ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам получения 6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
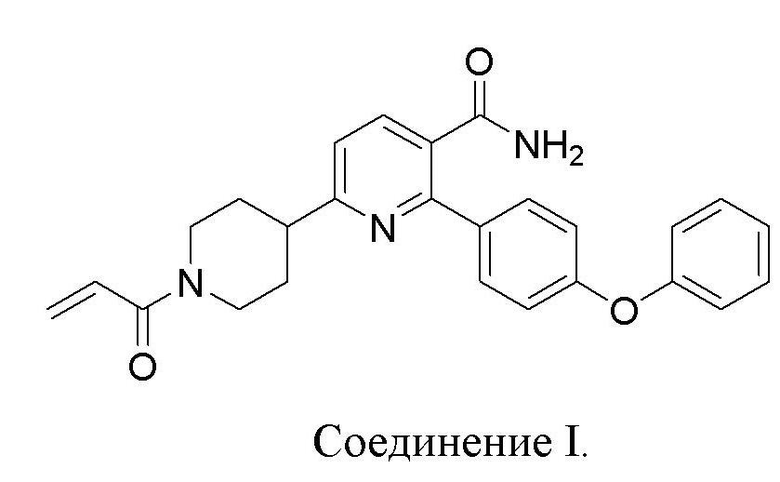
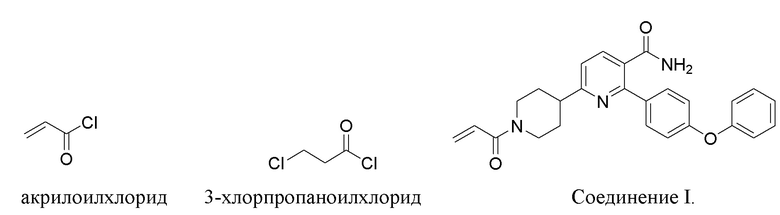
6-(1-Акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамид (Соединение I) представляет собой замещенный никотинамидный ингибитор Тирозинкиназы Брутона (BTK). Соединение I пригодно для лечения рака, воспаления и аутоиммунного заболевания (WO2015/028662). В WO2015/028662 раскрывается способ получения в количестве около 50 г Соединения I.
Существует необходимость в эффективных и контролируемых по чистоте способах получения Соединения I, особенно в крупных масштабах более 1 кг.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам получения 6-(1-Акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида (Соединения I) высокой чистоты и с хорошим выходом. Способ подходит для крупномасштабного производства (более 1 кг, предпочтительно более 2 кг, более 4 кг или более 10 кг). Способом обеспечивается чистота Соединения I ≥ 90%, или ≥ 95%, или ≥ 98%, или ≥ 99%.
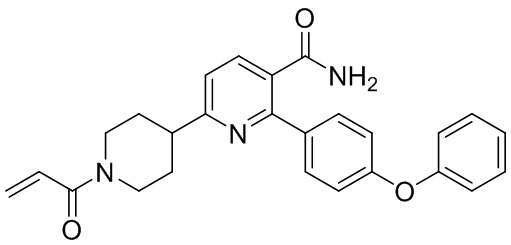
Соединение I
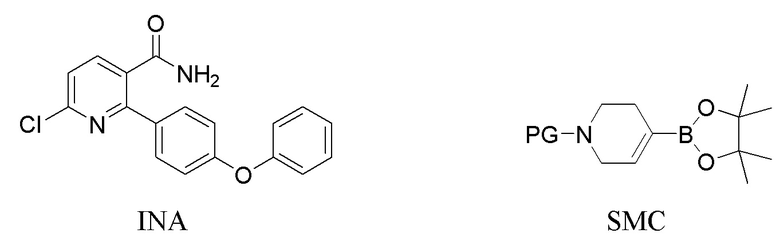
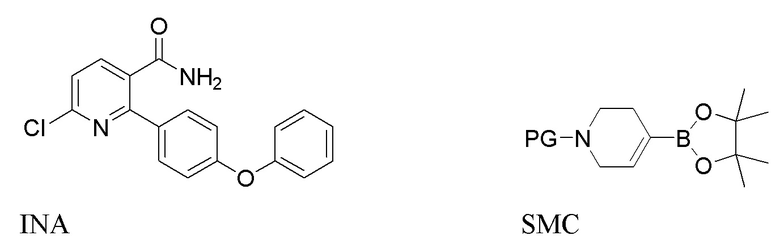
