ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицины и, в частности, относится к тетрациклическому пиримидиноновому соединению, способу его получения и его композиции, а также к их применению в медицине.
УРОВЕНЬ ТЕХНИКИ
Липопротеин-ассоциированная фосфолипаза A2 (Lp-PLA2) является членом суперсемейства фосфолипаз A2 (Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Chem Rev. 2011, 111, 6130-6185). Она в основном секретируется моноцитами, макрофагами, Т-лимфоцитами и главными клетками (Stafforini DM, Elstad MR, McIiterrre TM, Zimmerman GA, Prescott SM. J Biol Chem. 1990, 265: 9682-9687; Nakajima K, Murakami M, Yanoshita R, Samejima Y, Karasawa K, Setaka M, Nojima S Kudo I. J Biol Chem. 1997, 272, 19708-19713). Эфиры фосфатидилхолина sn-2 образуются во время окисления липопротеина низкой плотности (ЛПНП), и Lp-PLA2 отвечает за гидролиз окислительно-модифицированных эфиров фосфатидилхолина sn-2, а затем продуцирует окисленные жирные кислоты и лизофосфатидилхолин (LysoPC) (Caslake MJ, Packard CJ, Suckling KE, Holmes SD, Chamberlain P, Macphee CH. Atherosclerosis. 2000, 150, 413-419; MacPhee CH, Moores KE, Boyd HF, Dhanak D, Ife RJ, Leach CA, Leake DS, Milliner KJ, Patterson RA, Suckling KE, Tew DG, Hickey DM. Biochem J. 1999, 338, 479-487). Как окисленные жирные кислоты, так и LysoPC играют роль в активации макрофагов, увеличивая окислительный стресс, влияя на функцию Т-лимфоцитов и индуцируя воспалительные реакции (Quinn MT, Parthasarathy S, Steinberg D. Proc Natl Acad Sci U S A. 1988, 85, 2805-2809). Сообщалось, что LysoPC индуцирует высвобождение множественных цитотоксических воспалительных цитокинов (Shi, et al, Atherosclerosis, 2007, 191, 54-62). Кроме того, LysoPC также участвует в активации лейкоцитов, индукции апоптоза и опосредовании эндотелиальной дисфункции (Wilensky et al, Current Opinion in Lipidology, 2009, 20, 415-420).
Сообщалось, что уровень Lp-PLA2 в плазме связан с сердечно-сосудистыми заболеваниями (Fitzpatrick AL, Irizarry MC, Cushman M, Jenny NS, CHI GC, Koro C. Atherosclerosis. 2014, 235, 384-391), диабетическим макулярным отеком (DME) (Staurenghi G, Ye L, Magee MH, Danis RP, Wurzelmann J, Adamson P, McLaughlin MM, Darapladib DMESG. Ophthalmology. 2015, 122, 990-996) и раком предстательной железы (Bertilsson H, Tessem MB, Flatberg A, Viset T, Gribbestad I, Angelsen A, Halgunset J. Clin Cancer Res. 2012, 18, 3261-3269).
Болезнь Альцгеймера (БА) представляет собой хроническое нейродегенеративное заболевание, которое вызывает снижение когнитивных функций, перепады настроения, необратимую потерю памяти, дезориентацию, нарушение речи и потерю самосохранения (Hardy J, et al. Science 2002, 297, 353-356). Болезнь Альцгеймера обычно начинается медленно и прогрессирует с течением времени, что является причиной от 60 до 70 процентов случаев деменции и затрагивает около 6 процентов населения в возрасте старше 65 лет. Пациенты с БА постепенно отдаляются от семьи и общества, становятся все более и более зависимыми от помощи и в конечном итоге умирают. БА является одним из наиболее дорогостоящих заболеваний как в развитых странах, так и в других странах. Учитывая, что старение становится важной социальной проблемой, затраты будут быстро расти. Нет сомнений в том, что БА - это сложное заболевание, включающее множество факторов. Хотя патогенез БА не до конца установлен, ясно, что в развитии и прогрессировании заболевания участвует несколько факторов, включая агрегированные тау-белки и пептиды Aβ, окислительный стресс и нейровоспаление (Echeverria V, Yarkov A, Aliev G. Prog Neurobiol. 2016, 144, 142-157). Современная разработка лекарств от БА фокусируется в основном на мишенях Aβ-амилоидоза и тау (Chiang K, Koo EH. Annu Rev Pharmacol Toxicol. 2014, 54, 381-405; Awasthi M, Singh S, Pandey VP, Dwivedi UN. J Neurol Sci. 2016, 361, 256-271). Однако, несмотря на достоверные доклинические данные, результаты клинических исследований на поздней стадии до сих пор не продемонстрировали клинической эффективности. Эти разочаровывающие результаты позволяют предположить, что для лечения БА, возможно, придется изучить другие нейропатологические механизмы, такие как окислительный стресс и нейровоспаление.
Повышенные уровни Lp-PLA2 в плазме повышают риск деменции, включая БА (Van Oijen, et al. Annals of Neurology, 2006, 59,139). У пациентов с БА были обнаружены сосудистая деменция и смешанная деменция, а также высокие уровни окислительных ЛПНП (Maher-Edwards G, De’Ath J, Barnett C, Lavrov A, Lockhart A, Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2015, 1, 131-140; Kassner et al. Current Alzheimer Research, 2008, 5, 358-366; Dildar, et al., Alzheimer Dis Assoc Disord, 24, April-June (2010); Sinem, et al. Current Alzheimer Research, 2010, 7, 463-469). У пациентов с БА также было обнаружено нейровоспаление и множество повышающе-регулируемых воспалительных цитокинов (Colangelo, et al., Journal of Neuroscience Research, 2002, 70, 462-473; Wyss-Coray, Nature Medicine, 2006, 12, Sept.).
Учитывая все эти результаты, Lp-PLA2 является потенциальной мишенью для лечения БА, что дополнительно подтверждается клиническими результатами применения ингибитора Lp-PLA2 Рилапладиба у пациентов с БА (Maher-Edwards G, De'Ath J, Barnett C, Lavrov A, Lockhart A, Alzheimer' s & Dementia: Translational Research & Clinical Interventions. 2015, 1, 131-140).
Глаукома и возрастная макулярная дегенерация (ВМД) являются нейродегенеративными заболеваниями сетчатки. Buschini с соавторами сообщали, что воспаление, включая передачу сигналов ФНО-α, может играть важную роль в патогенезе глаукомы и ВМД (Buschini et al, Progress in Neurobology, 2011, 95, 14-25; Tezel, Progress in Brain Research, vol. 173, ISSN0079-6123, Chapter 28). Кроме того, Shi с соавторами продемонстрировали, что ингибитор Lp-PLA2 может блокировать высвобождение воспалительных цитокинов (Shi, et al, Atherosclerosis, 2007, 191, 54-62). Ингибирование Lp-PLA2 является потенциальной терапией глаукомы и ВМД.
Сообщалось о многих ингибиторах Lp-PLA2, включая β-лактамы (Tew DG, Boyd HF, Ashman S, Theobald C, Leach CA. Biochemistry. 1998, 37, 10087-10093), oximes (Jeong TS, Kim MJ, Yu H, Kim HS, Choi JK, Kim SS, Lee WS. Bioorg Med Chem Lett. 2005, 15, 1525-1527; Jeong HJ, Park YD, Park HY, Jeong IY, Jeong TS, Lee WS. Bioorg Med Chem Lett. 2006, 16, 5576-5579), amides of xanthuric acids (Lin EC, Hu Y, Amantea CM, Pham LM, Cajica J, Okerberg E, Brown HE, Fraser A, Du L, Kohno Y, Ishiyama J, Kozarich JW, Shreder KR. Bioorg Med Chem Lett. 2012, 22, 868-871; Hu Y, Lin EC, Pham LM, Cajica J, Amantea CM, Okerberg E, Brown HE, Fraser A, Du L, Kohno Y, Ishiyama J, Kozarich JW, Shreder KR. Bioorg Med Chem Lett. 2013, 23, 1553-1556), and urethane (Nagano JM, Hsu KL, Whitby LR, Niphakis MJ, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Hodder P, Srinivasan P, Gonzalez TD, Rosen H, Bahnson BJ, Cravatt BF. Bioorg Med Chem Lett. 2013, 23, 839-843).
Сообщалось, что ингибитор Lp-PLA2 дарапладиб является потенциальной терапией против атеросклероза и DME (Magrioti V, Kokotos G. Expert Opin Ther Pat. 2013; 23: 333-344).
КРАТКОЕ ОПИСАНИЕ
Авторы настоящего изобретения обнаружили, что ингибиторы Lp-PLA2 играют значительную роль в лечении связанных с нейродегенерацией заболеваний, таких как болезнь Альцгеймера (БА), глаукома и возрастная макулярная дегенерация (ВМД), или сердечно-сосудистых заболеваний, включая атеросклероз. С этой целью авторы настоящего изобретения поставили задачу разработать совершенно новый ингибитор Lp-PLA2, т.е. тетрациклическое пиримидиноновое соединение.
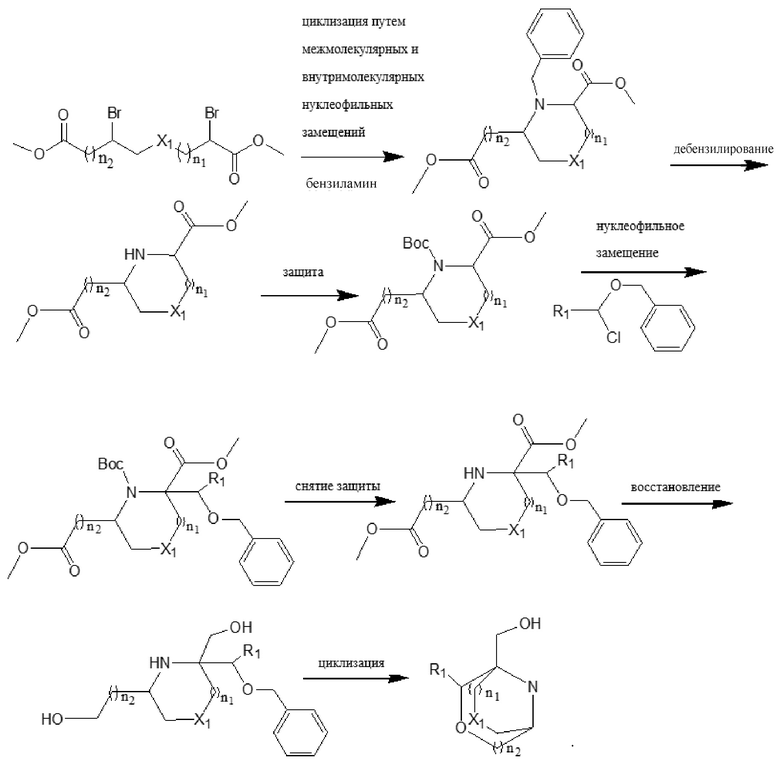
Указанное тетрациклическое пиримидиноновое соединение представляет собой соединение, имеющее структуру, представленную формулой (I),
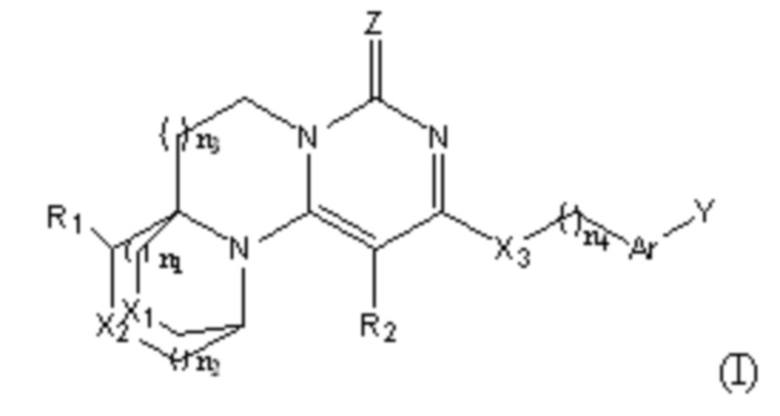
или его фармацевтически приемлемую соль, где:
n1, n2, n3, и n4 каждый независимо представляют собой 0, 1 или 2;
R1 и R2 каждый независимо выбран из: -H, гидроксила, циано, галогена, алкила, дейтероалкила, гидроксиалкила, галогеналкила, циклоалкила и алкоксила;
X1, X2, и X3 каждый независимо выбран из алкилена, -O-, -S-, или -NR'-;
R' выбран из -H, алкила, дейтероалкила или циклоалкила;
Ar представляет собой арилен или гетероарилен; и атомы водорода в указанном арилене или гетероарилене необязательно замещены одним или более заместителями, каждый из которых независимо выбран из галогена, алкила, галогеналкила, алкоксила, галогеналкоксила, дейтероалкила, дейтероалкоксила, гидроксила, гидроксиалкила, циано, амино, моноалкил- или диалкил-замещенного амино, нитро, карбоксила, альдегида, циклоалкила, гетероциклила, арила или гетероарила;
Y представляет собой -H, галоген, алкил, галогеналкил, галогеналкоксил, циклоалкил, алкоксил, дейтероалкил, дейтероалкоксил, -OAr', -SAr', -NH-Ar', -NMe-Ar', -NR'' или -R'''-Ar';
Ar' выбран из арила или гетероарила; и атомы водорода в указанном ариле или гетероариле необязательно замещены одним или более заместителями, каждый из которых независимо выбран из галогена, алкила, галогеналкила, алкоксила, гидроксила, гидроксиалкила, галогеналкоксила, дейтероалкила, дейтероалкоксила, циано, амино, нитро, карбоксила, альдегида, циклоалкила, гетероциклила, арила или гетероарила;
R'' представляет собой алкил;
R''' представляет собой алкилен; и
Z представляет собой O или S;
необязательно, где:
n1, n2, n3, и n4 каждый независимо составляет 0, 1 или 2;
R1 и R2 каждый независимо выбран из -H, гидроксила, циано, галогена, алкила, дейтероалкила, гидроксиалкила, галогеналкила, циклоалкила и алкоксила;
X1, X2 и X3 каждый независимо выбран из алкилена, -O-, -S- или -NR'-,
R' выбран из: -H, алкила, дейтероалкила или циклоалкила;
Ar представляет собой арилен или гетероарилен; и атомы водорода в указанном арилене или гетероарилене необязательно замещены одним или более заместителями, каждый из которых независимо выбран из галогена, алкила, галогеналкила, алкоксила, галогеналкоксила, гидроксила, гидроксиалкила, циано, амино, моноалкил- или диалкил-замещенного амино, нитро, карбоксила, альдегида, циклоалкила, гетероциклила, арила или гетероарила;
Y представляет собой -H, галоген, алкил, галогеналкил, галогеналкоксил, циклоалкил, алкоксил, -OAr', -SAr', -NH-Ar', -NMe-Ar', -NR'' или -R'''-Ar';
Ar' выбран из арила или гетероарила; и атомы водорода в указанном ариле или гетероариле необязательно замещены одним или более заместителями, каждый из которых независимо выбран из: галогена, алкила, галогеналкила, алкоксила, гидроксила, гидроксиалкила, галогеналкоксила, циано, амино, нитро, карбоксила, альдегида, циклоалкила, гетероциклила, арила или гетероарила;
R'' представляет собой алкил;
R''' представляет собой алкилен; и
Z представляет собой O или S.
