ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящее изобретение заявляет приоритет заявки на патент Китая №201710728132.Х, поданной в Государственное ведомство интеллектуальной собственности Китая 23 августа 2017 года, которая включена здесь посредством ссылки во всей ее полноте.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к аминопиразолопиримидин-содержащему макроциклическому соединению, способу его получения, фармацевтической композиции, содержащей это соединение, и ее применению при лечении заболевания, опосредованного Trk-киназой (тропомиозин-рецепторной киназой).
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
NTRK/TRK (тропомиозин-рецепторная киназа) представляет собой тирозинкиназный рецептор нейротрофического фактора и принадлежит к семейству рецепторных тирозинкиназ. Семейство Trk главным образом включает три члена, а именно NTRK1/TrkA, NTRK2/TrkB и NTRK3/TrkC, где NGF (фактор роста нервов) связывается с TrkA; BDNF (нейротрофический фактор мозга) связывается с TrkB; и NT3 (нейротрофический фактор 3) связывается с TrkC.
Trk-киназа играет важную физиологическую роль в развитии нервов. Большое количество исследований показали, что активация сигнального пути Trk также строго коррелирует с возникновением и развитием опухоли. Активированные Trk сигнальные белки обнаружены в нейробластоме, аденокарциноме легких, карциноме поджелудочной железы, карциноме молочной железы и так далее. Открытие различных слитых белков Trk за последние годы дополнительно подемонстрировало их биологическую функцию в стимулировании онкогенеза. Первый слитый белок ТРМ3-TrkA был обнаружен в клетках рака ободочной кишки. Позднее различные типы слитых белков Trk, такие как CD74-NTRK1, MPRIP-NTRK1, QKI-NTRK2, ETV6-NTRK3, BTB1-NTRK3 и так далее, были обнаружены в образцах различных типов опухолевых тканей пациентов, страдающих, например раком легких, раком головы и шеи, раком молочной железы, раком щитовидной железы, глиомой и так далее. Эти различные слитые белки NTRK как таковые находятся в состоянии, в котором киназа высокоактивирована без необходимости в связывании с лигандом, и поэтому могут непрерывно фосфорилировать нисходящие сигнальные пути, индуцировать клеточную пролиферацию и способствовать возникновению и развитию опухоли. Более того, ингибиторы Trk являются эффективными при ингибировании роста опухоли и предотвращении метастазирования опухоли в доклинической модели рака. Поэтому в последние годы слитые белки Trk стали эффективной мишенью для противораковой терапии. Например, в WO 2010048314, WO 2012116217, WO 2010033941, WO 2011146336, WO 2017035354 и так далее раскрыты ингибиторы Trk-киназы, обладающие разными структурами центральной части.
Принимая во внимание важные физиологические функции Trk-киназ, крайне важно найти эффективный ингибитор Trk-киназ.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
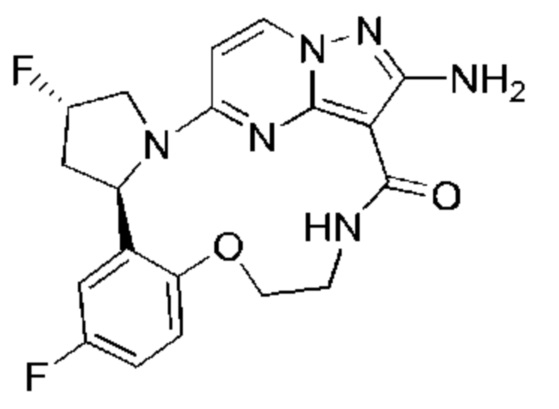
В одном аспекте настоящее изобретение относится к соединению формулы (I)
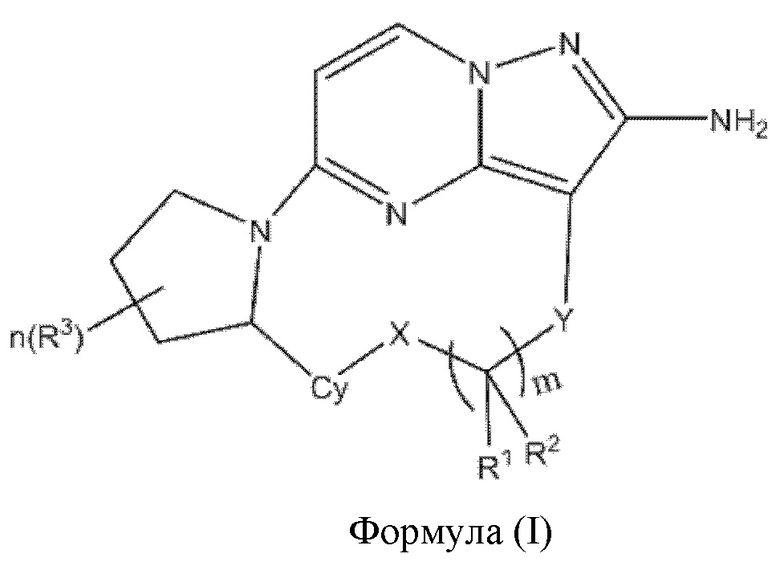
или его фармацевтически приемлемой соли, где
X выбран из группы, состоящей из связи, -О-, -S- и -NR4-;
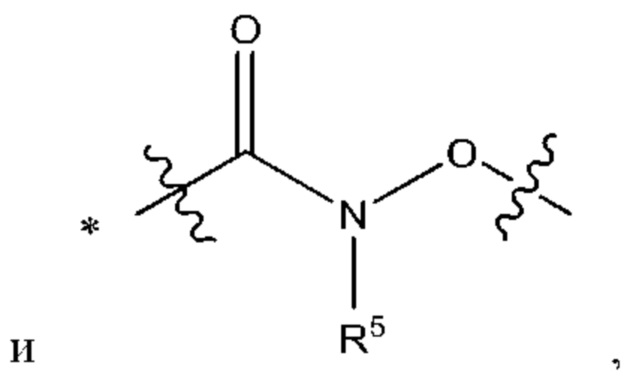
Y выбран из группы, состоящей из 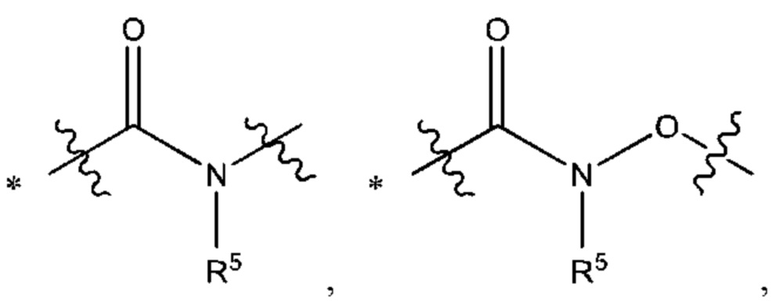
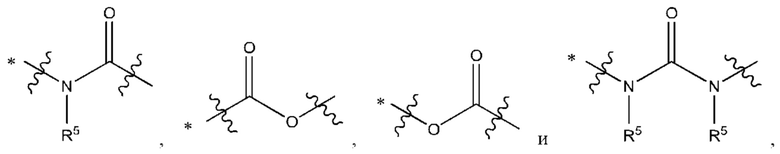
где "*" обозначает конец группы Y, присоединенный к аминопиразолопиримидиновому кольцу;
R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-С6алкила, C1-С6алкокси, галогена, нитро, гидрокси, циано и амино, где C1-С6алкил и C1-С6алкокси возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из галогена, нитро, гидрокси, циано и амино; или
R1 и R2 взятые вместе образуют
R3 выбран из группы, состоящей из C1-С6алкила, C1-С6алкокси, галогена, нитро, гидрокси, циано и амино, где C1-С6алкил и C1-С6алкокси возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из галогена, нитро, гидрокси, циано и амино;
R4 и R5 независимо выбраны из группы, состоящей из водорода и C1-С6алкила;
m выбран из 0, 1, 2, 3, 4, 5 или 6;
n выбран из 0, 1, 2, 3, 4, 5, 6 или 7;
Су выбран из группы, состоящей из 6-10-членного ароматического кольца, 5-10-членного ароматического гетероцикла, 3-10-членного алифатического гетероцикла и 3-10-членного циклоалкильного кольца, где 6-10-членное ароматическое кольцо, 5-10-членный ароматический гетероцикл, 3-10-членный алифатический гетероцикл или 3-10-членное циклоалкильное кольцо возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из C1-С6алкила, C1-С6алкокси,  галогена, нитро, гидрокси, циано и амино.
галогена, нитро, гидрокси, циано и амино.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) по настоящему изобретению или его фармацевтически приемлемую соль.
В другом аспекте настоящее изобретение относится к способу лечения заболевания, опосредованного Trk-киназой, у млекопитающего, включающему введение млекопитающему, предпочтительно человеку, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, или его фармацевтической композиции.
В другом аспекте настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли, или его фармацевтической композиции в изготовлении лекарственного средства для профилактики или лечения заболевания, опосредованного Trk-киназой.
В другом аспекте настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, или его фармацевтической композиции для применения в профилактике или лечении заболевания, опосредованного Trk-киназой.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы (I)
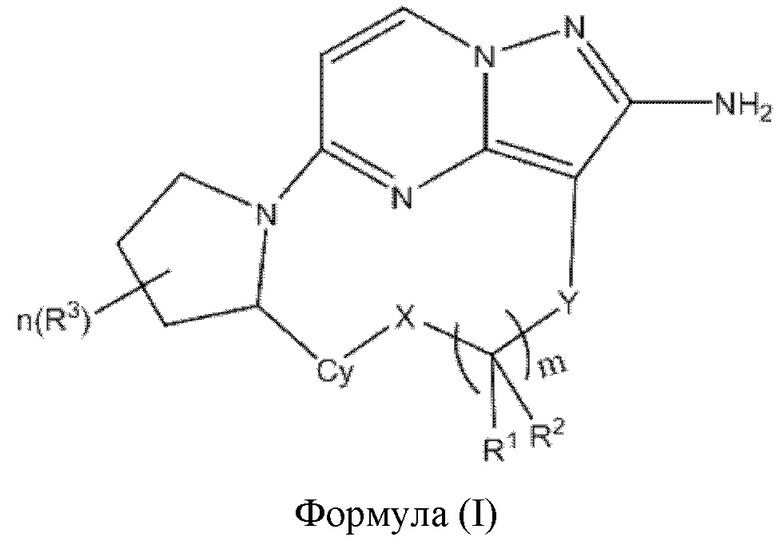
или его фармацевтически приемлемой соли, где
X выбран из группы, состоящей из связи, -О-, -S- и -NR4-;
Y выбран из группы, состоящей из 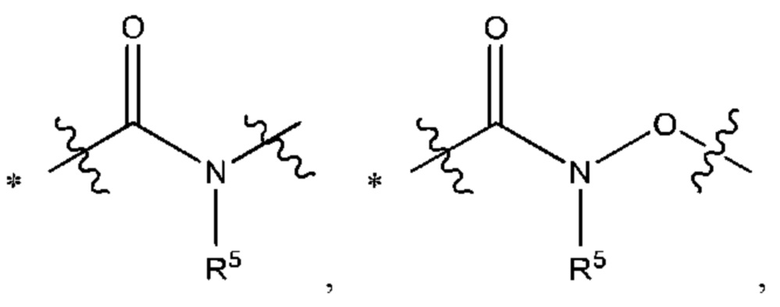
"*" обозначает конец группы Y, присоединенный к аминопиразолопиримидиновому кольцу;
R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-С6алкила, C1-С6алкокси, галогена, нитро, гидрокси, циано и амино, где C1-С6алкил и C1-С6алкокси возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из галогена, нитро, гидрокси, циано и амино; или
R1 и R2, взятые вместе, образуют 
R3 выбран из группы, состоящей из C1-С6алкила, C1-С6алкокси, галогена, нитро, гидрокси, циано и амино, где C1-С6алкил и C1-С6алкокси возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из галогена, нитро, гидрокси, циано и амино;
R4 и R5 независимо выбраны из группы, состоящей из водорода и C1-С6алкила;
m выбран из 0, 1, 2, 3, 4, 5 или 6;
n выбран из 0, 1, 2, 3, 4, 5, 6 или 7;
Су выбран из группы, состоящей из 6-10-членного ароматического кольца, 5-10-членного ароматического гетероцикла, 3-10-членного алифатического гетероцикла и 3-10-членного циклоалкильного кольца, где 6-10-членное ароматическое кольцо, 5-10-членный ароматический гетероцикл, 3-10-членный алифатический гетероцикл или 3-10-членное циклоалкильное кольцо возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из C1-С6алкила, C1-С6алкокси, 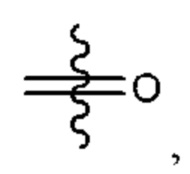 галогена, нитро, гидрокси, циано и амино.
галогена, нитро, гидрокси, циано и амино.
В некоторых воплощениях X выбран из группы, состоящей из связи и -О-;
В некоторых воплощениях R4 выбран из группы, состоящей из водорода и C1-С3алкила, предпочтительно водорода.
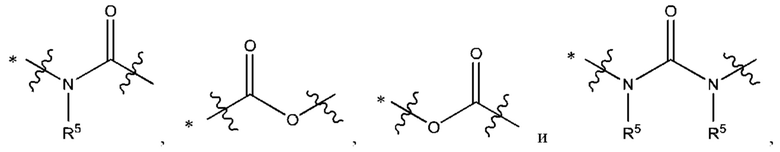
В некоторых воплощениях Y выбран из группы, состоящей из 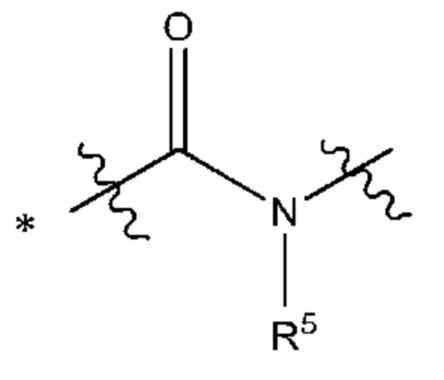
 где "*" обозначает конец группы Y, присоединенный к аминопиразолопиримидиновому кольцу.
где "*" обозначает конец группы Y, присоединенный к аминопиразолопиримидиновому кольцу.
В некоторых воплощениях R5 выбран из группы, состоящей из водорода и C1-С3алкила. В некоторых типичных воплощениях R5 выбран из группы, состоящей из водорода и метила.
В некоторых более типичных воплощениях Y выбран из группы, состоящей из *-CONH-, *-CON(CH3)- и *-CONHO-, где "*" обозначает конец группы Y, присоединенный к аминопиразолопиримидиновому кольцу.
В некоторых воплощениях R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-С3алкила, C1-С3алкокси, фтора, хлора, брома, иода, нитро, гидрокси, циано и амино, где C1-С3алкил и C1-С3алкокси возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из фтора, хлора, брома, иода, нитро, гидрокси, циано и амино.
В некоторых типичных воплощениях R1 и R2 независимо выбраны из группы, состоящей из водорода, фтора и C1-С3алкила. В некоторых более типичных воплощениях R1 и R2 независимо выбраны из группы, состоящей из водорода, фтора и метила.
В некоторых воплощениях m выбран из 1, 2, 3, 4 или 5. В некоторых типичных воплощениях m выбран из 2, 3 или 4.
В некоторых наиболее типичных воплощениях структурная единица 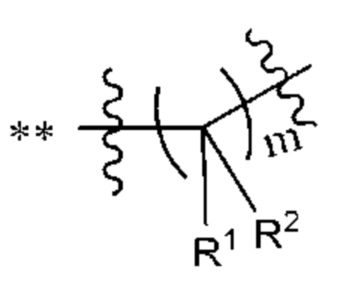 выбрана из группы, состоящей из
выбрана из группы, состоящей из 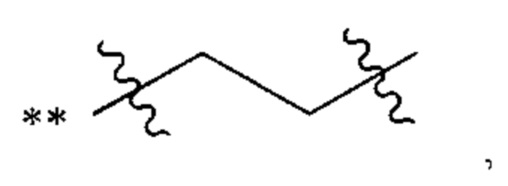

 где ** обозначает конец структурной единицы
где ** обозначает конец структурной единицы 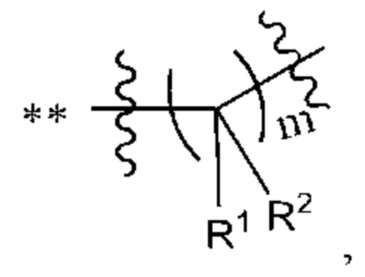 присоединенный к X.
присоединенный к X.
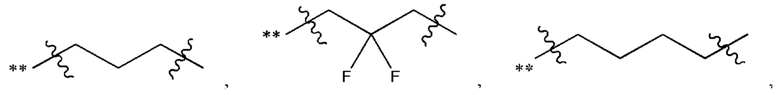
В некоторых наиболее типичных воплощениях структурная единица 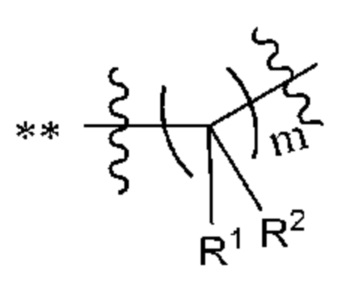 выбрана из группы, состоящей из
выбрана из группы, состоящей из 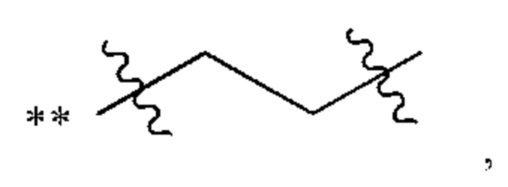
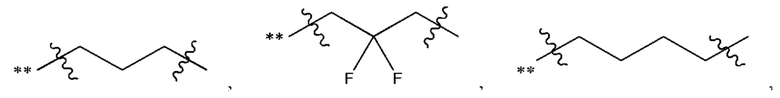
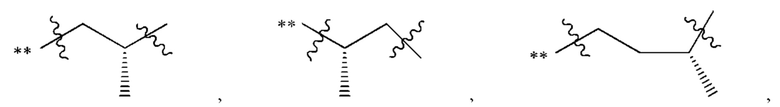
 где ** обозначает конец структурной единицы
где ** обозначает конец структурной единицы 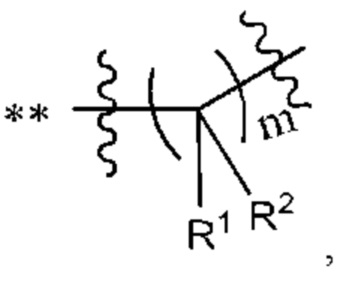 присоединенный к X.
присоединенный к X.
В некоторых воплощениях R3 выбран из группы, состоящей из C1-С3алкила, C1-С3алкокси, фтора, хлора, брома, иода, нитро, гидрокси, циано и амино, где C1-С3алкил и C1-С3алкокси возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из фтора, хлора, брома, иода, нитро, гидрокси, циано и амино.
