ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к классу производных пирролидина, являющихся агонистами PPAR, а также к их применению для лечения некоторых заболеваний, связанных с путями, ассоциированными с рецептором PPAR (такими как неалкогольный стеатогепатит и сопутствующий фиброз, инсулинорезистентность, первичный билиарный холангит, дислипидемия, гиперлипидемия, гиперхолестеринемия, атеросклероз, гипертриглицеридемия, сердечно-сосудистое заболевание, ожирение и т.д.). В частности, настоящее изобретение относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли.
УРОВЕНЬ ТЕХНИКИ
Неалкогольная жировая болезнь печени (НАЖБП), которая является наиболее распространенным заболеванием печени в развитых странах или регионах, соответствует накоплению избыточного жира в печени в форме триглицеридов (стеатоз > 5% гепатоцитной ткани). Помимо избытка жира у пациентов, страдающих от НАЖБП, может происходить повреждение гепатоцитов и воспаление (жировой гепатоз печени), причем последняя форма соответствует НАСГ (неалкогольный стеатогепатит). НАЖБП, сопровождающаяся только стеатозом, не связана с повышенной краткосрочной заболеваемостью или смертностью, но развитие до НАСГ значительно увеличивает риск цирроза печени, печеночной недостаточности и печеночноклеточной карциномы (НСС). Цирроз печени, вызванный НАСГ, является одной из причин роста потребности в трансплантации печени. У пациентов с НАСГ заболеваемость и смертность, вызванные заболеваниями печени, значительно увеличиваются, также НАСГ тесно связан с увеличением заболеваемости и смертности при сердечно-сосудистых заболеваниях. При диагностике не имеющих симптомов пациентов мужского пола среднего возраста было показано, что 46% пациентов имели неалкогольную жировую болезнь печени (НАЖБП), и 12,2% имели НАСГ. Большинство пациентов с НАЖБП являются пожилыми мужчинами, страдающими от гипертензии и диабета. 60-76% диабетиков имеют НАЖБП, и 22% имеют НАСГ. Число детей с НАЖБП также растет год за годом, и 38-53% детей с ожирением имеют НАЖБП. В Китае неалкогольная жировая болезнь печени по уровню заболеваемости находится на первом месте.
В настоящее время отсутствуют одобренные FDA лекарственные средства для лечения указанного заболевания. В Китае в клинической терапии обычно применяют лекарственные средства для защиты печени, такие как полиен-фосфатидилхолин, силимарин, урсодезоксихолиевая кислота, глицирризиновая кислота и т.д.
Рецепторы, активируемые пероксисомными пролифераторами (PPAR), которые являются членами суперсемейства ядерных рецепторов гормонов, представляют собой активируемые лигандами факторы транскрипции, которые регулируют экспрессию генов. PPAR, главным образом, включают три подтипа: РРАР-альфа, который в основном экспрессируется в бурой жировой ткани, печени, сердце и скелетных мышцах, и играет ключевую роль в метаболизме желчных кислот, липидов и сахаров; РРАР-дельта, который не имеет четкой специфической экспрессии и может оказывать противовоспалительное действие; и РРАР-гамма, который в определенной степени влияет на инсулинорезистентность. Этот рецептор связан с различными заболеваниями, включая дислипидемию, гиперлипидемию, гиперхолестеринемию, атеросклероз, атерогенез, гипертриглицеридемию, сердечную недостаточность, инфаркт миокарда, сосудистые заболевания, сердечно-сосудистые заболевания, гипертензию, ожирение, воспаление, артрит, рак, болезнь Альцгеймера, кожные заболевания, респираторные заболевания, нарушения глаз, IBD (воспалительное заболевание кишечника), язвенный колит и болезнь Крона. Так как PPAR имеют различные механизмы, благоприятно влияющие на функции печени, то агонисты PPAR являются одними из наиболее эффективных вероятных лекарственных средств для лечения жировой болезни печени.
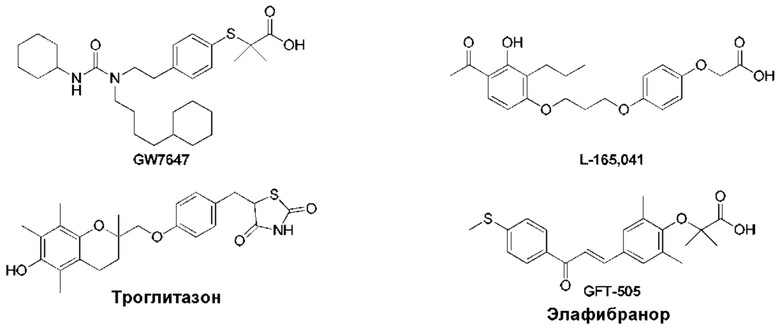
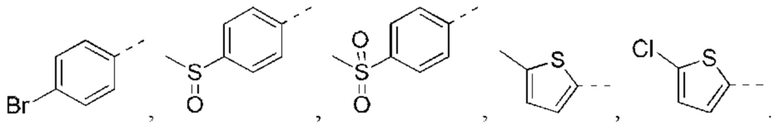
Следующие соединения представляют собой соединения-агонисты PPAR, описанные в литературе.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено соединение формулы (I)
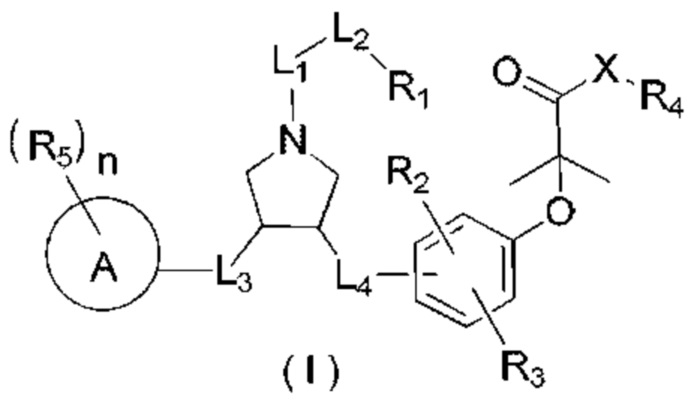
и его фармацевтически приемлемая соль,
где:
R1 выбран из Н, NH2 или C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, фенила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R;
R2, R3 независимо выбраны из Н, галогена, ОН, NH2 или С1-3 алкила, необязательно замещенного 1, 2 или 3 R;
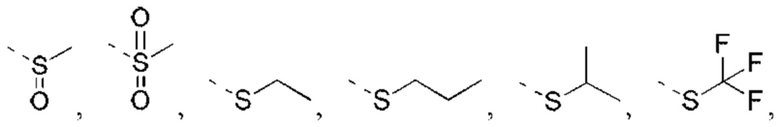
X выбран из NH, О и S;
если X выбран из О или S, то R4 выбран из Н или C1-6 алкила, необязательно замещенного 1, 2 или 3 R;
если X выбран из NH, то R4 выбран из Н, C1-6 алкила или C1-6 алкил-S(=O)2-, -C1-6 алкил-S(=O)2OH, каждый из которых необязательно замещен 1, 2 или 3 R;
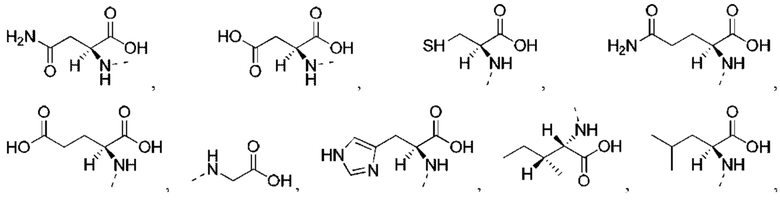
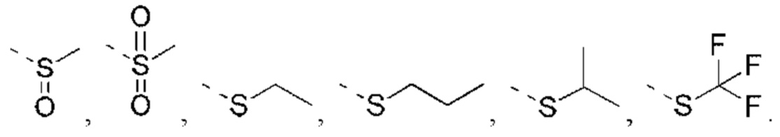
или структурное звено R4-X- выбрано из: 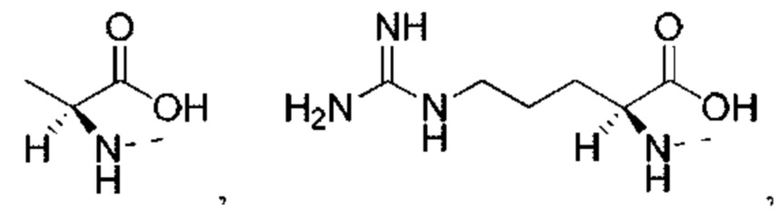
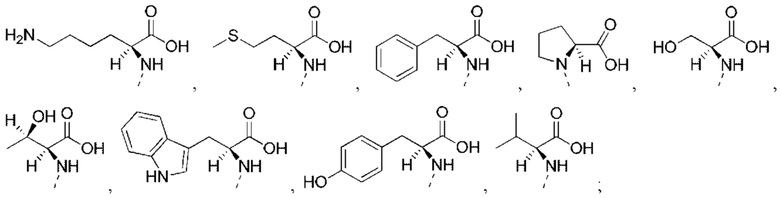
R5 выбран из Н, галогена, ОН, NH2, CN, СООН или C1-6 алкила, C1-6 алкил-S(=O)-, C1-6 алкил-S(=O)2-, С1-6 алкокси, C1-6 алкилтио, каждый из которых необязательно замещен 1, 2 или 3 группами R;
n выбран из 0, 1, 2 или 3;
кольцо А выбрано из фенила, нафтила, 5-6-членного гетероарила;
L1 выбран из простой связи, -С(=O)-, -О-, -NH-, -С(=O)O-, -C(=O)NH-, -S(=O)2-, -S(=O)-, -(CRR)1-3-;
L2 выбран из простой связи, -(CRR)1-3-, -С(=O)-, -О-, -S-, -NH-, -С(=O)O-, -C(=O)NH-, -S(=O)2-, -S(=O)-;
L3 выбран из -(CRR)-, -С(=O)-;
L4 выбран из простой связи, -(CRR)1-3-;
R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2 или С1-6 алкила, С1-6 гетероалкила, каждый из которых необязательно замещен 1, 2 или 3 R';
R' выбран из F, Cl, Br, I, ОН, CN, NH2, СООН, Me, Et, CF3, CHF2, CH2F, NHCH3, N(CH3)2;
"гетеро-" относится к гетероатому или гетероатомной группе и выбран из -C(=O)NH-, -NH-, -C(=NH)-, -S(=O)2 NH-, -S(=O) NH-, -O-, -S-,=O,=S, -O-N=, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -NHC(=O)NH-;
в любом из случаев, определенных выше, количество гетероатомов или гетероатомных групп независимо выбрано из 1, 2 или 3.
В некоторых вариантах реализации настоящего изобретения описанный выше R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2 или C1-3 алкила, C1-3 алкокси, C1-3 алкилтио, C1-3 алкиламино, N,N'-ди(C1-3 алкил)амино, каждый из которых необязательно замещен 1, 2 или 3 R'.
В некоторых вариантах реализации настоящего изобретения описанный выше R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2, Me, CF3, CHF2, CH2F, 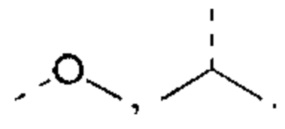
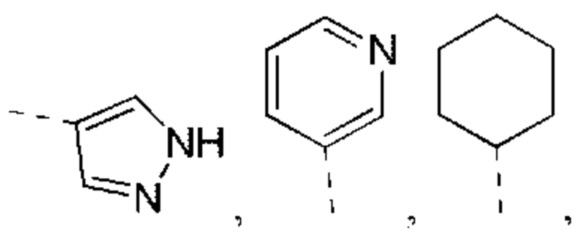
В некоторых вариантах реализации настоящего изобретения описанный выше R1 выбран из Н, NH2 или С1-6 алкила, циклопентила, азетидинила, пирролидинила, тетрагидротиенила, тетрагидрофуранила, пиперидила, морфолинила, пиперазинила, фенила, пиразолила, пиридила, циклогексила, каждый из которых необязательно замещен 1, 2 или 3 R.
В некоторых вариантах реализации настоящего изобретения описанный выше R1 выбран из Н, NH2 или Me, Et, 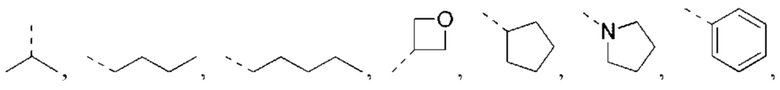
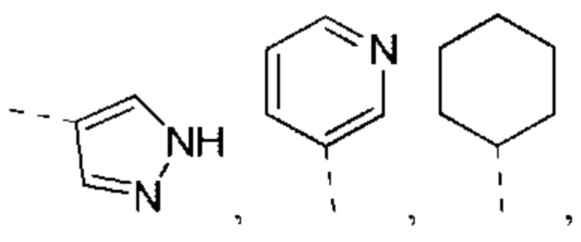 каждый из которых необязательно замещен 1, 2 или 3 R.
каждый из которых необязательно замещен 1, 2 или 3 R.
В некоторых вариантах реализации настоящего изобретения описанный выше R1 выбран из Н, NH2, Me, Et, 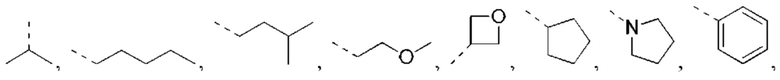
В некоторых вариантах реализации настоящего изобретения описанные выше R2, R3 независимо выбраны из Н, F, Cl, Br, I, ОН, NH2, или Me, Et, каждый из которых необязательно замещен 1, 2 или 3 R.
В некоторых вариантах реализации настоящего изобретения описанные выше R2, R3 независимо выбраны из Н, F, Cl, Br, I, ОН, NH2, Me, Et.
В некоторых вариантах реализации настоящего изобретения, если X выбран из О или S, как описано выше, то R4 выбран из Н или Me, Et, 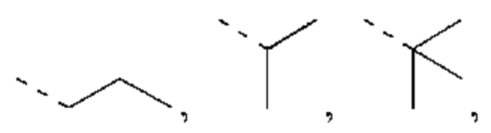 каждый из которых необязательно замещен 1, 2 или 3 R.
каждый из которых необязательно замещен 1, 2 или 3 R.
В некоторых вариантах реализации настоящего изобретения, если X выбран из О или S, как описано выше, то R4 выбран из Н, Me, Et, 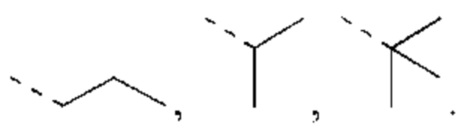
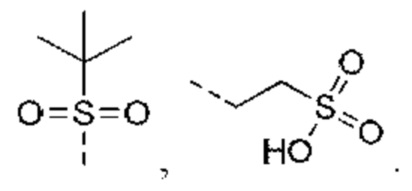
В некоторых вариантах реализации настоящего изобретения, если X выбран из NH, как описано выше, то R4 выбран из Н, C1-4 алкила или C1-4 алкил-S(=O)2-, -С1-3 алкил-S(=O)2OH, каждый из которых необязательно замещен 1, 2 или 3 R.
В некоторых вариантах реализации настоящего изобретения, если X выбран из NH, как описано выше, то R4 выбран из Н, Me, Et, 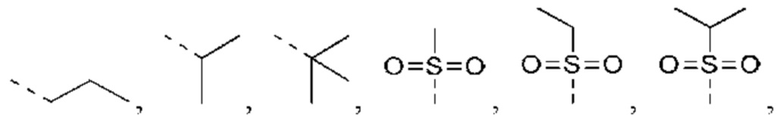
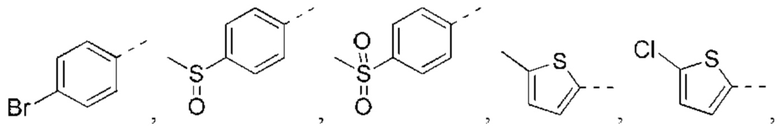
В некоторых вариантах реализации настоящего изобретения описанный выше R5 выбран из Н, F, Cl, Br, I, ОН, NH2, CN, СООН или Me, Et, 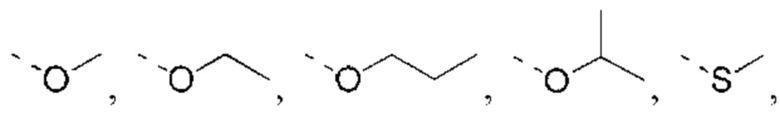
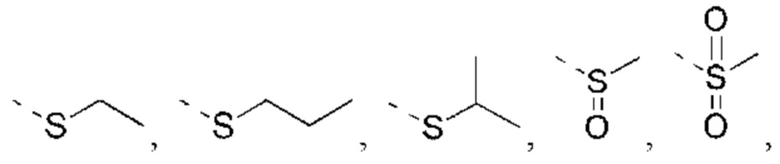 каждый из которых необязательно замещен 1, 2 или 3 R.
каждый из которых необязательно замещен 1, 2 или 3 R.
В некоторых вариантах реализации настоящего изобретения описанный выше R5 выбран из Н, F, Cl, Br, I, ОН, NH2, CN, СООН, CF3,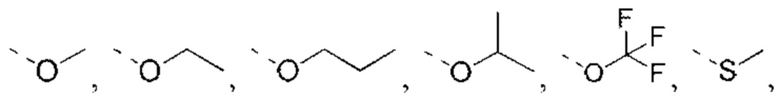
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено  выбрано из:
выбрано из: 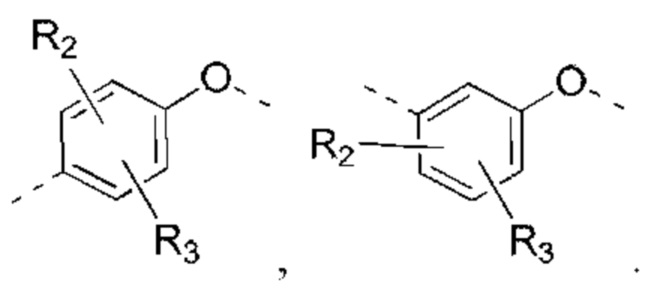
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено 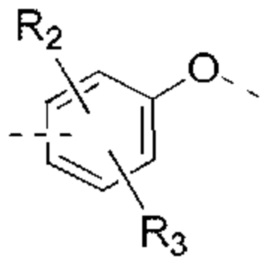 выбрано из:
выбрано из: 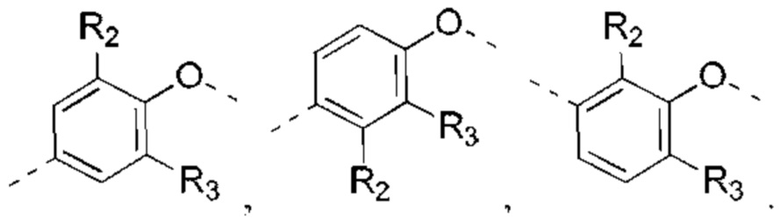
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено 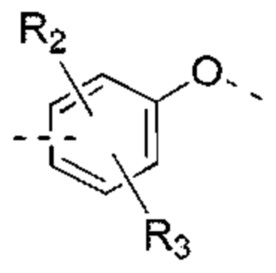 выбрано из:
выбрано из: 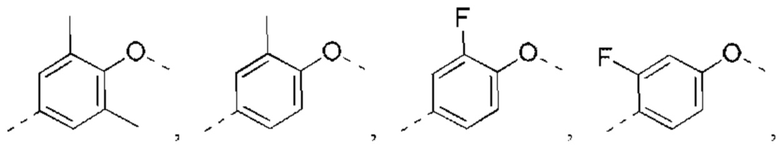
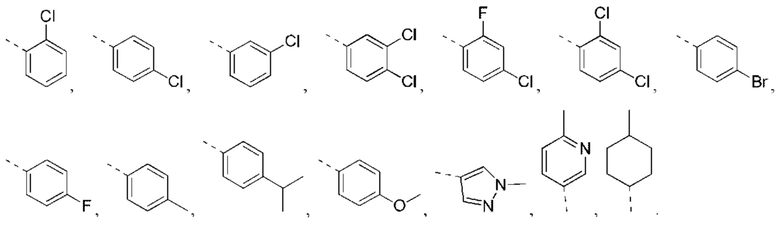
В некоторых вариантах реализации настоящего изобретения описанное выше кольцо А выбрано из: фенила, нафтила, пиридила, пиримидила, пиразинила, пиридазинила, тиенила, пиразолила, имидазолила, оксазолила, тиазолила, изоксазолила, изотиазолила.
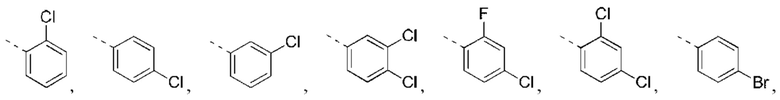
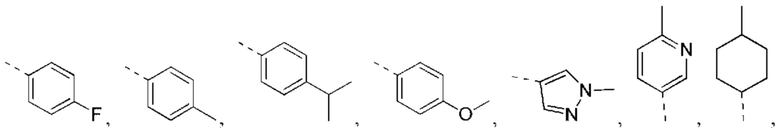
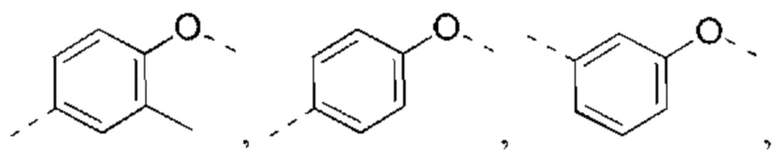
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено 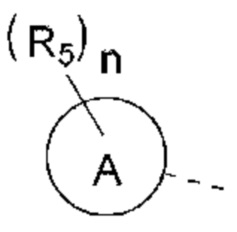 выбрано из:
выбрано из: 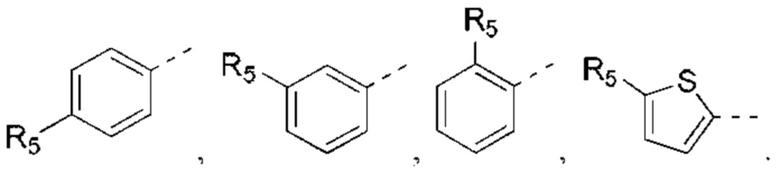
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено 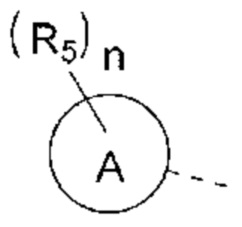 выбрано из
выбрано из 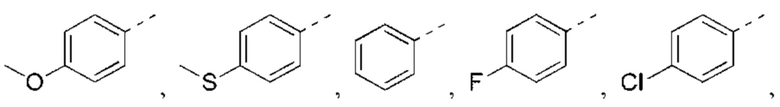
В некоторых вариантах реализации настоящего изобретения описанный выше L2 выбран из простой связи, -СН2-, -СН2СН2-, -С(=O)-, -О-, -S-, -NH-, -С(=O)O-, -C(=O)NH-, -S(=O)2-, -S(=O)-.
В некоторых вариантах реализации настоящего изобретения описанный выше L3 выбран из -СН2-, -С(=O)-.
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено -L1-L2- выбрано из: простой связи, -СН2-, -С(=O)-, -S(=O)2-, -С(=O)O-, -C(=O)NH-, -C(=O)-CH2-, -C(=O)O-CH2-, -СН2СН2О-.
В некоторых вариантах реализации настоящего изобретения описанный выше L4 выбран из простой связи, -СН2-, - СН2СН2-.
В некоторых вариантах реализации настоящего изобретения описанный выше R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2 или C1-3 алкила, C1-3 алкокси, C1-3 алкилтио, C1-3 алкиламино, N,N'-ди(C1-3 алкил)амино, каждый из которых необязательно замещен 1, 2 или 3 R', и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанный выше R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2, Me, CF3, CHF2, CH2F, 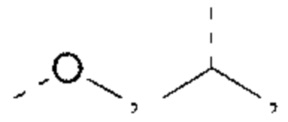 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанный выше R1 выбран из Н, NH2 или С1-6 алкила, циклопентила, азетидинила, пирролидинила, тетрагидротиенила, тетрагидрофуранила, пиперидила, морфолинила, пиперазинила, фенила, пиразолила, пиридила, циклогексила, каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанный выше R1 выбран из Н, NH2 или Me, Et, 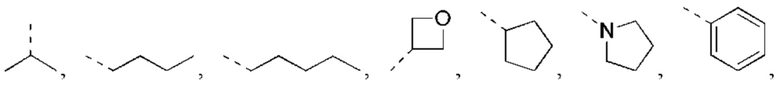
 каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные такие, как определено выше.
каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанный выше R1 выбран из Н, NH2, Me, Et, 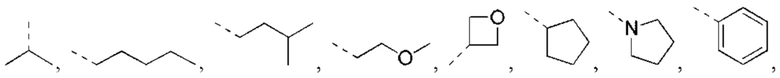
 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанные выше R2, R3 независимо выбраны из Н, F, Cl, Br, I, ОН, NH2 или Me, Et, каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанные выше R2, R3 независимо выбраны из Н, F, Cl, Br, I, ОН, NH2, Me, Et, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения, если X выбран из О или S, как описано выше, то R4 выбран из Н или Me, Et, 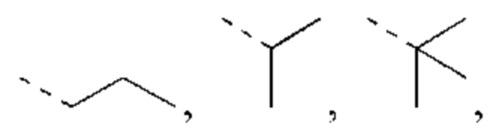 каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные такие, как определено выше.
каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения, если X выбран из О или S, как описано выше, то R4 выбран из Н, Me, Et, 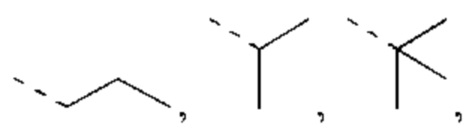 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения, если X выбран из NH, как описано выше, то R4 выбран из Н, С1-4 алкила или С1-4 алкил-S(=O)2-, -C1-3 алкил-S(=O)2OH, каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения, если X выбран из NH, как описано выше, то R4 выбран из Н, Me, Et, 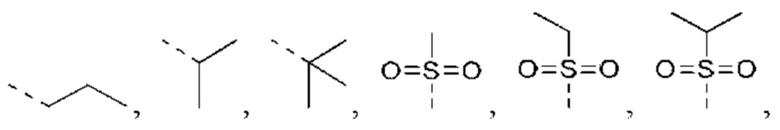
 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
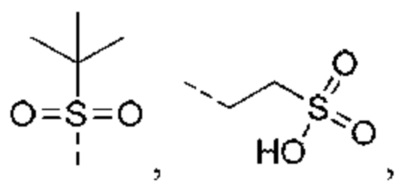
В некоторых вариантах реализации настоящего изобретения описанный выше R5 выбран из Н, F, Cl, Br, I, ОН, NH2, CN, СООН или Me, Et, 
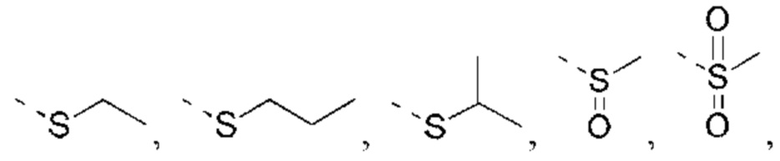 каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные такие, как определено выше.
каждый из которых необязательно замещен 1, 2 или 3 R, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанный выше R5 выбран из Н, F, Cl, Br, I, ОН, NH2, CN, СООН, CF3, 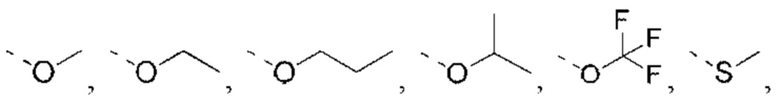
 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено 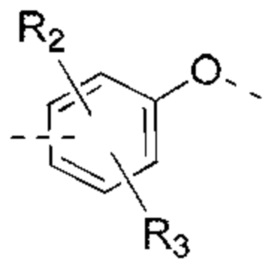 выбрано из:
выбрано из: 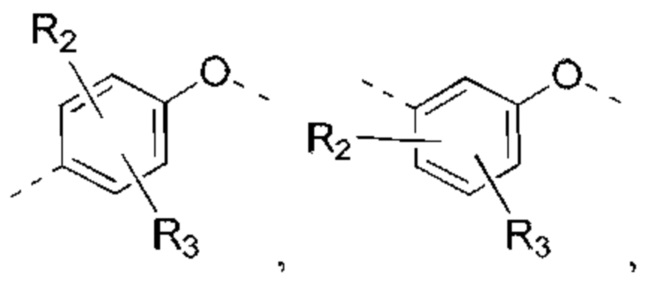 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено 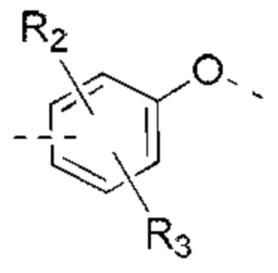 выбрано из:
выбрано из: 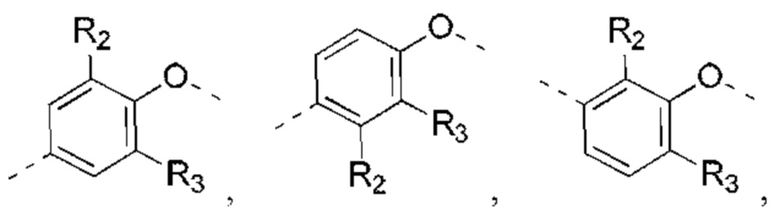 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено 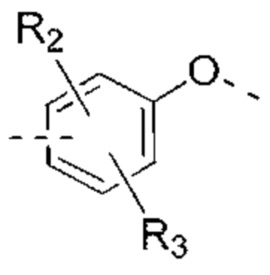 выбрано из:
выбрано из: 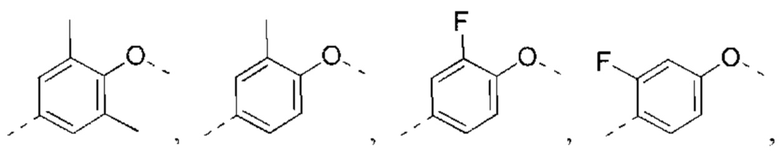
 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанное выше кольцо А выбрано из: фенила, нафтила, пиридила, пиримидила, пиразинила, пиридазинила, тиенила, пиразолила, имидазолила, оксазолила, тиазолила, изоксазолила, изотиазолила, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено 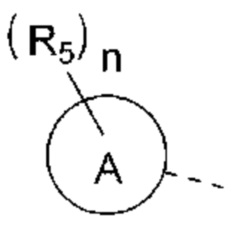 выбрано из:
выбрано из: 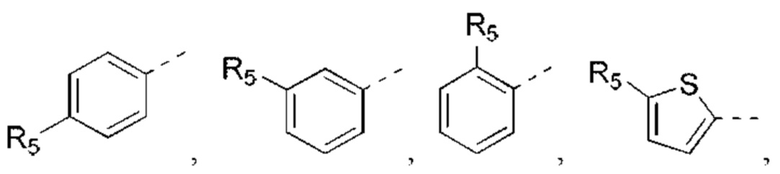 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено 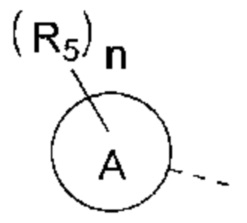 выбрано из
выбрано из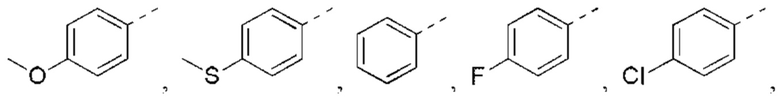
 и другие переменные такие, как определено выше.
и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанный выше L2 выбран из простой связи, -СН2-, -СН2СН2-, -С(=O)-, -О-, -S-, -NH-, -С(=O)O-, -C(=O)NH-, -S(=O)2-, -S(=O)-, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанный выше L3 выбран из -СН2-, -С(=O)-, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанное выше структурное звено -L1-L2- выбрано из: простой связи, -СН2-, -С(=O)-, -S(=O)2-, -С(=O)O-, -C(=O)NH-, -С(=O)-СН2-, -С(=O)O-СН2-, и другие переменные такие, как определено выше.
В некоторых вариантах реализации настоящего изобретения описанный выше L4 выбран из простой связи, -СН2-, -СН2СН2-, и другие переменные такие, как определено выше.
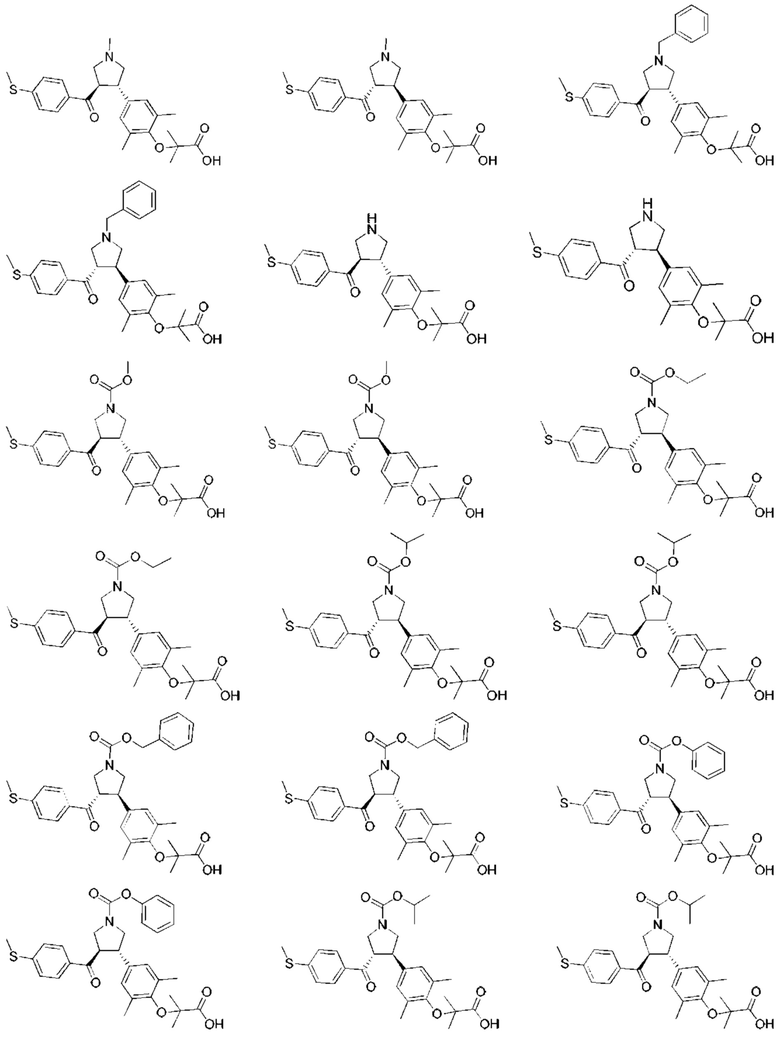
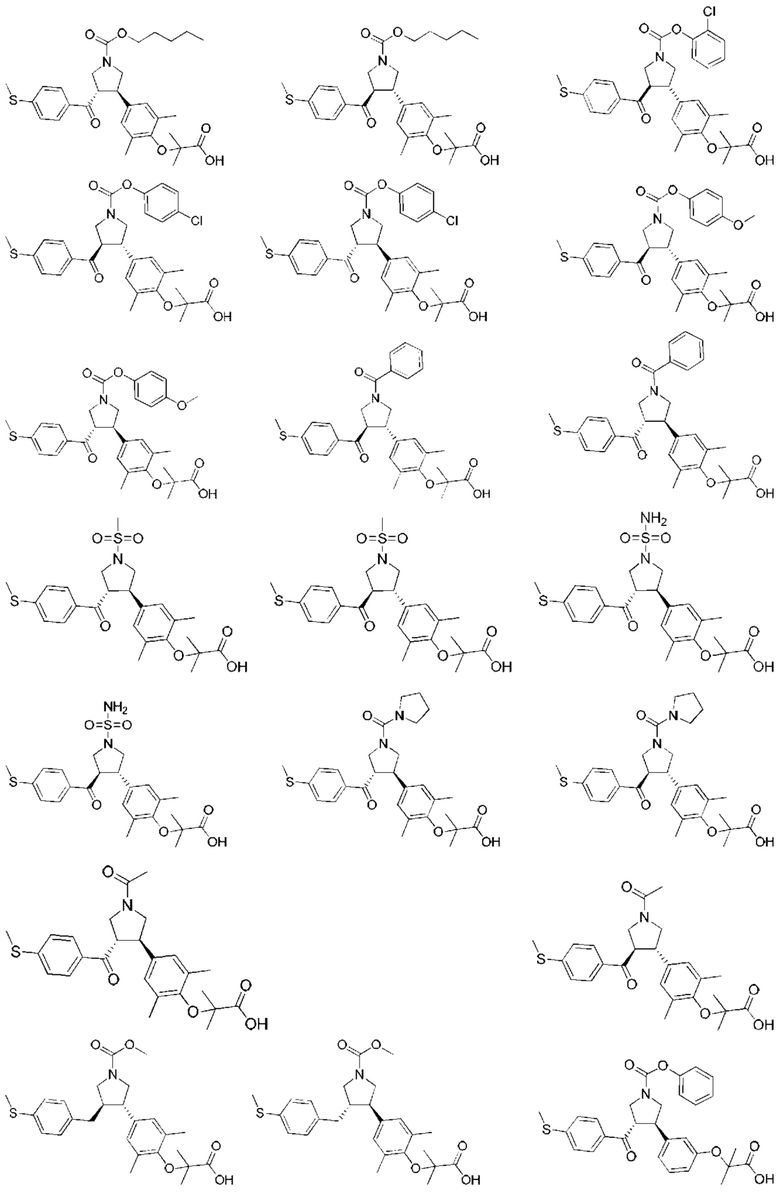
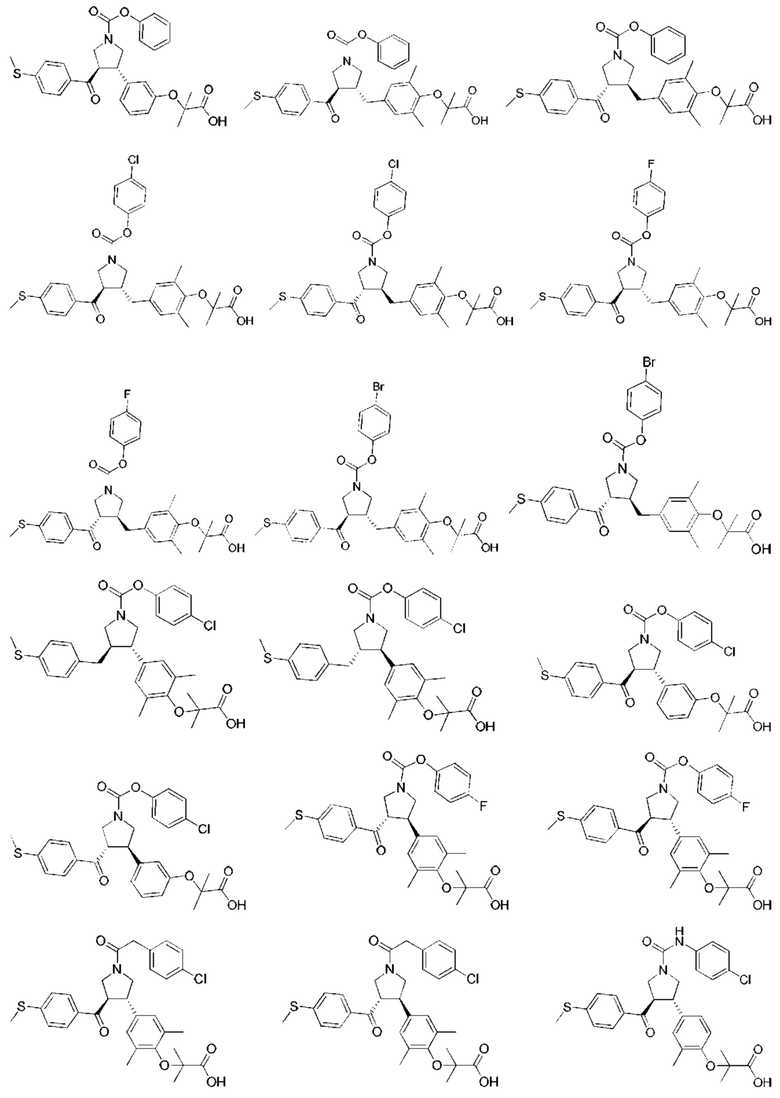
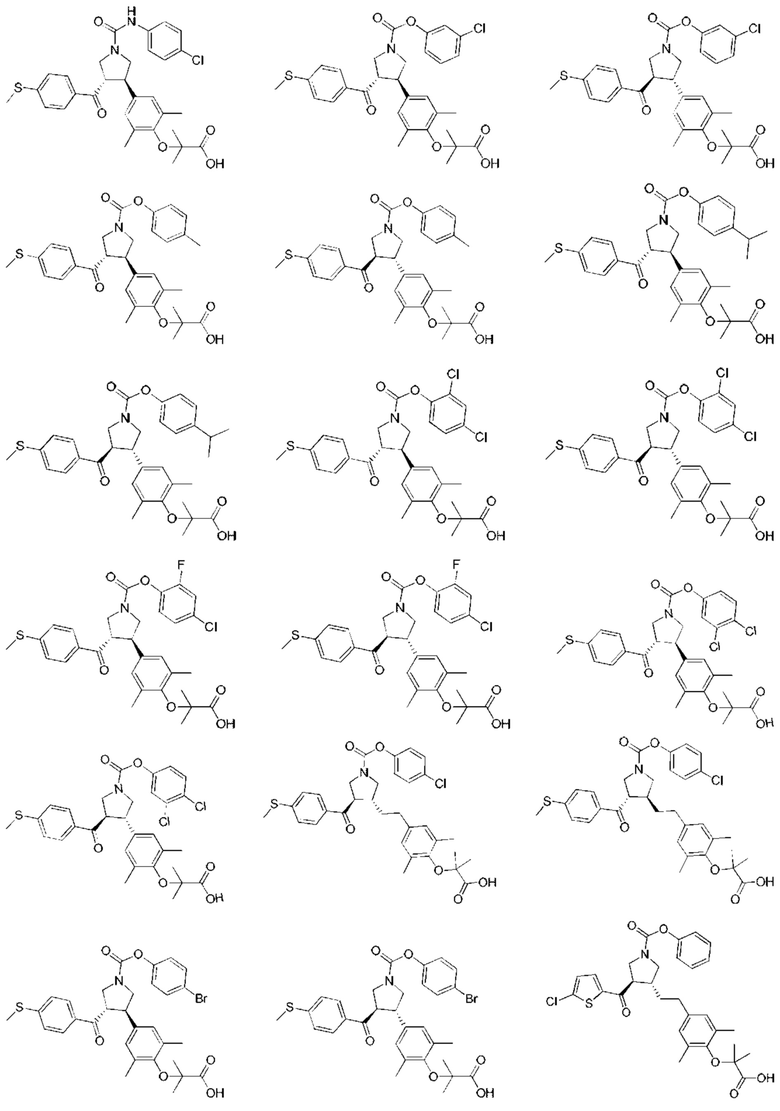
В некоторых вариантах реализации настоящего изобретения описанное выше соединение или его фармацевтически приемлемая соль выбрано из:
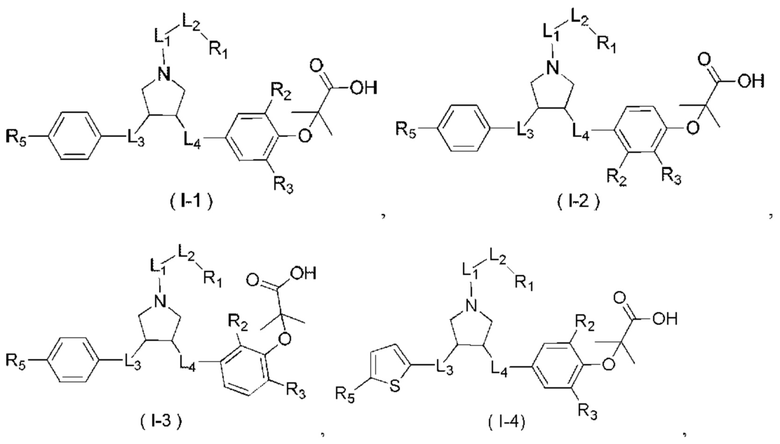
где R1, R2, R3, R5, L1, L2, L3, L4 такие, как определено выше.
В настоящем изобретении дополнительно предложено соединение, представленное формулой, выбранной из:
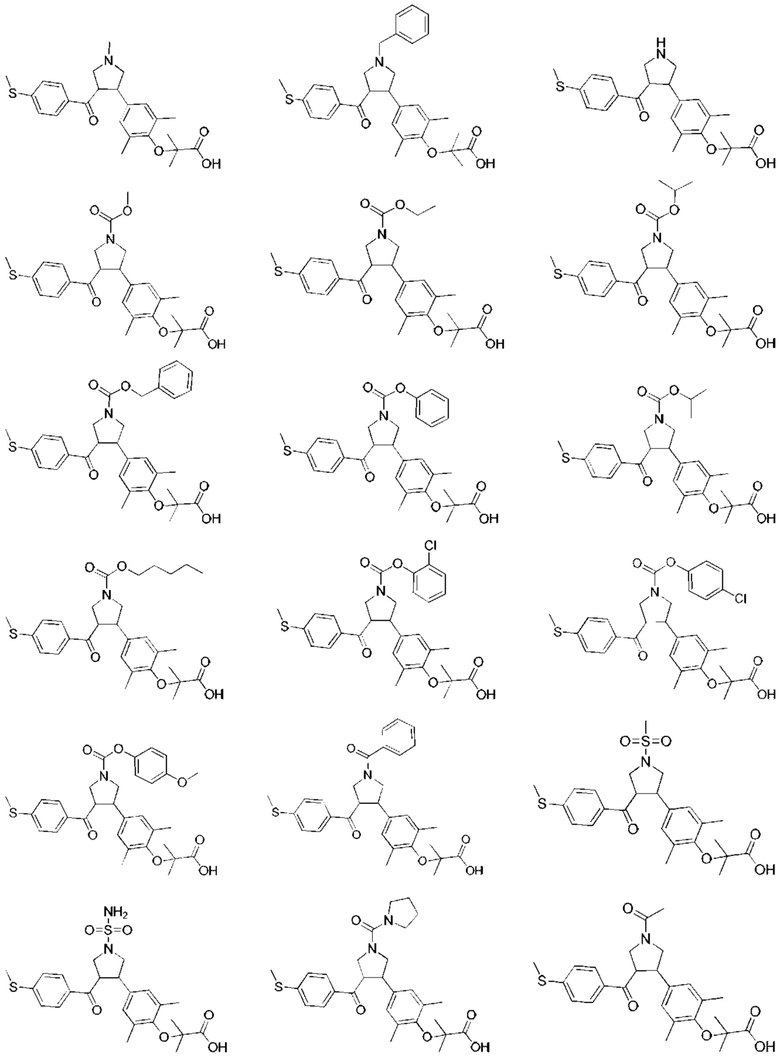
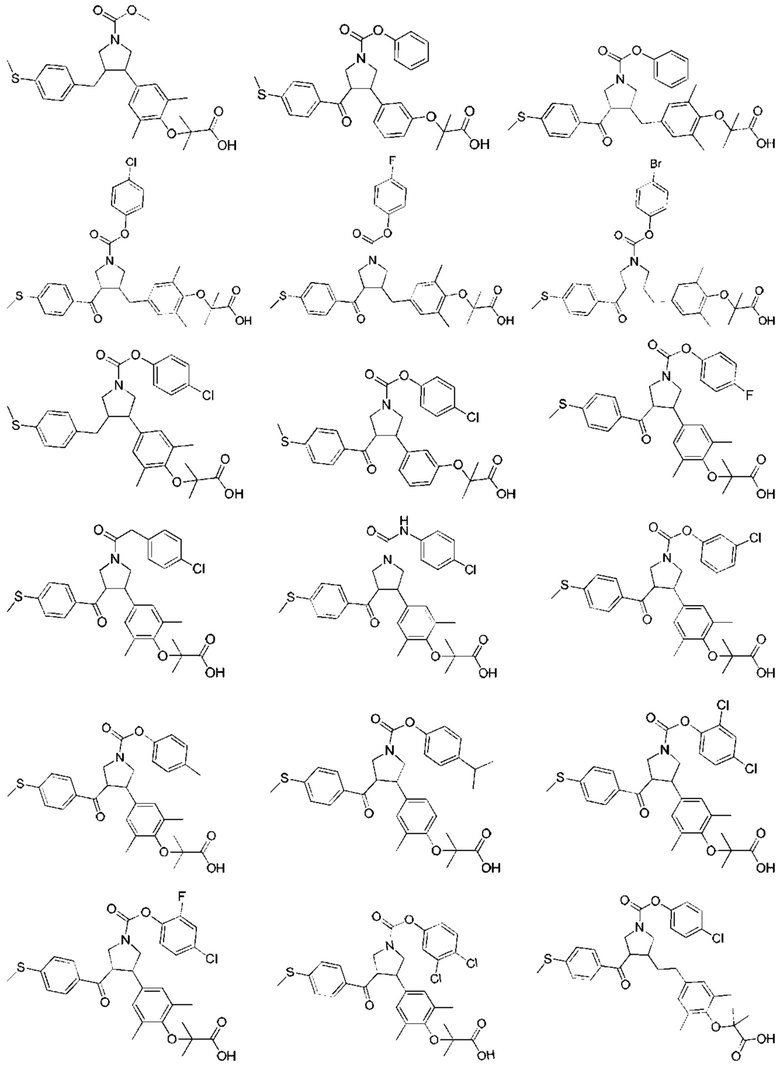
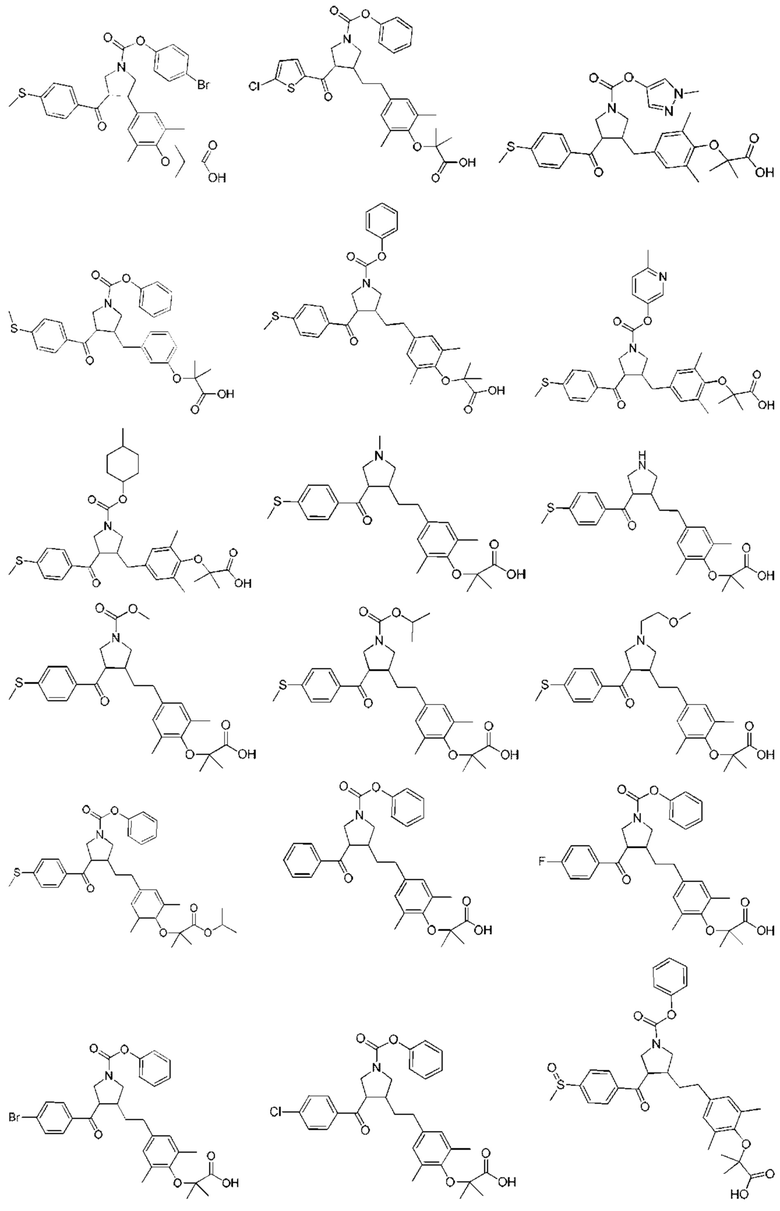
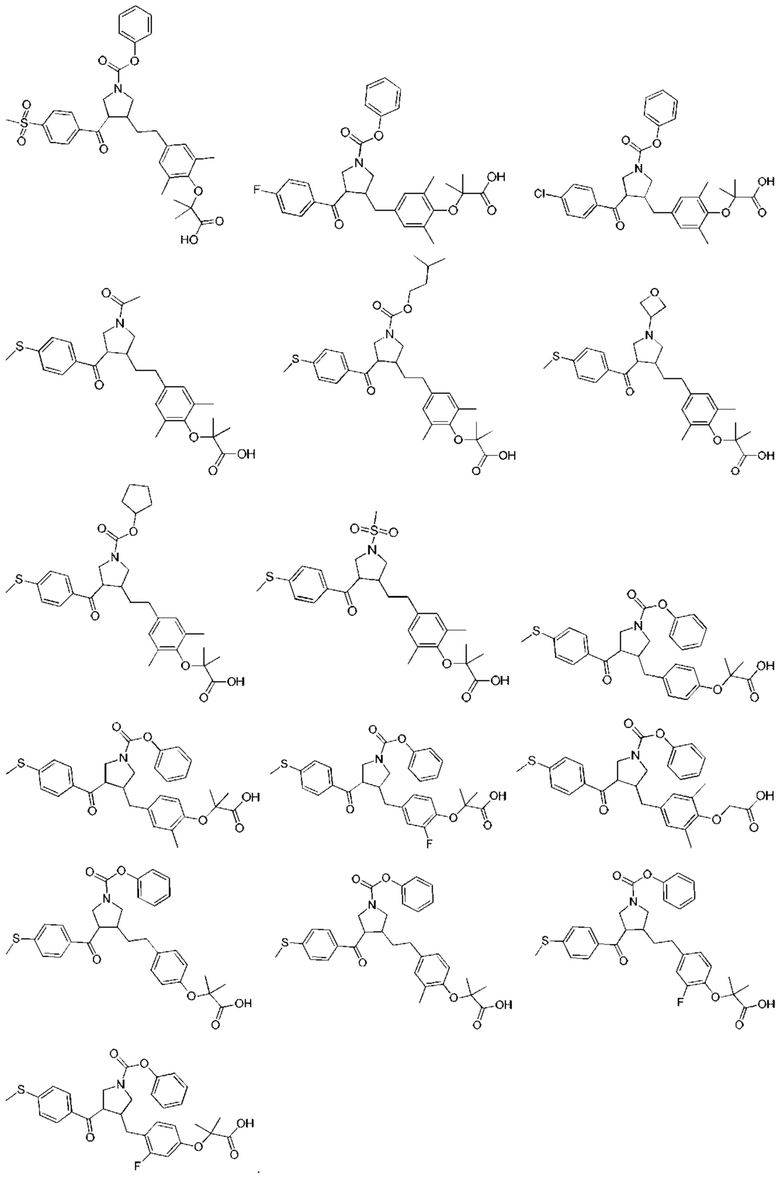
В некоторых вариантах реализации настоящего изобретения описанное выше соединение выбрано из:
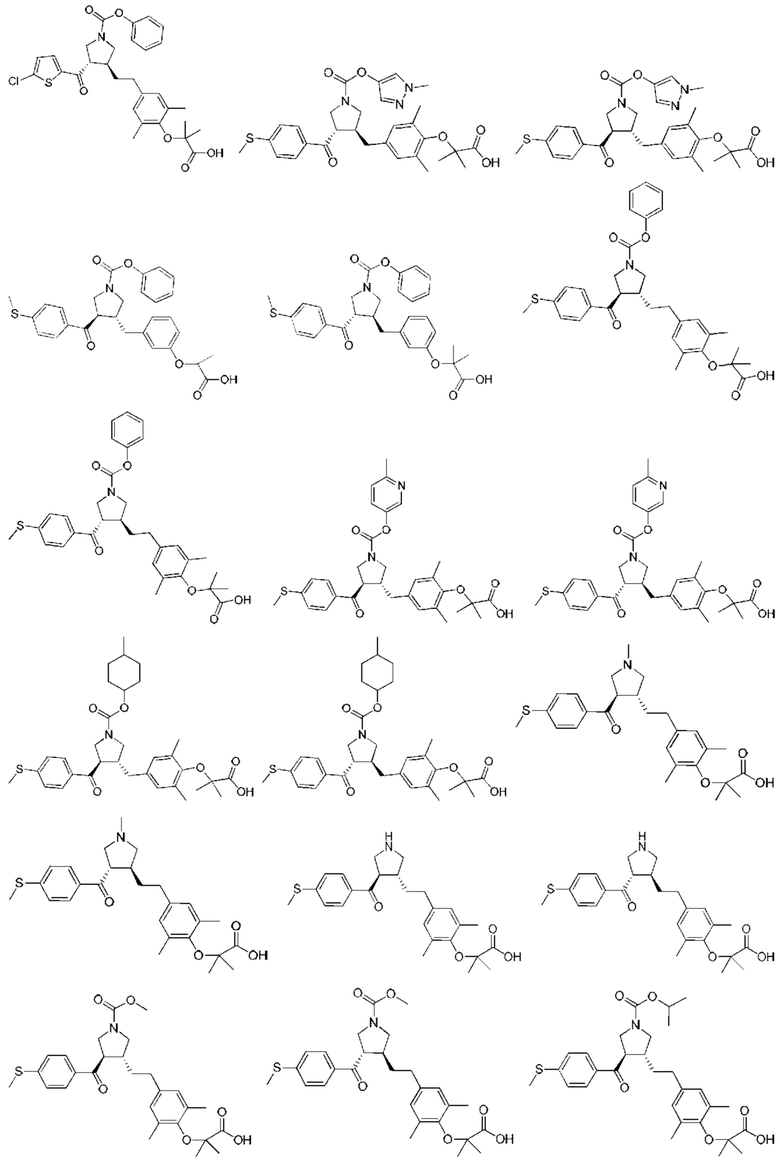
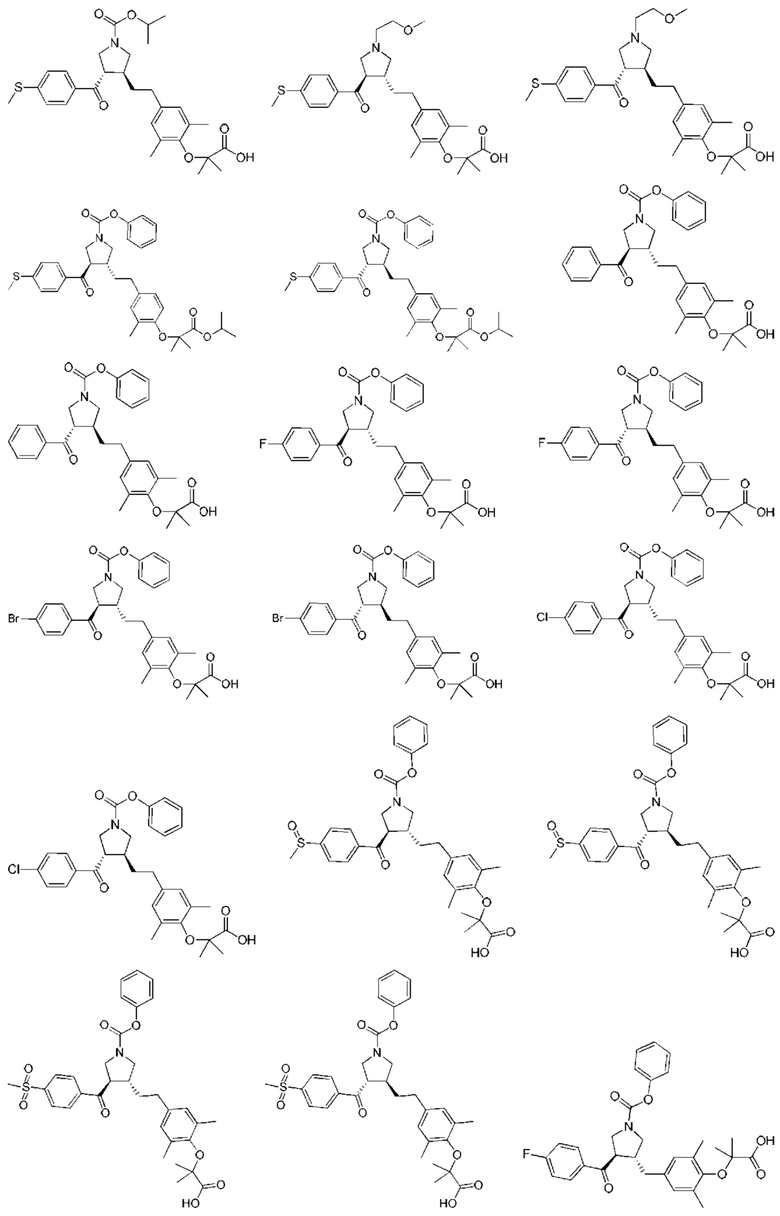
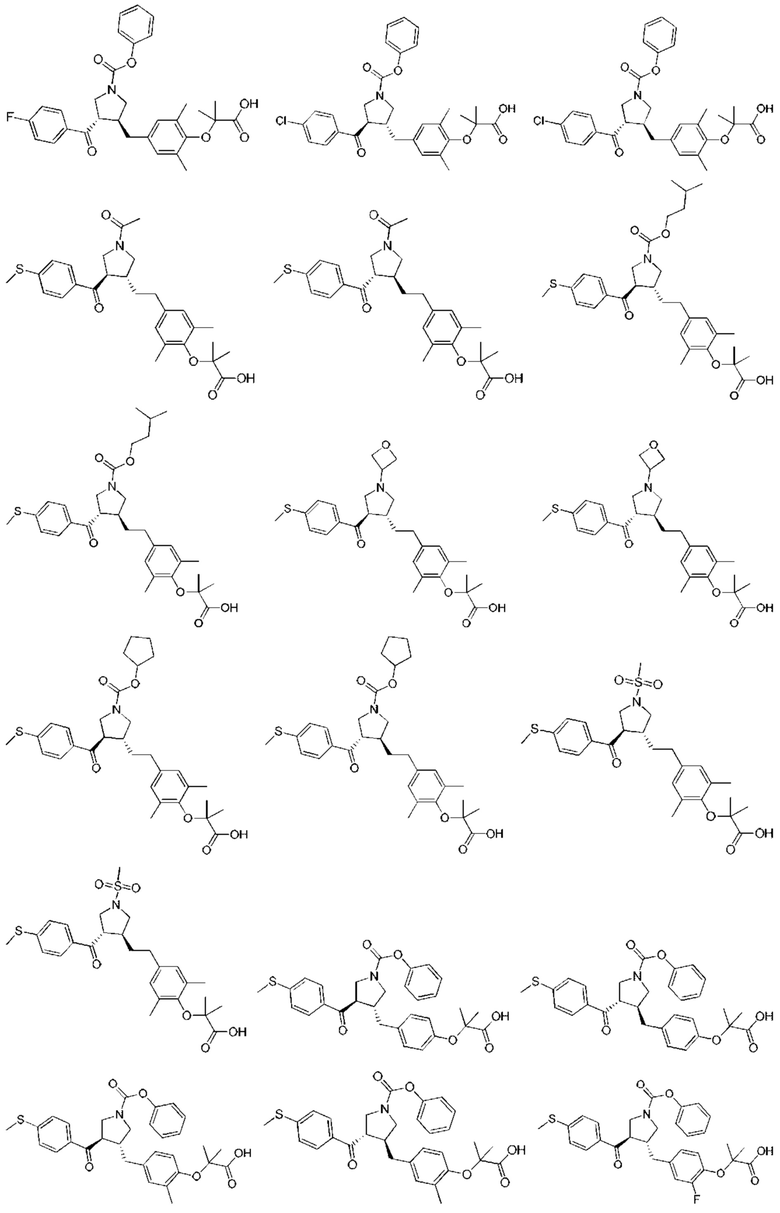
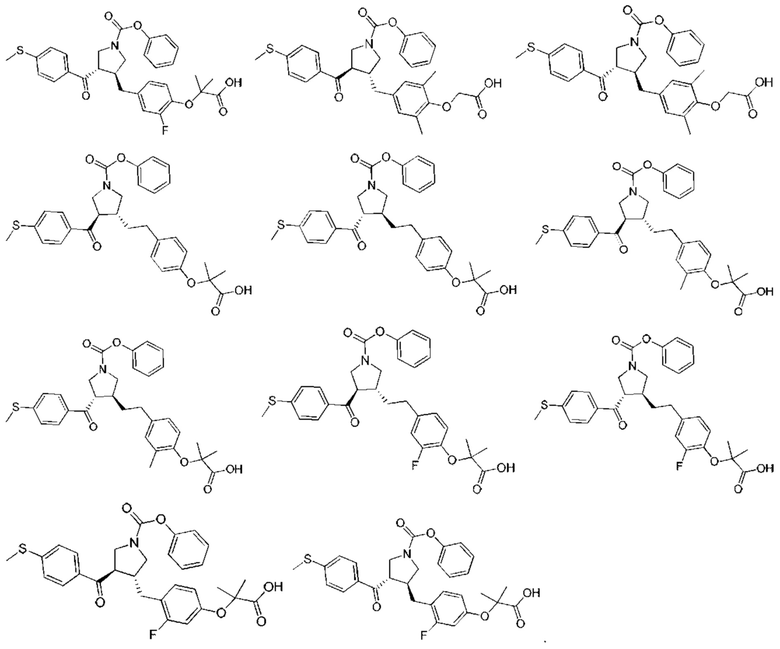
Настоящее изобретение может дополнительно включать некоторые варианты реализации, полученные из любой комбинации описанных выше переменных.