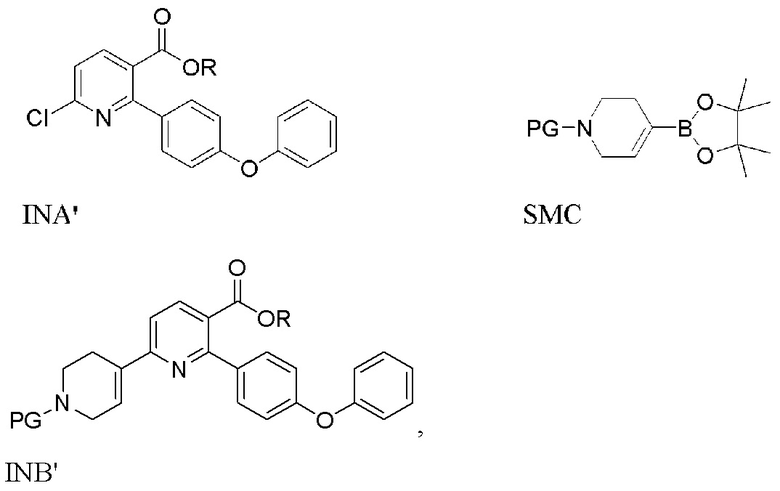
В первом варианте выполнения изобретения А Соединение I получают из 6-хлор-2-(4-феноксифенил)никотинамида (INA), и в способе используется трет-бутилоксикарбонил (t-Boc) в качестве защитной группы. Способ включает следующие стадии:
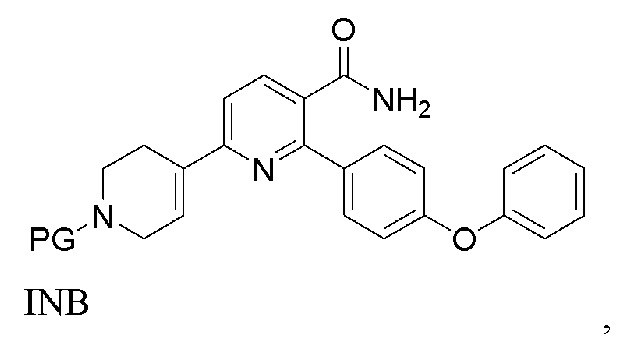
(a) Нагревание смеси INA, SMC и первого содержащего палладий катализатора при 60-140°C с получением INB,
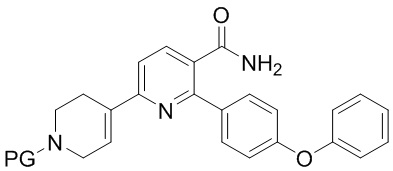
INB
где PG представляет собой защитную группу трет-бутилоксикарбонила;
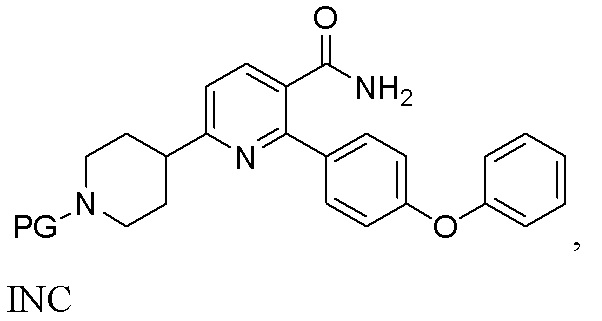
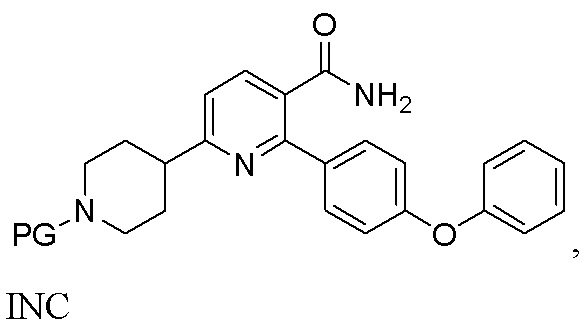
(b) Гидрирование INB под действием H2 в присутствии второго содержащего палладий катализатора с получением INC;
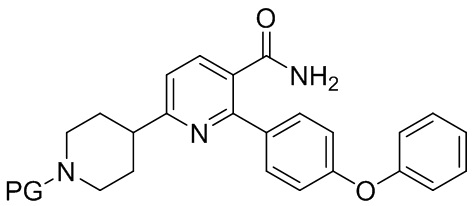
INC
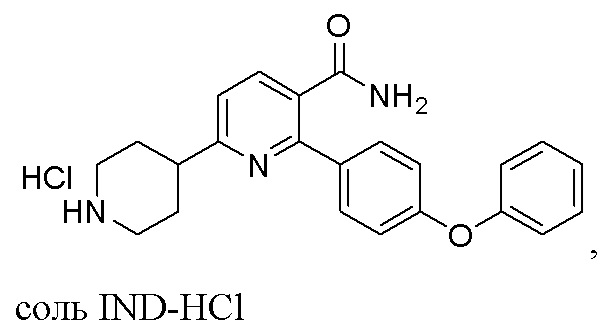
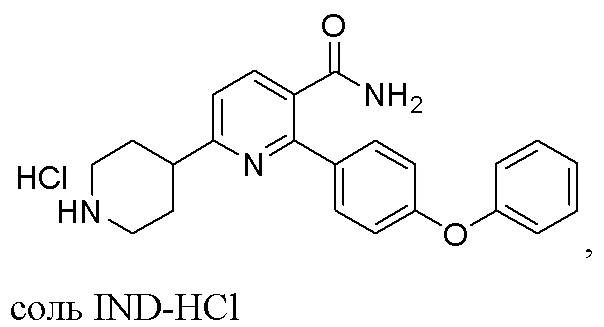
(c) Снятие защитной группы с INC раствором, содержащим HCl, с получением соли IND-HCl в твердой форме;
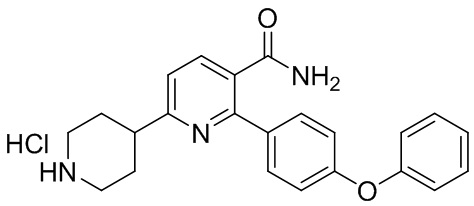
соль IND-HCl
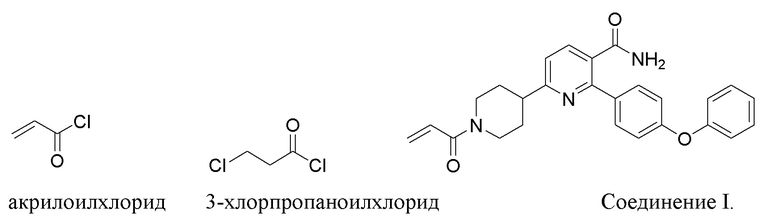
(d) Взаимодействие соли IND-HCl с акрилоилхлоридом или 3-хлорпропаноилхлоридом в основных условиях с получением 6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида.

