Ссылка на родственные заявки
[0001] По этой заявке испрашивается приоритет временной заявки на патент США № 62/772823, поданной 29 ноября 2018, которая в полном объеме включена в настоящее описание посредством ссылки.
Область изобретения
[0002] Настоящее изобретение относится к стандартной пероральной лекарственной форме, которая индуцирует иммунизацию слизистой оболочки. Более конкретно, настоящее изобретение относится к лиофилизованной перорально диспергируемой вакцине, содержащей виросомы. Основной проект, представленный в настоящей заявке, был создан на средства, полученные от компании European Union’s Horizon 2020 в соответствии с программой исследований и инноваций, проводимой на грант No. 646122.
Предпосылки к созданию изобретения
[0003] Вакцины традиционно вводят путем внутримышечной, интрадермальной или подкожной инъекции. Эти инъекции могут вызывать сильные системные иммунные ответы, в то время как эффективность для запуска иммунных ответов в слизистой варьируется и часто является слабой или недетектируемой, а в частности, для субъединичных вакцин. Из дренирующих лимфатических узлов, которые утилизуют инъецированную вакцину, антиген-специфические цитотоксические T-клетки (CTL) и антитела, продуцируемые В-клетками, могут мигрировать в различные органы в организме, но их миграция в различные ткани слизистой (например, половых органов, кишечника, дыхательных путей) часто ограничена или невозможна из-за неадекватного хоминга рецепторов слизистой и хемотаксиса. Однако способ интраназального введения, который также рассматривается как способ парентеральной иммунизации, может запускать эффективные иммунные ответы слизистой в дыхательных путях, половых органах и в кишечном тракте, которые иногда действуют сообща, что делает их более доступными, если вакцина доставляется в область слизистой. Поэтому, в некоторых случаях, такие парентеральные вакцины могут обеспечивать защиту против патогенов слизистой оболочки.
[0004] Поскольку большинство патогенов поступает в организм через ткани слизистой (ротовой полости, дыхательных путей, половых органов и кишечного тракта), и многие из них реплицируются только в тканях слизистой оболочки, то вакцинация слизистой может оптимально индуцировать защиту первой линии посредством индукции как врожденного иммунного ответа (например, NK-клеток), так и адаптивного иммунного ответа (T- и В-клетками) в локальном и дистальном участках слизистой оболочки.
[0005] При постоянной защите слизистой оболочки такая защита может осуществляться немедленно без задержки при рекрутинге клеток из периферических участков, что позволяет более эффективно блокировать события передачи и инфицирования на очень ранней стадии еще до распространения патогена, а в некоторых случаях, образование резервуара.
[0006] Ткани слизистых оболочек и лимфатические узлы содержат более 90% иммунных клеток. Кроме того, антитела слизистой оболочки составляют приблизительно 80% от всех антител, продуцируемых организмом. Таким образом, локальный иммунный ответ, продуцируемый антителами слизистых, может действовать как защита первой линии от инфекций слизистой оболочки (например, от ВИЧ-1, вирусов герпеса, ротавирусов и т.п.) и их проникновения через ткани слизистой в другие органы (например, в случае ВИЧ-1, гепатита В, туберкулеза). В отличие от этого, антитела крови могут действовать в качестве резервной защиты после того, как патогены будут преодолевать защиту слизистой оболочки или синергически действовать с антителами слизистой оболочки и средой слизи. Антитела крови действуют, главным образом, как эффективная защита первой линии для борьбы с патогенами, проникающими непосредственно в кровоток после укусов москитов (например, при инфицировании малярийным плазмодием, вирусом чикунгунья, вирусом денге, вирусом Зика, вирусом Западного Нила, вирусом желтой лихорадки и т.п.) или после случайных повреждений кожи/слизистой (например, бактериями Staphylococcus aureus, Pseudomonas aeruginosa).
[ 0007] Доставка вакцины через слизистую оболочку (через слизистую после трансбуккального, подъязычного, интраназального, перорального или вагинального введения) представляет все возрастающий интерес как средство индукции локального и дистального гуморального иммунного ответа, а также системного иммунного ответа. Кроме того, доставка вакцины в слизистую в виде твердых лекарственных форм (например, в виде таблеток для трансбуккального/подъязычного введения, таблеток или капсул для перорального ввдения, влагалищных прокладок) может давать некоторые преимущества, такие как возможность проведения массовой иммунизации, соблюдения пациентом режима и схемы лечения, удобство применения, стабильность продукта при хранении, возможность автономного применения независимо от холодовой цепи. Кроме того, доставка вакцины в слизистую оболочку может быть подходяей для пациентов, которые страдают фобией инъекции иглы, и пациент может самостоятельно вводить вакцину при наличии адекватных инструкций. Способ трансбуккального/подъязычного введения уже применяется в течение многих лет для доставки лекарственных средств и небольших молекул в кровоток, но его применение в качестве средства для доставки вакцин через слизистую почти не рассматривалось.
Краткое описание сущности изобретения
[0008] Везикулы на основе липидов могут быть использованы в качестве систем для доставки лекарственных средств, вакцин или адъюванта и их комбинаций. Везикулы на основе липидов могут состоять из одного или нескольких природных и/или синтетических липидов, образующих основную структуру (частицу), и дополнительных необязательных компонентов (пептидов, белков, углеводов, нуклеиновых кислот, небольших молекул). Везикулы на основе липидов могут иметь размер в диапазоне от наночастиц (приблизительно 20-200 нм) до субмикромикронных частиц (приблизительно 200-800 нм) и до микронных частиц (приблизительно 800 нм - 10 мкм).
[0009] Виросомы и липосомы относятся к системам везикул на основе липидов. Виросомы являются однослойными, а липосомы могут быть однослойными, двуслойными или многослойными. Виросомы представляют собой тип субъединичной вакцины, которая может содержать любой оболочечный белок, происходящий от вирусов, который используется в качестве исходного материала для образования виросомных частиц на основе липидов. Эти виросомы могут быть лишены генетического материала, а также не являются репликативными и не являются инфекционными, что делает их пригодными и безопасными для энтеральной и парентеральной иммунизации, при условии, что они могут быть получены в стабилизированной форме. Виросомы могут содержать: дополнительные молекулы, такие как гомологичные молекулы (от одного и того же патогенного источника) или гетерологичные молекулы-мишени (происходящие от патогена, отличающегося от исходного вирусного материала), в форме пептидов, белков и/или углеводов; нуклеиновые кислоты; адъюванты; специфические липиды и/или небольшие молекулы (лекарственные средства). Подобно виросомам, липосомы могут быть также использованы в качестве носителя для введения фармацевтических лекарственных средств, вакцин и адъювантов, но липидная мембрана не содержит каких-либо природных вирусных белков. В некоторых вариантах осуществления изобретения, используемые здесь липосомы могут представлять собой протеолипосомы (то есть, липосомы с белками).
[0010] В идеальном случае, стабилизация либо позволяетт хранить вакцину независимо от холодовой цепи, либо дает возможность поддерживать вакцину при высоких и низких температурных отклонениях за пределами рекомендуемых условий холодовой цепи без нарушения биологической активности продукта.
[0011] Заявителями была создана лиофилизованная перорально диспергируемая вакцина, содержащая виросомы, и был разработан способ получения такой вакцины, которая может сохранять стабильность виросом (физическую стабильность структуры частицы и химическую стабильность молекул-мишеней). Стабильность продукта может поддерживаться при хранении при температуре окружающей среды (например, приблизительно при 25°С) независимо от условий хранения в холодовой цепи, и может сохраняться при случайных условиях замораживания (например, в пределах приблизительно от -4°C и приблизительно до -17°С), а также при воздействии высоких температур, характерных для теплых стран (например, от 35°C до 45°С). Описанные здесь лиофилизованные лекарственные формы для подъязычного введения, которые содержат виросомную вакцину, могут индуцировать иммунитет слизистой оболочки и могут также вызывать системные иммунные ответы. В частности, комбинация композиции жидкого виросомного концентрата и его буфера, композиции жидкой основной матрицы для твердой лекарственной формы (за исключением виросомного концентрата) и условий производства для приготовления твердых лекарственных форм в виде лиофилизованных таблеток для подъязычного введения может обеспечивать получение твердой вакцинной формы, которая имела бы подходящие физические свойства для таблеток для подъязычного введения с сохранением виросом и структурной целостности молекулы-мишени и стабильность при хранении в различных условиях окружающей среды. Введение стабильных виросомных твердых лекарственных форм может оказаться подходящим способом иммунизации слизистой путем подъязычного введения.
[0012] Виросомы принадлежат к семейству оболочечных вирусоподобных частиц (VLP), поскольку они имеют липидные мембраны, содержащие белки вирусной мембраны, происходящие от исходных очищенных вирусов. В качестве VLP, виросомы могут точно имитировать архитектуру вирусных частиц, состав, презентацию антигена на поверхности мембраны и могут имитировать, а могут и не имитировать функциональную активность нативной вирусной оболочки. Виросомы могут содержать реконструированные вирусные мембраны, обычно получаемые путем очистки солюбилизированных мембранных белков и липидов от оболочечных вирусов под действием солюбилизирующего агента с последующим добавлением природных или синтетических липидов в присутствии или в отсутствии адъювантов, и удалением указанного солюбилизирующего агента из смеси, что будет приводить к образованию липидного бислоя с белками, выступающими из него. Антигены могут представлять собой нативные белки, а также рекомбинантные или синтетические белки или пептиды. Сначала могут быть также получены виросомы, а затем, они могут быть модифицированы путем ковалентной или нековалентной модификации их мембран так, чтобы они содержали адъюванты и другие молекулы. Характерным признаком виросом является то, что они могут точно имитировать состав, поверхностную архитектуру и функциональные активности нативной вирусной оболочки. В некоторых случаях, если индуцирование CD8+-T-клеток является частью стратегии вакцинации, то особенно важным свойством указанных виросом является сохранение связывания с рецептором гемагглютинина (НА) и активностью их гибридной мембраны. Если стратегия вакцинации фокусируется на индуцировании антигенспецифических антител, то активность связывания с НА не является абсолютно необходимой, однако, присутствие НА в качестве наполнителя все еще является важным условием, поскольку оно может обеспечивать помощь Т-клеткам, в частности, для небольших антигенов и пептидов, которые могут быть лишены таких свойств. Хотя в данном описании упоминаются специфические вакцины, однако, все вакцины, в которых используются виросомы в качестве носителя для доставки, входят в объем настоящего изобретения. Виросома гриппа, содержащая антигены ВИЧ (P1 и rgp41) и адъювант TLR 7/8, может быть использована в качестве моделей виросомных концентратов для вакцин, раскрытых в настоящей заявке. Было показано, что виросомы гриппа с антигеном Rgp41 ВИЧ были способны вызывать иммунные ответы в различных исследованиях на животных и в фазе 1 клинического исследования путем интраназального и внутримышечного введения. В таких исследованиях, виросомные препараты получают в жидкой форме, вводимой иглой или в виде интраназального жидкого спрея, и эти прерапаты имели ограниченную стабильность при хранении при 4°C вследствие химических модификаций активных фармацевтических ингредиентов («API»). Для предотвращения или уменьшения таких химических модификаций API, часто присутствующих в водных средах, разработка твердых форм вакцин с низким содержанием влаги была идентифицирована как перспективный подход.
[0013] Заявители смогли решить различные проблемы при получении твердой вакцины в виде лиофилизованных перорально диспергируемых вакцин, описанных в настоящей заявке. В частности, виросомные концентраты на основе вируса гриппа, которые содержат виросомы, несущие антигены и адъюванты, могут поставляться в виде суспензии в буферном солевом растворе. Для получения лиофилизованных вакцинных лекарственных форм в виде таблеток, эти таблетки должны иметь устойчивые и пористые структуры во время их изготовления, и эти структуры должны поддерживаться во время последующего хранения. Присутствие буферных солей в виросомных концентратах может создавать проблему при изготовлении такой устойчивой и пористой структуры. По существу, заявителями было получено соответствующее количество наполнителей в комбинации с соответствующими условиями их получения для обеспечения образования устойчивой лекарственной формы, которая не разрушается или разрушается лишь частично во время лиофилизации.
[0014] Кроме того, процесс замораживания, отжига и лиофилизации может также приводить к нарушению целостности виросомных частиц. Это повреждение виросом должно быть сведено к минимуму так, чтобы в лиофилизованном продукте сохранялось достаточное количество виросомных частиц, и чтобы это достаточное количество могло проходить через мембрану слизистой при подъязычном введении при обработке иммунными клетками для индукции иммунных ответов в слизистой оболочке, которые могут также поддерживаться системными иммунными ответами. По существу, заявители смогли сбалансировать использование наполнителя и процесс лиофилизации для поддержания как виросомной иммуногенности (например, интактных виросом с ограниченным присутствием кластеров), так и полезных свойств лекарственной формы (например, таблеток для подъязычного введения, распадающихся в ротовой полости).
[0015] В некоторых вариантах осуществления изобретения, пероральная твердая вакцинная лекарственная форма включает липидные везикулы, содержащие иммуногенное количество по меньшей мере одной молекулы-мишени; 5-20 масс. % по меньшей мере одного криолиопротектора; 25-40 масс. % вещества, образующего матрицу, и 40-55 масс. % вещества, формирующего структуру. В некоторых вариантах осуществления изобретения, везикулы на основе липидов представляют собой виросомы или протеолипосомы. В некоторых вариантах осуществления изобретения, лекарственная форма содержит 10-15 масс. % по меньшей мере одного криолиопротектора. В некоторых вариантах осуществления изобретения, по меньшей мере один криолиопротектор содержит трегалозу. В некоторых вариантах осуществления изобретения, лекарственная форма содержит 33-37 масс. % вещества, образующего матрицу. В некоторых вариантах осуществления изобретения, вещество, образующее матрицу, содержит желатин. В некоторых вариантах осуществления изобретения, желатин включает рыбий желатин. В некоторых вариантах осуществления изобретения, рыбий желатин представляет собой высокомолекулярный рыбий желатин. В некоторых вариантах осуществления изобретения, лекарственная форма содержит 45-50 масс.% вещества, формирующего структуру. В некоторых вариантах осуществления изобретения, вещество, формирующее структуру, содержит маннит. В некоторых вариантах осуществления изобретения, виросомы получают из мембраны вируса гриппа или других оболочечных вирусов. В некоторых вариантах осуществления изобретения, по меньшей мере одна молекула-мишень присутствует на виросоме. В некоторых вариантах осуществления изобретения, по меньшей мере одна молекула-мишень содержит антиген, происходящий от оболочки ВИЧ-1. В некоторых вариантах осуществления изобретения, антиген, происходящий от оболочки ВИЧ-1, содержит пептид PI ВИЧ-1 и/или рекомбинантный gp41 ВИЧ-1. В некоторых вариантах осуществления изобретения, виросомы содержат адъювант. В некоторых вариантах осуществления изобретения, лекарственная форма облегчает поглощение по меньшей мере одной молекулы-мишени в ротовой полости. В некоторых вариантах осуществления изобретения, лекарственная форма разлагается в течение 180 секунд после ее помещения в ротовую полость. В некоторых вариантах осуществления изобретения, лекарственная форма разлагается в течение 90 секунд после ее помещения в ротовую полость. В некоторых вариантах осуществления изобретения, лекарственная форма разлагается в течение 60 секунд после ее помещения в ротовую полость. В некоторых вариантах осуществления изобретения, лекарственная форма разлагается в течение 30 секунд после ее помещения в ротовую полость. В некоторых вариантах осуществления изобретения, иммунный ответ индуцируется при введении пациенту лекарственной формы в ротовую полость. В некоторых вариантах осуществления изобретения, помещение в ротовую полость осуществляют путем введения лекарственной формы на язык или под язык или в щечную область или в область глотки.
[0016] В некоторых вариантах осуществления изобретения, способ индукции иммунного ответа у пациента включает введение любой из описанных выше лекарственных форм в ротовую полость человека, нуждающегося в иммунном ответе. В некоторых вариантах осуществления изобретения, введение в ротовую полость осуществляют путем помещения лекарственной формы на язык или под язык или в щечную область или область глотки.
[0017] В некоторых вариантах осуществления изобретения, способ получения пероральной твердой вакцинной лекарственной формы включает дозирование жидкой виросомной композиции в предварительно сформованной форме, где виросомная композиция включает: липидные везикулы, содержащие иммуногенное количество по меньшей мере одной молекулы-мишени, 1-5 масс.% криолиопротектора, 4-8 масс.% вещества, образующего матрицу, и 5-10 масс.% вещества, формирующего структуру; замораживание дозированной виросомной композиции при температуре от -60°С до -90°С; отжиг замороженной виросомной композиции путем ее выдерживания при температуре ниже -15°С в течение 3-9 часов; и лиофилизацию гибридизованной виросомной композиции с образованием лекарственной формы. В некоторых вариантах осуществления изобретения, дозированную виросомную композицию замораживают при температуре от -60°С до -90°С в течение приблизительно 1-5 минут. В некоторых вариантах осуществления изобретения, лиофилизация гибридизованной виросомной композиции включает первую стадию выдерживания гибридизованной виросомной композиции при температуре от -10°С до -20°C в течение 20-28 часов и вторую стадию выдерживания гибридизованной виросомной композиции при температуре от -5°С до приблизительно -15°С в течение 14-22 часов. В некоторых вариантах осуществления изобретения, лиофилизацию осуществляют под давлением менее, чем 600 мбар. В некоторых вариантах осуществления изобретения, виросомная композиция имеет рН приблизительно 6,5-8. В некоторых вариантах осуществления изобретения, криолиопротектор содержит трегалозу. В некоторых вариантах осуществления изобретения, вещество, образующее матрицу, содержит желатин. В некоторых вариантах осуществления изобретения, желатин включает рыбий желатин. В некоторых вариантах осуществления изобретения, рыбий желатин представляет собой высокомолекулярный рыбий желатин. В некоторых вариантах осуществления изобретения, вещество, формирующее структуру, содержит маннит. В некоторых вариантах осуществления изобретения, липидные везикулы получают из вируса гриппа или респираторно-синцитиального вируса. В некоторых вариантах осуществления изобретения, по меньшей мере одна молекула-мишень содержит антиген, происходящий от оболочки ВИЧ-1. В некоторых вариантах осуществления изобретения, антиген, происходящий от оболочки ВИЧ-1, содержит пептид PI ВИЧ-1 или рекомбинантный gp41 ВИЧ-1. В некоторых вариантах осуществления изобретения, везикулы на основе липидов включают адъювант.