В настоящем изобретении дополнительно предложена фармацевтическая композиция, содержащая терапевтически эффективное количество описанного выше соединения или его фармацевтически приемлемой соли в качестве активного ингредиента, а также фармацевтически приемлемый носитель.
В настоящем изобретении дополнительно предложено применение описанного выше соединения или его фармацевтически приемлемой соли или описанной выше композиции для получения лекарственного средства для лечения нарушений, связанных с рецептором PPAR.
В настоящем изобретении дополнительно предложено применение описанного выше соединения или его фармацевтически приемлемой соли или описанной выше композиции для получения лекарственного средства для лечения неалкогольного стеатогепатита и сопутствующего фиброза, инсулинорезистентности, первичного билиарного холангита, дислипидемии, гиперлипидемии, гиперхолестеринемии, атеросклероза, гипертриглицеридемии, сердечно-сосудистого заболевания, ожирения.
Технический эффект
В настоящем изобретении описан класс производных пирролидина формулы (I) в качестве агонистов PPAR и способ их получения, где указанный класс соединений можно применять для лечения некоторых заболеваний, связанных с путями, ассоциированными с рецептором PPAR (таких как неалкогольный стеатогепатит и сопутствующий фиброз, инсулинорезистентность, первичный билиарный холангит, дислипидемия, гиперлипидемия, гиперхолестеринемия, атеросклероз, гипертриглицеридемия, сердечно-сосудистое заболевание, ожирение и т.д.). По сравнению с традиционными агонистами PPAR указанный класс агонистов имеет улучшенную активность, повышенную селективность и превосходную эффективность.
Определение и объяснение
Если не указано иное, приведенные далее термины и фразы при использовании в настоящем документе имеют следующие значения. Конкретный(-ую) термин или фразу, для которого(-ой) отсутствует конкретное определение, не следует рассматривать как неопределенный(-ую) или неоднозначный(-ую), но следует рассматривать в обычном значении. Торговая марка при использовании настоящем документе относится к соответствующему коммерчески доступному продукту или его активному ингредиенту. Термин «фармацевтически приемлемый» при использовании в настоящем документе означает что соединения, материалы, композиции и/или лекарственные формы можно применять в контакте с тканями человека и животного, что может быть определено в рамках клинически обоснованной экспертизы, в отсутствие избыточной токсичности, раздражения, аллергических реакций или других проблем или осложнений и при приемлемом отношении польза/риск.
Термин «фармацевтически приемлемая соль» относится к солям соединений согласно настоящему изобретению, соли получают из соединений, содержащих специфический(-е) заместитель(-и), и относительно нетоксичных кислот или щелочей. Если соединения согласно настоящему изобретению содержат относительно кислые группы, то можно получать соль присоединения щелочи путем приведения достаточного количества щелочи в контакт с нейтральной формой указанных соединений в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения щелочи включают соли натрия, калия, кальция, аммония, органического амина или магния и т.д. Если соединения согласно настоящему изобретению содержат относительно щелочные функциональные группы, то можно получать соль присоединения кислоты путем приведения достаточного количества кислоты в контакт с нейтральной формой указанных соединений в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли неорганических кислот, где неорганические кислоты включают, например, хлороводородную кислоту, бромоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонатный радикал, фосфорную кислоту, моногидрофосфатный радикал, дигидрофосфатный радикал, серную кислоту, гидросульфатный радикал, йодоводородную кислоту, фосфористую кислоту и т.д.; и соли органических кислот, где органические кислоты включают, например, уксусную кислоту, пропановую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфокислоту, п-толуолсульфокислоту, лимонную кислоту, винную кислоту, метансульфокислоту и т.д.; и соли аминокислот, таких как аргинин и т.д.; а также соли органических кислот, таких как глюкуроновая кислота и т.д. (см. Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторые конкретные соединения согласно настоящему изобретению содержат щелочные и кислые группы, и, таким образом, их можно превращать в соль присоединения щелочи или кислоты.
Предпочтительно нейтральная форма соединения может быть восстановлена традиционным способом путем приведения соли в контакт с щелочью или кислотой и последующего выделения исходного соединения. Исходная форма соединения отличается от соли определенными физическими свойствами, например, растворимостью в полярном растворителе.
Термин «фармацевтически приемлемая соль» в настоящем документе относится к производному соединения согласно настоящему изобретению, где исходное соединение модифицировано путем образования соли с кислотой или щелочью. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, соли неорганических или органических кислот с щелочными группами, такими как амины; соли щелочных металлов или органические соли с кислотными радикалами, такими как карбоновая кислота. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммонийные соли исходного соединения, такие как соли, полученные из нетоксичных неорганических или органических кислот. Обычные нетоксичные соли включают, но не ограничиваются ими, соли, полученные из неорганических и органических кислот, выбранных из 2-ацетоксибензойной кислоты, 2-гидроксиэтансульфокислоты, уксусной кислоты, аскорбиновой кислоты, бензолсульфокислоты, бензойной кислоты, гидрокарбонатного радикала, угольной кислоты, лимонной кислоты, эдетовой кислоты, этандисульфокислоты, этансульфокислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромоводородной кислоты, хлороводородной кислоты, гидроиодата, гидроксила, гидроксинафталина, изетионовой кислоты, молочной кислоты, лактозы, додецилсульфокислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфокислоты, азотной кислоты, щавелевой кислоты, памовой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, альдегида полигалактозы, пропановой кислоты, салициловой кислоты, стеариновой кислоты, фолината, янтарной кислоты, аминосульф окис лоты, п-аминобензолсульфокислоты, серной кислоты, таннина, винной кислоты и п-толуолсульфокислоты.
Фармацевтически приемлемые соли согласно настоящему изобретению можно синтезировать из исходного соединения, содержащего кислотную или щелочную группу, традиционным химическим способом. В общем случае, указанные соли получают путем взаимодействия указанных соединений в форме свободной кислоты или щелочи со стехиометрическим количеством подходящей щелочи или кислоты в воде или органическом растворителе или в их смеси. Как правило, предпочтительными являются неводные среды, например, диэтиловый эфир, этилацетат, этанол, изопропанол, ацетонитрил и т.д.
Помимо солевой формы соединения, предложенные в настоящем изобретении, могут иметь форму пролекарства. Пролекарства соединений, таких как описано в настоящем документе, могут легко претерпевать химическую конверсию до соединений согласно настоящему изобретению в физиологических условиях. Кроме того, пролекарства могут превращаться в соединения согласно настоящему изобретению in vivo посредством химических или биохимических процессов.
Некоторые соединения согласно настоящему изобретению могут присутствовать в несольватированной или сольватированной форме, включая форму гидрата. В общем случае несольватированная форма и сольватированная форма сравнимы друг с другом, и обе они включены в объем настоящего изобретения.
Некоторые соединения согласно настоящему изобретению могут содержать асимметрические атомы углерода (оптические центры) или двойные связи. Все рацематы, диастереомеры, геометрические изомеры и отдельные изомеры включены в объем настоящего изобретения.
Если не указано иное, клиновидную связь и пунктирную связь ( ) используют для указания абсолютной конфигурации стереоцентра; волнистую линию
) используют для указания абсолютной конфигурации стереоцентра; волнистую линию 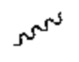 используют для указания клиновидной или пунктирной связи (
используют для указания клиновидной или пунктирной связи ( или
или  ), и
), и 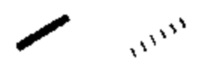 используют для указания относительной конфигурации стереоцентра. Если соединения, такие как описано в настоящем документе, содержат олефиновую двойную связь или другие центры геометрической асимметрии, и отсутствуют иные определения, то они могут включать Е-, Z-геометрические изомеры. Аналогично, все таутомеры включены в объем настоящего изобретения.
используют для указания относительной конфигурации стереоцентра. Если соединения, такие как описано в настоящем документе, содержат олефиновую двойную связь или другие центры геометрической асимметрии, и отсутствуют иные определения, то они могут включать Е-, Z-геометрические изомеры. Аналогично, все таутомеры включены в объем настоящего изобретения.
Соединения согласно настоящему изобретению могут присутствовать в специфической геометрической или стереоизомерной форме. Согласно настоящему изобретению можно ожидать, что все указанные соединения включают цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры и их рацемические смеси, а также другие смеси, такие как смеси, обогащенные энантиомером или диастереомером, все смеси включены в объем настоящего изобретения. Заместители, такие как алкил и т.д., могут содержать дополнительные асимметрические атомы углерода. Все указанные изомеры и их смеси включены в объем настоящего изобретения.
Оптические активные (R)- и (S)-изомеры и D- и L-изомеры можно получать путем хирального синтеза или с использованием хиральных реагентов или других традиционных технологий. Если требуется один из энантиомеров соединения согласно настоящему изобретению, то его можно получать путем асимметрического синтеза или дериватизации с использованием хирального вспомогательного вещества, после чего можно разделять полученную смесь диастереомеров и обеспечивать чистый желаемый энантиомер путем отщепления вспомогательной группы. В качестве альтернативы, если молекула содержит щелочную (например, амино) или кислую (например, карбоксил) функциональную группу, то можно проводить ее взаимодействие с подходящей оптически активной кислотой или щелочью для получения диастереомерных солей, которые в свою очередь подвергают диастереомерному разделению традиционным способом, хорошо известным в данной области техники, и затем выделяют для получения чистого энантиомера. Кроме того, выделение энантиомеров и диастереомеров обычно проводят путем хроматографии, в которой можно применять хиральную неподвижную фазу, необязательно в комбинации со способом химической дериватизации (например, путем получения карбамата из амина).
Соединения согласно настоящему изобретению могут содержать изотоп(-ы) одного или более атомов в соединении в отличных от природных отношениях. Например, соединения могут быть меченными радиоизотопом(-ами), таким(-ими) как тритий (3Н), йод-125 (125I) или С-14 (14С). Все изотопные формы соединений согласно настоящему изобретению включены в объем настоящего изобретения независимо от их радиоактивности.
Термин «фармацевтически приемлемый носитель» относится к любому препарату или среде-носителю, который(-ая) может доставлять эффективное количество активных веществ согласно настоящему изобретению в отсутствие отрицательного влияния на биоактивность активных веществ и токсических и побочных эффектов у субъектов или пациентов. Типовые носители включают воду, масла, растительные и минеральные, кремовую основу, основу лосьона, основу мази и т.д. Указанные материалы основы включают суспендирующие агенты, загустители, усилители чрескожного всасывания и т.д. Препараты хорошо известны специалистам в области косметики или местных лекарственных средств. Другую информацию о носителях можно найти в Remington: The Science and Practice of Pharmacy, 21-е изд., Lippincott, Williams & Wilkins (2005), содержание которой включено в настоящую заявку посредством ссылки.
Термин «вспомогательное вещество» в общем случае относится к носителям, разбавителям и/или средам, требуемым для получения эффективной фармацевтической композиции.
Если рассматривать лекарственное средство или фармацевтически активный агент, то термин «эффективное количество» или «терапевтически эффективное количество» относится к количеству лекарственного средства или медикамента, которое может обеспечивать желаемый эффект в отсутствие токсичности. Что касается пероральной лекарственной формы согласно настоящему изобретению, то «эффективное количество» одного активного вещества в композиции относится к количеству, требуемому для обеспечения желаемого эффекта при использовании в комбинации с другим активным веществом в композиции. Определяемое эффективное количество может быть различным от человека к человеку в зависимости от возраста и общего состояния здоровья субъекта и конкретного активного вещества. Соответствующее эффективное количество для каждого индивидуального случая может быть определено специалистом в данной области техники в рамках традиционной экспериментальной работы.
Термин «активный(-е) ингредиент(-ы)», «терапевтический(-е) агент(-ы)», «активное(-ые) вещество(-а)» или «активный(-е) агент(-ы)» относится к химической частице, которая может эффективно излечивать нарушения, заболевания или состояния у субъекта.
Фраза «необязательный» или «необязательно» означает, что описываемое далее явление или условие может происходить (или выполняться) не во всех случаях, то есть, описание включает случаи, где явление или условие происходит (или выполняется), и случаи, где явление или условие не происходит (или не выполняется).
Термин «замещенный» означает, что любые один или более атомов водорода при конкретном атоме заменены на заместитель, который может включать тяжелые формы водорода и водород, если валентность конкретного атома является нормальной, и замещенное соединений стабильно. Если заместитель представляет собой кетоновую группу (т.е. =O), это означает, что заменены два атома водорода. Кетоновое замещение при ароматической группе не происходит. Термин «необязательно замещенный» означает, что замещение может происходить или не происходить. Если отсутствуют иные определения, заместители могут иметь любой тип и присутствовать в любом количестве, если это допускается с химической точки зрения.
Если какая-либо переменная (например, R) встречается в составе или структуре соединения более одного раза, то ее определение в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, то группа может быть необязательно замещена не более чем 2 R, и каждый R в каждом случае выбран независимо. Кроме того, комбинация заместителей и/или их форм допустима, только если указанная комбинация обеспечивает получение стабильного соединения.
Если индекс при линкерной группе равен нулю, как в случае -(CRR)0-, это означает, что линкерная группа представляет собой простую связь.
Если переменная выбрана из простой связи, это означает, что две группы, связанные указанной переменной, связаны друг с другом непосредственно. Например, если L в структуре A-L-Z представляет собой простую связь, то указанная структура фактически представляет собой A-Z.
Если заместитель отсутствует, это означает, что заместитель не существует. Например, если X в структуре А-Х отсутствует, это означает, что структура фактически представляет собой А. Если заместитель может образовывать связь более чем с одним атомом в кольце, то заместитель может быть связан с любым атомом в кольце. Например, структурное звено 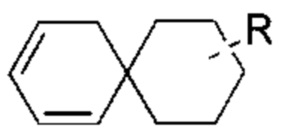 или
или 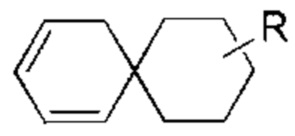 означает, что заместитель R может быть присоединен по любому участку циклогексила или циклогексадиена. Если не указано, по какому атому указанный заместитель присоединен к замещенной группе, то заместитель может быть присоединен по любому атому в указанной группе. Например, пиридил в качестве заместителя может быть присоединен к замещенной группе по любому атому углерода пиридильного кольца. Если направление связывания в указанной линкерной группе не указано, то направление связывания является случайным. Например, если линкерная группа L в
означает, что заместитель R может быть присоединен по любому участку циклогексила или циклогексадиена. Если не указано, по какому атому указанный заместитель присоединен к замещенной группе, то заместитель может быть присоединен по любому атому в указанной группе. Например, пиридил в качестве заместителя может быть присоединен к замещенной группе по любому атому углерода пиридильного кольца. Если направление связывания в указанной линкерной группе не указано, то направление связывания является случайным. Например, если линкерная группа L в 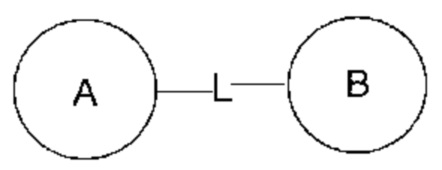 представляет собой -M-W-, то -M-W- может связывать кольцо А и кольцо В не только в направлении, идентичном порядку написания слева направо, то есть с образованием
представляет собой -M-W-, то -M-W- может связывать кольцо А и кольцо В не только в направлении, идентичном порядку написания слева направо, то есть с образованием 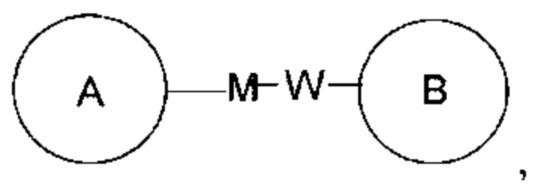 но также связывает кольцо А и кольцо В в направлении, противоположном порядку написания слева направо, то есть с образованием
но также связывает кольцо А и кольцо В в направлении, противоположном порядку написания слева направо, то есть с образованием 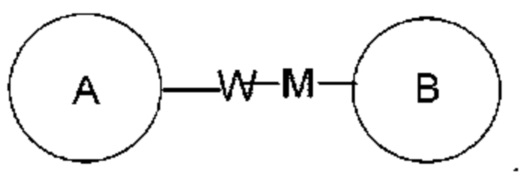 Комбинация линкерных групп, заместителей и/или их форм допустима, только если указанная комбинация обеспечивает получение стабильного соединения.
Комбинация линкерных групп, заместителей и/или их форм допустима, только если указанная комбинация обеспечивает получение стабильного соединения.
Если отсутствуют иные определения, термин «гетеро-» относится к гетероатому или гетероатомной группе (т.е. к группе атомов, содержащей гетероатом(-ы)), включая атом(-ы), отличные от углерода (С) и водорода (Н), а также к группе атомов, содержащей указанный(-е) гетероатом(-ы), например, кислород (О), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Al), бор (В), -О-, -S-, =O, =S, -С(=O)O-, -С(=O)-, -C(=S)-, -S(=O), -S(=O)2- и необязательно замещенный -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2 N(H)- или -S(=O)N(H)-.
Если отсутствуют иные определения, «кольцо» относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкилену, гетероциклоалкилену, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Указанное кольцо включает монокольцо, связанное кольцо, спирокольцо, конденсированное кольцо или мостиковое кольцо. Количество атомов в кольце, как правило, определяют как количество членов в кольце. Например, «5-7-членное кольцо» означает, что 5-7 атомов распределены по кругу. Если отсутствуют иные определения, кольцо необязательно содержит 1-3 гетероатомов. Таким образом, «5-7-членное кольцо» включает, например, фенил, пиридинил и пиперидил; и, с другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает пир ид ил и пиперидил, но не включает фенил. Термин «цикло-/кольцо» дополнительно включает систему колец, содержащую по меньшей мере одно кольцо, где каждое «кольцо» независимо удовлетворяет приведенному выше определению.
Если отсутствуют иные определения, термин «гетероцикл» или «гетероциклил» относится к стабильному моно-, двойному или тройному кольцу, содержащему гетероатом(-ы) или гетероатомную(-ые) группу(-ы), причем указанное кольцо может быть насыщенным, частично ненасыщенным или ненасыщенным (ароматическим) и может содержать атом(-ы) углерода и 1, 2, 3 или 4 гетероатома в кольце, независимо выбранные из N, О и S, где любой из описанных выше гетероциклов может быть конденсирован с фенильным кольцом с образованием двойного кольца. Гетероатомы азота и серы могут быть необязательно окисленными (т.е. NO и S(O)p, где р равен 1 или 2). Атом азота может быть замещенным или незамещенным (т.е. N или NR, где R представляет собой Н или другой заместитель, такой как определено в настоящем описании). Гетероцикл может быть присоединен к боковой группе через любой гетероатом или атом углерода с образованием стабильной структуры. Если полученное соединение является стабильным, то гетероцикл, такой как описано в настоящем документе, может быть замещен по атому углерода или атому азота. Атом азота в гетероцикле необязательно является четвертичным. В предпочтительном варианте реализации, если общее количество атомов S и О в гетероцикле больше одного, то указанные гетероатомы не находятся по соседству друг с другом. В другом предпочтительном варианте реализации общее количество атомов S и О в гетероцикле составляет не более 1. При использовании в настоящем описании термин «ароматический гетероциклил» или «гетероарил» относится к стабильному 5-, 6-, 7-членному моноциклическому или бициклическому или 7-, 8-, 9- или 10-членному бициклическому гетероциклильному ароматическому кольцу, содержащему атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, независимо выбранные из N, О и S. Атом азота может быть замещенным или незамещенным (т.е. N или NR, где R представляет собой Н или другой заместитель, такой как определено в настоящем описании). Гетероатомы азота и серы необязательно могут быть окисленными (т.е. NO и S(O)p, где р равен 1 или 2). Следует отметить, что общее количество атомов S и О в ароматическом гетероцикле составляет не более 1. Мостиковое кольцо также включено в определение гетероцикла. Мостиковое кольцо может быть образовано, если один или более атомов (т.е. С, О, N или S) соединяют два несоседних атома углерода или азота. Предпочтительно мостиковое кольцо включает, но не ограничивается ими, один атом углерода, два атома углерода, один атом азота, два атома азота и одну связь углерод-азот. Следует отметить, что один мостик всегда превращает моноцикл в тройной цикл. В мостиковом кольце заместитель(-и) при кольце также может(-гут) быть присоединен(-ы) к мостику.
Примеры гетероциклических соединений включают, но не ограничиваются ими: акридинил, азоцинил, бензимидазолил, бензофурил, бензотиофурил, бензотиофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хроменил, циннолинил, декагидрохинолил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуран, фурил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1Н-индазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изобензофурил, изоиндолил, изоиндолинил, изохинолил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксииндолил, пиримидил, фенантридинил, фенантролинил, феназинил, фенотиазинил, бензоксантинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидилин, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридил, пирролидинил, пирролинил, 2H-пирролил, пирролил, хиназолинил, хинолил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофурил, тетрагидроизохинолил, тетрагидрохинолил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолил, тиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил. Также включены конденсированные кольца и спирокольца.
Если отсутствуют иные определения, то термин «гидрокарбил» или его конкретные формы (например, алкил, алкенил, алкинил, арил и т.д.), как таковой или в составе другого заместителя, представляет собой линейный, разветвленный или циклический углеводородный радикал или их комбинацию и может являться: полностью насыщенным (например, алкил), моно- или полиненасыщенным (например, алкенил, алкинил, арил); моно- или полизамещенным; одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метин); и включает двухвалентную(-ые) или поливалентную(-е) группу(-ы) атомов, содержащую(-ие) указанное количество атомов углерода (например, С1-С12 соответствует 1-12 атомам углерода, С1-12 выбран из C1, С2, С3, С4, С5, С6, С7, С8, С9, С10, С11 и С12; С3-12 выбран из С3, С4, С5, С5, С7, C8, С9, С10, С11 и С12). Термин «гидрокарбил» включает, но не ограничивается ими, алифатический гидрокарбил и ароматический гидрокарбил, где алифатический гидрокарбил может быть линейным или циклическим и, в частности, включает, но не ограничивается ими, алкил, алкенил, алкинил; и ароматический гидрокарбил включает, но не ограничивается ими, 6-12-членные ароматические гидрокарбилы, такие как фенил, нафтил и т.д. В некоторых вариантах реализации термин «гидрокарбил» представляет собой линейные или разветвленные группы атомов или их комбинации и может быть полностью насыщенным, моно- или полиненасыщенным и может включать двухвалентную(-ые) и поливалентную(-ые) группу(-ы) атомов. Примеры насыщенной углеводородной группы атомов включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, изобутил, циклогексил, (циклогексил)метил, циклопропилметил и гомологи или изомеры н-пентила, н-гексила, н-гептила, н-октила и других групп атомов. Ненасыщенный гидрокарбил может содержать одну или более двойных связей или тройных связей, примеры включают, но не ограничиваются ими, этенил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и высшие гомологи и изомеры.
Если отсутствуют иные определения, то термин «гетерогидрокарбил» или его конкретные формы (например, гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т.д.), как таковой или в комбинации с другим термином, представляет собой стабильную линейную, разветвленную или циклическую углеводородную группу атомов или их комбинацию и состоит из атомов углерода и по меньшей мере одного гетероатома. В некоторых вариантах реализации термин «гетероалкил», как таковой или в комбинации с другим термином, представляет собой стабильную линейную, разветвленную углеводородную группу атомов или их комбинацию и состоит из атомов углерода и по меньшей мере одного гетероатома. В типовом варианте реализации гетероатом выбран из В, О, N и S, где атомы азота и серы необязательно являются окисленными, и гетероатом азота необязательно является четвертичным. Гетероатом или гетероатомная группа может быть расположен(-а) в любом участке гетерогидрокарбила, включая участок, через который гетерогидрокарбил присоединен к оставшемуся фрагменту молекулы. Тем не менее, термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкокси) являются традиционными и относятся к алкильным группам, присоединенным к оставшемуся фрагменту молекулы через один атом кислорода, амино или атом серы, соответственно. Примеры включают, но не ограничиваются ими, -СН2-СН2-О-СН3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -СН2-СН2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-СН3. Последовательно могут быть расположены не более двух гетероатомов, например, -CH2-NH-ОСН3.
Если отсутствуют иные определения, термин «циклогидрокарбил», «гетероциклогидрокарбил» или их конкретные формы (например, арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкилен, гетероциклоалкилен, циклоалкинил, гетероциклоалкинил и т.д.), как таковой или в комбинации с другими терминами, представляет собой циклизованный «гидрокарбил», «гетерогидрокарбил», соответственно. Кроме того, в случае гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкила, гетероциклоалкила) гетероатом(-ы) может(-гут) быть расположен(-ы) в участке, через который гетероциклил присоединен к оставшемуся фрагменту молекулы. Примеры циклогидрокарбила включают, но не ограничиваются ими, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т.д. Неограничивающие примеры гетероциклила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидил, 2-пиперидил, 3-пиперидил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофураноиндол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
Если отсутствуют иные определения, термин «алкил» относится к линейному или разветвленному насыщенному гидрокарбилу и может быть монозамещенным (например, -CH2F) или полизамещенным (например, -CF3) и может быть одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метин). Примеры алкила включают метил (Me), этил (Et), пропил (например, н-пропил и изопропил), бутил (например, н-бутил, изобутил, втор-бутил, трет-бутил), пентил (например, н-пентил, изопентил, неопентил) и т.д.
Если отсутствуют иные определения, «алкенил» относится к алкилу, содержащему одну или более углерод-углеродных двойных связей в любом участке цепи, и может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры алкенила включают этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил и т.д.
Если отсутствуют иные определения, «алкинил» относится к алкилу, содержащему одну или более углерод-углеродных тройных связей в любом участке цепи, и может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры алкинила включают этинил, пропинил, бутинил, пентинил и т.д.
Если отсутствуют иные определения, циклоалкил включает любой стабильный циклический или полициклический гидрокарбил, в котором все атомы углерода являются насыщенными, и может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры указанных циклоалкилов включают, но не ограничиваются ими, циклопропил, норборнанил, [2.2.2]бициклооктил, [4.4.0]бициклодеканил и т.д.
Если отсутствуют иные определения, то циклоалкилен включает любой стабильный циклический или полициклический гидрокарбил, который содержит одну или более ненасыщенных углерод-углеродных двойных связей в любом участке кольца(колец) и может быть монозамещенным или полизамещенным, может быть одновалентным, двухвалентным или поливалентным. Примеры указанных циклоалкиленов включают, но не ограничиваются ими, циклопентенил, циклогексенил и т.д.
Если отсутствуют иные определения, то циклоалкинил включает любой стабильный циклический или полициклический гидрокарбил, который содержит одну или более углерод-углеродных тройных связей в любом участке кольца(колец) и может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным.
Если отсутствуют иные определения, то термин «галоген-» или «галоген», как таковой или в составе другого заместителя, представляет собой атом фтора, хлора, брома или йода. Кроме того, термин «галогеналкил» включает моногалогеналкил и полигалогеналкил. Например, термин «галоген-(С1-С4) алкил» включает, но не ограничивается ими, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил и 3-бромпропил и т.д. Если отсутствуют иные определения, то примеры галогеналкила включают, но не ограничиваются ими, трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
«Алкокси» представляет собой описанный выше алкил, который присоединен через кислородный мостик и содержит определенное количество атомов углерода. Если отсутствуют иные определения, то C1-6 алкокси включает C1, С2, С3, С4, С5 и С6 алкокси. Примеры алкокси включают, но не ограничиваются ими, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и втор-пентилокси.
Если отсутствуют иные определения, то термин «арил» относится к полиненасыщенному ароматическому углеводородному заместителю, может быть моно- или полизамещенным, может быть одно-, двух- или поливалентным и может быть моно- или полициклическим (например, 1-3 кольцо(-а), среди которых по меньшей мере одно кольцо является ароматическим и которые конденсированы или ковалентно связаны друг с другом). Термин «гетероарил» относится к ар илу (или кольцу), содержащему 1-4 гетероатомов. В типовом варианте реализации гетероатом выбран из В, N, О и S, где атомы азота и серы необязательно являются окисленными, и атом азота необязательно является четвертичным. Гетероарил может быть присоединен через гетероатом к оставшемуся фрагменту молекулы. Неограничивающие примеры арила или гетероарила включают фенил, нафтил, бифенил, пирролил, пиразолил, имидазолил, пиразинил, оксазолил, фенилоксазолил, изоксазолил, тиазолил, фуранил, тиенил, пиридил, пиримидил, бензотиазолил, пуринил, бензимидазолил, индолил, изохинолил, хиноксалинил, хинолил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместитель любой из описанных выше ар ильных и гетероар ильных циклических систем выбран из приемлемых заместителей, таких как описано ниже.
Если отсутствуют иные определения, то при использовании в комбинации с другими терминами (например, арилокси, арилтио, аралкил) арил включает арильные и гетероарильные кольца, такие как определено выше. Таким образом, термин «аралкил» включает группы, в которых арил присоединен к алкилу (например, бензил, фенилэтил, пиридилметил и т.д.) и к алкильным группам, в которых атом(-ы) углерода (например, метилен) заменен(-ы) на атом(-ы) кислорода, такие как феноксиметил, 2-пиридилоксиметил, 3-(1-нафтилокси)пропил и т.д.
Термин «уходящая группа» относится к функциональной группе или атому, которая(-ый) может быть заменена(-ы) на другую функциональную группу или атом по реакции замещения (например, по реакции нуклеофильного замещения). Например, типовые уходящие группы включают трифторметансульфонат; хлорид, бромид, йодид; сульфонат, например, метансульфонат, тозилат, n-бромбензолсульфонат, n-толуолсульфонат и т.д.; ацилокси, например, ацетокси, трифторацетокси и т.д.
Термин «защитная группа» включает, но не ограничивается ими, «амино-защитную группу», «гидроксил-защитную группу» или «меркапто-защитную группу». Термин «амино-защитная группа» относится к защитной группе, предназначенной для предотвращения вторичной реакции по атому азота в аминогруппе. Типовые амино-защитные группы включают, но не ограничиваются ими: формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Boc); арилметоксикарбонил, такой как карбобензокси (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-ди-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и т.д. Термин «гидроксил-защитная группа» относится к защитной группе, предназначенной для предотвращения вторичной реакции по гидроксильной группе. Типовые гидроксил-защитные группы включают, но не ограничиваются ими,: алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), n-метоксибензил (РМВ), 9-флуоренилметил (Fm) и бензгидрил (дифенилметил, DPM); силил, такой как триметилсилил (TMS) и дареда-бутилдиметилсилил (TBS) и т.д.
Соединения согласно настоящему изобретению можно получать различными способами синтеза, хорошо известными специалистам в данной области техники, включая варианты реализации, перечисленные ниже, варианты реализации, образованные вариантами реализации, перечисленными ниже, в комбинации с другими химическими способами синтеза, а также эквивалент(-ы), хорошо известный(-е) специалистам в данной области техники. Предпочтительные варианты реализации включают, но не ограничиваются ими, примеры настоящего изобретения.
Растворитель, такой как применяют в настоящем изобретении, может быть коммерчески доступным. В настоящем изобретении используют следующие сокращения: водн. обозначает водный; HATU обозначает гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины; EDC обозначает гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида; мХПБК обозначает 3-хлорпероксибензойную кислоту; экв. обозначает эквивалент; CDI обозначает карбонилдиимидазол; ДХМ обозначает дихлорметан; ПЭ обозначает петролейный эфир; DIAD обозначает диизопропилазодикарбоксилат; ДМФ обозначает N,N-диметилформамид; ДМСО обозначает диметилсульфоксид; EtOAc обозначает этилацетат; EtOH обозначает этанол; MeOD обозначает метанол; CBz обозначает бензилоксикарбонил, амино-защитную группу; Boc обозначает трет-бутилоксикарбонил, амино-защитную группу; НОАс обозначает уксусную кислоту; NaCNBH3 обозначает цианоборгидрид натрия; КТ обозначает комнатную температуру; О/N обозначает на ночь; ТГФ обозначает тетрагидрофуран; Вос2О обозначает ди-трет-бутилдикарбонат; ТФУК обозначает трифторуксусную кислоту; DIPEA обозначает диизопропилэтиламин; SOCl2 обозначает хлорид сульфоксида; CS2 обозначает дисульфид углерода; TsOH обозначает n-толуолсульф окис лоту; NFSI обозначает N-фтор-N-(бензолсульфонил)бензолсульфонамид; NCS обозначает 1-хлорпирролидин-2,5-дион; n-Bu4NF обозначает фторид тетрабутиламмония; iPrOH обозначает 2-пропанол; Тпл обозначает температуру плавления; LDA обозначает диизопропиламид лития.
Названия соединений вводили вручную или при помощи программного обеспечения ChemDraw®, названия коммерчески доступных соединений указаны в соответствии с названиями из каталогов производителей.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ РЕАЛИЗАЦИИ
Пример 1: Соединение 1
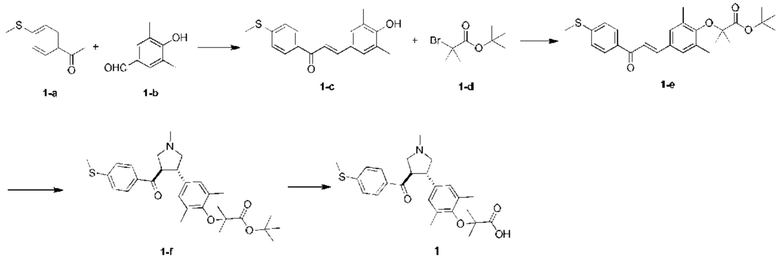
Этап 1: Соединение 1-c
Раствор соединения 1-а (5,00 г, 30,08 ммоль, 1,00 экв.) и соединения 1-b (4,52 г, 30,08 ммоль, 1,00 экв.) в HCl/МеОН (4 н., 40,01 мл, 5,32 экв.) перемешивали при 20°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт промывали раствором дихлорметан/МеОН (1:1,50 мл), в результате чего получали Соединение 1-c. МС: m/z (ионизация электрораспылением): 298,9 [М+1].
Этап 2: Соединение 1-е
Раствор соединения 1-c (2,00 г, 6,70 ммоль, 1,00 экв.), соединения 1-d (2,99 г, 13,40 ммоль, 2,49 мл, 2,00 экв.), карбоната калия (1,85 г, 13,40 ммоль, 2,00 экв.) и йодида калия (111,26 мг, 670,00 мкмоль, 0,10 экв.) в диметилсульфоксиде (30,00 мл) перемешивали в защитной атмосфере азота при 90°С в течение 12 ч. К реакционной смеси добавляли воду (30 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу промывали водой (20 мл × 2) и насыщенным солевым раствором (20 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали хроматографией на силикагеле (петролейный эфир/этилацетат = 3/1) с получением соединения 1-е.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,98-7,94 (m, 2Н), 7,71 (d, J=15,8 Гц, 1H), 7,42 (d, J=15,8 Гц, 1H), 7,34-7,29 (m, 2Н), 7,28 (s, 2Н), 2,55 (s, 3Н), 2,28 (s, 6Н), 1,52 (s, 9Н), 1,46 (s, 6Н).
Этап 3: Соединение 1-f
Раствор соединения 1-е (300,00 мг, 680,91 мкмоль, 1,00 экв.), 2-(метиламино)уксусной кислоты (151,65 мг, 1,70 ммоль, 2,50 экв.) и параформальдегида (368,02 мг, 4,09 ммоль, 6,00 экв.) в толуоле (10,00 мл) перемешивали в защитной атмосфере азота при 110°С в течение 12 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1), в результате чего получали Соединение 1-f.
МС: m/z (ионизация электрораспылением): 498,3 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,65 (d, J=8,5 Гц, 2H), 7,14 (d, J=8,5 Гц, 2H), 6,86 (s, 2Н), 3,93-3,85 (m, 1H), 3,66 (q, J=7,0 Гц, 1H), 3,03 (t, J=8,7 Гц, 2H), 2,97-2,91 (m, 1H), 2,80-2,73 (m, 1H), 2,48 (s, 3Н), 2,42 (s, 3Н), 2,18 (s, 6Н), 1,51 (s, 9Н), 1,41 (s, 6Н).
Этап 4: Соединение 1
В защитной атмосфере азота в раствор соединения 1-f (100,00 мг, 200,93 мкмоль, 1,00 экв.) в дихлорметане (5,00 мл) добавляли по каплям трифторуксусную кислоту (1,50 мл) при 0°С. Смесь перемешивали при 20°С в течение 1 ч. Смесь концентрировали при пониженном давлении. Остаток растворяли в этилацетате (30 мл). Органическую фазу промывали водой (10 мл × 2) и насыщенным солевым раствором (10 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат =1:2), в результате чего получали Соединение 1.
МС: m/z (ионизация электрораспылением): 442,1 [М+1].
1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 7,74 (d, J=8,5 Гц, 2Н), 7,27 (d, J=8,5 Гц, 2Н), 6,91 (s, 2Н), 4,02 (td, J=6,7, 9,0 Гц, 1H), 3,63 (q, J=7,4 Гц, 1H), 3,07 (t, J=9,3 Гц, 1H), 2,99 (t, J=8,4 Гц, 1H), 2,77 (dd, J=6,0, 9,3 Гц, 1H), 2,60 (t, J=8,3 Гц, 1H), 2,51 (br. s., 3Н), 2,31 (s, 3Н), 2,12 (s, 6Н), 1,34 (s, 6Н).
Пример 2: Соединение 2
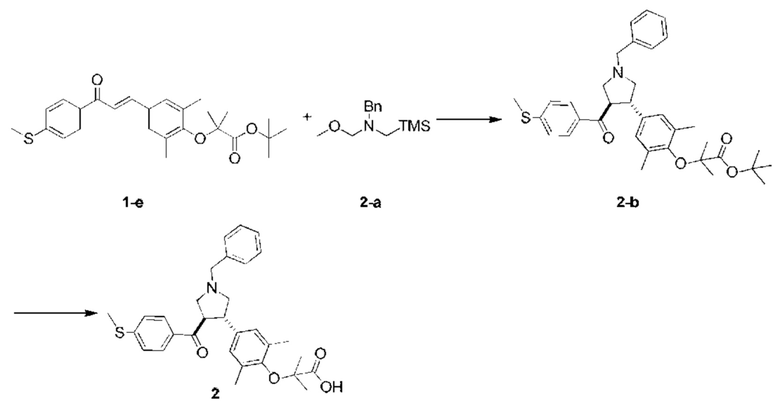
Этап 1: Соединение 2-b
В защитной атмосфере азота в раствор соединений 1-е (200,00 мг, 453,94 мкмоль, 1,00 экв.) и 2-а (129,32 мг, 544,73 мкмоль, 1,20 экв.) в толуоле (3,00 мл) добавляли по каплям трифторуксусную кислоту (50,00 мкл) при 0°С. Смесь перемешивали при 20°С в течение 1 ч. Смесь доводили насыщенным раствором бикарбоната натрия до рН~7. Органическую фазу концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат =5:1), в результате чего получали соединение 2-b.
МС: m/z (ионизация электрораспылением): 574,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,67 (d, J=8,5 Гц, 2H), 7,40-7,36 (m, 2Н), 7,32 (t, J=7,3 Гц, 2H), 7,26-7,22 (m, 1H), 7,14 (d, J=8,5 Гц, 2H), 6,88 (s, 2Н), 3,93-3,85 (m, 1H), 3,75-3,62 (m, 3Н), 3,11 (t, J=8,9 Гц, 1H), 3,01 (t, J=8,7 Гц, 1H), 2,89 (dd, J=6,9, 9,2 Гц, 1H), 2,82 (dd, J=6,3, 9,3 Гц, 1H), 2,48 (s, 3Н), 2,18 (s, 6Н), 1,50 (s, 9Н), 1,41 (s, 6Н).
Этап 2: Соединение 2
Раствор AHCl/этилацетат (4 N, 217,85 мкл, 5,00 экв.) добавляли в раствор соединения 2-b (100,00 мг, 174,28 мкмоль, 1,00 экв.) в этилацетате (2,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 8 ч. Смесь концентрировали при пониженном давлении, и очищали остаток препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 2.
МС: m/z (ионизация электрораспылением): 518,3 [М+1].
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,62-7,55 (m, 4H), 7,53-7,47 (m, 3Н), 7,14 (d, J=8,5 Гц, 2H), 6,94 (s, 2H), 4,49 (s, 2H), 4,46-4,40 (m, 1H), 3,90-3,81 (m, 1H), 3,79-3,71 (m, 2H), 3,61-3,50 (m, 2H), 2,49 (s, 3Н), 2,19 (s, 6H), 1,42 (s, 6H).
Пример 3: Соединение 3
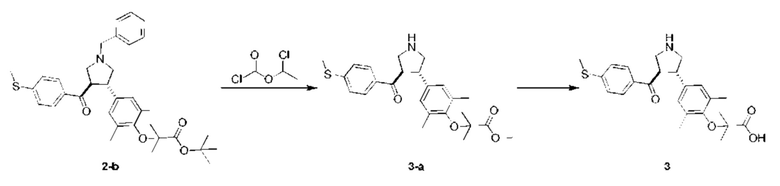
Этап 1
Соединение 3-а
Раствор соединения 2-b (1,00 г, 1,74 ммоль, 1,00 экв.) и 1-хлорэтилметилхлороформиата (746,30 мг, 5,22 ммоль, 3,00 экв.) в 1,2-дихлорэтане (10,00 мл) перемешивали при 80°С в течение 8 ч. Смесь концентрировали при пониженном давлении. Затем в остаток добавляли метанол (10,00 мл) добавляли, нагревали до 80°С и перемешивали в течение 1 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат =5/1), в результате чего получали соединение 3-а.
МС: m/z (ионизация электрораспылением): 442,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,57 (d, J=8,5 Гц, 2H), 7,09 (d, J=8,5 Гц, 2H), 6,91 (s, 2Н), 4,22 (q, J=8,2 Гц, 1H), 3,94-3,74 (m, 6Н), 3,71-3,52 (m, 2Н), 2,47 (s, 3Н), 2,11 (s, 6Н), 1,40 (d, J=4,8 Гц, 6H).
Этап 2: Соединение 3
Раствор гидроксида натрия (2 н., 339,69 мкл, 3,00 экв.) добавляли в раствор соединения 3-а (100,00 мг, 226,46 мкмоль, 1,00 экв.) в этаноле (3,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 1 ч. Смесь нейтрализовали раствором HCl (1 н.) до рН~7, а затем концентрировали при пониженном давлении. В остаток добавляли метанол (20 мл). Смесь перемешивали при 20°С в течение 10 мин, а затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 3.
МС: m/z (ионизация электрораспылением): 428,2 [М+1].
1H-ЯМР (400 МГц, MeOD-d4) δ ppm 7,64 (d, J=8,5 Гц, 2H), 7,19 (d, J=8,5 Гц, 2H), 6,93 (s, 2H), 4,40 (q, J=7,8 Гц, 1H), 3,87-3,68 (m, 3Н), 3,53-3,43 (m, 2H), 2,51 (s, 3Н), 2,20 (s, 6H), 1,39 (s, 6H).
Примеры 4 и 5: Соединения 4 и 5
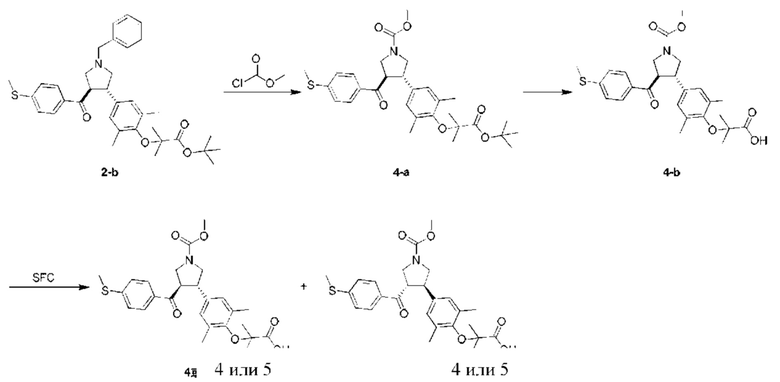
Этап 1: Соединение 4-а
В защитной атмосфере азота метилхлорформиат (12,35 мг, 130,71 мкмоль, 1,50 экв.) добавляли в раствор соединения 2-b (50,00 мг, 87,14 мкмоль, 1,00 экв.) в 1,2-дихлорэтане (10,00 мл) при 20°С. Смесь перемешивали при 80°С в течение 12 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат =5/1), в результате чего получали соединение 4-а.
МС: m/z (ионизация электрораспылением): 564,3 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,70 (t, J=8,9 Гц, 2H), 7,18 (d, J=8,0 Гц, 2H), 6,81 (s, 2Н), 4,07-3,82 (m, 3Н), 3,76-3,50 (m, 6Н), 2,50 (s, 3Н), 2,16 (s, 6Н), 1,49 (s, 9Н), 1,39-1,35 (m, 6Н).
Этап 2: Соединение 4-b
Трифторуксусную кислоту (500,00 мкл) добавляли в раствор соединения 4-а (100,00 мг, 184,60 мкмоль, 1,00 экв.) в дихлорметане (2,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 2 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан: метанол =20:1), в результате чего получали соединение 4-b.
MC: m/z (ионизация электрораспылением): 486,2 [М+1].
1H-ЯМР (400 МГц, MeOD-d4) δ ppm 7,76 (d, J=8,0 Гц, 2H), 7,23 (d, J=8,5 Гц, 2H), 6,93 (s, 2Н), 4,39-4,28 (m, 1H), 3,96-3,86 (m, 2Н), 3,74 (s, 3Н), 3,66 (dd, J=8,9, 10,7 Гц, 1H), 3,60-3,52 (m, 2Н), 2,52 (s, 3Н), 2,16 (s, 6Н), 1,38 (s, 6Н).
Этап 3: соединения 4 и 5
Соединение 4-b (35 мг) подвергали хиральному разделению, в результате чего получали соединение 4; Соединение 5.
Соединение 4:
MC: m/z (ионизация электрораспылением): 508,1 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,65 (br. s., 2H), 7,12 (d, J=8,03 Гц, 2H), 6,78 (s, 2H), 3,96-3,84 (m, 3Н), 3,66 (s, 3Н), 3,60-3,48 (m, 3Н), 2,42 (s, 3Н), 2,08 (br. s., 6H), 1,33 (br. s., 6H). Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2.
Время удерживания соединения 4: 4,766 мин (пик 1).
Соединение 5:
MC: m/z (ионизация электрораспылением): 508,1 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,65 (br. s., 2H), 7,11 (br. s., 2H), 6,78 (br. s., 2H), 3,93-3,84 (m, 3H), 3,65-3,55 (m, 6H), 2,42 (br. s., 3H), 2,08 (br. s., 6H), 1,34 (br. s., 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2.
Время удерживания соединения 5: 5,434 мин (пик 2).
Пример 6: Соединение 6
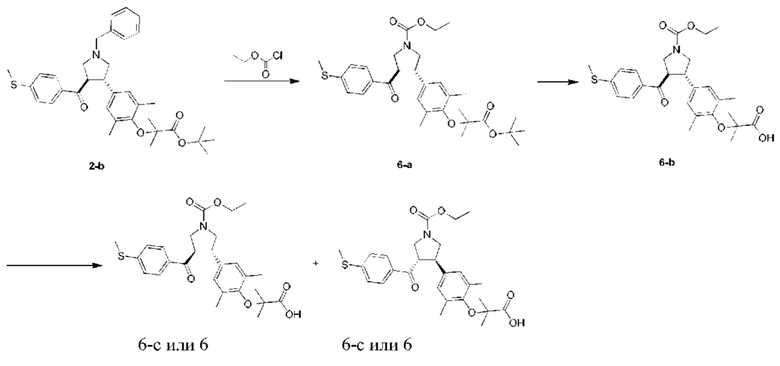
Этап 1: Соединение 6-а
Этилхлорформиат (378,26 мг, 3,49 ммоль, 331,81 мкл, 2,00 экв.) медленно добавляли в раствор соединения 2-b (1,00 г, 1,74 ммоль, 1,00 экв.) в дихлорметане (10,00 мл). Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении, и очищали остаток колоночной флэш-хроматографией, в результате чего получали соединение 6-а.
МС: m/z (ионизация электрораспылением): 578,2 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,70 (t, J=8,41 Гц, 2H), 7,17 (d, J=8,03 Гц, 2H), 6,81 (s, 2Н), 4,16-4,10 (m, 2Н), 3,99-3,91 (m, 3Н), 3,66-3,53 (m, 3Н), 2,49 (s, 3Н), 2,15 (br. s., 6H), 1,48 (s, 9H), 1,36 (br. s., 6H), 1,27-1,24 (m, 3H).
Этап 2: Соединение 6-b
Трифторуксусная кислоты (923,40 мг, 8,10 ммоль, 599,61 мкл, 11,54 экв.) медленно добавляли в раствор соединения 6-а (390,00 мг, 701,78 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл). Смесь перемешивали при 20°С в течение 3 ч. Смесь концентрировали при пониженном давлении, и очищали остаток препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 6-b.
МС: m/z (ионизация электрораспылением): 500,2 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,76-7,72 (m, 2Н), 7,20 (d, J=8,28 Гц, 2H), 6,88 (s, 2Н), 4,17 (q, J=7,03 Гц, 2H), 4,01-3,92 (m, 3Н), 3,61-3,53 (m, 1H), 2,50 (s, 3Н), 2,18 (br. s., 6H), 1,46 (s, 6H), 1,27 (t, J=7,03 Гц, 3Н).
Этап 3: Соединение 6
Соединение 6-b (100 мг) подвергали хиральному разделению, в результате чего получали соединение 6 (24,30 мг, Выход: 24,30%).
МС: m/z (ионизация электрораспылением): 522,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,72 (t, J=8,16 Гц, 2H), 7,19 (d, J=8,03 Гц, 2H), 6,86 (br. s., 2H), 4,17 (q, J=7,03 Гц, 2H), 4,01-3,91 (m, 3Н), 3,62-3,53 (m, 3Н), 2,50 (s, 3Н), 2,16 (br. s., 6H), 1,45 (br. s., 6H), 1,27 (t, J=7,03 Гц, 3Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2.
Время удерживания соединения 6: 5,198 мин (пик 2).
Пример 7: Соединение 7
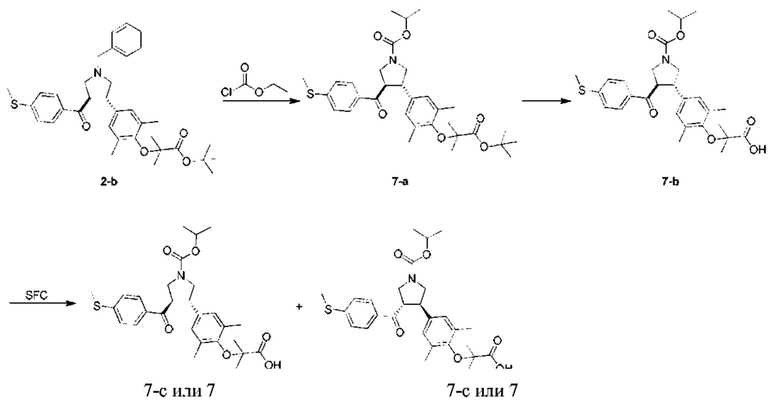
Этап 1: Соединение 7-а
В защитной атмосфере азота изопропилхлорформиат (426,47 мг, 3,48 ммоль, 484,63 мкл, 2,00 экв.) добавляли в раствор соединения 2-b (1,00 г, 1,74 ммоль, 1,00 экв.) в дихлорметане (10,00 мл) при 20°С. Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (30,6%, этилацетат/петролейный эфир), в результате чего получали соединение 7-а.
МС: m/z (ионизация электрораспылением): 592,2 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,73-7,68 (m, 2Н), 7,18 (d, J=8,28 Гц, 2H), 6,81 (s, 2Н), 4,95 (dt, J=12,30, 6,15 Гц, 1H), 4,05-3,88 (m, 3Н), 3,71-3,51 (m, 3Н), 2,50 (s, 3Н), 2,15 (br. s., 6H), 1,48 (s, 9H), 1,37 (d, J=2,76 Гц, 6H), 1,28-1,26 (m, 6H).
Этап 2: Соединение 7-b
Трифторуксусную кислоту (3,09 г, 27,09 ммоль, 2,01 мл, 19,29 экв.) добавляли в раствор соединения 7-а (800,00 мг, 1,40 ммоль, 1,00 экв.) в дихлорметане (10,00 мл). Смесь перемешивали при 25°С в течение 3 ч. Смесь концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 7-b.
МС: m/z (ионизация электрораспылением): 514,2 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,76-7,74 (m, 2Н), 7,20 (d, J=8,28 Гц, 2H), 6,88 (s, 2Н), 4,97-4,94 (m, 1H), 4,02-3,90 (m, 1H), 3,58-3,55 (m, 1H), 2,50 (s, 3Н), 2,17 (br. s., 6H), 1,46 (s, 6H), 1,25 (br. s., 6H).
Этап 3: Соединение 7
Соединение 7-b (100 мг) подвергали хиральному разделению, в результате чего получали соединение 7.
МС: m/z (ионизация электрораспылением): 536,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,74 (t, J=9,29 Гц, 2H), 7,21 (d, J=8,28 Гц, 2H), 6,88 (br. s., 2H), 4,97 (dt, J=12,30, 6,15 Гц, 1H), 4,07-3,92 (m, 3Н), 3,64-3,53 (m, 3Н), 2,52 (s, 3Н), 2,18 (d, J=4,27 Гц, 6H), 1,47 (br. s., 6H), 1,27 (br. s., 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2.
Время удерживания соединения 7: 4,639 мин (пик 2).
Пример 8: Соединение 8
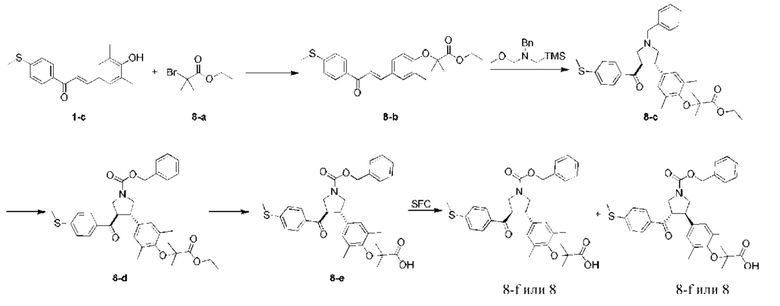
Этап 1: Соединение 8-b
Раствор соединения 1-е (7,00 г, 23,46 ммоль, 1,00 экв.), соединения 8-а (18,30 г, 93,84 ммоль, 13,76 мл, 4,00 экв.), карбоната калия (9,73 г, 70,38 ммоль, 3,00 экв.) и йодида калия (1,17 г, 7,04 ммоль, 0,30 экв.) в диметилсульфоксиде (30,00 мл) перемешивали в защитной атмосфере азота при 110°С в течение 16 ч. К реакционной смеси добавляли этилацетат (70,00 мл) и промывали водой (400 мл × 3). Органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат =15/1), в результате чего получали соединение 8-b.
МС: m/z (ионизация электрораспылением): 413,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,95 (d, J=8,28 Гц, 2H), 7,71 (d, J=15,56 Гц, 1H), 7,42 (d, J=15,56 Гц, 1H), 7,31-7,28 (m, 4H), 4,30 (q, J=7,28 Гц, 2H), 2,54 (s, 3Н), 2,25 (s, 6H), 1,50 (s, 6H), 1,36 (t, 1=7,15 Гц, 3Н).
Этап 2: Соединение 8-с
В защитной атмосфере азота трифторуксусную кислоту (1,53 г, 13,45 ммоль, 0,73 экв.) добавляли в раствор соединения 8-b (7,60 г, 18,42 ммоль, 1,00 экв.) и N-метоксиметил-1-фенил-N-(триметилсилилэтил)метиламина (5,25 г, 22,10 ммоль, 1,20 экв.) в дихлорметане (40,00 мл) при 0°С. Смесь перемешивали при 25°С в течение 16 ч. Смесь нейтрализовали насыщенным раствором бикарбоната натрия до рН=7, а затем обрабатывали водой и дихлорметаном (1:1,200 мл). Водную фазу экстрагировали дихлорметаном (200 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (23,1%, этилацетат/петролейный эфир), в результате чего получали соединение 8-с.
МС: m/z (ионизация электрораспылением): 546,6 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,67 (d, J=8,53 Гц, 2H), 7,39-7,37 (m, 2Н), 7,32 (t, J=7,53 Гц, 2H), 7,30-7,24 (m, 1H), 7,14 (d, J=8,53 Гц, 2H), 6,88 (s, 2Н), 4,28 (q, J=7,03 Гц, 2H), 3,90-3,88 (m, 1H), 3,71-3,65 (m, 3Н), 3,11 (t, J=9,03 Гц, 1H), 3,02 (t, J=8,78 Гц, 1H), 2,90 (dd, J=9,03, 7,03 Гц, 1H), 2,82 (dd, J=9,29, 6,27 Гц, 1H), 2,48 (s, 3Н), 2,15 (s, 6H), 1,45 (s, 6H), 1,35 (t, J=7,03 Гц, 3Н).
Этап 3: Соединение 8-d
Соединение 8-с (1,00 г, 1,83 ммоль, 1,00 экв.), 1,2-дихлорэтан (2,00 мл) и бензилхлорформиат (937,77 мг, 5,50 ммоль, 781,48 мкл, 3,00 экв.) добавляли в сухую колбу. Смесь перемешивали при 80°С в течение 16 ч, а затем концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (32,5%, этилацетат/петролейный эфир), в результате чего получали соединение 8-d.
МС: m/z (ионизация электрораспылением): 590,3 [М+1].
Этап 4: Соединение 8-е
Соединение 8-d (680,00 мг, 1,15 ммоль, 1,00 экв.) и 1,4-диоксан (5,00 мл) добавляли в сухую колбу. Затем добавляли гидроксид лития (82,63 мг, 3,45 ммоль, 3,00 экв.) и воду (1,00 мл) добавляли. Смесь перемешивали при 25°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=3, и обрабатывали водой и этилацетатом (1:1,50 мл). Водную фазу экстрагировали этилацетатом (20 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 8-е.
MC: m/z (ионизация электрораспылением): 562,2 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,75 (dd, J=15,18, 8,16 Гц, 2H), 7,40-7,35 (m, 5Н), 7,21 (t, J=7,65 Гц, 2H), 6,89 (s, 2Н), 5,19 (d, J=3,51 Гц, 2H), 4,06-3,98 (m, 2Н), 3,75-3,61 (m, 2Н), 2,52 (s, 3Н), 2,19 (s, 6Н), 1,48 (d, J=1,76 Гц, 6H).
Этап 5: Соединение 8
Соединение 8-е (50 мг) подвергали хиральному разделению, в результате чего получали соединение 8.
MC: m/z (ионизация электрораспылением): 584,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,72 (dd, J=15,56, 8,28 Гц, 2H), 7,37-7,33 (m, 5Н), 7,19 (t, J=7,40 Гц, 2H), 6,86 (s, 2Н), 5,17-5,13 (m, 2Н), 4,04-3,95 (m, 3Н), 3,76-3,59 (m, 3Н), 2,50 (s, 3Н), 2,16 (s, 6Н), 1,45 (br. s., 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 8: 4,956 мин (пик 2).
Пример 9: Соединение 9
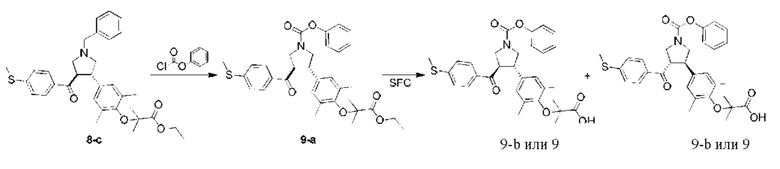
Этап 1: Соединение 9-а
В защитной атмосфере азота фенилхлорформиат (294,47 мг, 1,88 ммоль, 235,57 мкл, 2,00 экв.) медленно добавляли в раствор соединения 8-с (500,00 мг, 940,36 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл). Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (40,6%, этилацетат/петролейный эфир), в результате чего получали соединение 9-а.
MC: m/z (ионизация электрораспылением): 576,2 [М+1].
Этап 2: Соединение 9
Соединение 9-а (500,00 мг, 868,48 мкмоль, 1,00 экв.), этанол (3,00 мл) и 1,4-диоксан (1,00 мл) добавляли в колбу. Затем добавляли гидроксид лития (62,40 мг, 2,61 ммоль, 3,00 экв.) и Н2О (1,00 мл). Смесь перемешивали при 40°С в течение 16 ч. Смесь доводили при помощи 1н. разбавленной HCl до рН=2 и затем обрабатывали водой/этилацетат (1:1, 50 мл). Водную фазу экстрагировали этилацетатом (50 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметан) и подвергали продукт хиральному разделению, в результате чего получали соединение 9.
