Область техники, к которой относится изобретение
Настоящее изобретение относится к новой кристаллической форме соединения-блокатора проницаемости сосудов.
Уровень техники
Соединение, описывающее формулой 1, обладает названием (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат и является соединением, раскрытым в публикации выложенного патента Кореи № 10-2011-0047170 под кодовым обозначением SAC-1004.
[формула 1]
Соединение подавляет гибель клеток эндотелия сосудов, подавляет образование актиновых стрессорных волокон, индуцируемое VEGF, увеличивает структуру кортикального актинового кольца и повышает стабильность TJ (плотное соединение) между сосудистыми клетками, тем самым подавляя проницаемость сосудов. Соединение не только подавляет проницаемость кровеносных сосудов, но и обладает превосходной активностью по сохранению целостности поврежденных кровеносных сосудов. Поэтому известно, что соединение можно эффективно использовать для предупреждения или лечения разных заболеваний, вызванных проницаемостью сосудов.
В заявке на патент Кореи № 10-2019-0166864 раскрыт новый способ получения химического соединения, дающий возможность разделять и получать в больших количествах стереоизомеры соединения формулы 1 и получать соединения с высоким выходом.
С другой стороны, полиморфы означают кристаллические твердые вещества, которые обладают одинаковой структурой молекул, но структура кристалла которых изменена путем изменения упаковки и конформации молекул кристаллов.
Критерием выбора хорошей кристаллической формы являются наиболее важные физико-химические характеристики, необходимые для лекарственных средств. Выбор оптимизированной кристаллической формы может меняться в зависимости от назначения, например, выбор термодинамически наиболее стабильной формы, выбор формы, оптимизированной для получения фармацевтического сырья и готовых продуктов, улучшения растворимости и скорости растворения лекарственных средств, или изменения фармакокинетических характеристик.
Соответственно, авторы настоящего изобретения раскрыли первую кристаллическую форму (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, способ ее получения и физико-химические характеристики новой кристаллической формы.
Раскрытие
Техническая задача
Объектом настоящего изобретения является получение новой кристаллической формы соединения (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат.
Другим объектом настоящего изобретения является получение блокатора проницаемости сосудов, включающего новую кристаллическую форму соединения.
Техническое решение
Для решения указанных выше задач одним объектом настоящего изобретения является соединение, характеризующееся тем, что содержание α-изомера соединения, представляющего собой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, равно 85% или более и оно находится в твердом состоянии.
Другим объектом настоящего изобретения является соединение, представляющее собой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат.
Одним объектом настоящего изобретения является кристаллическая форма I (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, обладающая на порошковой рентгенограмме пиками при следующих углах дифракции 2Ɵ:
16,0°±0,2°, 19,5°±0,2°, 18,3°±0,2°, 15,1°±0,2°, 21,9°±0,2°.
Другим объектом настоящего изобретения является кристаллическая форма II (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, обладающая на порошковой рентгенограмме пиками при следующих углах дифракции 2Ɵ:
17,3°±0,2°, 11,4°±0,2°, 35,2°±0,2°, 19,0°±0,2°, 15,3°±0,2°, 5,7°±0,2°.
Другим объектом настоящего изобретения является кристаллическая форма III (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, обладающая на порошковой рентгенограмме пиками при следующих углах дифракции 2Ɵ:
15,6°±0,2°, 18,2°±0,2°, 14,4°±0,2°, 24,6°±0,2°, 16,9°±0,2°, 17,2°±0,2°.
Другим объектом настоящего изобретения является кристаллическая форма IV (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, обладающая на порошковой рентгенограмме пиками при следующих углах дифракции 2Ɵ:
15,3°±0,2°, 22,4°±0,2°, 17,2°±0,2°, 36,5°±0,2°, 24,8°±0,2°, 22,7°±0,2°, 11,7°±0,2°, 18,5°±0,2°, 21,8°±0,2°, 23,8°±0,2°, 16,3°±0,2°, 17,6°±0,2°.
Другим объектом настоящего изобретения является кристаллическая форма V (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, обладающая на порошковой рентгенограмме пиками при следующих углах дифракции 2Ɵ:
23,8°±0,2°, 16,2°±0,2°, 17,4°±0,2°, 35,3°±0,2°, 26,0°±0,2°, 36,6°±0,2°, 22,2°±0,2°, 20,7°±0,2°.
Другим объектом настоящего изобретения является фармацевтическая композиция для предупреждения или лечения заболевания проницаемости сосудов, содержащая любое из новых соединений и новых кристаллических форм I - V (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата.
Полезный эффект
Настоящее изобретение относится к новой кристаллической форме (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, соединения, описывающегося формулой 1, и способу его получения. Новая кристаллическая форма обладает высокой чистотой, превосходной стабильностью, превосходной длительной сохранностью и фармацевтической стабильностью и ее можно использовать в качестве блокатора проницаемости сосудов, так что она является весьма полезной для получения высококачественных лекарственных веществ.
Описание чертежей
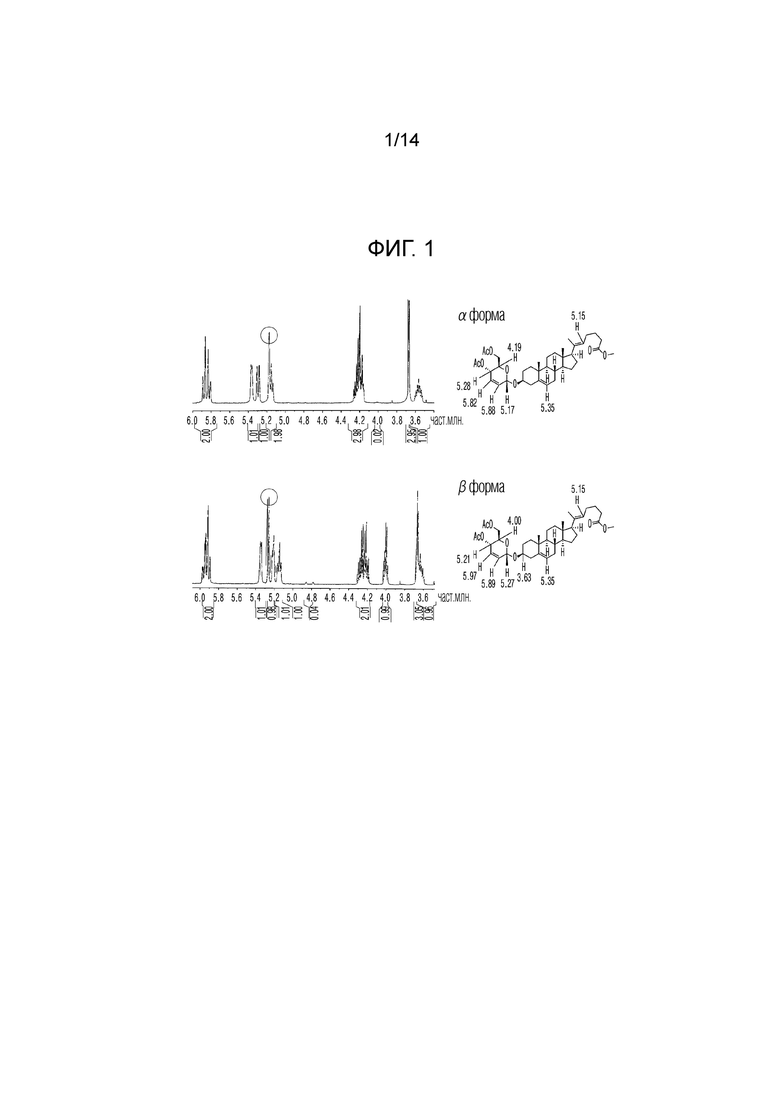
На фиг. 1 приведена диаграмма с результатами анализа посредством NMR для подтверждения структур α-изомера и β-изомера соединения 1, полученных в примере получения 1, и стереохимических структур α-изомера и β-изомера.
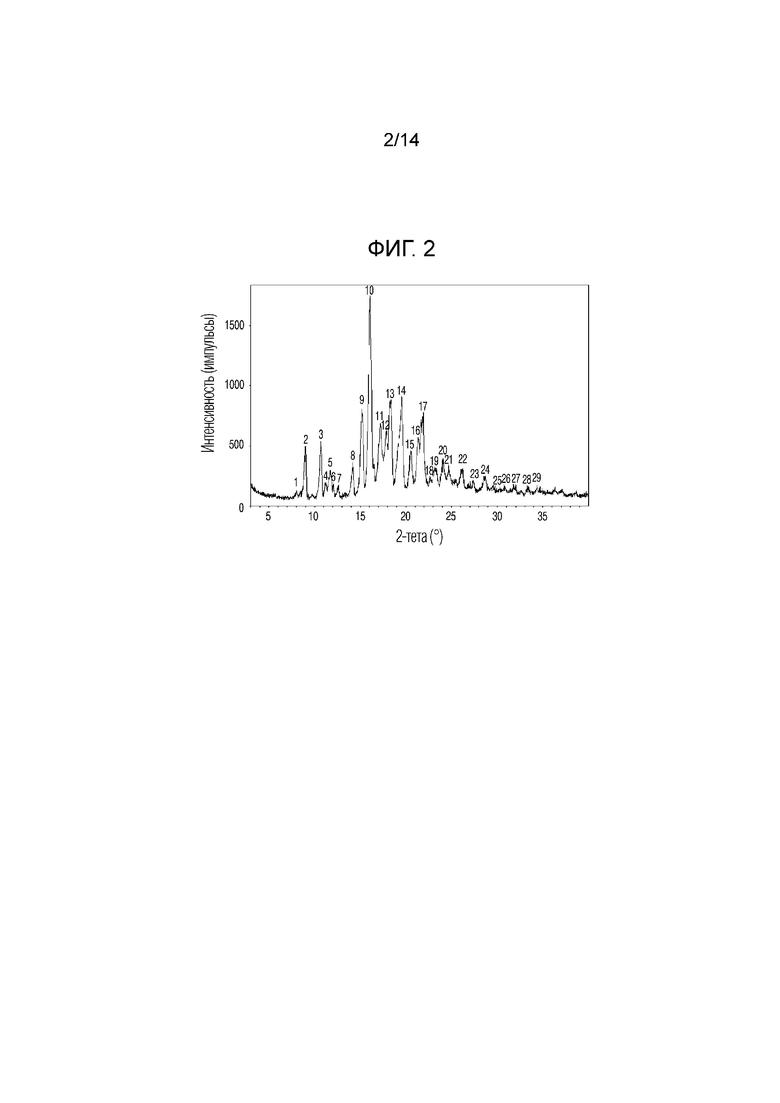
На фиг. 2 приведены данные порошковой рентгенографии (XRPD) кристаллической формы I, полученной в примере 1.
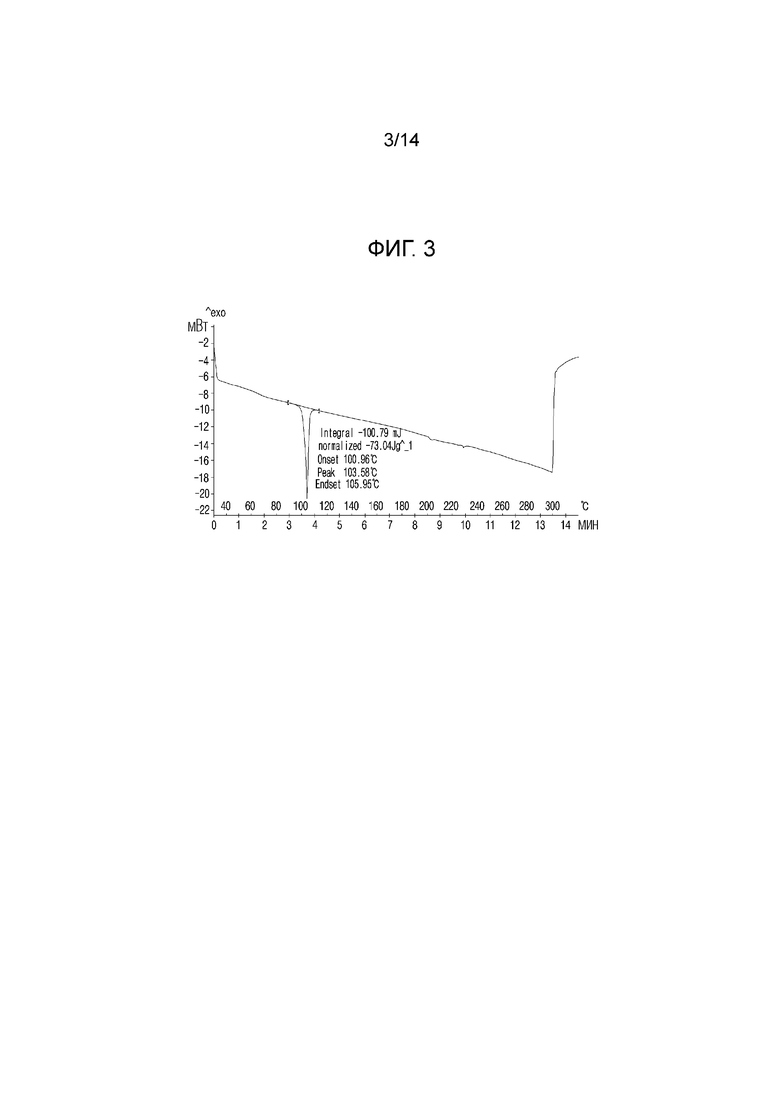
На фиг. 3 приведены данные дифференциальной сканирующей калориметрии (DSC) кристаллической формы I, полученной в примере 1.