В некоторых типичных воплощениях R3 выбран из группы, состоящей из фтора, хлора, брома, иода и гидрокси. В некоторых более типичных воплощениях R3 выбран из группы, состоящей из фтора и гидрокси.
В некоторых воплощениях n выбран из 0, 1, 2 или 3. В некоторых типичных воплощениях n выбран из 0 или 1.
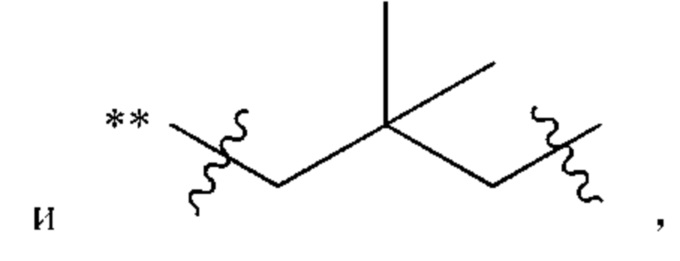
В некоторых воплощениях Су выбран из группы, состоящей из бензольного кольца, нафталинового кольца, пиррола, фурана, тиофена, имидазола, оксазола, пиразола, пиридина, пиримидина, пиразина, хинолина, изохинолина, бензофурана, бензотиофена, индола, изоиндола, оксирана, тетрагидрофурана, дигидрофурана, пирролидина, дигидропирролидина, 2H-пиридина, пиперидина, пиперазина, пиразолидина, тетрагидропирана, морфолина, тиоморфолина, тетрагидротиофена, циклопропана, циклопентана и циклогексана, каждый из которых возможно замещен одним или более чем одним заместителем, независимо выбранным из группы, состоящей из C1-С3алкила, C1-С3алкокси,  фтора, хлора, брома, иода, нитро, гидрокси, циано и амино.
фтора, хлора, брома, иода, нитро, гидрокси, циано и амино.
В некоторых типичных воплощениях Су выбран из группы, состоящей из бензольного кольца, пиридина и 1,2-2H-пиридина, каждый из которых возможно замещен одним или более чем одним заместителем, независимо выбранным из группы, состоящей из фтора и 
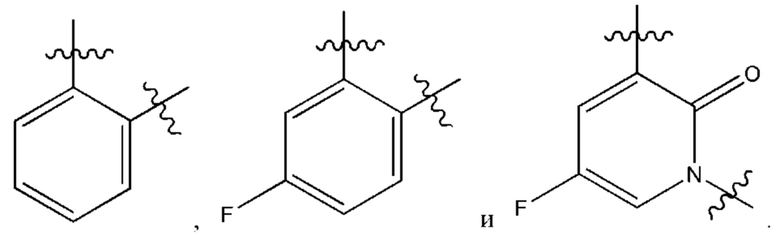
В некоторых более типичных воплощениях Су выбран из группы, состоящей из 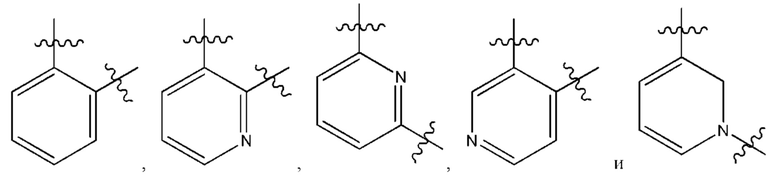 , каждый из которых возможно замещен одним или более чем одним заместителем, независимо выбранным из группы, состоящей из фтора и
, каждый из которых возможно замещен одним или более чем одним заместителем, независимо выбранным из группы, состоящей из фтора и 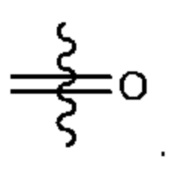
В некоторых более типичных воплощениях Су выбран из группы, состоящей из 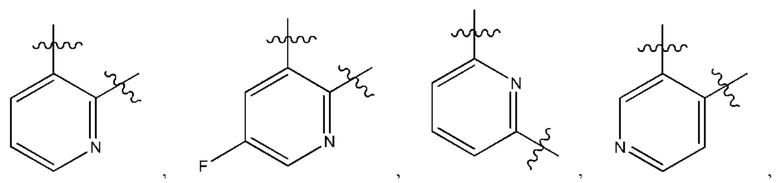
В некоторых воплощениях настоящего изобретения вышеупомянутое соединение формулы (I) выбрано из соединения формулы (II)
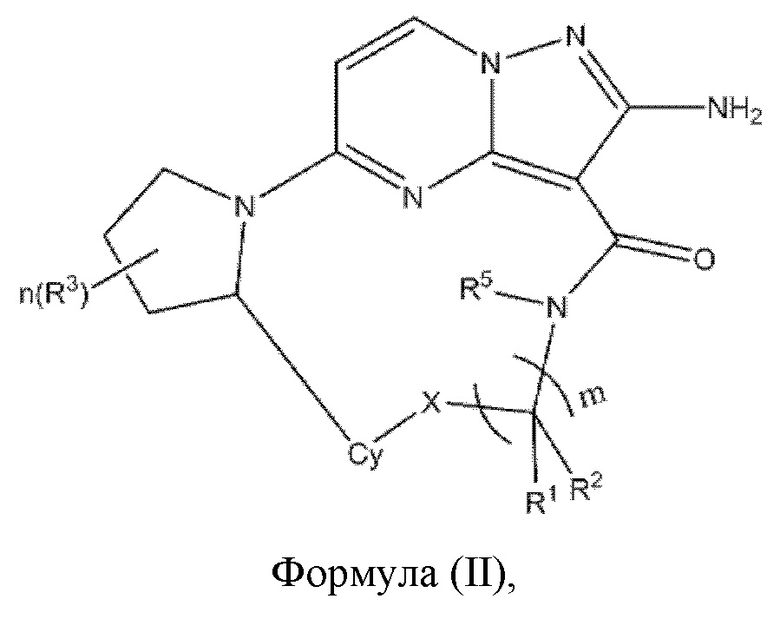
где X, R1, R2, R3, R5, Су, m и n являются такими, как определено в вышеупомянутом соединении формулы (I).
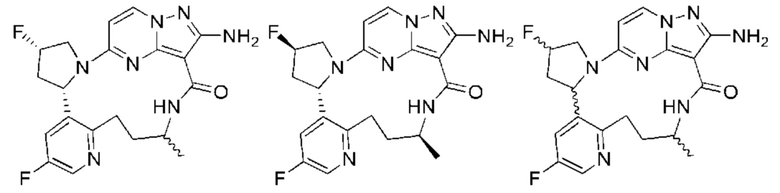
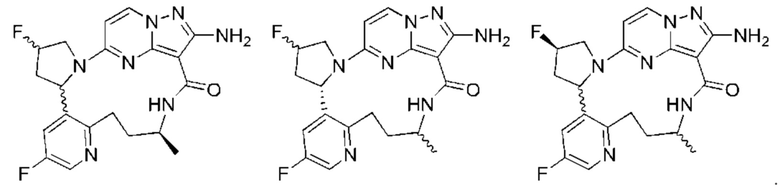
В некоторых воплощениях настоящего изобретения вышеупомянутое соединение формулы (I) или его фармацевтически приемлемая соль выбраны из группы, состоящей из
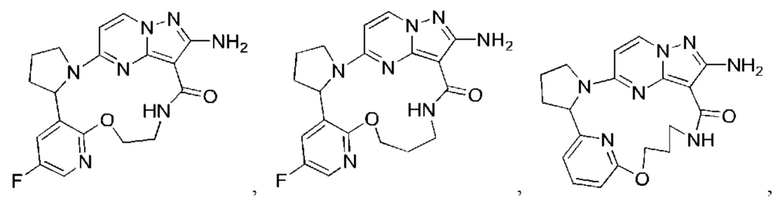
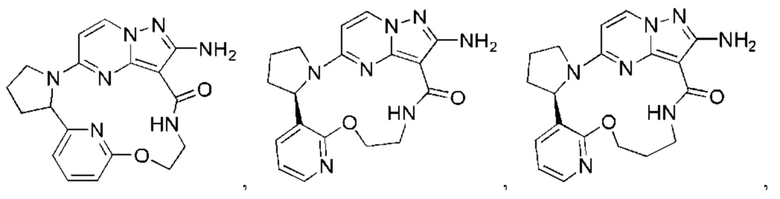
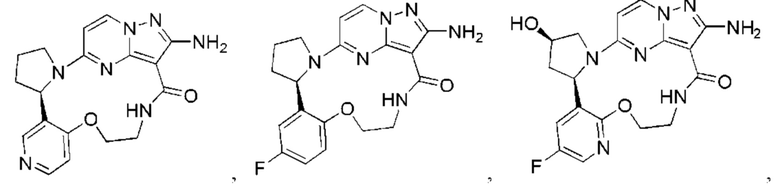
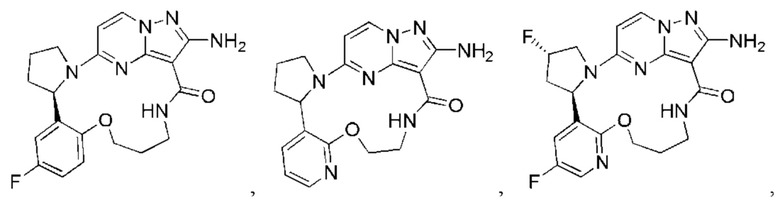

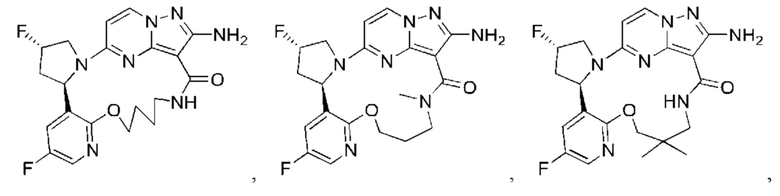
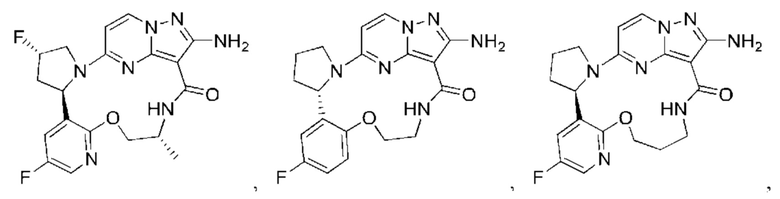

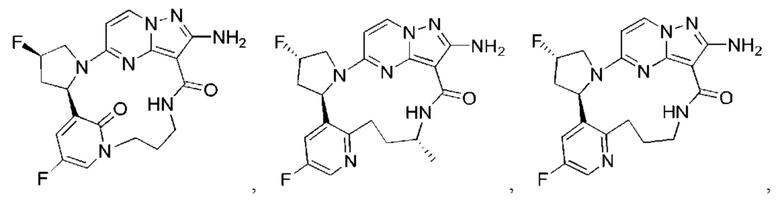
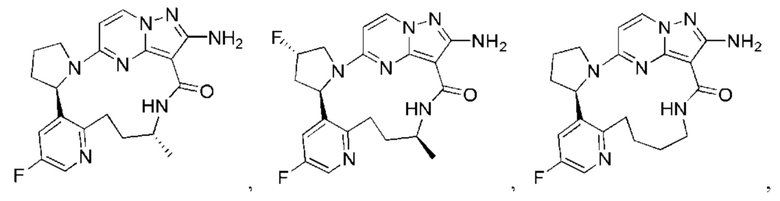
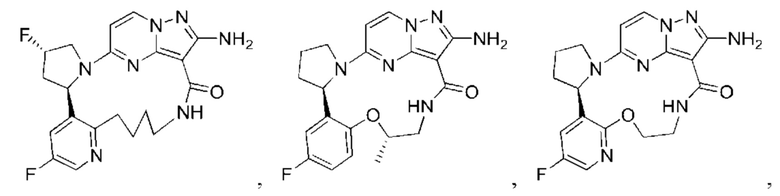
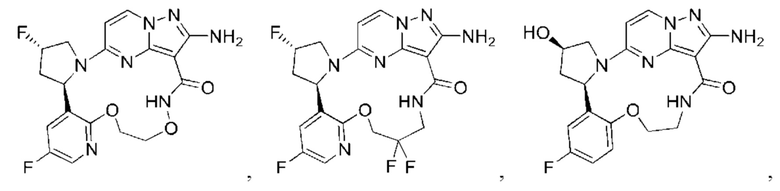
и их фармацевтически приемлемых солей.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) по настоящему изобретению или его фармацевтически приемлемую соль. В некоторых воплощениях фармацевтическая композиция по настоящему изобретению дополнительно содержит фармацевтически приемлемый эксципиент.
Фармацевтическая композиция по настоящему изобретению может быть приготовлена посредством объединения соединения формулы (I) по настоящему изобретению с соответствующим(и) фармацевтически приемлемым(и) эксципиентом (эксципиентами). Например фармацевтические композиции по настоящему изобретению могут быть приготовлены в виде твердых, полутвердых, жидких или газообразных препаратов, таких как таблетки, пилюли, капсулы, порошки, гранулы, мази, эмульсии, суспензии, суппозитории, инъекции, средства для ингаляции, гели, микросферы, аэрозоли и тому подобное.
Традиционные пути введения соединения формулы (I) по настоящему изобретению, или его фармацевтически приемлемой соли, или его фармацевтической композиции включают пероральное, ректальное, местное, посредством ингаляции, парентеральное, сублингвальное, интравагинальное, интраназальное, интраокулярное, интраперитонеальное, внутримышечное, подкожное и внутривенное введение, но не ограничиваются ими.
Фармацевтические композиции по настоящему изобретению могут быть приготовлены посредством использования способов, хорошо известных в данной области техники, таких как традиционный способ смешивания, способ растворения, способ гранулирования, способ производства драже, способ измельчения, способ эмульгирования, способ лиофилизации и тому подобных.
В некоторых воплощениях фармацевтическая композиция находится в пероральной форме. Для перорального введения фармацевтическая композиция может быть приготовлена в виде препарата посредством смешивания активного соединения (соединений) с фармацевтически приемлемым(и) эксципиентом(эксципиентами), хорошо известными в данной области техники. Такие эксципиенты дают возможность готовить соединения формулы (I) по настоящему изобретению в виде таблеток, пилюль, лепешек, драже, капсул, жидкостей, гелей, сиропов, суспензий и тому подобного для перорального введения пациентам.
Твердая фармацевтическая композиция для перорального применения может быть приготовлена посредством традиционного способа смешивания, наполнения или таблетирования. Например, она может быть получена посредством смешивания активного соединения с твердым эксципиентом, возможно измельчения полученной смеси, добавления других подходящих эксципиентов, если необходимо, и затем превращения смеси в гранулы с получением ядер таблеток или драже. Подходящие эксципиенты включают связующие вещества, разбавители, разрыхлители, смазывающие вещества, скользящие агенты, подсластители, ароматизаторы и тому подобные, но не ограничиваются ими.
Фармацевтическая композиция также подходит для парентерального введения, например стерильных растворов, суспензий или лиофилизированных продуктов в подходящей стандартной лекарственной форме.
В дополнительном аспекте настоящее изобретение относится к способу лечения заболевания, опосредованного Trk-киназой, у млекопитающего, включающему введение млекопитающему, предпочтительно человеку, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, или его фармацевтической композиции.
Суточная доза соединения формулы (I) при всех способах введения, которые описаны здесь, составляет от 0,01 мг/кг массы тела до 300 мг/кг массы тела, предпочтительно от 10 мг/кг массы тела до 300 мг/кг массы тела, и более предпочтительно от 25 мг/кг массы тела до 200 мг/кг массы тела, в виде однократной дозы или раздельными дозами.
В другом аспекте настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли, или его фармацевтической композиции при получении лекарственного средства для профилактики или лечения заболевания, опосредованного Trk-киназой.
В другом аспекте настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли, или его фармацевтической композиции в профилактике или лечении заболевания, опосредованного Trk-киназой.
В другом аспекте согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль, или его фармацевтическая композиция для применения в профилактике или лечении заболевания, опосредованного Trk-киназой.
Соединения по настоящему изобретению могут быть получены посредством различных способов синтеза, хорошо известных специалисту в данной области техники, включая конкретные воплощения, проиллюстрированные ниже, воплощения, полученные путем объединения таких конкретных воплощений с другими способами химического синтеза, и эквиваленты, хорошо известные специалисту в данной области техники. Предпочтительные воплощения включают рабочие примеры, продемонстрированные в настоящем изобретении, но не ограничиваются ими.
Химическое взаимодействие в конкретных воплощениях настоящего изобретения проводят в соответствующем растворителе, который должен быть подходящим для химического превращения (превращений) и требуемого реагента(реагентов) и вещества(веществ) в настоящему изобретению. Для получения соединений по настоящему изобретению специалисту в данной области техники иногда необходимо провести модификацию или выбор стадии (стадий) синтеза или схем(ы) реакций на основании существующих воплощений.
Важным вопросом, который следует рассмотреть при разработке пути синтеза в данной области техники, является выбор подходящей защитной группы для реакционноспособной функциональной группы, такой как аминогруппа, в настоящем изобретении. Например, может быть дана ссылка на Greene's Protective Groups in Organic Synthesis (4th Ed). Hoboken, New Jersey: John Wiley & Sons, Inc. Bee упомянутые здесь ссылки включены во всей их полноте.
В некоторых воплощениях соединение формулы (II) по настоящему изобретению может быть получено специалистом в области органического синтеза при использовании стандартного способа с помощью следующих иллюстративных схем синтеза, но не ограничиваясь ими:
Схема синтеза I:

где Ra выбран из галогена, предпочтительно фтора, хлора, брома и иода;
Rb выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно этила;
Rc выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно метила;
R1, R2, R3, R5, Су и m являются такими, как определено в вышеупомянутом соединении формулы (II).
Схема синтеза II:
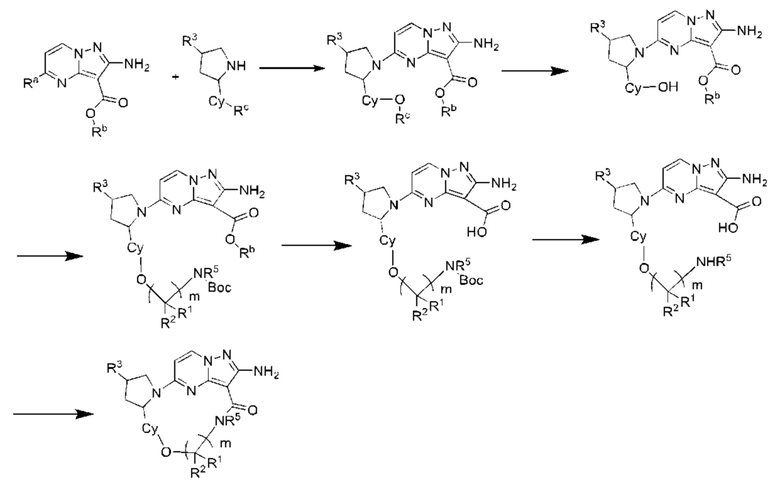
где Ra выбран из галогена, предпочтительно фтора, хлора, брома и иода;
Rb выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно этила;
Rc выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно метила;
R1, R2, R3, R5, Су и m являются такими, как определено в вышеупомянутом соединении формулы (II).