[0018] В некоторых вариантах осуществления изобретения, способ получения пероральной твердой вакцинной лекарственной формы включает: дозирование жидкой виросомной композиции в предварительно сформованную форму, где виросомная композиция включает: (1) 20-50 масс.% виросомного концентрата, где виросомный концентрат включает: виросомы, содержащие иммуногенное количество по меньшей мере одной молекулы-мишени; 2-10 масс. % криолиопротектора; и 60-200 мМ буферной системы; (2) 4-8 масс.% вещества, образующего матрицу; и (3) 5-10 масс. % вещества, формирующего структуру; замораживание дозированной виросомной композиции при температуре от -60°С до -90°С; отжиг замороженной виросомной композиции путем ее выдерживания при температуре ниже -15°С в течение 3-9 часов; лиофилизацию гибридизованной виросомной композиции с получением лекарственной формы. В некоторых вариантах осуществления изобретения, буферная система содержит HEPES-хлорид натрия.
[0019] Дополнительные преимущества будут совершенно очевидны специалистам в данной области из следующего подробного описания. Приведенные здесь примеры и описание следует рассматривать как иллюстративные, а не ограничивающие.
[0020] Все публикации, включая патентные документы, научные статьи и базы данных, упоминаемые в настоящей заявке, включены в настоящее описание посредством ссылки во всей своей полноте и во всех целях так, как если бы каждая отдельная публикация была отдельно включена посредством ссылки. Если приведенное здесь определение противоречит или как-либо иначе не совпадает с определениями, представленными в патентах, в заявках, в опубликованных заявках и в других публикациях, которые включены в настоящее описание посредством ссылки, то следует отдать предпочтение определениям, представленным в настоящей заявке, а не определениям, которые включены в настоящее описание посредством ссылки.
Краткое описание чертежей
[0021] Репрезентативные варианты осуществления изобретения описаны со ссылкой на прилагаемые чертежи, где:
[0022] На фигуре 1 представлена иллюстративная блок-схема получения раскрытой здесь вакцинной лекарственной формы.
[0023] На фигуре 2 показана блок-схема получения раскрытой здесь вакцинной лекарственной формы начиная от получения матрицы и до получения готовых таблеток для подъязычного введения.
[0024] На фигуре 3А схематически представлено изображение полностью увлажненной таблетки.
[0025] На фигуре 3В схематически представлено изображение таблеток с плотными комками.
[0026] На фигуре 3С схематически представлено изображение таблетки с пленкой дезинтегрированной матрицы для получения композиции, которая образуется на поверхности лиофилизованной таблетки (с оболочкой).
[0027] На фигуре 4 представлена фотография иммуноблотов, иллюстрирующих специфичные анти-Р1 и анти-rg41 антитела, связывающиеся с виросомами-Р1 и виросомами-rgp41, в разведенных таблетках для подъязычного введения Zydis®, хранящихся при 5°С, 25°С и 40°С в течение 1 и 3 месяцев (анализ, описанный в Примере 2).
[0028] На фигуре 5 проиллюстрирована иммуногенность антигенов PI и rg41 в жидких виросомных концентратах и лиофилизованных таблеток для подъязычного введения, содержащих виросомы, и хранящихся при 4°C и 40°С в течение определенного периода времени.
Подробное описание изобретения
[0029] В настоящей заявке описаны фармацевтические композиции везикул на основе липидов (например, виросом или VLP) и cпособы получения этих фармацевтических композиций. В частности, настоящее изобретение относится к лиофилизованным перорально диспергируемым или разлагающимся лекарственным формам, которые могут сохранять стабильность VLP (то есть, структурную целостность и химическую стабильность антигена), могут храниться независимо от условий хранения в холодовой цепи и могут также сохраняться в случайных условиях замораживания, а также под воздействием высоких температур, характерных для теплых стран. Лекарственные формы могут также сохранять физические и химические свойства VLP, что делает его подходящими для подъязычной доставки для индукции иммунизации слизистой.
[0030] Заявителями было получено и оптимизировано количество вещества, образующего матрицу, и вещества, формирующего структуру, которые позволяют получить композицию, допускающую использование высокой нагрузки виросомных концентратов, содержащих буферные системы и криолиопротекторов. В сочетании с коррекцией количества наполнителя были оптимизированы параметры производства лекарственных форм. В частности, время отжига было оптимизировано для максимизации кристаллизации маннита, которая придает стабильность лекарственной форме и сводит к минимуму повреждение виросом. Кроме того, условия лиофилизации были оптимизированы для минимизации повреждения виросомных частиц, а также сведения к минимуму разрушения структуры во время лиофилизации.
Системы везикул на основе липидов
[0031] Везикулы на основе липидов могут быть использованы в качестве систем для доставки лекарственных средств, вакцин или адъюванта и их комбинаций. Везикулярные системы на основе липидов могут состоять из одного или нескольких природных и/или синтетических липидов (например, фосфатидилэтаноламина, фосфатидилхолина, фосфатидилсерина, фосфатидилинозита, фосфатидилглицерина, холестерина, сфинголипидов и их производных), образующих везикулярную мембрану (частицы), и дополнительных необязательных компонентов (пептидов, белков, углеводов, небольших молекул). Везикулы на основе липидов могут иметь размер в диапазоне от наночастиц (приблизительно 20-200 нм) до субмикронных частиц (приблизительно 200-800 нм) и до микронных частиц (приблизительно 800 нм - 10 мкм).
[0032] Везикула/частица на основе липида могут содержать униламеллярную, биламеллярную или мультиламеллярную липидную бислойную везикулу. В некоторых вариантах осуществления изобретения, везикулы/частицы на основе липида могут включать липидный бислой, содержащий липиды, выбранные из природных и/или синтетических липидов. Такие липиды могут быть использованы для еще большей имитации патогенной мембраны и микродоменов липидного остова для улучшения заякоривания антигена на мембране, презентации антигена и/или укладки для оптимального воздействия эпитопа. Везикула/частица на основе липида может содержать заякоренный на мембране антиген и/или адъювант, расположенный на поверхности частицы или внутри частицы или имеющий произвольную ориентацию. Антиген и/или адъювант также могут быть инкапсулированы внутри просвета везикулы/частицы на основе липида.
[0033] Виросомы представляют собой везикулы на липидной основе, собранные in vitro в бесклеточной системе с образованием оболочечных VLP, которые принадлежат к категории субъединичных вакцин. Виросомные липидные мембраны, используемые в качестве носителя, могут быть происходить от любого оболочечного вируса и, следовательно, содержать по меньшей мере нативные вирусные мембранные белки исходного вируса. Эти виросомы лишены генетического вирусного материала, не могут реплицироваться и не являются инфекционными, что делает их пригодными и безопасными для системной иммунизации и иммунизации слизистой. Кроме того, виросомы могут содержать дополнительные молекулы, такие как антигены (например, пептиды, белки, углеводы, нуклеиновые кислоты), адъюванты, специфические липиды и/или небольшие молекулы (лекарственные средства), которые могут быть заякорены на поверхности виросом и/или заключены внутри просвета виросомы.
[0034] Липосомы также представляют собой везикулы на липидной основе, образующие тип субъединичной вакцины, которая может быть получена в виде везикул, имеющих по меньшей мере один липидный бислой, который не содержат белков, происходящих от природной вирусной мембраны оболочечных вирусов. Такие частицы на липидной основе, собранные in vitro, не содержат генетического материала и могут быть подходящими для системного введения и введения в слизистую. Кроме того, липосомы могут содержать дополнительные молекулы, такие как антигены (пептиды, белки и/или углеводы, нуклеиновые кислоты), адъюванты, специфические липиды и/или небольшие молекулы (лекарственные средства), которые могут быть заякорены на поверхности липосом и/или заключены внутри просвета липосом. В некоторых вариантах осуществления изобретения, липосомы, используемые в настоящем изобретении, могут представлять собой протеолипосомы (то есть, липосомы с белками).
[0035] Липиды, используемые в описанных здесь лекарственных формах, могут представлять собой катионные липиды, гликолипиды, фосфолипиды, глицерофосфолипиды, галактозилцерамиды, сфинголипиды, холестерин и их производные. Фосфолипиды могут включать, но не ограничиваются ими, фосфатидилхолин, сфингомиелин, фосфатидилэтаноламин, фосфатидилсерин, фосфатидилглицерин, фосфатидиновую кислоту, кардиолипин и фосфатидилинозит с различными составами жирных ацилов.
[0036] Кроме того, липиды могут быть выбраны из DOTMA (хлорида N-[1-(2,3-диолеилокси)пропил]-N, N,N-триметиламмония), DOTAP (хлорида N-[1-(2,3-диолеоилокси)пропил]-N, N,N-триметиламмония), DODAC (хлорида N, N-диолеил-N, N-диметиламмония), DDAB (бромида дидодецилдиметиламмония) и стеариламина или других алифатических аминов и т.п.
Виросомный препарат
Виросомный концентрат
[0037] На фигуре 1 представлена блок-схема способа 100 получения описанной здесь вакцинной лекарственной формы. На стадии 101, жидкий виросомный концентрат может быть смешан с предварительной полученной смесью основного матричного состава с образованием жидкой виросомной композиции, подходящей для лиофилизации. Заявителями было обнаружено, что при объединении композиции виросомного концентрата с конкретным составом основной матрицы и получении в комбинации с рядом условий приготовления, оптимизированных для сохранения достаточного количества виросом с требуемыми характеристиками частиц, можно поддерживать биологическую активность вакцины. Биоактивность виросомных частиц может зависеть от конформационной целостности частиц и качества антигенных молекул, которые связаны с его униламеллярной фосфолипидной мембраной.
[0038] В некоторых вариантах осуществления изобретения, виросомный концентрат может представлять собой жидкий виросомный концентрат. Виросомный концентрат включает по меньшей мере одну виросомную популяцию. Виросомная популяция может представлять собой виросомы, содержащие данное лекарственное средство, или виросомы, действующие в качестве носителя для доставки вакцины, содержащего вакцинные антигены и вирусные белки (например, НА, если виросомы происходят от вируса гриппа).
[0039] В некоторых вариантах осуществления изобретения, виросомы могут быть получены из вируса гриппа для продуцирования виросом вируса гриппа в качестве оболочечных VLP, действующих в качестве носителя для гетерологичных вакцинных антигенов (например, антигенов ВИЧ, заякоренных на виросомах, происходящих от вируса гриппа), или от другого оболочечного вируса, такого как респираторно-синцитиальный вирус («RSV»). В некоторых вариантах осуществления изобретения, оболочечные вирусы, такие как RSV, вирус Сендай, вирус лесов Семлики (SFV), вирус везикулярного стоматита (VSV) или вирус Синдбис могут быть использованы для получения соответствующих RSV-виросом, виросом Сендай, SFV-виросом, VSV-виросом или виросом Синдбис для представления гомологичного вакцинного антигена (например, нативных антигенов RSV на виросомах, происходящих от RSV). В некоторых вариантах осуществления изобретения, виросомы на основе вирусов могут происходить от любого оболочечного вируса. В некоторых вариантах осуществления изобретения, виросомы на основе вирусов могут быть получены из ДНК-вирусов, включая, но не ограничиваясь ими, герпесвирусы, поксвирусы и гепаднавирусы. В некоторых вариантах осуществления изобретения, виросомы на основе вирусов могут быть получены из РНК-вирусов, включая, но не ограничиваясь ими, флавивирус, тогавирус, коронавирус, вирус гепатита D, ортомиксовирус, парамиксовирус, рабдовирус, буньявирус и филовирус. В некоторых вариантах осуществления изобретения, виросомы на основе вирусов могут быть получены из ретровирусов.
[0040] В некоторых вариантах осуществления изобретения, виросома на основе вируса гриппа может быть полученв различными способами, описанными в публикации Influenza virosomes as vaccine adjuvant and carrier system; Moser C et al. Expert review, 779-791 (2013); WO 2004110486; WO 2004071492; WO 2007099446; WO 201603919; EP 2058002 и WO 2016039620, которые в полном объеме включены в настоящее описание посредством ссылки. Кроме того, различные патенты и другие публикации, перечисленные в ранее цитированных документах, также в полном объеме включены в настоящее описание посредством ссылки. По существу, настоящая заявка не ограничена конкретным способом получения виросом. Так, например, ссылка, приведенная выше, указывает просто на пример виросомного препарата. Данная заявка относится ко всем частицам на липидной основе, таким как, но не ограничивающимся ими, виросомы, VLP и препарат наночастиц, включая липосомы с антигенами.
[0041] Хотя в этом разделе описан жидкий виросомный концентрат, однако, виросома может быть заменена липосомой с образованием жидкого липосомного концентрата. Так, например, компоненты в виросомном концентрате (помимо самих виросом) также могут быть в равной степени применимы к липосомному концентрату.
[0042] Виросома (например, виросома, происходящая от вируса гриппа) может иметь вирусные белки на своей поверхности. В некоторых вариантах осуществления изобретения, белок может представлять собой гемагглютинин («HA») и/или нейраминидазу («NA»). В некоторых вариантах осуществления изобретения, жидкий виросомный концентрат включает белки вирусной мембраны (например, НА), присутствующие в концентрации, определяемой с помощью стандартных анализов. В некоторых вариантах осуществления изобретения, белки вирусной мембраны составляют приблизительно 10-300 мкг/мл или приблизительно 5-150 мкг/мл жидкого виросомного концентрата, если в качестве носителя гетерологических вакцинных антигенов используются виросомы вируса гриппа. Для вирососом гриппа, предназначенных для индукции CTL-ответов, концентрация НА может составлять в диапазоне приблизительно 150-800 мкг/мл или приблизительно 75-400 мкг/мл. В некоторых вариантах осуществления изобретения, концентрация вирусных мембранных белков может превышать концентрации, перечисленные выше в качестве примера.
[0043] Виросомный концентрат может также включать липиды. Липиды, используемые в виросомном концентрате, могут представлять собой катионные липиды, гликолипиды, фосфолипиды, глицерофосфолипиды, галактозилцерамиды, сфинголипиды, холестерин и их производные. Фосфолипиды могут включать, в частности, фосфатидилхолин, сфингомиелин, фосфатидилэтаноламин, фосфатидилсерин, фосфатидилглицерин, фосфатидиновую кислоту, кардиолипин и фосфатидилинозит с различными составами жирных ацилов. Кроме того, липиды могут быть выбраны из DOTMA (хлорида N-[1-(2,3-диолеилокси)пропил]-N, N,N-триметиламмония), DOTAP (хлорида N-[1-(2,3-диолеоилокси)пропил]-N, N,N-триметиламмония), DODAC (хлорида N, N-диолеил-N, N-диметиламмония), DDAB (бромида дидодецилдиметиламмония) и стеариламина или других алифатических аминов и т.п. В некоторых вариантах осуществления изобретения, жидкий виросомный концентрат может включать приблизительно 0,1-10 мг/мл, приблизительно 0,3-8 мг/мл или приблизительно 0,5-5 мг/мл липидов.
[0044] Виросомы могут содержать молекулы-мишени (например, вакцинные антигены) в присутствии или в отсутствии адъювантов. Антигены могут быть растворимыми и могут быть захвачены внутри просвета виросомы или ковалентно или нековалентно заякорены на виросомах, и могут представлять собой пептиды, белки, полисахариды, целые частицы или неполные фрагменты или экстракты бактериальных клеток, вирусные частицы или нуклеиновые кислоты, либо они могут быть получены от паразитов, таких как простейшие или черви, которые вызывают заболевание, или их комбинации, или от любых клеток или клеточных линий растений, животных или человека. Любой антиген, известный специалистам в данной области, может оказаться подходящим для его применения вместе с виросомами, включая коммерчески доступные виросомы, либо он может быть получен путем очистки препаратов патогена или раковой клетки или нетрансформированных клеток (нативных белков), рекомбинантно экспрессированных или полученных путем синтеза стандартным промышленным способом. Способы получения подходящих антигенов для их включения в виросомы известны специалистам в данной области, и любой из этих известных способов может быть применен как описано в настоящей заявке.
[0045] Молекула(ы)-мишень(и) включена(ы) в виросомы жидкого виросомного концентрата и в полученные затем лекарственные формы, описанные в настоящей заявке, в количестве, достаточном для сообщения ему иммуногенности при его введении в лекарственной форме. «Иммуногенное количество» определяется как количество, достаточное для стимуляции желаемого иммунного ответа. Специалист в данной области может легко определить иммуногенное количество для данного заболевания или инфекции в зависимости, среди прочих факторов, от способа иммунизации, возраста и массы пациента, которому будет введена лекарственная форма.
[0046] В некоторых вариантах осуществления изобретения, жидкий виросомный концентрат может включать приблизительно 1-2000 мкг/мл молекулы-мишени. Молекула-мишень может представлять собой пептид, белок, углевод, нуклеиновую кислоту или небольшую молекулу или их комбинацию. Молекула-мишень может функционировать в качестве антигена (например, вакцинного антигена), лекарственного средства, диагностической молекулы, аналитического сенсора или их комбинации. В некоторых вариантах осуществления изобретения, жидкий виросомный концентрат может включать приблизительно 1-1000 мкг/мл одной молекулы-мишени и приблизительно 1-1000 мкг/мл второй молекулы-мишени. После дальнейшей обработки с получением твердых лекарственных форм, каждая таблетка может содержать приблизительно от 0,01 до 250 мкг каждой молекулы-мишени.