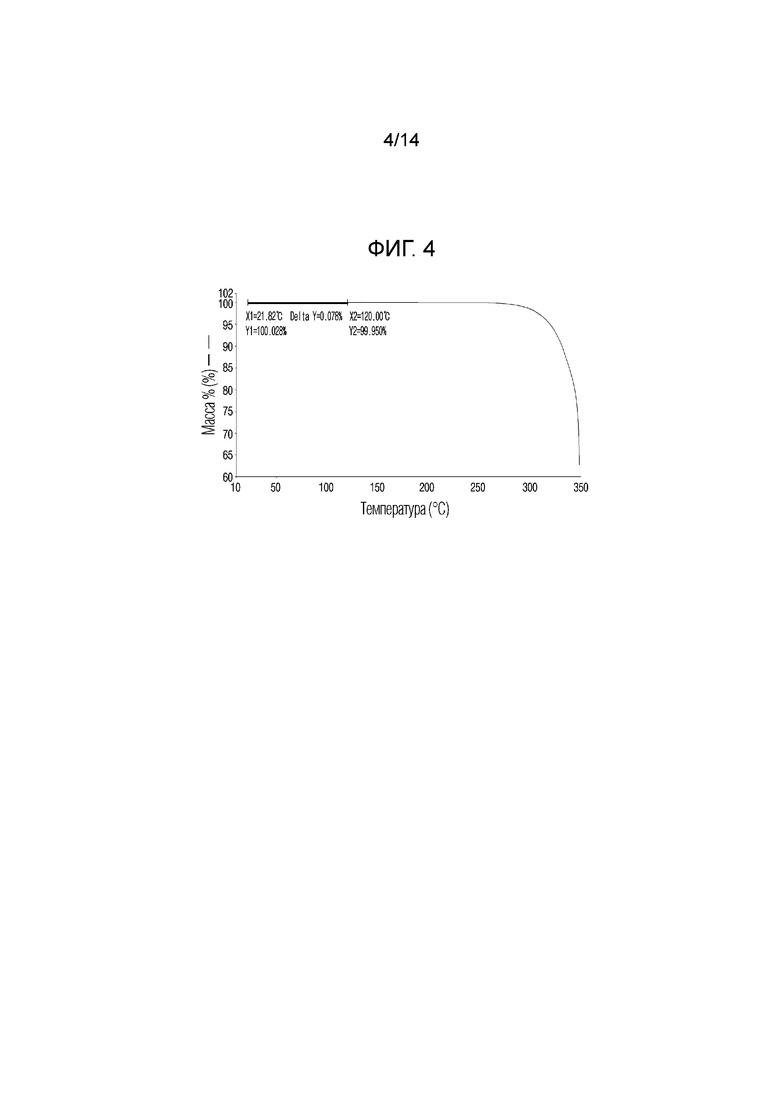
На фиг. 4 приведены данные термического гравиметрического анализа (TGA) кристаллической формы I, полученной в примере 1.
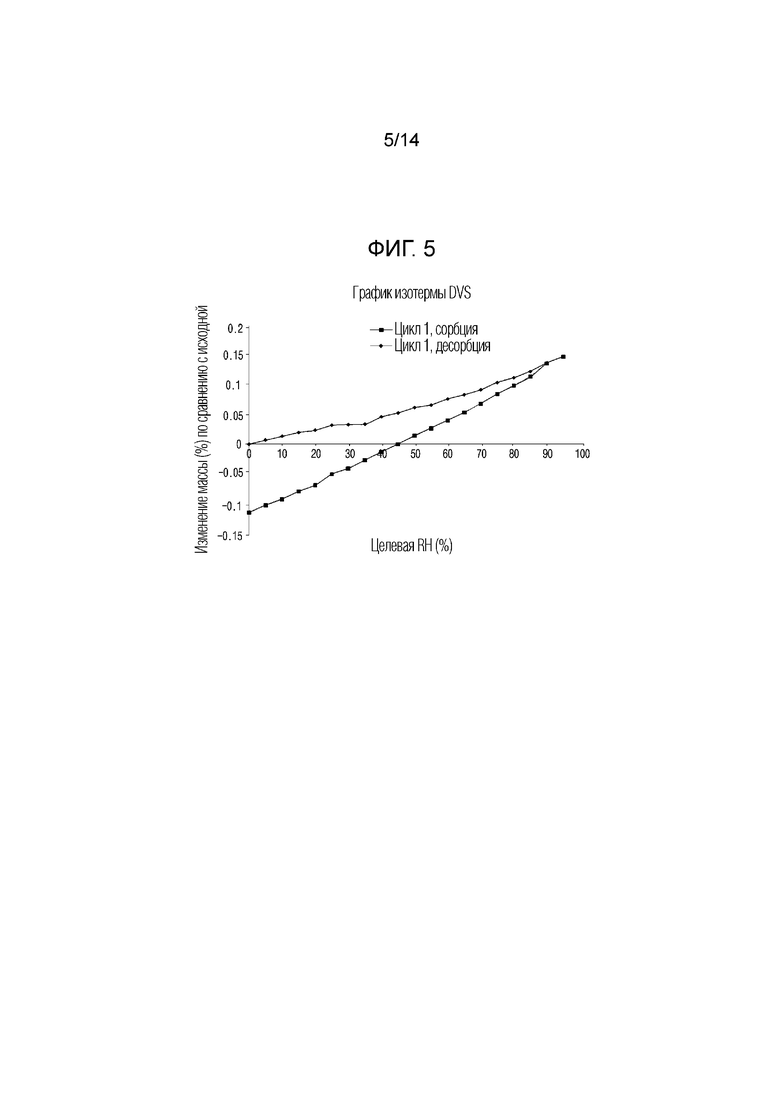
На фиг. 5 приведены данные динамической сорбции паров (DVS) кристаллической формы I, полученной в примере 1.
На фиг. 6 приведена фотография кристаллической формы I, полученной в примере 1, снятой в поляризационном микроскопе (PLM).
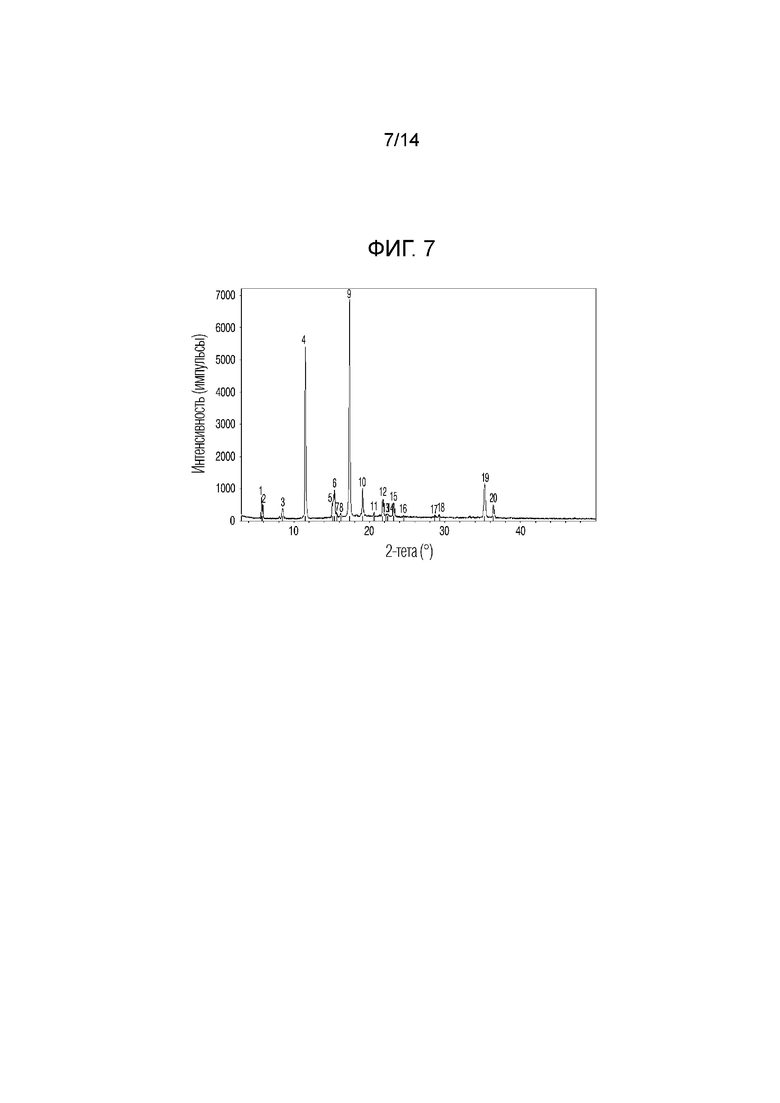
На фиг. 7 приведены данные порошковой рентгенографии (XRPD) кристаллической формы II, полученной в примере 2.
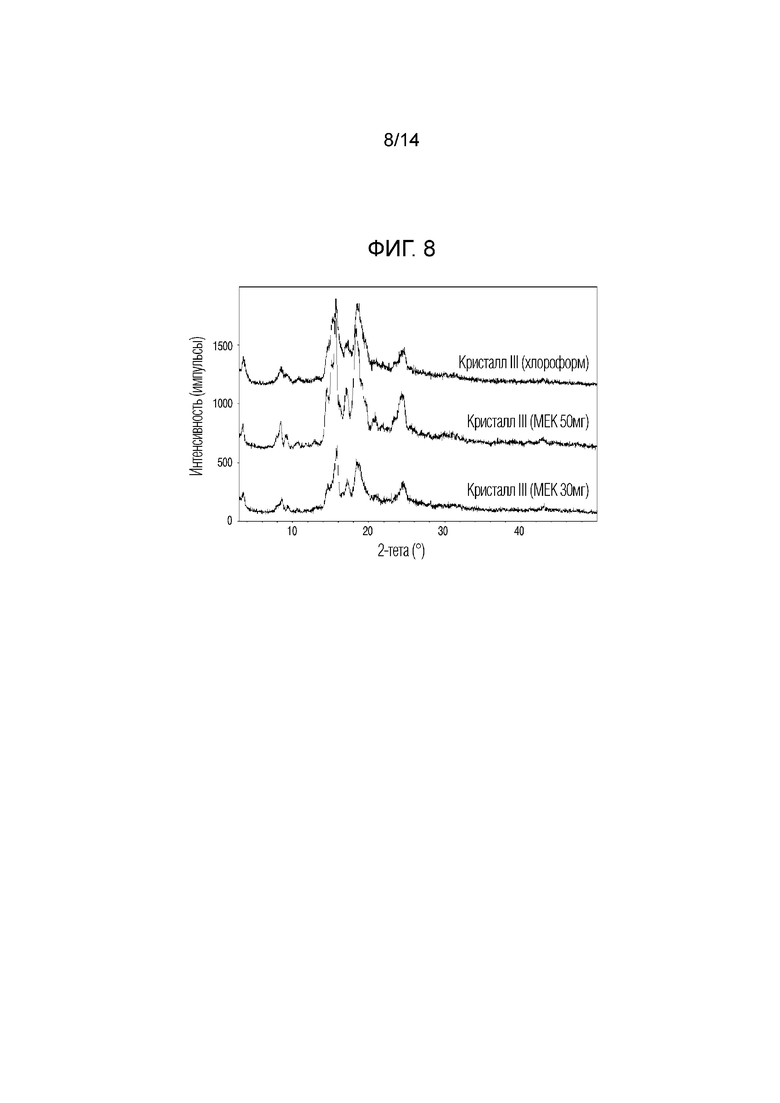
На фиг. 8 приведены данные порошковой рентгенографии (XRPD) кристаллической формы III, полученной в примере 3, полученной в растворителе MEK (30 мг, 50 мг) или хлороформе (50 мг).