Необязательно, атомы галогена в “галогене”, “галогеналкиле” и “галогеналкоксиле” каждый независимо выбраны из F, Cl, Br или I;
необязательно, алкилы в указанных “алкиле”, “дейтероалкиле”, “дейтероалкоксиле”, “гидроксиалкиле”, “галогеналкиле”, “галогеналкоксиле”, “алкоксиле”, “моноалкил- или диалкил-замещенном амино” каждый независимо представляет собой C1-C10 линейный или разветвленный алкил; необязательно, C1-C7 линейный или разветвленный алкил; необязательно, C1-C4 линейный или разветвленный алкил; и необязательно выбран из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, втор-бутила, н-пентила, 1-метилбутила, 2-метилбутила, 3-метилбутила, изопентила, 1-этилпропила, неопентила, н-гексила, 1-метилпентила, 2-метилпентила, 3-метилпентила, изогексила, 1,1-диметилбутила, 2,2-диметилбутила, 3,3-диметилбутила, 1,2-диметилбутила, 1,3-диметилбутила, 2,3-диметилбутила, 2-этилбутила, н-гептила, 2-метилгексила, 3-метилгексила, 2,2-диметилпентила, 3,3-диметилпентила, 2,3-диметилпентила, 2,4-диметилпентила, 3-этилпентила или 2,2,3-триметилбутила;
необязательно, “алкилены” каждый независимо представляет собой C1-C10 линейный или разветвленный алкилен; необязательно, C1-C7 линейный или разветвленный алкилен; необязательно, C1-C5 линейный или разветвленный алкилен; необязательно, выбранный из метилена, этилена, н-пропилидена, изопропилидена, н-бутилидена, изобутилидена, трет-бутилидена, втор-бутилидена, н-пентилидена, 1-метилбутилидена, 2-метилбутилидена, 3-метилбутилидена, изопентилидена, 1-этилпропилидена, неопентилидена, н-гексилидена, 1-метилпентилидена, 2-метилпентилидена, 3-метилпентилидена, изогексилидена, 1,1-диметилбутилидена, 2,2-диметилбутилидена, 3,3-диметилбутилидена, 1,2-диметилбутилидена, 1,3-диметилбутилидена, 2,3-диметилбутилидена, 2-этилбутилидена, н-гептилидена, 2-метилгексилидена, 3-метилгексилидена, 2,2-диметилпентилидена, 3,3-диметилпентилидена, 2,3-диметилпентилидена, 2,4-диметилпентилидена, 3-этилпентилидена или 2,2,3-триметилбутилидена;
необязательно, “циклоалкил” представляет собой C3-C10 моноциклический или бициклический циклоалкил, необязательно C3-C7 моноциклический циклоалкил и необязательно циклопропил, циклобутил, циклопентил, циклогексил или циклогептил;
необязательно, “гетероциклил” представляет собой 3-10-членное неароматическое гетероциклическое кольцо, содержащее 1, 2 или 3 гетероатома, выбранных из N, O и S, в кольце, при этом необязательно указанное гетероциклическое кольцо представляет собой 3-10-членное неароматическое кольцо, содержащее 1 или 2 гетероатома, выбранных из N и O в кольце; необязательно, указанное гетероциклическое кольцо представляет собой 3-6-членное неароматическое кольцо, содержащее 1 или 2 гетероатома, выбранных из N и O в кольце; необязательно, указанное гетероциклическое кольцо представляет собой 3-10-членное неароматическое кольцо, содержащее 1 или 2 гетероатома, выбранных из N и S в кольце; и необязательно, указанное гетероциклическое кольцо представляет собой 3-6-членное неароматическое кольцо, содержащее 1 или 2 гетероатома, выбранных из N и S в кольце;
необязательно, “арил” представляет собой 6-10-членный арил, необязательно фенил или нафтил и необязательно фенил, 1-нафтил или 2-нафтил;
необязательно, “арилен” представляет собой 6-10-членный арилен и необязательно фенилен или нафтилен;
необязательно, “гетероарил” представляет собой 5-10-членное гетероароматическое кольцо, содержащее 1-3 гетероатома, выбранных из N, O и S в кольце, и необязательно 5-10-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из N, O и S в кольце; необязательно, гетероароматическое кольцо выбрано из пиридинового кольца, пиррольного кольца, пиримидинового кольца, пиразинового кольца, пиридазинового кольца, тиофенового кольца и фуранового кольца; необязательно, указанный гетероарил выбран из пиридин-2-ила, пиридин-3-ила, пиридин-4-ила, пиридазин-3-ила, пиридазин-4-ила, пиримидин-2-ила, пиримидин-4-ила, пиримидин-5-ила, пиразин-2-ила, пиразин-3-ила, индолила, изоиндолила, индазолила, индолизинила, пуринила, хинолизинила, хинолинила, изохинолинила, цинолинила, фталазинила, нафтиридинила, хиназолинила, хиноксалинила, тиено[2,3-b]фурила, фуро[3,2-b]-пиранила, пиридо[2,3-d]оксазинила, пиразоло[4,3-d]оксазолила, имидазо[4,5-d]тиазолила, пиразино[2,3-d]пиридазинила, имидазо[2,1-b]тиазолила, имидазо[1,2-b][l,2,4]триазинила, бензотиенила, бензоксазолила, бензимидазолила, бензотиазолила, бензоксепинила, бензоксазинила, бензофуранила, бензотриазолила, пирроло[2,3-b]пиридила, пирроло[3,2-c]пиридила, пирроло[3,2-b]пиридила, имидазо[4,5-b]пиридила, имидазо[4,5-c]пиридила, пиразоло[4,3-d]пиридила, пиразоло[4,3-c]пиридила, пиразоло[3,4-c]пиридила, пиразоло[3,4-d]пиридила, пиразоло[3,4-b]пиридила, имидазо[1,2-a]пиридила, пиразоло[1,5-a]пиридила, пирроло[1,2-b]пиридазинила, имидазо[1,2-c]пиримидинила, пиридо[3,2-d]пиримидинила, пиридо[4,3-d]пиримидинила, пиридо[3,4-d]пиримидинила, пиридо[2,3-d]пиримидинила, пиридо[2,3-b]пиразинила, пиридо[3,4-b]пиразинила, пиримидо[5,4-d]пиримидинила, пиразоло[2,3-b]пиразинила и пиримидо[4,5-d]пиримидинила и необязательно пиридин-2-ила, пиридин-3-ила, пиридин-4-ила, пиримидин-2-ила, пиримидин-4-ила или пиримидин-5-ила;
необязательно, “гетероарилен” представляет собой 5-10-членное гетероариленовое кольцо, содержащее 1-3 гетероатома, выбранных из N, O и S в кольце, и необязательно 5-10-членное гетероариленовое кольцо, содержащее 1-2 гетероатома, выбранных из N, O и S в кольце; и необязательно, гетероариленовое кольцо выбрано из пиридилиденового кольца, пиррилиденового кольца, пиримидилиденового кольца, пиразинилиденового кольца, пиридазинилиденового кольца, тиенилиденового кольца, фурилиденового кольца;
необязательно, n1, n2, n3 и n4 каждый независимо 0, 1 или 2; необязательно, n1 равен 0, необязательно, n2 равен 0 или 1, необязательно, n3 равен 0, необязательно, n4 равен 1;
необязательно, R1 и R2 каждый независимо выбран из -H, F, Cl, Br, гидроксила, циано, C1-C7 алкила (метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, втор-бутила, н-пентила, 1-метилбутила, 2-метилбутила, 3-метилбутила, изопентила, 1-этилпропила, неопентила, н-гексила, 1-метилпентила, 2-метилпентила, 3-метилпентила, изогексила, 1,1-диметилбутила, 2,2-диметилбутила, 3,3-диметилбутила, 1,2-диметилбутила, 1,3-диметилбутила, 2,3-диметилбутила, 2-этилбутила, н-гептила, 2-метилгексила, 3-метилгексила, 2,2-диметилпентила, 3,3-диметилпентила, 2,3-диметилпентила, 2,4-диметилпентила, 3-этилпентила или 2,2,3-триметилбутила), C1-C3 дейтероалкила (-CD3, -C2D5, -C3D7), циклопропанила, циклобутанила и циклопентила; необязательно, R1 представляет собой -H, необязательно, R2 представляет собой -H;
необязательно, X1, X2 и X3 каждый независимо выбран из: C1-C7 алкилена (необязательно, -CH2-, этилена, н-пропилидена, изопропилидена, н-бутилидена или изобутилидена), -O-, -S- или -NR'-; необязательно, X1 представляет собой -CH2-; необязательно, X2 выбран из -CH2- или -O-; необязательно, X3 представляет собой -O-;
необязательно, R' выбран из -H, C1-C7 алкила (метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, втор-бутила, н-пентила, 1-метилбутила, 2-метилбутила, 3-метилбутила, изопентила, 1-этилпропила, неопентила, н-гексила, 1-метилпентила, 2-метилпентила, 3-метилпентила, изогексила, 1,1-диметилбутила, 2,2-диметилбутила, 3,3-диметилбутила, 1,2-диметилбутила, 1,3-диметилбутила, 2,3-диметилбутила, 2-этилбутила, н-гептила, 2-метилгексила, 3-метилгексила, 2,2-диметилпентила, 3,3-диметилпентила, 2,3-диметилпентила, 2,4-диметилпентила, 3-этилпентила или 2,2,3-триметилбутила;), дейтероалкила (необязательно -CD3, -C2D5, -C3D7) или C3-C6 циклоалкила (циклопропанила, циклобутанила, циклопентила или циклогексила);
необязательно, Ar представляет собой фенилен или пиридил, и атом водорода в указанном фенилене или пиридиле необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из: F, Cl, Br, I, -CN, -Me, -C2H5, циклопропила, -CD3, -OMe, или -OCF3; необязательно, Ar представляет собой фенилен, и атом водорода в указанном фенилене необязательно замещен 2 заместителями, представляющими собой F;
необязательно, Y представляет собой -H, -F, -Cl, -Br, метил, этил, н-пропил, изопропил, -CD3, -CF3, -CH2CF3, -OCF3, -OCHF2, циклопропил, -циклобутил, -циклопентил, -OCH3, -OC2H5, -OC3H7, или -OAr'; необязательно, Y представляет собой -H или -OAr';
необязательно, Ar' выбран из фенила, пиридила, пиримидинила или хинолинила, и атомы водорода в указанном фенильном, пиридильном, пиримидинильном или хинолинильном кольце каждый независимо необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из: F, Cl, Br, -CN, C1-C7 алкила (необязательно выбранного из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, втор-бутила, н-пентила, 1-метилбутила, 2-метилбутила, 3-метилбутила, изопентила, 1-этилпропила, неопентила, н-гексила, 1-метилпентила, 2-метилпентила, 3-метилпентила, изогексила, 1,1-диметилбутила, 2,2-диметилбутила, 3,3-диметилбутила, 1,2-диметилбутила, 1,3-диметилбутила, 2,3-диметилбутила, 2-этилбутила, н-гептила, 2-метилгексила, 3-метилгексила, 2,2-диметилпентила, 3,3-диметилпентила, 2,3-диметилпентила, 2,4-диметилпентила, 3-этилпентила или 2,2,3-триметилбутила), -CD3, C1-C6галогеналкила, -OCH3, -OC2H7, -OC3H7, C1-C6 галогеналкоксила или C3-C6циклоалкила (необязательно циклопропанила, циклобутанила, циклопентила или циклогексила); необязательно, Ar' выбран из фенила, пиридин-3-ила или пиридин-4-ила или пиримидин-5-ила, необязательно, Ar' замещен 1 или 2 заместителями, выбранными из Cl, -CH3, -CF3 или -OCF3; и
необязательно, Z представляет собой O.
Необязательно, соединение формулы (I) или его фармацевтически приемлемая соль, причем указанное соединение формулы (I) выбрано из следующих соединений:
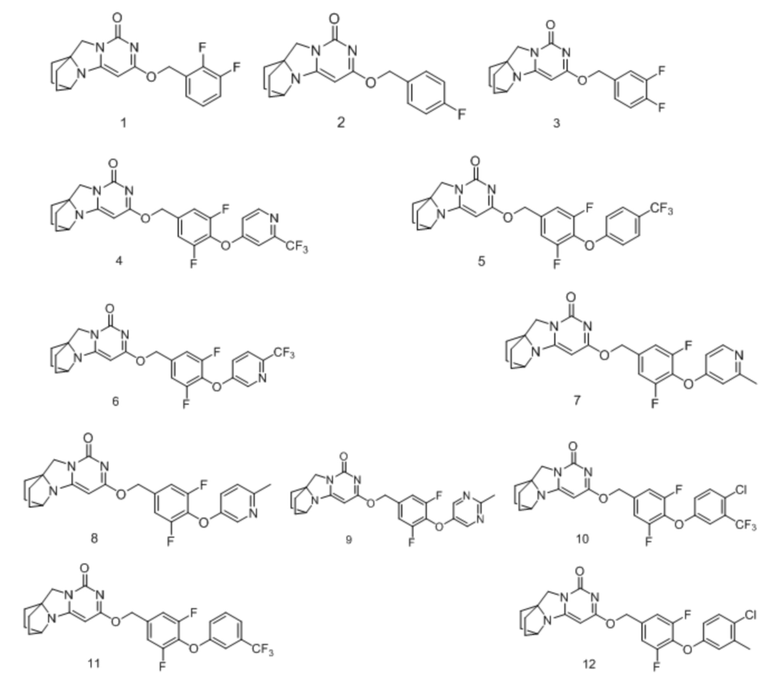
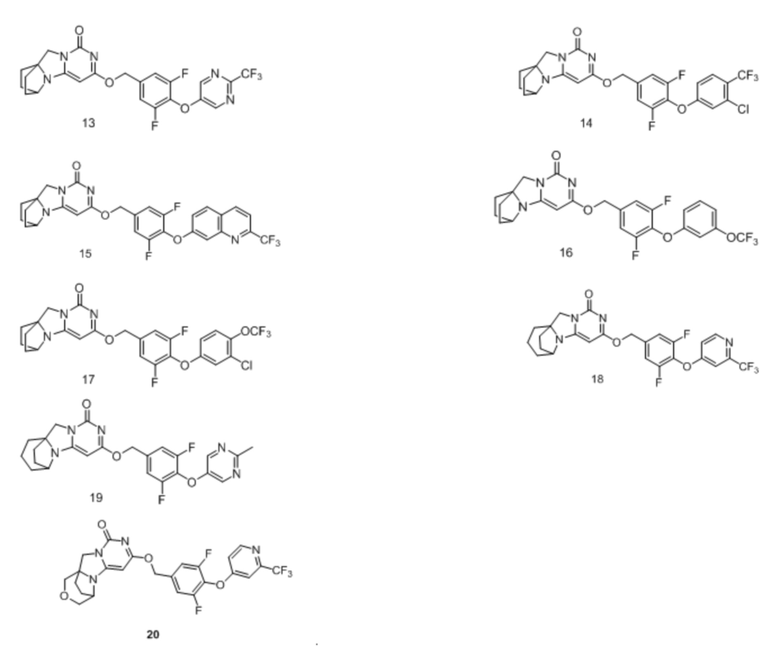
Необязательно, соединение формулы (I) или его фармацевтически приемлемая соль, причем указанная фармацевтически приемлемая соль включает анионную соль и катионную соль соединения формулы (I);
необязательно, фармацевтически приемлемая соль включает соль щелочного металла, соль щелочно-земельного металла и аммониевую соль соединения формулы (I); необязательно, щелочной металл включает натрий, калий, литий и цезий, а щелочно-земельный металл включает магний, кальций и стронций;
необязательно, фармацевтически приемлемая соль включает соль, образуемую соединением формулы (I) и органическим основанием;
необязательно, органическое основание включает триалкиламин, пиридин, хинолин, пиперидин, имидазол, пиколин, диметиламинопиридин, диметиланилин, N-алкилморфолин, 1,5-диазабицикло[4,3.0]нонен-5,1,8-диазабицикло[5,4.0]ундецен-7, и 1,4-диазабицикло[2,2.2]октан; необязательно, триалкиламин включает триметиламин, триэтиламин и N-этилдиизопропиламин; и необязательно, N-алкилморфолин включает N-метилморфолин;
необязательно, фармацевтически приемлемая соль включает соль, формируемую соединением формулы (I) и кислотой; и
необязательно, кислота включает неорганическую кислоту и органическую кислоту; необязательно, неорганическая кислота включает хлористоводородную кислоту, бромистоводородную кислоту, иодистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту, угольную кислоту; необязательно, органическая кислота включает муравьиную кислоту, уксусную кислоту, пропионовую кислоту, щавелевую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, малеиновую кислоту, молочную кислоту, яблочную кислоту, лимонную кислоту, лимонную кислоту, винную кислоту, угольную кислоту, пикриновую кислоту, метансульфоновую кислоту, этансульфоновую кислоту, п-толуолсульфоновую кислоту, глутаминовую кислоту и памовую кислоту.
