Область техники, к которой относится изобретение
Настоящее изобретение относится к области диагностической визуализации и к новым контрастным агентам, обладающим улучшенной релаксивностью. Более конкретно, оно относится к функционализированным макроциклам, способным к образованию хелатных соединений ионов парамагнитных металлов, их хелатным комплексам с ионами металлов и их использованию в качестве контрастных агентов при магнитно-резонансной томографии (МРТ).
Предшествующий уровень техники
Магнитно-резонансная томография (МРТ) является широко известной методикой диагностической визуализации, все больше используемой в клинической диагностике для растущего числа показаний.
Неоспоримый успех этой методики определяется преимуществами, которые она предлагает, включая превосходное временное и пространственное разрешение, выдающуюся способность дифференцировать мягкие ткани и ее безопасность вследствие ее неинвазивности и отсутствия какого-либо ионизирующего излучения, в отличие, например, от рентгеновского исследования, ПЭТ и SPECT.
При визуализации с помощью МРТ контраст в основном обусловлен различиями, существующими во времени продольной T1 и поперечной Т2 релаксации протонов воды в разных органах и тканях организма, что позволяет получать трехмерные изображения высокого разрешения распределения воды in vivo.
Интенсивность сигнала при визуализации с помощью МРТ с помощью ПРЭМ регистрируется по существу из локального значения скорости продольной релаксации 1/T1, и скорости поперечной, 1/T2 протонов воды, и увеличивается с увеличением значения 1/T1 (скорости продольной релаксации протонов воды), а уменьшается с увеличением 1/T2. Другими словами, чем короче T1, тем выше интенсивность регистрируемого сигнала при МРТ, и тем лучше диагностическое изображение.
Значительное распространение медицинской МРТ еще больше выиграло от разработки класса соединений, контрастных агентов для МРТ, которые действуют, вызывая резкое изменение скорости релаксации близлежащих протонов воды в тканях/органах/жидкостях, в которых они распределены, тем самым добавляя соответствующую физиологическую информацию к впечатляющему анатомическому разрешению, обычно получаемому при визуализации с помощью МРТ без контрастирования.
Контрастные агенты для применения при методике визуализации с помощью МРТ визуализации обычно включают ион парамагнитного металла, который образует комплекс с циклическим или ациклическим хелатирующим лигандом, более часто с полиаминополикарбоновым хелатором. Наиболее важный класс контрастных агентов для МРТ представлен хелатами Gd(III), которые в настоящее время применяют приблизительно в 1/3 клинических тестов. Действительно, Gd(III) является высоко парамагнитным с семью неспаренными электронами и длительным временем электронной релаксации, что делает его отличным кандидатом в качестве релаксационного агента. И наоборот, свободный ион металла [Gd(H2O)8]3+ является чрезвычайно токсичным для живого организма даже в низких дозах (10-20 микромоль/кг). Таким образом, для того, чтобы рассматривать его в качестве потенциально полезного контрастного агента для МРТ, комплекс Gd(III) должен демонстрировать высокую термодинамическую (и возможно, кинетическую) стабильность, предотвращающую высвобождение ионов токсичных металлов.
Кроме того, предпочтительный контрастный агент для МРТ должен демонстрировать оптимальную релаксивность. Релаксивность (r1p, r2p), выраженная в мМ-1с-1 и обычно измеряемая при 298 K и 20 мГц (прибл. 0,5 T), представляет собой свойство, присущее парамагнитному комплексу, которое характеризует его способность увеличивать скорость ядерно-магнитной релаксации, продольной (1/T1) и поперечной (1/T2), соответственно, вицинальных протонов воды и, таким образом, его эффективность в качестве агента, повышающего контрастность при МРТ. Общими словами, чем выше релаксивность контрастного агента для МРТ, тем больше его способность усиливать контрастность и сильнее контрастность, обеспечиваемая на регистрируемых изображениях, полученных с помощью МРТ.
В этой области известен ряд комплексов ионов парамагнитных металлов (для примера см.: Caravan P. et al. Chem. Rev. 1999, 99, 2293-2352 и US 4647447, US 4885363; US 4916246; US 5132409; US 6149890; DE19849465 и US 5980864).
Производные DO3A, имитирующие фосфолипиды, образующие супрамолекулярные структуры, раскрыты, например, в J. Chem. Soc. Perkin Trans. 2, 2001; 929-933.
Примеры коммерчески приемлемых контрастных агентов для МРТ включают комплексное соединение иона Gd3+ с лигандом DTPA, поставляемое на рынок под названием MAGNEVIST®; комплекс Gd3+ с лигандом DTPA-BMA, поставляемый на рынок под названием OMNISCAN®; комплекс Gd3+ BOPTA, известный как гадобенат димеглюмин и поставляемый на рынок под названием MultiHance™; комплекс Gd3+ лиганда DOTA, поставляемый на рынок под названием DOTAREM®; комплекс Gd3+ гидроксилированного тетраазамакроциклического лиганда, известный как HPDO3A, долгое время поставляемый на рынок под названием ProHance®, и комплекс соответствующего производного бутил-триола, известный как Гадобутрол и поставляемый на рынок под названием Gadavist®. Все перечисленные выше контрастные агенты являются неспецифическими агентами (NSA), разработанными для общего применения.
Хотя известные соединения обычно обеспечивают качество визуализации, соответствующее и удовлетворяющее нынешние потребности радиологов, что приводит к точной и подробной диагностической информации, тем не менее, все же имеется потребность в новых соединениях с улучшенными характеристиками контрастной визуализации, такими как повышенная релаксивность.
В частности, соединения с улучшенной релаксивностью могли бы уменьшить требуемую дозу парамагнитного контрастного агента и, возможно, сократить время экспозиции процесса визуализации.
Сущность изобретения
Настоящее изобретение в целом относится к новым макроциклическим хелатирующим лигандам, полезным для получения парамагнитных комплексов, имеющих особенно благоприятные характеристики, среди прочих с точки зрения улучшенной релаксивности.
Общими словами, аспект согласно настоящему изобретению относится к новым тетраазамакроциклическим лигандам, имеющим боковое плечо, связанное с атомом азота хелатирующей клетки, содержащее гидроксильный остаток и подходящую замещающую группу (группы). В частности, выбор подходящих заместителей на боковом плече предоставляет хелатные комплексы, имеющие улучшенную релаксивность.
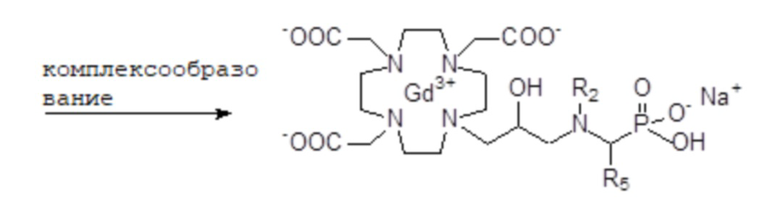
Кроме того, изобретение относится к соответствующим хелатным комплексам указанных хелатирующих лигандов с ионом парамагнитного металла, и особенно с Gd3+, или их физиологически приемлемым солям.
Дополнительный аспект изобретения относится к применению таких хелатных комплексов в качестве контрастных агентов, в частности для диагностической визуализации органов или тканей тела человека или животного посредством применения методики МРТ.
В дополнительном аспекте изобретение относится к способу изготовления для получения предоставленных лигандов, их комплексных соединений с ионом парамагнитного металла и их фармацевтически приемлемых солей, и их использования при получении диагностического агента.
Согласно другому аспекту, изобретение относится к фармацевтически приемлемой композиции, содержащей по меньшей мере одно парамагнитное комплексное соединение изобретения или его фармацевтически приемлемую соль в смеси с одним или несколькими физиологически приемлемыми носителями или эксципиентами. Указанные композиции полезны в частности в качестве контрастных сред при МРТ для обеспечения диагностически полезных изображений органов или тканей тела человека или животного.
Поэтому в другом аспекте настоящее изобретение относится к способу диагностической визуализации органа, ткани или области организма с использованием методики МРТ, который включает применение эффективной дозы соединения изобретения.
Подробное описание изобретения
Настоящее изобретение относится к хелатирующим лигандам формулы (I)
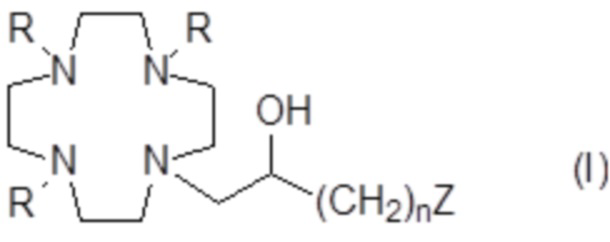
где:
R представляет собой -CH(R1)-COOH, где:
R1 представляет собой H или алкильную цепь C1-C3, которая необязательно замещена C1-C3 алкокси или C1-C3 гидроксиалкокси группой;
n составляет 1 или 2;
Z представляет собой аминопроизводное, выбранное из N(R2)(R3) и -NHR4; при этом:
R2 выбирают из группы, состоящей из: арильного кольца, циклоалкильного кольца и C1-C10 алкила, необязательно разделенного одним или более атомами кислорода и/или необязательно замещенного одной или более гидроксильными группами или арильным или циклоалкильным кольцом;
R3 выбирают из группы, состоящей из: C5-C12 гидроксиалкила, содержащего по меньшей мере 2 гидроксильные группы; C2-C10 гидроксиалкоксиалкилена формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH; и группы формулы -(CH2)sCH(R5)-G; в которой
s составляет 0, 1 или 2;
каждый r составляет независимо 1 или 2;
m составляет 1, 2 или 3; и в которой
R5 представляет собой H, или арилалкилен или циклоалкил-алкилен, имеющие до 3 атомов углерода в алкиленовой цепи;
G представляет собой группу, выбранную из -PO(OR6)2, -PO(R7)(OR6) и -COOH; в которых
R6 независимо друг от друга представляет собой H или C1-C5 алкил;
R7 представляет собой арильное или циклоалкильное кольцо или C1-C5 алкил, который необязательно замещен арильным или циклоалкильным кольцом;
или
R2 и R3 вместе с соединительным атомом азота образуют замещенное пяти- или шестичленное гетероциклическое кольцо;
R4 выбирают из группы, состоящей из: циклоалкильного кольца; циклоалкил-алкилена, имеющего до 3 атомов углерода в алкиленовой цепи; C5-C12 гидроксиалкила, содержащего по меньшей мере 2 гидроксильные группы; C2-C10 гидроксиалкоксиалкилена формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH; -(CH2)mPO(OR6)2; -(CH2)mPO(R7)(OR6); и -(CH2)sCH(R8)G; где:
R8 представляет собой арилалкилен или циклоалкил-алкилен, имеющий до 3 атомов углерода в алкиленовой цепи; и
m, r, s, R6, R7 и G соответствуют определению выше.
Предпочтительно в указанных выше соединениях формулы (I) R1 представляет собой H.
В настоящем описании, и если не предусмотрено иное, выражение алкил включает в свое смысловое значение любую линейную или разветвленную углеводородную цепь, полученную из соответствующего углеводорода посредством удаления одного атома водорода, предпочтительно содержащую до 12 атомов углерода. В частности, «C1-C10 алкил» включает в свое смысловое значение линейную или разветвленную углеводородную цепь, содержащую от 1 до 10 атомов углерода, например: метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, изопентил, трет-пентил, гексил, изогексил, гептил, изогептил, октил и т.п. Аналогичным образом, термин «C1-C3 алкил» включает в свое смысловое значение линейную или разветвленную углеводородную цепь, содержащую от 1 до 3 атомов углерода, такую как, например, метил, этил, пропил и изопропил; термин «C1-C5 алкил» включает в свое смысловое значение линейную или разветвленную углеводородную цепь, содержащую от 1 до 5 атомов углерода, например: метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил и т.п.; и термин «C5-C7 алкил» включает в свое смысловое значение любую линейную или разветвленную углеводородную цепь, содержащую от 5 до 7 атомов углерода, такую как пентил, изопентил, трет-пентил, гексил, изогексил, трет-гексил, гептил, изогептил и трет-гептил.
По аналогии, выражение «алкилен» включает в свое смысловое значение двухвалентную линейную или разветвленную углеводородную цепь, полученную из любой из соответствующих углеводородных цепей посредством удаления двух атомов водорода из разных атомов углерода, включая C1-C5 алкилен, такой как, например, метилен, этилен, (изо)пропилен и т.д.
Термин «гидроксиалкил» включает в свое смысловое значение любой из перечисленных выше соответствующих алкильных фрагментов, в которых один или более атомов водорода замещены гидроксильными группами. Подходящие примеры включают C1-C3 гидроксиалкил, например, гидроксиметил (-CH2OH), гидроксиэтил (-CH2CH2OH), гидроксипропил (-CH2CH2CH2OH), дигидроксипропил, (-CH(CH2OH)2 и CH2CH2OHCH2OH) и т.п., и полигидроксиалкилы или «полиолы», как взаимозаменяемо используется в данном документе, в которых по меньшей мере два, а предпочтительно три или более атомов водорода углеводородной цепи замещены гидроксильными группами.
Например, и если не предусмотрено иное, выражение «C5-C12 полиол» (или «C5-C12 полигидроксиалкил») включает в свое смысловое значение любой из соответствующего C5-C12 алкильного фрагмента, у которого 2 или более, например, от 2 до 11 атомов водорода, замещены гидроксильными группами. Среди них предпочтительными являются C5-C10 полиолы, а C5-C7 полиолы являются особенно предпочтительными. Примеры C5-C7 полиолов включают пентил-полиолы (или полигидроксипентилы), такие как пентил-диолы, пентил-триолы, пентил-тетраолы и пентил-пентаолы, соответственно содержащие от 2, 3, 4 и 5 гидроксильных групп на C5 алкильной цепи; гексил-полиолы (или полигидроксигексилы), аналогично содержащие от 2 до 6 гидроксильных групп на C6 алкильной цепи; и гептил-полиолы (или полигидроксигептилы), содержащие от 2 до 7 гидроксильных групп на C7 алкильной цепи.
Термин «алкокси» включает в свое смысловое значение алкильный фрагмент, как определено выше, дополнительно содержащий один или более атомов кислорода; примеры включают, например, алкил-окси (или -Oалкил) группы, такие как метокси, этокси, n-пропокси, изопропокси и т.п., алкил-(поли)окси, в которых алкильная цепь разделена одним или более, например, до трех, атомами кислорода.
Термин «гидроксиалкокси» включает в свое смысловое значение любой из перечисленных выше остатков алкокси, дополнительно содержащих один или более гидроксилов (-OH) в алкильной цепи, таких как, например, -OCH2OH, -OCH2CH2OH, -OCH2CH2CH2OH, -OCH2OCH2OH, -OCH2CH2OCH2CH2OH, -OCH2CH(OH)CH2-OCH2CH2OH и т.п.
Термин «гидроксиалкоксиалкилен» (или «гидроксиалкокси-алкилен») включает в свое смысловое значение любой из перечисленных выше гидроксиалкокси, у которых связующей группой остатка является алкиленовая цепь -(CH2)r-, включая C2-C10 гидроксиалкокси-алкилены формулы -(CH2)r- [(O-(CH2)r]m(CH2)sOH, в которой m, r и s соответствуют определению выше.
Термин «аминополиол» (или «аминополигидроксиалкил» или «полигидрокси-аминоалкил», как взаимозаменяемо используется в данном документе) включает в свое смысловое значение C5-C12 углеводородную цепь, например, содержащую от 5 до 12 атомов углерода, который замещен 2 или более, например, от 2 до 11 гидроксильными группами, и содержит аминогруппу, образующую мостик между полигидроксилированной цепью, или полиолом, с остатком макроциклической молекулы. Предпочтительными являются C5-C7 аминополиолы, например, содержащие углеводородную цепь, содержащую 5, 6 или 7 атомов углерода, который замещен 2 или более, например, 2, 3, 4, 5 или 6 гидроксильными группами, и образующую мостик аминогруппу, как указано выше. Предпочтительно, аминогруппа связана с 1-C атомом углерода полигидроксилированной цепи (полиола), приводя тем самым к соответствующим 1-амино(C5-C12)полиолам. Аминогруппа может представлять собой либо вторичную аминогруппу, т.е. -NH-[(C5-C12)полиол], либо третичную аминогруппу, у которой атом азота, кроме того, предпочтительно связан с алкильной цепью, предпочтительно с C1-C3 алкилом, т.е. аминоалкил-полиолом формулы -N[алкил][(C5-C12)полиол].
Подходящие примеры аминополиолов, согласно изобретению, таким образом, включают полигидроксилированные аминоалкильные группы формулы -N(R9)(R10), в которых:
R9 представляет собой H или C1-C3 алкил, например, пропил, этил или, предпочтительно, метил; а
R10 представляет собой C5-C12 полиол.
Согласно изобретению, предпочтительными являются аминополиолы указанной выше общей формулы, у которых R10 представляет собой C5-C7 полиол, выбранный из пентил(поли)олов (или полигидроксипентилов), содержащих по меньшей мере 2, и предпочтительно от 2 до 4 гидроксильных групп на C5 алкильной цепи; гексил(поли)олов, содержащих по меньшей мере 2, а предпочтительно от 2 до 5 гидроксильных групп на C6 алкильной цепи; и гептил(поли)олов, содержащих по меньшей мере 2, а предпочтительно от 3 до 6 гидроксильных групп на C7 алкильной цепи, а R9 представляет собой H или метильную группу.
Согласно изобретению, особенно предпочтительными являются аминополиолы, выбранные из группы, состоящей из 1-амино-1-дезокси-пентитолов формулы
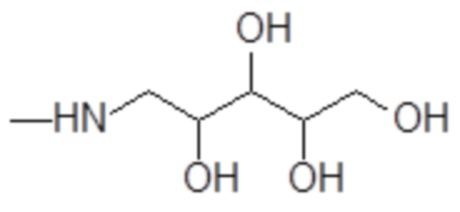
например, включая 1-амино-1-дезокси-D-рибит формулы
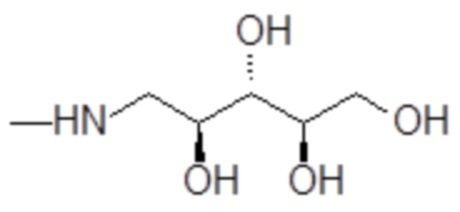 ,
,
и 1-амино-1-дезокси-D-ксилит формулы
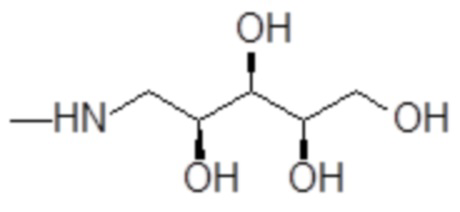 ;
;
1-амино-1-дезокси-гекситы формулы
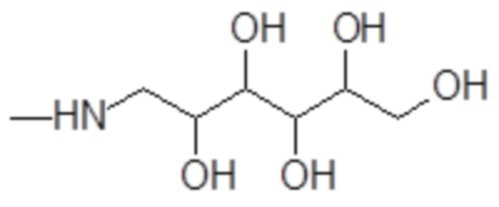
например, включая 1-амино-1-дезокси-D-глюцитол формулы
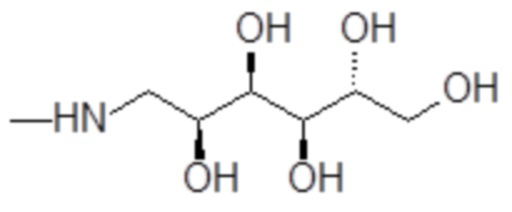 ,
,
1-амино-1-дезокси-D-галактитол формулы
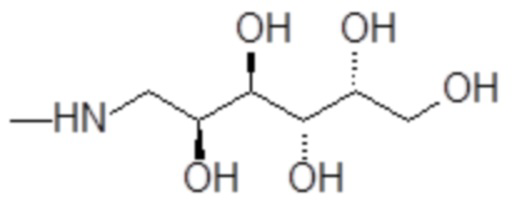 ,
,
1-амино-1-дезокси-D-идит формулы
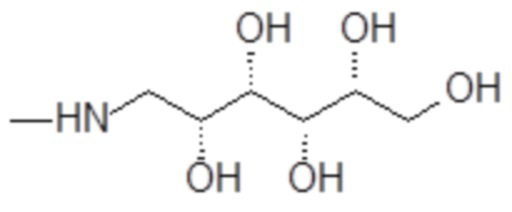 ,
,
и 1-амино-1-дезокси-D-маннит формулы
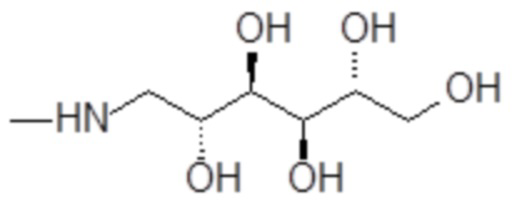 ,
,
а также соответствующих аминоалкил-полиолов, в которых атом водорода, связанный с азотом, замещен на R9 с указанными выше значениями.
Выражение «карбоксил» включает в свое смысловое значение остаток формулы -COOH, или содержащий указанный остаток -COOH, такой как группы формулы -(CH2)s-COOH или -[(O(CH2)n]s-COOH, в которых s и n соответствуют определению выше.
Термин «арил» или «арильное кольцо» относится к ароматическому углеводороду, а предпочтительно фенильному кольцу. Если конкретно не предусмотрено иное, арилы согласно изобретению могут быть либо незамещенными, либо замещенными одной или более, одинаковыми или отличающимися, замещающими группами, например, выбранными из гидроксила (OH), галогена, C1-C3 алкила, C1-C3 алкокси, C1-C3 гидроксиалкила, карбокси, карбамоила, нитро, -NH2, и C1-C3 алкил- или диалкиламино; предпочтительно из гидроксила, галогена, C1-C3 алкила или алкокси, и карбокси, а более предпочтительно из C1-C3 алкила или алкокси, -CH2COOH и -COOH.
Термин «циклоалкильное кольцо» в рамках изобретения относится к циклоалифатическому кольцу, а предпочтительно к C5-C7 карбоциклическому кольцу, например, циклогексильному кольцу. Если конкретно не предусмотрено иное, циклоалкилы согласно изобретению могут быть либо незамещенными, либо замещенными одной или более, одинаковыми или отличающимися, замещающими группами, например, выбранными из гидроксила, галогена, C1-C3 алкила, C1-C3 алкокси, C1-C3 гидроксиалкила, карбоксила, карбамоила, нитро, -NH2 и C1-C3 алкил- или диалкиламино; предпочтительно из гидроксила, галогена, C1-C3 алкила или алкокси, и карбокси, а более предпочтительно из C1-C3 алкила или алкокси, -CH2COOH и -COOH.
Термин «гетероциклическое кольцо» (или «гетероцикл») включает в свое смысловое значение 5- или 6-членный насыщенный циклический остаток, содержащий атом азота в циклической цепи, и, необязательно, еще один, такой же или другой, гетероатом, выбранный, например, из N, O и S. Подходящие примеры включают гетероциклы, такие как пирролидин, пиперазин, морфолин и пиперидин, при этом последний является особенно предпочтительным. Если конкретно не предусмотрено иное, азотсодержащие гетероциклы согласно изобретению содержат одну или более заместительных групп, связанных с атомом (атомами) углерода цикла, выбранных, например, из гидроксила, C1-C3 гидроксиалкила, C1-C3 алкокси, C1-C3 гидроксиалкокси, C1-C3 гидроксиалкокси-алкила и карбоксила, таких как -(CH2)s-COOH или -[(O(CH2)n]s-COOH, как определено выше.
Из всего вышеизложенного, после определения смыслового значения для алкила, алкилена, арила и циклоалкила, специалисту в данной области должно быть понятно любое составное название, такое как алкил-арил, арил-алкилен, циклоалкил-алкилен и тому подобное.
Например, термин алкиларил (или алкил-арил) включает в свое смысловое значение арильную группу, дополнительно замещенную алкилом, (например, p-этил-фенил; pC2H5-C6H5-), тогда как термин арилалкилен (или арил-алкилен) или циклоалкил-алкилен включает в свое смысловое значение алкил, дополнительно замещенный арилом (например, фенил-этилен=C6H5-C2H4-) или циклоалкилом (например, циклогексил-этилен=C6H11-C2H4-); и т.п.
В настоящем описании термин «защитная группа» обозначает защитную группу, приспособленную для защиты функции группы, с которой она связана. Конкретно, защитные группы используют для защиты функций амино, гидроксила или карбоксила. Соответствующие защищающие карбоксильные группы могут таким образом включать, например, бензил, алкил, например, трет-бутиловый или бензиловый сложные эфиры, или другие заместители, в основном используемые для защиты таких функций, которые хорошо известны специалистам в данной области техники [для общей справки см. T. W. Green и P. G. M. Wuts; Protective Groups in Organic Synthesis, Wiley, N.Y. 1999, third edition].
Кроме того, термины «фрагмент» или «фрагменты», «остаток» или «остатки» предназначены настоящим для определения остаточной части данной молекулы после правильного присоединения или конъюгирования либо непосредственно, либо посредством любого подходящего линкера, к остальной части молекулы.