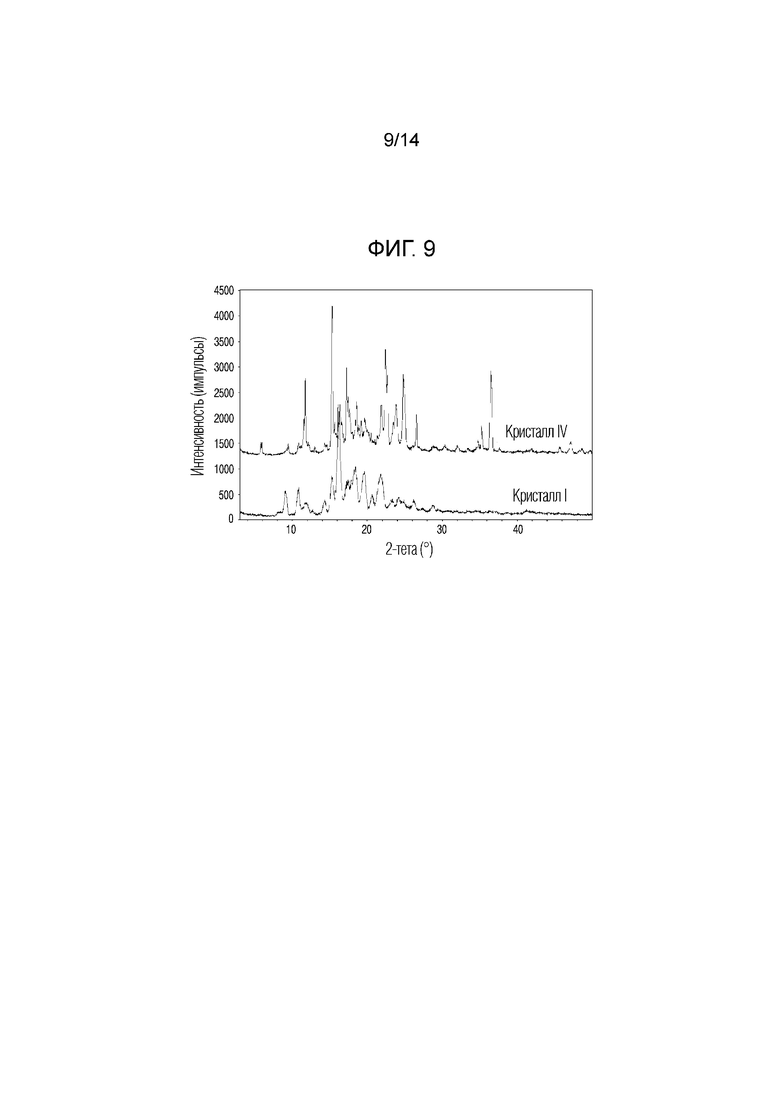
На фиг. 9 приведены данные порошковой рентгенографии (XRPD), на котором проведено сопоставление кристаллической формы IV, полученной в примере 4, с кристаллической формой I.
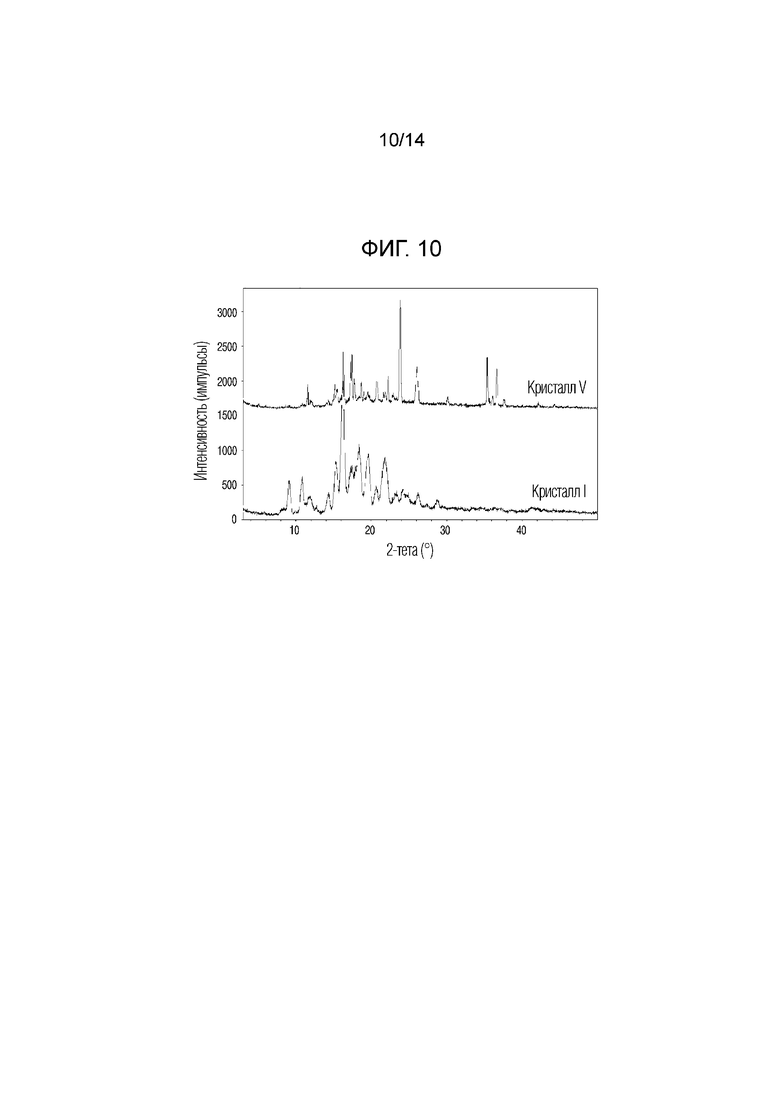
На фиг. 10 приведены данные порошковой рентгенографии (XRPD), на котором проведено сопоставление кристаллической формы V, полученной в примере 5, с кристаллической формой I.
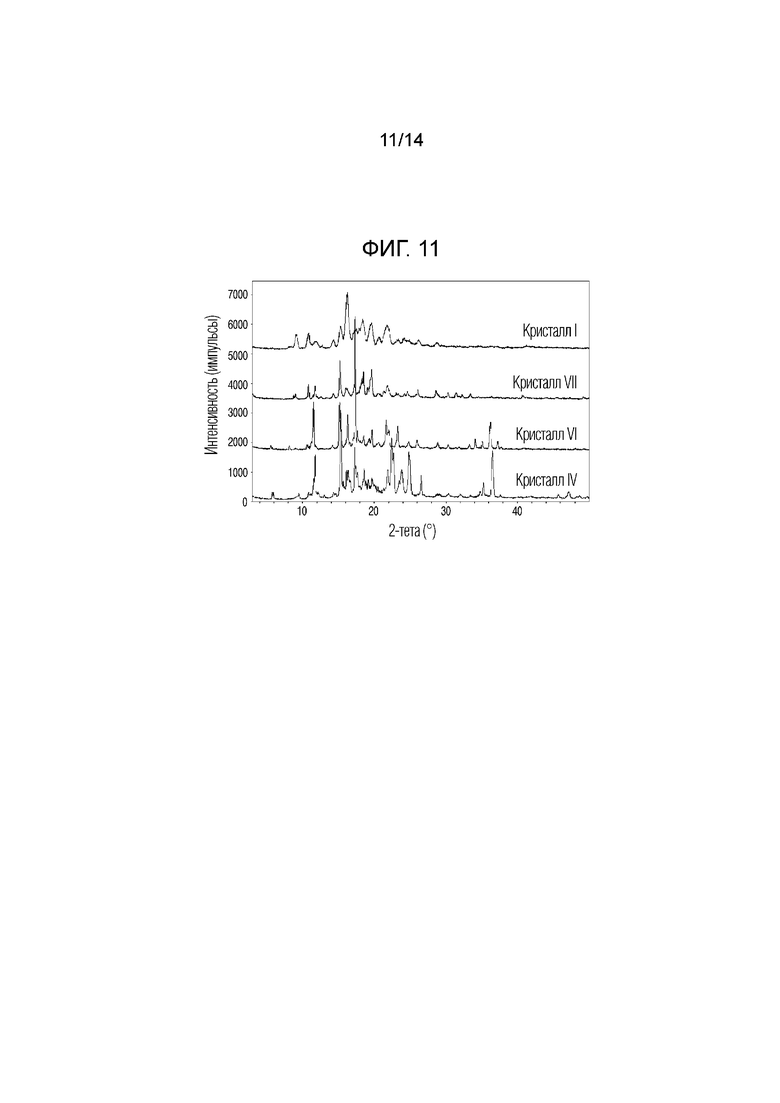
На фиг. 11 приведены данные порошковой рентгенографии (XRPD), на котором проведено сопоставление кристаллических форм VI и VII, полученных в примерах 6 и 7, с кристаллическими формами I и IV.
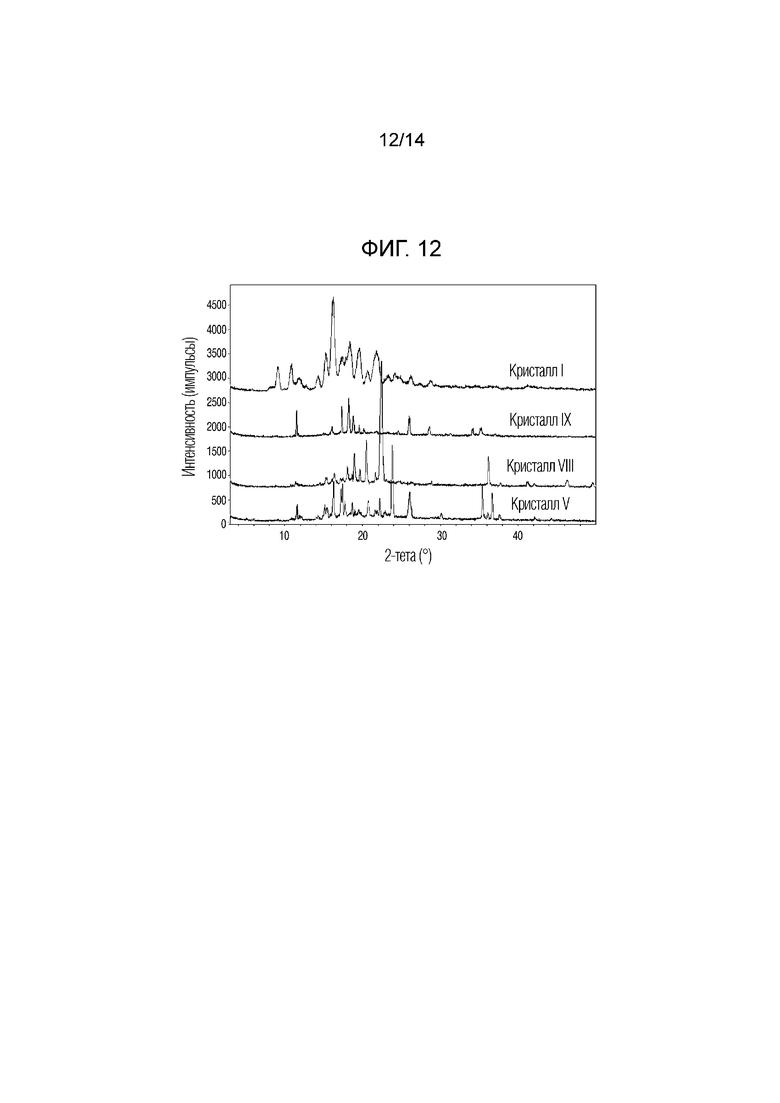
На фиг. 12 приведены данные порошковой рентгенографии (XRPD), на котором проведено сопоставление кристаллических форм VIII и IX, полученных в примерах 8 и 9, с кристаллическими формами I и V.
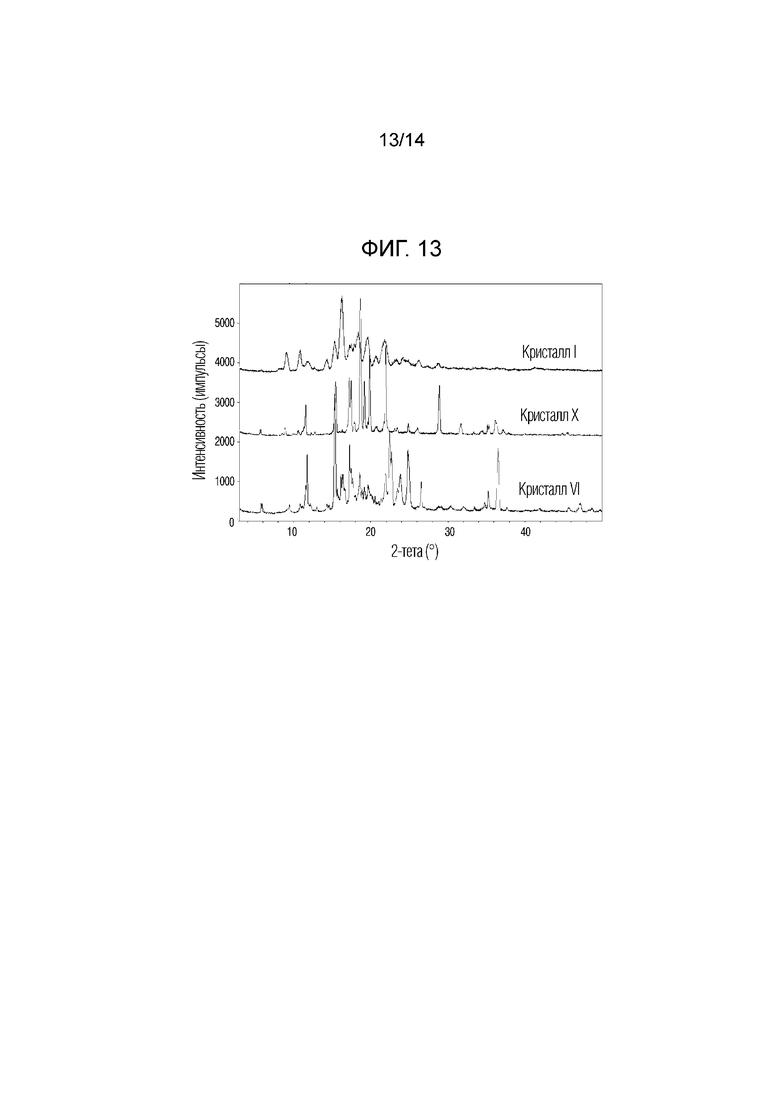
На фиг. 13 приведены данные порошковой рентгенографии (XRPD), на котором проведено сопоставление кристаллической формы X, полученной в примере 10, с кристаллическими формами I и VI.