На стадии (а) 6-хлор-2-(4-феноксифенил)никотинамид (INA), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (SMC) и подходящий содержащий палладий катализатор подвергают реакции в реакторе под азотом с низким содержанием кислорода (< 2%) в основной водно-органической смеси растворителей с получением неочищенного трет-бутил 5-карбамоил-6-(4-феноксифенил)-3',6'-дигидро-[2,4'-бипиридина]-1'(2'H)-карбоксилата (INB). Температура реакции составляет 60-140°C, предпочтительно 60-100°C, более предпочтительно 75-85°C. Время реакции обычно составляет 1-4 часа. Загрузка Pd (молярное соотношение катализатора Pd к реагенту INA) составляет 0,5-5%. Более высокая загрузка Pd ускоряет реакцию, но также увеличивает стоимость и количество примесей. Сырой INB может быть дополнительно очищен без колоночной хроматографии до чистоты по меньшей мере 90% путем перекристаллизации и/или растирания, например, в тетрагидрофуране и этилацетате.
Подходящие содержащие палладий катализаторы, используемые в этой заявке, включают органопалладиевые соединения, такие как трис(дибензилиденацетон)дипалладий(0) (Pd2(dba)3), тетракис(трифенилфосфин)палладий(0), бис(трифенилфосфин)палладий(II) дихлорид (Pd(PPh3)2Cl2), [1,1’-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2) и неорганические соединения палладия, включая Pd(OAc)2 с различными лигандами, например, Ph3P, Ph2Cy и Cy3P-HBF4.
На стадии (b) гидрируют органический раствор INB, катализируемый подходящим содержащим палладий катализатором в реакторе под действием водорода, с получением неочищенного трет-бутил 4-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пиперидин-1-карбоксилата (INC). Температура реакции составляет 15-50°C и предпочтительно 20-45°C. Время реакции обычно составляет 2-10 часов. Катализатором, содержащим палладий, может быть Pd/C или Pd(OH)2/C, предпочтительно Pd(OH)2/C, что более благоприятно для способа.
На стадии (c) к органическому раствору, содержащему INC, добавляют газ HCl или раствор HCl и перемешивают при 10-40°C в течение 4-9 часов для удаления защитной группы (t-Boc) и образования соли 2-(4-Феноксифенил)-6-(пиперидин-4-ил)никотинамида (IND)-HCl в твердой форме. IND-HCl осаждается из реакционной среды и может быть легко собран путем фильтрации или центрифугирования, что подходит для крупномасштабного производства. В одном варианте выполнения изобретения газ HCl барботируется в раствор INC. В других вариантах выполнения изобретения органический раствор HCl (напр., HCl в этилацетате или этаноле) добавляют к органическому раствору INC (напр., INC в этаноле или дихлорметане), или органический раствор INC добавляют к органическому раствору HCl. Твердое вещество IND-HCl может быть растерто до чистоты по меньшей мере 90%, предпочтительно по меньшей мере 95%, в этилацетате.
На стадии (d) соль IND-HCl взаимодействует с акрилоилхлоридом в основном растворе (рН 8-14, напр., растворе бикарбоната HCO3-, растворе карбоната CO3-2, растворе фосфата PO4-3 и растворе гидроксида OH-) при низкой температуре (0-8°C или 2-6°C) для уменьшения образования примесей и получения Соединения I. Растворителем раствора может быть органический растворитель (напр., THF или дихлорметан) с водой или без воды. Время реакции обычно составляет 1-5 часов или 1-3 часа. Альтернативно, на стадии (d) соль IND-HCl сначала реагирует с 3-хлорпропаноилхлоридом в умеренном основном растворе (рН 10-12, напр., растворе карбоната или растворе фосфата) с образованием промежуточного INE, а затем при увеличении рН до 14 до сильного основного раствора (напр., раствором гидроксида) с образованием Соединения I. Растворителем растворов может быть органический растворитель (напр., THF или дихлорметан) с водой или без воды.
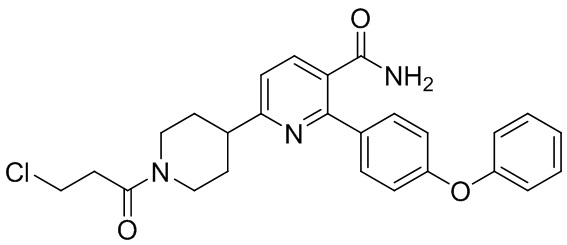
INE
Соединение I может быть дополнительно очищено путем растирания в THF/воде для повышения чистоты до по меньшей мере 90% чистоты, предпочтительно по меньшей мере 95% чистоты.
