 1,3-бутиленгликоль и фенол, высшие жирные кислоты и их эфиры, воска, высшие спирты, многоатомные спирты, углеводороды, такие как белый вазелин, жидкий парафин и парафин, гидрофильный вазелин, очищенный ланолин, абсорбирующаяся мазь, гидратированный ланолин, гидрофильная мазь, крахмал, пуллулан, аравийская камедь, трагакантовая камедь, желатин, декстран, производные целлюлозы, такие как метилцеллюлоза, карбоксиметилцеллюлоза, гидроксиэтилцеллюлоза и гидроксипропилцеллюлоза, синтетические полимеры, такие как карбоксивиниловый полимер, полиакрилат натрия, поливиниловый спирт и поливинилпирролидон, пропиленгликоль, макрогол, такой как Макрогол от 200 до 600, и комбинация двух или более из перечисленных.
Такой “консервант” включает, например, этил парагидроксибензоат, хлорбутанол, бензиловый спирт, дегидроацетат натрия и сорбиновую кислоту.
Такой “антиоксидант” включает, например, сульфит натрия и аскорбиновую кислоту.
Такой “краситель” включает, например, пищевые красители (например, Food Red No. 2 или No. 3, Food Yellow No. 4 или No. 5) и β-каротин.
Такой “подсластитель” включает, например, сахарин натрия, дикалия глицирризинат и аспартам.
Фармацевтическую композицию по настоящему изобретению можно вводить перорально или парентерально (например, наружно, ректально, внутривенно, внутримышечно и подкожно) людям, а также млекопитающим, отличным от человека, таким как мыши, крысы, хомяки, морские свинки, кролики, кошки, собаки, свиньи, коровы, лошади, овцы и обезьяны. Дозировка варьируется в зависимости от субъекта, которому осуществляется введение, от заболеваний, состояний, дозированных форм и способа введения. Например, суточная доза при пероральном введении взрослому пациенту обычно находится в диапазоне от примерно 0,01 до примерно 1 г действующего вещества, т.е. соединения, имеющего формулу [X]. Такую дневную дозу можно вводить за один раз или в несколько приемов.
Могут применяться также набор, такой как набор для введения, лечения и/или профилактики, упаковка, такая как упакованные изделия, и комплекс и/или коробка с лекарственным средством, которые включают фармацевтическую композицию, содержащую соединение, имеющее формулу [I], или его фармацевтически приемлемую соль в качестве действующего вещества или активного агента, и письменный материал о композиции, указывающий, что данная композиция может или должна быть использована для лечения и/или профилактики. Такой набор, упаковка и комплекс с лекарственным средством может содержать один или больше контейнеров, заполненных фармацевтической композицией или одним или больше действующими веществами и другими лекарственными средствами (или ингредиентами), применяющимися в композиции. Примеры такого набора, упаковки и комплекса с лекарственным средством включают коммерческие наборы, коммерческие упаковки и коммерческие медицинские комплексы для соответствующего применения в лечении и/или предотвращении определенных заболеваний. Письменный материал, содержащийся в таком наборе, упаковке или медицинском комплексе, включает предупреждение или вкладыш в форме, установленной правительственной организацией, регулирующей производство, применение или продажу фармацевтических или биологических продуктов, где указывается разрешение государственной организации на производство, применение или продажу продуктов для введения людям. Такой набор, упаковка или медицинский комплекс могут включать упакованные продукты, а также приспособления для соответствующих операций введения, сконструированные таким образом, чтобы обеспечить наиболее предпочтительное медицинское лечение и/или профилактику, включая лечение и/или профилактику определенных заболеваний.
Общий способ получения соединения, имеющего формулу [X], или его фармацевтически приемлемой соли проиллюстрирован ниже. Соединения, имеющие Формулы [1] - [20], включают их фармацевтически приемлемые соли, в случае если они могут существовать. Например, термин “соединение, имеющее Формулу [1]” включает соединение, имеющее формулу [1], и его фармацевтически приемлемую соль, если такая фармацевтически приемлемая соль существует.
В настоящем документе термин “комнатная температура” означает температуру в диапазоне, например, от примерно 15°C до примерно 30°C, предпочтительно от примерно 20°C до примерно 25°C.
Общий способ получения: Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль
Соединение, имеющее формулу [X], или его фармацевтически приемлемую соль можно получить, например, по следующим методикам.
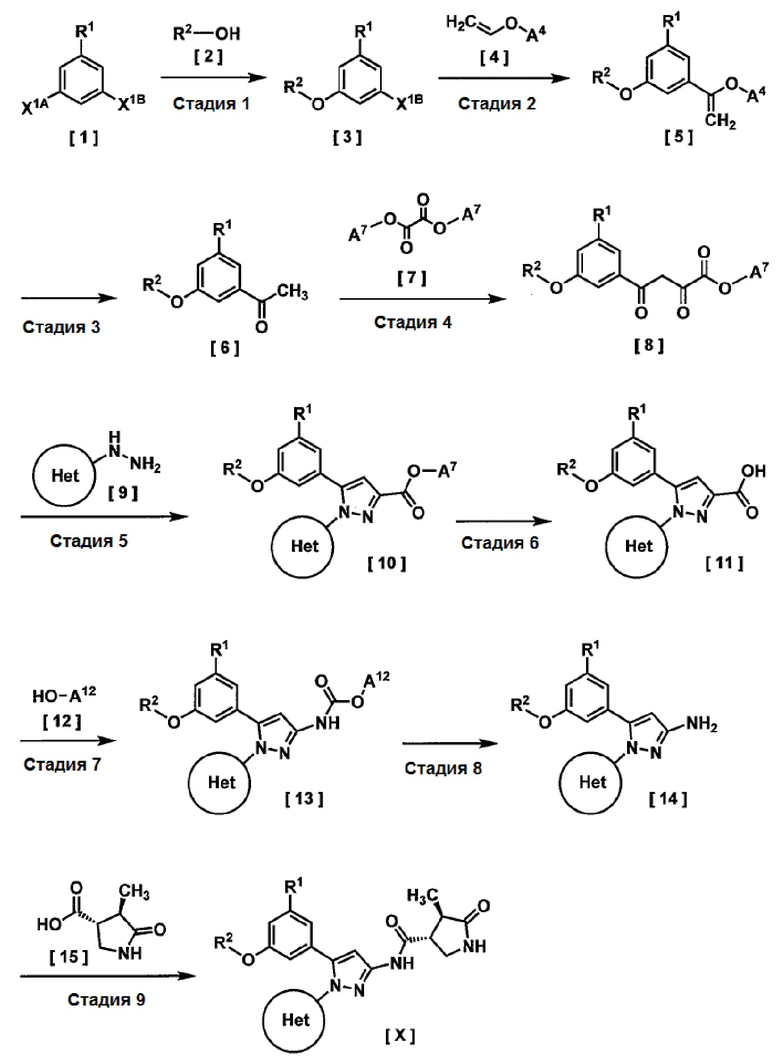
На этой схеме
R1, R2 и Кольцо Het имеют значения, указанные выше,
X1A и X1B каждый независимо представляет собой галоген, при условии, что X1A более реакционноспособен, чем X1B, на Стадии 1,
когда R1 представляет собой галоген, R1 предпочтительно представляет собой такой же галоген, что и X1A,
A4 представляет собой н-бутил,
A7 представляет собой C1-4 алкил или бензил, и
A12 представляет собой трет-бутил или бензил.
Стадия 1
Соединение, имеющее формулу [3], можно получить реакцией соединения, имеющего формулу [1], с соединением, имеющим формулу [2], в растворителе в присутствии основания.
Подходящий растворитель включает, например, простоэфирные растворители, такие как 1,2-диметоксиэтан; и полярные растворители, такие как N,N-диметилформамид, N-метилпирролидон, 1,3-диметил-2-имидазолидинон и N,N’-диметилпропиленмочевина. Предпочтительным растворителем в данном случае является 1,3-диметил-2-имидазолидинон.
Подходящее основание включает, например, карбонат цезия и гидрид натрия. Предпочтительным основанием в данном случае является гидрид натрия.
Температура реакции в данном случае находится в диапазоне, например, от 60°C до 170°C, предпочтительно от 100°C до 140°C.
Соединения, имеющие Формулы [1] и [2], оба могут быть коммерчески доступны или получены известными методами.
В альтернативном варианте, когда R2 представляет собой трифторметил, соединение, имеющее формулу [3], может быть коммерчески доступно.
Стадия 2
Соединение, имеющее формулу [5], можно получить по реакции Мизороки-Хека между соединением, имеющим формулу [3], и соединением, имеющим формулу [4]. Например, соединение, имеющее формулу [5], можно получить реакцией соединения, имеющего формулу [3], с соединением, имеющим формулу [4], в растворителе в присутствии палладиевого катализатора и основания.
Подходящий растворитель включает, например, спиртовые растворители, такие как этиленгликоль; и полярные растворители, такие как N,N-диметилформамид. Предпочтительным растворителем в данном случае является этиленгликоль.
Подходящий палладиевый катализатор включает, например, смесь ацетата палладия(II) с 1,1’-бис(дифенилфосфино)ферроценом или 1,3-бис(дифенилфосфино)пропаном. Предпочтительным палладиевым катализатором в данном случае является смесь ацетата палладия(II) с 1,1’-бис(дифенилфосфино)ферроценом.
Подходящее основание включает, например, органические основания, такие как триэтиламин. Предпочтительным основанием в данном случае является триэтиламин.
Температура реакции в данном случае находится в диапазоне, например, от 80°C до 150°C, предпочтительно от 100°C до 140°C.
Соединение, имеющее формулу [4], может быть коммерчески доступно или получено по известным методикам.
Стадия 3
Соединение, имеющее формулу [6], можно получить превращением группы “-C(=CH2)OA4” в группу “-C(=O)CH3” в соединении, имеющем формулу [5]. Например, соединение, имеющее формулу [6], можно получить реакцией соединения, имеющего формулу [5], в растворителе в присутствии кислоты.
Подходящий растворитель включает, например, кетонные растворители, такие как ацетон; спиртовые растворители, такие как этиленгликоль; простоэфирные растворители, такие как тетрагидрофуран и 1,4-диоксан; галогенированные углеводороды, такие как дихлорметан; полярные растворители, такие как N,N-диметилформамид; воду; и смесь любых из перечисленных растворителей. Предпочтительным растворителем в данном случае является смесь тетрагидрофурана и воды.
Подходящая кислота включает, например, соляную кислоту и трифторуксусную кислоту. Предпочтительной кислотой в данном случае является соляная кислота.
Температура реакции в данном случае находится в диапазоне, например, от 20°C до 50°C, и предпочтительно представляет собой комнатную температуру.
Стадия 4
Соединение, имеющее формулу [8], можно получить реакцией соединения, имеющего формулу [6], с соединением, имеющим формулу [7], в растворителе в присутствии основания.
Подходящий растворитель включает, например, простоэфирные растворители, такие как тетрагидрофуран, диэтиловый эфир и 1,2-диметоксиэтан; спиртовые растворители, такие как метанол и этанол; углеводороды, такие как толуол; полярные растворители, такие как N,N-диметилформамид; и смесь любых из перечисленных растворителей. Предпочтительным растворителем в данном случае является тетрагидрофуран.
Подходящее основание включает, например, трет-бутоксид лития, трет-бутоксид натрия, трет-бутоксид калия, метоксид натрия, этоксид натрия, диизопропиламид лития, гексаметилдисилазан лития и гидрид натрия. Предпочтительным основанием в данном случае является трет-бутоксид лития.
Температура реакции в данном случае находится в диапазоне, например, от -78°C до 110°C, предпочтительно от 0°C до комнатной температуры.
Соединение, имеющее формулу [7], может быть коммерчески доступно или получено по известным методикам.
Стадия 5
Соединение, имеющее формулу [10], можно получить реакцией соединения, имеющего формулу [8], с соединением, имеющим формулу [9], в растворителе в присутствии кислоты.
Подходящий растворитель включает, например, простоэфирные растворители, такие как тетрагидрофуран; спиртовые растворители, такие как метанол и этанол; и углеводороды, такие как толуол.
Подходящая кислота включает, например, соляную кислоту, серную кислоту, уксусную кислоту, трифторуксусную кислоту и п-толуолсульфокислоту. Предпочтительной кислотой в данном случае является уксусная кислота. Эти кислоты могут в данном случае применяться также в качестве растворителя.
Температура реакции находится в диапазоне, например, от 20°C до 130°C, предпочтительно от 80°C до 110°C.
Соединение, имеющее формулу [9], может быть коммерчески доступно или получено по известным методикам, или его можно получить общим способом, описанным ниже.
Стадия 6
Соединение, имеющее формулу [11], можно получить элиминированием группы “-A7” из соединения, имеющего формулу [10]. Реакцию элиминирования можно проводить в подходящих условиях в зависимости от A7. Например, когда A7 представляет собой этил, соединение, имеющее формулу [11], можно получить реакцией соединения, имеющего формулу [10], в растворителе в присутствии основания.
Подходящий растворитель включает, например, спиртовые растворители, такие как метанол и этанол; простоэфирные растворители, такие как тетрагидрофуран; воду; и смесь любых из перечисленных растворителей. Предпочтительным растворителем в данном случае является смесь двух или больше растворителей, выбранных из группы, состоящей из метанола, тетрагидрофурана и воды.
Подходящее основание включает, например, гидроксид лития, гидроксид натрия и гидроксид калия. Предпочтительным основанием в данном случае является гидроксид натрия.
Температура реакции в данном случае находится в диапазоне, например, от 0°C до 100°C, предпочтительно от комнатной температуры до 40°C.
Стадия 7
Соединение, имеющее формулу [13], можно получить перегруппировкой Курциуса из соединения, имеющего формулу [11], и соединения, имеющего формулу [12]. Например, соединение, имеющее формулу [13], можно получить реакцией соединения, имеющего формулу [11], с азидирующим агентом в присутствии основания в растворителе, и затем реакцией с соединением, имеющим формулу [12].
Подходящий растворитель включает, например, простоэфирные растворители, такие как тетрагидрофуран и 1,4-диоксан; и углеводороды, такие как толуол. В альтернативном варианте соединение, имеющее формулу [12], в данном случае можно использовать в качестве растворителя. Предпочтительным растворителем в данном случае является толуол или смесь толуола с соединением, имеющим формулу [12].
Подходящий азидируюший агент включает, например, дифенилфосфорил.
Подходящее основание включает, например, органические основания, такие как триэтиламин и N,N-диизопропилэтиламин. Предпочтительным основанием в данном случае является триэтиламин.
Температура реакции в данном случае находится в диапазоне, например, от 65°C до 130°C, предпочтительно от 90°C до 110°C.
Соединение, имеющее формулу [12], может быть коммерчески доступно или получено по известным методикам.
Стадия 8
Соединение, имеющее формулу [14], можно получить элиминированием группы “-C(=O)OA12” из соединения, имеющего формулу [13], в растворителе. Реакцию элиминирования можно проводить в подходящих условиях в зависимости от A12. Например, когда A12 представляет собой трет-бутил, соединение, имеющее формулу [14], можно получить реакцией соединения, имеющего формулу [13], в растворителе в присутствии кислоты.
Подходящий растворитель включает, например, сложноэфирные растворители, такие как этилацетат; спиртовые растворители, такие как метанол и этанол; простоэфирные растворители, такие как тетрагидрофуран и 1,4-диоксан; галогенированные углеводороды, такие как дихлорметан; воду; и смесь любых из перечисленных растворителей. Предпочтительным растворителем в данном случае является 1,4-диоксан.
Подходящая кислота включает, например, соляную кислоту, серную кислоту и трифторуксусную кислоту. Предпочтительной кислотой в данном случае является соляная кислота. Эти кислоты в данном случае можно также использовать в качестве растворителя.
Температура реакции в данном случае находится в диапазоне, например, от 0°C до 60°C, предпочтительно от 0°C до комнатной температуры.
Стадия 9
Соединение, имеющее формулу [X], можно получить конденсацией соединения, имеющего формулу [14], с соединением, имеющим формулу [15], в растворителе.
Подходящий растворитель включает, например, галогенированные углеводороды, такие как хлороформ; простоэфирные растворители, такие как тетрагидрофуран; полярные растворители, такие как пиридин, ацетонитрил и N,N-диметилформамид; и смесь любых из перечисленных растворителей. Предпочтительным растворителем в данном случае является пиридин.
Конденсирующий реагент включает, например, дициклогексилкарбодиимид (ДЦК), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (WSC.HCl), диизопропилкарбодиимид, 1,1’-карбонилдиимидазол (CDI), O-(7-азабензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфат (HATU), {{[(1-циано-2-этокси-2-оксоэтилиден)амино]окси}-4-морфолинометилен}диметиламмония гексафторфосфат (COMU), 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид н-гидрат (DMT-MM), (бензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат (PyBOP), дифенилфосфорилазид и безводная пропилфосфоновая кислота. Предпочтительным конденсирующим агентом в данном случае является 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (WSC.HCl).
Температура реакции в данном случае находится в диапазоне, например, от 0°C до 100°C, и предпочтительно представляет собой комнатную температуру.
Соединение, имеющее формулу [15], можно получить, например, способом из описанного ниже Сравнительного Примера A.
Общий способ получения: Соединение, имеющее формулу [9]
Соединение, имеющее формулу [9], можно получить, например, следующим способом.
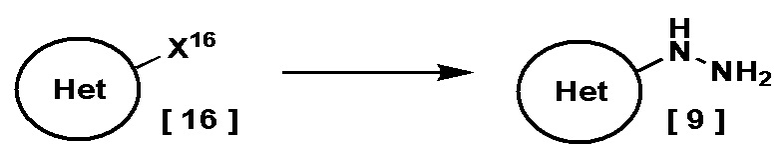
В этой схеме Кольцо Het имеет указанное выше значение, и X16 представляет собой галоген.
Соединение, имеющее формулу [9], можно получить реакцией соединения, имеющего формулу [16], с моногидратом гидразина в растворителе.
Подходящий растворитель включает, например, простоэфирные растворители, такие как тетрагидрофуран и 1,4-диоксан; спиртовые растворители, такие как этанол и 2-пропанол; галогенированные углеводороды, такие как дихлорметан; полярные растворители, такие как N,N-диметилформамид и пиридин; воду; и смесь любых из перечисленных растворителей. В альтернативном варианте моногидрат гидразина в данном случае можно использовать в качестве растворителя. Предпочтительным растворителем в данном случае является смесь 2-пропанола и моногидрата гидразина.
Температура реакции в данном случае находится в диапазоне, например, от комнатной температуры до 140°C, предпочтительно от 60°C до 100°C.
Соединение, имеющее формулу [16], может быть коммерчески доступно или получено по известным методикам.
Когда Кольцо Het представляет собой пиридил, замещенный R3, соединение, имеющее формулу [9] можно получить, например, в ходе следующего процесса.
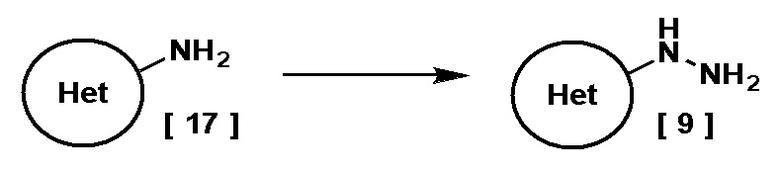
На этой схеме Кольцо Het представляет собой пиридил, замещенный R3, и R3 имеет указанное выше значение.
Соединение, имеющее формулу [9], можно получить диазотированием соединения, имеющего формулу [17], в присутствии кислоты в растворителе, с последующим восстановлением.
Подходящий растворитель включает, например, воду.
Подходящий диазотирующий агент включает, например, нитрит натрия.
Подходящая кислота включает, например, соляную кислоту и серную кислоту. Предпочтительной кислотой в данном случае является соляная кислота.
Подходящий восстановитель включает, например, хлорид олова (II) и сульфит натрия. Предпочтительным восстановителем в данном случае является хлорид олова (II).
Температура реакции диазотирования находится в диапазоне, например, от -20°C до 5°C, предпочтительно от -5°C до 0°C.
Температура реакции восстановления находится в диапазоне, например, от -5°C до комнатной температуры, предпочтительно от 0°C до комнатной температуры.
Соединение, имеющее формулу [17], может быть коммерчески доступно или получено по известным методикам.
В альтернативном варианте, когда Кольцо Het представляет собой (1) пиридил, замещенный R3, или (2) пиримидинил, необязательно замещенный R4, соединение, имеющее формулу [9], можно также получить, например, по следующей схеме.
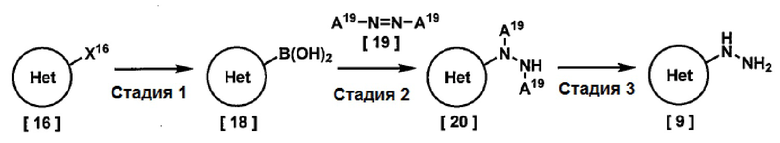
На этой схеме
Кольцо Het представляет собой (1) пиридил, замещенный R3, или (2) пиримидинил, необязательно замещенный R4,
R3, R4 и X16 имеют значения, указанные выше, и
A19 представляет собой трет-бутоксикарбонил или бензилоксикарбонил.
Стадия 1
Соединение, имеющее формулу [18], можно получить реакцией соединения, имеющего формулу [16], с основанием и эфиром бороновой кислоты в растворителе.
Подходящий растворитель включает, например, простоэфирные растворители, такие как тетрагидрофуран; углеводороды, такие как толуол; и смесь любых из перечисленных растворителей. Предпочтительным растворителем в данном случае является тетрагидрофуран.