[0047] В более предпочтительных вариантах осуществления изобретения, жидкий виросомный концентрат может включать приблизительно 25-500 мкг/мл, приблизительно 50-500 мкг/мл, приблизительно 25-225 мкг/мл, приблизительно 25-200 мкг/мл, приблизительно 50-450 мкг/мл, приблизительно 50-400, приблизительно 100-400 мкг/мл, приблизительно 100-200 мкг/мл или приблизительно 200-400 мкг/мл по меньшей мере одной молекулы-мишени (например, антигена). В некоторых вариантах осуществления изобретения, жидкий виросомный концентрат может включать приблизительно 1-500 мкг/мл, приблизительно 25-500 мкг/мл, приблизительно 50-450 мкг/мл, приблизительно 50-400 мкг/мл, приблизительно 25-250 мкг/мл, приблизительно 25-200 мкг/мл, приблизительно 50-250 мкг/мл, приблизительно 75-225 мкг/мл или приблизительно 100-200 мкг/мл одной молекулы-мишени и приблизительно 50-450 мкг/мл, приблизительно 50-400 мкг/мл, приблизительно 25-225 мкг/мл, приблизительно 25-200 мкг/мл, приблизительно 100-500 мкг/мл, приблизительно 150-450 мкг/мл, приблизительно 175-425 мкг/мл или приблизительно 200-400 мкг/мл второй молекулы-мишени.
[0048] Раскрытые здесь лекарственные формы могут быть использованы для доставки терапевтических или профилактических вакцин в целях профилактики или ослабления симптомов, ассоциированных с аллергиями или инфекцией, развитием и распространением опухоли, передачей патогенов, клеточной инфекцией, нагрузкой патогеном, после индукции В-клеток (антител) и/или соответствующих T-клеточных субпопуляций (например, Th1, Th2, Thl6, Thf, Tel, Tc2, Tc3, Treg и т.п.), в зависимости от состава виросомных композиций. Для этой цели, молекулы-мишени могут быть получены так, чтобы они обеспечивали защиту от перечисленных ниже заболеваний, список которых не являются исчерпывающим, таких как грипп, туберкулез, менингит, гепатит, коклюш, полиомиелит, столбняк, дифтерия, малярия, холера, герпес, тиф, ВИЧ/СПИД, корь, болезнь Лайма, диарея путешественников, гепатит А, В и С, отит среднего уха, лихорадка денге, бешенство, парагрипп, краснуха, желтая лихорадка, дизентерия, болезнь легионеров, токсоплазмоз, q-лихорадка, геморрагическая лихорадка, Аргентинская геморрагическая лихорадка, кариес, болезнь Шагаса, инфекция мочевых путей, вызываемая E. coli, пневмококковые заболевания, паротит, болезнь, вызываемая вирусом чикунгунья, рак, аллергии и их комбинации. Кроме того, молекулы-мишени могут обеспечивать защиту от заболевания, вызываемого нижеследующими микроорганизмами-возбудителями, входящими в нижеследующий неисчерпывающий список, такими как виды Vibrio, виды Salmonella, виды Bordetella, виды Haemophilus, Toxoplasmosis gondii, цитомегаловирус, виды Chlamydia, стрептококки, вирус Норуолк, Escherischia coli, Helicobacter pylori, ротавирус, Neisseria gonorrhae, Neisseria meningiditis, аденовирус, вирус Эпштейна-Барра, вирус японского энцефалита, Pneumocystis carini, вирус простого герпеса, виды Clostridia, респираторно-синцитиальный вирус, виды Klebsiella, виды Shigella, Pseudomonas aeruginosa, парвовирус, виды Camylobacter, виды Rickettsia, вирус ветряной оспы, виды Yersinia, вирус реки Росс, вирус J.C., Rhodococcus equi, Moraxella catarrhalis, Borrelia burgdorferi, Pasteurella haemolytica и их комбинации. Кроме того или альтернативно, молекула-мишень может обеспечивать защиту или лечение от аллергий (то есть, с использованием виросом, содержащих аллергены), рака (например, с использованием опухолевых антигенов, антител, противораковых лекарственных средств, нуклеиновой кислоты) и заболеваний других типов.
[0049] Также рассматривается применение настоящего изобретения в ветеринарии. Соответственно, молекулы-мишени могут обеспечивать защиту против заболеваний животных, указанных в нижеследующем неисчерпывающем списке, таких как кокцидиоз, ньюкаслская болезнь, энзоотическая пневмония, лейкоз кошек, атрофический ринит, рожа, ящур, болезни свиней, пневмонии и другие патологические состояния и другие инфекции, поражающие домашних питомцев и сельскохозяйственных животных, и их комбинации.
[0050] В некоторых вариантах осуществления изобретения, виросома содержит по меньшей мере один вакцинный антиген в дополнение к вирусным белкам, присутствующим в реконструированной мембране. В некоторых вариантах осуществления изобретения, вакцинным антигеном может быть пептид P1 ВИЧ-1 и/или рекомбинантный gp41 ВИЧ-1.
[0051] Как указано выше, виросомы жидкого виросомного концентрата могут также содержать адъювант или могут быть смешаны с адъювантом. Для улучшения иммунного ответа иожет потребоваться включение адъюванта в виросомы и другие субъединичные вакцины, что будет приводить к ускорению и усилению продуцирования антител и T-клеток с сохранением иммунологической памяти. Для обеспечения эффективности, предпочтительно, чтобы они продуцировали иммунный ответ, ассоциированный с вырабатыванием иммунологической памяти, который обеспечивает длительную защиту от конкретного заболевания. Адъювант также может снижать дозу антигена (обеспечивать щадящую дозу) и расширять спектр желаемого иммунного ответа. После воздействия антигенов, иммунная система может «запоминать» его, и во время повторного воздействия антигена, иммунный ответ будет вырабатываться гораздо быстрее. Эффективность адъюванта в усилении иммунного ответа может быть независимой от антигена, с которым он объединен, поскольку сам адъювант может запускать неспецифические иммунные ответы, которые могут приводить к аутоиммунным побочным эффектам, если сильная активация клеток достигается в отсутствие антигена. Однако, если антиген и адъювант физически связываются друг с другом, то все они могут мигрировать в один и тот же участок после инъекции, что будет способствовать антиген-специфической иммунной активации с более низкими неспецифическими воспалительными реакциями. Подходящие адъюванты включают, но не ограничиваются ими: агонисты ловушко-подобного рецептора (TLR), агонисты инфламмасомы, агонисты рецепторов, подобных доменам, связывающимся с нуклеотидом, и доменам олигомеризации (NOD)(NLR), а более конкретно, нетоксичные бактериальные фрагменты; холерный токсин (и его детоксифицированные формы и фракции); хитозан; термолабильный токсин E. coli (и его детоксифицированные формы и фракции); гомополимеры и сополимеры лактида/гликолида (PLA/GA); полиангидрид, например, тримеллитилимидо-L-тирозин, DEAE-декстран, сапонины, образующие комплекс с антигенами мембранного белка (иммуностимулирующие комплексы - ISCOM), бактериальные продукты, такие как липополисахарид (LPS) и мурамил-дипептид (MDP), липосомы, кохлеаты, протеоноиды, цитокины (интерлейкины, интерфероны), генетически сконструированные живые микробные векторы, неинфекционный коклюшный мутантный токсин, нейримидазу/галактозооксидазу, и аттенюированные бактериальные и вирусные токсины, происходящие от мутантных штаммов, и их комбинации. Подходящее количество адъюванта может быть легко определено специалистом в данной области.
[0052] В некоторых вариантах осуществления изобретения, виросомы могут содержать адъювант 3M-052 (агонист TLR7/8, поставляемый 3M). В некоторых вариантах осуществления изобретения, жидкий виросомный концентрат может содержать адъювант 3M-052 в количестве приблизительно 8-140 мкг/мл, приблизительно 4-70 мкг/мл, приблизительно 1-60 мкг/мл и от 0,01 до 16 мкг на таблетку.
[0053] В некоторых вариантах осуществления изобретения, жидкий виросомный концентрат может включать по меньшей мере две различных виросомных популяции, каждая из которых содержит по меньшей мере один антиген с адъювантом или без адъюванта. В некоторых вариантах осуществления изобретения, эти две различные виросомные популяции могут иметь различные антигены, но один и тот же адъювант (например, виросома-Р1/3M-052, смешанная с виросомами-rgp41/3M-052). В некоторых вариантах осуществления изобретения, эти две различные виросомные популяции могут иметь различные антигены и различные адъюванты (например, виросома-P1/адъювант А, смешанные с виросомами-rgp41/адъювант В).
[0054] В некоторых вариантах осуществления изобретения, виросомный концентрат может включать по меньшей мере два различных антигена на виросому с адъювантом или без адъюванта (например, виросому, содержащую антигены P1 и rgp41 с адъювантом или без адъюванта).
[0055] Жидкий виросомный концентрат может также включать буферную систему. В некоторых вариантах осуществления изобретения, виросомы могут быть суспендированы в буферной системе. Буферная система может сохранять физическую целостность и химическую стабильность виросом в виросомном концентрате. В некоторых вариантах осуществления изобретения, виросомы суспендируют в буферной системе для поддержания нужного рН приблизительно 6-9, приблизительно 6,5-8, приблизительно 7-8, приблизительно 7,2-7,6, приблизительно 7,3-7,5 или приблизительно 7,4. Кроме того, буферная система может стабилизировать виросомы, если она присутствует в жидкой форме при температуре хранения приблизительно 2-8°С.
[0056] Подходящие буферные системы включают, но не ограничиваются ими, буферы HEPES-хлорид натрия (HN), буферы HEPES-хлорид натрия-EDTA (HNE), фосфатные буферные системы (PBS) или их комбинации. В некоторых вариантах осуществления изобретения, буферная система может составлять приблизительно 5-1000 мМ, приблизительно 60-200 мМ, приблизительно 100-300 мМ, приблизительно 125-275 мМ, приблизительно 150-250 мМ, приблизительно 175-225 мМ, приблизительно 180-210 мМ, приблизительно 185-200 мМ, приблизительно 185-195 мМ или приблизительно 190-195 мМ в виросомном концентрате. Если буферная система представляет собой HEPES-хлорид натрия в виросомном концентрате, то хлорид натрия может составлять приблизительно 5-1000 мМ, приблизительно 50-150 мМ, приблизительно 125-175 мМ, приблизительно 130-160 мМ, приблизительно 130-150 мМ, приблизительно 135-145 мМ или приблизительно 140-145 мМ в виросомном концентрате. Если буферная система представляет собой HEPES-хлорид натрия в вировом концентрате, то HEPES может составлять приблизительно 1-200 мМ, приблизительно 10-75 мМ, приблизительно 10-50 мМ, приблизительно 25-75 мМ, приблизительно 30-70 мМ, приблизительно 40-60 мМ, приблизительно 45-55 мМ или приблизительно 48-52 мМ в виросомном концентрате.
[0057] Жидкий виросомный концентрат может также включать по меньшей мере один криолиопротектор. Виросомы могут быть повреждены во время стадий замораживания и/или лиофилизации, проводимых для получения лекарственных форм, описанных в настоящей заявке. Так, например, криолиопротектор может быть включен в виросомный концентрат для улучшения сохранения виросомы во время стадий замораживания и/или лиофилизации. Примеры криолиопротекторов включают, но не ограничиваются ими, полиолы, такие как трегалоза, сахара, такие как сахароза, и аминокислоты, такие как лизин, олигосахариды, такие как инулин (олигосахарид со средней длиной цепи), или их комбинации. Используемые здесь криолиопротекторы могут быть инертными по отношению к составу вакцины. Жидкий виросомный концентрат может включать приблизительно 1-20% масс./масс. приблизительно 1,5-10% масс./масс., приблизительно 2-10% масс./масс., приблизительно 4-10% масс./масс., приблизительно 2-9% масс./масс., приблизительно 2-5% масс./масс., приблизительно 3-8% масс./масс., приблизительно 3,5-8% масс./масс., приблизительно 3,5-7% масс./масс., приблизительно 4-8% масс./масс. или приблизительно 5-7% масс./масс. криолиопротектора.
[0058] Виросомный концентрат может составлять приблизительно 1-75% масс./масс. приблизительно 10-65% масс./масс. приблизительно 15-60% масс./масс. приблизительно 20-55% масс./масс. приблизительно 20-50% масс./масс. или приблизительно 25-50% масс./масс. виросомной композиции. В некоторых вариантах осуществления изобретения, виросомный концентрат может составлять приблизительно 15-35% масс./масс. приблизительно 20-30% масс./масс. приблизительно 23-27% масс./масс. или приблизительно 25% масс./масс. виросомной композиции.
Состав основной матрицы
[0059] Состав основной матрицы формирует структуру конечной лекарственной формы. Так, например, состав основной матрицы может включать вещество, образующее матрицу. Вещество, образующее матрицу, может обеспечивать сетевую структуру дозированной формы, которая придает прочность и упругость при работе с ней. Подходящие вещества, образующие матрицу, могут включать, но не ограничиваются ими, желатин, крахмал или их комбинации. Дополнительные вещества, образующие матрицу, можно найти в ЕР 2624815 В1, которая в полном объеме включена в настоящее описание посредством ссылки. Желатин может представлять собой рыбий желатин, бычий желатин, свиной желатин или их комбинацию. Каждый из этих желатинов может иметь различные гелеобразующие свойства. Степень образования геля из раствора желатина может зависеть от концентрации желатина и температуры раствора желатина. Раствор бычьего желатина имеет тенденцию к образованию геля при температурах ниже 18°С и таким образом может рассматриваться как гелеобразующий желатин. И напротив, рыбий желатин может оставаться в растворе при температурах по меньшей мере 10°С и таким образом может рассматриваться как негелеобразующий желатин. В некоторых вариантах осуществления изобретения, желатин может представлять собой желатин с низким содержанием эндотоксина, такой как желатин, происходящий от определенного источника или полученный способом, описанным в Предварительной заявке No 62/640394, которая в полном объеме включена в настоящее описание посредством ссылки. В некоторых вариантах осуществления изобретения, количество вещества, образующего матрицу, в виросомной композиции может составлять приблизительно 1-15% масс./масс., приблизительно 2-12% масс./масс., приблизительно 3-10% масс./масс. приблизительно 4-8% масс./масс. приблизительно 4-6%, приблизительно 5-7% масс./масс. или приблизительно 6% масс./масс.
[0060] Температура, при которой дозируется виросомная композиция, может составлять по меньшей мере 10-18°С. Так, например, композиция, в которой используется только бычий желатин, может не дозироваться при этих низких температурах. Однако, может быть использована комбинация бычьего желатина и желатина другого типа (например, рыбьего желатина). Заявителелями было обнаружено, что с использованием рыбьего желатина может быть получена лиофилизованная таблетка с прочной матричной структурой и временем дезинтеграции приблизительно 30-180 или 30-60 секунд, что желательно для обеспечения достаточного времени контактирования со слизистой оболочкой рта. Кроме того, с использованием рыбьего желатина могут быть изготовлены лиофилизованные лекарственные формы с хорошими физическими свойствами для получения составов препарата, которые имеют высокую загрузку растворимого компонента, такого как буферные соли, например, в описанных здесь количествах.
[0061] В некоторых вариантах осуществления изобретения, рыбий желатин может представлять собой высокомолекулярный рыбий желатин, рыбий желатин со стандартной молекулярной массой или их комбинации. Высокомолекулярный рыбий желатин определяется как рыбий желатин, в котором более 50% распределение молекулярной массы превышает 30000 дальтон. Рыбий желатин со стандартной молекулярной массой определяется как рыбий желатин, в котором более 50% распределение молекулярной массы составляет ниже 30000 дальтон.
[0062] В некоторых вариантах осуществления изобретения, вещество, образующее матрицу, может также служить в качестве стабилизатора для антигенов, а также мукоадгезива. Кроме того, крахмал может также служить в качестве иммуностимулирующего наполнителя.
[0063] Состав основной матрицы может также включать вещество, формирующее структуру. Подходящие вещества, формирующие структуру, могут включать сахара, включая, но не ограничиваясь ими, маннит, декстрозу, лактозу, галактозу, циклодекстрин или их комбинации. Вещества, формирующие структуру, могут быть использованы для лиофилизации в качестве наполнителя, если он кристаллизуется для обеспечения структурной устойчивости лиофилизованного продукта. Растворимые наполнители, такие как буферные соли и трегалоза в виросомной композиции, могут ингибировать ее кристаллизацию. Для обеспечения кристаллизации обычно проводят гибридизацию в течение продолжительного периода времени. Однако, присутствие этих растворимых наполнителей может также вызвать плавление замороженного продукта во время отжига. Так, например, заявителями был установлен баланс между количеством вещества, формирующего структуру, буферными солями и криолиопротектором и условиями отжига (то есть, температурой и временем). В некоторых вариантах осуществления изобретения, количество вещества, формирующего структуру, в виросомной композиции может составлять приблизительно 1-20% масс./масс. приблизительно 3-15% масс./масс. приблизительно 4,5-10% масс./масс. приблизительно 4,5-8% масс./масс. приблизительно 5-10% масс./масс. приблизительно 6-10% масс./масс. приблизительно 7-9% масс./масс. или приблизительно 8% масс./масс. Заявителями было обнаружено, что при значениях для вещества, формирующего структуру, ниже 4,5% масс./масс., во время лиофилизации может наблюдаться некоторое разрушение микроструктуры, что будет приводить к плохому диспергированию/плохой дезинтеграции лиофилизованной лекарственной формы. Так, например, было установлено, что большее количество вещества, формирующего структуру, будет минимизировать или устранять разрушение микроструктуры без какого-либо значительного воздействия на виросому.