В другом аспекте предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, включающий следующий путь проведения реакции:
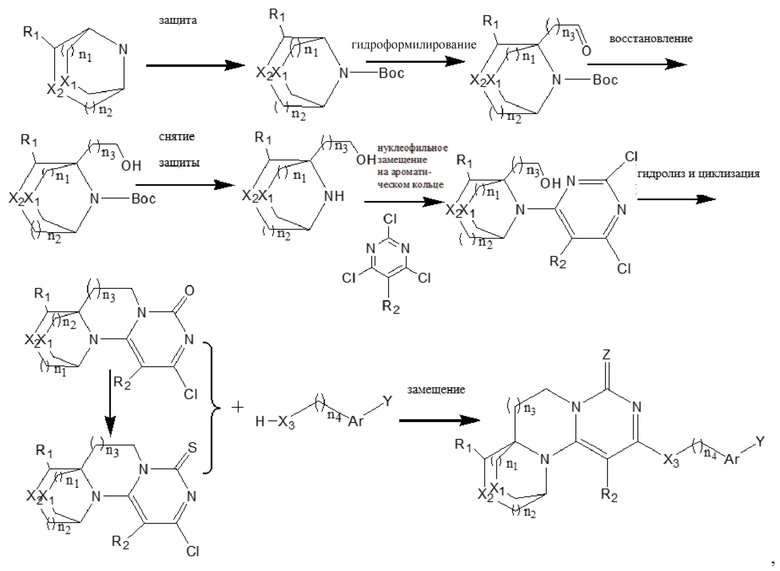
в соответствующих формулах n1, n2, n3, R1, R2, X1, X2, X3, Z, Ar и Y являются такими, как определено выше;
необязательно, когда X2 представляет собой -O-, а n3 представляет собой 0, дополнительно используют следующий путь синтеза:
Конкретные условия реакции для каждой из вышеупомянутых реакций конкретно не ограничены, и могут быть использованы существующие обычные условия реакции или стадии.
В другом аспекте предложена фармацевтическая композиция, содержащая терапевтически эффективную дозу одного или более из вышеупомянутого соединения формулы (I) или его фармацевтически приемлемой соли и, необязательно, фармацевтически приемлемый носитель.
необязательно, лекарственная форма фармацевтической композиции включает пероральный препарат, ректально вводимый препарат и парентерально вводимый препарат;
необязательно, препарат для перорального введения включает твердый препарат и жидкий препарат;
необязательно, твердый препарат включает таблетки, порошки, гранулы и капсулы;
необязательно, жидкий препарат включает водные или масляные суспензии и сиропы; и
необязательно, препарат для парентерального введения включает растворы для инъекций и водные или масляные суспензии.
В другом аспекте предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, описанных выше, или фармацевтической композиции, описанной выше, для получения ингибитора Lp-PLA2.
В другом аспекте предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как описано выше, или фармацевтической композиции, как описано выше, для получения лекарственного средства для лечения заболевания, связанного с нейродегенерацией; и
необязательно, заболевание, связанное с нейродегенерацией, включает болезнь Альцгеймера (БА), глаукому и возрастную макулярную дегенерацию (ВМД).
В другом аспекте предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как описано выше, или фармацевтической композиции, как описано выше, для получения лекарственного средства для лечения сердечно-сосудистых заболеваний, диабетического макулярного отека (DME) или заболевания предстательной железы; и
необязательно, сердечно-сосудистое заболевание включает атеросклероз.
Предпочтительные эффекты:
Соединение формулы (I) представляет собой тетрациклическое пиримидиноновое соединение, которое представляет собой совершенно новый ингибитор Lp-PLA2. Он подходит для лечения нейродегенеративных заболеваний, таких как болезнь Альцгеймера (БА), глаукома и возрастная дегенерация желтого пятна (ВМД), или сердечно-сосудистых заболеваний, включая атеросклероз.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет дополнительно объяснено со ссылкой на следующие примеры. Следует понимать, что примеры, описанные в настоящем документе, предназначены только для иллюстрации настоящего изобретения и никоим образом не ограничивают объем настоящего изобретения.
Исходные вещества для настоящего описания могут быть синтезированы способами, известными в данной области техники, или могут быть приобретены в компаниях ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc., Darui Chemicals и других компаниях.
Если в примерах не указано иное, раствор относится к водному раствору.
Если в примерах не указано иное, температура реакции представляет собой комнатную температуру, например, от 20°С до 30°С.
Пример 1.
Получение Соединения 1
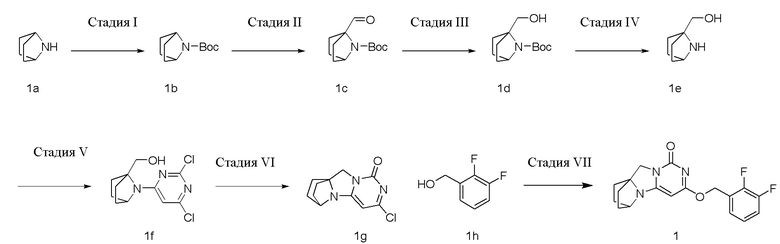
Стадия I: Получение Соединения 1b
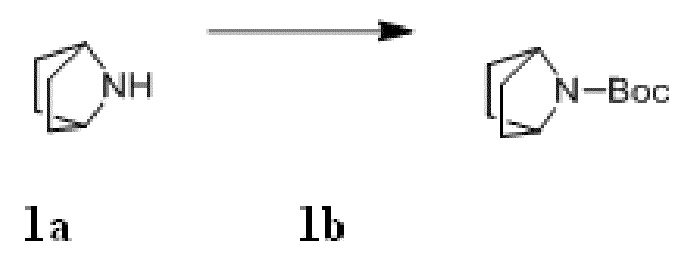
При комнатной температуре 7-азабицикло[2,2.1]гептан 1a (4 г, 41,1 ммоль) растворяли в 300 мл дихлорметана, затем добавляли триэтиламин (6,3 г, 61,8 ммоль) и ди-трет-бутилдикарбонат (13,5 г, 61,8 ммоль). Материалы перемешивали при комнатной температуре и проводили реакцию в течение 4 ч, и реагент подвергали экстракции дихлорметаном (100 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 20:1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 1b (10 г, выход: 100%). 1H ЯМР (400 МГц, CDCl3) δ 4,16 (s, 2H), 1,74 - 1,72 (m, 4H), 1,42 (s, 9H), 1,37-1,35 (m, 4H).
Стадия II: Получение соединения 1c
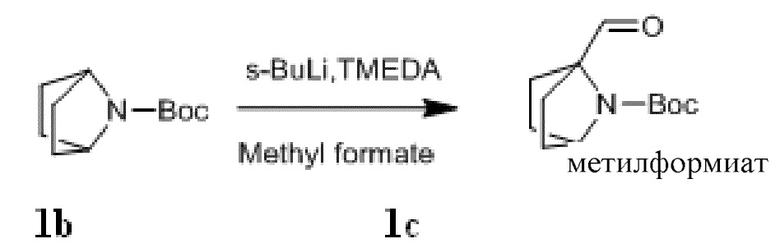
При комнатной температуре трет-бутил 7-азабицикло[2,2.1]гептан-7-карбоксилат 1b (10 г, 50,8 ммоль) и TMEDA (7,1 г, 60,9 ммоль) растворяли в 200 мл сухого эфира, затем по каплям добавляли раствор s-BuLi (1,3 M, 60,9 ммоль) в гексане при -65°C под азотной защитой. После того, как материалы перемешивали и реакция проходила в течение 30 мин, по каплям добавляли раствор метилформиата (3,65 г, 60,9 ммоль) в эфире. Эту смесь перемешивали и проводили реакцию при -65°C в течение 30 мин, нагревали до 0°C, и проводили реакцию в течение 2 ч. К реакционной смеси добавляли 100 мл насыщенного водного раствора хлорида аммония и экстрагировали эфиром (50 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 40:1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 1c (8,14 г, выход: 71%). 1H ЯМР (400 МГц, CDCl3) δ 9,93 (s, 1H), 4,29 (s, 1H), 2,03-1,88 (m, 4H), 1,65-1,46 (m, 4H), 1,42 (s, 9H).
Стадия III: Получение соединения 1d
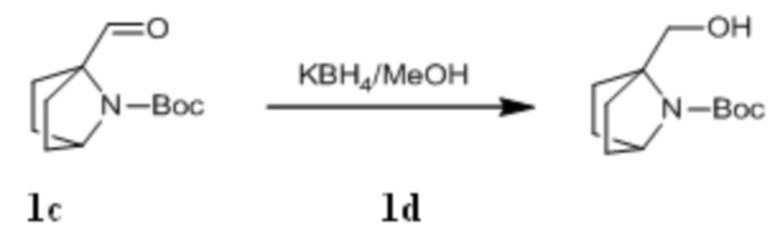
Трет-бутил 1-формил-7-азабицикло[2,2.1]гептан-7-карбоксилат 1c (8,14 г, 36,1 ммоль) растворяли в 300 мл метанола, к которому порциями добавляли KBH4 (3,03 г, 54,2 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 1 ч, реагент концентрировали при пониженном давлении и экстрагировали дихлорметаном (60 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 20/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 1d (6,53 г, выход: 79,5%). 1H ЯМР (400 МГц, CDCl3) δ4,96 (s, 1H), 4,24 (m, 1H), 3,90 (m, 2H), 1,89-1,77 (m, 4H), 1,48-1,35 (m, 13H).
Стадия IV: Получение соединения 1e
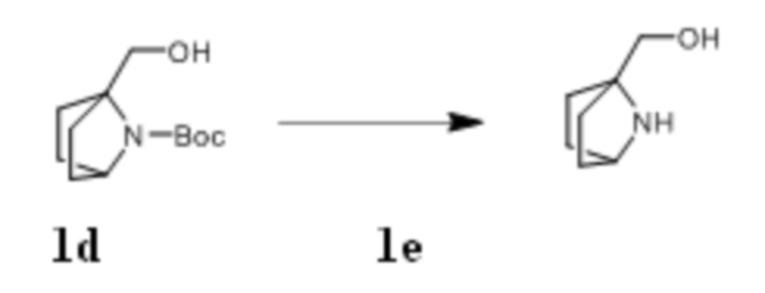
Трет-бутил 1-(гидроксиметил)-7-азабицикло[2,2.1]гептан-7-карбоксилат 1d (6,53 г, 28,7 ммоль) растворяли в 100 мл соляной кислоты/этанола (2M). После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 4 ч, реагент концентрировали при пониженном давлении , получая твердый продукт желтого цвета 1e (4,72 г, выход: 100%).
1H ЯМР (400 МГц, ДМСО) δ 9,04 (s, 2H), 3,98 (m, 1H), 3,73 (s, 2H), 1,98-1,82 (m, 4H), 1,69-1,53 (m, 4H).
Стадия V: Получение соединения 1f
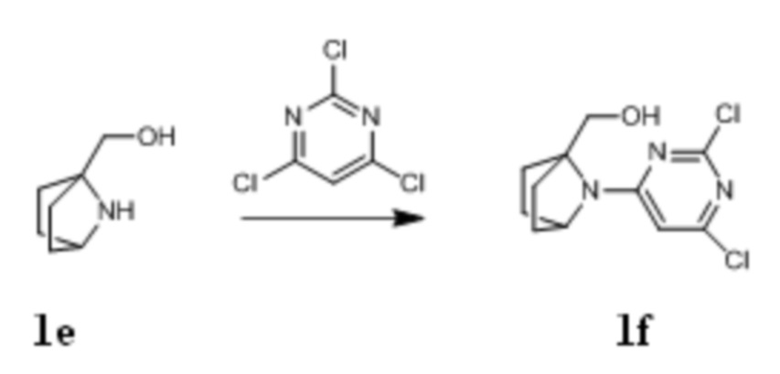
При комнатной температуре (7-азабицикло[2,2.1]гептан-1-ил)метанол 1e (4,5 г, 35,5 ммоль), 2,4,6-трихлорпиримидин (7,79 г, 42,6 ммоль), и диизопропилэтиламин (13,7 г, 106,5 ммоль) растворяли в 200 мл ацетонитрила, перемешивали в течение ночи, концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 5:1) посредством колоночной хроматографии на силикагеле, получая бесцветный твердый продукт 1f (5,2 г, выход: 53%).
1H ЯМР (400 МГц, CDCl3) δ 6,37 (s, 1H), 4,91 (m, 1H), 4,26 (m, 1H), 4,01 (m, 2H), 2,01-1,82 (m, 4H), 1,68-1,52 (m, 4H).
Стадия VI: Получение соединения 1g
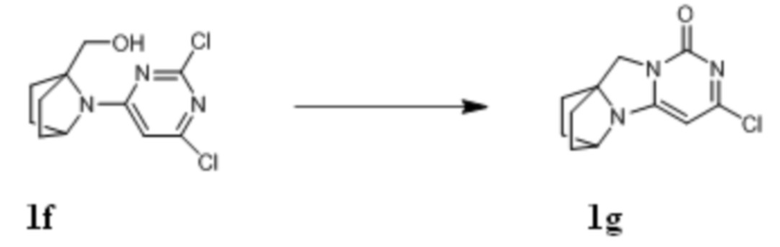
При комнатной температуре (7-(2,6-дихлорпиримидин-4-ил)-7-азабицикло[2,2.1]гептан-1-ил)метанол 1f (5,46 г, 19,9 ммоль) и триэтиламин (6,0 г, 59,7 ммоль) растворяли в 100 мл сухого дихлорметана, к по каплям добавляли которому MsCl (2,5 г, 21,9 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при 0°C в течение 1 ч, реагент концентрировали при пониженном давлении, и растворяли в 100 мл смешанного растворителя диоксан/вода (1:1), к которому добавляли карбонат калия (8,2 г, 59,7 ммоль), и перемешивали при 90°C в течение ночи. Затем реагент концентрировали при пониженном давлении и очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая бесцветный твердый продукт 1g (2,2 г, выход: 46%).
1H ЯМР (400 МГц, ДМСО) δ 6,10 (s, 1H), 4,42 (m, 1H), 3,95 (s, 2H), 1,94-1,89 (m, 2H), 1,78-1,69 (m, 6H).
Стадия VII: Получение соединения 1
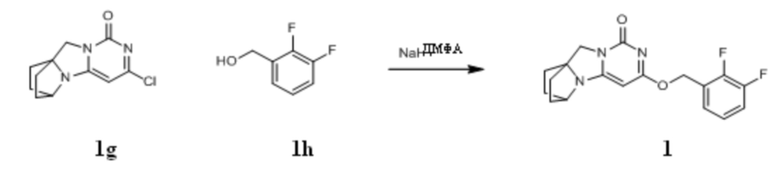
(2,3-Дифторфенил)метанол 1h (46 мг, 0,32 ммоль) растворяли в 2 мл сухого N,N-диметилформамида в течение 1 ч, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, Соединение 1g (50 мг, 0,21 ммоль) добавляли, перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 1 (31 мг, выход: 43%).
1H ЯМР (400 МГц, CDCl3) δ 7,24-7,03 (m, 3H), 5,45 (s, 2H), 5,15 (s, 1H), 4,09 (m, 1H), 3,99 (s, 2H), 2,14-1,99 (m, 2H), 1,78-1,66 (m, 6H). МС (ИЭР): m/z 346,0 [M+H]+.
Пример 2 Получение соединения 2
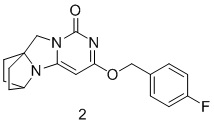
(4-Фторфенил)метанол (40 мг, 0,32 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 2 (40 мг, выход: 58%).
1H ЯМР (400 МГц, CDCl3) δ 7,43-7,40 (m, 2H), 7,08-7,04 (m, 2H), 5,37 (s, 2H), 5,17 (s, 1H), 4,11 (m, 1H), 4,01 (s, 2H), 2,10-2,06 (m, 2H), 1,82-1,67 (m, 6H). МС (ИЭР): m/z 328,0 [M+H]+.
Пример 3 Получение соединения 3
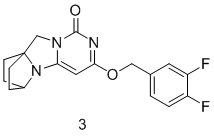
(3,4-дихлорфенил)метанол (46 мг, 0,32 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 3 (17 мг, выход: 23%).
1H ЯМР (400 МГц, CDCl3) δ 7,28-7,22 (m, 1H), 7,18-7,11 (m, 2H), 5,35 (s, 2H), 5,17 (s, 1H), 4,12 (m, 1H), 4,01 (s, 2H), 2,12- 2,07 (m, 2H), 1,83- 1,74 (m, 6H). МС (ИЭР): m/z 346,0 [M+H]+.
