Область техники, к которой относится настоящее изобретение
[0001] Настоящее изобретение относится к соединениям, фармацевтическим композициям, содержащим их, и их применению в качестве ингибиторов TRK. Более конкретно, настоящее изобретение относится к новым соединениям в качестве ингибиторов TRK, фармацевтическим композициям, содержащим такие соединения, и способам применения соединений для лечения или предупреждения опосредованных TRK заболеваний, таких как опухоли. Настоящее изобретение также относится к способам получения соединений, как описывается ниже.
Предшествующий уровень техники настоящего изобретения
[0002] TRK (киназа рецептора тропомиозина) представляет собой тирозинкиназу нейротрофического рецептора, представленного во многих тканях, и активирует множество последующих процессов в пролиферации и выживании клеток. В семействе протоонкогенов TRK имеется три представителя: TRK А, В и С, которые кодируются NTRK1, NTRK2 и NTRK3, соответственно. Связывание нейротрофических факторов и белков TRK приводит к димеризации рецепторов, фосфорилированию и активации нижестоящих сигнальных путей, таких как пути Ras/MAPK, PI3K/Akt и PLCγ, которые регулируют пролиферацию, дифференцировку, метаболизм и апоптоз клеток (Brodeur G.М., Minturn J. е., Но R, et al. Clinical cancer research, 2009, 15, 3244-50). Геномный анализ слияния киназ подтвердил, что слияние генов NTRK происходит при различных раковых заболеваниях, таких как глиома, гепатобилпарная карцинома, папиллярная карцинома щитовидной железы, рак толстой кишки, немелкоклеточный рак легкого, плоскоклеточный рак головы и шеи, рак поджелудочной железы, саркома и меланома (Khotskaya, Y.В. et al. Pharmacology & Therapeutics, 2017,173, 58-66). Ингибиторы TRK могут использоваться для лечения различных опухолей, вызванных белком слияния NTRK, а исследования и разработки ингибиторов TRK имеют большой потенциал и широкие рыночные перспективы. В ранних клинических испытаниях ингибитора TRK ларотректиниба (Loxo-101) у 38 больных (76%) были достигнуты объективные ответы, а у 6 больных (12%) наблюдалась полная ремиссия, и никакие опухоли не могли быть выявлены существующими методами. Из этих больных 30 больных находились в стадии ремиссии более года (American Society of Clinical Oncology annual meeting 2017). Однако целевая мутация из-за непрерывного введения лекарственного средства приводит к устойчивости к лекарственному средству. Клинически были выявлены случаи с мутациями NTRK, такими как мутации NTRK1 G595R и G667C (Russo, М. et al., Cancer discovery, 2016, 6 (1), 36-44). Следовательно, необходимо разработать более активные ингибиторы TRK с меньшим количеством побочных эффектов и все же эффективные при мутациях TRK.
Краткое раскрытие настоящего изобретения
[0003] Цель настоящего изобретения заключается в обеспечении соединения, показанного в формуле I, и его изомеров, про лекарств, сольватов, стабильных изотопных производных или фармацевтически приемлемых солей, которые могут быть применимы в качестве ингибиторов TRK:

при этом
L1 выбран из -NR6C(O)-, -NR6CON(R7)-, -NR6S(O)m- и -NR6S(O)mN(R7)-, в которых NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3; предпочтительно L1 выбран из -NR6C(O)- и -NR6CON(R7)-, в которых NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3; наиболее предпочтительно L1 выбран из NR6CON(R7)-, в котором NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3;
L2 выбран из С1-С8алкилена, С2-С8алкенилена, С2-С8алкинилен и С3-С8циклилена, при этом алкилен, алкенилен, алкинилен и циклилен могут быть необязательно замещены одним или несколькими G1; предпочтительно L2 выбран из С1-С6алкилена и С2-С6алкенилена, которые могут быть необязательно замещены одним или несколькими G1; более предпочтительно L2 выбран из С1-С4алкилена, который может быть необязательно замещен одним или несколькими G1;
L3 выбран из одинарной связи, -О- и -N(Rx)-; предпочтительно L3 выбран из одинарной связи и -О-; наиболее предпочтительно L3 представляет собой -О-;
каждый R1, R2, R3 независимо выбран из водорода, галогена, циано, С1-С8алкила, С3-С8циклила, 3-8-членного гетероциклила, арила, гетероарила, формила, -NR8R9, -C(O)R10, карбоксила, алкенила, алкинила, -OR10, -OC(O)NR8R9, -C(O)OR10, -C(O)NR8R9, -NR11C(O)R10, -NR11C(O)NR8R9, -S(O)mR10, -NR11S(O)mR10, -SR10, -S(O)mNR8R9 и -NR11S(O)mNR8R9, при этом алкил, циклил, гетероциклил, арил или гетероарил необязательно замещены одним или несколькими заместителями, выбранными из галогена, циано, С1-С8алкила, С3-С8циклила, 3-8-членного гетероциклила, -OR12, -NR13R14, -OC(O)NR13R14, -C(O)OR12, -C(O)R12, -C(O)NR13R14, -NR15C(O)R12, -NR15C(O)NR13R14, -S(O)mR12, -NR15S(O)mR12, -SR12, -S(O)mNR13R14 и -NR15S(O)mNR13R14; предпочтительно и R2, и R3 представляют собой водород; более предпочтительно все из R1, R2, R3 представляют собой водород;
R4 выбран из водорода, галогена, циано, С1-С8алкила, С3-С8циклила, 3-8-членного гетероциклила, арила, гетероарила, формила, -C(O)R10, карбоксила, алкенила, алкинила, -OR10, -NR8R9, -OC(O)NR8R9, -C(O)OR10, -C(O)NR8R9, -NR8C(O)R10, -NR10C(O)NR8R9, -S(O)mR10, -NR8S(O)mR10, -SR10, -S(O)mNR8R9 и -NR10S(O)mNR8R9; предпочтительно R4 выбран из водорода, галогена; более предпочтительно R4 представляет собой водород или фтор;
R5 выбран из водорода, галогена, циано, С1-С8алкила, С3-С8циклила, 3-8-членного гетероциклила, арила, гетероарила, формила, -C(O)R10, карбоксила, алкенила, алкинила, -OR10, -NR8R9, -OC(O)NR8R9, -C(O)OR10, -C(O)NR8R9, -NR8C(O)R10, -NR10C(O)NR8R9, -S(O)mR10, -NR8S(O)mR10, -SR10, -S(O)mNR8R9 и -NR10S(O)mNR8R9; предпочтительно R5 выбран из водорода, галогена, С1-С6алкила и С3-С6циклила; более предпочтительно R5 выбран из водорода, галогена, С1-С4алкила и С3-С6циклила; кроме того, предпочтительно R5 выбран из водорода и галогена; наиболее предпочтительно R5 представляет собой фтор;
каждый R6, R7, Rx независимо выбран из водорода, С1-С8алкила, С1-С8галогеналкила, гетероалкила, С3-С8циклила, 3-8-членного моноциклического гетероциклила, моноциклического арила, моноциклического гетероарила, алкенила и алкинила; предпочтительно каждый R6, R7, Rx независимо выбран из водорода, С1-С6алкила и C1-С6галогеналкила; более предпочтительно каждый R6, R7, Rx независимо выбран из водорода, С1-С4алкила и С1-С4галогеналкила; кроме того, предпочтительно каждый R6, R7, Rx независимо выбран из водорода и С1-С4алкила;
G1 выбран из галогена, циано, С1-С8алкила, С3-С8циклила, 3-8-членного гетероциклила, арила, гетероарила, формила, -NR8R9, -C(O)R10, карбоксила, алкенила, алкинила, -OR10, -OC(O)NR8R9, -C(O)OR10, -C(O)NR8R9, -NR11C(O)R10, -NR11C(O)NR8R9, -S(O)mR10, -NR11S(O)mR10, -SR10, -S(O)mNR8R9 и -NR11S(O)mNR8R9; предпочтительно G1 выбран из галогена, С1-С6алкила, -OR10, -NR8R9, более предпочтительно G1 выбран из галогена, С1-С4алкила, -OR10, -NR8R9, при этом ал кил необязательно замещен одним или несколькими заместителями, выбранными из галогена, -OR16, -NR13R14, если два G1 присоединяются к одному и тому же атому углерода или к двум соседним атомам углерода, то два G1 могут образовывать 3-8-членный циклил вместе с атомом(ами) углерода, соединенным(ыми) с ними, предпочтительно могут образовывать 3-6-членный циклоалкил, при этом образованный циклоалкил необязательно замещен одним или несколькими заместителями, выбранными из галогена, OR16 и -NR13R14;
каждый R8, R9, R10, R11, R12, R13, R14, R15, R16 независимо выбран из группы, состоящей из водорода, С1-С8алкила, С1-С8галогеналкила, гетероалкила, С3-С8циклила, 3-8-членного моноциклического гетероциклила, моноциклического гетероарила, моноциклического арила, алкенила и алкинила, при этом R8 и R9, R13 и R14 могут образовывать 3-7-членный гетероциклил;
и m равняется 1 или 2;
при этом следующие соединения (1)-(7) исключаются:


[0004] Согласно предпочтительным вариантам осуществления настоящего изобретения представлены соединение, показанное в приведенной выше общей формуле I, и его изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли, в которых
L1 выбран из -NR6C(O)-, -NR6CON(R7)-, -NR6S(O)m- и -NR6S(O)mN(R7)-, в которых NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3;
L2 выбран из С1-С6алкилена, С2-С6алкенилена, С2-С6алкинилена и С3-С6циклилена, при этом алкилен, алкенилен, алкинилен и цигсггилен могут быть необязательно замещены одним или несколькими G1;
L3 выбран из одинарной связи и -О-;
каждый R1, R2, R3 независимо выбран из водорода, галогена, С1-С6алкила, С3-С6циклила, 3-6-членного гетероциклила, арила и гетероарила, при этом алкил, циклил, гетероциклил, арил или гетероарил необязательно замещены одним или несколькими заместителями, выбранными из галогена, циано, С1-С6алкила, С3-С6циклила и 3-6-членного гетероциклила;
R4 выбран из водорода, галогена, -NR8R9, -OR10;
R5 выбран из водорода, галогена, С1-С6алкилаи С3-С6циклила;
R6, R7 каждый независимо выбран из водорода, С1-С6алкила, С1-С6галогеналкила;
G1 выбран из галогена, С1-С6алкила, -NR8R9, -OR10, при этом алкил необязательно замещен одним или несколькими заместителями, выбранными из галогена, -NR11R12, -OR16;
каждый R8, R9, R10, R11, R12 и R16 независимо выбран из водорода, С1-С6алкила и С1-С6галогеналкила;
и m равняется 1 или 2;
при этом следующие соединения (1)-(7) исключаются:


[0005] Согласно другому предпочтительному варианту осуществления настоящего изобретения представлены соединение, показанное в приведенной выше общей формуле I, и его изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли, в которых
L1 выбран из -NR6C(O)- и -NR6CON(R7)-, в которых NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3;
L2 выбран из С1-С6алкилена, С2-С6алкенилена, С2-С6алкинилена и С3-С6циклилена, при этом алкилен, алкенилен, алкинилен и циклилен могут быть необязательно замещены одним или несколькими G1;
L3 выбран из одинарной связи и -О-;
каждый R1, R2, R3 независимо выбран из водорода, галогена, С1-С4алкила, С4-С6циклила, 4-6-членного гетероциклила, при этом алкил, циклил и гетероциклил необязательно замещены одним или несколькими заместителями, выбранными из галогена;
R4 выбран из водорода, галогена, -NR8R9, -OR10;
R5 выбран из водорода, галогена, С1-С6алкила и С3-С6циклила;
каждый R6, R7 независимо выбран из водорода, С1-С6алкила, С1-С6галогеналкила;
G1 выбран из галогена, С1-С6алкила, -NR8R9, -OR10, при этом алкил необязательно замещен одним или несколькими заместителями, выбранными из галогена, -NR11R12, -OR16;
каждый R8, R9, R10, R11, R12 и R16 независимо выбран из водорода, С1-С6алкила и С1-С6галогеналкила;
при этом следующие соединения (1)-(7) исключаются:
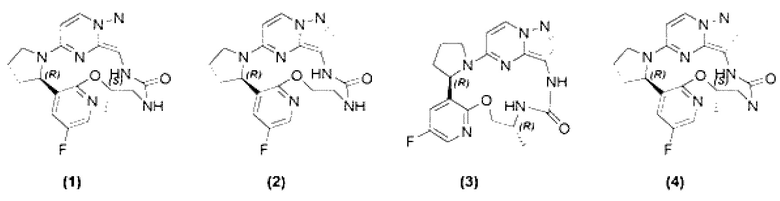

[0006] Согласно другому предпочтительному варианту осуществления настоящего изобретения представлены соединение, показанное в приведенной выше общей формуле I, и его изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли, в которых
L1 выбран из -NR6CON(R7)-, в котором NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3,
L2 выбран из С1-С4алкилена, С2-С4алкенилена, С2-С4алкинилена и С3-С4циклилена, при этом алкилен, алкенилен, алкинилен и циклилен могут быть необязательно замещены одним или несколькими G1;
L3 выбран из одинарной связи и -О-;
каждый R1, R2, R3 независимо выбран из водорода, галогена и С1-С4алкила, при этом алкил необязательно замещен одним или несколькими заместителями, выбранными из галогена,
R4 выбран из водорода, галогена, -NR8R9, -OR10;
R5 выбран из водорода, галогена, С1-С6алкилаи С3-С6циклила;
каждый R6, R7 независимо выбран из водорода, С1-С6алкила, С1-С6 галогеналкила;
G1 выбран из галогена, С1-С6алкила, -NR8R9, -OR10, при этом алкил необязательно замещен одним или несколькими заместителями, выбранными из галогена, -NR11R12, -OR16;
каждый R8, R9, R10, R11, R12 и R16 независимо выбран из группы, состоящей из водорода, С1-С6алкила и С1-С6галогеналкила;
при этом следующие соединения (1)-(5) исключаются:


[0007] Согласно другому предпочтительному варианту осуществления настоящего изобретения представлены соединение, показанное в приведенной выше общей формуле I, и его изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли, в которых
L1 выбран из -NR6CON(R7)-, в котором NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3;
L2 выбран из С1-С4алкилена и С2-С4алкенилена, при этом алкилен и алкенилен могут быть необязательно замещены одним или несколькими G1;
L3 выбран из одинарной связи и -О-;
каждый R1, R2, R3 независимо выбран из водорода, галогена и С1-С4алкила, при этом алкил необязательно замещен одним или несколькими заместителями, выбранными из галогена;
R4 выбран из водорода, галогена, -NR8R9, -OR10;
R5 выбран из водорода, галогена, С1-С4алкила и С3-С6циклила;
каждый R6, R7 независимо выбран из водорода, С1-С4алкила, С1-С4галогеналкила;
G1 выбран из галогена, С1-С4алкила, -NR8R9, -OR10, при этом алкил необязательно замещен одним или несколькими заместителями, выбранными из галогена, -NR11R12, -OR16;
каждый R8, R9, R10, R11, R12 и R16 независимо выбран из водорода, С1-С4алкила, С1-С4галогеналкила;
при этом следующие соединения (1)-(5) исключаются:
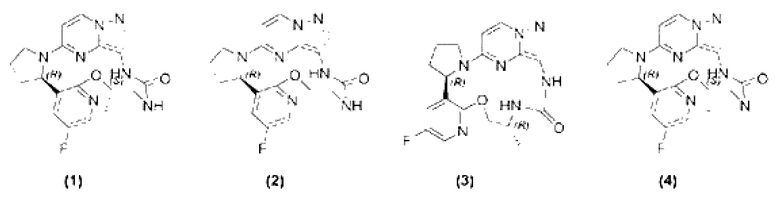
[0008] Согласно другому предпочтительному варианту осуществления настоящего изобретения представлены соединение, показанное в приведенной выше общей формуле I, и его изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли, в которых
L1 выбран из -NR6CON(R7)-, в котором NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3;
L2 выбран из С1-С4алкилена, при этом алкилен может быть необязательно замещен одним или несколькими G1;
L3 представляет собой -О-;
каждый R1, R2, R3 независимо выбран из водорода, галогена и С1-С4алкила, при этом алкил необязательно замещен одним или несколькими заместителями, выбранными из галогена;
R4 выбран из водорода, галогена, -NR8R9, -OR10;
R5 выбран из водорода, галогена и С1-С4алкила;
каждый R6, R7 независимо выбран из водорода, С1-С4алкила, С1-С4галогеналкила;
G1 выбран из галогена, С1-С4алкила, -NR8R9, -OR10, при этом алкил необязательно замещен одним или несколькими заместителями, выбранными из галогена, -NR11R12, -OR16;
каждый R8, R9, R10, R11, R12 и R16 независимо выбран из водорода, С1-С4алкила и С1-С4галогеналкила,
при этом следующие соединения (1)-(4) исключаются:

[0009] Согласно другому предпочтительному варианту осуществления настоящего изобретения представлены соединение, показанное в приведенной выше общей формуле I, и его изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли, в которых
L1 выбран из -NR6CON(R7)-, в котором NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3;
L2 выбран из С1-С4алкилена, при этом алкилен может быть необязательно замещен одним или несколькими G1;
L3 представляет собой -О-,
каждый R1, R2, R3 независимо выбран из водорода и галогена;
R4 выбран из водорода и галогена;
R5 выбран из водорода, галогена и С1-С4алкила и располагается в пара положении L3;
каждый R6, R7 независимо выбран из водорода и С1-С4алкила;
G1 выбран из галогена, С1-С4алкила, при этом алкил необязательно замещен одним или несколькими заместителями, выбранными из галогена, -NR11R12, -OR16;
каждый R11, R12 и R16 независимо выбран из водорода, С1-С4алкила и С1-С4галогеналкила;
при этом следующие соединения (1)-(4) исключаются:

[0010] Согласно другому предпочтительному варианту осуществления настоящего изобретения представлены соединение, показанное в приведенной выше общей формуле I, и его изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли, характеризующиеся тем, что соединения выбраны из:

[0011] Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей соединение согласно любому из вариантов осуществления, упомянутых выше, или его изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли и фармацевтически приемлемые носитель, разбавитель, вспомогательное средство.
[0012] Настоящее изобретение также относится к применению соединения согласно любому из вариантов осуществления, упомянутых выше, и его изомеров, пролекарств, сольватов, стабильных изотопных производных или фармацевтически приемлемых солей или к применению фармацевтической композиции в соответствии с настоящим изобретением в изготовлении медикамента для лечения или предупреждения опосредованных TRK заболеваний, таких как рак, особенно гематологические злокачественные заболевания, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы и глиома.
[0013] Настоящее изобретение также относится к способу лечения или предупреждения опосредованных TRK заболеваний (таких как опухоли, особенно гематологические злокачественные заболевания, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы, глиома), который предусматривает введение больному при необходимости этого терапевтически эффективного количества соединения согласно любому из вариантов осуществления, упомянутых выше, или его изомеров, пролекарств, сольватов, стабильных изотопных производных или фармацевтически приемлемых солей, или фармацевтической композиции в соответствии с настоящим изобретением.
[0014] Другой аспект настоящего изобретения относится к соединению, представленному общей формулой I, описываемой в любом из вариантов осуществления настоящего изобретения, или к его изомерам, пролекарствам, сольватам, стабильным изотопным производным или фармацевтически приемлемым солям, или к содержащей их фармацевтической композиции для применения в лечении или предупреждении опосредованных TRK заболеваний, таких как рак, особенно гематологические злокачественные заболевания, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы и глиома.
[0015] Другой аспект настоящего изобретения относится к соединению, представленному общей формулой I, описываемой в любом из вариантов осуществления настоящего изобретения, или его таутомерам, мезомерам, рацематам, энантиомерам, диастереомерам или их смеси, или его фармацевтически приемлемым солям, в качестве медикамента для лечения и/или предупреждения опухолей и других заболеваний.
[0016] Предпочтительные соединения в соответствии с настоящим изобретением включают в себя без ограничения

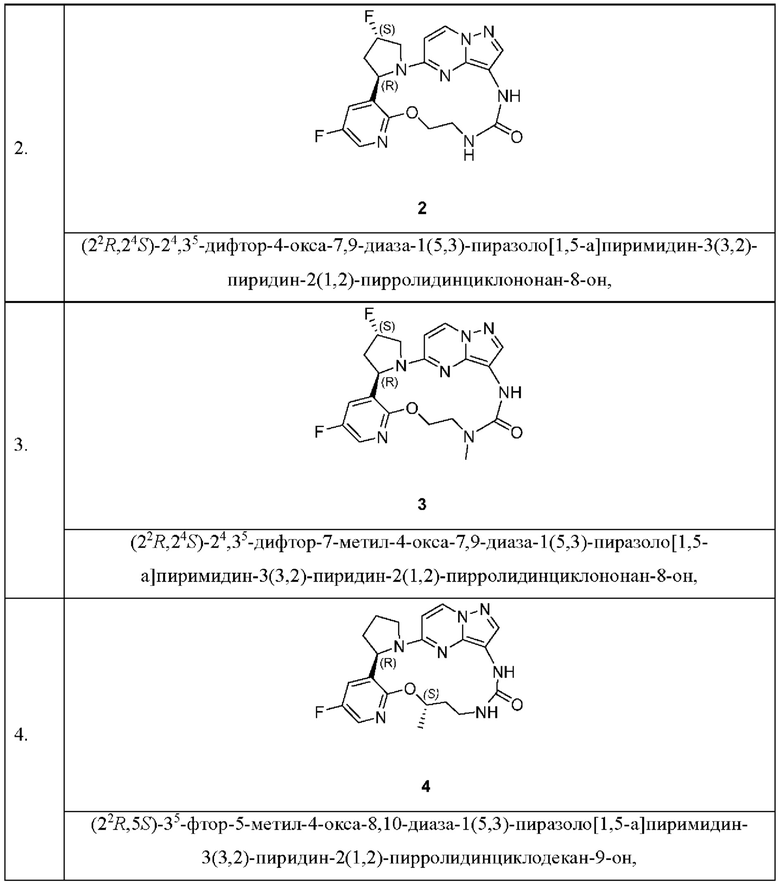
а также их изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли.
[0017] Соединение, представленное общей формулой I в соответствии с настоящим изобретением, представляет собой ингибитор TRK, поэтому соединение, представленное общей формулой I в соответствии с настоящим изобретением, можно применять для лечения или предупреждения опосредованных TRK заболеваний, таких как опухоли, особенно гематологические злокачественные заболевания, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы, глиома головного мозга.
[0018] Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей соединение, представленное общей формулой I в соответствии с настоящим изобретением, или его изомеры, пролекарства, сольваты, стабильные изотопные производные или фармацевтически приемлемые соли и фармацевтически приемлемые носители, разбавители или вспомогательные средства.
[0019] Другой аспект настоящего изобретения относится к применению соединения, представленного общей формулой I, описанной в любом из вариантов осуществления настоящего изобретения, или его изомеров, пролекарств, сольватов, стабильных изотопных производных или фармацевтически приемлемых солей, или фармацевтической композиции в соответствии с настоящим изобретением в изготовлении медикамента для лечения или предупреждения опосредованных TRK заболеваний, таких как опухоли, особенно гематологические злокачественные заболевания, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы, глиома.
[0020] Другой аспект настоящего изобретения относится к применению соединения, представленного общей формулой I, или его таутомеров, мезомеров, рацематов, энантиомеров, диастереомеров или их смеси и их фармацевтически приемлемых солей или содержащей их фармацевтической композиции в изготовлении медикамента для лечения и/или предупреждения опухолей.
[0021] В соответствии с настоящим изобретением медикамент может находиться в любой дозированной форме, включающей в себя без ограничения таблетки, капсулы, растворы, лиофилизированные препараты, инъекции.
[0022] Фармацевтический препарат в соответствии с настоящим изобретением может быть введен в форме единицы дозирования, содержащей предварительно определенное количество активного ингредиента на единицу дозирования. Такая единица может содержать, например, от 0,5 мг до 1 г, предпочтительно от 1 мг до 700 мг, особенно предпочтительно от 5 мг до 300 мг соединения в соответствии с настоящим изобретением, в зависимости от состояния, подлежащего лечению, способа введения, а также возраста, массы и общего состояния больного. Состав может быть введен в форме единицы дозирования, содержащей предварительно определенное количество активного ингредиента на единицу дозирования. Предпочтительными составами единицы дозирования являются такие, которые содержат активный ингредиент в суточных или поделенных дозах или их соответствующих фракциях, как указано выше. Кроме того, такие фармацевтические препараты могут быть получены с использованием способов, известных в фармацевтической области.
[0023] Фармацевтические препараты в соответствии с настоящим изобретением могут подходить для введения любым желаемым подходящим способом, таким как пероральный (в том числе пероральный или подъязычный), ректальный, назальный, местный (в том числе пероральный, подъязычный или трансдермальный), вагинальный или парентеральный (в том числе подкожный, внутримышечный, внутривенный или внутрикожный). Все способы, известные в фармацевтической области, можно применять для получения таких составов, например, путем объединения активного ингредиента с одним или несколькими вспомогательными средствами или с одним или несколькими адъювантами.
[0024] Настоящее изобретение также относится к способу лечения или предупреждения опосредованных TRK заболеваний (таких как опухоли, особенно гематологические злокачественные заболевания, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы, глиома), который предусматривает введение больному при необходимости терапевтически эффективного количества соединения в соответствии с настоящим изобретением или его изомеров, пролекарств, сольватов, стабильных изотопных производных или фармацевтически приемлемых солей, или фармацевтической композиции в соответствии с настоящим изобретением.
[0025] Другой аспект настоящего изобретения относится к соединению, представленному общей формулой I, или его изомерам, пролекарствам, сольватам, стабильным изотопным производным или фармацевтически приемлемым солям, или содержащей их фармацевтической композиции для применение в лечении или предупреждении опосредованных TRK заболеваний, таких как опухоли, особенно гематологические злокачественные заболевания, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы и глиома.
[0026] Другой аспект настоящего изобретения относится к соединению, представленному общей формулой I, или их таутомерам, мезомерам, рацематам, энантиомерам, диастереомерам или их смеси и их фармацевтически приемлемым солям для применения в лечении и/или предупреждении опухолей и других заболеваний.
[0027] Схемы получения
Настоящее изобретение, кроме того, относится к способам получения соединений.
[0028] Схема 1
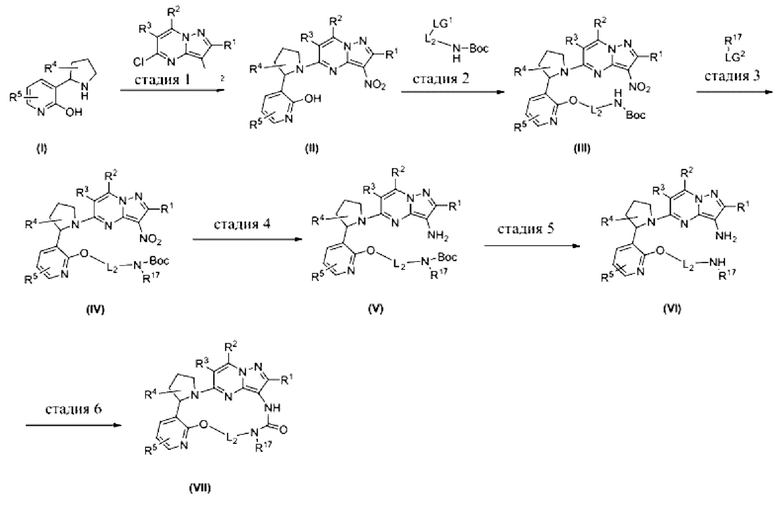
[0029] Соответственно, каждый заместитель определяется следующим образом:
каждый R1, R2, R3 независимо выбран из водорода, галогена и С1-С4алкила;
R4 выбран из водорода, галогена, -NR8R9, -OR10;
R5 выбран из водорода, галогена, С1-С4алкила и С3-С6циклила;
R17 выбран из водорода, С1-С4алкила;
L2 выбран из С1-С4алкилена, при этом алкилен может быть необязательно замещен одним или несколькими заместителями, выбранными из галогена, С1-С4алкила, -OR10, -NR8R9, при этом алкил может быть необязательно замещен одним или несколькими заместителями, выбранными из галогена, -NR11R12, -OR16;
каждый R8, R9, R10, R11, R12 и R16 независимо выбран из водорода, С1-С4алкила и С1-С4галогеналкила.
Стадия 1
Реакцию замещения выполняли в растворителе, таком как н-бутанол, или добавляли N,N-диметилацетамид, N,N-диизопропилэтиламин или 1,8-диазабициклоундец-7-ен (DBU) и т.д., реакцию выполняли при 60-80°С в условиях нагревания, например, с помощью микроволнового излучения или масляной бани, с получением соединения (II).
Стадия 2
LG1 представляет собой галоген, такой как Cl, Br, I, или уходящую группу, такую как OTf, ОТ, ОМ и т.д. Реакцию замещения выполняли в растворителе, таком как ацетонитрил, и добавляли основание, такое как карбонат цезия. Реакцию выполняли при 50-100°С в условиях нагревания, например, с помощью микроволнового излучения или масляной бани, с получением соединения (III).
Стадия 3
LG2 представляет собой галоген, такой как Cl, Br, I, или уходящую группу, такую как OTf, ОТ, ОМ и т.д. Реакцию замещения выполняли в растворителе, таком как N,N-диметилацетамид, одновременно добавляли основание, такое как гидрид натрия, и реакцию выполняли при 0-25°С с получением соединения (IV).
Стадия 4
Порошок цинка использовали в качестве восстановителя при восстановлении нитрогруппы; добавляли насыщенный раствор хлорида аммония и реакцию выполняли в растворителе, таком как дихлорметан, при 0-25°С с получением соединения (V).
Стадия 5
Трифторуксусную кислоту использовали в качестве кислоты в реакции снятия защитной группы трет-бутоксикарбонильной группы; реакцию выполняли в растворителе, таком как дихлорметан, при 0-25°С с получением соединения (VI).
Стадия 6
N,N'-Карбонилдиимидазол или N,N'-карбонилди(1,2,4-триазол) использовали в реакции образования мочевины из диамина (VI). Иногда добавляли основание, такое как триэтиламин, и реакцию выполняли в растворителе, таком как N,N-диметилформамид и т.д., при комнатной температуре или 20-50°С в условиях нагревания, например, с помощью масляной бани, с получением соединения (VII).
Варианты осуществления
[0030] Определения
Если не указано обратное, следующие используемые в настоящем описании и формуле изобретения термины имеют значения, изложенные ниже.
[0031] Используемое в настоящем документе выражение «Сх-Су» представляет диапазон числа атомов углерода, при этом и х, и у представляют собой целые числа. Например, С3-С8циклил представляет собой циклильную группу, имеющую от 3 до 8 атомов углерода, а С0-С2алкил представляет собой алкильную группу, имеющую от 0 до 2 атомов углерода, при этом С0алкил относится к одинарной связи.
[0032] Используемый в настоящем документе термин «алкил» относится к насыщенной алифатической углеводородной группе, включающей в себя линейные и разветвленные группы, имеющие от 1 до 20 атомов углерода, например, от 1 до 18 атомов углерода, от 1 до 12 атомов углерода, от 1 до 8 атомов углерода, от 1 до 6 атомов углерода или от 1 до 4 атомов углерода. Неограничивающие примеры включают в себя метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, а также их различные разветвленные изомеры и т.д. Алкил может быть замещенным или незамещенным.
[0033] Используемый в настоящем документе термин «алкенил» относится к линейной, разветвленной углеводородной группе, содержащей по меньшей мере одну углерод-углерод двойную связь, включающей в себя линейные и разветвленные группы, имеющие от 2 до 20 атомов углерода, например, от 2 до 18 атомов углерода, от 2 до 12 атомов углерода, от 2 до 8 атомов углерода, от 2 до 6 атомов углерода или от 2 до 4 атомов углерода. При этом от 1 до 3 углерод-углерод двойных связей могут присутствовать, и предпочтительно может присутствовать 1 углерод-углерод двойная связь. Термин «С2-4алкенил» относится к алкенилу имеющему от 2 до 4 атомов углерода, включающему в себя винил, пропенил, бутенил, бутен-2-ил, 2-метилбутенил. Алкенильная группа необязательно может быть замещенной.
[0034] Используемый в настоящем документе термин «алкинил» относится к линейной или разветвленной углеводородной группе, содержащей по меньшей мере одну углерод-углерод тройную связь, включающей в себя линейные и разветвленные группы, имеющие от 2 до 20 атомов углерода, например, линейные и разветвленные группы, имеющие от 2 до 18 атомов углерода, от 2 до 12 атомов углерода, от 2 до 8 атомов углерода, от 2 до 6 атомов углерода или от 2 до 4 атомов углерода. Среди них от 1 до 3 углерод-углерод тройных связей могут присутствовать, и предпочтительно может присутствовать 1 углерод-углерод тройная связь. Термин «С2-4алкинил» относится к алкинилу, имеющему от 2 до 4 атомов углерода, Неограничивающие примеры включают в себя ацетенил, пропинил, бутинил, бутин-2-ил и 3-метил-1-бутинил.
[0035] Используемые в настоящем документе термины «алкилен», «алкенилен» и «алкинилен», соответственно, относятся к замещенным или незамещенным алкильным, алкенильным и алкинильным группам, имеющим два концевых ядра из одновалентных групп, которые получаются путем удаления одного атома водорода от каждого из двух концевых атомов углерода; «алкилен», «алкенилен» и «алкинилен» обычно имеют 1-8 атомов углерода, предпочтительно 1-6 атомов углерода, более предпочтительно 1-4 атомов углерода. Неограничивающие примеры «алкилена» включают в себя замещенные или незамещенные метилен, этилен, пропилен, бутилен и т.д.; неограничивающие примеры «алкенилена» включают в себя замещенные или незамещенные винилен, пропенилен, бутенилен и т.д. Неограничивающие примеры «алкинилена» включают в себя замещенные или незамещенные этинилен, пропинилен, бутинилен и т.д.
Используемый в настоящем документе термин «циклил» относится ко всем углеродным насыщенным или частично ненасыщенным моноциклическим или полициклическим гидрокарбильным группам, содержащим от 3 до 12 циклических атомов углерода, например, от 3 до 12, от 3 до 10, от 3 до 8 или от 3 до 6 циклических атомов углерода, или они могут представлять собой 3, 4, 5, 6-членные кольца. Неограничивающие примеры моноциклического циклила включают в себя циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.д. Циклил может быть замещенным или незамещенным.
[0036] Используемый в настоящем документе термин «циклилен» относится к замещенной или незамещенной циклической группе, имеющей два концевых ядра из одновалентных групп, и циклическая группа определяется, как упомянуто выше. Неограничивающие примеры «циклилена» включают в себя циклопропилен, циклобутилен, циклопентилен, циклопентенилен, циклогексилен, циклогексенилен, циклогексадиенил, Циклогептилен, циклогептатриенилен, циклооктилен и т.д. Циклиленовая группа может быть замещенной или незамещенной.
[0037] Используемый в настоящем документе термин «гетероциклил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической гидрокарбильной группе, содержащей от 3 до 20 кольцевых атомов, например, от 3 до 16, от 3 до 12, от 3 до 10, от 3 до 8 или от 3 до 6 кольцевых атомов, при этом одним или несколькими кольцевых атомов представляют собой гетероатомы, выбранные из азота, кислорода или S(O)m (при этом m представляет собой целое число от 0 до 2), но за исключением кольцевых частей -О-О-, -O-S- или -S-S-, а остальные кольцевые атомы представляют собой углерод. Предпочтительно содержится от 3 до 12 кольцевых атомов (из которых от 1 до 4 являются гетероатомами); более предпочтительно гетероциклильное кольцо содержит от 3 до 10 кольцевых атомов, более предпочтительно от 3 до 8 кольцевых атомов, кроме того, предпочтительно от 3 до 6 кольцевых атомов; наиболее предпочтительными являются 5-членные кольца или 6-членные кольца, в которых от 1 до 4 членов представляют собой гетероатомы, более предпочтительно, в которых от 1 до 3 членов представляют собой гетероатомы, и наиболее предпочтительно от 1 до 2 членов представляют собой гетероатомы. Неограничивающие примеры моноциклического гетероциклила включают в себя пирролидинил, пиперидил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.д. Бициклические и полициклические гетероциклические группы включают в себя спироциклические, слитые и мостиковые циклические гетероциклические группы.
[0038] Используемый в настоящем документе термин «спирогетероциклил» относится к 5-20-членной полициклической гетероциклической группе с одним атомом (называемым спироатомом), общим для моноциклических колец, при этом один или несколько из кольцевых атомов представляют собой гетероатомы, выбранные из азота, кислорода или S(O)m (при этом m представляет собой целое число от 0 до 2), а остальные из кольцевых атомов представляют собой углерод. Они могут содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью конъюгированной системы π-электронов. Предпочтительно они являются 6-14-членными и более предпочтительно 7-10-членными. В соответствии с числом спироатомов, общих для колец, спирогетероциклические группы делят на моноспирогетероциклическую группу, биспирогетероциклическую группу или полиспирогетероциклическую группу, предпочтительно моноспироциклическую группу и биспироциклическую группу и более предпочтительно 4-членную/4-членную, 4-членную/5-членную, 4-членную/6-членную, 5-членную/5-членную или 5-членную/6-членную моноспироциклическую группу. Неограничивающие примеры спирогетероциклила включают в себя