[0064] Кроме того, состав основной матрицы может также включать криолиопротектор. Примеры криолиопротекторов включают, но не ограничиваются ими, полиолы, такие как трегалоза, сахара, такие как сахароза, и аминокислоты, такие как лизин, олигосахариды, такие как инулин (олигосахарид со средней длиной цепи), или их комбинации. Криолиопротектор может быть использован для защиты виросомы от повреждения во время последующего замораживания и лиофилизации. Однако, добавление криолиопротектора может вызывать разрушение микроструктуры матрицы лекарственной формы во время лиофилизации. Так, например, для сведения к минимуму разрушения микроструктуры и в то же время сохранения достаточного количества виросом для поддержания качества виросом в целях индукции иммунного ответа должен быть найден определенный баланс. Количество криолиопротектора в составе основной матрицы может составлять приблизительно 0,01-2% масс./масс., приблизительно 0,1-1,5% масс./масс., приблизительно 0,2-1% масс./масс. или приблизительно 0,25-0,75% масс./масс. Так, например, суммарное количество (далее называемое «общим количеством») криолиопротектора в виросомной композиции (то есть, жидкого виросомного концентрата плюс состава основной матрицы) может составлять приблизительно 0,5-6% масс./масс., приблизительно 0,5-5% масс./масс., приблизительно 0,5-4,5% масс./масс., приблизительно 1-4,5% масс./масс., приблизительно 1,5-4,5% масс./масс., приблизительно 1,5-3% масс./масс., приблизительно 1,5-2,5, приблизительно 2-3% масс./масс., приблизительно 2,5% масс./масс. или приблизительно 2% масс./масс. Заявителями было также обнаружено, что при этих уровнях, криолиопротектор может обеспечивать достаточную криолиозащиту и не приводить к нежелательному разрушению микроструктуры во время лиофилизации.
[0065] В некоторых вариантах осуществления изобретения, состав основной матрицы может также включать мукоадгезивный агент, такой как смола. Подходящие смолы включают, но не ограничиваются ими, аравийскую камедь, гуаровую камедь, агар, ксантан, геллан, карагенан, створаживающий агент, коньяк, бобы рожкового дерева, веллан, трагакантовую камедь, аравийскую камедь, камедь карайи, камедь гхатти, пектины, декстран, глюкоманнан и альгинаты, или их комбинации.
[0066] Состав основной матрицы может также содержать дополнительные фармацевтически приемлемые агенты или наполнители. Такие дополнительные фармацевтически приемлемые агенты или наполнители включают, но не ограничиваются ими, сахара, такие как маннит, декстроза и лактоза, неорганические соли, такие как хлорид натрия и силикаты алюминия, желатины млекопитающих, рыбий желатин, модифицированные крахмалы, консерванты, антиоксиданты, поверхностно-активные вещества, усилители вязкости, усилители проницаемости, красители, ароматизаторы, модификаторы рН, подсластители, агенты, маскирующие вкус, и их комбинации. Подходящие красящие агенты могут включать красный, черный и желтый оксиды железа и красители FD & C, такие как синий FD & C No 2 и красный FD & C No 40, и их комбинации. Подходящие ароматизаторы могут включать мятные, малиновые, лакричные, апельсиновые, лимонные, грейпфрутовые, карамельные, ванильные, вишневые и виноградные ароматизаторы и их комбинации. Подходящие модификаторы рН могут включать лимонную кислоту, винную кислоту, фосфорную кислоту, хлористоводородную кислоту, малеиновую кислоту, гидроксид натрия (например, 3% масс./масс. раствора гидроксида натрия) и их комбинации. В некоторых вариантах осуществления изобретения, состав основной матрицы и/или виросомная композиция содержит модификатор рН в количестве, достаточном для поддержания нужного рН приблизительно 6-9, приблизительно 7-8, приблизительно 7,2-7,6, приблизительно 7,3-7,5 или приблизительно 7,4. Подходящие подсластители могут включать аспартам, ацесульфам К и тауматин и их комбинации. Специалист в данной области может легко определить подходящие количества этих различных дополнительных наполнителей, если это необходимо.
[0067] Состав основной матрицы может также включать растворитель. В некоторых вариантах осуществления изобретения, растворителем может быть вода (например, очищенная вода). В некоторых вариантах осуществления изобретения, все остальное в составе основной матрицы и/или в виросомной композиции, представляет собой растворитель.
[0068] Состав основной матрицы может составлять приблизительно 25-99% масс./масс. приблизительно 35-90% масс./масс. приблизительно 40-85% масс./масс. приблизительно 45-80% масс./масс. или приблизительно 50-75% масс./масс. от виросомной композиции. В некоторых вариантах осуществления изобретения, состав основной матрицы может составлять приблизительно 65-85% масс./масс. приблизительно 70-80% масс./масс. приблизительно 73-77% масс./масс. или приблизительно 75% масс./масс. от виросомной композиции.
Получение лекарственной формы, содержащей виросомную композицию
[0069] Как указано выше, жидкий виросомный концентрат смешивают с составом основной матрицы для получения виросомной композиции на стадии 101, подходящей для проведения лиофилизации. На фигуре 2 представлено более подробное описание способа получения описанной здесь вакцинной лекарственной формы. В некоторых вариантах осуществления изобретения, состав основной матрицы может быть получен путем растворения вещества, образующего матрицу, и вещества, формирующего структуру, в растворителе с образованием премикса. Так, например, желатин и маннит могут быть растворены в воде как показано на стадии 201 на фигуре 2. Премикс может быть нагрет приблизительно до 40-−80°C, приблизительно до 50-70°C, приблизительно до 55-65°С или приблизительно до 60°С и может поддерживаться в течение приблизительно 45-75 минут, приблизительно 55-65 минут или приблизительно 60 минут. Как показано на стадии 202, премикс может быть нагрет до 60°С и может поддерживаться в течение 1 часа. Затем премикс может быть охлажден приблизительно до 30-50°С, приблизительно до 35-45°С или приблизительно до 40°С и просеян перед охлаждением приблизительно до 10-20°C или приблизительно до 15°С и может поддерживаться при этой температуре в течение остального времени проведения этого процесса. Как показано на стадии 203, премикс может быть охлажден до 40°С и просеян. Затем премикс может быть охлажден до 15°С, как показано на стадии 204.
[0070] Затем к премиксу может быть добавлен криолиопротектор. Так, например, к премиксу может быть добавлена трегалоза, как показано на стадии 205. Затем, рН может быть доведен приблизительно до 6-9, приблизительно до 7-8, приблизительно до 7,2-7,6, приблизительно до 7,3-7,5 или приблизительно до 7,4 с использованием модификатора рН. Так, например, рН может быть доведен до 7,4 с использованием раствора гидроксида натрия, как показано на стадии 206. После коррекции рН может быть добавлен жидкий виросомный концентрат. После добавления жидкого виросомного концентрата, рН может быть снова скорректирован (на стадии 207) и, если это необходимо, доведен приблизительно до 6,0-8,5, приблизительно до 7-8, приблизительно до 7,2-7,6, приблизительно до 7,3-7,5 или приблизительно до 7,4 с использованием дополнительного модификатора рН. Эта смесь может быть приготовлена до требуемого размера партии с растворителем (то есть, с получением виросомной композиции). Так, например, при необходимости, к смеси может быть добавлено некоторое количество воды, как показано на стадии 208.
[0071] На стадии 102, как показано на фигуре 1, жидкая виросомная композиция может быть дозирована в предварительно сформованной форме. Используемый здесь термин «дозированный» относится к осаждению предварительно определенной аликвоты раствора или суспензии. Используемый здесь термин «предварительно сформованная форма» относится к любому подходящему контейнеру или сосуду, в которых могут осаждаться водный раствор или суспензия, и в которых впоследствии может быть проведена лиофилизация. В некоторых вариантах осуществления изобретения, предварительно сформованная форма представляет собой блистерную упаковку с одним или более блистерными карманами. Предварительно определенные аликвоты в количестве приблизительно 150-1000 мг или приблизительно 500 мг массы приготовленной дозы во влажном состоянии (также упоминаемой здесь как масса приготовленной дозы) виросомной композиции могут быть приготовлены в виде предварительно сформованных форм. В некоторых вариантах осуществления изобретения, виросомная композиция может быть дозирована приблизительно при 10-20°C мли приблизительно при 15°С. Так, например, виросомная композиция может быть дозирована при 15°С с массой приготовленной дозы 500 мг, как показано на стадии 209.
[0072] На стадии 103, показанной на фигуре 1, дозированные виросомные композиции могут быть затем заморожены в предварительно сформованных формах. Дозированные виросомные композиции в предварительно сформованных формах могут быть заморожены любым способом, известным специалистам в данной области. Так, например, композиции могут быть пропущены через криогенную камеру (например, через туннель с жидким азотом). Температура во время замораживания может составлять приблизительно от -50 до -100°С, приблизительно от -60 до -90°С, приблизительно от -60 до −80°C, приблизительно от -65 до -75°С или приблизительно -70°C. Время замораживания может составлять приблизительно от 1,5 до 5 минут, приблизительно 2-4,5 минут, приблизительно 2,5-4 минут, приблизительно 3-4 минут, приблизительно 3-3,5 минут или приблизительно 3,25 минут. Так, например, дозированная виросомная композиция может быть заморожена при -70°C в течение 3 минут и 15 секунд, как показано на стадии 210.
[0073] На стадии 104, показанной на фигуре 1, замороженные унифицированные дозы в предварительно сформованных формах могут быть собраны и помещены в холодильник при температуре менее, чем приблизительно -25°С, приблизительно -20°C, приблизительно -15°С, приблизительно -10°С, приблизительно -5°С и подвергнуты отжигу (то есть, в замороженном состоянии) в течение периода времени, достаточного для кристаллизации вещества, формирующего структуру. В результате кристаллизации вещества, формирующего структуру, могут быть получены замороженные унифицированные дозы со структурной прочностью, подходящей для предотвращения разрушения замороженных унифицированных доз во время лиофилизации. Это может иметь важное значение для целостности виросомы. Время отжига может составлять приблизительно 3-9 часов, приблизительно 4-8 часов, приблизительно 5-7 часов или приблизительно 6 часов. Так, например, замороженные унифицированные дозы могут быть подвергнуты отжигу при температуре ниже -15°С в течение приблизительно 3-9 часов, как показано на стадии 211.
[0074] После отжига, гибридизованные замороженные унифицированные дозы могут быть лиофилизованы на стадии 105 с образованием лекарственной формы. Во время процесса лиофилизации, вода сублимируется из замороженных унифицированных доз. В некоторых вариантах осуществления изобретения, замороженные унифицированные дозы могут быть загружены на полки лиофилизатора. В некоторых вариантах осуществления изобретения, лиофилизатор может быть предварительно охлажден до температуры приблизительно от -15 до -35°C, приблизительно от -20 до -30°C или приблизительно -25°С. После помещения гибридизованных замороженных унифицированных доз в лиофилизатор может быть запущен цикл лиофилизации. В некоторых вариантах осуществления изобретения, после начала цикла лиофилизации, вакуум может быть сброшен, и температура на полке будет увеличиваться. Лиофилизатор может работать при низком давлении (то есть, в вакууме). В некоторых вариантах осуществления изобретения, лиофилизатор может работать под давлением приблизительно менее или равном 1000 мбар, приблизительно 900 мбар, приблизительно 800 мбар, приблизительно 700 мбар, приблизительно 600 мбар, приблизительно 500 мбар или приблизительно 400 мбар.
[0075] Заявителями было обнаружено, что двухстадийный цикл лиофилизации (стадия 212) может обеспечивать структурную устойчивость лекарственной формы с минимальным повреждением виросомы в лекарственной форме. Двухстадийный цикл лиофилизации может включать первую стадию выдерживания замороженных унифицированных доз при температуре приблизительно от -5°С до -25°С, приблизительно от -10°С до -20°C, приблизительно от -13°С до -17°С или приблизительно -15°С в течение приблизительно 12-36 часов, приблизительно 18-30 часов, приблизительно 20-28 часов или приблизительно 24 часов. Кроме того, двухстадийный цикл лиофилизации может включать вторую стадию, которая следует за первой стадией. Вторая стадия может включать выдерживание замороженных унифицированных доз при температуре приблизительно от 0°С до -20°C, приблизительно от -5°С до приблизительно -15°С, приблизительно от -8°С до приблизительно -12°С или приблизительно -10°С в течение приблизительно 6-30 часов, приблизительно 12-24 часов, приблизительно 14-22 часов или приблизительно 18 часов. По окончании двухстадийного цикла лиофилизации, температура лиофилизатора может быть повышена до температуры окружающей среды (то есть, приблизительно до 20-25°С или приблизительно 23°С).
[0076] В некоторых вариантах осуществления изобретения, двухстадийный способ лиофилизации может включать предварительное охлаждение лиофилизатора приблизительно до -25°С, повышение температуры лиофилизатора в течение 2 часов до -15°С, выдерживание лиофилизатора при -15°С в течение 24 часов, повышение температуры лиофилизатора до -10°С в течение 2 часов, выдерживание при -10°С в течение 18 часов, повышение температуры до 0°С в течение 15 минут и повышение температуры до 23°С в течение 15 минут в указанном порядке.
[0077] Лиофилизованные лекарственные формы могут быть удалены из лиофилизатора и оценены на какие-либо дефекты (путем проверки качества, как описано ниже) на стадии 213. Затем лекарственные формы могут быть помещены в шкаф для хранения при влажности воздуха приблизительно менее 35% RH до тех пор, пока лекарственные формы не будут герметизированы в предварительно сформованных формах. Процесс герметизации (стадия 214) может быть проведен путем их заворачивания в герметичную фольгу на предварительно отформованных формах с получением блистерных упаковок с лиофилизованными лекарственными формами.
[0078] Вода в лиофилизованных лекарственных формах может быть удалена путем сублимации во время лиофилизации. В соответствии с этим, остаток виросомного концентрата в лиофилизованной лекарственной форме, за исключением криолиопротекторов (то есть, виросом, антигенов, адъювантов и буферной системы, оставшихся в лиофилизованном виросомном концентрате), может составлять приблизительно 1-5 масс.%, приблизительно 2-4 масс.%, приблизительно 2,5-3,5 масс.%, приблизительно 2,6-3,4 масс.%, приблизительно 2,7-3,3 масс.%, приблизительно 2,8-3,2 масс.%, приблизительно 2,9-3,1 масс.% или приблизительно 3-3,1 масс.% лекарственной формы.
[0079] Как указано выше, молекула(ы)-мишень(и) включена(ы) в описанные здесь лекарственные формы в количестве, которое является достаточным для сообщения иммуногенности при ее введении в лекарственной форме. Специалист в данной области может легко определить иммуногенное количество для данного заболевания или инфекции исходя, среди прочих факторов, из пути введения, возраста и массы пациента, которому будет введена эта лекарственная форма. В некоторых вариантах осуществления изобретения, твердая лекарственная форма может содержать 0,01-250 мкг каждой молекулы-мишени (например, пептида P1 и/или rgp41 ВИЧ-1).
[0080] В некоторых вариантах осуществления изобретения, по меньшей мере один из криолиопротекторов в лиофилизованной лекарственной форме может составлять приблизительно 5-20 масс. %, приблизительно 8-18 масс. %, приблизительно 10-15 масс. %, приблизительно 11-15 масс. % или приблизительно 12-15 масс. % лекарственной формы. В некоторых вариантах осуществления изобретения, по меньшей мере один из криолиопротекторов в лиофилизованной лекарственной форме может составлять приблизительно 1-5 масс.%, приблизительно 1-4 масс.% или приблизительно 2-4 масс.% лекарственной формы.
[0081] В некоторых вариантах осуществления изобретения, количество вещества, образующего матрицу, в лекарственной форме может составлять приблизительно 20-50 масс. %, приблизительно 25-45 масс. %, приблизительно 25-40 масс. %, приблизительно 30-40 масс. %, приблизительно 33-37 масс. % или приблизительно 35-37 масс. %. В некоторых вариантах осуществления изобретения, количество вещества, формирующего структуру, в лекарственной форме может составлять приблизительно 27-65 масс. %, приблизительно 27-60 масс. %, приблизительно 40-55 масс. % или приблизительно 45-50 масс. %. В некоторых вариантах осуществления изобретения, остаточное количество модификатора рН в лиофилизованной дозе (например, гидроксида натрия) может составлять приблизительно 0,01-0,08 масс. %.
[0082] Лекарственные формы согласно изобретению представляют собой растворяющиеся лекарственные формы, и в соответствии с этим, имеют явное преимущество, заключающееся в более быстром времени дезинтеграции. Способ введения может быть пероральным, вагинальным или интраназальным, хотя предпочтительным является способ перорального введения (то есть, подъязычного и/или трансбуккального введения). Лекарственная форма, после ее помещения в ротовую полость и контактирования со слюной, может дезинтегрироваться в течение периода времени приблизительно от 1 до приблизительно 180 секунд, приблизительно от 1 до приблизительно 120 секунд, приблизительно от 1 до приблизительно 60 секунд, а предпочтительно, в течение приблизительно от 1 до приблизительно 30 секунд, более предпочтительно, в течение приблизительно от 1 до приблизительно 10 секунд, а наиболее предпочтительно, менее, чем приблизительно 5 секунд.