Схема синтеза III:
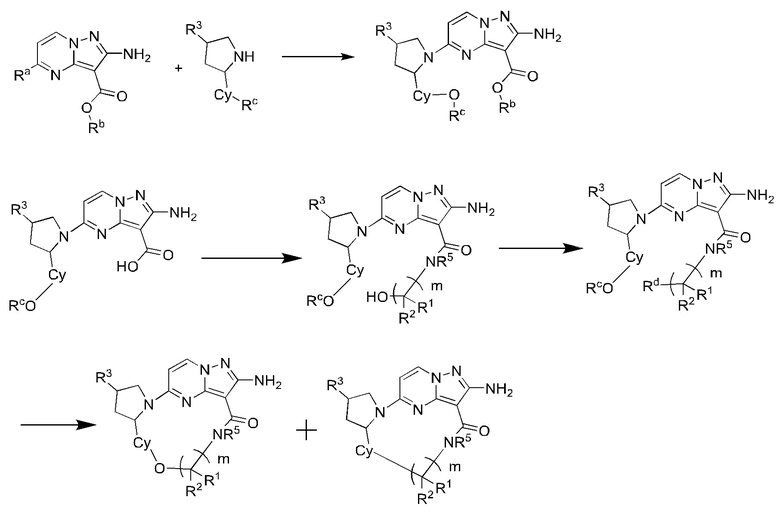
где Ra выбран из галогена, предпочтительно фтора, хлора, брома и иода;
Rb выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно этила;
Rc выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно метила;
Rd выбран из галогена, предпочтительно фтора, хлора, брома и иода;
R1, R2, R3, R5, Су и m являются такими, как определено в вышеупомянутом соединении формулы (II).
Схема синтеза IV:
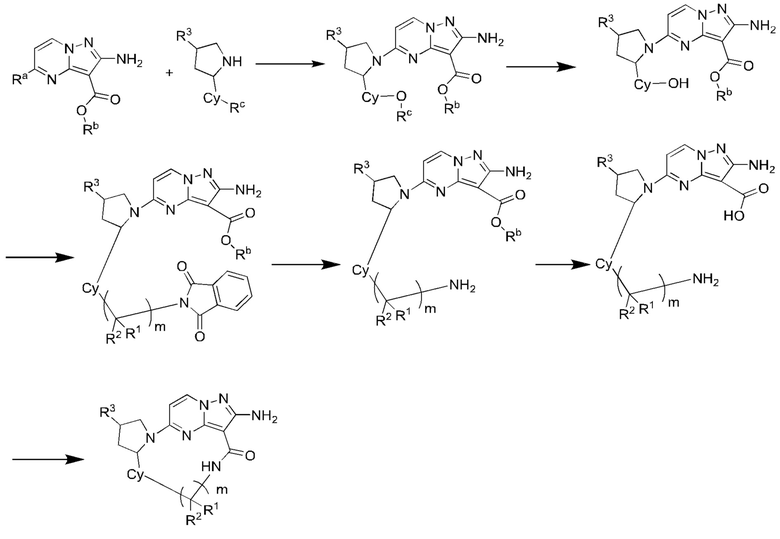
где Ra выбран из галогена, предпочтительно фтора, хлора, брома и иода;
Rb выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно этила;
Rc выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно метила;
R1, R2, R3, Су и m являются такими, как определено в вышеупомянутом соединении формулы (II).
Схема синтеза V:
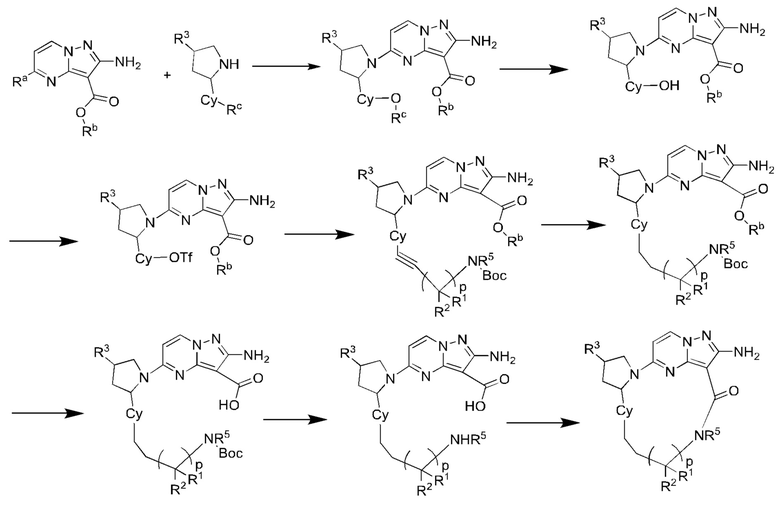
где Ra выбран из галогена, предпочтительно фтора, хлора, брома и иода;
Rb выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно этила;
Rc выбран из C1-С6алкила, предпочтительно C1-С3алкила, и более предпочтительно метила;
Р выбран из 0, 1, 2, 3 или 4.
R1, R2, R3, R5 и Су являются такими, как определено в вышеупомянутом соединении формулы (II).
Определения
Если не оговорено особо, следующие использованные здесь термины имеют следующие значения. Конкретный термин не следует считать неточным или неопределенным, когда он не определен особо. Его следует понимать в соответствии с его общим значением. Использованное здесь торговое наименование относится к соответствующему продукту или его активному ингредиенту.
Термин "замещенный" означает, что один или более чем один атом водорода по данному атому заменен заместителем при условии, что данный атом находится в нормальном валентном состоянии, а соединение после замещения стабильно. Когда заместитель представляет собой оксо (то есть =O), который означает, что заменены два атома водорода, замещение группой оксо по ароматической группе не будет происходить.
Термин "возможный" или "возможно" означает, что описанное далее событие или состояние может иметь место или может не иметь места, и что описание включает случаи, когда указанное событие или состояние имеет место, и случаи, когда указанное событие или состояние не имеет места. Например, этильная группа "возможно" замещена атомом (атомами) галогена, что означает, что этильная группа может быть незамещенной (СН2СН3), монозамещенной (такой как CH2CH2F), многократно замещенной (такой как CHFCH2F, CH2CHF2 и так далее) или полностью замещенной (CF2CF3). Специалисту в данной области техники будет понятно, что в отношении любой группы, содержащей один или более чем один заместитель, любое замещение или способ замещения, который пространственно невозможен и/или в результате которого не происходит синтез, не будет представлен.
Использованное здесь выражение Cm-Cn показывает, что эта группировка имеет целое число атомов углерода, находящееся в пределах данного диапазона. Например "С1-С6" означает, что эта группа может иметь 1 атом углерода, 2 атома углерода, 3 атома углерода, 4 атома углерода, 5 атомов углерода или 6 атомов углерода.
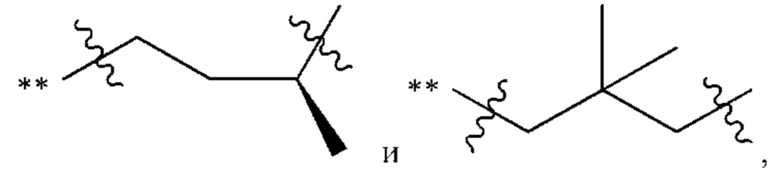
Когда любая переменная (такая как R) встречается больше одного раза в составе или структуре соединения, в каждом случае переменную определяют независимо. Поэтому, например, если группа замещена двумя Rs, тогда каждый R имеет независимое значение. В качестве другого примера, когда m не менее 2 в структурной единице 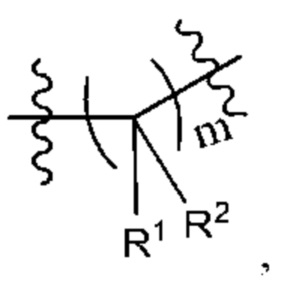 и R1, и R2 в каждой повторяющейся единице имеет независимое значение. В другом примере, когда n не менее 2 в структурной единице
и R1, и R2 в каждой повторяющейся единице имеет независимое значение. В другом примере, когда n не менее 2 в структурной единице 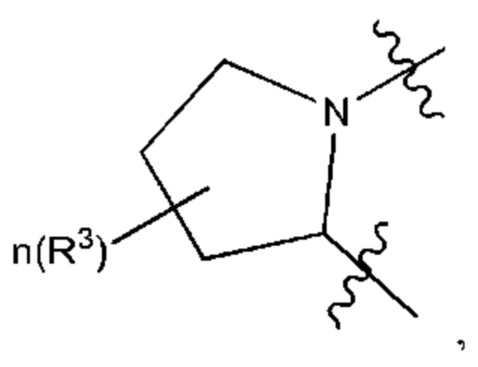 каждый R3 имеет независимое значение.
каждый R3 имеет независимое значение.
X представляет собой связь, что означает, что X в соединении формулы (I) отсутствует. То есть группа Су в соединении формулы (I) связана со структурной единицей 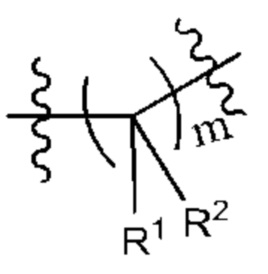 непосредственно через ковалентную связь.
непосредственно через ковалентную связь.
Термин "галоген" относится к фтору, хлору, брому и иоду.
Термин "гидрокси" относится к группе -ОН.
Термин "циано" относится к группе -CN.
Термин "амино" относится к группе -NH2.
Термин "нитро" относится к группе -NO2.
Термин "алкил" относится к гидрокарбильной группе формулы CnH2n+1. Алкильная группа может быть прямой или разветвленной. Например, термин "C1-С6алкил" относится к алкильной группе, имеющей от 1 до 6 атомов углерода (такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, неопентил, гексил, 2-метилпентил и так далее). Аналогично, алкильная группировка (то есть алкил) в алкоксигруппе, алкиламиногруппе, диалкиламиногруппе, алкилсульфонильной группе и алкилтиогруппе имеет такое же определение, как определено выше.
Термин "алкокси" относится к -О-алкилу.
Термин "циклоалкильное кольцо" относится к углеродному кольцу, которое является полностью насыщенным и может существовать в форме моноциклического кольца, мостикового кольца или спироциклического кольца. Если не оговорено особо, карбоцикл типично представляет собой 3-10-членное кольцо. Неограничивающие примеры циклоалкильного кольца включают циклопропан, циклобутан, циклопентан, циклогексан, бицикло[2.2.2]октан, адамантан и так далее, но не ограничиваются ими.
Термин "алифатический гетероцикл" относится к полностью насыщенному или частично ненасыщенному (но не к полностью ненасыщенному гетероароматическому) неароматическому кольцу, которое может существовать в форме моноциклического кольца, бициклического кольца или спироциклического кольца. Если не оговорено особо, гетероцикл типично представляет собой 3-6-членное кольцо, содержащее от 1 до 3 гетероатомов (предпочтительно 1 или 2 гетероатома), независимо выбранных из серы, кислорода и/или азота. Неограничивающие примеры алифатического гетероцикла включают оксиран, тетрагидрофуран, дигидрофуран, пирролидин, N-метилпирролидин, дигидропиррол, пиперидин, пиперазин, пиразолидин, 4Н-пиран, морфолин, тиоморфолин, тетрагидротиофен и так далее, но не ограничиваются ими.
Термин "ароматический гетероцикл" относится к моноциклической или конденсированной полициклической системе, содержащей по меньшей мере один кольцевой атом, выбранный из N, О и S, при этом остальные кольцевые атомы представляют собой С, и имеющей по меньшей мере одно ароматическое кольцо. Предпочтительный ароматический гетероцикл имеет одиночное 4-8-членное кольцо, особенно одиночное 5-8-членное кольцо, или имеет конденсированное полициклическое кольцо, содержащее от 6 до 14, особенно от 6 до 10 кольцевых атомов. Неограничивающие примеры ароматического гетероцикла включают пиррол, фуран, тиофен, имидазол, оксазол, пиразол, пиридин, пиримидин, пиразин, хинолин, изохинолин, бензофуран, бензотиофен, индол, изоиндол и так далее, но не ограничиваются ими.
Термин "лечение" или "лечить" относится к введению соединений или препаратов по настоящему изобретению для предупреждения, уменьшения интенсивности или элиминации заболеваний или одного или более чем одного симптома, связанного с заболеваниями, включая:
(I) профилактику возникновения заболеваний или состояний у млекопитающих, особенно когда млекопитающие чувствительны к состояниям, но эти состояния у них не диагностированы;
(II) ингибирование заболеваний или состояний, то есть ограничение их развития;
или
(III) ослабление заболеваний или состояний, то есть излечение заболеваний или состояний.
Термин "терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое (I) лечит или предупреждает конкретное заболевание, состояние или расстройство, (II) смягчает, уменьшает интенсивность или элиминирует один или более чем один симптом конкретного заболевания, состояния или расстройства, или (III) предупреждает или задерживает проявление одного или более чем одного симптома конкретного заболевания, состояния или расстройства, которое описано здесь. Количество соединений по настоящему изобретению, которое представляет собой так называемое "терапевтически эффективное количество", зависит от соединения, болезненного состояния и его тяжести, пути введения и возраста млекопитающего, подвергаемого лечению, но может быть легко определено специалистами в данной области техники на основании их знаний и данного раскрытия.
Термин "фармацевтически приемлемый" относится к соединению, веществу, композиции и/или лекарственной форме, которая приводится в контакт с тканями человека и животного, без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений с медицинской точки зрения, и в соответствии с допустимым соотношением риск/польза.
В качестве фармацевтически приемлемой соли могут быть упомянуты, например соль металла, аммонийная соль, соль органического основания, соль неорганической кислоты, соль органической кислоты, соль щелочной или кислой аминокислоты и так далее.
Термин "фармацевтическая композиция" относится к смеси одного или более чем одного соединения по настоящему изобретению или его соли и фармацевтически приемлемого эксципиента. Назначение фармацевтической композиции состоит в том, чтобы способствовать введению соединений по настоящему изобретению в организм.
Термин "фармацевтически приемлемый эксципиент" относится к таким эксципиентам, которые не вызывают значительной стимуляции организма и не будут отрицательно влиять на биологическую активность и свойства активного соединения. Подходящие эксципиенты хорошо известны специалистам в данной области техники, например углеводы, воски, водорастворимые и/или водонабухающие полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, вода и тому подобное.
Формулировку "содержать" и ее варианты, такие как "содержит" и "содержащий", следует понимать в широком и общем смысле, то есть как "включающий, но не ограниченный".
Если не оговорено особо, использованные здесь аббревиатуры имеют следующие значения:
мин означает минуту;
ч означает час;
°С означает градус Цельсия;
об./об. означает объемное соотношение;
DCM означает дихлорметан;
АС2О означает ангидрид уксусной кислоты;
ЕА означает этилацетат;
РЕ означает петролейный эфир;
МеОН означает метанол;
THF означает тетрагидрофуран;
ACN означает ацетонитрил;
толуол означает метилбензол;
DMF означает N,N-диметилформамид;
DMSO означает диметилсульфоксид;
TEA означает триэтиламин;
EDCI означает гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида;
НОВТ означает 1-гидроксибензотриазол;
Ti(OEt)4 означает тетраэтилтитанат;
DMAP означает 4-диметиламинопиридин;
DIAD означает диизопропилазодикарбоксилат;
PPh3 означает трифенилфосфин;
PD(PPh3)4 означает тетракис(трифенилфосфин)палладий;
PdCl2 означает хлорид палладия;
CuI означает иодид меди;
TFA означает трифторуксусную кислоту;
TBDMSCl означает дареда-бутилдиметилхлорсилан;
NaBH4 означает борогидрид натрия;
LiHMDS означает гексаметилдисилазид лития;
(ВОС)2О означает ди-дареда-бутилдикарбонат;
NBS означает N-бромсукцинимид;
Десс-Мартин означает периодинан Десса-Мартина;
DAST означает трифторид диэтиламиносеры;
HATU означает гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония;
DIEA означает N,N-диизопропилэтиламин;
FDPP означает пентафторфенилдифенилфосфат;
ЖХ-МС означает жидкостную хроматографию-масс-спектрометрию;
FIMPA означает гексаметилфосфортриамид;
Cs2CO3 означает карбонат цезия;
LiH означает гидрид лития;
ТСХ означает тонкослойную хроматографию;
М означает единицу молярной концентрации моль/л, например 2 М означает 2 моль/л;
мМ означает единицу молярной концентрации миллимоль на литр, например 2 мМ означает 2 ммоль/л;
н означает эквивалентную концентрацию, например 1 н HCl означает соляную кислоту с концентрацией 1 моль/л; 2 н NaOH означает гидроксид натрия с концентрацией 2 моль/л;
Ts означает пара-метилбензолсульфонил;
TsCl означает пара-толуолсульфонилхлорид;
Et означает этил;
Me означает метил;
Ас означает ацетил;
РМВ означает пара-метоксибензил;
Вос означает трет-бутоксикарбонил;
TBS означает трет-бутилдиметилсилил.
Промежуточные соединения и соединения по настоящему изобретению могут также существовать в форме различных таутомеров, и все такие формы включены в объем настоящего изобретения. Термин "таутомер" или "таутомерная форма" относится к структурным изомерам различных энергий, которые являются взаимопревращаемыми при переходе через низкоэнергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения посредством миграции протона, такие как кето-енольная и имин-енаминовая изомеризации. Конкретным примером протонных таутомеров является имидазольная группировка, в которой протон может мигрировать между двумя кольцевыми атомами азота. Валентные таутомеры включают взаимопревращения посредством перегруппировки некоторых из связывающих электронов. Неограничивающие примеры таутомеров включают, но не ограничиваются лишь
Соединения по настоящему изобретению также включают изотопно-меченые соединения по настоящему изобретению, которые идентичны приведенным здесь, за исключением того, что один или более чем один атом заменен на атом, имеющий атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, йода и хлора, такие как 2Н, 3Н, 11С, 13С, 14С, 13N, 15N, 15О, 17О, 18О, 31Р, 32Р, 35S, 18F, 123I, 125I и 36Cl, соответственно.
Некоторые изотопно-меченые соединения по настоящему изобретению (например, меченые 3Н и 14С) полезны в анализах тканевого распределения соединения и/или субстрата. Изотопы трития (то есть 3Н) и углерода-14 (то есть 14С) особенно предпочтительны ввиду легкости их получения и обнаружения. Позитронно-активные изотопы, такие как 15O, 13N, 11С и 18F, полезны для исследований при помощи позитронно-эмиссионной томографии (ПЭТ) для проверки степени занятости субстрата. Изотопно-меченые соединения по настоящему изобретению обычно могут быть получены посредством следующих способов, аналогичных описанным на схемах и/или в приведенных ниже примерах, путем замещения изотопно-меченого реагента на не меченый изотопом реагент.
