Настоящее изобретение относится к новому способу получения и очистки комплекса гадолиния и хелатирующего лиганда на основе PCTA, который позволяет предпочтительно получать стереоизомеры указанного комплекса, обладающие физико-химическими свойствами, которые являются особенно преимущественными для вариантов применения в качестве контрастного средства в области медицинской визуализации, в особенности для магнитно-резонансной томографии. Настоящее изобретение также относится к диастереоизомерно обогащенному комплексу как таковому, к композиции, содержащей указанный комплекс, а также к способу получения соответствующего хелатирующего лиганда путем распада комплекса указанного комплекса и к лиганду как таковому.
Известно множество контрастных средств на основе хелатов лантаноидов (парамагнитного металла), в частности гадолиния (Gd), например, описанных в US 4647447. Эти продукты часто объединяют под термином GBCA (контрастное средство на основе гадолиния). На рынке имеется несколько продуктов, среди которых макроциклические хелаты, такие как меглумина гадотерат на основе DOTA (1,4,7,10-тетраазациклододекан-N,N',N",N'"-тетрауксусной кислоты), гадобутрол на основе DO3A-бутрола, гадотеридол на основе HPDO3A, а также линейные хелаты, в особенности на основе DTPA (диэтилентриаминпентауксусной кислоты) или DTPA-BMA (гадодиамид в качестве лиганда).
Другие продукты, некоторые из которых находятся в процессе разработки, представляют собой новое поколение GBCA. По сути, они представляют собой комплексы макроциклических хелатов, таких как бициклополиазамакроциклокарбоновая кислота (ЕР 0438206) или производные PCTA (т.е. производные, характеризующиеся по меньшей мере химической структурой 3,6,9,15-тетраазабицикло[9,3,1]пентадека-1(15),11,13-триен-3,6,9-триуксусной кислоты),описанные в EP 1931673.
Примечательно, что комплексы хелатирующих лигандов на основе PCTA, описанные в EP 1931673, имеют преимущество, состоящее в том, что их относительно легко синтезировать химическим путем и, что еще важнее, они обладают большей релаксивностью, чем другие представленные в настоящее время на рынке GBCA (релаксивность r1, которая может составлять не более 11-12 ммоль-1.с-1 в воде), при этом эта релаксивность соответствует эффективности данных продуктов и, следовательно, их контрастирующей способности.
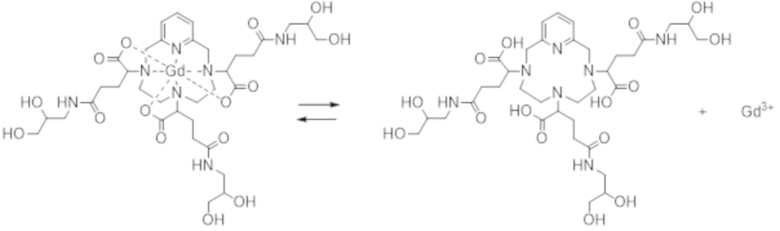
В организме хелаты (или комплексы) лантаноида и, в особенности, гадолиния, находятся в состоянии химического равновесия (характеризующегося его термодинамической константой Kтерм.), что может привести к нежелательному высвобождению указанного лантаноида (см. уравнение 1 ниже):
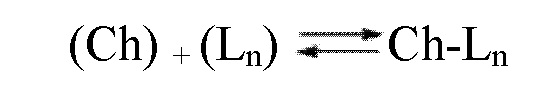
(уравнение 1)
Химическое равновесие комплексообразования между хелатом или лигандом (Ch) и лантаноидом (Ln) с образованием комплекса.
С 2006 года патология, называемая NSF (нефрогенным системным фиброзом или фиброгенной дермопатией) по меньшей мере частично была связана с высвобождением свободного гадолиния в организме. Данное заболевание привлекло внимание органов здравоохранения в отношении контрастных средств на основе гадолиния, продаваемых определенным категориям пациентов.
Таким образом, были разработаны стратегии для решения совершенно безопасным образом сложной проблемы переносимости пациентом и ограничения или даже устранения риска нежелательного высвобождения лантаноида после введения. Эта проблема является трудноразрешимой, поскольку введение контрастных средств часто повторяется либо во время диагностических обследований, либо при корректировке доз и мониторинге эффективности терапевтического лечения.
Кроме того, с 2014 года упоминалось о возможном отложении гадолиния в головном мозге после повторяемого введения продуктов на основе гадолиния, в частности линейных хелатов гадолиния, причем подобное отложение редко или вообще не было связано с макроциклическими хелатами гадолиния, такими как Dotarem®. В результате, различные страны решили либо отозвать большинство линейных хелатов с рынка, либо значительно ограничить их показания к применению, учитывая их устойчивость, которая считается недостаточной.
Таким образом, первая стратегия ограничения риска высвобождения лантаноидов в организме состоит в отдаче предпочтения комплексам, отличающимся термодинамической и/или кинетической устойчивостью, которая должна быть как можно более высокой. Это обусловлено тем, что чем стабильнее комплекс, тем больше будет ограничиваться количество высвобождаемого с течением времени лантаноида.
Другие подходы к повышению переносимости хелатов лантаноида (в особенности гадолиния) описаны в предшествующем уровне техники. В документе US 5876695, которому более 30 лет, представлены, например, составы, которые помимо хелата лантаноида содержат дополнительное комплексообразующее средство, предназначенное для предотвращения нежелательного высвобождения лантаноида in vivo путем комплексообразования выделяемого лантаноида (иона металла Gd3+). Дополнительное хелатирующее средство может быть введено в состав либо в его свободной форме, либо в форме слабого комплекса, обычно кальция, натрия, цинка или магния. Несмотря на то, что это средство, возможно, может отличаться от лиганда, составляющего активный комплекс, тем не менее важно, чтобы комплекс, который оно образует с высвобожденным лантаноидом, был менее стабильным, чем активный комплекс, чтобы предотвратить реакцию перелигандирования между активным комплексом и дополнительным хелатом, которая, в особенности, приведет к полному поглощению указанного дополнительного лиганда, который в таком случае больше не сможет улавливать выделяемый лантаноид. Этот риск поглощения дополнительного хелатирующего средства путем перелигандирования более выражен, когда оно добавляется в свободной форме, чем, например, в форме комплекса кальция.
Таким образом, в двух вышеописанных стратегиях важно, чтобы активный комплекс был как можно более стабильным.
Однако комплексы хелатирующих лигандов на основе PCTA, имеющие структуру типа пиклена, которая описана в EP 1931673, обладая хорошей кинетической устойчивостью, как правило, характеризуются термодинамической константой, которая ниже, чем у комплексов других макроциклов на основе циклена.
Это особенно верно в случае комплекса формулы (II), которая представлена ниже:
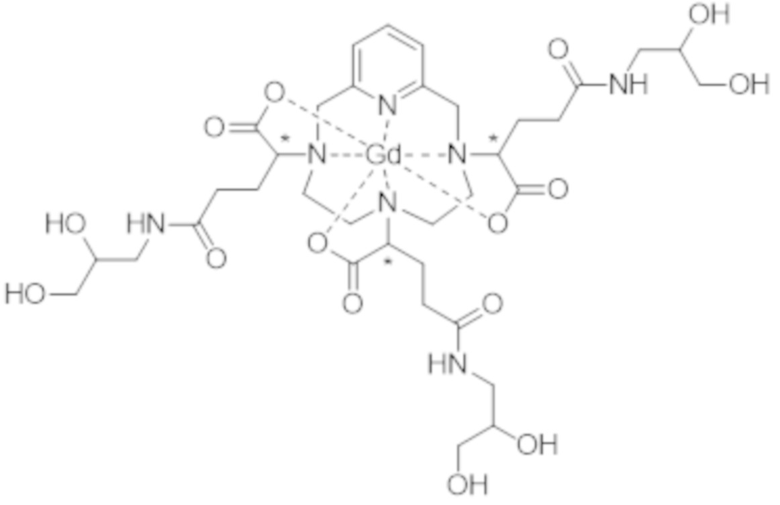 (II).
(II).
Более конкретно, в WO 2014/174120 подробно описано, что термодинамическая константа равновесия, соответствующая реакции образования комплекса формулы (II), которую также называют константой устойчивости, составляет 1014,9 (т.е. log (Kтерм.) = 14,9). Для сравнения, константа устойчивости комплекса гадолиния и 1,4,7,10-тетраазациклододекан-N,N',N",N"'-тетрауксусной кислоты (DOTA-Gd) составляет 1025,6 (т.е. log (Kтерм.) = 25,6).
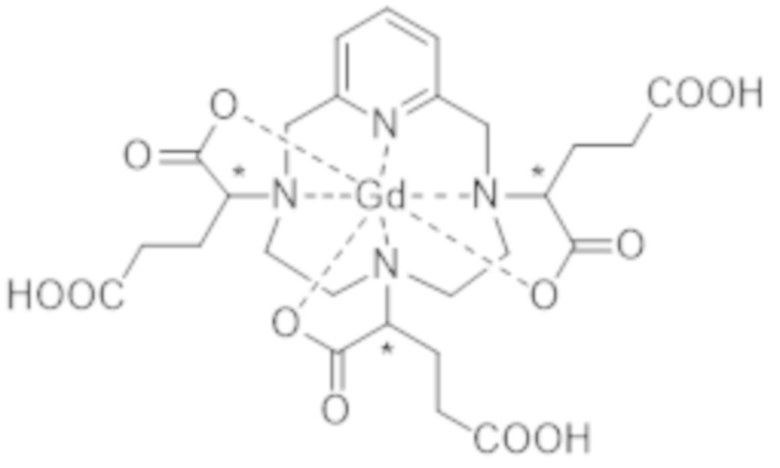
Однако следует отметить, что комплекс формулы (II) соответствует нескольким стереоизомерам, в особенности, из-за присутствия трех асимметрических атомов углерода, расположенных на боковых цепях комплекса в α-положении по отношению к атомам азота макроцикла, к которым привиты указанные боковые цепи. Эти три асимметрических атома углерода отмечены звездочкой (*) в формуле (II), которая представлена выше.
Таким образом, синтез комплекса формулы (II), описанный в ЕР 1931673, приводит к получению смеси стереоизомеров.
Аминопропандиольные группы боковых цепей комплекса формулы (II) также включают асимметрический атом углерода. Таким образом, комплекс формулы (II) содержит в целом шесть асимметрических атомов углерода и, таким образом, существует в виде 64 конфигурационных стереоизомеров. Однако в остальной части описания единственным источником стереоизомерии, рассматриваемым для данной боковой цепи, в целях простоты изложения будет тот, который соответствует асимметрическому атому углерода, несущему карбоксилатную группу, который отмечен звездочкой (*) в формуле (II), представленной выше.
Поскольку каждый из этих трех асимметрических атомов углерода может иметь абсолютную R- или S-конфигурацию, комплекс формулы (II) существует в виде восьми семейств стереоизомеров, упоминаемых далее в данном документе как II-RRR, II-SSS, II-RRS, II-SSR, II-RSS, II-SRR, II-RSR и II-SRS. А именно, согласно обычной стереохимической номенклатуре комплекс формулы (II) существует в виде восьми семейств диастереоизомеров.
Применение термина "семейство" оправдано тем, что каждое из этих семейств объединяет несколько стереоизомеров, в особенности, из-за присутствия асимметрического атома углерода в аминопропандиольной группе, как упомянуто ранее.
Тем не менее, поскольку в остальной части описания не будет рассматриваться стереоизомерия, связанная с асимметрическим атомом углерода данной аминопропандиольной группы, термины "изомеры", "стереоизомеры" или "диастереоизомеры" II-RRR, II-SSS, II-RRS, II-SSR, II-RSS, II-SRR, II-RSR и II-SRS будут применяться в одинаковой мере без указания соответствия каждого из них семейству стереоизомеров.
Авторам настоящего изобретения удалось разделить и идентифицировать с помощью высокоэффективной жидкостной хроматографии (HPLC) и ультра высокоэффективной жидкостной хроматографии (UHPLC) четыре неразрешенных пика или группы изомеров комплекса формулы (II), полученных согласно способу из предшествующего уровня техники, которые соответствуют четырем различным пикам элюирования, характеризующимся своим временем удерживания на хроматограмме, которые в остальной части описания настоящего изобретения будут упоминаться как iso1, iso2, iso3 и iso4. При осуществлении способа, описанного в EP 1931673, значения соответствующего содержания групп iso1, iso2, iso3 и iso4 в полученной смеси будет следующим: 20%, 20%, 40% и 20%.
Затем авторы настоящего изобретения обнаружили, что данные различные группы изомеров имеют разные физико-химические свойства, и определили, что группа изомеров, называемая iso4, которая включает смесь изомеров II-RRR и II-SSS формулы (II-RRR) и формулы (II-SSS), представленных ниже, показывает себя наиболее преимущественной в качестве контрастного средства для медицинской визуализации.
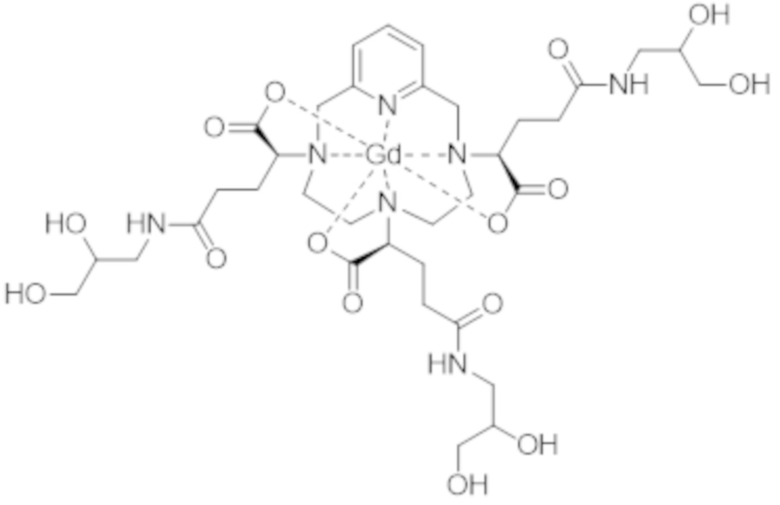 (II-SSS)
(II-SSS)
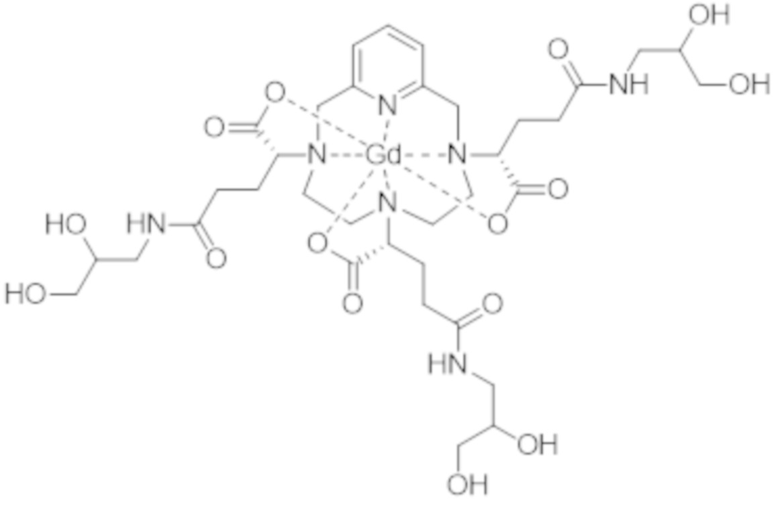 (II-RRR).
(II-RRR).
Таким образом, неожиданно iso4 отличается термодинамической устойчивостью, которая значительно превосходит термодинамическую устойчивость смеси диастереоизомеров, в форме которой комплекс формулы (II) получают путем осуществления способа, описанного в EP 1931673. Более конкретно, термодинамическая константа равновесия Kтерм. iso4 равняется 1018,7 (т.е. log (Kтерм. iso4) = 18,7), при этом это значение было определено по методу в Pierrard et al., Contrast Media Mol. Imaging, 2008, 3, 243-252 и Moreau et al., Dalton Trans., 2007, 1611-1620.
Более того, iso4 представляет собой группу изомеров, обладающую наилучшей кинетической инертностью (также называемой кинетической устойчивостью) среди четырех групп, выделенных авторами настоящего изобретения. Более конкретно, авторы настоящего изобретения оценили кинетическую инертность четырех групп изомеров, изучая их кинетику распада комплекса в кислом водном растворе (pH = 1,2) при 37°C. Значения периода полураспада (1/2), которые определяли для каждой из групп изомеров, указаны в таблице 1 ниже, при этом период полураспада соответствует времени, по истечении которого 50% первоначально присутствующего количества комплекса продиссоциировало в соответствии со следующей реакцией распада комплекса (уравнение 2):
(уравнение 2)
Таблица 1. Кинетика распада комплекса для групп изомеров iso1–iso4
Для сравнения, гадобутрол или гадотерат, представляющие собой макроциклические комплексы гадолиния, обладают кинетической инертностью 18 часов и 4 дня соответственно, при тех же условиях, тогда как линейные комплексы гадолиния, такие как гадодиамид или гадопентетат, диссоциируют мгновенно.
Кроме того, в особенности, iso4 химически более устойчива, чем iso3. Это обусловлено тем, что амидные функциональные группы комплекса формулы (II) склонны к гидролизу. Реакция гидролиза амидной функциональной группы (уравнение 3) приводит к образованию примеси, образующейся в результате двойного присоединения, которое сопровождается высвобождением 3-амино-1,2-пропандиола. Авторы настоящего изобретения изучили кинетику реакции гидролиза комплекса формулы (II) в водном растворе при pH 13 и обнаружили, что амидные функциональные группы в iso4 более устойчивы в отношении гидролиза, чем функциональные группы в iso3.
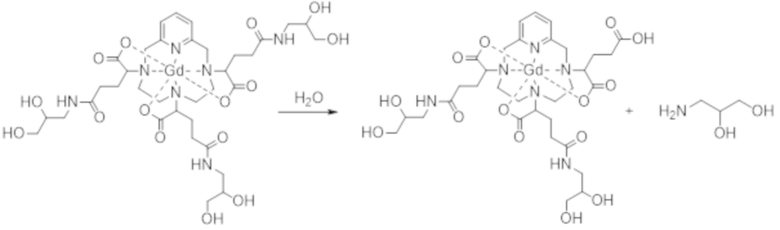
(уравнение 3)
Что касается релаксивности различных групп изомеров, т.е. их эффективности в качестве контрастного средства, проведенные измерения демонстрируют контрастирующую способность, которая относительно эквивалентна для групп iso1, iso2 и iso4, и пониженную эффективность для iso3 (см. таблицу 2).