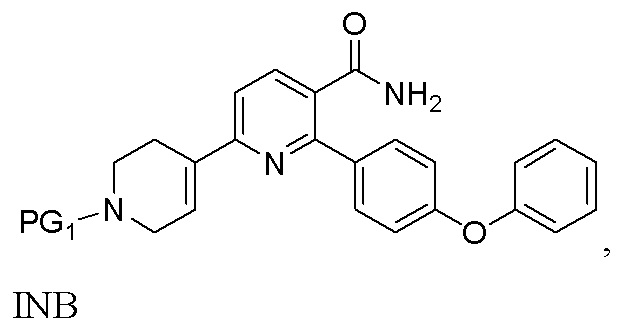
Во втором варианте выполнения изобретения В Соединение I получают из 6-хлор-2-(4-феноксифенил)никотиновой кислоты/сложного эфира (INA’), и в способе используется t-Boc в качестве защитной группы. Способ включает следующие стадии:
(a) Нагревание смеси INA’, SMC и первого содержащего палладий катализатора при 60-140°C с получением INB’,
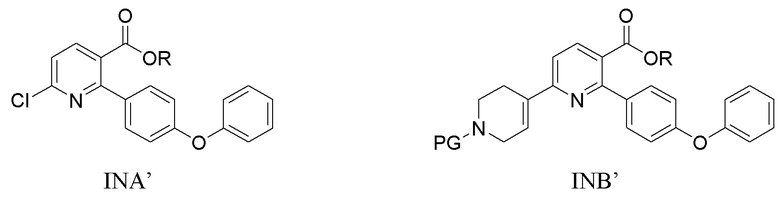
где R представляет собой Н или C1-4алкил,
PG представляет собой защитную группу трет-бутилоксикарбонила;
(b) Амидирование INB’ путем обработки сначала оксалилхлоридом, а затем аммиаком, когда R представляет собой H, или путем взаимодействия с аммиаком, когда R представляет собой C1-4алкил, с получением INB;
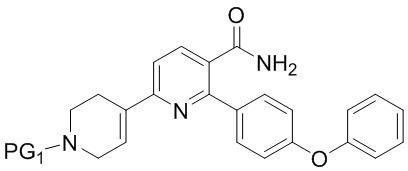
INB
(c) Гидрирование INB под действием H2 в присутствии второго содержащего палладий катализатора с получением INC;
(d) Снятие защитной группы с INC раствором, содержащим HCl, с получением соли IND-HCl в твердой форме; и
(e) Взаимодействие соли IND-HCl с акрилоилхлоридом или 3-хлорпропаноилхлоридом в основных условиях с получением Соединения I.
Вариант выполнения В аналогичен Варианту выполнения А за исключением следующего. В Варианте выполнения В исходным материалом является INA’ (кислота или сложный эфир), а не INA (амид). Продуктом на стадии (а) является INB’ (кислота или сложный эфир), который необходимо амидировать до INB перед гидрированием. На стадии (b), когда R представляет собой H, INB сначала обрабатывают оксалилхлоридом в органическом растворителе (напр., THF) при 15-50°C в течение 2-10 часов с получением промежуточного продукта ацилхлорида, который затем реагирует с 20-35% (мас./мас.) аммиаком в воде при 15-50°C в течение 1-5 часов. На стадии (b), когда R представляет собой C1-4алкил, INB’ в органическом растворе (напр., THF) обрабатывают 20-35% аммиаком (мас./мас.) в воде при 15-75°C в течение 4-20 часов.
В третьем варианте выполнения C Соединение I получают из 6-хлор-2-(4-феноксифенил)никотинамида (INA) и используют бензил или карбоксибензил в качестве защитной группы. Способ включает следующие стадии:
(a) Нагревание смеси INA, SMC’ и первого содержащего палладий катализатора при 60-140°C с получением INB,
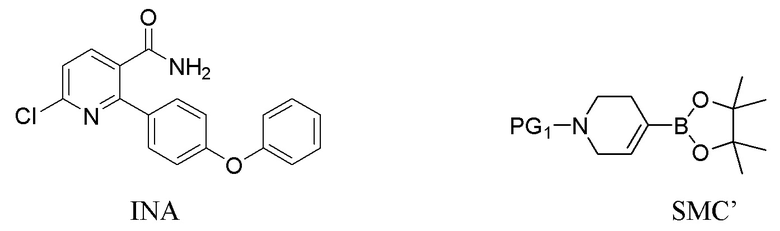
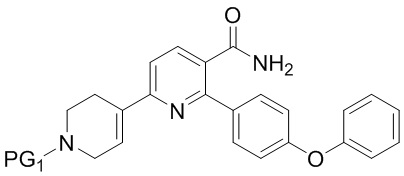
INB
где PG1 представляет собой защитную группу бензила или карбоксибензила;
(b) Гидрирование INB под действием H2 в присутствии второго содержащего палладий катализатора с получением IND;
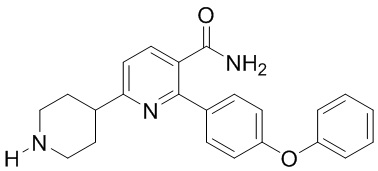
IND
(c) Взаимодействие IND с акрилоилхлоридом или 3-хлорпропаноилхлоридом в основных условиях с получением 6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида.
Вариант выполнения С аналогичен Варианту выполнения А за исключением того, что в Варианте выполнения С в качестве реагента на стадии (а) используется другая защитная группа бензила или карбоксибензила. Такая защитная группа не является стабильной во время гидрирования и снимается во время стадии (b) с образованием IND. Затем IND может вступать в реакцию с акрилоилхлоридом или 3-хлорпропаноилхлоридом в основном состоянии с образованием Соединения I. Необязательно после стадии (b) к IND можно добавить раствор, содержащий HCl, с образованием соли IND-HCl в твердой форме, а затем подвергнуть взаимодействию с акрилоилхлоридом или 3-хлорпропаноилхлоридом.