Пример 4 Получение соединения 4
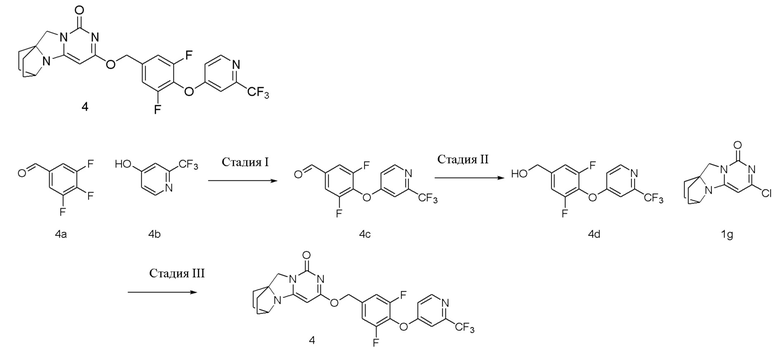
Стадия I: Получение соединения 4c
При комнатной температуре 2-(трифторметил)пиридин-4-ол 4b (0,85 г, 5,2 ммоль), 3,4,5-трифторбензальдегид (1 г, 6,2 ммоль) и карбонат калия (0,93 г, 6,76 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 1 ч. Реагент охлаждали до комнатной температуры, затем вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 5/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 4c (1,47 г, выход: 78,2%).
1H ЯМР (400 МГц, CDCl3) δ 9,97 (s, 1H), 8,65 (m, 1H), 7,63 (m, 2H), 7,27 (m, 1H), 7,01 (m, 1H).
Стадия II: Получение соединения 4d
При комнатной температуре 3,5-дифтор-4-((2-(трифторметил)пиридин-4-ил)окси)бензальдегид 4c (1,47 г, 4,85 ммоль) растворяли в 50 мл этанола, к добавляли которому NaBH4 (184 мг, 4,85 ммоль) при 0°C. Материалы перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 2:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 4d (1,04 г, выход: 70,3%).
1H ЯМР (400 МГц, CDCl3) δ 8,59 (m, 1H), 7,24 (m, 1H), 7,11 (m, 2H), 6,99 (m, 1H), 4,75 (m, 2H), 2,19 (m, 1H).
Стадия III: Получение соединения 4
(3,5-Дифтор-4-((2-(трифторметил)пиридин-4-ил)окси)фенил)метанол (77 мг, 0,25 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 5 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 4 (35 мг, выход: 33%).
1H ЯМР (400 МГц, CDCl3) δ 8,62 (m, 1H), 7,28 (s, 1H), 7,15 (m, 2H), 7,01 (m, 1H), 5,44 (s, 2H), 5,24 (s, 1H), 4,16 (m, 1H), 4,03 (s, 2H), 2,12-2,07 (m, 2H), 1,83-1,79 (m, 6H). МС (ИЭР): m/z 506,9 [M+H]+.
Пример 5 Получение соединения 5
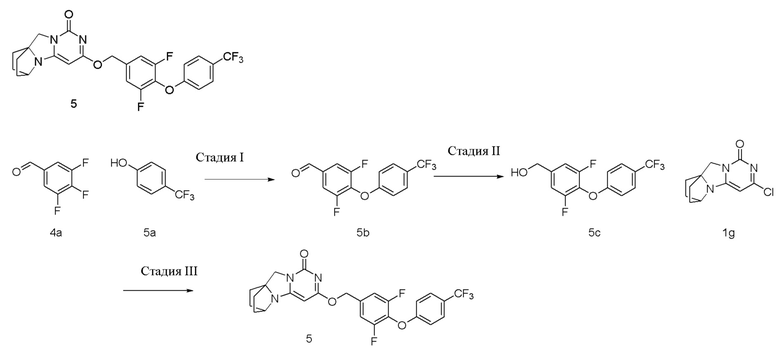
Стадия I: Получение соединения 5b
При комнатной температуре 4-(трифторметил)фенол 5a (0,84 г, 5,2 ммоль), 3,4,5-трифторбензальдегид 4a (1 г, 6,2 ммоль) и карбонат калия (0,93 г, 6,76 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 1 ч. Реагент охлаждали до комнатной температуры, затем вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 5/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 5b (1,33 г, выход: 71,0%).
1H ЯМР (400 МГц, CDCl3) δ 9,94 (m, 1H), 7,59 (m, 4H), 7,04 (m, 2H).
Стадия II: Получение соединения 5c
При комнатной температуре 3,5-дифтор-4-(4-(трифторметил)фенокси)бензальдегид 5b (1,33 г, 4,4 ммоль) растворяли в 50 мл метилэтанола, к которому добавляли NaBH4 (166 мг, 4,4 ммоль) при 0°C. Материалы перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 2/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 5c (0,85 г, выход: 63,6 %).
1H ЯМР (400 МГц, CDCl3) δ 7,57 (m, 2H), 7,09-7,00 (m, 4H), 4,72 (m, 2H), 2,03 (m, 1H).
Стадия III: Получение соединения 5
3,5-Дифтор-4-(4-(трифторметил)фенокси)фенил)метанол 5c (64 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 5 (17 мг, выход: 16%).
1H ЯМР (400 МГц, CDCl3) δ 7,59 (m, 2H), 7,12-7,02 (m, 4H), 5,42 (s, 2H), 5,23 (s, 1H), 4,15 (m, 1H), 4,03 (s, 2H), 2,12 (m, 2H), 1,87-1,74 (m, 6H). МС (ИЭР): m/z 506,0 [M+H]+.
Пример 6 Получение соединения 6
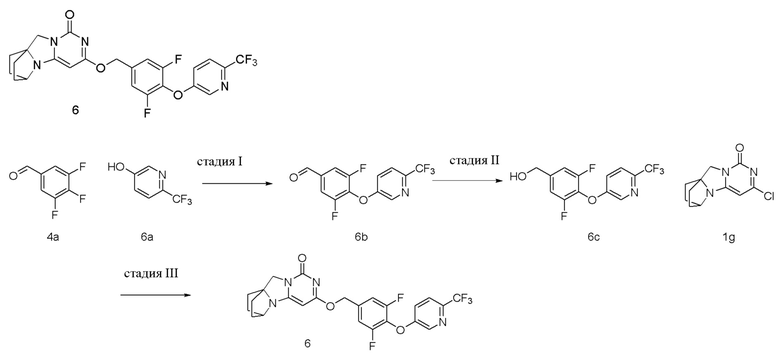
Стадия I: Получение соединения 6b
При комнатной температуре 6-(трифторметил)пиридин-3-ол 6a (0,85 г, 5,2 ммоль), 3,4,5-трифторбензальдегид 4a (1 г, 6,2 ммоль) и карбонат калия (0,93 г, 6,76 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 1 ч. Реагент охлаждали до комнатной температуры, затем вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 5/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 6b (1,34 г, выход: 84,6 %).
Стадия II: Получение соединения 6c
При комнатной температуре 3,5-дифтор-4-((6-(трифторметил)пиридин-3-ил)окси)бензальдегид 6b (1,34 г, 4,4 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (167 мг, 4,4 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 2:1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 6c (0,77 г, выход: 57,4 %).
1H ЯМР (400 МГц, CDCl3) δ 8,46 (s, 1H), 7,63 (m, 1H), 7,30 (m, 1H), 7,09 (m, 2H), 4,73 (m, 2H), 2,40 (m, 1H).
Стадия III: Получение соединения 6
(3,5-Дифтор-4-((6-(трифторметил)пиридин-3-ил)окси)фенил)метанол 6c (50 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 6 (31 мг, выход: 29%).
1H ЯМР (400 МГц, CDCl3) δ 8,51 (m, 1H), 7,65 (m, 1H), 7,31 (m, 1H), 7,13 (m, 2H), 5,42 (s, 2H), 5,23 (s, 1H), 4,15 (m, 1H), 4,03 (s, 2H), 2,12-2,06 (m, 2H), 1,85-1,70 (m, 6H). МС (ИЭР): m/z 507,0 [M+H]+.
Пример 7 Получение соединения 7
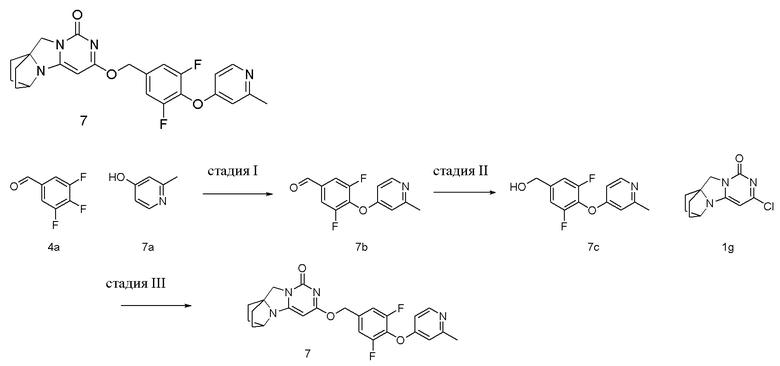
Стадия I: Получение соединения 7b
При комнатной температуре 6-метилпиридин-4-ол 7a (0,5 г, 4,6 ммоль), 3,4,5-трифторбензальдегид 4a (0,88 г, 5,5 ммоль) и карбонат калия (0,823 г, 5,95 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. Реагент охлаждали до комнатной температуры, затем вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 7b (0,4 г, выход: 33,8 %).
1H ЯМР (400 МГц, CDCl3) δ 9,94 (s, 1H), 8,39 (m, 1H), 7,62-7,56 (m, 2H), 6,70-6,66 (m, 2H), 2,52 (s, 3H).
Стадия II: Получение соединения 7c
При комнатной температуре 3,5-дифтор-4-((2-метилпиридин-4-ил)окси)бензальдегид 7b (0,4 г, 1,86 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (71 мг, 1,86 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 7c (0,40 г, выход: 85,7 %).
1H ЯМР (400 МГц, CDCl3) δ 8,29 (m, 1H), 7,07 (m, 2H), 6,70-6,64 (m, 2H), 4,73 (s, 2H), 3,20 (m, 1H), 2,50 (s, 3H).
Стадия III: Получение соединения 7
(3,5-Дифтор-4-((2-метилпиридин-4-ил)окси)фенил)метанол 7c (53 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 7 (17 мг, выход: 18%).
1H ЯМР (400 МГц, CDCl3) δ 8,38 (m, 1H), 7,10 (m, 2H), 6,69 (m, 2H), 5,42 (s, 2H), 5,23 (s, 1H), 4,15 (m, 1H), 4,03 (s, 2H), 2,53 (s, 3H), 2,21-2,07 (m, 2H), 1,88-1,70 (m, 6H). МС (ИЭР): m/z 453,0 [M+H]+.
Пример 8 Получение соединения 8
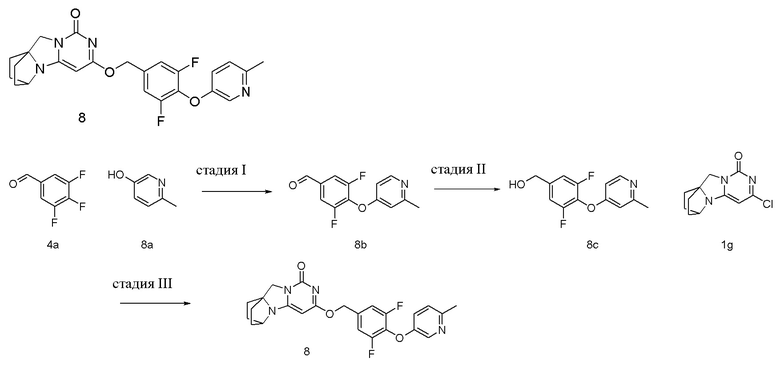
Стадия I: Получение соединения 8b
При комнатной температуре 6-метилпиридин-3-ол 8a (0,57 г, 5,2 ммоль), 3,4,5-трифторбензальдегид 4a (1 г, 6,2 ммоль) и карбонат калия (0,93 г, 6,76 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 1 ч. Реагент охлаждали до комнатной температуры, затем вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 8b (0,91 г, выход: 69,4 %).
1H ЯМР (400 МГц, CDCl3) δ 9,92 (s, 1H), 8,28 (s, 1H), 7,62-7,49 (m, 2H), 7,18-7,10 (m, 2H), 2,54 (s, 3H).
Стадия II: Получение соединения 8c
При комнатной температуре 3,5-дифтор-4-((2-метилпиридин-4-ил)окси)бензальдегид 8b (0,91 г, 4,3 ммоль) растворяли в 50 мл метанола, к которому NaBH4 (161 мг, 4,3 ммоль) добавляли при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 8c (0,89 г, выход: 82,5 %).
1H ЯМР (400 МГц, CDCl3) δ 8,20 (m, 1H), 7,16-6,98 (m, 4H), 4,69 (m, 2H), 2,88 (m, 1H), 2,50 (s, 3H).
Стадия III: Получение соединения 8
(3,5-Дифтор-4-((2-метилпиридин-4-ил)окси)фенил)метанол 8c (53 мг, 0,21 ммоль) растворяли в 2 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 8 (20 мг, выход: 21%).
1H ЯМР (400 МГц, CDCl3) δ 8,30 (m, 1H), 7,18-7,04 (m, 4H), 5,40 (s, 2H), 5,22 (s, 1H), 4,15 (m, 1H), 4,03 (s, 2H), 2,54 (s, 3H), 2,11 (m, 2H), 1,86-1,74 (m, 6H). МС (ИЭР): m/z 453,0 [M+H]+.
Пример 9 Получение соединения 9
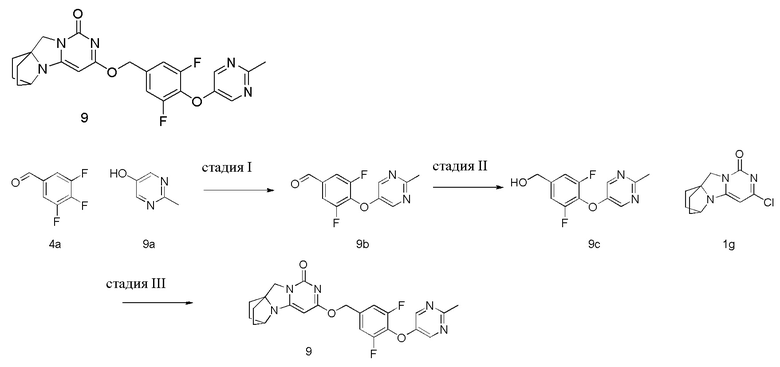
Стадия I: Получение соединения 9b
При комнатной температуре 2-метилпиримидин-5-ол 9a (0,25 г, 2,27 ммоль), 3,4,5-трифторбензальдегид 4a (0,44 г, 2,72 ммоль) и карбонат калия (0,41 г, 2,95 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. После охлаждения реагент вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 9b (0,24 г, выход: 34,8 %).
1H ЯМР (400 МГц, CDCl3) δ 9,93 (s, 1H), 8,39 (s, 2H), 7,64-7,54 (m, 2H), 2,72 (s, 3H).
Стадия II: Получение соединения 9c
При комнатной температуре 3,5-дифтор-4-((2-метилпиримидин-5-ил)окси)бензальдегид 9b (0,24 г, 0,79 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (30 мг, 0,79 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4:1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 9c (0,17 г, выход: 85,4 %).
1H ЯМР (400 МГц, CDCl3) δ 8,33 (s, 2H), 7,04 (m, 2H), 4,71 (m, 2H), 2,70 (s, 3H).
Стадия III: Получение соединения 9
(3,5-Дифтор-4-((2-метилпиримидин-5-ил)окси)фенил)метанол 9c (53 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 9 (18 мг, выход: 19%).
1H ЯМР (400 МГц, CDCl3) δ 8,34 (s, 2H), 7,09 (m, 2H), 5,37 (s, 2H), 5,20 (s, 1H), 4,12 (m, 1H), 3,99 (s, 2H), 2,69 (s, 3H), 2,13-2,03 (m, 2H), 1,81-1,71 (m, 6H). МС (ИЭР): m/z 454,0 [M+H]+.