Таблица 2. Релаксивность групп изомеров iso1–iso4 при 37°C
(ммоль-1.с-1)
(ммоль-1.с-1)
Авторам настоящего изобретения удалось разработать новый способ получения и очистки комплекса формулы (II), позволяющий предпочтительно получать диастереоизомеры II-RRR и II-SSS указанного комплекса, которые обладают особенно преимущественными физико-химическими свойствами. Способ в соответствии с настоящим изобретением включает стадию изомерного обогащения путем превращения наименее устойчивых стереоизомеров в наиболее устойчивые стереоизомеры, что неожиданно хотя и проводится для промежуточного комплекса с гексакислотой, а не с конечным комплексом, позволяет преимущественно получать наиболее устойчивые изомеры комплекса формулы (II).
Осуществление способа, позволяющего преимущественно получать представляющие интерес диастереоизомеры, безусловно преимущественно по сравнению с альтернативой, состоящей в получении смеси стереоизомеров с последующей попыткой разделить диастереоизомеры обычными методиками и, таким образом, выделить представляющие интерес изомеры с помощью любой методики разделения, которая хорошо известна в данной области. Более конкретно, не считая того, что легче осуществлять способ, не включающий стадию разделения диастереоизомеров в промышленном масштабе, отсутствие разделения, во-первых, обеспечивает значительную экономию времени, а во-вторых, позволяет повысить общий выход способа путем ограничения, по мере возможности, образования нежелательных диастереоизомеров, которые в конечном итоге будут отброшены. Более того, обычные методики разделения, как правило, включают обильное применение растворителей, что нежелательно по экологическим соображениям, не говоря уже о финансовых затратах. Кроме того, следует избегать, в частности хроматографии на диоксиде кремния, учитывая риски для здоровья, связанные с профессиональным воздействием диоксида кремния, которое классифицируется Международным агентством по изучению рака как канцерогенное для человека (группа 1).
Как указано выше, способ получения комплекса формулы (II), разработанный авторами настоящего изобретения, основан на стадии изомерного обогащения промежуточного комплекса гадолиния и гексакислоты формулы (I), которая представлена ниже:
 (I).
(I).
Комплекс формулы (I) соответствует нескольким стереоизомерам из-за присутствия трех асимметрических атомов углерода, расположенных на боковых цепях комплекса в α-положении по отношению к атомам азота макроцикла, к которому привиты указанные боковые цепи. Эти три асимметрических атома углерода отмечены звездочкой (*) в формуле (I), которая представлена выше.
Поскольку каждый из этих трех асимметрических атомов углерода, несущий карбоксилатную группу, может иметь абсолютную R- или S-конфигурацию, комплекс формулы (I) существует в виде восьми стереоизомеров, упоминаемых далее в данном документе как I-RRR, I-SSS, I-RRS, I-SSR, I-RSS, I-SRR, I-RSR и I-SRS. А именно, согласно обычной стереохимической номенклатуре, комплекс формулы (I) существует в виде четырех пар энантиомеров, являющихся диастереоизомерами по отношению друг к другу.
Авторам настоящего изобретения удалось разделить и идентифицировать с помощью высокоэффективной жидкостной хроматографии (HPLC) и ультра высокоэффективной жидкостной хроматографии (UHPLC) четыре неразрешенных пика или группы изомеров комплекса формулы (I), полученных согласно способу, описанному в EP 1931673, которые соответствуют четырем различным пикам элюирования, характеризующимся своим временем удерживания на хроматограмме, которые в остальной части описания настоящего изобретения будут упоминаться как isoA, isoB, isoC и isoD.
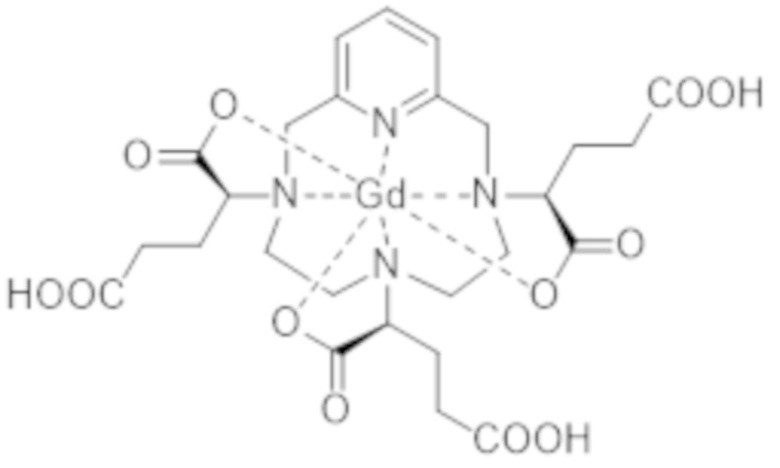
IsoD кристаллизуется из воды. С помощью рентгеноструктурного анализа авторы настоящего изобретения смогли определить кристаллическую структуру данной группы изомеров и, таким образом, обнаружить, что она включает диастереоизомеры I-RRR и I-SSS комплекса формулы (I), формулы (I-RRR) и формулы (I-SSS), представленных ниже.
 (I-SSS),
(I-SSS),
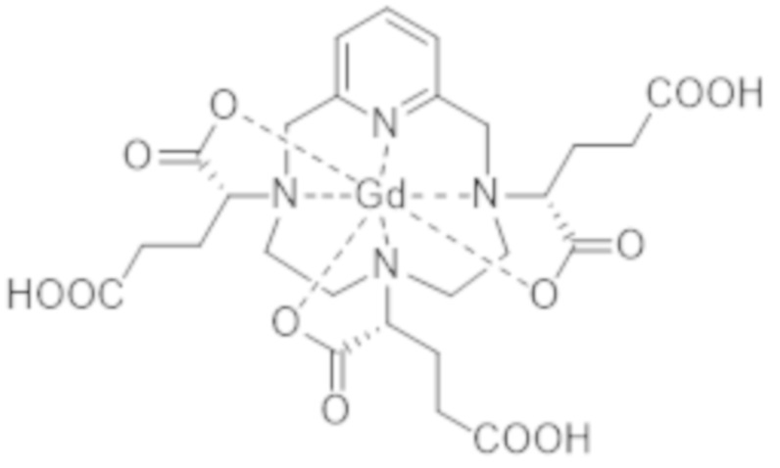 (I-RRR).
(I-RRR).
Следует отметить, что диастереоизомеры I-RRR и I-SSS комплекса формулы (I) являются энантиомерами друг друга.
Стадия изомерного обогащения способа по настоящему изобретению направлена на обогащение промежуточного комплекса гадолиния и гексакислоты формулы (I) с помощью isoD.
В частности, синтез комплекса формулы (II), включает превращение функциональных групп карбоновой кислоты промежуточного комплекса с гексакислотой формулы (I) в амидные функциональные группы. Данная реакция амидирования не изменяет абсолютную конфигурацию трех асимметрических атомов углерода комплекса формулы (I).
Таким образом, проведение реакции амидирования полученного ранее комплекса с гексакислотой формулы (I), обогащенного isoD, позволяет получать комплекс формулы (II), обогащенный iso4.
Более того, способ очистки, разработанный авторами настоящего изобретения, позволяет, при осуществлении способа получения комплекса вышеупомянутой формулы (II), получить комплекс формулы (II) с оптимизированным изомерным профилем, а также заметно улучшенным профилем содержания примесей.
В результате, данный диастереоизомерно обогащенный и очищенный комплекс с улучшенной устойчивостью можно составлять со свободным макроциклическим лигандом, таким как свободная DOTA, вместо кальциевого комплекса с DOTA, применение которой рекомендовалось в WO 2014/174120. В частности, применение свободной DOTA, имеет преимущество с точки зрения производства в промышленности в том смысле, что позволяет исключить из способа стадию синтеза состава, описанную в WO 2014/174120, а именно добавление CaCl2.
Комплекс формулы (II)
Таким образом, настоящее изобретение относится, прежде всего, к комплексу формулы (II):
 (II),
(II),
характеризующемуся по меньшей мере 80% диастереоизомерным избытком смеси изомеров II-RRR и II-SSS формул:
(II-SSS),
(II-RRR).
В контексте настоящего изобретения под термином "диастереоизомерный избыток" подразумевают указание в отношении комплекса формулы (II) того факта, что указанный комплекс преимущественно присутствует в виде изомера или группы изомеров, выбранных из диастереоизомеров II-RRR, II-SSS, II-RRS, II-SSR, II-RSS, II-SRR, II-RSR и II-SRS. Указанный диастереоизомерный избыток выражается в процентах и соответствует количеству, представленному преобладающим изомером или группой изомеров относительно общего количества комплекса формулы (II). Понятно, что это процентное содержание может быть молярным или массовым, поскольку изомеры по определению имеют одинаковую молярную массу.
В одном конкретном варианте осуществления комплекс формулы (II) в соответствии с настоящим изобретением характеризуется по меньшей мере 85%, в особенности по меньшей мере 90%, в частности по меньшей мере 92%, предпочтительно по меньшей мере 94%, преимущественно по меньшей мере 97%, более преимущественно по меньшей мере 99% диастереоизомерным избытком смеси изомеров II-RRR и II-SSS.
Предпочтительно указанный диастереоизомерный избыток предусматривает по меньшей мере 70%, в особенности по меньшей мере 80%, преимущественно по меньшей мере 90%, предпочтительно по меньшей мере 95% диастереоизомерный избыток смеси изомеров II-RRR и II-SSS.
Преимущественно указанный диастереоизомерный избыток предусматривает смесь изомеров II-RRR и II-SSS.
Термин "смесь изомеров II-RRR и II-SSS" также охватывает, в более широком смысле, случай, когда присутствует только один из изомеров: II-RRR или II-SSS. Однако термин "смесь изомеров II-RRR и II-SSS" предпочтительно обозначает все случаи, в которых каждый из изомеров II-RRR и II-SSS присутствует в различном количестве, отличном от нуля.
В предпочтительном варианте осуществления изомеры II-RRR и II-SSS присутствуют в указанной смеси в соотношении от 65/35 до 35/65, в особенности от 60/40 до 40/60, в частности от 55/45 до 45/55. Преимущественно изомеры II-RRR и II-SSS присутствуют в смеси в соотношении 50/50.
Более конкретно, диастереоизомерный избыток, как определено ранее, соответствует пику 4 на хроматограмме UHPLC (т.е. четвертому неразрешенному пику изомеров в порядке элюирования и соответствует iso4), характеризующемуся временем удерживания от 6,0 до 6,6 минуты, как правило, приблизительно 6,3 минуты, при этом указанную хроматограмму получали с применением методики UHPLC, описанной ниже.
Для целей настоящего изобретения термин "хроматограмма UHPLC" означает профиль концентраций, измеренных детектором после прохождения и разделения смеси соединений (в данном случае изомеров соединения) на неподвижной фазе, в зависимости от времени для данного состава и данной скорости потока элюента. Хроматограмма UHPLC состоит из различных пиков или неразрешенных пиков, характерных для соединения или смеси анализируемых соединений.
Методика UHPLC
- Колонка Waters Cortecs® UPLC T3, 150 x 2,1 мм - 1,6 мкм.
Она представляет собой колонку для UPLC с обращенной фазой со сферическими частицами, которая состоит из основы, которая предпочтительно является очень жесткой и сделана из диоксида кремния, окруженной пористым диоксидом кремния с трифункциональной C18-прививкой (октадецильной), и силанольные группы которой обработаны средствами для блокирования концевых групп (эндкепированы). Она также характеризуется длиной 150 мм, внутренним диаметром 2,1 мм, размером частиц 1,6 мкм, пористостью 120 Å и содержанием углерода 4,7%.
Предпочтительно применяемая неподвижная фаза должна быть совместима с водными подвижными фазами.
- Аналитические условия
- Градиент подвижной фазы (% об./об.)
(мин.)
(100%)
(0,0005% об./об. водный раствор)
Композиция, содержащая комплекс формулы (II)
Во-вторых, настоящее изобретение относится к композиции, содержащей
- комплекс формулы (II), характеризующийся по меньшей мере 80% диастереоизомерным избытком смеси изомеров II-RRR и II-SSS, и
- свободный макроциклический лиганд.
В настоящем описании термины "макроциклический лиганд" или "макроциклический хелат" могут применяться в одинаковой мере.
В контексте настоящего изобретения термин "макроцикл" обозначает кольцо, как правило, содержащее по меньшей мере девять атомов, независимо от того, являются ли они атомами углерода или гетероатомами, а термин "макроциклический лиганд" или "макроциклический хелат" представляет собой полидентатный, по меньшей мере бидентатный, лиганд.
Для целей настоящего изобретения, термин "свободный макроциклический лиганд" означает макроциклический лиганд в свободной форме, т.е. не образующий комплекс, в частности, с металлами, включая лантаноиды и актиноиды, или с катионами щелочноземельных металлов, такими как кальций или магний. В частности, свободный макроциклический лиганд не находится в форме комплекса с гадолинием и не вводится в композицию в форме слабого комплекса, как правило, кальция, натрия, цинка или магния, описанного в US 5876695, однако присутствие указанных катионов в следовых количествах в композиции и, следовательно, соответствующих комплексов не исключается.
Как обсуждалось ранее, получение состава на основе комплекса формулы (II) со свободным макроциклическим лигандом, а не слабого комплекса указанного макроциклического лиганда, как рекомендовано в EP 1931673, стало возможным благодаря улучшенной устойчивости диастереоизомерно обогащенного комплекса формулы (II) в соответствии с настоящим изобретением.
В предпочтительном варианте осуществления комплекс формулы (II), присутствующий в композиции в соответствии с настоящим изобретением, характеризуется по меньшей мере 85%, в особенности по меньшей мере 90%, в частности по меньшей мере 92%, более конкретно по меньшей мере 94%, предпочтительно по меньшей мере 97%, преимущественно по меньшей мере 99% диастереоизомерным избытком смеси изомеров II-RRR и II-SSS.
Предпочтительно указанный диастереоизомерный избыток предусматривает по меньшей мере 70%, в особенности по меньшей мере 80%, преимущественно по меньшей мере 90%, предпочтительно по меньшей мере 95% диастереоизомерный избыток смеси изомеров II-RRR и II-SSS.
Преимущественно указанный диастереоизомерный избыток предусматривает смесь изомеров II-RRR и II-SSS.
Термин "смесь изомеров II-RRR и II-SSS" также охватывает, в более широком смысле, случай, когда присутствует только один из изомеров: II-RRR или II-SSS. Однако термин "смесь изомеров II-RRR и II-SSS" предпочтительно обозначает все случаи, в которых каждый из изомеров II-RRR и II-SSS присутствует в различном количестве, отличном от нуля.
В предпочтительном варианте осуществления изомеры II-RRR и II-SSS присутствуют в указанной смеси в соотношении от 65/35 до 35/65, в особенности от 60/40 до 40/60, в частности от 55/45 до 45/55. Преимущественно изомеры II-RRR и II-SSS присутствуют в смеси в соотношении 50/50.
В одном преимущественном варианте осуществления композиция в соответствии с настоящим изобретением характеризуется концентрацией свободного гадолиния, составляющей менее 1 ppm (масса/объем), предпочтительно менее 0,5 ppm (масса/объем).
В настоящем описании, если не указано иное, термины "Gd", "гадолиний" и "Gd3+" применяются в одинаковой мере для обозначения иона Gd3+. В более широком смысле, также может предусматриваться источник свободного гадолиния, такой как хлорид гадолиния (GdCl3) или оксид гадолиния (Gd2O3).
В настоящем изобретении термин "свободный Gd" обозначает не образующие комплекс формы гадолиния, которые предпочтительно доступны для комплексообразования. Как правило, предусматривается ион Gd3+, растворенный в воде. В более широком смысле, также может предусматриваться источник свободного гадолиния, такой как хлорид гадолиния (GdCl3) или оксид гадолиния (Gd2O3).
Гадолиний в свободной форме, как правило, измеряют с помощью колориметрического анализа, применяя, как правило, ксиленоловый оранжевый или арсеназо (III). В отсутствие иона металла (например, гадолиния) данные индикаторы имеют определенный цвет: при кислом значении pH ксиленоловый оранжевый имеет желтый цвет, а арсеназо имеет розовый цвет. В присутствии гадолиния их цвет меняется на фиолетовый.
Визуальное определение изменения цвета раствора позволяет проверить наличие или отсутствие гадолиния в растворе.
Более того, можно количественно измерить содержание свободного гадолиния, который находится в растворе, с помощью обратного титрования, например, с применением EDTA в качестве "слабого" хелата гадолиния. В таком анализе цветной индикатор добавляют до получения фиолетового цвета. Затем к смеси по каплям добавляют EDTA, лиганд гадолиния. Поскольку EDTA является более сильным комплексообразующим средством, чем цветной индикатор, гадолиний меняет лиганд и покидает цветной индикатор, предпочтительно образуя комплекс с EDTA. Таким образом, цветной индикатор постепенно восстанавливает свою не образующую комплекс форму.
Когда количество добавленной EDTA равняется начальному количеству свободного Gd, цветной индикатор полностью находится в свободной форме, а цвет раствора "становится" желтым. Поскольку количество добавленной EDTA известно, это позволяет узнать начальное количество свободного Gd в растворе, подлежащем анализу.
Все эти способы хорошо известны специалистам в данной области техники и, в особенности, описаны в Barge et al. (Contrast Media and Molecular Imaging 1, 2006, 184-188).
Таким образом, эти колориметрические методы, как правило, применяют для раствора со значением pH от 4 до 8. Это обусловлено тем, что за пределами этих диапазонов значений pH точность измерения может быть нарушена из-за модификации (или даже подавления) изменения цвета.
Таким образом, при необходимости значение pH образца, подлежащего анализу, регулируют до 4-8. В особенности, если значение pH образца является кислым и, в частности, составляет менее 4, значение pH преимущественно регулируют путем добавления основания, а затем проводят измерение свободного Gd в образце при отрегулированном значении pH.
Таким образом, композиция в соответствии с настоящим изобретением обладает стабильностью с течением времени, т.е. ее состав продолжает соответствовать спецификациям в отношении концентрации свободного гадолиния (в частности, концентрация свободного Gd остается менее 1 ppm (масса/объем)) в течение периода, составляющего по меньшей мере 3 лет, предпочтительно по меньшей мере 4 лет или более предпочтительно по меньшей мере 5 лет, в особенности, в отношении содержания свободного парамагнитного металла. В соответствии с руководством ICH, наблюдение этой стабильности в течение шести месяцев при температуре 40°C считается хорошим показателем стабильности в течение 3 лет при 25°C.