В четвертом варианте выполнения D Соединение I получают из 6-хлор-2-(4-феноксифенил)никотиновой кислоты/сложного эфира (INA’) и используют бензил или карбоксибензил в качестве защитной группы. Способ включает следующие стадии:
(a) Нагревание смеси INA’, SMC’ и первого содержащего палладий катализатора при 60-140°C с получением INB’,
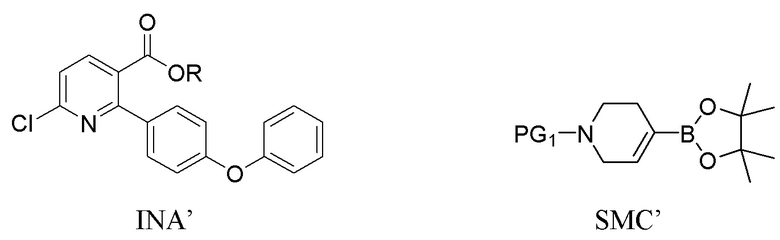
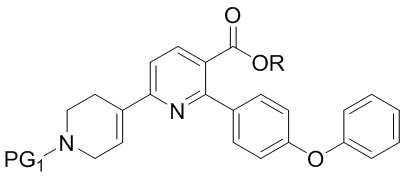
INB’
где R представляет собой Н или C1-4алкил,
PG1 представляет собой защитную группу бензила или карбоксибензила;
(b) Амидирование INB’ путем обработки сначала оксалилхлоридом, а затем аммиаком, когда R представляет собой H, или путем взаимодействия с аммиаком, когда R представляет собой C1-4алкил, с получением INB;
INB
(c) Гидрирование INB под действием H2 в присутствии второго содержащего палладий катализатора с получением IND;
(d) Взаимодействие IND с акрилоилхлоридом или 3-хлорпропаноилхлоридом в основных условиях с получением 6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида.
Вариант выполнения D аналогичен варианту выполнения C за исключением следующего. В варианте выполнения D исходным материалом является INA’ (кислота или сложный эфир), а не INA (амид). Продуктом на стадии (а) является INB’ (кислота или сложный эфир), который необходимо амидировать до INB перед гидрированием. На стадии (b), когда R представляет собой H, INB’ сначала обрабатывают оксалилхлоридом в органическом растворителе (напр., THF) при 15-50°C в течение 2-10 часов с получением промежуточного продукта ацилхлорида, который затем реагирует с 20-35% (мас./мас.) аммиаком в воде при 15-50°C в течение 1-5 часов. На стадии (b), когда R представляет собой C1-4алкил, INB’ в органическом растворе (напр., THF) обрабатывают 20-35% аммиаком (мас./мас.) в воде при 15-75°C в течение 4-20 часов.
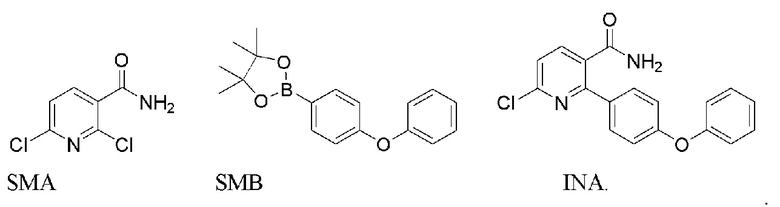
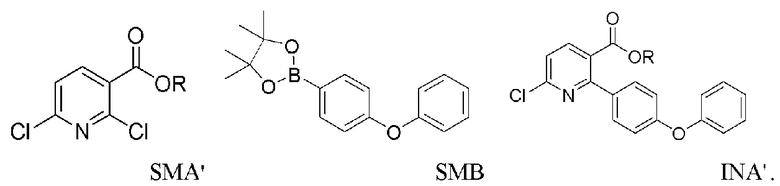
Исходный материал INA может быть получен путем нагревания смеси 2,6-дихлорникотинамида (SMA), SMB и содержащего палладий катализатора в основном растворителе или смеси растворителей (напр., карбонате натрия или калия, 1,4-диоксане, диметилацетамиде, этаноле/метаноле) при 60-140°C в течение 8-12 часов с получением INA.
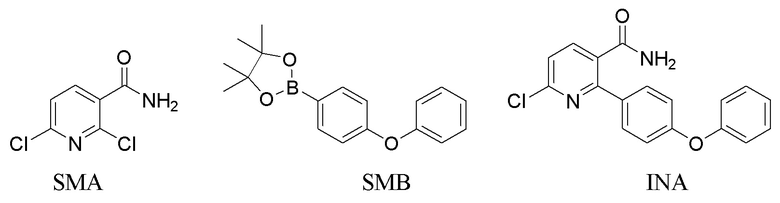
Исходный материал INA’ может быть получен путем нагревания смеси 2,6-дихлорникотиновой кислоты/сложного эфира (SMA’), SMB и содержащего палладий катализатора в основном растворителе или смеси растворителей (напр., карбонате натрия или калия, 1,4-диоксане, диметилацетамиде) при 60-140°C в течение 8-12 часов с получением INA’.
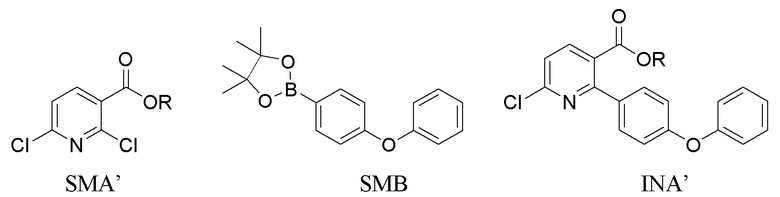
В способе получения INA или INA’ используется сложный борный эфир SMB. SMB работает лучше, чем соответствующая борная кислота, в том смысле, что SMB уменьшает побочные реакции ди-связывания и улучшает региоселективность хлора. Неочищенный INA может быть дополнительно растерт в тетрагидрофуране для повышения чистоты до по меньшей мере 90%.
2,6-Дихлорникотинамид (SMA) можно получить у коммерческого поставщика. Альтернативно, SMA может быть получен взаимодействием 2,6-дихлорникотиновой кислоты (SM1) с оксалилхлоридом (COCl)2 при 10-30°C в течение 4-12 часов с последующим взаимодействием с водным аммиаком в органическом растворителе при 10-30°C в течение 1-3 часов.
2,6-Дихлорникотиновая кислота/сложный эфир (SMA’) может быть получена у коммерческого поставщика.