Кроме того, замещение более тяжелыми изотопами (такими как дейтерий, то есть 2Н) может обеспечить некоторые терапевтические преимущества, являющиеся результатом большей метаболической стабильности, например увеличенного периода полувыведения in vivo или уменьшенной потребностью в дозах, и, следовательно, может быть предпочтительным в некоторых случаях, в которых дейтерирование может быть частичным или полным, и частичное дейтерирование означает, что по меньшей мере один атом водорода заменен по меньшей мере одним дейтерием. Неограничивающие примеры дейтерированных соединений включают, но не ограничиваются, следующие:
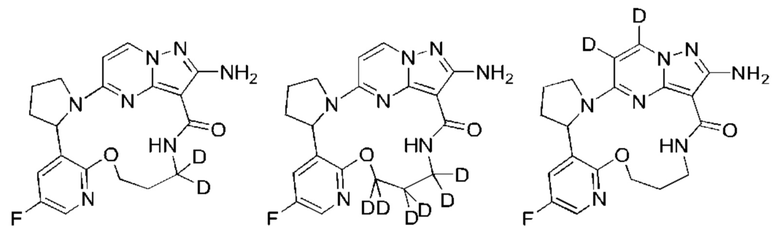
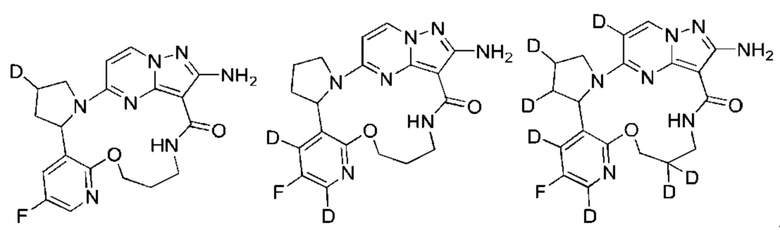
Соединения по настоящему изобретению могут быть асимметричными, например имеющими один или более чем один стереоизомер. Если не оговорено особо, включены все стереоизомеры, такие как энантиомеры и диастереомеры. Соединения, содержащие асимметричный(е) атом(ы) углерода, по настоящему изобретению могут быть выделены в оптически активной чистой форме или рацемической форме. Оптически активная чистая форма может быть выделена из рацемической смеси или синтезирована при использовании хирального исходного вещества (хиральных исходных веществ) или хирального реагента(хиральных реагентов). Неограничивающие примеры стереоизомеров включают, но не ограничиваются лишь
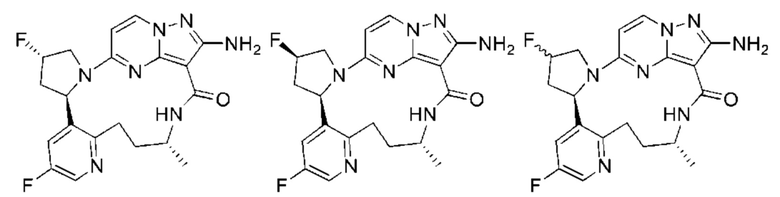
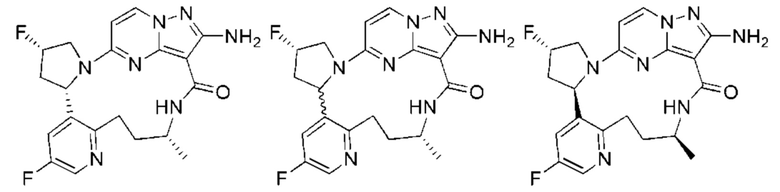

Для лучшего понимания настоящее изобретение дополнительно проиллюстрировано с помощью следующих примеров, но эти примеры не предназначены для ограничения объема настоящего изобретения. Все реагенты, использованные в настоящем изобретении, имеются в продаже и могут быть использованы без дополнительной очистки.
КОНКРЕТНЫЕ ПРИМЕРЫ
Получение промежуточных соединений
Промежуточное соединение 1: синтез 5-фтор-2-метокси-3-(пирролидин-2-ил)пиридина
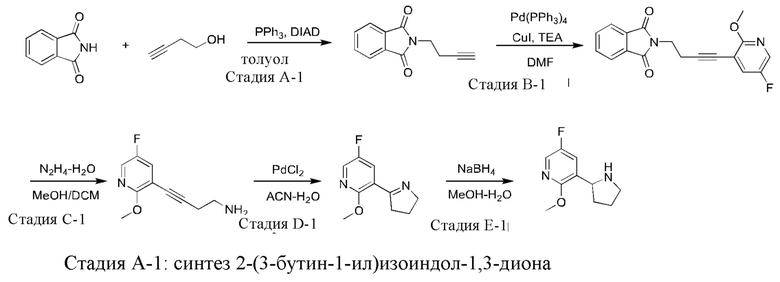
К раствору смеси фталимида (20,0 г), 3-бутин-1-ола (10,5 г) и трифенилфосфина (39,3 г) в толуоле (200 мл) медленно по каплям добавляли DIAD (34,0 г) при 0°С. После завершения добавления полученную смесь нагревали до комнатной температуры и непрерывно перемешивали в течение 1 часа. В реакционную смесь добавляли метанол (50 мл) и перемешивали в течение 1 часа. Большое количество белого твердого вещества осаждали и затем отфильтровали. Осадок на фильтре промывали метанолом с получением указанного в заголовке соединения (15,6 г). Фильтрат концентрировали, и остаток суспендировали в метаноле и затем фильтровали с получением указанного в заголовке соединения (7,95 г).
1Н ЯМР (ядерный магнитный резонанс) (400 МГц, CDCl3) δ 7.88-7.81 (m, 4Н), 3.69 (t, J=6,8 Гц, 2Н), 3.80 (t, J=2,8 Гц, 1H), 2.55-2.51 (m, 2Н).
Стадия В-1: синтез 2-(4-(5-фтор-2-метоксипиридин-3-ил)-3-бутин-1-ил)- изоиндол-1,3-диона
3-Бром-5-фтор-2-метоксипиридин (24,4 г), 2-(3-бутин-1-ил)изоиндол-1,3-дион (23,6 г) и триэтиламин (66 мл) растворяли в DMF (200 мл) при комнатной температуре. Газообразный азот барботировали через реакционную систему в течение 10 минут и затем туда добавляли тетракис(трифенилфосфин)палладий (7,0 г) и иодид меди (2,3 г). Смесь нагревали до 90°С и перемешивали в течение 2 часов в защитной атмосфере азота и затем охлаждали до комнатной температуры. Добавляли метанол (100 мл), и большое количество твердого вещества осаждали и затем отфильтровали. Осадок на фильтре промывали метанолом и сушили под вакуумом с получением указанного в заголовке соединения (38,3 г).
1H ЯМР (400 МГц, CDCl3) δ 7.89-7.86 (m, 3Н), 7.75-7.73 (m, 2Н), 7.35-7.30 (m, 1H), 3.99 (t, J=7,2 Гц, 2H), 3.85 (s, 3Н), 2.90 (t, J=7,2 Гц, 2H).
Стадия С-1: синтез 4-(5-фтор-2-метоксипиридин-3-ил)-3-бутин-1-амина К раствору смеси 2-(4-(5-фтор-2-метоксипиридин-3-ил)-3-бутин-1-ил)изоиндол-1,3-диона (38.3 г) в метаноле (120 мл) и дихлорметане (600 мл) медленно по каплям добавляли гидразин-гидрат (12,0 г; чистота 80%) при комнатной температуре, перемешивали при комнатной температуре в течение 12 часов и затем фильтровали. Осадок на фильтре промывали дихлорметаном. В фильтрат добавляли воду (500 мл), и затем полученную смесь разделяли. Органическую фазу сушили над безводным сульфатом натрия и затем фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (17,6 г).
Стадия D-1: синтез 3-(3,4-дигидро-2H-пиррол-5-ил)-5-фтор-2-метоксипиридина
При комнатной температуре 4-(5-фтор-2-метоксипиридин-3-ил)-3-бутин-1-амин (17,6 г) и хлорид палладия (178 мг) добавляли в смесь ацетонитрила (200 мл) и воды (70 мл), и полученный раствор смеси перемешивали при 80°С в течение 5 часов, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении для удаления ацетонитрила. Полученный остаток экстрагировали дихлорметаном (200 мл × 3), и объединенную органическую фазу сушили над безводным сульфатом натрия и затем фильтровали. Фильтрат концентрировали, и остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 20:1 (об./об.)) с получением указанного в заголовке соединения (11,2 г).
1H ЯМР (400 МГц, CDCl3) δ 8.04 (d, J=3,2 Гц, 1H), 7.92 (dd, J=8,4, 2,8 Гц, 1H), 4.05-3.96 (m, 5Н), 3.03-2.98 (m, 2Н), 2.04-1.96 (m, 2Н).
Стадия Е-1: 5-фтор-2-метокси-3-(пирролидин-2-ил)пиридин
К раствору смеси 3-(3,4-дигидро-2H-пиррол-5-ил)-5-фтор-2-метоксипиридина (11,2 г) в метаноле (100 мл) и воде (25 мл) порциями добавляли NaBH4 (4,4 г) при 0°С. После завершения добавления полученную смесь медленно нагревали до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь гасили 2 н водным раствором соляной кислоты и затем концентрировали при пониженном давлении для удаления метанола. Затем значение рН реакционной системы доводили до 8 насыщенным водным раствором гидроксида натрия и экстрагировали дихлорметаном (100 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и затем фильтровали, и фильтрат концентрировали с получением указанного в заголовке соединения (11,3 г).
1Н ЯМР (400 МГц, CDCl3) δ 7.83 (d, J=3,2 Гц, 1H), 7.56 (dd, J=8,8, 3,2 Гц, 1H), 4.28 (t, J=7,6 Гц, 1H), 3.92 (s, 3Н), 3.15-3.01 (m, 2Н), 2.28-2.19 (m, 1H), 1.95-1.78 (m, 3Н), 1.59-1.52 (m, 1H).
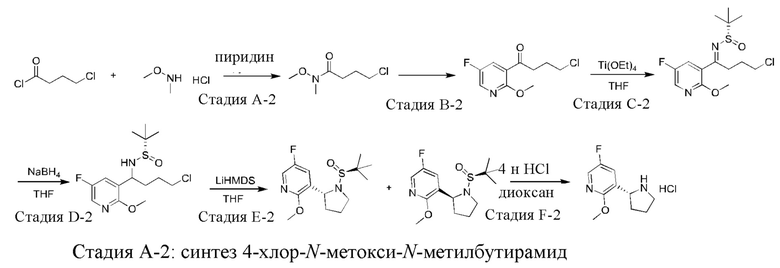
Промежуточное соединение 2: синтез гидрохлорида (R)-5-фтор-2-метокси-3-(пирролидин-2-ил)пиридина
К раствору гидрохлорида N,O-диметилгидроксиламина (100,0 г) в DCM (1500 мл) добавляли пиридин (250 мл) при 0°С при перемешивании и непрерывно перемешивали в течение 30 минут. К этой смеси затем по каплям добавляли 4-хлорбутирилхлорид (145,0 г). После завершения добавления реакционную смесь нагревали до комнатной температуры и непрерывно перемешивали в течение 2 часов. Реакционную смесь выливали в воду (250 мл) и затем экстрагировали дихлорметаном (100 мл × 3). Органическую фазу последовательно промывали 1 н соляной кислотой, водой и затем насыщенным рассолом, сушили над безводным сульфатом натрия и затем фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (125,1 г).
Стадия В-2: синтез 4-хлор-1-(5-фтор-2-метоксипиридин-3-ил)бутан-1-она
К раствору 3-бром-5-фтор-2-метоксипиридина (20,0 г) в THF (200 мл) по каплям добавляли н-бутиллитий (2,5 М раствор в гексане) (43 мл) при -90°С, при этом температуру реакционной системы поддерживали при -90°С. После завершения добавления по каплям полученную смесь перемешивали при -90°С в течение 2 часов, и затем в реакционную смесь по каплям добавляли раствор 4-хлор-N-метокси-N-метилбутирамида (17,7 г) в THF (100 мл), при этом температуру реакционной системы поддерживали при -90°С. После завершения добавления по каплям температуру реакции постепенно повышали до 10°С. Реакционную смесь гасили насыщенным водным раствором хлорида аммония и затем экстрагировали этилацетатом (100 мл × 3). Органическую фазу промывали водой и затем насыщенным рассолом. Затем органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 20:1 (об./об.)) с получением указанного в заголовке соединения (3,82 г).
1H ЯМР (400 МГц, CDCl3) δ 8.17 (d, J=3,2 Гц, 1H), 7.88 (dd, J=8,0, 3,2 Гц, 1H), 4.05 (s, 3Н), 3.66 (t, J=6,4 Гц, 2Н), 3.22 (t, J=7,2 Гц, 2Н), 4.19 (dd, J=13,2, 6,4 Гц, 2Н).
Стадия С-2: синтез (S,E)-N-(4-хлор-1-(5-фтор-2-метоксипиридин-3-ил)бутилен)-2-метилпропан-2-сульфинамида
К раствору 4-хлор-1-(5-фтор-2-метоксипиридин-3-ил)бутан-1-она (3,82 г) и (S)-2-метилпропан-2-сульфинамида (3,01 г) в THF (20 мл) добавляли тетраэтилтитанат (5,66 г) при комнатной температуре при перемешивании. Смесь непрерывно перемешивали при 70°С в течение 5 часов. Затем реакционную смесь охлаждали до комнатной температуры, гасили насыщенным водным раствором хлорида аммония и фильтровали, и осадок на фильтре промывали этилацетатом. Фильтрат разделяли, и органическую фазу промывали водой и затем насыщенным рассолом. Затем органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5,09 г).
Стадия D-2: синтез (S)-N-(4-хлор-1-(5-фтор-2-метоксипиридин-3-ил)бутил)-2-метилпропан-2-сульфинамида
К раствору (S,E)-N-(4-хлор-1-(5-фтор-2-метоксипиридин-3-ил)бутилен)-2-метилпропан-2-сульфинамида (5,09 г) в THF (20 мл) порциями добавляли NaBH4 (576 мг) при -78°С, при этом температуру реакционной системы поддерживали не выше -78°С. После завершения добавления порциями полученную смесь медленно нагревали до комнатной температуры и перемешивали в течение 1 часа. Затем реакционный раствор медленно выливали в ледяную воду для остановки реакции и экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5,12 г).
Стадия Е-2: синтез 3-((R)-1-((S)-трет-бутилсульфинил)пирролидин-2-ил)-5-фтор-2-метоксипиридина и 3-((S)-1-((S)-трет-бутилсульфинил)пирролидин-2-ил)-5-фтор-2-метоксипиридина
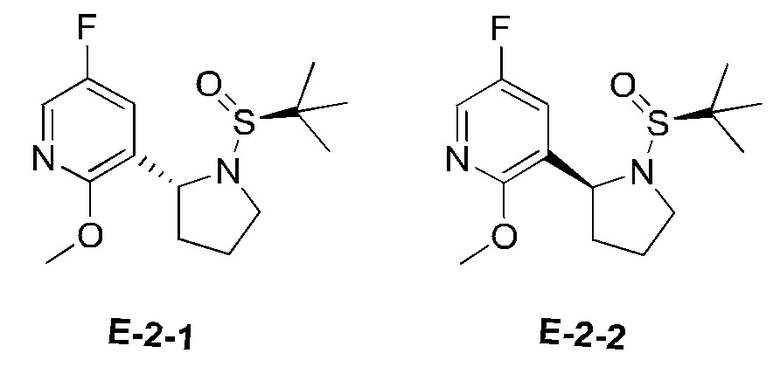
К раствору (S)-N-(4-хлор-1-(5-фтор-2-метоксипиридин-3-ил)бутил)-2-метилпропан-2-сульфинамида (5,12 г) в THF (30 мл) медленно по каплям добавляли LiHMDS (1 М раствор в THF) (23 мл) при -78°С, при этом температуру реакционной системы поддерживали не выше -78°С. После завершения добавления по каплям полученную смесь медленно нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором хлорида аммония и экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 20:1 (об./об.)) с получением двух указанных в заголовке соединений (1,1 г), соответственно.
Е-2-1: 1Н ЯМР (400 МГц, CDCl3) δ 7.88 (d, J=3,2 Гц, 1H), 7.32 (dd, J=8,4, 3,2 Гц, 1H), 4.96 (t, J=6,8 Гц, 1H), 3.94 (s, 3Н), 3.91-3.85 (m, 1H), 3.00-2.94 (m, 1H), 2.25-2.20 (m, 1H), 1.91-1.83 (m, 2Н), 1.75-1.68 (m, 1H), 1.18 (s, 9Н). m/z составляет 301 [М+1]+.
Е-2-2: 1Н ЯМР (400 МГц, CDCl3) δ 7.87 (d, J=2,8 Гц, 1H), 7.32 (dd, J=8,4, 2,8 Гц, 1H), 5.22 (d, J=8,0 Гц, 1H), 3.93 (s, 3Н), 3.65-3.54 (m, 2Н), 2.14-2.09 (m, 1H), 1.93-1.88 (m, 1H), 1.75-1.68 (m, 2Н), 1.18 (s, 9Н). m/z составляет 301 [М+1]+.
Стадия F-2: синтез гидрохлорида (R)-5-фтор-2-метокси-3-(пирролидин-2-ил)пиридина
Соединение Е-2-1 (2,25 г) в виде твердого вещества растворяли в дихлорметане (20 мл) при -10°С и туда медленно по каплям добавляли раствор HCl в 1,4-диоксане (4 М; 10 мл). После завершения добавления по каплям полученную смесь нагревали до комнатной температуры и перемешивали в течение 10 минут. Большое количество белого твердого вещества осаждали и отфильтровывали, и осадок на фильтре промывали дихлорметаном и затем сушили под вакуумом с получением указанного в заголовке соединения (1,75 г).
1Н ЯМР (400 МГц, DMSO-d6) δ 10.07 (ушир. s, 1H), 9.31 (ушир. s, 1H), 8.18 (d, J=3,2 Гц, 1H), 7.32 (dd, J=8,8, 3,2 Гц, 1H), 4.62 (t, J=7,6 Гц, 1H), 3.89 (s, 3Н), 3.28-3.24 (m, 2Н), 2.30-2.24 (m, 1H), 2.10-1.91 (m, 3Н). m/z составляет 197 [М+1]+.
Промежуточные соединения 3 и 4: синтез (R)-2-(5-фтор-2-метоксифенил)пирролидина (промежуточное соединение 3) и (S)-2-(5-фтор-2-метоксифенил)пирролидина (промежуточное соединение 4)
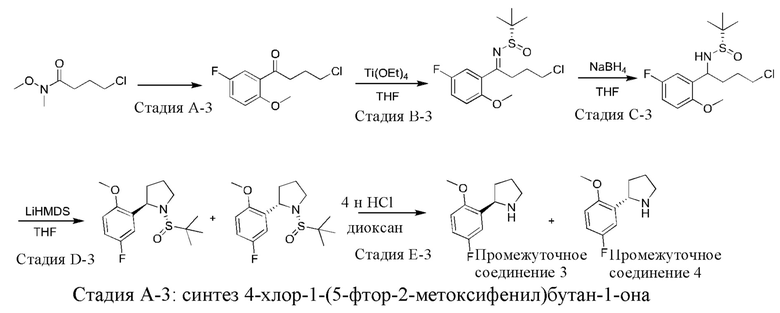
К раствору 2-бром-4-фторанизола (23,5 г) в THF (150 мл) по каплям добавляли раствор хлорида изопропилмагния (2 М) в THF (54 мл) при -50°С. После завершения добавления по каплям реакционную смесь нагревали до комнатной температуры и непрерывно перемешивали в течение 1 ч и затем снова охлаждали до -50°С. В реакционную смесь по каплям добавляли раствор 4-хлор-N-метокси-N-метилбутирамида (9,0 г) в THF (30 мл) при перемешивании. После завершения добавления по каплям полученную смесь постепенно нагревали до 30°С и непрерывно перемешивали при 30°С в течение 2 часов. Затем реакционную смесь гасили насыщенным водным раствором хлорида аммония и экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 20:1 (об./об.)) с получением указанного в заголовке соединения (7,6 г).