Подходящее основание включает, например, н-бутиллитий и изопропилмагний бромид. Предпочтительным основанием в данном случае является н-бутиллитий.
Подходящий эфир бороновой кислоты включает, например, триизопропилборат и триметилборат. Предпочтительным эфиром бороновой кислоты в данном случае является триизопропилборат.
Температура реакции в данном случае находится в диапазоне, например, от -78°C до комнатной температуры, предпочтительно от -78°C до 0°C.
Соединение, имеющее формулу [16], может быть коммерчески доступно или получено по известным методикам.
Стадия 2
Соединение, имеющее формулу [20], можно получить реакцией соединения, имеющего формулу [18], с соединением, имеющим формулу [19], в присутствии медного катализатора в растворителе.
Подходящий растворитель включает, например, простоэфирные растворители, такие как тетрагидрофуран; и спиртовые растворители, такие как метанол. Предпочтительным растворителем в данном случае является метанол.
Подходящий медный катализатор включает, например, ацетат меди (II).
Температура реакции в данном случае находится в диапазоне, например, от комнатной температуры до 100°C, предпочтительно от 45°C до 65°C.
Стадия 3
Соединение, имеющее формулу [9], можно получить элиминированием групп “-A19” от соединения, имеющего формулу [20]. Реакцию элиминирования можно проводить в подходящих условиях, в зависимости от A19. Например, когда A19 представляет собой трет-бутоксикарбонил, соединение, имеющее формулу [9], можно получить реакцией соединения, имеющего формулу [20], в присутствии кислоты в растворителе.
Подходящий растворитель включает, например, сложноэфирные растворители, такие как этилацетат; спиртовые растворители, такие как метанол и этанол; простоэфирные растворители, такие как тетрагидрофуран и 1,4-диоксан; галогенированные углеводороды, такие как дихлорметан; воду; и смесь любых из перечисленных растворителей. Предпочтительным растворителем в данном случае является 1,4-диоксан.
Подходящая кислота включает, например, соляную кислоту, серную кислоту и трифторуксусную кислоту. Предпочтительной кислотой в данном случае является соляная кислота.
Температура реакции в данном случае находится в диапазоне, например, от 0°C до 60°C, предпочтительно от 0°C до комнатной температуры.
Примеры
Настоящее изобретение более подробно проиллюстрировано с привлечением описанных ниже Сравнительных Примеров, Примеров, Примеров Тестов и Примеров препаратов, но не ограничивается только ими.
Применяющиеся в настоящем документе аббревиатуры имеют указанные ниже значения.
ДМФА: N,N-диметилформамид
ДМСО: Диметилсульфоксид
ТГФ: Тетрагидрофуран
CPME: Циклопентилметиловый эфир
WSC.HCl: 1-Этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид
1H-ЯМР спектры регистрировали в CDCl3 или ДМСО-d6 с тетраметилсиланом в качестве внутреннего стандарта, и все значения δ приведены в миллионных долях. Измерения проводили на ЯМР спектрометре с рабочей частотой 400 МГц, если не указано иное.
Символы в Примерах имеют указанные ниже значения.
с: синглет
д: дублет
т: триплет
кв: квадруплет
дд: дублет дублетов
ддд: дублет дублетов дублетов
ушир.с: уширенный синглет
м: мультиплет
J: константа спин-спинового взаимодействия
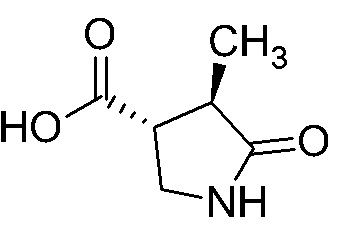
Сравнительный Пример A Получение (3R,4R)-4-метил-5-оксопирролидин-3-карбоновой кислоты
Стадия A-1 Получение диэтил 2-метил-3-метиленсукцината
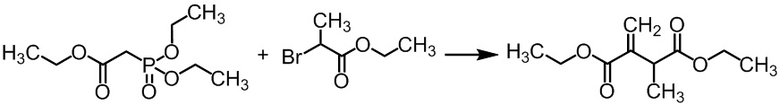
К трет-бутоксиду калия (180 г) добавляли ТГФ (2,55 л) при комнатной температуре в токе азота. В полученную смесь по каплям добавляли триэтил фосфоноацетат (314 г) при охлаждении льдом в течение 13 минут. Использовавшуюся капельную воронку промывали тетрагидрофураном (511 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали 2 часа 9 минут при охлаждении льдом. В смесь добавляли по каплям этил 2-бромпропионат (247 г) в течение 20 минут при охлаждении льдом. Использовавшуюся капельную воронку промывали тетрагидрофураном (79 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при комнатной температуре 22 часа 45 минут. В реакционную смесь добавляли карбонат калия (188 г) в течение 1 минуты при охлаждении льдом. В реакционную смесь добавляли по каплям 37%-ного водного раствора формальдегида (152 мл) в течение 10 минут при охлаждении льдом. Реакционную смесь перемешивали при комнатной температуре 19 часов 44 минут. В реакционную смесь добавляли воду (1,57 л) при комнатной температуре в течение 1 минуты. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа 48 минут. Реакционную смесь разделяли. Отделенный водный слой экстрагировали тетрагидрофураном (200 мл) два раза. Полученные органические слои объединяли и упаривали. В остаток добавляли толуол (471 мл) и насыщенный водный раствор хлорида натрия (471 мл). Реакционную смесь перемешивали и разделяли. Органический слой сушили над сульфатом натрия (63 г). Сульфат натрия отфильтровывали. Отдельно проводили такую же реакцию с триэтил фосфоноацетатом (300 г), получая фильтрат, который затем объединяли с полученным выше фильтратом, получая раствор указанного в заголовке соединения (эквивалент 2,66 моль) в толуоле (примерно 921 мл). Полученный раствор указанного в заголовке соединения в толуоле считали полученным с выходом 100% и использовали на следующей стадии. Образование указанного в заголовке соединения было подтверждено ВЭЖХ анализом.
Применялись следующие приборы и условия ВЭЖХ анализа:
Прибор: ВЭЖХ система от Shimadzu Corporation, High-Performance Liquid Chromatograph Prominence
Условия анализа:
Колонка: Kinetex C18: 2,6 мкм, 50 мм x 2,1 мм (Phenomenex)
Температура колонки: 40°C
Скорость потока: 0,4 мл/мин.
Время анализа: 10 мин.
Длина волны детектора: УФ (220 нм)
Подвижная фаза: (Раствор A) вода, (Раствор B) ацетонитрил.
Градиент подвижной фазы: Поддерживали соотношение (Раствор A/ Раствор B (об.%)) между Раствором A и Раствором B 80/20 с 0 минуты до 0,01 минуты после введения, линейно изменяли от 80/20 до 10/90 с 0,01 минуты до 7 минут, выдерживали 10/90 с 7 минут до 8 минут, линейно изменяли от 10/90 до 80/20 с 8 минут до 9 минут, и поддерживали соотношение 80/20 с 9 минут до 10 минут.
Время удерживания указанного в заголовке соединения составляло около 3,7 минут в применявшихся при ВЭЖХ-анализе условиях.
Стадия А-2 Получение смеси этил (цис)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоксилата и этил (транс)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоксилата
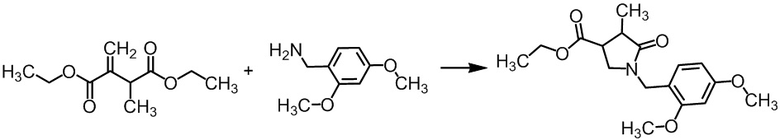
В раствор диэтил 2-метил-3-метиленсукцината (эквивалентно 2,66 моль), полученного на Стадии А-1, в толуоле (около 921 мл) добавляли по каплям 2,4-диметоксибензиламин (468 г) в течение 2 минут при комнатной температуре в токе азота. Реакционную смесь перемешивали при 120°C в течение 5 часов 45 минут. Реакционную смесь оставляли стоять на выходные при комнатной температуре. Реакционную смесь охлаждали льдом до внутренней температуры около 15°C. В реакционную смесь добавляли по каплям 2н. раствор соляной кислоты (1,33 л), и полученную смесь перемешивали. Реакционную смесь разделяли. Водный слой экстрагировали толуолом (150 мл). Органические слои объединяли, промывали смесью насыщенного водного раствора хлорида натрия и воды (600 мл, насыщенный водный раствор хлорида натрия/вода = 1/1), сушили над сульфатом натрия (120 г), упаривали, и сушили при пониженном давлении при комнатной температуре в течение ночи, получая сырое указанное в заголовке соединение (790 г; цис/транс = около 1/1, включая 5,5 мас.% толуола). Образование указанного в заголовке соединения было подтверждено методом ВЭЖХ-анализа.
Применялись следующие приборы и условия ВЭЖХ анализа:
Прибор: ВЭЖХ система от Shimadzu Corporation, High-Performance Liquid Chromatograph Prominence
Условия анализа:
Колонка: Atlantis T3: 5 мкм, 150 мм x 4,6 мм (Waters)
Температура колонки: 40°C
Скорость потока: 1,15 мл/мин.
Время анализа: 18 мин.
Длина волны детектора: УФ (220 нм)
Подвижная фаза: (Раствор A) 10 мМ (натрия) фосфатный буфер (pH = 2,6), (Раствор B) ацетонитрил.
Градиент подвижной фазы: Поддерживали соотношение (Раствор A/ Раствор B (об.%)) между Раствором A и Раствором B 60/40 с 0 минуты до 0,5 минуты после введения, линейно изменяли от 60/40 до 10/90 с 0,5 минуты до 8 минут, выдерживали 10/90 с 8 минут до 12,5 минут, линейно изменяли от 10/90 до 60/40 с 12,5 минут до 13,5 минут, и поддерживали соотношение 60/40 с 13,5 минут до 18 минут.
Время удерживания составляло около 6,6 минут для этил (цис)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоксилата и около 6,9 минут для этил (транс)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоксилата в применявшихся при ВЭЖХ-анализе условиях.
Стадия А-3 Получение (транс)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоновой кислоты
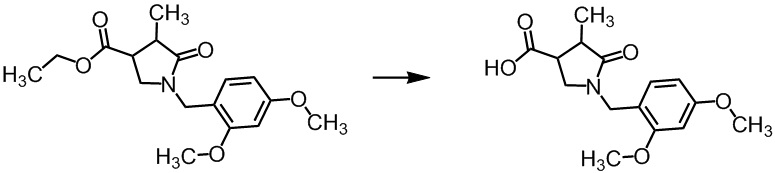
В неочищенную смесь (790 г, включая 5,5 мас.% толуола) этил (цис)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоксилата и этил (транс)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоксилата, полученную на Стадии А-2, добавляли этанол (1,15 л) при комнатной температуре в токе азота. В реакционную смесь добавляли по каплям этоксид натрия (20 мас.% раствор в этаноле, 1,15 л) при комнатной температуре в течение 31 минуты. Реакционную смесь перемешивали при комнатной температуре 2 часа 57 минут. Реакционную смесь охлаждали льдом и добавляли по каплям воду (1,84 л) в течение 33 минут. В реакционную смесь добавляли ЦПМЭ (1,8 л) и толуол (1,8 л) при комнатной температуре, и полученную смесь разделяли (органический слой 1). В водный слой добавляли ЦПМЭ (1,8 л), и полученную смесь разделяли (органический слой 2). Растворитель (1,8 л) удаляли из водного слоя упариванием. В водный слой добавляли по каплям 6н. раствор соляной кислоты (110 мл) при охлаждении льдом, и затем добавляли этилацетат (1,8 л). В полученную смесь добавляли по каплям 6н. раствор соляной кислоты (300 мл) при охлаждении льдом, и полученную смесь перемешивали около 10 минут. В смесь последовательно добавляли воду (2,2 л), 6н. раствор соляной кислоты (50 мл), воду (1,0 л), 10%-ный раствор гидросульфата натрия (300 мл) и этанол (300 мл) при охлаждении льдом. Смесь перемешивали при комнатной температуре в течение ночи. В полученную смесь добавляли этилацетат (600 мл), и полученную смесь разделяли. Водный слой экстрагировали этилацетатом (600 мл) два раза. Органические слои объединяли (за исключением органического слоя 1 и органического слоя 2) и промывали смесью насыщенного водного раствора хлорида натрия и воды (1 л, насыщенный водный раствор хлорида натрия/вода = 1/1). В полученный органический слой добавляли сульфат натрия (120 г) и активированный уголь (30 г), и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали через целит для удаления нерастворенных частиц. Нерастворенные частицы промывали этилацетатом (3 л). Фильтраты объединяли и упаривали, сушили при пониженном давлении при комнатной температуре в течение 3 часов, получая сырое указанное в заголовке соединение (561 г).
Отдельно объединяли и упаривали описанные выше органический слой 1 и органический слой 2. В остаток добавляли толуол (450 мл) и воду (450 мл), и полученную смесь разделяли. Водный слой промывали толуолом (450 мл) два раза. В водный слой добавляли этилацетат (450 мл). В полученную смесь добавляли по каплям 6н. раствор соляной кислоты (70 мл) при охлаждении льдом. В полученную смесь добавляли этилацетат (300 мл), и смесь разделяли. Водный слой экстрагировали этилацетатом (150 мл). Органические этилацетатные слои объединяли и промывали смесью насыщенного водного раствора хлорида натрия и воды (225 мл, насыщенный водный раствор хлорида натрия/вода = 1/1). В органический слой добавляли сульфат натрия (30 г) и активированный уголь (7,5 г), и смесь перемешивали при комнатной температуре в течение 1 часа. Полученную смесь фильтровали для удаления нерастворенных частиц. Нерастворенные частицы промывали этилацетатом (750 мл). Фильтраты объединяли и упаривали, и сушили при пониженном давлении при комнатной температуре в течение 3 часов, получая сырое указанное в заголовке соединение (87,3 г).
Этот сырой продукт объединяли с описанным выше сырым указанным в заголовке соединением, и затем добавляли ЦПМЭ (3 л) в токе азота. Смесь перемешивали при 120°C. Полученную смесь медленно охлаждали до комнатной температуры при перемешивании в течение 17 часов 34 минут. Смесь охлаждали льдом и перемешивали при внутренней температуре примерно 1°C в течение 3 часов. Осадок отфильтровывали и промывали холодным ЦПМЭ (900 мл). Осадок сушили при пониженном давлении при 50°C в течение ночи, получая указанное в заголовке соединение (585 г) с общим выходом 75% за 3 стадии. Формирование указанного в заголовке соединения было подтверждено методом ВЭЖХ анализа и ЯМР.
Прибор и условия проведения ВЭЖХ анализа такие же, как на Стадии А-2. Время удерживания указанного в заголовке соединения составляло около 3,1 минут в указанных условиях проведения ВЭЖХ анализа.
1H-ЯМР (CDCl3) δ: 1,33 (д, 3H, J = 6,5 Гц), 2,68-2,85 (м, 2H), 3,33-3,48 (м, 2H), 3,80 (с, 6H), 4,43 (с, 2H), 6,42-6,46 (м, 2H), 7,11-7,15 (м, 1H).
Стадия А-4 Получение диастереомерной соли (3R,4R)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоновой кислоты с (1R,2R)-(-)-2-амино-1-(4-нитрофенил)-1,3-пропандиолом
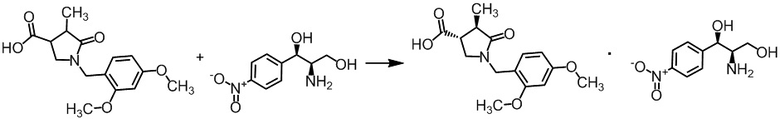
К (транс)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоновой кислоте (585 г), полученной на Стадии А-3, добавляли ацетонитрил (2,9 л) при комнатной температуре в токе азота. Смесь перемешивали при 85°C. В полученную смесь добавляли (1R,2R)-(-)-2-амино-1-(4-нитрофенил)-1,3-пропандиол (254 г) в течение 14 минут при 85°C. Реакционную смесь перемешивали при 90°C в течение 2 часов 48 минут. Реакционную смесь охлаждали до комнатной температуры при перемешивании в течение ночи. Осадок отфильтровывали и промывали ацетонитрилом (2,4 л). Осадок сушили при обычном давлении в течении 8,5 часов при комнатной температуре, получая неочищенные кристаллы указанного в заголовке соединения (516 г). К полученным неочищенным кристаллам добавляли ацетонитрил (2,5 л) и воду (0,5 л) при комнатной температуре в токе азота. Полученную смесь перемешивали при 100°C в течение 1 часа 14 минут. В смесь добавляли по каплям ацетонитрил (1,5 л) при t 100°C в течение 1 часа 7 минут. Смесь перемешивали при 100°C в течение 10 минут. Смесь охлаждали до комнатной температуры при перемешивании в течение 21 часа 10 минут. Смесь перемешивали в течение 3 часов 54 минут при охлаждении льдом. Осадок отделяли фильтрованием и промывали ацетонитрилом (1,5 л). Осадок сушили при атмосферном давлении при комнатной температуре 4 часа, получая указанное в заголовке соединение (448 г, 99,8% de) с выходом 45%. Формирование указанного в заголовке соединения было подтверждено методом ВЭЖХ анализа.
Применялись следующие приборы и условия ВЭЖХ анализа:
Прибор: ВЭЖХ система от Shimadzu Corporation, High-Performance Liquid Chromatograph Prominence
Условия анализа:
Колонка: CHIRAL PAK AD-3R: 3 мкм, 150 мм x 4,6 мм (Daicel)
Температура колонки: 40°C
Скорость потока: 0,50 мл/мин.
Время анализа: 10 мин.
Длина волны детектора: УФ (220 нм)
Подвижная фаза: (Раствор A) 10 мМ (натрия) фосфатный буфер (pH = 2,6), (Раствор B) ацетонитрил.
Режим подачи подвижной фазы: Поддерживали соотношение (Раствор A/ Раствор B (об.%)) между Раствором A и Раствором B 60/40.
Время удерживания составляло около 5,6 минут для (3R,4R)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоновой кислоты и около 6,5 минут для (3S,4S)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоновой кислоты в указанных условиях проведения ВЭЖХ анализа.
Конформацию указанного в заголовке соединения определяли методом рентгеновской кристаллографии на его монокристалле, полученном перекристаллизацией из метилизобутилкетона.
Диастереомерный избыток определяли по площадям ВЭЖХ пиков, и он составлял ((3R,4R)/(3S,4S) = 99,886%/0,114%).
Стадия А-5 Получение (3R,4R)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоновой кислоты
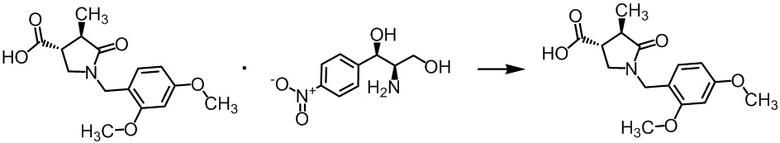
К диастереомерной соли (3R,4R)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоновой кислоты с (1R,2R)-(-)-2-амино-1-(4-нитрофенил)-1,3-пропандиолом (448 г), полученной на Стадии А-4, добавляли этилацетат (1,8 л) и воду (1,34 л) при комнатной температуре. В полученную смесь добавляли по каплям 6н. раствор соляной кислоты (168 мл) при комнатной температуре в течение 16 минут. Смесь разделяли. Водный слой экстрагировали этилацетатом (450 мл) три раза. Органические слои объединяли и промывали последовательно 2н. раствором соляной кислоты (224 мл) и насыщенным водным раствором хлорида натрия (224 мл), и затем сушили над сульфатом натрия (90 г) и упаривали. В остаток добавляли толуол (220 мл), и смесь упаривали. Остаток сушили при пониженном давлении при комнатной температуре, получая указанное в заголовке соединение (254 г) с выходом 98%.
1H-ЯМР (ДМСО-D6) δ: 1,15 (д, 3H, J = 7,2 Гц), 2,50-2,58 (м, 1H), 2,73-2,83 (м, 1H), 3,18-3,25 (м, 1H), 3,30-3,38 (м, 1H), 3,75 (с, 3H), 3,77 (с, 3H), 4,19-4,35 (м, 2H), 6,48 (дд, 1H, J = 8,4, 2,3 Гц), 6,56 (д, 1H, J = 2,3 Гц), 7,00 (д, 1H, J = 8,4 Гц), 12,61 (ушир.с, 1H).
Стадия A-6 Получение (3R,4R)-4-метил-5-оксопирролидин-3-карбоновой кислоты
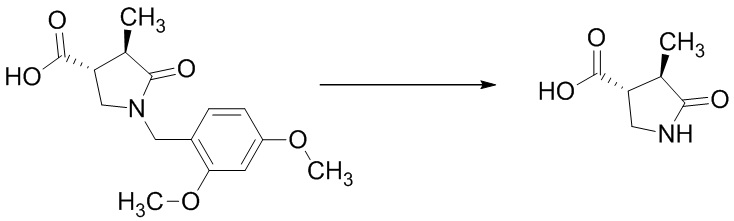
В смесь (3R,4R)-1-(2,4-диметоксибензил)-4-метил-5-оксопирролидин-3-карбоновой кислоты (254 г), полученной на Стадии A-5, и соединения (33 г), полученного по методике, аналогичной Стадии A-5, добавляли раствор анизола (160 мл) в трифторуксусной кислоте (1,44 л) при комнатной температуре в токе азота. Реакционную смесь перемешивали при 80°C в течение 4 часов 4 минут. Реакционную смесь охлаждали водой до комнатной температуры. Реакционную смесь упаривали. В остаток добавляли толуол (287 мл), и смесь упаривали. Остаток оставляли стоять при комнатной температуре на ночь. В остаток добавляли толуол (287 мл), и смесь упаривали. В остаток добавляли толуол (80 мл) при комнатной температуре. В смесь добавляли диизопропиловый эфир (2,9 л) при охлаждении водой. Смесь перемешивали при охлаждении водой. Выпавший из смеси осадок отделяли фильтрованием и промывали диизопропиловым эфиром (431 мл). Полученное твердое вещество сушили при атмосферном давлении при комнатной температуре, получая указанное в заголовке соединение (137 г) с выходом 98%.