MC: m/z (ионизация электрораспылением): 570,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,75 (t, J=7,78 Гц, 2H), 7,40-7,36 (m, 2Н), 7,24-7,16 (m, 5Н), 6,88 (d, J=6,78 Гц, 2H), 4,16-4,10 (m, 3Н), 3,88-3,71 (m, 3Н), 2,49 (br. s., 3Н), 2,12 (br. s., 6H), 1,33 (br. s., 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 9: 4,709 мин (пик 2).
Пример 10: Соединение 10
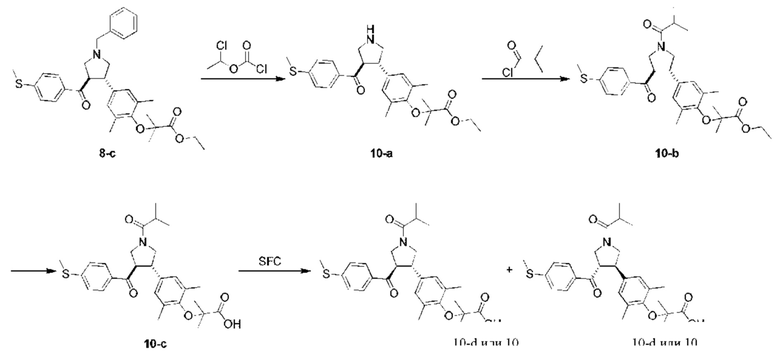
Этап 1: Соединение 10-а
1-хлорэтилхлорформиат (3,93 г, 27,48 ммоль, 3,00 экв.) добавляли в раствор соединения 8-с (5,00 г, 9,16 ммоль, 1,00 экв.) в толуоле (50,00 мл) при 25°С. Смесь перемешивали при 80°С в течение 16 ч. Затем концентрировали смесь при пониженном давлении. Остаток растворяли в метанол (50,00 мл). Раствор перемешивали при 80°С в течение 1 ч. Смесь сразу концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметан: метанол =100:0-85:15), в результате чего получали Соединение 10-а.
МС: m/z (ионизация электрораспылением): 456,3 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,59 (d, J=8,53 Гц, 2H), 7,12 (d, J=8,53 Гц, 2H), 6,93 (s, 2Н), 4,32-4,26 (m, 2Н), 4,23-4,21 (m, 1H), 3,91-3,65 (m, 5Н), 2,50 (s, 3Н), 2,15 (s, 6Н), 1,42 (d, J=5,52 Гц, 6H), 1,36 (t, J=7,03 Гц, 3Н).
Этап 2: Соединение 10-b
Изопропилхлорформиат (140,32 мг, 1,32 ммоль, 137,57 мкл, 1,50 экв.) и триэтиламин (177,68 мг, 1,76 ммоль, 243,39 мкл, 2,00 экв.) добавляли в раствор соединения 10-а (400,00 мг, 877,94 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл) при 0°С. Смесь перемешивали при 20°С в течение 2 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметан: метанол =100:0-80:20), в результате чего получали Соединение 10-b .
МС: m/z (ионизация электрораспылением): 526,2 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,68 (d, J=8,53 Гц, 1H), 7,58 (d, J=8,53 Гц, 1H), 7,11 (dd, J=8,53, 17,57 Гц, 2H), 6,75 (d, J=9,54 Гц, 2H), 4,11-3,41 (m, 7Н), 2,66-2,47 (m, 2Н), 2,43 (d, J=l,51 Гц, 3Н), 2,06 (d, J=12,05 Гц, 6H), 1,38-1,30 (m, 6Н), 1,27 (t, J=7,28 Гц, 3Н), 1,15-1,05 (m, 11Н).
Этап 3: Соединение 10-с
Гидроксид лития (13,67 мг, 570,67 мкмоль, 3,00 экв.) добавляли в раствор соединения 10-b (100,00 мг, 190,22 мкмоль, 1,00 экв.) в этаноле (5,00 мл) и воде (5,00 мл) при 0°С. Смесь перемешивали при 20°С в течение 2 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметан: метанол =100:0-80:20), в результате чего получали Соединение 10-с.
МС: m/z (ионизация электрораспылением): 498,1 [М+1].
Этап 4: Соединение 10
Соединение выделяли хиральной сверхкритической хроматографией, в результате чего получали Соединение 10.
МС: m/z (ионизация электрораспылением): 498,2 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,77 (d, J=8,28 Гц, 1H), 7,68 (d, J=8,53 Гц, 1H), 7,20 (dd, J=8,53, 17,57 Гц, 2H), 6,86 (d, J=12,30 Гц, 2H), 4,18-3,85 (m, 4H), 3,67-3,62 (m, 2H), 2,69 (qd, J=6,55, 12,99 Гц, 1H), 2,51 (d, J=2,01 Гц, 3Н), 2,17 (d, J=13,80 Гц, 6H), 1,46-1,43 (m, 6H), 1,18 (dd, J=6,78, 10,04 Гц, 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 10: 4,188 мин (пик 2).
Пример 11: Соединение 11
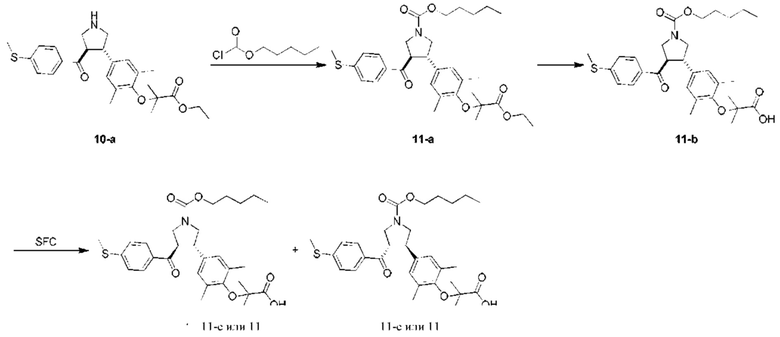
Этап 1: Соединение 11-а
Соединение 10-а (300,00 мг, 658,46 мкмоль, 1,00 экв.), триэтиламин (199,89 мг, 1,98 ммоль, 273,82 мкл, 3,00 экв.) и дихлорметан (10,00 мл) добавляли в высушенную круглодонную колбу объемом 100 мл. Затем добавляют н-пентилхлорформиат (247,91 мг, 1,65 ммоль, 2,50 экв.). Смесь перемешивали при 15°С в течение 2 ч. Смесь разбавляли дихлорметаном (20 мл), добавляли к ней насыщенный раствор бикарбоната натрия (10 мл), а затем перемешивали в течение 10 мин. Органическую фазу промывали насыщенным солевым раствором (10 мл × 2), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат =10:1-3:1), в результате чего получали Соединение 11-а.
МС: m/z (ионизация электрораспылением): 592,1 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ 7,63 (t, J=11,2 Гц, 2Н), 7,10 (d, J=7,6 Гц, 2Н), 6,75 (s, 2Н), 4,08-3,44 (m, 10Н), 2,43 (s, 3Н), 2,06 (br s, 6Н), 1,57 (t, J=6,8 Гц, 6H), 1,34-1,25 (m, 7Н), 0,83 (t, J=6,4 Гц, 3Н).
Этап 2: Соединение 11-b
Гидроксид лития (73,65 мг, 1,76 ммоль, 5,00 экв.) добавляли в раствор соединения 11-а (200,00 мг, 351,03 мкмоль, 1,00 экв.) в этаноле (2,00 мл), тетрагидрофуран (2,00 мл) и H2O (1,00 мл). Смесь перемешивали при 50°С в течение 3 ч. Смесь доводили насыщенным раствором бисульфата калия до рН=2-3, а затем добавляли к ней этилацетат (10 мл) и воду (10 мл). Водную фазу экстрагировали этилацетатом (10 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали препаративной тонкослойной хроматографией (петролейный эфир: этилацетат =15:1), в результате чего получали Соединение 11-b .
МС: m/z (ионизация электрораспылением): 564,1 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ 7,74 (t, J=8,8 Гц, 2Н), 7,20 (d, J=8,0 Гц, 2Н), 6,87 (d, J=2,8 Гц, 2Н), 4,13-3,14 (m, 8Н), 2,51 (s, 3Н), 2,18 (d, J=5,2 Гц, 6Н), 2,02 (s, 4Н), 1,67-1,64 (m, 2Н), 1,35-1,27 (m, 6Н), 0,92 (br.s., 3Н).
Этап 3: Соединение 11
Соединение выделяли хиральной сверхкритической хроматографией, в результате чего получали Соединение 11.
МС: m/z (ионизация электрораспылением): 536,1 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ 7,65 (br s, 2Н), 7,11 (br d, J=7,28 Гц, 2H), 6,78 (br s, 2H), 4,08-3,37 (m, 10H), 2,42 (s, 3H), 2,07 (br s, 6H), 1,56 (br d, J=7,04 Гц, 3Н), 1,31 (br s, 4H), 0,94-0,67 (m, 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2.
Время удерживания соединения 11: 5,294 мин (пик 2).
Пример 12: Соединение 12
Этап 1
Соединение 12-а
(2-хлорфенил)хлорформиат (83,85 мг, 438,97 мкмоль, 61,20 мкл, 2,00 экв.) и триэтиламин (44,42 мг, 438,97 мкмоль, 60,85 мкл, 2,00 экв.) медленно добавляли в раствор соединения 10-а (100,00 мг, 219,49 мкмоль, 1,00 экв.) в дихлорметане (5,00 мл). Смесь перемешивали при 25°С в течение 4 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (52,6%, этилацетат/петролейный эфир), в результате чего получали Соединение 12-а.
MC: m/z (ионизация электрораспылением): 632,1 [М+23].
Этап 2: Соединение 12
Соединение 12-а (180,00 мг, 295,00 мкмоль, 1,00 экв.) и этанол (6,00 мл) добавляли в колбу. Затем добавляли гидроксид лития (21,20 мг, 885,00 мкмоль, 3,00 экв.) и воду (2,00 мл) добавляли. Смесь перемешивали при 40°С в течение 5 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=3, и обрабатывали смесью вода/этилацетат (1:1, 20 мл). Водную фазу экстрагировали этилацетатом (20 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали Соединение 12.
MC: m/z (ионизация электрораспылением): 604,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,69 (dd, J=8,53, 6,78 Гц, 2H), 7,36 (dd, J=8,03, 1,25 Гц, 1H), 7,20 (s, 2Н), 7,16-7,13 (m, 3Н), 6,86 (d, J=7,28 Гц, 2H), 4,15-4,03 (m, 3Н), 3,79-3,65 (m, 3Н), 2,44 (s, 3Н), 2,12 (d, J=4,77 Гц, 6H), 1,40-1,39 (m, 6Н).
Примеры 13 и 14: Соединения 13 и 14
Этап 1: Соединение 13-а
В защитной атмосфере азота раствор трифосгена (97,70 мг, 329,23 мкмоль, 1,00 экв.) в тетрагидрофуране (3 мл) добавляли по каплям в раствор 4-хлорфенола (42,33 мг, 329,23 мкмоль, 32,31 мкл, 1,00 экв.) в тетрагидрофуране (15,00 мл) 0°С, и медленно добавляли в эту смесь триэтиламин (33,31 мг, 329,23 мкмоль, 45,63 мкл, 1,00 экв.) медленно добавляли. Смесь перемешивали при 25°С в течение 2 ч. После завершения реакции смесь фильтровали, остаток на фильтре промывали тетрагидрофураном (5 мл × 2). Соединение 10-а (150,00 мг, 329,23 мкмоль, 1,00 экв.) и триэтиламин (33,31 мг, 329,23 мкмоль, 45,63 мкл, 1,00 экв.) добавляли в фильтрат при 25°С. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (этилацетат/петролейный эфир = 22,7%), в результате чего получали Соединение 13-а.
МС: m/z (ионизация электрораспылением): 610,1 [М+1].
Этап 2: Соединение 13-b
Гидроксид лития (21,20 мг, 885,00 мкмоль, 3,00 экв.) и H2O (2,00 мл) добавляли в раствор соединения 13-а (255,00 мг, 417,92 мкмоль, 1,00 экв.) в этаноле (6,00 мл). Смесь перемешивали при 40°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=2, и обрабатывали смесью вода/этилацетат (1:1, 20 мл). Водную фазу экстрагировали этилацетатом (20 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали Соединение 13-b.
МС: m/z (ионизация электрораспылением): 604,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,69 (dd, J=8,53, 6,78 Гц, 2H), 7,36 (dd, J=8,03, 1,25 Гц, 1H), 7,20 (s, 2Н), 7,16-7,13 (m, 3Н), 6,86 (d, J=7,28 Гц, 2H), 4,15-4,02 (m, 3Н), 3,79-3,65 (m, 3Н), 2,44 (s, 3Н), 2,12 (d, J=4,77 Гц, 6H), 1,40-1,39 (m, 6Н).
Этап 3: Соединения 13 и 14
Соединение 13-b (30,00 мг, 51,54 мкмоль, 1,00 экв.) выделяли и очищали хиральной сверхкритической жидкостной хроматографией, в результате чего получали Соединение 13;
Соединение 14.
Соединение 13:
МС: m/z (ионизация электрораспылением): 582,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,67 (t, J=8,66 Гц, 2H), 7,26-7,24 (m, 2H), 7,13 (dd, J=8,53, 4,02 Гц, 2H), 7,03 (d, J=7,94 Гц, 2H), 6,83 (d, J=6,27 Гц, 2H), 4,07-4,00 (m, 3Н), 3,79-3,65 (m, 3Н), 2,43 (s, 3Н), 2,11 (d, J=5,77 Гц, 6H), 1,39 (br s, 6H).
Условия хирального разделения: хиральная колонка: Lux Cellulose-2 150×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,5 мл/мин; температура колонки: 40°С.
Время удерживания соединения 13: 5,715 мин (пик 1).
Соединение 14:
MC: m/z (ионизация электрораспылением): 582,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,68 (t, J=8,41 Гц, 2H), 7,27-7,24 (m, 2Н), 7,13 (dd, J=8,53, 4,02 Гц, 2H), 7,03 (d, J=7,94 Гц, 2H), 6,84 (d, J=6,27 Гц, 2H), 4,06-4,00 (m, 3Н), 3,79-3,63 (m, 3Н), 2,43 (s, 3Н), 2,12 (d, J=5,52 Гц, 6H), 1,39-1,38 (m, 6Н).
Условия хирального разделения: хиральная колонка: Lux Cellulose-2 150×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,5 мл/мин; температура колонки: 40°С.
Время удерживания соединения 14: 9,994 мин (пик 2).
Пример 15: Соединение 15
Этап 1: Соединение 15-а
Соединение 10-а (300,00 мг, 658,46 мкмоль, 1,00 экв.), ди-изо-пропилэтиламин (170,20 мг, 1,32 ммоль, 230,00 мкл, 2,00 экв.) и трихлорметан (5,00 мл) добавляли в 50 мл высушенную круглодонную колбу при 20°С. Затем добавляли p-метоксифенилхлорформиат (184,29 мг, 987,69 мкмоль, 147,43 мкл, 1,50 экв.). Смесь перемешивали при 20°С в течение 2 ч. В смесь добавляли насыщенный раствор бикарбоната натрия (5 мл) и перемешивали в течение 10 мин. Водную фазу экстрагировали дихлорметаном (10 мл × 3). Объединенную органическую фазу промывали насыщенным слоевым раствором, сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат = от 4:1 до 2:1), в результате чего получали Соединение 15-а.
МС: m/z (ионизация электрораспылением): 606,2 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,65 (dd, J=10,4, 9,2 Гц, 2H), 7,12 (dd, J=8,8, 4,8 Гц, 2H), 6,99 (d, J=8,8 Гц, 2H), 6,81-6,78 (m, 4Н), 4,08-3,99 (m, 5Н), 3,75-3,59 (m, 7Н), 2,43 (s, 3Н), 2,08 (s, 3Н), 2,07 (s, 3Н), 1,55 (b.r.s., 1Н), 1,35-1,34 (m, 6Н), 1,19 (t, J=6,8 Гц, 1H).
Этап 2: Соединение 15
Соединение 15-а (150,00 мг, 247,63 мкмоль, 1,00 экв.), гидроксид лития (59,31 мг, 2,48 ммоль, 10,00 экв.), этанол (2,00 мл), тетрагидрофуран (2,00 мл) и воду (1,00 мл) добавляли в реакционную колбу. Смесь перемешивали при 20°С в течение 20 ч. Добавляли гидроксид лития (59,31 мг, 2,48 ммоль, 10,00 экв.) и перемешивали смесь при 20°С в течение 30 ч. Смесь разбавляли водой (5 мл), доводили насыщенным раствором бисульфата калия до рН=2-3, а затем добавляли к ней этилацетат (10 мл × 3). Органическую фазу промывали насыщенным слоевым раствором, сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали препаративной тонкослойной хроматографией (этилацетат), в результате чего получали Соединение 15.
МС: m/z (ионизация электрораспылением): 578,2 [М+1].
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,64 (dd, J=8,4, 6,8 Гц, 2Н), 7,10 (d, J=8,4 Гц, 2Н), 6,94 (dd, J=10,0, 8,0 Гц, 2Н), 6,85-6,80 (m, 4Н), 4,31-4,00 (m, 1H), 3,86-3,85 (m, 1H), 3,84-3,50 (m, 7Н), 2,39 (s, 3Н), 2,06 (b.r.s, 6Н), 1,38-1,36 (m, 6Н).
Примеры 16 и 17: Соединения 16 и 17
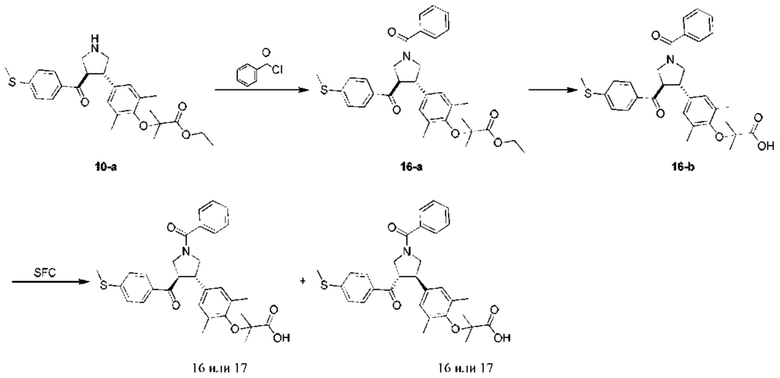
Этап 1: Соединение 16-а
Бензоилхлорид (185,12 мг, 1,32 ммоль, 152,99 мкл, 1,50 экв.) и триэтиламин (177,68 мг, 1,76 ммоль, 243,39 мкл, 2,00 экв.) добавляли в раствор соединения 10-а (400,00 мг, 877,94 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл) при 0°С. Смесь перемешивали при 20°С в течение 2 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметан: метанол =100:0-80:20), в результате чего получали Соединение 16-а.
МС: m/z (ионизация электрораспылением): 560,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,71 (d, J=8,53 Гц, 1H), 7,58-7,28 (m, 7Н), 7,17-6,99 (m, 2Н), 6,82-6,68 (m, 2Н), 4,19 (br d, J=7,28 Гц, 2H), 4,01-3,51 (m, 5H), 2,42 (d, J=18,57 Гц, 3Н), 2,05 (s, 6H), 1,36-1,30 (m, 6H), 1,29-1,23 (m, 3Н).
Этап 2: Соединение 16-b
Гидроксид лития (213,95 мг, 8,93 ммоль, 10,00 экв.) добавляли в раствор соединения 16-а (500,00 мг, 893,30 мкмоль, 1,00 экв.) в этаноле (10,00 мл) и Н2O (5,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 2 ч. Смесь экстрагировали дихлорметаном (20 мл × 3), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметан: метанол =100:0-80:20), в результате чего получали Соединение 16-b.
МС: m/z (ионизация электрораспылением): 532,1 [М+1].
Этап 3: Соединения 16 и 17
Соединение 18-b (400 мг, 752,36 мкмоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали Соединение 16; Соединение 17.
Соединение 16:
МС: m/z (ионизация электрораспылением): 532,5 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,80 (d, J=8,28 Гц, 1H), 7,62-7,55 (m, 3Н), 7,44-7,41 (m, 3Н), 7,23 (d, J=8,28 Гц, 1H), 7,11 (d, J=8,28 Гц, 1H), 6,89 (s, 1H), 6,82 (s, 1H), 4,31 (dd, J=11,92, 8,91 Гц, 1H), 4,18-4,15 (m, 1H), 3,99-3,93 (m, 3Н), 3,77-3,64 (m, 1H), 2,49 (d, J=19,32 Гц, 3Н), 2,15 (s, 6Н), 1,43 (br. s., 6Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 16: 6,673 мин (пик 2).
Соединение 17:
МС: m/z (ионизация электрораспылением): 532,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,80 (d, J=8,28 Гц, 1H), 7,63-7,54 (m, 3Н), 7,44-7,41 (m, 3Н), 7,24 (d, J=8,28 Гц, 1H), 7,12 (d, J=8,53 Гц, 1H), 6,87 (d, J=28 Гц, 2H), 4,32-4,15 (m, 2H), 3,96-3,93 (m, 2H), 3,78-3,66 (m, 2H), 2,50 (d, J=19,58 Гц, 3Н), 2,16 (s, 6H), 1,44 (br. s., 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 17: 4,992 мин (пик 1).
Пример 18: Соединение 18
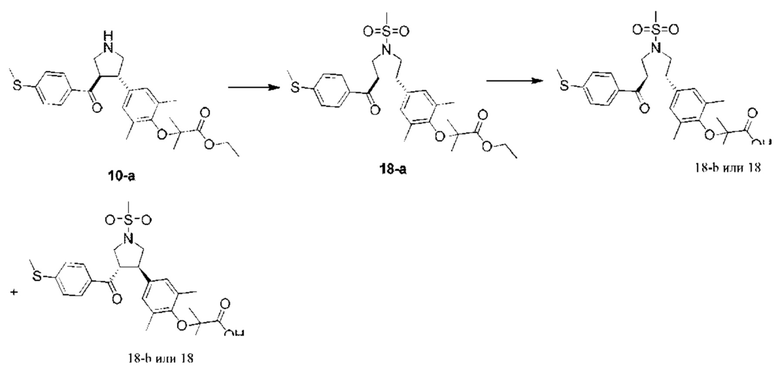
Этап 1: Соединение 18-а
Мезилхлорид (201,14 мг, 1,76 ммоль, 135,91 мкл, 2,00 экв.) и триэтиламин (177,68 мг, 1,76 ммоль, 243,40 мкл, 2,00 экв.) добавляли в раствор соединения 10-а (400,00 мг, 877,94 мкмоль, 1,00 экв.) в дихлорметане (5,00 мл). Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (46,1%, этилацетат/петролейный эфир), в результате чего получали Соединение 18-а.
МС: m/z (ионизация электрораспылением): 534,1 [М+1].
Этап 2: Соединение 18
Соединение 18-а (110,00 мг, 206,11 мкмоль, 1,00 экв.) и диоксан (1,00 мл) добавляли в круглодонную колбу. Затем добавляли гидроксид лития (14,81 мг, 618,33 мкмоль, 3,00 экв.) и воду (1,00 мл). Смесь перемешивали при 40°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=3, и добавляли в нее воду/этилацетат (1:1, 50 мл). Водную фазу экстрагировали этилацетатом (20 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией. Полученный продукт подвергали хиральному разделению, в результате чего получали Соединение 18.
MC: m/z (ионизация электрораспылением): 528,1 [М+23].
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,75 (d, J=8,53 Гц, 2H), 7,22 (d, J=8,28 Гц, 2H), 6,95 (s, 2Н), 4,62-4,35 (m, 1H), 3,86-3,82 (m, 2Н), 3,73-3,68 (m, 1H), 3,57-3,52 (m, 2Н), 3,04 (s, 3Н), 2,51 (s, 3Н), 2,22 (s, 6Н), 1,36 (d, J=5,27 Гц, 6H).
Условия хирального разделения: хиральная колонка: Chiralpak OJ-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2.
Время удерживания соединения 18: 5,853 мин (пик 2).
Пример 19: Соединение 19
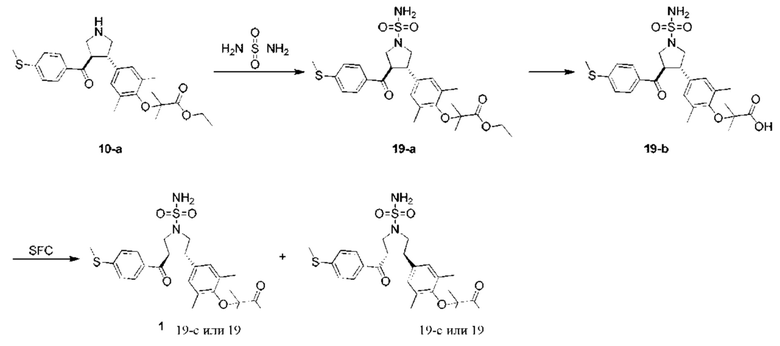
Этап 1: Соединение 19-а
Соединение 10-а (400,03 мг, 878,00 мкмоль, 1,00 экв.) добавляли в раствор сульфурилдиамида (421,90 мг, 4,39 ммоль, 262,05 мкл, 5,00 экв.) в диоксане (10,00 мл) при 25°С. Смесь перемешивали при 110°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметаг метанол =100:0-80:20), в результате чего получали Соединение 19-а.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,71 (d, J=8,04 Гц, 2H), 7,20 (d, J=8,04 Гц, 2H), 6,88 (s, 2Н): 4,72 (s, 2Н), 4,48-4,38 (m, 1H), 4,29 (q, J=7,04 Гц, 2H), 3,84-3,56 (m, 9Н), 2,52 (s, 3Н), 2,17 (s, 6Н), 1,45 (d, J=4,02 Гц, 6H), 1,37-1,34 (m, 3Н).
Этап 2: Соединение 19-b
Гидроксид лития (291,15 мг, 12,16 ммоль, 10,00 экв.) добавляли в раствор соединения 19-а (650,00 мг, 1,22 ммоль, 1,00 экв.) в этаноле (10,00 мл) и воде (5 мл) при 20°С. Смесь перемешивали при 20°С в течение 2 ч. Смесь экстрагировали дихлорметаном (20 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметан: метанол =100:0-80:20), в результате чего получали Соединение 19-b.
МС: m/z (ионизация электрораспылением): 507,1 [М+1].
Этап 3: Соединение 19
Соединение 19-b (400,00 мг, 789,51 мкмоль, 1,00 экв.) подвергали хиральному разделению, в результате чего получали Соединение 19.
МС: m/z (ионизация электрораспылением): 507,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,71 (d, J=8,04 Гц, 2H), 7,20 (d, J=8,04 Гц, 2H), 6,88 (s, 2Н), 4,72 (s, 2Н), 4,48-4,38 (m, 1H), 4,29 (q, J=7,04 Гц, 2H), 3,91-3,47 (m, 9Н), 2,52 (s, 3Н), 2,17 (s, 6Н), 1,45 (d, J=4,02 Гц, 6H), 1,38-1,32 (m, 3Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 19: 4,227 мин (пик 2).
Пример 20: Соединение 20
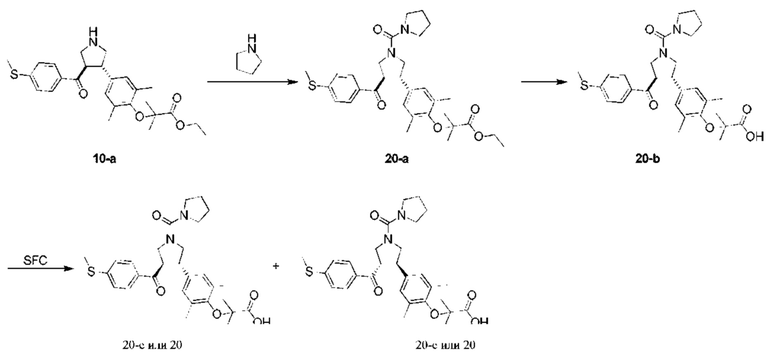
Этап 1: Соединение 20-а
Триэтиламин (177,68 мг, 1,76 ммоль, 243,39 мкл, 2,00 экв.) и карбонилдиимидазол (213,54 мг, 1,32 ммоль, 1,50 экв.) добавляли в раствор соединения 10-а (400,00 мг, 877,94 мкмоль, 1,00 экв.) в толуоле (10,00 мл) при 25°С. Смесь перемешивали при 80°С в течение 16 ч. Затем добавляли тетрагидропиррол (93,66 мг, 1,32 ммоль, 110,19 мкл, 1,50 экв.). Смесь перемешивали при 80°С в течение 2 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметан: метанол =100:0-75:25), в результате чего получали соединение 20-а.
МС: m/z (ионизация электрораспылением): 553,3 [М+1]
Этап 2: Соединение 20-b
Гидроксид лития (194,99 мг, 8,14 ммоль, 10,00 экв.) добавляли в раствор соединения 20-а (450,00 мг, 814,16 мкмоль, 1,00 экв.) в этаноле (5,00 мл) и воде (5 мл) при 20°С. Смесь перемешивали при 45°С в течение 16 ч. Смесь экстрагировали дихлорметаном (20 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (дихлорметан: метанол =100:0-75:25), в результате чего получали соединение 20-b.
МС: m/z (ионизация электрораспылением): 525,1 [М+1].
Этап 3: Соединение 20
Соединение 20-b (100,00 мг, 319,58 мкмоль, 1,00 экв.) подвергали хиральному разделению, в результате чего получали соединение 20-с (70,0 мг, Выход: 17,50%); Соединение 20.
1Н-ЯМР (400 МГц, CDCl3) δ 7,72 (d, J=8,54 Гц, 2Н), 7,18 (d, J=8,28 Гц, 2Н), 6,89 (s, 2Н), 4,03-3,85 (m, 3Н), 3,75-3,63 (m, 3Н), 3,39 (br dd, J=5,52, 10,78 Гц, 4H), 2,49 (s, 3Н), 2,15 (s, 6Н), 1,85 (br dd, J=5,90, 13,68 Гц, 4H), 1,43 (br s, 6H).
Условия хирального разделения: хиральная колонка: Chiralpak OJ-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 20: 3,362 мин (пик 2).
Пример 21: Соединение 21
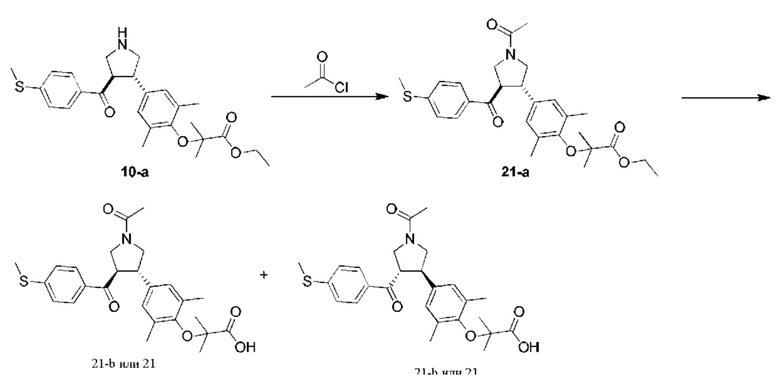
Этап 1: Соединение 21-а
Соединение 10-а (500,00 мг, 1,10 ммоль, 1,00 экв.), дихлорметан (5,00 мл), ацетилхлорид (86,15 мг, 1,10 ммоль, 78,32 мкл, 1,00 экв.) и триэтиламин (222,10 мг, 2,19 ммоль, 304,24 мкл, 2,00 экв.) добавляли в высушенную круглодонную колбу. Смесь перемешивали при 25°С в течение 2 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (этилацетат), в результате чего получали соединение 21-а.
МС: m/z (ионизация электрораспылением): 498,1 [М+1].
Этап 2: Соединение 21
Соединение 21-а (130,00 мг, 261,23 мкмоль, 1,00 экв.) и диоксан (1,00 мл) добавляли в круглодонную колбу. Затем добавляли гидроксид лития (18,77 мг, 783,68 мкмоль, 3,00 экв.) и воду (1,00 мл). Смесь перемешивали при 40°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=3, и добавляли к ней воду/этилацетат (1:1, 20 мл). Водную фазу экстрагировали этилацетатом (20 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (11,3%, дихлорметан/МеОН). Полученный продукт подвергали хиральному разделению, в результате чего получали соединение 21.
МС: m/z (ионизация электрораспылением): 470,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,68 (d, J=8,03 Гц, 1H), 7,61 (d, J=8,03 Гц, 1H), 7,15-7,09 (m, 2Н), 6,81-6,77 (m, 2Н), 4,11-4,06 (m, 1H), 3,95-3,91 (m, 1H), 3,81-3,79 (m, 2Н), 3,58-3,48 (m, 2Н), 2,44 (s, 3Н), 2,12-2,03 (m, 9Н), 1,40-1,38 (m, 6Н).
Условия хирального разделения: хиральная колонка: Chiralpak OJ-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 21: 3,430 мин (пик 2).
Примеры 22 и 23: соединения 22 и 23
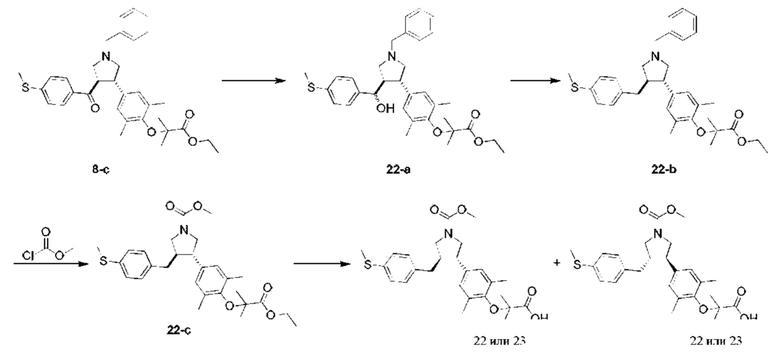
Этап 1: Соединение 22-а
Борогидрид натрия (34,66 мг, 916,20 мкмоль, 1,00 экв.) добавляли в раствор соединения 8-с (500,00 мг, 916,20 мкмоль, 1,00 экв.) в этаноле (5,00 мл) при 0°С. Смесь перемешивали при 50°С в течение 2 ч. В смесь добавляли смесь этилацетат/вода (1:1, 50 мл). Водную фазу экстрагировали этилацетатом (20 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 22-а.
МС: m/z (ионизация электрораспылением): 548,1 [М+1].
1H-ЯМР (400 МГц, MeOD-d4) δ ppm 7,34 (br d, J=4,27 Гц, 6H), 7,24 (br d, J=8,03 Гц, 2H), 7,18-7,14 (m, 3H), 4,30-4,23 (m, 3H), 3,32-3,18 (m, 3H), 3,03 (br d, J=9,03 Гц, 1H), 2,71-2,69 (m, 1H), 2,46 (s, 3H), 2,42-2,33 (m, 3H), 2,04 (s, 6H), 1,40 (s, 6H), 1,33 (t, J=7,15 Гц, 3Н).
Этап 2: Соединение 22-b
Триэтилсилан (636,86 мг, 5,48 ммоль, 872,41 мкл, 5,00 экв.) добавляли в раствор соединения 22-а (600,00 мг, 1,10 ммоль, 1,00 экв.) в дихлорметане (5,00 мл), а затем добавляли трифторуксусную кислоту (624,48 мг, 5,48 ммоль, 405,51 мкл, 5,00 экв.). Смесь перемешивали при 25°С в течение 3 ч. К смеси добавляли насыщенный раствор карбоната натрия (30 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией, в результате чего получали соединение 22-b.
МС: m/z (ионизация электрораспылением): 532,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,29-7,27 (m, 2Н), 7,24-7,17 (m, 3Н), 7,03 (d, J=8,28 Гц, 2H), 6,92 (d, J=8,03 Гц, 2H), 6,65 (s, 2Н), 4,21 (q, J=7,28 Гц, 2H), 3,63-3,50 (m, 2Н), 2,89-2,77 (m, 1H), 2,75-2,68 (m, 3Н), 2,58-2,55 (m, 2Н), 2,37 (s, 3Н), 2,07 (s, 6Н), 1,60 (br s, 2H), 1,38 (s, 6H), 1,30-1,27 (m, 3Н).
Этап 3: Соединение 22-с
Соединение 22-b (185,00 мг, 347,91 мкмоль, 1,00 экв.), дихлорметан (5,00 мл) и метилхлорформиат (164,39 мг, 1,74 ммоль, 134,75 мкл, 5,00 экв.) добавляли в высушенную круглодонную колбу. Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (34,6%, этилацетат/петролейный эфир), в результате чего получали соединение 22-с.
МС: m/z (ионизация электрораспылением): 522,1 [М+23].
Этап 4: соединения 22 и 23
Соединение 22-с (89,00 мг, 178,12 мкмоль, 1,00 экв.) и этанол (5,00 мл) добавляли в высушенную круглодонную колбу. Затем добавляли гидроксид лития (12,80 мг, 534,36 мкмоль, 3,00 экв.) и воду (1,00 мл). Смесь перемешивали при 40°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=3, а затем добавляли смесь вода/этилацетат (1: 1, 30 мл). Водную фазу экстрагировали этилацетатом (30 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией, в результате чего получали соединение, которое подвергали хиральному разделению, в результате чего получали соединение 22; Соединение 23. Соединение 22:
МС: m/z (ионизация электрораспылением): 472,1 [М+1].
1H-ЯМР (400 МГц, MeOD-d4) δ ppm 7,15 (d, J=8,03 Гц, 2H), 7,03 (br d, J=6,78 Гц, 2H), 6,93 (s, 2H), 3,79 (dd, J=8,28, 10,54 Гц, 1H), 3,68 (s, 3Н), 3,56-3,53 (m, 1H), 3,12 (t, J=10,16 Гц, 1H), 2,94-2,92 (m, 1H), 2,69 (br d, J=13,05 Гц, 1H), 2,45-2,43 (m, 5H), 2,27 (s, 6H), 1,43 (s, 6H). Соединение 23:
MC: m/z (ионизация электрораспылением): 494,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,09-7,06 (m, 2Н), 6,91 (br d, J=7,03 Гц, 2H), 6,79 (br d, J=2,76 Гц, 2H), 3,79-3,72 (m, 1H), 3,61 (s, 3H), 3,54-3,49 (m, 1H), 3,31-3,25 (m, 1H), 3,07-3,01 (m, 1H), 2,83-2,78 (m, 1H), 2,65 (br d, J=14,05 Гц, 1H), 2,39-2,32 (m, 5H), 2,16 (s, 6H), 1,45 (s, 6H).
Условия хирального разделения: хиральная колонка: Chiralpak OJ-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в CO2.
Время удерживания соединения 22: 4,092 мин (пик 1); время удерживания соединения 23:4,594 мин (пик 2)
Пример 24: Соединение 24
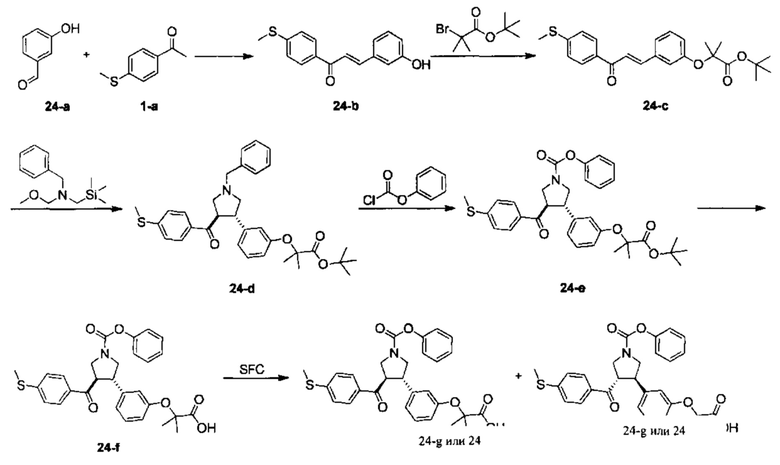
Этап 1: Соединение 24-b
Соединение 1-а (20,42 г, 122,83 ммоль, 1,00 экв.) добавляли в раствор соединения 24-а (15,00 г, 122,83 ммоль, 1,00 экв.) в HCl/МеОН (4 N, 100,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 2 ч. Смесь фильтровали, остаток на фильтре растворяли в этилацетате (500 мл), добавляли воду (1000 мл) и экстрагировали этилацетатом (200 мл × 3). Объединенную органическую фазу промывали водой (200 мл), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 24-b.
MC: m/z (ионизация электрораспылением): 270,9 [М+1].
1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,04 (d, J=8,54 Гц, 2Н), 7,71 (d, J=2,26 Гц, 2Н), 7,41 (d, J=8,54 Гц, 2Н), 7,32-7,21 (m, 2Н), 7,16 (s, 1H), 6,89 (d, J=7,78 Гц, 1H), 2,60-2,53 (s, 3Н).
Этап 2: Соединение 24-с
Смесь трет-бутил-бром-изопропионата (12,38 г, 55,47 ммоль, 3,00 экв.) и карбоната калия (7,67 г, 55,47 ммоль, 3,00 экв.) добавляли в раствор соединения 24-b (5,00 г, 18,49 ммоль, 1,00 экв.) в ацетонитриле (30,00 мл) при 20°С. Смесь перемешивали при 80°С в течение 16 ч. Смесь фильтровали, а фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат =100:0-80:20), в результате чего получали соединение 24-с.
МС: m/z (ионизация электрораспылением): 413,0 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ 7,96 (d, J=8,54 Гц, 2Н), 7,75 (d, J=15,56 Гц, 1H), 7,48 (d, J=15,56 Гц, 1H), 7,35-7,27 (m, 3Н), 7,26-7,23 (m, 1H), 7,16 (s, 1H), 6,91 (td, J=1,22, 7,84 Гц, 1H), 2,56 (s, 3Н), 1,61 (s, 6Н), 1,46 (s, 9Н).
Этап 3: Соединение 24-d
N-(метоксиметил)-1-фенил-N-(триметилсилилметил) метиламин (6,10 г, 25,70 ммоль, 2,00 экв.) и ТФУК (2,20 г, 19,28 ммоль, 1,43 мл, 1,50 экв.) добавляли в раствор соединения 24-с (5,30 г, 12,85 ммоль, 1,00 экв.) в дихлорметане (25,00 мл) при 0°С. Смесь перемешивали при 20°С в течение 16 ч. Смесь сразу концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир: этилацетат =100:0-60:40), в результате чего получали соединение 24-d.
МС: m/z (ионизация электрораспылением): 546,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ 7,69 (d, J=8,53 Гц, 2Н), 7,42-7,36 (m, 2Н), 7,32 (t, J=7,40 Гц, 2Н), 7,26-7,21 (m, 1H), 7,18-7,08 (m, 3Н), 6,94-6,87 (m, 2Н), 6,71-6,66 (m, 1H), 3,99-3,89 (m, 1H), 3,80-3,62 (m, 3Н), 3,14 (t, J=9,03 Гц, 1H), 3,02 (t, J=8,66 Гц, 1H), 2,89-2,80 (m, 2Н), 2,52-2,43 (m, 3Н), 1,55 (s, 6Н), 1,43 (s, 9Н).
Этап 4: Соединение 24-е
Фенилхлорформиат (3,44 г, 21,99 ммоль, 2,75 мл, 3,00 экв.) добавляли в раствор соединения 24-d (4,00 г, 7,33 ммоль, 1,00 экв.) в дихлорметане (30,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир: этилацетат =100:0-60:40), в результате чего получали соединение 24-е.
МС: m/z (ионизация электрораспылением): 598,0 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ 7,78 (dd, J=5,02, 8,54 Гц, 2H), 7,43-7,35 (m, 2Н), 7,26-7,12 (m, 6h), 6,96-6,82 (m, 2H), 6,72 (dd, J=2,14, 8,16 Гц, 1H), 4,25-4,04 (m, 3H), 3,97-3,67 (m, 3H), 2,52 (s, 3H), 1,55 (d, J=5,02 Гц, 6H), 1,44 (s, 9H).
Этап 5: Соединение 24-f
Трифторуксусную кислоту (11,88 г, 104,22 ммоль, 7,72 мл, 20,00 экв.) добавляли в раствор соединения 24-е (3,00 г, 5,21 ммоль, 1,00 экв.) в дихлорметане (20,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 3 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан: метанол =100:0-90:10), в результате чего получали соединение 24-f.
МС: m/z (ионизация электрораспылением): 520,0 [М+1].
Этап 6: Соединение 24
Соединение 24-f (1,00 г, 1,92 ммоль, 1,00 экв.) подвергали хиральному разделению, в результате чего получали соединение 24.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,70 (t, J=7,54 Гц, 2Н), 7,37-7,25 (m, 2Н), 7,18-7,05 (m, 6 h), 6,91 (d, J=7,78 Гц, 1H), 6,80 (s, 1H), 6,73 (t, J=5,90 Гц, 1H), 4,15-3,92 (m, 3Н), 3,88-3,58 (m, 3Н), 2,43 (s, 3Н), 1,55-1,40 (m, 6Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 24: 6,698 мин (пик 2)
Примеры 25, 26: соединения 25, 26
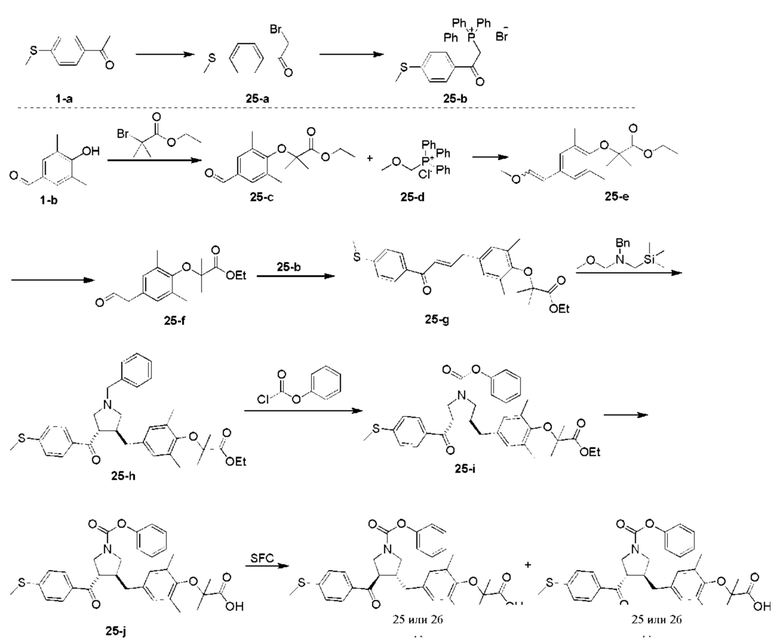
Этап 1: Соединение 25-а
Триметилхлорсилан (23,53 г, 216,55 ммоль, 27,36 мл, 1,20 экв.) добавляли в раствор соединения 1-а (30,00 г, 180,46 ммоль, 1,00 экв.) в дихлорметане (200,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 1 ч. Затем добавляли N-бромсукцинимид (38,54 г, 216,55 ммоль, 1,20 экв.). Смесь перемешивали при 20°С в течение 1 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат =100/1-100/30), в результате чего получали соединение 25-а.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,88-7,85 (m, 2Н), 7,27-7,24 (m, 2Н), 4,40 (s, 2Н), 2,52 (s, 3Н).
Этап 2: Соединение 25-b
Трифенилфосфин (32,10 г, 122,38 ммоль, 1,00 экв.) добавляли в раствор соединения 25-а (30,00 г, 122,38 ммоль, 1,00 экв.) в толуоле (250,00 мл) при 20°С. Смесь перемешивали при 80°С течение 17 ч. Смесь фильтровали, остаток на фильтре промывали этилацетатом (250 мл × 3).Органические фазы объединяли и концентрировали, в результате чего получали соединение 25-b.
1H-ЯМР (400 МГц, ДМСО-d6) δ ppm 7,99 (d, J=8,5 Гц, 2Н), 7,89-7,70 (m, 15Н), 7,44 (d, J=8,5 Гц, 2H), 6,17 (d, J=13,l Гц, 2H), 2,57 (s, 3Н).
Этап 3: Соединение 25-с
Карбонат калия (110,44 г, 799,05 ммоль, 3,00 экв.), этил-2-бром-изобутират (207,81 г, 1,07 моль, 156,25 мл, 4,00 экв.) и йодид калия (44,21 г, 266,35 ммоль, 1,00 экв.) добавляли в раствор соединения 1-b (40,00 г, 266,35 ммоль, 1,00 экв.) в диметилсульфоксиде (400,00 мл) при 20°С. Смесь перемешивали при 130°С в течение 4 ч. В смесь добавляли воду (1 л) добавляли и экстрагировали этилацетатом (200 мл × 6). Объединенную органическую фазу промывали водой (1 л) и насыщенным солевым раствором (1 л), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир: этилацетат =100:0-100:50), в результате чего получали соединение 25-с.
1H-ЯМР (400 МГц, CDCl3) δ ppm 9,85 (s, 1Н), 7,50 (s, 2Н), 4,31-4,23 (m, 2Н), 2,25 (s, 6Н), 1,45 (s,6H), 1,33 (t, J=7,2 Гц, 1H).
Этап 4: Соединение 25-е
Трет-бутоксид калия (19,10 г, 170,25 ммоль, 1,50 экв.) добавляли в раствор соединения 25-d (58,36 г, 170,25 ммоль, 1,50 экв.) в тетрагидрофуране (50,00 мл) при 0°С. Смесь перемешивали при 0°С в течение 30 мин. Затем добавляли Соединение 25-с (30,00 г, 113,50 ммоль, 1,00 экв.). Смесь перемешивали при 20°С в течение 17 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле петролейный эфир/этилацетат =100/1-100/20), в результате чего получали соединение 25-е.
Этап 5: Соединение 25-f
Касфорсульфоновую кислоту (17,88 г, 76,95 ммоль, 3,00 экв.) добавляли в раствор соединения 25-е (15 г, 51,30 ммоль, 1,00 экв.) в тетрагидрофуране (200,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 17 ч. В смесь добавляли насыщенный раствор карбоната натрия (500 мл, 4 н.) и экстрагировали этилацетатом (500 мл × 2). Объединенную органическую фазу промывали насыщенным солевым раствором (500 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 25-f.
1H-ЯМР (400 МГц, CDCl3) δ ppm 9,80-9,69 (m, 1H), 9,62 (s, 1H), 6,75 (s, 2H), 4,21 (br d, J=4,0 Гц, 2H), 4,20 (s, 1H), 3,25 (s, 2H), 2,12-2,10 (m, 6H), 1,40-1,38 (m, 6H), 1,28 (d, J=1,8 Гц, 2H), 1,28-1,27 (m, 1H).
Этап 6: Соединение 25-g
В защитной атмосфере азота трет-бутоксид калия (1,11 г, 9,85 ммоль, 1,00 экв.) добавляли в раствор соединения 25-b (5,00 г, 9,85 ммоль, 1,00 экв.) в тетрагидрофуране (50,00 мл). Смесь перемешивали при 20°С в течение 1 ч. Затем добавляли соединение 25-f (2,74 г, 9,85 ммоль, 1,00 экв.). Смесь перемешивали при 50°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат =100/1-100/30), в результате чего получали соединение 25-g.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,85 (d, J=8,5 Гц, 2H), 7,28-7,27 (m, 2Н), 7,22-7,14 (m, 1H), 7,02 (s, 1H), 6,83 (s, 2Н), 4,30 (s, 2Н), 3,53 (d, J=6,8 Гц, 2H), 2,55 (s, 3Н), 2,21 (s, 6Н), 1,49 (s, 6Н), 1,38 (s, 3Н).
Этап 7: Соединение 25-h
N-(метоксиметил)-1-фенил-N-(триметилсилил)метиламин (3,33 г, 14,04 ммоль, 6,00 экв.) и трифторуксусную кислоту (1,60 г, 14,04 ммоль, 1,04 мл, 6,00 экв.) добавляли в раствор соединения 25-g (1,00 г, 2,34 ммоль, 1,00 экв.) в дихлорметане (60,00 мл) при 20°С. Смесь перемешивали при 40°С в течение 17 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат =100/1-100/30), в результате чего получали соединение 25-h.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,71 (d, J=8,5 Гц, 1H), 7,31-7,20 (m, 9Н), 6,71 (s, 1H), 4,30-4,23 (m, 2Н), 3,76-3,36 (m, 6Н), 3,07-2,96 (m, 2Н), 2,68 (t, J=7,3 Гц, 2H), 2,51 (s, 3Н), 2,10-2.03 (m, 6Н), 1,36-1,22 (m, 9Н).
Этап 8: Соединение 25-i
Фенилхлорформиат (375,00 мг, 2,40 ммоль, 300,00 мкл, 3,12 экв.) добавляли в раствор соединения 25-h (430,00 мг, 768,19 мкмоль, 1,00 экв.) в дихлорметане (20,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 17 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = от 100/1 до 100/50), в результате чего получали соединение 25-i.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,66 (dd, J=4,0, 8,3 Гц, 2H), 7,41-7,33 (m, 2Н), 7,23-7,08 (m, 5Н), 6,79 (d, J=2,0 Гц, 2H), 4,12 (q, J=7,0 Гц, 2H), 3,93-3,60 (m, 4Н), 3,48-3,32 (m, 1H), 2,97-2,84 (m, 1H), 2,67 (d, J=7,8 Гц, 2H), 2,57-2,49 (m, 3Н), 2,25-2,13 (m, 6H), 1,46 (s, 6H), 1,36 (t, J=7,2 Гц, 3Н).
Этап 9: Соединение 25-j
Воду (8,00 мл) и гидроксид лития (81,22 мг, 3,39 ммоль, 20,00 экв.) добавляли в раствор соединения 25-i (100,00 мг, 169,57 мкмоль, 1,00 экв.) в этаноле (8,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 17 ч. В смесь добавляли разбавленную HCl (1 н., 10 мл) и экстрагировали дихлорметаном (50 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 25-j.
МС: m/z (ионизация электрораспылением): 584,1 [М+23].
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,65 (d, J=6,8 Гц, 2H), 7,43-7,37 (m, 2Н), 7,29-7,22 (m, 3Н), 7,16 (t, J=7,2 Гц, 2H), 6,87 (s, 2Н), 4,13-3,92 (m, 2Н), 3,89-3,80 (m, 1H), 3,69 (dd, J=5,4, 10,7 Гц, 1H), 3,58-3,49 (m, 1H), 2,81 (d, J=14,1 Гц, 2H), 2,71-2,63 (m, 1H), 2,55 (s, 3Н), 2,23 (s, 6Н), 1,43 (s,6H).
Этап 10: соединения 25 и 26
Соединение 25-j (25,00 мг, 44,51 мкмоль, 1,00 экв.) подвергали хиральному разделению, в результате чего получали соединение 25; Соединение 26.
Соединение 25: 1Н-ЯМР (400 МГц, CDCl3) δ 7,73 (d, J=8,28 Гц, 2Н), 7,41-7,33 (m, 2Н), 7,27-7,18 (m, 3Н), 7,15 (d, J=8,28 Гц, 2Н), 6,83 (d, J=5,52 Гц, 2Н), 4,02-3,64 (m, 4Н), 3,50-3,32 (m, 1H), 3,08-2,88 (m, 1H), 2,81-2,61 (m, 2Н), 2,55 (s, 3Н), 2,20 (d, J=3,51 Гц, 6Н).
Соединение 26: 1Н-ЯМР (400 МГц, CDCl3) δ 7,73 (d, J=8,28 Гц, 2Н), 7,42-7,34 (m, 2Н), 7,27-7,18 (m, 3Н), 7,15 (d, J=8,54 Гц, 2Н), 6,83 (d, J=5,78 Гц, 2Н), 4,05-3,64 (m, 4Н), 3,50-3,32 (m, 1H), 3,07-2,88 (m, 1H), 2,83-2,62 (m, 2Н), 2,55 (s, 3Н), 2,20 (d, J=3,52 Гц, 6Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 25: 2,018 мин (пик 1); Время удерживания соединения 26: 2,410 мин (пик 2).
Пример 27: Соединение 27
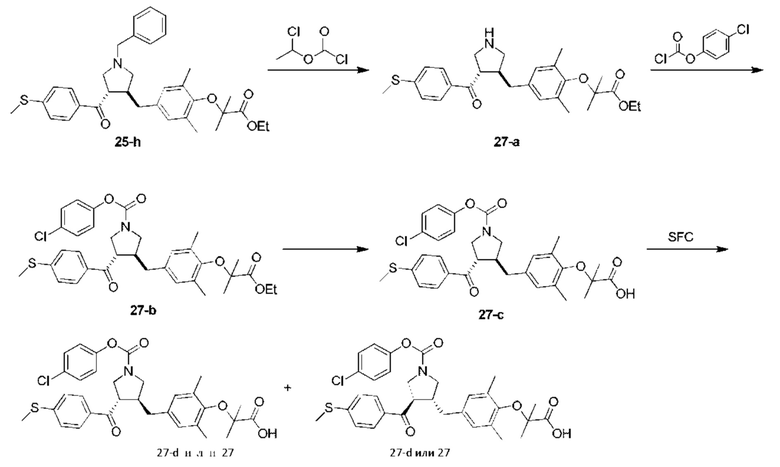
Этап 1: Соединение 27-а
Хлорметил-1-хлорформиат (3,32 г, 23,22 ммоль, 2,00 экв.) добавляли в раствор соединения 25-h (6,50 г, 11,61 ммоль, 1,00 экв.) в толуоле (60,00 мл) при 25°С. Смесь перемешивали в защитной атмосфере азота при 100°С в течение 8 ч. Смесь концентрировали при пониженном давлении. В остаток добавляли метанол (50,00 мл). Смесь перемешивали при 70°С в течение 1 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан: метанол =10:1), в результате чего получали соединение 27-а.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,57 (d, J=8,8 Гц, 2H), 7,15 (d, J=8,5 Гц, 2H), 6,77 (s, 2Н), 4,27 (q, J=7,2 Гц, 2H), 3,96-3,86 (m, 1H), 3,74 (dd, J=3,3, 7,0 Гц, 1H), 3,69-3,59 (m, 1H), 3,53-3,36 (m, 1H), 3,32-3,20 (m, 1H), 2,88 (br d, J=7,0 Гц, 1H), 2,76 (dd, J=2,6, 7,9 Гц, 2H), 2,52-2,49 (m, 3Н), 2,14-2,09 (m, 6H), 1,42 (d, J=2,8 Гц, 6H), 1,34 (t, J=7,2 Гц, 3Н).
Этап 2: Соединение 27-b
Раствор соединения 27-а (250,00 мг, 532,32 мкмоль, 1,00 экв.), ди-изо-пропилэтиламина (137,59 мг, 1,06 ммоль, 185,93 мкл, 2,00 экв.) и дихлорметана (5,00 мл) добавляли в 50 мл реакционную колбу. Затем добавляли (4-хлорфенил)-метилхлорформиат (152,52 мг, 798,48 мкмоль, 1,50 экв.). Смесь перемешивали при 25°С в течение 2 ч. В смесь добавляли насыщенный раствор бикарбоната натрия (5 мл). Водную фазу экстрагировали дихлорметаном (3×10 мл). Объединенную органическую фазу промывали насыщенным водным раствором бисульфата калия(10 мл) и насыщенным солевым раствором (10 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат =от 8:1 до 5:1), в результате чего получали соединение 27-b.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,56 (dd, J=8,8, 4,0 Гц, 2H), 7,24 (d, J=8,4 Гц, 2Н), 7,13 (d, J=8,4 Гц, 2Н), 7,02 (dd, J=9,2, 4,0 Гц, 2Н), 6,70 (d, J=2,4 Гц, 2Н), 4,22 (q, J=7,2 Гц, 2Н), 3,82-3,53 (m, 4Н), 3,38-3,30 (m, 1H), 2,85-2,77 (m, 1H), 2,59-2,46 (m, 2Н), 2,46 (s, 3Н), 2,10 (s, 6Н), 1,39 (s, 6Н), 1,29 (t, J=7,2 Гц, 3Н).
Этап 3: Соединение 27-с
Гидроксид лития (29,93 мг, 1,25 ммоль, 3,00 экв.) добавляли в раствор соединения 27-b (260,00 мг, 416,54 мкмоль, 1,00 экв.) в этаноле (2,00 мл), тетрагидрофуране (2,00 мл) и воде (1,00 мл). Смесь перемешивали при 25°С в течение 60 ч. Смесь доводили насыщенным водным раствором бисульфата калия до рН=2-3 и экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу промывали насыщенным слоевым раствором, сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат =1:2), в результате чего получали соединение 27-с.
МС: m/z (ионизация электрораспылением): 618,0 [М+23].
Этап 4: Соединение 27
Соединение 27-с (100,00 мг) подвергали хиральному разделению, в результате чего получали соединение 27.
МС: m/z (ионизация электрораспылением): 618,0 [М+23].
1H-ЯМР (400 МГц, MeOD-d4) δ ppm 7,65 (d, J=8,4 Гц, 2Н), 7,39 (d, J=8,8 Гц, 2Н), 7,13 (d, J=8,4 Гц, 2Н), 7,17 (t, J=8,8 Гц, 2Н), 6,87 (s, 2Н), 4,01-3,96 (m, 2Н), 3,82-3,81 (m, 1H), 3,70-3,67 (m, 1H), 3,53-3,51 (m, 1H), 2,82-2,68 (m, 3Н), 2,67 (s, 3Н), 2,24 (s, 6Н), 1,42 (s, 6Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 50×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 4 мл/мин; температура колонки: 40°С.
Время удерживания соединения 27: 1,148 мин (пик 1)
Пример 28: Соединение 28
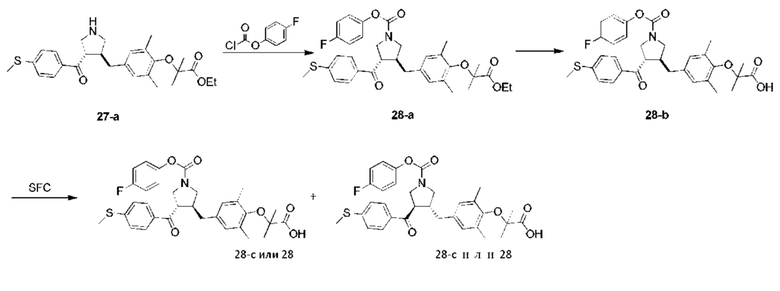
Этап 1: Соединение 28-а
Раствор соединения 27-а (250,00 мг, 532,32 мкмоль, 1,00 экв.), ди-изо-пропилэтиламина (137,59 мг, 1,06 ммоль, 185,93 мкл, 2,00 экв.) и дихлорметана (5,00 мл) добавляли в 50 мл реакционную колбу. Затем добавляли (4-фторфенил) метилхлорформиат (139,38 мг, 798,48 мкмоль, 104,80 мкл, 1,50 экв.). Смесь перемешивали при 25°С в течение 2 ч. В смесь добавляли насыщенный раствор бикарбоната натрия (5 мл). Водную фазу экстрагировали дихлорметаном (3×10 мл), промывали объединенную органическую фазу насыщенным водным раствором бисульфата калия (10 мл) и насыщенным солевым раствором (10 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат =10:1-5:1), в результате чего получали соединение 28-а.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,57 (dd, J=8,4, 4,4 Гц, 2H), 7,13 (d, J=8,0 Гц, 2H), 7,03-6,96 (m, 4Н), 6,71 (d, J=2,8 Гц, 2H), 4,22 (q, J=7,2 Гц, 2H), 3,83-3,66 (m, 4Н), 3,64-3,36 (m, 1H), 2,78-2,76 (m, 1H), 2,61-2,56 (m, 2Н), 2,46 (s, 3Н), 2,09 (s, 6Н), 1,39 (s, 6Н), 1,29 (t, J=7,2 Гц, 3Н).