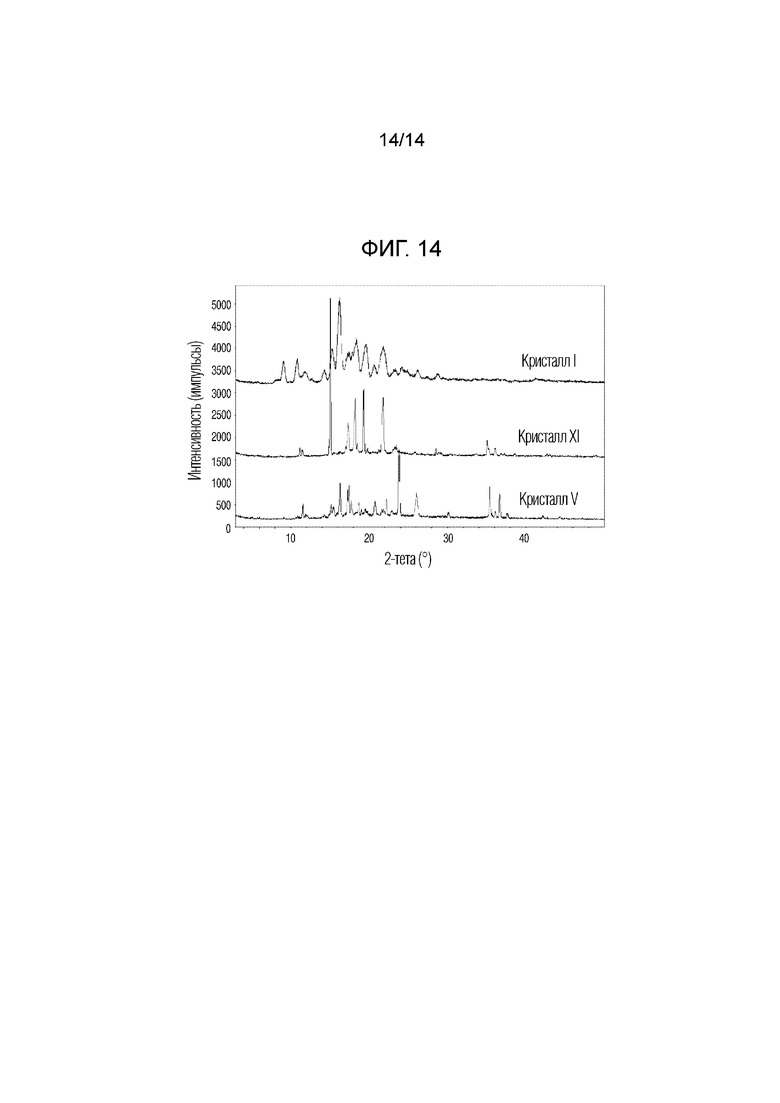
На фиг. 14 приведены данные порошковой рентгенографии (XRPD), на котором проведено сопоставление кристаллической формы XI, полученной в примере 11, с кристаллическими формами I и V.
Наилучший режим осуществления
Ниже настоящее изобретение описано подробно.
Варианты осуществления настоящего изобретения можно изменить с получением разных других вариантами осуществления и объем настоящего изобретения не ограничивается вариантами осуществления, описанными ниже. Специалисты с общей подготовкой в данной области техники полностью понимают, что варианты осуществления настоящего изобретения приведены для более точного описания настоящего изобретения. Кроме того, "включение" элемента в описание не исключает другие элементы, а может включать другие элементы, если специально не указано иное.
[формула 1]
(E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат
В настоящем изобретении из двух стереохимических структур соединения формулы 1, структурой, обладающей кристаллической формой, является (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат.
Одним объектом настоящего изобретения является соединение, отличающееся тем, что содержание α-изомера соединения, описывающееся формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, равно 85% или более и оно находится в твердом состоянии.
В настоящем изобретении содержание α-изомера может равняться 87% или более, 89% или более, 90% или более, 92% или более, 95% или более, 98% или более, или 99% или более.
Другим объектом настоящего изобретения является соединение, описывающееся формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат.
Другим объектом настоящего изобретения является новая кристаллическая форма I соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 16,0°±0,2°, 19,5°±0,2°, 18,3°±0,2°, 15,1°±0,2° и 21,9°±0,2°, на порошковой рентгенограмме (XRPD).
В настоящем изобретении кристаллическая форма I может дополнительно включать пики при углах дифракции 2Ɵ, равных 10,7°±0,2°, 8,9°±0,2°, 17,8°±0,2°, 21,3°±0,2° и 17,2°±0,2°.
Кроме того, кристаллическая форма I может дополнительно включать пики при углах дифракции 2Ɵ, равных 14,1°±0,2°, 20,5°±0,2° и 11,7°±0,2°.
Кроме того, кристаллическая форма I может включать пики при углах дифракции 2Ɵ, равных 24,0°±0,2°, 24,6°±0,2° и 26,1°±0,2°. Или кристаллическая форма I может дополнительно включать пики при углах дифракции 2Ɵ, равных 24,0°±0,2°, 24,6°±0,2° и 26,1°±0,2°.
Например, кристаллическая форма I может быть кристаллической формой, обладающей характеристическими пиками при углах дифракции 2Ɵ, равных 8,9°±0,2°, 10,7°±0,2°, 11,7°±0,2°, 14,1°±0,2°, 15,1°±0,2°, 16,0°±0,2°, 17,2°±0,2°, 17,8°±0,2°, 18,3°±0,2°, 19,5°±0,2°, 20,5°±0,2°, 21,3°±0,2°, 21,9°±0,2°, 24,0°±0,2°, 24,6°±0,2° и 26,1°±0,2°, обладающими относительной интенсивностью (I/I0), равной 10% или более.
Кроме того, настоящее изобретение относится к новой кристаллической форме, которая обладает эндотермическим пиком при 100,96°C±3°C по данным дифференциальной сканирующей калориметрии (DSC).
Одним объектом настоящего изобретения является новая кристаллическая форма II соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 17,3°±0,2°, 11,4°±0,2°, 35,2°±0,2°, 19,0°±0,2°, 15,3°±0,2° и 5,7°±0,2°, на порошковой рентгенограмме (XRPD).
Одним объектом настоящего изобретения является новая кристаллическая форма III соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 15,6°±0,2°, 18,2°±0,2°, 14,4°±0,2°, 24,6°±0,2°, 16,9°±0,2° и 17,2°±0,2°, на порошковой рентгенограмме (XRPD).
В настоящем изобретении кристаллическая форма III может дополнительно включать пики при углах дифракции 2Ɵ, равных 8,5°±0,2°, 24,3°±0,2°, 15,0°±0,2°, 3,5°±0,2°, 9,3°±0,2°, 20,6°±0,2°, 9,0°±0,2°, 24,0°±0,2°, 20,9°±0,2° и 20,7°±0,2°.
Например, кристаллическая форма III может быть кристаллической формой, обладающей характеристическими пиками при углах дифракции 2Ɵ, равных 3,5°±0,2°, 8,5°±0,2°, 9,0°±0,2°, 9,3°±0,2°, 14,4°±0,2°, 15,0°±0,2°, 15,6°±0,2°, 16,9°±0,2°, 17,2°±0,2°, 18,2°±0,2°, 20,6°±0,2°, 20,7°±0,2°, 20,9°±0,2°, 24,0°±0,2°, 24,3°±0,2° и 24,6°±0,2°, обладающими относительной интенсивностью (I/I0), равной 10% или более.
Одним объектом настоящего изобретения является новая кристаллическая форма IV соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 15,3°±0,2°, 22,4°±0,2°, 17,2°±0,2°, 36,5°±0,2°, 24,8°±0,2°, 22,7°±0,2°, 11,7°±0,2°, 18,5°±0,2°, 21,8°±0,2°, 23,8°±0,2°, 16,3°±0,2° и 17,6°±0,2°, на порошковой рентгенограмме (XRPD).
В настоящем изобретении кристаллическая форма IV может дополнительно включать пики при углах дифракции 2Ɵ, равных 26,5°±0,2°, 19,6°±0,2°, 19,1°±0,2°, 16,6°±0,2°, 23,4°±0,2°, 11,5°±0,2°, 18,8°±0,2°, 35,2°±0,2°, 15,6°±0,2°, 18,0°±0,2°, 20,1°±0,2°, 34,7°±0,2°, 47,1°±0,2°, 10,8°±0,2°, 5,9°±0,2° и 14,3°±0,2°.
Например, кристаллическая форма IV может быть кристаллической формой, обладающей характеристическими пиками при углах дифракции 2Ɵ, равных 5,9°±0,2°, 10,8°±0,2°, 11,5°±0,2°, 11,7°±0,2°, 14,3°±0,2°, 15,3°±0,2°, 15,6°±0,2°, 16,3°±0,2°, 16,6°±0,2°, 17,2°±0,2°, 17,6°±0,2°, 18,0°±0,2°, 18,5°±0,2°, 18,8°±0,2°, 19,1°±0,2°, 19,6°±0,2°, 20,1°±0,2°, 21,8°±0,2°, 22,4°±0,2°, 22,7°±0,2°, 23,4°±0,2°, 23,8°±0,2°, 24,8°±0,2°, 26,5°±0,2°, 34,7°±0,2°, 35,2°±0,2°, 36,5°±0,2° и 47,1°±0,2°, обладающими относительной интенсивностью (I/I0), равной 10% или более.
Одним объектом настоящего изобретения является новая кристаллическая форма V соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 23,8°±0,2°, 16,2°±0,2°, 17,4°±0,2°, 35,3°±0,2°, 26,0°±0,2°, 36,6°±0,2°, 22,2°±0,2° и 20,7°±0,2°, на порошковой рентгенограмме (XRPD).
В настоящем изобретении кристаллическая форма V может дополнительно включать пики при углах дифракции 2Ɵ, равных 17,7°±0,2°, 18,6°±0,2°, 11,5°±0,2°, 15,1°±0,2°, 15,4°±0,2°, 19,0°±0,2°, 19,4°±0,2°, 21,8°±0,2°, 21,6°±0,2°, 22,9°±0,2°, 36,0°±0,2°, 30,1°±0,2°, 18,4°±0,2°, 23,3°±0,2°, 37,5°±0,2° и 11,8°±0,2°.
Например, кристаллическая форма V может быть кристаллической формой, обладающей характеристическими пиками при углах дифракции 2Ɵ, равных 11,5°±0,2°, 11,8°±0,2°, 15,1°±0,2°, 15,4°±0,2°, 16,2°±0,2°, 17,4°±0,2°, 17,7°±0,2°, 18,4°±0,2°, 18,6°±0,2°, 19,0°±0,2°, 19,4°±0,2°, 20,7°±0,2°, 21,6°±0,2°, 21,8°±0,2°, 22,2°±0,2°, 22,9°±0,2°, 23,3°±0,2°, 23,8°±0,2°, 26,0°±0,2°, 30,1°±0,2°, 35,3°±0,2°, 36,0°±0,2°, 36,6°±0,2° и 37,5°±0,2°, обладающими относительной интенсивностью (I/I0), равной 10% или более.
Одним объектом настоящего изобретения является новая кристаллическая форма VI соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 17,3°±0,2°, 15,2°±0,2° и 11,5°±0,2°, на порошковой рентгенограмме (XRPD).
В настоящем изобретении кристаллическая форма VI может дополнительно включать пики при углах дифракции 2Ɵ, равных 16,2°±0,2°, 21,6°±0,2°, 36,1°±0,2°, 23,2°±0,2°, 19,6°±0,2°, 22,0°±0,2°, 17,6°±0,2° и 18,5°±0,2°.
Например, кристаллическая форма VI может быть кристаллической формой, обладающей характеристическими пиками при углах дифракции 2Ɵ, равных 11,5°±0,2°, 15,2°±0,2°, 16,2°±0,2°, 17,3°±0,2°, 17,6°±0,2°, 18,5°±0,2°, 19,6°±0,2°, 21,6°±0,2°, 22,0°±0,2°, 23,2°±0,2° и 36,1°±0,2°, обладающими относительной интенсивностью (I/I0), равной 10% или более.
Одним объектом настоящего изобретения является новая кристаллическая форма VII соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 15,2°±0,2°, 19,6°±0,2°, 18,5°±0,2°, 18,3°±0,2°, 17,2°±0,2°, 10,8°±0,2°, 21,8°±0,2°, 11,7°±0,2°, 18,1°±0,2°, 19,0°±0,2° и 16,0°±0,2°, на порошковой рентгенограмме (XRPD).