В одном конкретном варианте осуществления композиция в соответствии с настоящим изобретением характеризуется концентрацией вышеописанного комплекса формулы (II), составляющей от 0,01 до 1,5 моль.л-1, предпочтительно от 0,2 до 0,7 моль.л-1, более предпочтительно от 0,3 до 0,6 моль.л-1.
Комплекс формулы (II) анализируют с помощью способов, известных специалистам в данной области техники. В особенности, его можно анализировать после минерализации и анализа общего содержания гадолиния в композиции с помощью атомно-эмиссионной спектрометрии (также называемой ICP-AES или атомно-эмиссионной спектрометрией с ICP).
Содержание комплекса формулы (II) позволяет этой композиции обладать оптимальной контрастирующей способностью, при этом в то же время обладать удовлетворительной вязкостью. Более конкретно, при концентрации вышеописанного комплекса формулы (II) ниже 0,01 моль.л-1 эксплуатационные характеристики в качестве контрастного средства менее удовлетворительны, а при концентрации выше 1,5 моль.л-1 вязкость данной композиции становится слишком большой для удобного обращения.
В одном конкретном варианте осуществления композиция в соответствии с настоящим изобретением содержит от 0,002 до 0,4 моль/моль %, в особенности от 0,01 до 0,3 моль/моль %, предпочтительно от 0,02 до 0,2 моль/моль % и более предпочтительно от 0,05 до 0,15 моль/моль % свободного макроциклического лиганда относительно комплекса формулы (II).
Преимущественно макроциклический лиганд выбран из группы, состоящей из DOTA, NOTA, DO3A, BT-DO3A, HP-DO3A, PCTA, DOTA-GA и их производных.
Предпочтительно он представляет собой DOTA (1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусную кислоту).
Концентрацию свободной DOTA в композиции, как правило, измеряют с помощью обратного титрования с помощью меди, например, с применением сульфата меди в качестве источника ионов меди.
В данном методе, который хорошо известен специалистам в данной области техники, предпочтительно применяют раствор, содержащий известную начальную концентрацию Q0 сульфата меди, при этом эта концентрация превышает количество свободного лиганда в растворе. Раствор, подлежащий анализу, содержащий свободную DOTA в количестве Q1, которое необходимо определить, добавляют к данному раствору сульфата меди. DOTA является очень хорошим комплексообразующим средством для меди: таким образом, наблюдают образование комплекса DOTA-медь.
Обратное титрование меди, остающейся свободной в растворе, затем преимущественно проводят потенциометрическим методом. Для этого к смеси по каплям добавляют, например, EDTA. EDTA образует комплекс со свободной медью в растворе, не приводя к распаду комплекса DOTA-медь, поскольку DOTA является более сильным комплексообразующим средством, чем EDTA. Когда количество добавленной EDTA Q2 равняется количеству свободной меди в растворе, наблюдается резкое падение потенциала раствора.
Зная начальное количество меди Q0 и количество добавленной EDTA Q2, вычитание этих двух значений Q0 – Q2 дает количество свободной DOTA в растворе, подлежащем анализу, Q1.
В качестве альтернативы можно применять методики HPLC, в особенности методику HILIC LC-UV.
Данные методы измерения (в частности потенциометрические методы) применяют для растворов со значением pH преимущественно от 4 до 8. Таким образом, при необходимости значение pH образца, подлежащего анализу, регулируют до 4-8. В особенности, если значение pH образца является кислым и, в частности, составляет менее 4, значение pH преимущественно регулируют путем добавления основания, такого как меглумин, а затем проводят измерение свободной DOTA в образце при отрегулированном значении pH.
Предпочтительно пропорции, указанные в настоящем изобретении и, в частности, указанные выше, представляют собой пропорции до стерилизации композиции.
Преимущественно значение pH композиции составляет от 4,5 до 8,5, предпочтительно от 5 до 8, преимущественно от 6 до 8, в особенности от 6,5 до 8. В частности, данные диапазоны значений pH позволяют ограничить возникновение определенных примесей и способствовать комплексообразованию иона парамагнитного металла M.
В частности, композиция в соответствии с настоящим изобретением может быть забуферена, т.е. она может также содержать буфер, выбранный из общепринятых буферов с pH в диапазоне от 5 до 8, предпочтительно из лактатного, тартратного, малатного, малеатного, сукцинатного, аскорбатного, карбонатного, Tris (трис(гидроксиметил)аминометана), HEPES (2-[4-(2-гидроксиэтил)-1-пиперазин]этансульфоновой кислоты) и MES (2-морфолиноэтансульфоновой кислоты) буферов и их смесей, и предпочтительно буфер, выбранный из Tris, лактатного, тартратного, карбонатного и MES буферов и их смесей. Преимущественно композиция в соответствии с настоящим изобретением содержит Tris буфер.
Композиция, являющаяся предметом изобретения, предпочтительно стерильна.
Способ получения комплекса формулы (II)
Настоящее изобретение также относится к способу получения комплекса формулы (II), включающего следующие последовательные стадии:
a) обеспечение комплексообразования гексакислоты формулы (III), представленной ниже:
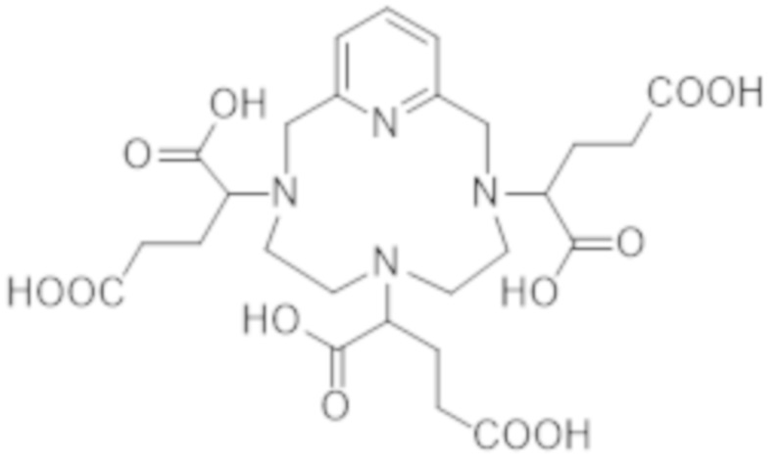 (III),
(III),
с гадолинием с получением комплекса гадолиния и гексакислоты формулы (I), определенной ранее,
b) обеспечение изомеризации путем нагревания комплекса гадолиния и гексакислоты формулы (I) в водном растворе при значении pH от 2 до 4 с получением диастереоизомерно обогащенного комплекса, характеризующегося по меньшей мере 80% диастереоизомерным избытком смеси изомеров I-RRR и I-SSS указанного комплекса гадолиния и гексакислоты формулы (I), и
c) образование комплекса формулы (II) посредством проведения реакции диастереоизомерно обогащенного комплекса, полученного на стадии b), с 3-амино-1,2-пропандиолом.
В настоящем описании, если не указано иное, термины "Gd", "гадолиний" и "Gd3+" применяются в одинаковой мере для обозначения иона Gd3+. В более широком смысле, также может предусматриваться источник свободного гадолиния, такой как хлорид гадолиния (GdCl3) или оксид гадолиния (Gd2O3).
В настоящем изобретении термин "свободный Gd" обозначает не образующие комплекс формы гадолиния, которые предпочтительно доступны для комплексообразования. Как правило, предусматривается ион Gd3+, растворенный в воде. В более широком смысле, также может предусматриваться источник свободного гадолиния, такой как хлорид гадолиния (GdCl3) или оксид гадолиния.
▪ Стадия a)
В ходе этой стадии происходит реакция комплексообразования между гексакислотой формулы (III) и гадолинием, что позволяет получить комплекс гадолиния и гексакислоты формулы (I), определенной ранее.
В соответствии с конкретным вариантом осуществления стадия а) включает проведение реакции между гексакислотой формулы (III) и источником свободного Gd в воде.
В предпочтительном варианте осуществления источником свободного Gd является GdCl3 или Gd2O3, предпочтительно Gd2O3.
Предпочтительно реагенты, применяемые на стадии а), т.е. источник гадолиния (как правило, оксид гадолиния), гексакислота формулы (III) и вода, должны быть как можно более чистыми, в особенности, в том, что касается примесей металлов.
Таким образом, источником гадолиния преимущественно будет представлять собой оксид гадолиния, предпочтительно с чистотой более 99,99% и еще более предпочтительно более 99,999%.
Вода, применяемая в способе, предпочтительно содержит менее 50 ppm кальция, более предпочтительно менее 20 ppm и наиболее предпочтительно менее 15 ppm кальция. Как правило, вода, применяемая в способе, представляет собой деионизированную воду, воду для инъекций (вода со степенью чистоты, подходящей для инъекций) или очищенную воду.
Преимущественно количества реагентов (гексакислоты формулы (III) и гадолиния), применяемых на этой стадии а), соответствуют стехиометрическим пропорциям или близки к ним согласно уравнению баланса реакции комплексообразования, которая происходит во время данной стадии.
Термин "близки к стехиометрическим пропорциям" означает, что разница между молярными пропорциями, в которых вводятся реагенты, и стехиометрическими пропорциями составляет менее 15%, в особенности менее 10%, предпочтительно менее 8%.
В частности, гадолиний можно вводить в небольшом избытке относительно стехиометрических пропорций. В таком случае, отношение количества материала, вводимого в виде гадолиния, к количеству материала, вводимого в виде гексакислоты формулы (III), составляет более 1, но, как правило, менее 1,15, в особенности менее 1,10, преимущественно менее 1,08. Другими словами, количество вводимого гадолиния составляет более 1 эквивалента (экв.), но, как правило, менее 1,15 экв., в особенности менее 1,10 экв., преимущественно менее 1,08 экв. относительно количества вводимой гексакислоты формулы (III), которое само по себе соответствует 1 эквиваленту. В предпочтительном варианте осуществления, в котором источником свободного гадолиния является Gd2O3, количество вводимого Gd2O3, как правило, составляет более 0,5 экв., но менее 0,575 экв., в особенности менее 0,55 экв., преимущественно менее 0,54 экв. относительно количества вводимой гексакислоты формулы (III) (1 экв.).
В соответствии с конкретным вариантом осуществления стадия а) включает следующие последовательные стадии:
a1) получение водного раствора гексакислоты формулы (III) и
a2) добавление к водному раствору, полученному на стадии а1), источника свободного гадолиния.
В данном варианте осуществления содержание гексакислоты формулы (III) в водном растворе, полученном на стадии а1), как правило, составляет от 10% до 60%, в особенности от 15% до 45%, предпочтительно от 20% до 35%, преимущественно от 25% до 35% и еще более преимущественно от 25% до 30% по весу относительно общего веса водного раствора.
Предпочтительно стадии а) и b) выполняют в соответствии с вариантом осуществления с однореакторным синтезом, т.е. в одном реакторе и без промежуточной стадии выделения или очистки.
Таким образом, в данном предпочтительном варианте осуществления комплекс гадолиния и гексакислоты формулы (I), образованный на стадии а), непосредственно подвергают стадии b) изомеризации без выделения или очистки и в том же реакторе, что и на стадии а).
▪ Стадия b)
Комплекс гадолиния и гексакислоты формулы (I), образованный посредством реакции комплексообразования между гексакислотой формулы (III) и гадолинием на стадии а), первоначально получают в форме смеси диастереоизомеров.
Стадия b) направлена на обогащение смеси диастереоизомеров изомерами I-RRR и I-SSS для получения диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I), характеризующегося по меньшей мере 85%, в особенности по меньшей мере 90%, в частности по меньшей мере 95%, предпочтительно по меньшей мере 97%, преимущественно по меньшей мере 98%, более преимущественно по меньшей мере 99% диастереоизомерным избытком смеси изомеров I-RRR и I-SSS.
В контексте настоящего изобретения под термином "диастереоизомерный избыток" подразумевают указание в отношении комплекса гадолиния и гексакислоты формулы (I) того факта, что указанный комплекс преимущественно присутствует в виде изомера или группы изомеров, выбранных из диастереоизомеров I-RRR, I-SSS, I-RRS, I-SSR, I-RSS, I-SRR, I-RSR и I-SRS. Указанный диастереоизомерный избыток выражается в процентах и соответствует количеству, представленному преобладающим изомером или группой изомеров относительно общего количества комплекса гадолиния и гексакислоты формулы (I). Понятно, что это процентное содержание может быть молярным или массовым, поскольку изомеры по определению имеют одинаковую молярную массу.
Предпочтительно указанный диастереоизомерный избыток предусматривает по меньшей мере 70%, в особенности по меньшей мере 80%, преимущественно по меньшей мере 90%, предпочтительно по меньшей мере 95% диастереоизомерный избыток смеси изомеров I-RRR и I-SSS.
Преимущественно указанный диастереоизомерный избыток предусматривает смесь изомеров I-RRR и I-SSS.
По сути, авторы настоящего изобретения обнаружили, что такие факторы, как значение pH и температура раствора комплекса гадолиния и гексакислоты формулы (I), полученного по завершении стадии а), влияют на соотношение, в котором различные изомеры комплекса формулы (I) присутствуют в смеси диастереоизомеров. С течением времени смесь имеет склонность к обогащению группой изомеров, включающей изомеры, которые неожиданно являются наиболее термодинамически устойчивыми, а также наиболее химически устойчивыми, в данном случае изомеры I-RRR и I-SSS.
Термин "смесь изомеров I-RRR и I-SSS" также охватывает, в более широком смысле, случай, когда присутствует только один из изомеров: I-RRR или I-SSS.
Однако в предпочтительном варианте осуществления изомеры I-RRR и I-SSS присутствуют в указанной смеси в соотношении от 65/35 до 35/65, в особенности от 60/40 до 40/60, в особенности от 55/45 до 45/55. Преимущественно смесь изомеров I-RRR/I-SSS представляет собой рацемическую смесь (50/50).
Стадию b) изомеризации комплекса гадолиния и гексакислоты формулы (I) в водном растворе, как правило, проводят при значении pH от 2 до 4, в особенности от 2 до 3, преимущественно от 2,2 до 2,8.
Значение pH предпочтительно регулируют с помощью кислоты, предпочтительно неорганической кислоты, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота или фосфорная кислота, например, с помощью хлористоводородной кислоты.
Совершенно удивительно, что при таких условиях pH происходит обогащение смеси конкретными изомерами, в данном случае изомерами I-RRR и I-SSS, поскольку в данной области техники известно, что хелаты гадолиния характеризуются низкой кинетической инертностью в кислой среде. Более конкретно, чем выше концентрация ионов H+ в среде, тем больше вероятность переноса протона на один из донорных атомов лиганда, что приводит к диссоциации комплекса. В результате, специалист в данной области техники мог бы ожидать, что помещение комплекса гадолиния и гексакислоты формулы (I) в водный раствор при значении pH от 2 до 4 приведет к диссоциации указанного комплекса, а не к его изомеризации в I-RRR и I-SSS.
Следует отметить, что диапазон значений pH, рекомендованный в EP 1931673 для комплексообразования гексакислоты формулы (III), а именно 5,0-6,5, не позволяет получать комплекс формулы (I), обогащенный его изомерами I-RRR и I-SSS.
Стадию b), как правило, проводят при температуре от 80°C до 130°C, в особенности от 90°C до 125°C, предпочтительно от 98°C до 122°C, преимущественно от 100°C до 120°C, как правило, в течение времени от 10 часов до 72 часов, в особенности от 10 часов до 60 часов, преимущественно от 12 часов до 48 часов.
Вопреки всем ожиданиям, такие температурные условия, которые в сочетании с вышеупомянутыми условиями pH должны способствовать неустойчивости хелата гадолиния, приводят не к распаду его комплекса или образованию любой другой примеси, а к его изомеризации в I-RRR и I-SSS.
В одном конкретном варианте осуществления водный раствор на стадии b) содержит уксусную кислоту. Стадию b) затем преимущественно проводят при температуре от 100°C до 120°C, в особенности от 110°C до 118°C, как правило, в течение времени от 12 часов до 48 часов, в особенности от 20 часов до 30 часов, в частности от 24 часов до 26 часов.
Уксусную кислоту предпочтительно добавляют перед нагреванием раствора комплекса гадолиния и гексакислоты формулы (I), любых примесей, присутствующих в водном растворе, которые могут возникнуть в результате предыдущих стадий, для получения на стадии а), в таком количестве, чтобы содержание уксусной кислоты составляло от 25% до 75%, в особенности от 40% до 50% по массе относительно массы гексакислоты формулы (III), применяемой на стадии а).
При нагревании водного раствора до температуры преимущественно от 100°C до 120°C, как правило, от 110°C до 118°C, уксусную кислоту добавляют постепенно по мере испарения воды, чтобы поддерживать постоянный объем раствора.
В соответствии с предпочтительным вариантом осуществления по завершении стадии b) диастереоизомерно обогащенный комплекс выделяют путем кристаллизации, предпочтительно путем кристаллизации с применением затравочных кристаллов.
В данном варианте осуществления стадия b) включает следующие последовательные стадии:
b1) обеспечение изомеризации путем нагревания комплекса гадолиния и гексакислоты формулы (I) в водном растворе при значении pH от 2 до 4 с получением диастереоизомерно обогащенного комплекса, характеризующегося по меньшей мере 80% диастереоизомерным избытком смеси изомеров I-RRR и I-SSS указанного комплекса гадолиния и гексакислоты формулы (I), и
b2) выделение путем кристаллизации указанного диастереоизомерно обогащенного комплекса, предпочтительно путем кристаллизации с применением затравочных кристаллов.
Стадия b2) кристаллизации направлена, во-первых, на удаление л обесцвеченного продукта более высокой чистоты в форме кристаллов, и, во-вторых, на продолжение диастереоизомерного обогащения комплекса гадолиния и гексакислоты формулы (I) для получения более высокого диастереоизомерного избытка смеси изомеров I-RRR и I-SSS указанного комплекса, чем полученный по завершении стадии b1). Более конкретно, изомеры I-RRR и I-SSS комплекса с гексакислотой формулы (I) кристаллизуются из воды. С другой стороны, комплекс гадолиния и гексакислоты формулы (I), не обогащенный указанными изомерами, не кристаллизуется.
Совершенно неожиданным результатом является то, что изомеры I-RRR и I-SSS, которыми комплекс имеет склонность обогащаться в ходе стадии b) (и, вопреки всем ожиданиям, ввиду условий, в которых она проводится), являются единственными изомерами комплекса, которые кристаллизуются из воды. Таким образом, изомеризация и кристаллизация синергетически способствуют обогащению изомерами I-RRR и I-SSS и, в результате, общей эффективности способа в соответствии с настоящим изобретением.