[0039] Используемый в настоящем документе термин «слитый гетероциклил» относится к 5-20-членной полициклической гетероциклической группе, при этом каждое кольцо в системе имеет общую пару соседних атомов с другими кольцами в системе, одно или несколько колец могут содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью конъюгированной системы π-электронов, при этом один или несколько кольцевых атомов представляют собой гетероатомы, выбранные из азота, кислорода или S(O)m (при этом m представляет собой целое число от 0 до 2), а остальные кольцевые атомы представляют собой углерод. Предпочтительно они являются 6-14-членными и более предпочтительно 7-10-членными. В соответствии с числом колец они могут быть разделены на бициклическую, трициклическую, тетрациклическую или полициклическую слитую гетероциклическую группу, и слитые гетероциклические группы предпочтительно являются бициклическими или трициклическими, более предпочтительно 5-членными/5-членными или 5-членными/6-членными бициклическими слитыми гетероциклическими группами. Неограничивающие примеры слитого гетероциклила включают в себя


[0040] Гетероциклическое кольцо может быть слитым с арилом, гетероарилом или циклическим кольцом, при этом кольцо, присоединенное к исходной структуре, представляет собой гетероциклическую группу, и неограничивающие примеры включают в себя
 и подобные.
и подобные.
Гетероциклил может быть замещенным или незамещенным.
[0041] Используемый в настоящем документе термин «арил» относится к 6-14-членной только из углеродов моноциклической или конденсированной полициклической (т.е. кольца, имеющие общие соседние пары атомов углерода) группе и полициклической (т.е. кольца, несущие соседние пары атомов углерода) группе, имеющей конъюгированную систему π-электронов. Арильные группы могут быть моноциклическими или полициклическими (т.е. могут содержать более чем одно кольцо). В случае полициклических ароматических колец только одно кольцо в полициклическое системе должно быть ароматическим кольцом, тогда как остальные кольца могут быть насыщенными, частично насыщенными или ненасыщенными кольцами. Арильная группа предпочтительно является 6-10-членной, например, фенилом и нафтилом, и наиболее предпочтительно фенилом. Арильное кольцо может быть слито с гетероарилом, гетероциклильным или циклильным кольцом, при этом кольцо, соединенное с исходной структурой, представляет собой арильное кольцо, и неограничивающие примеры включают в себя

Арил может быть замещенным или незамещенным.
[0042] Термин «арилен» относится к замещенной или незамещенной арильной группе, имеющей два ядра из одновалентных групп, соответственно, и определение арильной группы является таким же, как описано выше. Неограничивающими примерами ариленовых групп являются фенилен, нафтилен и т.п. Ариленовая группа может быть необязательно замещенной или незамещенной.
[0043] Используемый в настоящем документе термин «гетероарил» относится к гетероароматической системе, содержащей от 1 до 4 гетероатомов и от 5 до 14 кольцевых атомов, при этом гетероатомы включают в себя кислород, серу и азот. Гетероарил предпочтительно является 5-10-членным и более предпочтительно 5-членным или 6-членным, например, фурил, тиенил, пиридил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил, тетразил, оксазолил и изоксазолил и т.д. Гетероарильное кольцо может быть слитым с арильным, гетероциклильным или циклильным кольцом, при этом кольцо, соединенное с исходной структурой, является гетероарильным кольцом, и неограничивающие примеры включают в себя


