ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Настоящая заявка испрашивает преимущество приоритета по предварительной заявке на патент США № 62/896548, поданной 5 сентября 2019 г., предварительной заявке на патент США № 62/908555, поданной 30 сентября 2019 г., и предварительной заявке на патент США № 63/006651, поданной 7 апреля 2020 г. Содержание каждой из родственных заявок полностью включено в данный документ посредством ссылки.
Уровень техники
Область техники
[0002] Настоящее описание относится по существу к области генотипирования паралогов и, в частности, к генотипированию паралогов с использованием данных секвенирования.
Уровень техники
[0003] Генотипирование является сложной задачей. Например, спинальная мышечная атрофия вызвана потерей функциональности гена выживания моторного нейрона 1 (англ.: survival of motor neuron 1-SMN1), но сохранением паралогического гена SMN2. Из-за практически идентичных последовательностей SMN1 и его паралога SMN2 анализ этой области представляет собой сложную задачу. В качестве другого примера, CYP2D6 участвует в метаболизме 25% всех лекарственных средств. Генотипирование CYP2D6 является сложной задачей из-за его высокого полиморфизма, наличия общих структурных вариантов (SV) и высокого сходства последовательностей с паралогом псевдогена CYP2D7.
раскрытие сущности изобретения
[0004] В данном документе раскрыты способы определения количества копий гена выживания моторного нейрона 1 (SMN1). В некоторых вариантах осуществления способ определения количества копий гена SMN1 контролируется процессором (таким как аппаратный процессор или виртуальный процессор) и включает: получение данных секвенирования, содержащих множество прочтений последовательностей из образца от субъекта, выровненных с геном SMN1 или геном выживания моторных нейронов 2 (SMN2). Способ может включать: определение (i) первого количества прочтений последовательности из множества прочтений последовательностей, выровненных по первому участку гена SMN1 или SMN2, содержащим по меньшей мере один из первых 6 экзонов гена SMN1 или гена SMN2 соответственно, и (ii) второго количества прочтений последовательности из множества прочтений последовательностей, выровненных по второму участку SMN1 или SMN2, содержащим по меньшей мере экзон 7 или экзон 8 гена SMN1 или гена SMN2 соответственно. Способ может включать: определение (i) первого нормализованного количества прочтений последовательности, выровненной по первой области гена SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной по второй области SMN1 или SMN2, с применением (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2 соответственно. Способ может включать: определение (i) количества копий полноразмерных генов выживания моторных нейронов (SMN), каждый из которых является интактным геном SMN1, интактным геном SMN2, укороченным геном SMN1 или укороченным геном SMN2; и (ii) количества копий любых интактных генов SMN, каждый из которых является интактным геном SMN1 или интактным геном SMN2, с использованием модели смеси нормальных распределений, содержащей множество нормальных распределений, каждое из которых представляет разное целое количество копий, с учетом (i) первого нормализованного количества прочтений последовательности, выровненной по первой области SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной по второй области SMN1 или SMN2, соответственно. Способ может включать: для одного множества специфичных для гена SMN1 оснований, связанных с интактным геном SMN1, определение наиболее вероятной комбинации из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любых определенных интактных генов SMN, учитывая (a) количество прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат основание, специфичное для гена SMN1, и (b) количество прочтений последовательностей из множества прочтений последовательности с основаниями, которые содержат специфическое для гена SMN2 основание гена SMN2, соответствующее специфическому для гена SMN1 основанию. Способ может включать: определение количества копий гена SMN1 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенного для специфического для гена SMN1 основания.
[0005] В некоторых вариантах осуществления данные секвенирования содержат данные секвенирования целого генома (полногеномного секвенирования, whole genome sequencing, WGS) или данные WGS с помощью коротких прочтений. В некоторых вариантах осуществления субъект является субъектом-плодом, неонатальным субъектом, педиатрическим субъектом, субъектом-подростком или взрослым субъектом. Образец может содержать клетки или внеклеточную ДНК. Образец может содержать фетальные клетки или внеклеточную фетальную ДНК.
[0006] В некоторых вариантах осуществления прочтение последовательности из множества прочтений последовательностей, выравнивается с первой областью SMN1 или SMN2 или со второй областью SMN1 или SMN2 с показателем качества выравнивания, равным приблизительно нулю. Первый участок SMN1 или SMN2 может содержать экзон от 1 до 6 гена SMN1 или гена SMN2 соответственно и иметь длину примерно 22,2 т. п. н. Второй участок SMN1 или SMN2 может содержать экзон 7 и экзон 8 гена SMN1 или гена SMN2 соответственно и иметь длину примерно 6 т. п. н.
[0007] В некоторых вариантах осуществления определение (i) первого нормализованного количества прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной со второй областью, включает: определение (i) первого нормализованного количества прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной со второй областью SMN1 или SMN2, с применением (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2, соответственно, и (iii) глубины прочтения последовательности области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования. Определение (i) первого нормализованного количества прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной со второй областью SMN1 или SMN2, может включать: определение (i) нормализованного количества прочтений по длине участка SMN1 или SMN2 последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) нормализованного количества прочтений по длине участка SMN1 или SMN2 последовательности, выровненной со второй областью SMN1 или SMN2, с использованием (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2 соответственно. Определение (i) первого нормализованного количества прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной со второй областью SMN1 или SMN2, может включать: определение (i) первой нормализованной глубины прочтения последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) второй нормализованной глубины прочтения последовательности, выровненной со второй областью SMN1 или SMN2, на основе (i) первого нормализованного количества прочтений по длине участка SMN1 или SMN2 и (ii) второго нормализованного количества прочтений по длине участка SMN1 или SMN2, соответственно, при использовании интенсивности прочтения последовательности области генома субъекта, отличной от генетических локусов, содержащих ген SMN1 и ген SMN2, первое нормализованное количество прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и второе нормализованное количество прочтений последовательности, выровненной со второй областью SMN1 или SMN2, которые представляют собой первую нормализованную интенсивность и вторую нормализованную интенсивность соответственно.
[0008] В некоторых вариантах осуществления определение (i) первого нормализованного количества прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной со второй областью, включает: определение (i) первого нормализованного количества прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной со второй областью SMN1 или SMN2, с применением (i) содержания GC в первой области SMN1 или SMN2 и (ii) содержания GC в второй области SMN1 или SMN2, соответственно, и (iii) глубины прочтения последовательности области генома субъекта, отличной от генетических локусов, содержащих ген SMN1 и ген SMN2 по данным секвенирования, и (iv) содержания GC в области генома.
[0009] В некоторых вариантах осуществления глубина прочтения области представляет собой среднюю глубину или медианную глубину прочтений последовательности области генома субъекта, отличной от генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования. Область может содержать примерно 3000 предварительно выбранных областей длиной примерно 2 т. п. н. каждая в геноме субъекта. В некоторых вариантах осуществления (i) первое нормализованное количество прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и/или (ii) второе нормализованное количество прочтений последовательности, выровненной со второй областью SMN1 или SMN2, составляет от примерно 30 до примерно 40.
[0010] В некоторых вариантах осуществления модель смеси нормальных распределений представляет собой одномерную модель смеси нормальных распределений. Множество распределений модели смеси нормальных распределений могут представлять целые числа копий от 0 до 10. Среднее значение для каждого из множества нормальных распределений может представлять собой целое число копий, представленное нормальными распределениями.
[0011] В некоторых вариантах осуществления определение (i) количества копий всех генов SMN и (ii) количества копий любых интактных генов SMN включает определение (i) количества копий всех генов SMN и (ii) количества копий любых интактных генов SMN с использованием модели смеси нормальных распределений, и первый предварительно определенный порог апостериорной вероятности с учетом (i) первого нормализованного количества прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной со второй областью SMN1 или SMN2 соответственно. Первый предварительно определенный порог апостериорной вероятности может составлять 0,95.
[0012] В некоторых вариантах осуществления способ включает: определение количества копий укороченных генов SMN с использованием (i) определенного количества копий от общего числа генов SMN и (ii) определенного количества копий интактных генов SMN. Количество копий укороченных генов SMN может представлять собой разницу (i) общего количества копий определенных генов SMN и (ii) определенного количества копий интактных генов SMN.
[0013] В некоторых вариантах осуществления специфичное для гена SMN1 основание представляет собой энхансер сплайсинга. Специфичное для гена SMN1 основание может представлять собой основание в с.840 гена SMN1. В некоторых вариантах осуществления наиболее вероятная комбинация возможного количества копий гена SMN1 и возможного количества копий гена SMN2 связана с самой высокой апостериорной вероятностью, по сравнению с другими комбинациями множества комбинаций с заданным (a) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена SMN1 основание, и (b) количество прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат соответствующее специфическое для гена SMN2 основание.
[0014] В некоторых вариантах осуществления определение наиболее вероятной комбинации возможного количества копий гена SMN1 и возможной комбинации гена SMN2 включает: определение наиболее вероятной комбинации, из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любых определенных интактных генов SMN, с учетом соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат SMN2 ген-специфическое основание гена SMN2, соответствующее специфичному для гена SMN1 основанию. Определение наиболее вероятной комбинации возможного количества копий гена SMN1 и возможной комбинации гена SMN2 может включать: определение (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат SMN2 ген-специфическое основание гена SMN2 соответствующее специфичному для гена SMN1 основанию; определение соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN2 основание гена SMN2, соответствующее специфичному для гена SMN1 основанию; и определение наиболее вероятной комбинации, из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любого интактного гена SMN, определенным на основе соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат SMN2 ген-специфическое основание гена SMN2 соответствующее специфичному для гена SMN1 основанию.
[0015] В некоторых вариантах осуществления определение наиболее вероятной комбинации возможного количества копий гена SMN1 и возможной комбинации гена SMN2 включает: для каждого из множества ген-специфических оснований SMN1, определение наиболее вероятной комбинации из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любых определенных интактных генов SMN, которая связана с наибольшей апостериорной вероятностью, заданной (a) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат SMN2 ген-специфическое основание гена SMN2 соответствующее специфичному для гена SMN1 основанию. Определение количества копий гена SMN1 может включать: определение количества копий гена SMN1 на основе возможного количества копий гена SMN1 наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенного для каждого из множества специфичных для гена SMN1 оснований.
[0016] В некоторых вариантах осуществления специфичное для гена SMN1 основание имеет соответствие с каждым из множества характерных для конкретного гена SMN1 оснований, отличных от специфичного для гена SMN1 основания выше заранее определенного порога соответствия. Порог соответствия может составлять 97%. Множество оснований, специфичных для гена SMN1 может содержать 8 оснований, специфичных для гена SMN1. Каждое из множества оснований, специфичных для гена SMN1 может находиться на интроне 6, экзоне 7, интроне 7 или экзоне 8 гена SMN1. Множество оснований, специфичных для гена SMN1, если субъект принадлежит к первой расе, множество оснований, специфичных для гена SMN1, если субъект принадлежит к второй расе и множество оснований, специфичных для гена SMN1, если субъект принадлежит к неизвестной расе, могут различаться. Раса субъекта может быть неизвестной, а множество оснований, специфичных для гена SMN1 могут быть неспецифичными для расы. Раса субъекта может быть известна, и множество оснований, специфичных для гена SMN1 могут быть специфичными для расы субъекта. В некоторых вариантах осуществления способ включает получение информации о расе субъекта. Способ может включать: выбор множества оснований, специфичных для гена SMN1 из множества оснований, специфичных для гена SMN1 на основе полученной информации о расе.
[0017] В некоторых вариантах осуществления определение количества копий гена SMN1 включает: определение количества копий гена SMN1 и количества копий гена SMN2 с использованием наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенного для каждого из множества оснований, специфичных для гена SMN1. Определение количества копий может включать: определение количества копий гена SMN1 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенного для специфического для гена SMN1 основания и второго предварительно заданного порога апостериорной вероятности для комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2. Второй предварительно определенный порог апостериорной вероятности может составлять 0,6 или 0,8.
[0018] В некоторых вариантах осуществления большинство определенных возможных количеств копий гена SMN1 совпадают. Количество копий определенного гена SMN1 может быть согласованным с возможным количеством копий гена SMN1. Способ может включать: определение возможной комбинации, содержащей возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любого интактного гена SMN, определенных с учетом (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат любое из множества оснований, специфических для гена SMN1, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат любое из множества соответствующих оснований, специфических для гена SMN2. Способ может включать: определение возможного количества копий возможной комбинации представляет собой согласованное возможное количество копий гена SMN1.
[0019] В некоторых вариантах осуществления определение количества копий гена SMN1 включает: определение количества копий гена SMN1, равного нулю, единице или более единицы. В некоторых вариантах осуществления способ включает: определение статуса спинальной мышечной атрофии ( англ.: spinal muscular atrophy, SMA) у субъекта на основе количества копий гена SMN1. Статус SMA для субъекта может включать в себя SMA, носитель SMA/отсутствие SMA и не носитель SMA. В некоторых вариантах осуществления способ включает определение субъекта как молчащего носителя SMA с использованием ряда прочтений последовательности из множества прочтений последовательностей, выровненных с g.27134 гена SMN1, и на основе прочтений последовательностей, выровненных с g.27134 гена SMN1.
[0020] В некоторых вариантах осуществления способ включает: определение рекомендации по лечению для субъекта на основании определенного количества копий гена SMN1. Рекомендация по лечению может включать введение субъекту Nusinersen и/или Zolgensma.
[0021] В данном документе описаны способы генотипирования гена члена 6 подсемейства D семейства 2 цитохрома P450 (CYP2D6). В некоторых вариантах осуществления способ генотипирования гена CYP2D6 контролируется процессором (таким как аппаратный процессор или виртуальный процессор) и включает: получение данных секвенирования, содержащих множество прочтений последовательностей из образца от субъекта, выровненных с геном CYP2D6 или геном члена 7 подсемейства D семейства 2 цитохрома P450 (CYP2D7). Способ может включать: определение (i) первого количества прочтений последовательности из множества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7. Способ может включать: определение (i) первого нормализованного количества прочтений последовательности, выровненной с геном CYP2D6 или геном CYP2D7, с применением (i) длины гена CYP2D6 или гена CYP2D7 соответственно. Способ может включать: определение (i) общего количества копий гена CYP2D6 и гена CYP2D7 с помощью модели смеси нормальных распределений, содержащей множество нормальных распределений, каждое из которых представляет другое целое количество копий, на основании (i) первого нормализованного количества прочтений последовательности, выровненной с геном CYP2D6 или геном CYP2D7. Способ может включать: для одного из множества оснований, специфичных для гена CYP2D6 определение наиболее вероятной комбинации множества возможных комбинаций, каждая из которых содержит возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, суммированное до общего количества копий гена CYP2D6 и определенного гена CYP2D7, учитывая (a) прочтение множества последовательностей с основаниями, которые содержат специфическое для гена CYP2D6 основание, и (b) прочтение множества последовательностей с основаниями, которые содержат специфическое для гена CYP2D7 основание, соответствующее основанию, специфичному для гена CYP2D6. Способ может включать: определение аллеля гена CYP2D6, имеющегося у субъекта, с использованием наиболее вероятной комбинации возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7, определенного для основания, специфичного для гена CYP2D6.
[0022] В некоторых вариантах осуществления данные секвенирования содержат данные секвенирования целого генома (WGS) или данные WGS с помощью коротких прочтений. Субъект может являться субъектом-плодом, неонатальным субъектом, педиатрическим субъектом, субъектом-подростком или взрослым субъектом. Образец может содержать клетки или внеклеточную ДНК. Образец может содержать клетки или внеклеточную ДНК.
[0023] В некоторых вариантах осуществления прочтение последовательности из множестве прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, при этом показатель качества выравнивания составляет около нуля. В некоторых вариантах осуществления определение (i) первого количества прочтений последовательности из множества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, включает: определение (i) первого количества прочтений последовательности из множества прочтений последовательностей, выровненных с по меньшей мере одним экзоном или интроном гена CYP2D6 или по меньшей мере одним из экзонов или интронов гена CYP2D7.
[0024] В некоторых вариантах осуществления определение (i) первого нормализованного количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, включает: определение (i) первого нормализованного количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, с использованием (i) длины гена CYP2D6 или гена CYP2D7, соответственно, и (iii) глубины прочтения последовательности области генома субъекта, отличной от генетических локусов, содержащих ген CYP2D6 и ген CYP2D7 по данным секвенирования. Определение (i) первого нормализованного количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, и (ii) второго нормализованного количества прочтений последовательностей, выровненных со второй областью, может включать: определение (i) первого гена CYP2D6 или нормализованного по длине гена CYP2D7 количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, с применением (i) длины гена CYP2D6 или гена CYP2D7 соответственно. Определение (i) первого нормализованного количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, и (ii) второго нормализованного количества прочтений последовательностей, выровненных со второй областью, может включать: определение (i) первой нормализованной глубины прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, из (i) нормализованного количества по длине гена CYP2D6 или гена CYP2D7, с применением глубины прочтения последовательностей области генома субъекта, отличных от генетических локусов, содержащих ген CYP2D6 и CYP2D7, первая нормализованная глубина прочтения последовательности, выровненной с геном CYP2D6 или геном CYP2D7, представляет собой первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7 соответственно.
[0025] В некоторых вариантах осуществления определение (i) первого нормализованного количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, включает: определение (i) первого нормализованного количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, с применением (i) содержания GC в гене CYP2D6 или гене CYP2D7 и (iii) глубины прочтения последовательности области генома субъекта, отличных от генетических локусов, содержащих ген CYP2D6 и ген CYP2D7, по данным секвенирования и (iv) содержания GC в области генома. Глубина прочтения области может включать среднюю глубину или медианную глубину прочтений последовательности области генома субъекта, отличной от генетических локусов, содержащих ген CYP2D6 и ген CYP2D7, по данным секвенирования. Область может содержать примерно 3000 предварительно выбранных областей длиной примерно 2 т. п. н. каждая в геноме субъекта. В некоторых вариантах осуществления (i) первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, и/или (ii) второе нормализованное количество прочтений последовательностей, выровненных со второй областью, составляет от примерно 30 до примерно 40.
[0026] В некоторых вариантах осуществления модель смеси нормальных распределений представляет собой одномерную модель смеси нормальных распределений. Множество распределений модели смеси нормальных распределений могут представлять целые числа копий от 0 до 10. Среднее значение для каждого из множества нормальных распределений может представлять собой целое число копий, представленное нормальными распределениями.
[0027] В некоторых вариантах осуществления определение (i) общего количества копий гена CYP2D6 и гена CYP2D7 включает определение (i) общего количества копий гена CYP2D6 и гена CYP2D7 с помощью модели смеси нормальных распределений и первого предварительно заданного порога апостериорной вероятности, при условии, что (i) первое нормализованное количество прочтений последовательности совпадает с геном CYP2D6 или геном CYP2D7. Первый предварительно определенный порог апостериорной вероятности может составлять 0,95.
[0028] В некоторых вариантах осуществления наиболее вероятная комбинация возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7 связана с самой высокой апостериорной вероятностью, по сравнению с другими комбинациями множества комбинаций с заданным (a) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена CYP2D6 основание, и (b) количество прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат соответствующее специфическое для гена CYP2D7 основание.
[0029] В некоторых вариантах осуществления определение наиболее вероятной комбинации, содержащей возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, включает: определение наиболее вероятной комбинации, из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, суммированное с общим количеством копий гена CYP2D6 и определенным геном CYP2D7, учитывая соотношение (a) количества прочтений последовательности при прочтениях множества последовательностей с основаниями, которые содержат специфическое для гена CYP2D6 основание, и (b) количества прочтений последовательности при прочтениях множества последовательностей с основаниями, которые содержат специфическое для гена CYP2D7 основание, соответствующее специфичному для гена CYP2D6 основанию. Определение наиболее вероятной комбинации, содержащей возможное количество копий гена CYP2D6 и возможное количество копий, может включать: определение (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D6 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D7 основание, соответствующее специфичному для гена CYP2D6 основанию; определение соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D6 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D7 основание, соответствующее специфичному для гена CYP2D6 основанию; и определение наиболее вероятной комбинации множества возможных комбинаций, каждая из которых содержит возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, суммированное с общим количеством копий гена CYP2D6 и определенным геном CYP2D7, с учетом соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D6 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D7 основание, соответствующее специфичному для гена CYP2D6 основанию.
[0030] В некоторых вариантах осуществления определение аллеля гена CYP2D6, имеющегося у субъекта, включает: определение одного или более структурных вариантов гена CYP2D6 у субъекта с использованием наиболее вероятной комбинации возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7, определенного для специфичного для гена CYP2D6 основания. В некоторых вариантах осуществления определение наиболее вероятной комбинации возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7 включает определение наиболее вероятной комбинации для каждого из множества ген-специфических оснований CYP2D6, множества возможных комбинаций, каждая из которых содержит возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, суммированное с общим количеством копий определенного гена CYP2D6 и гена CYP2D7, которая связана с наибольшей апостериорной вероятностью, заданной (a) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D6 основание, и (b) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат CYP2D7 ген-специфическое основание гена CYP2D7 соответствующее специфичному для гена CYP2D6 основанию. Определение одного или более структурных вариантов гена CYP2D6, имеющегося у субъекта, может включать определение одного или более структурных вариантов с использованием наиболее вероятной комбинации возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7, определенного для каждого из множества специфических для гена CYP2D6 оснований. В некоторых вариантах осуществления определение одного или более структурных вариантов гена CYP2D6, имеющегося у субъекта, включает: определение одного или более структурных вариантов гена CYP2D6, у субъекта на основе количества копий гена CYP2D6 наиболее вероятных комбинаций, определенных для двух или более из множества различных оснований, специфичных для гена CYP2D6, и положений двух или более оснований, специфичных для гена CYP2D6.
[0031] В некоторых вариантах осуществления специфичное для гена CYP2D6 основание имеет соответствие с каждым из множества специфичных для гена CYP2D6 оснований, за исключением специфичного для гена CYP2D6 основания, при превышении заданного порогового значения соответствия. Порог соответствия может составлять 97%. Множество оснований, специфичных для гена CYP2D6 может содержать 118 оснований, специфичных для гена CYP2D6. Множество оснований, специфичных для гена CYP2D6, если субъект принадлежит к первой расе, множество оснований, специфичных для гена CYP2D6, если субъект принадлежит к второй расе, и множество оснований, специфичных для гена CYP2D6, если субъект принадлежит к неизвестной расе, могут различаться. Раса субъекта может быть неизвестной, а множество оснований, специфичных для гена CYP2D6 могут быть неспецифичными для расы. Раса субъекта может быть известна, и множество оснований, специфичных для гена CYP2D6 могут быть специфичными для расы субъекта. В некоторых вариантах осуществления способ включает получение информации о расе субъекта. Способ может включать: выбор множества оснований, специфичных для гена CYP2D6 из множества оснований, специфичных для гена CYP2D6 на основе полученной информации о расе.
[0032] В некоторых вариантах осуществления способ включает: определение (ii) второго количества прочтений последовательности из множества прочтений последовательностей, выровненных с областью спейсера между геном CYP2D7 и повторяющимся элементом REP7 ниже гена CYP2D7. Способ может включать: определение (ii) второго нормализованного количества прочтений последовательности, выровненной с областью спейсера, с использованием (ii) длины области спейсера. Способ может включать: определение (ii) количества копий области спейсера с использованием модели смеси нормальных распределений, заданной (ii) вторым нормализованным количеством прочтений последовательностей, выровненных с областью спейсера. Определение одного или более структурных вариантов гена CYP2D6, имеющегося у субъекта, может включать: определение одного или более структурных вариантов гена CYP2D6 у субъекта с использованием наиболее вероятной комбинации возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7, определенного для специфичного для гена CYP2D6 основания, и количества копий области спейсера. Один или более структурных вариантов могут содержать слитый аллель CYP2D6/CYP2D7 с спейсерной областью и повторяющимся элементом REP7 ниже слитого аллеля CYP2D6/CYP2D7.
[0033] В некоторых вариантах осуществления способ включает: определение одного или более малых вариантов гена CYP2D6 у субъекта с применением полученных данных секвенирования. В некоторых вариантах осуществления определение одного или более малых вариантов гена CYP2D6, имеющегося у субъекта, включает: для положения малого варианта гена CYP2D6, связанного с малым вариантом аллеля гена CYP2D6, определение наиболее вероятной комбинации возможного количества копий небольшого варианта аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6, суммированного с количеством копий гена CYP2D6 в положении малого варианта, учитывая (a) количество прочтений последовательности с основаниями, которые содержат малый вариантный аллель гена CYP2D6 в положении малого варианта, и (b) количество прочтений последовательности с основаниями, которые содержат эталонный аллель гена CYP2D6 в положении малого варианта, возможное количество копий малого варианта аллеля гена CYP2D6, наиболее вероятно, комбинации в малом варианте указывает на один или более малых вариантов гена CYP2D6. В некоторых вариантах осуществления определение одного или более малых вариантов гена CYP2D6, имеющегося у субъекта, включает в себя: для каждого из множества положений малых вариантов гена CYP2D6 положение малого варианта связано с малым вариантом аллеля гена CYP2D6, определение наиболее вероятной комбинации возможного количества копий малого аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6 в положении малого варианта, суммированного с количеством копий гена CYP2D6 в положении малого варианта, учитывая (a) количество прочтений последовательности с основаниями, которые содержат малый вариантный аллель гена CYP2D6 в положении малого варианта, и (b) количество прочтений последовательности с основаниями, которые содержат эталонный аллель гена CYP2D6 в положении малого варианта, возможное количество копий малых вариантных аллелей гена CYP2D6, наиболее вероятно, комбинации в множестве положений малых вариантов указывает на один или более малых вариантов гена CYP2D6.
[0034] В некоторых вариантах осуществления способ включает: для положения малого варианта гена CYP2D6, связанного с малым аллелем варианта гена CYP2D6, определение наиболее вероятной комбинации возможного количества копий малого аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6 в положении малого варианта, суммированного с количеством копий гена CYP2D6 в положении малого варианта, учитывая (a) количество прочтений последовательностей, выровненных с геном CYP2D6, перекрывающимся с положением малого варианта, и основания, которое содержит аллель малого варианта гена CYP2D6 в положении малого варианта и (b) количество прочтений последовательностей, выровненных с геном CYP2D6, перекрывающимся с положением малого варианта, и основания, которое содержит эталонный аллель гена CYP2D6 в положении малого варианта; и определение одного или более малых вариантов гена CYP2D6 с использованием возможного количества копий малого варианта аллеля гена CYP2D6 наиболее вероятной определенной комбинации. В некоторых вариантах осуществления способ включает: для каждого из множества положений малых вариантов гена CYP2D6 положение малого варианта связано с малым вариантом аллеля гена CYP2D6, определение наиболее вероятной комбинации возможного количества копий малого аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6 в положении малого варианта, суммированного с количеством копий гена CYP2D6 в положении малого варианта, учитывая (a) количество прочтений последовательностей, выровненных с геном CYP2D6, перекрывающимся с положением малого варианта, и основания, которое содержит аллель малого варианта гена CYP2D6 в положении малого варианта и (b) количество прочтений последовательностей, выровненных с геном CYP2D6, с положением малого варианта, и основания, которое содержит эталонный аллель гена CYP2D6 в положении малого варианта; и определение одного или более малых вариантов гена CYP2D6 с использованием возможного количества копий малых вариантов аллеля гена CYP2D6 наиболее вероятных комбинаций во множестве определенных положений малых вариантов.
[0035] В некоторых вариантах осуществления положение малого варианта находится в гомологичной области CYP2D6/CYP2D7, определение наиболее вероятной комбинации включает определение наиболее вероятной комбинации возможного количества копий малого варианта аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6 в положении малого варианта, суммированное с количеством копий гена CYP2D6 в положении малого варианта, учитывая (а) количество прочтений последовательности, выровненной с геном CYP2D6 или геном CYP2D7 с основанием, которое содержит малый вариант аллеля гена CYP2D6 в положении малого варианта, и/или (b) количество прочтений последовательности, выровненной с геном CYP2D6 или геном CYP2D7, с основанием, которое содержит эталонный аллель CYP2D6 в положении малого варианта. В некоторых вариантах осуществления положение малого варианта не находится в гомологичной области CYP2D6/CYP2D7, определение наиболее вероятной комбинации включает определение наиболее вероятной комбинации возможного количества копий малого варианта аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6 в положении малого варианта, суммированного с количеством копий гена CYP2D6 в положении малого варианта, учитывая (a) количество прочтений последовательностей, выровненных с геном CYP2D6, а не с геном CYP2D7, с основанием, которое содержит малый вариант аллеля гена CYP2D6 в положении малого варианта, и/ или (b) количество прочтений последовательностей, выровненных с геном CYP2D6, а не геном CYP2D7, с основанием, которое содержит эталонный аллель CYP2D6 в положении малого варианта.
[0036] В некоторых вариантах осуществления способ включает определение количества копий гена CYP2D6 в положении малого варианта. Количество копий гена CYP2D6 в положении малого варианта может включать количество копий гена CYP2D6. Количество копий гена CYP2D6 в положении малого варианта может включать количество копий гена CYP2D6 из возможных количеств копий гена CYP2D6 наиболее вероятных определенных комбинаций. Количество копий гена CYP2D6 в положении малого варианта может включать количество копий гена CYP2D6 из возможных количеств копий гена CYP2D6 наиболее вероятных определенных комбинаций и расположены ближе всего к положению малого варианта. Количество копий гена CYP2D6 в положении малого варианта может включать количество копий гена CYP2D6 в 5’-положении или в 3’-положении от положения малого варианта. В некоторых вариантах осуществления данного изобретения способ включает: (a) определение количества прочтений последовательностей с основаниями, которые содержат малый вариант аллеля гена CYP2D6; и (b) определение количества прочтений последовательностей с основаниями, которые содержат эталонный аллель гена CYP2D6.
[0037] В некоторых вариантах осуществления определение аллеля гена CYP2D6 у субъекта включает: определение аллелей (например, 2, 3, 4, 5 или более аллелей) гена CYP2D6, имеющихся у субъекта. В некоторых вариантах осуществления определение аллеля гена CYP2D6, имеющегося у субъекта, включает: определение звездчатого аллеля и/или гаплотипа гена CYP2D6 у субъекта с использованием одного или нескольких определенных структурных вариантов гена CYP2D6, и/или один или более малых вариантов гена CYP2D6, при этом необязательно звездчатый аллель связан с известной функцией.
[0038] В некоторых вариантах осуществления способ включает: определение уровня ферментативной активности CYP2D6 у субъекта с использованием определенного аллеля гена CYP2D6. Ферментативная активность может быть низкой, средней, нормальной или сверхбыстрой. В некоторых вариантах осуществления способ включает определение рекомендации по дозировке лечения и/или рекомендации по лечению субъекта на основании аллеля гена CYP2D6, имеющегося у субъекта.
[0039] В данном документе описаны для генотипирования паралогов. В некоторых вариантах осуществления система генотипирования паралогов содержит: энергонезависимую память, выполненную с возможностью хранения исполняемых команд и данных секвенирования, содержащих множество прочтений последовательностей, полученных из образца от субъекта, выровненных с первым или вторым паралогом. Система может содержать: процессор (такой как аппаратный процессор или виртуальный процессор), обменивающийся данными с энергонезависимой памятью, причем процессор запрограммирован с помощью исполняемых команд для выполнения: определение количества копий паралогов первого типа с помощью модели смеси нормальных распределений, содержащей множество распределений, каждое из которых представляет другое целое количество копий, выраженное (i) первым количеством прочтений последовательностей, выровненных с первой областью. Аппаратный процессор, запрограммированный исполняемыми командами для выполнения: определения наиболее вероятной комбинации для одного из множества оснований, специфичных для паралога, из множества возможных комбинаций, каждая из которых содержит возможное количество копий первого паралога первого типа и возможное количество копий второго паралога первого типа, суммированное с количеством копий определенных паралогов первого типа, при заданном (a) количестве прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат основание специфическое для первого паралога, и (b) количестве прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат основание специфическое для второго паралога, соответствующее основанию специфическому для первого паралога. Аппаратный процессор запрограммирован с помощью исполняемых команд для выполнения: определения количества копий или аллеля первого паралога с использованием наиболее вероятной комбинации возможного количества копий первого паралога и возможного количества копий второго паралога, определенного для основания специфического для первого паралога. В некоторых вариантах осуществления первый паралог и второй паралог имеют идентичность последовательности по меньшей мере 90%.
[0040] В некоторых вариантах осуществления аппаратный процессор с помощью исполняемых команд для выполнения: определения (i) первого количества прочтений последовательности из множества прочтений последовательностей в данных секвенирования, полученных из образца от субъекта, выровненных с первой областью. Способ может включать: определение (i) первого нормализованного количества прочтений последовательностей, выровненных с первой областью, с использованием (i) длины первой области, причем определение количества копий паралогов первого типа включает: определение количества копий паралогов первого типа с использованием модели смеси нормальных распределений, заданной (i) первым нормализованным количеством прочтений последовательностей, выровненных с первой областью. Аппаратный процессор может быть запрограммирован с помощью исполняемых команд для выполнения: может включать: прием данных секвенирования, содержащих множество прочтений последовательностей, выровненных с первой областью.
[0041] В некоторых вариантах осуществления аппаратный процессор запрограммирован с помощью исполняемых команд для выполнения: определения количества копий одного или более паралогов второго типа с использованием смеси нормальных распределений, заданной (ii) вторым количеством прочтений последовательностей, выровненных со второй областью. Определение количества копий или аллеля первого паралога может включать: определение количества копий или аллеля первого паралога с использованием наиболее вероятной комбинации возможного количества копий первого паралога и возможного количества копий второго паралога, определенного для основания, специфического для первого паралога, и количества копий одного или более паралогов второго типа. Способ может включать: определение количества копий паралогов третьего типа по количеству копий паралогов первого типа и количеству копий паралогов второго типа. Определение количества копий или аллеля первого паралога включает: определение количества копий или аллеля первого паралога с помощью наиболее вероятной комбинации возможного количества копий первого паралога и возможного количества копий второго паралога, определенного для основания, специфического для первого паралога,
[0042] В некоторых вариантах осуществления первым паралогом является ген выживания моторных нейронов 1 (SMN1). Вторым паралогом может быть ген выживания моторных нейронов 2 (SMN2). Первая область может содержать по меньшей мере один экзон от 1 до 6 гена SMN1 и по меньшей мере один экзон от 1 до 6 гена SMN2. Вторая область может содержать по меньшей мере экзон 7 или 8 гена SMN1 и по меньшей мере один из экзон 7 или 8 гена SMN2. Паралоги первого типа могут включать интактный ген SMN1 и интактный ген SMN2. Один или более паралогов второго типа могут включать интактный ген SMN1, интактный ген SMN2, укороченный ген SMN1 или укороченный ген SMN2. Количество копий первого паралога может включать количество копий гена SMN1.
[0043] В некоторых вариантах осуществления первый паралог представляет собой ген члена 6 подсемейства D семейства 2 цитохрома P450 (CYP2D6). Вторым паралогом может быть ген члена 7 подсемейства D семейства 2 цитохрома P450 (CYP2D7). Первая область может содержать ген CYP2D6 и ген CYP2D7. Вторая область может содержать спейсерную область между геном CYP2D7 и повторяющимся элементом REP7 ниже гена CYP2D7. Паралоги первого типа могут содержать ген CYP2D6 и ген CYP2D7. Один или более паралогов второго типа могут содержать слитый аллель CYP2D6/CYP2D7 с спейсерной областью и повторяющимся элементом REP7 ниже слитого аллеля CYP2D6/CYP2D7. Количество копий первого паралога может содержать аллель гена CYP2D6, имеющегося у субъекта, который представляет собой малый вариант или структурный вариант гена CYP2D6.
[0044] В данном документе описаны варианты осуществления системы (например, компьютерной системы), содержащей энергонезависимую память, выполненную с возможностью хранения исполняемых команд; и процессора (например, аппаратный процессор или виртуальный процессор), находящийся в соединении с энергонезависимой памятью, причем аппаратный процессор запрограммирован с помощью исполняемых команд для выполнения любого способа, описанного в данном документе. Описанные в данном документе варианты осуществления устройства (например, электронного устройства), содержащего энергонезависимую память, выполненную с возможностью хранения исполняемых команд; и процессора (например, аппаратный процессор или виртуальный процессор), находящийся в соединении с энергонезависимой памятью, причем аппаратный процессор запрограммирован с помощью исполняемых команд для выполнения любого способа, описанного в данном документе. Описанные в данном документе варианты осуществления машиночитаемого носителя, содержащего исполняемые команды, которые при исполнении процессором (например, аппаратным процессором или виртуальным процессором) системы или устройства приводят к выполнению аппаратным процессором любого способа, описанного в данном документе.
[0045] Подробное описание одного или более вариантов осуществления представлено в приведенных ниже сопроводительных графических материалах и описании. Прочие признаки, аспекты и преимущества станут очевидными из описания, рисунков и формулы изобретения. Ни это краткое изложение, ни последующее подробное описание не претендуют на определение или ограничение объема изобретения.
Краткое описание графических материалов
[0046] На ФИГ. 1A-1E представлены пояснения определения количества копий SMA и SMN в соответствии с одним вариантом осуществления способа, описанного в данном документе.
[0047] На ФИГ. 2A-2C показаны распределения популяции количества копий SMN1/2, определенные с помощью одного варианта осуществления способа, описанного в данном документе.
[0048] На ФИГ. 3 показан показатель SMA, идентифицированный в двух тройках в проекте Next Generation Children и подтвержденный с помощью MLPA.
[0049] На ФИГ. 4 показаны частоты популяций, определенные с использованием одного варианта осуществления способа, описанного в данном документе, согласованного с предыдущими исследованиями.
[0050] На ФИГ. 5 показан неограничивающий пример IGV снимка, показывающий, что CYP2D6 является высокополиморфным и расположен после CYP2D7, псевдогенного паралога CYP2D6.
[0051] На ФИГ. 6 показан неограничивающий пример схематической иллюстрации делеций, дупликаций и слитых генов CYP2D6/7.
[0052] На ФИГ. 7 показан неограничивающий пример графика, показывающего, что аллельные частоты, определенные способом, согласуются с базой данных PharmVar Database Pharmacogene Variation (PharmVar) Consortium.
[0053] На ФИГ. 8 представлена блок-схема, показывающая пример способа определения количества копий гена выживания моторных нейронов 1 (SMN1) с использованием данных секвенирования.
[0054] На ФИГ. 9 представлена блок-схема, показывающая пример способа генотипирования гена члена 6 подсемейства D семейства 2 цитохрома P450 (CYP2D6) с использованием данных секвенирования.
[0055] На ФИГ. 10 представлена блок-схема, показывающая пример способа генотипирования паралога с использованием данных секвенирования.
[0056] На ФИГ. 11 представлена блок-схема иллюстративной вычислительной системы, выполненной с возможностью реализации генотипирования паралога с использованием данных секвенирования.
[0057] На ФИГ. 12A и 12B показаны неограничивающие примеры графиков, иллюстрирующих общие ВКК, влияющие на локусы SMN1/SMN2. На ФИГ. 12A представлены профили глубины в областях SMN1/SMN2. Образцы с общим количеством копий SMN1+SMN2 2, 3, 4 и 5 показаны точками соответственно. Для каждой категории количества копий суммируют глубину 50 образцов. Каждая точка представляет нормированные значения глубины в окне длиной 100 п. н. Количество прочтений рассчитывали в каждом окне длиной 100 п. н., суммировали показания для SMN1 и SMN2 и нормализовали по глубине образцов дикого типа (CN=4). Экзоны SMN представлены в виде пурпурных прямоугольников. Две оси x показывают координаты в SMN1 (внизу) и SMN2 (вверху). На ФИГ. 12B показаны профили глубины, объединенные из 50 образцов, несущих делецию экзонов 7 и 8, показаны в виде точек. Значения глубины прочтения рассчитывали таким же образом, как показано на ФИГ. 12A.
[0058] На ФИГ. 13 показан неограничивающий пример диаграммы рассеяния общего количества копий SMN (SMN1+SMN2) (ось X, обозначает глубину прочтения экзонов 1-6) и количества копий интактного SMN (ось y, обозначает глубину прочтения экзонов 7-8).