Соединения указанной выше формулы (I) могут иметь один или более асимметричных атомов углерода, иначе называемых хиральными атомами углерода, и, таким образом, могут образовывать диастереомеры и оптические изомеры. Если не предусмотрено иное, настоящее изобретение дополнительно включает все такие возможные отдельные диастереомеры, а также их рацемические смеси, их по существу чистые разделенные энантиомеры, все возможные геометрические изомеры и их фармацевтически приемлемые соли.
Кроме того, настоящее изобретение относится к соединениям указанной выше формулы (I), в которых каждая из кислотных групп, содержащая либо карбоксильные группы R, связанные с атомами азота макроцикла, либо любую другую необязательную кислотную группу на гидроксилированном боковом плече, может быть в форме фармацевтически приемлемой соли или производного, кислотная группа которой соответствующим образом защищена соответствующей защитной группой (Pg), как указано выше, например, предпочтительно, C1-C5 алкилового сложного эфира, а более предпочтительно трет-бутилового сложного эфира, находящей, например, применение как таковой, или в качестве подходящего предшественника или промежуточного соединения при получении определенного соединения формулы (I) или подходящего парамагнитного комплекса или его соли.
В одном варианте осуществления соединения формулы (I) содержат аминопроизводное Z, связанное с атомом углерода, несущим гидроксильную группу посредством алкиленовой цепи, содержащей 1 или 2 атома углерода.
В предпочтительном варианте осуществления настоящее изобретение относится к соединениям формулы (I), в которых Z представляет собой производное третичного амина формулы -N(R2)(R3).
Подходящие примеры включают аминопроизводные формулы (II)
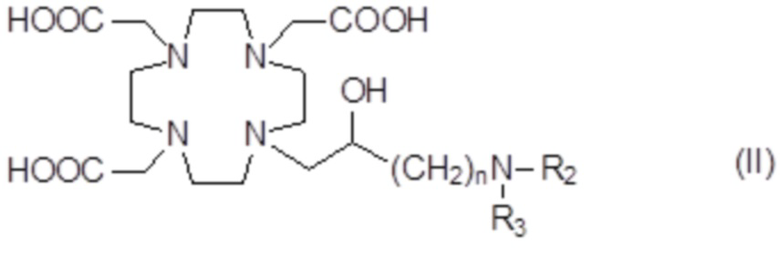
в которых n представляет собой целое число от 1 до 2, а R2 и R3 соответствуют определению для соединений формулы (I).
В одном варианте осуществления изобретение относится к соединениям согласно указанной выше формуле (II), в которой R3 представляет собой группу формулы -(CH2)sCH(R5)-G.
В частности, в одном варианте осуществления изобретение относится к соединениям формулы (II A)
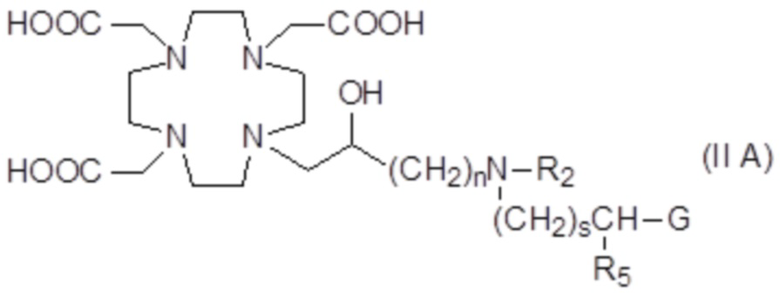
в которых n составляет 1 или 2, а предпочтительно составляет 1, а s, G, R2 и R5 соответствуют определению для соединений формулы (I).
Подходящие варианты осуществления включают соединения формулы (IIA), в которых:
n составляет 1;
G выбирают из групп формулы -PO(OR6)2, -COOH и -PO(R7)(OR6), в которых R6 представляет собой H или трет-бутил, а предпочтительно представляет собой H; и R7 необязательно замещен фенильным или циклогексильным кольцом, или C1-C5, а предпочтительно C1-C3 алкилом, таким как метил, этил или пропил, который замещен или не замещен арильным или циклоалкильным кольцом, например, предпочтительно, бензильной, фенил-этиленовой, циклогексил-метиленовой или циклогексил-этиленовой группой;
s составляет 0, 1 или 2, предпочтительно 0 или 1;
R5 представляет собой H или арилалкилен или циклоалкил-алкилен, имеющий до 3 атомов углерода в алкиленовой цепи; а
R2 соответствует определению для соединений формулы (I).
Предпочтительно в указанных выше соединениях формулы (II A) R2 представляет собой арильное или циклоалкильное кольцо, такое как фенильное или циклогексильное, которое может быть либо незамещенным, либо замещенным группой, например, выбранной из C1-C3 алкила, C1-C3 алкокси и -(CH2)sCOOH; или представляет собой C1-C10 алкил, который необязательно разделен 1, 2 или 3 атомами кислорода и/или необязательно замещен одной или более гидроксильными группами, например, 1, 2, 3, 4 или 5 гидроксильными группами, или необязательно замещен арильным или циклоалкильным кольцом.
Более предпочтительно, R2 представляет собой фенильное или циклогексильное кольцо, или C1-C7 алкил, который необязательно замещен одной или более гидроксильными группами или необязательно замещен фенильным или циклогексильным кольцом, таким как метильная, этильная, пропильная, изопропильная и трет-бутильная цепь, замещенная или не замещенная одной или более гидроксильными группами, например, включая гидроксиметил, гидроксиэтил, гидроксипропил, 1,3- и 2,3-дигидроксипропил и 2-(гидроксиметил)-1,3-дигидроксипропил, или фенильным или циклоалкильным кольцом, например, включая бензильную, фенил-этиленовую, циклогексил-метиленовую и циклогексил-этиленовую группу.
Особенно предпочтительными являются соединения формулы (II A), в которых:
n составляет 1;
R2 выбирают из группы, состоящей из: C1-C7 алкила, выбранного из метила, этила, пропила, изопропила и трет-бутила; их производных моно-, бис- и трис-гидроксиалкила, например, включая гидроксиметил, гидроксиэтил, гидроксипропил, 1,3- и 2,3-дигидроксипропилы и 2-(гидроксиметил)-1,3-дигидроксипропил; и арил-алкилена или циклоалкил-алкилена, предпочтительно содержащих до 3 атомов углерода в алкиленовой цепи, такого как бензил, фенил-этил, циклогексил-метил и циклогексил-этилен;
s составляет 0 или 1;
R5 представляет собой H или арилалкилен или циклоалкил-алкилен, выбранный из бензила, фенил-этилена, циклогексил-метилена и циклогексил-этила; а
G представляет собой -PO(OH)2 или -COOH.
В другом варианте осуществления изобретение относится к соединению аминов согласно указанной выше формуле (II), в которой R3 представляет собой C2-C10 гидроксиалкоксиалкилен формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH, а R2 соответствует определению для соединений формулы (I).
Более конкретно, в другом варианте осуществления изобретение относится к аминопроизводным формулы (II B)
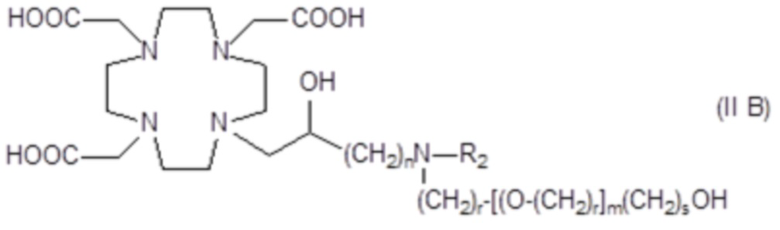
в которой:
n составляет 1 или 2, а предпочтительно 1;
каждый r составляет независимо 1 или 2;
m составляет 1, 2 или 3;
s составляет 0, 1 или 2; и
R2 соответствует определению для соединений формулы (I).
Подходящие примеры включают аминопроизводные формулы (II B), в которой R2 представляет собой C1-C10 алкил, необязательно разделенный одним или более атомами кислорода и/или необязательно замещенный одной или более гидроксильными группами, или арильное или циклоалкильное кольцо.
Предпочтительно, в указанных выше соединениях формулы (II B) R2 представляет собой C1-C10 алкильную цепь, замещенную одной или более, например, от 1 до 3 гидроксильными группами, и необязательно разделенную 1, 2 или 3 атомами кислорода.
В одном предпочтительном варианте осуществления изобретение относится к соединениям аминов указанной выше формулы (II B), в которой R2 представляет собой вторую гидроксиалкоксиалкиленовую цепь формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH, в которой r, s и m соответствуют сказанному выше. Предпочтительно, каждую цепь независимо выбирают из групп формулы -CH2(OCH2CH2)sOCH2OH, -(CH2)r-O(CH2)rOH и -CH2(CH2OCH2)rCH2OH, где m, r и s соответствуют сказанному.
Более предпочтительно, гидроксиалкоксиалкиленовые цепи, связанные с атомом азота, являются одинаковыми, и их выбирают из групп формулы -CH2(OCH2CH2)sOCH2OH и формулы -CH2(CH2OCH2)rCH2OH.
В особенно предпочтительном варианте осуществления изобретение относится к соединениям аминов формулы (II C)
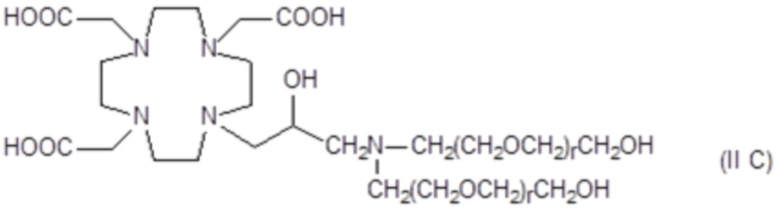
в которых r составляет 1 или 2, а предпочтительно 1.
Соединения формулы (I) согласно изобретению дополнительно включают аминопроизводные формулы (II), в которой R2 и R3 вместе с соединительным атомом азота образует замещенное пяти- или шести-членное насыщенное гетероциклическое кольцо.
Примеры указанных гетероциклических колец включают производные морфолина, пирролидина, а предпочтительно пиперидина, имеющие одну или более заместительных групп, связанных с атомом (атомами) углерода цикла.
В предпочтительном варианте осуществления изобретение относится к соединениям аминов формулы (I), в которой Z представляет собой производное пиперидина.
Подходящие примеры включают соединения формулы (III)
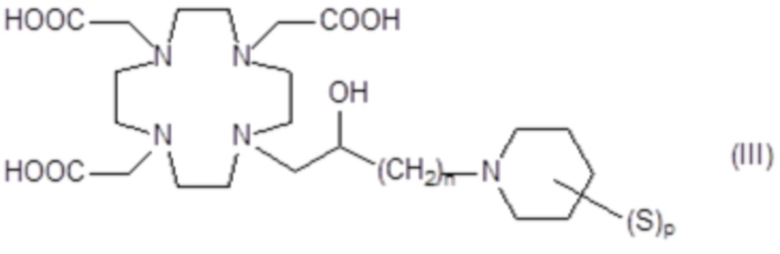
в которой:
n составляет 1 или 2, а предпочтительно 1;
p представляет собой целое число от 1 до 8; а
S представляет собой замещающую группу, связанную с атомом углерода пиперидинового кольца.
В одном варианте осуществления изобретение относится к соединениям формулы (III), в которой p составляет 1, а S представляет собой замещающую группу, выбранную из группы, состоящей из: гидроксила, C1-C3 гидроксиалкила, C1-C3 алкокси, C1-C3 гидроксиалкокси, C1-C3 гидроксиалкокси-алкилена и карбоксила, а предпочтительно из гидроксила, C1-C3 гидроксиалкила, C1-C3 гидроксиалкокси и карбоксила, такого как -(CH2)s-COOH или -OCH2-COOH.
Среди них предпочтительными являются соединения формулы (III), в которой S представляет собой замещающую группу, выбранную из гидроксила, -CH2OH и -COOH, который связан с C3 атомом углерода кольца.
В предпочтительном варианте осуществления изобретение относится к соединениям указанной выше формулы (III), в которой p представляет собой целое число от 2 до 8, которые содержат пиперидиновое кольцо, имеющее от 2 до 8, предпочтительно от 2 до 6, а более предпочтительно от 3 до 5, например, 3, 4 или 5 замещающих групп S, связанных с одним или более атомом (атомами) углерода кольца, каждую из которых независимо выбирают из гидроксила, C1-C3 гидроксиалкила, C1-C3 алкокси, C1-C3 гидроксиалкокси, C1-C3 гидроксиалкокси-алкилена и карбоксила, например, -(CH2)s-COOH или -(OCH2)s-COOH.
Предпочтительными среди них являются соединения формулы (III A)
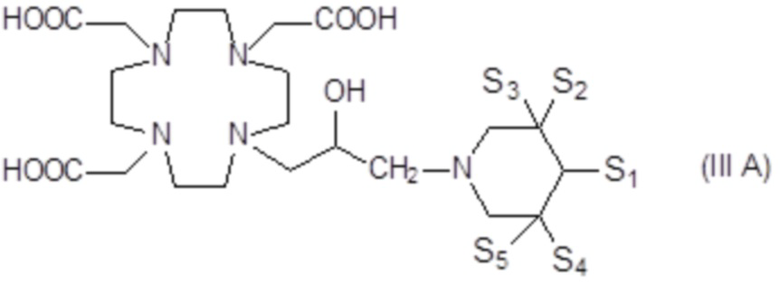
в которой каждую из замещающих групп S1-S5 независимо выбирают из группы, состоящей из: H, гидроксила, C1-C3 гидроксиалкила, C1-C3 гидроксиалкокси и C1-C3 гидроксиалкокси-алкилена, при условии, что по меньшей мере 3 из S1-S5 замещающих групп не являются H.
Подходящие примеры включают соединения формулы (III A), в которой замещенное пиридиновое кольцо представляет собой группу формулы:
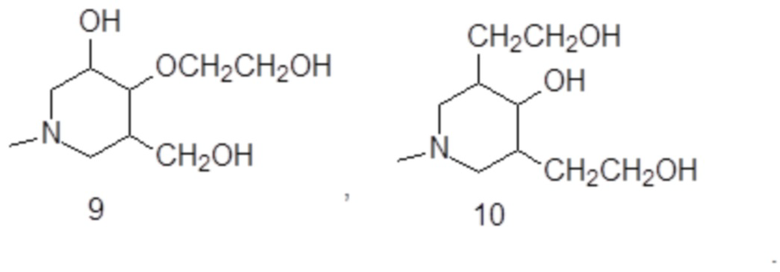
В особенно предпочтительном варианте осуществления изобретение относится к соединению формулы (III A), в которой S1 представляет собой гидроксильную группу, и S2-S4 представляют собой C1-C3 гидроксиалкилы, одинаковые или отличающиеся друг от друга.
В дополнительном варианте осуществления настоящее изобретение относится к соединениям аминов формулы (I), в которой Z представляет собой производное вторичных аминов формулы -NHR4.
Подходящие примеры включают аминопроизводное формулы (IV),
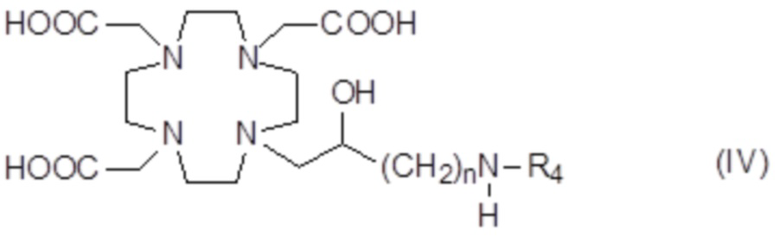
в которой n представляет собой целое число от 1 до 2, а R4 соответствует определению выше для соединений формулы (I).
Предпочтительно, R4 выбирают из: необязательно замещенного циклогексила или циклогексил-алкилена, имеющего до 3 атомов углерода в алкиленовой цепи, например, циклогексил-метилена или циклогексил-этилена; группы формулы -(CH2)mPO(R7)(OR6) или -(CH2)mPO(OR6)2, где m представляет собой целое число от 1 до 3, R6 представляет собой H или трет-бутил и предпочтительно G, а R7 представляет собой необязательно замещенный фенил или циклогексил или C1-C5, а предпочтительно C1-C3 алкил, такой как метил, этил или пропил, замещенный или незамещенный арильным или циклоалкильным кольцом, например, предпочтительно, бензильной, фенил-этиленовой, циклогексил-метиленовой или циклогексил-этиленовой группой; C2-C10 гидроксиалкоксиалкилена формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH, в которой r, s и m соответствуют сказанному выше для соединений формулы (I); и группы формулы -(CH2)sCH(R8)G, где s составляет 0, 1 или 2, и предпочтительно составляет 0 или 1, R8 представляет собой необязательно замещенный арилалкилен или циклоалкил-алкилен, имеющий до 3 атомов углерода в алкиленовой цепи, а G представляет собой группу, выбранную из -PO(OR6)2, -PO(R7)(OR6) и -COOH, где R6 и R7 соответствуют сказанному выше.
В частности, в одном предпочтительном варианте осуществления изобретение относится к аминопроизводным согласно указанной выше формуле (IV), в которой n составляет 1, а R4 представляет собой группу формулы -(CH2)mPO(R7)(OR6), а более предпочтительно формулы -(CH2)mPO(OR6)2, где m представляет собой целое число от 1 до 3, а предпочтительно составляет 1 или 2, R6 представляет собой H, а R7 представляет собой необязательно замещенный фенил или циклогексил или группу, выбранную из метила, этила, пропила, бензила, фенил-этилена, циклогексил-метилена и циклогексил-этилена.
В другом предпочтительном варианте осуществления изобретение относится к аминопроизводным согласно указанной выше формуле (IV), имеющим формулу (IV A)
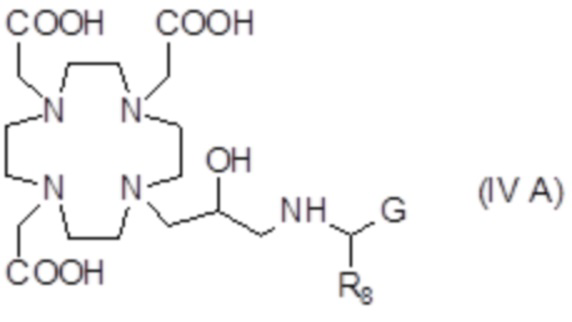
где R8 представляет собой необязательно замещенный арил-алкилен или циклогексил-алкилен, как указано выше, а предпочтительно выбран из бензила, фенил-этилена, циклогексил-метилена и циклогексил-этилена, и G представляет собой группу, выбранную из -PO(OR6)2, -PO(R7)(OR6) и -COOH, а более предпочтительно из -PO(OR6)2 и -COOH, где R6 предпочтительно представляет собой H.
В предпочтительном варианте осуществления изобретение относится к аминопроизводным согласно формуле (I), в которых Z представляет собой третичный или вторичный C5-C12 аминополиол.
Подходящие примеры включают соединения указанной выше формулы (I), в которых Z представляет собой аминопроизводное, выбранное из -N(R2)(R3) или -NHR4, в которых R3 и R4 представляют собой C5-C12 полиол, а R2 соответствует определению выше для соединений формулы (I).
Предпочтительными среди них являются производные аминополиола формулы (V)

в которых:
n составляет 1;
R9 представляет собой H или C1-C3 алкил, например, пропил, этил или, предпочтительно, метил; а
R10 представляет собой C5-C12 полиол.
Предпочтительными согласно изобретению являются производные аминополиолов указанной выше формулы (V), в которых R10 представляет собой C5-C7 полиол, например, выбранный из пентил(поли)олов (или полигидроксипентилов), содержащих по меньшей мере 2, а предпочтительно от 2 до 4 гидроксильных групп на C5 алкильной цепи; гексил(поли)олов, содержащих по меньшей мере 2, а предпочтительно от 2 до 5 гидроксильных групп на C6 алкильной цепи; и гептил(поли)олов, содержащих по меньшей мере 2, а предпочтительно от 3 до 6 гидроксильных групп на C7 алкильной цепи, а R9 представляет собой H или метильную группу.
Подходящие примеры включают пентил(поли)олы, такие как пентил-диолы, пентил-триолы, и пентил-тетраолы; гексил(поли)олы, такие как гексил-диолы, гексил-триолы, гексил-тетраолы и гексил-пентаолы; и гептил(поли)олы, такие как гептил-диолы, гептил-триолы, гептил-тетраолы, гептил-пентаолы и гептил-гексаолы.
Особенно предпочтительными согласно изобретению являются производные аминополиолов указанной выше формулы (V), в которых:
R9 представляет собой H или метил; а
R10 выбирают из пентил-тетраола формулы
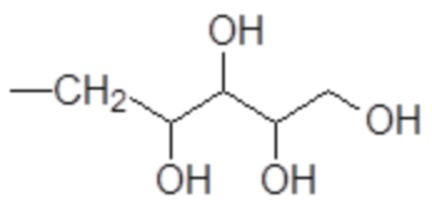 и
и
гексил-пентаола формулы
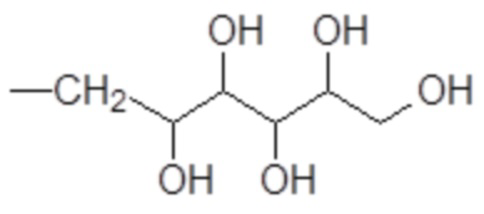 .
.
В частности, в предпочтительном варианте осуществления изобретение относится к соединениям согласно формуле (I), в которых Z представляет собой остаток аминополиола, выбранный из группы, состоящей из 1-амино-1-дезокси-пентитолов, таких как 1-амино-1-дезокси рибит, 1-амино-1-дезокси-ксилит и 1-амино-1-дезокси-арабит; 1-амино-1-дезокси-гекситы, такие как 1-амино-1-дезокси-глюцит, 1-амино-дезокси-галактитол, 1-амино-1-дезокси-аллит, 1-амино-1-дезокси-маннитол и 1-амино-1-дезокси-идитол; и 1-амино-1-дезокси-гептитолы, такие как 1-амино-1-дезокси-глицеро-манно-гептитол, а также их N-(C1-C3)алкил производных, предпочтительно N-метил.
Более предпочтительно, Z представляет собой остаток 1-амино-1-дезокси-гексита, например, выбранный из группы, состоящей из 1-амино-1-дезокси-глюцитола, 1-амино-дезокси-галактитола, 1-амино-1-дезокси-маннитола, 1-амино-1-дезокси-дитола, и их N-метил производных.
В особенно предпочтительном варианте осуществления изобретение относится к соединению формулы (I), в котором Z представляет собой 1-дезокси-1-амино-D-глюцитоловый или, в частности, 1-дезокси-1-(метиламино)-D-глюцитоловый остаток, имеющий, соответственно, формулу
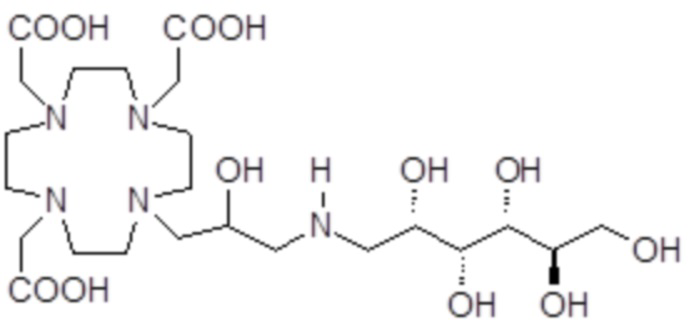
и формулу
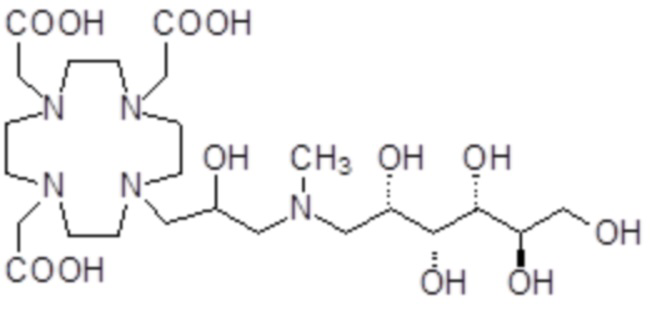 .
.