Этап 2: Соединение 28-b
Гидроксид лития (30,74 мг, 1,28 ммоль, 3,00 экв.) добавляли в раствор соединения 28-а (260,00 мг, 416,54 мкмоль, 1,00 экв.) в этаноле (2,00 мл), тетрагидрофуране (2,00 мл) и воде (1,00 мл). Смесь перемешивали при 25°С в течение 60 ч. Смесь доводили насыщенным водным раствором бисульфата калия до рН=2-3 и экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу промывали насыщенным слоевым раствором, сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат =1:2), в результате чего получали соединение 28-b.
МС: m/z (ионизация электрораспылением): 602,1 [М+23].
Этап 3: Соединение 28
Соединение 28-b (100,00 мг) подвергали хиральному разделению, в результате чего получали соединение 28.
МС: m/z (ионизация электрораспылением): 602,1 [М+23].
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,65 (dd, J=8,4, 2,4 Гц, 2H), 7,26 (d, J=8,4 Гц, 2H), 7,17-7,13 (m, 4Н), 6,88 (s, 2Н), 4,02-3,97 (m, 2Н), 3,82-3,80 (m, 1H), 3,67-3,65 (m, 1H), 3,53-3,50 (m, 1H), 2,84-2,67 (m, 3Н), 2,56 (s, 3Н), 2,19 (s, 6Н), 1,45 (s, 6Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 50×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 4 мл/мин; температура колонки: 40°С.
Время удерживания соединения 28: 1,867 мин. (пик 1)
Примеры 29 и 30: соединения 29 и 30
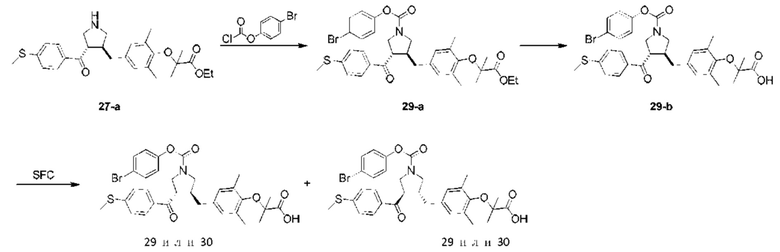
Этап 1: Соединение 29-а
Раствор соединения 27-а (250,00 мг, 532,32 мкмоль, 1,00 экв.), ди-изо-пропилэтиламина (137,60 мг, 1,06 ммоль, 2,00 экв.) и трихлорметана (5,00 мл) добавляли в 50 мл реакционную колбу. Затем добавляли (4-бром фенил) метилхлорформиат (188,02 мг, 798,50 мкмоль, 113,95 мкл, 1,50 экв.). Смесь перемешивали при 25°С в течение 18 ч. В смесь добавляли насыщенный раствор бикарбоната натрия (5 мл). Водную фазу экстрагировали дихлорметаном (2*10 мл) и промывали объединенную органическую фазу насыщенным водным раствором бисульфата калия (10 мл) и насыщенным солевым раствором (10 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат = 8:1), в результате чего получали соединение 29-а.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,55 (dd, J=8,8, 3,2 Гц, 2H), 7,39 (d, J=8,4 Гц, 2Н), 7,13 (d, J=8,8 Гц, 2Н), 6,71 (d, J=2,4 Гц, 2Н), 4,22 (q, J=7,6 Гц, 2Н), 3,82-3,66 (m, 4Н), 3,37-3,30 (m, 1H), 2,85-2,80 (m, 1H), 2,77 (d, J=6,4 Гц, 2Н), 2,45 (s, 3Н), 2,09 (s, 6Н), 1,39 (s, 6Н), 1,29 (t, J=7,6 Гц, 1H).
Этап 2: Соединение 29-b
Гидроксид лития (29,01 мг, 1,21 ммоль, 3,00 экв.) добавляли в раствор соединения 29-а (270,00 мг, 403,80 мкмоль, 1,00 экв.) в этаноле (2,00 мл), тетрагидрофуране (2,00 мл) и воде (1,00 мл). Смесь перемешивали при 25°С в течение 60 ч. Смесь доводили насыщенным водным раствором бисульфата калия до рН=2-3 и экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу промывали насыщенным слоевым раствором, сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат = 1:3), в результате чего получали соединение 29-b.
MC: m/z (ионизация электрораспылением): 664,0 [М+23].
Этап 3: соединения 29 и 30
Соединение 29-b (100,00 мг) подвергали хиральному разделению, в результате чего получали соединение 29; Соединение 30.
Соединение 29:
MC: m/z (ионизация электрораспылением): 664,0 [М+23].
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,65 (dd, J=8,0, 2,0 Гц, 2Н), 7,53 (d, J=9,2 Гц, 2Н), 7,26 (d, J=8,0 Гц, 2Н), 7,12 (dd, J=8,8, 2,4 Гц, 2Н), 6,88 (s, 2Н), 3,98-3,95 (m, 2Н), 3,84-3,80 (m, 1H), 3,69-3,67 (m, 1H), 3,53-3,51 (m, 1H), 2,82-2,66 (m, 3Н), 2,55 (s, 3Н), 2,23 (s, 6Н), 1,42 (s, 6Н).
Соединение 30:
MC: m/z (ионизация электрораспылением): 664,0 [М+23].
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,66 (d, J=6,4 Гц, 2Н), 7,54 (d, J=8,4 Гц, 2Н), 7,26 (d, J=8,4 Гц, 2Н), 7,12 (dd, J=8,8, 2,8 Гц, 2Н), 6,88 (s, 2Н), 4,02-3,84 (m, 2Н), 3,84-3,82 (m, 1H), 3,69-3,66 (m, 1H), 3,53-3,51 (m, 1H), 2,83-2,79 (m, 4Н), 2,67 (s, 3Н), 2,23 (s, 6Н), 1,43 (s, 6Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 50×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 4 мл/мин; температура колонки: 40°С.
Время удерживания соединения 29: 1,376 мин (пик 1); Время удерживания соединения 30: 2,462 мин (пик 2).
Примеры 31 и 32: соединения 31 и 32
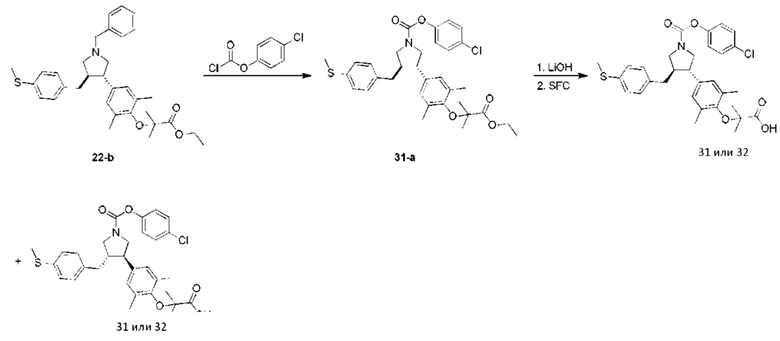
Этап 1: Соединение 31-а
Соединение 22-b (850,00 мг, 1,60 ммоль, 1,00 экв.) и дихлорметан (10,00 мл) добавляли в высушенную круглодонную колбу. Затем добавляли (4-хлорфенил) хлорформиат (610,66 мг, 3,20 ммоль, 445,74 мкл, 2,00 экв.). Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (40,6%, этилацетат/петролейный эфир), в результате чего получали соединение 31-а.
МС: m/z (ионизация электрораспылением): 596,1 [М+1].
Этап 2: соединения 31 и 32
Гидроксид лития (8,03 мг, 335,47 мкмоль, 1,00 экв.) и воду (1,00 мл) добавляли в раствор соединения 31-а (200,00 мг, 335,47 мкмоль, 1,00 экв.) в тетрагидрофуране (3 мл) и этаноле (2,00 мл). Смесь перемешивали при 25°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=2, а затем добавляли смесь вода/этилацетат (1:1, 50 мл). Водную фазу экстрагировали этилацетатом (50 мл × 2). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (50,7%, этилацетат/петролейный эфир) с получением соединения, которое подвергали хиральному разделению, в результате чего получали соединение 31; Соединение 32.
Соединение 31:
МС: m/z (ионизация электрораспылением): 590,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,24-7,21 (m, 2Н), 7,08 (t, J=6,7 Гц, 2H), 6,99-6,94 (m, 4Н), 6,83 (s, 2Н), 3,94-3,85 (m, 1H), 3,70-3,69 (m, 1H), 3,45-3,38 (m, 1H), 3,21-3,15 (m, 1H), 2,90-2,88 (m, 1H), 2,70-2,67 (m, 1H), 2,52-2,50 (m, 2Н), 2,39 (d, J=1,5 Гц, 3Н), 2,18 (s, 6Н), 1,45 (s, 6Н).
Соединение 32:
МС: m/z (ионизация электрораспылением): 590,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,23 (dd, J=2,0, 8,8 Гц, 2H), 7,08 (t, J=6,8 Гц, 2H), 6,99-6,94 (m, 4Н), 6,83 (s, 2Н), 3,94-3,85 (m, 1H), 3,70-3,69 (m, 1H), 3,45-3,39 (m, 1H), 3,21-3,15 (m, 1H), 2,90-2,88 (m, 1H), 2,70-2,67 (m, 1H), 2,52-2,50 (m, 2Н), 2,39 (d, J=1,5 Гц, 3Н), 2,18 (s, 6Н), 1,45 (s,6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 150×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 2,5 мл/мин; температура колонки: 40°С.
Время удерживания соединения 31: 2,644 мин (пик 1); Время удерживания соединения 32: 3,026 мин (пик 2).
Примеры 33 или 34: соединения 33 или 34
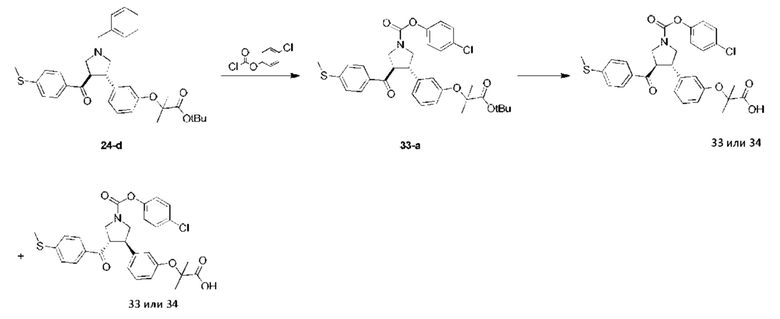
Этап 1: Соединение 33-а
В защитной атмосфере азота (4-хлорфенил) хлорформиат (5,25 г, 27,48 ммоль, 3,83 мл, 2,00 экв.) медленно добавляли в раствор соединения 2-b (7,50 г, 13,74 ммоль, 1,00 экв.) в дихлорметане (60,00 мл). Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (41,5%, этилацетат/петролейный эфир), в результате чего получали соединение 33-а.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,68 (dd, J=5,3, 8,5 Гц, 2H), 7,26-7,24 (m, 2Н), 7,15-7,03 (m, 5Н), 6,87-6,82 (m, 2Н), 6,63 (dd, J=2,3, 8,3 Гц, 1H), 4,11-3,96 (m, 3Н), 3,82-3,70 (m, 1H), 3,68-3,60 (m, 2Н), 2,43 (s, 3Н), 1,46 (d, J=4,8 Гц, 6H), 1,35 (s, 9Н)
Этап 2: соединения 33 и 34
В колбе, трифторуксусную кислоту (23,10 г, 202,60 ммоль, 15,00 мл, 16,94 экв.) медленно добавляли в раствор соединения 33-а (7,30 г, 11,96 ммоль, 1,00 экв.) в дихлорметане (60,00 мл). Смесь перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (40,6%, этилацетат/петролейный эфир) и подвергали полученный продукт хиральному разделению, в результате чего получали соединение 33, Выход: 3,65%; Соединение 34.
Соединение 33:
МС: m/z (ионизация электрораспылением): 576,0 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,78-7,75 (m, 2Н), 7,33-7,31 (m, 2Н), 7,23-7,12 (m, 5Н), 6,93-6,86 (m, 2Н), 6,79 (t, J=7,1 Гц, 1H), 4,08-3,97 (m, 3Н), 3,81-3,77 (m, 3Н), 2,50 (s, 3Н), 1,56-1,53 (m, 6Н)
Соединение 34:
МС: m/z (ионизация электрораспылением): 576,0 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,69 (dd, J=6,0, 8,3 Гц, 2H), 7,24 (d, J=8,8 Гц, 2H), 7,15-7,03 (m, 5Н), 6,83 (br t, J=6,5 Гц, 1H), 6,78 (s, 1H), 6,75-6,72 (m, 1H), 4,01-3,91 (m, 3Н), 3,73-3,69 (m, 3Н), 2,42 (s, 3Н), 1,47-1,45 (m, 6H)
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2.
Время удерживания соединения 33: 5,039 мин (пик 1); Время удерживания соединения 34: 6,986 мин (пик 2).
Пример 35: Соединение 35
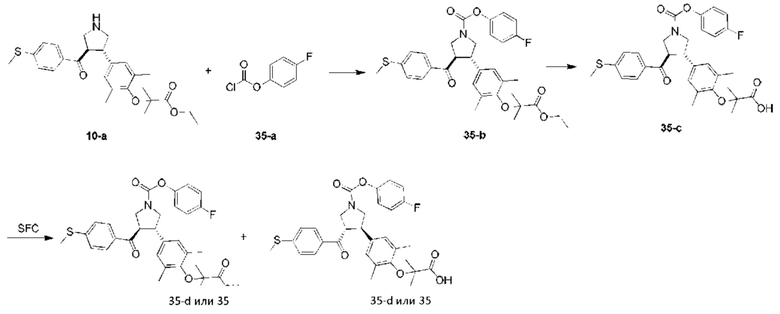
Этап 1: Соединение 35-b
В защитной атмосфере азота Соединение 10-а (150,00 мг, 329,23 мкмоль, 1,00 экв.), ди-изо-пропилэтиламин (85,10 мг, 658,46 мкмоль, 115,00 мкл, 2,00 экв.) и 35-а (68,96 мг, 395,07 мкмоль, 51,85 мкл, 1,20 экв.) перемешивали при 20°С в течение 12 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 35-b.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,77-7,72 (m, 2Н), 7,23-7,21 (m, 2Н), 7,13-7,12 (m, 2Н), 7,08-7,06 (m, 2Н), 6,88-6,87 (m, 2Н), 4,32-4,17 (m, 2Н), 4,15-4,09 (m, 3Н), 3,84-3,77 (m, 3Н), 2,53 (s, 3Н), 2,16 (t, J=6,53 Гц, 6 H), 1,44 (s, 6 Н), 1,38-1,34 (m, 3Н).
Этап 2: Соединение 35-с
Раствор соединения 35-b (180,00 мг, 303,18 мкмоль, 1,00 экв.) и гидроксид лития (127,22 мг, 3,03 ммоль, 10,00 экв.) в этаноле (9,00 мл) и воде (3,00 мл) перемешивали при 25°С в течение 12 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=3, и обрабатывали водой и этилацетатом (1:1, 50 мл). Водную фазу экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл × 1), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1), в результате чего получали соединение 35-с.
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,67-7,65 (m, 2Н), 7,45-7,39 (m, 1H), 7,13-7,02 (m, 5Н), 6,87-6,86 (m, 2Н), 4,35-4,31 (m, 1H), 4,06-4,03 (m, 1H), 3,88-3,78 (m, 2Н), 3,54-3,51 (m, 2Н), 2,40 (s, 3Н), 2,06-2,05 (m, 6Н), 1,26 (d, J=2,80 Гц, 6H).
Этап 3: Соединение 35
Соединение 35-с (120 мг) подвергали хиральному разделению, в результате чего получали соединение 35.
МС: m/z (ионизация электрораспылением): 588,1 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,68 (t, J=8,41 Гц, 2H), 7,13 (dd, J=4,02, 8,53 Гц, 2H), 7,03-7,02 (m, 2Н), 6,99-6,97 (m, 2Н), 6,84 (d, J=6,53 Гц, 2H), 4,08-4,01 (m, 3Н), 3,70-3,65 (m, 3Н), 2,44 (s, 3Н), 2,12 (d, J=5,52 Гц, 6H), 1,40 (s, 6 Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 35: 2,429 мин (пик 2).
Пример 36: Соединение 36
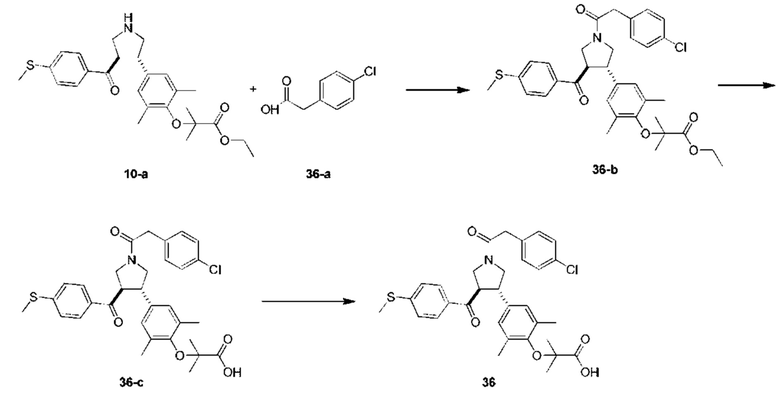
Этап 1: Соединение 36-b
В защитной атмосфере азота раствор соединения 10-а (150,00 мг, 329,23 мкмоль, 1,00 экв.), триэтиламина (66,63 мг, 658,46 мкмоль, 91,27 мкл, 2,00 экв.), окси-(7-азабензотриазол-1-ил)-N,N,N',N'-третраметилмочевинагексафторфосфоната (187,77 мг, 493,85 мкмоль, 1,50 экв.) и 36-а (84,25 мг, 493,85 мкмоль, 1,50 экв.) в дихлорметане (10,00 мл) перемешивали при 20°С в течение 12 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 36-b.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,65 (d, J=8,4 Гц, 2H), 7,53-7,52 (m, 1H), 7,24-7,09 (m, 6 H), 6,67 (d, J=4,8 Гц, 2H), 3,97-3,89 (m, 3Н), 3,60-3,58 (m, 5H), 2,43 (s, 3Н), 2,03 (d, J=8,0 Гц, 6 H), 1,34-1,31 (m, 6 H), 1,29-1,25 (m, 3Н).
Этап 2: Соединение 36-с
Раствор соединения 36-b (120,00 мг, 197,31 мкмоль, 1,00 экв.) и гидроксид лития (82,79 мг, 1,97 ммоль, 10,00 экв.) в этаноле (9,00 мл) и воде (3,00 мл) перемешивали при 25°С в течение 12 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=3 и обрабатывали водой и этилацетатом (1:1, 50 мл). Водную фазу экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл × 1), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1), в результате чего получали соединение 36-с.
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,78-7,75 (m, 2 Н), 7,36-7,31 (m, 4 Н), 7,24-7,21 (m, 2 Н), 6,89 (s, 2 Н), 4,09-3,91 (m, 3 Н), 3,77-3,50 (m, 5 Н), 2,51 (d, J=2,8 Гц, 3 H), 2,16 (s, 6 Н), 1,36 (s, 6 H).
Этап 3: Соединение 36
Соединение 36-с (80 мг) отделяли высокоэффективной жидкостной хроматографией, в результате чего получали соединение 36.
МС: m/z (ионизация электрораспылением): 602,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,67 (d, J=8,53 Гц, 1H), 7,56 (d, J=8,53 Гц, 1H), 7,22-7,21 (m, 2Н), 7,18-7,09 (m, 4Н), 6,70 (d, J=5,02 Гц, 2H), 4,03-3,98 (m, 1H), 3,91-3,89 (m, 1H), 3,80 (d, J=8,03 Гц, 1H), 3,72 (d, J=7,78 Гц, 1H), 3,61-3, 60 (m, 3Н), 3,59-3,56 (m, 1H), 2,43 (s, 3Н), 2,07 (d, J=9,54 Гц, 6H), 1,39-1,35 (m, 6H).
Пример 37: Соединение 37
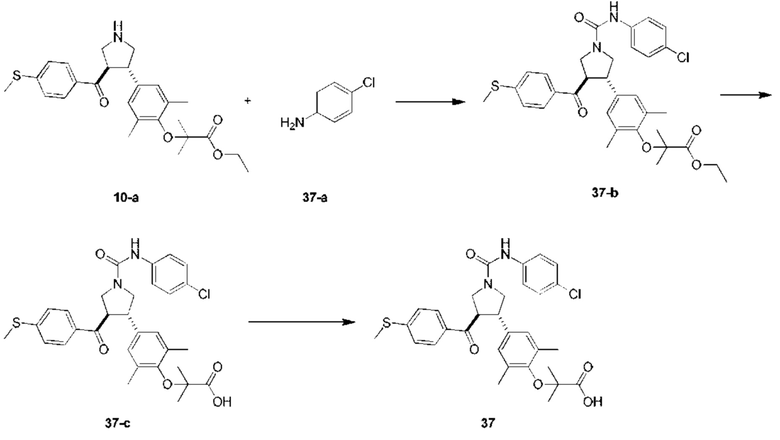
Этап 1: Соединение 37-b
В защитной атмосфере азота раствор соединения 10-а (400,00 мг, 877,94 мкмоль, 1,00 экв.), трифосгена (208,42 мг, 702,35 мкмоль, 0,80 экв.), ди-изо-пропилэтиламина (226,93 мг, 1,76 ммоль, 306,66 мкл, 2,00 экв.) и 37-а (134,40 мг, 1,05 ммоль, 1,20 экв.) в тетрагидрофуране (20,00 мл) перемешивали при 25°С в течение 12 ч. Реакционную смесь концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 37-b.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,65-7,62 (m, 2Н), 7,30-7,27 (m, 3Н), 7,17-7,10 (m, 3Н), 6,79 (d, J=6,4 Гц, 2H), 4,22-4,05 (m, 5 Н), 3,93-3,58 (m, 3 Н), 2,43 (s, 3 Н), 2,05 (s, 6 Н), 1,33-1,25 (m, 8 H).
Этап 2: Соединение 37-с
Раствор соединения 37-b (200,00 мг, 328,32 мкмоль, 1,00 экв.) и гидроксида лития (13,78 мг, 328,32 мкмоль, 1,00 экв.) в этаноле (6,00 мл) и воде (3,00 мл) перемешивали при 25°С в течение 12 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=3 и обрабатывали водой и этилацетатом (1:1,50 мл). Водную фазу экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл × 1), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1), в результате чего получали соединение 37-с.
1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 7,82 (d, J=8,8 Гц, 2H), 7,54 (d, J=8,8 Гц, 2 Гц), 7,46 (d, J=8,8 Гц, 1H), 7,32-7,27 (m, 4Н), 6,97 (s, 1H), 4,49-4,45 (m, 1H), 4,02-3,93 (m, 2Н), 3,62-3,60 (m, 1H), 3,49-3,45 (m, 2Н), 2,56 (s, 3Н), 2,08 (s, 6Н), 1,25 (d, J=3,6 Гц, 6H).
Этап 3: Соединение 37
Соединение 37-с (110 мг) отделяли путем ВЭЖХ, в результате чего получали соединение 37.
МС: m/z (ионизация электрораспылением): 603,0 [М+23].
1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8,35 (s, 1H), 7,82 (d, J=8,53 Гц, 2Н), 7,54 (d, J=7,70 Гц, 2Н), 7,28-7,23 (m, 4Н), 6,96 (s, 2Н), 4,47-4,43 (m, 3H), 3,98-3,90 (m, 3Н), 2,48 (br s, 3Н), 2,08 (s, 6 Н), 1,26 (d, J=3,51 Гц, 6H).
Пример 38: Соединение 38
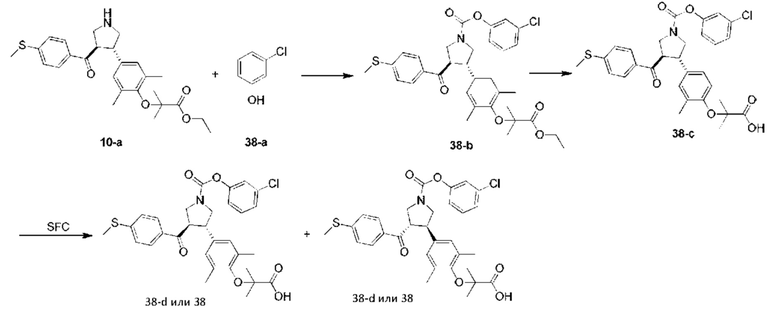
Этап 1: Соединение 38-а
В защитной атмосфере азота раствор соединения 38-а (1,00 г, 7,78 ммоль, 819,67 мкл, 1,00 экв.), трифосгена (1,85 г, 6,22 ммоль, 0,80 экв.) и триэтиламина (787,26 мг, 7,78 ммоль, 1,08 мл, 1,00 экв.) в тетрагидрофуране (20,00 мл) перемешивали при 0°С в течение 15 мин, нагревали до 20°С, и перемешивали в течение 1 ч. Реакционный раствор фильтровали и концентрировали, и растворяли полученный неочищенный продукт в тетрагидрофуране (5,00 мл), а затем добавляли 10-а (1,00 г, 2,19 ммоль, 1,00 экв.) и ди-изо-пропилэтиламин (424,55 мг, 3,29 ммоль, 573,72 мкл, 1,50 экв.) и перемешивали при 20°С в течение 1 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 38-b.
МС: m/z (ионизация электрораспылением): 610,2 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,78 (dd, J=3,52, 8,54 Гц, 2H), 7,44-7,38 (m, 1H), 7,32-7,24 (m, 4H), 7,18-7,12 (m, 1H), 6,97 (d, J=2,76 Гц, 2H), 4,55-4,38 (m, 1H), 4,13 (q, J=7,28 Гц, 2H), 3,97-3,84 (m, 1H), 3,67-3,43 (m, 3Н), 2,50 (s, 3Н), 2,04 (s, 6H), 1,28 (s, 6 H), 1,20 (t, J=7,04 Гц, 3Н)
Этап 2: Соединение 38-с
Раствор соединения 38-b (1,00 г, 1,64 ммоль, 1,00 экв.) и гидроксида лития (117,83 мг, 4,92 ммоль, 3,00 экв.) в этаноле (20,00 мл) и воде (5,00 мл) перемешивали при 20°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=4 и обрабатывали водой и этилацетатом (1:1,50 мл). Водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1), в результате чего получали соединение 38-с.
МС: m/z (ионизация электрораспылением): 582,2 [М+1].
Этап 3: Соединение 38
Соединение 38-с (200 мг) подвергали хиральному разделению, в результате чего получали соединение 38.
МС: m/z (ионизация электрораспылением): 604,0 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,74 (t, J=8,66 Гц, 2H), 7,32-7,27 (m, 1H), 7,23-7,15 (m, 4Н), 7,09-7,03 (m, 1H), 6,90 (br d, J=5,52 Гц, 2H), 4,19-4,02 (m, 3Н), 3,90-3,64 (m, 3Н), 2,50 (s, 3Н), 2,19 (br d, J=5,52 Гц, 6H), 1,46 (br s, 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 50×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 4,0 мл/мин; температура колонки: 40°С.
Время удерживания соединения 38: 1,667 мин (пик 2).
Пример 39: Соединение 39
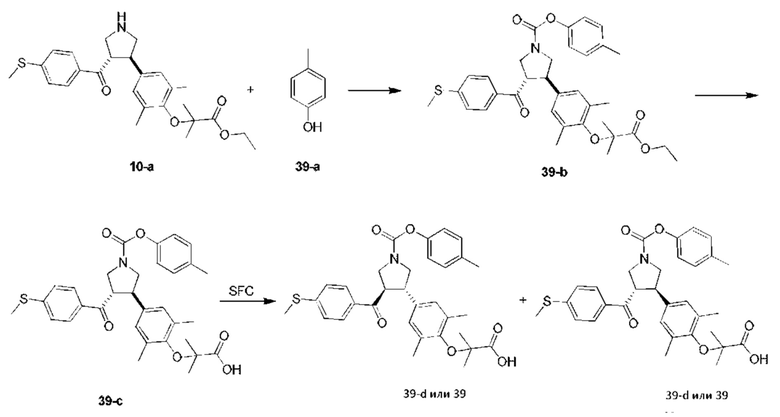
Этап 1: Соединение 39-b
В защитной атмосфере азота раствор соединения 39-а (1,00 г, 9,25 ммоль, 1,00 экв.), трифосгена (2,20 г, 7,40 ммоль, 0,80 экв.) и триэтиламина (1,12 г, 11,10 ммоль, 1,54 мл, 1,20 экв.) в тетрагидрофуране (20,00 мл) перемешивали при 0°С в течение 15 мин, нагревали до 20°С и перемешивали в течение 1 ч. Реакционный раствор фильтровали и концентрировали, и растворяли полученный неочищенный продукт в тетрагидрофуране (20,00 мл), 10-а (1,00 г, 2,19 ммоль, 1,00 экв.), добавляли ди-изо-пропилэтиламин (424,55 мг, 3,29 ммоль, 573,72 мкл, 1,50 экв.) и перемешивали при 20°С в течение 1 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 39-b.
МС: m/z (ионизация электрораспылением): 612,1 [М+23].
Этап 2: Соединение 39-с
Раствор соединения 39-b (200,00 мг, 339,13 мкмоль, 1,00 экв.) и гидроксида лития (81,22 мг, 3,39 ммоль, 10,00 экв.) в этаноле (20,00 мл) и воде (5,00 мл) перемешивали при 20°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=4 и обрабатывали водой и этилацетатом (1:1, 50 мл). Водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1), в результате чего получали соединение 39-с.
МС: m/z (ионизация электрораспылением): 562,2 [М+1].
Этап 3: Соединение 39
Соединение 39-с (200 мг) подвергали хиральному разделению, в результате чего получали соединение 39.
МС: m/z (ионизация электрораспылением): 562,2 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,77 (t, J=8,16 Гц, 2H), 7,25-7,15 (m, 4Н), 7,04 (d, J=8,28 Гц, 2H), 6,93 (d, J=6,54 Гц, 2H), 4,21-4,05 (m, 3Н), 3,92-3,65 (m, 3Н), 2,53 (s, 3Н), 2,21 (br d, J=5,52 Гц, 6 H), 1,49 (s, 6 H)
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 39: 3,697 мин (пик 2).
Пример 40: Соединение 40
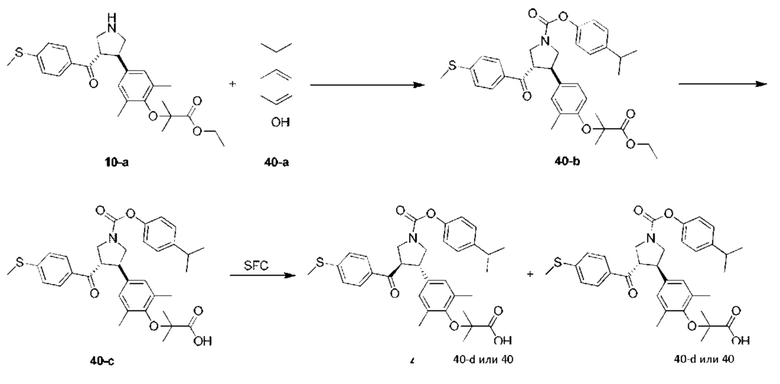
Этап 1: Соединение 40-а
В защитной атмосфере азота раствор соединения 40-а (1,00 г, 7,34 ммоль, 1,00 экв.), трифосгена (1,74 г, 5,87 ммоль, 0,80 экв.) и триэтиламина (891,28 мг, 8,81 ммоль, 1,22 мл, 1,20 экв.) в тетрагидрофуране (20,00 мл) перемешивали при 0°С в течение 15 мин, нагревали до 20°С и перемешивали в течение 1 ч. Реакционный раствор фильтровали и концентрировали и растворяли полученный неочищенный продукт в тетрагидрофуране (20,00 мл), а затем добавляли 10-а (1,00 г, 2,19 ммоль, 1,00 экв.) и ди-изо-пропилэтиламин (424,55 мг, 3,29 ммоль, 573,72 мкл, 1,50 экв.) и перемешивали при 20°С в течение 1 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 40-b.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,81 (d, J=7,54 Гц, 2H), 7,26 (dd, J=8,54, 13,30 Гц, 4H), 7,11-6,94 (m, 4Н), 4,58-4,41 (m, 1H), 4,20-4,09 (m, 2Н), 3,99-3,82 (m, 1H), 3,43-3,72 (m, 3Н), 2,89 (q, J=6,74, 13,69 Гц, 1H), 2,50-2,48 (m, 3Н), 2,14-2,01 (m, 6 Н), 1,30 (s, 6 Н), 1,25-1,18 (m, 9Н).
Этап 2: Соединение 40-с
Раствор соединения 40-b (1,00 г, 1,62 ммоль, 1,00 экв.) и гидроксида лития (387,67 мг, 16,20 ммоль, 10,00 экв.) в этаноле (20,00 мл) и воде (5,00 мл) перемешивали при 20°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=4 и обрабатывали водой и этилацетатом (1:1,50 мл). Водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1), в результате чего получали соединение 40-с.
МС: m/z (ионизация электрораспылением): 612,2 [М+23].
Этап 3: Соединение 40
Соединение 40-с (200 мг) подвергали хиральному разделению, в результате чего получали соединение 40.
МС: m/z (ионизация электрораспылением): 612,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,66 (br t, J=8,78 Гц, 2H), 7,15-7,08 (m, 4H), 6,98 (d, J=8,54 Гц, 2H), 6,82 (br d, J=7,28 Гц, 2H), 4,13-3,95 (m, 3H), 3,84-3,56 (m, 3H), 2,88-2,77 (m, 1H), 2,41 (s, 3H), 2,10 (br d, J=6,54 Гц, 6 H), 1,37 (br s, 6 H), 1,16 (d, J=6,78 Гц, 6 H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% изо-пропанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 40: 2,240 мин (пик 2).
Пример 41: Соединение 41
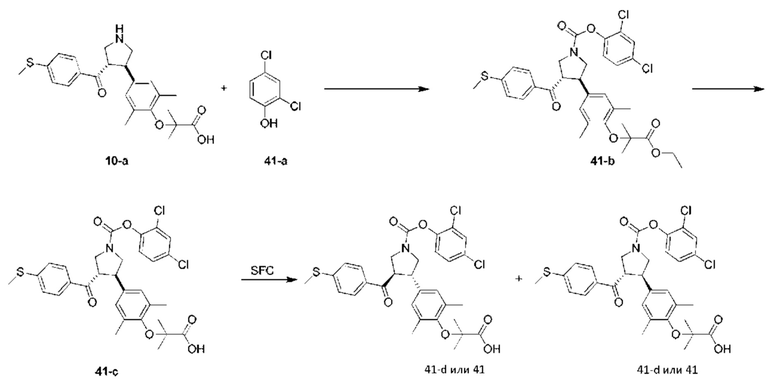
Этап 1: Соединение 41-b
В защитной атмосфере азота раствор соединения 41-а (1,00 г, 6,13 ммоль, 1,00 экв.), трифосгена (1,46 г, 4,90 ммоль, 0,80 экв.) и триэтиламина (620,80 мг, 6,13 ммоль, 850,41 мкл, 1,00 экв.) в тетрагидрофуране (20,00 мл) перемешивали при 0°С в течение 15 мин, нагревали до 20°С и перемешивали в течение 1 ч. Реакционный раствор фильтровали и концентрировали и растворяли полученный неочищенный продукт в тетрагидрофуране (20,00 мл), а затем добавляли 10-а (1,00 г, 2,19 ммоль, 1,00 экв.) и ди-изо-пропилэтиламин (424,55 мг, 3,29 ммоль, 573,72 мкл, 1,50 экв.) и перемешивали при 20°С в течение 1 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 41-b.
МС: m/z (ионизация электрораспылением): 644,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,82-7,73 (m, 3Н), 7,45 (dt, J=2,51, 8,66 Гц, 1H), 7,40-7,36 (m, 1H), 7,27 (d, J=8,28 Гц, 2H), 6,97 (d, J=3,52 Гц, 2H), 4,56-4,42 (m, 1H), 4,13 (q, J=7,04 Гц, 2H), 3,96-3,83 (m, 1H), 3,42-3,79 (m, 4Н), 2,06-2,02 (m, 6 Н), 1,28 (d, J=1,76 Гц, 6 H), 1,20 (t, 1=7,16 Гц, 3Н)
Этап 2: Соединение 41-с
Раствор соединения 41-b (1,00 г, 1,55 ммоль, 1,00 экв.) и гидроксид лития (111,46 мг, 4,65 ммоль, 3,00 экв.) в этаноле (20,00 мл) и воде (5,00 мл) перемешивали при 20°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=4, и обрабатывали водой и этилацетатом (1:1, 50 мл). Водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1), в результате чего получали соединение 41-с.
МС: m/z (ионизация электрораспылением): 616,1 [М+1].
Этап 3: Соединение 41
Соединение 41-с (200 мг) подвергали хиральному разделению, в результате чего получали соединение 41.
МС: m/z (ионизация электрораспылением): 616,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,69 (t, J=8,16 Гц, 2H), 7,37 (d, J=2,26 Гц, 1H), 7,18 (d, J=2,26 Гц, 1H), 7,16-7,11 (m, 3Н), 6,84 (d, J=7,28 Гц, 2H), 4,15-3,62 (m, 6 h), 2,44 (s, 3Н), 2,12 (d, J=5,02 Гц, 6 H), 1,44-1,35 (m, 6 H)
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 50×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% изо-пропанола (0,05% диэтаноламина) в СО2; скорость потока: 4,0 мл/мин; температура колонки: 40°С.
Время удерживания соединения 41: 1,899 мин (пик 2).
Пример 42: Соединение 42
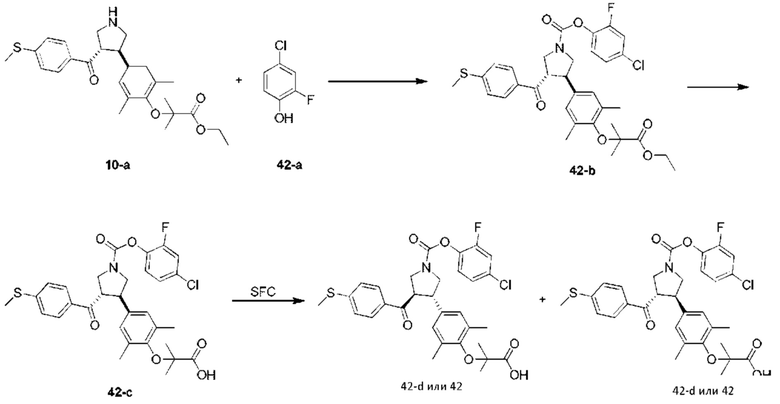
Этап 1: Соединение 42-b
В защитной атмосфере азота раствор соединения 42-а (1,00 г, 6,82 ммоль, 1,00 экв.), трифосгена (1,62 г, 5,46 ммоль, 0,80 экв.) и триэтиламина (690,48 мг, 6,82 ммоль, 945,86 мкл, 1,00 экв.) в тетрагидрофуране (20,00 мл) перемешивали при 0°С в течение 15 мин, нагревали до 20°С и перемешивали в течение 1 ч. Реакционный раствор фильтровали и концентрировали и растворяли полученный неочищенный продукт в тетрагидрофуране (20,00 мл), а затем добавляли 10-а (1,00 г, 2,19 ммоль, 1,00 экв.) и ди-изо-пропилэтиламин (424,55 мг, 3,29 ммоль, 573,72 мкл, 1,50 экв.) и перемешивали при 20°С в течение 1 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 42-b.
МС: m/z (ионизация электрораспылением): 628,1 [М+1].
1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 7,78 (d, J=7,28 Гц, 2Н), 7,60 (dd, J=2,38, 10,16 Гц, 1H), 7,41-7,21 (m, 5Н), 6,97 (d, J=3,76 Гц, 2Н), 4,56-4,41 (m, 1H), 4,13 (q, J=7,18 Гц, 2Н), 3,97-3,81 (m, 1H), 3,74-3,40 (m, 4Н), 2,51-2,50 (m, 3Н), 2,06-2,01 (m, 6 Н), 1,28 (d, J=1,52 Гц, 6Н), 1,20 (t, J=7,16 Гц, 3Н).
Этап 2: Соединение 42-с
Раствор соединения 42-b (1,00 г, 1,59 ммоль, 1,00 экв.) и гидроксид лития (114,24 мг, 4,77 ммоль, 3,00 экв.) в этаноле (20,00 мл) и воде (5,00 мл) перемешивали при 20°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=4, и обрабатывали водой и этилацетатом (1:1,50 мл). Водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1), в результате чего получали соединение 42-с.
МС: m/z (ионизация электрораспылением): 622,1 [М+23].
Этап 3: Соединение 42
Соединение 42-с (200 мг) подвергали хиральному разделению, в результате чего получали соединение 42.
МС: m/z (ионизация электрораспылением): 600,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,72 (d, J=7,54 Гц, 2H), 7,21-7,12 (m, 5Н), 6,89 (s, 2Н), 4,21-4,00 (m, 3Н), 3,83-3,64 (m, 3Н), 2,49 (s, 3Н), 2,18 (s, 6 Н), 1,25 (s, 6 Н)
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% изо-пропанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 42: 3,045 мин (пик 2).
Пример 43: Соединение 43
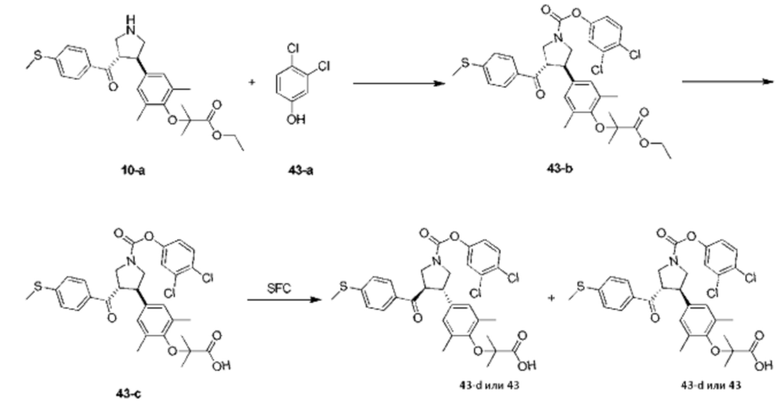
Этап 1: Соединение 43-b
В защитной атмосфере азота раствор соединения 43-а (1,00 г, 6,13 ммоль, 1,00 экв.), трифосгена (1,46 г, 4,90 ммоль, 0,80 экв.) и триэтиламина (620,80 мг, 6,13 ммоль, 850,41 мкл, 1,00 экв.) в тетрагидрофуране (20,00 мл) перемешивали при 0°С в течение 15 мин, нагревали до 20°С и перемешивали в течение 1 ч. Реакционный раствор фильтровали и концентрировали и растворяли полученный неочищенный продукт в тетрагидрофуране (20,00 мл), а затем добавляли 10-а (1,00 г, 2,19 ммоль, 1,00 экв.) и ди-изо-пропилэтиламин (424,55 мг, 3,29 ммоль, 573,72 мкл, 1,50 экв.) и перемешивали при 20°С в течение 1 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 43-b.
МС: m/z (ионизация электрораспылением): 644,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δppm 7,78 (dd, J=4,02, 8,54 Гц, 2H), 7,65 (d, J=8,78 Гц, 1H), 7,55 (d, J=2,52 Гц, 1H), 7,38 (d, J=8,78 Гц, 1H), 7,29-7,18 (m, 3Н), 6,96 (d, J=2,52 Гц, 3Н), 6,75 (dd, J=2,76, 8,78 Гц, 1H), 4,55-4,39 (m, 1H), 4,12 (q, J=7,20 Гц, 2H), 3,84-3,97 (m, 1H), 3,45-3,67 (m, 3Н), 2,50-2,50 (m, 3Н), 2,02-2,07 (m, 6 Н), 1,28 (s, 6Н), 1,20 (t, J=7,04 Гц, 3Н)
Этап 2: Соединение 43-с
Раствор соединения 43-b (1,00 г, 1,55 ммоль, 1,00 экв.) и гидроксида лития (111,37 мг, 4,65 ммоль, 3,00 экв.) в этаноле (20,00 мл) и воде (5,00 мл) перемешивали при 20°С в течение 16 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=4 и обрабатывали водой и этилацетатом (1:1,50 мл). Водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1), в результате чего получали соединение 43-с.
МС: m/z (ионизация электрораспылением): 616,1 [М+1].
Этап 3: Соединение 43
Соединение 43-с (200 мг) подвергали хиральному разделению, в результате чего получали соединение 43.
МС: m/z (ионизация электрораспылением): 616,0 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δppm 7,71-7,62 (m, 2Н), 7,35 (d, J=8,78 Гц, 1H), 7,24 (br s, 1H), 7,12 (br s, 2H), 6,97 (br d, J=8,78 Гц, 1H), 6,81 (br s, 2H), 4,01 (br s, 3H), 3,81-3,56 (m, 3H), 2,43 (s, 3H), 2,11 (br s, 6 H), 1,18 (s, 6 H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 50×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 4,0 мл/мин; температура колонки: 40°С.
Время удерживания соединения 43: 4,876 мин (пик 2).
Примеры 44 и 45: соединения 44 и 45
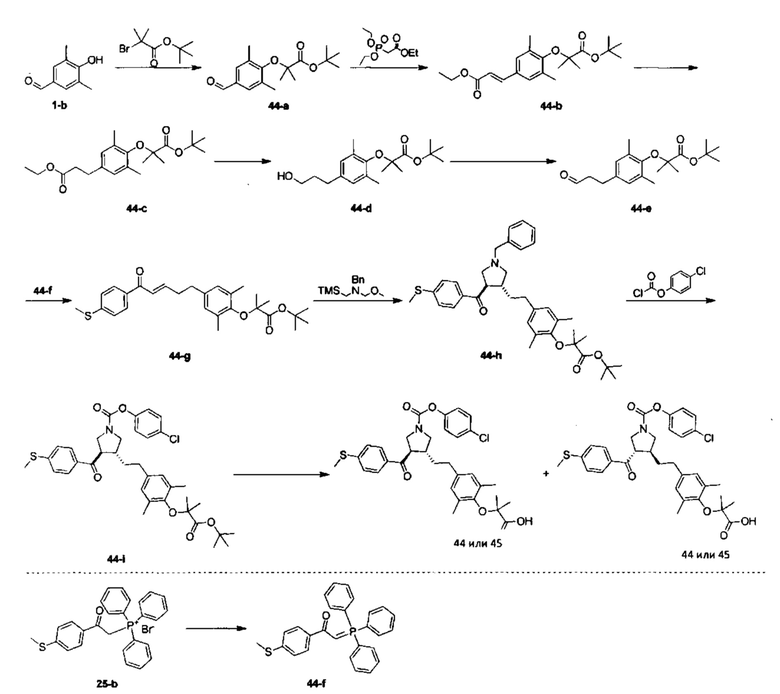
Этап 1: Соединение 44-а
В защитной атмосфере азота раствор соединения 1-b (20,00 г, 133,18 ммоль, 1,00 экв.), трет-бутил 2-бром-изобутирата (118,86 г, 532,72 ммоль, 99,05 мл, 4,00 экв.), карбоната калия (55,22 г, 399,54 ммоль, 3,00 экв.) и йодида калия (2,21 г, 13,32 ммоль, 0,10 экв.) в диметилсульфоксиде (250,00 мл) перемешивали при 110°С в течение 16 ч. Смесь фильтровали, и добавляли в фильтрат смесь этилацетат/вода (1:1, 300 мл). Органическую фазу промывали водой (2×300 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (11,8%, этилацетат/петролейный эфир), в результате чего получали соединение 44-а.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 9,89 (s, 1Н), 7,48 (s, 2H), 2,26 (s, 6 H), 1,47 (s, 9H), 1,42 (s, 6 H).
Этап 2: Соединение 44-b
Раствор триэтилфосфоноацетата (2,30 г, 10,26 ммоль, 2,04 мл, 1,50 экв.) в тетрагидрофуране (5 мл) добавляли в раствор гидрида натрия (547,25 мг, 13,68 ммоль, чистота 60%, 2,00 экв.) в тетрагидрофуране (15,00 мл) при 0°С. Смесь перемешивали при 25°С в течение 1 ч, а затем добавляли раствор соединения 44-а (2,00 г, 6,84 ммоль, 1,00 экв.) в тетрагидрофуране (10 мл). Смесь перемешивали в течение еще 4 ч. В смесь добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 44-b.
МС: m/z (ионизация электрораспылением): 363,0 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,50 (d, J=15,81 Гц, 1H), 7,09 (s, 2Н), 6,25 (d, J=15,81 Гц, 1H), 4,11-4,05 (m, 2Н), 2,17 (s, 6 h), 1,44 (s, 9H), 1,37 (s, 6 H), 1,27-1,25 (m, 3H).
Этап 3: Соединение 44-c
Палладий на угле (300,00 мг, чистота 10%) и безводный этанол (50 мл) добавляли в колбу для гидрирования в защитной атмосфере аргона. Затем добавляли раствор соединения 44-b (2,00 г, 5,52 ммоль, 1,00 экв.) в этаноле (50 мл). Смесь перемешивали при 25°С в течение 4 ч в атмосфере водорода (50 фунтов/кв. дюйм). Смесь фильтровали и концентрировали фильтрат при пониженном давлении, в результате чего получали соединение 44-с.
МС: m/z (ионизация электрораспылением): 387,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δppm 6,71 (s, 2Н), 4,03 (q, J=7,19 Гц, 2H), 2,74 (br t, J=7,78 Гц, 2H), 2,50-2,46 (m, 2H), 2,12 (s, 6 H), 1,43 (s, 9H), 1,34 (s, 6 H), 1,27 (t, J=7,03 Гц, 6 H), 1,21 (br t, J=7,03 Гц, 3Н).
Этап 4: Соединение 44-d
В защитной атмосфере азота раствор соединения 44-с (200,00 мг, 548,73 мкмоль, 1,00 экв.) в тетрагидрофуране (5 мл) добавляли в раствор алюмогидрида лития (41,65 мг, 1,10 ммоль, 2,00 экв.) в тетрагидрофуране (5,00 мл) при 0°С. Смесь перемешивали при 0°С в течение 3 ч. В реакционную смесь добавляли воду (100 мл) и экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 44-d.
МС: m/z (ионизация электрораспылением): 345,0 [М+23].
Этап 5: Соединение 44-е
Оксалилхлорид (1,42 г, 11,16 ммоль, 976,92 мкл, 2,00 экв.) добавляли в раствор диметилсульфоксида (1,74 г, 22,32 ммоль, 1,74 мл, 4,00 экв.) в дихлорметане (15,00 мл) при -78°С. Смесь перемешивали при -78°С в течение 5 мин. Затем добавляли соединение 44-d (1,80 г, 5,58 ммоль, 1,00 экв.) и перемешивали смесь при -78°С в течение 40 мин. Затем добавляли триэтиламин (3,39 г, 33,48 ммоль, 4,64 мл, 6,00 экв.) при -78°С и перемешивали смесь при 0°С в течение 30 мин. Смесь нейтрализовали водой (50 мл), и экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (24,3%, этилацетат/петролейный эфир), в результате чего получали соединение 44-е.
МС: m/z (ионизация электрораспылением): 343,2 [М+23].
Этап 6: Соединение 44-f
Трет-бутоксид натрия (1,36 г, 14,18 ммоль, 1,20 экв.) добавляли в раствор соединения 25-b (6,00 г, 11,82 ммоль, 1,00 экв.) в тетрагидрофуране (50,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 30 мин. В реакционную смесь добавляли воду (50 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 44-f.
Этап 7: Соединение 44-g
Соединение 44-f (2,06 г, 4,84 ммоль, 1,00 экв.) добавляли в раствор соединения 44-е (1,55 г, 4,84 ммоль, 1,00 экв.) в тетрагидрофуране (10 мл). Смесь перемешивали при 50°С в течение 16 ч. В смесь добавлял смесь вода/этилацетат (1:1, 50 мл) и экстрагировали водную фазу этилацетатом (50 мл × 2). Объединенную органическую фазу концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (16,5%, этилацетат/петролейный эфир), в результате чего получали соединение 44-g.
Этап 8: Соединение 44-h
В защитной атмосфере азота трифторуксусную кислоту (1,00 мл) добавляли в раствор соединения 44-g (1,60 г, 3,41 ммоль, 1,00 экв.) и N-(метоксиметил)-1-фенил-N-(триметилсилилметил)метиламина (972,64 мг, 4,10 ммоль, 1,20 экв.) в дихлорметане (150,00 мл) при 0°С. Смесь перемешивали при 25°С в течение 16 ч. В смесь добавляли смесь вода/дихлорметан (1:1, 100 мл) и экстрагировали водную фазу дихлорметаном (100 мл × 2). Объединенную органическую фазу концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (27,3%, этилацетат/петролейный эфир), в результате чего получали соединение 44-h.
МС: m/z (ионизация электрораспылением): 602,3 [М+1].
Этап 9: Соединение 44-i
(4-хлорфенил) метилхлорформиат (952,13 мг, 4,98 ммоль, 694,99 мкл, 2,00 экв.) медленно добавляли в раствор соединения 44-h (1,50 г, 2,49 ммоль, 1,00 экв.) в дихлорметане (15,00 мл). Смесь перемешивали при 25°С в течение 20 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (25,6%, этилацетат/петролейный эфир), в результате чего получали соединение 44-i.
МС: m/z (ионизация электрораспылением): 610,2 [М+1].
Этап 10: соединения 44 и 45
Раствор соединения 44-i (220,00 мг, 330,20 мкмоль, 1,00 экв.) и трифторуксусной кислоты (1,54 г, 13,51 ммоль, 1,00 мл, 40,90 экв.) в дихлорметане (5,00 мл) перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (54,2%, этилацетат/петролейный эфир), и подвергали полученный продукт хиральному разделению, в результате чего получали соединение 44;
Соединение 45.
Соединение 44:
МС: m/z (ионизация электрораспылением): 632,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,86 (dd, J=5,8, 8,5 Гц, 2H), 7,31-7,27 (m, 4Н), 7,09-7,05 (m, 2Н), 6,76 (d, J=5,3 Гц, 2H), 3,96-3,91 (m, 1H), 3,77-3,59 (m, 3Н), 3,20-3,16 (m, 1H), 2,80-2,67 (m, 1H), 2,55-2,52 (m, 5Н), 2,19 (d, J=4,5 Гц, 6 Н), 1,93-1,85 (m, 1H), 1,74-1,63 (m, 1H), 1,51-1,47 (m, 6 Н).
Соединение 45:
МС: m/z (ионизация электрораспылением): 632,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δppm 7,86 (dd, J=5,6, 8,4 Гц, 2H), 7,33-7,26 (m, 4Н), 7,07-7,05 (m, 2Н), 6,77-6,75 (m, 2Н), 3,93 (dt, J=2,6, 10,6 Гц, 1H), 3,80-3,59 (m, 3Н), 3,39-3,20 (m, 1H), 2,84-2,64 (m, 1H), 2,55-2,51 (m, 5Н), 2,18 (d, J=3,5 Гц, 6 H), 1,95-1,85 (m, 1H), 1,70 (br dd, J=3,8, 8,8 Гц, 1H), 1,51-1,47 (m, 6 H).
Условия хирального разделения: хиральная колонка: Chiralpak IC-3 150×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 2,5 мл/мин; температура колонки: 40°С.
Время удерживания соединения 44: 5,592 мин (пик 1); Время удерживания соединения 45: 7,585 мин (пик 1)
Пример 46: Соединение 46
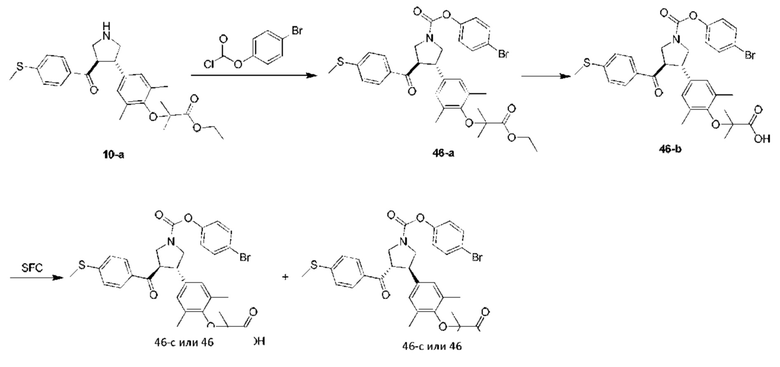
Этап 1: Соединение 46-а
В защитной атмосфере азота раствор соединения 10-а (150,00 мг, 329,23 мкмоль, 1,00 экв.), ди-изо-пропилэтиламина (85,10 мг, 658,46 мкмоль, 115,00 мкл, 2,00 экв.) и 4-бромфенилхлорформиата (93,02 мг, 395,08 мкмоль, 56,38 мкл, 1,20 экв.) в дихлорметане (10 мл) перемешивали при 20°С в течение 12 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1), в результате чего получали соединение 46-а.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,76-7,71 (m, 2 Н), 7,49 (d, J=8,8 Гц, 2 H), 7,21 (d, J=5,2 Гц, 2 H), 7,07 (d, J=8,8 Гц, 2 H), 6,86 (d, J=5,6 Гц, 2 H), 4,17-4,09 (m, 5 Н), 3,84-3,77 (m, 3 Н), 2,52 (s, 3 Н), 2,15 (s, 6 Н), 1,44-1,42 (m, 6 Н), 1,38-1,36 (m, 3 Н).
Этап 2: Соединение 46-b
Раствор соединения 46-а (140,00 мг, 213,87 мкмоль, 1,00 экв.) и гидроксида лития (89,74 мг, 2,14 ммоль, 10,00 экв.) в этаноле (9,00 мл) и воде (3,00 мл) перемешивали при 25°С в течение 12 ч. Смесь доводили при помощи 1 н. разбавленной HCl до рН=3, и обрабатывали водой и этилацетатом (1:1, 50 мл). Водную фазу экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл × 1), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1), в результате чего получали соединение 46-b.
1Н-ЯМР (400 МГц, MeOD-d4) δ ppm 7,78 (d, J=8,0 Гц, 2 H), 7,55 (d, J=8,0 Гц, 2 H), 7,23 (d, J=8,8 Гц, 2 H), 7,15-7,12 (m, 2 Н), 6,98 (d, J=4,0 Гц, 2 H), 4,17-4,14 (m, 1 Н), 4,00-3,97 (m, 2 Н), 3,92-3,90 (m, 1 Н), 3,66-3,64 (m, 2 Н), 2,52 (s, 3 Н), 2,17 (d, J=5,6 Гц, 6 H), 1,38 (s, 6 Н).
Этап 3: Соединение 46
Соединение 46-b (50 мг) подвергали хиральному разделению, в результате чего получали соединение 46.
МС: m/z (ионизация электрораспылением): 650,0 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,67 (t, J=8,5 Гц, 2H), 7,41-7,39 (m, 2Н), 7,13 (dd, J=4,3, 8,5 Гц, 2H), 6,99-6,97 (m, 2Н), 6,83 (d, J=6,3 Гц, 2H), 4,09-4,00 (m, 3Н), 3,70-3,60 (m, 3Н), 2,43 (s, 3Н), 2,12 (d, J=5,5 Гц, 6H), 1,39-1,38 (m, 6Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С.
Время удерживания соединения 46: 7,984 мин (пик 2).
Пример 47: Соединение 47
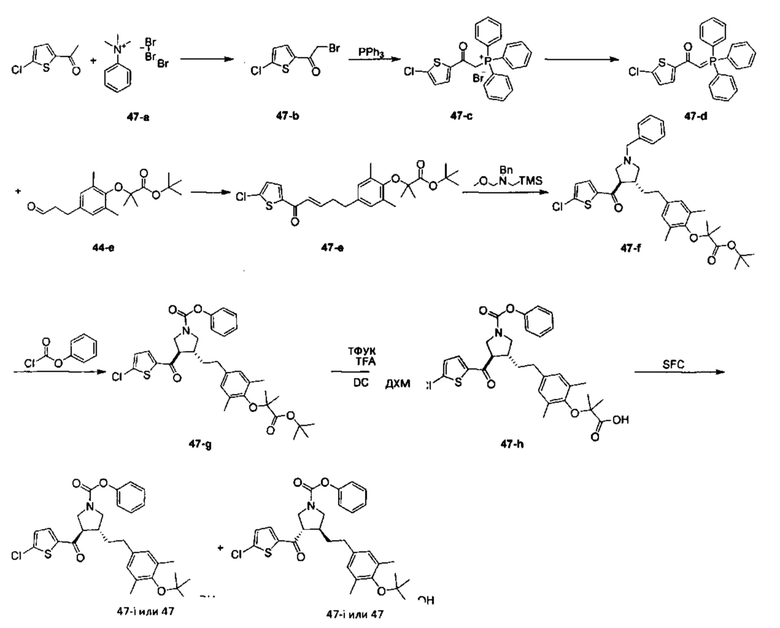
Этап 1: Соединение 47-b
Соединение 1-(5-хлоротиен-2-ил)-этанон (10,00 г, 62,26 ммоль, 1,00 экв.), 47-а (23,40 г, 62,26 ммоль, 1,00 экв.) и дихлорметан (200,00 мл) добавляли в высушенную круглодонную колбу и перемешивали полученный прозрачный раствор при 20°С в течение 16 ч. Смесь концентрировали при пониженном давлении, в результате чего получали остаток. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали 47-b.
МС: m/z (ионизация электрораспылением): 240,7 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,60 (d, J=4,0 Гц, 1H), 7,00 (d, J=4,0 Гц, 1H), 4,28 (s, 1H).
Этап 2: Соединение 47-c
При 20°C соединение 47-b (13,00 г, 54,28 ммоль, 1,00 экв.), трифенилфосфин (14,24 г, 54,28 ммоль, 1,00 экв.) и толуол (100,00 мл) добавляли в высушенную круглодонную колбу, полученную суспензию перемешивали при 20°С в течение 2 ч. Реакционную систему охлаждали до комнатной температуры, в результате чего получали желтый осадок, который фильтровали. Осадок на фильтре промывали этилацетатом (3×50 мл), в результате чего получали соединение 47-с.
1Н-ЯМР (CDCl3) δ ppm 8,91 (d, J=4,0 Гц, 1H), 7,92-7,87 (m, 6Н), 7,75-7,72 (m, 2Н), 7,66-7,60 (m, 6Н), 7,00 (d, J=4,0 Гц, 1H), 6,13 (d, J=12,8 Гц, 1H).
Этап 3: Соединение 47-d
При 20°С соединение 47-с (5,00 г, 10,27 ммоль, 1,00 экв.), трет-бутоксид калия (1,73 г, 15,41 ммоль, 1,50 экв.) и тетрагидрофуран (30,00 мл) добавляли в высушенную круглодонную колбу и перемешивали полученную суспензию при 20°С в течение 1 ч. Реакционную систему охлаждали до комнатной температуры, и разбавляли водой (50 мл) и этилацетат (20 мл). После разделения фаз органическую фазу собирали, а водную фазу экстрагировали этилацетатом (3×20 мл). Органические фазы объединяли, сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток с получением соединения 47-d.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,71-7,68 (m, 6Н), 7,66-7,63 (m, 3Н), 7,51-7,48 (m, 7Н), 7,26 (s, 1H), 6,82 (d, J=4,0 Гц, 1H).