[0059] На ФИГ. 14A-14D показаны распределения количества копий SMN1/SMN2/SMN* в популяции. На ФИГ. 14A представлен неограничивающий пример иллюстративного графика, иллюстрирующий процентную долю образцов, показывающих согласование определения количества копий с c.840C>T по 16 сайтам различия оснований SMN1-SMN2 в африканских и неафриканских популяциях. Сайт 13* представляет собой сплайс-вариантный сайт c.840C>Т. Черной горизонтальной линией обозначено 85% совпадения. На ФИГ. 14B показаны неограничивающие примеры гистограмм распределений количества копий SMN1, SMN2 и SMN* по пяти популяциям в 1kGP и когорте NIHR BioResource (числа приведены в таблице 15). На ФИГ. 14C показан неограничивающий пример графика зависимости количества копий SMN1 от общего количества копий SMN2 (интактный SMN2 + SMN*). На ФИГ. 14D показаны два трио с пробандом SMA, обнаруженным специалистом и ортогонально подтвержденным в когорте NIHR BioResource. Количество копий на аллель SMN1, SMN2 и SMN* фазировано и помечено для каждого члена трио.
[0060] На ФИГ. 15 показаны неограничивающие примеры графиков, каждый из которых иллюстрирует распределение апостериорной вероятности для моделирования количества копий SMN1 с использованием одного сайта при разных глубинах прочтения и комбинаций количества копий SMN1:SMN2
[0061] На ФИГ. 16 показан неограничивающий пример IGV снимка области SMN2 в образце с делецией в экзоне 7-8. Горизонтальные линии соединяют два прочтения в пару на центральной дорожке выравнивания. Результаты BLAT для двух разделенных прочтений, охватывающих точку разрыва, показаны в нижней дорожке, показывая два сегмента одного и того же выравнивания для прочтения с каждой стороны от точки разрыва делеции.
[0062] На ФИГ. 17 показаны неограничивающие примеры графиков, иллюстрирующих корреляцию между необработанными количествами копий SMN1 при 15 различиях оснований вблизи с840.C>T и необработанными количествами копий SMN1 на сайте с840.C>T. Необработанное количество копий SMN1 в каждом сайте рассчитывали как количество копий интактного SMN, умноженное на долю SMN1, поддерживающие количество прочтений SMN1 + SMN2 из количества прочтений. Коэффициенты корреляции приведены в заголовке каждого графика.
[0063] На ФИГ. 18A и 18B показаны неограничивающие примеры графиков с гаплотипами SMN1/SMN2 в образцах с SMN1: 2 SMN2: 0 и SMN1: 2 SMN2: 1 в 1kGP. По оси y показаны необработанные количества копий SMN1, как показано на ФИГ. 16. По оси X показаны 16 сайтов, индексы которых перечислены и объяснены в таблице 8. Индекс № 13 представляет сайт c840.C>T. Образцы с SMN1:2 SMN2: 0 показаны вместе на верхнем левом графике. Образцы с SMN1:2 SMN2:1 показаны в виде 5 кластеров. ФИГ. 18A Неафриканская популяция ФИГ. 18B Африканская популяция
[0064] На ФИГ. 19 показан неограничивающий пример IGV снимка, показывающего делецию 1,9 т. п. н. в SMN1 в MB509.
[0065] На ФИГ. 20 показан неограничивающий иллюстративный график, иллюстрирующий количество копий SMN1/SMN2/SMN* в когортах 1kGP и NIHR.
[0066] На ФИГ. 21A и 21B показаны расхождения и отсутствие определений в проверочных выборках.
[0067] На ФИГ. 22 представлены определения количества копий, полученные из BWA и Isaac BAM.
[0068] На ФИГ. 23 представлен неограничивающий пример графика, на котором показано качество данных WGS в области CYP2D6/7. Среднее качество картирования для выборок 1kGP нанесено на график для каждого положения в области CYP2D6/7. В окне длиной 200 п. о. применяют медианный фильтр. REP6, REP7 и экзоны 9 CYP2D6/7 показаны в виде прямоугольников слева (CYP2D6) и справа (CYP2D7). Две области повтора длиной 2,8 т. п. н. ниже CYP2D6 (REP6) и CYP2D7 (REP7) идентичны и по существу несовместимы. Пунктирной рамкой обозначена область спейсера между CYP2D7 и REP7. Две основные гомологичные области в генах заштрихованы.
[0069] На ФИГ. 24 показаны структурные варианты, подтвержденные прочтением PacBio CCS. Прочтение PacBio подтверждает делецию (*5), дупликацию и слияние (*36, *68 и *13). Графики получали с использованием sv-viz2 (zotero.org/google-docs/?xAunA6). Для делеций и дупликаций из-за значительной гомологии в регионах REP точное положение точек разрыва в REP недоступно. Точки разрыва в А и В приведены только для иллюстрации.
[0070] На ФИГ. 25 показан неограничивающий пример графика, на котором показаны частоты аллелей CYP2D6 для пяти этнических популяциях для десяти наиболее распространенных гаплотипов с измененной функцией CYP2D6. Один гаплотип (*2x2) характеризуются повышенной функцией, два гаплотипа (*4 и *4 + *68) - отсутствием функции, а остальные гаплотипы - сниженной функцией.
[0071] На ФИГ. 26 показано, что сайты из различием оснований CYP2D6/CYP2D7 отличаются высокой вариабельностью в популяции. На оси y показана частота образцов, в которых CN для основания CYP2D6 определены в 2 из всех образцов, имеющих общее CN CYP2D6 + CYP2D7, равное 4. По оси X показаны координаты генома в hg38. Экзоны CYP2D6 показаны серыми прямоугольниками над графиком. Черной горизонтальной линией обозначено отсечение 98%.
[0072] На ФИГ. 27 показаны необработанные CNCYP2D6 в сайтах дифференцировки CYP2D6/7 в примерах с SV. Необработанные CN CYP2D6 рассчитывали как общее CN CYP2D6+CYP2D7, умноженное на соотношение CYP2D6 поддерживающих прочтений из CYP2D6 и CYP2D7 поддерживающих прочтений. Большой ромб обозначает количество копий генов, полученных из CYP2D6 на конце гена (может представлять собой полный ген CYP2D6 или слитый ген, заканчивающийся CYP2D6), вычисленное как общее CN CYP2D6+CYP2D7 минус CN спейсерной области CYP2D7 (см. ФИГ. 23). Для обнаружения SV в каждом сайте определяли CN CYP2D6, и изменение CN CYP2D6 в гене указывало на присутствие SV. Например, в HG01161 CN CYP2D6 изменился с 2 на 1 между экзоном 7 и экзоном 9, что указывает на гибридный ген CYP2D7-CYP2D6. В HG00553 CN CYP2D6 изменилось с 2 на 3 между экзоном 1 и экзоном 2, что указывает на гибридный ген CYP2D6-CYP2D7.
[0073] На ФИГ. 28 показано, что данные PacBio подтверждают слияние *10D в HG00421. Для сравнения показан образец с *36 (HG00612). прочтения PacBio, содержащие слияния, это прочтения с заштрихованными основаниями, основаниями, которые представляют собой программное сшивание, сделанное выравнивателем, и были получены из части слияния CYP2D7. Точки разрыва слияний близки друг к другу, но точка разрыва для *36 расположена выше от различий оснований в экзоне 9 (находящихся внутри черного блока), а точка разрыва для *10D расположена ниже, оставляя ген CYP2D6 интактным.
[0074] На ФИГ. 29 показано, что данные PacBio имели ложный *61 (гибрид CYP2D6/CYP2D7), полученный Aldy в HG02622. Ожидаемый генотип представлял собой *17/*45, но Aldy вызвал *61-подобный/*78 (оба *61 и *78 представляют собой звездчатые аллели с SV). Данные PacBio показали отсутствие структурного варианта в этой области (каждое прочтение полностью выровнено, без каких-либо мягких сшиваний, указывающих на не выровненные части).
[0075] На ФИГ. 30A и 30B показан новый гаплотип *10+*36+*36+*83 в HG00597. ФИГ. 30A График глубины, представленный на ФИГ. 27, показывает, что HG00597 имел три копии *36-подобных слияний, все из которых имели точку разрыва в гомологичной области между экзоном 7 и экзоном 9. ФИГ. 30B снимок экрана IGV с данными PacBio, демонстрирующий все прочтения, содержащие слияния, то есть те, которые выровнены мягким сшиванием. Одна копия слитого гена не имела g.42130692G>A, SNP, который находился в *36, но не в *83, как показано в области, фланкированной двумя черными вертикальными линиями. Эта копия была *83, и в отличие от того, что сообщалось в PharmVar, это был гибридный ген с REP7, а не с REP6, в противном случае количество копий области ниже экзона 9 было бы равно 3 вместо 2 на ФИГ. 30A.
[0076] На ФИГ. 31A и 31B сравнивали частоты 1kGP и pharmGKB. Каждая точка представляет гаплотип с частотой, которая больше или равна 0,5% либо в 1kGP, либо в pharmGKB. Отмечены связанные с SV гаплотипы, включая два гаплотипа с наибольшим отклонением (*10+*36 у жителей Восточной Азии и *4+*68 у европейцев). Другие гаплотипы с отклоненными значениями помечены (*2, *41, *34, *39, *2 и *29). Для каждой панели проводят диагональную линию. Коэффициенты корреляции приведены для каждой популяции (*10+*36 исключено для жителей Восточной Азии и *4+*68 исключено для европейцев для расчета). На ФИГ. 31B показаны значения в нижнем диапазоне значений (<5%).
[0077] На ФИГ. 32 показан неограничивающий пример снимка IGV, показывающий сборку de novo результатов прочтения PacBio в HG00733, не включающую слияние *68.
[0078] На всех чертежах ссылочные номера можно использовать повторно для указания соответствия между элементами ссылки. Рисунки представлены для иллюстрации примеров осуществления, описанных в данном документе, и не предназначены для ограничения объема описания.
Подробное описание сущности изобретения
[0079] В приведенном ниже подробном описании содержатся ссылки на соответствующие рисунки, которые являются частью настоящего документа. В графических материалах аналогичные символы, как правило, обозначают аналогичные компоненты, если иное не следует из контекста. Предполагается, что иллюстративные варианты осуществления, описанные в подробном описании, графических материалах и пунктах формулы изобретения, не имеют ограничительного характера. Допускается использовать другие варианты осуществления и вносить другие изменения без отступления от сущности или объема заявленного объекта изобретения, представленного в настоящем документе. Следует понимать, что аспекты данного описания, в общем и целом представленные в данном документе и проиллюстрированные на фигурах, можно перераспределять, заменять, комбинировать, разделять и конструировать в широком спектре различных конфигураций, все из которых явным образом предусмотрены данным описанием и являются частью описания в данном документе.
[0080] Все патенты, опубликованные заявки на патенты, другие публикации и последовательности из GenBank и других баз данных, упомянутые в данном документе, полностью включены в данный документ посредством ссылки в отношении соответствующей технологии.
[0081] В данном документе описаны способы определения количества копий гена выживания моторных нейронов 1 (SMN1) и/или гена выживания моторных нейронов 2 (SMN2). В некоторых вариантах осуществления способ определения количества копий гена SMN1 и/или гена SMN2 контролируется процессором (таким как аппаратный процессор или виртуальный процессор) и включает: получение данных секвенирования, содержащих множество прочтений последовательностей из образца от субъекта, выровненных с геном SMN1 или геном SMN2. Способ может включать: определение (i) первого количества прочтений последовательности из множества прочтений последовательностей, выровненных по первому участку гена SMN1 или SMN2, содержащим по меньшей мере один из первых 6 экзонов гена SMN1 или гена SMN2 соответственно, и (ii) второго количества прочтений последовательности из множества прочтений последовательностей, выровненных по второму участку SMN1 или SMN2, содержащим по меньшей мере экзон 7 или экзон 8 гена SMN1 или гена SMN2 соответственно. Способ может включать: определение (i) первого нормализованного количества прочтений последовательности, выровненной по первой области гена SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной по второй области SMN1 или SMN2, с применением (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2 соответственно. Способ может включать: определение (i) количества копий полноразмерных генов выживания моторных нейронов (SMN), каждый из которых является интактным геном SMN1, интактным геном SMN2, укороченным геном SMN1 или укороченным геном SMN2; и (ii) количества копий любых интактных генов SMN, каждый из которых является интактным геном SMN1 или интактным геном SMN2, с использованием модели смеси нормальных распределений, содержащей множество нормальных распределений, каждое из которых представляет разное целое количество копий, с учетом (i) первого нормализованного количества прочтений последовательности, выровненной по первой области SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненной по второй области SMN1 или SMN2, соответственно. Способ может включать: для одного множества специфичных для гена SMN1 оснований, связанных с интактным геном SMN1, определение наиболее вероятной комбинации из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любых определенных интактных генов SMN, учитывая (a) количество прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат основание, специфичное для гена SMN1, и (b) количество прочтений последовательностей из множества прочтений последовательности с основаниями, которые содержат специфическое для гена SMN2 основание гена SMN2, соответствующее специфическому для гена SMN1 основанию. Способ может включать: определение количества копий гена SMN1 и/или гена SMN2 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенного для специфического для гена SMN1 основания.
[0082] В данном документе описаны способы генотипирования гена члена 6 подсемейства D семейства 2 цитохрома P450 (CYP2D6). В некоторых вариантах осуществления способ генотипирования гена CYP2D6 контролируется процессором (таким как аппаратный процессор или виртуальный процессор) и включает: получение данных секвенирования, содержащих множество прочтений последовательностей из образца от субъекта, выровненных с геном CYP2D6 или геном члена 7 подсемейства D семейства 2 цитохрома P450 (CYP2D7). Способ может включать: определение (i) первого количества прочтений последовательности из множества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7. Способ может включать: определение (i) первого нормализованного количества прочтений последовательности, выровненной с геном CYP2D6 или геном CYP2D7, с применением (i) длины гена CYP2D6 или гена CYP2D7 соответственно. Способ может включать: определение (i) общего количества копий гена CYP2D6 и гена CYP2D7 с помощью модели смеси нормальных распределений, содержащей множество нормальных распределений, каждое из которых представляет другое целое количество копий, на основании (i) первого нормализованного количества прочтений последовательности, выровненной с геном CYP2D6 или геном CYP2D7. Способ может включать: для одного из множества оснований, специфичных для гена CYP2D6 определение наиболее вероятной комбинации множества возможных комбинаций, каждая из которых содержит возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, суммированное до общего количества копий гена CYP2D6 и определенного гена CYP2D7, учитывая (a) прочтение множества последовательностей с основаниями, которые содержат специфическое для гена CYP2D6 основание, и (b) прочтение множества последовательностей с основаниями, которые содержат специфическое для гена CYP2D7 основание, соответствующее основанию, специфичному для гена CYP2D6. Способ может включать: определение аллеля гена CYP2D6, имеющегося у субъекта, с использованием наиболее вероятной комбинации возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7, определенного для основания, специфичного для гена CYP2D6.
[0083] В данном документе описаны способы генотипирования паралогов. В некоторых вариантах осуществления способ генотипирования паралогов контролируется процессором (таким как аппаратный процессор или виртуальный процессор) и включает: получение данных секвенирования, содержащих множество прочтений последовательностей из образца от субъекта, выровненных с первым паралогом или вторым паралогом. Способ может включать: определение количества копий паралогов первого типа с помощью модели смеси нормальных распределений, содержащей множество распределений, каждое из которых представляет различное целое число копий, заданное (i) первым количеством прочтений последовательностей, выровненных с первой областью. Способ может включать: определения наиболее вероятной комбинации для одного из множества оснований, специфичных для первого паралога, из множества возможных комбинаций, каждая из которых содержит возможное количество копий первого паралога первого типа и возможное количество копий второго паралога первого типа, суммированное с количеством копий определенных паралогов первого типа, при заданном (a) количестве прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат основание специфическое для первого паралога, и (b) количестве прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат основание специфическое для второго паралога, соответствующее основанию специфическому для первого паралога. Способ может включать: определения количества копий или аллеля первого паралога с использованием наиболее вероятной комбинации возможного количества копий первого паралога и возможного количества копий второго паралога, определенного для основания специфического для первого паралога.
[0084] В данном документе описаны варианты осуществления системы (например, компьютерной системы), содержащей энергонезависимую память, выполненную с возможностью хранения исполняемых команд; и процессора (например, аппаратный процессор или виртуальный процессор), находящийся в соединении с энергонезависимой памятью, причем аппаратный процессор запрограммирован с помощью исполняемых команд для выполнения любого способа, описанного в данном документе. Описанные в данном документе варианты осуществления устройства (например, электронного устройства), содержащего энергонезависимую память, выполненную с возможностью хранения исполняемых команд; и процессора (например, аппаратный процессор или виртуальный процессор), находящийся в соединении с энергонезависимой памятью, причем аппаратный процессор запрограммирован с помощью исполняемых команд для выполнения любого способа, описанного в данном документе. Описанные в данном документе варианты осуществления машиночитаемого носителя, содержащего исполняемые команды, которые при исполнении процессором (например, аппаратным процессором или виртуальным процессором) системы или устройства приводят к выполнению аппаратным процессором любого способа, описанного в данном документе.
Диагностика спинальной мышечной атрофии и исследование на носительство на основе данных секвенирования всего генома
[0085] Спинальная мышечная атрофия (SMA) характеризуется ослаблением скелетных мышц и является ведущей генетической причиной смерти в раннем детском возрасте с частотой 1 на 6000-10000 живорождений и частотой носительства 1:40-8012. SMA вызывается мутациями в гене SMN1 (ген выживания моторных нейронов 1) (ФИГ. 1A). Дублированный ген SMN2 отличается от SMN1 всего несколькими парами оснований, одна из которых, вариант сплайсинга c.840C>T в экзоне 7, имеет функциональные последствия. При прерывании энхансера сплайсинга мутация c.840C>T приводит к усилению пропуска экзона 7 и снижению полноразмерных транскриптов в SMN23 (ФИГ. 1B-1D). Геномная область подвержена неравному кроссинговеру и генной конверсии, что приводит к вариабельному количеству копий SMN1 и SMN2 (ФИГ. 1B). Из-за высокой частоты возникновения и тяжести заболевания рекомендуется проводить обширный скрининг на SMA, и ключевым фактором для данного скрининга является определение количества копий SMN1 для диагностики SMA и тестирования на носительство. Кроме того, количество копий SMN2 определяет степень тяжести SMA и важно для клинической классификации и прогноза.
[0086] В стандартных тестах на носительство SMA используются способы на основе ПЦР, такие как мультиплексная амплификация лигированных зондов (MLPA), количественная ПЦР (кПЦР) и цифровая ПЦР. Эти способы в основном нацелены на сайт c.840C>T. Включение SMA-скрининга в высокопроизводительные тесты на основе NGS, которые могут одновременно профилировать большое количество генов или даже весь геном, может быть полезным. Почти идеальная идентичность последовательностей между SMN1 и SMN2 затрудняет выявление вариантов стандартными способами на основе NGS.
[0087] В данном документе описан определитель количества копий SMN на основе способа биоинформатики, который определяет количество копий SMN1 и SMN2 с данными полногеномного секвенирования (WGS) (ФИГ. 1E). Способ может включать определение количества копий SMN1 + SMN2 в двух областях, экзонах 1-6 и экзонах 7-8, путем суммирования прочтений в SMN1 и SMN2. Способ может включать дифференцировку SMN1 от SMN2 с использованием количества прочтений при фиксированных различиях оснований. В некоторых вариантах осуществления способ не включает повторное выравнивание выровненных последовательностей по модифицированному эталонному образцу. Способ представляет собой первый инструмент определения количества копий SMN, который может идентифицировать как пациентов с SMA, так и носителей по данным WGS. Некоторые варианты осуществления способа не ограничены экзонами 7 и 8 и не сосредоточены главным образом на c.840C>T. Этот метод использует общегенный подход и обеспечивает наиболее полный набор определений, включая количество копий полноразмерного SMN1 и SMN2, а также укороченную форму SMN с делецией экзонов 7 и 8. Этот способ можно легко применить к любым данным WGS и будет полезным инструментом для диагностики SMA и скрининга носителей для включения в высокопроизводительный скрининг WGS в масштабах всей популяции.
[0088] На ФИГ. 1A-1E показаны пояснения определения количества копий SMA SMN в соответствии с одним вариантом осуществления способа биоинформатики, описанного в данном документе. В таблице 1 показана дифференцировка SMN1 от SMN2 на основе фиксированного однонуклеотидного полиморфизма (SNP) в соответствии с вариантом осуществления способа. Определение количества копий SMN1 производится в 16 сайтах вблизи c.840C>T. Девять сайтов с высоким процентом совпадения с c.840C> T выбраны для совместного определения количества копий SMN1. На ФИГ. 2A-2C и в таблице 2 показано популяционное распределение определенного количества копий SMN1/2. Большее количество копий SMN1 наблюдали при меньшем количестве копий SMN2, что указывает на генную конверсию как механизм вариабельности количества копий SMN1 и SMN2. В табл. 3 показана проверка количества копий, определенных с использованием способа биоинформатики, в сравнении с количеством копий, определенным методом цифровой ПЦР. Проверка по сравнению с цифровой ПЦР показала 100% совпадение количества копий в SMN1 и 98% в SMN2. На ФИГ. 3 показан показатель SMA, идентифицированный в двух тройках в проекте Next Generation Children и подтвержденный с помощью MLPA. На Фиг. 4 и в таблице 4 показаны частоты в популяциях, определенные с помощью способа биоинформатики, согласующиеся с предыдущими исследованиями.
Таблица 1. Дифференциация SMN1 от SMN2 на основании фиксированного однонуклеотидного полиморфизма (SNP)
(c.840 C>T)
Таблица 2. Распределение в популяциях количества копий SMN1/2
Таблица 3. Проверка количества копий, определенных с использованием способа биоинформатики, в сравнении с количеством копий, определенным методом цифровой ПЦР
Таблица 4. Частоты в популяциях, определенные с помощью способа биоинформатики, согласующиеся с предыдущими исследованиями
aHendrickson et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet. 2009;46(9):641-644. doi:10.1136/jmg.2009.066969.
bSugarman et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72 400 specimens. Eur J Hum Genet. 2012; 20(1): 27-32.doi: 10.1038/Ejhg.2011.134.
*Афроамериканцы
Характеристика пригодных для медицинского применения вариантов из 2500 общедоступных высокоглубинных геномов различного происхождения
[0089] Данные о популяционном секвенировании всего генома (WGS) становятся все более доступными. Например, доступны общедоступные данные секвенирования, такие как данные WGS с большой глубиной (>30X) для >2500 образцов из проекта 1000 Genomes (1kGP). Это значительно улучшило клиническую интерпретацию простых однонуклеотидных вариантов (SNV) и вставок/делеций (индексов). Однако многие важные с медицинской точки зрения области и варианты, такие как повторы триплетов и гомологи, не включены в базы данных на основе WGS, поскольку аннотирование этих областей и вариантов требует применения специализированных биоинформатических способов. В связи с этим необходима характеристика известных клинических вариантов на уровне популяции, чтобы максимизировать влияние экспериментов по секвенированию популяции. В некоторых вариантах осуществления способы, описанные в данном документе, устраняют три недостатка процессов стандартного вторичного анализа: 1) обнаружение спинальной мышечной атрофии (SMA) и скрининг носителей, 2) генотипирование CYP2D6 для фармакогеномного применения и 3) обнаружение экспансии повторных триплетов. Способы можно применять для определения количества копий SMN1/2, звездчатых аллелей CYP2D6, экспансии повторов в популяции 1kGP и количественного определения различий между субпопуляциями. В данном документе описаны распределения частот по субпопуляции и перпендикулярная проверка этих способов с использованием подтверждающих данных, сгенерированных из высококачественных длинных прочтений.
CYP2D6
[0090] CYP2D6 является важным ферментом, метаболизирующим лекарственные средства, который является высокополиморфным (ФИГ. 5). Последовательность CYP2D6 имеет высокое сходство со своим псевдогенным паралогом (CYP2D7). Генотипирование CYP2D6 при помощи WGS является сложной задачей из-за общих конверсий генов между CYP2D6 и CYP2D7 (далее именуемых CYP2D6/7), общих SV (делеции генов, дупликации и гибридные гены CYP2D6/7; См. ФИГ. 6 для иллюстрации), а также сходство последовательностей CYP2D/7, что приводит к неоднозначному выравниванию прочтения для любого из генов (ФИГ. 5). В данном документе описан определитель CYP2D6, основанный на способе биоинформатики, который способен определить (например, точно определить) диплотипы, нацеленные на звездчатые аллели (например, все звездчатые аллели) с известными функциями. В некоторых вариантах осуществления способ включает следующие действия:
[0091] 1. Определение общего количества копий CYP2D6+CYP2D7.
[0092] 2. Определение CNV/гибридов на основании определений количества копий на сайтах дифференциации CYP2D6/CYP2D7.
[0093] 3. Определение 56 SNP/индексов из BAM (или другого файла, содержащего прочтение последовательностей).
- Использование информации о количестве копий.
- прочтение результатов в обоих положениях CYP2D6 и CYP2D7 в гомологичных областях.
[0094] 4. Определение звездчатых аллелей и диплотипов на основе всех определенных вариантов.
[0095] В таблице 5 показаны результаты проверки определений звездчатого аллеля CYP2D6, выполненных этим способом. Определения звездчатого аллеля CYP2D6, полученные способом для 92 из 96 образцов, согласуются с определениями консенсуса GeT-RM из множества платформ. Этот способ превзошел по эффективности такие определители, как Aldy (определение звездчатого аллеля CYP2D6 для 89 из 96 образцов согласуется с консенсусом GeT-RM) и Stargazer (определение звездчатого аллеля CYP2D6 для 83 из 96 образцов согласуется с консенсусом GeT-RM).
Таблица 5. Проверка определителя CYP2D6.
[0096] На ФИГ. 7 показано, аллельные частоты, определенные способом, согласуются с базой данных PharmVar Database Pharmacogene Variation (PharmVar) Consortium.
Определение количества копий гена выживания моторных нейронов 1 с использованием данных секвенирования
[0097] На ФИГ. 8 представлена блок-схема, показывающая пример способа 800 определения количества копий выживания гена моторных нейронов 1 с использованием данных секвенирования, таких как данные секвенирования целого генома. Способ 800 может быть реализован в виде набора исполняемых программных команд, хранящихся на машиночитаемом носителе, таком как один или более дисководов вычислительной системы. Например, вычислительная система 1100, показанная на ФИГ. 11 и более подробно описанная ниже, может выполнять набор исполняемых программных команд для реализации способа 800. При инициировании способа 800 исполняемые программные команды могут быть загружены в запоминающее устройство, такое как RAM, и выполнены одним или более процессорами вычислительной системы 1100. Хотя способ 800 описан в отношении вычислительной системы 1100, показанной на ФИГ. 11, описание приводится только в качестве иллюстрации и не носит ограничительного характера. В некоторых вариантах осуществления способ 800 или его фрагменты могут быть выполнены последовательно или параллельно множеством вычислительных систем.
[0098] После того, как способ 800 начинается на этапе 804, способ 800 переходит к этапу 808, где вычислительная система (такая как вычислительная система 1100, описанная со ссылкой на ФИГ. 11) определяет (i) первое количество прочтений последовательности из множества прочтений последовательностей, выровненных с первой областью гена выживания моторных нейронов 1 (SMN1) или выживания моторных нейронов 2 (SMN2), содержащим по меньшей мере один из 1-6 экзонов гена SMN1 или гена SMN2, соответственно, и (ii) второе количество прочтений последовательности из множества прочтений последовательностей, выровненных со второй областью гена SMN1 или SMN2, содержащим по меньшей мере экзон 7 или экзон 8 гена SMN1 или гена SMN2 соответственно. Первое количество прочтений последовательностей, выровненных с первой областью SMN1 или SMN2 (или второе количество прочтений последовательностей, выровненных со второй областью SMN1 или SMN2), может быть или примерно равно, например, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 2000, 3000, 4000, 5000, 6000, 7000, 8000, 9000, 10000 или более.
[0099] По меньшей мере один из экзонов 1-6 гена SMN1 может содержать экзон 1, экзон 2, экзон 3, экзон 4, экзон 5 и/или экзон 6 гена SMN1. По меньшей мере один из экзонов 1-6 гена SMN2 может содержать экзон 1, экзон 2, экзон 3, экзон 4, экзон 5 и/или экзон 6 гена SMN2. Первая область SMN1 или SMN2 может содержать экзон от 1 до 6 гена SMN1 или гена SMN2 соответственно и может иметь длину примерно 22,2 т. п. н. Вторая область SMN1 или SMN2 может содержать экзон 7 и экзон 8 гена SMN1 или гена SMN2 соответственно и может иметь длину примерно 6 т. п. н.
[0100] В некоторых вариантах осуществления вычислительная система получает данные секвенирования, содержащие множество прочтений последовательностей, полученных из образца от субъекта, выровненных с геном SMN1 или геном SMN2. Данные секвенирования могут включать в себя данные секвенирования целого генома (WGS) или данные WGS с помощью коротких прочтений. В некоторых вариантах осуществления субъект является субъектом-плодом, неонатальным субъектом, педиатрическим субъектом, субъектом-подростком или взрослым субъектом. Образец может содержать клетки или внеклеточную ДНК. Образец может содержать фетальные клетки или внеклеточную фетальную ДНК.
[0101] В некоторых вариантах осуществления прочтение последовательности из множества прочтений последовательностей, выравнивается с первой областью SMN1 или SMN2 или со второй областью SMN1 или SMN2 с показателем качества выравнивания, равным приблизительно нулю. Качество выравнивания может составлять, например, 0, 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,10 или более (по шкале от 0 до 1 от оценки выравнивания).
[0102] Способ 800 переходит от этапа 808 к этапу 812, где вычислительная система определяет (i) первое нормализованное количество прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и (ii) второе нормализованное количество прочтений последовательности, выровненной со второй областью SMN1 или SMN2, с применением (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2 соответственно. Первое нормализованное количество прочтений последовательностей, выровненных с первой областью SMN1 или SMN2 (или второе нормализованное количество прочтений последовательностей, выровненных со второй областью SMN1 или SMN2), может быть или примерно равно, например, 1, 2, 3, 4, 5, 6, 7, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Длина первой области SMN1 или SMN2 может быть или примерно равно, например, 3 т. п. н., 6 т. п. н., 9 т. п. н., 12 т. п. н., 15 т. п. н., 18 т. п. н., 21 т. п. н., 22,2 т. п. н., 24 т. п. н. или более. Длина второй области SMN1 или SMN2 может быть или примерно равно, например, 3 т. п. н., 6 т. п. н. или более.
[0103] В некоторых вариантах осуществления для определения (i) первого нормализованного количества прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательностей, выровненных со второй областью, вычислительная система может определять (i) первое нормализованное количества прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) второе нормализованное количества прочтений последовательностей, выровненных со второй областью SMN1 или SMN2, с применением (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2, соответственно, и (iii) глубину прочтений последовательности области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования. Глубина прочтения последовательности области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования может быть или примерно равно, например, 3, 4, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более.
[0104] Для определения (i) первого нормализованного количества прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательностей, выровненных со второй областью SMN1 или SMN2, вычислительная система определяет (i) нормализованное количество прочтений последовательностей по длине первой области SMN1 или SMN2, выровненных с первой областью SMN1 или SMN2, и (ii) нормализованное количество прочтений последовательностей по длине второй области SMN1 или SMN2, выровненных со второй областью SMN1 или SMN2, с применением (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2, соответственно. Вычислительная система может определять (i) первую нормализованную глубину прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) вторую нормализованную глубину прочтений последовательностей, выровненных со второй областью SMN1 или SMN2, на основе (i) нормализованного количества прочтений последовательностей по длине первой области SMN1 или SMN2 и (ii) нормализованного количества прочтений последовательностей по длине второй области SMN1 или SMN2, соответственно, при использовании глубины прочтения последовательности области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2. Первое нормализованное количество прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и второе нормализованное количество прочтений последовательностей, выровненных со второй областью SMN1 или SMN2, могут представлять собой первую нормализованную глубину и вторую нормализованную глубину соответственно.
[0105] В некоторых вариантах осуществления для определения (i) первого нормализованного количества прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательностей, выровненных со второй областью, вычислительная система может определять (i) первое нормализованное количество прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) второе нормализованное количество прочтений последовательностей, выровненных со второй областью SMN1 или SMN2, с применением (i) содержания GC в первой области SMN1 или SMN2 и (ii) содержания GC во второй области SMN1 или SMN2, соответственно, и (iii) глубину прочтения последовательности области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования и (iv) содержание GC в области генома. Содержание GC в первой области SMN1 или SMN2 (или содержание GC во второй области SMN1 или SMN2) может быть или примерно равно, например, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59% или 60%. Глубина прочтения последовательности области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования может быть или примерно равно, например, 3, 4, 5, 10, 20, 30, 40, 50, 100 или более. Содержание GC в области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования может быть или примерно равно, например, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59% или 60%.
[0106] В некоторых вариантах осуществления глубина области включает среднюю глубину прочтения последовательности области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования. Глубина области может включать в себя медианную глубину прочтения последовательности области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования. Глубина области может быть или примерно равно, например, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Длина области может содержать примерно 500, 1000, 1500, 2000, 2500, 3000, 3500, 4000 или более предварительно выбранных областей длиной примерно 0,5 т. п. н., 1 т. п. н., 1,5 т. п. н., 2 т. п. н., 2,5 т. п. н. или 3 т. п. н. в каждой по всему геному субъекта. Например, область может содержать примерно 3000 предварительно выбранных областей длиной примерно 2 т. п. н. в каждой по всему геному субъекта.
[0107] В некоторых вариантах осуществления первое нормализованное количество прочтений последовательностей, выровненных с первой областью SMN1 или SMN2 (или второе нормализованное количество прочтений последовательностей, выровненных со второй областью SMN1 или SMN2), составляет или составляет примерно 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Например, (i) первое нормализованное количество прочтений последовательности, выровненной с первой областью SMN1 или SMN2, и/или (ii) второе нормализованное количество прочтений последовательности, выровненной со второй областью SMN1 или SMN2, составляет от примерно 30 до примерно 40.
[0108] Способ 800 переходит от этапа 812 к этапу 816, где вычислительная система определяет (i) количество копий общих генов выживания моторных нейронов (SMN) и (ii) количество копий любого(-ых) интактного(-ых) гена(-ов) SMN с использованием модели смеси нормальных распределений, содержащей множество распределений, каждое из которых представляет другое целое число копий, заданное (i) первым нормализованным количеством прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) второе нормализованное количество прочтений последовательностей, выровненных со второй областью SMN1 или SMN2 соответственно. Общие гены выживания моторных нейронов могут содержать интактный ген SMN1, интактный ген SMN2, укороченный ген SMN1 и/или укороченный ген SMN2. Любой(-ые) интактный(-ые) ген(-ы) SMN может (могут) содержать интактный ген SMN1 и/или интактный ген SMN2. Количество копий общего (-их) гена(-ов) SMN (или любого(-ых) гена(-ов) данного описания) может составлять или примерно составлять, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более. Количество копий любого(-ых) интактного(-ых) гена(-ов) SMN (или любого(-ых) гена(-ов) данного описания) может составлять или примерно составлять, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более.
[0109] В некоторых вариантах осуществления модель смеси нормальных распределений представляет собой одномерную модель смеси нормальных распределений. Множество распределений модели смеси нормальных распределений могут представлять целые числа копий, например от 0 до 5, от 0 до 6, от 0 до 7, от 0 до 8, от 0 до 9, от 0 до 10, от 0 до 11, от 0 до 12, от 0 до 13, от 0 до 14 или от 0 до 15. Например, множество распределений модели смеси нормальных распределений могут представлять целые числа копий от 0 до 10. Среднее значение (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более) каждого из множества распределений может представлять собой целое количество копий, представленное распределением (например, количество копий 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более). Стандартное отклонение распределений может составлять или примерно составлять, например, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1 или более.
[0110] В некоторых вариантах осуществления для определения (i) количества копий общего(-ых) гена(-ов) SMN и (ii) количества копий любого(-ых) интактного(-ых) гена(-ов) SMN вычислительная система может определять (i) количество копий общего(-их) гена(-ов) SMN и (ii) количество копий любого(-ых) интактного(-ых) гена(-ов) SMN с использованием модели смеси нормальных распределений, и первый предварительно определенный порог апостериорной вероятности с учетом (i) первого нормализованного количества прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательностей, выровненных со второй областью SMN1 или SMN2 соответственно. Первый заданный порог апостериорной вероятности (или любой заданный порог апостериорной вероятности данного описания) может составлять или примерно составлять, например, 0,80, 0,81, 0,82, 0,83, 0,84, 0,85, 0,86, 0,87, 0,88, 0,89, 0,90, 0,91, 0,92, 0,93, 0,94, 0,95, 0,96, 0,97, 0,98, 0,99 или более. Например, первый предварительно определенный порог апостериорной вероятности может составлять 0,95.
[0111] Способ 800 переходит от этапа 816 к этапу 820, где вычислительная система определяет для одного из множества, оснований, специфичных для гена SMN1 (также называемых в данном документе дифференцирующими основаниями SMN), связанных с интактным геном SMN1, наиболее вероятная комбинация из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любого(-ых) интактного(-ых) гена(-ов) SMN, учитывая (a) количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей) из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей) из множества прочтений последовательности с основаниями, которые содержат специфическое для гена SMN2 основание гена SMN2, соответствующее специфическому для гена SMN1 основанию. Возможное количество копий гена SMN1 может составлять или примерно составлять, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более. Возможное количество копий гена SMN2 может составлять или примерно составлять, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более.
[0112] В некоторых вариантах осуществления наиболее вероятная комбинация возможного количества копий гена SMN1 и возможного количества копий гена SMN2 связана с самой высокой апостериорной вероятностью, по сравнению с другими комбинациями множества комбинаций с заданным (a) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена SMN1 основание, и (b) количество прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат соответствующее специфическое для гена SMN2 основание. Самая высокая апостериорная вероятность (или любая вероятность данного описания) может составлять или примерно составлять, например, 60%, 61%, 62%, 63%, 64%, 65%, 66%, 67%, 68%, 69%, 70%, 71%, 72%, 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или более. Различие в апостериорной вероятности (или любой вероятности данного описания) может составлять или примерно составлять, например, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24%, 25%, 26%, 27%, 28%, 29%, 30% или более.
[0113] В некоторых вариантах осуществления для определения наиболее вероятной комбинации возможного количества копий гена SMN1 и возможной комбинации гена SMN2 компьютерная система может определять наиболее вероятную комбинацию из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любых определенных интактных генов SMN, учитывая соотношение (a) количества прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена SMN1 основание, и (b) количества прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат SMN2 ген-специфическое основание гена SMN2, соответствующее специфичному для гена SMN1 основанию. Для определения наиболее вероятной комбинации возможного количества копий гена SMN1 и возможной комбинации гена SMN2; вычислительная система может определять (a) количество прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количество прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат SMN2 ген-специфическое основание гена SMN2 соответствующее специфичному для гена SMN1 основанию. Вычислительная система может определять соотношение (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат SMN2 ген-специфическое основание гена SMN2 соответствующее специфичному для гена SMN1 основанию. Вычислительная система может определить наиболее вероятную комбинацию, из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любого интактного гена SMN, определенным на основе соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат SMN2 ген-специфическое основание гена SMN2 соответствующее специфичному для гена SMN1 основанию.
[0114] В некоторых вариантах осуществления для определения наиболее вероятной комбинации возможного количества копий гена SMN1 и возможной комбинации гена SMN2 вычислительная система определяет для каждого из множества ген-специфических оснований SMN1: наиболее вероятную комбинацию из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любого определенного интактного гена SMN, которая связана с наибольшей апостериорной вероятностью, заданной (a) количество прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количество прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат SMN2 ген-специфическое основание гена SMN2 соответствующее специфичному для гена SMN1 основанию. Количество прочтений последовательностей, выровненных с специфичным для гена SMN1 основанием (или специфичным для гена SMN2 основанием), может составлять или примерно составлять, например, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Для определения количества копий гена SMN1 вычислительная система может определять количество копий гена SMN1 на основе возможного количества копий гена SMN1 наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенного для каждого из множества оснований, специфических для гена SMN1.
[0115] В некоторых вариантах осуществления специфичное для гена SMN1 основание представляет собой энхансер сплайсинга. Специфичное для гена SMN1 основание может представлять собой основание в с.840 гена SMN1. В некоторых вариантах осуществления специфичное для гена SMN1 основание имеет соответствие с каждым из множества характерных для конкретного гена SMN1 оснований, отличных от специфичного для гена SMN1 основания выше заранее определенного порога соответствия. Заданное пороговое значение соответствия (или любое пороговое значение данного описания) может составлять или примерно составлять, например, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или более. Например, пороговое значение соответствия может составлять 97%. Множество оснований, специфичных для гена SMN1 может содержать или примерно содержать 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 или более специфических для гена SMN1 оснований. Например, множество оснований, специфичных для гена SMN1 может содержать 8 оснований, специфичных для гена SMN1. Каждое из множества оснований, специфичных для гена SMN1 может находиться на интроне 6, экзоне 7, интроне 7 или экзоне 8 гена SMN1.