Пример 10 Получение соединения 10
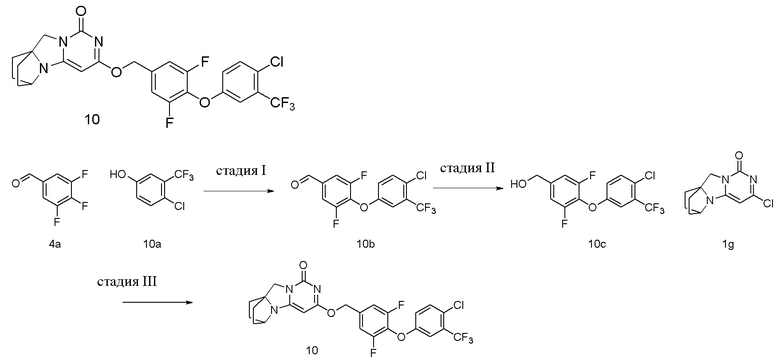
Стадия I: Получение соединения 10b
При комнатной температуре 4-хлор-3-(трифторметил)фенол 10a (0,5 г, 2,54 ммоль), 3,4,5-трифторбензальдегид 4a (0,45 г, 2,8 ммоль) и карбонат калия (0,46 г, 3,3 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. После охлаждения реагент вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 10b (0,6 г, выход: 70,1 %).
1H ЯМР (400 МГц, CDCl3) δ 9,94 (s, 1H), 7,64-7,55 (m, 2H), 7,45 (m, 1H), 7,31 (m, 1H), 7,05 (m, 1H).
Стадия II: Получение соединения 10c
При комнатной температуре 4-(4-хлор-3-(трифторметил)фенокси)-3,5-дифторбензальдегид 10b (0,6 г, 1,78 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (67 мг, 1,78 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 10c (0,28 г, выход: 46,4 %).
1H ЯМР (400 МГц, CDCl3) δ 7,41 (m, 1H), 7,28 (m, 1H), 7,08-7,00 (m, 3H), 4,73 (m, 2H), 1,94 (m, 1H).
Стадия III: Получение соединения 10
(4-(4-Хлор-3-(трифторметил)фенокси)-3,5-дифторфенил)метанол 10c (71 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 10 (28 мг, выход: 25%).
1H ЯМР (400 МГц, CDCl3) δ 7,41 (m, 1H), 7,29 (m, 1H), 7,08 (m, 2H), 7,01 (m, 1H), 5,38 (s, 2H), 5,20 (s, 1H), 4,13 (m, 1H), 4,00 (s, 2H), 2,19-2,08 (m, 2H), 1,84-1,74 (m, 6H). МС (ИЭР): m/z 539,9 [M+H]+.
Пример 11 Получение соединения 11
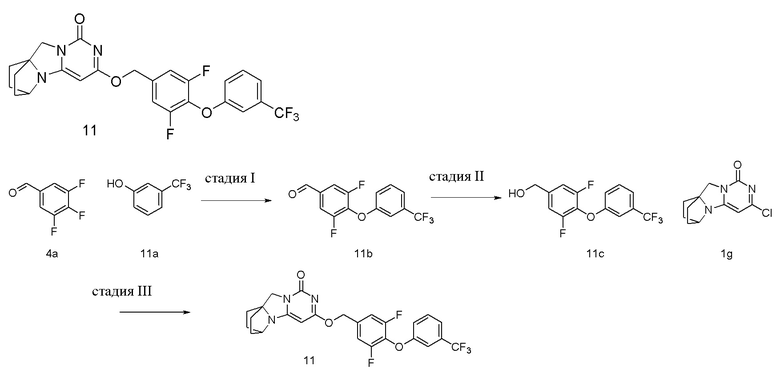
Стадия I: Получение соединения 11b
При комнатной температуре 3-(трифторметил)фенол 11a (1 г, 6,17 ммоль), 3,4,5-трифторбензальдегид 4a (1,09 г, 6,79 ммоль) и карбонат калия (1,1 г, 8,02 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. После охлаждения реагент вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 11b (1,7 г, выход: 82,5 %).
1H ЯМР (400 МГц, CDCl3) δ 9,94 (s, 1H), 7,63-7,55 (m, 2H), 7,46 (m, 1H), 7,39 (m, 1H), 7,21 (s, 1H), 7,13 (m, 1H).
Стадия II: Получение соединения 11c
При комнатной температуре 3,5-дифтор-4-(3-(трифторметил)фенокси)бензальдегид 11b (1,7 г, 5,6 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (213 мг, 5,6 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 11c (1,27 г, выход: 74,5 %).
Стадия III: Получение соединения 11
(3,5-Дифтор-4-(3-(трифторметил)фенокси)фенил)метанол 11c (71 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 11 (33 мг, выход: 31%).
1H ЯМР (400 МГц, CDCl3) δ 7,44 (m, 1H), 7,36 (m, 1H), 7,22 (s, 1H), 7,11 (m, 3H), 5,42 (s, 2H), 5,23 (s, 1H), 4,15 (m, 1H), 4,03 (s, 2H), 2,12 (m, 2H), 1,83-1,77 (m, 6H). МС (ИЭР): m/z 505,9 [M+H]+.
Пример 12 Получение соединения 12
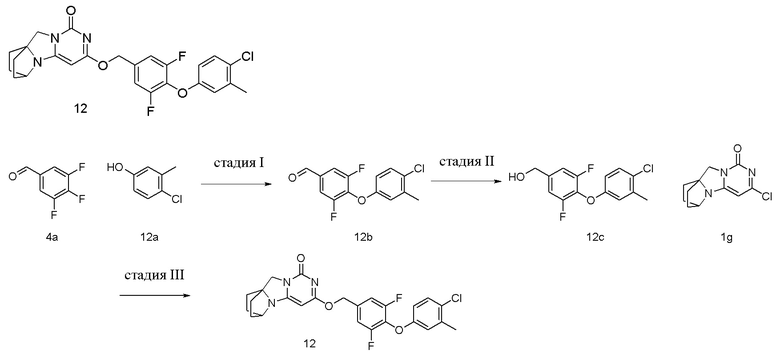
Стадия I: Получение соединения 12b
При комнатной температуре 4-хлор-3-метилфенол 12a (1 г, 7,0 ммоль), 3,4,5-трифторбензальдегид 4a (1,2 г, 7,7 ммоль) и карбонат калия (1,3 г, 9,1 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. После охлаждения реагент вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 12b (1,2 г, выход: 54,5 %).
1H ЯМР (400 МГц, CDCl3) δ 9,92 (s, 1H), 7,61-7,51 (m, 2H), 7,30-7,23 (m, 1H), 6,85 (m, 1H), 6,73 (m, 1H), 2,34 (s, 3H).
Стадия II: Получение соединения 12c
При комнатной температуре 4-(4-хлор-3-метилфенокси)-3,5-дифторбензальдегид 12b (1,2 г, 4,2 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (161 мг, 4,2 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 12c (0,89 г, выход: 74,4 %).
Стадия III: Получение соединения 12
4-(4-Хлор-3-метилфенокси)-3,5-дифторфенил)метанол 12c (59 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 12 (29 мг, выход: 31%).
1H ЯМР (400 МГц, CDCl3) δ 7,25 (m, 1H), 7,08 (m, 2H), 6,84 (m, 1H), 6,73 (m, 1H), 5,40 (s, 2H), 5,22 (s, 1H), 4,15 (m, 1H), 4,03 (s, 2H), 2,35 (s, 3H), 2,12 (m, 2H), 1,87-1,74 (m, 6H). МС (ИЭР): m/z 486,0 [M+H]+.
Пример 13: Получение соединения 13
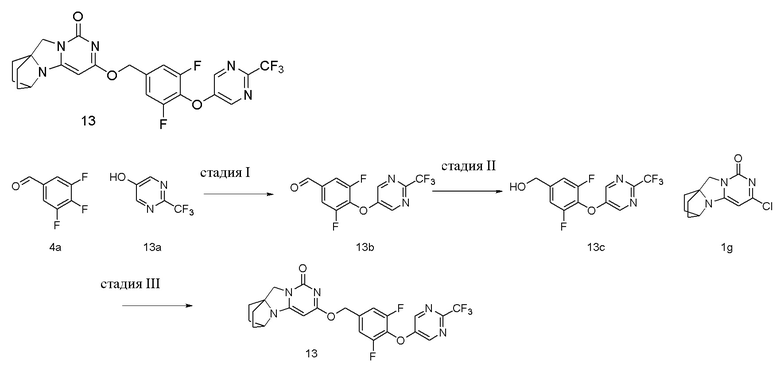
Стадия I: Получение соединения 13b
При комнатной температуре 2-(трифторметил)пиримидин-5-ол 13a (0,25 г, 1,52 ммоль), 3,4,5-трифторбензальдегид 4a (0,29 г, 1,83 ммоль) и карбонат калия (0,27 г, 1,98 ммоль) растворяли в 20 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. После охлаждения реагент вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 13b (0,24 г, выход: 52,0 %).
1H ЯМР (400 МГц, CDCl3) δ 9,97 (s, 1H), 8,59 (s, 2H), 7,69-7,54 (m, 2H).
Стадия II: Получение соединения 13c
При комнатной температуре 3,5-дифтор-4-((2-(трифторметил)пиримидин-5-ил)окси)бензальдегид 13b (0,24 г, 0,79 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (30 мг, 0,79 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 13c (0,12 г, выход: 49,6 %).
1H ЯМР (400 МГц, CDCl3) δ 8,54 (s, 2H), 7,12 (m, 2H), 4,74 (m, 2H), 2,23 (m, 1H).
Стадия III: Получение соединения 13
(3,5-Дифтор-4-((2-(трифторметил)пиримидин-5-ил)окси)фенил)метанол 13c (64 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 13 (18 мг, выход: 17%).
1H ЯМР (400 МГц, CDCl3) δ 8,58 (s, 2H), 7,17 (m, 2H), 5,43 (s, 2H), 5,24 (s, 1H), 4,16 (m, 1H), 4,03 (s, 2H), 2,13 (m, 2H), 1,88-1,76 (m, 6H).
МС (ИЭР): m/z 508,1 [M+H]+.
Пример 14: Получение соединения 14
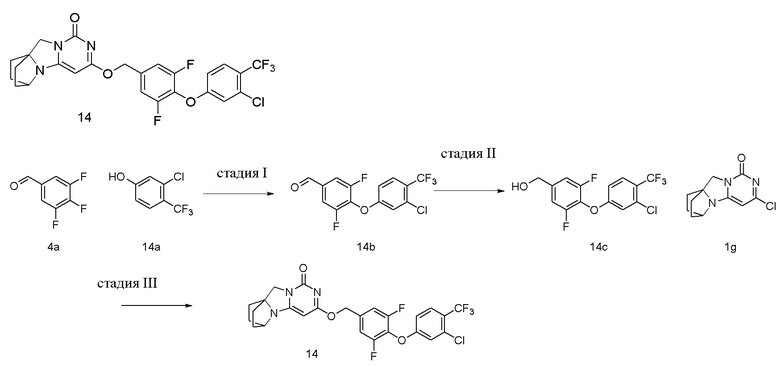
Стадия I: Получение соединения 14b
При комнатной температуре 3-хлор-4-(трифторметил)фенол 14a (0,25 г, 1,27 ммоль), 3,4,5-трифторбензальдегид 4a (0,22 г, 1,4 ммоль) и карбонат калия (0,23 г, 1,65 ммоль) растворяли в 20 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. После охлаждения реагент вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 14b (0,32 г, выход: 74,0 %).
1H ЯМР (400 МГц, CDCl3) δ 9,95 (s, 1H), 7,69-7,56 (m, 3H), 7,10 (m, 1H), 6,92 (m, 1H).
Стадия II: Получение соединения 14c
При комнатной температуре 4-(3-хлор-4-(трифторметил)фенокси)-3,5-дифторбензальдегид 14b (0,32 г, 0,94 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (36 мг, 0,94 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 14c (0,15 г, выход: 47,1 %).
1H ЯМР (400 МГц, CDCl3) δ 7,62 (m, 1H), 7,13-7,00 (m, 3H), 6,90 (m, 1H), 4,74 (m, 2H), 1,88 (m, 1H).
Стадия III: Получение соединения 14
(4-(3-Хлор-4-(трифторметил)фенокси)-3,5-дифторфенил)метанол 14c (71 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 14 (12 мг, выход: 10%).
1H ЯМР (400 МГц, CDCl3) δ 7,64 (m, 1H), 7,10 (m, 3H), 6,91 (m, 1H), 5,41 (s, 2H), 5,23 (s, 1H), 4,15 (m, 1H), 4,03 (s, 2H), 2,19-2,04 (m, 2H), 1,83-1,75 (m, 6H). МС (ИЭР): m/z 539,9 [M+H]+.
Пример 15 Получение соединения 15
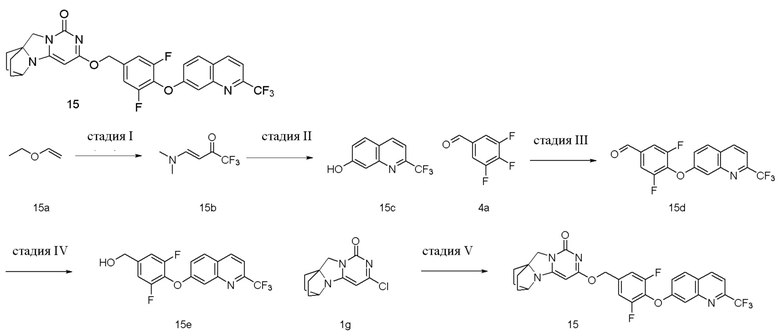
Стадия I: Получение соединения 15b
Этоксиэтилен 15a (1 г, 13,9 ммоль) и пиридин (1,65 г, 20,9 ммоль) растворяли в 20 мл дихлорметана, и добавляли трифторуксусный ангидрид при 0°C под азотной защитой. Материалы перемешивали и проводили реакцию в течение 20 мин, нагревали до комнатной температуры, и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до -20°C, к которой по каплям добавляли диметиламин в течение 10 мин, нагревали снова до комнатной температуры, и проводили реакцию в течение 2 ч. Реакцию гасили водой. Реагент подвергали экстракции дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 15b (540 мг, выход: 23,1%).
1H ЯМР (400 МГц, CDCl3) δ 7,85 (m, 1H), 5,26 (m, 1H), 3,21 (s, 3H), 2,95 (s, 3H).
Стадия II: Получение соединения 15c
При комнатной температуре (E)-4-(диметиламино)-1,1,1-трифторбут-3-ен-2-он 15b (0,54 г, 2,98 ммоль) растворяли в 20 мл дихлорэтана, и раствор Tf2O в дихлорэтане (5 мл) по каплям добавляли при 0°C, с последующим добавлением раствора 3-аминофенол в дихлорэтане (10 мл). Материалы перемешивали и проводили реакцию при 40°C в течение 2 ч и фильтровали , получая твердый продукт белого цвета 15c (275 мг, 57,4%).
1H ЯМР (400 МГц, MeOD) δ 8,43 (m, 1H), 7,90 (m, 1H), 7,62 (m, 1H), 7,40 (m, 1H), 7,32 (m, 1H).
Стадия III: Получение соединения 15d
При комнатной температуре 2-(трифторметил)хинолин-7-ол 15c (0,37 г, 1,71 ммоль), 3,4,5-трифторбензальдегид 4a (0,28 г, 1,71 ммоль) и карбонат калия (0,71 г, 5,13 ммоль) растворяли в 20 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. После охлаждения реагент вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 5/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 15d (310 мг, 51,5%).
1H ЯМР (400 МГц, CDCl3) δ 9,97 (s, 1H), 8,36 (m, 1H), 7,96 (m, 1H), 7,69 (m, 1H), 7,62 (m, 3H), 7,40 (s, 1H).
Стадия IV: Получение соединения 15e
При комнатной температуре 3,5-дифтор-4-((2-(трифторметил)хинолин-7-ил)окси)бензальдегид 15d (0,31 г, 0,88 ммоль) растворяли в 20 мл метанола, к которому добавляли NaBH4 (33 мг, 0,88 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 15e (230 мг, 74,6%).
Стадия V: Получение соединения 15
(3,5-Дифтор-4-((2-(трифторметил)хинолин-7-ил)окси)фенил)метанол 15e (75 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции и проводили очистку с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 15 (18 мг, выход: 17%).