1H ЯМР (400 МГц, CDCl3) δ 7.33 (dd, J=9,0, 3,2 Гц, 1H), 7.07-7.02 (m, 1H), 6.83 (dd, J=9,0, 4,0 Гц, 1H), 3.81 (s, 3Н), 3.55 (t, J=6,4 Гц, 2Н), 3.07 (t, J=7,0 Гц, 2Н), 2.12-2.15 (m, 2Н).
Стадии В-3, С-3 и D-3 проводили последовательно в соответствии со стадиями С-2, D-2 и Е-2, которые показаны в способе синтеза промежуточного соединения 2.
Стадия Е-3: синтез (R)-2-(5-фтор-2-метоксифенил)пирролидина
К раствору (R)-1-((S)-трет-бутилсульфинил)-2-(5-фтор-2-метоксифенил)пирролидина (2,8 г) в 1,4-диоксане (25 мл) медленно по каплям добавляли раствор HCl в 1,4-диоксане (4 М; 14 мл) при 0°С. После завершения добавления по каплям полученную смесь нагревали до комнатной температуры и непрерывно перемешивали в течение 1 часа, и затем значение рН доводили до 8 водным раствором NaOH и экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением промежуточного соединения 3 (1,8 г).
Промежуточное соединение 3: 1Н ЯМР (400 МГц, CDCl3) δ 7.19 (dd, J=9,6, 3,0 Гц, 1H), 6.92-6.83 (m, 1H), 6.75 (dd, J=8,8, 4,4 Гц, 1H), 4.38 (t, J=7,6 Гц, 1H), 3.80 (s, 3Н), 3.19-3.14 (m, 1H), 3.10-2.98 (m, 1H), 2.89 (ушир. s, 1H), 2.34-2.12 (m, 1H), 1.96-1.76 (m, 2Н), 1.72-1.52 (m, 1H).
Промежуточное соединение 4: в соответствии со способом стадии Е-3 промежуточное соединение 4 получали при использовании (S)-1-((S)-трет-бутилсульфинил)-2-(5-фтор-2-метоксифенил)пирролидина в качестве исходного вещества.
1Н ЯМР (400 МГц, CDCl3) δ 7.19 (dd, J=9,6, 3,0 Гц, 1H), 6.92-6.83 (m, 1H), 6.75 (dd, J=8,8, 4,4 Гц, 1H), 4.38 (t, J=7,6 Гц, 1H), 3.80 (s, 3Н), 3.19-3.14 (m, 1H), 3.10-2.98 (m, 1H), 2.89 (ушир. s, 1H), 2.34-2.12 (m, 1H), 1.96-1.76 (m, 2Н), 1.72-1.52 (m, 1H).
Промежуточное соединение 5: синтез (R)-2-метокси-3-(пирролидин-2-ил)пиридина

Синтез промежуточного соединения 5 проводили при использовании 2-метокси-3-бромпиридина в качестве исходного вещества в соответствии со способами синтеза промежуточного соединения 3 и промежуточного соединения 4.
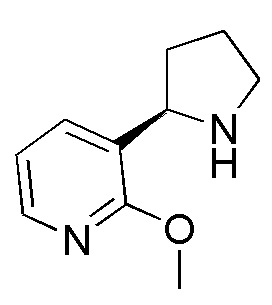
Промежуточное соединение 5: 1Н ЯМР (400 МГц, CDCl3) δ 8.03 (d, J=4,0 Гц, 1H), 7.69 (d, J=7,2 Гц, 1H), 6.85 (dd, J=7,2, 5,2 Гц, 1H), 4.28 (t, J=7,6 Гц, 1H), 3.96 (s, 3Н), 3.14-3.19 (m, 1H), 3.06-3.00 (m, 1H), 2.16-2.24 (m, 2Н), 1.82-1.89 (m, 2Н), 1.60-1.65 (m, 1H).
Промежуточное соединение 6: синтез (R,S)-2-метокси-3-(пирролидин-2-ил)пиридина
Синтез промежуточного соединения 6 проводили при использовании рацемического продукта, полученного на стадии D-5 синтеза промежуточного соединения 5, в качестве исходного вещества в соответствии со стадией Е-5 синтеза.

1Н ЯМР (400 МГц, CDCl3) δ 8.03 (d, J=4,0 Гц, 1H), 7.69 (d, J=7,2 Гц, 1H), 6.85 (dd, J=7,2, 5,2 Гц, 1H), 4.28 (t, J=7,6 Гц, 1H), 3.96 (s, 3Н), 3.14-3.19 (m, 1H), 3.06-3.00 (m, 1H), 2.16-2.24 (m, 2Н), 1.82-1.89 (m, 2Н), 1.60-1.65 (m, 1H).
Промежуточное соединение 7: синтез 2-метокси-6-(пирролидин-2-ил)пиридина
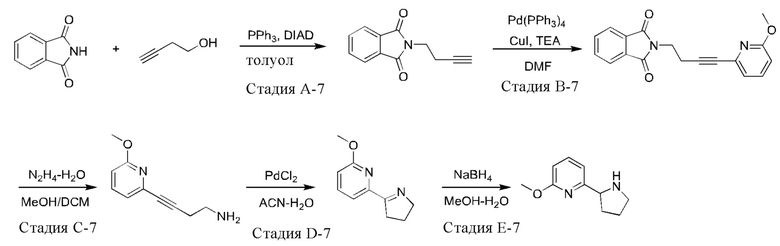
Синтез промежуточного соединения 7 проводили при использовании 2-метокси-6-бромпиридина в качестве исходного вещества в соответствии со способом синтеза промежуточного соединения 1.

Промежуточное соединение 7: 1Н ЯМР (400 МГц, CDCl3) δ 7.50 (t, J=8,0 Гц, 1H), 6.85 (d, J=7,6 Гц, 1H), 6.58 (d, J=8,4 Гц, 1H), 4.14 (t, J=7,2 Гц, 1H), 3.92 (s, 3Н), 3.26-3.20 (m, 1H), 3.01-2.95 (m, 1H), 2.48 (ушир. s, 1H), 2.18-2.13 (m, 1H), 1.89-1.74 (m, 3Н).
Промежуточное соединение 8: синтез (R)-4-метокси-3-(пирролидин-2-ил)пиридина
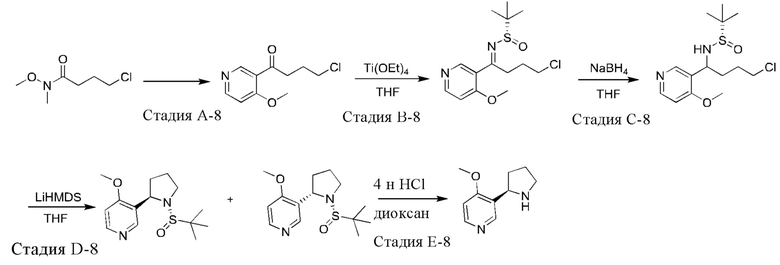
Синтез промежуточного соединения 8 проводили при использовании 3-бром-4-метоксипиридина в качестве исходного вещества в соответствии со способами синтеза промежуточного соединения 3 и промежуточного соединения 4.

Промежуточное соединение 8: 1Н ЯМР (400 МГц, CDCl3) δ 8.51 (s, 1H), 8.40 (d, J=5,6 Гц, 1H), 6.76 (d, J=6,0 Гц, 1H), 4.33 (t, J=7,6 Гц, 1H), 3.88 (s, 3Н), 3.71 (ушир. s, 1H), 3.22-3.16 (m, 1H), 3.05-2.99 (m, 1H), 2.21-2.14 (m, 2Н), 1.91-1.85 (m, 1H), 1.75-1.68 (m, 1H).
Промежуточное соединение 9: синтез 5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)-2-метоксипиридина
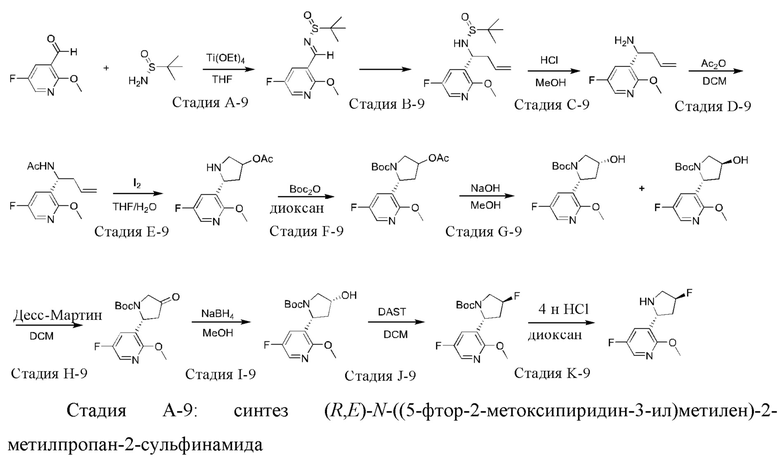
К раствору 5-фтор-2-метоксиникотинальдегида (54 г) в тетрагидрофуране (250 мл) добавляли (R)-трет-бутилсульфинамид (54,8 г) при 0°С и затем в реакционную систему по каплям добавляли тетраэтилтитанат (103,2 г). После завершения добавления по каплям полученную смесь нагревали до комнатной температуры и непрерывно перемешивали в течение 3 часов. После охлаждения реакционной системы до 0°С туда по каплям добавляли насыщенный рассол (80 мл), и полученную смесь непрерывно перемешивали в течение 20 минут и затем фильтровали. Осадок на фильтре промывали дихлорметаном, и промывки и фильтрат объединяли и затем разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 20:1 (об./об.)) с получением указанного в заголовке соединения (88,0 г).
1Н ЯМР (400 МГц, CDCl3) δ 8.89 (d, J=2,4 Гц, 1H), 8.15 (d, J=3,2 Гц, 1H), 7.98 (dd, J=8,0, 2,8 Гц, 1H), 4.01 (s, 3Н), 1.27 (s, 9Н).
Стадия В-9: синтез (R)-N-((R)-1-(5-фтор-2-метоксипиридин-3-ил)бут-3-енил)-2-метилпропан-2-сульфинамида
К раствору (R,E)-N-((5-фтор-2-метоксипиридин-3-ил)метилен)-2-метилпропан-2-сульфинамида (88,0 г) в гексаметилфосфортриамиде (400 мл) последовательно добавляли 3-бромпропилен (59,2 мл), цинковый порошок (44,7 г) и воду (6,15 мл) при 0°С, и после завершения добавления ледяную баню убирали. Полученную смесь нагревали до 25°С и перемешивали в течение ночи, и затем охлаждали до 0°С. В реакционную систему добавляли воду (500 мл) и перемешивали в течение 20 минут. Затем туда добавляли метил-трет-бутиловый эфир (500 мл) и 10% раствор лимонной кислоты (100 мл) и непрерывно перемешивали в течение 30 минут. Полученную смесь фильтровали посредством отсасывания, и фильтрат разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 20:1 (об./об.)) с получением указанного в заголовке соединения (48,4 г).
1Н ЯМР (400 МГц, CDCl3) δ 7.90 (d, J=3,2 Гц, 1H), 7.34 (dd, J=8,0, 2,8 Гц, 1H), 5.69-5.60 (m, 1H), 5.08-5.03 (m, 2Н), 4.50 (dd, J=14,8, 7,2 Гц, 1H), 4.07 (d, J=8,0 Гц, 1H), 3.97 (s, 3Н), 2.65-1.72 (m, 2Н), 1.21 (s, 9Н).
Стадия С-9: синтез (R)-1-(5-фтор-2-метоксипиридин-3-ил)бут-3-ен-1-амин
К раствору (R)-N-((R)-1-(5-фтор-2-метоксипиридин-3-ил)бут-3-енил)-2-метилпропан-2-сульфинамида (48,4 г) в метаноле (250 мл) добавляли раствор HCl в 1,4-диоксане (4 М; 67,5 мл) при комнатной температуре, перемешивали в течение 2 часов и затем концентрировали при пониженном давлении. Остаток выливали в воду (250 мл), и полученную смесь доводили до значения pH 8 насыщенным водным раствором бикарбоната натрия и затем экстрагировали этилацетатом (200 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (40,0 г).
Стадия D-9: синтез (R)-N-(1-(5-фтор-2-метоксипиридин-3-ил)бут-3-енил)-ацетамида К раствору (R)-1-(5-фтор-2-метоксипиридин-3-ил)-бут-3-ен-1-амина (40,0 г) в дихлорметане (200 мл) добавляли пиридин (19,5 мл) и ангидрид уксусной кислоты (16 мл) при 0°C. Полученную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционный раствор выливали в насыщенный водный раствор бикарбоната натрия (500 мл) и экстрагировали дихлорметаном (200 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (37,9 г).
1H ЯМР (400 МГц, CDCl3) δ 7.90 (d, J=2,8 Гц, 1H), 7.30 (dd, J=8,0, 2,8 Гц, 1H), 6.22 (d, J=8,0 Гц, 1H), 5.67-5.58 (m, 1H), 5.13-5.04 (m, 3H), 3.98 (s, 3H), 2.56-2.52 (m, 2H), 2.00 (s, 3H).
Стадия E-9: синтез (5R)-5-(5-фтор-2-метоксипиридин-3-ил)пирролидин-3-ил-ацетата
К раствору (R)-N-(1-(5-фтор-2-метоксипиридин-3-ил)бут-3-енил)ацетамида (37,6 г) в тетрагидрофуране (360 мл) и воде (84 мл) добавляли иод (113,7 г) при комнатной температуре и перемешивали в течение ночи. В реакционный раствор добавляли насыщенный водный раствор бикарбоната натрия (100 мл) и насыщенный водный раствор сульфита натрия (100 мл), и полученную смесь экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (43,0 г).
Стадия F-9: синтез трет-бутил-(2R)-4-ацетокси-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-карбоксилата
К раствору (5R)-5-(5-фтор-2-метоксипиридин-3-ил)пирролидин-3-илацетата (43,0 г) в 1,4-диоксане (250 мл) по каплям добавляли Boc-ангидрид (56,0 мл) и затем водный раствор гидроксида натрия (50 мл) со значением pH 9 при комнатной температуре и перемешивали в течение 3 часов. В реакционную систему добавляли 1 л воды, и полученную смесь экстрагировали этилацетатом (100 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (45,0 г).
Стадия G-9: синтез трет-бутил-(2R,4RS)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата
К раствору трет-бутил-(2R)-4-ацетокси-2-(5-фтор-2-метоксипиридин-3-ил)-пирролидин-1-карбоксилата (45,0 г) в метаноле (500 мл) добавляли раствор гидроксида натрия (2 н; 88 мл) при комнатной температуре и перемешивали в течение 1 часа. Полученную смесь концентрировали при пониженном давлении для удаления растворителя, и к остатку добавляли соляную кислоту (1 н; 180 мл). Затем полученную смесь экстрагировали дихлорметаном (150 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 10:1 (об./об.)) с получением указанного в заголовке соединения (34,0 г).
Стадия Н-9: синтез трет-бутил-(R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-оксопирролидин-1-карбоксилата
трет-Бутил-(2R,4RS)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат (15,1 г) растворяли в дихлорметане (200 мл) и добавляли туда бикарбонат натрия (4,06 г) и затем периодинан Десса-Мартина (92 г) при комнатной температуре и перемешивали в течение ночи. В реакционный раствор добавляли насыщенный водный раствор бикарбоната натрия для доведения значения рН до 7, и полученную смесь экстрагировали дихлорметаном (150 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 10:1 (об./об.)) с получением указанного в заголовке соединения (10,1 г).
1Н ЯМР (400 МГц, CDCl3) δ 7.92 (ушир. s, 1H), 7.30 (ушир. s, 1H), 5.30-5.16 (m, 1H), 4.09-3.88 (m, 5Н), 3.06 (dd, J=18,4, 10,8 Гц, 1H), 2.56 (d, J=18,0 Гц, 1H), 1.47-1.29 (m, 9Н).
Стадия 1-9: синтез трет-бутил-(2R,4R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата
К раствору трет-бутил-(R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-оксопирролидин-1-карбоксилата (14 г) в метаноле (100 мл) порциями добавляли борогидрид натрия (1,42 г) при 0°С, и полученную смесь поддерживали при 0°С и перемешивали в течение 45 минут. В реакционный раствор добавляли насыщенный водный раствор хлорида аммония (100 мл), и полученную смесь постепенно нагревали до комнатной температуры и затем экстрагировали дихлорметаном (150 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 10:1 (об./об.)) с получением указанного в заголовке соединения (13,6 г).
1Н ЯМР (400 МГц, CDCl3) δ 7.84 (ушир. s, 1H), 7.35-7.19 (m, 1H), 5.09-4.92 (m, 1H), 4.44 (d, J=2,8 Гц, 1H), 3.91 (s, 3Н), 3.73-3.70 (m, 1H), 3.62-3.55 (m, 1H), 2.50 (ушир. s, 1H), 1.95 (dd, J=14,0, 1,2 Гц, 1H), 1.57-1.18 (m, 10Н).
Стадия J-9: синтез трет-бутил-(2R,4S)-4-фтор-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-карбоксилата
К раствору трет-бутил-(2R,4R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата (5,1 г) в дихлорметане (50 мл) добавляли трифторид диэтиламиносеры (5,27 г) при -78°С. Полученную смесь поддерживали при -78°С и перемешивали в течение 20 минут и затем постепенно нагревали до комнатной температуры и перемешивали в течение ночи. В реакционную систему добавляли насыщенный водный раствор бикарбоната натрия (100 мл), и полученную смесь экстрагировали дихлорметаном (100 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 10:1 (об./об.)) с получением указанного в заголовке соединения (3,71 г).
1Н ЯМР (400 МГц, CDCl3) δ 7.88 (ушир. s, 1H), 7.23-2.16 (m, 1H), 5.29-5.02 (m, 2Н), 4.13-4.04 (m, 1H), 3.94 (s, 3Н), 3.71-3.58 (m, 1H), 2.84-2.61 (m, 1H), 2.09-1.80 (m, 1H), 1.50-1.12 (m, 9Н).
Стадия К-9: синтез 5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)-2-метоксипиридина
К трет-бутил-(2R,4S)-4-фтор-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-карбоксилату (3,71 г) добавляли раствор хлористого водорода в 1,4-диоксане (4 М; 25 мл), и перемешивали при комнатной температуре в течение 30 минут. Полученную смесь доводили до значения рН 8 насыщенным водным раствором гидроксида натрия и затем экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (3,0 г).