1H-ЯМР (ДМСО-D6) δ: 1,10 (д, 3H, J = 7,2 Гц), 2,35-2,44 (м, 1H), 2,79-2,87 (м, 1H), 3,19-3,25 (м, 1H), 3,34-3,40 (м, 1H), 7,64 (с, 1H), 12,56 (с, 1H).
Сравнительный Пример B Получение 5-гидразинил-2-(трифторметил)пиримидина
Стадия B-1 Получение 5-гидразинил-2-(трифторметил)пиримидина
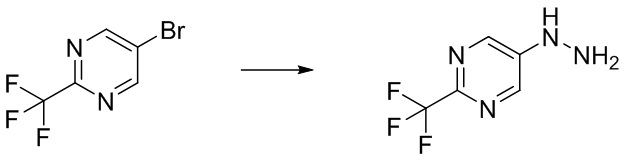
В 5-бром-2-(трифторметил)пиримидин (2 г) добавляли гидразин моногидрат (4,27 мл) и 2-пропанол (1 мл) в атмосфере аргона. Реакционную смесь перемешивали при 95°C в течение 22 часов с взрывозащитными экранами. Реакционную смесь охлаждали до комнатной температуры. В смесь добавляли воду и насыщенный водный раствор гидрокарбоната натрия, и смесь экстрагировали 5 раз этилацетатом. Полученные органические слои объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. В остаток добавляли смесь н-гексан/этилацетат (3/1) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре. Твердый осадок отфильтровывали из суспензии и промывали смесью н-гексан/этилацетат (3/1). Полученное твердое вещество сушили при пониженном давлении при комнатной температуре, получая указанное в заголовке соединение (647 мг) с выходом 41%.
1H-ЯМР (ДМСО-D6) δ: 4,43 (ушир.с, 2H), 7,94 (ушир.с, 1H), 8,33 (с, 2H).
Пример 1 Синтез (3R,4R)-N-(5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-ил)-4-метил-5-оксопирролидин-3-карбоксамида.
Стадия 1-1 Получение 1-бром-3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)бензола
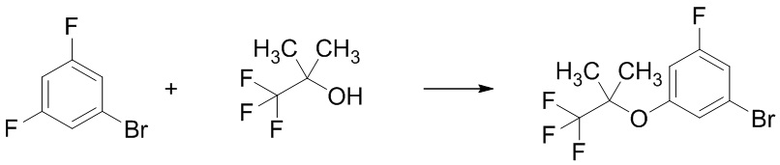
В раствор 1-бром-3,5-дифторбензола (5,97 мл) в 1,3-диметил-2-имидазолидиноне (10 мл) добавляли гидрид натрия (4,14 г) при комнатной температуре в токе азота. В смесь добавляли по каплям 1,1,1-трифтор-2-метилпропан-2-ол (8 мл) при охлаждении водой. В реакционную смесь добавляли 1,3-диметил-2-имидазолидинон (2 мл) при комнатной температуре. В реакционную смесь добавляли по каплям 1,1,1-трифтор-2-метилпропан-2-ол (3,16 мл) при комнатной температуре. Общее время прикапывания этих спиртов составило 45 минут. Реакционную смесь перемешивали при комнатной температуре в течение 20 минут, при 80°C в течение 20 минут, при 100°C в течение 20 минут, и при 130°C в течение 20 часов 40 минут. В реакционную смесь добавляли воду при охлаждении льдом. Смесь экстрагировали 3 раза н-гексаном. Полученные органические слои объединяли, промывали 3 раза водой, промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали при пониженном давлении 140 мм рт.ст. при 35°C. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 100/0 до 0/100), получая указанное в заголовке соединение (8,31 г; включая 12в ес.% н-гексана) с выходом 47%.
1H-ЯМР (ДМСО-D6) δ: 1,46 (с, 6H), 7,08 (дт, 1H, J = 10,2, 2,1 Гц), 7,18 (с, 1H), 7,39-7,45 (м, 1H).
Стадия 1-2 Получение 1-(1-бутоксивинил)-3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)бензола
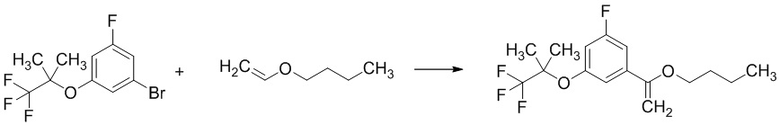
В раствор смеси 1-бром-3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)бензола (2,86 г; включая 12мас.% н-гексана), полученной на Стадии 1-1, и соединения, полученного по методике, аналогичной Стадии 1-1, в этиленгликоле(69 мл) добавляли бутилвиниловый эфир (19,77 мл), триэтиламин (10,65 мл), 1,1'-бис(дифенилфосфино)ферроцен (1,271 г) и ацетат палладия(II) (0,257 г) при комнатной температуре. Реакционную смесь перемешивали при 110°C в атмосфере аргона 19 часов. Реакционную смесь охлаждали до комнатной температуры. В смесь добавляли воду и н-гексан. Смесь фильтровали через целит. Фильтрат экстрагировали два раза н-гексаном. Полученные органические слои объединяли, промывали водой (два раза), насыщенным водным раствором хлорида натрия, сушили над сульфатом магния и упаривали при пониженном давлении 140 мм рт.ст. при 35°C. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 100/0 до 95/5), получая указанное в заголовке соединение (6,39 г; включая 15мас.% н-гексана) с выходом 44%.
1H-ЯМР (ДМСО-D6) δ: 0,95 (т, 3H, J = 7,3 Гц), 1,40-1,51 (м, 2H), 1,44 (с, 6H), 1,69-1,76 (м, 2H), 3,84 (т, 2H, J = 6,3 Гц), 4,39 (д, 1H, J = 3,0 Гц), 4,90 (д, 1H, J = 3,0 Гц), 6,96-7,01 (м, 1H), 7,12 (с, 1H), 7,24-7,29 (м, 1H).
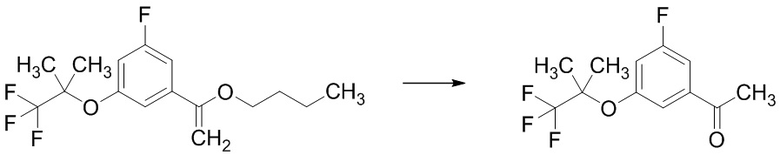
Стадия 1-3 Получение 1-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)этан-1-она
В раствор 1-(1-бутоксивинил)-3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)бензола (6,39 г; включая 15мас.% н-гексана), полученного на Стадии 1-2, в ТГФ (25 мл) добавляли 2н. раствор соляной кислоты (12,71 мл) при 0°C. Реакционную смесь перемешивали 1 час 10 минут при комнатной температуре. Реакционную смесь доводили до pH 12 2н. водного раствора гидроксида натрия при охлаждении льдом. Смесь экстрагировали два раза н-гексаном. Полученные органические слои объединяли и промывали два раза насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали при пониженном давлении 120 мм рт.ст. при 35°C. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 98/2 до 85/15), получая указанное в заголовке соединение (4,09 г; включая 6 мас.% н-гексана) с выходом 86%.
1H-ЯМР (ДМСО-D6) δ: 1,47 (с, 6H), 2,60 (с, 3H), 7,32 (дт, 1H, J = 9,7, 2,3 Гц), 7,42-7,43 (м, 1H), 7,58-7,62 (м, 1H).
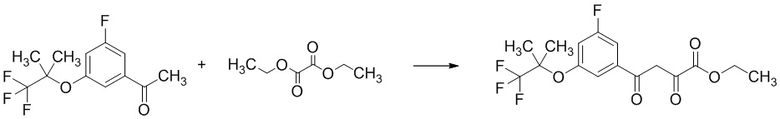
Стадия 1-4 Получение этил 4-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-2,4-диоксобутаноата
В раствор 1-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)этан-1-она (4,09 г; включая 6 мас.% н-гексана), полученного на Стадии 1-3, в ТГФ (38,4 мл) добавляли диэтилоксалат (2,171 мл) в атмосфере аргона. В смесь добавляли трет-бутоксид лития (1,396 г) при 0°C. Реакционную смесь перемешивали при 0°C в течение 3 часов 10 минут. Реакционную смесь доводили до pH 1 добавлением 1н. раствора соляной кислоты при охлаждении льдом. В смесь добавляли воду, и смесь экстрагировали два раза этилацетатом. Полученные органические слои промывали два раза насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Органический слой упаривали, получая указанное в заголовке соединение (5,53 г; включая 4 мас.% диэтилоксалата и 6 мас.% этилацетата) с выходом 94%.
1H-ЯМР (CDCl3) δ: 1,42 (т, 3H, J = 7,5 Гц), 1,50 (с, 6H), 4,42 (кв, 2H, J = 7,5 Гц), 6,97 (с, 1H), 7,01 (дт, 1H, J = 9,3, 2,2 Гц), 7,42-7,45 (м, 1H), 7,48 (дт, 1H, J = 8,8, 2,2 Гц), 15,02 (ушир.с, 1H).
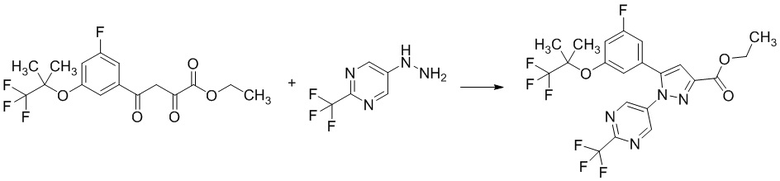
Стадия 1-5 Получение этил 5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-карбоксилата
В раствор этил 4-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-2,4-диоксобутаноата (500 мг; включая 4 мас.% диэтилоксалата и 6 мас.% этилацетата), полученного на Стадии 1-4, в уксусной кислоте (2,25 мл) добавляли 5-гидразинил-2-(трифторметил)пиримидин (242 мг), полученный на Стадии B-1, в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 21 часов и 30 минут. Реакционную смесь оставляли стоять при комнатной температуре на выходные. Реакционную смесь упаривали. Уксусную кислоту подвергали трехкратной азеотропной отгонке с толуолом. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 75/25 до 0/100), получая сырое указанное в заголовке соединение. В полученный сырой продукт добавляли смесь н-гексан/этилацетат (20/1) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре. Полученное твердое вещество выделяли из суспензии фильтрованием и промывали смесью н-гексан/этилацетат (20/1). Твердый продукт сушили при пониженном давлении при комнатной температуре, получая указанное в заголовке соединение (541 мг) с выходом 86%.
1H-ЯМР (ДМСО-D6) δ: 1,29 (с, 6H), 1,33 (т, 3H, J = 7,1 Гц), 4,38 (кв, 2H, J = 7,1 Гц), 6,83-6,84 (м, 1H), 7,13 (дт, 1H, J = 10,0, 2,3 Гц), 7,31-7,35 (м, 1H), 7,39 (с, 1H), 9,12 (с, 2H).
Стадия 1-6 Получение 5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-карбоновой кислоты.
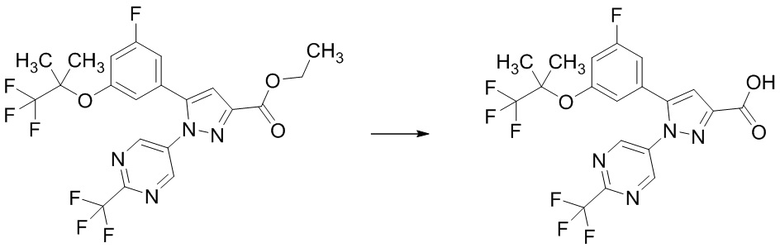
В раствор этил 5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-карбоксилата (541 мг), полученного на Стадии 1-5, в смеси ТГФ (1,623 мл)/метанол (3,246 мл) добавляли 2н. водный раствор гидроксида натрия (1,068 мл) при комнатной температуре. В реакционную смесь добавляли метанол (4 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре 25 часов 30 минут. Реакционную смесь доводили до pH 1 добавлением 1н. раствора соляной кислоты при охлаждении льдом. В смесь добавляли воду, и смесь экстрагировали два раза этилацетатом. Полученные органические слои объединяли, промывали два раза насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Органический слой упаривали, получая указанное в заголовке соединение (504 мг) с выходом 99%.
1H-ЯМР (ДМСО-D6) δ: 1,29 (с, 6H), 6,84 (с, 1H), 7,11-7,15 (м, 1H), 7,30-7,34 (м, 2H), 9,10 (с, 2H), 13,35 (ушир.с, 1H).
Стадия 1-7 Получение трет-бутил (5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-ил)карбамата
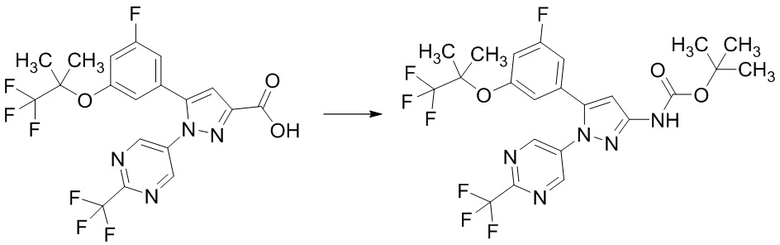
В смесь 5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-карбоновой кислоты (495 мг), полученной на Стадии 1-6, в толуоле (4,95 мл) добавляли триэтиламин (0,346 мл) и дифенилфосфорилазид (0,267 мл) в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре 1 час. В реакционную смесь добавляли трет-бутанол (4,26 мл) при комнатной температуре. Смесь перемешивали при 100°C в течение 27 часов 15 минут. Реакционную смесь упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 99/1 до 50/50), получая указанное в заголовке соединение (315 мг) с выходом 55%.
1H-ЯМР (ДМСО-D6) δ: 1,32 (с, 6H), 1,48 (с, 9H), 6,85 (с, 1H), 6,92 (с, 1H), 7,09-7,14 (м, 1H), 7,27-7,31 (м, 1H), 8,90 (с, 2H), 10,18 (ушир.с, 1H).
Стадия 1-8 Получение 5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-амина
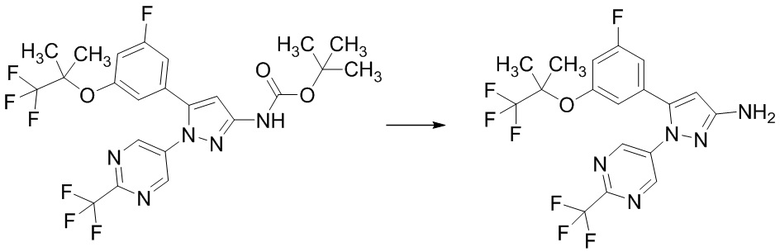
В трет-бутил (5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-ил)карбамат (315 мг), полученный на Стадии 1-7, добавляли 4н.раствор хлороводорода в 1,4-диоксане (1,575 мл) при 0°C в атмосфере аргона. Реакционную смесь перемешивали при 0°C в течение 10 минут и перемешивали при комнатной температуре 27 часов 40 минут. Реакционную смесь упаривали. В остаток добавляли насыщенный водный раствор гидрокарбоната натрия, и смесь экстрагировали два раза этилацетатом. Полученные органические слои объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 90/10 до 50/50), получая твердый продукт. В полученный твердый продукт добавляли смесь н-гексан/этилацетат (10/1) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре. Твердое вещество выделяли из суспензии фильтрованием и промывали смесью н-гексан/этилацетат (10/1). Твердый продукт сушили при пониженном давлении при комнатной температуре, получая указанное в заголовке соединение (224 мг) с выходом 87%.
1H-ЯМР (ДМСО-D6) δ: 1,34 (с, 6H), 5,50 (ушир.с, 2H), 6,11 (с, 1H), 6,82-6,85 (м, 1H), 7,10 (дт, 1H, J = 10,1, 2,3 Гц), 7,21-7,26 (м, 1H), 8,76 (с, 2H).
Стадия 1-9 Получение (3R,4R)-N-(5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-ил)-4-метил-5-оксопирролидин-3-карбоксамида
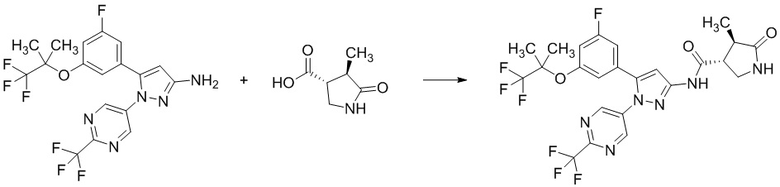
В раствор 5-(3-фтор-5-((1,1,1-трифтор-2-метилпропан-2-ил)окси)фенил)-1-(2-(трифторметил)пиримидин-5-ил)-1H-пиразол-3-амина (60 мг), полученного на Стадии 1-8, и (3R,4R)-4-метил-5-оксопирролидин-3-карбоновой кислоты (21,0 мг), полученной по методике, аналогичной Стадии A-6, в пиридине (1 мл) добавляли WSC.HCl (28,2 мг) в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре 29 часов. Реакционную смесь упаривали. В остаток добавляли воду, и смесь экстрагировали этилацетатом. Органический слой последовательно промывали 1н. раствором соляной кислоты (два раза), водой, насыщенным водным раствором гидрокарбоната натрия, насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. Остаток очищали методом тонкослойной хроматографии на силикагеле (элюент: этилацетат/метанол = 50/1), получая указанное в заголовке соединение (69 мг; включая 4мас.% этилацетата и 1мас.% н-гексана) с выходом 86%.
1H-ЯМР (ДМСО-D6) δ: 1,09 (д, 3H, J = 7,2 Гц), 1,32 (с, 6H), 2,50-2,59 (м, 1H), 3,03-3,11 (м, 1H), 3,20-3,27 (м, 1H), 3,43-3,50 (м, 1H), 6,85-6,87 (м, 1H), 7,13 (дт, 1H, J = 9,9, 2,3 Гц), 7,17 (с, 1H), 7,27-7,32 (м, 1H), 7,68 (с, 1H), 8,95 (с, 2H), 11,20 (ушир.с, 1H).
МС (M+H) 575, МС (M-H) 573.
Сравнительный Пример C Получение 3-гидразинил-5-(трифторметил)пиридина
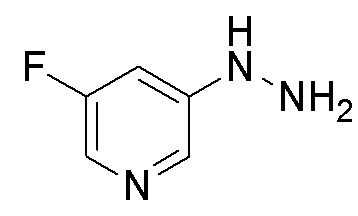
Стадия C-1 Получение 3-фтор-5-гидразинилпиридина
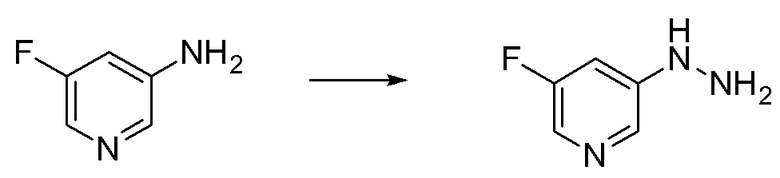
В раствор 5-фторпиридин-3-амина (1,5 г) в 6н. соляной кислоте (15 мл) добавляли раствор нитрита натрия (0,923 г) в воде (7,5 мл) по каплям в течение 2 минут при 0°C. Реакционную смесь перемешивали при 0°C 1 час 7 минут. В смесь добавляли суспензию хлорида олова(II) (6,34 г) в 6н. соляной кислоте (15 мл) по каплям в течение 3 минут при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 минут и при комнатной температуре 23 часа. В смесь добавляли по каплям 8н. водный раствор гидроксида натрия (примерно 34 мл) при 0°C. Смесь перемешивали при 0°C. Смесь экстрагировали 8 раз этилацетатом. Полученные органические слои объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. В остаток добавляли смесь метил-трет-бутиловый эфир (6 мл)/н-гексан (36 мл) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре. Твердое вещество выделяли из суспензии фильтрованием и промывали н-гексаном. Полученное твердое вещество сушили при пониженном давлении при 60°C, получая указанное в заголовке соединение (965,8 мг) с выходом 57%.
1H-ЯМР (CDCl3) δ: 3,64 (ушир.с, 2H), 5,41 (ушир.с, 1H), 6,99 (дт, 1H, J = 10,8, 2,5 Гц), 7,89 (д, 1H, J = 2,5 Гц), 7,97-7,99 (м, 1H).