Гетероарил может быть замещенным или незамещенным.
[0044] Используемый в настоящем документе термин «гетероарилен» относится к замещенной или незамещенной гетероарильной группе, имеющей два ядра из одновалентных групп, соответственно, и определение гетероарильной группы является таким же, как описано выше. Неограничивающими примерами гетероариленовых групп являются фуранилен, тиенилен, пиридилен, пирролилен, N-алкилпирролилен, пиримидинилен, пиразинилен, имидазолилиден, тетразолилиден, оксазолилиден, изоксазолилиден и т.д. Гетероариленовая группа может быть необязательно замещенной или незамещенной.
[0045] Используемый в настоящем документе термин «галоген» относится к фтору, хлору, брому или йоду.
[0046] Термин «галогеналкил» относится к алкильному заместителю, в котором по меньшей мере один водород заменяется галогеновой группой. Типичные галогеновые группы включают в себя хлор, фтор, бром и йод. Примеры галогеналкильных групп включают в себя фторметил, фторэтил, хлорметил, хлорэтил, 1-бромэтил, дифторметил, трифторметил и 1,1,1-трифторэтил. Следует учитывать, что, если заместитель является замещенным более чем одной галогеновой группой, эти галогеновые группы могут быть одинаковыми или разными (если не указано иное).
[0047] Используемый в настоящем документе термин «формил» относится к -СНО.
[0048] Используемый в настоящем документе термин «карбоксил» относится к -СООН.
[0049] Используемый в настоящем документе термин «циано» относится к -CN.
[0050] Используемый в настоящем документе термин «гетероалкил» относится к стабильной с прямой цепью или с разветвленной цепью углеводородной группой, состоящей из определенного числа атомов углерода и по меньшей мере одного гетероатома, выбранного из кислорода, азота и серы. Среди них атомы азота и серы могут быть необязательно окислены, атомы азота могут быть необязательно кватернизированы, и гетероатомы, такие как кислород, азот и сера, могут быть расположены в любом внутреннем положении гетероалкильной группы или в положении, в котором алкильная группа соединена с остальной частью молекула. Более чем два гетероатома могут быть независимыми или непрерывными.
[0051] Используемые в настоящем документе термины «необязательный» и «необязательно» означают, что событие или окружающее условие, описанное далее, необязательно может произойти или может не произойти, включая случаи, когда событие или окружающее условие происходит или не происходит. Например, термин «гетероциклил, необязательно замещенный алкилом» означает, что алкил необязательно может существовать или может не существовать, включая случаи, когда гетероциклил замещен алкилом и не замещен алкилом.
[0052] Используемый в настоящем документе термин «замещенный» относится к тому, что один или несколько атомов водорода, предпочтительно максимум 5 и более предпочтительно от 1 до 3 атомов водорода, в группе независимо заменяются соответствующим числом заместителей. Разумеется, что заместители расположены только в своих возможных химических положениях, и специалист в данной области без больших усилий может определить (экспериментально или теоретически) возможные или невозможные замещения. Например, амино или гидроксигруппы, имеющие свободный водород, могут быть нестабильными в сочетании с атомами углерода, имеющими ненасыщенные (например, олефиновые) связи.
[0053] Заместитель(и) включает(включают) в себя без ограничения группы, описываемые выше.
[0054] Используемый в настоящем документе термин «фармацевтическая композиция» представляет собой смесь одного или нескольких соединений, описываемых в настоящем документе, или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами, а также другими компонентами, такими как физиологически/фармацевтически приемлемые носители и вспомогательные средства. Целью фармацевтических композиций является обеспечение введения лекарственных средств в организмы, облегчение абсорбции активных ингредиентов и, таким образом, проявление биологической активности.
[0055] Термин «комнатная температура» в настоящем изобретении относится к 15-30°С.
[0056] Соединения в соответствии с настоящим изобретением также могут существовать в виде их изомера, пролекарства, комплекса с растворителем или стабильного изотопного производного. Специалистам в данной области будет понятно, что эти изомеры, пролекарства, комплексы с растворителем или стабильные изотопные производные, как правило, характеризуются активностями, подобными таковым соединений в соответствии с настоящим изобретением или их фармацевтически приемлемых солей, и, поэтому, попадают в объем защиты настоящего изобретения.
[0057] Используемые в настоящем документе термины «стабильное изотопное производное» и «стабильные изотопные производные» включают в себя производные, замещенные изотопами, такие как производные, полученные путем замещения любого атома водорода в формуле I 1-5 атомами дейтерия, производные, замещенные изотопами, полученными путем замещения любого атома углерода в формуле I 1-3 атомами углерода-14, или производные, замещенные изотопами, полученными путем замещения любого атома кислорода в формуле I 1-3 атомами кислорода-18.
[0058] Термин «фармацевтически приемлемые соли», описываемый в настоящем изобретении, обсуждался в работе Berge, et al., "Pharmaceutically acceptable salts," J. Pharm. Sci., 66, 1-19 (1977), и для химиков-фармацевтов очевидно, что указанные соли по сути нетоксичны и могут обеспечивать желаемые фармакокинетические свойства, вкусовые качества, абсорбцию, распределение, метаболизм или выведение и т.п.
[0059] «Фармацевтически приемлемые соли» в соответствии с настоящим изобретением могут быть синтезированы посредством общего химического способа.
[0060] Как правило, получение солей может быть достигнуто путем осуществления реагирования соединений в форме свободных основания или кислоты с равными химическими эквивалентами или избыточными количествами кислот (неорганических или органических кислот) или оснований в подходящих растворителях или композициях растворителей.
[0061] Описываемый в настоящем изобретении термин «пролекарство» относится к соединению, которое может быть превращено в оригинальное активное соединение после его метаболизма in vivo. В целом, пролекарства представляют собой неактивные вещества или характеризуются активностью ниже, чем у активных исходных соединений, но могут обеспечивать удобное действие, введение или улучшение метаболических свойств.
[0062] Термин «изомер» в соответствии с настоящим изобретением означает, что соединение формулы I в соответствии с настоящим изобретением может иметь один или несколько центров асимметрии и может представлять собой рацемат, рацемическую смесь и отдельный диастереоизомер. Изомеры, такие как стереоизомеры и геометрические изомеры, включены в настоящее изобретение. Геометрические изомеры включают в себя цис- и транс-изомеры.
[0063] Настоящее изобретение включает в себя любой полиморф соединения или иго солей, а также любой вид гидрата или другого сольвата.
[0064] Используемый в настоящем документе термин «опухоль» включает в себя доброкачественные опухоли и злокачественные опухоли, такие как рак.
[0065] Используемый в настоящем документе термин «рак» включает в себя различные опухоли, опосредованные TRK, в том числе без ограничения гематологические злокачественные опухоли, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы и глиому.
[0066] Термин «терапевтически эффективное количество» относится к количеству соединения в соответствии с настоящим изобретением, которое может эффективно лечить или предупреждать соответствующие заболевания, опосредованные TRK.
Примеры
[0067] Далее настоящее изобретение будет иллюстрироваться посредством приведенных ниже примеров, но его объем не ограничивается описываемыми примерами. В следующих примерах выбраны экспериментальные способы без указания конкретных условий в соответствии с традиционными способами и условиями или в соответствии с инструкциями по применению.
[0068] Структуры всех соединений в соответствии с настоящим изобретением можно идентифицировать с помощью ядерного магнитного резонанса (1Н ЯМР) и/или масс-спектрометрического (MS) выявления.
[0069] Химический сдвиг (δ) 1Н ЯМР регистрируют в миллионных долях (единица: 10-6 миллионных долей). ЯМР выполняют на спектрометре Bruker AVANCE-400. Подходящие растворители включают в себя дейтерированный хлороформ (CDCl3), дейтерированный метанол (CD3OD) и дейтерированный диметилсульфоксид (DMSO-d6) с тетраметилсиланом в качестве внутреннего стандарта (TMS).
[0070] Масс-спектрограмму низкого разрешения (MS) получают с помощью масс-спектрометра Agilent 1260HPLC/6120 с использованием Agilent ZORBAX XDB-C18, 4,6×50 мм, 3,5 мкм, в условиях градиентного элюирования I: 0: 95% растворителя А1 и 5% растворителя В1, 1-2: 5% растворителя А1 и 95% растворителя В1; 2,01-2,50: 95% растворителя А1 и 5% растворителя В1. Процент представляет собой объемный процент определенного растворителя от общего объема растворителя. Растворитель А1: 0,01% водный раствор муравьиной кислоты; растворитель В1: 0,01% раствор муравьиной кислоты в ацетонитриле; и процент представляет собой объемный процент растворенного вещества в расчете на раствор.
[0071] Пластина с тонким слоем силикагеля представляет собой пластину с силикагелем Yantai Yellow Sea HSGF254 или Qingdao Haiyang GF254. Силикагель Yantai Yellow Sea 100-200 или 200-300 мэш обычно используют в качестве подложки в колоночной хроматографии.
[0072] Используемая препаративная жидкостная хроматография (препаративная HPLC) представляет собой сепаратор для жидкостной хроматографии высокого давления с масс-спектрометрическим контролем Waters SQD2, XBridge-C18; препаративная колонка 30×150 мм, 5 мкм; способ 1: ацетонитрил-вода (0,2% муравьиная кислота), скорость потока: 25 мл/минута; способ 2: ацетонитрил-вода (0,8% бикарбонат аммония), скорость потока: 25 мл/минута. Известные исходные сырьевые материалы в соответствии с настоящим изобретением могут быть синтезированы согласно способам, известным в уровне техники, или в соответствии с ними, или их можно приобрести у таких компаний, как Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc., Shanghai Bide Pharmatech, Shanghai Aladdin Chemistry, Shanghai Meryer Chemistry, Accelerating Chemistry, Energy Chemistry и т.п.
[0073] В примерах, если не указано иное, все растворители, используемые в реакции, представляют собой безводные растворители. Безводный тетрагидрофуран получали обработкой коммерчески доступного тетрагидрофурана кусочками металлического натрия в качестве дегидранта, бензофеноном в качестве индикатора. Раствор кипятили с обратным холодильником в газообразном аргоне до тех пор, пока он не приобретал голубовато-фиолетовый цвет, затем безводный тетрагидрофуран перегоняли, собирали и хранили при комнатной температуре под защитой газообразного азота. Остальные безводные растворители приобретали в компаниях Energy Chemistry и Accelerating Chemistry. Перенос и использование всех безводных растворителей следует проводить в атмосфере аргона, если не указано иное.
[0074] В примерах все реакции проводили в атмосфере аргона или атмосфере азота, если не указано иное.
[0075] Атмосфера аргона или атмосфера азота означает, что реакционная колба соединена с баллоном с аргоном или азотом объемом приблизительно 1 л.
[0076] Атмосфера водорода означает, что реакционная колба соединена с баллоном с водородом объемом приблизительно 1 л.
[0077] Для реакции гидрогенизации обычно требуется вакуумирование реакционного контейнера и заполнение его газообразным водородом, и такую операцию следует повторить 3 раза.
[0078] Реакцию проводили при комнатной температуре, и диапазон температур составлял от 15°С до 30°С, если не указано иное.
[0079] Тонкослойную хроматографию (TLC) использовали для мониторинга процесса реакции в примерах. Система проявителя, используемая для реакции, включала в себя А - систему дихлорметана и метанола, и В - систему петролейного эфира и этилацетата, и объемное соотношение растворителей регулировали в соответствии с полярностью соединения.
[0080] Элюентная система для колоночной хроматографии и система проявителя для тонкослойной хроматографии, используемые для очистки соединений, включали в себя А - систему дихлорметана и метанола, и В - систему петролейного эфира и этилацетата, объемное соотношение растворителей регулировали в соответствии с полярностью соединения, и для регулировки также может быть добавлено небольшое количество триэтиламина, кислотных или основных реагентов и т.п.
[0081] Синтез промежуточного соединения I (INT 1)