[0116] Множество оснований, специфичных для гена SMN1, если субъект принадлежит к первой расе (или этнической принадлежности), множество оснований, специфичных для гена SMN1, если субъект принадлежит к второй расе (или этнической принадлежности), и оснований, специфичных для гена SMN1, если субъект принадлежит к неизвестной расе, могут различаться. Расой может быть, например, европеоид, африканец, афроамериканец, американский индеец, коренной житель Аляски, азиат, южноазиатский житель, восточноазиатский житель, коренной житель Гавайских островов, выходец с островов Тихого океана или их комбинация. Расовая принадлежность (или этническая принадлежность) субъекта может быть неизвестной, а множество оснований, специфичных для гена SMN1, может быть неспецифичным для расы (или этнической принадлежности). Раса (или этническая принадлежность) субъекта может быть известна, и множество оснований, специфичных для гена SMN1, могут быть специфичными для расы (или этнической принадлежности) субъекта. В некоторых вариантах осуществления вычислительная система может принимать информацию о расе (или этнической принадлежности) субъекта. На основе принятой информации о расе (или этнической принадлежности) вычислительная система может выбирать множества оснований, специфичных для гена SMN1 из множества оснований, специфичных для гена SMN1.
[0117] Способ 800 переходит от этапа 820 к этапу 824, где вычислительная система определяет количество копий гена SMN1 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенного для специфического для гена SMN1 основания. В альтернативном или дополнительном варианте осуществления вычислительная система определяет количество копий гена SMN2 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенного для специфического для гена SMN1 основания.
[0118] В некоторых вариантах осуществления для определения количества копий гена SMN1 компьютерная система может определять количество копий гена SMN1 и количество копий гена SMN2 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенного для каждого из множества оснований, специфичных для гена SMN1. Для определения количества копий вычислительная система может определять количество копий гена SMN1, используя наиболее вероятную комбинацию возможного количества копий гена SMN1 и возможного количества копий определенного гена SMN2 основания, специфичного для гена SMN1 и второго предварительно заданного порога апостериорной вероятности для комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2. Второй предварительно заданный порог апостериорной вероятности (или любой заданный порог апостериорной вероятности данного описания) может составлять или примерно составлять, например, 0,50, 0,51, 0,52, 0,53, 0,54, 0,55, 0,56, 0,57, 0,58, 0,59, 0,60, 0,61, 0,62, 0,63, 0,64, 0,65, 0,66, 0,67, 0,68, 0,69, 0,70, 0,71, 0,72, 0,73, 0,74, 0,75, 0,76, 0,77, 0,78, 0,79, 0,80, 0,81, 0,82, 0,83, 0,84, 0,85, 0,86, 0,87, 0,88, 0,89, 0,90, 0,91, 0,92, 0,93, 0,94, 0,95, 0,96, 0,97, 0,98, 0,99 или более. Например, второй предварительно заданный порог апостериорной вероятности может составлять 0,6 или 0,8.
[0119] В некоторых вариантах осуществления большинство определенных возможных количеств копий гена SMN1 совпадают. Количество копий определенного гена SMN1 может быть согласованным с возможным количеством копий гена SMN1. Вычислительная система может определить возможную комбинацию, содержащую возможное количество копий гена SMN1 и возможное количество копий гена SMN2, суммированное с количеством копий любого интактного гена SMN, определенных с учетом (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат любое из множества оснований, специфических для гена SMN1, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат любое из множества соответствующих оснований, специфических для гена SMN2. Вычислительная система может определить возможное количество копий возможной комбинации - согласованное возможное количество копий гена SMN1.
[0120] В некоторых вариантах осуществления для определения количества копий гена SMN1 вычислительная система может определять количество копий гена SMN1, равное нулю, единице или более единицы. В некоторых вариантах осуществления вычислительная система может определить статус спинальной мышечной атрофии (SMA) у субъекта на основе количества копий гена SMN1. Статус SMA для субъекта может включать в себя SMA, носитель SMA/отсутствие SMA и не носитель SMA. В некоторых вариантах осуществления вычислительная система может определить субъекта как молчащего носителя SMA с использованием ряда прочтений последовательности из множества прочтений последовательностей, выровненных с g.27134 гена SMN1, и на основе прочтений последовательностей, выровненных с g.27134 гена SMN1.
[0121] Например, вычислительная система на этапе 820 может определить соотношение количества прочтений SMN1 к SMN2, перекрывающихся местоположений, где гены имеют разные основания последовательности. Для позиций, в которых SMN1 отличается от SMN2, вычислительная система может проводить прочтения, которые перекрываются либо на основании SMN1, либо SMN2. На основе этих прочтений вычислительная система может подсчитывать количество специфичных для SMN1 оснований и количество специфичных для SMN2 оснований. Вычислительная система может определить долю прочтений SMN1 или SMN2. Вычислительная система может вычислить количество копий SMN1 и SMN2 в позициях, в которых SMN1 отличается от SMN2. Вычислительная система может комбинировать количество полноразмерных копий с соотношением SMN1 к SMN2 для определения количества копий SMN1 и SMN2. Вычислительная система на этапе 824 может комбинировать количество копий из множества фиксированных различий между SMN1 и SMN2 для получения точного количества копий SMN1 и SMN2.
[0122] Для определения SMA/отсутствия SMA или носительства/отсутствия носительства. В некоторых вариантах осуществления вычислительная система может определять количество копий укороченного(-ых) гена(-ов) SMN с использованием (i) количества копий общего(-ых) определенного(-ых) гена(-ов) SMN и (ii) количества копий интактного(-ых) гена(-ов) SMN. Количество копий укороченного(-ых) гена(-ов) SMN может представлять собой разницу (i) количества копий общего(-ых) гена(-ов) SMN и (ii) количества копий интактного(-ых) гена(-ов) SMN.
[0123] Обработка. В некоторых вариантах осуществления вычислительная система может определять рекомендацию по лечению для субъекта на основании определенного количества копий гена SMN1. Рекомендация по лечению может включать введение субъекту Nusinersen и/или Zolgensma.
[0124] Способ 800 заканчивается этапом 828.
Генотипирование гена члена 6 подсемейства D семейства 2 цитохрома Р450 с использованием данных секвенирования
[0125] На ФИГ. 9 представлена блок-схема, показывающая пример способа 900 генотипирования гена члена 6 подсемейства D семейства 2 цитохрома Р450 с использованием данных секвенирования, таких как данные секвенирования целого генома. Способ 900 может быть реализован в виде набора исполняемых программных команд, хранящихся на машиночитаемом носителе, таком как один или более дисководов вычислительной системы. Например, вычислительная система 1100, показанная на ФИГ. 11 и более подробно описанная ниже, может выполнять набор исполняемых программных команд для реализации способа 900. При инициировании способа 900 исполняемые программные команды могут быть загружены в запоминающее устройство, такое как RAM, и выполнены одним или более процессорами вычислительной системы 1100. Хотя способ 900 описан в отношении вычислительной системы 1100, показанной на ФИГ. 11, описание приводится только в качестве иллюстрации и не носит ограничительного характера. В некоторых вариантах осуществления способ 900 или его фрагменты могут быть выполнены последовательно или параллельно множеством вычислительных систем.
[0126] Количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей), сопоставленных с геном CYP2D6 или геном CYP2D7, можно использовать для определения общего количества копий (CN) гена CYP2D6 и гена CYP2D7, с использованием модели смеси нормальных распределений. Общее количество копий гена CYP2D6 и гена CYP2D7 можно использовать для определения количества копий CYP2D6 при различных дифференцирующих основаниях CYP2D6/CYP2D7 (в данном документе также называемых основаниями, специфичными для гена CYP2D6) путем повторения всех возможных комбинаций количества копий CYP2D6 и CYP2D7 при дифференцирующих основаниях CYP2D6/CYP2D7. Для обозначения структурных вариантов можно использовать количество копий CYP2D6 при различных дифференцирующих основаниях CYP2D6/CYP2D7. Например, в каждом из дифференцирующих оснований CYP2D6/CYP2D7 (также называемых в данном документе основаниями, специфичными для гена CYP2D6) число хромосом, несущих ген CYP2D6, и число хромосом, несущих ген CYP2D7, можно определять путем комбинирования общего количества копий гена CYP2D6 и гена CYP2D7 с количеством прочтений, поддерживающим основание, специфичное для каждого гена. На основе определенного общего количества копий все возможные комбинации количества копий CYP2D6 и CYP2D7 могут быть повторены, чтобы получить комбинацию, которая дает самую высокую апостериорную вероятность для наблюдаемого количества прочтений CYP2D6 и CYP2D7. Структурные варианты можно определить путем идентификации оснований, в которых изменяется количество копий гена CYP2D6.
[0127] Можно определить один или более малых вариантов. Малые варианты могут быть определены, для каждого положения малого варианта малого варианта путем повторения всех возможных комбинаций количества копий вариантного аллеля и эталонного (невариантного) аллеля, чтобы определить наиболее вероятное количество копий аллеля, используя прочтение последовательности с положением малого варианта в гене CYP2D6 или CYP2D7. Например, при наличии всего трех копий гена CYP2D6 и 10 прочтений вариантного аллеля, и 20 прочтений эталонного аллеля, можно определить, что количество копий вариантного аллеля может быть определено как одно, т. е. существует одна копия гена CYP2D6, несущего малый вариант. Например, можно выполнять поиск малых вариантов, определяющих звездчатые аллели в данных секвенирования (например, в файле BAM). Интересующие малые варианты можно разделить на варианты, которые попадают в гомологичные области CYP2D6/CYP2D7, и варианты, которые не попадают. Для первых, прочтение варианта, выровненного с геном CYP2D6 или геном CYP2D7, перекрывающие каждое положение малого варианта интересующего гена CYP2D6 или соответствующее положение в гене CYP2D7, могут быть найдены. В последнем случае прочтения, выровненные с геном CYP2D6 и перекрывающие положение малого варианта интересующего гена CYP2D6, могут быть найдены. Количество копий определенное в области, также может быть учтено при определении малых вариантов. Определенные структурные варианты и малые варианты могут быть сопоставлены с определением звездчатых аллелей, чтобы определить звездчатые аллели, которые можно дополнительно группировать в гаплотипы.
[0128] После того как способ 900 начинается с этапа 904, способ 900 переходит к этапу 908, где вычислительная система (например, вычислительная система 1100, описанная со ссылкой на ФИГ. 11) определяет (i) первое количество прочтений последовательностей из множества прочтений последовательностей, выровненных с геном члена 6 подсемейства D семейства 2 цитохрома P450 (CYP2D6) или геном члена 7 подсемейства D семейства 2 цитохрома P450 (CYP2D7). Первое количество прочтений последовательностей, выровненных с первым геном CYP2D6 или геном CYP2D7 (или любым геном данного описания), может составлять или примерно составлять, например, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 2000, 3000, 4000, 5000, 6000, 7000, 8000, 9000, 10000 или более).
[0129] Вычислительная система может принимать данные секвенирования, содержащие множество прочтений последовательностей, полученных от образца от субъекта, выровненных с геном CYP2D6 или геном CYP2D7. В некоторых вариантах осуществления данные секвенирования содержат данные секвенирования целого генома (WGS) или данные WGS с помощью коротких прочтений. Субъект может являться субъектом-плодом, неонатальным субъектом, педиатрическим субъектом, субъектом-подростком или взрослым субъектом. Образец может содержать клетки или внеклеточную ДНК. Образец может содержать клетки или внеклеточную ДНК.
[0130] В некоторых вариантах осуществления прочтение последовательности из множестве прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, при этом показатель качества выравнивания составляет около нуля. Качество выравнивания может составлять, например, 0, 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,10 или более (по шкале от 0 до 1 от оценки выравнивания).
[0131] В некоторых вариантах осуществления для определения (i) первого количества прочтений последовательностей из множества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, вычислительная система может определять (i) первое количество прочтений последовательностей из множества прочтений последовательностей, выровненных по меньшей мере с одним экзоном или интроном гена CYP2D6 (например, одним из экзонов 1-9 или одним из интронов 1-8 гена CYP2D6) и/или по меньшей мере с одним из экзонов или интронов гена CYP2D7 (например, одним из экзонов 1-9 или одним из интронов 1-8 гена CYP2D7).
[0132] Способ 900 переходит от этапа 908 к этапу 912, где вычислительная система определяет (i) первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, с применением (i) длины гена CYP2D6 или гена CYP2D7 соответственно. Первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7 (или любым геном данного описания), может составлять или примерно составлять, например, 1, 2, 3, 4, 5, 6, 7, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Длина гена CYP2D6 может составлять или примерно составлять, например, 4,4 т. п. н. Длина гена CYP2D7 может составлять или примерно составлять, например, 4,9 т. п. н.
[0133] В некоторых вариантах осуществления для определения (i) первого нормализованного количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, вычислительная система может определять (i) первое нормализованное количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, с использованием (i) длины гена CYP2D6 или гена CYP2D7, соответственно, и (iii) глубину прочтения последовательности области генома субъекта, отличной от генетических локусов, содержащих ген CYP2D6 и ген CYP2D7 по данным секвенирования. Глубина прочтения последовательностей области генома субъекта, за исключением генетических локусов, содержащих ген CYP2D6 и ген CYP2D7 (или любые гены данного описания), в данных секвенирования может составлять или примерно составлять, например, 3, 4, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более.
[0134] Для определения (i) первого нормализованного количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, и (ii) второго нормализованного количества прочтений последовательностей, выровненных со второй областью, вычислительная система может определять (i) первое нормализованное количество прочтений по длине гена CYP2D6 или гена CYP2D7 последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, с применением (i) длины гена CYP2D6 или гена CYP2D7 соответственно. Вычислительная система может определять (i) первую нормализованную глубину прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, из (i) прочтения нормализованного по длине гена CYP2D6 или CYP2D7, с применением глубины прочтения последовательностей области генома субъекта, отличных от генетических локусов, содержащих ген CYP2D6 и CYP2D7. Первая нормализованная глубина прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, может представлять собой первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7 соответственно.
[0135] В некоторых вариантах осуществления для определения (i) первого нормализованного количества прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, вычислительная система может определять (i) первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, с применением(i) содержания GC в гене CYP2D6 или гене CYP2D7 и (iii) глубины прочтения последовательностей в области генома субъекта, за исключением генетических локусов, содержащих ген CYP2D6 и ген CYP2D7, по данным секвенирования, и (iv) содержание GC в области генома. Содержание GC в гене CYP2D6 или гене CYP2D7 (или любом гене данного описания) может составлять или примерно составлять, например, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59% или 60%. Глубина прочтения последовательности области генома субъекта, за исключением генетических локусов, содержащих ген CYP2D6 и ген CYP2D7, по данным секвенирования может быть или примерно равно, например, 3, 4, 5, 10, 20, 30, 40, 50, 100 или более. Содержание GC в области генома субъекта, за исключением генетических локусов, содержащих ген CYP2D6 и ген CYP2D7 (или любые гены данного описания), по данным секвенирования может составлять или примерно составлять, например, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59% или 60%.
[0136] Глубина области может представлять собой среднюю глубину прочтения последовательностей области генома субъекта, за исключением генетических локусов, содержащих ген CYP2D6 и ген CYP2D7, по данным секвенирования. Глубина области может включать медианную глубину прочтения последовательностей области генома субъекта, за исключением генетических локусов, содержащих ген CYP2D6 и ген CYP2D7, по данным секвенирования. Глубина области может составлять или примерно составлять приблизительно 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Длина области может содержать примерно 500, 1000, 1500, 2000, 2500, 3000, 3500, 4000 или более предварительно выбранных областей длиной примерно 0,5 т. п. н., 1 т. п. н., 1,5 т. п. н., 2 т. п. н., 2,5 т. п. н. или 3 т. п. н. в каждой по всему геному субъекта. Например, область может содержать примерно 3000 предварительно выбранных областей длиной примерно 2 т. п. н. в каждой по всему геному субъекта.
[0137] В некоторых вариантах осуществления (i) первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, и/или (ii) второе нормализованное количество прочтений последовательностей, выровненных со второй областью, составляет или составляет примерно 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Например, (i) первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, и/или (ii) второе нормализованное количество прочтений последовательностей, выровненных со второй областью, составляет от примерно 30 до примерно 40.
[0138] Способ 900 переходит от этапа 912 к этапу 916, где вычислительная система определяет (i) общее количество копий гена CYP2D6 и гена CYP2D7 с использованием модели смеси нормальных распределений, содержащей множество распределений, каждое из которых представляет разное целое число копий, учитывая (i) первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7. Общее количество копий гена CYP2D6 и гена CYP2D7 (или любых генов данного описания) может составлять или составлять примерно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более.
[0139] В некоторых вариантах осуществления модель смеси нормальных распределений представляет собой одномерную модель смеси нормальных распределений. Множество распределений модели смеси нормальных распределений могут представлять целые числа копий, например от 0 до 5, от 0 до 6, от 0 до 7, от 0 до 8, от 0 до 9, от 0 до 10, от 0 до 11, от 0 до 12, от 0 до 13, от 0 до 14 или от 0 до 15. Например, множество распределений модели смеси нормальных распределений могут представлять целые числа копий от 0 до 10. Среднее значение (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более) каждого из множества распределений может представлять собой целое количество копий, представленное распределением (например, количество копий 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более). Стандартное отклонение распределений может составлять или примерно составлять, например, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1 или более.
[0140] В некоторых вариантах осуществления для определения (i) общего количества копий гена CYP2D6 и гена CYP2D7 вычислительная система может определять (i) общее количество копий гена CYP2D6 и гена CYP2D7 с помощью модели смеси нормальных распределений и первого предварительно заданного порога апостериорной вероятности, учитывая (i) первое нормализованное количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7. Первый заданный порог апостериорной вероятности (или любой заданный порог апостериорной вероятности данного описания) может составлять или примерно составлять, например, 0,80, 0,81, 0,82, 0,83, 0,84, 0,85, 0,86, 0,87, 0,88, 0,89, 0,90, 0,91, 0,92, 0,93, 0,94, 0,95, 0,96, 0,97, 0,98, 0,99 или более. Например, первый предварительно определенный порог апостериорной вероятности может составлять 0,95.
[0141] Способ 900 переходит от этапа 916 к этапу 920, где вычислительная система определяет для одного из множества оснований, специфичных для гена CYP2D6 (также называемых в данном документе дифференцирующими основаниями CYP2D6/CYP2D7) наиболее вероятную комбинацию из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, суммированное с общим количеством копий гена CYP2D6 и определенным геном CYP2D7, учитывая (a) количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей) из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D6 основание, и (b) количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей) из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D7 основание, соответствующее специфичному для гена CYP2D6 основанию. Возможное количество копий гена CYP2D6 может составлять или составлять примерно, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более. Возможное количество копий гена CYP2D7 может составлять или составлять примерно, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более.
[0142] В некоторых вариантах осуществления наиболее вероятная комбинация возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7 связана с самой высокой апостериорной вероятностью, по сравнению с другими комбинациями множества комбинаций с заданным (a) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена CYP2D6 основание, и (b) количество прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат соответствующее специфическое для гена CYP2D7 основание. Самая высокая апостериорная вероятность (или любая вероятность данного описания) может составлять или примерно составлять, например, 60%, 61%, 62%, 63%, 64%, 65%, 66%, 67%, 68%, 69%, 70%, 71%, 72%, 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или более. Различие в апостериорной вероятности (или любой вероятности данного описания) может составлять или примерно составлять, например, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24%, 25%, 26%, 27%, 28%, 29%, 30% или более.
[0143] В некоторых вариантах осуществления для определения наиболее вероятной комбинации, содержащей возможное количество копий гена CYP2D6 и возможное количество копий, вычислительная система может определять наиболее вероятную комбинацию из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, суммированное с общим количеством копий гена CYP2D6 и определенным геном CYP2D7, учитывая соотношение (a) количества прочтений последовательности при прочтениях множества последовательностей с основаниями, которые содержат специфическое для гена CYP2D6 основание, и (b) количества прочтений последовательности при прочтениях множества последовательностей с основаниями, которые содержат специфическое для гена CYP2D7 основание, соответствующее специфичному для гена CYP2D6 основанию. Чтобы определить наиболее вероятную комбинацию, включающую возможное количество копий гена CYP2D6 и возможное количество копий, вычислительная система может определить (а) количество прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена CYP2D6 основание, и (b) из множества прочтений последовательностей с основаниями, которые содержат специфическое для гена CYP2D7 основание, соответствующее специфичному для гена CYP2D6 основанию. Вычислительная система может определять соотношение (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D6 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D7 основание, соответствующее специфичному для гена CYP2D6 основанию. Вычислительная система может определять наиболее вероятную комбинацию из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, суммированное с общим количеством копий гена CYP2D6 и определенным геном CYP2D7, с учетом соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D6 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D7 основание, соответствующее специфичному для гена CYP2D6 основанию.
[0144] В некоторых вариантах осуществления для определения наиболее вероятной комбинации возможного количества копий гена CYP2D6 и возможной комбинации гена CYP2D7 вычислительная система определяет для каждого из множества оснований, специфичных для гена CYP2D6: наиболее вероятная комбинация из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена CYP2D6 и возможное количество копий гена CYP2D7, суммированное с общим количеством копий определенного гена CYP2D6 и гена CYP2D7, которая связана с наибольшей апостериорной вероятностью, заданной (a) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена CYP2D6 основание, и (b) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат CYP2D7 ген-специфическое основание гена CYP2D7 соответствующее специфичному для гена CYP2D6 основанию. Количество прочтений последовательностей, выровненных с специфичным для гена SMN1 основанием (или специфичным для гена SMN2 основанием), может составлять или примерно составлять, например, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Для определения аллеля гена CYP2D6 у субъекта вычислительная система может определить аллель гена CYP2D6, который у субъекта имеется малый вариант или структурный вариант гена CYP2D6, или ни один из них, используя наиболее вероятную комбинацию возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7, определенного для каждого из множества оснований, специфических для гена CYP2D6.
[0145] В некоторых вариантах осуществления специфичное для гена CYP2D6 основание имеет соответствие с каждым из множества специфичных для гена CYP2D6 оснований, за исключением специфичного для гена CYP2D6 основания, при превышении заданного порогового значения соответствия. Порог соответствия (или любой порог данного описания) может составлять или составлять примерно, например, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или более. Например, предварительно заданное пороговое значение соответствия может составлять 97%. Множество оснований, специфичных для гена CYP2D6 может содержать или содержать примерно, например, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 118, 120, 130, 140, 150, 160, 170 или более специфических для гена CYP2D6 оснований. Например, множество оснований, специфичных для гена CYP2D6 может содержать 118 оснований, специфичных для гена CYP2D6.
[0146] Способ 900 переходит от этапа 920 к этапу 924, в котором вычислительная система определяет один или более структурных вариантов гена CYP2D6, имеющегося у субъекта, используя наиболее вероятную комбинацию возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7, определенного для основания, специфичного для гена CYP2D6. Например, вычислительная система может определить соотношение количества прочтений CYP2D6 к CYP2D7, перекрывающихся местоположений, где гены имеют разные основания последовательности Для позиций, в которых CYP2D6 отличается от CYP2D7, вычислительная система может проводить прочтения, которые перекрываются либо на основании CYP2D6, либо CYP2D7. На основе этих прочтений вычислительная система может подсчитывать количество специфичных для CYP2D6 оснований и количество специфичных для CYP2D7 оснований. Вычислительная система может определять долю прочтений CYP2D6 или CYP2D7. Вычислительная система может вычислить количество копий CYP2D6 и CYP2D7 в позициях, в которых CYP2D6 отличается от CYP2D7. Вычислительная система может комбинировать общее количество копий CYP2D6 и CYP2D7 с соотношением CYP2D6 и CYP2D7, чтобы определить количество копий CYP2D6 и CYP2D7. Вычислительная система может определять малые варианты, используя количество копий CYP2D6 и CYP2D7 при одном или более фиксированных различиях между CYP2D6 и CYP2D7. Вычислительная система может проводить определение структурных вариантов путем комбинирования количества копий CYP2D6 и CYP2D7 при множественных фиксированных различиях между CYP2D6 и CYP2D7, чтобы определить наличие перехода между количеством копий CYP2D6 и CYP2D7, который определяет тип структурного варианта, находящегося в образце.
[0147] REP-содержащие гены слияния. В некоторых вариантах осуществления вычислительная система может определять (ii) второе количество прочтений последовательностей из множества прочтений последовательностей, выровненных с областью спейсера между геном CYP2D7 и повторяющимся элементом REP7 ниже гена CYP2D7. Второе количество прочтений последовательностей из множества прочтений последовательностей, выровненных с областью спейсера между геном CYP2D7 и повторяющимся элементом REP7 ниже гена CYP2D7, может составлять или составлять примерно, например, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 2000, 3000, 4000, 5000, 6000, 7000, 8000, 9000, 10000 или более. Вычислительная система может определять (ii) второе нормализованное количество прочтений последовательности, выровненной с областью спейсера, с использованием (ii) длины области спейсера. Второе нормализованное количество прочтений последовательностей, выровненных с областью спейсера, может составлять или составлять примерно, например, 1, 2, 3, 4, 5, 6, 7, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Длина области спейсера может составлять или составлять примерно, например, 1,5 т. п. н. Вычислительная система может определять (ii) количество копий области спейсера с использованием модели смеси нормальных распределений, заданной (ii) вторым нормализованным количеством прочтений последовательностей, выровненных с областью спейсера. Количество копий области спейсера может составлять или составлять примерно, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более. Для определения аллеля гена CYP2D6, имеющегося у субъекта, вычислительная система может определить аллель гена CYP2D6, имеющегося у субъекта, малый вариант или структурный вариант гена CYP2D6,или ни один из них, используя комбинацию возможного количества копий гена CYP2D6 и возможного количества копий гена CYP2D7, определенного для основания, специфичного для гена CYP2D6, и количества копий спейсерной области. Структурный вариант может содержать слитый аллель CYP2D6/CYP2D7 с областью спейсера и повторяющимся элементом REP7 ниже слитного аллеля CYP2D6/CYP2D7.
[0148] Способ 900 переходит от этапа 924 к этапу 928, в котором вычислительная система может для положения малого варианта гена CYP2D6, связанного с малым аллелем варианта гена CYP2D6, определение наиболее вероятной комбинации возможного количества копий малого аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6 в положении малого варианта, суммированного с количеством копий гена CYP2D6 в положении малого варианта, учитывая (a) количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей), выровненных с геном CYP2D6, перекрывающимся с положением малого варианта, и основания, которое содержит аллель малого варианта гена CYP2D6 в положении малого варианта и (b) количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей), выровненных с геном CYP2D6, перекрывающимся с положением малого варианта, и основания, которое содержит эталонный аллель гена CYP2D6 в положении малого варианта. Возможное количество копий малого варианта аллеля гена CYP2D6, наиболее вероятной комбинации в положении малого варианта может указывать на один или более малых вариантов гена CYP2D6.
[0149] Вычислительная система может определять для каждого из множества положений малых вариантов гена CYP2D6 положение малого варианта связано с малым вариантом аллеля гена CYP2D6, определение наиболее вероятной комбинации возможного количества копий малого аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6 в положении малого варианта, суммированного с количеством копий гена CYP2D6 в положении малого варианта, учитывая (a) количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей), выровненных с геном CYP2D6, перекрывающимся с положением малого варианта, и основания, которое содержит аллель малого варианта гена CYP2D6 в положении малого варианта и (b) количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей), выровненных с геном CYP2D6, перекрывающимся с положением малого варианта, и основания, которое содержит эталонный аллель гена CYP2D6 в положении малого варианта. Возможное количество копий малых вариантных аллелей гена CYP2D6, наиболее вероятной комбинаций во множестве положений малых вариантов может указывать на один или более малых вариантов гена CYP2D6.
[0150] В некоторых вариантах осуществления вычислительная система может определять количество копий гена CYP2D6 в положении малого варианта. Количество копий гена CYP2D6 в положении малого варианта может включать количество копий гена CYP2D6. Количество копий гена CYP2D6 в положении малого варианта может включать количество копий гена CYP2D6 из возможных количеств копий гена CYP2D6 наиболее вероятных определенных комбинаций. Количество копий гена CYP2D6 в положении малого варианта может включать количество копий гена CYP2D6 из возможных количеств копий гена CYP2D6 наиболее вероятных определенных комбинаций и расположены ближе всего к положению малого варианта. Количество копий гена CYP2D6 в положении малого варианта может включать количество копий гена CYP2D6 в 5’-положении или в 3’-положении от положения малого варианта.
[0151] В некоторых вариантах осуществления вычислительная система может (a) определять количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей) с основаниями, которые содержат малый вариантный аллель гена CYP2D6. Вычислительная система может (b) определять количество прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей) с основаниями, которые содержат эталонный аллель гена CYP2D6.
[0152] Способ 900 переходит от этапа 928 к этапу 932, где вычислительная система определяет один или более малых вариантов гена CYP2D6 с применением возможного количества копий малого варианта аллеля гена CYP2D6 наиболее вероятной определенной комбинации. Вычислительная система может определять один или более малых вариантов гена CYP2D6 с помощью возможного количества копий малых вариантных аллелей гена CYP2D6, наиболее вероятных комбинаций во множестве определенных положений малых вариантов.
[0153] В некоторых вариантах осуществления положение малого варианта находится в гомологичной области CYP2D6/CYP2D7. Для определения наиболее вероятной комбинации вычислительная система может определить наиболее вероятную комбинацию возможного количества копий малого варианта аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6 в положении малого варианта, суммированное с количеством копий гена CYP2D6 в положении малого варианта, учитывая (а) количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, с основанием, которое содержит малый вариант аллеля гена CYP2D6 в положении малого варианта, и/или (b) количество прочтений последовательностей, выровненных с геном CYP2D6 или геном CYP2D7, с основанием, которое содержит эталонный аллель CYP2D6 в положении малого варианта. В некоторых вариантах осуществления положение малого варианта не находится в гомологичной области CYP2D6/CYP2D7. Для определения наиболее вероятной комбинации вычислительная система может определить наиболее вероятную комбинацию возможного количества копий малого варианта аллеля гена CYP2D6 в положении малого варианта и возможного количества копий эталонного аллеля гена CYP2D6 в положении малого варианта, суммированного с количеством копий гена CYP2D6 в положении малого варианта, учитывая (a) количество прочтений последовательностей, выровненных с геном CYP2D6, а не с геном CYP2D7, с основанием, которое содержит малый вариант аллеля гена CYP2D6 в положении малого варианта, и/ или (b) количество прочтений последовательностей, выровненных с геном CYP2D6, а не геном CYP2D7, с основанием, которое содержит эталонный аллель CYP2D6 в положении малого варианта.
[0154] Например, вычислительная система может сначала определить характер и точки разрыва SV (структурный вариант, например, делецию или дупликацию) на основе количества копий оснований, специфичных для паралога. Дополнительно или в качестве альтернативы вычислительная система может впоследствии идентифицировать заранее определенное множество малых вариантов (это варианты, специфичные для интересующего гена, например, CYP2D6, и они отличаются от дифференцирующих оснований паралогов), на основе выравнивания прочтения, общего количества копий, а также (иногда) характера SV и точек разрыва, определенных на первом этапе. Поскольку выравнивание не всегда является точным, вычислительная система может извлечь интересующие основания из прочтений, которые соответствуют любому паралогу.
[0155] Способ 900 переходит от этапа 932 к этапу 936, где вычислительная система может определять звездчатый аллель и/или гаплотип гена CYP2D6, имеющегося у субъекта, используя один или более определенных структурных вариантов гена CYP2D6, и/или один или более малых вариантов гена CYP2D6. Звездчатый аллель может быть связан с известной функцией. Звездчатый аллель и/или гаплотип гена CYP2D6 может включать, например, CYP2D6*1, *2, *3, *4, *5, *6, *7, *9, *10, *11, *13, *14, *15, *17, *21, *22, *28, *29, *31, *33, *34, *35, *36, *37, *38, *39, *40, *41, *43, *45, *46, *47, *49, *52, *54, *56, *57, *59, *64, *65, *68, *71, *72, *82, *84, *86, *94, *95, *99, *100, *101, *106, *108, *111, *112, *113 или их комбинации.
[0156] Ферментативная активность. В некоторых вариантах осуществления вычислительная система может определять уровень ферментативной активности CYP2D6 у субъекта, используя определенный аллель гена CYP2D6. Ферментативная активность может быть низкой, средней, нормальной или сверхбыстрой. Вычислительная система может определить рекомендацию по дозировке лечения и/или рекомендацию по лечению для субъекта на основании одного или более малых вариантов и/или одного или более структурных вариантов.
[0157] Способ 900 заканчивается этапом 940.
Генотипирование паралогов с использованием данных секвенирования
[0158] На ФИГ. 10 представлена блок-схема, показывающая пример способа 1000 генотипирования паралога с использованием данных секвенирования, таких как данные секвенирования целого генома. Способ 1000 может быть реализован в виде набора исполняемых программных команд, хранящихся на машиночитаемом носителе, таком как один или более дисководов вычислительной системы. Например, вычислительная система 1100, показанная на ФИГ. 11 и более подробно описанная ниже, может выполнять набор исполняемых программных команд для реализации способа 1000. При инициировании способа 1000 исполняемые программные команды могут быть загружены в запоминающее устройство, такое как RAM, и выполнены одним или более процессорами вычислительной системы 1100. Хотя способ 1000 описан в отношении вычислительной системы 1100, показанной на ФИГ. 11, описание приводится только в качестве иллюстрации и не носит ограничительного характера. В некоторых вариантах осуществления способ 1000 или его фрагменты могут быть выполнены последовательно или параллельно множеством вычислительных систем.
[0159] После того как способ 1000 начинается с этапа 1004, способ 1000 переходит к этапу 1008, где вычислительная система (например, вычислительная система 1100, описанная со ссылкой на ФИГ. 11) принимает данные секвенирования, содержащие множество прочтений последовательностей, полученных из образца от субъекта, выровненных с первым паралогом или вторым паралогом. Техники создания прочтений последовательностей включают секвенирование путем синтеза с использованием, например, инструментов секвенирования MINISEQ, MISEQ, NEXTSEQ, HISEQ и NOVASEQ от компании Illumina, Inc. (Сан-Диего, Калифорния).
[0160] Способ 1000 переходит от этапа 1008 к этапу 1012, где вычислительная система определяет количество копий паралогов первого типа с применением модели смеси нормальных распределений, содержащей множество распределений, каждое из которых представляет другое целое количество копий, заданное (i) первым количеством прочтений последовательностей, выровненных с первой областью. Количество копий паралогов первого типа (или любого типа данного описания) может составлять или составлять примерно, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более.
[0161] Множество распределений модели смеси нормальных распределений могут представлять целые числа копий, например от 0 до 5, от 0 до 6, от 0 до 7, от 0 до 8, от 0 до 9, от 0 до 10, от 0 до 11, от 0 до 12, от 0 до 13, от 0 до 14 или от 0 до 15. Например, множество распределений модели смеси нормальных распределений могут представлять целые числа копий от 0 до 10. Среднее значение (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более) каждого из множества распределений может представлять собой целое количество копий, представленное распределением (например, количество копий 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более). Стандартное отклонение распределений может составлять или примерно составлять, например, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1 или более.
[0162] В некоторых вариантах осуществления вычислительная система может определять (i) первое количество прочтений последовательностей из множества прочтений последовательностей в данных секвенирования, полученных из образца от субъекта, выровненных с первой областью. Первое количество прочтений последовательностей из множества прочтений последовательностей в данных секвенирования, полученных из образца от субъекта, выровненных с первой областью (или любой областью данного описания), может составлять или примерно составлять, например, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 2000, 3000, 4000, 5000, 6000, 7000, 8000, 9000, 10000 или более. Вычислительная система может определять (i) первое нормализованное количество прочтений последовательностей, выровненных с первой областью, с использованием (i) длины первой области. Первое нормализованное количество прочтений последовательностей, выровненных с первой областью (или любой областью данного описания), может составлять или составлять примерно, например, 1, 2, 3, 4, 5, 6, 7, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более. Длина первой области может составлять или составлять примерно, например, 1 т. п. н., 2 т. п. н., 3 т. п. н., 4 т. п. н., 5 т. п. н., 6 т. п. н., 7 т. п. н., 8 т. п. н., 9 т. п. н., 10 т. п. н., 11 т. п. н., 12 т. п. н., 13 т. п. н., 14 т. п. н., 15 т. п. н., 16 т. п. н., 17 т. п. н., 18 т. п. н., 19 т. п. н., 20 т. п. н., 21 т. п. н., 22 т. п. н., 23 т. п. н., 24 т. п. н., 25 т. п. н., 26 т. п. н., 27 т. п. н., 28 т. п. н., 29 т. п. н., 30 т. п. н. или более. Для определения количества копий паралогов первого типа вычислительная система может определять количество копий паралогов первого типа с помощью модели смеси нормальных распределений, заданной (i) первым нормализованным количеством прочтений последовательностей, выровненных с первой областью.
[0163] В некоторых вариантах осуществления вычислительная система может определять количество копий одного или более паралогов второго типа с использованием смеси нормальных распределений, заданной (ii) вторым количеством прочтений последовательностей, выровненных со второй областью. Количество копий одного или более паралогов второго типа (или любого типа данного описания) может составлять или составлять примерно, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более. Для определения количества копий или аллеля первого паралога вычислительная система может определять количество копий или аллель первого паралога с помощью наиболее вероятной комбинации возможного количества копий первого паралога и возможного количества копий второго паралога, определенного для основания, специфического для первого паралога, и количества копий одного или более паралогов второго типа. Вычислительная система может определять количество копий паралогов третьего типа по количеству копий паралогов первого типа и количеству копий паралогов второго типа. Количество копий паралогов третьего типа (или любого типа данного описания) может составлять или составлять примерно, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более. Для определения количества копий или аллеля первого паралога вычислительная система может определять количество копий или аллель первого паралога с помощью наиболее вероятной комбинации возможного количества копий первого паралога и возможного количества копий второго паралога, определенного для основания, специфического для первого паралога.
[0164] В способах выравнивания последовательностей с эталонной геномной последовательностью можно использовать такие преобразователи, как преобразователь Барроуза-Уилера (BWA) и iSAAC. Другие способы выравнивания включают BarraCUDA, BFAST, BLASTN, BLAT, Bowtie, CASHX, Cloudburst, CUDA-EC, CUSHAW, CUSHAW2, CUSHAW2-GPU, drFAST, ELAND, ERNE, GNUMAP, GEM, GensearchNGS, GMAP и GSNAP, Geneious Assembler, LAST, MAQ, mrFAST and mrsFAST, MOM, MOSAIK, MPscan, Novoaligh & NovoalignCS, NextGENe, Omixon, PALMapper, Partek, PASS, PerM, PRIMEX, QPalma, RazerS, REAL, cREAL, RMAP, rNA, RT Investigator, Segemehl, SeqMap, Shrec, SHRiMP, SLIDER, SOAP, SOAP2, SOAP3 и SOAP3-dp, SOCS, SSAHA и SSAHA2, Stampy, SToRM, Subread и Subjunc, Taipan, UGENE, VelociMapper, XpressAlign и ZOOM.
[0165] Способ 1000 переходит от этапа 1012 к этапу 1016, где вычислительная система определяет для одного множества оснований, специфичных для первого паралога, из множества возможных комбинаций, каждая из которых содержит возможное количество копий первого паралога первого типа и возможное количество копий второго паралога первого типа, суммированное с количеством копий определенных паралогов первого типа, при заданном (a) количестве прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей) из множества прочтений последовательностей с основаниями, которые содержат основание специфическое для первого паралога, и (b) количестве прочтений последовательностей (например, ненормализованное или нормализованное количество прочтений последовательностей) из множества прочтений последовательностей с основаниями, которые содержат основание специфическое для второго паралога, соответствующее основанию специфическому для первого паралога.
[0166] Способ 1000 переходит от этапа 1016 к этапу 1020, где вычислительная система определяет количество копий или аллель первого паралога с помощью наиболее вероятной комбинации возможного количества копий первого паралога и возможного количества копий второго паралога, определенного для основания, специфичного для первого паралога.