Более того, следует отметить, что кристаллизация из воды представляющих интерес изомеров комплекса гадолиния и гексакислоты формулы (I) позволяет избежать добавления растворителя, как описано в примере 7 в ЕР 1931673, который включает стадию осаждения из этанола тринатриевой соли указанного комплекса.
Стадию b2) преимущественно проводят при температуре от 10°C до 70°C, в особенности от 30°С до 65°С, в частности от 35°С до 60°С.
В соответствии с одним вариантом осуществления после понижения температуры водного раствора так, чтобы она находилась в пределах указанных выше диапазонов, процесс кристаллизации инициируют путем применения затравочных кристаллов. "Кристаллизация с применением затравочных кристаллов", также называемая "кристаллизация с применением затравки", предусматривает введение в реактор, в котором проводят кристаллизацию (также называемый кристаллизационным сосудом), известного количества кристаллов, называемых "затравочные кристаллы" или "затравка". Это позволяет сократить время кристаллизации. Кристаллизация с применением затравочных кристаллов хорошо известна специалистам в данной области техники. В способе согласно настоящему изобретению введение затравочных кристаллов с применением затравки, в данном случае кристаллов диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I), добавляемых к водному раствору диастереоизомерно обогащенного комплекса, температуру которого заранее понизили, позволяет обеспечивать зародышеобразование и, таким образом, инициировать кристаллизацию. Продолжительность кристаллизации с применением затравочных кристаллов преимущественно составляет от 2 часов до 20 часов и предпочтительно от 6 часов до 18 часов; как правило, составляет 16 часов.
Кристаллы диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I) затем, как правило, выделяют путем фильтрования и высушивания с помощью любой методики, хорошо известной специалистам в данной области техники.
Преимущественно степень чистоты диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I), выделенного по завершении стадии b2), составляет более 95%, в особенности более 98%, преимущественно более 99%, при этом указанная степень чистоты выражается в виде массовой доли комплекса формулы (I) относительно общей массы, полученной по завершении стадии b2).
В конкретном варианте осуществления диастереоизомерно обогащенный комплекс со стадии b), выделенный путем кристаллизации, снова очищают путем перекристаллизации с получением диастереоизомерно обогащенного и очищенного комплекса.
В данном варианте осуществления стадия b) включает, помимо последовательных стадий b1) и b2), описанных ранее, стадию b3) очистки путем перекристаллизации выделенного диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I).
Стадия b3) перекристаллизации, как и стадия b2) кристаллизации, направлена, во-первых, на получение продукта более высокой чистоты, а, во-вторых, на продолжение диастереоизомерного обогащения комплекса гадолиния и гексакислоты формулы (I) для получения более высокого диастереоизомерного избытка смеси изомеров I-RRR и I-SSS указанного комплекса, чем полученный по завершении стадии b2).
Стадия b3), как правило, включает следующие последовательные подстадии:
• суспендирование диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I), выделенного на стадии b2), в водном растворе, предпочтительно в воде,
• растворение указанного комплекса путем нагревания до температуры преимущественно от 80°C до 120°C, например, до 100°C,
• перекристаллизация, предпочтительно путем применения затравочных кристаллов, при температуре преимущественно от 10°C до 90°C, в особенности от 20°C до 87°C, в частности от 55°C до 85°C, как правило, в течение времени от 2 часов до 20 часов, а именно от 6 часов до 18 часов, и
• выделение кристаллов диастереоизомерно обогащенного и очищенного комплекса гадолиния и гексакислоты формулы (I), например, путем фильтрования и высушивания.
Степень чистоты очищенного диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I), выделенного по завершении стадии b3), как правило, составляет более 98%, в особенности более 99%, преимущественно более 99,5%, при этом указанная степень чистоты выражается в виде массовой доли комплекса формулы (I) относительно общей массы, полученной по завершении стадии b2).
В другом варианте осуществления диастереоизомерно обогащенный комплекс со стадии b) дополнительно обогащают путем обеспечения селективного распада комплексов диастереоизомеров комплекса формулы (I), отличных от диастереоизомеров I-RRR и I-SSS, т.е. путем обеспечения селективного распада комплексов диастереоизомеров I-RSS, I-SRR, I-RSR, I-SRS, I-RRS и I-SSR.
В данном варианте осуществления стадия b) включает, помимо последовательных стадий b1) и b2), описанных ранее, стадию b4) обеспечения селективного распада комплексов диастереоизомеров комплекса формулы (I), отличных от диастереоизомеров I-RRR и I-SSS. В данном варианте осуществления стадия b) может также включать стадию b3), описанную ранее, при этом указанную стадию b3) проводят между стадиями b2) и b4) или после стадии b4).
Стадия обеспечения селективного распада комплекса b4) направлена на продолжение диастереоизомерного обогащения комплекса гадолиния и гексакислоты формулы (I) для получения более высокого диастереоизомерного избытка смеси изомеров I-RRR и I-SSS указанного комплекса, чем полученный по завершении стадии b2) или по завершении стадии b3), при проведении указанной стадии до стадии b4).
Стадия b4), как правило, включает следующие последовательные подстадии:
• суспендирование диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I), выделенного на стадии b2) или на стадии b3), в воде,
• добавление основания, например, гидроксида натрия,
• нагревание до температуры преимущественно от 30°C до 60°C, в особенности от 35°C до 55°C, например, при 40°C, как правило, в течение времени от 2 часов до 20 часов, в особенности от 10 часов до 18 часов,
• охлаждение до температуры преимущественно от 10°C до 30°C, например до 30°C, и
• выделение диастереоизомерно обогащенного и очищенного комплекса гадолиния и гексакислоты формулы (I), например, путем фильтрования и высушивания.
Проведение стадии b4) стало возможным благодаря тому, что изомеры I-RRR и I-SSS являются наиболее устойчивыми в основной среде. Такие основные условия способствуют образованию гидроксида гадолиния и, в результате, распаде комплексов наименее устойчивых изомеров. Таким образом, следует отметить, что неожиданно изомеры I-RRR и I-SSS более устойчивы как в кислой среде, что позволяет проводить стадию b1) изомеризации, так и в основной среде, что позволяет проводить стадию b4) обеспечения селективного распада комплекса.
В предпочтительном варианте осуществления диастереоизомерно обогащенный комплекс, полученный по завершении стадии b) в соответствии с любым из вышеописанных вариантов осуществления, характеризуется по меньшей мере 85%, в особенности по меньшей мере 90%, в частности по меньшей мере 95%, предпочтительно по меньшей мере 97%, преимущественно по меньшей мере 98%, более преимущественно по меньшей мере 99% диастереоизомерным избытком смеси изомеров I-RRR и I-SSS.
Предпочтительно указанный диастереоизомерный избыток предусматривает по меньшей мере 70%, в особенности по меньшей мере 80%, преимущественно по меньшей мере 90%, предпочтительно по меньшей мере 95% диастереоизомерный избыток смеси изомеров I-RRR и I-SSS.
Преимущественно указанный диастереоизомерный избыток предусматривает смесь изомеров I-RRR и I-SSS.
Термин "смесь изомеров I-RRR и I-SSS" также охватывает, в более широком смысле, случай, когда присутствует только один из изомеров: I-RRR или I-SSS. Однако термин "смесь изомеров I-RRR и I-SSS" предпочтительно обозначает все случаи, в которых каждый из изомеров I-RRR и I-SSS присутствует в различном количестве, отличном от нуля.
В предпочтительном варианте осуществления изомеры I-RRR и I-SSS присутствуют в указанной смеси в соотношении от 65/35 до 35/65, в особенности от 60/40 до 40/60, в частности от 55/45 до 45/55. Преимущественно смесь изомеров I-RRR/I-SSS представляет собой рацемическую смесь (50/50).
▪ Стадия c)
Стадия c) направлена на образование комплекса формулы (II) из его предшественника, диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I), полученного на стадии b).
Во время данной стадии три функциональные группы карбоновой кислоты комплекса гадолиния и гексакислоты формулы (I), переносимые атомами углерода, которые расположены на боковых цепях комплекса в γ-положении по отношению к атомам азота макроцикла, к которому привиты указанные боковые цепи, превращают в амидные функциональные группы посредством реакции амидирования с 3-амино-1,2-пропандиолом, в рацемической или энантиомерно чистой форме, предпочтительно в рацемической форме.
Данная реакция амидирования не изменяет абсолютную конфигурацию трех асимметрических атомов углерода, расположенных на боковых цепях в α-положении по отношению к атомам азота макроцикла, к которому привиты указанные боковые цепи. В результате, стадия c) позволяет получать комплекс формулы (II) с диастереоизомерным избытком смеси изомеров II-RRR и II-SSS, идентичным диастереоизомерному избытку изомеров I-RRR и I-SSS, с которым получают диастереоизомерно обогащенный комплекс гадолиния и гексакислоты формулы (I), полученный по завершении стадии b), который составляет по меньшей мере 80%.
В предпочтительном варианте осуществления комплекс формулы (II), полученный по завершении стадии c), характеризуется по меньшей мере 85%, в особенности по меньшей мере 90%, в частности по меньшей мере 92%, предпочтительно по меньшей мере 94%, преимущественно по меньшей мере 97%, более преимущественно по меньшей мере 99% диастереоизомерным избытком смеси изомеров II-RRR и II-SSS.
Предпочтительно указанный диастереоизомерный избыток предусматривает по меньшей мере 70%, в особенности по меньшей мере 80%, преимущественно по меньшей мере 90%, предпочтительно по меньшей мере 95% диастереоизомерный избыток смеси изомеров II-RRR и II-SSS.
Преимущественно указанный диастереоизомерный избыток предусматривает смесь изомеров II-RRR и II-SSS.
Термин "смесь изомеров II-RRR и II-SSS" также охватывает, в более широком смысле, случай, когда присутствует только один из изомеров: II-RRR или II-SSS. Однако термин "смесь изомеров II-RRR и II-SSS" предпочтительно обозначает все случаи, в которых каждый из изомеров II-RRR и II-SSS присутствует в различном количестве, отличном от нуля.
В предпочтительном варианте осуществления изомеры II-RRR и II-SSS присутствуют в указанной смеси в соотношении от 65/35 до 35/65, в особенности от 60/40 до 40/60, в частности от 55/45 до 45/55. Преимущественно изомеры II-RRR и II-SSS присутствуют в смеси в соотношении 50/50.
Реакция амидирования может быть проведена согласно любого способа, который хорошо известен специалистам в данной области техники, в особенности, в присутствии реагента для активации функциональных групп карбоновой кислоты и/или путем кислотного катализа.
В особенности, она может быть проведена в соответствии со способами, описанными в EP 1931673, в особенности, в абзаце [0027] указанного патента.
В одном конкретном варианте осуществления стадия c) включает активацию функциональных групп карбоновой кислоты (-COOH) комплекса с гексакислотой формулы (I), переносимых атомами углерода, которые расположены на боковых цепях комплекса в γ-положении по отношению к атомам азота макроцикла, к которому привиты указанные боковые цепи, в форме производных функциональных групп, включающих карбонильную группу (C=O), в которых атом углерода карбонильной группы является более электрофильным, чем атом углерода карбонильной группы в составе функциональных групп карбоновой кислоты. Таким образом, в соответствии с данным конкретным вариантом осуществления указанные функциональные группы карбоновой кислоты могут быть, в особенности, активированы в форме сложноэфирных, ацилхлоридных или ангидридных функциональных групп, или в любой активированной форме, которая может приводить к образованию амидной связи. Активированные формы, которые могут приводить к образованию амидной связи, хорошо известны специалистам в данной области техники и могут быть получены, например, с помощью набора способов создания пептидной связи, известных в химии пептидов. Примеры таких способов приведены в публикации Synthesis of peptides and peptidomimetics, volume E22a, pages 425-588, Houben-Weyl et al., Goodman Editor, Thieme-Stuttgart-New York (2004), и среди этих примеров можно в особенности указать способы активации карбоновых кислот посредством образования азида (ацилазида), например, посредством действия реагента, такого как дифенилфосфорилазид (обычно обозначаемый сокращением DPPA), применения карбодиимидов отдельно или в присутствии катализаторов (например, N-гидроксисукцинимида и его производных), применения карбонилдиимидазола (1,1'-карбонилдиимидазола, CDI), применения солей фосфония, таких как бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфат (обычно обозначаемый сокращением BOP), или урония, таких как 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (обычно обозначаемый сокращением HBTU).
Предпочтительно стадия c) включает активацию вышеупомянутых функциональных групп карбоновой кислоты (-COOH) в форме сложноэфирных, ацилхлоридных или ангидридных функциональных групп.
Данный вариант осуществления является более предпочтительным по сравнению с проведением реакции образования пептидной связи путем активации функциональной группы карбоновой кислоты с применением вещества для сочетания, такого как EDCI/HOBT, как описано в EP 1931673. Более конкретно, такая реакция сочетания приводит к образованию одного эквивалента 1-этил-3-[3–(диметиламино)пропил]мочевины, которую необходимо удалить, в особенности, с помощью хроматографии на диоксиде кремния или жидкофазной экстракцией путем добавления растворителя. Независимо от увеличения сложности способа, вызванного такой дополнительной стадией, применение таких способов очистки нежелательно, как обсуждалось ранее. Кроме того, применение HOBT само по себе проблематично, поскольку он представляет собой взрывоопасный продукт.
Для целей настоящего изобретения под термином "сложноэфирная функциональная группа" подразумевают обозначение группы -C(O)O-. В частности, она может представлять собой группу -C(O)O-R1, в которой R1 соответствует (C1-C6)алкильной группе.
Для целей настоящего изобретения термин "(C1-C6)алкильная группа" означает линейную или разветвленную цепь на основе насыщенного углеводорода, содержащую от 1 до 6 и предпочтительно от 1 до 4 атомов углерода. Примеры, которые можно указать, включают метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, пентильную и гексильную группы.
Для целей настоящего изобретения под термином "ацилхлоридная функциональная группа", также известным как "хлорангидридная функциональная группа", подразумевают обозначение группы -CO-Cl.
Для целей настоящего изобретения под термином "ангидридная функциональная группа" подразумевают обозначение группы -CO-O-CO-. В частности, она может представлять собой группу -CO-O-CO-R2, где R2 соответствует (C1-C6)алкильной группе.
Реакции превращения функциональной группы карбоновой кислоты в сложноэфирную, ацилхлоридную или ангидридную функциональные группы хорошо известны специалисту в данной области техники, который сможет провести их любым знакомым ему обычным способом.
Затем комплекс формулы (II) получают посредством аминолиза функциональных групп карбоновой кислоты, активированных в форме сложноэфирных, ацилхлоридных или ангидридных функциональных групп, в особенности, сложноэфирных или ангидридных функциональных групп, предпочтительно сложноэфирных функциональных групп, посредством проведения реакции с 3-амино-1,2-пропандиолом в рацемической или энантиомерно чистой форме, предпочтительно в рацемической форме.
Предпочтительно стадии активации функциональных групп карбоновой кислоты и аминолиза проводят в соответствии с вариантом осуществления с однореакторным синтезом, т.е. в одном реакторе и без промежуточной стадии выделения или очистки промежуточного продукта, включающего функциональные группы карбоновой кислоты, активированные в форме сложноэфирных, ацилхлоридных или ангидридных функциональных групп, в особенности сложноэфирных или ангидридных функциональных групп, предпочтительно сложноэфирных функциональных групп.
В соответствии с конкретным вариантом осуществления стадия c) включает следующие последовательные стадии:
c1) образование активированного комплекса формулы (VII),
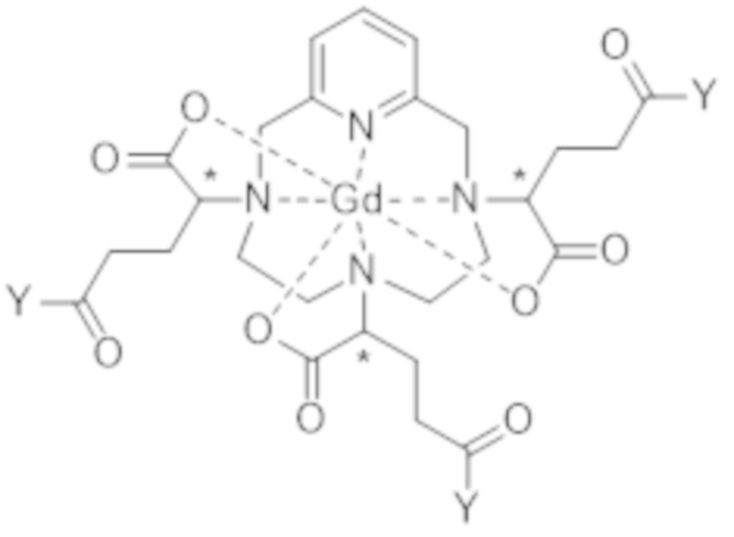 (VII),
(VII),
где Y представляет собой атом хлора, группу -OR1 или -O-C(O)-R2; предпочтительно Y представляет собой группу -OR1 или -O-C(O)-R2, при этом R1 и R2 независимо друг от друга соответствуют (C1-C6)алкильной группе, и
c2) обеспечение аминолиза активированного комплекса формулы (VII) с 3-амино-1,2-пропандиолом.
Как станет ясно специалисту в данной области техники, реакция образования активированного комплекса формулы (VII) не изменяет абсолютную конфигурацию трех асимметрических атомов углерода, расположенных на боковых цепях в α-положении по отношению к атомам азота макроцикла, к которому привиты указанные боковые цепи. В результате, стадия c1) позволяет получать активированный комплекс формулы (VII) с диастереоизомерным избытком смеси изомеров VII-RRR и VII-SSS формулы (VII-RRR) и формулы (VII-SSS), представленных ниже, идентичным диастереоизомерному избытку смеси изомеров I-RRR и I-SSS, с которым получают диастереоизомерно обогащенный комплекс гадолиния и гексакислоты формулы (I), полученный по завершении стадии b), который составляет по меньшей мере 80%.