Стадия 1
(S)-N-((R)-1-(5-Фтор-2-метоксипиридин-3-ил)бутан-3-ен-1-ил)-2-метилпропан-2-сульфинамид 1-3
Растворяли 5-фтор-2-метоксиникотинальдегид 1-1 (2,50 г, 16,10 ммоль) в тетрагидрофуране (35 мл), последовательно при перемешивании добавляли порошок индия (2,40 г, 20,90 ммоль), (S)-метилпропан-2-сульфаниламид (2,33 г, 19,30 ммоль) и тетраэтоксититан (5,50 г, 24,20 ммоль), перемешивали при 70°С в течение 2 часов, охлаждали до 0°С, а затем добавляли 3-бромпропен 1-2 (3,10 г, 26,00 ммоль), обеспечивали реагирование при 70°С в течение 16 часов после добавления. Охлаждали на ледяной бане, гасили добавлением 150 мл воды, фильтровали, фильтрат экстрагировали дихлорметаном (200 мл × 3), органическую фазу сушили безводным сульфатом натрия, фильтровали, центрифугировали и очищали на колонке с силикагелем (петролейный эфир : этилацетат = 1:0~1:1) с получением ((S)-N-((R)-1-(5-фтор-2-метоксипиридин-3-ил)бутан-3-ен-1-ил)-2-метилпропан-2-сульфинамида 1-3 (4,60 г, желтое масло), выход 95,2%. 5-Фтор-2-метоксиникотинальдегид 1-1 (2,50 г, 16,10 ммоль) растворяли в тетрагидрофуране (35 мл). Последовательно при перемешивании добавляли порошок индия (2,40 г, 20,90 ммоль), (S)-2-метилпропан-2-сульфинамид (2,33 г, 19,30 ммоль) и тетраэтоксититан (5,50 г, 24,20 ммоль). Реакционную смесь взбалтывали при 70°С в течение 2 часов, охлаждали до 0°С, затем добавляли 3-бромпропен 1-2 (3,10 г, 26,00 ммоль). Реакцию продолжали в течение 16 часов при 70°С. Затем реакционную смесь охлаждали в ледяной бане и добавляли 150 мл воды для прекращения реакции. Смесь фильтровали и фильтрат экстрагировали дихлорметаном (200 мл × 3). Органическую фазу сушили безводным сульфатом натрия, фильтровали и очищали с помощью колонки с силикагелем (петролейный эфир: этилацетат = 1:0~1:1) с получением (S)-N-((R)-1-(5-фтор-2-метоксипиридин-3-ил)бутан-3-ен-1-ил)-2-метилпропан-2-сульфинамида 1-3 (4,60 г, желтое масло). Выход: 95,2%.
MS масса/заряд (ESI): 301 [М+1].
1Н ЯМР (400 МГц, CDC13) δ 7,91 (d, J=3,2 Гц, 1Н), 7,36-7,33 (m, 1Н), 5,74-5,66 (m, 1H), 5,20-5,15 (m, 2Н), 4,77-4,73 (m, 1H), 3,95 (s, 3Н), 3,78-3,75 (m, 1H), 2,70-2,65 (m, 1H), 2,48-2,43 (m, 1H), 1,23 (s, 9Н).
Стадия 2
(R)-1-(5-Фтор-2-метоксифенил)бут-3-ен-1-амина гидрохлорид 1-4
(S)-N-((R)-1-(5-Фтор-2-метоксипиридин-3-ил)бутан-3-ен-1-ил)-2-метилпропан-2-сульфинамид 1-3 (4,60 г, 15,30 ммоль) растворяли в растворе хлороводорода в диоксане (4 М, 25 мл) и метаноле (25 мл). Реакционную смесь взбалтывали при комнатной температуре в течение 2 часов до завершения реакции. Растворитель удаляли при пониженном давлении с получением (R)-1-(5-фтор-2-метоксифенил)бут-3-ен-1-амина гидрохлорида 1-4 (5 г, белое твердое вещество) в виде неочищенного продукта, теоретический выход: 3,56 г.
MS масса/заряд (ESI): 197 [М+1].
Стадия 3
(R)-N-(1-(5-Фтор-2-метоксипиридин-3-ил)бут-3-ен-1-ил)ацетамид 1-5
(R)-1-(5-Фтор-2-метоксифенил)бут-3-ен-1-амингидрохлорид 1-4 (3,56 г, 15,30 ммоль) растворяли в дихлорметане (50 мл) и добавляли триэтиламин (3,86 г, 38,00 ммоль) при взбалтывании. Смесь взбалтывали при комнатной температуре в течение 5 минут, затем добавляли ангидрид уксусной кислоты (2,34 г, 23,00 ммоль) и взбалтывание продолжали при комнатной температуре в течение 3 часов. Насыщенный водный раствор бикарбоната натрия (30 мл) добавляли для прекращения реакции и смесь экстрагировали дихлорметаном (50 мл × 3). Органический слой промывали соляным раствором (50 мл), сушили безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением (R)-N-(1-(5-фтор-2-метоксипиридин-3-ил)бут-3-ен-1-ил)ацетамида 1-5 (3,46 г, желтое твердое вещество), выход: 95,0%.
MS масса/заряд (ESI): 239 [М+1].
1Н ЯМР (400 МГц, CDCl3) δ 7,90 (d, J=2,8 Гц, 1H), 7,23-7,21 (m, 1H), 6,19 (d, J=7,6 Гц, 1H), 5,68-5,58 (m, 1Н), 5,13-5,07 (m, 2H), 5,06-5,04 (m, 1H), 3,98 (s, 3Н), 2,56-2,53 (m, 2H), 2,01 (s, 3Н).
Стадия 4
(5R)-5-(5-Фтор-2-метоксипиридин-3-ил)пирролидин-3-ил-ацетат 1-6
(R)-N-(1-(5-Фтор-2-метоксипиридин-3-ил)бут-3-ен-1-ил)ацетамид 1-5 (3,46 г, 14,50 ммоль) растворяли в тетрагидрофуране (80 мл) и воде (20 мл), добавляли йод (11,08 г, 43,60 ммоль) при взбалтывании. Реакционную смесь взбалтывали при комнатной температуре в течение 18 часов. Добавляли насыщенный водный раствор сульфита натрия и бикарбоната натрия (100 мл) и взбалтывали в течение 0,5 часа. Смесь экстрагировали этилацетатом (100 мл × 3) и объединенный органический слой промывали соляным раствором (100 мл). Органическую фазу сушили безводным сульфатом натрия, фильтровали и фильтрат концентрировали с получением (5R)-5-(5-фтор-2-метоксипиридин-3-ил)пирролидин-3-ил-ацетата 1-6 (3,68 г, желтое твердое вещество) в виде неочищенного продукта.
MS масса/заряд (ESI): 255 [М+1].
Стадия 5
Трет-бутил-(2R)-4-ацетокси-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-карбоксилат 1-7
(5R)-5-(5-Фтор-2-метоксипиридин-3-ил)пирролидин-3-ил-ацетат 1-6 (3,68 г, 14,50 ммоль) растворяли в тетрагидрофуране (15 мл) и воде (15 мл). Добавляли водный раствор гидроксида натрия (1 М, 10 мл) и ди-трет-бутил-бикарбоната (4,16 г, 18,90 ммоль) при взбалтывании. Реакционную смесь взбалтывали при комнатной температуре в течение 18 часов и добавляли воду (100 мл) для разбавления смеси. Реакционную смесь экстрагировали этилацетатом (80 мл × 3) и объединенный органические слой промывали водой (100 мл × 2) и солевым раствором (100 мл × 2). Органическую фазу сушили сульфатом натрия, фильтровали, фильтрат концентрировали с получением трет-бутил-(2R)-4-ацетокси-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-карбоксилата 1-7 (6,0 г, желтое твердое вещество) в виде неочищенного продукта.
MS масса/заряд (ESI): 377 [М+23].
Стадия 6
Трет-бутил-(2R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат 1-8
Трет-бутил-(2R)-4-ацетокси-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-карбоксилат 1-7 (5,13 г, 14,50 ммоль) растворяли в метаноле (40 мл) и добавляли водный раствор гидроксида натрия (1 М, 20 мл) при взбалтывании. Реакционную смесь взбалтывали при комнатной температуре в течение 2 часов. Добавляли воду (100 мл) для разбавления реакционной смеси, затем ее экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу промывали водой (100 мл × 2) и солевым раствором (100 мл × 2). Органическую фазу сушили сульфатом натрия и фильтровали. Фильтрат концентрировали и очищали с помощью колонки с силикагелем (петролейный эфир : этилацетат = 1:0~1:1) с получением трет-бутил-(2R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата 1-8 (2,45 г, желтое твердое вещество), суммарный выход трех стадий составляет 54,1%.
MS масса/заряд (ESI): 335 [М+23].
1H ЯМР (400 МГц, CDC13) δ 7,87 (d, J=7,6 Гц, 1H), 7,38-7,14 (m, 1H), 5,12-4,93 (m, 1H), 4,49-4,41 (m, 1H), 3,93 (s, 3H), 3,75-3,52 (m, 2H), 2,53-2,41 (m, 1H), 1,98-1,91 (m, 1H), 1,46 (s, 4H), 1,18 (s, 5H).
Стадия 7
Трет-бутил-(R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-оксопирролидин-1-карбоксилат 1-9
Трет-бутил-(2R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат 1-8 (2,45 г, 7,85 ммоль) растворяли в дихлорметане (40 мл), добавляли периодинан Десса-Мартина (4,16 г, 9,82 ммоль) при комнатной температуре. Реакционную смесь взбалтывали при комнатной температуре в течение 18 часов и для разбавления ее добавляли дихлорметан (50 мл). Смесь последовательно промывали насыщенным водным раствором сульфита натрия (30 мл) и солевым раствором (50 мл). Органическую фазу сушили безводным сульфатом натрия, фильтровали и фильтрат концентрировали с получением трет-бутил-(R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-оксопирролидин-1-карбоксилата 1-9 (2,40 г, желтое масло), выход: 99,0%.
MS масса/заряд (ESI): 333 [М+23].
1Н ЯМР (400 МГц, CDC13) δ 7,93 (s, 1H), 7,27-7,25 (m, 1H), 5,27-5,17 (m, 1Н), 4,00-3,85 (m, 2Н), 3,89 (s, 3Н), 3,09-3,02 (m, 1H), 2,58-2,54 (m, 1Н), 1,45 (s, 4Н), 1,38 (s, 5Н).
Стадия 8
Трет-бутил-(2R,4R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат 1-10
Трет-бутил-(R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-оксопирролидин-1-карбоксилат 1-9 (2,40 g, 7,70 ммоль) растворяли в метаноле (15 мл) и добавляли борогидрид натрия (0,29 г, 7,70 ммоль) при -78°С. Реакционную смесь взбалтывали при -78°С в течение 1 часа. Насыщенный водный раствор хлорида аммония (10 мл) добавляли для прекращения реакции и смесь экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу промывали водой (100 мл × 2) и солевым раствором (100 мл × 2). Органическую фазу сушили безводным сульфатом натрия, фильтровали и фильтрат концентрировали с получением трет-бутил-(2R,4R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата 1-10 (2,27 г, желтое масло), выход: 93,7%.
MS масса/заряд (ESI): 335 [М+23].
1Н ЯМР (400 МГц, CDCl3) δ 7,85 (s, 1H), 7,35-7,27 (m, 1H), 5,06-4,98 (m, 1H), 4,50-4,47 (m, 1Н), 3,93 (s, 3Н), 3,76-3,75 (m, 1H), 3,62-3,59 (m, 1H), 2,55-2,53 (m, 1Н), 1,97-1,94 (m, 1H), 1,96-1,92 (m, 1Н), 1,47 (s, 4Н), 1,24 (s, 5Н).
Стадия 9
Трет-бутил-(2R,4S)-4-фтор-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-карбоксилат 1-11
Трет-бутил-(2R,4R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат 1-10 (2,27 г, 7,30 ммоль) растворяли в дихлорметане (45 мл), добавляли трифторид диэтиламиносеры (2,94 г, 18,25 ммоль) при -78°С. Реакционную смесь медленно нагревали до комнатной температуры, а затем взбалтывали в течение 16 часов. Добавляли насыщенный водный раствор бикарбоната натрия (15 мл) для прекращения реакции и смесь экстрагировали дихлорметаном (50 мл × 3). Объединенную органическую фазу промывали водой (100 мл × 2) и солевым раствором (100 мл × 2). Органический слой сушили безводным сульфатом натрия, фильтровали, фильтрат концентрировали. Остаток очищали с помощью колонки с силикагелем (петролейный эфир : этилацетат = 1:0~15:1) с получением трет-бутил-(2R,4S)-4-фтор-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-карбоксилата 1-11 (1,40 г, бесцветное масло), выход: 51,8%.
MS масса/заряд (ESI): 337 [М+23].
1Н ЯМР (400 МГц, CDCl3) δ 7,89 (s, 0,6Н), 7,87 (s, 0,4Н), 7,26-7,21 (m, 1H), 5,28-5,03 (m, 2Н), 4,13-4,08 (m, 1H), 3,95 (s, 3Н), 3,71-3,59 (m, 1H), 2,78-2,73 (m, 1H), 1,98-1,81 (m, 1H), 1,46 (s, 3Н), 1,21 (s, 6Н).
Стадия 10
5-Фтор-3-((2R,4S)-4-фторпирролидин-2-ил)-2-метоксипиридина гидрохлорид 1-12
Трет-бутил-(2R,4S)-4-фтор-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-карбоксилат 1-11 (1,40 г, 4,46 ммоль) растворяли в растворе хлороводорода в метаноле (4 М, 20 мл) и реакционную смесь взбалтывали при комнатной температуре на протяжении ночи. Растворитель удаляли при пониженном давлении с получением целевого соединения 5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)-2-метоксипиридина гидрохлорида 1-12 (1,12 г, 4,46 ммоль, желтое твердое вещество), выход: 100%.
MS масса/заряд (ESI):215 [М+1].
Стадия 11
5-Фтор-3-((2R,4S)-4-фторпирролидин-2-ил)пиридин-2-ола гидрохлорид INT1
5-Фтор-3-((2R,4S)-4-фторпирролидин-2-ил)-2-метоксипиридина гидрохлорид 1-12 (1,12 г, 4,46 ммоль) растворяли в ацетонитриле (40 мл), медленно добавляли йодтриметилсилан (1,8 г, 9,92 ммоль). Реакционную смесь взбалтывали при 50°С в течение 1 часа. Растворитель удаляли при пониженном давлении и добавляли воду для прекращения реакции. Водную фазу промывали этилацетатом (30 мл × 2) для удаления примеси и полученную в результате водную фазу концентрировали с получением 5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)пиридин-2-ола гидрохлорида INT1 (0,95 г, 4,02 ммоль, красновато-коричневое масло), выход: 90,1%.
MS масса/заряд (ESI): 201 [М+1].
1Н ЯМР (400 МГц, CD3OD) δ 7,83 (s, 1H), 7,62 (s, 1H), 4,94-4,86 (m, 1H), 3,82-3,86 (m, z, 1H), 3,65-3,70 (m, 2Н), 2,63-2,47 (m, 2Н).
[0082] Пример 1
(22R,24S,5S)-24,35-Дифтор-5-метил-4-окса-7,9-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклононан-8-он


Стадия 1
Трет-бутил-(R)-(2-гидроксипропил)карбамат
(R)-1-Аминопропан-2-ол 1с-1 (1,11 г, 14,80 ммоль) растворяли в тетрагидрофуране (80 мл) медленно добавляли и ди-трет-бутилбикарбонат (3,55 г, 16,30 ммоль) в упомянутый выше раствор. Реакционную смесь взбалтывали при комнатной температуре в течение 30 минут после добавления. После завершения реакции смесь концентрировали с непосредственным получением трет-бутил-(R)-(2-гидроксипропил)карбамата 1с-2 (2,60 г, бесцветная жидкость) в виде неочищенного продукта
1Н ЯМР (400 МГц, CDC13) δ 3,28-3,25 (m, 1H), 3,04-2,97 (m, 1H), 2,29-2,27 (m, 1H), 1,45 (s, 9Н), 1,18 (d,J=6,4 Гц, 3Н).
Стадия 2
(R)-1-((Трет-бутоксикарбонил)амино)пропан-2-ил-4-метилбензолсульфонат
Трет-бутил-(R)-(2-гидроксипропил)карбамат 1с-2 (3,90 г, 22,00 ммоль) растворяли в дихлорметане (40 мл), добавляли триэтиламин (3,50 г, 34,50 ммоль), 4-метилбензолсульфонилхлорид (4,18 г, 22,00 ммоль) и 4-(диметиламино)пиридин (0,39 г, 3,20 ммоль) в реакционную смесь, затем реакционную смесь взбалтывали при 30°С в течение 18 часов. Реакционную смесь промывали соляным раствором (30 мл × 3), сушили безводным сульфатом натрия и концентрировали с получением (R)-1-((трет-бутоксикарбонил)амино)пропан-2-ил-метилбензолсульфоната 1с (7,00 г, желтое масло, желтое твердое вещество после охлаждения) в виде неочищенного продукта.
MS масса/заряд (ESI): 352 [М+23].
Стадия 3
5-Фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ол 1b
5-Фтор-3-((2R,4S)-4-фторпирролидин-2-ил)пиридин-2-ола гидрохлорид INT1 (0,50 г, 2,00 ммоль) и 5-хлор-3-нитропиразоло[1,5-а]пиримидин 1а (0,40 г, 2,00 ммоль) растворяли в бутан-1-оле (25 мл) и добавляли N,N-диизопропилэтанамин (0,78 г, 6,00 ммоль). Реакционную смесь взбалтывали при 40°С в течение 3 часов, охлаждали до комнатной температуры, фильтровали. Твердое вещество сушили с получением 5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ола 1b (0,66 г, 1,82 ммоль, желтое твердое вещество), выход: 91%.
MS масса/заряд (ESI): 363 [М+1].
Стадия 4
Трет-бутил-((S)-2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)изопропил)карбамат 1d
5-Фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ол 1b (0,25 г, 0,69 ммоль) и (R)-1-((трет-бутоксикарбонил)амино)пропан-2-ил-4-метилбензолсульфонат (0,68 г, 2,10 ммоль) растворяли в ацетонитриле (15,0 мл), добавляли карбонат цезия (0,68 г, 2,10 ммоль). Реакционную смесь взбалтывали при 80°С в течение 3 часов и охлаждали до комнатной температуры. Растворитель удаляли при пониженном давлении и остаток очищали с помощью препаративной TLC (этилацетат : петролейный эфир = 1:1) с получением целевого соединения трет-бутил-((S)-2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)пропил)карбамата 1d (0,10 г, 0,19 ммоль, желтое твердое вещество), выход: 28%.
MS масса/заряд (ESI): 542 [М+23].
1H ЯМР (400 МГц, CDCl3) δ 8,50 (s, 0,5Н), 8,41 (s, 0,5Н), 8,29 (d, J=8,0 Гц, 0,5Н), 8,21 (d, J=8,0 Гц, 0,5Н), 7,92 (s, 0,5Н), 7,84 (s, 0,5Н), 7,77-7,74 (m, 0,5Н), 7,12-7,11 (m, 0,5Н), 6,36 (d, J=8,0 Гц, 0,5Н), 6,20 (d, J=8,0 Гц, 0,5Н), 5,40-5,55 (m, 1H), 5,42-5,27 (m, 1Н), 4,79-4,76 (m, 1H), 4,21-4,11 (m, 1H), 4,04-3,95 (m, 1H), 3,94-3,90 (m, 1H), 3,51-3,48 (m, 1H), 2,70-2,52 (m, 1H), 2,26-2,06 (m, 1H), 1,44 (d, J=6,0 Гц, 3Н).
Стадия 5
Трет-бутил-((S)-2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)изопропил)карбамат le
Терт-бутил-((S)-2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)изопропил)карбамат 1d (0,10 г, 0,19 ммоль) растворяли в метаноле/дихлорметане (5,0 мл/5,0 мл), добавляли насыщенный раствор хлорида аммония (5,0 мл) и порошок цинка (0,18 г, 2,80 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 1 часа, экстрагировали дихлорметаном (10 мл × 3) и органические слой сушили безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением трет-бутил-((S)-2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)изопропил)карбамата 1е (93 мг, 0,19 ммоль, желтое масло), выход: 99%.
MS масса/заряд (ESI): 490 [М+1].
Стадия 6
5-((2R,4S)-2-(2-(((S)-1-Аминопропан-2-ил)окси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амин 1f
Трет-бутил-((S)-2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)изопропил)карбамат 1е (93 мг, 0,19 ммоль) растворяли в дихлорметане (5,0 мл), добавляли 2,2,2-трифторуксусную кислоту (2,0 мл) при комнатной температуре. Реакционную смесь взбалтывали в течение 1 часа при комнатной температуре. Растворитель удаляли при пониженном давлении и смесь разбавляли дихлорметаном (5,0 мл). Триэтиламин добавляли для нейтрализации реакционной системы. Растворитель удаляли при пониженном давлении с получением целевого соединения 5-((2R,4S)-2-(2-(((S)-1-аминопропан-2-ил)окси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амина 1f (73 мг, 0,19 ммоль, желтое масло) в виде неочищенного продукта.
MS масса/заряд (ESI): 390 [М+1].
Стадия 7
(22R,24S,5S)-24,35-Дифтор-5-метил-4-окса-7,9-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклононан-8-он
5-((2R,4S)-2-(2-(((S)-1-Аминопропан-2-ил)окси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амин 1f (73 мг, 0,19 ммоль) растворяли в N,N-диметилформамиде (3,0 мл) и добавляли N,N-карбонилдиимидазол (62 мг, 0,38 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении и остаток очищали с помощью препаративной TLC (дихлорметан : метанол = 20:1) с получением целевого соединения (22R,24S,5S)-24,35-дифтор-5-метил-4-окса-7,9-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклононан-8-она 1 (11 мг, 0,03 ммоль, желтое твердое вещество), выход: 14%.
MS масса/заряд (ESI): 41 6 [М+1].
1Н ЯМР (400 МГц, CDC13) δ 8,25 (d, J=8,0 Гц, 1Н), 7,88 (s, 1H), 7,64-7,62 (m, 1H), 7,59 (s, 1H), 7,14-7,11 (m, 1H), 6,35 (s, 1H), 6,20 (d, J=8,0 Гц, 1H), 5,68-5,61 (m, 1H), 5,60-5,52 (m, 1H), 4,23-4,14 (m, 1H), 4,04-3,98 (m, 1H), 3,88-3,83 (m, 1H), 3,03-2,84 (m, 2H), 2,26-2,06 (m, 2H), 1,44 (d, J=6,4 Гц, 3Н).
[0083] Пример 2
(22R,24S)-24,35-Дифтор-4-окса-7,9-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклононан-8-он
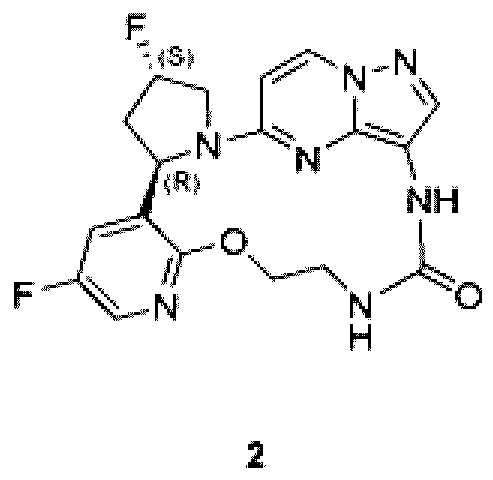