1H ЯМР (400 МГц, CDCl3) δ 8,33 (m, 1H), 7,92 (m, 1H), 7,65 (m, 1H), 7,60 (m, 1H), 7,36 (s, 1H), 7,13 (m, 2H), 5,40 (s, 2H), 5,23 (s, 1H), 4,14 (m, 1H), 4,02 (s, 2H), 2,11 (m, 2H), 1,79 (m, 6H). МС (ИЭР): m/z 557,0 [M+H]+.
Пример 16 Получение соединения 16
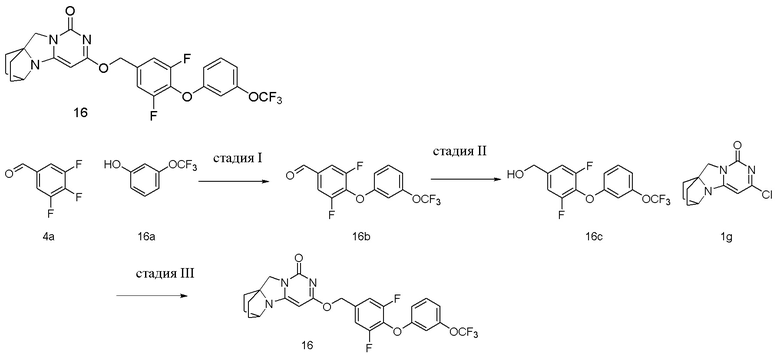
Стадия I: Получение соединения 16b
При комнатной температуре 3-(трифторметокси)фенол 16a (0,50 г, 2,8 ммоль), 3,4,5-трифторбензальдегид 4a (0,5 г, 2,8 ммоль) и карбонат калия (0,5 г, 3,64 ммоль) растворяли в 30 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. После охлаждения реагент вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 16b (0,73 г, выход: 81,4 %).
1H ЯМР (400 МГц, CDCl3) δ 9,94 (s, 1H), 7,64-7,54 (m, 2H), 7,34 (m, 1H), 7,00 (m, 1H), 6,87 (m, 2H).
Стадия II: Получение соединения 16c
При комнатной температуре 4-(3-(трифторметокси)фенокси)-3,5-дифторбензальдегид 16b (0,73 г, 2,28 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (86 мг, 2,28 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 16c (0,57 г, выход: 71,2 %).
1H ЯМР (400 МГц, CDCl3) δ 7,30 (m, 1H), 7,06 (m, 2H), 6,94 (m, 1H), 6,85 (m, 1H), 6,81 (s, 1H), 4,72 (m, 2H), 1,94 (m, 1H).
Стадия III: Получение соединения 16
4-(3-(Трифторметокси)фенокси)-3,5-дифторфенил)метанол 16c (67 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 16 (25 мг, выход: 23%).
1H ЯМР (400 МГц, CDCl3) δ 7,29 (m, 1H), 7,07 (m, 2H), 6,93 (m, 1H), 6,83 (m, 2H), 5,38 (s, 2H), 5,20 (s, 1H), 4,12 (m, 1H), 4,00 (s, 2H), 2,08 (m, 2H), 1,83-1,73 (m, 6H). МС (ИЭР): m/z 522,0 [M+H]+.
Пример 17 Получение соединения 17
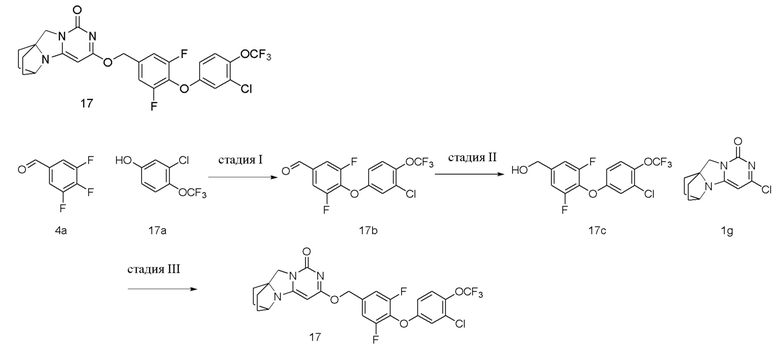
Стадия I: Получение соединения 17b
При комнатной температуре 3-хлор-4-(трифторметокси)фенол 17a (0,50 г, 2,34 ммоль), 3,4,5-трифторбензальдегид 4a (0,41 г, 2,58 ммоль) и карбонат калия (0,42 г, 3,04 ммоль) растворяли в 20 мл N,N-диметилформамида, перемешивали и проводили реакцию при 90°C в течение 2 ч. После охлаждения реагент вливали в 100 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт желтого цвета 17b (0,62 г, выход: 74,4 %).
1H ЯМР (400 МГц, CDCl3) δ 9,94 (s, 1H), 7,63-7,54 (m, 2H), 7,29 (m, 1H), 7,07 (m, 1H), 6,90 (m, 1H).
Стадия II: Получение соединения 17c
При комнатной температуре 4-(3-хлор-4-(трифторметокси)фенокси)-3,5-дифторбензальдегид 17b (0,62 г, 1,94 ммоль) растворяли в 50 мл метанола, к которому добавляли NaBH4 (62 мг, 1,94 ммоль) при 0°C, перемешивали и проводили реакцию при комнатной температуре в течение 0,5 ч, концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 17c (0,53 г, выход: 77,0 %).
1H ЯМР (400 МГц, CDCl3) δ 7,25 (m, 1H), 7,06 (m, 2H), 7,01 (m, 1H), 6,87 (m, 1H), 4,72 (s, 2H), 2,04 (m, 1H).
Стадия III: Получение соединения 17
(4-(3-Хлор-4-(трифторметокси)фенокси)-3,5-дифторфенил)метанол 17c (75 мг, 0,21 ммоль) растворяли в 5 мл сухого N,N-диметилформамида, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,42 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 1g (50 мг, 0,21 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции и очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 17 (23 мг, выход: 20%).
1H ЯМР (400 МГц, CDCl3) δ 7,23 (s, 1H), 7,09 (m, 2H), 7,03 (m, 1H), 6,87 (m, 1H), 5,39 (s, 2H), 5,21 (s, 1H), 4,13 (m, 1H), 4,01 (s, 2H), 2,10 (m, 2H), 1,83-1,71 (m, 6H). МС (ИЭР): m/z 555,9 [M+H]+.
Пример 18 Получение соединения 18
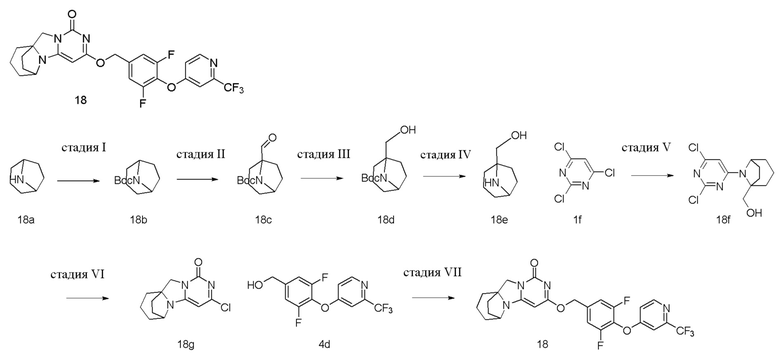
Стадия I: Получение соединения 18b
При комнатной температуре 8-азабицикло[3,2.1]октан 18a (5 г, 33,9 ммоль) и триэтиламин (5,2 г, 50,8 ммоль) растворяли в 300 мл дихлорметана, и (добавляли Boc)2O (11,1 г, 50,8 ммоль). Материалы перемешивали и проводили реакцию в течение 4 ч и экстрагировали дихлорметаном (100 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая бесцветный жидкий продукт 18b (9,4 г, 100%).
1H ЯМР (400 МГц, CDCl3) δ 4,14 (m, 2H), 1,97-1,86 (m, 2H), 1,81-1,53 (m, 6H), 1,46 (s, 9H), 1,41-1,35 (m, 2H).
Стадия II: Получение соединения 18c
При комнатной температуре трет-бутил 8-азабицикло[3,2.1]октан-8-карбоксилат 18b (7,2 г, 34,1 ммоль) и TMEDA (4,7 г, 40,9 ммоль) растворяли в 150 мл сухого эфира, и по каплям добавляли s-BuLi (1,3 M in гексан, 31,5 мл, 40,9 ммоль) при -65°C под азотной защитой. После перемешивания и проведения реакции при -65°C в течение 30 мин, по каплям добавляли раствор метилформиата (2,45 г, 40,9 ммоль) в эфире (20 мл). После перемешивания и проведения реакции при -65°C в течение 30 мин, реагент нагревали до 0°C, перемешивали и проводили реакцию в течение 2 ч, добавляли насыщенный водный раствор хлорида аммония (100 мл) и экстрагировали эфиром (50 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10:1) посредством колоночной хроматографии на силикагеле, получая бесцветный жидкий продукт 18c (3,13 г, 38,4%).
1H ЯМР (400 МГц, CDCl3) δ 9,45 (s, 1H), 4,24 (m, 1H), 2,15 (s, 1H), 1,97 (m, 1H), 1,85 (m, 2H), 1,82-1,60 (m, 6H), 1,45 (s, 9H).
Стадия III: Получение соединения 18d
При комнатной температуре трет-бутил 1-формил-8-азабицикло[3,2.1]октан-8-карбоксилат 18c (3,13 г, 13,1 ммоль) растворяли в 100 мл сухого метанола, и порциями добавляли KBH4 (1,1 г, 19,6 ммоль) при 0°C. Материалы перемешивали и проводили реакцию при комнатной температуре в течение 1 ч, добавляли 40 мл воды и экстрагировали дихлорметаном (60 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая бесцветный жидкий продукт 18d (3,45 г, 100%).
1H ЯМР (400 МГц, CDCl3) δ 5,37 (m, 1H), 4,21 (m, 1H), 3,72-3,49 (m, 2H), 2,00-1,73 (m, 4H), 1,61 (m, 6H), 1,46 (s, 9H).
Стадия IV: Получение соединения 18e
При комнатной температуре трет-бутил 1-(гидроксиметил)-8-азабицикло[3,2.1]октан-8-карбоксилат 18d (3,13 г, 13,1 ммоль) растворяли в 60 мл смешанного растворителя HCl/этанол, перемешивали и проводили реакцию в течение 4 ч, и концентрировали при пониженном давлении, получая твердый продукт желтого цвета 18e (2,45 г, 100%).
1H ЯМР (400 МГц, ДМСО) δ 8,94 (s, 2H), 5,55 (s, 1H), 3,80 (m, 1H), 3,58-3,48 (m, 2H), 2,04 (m, 1H), 1,86-1,40 (m, 9H).
Стадия V: Получение соединения 18f
При комнатной температуре (8-азабицикло[3,2.1]окт-1-ил)метанол 18e (2,45 г, 17,35 ммоль), 2,4,6-трихлорпиримидин (3,49 г, 19,1 ммоль) и DIPЭА (0,42 г, 52,05 ммоль) растворяли в 100 мл ацетонитрила, перемешивали и проводили реакцию в течение ночи, концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 4:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 18f (1 г, 20,1%).
1H ЯМР (400 МГц, CDCl3) δ 6,41 (s, 1H), 5,43 (m, 1H), 4,30 (m, 1H), 3,83 (m, 1H), 3,72 (m, 1H), 2,13-1,52 (m, 10H).
Стадия VI: Получение соединения 18g
18f (1 г, 3,1 ммоль) и триэтиламин (0,94 г, 1,3 ммоль) растворяли в 50 мл сухого дихлорметана, и по каплям добавляли MsCl (0,4 г, 3,4 ммоль) при 0°C. После перемешивания и проведения реакции при 0°C в течение 1 ч, реагент концентрировали при пониженном давлении , получая a бесцветное маслянистое промежуточное соединение (1,1 г, 100%). Этот необработанный продукт и карбонат калия (1,28 г, 9,3 ммоль) растворяли в 60 мл смешанного раствора диоксан/вода (1/1), перемешивали и проводили реакцию при 90°C в течение ночи, концентрировали при пониженном давлении и очищали с помощью элюентной системы (дихлорметан/метанол = 20/1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 18g (0,4 г, 51%).
1H ЯМР (400 МГц, 1H ЯМР (400 МГц, CDCl3) δ 5,66 (s, 1H), 4,25 (m, 1H), 4,11 (m, 1H), 3,70 (m, 1H), 2,05-1,88 (m, 4H), 1,85-1,71 (m, 6H).
Стадия VII: Получение соединения 18
(3,5-Дифтор-4-((2-(трифторметил)пиридин-4-ил)окси)фенил)метанол 4d (92 мг, 0,3 ммоль) растворяли в 5 мл сухого ДМФА, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,4 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 18g (50 мг, 0,2 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 18 (49 мг, выход: 47%).
1H ЯМР (400 МГц, CDCl3) δ 8,60 (m, 1H), 7,26 (s, 1H), 7,13 (m, 2H), 6,98 (m, 1H), 5,41 (s, 2H), 5,19 (s, 1H), 4,24 (m, 1H), 4,07 (m, 1H), 3,70 (m, 1H), 2,05-1,87 (m, 3H), 1,73 (m, 7H). МС (ИЭР): m/z 521,2 [M+H]+.
Пример 19 Получение соединения 19
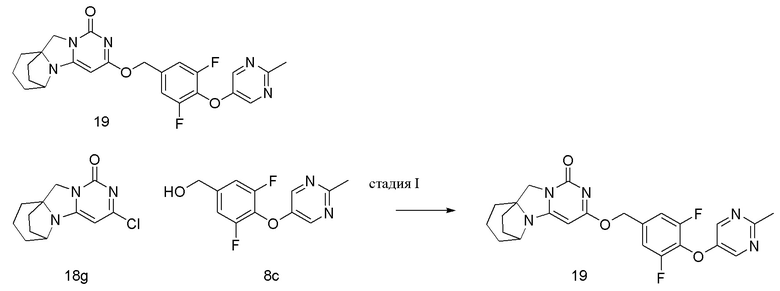
Стадия I: Получение соединения 19
(3,5-Дифтор-4-((2-метилпиридин-4-ил)окси)фенил)метанол 8c (53 мг, 0,21 ммоль) растворяли в 5 мл сухого ДМФА, к которому добавляли гидрид натрия (60% в минеральном масле, 17 мг, 0,4 ммоль) при 0°C. После того, как материалы перемешивали и проводили реакцию при комнатной температуре в течение 20 мин, добавляли Соединение 18g (50 мг, 0,2 ммоль), перемешивали и проводили реакцию в течение 1 ч. После этого добавляли небольшое количество воды для гашения реакции. Реагент очищали с помощью элюентной системы (дихлорметан/метанол = 20:1) посредством колоночной хроматографии на силикагеле, получая твердый продукт белого цвета 19 (52 мг, выход: 55,5%).
1H ЯМР (400 МГц, CDCl3) δ 8,38 (s, 2H), 7,10 (m, 2H), 5,41 (s, 2H), 5,20 (s, 1H), 4,23 (m, 1H), 4,06 (m, 1H), 3,68 (m, 1H), 2,70 (s, 3H), 2,00-1,90 (m, 3H), 1,85-1,69 (m, 7H). МС (ИЭР): m/z 468,2 [M+H]+.
Пример 20 Получение соединения 20
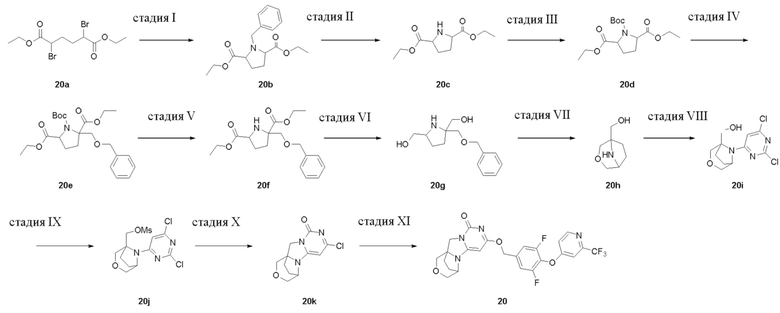
Стадия I: Получение соединения 20b
При комнатной температуре соединение диэтилмезо-2,5-дибромадипат (10,0 г, 27,8 ммоль) и карбонат калия (4,60 г, 33,3 ммоль) растворяли в толуоле и воде (200 мл, об./об. = 4:1) и добавляли фенилметиламин (2,99 г, 27,8 ммоль). Реакционную смесь нагревали до 80°C и проводили реакцию в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении с удалением большей части растворителя, добавляли воду и экстрагировали этилацетатом (80 мл × 3). Органический слои объединяли, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 10/1) посредством колоночной хроматографии на силикагеле, получая маслянистый продукт 20b (6,6 г, 77,7%).