Пример 2 Синтез (3R,4R)-N-(5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-ил)-4-метил-5-оксопирролидин-3-карбоксамида
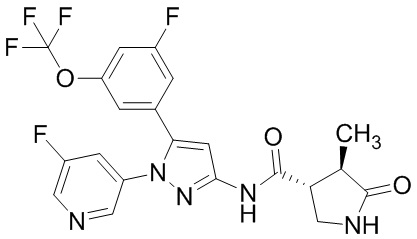
Стадия 2-1 Получение бензил 4-(3-фтор-5-(трифторметокси)фенил)-2,4-диоксобутаноата
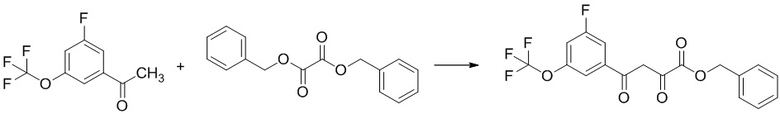
В раствор 1-(3-фтор-5-(трифторметокси)фенил)этан-1-она (5 г) и дибензилоксалата (6,69 г) в ТГФ (50 мл) добавляли трет-бутоксид лития (1,982 г) при охлаждении льдом в атмосфере аргона. Реакционную смесь перемешивали при охлаждении льдом 1 час. В смесь добавляли 2н. соляную кислоту (12,5 мл), этилацетат и воду при охлаждении льдом. Смесь разделяли. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Органический слой упаривали, получая сырое указанное в заголовке соединение (11,7 г).
Стадия 2-2 Получение бензил 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-карбоксилата
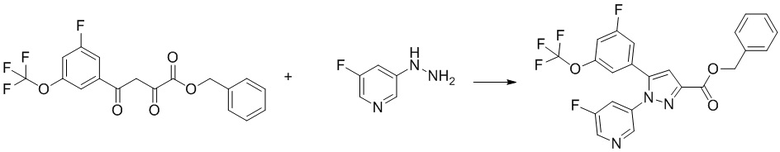
В раствор сырого (800 мг) бензил 4-(3-фтор-5-(трифторметокси)фенил)-2,4-диоксобутаноата, полученного на Стадии 2-1, в уксусной кислоте (6 мл) добавляли 3-фтор-5-гидразинилпиридин (218 мг), полученный на Стадии C-1, в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 19 часов 42 минут. Реакционную смесь охлаждали до комнатной температуры и упаривали. В остаток добавляли толуол, и смесь упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 90/10 до 69/31), получая указанное в заголовке соединение (589,5 мг) с выходом 79% за 2 стадии.
1H-ЯМР (CDCl3) δ: 5,44 (с, 2H), 6,83-6,86 (м, 1H), 6,93 (ддд, 1H, J = 8,4, 2,3, 1,6 Гц), 6,99-7,03 (м, 1H), 7,12 (с, 1H), 7,34-7,42 (м, 3H), 7,46-7,50 (м, 2H), 7,60 (ддд, 1H, J = 8,6, 2,5, 1,8 Гц), 8,32 (д, 1H, J = 1,8 Гц), 8,53 (д, 1H, J = 2,5 Гц).
Стадия 2-3 Получение 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-карбоновой кислоты
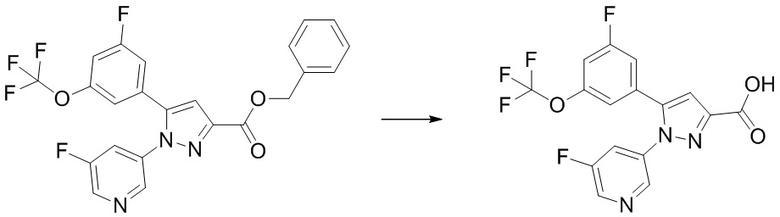
В раствор бензил 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-карбоксилата (589,5 мг), полученного на Стадии 2-2, в этилацетате (5,90 мл) добавляли 5 мас.% палладия на угле (88 мг) в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали в токе водорода (1 атм) при комнатной температуре 2 часа. После замены водородной атмосферы на азот, палладий на угле удаляли из реакционной смеси фильтрованием через целит. Использованный целит промывали смесью этилацетат/метанол (9/1). Фильтраты объединяли и упаривали. В остаток добавляли толуол, и смесь упаривали. Остаток сушили при пониженном давлении при комнатной температуре, получая указанное в заголовке соединение (425,9 мг) с выходом 89%.
1H-ЯМР (ДМСО-D6) δ: 7,06-7,09 (м, 1H), 7,33 (с, 1H), 7,45 (ддд, 1H, J = 9,2, 2,4, 1,5 Гц), 7,47-7,52 (м, 1H), 7,96 (ддд, 1H, J = 9,2, 2,5, 2,1 Гц), 8,44-8,47 (м, 1H), 8,73 (д, 1H, J = 2,5 Гц), 13,23 (ушир.с, 1H).
Стадия 2-4 Получение трет-бутил (5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-ил)карбамата
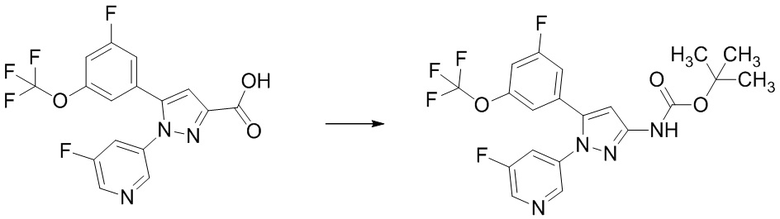
В раствор 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-карбоновой кислоты (425,9 мг), полученной на Стадии 2-3, и триэтиламина (0,370 мл) в смеси трет-бутанол (4,26 мл)/толуол (8,52 мл) добавляли дифенилфосфорилазид (0,286 мл) в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали при 110°C в течение 14 часов 50 минут. Реакционную смесь охлаждали до комнатной температуры и упаривали. В остаток добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. В остаток добавляли смесь н-гексан/этилацетат (1/1) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре. Нерастворенные частицы отфильтровывали и промывали смесью н-гексан/этилацетат (1/1). Фильтраты объединяли и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 90/10 до 69/31), получая указанное в заголовке соединение (207,4 мг) с выходом 41%.
1H-ЯМР (ДМСО-D6) δ: 1,48 (с, 9H), 6,90 (с, 1H), 7,06 (с, 1H), 7,40 (ддд, 1H, J = 9,1, 2,4, 1,5 Гц), 7,44-7,49 (м, 1H), 7,73 (ддд, 1H, J = 9,5, 2,5, 2,1 Гц), 8,32-8,34 (м, 1H), 8,61 (д, 1H, J = 2,3 Гц), 10,05 (ушир.с, 1H).
Стадия 2-5 Получение 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-амина
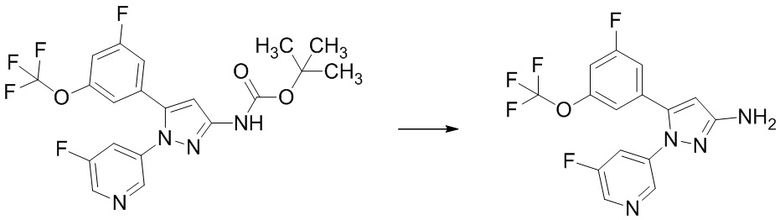
В трет-бутил (5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-ил)карбамат (207,4 мг), полученный на Стадии 2-4, добавляли трифторуксусную кислоту (2,07 мл) в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре 22 часа 40 минут. В смесь добавляли воду при 0°C. В смесь добавляли по каплям 8н. водный раствор гидроксида натрия (около 3,36 мл) при 0°C. В смесь добавляли насыщенный водный раствор гидрокарбоната натрия при 0°C. Смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 64/36 до 43/57), получая твердое вещество. К полученному твердому веществу добавляли н-гексан при комнатной температуре. Образовавшуюся суспензию перемешивали при комнатной температуре. Полученное твердое вещество выделяли из суспензии фильтрованием и промывали н-гексаном. Твердый продукт сушили при пониженном давлении при 60°C, получая указанное в заголовке соединение (100,0 мг; включая 0,21мас.% этилацетата) с выходом 62%.
1H-ЯМР (CDCl3) δ: 3,89 (ушир.с, 2H), 6,00 (с, 1H), 6,86-6,89 (м, 1H), 6,93 (ддд, 1H, J = 8,6, 2,3, 1,4 Гц), 6,96-7,00 (м, 1H), 7,43 (дт, 1H, J = 9,2, 2,5 Гц), 8,20-8,22 (м, 1H), 8,36 (д, 1H, J = 2,5 Гц).
Стадия 2-6 Получение ((3R,4R)-N-(5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-ил)-4-метил-5-оксопирролидин-3-карбоксамида
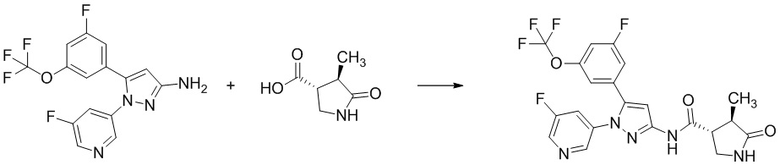
В раствор 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-амина (38 мг; включая 0,21мас.% этилацетата), полученного на Стадии 2-5, и (3R,4R)-4-метил-5-оксопирролидин-3-карбоновой кислоты (18,3 мг), полученной по методике, аналогичной Стадии A-6, в пиридине (0,380 мл) добавляли WSC.HCl (24,5 мг) в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре 2 часа 54 минуты. В реакционную смесь добавляли (3R,4R)-4-метил-5-оксопирролидин-3-карбоновую кислоту (18 мг), полученную по методике, аналогичной Стадии A-6, и WSC.HCl (25 мг) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи. В реакционную смесь добавляли 10 мас.% водный раствор лимонной кислоты при комнатной температуре, и смесь экстрагировали этилацетатом. Органический слой промывали водой, насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. Остаток очищали методом тонкослойной хроматографии на силикагеле (элюент: этилацетат/метанол = 97/3), получая указанное в заголовке соединение. К этому соединению добавляли смесь н-гексан/этилацетат при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре. Твердое вещество выделяли из суспензии фильтрованием и промывали н-гексаном. Твердый продукт сушили при пониженном давлении при 70°C, получая указанное в заголовке соединение (46,6 мг; включая 3,5 мас.% н-гексана) с выходом 87%.
1H-ЯМР (ДМСО-D6) δ: 1,09 (д, 3H, J = 7,4 Гц), 2,49-2,59 (м, 1H), 3,01-3,10 (м, 1H), 3,20-3,26 (м, 1H), 3,42-3,49 (м, 1H), 7,06-7,08 (м, 1H), 7,16 (с, 1H), 7,42 (ддд, 1H, J = 9,2, 2,3, 1,4 Гц), 7,46-7,51 (м, 1H), 7,68 (ушир.с, 1H), 7,77 (ддд, 1H, J = 9,7, 2,5, 2,1 Гц), 8,36-8,39 (м, 1H), 8,63 (д, 1H, J = 2,5 Гц), 11,10 (ушир.с, 1H).
МС (M+H) 482, МС (M-H) 480.
Стадия 2-7 Получение моногидрата ((3R,4R)-N-(5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-ил)-4-метил-5-оксопирролидин-3-карбоксамида
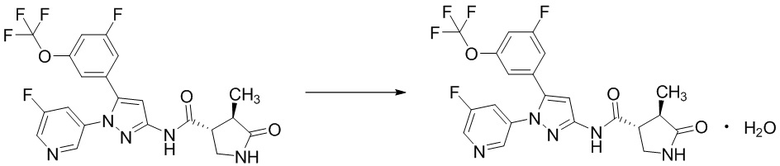
К ((3R,4R)-N-(5-(3-фтор-5-(трифторметокси)фенил)-1-(5-фторпиридин-3-ил)-1H-пиразол-3-ил)-4-метил-5-оксопирролидин-3-карбоксамиду (200 мг) добавляли этанол (0,6 мл), и смесь нагревали при 60°C до получения раствора. Раствор охлаждали до комнатной температуры. В раствор добавляли по каплям воду (1,2 мл) при комнатной температуре, и смесь перемешивали 4 часа. Выпавший осадок отделяли фильтрованием и промывали смесью этанол/вода (= 1/2). Твердый продукт сушили при пониженном давлении при 40°C, получая указанное в заголовке соединение (192 мг) с выходом 92%.
1H-ЯМР (ДМСО-D6) δ: 1,09 (д, 3H, J = 7,2 Гц), 2,48-2,60 (м, 1H), 3,00-3,10 (м, 1H), 3,20-3,27 (м, 1H), 3,41-3,49 (м, 1H), 7,05-7,09 (м, 1H), 7,16 (с, 1H), 7,42 (ддд, 1H, J = 9,2, 2,3, 1,4 Гц), 7,47-7,52 (м, 1H), 7,69 (ушир.с, 1H), 7,77 (ддд, 1H, J = 9,6, 2,3, 2,1 Гц), 8,36-8,40 (м, 1H), 8,64 (д, 1H, J = 2,3 Гц), 11,11 (ушир.с, 1H).
Элементный анализ
Вычислено: C 50,51 мас.%, H 3,63 мас.%, N 14,02 мас.%.
Найдено: C 50,61 мас.%, H 3,46 мас.%, N 13,95 мас.%.
Сравнительный Пример D Получение 3-гидразинил-5-(трифторметил)пиридина
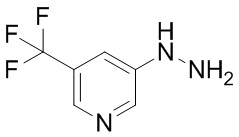
Стадия D-1 Получение 3-гидразинил-5-(трифторметил)пиридина
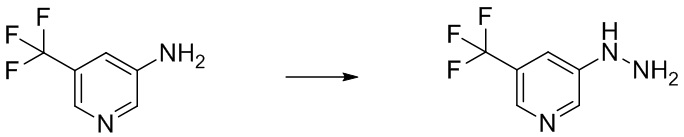
В раствор 5-(трифторметил)пиридин-3-амина (3 г) в 6н. растворе соляной кислоты (30 мл) по каплям добавляли раствор нитрита натрия (1,277 г) в воде (15 мл) в течение 2 минут при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа. В реакционную смесь по каплям добавляли суспензию хлорида олова(II) (8,77 г) в 6н. растворе соляной кислоты (30 мл) в течение 3 минут при 0°C. Реакционную смесь перемешивали при 0°C в течение 28 минут и при комнатной температуре 20 часов 9 минут. В реакционную смесь по каплям добавляли 8н. раствор гидроксида натрия (около 68 мл) при 0°C. Смесь перемешивали при 0°C. Смесь экстрагировали три раза этилацетатом. Полученные органические слои объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. В остаток добавляли затравочный кристалл указанного в заголовке соединения, отдельно полученного по аналогичной методике. В смесь добавляли смесь диизопропилового эфира (2 мл) и н-гексана (30 мл) при комнатной температуре. Образовавшуюся суспензию перемешивали при комнатной температуре. Твердое вещество выделяли из суспензии фильтрованием и промывали н-гексаном. Полученное твердое вещество сушили при пониженном давлении при комнатной температуре, получая указанное в заголовке соединение (2,8464 г) с выходом 87%.
1H-ЯМР (CDCl3) δ: 3,69 (ушир.с, 2H), 5,49 (ушир.с, 1H), 7,43-7,45 (м, 1H), 8,28-8,30 (м, 1H), 8,34 (д, 1H, J = 2,8 Гц).
Затравочный кристалл целевого соединения, использовавшийся на Стадии D-1, получали очисткой остатка, полученного по методике, аналогичной Стадии D-1, методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = 1/1).
Пример 3 Синтез ((3R,4R)-N-(5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил)пиридин-3-ил)-1H-пиразол-3-ил)-4-метил-5-оксопирролидин-3-карбоксамида
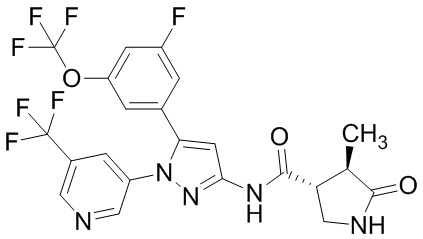
Стадия 3-1 Получение бензил 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил)пиридин-3-ил)-1H-пиразол-3-карбоксилата
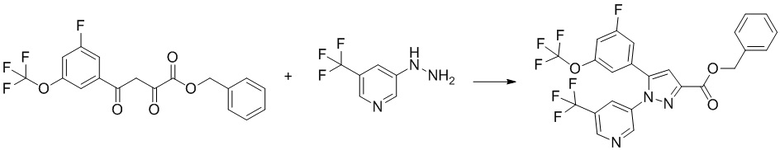
В раствор сырого (800 мг) бензил 4-(3-фтор-5-(трифторметокси)фенил)-2,4-диоксобутаноата, полученного на Стадии 2-1, в уксусной кислоте (6 мл) добавляли 3-гидразинил-5-(трифторметил)пиридин (304 мг), полученный на Стадии D-1, в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 22 часов 30 минут. Реакционную смесь охлаждали до комнатной температуры и упаривали. В остаток добавляли толуол и упаривали. Эту процедуру повторяли. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 97/3 до 70/30), получая указанное в заголовке соединение (640 мг) с выходом 78% за 2 стадии.
1H-ЯМР (CDCl3) δ: 5,45 (с, 2H), 6,80-6,83 (м, 1H), 6,94 (ддд, 1H, J = 8,3, 2,3, 1,6 Гц), 7,00-7,05 (м, 1H), 7,14 (с, 1H), 7,33-7,42 (м, 3H), 7,46-7,50 (м, 2H), 8,04-8,07 (м, 1H), 8,69 (д, 1H, J = 2,5 Гц), 8,88-8,92 (м, 1H).
Стадия 3-2 Получение 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил)пиридин-3-ил)-1H-пиразол-3-карбоновой кислоты
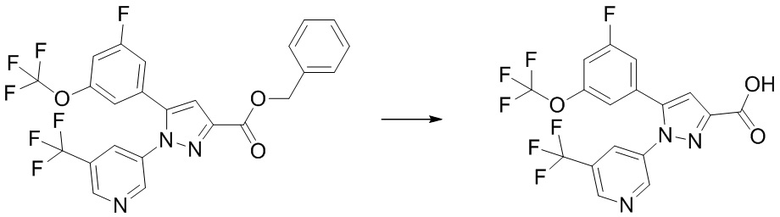
В раствор бензил 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил) пиридин-3-ил)-1H-пиразол-3-карбоксилата (640 мг), полученного на Стадии 3-1, в этилацетате (6,4 мл) добавляли 5 мас.% палладия на угле (32 мг) при комнатной температуре. Реакционную смесь перемешивали в атмосфере водорода (1 атм) 2 часа. После замены водородной атмосферы на азот, добавляли в реакционную смесь ТГФ. Палладий на угле удаляли из реакционной смеси фильтрованием через целит. Использовавшийся целит промывали тетрагидрофураном. Фильтраты объединяли и упаривали. В остаток добавляли н-гексан, и смесь упаривали. Эту процедуру повторяли. Остаток сушили при пониженном давлении при комнатной температуре, получая неочищенное указанное в заголовке соединение (525 мг).
Стадия 3-3 Получение трет-бутил (5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил)пиридин-3-ил)-1H-пиразол-3-ил)карбамата
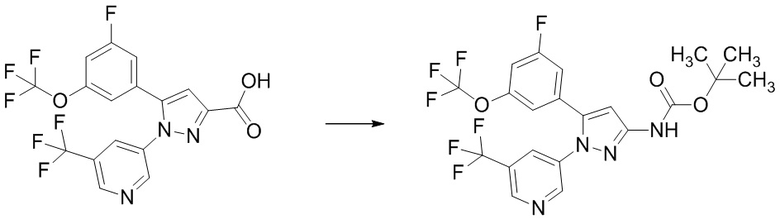
В раствор сырой (525 мг) 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил)пиридин-3-ил)-1H-пиразол-3-карбоновой кислоты, полученной на Стадии 3-2, и триэтиламина (0,403 мл) в смеси трет-бутанол (5 мл)/толуол (10 мл) добавляли дифенилфосфорилазид (0,311 мл) в атмосфере аргона при комнатной температуре. Смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 97/3 до 70/30), получая указанное в заголовке соединение (420 мг) с выходом 68% за 2 стадии. Формирование целевого соединения подтверждалось тонкослойной хроматографией (элюент: н-гексан/этилацетат = 4/1, Rf = 0,46).
Стадия 3-4 Получение 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил)пиридин-3-ил)-1H-пиразол-3-амина
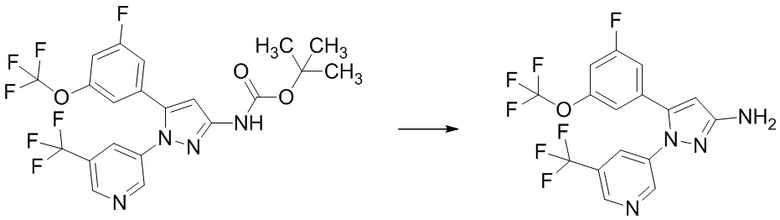
К трет-бутил (5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил)пиридин-3-ил)-1H-пиразол-3-ил)карбамату (420 мг), полученному на Стадии 3-3, добавляли трифторуксусную кислоту (3 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре 1 час 30 минут. Смесь упаривали. В остаток добавляли толуол, и смесь упаривали. Эту процедуру повторяли. В остаток добавляли этилацетат и насыщенный водный раствор гидрокарбоната натрия. Полученную смесь разделяли. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/этилацетат = от 92/8 до 20/80), получая указанное в заголовке соединение (313 мг) с выходом 93%.
1H-ЯМР (CDCl3) δ: 3,92 (ушир.с, 2H), 6,03 (с, 1H), 6,84-6,87 (м, 1H), 6,94 (ддд, 1H, J = 8,4, 2,2, 1,3 Гц), 6,97-7,02 (м, 1H), 7,87-7,90 (м, 1H), 8,57 (д, 1H, J = 2,4 Гц), 8,71-8,74 (м, 1H).