В настоящем изобретении кристаллическая форма VII может дополнительно включать пики при углах дифракции 2Ɵ, равных 26,0°±0,2°, 28,5°±0,2°, 21,3°±0,2°, 24,6°±0,2°, 30,3°±0,2°, 23,0°±0,2°, 31,4°±0,2°, 20,4°±0,2°, 14,3°±0,2°, 33,4°±0,2°, 23,3°±0,2°, 9,0°±0,2°, 25,8°±0,2°, 32,3°±0,2°, 11,4°±0,2°, 8,7°±0,2°, 40,7°±0,2°, 36,4°±0,2°, 12,6°±0,2°, 49,1°±0,2°, 34,6°±0,2°, 37,6°±0,2° и 24,2°±0,2°.
Например, кристаллическая форма VII может быть кристаллической формой, обладающей характеристическими пиками при углах дифракции 2Ɵ, равных 8,7°±0,2°, 9,0°±0,2°, 10,8°±0,2°, 11,4°±0,2°, 11,7°±0,2°, 12,6°±0,2°, 14,3°±0,2°, 15,2°±0,2°, 16,0°±0,2°, 17,2°±0,2°, 18,1°±0,2°, 18,3°±0,2°, 18,5°±0,2°, 19,0°±0,2°, 19,6°±0,2°, 20,4°±0,2°, 21,3°±0,2°, 21,8°±0,2°, 23,0°±0,2°, 23,3°±0,2°, 24,2°±0,2°, 24,6°±0,2°, 25,8°±0,2°, 26,0°±0,2°, 28,5°±0,2°, 30,3°±0,2°, 31,4°±0,2°, 32,3°±0,2°, 33,4°±0,2°, 34,6°±0,2°, 36,4°±0,2°, 37,6°±0,2°, 40,7°±0,2° и 49,1°±0,2°, обладающими относительной интенсивностью (I/I0), равной 10% или более.
Одним объектом настоящего изобретения является новая кристаллическая форма VIII соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 22,4°±0,2° и 20,5°±0,2°, на порошковой рентгенограмме (XRPD).
В настоящем изобретении кристаллическая форма VIII может дополнительно включать пики при углах дифракции 2Ɵ, равных 16,3°±0,2°, 18,0°±0,2°, 18,9°±0,2°, 19,6°±0,2°, 21,6°±0,2 и 36,1°±0,2.
Например, кристаллическая форма VIII может быть кристаллической формой, обладающей характеристическими пиками при углах дифракции 2Ɵ, равных 16,3°±0,2°, 18,0°±0,2°, 18,9°±0,2°, 19,6°±0,2°, 20,5°±0,2°, 21,6°±0,2°, 22,4°±0,2° и 36,1°±0,2°, обладающими относительной интенсивностью (I/I0), равной 10% или более.
Одним объектом настоящего изобретения является новая кристаллическая форма IX соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 18,2°±0,2°, 17,3°±0,2°, 11,5°±0,2°, 18,8°±0,2°, 26,0°±0,2°, 19,5°±0,2° и 16,0°±0,2°, на порошковой рентгенограмме (XRPD).
В настоящем изобретении кристаллическая форма IX может дополнительно включать пики при углах дифракции 2Ɵ, равных 35,2°±0,2°, 34,1°±0,2°, 20,1°±0,2°, 24,6°±0,2°, 21,8°±0,2°, 23,2°±0,2°, 14,9°±0,2°, 11,7°±0,2°, 34,5°±0,2°, 31,3°±0,2°, 37,0°±0,2°, 27,8°±0,2°, 28,6°±0,2°, 14,2°±0,2°, 33,1°±0,2°, 5,5°±0,2°, 37,5°±0,2°, 8,9°±0,2° и 12,4°±0,2°.
Например, кристаллическая форма IX может быть кристаллической формой, обладающей характеристическими пиками при углах дифракции 2Ɵ, равных 5,5°±0,2°, 8,9°±0,2°, 11,5°±0,2°, 11,7°±0,2°, 12,4°±0,2°, 14,2°±0,2°, 14,9°±0,2°, 16,0°±0,2°, 17,3°±0,2°, 18,2°±0,2°, 18,8°±0,2°, 19,5°±0,2°, 20,1°±0,2°, 21,8°±0,2°, 23,2°±0,2°, 24,6°±0,2°, 26,0°±0,2°, 27,8°±0,2°, 28,6°±0,2°, 31,3°±0,2°, 33,1°±0,2°, 34,1°±0,2°, 34,5°±0,2°, 35,2°±0,2°, 37,0°±0,2° и 37,5°±0,2°, обладающими относительной интенсивностью (I/I0), равной 10% или более.
Одним объектом настоящего изобретения является новая кристаллическая форма X соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 18,6°±0,2°, 21,9°±0,2°, 19,8°±0,2°, 17,2°±0,2°, 17,4°±0,2°, 19,1°±0,2°, 15,4°±0,2° и 28,9°±0,2°, на порошковой рентгенограмме (XRPD).
В настоящем изобретении кристаллическая форма X может дополнительно включать пики при углах дифракции 2Ɵ, равных 11,5°±0,2°, 36,1°±0,2°, 17,8°±0,2°, 24,8°±0,2° и 31,6°±0,2°.
Например, кристаллическая форма X может быть кристаллической формой, обладающей характеристическими пиками при углах дифракции 2Ɵ, равных 11,5°±0,2°, 15,4°±0,2°, 17,2°±0,2°, 17,4°±0,2°, 17,8°±0,2°, 18,6°±0,2°, 19,1°±0,2°, 19,8°±0,2°, 21,9°±0,2°, 24,8°±0,2°, 28,9°±0,2°, 31,6°±0,2° и 36,1°±0,2°, обладающими относительной интенсивностью (I/I0), равной 10% или более.
Одним объектом настоящего изобретения является новая кристаллическая форма XI соединения, описывающегося формулой (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат, обладающая пиками при углах дифракции 2Ɵ, равных 15,0°±0,2°, 19,3°±0,2°, 21,8°±0,2°, 18,2°±0,2°, 17,3°±0,2° и 35,0°±0,2°, на порошковой рентгенограмме (XRPD).
Другим объектом настоящего изобретения является фармацевтическая композиция для предупреждения или лечения заболевания проницаемости сосудов, содержащая любое из соединений и новых кристаллических формы I - V соединения. Или настоящее изобретение относится к фармацевтической композиции для предупреждения или лечения заболевания проницаемости сосудов, содержащей любую из кристаллических форм I - XI соединения.
В настоящем изобретении заболеванием проницаемости сосудов может быть диабет, воспаление, ретинопатия, диабетическая ретинопатия, дегенерация желтого пятна, глаукома, стеноз, рестеноз, артериосклероз, атеросклероз, отек головного мозга, артрит, артропатия, увеит, воспалительная болезнь кишечника, отек желтого пятна, рак, иммунный противораковый адъювант, наследственный ангионевротический отек (HAE), гиперлипидемия, ишемическая болезнь, диабетическое изъязвление стопы, легочная гипертензия, острое поражение легких, ишемия миокарда, сердечная недостаточность, острая ишемия нижних конечностей, инфаркт миокарда, удар, ишемия или реперфузионное поражение, VLS (синдром проницаемости сосудов), отек, отторжение трансплантата, ожоги, острый или респираторный дистресс-синдром у взрослых (ARDS), сепсис или аутоиммунное заболевание.
Осуществление настоящего изобретения
Ниже настоящее изобретение подробно описано с помощью следующих примеров и экспериментальных примеров.
Однако следующие примеры и экспериментальные примеры приведены только для иллюстрации настоящего изобретения и ими не ограничивается содержание настоящего изобретения.
Экспериментальные методики
1. Рентгеновский порошковый дифрактометр (XRPD)
Порошковую рентгенограмму (XRPD) получали на приборе Shimadzu XRD-6000.
- Установки
Трубка: Cu: K-α (λ=1,54056 Å)
Генератор: напряжение: 40 кВ; ток: 30 мА
Диапазон сканирования: 3~50 градусов. Скорость сканирования: 5 градусов/мин
2. Дифференциальный сканирующий калориметр (DSC)
Образцы соединений (~1 мг) исследовали в алюминиевой чашке с точечным отверстием при продувке азотом.
- Установки
Линейная скорость повышения температуры: 20°C/мин в диапазоне 30°C~300°C
Продувка азотом: 50 мл/мин
Образцы: Примерно 1 мг
3. Термический гравиметрический анализ (TGA)
Образцы соединений (4~6 мг) отвешивали чашку и нагревали при продувке азотом.
- Установки
Линейная скорость повышения температуры: 20°C/мин в диапазоне 30°C~300°C
Продувка азотом: 50 мл/мин
Образцы: Примерно 5 мг
4. Динамическая сорбция паров (DVS)
Примерно 10 мг образцов использовали для исследования их профилей сорбции/десорбции воды.
- Установки
Температура: 25°C
Равновесие: dm/dt: 0,01%/мин
Диапазон измерения относительной влажности (RH): 0%~95%~0% RH
Шаг измерения относительной влажности (RH): 5% RH
Образцы: 10~20 мг
- Классификация гигроскопичности
Гигроскопичность: Критерий сорбции воды*
Расплывающийся за счет поглощения воды: Абсорбируется достаточное количество воды в форме жидкости
Очень гигроскопичный: W%≥15%
Гигроскопичный: W%≥2%
Немного гигроскопичный: W%≥0,2%
Негигроскопичный: W%<0,2%
*25±1°C, 80±2%RH
5. Поляризационный оптический микроскоп (PLM)
Образцы, диспергированные в силиконовом масле, изучали с помощью окуляра: 10× и объектива: 10× или 5× в скрещенных поляризаторах регистрировали фотокамерой/компьютерной системой со шкалой увеличения.
6. Высокоэффективная жидкостная хроматография (HPLC)
Колонка: Agilent Zorbax Rx C8, 4,6 мм×250 мм, 5,0 мкм
Подвижная фаза: 85% ацетонитрила
Скорость потока: 1,0 мл/мин
Длина волны: 205 нм
Инжектируемый объем: 10 мкл
Температура колонки: 30°C
Разбавитель: 100% ацетонитрила
<Пример получения 1> Получение соединения 1
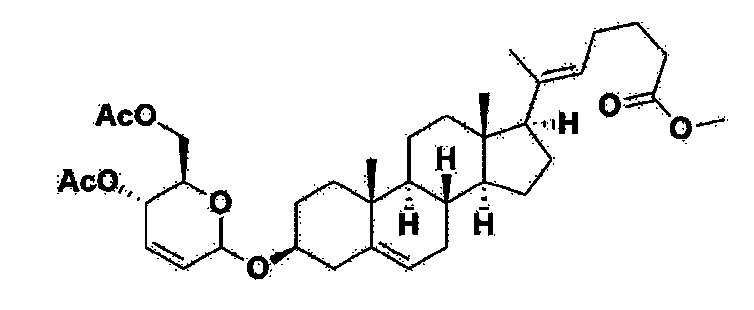
Соединение 1, описывающееся следующей формулой 1, можно получить способом получения, описанным в заявке на патент Кореи № 10-2019-0166864 (неопубликованная). В частности, соединение можно получить способом, соответствующим следующей схеме реакции 1 или 2.
[формула 1]
[Схема реакции 1]
Как показано на схеме реакции 1, соединение 1, описывающееся формулой 1, можно получить способом, включающим следующие стадии:
получение соединения, описывающегося формулой 6 по реакции прегненолона с соединением, описывающимся формулой 8 (стадия 1);
получение соединения, описывающегося формулой 4, по реакции соединения, описывающегося формулой 6, полученного на стадии 1 выше, с соединением, описывающимся формулой 7, с последующей обработкой в кислой среде (стадия 2);
получение соединения, описывающегося формулой 2 по реакции соединения, описывающегося формулой 4, полученного на стадии 2 выше, с соединением, описывающимся формулой 5 (стадия 3); и
получение соединения, описывающегося формулой 1 по реакции соединения, описывающегося формулой 2, полученного на стадии 3 выше, с соединением, описывающимся формулой 3, в присутствии катализатора (стадия 4).
В настоящем изобретении R1 на схеме реакции 1 означает линейный или разветвленный C1-10 алкил.