Композиции, методы испытаний и примеры
[0083] В качестве примеров был использован жидкий виросомный концентрат, полученный из вируса гриппа. Виросомы содержали HA вируса гриппа, а также добавленные антигены и адъюванты. Было получено два виросомных препарата, каждый из которых содержит один антиген, происходящий от гликопротеина оболочки ВИЧ. Жидкий виросомный концентрат представляет собой смесь двух виросомных препаратов. Два антигена, происходящие от gp41 ВИЧ, представляют собой пептид P1, имеющий последние 35 C-концевых эктодоменных остатков и усеченный rgр41, не содержащий кластера I и последнего 21-го C-концевого эктодоменного остатка. Адъювант 3M-052 либо присутствовал, либо отсутствовал в любом виросомном препарате. Виросомы суспендировали в буфере HEPES- хлорид натрия, содержащем 142,5 мМ хлорида натрия и 50 мМ HEPES при рН 7,4. Кроме того, трегалоза (крилиопротектор) была протестирована в концентрации 0-10% масс./масс. жидкого виросомного концентрата. В нижеследующей Таблице 1 систематизированы целевые композиции жидкого виросомного концентрата ВИЧ-1, используемого в некоторых проведенных авторами экспериментах, во время которых аликвоту 500 мг (массы приготовленной дозы) водной виросомной композиции отмеряли и помещали в карманы предварительно сформованной блистерной упаковки с последующим замораживанием и лиофилизацией. Масса приготовленной дозы может составлять в пределах от 150 мг до 1000 мг, а композиции жидкого виросомного концентрата ВИЧ-1 могут быть скорректированы так, чтобы они соответствовали требуемой целевой дозе. Следует отметить, что нижеследующие целевые интервалы могут зависеть от цели и последующего использования, например, для исследований на животных или для исследований с участием человека. Таким образом, для других целей могут быть использованы другие нужные концентрации.
Таблица 1
- Молекула-мишень (антиген (rgp41): 50-400 мкг/мл
- Наполнитель HA: 10-160 мкг/мл
- Адъювант (например, 3M-052): 8-140 мкг/мл
- Фосфолипиды: 0,5-5 мг/мл
- Хлорид натрия: 50-150 мМ
- HEPES 10-50 мМ
- Трегалоза: 4-10% масс./масс.
- pH 6,5-8,0
- Молекула-мишень (антиген (rgp41): 25-200 мкг/мл
- Наполнитель HA: 5-80 мкг/мл
- Адъювант (например, 3M-052): 4-70 мкг/мл
- Фосфолипиды: 0,5-5 мг/мл
- Хлорид натрия: 50-150 мМ
- HEPES 10-50 мМ
- Трегалоза: 2-5% масс./масс.
- pH 6,5-8,0
- Адъювант (например, 3M-052): 8-140 мкг/мл
- Фосфолипиды: 0,5-5 мг/мл
- Хлорид натрия: 50-150 мМ
- HEPES 10-50 мМ
- Трегалоза: 4-10% масс./масс.
- pH 6,5-8,0
[0084] В некоторых из проведенных авторами экспериментов, целевая доза для HA и антигенов ВИЧ-1 для каждой таблетки составляла 20 мкг HA, 25 мкг P1 и 50 мкг rgp41. Для получения этих доз требуется высокая полезная нагрузка жидких виросомных концентратов в комбинации с большой массой приготовленной дозы во влажном состоянии. В Таблице 2 приведены различные комбинации загрузки жидкого виросомного концентрата (в пределах от 25 до 50% масс./масс.) с массой приготовленной дозы во влажном состоянии (в пределах от 250 до 1000 мг). Масса приготовленной дозы во влажном состоянии представляет собой количество аликвоты виросомной композиции, которое было отмерено на дозу перед лиофилизацией.
Таблица 2
100 мкг/мл P1
200 мкг/мл rgp41
200 мкг/мл P1
400 мкг/мл rgp41
[0085] 25%-ная загрузка жидкого виросомного концентрата может быть добавлена к составу основной матрицы. Масса приготовленной дозы может составлять 500 мг. В Таблице 3 указаны интервалы оцененного содержания НА и антигена.
Таблица 3
P1: 50-450 мкг/мл
rgp41: 50-400 мкг/мл
3M-052: 8-140 мкг/мл
P1: 40-100 мкг/мл
rgp41: 70-230 мкг/мл
3M-052: 16-65 мкг/мл
P1: 10-25 мкг/мл
rgp41: 17,5-57,5 мкг/мл
3M-052: 4-16,3 мкг/мл
[0086] Присутствие буфера в водной композиции может снижать температуру замерзания, что затрудняет замораживание состава композиции и сохранение ее в замороженном состоянии. Кроме того, разрушение матричной структуры таблетки может также происходить во время лиофилизации, поскольку буферные соли могут подавлять кристаллизацию маннита во время отжига. Для обеспечения прочности и целостности структуры матрицы таблеток в целях предотвращения разрушения структуры требуется кристаллизация маннита. Однако кристаллизация маннита может повреждать виросомные частицы во время замораживания, отжига и лиофилизации. Более низкая процентная загрузка жидкого виросомного концентрата (например, 25% загрузка) приводит к уменьшению такого повреждения. Может быть также рассмотрена комбинация более низкого процентного содержания жидкого виросомного концентрата и большей массы приготовленной дозы.
[0087] Высокое содержание приготовленной дозы виросомной композиции во влажном состоянии в комбинации с композицией препарата с высоким содержанием буфера может также еще больше затруднять замораживание и сохранение структуры для минимизации разрушения во время лиофилизации. Однако, более крупные таблетки (например, с массай приготовленной дозы 1000 мг) могут покрывать большую площадь поверхности и потенциально могут улучшать прохождение виросомы. Если требуется высокая масса приготовленной дозы во влажном состоянии, то предпочтительно, чтобы композиция препарата имела низкое содержание буфера.
[0088] В Таблице 4 (Композиция 1) систематизированы водные композиции виросомного препарата и соответствующая композиция для оцененной здесь таблетированной виросомы. Нижеследующие композиции и таблетки получали в соответствии со стадиями, показанными и описанными на фигуре 2. Кроме того, замороженные композиции подвергали двухстадийному процессу лиофилизации в вакууме под давлением 500 мбар, включающему: (a) предварительное охлаждение до -25°С; (b) увеличение температуры до -15°С в течение 2 часов; (с) выдерживание при -15°С в течение 24 часов; (d) увеличение температуры до -10°С в течение 2 часов; (е) выдерживание при -10°С в течение 18 часов; (f) увеличение температуры до 0°C в течение 15 минут; (g) увеличение температуры до 23°C в течение 15 минут. Затем вакуум сбрасывали и давление в лиофилизаторе снова доводили до атмосферного давления.
[0089] Концентрация каждого ингредиента (% масс./масс.) представляет собой количество до удаления воды, присутствующей в жидком виросомном концентрате, раствора гидроксида натрия и воды, используемой для приготовления препарата посредством сублимации во время лиофилизации. Кроме того, в нижеследующей таблице указаны возможные интервалы количеств для каждого ингредиента.
Таблица 4
масс. оцененного интервала
масс. композиции 1/2
(дост. кол-во до pH 7,4)
[0090] Как указано выше, жидкий виросомный концентрат включает виросомы, суспендированные в буферной системе, состоящей из 142,5 мМ NaCl и 50 мМ HEPES с 5% масс./масс. трегалозы. Для 25% загрузки виросомной композиции с массой приготовленной дозы 500 мг (то есть, 125 мг жидкого виросомного концентрата), количество сухого вещества с водой, сублимированной из виросом, антигенов, адъювантов, NaCl и HEPES (за исключением трегалозы) составляет приблизительно 2,5-2,6 мг.
[0091] ** Означает суммарное количество трегалозы в виросомной композиции и в лекарственной форме. Так, например, такое количество включает количество трегалозы в жидком виросомном концентрате, а также количество трегалозы, добавленной из состава основной матрицы.
Свойства лиофилизованных лекарственных форм:
[0092] Лиофилизованные лекарственные формы могут быть стабильными по своим физическим свойствам и качеству виросом (по гранулометрическим свойствам и содержанию антигена) и могут храниться независимо от условий хранения в холодовой цепи. Кроме того, лиофилизованные лекарственные формы могут сообщать виросомам резистентность к случайному воздействию условий хранения при температуре ниже нуля во время хранения или транспортировки.
Физические свойства лекарственной Формы
[0093] Были приготовлены лиофилизованные таблетки с приемлемыми физическими свойствами. Физические свойства таблеток включают внешний вид, дисперсионный свойства, время дезинтеграции и содержание влаги.
[0094] Был протестирован внешний вид десяти лиофилизованных таблеток. Каждая таблетка была взята из блистерной упаковки. Был проведен визуальный осмотр каждой таблетки на дефекты поверхности и основания таблетки. В качестве критерия служит хороший внешний вид лиофилизованной таблетки и отсутствие каких-либо дефектов на ее поверхности. Кроме того, таблетки должны обладать достаточной прочностью при их удалении из блистерного кармана без повреждения.
[0095] Дисперсионные свойства (тест in vitro). Было протестировано минимум 5 таблеток. Сначала приготавливали химический стакан, содержащий приблизительно 200 мл очищенной воды при 20 ± 0,5°С. Каждую таблетку затем удаляли из блистерной упаковки и помещали на поверхность воды основанием вниз. Время определено как время, за которое каждая таблетка становилась полностью влажной или диссоциировалась. Смачивавание означает период времени, за который таблетка становилась полностью влажной. Смачивание таблетки может происходить в определенных участках, которые, в конечном счете, сливаются друг с другом, в результате чего вся таблетка становится влажной. Тест на диспергирование считается завершенным, если центр таблетки представляет собой увлажненную массу. Таким образом, время смачивания определяется с того момента, когда смачивается центр таблетки, поскольку эта часть таблетки является самой толстой. Время смачивания регистрировали для каждой из пяти таблеток. Максимальное время для каждого теста составляло 60 секунд. Более длительное время может быть зарегистрировано просто как время, составляющее более 60 секунд. Время диссоциации означает время, необходимое для полного разрушения таблетки. Это время может быть зарегистрировано после того, как таблетка начнет распадаться по краям. Время диссоциации регистрировали для каждой из пяти таблеток. Максимальное время для каждого теста составляло 60 секунд. Более длительное время может быть зарегистрировано как время, составляющее более 60 секунд. Иногда таблетка не будет полностью смачиваться или полностью диссоциироваться в этом временном интервале. Иногда таблетка может иметь плотные комки; а иногда, она может вообще не смачиваться на поверхности. Кроме того, вся таблетка может быть покрыта твердой оболочкой. Следует отметить, что если это происходит, то в данном описании приводится указание на «плотные комки» или «остатки оболочки». Образование «плотных комков» и/или «оболочки» может быть показателем разрушения микроструктуры во время лиофилизации. На фигурах 3A-C показано упрощенное представление трех возможных недиспергированных состояний таблеток с видом сбоку и видом сверху, когда они появляются в воде. На фотографиях фигур 3A-C показаны некоторые репрезентативные таблетки для тех же категорий. Критерием для прохождения теста на дисперсионные свойства является то, что 5 таблеток будут быть полностью увлажнены и/или диссоциированы с образованием пальпируемой массы без плотных комков и/или оболочки в течение 60 секунд или менее. В некоторых вариантах осуществления изобретения, описанные здесь лекарственные формы могут быть полностью увлажнены и/или диссоциированы с образованием пальпируемой массы без плотных комков и/или оболочки в течение 60 секунд или менее.
[0096] Время дезинтеграции (тест in vitro): Для этого теста использовали шесть таблеток. Шесть химических стаканов заполняли очищенной водой и помещали на водяную баню, регулируемую при 37°C ± 0,5°С. Затем каждую таблетку удаляли из блистерной упаковки. Затем на каждую из шести таблеток осторожно помещали проволочный зажим. При этом должно гарантироваться, что этот зажим будет захватывать таблетку, не вызывая повреждений. Затем проводили тест, как описано в Фармакопее. Примером такого теста является тест на дезинтеграцию в соответствии с Фармакопеей США (701). Максимальное время дезинтеграции регистрировали для каждой таблетки. Критерием для теста на время дезинтеграции является то, что время дезинтеграции не должно превышать 60 секунд для каждой из шести таблеток. В некоторых вариантах осуществления изобретения, описанные здесь лекарственные формы могут иметь время дезинтеграции менее 60 секунд.
[0097] Содержание влаги. Для определения содержания воды в таблетке использовали кулонометр Карла Фишера Methron 831 с процессором для подачи образца 744 в печь (Metrohm, Herisau, Switzerland). Таблетку точно взвешивали и помещали в стеклянный сосуд. Сосуд сразу герметично закрывали гофрированной крышкой для гарантии отсутствия проникновения влаги. Затем сосуд с образцом помещали в процессор для образцов 744, загружаемый в печь, и устанавливали температуру 102°С. Испарившуюся влагу титровали с использованием реагента Hydranal Coulometric AG Oven для определения количества высвобождаемой воды. Тест проводили с тремя повторностями и регистрировали среднее значение. Критерием для оценки содержания влаги является содержание влаги в лиофилизованной лекарственной форме, составляющее менее, чем приблизительно 8%, предпочтительно менее, чем 6%, а более предпочтительно менее, чем 4%. В некоторых вариантах осуществления изобретения, описанные здесь лекарственные формы могут иметь содержание влаги менее, чем приблизительно 8%, предпочтительно менее, чем 6%, а более предпочтительно менее, чем 4%.
Свойства виросом:
[0098] Лиофилизованные лекарственные формы, которые содержат виросомы, могут быть в достаточной степени сохранены с точки зрения содержания интактных виросом в исходной жидкой виросомной популяции, их размера частиц и содержания поверхностных антигенов, необходимых для сообщения виросомамиммуногенности и иммунологической эффективности. Виросомная структура может быть разрушена посредством лиофилизации, проводимой для получения лекарственных форм. Так, например, свойства виросомных частиц оценивают в растворе разведенной лиофилизованной таблетки. Виросомы могут существовать в виде интактных отдельных частиц и в виде кластеров различных размеров, причем, все они являются иммуногенными, но подвержены различному воздействию на них антигена/эпитопа и обладают различной способностью к переходу через подъязычный барьер. Виросомы могут быть охарактеризованы с точки зрения (а) среднего размера частиц и (b) степени сохранения виросом (интактных виросом).
[0099] Оценку размера частиц и концентрации частиц виросом (количества частиц виросом на мл) проводили методом NTA: анализ путем мониторинга пути наночастиц (NTA; на приборе NanoSight NS300) представляет собой чувствительный метод, который может идентифицировать различные размеры частиц, присутствующих в растворе в пределах 30-1000 нм. Частицы в растворе образца могут быть индивидуально отслежены и одновременно проанализированы путем непосредственного наблюдения. Наночастицы перемещаются посредством броуновского движениям из-за случайного перемещения молекул воды, окружающих эти частицы. Небольшие частицы движутся быстрее, чем крупные частицы. Броуновское движение каждой частицы наблюдают в режиме реального времени с помощью видео, и NTA позволяет анализировать броуновское движение для определения размера частиц. Коэффициент диффузии может быть рассчитан путем отслеживания движения каждой частицы, а затем размер частиц может быть вычислен по уравнению Стокса-Эйнштейна. Эта методика «взаимодействия частиц» дает результаты с высоким разрешением для распределения и концентрации частиц по размерам (то есть, количества частиц в известном объеме жидкости). Целевые значения для оптимального детектирования, применяемые для детектирования на приборе, систематизированы в нижеследующей Таблице 5.
Таблица 5
[0100] Тестируемый образец должен находиться в жидкой форме. Для проведения этого теста, виросомную вакцину подают в жидкой форме. Для тестируемых образцов описанных здесь вакцинных композиций, смесь подают в жидкой форме, замороженную таблетку оттаивают до образования жидкой формы перед тестированием, и лиофилизованную лекарственную форму разводят водным буфером до жидкой формы. Тестируемый образец не должен быть слишком концентрированным. Жидкий тестируемый образец может быть дополнительно разведен HN-буфером, если это необходимо для оптимизации детектирования. Буфер для разведения может иметь высокую степень чистоты и может быть отфильтрован по меньшей мере через 0,22 мкм-фильтр перед его использованием. Экспериментальные параметры, установленные для анализа NTA, систематизированы ниже в Таблице 6.
Таблица 6
[0101] Образцы измеряли с помощью NTA, как известно специалистам в данной области. В некоторых вариантах осуществления изобретения, диапазон размеров виросомных частиц, включая описанные здесь фрагменты и кластеры, может составлять приблизительно 50-500 нм. В некоторых вариантах осуществления изобретения, частицы виросом, которые являются интактными виросомами, могут составлять в пределах приблизительно 70-400 нм или приблизительно 70-200 нм (главный пик). В некоторых вариантах осуществления изобретения, средний диаметр виросомных частиц может составлять приблизительно 70-200 нм, приблизительно 100-175 нм и приблизительно 125-155 нм. Для детектирования, концентрация частиц в популяции виросом в образце может составлять по меньшей мере 1010 единиц/мл.
[0102] Количественная оценка степени сохранения виросом с помощью проточной цитометрии. Для этой оценки проводили проточную цитометрию. Сначала, исходные жидкие виросомные частицы метили (контрольный образец представляет 100% исходного материала) путем введения липофильного красителя Dil (длинноцепочечного диаллилкарбоцианина) в липидный бислой виросом (мечение красителем Dil не оказывает заметного влияния на размер частиц). Затем, лиофилизованную таблетку разводят, и виросомы в лиофилизованной таблетке метят Dil (тестируемый образец). Затем образцы анализируют с помощью визуализирующего проточного цитометра AMNIS и проводят сравнение между контрольными образцами (жидкими виросомами перед лиофилизацией) и тестируемыми образцами (лиофилизованными виросомами) для оценки степени сохранения виросом после лиофилизации по: (а) проценту выделения виросом и (b) проценту виросомных кластеров. В некоторых вариантах осуществления изобретения, процент выделения виросомных частиц в виде отдельных частиц может составлять приблизительно 20-50%, приблизительно 30-50% или приблизительно 40-50% от исходного материала. В некоторых вариантах осуществления изобретения, процент виросомных кластеров (дуплетов, триплетов или форм с более высоким числом) может составлять приблизительно менее 50%, приблизительно 25%, приблизительно 10% или приблизительно 5%.