Настоящий способ обеспечивает хороший выход Соединения I. Общий выход из INA или INA’ в Соединение I составляет около 55-70%, а из SMA или SMA в Соединение I - около 30-50%.
Следующие примеры дополнительно иллюстрируют настоящее изобретение. Эти примеры предназначены только для иллюстрации настоящего изобретения и не должны толковаться как ограничивающие.
ПРИМЕРЫ
Примеры 1-6 обобщены на Схеме A.
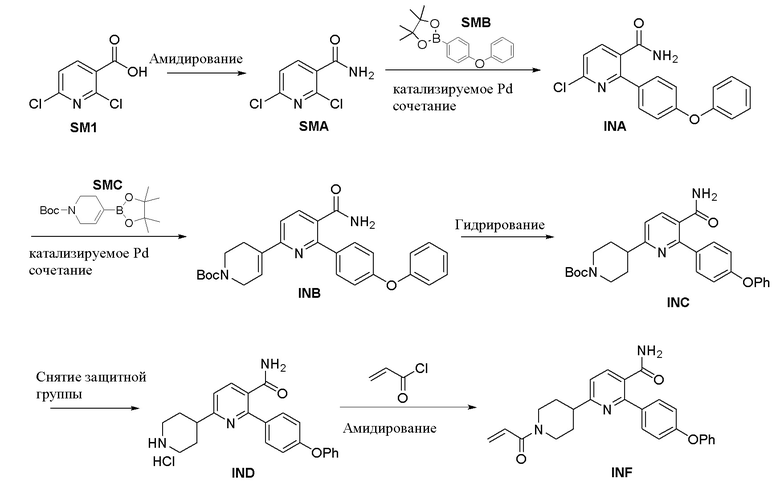
Снятие защитной группы
Амидирование
Пример 1. Получение 2,6-дихлорникотинамида (SMA)
В реактор под азотом загружали 2,6-дихлорникотиновую кислоту (SM1, 39,8 кг, 207 моль), DMF (1,9 кг, 26 моль) и THF (189,9 кг). В вышеуказанный раствор медленно добавляли оксалилхлорид (36,0 кг, 283 моль) при поддержании температуры от 15 до 25°C. После добавления реакцию перемешивали при той же температуре в течение 8 ч. При поддержании температуры ниже 50°C смесь концентрировали при пониженном давлении до тех пор, пока объем не уменьшился в 2,5 - 3,5 Об. Затем смесь охлаждали до 20 ~ 30°C и добавляли к перемешиваемому раствору THF (106,1 кг) и водному аммиаку (180,0 кг), охлажденному при 0 ~ 10°C. После добавления реакционную смесь нагревали до 20 ~ 30°C и перемешивали в течение дополнительных 1,5 ч. Затем смесь концентрировали при пониженном давлении ниже 50°C до тех пор, пока не вышел дистиллят. Полученную суспензию охлаждали до 15 ~ 25°C, перемешивали в течение 2 ~ 4 ч и центрифугировали. Твердый осадок промывали водой (40,4 кг) и смешивали с метил-трет-бутиловым эфиром (148,5 кг). Полученную смесь нагревали до 45 ~ 55°C и перемешивали в течение 3 ~ 5 ч. Затем её охлаждали до 20 ~ 30°C, перемешивали в течение 1 ~ 3 ч и центрифугировали. Твердый осадок промывали метил-трет-бутиловым эфиром (6,8 кг) и сушили в течение 16 ч при пониженном давлении (ниже -0,080 МПа) при 35 ~ 45°C с получением SMA (30,48 кг, выход 77%, чистота 99,6%).
Пример 2. Получение 6-хлор-2-(4-феноксифенил)никотинамида (INA)
К раствору SMB (58,7 кг, 198,2 моль) в этаноле (120,0 кг) и метаноле (36,3 кг) добавляли SMA (30,0 кг, 157 моль) и Na2CO3 (66,6 кг, 628 моль), и полученную смесь трижды продували N2. Затем добавляли Pd2(dba)3 (трис(дибензилиденацетон)дипалладий(0)), 4,32 кг, 4,7 моль) и барботировали N2 в течение 2 ~ 5 мин. Реакционную смесь нагревали до 75 ~ 85°C и перемешивали в течение 12 ч. Затем смесь охлаждали до 15 ~ 25°C, перемешивали в течение 4 ~ 6 ч и фильтровали. Осадок с фильтра замачивали в этаноле (30,3 кг) в течение 10 ~ 20 мин и фильтровали. Осадок с фильтра растворяли в THF (268,4 кг), нагревали до 45 ~ 55°C и перемешивали в течение 2 ~ 4,5 ч. Смесь охлаждали до 20 ~ 30°C и фильтровали. Собирали фильтрат. Осадок с фильтра замачивали в THF (62,4 кг) в течение 10 ~ 20 минут и фильтровали. Собирали фильтрат. Осадок с фильтра дополнительно замачивали в THF (60,3 кг) в течение 10 ~ 20 минут и фильтровали. Собирали фильтрат, объединяли с двумя вышеуказанными партиями и концентрировали при пониженном давлении ниже 50°C. В остаток добавляли этанол (60,1 кг) и концентрировали при пониженном давлении ниже 50°C. В остаток добавляли другую порцию этанола (60,4 кг) и концентрировали при пониженном давлении ниже 50°C. В остаток добавляли этанол (90,1 кг), нагревали до 75 ~ 85°C и перемешивали в течение 1,5 ~ 2,5 ч. Полученную смесь охлаждали до 15 ~ 25°C, перемешивали в течение 4 ~ 6 ч и центрифугировали. Твердый осадок промывали этанолом (30,1 кг) и сушили в течение 16 ч при пониженном давлении (ниже -0,080 МПа) при 35 ~ 45°C с получением INA (39,23 кг, выход 77%, чистота 93,1%).