Стадия 3-5 Получение ((3R,4R)-N-(5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил)пиридин-3-ил)-1H-пиразол-3-ил)-4-метил-5-оксопирролидин-3-карбоксамида
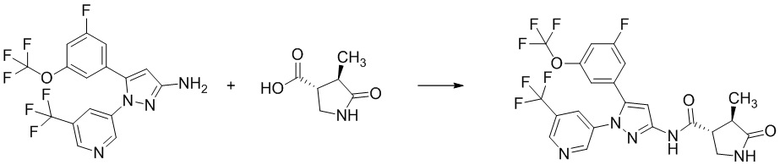
В раствор 5-(3-фтор-5-(трифторметокси)фенил)-1-(5-(трифторметил)пиридин-3-ил)-1H-пиразол-3-амина (60 мг), полученного на Стадии 3-4, и (3R,4R)-4-метил-5-оксопирролидин-3-карбоновой кислоты (23,3 мг), полученной по методике, аналогичной Стадии A-6, в пиридине (1 мл) добавляли WSC.HCl (31,1 мг) при комнатной температуре. Реакционную смесь перемешивали 15 часов 30 минут при комнатной температуре. В смесь добавляли воду и этилацетат при комнатной температуре. Полученную смесь разделяли. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали. В остаток добавляли толуол, и смесь упаривали. Эту процедуру повторяли. Остаток очищали методом тонкослойной хроматографии на силикагеле (элюент: этилацетат), получая указанное в заголовке соединение (75 мг) с выходом 96%.
1H-ЯМР (CDCl3) δ: 1,35 (д, 3H, J = 6,9 Гц), 2,85-2,95 (м, 1H), 2,99-3,09 (м, 1H), 3,55-3,68 (м, 2H), 6,51 (ушир.с, 1H), 6,87 (с, 1H), 6,96-7,06 (м, 2H), 7,23 (с, 1H), 7,80-7,85 (м, 1H), 8,73 (д, 1H, J = 2,3 Гц), 8,81-8,85 (м, 1H), 9,13 (ушир.с, 1H).
МС (M+H) 532, МС (M-H) 530.
Другие примеры соединений получали по методикам, аналогичным описанным выше общим способам получения и примерам и, при необходимости, другими способами. В следующих далее таблицах приведены структуры и физические характеристики соединений из Примеров 1 - 38.
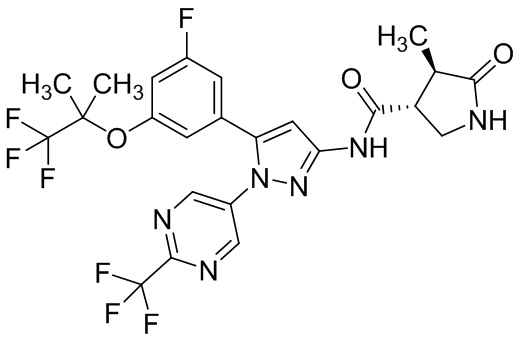
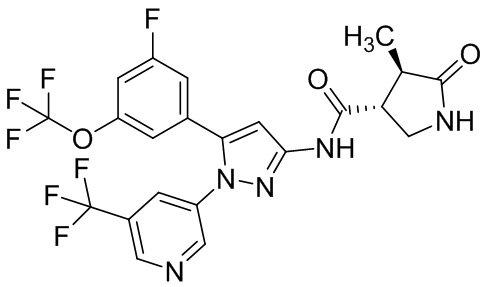
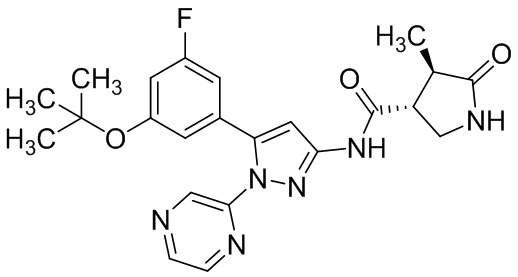
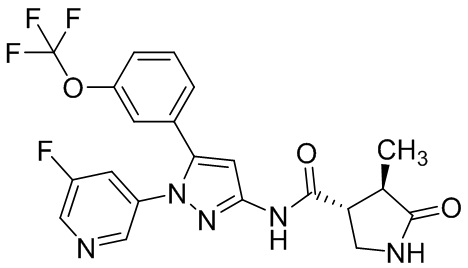
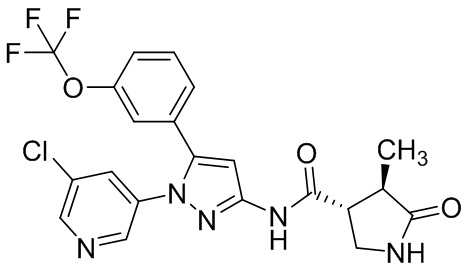
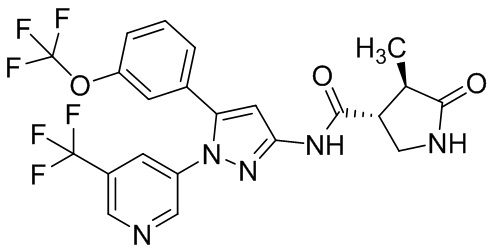
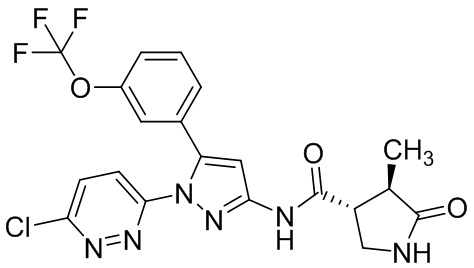
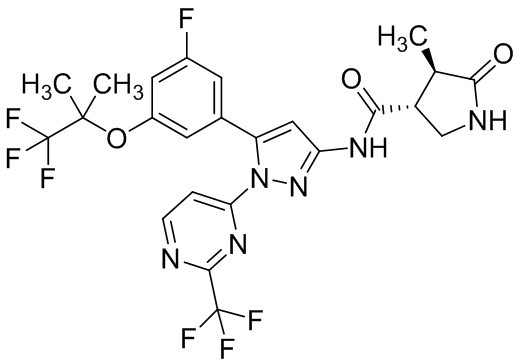
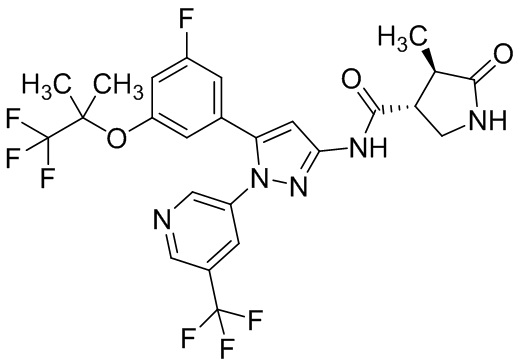
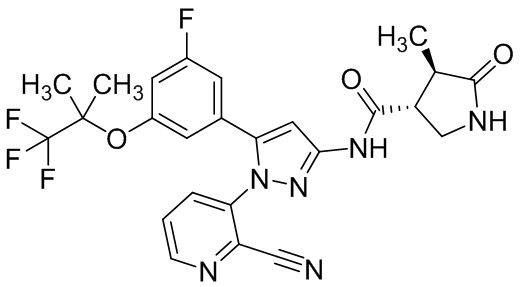
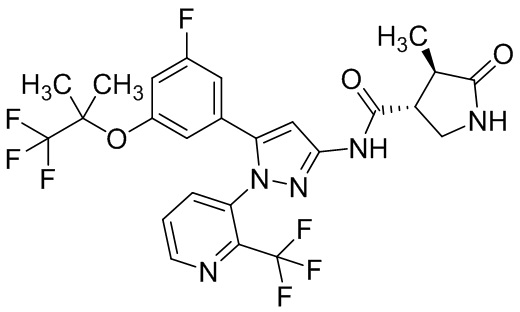
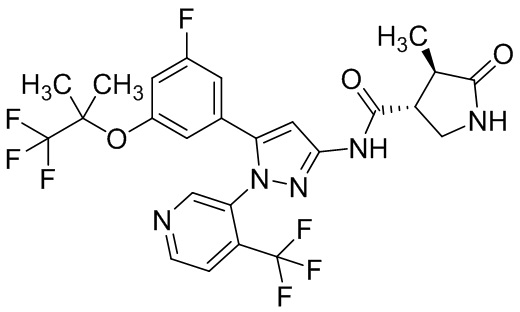
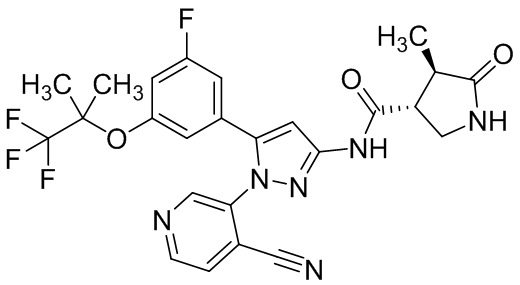
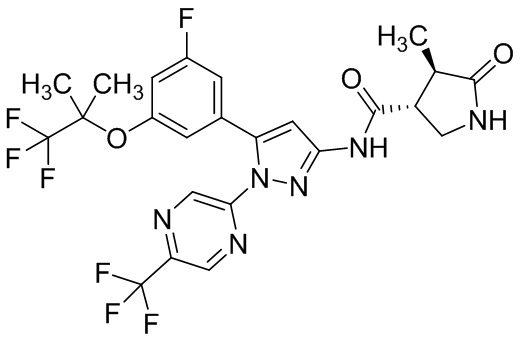
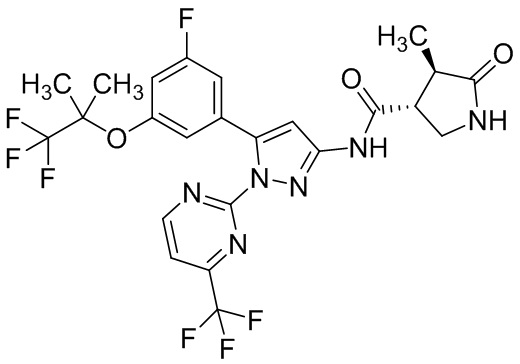
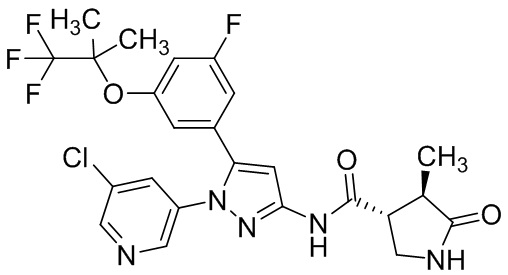
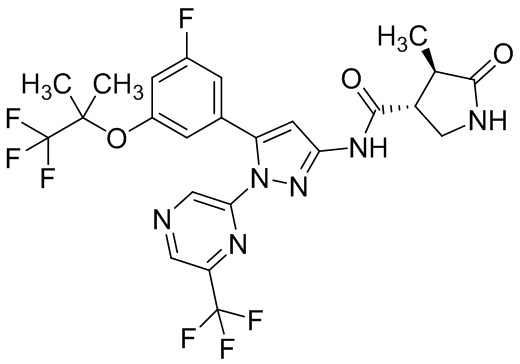
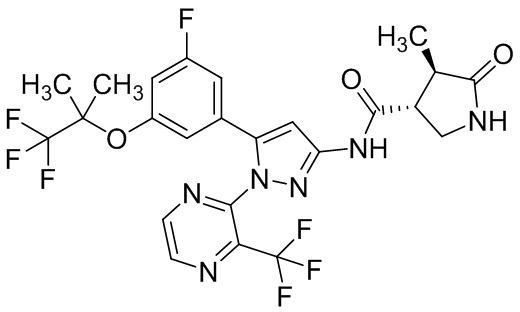
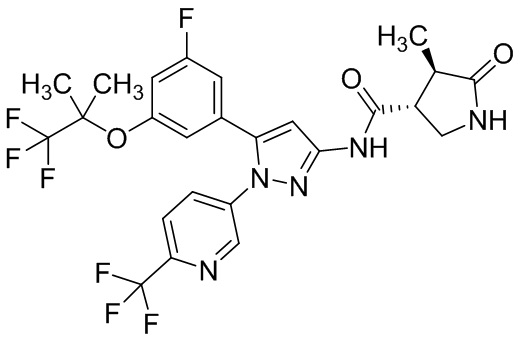
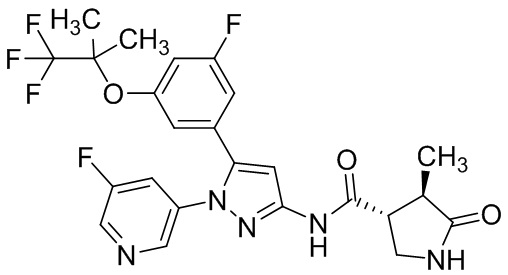
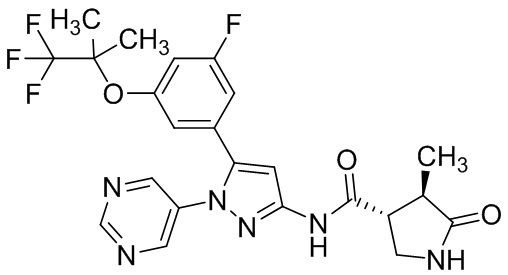
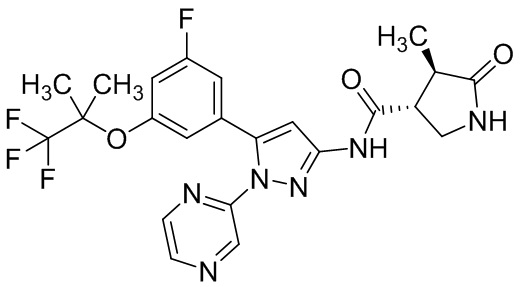
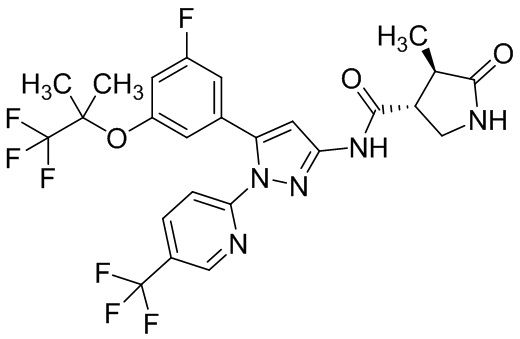
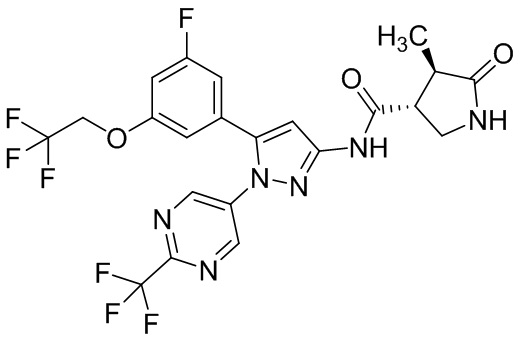
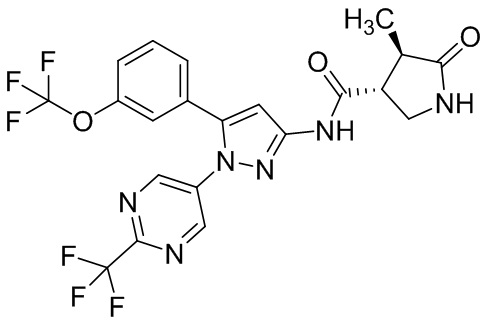
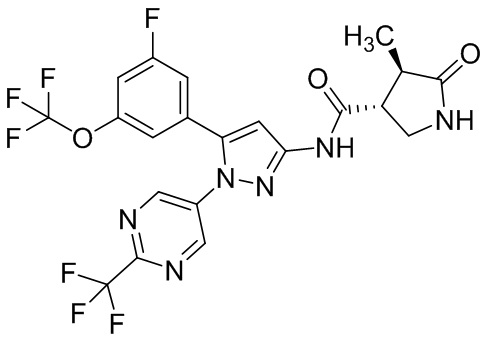
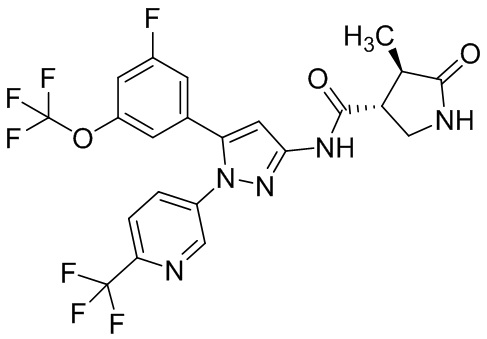
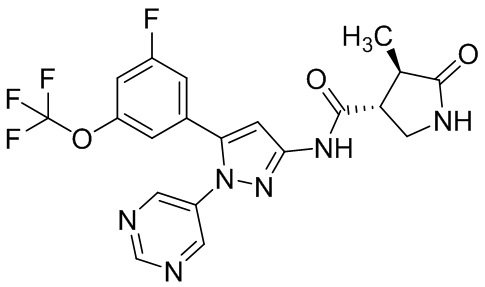
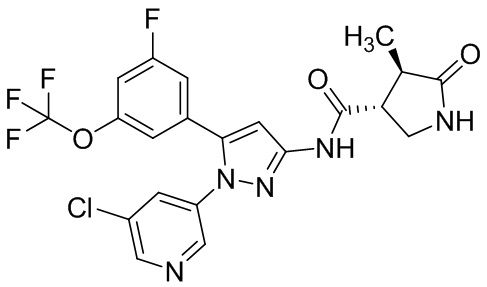
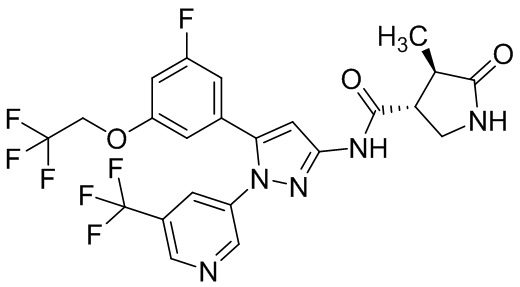
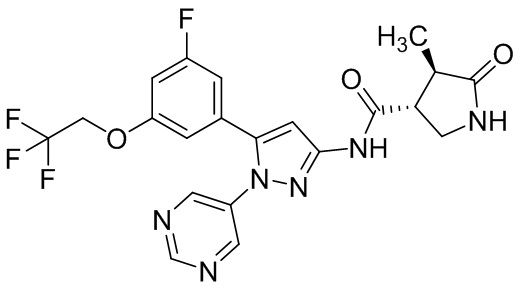
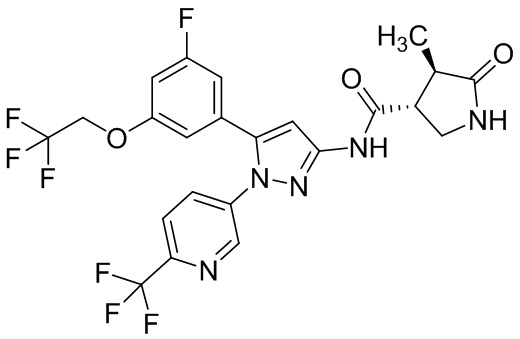
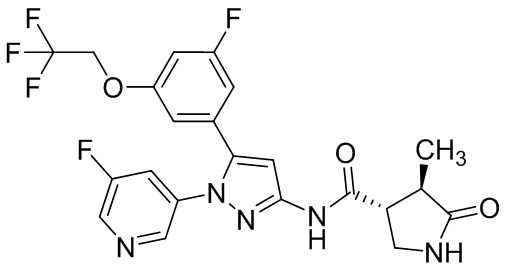
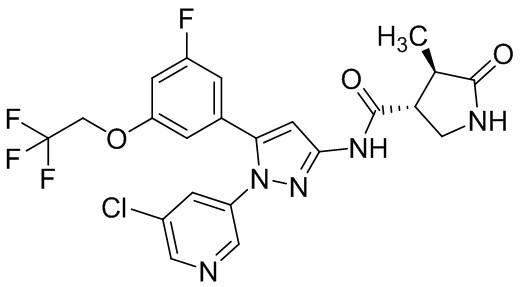
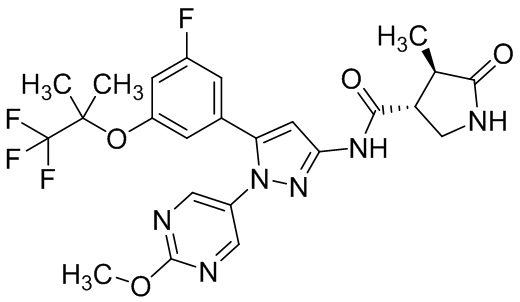
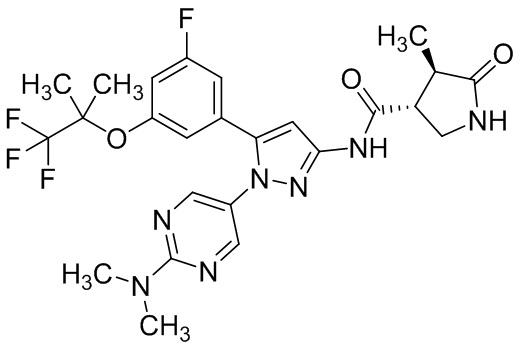
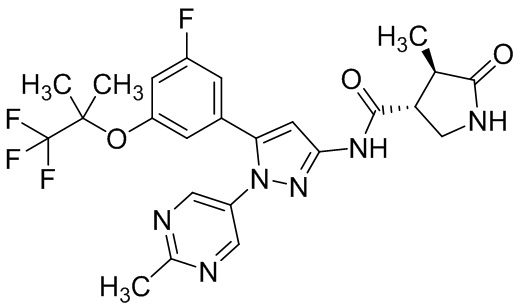
(M+H)
(M-H)
Изображенные ниже в таблице Соединения A - E были получены по методикам, описанным в WO 2013/031922.