Содержание гемагглютинина (НА), антигена Р1 ВИЧ-1, антигенов ВИЧ-1 и адъюванта в виросоме
[0103] Содержание HA вируса гриппа, антигенов ВИЧ-1 и адъюванта в виросоме может быть количественно оценено различными методами. Эти результаты представлены ниже в Таблице 7.
Таблица 7
Анализ методом светового рассеяния (SRID)
ELISA
ВЭЖХ-анализ
ВЭЖХ-анализ
ELISA
ВЭЖХ-анализ
[0104] Метод иммуноблот-анализа для HA, PI и rgp41: В этом анализе, композиция (жидкие виросомные концентраты или восстановленные лиофилизованные лекарственные формы, содержащие виросомы) могут быть абсорбированы на нитроцеллюлозной мембране, и антигены могут поддерживаться в их нативном состоянии благодаря отсутствию нагревания, и отсутствию денатурирующих или восстанавливающих агентов. Этот анализ позволяет выявить все антигены, доступные для специфических антител, и указывает на разложение или денатурацию основного антигена, которая нарушает или блокирует доступ к специфическому эпитопу. Этот анализ также показывает, могут ли специфические наполнители предотвращать связывание антитела с антигеном. Для получения лиофилизованных лекарственных форм для анализа (тестируемых образцов), каждую таблетку растворяют в 0,5 мл воды (то есть, лиофилизованную таблетку разводят снова до получения композиции виросомного препарата перед лиофилизацией). Для получения позитивного контроля, образец жидкого виросомного концентрата разводят сверхчистой водой (4-кратно). В идеальном случае, жидкий виросомный концентрат представляет собой ту же партию, которая используется при получении образца для испытания лекарственной формы. Затем получают серийные 2-кратные разведения всех тестируемых образцов и позитивного контроля. Также могут быть использованы дополнительные позитивные контроли, такие как очищенный HA и Rgp41 и синтетический P1. 1,5 мкл каждого образца и позитивного контроля наносят пятнами на сухую нитроцеллюлозную мембрану (с увеличением разведения: слева направо - неразведенные, 1/2, 1/4, 1/8, 1/16, 1/32, 1/64, 1/128). Сухую мембрану блокируют 1% (масс./об.) казеином и инкубируют с раствором специфического антитела, то есть, с человеческим моноклональным антителом (mab) 2F5, специфичным к антигену P1 ВИЧ-1, и с кроличьей антисывороткой против rgp41, специфичной к антигену rgp41 ВИЧ-1. После инкубирования, нитроцеллюлозную мембрану промывают. Затем специфические антитела, связанные с флуоресцентно меченными вторыми антителами (античеловеческими или антикроличьими), детектируют с использованием сканера, флуоресцирующего в дальнем красном диапазоне спектра, что позволяет одновременно детектировать 2 различных флуоресцентных метки на 700 нм и 800 нм. Флуоресцентный сигнал для исходных данных для каждого пятна образца (от самого низкого до самого высокого) сравнивают с соответствующим разведением пятна, используемого в качестве позитивного контроля (в % от позитивного контроля). Затем вычисляют среднее арифметическое значение процентного содержания образца.
[0105] УФ-спектроскопический анализ для 3M-052: Молекула адъюванта 3M-052 имеет несколько пиков кривой УФ-адсорбции. Некоторые из этих пиков перекрываются с поглощением липидов и белков. Анализ проводили с помощью УФ-спектроскопии на 320 нм.
[0106] ВЭЖХ-анализ на P1 и rgp41: Антигены P1 и rgp41 ВИЧ разделяли с помощью обращенно-фазовой жидкостной хроматографии высокого давления на колонке Cl8, с использованием градиента воды и ацетонитрила. Анализ проводили с помощью УФ-спектроскопии на 280 нм, и площади пиков количественно определяли с помощью программного обеспечения для системы ВЭЖХ.
[0107] ELISA-титр антител в конечной точке: 96-луночный планшет Maxisorp (плоскодонный планшет Nunc) и планшеты Polysorp соответственно покрывали при 4°C в течение 16 часов 0,1 мл пептида rgp41 или P1 (2 мкг/мл), приготовленного в PBS pH 7,4. Планшеты промывали 3 раза PBS с 0,05% об./об. Твина 20 (PBST), а затем в каждую лунку добавляли блокирующий раствор 1% BSA, приготовленный в PBST, и инкубировали в течение 2 часов при комнатной температуре (КТ). Планшеты три раза промывали PBST, а затем добавляли 0,1 мл на лунку неиммунной сыворотки, разведенной 1/1000, или серийные разведения иммунных сывороток (от 1/1000 до 1/64000), приготовленных в 0,1% BSA в PBST, и инкубировали в течение 2 часов при комнатной температуре. Планшеты три раза промывали PBST и инкубировали в течение 2 часов при комнатной температуре с ПХ-конъюгированным козьим антителом против крысиных IgG, разведенным 1:4000 в 0,1% BSA в PBST. Планшеты снова промывали, а затем добавляли 0,1 мл колориметрического субстрата о-фенилендиамина (OPD), и реакцию прекращали добавлением 2М H2SO4 с последующим считыванием планшетов на 492 нм.
Пример 1: Использование высокого уровня маннита (8% масс./масс.) в комбинации с проведением низкотемпературного цикла лиофилизации для уменьшения разрушения микроструктуры во время лиофилизации без нарушения целостности виросомы.
[0108] Маннит используют в лекарственных формах для повышения структурной устойчивости. Из-за присутствия высоких уровней буферов и добавления трегалозы для защиты виросомных частиц, применение маннита при обычном уровне 4,5% масс./масс. не давало возможность обеспечивать достаточное сохранение структуры во время лиофилизации. Таким образом, происходит разрушение микроструктуры.
[0109] Однако, заявителями было обнаружено, что при использовании комбинации более высокого уровня маннита и проведения низкотемпературных циклов лиофилизации может быть получена лиофилизованная таблетка с более прочной структурой. В этом примере представлены данные сравнения композиции, содержащей 4,5% масс./масс. маннита, с композицией, содержащей 8% масс./масс. маннита. В обеих виросомных композициях использовали 25% масс./масс. загрузку жидких виросомных концентратов MYM-201 партии 160125-1, поставляемой Mymetics, и содержащей 70-80 мкг/мл HA, 40-50 мкг/мл P1, 70-80 мкг/мл rgp41 (A/Brisbane/59/2007 (H1N1) в HN-буфере, рН 7,4 (50 мМ HEPES, 142,5 мМ NaCl). В виросомной композиции, содержание рыбьего желатина и чистой трегалозы поддерживали на уровне 6% масс./масс. и 2% масс./масс. Как показано на фигуре 2, композиции получали в виде доз с массой приготовленной дозы в аликвотах по 500 мг при 15°С в блистерных упаковках, содержащихся в карманах алюминиевых лотков. Лотки, содержащие дозированную водную вакцинную смесь, замораживали путем пропускания алюминиевых лотков через морозильную камеру, установленную на -70°C в течение 3 минут 15 секунд. Алюминиевые лотки, содержащие замороженные продукты, собирали и помещали в холодильник при температуре <-15°С° и гибридизовали в течение 6 часов в замороженном состоянии перед лиофилизацией. Затем проводили 2-стадийный FD-цикл при -15°С в течение 24 часов, а затем при -10°С в течение 18 часов. Блистерные упаковки с лиофилизованными таблетками герметично упаковывали в саше и хранили в условиях окружающей среды.
[0110] Физические свойства лиофилизованных таблеток (внешний вид и время диспергирования) оценивали в соответствии с испытаниями, описанными выше. Кроме того, размер виросомных частиц в лиофилизованных таблетках охарактеризовывали в соответствии с испытаниями, описанными выше. Результаты систематизированы ниже в Таблице 8.
Таблица 8
Время диспергирования (n=8)
Смачивание: > 60 сек. (2 таблетки с оболочкой, 5 таблеток с плотными комками
Диссоциация: > 60 сек. (полностью не диспергировались)
Анализ NTA
Лиофилизованныая таблетка: 136 нм (главный пик)
% виросомных частиц (<200 нм): >50%
Время диспергирования (n=8)
Смачивание: < 28 сек.
Диссоциация: <36 сек.
Анализ NTA
Лиофилизованная таблетка: 140 нм (главный пик)
% виросомных частиц (< 200 нм): >50%
[0111] Все таблетки имели хороший внешний вид. Композиция с 4,5% масс./масс. маннита имеет плохие дисперсионные свойства с оболочкой и/или плотными комками, наблюдаемыми в таблетках из-за разрушения микроструктуры. Увеличение маннита в этой композиции до 8% масс./масс. способствовало кристаллизации маннита и улучшало структуру лиофилизованной таблетки. Композиция с 8% масс./масс. маннита имела хорошие дисперсионные свойства и мягкую и пальпируемую массу. В комбинации с проведением цикла лиофилизации при низкой температуре, виросомные частицы в лиофилизованной таблетке не подвергались какому-либо воздействию. Размер виросомных частиц в лиофилизованных таблетках был, в основном, сравнимым во всех композициях.
Пример 2: Данные стабильности лиофилизованных вакцинных лекарственных форм (композиция 1), хранящихся в течение 3 месяцев в условиях ICH
[0112] Жидкие виросомные концентраты обычно стабильны при 2-8°С, но жидкая виросомная ВИЧ-вакцина имеет ограниченный срок хранения в несколько месяцев, что обусловлено важными химическими модификациями, происходящими на антигенах, и при этом не агрегируется. Предполагается, что при использовании твердой вакцинной формы с низким содержанием влаги, такие модификации замедляются и со временем становятся минимальными, что продлевает срок хранения на >1 год. В этом примере проиллюстрирована стабильность виросомной вакцины в форме лиофилизованной таблетки 1. Для получения лиофилизованных вакцинных таблеток была закуплена партия жидких виросомных концентратов MYM-201, партия 170130-1, поставляемая Mymetics, и содержащая приблизительно 100 мкг/мл P1 ВИЧ-1, 230 мкг/мл rgp41 ВИЧ-1, 130 мкг/мл НА (A/Brisbane/59/2007 (H1N1), 65 мкг/мл адъюванта 3М-052 в HN-буфере, рН 7,4 (50 мМ HEPES, 142,5 мМ NaCl) с 7% масс./масс. трегалозы (партия Z33787A101).
[0113] Для получения вакцинной таблетки сначала приготавливают смесь жидкой виросомной композиции. Эта смесь содержит 25% масс./масс. партии жидкого виросомного концентрата MYM V202 170130-1, 6% масс./масс. рыбьего желатина, 4,5% масс./масс. маннита, 2% масс./масс. (чистой) трегалозы, раствор гидроксида натрия (достаточное количество) до рН 7,4 и очищенную воду (достаточное количество) до 100% масс./масс.
[0114] Смесь жидкой виросомной композиции получали в виде доз с массой приготовленной дозы в аликвотах по 500 мг при 15°С в блистерных карманах, содержащихся в алюминиевых лотках. Лотки, содержащие дозированную водную вакцинную смесь, замораживали путем пропускания алюминиевых лотков через морозильную камеру, установленную на -70°C в течение 3 минут 15 секунд. Алюминиевые лотки, содержащие замороженные продукты, собирали и помещали в холодильник при температуре <-15°С. Затем проводили 2-стадийный FD-цикл при -15°С в течение 24 часов, а затем при -10°С в течение 18 часов.
[0115] Блистерные упаковки лиофилизованных таблеток герметично упаковывали в саше и помещали на хранение на 3 месяца при 5°С, 25°С/60% относительной влажности и 40°С/75% относительной влажности. Результаты начальных испытаний и испытаний, проводимых через 3 месяца, систематизированы в Таблице 9.
Таблица 9
[0116] Данные по стабильности показали, что внешний вид таблеток был хорошим и одинаковым для всех партий. Содержание влаги составляло 5-6% масс./масс. с небольшими различиями в различных условиях стабильности в течение 3-месячного периода хранения. Поскольку эта композиция содержит 4,5% масс./масс. маннита, то во время лиофилизации наблюдалось некоторое разрушение микроструктуры, на что указывает изменение времени дезинтеграции. Что касается размера виросомных частиц, то данные DLS и NTA не показали заметного различия в размерах виросомных частиц между таблетками, хранящимися в различных условиях стабильности. Исходные жидкие виросомные концентраты имели >50% частиц размером <200 нм. Результаты показали, что виросома размером <200 нм, выделенная из разведенной таблетки, по существу, сохраняется до величины того же порядка.
[0117] С использованием полуколичественных иммуноблот-анализов был проведен мониторинг антигенов P1 и rgp41 ВИЧ-1 на разложение в течение 3 месяцев. Все образцы были предварительно 2-кратно разведены. Для этой цели использовали человеческое mAb 2F5, специфичное для P1, и кроличью антисыворотку против rgp41. Жидкий виросомный концентрат партии MYM-V202 170130-1, который был использован для получения партии вакцин, 8-кратно разводили и использовали в качестве позитивного контроля. На фигуре 4 представлена фотография иммуноблот-анализа. Для образца Z33787A101, таблетки, хранящиеся при 5°С, показали отсутствие или только минимальное снижение сигнала антигена rgp41 после хранения в течение 1 месяца и 3 месяцев по сравнению с исходным образцом (t=0). Аналогичным образом, наблюдалось лишь минимальное различие в интенсивности сигнала для таблеток, хранившихся при 25°С, для антигена rgp41, и небольшое снижение для антигена P1. Различие для таблеток, хранящихся при 40°С, было более выраженным, хотя это различие может быть в пределах точности анализа на 10-20%.
[0118] Количественная оценка данных флуоресценции для rgp41 и P1 представлена в Таблицах 10 и 11, соответственно. В верхней части каждой таблицы показаны исходные данные для флуоресцентных сигналов для каждого пятна (от самого низкого и до самого высокого разведения) для указанного образца, оцениваемого на стабильность. В нижней части каждой таблицы показаны величины для пятна каждого образца по сравнению с соответствующим разведением позитивного контрольного пятна (в % от позитивного контроля). Нижняя линия соответствует среднему арифметическому для всех процентных содержаний образцов. Из вычисления среднего исключались очевидные «выбросы» измерений.
Таблица 10
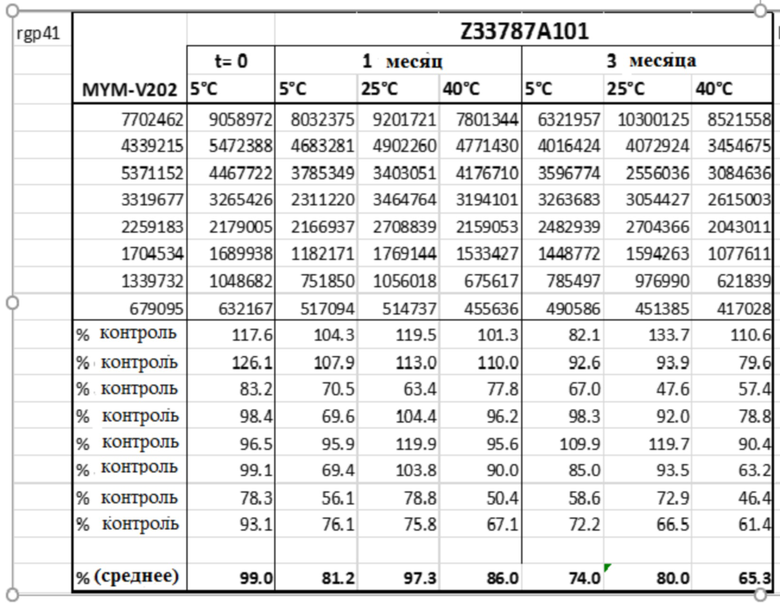
Таблица 11
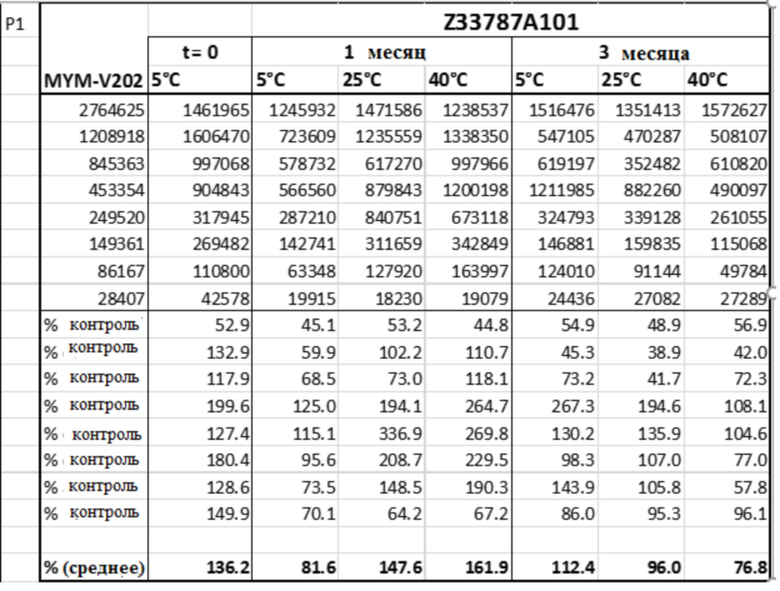
[0119] Для вакцинных таблеток партии Z33787A101, таблетки, хранящиеся при 5°С, показали отсутствие или только минимальное снижение сигнала антигенов rgp41 и Р1 после хранения в течение 1 месяца и 3 месяцев по сравнению с исходным образцом (t=0). Аналогичным образом, наблюдалось лишь минимальное различие в интенсивности сигнала для таблеток, хранившихся при 25°С, для антигена rgp41, и небольшое снижение для антигена P1. Различие для таблеток, хранящихся при 40°С, было более выраженным для антигенов Р1 и rgp41, хотя это различие может быть в пределах точности анализа, особенно поскольку изменения наблюдались снова в образцах с серийным разведением.