Пример 3. Получение трет-Бутил 5-карбамоил-6-(4-феноксифенил)-3',6'-дигидро-[2,4'-бипиридин]-1'(2'Н)-карбоксилата (INB)
В реактор под азотом загружали INA (31,7 кг, 90,7 моль, чистота 93,1%), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (30,8 кг, 99,7 моль), Na2CO3 (25,1 кг, 237 моль), EtOH (118,7 кг) и Н2О (148,2 кг). Реактор барботировали N2 до тех пор, пока содержание кислорода не стало менее чем 1,0%. Затем добавляли Pd(PPh3)2Cl2 (1,18 кг, 1,68 моль). Реакционную смесь нагревали до 75 ~ 85°C и перемешивали в течение 2,5 ч. После охлаждения до 15 ~ 25°C реакционную смесь перемешивали в течение 1 ~ 3 ч и фильтровали. Осадок с фильтра промывали этанолом (26,2 кг) и Н2О (35,8 кг) последовательно, растворяли в THF (261,7 кг) и перемешивали при 45 ~ 55°C в течение 2 ~ 4 ч. Затем раствор охлаждали до 20 ~ 30°C и фильтровали. Собирали фильтрат. Осадок с фильтра дважды промывали THF (использовали 59,3 кг THF и 59,7 кг THF соответственно) и собирали фильтрат. Три партии фильтрата объединяли и концентрировали при температуре ниже 60°C. В остаток добавляли этилацетат (148,1 кг) и концентрировали при температуре ниже 60°C. Затем добавляли еще одну партию этилацетата (147,5 кг), и смесь концентрировали при температуре ниже 60°C. В полученный остаток добавляли этилацетат (147,5 кг), нагревали до 65 ~ 75°C и перемешивали в течение 2 ~ 4 ч. Затем смесь охлаждали до 15 ~ 25°C, перемешивали в течение 2 ~ 4 ч и центрифугировали. Твердый осадок промывали этилацетатом (59,0 кг) и сушили в течение 12 ~ 16 ч при пониженном давлении (ниже -0,080 МПа) при 35 ~ 45°C с получением INB (30,44 кг, выход 71%, чистота 98,7%).
Пример 4. Получение трет-Бутил 4-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пиперидин-1-карбоксилата (INC)
В реактор под азотом загружали Pd(OH)2/C (2,03 кг, 20%) и THF (32,0 кг) и перемешивали в течение 5 ~ 10 мин. Затем добавляли раствор INB (14,85 кг, 31,5 моль) в THF (47 кг). Было добавлено больше THF (32,0 кг), и реакционную смесь продували N2 3 раза, а затем продували H2 3 раза при поддержании температуры системы от 20 до 25°C. Реакционную смесь нагревали до 35 ~ 45°C и перемешивали в течение 4 ч при давлении H2 0,10 ~ 0,20 МПа. Затем смесь фильтровали и осадок с фильтра дважды промывали THF (30 кг ×2). Три партии фильтрата объединяли с получением раствора неочищенного INC (181,5 кг). Две отдельные реакционные партии (в общей сложности использовалось 30,5 кг чистого INB) объединяли с получением раствора INC в THF (365,3 кг), в который добавляли активированный уголь (3,00 кг), нагревали до 45 ~ 55°C и перемешивали в течение 1 ~ 3 ч. Затем смесь охлаждали до 20 ~ 30°C, фильтровали диатомитом (15,2 кг) и дважды промывали THF (использовали 62,2 кг и 61,1 кг THF соответственно). Три партии фильтрата объединяли и концентрировали при пониженном давлении ниже 50°C. В остаток добавляли этанол (244,0 кг) и концентрировали при пониженном давлении ниже 50°C. Было добавлено больше этанола (246,1 кг), и полученную смесь концентрировали при пониженном давлении ниже 50°C. В остаток добавляли этилацетат (262,3 кг) и этанол (43,5 кг) и перемешивали при 20 ~ 55°C до тех пор, пока все твердое вещество не растворялось с получением раствора INC.
Пример 5. Получение 2-(4-Феноксифенил)-6-(пиперидин-4-ил)никотинамида (IND)
Вышеуказанный раствор INC в этилацетате (262,3 кг) и этаноле (43,5 кг) трижды продували N2. Затем в раствор барботировали HCl (газ, 11,7 кг) и реакционную смесь перемешивали при 10 ~ 40°C в течение 7 ч. Смесь охлаждали до 15 ~ 25°C, перемешивали в течение 1 ~ 3 ч и центрифугировали. Твердый осадок промывали этилацетатом (60,8 кг) и сушили в течение 24 ч при пониженном давлении (ниже -70 кПа) при 30 ~ 50°C с получением IND (26,3 кг, соль HCl, выход 99%, чистота 99,4%).