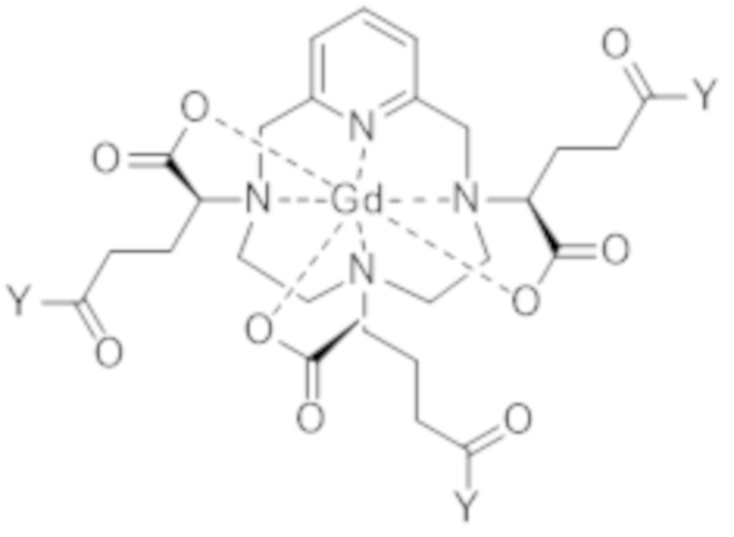 (VII-SSS),
(VII-SSS),
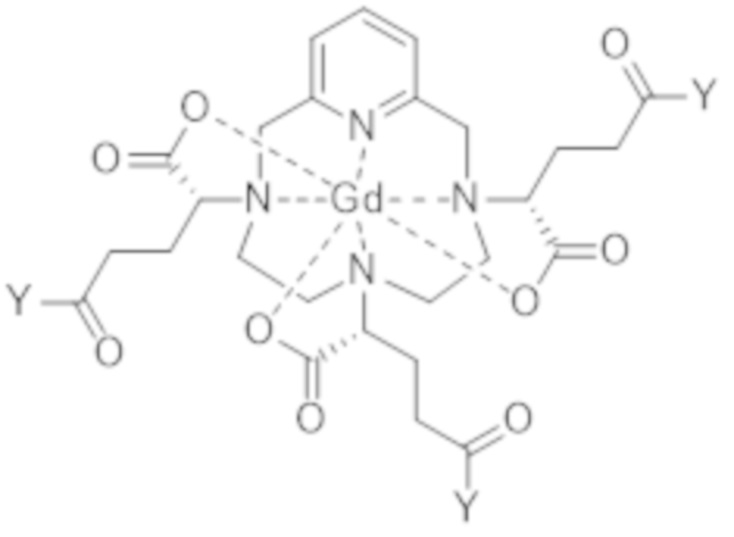 (VII-RRR).
(VII-RRR).
В случае, если Y представляет собой атом хлора, стадию c1), как правило, проводят посредством проведения реакции между диастереоизомерно обогащенным комплексом гадолиния и гексакислоты формулы (I), полученным на стадии b), и тионилхлоридом (SOCl2).
В случае, если Y представляет собой группу -O-C(O)-CH3, стадию c1), как правило, проводят посредством проведения реакции между диастереоизомерно обогащенным комплексом гадолиния и гексакислоты формулы (I), полученным на стадии b), и ацетилхлоридом.
В преимущественном варианте осуществления стадия c) включает активацию вышеупомянутых функциональных групп карбоновой кислоты (-COOH) в форме сложноэфирных функциональных групп.
В соответствии с данным вариантом осуществления стадия c) может, в частности, включать следующие последовательные стадии:
c1) образование сложного триэфира формулы (VIII),
 (VIII),
(VIII),
где R1 представляет собой (C1-C6)алкильную группу, и
c2) обеспечение аминолиза сложного триэфира формулы (VIII) с 3-амино-1,2-пропандиолом.
Стадию c1), как правило, проводят в спирте формулы R1OH, который выступает в качестве растворителя и реагента в присутствии кислоты, такой как хлористоводородная кислота.
Стадию c2), как правило, также проводят в спирте формулы R1OH в присутствии кислоты, такой как хлористоводородная кислота.
На первой стадии в реактор помещают комплекс гадолиния и гексакислоты формулы (I) и спирт R1OH. Затем реакционную среду охлаждают до температуры ниже 10°C, в особенности ниже 5°C, как правило, до 0°C, и затем постепенно добавляют кислый раствор спирта R1OH, как правило, раствор хлористоводородной кислоты в R1OH. Реакционную среду продолжают перемешивать при комнатной температуре (т.е. при температуре от 20 до 25°C) в течение времени, как правило, более 5 часов, предпочтительно от 10 часов до 20 часов. Реакционную среду охлаждают до температуры ниже 10°C, в особенности от 0°C до 5°C, перед проведением стадии c2).
Таким образом, стадии c1) и c2) могут быть легко проведены в соответствии с вариантом осуществления с однореакторным синтезом. Преимущественно сложный триэфир формулы (VII) не выделяют между стадиями с1) и с2).
Однако для ускорения реакции аминолиза на стадии c2) спирт формулы R1OH предпочтительно удаляют с помощью вакуумной перегонки.
Для целей настоящего изобретения термин "вакуумная перегонка" означает перегонку смеси, проводимую при давлении от 10 до 500 мбар, в особенности от 10 до 350 мбар, предпочтительно от 10 до 150 мбар, в частности от 50 до 100 мбар.
Аналогичным образом, чтобы ускорить реакцию аминолиза на стадии c2) 3-амино-1,2-пропандиол вводят в большом избытке. Как правило, количество вводимого материала 3-амино-1,2-пропандиола составляет более 4 экв., в особенности более 7 экв., преимущественно более 10 экв. относительно количества материала диастереоизомерно обогащенного комплекса гадолиния и гексакислоты формулы (I), первоначально вводимого на стадии c), которое само по себе соответствует 1 эквиваленту.
Неожиданно, несмотря на кислые условия, как правило, применяемые на стадиях c1) и c2), которые должны увеличивать кинетическую неустойчивость комплексов гадолиния, распада комплекса или изомеризации сложного триэфира формулы (VIII) не наблюдается. Необходимый триамид получают с очень хорошей степенью превращения и сохранением абсолютной конфигурации трех асимметрических атомов углерода, расположенных на боковых цепях в α-положении по отношению к атомам азота макроцикла.
Более того, следует отметить, что, как правило, реакции амидирования посредством непосредственной реакции между сложным эфиром и амином почти не описаны в литературе (см. по данному вопросу K.C. Nadimpally et al., Tetrahedron Letters, 2011, 52, 2579-2582).
В предпочтительном варианте осуществления стадия c) включает следующие последовательные стадии:
c1) образование метилового сложного триэфира формулы (IV),
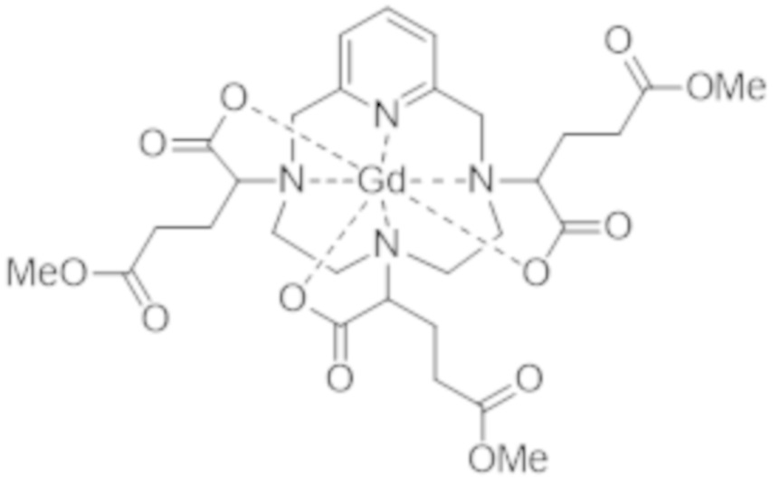 (IV),
(IV),
в частности, посредством проведения реакции в метаноле в присутствии кислоты, такой как хлористоводородная кислота, и
c2) обеспечение аминолиза метилового сложного триэфира формулы (IV) с 3-амино-1,2-пропандиолом, в особенности, в метаноле в присутствии кислоты, такой как хлористоводородная кислота.
Преимущественно метиловый сложный триэфир формулы (IV) не выделяют между стадиями с1) и с2).
В предпочтительном варианте осуществления на стадии c2) метанол удаляют с помощью вакуумной перегонки до тех пор, пока температура, как правило, не превысит 55°C, в особенности, не превысит от 60°C до 65°C, и реакционную среду поддерживают при данной температуре под вакуумом в течение времени, как правило, более 5 часов, в особенности от 10 часов до 20 часов, прежде чем охладить до комнатной температуры и разбавить водой.
Настоящее изобретение охватывает все комбинации конкретных, преимущественных или предпочтительных вариантов осуществления, описанных выше в связи с каждой стадией способа.
▪ Получение гексакислоты формулы (III)
Гексакислота формулы (III), которая участвует в стадии а) способа получения комплекса формулы (II) в соответствии с настоящим изобретением, может быть получена любым уже известным способом, в особенности, способами, описанными в ЕР 1931673.
Однако в соответствии с предпочтительным вариантом осуществления гексакислоту формулы (III) получают алкилированием пиклена формулы (V):
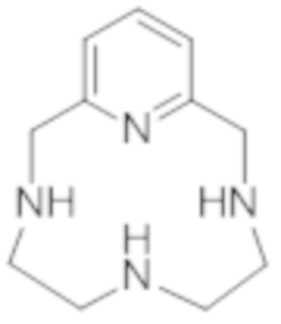 (V),
(V),
соединением формулы R3OOC-CHGp-(CH2)2-COOR4 (IX),
где
- R3 и R4 независимо друг от друга представляют собой (C3-C6)алкильную группу, в особенности (C4-C6)алкильную группу, такую как бутильная, изобутильная, втор-бутильная, трет-бутильная, пентильная или гексильная группа, и
- Gp представляет собой уходящую группу, такую как тозилатная или трифлатная группа, или атом галогена, предпочтительно атом брома,
с получением сложного гексаэфира формулы (X),
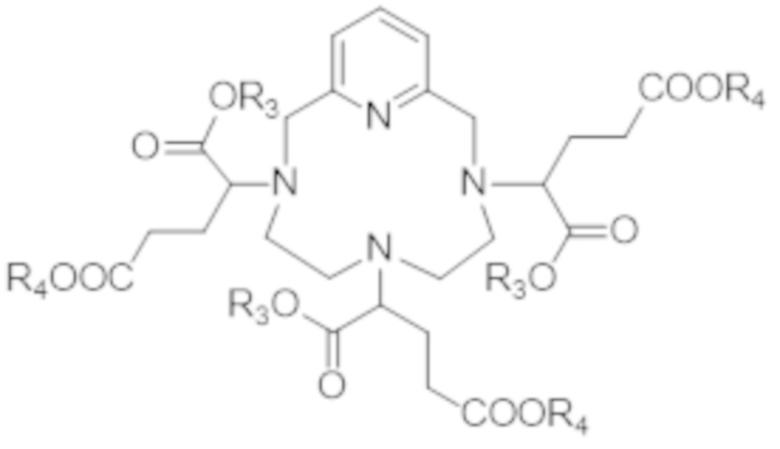 (X),
(X),
с последующей стадией гидролиза, приводящей к получению указанной гексакислоты формулы (III).
В предпочтительном варианте осуществления R3 и R4 идентичны.
В соответствии с предпочтительным вариантом осуществления гексакислоту формулы (III) получают алкилированием пиклена формулы (V):
(V),
дибутил-2-бромглутаратом с получением бутилового сложного гексаэфира формулы (VI):
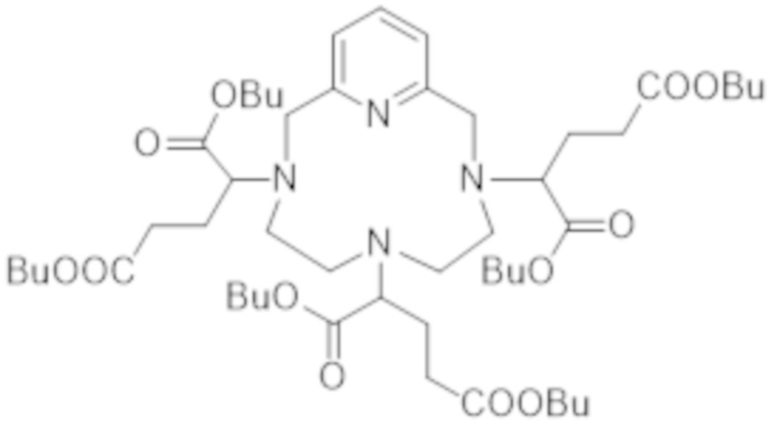 (VI),
(VI),
с последующей стадией гидролиза, приводящей к получению указанной гексакислоты формулы (III).
Применяемый дибутил-2-бромглутарат находится в рацемической или энантиомерно чистой форме, предпочтительно в рацемической форме.
Применение дибутил-2-бромглутарата особенно преимущественно по сравнению с применением этил-2-бромглутарата, описанного в EP 1931673. Более конкретно, коммерческий диэтил-2-бромглутарат представляет собой относительно неустойчивое соединение, которое разлагается с течением времени и под действием температуры. А именно, этот сложный эфир имеет склонность к гидролизу или циклизации и, таким образом, к потере его атома брома. Попытки очистить коммерческий диэтил-2-бромглутарат или разработать новые пути синтеза для его получения с улучшенной чистотой и, таким образом, предотвратить его разложение оказались безуспешными.
Реакцию алкилирования, как правило, проводят в полярном растворителе, предпочтительно в воде, в частности в деионизированной воде, преимущественно в присутствии основания, такого как карбонат калия или натрия.
В особенности, применение воды является более предпочтительным по сравнению с применением ацетонитрила, описанным в EP 1931673, по понятным причинам.
Реакцию преимущественно проводят при температуре от 40°C до 80°C, как правило, от 50°C до 70°C и, в особенности от 55°C до 60°C, в течение времени от 5 часов до 20 часов, в особенности от 8 часов до 15 часов.
Стадию гидролиза преимущественно проводят в присутствии кислоты или основания, преимущественно основания, такого как гидроксид натрия. Растворителем для гидролиза может быть вода, спирт, такой как этанол, или смесь вода/спирт. Данную стадию преимущественно проводят при температуре от 40°C до 80°C, как правило, от 40°C до 70°C и, в особенности, от 50°C до 60°C, как правило, в течение времени от 10 часов до 30 часов, в особенности от 15 часов до 25 часов.
Способ очистки комплекса формулы (II)
Настоящее изобретение также относится к способу очистки комплекса формулы (II), представленной ниже:
(II),
характеризующегося по меньшей мере 80% диастереоизомерным избытком смеси изомеров II-RRR и II-SSS формул:
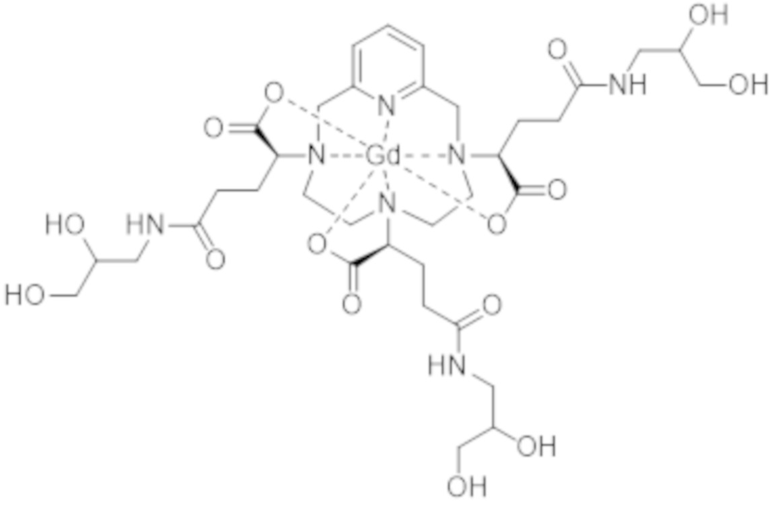 (II-SSS),
(II-SSS),
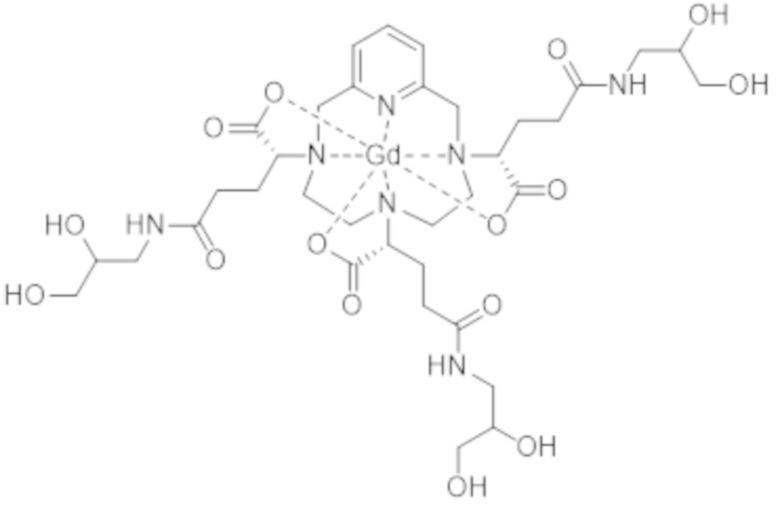 (II-RRR),
(II-RRR),
включающему
1) комбинацию следующих двух стадий:
1b) пропускания через ионообменную(-ые) смолу(-ы) и
1c) ультрафильтрации указанного комплекса, и
2) выделение полученного таким образом очищенного комплекса в твердой форме.
Преимущественно указанный комплекс формулы (II), характеризующийся по меньшей мере 80%, предпочтительно по меньшей мере 85%, в особенности по меньшей мере 90%, в частности по меньшей мере 95%, более конкретно по меньшей мере 97%, предпочтительно по меньшей мере 98% и преимущественно по меньшей мере 99% диастереоизомерным избытком смеси изомеров II-RRR и II-SSS, получали ранее в соответствии со способом получения, описанным ранее.
В предпочтительном варианте осуществления диастереоизомерно обогащенный комплекс, в отношении которого проводят стадию очистки, характеризуется по меньшей мере 85%, в особенности по меньшей мере 90%, в частности по меньшей мере 92%, предпочтительно по меньшей мере 94%, преимущественно по меньшей мере 97%, более преимущественно по меньшей мере 99% диастереоизомерным избытком смеси изомеров II-RRR и II-SSS.
Предпочтительно указанный диастереоизомерный избыток предусматривает по меньшей мере 70%, в особенности по меньшей мере 80%, преимущественно по меньшей мере 90%, предпочтительно по меньшей мере 95% диастереоизомерный избыток смеси изомеров II-RRR и II-SSS.
Преимущественно указанный диастереоизомерный избыток предусматривает смесь изомеров II-RRR и II-SSS.