Этап 4: Соединение 47-е
При 20°С соединение 44-е (2,00 г, 6,24 ммоль, 1,00 экв.), 47-d (2,63 г, 6,24 ммоль, 1,00 экв.) и тетрагидрофуран (20,00 мл) добавляли в высушенную круглодонную колбу, полученный прозрачный раствор перемешивали при 65°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали остаток. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 47-е.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,45 (d, J=4,0 Гц, 1H), 7,10 (td, J=6,8, 15,2 Гц, 1H), 6,98-6,94 (m, 1H), 6,78 (br s, 2H), 6,66 (d, J=15,3 Гц, 1H), 2,83 (s, 2H), 2,72 (s, 2H), 2,21-2,20 (m, 6H), 1,51-1,50 (m, 9H), 1,41-1,40 (m, 6H).
Этап 5: Соединение 47-f
При 20°С соединение 47-е (1,00 г, 2,16 ммоль, 1,00 экв.), трифторуксусную кислоту (12,31 мг, 108,00 мкмоль, 7,99 мкл, 0,05 экв.) добавляли в высушенную круглодонную колбу. Реакционную смесь нагревали до 80°С и медленно по каплям добавляли в реакционную систему N-бензил-1-метокси-N-((триметилсилил)метил)метиламин (1,54 г, 6,48 ммоль, 1,66 мл, 3,00 экв.), и перемешивали реакционную систему при 80°С в течение 30 мин. Реакционную смесь концентрировали при пониженном давлении и очищали остаток колоночной флэш-хроматографией (градиент элюирования: петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 47-f.
МС: m/z (ионизация электрораспылением): 596,3 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,38-7,33 (m, 2Н), 7,38-7,21 (m, 4Н), 6,78 (s, 2Н), 4,41-4,28 (m, 2Н), 4,08-3,94 (m, 1H), 3,74-3,69 (m, 2Н), 3,08 (dd, J=6,5, 11,0 Гц, 1H), 2,77-2,48 (m, 3Н), 2,20 (s, 6Н), 1,96-1,84 (m, 1H), 1,81-1,66 (m, 2Н), 1,53-1,51 (m, 9Н), 1,41 (s, 6 Н).
Этап 6: Соединение 47-g
При 20°С соединение 47-f (1,10 г, 1,84 ммоль, 1,00 экв.), фенилхлорформиат (1,44 г, 9,20 ммоль, 1,15 мл, 5,00 экв.) и хлороформ (10,00 мл) добавляли в высушенную круглодонную колбу, полученный прозрачный раствор перемешивали при 80°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 47-g.
МС: m/z (ионизация электрораспылением): 648,1 [М+23].
Этап 7: Соединение 47-h
Соединение 47-g (220,00 мг, 348,20 мкмоль, 1,00 экв.) и дихлорметан (6,00 мл), а затем трифторуксусную кислоту (397,02 мг, 3,48 ммоль, 257,81 мкл, 10,00 экв.) добавляли в 100 мл реакционную колбу. Смесь перемешивали при 25°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Остаток очищали путем тонкослойной хроматографии на силикагелевой пластине (дихлорметан : метанол = 10:1), в результате чего получали соединение 47-h.
Этап 8: Соединение 47
Соединение 47-h (17,00 мг, 29,82 мкмоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 47.
МС: m/z (ионизация электрораспылением): 570,3 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,55 (br d, J=6,0 Гц, 1H), 7,38-7,33 (m, 2H), 7,21 (br d, J=6,5 Гц, 1H), 7,15-7,11 (m, 2H), 7,02 (br d, J=4,5 Гц, 1H), 6,78 (br d, J=5,0 Гц, 2H), 3,97 (br d, J=7,5 Гц, 2H), 3,73 (br d, J=8,0 Гц, 1H), 3,39-3,34 (m, 1H), 3,16 (br d, J=10,0 Гц, 1H), 2,54 (br s, 2H), 2,21 (br d, J=4,5 Гц, 6 h), 1,86 (br s, 1H), 1,68 (br s, 2H), 1,51 (br d, J=19,6 Гц, 6H)
Условия хирального разделения: хиральная колонка: OJ (250 мм × 30 мм, 10 мкм); подвижная фаза: 50% метанола (0,05% диэтаноламина) в СО2; скорость потока: 80 мл/мин; температура колонки: 40°С.
Время удерживания соединения 47: 3,760 мин (пик 1).
Пример 48: Соединение 48
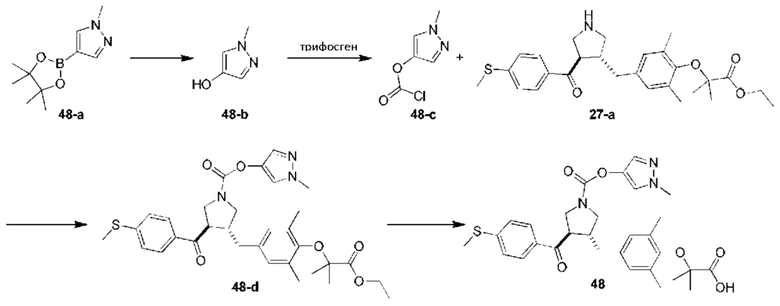
Этап 1: Соединение 48-b
При 20°С перекись водорода (599,71 мг, 5,29 ммоль, 508,23 мкл, чистота 30%, 1,10 экв.) и гидроксид натрия (211,64 мг, 5,29 ммоль, 1,10 экв.) добавляли в раствор соединения 48-а (1,00 г, 4,81 ммоль, 1,00 экв.) в тетрагидрофуране (30,00 мл). Смесь перемешивали при 20°С в течение 2 ч. Реакционную смесь гасили добавлением насыщенного раствора сульфита натрия (10 мл) и экстрагировали этилацетатом (5 мл × 3). Объединенную органическую фазу промывали водой (5 мл), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 48-b.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,13 (s, 1 Н) 7,03 (s, 1 Н) 3,78 (s, 3 Н) 1,25 (s, 8 Н).
Этап 2: Соединение 48-с
При 0°С соединение 48-b (400,00 мг, 4,08 ммоль, 1,00 экв.) и триэтиламин (412,60 мг, 4,08 ммоль, 565,20 мкл, 1,00 экв.) добавляли в раствор трифосгена (967,99 мг, 3,26 ммоль, 0,80 экв.) в тетрагидрофуране (10,00 мл). Смесь перемешивали при 20°С в течение 2 ч. После фильтрации фильтрат концентрировали при пониженном давлении, в результате чего получали соединение 48-с.
Этап 3: Соединение 48-d
При 20°С Соединение 27-а (1,00 г, 2,13 ммоль, 1,00 экв.) и DIEA (диизопропилэтиламин) (550,38 мг, 4,26 ммоль, 743,76 мкл, 2,00 экв.) добавляли в раствор 48-с (410,25 мг, 2,56 ммоль, 1,20 экв.) в дихлорметане (50,00 мл). Смесь перемешивали при 20°С в течение 1 ч. Смесь концентрировали при пониженном давлении и очищали остаток колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100/0-40/60), в результате чего получали соединение 48-d.
МС: m/z (ионизация электрораспылением): 594,3 [М+1].
Этап 4: Соединение 48
При 20°С гидроксид лития (201,69 мг, 8,42 ммоль, 10,00 экв.) добавляли в раствор соединения 48-d (500,00 мг, 842,13 мкмоль, 1,00 экв.) в этаноле (20,00 мл) и воде (5,00 мл). Смесь перемешивали при 20°С в течение 16 ч. Смесь подкисляли 1 н. разбавленной HCl (5 мл), а затем экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали при пониженном давлении и концентрировали при пониженном давлении. Остаток отделяли высокоэффективной жидкостной хроматографией, в результате чего получали соединение 48.
МС: m/z (ионизация электрораспылением): 566,4 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,61 (br t, J=7,92 Гц, 2 H) 7,41 (s, 1 H) 7,29 (s, 1 H) 7,10-7,17 (m, 2 H) 6,72 (br s, 2 H) 3,77 (s, 3 H) 3,67 (br d, J=11,54 Гц, 2 H) 3,28 (br s, 2 H) 2,84 (br d, J=14,56 Гц, 2 H) 2,59 (br s, 2 H) 2,45 (s, 3 H) 2,10 (br s, 6 H) 1,41 (br s, 6 H).
Примеры 49 и 50: соединения 49 и 50
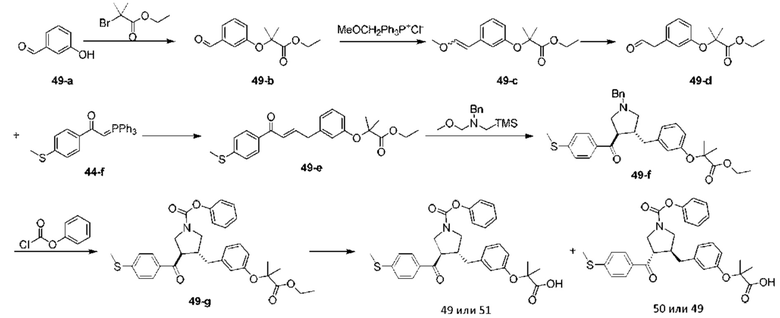
Этап 1: Соединение 49-b
Раствор этоксида натрия (9,19 г, 135,11 ммоль, 1,10 экв.) в этаноле (50,00 мл) добавляли в раствор соединения 49-а (15,00 г, 122,83 ммоль, 1,00 экв.) и этил-2-бром-изобутирата (31,15 г, 159,68 ммоль, 23,42 мл, 1,30 экв.) в этаноле (100,00 мл). В защитной атмосфере азота смесь перемешивали при 90°С в течение 8 ч. Смесь подкисляли разбавленной HCl (1 н.) до рН=6-7, а затем экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу промывали водой (100 мл × 3) и насыщенным солевым раствором (100 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали отсасыванием и концентрировали при пониженном давлении. Остаток выделяли хроматографией на колонке с силикагелем (петролейный эфир : этилацетат = 20/1), в результате чего получали соединение 49-b.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 9,95 (s, 1H), 7,52 (d, J=6,4 Гц, 1H), 7,43 (t, J=8,0 Гц, 1H), 7,34 (t, J=1,2 Гц, 1H), 7,15 (s, 1H), 4,29-4,24 (q, J=7,2 Гц, 2H), 1,64 (s, 6H), 1,27 (t, J=7,2 Гц, 3Н).
Этап 2: Соединение 49-с
В защитной атмосфере азота трет-бутоксид калия (8,55 г, 76,16 ммоль, 1,20 экв.) добавляли в раствор соединеия метоксиметилтрифенилфосфинхлорида (28,29 г, 82,54 ммоль, 1,30 экв.) в тетрагидрофуране (100,00 мл) при 0°С и перемешивали смесь при 0°С в течение 30 мин. Затем добавляли по каплям раствор соединения 49-b (15,00 г, 63,49 ммоль, 1,00 экв.) в тетрагидрофуране (50,00 мл). Смесь гасили добавлением воды (100 мл), а затем экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу промывали водой (100 мл) и насыщенным солевым раствором (100 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали отсасыванием и концентрировали при пониженном давлении. Остаток выделяли хроматографией на колонке с силикагелем (петролейный эфир : этилацетат = 20/1) с получением соединения 49-с.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,10-7,04 (m, 2Н), 6,93 (d, J=13,2 Гц, 1H), 6,67-6,56 (m, 1H), 6,04 (d, J=6,8 Гц, 1H), 5,68-5,07 (m, 1H), 4,19-4,13 (q, J=7,2 Гц, 2H), 3,70-3,60 (m, 3Н), 1,52 (s, 6 Н), 1,20-1,15 (m, 3Н)
Этап 3: Соединение 49-d
В защитной атмосфере азота оксалилхлорид (19,10 г, 150,50 ммоль, 13,17 мл, 2,00 экв.), а затем этанол (6,93 г, 150,50 ммоль, 8,77 мл, 2,00 экв.) и воду (2,71 г, 150,50 ммоль, 2,00 экв.) добавляли в раствор соединения 49-с (22,00 г, 75,25 ммоль, 1,00 экв.) в хлороформе (200,00 мл) при 0°С. Смесь перемешивали 0°С в течение 0,5 ч. Смесь доводили насыщенным раствором карбоната натрия до рН 7-8. Органическую фазу промывали водой (50 мл × 2) и насыщенным солевым раствором (50 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток выделяли хроматографией на колонке с силикагелем (петролейный эфир : этилацетат = 30/1-10/1), в результате чего получали соединение 49-d.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 9,64 (t, J=2,3 Гц, 1H), 7,16 (t, J=7,8 Гц, 1H), 6,77 (d, J=7,5 Гц, 1H), 6,70-6,66 (m, 2Н), 4,32-4,26 (m, 2Н), 3,55 (d, J=2,3 Гц, 2H), 1,53 (s, 6 Н), 1,31 (t, J=7,2 Гц, 3Н).
Этап 4: Соединение 49-е
В защитной атмосфере азота раствор соединения 49-d (22,00 г, 87,90 ммоль, 1,00 экв.) и 44-f (37,49 г, 87,90 ммоль, 1,00 экв.) в тетрагидрофуране (400,00 мл) перемешивали при 50°С в течение 12 ч. Смесь концентрировали при пониженном давлении. Остаток очищали при помощи колонки с силикагелем (хроматография, петролейный эфир : этилацетат = 30/1-10/1), в результате чего получали соединение 49-е.
МС: m/z (ионизация электрораспылением): 399,2 [М+1].
Этап 5: Соединение 49-f
В защитной атмосфере азота N-метоксиметил-1-фенил-N-(триметилдисиланил)метиламин (8,94 г, 37,64 ммоль, 1,50 экв.) добавляли по каплям в раствор соединения 49-е (10,00 г, 25,09 ммоль, 1,00 экв.) и трифторуксусной кислоты (143,05 мг, 1,25 ммоль, 92,89 мкл, 0,05 экв.) в диоксане (50,00 мл) при 90°С. Смесь перемешивали при 90°С в течение 0,5 ч. Смесь разбавляли водой (500 мл) и экстрагировали этилацетатом (300 мл × 3). Объединенную органическую фазу промывали водой (300 мл × 3) и насыщенным солевым раствором (300 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 20/1-5/1), в результате чего получали соединение 49-f.
МС: m/z (ионизация электрораспылением): 532,2 [М+1].
Этап 6: Соединение 49-g
В защитной атмосфере азота раствор соединения 49-f (6,00 г, 11,28 ммоль, 1,00 экв.) и фенилхлорформиата (3,53 г, 22,57 ммоль, 2,83 мл, 2,00 экв.) в хлороформе (40,00 мл) перемешивали при 70°С в течение 3 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 5/1), в результате чего получали соединение 49-g.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,66 (d, J=8,5 Гц, 1H), 7,61 (br d, J=8,5 Гц, 1H), 7,35 (t, J=7,6 Гц, 2H), 7,27-7,11 (m, 6H), 6,83 (br d, J=7,5 Гц, 1H), 6,78-6,66 (m, 2H), 4,26-4,17 (m, 2H), 3,95-3,57 (m, 4H), 3,52-3,35 (m, 1H), 2,98-2,65 (m, 3H), 2,52 (s, 3H), 1,59 (d, J=2,3 Гц, 6H), 1,26-1,21 (m, 3H).
Этап 7: соединения 49 и 50
При 25°С раствор моногидрата гидроксида лития (448,97 мг, 10,70 ммоль, 5,00 экв.) в воде (2,00 мл) добавляли в раствор соединения 49-g (1,20 г, 2,14 ммоль, 1,00 экв.) в этаноле (5,00 мл) и тетрагидрофуран (5,00 мл). Смесь перемешивали при 25°С в течение 8 ч. Смесь нейтрализовали разбавленной HCl (1 н.) до рН=5-6, а затем экстрагировали этилацетатом (20 мл × 3). Объединенную органическую фазу промывали водой (20 мл × 3) и насыщенным солевым раствором (20 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 3/1-1/1), а затем очищенный продукт подвергали хиральному разделению, в результате чего получали соединение 49;
Соединение 50.
Соединение 49: МС m/z (ионизация электрораспылением): 534,3 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,70-7,55 (m, 2Н), 7,27 (d, J=8,0 Гц, 2H), 7,20-7,04 (m, 6 Н), 6,79-6,63 (m, 3Н), 3,93-3,26 (m, 5Н), 2,88-2,49 (m, 3Н), 2,47-2,40 (m, 3Н), 1,56-1,45 (m, 6 h).
Соединение 50: МС: m/z (ионизация электрораспылением): 534,3 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,72-7,59 (m, 2Н), 7,30-7,07 (m, 9Н), 6,80-6,60 (m, 3Н), 3,91-3,73 (m, 3Н), 3,42-3,27 (m, 1H), 2,81-2,54 (m, 4Н), 2,47-2,44 (m, 3Н), 1,55-1,50 (m, 6 Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-50 мм × 4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% этанола (0,05% диэтаноламина) в СО2; скорость потока: 4 мл/мин; температура колонки: 40°С.
Время удерживания соединения 49: 1,904 мин (пик 1); Время удерживания соединения 50: 2,071 мин (пик 2).
Примеры 51 и 52: соединения 51 и 52
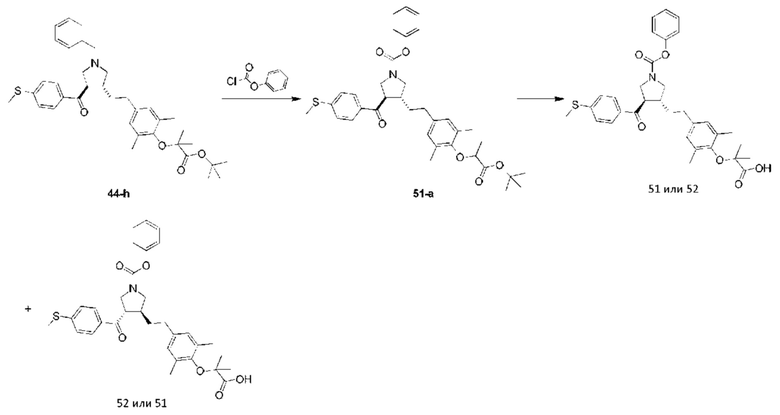
Этап 1: Соединение 51-а
Фенилхлорформиат (156,09 мг, 996,94 мкмоль, 124,87 мкл, 1,00 экв.) медленно добавляли в раствор соединения 44-h (600,00 мг, 996,94 мкмоль, 1,00 экв.) в хлороформе (10,00 мл). Смесь перемешивали при 70°С в течение 4 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (51,6%, этилацетат/петролейный эфир), в результате чего получали соединение 51-а.
МС: m/z (ионизация электрораспылением): 654,4 [М+23].
Этап 2: соединения 51 и 52
Раствор соединения 51-а (360,00 мг, 569,78 мкмоль, 1,00 экв.) и трифторуксусной кислоты (2,66 г, 23,30 ммоль, 1,73 мл, 40,90 экв.) в дихлорметане (5,00 мл) перемешивали при 25°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (54,2%, этилацетат/петролейный эфир), и подвергали полученный продукт хиральному разделению, в результате чего получали соединение 51;
Соединение 52.
Соединение 51: МС m/z (ионизация электрораспылением): 598,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,80 (dd, J=6,3, 8,3 Гц, 2H), 7,29-7,20 (m, 4Н), 7,05-7,03 (m, 3Н), 6,69 (d, J=5,3 Гц, 2H), 3,89-3,86 (m, 1H), 3,70-3,63 (m, 2Н), 3,51-3,31 (m, 1H), 3,12 (br dd, J=8,5, 10,8 Гц, 1H), 2,80-2,60 (m, 1H), 2,49-2,45 (m, 5H), 2,12 (d, J=4,8 Гц, 6H), 1,80-1,78 (m, 1H), 1,61-1,60 (m, 1H), 1,41 (br d, J=16,8 Гц, 6H).
Соединение 52: МС m/z (ионизация электрораспылением): 598,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,80 (dd, J=6,0, 8,3 Гц, 2H), 7,29-7,20 (m, 4Н), 7,07-7,05 (m, 3Н), 6,70 (d, J=5,3 Гц, 2H), 3,89-3,86 (m, 1H), 3,70-3,63 (m, 2Н), 3,52-3,24 (m, 1H), 3,12 (dd, J=8,5, 11,0 Гц, 1H), 2,77-2,62 (m, 1H), 2,49-2,45 (m, 5Н), 2,12 (d, J=4,8 Гц, 6 H), 1,80-1,78 (m, 1H), 1,61-1,52 (m, 1H), 1,42 (d, J=15,8 Гц, 6 H).
Условия хирального разделения: хиральная колонка: Lux Cellulose-2 150×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в CO2; скорость потока: 2,5 мл/мин; температура колонки: 40°С.
Время удерживания соединения 51: 8,092 мин (пик 1); Время удерживания соединения 52: 13,834 мин (пик 2).
Пример 53: Соединение 53
Этап 1: Соединение 53-b
В защитной атмосфере азота трифосген (2,17 г, 7,33 ммоль, 0,80 экв.) медленно добавляли в раствор соединения 53-а (1,00 г, 9,16 ммоль, 1,00 экв.) и триэтиламина (926,90 мг, 9,16 ммоль, 1,27 мл, 1,00 экв.) в тетрагидрофуране (20,00 мл) при 0°С. Смесь перемешивали при 20°С в течение 1 ч. Смесь фильтровали, а фильтрат концентрировали при пониженном давлении, в результате чего получали остаток. Соединение 27-а (1,00 г, 2,13 ммоль, 1,00 экв.) и N,N-ди-изо-пропилэтиламин (550,38 мг, 4,26 ммоль, 743,76 мкл, 2,00 экв.) добавляли в раствор остатка (365,47 мг, 2,13 ммоль, 1,00 экв.) в дихлорметане (50,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 1 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100/0-40/60), в результате чего получали соединение 53-b.
МС: m/z (ионизация электрораспылением): 605,3 [М+1].
Этап 2: Соединение 53
При 20°С гидроксид лития (118,81 мг, 4,96 ммоль, 10,00 экв.) добавляли в раствор соединения 53-b (300,00 мг, 496,06 мкмоль, 1,00 экв.) в этаноле (20,00 мл) и воде (5,00 мл). Смесь перемешивали при 20°С в течение 16 ч. Смесь подкисляли путем добавления 1 н. разбавленной HCl (5 мл), а затем экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток отделяли высокоэффективной жидкостной хроматографией, в результате чего получали соединение 53
МС: m/z (ионизация электрораспылением): 577,4 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 8,32 (t, J=2,76 Гц, 1 H), 7,59 (dd, J=8,54, 2,76 Гц, 2 H), 7,38-7,45 (m, 1 Н), 7,06-7,16 (m, 3 Н), 6,73 (s, 2 Н), 3,58-3,88 (m, 5 Н), 3,27-3,40 (m, 1 Н), 2,58-2,64 (m, 2 Н), 2,50 (s, 3 Н), 2,45 (s, 3 Н), 2,14 (br d, J=2,26 Гц, 6 H), 1,42 (br d, J=3,26 Гц, 6 H).
Пример 54: Соединение 54
Этап 1: Соединение 54-b
В защитной атмосфере азота трифосген (2,08 г, 7,01 ммоль, 0,80 экв.) медленно добавляли по каплям в раствор соединения 54-а (1,00 г, 8,76 ммоль, 1,08 мл, 1,00 экв.) и триэтиламина (886,42 мг, 8,76 ммоль, 1,21 мл, 1,00 экв.) в тетрагидрофуране (10,00 мл) при 0°С. Смесь перемешивали при 20°С в течение 2 ч. Смесь фильтровали, фильтрат концентрировали при пониженном давлении, в результате чего получали остаток. Остаток (752,49 мг, 4,26 ммоль, 2,00 экв.) и N,N-ди-изо-пропилэтиламин (550,38 мг, 4,26 ммоль, 743,76 мкл, 2,00 экв.) добавляли в раствор соединения 27-а (1,00 г, 2,13 ммоль, 1,00 экв.) в дихлорметане (50,00 мл) при 20°С. Смесь перемешивали при 20°С в течение 1 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100/0-40/60), в результате чего получали соединение 54-b.
МС: m/z (ионизация электрораспылением): 610,3 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,61-7,71 (m, 2 Н), 7,22 (br dd, J=5,28, 2,76 Гц, 2 H), 6,72-6,80 (m, 2 H), 4,50-4,64 (m, 1 H), 4,25-4,37 (m, 2 H), 3,43-3,79 (m, 4 H), 3,17-3,31 (m, 1 H), 2,84 (br s, 1 H), 2,58-2,67 (m, 2 H), 2,54 (s, 3 H), 2,16 (br d, J=4,02 Гц, 6 H), 1,69-2,03 (m, 4 H), 1,46 (s, 6 H), 1,37 (t, J=7,16 Гц, 3 H).
Этап 2: Соединение 54
При 20°C гидроксид лития (479,14 мг, 20,01 ммоль, 10,00 экв.) добавляли в раствор соединения 54-b (1,22 г, 2,00 ммоль, 1,00 экв.) в этаноле (20,00 мл) и воде (4,00 мл). Смесь перемешивали при 20°С в течение 2 ч. Смесь подкисляли путем добавления 1 н. разбавленной HCl (5 мл), а затем экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток отделяли высокоэффективной жидкостной хроматографией, в результате чего получали соединение 54.
МС: m/z (ионизация электрораспылением): 582,4 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,56-7,65 (m, 2Н), 7,09-7,17 (m, 2Н), 6,67 (br s, 2H), 3,34-3,78 (m, 5H), 3,08-3,23 (m, 1H), 2,77 (br s, 1H), 2,49-2,59 (m, 2H), 2,43 (br s, 3H), 2,06 (br s, 6 Н), 1,90 (br s, 1H), 1,77 (br d, J=10,04 Гц, 1 H), 1,63 (br d, J=13,06 Гц, 1 H), 1,36 (br s, 6 H), 1,06-1,26 (m, 4H), 0,89-1,02 (m, 2H), 0,80 (br d, J=6,54 Гц, 3Н).
Пример 55: Соединение 55
Этап 1: Соединение 55-а
Раствор соединения 44-g (900,00 мг, 1,92 ммоль, 1,00 экв.), саркозина (427,72 мг, 4,80 ммоль, 2,50 экв.) и параформальдегида (1,04 г, 11,52 ммоль, 6,00 экв.) в толуоле (10,00 мл) перемешивали при 110°С в течение 16 ч в защитной атмосфере азота. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (70,2%, этилацетат/петролейный эфир), в результате чего получали соединение 55-а.
МС: m/z (ионизация электрораспылением): 526,2 [М+1].
Этап 2: Соединение 55
Трифторуксусную кислоту (1,30 г, 11,41 ммоль, 844,16 мкл, 30,00 экв.) добавляли в раствор соединения 55-а (200,00 мг, 380,42 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл). Смесь перемешивали при 25°С в течение 4 ч. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (80,3%, этилацетат/петролейный эфир), а затем выделяли высокоэффективной жидкостной хроматографией, в результате чего получали соединение 55.
МС: m/z (ионизация электрораспылением): 470,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,95 (d, J=8,8 Гц, 2H), 7,29 (d, J=8,5 Гц, 2H), 6,67 (s, 2Н), 4,06-3,99 (m, 2Н), 3,44 (dd, J=3,5, 10,8 Гц, 1H), 2,92-2,83 (m, 3Н), 2,72 (s, 3Н), 2,51 (s, 3Н), 2,49-2,29 (m, 2Н), 2,15 (s, 6 h), 1,83-1,80 (m, 2H), 1,42 (d, J=8,3 Гц, 6H)
Пример 56: Соединение 56
Этап 1: Соединение 56-а
α-хлорэтилхлорформиат (3,56 г, 24,93 ммоль, 3,00 экв.) добавляли в раствор соединения 44-h (5,00 г, 8,31 ммоль, 1,00 экв.) в толуоле (50,00 мл). Реакционный раствор перемешивали при 80°С в течение 16 ч. Реакционный раствор концентрировали, и метанол (50,00 мл) добавляли и перемешивали при 80°С еще 1 ч. Реакционную смесь концентрировали при пониженном давлении, неочищенный продукт очищали колоночной флэш-хроматографией (76,3%, этилацетат/петролейный эфир) с получением соединения 56-а.
1Н-ЯМР (400 МГц, CDCl3) δppm 7,39 (br s, 2H), 7,22 (br d, J=6,5 Гц, 2H), 6,63 (br d, J=11,0 Гц, 2H), 4,38-4,19 (m, 2H), 3,87 (br s, 1H), 3,78 (s, 3H), 3,69 (br s, 1H), 3,34 (br s, 1H), 3,18-3,02 (m, 2H), 2,65 (br s, 1H), 2,49 (d, J=3,5 Гц, 3Н), 2,42-2,35 (m, 2H), 2,06 (d, J=7,5 Гц, 6H), 1,38 (d, J=4,8 Гц, 6H).
Этап 2: Соединение 56
Соединение 56-а (500,00 мг, 1,06 ммоль, 1,00 экв.), гидроксид лития (76,16 мг, 3,18 ммоль, 3,00 экв.), водй (2,00 мл) и метанол (6,00 мл) добавляли в высушенную реакционную колбу. Реакционный раствор перемешивали при 40°С в течение 16 ч. Реакционный раствор доводили разбавленной 1 н. HCl до рН=6 и обрабатывали водой и этилацетатом (1:1, 20 мл), водную фазу экстрагировали этилацетатом (2*20 мл). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали, концентрировали, в результате чего получали неочищенный продукт. Неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 56.
МС: m/z (ионизация электрораспылением): 456,1 [М+1].
1Н-ЯМР (400 МГц, MeOD-d4) δppm 8,52 (s, 1H), 7,89 (d, J=8,5 Гц, 2H), 7,35 (d, J=8,5 Гц, 2H), 6,74 (s, 2Н), 4,06-4,04 (m, 1H), 3,61-3,56 (m, 2Н), 3,40 (dd, J=7,3, 11,5 Гц, 1H), 3,14-3,12 (m, 1H), 2,56-2,45 (m, 6Н), 2,19 (s, 6Н), 1,87-1,83 (m, 1H), 1,75-1,70 (m, 1H), 1,37 (d, J=12,0 Гц, 6H).
Пример 57: Соединение 57
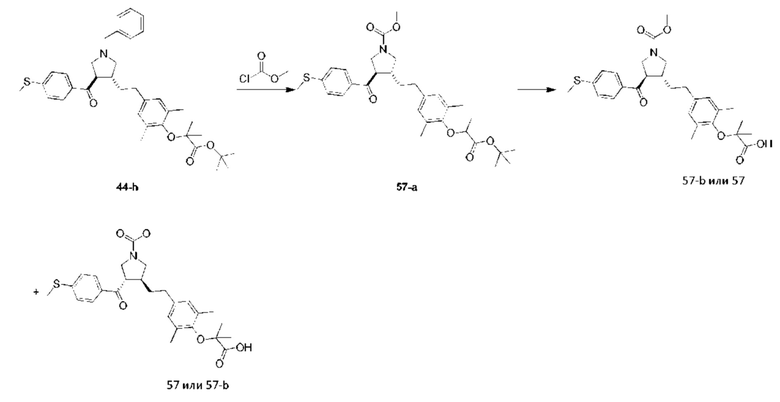
Этап 1: Соединение 57-а
Метилхлорформиат (785,10 мг, 8,31 ммоль, 643,52 мкл, 10,00 экв.) медленно добавляли в раствор соединения 44-h (500,00 мг, 830,79 мкмоль, 1,00 экв.) в хлороформе (10 мл). Смесь перемешивали при 70°С в течение 32 ч. Реакционный раствор концентрировали при пониженном давлении, неочищенный продукт очищали колоночной флэш-хроматографией (26,6%, этилацетат/петролейный эфир), в результате чего получали соединение 57-а.
МС: m/z (ионизация электрораспылением): 592,2 [М+23].
Этап 2: Соединение 57
Трифторуксусную кислоту (1,86 г, 16,32 ммоль, 1,21 мл, 30,00 экв.) добавляли в раствор соединения 57-а (310,00 мг, 544,10 мкмоль, 1,00 экв.) в дихлорметане (30,00 мл). Реакционный раствор перемешивали при 25°С в течение 4 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (73,3%, этилацетат/петролейный эфир), в результате чего получали продукт. Продукт подвергали хиральному разделению, в результате чего получали соединение 57.
МС: m/z (ионизация электрораспылением): 536,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δppm 7,77 (d, J=8,3 Гц, 2H), 7,21-7,19 (m, 2Н), 6,67 (s, 2Н), 3,79-3,55 (m, 6Н), 3,37-3,35 (m, 1H), 3,13-2,98 (m, 1H), 2,70-2,51 (m, 1H), 2,46-2,41 (m, 5Н), 2,11 (br d, J=7,0 Гц, 6H), 1,73 (br s, 1H), 1,57-1,53 (m, 1H), 1,45-1,40 (m, 6H).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 5 мкм); подвижная фаза: 40% метанола (0,1% NH3H2O) в СО2; скорость потока: 60 мл/мин; температура колонки: 40°С.
Время удерживания соединения 57: 2,964 мин (пик 1).
Пример 58: Соединение 58
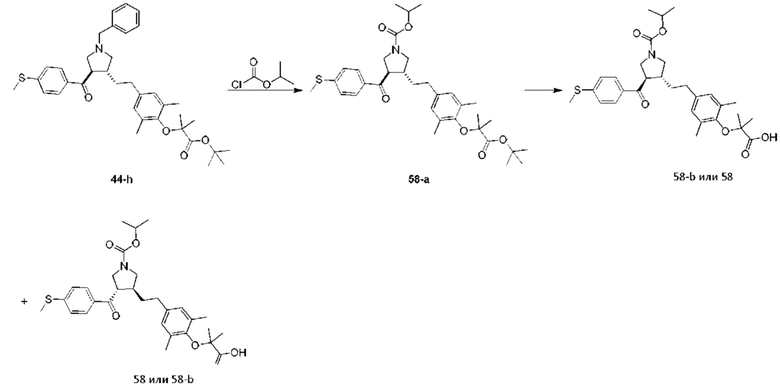
Этап 1: Соединение 58-а
Изопропилхлорформиат (1,02 г, 8,31 ммоль, 1,16 мл, 10,00 экв.) медленно добавляли в раствор соединения 44-h (500,00 мг, 830,79 мкмоль, 1,00 экв.) в хлороформе (10,00 мл). Реакционный раствор перемешивали при 70°С в течение 32 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (47,6%, этилацетат/петролейный эфир), в результате чего получали соединение 58-а.
МС: m/z (ионизация электрораспылением): 620,3 [М+23].
Этап 2: Соединение 58
Трифторуксусную кислоту (801,08 мг, 7,03 ммоль, 520,18 мкл, 30,00 экв.) добавляли в раствор соединения 58-а (140,00 мг, 234,19 мкмоль, 1,00 экв.) в дихлорметане (30,00 мл). Реакционный раствор перемешивали при 25°С в течение 4 ч. Реакционный раствор концентрировали при пониженном давлении, неочищенный продукт очищали колоночной флэш-хроматографией (73,3%, этилацетат/петролейный эфир), в результате чего получали продукт. Продукт подвергали хиральному разделению, в результате чего получали соединение 58.
МС: m/z (ионизация электрораспылением): 564,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,78 (br d, J=8,3 Гц, 2H), 7,23-7,21 (m, 2H), 6,68 (s, 2H), 4,85-4,82 (m, 1H), 3,82-3,59 (m, 2H), 3,44-3,29 (m, 2H), 3,11-2,89 (m, 1H), 2,76-2,54 (m, 1H), 2,47-2,40 (m, 5H), 2,14-2,11 (m, 6H), 1,74 (br s, 1H), 1,56-1,41 (m, 7H), 1,20-1,12 (m, 6 H).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 5 мкм); подвижная фаза: 40% метанола (0,1% NH3H2O) в СО2; скорость потока: 60 мл/мин; температура колонки: 40°С.
Время удерживания соединения 58: 6,217 мин (пик 1).
Примеры 59 и 60: соединения 59 и 60
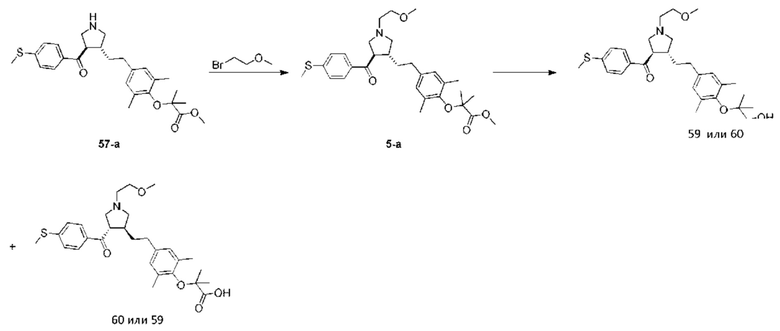
Этап 1: Соединение 59-а
1-бром-2-метоксиэтан (441,99 мг, 3,18 ммоль, 298,64 мкл, 3,00 экв.) и триэтиламин (321,78 мг, 3,18 ммоль, 440,79 мкл, 3,00 экв.) добавляли в раствор соединения 57-а (500,00 мг, 1,06 ммоль, 1,00 экв.) в ацетонитриле (10,00 мл). Реакционный раствор перемешивали при 25°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении, неочищенный продукт очищали колоночной флэш-хроматографией (80,3%, этилацетат/петролейный эфир), в результате чего получали соединение 59-а.
МС: m/z (ионизация электрораспылением): 528,4 [М+1].
Этап 2: соединения 59 и 60
Гидроксид лития (21,10 мг, 881,16 мкмоль, 3,00 экв.) и воду (2,00 мл) добавляли в раствор соединения 59-а (155,00 мг, 293,72 мкмоль, 1,00 экв.) в метаноле (6,00 мл). Реакционный раствор перемешивали при 40°С в течение 16 ч. Реакционный раствор доводили разбавленной 1 н. HCl до рН=6, и обрабатывали водой и этилацетатом (1:1, 15 мл). После разделения фаз водную фазу экстрагировали этилацетатом (2×15 мл). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали, концентрировали, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (82,3%, этилацетат/петролейный эфир) с получением продукта. Продукт подвергали хиральному разделению, в результате чего получали соединение 59 и Соединение 60.
Соединение 59:
1Н-ЯМР (400 МГц, CDCl3) δppm 7,99 (br d, J=8,3 Гц, 2H), 7,29 (br d, J=8,5 Гц, 2H), 6,67 (s, 2H), 3,95 (br s, 2H), 3,62 (br s, 1H), 3,38-3,29 (m, 5H), 3,07 (br s, 1H), 2,98 (br t, J=9,4 Гц, 2H), 2,86 (br s, 1H), 2,75 (s, 1H), 2,51 (s, 3H), 2,39 (br dd, J=5,1, 11,9 Гц, 1H), 2,35-2,26 (m, 1H), 2,17 (s, 6H), 1,84-1,70 (m, 2H), 1,47-1,38 (m, 6H).
Соединение 60:
1Н-ЯМР (400 МГц, CDCl3) δppm 7,99 (d, J=8,5 Гц, 2H), 7,29 (d, J=8,5 Гц, 2H), 6,67 (s, 2H), 3,97 (br s, 2H), 3,62 (br d, J=4,5 Гц, 1H), 3,40-3,28 (m, 5H), 3,15-3,07 (m, 1H), 2,98 (br t, J=10,2 Гц, 2H), 2,87 (br s, 1H), 2,79-2,69 (m, 1H), 2,51 (s, 3H), 2,45-2,35 (m, 1H), 2,34-2,23 (m, 1H), 2,17 (s, 6H), 1,89-1,68 (m, 2H), 1,48-1,37 (m, 6H).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 5 мкм); подвижная фаза: 40% EtOH (0,1% NH3H2O) в СО2; скорость потока: 60 мл/мин; температура колонки: 40°С.
Время удерживания соединения 59: 5,564 мин (пик 1); Время удерживания соединения 60: 5,981 мин (пик 2).
Пример 61: Соединение 61
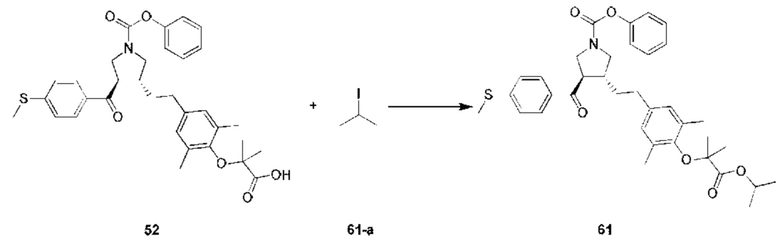
Этап 1: Соединение 61
При 20°С соединение 61-а (59,05 мг, 347,40 мкмоль, 34,74 мкл, 2,00 экв.) и безводный карбонат калия (72,02 мг, 521,10 мкмоль, 3,00 экв.) добавляли в раствор соединения 52 (100,00 мг, 173,70 мкмоль, 1,00 экв.) в N,N-диметилформамиде (10,00 мл). Смесь перемешивали при 20°С в течение 16 ч. Смесь разбавляли водой (10 мл), а затем экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт отделяли высокоэффективной жидкостной хроматографией, в результате чего получали соединение 61.
МС: m/z (ионизация электрораспылением): 618,2 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δppm 7,79 (dd, J=3,26, 8,53 Гц, 2H), 7,32-7,24 (m, 2H), 7,23-7,18 (m, 2H), 7,14-7,09 (m, 1H), 7,06 (dd, J=4,89, 7,40 Гц, 2H), 6,65 (d, J=6,27 Гц, 2H), 5,04 (spt, J=6,27 Гц, 1H), 3,96-3,47 (m, 4H), 3,33-3,19 (m, 1H), 2,79-2,57 (m, 1H), 2,53-2,34 (m, 5H), 2,08 (s, 6H), 1,85-1,71 (m, 1H), 1,66-1,60 (m, 1H), 1,34 (d, J=3,26 Гц, 6H), 1,24 (d, J=6,27 Гц, 6 H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 50*4,6 мм внутр. диам., 3 мкм; подвижная фаза: этанол (0,05% диэтаноламина); скорость потока: 4 мл/мин; температура колонки: 40°С.
Время удерживания соединения 61: 1,790 мин (пик 1).
Пример 62: Соединение 62
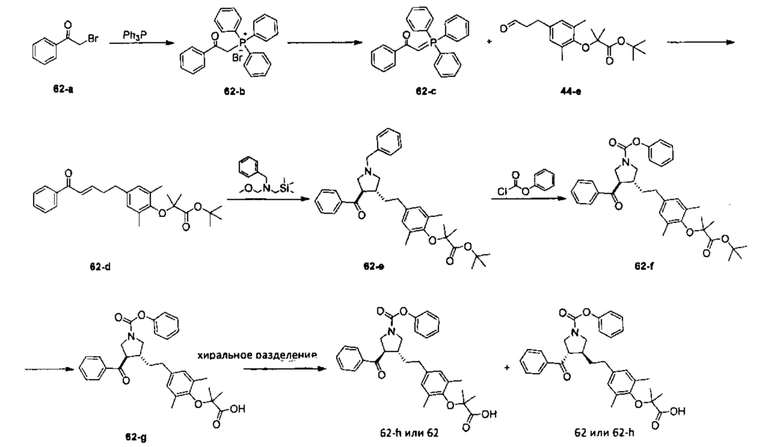
Этап 1: Соединение 62-b
При 25°С трифенилфосфин (37,37 г, 142,48 ммоль, 1,00 экв.) добавляли в раствор соединения 62-а (28,36 г, 142,48 ммоль, 1,00 экв.) в толуоле (300,00 мл). Смесь перемешивали при 25°С в течение 4 ч. Реакционный раствор фильтровали, и промывали осадок на фильтре дихлорметаном (200 мл) и сушили при пониженном давлении, в результате чего получали соединение 62-b.
1Н-ЯМР (400 МГц, CDCl3) δppm 8,09 (d, J=7,6 Гц, 2H), 7,88-7,86 (m, 9Н), 7,83-7,82 (m, 7Н), 7,78-7,76 (m, 2Н), 6,22 (d, J=13,2 Гц, 2H).
Этап 2: Соединение 62-с
При 20°С трет-бутоксид калия (3,65 г, 32,52 ммоль, 1,50 экв.) добавляли в раствор соединения 62-b (10,00 г, 21,68 ммоль, 1,00 экв.) в тетрагидрофуране (100 мл). Смесь перемешивали при 20°С в течение 0,5 ч. Реакционный раствор фильтровали, а фильтрат экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу промывали водой (100 мл), сушили при помощи безводного сульфата натрия, фильтровали, и концентрировали при пониженном давлении, в результате чего получали соединение 62-с.
МС: m/z (ионизация электрораспылением): 381,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 8,01 (d, J=7,2 Гц, 2H), 7,83 (t, J=5,2 Гц, 4H), 7,51-7,49 (m, 2Н), 7,48-7,38 (m, 13Н).
Этап 3: Соединение 62-d
При 20°С соединение 44-е (850 мг, 2,65 ммоль, 1,00 экв.) медленно добавляли в раствор соединения 62-с (1,01 г, 2,65 ммоль, 1,00 экв.) в тетрагидрофуране (10,00 мл). Реакционный раствор перемешивали при 50°С в течение 5 ч. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир : этилацетат = 100:0-70:30), в результате чего получали соединение 62-d
МС: m/z (ионизация электрораспылением): 445,2 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 9,80 (t, J=1,4 Гц, 2H), 7,90-7,83 (m, 1H), 7,61-7,41 (m, 2Н), 7,09-6,77 (m, 4Н), 6,77-6,76 (m, 2Н), 2,83-2,81 (m, 2Н), 2,72 (s, 2Н), 2,20 (br s, 6H), 1,51-1,50 (m, 9H), 1,42-1,41 (m, 6H)
Этап 4: Соединение 62-е
При 0°С N-(метоксиметил)-1-фенил-N-(триметилсилилметил) метиламин (264,06 мг, 1,11 ммоль, 1,00 экв.) и трифторуксусную кислоту (12,68 мг, 111,23 мкмоль, 0,10 экв.) добавляли в раствор соединения 62-d (470,00 мг, 1,11 ммоль, 1,00 экв.) в дихлорметане (20,00 мл). Реакционный раствор перемешивали при 25°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-40:60), в результате чего получали соединение 62-е.
МС: m/z (ионизация электрораспылением): 556,3 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δppm 7,95 (d, J=7,2 Гц, 2H), 7,49 (d, J=7,6 Гц, 2H), 7,45-7,31 (m, 6Н), 6,66 (s, 2Н), 4,41-4,33 (m, 1H), 3,74-3,72 (m, 1H), 2,81-2,70 (m, 4Н), 2,48-2,38 (m, 4Н), 2,14 (s, 6Н), 2,13-2,10 (m, 2Н), 1,51 (s, 9Н), 1,38 (s, 6Н).
Этап 5: Соединение 62-f
Метилфенилхлорформиат (394,42 мг, 2,52 ммоль, 5,00 экв.) медленно добавляли в раствор соединения 62-е (280,00 мг, 503,82 мкмоль, 1,00 экв.) в хлороформе (30,00 мл). Реакционный раствор перемешивали при 70°С в течение 4 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-30:70), в результате чего получали соединение 62-f.
МС: m/z (ионизация электрораспылением): 608,4 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δppm 7,91-7,89 (m, 2Н), 7,44-7,42 (m, 2Н), 7,30-7,27 (m, 3Н), 7,09-7,06 (m, 3Н), 6,66 (d, J=7,2 Гц, 2H), 3,92-3,80 (m, 2Н), 3,35-3,32 (m, 2Н), 2,95-2,83 (m, 2Н), 2,50-2,46 (m, 2Н), 2,16 (s, 6Н), 1,68-1,63 (m, 2Н), 1,44 (s, 9Н), 1,36 (s, 6Н).
Этап 6: Соединение 62-g
При 20°С трифторуксусную кислоту (778,65 мг, 6,83 ммоль, 505,62 мкл, 40,00 экв.) добавляли в раствор соединения 62-f (100,00 мг, 170,73 мкмоль, 1,00 экв.) в дихлорметане (20,00 мл). Смесь перемешивали при 20°С в течение 1 ч. Реакционный раствор концентрировали, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 62-g.
МС: m/z (ионизация электрораспылением): 530,4 [М+1].
Этап 7: Соединение 62
Соединение 62-g (20,00 мг) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 62.
МС: m/z (ионизация электрораспылением): 530,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δppm 7,96-7,76 (m, 1H), 7,57-7,15 (m, 5Н), 7,03 (br d, J=11,54 Гц, 1H), 6,24 (s, 1H), 5,29-5,16 (m, 6H), 3,42 (s, 1H), 2,13-1,48 (m, 10Н), 1,33 (br t, J=7,15 Гц, 1H), 1,21-1,17 (m, 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 50*4,6 мм внутр. диам., 3 мкм; подвижная фаза: этанол (0,05% диэтаноламина); скорость потока: 4 мл/мин; температура колонки: 40°С.
Время удерживания соединения 62: 1,651 мин (пик 1).
Пример 63: Соединение 63
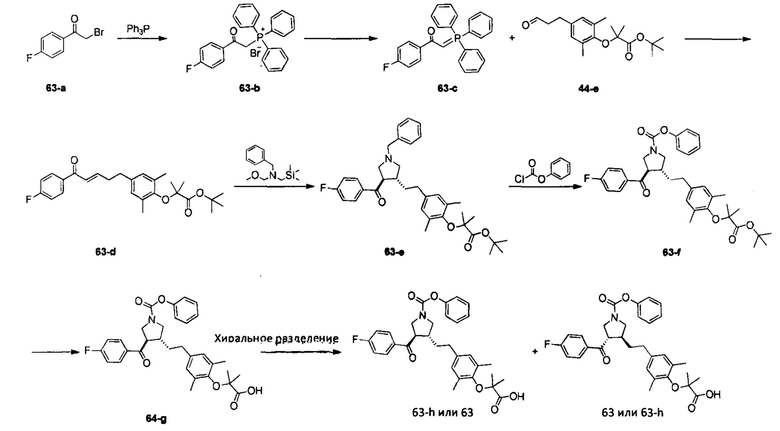
Этап 1: Соединение 63-b
При 25°С трифенилфосфин (60,43 г, 230,38 ммоль, 1,00 экв.) добавляли в раствор соединения 63-а (50,00 г, 230,38 ммоль, 1,00 экв.) в толуоле (300,00 мл). Смесь перемешивали при 25°С в течение 4 ч. Реакционный раствор фильтровали, остаток на фильтре промывали дихлорметаном (300 мл), а затем сушили при пониженном давлении, в результате чего получали соединение 63-b.
МС: m/z (ионизация электрораспылением); 399,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δppm 8,51 (dd, J=5,3, 8,8 Гц, 2H), 7,96 (dd, J=7,3, 13,3 Гц, 6 Н), 7,82-7,76 (m, 3H), 7,72-7,64 (m, 6H), 7,20 (t, J=8,5 Гц, 2H), 6,38 (d, J=12,0 Гц, 2H).
Этап 2: Соединение 63-с
При 20°С трет-бутоксид калия (3,51 г, 31,29 ммоль, 1,50 экв.) добавляли в раствор соединения 63-b (10,00 г, 20,86 ммоль, 1,00 экв.) в тетрагидрофуране (100,00 мл). Смесь перемешивали при 20°С в течение 0,5 ч. Реакционный раствор фильтровали, фильтрат экстрагировали этилацетатом (100 мл × 3). Органическую фазу промывали водой (100 мл), сушили при помощи безводного сульфата натрия, фильтровали, и концентрировали при пониженном давлении, в результате чего получали соединение 63-с.
МС: m/z (ионизация электрораспылением): 399,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 8,00-7,94 (m, 2H), 7,77-7,68 (m, 6H), 7,59-7,44 (m, 10H), 7,06-6,98 (m, 2H).
Этап 3: Соединение 63-d
При 50°С соединение 44-е (1,00 г, 3,26 ммоль, 1,00 экв.) медленно добавляли в раствор соединения 63-с (1,30 г, 3,26 ммоль, 1,00 экв.) в тетрагидрофуране (20,00 мл). В защитной атмосфере азота реакционный раствор перемешивали при 50°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир : этилацетат = 70:1), в результате чего получали соединение 63-d.
МС: m/z (ионизация электрораспылением): 463,2 [М+23].
1H-ЯМР (400 МГц, CDCl3) δppm 7,85-7,80 (m, 2H), 7,07-7,02 (m, 2H), 6,73-6,69 (m, 4H), 2,78-2,73 (m, 2H), 2,55-2,48 (m, 2H), 2,13 (s, 6 H), 1,43 (s, 9H), 1,34 (s, 6 H)
Этап 4: Соединение 63-е
При 0°С N-(метоксиметил)-1-фенил-N-(триметилсилилметил) метиламин (538,93 мг, 2,27 ммоль, 1,00 экв.) и трифторуксусная кислоты (258,83 мг, 2,27 ммоль, 168,07 мкл, 1,00 экв.) добавляли в раствор соединения 63-d (999,00 мг, 2,27 ммоль, 1,00 экв.) в дихлорметане (20,00 мл). Реакционный раствор перемешивали при 25°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-70:30), в результате чего получали соединение 63-е
МС: m/z (ионизация электрораспылением): 574,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 8,04-7,98 (m, 2H), 7,45-7,42 (m, 5H), 7,21-7,16 (m, 1H), 7,18 (t, J=8,5 Гц, 1H), 6,63 (s, 2H), 4,37-4,30 (m, 1H), 4,28-4,22 (m, 1H), 3,92 (br s, 1H), 3,50 (br s, 1H), 3,15 (br s, 2H), 2,51-2,30 (m, 4H), 2,14 (s, 5H), 1,87 (br d, J=7,0 Гц, 1H), 1,92-1,85 (m, 1H), 1,52-1,50 (m, 9H), 1,38 (s, 6H)
Этап 5: Соединение 63-f
При 20°С метилфенилхлорформиат (559,43 мг, 3,57 ммоль, 447,55 мкл, 5,00 экв.) медленно добавляли в раствор соединения 63-е (410,00 мг, 714,61 мкмоль, 1,00 экв.) в хлороформе (30,00 мл). Реакционный раствор перемешивали при 70°С в течение 4 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-30:70), в результате чего получали соединение 63-f.
МС: m/z (ионизация электрораспылением): 548,3 [М-56].
1H-ЯМР (400 МГц, ДМСО-d6) δppm 8,15 (dd, J=5,77, 7,78 Гц, 2Н), 7,44-7,37 (m, 5H), 7,16 (br d, J=8,03 Гц, 2Н), 6,82 (s, 2Н), 4,14 (br dd, J=7,78, 15,81 Гц, 2Н), 3,84-3,76 (m, 2Н), 3,68-3,57 (m, 4H), 2,12 (s, 6 H), 1,87-1,68 (m, 2Н), 1,45 (d, J=1,51 Гц, 9H), 1,33 (d, J=5,02 Гц, 6Н).
Этап 6: Соединение 63-g
При 25°С трифторуксусную кислоту (1,51 г, 13,25 ммоль, 980,52 мкл, 40,00 экв.) добавляли в раствор соединения 63-f (200,00 мг, 331,28 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл). Реакционный раствор перемешивали при 25°С в течение 4 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-40:60), в результате чего получали соединение 63-g.
МС: m/z (ионизация электрораспылением): 548,3 [М+1].
Этап 7: Соединение 63
Соединение 63-g (85 мг) подвергали хиральному разделению, в результате чего получали соединение 63.
МС: m/z (ионизация электрораспылением): 548,0 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 8,01 (td, J=6,0, 8,7 Гц, 2Н), 7,36 (q, J=8,0 Гц, 2Н), 7,24-7,10 (m, 5H), 6,78 (brd, J=5,5 Гц, 2Н), 4,02-3,93 (m, 1H), 3,57 (dd, J=8,0, 11,0 Гц, 1Н), 3,39 (dd, J=8,0, 10,5 Гц, 1H), 3,23-3,18 (m, 1H), 2,83 (br dd, J=7,8, 13,3 Гц, 1H), 2,73 (td, J=4,4, 8,3 Гц, 1H), 2,55 (br t, J=7,8 Гц, 2Н), 2,20 (br d, J=3,5 Гц, 6Н), 1,92-1,81 (m, 1H), 1,75-1,64 (m, 1H), 1,55-1,41 (m, 6H).
Условия хирального разделения: хиральная колонка: Lux Cellulose-2 150×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% метанола (0,05% диэтаноламина) в CO2; скорость потока: 2,5 мл/мин; температура колонки: 40°С.
Время удерживания соединения 63: 3,626 мин (пик 1).
Пример 64: Соединение 64
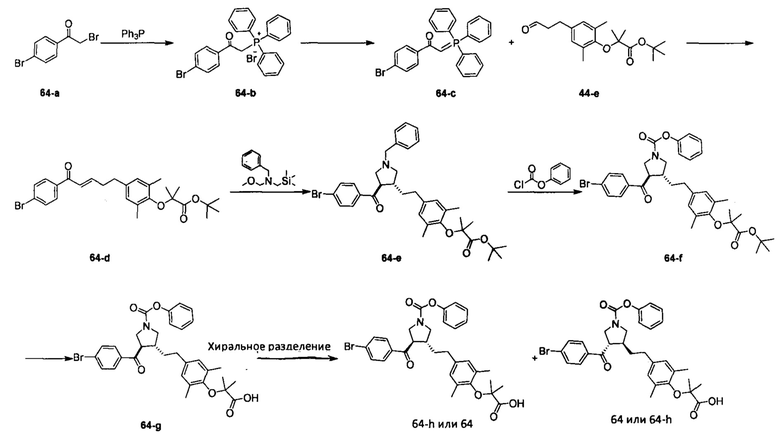
Этап 1
Соединение 64-b
При 25°С трифенилфосфин (49,54 г, 188,88 ммоль, 1,05 экв.) добавляли в раствор соединения 64-а (50,00 г, 179,89 ммоль, 1,00 экв.) в толуоле (500,00 мл). Реакционный раствор перемешивали при 25°С в течение 12 ч. Реакционный раствор фильтровали, остаток на фильтре промывали дихлорметаном (200 мл), а затем сушили при пониженном давлении, в результате чего получали соединение 64-b.
МС: m/z (ионизация электрораспылением): 461 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δppm 8,01 (d, J=8,5 Гц, 2H), 7,88-7,84 (m, 11H), 7,82-7,76 (m, 6H), 6,23 (d, J=13,1 Гц, 2H).
Этап 2: Соединение 64-с
При 20°С трет-бутоксид калия (3,12 г, 27,77 ммоль, 1,50 экв.) добавляли в раствор соединения 64-b (10,00 г, 18,51 ммоль, 1,00 экв.) в тетрагидрофуране (100,00 мл). Реакционный раствор перемешивали при 20°С в течение 0,5 ч. Реакционный раствор экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (200 мл), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 64-с.
1Н-ЯМР (400 МГц, CDCl3) δppm 7,90-7,77 (m, 4H), 7,73-7,68 (m, 4H), 7,59-7,54 (m, 4H), 7,51-7,45 (m, 8H).
Этап 3: Соединение 64-d
При 50°С соединение 44-е (1,00 г, 3,12 ммоль, 1,00 экв.) медленно добавляли в раствор соединения 64-с (1,43 г, 3,12 ммоль, 1,00 экв.) в тетрагидрофуране (20,00 мл). Реакционный раствор перемешивали при 50°С в течение 24 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-90:10), в результате чего получали соединение 64-d.
1H-ЯМР (400 МГц, CDCl3) δppm 7,79-7,72 (m, 2H), 7,61 (d, J=8,5 Гц, 2Н), 6,84-6,81 (m, 2H), 6,80-6,79 (m, 2H), 2,87-2,85 (m, 2H), 2,74-2,72 (m, 2H), 2,22 (s, 6H), 1,53 (s, 9H), 1,44 (s, 6H).
Этап 4: Соединение 64-е
При 0°С N-(метоксиметил)-1-фенил-N-(триметилсилилметил) метиламин (467,22 мг, 1,97 ммоль, 1,20 экв.) и трифторуксусную кислоту (187,00 мг, 1,64 ммоль, 121,43 мкл, 1,00 экв.) добавляли в раствор соединения 64-d (824,00 мг, 1,64 ммоль, 1,00 экв.) в дихлорметане (20,00 мл). Реакционный раствор перемешивали при 25°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-20:80), в результате чего получали соединение 64-е.
1H-ЯМР (400 МГц, CDCl3) δppm 7,81 (br d, J=8,3 Гц, 2H), 7,65 (d, J=8,5 Гц, 2H), 7,45-7,40 (m, 5H), 6,62 (s, 2H), 4,35-4,23 (m, 2H), 3,16 (br s, 2H), 2,66 (br s, 2H), 2,47-2,35 (m, 4H), 2,13 (s, 6H), 1,94-1,83 (m, 2H), 1,50 (s, 9H), 1,37 (d, J=3,0 Гц, 6 Н).
Этап 5: Соединение 64-f
При 25°С фенилхлорформиат (356,49 мг, 2,28 ммоль, 285,19 мкл, 5,00 экв.) добавляли в раствор соединения 64-е (289,00 мг, 455,38 мкмоль, 1,00 экв.) в хлороформе (20,00 мл). Реакционный раствор нагревали до 70°С и перемешивали в течение 48 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-40:60), в результате чего получали соединение 64-f.
1H-ЯМР (400 МГц, CDCl3) δppm 7,78-7,72 (m, 2H), 7,57 (dd, J=3,0, 8,5 Гц, 2H), 7,30 (br d, J=2,0 Гц, 2H), 7,11-7,03 (m, 3H), 6,66 (d, J=6,5 Гц, 2H), 3,85-3,55 (m, 4H), 3,40-3,25 (m, 1H), 2,76-2,58 (m, 1H), 2,56-2,36 (m, 2H), 2,12 (s, 6H), 1,79-1,62 (m, 2H), 1,44 (s, 9H), 1,36-1,33 (m, 6H).
Этап 6: Соединение 64-g
При 25°С трифторуксусную кислоту (686,22 мг, 6,02 ммоль, 445,60 мкл, 40,00 экв.) добавляли в раствор соединения 64-f (100,00 мг, 150,46 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл). Реакционный раствор перемешивали при 25°С в течение 1 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-40:60), в результате чего получали соединение 64-g.
МС: m/z (ионизация электрораспылением): 610,1 [М+1].
Этап 7: Соединение 64
Соединение 64-g (85,00 мг, 155,22 мкмоль, 1,00 экв.) подвергали хиральному разделению, в результате чего получали соединение 64.
МС: m/z (ионизация электрораспылением): 610,0 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 7,86-7,78 (m, 2H), 7,64 (dd, J=5,3, 8,3 Гц, 2H), 7,35 (q, J=8,0 Гц, 2H), 7,23-7,17 (m, 1H), 7,12 (brt, J=8,8 Гц, 2H), 6,77 (d, J=5,5 Гц, 2H), 4,02-3,88 (m, 2H), 3,81-3,67 (m, 2H), 3,43-3,34 (m, 1H), 3,23-3,15 (m, 1H), 2,54 (brt, J=7,8 Гц, 2H), 2,19 (d, J=5,0 Гц, 6 Н), 1,86 (br d, J=7,5 Гц, 2H), 1,50 (d, J=17,6 Гц, 6Н).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% NH3H2O, изопропиловый спирт; скорость потока: 60 мл/мин; температура колонки: 40°С.
Время удерживания соединения 64: 1,915 мин (пик 1).