1Н ЯМР (400 МГц, CDCl3) δ 7.85 (d, J=3,2 Гц, 1H), 7.62 (dd, J=8,8, 3,2 Гц, 1H), 5.33-5.17 (m, 1H), 4.61 (t, J=7,2 Гц, 1H), 3.94 (s, 3Н), 3.35 (dd, J=25,6, 13,6 Гц, 1H), 3.23-3.10 (m, 1H), 3.71-2.60 (m, 2Н), 1.74-0.96 (m, 1H).
Промежуточное соединение 10: синтез этил-2-амино-5-хлорпиразоло[1,5-a]пиримидин-3-карбоксилата

К раствору этилцианоацетата (41,22 г) и трихлорацетонитрила (100 г) в этаноле (120 мл) по каплям добавляли триэтиламин (2,0 г) при 0°С. Полученную смесь перемешивали при 0°С в течение 2 часов и затем медленно нагревали до комнатной температуры и непрерывно перемешивали в течение 30 минут. Полученную смесь концентрировали для удаления растворителя, и остаток очищали посредством колоночной хроматографии на силикагеле (элюирование дихлорметаном) с получением указанного в заголовке соединения (93,0 г).
1Н ЯМР (400 МГц, CDCl3) δ 10.20 (ушир. s, 1H), 6.93 (ушир. s, 1H), 4.30 (q, J=7,2 Гц, 2Н), 1.33 (t, J=7,2 Гц, 3Н).
Стадия В-10: синтез этил-3,5-диамино-1H-пиразол-4-карбоксилата
К раствору этил-(Z)-3-амино-4,4,4-трихлор-2-циано-бутеноата (92,1 г) в DMF (250 мл) медленно по каплям добавляли гидразин-гидрат (50 г; концентрация 80%). Реакционную смесь нагревали до 100°С и перемешивали в течение 1,5 часа и затем концентрировали для удаления растворителя. Остаток суспендировали с дихлорметаном и затем фильтровали посредством отсасывания. Твердое вещество промывали дихлорметаном и сушили с получением указанного в заголовке соединения (41,0 г).
1Н ЯМР (400 МГц, DMSO-d6) δ 10.4 (ушир. s, 1H), 5.35 (ушир. s, 4Н), 4.13 (q, J=7,2 Гц, 2Н), 1.24 (t, J=7,2 Гц, 3Н).
Стадия С-10: синтез этил-2-амино-5-оксо-4,5-дигидропиразоло[1,5-a]пиримидин-3-карбоксилата
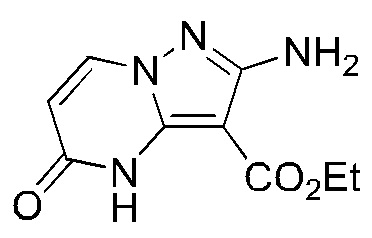
К раствору этилата натрия (33,2 г) в этаноле (500 мл) последовательно добавляли этил-3,5-диамино-1H-пиразол-4-карбоксилат (20,8 г) и 1,3-диметилпиримидин-2,4(1H,3H)-дион (17,0 г) при комнатной температуре. Реакционный раствор перемешивали при 90°С в течение 12 часов, охлаждали до комнатной температуры, доводили до значения рН 7 1 н соляной кислотой и затем фильтровали. Твердое вещество промывали этанолом и затем сушили с получением указанного в заголовке соединения (18,4 г).
1Н ЯМР (400 МГц, DMSO-d6) δ 11.17 (ушир. s, 1H), 8.24 (d, J=8,0 Гц,1Н), 5.93 (s, 2Н), 5.90 (d, J=8,0 Гц,1Н), 4.26 (q, J=7,2 Гц, 2Н), 1.27 (t, J=7,2 Гц, 3Н).
Стадия D-10: синтез этил-2-амино-5-хлорпиразоло[1,5-a]пиримидин-3-карбоксилата
К раствору этил-2-амино-5-оксо-4,5-дигидропиразоло[1,5-a] пиримидин-3-карбоксилата (33,6 г) в ацетонитриле (500 мл) добавляли хлорангидрид фосфорной кислоты (110 мл) при комнатной температуре. Полученную смесь нагревали до 40°С, и непрерывно перемешивали в течение 5 часов, и затем охлаждали до комнатной температуры, и концентрировали при пониженном давлении. К остатку добавляли насыщенный водный раствор бикарбоната натрия (250 мл), и полученную смесь экстрагировали этилацетатом (200 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : этилацетат : петролейный эфир, 2:1 (об./об.)) с получением указанного в заголовке соединения (4,5 г).
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, J=7,2 Гц, 1H), 6.80 (d, J=7,2 Гц, 1H), 5.51 (ушир. s, 2Н), 4.43 (q, J=7,2 Гц, 2Н), 1.44 (г, J=7,2 Гц, 3Н).
Промежуточное соединение 11: синтез 5-фтор-3-((2R,4R)-4-фторпирролидин-2-ил)-2-метоксипиридина
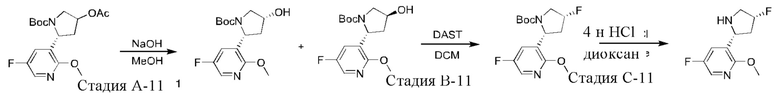
Промежуточное соединение 11 синтезировали при использовании трет-бутил-(2R,4S)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата, полученного на стадии G-9 синтеза промежуточного соединения 9, в качестве исходного вещества в соответствии со стадией J-9 и стадией К-9 синтеза промежуточного соединения 9.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.45 (ушир. s, 1H), 8.13 (d, J=2,8 Гц, 1H), 7.93 (dd, J=9,2, 2,8, Гц, 1H), 5.60-5.42 (m, 1H), 4.87 (d, J=8,4 Гц, 1H), 3.90 (s, 3Н), 3.58-3.36 (m, 2Н), 2.77-2.60 (m, 1H), 2.49-2.36 (m, 1H).
Промежуточное соединение 12: синтез (3R,5R)-5-(5-фтор-2-метоксипиридин-3-ил)-пирролидин-3-ола
Промежуточное соединение 12 синтезировали при использовании трет-бутил-(2R,4R)-2-(5-фтор-2-метоксипиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата, полученного на стадии G-9 синтеза промежуточного соединения 9, в качестве исходного вещества в соответствии со стадией К-9 синтеза промежуточного соединения 9.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.20 (s, 1H), 9.28 (s, 1H), 8.21 (d, J=3,2 Гц, 1H), 8.01 (dd, J=3,2, 9,2 Гц, 1H), 4.80 (t, J=7,2 Гц, 1H), 4.48-4.53(m, 1H), 3.91 (s, 3Н), 3.29-3.34 (m, 1H), 3.01-3.18 (m, 1H), 2.46-2.55 (m, 1H), 2.07-2.14 (m, 1H).
Промежуточное соединение 13: синтез 4-фтор-2-((2R,4S)-4-фторпирролидин-2-ил)фенола
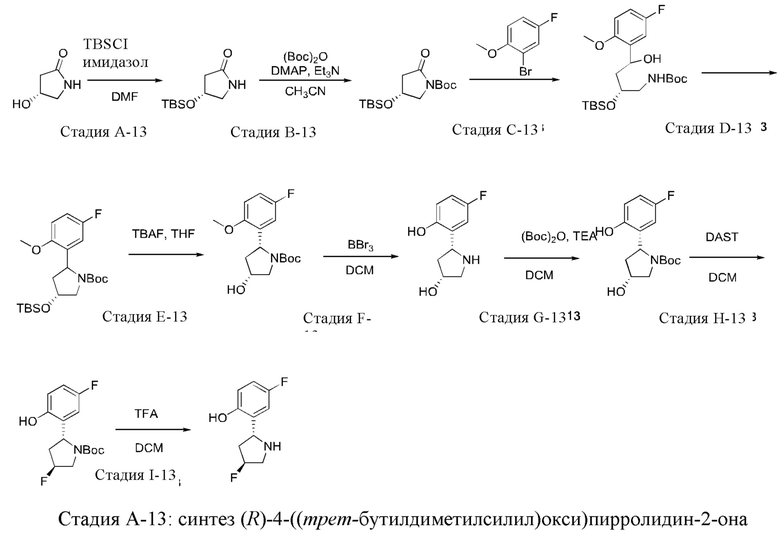
(R)-4-Гидрокси-2-пирролидон (6,0 г) растворяли в DMF (60 мл) и туда добавляли TBDMSCI (9,8 г) и имидазол (6,05 г) при 0°С. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. После контроля завершения взаимодействия в реакционную систему добавляли воду. Твердое вещество осаждали, фильтровали и сушили в течение ночи под лампой инфракрасного излучения с получением указанного в заголовке соединения (10,7 г).
1Н ЯМР (400 МГц, CDCl3) δ 7.45 (s, 1H), 4.44 (m, 1H), 3.42 (m, 1H), 2.93 (m, 1H), 2.40 (m, 1H), 1.85 (m, 1H), 0.79 (s, 9Н), 0.00 (s, 6Н).
Стадия В-13: синтез трет-бутил-(R)-4-((трет-бутилдиметилсилил)окси)-2-оксопирролидин-1-карбоксилата
К раствору (R)-4-((трет-бутилдиметилсилил)окси)пирролидин-2-она (10,67 г) в ацетонитриле добавляли триэтиламин (8,26 мл) и DMAP (3,0 г) при 0°С и затем по каплям добавляли (Вос)2О (15 мл) в защитной атмосфере азота. После завершения добавления, реакционную смесь перемешивали в течение 5 минут и затем нагревали до комнатной температуры и непрерывно перемешивали в течение ночи. Реакционную систему выливали в воду, и полученную смесь экстрагировали этилацетатом и затем очищали посредством колоночной хроматографии (РЕ/ЕА, 10/1) с получением указанного в заголовке соединения (14,5 г).
1Н ЯМР (400 МГц, CDCl3) δ 4.38-4.40 (m, 1H), 3.86 (dd, J=11,4, 5,6 Гц, 1H), 3.62 (dd, J=11,4, 3,2 Гц, 1H), 2.71 (dd, J=15,6, 5,6 Гц, 1H), 2.48 (dd, J=3,4, 5,6 Гц, 1H), 1.56 (s, 9Н), 0.89 (m, 9Н), 0.08 (m, 6Н).
Стадия С-13: синтез трет-бутил-(2R)-2-((трет-бутилдиметилсилил)окси)-4-(5-фтор-2-метоксифенил)-4-гидроксибутилкарбамата
5-Фтор-2-метокси-бромбензол (9,75 г) растворяли в безводном тетрагидрофуране и охлаждали до 0°С, и затем туда добавляли хлорид изопропилмагния (2 М; 23,5 мл). Реакционную систему нагревали до 70°С и перемешивали в течение 2 часов, и затем снова охлаждали до 0°С. В реакционную систему добавляли раствор трет-бутил-(R)-4-((трет-бутилдиметилсилил)окси)-2-оксопирролидин-1-карбоксилата (15,0 г) в тетрагидрофуране, снова нагревали до комнатной температуры и перемешивали в течение 2 часов. Добавляли метанол и затем борогидрид натрия (1,78 г) и перемешивали в течение 2 часов. После завершения взаимодействия реакционную смесь гасили насыщенным раствором хлорида аммония. Полученную смесь экстрагировали этилацетатом и затем очищали посредством колоночной хроматографии (РЕ/ЕА, 13/1) с получением указанного в заголовке соединения (4,2 г).
Стадия D-13: синтез трет-бутил-(4R)-4-(трет-бутилдиметилсилилокси)-2-(5-фтор-2-метоксифенил)пирролидин-1-карбоксилата
Трет-бутил-(2R)-2-((трет-бутилдиметилсилил)окси)-4-(5-фтор-2-метоксифенил)-4-гидроксибутилкарбамат (4,2 г) растворяли в дихлорметане и охлаждали до -60°С, и туда по каплям добавляли триэтиламин (3,95 мл) и метилсульфонилхлорид (0,807 мл), и перемешивали в течение 1 часа при поддержании той же температуры. Затем добавляли DBU (2,1 мл), и полученную смесь нагревали до комнатной температуры и непрерывно перемешивали в течение 3 часов. После контроля завершения взаимодействия реакционную систему выливали в воду и экстрагировали дихлорметаном (50 мл × 3). Органическую фазу промывали насыщенным рассолом, сушили над сульфатом натрия и затем очищали посредством колоночной хроматографии (РЕ/ЕА, 15/1) с получением указанного в заголовке соединения (3,18 г).
Стадия Е-13: синтез трет-бутил-(2R,4R)-2-(5-фтор-2-метоксифенил)-4-гидроксипирролидин-1-карбоксилата
трет-Бутил-(4R)-4-(трет-бутилдиметилсилилокси)-2-(5-фтор-2-метоксифенил)-пирролидин-1-карбоксилат (3,18 г) растворяли в тетрагидрофуране, и затем туда добавляли тригидрат фторида тетрабутиламмония (3,5 г) при 0°С, и перемешивали в течение 1 часа. После контроля завершения взаимодействия реакционную систему выливали в ледяную воду, и полученную смесь экстрагировали этилацетатом (×2) и затем очищали посредством колоночной хроматографии (градиентное элюирование элюентом: РЕ/ЕА, 20/1-10/1-1/1 (об./об.)) с получением указанного в заголовке соединения (1,2 г).
Стадия F-13: синтез (3R,5R)-5-(5-фтор-2-гидроксифенил)пирролидин-3-ола
К раствору трет-бутил-(2R,4R)-2-(5-фтор-2-метоксифенил)-4-гидроксипирролидин-1-карбоксилата (600 мг) в дихлорметане по каплям добавляли раствор трибромида бора (0,746 мл) при 0°С, нагревали до комнатной температуры и перемешивали в течение ночи. После контроля завершения взаимодействия посредством ЖХ-МС и ТСХ реакционную систему выливали в ледяную воду и затем экстрагировали смешанным растворителем DCM/iPrOH, 3/1 (v/v) (150 мл × 3). Органическую фазу сушили над сульфатом натрия и фильтровали. Растворитель удаляли из фильтрата с получением (3R,5R)-5-(5-фтор-2-гидроксифенил)пирролидин-3-ола (391 мг).
1Н ЯМР (400 МГц, CDCl3) δ 6.83-6.95 (m, 2Н), 6.63-6.66 (m, 1H), 4.26-4.33 (m, 2Н), 3.32 (s, 1H), 3.01-3.04 (m, 1H), 2.76-2.79 (m, 1H), 2.38-2.45 (m, 1H), 1.55-1.62 (m, 1H).
Стадия G-13: синтез трет-бутил-(2R,4R)-2-(5-фтор-2-гидроксифенил)-4-гидроксипирролидин-1-карбоксилата
К раствору (3R,5R)-5-(5-фтор-2-гидроксифенил)пирролидин-3-ола (391 мг) в дихлорметане по каплям добавляли Boc2O (476 мг) и триэтиламин (602 мг) при комнатной температуре и перемешивали в течение ночи при комнатной температуре. После контроля завершения взаимодействия растворитель удаляли, и остаток очищали посредством колоночной хроматографии (градиентное элюирование элюентом: РЕ/ЕА, 3/1-1/1 (v/v)) с получением указанного в заголовке соединения (330 мг).
1Н ЯМР (400 МГц, CDCl3) δ 8.60 (s, 1H), 7.16 (s, 1H), 6.72-6.76 (m, 1H), 6.62-6.66 (m, 1H), 5.15 (s, 1H), 4.55 (s, 1H), 3.77-3.81 (m, 1H), 3.53-3.56 (m, 1H), 2.58-2.66 (m, 1H), 2.04-2.15 (m, 1H), 1.41 (s, 9H).
Стадия Н-13: синтез трет-бутил-(2R,4S)-4-фтор-2-(5-фтор-2-гидроксифенил)-пирролидин-1-карбоксилата
К раствору трет-бутил-(2R,4R)-2-(5-фтор-2-гидроксифенил)-4-гидроксипирролидин-1-карбоксилата (330 мг) в дихлорметане по каплям добавляли реагент DAST (359 мг) при -78°С и перемешивали в течение 2 часов при поддержании той же температуры, и затем постепенно нагревали до комнатной температуры, и перемешивали в течение ночи. Реакцию останавливали посредством добавления насыщенного раствора бикарбоната натрия при 0°С, и полученную смесь экстрагировали дихлорметаном (200 мл × 2). Органическую фазу промывали насыщенным рассолом, сушили над сульфатом натрия и затем очищали посредством колоночной хроматографии (элюент: РЕ/ЕА, 7/1 (v/v)) с получением указанного в заголовке соединения (145 мг).
1Н ЯМР (400 МГц, CDCl3) δ 6.82-7.02 (m, 1H), 6.62-6.80 (m, 2Н), 5.10-5.40 (m, 2Н), 3.90-4.20 (m, 2Н), 3.81-3.50 (m, 1H), 3.60-3.78 (m, 1H), 2.18-2.6 (m, 1H), 1.35 (s, 9Н).
Стадия I-13: синтез 4-фтор-2-((2R,4S)-4-фторпирролидин-2-ил)фенола
К раствору трет-бутил-(2R,4S)-4-фтор-2-(5-фтор-2-гидроксифенил)-пирролидин-1-карбоксилата (145 мг) в дихлорметане добавляли 4 н раствор хлористого водорода в 1,4-диоксане (3 мл) при комнатной температуре и перемешивали в течение 1 часа. После контроля завершения взаимодействия растворитель удаляли с получением 4-фтор-2-((2R,4S)-4-фторпирролидин-2-ил)фенола (100 мг) без дальнейшей очистки.