[0167] В некоторых вариантах осуществления первым паралогом является ген выживания моторных нейронов 1 (SMN1). Вторым паралогом может быть ген выживания моторных нейронов 2 (SMN2). Первая область может содержать по меньшей мере один экзон от 1 до 6 гена SMN1 и по меньшей мере один экзон от 1 до 6 гена SMN2. Вторая область может содержать по меньшей мере экзон 7 или 8 гена SMN1 и по меньшей мере один из экзон 7 или 8 гена SMN2. Паралоги первого типа могут включать интактный ген SMN1 и интактный ген SMN2. Один или более паралогов второго типа могут включать интактный ген SMN1, интактный ген SMN2, укороченный ген SMN1 или укороченный ген SMN2. Количество копий первого паралога может включать количество копий гена SMN1. Вычислительная система может определять количество копий гена SMN1, реализуя способ 800 (или его часть), описанный со ссылкой на ФИГ. 8.
[0168] В некоторых вариантах осуществления первый паралог представляет собой ген члена 6 подсемейства D семейства 2 цитохрома P450 (CYP2D6). Вторым паралогом может быть ген члена 7 подсемейства D семейства 2 цитохрома P450 (CYP2D7). Первая область может содержать ген CYP2D6 и ген CYP2D7. Вторая область может содержать спейсерную область между геном CYP2D7 и повторяющимся элементом REP7 ниже гена CYP2D7. Паралоги первого типа могут содержать ген CYP2D6 и ген CYP2D7. Один или более паралогов второго типа могут содержать слитый аллель CYP2D6/CYP2D7 с спейсерной областью и повторяющимся элементом REP7 ниже слитого аллеля CYP2D6/CYP2D7. Аллель первого паралога может представлять собой аллель гена CYP2D6, имеющегося у субъекта, который представляет собой малый вариант или структурный вариант гена CYP2D6. Вычислительная система может определять аллель гена CYP2D6, реализуя способ 900 (или его часть), описанный со ссылкой на ФИГ. 9.
[0169] В различных вариантах осуществления первый и второй паралоги могут отличаться друг от друга. Примеры первого и второго паралогов включают, без ограничений, ген SMN1 и ген SMN2; Ген CYP2D6 и ген CYP2D7; ген double homeobox 4 (DUX4), ген DUX4c, ген DUX4-подобного белка 2 (DUX4L2), ген DUX4-подобного белка 3 (DUX4L3), ген DUX4-подобного белка 4 (DUX4L4), ген DUX4-подобного белка 5 (DUX4L5), ген DUX4-подобного белка 6 (DUX4L6), ген DUX4-подобного белка 7 (DUX4L7) и ген double homeobox 2 (DUX2); и ген рибосомального белка S17 (RpS17) и ген RpS17-подобного белка (RpS17L). В некоторых вариантах осуществления вычислительная система может определять количество копий или аллель первого паралога, реализуя способ 800 (или его часть), описанный со ссылкой на ФИГ. 8, и/или способ 900 (или его часть), описанный со ссылкой на ФИГ. 9.
[0170] В некоторых вариантах осуществления первый паралог и второй паралог имеют идентичность последовательности или примерно 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или более. Например, первый паралог и второй паралог имеют идентичность последовательности по меньшей мере 90%.
[0171] Способ 1000 заканчивается этапом 1024.
Условия выполнения
[0172] На ФИГ. 11 изображена общая архитектура примерного вычислительного устройства 1100, выполненного с возможностью генотипирования паралога. Общая архитектура вычислительного устройства 1100, показанного на ФИГ. 11, включает в себя расположение компьютерных аппаратных и программных компонентов. Вычислительное устройство 1100 может включать в себя намного больше (или меньше) элементов, чем показанные на ФИГ. 11. Однако необязательно чтобы все эти в общем обычные элементы были показаны, чтобы обеспечить описание изобретения. Как проиллюстрировано, вычислительное устройство 1100 включает в себя блок 1110 обработки данных, сетевой интерфейс 1120, привод машиночитаемого носителя 1130, интерфейс устройства ввода/вывода 1140, дисплей 1150 и устройство ввода 1160, все из которых могут обмениваться данными друг с другом посредством шины связи. Сетевой интерфейс 1120 может обеспечивать возможность подключения к одной или более сетям или вычислительным системам. Таким образом, блок 1110 обработки данных может принимать информацию и инструкции от других вычислительных систем или сервисов посредством сети. Блок 1110 обработки данных также может обмениваться данными с памятью 1170 и из нее и дополнительно предоставлять выходную информацию на необязательный дисплей 1150 через интерфейс устройства ввода/вывода 1140. Интерфейс устройства ввода/вывода 1140 может также принимать ввод от необязательного устройства ввода 1160, такого как клавиатура, мышь, цифровая ручка, микрофон, сенсорный экран, система распознавания жестов, система распознавания голоса, геймпад, акселерометр, гироскоп или другое устройство ввода.
[0173] Память 1170 может содержать команды компьютерной программы (сгруппированные как модули или компоненты в некоторых вариантах осуществления), которые выполняет блок 1110 обработки данных для реализации одного или более вариантов осуществления. Память 1170 по существу включает в себя RAM, ROM и/или другие постоянные, вспомогательные или энергонезависимые машиночитаемые носители. В запоминающем устройстве 1170 может храниться операционная система 1172, которая обеспечивает команды компьютерной программы для использования блоком 1110 обработки данных при общем введении и эксплуатации вычислительного устройства 1100. Запоминающее устройство 1170 может дополнительно включать команды компьютерной программы и другую информацию для реализации аспектов данного описания.
[0174] Например, в одном варианте осуществления память 1170 включает в себя модуль генотипирования паралогов 1174 для генотипирования одного или более паралогов с использованием данных секвенирования, таких как способ 1000, описанный со ссылкой на ФИГ. 10. В качестве альтернативы или дополнительно, модуль генотипирования паралогов 1174 может представлять собой или может включать в себя модуль для определения количества копий SMN1 с использованием данных секвенирования, такой как способ 800, описанный со ссылкой на ФИГ. 8. В качестве альтернативы или дополнительно, модуль генотипирования паралогов 1174 может представлять собой или может включать в себя модуль для генотипирования гена CYP2D6 с использованием данных секвенирования, такой как способ 900, описанный со ссылкой на ФИГ. 9. Кроме того, память 1170 может включать в себя или обмениваться данными с хранилищем данных 1190 и/или одним или более другими хранилищами данных, в которых хранятся данные секвенирования и/или результаты генотипирования одного или более паралогов.
Примеры
[0175] В следующих примерах более подробно описаны некоторые аспекты описанные выше, которые никоим образом не предназначены для ограничения объема данного раскрытия.
Пример 1
Диагностика спинальной мышечной атрофии и скрининг носителей на основе данных секвенирования целого генома
[0176] Спинальная мышечная атрофия (SMA), вызванная потерей функционального гена SMN1, но сохранением паралогического гена SMN2, является основной генетической причиной смерти в раннем детском возрасте. Из-за практически идентичных последовательностей SMN1 и его паралога SMN2 анализ этой области с использованием анализов на основе секвенирования следующего поколения (NGS) является сложной задачей. Американский колледж медицинской генетики рекомендует скрининг потенциальных родителей на SMA до зачатия для определения количества копий (CN) SMN1.
[0177] В данном примере описан способ биоинформатики, который точно идентифицирует количество копий SMN1 и SMN2 с использованием данных секвенирования целого генома (WGS). Способ рассчитывает количество копий SMN1 и SMN2 с использованием глубины прочтения и восьми информативных эталонных геномных различий между SMN1 и SMN2.
[0178] Были охарактеризованы статусы SMN1/2 в 12747 короткочитаемых целых геномах, секвенированных на большую глубину (> 30x) в пяти этнических популяциях. По этим образцам определили всего 251 (1317) образцов с полной потерей гена (конверсия) SMN1 и 6241 (374) образцов с потерей гена (конверсия) SMN2. Была рассчитана панэтническая частота носительства в 2%, что согласуется с предыдущими исследованиями. Кроме того, определенные количества копий были подтверждены, и все определения количества копий (48/48) SMN1 и 98% (47/48) SMN2 соответствовали определениям, измеренным с помощью цифровой ПЦР.
[0179] Данный способ определения количества копий SMN на основе WGS можно использовать для идентификации определения как носителя, так и подверженного воздействию статуса SMA, что позволяет предлагать тестирование на SMA в качестве комплексного теста в неонатальной диагностике, а также точного инструмента скрининга статуса носителя в крупномасштабных проектах секвенирования WGS.
Введение
[0180] Благодаря последним достижениям в секвенировании следующего поколения (NGS) теперь можно профилировать большое количество генов или даже весь геном с высокой пропускной способностью и в клинически значимые временные рамки. Исходя из этих достижений, во многих странах предпринимаются усилия по широкомасштабному секвенированию популяций, в которых тестирование на редкие генетические нарушения, включая статус носителя, будет одним из основных факторов. Спинальная мышечная атрофия (SMA), аутосомное рецессивное нейромышечное расстройство, характеризующееся потерей альфа-моторных нейронов, вызывает сильную мышечную слабость и атрофию, представляющую собой атрофию, проявляющуюся во время или вскоре после рождения. SMA является основной генетической причиной детской смертности после муковисцидоза. Частота возникновения SMA составляет 1 случай на 6000-10000 живорожденных, а частота носительства составляет 1:40-80 среди различных этнических групп. Четыре клинических типа СМА классифицируются в зависимости от возраста начала и тяжести заболевания: очень слабые младенцы, неспособные сидеть без поддержки (тип I), слабосидящие, но неспособные стоять (тип II), амбулаторные пациенты с более слабыми ногами, чем руки (тип III), и проявление SMA у взрослых пациентов, являющиеся довольно доброкачественным (тип IV). Раннее выявление SMA может иметь решающее значение для долгосрочного качества жизни из-за доступности двух ранних видов лечения, Nusinersen и Zolgensma, которые получили одобрение FDA для облегчения SMA.
[0181] Область SMN включает два паралогических гена: SMN1 и SMN2. SMN2 находится на расстоянии 875 т. п. н. от SMN1 на хромосоме 5q и вызван дупликацией предкового гена, уникальной для человеческой линии. Геномная область вокруг SMN1/2 подвергается неравномерному перекрестному сшиванию и генной конверсии, что приводит к вариабельному количеству копий (КК) SMN1 и SMN2. SMN2 имеет более чем 99,9% идентичности последовательности с SMN1, и одно из различий оснований, c.840C> T в экзоне 7, имеет критическое функциональное значение. Путем прерывания энхансера сплайсинга c.840T способствует пропуску экзона 7, в результате чего подавляющее большинство транскриптов, происходящих из SMN2 (70-85%, в зависимости от ткани), являются нестабильными и не полностью функциональными. Приблизительно 95% случаев SMA являются результатом биаллельного отсутствия функционального нуклеотида c.840C, вызванного делецией SMN1 или генной конверсией в SMN2 (c.840T). В остальных 5% случаев пациенты с SMA имеют другие патогенные варианты в SMN1 в транс-конфигурации с отсутствием аллеля c.840C. SMN2 может продуцировать небольшое количество функционального белка, а количество копий SMN2 у индивида изменяет тяжесть заболевания и сильно коррелирует с описанными выше клиническими типами.
[0182] Из-за высокой частоты возникновения и тяжести заболевания, обширный скрининг на SMA рекомендуется Американским колледжем медицинской генетики. Польза скрининга носителей среди населения была продемонстрирована в пилотных исследованиях. Скрининг на SMA включает: 1) определение количества копий SMN1 для диагностики SMA и тестирования носителя и 2) определение количества копий SMN2 для клинической классификации и прогноза. Традиционно тестирование на SMA и тестирование носителя проводят с использованием анализов на основе полимеразной цепной реакции (ПЦР), таких как количественная ПЦР (кПЦР), мультиплексная амплификация лигированных зондов (MLPA) и цифровая ПЦР. Эти способы в основном определяют количество копий SMN1 на основе сайта c.840C>T, который отличается между SMN1 и SMN2. Этот пример демонстрирует, что WGS может соответствовать или превосходить производительность этих тестов и указывает на то, что как текущие, так и будущие инициативы в области точной медицины могут использовать данные генома для скрининга на уровне населения.
[0183] Воспроизведение текущего режима тестирования на SMA представляет собой проблему для высокопроизводительных WGS из-за почти идеальной идентичности последовательностей между SMN1 и SMN2. Кроме того, считается, что часто встречающаяся генная конверсия между SMN1 и SMN2 приводит к образованию гибридных генов. Эти проблемы требуют применения способа биоинформатики, позволяющего преодолеть трудности в этой области. Представлены два теста на основе NGS для обнаружения носителя SMA. В публикации Larson et al. (Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project. BMC Med Genet. 2015;16:100. doi:10.1186/s12881-015-0246-2) использовали байесовскую иерархическую модель для расчета вероятности того, что доля прочтений, полученных из SMN1, равна или меньше 1/3 при трех различиях оснований между SMN1 и SMN2. Способ, описанный в публикации Larson, позволяет проводить тестирование на SMA; хотя, поскольку способ не выполняет определение количества копий, он не является идеальным решением для скрининга носителей. И наоборот, в публикации Feng et al. (The next generation of population-based spinal muscular atrophy carrier screening: comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Genet Med Off J Am Coll Med Genet. 2017; 19(8): 936-944. doi: 10.1038/gim.2016.215) описано определение количества копий как для SMN1, так и для SMN2 на основе данных целевого секвенирования, которые точно имитируют текущий способ кПЦР. Способ Feng разработан для целевого секвенирования и, следовательно, требует специальной нормализации, которая ограничивает способ одним анализом в одном сайте. Способ высчитывает общее количество копий SMN (включая как SMN1, так и SMN2) из покрытия прочтением в экзоне 7 и вычисляет соотношение SMN1: SMN2 на основе количества прочтений, поддерживающих SMN1 и SMN2, на сайте c.840 C>T. При использовании полного покрытия и соотношения SMN1: SMN2 в способе определяют абсолютное количество копий SMN1 и SMN2. Поскольку данный способ основан только на одном локусе, он является ненадежным для данных WGS, где вариабельность глубины для каждого локуса может быть очень высокой.
[0184] По сравнению с целевым секвенированием WGS обеспечивает гораздо более равномерное покрытие генома и обеспечивает подход с меньшим смещением для обнаружения количества копий вариантов (ККВ). Кроме того, WGS дает возможность всесторонне профилировать спектр изменчивости популяции в области SMN, понимание которой на уровне последовательностей оставляет желать лучшего. В этом примере описан новый способ, который обнаруживает количество копий как SMN1, так и SMN2 с использованием данных WGS. В то время как большинство традиционных анализов проверяют только отсутствие c.840C в качестве заместителя для «делеции экзона 7», в этом примере описан способ, который может более полно охарактеризовать вариабельность в области, включая: 1) Делеции ДНК, включая полную делецию/дупликацию гена и частичную делецию области, которая включает экзоны 7 и 8; и 2) обнаружение малых вариантов, включая g.27134T>G SNP, которые коррелирует с « молчащими » носителями SMA (две копии SMN1 на одном и том же гаплотипе). Точность этого способа была продемонстрирована путем сравнения определений количества копий с применением цифровой ПЦР с определениями на основе WGS из примера. Было показано соответствие 100% (48/48) для SMN1 и 98% (47/48) для SMN2. Кроме того, данный способ применяли к 2504 неродственным образцам из проекта 1000 Genomes и 10243 неродственным образцам из проекта NIHR BioResource, чтобы сообщить о распределении количества копий SMN1 и SMN2. Несущие частоты для SMA, определенные с использованием способа, описанным в примере, согласуются с данными, полученными в предыдущих исследованиях на основе ПЦР. Помимо демонстрации точности способа количественной оценки вариантов в области SMN, этот пример подчеркивает важность использования этнически разнообразных популяций при разработке новых информационных способов для определения сложных клинически значимых областей генома.
Материалы и способы
Образцы и обработка данных
[0185] Образцы, проверенные с помощью цифровой ПЦР, были получены из коллекции Лаборатории исследования заболеваний двигательных нейронов (Nemours Alfred I. duPont Hospital for Children) и получали из клеточных линий, как описано выше. Данная когорта содержала 29 образцов SMA (14 SMA типа I, 1 SMA типа I/II, 10 SMA типа II, 3 SMA типа III и 1 SMA с неизвестной клинической степенью), шесть образцов с нервно-мышечными заболеваниями, не относящихся к SMA (включая наследственную сенсорную и вегетативную нейропатию 3, миотоническую дистрофию типа I, дистальную наследственную моторную нейропатию типа I и периферическую нейропатию типа I и сенсомоторную нейропатию Шарко-Мари-Тута типа IA), а также 13 нормальных образцов. WGS выполняли с использованием набора для приготовления образцов TruSeq DNA PCR-free sample preparation, используя парные прочтения длиной 150 п. о., секвенированными на приборах HiSeq X компании Illumina (Сан-Диего, Калифорния, США). Для выравнивания прочтения использовали сборку генома GRCh37.
[0186] В рамках популяционных исследований были задействованы 13343 субъекта из проекта NIHR BioResource Rare Diseases (EGAS00001001012), которым выполняли WGS для людей с редкими заболеваниями и их близким родственникам. Также были исследованы дополнительные субъекты (n = 840) из проекта Next Generation Children (EGAD00001004357), которым выполняли диагностическое трио WGS на пациентах и их родителях из неонатальных и педиатрических отделений интенсивной терапии в Великобритании. WGS для этих исследований выполняли с использованием набора для приготовления образцов TruSeq DNA PCR-Free Preparation kit компании Illumina с парными прочтениями 100 или 125 пар оснований, секвенированных на приборе HiSeq 2500 компании Illumina или парных прочтений 150 п. о., секвенированных на приборе HiSeq X. Для выравнивания прочтения использовали сборку генома GRCh37. При проведении популяционного анализа были исключены родственные субъекты и субъекты неизвестного происхождения, в результате чего осталось 10243 неродственных субъектов.
[0187] Для данных из проекта 1000 Genomes (1kGP) WGS BAM загружали из ncbi.nlm.nih.gov/bioproject/PRJEB31736/. Эти файлы BAM получали путем секвенирования прочтений 2 x 150 п.о. на приборах NovaSeq 6000 компании Illumina из библиотек без проведения ПЦР, секвенированных на среднюю глубину по меньшей мере 30 раз, и выравнивания их с эталоном человека hs38 DH с использованием BWA-MEM v0.7.15 (среднее покрытие генома более 30 раз).
Анализ количества копий SMN ортогональными способами
[0188] Для проверочных образцов количество копий SMN1 и SMN2 измеряли с помощью системы цифровой ПЦР QuantStudio 3 D (Life Technologies, Карлсбад, Калифорния) с использованием аллель-специфических зондов экзона 7, как описано выше. Количество копий SMN1 и SMN2 нормировали относительно количества копий RPPH1 (РНКаза P). Обнаруженные образцы SMA в проекте Next Generation Children были подтверждены с использованием стандартной MLPA (SALSA MLPA P060 SMA Carrier Probemix, MRC-Holland).
Определение количества копий для интактного и укороченного SMN
[0189] На локусы SMN1 и SMN2 влияют количества копий двух общих вариантов, количества копий вариантов всего генома и частичная делеция экзонов 7 и 8 (см. результаты данного примера). Укороченная форма SMN с частичной делецией экзонов 7 и 8 была названа SMN*. Способ определения количества копий интактных генов SMN1 + SMN2 (далее именуемых SMN) и укороченных генов SMN (SMN*) с использованием следующих этапов.
[0190] Выявление и подсчет прочтений SMN1 и SMN2 : Количество прочтений рассчитывали непосредственно из файла BAM, выровненного по WGS на основе всех прочтений, сопоставленных с SMN1 или SMN2, включая прочтение с нулевым качеством сопоставления. Часто считываемые данные будут выравниваться с этими областями с нулевым качеством картирования, поскольку последовательность двух областей является идентичной. Эти два гена имеют одинаковую последовательность только друг с другом, а не с другими областями генома. Количество прочтений в области 22,2 т. п. н., включающей от экзоны от 1 до 6, использовали для расчета общего количества копий SMN (SMN1, SMN2 и SMN*), а число прочтений в области 6 т. п. н., включающей экзон 7 и экзон 8, использовали для расчета количества копий интактного SMN (SMN1 и SMN2).
[0191] Расчет нормализованной глубины областей SMN : Количество прочтений двух описанных выше областей было нормализовано по длине области и дополнительно нормализовано путем деления на среднюю глубину 3000 предварительно выбранных областей размером 2 т. п. н. из геному.
[0192] Преобразование нормализованной глубины в количество копий: Нормированные значения глубины по популяции моделировали с использованием одномерной смеси 11 распределений, которые сосредоточены вокруг каждого целочисленного значения количества копий, представляющего состояния количества копий в диапазоне от 0 до 10. Количество копий общего SMN и интактного SMN было определено с помощью модели смеси нормальных распределений (GMM) с порогом апостериорной вероятности 0,95.
[0193] Расчет количества копий интактного и укороченного SMN : Количество копий интактного SMN определяли как количество копий области 6,3 т. п. н., охватывающей экзоны 7 и 8. Количество копий укороченного SMN (SMN*) получали путем вычитания количества копий интактного SMN из общего количества копий SMN, рассчитанного из области размером 22,2 т.п.н., содержащей экзоны 1-6.
Генотипирование количества копий аллелей по отдельных основаниях
[0194] Количество хромосом, несущих основания SMN1 и SMN2, определяли путем комбинирования общего количества копий SMN с количеством прочтений, поддерживающим каждое из ген-специфических оснований. На основе определения количества копий интактного SMN в каждом положении способ повторял все возможные комбинации количества копий SMN1 и SMN2, выводил комбинацию, которая дает самую высокую апостериорную вероятность для наблюдаемого количества прочтений, поддерживающих SMN1 и SMN2. Помимо определения количества копий оснований, специфичных для SMN1 или SMN2, этот способ можно применять в положениях вариантов для определения количества копий SNP, которые, как известно, являются специфичными для одного из двух генов, например g27134T>G, как описано ниже.
Количество копий SMN1 и SMN2
[0195] Для 16 положений (локализованных от интрона 6 до экзона 8), отличающихся между SMN1 и SMN2 в эталонном геноме, исследовали, действительно ли эти сайты были фиксированными в популяции путем сравнения определенных количеств копий аллелей SMN1 для этих положений с определенным количеством копий сплайс-вариантной формой основания SMN1 c.840C. Восемь положений, включая c.840C>T, где основания SMN1 фиксированы или фиксированию в популяции, были идентифицированы на основе соответствия с сплайс-вариантной формой основания (см. раздел « Результаты » в данном примере, ФИГ. 14A). Остальные сайты могут быть полиморфными в популяции и могут быть ненадежными для использования при определении количества копий.
[0196] Для получения окончательного определения количества копий этим способом необходимо выполнить следующие действия: 1) определение количества копий SMN1 согласуются по крайней мере на 5 из 8 сайтов при отсечении апостериорной вероятности 0,8 или 2) по меньшей мере на 5 из 8 сайтов (с апостериорной вероятностью > 0,6) согласуются с количеством копий, полученным из перекрывающихся прочтений всех 8 сайтов (с апостериорной вероятностью > 0,9). В противном определение количества копий как для SMN1, так и для SMN2 не проводилось. Образцы SMA идентифицировали как имеющие нулевую копию интактного SMN1, а образцы носителей идентифицировали как имеющие одну копию интактного SMN1.
[0197] При более высоких значениях количества копий можно ожидать большую вариабельность глубины, прочтения что приведет к менее конфиденциальному определению количества копий (с более низкой апостериорной вероятностью) на отдельных сайтах и большему расхождению между сайтами. В результате вероятность отсутствия определения была выше в выборках с большим количеством копий SMN1/SMN2, т. е. для обоих значений, которые больше или равны двум (см. ФИГ. 15). Однако в таких выборках по-прежнему можно достоверно определить, является ли количество копий SMN1 равно 0 (SMA) или 1 (носитель), что позволило осуществить определение SMA/отсутствие SMA или носитель/не носитель. Если количество копий SMN1 не определялось, если по меньшей мере семь из определений количества копий SMN1 были достоверно больше нуля, то образец обозначали как «отсутствие SMA». Аналогичным образом, если по меньшей мере семь определений количества копий SMN1 были достоверно больше единицы, образец обозначали как «не носитель». Кроме того, если количество копий SMN1 не определялось, непосредственно тестировали отсутствие аллеля c.840C, который указывал бы на SMA. Это было сделано путем проверки того, является ли количество прочтений, поддерживающих основание SMN1 (c.840C), более вероятным при отсутствии SMN1 или его одной копии.
Результаты
Общее количество копий варианта, влияющее на локусы SMN1/SMN2
[0198] Гены SMN1 и SMN2 находятся в области~ 2 млн п. н. в эталонном геноме с большим количеством сложных сегментных и инвертированных сегментных дупликаций. Хотя существующие способы (например, способы на основе ПЦР) сосредоточены главным образом на сайте c.840C>T, данный пример иллюстрирует подход с определением количества копий, основанный на данных секвенирования для полных генов. Количество копий SMN1 определяли как количество генов SMN, несущих аллель c.840C, а количество копий SMN2 определяли как количество генов SMN с аллелем c.840T. Анализ последовательности проводили с использованием данных WGS высокой глубины (>30X) 2504 образцов из проекта 1000 Genomes (1kGP), а также 10243 неродственных образцов из проекта NIHR BioResource (см. способы данного примера).
[0199] Чтобы сформулировать стратегию определения количества копий сначала были охарактеризованы количества копий двух общих вариантов, которые приводили к делециям ДНК. Первичная оценка количества копий вариантов включает всю область гена SMN1/SMN2. Была исследована глубина прочтения в гомологичной области размером~ 30 т.п.н., содержащей гены SMN1 и SMN2. На ФИГ. 12A показаны нормализованные глубины прочтения в скользящих окнах из 100 п. н. в образцах с различными количествами копий вариантов SMN1+SMN2 в данной области (представляющих как SMN1, так и SMN2). Профиль глубины показывает, что вся область была удалена или дублирована в этих образцах. Ожидалось, что точные точки разрыва данного количества копий вариантов у разных образцов будут различаться из-за обширной гомологии последовательности в пределах и за пределами данной области, и их можно различить только при высоком разрешении с длинным прочтением. Для тестирования на SMA анализ был ограничен (~ 30 т.п.н.) областями, включающими гены SMN (SMN1 или SMN2).
[0200] Кроме количества копий вариантов всего гена была обнаружена частичная делеция 6.3 т. п. н. гена, охватывающая оба экзона 7 и 8 (ФИГ. 12B, ФИГ. 16). Последовательности в точке разрыва идентичны между SMN1 и SMN2, поэтому эта делеция происходит в любой из chr5: 70244114 - 70250420 в SMN1 или chr5: 69368689 - 69375000 в SMN2 (ФИГ. 16, hg19). Однако около 500 п. н. ниже точки разрыва, определяющей конец этой делеции, существуют три различия в основаниях между локусами SMN1 и SMN2 (70250881A>69375425C, 70250981A>69375525G, 70250991A>69375535G). Среди образцов, содержащих эту делецию, были выявлены 245 пар прочтений из 237 образцов, где одно прочтение охватывало точку разрыва, а другое охватывало по меньшей мере два из трех дифференцирующих оснований SMN. Анализ этих пар прочтения показал, что 100% соответствовали делеции, происходящей на фоне последовательности SMN2. Такая укороченная форма SMN2 была названа SMN*, и поскольку оба экзона 7 и 8 удалены, SMN*, вероятно, имеет ограниченную биологическую функцию или не имеет ее вовсе. Таким образом, SMN* является важным вариантом, который следует учитывать при любом определении количества копий SMN.
[0201] На ФИГ. 12A и 12B показаны неограничивающие примеры графиков, иллюстрирующих общие ВКК, влияющие на локусы SMN1/SMN2. На ФИГ. 12A представлены профили глубины в областях SMN1/SMN2. Образцы с общим количеством копий SMN1+SMN2 2, 3, 4 и 5 показаны точками соответственно. Для каждой категории количества копий суммируют глубину 50 образцов. Каждая точка представляет нормированные значения глубины в окне длиной 100 п. н. Количество прочтений рассчитывали в каждом окне длиной 100 п. н., суммировали показания для SMN1 и SMN2 и нормализовали по глубине образцов дикого типа (CN=4). Экзоны SMN представлены в виде пурпурных прямоугольников. Две оси x показывают координаты в SMN1 (внизу) и SMN2 (вверху). На ФИГ. 12B показаны профили глубины, объединенные из 50 образцов, несущих делецию экзонов 7 и 8, показаны в виде точек. Значения глубины прочтения рассчитывали таким же образом, как показано на ФИГ. 12A.
[0202] После поиска аномальных пар прочтения, других общих количеств копий вариантов в области SMN не было обнаружено. Объединив эту информацию вместе, количество копий генов SMN было призвано специально идентифицировать количество интактных и укороченных форм путем разделения генов на две области: область из 6,3 т.п.н., содержащая экзоны 7-8, и область из 22,2 т.п.н., содержащая экзоны 1-6. Количество копий этих двух областей рассчитывали по глубине прочтения, как описано в разделе «Способы» данного примера. Количество копий рассчитанное на основе области экзонов 7-8, обеспечивала количество интактных генов SMN. Образцы с SMN* имели более высокий уровень определения количества копий из области экзона 1-6 по сравнению со определением количества копий из области экзона 7-8, и их различие представляло собой количество копий SMN*. На ФИГ. 13 показаны результаты такого расчета для 12747 образцов когорты, где было определено 2144 экземпляров SMN*, включая 140 образцов с двумя копиями SMN* и один образец с тремя копиями SMN*.
[0203] На ФИГ. 13 показан неограничивающий пример диаграммы рассеяния общего количества копий SMN (SMN1+SMN2) (ось X, обозначает глубину прочтения экзонов 1-6) и количества копий интактного SMN (ось y, обозначает глубину прочтения экзонов 7-8).
Дифференциация количества копий SMN1 от SMN2
[0204] После расчета общего количества копий генов SMN SMN1 и SMN2 дифференцировали, как описано ниже. Поскольку c.840C>T является наиболее важным функциональным различием между SMN1 и SMN2, абсолютное количество копий этих двух генов можно теоретически получить при помощи соотношения между количеством прочтений, поддерживающих SMN1 и SMN2 на этом сайте. Однако глубина прочтения в одном диплоидном положении обычно составляет 30 - 40X для набора данных WGS и иногда не обеспечивает достаточной мощности для четкого различения между различными состояниями количества копий (см. ФИГ. 15). Таким образом, при выполнении определения количества копий использовали дополнительные различия оснований вблизи c.840C>T, чтобы информация на этих сайтах могла быть скомбинирована с c.840C>T. Поскольку желательно дифференцировать интактный SMN1 от SMN2, были рассмотрены варианты, которые встречаются в пределах делеции 6,3 т. п. н. SNP в гомополимерах и коротких тандемных повторах (TRs), которые могут быть более подвержены ошибкам, были исключены, что привело к различиям в 16 основаниях между SMN1 и SMN2 (таблица 8).
[0205] Для этих 16 различий оснований независимо определяли количество копий аллелей SMN1 и SMN2 (см. раздел «Способы» данного примера) и сравнивали определения количества копий для каждого положения с определениями количества копий в сплайс-вариантном сайте (ФИГ. 14A, ФИГ. 17). Наблюдалось заметное различие между соответствием определений в африканской и неафриканской популяциях (ФИГ. 14A). Для образцов неафриканцев обнаружено 13 сайтов с большим (>85%) количеством копий в соответствии с сайтом сплайсинга. И наоборот, для образцов африканцев обнаружено только семь участков с большим количеством копий в соответствии с сайтом сплайсинга, а значения соответствия были ниже, чем в неафриканских популяциях. Это согласуется с внутригенными вариациями во многих из этих положений и более высокими частотами для этих неэталонных аллелей в неафриканских популяциях. Вариант сплайсинга и семь позиций, которые были высоко согласованы с вариантом сплайсинга как в африканских, так и в неафриканских популяциях, были выбраны для определения количества копий SMN1 и SMN2. Ограничиваясь двумя состояниями количества копий, которые позволяют легко идентифицировать гибридные аллели (SMN1=CN2 и SMN2=CN0 или SMN1=CN2 и SMN2=CN1), стала возможной оценка частот аллелей этих сайтов в генах SMN1 и SMN2 (таблица 9, ФИГ. 18A и 18B). На основе этого анализа по всем этим восьми позициям было оценено, что до 0,5% генов SMN1 содержат аллель SMN2. И наоборот, по оценкам, до 0,9% генов SMN2 являются носителями аллеля SMN1. Эти наблюдения могут быть результатом генной конверсии или того, что многие из этих восьми сайтов полиморфны в популяции. Большая часть этих гибридных аллелей происходит из африканских популяций (таблица 9).
[0206] На ФИГ. 14A-14D показаны распределения количества копий SMN1/SMN2/SMN* в популяции. На ФИГ. 14A представлен неограничивающий пример иллюстративного графика, иллюстрирующий процентную долю образцов, показывающих согласование определения количества копий с c.840C>T по 16 сайтам различия оснований SMN1-SMN2 в африканских и неафриканских популяциях. Сайт 13* представляет собой сплайс-вариантный сайт c.840C>Т. Черной горизонтальной линией обозначено 85% совпадения. На ФИГ. 14B показаны неограничивающие примеры гистограмм распределений количества копий SMN1, SMN2 и SMN* по пяти популяциям в 1kGP и когорте NIHR BioResource (числа приведены в таблице 15). На ФИГ. 14C показан неограничивающий пример графика зависимости количества копий SMN1 от общего количества копий SMN2 (интактный SMN2 + SMN*). На ФИГ. 14D показаны два трио с пробандом SMA, обнаруженным специалистом и ортогонально подтвержденным в когорте NIHR BioResource. Количество копий на аллель SMN1, SMN2 и SMN* фазировано и помечено для каждого члена трио.
[0207] Введение большего количества различий оснований повышало способность дифференцировать SMN1 от SMN2. Однако поскольку данные сайты действительно не являются инвариантными в соответствующих генах, и определение количества копий в отдельных сайтах может быть вызвано ошибкой, вероятность того, что одно из отдельных определений будет отличаться от истинного состояния количества копий, будет увеличиваться. Чтобы сделать конечное определение было необходимо, чтобы определения количества копий SMN1 согласовывались друг с другом на 5 или более из 8 сайтов (полное описание правил определения количества копий см. в разделе «Способы» данного примера). С отсечением апостериорной вероятности 0,8 большинство образцов имели согласованные определения по меньшей мере в пяти из восьми сайтов, и только 1,4% образцов имели менее 5 согласованных сайтов (таблица 10). В 80% из этих образцов было проведено достоверное определение количества копий на основе второго правила консенсуса (требующего согласования с определением количества копий, сделанным суммированием прочтений на всех 8 сайтах). «Несогласующиеся » сайты чаще не были выявлены из-за низкой апостериорной вероятности, а не из-за несоответственных определений, и только 15,3% из них были достоверными определениями, которые не соответствовали с консенсусом других сайтов. Опять же, значительная часть рассогласований определялась в африканских популяциях (Таблица 10). Использование меньшего количества сайтов для большинства правил позволило получить большее количество неопределений и неверных определений по сравнению с использованием восьми сайтов (таблица 11).
Проверка определения количества копий SMN
[0208] Для тестирования данного способа секвенировали 48 образцов с известными количествами копий SMN1 и SMN2, включая 29 пробандов SMA, 6 носителей SMA и 13 образцов с количеством копий SMN1 больше 1. Результаты определения количества копий SMN1 согласуются с результатами цифровой ПЦР для всех 48 случаев, а результаты определения количества копий SMN2 согласуются для 47 (97,9%) из 48 случаев (таблицы 6A и 6B). В этом единственном несоответствующем случае (MB509) способ определили 3 копии SMN2, в то время как цифровая ПЦР показала 2 копии SMN2 (таблица 12). При более точном изучении обнаружили делецию 1884 п. н. в SMN1 (chr5:70247145-70249029, hg19) в этом образце (ФИГ. 19). Делеция невелика (значительно не изменяет глубину в области 6 т. п. н., используемой для определения количества копий интактного SMN) и ранее не сообщалась (и не обнаруживалась в данных популяции), поэтому способ не был предназначен для ее обнаружения. В результате этого данный образец был правильно идентифицирован как SMA, но количество копий SMN2 было завышено на единицу. Делеция согласуется с определениями количества копий, выполненными на 8 сайтах различия SMN1-SMN2, причем первые 2 сайта не находятся в делеции и количество копий SMN1 определялось как CN=1, а следующие 6 сайтов находятся в делеции и количество копий SMN1 определялось как CN=0.
[0209] Была проанализирована согласованность определений количества копий SMN1/SMN2/SMN* в 258 трио из когорты проекта Next Generation Children (см. раздел «Способы» данного примера). Ни в одном из определений не было менделевской ошибки (таблица 13).
Таблица 6A. Проверка относительно образцов с известными количествами копий SMN1/SMN2
Таблица 6B. Проверка относительно образцов с известными количествами копий SMN1/SMN2
Количество копий SMN1, SMN2 и SMN* по популяции
[0210] Учитывая высокую точность, продемонстрированную проверкой результатов цифровой ПЦР, способ применяли к данным WGS большой глубины (>30X) для 12747 неродственных образцов из 1kGP и NIHR BioResource (таблица 14). Распределение количества копий было проанализировано по популяциям (европейцы, африканцы, выходцы из Восточной Азии, Южной Азии и смешанные американцы, состоящие из колумбийцев, мексиканцев, перуанцев и пуэрториканцев). На ФИГ. 14B показана гистограмма количества индивидуумов с различным количеством копий интактного SMN1, интактного SMN2 и SMN*. Распределения аналогичны между образцами 1kGP и образцами NIHR BioResource (ФИГ. 20). В целом, у индивидов имелось больше копий SMN1, чем SMN2. Наиболее распространенными комбинациями количества копий SMN1/SMN2 были 2/2 (44,9%) и 2/1 (33,4%). За исключением африканцев, которые демонстрировали более высокую вариабельность как количества копий SMN1, так и SMN2, вариабельность количества копий SMN1 была намного ниже, чем количества копий SMN2. И наоборот, 54,7% африканцев имели три или более копий SMN1, что было более чем в два раза выше, чем наблюдалось у любой из четырех других популяциях (ФИГ. 14B, таблица 7). Существует обратная зависимость между количеством копий SMN1 и SMN2, количество копий SMN2 была ниже с увеличением количества копий SMN1 (ФИГ. 14C, коэффициент корреляции -0,344, p-значение < 2.2e-16). Это наблюдение согласуется с механизмом, при котором генная конверсия происходит между SMN1 и SMN2. Наблюдаемое более большое количество копий SMN1 по сравнению с количеством копий SMN2 может быть результатом смещения в сторону конверсии SMN2 в SMN1 или выбора к низкому количеству копий SMN1. У африканцев количество копий SMN2 значительно ниже, чем в других популяциях.
[0211] Количество носителей SMA, выявленных в разных популяциях, суммировано в таблице 7 и таблице 15. Из 12683 людей с достоверными определениями количества копий SMN1/SMN2, европейцы имели самую высокую частоту носительства - 2,2%, за ними следовали смешанные американцы (2,05%), выходцы из Восточной Азии (1,35%) и выходцы из Южной Азии (1,67%). Африканцы имели самую низкую частоту носительства (0,44%). Распределения частот количества копий, наблюдаемые в данном примере, согласуются с предыдущими исследованиями распределений количества копий SMN1/SMN2 в общей популяции. Кроме того, определяли частоту делеции в экзоне 7-8 (SMN*) в разных популяциях: 21,2% европейцев и 11,5% смешанных американцев имели хотя бы одну копию SMN*, в то время как частота была ниже у выходцев из Южной Азии (3,35%), африканцев (1,1%) и выходцев из Восточной Азии (0,34%).
[0212] В когорте проекта Next Generation Children (см. раздел «Способы» данного примера) идентифицировали SMA у двух неонатальных пробандов из трех анализов, которые были подтверждены независимо. Кроме того, количество копий SMN1, SMN2 и SMN* было фазировано для каждого члена трио (ФИГ. 14D).
Моделирование определения количества копий одного сайта
[0213] Количество прочтений на одном сайте на медианной глубине выборки 30X, 35X и 40X моделировали на основе распределения Пуассона, а прочтения, поддерживающие SMN1, отбирали на основе биномиальной модели со всеми возможными комбинациями количества копий SMN1 и SMN2, причем общее количество копий SMN составляло от 2 до 6. С учетом количества поддерживающих прочтений SMN1 и SMN2 была получена апостериорная вероятность смоделированного количества копий SMN1 (см. раздел «Способы» в данном примере). Апостериорная вероятность была высокой (больше 0,9), когда по крайней мере одно значение количества копий SMN1 или SMN2 было низким (меньше или равно 1) (ФИГ. 16). Когда оба значения были больше 2, т. е. в комбинациях SMN1:SMN2 2:2, 2:3, 2:4, 3:2, 3:3 и 4:2, апостериорная вероятность часто становилась низкой и опускалась ниже 0,9. Это связано с большей изменчивостью глубины прочтения, когда ожидаемое количество копий выше. Таким образом, в этих сценариях, создающих определения количества копий SMN1 и SMN2 с использованием одного сайта, могут быть менее точными.