1H ЯМР (400 МГц, ДМСО) δ 7,31-7,18 (m, 5H), 3,96-3,86 (m, 4H), 3,83 (s, 2H), 3,38 (m, 2H), 2,08-1,98 (m, 2H), 1,94-1,83 (m, 2H), 1,08 (m, 6H).
Стадия II: Получение соединения 20c
При комнатной температуре соединение диэтил N-бензил-2,5-пиррол дикарбоксилат (6,6 г, 21,6 ммоль) растворяли в метаноле (100 мл), и добавляли палладий на угле (20%, 1,3 г). Реакционную смесь подвергали реакции в течение 7 ч при комнатной температуре в условиях H2. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении , получая бесцветное маслянистое Соединение 20c (4,4 г, 94,4%).
1H ЯМР (400 МГц, CDCl3) δ 4,23-4,11 (m, 4H), 3,81-3,71 (m, 2H), 2,18-2,06 (m, 2H), 1,97-1,83 (m, 2H), 1,26 (m, 6H).
Стадия III: Получение соединения 20d
При комнатной температуре соединение диэтил-2,5-пирролидинкарбоксилат (4,4 г, 19,5 ммоль) растворяли в толуоле и добавляли ди-трет-бутилдикарбонат (5,35 г, 24,5 ммоль). Реакционную смесь нагревали до 95°C и проводили реакцию в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 5/1) посредством колоночной хроматографии на силикагеле, получая бесцветный маслянистый продукт 20d (4,3 г, 70,0 %).
1H ЯМР (400 МГц, CDCl3) δ 4,38 (s, 1H), 4,27 (s, 1H), 4,24-4,14 (m, 4H), 2,16 (m, 4H), 1,42 (s, 9H), 1,31-1,21 (m, 6H).
Стадия IV: Получение соединения 20e
При комнатной температуре соединение диэтил-N-трет-бутоксикарбонил-2,5-пиррол дикарбоксилат (2,7 г, 8,6 ммоль) растворяли в сухом тетрагидрофуране (80 мл). По каплям добавляли раствор диизопропиламида лития (2 M, 8,2 мл, 16,3 ммоль) в тетрагидрофуране при -78°C под азотной защитой. Добавляли бензилхлорметиловый эфир(1,49 г, 9,5 ммоль). Смесь перемешивали и проводили реакцию при -78°C в течение 1 ч, и нагревали до комнатной температуры и проводили реакцию в течение 2 ч. К реакционной смеси добавляли 100 мл насыщенного водного раствора хлорида аммония и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 5/1) посредством колоночной хроматографии на силикагеле, получая маслянистый продукт желтого цвета 20e (2,3 г, 61,4%).
1H ЯМР (400 МГц, CDCl3) δ 7,41-7,20 (m, 5H), 4,71-4,52 (m, 2H), 4,25-4,07 (m, 4H), 2,49 (m, 1H), 2,28 (m, 1H), 2,15-2,01 (m, 2H), 1,37 (s, 9H), 1,29-1,17 (m, 6H).
Стадия V: Получение соединения 20f
При комнатной температуре соединение диэтил-N-трет-бутоксикарбонил-2,5-(2-феноксиметил)пирролдикарбоксилат растворяли в диоксане гидрохлориде (4 н., 20 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, получая коричневое маслянистое необработанное Соединение 20f (1,44 г, 50,0%), которое непосредственно использовали на следующей стадии без очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,39-7,27 (m, 5H), 4,60-4,46 (m, 2H), 3,71 (m, 1H), 3,55 (m, 2H), 3,44-3,27 (m, 3H), 1,89-1,76 (m, 1H), 1,76-1,45 (m, 3H).
Стадия VI: Получение соединения 20g
Сухой тетрагидрофуран (40 мл) добавляли в реакционную колбу, и добавляли литийалюминийгидрид (0,33 г, 8,6 ммоль) в ледяной бане и при азотной защите. Спустя десять минут раствор по каплям добавляли [6-(бензилоксиметил)-6-(гидроксиметил)-2-пиперидинил]метанол в тетрагидрофуране, и затем доводили до комнатной температуры и проводили реакцию в течение 1 ч. Реакцию снова выполняли в ледяной бане и добавляли раствор гидроксида натрия (2M, 2 мл), получая твердое вещество белого цвета. Указанное твердое вещество белого цвета фильтровали и промывали тетрагидрофураном. Фильтрат сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении , получая желтоватое маслянистое необработанное Соединение 20g (1,0 г, 92,5 %) которое непосредственно использовали на следующей стадии без очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,39-7,27 (m, 5H), 4,57-4,46 (m, 2H), 3,71 (m, 1H), 3,55 (m, 2H), 3,44-3,31 (m, 3H), 1,88-1,78 (m, 1H), 1,75-1,58 (m, 2H), 1,52 (m,1H).
Стадия VII: Получение соединения 20h
При комнатной температуре соединение (2-((бензилокси)метил)пирролидин-2,5-диyl)диметанол (1,0 г, 3,98 ммоль) растворяли в метансульфоновой кислоте под защитой азота. Реакционную смесь нагревали до 140°C и проводили реакцию в течение 8 ч. После охлаждения до комнатной температуры, реакционный раствор вливали в ледяную воду (20 мл), добавляли 24 мл 50% гидроксида натрия при 0°C. После концентрировали при пониженном давлении, смесь растворяли в метаноле и фильтровали, и фильтрат концентрировали при пониженном давлении , получая желтоватое маслянистое необработанное целевое Соединение 20h (0,3 г, 52,8%).
Стадия VIII: Получение соединения 20i
При комнатной температуре соединение 3-окса-8-азабицикло[3,2.1]окт-5-илметанола (0,3 г, 2,1 ммоль), 2,4,6-трихлорпиримидина (0,46 г, 2,5 ммоль) и диизопропилэтиламина (0,81 г, 6,3 ммоль) растворяли в ацетонитриле (20 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении и очищали с помощью элюентной системы (петролейный эфир/этилацетат = 2:1) посредством колоночной хроматографии на силикагеле, получая белое твердое соединение 20i (0,15 г, 24,8%).
1H ЯМР (400 МГц, CDCl3) δ 6,39 (s, 1H), 5,25 (m, 1H), 4,22 (m, 1H), 3,98-3,76 (m, 3H), 3,67 (m, 3H), 2,23-2,14 (m, 1H), 2,14-1,96 (m, 2H), 1,78 (m, 1H).
Стадии IX & X: Получение соединения 20k
При комнатной температуре соединение (8-(2,6-дихлорпиримидин-4-ил)-3-окса-8-азабицикло[3,2.1]окт-1-ил)метанол (0,15, 24,8%) растворяли в сухом дихлорметане, добавляли триэтиламин (158 мг, 1,56 ммоль), и помещали в ледяную баню. Медленно добавляли метансульфонилхлорид (66 мг, 0,57 ммоль). Реакционную смесь подвергали реакции в ледяной бане в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении , получая белое твердое соединение, который применяли непосредственно на следующей стадии без очистки. Указанный необработанный продукт и карбонат калия (215 мг, 1,56 ммоль) растворяли в диоксане и воде (40 мл, 1/1). Реакционную смесь подвергали реакции при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении и очищали с помощью элюентной системы (дихлорметан/метанол = 20/1) посредством колоночной хроматографии на силикагеле, получая белое твердое Соединение 20k (40 мг, 30,8%).
1H ЯМР (400 МГц, CDCl3) δ 5,72 (s, 1H), 4,29 (m, 1H), 4,08 (m, 1H), 3,97 (m, 1H), 3,77 (m, 4H), 2,46-2,27 (m, 1H), 2,14 (m, 1H), 1,96 (m, 1H), 1,77 (m, 1H).
Стадия XI: Получение соединения 20
При комнатной температуре соединение (3,5-дифтор-4-((2-(трифторметил)пиридин-4-ил)окси)фенил)метанол (58, 0,19 ммоль) растворяли в сухом N,N-диметилформамиде (5 мл), и добавляли гидрид натрия (60% в минеральном масле, 13 мг, 0,32 ммоль) в ледяной бане. 15 мин спустя добавляли 7-Хлор-3,4-дигидро-1H-4,11a-этанопиримидо[6',1':2,3]имидазо[5,1-c][1,4]азинил-9(11H)-он (40 мг, 0,16 моль), и проводили реакцию при комнатной температуре в течение 1 ч. Реакционную смесь гасили путем добавления ледяной воды и экстрагировали этилацетатом (20 мл×3). Органический слои объединяли, сушили над безводным сульфатом натрия и фильтровали с удалением высушивающего агента. Фильтрат концентрировали при пониженном давлении и очищали с помощью элюентной системы (дихлорметан/метанол = 20/1) посредством колоночной хроматографии на силикагеле, получая белое твердое соединение 20 (35 мг, 35,3 %).
1H ЯМР (600 МГц, CDCl3) δ 8,61 (m, 1H), 7,26 (s, 1H), 7,14 (m, 2H), 6,99 (m, 1H), 5,43 (s, 2H), 5,22 (s, 1H), 4,26 (m, 1H), 4,00 (m, 1H), 3,93 (m, 1H), 3,78-3,68 (m, 4H), 2,33-2,26 (m, 1H), 2,08 (m, 1H), 1,99-1,92 (m, 1H), 1,73 (m, 1H). m/z 523,0 [M+H]+.
Биологическая оценка
Биологическая активность соединения может быть определена с использованием любого подходящего анализа, а также модели ткани и модели in vivo для определения активности соединения в качестве ингибитора LpPLA2.
(1) Рекомбинантный анализ Lp-PLA2 человека (rhLp-PLA2), также называемый анализом PED6
PED6 представлял собой флуоресцентно-меченный фосфолипид, который может быть приобретен напрямую в Invitogene или Molecular Probes. Он имел флуоресцентно-гасящую п-нитрофенильную группу в положении Sn3 и группу флуоресцеина Bodipy (FL) в положении sn2. После расщепления ферментом Lp-PLA2 он высвобождает группу FL, что приводит к усилению флуоресценции. Однако ингибитор Lp-PLA2 может предотвращать возникновение такого расщепления, так что усиленная флуоресценция не может наблюдаться.
Метод анализа: Испытуемое соединение (как показано в таблице 1) смешивали с раствором ДМСО в объемном отношении 1:3 и разбавляли с получением исходного планшета из 384-луночного микропланшета. Затем 0,01 мкл соединения переносили через дозатор жидкости ECHO из исходного планшета в 384-луночный планшет Greiner 784076 и в каждую лунку планшета добавляли 5 мкл буфера, состоящего из 50 мМ HEPES, pH 7,4, 150 мМ NaCl и 1 мМ CHAP (буферный раствор, содержащий рекомбинантный фермент Lp-PLA2 человека в концентрации 4 нМ или 110 пМ). Планшет центрифугировали при 500 об/мин в течение 10 секунд. После 30-минутной предварительной инкубации 5 мкл вышеупомянутого буфера добавляли в 384-луночный планшет Greiner 784076, планшет центрифугировали при 500 об/мин в течение 10 секунд. После того, как планшет инкубировали при комнатной температуре в течение 20 мин в темном месте, интенсивность флуоресценции считывали при возбуждении 480/эмиссии 540 с помощью визуализатора планшета ViewLux, и использовали модель подгонки XL в Excel для выполнения анализа кривой и анализа КК для расчета pIC50. Результаты приведены в таблице 1.
(2) Анализ Lp-PLA2 в плазме человека (также называемый анализом Thio-PAF)
Анализ плазмы человека проводили с использованием сульфатидного аналога PAF (фосфатидилхолина). После гидролиза получали фосфолипиды, содержащие свободные сульфгидрильные группы, которые подвергали реакции присоединения по Михаэлю с помощью CPM для получения малеимида, усиливающего флуоресценцию. Непрерывный количественный анализ тиола может быть проведен путем определения интенсивности флуоресценции. Этот анализ может быть использован для обнаружения ингибирующей активности ингибитора Lp-PLA2 на фермент Lp-PLA2 в плазме человека.
Метод анализа: Испытуемое соединение (как показано в таблице 2) смешивали с раствором ДМСО в объемном отношении (1:3) и разбавляли с получением исходного планшета из 384-луночного микропланшета. Затем 0,01 мкл соединения переносили через дозатор эхо-жидкости из исходного планшета в 384-луночный планшет Greiner 784076, а затем добавляли 8 мкл предварительно аликвотированной и замороженной смешанной человеческой плазмы. Планшет центрифугировали при 500 об/мин в течение 10 секунд. После 30-минутной предварительной инкубации 2 мкл раствора субстрата и буфера, содержащего 2,5 мМ 2-тио-PAF (раствор в этаноле), 32 мкМ CPM (раствор в ДМСО) и 3,2 мМ N-этилмалеимида (NEM) (буферный раствор, состоящий из 50 мМ HEPES, pH 7,4, 150 мМ NaCl, 1 мМ CHAPS), вносили с помощью станции для обработки жидкости BRAVO в 384-луночный планшет Greiner 784076 малого объема. Через две минуты реакционную смесь гасили 5 мкл 5% трифторуксусной кислоты. После того, как планшет инкубировали при комнатной температуре в течение 40 мин в темном месте, интенсивность флуоресценции считывали при возбуждении 380/ эмиссии 485 с помощью считывателя планшетов Envision, и модель подгонки XL в Excel использовали для анализа кривой и анализа КК для расчета pIC50. Результаты представлены в таблице 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ТРИЦИКЛИЧЕСКОГО ПИРИМИДИНОНОВОГО СОЕДИНЕНИЯ И ЕГО КОМПОЗИЦИИ | 2021 |
|
RU2838186C1 |
| ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ТРИЦИКЛИЧЕСКОГО ПИРИМИДИНОНОВОГО СОЕДИНЕНИЯ И ЕГО КОМПОЗИЦИИ | 2021 |
|
RU2820903C1 |
| АГОНИСТ S1P1 И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2754845C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2673458C1 |
| ТЕТРАГИДРОИМИДАЗО[1,5-d][1,4]ОКСАЗЕПИНОВОЕ ПРОИЗВОДНОЕ | 2014 |
|
RU2659219C2 |
| ИНГИБИТОР PDE4 | 2017 |
|
RU2743126C2 |
| ПИРАНОДИПИРИДИНОВОЕ СОЕДИНЕНИЕ | 2016 |
|
RU2743998C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| ПЕНТАЦИКЛИЧЕСКОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2020 |
|
RU2815382C2 |
| ИНГИБИТОР LSD1, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2763898C2 |
Группа изобретений относится к соединению формулы (I) или его фармацевтически приемлемой соли
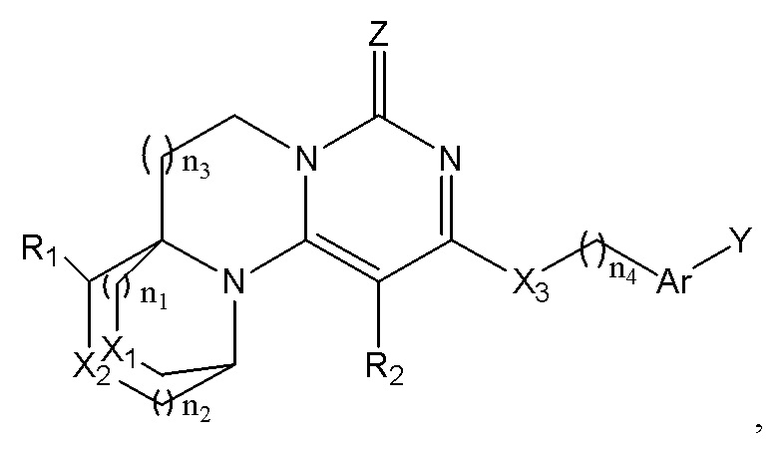
также относится к способу получения соединения формулы (I) или его фармацевтически приемлемой соли, также относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, обладающей свойством ингибитора липопротеин-ассоциированной фосфолипазы А2 (Lp-PLA2), также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции для получения ингибитора Lp-PLA2, также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции для получения лекарственного средства для лечения связанного с нейродегенерацией заболевания, также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции для получения лекарственного средства для лечения сердечно-сосудистого заболевания, диабетического макулярного отека (DME) или заболевания предстательной железы. Группа изобретений обеспечивает разработку синтеза новых тетрациклических пиримидиноновых соединений в качестве альтернативных ингибиторов липопротеин-ассоциированной фосфолипазы А2 (Lp-PLA2). 6 н. и 21 з.п. ф-лы, 2 табл., 20 пр.