Стадия 1
Терт-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)карбамат 2b
5-Фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ол 1b (0,25 г, 0,70 ммоль) и карбонат цезия (0,65 г, 2,00 ммоль) добавляли в ацетонитрил (10 мл). Реакционную смесь взбалтывали при 50°С в масляной бане в течение 10 минут, затем добавляли терт-бутил-(2-бромэтил)карбамат 2а (0,22 г, 1,00 ммоль). Смесь нагревали до 70°С и взбалтывали на протяжении ночи. Реакционную смесь охлаждали, разбавляли водой (30 мл) и экстрагировали дихлорметаном (50 мл × 3). Объединенную органическую фазу сушили безводным сульфатом натрия, фильтровали для удаления осушающего средства и фильтрат выпаривали при пониженном давлении. Очистка остатка с помощью колонки с силикагелем (петролейный эфир : этилацетат = 19:1~3:2) давали целевое соединение трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)карбамат 2b (0,11 г, 0,20 ммоль, желтое твердое вещество), выход: 30%.
MS масса/заряд (ESI): 506 [М+1].
1H ЯМР (400 МГц, CDCl3) δ 8,51 (s, 0,5Н), 8,42 (s, 0,5Н), 8,28-8,27 (m, 1H), 7,94 (s, 0,5Н), 7,84 (s, 0,5Н), 7,75-7,74 (m, 0,5Н), 7,14-7,12 (m, 0,5Н), 6,39-6,38 (m, 0,5Н), 6,10-6,08 (m, 0,5Н), 5,57 (s, 1H), 5,52-5,51 (m, 0,5Н), 5,31-5,29 (m, 0,5Н), 4,92-4,76 (m, 1,5Н), 4,53-4,34 (m, 2,5Н), 4,15-3,95 (m, 2Н), 3,15-3,12 (m, 0,5Н), 2,75-2,75 (m, 0,5Н), 2,68-2,55 (m, 1H), 1,55 (s, 9Н).
Стадия 2
Трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)карбамат 2с
Смешивали трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)карбамат 2b (40 мг, 0,08 ммоль), дихлорметан (2,0 мл), метанол (2,0 мл), насыщенный водный хлорид аммония (4,0 мл) и порошок цинка (65 мг, 1,00 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 30 минут и экстрагировали дихлорметаном (30 мл × 2). Органическую фазу промывали водой (30 мл × 3), сушили безводным сульфатом натрия и выпаривали с получением целевого соединения трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)карбамата 2с (40 мг, 0,08 ммоль, желтое твердое вещество) в виде неочищенного продукта.
MS масса/заряд (ESI): 476 [М+1].
Стадия 3
5-((2R,4S)-2-(2-(2-Аминоэтокси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амин-2,2,2-трифторацетат 2d
Трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)карбамат 2с (40 мг, 0,08 ммоль) растворяли в дихлорметане (1,0 мл) и добавляли трифторуксусную кислоту (1,0 мл). Реакционную смесь взбалтывали при комнатной температуре в течение 1 часа и выпаривали при пониженном давлении с получением целевого соединения 5-((2R,4S)-2-(2-(2-аминоэтокси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амин-2,2,2-трифторацетата 2d (30 мг, 0,08 ммоль, желтое твердое вещество) в виде неочищенного продукта.
MS масса/заряд (ESI): 376 [М+1].
Стадия 4
(22R,24S)-24,35-Дифтор-4-окса-7,9-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклононан-8-он
5-((2R,4S)-2-(2-(2-Аминоэтокси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амин-2,2,2-трифторацетат 2d (30 мг, 0,08 ммоль растворяли в N,N-диметилформамиде (1 мл), добавляли триэтиламин (0,2 мл) и N,N'-карбонилдиимидазол (10 мг, 0,06 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 1 часа, экстрагировали этилацетатом (50 мл × 3) и промывали солевым раствором (5 мл × 3). Органическую фазу концентрировали при пониженном давлении и очищали с помощью препаративной TLC (дихлорметан : метанол = 10:1) с получением целевого соединения (22R,24S)-24,35-дифтор-4-окса-7,9-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклононан-8-она 2 (8,0 мг, желтое твердое вещество), выход: 25%.
MS масса/заряд (ESI): 402 [М+1].
1Н ЯМР (400 МГц, CDCl3) δ 8,24 (d, J=7,6 Гц, 1H), 7,92-7,88 (m, 1H), 7,58 (s, 1H), 7,41-7,39 (m, 1H), 7,15-7,13 (m, 1H), 6,21 (d, J=7,6 Гц, 1H), 6,20-6,16 (m, 1H), 5,67-5,42 (m, 2Н), 4,18-3,88 (m, 5Н), 3,29-3,28 (m, 1H), 2,89-2,86 (m, 1H), 2,06-2,03 (m, 1H).
[0084] Пример 3
(22R,24S)-24,35-Дифтор-7-метил-4-окса-7,9-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклононан-8-он


Стадия 1
Трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)(метил)карбамат 3b
Трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)карбамат 2b (20 мг, 0,14 ммоль) растворяли в N,N-диметилформамиде (2 мл) и добавляли гидрид натрия (12 мг, 0,3 ммоль, 60%, диспергированный в минеральном масле). Реакционную смесь взбалтывали при комнатной температуре в течение 10 минут. Добавляли метилйодид 3а (42 мг, 0,3 ммоль) и смесь взбалтывали при комнатной температуре в течение 30 минут, экстрагировали этилацетатом (10 мл × 3), промывали водой (10 мл × 3) и органическую фазу сушили безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)(метил)карбамата 3b (61 мг, 0,12 ммоль, желтое твердое вещество). Продукт использовали непосредственно на следующей стадии реакции без дополнительной очистки.
MS масса/заряд (ESI): 520 [М+1].
Стадия 2
Трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)(метил)карбамат 3с
Смешивали трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)(метил)карбамат 3b (60 мг, 0,12 ммоль), дихлорметан (2,0 мл), метанол (2,0 мл), насыщенный водный раствор хлорида аммония (4,0 мл) и порошок цинка (0,13 г, 2,00 ммоль) и взбалтывали при комнатной температуре в течение 30 минут. Реакционную смесь экстрагировали дихлорметаном (30 мл × 2), и органическую фазу промывали водой (30 мл × 3), сушили безводным сульфатом натрия и концентрировали при пониженном давлении с получением целевого соединения терт-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)(метил)карбамата Зс (60 мг, 0,12 ммоль, желтое твердое вещество) в виде неочищенного продукта.
[0085] MS масса/заряд (ESI): 490 [М+1].
1Н ЯМР (400 МГц, CDC13) δ 8,03-8,02 (m, 1Н), 7,82-7,78 (m, 1H), 7,56 (s, 1H), 7,11-7,08 (m, 1H), 5,72-5,67 (m, 1H), 5,31-5,22 (m, 2Н), 4,15-3,48 (m, 6Н), 2,88 (s, 3Н), 2,82-2,78 (m, 1H), 2,18-2,13 (m, 1H), 1,25 (s, 9Н).
Стадия 3
5-((2R,4S)-4-Фтор-2-(5-фтор-2-(2-(метиламино)этокси)пиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амин 2,2,2-трифторацетат 3d
Трет-бутил-(2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)этил)(метил)карбамат 3с (60 мг, 0,12 ммоль) растворяли в дихлорметане (1,0 мл) и добавляли трифторуксусную кислоту (1,0 мл). Смесь взбалтывали при комнатной температуре в течение 1 часа и концентрировали при пониженном давлении с получением целевого соединения 5-((2R,4S)-4-фтор-2-(5-фтор-2-(2-(метиламино)этокси)пиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амин-2,2,2-трифторацетата 3d (40 мг, 0,10 ммоль, желтое твердое вещество) в виде неочищенного продукта.
MS масса/заряд (ESI): 390 [М+1].
Стадия 4
(22R,24S)-24,35-Дифтор-7-метил-4-окса-7,9-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклононан-8-он 3
5-((2R,4S)-4-Фтор-2-(5-фтор-2-(2-(метиламино)этокси)пиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амин-2,2,2-трифторацетат 3d (40 мг, 0,10 ммоль) растворяли в N,N-диметилформамиде (1 мл), добавляли триэтиламин (0,2 мл) и N,N-карбониддиимидазол (16 мг, 0,10 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 1 часа, экстрагировали этилацетатом (50 мл × 3) и промывали насыщенным раствором хлорида натрия (50 мл × 3). Органическую фазу концентрировали при пониженном давлении. Остаток очищали с помощью препаративной TLC (дихлорметан : метанол = 10:1) с получением целевого соединения (22R,24S)-24,35-дифтор-7-метил-4-окса-7,9-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклононан-8-она 3 (6,7 мг, белое твердое вещество), выход: 21%.
MS масса/заряд (ESI): 416 [M+1].
1H ЯМР (400 МГц, CDC13) δ 8,22 (d, J=7,6 Гц, 1H), 7,82-7,80 (m, 2Н), 7,16-7,14 (m, 1H), 6,14 (d, J=7,6 Гц, 1H), 5,74-5,52 (m, 3Н), 4,15-3,48 (m, 6Н), 3,08 (s, 3Н), 2,82-2,78 (m, 1H), 2,27-2,23 (m, 1H).
[0086] Пример 4
(22R,5S)-35-Фтор-5-метил-4-окса-8,10-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклодекан-9-он 4