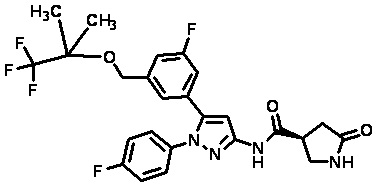
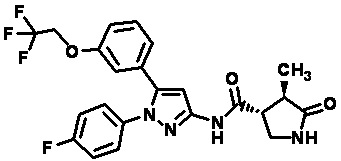
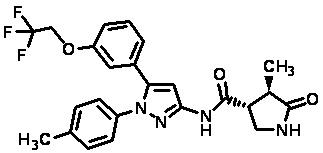
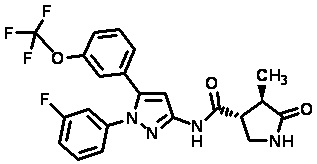
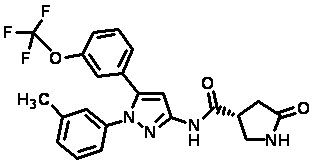
Метаболиты 1 - 3 (т.е., метаболиты соединений из Примеров 1 - 3) и Метаболиты A - E (т.е., метаболиты Соединений A - E), которые все изображены ниже в таблице, были получены согласно описанным выше Примерам 1 - 3 и методикам из WO 2013/031922.
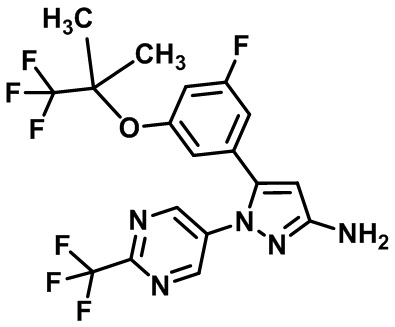
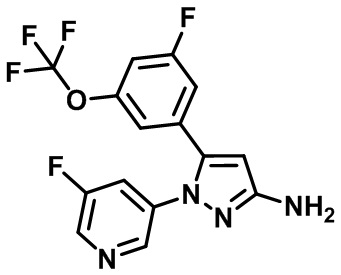
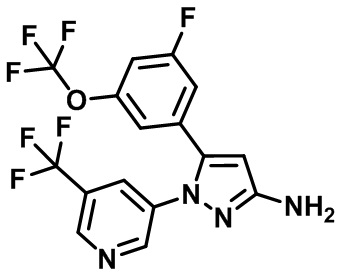
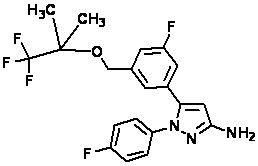
Пример Теста 1
Активность тестируемых соединений в качестве ингибиторов SGLT1 (значения IC50) рассчитывали по величине клеточного захвата меченого α-метил-D-глюкопиранозида (14C-AMG), переносимого SGLT1.
1) Формирование плазмиды, экспрессирующей человеческий SGLT1
Фрагмент ДНК, содержащий ген человеческого SGLT1, амплифицировали методом ПЦР (полимеразной цепной реакции) с использованием pCMV6-hSGLT1 (OriGene) в качестве темплата. В человеческий SGLT1 добавляли последовательность распознавания и расщепления NheI выше от консенсусной последовательности Kozac вектора, и добавляли стоп-кодон, TAG и последовательность распознавания и расщепления SalI сразу после белок-транслирующего участка человеческого SGLT1. Очищенный фрагмент ДНК расщепляли рестрикционными ферментами NheI и SalI, затем лигировали посредством pcDNA3,1 (+), который расщепляли с помощью NheI и XhoI, формируя тем самым экспрессирующую человеческий SGLT1 плазмиду, pcDNA-hSGLT1. Последовательность нуклеиновых кислот человеческого SGLT1, вставленная в вектор, была полностью идентична белок-транслирующему участку последовательности человеческого SGLT1 (Accession number NM_000343), зарегистрированному в GenBank, и последовательность участка, соединенного с вектором, была такой как ожидалось.
2) Формирование линий клеток, устойчиво экспрессирующих человеческий SGLT1
Плазмиду, экспрессирующую человеческий SGLT, pcDNA-hSGLT1, трансфицировали в каждую клетку CHO-K1 с применением Lipofectamine 2000 (Invitrogen) и выращивали в присутствии G418 (Nacalai Tesque), чтобы отобрать линии лекарственноустойчивых клеток. Линию клеток, имеющую наивысшее соотношение (соотношение S/B) величины клеточного захвата 14C-AMG на клетку к величине клеточного захвата 14C-AMG после воздействия ингибитора SGLT, флоризина, выбирали в качестве линии клеток, устойчиво экспрессирующей человеческий SGLT1, из числа линий лекарственноустойчивых клеток.
3) Оценка SGLT1-ингибирующей активности
Клетки, устойчиво экспрессирующие человеческий SGLT1, высевали в количестве 5 x 104 клеток на лунку в BioCoat™ Poly-D-Lysine 96-луночные планшеты с крышкой (Becton, Dickinson и Company) и выращивали при 37°C в атмосфере с 5% CO2 в течение ночи. Среду заменяли на 100 мкл/лунку Na(-) буфера (140 мМ холин хлорид, 2 мМ KCl, 1 мМ MgCl2, 1 мМ CaCl2, 10 мМ HEPES, 5 мМ Tris, pH 7,4), и смесь оставляли при 37°C в атмосфере с 5% CO2 на 20 минут. После удаления Na(-) буфера, добавляли 40 мкл/лунку раствора тестируемого соединения, приготовленного в Na(+) буфере (140 мМ NaCl, 2 мМ KCl, 1 мМ MgCl2, 1 мМ CaCl2, 10 мМ HEPES, 5 мМ Tris, pH 7,4), содержащем БСА (бычий сывороточный альбумин). Затем добавляли 40 мкл/лунку Na(+) буфера, содержащего 8 кБк 14C-AMG и 2 мМ AMG, и полученную смесь хорошо перемешивали. В контрольном образце добавляли 40 мкл/лунку Na(-) буфера, содержащего БСА, и кроме того добавляли 40 мкл/лунку Na(-) буфера, содержащего 8 кБк 14C-AMG и 2 мМ AMG, и полученную смесь хорошо перемешивали. После инкубации в течение 1 часа при 37°C в атмосфере с 5% CO2, клетки промывали два раза по 100 мкл на лунку ледяного промывного буфера (100 мМ AMG, 140 мМ холин хлорид, 2 мМ KCl, 1 мМ MgCl2, 1 мМ CaCl2, 10 мМ HEPES, 5 мМ Tris, pH 7,4) для остановки реакции. Клеточный лизат готовили посредством добавления 50 мкл/лунку 0,2н. водного раствора NaOH. Для оценки способности поглощения 14C-AMG, все количество клеточного лизата переносили в OptiPlate 96 (Perkin-Elmer), диспендируя с 100 мкл/лунку MicroScint-40 (Perkin-Elmer), и измеряли CPM (число импульсов в минуту) 14C с помощью TOPCOUNT NXT (Perkin-Elmer).
Вычисляли результат посредством вычитания среднего значения CPM для контрольного образца из среднего значения CPM для каждой обработанной ячейки. Степень ингибирования для каждого протестированного соединения в каждой концентрации вычисляли по следующему уравнению:
[(A-B)/A] x 100
где A это результат для контрольного образца, а B это результат при обработке каждым тестируемым соединением.
Каждое значение IC50 (50%-ингибирующая концентрация) для каждого тестируемого соединения вычисляли по двум концентрациям выше и ниже 50%-ного ингибирования, и по значению ингибирования. Результаты, полученные для соединений из Примеров, приведены ниже в таблице.
Пример Теста 2
Тест Эймса (тест обратной мутации)