Примеры 65 и 66: соединения 65 и 66
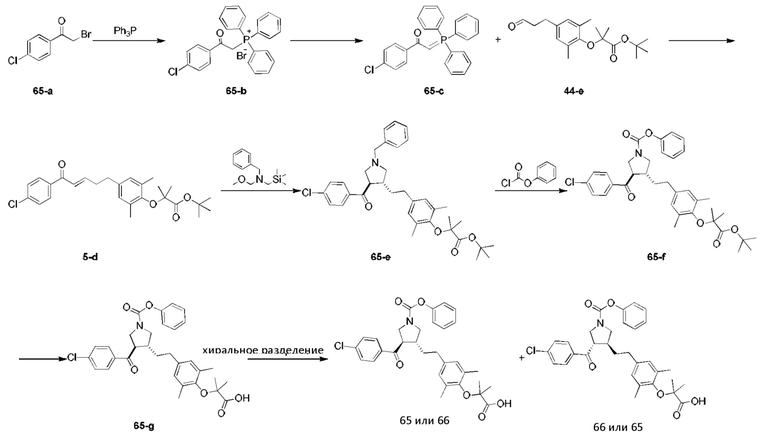
Этап 1: Соединение 65-b
При 25°С трифенилфосфин (58,97 г, 224,85 ммоль, 1,05 экв.) добавляли в раствор соединения 65-а (50,00 г, 214,14 ммоль, 1,00 экв.) в толуоле (500,00 мл). Реакционный раствор перемешивали при 25°С в течение 48 ч в защитной атмосфере азота. Реакционный раствор фильтровали, остаток на фильтре промывали дихлорметаном (200 мл), и сушили при пониженном давлении, в результате чего получали соединение 65-b.
МС: m/z (ионизация электрораспылением): 415,0 [М+1].
1H-ЯМР (400 МГц, ДМСО-d6) δppm 8,10 (d, J=8,8 Гц, 2Н), 7,91-7,81 (m, 9Н), 7,80-7,74 (m, 6 Н), 7,72 (d, J=8,8 Гц, 2Н), 6,20 (d, J=13,1 Гц, 2Н).
Этап 2: Соединение 65-с
При 20°С трет-бутоксид кальция (3,40 г, 30,26 ммоль, 1,50 экв.) добавляли в раствор соединения 65-b (10,00 г, 20,17 ммоль, 1,00 экв.) в тетрагидрофуране (100,00 мл). Реакционный раствор перемешивали при 20°С в течение 0,5 ч. Реакционный раствор экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли и промывали насыщенным слоевым раствором, сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 65-с.
1H-ЯМР (400 МГц, CDCl3) δppm 7,92-7,87 (m, 2Н), 7,84-7,76 (m, 2Н), 7,74-7,67 (m, 6 Н), 7,57-7,54 (m, 2Н), 7,51-7,48 (m, 6H), 7,33-7,29 (m, 2H).
Этап 3: Соединение 65-d
При 50°С соединение 44-е (1,00 г, 3,12 ммоль, 1,00 экв.) медленно добавляли в раствор соединения 65-с (1,29 г, 3,12 ммоль, 1,00 экв.) в тетрагидрофуране (20,00 мл). Реакционный раствор перемешивали при 50°С в течение 24 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-90:10), в результате чего получали соединение 65-d.
1H-ЯМР (400 МГц, CDCl3) δppm 7,76-7,71 (m, 2Н), 7,35 (d, J=8,5 Гц, 2Н), 6,77-6,74 (m, 2H), 6,72 (s, 2H), 2,78-2,76 (m, 2H), 2,68-2,67 (m, 2H), 2,13 (s, 6 Н), 1,44 (s, 9H), 1,35 (s, 6H)
Этап 4: Соединение 65-е
При 0°С N-(метоксиметил)-1-фенил-N-(триметилсилилметил)метиламин (441,99 мг, 1,86 ммоль, 1,20 экв.) и трифторуксусную кислоту (176,89 мг, 1,55 ммоль, 114,87 мкл, 1,00 экв.) добавляли в раствор соединения 65-d (709,00 мг, 1,55 ммоль, 1,00 экв.) в дихлорметане (20,00 мл). Реакционный раствор нагревали до 25°С и перемешивали в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-20:80), в результате чего получали соединение 65-е.
1H-ЯМР (400 МГц, CDCl3) δppm 7,89 (br d, J=8,3 Гц, 2H), 7,49-7,42 (m, 7H), 6,62 (s, 2H), 4,38-4,22 (m, 2H), 3,15 (br d, J=8,8 Гц, 2H), 2,67 (br s, 2H), 2,50-2,28 (m, 4H), 2,13 (s, 4H), 2,14-2,11 (m, 1H), 2,14-2,11 (m, 1H), 1,93-1,82 (m, 2H), 1,50 (s, 9H), 1,37 (s, 6 h).
Этап 5: Соединение 65-f
При 25°С фенилхлорформиат (332,94 мг, 2,13 ммоль, 266,35 мкл, 5,00 экв.) добавляли в раствор соединения 65-е (251,00 мг, 425,29 мкмоль, 1,00 экв.) в хлороформе (20,00 мл). Реакционный раствор нагревали до 70°С и перемешивали в течение 48 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-40:60), в результате чего получали соединение 65-f.
МС: m/z (ионизация электрораспылением): 642,4 [М+1].
Этап 6: Соединение 65-g
При 25°С трифторуксусную кислоту (588,33 мг, 5,16 ммоль, 382,03 мкл, 40,00 экв.) добавляли в раствор соединения 65-f (80,00 мг, 129,00 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл). Реакционный раствор перемешивали при 25°С в течение 1 ч. Реакционный раствор концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-50:50), в результате чего получали соединение 65-g.
МС: m/z (ионизация электрораспылением): 564,1 [М+1].
Этап 7: соединения 65 и 66
Соединение 65-g (60,00 мг, 106,37 мкмоль, 1,00 экв.) подвергали хиральному разделению, в результате чего получали соединение 65 и Соединение 66.
Соединение 65:
МС: m/z (ионизация электрораспылением): 564,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 7,90 (dd, J=6,0, 8,5 Гц, 2H), 7,48 (dd, J=5,5, 8,5 Гц, 2H), 7,40-7,31 (m, 2H), 7,20 (q, J=7,5 Гц, 1H), 7,12 (br t, J=8,8 Гц, 2H), 6,77 (d, J=6,0 Гц, 2H), 4,03-3,89 (m, 2H), 3,81-3,65 (m, 2H), 3,41-3,35 (m, 1H), 3,22-3,15 (m, 1H), 2,54 (br t, J=7,5 Гц, 2Н), 2,19 (d, J=5,0 Гц, 6Н), 1,90-1,81 (m, 2H), 1,50 (d, J=17,6 Гц, 6Н).
Соединение 66:
МС: m/z (ионизация электрораспылением): 564,1 [M+1].
1H-ЯМР (400 МГц, CDCl3) δppm 7,90 (dd, J=6,0, 8,5 Гц, 2Н), 7,48 (dd, J=5,0, 8,5 Гц, 2Н), 7,35 (q, J=8,0 Гц, 2Н), 7,19 (q, J=7,4 Гц, 1Н), 7,12 (t, J=8,8 Гц, 2Н), 6,77 (d, J=5,5 Гц, 2Н), 4,04-3,89 (m, 2Н), 3,80- 3,64 (m, 2Н), 3,38 (dd, J=7,8, 10,8 Гц, 1Н), 3,19 (dd, J=8,0, 11,0 Гц, 1Н), 2,60-2,48 (m, 2Н), 2,19 (d, J=5,0 Гц, 6Н), 1,91-1,80 (m, 2Н), 1,49 (d, J=16,6 Гц, 6Н).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% NH3H2O, изопропиловый спирт; скорость потока: 60 мл/мин; температура колонки: 40°С.
Время удерживания соединения 65: 1,835 мин (пик 1). Время удерживания соединения 66: 1,905 мин (пик 2).
Пример 67: Соединение 67
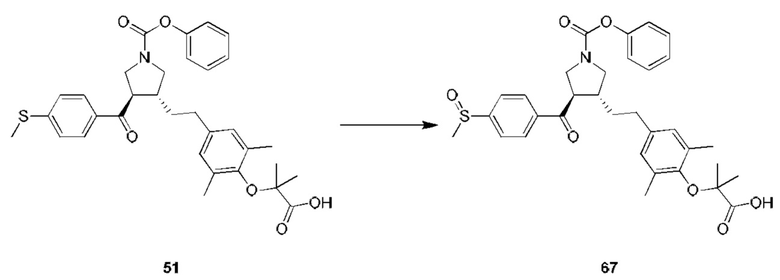
Этап 1: Соединение 67
Соединение 51 (50,00 мг, 86,85 мкмоль, 1,00 экв.) и хлороформ (5,00 мл) добавляли в высушенную реакционную колбу. Смесь охлаждали до 0°С в водно-ледяной ванне, а затем медленно добавляли m-хлорпербензойную кислоту (14,11 мг, 69,48 мкмоль, 0,80 экв.). После удаления водно-ледяной ванны смеси давали нагреться естественным путем до 20°С и перемешивали в течение 1 ч. Добавляли в реакционную систему по каплям насыщенный раствор тиосульфата натрия, за реакционной системой следили при помощи тестовой бумаги с йодидом калия до тех пор, пока тестовая бумага не переставала менять цвет. Затем смесь экстрагировали этилацетатом (20 мл). Органические фазы собирали, сушили при помощи безводного сульфата натрия, фильтровали и концентрировали, в результате чего получали неочищенный продукт. Неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией с получением соединения 67.
МС: m/z (ионизация электрораспылением): 592,2 [M+1].
1H-ЯМР (400 МГц, CDCl3) δppm 8,00-7,92 (m, 2Н), 7,71 (t, J=8,5 Гц, 2Н), 7,40-7,33 (m, 2H), 7,24-7,17 (m, 1Н), 7,14 (br dd, J=5,5, 7,0 Гц, 2Н), 6,78-6,72 (m, 2Н), 3,99-3,61 (m, 4H), 3,44 (ddd, J=3,8, 6,9, 10,9 Гц, 0,5Н), 3,34-3,26 (m, 0,5H), 2,83-2,79 (m, 3H), 2,68-2,61 (m, 1H), 2,56-2,45 (m, 1H), 2,21-2,11 (m, 6H), 1,93-1,78 (m, 2H), 1,57-1,41 (m, 6H).
Пример 68: Соединение 68
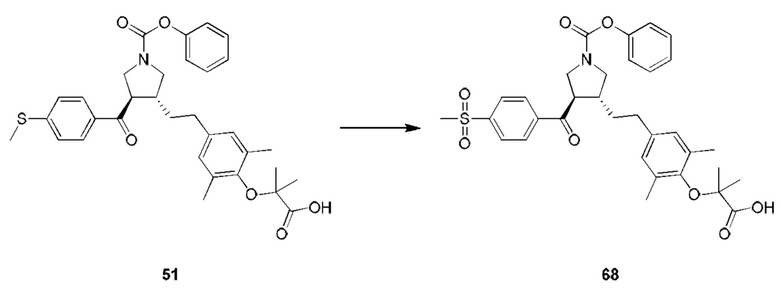
Этап 1: Соединение 68
Соединение 51 (50,00 мг, 86,85 мкмоль, 1,00 экв.) и хлороформ (5,00 мл) добавляли в высушенную реакционную колбу. Затем медленно добавляли m-хлорпербензойную кислоту (35,26 мг, 173,70 мкмоль, 2,00 экв.). Смесь перемешивали при 20°С в течение еще 1 ч. В реакционную систему по каплям добавляли насыщенный раствор тиосульфата натрия, за реакционной системой следили при помощи тестовой бумаги с йодидом калия до тех пор, пока тестовая бумага не переставала менять цвет. Затем смесь экстрагировали этилацетатом (3×20 мл). Органические фазы собирали и сушили при помощи безводного сульфата натрия, фильтровали и концентрировали, в результате чего получали неочищенный продукт. Неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией с получением соединения 68.
МС: m/z (ионизация электрораспылением): 608,2 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 8,05 (d, J=4,5 Гц, 4Н), 7,36 (q, J=7,5 Гц, 2H), 7,24-7,17 (m, 1H), 7,13 (t, J=7,8 Гц, 2H), 6,77 (d, J=3,5 Гц, 2H), 4,02-3,91 (m, 1H), 3,85-3,70 (m, 2,5H), 3,59 (dd, J=7,5, 11,0 Гц, 0,5Н), 3,43 (dd, J=7,5, 10,5 Гц, 0,5Н), 3,28 (dd, J=8,0, 11,0 Гц, 0,5Н), 3,10 (s, 3H), 2,87-2,68 (m, 1H), 2,59-2,49 (m, 1H), 2,17 (d, J=4,5 Гц, 6H), 1,93-1,72 (m, 2H), 1,48 (d=13,6 Гц, 6Н).
Пример 69: Соединение 69
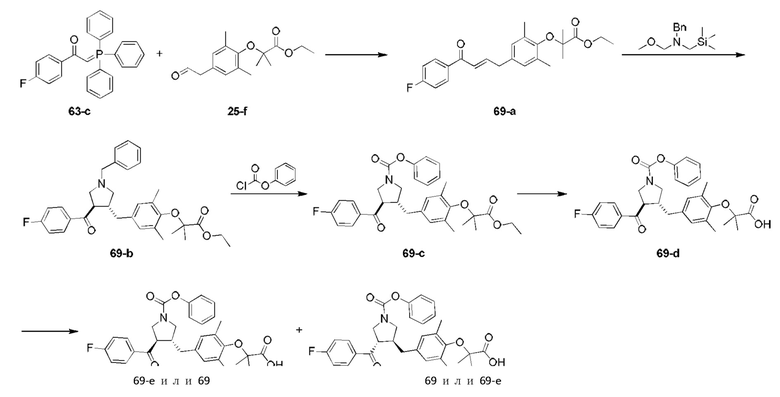
Этап 1: Соединение 69-а
Соединение 25-f (8,70 г, 31,26 ммоль, 1,00 экв.) и тетрагидрофуран (100,00 мл) добавляли в предварительно высушенную колбу объемом 250 мл. Затем в реакционную систему добавляли 63-с (12,45 г, 31,26 ммоль, 1,00 экв.). Смесь перемешивали при 50°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, в результате чего получали остаток. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-20:80) с получением число го соединения 69-а.
1H-ЯМР (400 МГц, CDCl3) δppm 7,96-7,92 (m, 2H), 7,17-7,11 (m, 3H), 7,01 (s, 1H), 6,81 (s, 2H), 4,37-4,34 (m, 2H), 3,52 (dd, J=1,0, 6,8 Гц, 2H), 2,19 (s, 6 H), 1,48-1,47 (m, 6 Н), 1,38-1,36 (m, 3H).
Этап 2: Соединение 69-b
Соединение 69-а (3,30 г, 8,28 ммоль, 1,00 экв.) и диоксан (100,00 мл) добавляли в предварительно высушенную колбу объемом 1000 мл, продутую газообразным азотом три раза, а затем добавляли трифторуксусную кислоту (83,01 мг, 728,00 мкмоль, 53,90 мкл, 0,05 экв.). Затем полученный прозрачный реакционный раствор нагревали и перемешивали при 80°С в течение 5 мин, а затем медленно добавляли раствор N-(метоксиметил)-1-фенил-N-(триметилсилилметил)метиламина (5,90 г, 24,84 ммоль, 3,00 экв.) в диоксане (10,00 мл), поддерживая температуру системы на уровне 80°С. Через 30 минут подачу завершали, реакционную систему перемешивали при 80°С в течение 1 ч. Реакционную систему охлаждали до комнатной температуры и концентрировали при пониженном давлении, в результате чего получали остаток. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-20:80), в результате чего получали продукт, чистое Соединение 69-b.
1H-ЯМР (400 МГц, CDCl3) δppm 7,75 (dd, J=5,5, 9,0 Гц, 2Н), 7,23-7,12 (m, 5H), 7,02-6,94 (m, 2H), 6,64 (s, 2Н), 4,19 (q, J=7,4 Гц, 2Н), 3,63-3,54 (m, 2Н), 3,04-2,94 (m, 2H), 2,66-2,61 (m, 2Н), 2,17-2,06 (m, 2H), 2,01-1,98 (m, 6H), 1,60-1,52 (m, 2H), 1,36 (s, 6H), 0,84-0,77 (m, 3H).
Этап 3: Соединение 69-с
Соединение 69-b (1,53 г, 2,88 ммоль, 1,00 экв.) и хлороформ (10,00 мл) добавляли в предварительно высушенную круглодонную колбу объемом 50 мл. Затем добавляли фенилхлорформиат (2,25 г, 14,39 ммоль, 1,80 мл, 5,00 экв.) добавляли. В защитной атмосфере азота реакционный сосуд помещали в масляную ванну при 70°С и перемешивали в течение 6 ч. Реакционную систему охлаждали до комнатной температуры и концентрировали при пониженном давлении, в результате чего получали остаток. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-90:10), в результате чего получали продукт, чистое соединение 69-с.
1H-ЯМР (400 МГц, CDCl3) δppm 7,76 (td, J=5,9, 8,3 Гц, 2H), 7,38-7,34 (m, 2H), 7,21-7,07 (m, 5H), 6,79 (s, 2H), 4,30 (q, J=7,0 Гц, 2H), 3,97-3,83 (m, 2H), 3,77-3,62 (m, 2H), 3,51-3,36 (m, 1Н), 2,97-2,83 (m, 1Н), 2,73-2,60 (m, 2H), 2,17 (s, 6H), 1,47 (s, 6H), 1,37 (t, J=6,8 Гц, 3H)
Этап 4: Соединение 69-d
Соединение 69-с (1,24 г, 2,21 ммоль, 1,00 экв.) добавляли в предварительно высушенную колбу объемом 100 мл, а затем растворяли путем добавления этанола (9,00 мл) и воды (3,00 мл). Затем в реакционную систему добавляли гидроксид лития (925,35 мг, 38,64 ммоль, 17,50 экв.) и перемешивали смесь при 40°С в течение 16 ч. В реакционную систему по каплям добавляли насыщенный водный раствор бисульфата натрия до рН=5-6. Затем смесь экстрагировали этилацетатом (3×10 мл). Органические фазы объединяли и последовательно промывали водой (20 мл) и насыщенным солевым раствором (20 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-50:50), в результате чего получали неочищенный продукт. Неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 69-d
МС: m/z (ионизация электрораспылением): 534,3 [М+1].
Этап 5: Соединение 69
Соединение 69-d (145,00 мг, 271,74 ммоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 70.
МС: m/z (ионизация электрораспылением): 534,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 7,75 (dd, J=5,5, 8,5 Гц, 2Н), 7,32- 7,25 (m, 2H), 7,16-7,00 (m, 5H), 6,75 (d, J=5,0 Гц, 2H), 3,95-3,81 (m, 1H), 3,79-3,64 (m, 2H), 3,59 (br dd, J=7,5, 11,0 Гц, 1H), 3,42-3,26 (m, 1H), 2,96-2,82 (m, 1H), 2,69-2,55 (m, 2H), 2,12 (d, J=3,0 Гц, 6Н), 1,43 (s, 6H).
Условия хирального анализа: хиральная колонка: AS (250 мм × 30 мм, 10 мкм); подвижная фаза: 0,1% NH3H2O, EtOH; скорость потока: 80 мл/мин; температура колонки: 40°С.
Время удерживания соединения 70: 3,561 мин (пик 1).
Пример 70: Соединение 70
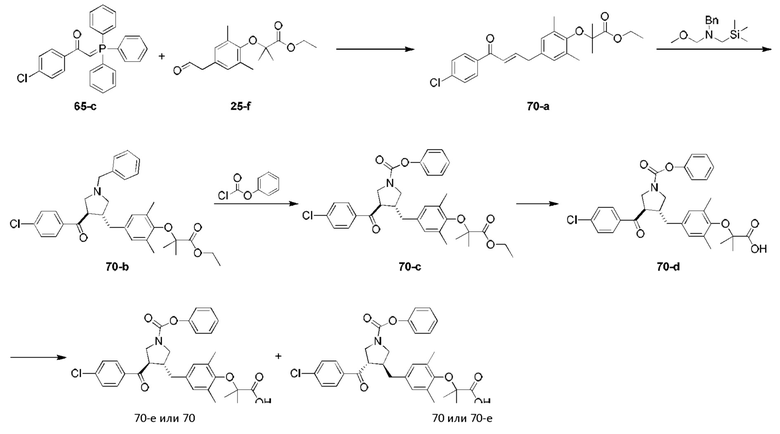
Этап 1: Соединение 70-а
Соединение 25-f (18,00 г, 64,67 ммоль, 1,00 экв.) и тетрагидрофуран (100,00 мл) добавляли в предварительно высушенную колбу объемом 250 мл. Затем в реакционную систему добавляли 65-с (26,83 г, 64,67 ммоль, 1,00 экв.) и перемешивали смесь при 50°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, в результате чего получали остаток. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-90:10), в результате чего получали соединение 70-а.
1H-ЯМР (400 МГц, CDCl3) δppm 7,86 (d, J=8,5 Гц, 2Н), 7,44 (d, J=8,5 Гц, 2Н), 7,28 (s, 0,5H), 7,24-7,15 (m, 1H), 7,02 (s, 0,5H), 6,82 (s, 2Н), 4,41-4,35 (m, 2Н), 3,53 (d, J=6,5 Гц, 2Н), 2,20 (s, 6H), 1,49 (s, 6H), 1,39-1,37 (m, 3H).
Этап 2: Соединение 70-b
Соединение 70-а (4,00 г, 9,64 ммоль, 1,00 экв.) и диоксан (120,00 мл) добавляли в сухую колбу, а затем трифторуксусная кислоты (83,01 мг, 728,00 мкмоль, 53,90 мкл, 0,05 экв.) добавляли. Раствор нагревали и перемешивали при 80°С в течение 5 мин, а затем медленно по каплям добавляли раствор N-(метоксиметил)-1-фенил-N-(триметилсилилметил)метиламина (6,87 г, 28,92 ммоль, 3,00 экв.) в диоксане (120,00 мл). После завершения добавления реакционную смесь перемешивали при 80°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-90:10), в результате чего получали соединение 70-b.
1H-ЯМР (400 МГц, CDCl3) δppm 7,66 (d, J=8,5 Гц, 2Н), 7,26-7,23 (m, 7H), 6,63 (s, 2Н), 4,22-4,18 (m, 2Н), 3,71-3,61 (m, 2Н), 3,54-3,44 (m, 2Н), 3,05-2,96 (m, 2Н), 2,69-2,59 (m, 2H), 2,11 (d, J=12,0 Гц, 2Н), 2,02-1,97 (m, 6 Н), 1,41-1,34 (m, 6 Н), 0,93-0,75 (m, 3H)
Этап 3: Соединение 70-с
Соединение 70-b (1,00 г, 1,82 ммоль, 1,00 экв.) и хлороформ (10,00 мл) добавляли в предварительно высушенную круглодонную колбу объемом 50 мл. Затем добавляли фенилхлорформиат (1,43 г, 9,12 ммоль, 1,14 мл, 5,00 экв.). В защитной атмосфере азота реакционный сосуд помещали в масляной ванне при 70°С и перемешивали в течение 6 ч. Реакционную систему охлаждали до комнатной температуры и концентрировали при пониженном давлении, в результате чего получали остаток. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-90:10), в результате чего получали соединение 70-с.
1H-ЯМР (400 МГц, CDCl3) δppm 7,68 (dd, J=6,3, 8,3 Гц, 2Н), 7,43-7,35 (m, 4H), 7,25-7,13 (m, 3H), 6,80 (s, 2Н), 4,32 (q, J=7,0 Гц, 2Н), 3,98-3,68 (m, 4H), 3,52-3,38 (m, 1H), 2,98-2,84 (m, 1H), 2,75-2,63 (m, 2Н), 2,20-2,16 (m, 6 Н), 1,59 (d, J=1,0 Гц, 6 Н), 1,42-1,35 (m, 3H).
Этап 4: Соединение 70-d
Соединение 70-с (560,00 мг, 968,69 мкмоль, 1,00 экв.) добавляли в сухую колбу, а затем растворяли путем добавления этанола (6,00 мл) и воды (2,00 мл). Затем в реакционную систему добавляли гидроксид лития (406,46 мг, 16,97 ммоль, 17,52 экв.) и перемешивали смесь при 40°С в течение 16 ч. В реакционную систему по каплям добавляли насыщенный водный раствор бисульфата натрия до рН=5-6. Затем смесь экстрагировали этилацетатом (3×10 мл). Органические фазы объединяли, и последовательно промывали водой (20 мл) и насыщенным солевым раствором (20 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-50:50) с получением неочищенного продукта. Неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 70-d.
МС: m/z (ионизация электрораспылением): 550,3 [М+1].
Этап 5: Соединение 70
Соединение 70-d (100,00 мг, 181,80 мкмоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 70.
МС: m/z (ионизация электрораспылением): 572,3 [М+23].
1H-ЯМР (400 МГц, CDCl3) δppm 7,66 (d, J=8,5 Гц, 2Н), 7,34 (d, J=8,5 Гц, 2Н), 7,32-7,26 (m, 2H), 7,16-7,09 (m, 1H), 7,06 (br d, J=7,5 Гц, 2Н), 6,75 (d, J=5,0 Гц, 2Н), 3,95-3,82 (m, 1H), 3,76-3,65 (m, 2Н), 3,60 (br dd, J=7,0, 11,0 Гц, 1H), 3,39-3,27 (m, 1H), 2,94-2,81 (m, 1H), 2,65-2,57 (m, 2Н), 2,12 (d, J=3,0 Гц, 6Н), 1,43 (s, 6H).
Условия хирального анализа: хиральная колонка: AS (250 мм × 30 мм, 10 мкм); подвижная фаза: 0,1% NH3H2O EtOH; скорость потока: 80 мл/мин; температура колонки: 40°С.
Время удерживания соединения 70: 3,827 мин (пик 1).
Пример 71: Соединение 71
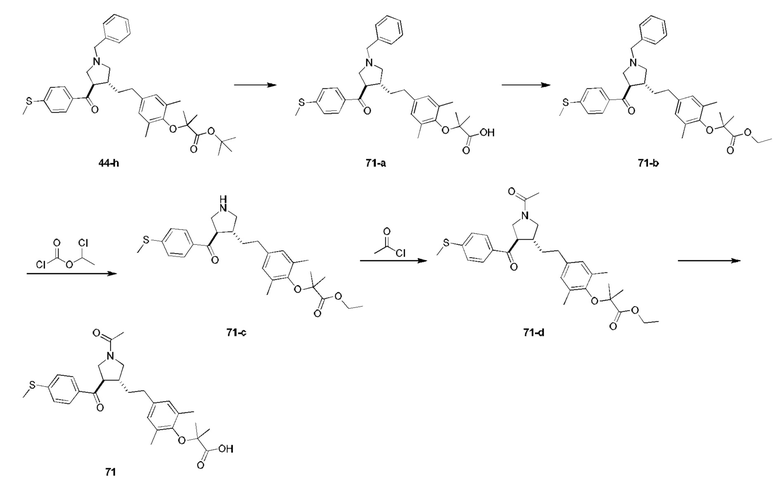
Этап 1: Соединение 71-а
Соединение 44-h (5,00 г, 8,31 ммоль, 1,00 экв.), трифторуксусную кислоту (9,47 г, 83,08 ммоль, 6,15 мл, 10,00 экв.) и дихлорметан (50,00 мл) добавляли в высушенную круглодонную колбу, и полученный прозрачный раствор перемешивали при 20°С в течение 3 ч. Реакционный раствор концентрировали при пониженном давлении. Остаток выделяли колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-10:90), в результате чего получали соединение 71-а.
МС: m/z (ионизация электрораспылением): 546,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 7,87 (d, J=8,4 Гц, 2H), 7,49-7,42 (m, 6H), 7,31 (s, 1H), 6,69 (s, 2H), 4,46-4,22 (m, 2H), 3,89 (s, 1H), 3,46 (s, 1H), 3,17-3,10 (m, 2H), 2,72-2,70 (m, 1H), 2,54 (s, 3H), 2,48-2,44 (m, 2H), 2,17 (s, 6H), 1,92-1,87 (m, 2H), 1,51 (s, 6H).
Этап 2: Соединение 71-b
71-а (4,10 г, 7,51 ммоль, 1,00 экв.), оксалилхлорид (9,53 г, 75,10 ммоль, 6,57 мл, 10,00 экв.) и дихлорметан (40,00 мл) добавляли в высушенную круглодонную колбу, и полученный прозрачный раствор перемешивали при 20°С в течение 1 ч. Реакционную систему концентрировали при пониженном давлении, а затем добавляли этанол (20,00 мл). Раствор перемешивали при 20°С в течение 1 ч. В реакционную систему по каплям добавляли насыщенный водный раствор карбоната натрия до рН=8-9. Реакционную систему экстрагировали этилацетатом (100 мл) и водой (100 мл). После разделения фаз органические фазы собирали и сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали соединение 71-b.
МС: m/z (ионизация электрораспылением): 574,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 7,85 (t, J=8,4 Гц, 2H), 7,31-7,22 (m, 7H), 6,68 (s, 2H), 4,31-4,24 (m, 2H), 3,59-3,53 (m, 2H), 2,99-2,65 (m, 4Н), 2,51 (s, 3H), 2,49-2,39 (m, 3H), 2,11 (s, 6Н), 1,76-1,74 (m, 3H), 1,42 (s, 6Н), 1,34 (t, J=7,2 Гц, 3Н).
Этап 3: Соединение 71-с
При 20°С α-хлорэтилхлорформиат (7,48 г, 52,30 ммоль, 10,00 экв.), соединение 71-b (3,00 г, 5,23 ммоль, 1,00 экв.) и безводный толуол (30,00 мл) добавляли в предварительно высушенную круглодонную колбу объемом 100 мл и перемешивали смесь при 80°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении и добавляли в него метанол (30,00 мл) и перемешивали при 80°С в тчение часа. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-10:90), в результате чего получали соединение 71-с.
МС: m/z (ионизация электрораспылением): 484,2 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 7,84 (d, J=4,8 Гц, 2H), 7,28-7,25 (m, 2H), 6,66 (d, J=12,8 Гц, 2H), 4,29-4,24 (m, 2H), 3,89-3,51 (m, 4Н), 3,18-3,02 (m, 1H), 2,53 (s, 1H), 2,49-2,46 (m, 2H), 2,12-2,08 (d, J=7,5 Гц, 6 Н), 1,40-1,32 (d, J=4,8 Гц, 6 Н).
Этап 4: Соединение 71-d
При 20°С соединение 71-с (100,00 мг, 206,76 мкмоль, 1,00 экв.) добавляли в предварительно высушенную круглодонную колбу объемом 100 мл, а затем добавляли ацетилхлорид (12,98 мг, 165,41 мкмоль, 11,80 мкл, 0,80 экв.) и триэтиламин (20,92 мг, 206,76 мкмоль, 28,66 мкл, 1,00 экв.). Реакционная система давала значительное количество белого дыма. Реакционный раствор перемешивали при 20°С в течение 3 ч. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией (дихлорметан : метанол = 100:0-30:70), в результате чего получали соединение 71-d.
МС: m/z (ионизация электрораспылением): 526,3 [М+1].
Этап 5: Соединение 71
71-d (50,00 мг, 95,11 мкмоль, 1,00 экв.), а затем этанол (6,00 мл) добавляли в предварительно высушенную колбу объемом 50 мл. Затем в реакционную систему добавляли гидроксид лития (22,78 мг, 951,10 мкмоль, 10,00 экв.) и воду (2,00 мл) добавляли. Реакционный раствор перемешивали при 40°С в течение 16 ч. В реакционную систему по каплям добавляли насыщенный водный раствор бисульфата калия до рН=6. Реакционную смесь экстрагировали этилацетатом и водой (1:1,20 мл). После разделения фаз органические фазы собирали, и водную фазу экстрагировали этилацетатом (310 мл). Органические фазы объединяли и сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток. Остаток очищали тонкослойной хроматограыией (дихлорметан : метанол = 10:1), в результате чего получали неочищенный продукт, который затем очищали препаративной высокоэффективной жидкостной хроматографией с получением соединения 71.
МС: m/z (ионизация электрораспылением): 498,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δppm 7,85 (br d, J=4,8 Гц, 2Н), 7,30 (br d, J=8,3 Гц, 2Н), 6,75 (br d, J=10,8 Гц, 2Н), 3,83-3,70 (m, 3Н), 3,50 (br s, 2Н), 2,54 (d, J=6,0 Гц, 3Н), 2,46 (br s, 3H), 2,20 (br d, J=16,8 Гц, 6H), 2,02 (br s, 3Н), 1,83 (br s, 2Н), 1,64-1,41 (m, 6H).
Пример 72: Соединение 72
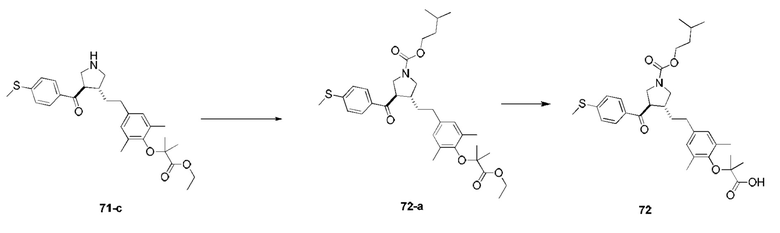
Этап 1: Соединение 72-а
Изо-пентилхлорформиат (31,14 мг, 206,76 мкмоль, 1,00 экв.) и триэтиламин (41,84 мг, 413,52 мкмоль, 57,32 мкл, 2,00 экв.) добавляли в раствор соединения 71-с (100,00 мг, 206,76 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл) Полученный прозрачный раствор перемешивали при 20°С в течение 1 ч. Реакционный раствор концентрировали при пониженном давлении. Остаток выделяли и очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-50:50), в результате чего получали соединение 72-а.
Этап 2: Соединение 72
Гидроксид лития (20,03 мг, 836,40 мкмоль, 10,00 экв.) и воду (2,00 мл) добавляли в раствор соединения 72-а (50,00 мг, 83,64 мкмоль, 1,00 экв.) в этаноле (6,00 мл). Смешанный раствор перемешивали при 40°С в течение 16 ч. Смесь доводили насыщенным водным раствором бисульфата калия до рН=6 и обрабатывали водой и этилацетатом (1:1,20 мл). Водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали остаток. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан : метанол = 10/1), в результате чего получали неочищенный продукт, который затем выделяли высокоэффективной жидкостной хроматографией, в результате чего получали соединение 72.
МС: m/z (ионизация электрораспылением): 570,4 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,88-7,84 (m, 2H), 7,30 (br s, 2H), 6,74 (br s, 2H), 3,96 (br s, 2H), 3,77-3,62 (m, 4H), 2,98 (br d, J=9,3 Гц, 1Н), 2,54 (s, 3H), 2,50 (br s, 2H), 2,19 (br d, J=13,1 Гц, 6Н), 2,03 (br s, 2H), 1,53 (br s, 6H), 1,45 (br s, 4H), 0,89 (br d, J=3,8 Гц, 6Н)
Пример 73: Соединение 73
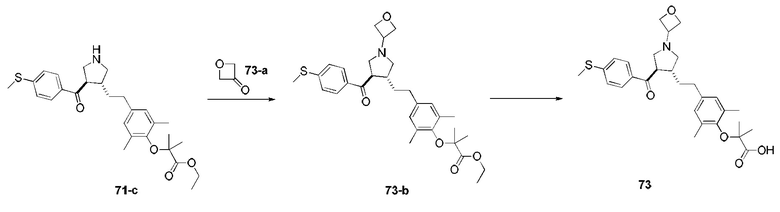
Этап 1: Соединение 73-b
73-а (22,35 мг, 310,14 мкмоль, 1,50 экв.) и уксусную кислоту (1,24 мг, 20,68 мкмоль, 1,18 мкл, 0,10 экв.) добавляли в раствор соединения 71-с (100,00 мг, 206,76 мкмоль, 1,00 экв.) в метаноле (10,00 мл). Полученный прозрачный раствор перемешивали при 20°С в течение 1 ч. Затем в реакционную систему добавляли цианоборогидрид (19,49 мг, 310,14 мкмоль, 1,50 экв.) и перемешивали при 20°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении. Остаток выделяли и очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-10:90), в результате чего получали соединение 73-b
МС: m/z (ионизация электрораспылением): 540,3 [М+1].
Этап 2: Соединение 73
Гидроксид лития (124,25 мг, 5,19 ммоль, 20,00 экв.) добавляли в смешанный раствор соединения 73-b (140,00 мг, 259,39 мкмоль, 1,00 экв.) в этаноле (10,00 мл) и воде (5,00 мл).
Полученный прозрачный раствор перемешивали при 50°С в течение 16 ч. В реакционную систему по каплям добавляли 1 н. водный раствор HCl до рН=6. Реакционную систему разбавляли смесью 10 мл этилацетата/10 мл воды. После разделения фаз органические фазы собирали и экстрагировали водную фазу этилацетатом (10 мл × 3). Органические фазы объединяли и сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток. Остаток очищали путем тонкослойной хроматографии на силикагелевой пластине (дихлор метан : метанол = 10:1), в результате чего получали неочищенный продукт в виде бесцветного масла. Неочищенный продукт выделяли и очищали препаративной высокоэффективной жидкостной хроматографией с получением соединения 73.
МС: m/z (ионизация электрораспылением): 512,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,92 (d, J=8,5 Гц, 2Н), 7,29 (d, J=8,5 Гц, 2Н), 6,72 (s, 2H), 4,79-4,71 (m, 4H), 3,97 (br s, 1H), 3,83 (br s, 1H), 3,54 (br s, 1H), 2,96 (br s, 1H), 2,84 (br s, 2Н), 2,71-2,61 (m, 1H), 2,53 (s, 3H), 2,41-2,33 (m, 1H), 2,17 (s, 6H), 1,81 (br d, J=5,3 Гц, 3Н), 1,48 (d, J=8,8 Гц, 6H)
Пример 74: Соединение 74
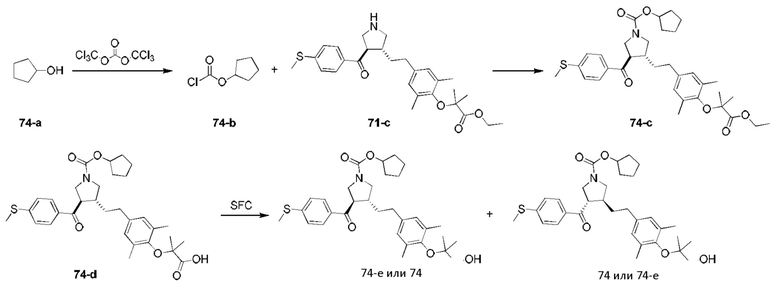
Этап 1: Соединение 74-b
При 20°С соединение 74-а (200,00 мг, 2,32 ммоль, 210,53 мкл, 1,00 экв.), триэтиламин (704,91 мг, 6,97 ммоль, 965,63 мкл, 3,00 экв.) и тетрагидрофуран (10,00 мл) добавляли в высушенную круглодонную колбу, а затем добавляли трифосген (551,26 мг, 1,86 ммоль, 0,80 экв.). Полученную суспензию перемешивали при 20°С в течение 1 ч. Смесь фильтровали, а фильтрат концентрировали при пониженном давлении, в результате чего получали соединение 74-b.
Этап 2: Соединение 74-с
При 20°С соединение 74-b (46,08 мг, 310,14 мкмоль, 1,50 экв.), соединение 71-с (100,00 мг, 206,76 мкмоль, 1,00 экв.), триэтиламин (41,84 мг, 413,52 мкмоль, 57,32 мкл, 2,00 экв.) и дихлорметан (10,00 мл) добавляли в высушенную круглодонную колбу и перемешивали полученный прозрачный раствор при 20°С в течение 1 ч. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 74-с.
МС: m/z (ионизация электрораспылением): 596,4 [М+1].
Этап 3: Соединение 74-d
Соединение 74-с (100,00 мг, 167,84 мкмоль, 1,00 экв.) и этанол (6,00 мл) добавляли в реакционную колбу а затем добавляли гидроксид лития (40,20 мг, 1,68 ммоль, 10,00 экв.) и воду (2,00 мл). Смесь перемешивали при 40°С в течение 16 ч. В реакционную систему добавляли по каплям 1 н. водный раствор HCl до рН=6. Реакционную систему экстрагировали этилацетатом (20 мл) и водой (20 мл). После разделения фаз органические фазы собирали, и экстрагировали водную фазу этилацетатом (3×10 мл). Объединенную органическую фазу сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали путем тонкослойной хроматографии на силикагелевой пластине (дихлорметан : метанол = 10:1), в результате чего получали соединение 74-d.
МС: m/z (ионизация электрораспылением): 590,2 [М+23].
Этап 4: Соединение 74
Соединение 74-d (14,00 мг, 24,66 мкмоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 74.
МС: m/z (ионизация электрораспылением): 568,4 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,85 (br s, 2H), 7,28-7,27 (m, 2H), 6,64 (br s, 2H), 3,89 (s, 1H), 3,70 (br s, 2H), 3,41 (br s, 1H), 3,12 (br s, 1H), 2,62 (s, 1H), 2,51 (br s, 5H), 2,03 (br s, 6 Н), 1,76-1,65 (m, 11H), 1,35-1,28 (m, 6 Н).
Хиральная колонка: AS (250 мм × 30 мм, 10 мкм); подвижная фаза: 35% метанола (0,05% диэтаноламина) в CO2; скорость потока: 80 мл/мин; температура колонки: 40°С.
Время удерживания соединения 74: 4,120 мин (пик 1).
Пример 75: Соединение 75
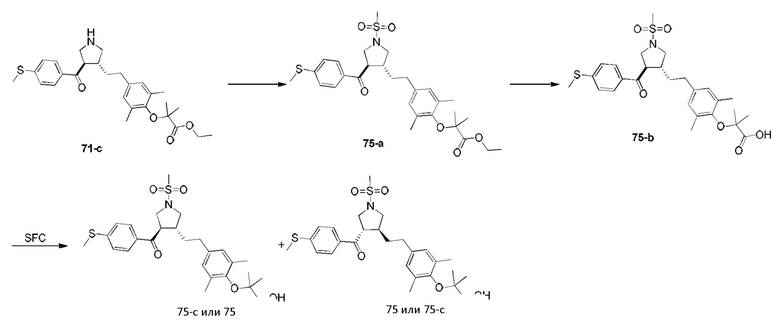
Этап 1: Соединение 75-а
При 20°С соединение 71-с (200,00 мг, 413,51 мкмоль, 1,00 экв.), мезилхлорид (71,05 мг, 620,27 мкмоль, 48,01 мкл, 1,50 экв.), триэтиламин (125,53 мг, 1,24 ммоль, 171,96 мкл, 3,00 экв.) и дихлорметан (10,00 мл) добавляли в высушенную круглодонную колбу и перемешивали полученный прозрачный раствор при 20°С в течение 1 ч. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 75-а.
МС: m/z (ионизация электрораспылением): 584,2 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,89-7,81 (m, 2H), 7,30 (s, 2H), 6,74 (s, 2H), 4,33-4,27 (m, 2H), 3,84-3,77 (m, 2H), 3,56-3,50 (m, 1H), 3,33-3,31 (m, 2H), 2,96 (s, 3H), 2,65-2,47 (m, 6H), 2,19 (s, 6H), 1,90-1,79 (m, 2H), 1,49 (d, J=2,0 Гц, 6H), 1,37-1,35 (m, 3H).
Этап 2: Соединение 75-b
Соединение 75-а (51,00 мг, 90,79 мкмоль, 1,00 экв.) и этанол (6,00 мл) добавляли в реакционную колбу объемом 100 мл, а затем добавляли гидроксид лития (21,74 мг, 907,88 мкмоль, 10,00 экв.) и воду (2,00 мл). Смесь перемешивали при 40°С в течение 16 ч. В реакционную систему по каплям добавляли насыщенный водный раствор бисульфата натрия до рН=6. Реакционную систему экстрагировали этилацетатом (20 мл) и водой (20 мл). После разделения фаз органические фазы собирали и экстрагировали водную фазу этилацетатом (3×10 мл). Объединенную органическую фазу сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали путем тонкослойной хроматографии на силикагелевой пластине (дихлорметан : метанол = 10:1), в результате чего получали соединение 75-b.
MC: m/z (ионизация электрораспылением): 534,3 [M+1].
Этап 3: Соединение 75
Соединение 75-b (16,00 мг, 29,98 мкмоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 75.
MC: m/z (ионизация электрораспылением): 534,3 [M+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,81 (d, J=8,5 Гц, 2H), 7,29 (s, 2H), 6,77 (s, 2H), 3,80-3,71 (m, 2H), 3,55- 3,50 (m, 1H), 3,40 (dd, J=6,8, 9,8 Гц, 1Н), 3,28 (dd, J=5,3, 9,8 Гц, 1Н), 2,94 (s, 3Н), 2,65-2,56 (m, 2H), 2,55 (s, 3H), 2,54-2,47 (m, 1Н), 2,19 (s, 6H), 1,90-1,79 (m, 2H), 1,49 (d, J=2,0 Гц, 6 Н)
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 5 мкм); подвижная фаза: 40% метанола (0,05% диэтаноламина) в CO2; скорость потока: 60 мл/мин; температура колонки: 40°С.
Время удерживания соединения 75: 4,725 мин (пик 1).
Пример 76: Соединение 76
Этап 1: Соединение 76-b
Карбонат цезия (388,20 г, 1,19 моль, 3,00 экв.) и этил-2-бром-изобутират (154,93 г, 794,30 ммоль, 2,00 экв.) добавляли в раствор соединения 76-а (48,50 г, 397,15 ммоль, 1,00 экв.) в 1,4-диоксане (500,00 мл). Смесь перемешивали при 90°С в течение 1 ч. Реакционную смесь фильтровали, остаток на фильтре промывали этанолом (200 мл × 3). Объединенный фильтрат концентрировали при пониженном давлении, в результате чего получали соединение 76-b.
1H-ЯМР (400 МГц, CDCl3) δppm 9,89 (s, 1H), 7,83-7,78 (m, 2H), 6,94-6,89 (m, 2H), 4,25-4,21 (m, 2H), 1,68 (s, 6H), 1,22 (t, J=7,2 Гц, 3Н).
Этап 2: Соединение 76-с
Метоксиметилтрифенилфосфина хлорид (28,29 г, 82,53 ммоль, 1,50 экв.) и тетрагидрофуран (200,00 мл) добавляли в высушенную реакционную колбу, а затем партиями добавляли трет-бутоксид калия (10,69 г, 95,27 ммоль, 1,73 экв.) при 20°С. После проведения реакции в течение 1 ч, в реакционный раствор добавляли соединение 76-b (13,00 г, 55,02 ммоль, 1,00 экв.). Смесь перемешивали при 20°С в течение 1 ч и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-70:30), в результате чего получали соединение 76-с.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,48-7,44 (m, 1H), 7,36-7,35 (m, 1H), 7,14-6,91 (m, 2H), 6,80-6,77 (m, 1H), 6,09-5,73 (m, 1H), 4,16-4,10 (m, 2H), 3,77-3,66 (m, 3Н), 1,58 (s, 6H), 1,29-1,27 (m, 3H).
Этап 3: Соединение 76-d
Оксалилхлорид (15,99 г, 125,98 ммоль, 11,03 мл, 2,00 экв.) медленно добавляли в раствор соединения 76-с (16,65 г, 62,99 ммоль, 1,00 экв.) в хлороформе (200,00 мл) при 0°С, а затем добавляли этанол (5,80 г, 125,98 ммоль, 7,34 мл, 2,00 экв.) и воду (2,27 г, 125,98 ммоль, 2,27 мл, 2,00 экв.). Смешанный раствор перемешивали при 0°С в течение 1 ч. В реакционную систему по каплям добавляли насыщенный водный раствор карбоната натрия до рН7-8. Реакционную систему экстрагировали дихлорметаном (20 мл) и водой (20 мл). После разделения фаз органические фазы собирали и экстрагировали водную фазу этилацетатом (3×20 мл). Органические фазы объединяли и последовательно промывали водой (3×20 мл) и насыщенным солевым раствором (3×20 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали соединение 76-d.
1H-ЯМР (400 МГц, CDCl3) δ ppm 9,71 (t, J=2,4 Гц, 1H), 7,08 (d, J=8,5 Гц, 2H), 6,87-6,81 (m, 2H), 4,23 (q, J=7,1 Гц, 2H), 3,92-3,90 (m, 2H), 1,59 (s, 6H), 1,40-1,37 (m, 3H).
Этап 4: Соединение 76-е
Соединение 44-f (23,79 г, 55,78 ммоль, 1,00 экв.) добавляли в раствор соединения 76-d (13,96 г, 55,78 ммоль, 1,00 экв.) в тетрагидрофуране (150,00 мл). Смесь перемешивали при 50°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 76-е.
МС: m/z (ионизация электрораспылением): 399,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,93-7,83 (m, 2H), 7,30-7,27 (m, 3H), 7,21-7,06 (m, 2H), 6,85-6,76 (m, 3H), 4,27-4,21 (m, 2H), 3,84 (dd, J=1,0, 6,8 Гц, 1Н), 3,57 (d, J=5,8 Гц, 1Н), 2,53 (d, J=2,0 Гц, 3H), 1,59 (d, J=1,5 Гц, 6Н), 1,27-1,25 (m, 3H)
Этап 5: Соединение 76-f
Трифторуксусную кислоту (90,27 мг, 791,50 мкмоль, 58,62 мкл, 0,05 экв.) добавляли в раствор соединения 76-е (6,31 г, 15,83 ммоль, 1,00 экв.) в 1,4-диоксане (350,00 мл), а затем добавляли N-бензил-1-метокси-N-((триметилсилил)метил)метиламин (11,28 г, 47,49 ммоль, 3,00 экв.) медленно. Смесь перемешивали при 80°С в течение 2 ч. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 76-f.
МС: m/z (ионизация электрораспылением): 532,2 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,89-7,83 (m, 1Н), 7,70 (d, J=8,8 Гц, 1Н), 7,33-7,29 (m, 5H), 7,22-7,18 (m, 2H), 6,99 (d, J=8,3 Гц, 2H), 6,72-6,66 (m, 2H), 4,21-4,15 (m, 2H), 4,09-4,00 (m, 1Н), 3,67-3,62 (m, 2H), 3,11-2,99 (m, 2H), 2,88-2,82 (m, 1Н), 2,75-2,57 (m, 4H), 2,52-2,50 (m, 3H), 1,55-1,50 (m, 6 Н), 1,23-1,18 (m, 3H)
Этап 6: Соединение 76-g
Фенилхлорформиат (6,37 г, 40,70 ммоль, 5,10 мл, 5,00 экв.) медленно добавляли в раствор соединения 76-f (4,33 г, 8,14 ммоль, 1,00 экв.) в хлороформе (50,00 мл) и перемешивали смесь при 70°С в течение 16 ч. Смесь концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 76-g.
МС: m/z (ионизация электрораспылением): 562,0 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,92-7,82 (m, 1Н), 7,65-7,62 (m, 1Н), 7,39-7,29 (m, 3H), 7,26-7,11 (m, 6Н), 7,08-7,05 (m, 1Н), 6,81-6,78 (m, 1Н), 4,29-4,20 (m, 2H), 4,00-3,90 (m, 1H), 3,88-3,80 (m, 1H), 3,77-3,70 (m, 2H), 3,49-3,35 (m, 1H), 2,95-2,80 (m, 1H), 2,74 (dd, J=2,6, 7,7 Гц, 1H), 2,55-2,53 (m, 3H), 2,51-2,49 (m, 1H), 1,61-1,59 (m, 6H), 1,27-1,25 (m, 3H).
Этап 7: Соединение 76-h
Гидроксид лития (766,40 мг, 32,00 ммоль, 10,00 экв.) и воду (5,00 мл) добавляли в раствор соединения 76-g (1,80 г, 3,20 ммоль, 1,00 экв.) в этаноле (15,00 мл) и перемешивали смесь при 40°С в течение 4 ч. В реакционную систему по каплям добавляли насыщенный водный раствор карбоната натрия до рН=6. Реакционную систему экстрагировали этилацетатом (20 мл) и водой (20 мл). После разделения фаз органические фазы собирали. Водную фазу экстрагировали этилацетатом (3×10 мл). Органические фазы объединяли и сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-0:100), в результате чего получали неочищенный продукт, который затем очищали высокоэффективной жидкостной хроматографией с получением соединения 76-h.
МС: m/z (ионизация электрораспылением): 534,2 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,68 (d, J=7,5 Гц, 2H), 7,39-7,33 (m, 2H), 7,26-7,18 (m, 3H), 7,16-7,09 (m, 4H), 6,91-6,85 (m, 2H), 4,00-3,87 (m, 1H), 3,84-3,73 (m, 2H), 3,67 (dd, J=6,5, 10,8 Гц, 1H), 3,51-3,35 (m, 1H), 3,02-2,91 (m, 1H), 2,78 (td, J=6,8, 13,9 Гц, 2Н), 2,53 (s, 3H), 1,59 (br s, 6H).
Этап 8: Соединение 76
Соединение 76-h (200,00 мг, 374,78 мкмоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 76.
МС: m/z (ионизация электрораспылением): 534,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,65 (br dd, J=4,9, 7,9 Гц, 2H), 7,38-7,32 (m, 2H), 7,23-7,17 (m, 3H), 7,15-7,11 (m, 2H), 7,08-7,01 (m, 2H), 6,84 (br s, 2H), 3,98-3,85 (m, 1H), 3,84-3,69 (m, 2H), 3,68-3,60 (m, 1H), 3,47-3,30 (m, 1H), 2,95-2,81 (m, 1H), 2,80-2,65 (m, 2H), 2,51 (s, 3H), l,53 (br s, 6H).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: 55% метанола (0,05% диэтаноламина) в CO2; скорость потока: 80 мл/мин; температура колонки: 40°С
Время удерживания соединения 76: 0,729 мин (пик 1).
Пример 77: Соединение 77
Этап 1: Соединение 77-b
Карбонат цезия (179,48 г, 550,86 ммоль, 3,00 экв.) и этил-2-бром-изобутират (71,63 г, 367,24 ммоль, 53,86 мл, 2,00 экв.) добавляли в раствор соединения 77-а (25,00 г, 183,62 ммоль, 1,00 экв.) в 1,4-диоксане (500,00 мл), и перемешивали при 90°С в течение 1 ч. Реакционную систему разбавляли этилацетатом (500 мл) и водой (500 мл). Органическую фазу промывали насыщенным солевым раствором (3×500 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали соединение 77-b.
1H-ЯМР (400 МГц, CDCl3) δppm 9,83 (s, 1H), 7,68 (d, J=1,5 Гц, 1Н), 7,58 (dd, J=2,0, 8,5 Гц, 1H), 6,66 (d, J=8,3 Гц, 1H), 4,23-4,17 (m, 2H), 2,27 (s, 3Н), 1,66 (s, 6H), 1,20 (t, J=7,2 Гц, 3Н).
Этап 2: Соединение 77-с
Метоксиметилтрифенилфосфинхлорид (35,61 г, 103,88 ммоль, 1,30 экв.) и тетрагидрофуран (200,00 мл) добавляли в высушенную реакционную колбу, а затем порциями добавляли трет-бутоксид калия (13,45 г, 119,87 ммоль, 1,50 экв.) при 20°С. После проведения реакции в течение 1 ч в реакционный раствор добавляли соединение 77-b (20,00 г, 79,91 ммоль, 1,00 экв.). Смесь перемешивали при 20°С в течение 1 ч и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-15:1), в результате чего получали соединение 77-с.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,30-7,18 (m, 1H), 6,98-6,81 (m, 2H), 6,53 (dd, J=5,6, 8,4 Гц, 1H), 5,99-5,63 (m, 1H), 4,17 (dq, J=1,3, 7,1 Гц, 2H), 3,69-3,57 (m, 3H), 2,14 (d, J=3,5 Гц, 3Н), 1,50 (s, 6H), 1,21-1,17 (m, 3H)
Этап 3: Соединение 77-d
Оксалилхлорид (12,51 г, 98,58 ммоль, 8,63 мл, 2,00 экв.) медленно добавляли в раствор соединения 77-с (13,72 г, 49,29 ммоль, 1,00 экв.) в хлороформе (150,00 мл) при 0°С, а затем добавляли этанол (4,54 г, 98,58 ммоль, 5,75 мл, 2,00 экв.) и воду (1,78 г, 98,58 ммоль, 1,78 мл, 2,00 экв.). Смешанный раствор перемешивали 0°С в течение 1 ч. В реакционную систему по каплям добавляли насыщенный водный раствор карбоната натрия до рН 7-8. Добавляли дихлорметан (20 мл) и воду (20 мл). После разделения фаз органические фазы собирали и экстрагировали водную фазу этилацетатом (3×20 мл). Органические фазы объединяли и последовательно промывали водой (3×20 мл) и насыщенным солевым раствором (3×20 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали соединение 77-d.
1H-ЯМР (400 МГц, CDCl3) δ ppm 9,69 (t, J=2,5 Гц, 1H), 6,99 (d, J=2,0 Гц, 1H), 6,89 (dd, J=2,0, 8,3 Гц, 1H), 6,64 (d, J=8,3 Гц, 1H), 4,24 (q, J=7,2 Гц, 2H), 3,56 (d, J=2,5 Гц, 2H), 2,23 (s, 3Н), 1,59 (s, 6H), 1,27-1,24 (m, 3H).
Этап 4: Соединение 77-е
Соединение 44-f (21,75 г, 51,00 ммоль, 1,00 экв.) добавляли в раствор соединения 77-d (13,48 г, 51,00 ммоль, 1,00 экв.) в тетрагидрофуране (150,00 мл). Смесь перемешивали при 50°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении и очищали неочищенный продукт колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 77-е.
МС: m/z (ионизация электрораспылением): 413,0 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,78-7,74 (m, 2H), 7,21-7,17 (m, 2H), 7,08 (td, J=6,8, 15,3 Гц, 1H), 6,91 (d, J=1,8 Гц, 1H), 6,83-6,72 (m, 2H), 6,56 (d, J=8,3 Гц, 1Н), 4,18 (q, J=7,0 Гц, 2H), 3,45 (d, J=6,8 Гц, 2H), 2,44 (s, 3Н), 2,14 (s, 3Н), 1,51 (s, 6H), 1,20-1,17 (m, 3H).
Этап 5: Соединение 77-f
Трифторуксусную кислоту (141,37 мг, 1,24 ммоль, 91,80 мкл, 0,05 экв.) добавляли в раствор соединения 77-е (10,23 г, 24,80 ммоль, 1,00 экв.) в 1,4-диоксане (350,00 мл), а затем медленно по каплям добавляли N-бензил-1-метокси-N-((триметилсилил)метил)метиламин (17,66 г, 74,39 ммоль, 3,00 экв.). Смесь перемешивали при 80°С в течение 2 ч. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 77-f.
МС: m/z (ионизация электрораспылением): 546,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,27-7,20 (m, 3H), 7,17-7,15 (m, 3H), 7,10-7,07 (m, 4H), 6,58 (td, J=6,8, 10,7 Гц, 2H), 5,85 (d, J=10,5 Гц, 2H), 5,23 (s, 2H), 4,63-4,36 (m, 8H), 3,33 (br s, 3H), 2,94 (br s, 3H), 1,39-1,38 (m, 6H), 1,35-1,35 (m, 3H).
Этап 6: Соединение 77-g
Фенилхлорформиат (4,30 г, 27,49 ммоль, 3,44 мл, 5,00 экв.) медленно добавляли в раствор соединения 77-f (3,00 г, 5,50 ммоль, 1,00 экв.) в хлороформе (50,00 мл) и перемешивали смесь при 70°С в течение 4 ч. Смесь концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 77-g.
МС: m/z (ионизация электрораспылением): 576,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,61 (t, J=8,9 Гц, 2H), 7,38-7,33 (m, 2H), 7,22-7,18 (m, 3H), 7,16-7,12 (m, 2H), 6,96 (br s, 1H), 6,87 (br d, J=8,3 Гц, 1Н), 6,61 (dd, J=2,0, 8,3 Гц, 1Н), 4,25 (dq, J=2,0, 7,1 Гц, 2H), 3,94-3,88 (m, 1Н), 3,86-3,78 (m, 1Н), 3,76-3,58 (m, 2H), 3,49-3,36 (m, 1Н), 2,91-2,78 (m, 1Н), 2,72-2,67 (m, 2H), 2,52 (s, 3H), 2,19 (d, J=2,0 Гц, 3H), 1,60 (s, 6H), 1,26 (s, 3H).
Этап 7: Соединение 77-h
Гидроксид лития (1,04 г, 43,40 ммоль, 10,00 экв.) и воду (7,00 мл) добавляли в раствор соединения 77-g (2,50 г, 4,34 ммоль, 1,00 экв.) в этаноле (21,00 мл) и перемешивали смесь при 30°С в течение 1 ч. В реакционную систему по каплям добавляли насыщенный водный раствор карбоната натрия до рН=6. Реакционную систему экстрагировали этилацетатом (20 мл) и водой (20 мл). После разделения фаз собирали органические фазы. Водную фазу экстрагировали этилацетатом (3×10 мл). Органические фазы объединяли и сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток в количестве 1,5 г. Из остатка брали 0,5 г неочищенного продукта и очищали высокоэффективной жидкостной хроматографией, в результате чего получали соединение 77-h.
МС: m/z (ионизация электрораспылением): 548,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,59 (br d, J=7,8 Гц, 2Н), 7,27 (br d, J=7,3 Гц, 2Н), 7,14 (br d, J=7,8 Гц, 3Н), 7,06 (br d, J=7,3 Гц, 2Н), 6,92 (br s, 1H), 6,82 (br s, 1H), 6,69 (br s, 1H), 3,91-3,80 (m, 1H), 3,73 (br s, 1H), 3,68-3,55 (m, 2Н), 3,40-3,26 (m, 1H), 2,92-2,79 (m, 1H), 2,63 (br s, 2Н), 2,45 (s, 3Н), 2,11 (br s, 3Н), 1,51 (br s, 6H).
Этап 8: Соединение 77
Соединение 77-h (100,00 г, 182,60 ммоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 77.
МС: m/z (ионизация электрораспылением): 548,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,65 (br s, 2Н), 7,33 (br d, J=7,5 Гц, 2Н), 7,18 (br s, 3Н), 7,13 (br s, 2Н), 7,06-7,01 (m, 1H), 6,95 (br s, 1H), 6,85 (br s, 1H), 3,88-3,54 (m, 5H), 2,84 (br s, 1H), 2,68 (br s, 2Н), 2,50 (br s, 3Н), 2,15 (br s, 3Н), 1,52 (br s, 6H).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: 50% метанола (0,05% диэтаноламина) в CO2; скорость потока: 80 мл/мин; температура колонки: 40°С.
Время удерживания соединения 77: 0,564 мин (пик 1).
Пример 78: Соединение 78
Этап 1: Соединение 78-b
Карбонат цезия (345,33 г, 10,71 ммоль, 3,00 экв.) и этил-2-бром-изобутират (137,82 г, 706,58 ммоль, 2,00 экв.) добавляли в раствор соединения 78-а (49,5 г, 353,29 ммоль, 1,00 экв.) в 1,4-диоксане (500,00 мл) и N,N-диметилформамиде (200 мл) при 25°С. Смесь перемешивали при 90°С в течение 16 ч. В реакционную смесь добавляли воду (800 мл) и экстрагировали этилацетатом (300 мл × 3). Объединенную органическую фазу промывали водой (300 мл × 3) и насыщенным солевым раствором (300 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, в результате чего получали соединение 78-b.