Соединения по примерам
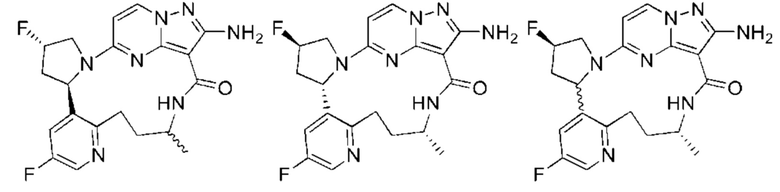
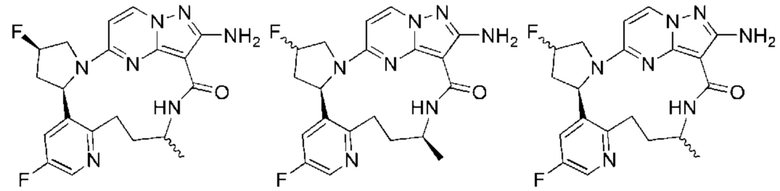
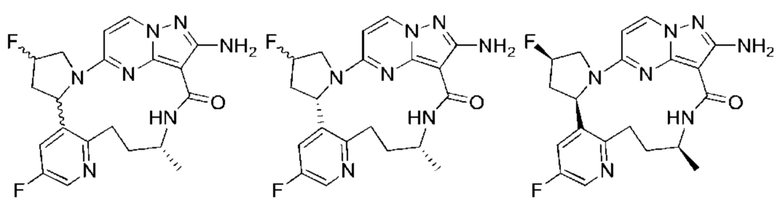
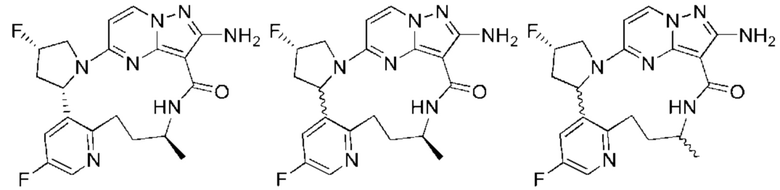
Пример 1: синтез (13E,14E)-12-амино-35-фтор-4-окса-7-аза-1(5,3)-пиразоло[1,5-a]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидилциклооктанфан-8-она
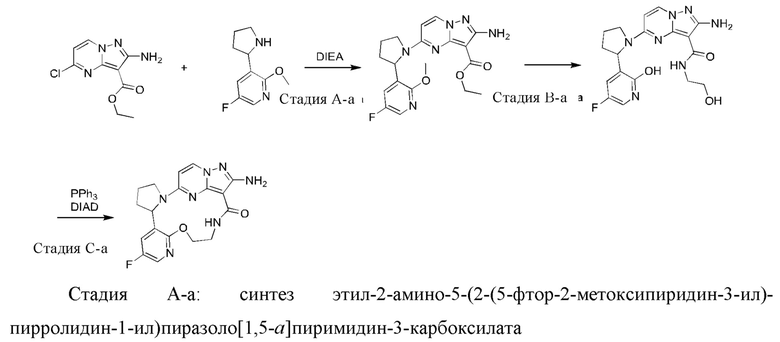
К раствору этил-2-амино-5-хлорпиразоло[1,5-a]пиримидин-3-карбоксилата (3,3 г) и 2-(2-метокси-5-фторпиридил)пирролидина (3,0 г) в н-бутаноле (50 мл) в реакционной пробирке добавляли DIEA (5,39 г), и пробирку герметизировали. Реакционную смесь нагревали до 160°С и подвергали взаимодействию в течение 5 часов, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении для удаления растворителя. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : петролейный эфир : этилацетат, 4:1 (об./об.)) с получением указанного в заголовке соединения (5,2 г).
1Н ЯМР (400 МГц, CDCl3) δ 7.89-8.07 (m, 2Н), 7.04 (s, 1H), 5.63 (s, 1H), 5.22-5.30 (m, 2Н), 5.03 (m, 1H), 3.50-4.50 (m, 7Н), 2.45 (s, 1H), 1.86-2.04 (m, 3Н), 1.40-1.48 (m, 2Н), 1.13-1.20 (m, 1H).
Стадия В-а: синтез 2-амино-5-(2-(5-фтор-2-гидроксипиридин-3-ил)пирролидин-1-ил)-N-(2-гидроксиэтил)пиразоло[1,5-a]пиримидин-3-карбоксамида
К раствору этил-2-амино-5-(2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-ил)-пиразоло[1,5-a]пиримидин-3-карбоксилата (600 мг) в н-бутаноле (3 мл) в реакционной пробирке добавляли этаноламин (4 мл), и пробирку герметизировали. Реакционную смесь нагревали до 160°С и подвергали взаимодействию в течение 16 часов и затем концентрировали при пониженном давлении для удаления растворителя. Остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (263 мг).
Стадия С-а: (13Е,14E)-12-амино-35-фтор-4-окса-7-аза-1(5,3)-пиразоло[1,5-a]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидилциклооктанфан-8-она
К раствору трифенилфосфина (344 мг) и 2-амино-5-(2-(5-фтор-2-гидроксипиридин-3-ил)пирролидин-1-ил)-N-(2-гидроксиэтил)пиразоло[1,5-a]пиримидин-3-карбоксамид а (263 мг) в тетрагидрофуране (10 мл) добавляли диизопропилазодикарбоксилат (265 мг) при 0°С нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан: метанол, 20:1) с получением указанного в заголовке соединения (66,0 мг).
1Н ЯМР (400 МГц, CDCl3) δ 8.85-8.93 (ушир. s, 1H), 7.99 (d, J=7,6 Гц, 1H), 7.85 (d, J=3,2 Гц, 1H), 7.23 (dd, J=8,4, 2,8 Гц, 1H), 6.02 (d, J=7,2 Гц, 1H), 5.66-5.70 (m, 1H), 5.12-5.21 (ушир. s, 2Н), 5.06-5.11 (m, 1H), 4.32-4.38 (m, 1H), 3.83-3.95 (m 2Н), 3.64-3.69 (m 2Н), 2.35-2.57 (m, 2Н), 2.16-2.27 (m, 1H), 1.91-1.99 (m 1H).
Следующие соединения по примерам синтезировали в соответствии со способом, который описан в примере 1.


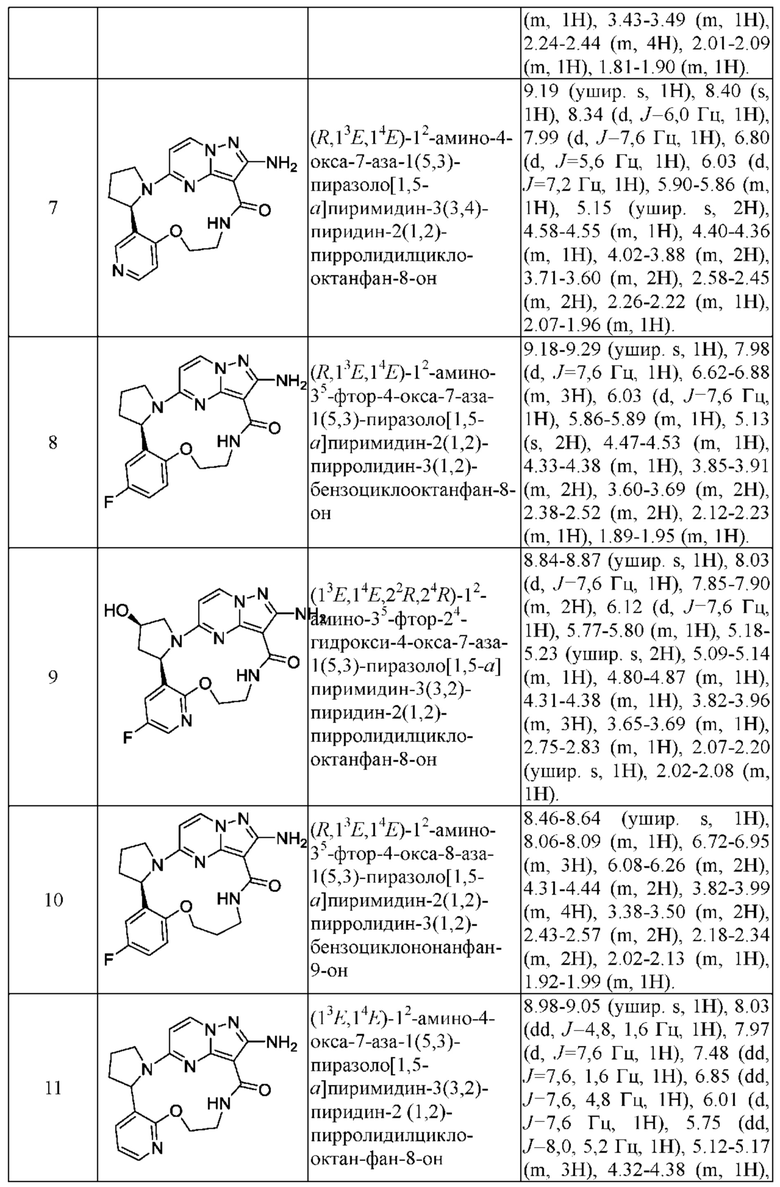

Пример 12: синтез (13E,14E,22R,24S)-12-амино-24,35-дифтор-4-окса-8-аза-1(5,3)-пиразоло [1,5-a]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидилциклононанфан-9-она
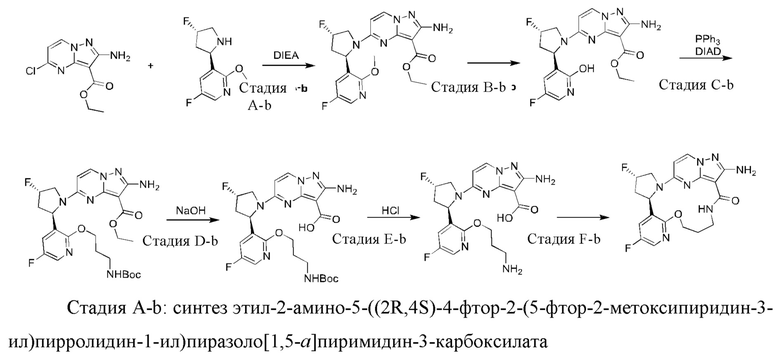
Проводили в соответствии со стадией А-а примера 1, но 2-(2-метокси-5-фторпиридил)пирролидин был заменен на 5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)-2-метоксипиридин.
Стадия В-b: синтез этил-2-амино-5-((2R,4S)-4-фтор-2-(5-фтор-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата
К этил-2-амино-5-((2R,4S)-4-фтор-2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоксилату (1,0 г) добавляли раствор HCl в 1,4-диоксане (3,5 М; 4 мл). Реакционную смесь нагревали до 100°С и проводили взаимодействие в герметичной пробирке в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением указанного в заголовке соединения (0,6 г).
Стадия С-b: синтез этил-2-амино-5-((2R,4S)-2-(2-(3-(трет-бутоксикарбониламино)пропокси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоксилата
К раствору трифенилфосфина (778 мг) в тетрагидрофуране (15 мл) добавляли диизопропилазодикарбоксилат (600 мг) при 0°С и перемешивали в течение 20 минут, и затем туда добавляли этил-2-амино-5-((2R,4S)-4-фтор-2-(5-фтор-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоксилат (400 мг). Полученную смесь нагревали до комнатной температуры и перемешивали в течение ночи, и затем концентрировали при пониженном давлении для удаления растворителя. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент : дихлорметан : метанол, 20:1 (об./об.)) с получением указанного в заголовке соединения (250 мг).
Стадия D-b: синтез 2-амино-5-((2R,4S)-2-(2-(3-(трет-бутоксикарбонил)пропокси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты
К раствору этил-2-амино-5-((2R,4S)-2-(2-(3-(трет-бутоксикарбониламино)пропил)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (375 мг) в метаноле (10 мл) добавляли насыщенный водный раствор гидроксида натрия (2 мл), нагревали до 80°С и перемешивали в течение 3 часов. После удаления метанола полученную смесь доводили до значения рН меньше 5 разбавленной соляной кислотой и затем экстрагировали смешанным растворителем дихлорметан/изопропанол (v/v, 3/1) (50 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (310 мг).
Стадия Е-b: синтез 2-амино-5-((2R,4S)-2-(2-(3-аминопропокси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоновой кислоты
2-Амино-5-((2R,4S)-2-(2-(3-(трет-бутоксикарбонил)пропил)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоновую кислоту (310 мг) растворяли в дихлорметане (100 мл) и затем туда добавляли раствор HCl в 1,4-диоксане (4 М; 30 мл). Полученную смесь подвергали взаимодействию в течение 10 минут и затем концентрировали при пониженном давлении с получением указанного в заголовке соединения (205 мг).
Стадия F-b: синтез (13E,14E,22R,24S)-12-амино-24,35-дифтор-4-окса-8-аза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидилциклононанфан-9-она
2-Амино-5-((2R,4S)-2-(2-(3-аминопропокси)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоновую кислоту (20 мг) растворяли в DMF (2 мл) и затем туда добавляли пентафторфенилдифенилфосфат (20 мг) и диизопропилэтиламин (31 мг) при 0°С. Полученную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционный раствор выливали в воду (50 мл) и затем экстрагировали этилацетатом (25 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан : метанол, 20:1) с получением указанного в заголовке соединения (8,2 мг).
1Н ЯМР (400 МГц, CDCl3) δ 8.05 (d, J=7,6 Гц, 1H), 7.91-7.98 (m, 1H), 7.86 (d, J=2,8 Гц, 1H), 7.13 (dd, J=8,4, 2,8 Гц, 1H), 6.02 (d, J=7,6 Гц, 1H), 5.83-5.88 (m, 1H), 5.41-5.55 (m, 3Н), 5.23-5.28 (m, 1H), 3.97-4.24 (m, 3Н), 3.40-3.48 (m, 1H), 2.80-2.91 (m, 1H), 2.26-2.36 (m, 1H), 1.90-2.09 (m, 3Н).
Следующие соединения по примерам синтезировали в соответствии со способом, который описан в примере 12.
Пример 20: синтез (S,13E,14E)-12-амино-35-фтор-4-окса-7-аза-1(5,3)-пиразоло[1,5-а]пиримидин-2(1,2)-пирролидин-3(1,2)-бензоциклооктанфан-8-она
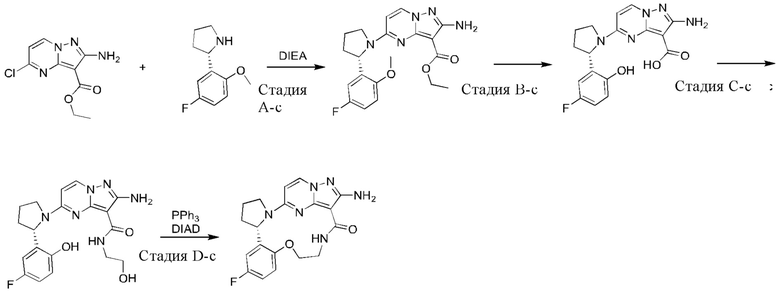
Стадия А-с: синтез этил-(S)-2-амино-5-(2-(5-фтор-2-метоксифенил)пирролидин-1-ил)-пиразоло[1,5-а] пиримидин-3-карбоксилата
Стадию А-с проводили в соответствии со стадией А-а примера 1.
1Н ЯМР (400 МГц, CDCl3) δ 7.81 (d, J=6,4, 1H), 6.91-6.71 (m, 3Н), 5.64 (d, J=6,4, 1H), 5.20-5.14 (m, 3Н), 4.38 (m, 2Н), 4.06-3.94 (m, 2Н), 3.88 (s, 3Н), 2.44 (m, 1H), 2.05-1.96 (m, 3Н), 1.46 (m, 3Н).
Стадия В-с: синтез (S)-2-амино-5-(2-(5-фтор-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты
К раствору этил-(S)-2-амино-5-(2-(5-фтор-2-метоксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (160 мг) в дихлорметане (10 мл) добавляли трибромид бора (193 мкл) при 0°С, нагревали до комнатной температуры и перемешивали в течение ночи. Смесь доводили до основных значений рН (рН 8) насыщенным водным раствором бикарбоната натрия, и затем доводили до значения рН 4 1 н раствором соляной кислоты, и затем экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (95,3 мг).
Стадия С-с: синтез (S)-2-амино-5-(2-(5-фтор-2-гидроксифенил)пирролидин-1-ил)-N-(2-гидроксиэтил)пиразоло[1,5-а]пиримидин-3-карбоксамида
К раствору (S)-2-амино-5-(2-(5-фтор-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (160 мг) и этаноламина (72 мг) в DMF (8 мл) добавляли EDCI (224 мг), HOBt (158 мг) и триэтиламин (272 мкл) при 0°С, нагревали до комнатной температуры и перемешивали в течение ночи. Затем добавляли воду (10 мл), и полученную смесь экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: дихлорметан : метанол, 20:1 (об./об.)) с получением указанного в заголовке соединения (66 мг).
Стадия D-c: синтез (S,13E,14E)-12-амино-35-фтор-4-окса-7-аза-1(5,3)-пиразоло[1,5-а]пиримидин-2(1,2)-пирролидин-3(1,2)-бензоциклооктанфан-8-она

Стадию D-c проводили в соответствии со стадией С-а примера 1.
1Н ЯМР (400 МГц, CDCl3) δ 9.13-9.20 (ушир. s, 1H), 7.92 (d, J=7,6 Гц, 1H), 6.75-6.81 (m, 3Н), 5.96 (d, J=7,2 Гц, 1H), 5.80-5.83 (m, 1H), 5.07 (s, 2Н), 4.41-4.47 (m, 1H), 4.26-4.31 (m, 1H), 3.78-3.85 (m, 2Н), 3.55-3.61 (m, 2Н), 2.33-2.43 (m, 2Н), 2.08-2.15 (m, 1H), 1.83-1.89 (m, 1H).
Синтез соединений по примеру 21 и примеру 22
Пример 21: (R,13E,14E)-12-амино-35-фтор-4-окса-8-аза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидилциклононанфан-9-он
Пример 22: (13Е,14Е,22R)-12-амино-35-фтор-31,32-дигидро-7-аза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,1)-пиридин-2(1,2)-пирролидилциклооктанфан-32,8-дион
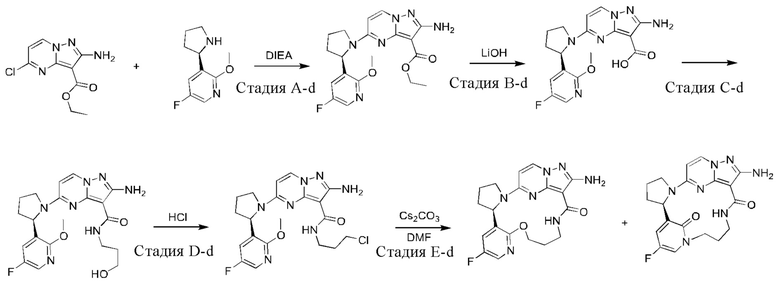
Стадия A-d: синтез этил-(R)-2-амино-5-(2-(5-фтор-2-метоксипиридин-3-ил)-пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата
Стадию A-d проводили в соответствии со стадией А-а примера 1.
1Н ЯМР (400 МГц, CDCl3) δ 8.20-7.81 (m, 2Н), 7.05 (ушир. s, 1H), 6.22-5.48 (m, 1H), 5.23 (ушир. s, 2Н), 5.03 (ушир. s, 1H), 4.45-4.26 (m, 1H), 4.20-3.42 (m, 6Н), 2.45 (ушир. s, 1H), 2.10-1.81 (m, 3Н), 1.45 (ушир. s, 2Н), 1.23 (ушир. s, 1H). m/z составляет 401 [М+1]+.
Стадия B-d: синтез (R)-2-амино-5-(2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-a] пиримидин-3 -карбоновой кислоты
В смесь этил-(R)-2-амино-5-(2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (896 мг), воды (2 мл) и метанола (10 мл) добавляли моногидрат гидроксида лития (500 мг) при комнатной температуре, нагревали до 80°С и нагревали с обратным холодильником в течение 5 часов. Полученную смесь концентрировали при пониженном давлении для удаления метанола и к остатку добавляли воду (20 мл). Полученную смесь доводили до значения рН 2 1 н водным раствором соляной кислоты, и большое количество белого твердого вещества осаждали и затем фильтровали. Твердое вещество сушили под вакуумом с получением указанного в заголовке соединения (656 мг).