Особенно предпочтительными соединениями являются те соединения формулы (I) или их соли, которые выбраны из группы, состоящей из:
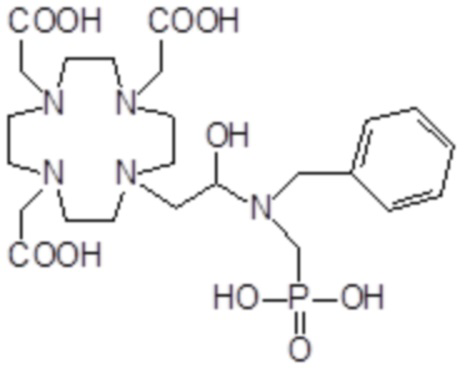 ,
, 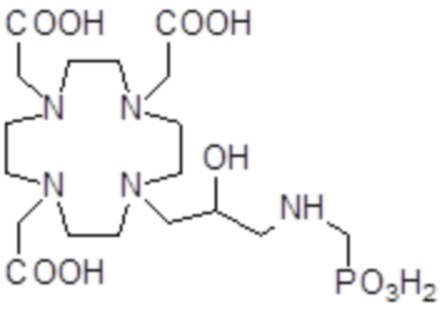 ,
,
Соединения 1 Соединения 2
, 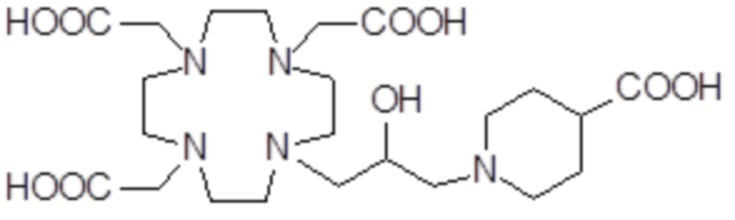 ,
, 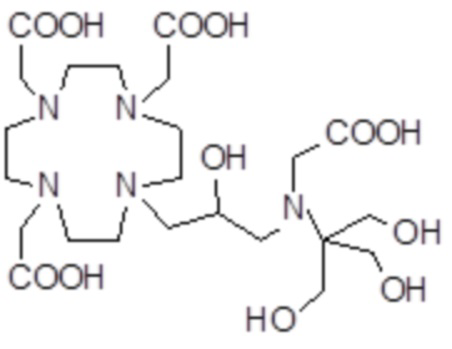 ,
,
Соединения 3 Соединения 4
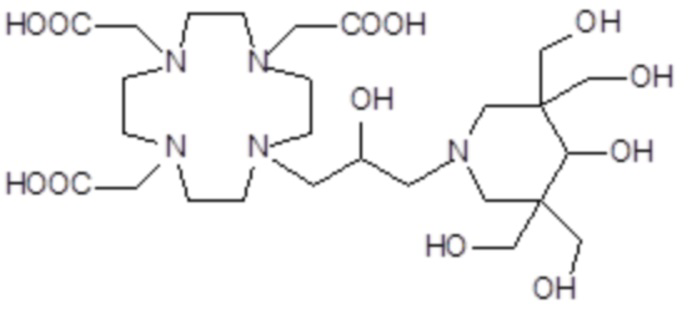 ,
, 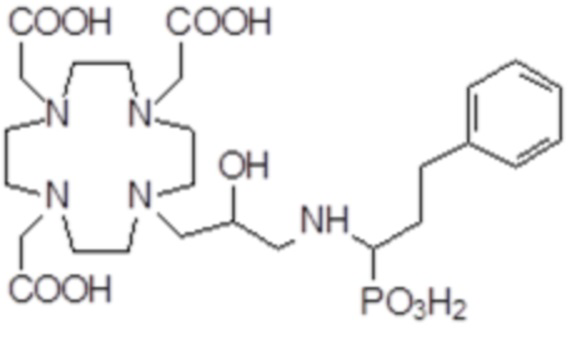
Соединения 5 Соединения 6
 ,
, 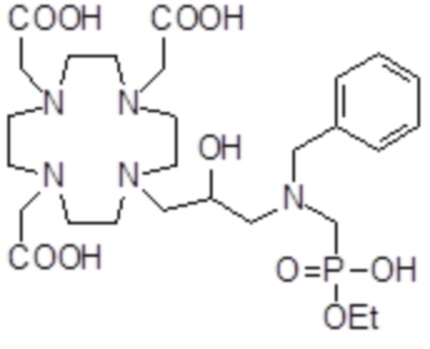 ,
, 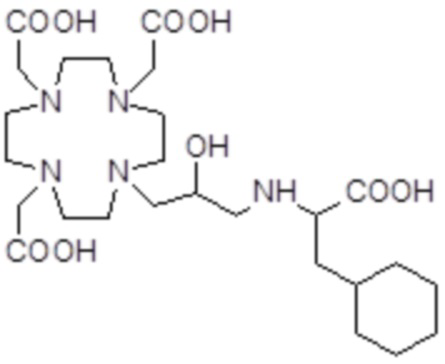 ,
,
Соединения 7 Соединения 8 Соединения 9
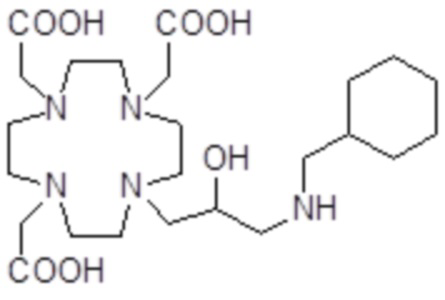 ,
, 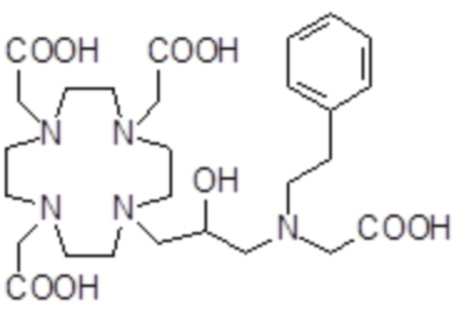 ,
, 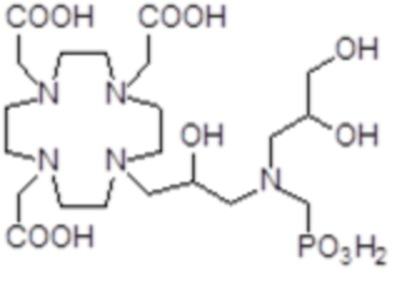 ,
,
Соединения 10 Соединения 11 Соединения 12
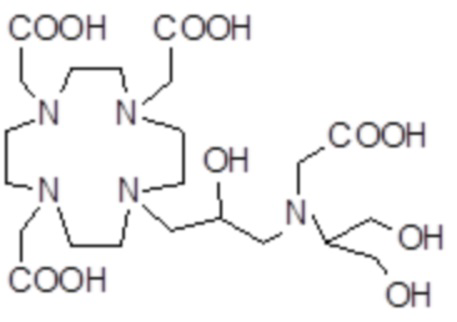 ,
, 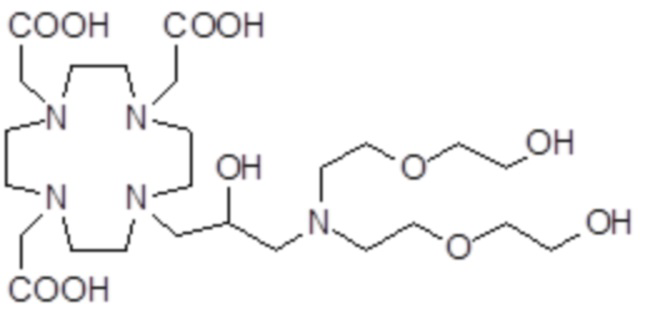 ,
,
Соединения 13 Соединения 14
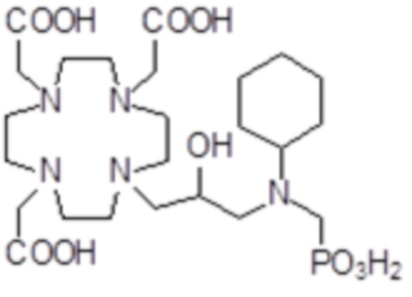 ,
, 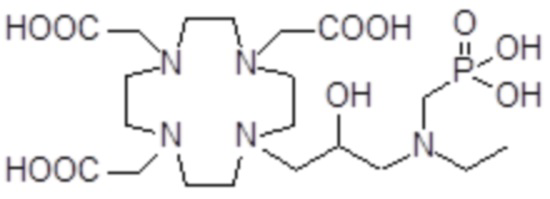 ,
, 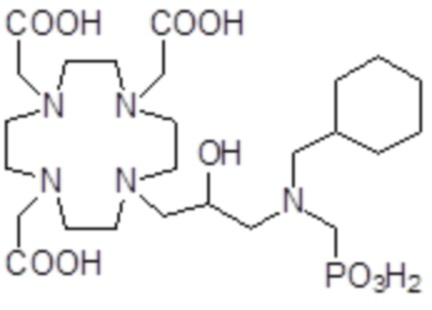 ,
,
Соединения 15 Соединения 16 Соединения 17
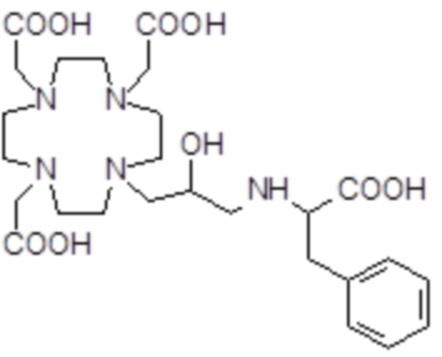 ,
, 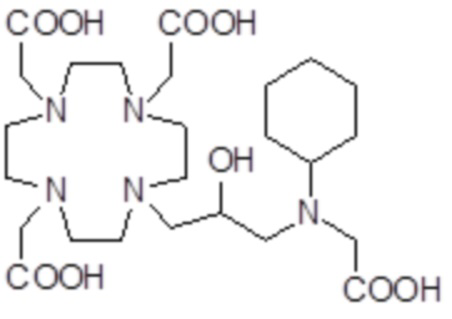 ,
,
Соединения 18 Соединения 19
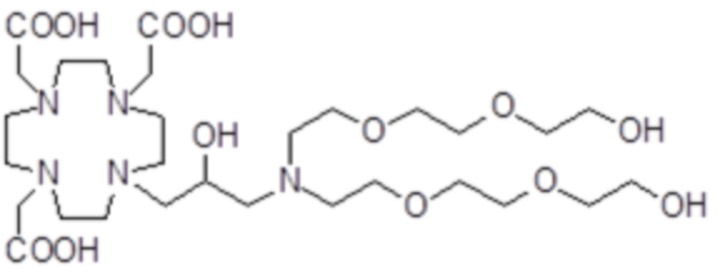 ,
, 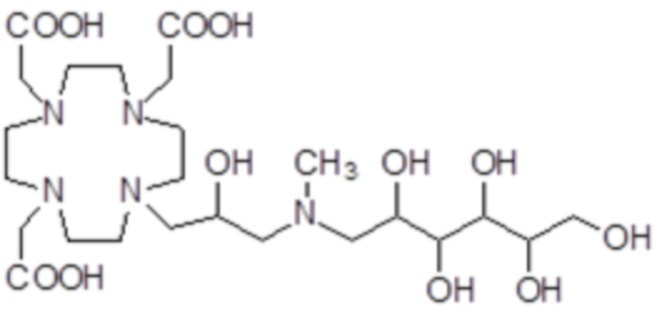
Соединения 20 Соединения 21
В дополнительном аспекте изобретение относится к хелатным комплексам соединений формулы (I), следовательно, охватывая соединения формул от (II) до (V), с ионом парамагнитного металла, или радионуклидом, или их подходящих солей.
Предпочтительно, ион парамагнитного металла выбирают из группы, состоящей из Fe2+, Fe3+, Cu2+, Cr3+, Gd3+, Eu3+, Dy3+, La3+, Yb3+ или Mn2+. Более предпочтительно, ион парамагнитного металла представляет собой Gd3+.
Предпочтительные радионуклиды согласно изобретению, обеспечивающие комплексы для применения в радиотерапии или радиодиагностике, включают 105Rh, 117mSn, 99mTc, 94mTc, 203Pb, 67Ga, 68Ga, 44Sc, 72As, 110In, 111In, 113In, 90Y, 97Ru, 60Cu, 62Cu, 64Cu, 52Fe, 51Mn, 140La, 175Yb, 153Sm, 166 чo, 149Pm, 177Lu, 186/188Re, 165Dy, 166Dy, 142Pr, 159Gd, 211Bi, 212Bi, 213Bi, 214Bi, 149Pm, 67Cu, 198Au, 199Au, 161Tb, 167Tm и 51Cr.
Как соединения формулы (I), включающие таким образом соединения формулы (II)-(V), так и их парамагнитные хелаты также могут быть в форме фармацевтически приемлемой соли, в частности в виде соли присоединения с физиологически совместимым основанием или кислотой.
Термин «фармацевтически приемлемая соль», в рамках изобретения, относится к производным соединений изобретения, при этом исходное соединение надлежащим образом модифицируют путем преобразования любой из групп свободных кислот или оснований, если имеются, в соответствующую соль присоединения с любым основанием или кислотой, традиционно предназначаемых в качестве фармацевтически приемлемых.
Предпочтительные катионы неорганических оснований, которые можно надлежащим образом использовать для получения соли комплексов или лигандов изобретения, включают, например, ионы щелочных или щелочноземельных металлов, таких как калий, натрий, кальций или магний.
Предпочтительные катионы органических оснований включают, например, катионы первичных, вторичных и третичных аминов, таких как, например, этаноламин, диэтаноламин, морфолин, глюкамин, N-метилглюкамин, N,N-диметилглюкамин.
Предпочтительные анионы неорганических кислот, которые можно надлежащим образом использовать для получения солей комплексов изобретения, включают ионы галокислот, например, хлоридов, бромидов или йодидов, а также других подходящих ионов, таких как сульфат.
Предпочтительные анионы органических кислот включают анионы, обычно используемые в фармацевтических методиках для получения методом солеобразования солей основных веществ, таких как, например, ацетат, сукцинат, цитрат, фумарат, малеат или оксалат.
Предпочтительные катионы и анионы аминокислот включают, например, катионы и анионы таурина, глицина, лизина, аргинина, орнитина или аспарагиновой и глютаминовой кислот.
Получение хелатирующих соединений изобретения и их хелатных комплексов, либо как таковых, либо в форме физиологически приемлемых солей, представляет дополнительную цель изобретения. Если дальше не указано иное, в частности при ссылке на общие способы получения, способы применения или фармацевтические готовые формы, необходимо понимать, что термин «соединения формулы (I)» охватывает также соединения формул (II)-(V), а также конкретные хелатирующие соединения изобретения, раскрытые в настоящем описании.
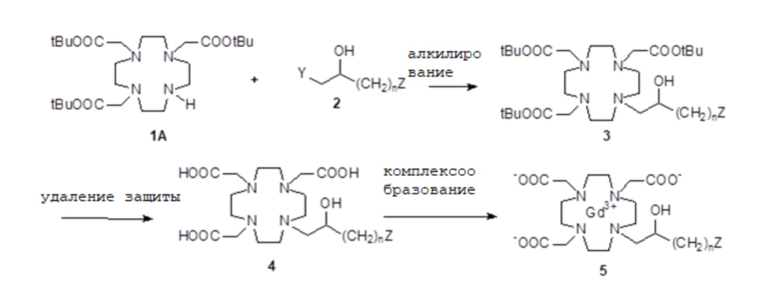
Соединения формулы (I) и их хелатные комплексы, и их соли, могут быть получены посредством общего способа синтеза, включающего следующие стадии:
a) Получение макроциклического субстрата 1 в подходящей защищенной форме, например, в которой карбоксильные группы субстрата защищены в виде трет-бутиловых сложных эфиров;
b) Получение алкилирующей молекулы 2, в которой любая необязательная функциональная группа (группы), не вовлеченная в реакцию связывания с субстратом 1, необязательно защищена надлежащим образом;
с) Связывание защищенного субстрата 1 с алкилирующей молекулой 2 с получением требуемого соединения формулы (I) в надлежащим образом защищенной форме или, в качестве альтернативы, его промежуточного соединения 3;
d) Необязательно преобразование полученного промежуточного соединения в надлежащим образом защищенном соединении формулы (I);
е) Удаление любой защитной группы и выделение хелатирующего лиганда формулы (I); и
f) Комплексообразование полученного лиганда с подходящим ионом парамагнитного металла и выделение хелатного комплекса или его соли.
В этом смысле, и если не указано иное, термин «промежуточное» (например, со ссылкой на соединение 3, получаемое в результате реакции макроциклического субстрата 1 с алкилирующей молекулой 2) относится к молекуле, которая требует одну (или более) дополнительных реакций, например, восстановления, дополнительного алкилирования и т.д., для получения требуемого продукта, т.е. в конкретном случае указанной выше общей схемы, в надлежащим образом защищенном соединении формулы (I) согласно стадии d). Отдельные стадии указанного выше общего способа, исчерпывающего в любом его варианте, особенно при ссылке на стадии защиты/снятия защиты и активации известных функциональных групп, могут выполняться согласно обычным способам, известным в данной области.
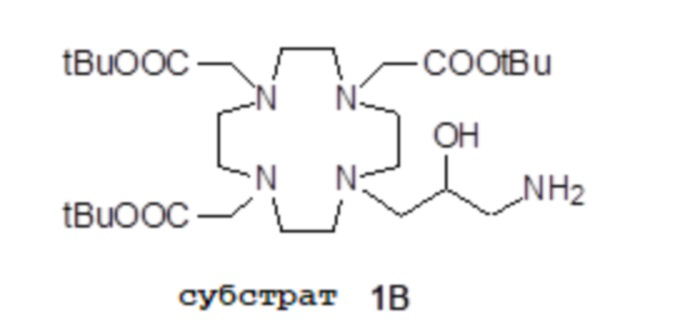
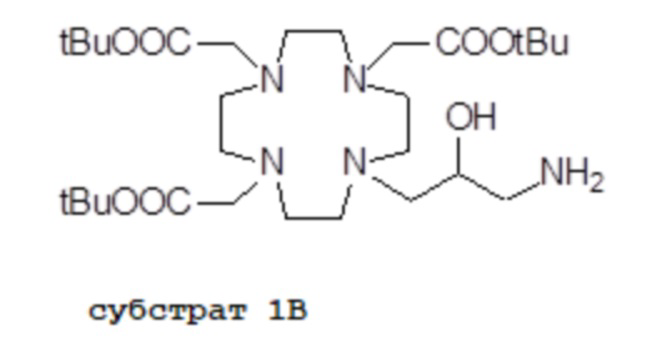
Например, подходящие субстраты 1A и 1B согласно стадии a) способа изобретения, формулы
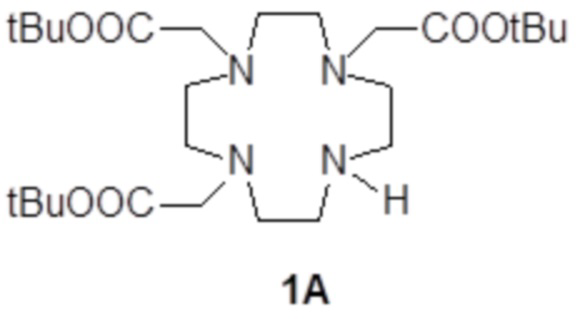 и
и 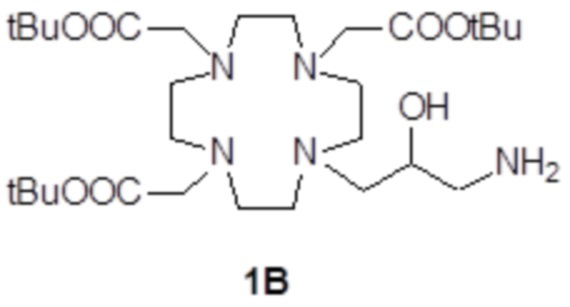 ,
,
в которых все карбоксильные группы надлежащим образом защищены в виде трет-бутиловых сложных эфиров и могут быть получены, например, как раскрыто в Org. Synth. 2008, 85, 10 и US7208140, соответственно.
Соответствующие алкилирующие молекулы 2 для применения изобретения коммерчески доступны или могут быть легко получены согласно методикам, известным специалистам в соответствующей области. Примеры конкретных методик для получения защищенных алкилирующих молекул 2, их связывания с соответствующей молекулой 1 субстрата и необязательного преобразования полученных промежуточных соединений в требуемое соединение формулы (I) приведены в экспериментальном разделе вместе с соответствующими рабочими подробностями.
В качестве общей ссылки на возможные защитные группы и условия расщепления, например, для выполнения стадии e) указанного выше общего метода синтеза, см. цитированное выше «T. W. Green and P. G. M. Wuts; Protective groups in organic synthesis» Wiley 3rd Ed. Главы 5 и 7.
Комплексообразование соединений формулы (I), например, полученных на стадии f) предыдущей общей схемы получения, с парамагнитным ионом, и особенно с гадолинием, можно проводить, например, путем стехиометрического добавления подходящего производного Gd(III), особенно соли или оксида Gd(III), в раствор лиганда, например, работая согласно хорошо известным экспериментальным методам, например, как сообщалось в EP 230893.
В заключение, необязательное солеобразование соединений изобретения можно выполнять путем правильного преобразования любой из свободных кислотных групп (например, карбоновой, фосфоновой или фосфиновой) или свободных аминогрупп в соответствующие фармацевтически приемлемые соли. В этом случае также, рабочие условия, используемые для необязательного солеобразования соединений изобретения, все находятся в пределах обычных знаний специалиста.
Ниже в настоящем описании схематично изложено иллюстративное исполнение указанной выше общей методики, ведущее к соединениям формулы (I) и их хелатных комплексов.
Например, соединения формулы (I) могут быть получены путем использования метода синтеза, схематично показанного в следующей схеме 1
Схема 1
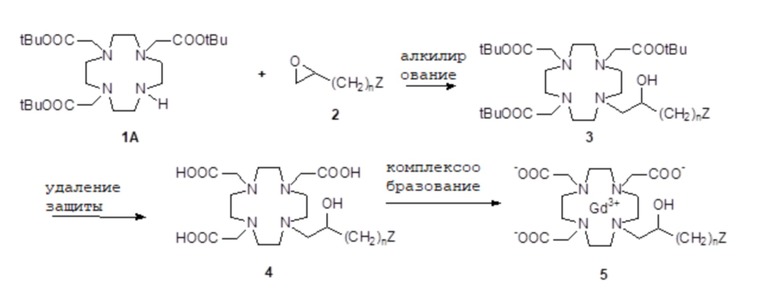
в которой подходящее эпоксипроизводное 2 выбранной группы Z реагирует с защищенным субстратом 1A DO3A с получением защищенного лиганда формулы (I), который после расщепления защитных групп образует комплекс с ионом металла гадолиния с получением требуемого Gd комплекса формулы (I).
Соединения формулы (I), в которых Z представляет собой, например, надлежащим образом замещенное гетероциклическое кольцо, например, производное пиперидина, как в соединениях формулы (III), в качестве альтернативы могут быть получены путем использования методики следующей общей схемы 2, в которой S представляет собой замещающую группу на гетероциклическом кольце.
Схема 2
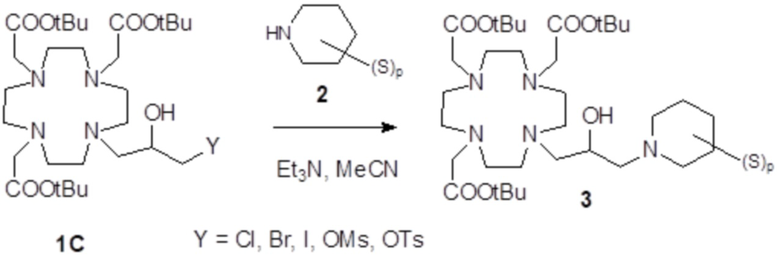
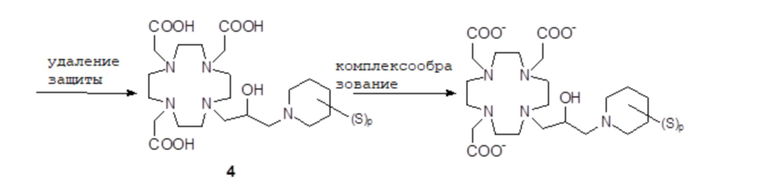
Согласно этому подходу, сначала получают надлежащим образом защищенный субстрат 1C, где Y представляет собой замещаемую группу, например, выбираемую из брома, хлора, йода и эфира арил/алкилсульфоновой кислоты, а более часто представляет собой атом хлора, например, как подробно описано в экспериментальном разделе. Затем получают промежуточное соединение 3 путем связывания субстрата 1C с подходящим производным 2 пиперидина, который после расщепления защитных групп образует комплекс с ионом металла гадолиния с получением требуемого Gd комплекса формулы (I), как обсуждалось выше.
Соединения формулы (I) согласно изобретению, в которых Z представляет собой аминопроизводное формулы -N(R2)(R3) или -NH(R4), как обсуждалось выше, могут быть получены иным образом путем использования методики, схематично показанной на следующей общей схеме 3, в которой -CH2Rx представляет собой группу со смысловыми значениями R2 или R4.