Термин "смесь изомеров II-RRR и II-SSS" также охватывает, в более широком смысле, случай, когда присутствует только один из изомеров: II-RRR или II-SSS. Однако термин "смесь изомеров II-RRR и II-SSS" предпочтительно обозначает все случаи, в которых каждый из изомеров II-RRR и II-SSS присутствует в различном количестве, отличном от нуля.
В предпочтительном варианте осуществления изомеры II-RRR и II-SSS присутствуют в указанной смеси в соотношении от 65/35 до 35/65, в особенности от 60/40 до 40/60, в частности от 55/45 до 45/55. Преимущественно изомеры II-RRR и II-SSS присутствуют в смеси в соотношении 50/50.
▪ Комбинация стадий 1b) и 1c)
Стадии 1b) и 1c) направлены на очистку комплекса формулы (II) путем удаления примесей, которые могут присутствовать в связи со способом его получения.
Указанные примеси могут, в особенности, включать 3-амино-1,2-пропандиол и/или примесь, образованную в результате двойного присоединения.
Более конкретно, 3-амино-1,2-пропандиол может присутствовать в конечном продукте, полученном во время осуществления способа получения комплекса формулы (II), как правило, при получении комплекса формулы (II) амидированием комплекса формулы (I) посредством проведения реакции с 3-амино-1,2-пропандиолом. Это особенно верно в случае способа получения комплекса формулы (II) в соответствии с настоящим изобретением. Как отмечалось ранее, реакция амидирования может включать активацию трех функциональных групп карбоновой кислоты, переносимых атомами углерода, которые расположены на боковых цепях комплекса формулы (I) в γ-положении по отношению к атомам азота макроцикла, к которому привиты указанные боковые цепи, с последующим аминолизом активированных функциональных групп карбоновой кислоты посредством реакции с 3-амино-1,2-пропандиолом. Затем 3-амино-1,2-пропандиол преимущественно применяют в избытке, чтобы обеспечить хорошую степень превращения трех активированных функциональных групп карбоновой кислоты в амидные функциональные группы.
Под термином "примесь, образованную в результате двойного присоединения" подразумевают обозначение комплекса формул (II-dc-a), (II-dc-b), (II-dc-c), представленных ниже, или их смеси:
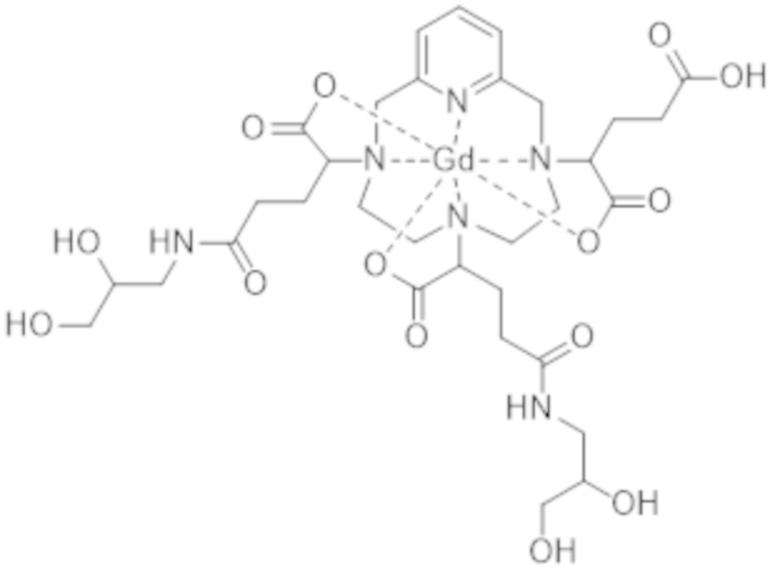 (II-dc-a),
(II-dc-a),
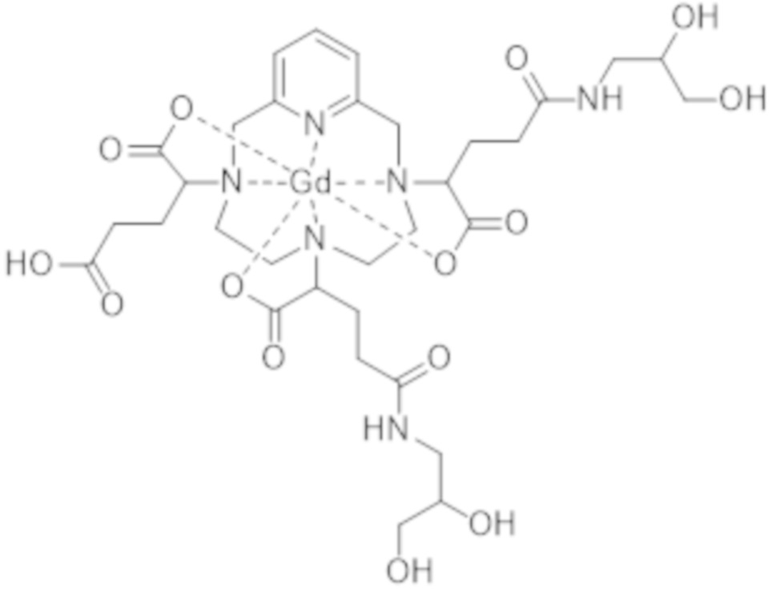 (II-dc-b),
(II-dc-b),
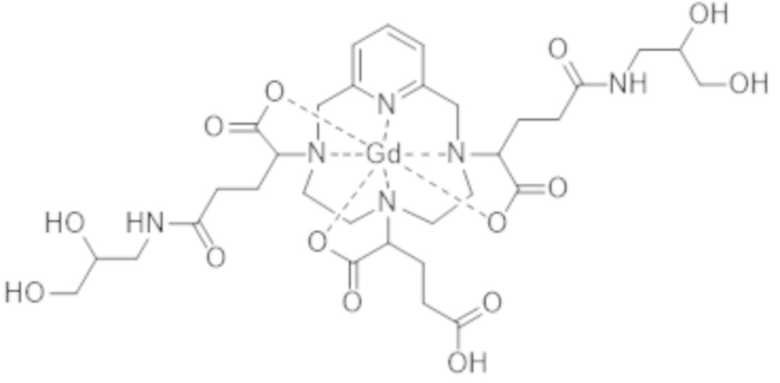 (II-dc-c).
(II-dc-c).
Примесь, образованную в результате двойного присоединения, может, в особенности, возникнуть в результате реакции гидролиза амидной функциональной группы комплекса формулы (II). Она также может возникнуть в результате неполной активации функциональных групп карбоновой кислоты комплекса формулы (I) (активации двух из трех функциональных групп) или неполного аминолиза активированных функциональных групп карбоновой кислоты (аминолиза двух из трех функциональных групп), если способ получения комплекса формулы (II) включает такие стадии. Это особенно верно в случае способа получения комплекса формулы (II) в соответствии с настоящим изобретением.
▪ Стадия 1b) соответствует пропусканию диастереоизомерно обогащенного комплекса формулы (II), описанного ранее, через ионообменную(-ые) смолу(-ы).
Для целей настоящего изобретения термин "ионообменная смола" означает твердый материал, как правило, в форме гранул, состоящий из полимерной матрицы, к которой привиты положительно заряженные функциональные группы (анионообменная смола) или отрицательно заряженные функциональные группы (катионообменная смола), что позволяет, соответственно, улавливать анионы или катионы путем адсорбции. Адсорбция анионов или катионов на смоле происходит посредством ионного обмена между противоионами функциональных групп, изначально присутствующими для обеспечения электронейтральности смолы, и анионами или катионами, которые нужно уловить.
Стадия 1b) включает приведение водного раствора диастереоизомерно обогащенного комплекса формулы (II) в контакт с сильной анионообменной смолой. Применяемая вода предпочтительно представляет собой очищенную воду.
Указанная сильная анионообменная смола, как правило, включает в качестве обменивающихся функциональных групп аммониевые группы (N(RR’R’’)+, где R, R’ и R’’ представляют собой идентичные или различные (C1-C6)алкильные группы). В особенности, можно указать смолу Amberlite® FPA900, продаваемую Dow Chemical, преимущественно в HO- форме.
Пропускание через сильную анионообменную смолу позволяет по меньшей мере частично удалить примеси, образованные в результате двойного присоединения
Стадия 1b) может также включать приведение водного раствора диастереоизомерно обогащенного комплекса формулы (II) в контакт со слабой катионообменной смолой. Применяемая вода предпочтительно представляет собой очищенную воду.
Указанная слабая катионообменная смола, как правило, включает в качестве обменивающихся функциональных групп карбоксилатные группы (CO2-). В особенности, можно указать смолу IMAC® HP336FPA900 продаваемую Dow Chemical, преимущественно в H+ форме.
Пропускание через слабую катионообменную смолу позволяет по меньшей мере частично удалить 3-амино-1,2-пропандиол и возможные остатки Gd3+.
Следует отметить, что проведение стадии 1b) пропускания через ионообменную(-ые) смолу(-ы) стало возможным благодаря улучшенной устойчивости диастереоизомерно обогащенного комплекса формулы (II) в соответствии с настоящим изобретением, структура которого, в результате, сохраняется во время этой стадии.
▪ Стадия 1c) соответствует ультрафильтрации диастереоизомерно обогащенного комплекса формулы (II), описанного ранее.
В настоящем изобретении термин "ультрафильтрация" означает способ фильтрации через мезопористую полупроницаемую мембрану, поры которой, как правило, характеризуются диаметром от 1 до 100 нм, в частности от 2 до 50 нм, в особенности от 10 до 50 нм (мезопоры), под действием сил, таких как градиенты давления, как правило, от 1 до 10 бар, и необязательно градиенты концентрации. Таким образом, он представляет собой способ мембранного разделения, посредством которого частицы в растворе или суспензии, размер которых превышает размер пор, удерживаются мембраной и отделяются от содержащей их жидкой смеси.
В контексте способа очистки в соответствии с настоящим изобретением ультрафильтрация особенно выгодна для удаления эндотоксинов.
Преимущественно ультрафильтрационная мембрана, применяемая на стадии 1c), характеризуется порогом отсечения менее 100 кДа, в особенности менее 50 кДа, в частности менее 25 кДа, как правило, характеризуется порогом отсечения 10 кДа.
Предпочтительно на стадии 1с) трансмембранное давление составляет от одного до 5 бар, в частности от 2,25 до 3,25 бар.
▪ В одном конкретном варианте осуществления стадии 1b) и 1c) также объединяют со стадией 1a) нанофильтрации.
В настоящем изобретении термин "нанофильтрация" означает способ фильтрации через пористую полупроницаемую мембрану, поры которой, как правило, характеризуются диаметром от 0,1 до 100 нм, в частности от 0,1 до 20 нм, в особенности от 1 до 10 нм, под действием сил, таких как градиенты давления, как правило, от 1 до 50 бар, и необязательно градиенты концентрации. Таким образом, он представляет собой способ мембранного разделения, посредством которого частицы в растворе или суспензии, размер которых превышает размер пор, удерживаются мембраной и отделяются от содержащей их жидкой смеси.
Стадия 1а) нанофильтрации позволяет удалить большую часть избытка 3-амино-1,2-пропандиола (необязательно в форме соли, в особенности в форме гидрохлорида, или в форме производных, в особенности в форме ацетамидного производного) и минеральные соли.
В данном конкретном варианте осуществления стадия нанофильтрации может быть проведена непосредственно в отношении неочищенного диастереоизомерно обогащенного комплекса формулы (II), полученного в соответствии со способом получения, описанным ранее. В частности, нет необходимости осаждать полученный ранее диастереоизомерно обогащенный комплекс формулы (II) путем добавления растворителя.
Преимущественно нанофильтрационная мембрана, применяемая на стадии 1a), характеризуется порогом отсечения менее 1 кДа, в особенности менее 500 дальтон, в частности менее 300 дальтон, как правило, характеризуется порогом отсечения 200 дальтон.
Предпочтительно на стадии 1a) трансмембранное давление составляет от 10 до 40 бар, в частности от 2 до 30 бар.
В частности, температура раствора комплекса формулы (II), подвергнутого ультрафильтрации на стадии 1а), составляет от 20 до 40°C, в особенности от 25 до 35°C.
В одной альтернативе данного конкретного варианта осуществления стадия 1b) не включает приведение водного раствора диастереоизомерно обогащенного комплекса формулы (II) в контакт со слабой катионообменной смолой.
В одном конкретном варианте осуществления стадии 1a), если она имеется, 1b и 1c проводят в данном порядке. Данный предпочтительный вариант осуществления, в особенности, позволяет свести к минимуму количества применяемых смол и, таким образом, стоимость промышленного производства.
▪ Стадия 2)
Стадия 2) направленана выделение в твердой форме очищенного комплекса формулы (II), полученного по завершении комбинации стадий 1b) и 1c), и, необязательно также в комбинации со стадией 1a).
Данная стадия выделения в твердой форме может быть проведена любым способом, который хорошо известен специалистам в данной области техники, в особенности, путем распыления, осаждения, лиофилизации или центрифугирования, преимущественно путем распыления.
В предпочтительном варианте осуществления стадия 2) предусматривает распыление.
Более конкретно, выделение в твердой форме очищенного комплекса формулы (II) путем распыления позволяет, в особенности, отказаться от применения растворителей для осаждения.
В таком случае, температура воздуха на входе в распылитель, как правило, составляет от 150°C до 180°C, в особенности от 160°C до 175°C, преимущественно от 165°C до 170°C. Температура на выходе, как правило, составляет от 90°C до 120°C, предпочтительно от 105°C до 110°C.
Преимущественно степень чистоты очищенного комплекса формулы (II), диастереоизомерно обогащенного смесью изомеров II-RRR и II-SSS, выделенного по завершении стадии 2), составляет более 95%, в особенности более 97%, предпочтительно более 97,5%, более предпочтительно более 98%, преимущественно более 99%, при этом указанная степень чистоты выражается в виде массовой доли комплекса формулы (II) относительно общей массы, полученной по завершении стадии 2).
Настоящее изобретение также относится к диастереоизомерно обогащенному и очищенному комплексу формулы (II), который может быть получен согласно способу очистки в соответствии с настоящим изобретением.
Предпочтительно комплекс формулы (II), включенный в композицию в соответствии с настоящим изобретением, описанную ранее, представляет собой диастереоизомерно обогащенный и очищенный комплекс формулы (II), который может быть получен согласно способу очистки в соответствии с настоящим изобретением.
Примеры
Приведенные ниже примеры представлены в виде иллюстраций, не ограничивающих настоящее изобретение.
Разделение групп изомеров iso1, iso2, iso3 и iso4 комплекса формулы (II) с помощью UHPLC
Применяют систему UHPLC, состоящую из насоса, инжектора, хроматографической колонки, УФ-детектора и системы сбора и обработки данных. Применяемая хроматографическая колонка представляет собой колонку UHPLC 150 × 2,1 мм - 1,6 мкм (колонка Waters Cortecs® UPLC T3).
- Подвижная фаза
Фаза A: 100% ацетонитрил, и фаза B: 0,0005% об./об. водный раствор H2SO4 (96%)
- Получение испытуемых растворов
Раствор комплекса формулы (II) в очищенной воде с концентрацией 2 мг/мл
- Аналитические условия
- Градиент
Получают четыре основных пика. Пик 4 на хроматограмме UHPLC, а именно iso4, соответствует времени удерживания 6,3 минуты.
Получение бутилового сложного гексаэфира формулы (VI)
В реакторе смешивают 184 кг (570 моль) дибутил-2-бромглутарата и 89 кг (644 моль) карбоната калия и нагревают до 55-60°C. К предыдущей смеси добавляют водный раствор 29,4 кг (143 моль) пиклена в 24 кг воды. Реакционную смесь выдерживают при 55-60°C, а затем нагревают с обратным холодильником в течение приблизительно 10 часов. После реакции среду охлаждают, разбавляют с помощью 155 кг толуола и затем промывают с помощью 300 литров воды. Бутиловый сложный гексаэфир экстрагируют водной фазой, состоящей из 175 кг (1340 моль) фосфорной кислоты (75%). Затем полученное трижды промывают с помощью 150 кг толуола. Бутиловый сложный гексаэфир повторно экстрагируют толуольной фазой путем разбавления с помощью 145 кг толуола и 165 кг воды с последующим подщелачиванием с помощью 30% раствора гидроксида натрия (масса/масса) с достижением pH 5-5,5. Нижнюю водную фазу удаляют. Бутиловый сложный гексаэфир получают посредством концентрирования до сухого состояния в вакууме при 60°C с выходом приблизительно 85%.
Получение гексакислоты формулы (III)
В реактор помещают 113 кг (121 моль) бутилового сложного гексаэфира вместе с 8 кг этанола. Среду доводят до 55 ± 5°C и затем в течение 3 часов добавляют 161 кг (1207,5 моль) 30% раствора гидроксида натрия (масса/масса). Реакционную смесь выдерживают при этой температуре в течение приблизительно 20 часов. Затем бутанол удаляют посредством декантации реакционной среды. Гексакислоту формулы (III), полученную в форме натриевой соли, разбавляют водой с получением водного раствора с концентрацией приблизительно 10% (масса/масса). Данный раствор обрабатывают на кислой катионообменной смоле. Водный раствор гексакислоты формулы (III) получают с выходом приблизительно 90% и чистотой 95%.
Получение комплекса гадолиния и гексакислоты формулы (I)
▪ Описание эксперимента
• Комплексообразование и изомеризация
- Без уксусной кислоты
В реактор помещают 418 кг (117 кг чистой гексакислоты формулы (III)/196 моль) водного раствора гексакислоты формулы (III) с концентрацией 28% по весу. Значение рH раствора регулируют до 2,7 путем добавления хлористоводородной кислоты, а затем добавляют 37 кг (103,2 моль) оксида гадолиния. Реакционную среду нагревают при 100-102°C в течение 48 часов для достижения ожидаемого изомерного распределения гексакислоты формулы (III).
- С уксусной кислотой
Суспендируют оксид гадолиния (0,525 молярного экв.) в растворе гексакислоты формулы (III) с концентрацией 28,1% по массе.
99-100% уксусную кислоту (50% по массе/чистая гексакислота формулы (III)) выливают в среду при комнатной температуре.
Среду нагревают с обратным холодильником с последующей перегонкой до 113°C по массе, постепенно доливая в среду уксусную кислоту по мере удаления воды. По достижении температуры 113°C добавляют достаточное количество уксусной кислоты, чтобы достичь начального объема.
Среду выдерживают при температуре 113°C в течение ночи.
• Кристаллизация, перекристаллизация
- Кристаллизация
Раствор комплекса гадолиния и гексакислоты формулы (I) охлаждают до 40°C, добавляют затравку и оставляют в контакте друг с другом в течение по меньшей мере 2 часов. Затем продукт выделяют путем фильтрования при 40°C и промывают очищенной с помощью осмоса водой.