Расхождения в проверочных образцах
[0214] Был взят один образец MB509, у которого наблюдалось расхождение между нашим определением количества копий и результатами цифровой ПЦР. При дальнейшем анализе данный образец имел две копии SMN2 и одну копию SMN1 с делецией 1884 п. н. (chr5:70247145-70249029, hg19, ФИГ. 20). Хотя выравнивание прочтения в области SMN1/2 не всегда является точным, тщательный анализ результатов разделенных прочтений показал, что прочтения или их партнеры перекрывали основания, специфичные для SMN1. Не ограничиваясь какой-либо теорией, было выдвинуто предположение, что эта делеция была правильно размещена на SMN1. Делеция невелика (значительно не изменяет глубину в области 6,3 т. п. н., используемой для определения количества копий интактного SMN) и ранее не сообщалась (и не обнаруживалась в образцах 1kGP, что является очень редким вариантом), поэтому способ не был предназначен для обнаружения делеции. В результате этого способ определил общее количество копий SMN1+SMN2 как 3. Делеция согласуется с определениями количества копий, выполненными на 8 сайтах различия SMN1-SMN2, причем первые 2 сайта не находились в делеции и количество копий SMN1 определялось как CN=1, а следующие 6 сайтов находились в делеции и количество копий SMN1 определялось как CN=0 (ФИГ. 21A). На основе большинства правил способ определял количество копий SMN1 как 0, правильно идентифицируя образец как SMA. Количество копий SMN2 рассчитывали как общее количество копий за вычетом количества копий SMN1, поэтому способ определил количество копий SMN2 как 3, завышая его на 1.
[0215] Четыре других образца, MB231, MB367, MB383 и LP2101748, имели расхождения между выполненными определениями количества копий и результатами цифровой ПЦР или MLPA. Количество прочтений и нормализованные значения глубины (количество прочтений, разделенное на глубину гаплоидных образцов) на 8 сайтах различия оснований поддерживали наши определения количества копий (ФИГ. 21A), и расхождение, вероятно, было вызвано ошибками в ортогональных способах. В двух образцах определение геномным секвенированием (GS) и определение цифровой ПЦР отличались в два раза (MB231: GS-0,2, PCR-0,4 и MB383: GS-3,1, PCR-6,2). При использовании цифровой ПЦР может возникнуть проблема нормализации, приводящая к завышению количества копий в два раза.
[0216] При сравнении определений количества копий, полученных с помощью MLPA, с 1109 образцов 1kGP был исключен один образец, в котором не было определено SMN2 Δ 7-8 из-за низкой апостериорной вероятности для всего количества копий SMN, а также три образца, где не было определено количество копий SMN1 и SMN2 из-за расхождения в определениях количества копий по 8 выбранным сайтам, которые не соответствовали согласованным правилам (ФИГ. 22B).
Обнаружение «молчащих» носителей
[0217] g.27134T>G SNP может быть связан со статусом молчащего носителя 2+0 SMA, где одна хромосома несет две копии SMN1 (либо путем дублирования SMN1, либо путем генной конверсии SMN2 в SMN1), а другая хромосома не имеет копий SMN1. Способ данного примера также может определять наличие этого SNP и, таким образом, может быть использован для скрининга потенциальных «молчащих» носителей. Этот SNP наиболее тесно связан с двухкопийными аллелями SMN1 у африканцев, где 84,5% субъектов с тремя копиями SMN1 и 92,6% субъектов с четырьмя копиями SMN1 имеют SNP g.27134T>G (таблица 7). Определение этого SNP значительно увеличил частоту обнаружения носителей у африканцев, поскольку африканцы имеют более высокую частоту аллелей, несущих две копии SMN1 (таблица 17 и таблица 18). Однако 33% субъектов с двумя копиями SMN1 также имели g.27134T>G SNP, что указывает на то, что значительная часть синглетных аллелей SMN1 также несет этот SNP. Рассчитывали оценки максимальной вероятности для процентных долей одно-и двухкопийных аллелей SMN1, несущих g.27134T>G (таблица 17), и остаточных рисков для комбинации определения количества копий и SNP (таблица 18). Рассчитанные оценки аналогичны предыдущим исследованиям, хотя есть значительные различия во всех этих оценках. Эта изменчивость может быть обусловлена вариативностью популяции: например, африканцы (этот пример) по сравнению с афроамериканцами (предыдущие исследования) и северные европейцы (чрезмерно представлены в этом примере) по сравнению с более разнообразной выборкой европеоидов (предыдущие исследования).
Таблица 7. Количество копий SMN1 и частоты g.27134T>G в зависимости от популяции
>G+
>G+
>G+
>G+
Сравнение между двумя преобразователями, BWA и Isaac
[0218] Способ, приведенный в данном примере, чрезмерно проанализировал прочтение как SMN1, так и SMN2, и, следовательно, был нечувствительным к тому, как преобразователь различает эти два гена. Таким образом, использование различных преобразователей должно давать аналогичные результаты. Данные BAM, проанализированные в данном примере, были получены с использованием двух различных преобразователей: BWA для данных 1kGP и различные версии Isaac для остальных. Согласованные распределения количества копий SMN1/2 между образцами 1kGP и NIHR (таблица 19, ФИГ. 20) указывают на то, что наш способ нечувствителен к преобразователю. Кроме того, способ испытывали на согласованность путем выравнивания 117 образцов как с BWA, так и с Isaac, включая 5 образцов SMA и 3 носителей. Все 117 образцов имели совершенно одинаковые определения количества копий (SMN1/SMN2/SMN2Δ7-8) в соответствии со способом из данного примера и нормализованные глубины как для экзонов 1-6, так и для экзонов 7-8 были практически идентичными (r > 0,999, ФИГ. 22).
Сравнение между определениями носителя в данном исследовании и Larson et al.
[0219] Определения носителя, полученные в образцах 1kGP в данном примере (N=37), сравнивали с теми, о которых сообщали Larson et al. (N=36) и обнаружили 26 перекрывающихся определений (таблица 15). Предполагается, что определения, полученные способом по данному примеру, являются правильными, Larson et al. сделали 10 ложноположительных (FP) и 11 ложноотрицательных определений (FN). Larson et al. идентифицировали носители путем определения того, была ли доля прочтений, поддерживающих SMN1 меньше или равна 1/3. В этом исследовании использовали данные секвенирования малой глубины, которые, как ожидается, приводят к некоторым ошибкам, но, что более важно, их подход имеет тенденцию к ошибкам без определения общего количества копий. Например, образец с одной копией SMN1 и одной копией SMN2 будет определятся как не носитель (фракция SMN1 1/2), а образец с двумя копиями SMN1 и четырьмя копиями SMN2 будет определятся как носитель (фракция SMN1 1/3) с получением ложноположительных и ложноотрицательных результатов (таблица 16).
Дополнительные фигуры и таблицы
[0220] На ФИГ. 15 показаны неограничивающие примеры графиков, каждый из которых иллюстрирует распределение апостериорной вероятности для моделирования количества копий SMN1 с использованием одного сайта при разных глубинах прочтения и комбинаций количества копий SMN1:SMN2
[0221] На Фиг. 16 показан неограничивающий пример IGV снимка области SMN2 в образце с делецией в экзоне 7-8. Горизонтальные линии соединяют два прочтения в пару на центральной дорожке выравнивания. Результаты BLAT для двух разделенных прочтений, охватывающих точку разрыва, показаны в нижней дорожке, показывая два сегмента одного и того же выравнивания для прочтения с каждой стороны от точки разрыва делеции.
[0222] На ФИГ. 17 показаны неограничивающие примеры графиков, иллюстрирующих корреляцию между необработанными количествами копий SMN1 при 15 различиях оснований вблизи с840.C>T и необработанными количествами копий SMN1 на сайте с840.C>T. Необработанное количество копий SMN1 в каждом сайте рассчитывали как количество копий интактного SMN, умноженное на долю SMN1, поддерживающие количество прочтений SMN1 + SMN2 из количества прочтений. Коэффициенты корреляции приведены в заголовке каждого графика.
[0223] На ФИГ. 18 и 18B показаны неограничивающие примеры графиков с гаплотипами SMN1/SMN2 в образцах с SMN1:2 SMN2:0 и SMN1:2 SMN2:1 в 1kGP. По оси y показаны необработанные количества копий SMN1, как показано на ФИГ. 16. По оси X показаны 16 сайтов, индексы которых перечислены и объяснены в таблице 8. Индекс № 13 представляет сайт c840.C>T. Образцы с SMN1:2 SMN2: 0 показаны вместе на верхнем левом графике. Образцы с SMN1:2 SMN2:1 показаны в виде 5 кластеров. ФИГ. 18A Неафриканская популяция ФИГ. 18B Африканская популяция
[0224] На ФИГ. 19 показан неограничивающий пример IGV снимка, показывающего делецию 1,9 т. п. н. в SMN1 в MB509.
[0225] На ФИГ. 20 показан неограничивающий иллюстративный график, иллюстрирующий количество копий SMN1/SMN2/SMN* в когортах 1kGP и NIHR.
[0226] На ФИГ. 21A и 21B показаны расхождения и отсутствие определений в проверочных выборках. На фиг. 21 A показаны пять образцов с расхождениями между определениями GS и цифровой ПЦР или MLPA. По оси X показаны 16 сайтов, индексы которых перечислены и объяснены в таблице 8. Индекс № 13 представляет сайт c840.C>T. Левая ось y для столбцов показывает количество прочтений, поддерживающее SMN1 и SMN2. Правая ось y для линий показывает нормализованную глубину прочтения, прокси для количества копий SMN1 и SMN2 (количество прочтений, деленное на глубину гаплоида). В заголовке каждой панели показаны определения с помощью GS и цифровой ПЦР/MLPA для каждого образца для SMN1 и SMN2, разделенные запятой. На ФИГ. 21B представлены три проверочных образца 1kGP, где определитель SMN не определил количество копий SMN1 и SMN2 из-за расхождения между сайтами различий оснований SMN1/SMN2. Для консенсусных правил способа используются восемь сайтов: № 7-8 и № 10-15. По оси y показаны необработанные количества копий SMN1, как показано на ФИГ. 17.
[0227] На ФИГ. 22 представлены определения количества копий, полученные из BWA и Isaac BAM.
Таблица 8. Геномные координаты различий оснований между SMN1 и SMN2
(c.840 C>T)
Таблица 9. Частоты гаплотипов SMN1 с аллелем SMN2 и гаплотипов SMN2с аллелем SMN1 в двух простых состояниях количества копий (SMN1=CN2 и SMN2=CN0 или SMN1=CN2 и SMN2=CN1)
(Цифры в скобках указывают на вклад представителей африканской популяции.)
Таблица 10. Количество образцов с разным количеством согласованных сайтов на 8 сайтах SNP
(Цифры в скобках указывают на вклад представителей африканской популяции.)
* Определения сделаны в этих образцах на основе второго правила большинства (см. «Способы»).
Таблица 11. Количество неопределений, обусловленных несоответствием, и определение несоответствия, сделанное при сокращенном количестве сайтов.
(Требуется 5 для согласования)
(4)
(3)
(2)
(1)
Таблица 12. Проверочные образцы
Количество копий SMN1
Количество копий SMN2
Таблица 13. Количество копий SMN1, SMN2 и SMN* определяли в 258 трио в когорте проекта Next Generation Children
Таблица 14. Количество образцов по популяции в когортах 1kGP и NIHR BioResource
Таблица 15. Частоты количества копий SMN1, SMN2 и SMN* по популяциях
(0,44%)
(44,79%)
(41,35%)
(13,41%)
(25,06%)
(49,78%)
(23,73%)
(1,44%)
(0,0%)
(98,89%)
(1,0%)
(0,11%)
(2,2%)
(92,24%)
(5,43%)
(0,13%)
(8,63%)
(39,9%)
(48,37%)
(2,89%)
(0,2%)
(78,74%)
(19,83%)
(1,42%)
(1,67%)
(80,48%)
(16,26%)
(1,58%)
(6,51%)
(33,39%)
(57,26%)
(2,42%)
(0,42%)
(96,65%)
(3,35%)
(0,0%)
(1,35%)
(93,09%)
(5,56%)
(0,0%)
(4,72%)
(35,58%)
(57,34%)
(2,02%)
(0,34%)
(99,66%)
(0,34%)
(0,0%)
(2,05%)
(86,8%)
(10,56%)
(0,59%)
(8,8%)
(39,88%)
(47,51%)
(3,23%)
(0,59%)
(88,56%)
(10,85%)
(0,59%)
Таблица 16. Сравнение определений носителя, полученных в образцах 1kGP в данном примере и в публикации Larson et al.
Таблица 17. Оценки максимальной вероятности для процентного содержания одно- и двухкопийных аллелей SMN1, несущих g.27134T>G
* (1kGP Европейцы: 0,11%)
* (1kGP Европейцы: 10,0%)
* Когорта NIHR BioResource, которая занимает большую часть европейской популяции, анализируемой в этом примере из-за большого размера выборки, включает образцы из Северной Европы, которые несут более низкую частоту SNP g.27134T>G, чем более разнообразные европейские выборки из проекта 1000 Genomes.
Таблица 18. Обнаружение носителя SMA и оценки остаточного риска из этого примера
(Афроамериканец)
(Афроамериканец)
(1kGP Европейцы
1 из 846
(1kGP Европейцы 1 из 27)
(испанцы)
(испанцы)
(испанцы)
a Количества и частоты аллелей SMN1 для расчета остаточного риска, взято у Sugarman et al. Панэтнический скрининг носителей и пренатальная диагностика спинальной мышечной атрофии: клинический лабораторный анализ> 72 400 образцов. Eur J Hum Genet. 2012;20(1):27-32. doi:10.1038/ejhg.2011.134;
b Включает выходцев из Южной и Восточной Азии;
с Luo et al. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet Med Off J Am Coll Med Genet. 2014;16(2):149-156. doi:10.1038/gim.2013.84;
d Feng et al. The next generation of population-based spinal muscular atrophy carrier screening: comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Genet Med Off J Am Coll Med Genet. 2017;19(8):936-944. doi:10.1038/gim.2016.215;
e Alias et al. Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling. Eur J Hum Genet. 2018;26(10):1554. doi:10.1038/s41431-018-0193-4.
Таблица 19. Количество копий SMN1/SMN2/SMN2 Δ 7-8 в когортах 1kGP и NIHR
Обсуждение
[0228] Из-за высокой гомологии последовательностей между SMN1 и SMN2 область SMN трудно определить путем как короткого, так и длинного прочтения секвенирования, и до сих пор эта важная область была исключена из стандартного анализа WGS. В данном примере продемонстрирован способ, который позволяет независимо определить количество копий SMN1 и SMN2, используя данные WGS с коротким прочтением, заполняя важный пробел в диагностике SMA и скрининге носителей для инициатив в области точной медицины. Точное измерение количества копий SMN1 и SMN2 важно не только для диагностики SMA, но также является прогностическим индикатором и основой терапевтических вариантов. Количество копий SMN2 применяли в качестве критерия для многих клинических испытаний SMA, включая Nusinersen и Zolgensma.
[0229] Для демонстрации данного способа определяли количество копий SMN1 и SMN2 с использованием данных секвенирования 12747 образцов, охватывающих пять различных субпопуляций. Были идентифицировано следующее: 251 образцов с потерей целого гена (менее двух копий) и 1317 с приростом целого гена (более двух копий) SMN1; 6241 образцов с потерей целого гена и 1274 с приростом целого гена SMN2; 2144 образцов, несущих одну или более копий укороченной формы SMN*. Невозможно точно определить роль, которую выполняют делеции, дупликации или генная конверсия, приводящая к изменениям количества копий в данной области. Доказательства, подтверждающие все три механизма, включают: 1) 3853 образцов с общим количеством копий (SMN1+SMN2) CN<4 (делеции), 2) 670 образцов с общим количеством CN>4 (дубликатов) и 3) сильная обратная корреляция между количеством копий SMN1 и SMN2 (генная конверсия, ФИГ. 14C). Кроме того, была определена частота носительства от 1:42 до 1:101, в зависимости от предковой популяции (таблица 7). Частоты количества копий по популяции сильно различались, и результаты по популяции в этом примере согласуются с предыдущими популяционными исследованиями. Хотя согласование дает качественную поддержку точности способа, точность способа непосредственно оценивали путем сравнения определений количества копий, полученных способом, с результатами цифровой ПЦР. При таком прямом сравнении все определения количества копий (48/48) SMN1 и 98% (47/48) SMN2 согласуются с результатами на основе цифровой ПЦР. Одно из расхождений было связано с удалением 2 т. п. н., на которое не был нацелен способ, и, что важно, способ правильно идентифицировал статус SMA этого образца.
[0230] В данном примере определение количества копий было оптимизировано для людей любого происхождения и, таким образом, ограничил дифференциацию SMN1/2 до функционально значимого варианта сплайсинга и семи сайтов высокой степени соответствия варианту сплайсинга во всех популяциях (ФИГ. 14A). Путем количественного определения соответствия между всеми эталонными различиями и вариантом сплайсинга способ позволял выявить вариации этих фиксированных различий, которые, если они не учитывались надлежащим образом (например, удалены из нашего анализа) могли бы привести к ошибкам в наших определениях количества копий. Отсутствие фиксированных различий было бы особенно проблематичным при анализе африканцев, поскольку они обладают более разнообразными гаплотипами. Популяционные генетические исследования, например, в том числе с использованием длинного секвенирования, могут помочь более прямо профилировать гаплотипическое разнообразие в популяциях и идентифицировать новые варианты сайтов, которые могут дополнительно повысить точность дифференциации SMN1/SMN2.
[0231] Один тип «молчащего» носителя возникает, когда субъект имеет две копии гена SMN1, но они обе принадлежат к одному и тому же гаплотипу. SNP (g.24134T>G) использовали для идентификации субъектов, которые подвержены повышенному риску стать носителями, если значение количества копий SMN1 равно двум, но риск, связанный с этим SNP, может сильно различаться в разных исследованиях и популяциях (таблица 17). Если у субъекта имеется только одна копия SMN1, то субъект можно определенно идентифицировать как носитель, но этот вариант указывает только на вероятность того, что 2-8% быть носителем, если количество копий SMN1 равно двум. Благодаря WGS возможны различные варианты, которые встречаются с различными комбинациями количества копий SMN1 и SMN2, и идентификация дополнительных маркеров, которые могут быть использованы для улучшения нашей способности идентифицировать эти «молчащие» носители. Кроме того, потеря варианта сплайсинга c.840C>T в настоящее время объясняет примерно 95% случаев SMA, а остальные случаи включают другие патогенные варианты. Эти другие патогенные варианты представляют собой другой тип «молчащего» носителя. Способ может непосредственно генотипировать эти другие патогенные варианты как часть процесса тестирования, дополнительно улучшая возможность выявления носителей и случаев SMA.
[0232] Хотя в геноме существуют сложные области, в которых нормальные конвейеры WGS не определяют варианты, этот пример демонстрирует возможность применения WGS в комбинации целевым биоинформатическим подходом для определения одной такой сложной области. Эту нацеленную стратегию (WGS + специализированную биоинформатику) можно применять к ряду сложных вариантов, таких как экспансия повторов и CYP2D6, описанные в данном документе. Традиционно проведение всех известных генетических тестов и скрининг носителей для каждого отдельного человека было экономически эффективным, поэтому кандидаты на конкретный генетический тест были идентифицированы с использованием такой информации, как частота носительства и семейный анамнез. Однако этот процесс означает, что многие люди без семейного анамнеза, которые могли бы получить пользу от знания о статусе SMA, не имели доступа к этим данным в обычном порядке. Как только анализ WGS сможет точно обнаружить все SNV и CNV во всех клинически значимых генах, тогда станет возможной более общая и популяционная стратегия генетического тестирования с помощью одного теста. Улучшение WGS в качестве замены одного существующего генетического теста поможет упростить интеграцию большего количества генетических тестов и скрининговых тестов в WGS, обеспечивая более общий доступ к генетическому тестированию в масштабах всей популяции. WGS обеспечивает ценную возможность оценки всего генома на предмет генетической вариации, и дальнейшая разработка более целенаправленных биоинформатических решений для сложных областей с данными WGS поможет приблизить перспективу персонализированной медицины на один шаг к реальности.
Пример 2
Точное генотипирование CYP2D6 с использованием данных секвенирования целого гена
[0233] В данном примере и приложении A описано генотипирование CYP2D6 с использованием данных секвенирования целого генома. Содержание приложения А полностью включено в данный документ посредством ссылки.
[0234] CYP2D6 участвует в метаболизме 25% всех лекарственных средств и является ключевой мишенью для персонализированной медицины. Генотипирование CYP2D6 является сложной задачей из-за его высокого полиморфизма, наличия общих структурных вариантов (SV) и высокого сходства последовательностей с паралогом псевдогена CYP2D7. В данном документе описан биоинформатический способ, также называемый в данном документе Cyrius, который может точно генотипировать CYP2D6, используя данные секвенирования целого генома (WGS). Этот способ показал превосходные характеристики (97,9% соответствия истине) по сравнению с другими способами (85,6-88,8%) в 138 образцах с консенсусными определениями с помощью GeT-RM и в 50 дополнительных образцах с помощью Pacific Biosciences, California, Inc. (Менло-Парк, Калифорния), также известный как PacBio, данные секвенирования. Конкретным отличительным признаком способа является способность определить структурные варианты звездчатых аллелей. Способ правильно идентифицировал 97,2% (70/72) структурных вариантов звездчатых аллелей по сравнению с 77,8-88,9% (56/72 и 64/72) для других способов. При применении способа к 2504 образцам из проекта 1000 Genomes (1kGP) было установлено, что звездчатые аллели CYP2D6, включающие SV, встречаются на 32,2% чаще, чем сообщалось ранее, для некоторых популяций. В этом примере представлены сравнительного анализа с самым большим набором данных проверки. В некоторых вариантах осуществления способ представляет собой полезный инструмент для фармакогеномного применения с WGS. Способ может помочь приблизить перспективу точной медицины на шаг ближе к реальности.
Введение
[0235] Существуют значительные вариации в реакции субъектов большое количество назначенных в клинике лекарственных средств. Существенным фактором, влияющим на этот дифференцированный ответ на лекарства, является генетический состав генов, метаболизирующих лекарственные средства. Для точной медицины требуется генотипирование фармакогенов, чтобы сделать возможным индивидуальное лечение. Цитохром P450 2D6 (CYP2D6) является одним из наиболее важных генов, метаболизирующих лекарственное средство, и он участвует в метаболизме 25% лекарственных средств. Ген CYP2D6 является высокополиморфным, 106 звездчатыми аллелями, определенными Pharmacogene Variation (PharmVar) Consortium (pharmvar.org/gene/CYP2D6). Звездчатые аллели CYP2D6 представляют собой копии гена CYP2D6, определяемые комбинацией малых вариантов (таких как однонуклеотидные варианты (SNV) и вставок/делеций (инделы)) и структурных вариантов (SV), и соответствуют разным уровням ферментативной активности CYP2D6, таким как слабый, промежуточный, нормальный или сверхбыстрый метаболизм.
[0236] Генотипирование CYP2D6 осложняется наличием нефункционального паралога CYP2D7, который расположен выше CYP2D6 и имеет 94% сходства последовательностей с несколькими почти идентичными областями. Распространены делеции и дупликации CYP2D6 и слияния CYP2D6 с его псевдогенным паралогом CYP2D7. Традиционно генотипирование CYP2D6 проводили с помощью чипов или способов, основанных на полимеразной цепной реакции (ПЦР), таких как анализы TaqMan, цифровая капельная ПЦР (ddPCR) и ПЦР длинных фрагментов. Данные анализы отличаются количеством звездчатых аллелей (вариантов), на которые они нацелены, что приводит к вариабельности результатов генотипирования в разных анализах. Общим ограничением этих способов является: 1) аллель дикого типа *1 часто определяется по умолчанию, когда ни один из целевых вариантов не обнаружен, или 2) родительский аллель, такой как *2, определяющий истинный звездчатый аллель, не тестируется. Данные анализы имеют низкую пропускную способность и часто затрудняют обнаружение структурных вариантов.
[0237] Профилирование всего генома с высокой пропускной способностью и в клинически значимый временной интервал возможно при секвенировании следующего поколения (NGS). Предпринимаются широкомасштабные усилия по секвенированию популяции, и желательной целью может быть фармакогеномическое тестирование. Генотипирование CYP2D6 с помощью NGS особенно сложно из-за общих конверсий генов между CYP2D6 и CYP2D7 (далее именуемых CYP2D6/7), общих SV (делеции генов, дупликации и слитные гены CYP2D6/7), а также из-за сходства последовательностей CYP2D/7, что приводит к неоднозначному выравниванию прочтения для любого из генов. Некоторые существующие определители не могут обнаруживать сложные структурные варианты, и было показано, что они имеют низкую эффективность. Другие существующие определители, такие как Aldy (Numanagic et al. Allelic decomposition and exact genotyping of highly polymorphic and structurally variant genes. Nat Commun. 2018;9(1):1-11. Doi:10.1038/s41467-018-03273-1) и Stargazer (Lee et al. Stargazer: a software tool for calling star alleles from next-generation sequencing data using CYP2D6 as a model. Genet Med. 2019;21(2):361. Doi: 10.1038/s41436-018-0054-0), основывается на точном выравнивании прочтения при прочтении последовательностей CYP2D6 для обнаружения SV на основе глубины и получения конфигураций гаплотипов на основе наблюдаемых малых вариантов и SV. Однако точное прочтение последовательностей CYP2D6 часто невозможно во многих положениях во всем гене, поскольку последовательность очень похожа на CYP2D7 или даже неотличима из-за генной конверсии. В результате этого шаблоны глубины могут быть неоднозначными, и определители могут определить ложноположительные/отрицательные малые варианты. Некоторые определители не поддерживают hg38, поэтому многие исследования потребуют повторного выравнивания до hg37, чтобы использовать эти инструменты.
[0238] Доступность панели эталонных образцов по программе CDC Genetic Testing Reference Material Program (GeT-RM; Gaedigk et al. Characterization of Reference Materials for Genetic Testing of CYP2D6 Alleles: A GeT-RM Collaborative Project. J Mol Diagn JMD. August 2019. Doi: 10.1016/j.jmoldx.2019.06.007), где консенсусные генотипы основных фармакогенетических генов получены с использованием множества платформ генотипирования, позволили оценить точность генотипирования для вновь разработанных способов. GeT-RM охватывает 43 из 106 звездчатых аллелей CYP2D6. Кроме того, многие из способов, используемых для этих консенсусных генотипов, могут быть склонны к ошибке, что приводит к конфликтам между способами. Доступность высококачественных длинных прочтений позволяет получить полную картину CYP2D6 для лучшей проверки сложных вариантов и гаплотипов. В данном документе описан Cyrius, способ генотипирования CYP2D6 на основе WGS, позволяющий преодолеть проблемы с CYP2D6 и CYP2D7 (в данном документе именуемые CYP2D6/7). Cyrius обладает превосходной точностью генотипирования по сравнению с Aldy и Stargazer в 138 эталонных образцах GeT-RM и 50 образцах с данными секвенирования целого генома PacBio, охватывая 41 из 106 известных звездчатых аллелей. Способ применяли к данным секвенирования с большой глубиной для 2504 неродственных образцов из проекта 1000 Genomes (1kGP), чтобы сообщить о распределении звездчатых аллелей в пяти этнических популяциях. Данный анализ демонстрирует различия с частотами в PharmGKB, подчеркивая потенциальные ошибки, связанные с объединением ограниченных определений звездчатых аллелей, созданных с использованием различных технологий, разработанных для идентификации конкретных подмножеств известных звездчатых аллелей. Данный анализ расширяет текущее понимание генетического разнообразия CYP2D6, особенно сложных звездчатых аллелей с SV.
Материалы и способы
Образцы
[0239] Было проанализировано следующее: Данные WGS для 138 эталонных образцов GeT-Rm, включая 96 образцов, которые были генотипированы в начальном исследовании GeT-RM и обновлены в последней версии GeT-RM, а также 42 дополнительных образца, которые были недавно добавлены в последней версии GeT-RM. Для первой партии из 96 образцов выполняли WGS с использованием набора TruSeq DNA PCR-free sample preparation, используя парные прочтения длиной 150 п. о., секвенированными на приборах HiSeq X компании Illumina, Inc.(Сан-Диего, Калифорния, США). Для выравнивания прочтения использовали сборку генома GRCh37. Данные секвенирования для 70 этих образцов загружали из ebi.ac.uk/ena/data/view/PRJEB19931. Данные WGS для второй партии из 42 образцов загружали из NYGC в рамках проекта 1000 Genomes (см. ниже).
[0240] Для популяционных исследований использовали данные из проекта 1000 Genomes (1kGP), для которых WGS BAM для 2504 образцов были загружены с ncbi.nlm.nih.gov/bioproject/PRJEB31736/. Данные файлы BAM получали путем секвенирования прочтений 2 x 150 п.о. на приборах NovaSeq 6000 компании Illumina из библиотек без проведения ПЦР и их выравнивания с человеческим эталонным образцом hs38DH. Данные WGS для 70 образцов GeT-RM были загружены из ebi.ac.uk/ena/data/view/PRJEB19931.
Секвенирование PacBio
[0241] Образцы гДНК были приобретены в Coriell Institute for Medical Research (Кориeл, Нью-Джерси, США). Качество образцов гДНК оценивали с помощью Nanodrop (ThermoFisher, MA, США). Соотношение A280/A260 должно находиться в диапазоне от 1,8 до 2,0, а соотношение A260/230 составляет ≥ 2,0. Молекулярную массу гДНК оценивали с помощью системы Femto Pulse (Agilent CA, США). Размер большинства фрагментов ДНК должен составлять > 40 т. п. н. Если качество образца гДНК из Coriell ниже, чем требование к протоколу, свежую ДНК экстрагировали из B-лимфоцитов (Кориeл, Нью-Джерси, США) при помощи набора для экстракции ДНК Qiagen (Qiagen, Калифорния, США).
[0242] Фрагментировали 10 мкг гДНК до 15 т. п. н. с использованием г-пробирок Covaris в соответствии с инструкциями производителя (Covaris, Массачусетс, США). ДНК очищали с использованием 0,45x гранул AMPure XP (Beckman Coulter, IN, США) в соответствии с инструкциями производителя. Размер разрезанной ДНК подтверждали с помощью Femto Pulse System (Agilent, Калифорния, США).
[0243] Библиотеки конструировали в соответствии с протоколом PacBio «Preparing HiFi SMRTbell® Libraries using SMRTbell Template Prep Kit 1.0» или «HiFi SMRTbell® Libraries using SMRTbell Express Template Prep Kit 2.0» (PacBio, Калифорния, США). Размер библиотеки выбирали для 15~ 20 т. п. н. с использованием прибора Sage Elf с 0,75% агарозой (Sage Science, MA, США). Контроль качества всех библиотек выполняли с помощью Qubit (Life Technologies, Калифорния, США) и Femto Pulse (Agilent, Калифорния, США).
Для секвенирования использовали платформу для секвенирования PacBio Sequel II. Данные WGS с 20-кратным покрытием по существу получены из 2 ~ 3 клеток SMRT (Pacific Biosciences, Калифорния, США). Способ генотипирования CYP2D6
[0244] Способ, описанный в данном примере Cyrius, сначала определяет сумму количества копий (CN) CYP2D6/7, в соответствии со способом, аналогичным описанному в примере 1. Количество прочтений рассчитывали непосредственно из файла BAM, выровненного по WGS на основе всех прочтений, сопоставленных с CYP2D6 или CYP2D7, включая прочтение с нулевым качеством картирования, чтобы учесть области с высокой гомологией последовательности. Суммарное количество прочтений нормализовали по длине области. Затем выполняли коррекцию GC против 3000 предварительно выбранных по всему геному областей размером 2 т. п. н. Эти 3000 областей нормализации были случайным образом выбраны из генома для стабильного покрытия образцов популяции для выявления глубины секвенирования и фиксирования систематической ошибки GC. Нормированные значения глубины по популяции моделировали с использованием одномерной смеси 11 распределений, которые сосредоточены вокруг каждого целочисленного значения количества копий, представляющего состояния количества копий в диапазоне от 0 до 10. ЦНС CYP2D6+CYP2D7 было определено с помощью модели смеси нормальных распределений (GMM) с порогом апостериорной вероятности 0,95. Тот же подход использовали для определения количества копий спейсерной области размером 1,5 т. п. н. между повтором REP7 и CYP2D7, чтобы выявить количество копий слитых генов, содержащих REP7 (ФИГ. 23).
[0245] На ФИГ. 23 представлен неограничивающий пример графика, на котором показано качество данных WGS в области CYP2D6/7. Среднее качество картирования для выборок 1kGP нанесено на график для каждого положения в области CYP2D6/7. В окне длиной 200 п. о. применяют медианный фильтр. REP6, REP7 и экзоны 9 CYP2D6/7 показаны в виде прямоугольников слева (CYP2D6) и справа (CYP2D7). Две области повтора длиной 2,8 т. п. н. ниже CYP2D6 (REP6) и CYP2D7 (REP7) идентичны и по существу несовместимы. Пунктирной рамкой обозначена область спейсера между CYP2D7 и REP7. Две основные гомологичные области в генах заштрихованы.
[0246] С помощью способа идентифицировано 118 дифференцирующих оснований CYP2D6/CYP2D7(см. дополнительную информацию к этому примеру, ФИГ. 26). В каждом из этих положений дифференцирующихся оснований Cyrius определял количество хромосом, несущих CYP2D6, и количество, несущих CYP2D7, путем комбинирования общего количества копий CYP2D6+CYP2D7 с количеством прочтений, поддерживающих каждое из ген-специфических оснований. На основе определенного общего количества копий Cyrius перебрал все возможные комбинации количества копий CYP2D6 и CYP2D7 и получил комбинацию, которая дает самую высокую апостериорную вероятность для наблюдаемого количества прочтений CYP2D6 и CYP2D7. Слияние генов определяли путем идентификации оснований, когда количество копий CYP2D6 менялось (ФИГ. 27).
[0247] Cyrius анализировал выравнивания для прочтения для выявления малых вариантов, которые определяют звездчатые аллели. Представляющие интерес варианты были разделены на те, которые попадали в гомологичные области CYP2D6/CYP2D7 (т. е. области низкого качества картирования на ФИГ. 23) и варианты, которые встречаются в уникальных областях CYP2D6. Для первого варианта Cyrius искал вариант прочтения CYP2D6 и соответствующий ему сайт CYP2D7. В последнем случае Cyrius использовал прочтения, выровненные с CYP2D6. Количество копий определенное в области, также было учтено при определении малых вариантов. Например, где было идентифицировано слияние дупликаций * 68, один гаплотип должен иметь интактную копию CYP2D6 плюс копию* 68, а другой гаплотип должен иметь интактную копию CYP2D6 и, следовательно, количество копий CYP2D6 должно располагаться на 3 выше от экзона 2 и на 2 ниже от экзона 2.
[0248] Наконец, Cyrius сопоставил определенные структурные варианты и малые варианты в соответствии с определением звездчатых аллелей (загружено и проанализировано с PharmVar, pharmvar.org/gene/CYP2D6, последний доступ в марте 2019 г.) для обозначения звездчатых аллелей, которые дополнительно группировали в гаплотипы, когда, например, было более двух копий CYP2D6. Для этого была включена предварительная информация для определения точных гаплотипов, например, *68 был в том же гаплотипе, что и *4, а *36 был в том же гаплотипе, что и *10). Эти предварительные значения были созданы на основе шаблонов тандемного расположения, описанных в PharmVar, и также подтверждаются нашими достоверными данными (12/12 для *68 и 25/25 для *36). Была доступна опция для сопоставления только определенных структурных вариантов и малых вариантов с звездчатыми аллелями с известными функциями.
[0249] Из 131 звездчатых аллелей, определенных в PharmVar (последний доступ в марте 2020 г.), 25 все еще ожидают отверждения, поэтому в примере они исключены и внимание сосредоточено на 106 отобранных аллелях (в Cyrius есть еще один вариант для включения этих неотверждаемых аллелей). Из этих 106 звездчатых аллелей четыре из нашего перечня мишеней были удалены, ни один из которых не находился в GeT-RM. Удаленные звездчатые аллели включают *61 и *63 (оба с неизвестными функциями), которые представляют собой гибридные гены CYP2D6/7, очень похожие на *36, с точкой разрыва слияния немного выше. Поскольку было невозможно различить область экзона 7-экзона 8 между CYP2D6/7 (ФИГ. 26), эти два звездчатые аллеля нельзя отличить от *36, и они будут обозначаться Cyrius как *36. Кроме того, удаляли *27 (нормальная функция) и *32 (неизвестная функция); *27 и *32 имеют общие g.42126938C>T, вариант генной конверсии в высокогомологичной области (идеально считанный вариант будет выровнен с CYP2D7. При подсчете результатов прочтения, поддерживающих CYP2D6 и CYP2D7, на одном сайте, может быть сложно точно отличить 1 копию CYP2D6 и 3 копии CYP2D7 от 2 копий каждая по 20. Таким образом,*27 будет называться *1, а *32 будет называться *41.
Проверка истинности результатов GeT-RM и долгому прочтении
[0250] При сравнении определений CYP2D6, сделанных Cyrius, Aldy и Stargazer, с согласованными генотипами, предоставленными GeT-RM, генотип считался совпадающим при условии, что присутствуют все звездчатые аллели в истинном генотипе, назначение гаплотипа было другим. Пример этого происходит в нескольких образцах, перечисленных в GeT-RM как *1/*10+*36+*36, но именуемых Aldy как *1+*36/*10+*36.
[0251] При проверке определений генотипа по данным PacBio анализировали прочтения PacBio, охватывающие весь ген CYP2D6, для выявления малых вариантов, которые, как известно, определяют звездчатые аллели. Длинные (~ 10 т. п. н.) прочтения позволяют полностью поэтапно преобразовать данные варианты в гаплотипы, и данные гаплотипы сопоставляются с таблицей звездчатых аллелей, чтобы определить, какой из звездчатых аллелей считан каждый раз. прочтения, несущие структурные вариации, определяли путем сопоставления прочтений с набором контрольных контигов, которые были сконструированы для представления известных структурных вариантов (*5/*13/*36/*68/дубликации).
Применение Aldy и Stargazer
[0252] Aldy v2.2.5 запускали с использованием команды « aldy genotype -p lumina -g CYP2D6 ».
[0253] Stargazer v1.0.7 был проведен для генотипа CYP2D6, используя VDR в качестве контрольного гена, с файлами GDF и VCF в качестве входных данных.
[0254] Поскольку Aldy and Stargazer поддерживают только GRCh37, с помощью Isaac были изначально выровнены образцы 1kGP с hs38DH, а с помощью Isaac была выполнена переориентация относительно GRCh37.
Результаты
Проверка и сравнение характеристик
[0255] Определения CYP2D6, сделанные Cyrius, Aldy и Stargazer, по 188 образцам, по которым была получена достоверная высококачественная информация. Сравнивали результаты полногеномного секвенирования этих 188 образцов, включая 138 образцов GeT-RM и 50 образцов с достоверностью PacBio (таблица 20, таблица 21). Данные PacBio CCS позволили обнаружить и визуализировать точки разрыва общих и редких структурных вариантов в области (ФИГ. 24) и, таким образом, служили ценным ресурсом для изучения сложных звездчатых аллелей и подтвердили фазирование вариантов звездчатых аллелей. При коротких прочтениях эти образцы с SV демонстрировали различные сигналы глубины, которые точно позволяли определять SV (ФИГ. 27).
Таблица 20. Сводные данные по результатам сравнения с достоверностью
N=18
N=14
N=40
N=116
* После поиска трех расходящихся образцов были сделаны усовершенствования Cyrius, и Cyrius мог точно определить 187 из 188 этих образцов.