Схема 3
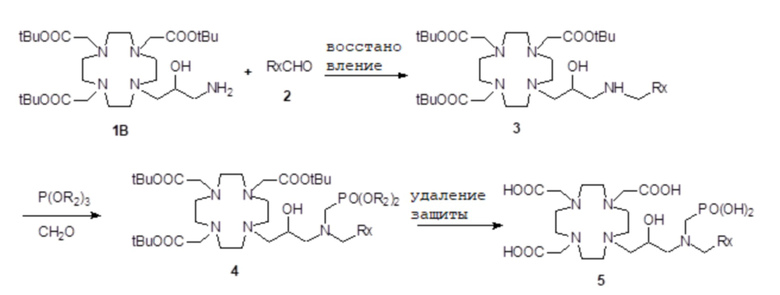
Согласно этому подходу, сначала получают надлежащим образом защищенный субстрат 1B,
например, как подробно описано в экспериментальном разделе, или как раскрыто в US 7208140, как указано выше, который затем преобразуют в требуемые бис-алкилированные производные формулы (X) путем алкилирования.
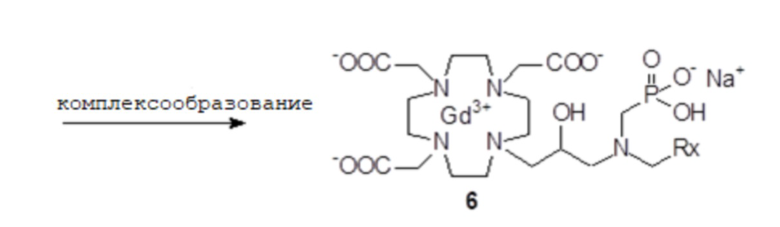
В частности, как показано на схеме 3 синтеза, субстрат 1B сначала реагирует с альдегидом формулы RxCHO с получением соответствующего имино-производного, что при восстановлении приводит к соответствующему защищенному лиганду формулы (IV) или моно-алкилированному промежуточному соединению 3, имеющему группу R2, соединенную с аминогруппой субстрата 1B. Затем полученное промежуточное соединение 3 дополнительно реагирует, например, с подходящим фосфитом, например, три(трет-бутил)фосфитом, полученным, например, как раскрыто в Tetrahedron Lett. 2005, 46, 4707-4710, с получением соответствующего производного 4 фосфоната, в котором кислотные группы находятся в защищенной форме. За счет снятия защиты всех защищенных групп затем получают соединение формулы (II), которое может образовать комплекс с ионом металла гадолиния (Gd3+), как обсуждалось ранее выше, и выделяют в виде соли, как представлено более подробно в следующем экспериментальном разделе.
В качестве альтернативы соединения формулы (II A) могут быть получены путем использования следующей схемы 4 синтеза
Схема 4
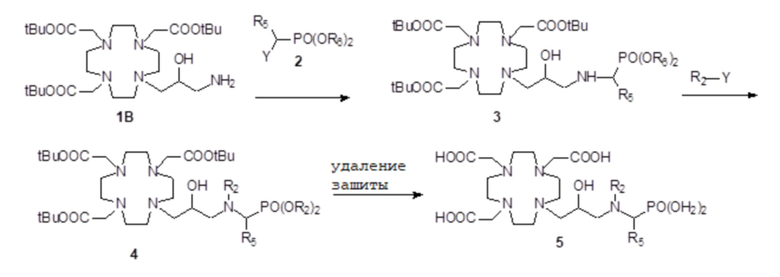
в которой субстрат 1B сначала реагирует с надлежащим образом защищенным фосфонатом формулы Y-CH(R5)-PO(OR6)2. Затем полученное соединение может дополнительно реагировать с подходящим производным R2, например, с Y-R2, в котором Y представляет собой, в обоих случаях, подходящую замещаемую группу, например, выбранную из Cl, Br, I, OMs, OTs, как указано выше.
Предпочтительные соединения согласно изобретению, в которых Z представляет собой остаток аминополиола, могут быть получены иным образом, например, путем использования методики следующей общей схемы 5
Схема 5
в которой подходящее производное 2 выбранного аминополиола, где Y представляет собой замещаемую группу, как указано, реагирует с защищенным субстратом 1A DO3A с получением соответствующего защищенного лиганда формулы (I), который после расщепления защитных групп образует комплекс с ионом металла гадолиния с получением требуемого Gd комплекса формулы (I).
Кроме того, конкретные примеры получения предпочтительных соединений формулы (I) согласно изобретению предоставлены в следующем экспериментальном разделе, составляющем общую ссылку на рабочие условия, используемые в указанных выше способах.
Макроциклические соединения формулы (I) согласно настоящему изобретению включают гидроксильный (OH) остаток вместе с аминопроизводным Z на боковом плече макроцикла.
Хотя и не желая связывать себя какой-либо конкретной теорией, заявитель считает, что релаксивность парамагнитных комплексов соединений формулы (I) может быть значительно улучшена в результате комбинированного эффекта, стимулируемого этими специфическими структурными компонентами.
Измеренная релаксивность особенно увеличивается относительно релаксивности, демонстрируемой в тех же самых условиях известными МРТ контрастными агентами, используемыми в настоящее время в диагностической ежедневной практике, например, включая Gd-DOTA, поставляемым на рынок под названием DOTAREM®, и Gd-HPDO3A, поставляемого на рынок под названием ProHance®, имеющими аналогичные макроциклические хелатирующие лиганды и сопоставимую молекулярную массу. В самом деле, как показано в таблице A экспериментального раздела, парамагнитные комплексные соединения изобретения показывают значения r1p релаксивности, которые приблизительно от 1,5 и до 2 раз выше, чем r1p значения, демонстрируемые аналогичными макроциклическими коммерческими контрастными агентами (такими как упомянутые выше DOTAREM® и ProHance®), которые, однако, лишены комбинированных структурных компонентов на боковом плече макроцикла.
В частности, парамагнитные комплексные соединения формулы (I) изобретения демонстрируют значение r1p релаксивности, измеренное в плазме человека при 37°C и прибл. 1,4 T, которое составляет по меньшей мере приблизительно 5,5, предпочтительно выше чем 6, и более предпочтительно, выше чем 7 мМ-1с-1.
Эту неожиданно высокую релаксивность можно дополнительно наблюдать, например, путем сравнения релаксивности, демонстрируемой комплексом гадолиния формулы
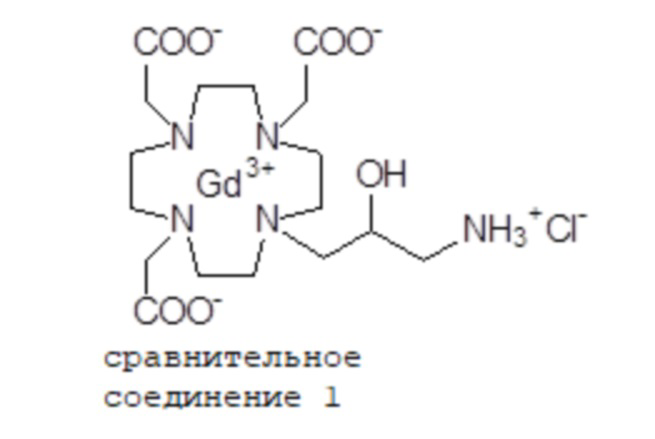
полученного посредством снятия защиты и комплексообразования субстрата 1B, и в настоящем описании используемого в качестве сравнительного соединения (сравнительного соединения 1), с релаксивностью, измеренной для его моно- и бис-алкилированного производного согласно формуле (II) изобретения. В самом деле, хотя значение r1p релаксивности, равное 5,3 мМ-1с-1, получают для сравнительного соединения 1 в плазме человека при 37°C и 1,41 T, что соответствует аналогично измеренным значениям в тех же самых условиях, для DOTAREM® и ProHance®, соответственно, равным 3,6 и 4,15, для моно-алкилированного производного (например, хелатного комплекса 2) наблюдается значительное увеличение до значения r1p, равного 7,5 (в плазме человека), которое еще увеличивается для бис-алкилированных производных, например, до 9,5, для хелатного комплекса 1.
Кроме того, было подтверждено, что парамагнитные комплексные соединения изобретения демонстрируют низкое, если не ничтожно малое, связывание белка с белками плазмы человека, включая, например, HSA.
В частности парамагнитный комплекс формулы (I) обычно отображает связывание белка с HSA ниже, чем 30%, предпочтительно, чем 25, а более предпочтительно, чем 20%.
Эти результаты позволяют предложить парамагнитные комплексные соединения изобретения в качестве неспецифических контрастных агентов, т.е. в качестве контрастных агентов для МРТ, подходящих для общего применения, таких как контрастные агенты на рынке наподобие Dotarem®, ProHance® и Magnevist®.
Кроме того, заявитель заметил, что присутствие остатка полиола или аминополиола на гидроксилированном боковом плече макроциклических соединений изобретения наряду с комплексными соединениями, имеющими благоприятную релаксивность и термическую стабильность, также может способствовать получению водных растворов соответствующего парамагнитного комплекса, наделенных оптимизированной вязкостью.
Предпочтительно, высокая релаксивность, демонстрируемая агентами изобретения, может кроме того обеспечивать уменьшение их диагностически эффективной дозы по сравнению с контрастными агентами, используемыми в настоящее время. Таким образом, парамагнитные комплексы, и особенно гадолиниевые комплексы соединений формулы (I) или их фармацевтически приемлемых солей находят преимущественное применение при получении фармацевтических готовых форм, предназначенных для общего применения при диагностической визуализации органов, ткани или области тела людей или животных либо in vivo или in vitro, ex vivo.
Согласно дополнительному аспекту, изобретение относится к применению соединений формулы (I) в форме комплексов с ионом парамагнитного металла, и особенно гадолиния, или их фармацевтических приемлемых солей для получения фармацевтической готовой формы для применения при диагностической визуализации, либо in vivo либо in vitro, ex vivo, органов, ткани или области тела людей или животных или биологического образца, включая клетки, биологические текучие среды и биологические ткани, происходящие из живого пациента-млекопитающего, а предпочтительно, пациента-человека, путем применения методики МРТ.
Дополнительный аспект изобретения относится к фармацевтической композиции для диагностического применения, содержащей соединение формулы (I) в форме комплекса с парамагнитным металлом, или его фармацевтически приемлемую соль в смеси с одним или более физиологически приемлемыми эксципиентами, разбавителями или растворителями. Предпочтительно, фармацевтической композицией является композиция для получения контрастного агента, а более предпочтительно создающая композиция для получения контрастного агента для МРТ, содержащая по меньшей мере один Gd-комплекс согласно изобретению.
В дополнительном аспекте изобретение относится к контрастному веществу для МРТ, содержащему эффективное количество по меньшей мере одного хелатного соединения согласно изобретению, и особенно гадолиниевого комплекса формулы (I), или их фармацевтических приемлемых солей, в комбинации с одним или более фармацевтически приемлемыми эксципиентами, разбавителями или растворителями.
В этом смысле, и если не предусмотрено иное, термин «эффективное количество» или «эффективная доза», в рамках изобретения, относится к любому количеству парамагнитного хелатного комплекса формулы (I) согласно изобретению или его фармацевтической композиции, которое является достаточным для выполнения предназначенной для него диагностической цели (целей): т.е., например, для визуализации ex vivo биологического элемента, включая клетки, биологические текучие среды и биологические ткани или для диагностической визуализации in vivo органов, тканей или областей пациента.
Если не указано иное, к «отдельному пациенту» или «пациенту» в рамках изобретения относится живой пациент-человек или животное, а предпочтительно человек, подвергаемый диагностической оценке МРТ.
В данной области известны подробности, касающиеся дозировок, лекарственных форм, режимов введения, фармацевтически приемлемых носителей, эксципиентов, разбавителей, адъювантов и т.п.
Интересно, и как обсуждалось выше, что подходящая дозировка парамагнитных комплексов согласно изобретению, т.е. позволяющая получить диагностически эффективную визуализацию органа, ткани или области тела, по меньшей мере сопоставимую с визуализацией, получаемой в ежедневной практике с имеющимися на рынке контрастными агентами МРТ, может содержать количество парамагнитного комплекса ниже, чем количество, используемое в настоящее время с неспецифическими контрастными агентами, имеющимися на рынке.
Например, удовлетворительные диагностические изображения МРТ, обеспечивающие врачу адекватную диагностическую поддержку, могут быть получены с дозами соединений с гадолиниевым комплексом, идентифицируемыми в настоящем изобретении, как приблизительно 90%, более предпочтительно 80%, и до 60% дозы контрастного агента МРТ, используемой в ежедневной практике, которая для взрослых пациентов обычно составляет приблизительно 0,1 ммоль/кг массы тела пациента.
Из всего вышеизложенного легко можно представить, что выбор парамагнитных комплексных соединений формулы (I), идентифицированных в настоящем изобретении, имеет широкий диапазон применения, так как они могут использоваться для внутрисосудистого (например, внутривенного, внутриартериального, внутрикоронарного, внутрижелудочкового введения и т.п.), внутриоболочечного, внутрибрюшинного, внутрилимфатического и внутриполостного введений. Кроме того, они подходят для перорального или парентерального введения и, вследствие этого, специально для визуализации желудочно-кишечного тракта.
Например, для парентерального введения они могут быть предпочтительно составлены в виде стерильных водных растворов или суспензий, pH которых может колебаться от 6,0 до 8,5.
Эти готовые формы могут быть лиофилизированы и поставляться в том виде, как они есть, для восстановления перед применением.
Для желудочно-кишечного применения или для инъекции в полости тела эти агенты могут быть составлены в виде раствора или суспензии, необязательно содержащей подходящие эксципиенты, например, для того чтобы регулировать вязкость.
Для перорального введения они могут быть составлены согласно способам получения, обычно используемым в фармацевтической технологии или в виде готовых форм с покрытием для получения дополнительной защиты против кислого pH желудка, предотвращая таким образом в случае хелатных ионов металлов их высвобождение, которое может произойти особенно при типичных значения pH желудочных текучих сред.
Другие эксципиенты, например, включая подсластители и/или ароматизирующие агенты, также можно добавлять согласно известным методикам фармацевтических готовых форм.
Растворы или суспензии соединений этого изобретения также могут быть составлены в виде аэрозоля, подлежащего использованию при аэрозольной бронхографии и инстилляции.
Например, они могут быть также заключены в липосомы или даже составлять сами липосомы, как изложено выше, и таким образом могут использоваться в виде моно- или мультиламеллярных везикул.
В предпочтительном аспекте фармацевтические композиции согласно изобретению правильно составляют в изотонических стерильных водных, необязательно забуференных растворах для парентерального введения, и наиболее предпочтительно для внутривенного или внутриартериального введения.
Более предпочтительно, указанная диагностическая композиция имеет концентрацию парамагнитного комплекса формулы (I) от 0,002 до 1,0 М и поставляется, например, в виде болюса, или в виде двух или более доз, разделенных во времени, или в виде инфузии с постоянным или нелинейным потоком.
В дополнительном аспекте изобретение относится к применению фармацевтической композиции, содержащей парамагнитный хелатный комплекс формулы (I) или его фармацевтическую приемлемую соль для диагностической визуализации патологических систем как in vitro (ex vivo), так и in vivo,, включая клетки, биологические текучие среды и биологические ткани, происходящие из живого пациента-млекопитающего, а предпочтительно, пациента-человека, а также органа, областей или тканей тела человека, включая опухоли или раковые ткани, воспаления, а также для мониторинга течения и результатов терапевтического лечения указанных патологий.
В дополнительном аспекте настоящее изобретение относится к способу визуализации in vivo органа, ткани или области тела путем применения методики МРТ, причем указанный способ включает усиление сигнала, генерируемого протонами воды путем применения парамагнитного хелатного комплекса формулы (I) согласно изобретению или его физиологической приемлемой соли.
В одном варианте осуществления указанный способ включает введение пациенту, человеку или животному, подлежащему лучевому исследованию, диагностически эффективного количества композиции изобретения, содержащей соединение формулы (I) в форме комплекса с ионом парамагнитного металла, а предпочтительно с ионом металла Gd3+, а затем подвергание пациента с введенной композицией диагностической визуализации путем применения методики МРТ.
Согласно особенно предпочтительному варианту осуществления, приведенный выше способ МРТ вместо этого проводят на организмах человека или животного с надлежащим образом проведенным введением диагностически эффективного количества композиции изобретения, как определено выше.
Более конкретно, согласно предпочтительному варианту осуществления, настоящее изобретение относится к способу визуализации in vivo органа или ткани тела человека или животного путем применения методики МРТ, который включает стадии:
a) подвергания человека или животного с предварительно введенной композицией изобретения, содержащей соединение формулы (I) в форме парамагнитного комплекса или его фармацевтически приемлемую соль, и размещенного в системе визуализации МРТ, излучению с частотой, выбранной для возбуждения ядер с ненулевыми спинами протонов активного парамагнитного субстрата; и
b) регистрации МР сигнала из указанных возбужденных ядер.
В еще одном аспекте изобретение предоставляет способ визуализации in vitro (ex vivo) биологических образцов, включая клетки, биологические текучие среды и биологические ткани, происходящие из живого пациента-млекопитающего, и предпочтительно, пациента-человека, путем применения методики МРТ, который включает контакт эффективного количества парамагнитного комплексного соединения формулы (I) или его физиологически приемлемой соли с интересующим биологическим образцом, а затем получение МРТ сигналов от указанных образцов путем применения методики МРТ.
Неограничивающие примеры предпочтительных соединений изобретения и промежуточных соединений для их получения излагаются в следующем разделе, нацеленном на более подробном пояснении изобретения без ограничения его объема.
Экспериментальная часть
Пример 1: получение субстрата 1B
Это соединение было получали путем использования метода синтеза, показанного на схеме 6:
Схема 6
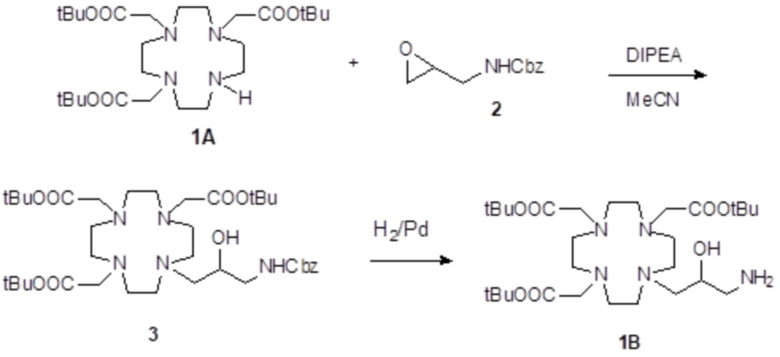
включающего:
а) Получение соединения 3
Раствор DO3A трис(t-бутилового сложного эфира) 1 (Org. Synth. 2008, 85, 10) (61,7 г; 0,12 моль), промежуточного соединения 2 (полученного, как сообщалось в WO2008/126034, страница 102) (30,0 г; 0,15 моль) и N,N-диизопропилэтиламина (DIPEA) (61,8 г; 0,48 моль) в ацетонитриле (300 мл) встряхивали при 60°C в течение 48 ч. Смесь выпаривали до остатка, который растворяли в EtOAc (300 мл). Раствор промывали водой (4×50 мл), рассолом (4×50 мл), фильтровали и выпаривали до остатка, который очищали посредством флэш-хроматографии (элюент: EtOAc/MeOH=1:1). Фракции, содержащие требуемый продукт, объединяли и выпаривали до остатка, который обрабатывали этиловым эфиром (200 мл). Промежуточное соединение 3 осаждалось в виде твердого вещества, которое фильтровали (48,2 г). Выход 56%.
т.п.=168°C
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой
b) Получение соединения субстрата 1B
Палладий 5% на угле (смоченный приблизительно 50% воды) (2,5 г) добавляли в раствор промежуточного соединения 3 (60 г; 77 ммоль) в MeOH (280 мл). Смесь встряхивали и гидрировали при комнатной температуре и атмосферном давлении в течение 5 ч. Смесь фильтровали и выпаривали. Остаток растворяли в диэтиловом эфире (400 мл), фильтровали и выпаривали с получением соединения 1A в виде стекловидного твердого вещества (45,2 г). Выход 91%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
Пример 2: получение субстрата 1C
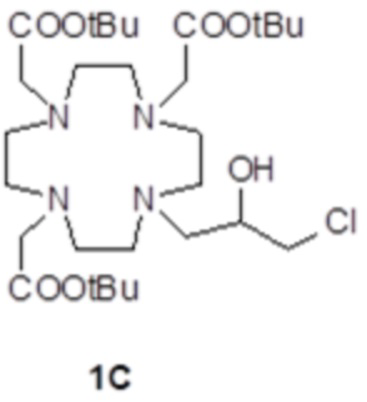
Это соединение было получали путем использования метода синтеза, показанного на схеме 7:
Схема 7
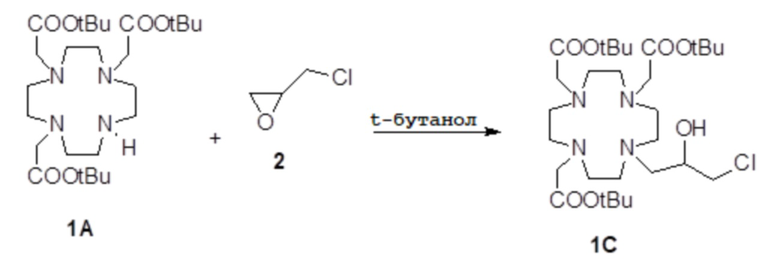
включающего:
а) Получение соединения 1C.
Коммерчески доступный эпихлоридрин 2 (10,5 мл; 137 ммоль) растворяли в ацетонитриле (300 мл), а полученный в результате раствор медленно добавляли при комнатной температуре в раствор DO3A трис-t-бутилового сложного эфира 1A (Org. Synth. 2008, 85, 10) (14,1 г; 27,4 ммоль) в ацетонитриле (100 мл). Смесь встряхивали в течение 24 ч, затем еще добавляли эпихлоридрин 2 (5,2 мл; 68 ммоль). Спустя 24 ч смесь выпаривали, а остаток очищали посредством хроматографии на силикагеле (элюент: CH2Cl2/MeOH=50:1ƒ1:1) с получением соединения 1C (10,6 г). Выход 64%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
Пример 3: получение сравнительного соединения 1
Препарат сравнительного соединения 1 получали, как показано на следующей схеме 8:
Схема 8
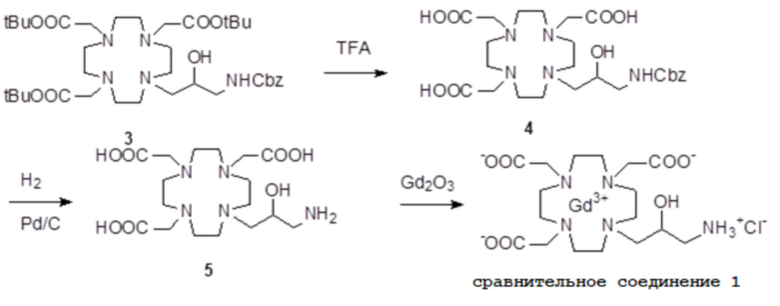
включающей:
а) Получение промежуточного соединения 4
Трифторуксусную кислоту (TFA) (130 мл) добавляли в соединение 3 (полученное, как описано выше в примере 1) (48,0 г; 0,066 моль), охлаждали в ванне со льдом. После встряхивания смеси в течение 24 ч, в неочищенную реакционную смесь добавляли этиловый эфир (800 мл), что приводило к образованию твердого осадка, который фильтровали, промывали этиловым эфиром и сушили с получением неочищенного продукта, который растворяли в воде (100 мл) и очищали посредством хроматографии на Amberchrome CG161M. Путем концентрирования чистых фракций получали требуемое промежуточное соединение 4 в виде стекловидного остатка (20,3 г). Выход 55%.