1H-ЯМР (400 МГц, CDCl3) δ ppm 9,79 (d, J=2,0 Гц, 1Н), 7,54 (dd, J=2,0, 10,8 Гц, 1Н), 7,51-7,44 (m, 1Н), 7,51-7,44 (m, 1Н), 6,89 (t, J=8,0 Гц, 1Н), 4,19-4,15 (m, 2H), 1,61 (s, 6H), 1,18 (t, J=7,2 Гц, 3Н)
Этап 2: Соединение 78-с
В защитной атмосфере азота метоксиметилтрифенилфосфинхлорид (26,29 г, 76,70 ммоль, 1,30 экв.) и тетрагидрофуран (120 мл) добавляли в реакционную колбу, а затем партиями добавляли трет-бутоксид калия (7,94 г, 70,80 ммоль, 1,20 экв.) при 0°С. Реакционную систему перемешивали при 0°С в течение 1 ч. Затем в реакционный раствор по каплям добавляли раствор соединения 78-b (15,00 г, 59,00 ммоль, 1,00 экв.) в тетрагидрофуране (30 мл). Реакционную систему перемешивали при 0°С в течение еще 30 мин. Реакционную смесь гасили насыщенным раствором хлорида аммония (30 мл) и экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу промывали водой (100 мл × 3) и насыщенным солевым раствором (100 мл × 3), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (градиент элюирования: петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 78-с.
МС: m/z (ионизация электрораспылением): 283,0 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,36 (dd, J=2,1, 12,9 Гц, 1Н), 7,02 (d, J=8,5 Гц, 1Н), 7,05-6,75 (m, 3Н), 6,04 (d, J=6,8 Гц, 1Н), 5,63 (d, J=13,1 Гц, 1Н), 5,06 (d, J=7,0 Гц, 1Н), 4,21-4,14 (m, 2H), 3,72-3,56 (m, 3Н), 1,49 (s, 6H), 1,22 (dt, J=1,3, 7,2 Гц, 3Н)
Этап 3: Соединение 78-d
Оксалилхлорид (10,79 г, 7,44 мл, 85,02 ммоль, 2,00 экв.) медленно добавляли по каплям в раствор соединения 78-с (12 г, 42,51 ммоль, 1,00 экв.) в хлороформе (100 мл) при 15°С, а затем последовательно по каплям добавляли этанол (3,92 г, 4,96 мл, 85,02 ммоль, 2,00 экв.) и воду (1,53 г, 85,02 ммоль, 2,00 экв.). Реакционную смесь перемешивали при 15°С в течение еще 30 мин. Смесь доводили насыщенным раствором бикарбоната натрия до рН=7-8. Органическую фазу промывали водой (50 мл × 2) и насыщенным солевым раствором (50 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, в результате чего получали соединение 78-d.
1H-ЯМР (400 МГц, CDCl3) δ ppm 9,66 (t, J=2,3 Гц, 1H), 6,93-6,85 (m, 2H), 6,85-6,72 (m, 1H), 4,18 (q, J=7,1 Гц, 2H), 3,56 (d, J=2,0 Гц, 2H), 1,51 (s, 6H), 1,21 (t, J=7,2 Гц, 3Н)
Этап 4: Соединение 78-е
Соединение 44-f (19,08 г, 44,73 ммоль, 1,00 экв.) добавляли в раствор соединения 78-d (12 г, 44,73 ммоль, 1,00 экв.) в тетрагидрофуране (120 мл). Смесь перемешивали при 55°С в течение 12 ч. Смесь концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Остаток очищали колоночной флэш-хроматографией (градиент элюирования: петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 78-е.
МС: m/z (ионизация электрораспылением): 417,0 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,85-7,73 (m, 2H), 7,23-7,17 (m, 2H), 7,11-6,29 (m, 5H), 4,18 (dq, J=3,6, 7,2 Гц, 2H), 3,77 (d, J=6,3 Гц, 1H), 3,49 (d, J=6,0 Гц, 1H), 2,45 (d, J=1,5 Гц, 3Н), 1,50 (d, J=1,8 Гц, 6H), 1,21 (dt, J=4,5, 7,2 Гц, 3Н).
Этап 5: Соединение 78-f
Трифторуксусную кислоту (134,14 мг, 1,18 ммоль, 0,05 экв.) добавляли в раствор соединения 78-е (9,8 г, 23,53 ммоль, 1,00 экв.) в 1,4-диоксане (250 мл), а затем нагревали реакционную систему до 80°С Затем в реакционный раствор по каплям добавляли раствор N-бензил-1-метокси-N-((триметилсилил) метил)метиламина (16,76 г, 70,59 ммоль, 3,00 экв.) в 1,4-диоксане (50 мл). Реакционную смесь перемешивали при 80°С в течение еще 30 мин. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (градиент элюирования : петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 78-f.
МС: m/z (ионизация электрораспылением): 550,1 [М+1].
Этап 6: Соединение 78-g
Фенилхлорформиат (3,85 г, 24,57 ммоль, 3,00 экв.) добавляли в раствор соединения 78-f (4,5 г, 8,19 ммоль, 1,00 экв.) в хлороформе (50 мл) при 15°С. Реакцию в реакционной системе проводили при 70°С в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Остаток очищали колоночной флэш-хроматографией (градиент элюирования : петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 78-g.
МС: m/z (ионизация электрораспылением): 580,1 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,62 (dd, J=6,4, 8,4 Гц, 2Н), 7,31-7,26 (m, 2H), 7,19-7,04 (m, 5H), 6,89-6,73 (m, 3H), 4,18 (dq, J=1,8, 7,1 Гц, 2H), 3,93-3,83 (m, 1H), 3,78-3,54 (m, 3H), 3,41-3,22 (m, 1H), 2,98-2,77 (m, 1H), 2,75-2,55 (m, 2H), 2,46 (s, 3H), 1,50 (s, 6 H), 1,22-1,19 (m, 3H).
Этап 7: Соединение 78-h
Раствор моногидрата гидроксида лития (434,29 мг, 10,35 ммоль, 3,00 экв.) в воде (4,00 мл) медленно добавляли в раствор соединения 78-g (2,00 г, 3,45 ммоль, 1,00 экв.) в тетрагидрофуране (10 мл) и этанол (10 мл). Реакционную смесь перемешивали при 25°С в течение 6 ч. Реакционную систему доводили насыщенным водным раствором бисульфата калия до рН=5-6 и экстрагировали этилацетатом (20 мл × 3). Объединенную органическую фазу промывали водой (10 мл × 3) и насыщенным солевым раствором (10 мл × 3), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 78-h.
МС: m/z (ионизация электрораспылением): 552,1 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,66 (d, J=8,5 Гц, 2H), 7,33- 7,24 (m, 2H), 7,19-7,16 (m, 2H), 7,15-7,09 (m, 1H), 7,05 (d, J=7,5 Гц, 2H), 6,96-6,85 (m, 2H), 6,84-6,78 (m, 1H), 3,96-3,84 (m, 1H), 3,79-3,51 (m, 3H), 3,44-3,23 (m, 1H), 3,00-2,82 (m, 1H), 2,78-2,69 (m, 1H), 2,65-2,57 (m, 1H), 2,46 (s, 3H), 1,51-1,46 (m, 6 H).
Этап 8: Соединение 78
Соединение 78-h (220 мг, 398,83 ммоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 78.
МС: m/z (ионизация электрораспылением): 574,1 [М+23].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,65 (dd, J=2,5, 8,5 Гц, 2H), 7,31-7,23 (m, 2H), 7,19-7,08 (m, 3H), 7,04 (d, J=7,8 Гц, 2H), 6,96-6,70 (m, 3H), 3,99-3,81 (m, 1H), 3,77-3,47 (m, 3H), 3,39-3,18 (m, 1H), 2,99-2,81 (m, 1H), 2,76-2,52 (m, 2H), 2,45 (s, 3H), 1,44 (br s, 6 H).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1% NH3H2O EtOH]; скорость потока: 80 мл/мин; температура колонки: 40°С.
Время удерживания соединения 78: 0,651 мин (пик 1).
Пример 79: Соединение 79
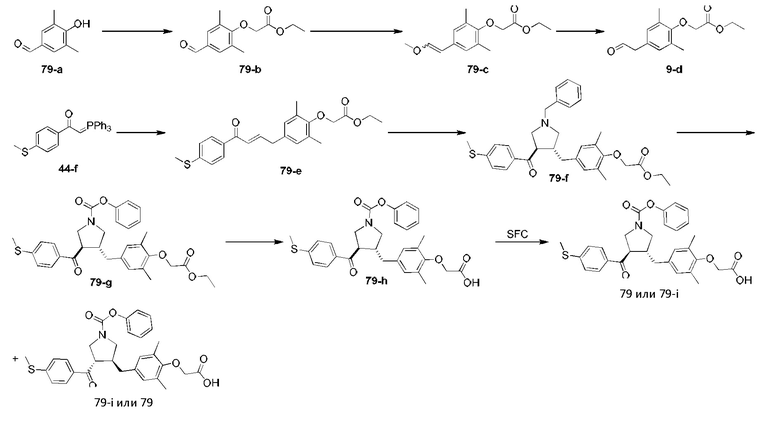
Этап 1: Соединение 79-b
Карбонат цезия (65,08 г, 10,71 ммоль, 1,50 экв.) и бромэтилацетат (33,36 г, 199,75 ммоль, 1,50 экв.) добавляли в раствор соединения 79-а (20 г, 133,17 ммоль, 1,00 экв.) в ацетоне (200,00 мл). Смесь перемешивали при 60°С в течение 1 ч. Реакционную смесь фильтровали, а фильтрат концентрировали при пониженном давлении, в результате чего получали соединение 79-b.
1H-ЯМР (400 МГц, CDCl3) δ ppm 9,91-9,85 (m, 1H), 7,56 (s, 2H), 4,46 (s, 2H), 4,34-4,27 (m, 2H), 2,37 (s, 6H), 1,33 (t, J=7,2 Гц, 3Н).
Этап 2: Соединение 79-с
Этоксид натрия (5,62 г, 82,54 ммоль, 1,30 экв.) партиями добавляли в раствор метоксиметилтрифенилфосфина хлорида (32,65 г, 95,23 ммоль, 1,50 экв.) в тетрагидрофуране (120 мл) при 0°С, а затем раствор соединения 79-b (15,00 г, 63,49 ммоль, 1,00 экв.) в тетрагидрофуране (30 мл) добавляли по каплям. Реакционную смесь перемешивали при 20°С в течение еще 12 ч, а затем гасили водой (100 мл), экстрагировали этилацетатом (200 мл × 3). Объединенную органическую фазу промывали водой (100 мл × 3) и насыщенным солевым раствором (100 мл × 3), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 79-с.
МС: m/z (ионизация электрораспылением): 265,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,25 (s, 1H), 6,97 (d, J=13,1 Гц, 1H), 6,90 (s, 1H), 6,09 (d, J=7,0 Гц, 1H), 5,73 (d, J=13,1 Гц, 1H), 5,13 (d, J=7,0 Гц, 1H), 4,40 (s, 2H), 4,32 (q, J=7,0 Гц, 2H), 3,80-3,67 (m, 3Н), 2,29 (d, J=3,5 Гц, 6Н), 1,36 (t, J=7,2 Гц, 3Н).
Этап 3: Соединение 79-d
Оксалилхлорид (3,36 г, 2,32 мл, 26,48 ммоль, 2,00 экв.) добавляли по каплям в раствор соединения 79-с (3,5 г, 13,24 ммоль, 1,00 экв.) в хлороформе (35 мл) при 0°С. После завершения добавления в реакционный раствор последовательно добавляли по каплям этанол (1,22 г, 1,54 мл, 26,48 ммоль, 2,00 экв.) и воду (477,22 мг, 26,48 ммоль, 2,00 экв.). Смесь перемешивали при 0°С в течение еще 30 мин. Смесь доводили насыщенным раствором бикарбоната натрия до рН=7-8. Органическую фазу промывали водой (200 мл × 2) и насыщенным солевым раствором (200 мл × 2), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, и таким образом получали соединение 79-d.
1H-ЯМР (400 МГц, CDCl3) δ ppm 9,64 (t, J=2,4 Гц, 1H), 6,79 (s, 2H), 4,33 (s, 2H), 4,23 (q, J=7,0 Гц, 2H), 3,50 (d, J=2,3 Гц, 2H), 2,22 (s, 6Н), 1,31 (t, J=7,2 Гц, 3Н)
Этап 4: Соединение 79-е
1-(4-(метилтио)фенил-2-(трифенилфосфоранил))этанон 44-f (5,96 г, 12,57 ммоль, 1,00 экв.) добавляли в раствор соединения 79-d (3,50 г, 13,98 ммоль, 1,00 экв.) в тетрагидрофуране (50 мл). Смесь перемешивали при 55°С в течение 12 ч. Смесь концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 79-е.
МС: m/z (ионизация электрораспылением): 399,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,88-7,73 (m, 2H), 7,24-7,18 (m, 2H), 7,11-7,03 (m, 0,5H), 6,83-6,73 (m, 2H), 6,39-6,21 (m, 0,5H), 4,33-4,29 (m, 2H), 4,27-4,18 (m, 2H), 2,45 (s, 2H), 2,23-2,19 (m, 6Н), 1,29-1,23 (m, 3H).
Этап 5: Соединение 79-f
Трифторуксусную кислоту (48,64 мг, 426,58 мкмоль, 0,05 экв.) добавляли в раствор соединения 79-е (3,4 г, 8,53 ммоль, 1,00 экв.) в 1,4-диоксане (170 мл) и нагревали до 80°С. Затем в реакционную систему медленно по каплям добавляли раствор N-бензил-1-метокси-N-((триметилсилил)метил)метиламина (5,68 г, 25,59 ммоль, 3,00 экв.) в 1,4-диоксане (30 мл). Смесь перемешивали в течение еще 30 мин. Смесь концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 79-f.
МС: m/z (ионизация электрораспылением): 532,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,55 (d, J=8,5 Гц, 2Н), 7,19-7,14 (m, 5H), 7,04 (d, J=8,5 Гц, 2H), 6,59 (s, 2Н), 4,14 (q, J=7,2 Гц, 2Н), 4,10 (s, 2Н), 3,58-3,45 (m, 3H), 2,99-2,79 (m, 2Н), 2,59-2,41 (m, 5H), 2,37 (s, 3H), 2,00 (s, 6H), 1,18 (t, J=7,2 Гц, 3H)
Этап 6: Соединение 79-g
Фенилхлорформиат (2,30 г, 14,67 ммоль, 3,00 экв.) добавляли в раствор соединения 79-f (2,60 г, 4,89 ммоль, 1,00 экв.) в хлороформе (30 мл) и осуществляли реакцию в реакционной системе при 70°С в течение 1 ч. Смесь концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Остаток очищали колоночной флэш-хроматографией (градиент элюирования : петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 79-g.
МС: m/z (ионизация электрораспылением): 562,3 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,59 (dd, J=5,5, 8,5 Гц, 2Н), 7,32-7,24 (m, 2Н), 7,16-7,09 (m, 3H), 7,09-7,03 (m, 2Н), 6,73 (d, J=4,8 Гц, 2Н), 4,29 (d, J=2,3 Гц, 2Н), 4,23 (dq, J=1,1, 7,2 Гц, 2Н), 3,91-3,80 (m, 1H), 3,78-3,72 (m, 1H), 3,61-3,55 (m, 1H), 3,40-3,25 (m, 1H), 2,93-2,76 (m, 1H), 2,60 (br d, J=7,3 Гц, 1H), 2,45 (s, 3H), 2,17 (d, J=3,0 Гц, 6H), 1,28-1,24 (m, 3H).
Этап 7: Соединение 79-h
Раствор моногидрата гидроксида лития (402,82 мг, 10,35 ммоль, 3,00 экв.) в воде (4,00 мл) добавляли в раствор соединения 79-g (1,80 г, 3,20 ммоль, 1,00 экв.) в тетрагидрофуране (10 мл) и этанол (10 мл) при 0°С. Реакционную смесь перемешивали при 25°С в течение 2 ч. Реакционную систему доводили насыщенным водным раствором бисульфата калия до рН=5-6 и экстрагировали этилацетатом (20 мл × 3). Объединенную органическую фазу промывали водой (10 мл × 3) и насыщенным солевым раствором (10 мл × 3), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 79-h.
МС: m/z (ионизация электрораспылением): 534,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,63 (dd, J=1,5, 8,5 Гц, 2Н), 7,33-7,25 (m, 2H), 7,18-7,02 (m, 5H), 6,75 (d, J=6,3 Гц, 1H), 6,79-6,72 (m, 1H), 4,34 (d, J=3,0 Гц, 2H), 3,93-3,83 (m, 1H), 3,75-3,56 (m, 3H), 3,40-3,23 (m, 1H), 2,98-2,78 (m, 1H), 2,71-2,52 (m, 2H), 2,46 (s, 3H), 2,16 (d, J=4,3 Гц, 6Н).
Этап 8: Соединение 79
Неочищенный материал 79-h (250 мг, 468,48 ммоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 79.
МС: m/z (ионизация электрораспылением): 534,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,62 (d, J=8,5 Гц, 2H), 7,30-7,22 (m, 2H), 7,16-7,08 (m, 3H), 7,04 (br d, J=8,0 Гц, 2H), 4,19 (br s, 1H), 4,27-4,07 (m, 1H), 3,93-3,78 (m, 1H), 3,74-3,48 (m, 3H), 3,37-3,17 (m, 1H), 2,95-2,74 (m, 1H), 2,68-2,46 (m, 2H), 2,43 (s, 3H), 2,09 (br d, J=5,0 Гц, 6Н).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1% NH3H2O EtOH]; скорость потока: 80 мл/мин; температура колонки: 40°С.
Время удерживания соединения 79: 0,600 мин (пик 1).
Пример 80: Соединение 80
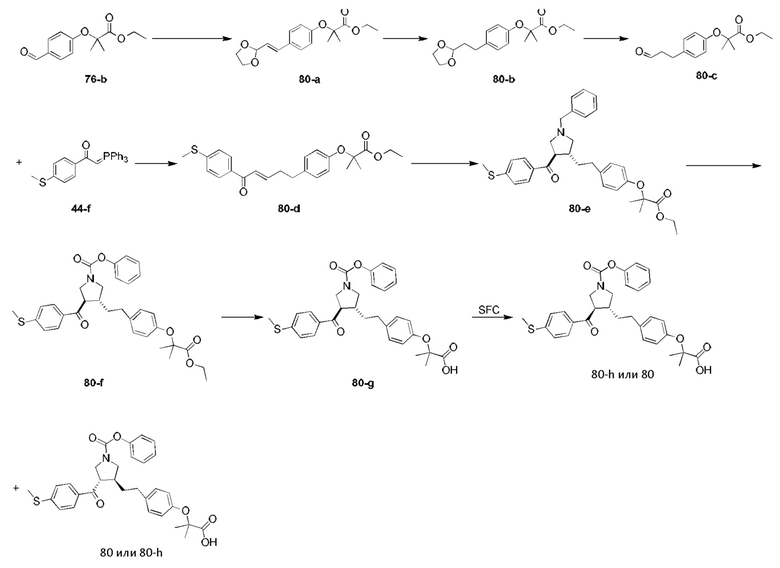
Этап 1: Соединение 80-а
Насыщенный водный раствор карбоната калия (63,49 ммоль, 100,00 мл, 1,00 экв.) добавляли в раствор соединения 76-b (15,00 г, 63,49 ммоль, 1,00 экв.), ((1,3-диоксолан-2-ил) метил)трифенилфосфина бромида (35,43 г, 82,54 ммоль, 1,30 экв.), 2-(2-метоксиэтокси)-N,N-ди[2-(2-метоксиэтокси)этил]этиламина (14,37 г, 44,44 ммоль, 0,70 экв.) в дихлорметане (100,00 мл). Реакционную систему перемешивали при 40°С в течение 20 ч. Реакционную систему разбавляли смесью дихлорметан/вода (1:1, 200 мл). Водную фазу экстрагировали дихлорметаном (300 мл × 3), и промывали объединенную органическую фазу насыщенным солевым раствором (300 мл × 1), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 80-а.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,24-7,17 (m, 2H), 6,75-6,69 (m, 2H), 6,67-6,59 (m, 1H), 6,00-5,53 (m, 1H), 5,47-5,30 (m, 1H), 4,15 (dq, J=4,0, 7,1 Гц, 2H), 4,07-3,82 (m, 4H), 1,53 (d, J=2,8 Гц, 6Н), 1,20-1,12 (m, 3H).
Этап 2: Соединение 80-b
В защитной атмосфере аргона гидроксид палладия (458,43 мг, 653,00 мкмоль, чистота 20%, 0,05 экв.) добавляли в раствор соединения 80-а (4,00 г, 13,06 ммоль, 1,00 экв.) в этаноле (50,00 мл). Реакционную систему перемешивали при 50°С в течение 5 ч в атмосфере водорода (50 фунтов/кв. дюйм). Реакционную смесь фильтровали, остаток на фильтре промывали этилацетатом (3×100 мл). Объединенную органическую фазу концентрировали при пониженном давлении, в результате чего получали соединение 80-b.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,06 (d, J=8,5 Гц, 2Н), 6,79-6,74 (m, 2H), 4,23 (q, J=7,3 Гц, 2H), 4,03-3,83 (m, 4H), 2,72-2,64 (m, 2H), 1,98-1,88 (m, 2H), 1,57 (s, 6H), 1,25 (t, J=7,2 Гц, 3Н).
Этап 3: Соединение 80-с
2 н. водный раствор HCl (18,00 мл) добавляли в раствор соединения 80-b (3,00 г, 9,73 ммоль, 1,00 экв.) в тетрагидрофуране (18,00 мл). Реакционную систему перемешивали при 70°С в течение 3 ч. В смесь добавляли воду(50 мл) и экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу промывали водой (100 мл × 3) и насыщенным солевым раствором (100 мл × 3), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 80-с.
1H-ЯМР (400 МГц, CDCl3) δ ppm 9,73 (s, 1Н), 7,00-6,96 (m, 2H), 6,72-6,68 (m, 2H), 4,16 (q, J=7,0 Гц, 2H), 2,86-2,78 (m, 2H), 2,70-2,62 (m, 2H), 1,50 (s, 6H), 1,18 (t, J=7,2 Гц, 3Н).
Этап 4: Соединение 80-d
Соединение 44-f (3,07 г, 7,19 ммоль, 1,00 экв.) добавляли в раствор соединения 80-с (1,90 г, 7,19 ммоль, 1,00 экв.) в тетрагидрофуране (20,00 мл) и перемешивали смесь при 50°С в течение 16 ч. Смесь концентрировали при пониженном давлении, и очищали остаток колоночной флэш-хроматографией (градиент элюирования : петролейный эфир : этилацетат = 100:0-90:10), в результате чего получали соединение 80-d.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,85-7,79 (m, 2H), 7,29-7,26 (m, 2H), 7,10-7,02 (m, 3H), 6,87-6,77 (m, 3Н), 4,26-4,20 (m, 2H), 2,81-2,76 (m, 2H), 2,64-2,57 (m, 2H), 2,53 (s, 3Н), 1,58 (s, 6H), 1,25 (t, J=7,2 Гц, 3Н).
Этап 5: Соединение 80-е
Трифторуксусную кислоту (23,49 мг, 206,04 мкмоль, 15,26 мкл, 0,05 экв.) добавляли в раствор соединения 80-d (1,7 г, 4,12 ммоль, 1,00 экв.) и 1,4-диоксан (100 мл) и нагревали до 80°С. Затем в реакционный раствор медленно по каплям добавляли раствор N-бензил-1-метокси-N-((триметилсилил)метил)метиламина (2,93 г, 12,36 ммоль, 3,00 экв.) в 1,4-диоксане (10 мл). После завершения добавления реакционный раствор перемешивали при 80°С в течение 1 ч. Смесь концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 80-е.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,87 (d, J=8,8 Гц, 2H), 7,34-7,29 (m, 5H), 7,26-7,24 (m, 2H), 6,96 (d, J=8,5 Гц, 2H), 6,72 (d, J=8,5 Гц, 2H), 4,13 (q, J=7,0 Гц, 2H), 3,71-3,53 (m, 4H), 3,05-2,98 (m, 1H), 2,92-2,78 (m, 2H), 2,69-2,61 (m, 1H), 2,52 (s, 3H), 2,48-2,40 (m, 2H), 1,81-1,71 (m, 2H), 1,56 (s, 6H), 1,26-1,22 (m, 3H).
Этап 6: Соединение 80-f
Фенилхлорформиат (2,30 г, 14,66 ммоль, 1,84 мл, 5,00 экв.) добавляли в раствор соединения 80-е (1,60 г, 2,93 ммоль, 1,00 экв.) в хлороформе (20,00 мл) и перемешивали при 70°С в течение 16 ч. Смесь концентрировали при пониженном давлении и очищали неочищенный продукт колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 80-f.
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,88 (br dd, J=3,4, 8,4 Гц, 2H), 7,43-7,28 (m, 4H), 7,25-7,11 (m, 3H), 7,06-6,98 (m, 2H), 6,82-6,74 (m, 2H), 4,28-4,19 (m, 2H), 4,03-3,54 (m, 4H), 3,43-3,27 (m, 1H), 2,89-2,71 (m, 1H), 2,67-2,51 (m, 5H), 1,93-1,65 (m, 2H), 1,60-1,54 (m, 6 Н), 1,30-1,26 (m, 3H).
Этап 7: Соединение 80-g
Раствор гидроксида лития (270,40 мг, 11,29 ммоль, 5,00 экв.) в воде (2,00 мл) добавляли в раствор соединения 80-f (1,30 г, 2,26 ммоль, 1,00 экв.) в этаноле (6,00 мл) и тетрагидрофуране (6,00 мл). Смесь перемешивали при 40°С в течение 4 ч. Реакционную систему доводили насыщенным водным раствором бисульфата калия до рН=5-6, а затем экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным солевым раствором (20 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток. Неочищенный продукт очищали высокоэффективной жидкостной хроматографией с получением соединения 80-g.
МС: m/z (ионизация электрораспылением): 548,1 [М+1].
Этап 8: Соединение 80
Соединение 80-g (400,00 мг, 0,73 ммоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 80.
МС: m/z (ионизация электрораспылением): 548,1 [М+1].
1H-ЯМР (400 МГц, CDCl3) δ ppm 7,87 (dd, J=5,4, 8,4 Гц, 2H), 7,39-7,27 (m, 4H), 7,23-7,10 (m, 3Н), 7,08-7,02 (m, 2H), 6,86 (d, J=8,3 Гц, 2H), 4,03-3,53 (m, 4H), 3,42-3,15 (m, 1H), 2,87-2,66 (m, 1H), 2,64-2,56 (m, 2H), 2,53 (d, J=1,8 Гц, 3Н), 1,93-1,62 (m, 2H), 1,57 (d, J=9,3 Гц, 6H).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1%NH3H2O EtOH]; скорость потока: 80 мл/мин; температура колонки: 40°С.
Время удерживания соединения 80: 2,054 мин (пик 1).
Пример 81: Соединение 81
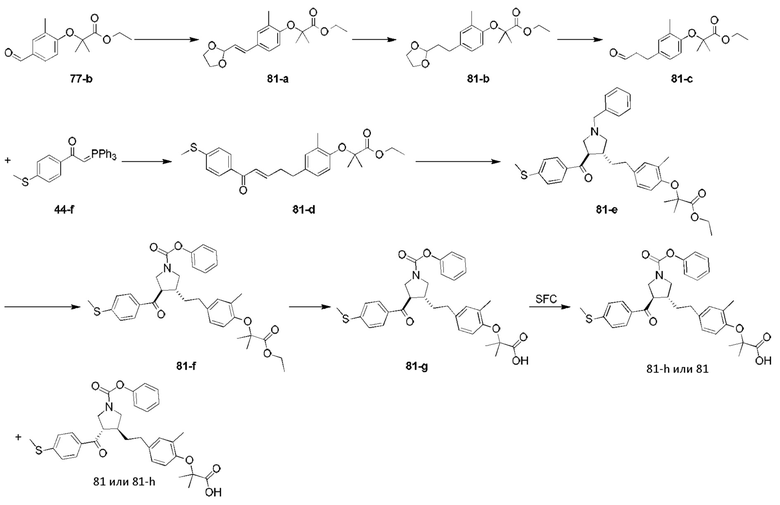
Этап 1: Соединение 81-а
Насыщенный водный раствор карбоната калия (79,91 ммоль, 100,00 мл, 1,00 экв.) добавляли в раствор соединения 77-b (20,00 г, 79,91 ммоль, 1,00 экв.), ((1,3-диоксолан-2-ил)метил)трифенилфосфина бромида (44,59 г, 103,88 ммоль, 1,30 экв.) и 2-(2-метоксиэтокси)-N,N-ди[2-(2-метоксиэтокси)этил]этиламина (8,09 г, 55,94 ммоль, 17,91 мл, 0,70 экв.) в дихлорметане (100,00 мл). Реакционную систему перемешивали при 40°С в течение 20 ч. Реакционную систему разбавляли (дихлорметан/вода = 1:1, 200 мл). Водную фазу экстрагировали дихлорметаном (300 мл × 3) и промывали объединенную органическую фазу насыщенным солевым раствором (300 мл × 1), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 81-а.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,24-7,08 (m, 2Н), 6,73-6,57 (m, 2Н), 6,11-5,35 (m, 2Н), 4,30-4,22 (m, 2Н), 4,07-3,91 (m, 4Н), 2,22 (d, J=5,0 Гц, 3Н), 1,60 (d, J=2,8 Гц, 6H), 1,29-1,24 (m, 3Н).
Этап 2: Соединение 81-b
В защитной атмосфере аргона, гидроксид палладия (657,53 мг, 936,39 мкмоль, чистота 20%, 0,05 экв.) добавляли в раствор соединения 81-а (6,00 г, 18,73 ммоль, 1,00 экв.) в этаноле (50,00 мл). Реакционную систему перемешивали при 50°С в течение 5 ч в атмосфере водорода (50 фунтов/кв.дюйм). Реакционную смесь фильтровали, остаток на фильтре промывали этилацетатом (3×100 мл). Объединенную органическую фазу концентрировали при пониженном давлении, в результате чего получали соединение 81-b.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 6,98 (d, J=1,8 Гц, 1H), 6,87 (dd, J=2,0, 8,3 Гц, 1H), 6,59 (d, J=8,3 Гц, 1H), 4,87 (t, J=4,8 Гц, 1H), 4,25 (q, J=7,1 Гц, 2H), 4,02-3,84 (m, 4Н), 2,68-2,61 (m, 2Н), 2,21 (s, 3Н), 1,96-1,90 (m, 2Н), 1,57 (s, 6Н), 1,26 (t, J=7,2 Гц, 3Н).
Этап 3: Соединение 81-с
Водный раствор HCl (2 М, 24,00 мл) добавляли в раствор соединения 81-b (4,50 г, 13,96 ммоль, 1,00 экв.) в тетрагидрофуране (24,00 мл) при 25°С. Смесь перемешивали при 70°С в течение 2 ч. В реакционную систему добавляли воду (50 мл) и экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу промывали водой (100 мл) и насыщенным солевым раствором (100 мл), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 81-с.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 9,81 (t, J=1,4 Гц, 1H), 6,97 (d, J=2,0 Гц, 1H), 6,85 (dd, J=2,0, 8,3 Гц, 1H), 6,59 (d, J=8,3 Гц, 1H), 4,28-4,23 (m, 2Н), 2,89-2,82 (m, 2Н), 2,76-2,70 (m, 2Н), 2,21 (s, 3Н), 1,58 (s, 6Н), 1,26 (t, J=7,2 Гц, 3Н).
Этап 4: Соединение 81-d
Соединение 44-f (4,75 г, 11,14 ммоль, 1,00 экв.) добавляли в раствор соединения 81-с (3,10 г, 11,14 ммоль, 1,00 экв.) в тетрагидрофуране (30,00 мл), а затем перемешивали при 50°С в течение 16 ч. Смесь концентрировали при пониженном давлении, и очищали остаток колоночной флэш-хроматографией (петролейный эфир: этилацетат =100: 0-90: 10), в результате чего получали соединение 81-d.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,74 (d, J=8,3 Гц, 2H), 7,21-7,17 (m, 2H), 7,02-6,89 (m, 2H), 6,82-6,72 (m, 2H), 6,53 (d, J=8,3 Гц, 1H), 4,16 (q, J=7,1 Гц, 2H), 2,70-2,62 (m, 2H), 2,51 (q, J=7,3 Гц, 2H), 2,45 (s, 3Н), 2,14 (s, 3Н), 1,50 (s, 6H), 1,20-1,16 (m, 3Н).
Этап 5: Соединение 81-е
Трифторуксусную кислоту (40,09 мг, 351,50 мкмоль, 26,03 мкл, 0,05 экв.) добавляли в раствор соединения 81-d (3,00 г, 7,03 ммоль, 1,00 экв.) в 1,4-диоксане (200 мл) и нагревали до 80°С. Затем медленно по каплям добавляли раствор N-бензил-1-метокси-N-((триметилсилил)метил)метиламина (5,01 г, 21,09 ммоль, 3,00 экв.) в 1,4-диоксане (20 мл) и перемешивали при 80°С в течение еще 1 ч. Реакционную смесь концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 81-е.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,81 (d, J=8,5 Гц, 2H), 7,27-7,18 (m, 7Н), 6,82 (d, J=1,8 Гц, 1H), 6,71 (dd, J=2,0, 8,3 Гц, 1H), 6,49 (d, J=8,3 Гц, 1H), 4,19 (q, J=7,0 Гц, 2H), 3,73-3,53 (m, 4Н), 3,00-2,93 (m, 1H), 2,85-2,70 (m, 2Н), 2,60 (dd, J=7,0, 9,3 Гц, 1H), 2,50-2,39 (m, 5Н), 2,11 (s, 3Н), 1,75-1,67 (m, 2Н), 1,50 (s, 6Н), 1,26-1,20 (m, 3Н).
Этап 6: Соединение 81-f
Фенилхлорформиат (5,03 г, 32,15 ммоль, 4,02 мл, 5,00 экв.) добавляли в раствор соединения 81-е (3,60 г, 6,43 ммоль, 1,00 экв.) в хлороформе (40,00 мл) и перемешивали при 70°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир: этилацетат =100: 0-80: 20), в результате чего получали соединение 81-f.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,87 (dd, J=3,3, 8,5 Гц, 2H), 7,40-7,33 (m, 2Н), 7,31-7,27 (m, 2Н), 7,24-7,11 (m, 3Н), 6,94-6,89 (m, 1H), 6,84-6,78 (m, 1H), 6,57 (dd, J=2,8, 8,3 Гц, 1H), 4,29-4,20 (m, 2Н), 4,03-3,55 (m, 4Н), 3,46-3,27 (m, 1H), 2,89-2,69 (m, 1H), 2,62-2,47 (m, 5Н), 2,19 (s, 3Н), 1,91-1,64 (m, 2Н), 1,56 (s, 6Н), 1,29-1,26 (m, 3Н).
Этап 7: Соединение 81-g
Раствор гидроксида лития (467,03 мг, 19,50 ммоль, 5,00 экв.) в воде (3,00 мл) добавляли в раствор соединения 81-f (2,30 г, 3,90 ммоль, 1,00 экв.) в этаноле (9,00 мл) и тетрагидрофуран (9,00 мл), а затем перемешивали при 40°С в течение 4 ч. Реакционную систему доводили насыщенным водным раствором бисульфата калия до рН=5-6, а затем смесь экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным солевым раствором (20 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток. Неочищенный продукт очищали высокоэффективной жидкостной хроматографией с получением соединения 81-g. МС: m/z (ионизация электрораспылением): 562,1 [М+1].
Этап 8: Соединение 81
Соединение 81-g (400,00 мг, 712,14 мкмоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 81. МС: m/z (ионизация электрораспылением): 562,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,91-7,85 (m, 2Н), 7,40-7,28 (m, 4Н), 7,24-7,08 (m, 3Н), 6,99-6,73 (m, 3Н), 4,01-2,98 (m, 6Н), 2,87-2,61 (m, 2Н), 2,54 (d, J=1,5 Гц, 3Н), 2,21 (d, J=3,5 Гц, 3Н), 1,94-1,67 (m, 2Н), 1,65-1,54 (m, 6Н).
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1%NH3H2O EtOH]; скорость потока: 60 мл/мин; температура колонки: 40°С.
Время удерживания соединения 81: 1,187 мин (пик 1).
Пример 82: Соединение 82
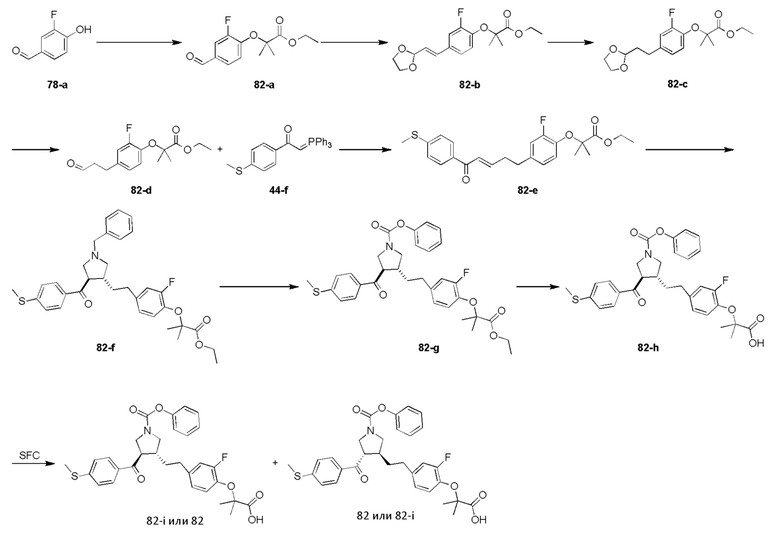
Этап 1: Соединение 82-а
Насыщенный раствор карбоната калия (83,00 мл) добавляли в раствор соединения 78-а (40,00 г, 157,33 ммоль, 1,00 экв.), ((1,3-диоксолан-2-ил)метил)трифенилфосфина бромида (33,77 г, 78,67 ммоль, 0,50 экв.) и 2-(2-метоксиэтокси)-N,N-ди[2-(2-метоксиэтокси)этил]этиламина (17,81 г, 55,07 ммоль, 17,63 мл, 0,35 экв.) в дихлорметане (83,00 мл). Смесь перемешивали при 40°С в течение 20 ч. Реакционную систему разбавляли (дихлорметан/вода = 1:1, 500 мл). Водную фазу экстрагировали дихлорметаном (100 мл × 3) и промывали объединенную органическую фазу насыщенным солевым раствором (100 мл × 1), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-80:20), в результате чего получали соединение 82-а.
МС: m/z (ионизация электрораспылением): 325,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ 7,04-7,11 (m, 1H), 6,93-6,98 (m, 1H), 6,84 (q, J=8,28 Гц, 1H), 6,54-6,63 (m, 1H), 5,57-6,01 (m, 1H), 5,29-5,44 (m, 1H), 4,17 (dq, J=3,51, 7,11 Гц, 2Н), 3,95-4,01 (m, 2Н), 3,83-3,89 (m, 2Н), 1,52 (d, J=3,51 Гц, 6Н), 1,19-1,24 (m, 3Н)
Этап 2: Соединение 82-b
В защитной атмосфере аргона гидроксид палладия (1,30 г, 1,85 ммоль, чистота 20%, 0,10 экв.) добавляли в раствор соединения 82-а (6,00 г, 18,50 ммоль, 1,00 экв.) в этаноле (30,00 мл). Смесь перемешивали при 50°С в течение 16 ч в атмосфере водорода (50 фунтов/кв. дюйм). Реакционную смесь фильтровали, остаток на фильтре промывали этилацетатом (3×10 мл). Объединенный фильтрат концентрировали при пониженном давлении, в результате чего получали соединение 82-b.
МС: m/z (ионизация электрораспылением): 349,1 [М+23].
1Н-ЯМР (400 МГц, CDCl3) δ 6,72-6,89 (m, 3Н), 4,80 (t, J=4,64 Гц, 1H), 4,17 (q, J=7,28 Гц, 2Н), 3,88-3,93 (m, 2Н), 3,76-3,82 (m, 2Н), 2,56-2,66 (m, 2Н), 1,81-1,91 (m, 2Н), 1,43-1,53 (m, 6Н), 1,21 (t, J=7,03 Гц, 3Н).
Этап 3: Соединение 82-с
Водный раствор HCl (2 М, 28,34 мл, 3,70 экв.) добавляли в раствор соединения 82-b (5,00 г, 15,32 ммоль, 1,00 экв.) в тетрагидрофуране (28,34 мл). Смесь перемешивали при 70°С в течение 3 ч. В смесь добавляли воду (50 мл) и экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу промывали водой (100 мл) и насыщенным солевым раствором (100 мл), сушили при помощи безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, в результате чего получали соединение 82-с.
1Н-ЯМР (400 МГц, CDCl3) δ 9,73 (t, J=1,13 Гц, 1H), 6,79-6,87 (m, 2Н), 6,73-6,77 (m, 1H), 4,17 (q, J=7,11 Гц, 2H), 2,79-2,84 (m, 2Н), 2,66-2,72 (m, 2Н), 1,49 (s, 6Н), 1,19-1,24 (m, 3Н).
Этап 4: Соединение 82-d
Соединение 44-f (6,65 г, 15,59 ммоль, 1,00 экв.) добавляли в раствор соединения 82-с (4,40 г, 15,59 ммоль, 1,00 экв.) в тетрагидрофуране (40,00 мл), а затем перемешивали при 50°С в течение 5 ч. Реакционную смесь концентрировали при пониженном давлении, и очищали остаток колоночной флэш-хроматографией (петролейный эфир: этилацетат =100: 0-60: 40), в результате чего получали соединение 82-d.
МС: m/z (ионизация электрораспылением): 431,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ 7,70-7,77 (m, 2Н), 7,15-7,22 (m, 2Н), 6,96 (td, J=6,78, 15,31 Гц, 1H), 6,72-6,88 (m, 4Н), 4,17 (q, J=7,28 Гц, 2Н), 2,66-2,72 (m, 2Н), 2,48-2,57 (m, 2Н), 2,45 (s, 3Н), 1,49 (s, 6Н), 1,18-1,23 (m, 3Н).
Этап 5: Соединение 82-е
Трифторуксусную кислоту (58,26 мг, 511,00 мкмоль, 37,83 мкл, 0,05 экв.) добавляли в раствор соединения 82-d (4,40 г, 10,22 ммоль, 1,00 экв.) в 1,4-диоксане (120 мл) и нагревали до 80°С. Затем в реакционную смесь по каплям добавляли раствор N-бензил-1-метокси-N-((триметилсилил)метил)метиламина (7,28 г, 30,66 ммоль, 7,84 мл, 3,00 экв.), а затем перемешивали в течение еще 30 мин. Смесь концентрировали при пониженном давлении, и очищали остаток колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 82-е.
МС: m/z (ионизация электрораспылением): 564,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ 7,72 (d, J=8,53 Гц, 2Н), 7,01-7,17 (m, 7Н), 6,53-6,76 (m, 3Н), 4,09 (q, J=7,19 Гц, 2Н), 3,37-3,46 (m, 2Н), 2,83-2,91 (m, 1H), 2,62-2,74 (m, 2Н), 2,49 (dd, J=7,15, 9,16 Гц, 1H), 2,36-2,40 (m, 1H), 2,37 (s, 2Н), 2,22-2,34 (m, 2Н), 1,58-1,66 (m, 2Н), 1,36-1,42 (m, 6Н), 1,14 (t, J=7,15 Гц, 3Н).
Этап 6: Соединение 82-f
Фенилхлорформиат (1,25 г, 7,98 ммоль, 999,54 мкл, 3,00 экв.) добавляли в раствор соединения 82-е (1,50 г, 2,66 ммоль, 1,00 экв.) в хлороформе (10,00 мл), а затем перемешивали при 70°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении и очищали остаток колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-70:30), в результате чего получали соединение 82-f.
МС: m/z (ионизация электрораспылением): 594,2 [М+1].
Этап 7: Соединение 82-g
Соединение 82-f (1,10 г, 1,85 ммоль, 1,00 экв.), гидроксид лития (443,08 мг, 18,50 ммоль, 10,00 экв.) и этанол (10,00 мл), воду (5,00 мл) добавляли в круглодонную колбу при 20°С, а затем перемешивали при 30°С в течение 3 ч. В реакционную систему по каплям добавляли насыщенный водный раствор бисульфата калия до рН=5. В смесь добавляли воду (10 мл) и экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли и промывали насыщенным солевым раствором (10 мл × 3), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией, в результате чего получали соединение 82-g.
МС: m/z (ионизация электрораспылением): 566,2 [М+1].
Этап 8: Соединение 82
Соединение 82-g (500,00 мг, 883,94 мкмоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 82.
МС: m/z (ионизация электрораспылением): 566,0 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ 7,74-7,83 (m, 2Н), 6,98-7,32 (m, 8Н), 6,63-6,90 (m, 3Н), 3,61-3,95 (m, 3Н), 3,39-3,60 (m, 1H), 3,04-3,32 (m, 1H), 2,56-2,79 (m, 1H), 2,37-2,53 (m, 5Н), 1,49-1,82 (m, 2Н), 1,39 (br d, J=7,53 Гц, 6H).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм внутр. диам., 3 мкм; подвижная фаза: 40% изо-пропанола (0,05% диэтаноламина) в СО2; скорость потока: 2,8 мл/мин; температура колонки: 40°С. Время удерживания соединения 82: 2,772 мин (пик 1).
Пример 83: Соединение 83
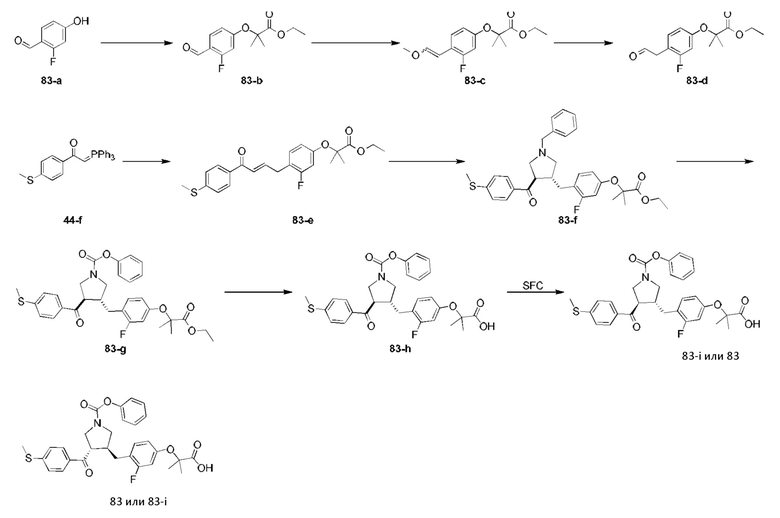
Этап 1: Соединение 83-b
Карбонат цезия (52,32 г, 160,59 ммоль, 1,50 экв.) и этил-2-бром-изобутират (52,21 г, 267,65 ммоль, 39,25 мл, 2,50 экв.) добавляли в раствор соединения 83-а (15,00 г, 107,06 ммоль, 1,00 экв.) в N,N-диметилформамиде (150,00 мл). Смесь перемешивали при 90°С в течение 2 ч. Реакционную смесь экстрагировали этилацетатом (200 мл) и водой (300 мл). После разделения фаз органические фазы собирали, водную фазу экстрагировали этилацетатом (100 мл × 3), и промывали объединенную органическую фазу 1 н. раствором гидроксида натрия (100 мл) и насыщенным водным раствором бисульфата калия (50 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали соединение 83-b.
МС: m/z (ионизация электрораспылением): 254,9 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 10,20 (s, 1H), 7,77 (t, J=8,5 Гц, 1H), 6,66 (dd, J=2,5, 8,8 Гц, 1H), 6,55 (dd, J=2,3, 12,3 Гц, 1H), 4,24 (q, J=7,0 Гц, 2H), 1,67 (s, 6Н), 1,23 (t, J=7,2 Гц, 3Н)
Этап 2: Соединение 83-с
Трет-бутоксид калия (10,29 г, 91,74 ммоль, 1,50 экв.) партиями добавляли в раствор метоксиметилтрифенилфосфина хлорида (27,26 г, 79,51 ммоль, 1,30 экв.) в тетрагидрофуране (200,00 мл) при 20°С. Реакцию смешанного раствора проводили в течение 1 ч и добавляли соединение 83-b (15,55 г, 61,16 ммоль, 1,00 экв.). Смесь перемешивали при 20°С в течение 1 ч. В реакционную систему добавляли борогидрид натрия (2,00 г, 52,87 ммоль, 0,86 экв.), а затем реакционную систему гасили добавлением воды(50 мл). Затем реакционную систему экстрагировали этилацетатом (100 мл) и водой (100 мл). После разделения фаз органические фазы собирали и экстрагировали водную фазу этилацетатом (3 × 50 мл). Органические фазы объединяли и сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-70:30), в результате чего получали соединение 83-с.
МС: m/z (ионизация электрораспылением): 282,9 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,95-7,32 (m, 1H), 7,15-7,03 (m, 1H), 6,61-6,15 (m, 2Н), 5,81-5,36 (m, 1H), 4,24 (dq, J=2,1, 7,2 Гц, 2H), 3,79-3,67 (m, 3Н), 1,59 (s, 6Н), 1,26 (dt, J=1,5, 7,2 Гц, 3Н).
Этап 3: Соединение 83-d
Соединение 83-с (8,84 г, 31,31 ммоль, 1,00 экв.) добавляли в сухую реакционную колбу, а затем добавляли хлороформ (100,00 мл). Затем в реакционную систему медленно добавляли оксалилхлорид (7,95 г, 62,62 ммоль, 5,48 мл, 2,00 экв.) в защитной атмосфере азота при 0°С, а затем в этот смешанный раствор добавляли этанол (2,88 г, 62,62 ммоль, 3,65 мл, 2,00 экв.), воду (1,13 г, 62,62 ммоль, 1,13 мл, 2,00 экв.). Смешанный раствор перемешивали 0°С в течение 1 ч. В реакционную систему по каплям добавляли насыщенный водный раствор карбоната натрия до рН=7-8. Реакционную систему экстрагировали дихлорметаном (100 мл) и водой (100 мл). После разделения фаз органические фазы собирали и экстрагировали водную фазу этилацетатом (3×50 мл). Органические фазы объединяли и последовательно промывали водой (3×50 мл) и насыщенным солевым раствором (3×50 мл), сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали соединение 83-d.
1Н-ЯМР (400 МГц, CDCl3) δ ppm 9,71 (q, J=1,8 Гц, 1H), 7,72-7,42 (m, 1H), 7,07-7,00 (m, 1H), 6,65-6,61 (m, 1H), 4,24 (q, J=7,1 Гц, 2H) 3,65 (s, 2Н), 1,61 (s, 6 Н), 1,25 (t, J=7,2 Гц, 3Н)
Этап 4: Соединение 83-е
Соединение 83-d (6,86 г, 25,57 ммоль, 1,00 экв.), соединение 44-f (10,91 г, 25,57 ммоль, 1,00 экв.) добавляли в высушенную реакционную колбу, а затем добавляли тетрагидрофуран (100,00 мл). Смесь перемешивали при 50°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100: 0-60: 40) с получением соединения 83-е.
МС: m/z (ионизация электрораспылением): 417,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,85-7,72 (m, 2Н), 7,29-7,16 (m, 3Н), 7,08-6,94 (m, 1H), 6,76 (d, J=15,3 Гц, 1H), 6,57-6,47 (m, 2Н), 4,19-4,14 (m, 2Н), 3,82-3,77 (m, 1H), 3,50 (d, J=6,5 Гц, 1H), 2,46-2,43 (m, 3Н), 1,54-1,51 (m, 6Н), 1,21-1,16 (m, 3Н).
Этап 5: Соединение 83-f
Соединение 83-е (3,69 г, 8,86 ммоль, 1,00 экв.) добавляли в высушенную реакционную колбу, а затем добавляли 1,4-диоксан (150,00 мл). Затем в реакционную систему добавляли трифторуксусную кислоту (50,51 мг, 443,00 мкмоль, 0,05 экв.), а затем в смешанный раствор добавляли N-бензил-1-метокси-N-((триметилсилил)метил метиламин (6,31 г, 26,58 ммоль, 3,00 экв.), растворенный в 1,4-диоксане (30,00 мл), со скоростью 1 капля в две секудны. Смешанный раствор перемешивали при 100°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 83-f.
МС: m/z (ионизация электрораспылением): 550,1 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,65 (d, J=8,5 Гц, 2H), 7,25 (br d, J=2,0 Гц, 2H), 7,19-7,11 (m, 5H), 6,93-6,84 (m, 1H), 6,41-6,35 (m, 2H), 4,05 (q, J=7,0 Гц, 2H), 3,61-3,52 (m, 3Н), 3,04-2,95 (m, 2H), 2,71-2,59 (m, 3Н), 2,55-2,46 (m, 2H), 2,43 (s, 3Н), 1,44 (d, J=4,0 Гц, 6H), 1,20-1,17 (m, 3Н).
Этап 6: Соединение 83-g
Соединение 83-f (1,85 г, 3,37 ммоль, 1,00 экв.) и хлороформ (30,00 мл) добавляли в высушенную реакционную колбу. Затем медленно добавляли фенилхлорформиат (2,64 г, 16,85 ммоль, 2,11 мл, 5,00 экв.). Смешанный раствор перемешивали при 70°С в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир: этилацетат =100: 0-85: 15), в результате чего получали соединение 83-g.
МС: m/z (ионизация электрораспылением): 580,2 [М+1].
1Н-ЯМР (400 МГц, CDCl3) δ ppm 7,68 (t, J=7,8 Гц, 2H), 7,39-7,33 (m, 2Н), 7,25-7,13 (m, 5Н), 7,04 (t, J=8,8 Гц, 1H), 6,61-6,53 (m, 2Н), 4,24 (dq, J=2,0, 7,1 Гц, 2H), 3,99-3,90 (m, 1H), 3,85-3,61 (m, 3Н), 3,51-3,36 (m, 1H), 3,01-2,86 (m, 1H), 2,79 (br d, J=5,8 Гц, 2H), 2,53 (s, 3H), 1,60 (d, J=4,3 Гц, 6H), 1,26 (s, 3H)
Этап 7: Соединение 83-h
Соединение 83-g (328,00 мг, 565,83 мкмоль, 1,00 экв.) добавляли в высушенную реакционную колбу, а затем добавляли этанол (15,00 мл). В реакционную систему добавляли гидроксид лития (135,52 мг, 5,66 ммоль, 10,00 экв.) и воду (5,00 мл) добавляли и перемешивали смешанный раствор при 20°С в течение 16 ч. В реакционную систему по каплям добавляли насыщенный водный раствор карбоната натрия до рН=6. Реакционную смесь экстрагировали этилацетатом (20 мл) и водой (20 мл). После разделения фаз собирали органические фазы и экстрагировали водную фазу этилацетатом (3×10 мл). Органические фазы объединяли и сушили при помощи безводного сульфата натрия и концентрировали при пониженном давлении, в результате чего получали остаток. Неочищенный продукт очищали колоночной флэш-хроматографией (петролейный эфир : этилацетат = 100:0-60:40), в результате чего получали соединение 83-h.
МС: m/z (ионизация электрораспылением): 552,1 [М+1].
Этап 8: Соединение 83
Соединение 83-h (195,00 мг, 353,50 мкмоль, 1,00 экв.) выделяли хиральной сверхкритической хроматографией, в результате чего получали соединение 83.
МС: m/z (ионизация электрораспылением): 552,1 [М+1].
1ЯМР (400 МГц, CDCl3) δ ppm 7,66 (br dd, J=5,4, 8,2 Гц, 2H), 7,39-7,32 (m, 2H), 7,25-7,18 (m, 3Н), 7,13 (br d, J=7,8 Гц, 2H), 7,08-7,00 (m, 1H), 6,63 (br s, 2H), 4,00-3,62 (m, 4H), 3,51-3,32 (m, 1H), 2,97-2,86 (m, 1H), 2,81-2,69 (m, 2H), 2,51 (s, 3H), 1,57 (br s, 6H)
Условия хирального разделения: хиральная колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: 40% метанола (0,05% диэтаноламина) в СО2; скорость потока: 80 мл/мин; температура колонки: 40°С.
Время удерживания соединения 83: 0,546 мин (пик 1).
Экспериментальный пример 1: Оценка In Vitro
Исследование In Vitro. Основы активности агониста PPAR
Клеточное исследование ядерного рецептора гормонов (NHR)
Исследование PathHunter взаимодействия белков и ядерной транслокации NHR использовали для определения возможности активации клеточных ядерных рецепторов гормонов в унифицированных экспериментах без визуализации. Указанная технология, которую называют Комплементацией фрагментированных ферментов (англ. Enzyme Fragmentation Complementation (ETC)), была разработана в DiscoverX.
Исследование белка NHR основано на обнаружении белок-белкового взаимодействия между белком NHR стандартной длины в активированном состоянии и ядерного гибридного белка, содержащего участок пептидного коактиватора стероидных рецепторов (SRCP) и одну или более стандартных активных последовательностей LXXLL.
В NHR вводят метку в компоненте ProLinkTM исследовательской системы EFC, в то же время проводят слияние участка SRCP и компонента-акцептора фермента (ЕА) и экспрессию в ядро. При связывании с лигандом происходит транслокация NHR в ядро, в результате чего образуется участок SRCP, в котором обеспечен комплементарный эффект, что приведет к активации одного эквивалента галактозидазы (-Gal), а также появлению сигнала хемилюминесценции. Преимущества такого подхода включают сокращение продолжительности инкубации соединений, прямое исследование мишеней NHR, использование последовательностей человеческого NHR со стандартной длиной и возможность выбора некоторых новых типов соединений на основании данных о нарушении белок-белкового взаимодействия.
В исследовании NHR NT определяют транслокацию NHR между цитоплазмой и ядром. В рецептор вводят метку в компоненте ProLinkTM исследовательской системы EFC, в то же время проводят слияние ЕА с ядерными последовательностями, что, тем самым, ограничивает экспрессию ЕА в ядрах. Транслокация NHR в ядро приводит к комплементации с ЕА, что обеспечивает активацию одного эквивалента галактозидазы, а также выработку сигнала хемилюминесценции.
Обработка клеток:
1. Штамм клеток PathHunter NHR выращивали из замороженной клеточной линии согласно стандартной процедуре.
2. Клетки высевали в белый 384-луночный планшет в количестве 20 мкл/лунка и инкубировали при 37°С в течение надлежащего периода времени, после чего проводили исследование. Питательная среда содержала активированный уголь/декстран, из нее была отфильтрована и удалена сыворотка для уменьшения уровня экспрессии гормонов.
Процедура эксперимента в режиме агониста:
1. Для исследования активности агониста требовалась инкубация клеток совместно с соединением для индуцирования ответа.
2. Соединение вводили в буферный раствор для получения маточного раствора, который необходимо разбавлять 5х.
3. 5 мкл разбавленного 5х раствора соединения добавляли к клеткам и затем инкубировали клетки при 37°С (или при комнатной температуре) в течение 3-16 часов. Необходимо убедиться, что конечная концентрация среды составляет 1%.
Процедура эксперимента в режиме ингибитора:
1. Для исследования активности ингибитора требовалась предварительная инкубация клеток совместно с антагонистом, а затем воздействие агониста в концентрации ECso.
2. Соединение вводили в буферный раствор для получения маточного раствора, который необходимо разбавлять 5х.
3. 5 мкл разбавленного 5х раствора соединения добавляли к клеткам и затем инкубировали клетки при 37°С (или при комнатной температуре) в течение 60 минут. Необходимо убедиться, что конечная концентрация среды составляет 1%.
4. В клетки добавляли 5 мкл агониста в концентрации EC80, разбавленного 6х в буферном растворе, и инкубировали при 37°С (или при комнатной температуре) в течение 3-16 часов.
Исследование сигналов:
1. Экспериментальные сигналы получали при помощи 12,5 мкл или 15 мкл (50% (об./об.)) смеси реагента для исследования PathHunter, которую добавляли за один раз и затем инкубировали при комнатной температуре в течение 1 часа.
2. Сигналы хемилюминесценции, полученные в микропланшете, детектировали на оборудовании PerkinElmer Envision Instrument.
Анализ данных:
1. Активность соединений анализировали при помощи программного обеспечения для анализа данных CIBS Data Analysis Software (ChemInnovation, CA).