[Схема реакции 2]
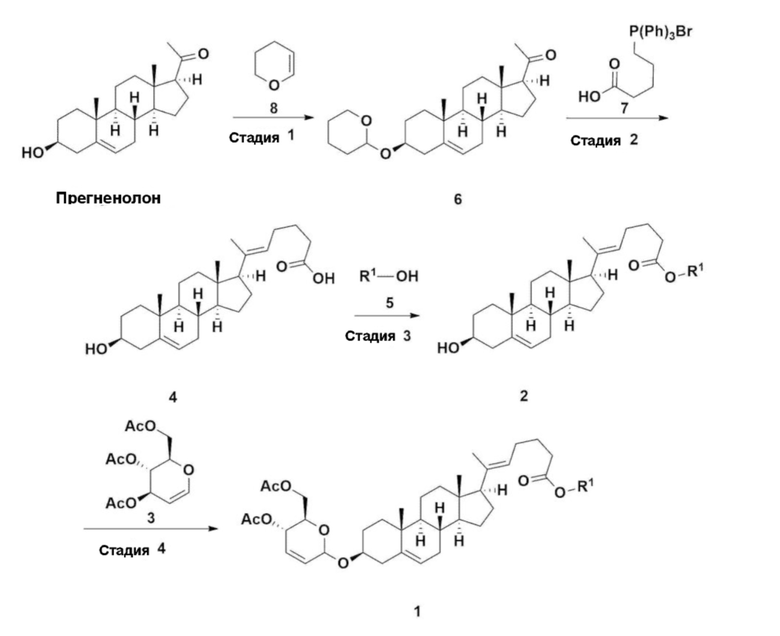
Как показано на схеме реакции 2, соединение 1, описывающееся формулой 1, можно получить способом, включающим следующие стадии:
получение соединения, описывающегося формулой 6, по реакции прегненолона с соединением, описывающимся формулой 8 (стадия 1);
получение соединения, описывающегося формулой 10, по реакции соединения, описывающегося формулой 6, соединения, описывающегося формулой 7, и соединения, описывающегося формулой 9 (стадия 2);
получение соединения, описывающегося формулой 2 по реакции соединения, описывающегося формулой 10, полученного на стадии 2 выше, с кислотой (стадия 3); и
получение соединения, описывающегося формулой 1, по реакции соединения, описывающегося формулой 2, полученного на стадии 3 выше, с соединением, описывающимся формулой 3, в присутствии катализатора (стадия 4).
В настоящем изобретении R1 означает линейный или разветвленный C1-10 алкил; и X1 означает галоген.
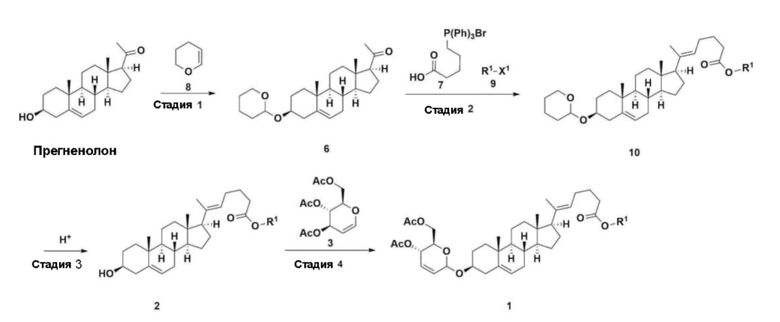
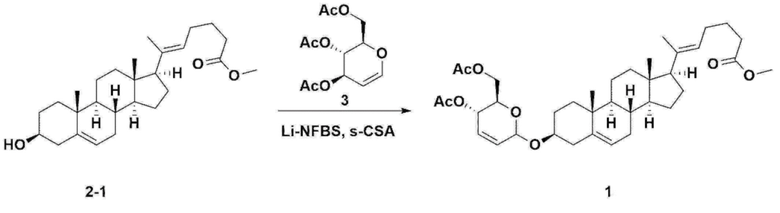
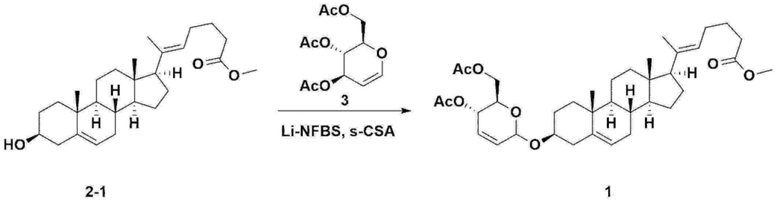
С другой стороны, соединение 1, описывающееся формулой 1, использующееся в следующих примерах и экспериментальных примерах настоящего изобретения, получали по методике, приведенной на схеме реакции 3 ниже.
[Схема реакции 3]
Конкретная методика получения является следующей.
Стадия 1: Получение соединения 6-1
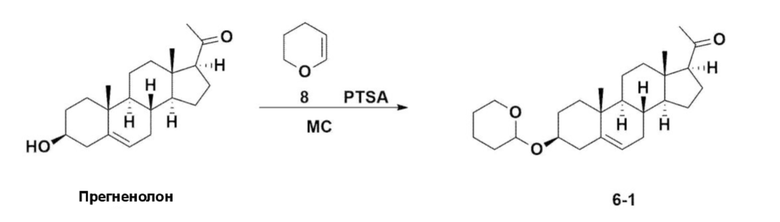
Прегненолон (200 г, 0,632 моля) добавляли к 2000 мл дихлорметана, который помещали в колбу объемом 5 л, снабженную термометром, в которую добавляли 173 мл (1,896 моля) 3,4-дигидро-2H-пирана. После снижения температуры до 0-5°C 3,0 г (15,8 ммоля) моногидрата п-толуолсульфоновой кислоты растворяли в 50 мл тетрагидрофурана (THF) и по каплям добавляли, затем перемешивали при 0°C в течение 1,5 ч. К реакционной смеси добавляли 800 мл насыщенного водного раствора бикарбоната натрия и 10 мл триэтиламина (TEA) при 0°C, затем перемешивали. После разделения слоев органический слой промывали с помощью 800 мл рассола и водные слои повторно экстрагировали с помощью 200 мл дихлорметана, объединяли с органическим слоем, сушили над 200 г безводного сульфата натрия, фильтровали и перегоняли при пониженном давлении. К полученному остатку добавляли 1000 мл MeOH и 5 мл TEA, нагревали до полного растворения и температуру снижали, затем перемешивали при -5°C в течение 1 ч. Полученное твердое вещество отфильтровывали и промывали с помощью 200 мл MeOH и получали 232,0 г (0,579 моля) соединения 6-1 (THP-прегненолон) в виде чистого белого твердого вещества (выход: 91,6%).
1H-NMR (400 MHz, CDCl3): δ 5,33-5,36 (m, 1H), 4,71-4,72 (m, 1H), 3,85-3,94 (m, 1H), 3,46-3,56 (m, 2H), 1,00-2,55 (m, 32H), 0,62 (s, 3H).
Стадия 2: Получение соединения 4-2
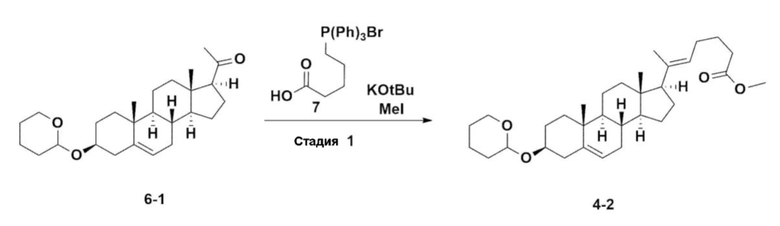
После установки холодильника, нагревательного кожуха и механической мешалки в реактор объемом 5 л его нагревали при 119°C (наружная температура) и охлаждали до комнатной температуры, пропуская азот в течение 5 мин и в него добавляли 332,5 г (0,75 моля) 4-(карбоксибутил)трифенилфосфонийбромида, 168,1 г (1,50 моля) трет-бутоксида калия, 2000 мл безводного толуола и 750 мл безводного тетрагидрофурана, затем перемешивали в течение примерно 2 ч при нагревании при 119°C (наружная температура, слабое внутреннее кипение).
Соединение 6-1 (100,0 г, 0,250 моля) растворяли в 500 мл безводного толуола и добавляли к раствору реакционной смеси, затем проводили реакцию в течение примерно 20 ч.
После завершения реакции реакционную смесь охлаждали до комнатной температуры, к ней добавляли 320 мл (5,14 моля) метилйодида и 1000 мл ацетона, затем перемешивали при комнатной температуре в течение 15 ч. Реакционную смесь перегоняли при пониженном давлении для удаления большей части органического растворителя, к ней добавляли 1500 мл этилацетата для растворения, затем промывали с помощью 1000 мл насыщенного водного раствора хлорида аммония . Органический слой дважды промывали с помощью 1000 мл воды и 1000 мл рассола, сушили над 100 г сульфата натрия, фильтровали через 80 г целита и концентрировали.
Полученный остаток растворяли в 2000 мл метанола, затем перемешивали в течение 13 ч при 10°C и 1 ч при 4-5°C. Полученное твердое вещество отфильтровывали, промывали с помощью 200 мл метанола и сушили в вакууме и получали 66,2 г соединения 4-2 в виде белого твердого вещества (выход: 53,2%).
1H NMR(400MHz, CDCl3 ): δ 5,36(t, J=5,80 Hz, 1H), 5,16(t, J=7,00 Hz, 1H), 4,71(m, 1H), 3,93(m, 1H), 3,66(s, 3H), 3,56(m, 2H), 2,37-0,88(m, 38H), 0,54(s, 3H).
Стадия 3: Получение соединения 2-1
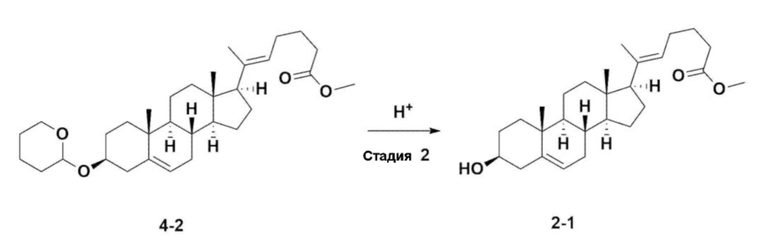
Соединение 4-2 (100 г, 0,200 моля), 1000 мл метанола и 3,82 г (0,020 моля) моногидрата п-толуолсульфоновой кислоты добавляли в колбу объемом 2 л, снабженную термометром, затем перемешивали при 60°C в течение 3 ч.
После завершения реакции раствор реакционной смеси перемешивали при комнатной температуре. После образования твердого вещества смесь перемешивали при комнатной температуре в течение 30 мин и перемешивали при 10°C в течение 1 ч. Раствор реакционной смеси фильтровали, промывали с помощью 100 мл охлажденного метанола и сушили в вакууме и получали 61,2 г (0,150 моля) соединения 2-1 в виде белого твердого вещества (выход: 75,0%).
1H-NMR (400 MHz, CDCl3): δ 5,33-5,35(m, 1H), 5,12-5,16 (m, 1H), 3,66 (s, 3H), 3,50-3,52 (m, 1H), 0,98-2,32 (m, 33H), 0,53 (s, 3H).
Стадия 4: Получение соединения 1
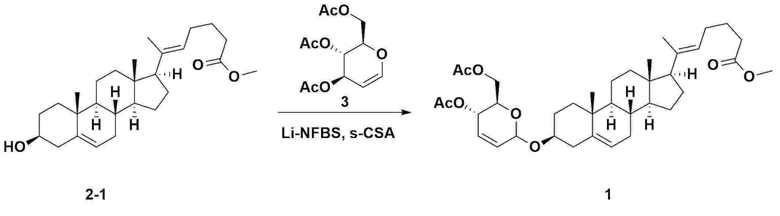
Соединение 2-1 (42,0 г, 0,101 моля) и три-изо-O-ацетил-D-глюкаль (34,5 г, 0,126 моля) растворяли в 126 мл безводного толуола и 252 мл ацетонитрила, затем добавляли в колбу объемом 1 л, снабженную термометром и водяной баней. При поддержании температуры, равной 30-35°C, добавляли нонафтор-1-бутилсульфонат лития (3,87 г, 0,0130 моля) и (s)-камфорсульфоновую кислоту (0,117 г, 0,0005 моля), затем перемешивали в течение 2 ч. После завершения реакции реакцию останавливали с помощью 504 мл насыщенного водного раствора бикарбоната натрия и экстрагировали с помощью 630 мл гептана. Органический слой дважды промывали с помощью 504 мл насыщенного водного раствора бикарбоната натрия и 504 мл рассола. Органический слой перемешивали после добавления 42 г безводного сульфата натрия и 34 г древесного угля, фильтровали через 34 г целита, промывали с помощью 210 мл метиленхлорида, объединяли с фильтратом, концентрировали и сушили в вакууме.
1H-NMR (400 MHz, CDCl3): δ 5,79-5,88 (m, 2H), 5,35-5,36 (m, 1H), 5,27-5,29 (m, 1H), 5,12-5,16 (m, 2H), 4,15-4,24 (m, 3H), 3,66 (s, 3H), 3,54-3,57 (m, 1H), 0,91-2,32 (m, 38H), 0,54 (s, 3H).
Стереохимический анализ соединения 1
Соединение 1, полученное по схеме реакции 3, содержится в виде смеси α- и β-изомеров. В настоящем изобретении маслообразный остаток нагревали и растворяли в 336 мл этанола при 50-55°C, затем перемешивали при 30°C. После образования твердого вещества температуру снижали до 0°C, затем перемешивали в течение 30 мин. Твердое вещество отфильтровывали при пониженном давлении и сушили в вакууме для отделения α-изомера в виде кристаллов и фильтрат обрабатывали с помощью колоночной хроматографии для отделения β-изомера. В настоящем изобретении α-изомер получали в твердом состоянии и чистота α-изомера составляла 93,0%.