ВЭЖХ 94% (площадь%)
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение лиганда 5
В раствор промежуточного соединения 4 (19 г; 0,034 моль) в воде (100 мл) и THF (100 мл) добавляли 5% палладий на угле (смоченный приблизительно 50% воды) (4,0 г) и гидрировали при комнатном давлении и температуре в течение 3 ч. Катализатор фильтровали, а раствор выпаривали до остатка. Этот последний затем растворяли в воде и два раза выпаривали, затем лиофилизировали в твердый остаток. Этот последний растворяли в воде (60 мл), а pH полученного раствора корректировали до 8,0 смолой Duolite 3ASFB (образование OH-). Затем смолу фильтровали, промывали водой, что приводило к водному раствору лиганда 5, который лиофилизировали до твердого остатка (10,6 г). Выход 74%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой
c) Комплексообразование
Оксид гадолиния (2,72 г; 0,0075 моль) добавляли в раствор лиганда 5 (6,85 г; 0,016 моль) в воде (100 мл), и полученную смесь встряхивали и нагревали до 90°C. Спустя 1 ч мутный раствор фильтровали на Millipore HA 0,45 мкм, и фильтрат доводили до нейтрального pH с помощью 1 N HCl. Раствор лиофилизировали, что приводило к требуемому контрольному соединению 1 в виде твердого вещества (9,8 г). Выход 98%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой
Пример 4: получение хелатного комплекса 1
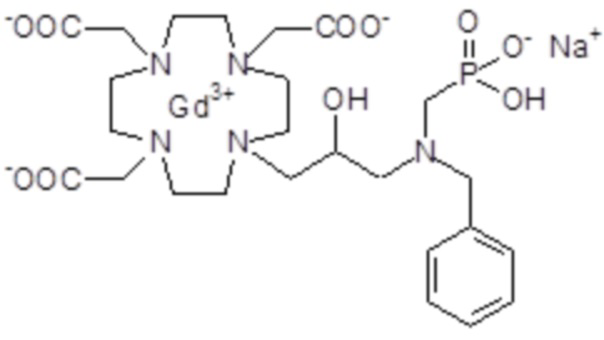
Это соединение получали с использованием метода синтеза, показанного на следующей схеме 9:
Схема 9
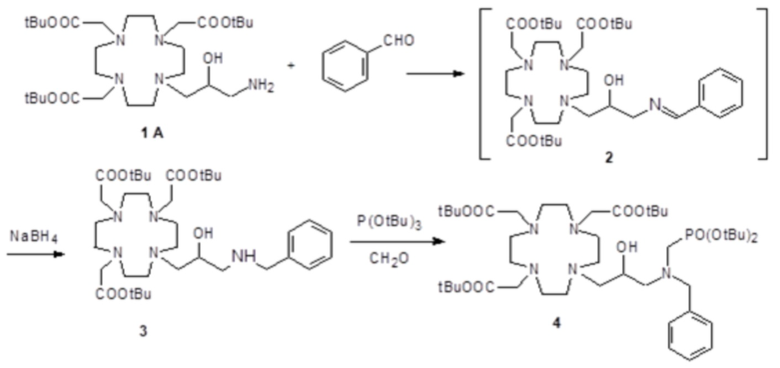
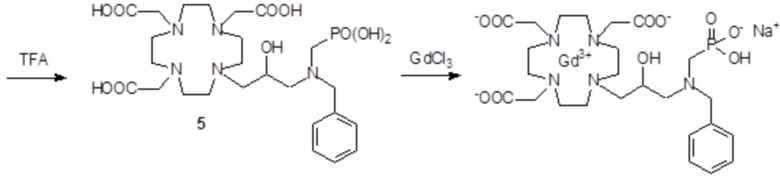
включающей в виде главных стадий:
а) Получение промежуточного соединения 3
Бензальдегид (3,18 г; 0,03 моль) и уксусную кислоту (9 мл) добавляли в раствор субстрата A (19,4 г; 0,03 моль) в EtOH (100 мл), и полученную реакционную смесь встряхивали в течение 16 ч. Затем раствор охлаждали до 0-5°C и небольшими частями добавляли натрий борогидрид (7,5 г; 0,21 моль). Реакцию поддерживали при комнатной температуре в течение 2 ч, затем охлаждали и разбавляли водой (200 мл). Органический растворитель выпаривали, а pH остального водного раствора повышали до pH 11 с помощью 2N NaOH (30 мл), затем проводили экстракцию дихлорметаном. После выпаривания органического растворителя в качестве остатка получали моноалкилированное промежуточное соединение 3 (17 г). Выход 84%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение промежуточного соединения 4
Параформальдегид (1,17 г; 0,039 моль) и трис(t-бутил) фосфит (10,3 г; 0,034 моль) (Tetrahedron Lett. 2005, 46, 4707-4710) добавляли в промежуточное соединение 3 (23,4 г; 0,034 моль), и полученную смесь нагревали при 70°C в течение 3 ч. В течение этого времени еще добавляли трис(t-бутил) фосфит, спустя 1 ч (3 г) и спустя 2 ч (1,5 г). Смесь выпаривали в вакууме с получением остатка (35,3 г), который растворяли в дихлорметане и очищали посредством флэш-хроматографии (элюент: дихлорметан/MeOH=4:1) для получения промежуточного соединения 4 (23,9 г). Выход 78%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
С) Получение лиганда 5
Трифторуксусную кислоту (30 мл) добавляли в раствор промежуточного соединения 4 (16,3 г; 0,018 моль) в дихлорметане (150 мл). Смесь выпаривали, остаток солюбилизировали в TFA (60 мл) и добавляли триизопропилсилан (0,1 мл). Полученную смесь содержали при встряхивании в течение 72 ч, затем разбавляли этиловым эфиром (450 мл) с получением осадка твердого вещества, который фильтровали и очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/MeCN) с получением требуемого лиганда 5 (5,3 г). Выход 49%.
ВЭЖХ соединения, указанного в заголовке, 97,3% (площадь%)
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Комплексообразование
Гексагидрат хлорида гадолиния (0,93 г, 2,5 ммоль) добавляли в раствор лиганда 5 (1,6 г; 2,54 ммоль) в воде (20 мл), и pH полученного раствора медленно увеличивали до pH 6,5-7 с помощью 2 N NaOH. полученный раствор встряхивали при комнатной температуре в течение 4 ч, затем фильтровали на Millipore HA 0,45 мкм, концентрировали и очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/MeCN) с получением 1,55 г требуемого гадолиниевого комплекса. Выход 80%.
ВЭЖХ соединения, указанного в заголовке, 99% (площадь%)
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
За счет применения такой же стратегии синтеза, как в схеме 9, хелатный комплекс 17 получали аналогичным образом посредством применения циклогексанкарбоксальдегида (поставляемого на рынок, например, Sigma-Aldrich).
Пример 5: получение хелатного комплекса 2
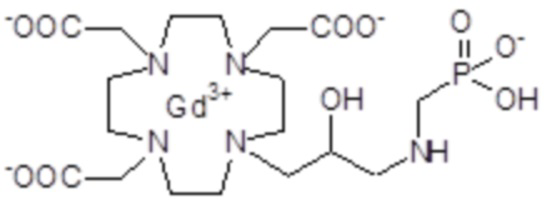
Это комплексное соединение получали путем восстановления хелатного комплекса 1 с помощью H2 и Pd/C. В частности:
Палладий 5% на угле (420 мг) добавляли в раствор гадолиниевого комплекса 1 (1,70 г; 2,176 ммоль) в воде (20 мл) и тетрагидрофуране (20 мл). Реакцию гидрогенизации проводили в течение 2 ч (комнатная температура, 1 атм.), затем катализатор фильтровали и промывали водой. Органический раствор концентрировали для удаления органического растворителя, фильтровали на Millipore HA 0,45 мкм и лиофилизировали с получением комплексного соединения 2 в виде твердого вещества (1,3 г). Выход 84%.
ВЭЖХ соединения, указанного в заголовке, 98% (площадь%)
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
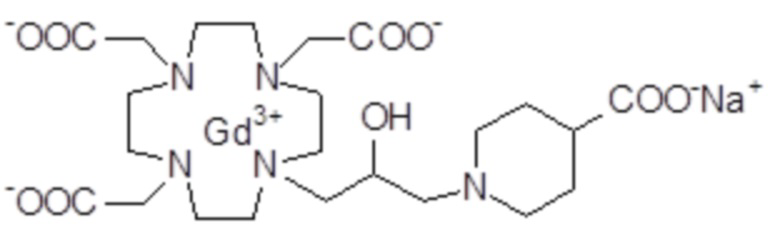
Пример 6: получение хелатного комплекса 3
Это соединение получали с использованием подхода синтеза, показанного на следующей схеме 10:
Схема 10
включающей в виде главных стадий:
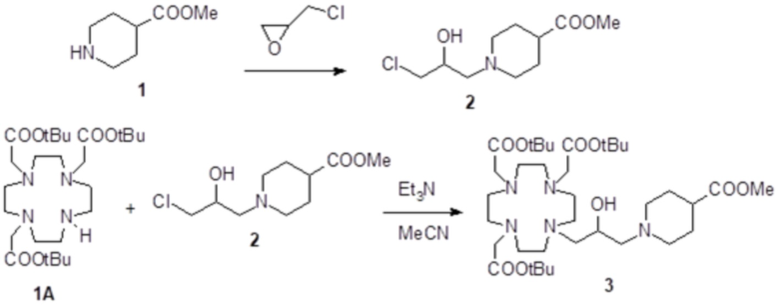
a) Получение алкилирующей молекулы 2
Коммерчески доступный эпихлоридрин (2,57 г; 27,8 ммоль) добавляли в раствор метил 4-пиперидинкарбоксилата 1 (коммерчески доступного) (2,65 г; 18,5 ммоль) в MeOH (50 мл). Смесь встряхивали при комнатной температуре в течение 18 ч, затем растворитель выпаривали для получения соединения 2 (4,24 г; выход: 87%), который использовали без всякой дополнительной очистки.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение промежуточного соединения 3
Раствор DO3A трис-t-бутилового сложного эфира 1A (Org. Synth. 2008, 85, 10) (8,15 г; 15,8 ммоль), алкилирующего агента 2 (4,15 г; 17,6 ммоль), Et3N (5 мл) и ацетонитрила (40 мл) нагревали при 55°C и встряхивали в течение 24 ч. Смесь выпаривали, а остаток растворяли в EtOAc (80 мл) и промывали водой (80 мл), затем соляным раствором (4×80 мл). Органическую фазу выпаривали, а остаток очищали посредством хроматографии на силикагеле (элюент: CH2Cl2/MeOH=100:1ƒ5:1). Фракции, содержащие чистый продукт, объединяли и выпаривали с получением промежуточного соединения 3 (5,2 г). Выход 46%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
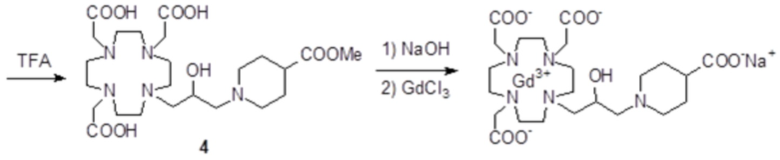
c) Получение промежуточного соединения 4
Трифторуксусную кислоту (4 мл) добавляли в раствор промежуточного соединения 3 (5,5 г; 7,7 ммоль) в дихлорметане (20 мл). Смесь встряхивали в течение 15 мин, затем выпаривали. Остаток растворяли в TFA (30 мл) и добавляли триизопропилсилан (0,1 мл). Смесь содержали при встряхивании в течение 40 ч, затем выпаривали, а остаток очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/MeCN) с получением промежуточного соединения 4 (3,1 г). Выход 74%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Гидролиз и комплексообразование
Промежуточное соединение 4 (5,6 г; 10,3 ммоль) растворяли в воде (100 мл) и добавляли 2 M NaOH до pH 10. Раствор нагревали до 45°C в течение 8 ч, сохраняя pH 10. Раствор охлаждали до комнатной температуры, pH регулировали до 7 путем добавления 1 M HCl и добавляли гексагидрат хлорида гадолиния (3,86 г; 10,3 ммоль). Смесь встряхивали при комнатной температуре в течение 18 ч. Затем раствор фильтровали на фильтрах Millipore HA 0,25 мкм и выпаривали при пониженном давлении. Неочищенный продукт очищали в колонке Amberchrome CG161M (элюент: градиент воды/ацетонитрила). Фракции, содержащие чистый продукт, объединяли и выпаривали. Твердый продукт сушили в вакууме с получением гадолиниевого комплекса в виде белого порошка (6,3 г). Выход 86%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
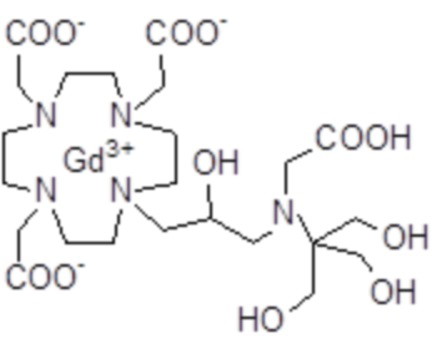
Пример 7: получение хелатного комплекса 4
Это комплексное соединение получали путем использования методики, показанной на схеме 11:
Схема 11
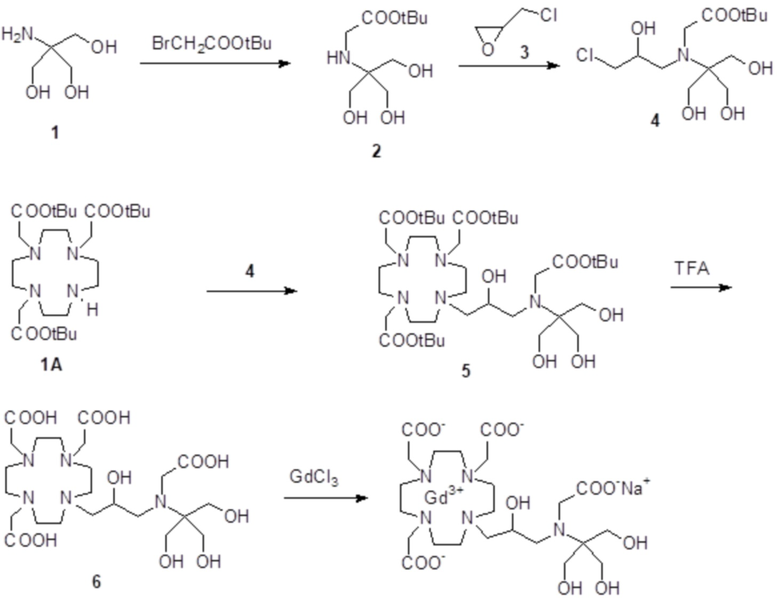
Включая:
a) Получение 2
Раствор t-бутил бромацетата (19,3 г; 99 ммоль) в ацетонитриле (50 мл) добавляли в раствор 2-амино-2-(гидроксиметил)-1,3-пропандиола 1 (коммерчески доступный) (20 г; 165 ммоль) в DMSO (70 мл). Смесь встряхивали при комнатной температуре в течение 72 ч. Добавляли воду (300 мл) и смесь экстрагировали дихлорметаном (4×300 мл). Органическую фазу промывали водой, затем все водные фазы объединяли и очищали посредством хроматографии в колонке Amberlite XE 750 (элюент: градиент воды/MeOH) с получением соединения 2 в виде белого твердого вещества (13,4 г). Выход 58%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение 4
Эпихлоргидрин 3 (19,8 г; 214 ммоль) добавляли в раствор соединения 2 (10,1 г; 43 ммоль) в MeOH (150 мл). Смесь нагревали при 55°C и встряхивали в течение 48 ч. Смесь выпаривали, а остаток очищали посредством хроматографии на силикагеле (элюент: CH2Cl2/MeOH=14:1ƒ1:1) с получением соединения 4 (11,8 г). Выход 84%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Получение 5
Смесь субстрата 1A (Org. Synth. 2008, 85, 10) (4 г; 7,8 ммоль), соединения 4 (4 г; 12,2 ммоль), K2CO3 (2,2 г; 15,8 ммоль) и ацетонитрила (70 мл) нагревали при 78°C и встряхивали в течение 20 ч. Смесь фильтровали, выпаривали, а остаток очищали посредством хроматографии на силикагеле (элюент: CH2Cl2/MeOH=50:1ƒ1:1) с получением промежуточного соединения 5 (3,6 г). Выход 57%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Получение 6
Трифторуксусную кислоту (4,5 мл) добавляли в раствор промежуточного соединения 5 (9,4 г; 12,5 ммоль) в дихлорметане (30 мл). Смесь встряхивали в течение 30 мин, затем выпаривали. Остаток растворяли в TFA (30 мл) и добавляли триизопропилсилан (0,1 мл). Полученную смесь содержали при встряхивании в течение 20 ч, затем выпаривали, а остаток очищали посредством хроматографии в колонке Amberlite XE 750 (элюент: градиент воды/MeCN) с получением требуемого лиганда 6 (5,7 г). Выход 78%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
e) Комплексообразование
Лиганд 6 (4 г; 6,9 ммоль) растворяли в воде (50 мл) и добавляли гексагидрат хлорида гадолиния (2,55 г; 6,9 ммоль). Смесь встряхивали при комнатной температуре в течение 6 ч. Затем раствор фильтровали на фильтрах Millipore HA 0,25 мкм и выпаривали при пониженном давлении. Неочищенный продукт очищали в колонке Amberchrome CG161M (элюент: градиент воды/ацетонитрила). Фракции, содержащие чистый продукт, объединяли и выпаривали. Твердый продукт сушили в вакууме с получением гадолиниевого комплекса в виде белого порошка (2,7 г). Выход 52%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
За счет применения такой же стратегии синтеза, как в схеме 11, и использования 2-амино-1,3-пропанолдиола (поставляемого на рынок, например, Sigma-Aldrich), хелатный комплекс 13 получали аналогичным образом.
Пример 8: получение хелатного комплекса 7
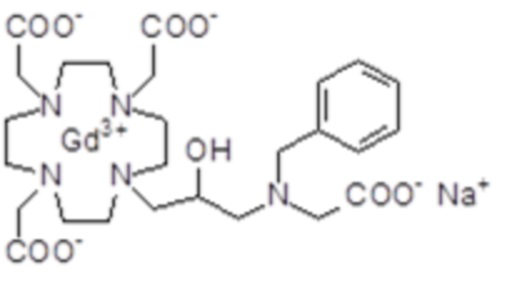
Это комплексное соединение получали путем использования методики, показанной на схеме 12:
Схема 12
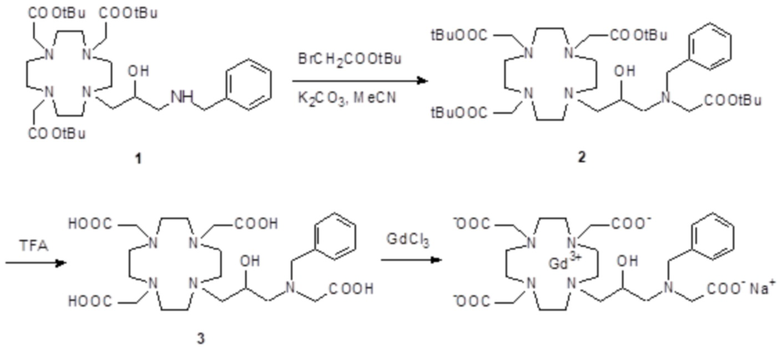
включающей в виде главных стадий:
a) Получение 2
Смесь соединения 1 (полученного, как сообщалось выше в примере 4) (9 г; 13,3 ммоль), t-бутил бромацетата (2,6 г; 13,3 ммоль) и K2CO3 (2,2 г; 16 ммоль) в ацетонитриле (100 мл) встряхивали при комнатной температуре в течение 20 ч, затем при 40°C в течение 4 ч. Добавляли еще t-бутил бромацетат (0,52 г; 2,7 ммоль) и K2CO3 (0,45 г; 3,3 ммоль) и смесь встряхивали при 40°C в течение 3 ч, затем при 55°C в течение 2 ч. Смесь фильтровали и раствор выпаривали с получением масла, которое растворяли с помощью CH2Cl2 (100 мл). Раствор промывали водой (3×100 мл), рассолом (100 мл) и выпаривали. Неочищенный маслянистый остаток очищали посредством флэш-хроматографии на силикагеле (элюент: CH2Cl2/MeOH=10:1) с получением промежуточного соединения 2 в виде масла (7,4 г). Выход 70%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение лиганда 3
Трифторуксусную кислоту (3,6 мл) добавляли в раствор промежуточного соединения 2 (7,4 г; 9 ммоль) в дихлорметане (10 мл) при 0°C. Затем смесь выпаривали, остаток солюбилизировали в TFA (40 мл) и добавляли триизопропилсилан (0,1 мл). Смесь встряхивали в течение 24 ч при комнатной температуре, затем выпаривали, а остаток очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/EtOH) с получением требуемого хелатирующего лиганда 3 (4,14 г). Выход 81%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Комплексообразование
Гексагидрат хлорида гадолиния (6,8 г, 18,3 ммоль) добавляли в перемешиваемый раствор хелатирующего лиганда 3 (10,4 г; 18,3 ммоль) в воде (400 мл), и pH смеси медленно увеличивали до pH 6,5-7 с помощью 1 N NaOH. Полученный раствор встряхивали при комнатной температуре в течение 5 ч, затем фильтровали на Millipore HA 0,45 мкм, концентрировали и очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/EtOH) с получением 13 г гадолиниевого комплекса. Выход 95%.