Таблица 21. Результаты Cyrius/Aldy/Stargazer в отношении достоверности данных GeT-RM и PacBio
(обновлено до *56/*5)
(+гибрид)
+*2
+*36
+*10
+*10
+*36
+*10
+*10
+*36
+*69
+*10
+*10
+*10
+*10
+*10
+*10
+*36
+*10
+*90
+*90
+*10
+*10
+*10
+*10
+*10
*52
+*36
+*52
+*10
+*10x2
+*10
+*10
+*10
+*36+*83
+*36+*83
/*36+*39
+*36
+*10
+*10
+*10
+*36
+*10
+*10
+*10
(NA24631)
(обновлено до
*10x2/*2)
(обновлено до *1/*40)
(NA24385)
[0256] Путем сравнения с образцами GeT-RM были обнаружены три образца, в которых определения всех трех определителей согласуются, но не согласуются с консенсусом GeT-RM. Секвенирование целого генома PacBio подтвердило, что три определения были правильными и необходимо обновить консенсус GeT-RM (ФИГ. 24).
[0257] На ФИГ. 24 показаны структурные варианты, подтвержденные прочтением PacBio CCS. прочтение PacBio подтверждает делецию (*5), дупликацию и слияние (*36, *68 и *13). Графики получали с использованием SV-VIZ2 (zotero.org/google-docs/?xAunA6). В случае делеций и дупликаций из-за идентичной последовательности в областях REP6/7 точные положения точек разрыва в пределах REP6/7 были недоступны. Точки разрыва в А и В приведены только для иллюстрации. Генотипами образцов в панели A-E являются *2/*5, *17/*2x2, *10/*10+*36, *29/*4+*68 and *1/*2+*13 соответственно.
[0258] Cyrius первоначально сделал четыре определения, расходящихся от достоверного GeT-RM, показывающих чувствительность 97,9%. Среди этих расхождений был включен образец NA19908 (GeT-RM определен *1/*46), в котором Cyrius определил 1/*46 и *43/*45 в качестве двух возможных диплотипов. Обе эти две комбинации звездчатых аллелей звезды дают один и тот же набор вариантов. Ни анализ фаз прочтения, ни частотный анализ популяции не могут исключить любую комбинацию генотипов. Результаты генотипирования в результате различных анализов, которые позволили получить консенсус GeT-RM для данного образца, также показали расхождение между *1/*46 и* *43/*45, что указывает на сложность этих комбинаций (таблица 22). Будущее секвенирование большего количества образцов любого из диплотипов может помочь идентифицировать новые варианты, которые их различают.
Таблица 22. Результаты GeT-RM для образца NA19908
+CNV
+XL-ПЦР
v.r6
пользовательская
v.r6+20180103
CYP2D6 V1.1
CYP2D6
V1.1
+индивидуальная панель и VeriDose
[0259] В оставшихся трех образцах, в которых Cyrius не соответствовал достоверности, были выявлены ошибки, а Cyrius был улучшен для определения правильных генотипов. Во-первых, в NA23275 (*1/*40) вставку из 18 п. н., определяющую *40, первоначально пропустили, поскольку прочтение, содержащие вставку, часто выравнивались не как имеющие вставку, а как мягкие сшивания. Определитель было улучшено для учета мягких сшиваний при поиске варианта. Во-вторых, в HG03225 (*5/*56) прочтения, полученные из CYP2D7, выравнивали с CYP2D6, предотвращая определение определяющего варианта* 56. Определитель был улучшен, чтобы он был более чувствительным к прочтениям вариантов в данной области. Наконец, в HG00421 (*10x2/*2) слияние было ошибочно обозначено как *36, как и двумя другими определителями. Более тщательное исследование этого образца по данным PacBio показало другое слияние, *10D, причем точка разрыва слияния находилась ниже экзона 9 (ФИГ. 28). Данное слияние выполняет ту же функцию, что и *10 (сниженная функция), а *36 не функционирует благодаря экзону 9, полученному из CYP2D7. Определитель был улучшен для возможности определения *10D. Хотя в данном примере эти три образца были обработаны как ошибочные определения, улучшения, внесенные в Cyrius после поиска этих трех образцов, позволили точно указать 187 из 188 образцов, что подчеркивает, как большее количество достоверных данных и большее количество данных по популяциям может выявить ограничения, которые могут обеспечить усовершенствования определителя для последующих образцов.
[0260] Напротив, оба других определителя CYP2D6 имели чувствительность менее 90% по сравнению с этими образцами. Aldy имел чувствительность 88,8%. В частности, он переопределил слияния CYP2D6/CYP2D7, такие как *61, *63, *78 и *83 (определили 8 из 21 расходящихся образцов, таблица 21). Слияние определенное Aldy может быть опровергнуто данными PacBio на ФИГ. 29. Stargazer имел чувствительность 85,6% и был наиболее подвержен ошибкам в присутствии SV. Чувствительность образцов с SV составляла только 77,8%, и 16 из 27 расходящихся определений находились в образцах со структурными вариантами. Следует отметить, что он ошибочно определили NA19317 (*5/*5) как *2/*2, при этом двойная делеция полностью отсутствует. Stargazer не смог генотипировать два образца с помощью слияния *13 (Таблица 21). Кроме того, Stargazer показал высокий уровень ошибок при слиянии *36 (7 неверных определений из 25 всех образцов со *36). В частности, Stargazer неправильно определил все 5 образцов, в которых в одном гаплотипе имеется более одной копии *36.
[0261] В совокупности 188 проверочных образцов, использованных в данном примере, подтвердили точность определения Cyrius CYP2D6 в 48 различных гаплотипах (таблица 23), включая 41 звездчатых аллелей, а также несколько общих и редких структур SV, таких как дупликации, *2+*13, *4+*68, *10+*36, *10+*36+*36 and *10+*36+*36+*83 (новый гаплотип, о котором ранее не сообщалось, см. ФИГ. 30A и 30B). Эти 41 звездчатые аллели, которые были протестированы при проверке данных, представляют 38,7% от 106 курируемых звездчатых аллелей, которые в настоящее время перечислены в PharmVar, и 53,4% (31 из 58) от тех, которые имеют известную функцию. Они перекрывают 96,4% гаплотипов определенных Cyrius в образцах 1kGP (таблица 23, также см. следующий раздел).
Таблица 23. Гаплотипы, подтвержденные в данном примере, и их частота в 1kGP
этническая принадлежность
Восточной Азии
Частота гаплотипов CYP2D6 в пяти этнических популяциях
[0262] Учитывая высокую точность, приведенную в предыдущем разделе, для исследования CYP2D6 в общей популяции использовали Cyrius за пределами проверочных выборок. Анализировали распределение гаплотипов по популяциям (европейцы, африканцы, выходцы из Восточной Азии, Южной Азии и смешанные американцы, состоящие из колумбийцев, мексиканцев, перуанцев и пуэрториканцев) в 2504 образцах 1kGP (ФИГ. 25, таблица 23). Cyrius окончательно определил диплотипы в 2445 (97,6%) из 2504 образцов, включая 46 отдельных звездчатых аллелей, при этом 41 звездчатых аллелей перекрывались с теми, которые были включены в данные проверки. Эти 41 подтвержденных ранее звездчатых аллелей представляют 96,5% от всех звездчатых аллелей, которые определялись в образцах 1kGP (таблица 23).
[0263] На ФИГ. 25 показан неограничивающий пример графика, на котором показаны частоты аллелей CYP2D6 для пяти этнических популяциях для десяти наиболее распространенных гаплотипов с измененной функцией CYP2D6. Один гаплотип (*2x2) характеризуются повышенной функцией, два гаплотипа (*4 и *4 + *68) - отсутствием функции, а остальные гаплотипы - сниженной функцией.
[0264] В 59 образцах, где Cyrius не сделал окончательного определения диплотипа, 10 образцов имели неокончательное определение SV, 30 образцов имели варианты определения, не соответствующие ни одному из известных звездчатых аллелей, четыре образца имели одинаковую неоднозначность между *1/*46 и *43/*45, как описано выше для проверочного образца NA19908, и 15 образцов имели окончательные определения звездчатых аллелей, которые Cyrius не мог однозначно преобразовать в диплотипы.
[0265] В большинстве случаев частоты гаплотипа согласуются с pharmGKB (ФИГ. 31A и 31B, таблица 24). Например, африканцы имеют высокую частоту *17 (~20%) и *29 (~ 10%), выходцы из Южной Азии имеют высокую частоту *41 (~ 12%), европейцы имеют высокую частоту *4 (18-20%, включая *4+*68), а выходцы из Восточной Азиии имеют высокую частоту *10 (40-50%, включая *10+*36). Повышенная чувствительность Cyrius к структурным вариантам позволяет получить более полную картину частот структурных вариантов в популяциях. Среди них гаплотип, содержащий слияние *10 + *36, очень распространен у выходцев из Восточной Азии (> 30% по сравнению с 1-2%, о которых сообщается в PharmGKB, ФИГ. 31A и 31B), а также другой гаплотип, содержащий слияние *4 +. *68 также довольно часто встречается у европейцев (> 5%, данные отсутствуют в PharmGKB, ФИГ. 31A и 31B). В совокупности частота гаплотипов с учетом SV была на 32,2%, 5,57%, 1,47%, 1,34% и 0,45% больше, чем сообщается в PharmGKB у выходцев из Восточной Азии, Европы, Америки, Африки и Южной Азии, соответственно (общая частота в PharmGKB составляет 7,48%, 5,33%, 5,17%, 9,9% и 6,19% соответственно).
[0266] Существует несколько других гаплотипов, для которых была отмечена более низкая частота, чем в PharmGKB (ФИГ. 31A и 31B), подчеркивая сложность объединения данных из множества исследований с использованием различных технологий. Они включают в себя *2 у выходцев из Африки и Южной Азии. Поскольку * 2 является присвоением по умолчанию, если некоторые другие звездчатые аллели не тестируются, его частота может быть завышена в PharmGKB. Определена более низкая частота *41 у африканцев. В соответствии с PharmGKB, *41 не всегда последовательно определялся с помощью определяющего SNP в исследованиях, что привело к переоценке частоты *41, особенно у лиц африканского происхождения. Гораздо более высокая частота *29 у выходцев из Южной Азии в PharmGKB (6% против 0%, оцененных в этом примере) была вызвана ошибкой в PharmGKB: 0,2% в публикации Sistonen et al. (CYP2D6 worldwide genetic variation shows high frequency of altered activity variants and no continental structure. Pharmacogenet Genomics. 2007;17(2):93-101. doi:10.1097/01.fc.0000239974.69464.f2) был ошибочно включен в PharmGKB в виде 20%. У европейцев частота *34 и *39 была гораздо ниже. *34 и *39 определяются одним из двух вариантов, которые определяют 2, поэтому оба этих двух варианта должны были быть протестированы в любом исследовании, в котором описывается CYP2D6. *34 и *39 представлены на уровне> 1% только в 3 из 91 исследования европейцев в PharmGKB, среди которых Wesmiller et al.(The Association of CYP2D6 Genotype and Postoperative Nausea and Vomiting in Orthopedic Trauma Patients. Biol Res Nurs. 2013; 15(4): 382-389. doi: 10.1177/1099800412449181), которые сообщили только о *39 и имели ограниченный размер выборки (N=112), Kapedanovska Nestorovska (Distribution of the most Common Genetic Variants Associated with a Variable Drug Response in the Population of the Republic of Macedonia. Balk J Med Genet BJMG. 2014; 17(2): 5-14. doi: 10,2478/BJMG-2014-0069) сообщали о обоих *34 и *39 и относились к конкретной стране Македонии, а также имели небольшой размер выборки (N=184) и Del Tredici et al. (Frequency of CYP2D6 Alleles Including Structural Variants in the United States. Front Pharmacol. 2018; 9. doi: 10.3389/far.2018.00305), не сообщали о *34 или *39, но PharmGKB, возможно, ошибочно принял частоту, указанную для *35, за частоту *34.
Анализ дифференцирующих оснований CYP2D6/CYP2D7
[0267] Всего из эталонного генома было извлечено 208 однонуклеотидных различий между CYP2D6/7. В образцах 1kGP, где общее количество копий CYP2D6+CYP2D7 равно 4, т.е. не было выявлено никаких структурных изменений, было запрошено процентное содержание образцов, в которых количество копий основания CYP2D6 определено как 2 на 208 сайтах (ФИГ. 26). Во многих сайтах наблюдался небольшой процент образцов с двумя копиями основания CYP2D6, что свидетельствует о том, что разница оснований CYP2D6/CYP2D7 не фиксируется в популяции, поэтому различия в основаниях нельзя использовать для различения двух генов. Использование выравнивания прочтения сайтов при прочтении может привести к значительному шуму при дифференцировке двух генов. Всего выбрали 118 высокостабильных сайтов, где >98% образцов показали две копии оснований CYP2D6 для дифференцировки CYP2D6/CYP2D7, что позволило получить чистый сигнал для определения SV.
Дополнительные фигуры и таблицы
[0268] На ФИГ. 26 показано, что сайты из различием оснований CYP2D6/CYP2D7 отличаются высокой вариабельностью в популяции. На оси y показана частота образцов, в которых CN для основания CYP2D6 определены в 2 из всех образцов, имеющих общее CN CYP2D6 + CYP2D7, равное 4. По оси X показаны координаты генома в hg38. Экзоны CYP2D6 показаны серыми прямоугольниками над графиком. Черной горизонтальной линией обозначено отсечение 98%.
[0269] На ФИГ. 27 показаны необработанные CNCYP2D6 в сайтах дифференцировки CYP2D6/7 в примерах с SV. Необработанные CN CYP2D6 рассчитывали как общее CN CYP2D6+CYP2D7, умноженное на соотношение CYP2D6 поддерживающих прочтений из CYP2D6 и CYP2D7 поддерживающих прочтений. Большой ромб обозначает количество копий генов, полученных из CYP2D6 на конце гена (может представлять собой полный ген CYP2D6 или слитый ген, заканчивающийся CYP2D6), вычисленное как общее CN CYP2D6+CYP2D7 минус CN спейсерной области CYP2D7 (см. ФИГ. 23). Для обнаружения SV в каждом сайте определяли CN CYP2D6, и изменение CN CYP2D6 в гене указывало на присутствие SV. Например, в HG01161 CN CYP2D6 изменился с 2 на 1 между экзоном 7 и экзоном 9, что указывает на гибридный ген CYP2D7-CYP2D6. В HG00553 CN CYP2D6 изменилось с 2 на 3 между экзоном 1 и экзоном 2, что указывает на гибридный ген CYP2D6-CYP2D7.
[0270] На ФИГ. 28 показано, что данные PacBio подтверждают слияние *10D в HG00421. Для сравнения показан образец с *36 (HG00612). Прочтения PacBio, содержащие слияния, это прочтения с заштрихованными основаниями, основаниями, которые представляют собой программное сшивание, сделанное выравнивателем, и были получены из части слияния CYP2D7. Точки разрыва слияний близки друг к другу, но точка разрыва для *36 расположена выше от различий оснований в экзоне 9 (находящихся внутри черного блока), а точка разрыва для *10D расположена ниже, оставляя ген CYP2D6 интактным.
[0271] На ФИГ. 29 показано, что данные PacBio имели ложный *61 (гибрид CYP2D6/CYP2D7), полученный Aldy в HG02622. Ожидаемый генотип представлял собой *17/*45, но Aldy вызвал *61-подобный/*78 (оба *61 и *78 представляют собой звездчатые аллели с SV). Данные PacBio показали отсутствие структурного варианта в этой области (каждое прочтение полностью выровнено, без каких-либо мягких сшиваний, указывающих на не выровненные части).
[0272] На ФИГ. 30A и 30B показан новый гаплотип *10+*36+*36+*83 в HG00597. ФИГ. 30A График глубины, представленный на ФИГ. 27, показывает, что HG00597 имел три копии *36-подобных слияний, все из которых имели точку разрыва в гомологичной области между экзоном 7 и экзоном 9. ФИГ. 30B Снимок экрана IGV с данными PacBio,, демонстрирующий все прочтения, содержащие слияния, то есть те, которые выровнены мягким сшиванием. Одна копия слитого гена не имела g.42130692G>A, SNP, который находился в *36, но не в *83, как показано в области, фланкированной двумя черными вертикальными линиями. Эта копия была *83, и в отличие от того, что сообщалось в PharmVar, это был гибридный ген с REP7, а не с REP6, в противном случае количество копий области ниже экзона 9 было бы равно 3 вместо 2 на ФИГ. 30A.
[0273] На ФИГ. 31A и 31B сравнивали частоты 1kGP и pharmGKB. Каждая точка представляет гаплотип с частотой >=0,5% для 1kGP или pharmGKB. Отмечены связанные с SV гаплотипы, включая два гаплотипа с наибольшим отклонением (*10+*36 у жителей Восточной Азии и *4+*68 у европейцев). Другие гаплотипы с отклоненными значениями помечены (*2, *41, *34, *39, *2 и *29). Для каждой панели проводят диагональную линию. Коэффициенты корреляции приведены для каждой популяции (*10+*36 исключено для жителей Восточной Азии и *4+*68 исключено для европейцев для расчета). На ФИГ. 31B показаны значения в нижнем диапазоне значений (<5%).
[0274] На ФИГ. 32 показан неограничивающий пример снимка IGV, показывающий сборку de novo результатов прочтения PacBio в HG00733, не включающую слияние *68.
Таблица 24. Сравнение так частот гаплотипа определенных Cyrius и частот pharmGKB
Обсуждение
[0275] Этот пример описывает Cyrius, способ, позволяющий точно диплотипировать сложную область CYP2D6. Уникальная особенность данного примера состоит в том, что для подтверждения как гаплотипов, так и SV использовали долгое прочтение данных. Длинные прочтения дают уникальную возможность подтвердить области точек разрыва общих SV (делеции и дупликации CYP2D6, а также слитые гены CYP2D6/7) и подтвердить фазирование гена CYP2D6. При использовании 188 образцов, включая 50 с данными проверки долгого прочтения, в качестве набора ортогональных данных проверки, было показано что Cyrius превосходит другие генотипы CYP2D6, достигая 97,9% точности по сравнению с 88,8% для Aldy и 85,6% для Stargazer. В частности, по сравнению с этими существующими определителями CYP2D6Cyrius позволил провести прочтение в областях, где CYP2D6/7 имеют высокое сходство. Неоднозначные выравнивания прочтения в данных областях могут привести к неправильной оценке количества копий и ошибкам при определении малых вариантов. За счет учета возможных невыровненных прочтений и выбора набора надежных сайтов дифференциации CYP2D6/7 Cyrius может намного лучше определять звездчатые аллели с помощью SV, достигая точности 97,2% по сравнению с 88,9% для Aldy и 77,8% для Stargazer.
[0276] Во всех 188 проверочных образцах было подтверждено всего 41 разных звездчатых аллелей, которые представляли (38,7%) всех звездчатых аллелей, перечисленных в PharmGKB, включая 53,4% аллелей с известным функциональным статусом. Хотя на основе анализа образцов 1kGP в данном примере, набор для проверки включал только 38,7% всех известных звездчатых аллелей, по оценкам, они представляли 96,5% звездчатых аллелей в пангеномной популяции. Как правило, частоты аллелей, рассчитанные для 2504 образцов 1kGP из пяти этнических популяций, согласовывались с предыдущими исследованиями простых звездчатых аллелей. И наоборот, для некоторых звездчатых аллелей, которые определялись наличием SV, были выявлены совершенно разные частоты, вероятно, потому что многие из звездчатых аллелей, подвергнутых SV-воздействию, сложно определить с помощью обычных анализов. Это подчеркивает неотъемлемые ошибки объединения результатов исследований, в которых использовали множество различных анализов CYP2D6, некоторые из которых могут быть разработаны для простого определения подмножества звездчатых аллелей. Например, из 5 анализов, использованных для создания консенсусных генотипов GeT-RM, индивидуальная точность варьировала от 47,1% до 75,2% по сравнению с консенсусом (таблица 25). Единый способ, позволяющий определить все известные звездчатые алели одним анализом, является лучший выбор для создания базы данных популяционного уровня.
Таблица 25. Точность отдельных анализов GeT-RM
+CNV
+XL-ПЦР
v.r6
пользовательская
v.r6+20180103
CYP2D6 V1.1
CYP2D6
V1.1
+индивидуальная панель и VeriDose
[0277] Кроме того, для анализа 2504 образцов 1kGP из пяти этнических популяций использовали Cyrius для определения частот звездчатых аллелей. Вычисленные частоты аллелей согласуются с предыдущими исследованиями простых звездчатых аллелей, а Cyrius значительно улучшил оценки частоты звездчатых аллелей с вовлечением структурных вариантов, обнаружение которых может быть затруднено обычными способами.
[0278] Некоторые существующие способы основаны на точном выравнивании прочтений для различения CYP2D6 и CYP2D7, которые могут быть склонны к ошибкам из-за нескольких областей с высоким сходством последовательностей между двумя генами, в частности между интроном 1-экзоном 2 и экзоном 7-экзоном 9. Неоднозначное выравнивание может приводить к шуму в профилях глубины, что приводит к ложным определениям CNV. Кроме того, ошибочные выравнивания при прочтении могут приводить к ложноположительным или ложноотрицательным определениям вариантов. Напротив, Cyrius впервые определили общее количество копий CYP2D6+CYP2D7 путем подсчета всех прочтений, которые совпадают с любым из генов, а общее количество копий, не равное 4, четко указывает на наличие SV. Для определения точного положения SV использовали не все отличия на основе эталонного генома. Многие различия оснований CYP2D6/CYP2D7 не фиксированы, поэтому не все эти положения можно использовать для надежного отличия CYP2D6 от CYP2D7 (ФИГ. 26). Cyrius использовал 118 позиций дифференциации CYP2D6/CYP2D7, выбранных для определения точного положения SV. Определив сначала общее количество копий, а затем дифференцируя их с помощью подмножества подходящих дифференцирующих оснований, Cyrius смог достичь более точных определений SV. Для определения малых вариантов Cyrius преодолевает зависимость от однозначного выравнивания, ища варианты прочтения как в положении CYP2D6, так и в соответствующем положении CYP2D7, таким образом получая наиболее точные определения малых вариантов.
[0279] В примере для подтверждения как гаплотипов, так и определений SV использовали долгое прочтение данных. Данные PacBio в этом примере обеспечивают четкую картину области CYP2D6 - CYP2D7 с высоким качеством длинных прочтений (10 - 20 т. п. н.). В частности, данные PacBio позволяют определить области точек разрыва для общих структурных вариантов (делеции и дупликации CYP2D6, а также слитые гены CYP2D6-CYP2D7). Даже при прочтениях PacBio генотипирование CYP2D6 может быть непростым и может потребовать проведения направленного анализа, особенно при структурных вариантах, включающих дублирование (CYP2D6 и дублирование CYP2D6-CYP2D7), где дублированная область содержит >10 т. п. н. Например, подход сборки de novo не смог уловить слияние * 68 в образце HG00733 (Фиг. 31A и 31B). Кроме того, прочтения PacBio являются недостаточно длинными, чтобы покрыть более одной копии дублированной последовательности, а прочтения PacBio являются слишком длинными для определения количества копий при подсчете количества прочтений (для коротких прочтений), поэтому расчет количества копий затруднен. Секвенирование всего генома с короткими прочтениями обеспечивает наиболее точное решение для генотипирования CYP2D6.
[0280] При анализе образцов 1kGP Cyrius смог определить окончательный генотип более 97,6% образцов. В некоторых вариантах осуществления Cyrius смог определить оставшиеся 2,4% образцов. Например, в образцах, где возможны множественные конфигурации гаплотипов, может оказаться полезным использование вероятностного подхода для получения наиболее вероятного генотипа с учетом наблюдаемых вариантов. Кроме того, продолжение секвенирования и тестирования большего количества образцов поможет подтвердить способность генотипировать редкие звездчатые аллели, а также выявить новые варианты, которые можно использовать для различения неоднозначных диплотипов. Этот процесс был продемонстрирован в данном примере, где были внесены усовершенствования для лучшего определения трех звездчатых аллелей, которые первоначально неправильно определялись в 188 проверочных образцах. Улучшения были полезны для генотипирования на уровне популяции, поскольку три звездчатые аллеля обнаружены почти в 1% (23 из 2504) образцов 1kGP.
[0281] По мере выявления новых звездчатых аллелей новые звездчатые аллели могут быть добавлены в базу данных Cyrius. Одним из соображений при добавлении новых звездчатых аллелей, которые определяются новыми вариантами, является то, что данные варианты вряд ли будут рассмотрены в предыдущих определениях звездчатых аллелей. В результате могут существовать новые комбинации новых и существующих вариантов, которые не могут соответствовать ни одной из известных комбинаций, что приводит к отсутствию определений. Например, Cyrius включает возможность генотипирования по 25 новым звездчатым аллелям, добавленным в PharmVar v4 (не включенным в GeT-RM, Aldy или Stargazer). Однако пять (*119, *122, *135, *136, *139) из 25 новых звездчатых аллелей имеют новые варианты, которые при включении привели к отсутствию определений в образцах, которые могли бы быть определены ранее, что указывает на существование общих новых звездчатых аллелей с комбинацией вариантов, не отраженных в PharmVar. В результате эти пять звездчатых аллелей были удалены вместе с двумя другими (*127, с вариантом генной конверсии в гомологичной области и *131 вариантом в сайте с помехами) с сохранением оставшихся 18. Новые звездчатые аллели могут быть возможны по мере выявления новых вариантов/звездчатых аллелей. Общедоступные наборы данных WGS, такие как 2504 образцов 1kGP, проанализированные в данном документе, могут быть важным компонентом интеграции новых вариантов в определения звездчатых аллелей, поскольку эти данные позволяют быстро оценивать варианты во многих образцах с различными генотипами.
[0282] WGS предоставляет ценную возможность профилировать все генетические вариации всего генома, но многие из клинически важных областей/вариантов находятся за пределами возможностей большинства конвейеров вторичного анализа. CYP2D6 относится к сложным областям генома, которые одновременно являются клинически важными и также требуют целевых биоинформатических решений в дополнение к нормальным конвейерам WGS. Такие нацеленные способы уже успешно применяются к некоторым сложным областям, таким как ген SMN1, ответственный за спинальную мышечную атрофию, как показано в примере 1. Более целевые способы, такие как Cyrius, могут ускорить развитие фармакогеномики и сделать возможным персонализированную медицину.
Дополнительные соображения
[0283] По меньшей мере в некоторых из ранее описанных вариантов осуществления один или более элементов, используемых в варианте осуществления, можно взаимозаменяемо использовать в другом варианте осуществления, если такая замена технически не осуществима. Специалистам в данной области будет понятно, что в описанные выше способы и структуры могут быть внесены различные другие опущения, добавления и модификации без отступления от объема заявленного объекта изобретения. Предполагается, что все такие модификации и изменения включены в объем объекта изобретения, как определено в прилагаемой формуле изобретения.
[0284] Специалисту в данной области будет понятно, что для этого и других процессов и способов, описанных в данном документе, функции, выполняемые в процессах и способах, могут быть реализованы в другом порядке. Кроме того, описанные этапы и операции приведены только в качестве примеров, и некоторые этапы и операции могут быть необязательными, объединены в меньшее число этапов и операций или могут быть расширены в дополнительные этапы и операции без ущерба для сущности описанных вариантов осуществления.
[0285] В отношении применения по существу любых множественных и/или единственных терминов в данном документе специалисты в данной области могут изменять множественное число на единственное и/или единственное число на множественное в соответствии с требованиями контекста и/или сферой применения. В данном документе различные комбинации единственного/множественного числа для ясности могут быть указаны явным образом. Применяемые в данном описании и приложенной формуле изобретения формы единственного числа включают упоминания форм множественного числа, если в контексте явно не указано иное. Соответственно, предполагается, что такие фразы, как «устройство, выполненное для», включают в себя одно или более упомянутых устройств. Такое одно или более упомянутых устройств также могут быть в совокупности выполнены с возможностью осуществления упомянутых выше перечислений. Например, «процессор, выполненный с возможностью выполнения изложений A, B и C, может включать в себя первый процессор, выполненный с возможностью выполнения изложения A, работающий совместно со вторым процессором, выполненным с возможностью выполнения изложений B и C. Любая ссылка на «или» в данном документе подразумевает включение «и/или», если не указано иное.
[0286] Специалистам в данной области будет понятно, что в целом термины, используемые в данном документе, и в особенности в прилагаемой формуле изобретения (например, основной части прилагаемой формулы изобретения), по существу считаются «неограничивающими» терминами (например, термин «включающий» следует интерпретировать как «включающий без ограничений», термин « имеющий » следует интерпретировать как «имеющий по меньшей мере», термин «включает» следует интерпретировать как « включает без ограничений» и т. д.). Кроме того, специалистам в данной области техники будет понятно, что если в конкретной мере подразумевается использование ссылки на представленный пункт формулы изобретения, то такое намерение будет явно указано в пункте формулы изобретения, а в отсутствии такой ссылки такое намерение отсутствует. Например, для облегчения понимания нижеследующая прилагаемая формула изобретения может содержать вводные фразы «по меньшей мере один» и «один или более» для введения перечисления пунктов формулы изобретения. Однако, использование таких фраз не должно подразумевать, что введение перечисления в пункте формулы изобретения с использованием формы единственного числа ограничивает любой конкретный пункт формулы изобретения, содержащий такое введенное перечисление в пункте формулы изобретения вариантами осуществления, содержащими только одно такое перечисление, даже если та же формула изобретения включает вводные фразы «один или более» или «по меньшей мере один» и неопределенные артикли, такие как «некоторый » (например, форму единственного числа следует интерпретировать как «по меньшей мере один» или «один или более»); то же самое справедливо в отношении использования определенных артиклей, используемых для введения перечислений формулы изобретения. Кроме того, даже если определенное количество представленного изложения пункта формулы изобретения указано явным образом, специалистам в данной области техники будет понятно, что такое перечисление следует интерпретировать как означающее, по меньшей мере, указанное количество (например, простое цитирование фразы «два изложения » без других модификаторов означает, по меньшей мере, два изложения или два или более изложений). Кроме того, в случаях использования правила, аналогичного правилу «по меньшей мере одно из A, B, C и т. д. », в общем случае такая конструкция предназначена для специалиста в данной области для понимания правила (e.g., «Система, содержащая по меньшей мере один из A, B и C» будет включать в себя, без ограничений, системы, которые имеют только A, только B, только C, одновременно A и B, одновременно A и C, одновременно B и C и/или одновременно A, B и C и т. д.). В случаях использования правила, аналогичного правилу «по меньшей мере одно из A, B, C и т. д.», в общем случае такая конструкция предназначена для специалиста в данной области техники для понимания правила (например, «Система, содержащая по меньшей мере одно из A, B или C» будет включать в себя, без ограничений, системы, которые имеют только A, только B, только C, одновременно A и B, одновременно A и C, одновременно B и C и/или одновременно A, B и C и т. д.). Кроме того, специалистам в данной области будет понятно, что практически любое разделительное слово и/или фраза, представляющие два или более альтернативных терминов, будь то в описании, формуле изобретения или на чертежах, следует понимать как предполагающую возможность включения одного из терминов, любого из терминов или обоих терминов. Например, фразу «A или B» следует понимать как включающую возможности «A», или «B», или «A и B».
[0287] Кроме того, если признаки или аспекты раскрытия описаны в терминах групп Маркуша, специалистам в данной области будет понятно, что описание, таким образом, также описано в терминах любого отдельного члена или подгруппы членов группы Маркуша.
[0288] Как будет понятно специалисту в данной области, для любых и всех целей, например с точки зрения обеспечения письменного описания, все диапазоны, описанные в данном документе, также охватывают все их возможные поддиапазоны и комбинации поддиапазонов. Любой перечисленный диапазон можно легко распознать как достаточно описывающий и позволяющий разбить один и тот же диапазон, по крайней мере, на равные половины, трети, четверти, пятые, десятые и т.д. в качестве неограничивающего примера каждый диапазон, описанный в данном документе, можно легко разбить на нижнюю треть, среднюю треть и верхнюю треть и т.д. Как также будет понятно специалисту в данной области техники, все выражения, такие как «до», «по меньшей мере», «больше чем», «меньше чем» и т.п., включают в себя перечисленное число и относятся к диапазонам, которые могут быть впоследствии разбиты на поддиапазоны, как обсуждалось выше. Наконец, как будет понятно специалисту в данной области, диапазон включает в себя каждый отдельный элемент. Таким образом, например, группа, имеющая 1-3 пункта, относится к группам, имеющим 1, 2 или 3 пункта. Аналогично группа, содержащая 1-5 пунктов, относится к группам, содержащим 1, 2, 3, 4 или 5 пунктов, и т.п.
[0289] Следует понимать, что в данном документе описаны различные варианты осуществления данного описания для целей иллюстрации и что различные модификации могут быть внесены без отступления от объема и сущности данного описания. Соответственно, различные варианты осуществления, описанные в данном документе, не имеют ограничительного характера, и объем и сущность представлены в следующих пунктах формулы изобретения.
[0290] Следует понимать, что не обязательно все объекты или преимущества могут быть достигнуты в соответствии с любым конкретным вариантом осуществления, описанным в данном документе. Таким образом, например, специалистам в данной области будет понятно, что некоторые варианты осуществления могут быть выполнены с возможностью функционирования таким образом, чтобы обеспечить или оптимизировать одно преимущество или группу преимуществ, описанных в данном документе, без необходимости достижения других объектов или преимуществ, которые могут быть описаны или предложены в данном документе.
[0291] Все процессы, описанные в данном документе, могут быть реализованы и полностью автоматизированы с помощью модулей программного кода, исполняемых компьютерной системой, которая включает один или более компьютеров или процессоров. Программные модули могут храниться на энергонезависимом машиночитаемом носителе любого типа или другом компьютерном устройстве хранения данных. Некоторые или все способы могут быть реализованы на специализированном компьютерном оборудовании.
[0292] Из данного описания будут очевидны многие другие варианты, отличные от описанных в данном документе. Например, в зависимости от варианта осуществления определенные действия, события или функции любого из алгоритмов, описанных в данном документе, могут выполняться в другой последовательности, могут быть добавлены, объединены или удалены вообще (например, не все описанные действия или события необходимы для практической реализации алгоритмов). Более того, в определенных вариантах осуществления действия или события могут выполняться одновременно, например, посредством многопоточной обработки, прерывания обработки, или нескольких процессоров, или ядер процессоров, или с другими параллельными архитектурами, а не последовательно. Кроме того, разные задачи или процессы могут выполняться разными машинами и/или вычислительными системами, которые могут функционировать вместе.
[0293] Различные иллюстративные логические блоки и модули, описанные в связи с раскрытыми в данном документе вариантами осуществления, могут быть реализованы или выполнены машиной, такой как блок обработки данных или процессор для цифровой обработки сигналов (DSP), заказная специализированная интегральная схема (ASIC), программируемая пользователем матрица логических элементов (FPGA) или другое программируемое логическое устройство, логический элемент на дискретных компонентах или транзисторные логические схемы, дискретные аппаратные компоненты или любая их комбинация, предназначенная для выполнения функций описанных в данном документе. Процессор может представлять собой микропроцессор, но в альтернативном варианте осуществления процессор может представлять собой контроллер, микроконтроллер или машину состояний, их комбинации и т.п. Процессор может включать в себя электрическую схему, выполненную с возможностью обработки исполняемых компьютером команд. В другом варианте осуществления процессор включает в себя FPGA или другое программируемое устройство, которое выполняет логические операции без обработки исполняемых компьютером команд. Процессор также может быть реализован в виде комбинации вычислительных устройств, например, комбинации DSP и микропроцессора, множества микропроцессоров, одного или более микропроцессоров, связанных с ядром DSP, или любой другой такой конфигурации. Хотя в данном документе описано главным образом в отношении цифровой технологии, процессор может также включать в себя главным образом аналоговые компоненты. Например, некоторые или все алгоритмы обработки сигналов, описанные в данном документе, могут быть реализованы в аналоговой схеме или в смешанной аналоговой и цифровой схеме. Вычислительная среда может включать в себя компьютерную систему любого типа, включая, без ограничений, компьютерную систему, основанную на микропроцессоре, системном компьютере, процессоре цифровой обработки сигналов, портативном вычислительном устройстве, контроллере устройства или вычислительном двигателе внутри прибора и многие другие.
[0294] Любые описания процессов, элементы или блоки в блок-схемах, описанных в данном документе и/или показанных на прилагаемых фигурах, следует понимать как потенциально представляющие модули, сегменты или части кода, которые включают одну или более исполняемых команд для реализации конкретных логических функций или элементов в процессе. Альтернативные варианты осуществления включены в объем вариантов осуществления, описанных в данном документе, в которых элементы или функции можно удалить, выполнить в порядке, отличном от показанных или описанных, включая по существу одновременно или в обратном порядке, в зависимости от задействованных функциональных возможностей, как будет понятно специалистам в данной области.