Стадия 1
(R)-3-Гидроксибутил 4-метилбензолсульфонаг 4g-2
(3R)-Бутан-1,3-диол 4g-1 (2,00 г, 22,22 ммоль) растворяли в дихлорметане (20 мл) и добавляли триэтиламин (3,37 г, 33,33 ммоль). Затем медленно добавляли 4-метилбензолсульфонилхлорид (4,46 г, 23,33 ммоль) при -20°С и реакционную смесь взбалтывали при комнатной температуре в течение 16 часов после добавления. Реакционную смесь разбавляли водой (50 мл) и экстрагировали дихлорметаном (30 мл × 3). Органическую фазу промывали соляным раствором (30 мл × 3), сушили безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии (петролейный эфир : этилацетат = 100:0~1:1) с получением (R)-3-гидроксибутил-4-метилбензолсульфоната 4g-2 (3,80 г, желтая жидкость), выход: 70%.
1H ЯМР (400 МГц, CDCl3) δ 7,87-7,76 (m, 2Н), 7,44-7,30 (m, 2Н), 4,33-4,16 (m, 1H), 4,15-4,07 (m, 1Н), 4,03-3,88 (m, 1H), 2,45 (s, 3Н), 1,90-1,78 (m, 1H), 1,76-1,63 (m, 1H), 1,22 (d, J=5,2 Гц, 3Н).
Стадия 2
(R)-4-Аминобутан-2-ол 4g-3
(R)-3-Гидроксибутил-4-метилбензолсульфонат 4g-2 (3,50 г, 14,34 ммоль) растворяли в водном аммиаке (25%, 50 мл) и полученную в результате смесь взбалтывали при 100°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали с получением (R)-4-аминобутан-2-ола 4g-3 (1,28 г, 14,34 ммоль) в виде неочищенного продукта.
MS масса/заряд (ESI): 90 [М+1].
Стадия 3
Трет-бутил-(R)-(3-гидроксибутил)карбамат 4g-4
(R)-4-Аминобутан-2-ол 4g-3 (1,28 г, 14,34 ммоль) растворяли в 30 мл тетрагидрофурана, добавляли триэтиламин (2,20 г, 21,51 ммоль), затем медленно добавляли ди-трет-бутилдикарбонат (3,30 г, 15,06 ммоль). Реакционную смесь взбалтывали в течение 1 часа при комнатной температуре после добавления. После завершения реакции реакционную смесь концентрировали непосредственно с получением трет-бутил-(R)-(3-гидроксибутил)карбамата 4g-4 (2,71 г, бесцветная жидкость) в виде неочищенного продукта.
1Н ЯМР (400 МГц, CDC13) δ 4,95-4,82 (m, 1H), 3,93-3,76 (m, 1H), 3,55-3,34 (m, 1H), 3,21-2,97 (m, 2H), 1,68-1,48 (m, 2H), 1,44 (s, 9H), 1,23 (d, J=5,2 Гц, 3Н).
Стадия 4
(R)-4-((Трет-бутоксикарбонил)амино)бутан-2-ил-4-метилбензолсульфонат 4g
Трет-бутил-(R)-(3-гидроксибутил)карбамат 4g-4 (0,38 г, 2,01 ммоль) растворяли в дихлорметане (5 мл), добавляли триэтиламин (0,30 г, 3,01 ммоль), 4-метилбензолсульфонилхлорид (0,36 г, 1,91 ммоль) и N,N-диметил-4-аминопиридин (25 мг, 0,20 ммоль). Реакционную смесь взбалтывали при 30°С в течение 18 часов после добавления, затем разбавляли дихлорметаном (50 мл), промывали солевым раствором (30 мл × 3) и сушили безводным сульфатом натрия. Остаток очищали с помощью колоночной хроматографии (петролейный эфир : этил ацетат = 100:0~65:35) с получением (R)-4-((трет-бутоксикарбонил)амино)бутан-2-ил-4-метилбензолсульфоната 4g (0,10 г, желтое масло), выход: 15%.
MS масса/заряд (ESI): 366 [М+Na].
Стадия 5
(R)-N-((5-Фтор-2-метоксипиридин-3-ил)метилен)-2-метилпропан-2-сульфинамид 4b 5-Фтор-2-метоксипиридин-3-карбальдегид 4а (10,00 г, 64,5 ммоль) растворяли в дихлорметане (120 мл), добавляли карбонат цезия (42,00 г, 129,00 ммоль) и (R)-2-метилпропан-2-сульфинамид (8,26 г, 67,70 ммоль). Реакционную смесь взбалтывали при 30°С в течение 4 часов после добавления и фильтровали после завершения реакции. Фильтрат концентрировали непосредственно с получением (R)-N-((5-фтор-2-метоксипиридин-3-ил)метилен)-2-метилпропан-2-сульфинамида 4b (17,50 г, желтое масло), его использовали непосредственно для следующей стадии без дополнительной очистки.
MS масса/заряд (ESI): 259 [М+1].
1H ЯМР (400 МГц, CDC13) δ 8,88 (s, 1H), 8,16 (s, 1H), 7,98 (d, J=8,0 Гц, 1H), 4,01 (s, 3Н), 1,27 (s, 9H).
Стадия 6
(R)-N-((R)-3-(1,3-Диоксан-2-ил)-1-(5-фтор-2-метоксипиридин-3-ил)пропил)-2-метилпропан-2-сульфинамид 4с
Кусочки магния (3,30 г, 136,00 ммоль) добавляли в сухой тетрагидрофуран (200 мл) и добавляли диизобутилалюминия гидрид (0,3 мл, 1 М раствор тетрагидрофурана). Смесь взбалтывали при 50°С в течение 15 минут, затем в смесь каплями добавляли раствор 2-(2-бромэтил)-1,3-диоксана (26,50 г, 60 ммоль) в тетрагидрофуране (50 мл) и полученную в результате смесь взбалтывали при 50°С в течение 1 часа. Далее реакционную смесь медленно охлаждали до -40°С, каплями добавляли раствор (R)-N-((5-фтор-2-метоксипиридин-3-ил)метилен)-2-метилпропан-2-сульфинамида 4b (17,50 г, 68,00 ммоль) в тетрагидрофуране (50 мл) и взбалтывали при -40°С в течение 1 часа, медленно нагревали до комнатной температуры, взбалтывали еще 1 час, и останавливали добавлением водного раствора лимонной кислоты (10%), затем смесь экстрагировали метил-трет-бутиловым эфиром (400 мл). Органическую фазу промывали насыщенным водным раствором бикарбоната натрия и водой, сушили, фильтровали и фильтрат концентрировали с получением (R)-N-((R)-3-(1,3-диоксан-2-ил)-1-(5-фтор-2-метоксипиридин-3-ил)пропил)-2-метилпропан-2-сульфинамида 4с (20,0 г, белое твердое вещество), выход: 79%.
MS масса/заряд (ESI): 375 [М+1].
1H ЯМР (400 МГц, CDCl3) δ 7,89 (s, 1H), 7,30 (d, J=8,0 Гц, 1H), 4,52-4,50 (m, 1H), 4,35-4,33 (m, 1H), 4,18-4,16 (m, 1H), 4,10-4,08 (m, 2H), 3,95 (s, 3H), 3,77-3,70 (m, 2H), 2,07-2,04 (m, 2H), 1,90-1,85 (m, 1H), 1,73-1,69 (m, 1H), 1,55-1,52 (m, 1H), 1,35-1,32 (m, 1H), 1,21 (s, 9H).
Стадия 7
(R)-5-Фтор-2-метокси-3-(пирролидин-2-ил)пиридин 4d
(R)-N-((R)-3-(1,3-Диоксан-2-ил)-1-(5-фтор-2-метоксипиридин-3-ил)пропил)-2-метилпропан-2-сульфинамид 4c (20,00 г, 53,48 ммоль) растворяли в трифторуксусной кислоте (100 мл) и воде (10 мл). Реакционную смесь взбалтывали при 20°С в течение 1 часа. Затем добавляли триэтилсилан (80 мл) и взбалтывание продолжали при 20°С в течение 16 часов. Растворитель выпаривали и остаток растворяли в воде (300 мл), экстрагировали метил-терт-бутиловым эфиром (300 мл). Значение рН водной фазы доводили до приблизительно 13 с помощью 40% водного раствора гидроксида натрия и смесь экстрагировали дихлорметаном (200 мл × 3). Органическую фазу промывали соляным раствором (100 мл), сушили безводным сульфатом натрия, фильтровали и фильтрат выпаривали с получением (R)-5-фтор-2-метокси-3-(пирролидин-2-ил)пиридина 4d (9,00 г, ярко-желтое масло), выход: 86%.
MS масса/заряд (ESI): 197 [М+1].
1H ЯМР (400 МГц, CDCl3) δ 7,83 (s, 1H), 7,57 (d, J=8,8 Гц, 1H), 4,31-4,29 (m, 1H), 3,93 (s, 3Н), 3,15-3,11 (m, 1H), 3,07-3,00 (m, 1H), 2,28-2,25 (m, 1H), 1,85-1,80 (m, 2H), 1,57-1,55(m, 1H).
Стадия 8
(R)-5-Фтор-3-(пирролидин-2-ил)пиридин-2-ол 4e
(R)-5-Фтор-2-метокси-3-(пирролидин-2-ил)пиридин 4d (1,20 г, 11,0 ммоль) растворяли в ацетонитриле (20 мл), добавляли йодид калия (3,70 г, 44,0 ммоль), затем каплями добавляли триметилхлорсилан (2,30 г, 22,0 ммоль). Реакционную смесь взбалтывали при 50°С в течение 24 часов, затем фильтровали и фильтрат концентрировали с получением твердого вещества, которое промывали дихлорметаном : метанолом = 5:1, фильтровали и фильтрат концентрировали с получением (R)-5-фтор-3-(пирролидин-2-ил)пиридин-2-ола 4е (1,6 г, неочищенный продукт, желтое твердое вещество).
MS масса/заряд (ESI): 183 [М+1].
1H ЯМР (400 МГц, DMSO-d6) δ 9,23 (s, 1H), 8,61 (s, 1H), 7,83-7,81 (m, 1H), 7,76 (d, J=2,0 Гц, 1H), 4,48-4,44 (m, 1H), 3,25-3,17 (m, 2H), 2,22-2,18 (m, 1H), 2,09-2,07 (m, 2H), 1,93-1,91 (m, 1H).
Стадия 9
(R)-5-Фтор-3-(1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ол 4f
С использованием процедуры, подобной описанной для первой стадии примера 1, используя (R)-5-фтор-3-(пирролидин-2-ил)пиридин-2-ол 4е вместо 5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)пиридин-2-ола гидрохлорида INT1, получали целевой продукт (R)-5-фтор-3-(1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ол 4f.
MS масса/заряд (ESI): 345 [М+1].
1H ЯМР (400 МГц, DMSO-d6) δ 8,82 (d, J=8,0 Гц, 0,44Н), 8,64 (d, J=8,0 Гц, 0,56Н), 8,63 (s, 0,56Н), 8,56 (s, 0,44Н), 7,56-7,55 (m, 0,56Н), 7,45-7,44 (m, 0,44Н), 7,29-7,27 (m, 1H), 6,75 (d, J=8,0 Гц, 0,44H), 6,14 (d, J=8,0 Гц, 0,56H), 5,40 (d, J=8,0 Гц, 0,44H), 5,07 (d, J=8,0 Гц, 0,56H), 4,05-3,95 (m, 2H), 3,81-3,75 (m, 1H), 3,62-3,55 (m, 1H), 2,41-2,35 (m, 1H), 2,26-2,15 (m, 1H), 1,95-1,90 (m, 1H).
Стадия 10
Трет-бутил-((S)-3-((5-фтор-3-((R)-1-(3-нитро-6,7-дигидропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)бутил)карбамат 4h
С использованием процедуры, подобной описанной для второй стадии примера 1, используя (R)-5-фтор-3-(1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ол 4f вместо 5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ола 1b, получали целевой продукт трет-бутил-((S)-3-((5-фтор-3-((R)-1-(3-нитро-6,7-дигидропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)бутил)карбамат 4h.
MS масса/заряд (ESI): 516 [М+1].
Стадия 11
Трет-бутил-((S)-3-((5-фтор-3-((R)-1-(3-амино-6,7-дигидропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)бутил)карбамат
С использованием процедуры, подобной описанной для третьей стадии примера 1, используя трет-бутил-((S)-3-((5-фтор-3-((R)-1-(3-нитро-6,7-дигидропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)бутил)карбамат 4h вместо трет-бутил-((S)-2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-нитропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)пропил)карбамата 1с, получали целевой продукт трет-бутил-((S)-3-((5-фтор-3-((R)-1-(3-амино-6,7-дигидропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)бутил)карбамат 4i.
MS масса/заряд (ESI): 488 [М+1].
Стадия 12
5-((R)-2-(2-(((S)-4-Аминобутан-2-ил)окси)-5-фторпиридин-3-ил)пирролидин-1-ил)-6,7-дигидропиразоло[1,5-а]пиримидин-3-аминтрифторацетат 4j
С использованием процедуры, подобной описанной для четвертой стадии примера 1, используя трет-бутил-((S)-3-((5-фтор-3-((R)-1-(3-амино-6,7-дигидропиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)бутил)карбамат 4i вместо трет-бутил-((S)-2-((5-фтор-3-((2R,4S)-4-фтор-1-(3-аминопиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)пиридин-2-ил)окси)изопропил)карбамата 1d, получали целевой продукт 5-((R)-2-(2-(((S)-4-аминобутан-2-ил)окси)-5-фторпиридин-3-ил)пирролидин-1-ил)-6,7-дигидропиразоло[1,5-а]пиримидин-3-аминтрифторацетат 4j.
MS масса/заряд (ESI): 386 [М+1].
Стадия 13
(22R,5S)-35-Фтор-5-метил-4-окса-8,10-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклодекан-9-он 4
С использованием процедуры, подобной описанной для пятой стадии примера 1, используя 5-((R)-2-(2-(((S)-4-аминобутан-2-ил)окси)-5-фторпиридин-3-ил)пирролидин-1-ил)-6,7-дигидропиразоло[1,5-а]пиримидин-3-аминтрифторацетат 4j вместо 5-((2R,4S)-2-(2-(((S)-1-аминопропан-2-ил)окси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-амина 1е, получали целевой продукт (22R,5S)-35-фтор-5-метил-4-окса-8,10-диаза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинциклодекан-9-он 4 (11,8 мг, 0,031 ммоль, бледно-желтое твердое вещество); выход: 31%.
MS масса/заряд (ESI): 412 [М+1].
1Н ЯМР (400 МГц, CDC13) δ 8,18 (d, J=7,8 Гц, 1H), 7,93-7,82 (m, 1H), 7,53 (s, 1H), 7,04-6,95 (m, 1H), 6,21 (d, J=7,8 Гц, 1H), 5,83 (s, 1H), 5,41-5,23 (m, 2Н), 4,00-3,84 (m, 1H), 3,76-3,60 (m, 1Н), 3,57-3,45 (m, 1H), 3,29-3,16 (m, 1H), 2,56-2,35 (m, 2H), 2,32-2,13 (m, 2H), 2,09-1,97 (m, 1H), 1,96-1,82 (m, 2H), 1,50 (d, J=5,2 Гц, 3Н).
[0087] Биологические эксперименты
Тест ингибирования активности TRKA
Эффект соединений в соответствии с настоящим изобретением в отношении активности TRKA оценивали с помощью анализов киназы in vitro. Далее кратко описываются экспериментальные способы.
In vitro активность TRKA измеряли путем применения набора для анализа киназы с использованием гомогенной флуоресценции с временным разрешением (HTRF) (Cisbio, №62ТК0РЕС) посредством выявления уровня фосфорилирования субстрата в киназной реакции. Реакционный буфер содержал следующие компоненты: реакционный буфер фермента в наборе (1×), 5 мМ MgCl2, 1 мМ DTT; гуманизированный ре комбинированный белок TRKA экспрессировали и очищали на платформе для очистки и идентификации Университета Цинхуа и разбавляли в растворе киназы (3 нг/мкл) реакционным буфером. Реакционный раствор субстрата содержал 0,23 мкМ меченного биотином субстрата тирозинкиназы и 8,4 мкМ АТФ, который разбавляли реакционным буфером. Буфер для выявления содержал 0,1 нг/мкл антитела против CAGE, меченного Eu3+, и 14,375 нМ XL665, меченного стрептавидином, которые разбавляли реакционным буфером.
[0088] Соединение растворяли в 100% DMSO и разбавляли до 100 или 10 мкМ, затем разбавляли с помощью DMSO в 4-кратном серийном разведении до минимальной концентрации 6,1 или 0,61 нМ и каждую точку концентрации дополнительно разбавляли в 40 раз реакционным буфером.
[0089] В 384-луночный планшет (Corning, №4512) добавляли 4 мкл раствора тестируемого соединения и 2 мкл раствора TRKA киназы, соответственно, затем тщательно перемешивали и инкубировали при комнатной температуре в течение 15 минут. Затем добавляли 4 мкл раствора субстрата и реакционную смесь инкубировали при комнатной температуре в течение 60 минут. Затем добавляли 10 мкл буфера для выявления того же объема, что и реакционная смесь, смесь хорошо перемешивали и оставляли на 30 минут при комнатной температуре. Процесс реакции контролировали на длинах волн 620 нм и 665 нм с помощью считывающего устройства Envision (Perkin Elmer). Отношение 665/620 положительно коррелировало с уровнем фосфорилирования субстрата, затем определяли активность TRKA киназы. В этом эксперименте группу без TRKA киназы использовали в качестве отрицательного контроля, группу с TRKA киназой, но без тестируемого соединения использовали в качестве положительного контроля (0% ингибирования). Процент ингибирования тестируемым соединением активности TRKA можно рассчитать с помощью следующей формулы.
Значение IC50 соединения вычисляли по 8 точкам концентрации путем применения программного обеспечения XLfit (ID Business Solutions Ltd. UK) с помощью следующей формулы:
Y=Низ+(Верх-Низ)/(1+10^((logIC50-X)*фактор наклона)),
в которой Y представляет собой процент ингибирования; X представляет собой логарифм концентрации соединения, подлежащего тестированию; Низ представляет собой максимальный процент ингибирования; Верх представляет собой минимальный процент ингибирования; фактор наклона представляет собой коэффициент наклона кривой.
[0090] Тест ингибирования активности TRKAG595R
Эффект соединений в соответствии с настоящим изобретением в отношении активности TRKA G595R оценивали с помощью анализов киназы in vitro.
Ниже кратко описываются экспериментальные способы.
In vitro активность TRKA G595R измеряли с использованием набора для анализа киназы с использованием HTRF (Cisbio, №62ТК0РЕС) посредством выявления уровня фосфорилирования субстрата в киназной реакции. Реакционный буфер содержал следующие компоненты: реакционный буфер фермента в наборе (1×), 5 мМ MgCl2, 1 мМ DTT. Гуманизированный рекомбинированный белок TRKA G595R (№N16-12BG, приобретенный в компании Signal Chem Life Sciences) разбавляли в растворе киназы 0,25 нг/мкл реакционным буфером. Реакционный раствор субстрата содержал 0,51 мкМ меченного биотином субстрата тирозинкиназы и 2,9 мкМ АТФ, который разбавляли реакционным буфером. Буфер для выявления содержал 0,15 нг/мкл антитела против CAGE, меченного Eu3+, и 31,875 нМ XL665, меченного стрептавидином, которые разбавляли реакционным буфером.
[0091] Соединение растворяли в 100% DMSO и разбавляли до 1 мМ или 100 мкМ, затем разбавляли с помощью DMSO в 4-кратном серийном разведении до минимальной концентрации 61 или 6,1 нМ и каждую точку концентрации дополнительно разбавляли в 40 раз реакционным буфером.
[0092] В 384-луночный планшет (Corning, №4512) добавляли 4 мкл раствора тестируемого соединения и 2 мкл раствора TRKA G595R киназы, тщательно перемешивали и инкубировали при комнатной температуре в течение 15 минут.Затем добавляли 4 мкл раствора субстрата и реакционную смесь инкубировали при комнатной температуре в течение 60 минут. Затем добавляли 10 мкл буфера для выявления того же объема, что и реакционная смесь, смесь хорошо перемешивали и оставляли на 30 минут при комнатной температуре. Процесс реакции контролировали на длинах волн 620 нм и 665 нм с помощью считывающего устройства Envision (Perkin Elmer). Отношение 665/620 положительно коррелировало с уровнем фосфорилирования субстрата, затем определяли активность TRKA G595R киназы. В этом эксперименте группу без TRKA G595R киназы использовали в качестве отрицательного контроля (100% ингибирование), группу с TRKA G595R, но без тестируемого соединения использовали в качестве положительного контроля (0% ингибирования). Процент ингибирования тестируемым соединением активности TRKA G595R можно рассчитать с помощью следующей формулы:
Процент ингибирования = 100-100 * (значение сигнала тестируемого соединения при определенной концентрации - значение сигнала отрицательного контроля)/(значение сигнала положительного контроля - значение сигнала отрицательного контроля).
Значение IC50 тестируемого соединения вычисляли по 8 точкам концентрации путем применения программного обеспечения XLfit (ID Business Solutions Ltd. UK) с помощью следующей формулы:
Y=Низ+(Верх-Низ)/(1+10^((logIC50-X)*фактор наклона)), в которой Y представляет собой процент ингибирования; X представляет собой логарифм концентрации соединения, подлежащего тестированию; Низ представляет собой максимальный процент ингибирования; Верх представляет собой минимальный процент ингибирования; фактор наклона представляет собой коэффициент наклона кривой.
[0093] Тест ингибирования активности TRKA G667C
Эффект соединений в соответствии с настоящим изобретением в отношении активности TRKA G667C оценивали с помощью анализов киназы in vitro.
Ниже кратко описываются экспериментальные способы.
In vitro активность TRKA G667C измеряли с использованием набора для анализа киназы с использованием HTRF (Cisbio, №62ТК0РЕС) посредством выявления уровня фосфорилирования субстрата в киназной реакции. Реакционный буфер содержал следующие компоненты: реакционный буфер фермента в наборе (1×), 5 мМ MgCl2, 1 мМ DTT. Гуманизированный рекомбинированный белок TRKA G667C (№N16-12 CG, Signal Chem Life Sciences) разбавляли в растворе киназы 0,09 нг/мкл реакционным буфером. Реакционный раствор субстрата содержал 0,21 мкМ меченного биотином субстрата тирозинкиназы и 2,7 мкМ АТФ, который разбавляли реакционным буфером. Буфер для выявления содержал 0,1 нг/мкл антитела против CAGE, меченного Eu3+, и 13,125 нМ XL665, меченного стрептавидином, которые разбавляли реакционным буфером.
[0094] Соединение растворяли в 100% DMSO и разбавляли до 200 мкМ, затем разбавляли с помощью DMSO в 4-кратном серийном разведении до минимальной концентрации 12,2 нМ и каждую точку концентрации дополнительно разбавляли в 40 раз реакционным буфером.
[0095] В 384-луночный планшет (Corning, №4512) добавляли 4 мкл раствора тестируемого соединения и 2 мкл раствора TRKA G667C киназы, тщательно перемешивали и инкубировали при комнатной температуре в течение 15 минут. Затем добавляли 4 мкл раствора субстрата и реакционную смесь инкубировали при комнатной температуре в течение 60 минут. Затем добавляли 10 мкл буфера для выявления того же объема, что и реакционная смесь, смесь хорошо перемешивали и оставляли на 30 минут при комнатной температуре. Процесс реакции контролировали на длинах волн 620 нм и 665 нм с помощью считывающего устройства Envision (Perkin Elmer). Отношение 665/620 положительно коррелировало с уровнем фосфорилирования субстрата, затем определяли активность TRKA G667C киназы. В этом эксперименте группу без TRKA G667C киназы использовали в качестве отрицательного контроля (100% ингибирование), группу с TRKA G667C киназой, но без тестируемого соединения использовали в качестве положительного контроля (0% ингибирования). Процент ингибирования тестируемым соединением активности TRKA G667C можно рассчитать с помощью следующей формулы:
Процент ингибирования = 100-100 * (значение сигнала тестируемого соединения при определенной концентрации значение сигнала отрицательного контроля)/(значение сигнала положительного контроля - значение сигнала отрицательного контроля).
Значение IC50 соединения вычисляли по 8 точкам концентрации путем применения программного обеспечения XLfit (ID Business Solutions Ltd. UK) с помощью следующей формулы:
Y=Низ+(Верх-Низ)/(1+10^((logIC50-X)*фактор наклона)),
в которой Y представляет собой процент ингибирования; X представляет собой логарифм концентрации соединения, подлежащего тестированию; Низ представляет собой максимальный процент ингибирования; Верх представляет собой минимальный процент ингибирования; фактор наклона представляет собой коэффициент наклона кривой
[0096] Определение средней эффективной концентрации ингибирования GI50 в клетках KM12
Эффект соединений в соответствии с настоящим изобретением в отношении пролиферации клеток KM12 оценивали с помощью теста жизнеспособности клеток с помощью люминесценции.
[0097] Далее кратко описываются экспериментальные способы.
Использовали набор для выявления CellTilter-Glo (CTG). Уникальную и стабильную люциферазу использовали для выявления АТФ, индикатора метаболизма жизнеспособных клеток. Сигнал люминесценции, генерируемый в эксперименте, был пропорционален количеству жизнеспособных клеток в культуральной среде, таким образом, выявляли пролиферацию клеток KM12.
[0098] Клетку KM12 (приобретенную в компании Shanghai Xinyu Biological Co.) культивировали в полной среде IMDM (Thermo Fisher, 12440053), содержащей 10% FBS (GBICO, 10099-141) и 100 единиц/мл смеси пенициллина и стрептомицина (Thermofisher, 15140122). Когда скорость покрытия клетками в культуральном контейнере достигала 80-90%, использовали 0,25% трипсина (содержащего EDTA) (Thermofisher, 25200056) для отщепления и диспергирования клеток, затем их высевали в белый 384-луночный планшет (164610). В каждую лунку, содержащую 1000 клеток, добавляли 27 мкл полной среды IMDM. Затем 384-луночный планшет культивировали на протяжении ночи в инкубаторе, содержащем 5% СО2, при 37°С. Соединение растворяли в 100% DMSO и разбавляли до 1 мМ, затем разбавляли с помощью DMSO в 4-кратном серийном разведении до самой низкой концентрации 0,061 мкМ. Каждую точку концентрации дополнительно разбавляли в 50 раз средой IMDM. Если значение IC50 соединения было очень низким, то начальная концентрация соединения могла быть снижена. Добавляли 3 мкл разбавленного соединения в каждую лунку, центрифугировали и осторожно, но тщательно перемешивали. Среду без клеток использовали в качестве отрицательного контроля (100% ингибирование), и группу с 0,2% DMSO использовали в качестве положительного контроля (0% ингибирование).
384-луночный планшет помещали в инкубатор при 37°С и 5% СО2 для дальнейшей инкубации. Через 96 часов планшет извлекали и оставляли при комнатной температуре на 30 минут. Реагент CTG также извлекали и уравновешивали до комнатной температуры. Добавляли 15 мкл реагента CTG в каждую лунку и осторожно перемешивали в течение 5 минут на шейкере для гарантии достаточного клеточного лизиса. После 10 минут отстаивания сигнал люминесцентного излучения был стабильным. Затем сигнал люминесцентного излучения считывали с помощью Envision (Perkin Elmer). Кроме того, для корректировки количества клеток одновременно устанавливали контроль Т0, в том числе пустой контроль, содержащий только культуральную среду, и контроль с добавленными клетками. Разницу между ними устанавливали как контроль Т0, который получали добавлением реагента CTG перед добавлением соединений.
[0099] Процент ингибирования пролиферации клеток KM12 соединениями может быть вычислен с помощью следующей формулы:
Процент ингибирования=100-100*{[(Сигналсоединение-Сигналотрицательный констроль)-Т0 контроль]/[(Сигналположительный контроль-Сигналотрицательный констроль)-Т0 контроль]}.
Значения IC50 соединений вычисляли с помощью программного обеспечения XLfit (ID Business Solutions Ltd. UK) в 8 точках концентрации с помощью следующей формулы:
Y=Низ+(Верх-Низ)/(1+10^((logIC50-X)*фактор наклона)),
в которой Y представляет собой процент ингибирования, Низ представляет собой нижнее плато кривой (значение нижней платформы кривой S), Верх представляет собой верхнее плато кривой (значение верхней платформы кривой S), X представляет собой логарифм концентрации соединения, подлежащего измерению.
[0100] Результаты биологического эксперимента показаны в таблице I.
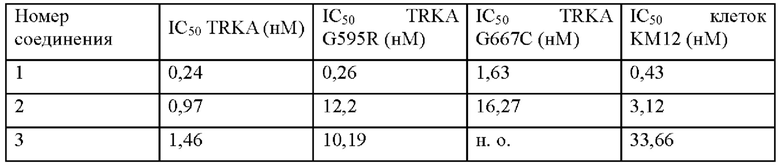