ВЭЖХ соединения, указанного в заголовке, 99% (площадь%)
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Пример 9: получение хелатного комплекса 8
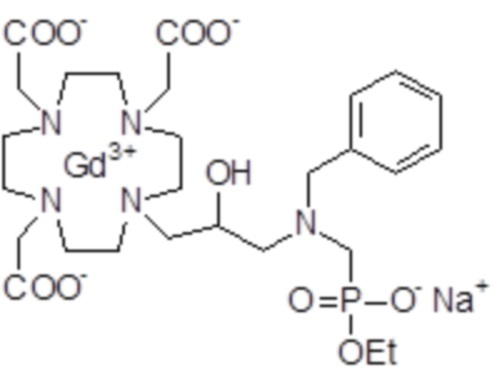
Это комплексное соединение получали путем использования методики, схематично показанной на следующей схеме 13:
Схема 13
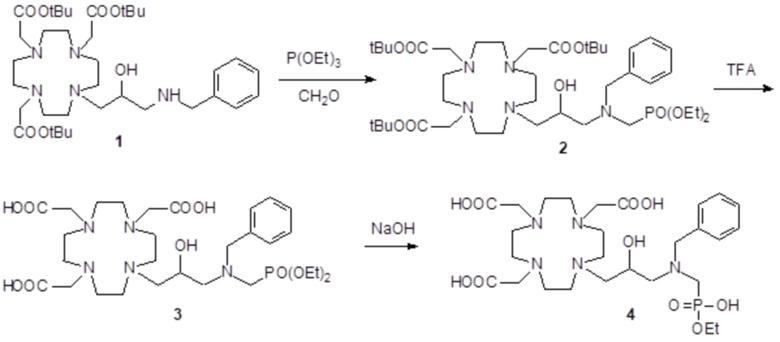
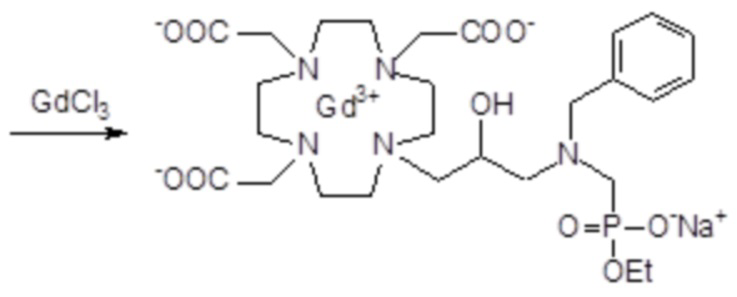
включающей в виде главных стадий:
a) Получение промежуточного соединения 2
Смесь соединения 1 (полученного, как сообщалось выше в примере 4) (10,6 г; 15,6 ммоль), параформальдегида (0,733 г; 24,4 ммоль), триэтилфосфита (4,27 г; 25,7 ммоль) и ацетонитрила (10 мл) встряхивали при 70-75°C в течение 32 ч. Смесь выпаривали, а остаток очищали посредством флэш-хроматографии на силикагеле (элюент: CH2Cl2/MeOH=30:1ƒ1:1) с получением промежуточного соединения 2 в виде оранжевого масла (6,6 г). Выход 51%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение промежуточного соединения 3
Трифторуксусную кислоту (2,9 мл) добавляли в раствор промежуточного соединения 2 (6,1 г; 7,6 ммоль) в дихлорметане (15 мл) при 0°C. Затем смесь выпаривали, остаток растворяли в TFA (25 мл) и добавляли триизопропилсилан (0,1 мл). Смесь встряхивали при комнатной температуре в течение 48 ч, затем выпаривали, а остаток очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/MeCN) с получением промежуточного соединения 3 (3,19 г). Выход 63%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Получение лиганда 4
Промежуточное соединение 3 (3,45 г; 5,2 ммоль) растворяли в воде (35 мл), и добавляли 1 M NaOH (31,4 мл; 31,4 ммоль). Раствор встряхивали при комнатной температуре в течение 48 ч, подкисляли конц. HCl до pH 1,5 и очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/MeCN) с получением хелатирующего лиганда 4 (2,54 г). Выход 77%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Комплексообразование
Гексагидрат хлорида гадолиния (1,18 г, 3,2 ммоль) добавляли в перемешиваемый раствор хелатирующего лиганда 4 (2 г; 3,2 ммоль) в воде (40 мл) и pH смеси медленно увеличивали до pH 6,5-7 с помощью 1 N NaOH. Спустя 2 ч полученный раствор фильтровали на Millipore HA 0,45 мкм, концентрировали, а затем очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/MeCN) с получением 2,49 г гадолиниевого комплекса. Выход 96%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Пример 10: получение хелатного комплекса 5
Это соединение получали с использованием методики следующей общей схемы 14:
Схема 14
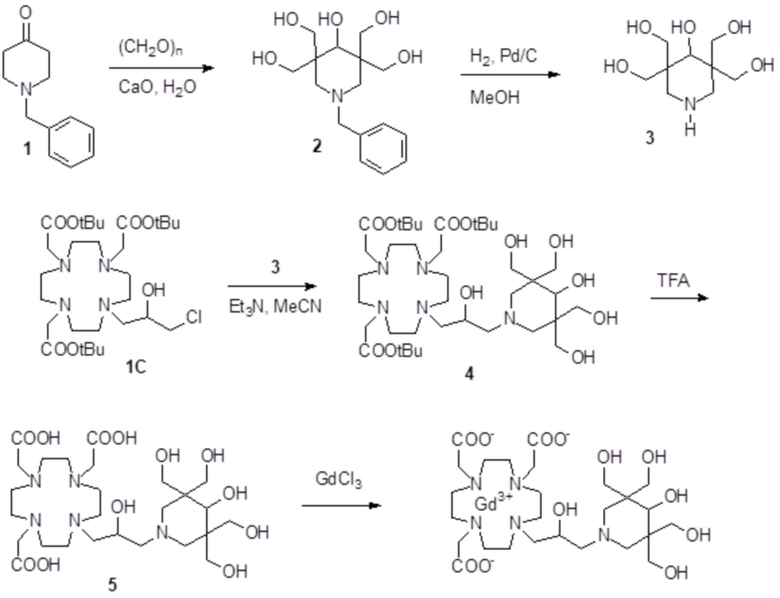
включающей в виде главных стадий
a) Получение 2
Оксид кальция (3,37 г; 60 ммоль) добавляли небольшими частями в смесь 1-бензил-4-пиперидона 1 (коммерчески доступного) (18 г; 95 ммоль) и параформальдегида (15,7 г; 523 ммоль) в воде (180 мл). Смесь встряхивали при 40°C в течение 24 ч, затем выпаривали. Остаток очищали посредством хроматографии на силикагеле (элюент: CH2Cl2/MeOH=100:1ƒ1:1) с получением промежуточного соединения 2 в виде белого липкого твердого вещества (4,15 г). Выход 14%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение производного пиперидина 3
В раствор промежуточного соединения 2 (4 г; 12,8 ммоль) в метаноле (50 мл) добавляли 5% палладий на угле (смоченный приблизительно 50% воды) (2,1 г) и гидрировали при комнатном давлении и 45°C в течение 5 ч. Катализатор фильтровали, а раствор выпаривали с получением промежуточного соединения 3 в виде белого твердого вещества (2,54 г). Выход 89%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Получение 4
Раствор субстрата 1C (5 г; 8,2 ммоль) в ацетонитриле (10 мл), полученного, как раскрыто в примере 2, добавляли в смесь производного пиперидина 3 (2,3 г; 10,4 ммоль), Et3N (2 мл) и ацетонитрила (25 мл) при 55°C. Спустя 7 ч добавляли DMSO (10 мл) и смесь нагревали при 65°C в течение 20 ч. Смесь выпаривали, а остаток растворяли с помощью CH2Cl2 (100 мл) и промывали соляным раствором (3×100 мл). Органическую фазу выпаривали с получением промежуточного соединения 4 в виде белого твердого вещества (3,8 г). Выход 58%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Получение лиганда 5
Трифторуксусную кислоту (10 мл) добавляли в раствор промежуточного соединения 4 (4,84 г; 6,1 ммоль) в дихлорметане (50 мл). Смесь встряхивали в течение 30 мин, затем выпаривали. Остаток растворяли в TFA (20 мл) и добавляли триизопропилсилан (0,1 мл). Полученную смесь содержали при встряхивании в течение 24 ч, затем выпаривали, а остаток очищали посредством хроматографии в колонке Amberlite XE 750 (элюент: градиент воды/MeCN) с получением требуемого лиганда 5 (2,7 г). Выход 71%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
e) Комплексообразование
Лиганд 5 (2,2 г; 3,5 ммоль) растворяли в воде (20 мл) и добавляли гексагидрат хлорида гадолиния (1,31 г; 3,5 ммоль). Смесь встряхивали при комнатной температуре в течение 6 ч. Затем раствор фильтровали на фильтрах Millipore HA 0,25 мкм и выпаривали при пониженном давлении. Неочищенный продукт очищали в колонке Amberchrome CG161M (элюент: градиент воды/ацетонитрила). Фракции, содержащие чистый продукт, объединяли и выпаривали. Твердый продукт сушили в вакууме с получением гадолиниевого комплекса в виде белого порошка (1,54 г). Выход 56%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Пример 11: получение хелатного комплекса 18
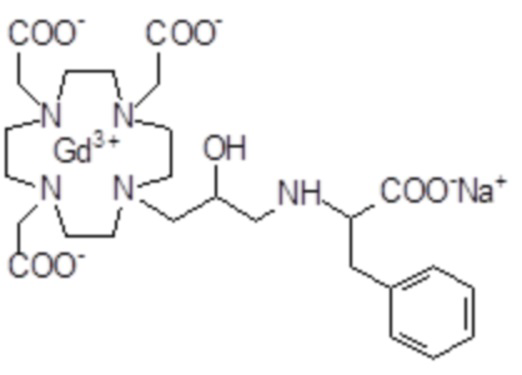
Это соединение получали с использованием методики следующей общей схемы 15:
Схема 15
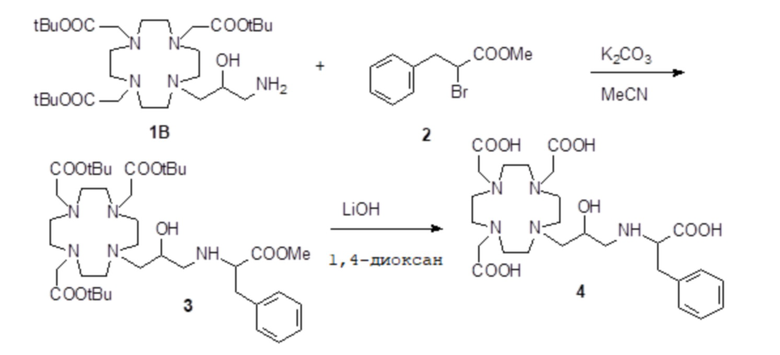
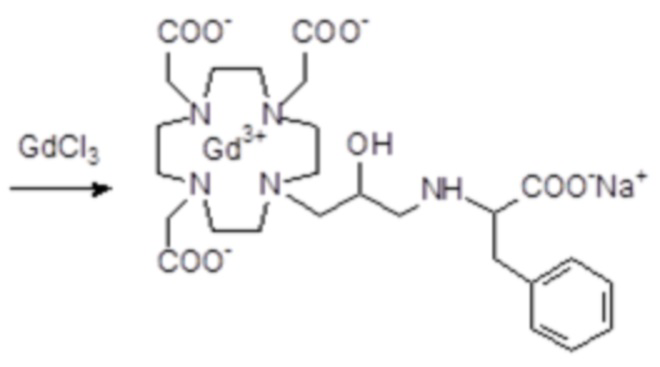
включающей следующие главные стадии
а) Получение промежуточного соединения 3
В смесь субстрата 1B (17,6 г; 30 ммоль) и K2CO3 (4,1 г; 30 ммоль) в MeCN (100 мл) добавляли метил 2-бром-3-фенилпропаноат (полученный, как сообщалось в Eur. J. Org. Chem. 2011, 1300) (2,43 г; 10 ммоль). Реакционную смесь встряхивали при комнатной температуре в течение 72 ч, затем фильтровали и выпаривали. Остаток растворяли с помощью CH2Cl2 (200 мл). Раствор промывали водой (3×100 мл), соляным раствором (100 мл) и выпаривали. Остаток очищали посредством флэш-хроматографии на силикагеле (элюент: CH2Cl2/MeOH) с получением промежуточного соединения 1 (4,3 г). Выход 57%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение лиганда 4
В раствор промежуточного соединения 3 (4 г; 5,3 ммоль) в 1,4-диоксане (50 мл) добавляли водный раствор 2 M LiOH (53 мл; 106 ммоль). Смесь встряхивали в течение 72 ч, затем подкисляли до pH 6 посредством медленного добавления 37% HCl. Раствор выпаривали, сухой остаток очищали посредством хроматографии на Amberlite XAD 16,00 колонка (элюент: градиент воды/MeCN) с получением хелатирующего лиганда 2 (2,7 г). Выход 90%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Комплексообразование
В раствор хелатирующего лиганда 2 (2,5 г; 4,4 ммоль) в воде (100 мл) добавляли гексагидрат хлорида гадолиния (1,64 г, 4,4 ммоль) и pH смеси медленно увеличивали до pH 6,5-7 с помощью 1 N NaOH. Полученный раствор встряхивали при комнатной температуре, затем фильтровали на Millipore HA 0,45 мкм, концентрировали и очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/EtOH) с получением 3,1 г гадолиниевого комплекса. Выход 94%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Применяя такую же стратегию синтеза и используя метил 2-бром-3-циклогексилпропаноат (полученный, как сообщалось в Eur. J. Org. Chem. 2011, 1300), получали хелатный комплекс 9.
Применяя такую же стратегию синтеза и используя сложный диэтиловый эфир 2-фенил-1-(метансульфонилокси) этилфосфоновой кислоты (полученный, как сообщалось в Synthesis 1996, 507), получали хелатный комплекс 6.
Пример 12: получение хелатного комплекса 12.
Это соединение получали с использованием методики следующей общей схемы 16:
Схема 16
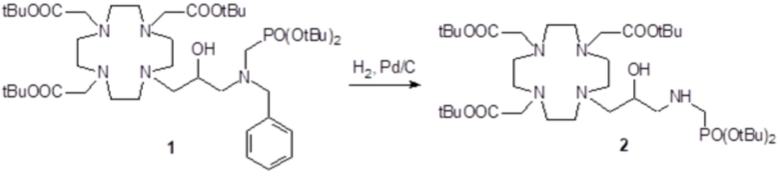
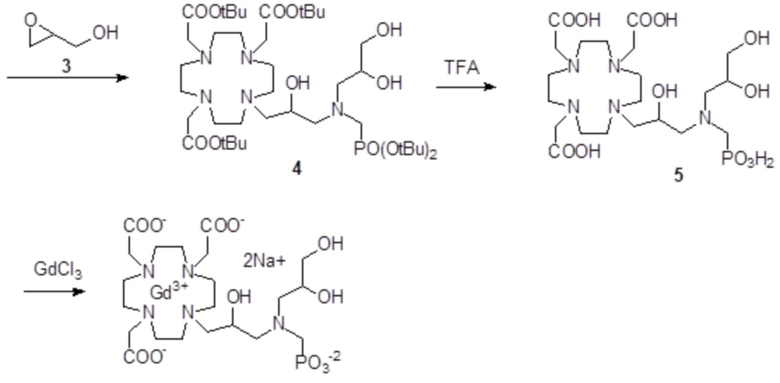
включающей:
a) Получение 2
В раствор промежуточного соединения 1 (полученного, как сообщалось в схеме 8) (20 г; 22,6 ммоль) в MeOH (250 мл) добавляли палладий 5% на угле (смоченный приблизительно 50% воды) (4 г). Смесь встряхивали и гидрировали при комнатной температуре и атмосферном давлении в течение 8 ч. Смесь фильтровали и выпаривали с получением промежуточного соединения 2 (17,6 г). Выход 98%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение 4
В раствор промежуточного соединения 2 (15 г; 18,9 ммоль) в ацетонитриле (150 мл) добавляли 2,3-Эпокси-1-пропанол 3 (коммерчески доступный) (1,5 г; 20,2 ммоль). Раствор нагревали с обратным холодильником в течение 16 ч, затем выпаривали. Остаток очищали посредством флэш-хроматографии на силикагеле (элюент: градиент EtOAc/MeOH) с получением промежуточного соединения 4 (9,52 г). Выход 58%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Получение хелатирующего лиганда 5
В раствор промежуточного соединения 4 (8,7 г; 10 ммоль) в дихлорметане (50 мл) при 0°C добавляли трифторуксусную кислоту (10 мл). Смесь встряхивали в течение 8 ч, затем выпаривали, а остаток растворяли в TFA (50 мл). Смесь встряхивали при комнатной температуре в течение 16 ч, затем выпаривали. Остаток очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/MeCN) с получением хелатирующего лиганда 5 (5,3 г). Выход 90%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Комплексообразование
В раствор хелатирующего лиганда 5 (5 г; 8,5 ммоль) в воде (100 мл) добавляли гексагидрат хлорида гадолиния (3,16 г, 8,5 ммоль) и pH смеси медленно увеличивали до pH 6,5-7 с помощью 1 N NaOH. Полученный раствор фильтровали на Millipore HA 0,45 мкм, концентрировали и затем очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/MeCN) с получением 6,48 г соответствующего гадолиниевого комплекса. Выход 97%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Пример 13: получение хелатного комплекса 19.
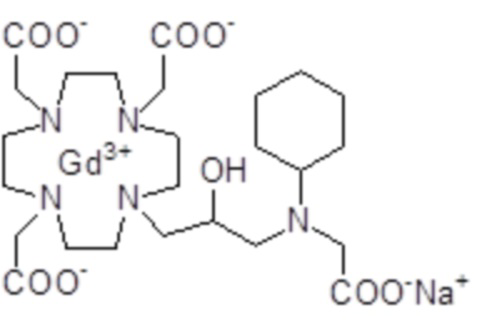
Это соединение получали с использованием методики следующей общей схемы 17:
Схема 17
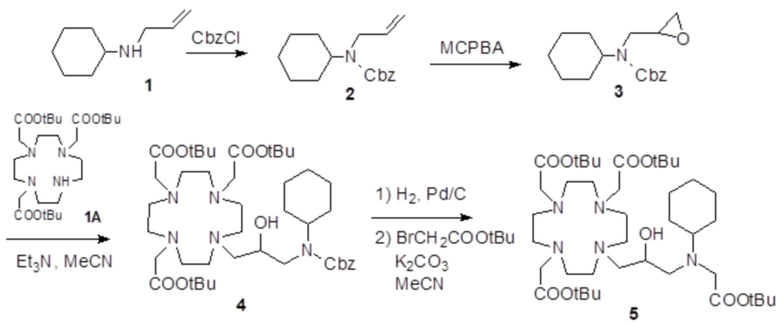
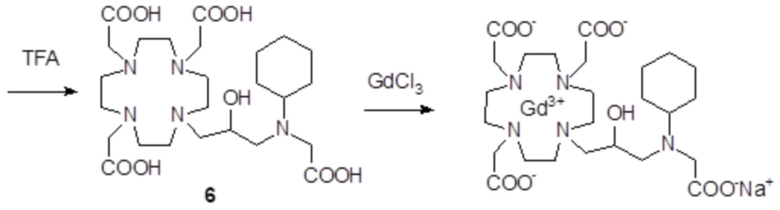
включающей:
a) Получение промежуточного соединения 2
В смесь аллилциклогексиламина 1 (коммерчески доступный) (13,9 г; 100 ммоль), K2CO3 (27,6 г; 200 ммоль), воды (150 мл) и EtOAc (200 мл) при 0°C в течение 1 ч добавляли бензил хлорформат (95%; 19,75 г; 110 ммоль). После встряхивания в течение 6 ч органическую фазу разделяли и промывали 1 N HCl (2×100 мл), водой (100 мл) и рассолом (100 мл). Органическую фазу сушили (Na2SO4) и выпаривали с получением промежуточного соединения 2 (26,2 г). Выход 96%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение промежуточного соединения 3
В раствор промежуточного соединения 2 (13,7 г; 50 ммоль) в дихлорметане (100 мл) каплями добавляли раствор 3-хлорпербензойной кислоты (MCPBA) (75%; 23 г; 100 ммоль) в дихлорметане (100 мл). Раствор встряхивали при комнатной температуре в течение 16 ч. Смесь фильтровали, промывали 10% водн. Na2SO3 (2×100 мл), 5% водн. NaHCO3 (4×100 мл), H2O (100 мл) и рассолом (100 мл). Органическую фазу разделяли, выпаривали, а остаток очищали посредством хроматографии на силикагеле (элюент: градиент n-гептан/EtOAc) для получения промежуточного соединения 3 (13,0 г). Выход 90%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Получение промежуточного соединения 4
Раствор эпоксида 3 (10 г, 34,6 ммоль), DO3A три-t-бутилового эфира 1A (Org. Synth. 2008, 85, 10) (15,44 г; 30 ммоль) и Et3N (3,54 г, 35 ммоль) в MeCN (200 мл) нагревали при 60°C и встряхивали в течение 24 ч. Реакционную смесь выпаривали, а остаток очищали посредством флэш-хроматографии на силикагеле (элюент: градиент CH2Cl2/MeOH) с получением соединения 4 (16,1 г) Выход 67%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Получение промежуточного соединения 5
В раствор промежуточного соединения 4 (15 г; 18,7 ммоль) в MeOH (200 мл) добавляли палладий 5% на угле (смоченный приблизительно 50% воды) (3 г). Смесь встряхивали и гидрировали при комнатной температуре и атмосферном давлении в течение 16 ч. Смесь фильтровали и выпаривали с получением остатка, который растворяли в ацетонитриле (200 мл), затем добавляли t-бутил бромацетат (4,38 г; 22,4 ммоль) и K2CO3 (6,9 г; 50 ммоль). Смесь встряхивали в течение 24 ч при комнатной температуре, затем фильтровали и выпаривали. Остаток растворяли в EtOAc (100 мл) и раствор промывали H2O (2×100 мл) и рассолом (100 мл). Органическую фазу выпаривали, а остаток очищали посредством колоночной хроматографии на силикагеле (элюент: градиент CH2Cl2/MeOH) с получением промежуточного соединения 5 (8,06 г). Выход 55%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
e) Получение хелатирующего лиганда 6
В раствор промежуточного соединения 5 (7,84 г; 10 ммоль) в дихлорметане (50 мл) при 0°C добавляли трифторуксусную кислоту (10 мл). Смесь встряхивали в течение 8 ч, затем выпаривали, а остаток растворяли в TFA (50 мл). Смесь встряхивали при комнатной температуре в течение 24 ч, затем выпаривали. Остаток очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/MeCN) с получением хелатирующего лиганда 6 (5,2 г). Выход 93%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
f) Комплексообразование
В раствор хелатирующего лиганда 5 (4,5 г; 8 ммоль) в воде (80 мл) добавляли гексагидрат хлорида гадолиния (2,97 г, 8 ммоль), и pH смеси медленно увеличивали до pH 6,5-7 с помощью 1 N NaOH. Полученный раствор фильтровали на Millipore HA 0,45 мкм, концентрировали и затем очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/MeCN), получая 5,65 г соответствующего гадолиниевого комплекса. Выход 96%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Используя такую же стратегию синтеза и используя трифлат гидроксиметилфосфонат ди-t-бутилового эфира (синтезированного, как сообщалось в US2014/0086846, страница 33) получали хелатный комплекс 15.
Пример 14: получение хелатного комплекса 10.
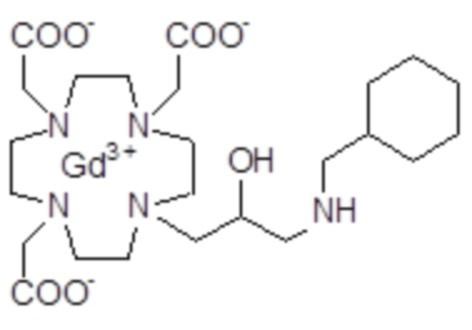
Это соединение получали с использованием методики следующей общей схемы 18:
Схема 18
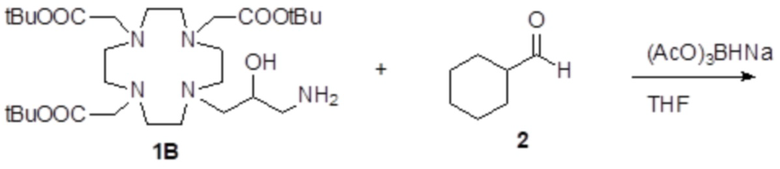
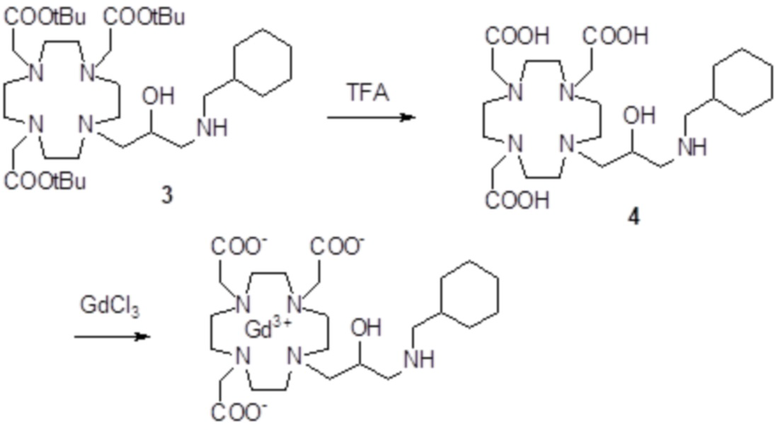
включающей:
a) Получение промежуточного соединения 3
В раствор субстрата 1B (9,7 г; 15 ммоль), циклогексанкарбоксальдегида (1,68 г; 15 ммоль) и AcOH (1,8 г; 30 ммоль) в THF (80 мл) добавляли натрий триацетоксиборогидрид (4,77 г; 22,5 ммоль). Реакционную смесь встряхивали в течение 24 ч, затем разбавляли водой (100 мл). Органический растворитель выпаривали, а pH остального водного раствора повышали до pH 11 с помощью 2N NaOH, затем проводили экстракцию дихлорметаном. После выпаривания органического растворителя промежуточное соединение 3 получали в качестве остатка (8,2 г). Выход 80%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение хелатирующего лиганда 4
В раствор промежуточного соединения 3 (8 г; 11,7 ммоль) в дихлорметане (50 мл) при 0°C добавляли трифторуксусную кислоту (15 мл). Смесь встряхивали в течение 8 ч, затем выпаривали, а остаток растворяли в TFA (50 мл). Смесь встряхивали при комнатной температуре в течение 24 ч, затем выпаривали. Остаток очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/MeCN) с получением хелатирующего лиганда 4 (5,5 г). Выход 91%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Комплексообразование
В раствор хелатирующего лиганда 4 (5 г; 9,7 ммоль) в воде (100 мл) добавляли гексагидрат хлорида гадолиния (3,6 г, 9,7 ммоль), и pH смеси медленно увеличивали до pH 6,5-7 с помощью 1 N NaOH. Полученный раствор фильтровали на Millipore HA 0,45 мкм, концентрировали и затем очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/MeCN) с получением 5,98 г соответствующего гадолиниевого комплекса. Выход 92%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Пример 15: получение хелатного комплекса 13
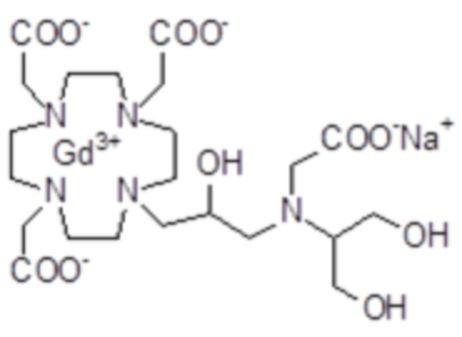
Это комплексное соединение получали путем использования методики, показанной на схеме 19:
Схема 19
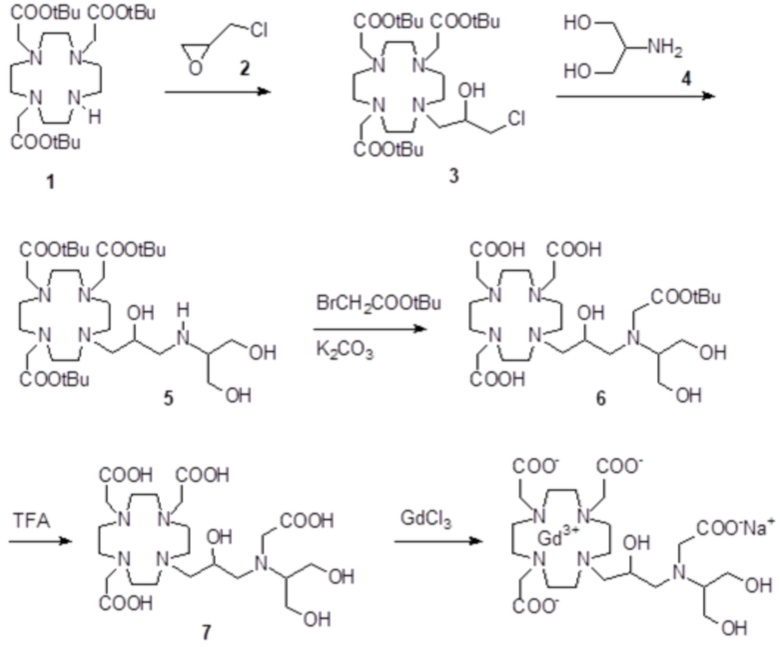
включающей:
а) Получение 3
Эпихлоргидрин 2 (10,5 мл; 137 ммоль) растворяли в ацетонитриле (300 мл), а полученный в результате раствор медленно добавляли при комнатной температуре в раствор DO3A трис-t-бутилового сложного эфира 1A (Org. Synth. 2008, 85, 10) (14,1 г; 27,4 ммоль) в ацетонитриле (100 мл). Смесь встряхивали в течение 24 ч, затем еще добавляли эпихлоридрин 5 (5,2 мл; 68 ммоль). Спустя 24 ч смесь выпаривали, а остаток очищали посредством хроматографии на силикагеле (элюент: CH2Cl2/MeOH=50:1ƒ1:1) с получением промежуточного соединения 3 (10,6 г). Выход 64%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение 5
В раствор соединения 3 (9 г; 14,8 ммоль) в ацетонитриле (100 мл) добавляли раствор серинола 4 (10,8 г; 118 ммоль) в DMSO (30 мл). Смесь нагревали при 75°C в течение 72 ч, затем растворитель выпаривали. Остаток растворяли в дихлорметане (100 мл), и раствор промывали водой (4×100 мл), затем рассолом (3×100 мл). Органическую фазу выпаривали, а остаток очищали посредством флэш-хроматографии на силикагеле (элюент: CH2Cl2/MeOH=200:1ƒ1:1) с получением соединения 5 (6,6 г). Выход 67%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Получение 6
Смесь соединения 5 (5,21 г; 8 ммоль), t-бутил бромацетата (1,85 г; 9,5 ммоль), K2CO3 (2,2 г; 15,8 ммоль) и ацетонитрила (100 мл) встряхивали при 45°C в течение 24 ч. Еще добавляли t-бутил бромацетат (0,9 г; 4,75 ммоль), и смесь встряхивали при 45°C дополнительно в течение 12 ч. Смесь фильтровали, и раствор выпаривали. Остаток растворяли в дихлорметане (100 мл), и раствор промывали водой (100 мл), затем соляным раствором (3×100 мл). Органическую фазу выпаривали, а остаток очищали посредством флэш-хроматографии на силикагеле (элюент: CH2Cl2/MeOH=100:1ƒ1:1)) с получением соединения 6 (5,42 г). Выход 89%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Получение лиганда 7
В раствор соединения 6 (3,8 г, 4,9 ммоль) в дихлорметане (20 мл) добавляли трифторуксусную кислоту (2 мл; 26 ммоль). Смесь встряхивали в течение 30 мин, затем выпаривали. Остаток растворяли в TFA (26 мл) и добавляли триизопропилсилан (0,1 мл). Полученную смесь содержали при встряхивании в течение 24 ч, затем выпаривали. Остаток очищали посредством хроматографии в колонке Amberchrome CG161M (элюент: градиент воды/ацетонитрила) с получением требуемого лиганда 7 (2,3 г). Выход 85%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
e) Комплексообразование
Лиганд 7 (2,03 г; 3,7 ммоль) растворяли в воде (15 мл), и 2 M NaOH (7 мл) добавляли до pH 10. Раствор встряхивали при 45°C в течение 4 ч, сохраняя pH при 10 путем добавления 2 M NaOH. Раствор охлаждали до комнатной температуры, добавляли 2M HCl до pH 8 и добавляли гексагидрат хлорида гадолиния (1,37 г; 3,7 ммоль). Суспензию встряхивали при 50°C в течение 6 ч. Затем раствор фильтровали на фильтрах Millipore HA 0,25 мкм и выпаривали при пониженном давлении. Неочищенный продукт очищали в колонке Amberchrome CG161M (элюент: градиент воды/ацетонитрила). Фракции, содержащие чистый продукт, объединяли и выпаривали. Твердый продукт сушили в вакууме с получением гадолиниевого комплекса в виде белого порошка (1,56 г). Выход 58%.