[0295] Следует подчеркнуть, что в описанные выше варианты осуществления можно вносить множество вариаций и модификаций, элементы которых следует понимать как относящиеся к другим приемлемым примерам. Предполагается, что все такие модификации и вариации включены в объем данного описания и защищены следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ И СПОСОБЫ МОДУЛЯЦИИ SMN2 СПЛАЙСИНГА У СУБЪЕКТА | 2019 |
|
RU2793459C2 |
| КОМПОЗИЦИИ И СПОСОБЫ МОДУЛЯЦИИ SMN2 СПЛАЙСИНГА У СУБЪЕКТА | 2010 |
|
RU2566724C9 |
| Способ определения делеции 7 экзона в гене SMN1 с определением количества копий гена SMN2 в образцах ДНК с помощью цифровой полимеразной цепной реакции | 2024 |
|
RU2838673C1 |
| КОМПОЗИЦИИ И СПОСОБЫ МОДУЛЯЦИИ SMN2 СПЛАЙСИНГА У СУБЪЕКТА | 2010 |
|
RU2683772C2 |
| Молекулярно-генетическая система детекции делеции экзона 7 гена SMN1, пригодная для проведения неонатального скрининга | 2021 |
|
RU2796350C1 |
| Способ преимплантационной генетической диагностики спинальной мышечной атрофии типа 1 | 2017 |
|
RU2671156C1 |
| АДЕНОАССОЦИИРОВАННЫЙ ВИРУСНЫЙ ВЕКТОР, СОСТОЯЩИЙ ИЗ БЕЛКОВ КАПСИДА РНР.В, НУКЛЕИНОВОЙ КИСЛОТЫ, КОДИРУЮЩЕЙ БЕЛОК SMN, И ЕГО ПРИМЕНЕНИЕ | 2022 |
|
RU2833225C2 |
| ИМИДАЗО[1,2-а]ПИРАЗИН-1-ИЛ-БЕНЗАМИДЫ ДЛЯ ЛЕЧЕНИЯ СПИНАЛЬНОЙ МЫШЕЧНОЙ АТРОФИИ | 2015 |
|
RU2725979C2 |
| Генетическая конструкция, содержащая последовательности химерных направляющих РНК для делеции гена SMN1 человека в культурах клеток человека | 2022 |
|
RU2816897C2 |
| Синергетическое действие SMN1 и miR-23a при лечении спинальной мышечной атрофии | 2021 |
|
RU2839898C2 |
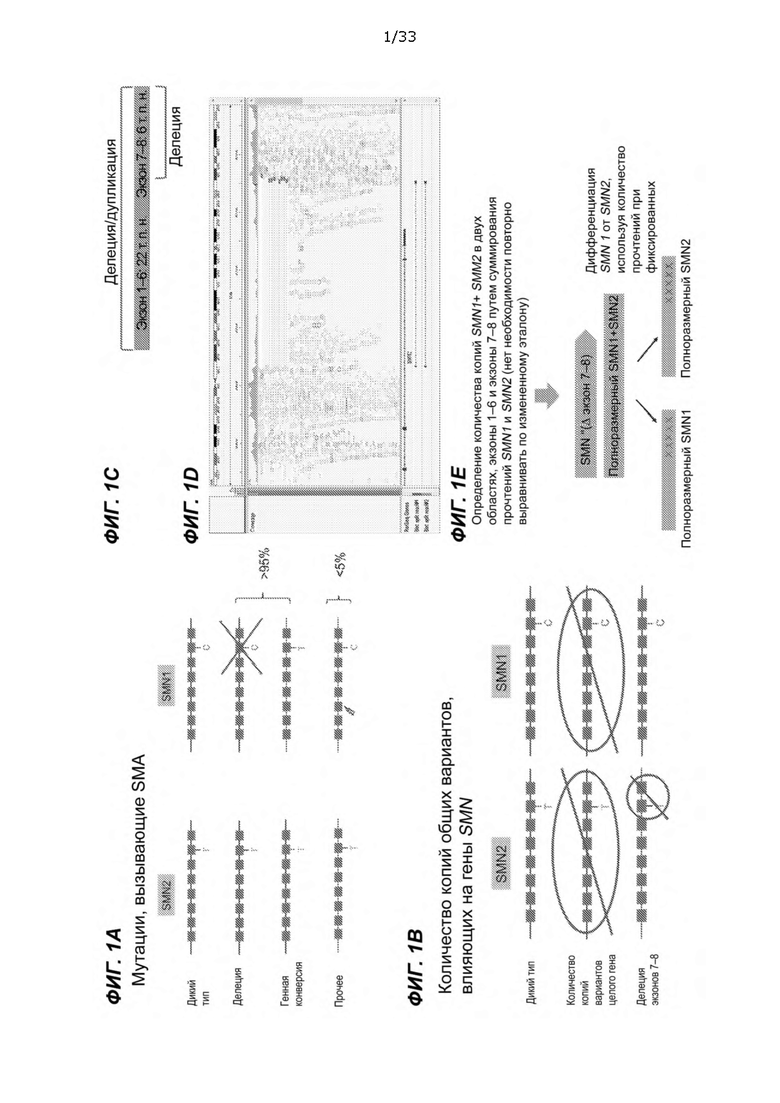
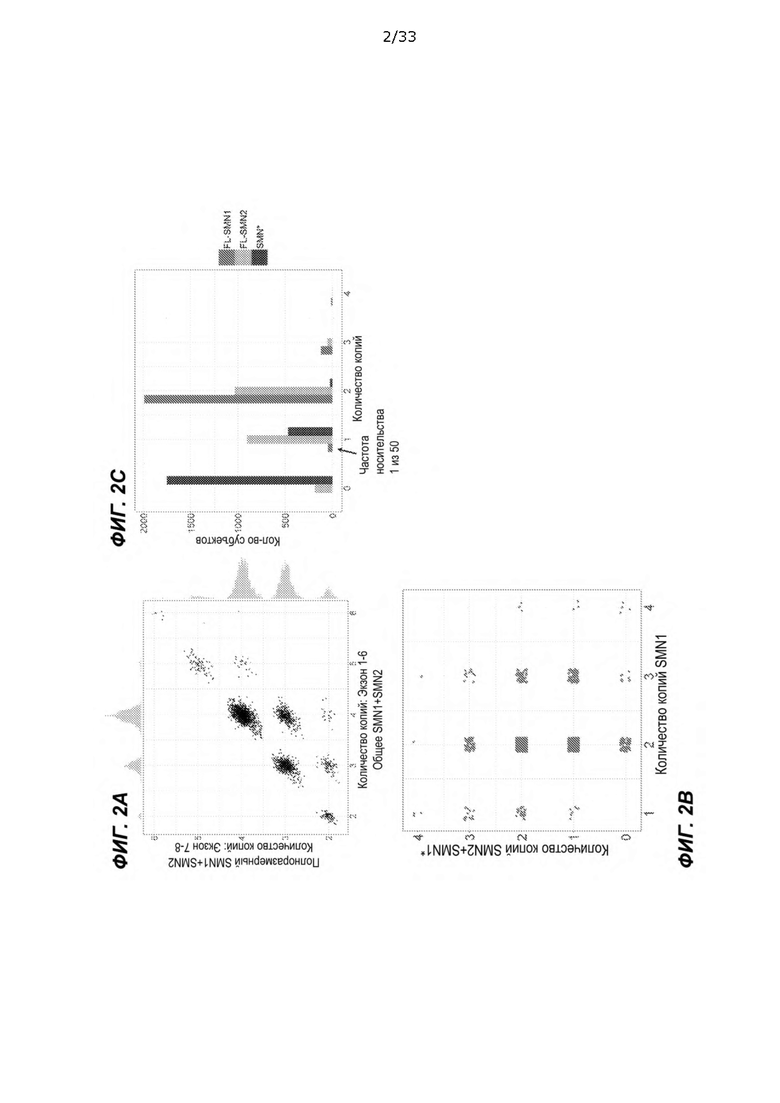
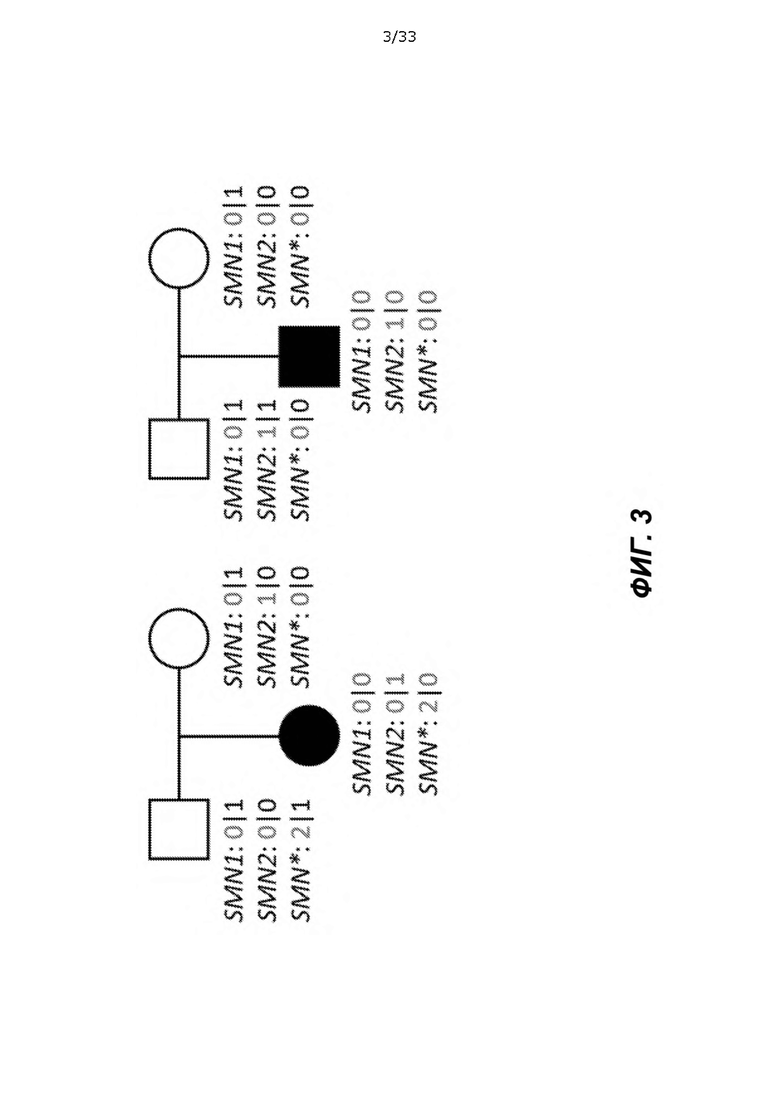
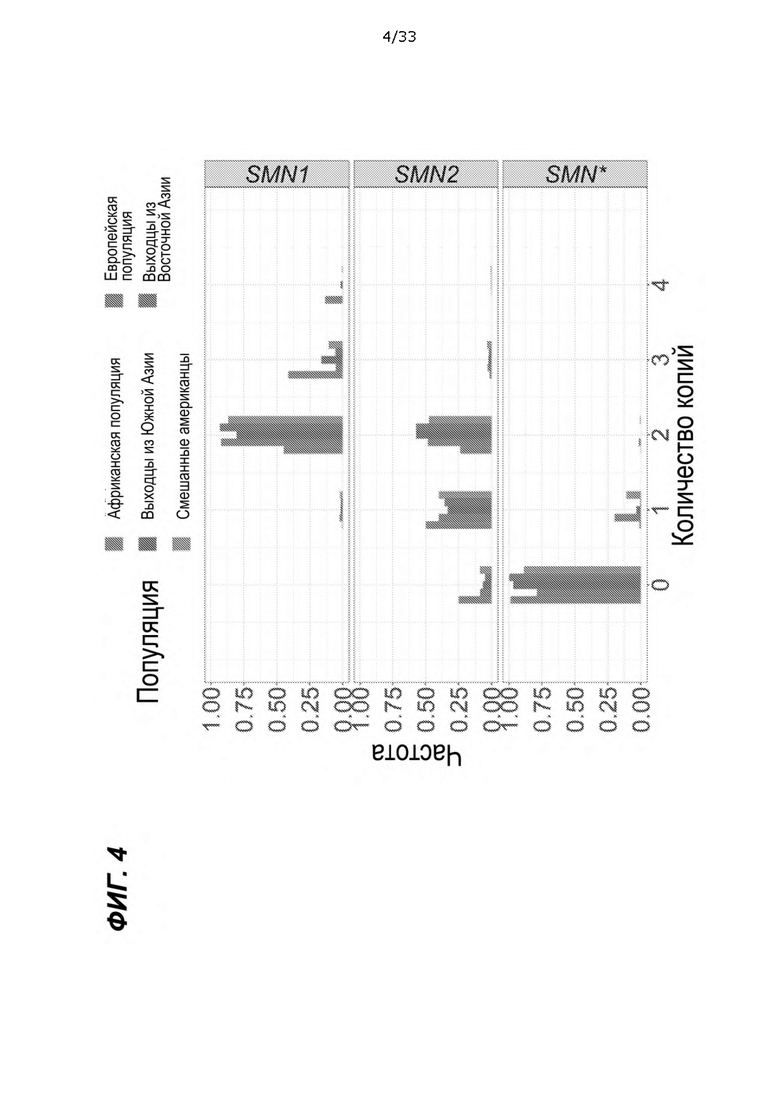
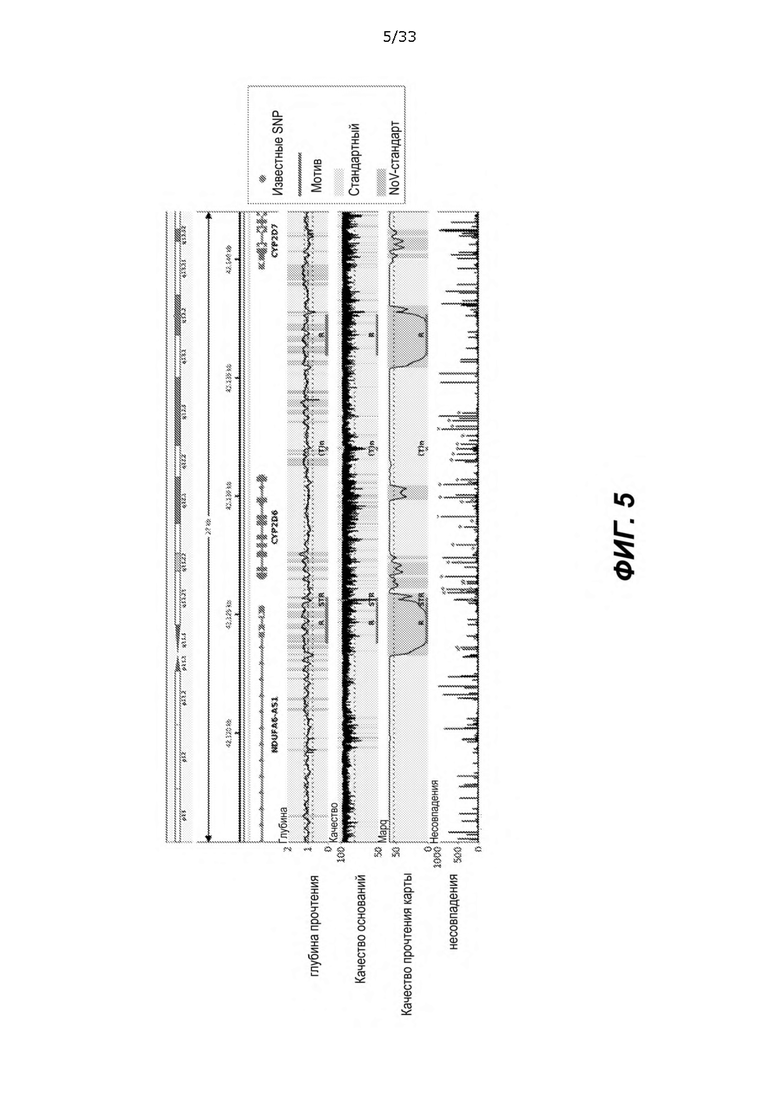
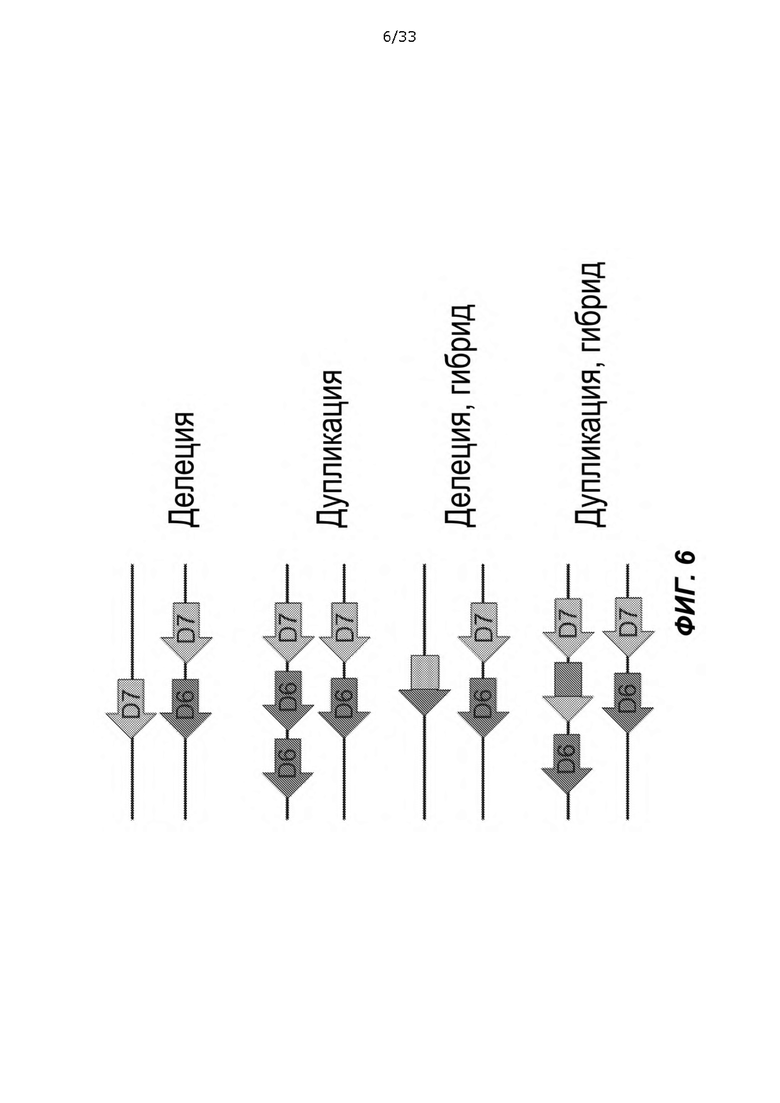
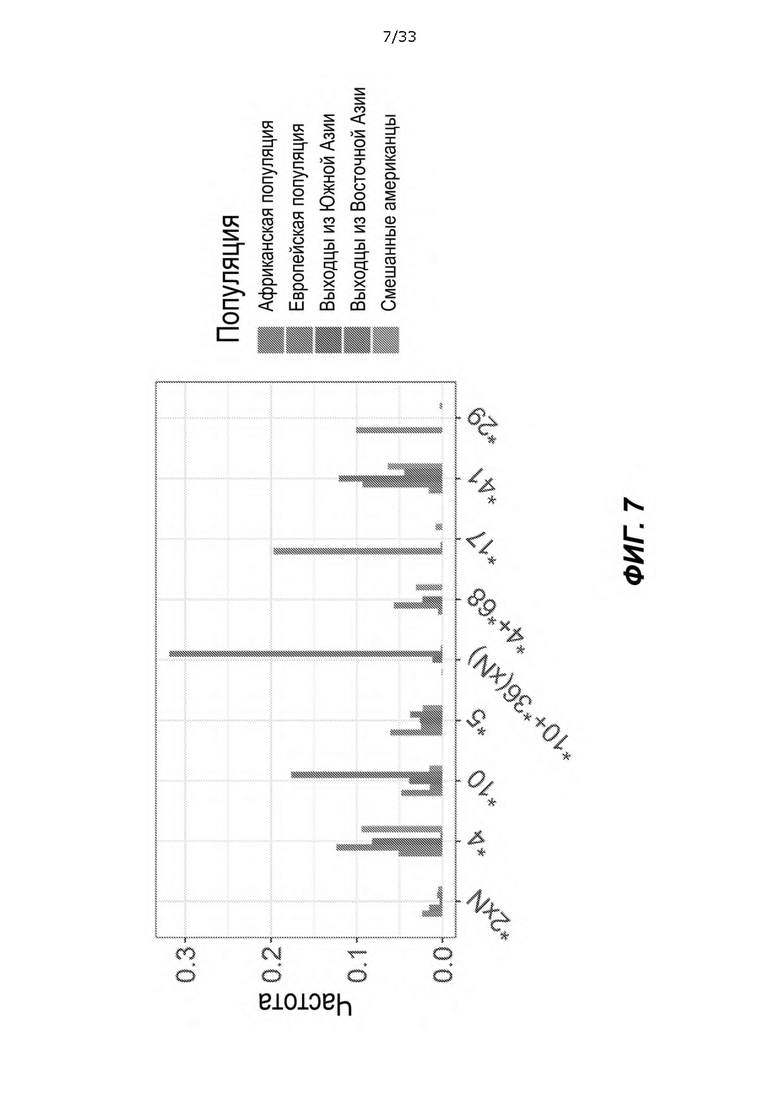
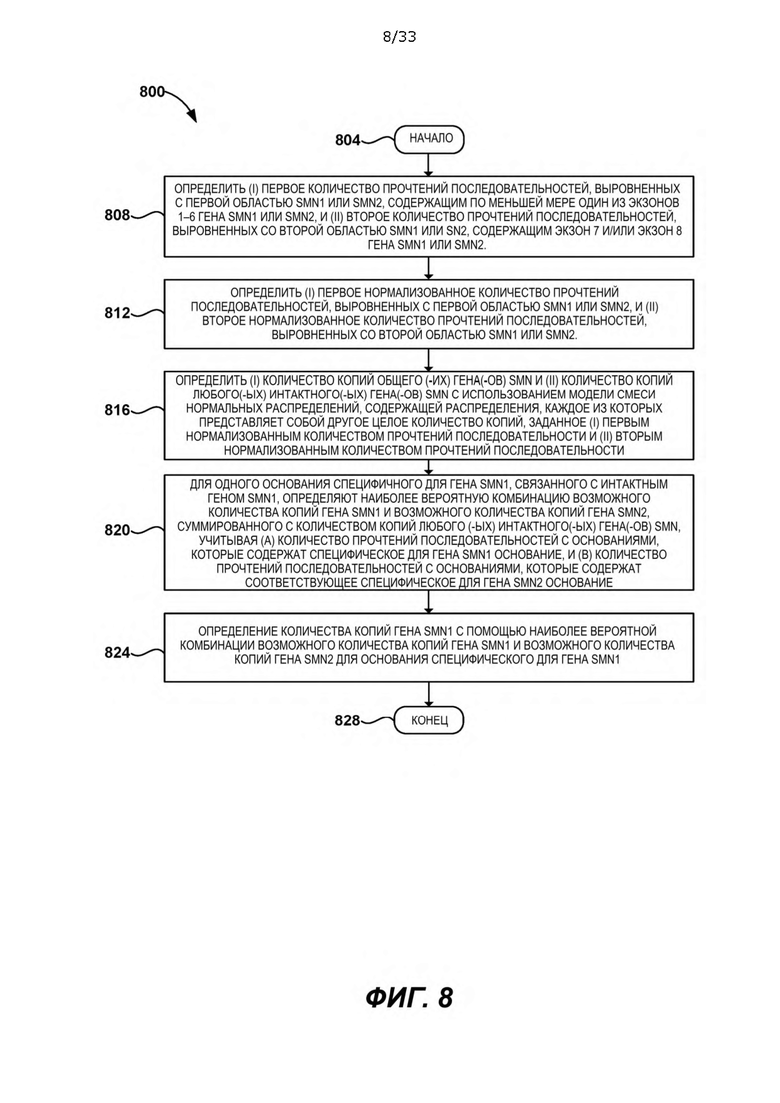
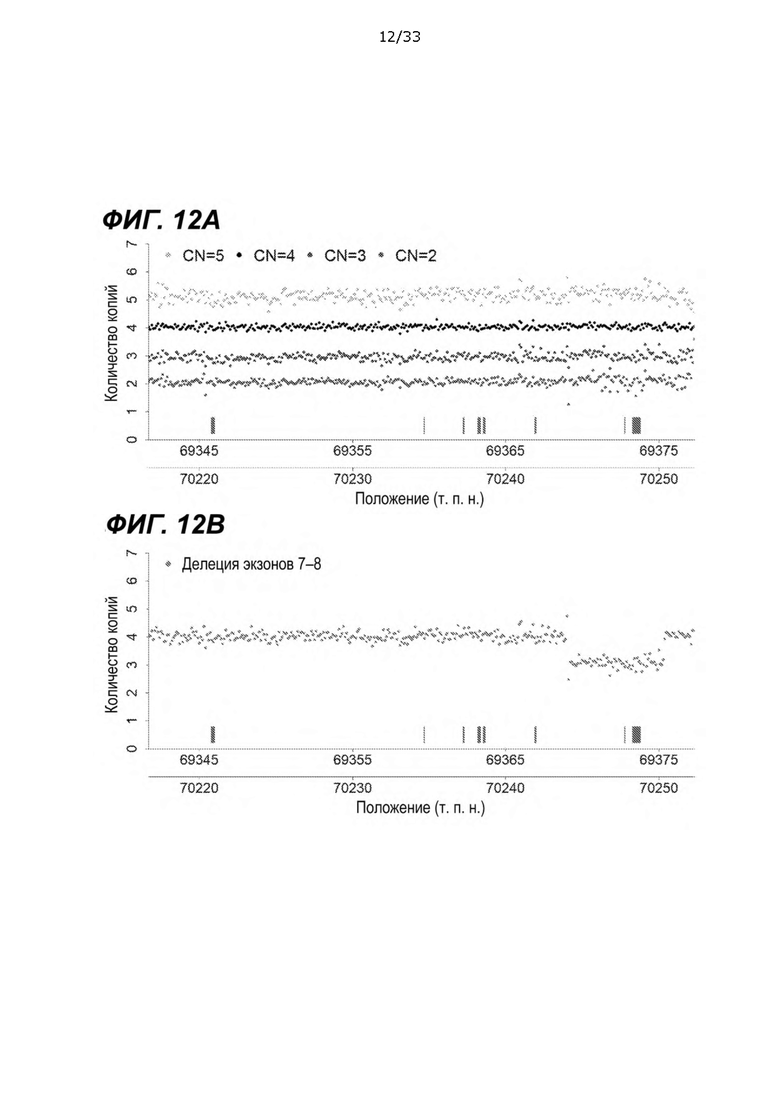
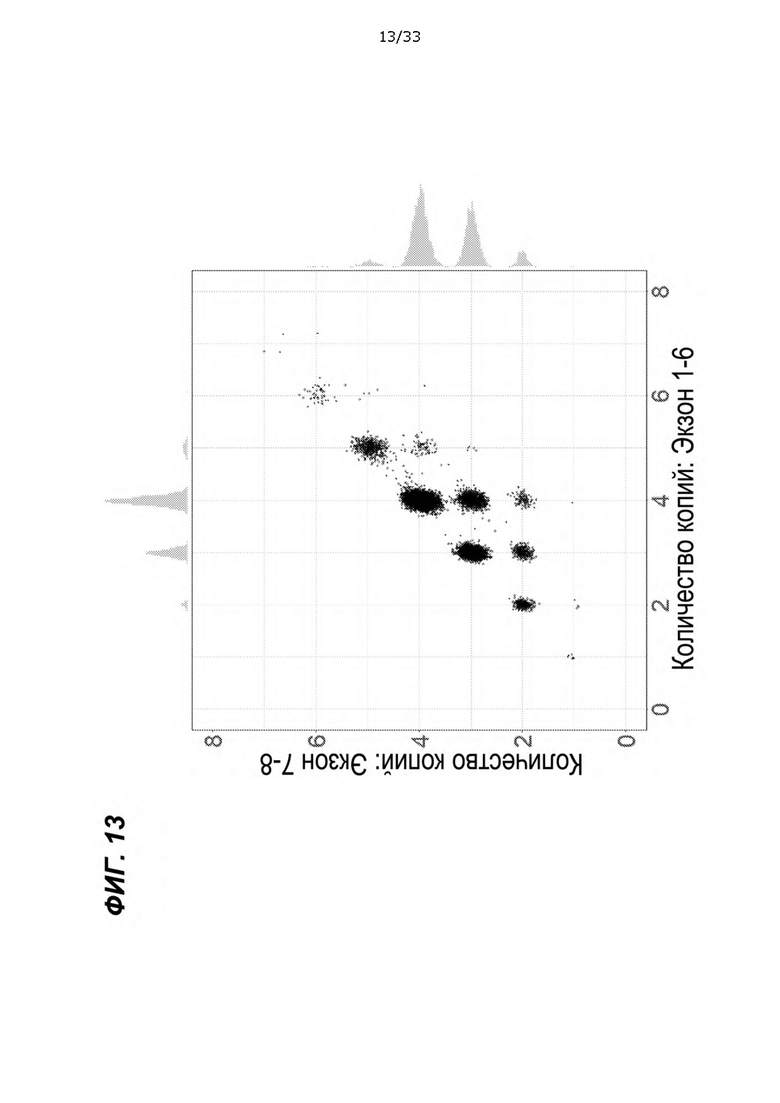
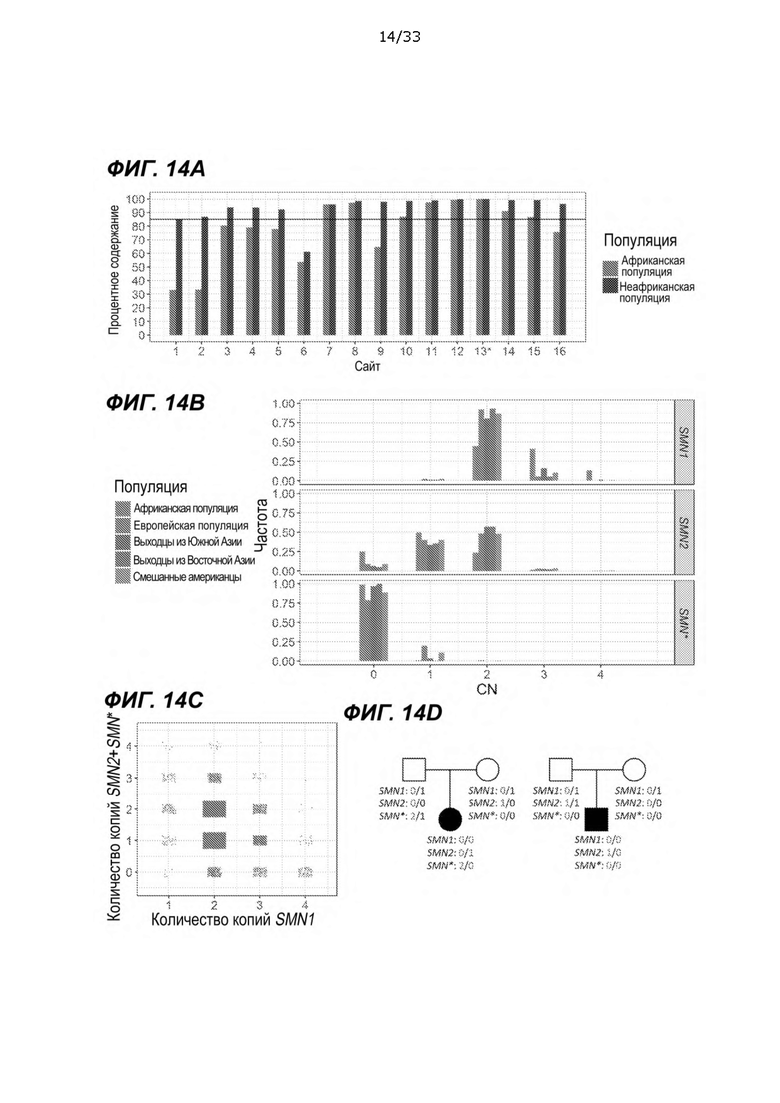


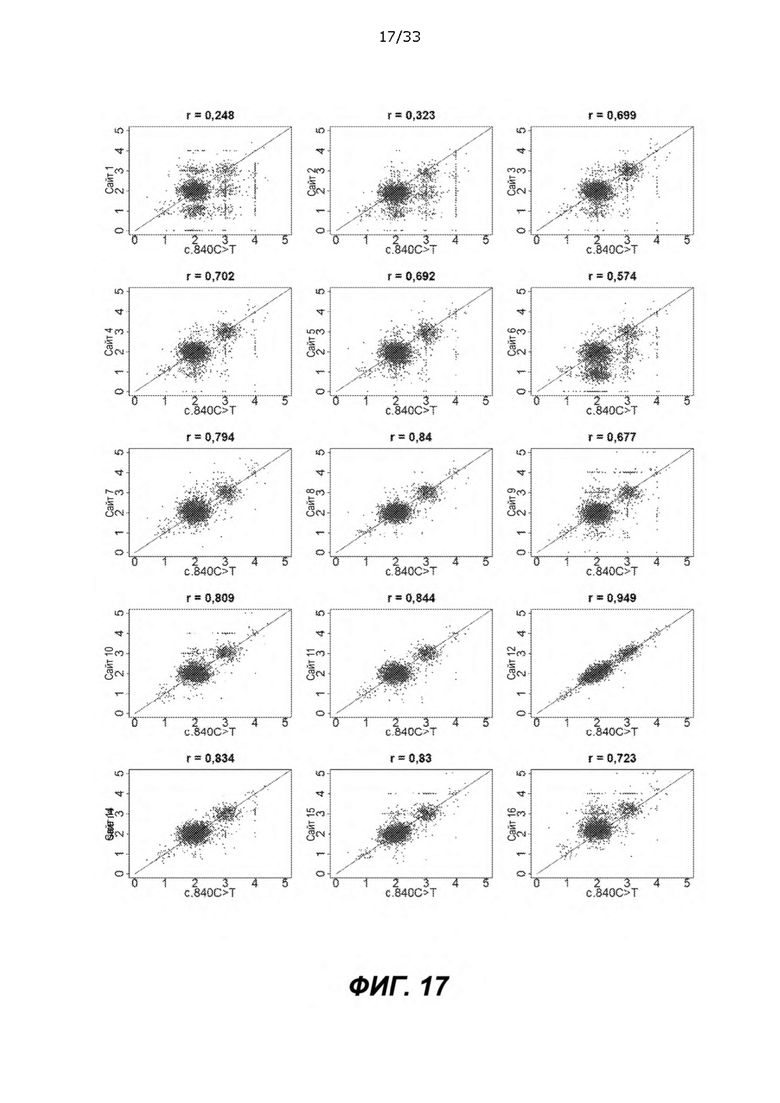
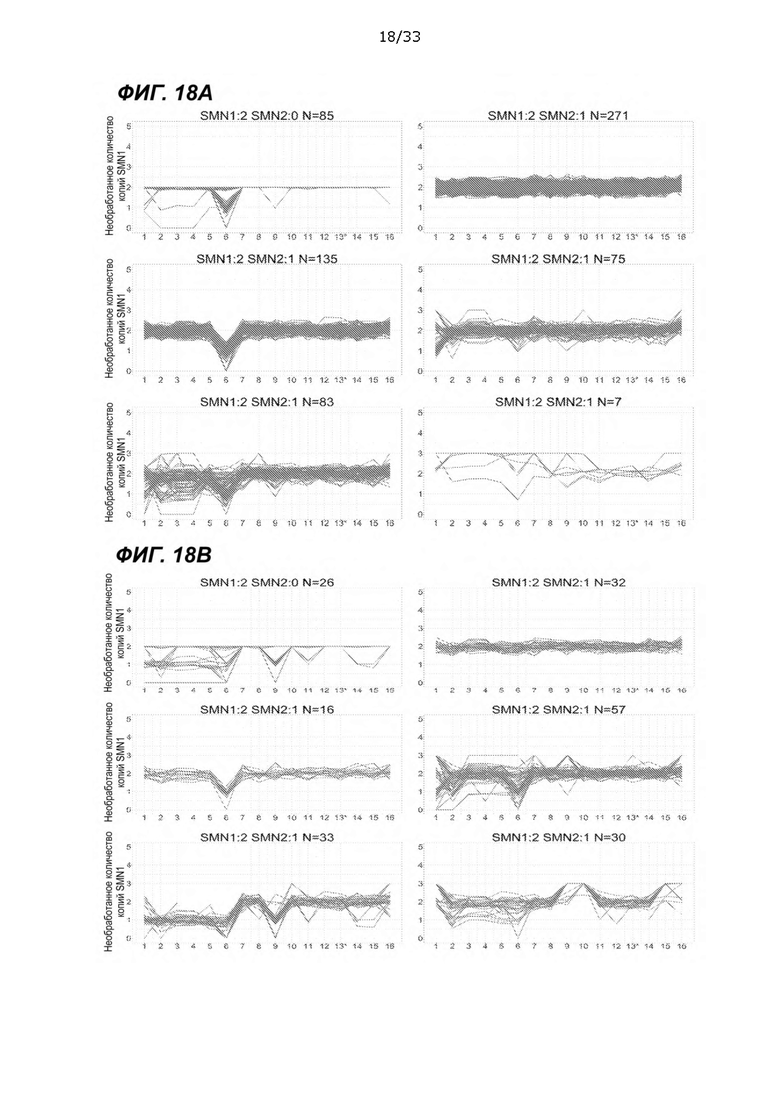

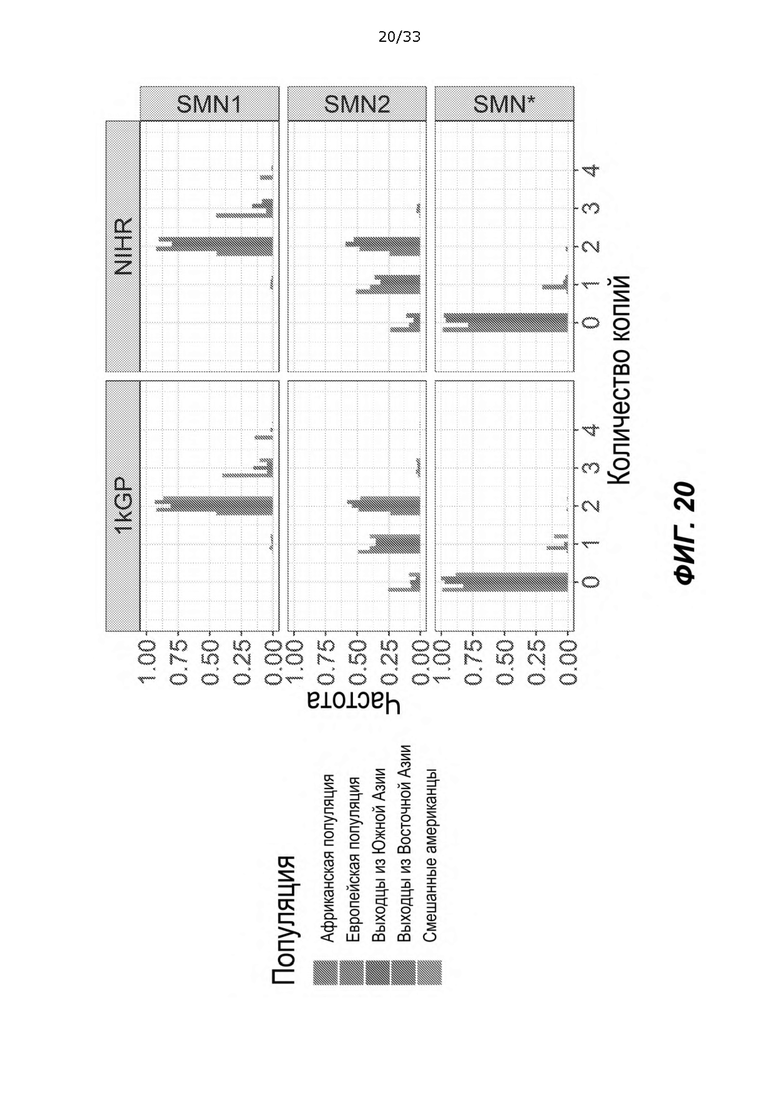
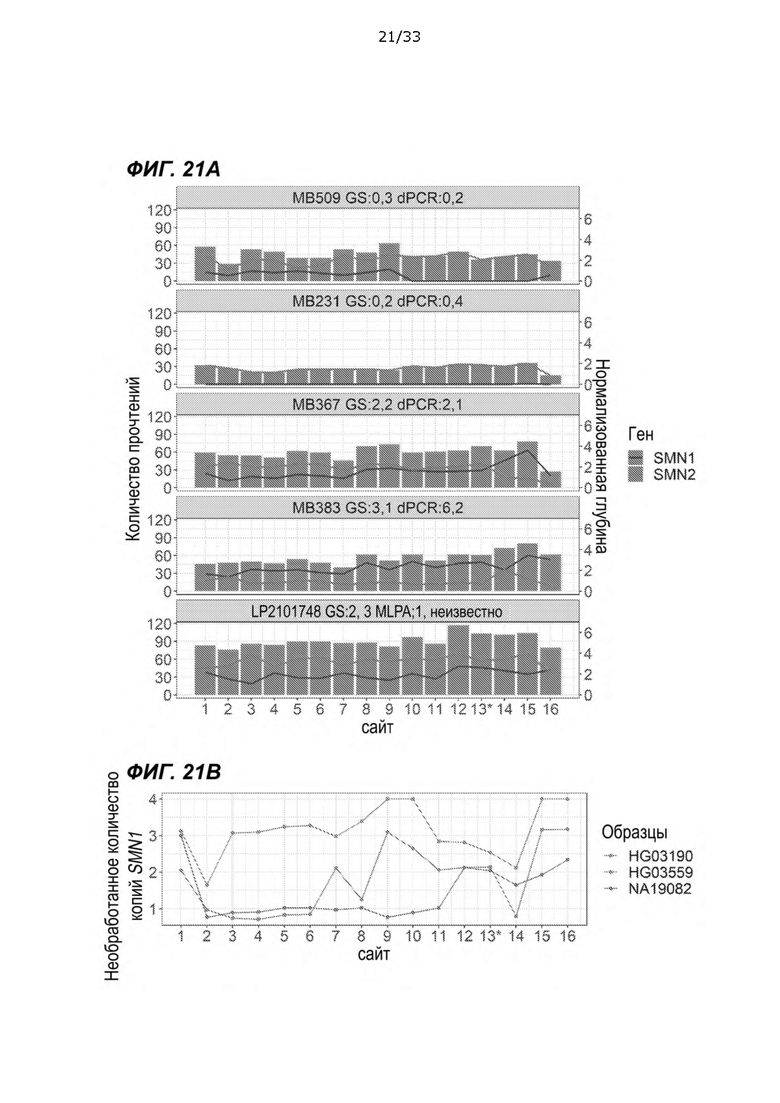
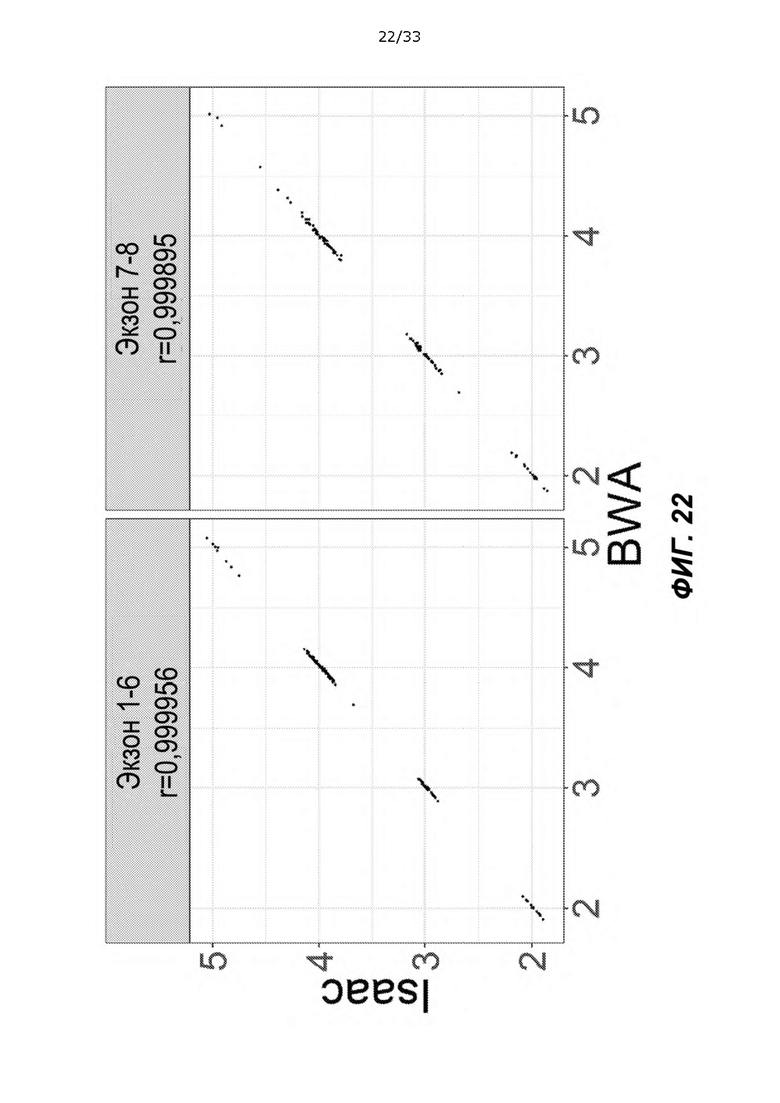
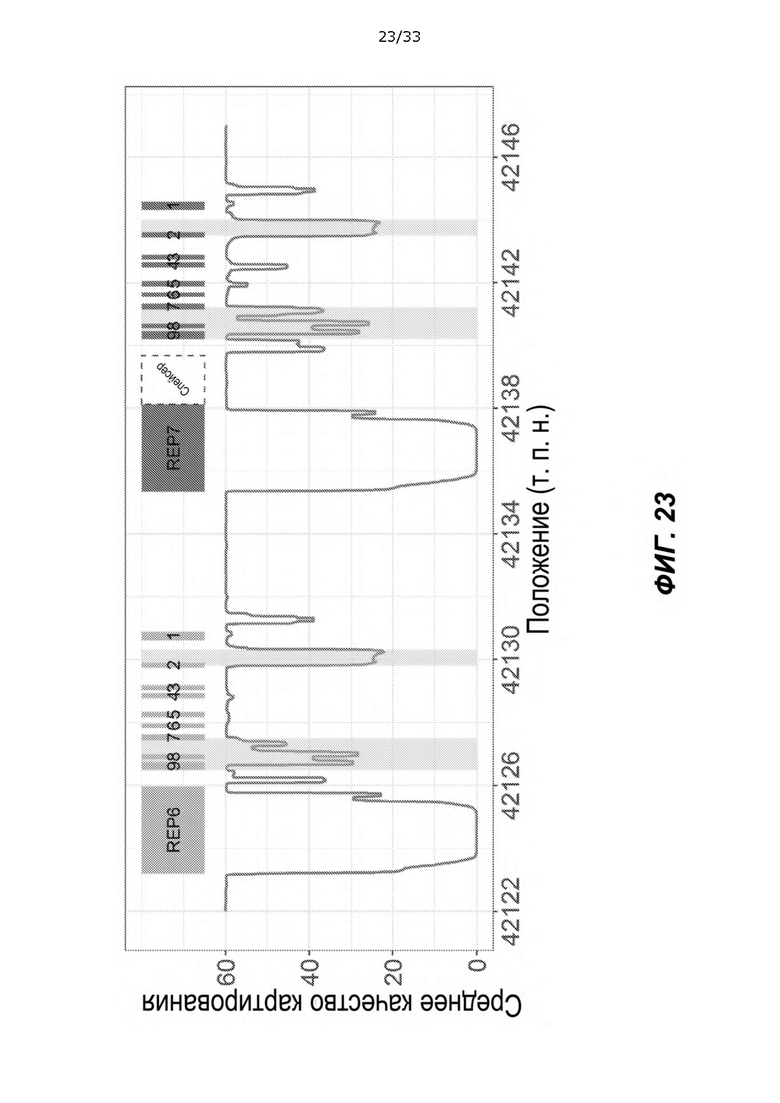

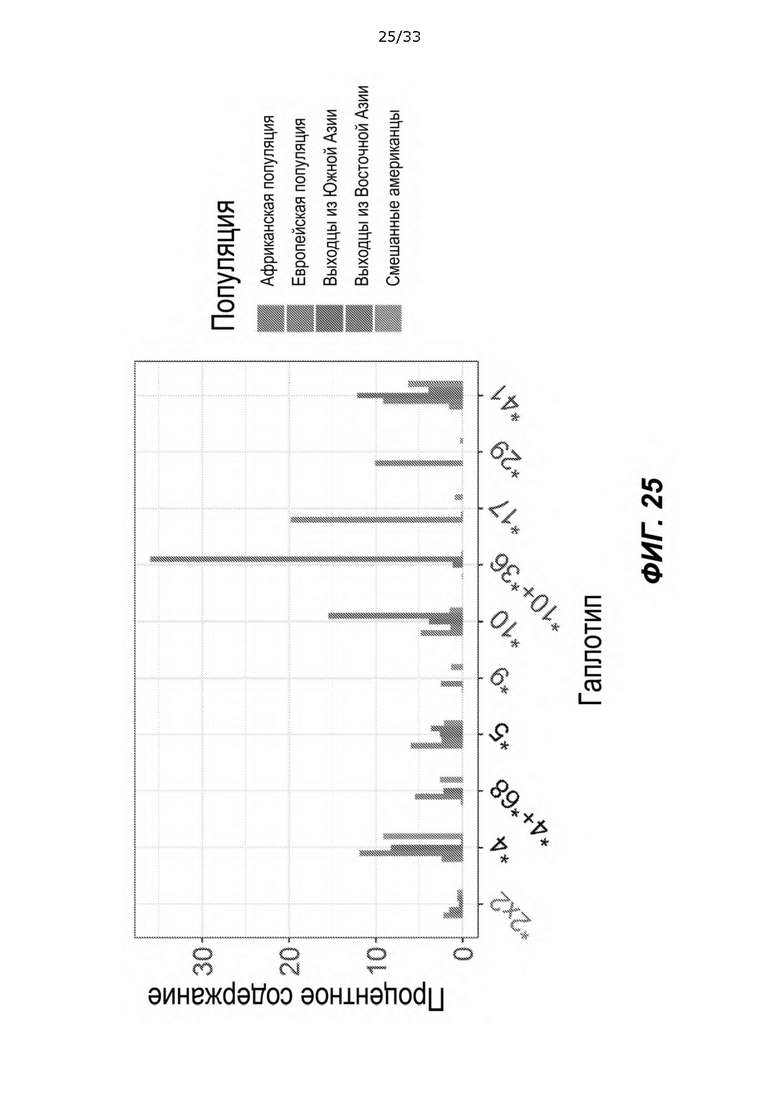
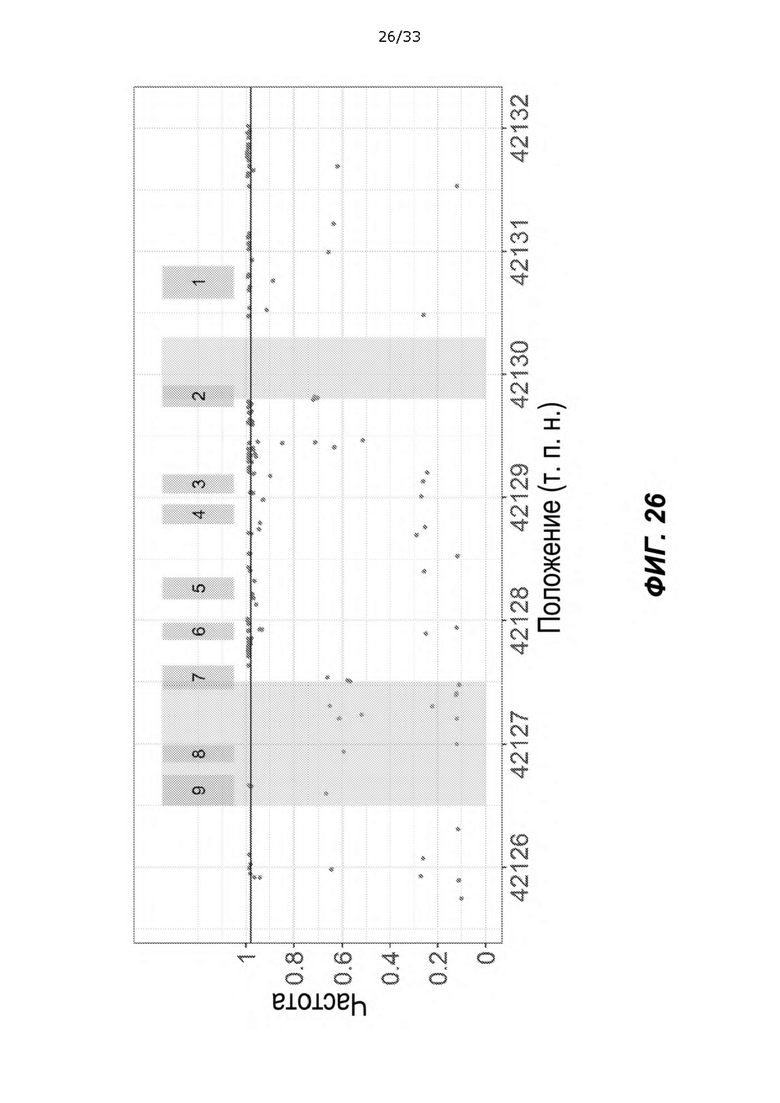
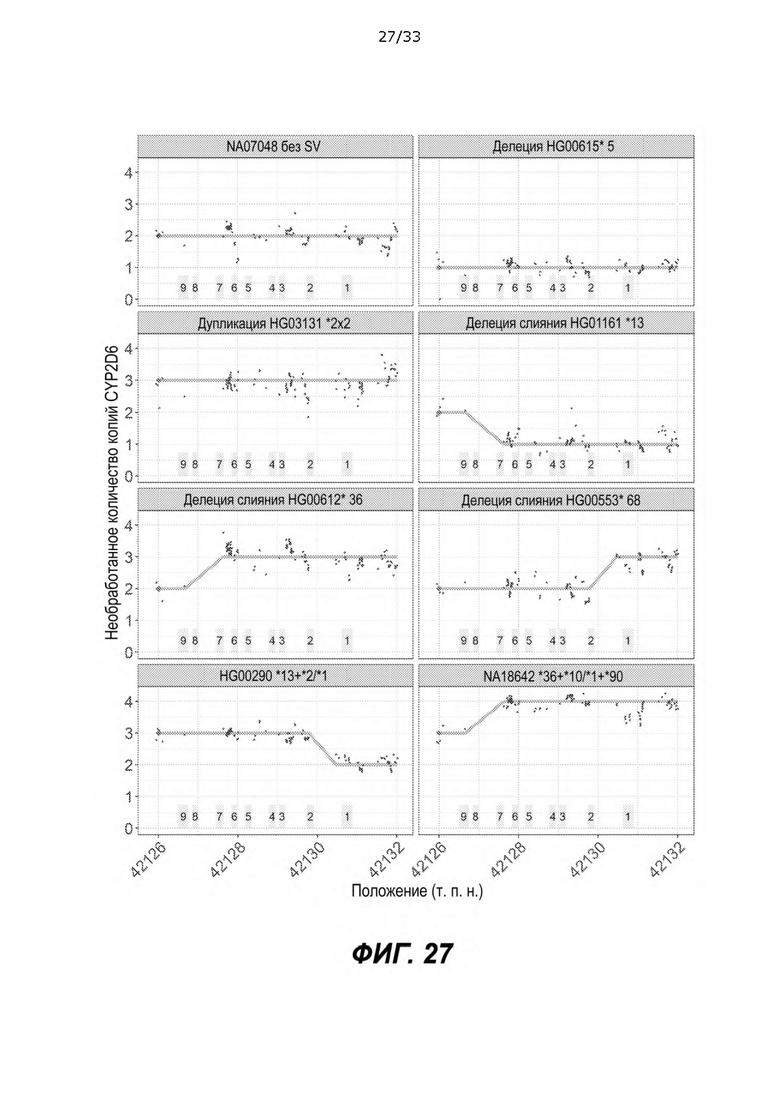
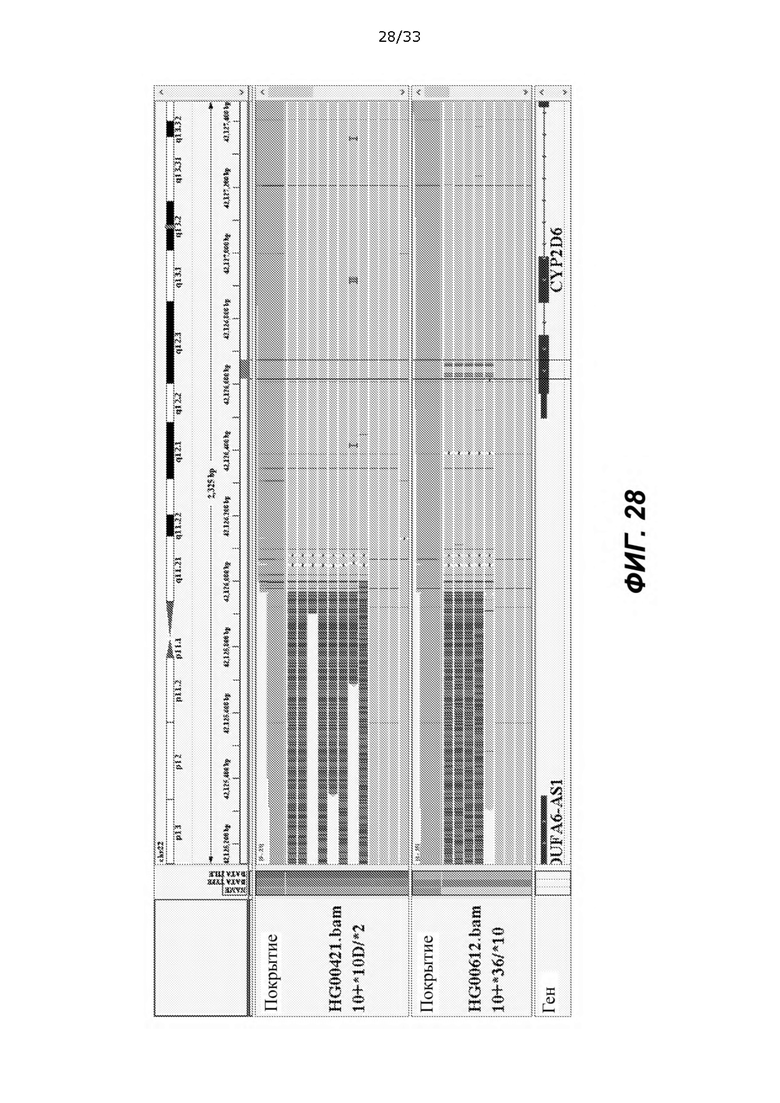
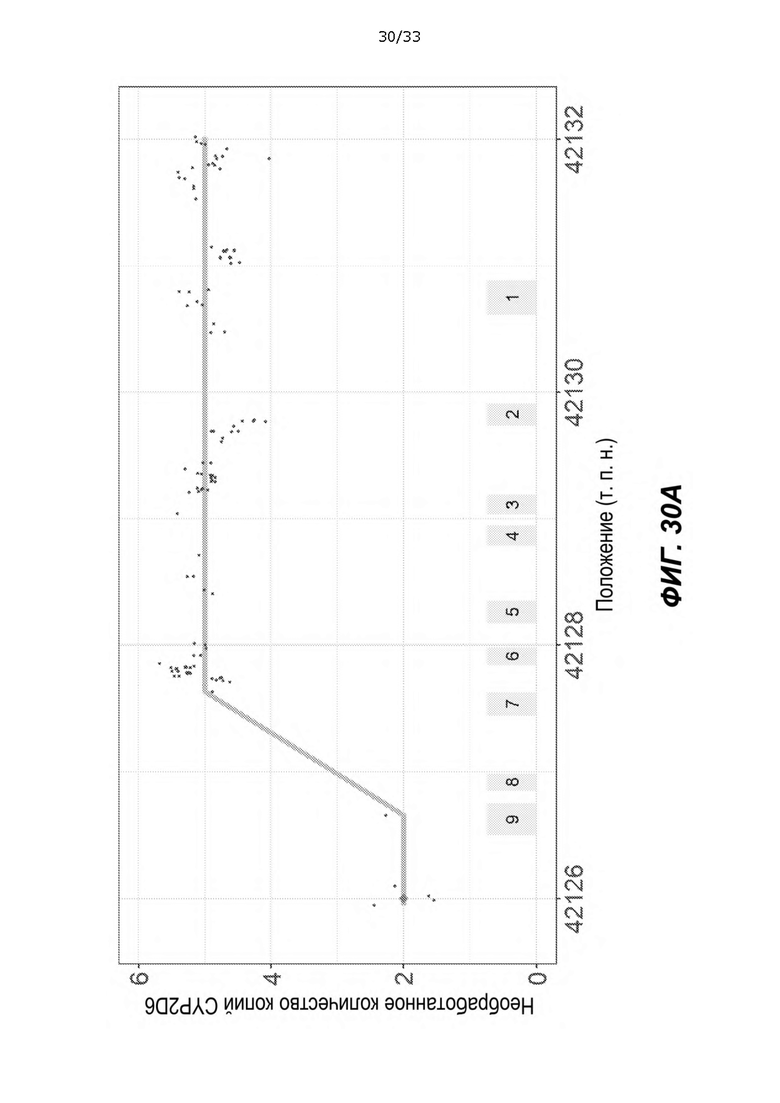
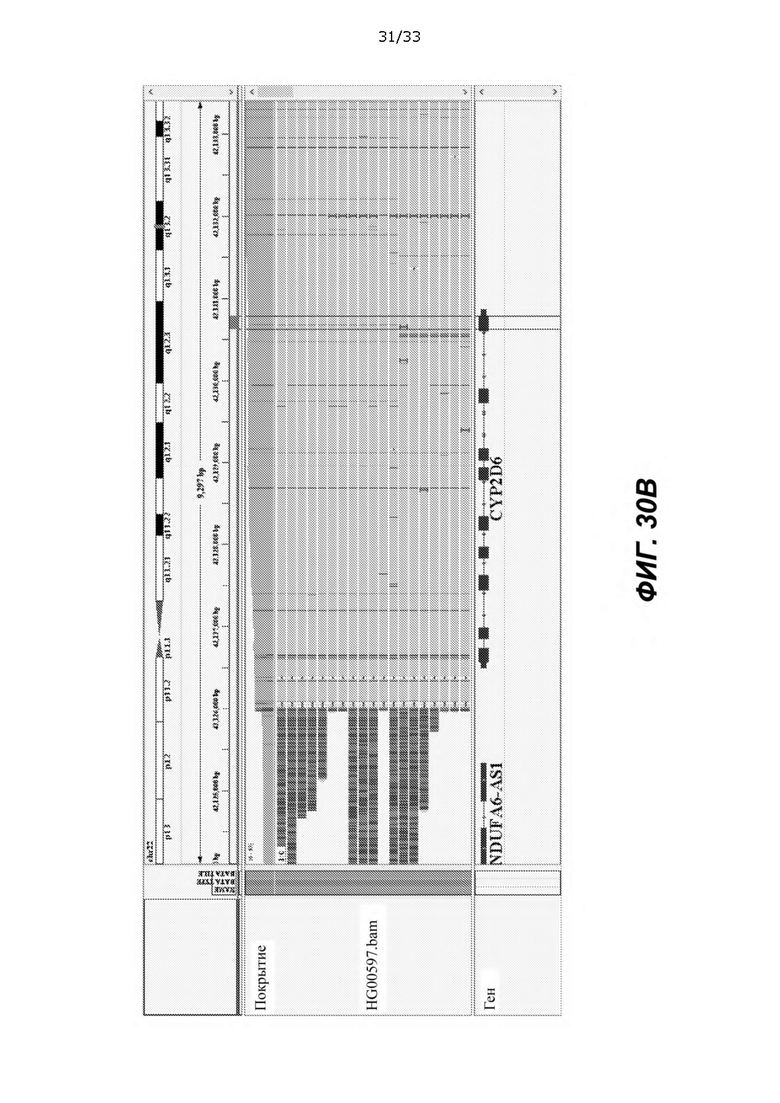
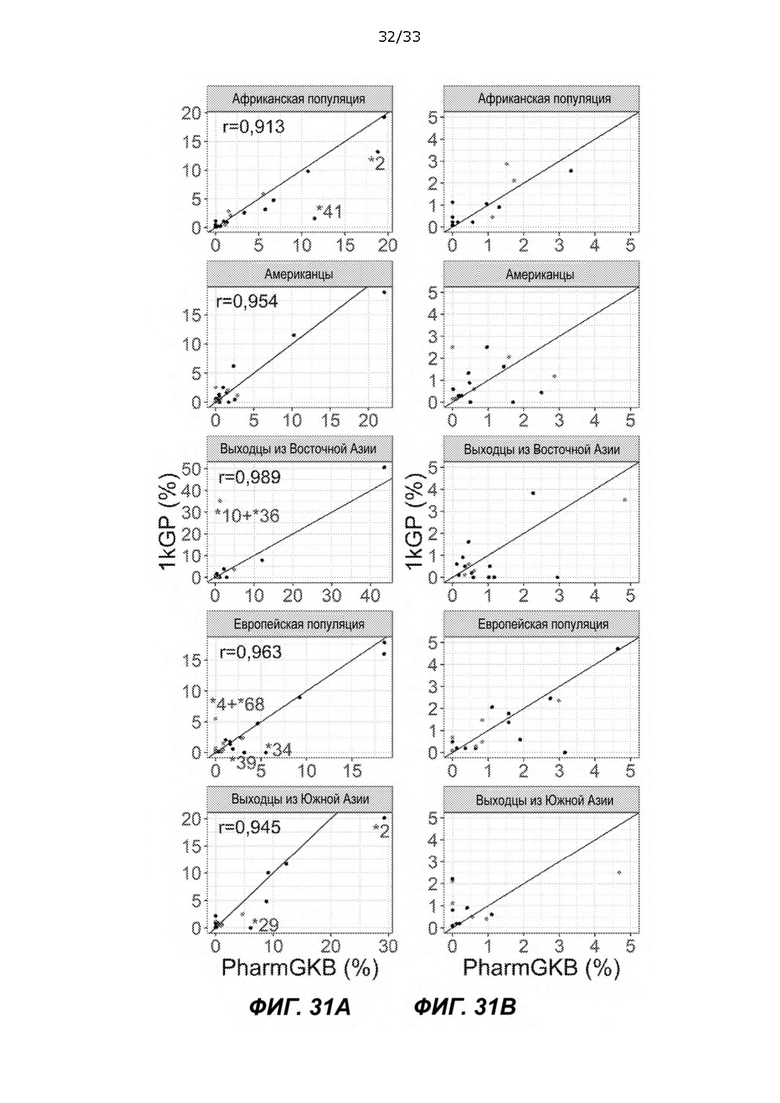
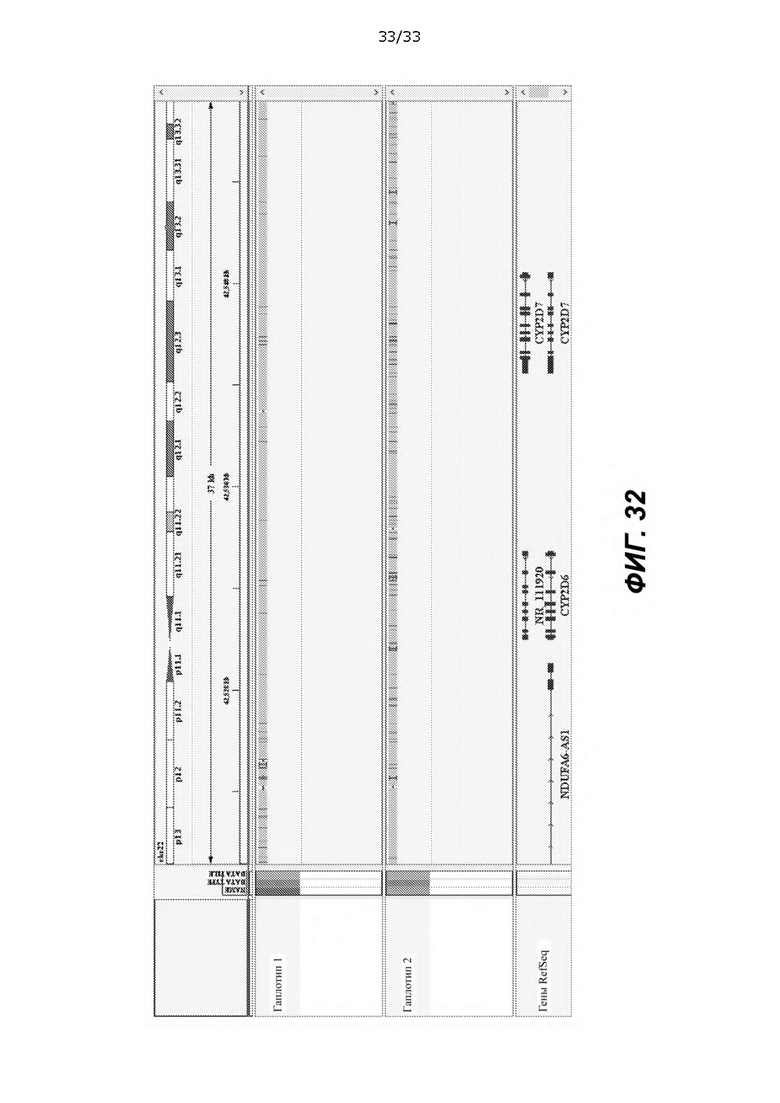
Изобретение относится к биоинформатике. Описан способ определения количества копий гена выживания моторных нейронов 1 (SMN1). Под управлением аппаратного процессора получают данные секвенирования, содержащие множество прочтений последовательностей, полученных из образца от субъекта, выровненных с геном выживания моторных нейронов 1 (SMN1) или геном выживания моторных нейронов 2 (SMN2). Определяют первое количество прочтений последовательности из множества прочтений последовательностей, выровненных по первой области SMN1 или SMN2, содержащей по меньшей мере 1 из первых 6 экзонов гена SMN1 или гена SMN2 соответственно, и второе количество прочтений последовательности из множества прочтений последовательностей, выровненных по второй области SMN1 или SMN2, содержащей по меньшей мере экзон 7 или экзон 8 гена SMN1 или гена SMN2. Определяют первое нормализованное количество прочтений последовательности и второе нормализованное количество прочтений последовательности. Определяют число копий полноразмерных генов выживания моторных нейронов (SMN) и число копий любых интактных генов SMN. Для одного множества специфичных для гена SMN1 оснований, связанных с интактным геном SMN1, определяют наиболее вероятную комбинацию из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, которые дают в сумме количество копий любых определенных интактных генов SMN. Определяют количество копий гена SMN1 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенных для специфичного для гена SMN1 основания. Изобретение позволяет определить количества копий гена SMN1. 44 з.п. ф-лы, 32 ил., 25 табл., 2 пр.
1. Способ определения количества копий гена выживания моторных нейронов 1 (SMN1), включающий
под управлением аппаратного процессора:
получение данных секвенирования, содержащих множество прочтений последовательностей, полученных из образца от субъекта, выровненных с геном выживания моторных нейронов 1 (SMN1) или геном выживания моторных нейронов 2 (SMN2);
определение (i) первого количества прочтений последовательности из множества прочтений последовательностей, выровненных по первой области SMN1 или SMN2, содержащей по меньшей мере 1 из первых 6 экзонов гена SMN1 или гена SMN2 соответственно, и (ii) второго количества прочтений последовательности из множества прочтений последовательностей, выровненных по второй области SMN1 или SMN2, содержащей по меньшей мере экзон 7 или экзон 8 гена SMN1 или гена SMN2 соответственно;
определение (i) первого нормализованного количества прочтений последовательности, выровненных по первой области гена SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненных по второй области SMN1 или SMN2, с применением (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2 соответственно;
определение (i) числа копий общего количества генов выживания моторных нейронов (SMN), каждый из которых является интактным геном SMN1, интактным геном SMN2, укороченным геном SMN1 или укороченным геном SMN2; и (ii) числа копий любых интактных генов SMN, каждый из которых является интактным геном SMN1 или интактным геном SMN2, с использованием модели смеси нормальных распределений, содержащей множество нормальных распределений, каждое из которых представляет разное целое число копий с учетом (i) первого нормализованного числа прочтений последовательности, выровненных по первой области SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненных по второй области SMN1 или SMN2 соответственно;
для одного множества специфичных для гена SMN1 оснований, связанных с интактным геном SMN1, определение наиболее вероятной комбинации из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, которые дают в сумме количество копий любых определенных интактных генов SMN с учетом (a) количества прочтений последовательностей из множества прочтений последовательностей с основаниями, которые содержат основание, специфичное для гена SMN1, и (b) количества прочтений последовательностей из множества прочтений последовательности с основаниями, которые содержат специфическое для гена SMN2 основание гена SMN2, соответствующее специфическому для гена SMN1 основанию; и
определение количества копий гена SMN1 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенных для специфичного для гена SMN1 основания.
2. Способ по п. 1, в котором данные секвенирования содержат данные полногеномного секвенирования (WGS) или данные WGS с помощью коротких прочтений.
3. Способ по пп. 1, 2, в котором субъект является субъектом-плодом, неонатальным субъектом, субъектом детского возраста, субъектом-подростком или взрослым субъектом.
4. Способ по любому из пп. 1-3, в котором образец содержит клетки или внеклеточную ДНК.
5. Способ по любому из пп. 1-4, в котором образец содержит фетальные клетки или внеклеточную фетальную ДНК.
6. Способ по любому из пп. 1-5, в котором прочтение последовательности из множества прочтений последовательностей выравнивается с первой областью SMN1 или SMN2 или со второй областью SMN1 или SMN2 с показателем качества выравнивания, равным приблизительно нулю.
7. Способ по любому из пп. 1-6, в котором первая область SMN1 или SMN2 содержит экзон от 1 до 6 гена SMN1 или гена SMN2 соответственно и имеет длину примерно 22,2 т. п. н., а вторая область SMN1 или SMN2 содержит экзон 7 и экзон 8 гена SMN1 или гена SMN2 соответственно и имеет длину примерно 6 т. п. н.
8. Способ по любому из пп. 1-7, в котором определение (i) первого нормализованного количества прочтений последовательности, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненных со второй областью, включает: определение (i) первого нормализованного количества прочтений последовательности, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненных со второй областью SMN1 или SMN2, с применением (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2 соответственно и (iii) глубины прочтений последовательности области генома субъекта, за исключением генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования.
9. Способ по п. 8, в котором определение (i) первого нормализованного количества прочтений последовательности, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненных со второй областью SMN1 или SMN2, включает:
определение (i) нормализованного по длине области SMN1 или SMN2 количества прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) нормализованного по длине области SMN1 или SMN2 количества прочтений последовательностей, выровненных со второй областью SMN1 или SMN2, с использованием (i) длины первой области SMN1 или SMN2 и (ii) длины второй области SMN1 или SMN2 соответственно; и
определение (i) первой нормализованной глубины прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и (ii) второй нормализованной глубины прочтений последовательностей, выровненных со второй областью SMN1 или SMN2, на основе (i) первого нормализованного по длине области SMN1 или SMN2 количества и (ii) второго нормализованного по длине области SMN1 или SMN2 количества соответственно, при использовании глубины прочтений последовательности области генома субъекта, отличной от генетических локусов, содержащих ген SMN1 и ген SMN2; первое нормализованное количество прочтений последовательностей, выровненных с первой областью SMN1 или SMN2, и второе нормализованное количество прочтений последовательностей, выровненных со второй областью SMN1 или SMN2, которые представляют собой первую нормализованную глубину и вторую нормализованную глубину соответственно.
10. Способ по любому из пп. 1-9, в котором определение (i) первого нормализованного количества прочтений последовательности, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненных со второй областью, включает: определение (i) первого нормализованного количества прочтений последовательности, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненных со второй областью SMN1 или SMN2, с применением (i) содержания GC в первой области SMN1 или SMN2 и (ii) содержания GC во второй области SMN1 или SMN2 соответственно, и (iii) глубины прочтений последовательности области генома субъекта, отличной от генетических локусов, содержащих ген SMN1 и ген SMN2 по данным секвенирования и (iv) содержания GC в области генома.