Данные NMR, проведенного для подтверждения структур α- и β-изомеров приведены на фиг. 1. Как показано на фиг. 1, подтверждено, что полученное неочищенное соединение 1 содержит α-изомер или β-изомер, обладающие разными стереохимическими структурами.
С другой стороны, неочищенное соединение, полученное способом получения, раскрытом в публикации выложенного патента Кореи № 10-2011-0047170, являлось масляной фазой, в которой смешаны α-изомер и β-изомер, тогда как соединение, полученное по методике получения, раскрытой в настоящей заявке, являлось твердой фазой α-изомера.
<Пример 1> Получение кристаллической формы I соединения 1
Примерно 50 мг соединения 1, полученного в примере получения 1, диспергировали в растворителе, обладающем относительно низкой растворяющей способностью (вода, метанол или этанол), затем перемешивали при комнатной температуре (25°C) в течение 3 дней. Полученное влажное твердое вещество отделяли центрифугированием. Твердое вещество сушили при комнатной температуре в течение 24 ч при пониженном давлении. Для анализа характеристик кристаллической формы высушенного твердого вещества проводили следующие эксперименты.
Анализ характеристик
Для анализа характеристик кристаллической формы, полученной в примере 1, кристаллическую форму в виде белого порошкообразного вещества исследовали с помощью определения растворимости, порошковой рентгенографии (XRPD), дифференциальной сканирующей калориметрии (DSC), термического гравиметрического анализа (TGA), динамической сорбции паров (DVS) и поляризационной микроскопии (PLM) по методикам, описанным в экспериментальных методиках. Результаты приведены на фиг. 2-6 соответственно.
Определение растворимости
Определение растворимости проводили путем осмотра и проводимого вручную разбавления при комнатной температуре. В частности, примерно 4,0 мг соединения 1, полученного в примере получения 1 отвешивали в стеклянный сосуд объемом 4,0 мл, в который последовательно добавляли растворитель, пока невооруженным глазом не переставали обнаруживаться частицы (т. е. все растворилось) или частицы оставались (т. е. не происходило растворение) даже после добавления 4 мл растворителя (например, полный объем равнялся 200, 500, 1000, 2000 или 4000 мкл). Для расчета растворимости в каждом растворителе регистрировали полное количество растворителя. Результаты приведены в таблице 1 ниже.
Таблица 1
Порошковая рентгенография (XRPD)
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, в спектре XRPD кристаллической формы, приведенном на фиг. 2, приведены в таблице 2 ниже.
Таблица 2
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Обычно диапазон погрешности угла дифракции (2Ɵ) в порошковой рентгенографии составляет порядка ±0,2°. Поэтому, следует понимать, что значения угла дифракции также включают значения в диапазоне погрешности примерно ±0,2°. Соответственно, настоящее изобретение включает не только кристаллы, обладающие одинаковыми углами дифракции и пиками в порошковой рентгенографии, но и кристаллы, обладающие углами дифракции, отличающимися на величину, согласующуюся с диапазоном погрешности, равным ±0,2°.
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 8,9°, 10,7°, 11,7°, 14,1°, 15,1°, 16,0°, 17,2°, 17,8°, 18,3°, 19,5°, 20,5°, 21,3°, 21,9°, 24,0°, 24,6° и 26,1° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой I.
Динамическая сорбция паров (DVS)
Результаты анализа зависимости DVS для кристаллической формы I приведены в таблице 3 ниже.
Таблица 3
По приведенным выше данным XRPD было подтверждено, что кристаллическая форма I соединения 1, полученная в примере 1 настоящего изобретения, обладала превосходной кристалличностью. По данным TGA было подтверждено, что кристаллическая форма I соединения 1 характеризовалась потерей массы, равной 0,078% при нагревании от комнатной температуры до 120°C. По данным DSC был подтвержден один эндотермический пик (100,96°C), который соответствовал температуре плавления кристаллической формы I соединения 1. По данным DVS была подтверждена сорбция воды равная 0,11% 80% RH и это показывало, что кристаллическая форма I соединения 1 является негигроскопичной.
<Пример 2> Получение кристаллической формы II соединения 1
Примерно 50 мг соединения 1, полученного в примере получения 1, диспергировали в изопропаноле (1 мл) в стеклянном сосуде объемом 4,0 мл, затем перемешивали при высокой температуре (50°C) в течение 3 дней. Затем стеклянный сосуд охлаждали в холодильнике при 4°C в течение одного дня и не наблюдали осадок твердого вещества. Стеклянный сосуд постоянно хранили в холодильнике при -20°C в течение 7 дней и осадок твердого вещества отделяли центрифугированием (12000 об/мин, 5 мин). Затем осадок сушили при комнатной температуре в течение 24 ч в вакуумном сушильном шкафу при пониженном давлении. В результате получали тонкое пластинчатое многослойное твердое вещество или чешуйчатое твердое вещество. Для анализа характеристик кристаллической формы полученного твердого вещества XRPD проводили по методике, описанной в экспериментальной методике 1. Порошковый дифрактометр (XRPD). Результаты приведены в таблице 4 и на фиг. 7.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, в спектре XRPD кристаллической формы, приведенном на фиг. 7 приведены в таблице 4 ниже.
Таблица 4
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 5,7°, 11,4°, 15,3°, 17,3°, 19,0° и 35,2° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой II.
<Пример 3> Получение кристаллической формы III соединения 1
Примерно 50 мг соединения 1, полученного в примере получения 1, полностью растворяли в MEK или хлороформе, относительно хорошем растворителе, при комнатной температуре, и затем сосуд для проведения реакции закрывали алюминиевой фольгой с небольшим отверстием. Затем смеси давали естественным образом испаряться в течение 1 недели при комнатной температуре и в результате осаждалось небольшое количество твердого вещества. Его дополнительно сушили при 40°C в течение 24 ч при пониженном давлении. Для анализа характеристик кристаллической формы полученного твердого вещества XRPD проводили по методике, описанной в экспериментальной методике 1. Порошковый дифрактометр (XRPD). Результаты приведены на фиг. 8. Кроме того, кристаллическую форму, полученную при таких же условиях, с тем отличием, что использовали примерно 30 мг растворителя MEK, исследовали с помощью XRPD и результаты приведены на фиг. 8.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, в спектре XRPD (растворитель: 50 мг MEK) кристаллической формы, приведенной на фиг. 8, приведены в таблице 5 ниже.
Таблица 5
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 3,5°, 8,5°, 9,0°, 9,3°, 14,4°, 15,0°, 15,6°, 16,9°, 17,2°, 18,2°, 20,6°, 20,7°, 20,9°, 24,0°, 24,3° и 24,6° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой III.
<Пример 4> Получение кристаллической формы IV соединения 1
Примерно 50 мг соединения 1, полученного в примере получения 1, полностью растворяли в EtOH (0,8 мл) при 55°C для получения насыщенного раствора, затем непрерывно перемешивали в течение 30 мин. Затем раствор быстро охлаждали до низкой температуры, равной -20°C, в холодильнике и хранили в течение 3 дней. После отделения полученного твердого вещества центрифугированием его сушили в вакууме при 40°C в течение ночи. Для анализа характеристик кристаллической формы полученного твердого вещества XRPD проводили по методике, описанной в экспериментальной методике 1. Порошковый дифрактометр (XRPD). Результаты приведены в таблице 6 и на фиг. 9.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, в спектре XRPD кристаллической формы, приведенном на фиг. 9, приведены в таблице 6 ниже.
Таблица 6
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 5,9°, 10,8°, 11,5°, 11,7°, 14,3°, 15,3°, 15,6°, 16,3°, 16,6°, 17,2°, 17,6°, 18,0°, 18,5°, 18,8°, 19,1°, 19,6°, 20,1°, 21,8°, 22,4°, 22,7°, 23,4°, 23,8°, 24,8°, 26,5°, 34,7°, 35,2°, 36,5° и 47,1° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой IV.
<Пример 5> Получение кристаллической формы V соединения 1
Примерно 50 мг соединения 1, полученного в примере получения 1, полностью растворяли в н-пропаноле (0,8 мл) при 55°C для получения насыщенного раствора, затем непрерывно перемешивали в течение 30 мин. Затем раствор быстро охлаждали до низкой температуры, равной -20°C, в холодильнике и хранили в течение 3 дней. После отделения полученного твердого вещества центрифугированием его сушили в вакууме при 40°C в течение ночи. Для анализа характеристик кристаллической формы полученного твердого вещества XRPD проводили по методике, описанной в экспериментальной методике 1. Порошковый дифрактометр (XRPD). Результаты приведены в таблице 7 и на фиг. 10.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, в спектре XRPD кристаллической формы, приведенном на фиг. 10, приведены в таблице 7 ниже.
Таблица 7
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 11,5°, 11,8°, 15,1°, 15,4°, 16,2°, 17,4°, 17,7°, 18,4°, 18,6°, 19,0°, 19,4°, 20,7°, 21,6°, 21,8°, 22,2°, 22,9°, 23,3°, 23,8°, 26,0°, 30,1°, 35,3°, 36,0°, 36,6° и 37,5° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой V.
<Пример 6> Получение кристаллической формы VI соединения 1
Для установления того, изменилась ли кристаллическая форма IV соединения 1, полученная в примере 4, после хранения в течение 2 месяцев при комнатной температуре, исследовали характеристики кристаллической формы IV, которую хранили при комнатной температуре в течение 2 месяцев. В частности, XRPD проводили по методике, описанной в экспериментальной методике 1. Порошковый дифрактометр (XRPD). В результате была подтверждена новая кристаллическая форма, обладающая рентгенограммой XRPD, отличающейся от рентгенограммы кристаллической формы IV. Результаты приведены в таблице 8 и на фиг. 11.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, для кристаллов после хранения в течение 2 месяцев кристаллической формы IV, в спектре XRPD кристаллической формы, приведенном на фиг. 11, приведены в таблице 8 ниже.
Таблица 8
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 11,5°, 15,2°, 16,2°, 17,3°, 17,6°, 18,5°, 19,6°, 21,6°, 22,0°, 23,2° и 36,1° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой VI.
<Пример 7> Получение кристаллической формы VII соединения 1
Эксперимент проводили таким же образом, как описано в примере 6, с тем отличием, что кристаллическую форму IV соединения 1, полученную в примере 4, хранили при комнатной температуре в течение 3 месяцев. В результате была подтверждена новая кристаллическая форма, обладающая рентгенограммой XRPD, отличающейся от рентгенограммы кристаллической формы IV. Результаты приведены в таблице 9 и на фиг. 11.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, для кристаллов после хранения в течение 3 месяцев кристаллической формы IV, в спектре XRPD кристаллической формы, приведенном на фиг. 11, приведены в таблице 9 ниже.
Таблица 9
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 8,7°, 9,0°, 10,8°, 11,4°, 11,7°, 12,6°, 14,3°, 15,2°, 16,0°, 17,2°, 18,1°, 18,3°, 18,5°, 19,0°, 19,6°, 20,4°, 21,3°, 21,8°, 23,0°, 23,3°, 24,2°, 24,6°, 25,8°, 26,0°, 28,5°, 30,3°, 31,4°, 32,3°, 33,4°, 34,6°, 36,4°, 37,6°, 40,7° и 49,1° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой VII.
<Пример 8> Получение кристаллической формы VIII соединения 1
Для установления того, изменилась ли кристаллическая форма V соединения 1, полученная в примере 5, после хранения в течение 2 месяцев при комнатной температуре, исследовали характеристики кристаллической формы V, которую хранили при комнатной температуре в течение 2 месяцев. В частности, XRPD проводили по методике, описанной в экспериментальной методике 1. Порошковый дифрактометр (XRPD). В результате была подтверждена новая кристаллическая форма, обладающая рентгенограммой XRPD, отличающейся от рентгенограммы кристаллической формы V. Результаты приведены в таблице 10 и на фиг. 12.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, для кристаллов после хранения в течение 2 месяцев кристаллической формы V, в спектре XRPD кристаллической формы, приведенном на фиг. 12, приведены в таблице 10 ниже.
Таблица 10
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 16,3°, 18,0°, 18,9°, 19,6°, 20,5°, 21,6°, 22,4° и 36,1° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой VIII.
<Пример 9> Получение кристаллической формы IX соединения 1
Эксперимент проводили таким же образом, как описано в примере 8, с тем отличием, что кристаллическую форму V соединения 1, полученную в примере 5, хранили при комнатной температуре в течение 3 месяцев. В результате была подтверждена новая кристаллическая форма, обладающая рентгенограммой XRPD, отличающейся от рентгенограммы кристаллической формы V. Результаты приведены в таблице 11 и на фиг. 12.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, для кристаллов после хранения в течение месяцев кристаллической формы V, в спектре XRPD кристаллической формы, приведенном на фиг. 12, приведены в таблице 11 ниже.
Таблица 11
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 5,5°, 8,9°, 11,5°, 11,7°, 12,4°, 14,2°, 14,9°, 16,0°, 17,3°, 18,2°, 18,8°, 19,5°, 20,1°, 21,8°, 23,2°, 24,6°, 26,0°, 27,8°, 28,6°, 31,3°, 33,1°, 34,1°, 34,5°, 35,2°, 37,0° и 37,5° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой IX.