Пример 16: получение хелатного комплекса 14
Это соединение получали с использованием методики следующей общей схемы 20
Схема 20
включающей:
a) Получение 3
Субстрат 1C (25,2 г), полученный, как сообщалось в примере 2, растворяли в DMSO (10 мл) и добавляли 3,9-диокса-6-азаундекан-1,11-диол 2 (полученный, как сообщалось в J. Org. Chem. 1995, 60, 6097-6102) (20 г; 100 ммоль). Смесь нагревали при 80°C в течение 8 ч, затем растворитель выпаривали, а остаток очищали посредством флэш-хроматографии на силикагеле (элюент: CH2Cl2/MeOH=98:2) с получением соединения 3 в виде бледно-желтого масла (14 г). Выход 45%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение лиганда 4
В раствор соединения 3 (13,5 г, 18 ммоль) в дихлорметане (15 мл) каплями добавляли трифторуксусную кислоту (40 мл; 235 ммоль). Раствор встряхивали при комнатной температуре в течение ночи. Растворитель выпаривали, а остаток растворяли в MeOH (5 мл), затем добавляли диэтиловый эфир (50 мл). Осажденное твердое вещество выделяли с помощью центрифугирования, маточный раствор удаляли, а осадок тщательно промывали диэтиловым эфиром (35 мл). Полученное липкое светло коричневое твердое вещество очищали посредством элюирования в колонке с ионообменной смолой (Amberlite IR 120, H-form). Свободный лиганд задерживался на смоле, а включения вымывали водой. Продукт элюировали, добавляя водный раствор NH4OH (2N), а кислотную фракцию собирали и выпаривали. Полученное аморфное твердое вещество растворяли в воде (2 мл) и осаждали путем добавления ацетона (40 мл). Лиганд 6 получали в виде липкого белого твердого вещества (3,1 г). Выход 29%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Комплексообразование
Хелатирующий лиганд 4 (2,3 г; 3,8 ммоль) суспендировали в воде (50 мл) и добавляли гексагидрат хлорида гадолиния (1,4 г; 3,8 ммоль). Суспензию встряхивали при 60°C в течение 6 ч. Затем раствор фильтровали на фильтрах Millipore HA 0,25 мкм и выпаривали при пониженном давлении. Неочищенный продукт очищали на смоле Amberlite XAD 1600 (элюент: вода). Фракции, содержащие чистый продукт, объединяли и выпаривали. Твердый продукт сушили в вакууме с получением гадолиниевого комплекса в виде белого порошка (1,1 г). Выход 38%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Хелатный комплекс 20 синтезировали, используя такую же методику и 3,6,12,15-тетраокса-9-азагептадекан-1,17-диол (полученный, как сообщалось в Tetrahedron 1982, 38, 3359-3362).
Пример 17: получение хелатного комплекса 21
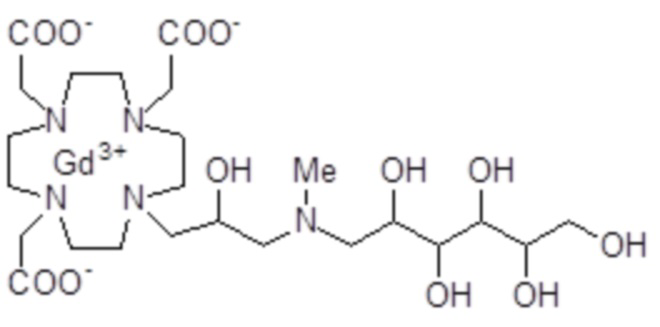
Это соединение получали с использованием методики следующей общей схемы 21:
Схема 21
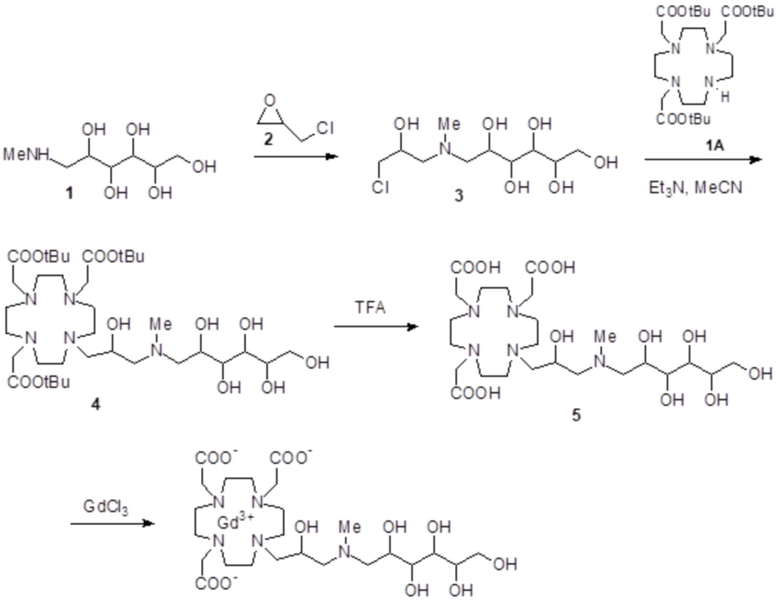
включающей:
a) Получение алкилирующей молекулы 3
В суспензию N-метил-D-глюкамина 1 (2,5 г; 12,8 ммоль) в метаноле (150 мл) добавляли эпихлоргидрин 2 (2,96 г; 32 ммоль). Смесь встряхивали при комнатной температуре в течение 72 ч, затем выпаривали в вакууме с получением алкилирующей молекулы 3 (3,7 г) в виде бесцветного масла. Количественный выход.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение промежуточного соединения 4
Раствор алкилирующей молекулы 3 (3,59 г; 12,5 ммоль) в DMSO (20 мл) добавляли в раствор субстрата 1A (2,83 г; 5,5 ммоль) и Et3N (3 мл) в ацетонитриле (10 мл). Смесь встряхивали при 65°C в течение 72 ч, затем выпаривали до остатка, который растворяли в воде (20 мл). Раствор очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/MeCN) с получением промежуточного соединения 4 (3,8 г). Выход 90%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Получение лиганда 5
Раствор промежуточного соединения 4 (4 г; 5,2 ммоль) в трифторуксусной кислоте (40 мл) встряхивали при комнатной температуре в течение 24 ч. Добавляли диэтиловый эфир (100 мл) и суспензию встряхивали в течение 2 ч, затем фильтровали. Твердое вещество растворяли в воде (20 мл), а раствор очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/ацетон) с получением лиганда 5 (2,7 г). Выход 87%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Комплексообразование
Гексагидрат хлорида гадолиния (2,3 г, 6,2 ммоль) добавляли в раствор хелатирующего лиганда 5 (3,7 г; 6,2 ммоль) в воде (50 мл), и pH смеси медленно увеличивали до pH 7 с помощью 1 N NaOH. Полученный раствор встряхивали при комнатной температуре в течение 4 ч, затем фильтровали на Millipore HA 0,45 мкм, концентрировали и очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/ацетонитрила) с получением 4 г гадолиниевого комплекса. Выход 86%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Пример 18: получение хелатного комплекса 11
Это соединение получали с использованием методики следующей общей схемы 22:
Схема 22
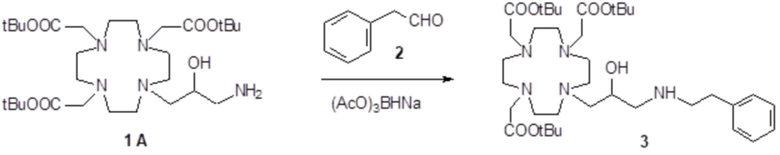
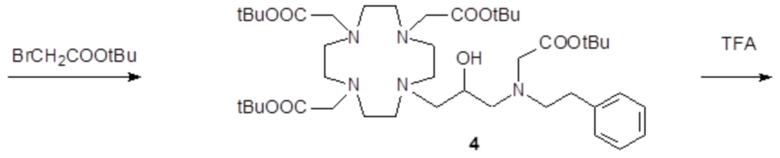
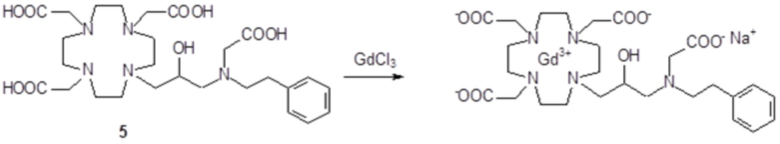
включающей в виде главных стадий:
а) Получение промежуточного соединения 3
В раствор субстрата 1A (70 г; 0,102 моль) в THF (600 мл) добавляли фенилацетальдегид (11,6 г; 0,097 моль) и уксусную кислоту (12 мл), и реакционную смесь встряхивали в течение 2 ч. Затем раствор охлаждали до 0°C и небольшими частями добавляли натрий триацетоксиборогидрид (32,4 г; 0,153 моль). Реакцию поддерживали при комнатной температуре в течение 16 ч, затем добавляли воду (150 мл). Органический растворитель выпаривали, а pH остального водного раствора повышали до pH 11 с помощью 2N NaOH, затем проводили экстракцию дихлорметаном (5×200 мл). После выпаривания органического растворителя в качестве остатка (54 г) получали моноалкилированное промежуточное соединение 3. Выход 80%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение промежуточного соединения 4
Смесь соединения 3 (54 г; 0,078 моль), t-бутил бромацетата (19,8 г; 0,101 моль) и K2CO3 (21,6 г; 0,156 моль) в ацетонитриле (350 мл) встряхивали при комнатной температуре в течение 48 ч. Смесь фильтровали, и раствор выпаривали с получением масла, которое растворяли с помощью EtOAc (400 мл). Раствор промывали водой (3×150 мл), соляным раствором (100 мл) и выпаривали. Неочищенный маслянистый остаток очищали посредством флэш-хроматографии на силикагеле (элюент: CH2Cl2/MeOH=100:1ƒ1:1). Фракции, содержащие продукт, объединяли и выпаривали с получением промежуточного соединения 4 (42,9 г). Выход 68%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Получение лиганда 5
В раствор промежуточного соединения 4 (42,5 г; 0,049 моль) в CH2Cl2 (300 мл) медленно добавляли трифторуксусную кислоту (10 мл), и смесь встряхивали при комнатной температуре в течение 1 ч. Растворитель выпаривали, а остаток растворяли в трифторуксусной кислоте (200 мл) и добавляли триизопропилсилан (2 мл). Полученную смесь содержали при встряхивании в течение 48 ч, затем выпаривали. Добавляли диэтиловый эфир (500 мл), а суспензию встряхивали в течение 2 ч, затем фильтровали. Твердое вещество растворяли в воде (20 мл), а раствор очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/ацетонитрила) с получением лиганда 5 (16,1 г). Выход 56%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
d) Комплексообразование
В раствор хелатирующего лиганда 5 (12,9 г; 0,222 моль) в воде (400 мл) добавляли гексагидрат хлорида гадолиния (8,25 г, 0,222 моль), и pH смеси медленно увеличивали до pH 7 с помощью 2 N NaOH. Полученный раствор встряхивали при комнатной температуре в течение 8 ч, затем фильтровали на Millipore HA 0,45 мкм, концентрировали и очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/ацетонитрила) с получением 16 г гадолиниевого комплекса. Выход 95%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Пример 19: получение хелатного комплекса 6
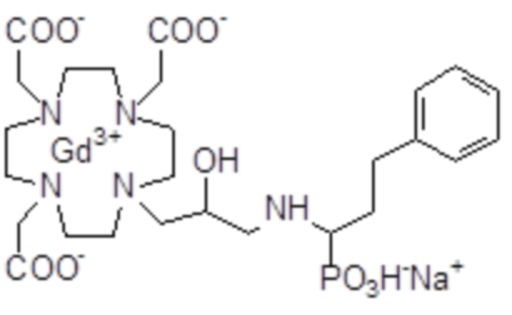
Это соединение получали с использованием методики следующей общей схемы 23:
Схема 23
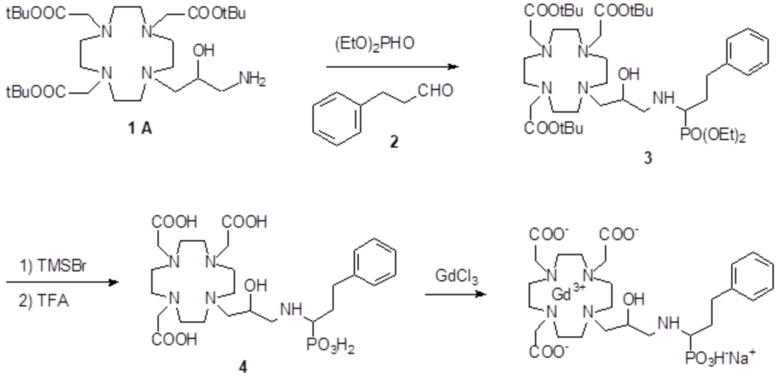
включающей в виде главных стадий:
a) Получение промежуточного соединения 3
Смесь субстрата 1A (15 г; 0,025 моль), 3-фенилпропиональдегида 2 (3,3 мл; 0,026 моль) и диэтилфосфита (3,9 мл; 0,030 моль) нагревали при 80°C в течение 8 ч. Неочищенную реакционную смесь очищали посредством флэш-хроматографии на силикагеле (элюент: CH2Cl2/2-пропанол=95/5). Фракции, содержащие продукт, собирали и выпаривали с получением промежуточного соединения 3 в виде бесцветного масла (17 г) Выход 80%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение промежуточного соединения 4
Раствор бромтриметилсилана (25,9 мл; 196 ммоль) в CH2Cl2 (50 мл) медленно добавляли в раствор соединения 3 (16,5 г; 19,6 ммоль) в CH2Cl2 (100 мл). Смесь встряхивали при комнатной температуре в течение 16 ч, затем выпаривали растворитель. Остаток обрабатывали трифторуксусной кислотой (50 мл), и смесь встряхивали в течение 12 ч. Растворитель выпаривали, а остаток очищали посредством хроматографии в колонке Amberlite XAD 1600 (элюент: градиент воды/MeOH) с получением лиганда 4 (5,5 г). Выход 45%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Комплексообразование
В раствор хелатирующего лиганда 4 (5,2 г; 5,2 ммоль) в воде (40 мл) добавляли гексагидрат хлорида гадолиния (1,93 г, 5,2 ммоль), и pH смеси медленно увеличивали до pH 7 с помощью 2 N NaOH. Полученный раствор встряхивали при 80°C в течение 24 ч, затем фильтровали на Millipore HA 0,45 мкм, концентрировали и очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/ацетонитрила) с получением 2,3 г гадолиниевого комплекса. Выход 54%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Пример 20: получение хелатного комплекса 16
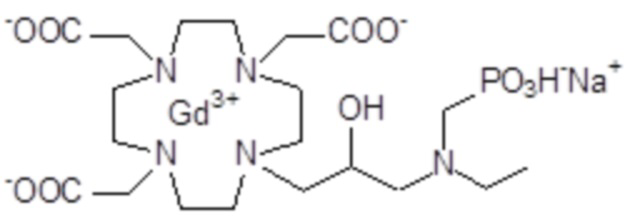
Это соединение получали с использованием методики следующей общей схемы 24:
Схема 24
включающей в виде главных стадий:
a) Получение промежуточного соединения 2
Смесь соединения 1 (полученного, как сообщалось в примере 12) (25 г; 31,5 ммоль) и иодоэтана (5,5 г; 35 ммоль) в DMF (200 мл) нагревали при 50°C и встряхивали в течение 24 ч. Растворитель выпаривали, а остаток очищали посредством флэш-хроматографии на силикагеле (элюент: CH2Cl2/MeOH=100:1ƒ1:1).). Фракции, содержащие продукт, собирали и выпаривали с получением промежуточного соединения 2 (17,3 г) Выход 67%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
b) Получение лиганда 3
В раствор промежуточного соединения 2 (15 г; 18 ммоль) в CH2Cl2 (100 мл) медленно добавляли трифторуксусную кислоту (10 мл), и смесь встряхивали при комнатной температуре в течение 1 ч. Растворитель выпаривали, а остаток растворяли в трифторуксусной кислоте (50 мл) и добавляли триизопропилсилан (0,1 мл). Полученную смесь содержали при встряхивании в течение 24 ч, затем выпаривали. Остаток очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/ацетонитрила) с получением лиганда 3 (6,1 г). Выход 62%.
1H-ЯМР, 13C-ЯМР и масс-спектр согласовались с ожидаемой структурой.
c) Комплексообразование
В раствор хелатирующего лиганда 3 (5,4 г; 10 ммоль) в воде (50 мл) добавляли гексагидрат хлорида гадолиния (3,7 г, 10 ммоль), и pH смеси медленно увеличивали до pH 7 с помощью 2 N NaOH. Раствор встряхивали при 80°C в течение 24 ч, затем фильтровали на Millipore HA 0,45 мкм, концентрировали и очищали посредством хроматографии в колонке Amberlite XE750 (элюент: градиент воды/ацетонитрила) с получением 5,7 г гадолиниевого комплекса. Выход 79%.
Масс-спектр и элементный анализ согласовались с ожидаемой структурой.
Пример 21: релаксометрические свойства
Релаксометрические свойства некоторых типичных комплексных соединений согласно изобретению определяли при различной напряженности магнитного поля, например, включая 0,47 и 1,41 T, при 37°C и в разных средах (физиологический раствор и плазма человека) и сравнивали с измеренными значениями релаксивности, в таких же условиях, для некоторых Gd-комплексов на рынке, имеющих аналогичный циклический координационный каркас.
Материалы
Устройство
Скорость продольной релаксации протонов воды (R1=1/T1) измеряли при 0,47 T спектрометром Minispec MQ-20 (Bruker Biospin, Germany), работающим при ларморовой частоте протонов, равной 20 мГц; МР эксперименты при 1,41 T проводили с использованием спектрометра Minispec MQ-60 (Bruker Biospin, Germany), работающего при ларморовой частоте протонов, равной 60 мГц.
Способы
Получение Образцов
Все опытные образцы использовали в поставляемом виде и разводили в выбранной среде (физиологическом растворе или плазме человека) путем взвешивания требуемого количества парамагнитного хелатного комплекса с получением 5 или 10 мМ исходного раствора.
Измерения Релаксивности
Пять образцов с разными концентрациями (0,1, 0,25, 0,5, 0,75 и 1 мМ) для каждой среды было получено путем дополнительного разведения исходного 5 или 10 мМ раствора.
Измерение Релаксации
Измерения релаксивности проводили при 0,47 T и 1,41 T на образце с заданной температурой, равной 37°C, поддерживаемой постоянной посредством термостатической ванны, связанной с держателем образцов спектрометра. Пять растворов образцов предварительно нагревали при 37°C во внешней термостатической ванне, а затем оставляли на 10 минут внутри ванны, обеспечивая стабилизацию температуры. Время T1 продольной релаксации измеряли посредством стандартной последовательности инверсия-восстановление, где время инверсии (TI) изменяли от 10 мс до по меньшей мере до 5 T1 за 15 шагов. Статистический анализ (моноэкспоненциальная подгонка для измерения T1, линейная подгонка для оценки продольной релаксивности) проводили с помощью Mathematica® (Wolfram, USA). Погрешности оцениваемых параметров оценивали с помощью методики подгонки.
Результаты
Значения r1p релаксивности, полученные из некоторых типичных соединений согласно изобретению, как в физиологическом растворе, так и в плазме человека, при 37°C, кратко изложены в следующей Таблице A вместе со структурой тестируемых соединений и напряженностью приложенного магнитного поля (в T) в сравнении с соответствующими значениями, измеренными для некоторых коммерческих контрастных агентов в клинической практике.