В этом тесте исследовали Метаболиты 1-3 и Метаболиты A-E. Целью этого теста была оценка потенциала каждого метаболита вызывать обратные мутации в стандартных линиях Salmonella typhimurium (TA98, TA1537, TA100 и TA1535) и Escherichia coli (WP2uvrA), в присутствии или в отсутствие системы метаболической активации печени крыс (S9 mix).
Растворитель, применяющийся в данном случае, представляет собой диметилсульфоксид (ДМСО, 100 мкл/лунку).
Тест проводили методом предварительной инкубации с S9 mix или без него. Когда тест проводили без S9 mix, добавляли натрий-фосфатный буферный раствор (pH 7,4).
0,5 мл S9 mix или 0,5 мл 0,1 моль/л натрий-фосфатного буферного раствора (pH 7,4), и 0,1 мл раствора бактериальной культуры добавляли в пробирку, содержащую 0,1 мл отрицательного контроля (только ДМСО), метаболита или положительного контроля. Смеси предварительно инкубировали при 37°C в течение 20 минут при встряхивании. После предварительной инкубации добавляли 2 мл агара для верхнего слоя, и смеси перемешивали на вихревой мешалке и распределяли в планшетах. Использовали два планшета на каждый испытуемый образец. Планшеты инкубировали при 37 ± 1°C в течение 48 часов или больше, и подсчитывали число ревертантных колоний. Вычисляли среднее число ревертантных колоний для каждого испытуемого образца. Наличие или отсутствие ингибирования роста вследствие антибактериального действия испытуемых образцов, а также выпадение осадка оценивали визуально или с помощью стереомикроскопа. Результаты оценивались как положительные, если среднее число ревертантных колоний показывало дозозависимый рост, достигавший 2-кратного в сравнении с отрицательным контролем, в одной или больше дозировках. Оценку проводили по средним значениям без применения статистического сравнения.
Результаты теста приведены в следующих далее таблицах (таблицы от 1-1 до 3-2 и таблицы от А-1 до Е-2). В заключение необходимо отметить, что Метаболиты 1-3 не обладают потенциалом вызывать обратные мутации ни у одной из протестированных линий бактерий, в то время как Метаболиты А-Е обладают потенциалом вызывать обратные мутации по меньшей мере у одной из протестированных линий бактерий в присутствии и/или в отсутствие S9 mix.
Метаболит A обладал потенциалом вызывать обратные мутации в протестированных линиях бактерий TA98 и TA1537 в присутствии S9 mix.
Метаболит B обладал потенциалом вызывать обратные мутации в протестированных линиях бактерий TA98, TA1537, TA100 и TA1535 в присутствии S9 mix и TA1537 без S9 mix.
Метаболит C обладал потенциалом вызывать обратные мутации в протестированных линиях бактерий TA98, TA1537 и TA100 в присутствии S9 mix и WP2uvrA без S9 mix.
Метаболит D обладал потенциалом вызывать обратные мутации в протестированных линиях бактерий TA100 в присутствии S9 mix и TA1535 без S9 mix.
Метаболит E обладал потенциалом вызывать обратные мутации в протестированных линиях бактерий TA98, TA1537 и TA100 в присутствии S9 mix.
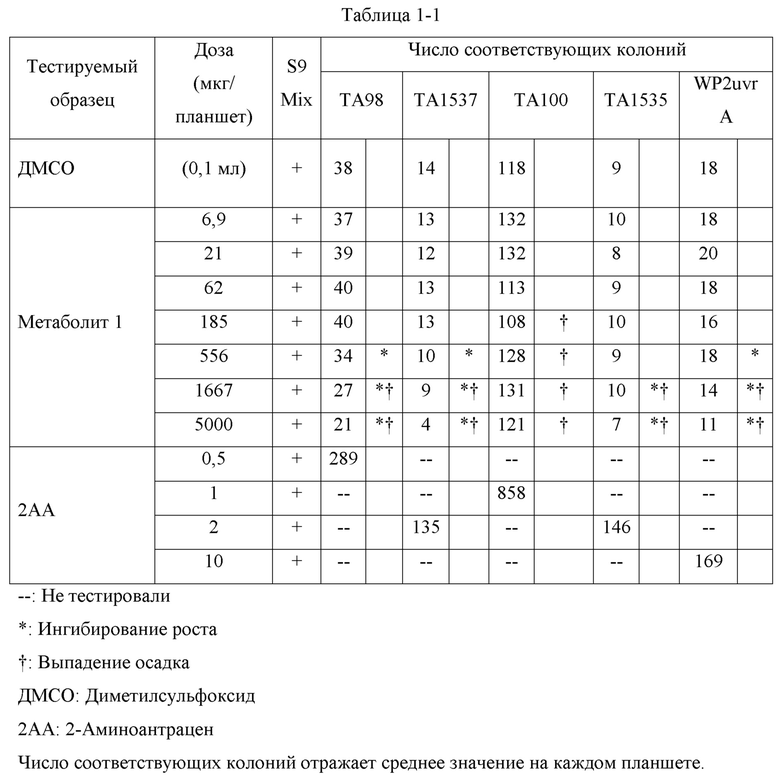
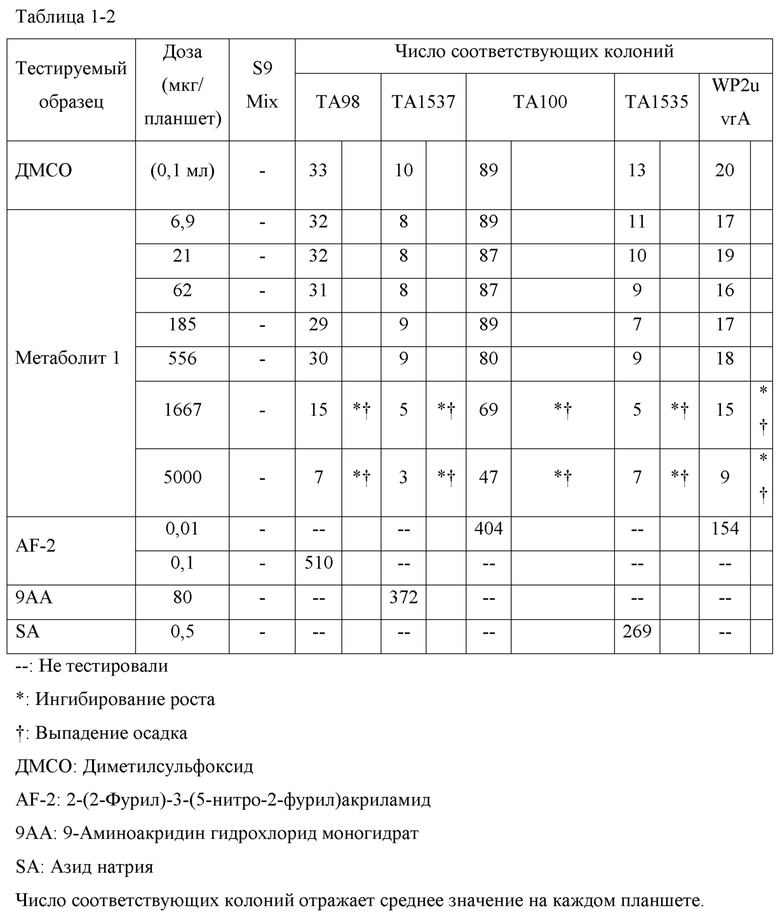
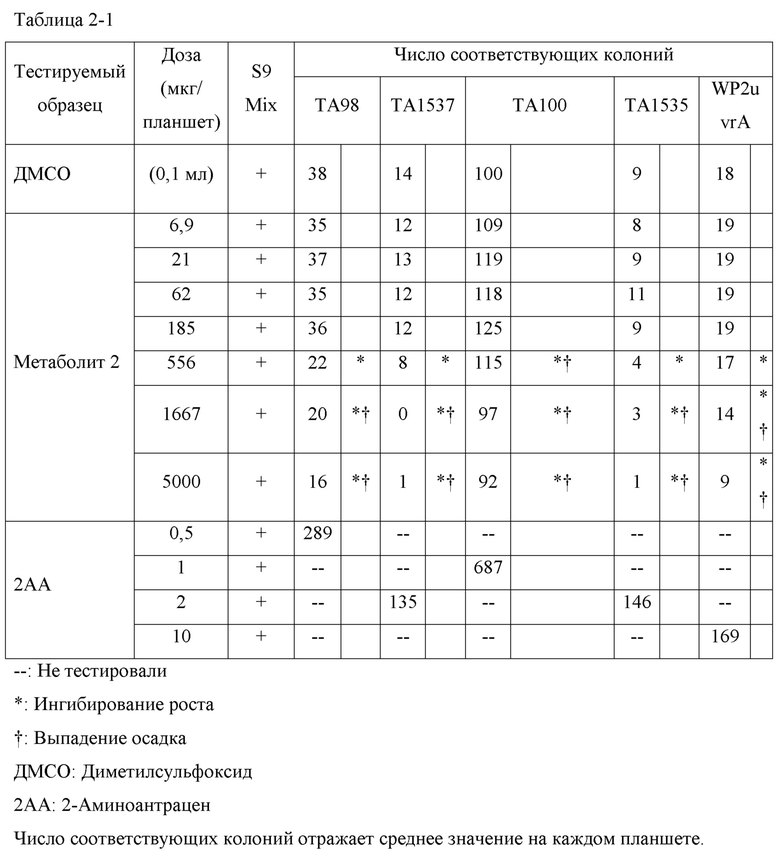
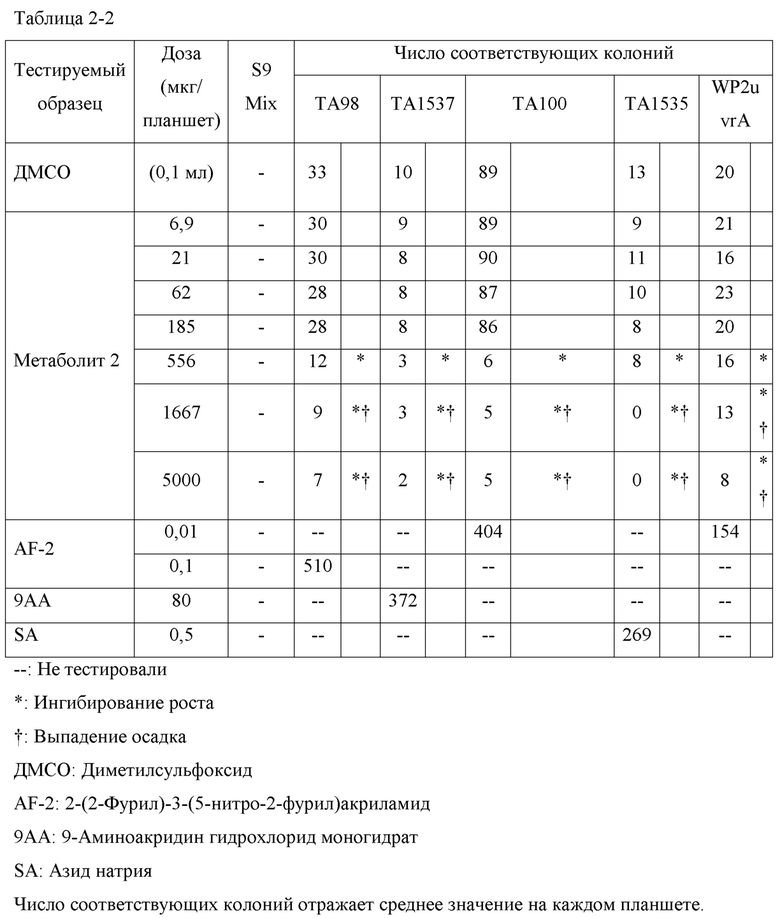
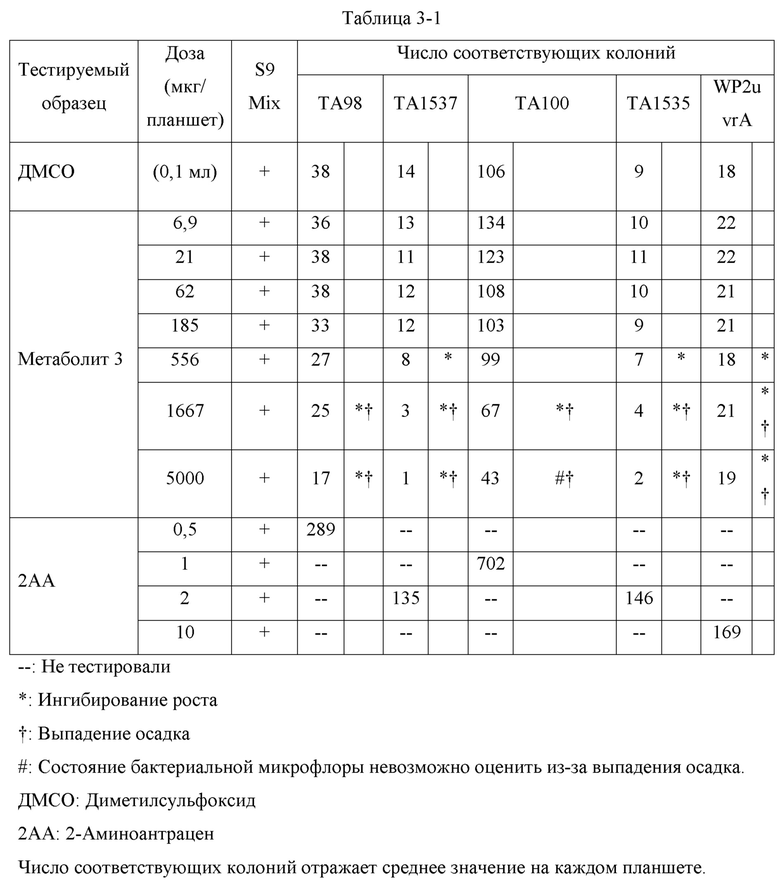
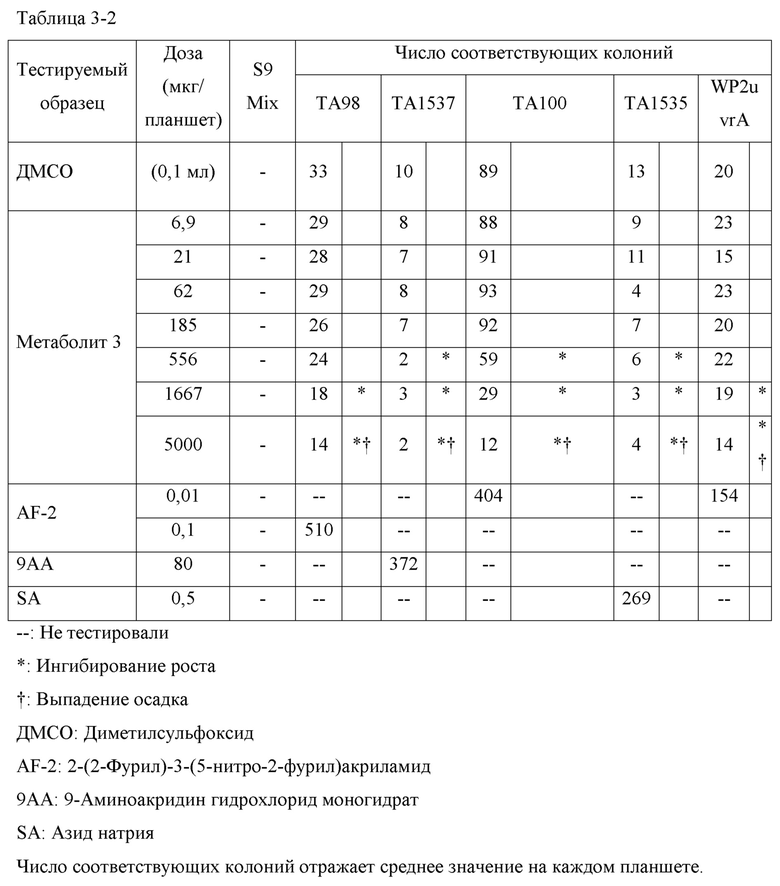
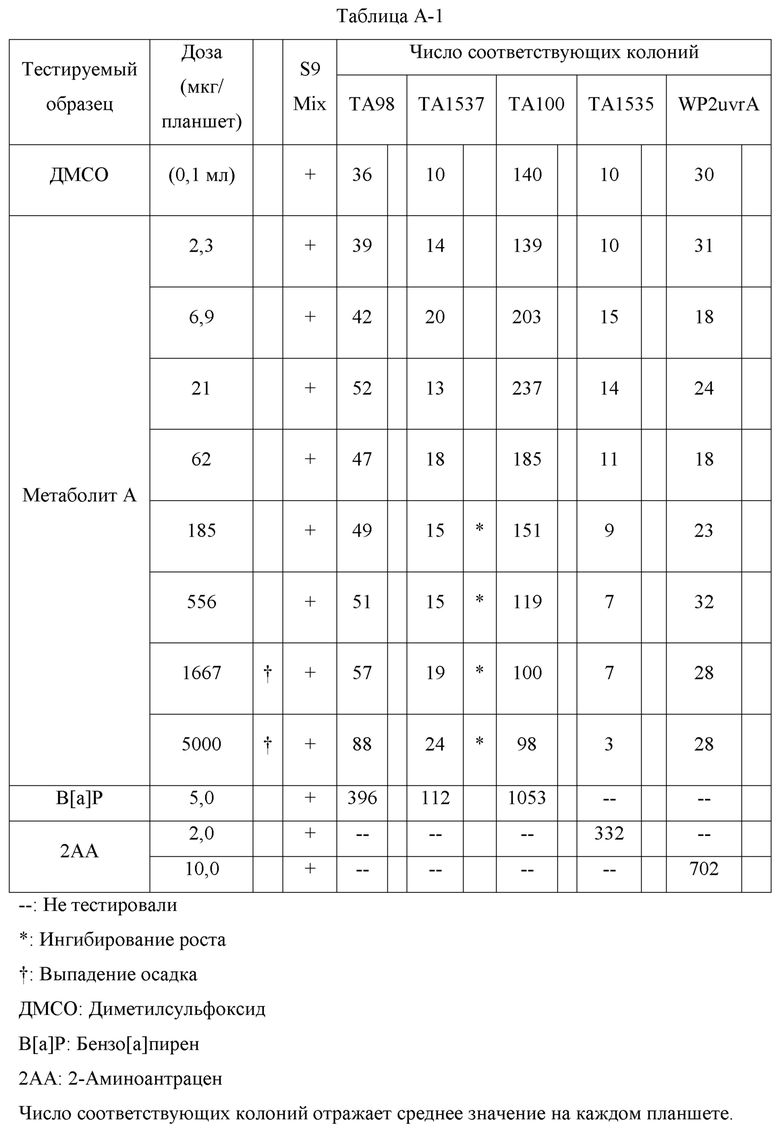
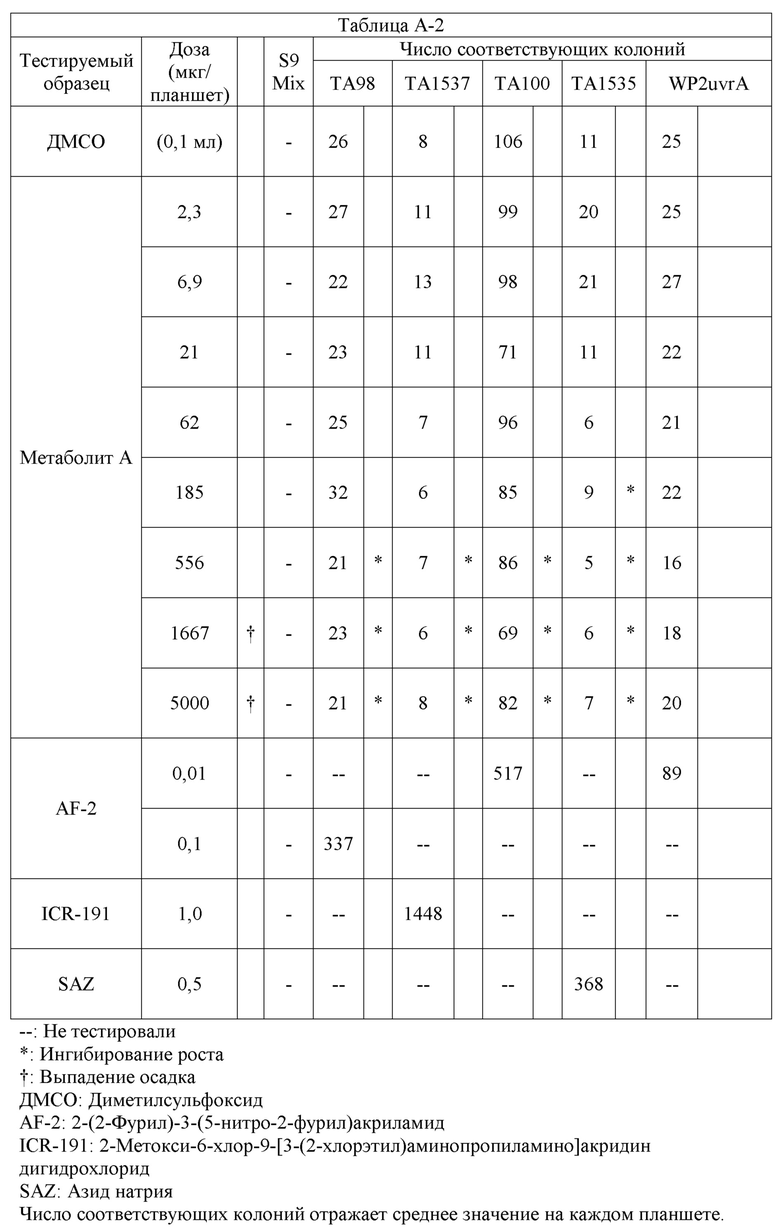
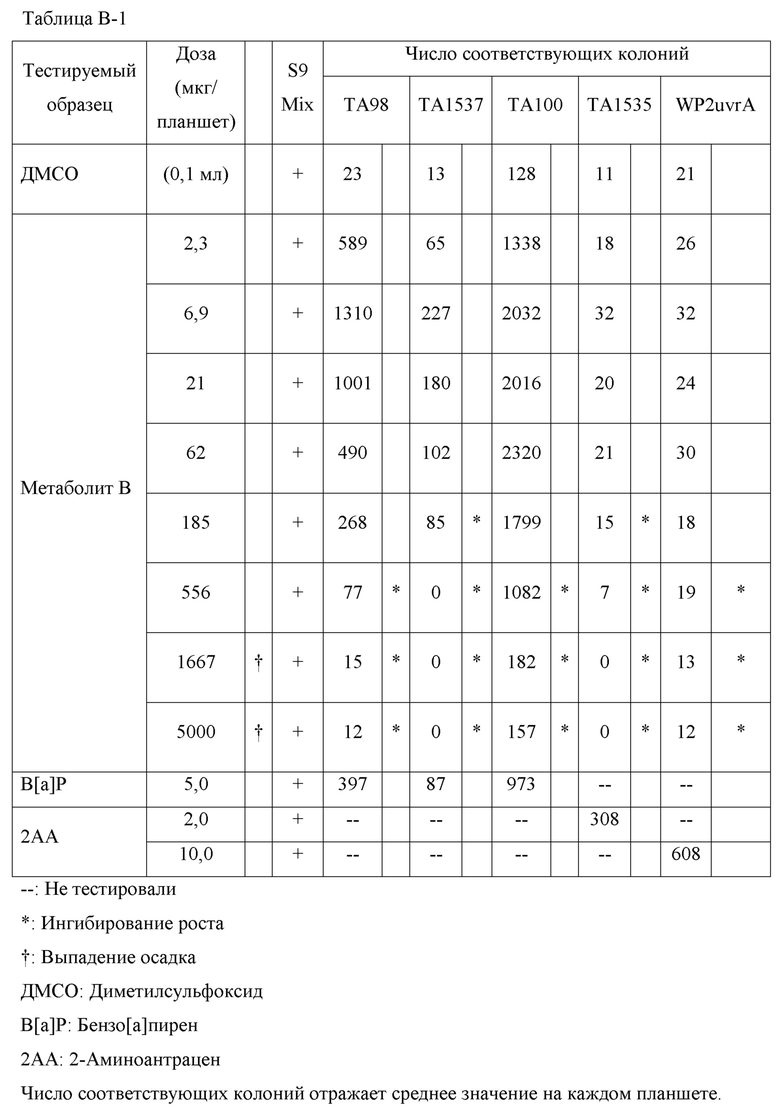
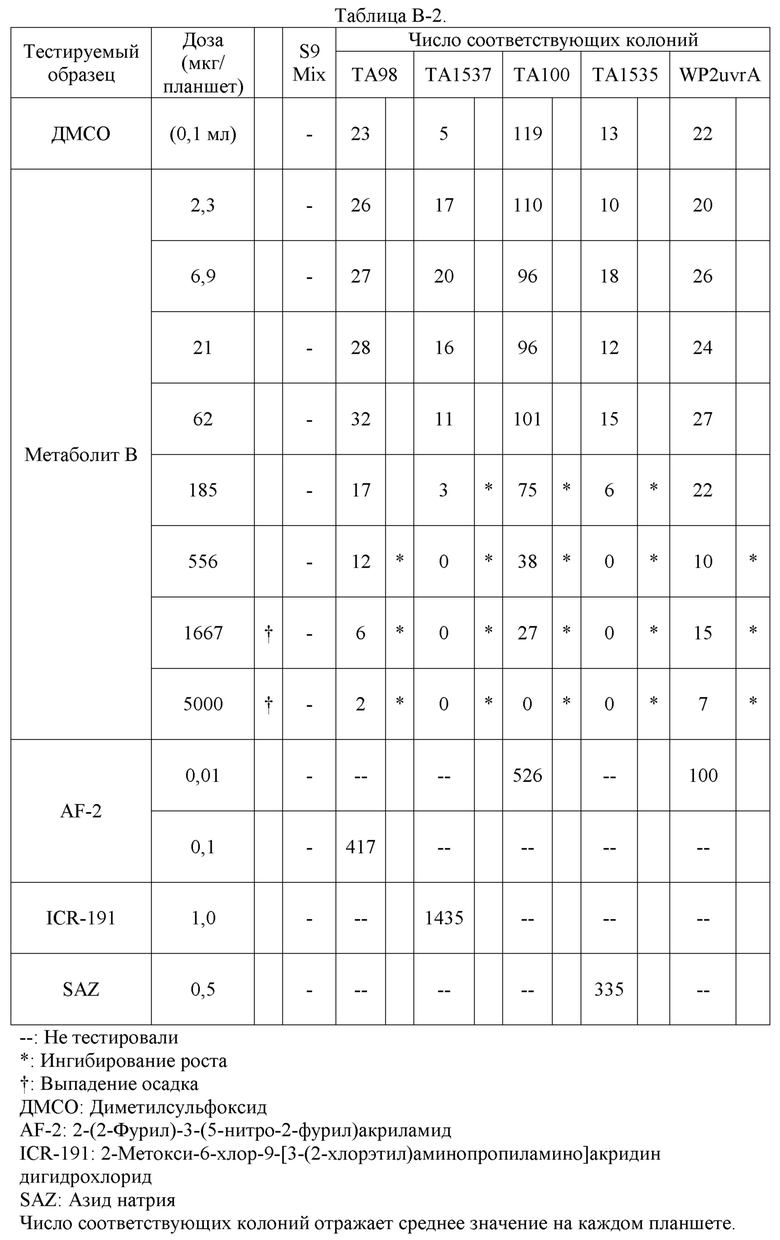
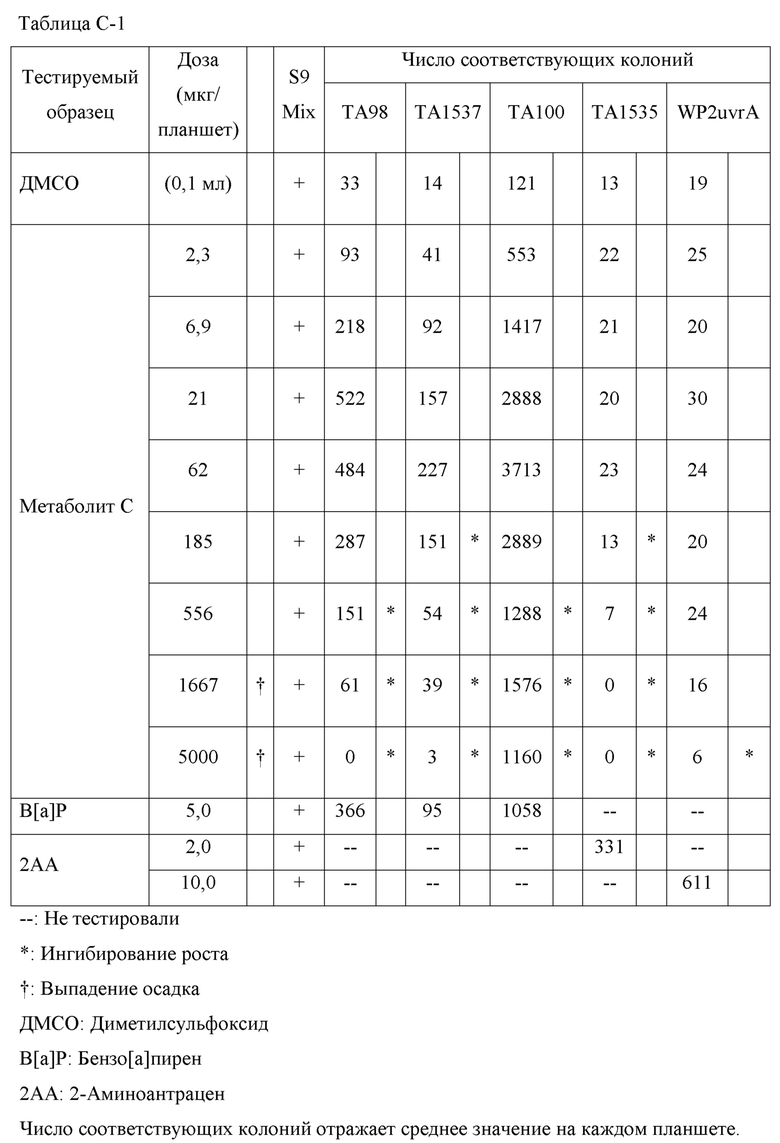
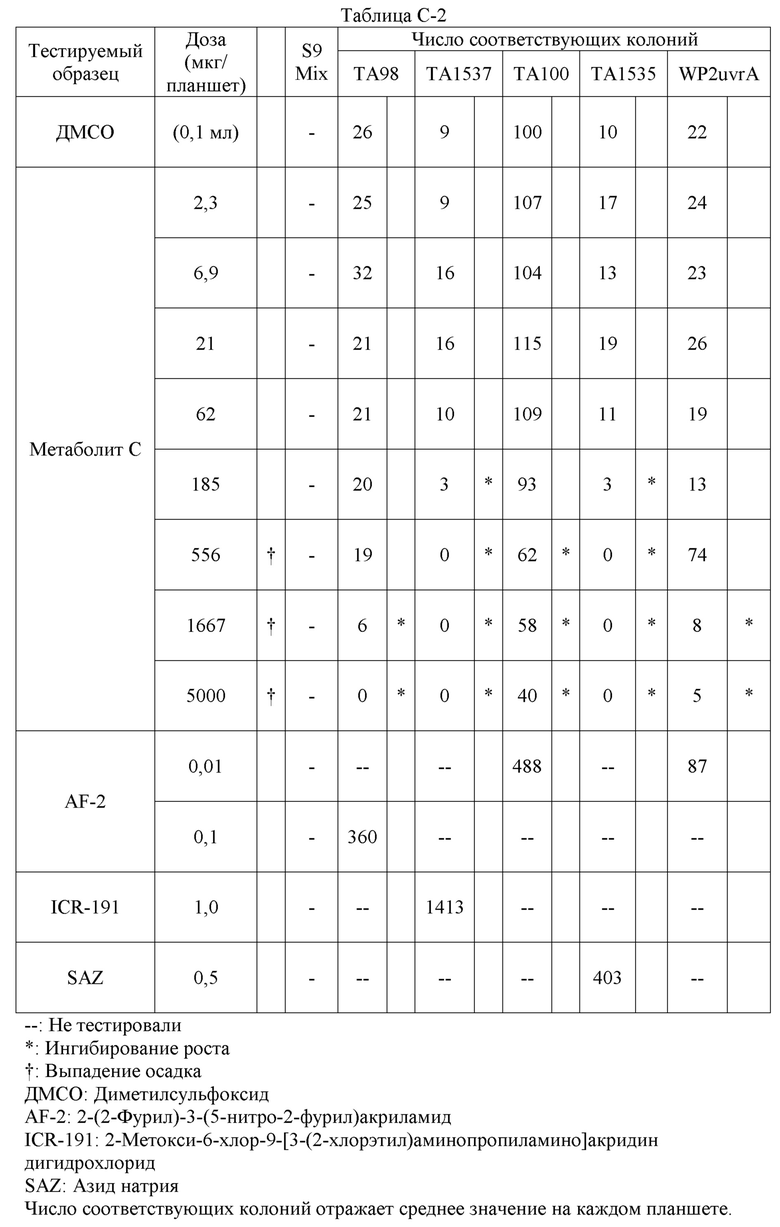
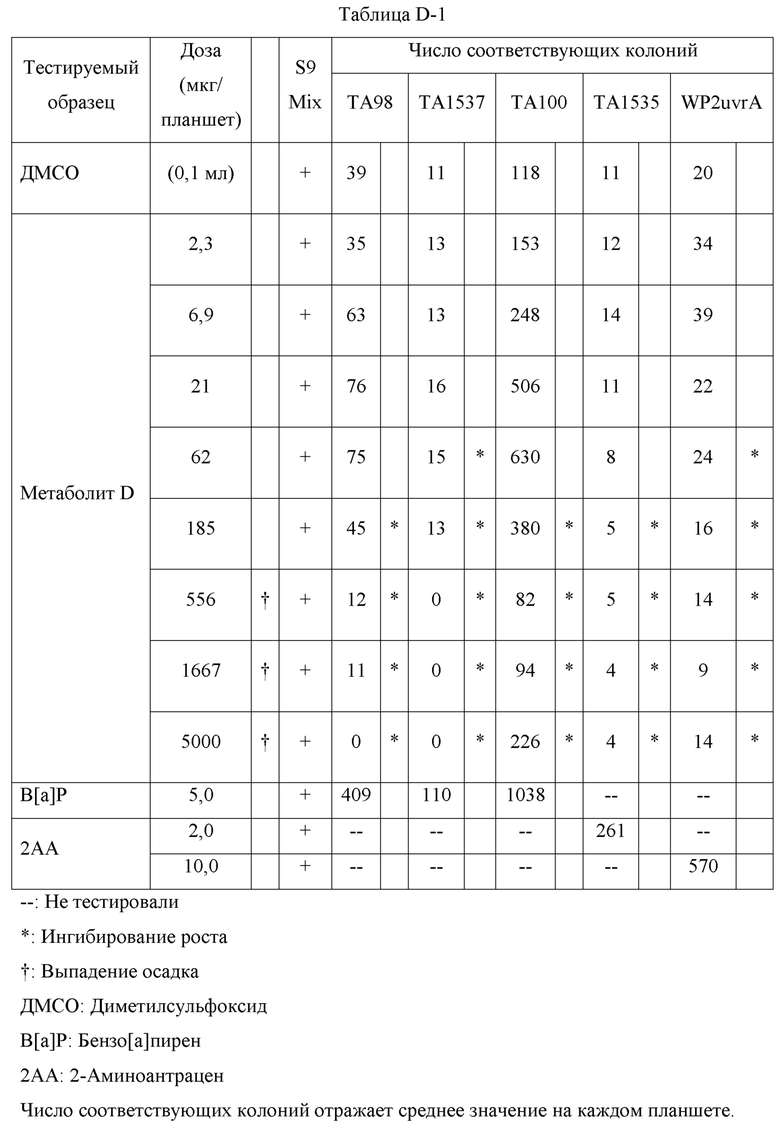
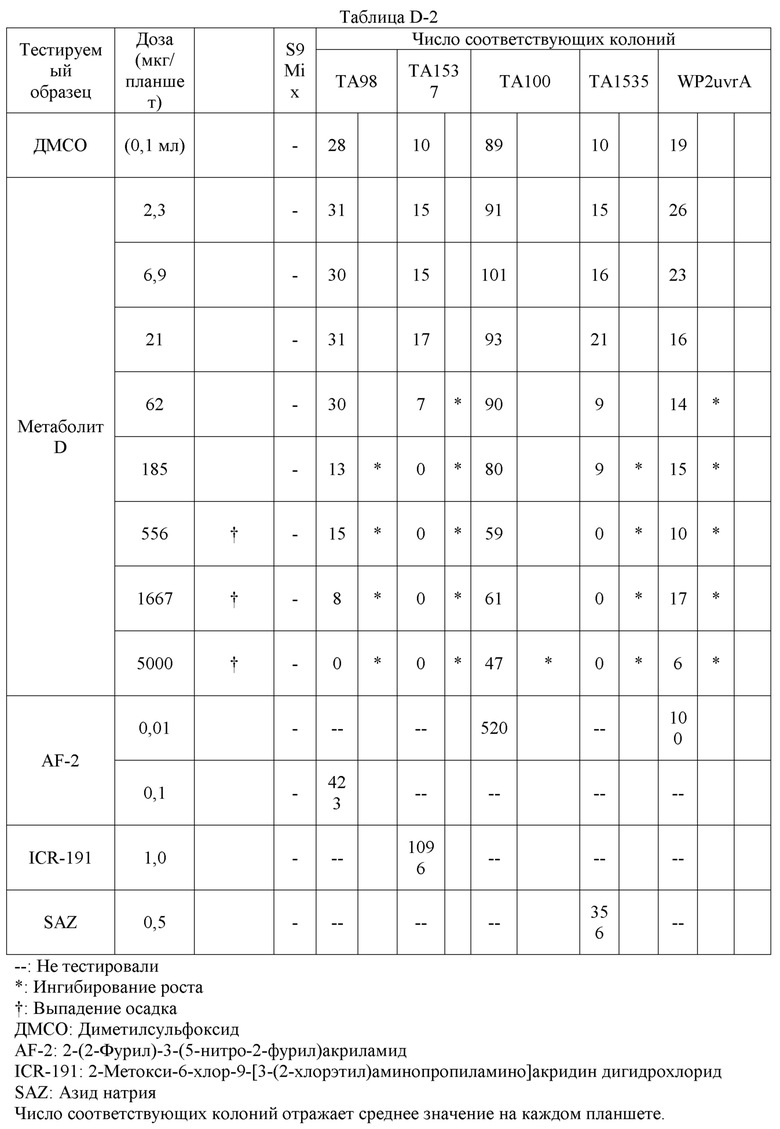
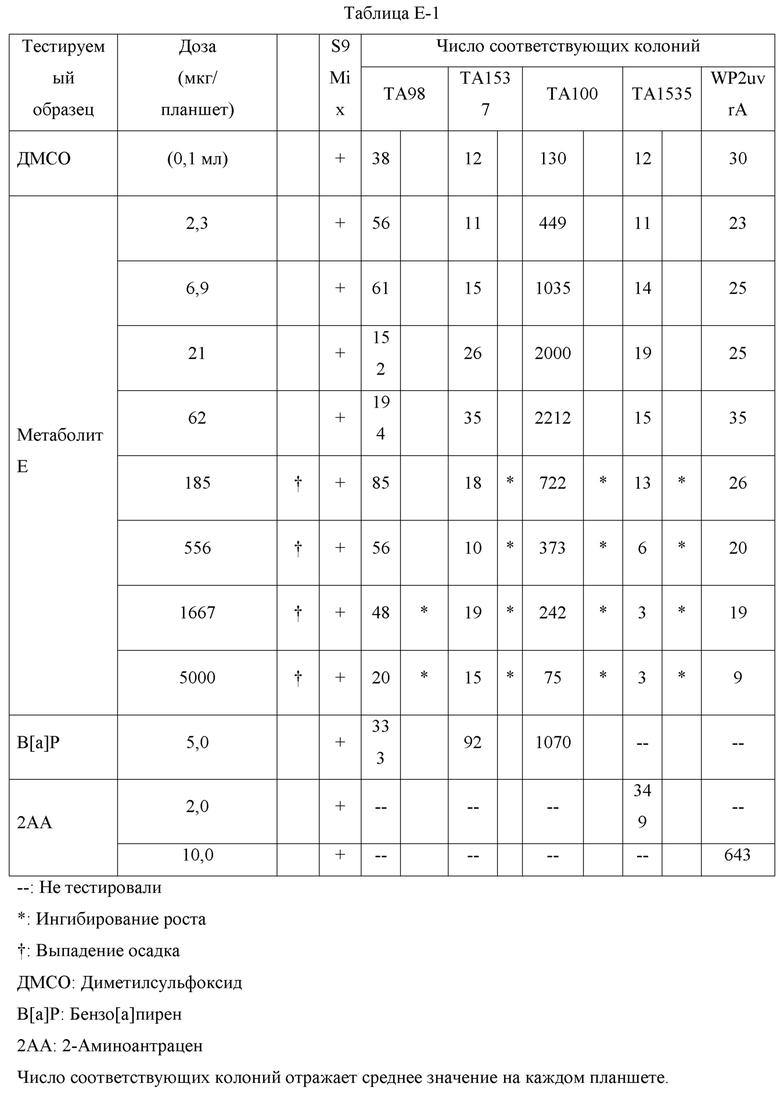
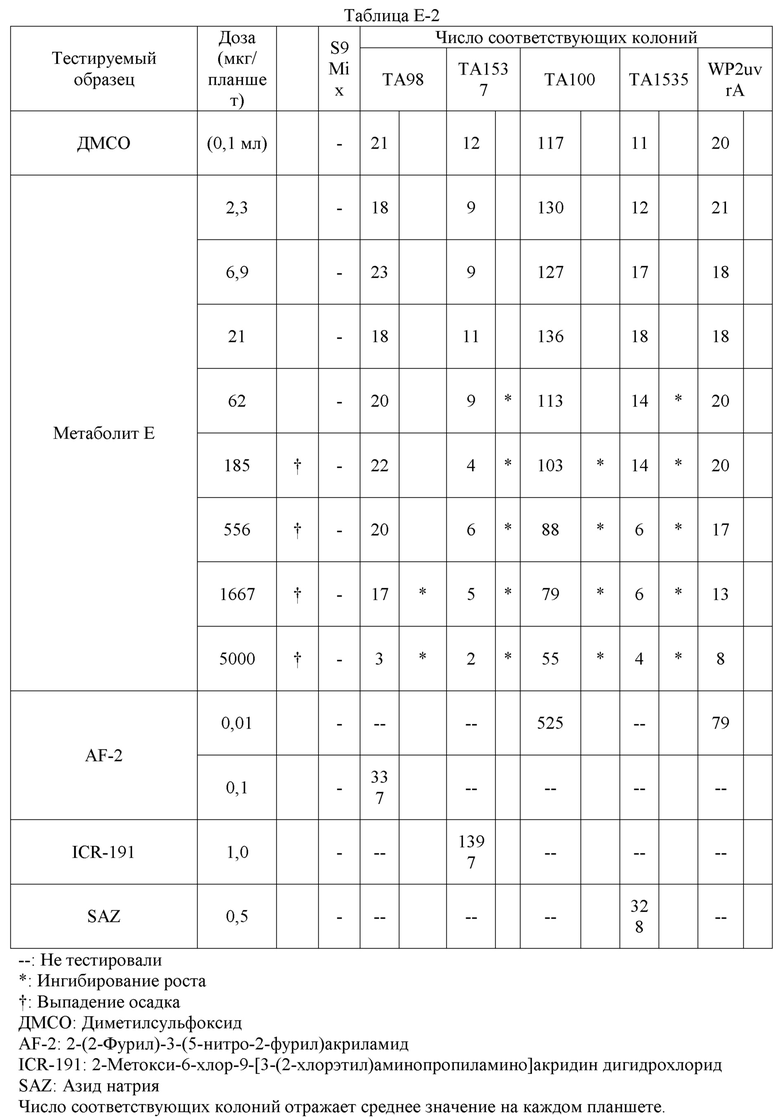
Примеры препаратов
Примеры препаратов с соединением по настоящему изобретению включают, например, описанные ниже препараты. Однако настоящее изобретение не ограничивается только эти примерами препаратов.
Пример препарата 1 (капсула)
Ингредиенты (1), (2), (3) и (4) смешивают для последующего заполнения в желатиновую капсулу.
Пример препарата 2 (таблетка)
Все количество ингредиентов (1), (2) и (3) и 30 г ингредиента (4) объединяют с водой, сушат в вакууме и затем гранулируют. Полученные гранулы смешивают с 14 г ингредиента (4) и 1 г ингредиента (5), и таблетируют на таблеточном прессе. Таким образом получают 1000 таблеток, содержащих 10 мг соединения из Примера 1 в каждой таблетке.
Промышленная применимость
Соединение, имеющее формулу [X], или его фармацевтически приемлемая соль имеют SGLT1-ингибирующую активность и поэтому могут применяться для лечения и/или предотвращения различных заболеваний или состояний, для которых ожидается улучшение при регулировании активности SGLT1.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СОДЕРЖАЩАЯ ЕГО ЛЕЧЕБНАЯ КОМПОЗИЦИЯ И ЕЕ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2006 |
|
RU2418803C2 |
| ПРОИЗВОДНЫЕ 4-ИЗОПРОПИЛФЕНИЛГЛЮЦИТА В КАЧЕСТВЕ ИНГИБИТОРОВ SGLT1 | 2010 |
|
RU2518896C2 |
| ПРОИЗВОДНОЕ (АЗА)ИНДОЛА И ЕГО ПРИМЕНЕНИЕ В ЛЕЧЕБНЫХ ЦЕЛЯХ | 2008 |
|
RU2477274C2 |