<Пример 10> Получение большего количества кристаллической формы IV и получение кристаллической формы X
Получение большего количества кристаллической формы IV, полученной в примере 4 проводили следующим образом. Примерно 100 мг соединения, полученного в примере получения 1, полностью растворяли в EtOH (1,6 мл) при 55°C для получения насыщенного раствора, затем непрерывно перемешивали в течение 30 мин. Затем раствор быстро охлаждали до низкой температуры, равной -20°C, в холодильнике и хранили в течение 3 дней. После отделения полученного твердого вещества центрифугированием его сушили в вакууме при 40°C в течение ночи. Для анализа характеристик кристаллической формы полученного твердого вещества XRPD проводили по методике, описанной в экспериментальной методике 1. Порошковый дифрактометр (XRPD). Результаты приведены в таблице 12 и на фиг. 13.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, в спектре XRPD кристаллической формы, приведенном на фиг. 13, приведены в таблице 12 ниже.
Таблица 12
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 11,5°, 15,4°, 17,2°, 17,4°, 17,8°, 18,6°, 19,1°, 19,8°, 21,9°, 24,8°, 28,9°, 31,6° и 36,1° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой X.
<Пример 11> Получение большего количества кристаллической формы V и получение кристаллической формы XI
Получение большего количества кристаллической формы V, полученной в примере 5 проводили следующим образом. Примерно 100 мг соединения 1, полученного в примере получения 1, полностью растворяли в н-бутаноле (1,6 мл) при 55°C для получения насыщенного раствора, затем непрерывно перемешивали в течение 30 мин. Затем раствор быстро охлаждали до низкой температуры, равной -20°C, в холодильнике и хранили в течение 3 дней. После отделения полученного твердого вещества центрифугированием его сушили в вакууме при 40°C в течение ночи. Для анализа характеристик кристаллической формы полученного твердого вещества XRPD проводили по методике, описанной в экспериментальной методике 1. Порошковый дифрактометр (XRPD). Результаты приведены в таблице 13 и на фиг. 14.
Пики, обладающие относительной интенсивностью (I/I0), равной 10% или более, в спектре XRPD кристаллической формы, приведенном на фиг. 14, приведены в таблице 13 ниже.
Таблица 13
2Ɵ: угол дифракции, d: межплоскостное расстояние для кристалла, I/I0 (%): относительная интенсивность (I: интенсивность каждого пика; I0: интенсивность наиболее интенсивного пика)
Если относительная интенсивность (I/I0) пика равнялась 10% или более, пик обладал углами дифракции, равными 15,0°, 17,3°, 18,2°, 19,3°, 21,8° и 35,0° (2Ɵ ± 0,2°). Кристаллическую форму соединения 1, обладающего такой кристаллической формой, называют кристаллической формой XI.
<Экспериментальный пример 1> Определение чистоты кристаллической формы
Примерно 5 мг каждой из кристаллических форм, полученных в примере получения 1 и примерах 1-5, отвешивали в мерную колбу объемом 10 мл и исследовали по следующей методике определения чистоты. 5 мг Каждой кристаллической формы в стабильном состоянии растворяли в 10 мл ацетонитрила и 10 мкл смеси инжектировали в прибор для HPLC. Условия проведения HPLC были такими же, как в экспериментальной методике 6. Высокоэффективная жидкостная хроматография (HPLC). Результаты приведены в таблице 14.
Таблица 14
<Экспериментальный пример 2> Ускоренное исследование стабильности
Исследование стабильности являются одним из важных для сохранения соответствующей стабильности и промышленного выпуска лекарственных средств, поскольку значительность изменения и срок годности устанавливаются на основе описанной методики определения стабильности для определения методики хранения и периода использования лекарственных средств и т. п.
Для подтверждения стабильности новой кристаллической формы, предлагаемой в настоящем изобретении, стабильность кристаллической формы I, полученной в примере 1, исследована с помощью жидкостной хроматографии при условиях проведения анализа, приведенных в экспериментальной методике 6. Высокоэффективная жидкостная хроматография (HPLC). Результаты ускоренного исследования стабильности для хранения в течение от 1 месяца до 6 месяцев при следующих условиях: 40±2°C и 75±5 RH приведены в таблице 15 ниже.
Таблица 15
В результате было подтверждено, что кристаллическая форма I примера 1 в настоящем изобретении сохраняла стабильность в течение до 6 месяцев без изменения чистоты.
Как отмечено выше, настоящее изобретение подробно описано в предпочтительных примерах получения, примерах и экспериментальных примерах, но объем настоящего изобретения не ограничивается конкретными примерами и должен определяться прилагаемой формулой изобретения. Кроме того, специалисты с общей подготовкой в данной области техники должны понимать, что возможны многие изменения и варианты без отклонения от объема настоящего изобретения.
Промышленное применение
Настоящее изобретение относится к кристаллической форме (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата и содержащему ее блокатору проницаемости сосудов. Новая кристаллическая форма обладает высокой чистотой, превосходной стабильностью, превосходной длительной сохранностью и фармацевтической стабильностью и ее можно использовать в качестве блокатора проницаемости сосудов, так что она является весьма полезной для получения высококачественных лекарственных веществ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ ПРОИЗВОДНЫХ 4-ПРЕГЕНЕН-11β-17-21-ТРИОЛ-3,20-ДИОНА | 2012 |
|
RU2683775C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2629957C2 |
| ПРОИЗВОДНЫЕ 4-ПРЕГЕНЕН-11β-17-21-ТРИОЛ-3,20-ДИОНА ДЛЯ ЛЕЧЕНИЯ ГЛАЗНЫХ БОЛЕЗНЕЙ | 2012 |
|
RU2688159C2 |
| РЕГУЛЯТОРЫ ПРОИЗВОДНЫХ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2803499C1 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ НУКЛЕИНОВЫХ КИСЛОТ | 2011 |
|
RU2582235C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| Средства на основе дегидроэпиандростерона, обладающие избирательной цитопротекторной, противовоспалительной, иммуномодулирующей активностями, и способы их получения | 2024 |
|
RU2837885C1 |
| САМОЭМУЛЬГИРУЮЩИЕСЯ СИСТЕМЫ ДОСТАВКИ ЛЕКАРСТВ (SEDDS) ДЛЯ ДОСТАВКИ ОФТАЛЬМОЛОГИЧЕСКОГО ЛЕКАРСТВА | 2016 |
|
RU2746083C2 |
| ПРЕГНАНОВЫЕ СТЕРОИДЫ, ОБЛАДАЮЩИЕ ЦИТОТОКСИЧЕСКОЙ АКТИВНОСТЬЮ, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2710964C1 |
Изобретение относится к способу получения кристаллической формы (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, характеризующейся порошковой рентгенограммой, содержащей пики при следующих углах дифракции 2Ɵ: 16,0°±0,2°, 19,5°±0,2°, 18,3°±0,2°, 15,1°±0,2°, 21,9°±0,2°. Способ получения включает получение соединения, представленного формулой 1, путем взаимодействия соединения формулы 2-1 в присутствии катализатора, содержащего нонафтор-1-бутилсульфонат лития (Li-NFBS) и (s)-камфорсульфоновую кислоту, в соответствии с приведенной ниже формулой реакции (1). Затем получают (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, представляющего собой ɑ-изомер соединения, представленного формулой 1, в твердой форме. Диспергируют полученный (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноат в растворителе с относительно низкой растворяющей способностью, выбранном из воды, метанола или этанола; перемешивают полученную дисперсию; отделяют полученное влажное твердое вещество; и сушат полученное твердое вещество. Также изобретение относится к способу получения (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, в соответствии с приведенной ниже формулой реакции (1). Технический результат – получение (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, в кристаллической форме, обладающей высокой чистотой и стабильностью. 3 н. и 9 з.п. ф-лы, 14 ил., 15 табл., 13 пр.
1. Способ получения кристаллической формы (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, характеризующейся порошковой рентгенограммой, содержащей пики при следующих углах дифракции 2Ɵ:
16,0°±0,2°, 19,5°±0,2°, 18,3°±0,2°, 15,1°±0,2°, 21,9°±0,2°,
включающий следующие стадии:
получение соединения, представленного формулой 1, путем взаимодействия соединения формулы 2-1 в присутствии катализатора, содержащего нонафтор-1-бутилсульфонат лития (Li-NFBS) и (s)-камфорсульфоновую кислоту, в соответствии с приведенной ниже формулой реакции,
[Формула реакции]
получение (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, представляющего собой ɑ-изомер соединения, представленного формулой 1, в твердой форме;
диспергирование полученного (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата в растворителе с относительно низкой растворяющей способностью, выбранном из воды, метанола или этанола;
перемешивание полученной дисперсии;
отделение полученного влажного твердого вещества; и
сушка полученного твердого вещества.
2. Способ по п. 1, где кристаллическая форма дополнительно характеризуется пиками при следующих углах дифракции 2Ɵ:
10,7°±0,2°, 8,9°±0,2°, 17,8°±0,2°, 21,3°±0,2°, 17,2°±0,2°.
3. Способ по п. 1, где кристаллическая форма дополнительно характеризуется пиками при следующих углах дифракции 2Ɵ:
14,1°±0,2°, 20,5°±0,2°, 11,7°±0,2°.
4. Способ по п. 1, где кристаллическая форма дополнительно характеризуется пиками при следующих углах дифракции 2Ɵ:
24,0°±0,2°, 24,6°±0,2°, 26,1°±0,2°.
5. Способ по п. 1, включающий следующие стадии:
диспергирование (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата в растворителе с относительно низкой растворяющей способностью, выбранном из воды, метанола или этанола;
перемешивание полученной дисперсии при комнатной температуре (25°C) в течение 3 дней;
отделение полученного влажного твердого вещества центрифугированием; и
сушка полученного твердого вещества при комнатной температуре в течение 24 ч при пониженном давлении.
6. Способ получения (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, включающий:
получение соединения, представленного формулой 1, путем взаимодействия соединения формулы 2-1 в присутствии катализатора, содержащего нонафтор-1-бутилсульфонат лития (Li-NFBS) и (s)-камфорсульфоновую кислоту, в соответствии с приведенной ниже формулой реакции,
[Формула реакции]
получение (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, представляющего собой α-изомер соединения, представленного формулой 1, в твердой форме.
7. Способ получения кристаллической формы (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, характеризующейся порошковой рентгенограммой, содержащей пики при следующих углах дифракции 2Ɵ:
16,0°±0,2°, 19,5°±0,2°, 18,3°±0,2°, 15,1°±0,2°, 21,9°±0,2°,
включающий следующие стадии:
диспергирование (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата в растворителе с относительно низкой растворяющей способностью, выбранном из воды, метанола или этанола;
перемешивание полученной дисперсии;
отделение полученного влажного твердого вещества; и
сушка полученного твердого вещества.
8. Способ по п. 7, где кристаллическая форма дополнительно характеризуется пиками при следующих углах дифракции 2Ɵ:
10,7°±0,2°, 8,9°±0,2°, 17,8°±0,2°, 21,3°±0,2°, 17,2°±0,2°.
9. Способ по п. 7, где кристаллическая форма дополнительно характеризуется пиками при следующих углах дифракции 2Ɵ:
14,1°±0,2°, 20,5°±0,2°, 11,7°±0,2°.
10. Способ по п. 7, где кристаллическая форма дополнительно характеризуется пиками при следующих углах дифракции 2Ɵ:
24,0°±0,2°, 24,6°±0,2°, 26,1°±0,2°.
11. Способ по п. 7, включающий следующие стадии:
диспергирование (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата в растворителе с относительно низкой растворяющей способностью, выбранном из воды, метанола или этанола;
перемешивание полученной дисперсии при комнатной температуре (25°C) в течение 3 дней;
отделение полученного влажного твердого вещества центрифугированием; и
сушка полученного твердого вещества при комнатной температуре в течение 24 ч при пониженном давлении.
12. Способ по п. 7, дополнительно включающий:
получение соединения, представленного формулой 1, путем взаимодействия соединения формулы 2-1 в присутствии катализатора, содержащего нонафтор-1-бутилсульфонат лития (Li-NFBS) и (s)-камфорсульфоновую кислоту, в соответствии с приведенной ниже формулой реакции,
[Формула реакции]
получение (E)-метил-6-((3S,8S,9S,10R,13S,14S,17R)-3-(((2S,5S,6R)-5-ацетокси-6-(ацетоксиметил)-5,6-дигидро-2H-пиран-2-ил)окси)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[a]фенантрен-17-ил)гепт-5-еноата, представляющего собой ɑ-изомер соединения, представленного формулой 1, в твердой форме.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Haiying Zhang, Joon Ha Park, et al.: "Sac-1004, a vascular leakage blocker, reduces cerebral ischemia—reperfusion injury by suppressing blood-brain barrier disruption and inflammation", Journal of Neuroinflammation, 2017, vol.14(122), p.1-15 | |||
| Maharjan, Sony; Kim, Kyeojin; et al.: " | |||