Таблица A
Doratem®
ProHance®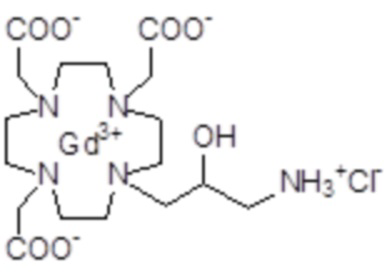
Сравнительное соединение 1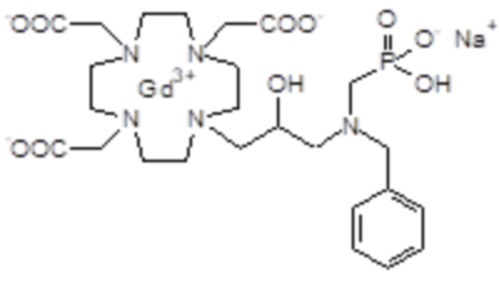
Хелатный Комплекс 1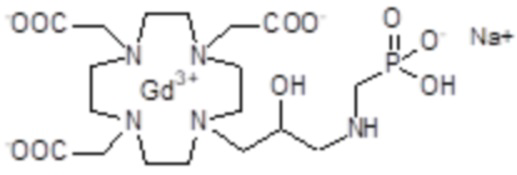
Хелатный Комплекс 2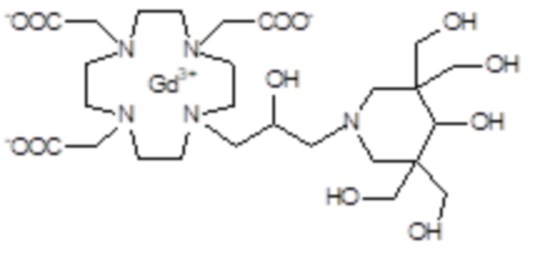
Хелатный Комплекс 5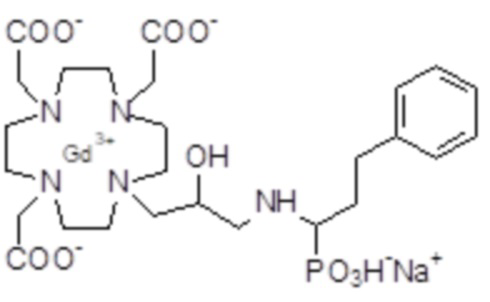
Хелатный Комплекс 6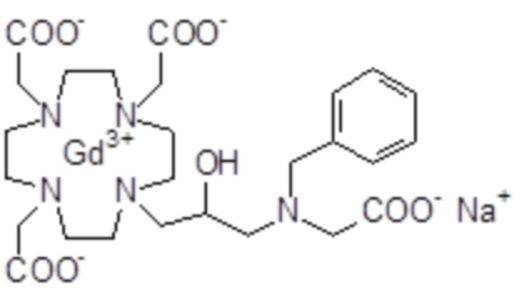
Хелатный Комплекс 7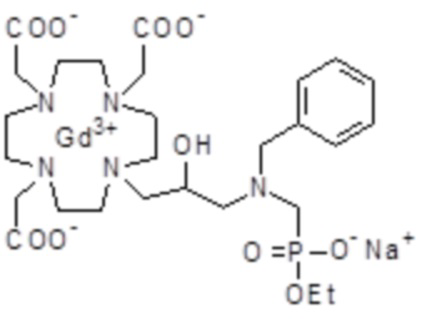
Хелатный Комплекс 8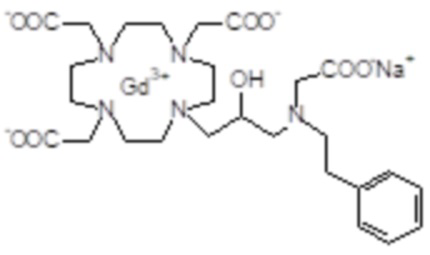
Хелатный Комплекс 11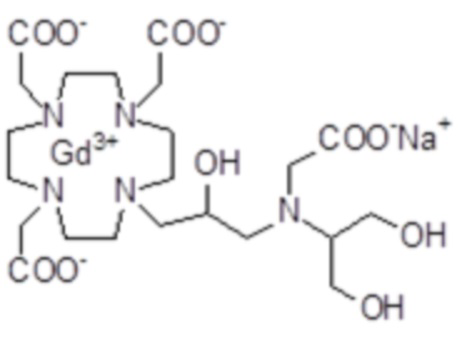
Хелатный Комплекс 13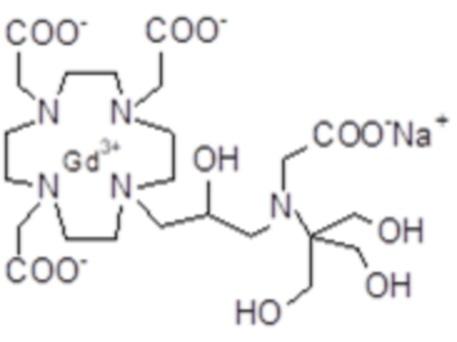
Хелатный Комплекс 4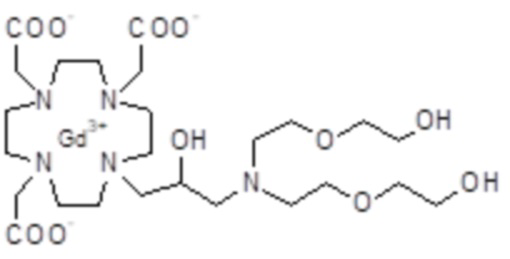
Хелатный Комплекс 14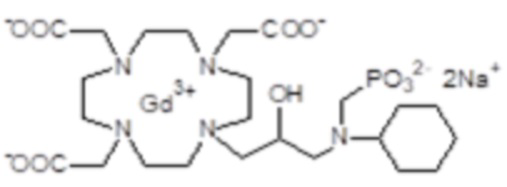
хелатный комплекс 15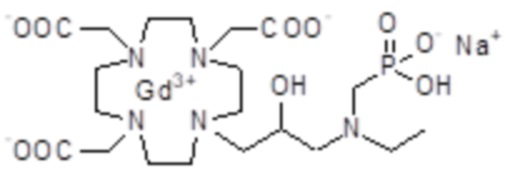
Хелатный Комплекс 16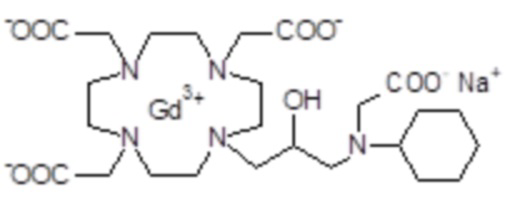
Хелатный Комплекс 19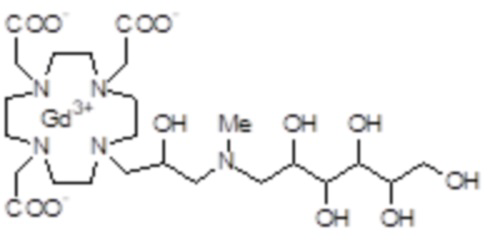
Хелатный Комплекс 21
Заключение
Релаксивность исследованных контрастных агентов колеблется между 4,3 (для незамещенного сравнительного соединения 1) и 8,3 (для хелатного комплекса 6) мМ-1с-1 при 0,47 T в физиологическом растворе, и от 6,25 до 13,8 мМ-1с-1 в плазме, одинаковое магнитное поле. Как ожидалось, эти значения уменьшаются с увеличением напряженности магнитного поля. Эти результаты подтверждают, что особый выбор, предоставляемый парамагнитными комплексами, и особенно Gd3+ комплексами соединений формулы (I) изобретения, показывают увеличенную релаксивность r1p, которая по меньшей мере приблизительно от 1,5 до 2 раза больше релаксивности, показанной в таких же условиях (т.е. в соляном растворе или в плазме человека, при 37°C) неспецифическими контрастными агентами, применяемыми в настоящее время в повседневной диагностической практике, такими как Dotarem® и ProHance®.
Изобретение относится к соединениям формулы (I) и их физиологически приемлемым солям, где в формуле (I) R представляет собой -CH(R1)-COOH, где R1 и R5 представляют собой H; n составляет 1; Z выбран из -N(R2)(R3) и -NHR4; R2 выбирают из группы, состоящей из C5-C7циклоалкильного кольца, цепи формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH и C1-C7алкила, необязательно разделенного одним-тремя атомами кислорода и/или необязательно замещенного одной-тремя гидроксильными группами или фенильным или циклогексильным кольцом; R3 выбирают из группы, состоящей из C5-C12гидроксиалкила, содержащего 3-6 гидроксильных групп; C2-C10гидроксиалкоксиалкилена формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH; и группы формулы -(CH2)sCH(R5)-G; s составляет 0 или 1; r и m составляют независимо друг от друга 1 или 2; G выбран из -PO(OR6)2 и -COOH; где R6 независимо друг от друга представляет собой H или C1-C5 алкил; или R2 и R3 вместе с соединительным атомом азота образуют пиперидиновое кольцо, включающее от 2 до 6 заместителей S, связанных с одним или несколькими атомами углерода пиперидинового кольца, каждая из которых независимо выбрана из гидроксила, C1-C3гидроксиалкила и карбоксила; R4 выбирают из группы, состоящей из C5-C12 гидроксиалкила, содержащего 3-6 гидроксильных групп; -(CH2)mPO(OR6)2 и -(CH2)sCH(R8)G; R8 представляет собой фенил-алкилен или циклогексил-алкилен, имеющий 1-3 атомов углерода в алкиленовой цепи. Также изобретение относится к хелатному комплексу соединения формулы (I) с ионом Gd3+; фармацевтической композиции, содержащей соединение формулы (I) или его хелатный комплекс; соединениям формулы (I), где R представляет собой -CH(R1)-COOH, и каждая карбоксильная группа указанных R групп находится в защищенной форме в виде трет-бутилового сложного эфира. Технический результат - соединения формулы (I) для диагностической визуализации органов или тканей тела человека или животного посредством применения методики МРТ. 4 н. и 15 з.п. ф-лы, 1 табл., 21 пр.
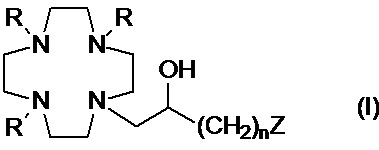
1. Соединение формулы (I)
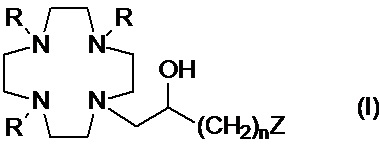 ,
,
где
R представляет собой -CH(R1)-COOH, где
R1 представляет собой H;
n составляет 1;
Z представляет собой аминопроизводное, выбранное из -N(R2)(R3) и -NHR4;
при этом
R2 выбирают из группы, состоящей из C5-C7 циклоалкильного кольца, гидроксиалкоксиалкиленовой цепи формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH и C1-C7 алкила, необязательно разделенного одним-тремя атомами кислорода и/или необязательно замещенного одной-тремя гидроксильными группами или фенильным или циклогексильным кольцом;
R3 выбирают из группы, состоящей из C5-C12 гидроксиалкила, содержащего 3-6 гидроксильных групп; C2-C10 гидроксиалкоксиалкилена формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH; и группы формулы -(CH2)sCH(R5)-G; где
s составляет 0 или 1;
r составляет независимо друг от друга 1 или 2;
m составляет 1 или 2; и где
R5 представляет собой H;
G представляет собой группу, выбранную из -PO(OR6)2 и -COOH; в которой
R6 независимо друг от друга представляет собой H или C1-C5 алкил;
или
R2 и R3 вместе с соединительным атомом азота образуют шестичленное насыщенное кольцо, представляющее собой пиперидиновое кольцо, которое включает от 2 до 6 групп заместителей S, связанных с одним или несколькими атомами углерода пиперидинового кольца, каждая из которых независимо выбрана из группы, состоящей из гидроксила, C1-C3 гидроксиалкила и карбоксила;
R4 выбирают из группы, состоящей из C5-C12 гидроксиалкила, содержащего 3-6 гидроксильных групп; -(CH2)mPO(OR6)2 и -(CH2)sCH(R8)G; где
R8 представляет собой фенил-алкилен или циклогексил-алкилен, имеющий 1-3 атомов углерода в алкиленовой цепи; и
m, s, r и R6 соответствуют сказанному выше,
или его физиологически приемлемая соль.
2. Соединение по п.1 формулы (II)
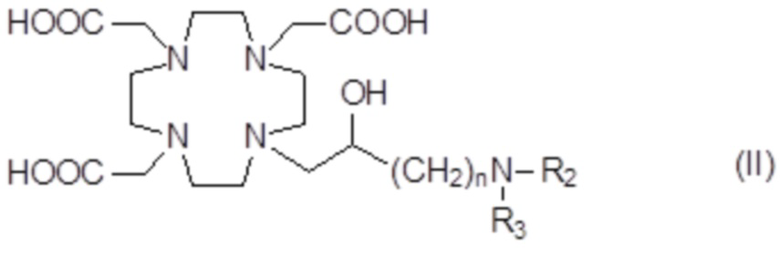
при этом n, R2 и R3 соответствуют определению по п.1.
3. Соединение по п.2 формулы (II A)
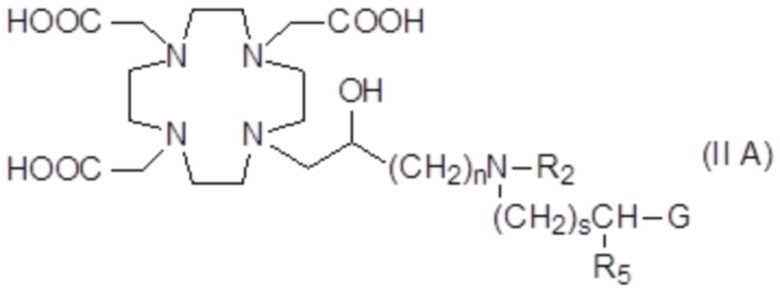 ,
,
в которой
n составляет 1;
s составляет 0 или 1;
G представляет собой группу формулы -PO(OR6)2 или -COOH, в которой R6 независимо друг от друга представляет собой H или C1-C5 алкил;
R5 представляет собой H; и
R2 представляет собой циклогексильное кольцо или C1-C5 алкил, который необязательно замещен одной-тремя гидроксильными группами или фенильным или циклогексильным кольцом.
4. Соединение по п.2 формулы (II B)
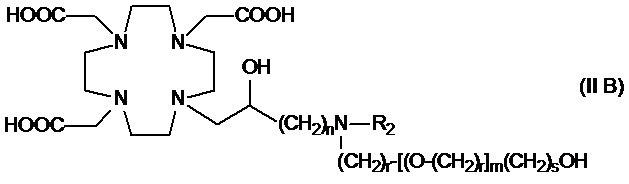 ,
,
в которой
n составляет 1;
каждый r составляет независимо 1 или 2;
m составляет 1 или 2;
s составляет 0 или 1; и
R2 представляет собой вторую гидроксиалкоксиалкиленовую цепь формулы -(CH2)r-[(O-(CH2)r]m(CH2)sOH.
5. Соединение по п.4 формулы (II C)
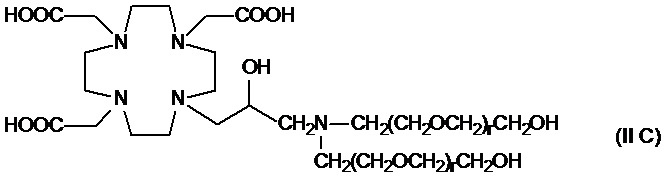 ,
,
в которой r составляет 1 или 2.
6. Соединение по п.2, в котором R2 и R3 вместе с соединительным атомом азота образует замещенное пиперидиновое кольцо, формулы (III)
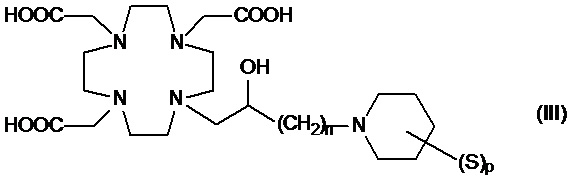 ,
,
в которой
n составляет 1;
p представляет собой целое число от 2 до 6; и
S представляет собой замещающую группу, связанную с атомом углерода пиперидинового кольца, выбранную из группы, состоящей из: гидроксила, C1-C3 гидроксиалкила и карбоксила.
7. Соединение по п.6 формулы (III A)
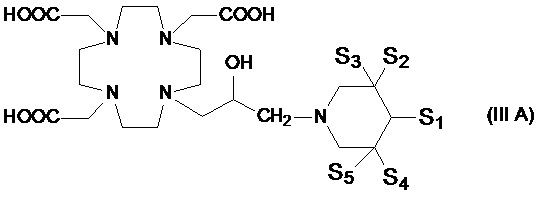 ,
,
в которой замещающие группы S1-S5 каждую независимо выбирают из группы, состоящей из: H, гидроксила, карбоксила и C1-C3 гидроксиалкила, при условии, что по меньшей мере 3 из S1-S5 замещающих групп не являются H.
8. Соединение по п.7, в котором в формуле (III A) замещенное пиперидиновое кольцо представляет собой
9. Соединение по п.1 формулы (IV)
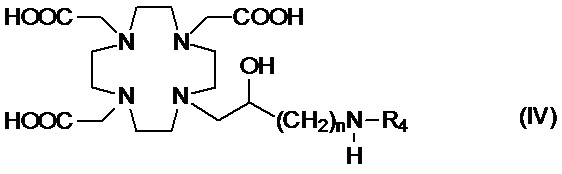 ,
,
в которой n составляет 1, и R4 выбирают из групп формулы -(CH2)mPO(OR6)2 и -(CH2)sCH(R8)G; где
m составляет 1 или 2;
s составляет 0 или 1;
R6 независимо друг от друга представляет собой H или C1-C5 алкил;
R8 представляет собой фенил-алкилен или циклоалкил-алкилен, имеющий 1-3 атомов углерода в алкиленовой цепи; а
G представляет собой группу, выбранную из -PO(OR6)2 и -COOH, где R6 соответствует сказанному выше.
10. Соединение по п.9 формулы (IV A)
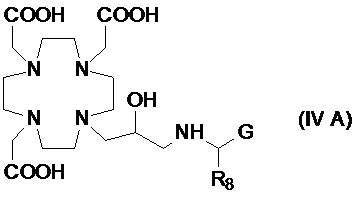 ,
,
в которой
R8 выбирают из группы, состоящей из бензила, фенил-этилена, циклогексил-метилена и циклогексил-этилена; и
G представляет собой группу, выбранную из -PO(OR6)2 и -COOH, где R6 представляет собой H или трет-бутил.
11. Соединение по п.1, в котором в формуле (I) Z представляет собой аминопроизводное, выбранное из N(R2)(R3) и -NHR4, в котором:
R2 представляет собой C1-C5 алкил; и
R3 и R4 представляют собой C5-C12 гидроксиалкил, содержащий 3-6 гидроксильных групп.
12. Соединение по п. 11 формулы (V)
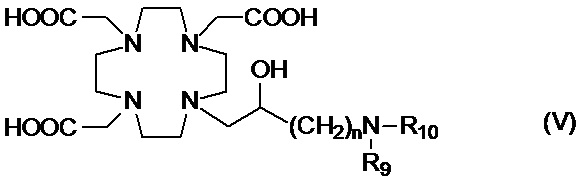 ,
,
в которой
n составляет 1;
R9 представляет собой H или метил; и
R10 представляет собой C5-C12 полиол, выбранный из группы, состоящей из пентил(поли)олов, содержащих от 3 до 4 гидроксильных групп в С5-алкильной цепи; гексил(поли)олов, содержащих от 3 до 5 гидроксильных групп в C6-алкильной цепи; и гептил(поли)олов, содержащих от 3 до 6 гидроксильных групп C7-алкильной цепи.
13. Соединение по любому одному из пп. 11, 12, имеющее формулу
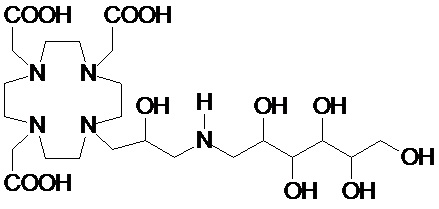
или формулу
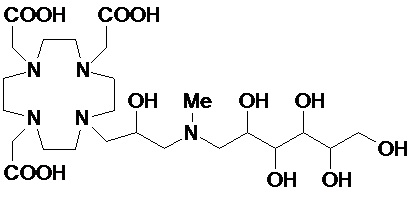 .
.
14. Соединение по п. 1, выбранное из:
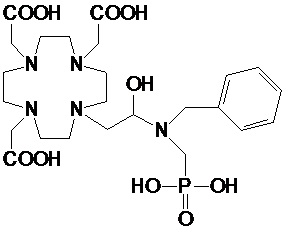
Соединения 1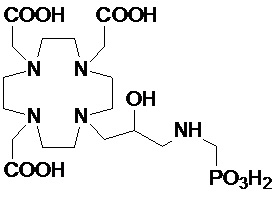 ,
,
Соединения 2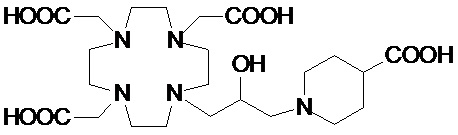
Соединения 3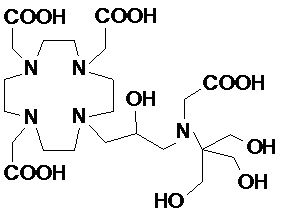
Соединения 4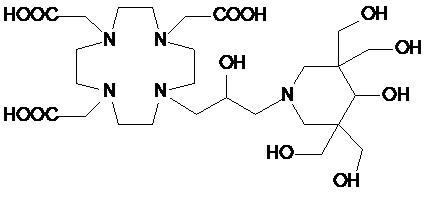
Соединения 5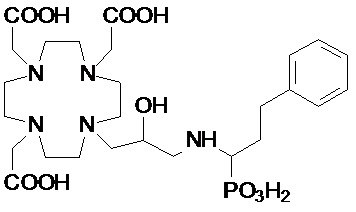
Соединения 6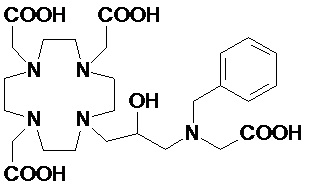
Соединения 7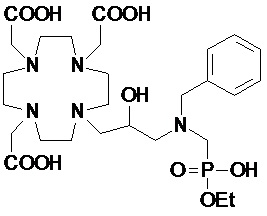
Соединения 8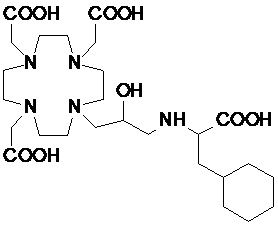
Соединения 9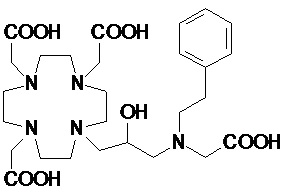
Соединения 11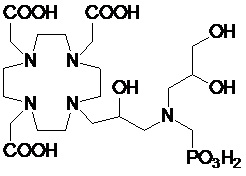
Соединения 12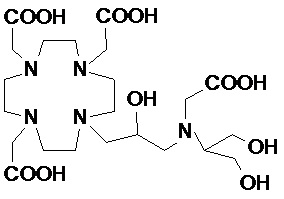
Соединения 13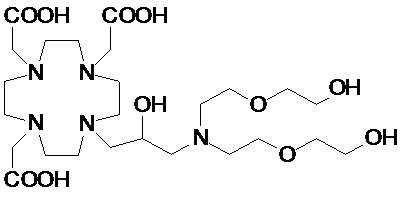
Соединения 14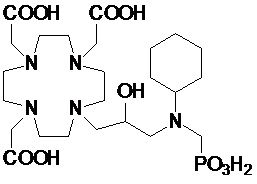
Соединения 15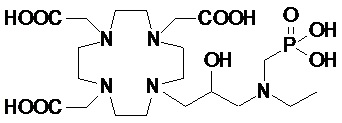
Соединения 16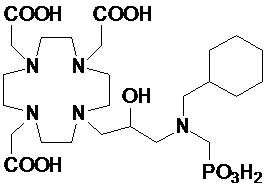
Соединения 17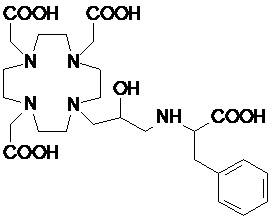
Соединения 18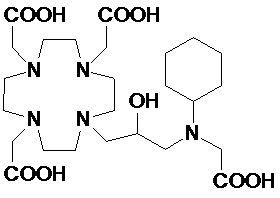
Соединения 19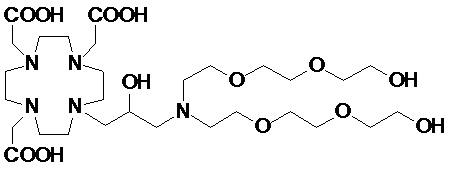 Соединения 20
Соединения 20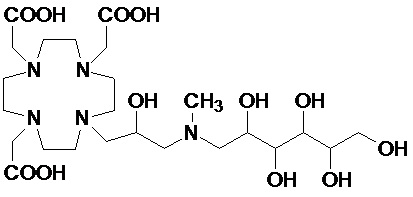 Соединения 21
Соединения 21
15. Хелатный комплекс соединения по любому одному из пп.1-14 с ионом парамагнитного металла, где ион парамагнитного металла представляет собой Gd3+, или его физиологически приемлемой соли.
16. Соединение по любому одному из пп.1-14 или хелатный комплекс по п.15, при этом физиологически приемлемой солью является катион (i) неорганического основания, выбранного из щелочного или щелочноземельного металла, (ii) органического основания, выбранного из этаноламина, диэтаноламина, морфолина, глюкамина, N-метилглюкамина, N,N-диметилглюкамина, или (iii) аминокислоты, выбранной из лизина, аргинина и орнитина.
17. Хелатный комплекс по любому одному из пп.15 или 16 для применения в качестве контрастного агента для МРТ.
18. Фармацевтическая композиция для диагностической визуализации органов или тканей тела человека или животного посредством применения методики МРТ, содержащая эффективное количество хелатного комплекса по любому одному из пп.15 или 16 в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
19. Соединение формулы (I)
,
где
R представляет собой -CH(R1)-COOH, где R1 представляет собой H;
n составляет 1, и Z является таким, как определено в п.1;
в котором каждая карбоксильная группа указанных R групп, связанная с атомом азота макроцикла, находится в защищенной форме в виде трет-бутилового сложного эфира.
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| HOVLAND R | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| LETCHWORTH, GB, 2001, no.6, pp.929-933 | |||
| CHRISTIAN GLOGARD et al.: "Novel radical-responsive MRI | |||