Пример 6. Получение 6-(1-Акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида (INF)
В реактор под азотом загружали IND (10,0 кг, 24,4 моль), THF (105,5 кг) и H2O (125,4 кг), и смесь перемешивали в течение 20 ~ 40 мин при 15 ~ 25°C. Затем через 20 ~ 40 мин добавляли NaHCO3 (10,3 кг, 123 моль). Реакционную смесь охлаждали до -2 ~ 6°C, добавляли акрилоилхлорид (3,8 кг, 42,0 моль) и перемешивали в течение 1 ~ 3 ч. ВЭЖХ показало, что IND был израсходован не полностью, поэтому было добавлено больше акрилоилхлорида, и реакционную смесь продолжали перемешивать до завершения реакции (0,68 кг акрилоилхлорида добавляли тремя партиями и реакция длилась 16 ч). Затем смесь нагревали до 5 ~ 20°C, добавляли воду (98,9 кг), перемешивали в течение 20 ~ 40 мин и фильтровали. Осадок с фильтра промывали водой (20 кг), смешивали с другой порцией воды (100,8 кг) и перемешивали в течение 1 ~ 2 ч при 15 ~ 25°C. Смесь фильтровали. Осадок с фильтра последовательно промывали водой (21,2 кг) и этанолом (5,0 кг). Затем осадок с фильтра растворяли в дихлорметане (200,6 кг), нагревали до 40 ~ 45°C и перемешивали до тех пор, пока раствор не стал прозрачным. После охлаждения до 15 ~ 25°C к раствору добавляли раствор HCl (300,7 кг, 0,18%) и перемешивали в течение 10 ~ 20 мин. Два слоя разделяли. Органический слой дважды последовательно промывали водой (использовали 51,0 кг воды и 50,2 кг воды соответственно), водным раствором Na2CO3 (0,42%, для регулирования рН = 8 ~ 9) и водой (50,0 кг). Органическую фазу концентрировали при пониженном давлении. В остаток добавляли этанол (100,0 кг) и концентрировали при пониженном давлении ниже 45°C. В остаток дополнительно добавляли этанол (80,0 кг), перемешивали в течение 1 ~ 2 ч при 15 ~ 25°C и фильтровали. Осадок с фильтра промывали этанолом и сушили в течение 14 ч при пониженном давлении при 20 ~ 30°C с получением INF (7,47 кг, выход 72%, чистота 99,5%).
Изобретение, а также манера и способ его изготовления и применения далее описаны в таких полных, ясных, кратких и точных терминах так, чтобы любой специалист в области техники, к которой оно относится, мог сделать и использовать то же самое. Следует понимать, что вышеизложенное описывает предпочтительные варианты выполнения настоящего изобретения и что в них могут быть внесены изменения, не выходящие за рамки настоящего изобретения, как указано в формуле изобретения. Для того чтобы особо указать и четко заявить сущность, рассматриваемую как изобретение, следующая формула изобретения завершает спецификацию.
Группа изобретений относится к способам получения 6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида (Соединение I). Способы осуществляют согласно стадиям, условиям и промежуточным продуктам, указанным в формуле изобретения. Технический результат – усовершенствованные способы получения Соединения I для крупномасштабного производства, обеспечивающие высокий выход и чистоту продукта. 2 н. и 3 з.п. ф-лы, 6 пр.
1. Способ получения 6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида (Cоединение I), включающий:
(a) нагревание смеси INA, SMC и первого содержащего палладий катализатора при 60-140°C с получением INB
где PG представляет собой защитную группу трет-бутилоксикарбонила;
(b) гидрирование INB под действием H2 в присутствии второго содержащего палладий катализатора с получением первого раствора, содержащего сырой INC
(c) добавление активированного угля для очистки сырого INC и затем концентрирование и растворение очищенного INC в этилацетате и этаноле с получением второго раствора, содержащего очищенный INC,
(d) добавление газа HCl во второй раствор для снятия защитной группы с INC и получения соли IND-HCl в твердой форме
(е) добавление THF, H2O и NaHCO3 для растворения и нейтрализации соли IND-HCl,
(f) взаимодействие соли IND-HCl с акрилоилхлоридом или 3-хлорпропаноилхлоридом в основных условиях с получением 6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида
 .
.
2. Способ получения 6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида (Cоединение I), включающий:
(a) нагревание смеси INA', SMC и первого содержащего палладий катализатора при 60-140°C с получением INB'
где R представляет собой Н или C1-4алкил;
PG представляет собой защитную группу трет-бутилоксикарбонила,
(b) амидирование INB' путем обработки сначала оксалилхлоридом, а затем аммиаком, когда R представляет собой H, или путем взаимодействия с аммиаком, когда R представляет собой C1-4алкил, с получением INB
(c) гидрирование INB под действием H2 в присутствии второго содержащего палладий катализатора с получением первого раствора, содержащего сырой INC
(d) добавление активированного угля для очистки сырого INC и затем концентрирование и растворение очищенного INC в этилацетате и этаноле с получением второго раствора, содержащего очищенный INC,
(e) добавление газа HCl во второй раствор для снятия защитной группы с INC и получения соли IND-HCl в твердой форме
(f) добавление THF, H2O и NaHCO3 для растворения и нейтрализации соли IND-HCl,
(g) взаимодействие соли IND-HCl с акрилоилхлоридом или 3-хлорпропаноилхлоридом в основных условиях с получением 6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида
3. Способ по п. 1, дополнительно включающий стадию нагревания смеси 2,6-дихлорникотинамида (SMA), SMB и содержащего палладий катализатора в основном растворителе или смеси растворителей при 60-140°C в течение 8-12 часов с получением INA
4. Способ по п. 3, дополнительно включающий стадию взаимодействия 2,6-дихлорникотиновой кислоты с оксалилхлоридом (COCl)2 с последующим взаимодействием с водным аммиаком в органическом растворителе с получением SMA.
5. Способ по п. 2, дополнительно включающий стадию нагревания смеси 2,6-дихлорникотиновой кислоты/сложного эфира (SMA'), SMB и содержащего палладий катализатора в основном растворителе или смеси растворителей при 60-140°C в течение 8-12 часов с получением INA'
| WO 2015048662 A2, 02.04.2015 | |||
| WO 2009111676 A2, 11.09.2009 | |||
| CN 101460466 B, 13.06.2012 | |||
| CN 107226805 A, 03.10.2017 | |||
| RU 2016119519 A, 28.11.2017. |