[0120] Анализ 3M-052 проводили с помощью УФ-спектроскопии при 320 нм для всех вакцинных таблеток, хранящихся при различных температурах в течение более 3 месяцев. Значения представлены ниже в Таблице 12.
Таблица 12
[0121] Между этими образцами не наблюдалось каких-либо заметных различий, и, следовательно, концентрации 3M-052 рассматривались как стабильные во всех образцах в пределах точности этого анализа. В целом, HA, P1, rpg41 и 3M-052 были стабильными в лиофилизованных таблетках в различных условиях хранения с небольшими изменениями во времени, как показано ниже в Таблице 13.
Таблица 13
Пример 3: Данные стабильности для лиофилизованных вакцинных лекарственных форм (композиция 2), хранящихся в условиях ICH
[0122] В этом примере проиллюстрирована стабильность виросомной вакцины в форме лиофилизованной таблетки для композиции 2. Для получения лиофилизованных вакцинных таблеток была закуплена партия жидких виросомных концентратов MYM-V202, партия 17MYM002/F17255, поставляемая Mymetics, и содержащая приблизительно 121 мкг/мл P1 ВИЧ-1, 67 мкг/мл rgp41 ВИЧ-1, 41 мкг/мл НА (A/Brisbane/59/2007 (H1N1), 39 мкг/мл адъюванта 3М-052 в HN-буфере, рН 7,4 (50 мМ HEPES, 142,5 мМ NaCl) с 7% масс./масс. трегалозы (партия MYM-212, 1690747).
[0123] Для получения вакцинной таблетки сначала приготавливали смесь жидкой виросомной композиции. Эта смесь содержит 25% масс./масс. партии жидкого виросомного концентрата MYM V202 17MYM002/F17255, 6% масс./масс. рыбьего желатина, 8% масс./масс. маннита, 2% масс./масс. (чистой) трегалозы, раствор гидроксида натрия (достаточное количество) до рН 7,4 и очищенную воду (достаточное количество) до 100% масс./масс.
[0124] Смесь жидкой виросомной композиции получали в виде доз с массой приготовленной дозы в аликвотах по 500 мг при 15°С в блистерных упаковках, содержащихся в блистерных карманах алюминиевых лотков. Лотки, содержащие дозированную водную вакцинную смесь, замораживали путем пропускания лотков через морозильную камеру, установленную на -70°C в течение 3 минут 15 секунд. Алюминиевые лотки, содержащие замороженные продукты, собирали и помещали в холодильник при температуре <-15°С. Затем проводили 2-стадийный FD-цикл при -15°С в течение 24 часов, а затем при -10°С в течение 18 часов.
[0125] Блистерные упаковки лиофилизованных таблеток герметично упаковывали в саше и помещали на хранение на 3 месяца при 5°С, 25°С/60% относительной влажности и 40°С/75% относительной влажности. Результаты начальных испытаний и испытаний, проводимых через 6 месяцев, систематизированы ниже в Таблице 14.
Таблица 14
[0126] Данные по стабильности показали, что внешний вид таблеток был хорошим и одинаковым для всех партий. Содержание влаги составляло 3,6-4,0% масс./масс. с небольшими различиями в различных условиях стабильности в течение 6-месячного периода хранения. Поскольку эта композиция содержит 8% масс./масс. маннита, то во время лиофилизации наблюдалось меньшее разрушение микроструктуры, на что указывает более короткое и более стабильное время дезинтеграции.
[0127] Был проведен мониторинг стабильности содержания антигенов (антигена P1 и rgp41 ВИЧ-1) на разложение в течение 3 месяцев с помощью ВЭЖХ-анализа. Было обнаружено, что для жидкой вакцины MYM V202 (исходный материал), содержание P1 снижалось на 4% и 7% при хранении при 2-8°С (в условиях хранения в холодильнике) в течение 1 месяца и 3 месяцев, соответственно. Для rgp41, такое содержание снижалось на 16% и 25% через 1 и 3 месяца, соответственно, в условиях хранения в холодильнике. Если жидкую виросому хранили при 25°С и 40°С, то P1 и rgp41 больше не обнаруживались через 1 месяц и 3 месяца.
[0128] Результаты оценки содержания антигена в лиофилизированной таблетке при хранении представлены в Таблице 15 и в Таблице 16 для антигена P1 и антигена rgp41, соответственно. В лиофилизированной таблетированной форме, антиген P1 оставался достаточно стабильным без наблюдаемого снижения его содержания через 3 месяца при 2-8°С (в условиях холодовой цепи), а также оставался неизменным в течение 1 и 3 месяцев хранения вне условий холодовой цепи. Для антигена rgp41 наблюдалось снижение приблизительно на 5%, 10% и 20% через 1 месяц при 2-8°С, 25°С и 40°С, соответственно. При хранении в течение 3 месяцев наблюдалось снижение приблизительно на 13%, 15% и 19%, соответственно. Принимая во внимание точность метода ВЭЖХ, наблюдаемая разница в снижении концентраций менее 15% не является значимой. Кроме того, наблюдаемая постепенная потеря или снижение уровня антигена связана с химическими модификациями, а не с усиленным разложением в результате расщепления структуры, потери аминокислот или агрегации. Между тем, химическая(ие) модификация(и) в данном эпитопе может (могут) потенциально изменить его распознавание, уменьшая или увеличивая уровень антител, связывающихся с этой областью, в то время как другие области остаются столь же хорошо узнаваемыми.
[0129] На время 0, антигены P1 и rgp41 имеют сходный профиль миграции в ДСН-ПААГ и все еще распознаются специфическими моноклональными антителами в таблетках для подъязычного введения, а сывороточные антитела против P1 и rgp41 все еще реагируют на различные пептиды P1 и gp41, несущие ключевые эпитопы. Эти анализы позволяют предположить, что, в целом, P1 и rgp41 сохраняют большую часть своей антигенности и иммуногенности во время процесса приготовления таблеток (данные не показаны).
Таблица 15
Таблица 16
Пример 4: Стабильность лиофилизованных таблеток, хранящихся в условиях хранения при температуре ниже нуля.
[0130] Данные Примера 4 показали стабильность физических свойств таблеток виросомной вакцины и виросомных частиц при хранении при температуре ниже нуля. Смесь жидкой виросомной композиции, содержащей 25% масс./масс. загрузку жидкого виросомного концентрата MYM-201, партия 160125-1, поставляемого Mymetics (содержащего 70-80 мкг/мл HA, 40-50 мкг/мл P1, 70-80 мкг/мл rgp41 (A/Brisbane/59/2007 (H1N1) в HN-буфере pH 7,4 (50 мМ HEPES, 142,5 мМ NaCl), 6% масс./масс. рыбьего желатина, 8% масс./масс. маннита и 2% масс./масс. (чистой) трегалозы получали в виде дозы с массой приготовленной дозы 500 мг и лиофилизовали. Композиции получали в виде доз с массой приготовленной дозы в аликвотах по 500 мг при 15°С в блистерных карманах, содержащихся в алюминиевых лотках. Лотки, содержащие дозированную водную вакцинную смесь, замораживали путем пропускания алюминиевых лотков через морозильную камеру, установленную на -70°C в течение 3 минут 15 секунд. Алюминиевые лотки, содержащие замороженные продукты, собирали и помещали в холодильник при температуре <-15°С, и подвергали отжигу в течение 6 часов в замороженном виде до лиофилизации. Затем проводили 2-стадийный FD-цикл при -15°С в течение 24 часов, а затем при -10°С в течение 18 часов. Блистерные упаковки лиофилизованных таблеток герметично запечатывали в саше и помещали на хранение в холодильник при температуре -15°С на 1 неделю. Соответствующий набор блистерных упаковок с таблетками герметично запечатывали в саше и хранили в условиях окружающей среды в течение тех же самых периодов времени. Эти таблетки оценивали на внешний вид, дисперсионные свойства (время смачивания и время диссоциации) и гранулометрическое распределение виросомных частиц, как описано выше в соответствии с методами испытаний.
[0131] Было обнаружено, что все таблетки имели хороший внешний вид после хранения в обоих условиях. Данные показали, что хранение при температуре ниже нуля оказывает незначительное влияние на дисперсионные свойства таблетки (время смачивания и диссоциации) (Таблица 17) и на гранулометрическое распределение виросомных частиц (Таблица 18).
Таблица 17
Таблица 18
Пример 5: Оценка виросомных частиц в разведенных лиофилизованных таблетках с помощью проточной цитометрии
[0132] Оценки количества виросом, сохранившихся в лиофилизованных образцах таблеток и полученных разработанным способом, систематизированы ниже. Значения, представленные в Таблице 19, были получены исходя из данных проточной цитометрии AMNIS путем оценки событий в фокусной зоне, соответствующей виросомному стробированию.
Таблица 19
[0133] Данные показали, что 24-40% исходной виросомы сохраняется в лиофилизованных таблетках, из которых от 10 до 25% этих виросом находятся в кластерах, в основном, в виде дублетов и триплетов.
Пример 6. Оценка иммуногенности антигенов P1 и rgp41 в жидкой виросомной композиции и в разведенных лиофилизованных таблетках для подъязычного введения, содержащих виросому.
[0134] Жидкие виросомные концентраты (жидкая вакцина MYM-V202) и лиофилизованные таблетки для подъязычного введения, содержащие виросому Примера 3, помещали на хранение при 4°C и 40°С на три месяца для оценки иммуногенности. После хранения в течение 1 месяца, образцы жидкой вакцины и таблеток для подъязычного введения, которые хранились при 40°С, помещали в шкаф для хранения для оценки иммуногенности. После хранения в течение 3 месяцев, образцы жидкой вакцины и таблеток для подъязычного введения, которые хранились при 4°C и 40°С, брали для оценки иммуногенности.
[0135] Для оценки иммуногенности использовали крыс Wistar (n=10 на группу), по 50% каждого пола. Крыс иммунизировали на день 0, 28 и 56. Жидкая вакцина содержала 3,9 мкг P1, 2,2 мкг rgp41 и 1,3 мкг 3M-052 TLR7/8 (адъюванта) в 0,1 мл и была использована для подкожной инъекции. Для таблеток, вводимых подъязычно, адекватное количество таблеток для подъязычного введения растворяли в стерильной воде с получением приблизительно 3 мкг P1, 1,7 мкг rgp41 и 1 мкг 3M-052 TLR7/8 (адъюванта) в 0,1 мл для введения путем подкожной инъекции. Неиммунные сыворотки собирали на день 0, а иммунные сыворотки собирали на 65-й день для определения конечных титров антител в сыворотках каждого животного и в пуле сывороток.
[0136] На Фигуре 5 показана иммуногенность P1 и rgp41 в жидкой вакцине и в таблетках для подъязычного введения, хранящихся при различных температурах. Жидкая вакцинная композиция MYM-V202 с адъювантом, содержащая антигены P1 и rgp41, была термочувствительной и служила в качестве эталона для сравнения с иммуногенностью таблетированной вакцины для подъязычного введения с улучшенной термостабильностью. Черная линия соответствует вакцинам, хранящимся в течение 3 месяцев при 4°C; черная пунктирная линия соответствует вакцинам, хранящимся в течение 1 месяца при 40°С; а серая линия соответствует вакцинам, хранящимся в течение 3 месяцев при 40°С. Полученные данные указывают на сохранение иммуногенности антигенов для лиофилизованных таблеток. На каждой панели также указаны титры антител в конечной точке. Титр в конечной точке соответствует последнему разведению сыворотки, дающему значение OD, которое более, чем в 2 раза превышает значение неиммунного фона.
Дополнительные определения
[0137] Если это не оговорено особо, то все термины, известные специалистам в данной области, обозначения и другие технические и научные термины или терминология, используемые в настоящей заявке, имеют свое общепринятое значение, известное специалистам в данной области, которое относится к заявленному предмету изобретения. В некоторых случаях, термины с общепринятыми значениями определены здесь для ясности и/или для удобства ссылок, и включение таких определений в настоящую заявку не обязательно должно быть истолковано как представляющее существенное отличие от определений, по существу, известных специалистам.
[0138] Термин «приблизительно», относящийся к используемым здесь значениям или параметрам включает (и описывает) изменения, которые относятся к этим значениям или параметрам per se. Так, например, выражение «приблизительно X», включает описание «X». Кроме того, слова «менее, чем», «более, чем», «максимум», «минимум», «меньше или равно», «больше или равно» или другие аналогичные выражения, за которыми следует слова «величина» или «параметр», означают каждую величину или параметр, упоминаемые в ряде таких величин или параметров. Так, например, указание на то, что раствор имеет концентрацию по меньшей мере приблизительно 10 мМ, приблизительно 15 мМ или приблизительно 20 мМ, означает, что раствор имеет концентрацию по меньшей мере приблизительно 10 мМ, по меньшей мере приблизительно 15 мМ или по меньшей мере приблизительно 20 мМ.
[0139] Используемые здесь артикли «а» и «an» и «the», употребляемые с существительными в формах единственного числа, могут относиться и к существительным во множественном числе, если из контекста описания не следует иное. Следует также отметить, что используемое здесь словосочетание «и/или» охватывает любые и все возможные комбинации одного или более из перечисленных здесь связанных элементов. Кроме того, следует отметить, что термины «включает», «включающий», «содержит» и/или «содержащий», если они используются в настоящей заявке, указывают на наличие определенных признаков, целых чисел, стадий, процедур, элементов, компонентов и/или единиц, но не исключают наличие или добавление одного или более других признаков, целых чисел, стадий, процедур, элементов, компонентов, единиц и/или их групп.
[0140] В настоящей заявке раскрывается несколько числовых интервалов, представленных в тексте и на чертежах. Раскрытые числовые интервалы, по своей сути, означают любые интервалы или значения в раскрытых числовых диапазонах, включая конечные точки, даже несмотря на то, что точные границы данных интервалов не указаны дословно в данном описании, поскольку такое раскрытие может применяться на практике для всех указанных числовых интервалов.
[0141] Приведенное выше описание дает возможность специалисту в данной области осуществить и применить настоящее изобретение в соответствии с конкретными целями и требованиями. В предпочтительные варианты осуществления изобретения могут быть внесены различные модификации, которые будут очевидны для специалиста в данной области, и общие принципы, определенные в настоящей заявке могут быть применены и к другим вариантам и целям его осуществления, не выходящим за рамки существа и объема изобретения. Таким образом, настоящее изобретение не ограничивается указанными здесь вариантами его осуществления, но оно должно соответствовать самому широкому объему, в который входят раскрытые здесь принципы и признаки.