1. Соединение, представленное формулой (I)
 (I),
(I),
или его фармацевтически приемлемая соль,
где n1, n3, каждый независимо, представляют собой 0, 1 или 2;
n2 представляет собой 0 или 1;
n4 представляет собой 1 или 2;
R1 и R2, каждый независимо, выбраны из -H;
X1 выбран из алкилена;
X2 представляет собой алкилен, -O- или -S-;
X3 представляет собой -O- или –S-;
Ar представляет собой арилен; и атомы водорода в указанном арилене необязательно замещены одним или более заместителями, каждый из которых независимо выбран из галогена;
Y представляет собой -H, галоген или -OAr';
Ar' выбран из арила или гетероарила; и атомы водорода в указанном ариле или гетероариле необязательно замещены одним или более заместителями, каждый из которых независимо выбран из галогена, алкила, галогеналкила, алкоксила, галогеналкоксила, дейтероалкила или дейтероалкоксила;
Z представляет собой O или S;
атомы галогена в указанном галогене, галогеналкиле и галогеналкоксиле, каждый независимо, выбраны из F, Cl, Br или I;
алкилы в указанных алкиле, дейтероалкиле, дейтероалкоксиле, галогеналкиле, галогеналкоксиле и алкоксиле, каждый независимо, представляют собой C1-C4 неразветвленный или разветвленный алкил;
указанные алкилены, каждый независимо, представляют собой C1-C5 неразветвленный или разветвленный алкилен;
указанный арил представляет собой 6-10-членный арил;
указанный арилен представляет собой 6-10-членный арилен;
указанный гетероарил представляет собой 5-10-членное гетероароматическое кольцо, содержащее 1-2 гетероатома в кольце, выбранных из N, O и S.
2. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
алкилы в указанном алкиле, дейтероалкиле, дейтероалкоксиле, галогеналкиле, галогеналкоксиле, алкоксиле, каждый независимо, выбраны из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, втор-бутила.
3. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
указанные алкилены, каждый независимо, выбраны из метилена, этилена, н-пропилидена, изопропилидена, н-бутилидена, изобутилидена, трет-бутилидена, втор-бутилидена, н-пентилидена, 1-метилбутилидена, 2-метилбутилидена, 3-метилбутилидена, изопентилидена, 1-этилпропилидена, неопентилидена.
4. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
указанный арил представляет собой фенил или нафтил.
5. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
указанный арил представляет собой фенил, 1-нафтил или 2-нафтил.
6. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
указанный арилен представляет собой фенилен или нафтилен.
7. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
указанный гетероарил представляет собой 5-10-членное гетероароматическое кольцо, выбранное из пиридинового кольца, пиррольного кольца, пиримидинового кольца, пиразинового кольца, пиридазинового кольца, тиофенового кольца и фуранового кольца.
8. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
указанный гетероарил выбран из пиридин-2-ила, пиридин-3-ила, пиридин-4-ила, пиридазин-3-ила, пиридазин-4-ила, пиримидин-2-ила, пиримидин-4-ила, пиримидин-5-ила, пиразин-2-ила, пиразин-3-ила, индолила, изоиндолила, индазолила, индолизинила, хинолизинила, хинолинила, изохинолинила, цинолинила, фталазинила, нафтиридинила, хиназолинила, хиноксалинила, тиено[2,3-b]фурила, фуро[3,2-b]-пиранила, бензотиенила, бензоксазолила, бензимидазолила, бензотиазолила, бензоксазинила, бензофуранила, пирроло[2,3-b]пиридила, пирроло[3,2-c]пиридила, пирроло[3,2-b]пиридила.
9. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
X1 представляет собой -CH2-.
10. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
X2 выбран из -CH2- или -O-.
11. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
X3 представляет собой -O-.
12. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
Ar представляет собой фенилен и атом водорода в указанном фенилене необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из F, Cl, Br, I.
13. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
Y представляет собой -H или -OAr';
Ar' выбран из фенила, пиридила, пиримидинила или хинолинила, и атомы водорода в указанном фенильном, пиридильном, пиримидинильном или хинолинильном кольце, каждый независимо, необязательно замещены 1, 2 или 3 заместителями, каждый из которых независимо выбран из F, Cl, Br, метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, втор-бутила, -CD3, C1-C4 галогеналкила, -OCH3, -OC2H7, -OC3H7 или C1-C6 галогеналкоксила.
14. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
Ar' выбран из фенила, пиридин-3-ила, или пиридин-4-ила, или пиримидин-5-ила, при этом необязательно указанный Ar' замещен 1 или 2 заместителями, выбранными из Cl, -CH3, -CF3 или -OCF3.
15. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где указанное соединение формулы (I) выбрано из следующих соединений:
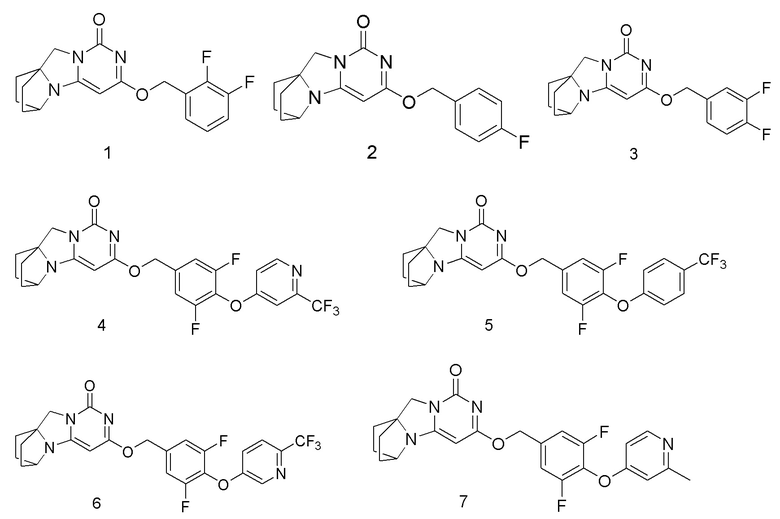
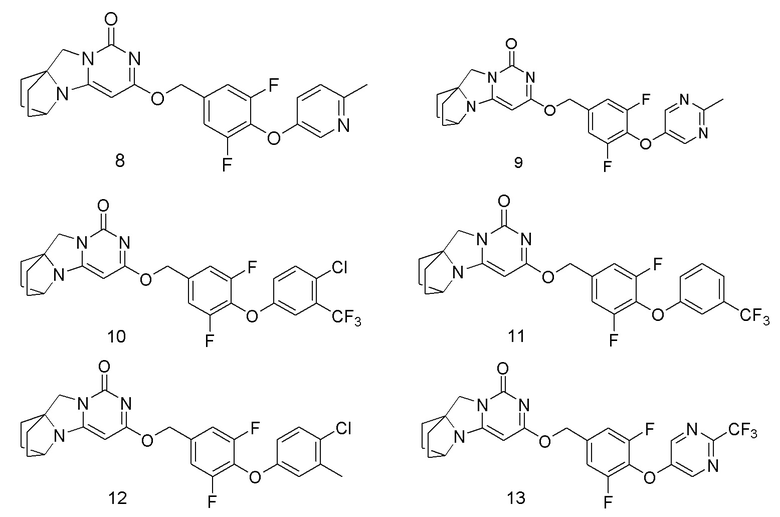
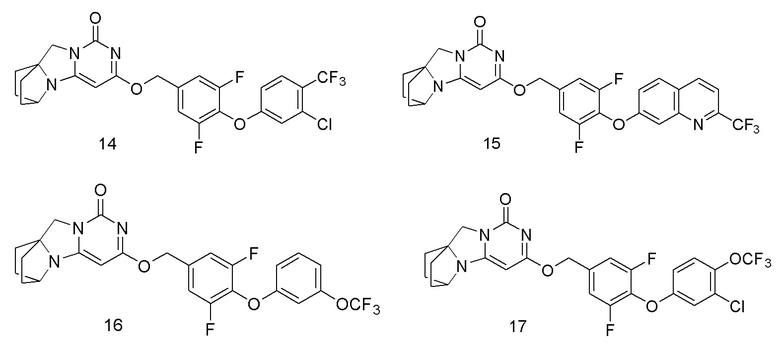

16. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-15, где указанная фармацевтически приемлемая соль включает анионную соль и катионную соль соединения формулы (I).
17. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-15, где указанная фармацевтически приемлемая соль включает соль щелочного металла, соль щелочно-земельного металла и аммониевую соль соединения формулы (I); где указанный щелочной металл включает натрий, калий, литий и цезий и указанный щелочно-земельный металл включает магний, кальций и стронций.
18. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-15, где указанная фармацевтически приемлемая соль включает соль, образуемую соединением формулы (I) и органическим основанием;
где указанное органическое основание включает триалкиламин, пиридин, хинолин, пиперидин, имидазол, пиколин, диметиламинопиридин, диметиланилин, N-алкилморфолин, 1,5-диазабицикло[4,3.0]нонен-5, 1,8-диазабицикло[5,4.0]ундецен-7 и 1,4-диазабицикло[2,2.2]октан.
19. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 18, где указанный триалкиламин включает триметиламин, триэтиламин и N-этилдиизопропиламин; и указанный N-алкилморфолин включает N-метилморфолин.
20. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-15, где указанная фармацевтически приемлемая соль включает соль, образуемую соединением формулы (I) и кислотой; и
указанная кислота включает неорганическую кислоту и органическую кислоту; где указанная неорганическая кислота включает хлористоводородную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту, угольную кислоту; где указанная органическая кислота включает муравьиную кислоту, уксусную кислоту, пропионовую кислоту, щавелевую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, малеиновую кислоту, молочную кислоту, яблочную кислоту, лимонную кислоту, винную кислоту, пикриновую кислоту, метансульфоновую кислоту, этансульфоновую кислоту, п-толуолсульфоновую кислоту, глутаминовую кислоту и памовую кислоту.
21. Способ получения соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-20, включающий следующий путь синтеза:
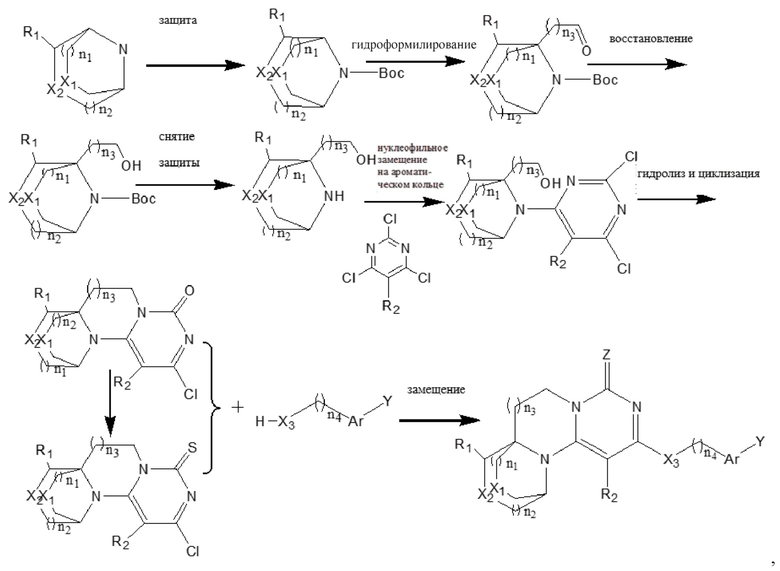
где в соответствующих формулах n1, n2, n3, R1, R2, X1, X2, X3, Z, Ar и Y являются такими, как определено в любом из пп. 1-20.
22. Способ по п. 21, где,
когда X2 представляет собой -O- и n3 равен 0, дополнительно применяют следующий путь синтеза:
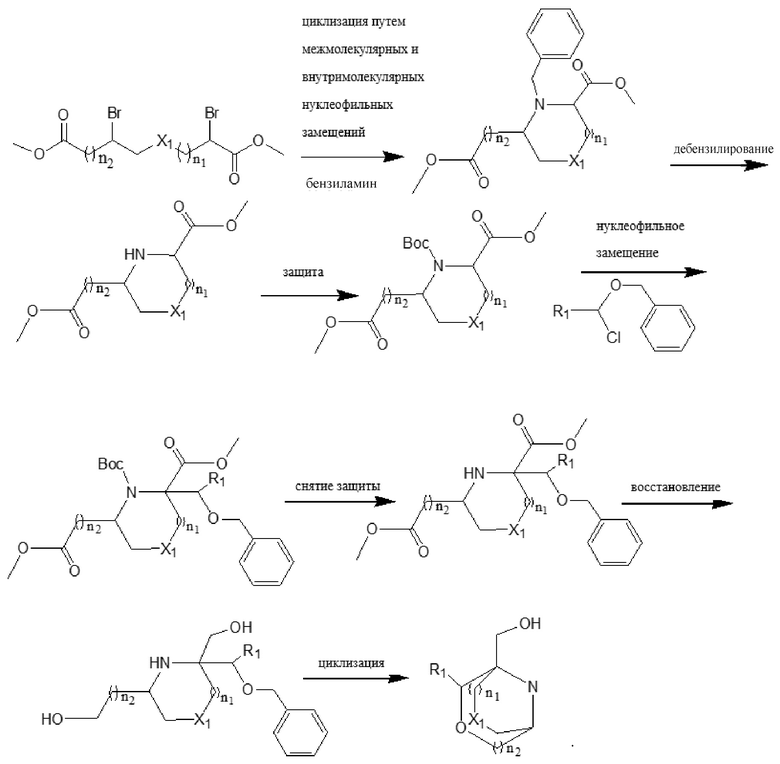
23. Фармацевтическая композиция, обладающая свойством ингибитора липопротеин-ассоциированной фосфолипазы А2 (Lp-PLA2), содержащая одно или более соединение формулы (I) или его фармацевтически приемлемую соль по любому из пп. 1-20 в эффективном количестве и, необязательно, фармацевтически приемлемый носитель.
24. Фармацевтическая композиция по п. 23, где лекарственная форма фармацевтической композиции включает пероральный препарат, ректально вводимый препарат и парентерально вводимый препарат; при этом:
необязательно, пероральный препарат включает твердый препарат и жидкий препарат;
необязательно, твердый препарат включает таблетки, порошки, гранулы и капсулы;
необязательно, жидкий препарат включает водные или масляные суспензии и сиропы; и
необязательно, парентерально вводимый препарат включает растворы для инъекций и водные или масляные суспензии.
25. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-20 или фармацевтической композиции по п. 23 или 24 для получения ингибитора Lp-PLA2.
26. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-20 или фармацевтической композиции по п. 23 или 24 для получения лекарственного средства для лечения связанного с нейродегенерацией заболевания; при этом,
необязательно, связанное с нейродегенерацией заболевание включает болезнь Альцгеймера (БА), глаукому и возрастную макулярную дегенерацию (AMD).
27. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-20 или фармацевтической композиции по п. 23 или 24 для получения лекарственного средства для лечения сердечно-сосудистого заболевания, диабетического макулярного отека (DME) или заболевания предстательной железы; при этом,
необязательно, указанное сердечно-сосудистое заболевание включает атеросклероз.
| RU 2017105353 A, 22.08.2018 | |||
| CN 106536521 A, 22.03.2017 | |||
| CN 103827118 A, 28.05.2014 | |||
| CN 103827116 A, 28.05.2014 | |||
| CN 105008365 A, 28.10.2015 | |||
| CN 104968665 A, 07.10.2015 | |||
| AU 2012288865 A1, 13.02.2014. |