- Перекристаллизация
Суспендируют 180 кг комплекса гадолиния и гексакислоты формулы (I), полученного ранее (содержание сухого вещества приблизительно 72%), в 390 кг воды. Среду нагревают до 100°C для растворения продукта, а затем охлаждают до 80°C и добавляют небольшое количество затравки. После охлаждения до комнатной температуры комплекс гадолиния и гексакислоты формулы (I) выделяют путем фильтрования и высушивания.
• Селективный распад комплекса
Помещают сухой продукт в реактор с очищенной с помощью осмоса водой при 20°C. Масса добавленной воды равна удвоенной теоретической массе комплекса гадолиния и гексакислоты формулы (I). Выливают 30,5% раствор гидроксида натрия (масса/масса) (6,5 экв.) в среду при 20°C. По окончании добавления NaOH среду оставляют в контакте друг с другом при 50°C в течение 16 часов. Среду охлаждают до 25°C и продукт отфильтровывают через слой Clarcel.
▪ Содержание смеси диастереоизомеров I-RRR и I-SSS
Соотношение, в котором различные изомеры комплекса формулы (I) присутствуют в смеси диастереоизомеров, зависит от условий, при которых проводят стадии комплексообразования и изомеризации, как показано в таблице 3 ниже.
Таблица 3. Содержание смеси I-RRR и I-SSS в зависимости от условий комплексообразования/изомеризации
I-RRR и I-SSS
Дополнительные стадии перекристаллизации и обеспечения селективного распада комплекса позволяют увеличить диастереоизомерный избыток смеси I-RRR и I-SSS (см. таблицу 4).
Таблица 4. Содержание смеси I-RRR и I-SSS после кристаллизации/перекристаллизации/селективного распада комплекса
I-RRR и I-SSS
Получение комплекса формулы (II)
В реактор помещают 90 кг (119 моль) комплекса с гексакислотой формулы (I) и 650 кг метанола. Смесь охлаждают до приблизительно 0°C и затем выливают в нее 111 кг (252 моль) метанольного раствора хлористоводородной кислоты (8,25% HCl в метаноле), поддерживая температуру при 0°C. Реакционную среду доводят до комнатной температуры и затем продолжают перемешивание в течение 16 часов. После охлаждения до 0-5°C добавляют 120 кг (1319 моль) 3-амино-1,2-пропандиола. Затем реакционную среду нагревают, отгоняя метанол в вакууме, до достижения температуры 60-65°C. Концентрат выдерживают в течение 16 часов при этой температуре под вакуумом. По окончании реакции среду разбавляют с помощью 607 кг воды при охлаждении до комнатной температуры. Раствор неочищенного комплекса формулы (II) нейтрализуют с помощью 20% раствора хлористоводородной кислоты (масса/масса). Таким образом, получают 978,6 кг раствора с концентрацией 10,3%, что составляет 101 кг материала. Полученный выход составляет 86,5%, чистота комплекса формулы (II) составляет 92,3% (HPLC s/s). Количество примесей, образованных в результате двойного присоединения, составляет 6,4% (HPLC s/s).
Очистка комплекса формулы (II)
• Нанофильтрация
Применяемая нанофильтрационная мембрана характеризуется порогом отсечения 200 дальтон (Koch Membran System SR3D). Данную обработку проводят следующим образом.
Раствор неочищенного комплекса формулы (II) нагревают до 30°C. Заполняют нанофильтр указанным раствором. Насос сначала включают с низкой скоростью для продувки системы, затем скорость насоса нанофильтра постепенно увеличивают до требуемой скорости рециркуляции (1,0 м3/ч. для мембраны 2,5 × 40 дюймов). Затем систему помещают в режим полной рециркуляции при 30°C не менее чем на 2 часа для создания поляризационного слоя. Затем среду подвергают диафильтрации при 30°C и давлении 2,5 бар, поддерживая постоянный объем путем добавления чистой воды до тех пор, пока не будет достигнута проводимость ретентата менее 1000 мкСм. В конце диафильтрации среду концентрируют с получением концентрации приблизительно 40% (масса/масса).
• Обработка на смолах
Раствор комплекса формулы (II), полученный в результате нанофильтрации, разбавляют очищенной водой при перемешивании с получением 15% раствора (масса/масса). Данный раствор элюируют последовательно на 50 литрах сильных анионообменных смол (FPA900) в ОН- форме, а затем на 50 литрах слабых катионообменных смол (HP336) в Н+ форме при средней скорости элюирования 2 об./об./ч. (2 объема раствора на один объем смолы в час). Затем смолы промывают с помощью приблизительно 450 литров очищенной воды до достижения показателя преломления менее 1,3335.
Затем раствор комплекса формулы (II) концентрируют путем нагревания до 50-60°C при вакууме 20 мбар до концентрации 35% (масса/масса).
• Ультрафильтрация
Ультрафильтрационная мембрана представляет собой мембрану UF 10KD Koch Spiral.
В ультрафильтр подают предыдущий раствор комплекса формулы (II) с концентрацией 35%, нагретый до 40°C. Проводят ультрафильтрацию при скорости потока 3 м3/ч. с трансмембранным давлением 2,5-3 бар. Систему несколько раз промывают с помощью 13 литров апирогенной очищенной воды до достижения конечного разбавления комплекса формулы (II) 25% (масса/масса).
• Распыление
Комплекс формулы (II) получают в форме порошка путем распыления предыдущего раствора комплекса формулы (II), концентрированного до 25%.
Распыление проводят следующим образом.
Распылитель уравновешивают апирогенной чистой водой, устанавливая температуру на входе 165-170°C и регулируя скорость подачи таким образом, чтобы температура на выходе составляла 105-110°C.
Затем добавляют концентрированный раствор комплекса формулы (II) и регулируют скорость потока так, чтобы сохранить вышеуказанные параметры.
Данные рабочие параметры поддерживают на протяжении всего процесса распыления, обеспечивая при этом хорошие характеристики порошка в распылительной камере и на выходе из распылителя. В частности, следует убедиться, что продукт не прилипает.
В конце подачи раствора в распылитель, контейнер с данным комплексом формулы (II) и распылитель промывают апирогенной чистой водой до достижения максимальной степени извлечения порошка.
Получают комплекс формулы (II) с чистотой 99,6%.
Данная степень чистоты была определена с помощью жидкостной хроматографии с обращенной фазой.
Композиция в соответствии с настоящим изобретением и результаты исследований по ней
• Пример способа изготовления в соответствии с настоящим изобретением
Способ изготовления композиции в соответствии с настоящим изобретением выполняют в соответствии со следующими стадиями.
а) Растворяют 485,1 г (т.е. 0,5 М) комплекса формулы (II) в воде (необходимое количество до 1 литра), нагревают сосуд до температуры от 39°C до 48°C и интенсивно перемешивают раствор до полного растворения данного комплекса в воде. Затем раствор охлаждают до приблизительно 30°C.
b) Добавляют 0,404 г (т.е. 0,2 моль/моль % относительно доли комплекса, добавленного на стадии а)) DOTA (Simafex, Франция) при перемешивании к раствору, полученному на стадии а) через раствор DOTA с концентрацией 10% масса/объем
c) Добавляют трометамол (Tris) к раствору, полученному на стадии b), при перемешивании. Затем регулируют pH до значения от 7,2 до 7,7 путем добавления раствора хлористоводородной кислоты при перемешивании.
d) Получают целевую концентрацию (0,5 моль/л) путем добавления воды для инъекций в две стадии до достижения значения плотности от 1,198 до 1,219 г/мл.
Затем фильтруют жидкую композицию через полиэфирсульфоновую мембрану и помещают в ее конечный контейнер, который, наконец, стерилизуют при 121°C в течение 15 минут.
• Пример композиции в соответствии с настоящим изобретением.
Следующий состав получают с помощью способа, описанного выше.
* Измерение проводили с помощью колориметрического способа с применением ксиленолового оранжевого
**в пересчете на безводное и чистое вещество
• Проведенные исследования в отношении состава
Исследовали различные концентрации трометамола от 0 до 100 мМ. Результаты данных исследований показали, что содержание 10 мМ (0,12% масса/объем) было достаточным для обеспечения стабильности состава при определенном pH при одновременном ограничении образования примесей в результате разложения.
Исследовали различные концентрации DOTA от 0 до 2,5 мМ. Результаты данных исследований показали, что содержание 1 мМ, соответствующее 0,04% масса/объем или 0,2 моль/моль %, обеспечивает отсутствие высвобождения свободного Gd при получении и в течение срока годности продукта.
• Ускоренные испытания стабильности композиции в соответствии с настоящим изобретением
Состав из предыдущего примера анализируют сразу после его изготовления (T0) и после хранения при 40°C в течение 6 месяцев после его изготовления (T+6 месяцев).
В точке T0:
- Оценивают чистоту с помощью хроматографии*: 99,6%.
- Концентрация Gd-DOTA: 0,007% (масса/объем).
- Концентрация Gd: ниже 0,0001% (масса/объем).
- Значение pH: 7,5.
В точке T+6 месяцев:
- Оценивают чистоту с помощью хроматографии*: 97,2%.
- Концентрация Gd-DOTA: 0,014% (масса/объем) - 0,25 мМ.
- Концентрация Gd: ниже 0,0001% (масса/объем).
- Значение pH: 7,5.
* жидкостная хроматография с обращенной фазой
Данные результаты демонстрируют, что этот состав имеет хорошую стабильность с течением времени.
• Сравнительные испытания стабильности
Стабильность представленных ниже композиций оценивали с течением времени. Термин "неоптимизированное AP" обозначает активное вещество, а именно комплекс формулы (II), полученный в соответствии со способом, описанным в EP 1931673. Термин "оптимизированное AP" обозначает диастереоизомерно обогащенный и очищенный комплекс формулы (II), полученный по способу в соответствии с настоящим изобретением.
(0,5 М)
моль/моль %
мМ
(ксиленол)
(LC с применением формиата*)
* LC с применением формиата: метод хроматографии с флуориметрическим детектированием. Разделение проводят на С18-привитой хроматографической колонке с обращенной фазой с элюированием в градиентном режиме.
Приведенные выше результаты указывают на то, что получение состава на основе неоптимизированного AP со свободной DOTA не представляется возможным. Это обусловлено тем, что хелатирующее вспомогательное средство полностью расходуется в реакции перелигандирования между комплексом формулы (II) и DOTA и, в результате, больше не может улавливать выделяемый Gd3+.
С другой стороны, диастереоизомерно обогащенный и очищенный комплекс формулы (II), полученный по способу в соответствии с настоящим изобретением, может быть составлен со свободной DOTA. Более конкретно, наблюдается отсутствие свободного Gd в композиции через 6 месяцев при 40°C, независимо от pH состава и наличия или отсутствия буферных веществ. Кроме того, расход хелатирующего вспомогательного средства очень низкий, поскольку он не превышает 0,08 моль/моль %.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИАСТЕРЕОИЗОМЕРНО ОБОГАЩЕННЫЙ КОМПЛЕКС ГАДОЛИНИЯ И ХЕЛАТИРУЮЩЕГО ЛИГАНДА НА ОСНОВЕ PCTA И СПОСОБ СИНТЕЗА | 2020 |
|
RU2810975C2 |
| Составы металлических комплексов | 2015 |
|
RU2710360C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ КОМПЛЕКСЫ МЕТАЛЛОВ РЯДА ЛАНТАНОИДОВ | 2015 |
|
RU2707070C2 |
| ДИМЕРНЫЕ КОНТРАСТНЫЕ СРЕДСТВА | 2016 |
|
RU2739834C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАЦЕМИЧЕСКОГО НЕБИВОЛОЛА | 2006 |
|
RU2392277C2 |
| CEST-СИСТЕМЫ, ПРОЯВЛЯЮЩИЕ НЕ ЗАВИСЯЩУЮ ОТ КОНЦЕНТРАЦИИ ЧУВСТВИТЕЛЬНОСТЬ | 2011 |
|
RU2605823C2 |
| СОСТАВ КОНТРАСТНЫХ СРЕДСТВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2815053C2 |
| КОНТРАСТНЫЕ АГЕНТЫ | 2016 |
|
RU2743167C2 |
| НОВЫЕ ХЕЛАТНЫЕ СОЕДИНЕНИЯ ГАДОЛИНИЯ С ВЫСОКОЙ РЕЛАКСИВНОСТЬЮ ДЛЯ ПРИМЕНЕНИЯ В МАГНИТНО-РЕЗОНАНСНОЙ ВИЗУАЛИЗАЦИИ | 2017 |
|
RU2755181C2 |
| КОНЪЮГИРОВАННЫЕ БИСФОСФОНАТЫ ДЛЯ ДИАГНОСТИКИ И ТЕРАПИИ ЗАБОЛЕВАНИЙ КОСТЕЙ | 2015 |
|
RU2742660C2 |
Настоящее изобретение относится к композиции для применения в качестве контрастного агента. Предложенная композиция содержит: 1) комплекс формулы (II), имеющий диастереоизомерный избыток по меньшей мере 80% смеси изомеров II-RRR и II-SSS формул, и 2) свободный макроциклический лиганд, где композиция имеет концентрацию свободного гадолиния, составляющую менее 1 ppm (масса/объем). При этом композиция содержит от 0,002 до 0,4 моль/моль % свободного макроциклического лиганда относительно комплекса формулы (II). Изобретение также относится к способу очистки указанного комплекса формулы (II). Композиция в соответствии с настоящим изобретением обладает стабильностью с течением времени. 2 н. и 19 з.п. ф-лы, 4 табл., 2 пр.
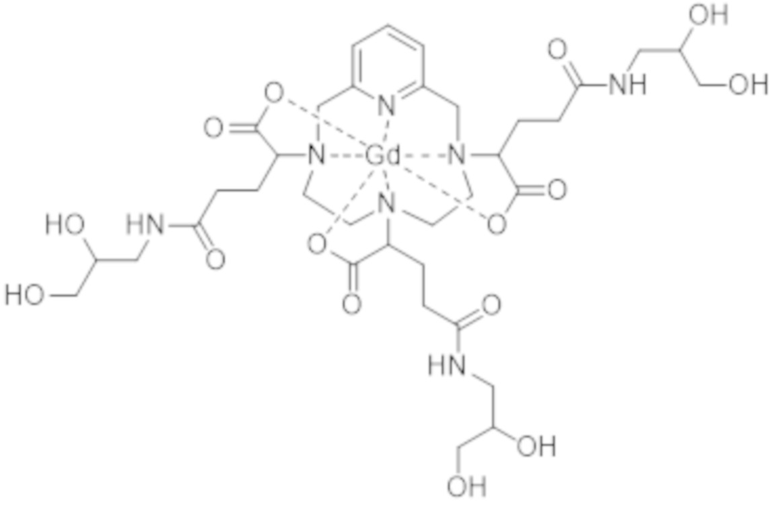 (II)
(II)
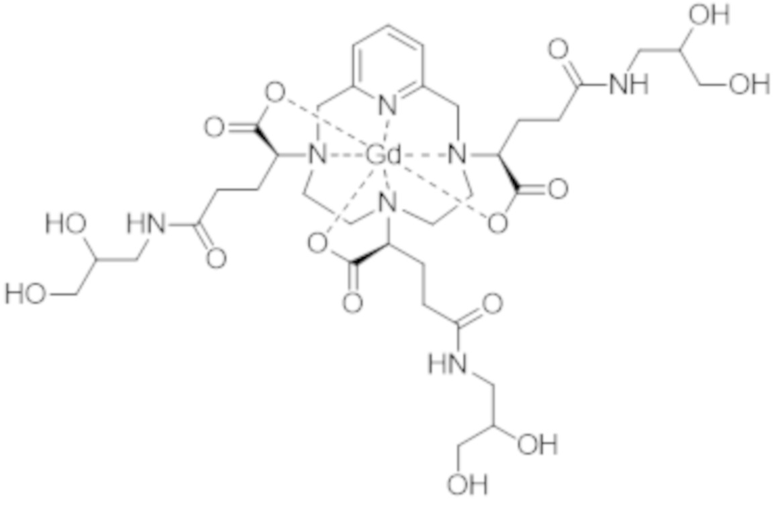 (II-SSS)
(II-SSS)
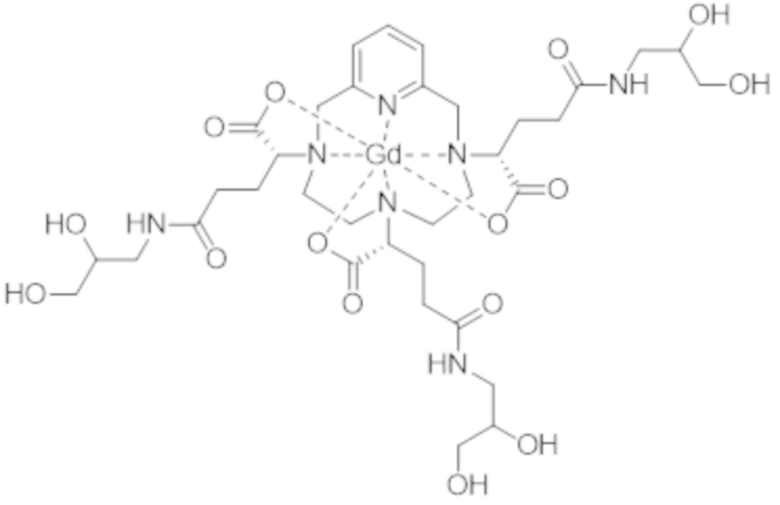 (II-RRR)
(II-RRR)
1. Композиция для применения в качестве контрастного агента, содержащая:
1) комплекс формулы (II), представленной ниже:
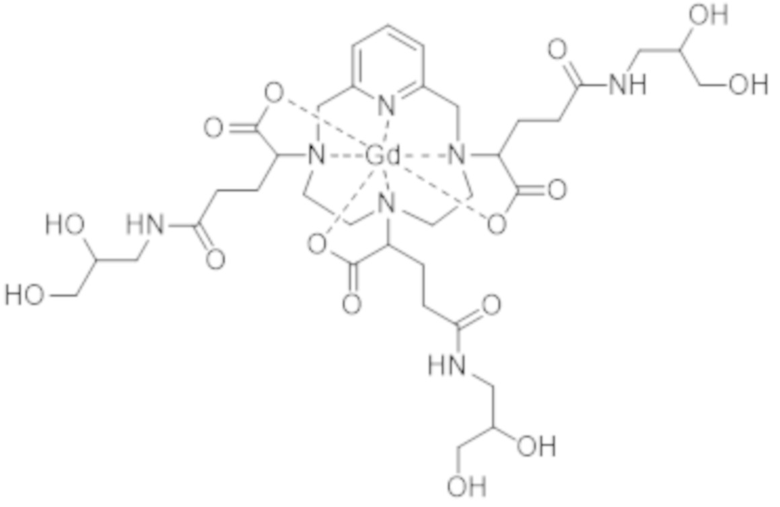 (II),
(II),
имеющий диастереоизомерный избыток по меньшей мере 80% смеси изомеров II-RRR и II-SSS формул:
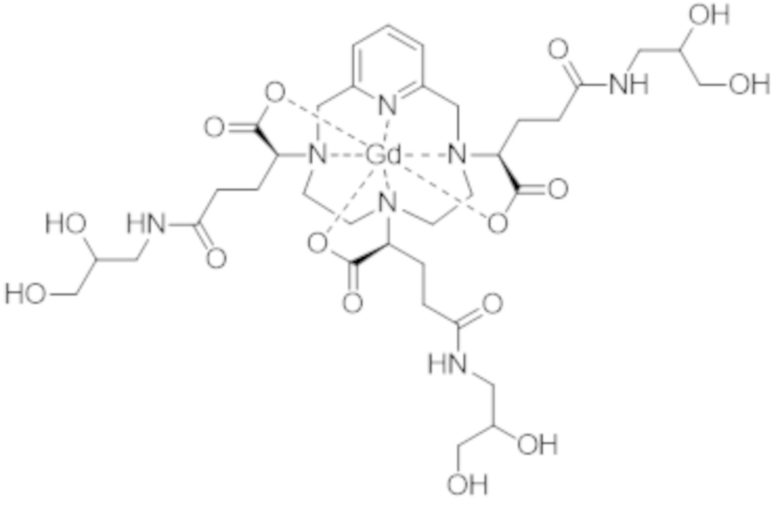 (II-SSS),
(II-SSS),
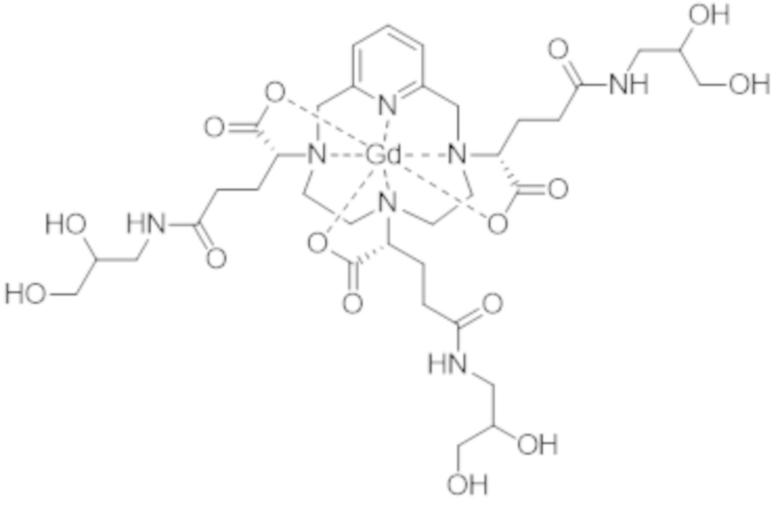 (II-RRR)
(II-RRR)
и
2) свободный макроциклический лиганд, где композиция имеет концентрацию свободного гадолиния, составляющую менее 1 ppm (масса/объем),
и
где композиция содержит от 0,002 до 0,4 моль/моль % свободного макроциклического лиганда относительно комплекса формулы (II).