11. Способ по любому из пп. 8-10, в котором глубина прочтений области включает среднюю глубину или медианную глубину прочтений последовательности области генома субъекта, отличной от генетических локусов, содержащих ген SMN1 и ген SMN2, по данным секвенирования.
12. Способ по п. 11, в котором область включает примерно 3000 предварительно выбранных областей длиной примерно 2 т. п. н. каждая в геноме субъекта.
13. Способ по любому из пп. 1-12, в котором (i) первое нормализованное количество прочтений последовательности, выровненных с первой областью SMN1 или SMN2, и/или (ii) второе нормализованное количество прочтений последовательности, выровненных со второй областью SMN1 или SMN2, составляет от примерно 30 до примерно 40.
14. Способ по любому из пп. 1-13, в котором модель смеси нормальных распределений включает одномерную модель смеси нормальных распределений.
15. Способ по любому из пп. 1-14, в котором множество нормальных распределений модели смеси нормальных распределений представляет целое число копий от 0 до 10.
16. Способ по любому из пп. 1-15, в котором среднее значение для каждого из множества нормальных распределений представляет собой целое количество копий, представленное нормальным распределением.
17. Способ по любому из пп. 1-16, в котором определение (i) числа копий всех генов SMN и (ii) числа копий любых интактных генов SMN включает определение (i) числа копий всех генов SMN и (ii) числа копий любых интактных генов SMN с использованием модели смеси нормальных распределений, и первого предварительно определенного порога апостериорной вероятности с учетом (i) первого нормализованного количества прочтений последовательности, выровненных с первой областью SMN1 или SMN2, и (ii) второго нормализованного количества прочтений последовательности, выровненных со второй областью SMN1 или SMN2 соответственно.
18. Способ по п. 17, в котором первый определенный порог апостериорной вероятности равен 0,95.
19. Способ по любому из пп. 1-18, включающий определение количества копий укороченных генов SMN с использованием (i) определенного количества копий общего количества генов SMN и (ii) определенного количества копий интактных генов SMN.
20. Способ по п. 19, в котором количество копий укороченных генов SMN представляет собой разницу (i) определенного общего количества копий генов SMN и (ii) определенного количества копий интактных генов SMN.
21. Способ по любому из пп. 1-20, в котором специфичное для гена SMN1 основание представляет собой энхансер сплайсинга.
22. Способ по любому из пп. 1-21, в котором специфичное для гена SMN1 основание представляет собой основание в c.840 гена SMN1.
23. Способ по любому из пп. 1-22, в котором наиболее вероятная комбинация возможного количества копий гена SMN1 и возможного количества копий гена SMN2 связана с самой высокой апостериорной вероятностью, по сравнению с другими комбинациями множества комбинаций с заданным (a) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количеством прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат соответствующее специфичное для гена SMN2 основание.
24. Способ по любому из пп. 1-23, в котором определение наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2 включает: определение наиболее вероятной комбинации из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, которые дают в сумме определенное количество копий любых интактных генов SMN с учетом соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN2 основание гена SMN2, соответствующее специфичному для гена SMN1 основанию.
25. Способ по любому из пп. 1-24, в котором определение наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2 включает:
определение (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN2 основание гена SMN2, соответствующее специфичному для гена SMN1 основанию;
определение соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN2 основание гена SMN2, соответствующее специфичному для гена SMN1 основанию; и
определение наиболее вероятной комбинации из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, которые дают в сумме определенное количество копий любого интактного гена SMN на основе соотношения (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN2 основание гена SMN2, соответствующее специфичному для гена SMN1 основанию.
26. Способ по любому из пп. 1-25,
в котором определение наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2 включает: для каждого из множества специфичных для гена SMN1 оснований, определение наиболее вероятной комбинации из множества возможных комбинаций, каждая из которых содержит возможное количество копий гена SMN1 и возможное количество копий гена SMN2, которые дают в сумме определенное количество копий любых интактных генов SMN, которая связана с наибольшей апостериорной вероятностью с учетом (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN1 основание, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат специфичное для гена SMN2 основание гена SMN2, соответствующее специфичному для гена SMN1 основанию, и
в котором определение количества копий гена SMN1 включает в себя: определение количества копий гена SMN1 на основе возможного количества копий гена SMN1 наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенных для каждого из множества специфичных для гена SMN1 оснований.
27. Способ по п. 26, в котором специфичное для гена SMN1 основание имеет соответствие с каждым из множества характерных для конкретного гена SMN1 оснований, отличных от специфичного для гена SMN1 основания, выше заранее определенного порога соответствия.
28. Способ по п. 27, в котором порог соответствия составляет 97%.
29. Способ по любому из пп. 26-28, в котором множество специфичных для гена SMN1 оснований включает 8 специфичных для гена SMN1 оснований.
30. Способ по любому одному из пп. 26-29, в котором каждое из множества специфичных для гена SMN1 оснований находится в интроне 6, экзоне 7, интроне 7 или экзоне 8 гена SMN1.
31. Способ по любому из пп. 26-30, в котором множество оснований, специфичных для гена SMN1, если субъект принадлежит к первой расе, множество оснований, специфичных для гена SMN1, если субъект принадлежит ко второй расе, и множество оснований, специфичных для гена SMN1, если субъект принадлежит к неизвестной расе, различаются.
32. Способ по любому из пп. 26-31, в котором раса субъекта неизвестна, и при этом множество оснований, специфичных для гена SMN1, не является специфичным для расы.
33. Способ по любому из пп. 26-31, в котором раса субъекта известна, и при этом множество оснований, специфичных для гена SMN1, специфично для расы субъекта.
34. Способ по любому из пп. 26-33, дополнительно включающий:
получение информации о расе субъекта; и
на основе полученной информации о расе выбирают множество оснований, специфичных для гена SMN1, из множества оснований, специфичных для гена SMN1.
35. Способ по любому из пп. 1-34, в котором определение количества копий гена SMN1 включает: определение количества копий гена SMN1 и количества копий гена SMN2 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенных для каждого из множества оснований, специфичных для гена SMN1.
36. Способ по любому одному из пп. 1-35, в котором определение количества копий гена SMN1 включает: определение количества копий гена SMN1 с помощью наиболее вероятной комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2, определенных для основания, специфичного для гена SMN1, и второго предварительно определенного порога апостериорной вероятности для комбинации возможного количества копий гена SMN1 и возможного количества копий гена SMN2.
37. Способ по п. 36, в котором второй предварительно определенный порог апостериорной вероятности представляет собой 0,6 или 0,8.
38. Способ по любому из пп. 26-37, в котором большинство возможных значений количества копий гена SMN1 согласуется, и причем определенное количество копий гена SMN1 представляет собой согласованное возможное количество копий гена SMN1.
39. Способ по п. 38, включающий:
определение возможной комбинации, содержащей возможное количество копий гена SMN1 и возможное количество копий гена SMN2, которые дают в сумме определенное количество копий любого интактного гена SMN с учетом (a) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат любое из множества оснований, специфичных для гена SMN1, и (b) количества прочтений последовательности из множества прочтений последовательностей с основаниями, которые содержат любое из множества соответствующих оснований, специфичных для гена SMN2; и
определение возможного количества копий возможной комбинации представляет собой согласованное возможное количество копий гена SMN1.
40. Способ по любому одному из пп. 1-39, в котором определение количества копий гена SMN1 включает определение количества копий гена SMN1, равного нулю, единице или более единицы.
41. Способ по любому из пп. 1-40, включающий определение статуса спинальной мышечной атрофии (SMA) субъекта на основе количества копий гена SMN1.
42. Способ по п. 41, в котором статус SMA субъекта включает SMA, носитель SMA/отсутствие SMA и не носитель SMA.
43. Способ по любому из пп. 1-42, включающий определение субъекта как молчащего носителя SMA с использованием ряда прочтений последовательности из множества прочтений последовательностей, выровненных с g.27134 гена SMN1, и оснований прочтений последовательностей, выровненных с g.27134 гена SMN1.
44. Способ по одному из пп. 1-43, включающий определение рекомендации по лечению для субъекта на основе определенного количества копий гена SMN1.
45. Способ по п. 44, в котором рекомендация по лечению включает в себя введение субъекту Nusinersen и/или Zolgensma.
| WO 2017136059 A1, 10.08.2017 | |||
| Способ преимплантационной генетической диагностики спинальной мышечной атрофии типа 1 | 2017 |
|
RU2671156C1 |
| YANMING FENG et al., "The next generation of population-based spinal muscular atrophy carrier screening: comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing", GENETICS IN MEDICINE,Vol | |||
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