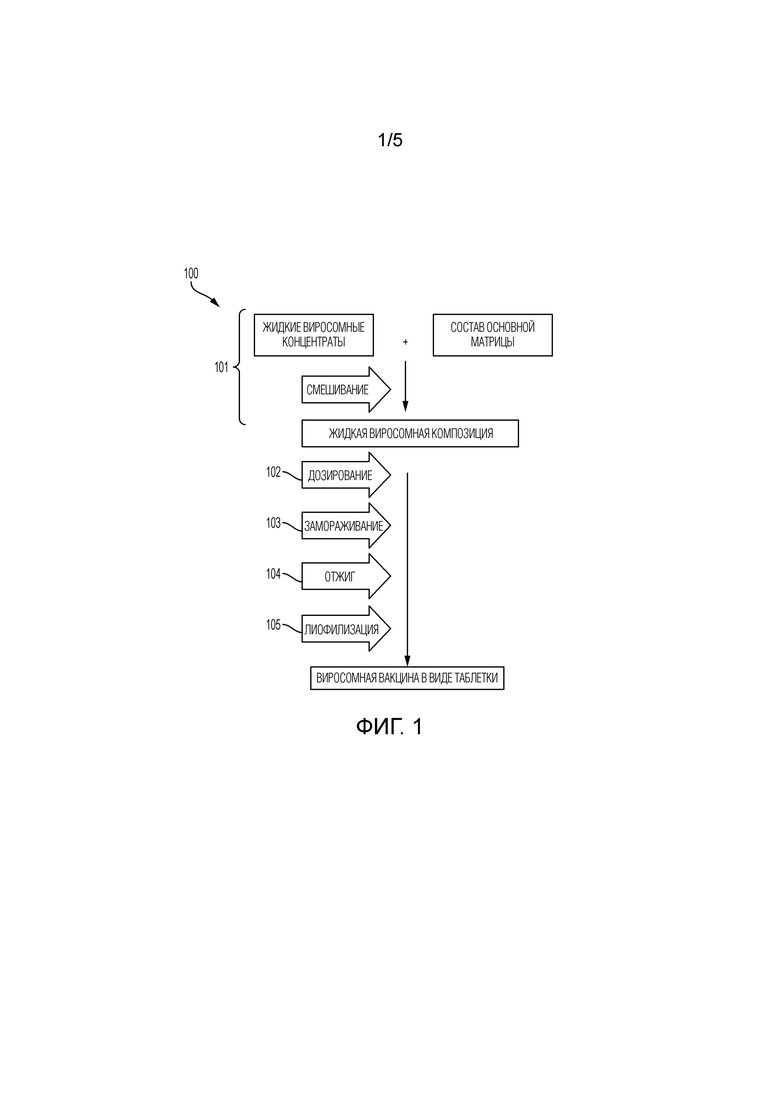
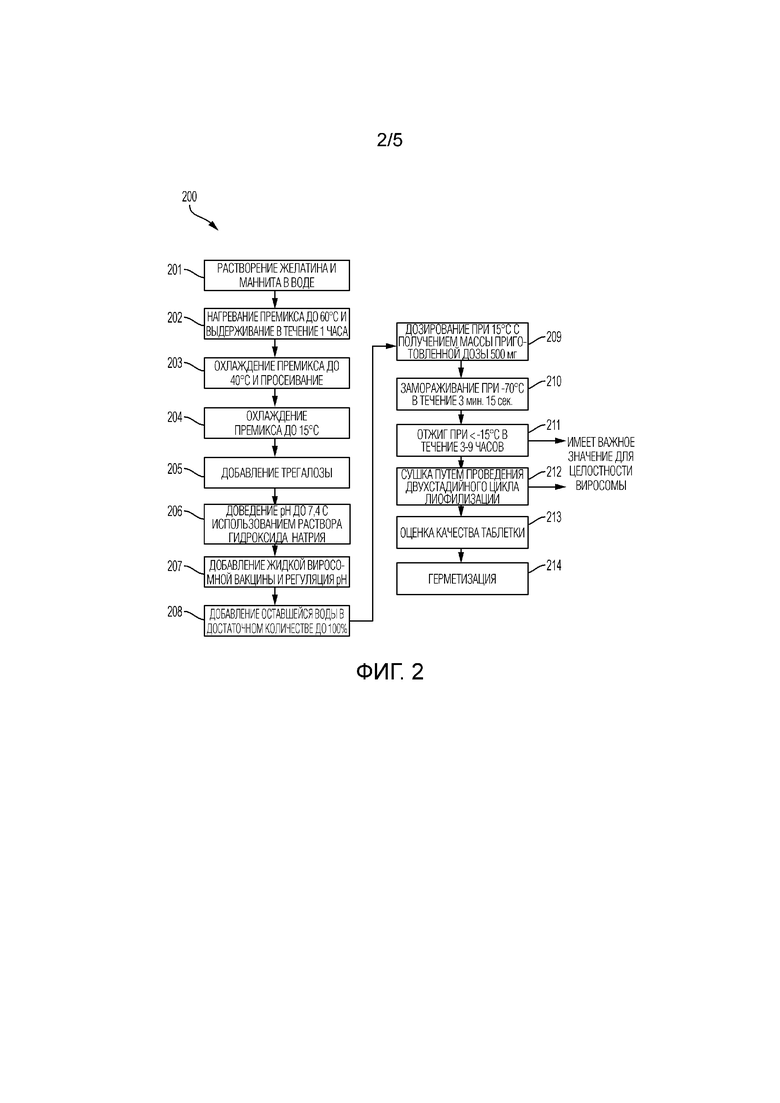
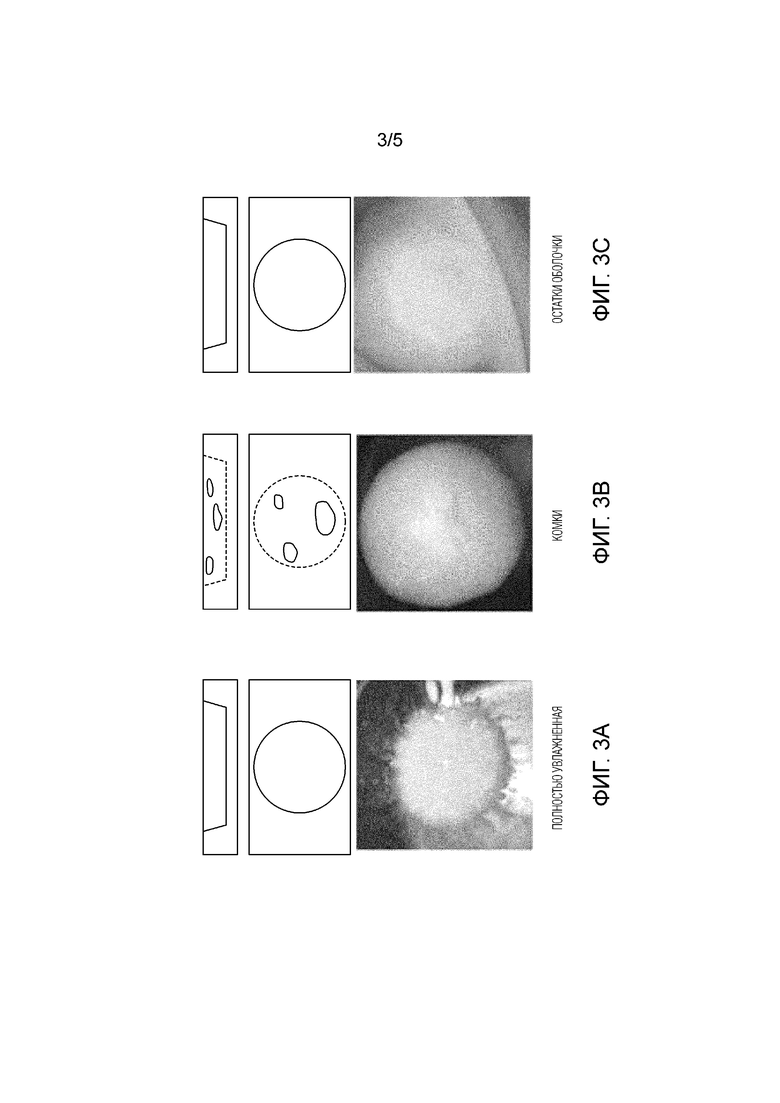

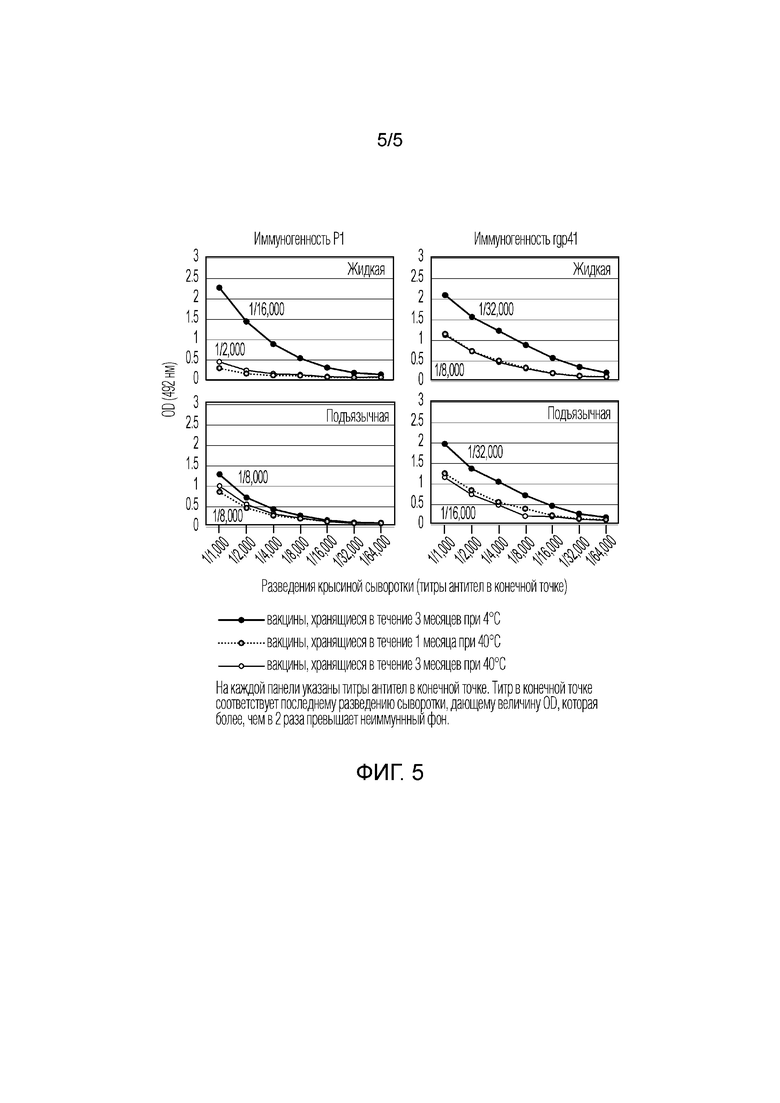
Изобретение относится к биотехнологии. Описаная твердая стандартная лекарственная форма для перорального введения. Композиция содержит: везикулы на основе липидов, содержащие иммуногенное количество по меньшей мере одного вакцинного антигена; 5-20 масс.% по меньшей мере одного криолиопротектора; 25-40 масс.% вещества, образующего матрицу; и 40-55 масс.% вещества, формирующего структуру. Представлен способ индукции иммунного ответа у пациента, включающий введение стандартной лекарственной формы в ротовую полость человека. Описан способ получения твердой стандартной лекарственной формы для перорального введения. Дозируют жидкую виросомную композицию в предварительно сформованную форму. При этом виросомная композиция включает: везикулы на основе липидов, содержащие иммуногенное количество по меньшей мере одного вакцинного антигена; 1-5 масс.% криолиопротектора; 4-8 масс.% вещества, образующего матрицу; и 5-10 масс.% вещества, формирующего структуру. Замораживают дозированную виросомную композицию при температуре от -60°С до -90°С. Отжигают замороженную виросомную композицию путем ее выдерживания при температуре ниже -15°С в течение 3-9 часов. Лиофилизируют гибридизованную виросомную композицию с образованием лекарственной формы. Также описан способ получения твердой стандартной лекарственной формы для перорального введения. Дозируют жидкую виросомную композицию в предварительно сформованную форму. При этом виросомная композиция включает: 20-50 масс.% виросомного концентрата, где виросомный концентрат содержит: виросомы, содержащие иммуногенное количество по меньшей мере одного вакцинного антигена; 2-10 масс.% криолиопротектора; и 60-200 мМ буферной системы; 4-8 масс.% вещества, образующего матрицу; и 5-10 масс.% вещества, формирующего структуру. Замораживают дозированную виросомную композицию при температуре от -60°С до -80°С. Отжигают замороженную виросомную композицию путем ее выдерживания при температуре ниже -15°С в течение 3-9 часов. Лиофилизируют гибридизованную виросомную композиции с образованием лекарственной формы. Изобретение позволяет получать виросомные композиции для подъязычного введения, обладающие физической устойчивостью, целостностью и стабильностью частиц и антигенов. 4 н. и 36 з.п. ф-лы, 5 ил., 17 табл., 6 пр.
1. Твердая стандартная лекарственная форма для перорального введения, содержащая:
везикулы на основе липидов, содержащие иммуногенное количество по меньшей мере одного вакцинного антигена;
5-20 масс.% по меньшей мере одного криолиопротектора;
25-40 масс.% вещества, образующего матрицу; и
40-55 масс.% вещества, формирующего структуру.
2. Стандартная лекарственная форма по п. 1, где везикулы на основе липидов представляют собой виросомы или протеолипосомы.
3. Стандартная лекарственная форма по любому из пп. 1, 2, где лекарственная форма содержит 10-15 масс.% по меньшей мере одного криолиопротектора.
4. Стандартная лекарственная форма по любому из пп. 1-3, где по меньшей мере один криолиопротектор содержит трегалозу.
5. Стандартная лекарственная форма по любому из пп. 1-4, где лекарственная форма содержит 33-37 масс.% вещества, образующего матрицу.
6. Стандартная лекарственная форма по любому из пп. 1-5, где вещество, образующее матрицу, содержит желатин.
7. Стандартная лекарственная форма по п. 6, где желатин включает рыбий желатин.
8. Стандартная лекарственная форма по п. 7, где рыбий желатин представляет собой высокомолекулярный рыбий желатин.
9. Стандартная лекарственная форма по любому из пп. 1-8, где лекарственная форма содержит 45-50 масс.% вещества, формирующего структуру.
10. Стандартная лекарственная форма по любому из пп. 1-9, где вещество, формирующее структуру, содержит маннит.
11. Стандартная лекарственная форма по любому из пп. 1-10, где виросомы происходят из мембраны вируса гриппа или других оболочечных вирусов.
12. Стандартная лекарственная форма по любому из пп. 1-11, где по меньшей мере один вакцинный антиген присутствует на виросоме.
13. Стандартная лекарственная форма по любому из пп. 1-12, где по меньшей мере один вакцинный антиген содержит антиген, происходящий из оболочки ВИЧ-1.
14. Стандартная лекарственная форма по п. 13, где антиген, происходящий из оболочки ВИЧ-1, содержит пептид P1 ВИЧ-1 и/или рекомбинантный gp41 ВИЧ-1.
15. Стандартная лекарственная форма по любому из пп. 1-14, где виросомы содержат адъювант.
16. Стандартная лекарственная форма по любому из пп. 1-15, где лекарственная форма способствует поглощению по меньшей мере одного вакцинного антигена в ротовой полости.
17. Стандартная лекарственная форма по п. 16, где лекарственная форма разлагается в течение 180 секунд после ее введения в ротовую полость.
18. Стандартная лекарственная форма по п. 16, где лекарственная форма разлагается в течение 90 секунд после ее введения в ротовую полость.
19. Стандартная лекарственная форма по п. 16, где лекарственная форма разлагается в течение 60 секунд после ее введения в ротовую полость.
20. Стандартная лекарственная форма по п. 16, где лекарственная форма разлагается в течение 30 секунд после ее введения в ротовую полость.
21. Стандартная лекарственная форма по п. 16, где иммунный ответ индуцируется при ее введении пациенту в ротовую полость.
22. Стандартная лекарственная форма по п. 21, где введение в ротовую полость представляет собой помещение на язык или под язык или в щечную или в глоточную область.
23. Способ индукции иммунного ответа у пациента, включающий введение стандартной лекарственной формы по любому из пп. 1-22 в ротовую полость человека, нуждающегося в иммунном ответе.
24. Способ по п. 23, где введение в ротовую полость представляет собой помещение на язык или под язык или в щечную или в глоточную область.
25. Способ получения твердой стандартной лекарственной формы для перорального введения, включающий:
дозирование жидкой виросомной композиции в предварительно сформованную форму, где виросомная композиция включает:
везикулы на основе липидов, содержащие иммуногенное количество по меньшей мере одного вакцинного антигена;
1-5 масс.% криолиопротектора;
4-8 масс.% вещества, образующего матрицу; и
5-10 масс.% вещества, формирующего структуру;
замораживание дозированной виросомной композиции при температуре от -60°С до -90°С;
отжиг замороженной виросомной композиции путем ее выдерживания при температуре ниже -15°С в течение 3-9 часов; и
лиофилизацию гибридизованной виросомной композиции с образованием лекарственной формы.
26. Способ по п. 25, где дозированную виросомную композицию замораживают при температуре от -60°С до -90°С в течение приблизительно 1-5 минут.
27. Способ по любому из пп. 25, 26, где лиофилизация гибридизованной виросомной композиции включает первую стадию выдерживания гибридизованной виросомной композиции при температуре от -10°С до -20°C в течение 20-28 часов и вторую стадию выдерживания гибридизованной виросомной композиции при температуре от -5°С до приблизительно -15°С в течение 14-22 часов.
28. Способ по любому из пп. 25-27, где лиофилизацию осуществляют под давлением менее, чем 600 мбар.
29. Способ по любому из пп. 25-28, где виросомная композиция имеет рН приблизительно 6,5-8.
30. Способ по любому из пп. 25-29, где криолиопротектор включает трегалозу.
31. Способ по любому из пп. 25-30, где вещество, образующее матрицу, содержит желатин.
32. Способ по п. 31, где желатин включает рыбий желатин.
33. Способ по п. 32, где рыбий желатин представляет собой высокомолекулярный рыбий желатин.
34. Способ по любому из пп. 25-33, где вещество, формирующее структуру, включает маннит.
35. Способ по любому из пп. 25-34, где везикулы на основе липидов происходят из вируса гриппа или респираторно-синцитиального вируса.
36. Способ по любому из пп. 25-35, где по меньшей мере один вакцинный антиген содержит антиген, происходящий из оболочки ВИЧ-1.
37. Способ по п. 36, где антиген, происходящий из оболочки ВИЧ-1, содержит пептид P1 ВИЧ-1 и/или рекомбинантный gp41 ВИЧ-1.
38. Способ по любому из пп. 25-37, где везикулы на основе липидов содержат адъювант.
39. Способ получения твердой стандартной лекарственной формы для перорального введения, включающий:
дозирование жидкой виросомной композиции в предварительно сформованную форму, где виросомная композиция включает:
20-50 масс.% виросомного концентрата, где виросомный концентрат содержит:
виросомы, содержащие иммуногенное количество по меньшей мере одного вакцинного антигена;
2-10 масс.% криолиопротектора; и
60-200 мМ буферной системы;
4-8 масс.% вещества, образующего матрицу; и
5-10 масс.% вещества, формирующего структуру;
замораживание дозированной виросомной композиции при температуре от -60°С до -80°С;
отжиг замороженной виросомной композиции путем ее выдерживания при температуре ниже -15°С в течение 3-9 часов; и
лиофилизацию гибридизованной виросомной композиции с образованием лекарственной формы.
40. Способ по п. 39, где буферная система содержит HEPES-хлорид натрия.
| WO 2017047089 A1, 23.03.2017 | |||
| БЫСТРОРАСТВОРЯЮЩАЯСЯ ЛЕКАРСТВЕННАЯ ФОРМА ПЕРОРАЛЬНОЙ ВАКЦИНЫ, В КОТОРОЙ ИСПОЛЬЗУЮТ КРАХМАЛ | 2011 |
|
RU2639447C2 |
| WO 1997013531 A1, 17.04.1997 | |||
| WO 2011124876 A2, 13.10.2011 | |||
| WO 2008042789 A1, 10.04.2008 | |||
| "Устройство "Челнок" для нанесения покрытия" | 1988 |
|
SU1636063A1 |