2. В экспериментах в режиме агониста активность в процентах вычисляли согласно следующему уравнению:
Активность, %=100% × (среднее в ОЕЛ для исследуемого соединения - средний фон в ОЕЛ для среды)/(максимальное среднее для контроля с лигандом - средний фон в ОЕЛ для среды)
3. В экспериментах в режиме антагониста активность в процентах вычисляли согласно следующему уравнению:
Ингибирование, % = 100% × (1-(среднее в ОЕЛ для исследуемого соединения - средний фон в ОЕЛ для среды)/(среднее в ОЕЛ для EC80 контрольного соединения - средний фон в ОЕЛ для среды))
4. Следует отметить, что ответ лиганда приводит к уменьшению активности рецептора (обратные агонисты, имеющие постоянно активную мишень). Активность указанных обратных агонистов вычисляют согласно следующему уравнению:
Активность обратного агониста, % = 100% × ((средний фон в ОЕЛ для среды - среднее в ОЕЛ для исследуемого соединения)/(средний фон в ОЕЛ для среды - максимальное среднее в ОЕЛ для контроля с лигандом))
Результаты эксперимента показаны в таблице 1:
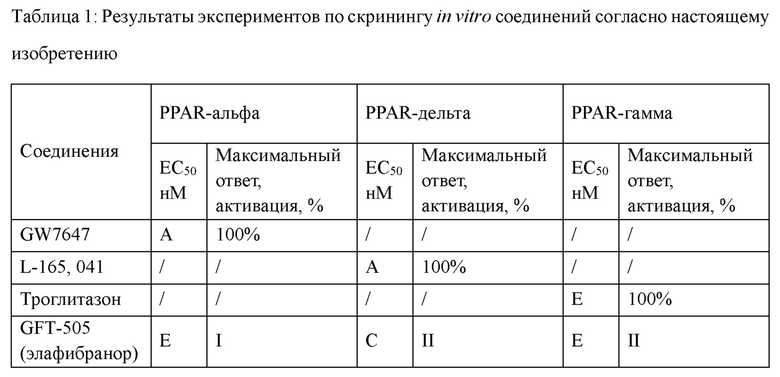
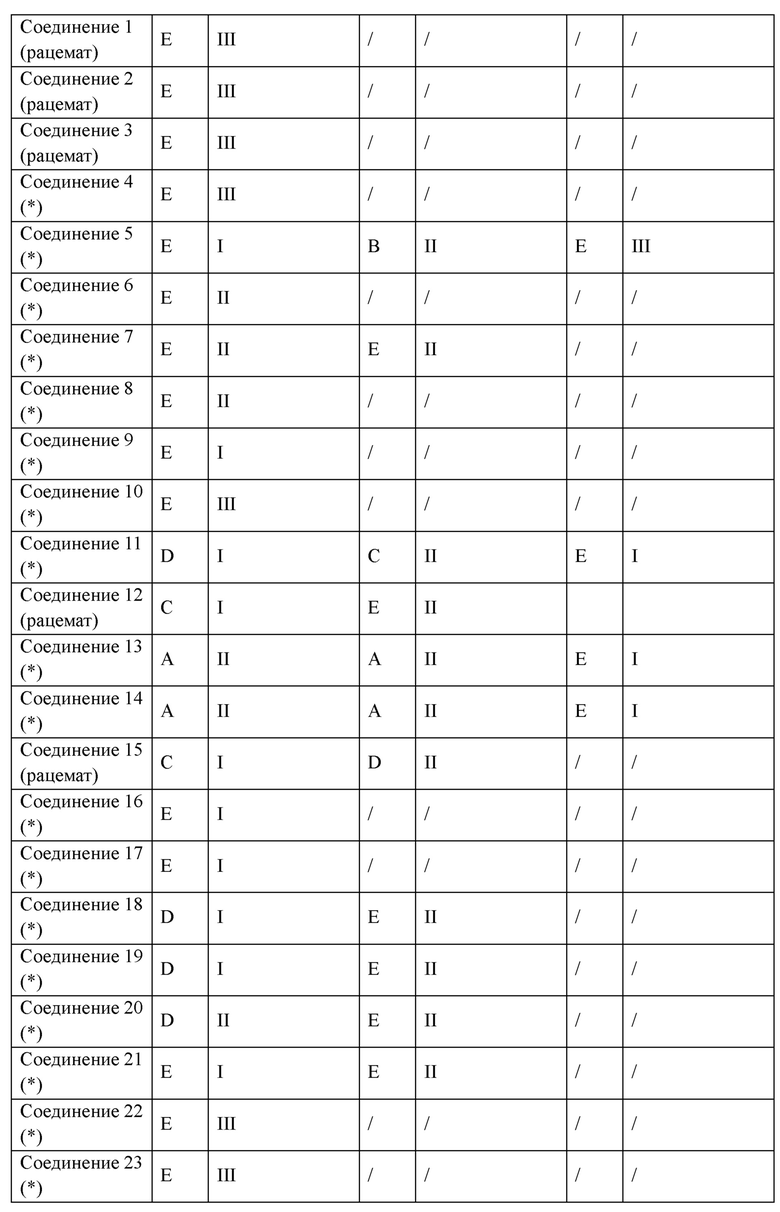
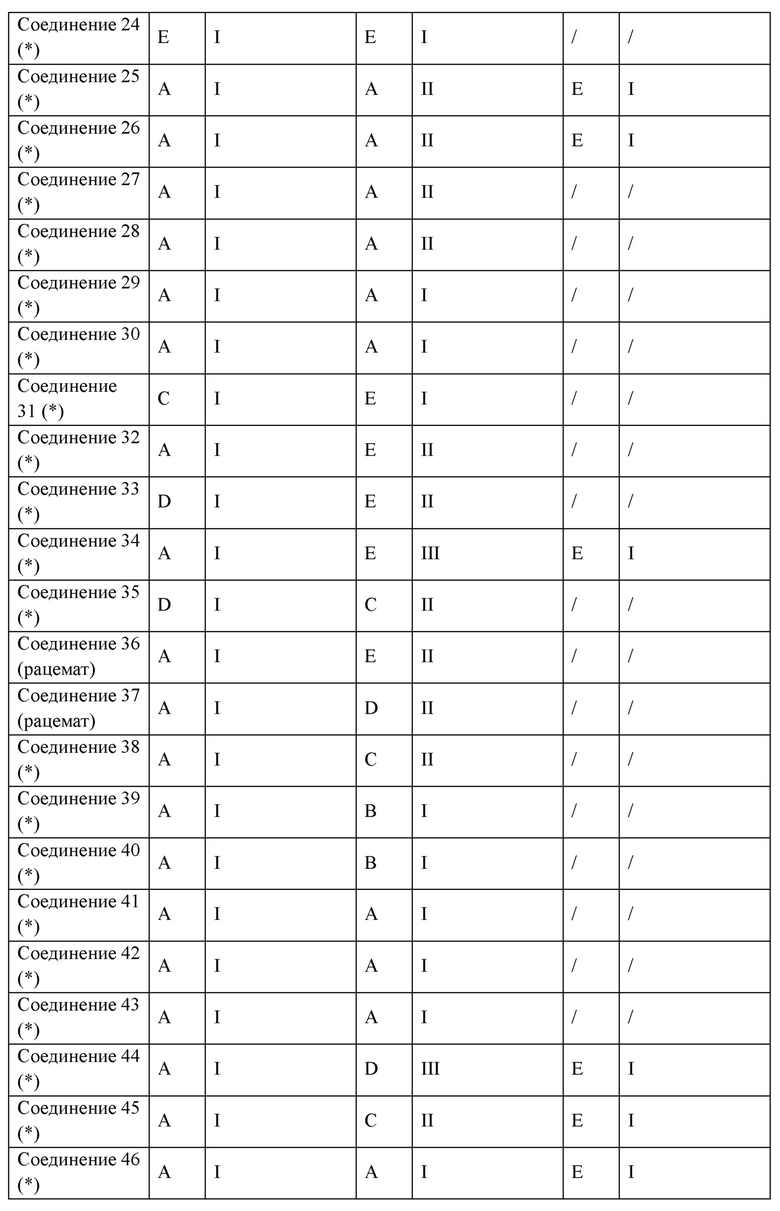
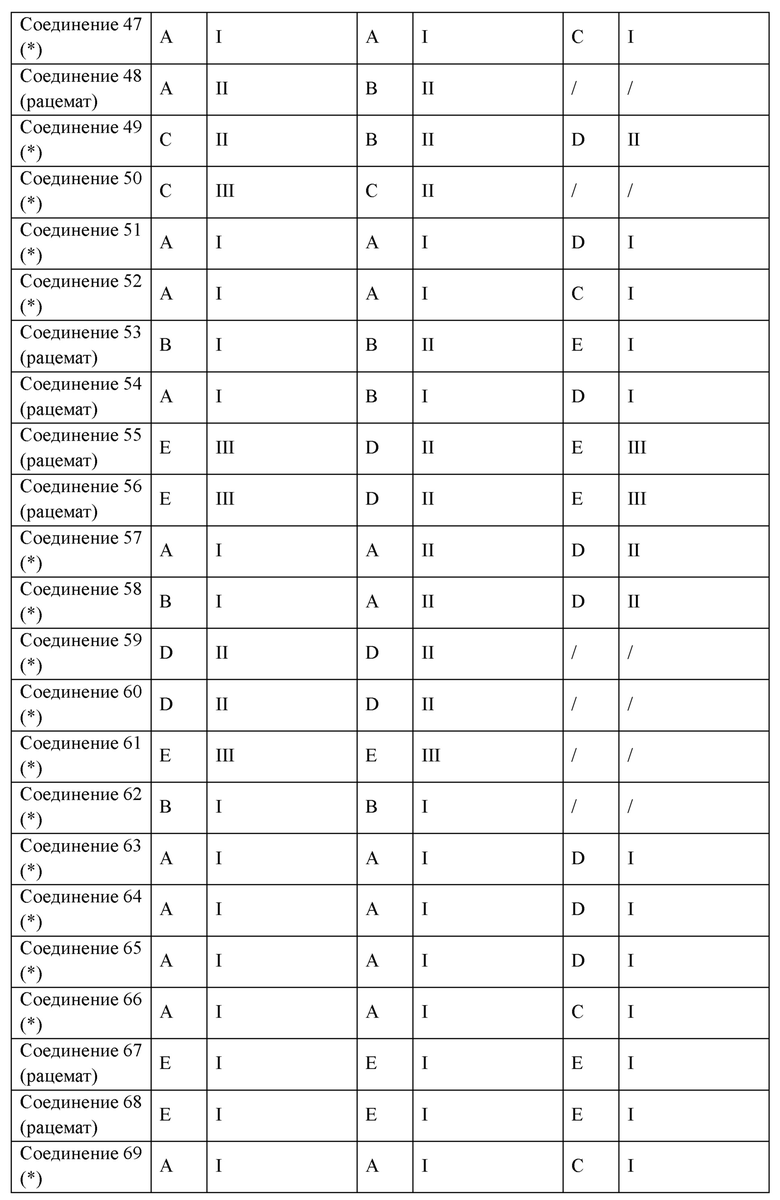
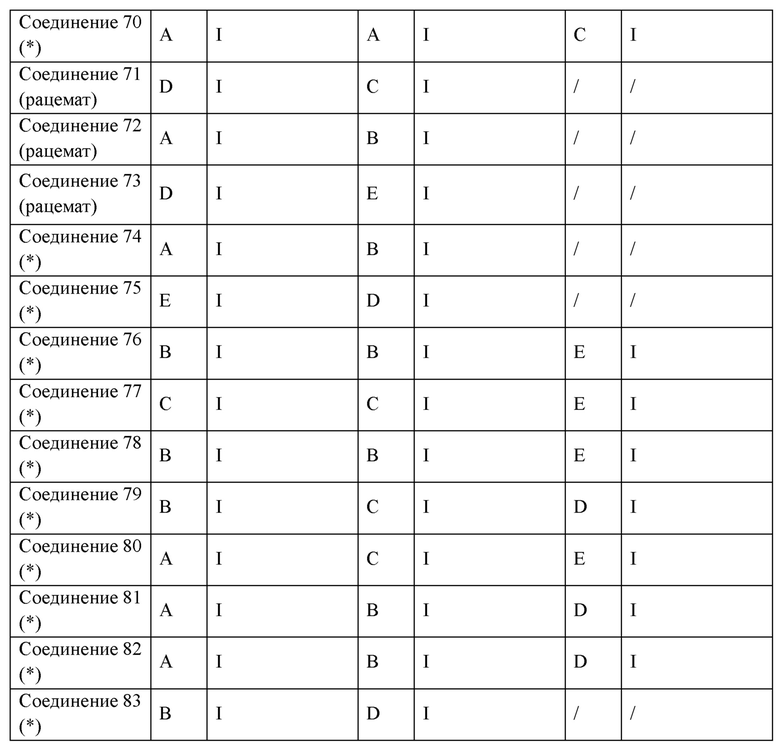
Примечание 1: Значения максимального ответа при активации, определенные на платформе in vitro, для известного агониста PPARα GW7647, агониста PPARδ L-165,041 и агониста PPARγ троглитазона принимали за 100%. Значения максимального ответа для других соединений сравнивали с максимальным ответом при активации для известных агонистов и получали соответствующие значения максимального ответа при активации. В общем случае, соединение, имеющее значение максимального ответа при активации более 80% рассматривали как полный агонист, соединение, имеющее значение максимального ответа при активации более 50% и менее 80% рассматривали как частичный агонист, и соединение, имеющее значения максимального ответа при активации менее 50% рассматривали как недостаточно эффективный агонист.
Примечание 2: А ≤ 100 нМ; 100 нМ < В ≤ 150 нМ; 150 нМ < С ≤ 200 нМ; 200 нМ < D ≤ 250 нМ; Е > 250 нМ.
Примечание 3: 100% ≥ I ≥ 80%; 80% ≥ II ≥ 50%; III 50%.
Примечание 4: «*» относится к оптически чистым соединениям.
Примечание 5: «рацемат» относится к транс-рацемическим соединениям.
Вывод: соединения согласно настоящему изобретению в значительной степени активируют рецепторы PPAR-альфа и -дельта и селективно активируют рецептор PPAR-гамма.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| Производное индола, используемое в качестве ингибитора CRTH2 | 2017 |
|
RU2756270C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ИХ ПРИМЕНЕНИЕ ПРОТИВ МИКОБАКТЕРИЙ | 2015 |
|
RU2664587C1 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
| АГОНИСТ S1P1 И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2754845C2 |
| ПИРИДОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, ВЫПОЛНЯЮЩИЕ ФУНКЦИЮ ДВОЙНЫХ ИНГИБИТОРОВ mTORC 1/2 | 2018 |
|
RU2771201C2 |
Изобретение относится к области органической химии, а именно к соединению формулы (I) 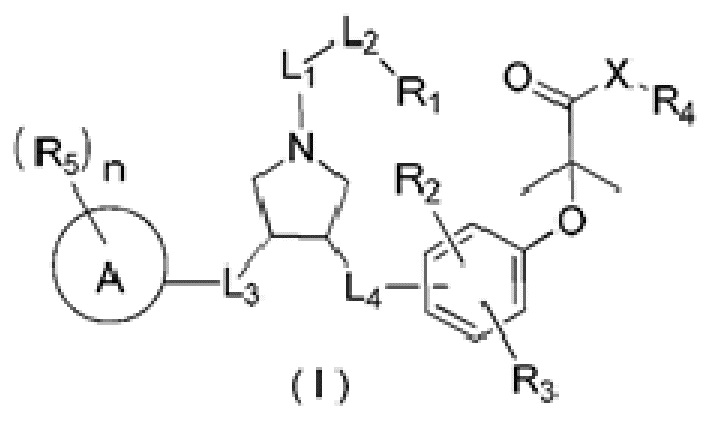 и его фармацевтически приемлемой соли. В соединении формулы (I): R1 выбран из Н, NH2 или С1-6 алкила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, фенила, 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R; R2, R3 независимо выбраны из Н, галогена или С1-3 алкила, необязательно замещенного 1, 2 или 3 R; X выбран из О или S; если X выбран из О или S, то R4 выбран из Н или С1-3 алкила, необязательно замещенного 1, 2 или 3 R; R5 выбран из Н, галогена или СН3, CH3-S(=O)-, CH3-S(=O)2-, CH3S, каждый из которых необязательно замещен 1, 2 или 3 R; n выбран из 0, 1, 2 или 3; кольцо А выбрано из фенила или 5-6-членного гетероарила; L1 выбран из простой связи, -С(=O)-, -О-, -NH-, -С(=O)O-, -C(=O)NH-, -S(=O)2-, -S(=O)-, -(CRR)1-3-; L2 выбран из простой связи, -(CRR)1-3-, -О-, -S-; L3 выбран из -(CRR)-, -С(=O)-; L4 выбран из простой связи, -(CRR)1-3-; R выбран из Н, F, Cl, Br, I или С1-3 алкила,
и его фармацевтически приемлемой соли. В соединении формулы (I): R1 выбран из Н, NH2 или С1-6 алкила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, фенила, 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R; R2, R3 независимо выбраны из Н, галогена или С1-3 алкила, необязательно замещенного 1, 2 или 3 R; X выбран из О или S; если X выбран из О или S, то R4 выбран из Н или С1-3 алкила, необязательно замещенного 1, 2 или 3 R; R5 выбран из Н, галогена или СН3, CH3-S(=O)-, CH3-S(=O)2-, CH3S, каждый из которых необязательно замещен 1, 2 или 3 R; n выбран из 0, 1, 2 или 3; кольцо А выбрано из фенила или 5-6-членного гетероарила; L1 выбран из простой связи, -С(=O)-, -О-, -NH-, -С(=O)O-, -C(=O)NH-, -S(=O)2-, -S(=O)-, -(CRR)1-3-; L2 выбран из простой связи, -(CRR)1-3-, -О-, -S-; L3 выбран из -(CRR)-, -С(=O)-; L4 выбран из простой связи, -(CRR)1-3-; R выбран из Н, F, Cl, Br, I или С1-3 алкила,  каждый из которых необязательно замещен 1, 2 или 3 R'; R' выбран из F, Cl, Br, I; «гетеро-» относится к гетероатому или гетероатомной группе и выбран из -NH-, -О-, -S-; в любом из случаев, определенных выше, количество гетероатомов или гетероатомных групп независимо выбрано из 1, 2 или 3. Также предложены конкретные соединения, фармацевтическая композиция и применение соединения формулы (I). Технический результат: получены новые соединения, производные пирролидина, в качестве агонистов PPAR и их применение для лечения заболеваний, связанных с путями, ассоциированными с рецептором PPAR. 5 н. и 23 з.п. ф-лы, 1 табл., 84 пр.
каждый из которых необязательно замещен 1, 2 или 3 R'; R' выбран из F, Cl, Br, I; «гетеро-» относится к гетероатому или гетероатомной группе и выбран из -NH-, -О-, -S-; в любом из случаев, определенных выше, количество гетероатомов или гетероатомных групп независимо выбрано из 1, 2 или 3. Также предложены конкретные соединения, фармацевтическая композиция и применение соединения формулы (I). Технический результат: получены новые соединения, производные пирролидина, в качестве агонистов PPAR и их применение для лечения заболеваний, связанных с путями, ассоциированными с рецептором PPAR. 5 н. и 23 з.п. ф-лы, 1 табл., 84 пр.
1. Соединение формулы (I)
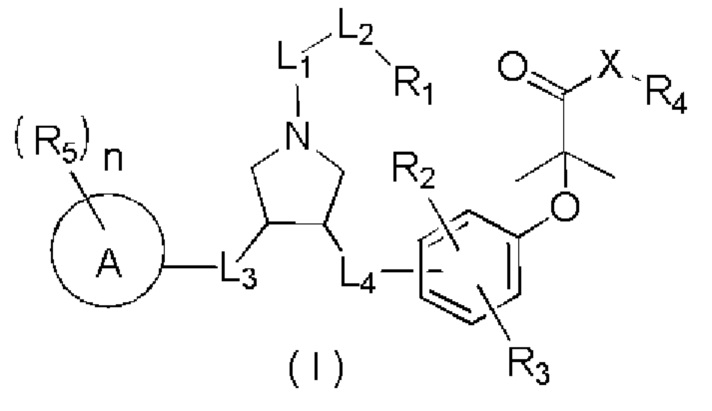
или его фармацевтически приемлемая соль,
где
R1 выбран из Н, NH2 или С1-6 алкила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, фенила, 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R;
R2, R3 независимо выбраны из Н, галогена или С1-3 алкила, необязательно замещенного 1, 2 или 3 R;
X выбран из О или S;
если X выбран из О или S, то R4 выбран из Н или С1-3 алкила, необязательно замещенного 1, 2 или 3 R;
R5 выбран из Н, галогена или СН3, CH3-S(=O)-, CH3-S(=O)2-, CH3S, каждый из которых необязательно замещен 1, 2 или 3 R;
n выбран из 0, 1, 2 или 3;
кольцо А выбрано из фенила или 5-6-членного гетероарила;
L1 выбран из простой связи, -С(=O)-, -О-, -NH-, -С(=O)O-, -C(=O)NH-, -S(=O)2-, -S(=O)-, -(CRR)1-3-;
L2 выбран из простой связи, -(CRR)1-3-, -О-, -S-;
L3 выбран из -(CRR)-, -С(=O)-;
L4 выбран из простой связи, -(CRR)1-3-;
R выбран из Н, F, Cl, Br, I или С1-3 алкила, 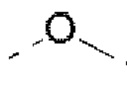 каждый из которых необязательно замещен 1, 2 или 3 R';
каждый из которых необязательно замещен 1, 2 или 3 R';
R' выбран из F, Cl, Br, I;
«гетеро-» относится к гетероатому или гетероатомной группе и выбран из -NH-, -О-, -S-;
в любом из случаев, определенных выше, количество гетероатомов или гетероатомных групп независимо выбрано из 1, 2 или 3.
2. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что R выбран из Н, F, Cl, Br, I или C1-3 алкила, .
3. Соединение или его фармацевтически приемлемая соль по п. 2, отличающееся тем, что R выбран из Н, F, Cl, Br, I, Me,  .
.
4. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что R1 выбран из Н, NH2 или C1-6 алкила, циклопентила, пирролидинила, фенила, пиразолила, пиридила, циклогексила, каждый из которых необязательно замещен 1, 2 или 3 R.
5. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что R1 выбран из Н, NH2 или Me, Et, 
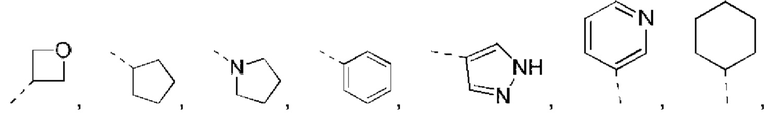 каждый из которых необязательно замещен 1, 2 или 3 R.
каждый из которых необязательно замещен 1, 2 или 3 R.
6. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что R1 выбран из Н, NH2, Me, Et, 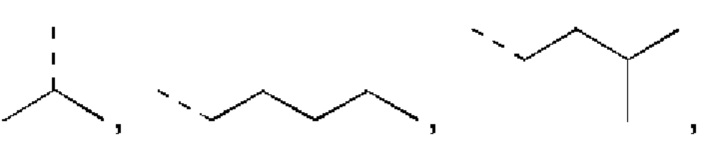
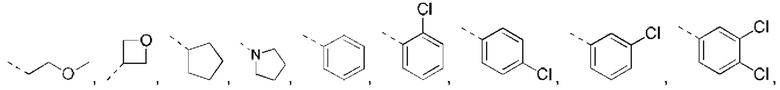
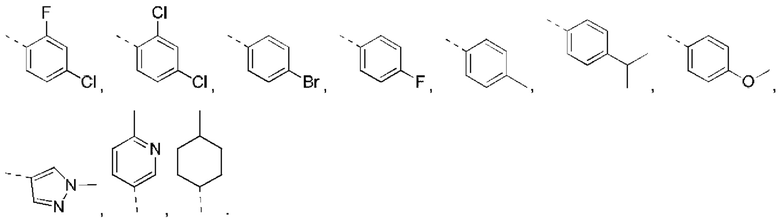
7. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что R2, R3 независимо выбраны из Н, F, Cl, Br, I или Me, Et, каждый из которых необязательно замещен 1, 2 или 3 R.
8. Соединение или его фармацевтически приемлемая соль по п. 7, отличающееся тем, что R2, R3 независимо выбраны из Н, F, Cl, Br, I, Me, Et.
9. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что если X выбран из О или S, то R4 выбран из Н или Me, Et,  , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
10. Соединение или его фармацевтически приемлемая соль по п. 9, отличающееся тем, что если X выбран из О или S, то R4 выбран из Н, Me, Et,  .
.
11. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что R5 выбран из Н, F, Cl, Br, I или Me, 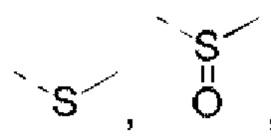 , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
12. Соединение или его фармацевтически приемлемая соль по п. 11, отличающееся тем, что R5 выбран из Н, F, Cl, Br, I, 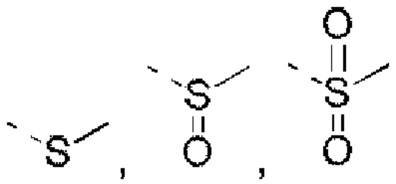 .
.
13. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что структурное звено 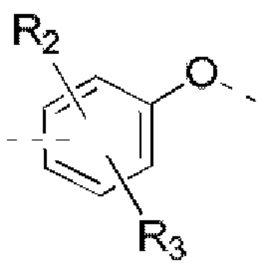 выбрано из:
выбрано из: 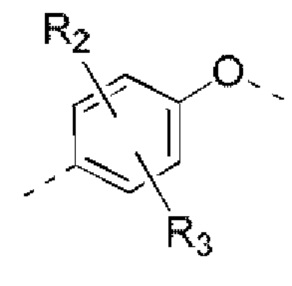 ,
, 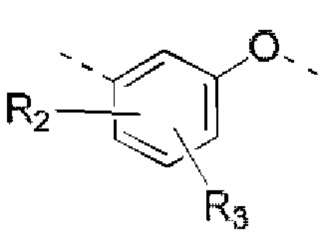 .
.
14. Соединение или его фармацевтически приемлемая соль по п. 13, отличающееся тем, что структурное звено 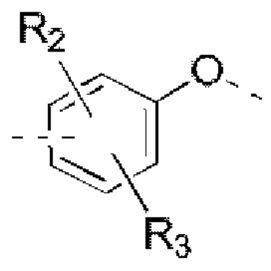 выбрано из:
выбрано из: 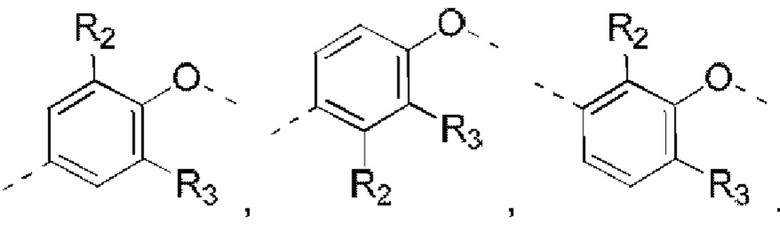
15. Соединение или его фармацевтически приемлемая соль по п. 14, отличающееся тем, что структурное звено 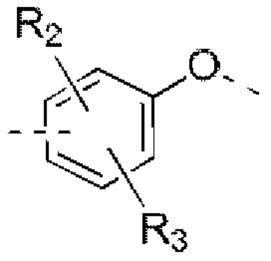 выбрано из:
выбрано из: 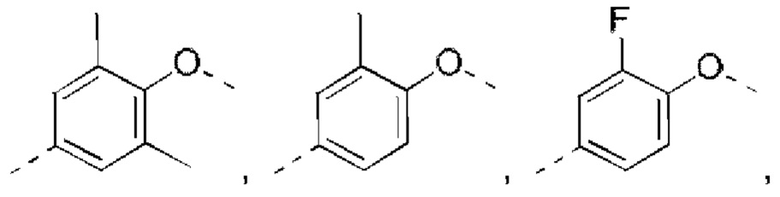
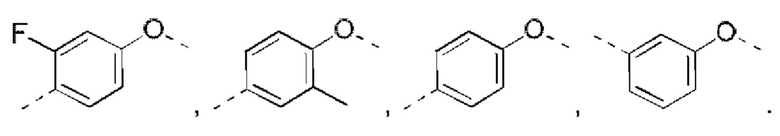
16. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что указанное кольцо А выбрано из: фенила, пиридила, пиримидила, пиразинила, пиридазинила, тиенила, пиразолила, имидазолила, оксазолила, тиазолила, изоксазолила, изотиазолила.
17. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что структурное звено 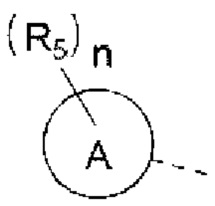 выбрано из:
выбрано из: 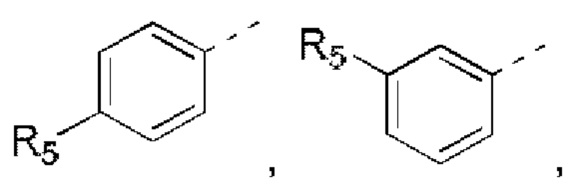
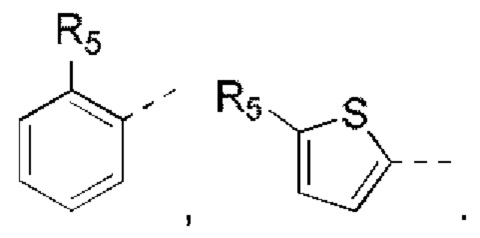
18. Соединение или его фармацевтически приемлемая соль по п. 17, отличающееся тем, что структурное звено 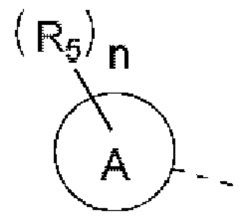 выбрано из
выбрано из 
19. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что L2 выбран из простой связи, -СН2-, -СН2СН2-, -О-, -S-.
20. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что L3 выбран из -СН2-, -С(=O)-.
21. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что структурное звено -L1-L2- выбрано из: простой связи, -СН2-, -С(=O)-, -S(=O)2, -С(=O)O-, -C(=O)NH-, -C(=O)-CH2-, -С(=O)O-СН2-, -СН2СН2О-.
22. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что L4 выбран из простой связи, -СН2-, -СН2СН2-.
23. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-12 и 19-22, выбранное из:
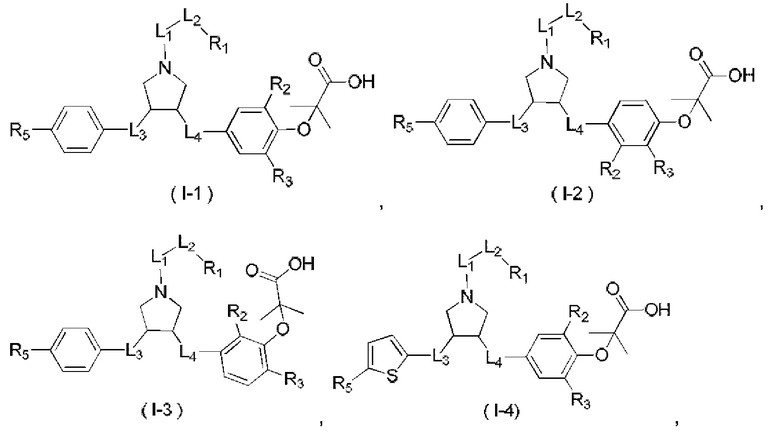
где R1, R2, R3, R5, L1, L2, L3, L4 такие, как определено по пп. 1-22.
24. Соединение, представленное формулой, выбранной из:
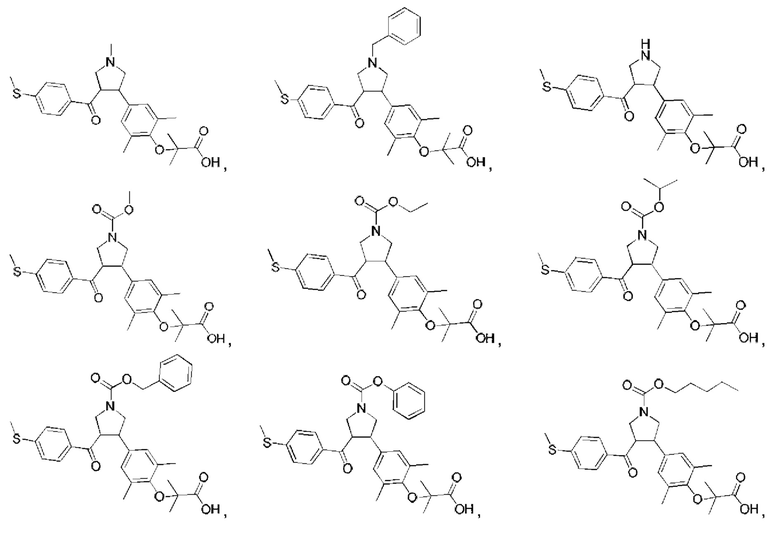
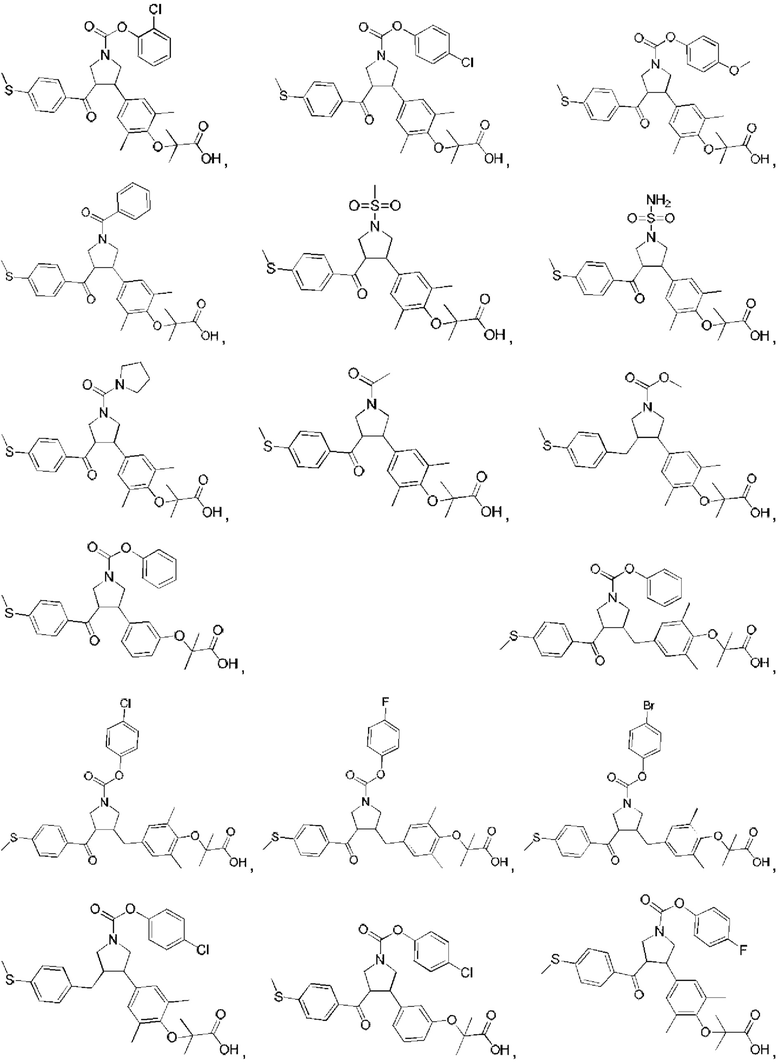
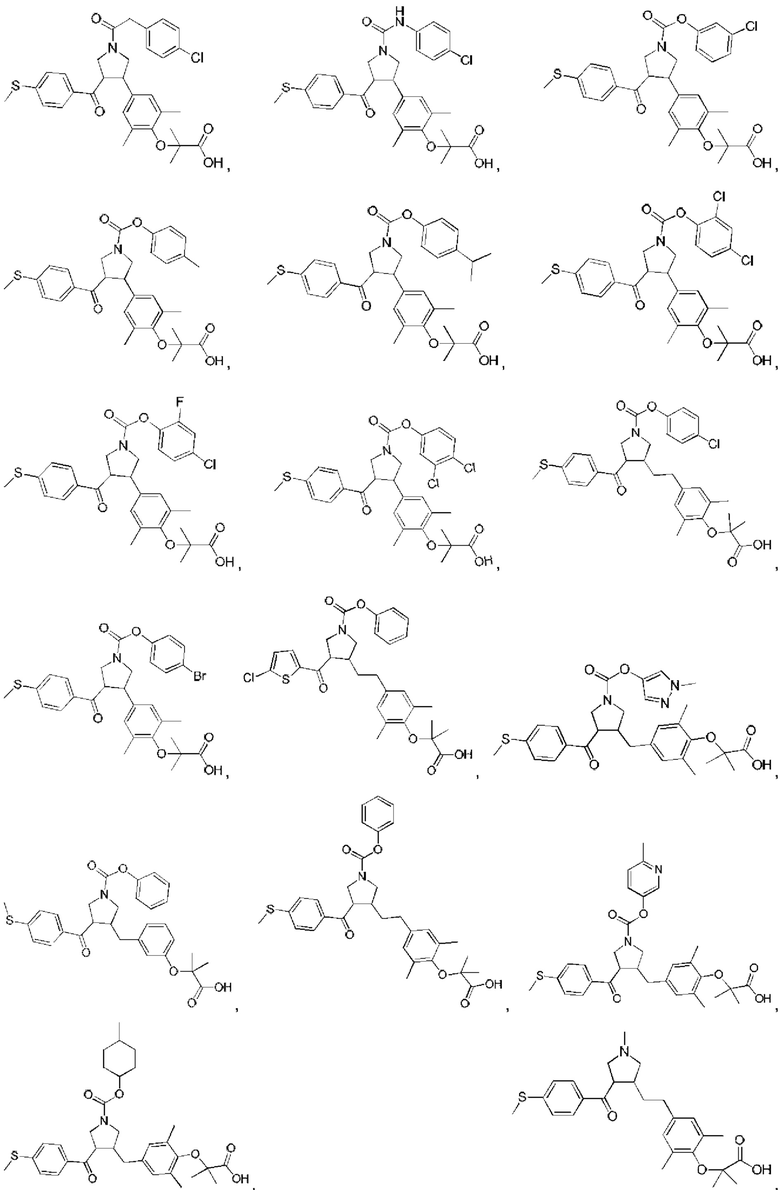
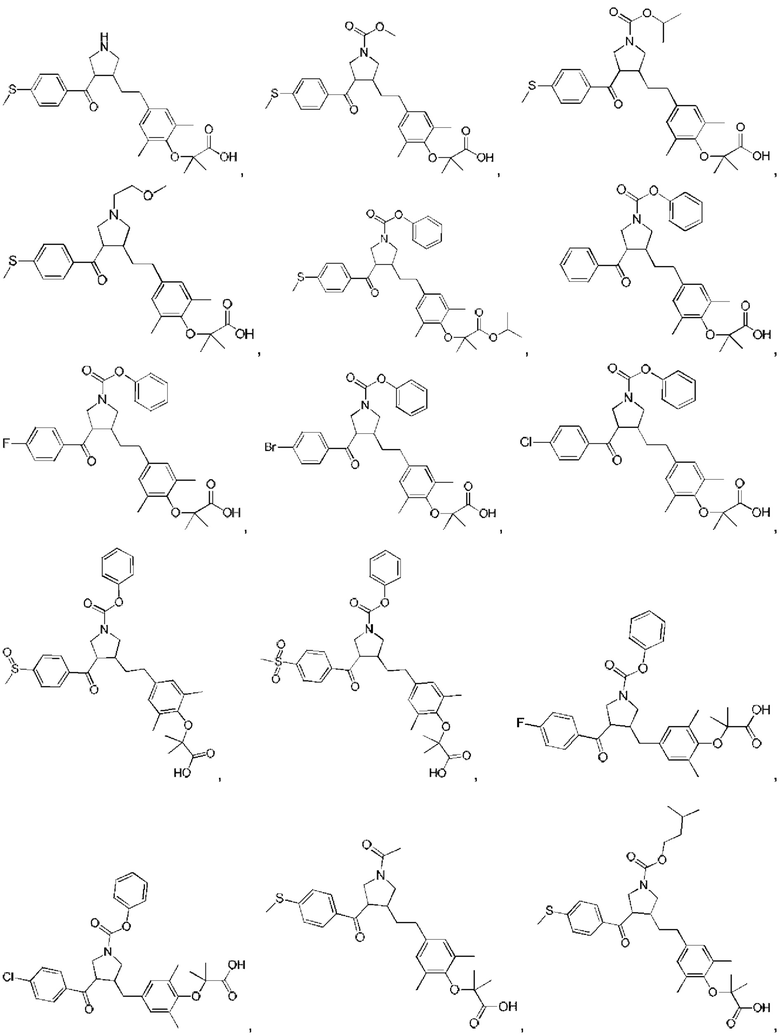
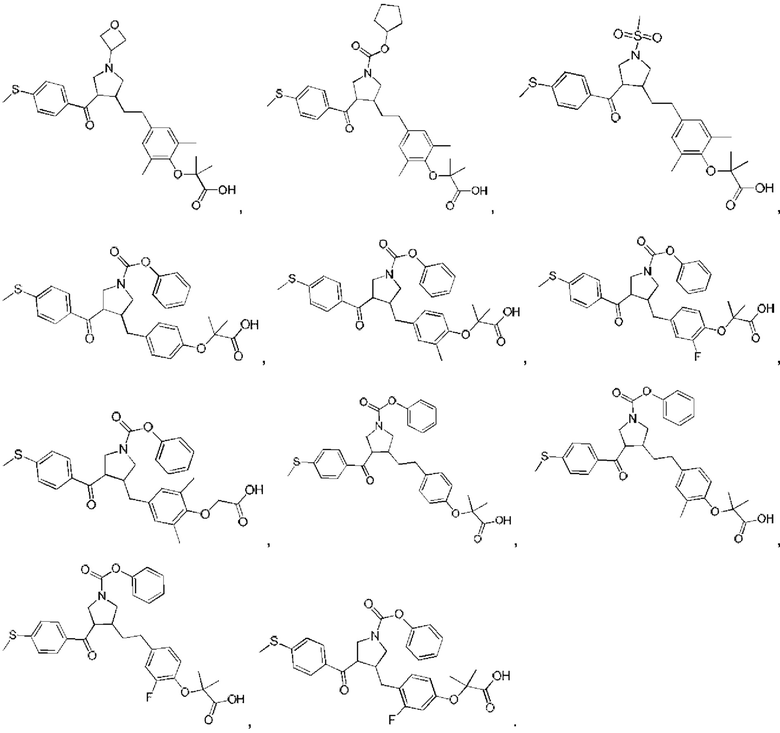
25. Соединение по п. 24, выбранное из:
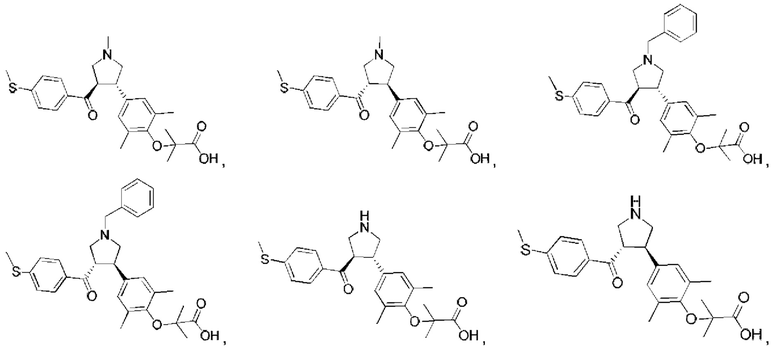
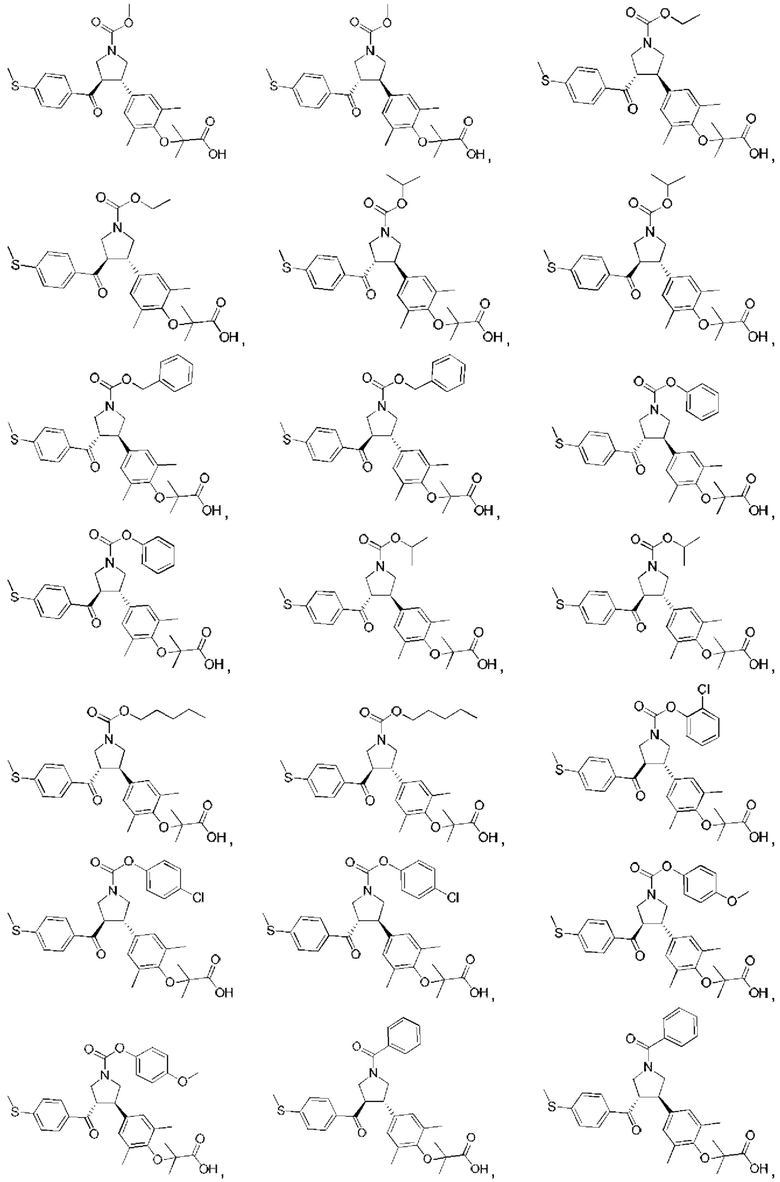
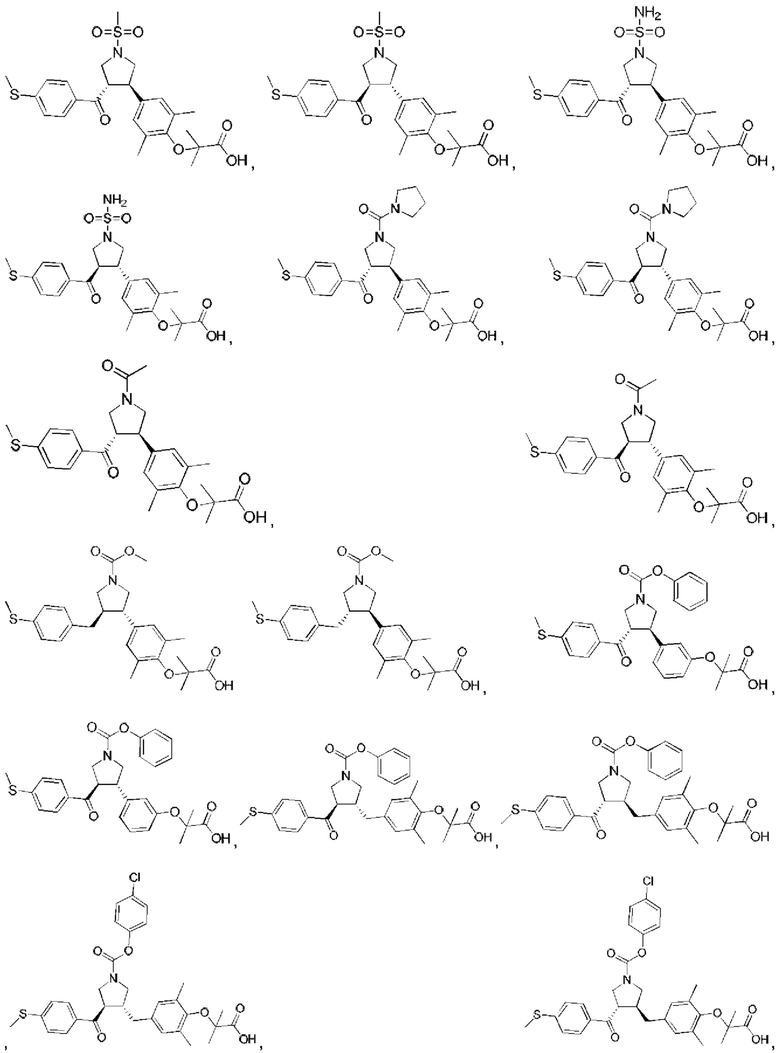
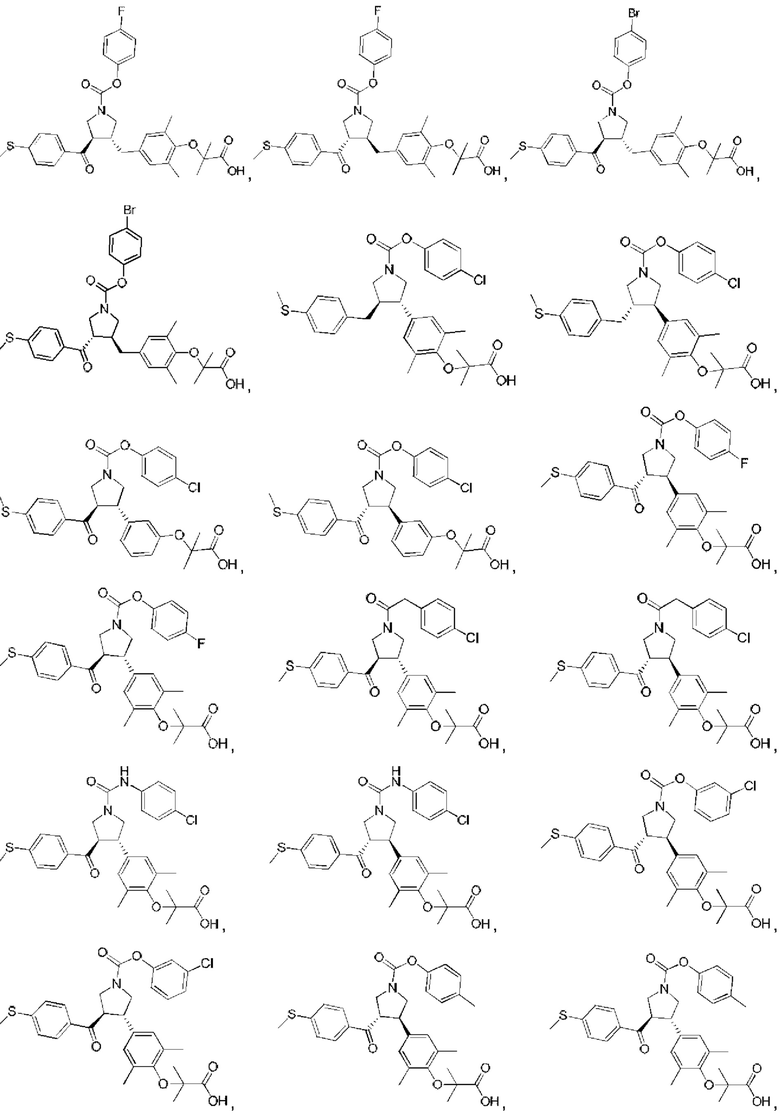
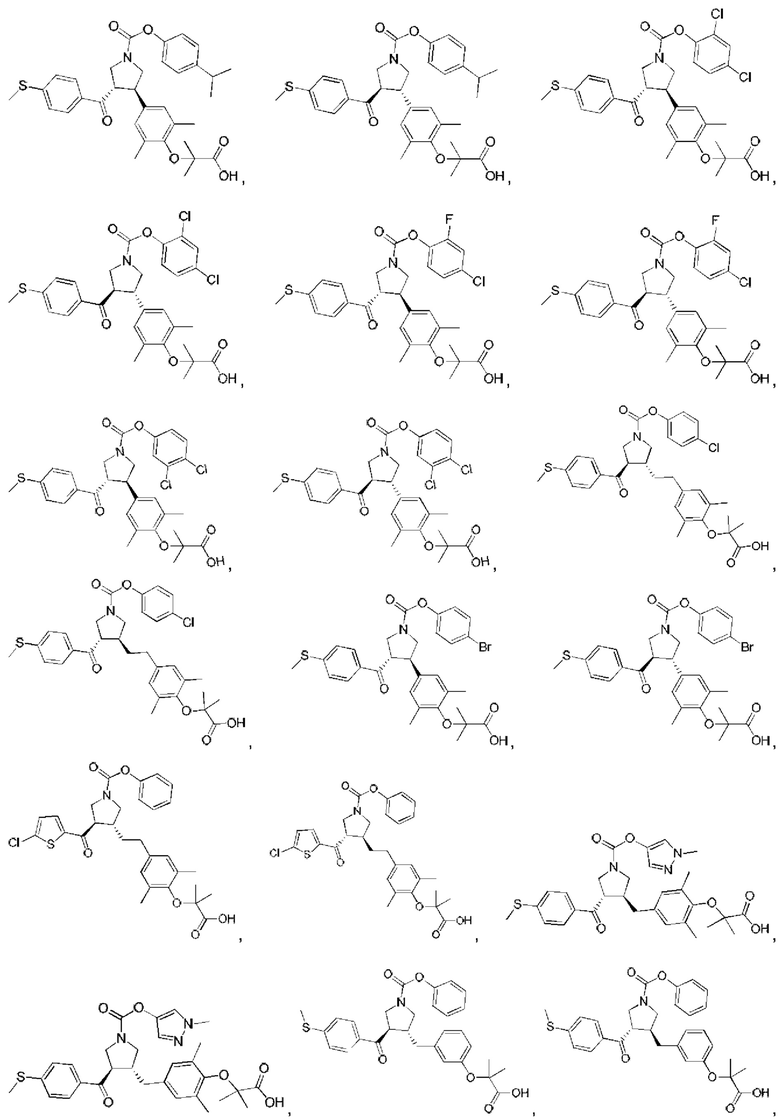
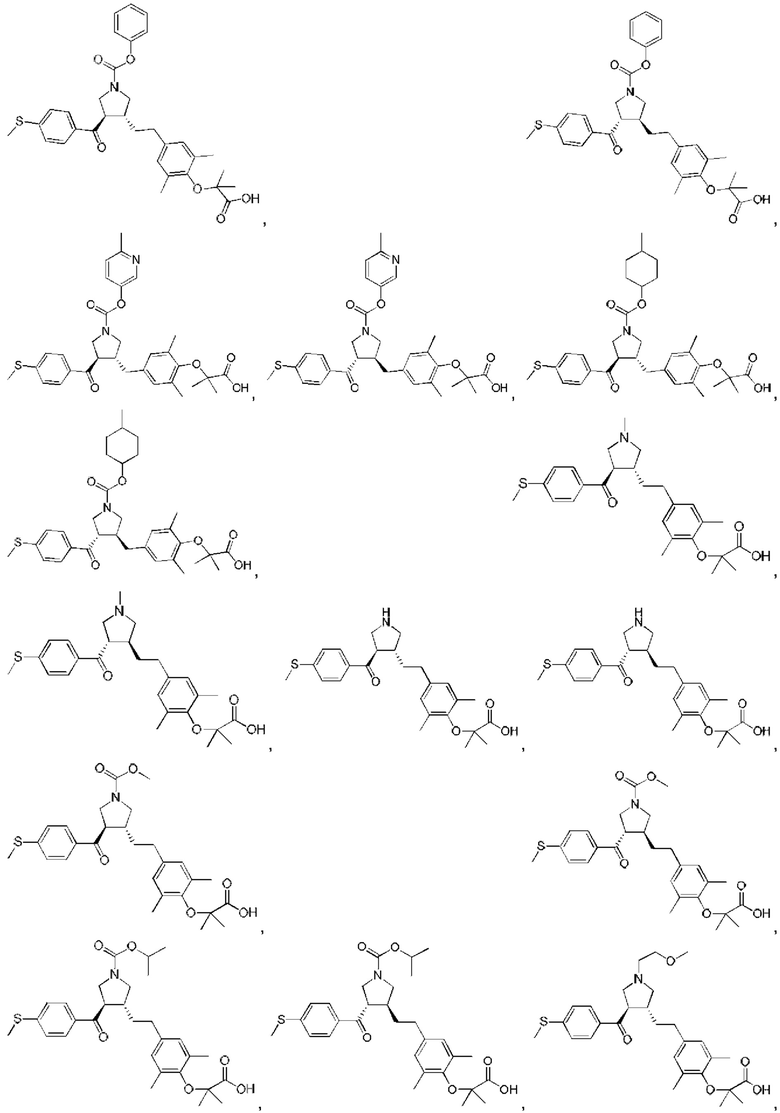
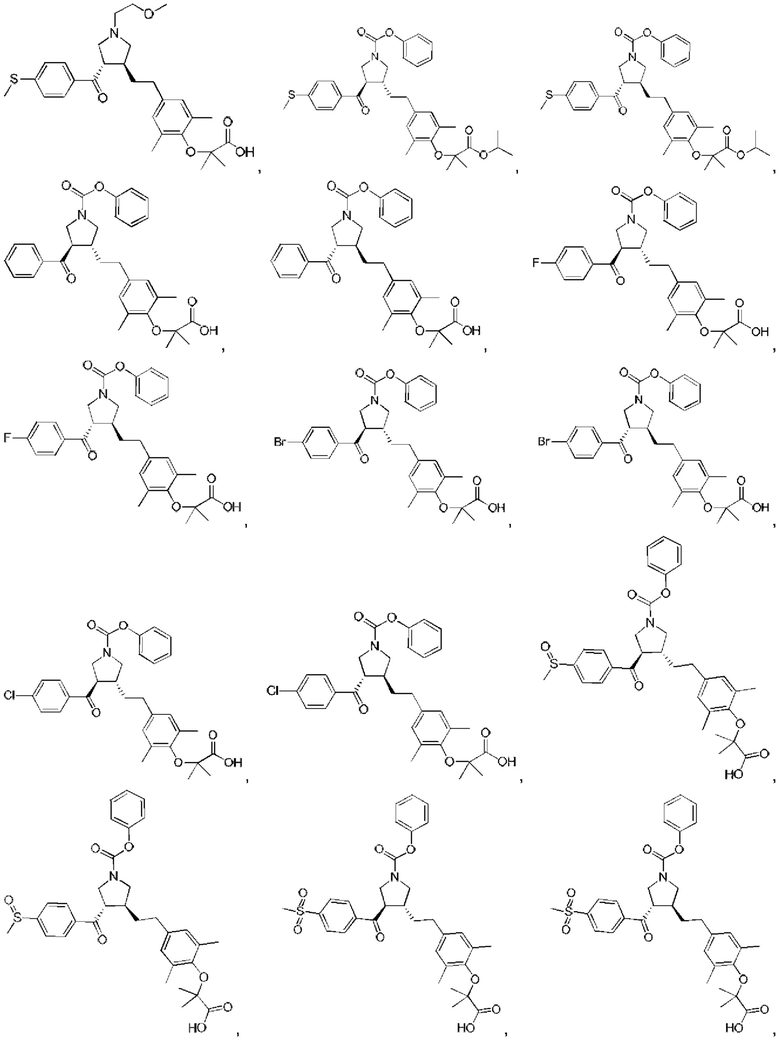
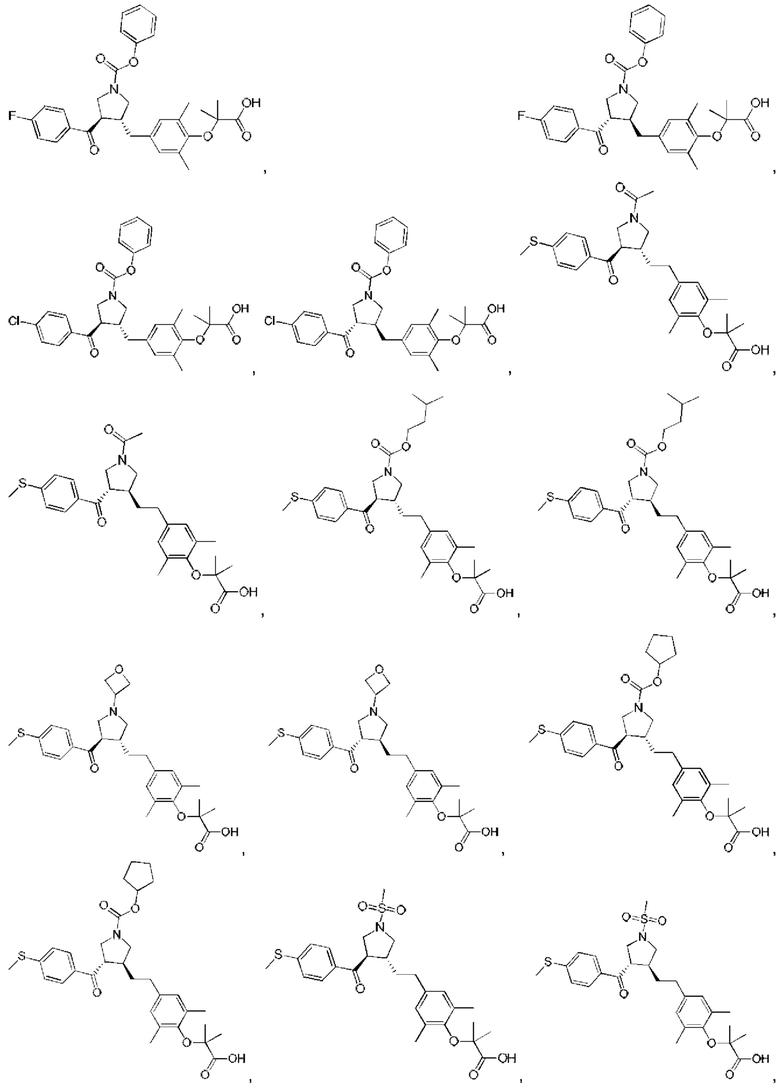
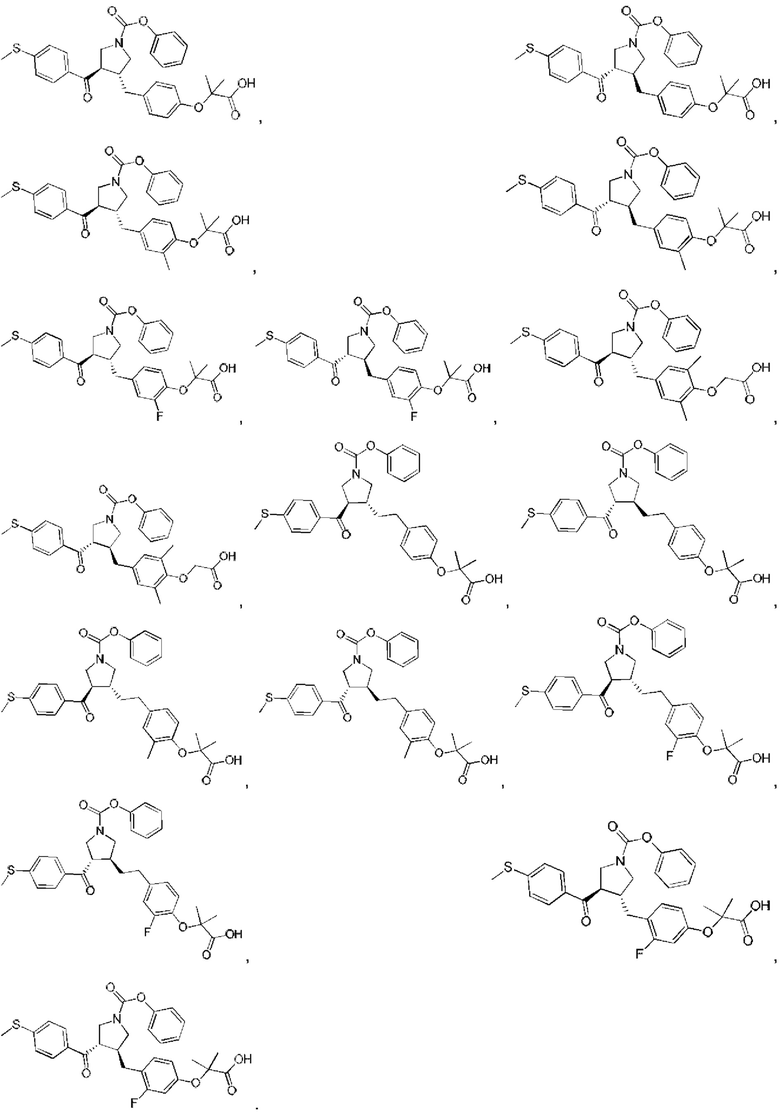
26. Фармацевтическая композиция для лечения нарушений, связанных с рецептором PPAR, содержащая терапевтически эффективное количество соединения по любому из пп. 1-25 или его фармацевтически приемлемую соль в качестве активного ингредиента, а также фармацевтически приемлемый носитель.
27. Применение соединения по любому из пп. 1-25 или его фармацевтически приемлемой соли или композиции по п. 26 для получения лекарственного средства для лечения нарушений, связанных с рецептором PPAR.
28. Применение соединения по любому из пп. 1-25 или его фармацевтически приемлемой соли или композиции по п. 26 для получения лекарственного средства для лечения неалкогольного стеатогепатита и сопутствующего фиброза, инсулинорезистентности, первичного билиарного холангита, дислипидемии, гиперлипидемии, гиперхолестеринемии, атеросклероза, гипертриглицеридемии, сердечно-сосудистого заболевания, ожирения.
| МАГНИТНАЯ РАЗНОИМЕННОПОЛЮСНАЯ ПЛИТА | 0 |
|
SU217912A1 |
| WO 2007053819 A3, 10.05.2007 | |||
| АМИДЫ АМИНОАЛКИЛЗАМЕЩЕННЫХ АЗЕТИДИНОВ, ПИРРОЛИДИНОВ, ПИПЕРИДИНОВ И АЗЕПАНОВ | 2003 |
|
RU2366652C2 |