| НОВОЕ БИЦИКЛИЧЕСКОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2009 |
|
RU2518073C2 |
| СОЕДИНЕНИЕ ДИКАРБОНОВОЙ КИСЛОТЫ | 2014 |
|
RU2662817C2 |
| ПИРАЗОЛОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ | 2012 |
|
RU2605096C2 |
| ПРОИЗВОДНОЕ 6-(ГЕТЕРОЦИКЛЗАМЕЩЕННЫЙ БЕНЗИЛ)-4-ОКСОХИНОЛИНА И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ИНТЕГРАЗЫ ВИЧ | 2007 |
|
RU2399616C1 |
| НОВОЕ СОЕДИНЕНИЕ ИНДОЛИНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2003 |
|
RU2318808C2 |
| НОВОЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ БЕНЗИМИДАЗОЛА | 2004 |
|
RU2329261C2 |
| СОЕДИНЕНИЕ С МЕТИЛЛАКТАМНЫМ КОЛЬЦОМ И ЕГО ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2019 |
|
RU2809634C2 |
Настоящее изобретение касается пиразольных соединений, замещенных гетероарилом, или их фармацевтически приемлемых солей, которые обладают SGLT1-ингибирующей активностью, содержащих их фармацевтических композиций и их применения в фармацевтике. Описано соединение, имеющее формулу [X], где R1 представляет собой атом водорода или галоген; R2 представляет собой C1-6 алкил или галоген-C1-6 алкил; Кольцо Het представляет собой: (1) пиридил, замещенный R3; или (2) пиримидинил, необязательно замещенный R4; R3 представляет собой циано-группу, галоген или галоген-C1-3 алкил; R4 представляет собой галоген, гидрокси-группу, C1-3 алкил, галоген-C1-3 алкил, C1-3 алкокси-группу или -N(R5)(R6); и R5 и R6 каждый независимо представляет собой атом водорода или C1-3 алкил, или его фармацевтически приемлемая соль. Также описаны соединения, имеющие формулы [I], [II], [III], [IV] или их фармацевтически приемлемые соли, фармацевтическая композиция, обладающая ингибирующей активностью в отношении SGLT1, содержащая любое из вышеописанных соединений или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, и SGLT1 ингибитор, содержащий любое из вышеописанных соединение или его фармацевтически приемлемую соль. В заявленном изобретении раскрываются применение любого из вышеописанных соединения или его фармацевтически приемлемой соли для производства ингибитора SGLT1, и применение любого из вышеописанных cоединения или его фармацевтически приемлемой соли для ингибирования SGLT1. Технический результат – получение пиразольных соединений, которые обладают SGLT1-ингибирующей активностью. 9 н. и 4 з.п. ф-лы, 16 табл., 20 пр.
1. Соединение, имеющее формулу [X]
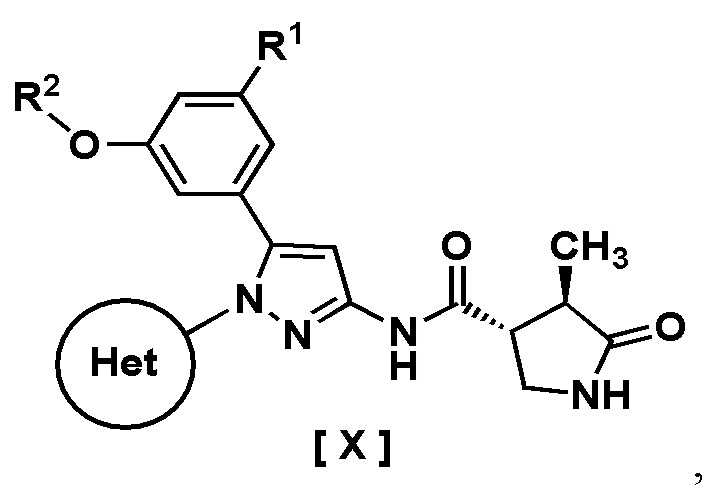
где R1 представляет собой атом водорода или галоген;
R2 представляет собой C1-6 алкил или галоген-C1-6 алкил;
Кольцо Het представляет собой:
(1) пиридил, замещенный R3; или
(2) пиримидинил, необязательно замещенный R4;
R3 представляет собой циано-группу, галоген или галоген-C1-3 алкил;
R4 представляет собой галоген, гидрокси-группу, C1-3 алкил, галоген-C1-3 алкил, C1-3 алкокси-группу или -N(R5)(R6); и
R5 и R6 каждый независимо представляет собой атом водорода или C1-3 алкил,
или его фармацевтически приемлемая соль.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой галоген.
3. Соединение или его фармацевтически приемлемая соль по любому из пп. 1 или 2, где R2 представляет собой галоген-C1-6 алкил.
4. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, где Кольцо Het представляет собой пиридил, замещенный R3.
5. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, где Кольцо Het представляет собой пиримидинил, необязательно замещенный R4.
6. Соединение, имеющее формулу [I]
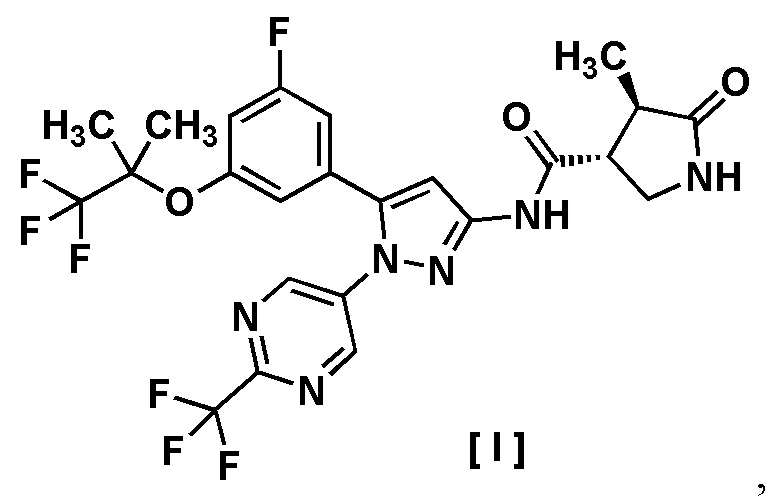
или его фармацевтически приемлемая соль.
7. Соединение, имеющее формулу [II]
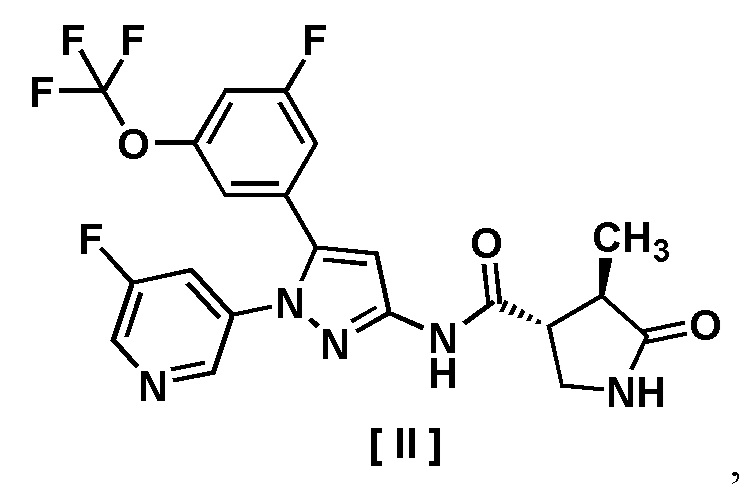
или его фармацевтически приемлемая соль.
8. Соединение, имеющее формулу [III]
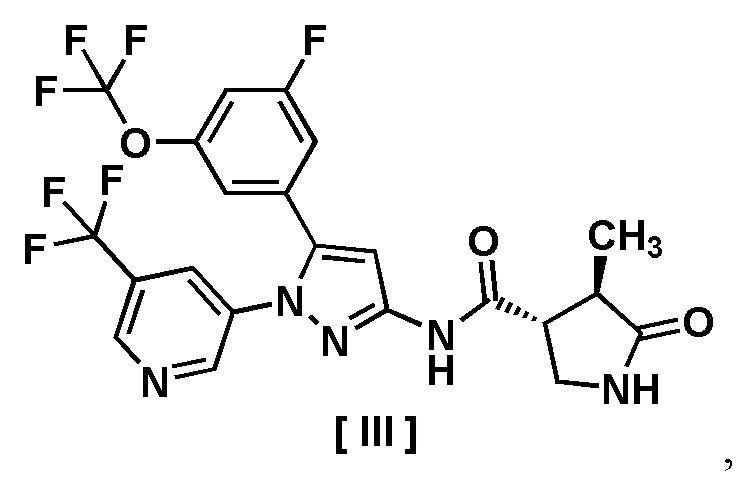
или его фармацевтически приемлемая соль.
9. Соединение, имеющее формулу [IV]
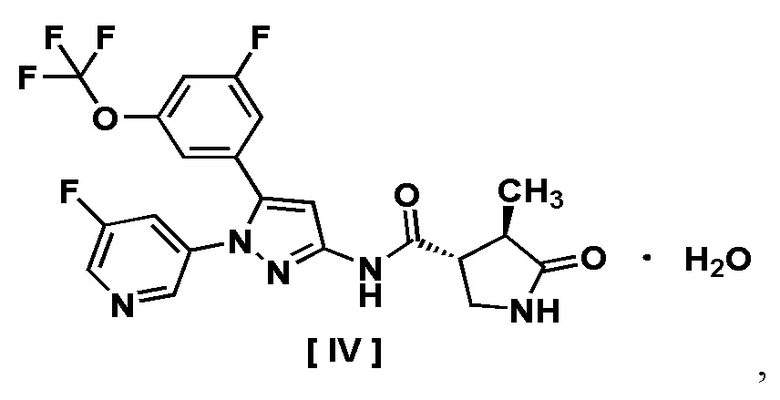
или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении SGLT1, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1-9 и фармацевтически приемлемый носитель.
11. SGLT1 ингибитор, содержащий соединение или его фармацевтически приемлемую соль по любому из пп. 1-9.
12. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-9 для производства ингибитора SGLT1.
13. Применение cоединения или его фармацевтически приемлемой соли по любому из пп. 1-9 для ингибирования SGLT1.
| WO 2013031922 A1, 07.03.2013 | |||
| WO 2008045484 A1, 17.04.2008 | |||
| US 20060111422 A1, 25.05.2006 | |||
| EP 2850084 A1, 25.03.2015 | |||
| НОВЫЕ ФТОРГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ ПИРАЗОЛОВ, СОДЕРЖАЩЕЕ ИХ ЛЕКАРСТВЕННОЕ СРЕДСТВО И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2370499C2 |
| KOJI OHSUMI et al | |||
| Pyrazole-O-Glucosides as Novel Na+-Glucose Contransporter (SGLT) Inhibitors | |||
| Насос | 1917 |
|
SU13A1 |
| Прибор для равномерного смешения зерна и одновременного отбирания нескольких одинаковых по объему проб | 1921 |
|
SU23A1 |
| Подводный аппарат для наблюдения за подводными работами | 1925 |
|
SU2269A1 |