2. Композиция для применения в качестве контрастного агента по п. 1, где композиция содержит от 0,01 до 0,3 моль/моль % свободного макроциклического лиганда относительно комплекса формулы (II).
3. Композиция для применения в качестве контрастного агента по п. 1 или 2, где комплекс формулы (II) имеет диастереоизомерный избыток по меньшей мере 90%.
4. Композиция для применения в качестве контрастного агента по любому из пп. 1-3, где комплекс формулы (II) имеет диастереоизомерный избыток по меньшей мере 92%.
5. Композиция для применения в качестве контрастного агента по любому из пп. 1-4, где композиция имеет концентрацию комплекса формулы (II) от 0,01 до 1,5 моль⋅л-1.
6. Композиция для применения в качестве контрастного агента по любому из пп. 1-5, где свободный макроциклический лиганд выбран из группы, состоящей из DOTA, NOTA, DO3A, BT-DO3A, HP-DO3A, PCTA, DOTA-GA и их производных.
7. Композиция для применения в качестве контрастного агента по любому из пп. 1-6, где рН композиции составляет от 4,5 до 8,5.
8. Композиция для применения в качестве контрастного агента по любому из пп. 1-7, где композиция дополнительно содержит буфер, выбранный из группы, состоящей из лактатного, тартратного, малатного, малеатного, сукцинатного, аскорбатного, карбонатного, Tris (трис(гидроксиметил)аминометана), HEPES (2-[4-(2-гидроксиэтил)-1-пиперазин]этансульфоновой кислоты) и MES (2-морфолиноэтансульфоновой кислоты) буферов и их смесей.
9. Композиция для применения в качестве контрастного агента по п. 1, где комплекс формулы (II) имеет диастереоизомерный избыток по меньшей мере 90% и свободный макроциклический лиганд представляет собой DOTA (1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусную кислоту).
10. Композиция для применения в качестве контрастного агента по п. 9, где композиция имеет концентрацию комплекса формулы (II) от 0,3 до 0,6 моль⋅л-1 и дополнительно содержит буфер Tris (трис(гидроксиметил)аминометан) и имеет рН от 6,5 до 8.
11. Способ очистки комплекса формулы (II), представленной ниже:
(II),
имеющего диастереоизомерный избыток по меньшей мере 80% смеси изомеров II-RRR и II-SSS формул:
(II-SSS),
(II-RRR),
включающий:
1) очистку комплекса формулы (II) путем:
1b) пропускания комплекса формулы (II) через ионообменную смолу и
1c) ультрафильтрации комплекса формулы (II), и
2) выделение очищенного комплекса формулы (II) в твердой форме.
12. Способ по п. 11, где стадия 1b) включает приведение водного раствора комплекса формулы (II) в контакт с сильной анионообменной смолой.
13. Способ по п. 12, где стадия 1b) дополнительно включает приведение водного раствора комплекса формулы (II) в контакт со слабой катионообменной смолой.
14. Способ по любому из пп. 11-13, где стадия 1 дополнительно включает стадию 1a) нанофильтрации комплекса формулы (II).
15. Способ по п. 14, где стадию 1 проводят в порядке 1а), 1b) и 1c).
16. Способ по любому из пп. 11-15, где стадия 2) включает распыление комплекса формулы (II), полученного на стадии 1).
17. Способ по любому из пп. 11-16, где комплекс формулы (II), в отношении которого проводят очистку, предварительно получают с помощью следующих последовательных стадий:
a) комплексообразования гексакислоты формулы (III), представленной ниже:
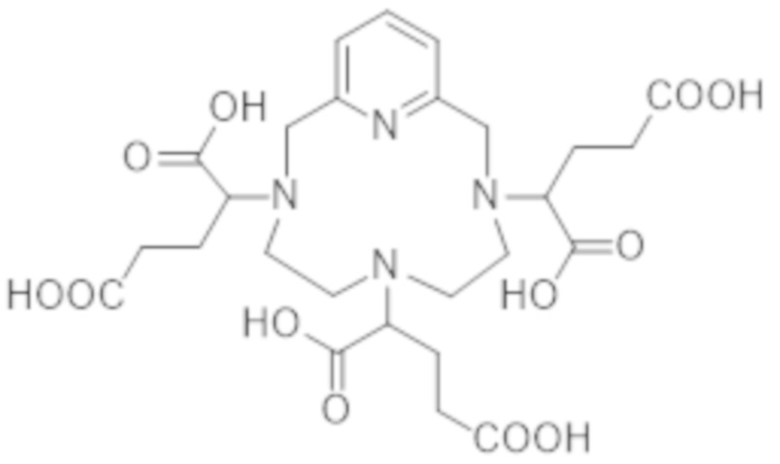 (III),
(III),
с гадолинием с получением комплекса гадолиния и гексакислоты формулы (I), представленной ниже:
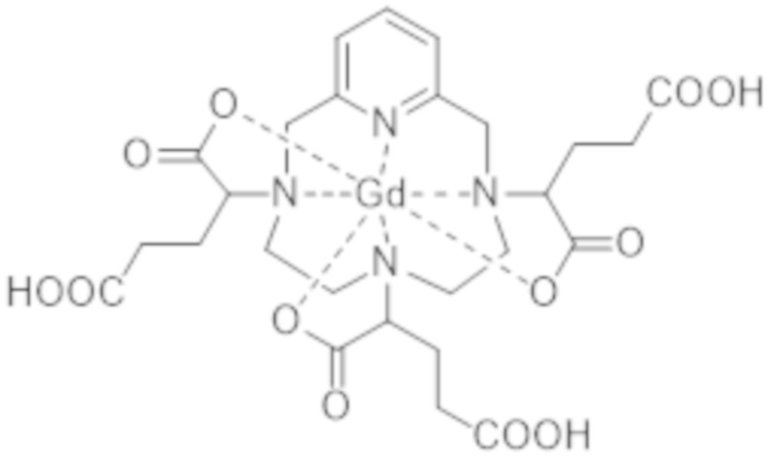 (I),
(I),
b) изомеризации путем нагревания комплекса гадолиния и гексакислоты формулы (I) в водном растворе при pH от 2 до 4 с получением диастереоизомерно обогащенного комплекса, имеющего диастереоизомерный избыток по меньшей мере 80% смеси изомеров I-RRR и I-SSS
формул:
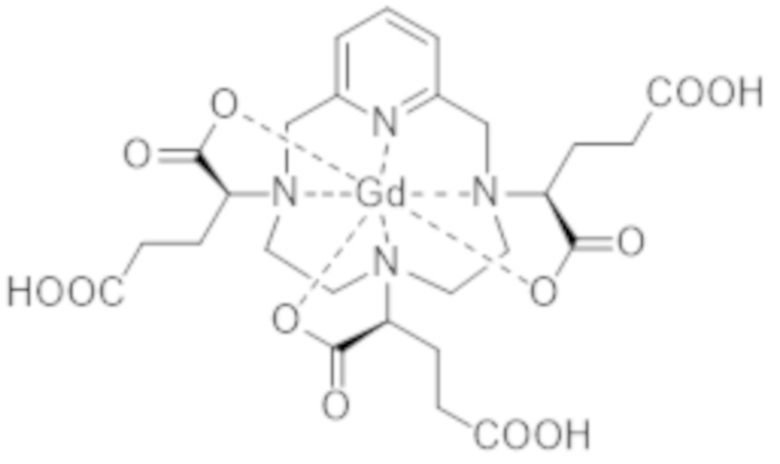 (I-SSS),
(I-SSS),
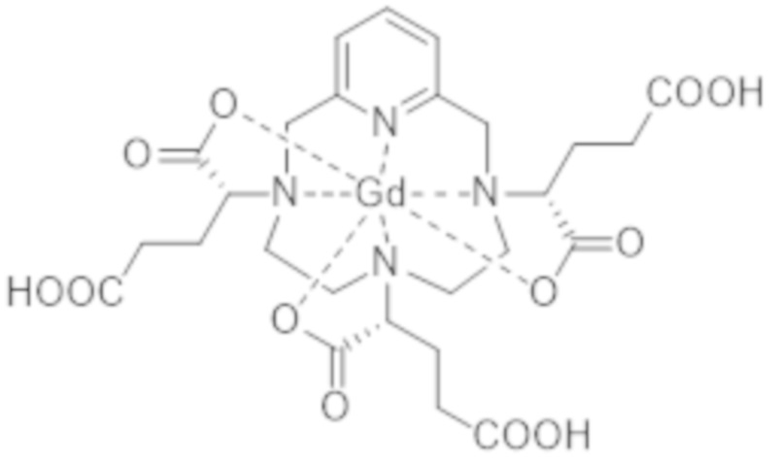 (I-RRR),
(I-RRR),
и
c) взаимодействие диастереоизомерно обогащенного комплекса с 3-амино-1,2-пропандиолом с образованием комплекса формулы (II).
18. Способ по п. 17, где по завершении стадии b) диастереоизомерно обогащенный комплекс выделяют путем кристаллизации и очищают путем перекристаллизации.
19. Способ по п. 17 или 18, где
стадия c) включает следующие последовательные стадии:
c1) взаимодействие диастереоизомерно обогащенного комплекса, полученного на стадии b), со спиртом формулы R1OH в присутствии кислоты с образованием сложного триэфира формулы (VIII), представленной ниже:
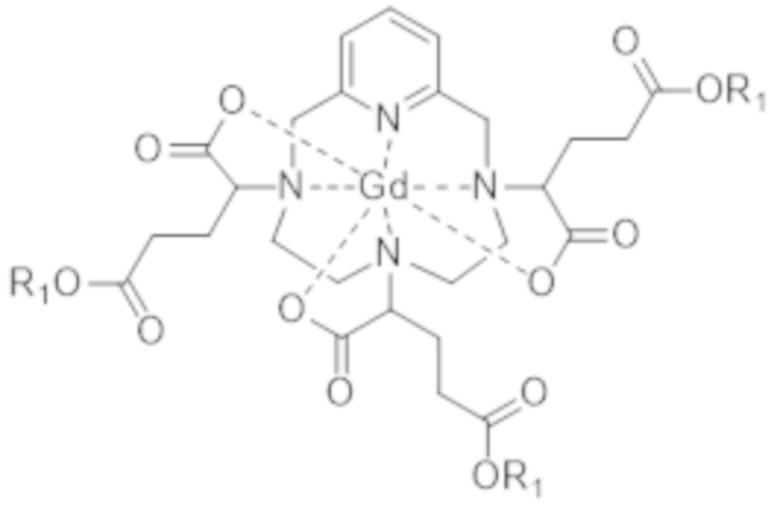 (VIII),
(VIII),
где R1 представляет собой (C1-C6)алкильную группу,
и
c2) аминолиз сложного триэфира формулы (VIII) с 3-амино-1,2-пропандиолом,
в присутствии кислоты.
20. Способ по п. 19, где стадия c) включает следующие последовательные стадии:
c1) взаимодействие диастереоизомерно обогащенного комплекса, полученного на стадии b), с метанолом в присутствии кислоты с образованием сложного метилового триэфира формулы (IV), представленной ниже:
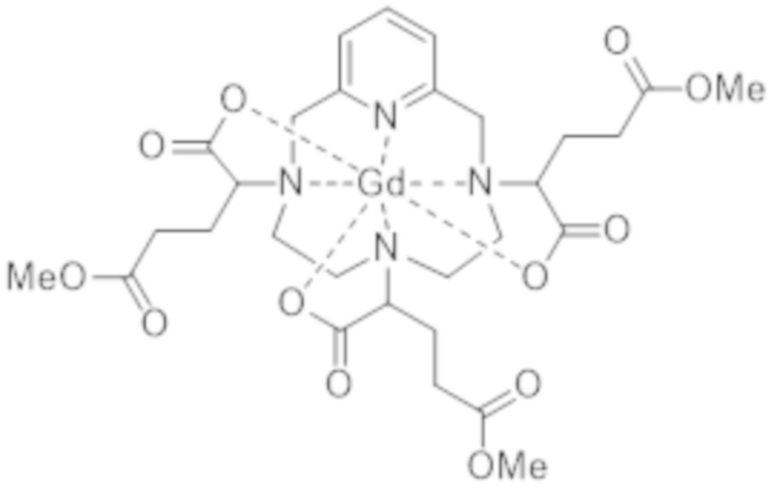
(IV)
и
c2) аминолиз сложного метилового триэфира формулы (IV) с 3-амино-1,2-пропандиолом в метаноле в присутствии кислоты, при этом метанол удаляют вакуумной перегонкой до достижения температуры по меньшей мере около 55°C, реакционную среду выдерживают при этой температуре под вакуумом в течение времени, превышающего 5 часов, перед охлаждением до комнатной температуры и разбавлением водой,
при этом сложный триэфир формулы (IV) не выделяют между стадиями с1) и с2).
21. Способ по любому из пп. 17-20, где гексакислоту формулы (III) получают путем алкилирования пиклена формулы (V):
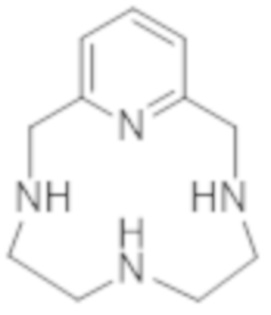 (V)
(V)
с дибутил-2-бромглутаратом с получением бутилового гексаэфира формулы (VI):
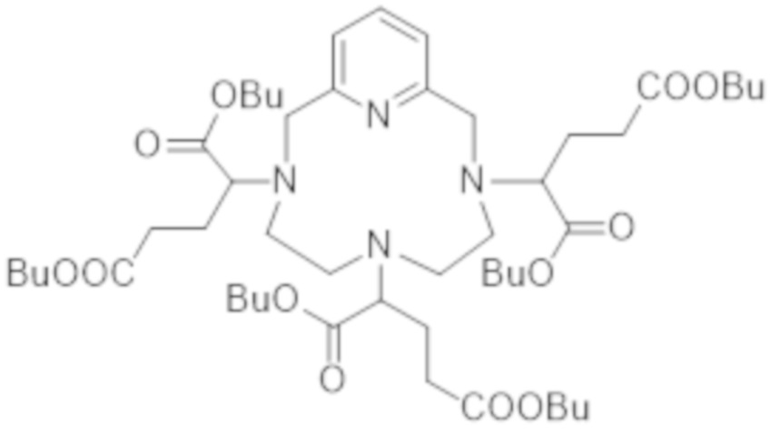 (VI),
(VI),
и
гидролиз бутилового гексаэфира формулы (VI) с получением гексакислоты формулы (III).
| WO 2007042506 A1, 19.04.2007 | |||
| US 20160101196 А1, 14.04.2016 | |||
| ГОРДАДЗЕ Г.М | |||
| и др., Органическая химия углеводородов Книга 1, Учебное пособие, РГУ нефти и газа имени И.М | |||
| Губкина, 2012, стр | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| КОМПЛЕКСЫ МЕТАЛЛОВ С БИЦИКЛИЧЕСКИМИ ПОЛИАМИНОКИСЛОТАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ ДЛЯ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЯ | 2000 |
|
RU2232763C2 |
| Предохранительный щит для огнестрельного ручного оружия | 1930 |
|
SU26572A1 |