[0101] Из приведенных выше экспериментальных результатов можно видеть, что соединения примеров в настоящем изобретении могут эффективно ингибировать активность TRKA киназы и TRKA киназы с мутациями G595R и G667C и могут использоваться для лечения ряда раковых заболеваний, вызванных слиянием генов NTRK, таких как глиома, гепатобилиарная карцинома, папиллярная карцинома щитовидной железы, рак толстой кишки, не мелкоклеточный рак легкого, плоскоклеточный рак головы и шеи, карцинома поджелудочной железы, саркома и меланома (Khotskaya, Y.В. et at. Pharmacology & Therapeutics, 2017, 173, 58-66). Некоторые соединения могут также ингибировать пролиферацию клеток рака толстой кишки KM12. Они оказывают сильное ингибирующее действие на рак толстой кишки, вызванный слиянием генов NTRK.
[0102] Для специалистов в данной области очевидно, что настоящее раскрытие не ограничивается примерами, описываемыми выше, и может быть реализовано в других конкретных формах без отклонения от существенных характеристик настоящего раскрытия. Следовательно, предполагается, что эти варианты осуществления являются иллюстративными и неограничивающими во всех аспектах, и следует делать ссылку на прилагаемую формулу изобретения, а не на упомянутые выше варианты осуществления, и, таким образом, все изменения в пределах эквивалента и объема формулы изобретения предусматриваются ею.
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОЦИКЛ, СОДЕРЖАЩИЙ АМИНОПИРАЗОЛ И ПИРИМИДИН, И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2779497C2 |
| Макроциклический ингибитор тирозинкиназы и его применение | 2019 |
|
RU2798231C2 |
| КОНДЕНСИРОВАННОЕ АЗА-ГЕТЕРОЦИКЛИЧЕСКОЕ АМИДНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2811975C1 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| КОНДЕНСИРОВАННЫЕ КОЛЬЦЕВЫЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ TRK | 2015 |
|
RU2708674C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2011 |
|
RU2548684C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРАЗОЛО[1,5-a]ПИРИМИДИНА КАК ИНГИБИТОРЫ КИНАЗЫ TRK | 2010 |
|
RU2584157C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ КИНАЗ TRK | 2019 |
|
RU2778294C2 |
Изобретение относится к соединению формулы I и его фармацевтически приемлемым солям, где L1 выбран из -NR6CON(R7)-, в котором NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3; L2 выбран из C1-C4алкилена, при этом алкилен может быть необязательно замещен одним или несколькими G1; L3 представляет собой -O-; каждый R1, R2, R3 независимо выбран из водорода; R4 выбран из водорода и галогена; R5 выбран из водорода и галогена; каждый R6, R7 независимо выбран из водорода, C1-C4алкила; G1 выбран из C1-C4алкила. Изобретение также относится к фармацевтической композиции для лечения или предупреждения опосредованных TRK заболеваний, содержащей терапевтически эффективное количество соединения по изобретению или его фармацевтически приемлемых солей и фармацевтически приемлемые носители, разбавители и вспомогательные средства. Соединения по изобретению или его фармацевтически приемлемые соли или фармацевтическую композицию на их основе применяют в получении медикамента для лечения или предупреждения опосредованных TRK заболеваний, таких как рак, особенно гематологические злокачественные заболевания, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы и глиома. Технический результат – соединения в качестве ингибиторов TRK. 3 н. и 1 з.п. ф-лы, 1 табл., 4 пр.

1. Соединение, показанное в формуле I, и его фармацевтически приемлемые соли,

где
L1 выбран из -NR6CON(R7)-, в котором NR6 соединен с содержащим азот гетероарилом, замещенным R1, R2, R3;
L2 выбран из C1-C4алкилена, при этом алкилен может быть необязательно замещен одним или несколькими G1;
L3 представляет собой -O-;
каждый R1, R2, R3 независимо выбран из водорода;
R4 выбран из водорода и галогена;
R5 выбран из водорода и галогена;
каждый R6, R7 независимо выбран из водорода, C1-C4алкила;
G1 выбран из C1-C4алкила,
при этом следующие соединения (1) - (4) исключаются:
 .
.
2. Соединение по п. 1 и его фармацевтически приемлемые соли, отличающиеся тем, что соединения выбраны из:
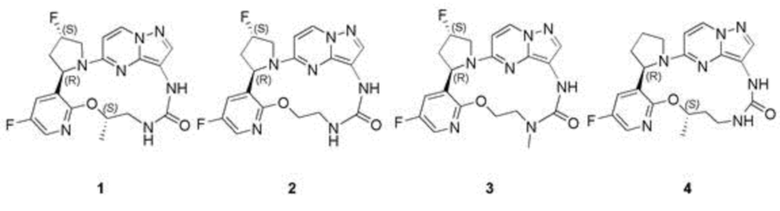 .
.
3. Фармацевтическая композиция для лечения или предупреждения опосредованных TRK заболеваний, содержащая терапевтически эффективное количество соединения по любому из пп. 1, 2 или его фармацевтически приемлемые соли и фармацевтически приемлемые носители, разбавители и вспомогательные средства.
4. Применение соединения по любому из пп. 1, 2 или его фармацевтически приемлемых солей или фармацевтической композиции по п. 3 в получении медикамента для лечения или предупреждения опосредованных TRK заболеваний, таких как рак, особенно гематологические злокачественные заболевания, рак легкого, рак молочной железы, рак яичника, рак предстательной железы, рак поджелудочной железы и глиома.
| WO 2011146336 A1, 24.11.2011 | |||
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ КИНАЗЫ LRRK2 | 2012 |
|
RU2622104C2 |
| WO 2010048314 A1, 29.04.2010 | |||
| WO 2017075107 A9, 02.08.2018. | |||