Стадия C-d: синтез (R)-2-амино-5-(2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-(3-гидроксипропил)пиразоло[1,5-а]пиримидин-3-карбоксамида
К раствору (R)-2-амино-5-(2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (325 мг) и 3-аминопропанола (100 мг) в безводном DMF (9 мл) последовательно добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (502 мг), 1-гидроксибензотриазол (354 мг) и триэтиламин (441 мг) в защитной атмосфере азота. После завершения добавления полученную смесь перемешивали при комнатной температуре в течение ночи, и затем туда добавляли воду (20 мл) и этилацетат (100 мл), и перемешивали в течение 5 минут. Затем полученную смесь разделяли, и органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения (137 мг).
Стадия D-d: синтез (R)-2-амино-N-(3-хлорпропил)-5-(2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида
Смесь (R)-2-амино-5-(2-(5-фтор-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-(3-гидроксипропил)пиразоло[1,5-а]пиримидин-3-карбоксамида (130 мг) и раствор HCl в 1,4-диоксане (4 М; 6 мл) герметизировали в пробирке под давлением, нагревали до 100°С и проводили взаимодействие непрерывно в течение 1 часа. Полученную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении для удаления 1, 4-диоксана с получением указанного в заголовке соединения (110 мг).
Стадия E-d: синтез (R,13E,14E)-12-амино-35-фтор-4-окса-8-аза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидилциклононанфан-9-она и (13E,14E,22R)-12-амино-35-фтор-31,32-дигидро-7-аза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,1)-пиридин-2(1,2)-пирролидилциклооктанфан-32,8-диона
Смесь (R)-2-амино-N-(3-хлорпропил)-5-(2-(5-фтор-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (110 мг), карбоната цезия (414 мг) и DMF (6 мл) нагревали до 80°С, и перемешивали в течение 6 часов, и туда добавляли воду (20 мл) и этилацетат (100 мл), и перемешивали в течение 5 минут. Полученную смесь разделяли, и органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством колоночной хроматографии на силикагеле (элюент: дихлорметан : метанол, 20:1 (об./об.)) с получением двух указанных в заголовке соединений (15 мг), соответственно.
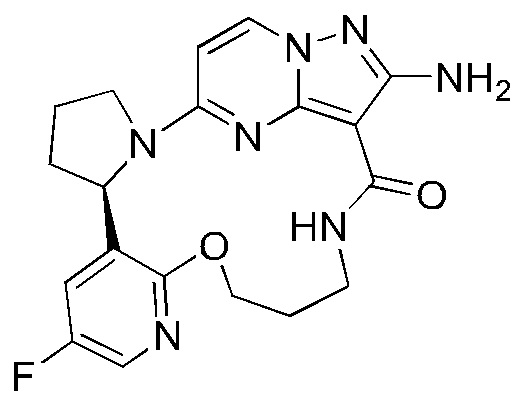
1H ЯМР (400 МГц, CDCl3) δ 7.99 (d, J=7,6 Гц, 2Н), 7.84 (ушир. s, 1H), 7.14 (d, J=8,4 Гц, 1H), 6.02 (d, J=7,6 Гц, 1H), 5.76-5.74 (m, 1H), 5.41 (ушир. s, 2Н), 5.24-5.20 (m, 1H), 4.20 (d, J=8,8 Гц, 1H), 4.02-3.95 (m, 1H), 3.92-3.86 (m, 1H), 3.65 (dd, J=17,2, 7,6 Гц, 1H), 3.46 (t, J=6,8 Гц, 1H), 2.49-2.19 (m, 4Н), 2.061.95 (m, 1H), 1.90-1.83 (m, 1H). m/z составляет 398 [М+1]+.
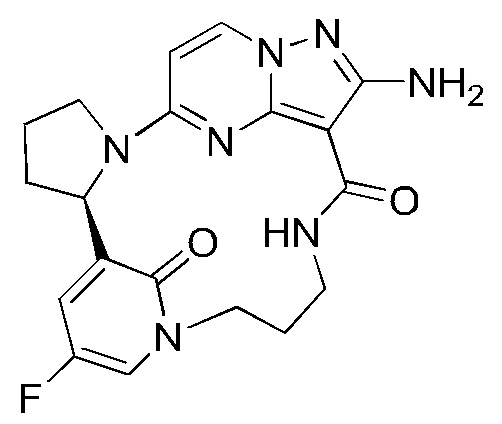
1Н ЯМР (400 МГц, CDCl3) δ 7.97 (d, J=7,6 Гц, 1H), 7.37 (ушир. s, 1H), 7.05-6.98 (ушир. s, 2Н), 6.01 (d, J=7,6 Гц, 1H), 5.51 (t, J=7,2 Гц, 1H), 5.41 (ушир. s, 2Н), 5.07 (t, J=12,4 Гц, 1H), 4.39-4.31 (m, 1H), 3.81-3.65 (m, 2Н), 3.43-3.31 (m, 2Н), 2.56-2.53 (m, 1H), 2.39-1.98 (m, 3Н), 1.94-1.76 (m, 2Н). m/z составляет 398 [М+1]+.
Пример 23: синтез (13E,14E,22R,24S)-12-амино-24,35-дифтор-31,32-дигидро-7-аза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,1)-пиридин-2(1,2)-пирролидилциклооктанфан-32,8-диона
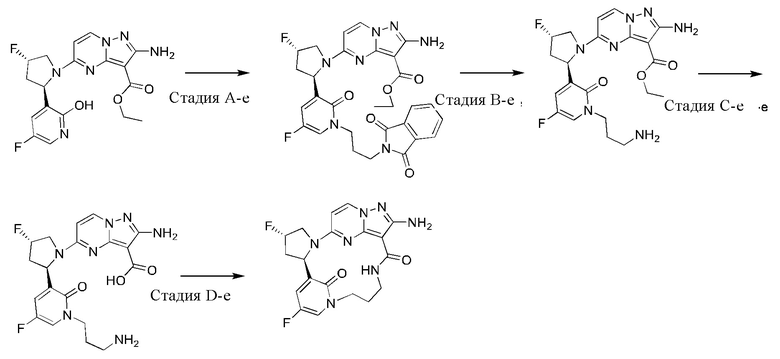
Стадия А-е: синтез этил-2-амино-5-((2R,4S)-2-(1-(3-(1,3-диоксоизоиндолин-2-ил)пропил)-5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло-[1,5-а]пиримидин-3-карбоксилата
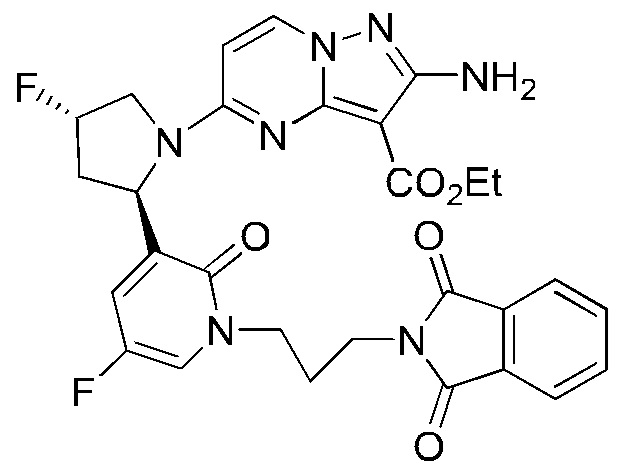
К раствору этил-2-амино-5-((2R,4S)-4-фтор-2-(5-фтор-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (380 мг) в DMF (8 мл) добавляли гидрид лития (12 мг) при 0°С, и перемешивали в течение 5 минут, и затем туда добавляли N-бромпропилфталамид (500 мг). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 4 часов. Добавляли воду (20 мл), и полученную смесь экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении для удаления растворителя с получением неочищенного продукта, который использовали на следующей стадии без очистки.
Стадия В-е: синтез этил-2-амино-5-((2R,4S)-2-(1-(3-аминопропил)-5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата
К раствору этил-2-амино-5-((2R,4S)-2-(1-(3-(1,3-диоксоизоиндолин-2-ил)пропил)-5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (120 мг) в метаноле (8 мл) добавляли гидразин-гидрат (0,5 мл; концентрация: 80%) при комнатной температуре, нагревали до 50°С и перемешивали в течение 5 часов. Полученную смесь концентрировали при пониженном давлении для удаления растворителя с получением неочищенного продукта, который использовали на следующей стадии без очистки.
Стадия С-е: синтез 2-амино-5-((2R,4S)-2-(1-(3-аминопропил)-5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты
К раствору этил-2-амино-5-((2R,4S)-2-(1-(3-аминопропил)-5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата в метаноле (8 мл) добавляли гидроксид натрия (300 мг) при комнатной температуре, нагревали до 70°С и перемешивали в течение 1 часа. Полученную смесь доводили до значения рН 5 концентрированной соляной кислотой и затем фильтровали. Твердое вещество сушили с получением неочищенного продукта, который использовали на следующей стадии без очистки.
Стадия D-e: синтез (13E,14E,22R,24S)-12-амино-24,35-дифтор-31,32-дигидро-7-аза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,1)-пиридин-2(1,2)-пирролидилциклооктанфан-32,8-диона
2-Амино-5-((2R,4S)-2-(1-(3-аминопропил)-5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновую кислоту, N,N-диизопропилэтиламин, гидрохлорид 1-этил-(3-диметиламинопропил)карбодиимида и 1-гидроксибензотриазол растворяли в смешанном растворителе N,N-диметилформамид/дихлорметан (50 мл; об./об., 1:1) и перемешивали при комнатной температуре в течение 4 часов. После удаления растворителя полученную смесь очищали посредством колоночной хроматографии (элюент: дихлорметан : метанол, 20:1 (об./об.)) с получением целевого продукта (2 мг).
1Н ЯМР (400 МГц, CD3OD) δ 8.15 (d, J=7,6 Гц, 1H), 7.62-7.64 (m, 1H), 7.50-7.52 (m, 1H), 7.44-7.46 (m, 1H), 6.29 (d, J=7,6 Гц, 1H), 5.43-5.62 (m, 2Н), 4.92-4.98 (m, 1H), 4.11-4.25 (m, 2Н), 3.91-4.00 (m, 1H), 3.64-3.68 (m, 1H), 3.30-3.37 (m, 1H), 2.78-2.80 (m, 1H), 2.05-2.23 (m, 2Н), 1.78-1.81 (m, 1H). m/z составляет 416 [М+1]+.
Следующие соединения по примерам синтезировали в соответствии со способом, который описан в примере 23.
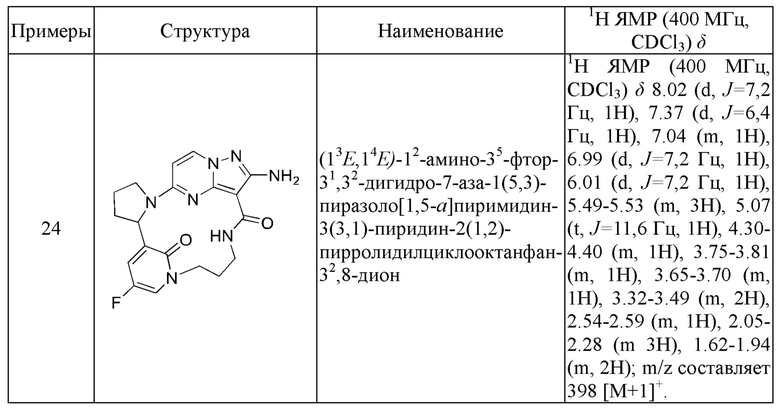
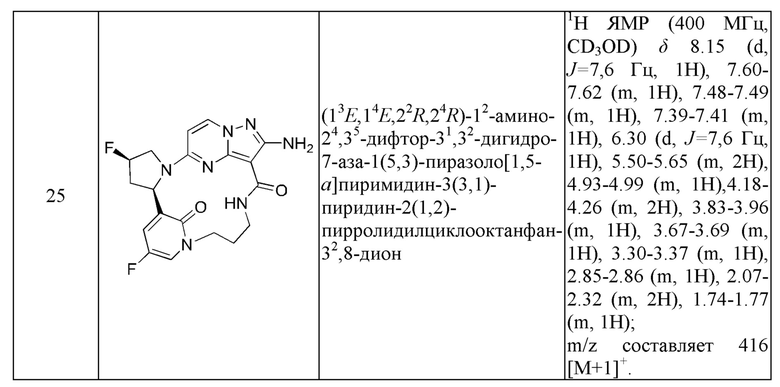
Пример 26: синтез (13Е,14Е,22R,24S,6R)-12-амино-24,35-дифтор-6-метил-7-аза-1(5,3)-пиразоло[1,5-а]пиримидин-3(3,2)-пиридин-2(1,2)-пирролидинилциклооктанфан-8-она
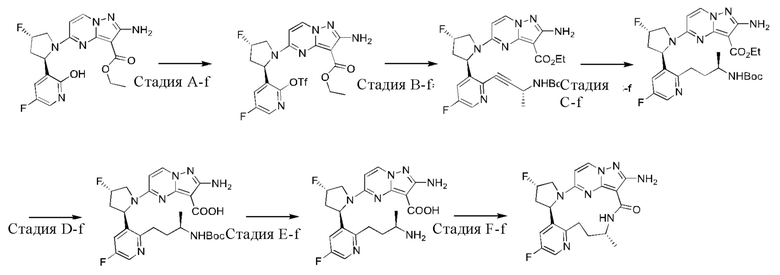
Стадия A-f: синтез этил-2-амино-5-((2R,4S)-4-фтор-2-(5-фтор-2-(трифторметил-сульфонилокси)пиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата
К раствору этил-2-амино-5-((2R,4S)-4-фтор-2-(5-фтор-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (1,8 г) и триэтиламина (1,43 мл) в DMF (15 мл) добавляли N-фенилбис(трифторметан)сульфонимид (1,76 г) и перемешивали при комнатной температуре в течение ночи. Реакцию останавливали посредством добавления воды (100 мл), и полученную смесь экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: дихлорметан : метанол, 20:1 (об./об.)) с получением указанного в заголовке соединения (1,6 г).
Стадия B-f: синтез этил-2-амино-5-((2R,4S)-2-(2-((R)-3-(трет-бутоксикарбониламино)бут-1-ин-1-ил)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксилата
К раствору этил-2-амино-5-((2R,4S)-4-фтор-2-(5-фтор-2-(трифторметил-сульфонилокси)пиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (600 мг) и трет-бутил-N-[(1R)-1-метил-2-пропин-1-ил]карбамата (378 мг) в DMF (8 мл) добавляли иодид меди (43 мг), дихлорид бис(трифенилфосфин)палладия (157 мг) и диизопропилэтиламин (417 мкл) при комнатной температуре в защитной атмосфере азота, и нагревали до 65°С, и перемешивали в течение 9 часов. Полученную смесь концентрировали при пониженном давлении для удаления DMF, и остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан : метанол, 20:1) с получением указанного в заголовке соединения (563 мг).
1Н ЯМР (400 МГц, CDCl3) δ 8.37 (s, 1H), 8.04-8.01 (m, 1H), 7.28-7.23 (m, 1H), 5.80-5.30 (m, 4Н), 4.85-4.79 (m, 3Н), 4.33 (m, 2Н), 4.13-4.10 (m, 2Н), 3.05 (m, 1H), 2.05 (m, 1H), 1.54-1.44 (m, 15Н). m/z составляет 556 [М+1]+.
Стадия C-f: синтез этил-2-амино-5-((2R,4S)-2-(2-((R)-3-(трет-бутоксикарбониламино)бутил)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата
К раствору этил-2-амино-5-((2R,4S)-2-(2-((R)-3-(трет-бутоксикарбониламино)бут-1-ин-1-ил)-5-фторпиридин-3-ил)-4-фторпирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (563 мг) в метаноле (100 мл) добавляли гидроксид палладия на угле (20%) (860 мг). Полученную смесь три раза продували газообразным водородом, перемешивали при комнатной температуре в течение ночи в защитной атмосфере водорода и затем фильтровали посредством отсасывания. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения.
Стадии D-f, E-f и F-f проводили в соответствии со стадиями D-b, E-b и F-b примера 12.
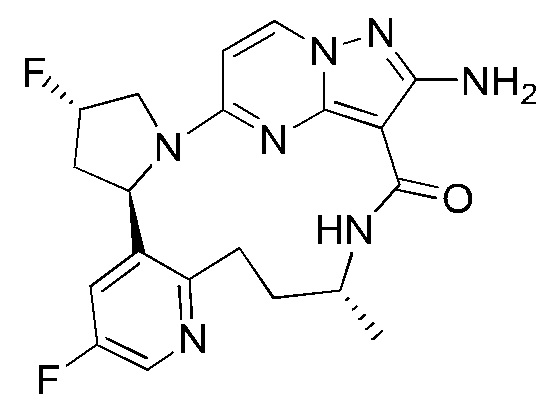
1Н ЯМР (400 МГц, CDCl3) δ 8.35 (d, J=2,8 Гц, 1H), 8.09 (d, J=7,6 Гц, 1H), 7.43 (d, J=7,2 Гц, 1H), 7.19 (dd, J=9,2, 2,8 Гц, 1H), 6.06 (d, J=7,6 Гц, 1H), 5.62-5.67 (m, 1H), 5.47 (dt, J=52,0, 2,8 Гц, 1H), 5.28-5.39 (ушир. s, 2Н), 4.27-4.33 (m, 1H), 3.98-4.18 (m, 2Н), 3.67-3.73 (m, 1H), 2.95-3.00 (m, 1H), 2.66-2.84 (m, 2Н), 2.32-2.40 (m, 1H), 1.91-2.07 (m, 1H), 1.28 (d, J=6,8 Гц, 3Н).
Следующие соединения по примерам синтезировали в соответствии со способом, который описан в примере 26.
Следующие соединения по примерам синтезировали в соответствии со способом, который описан в примере 1.

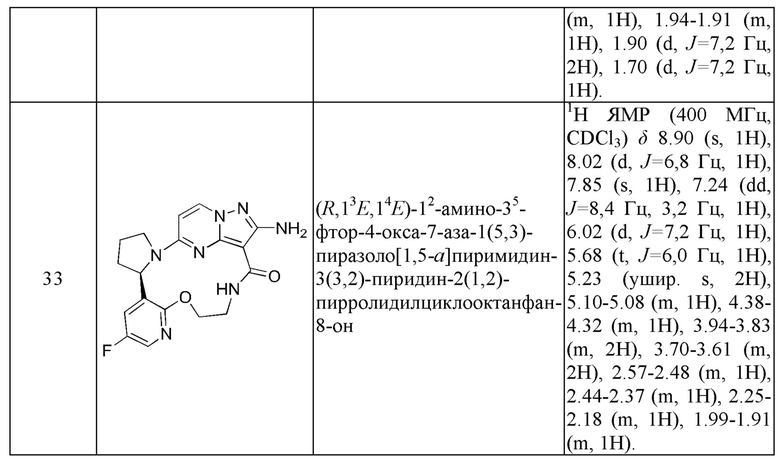
Следующие соединения по примерам синтезировали в соответствии со способом, который описан в примере 12.
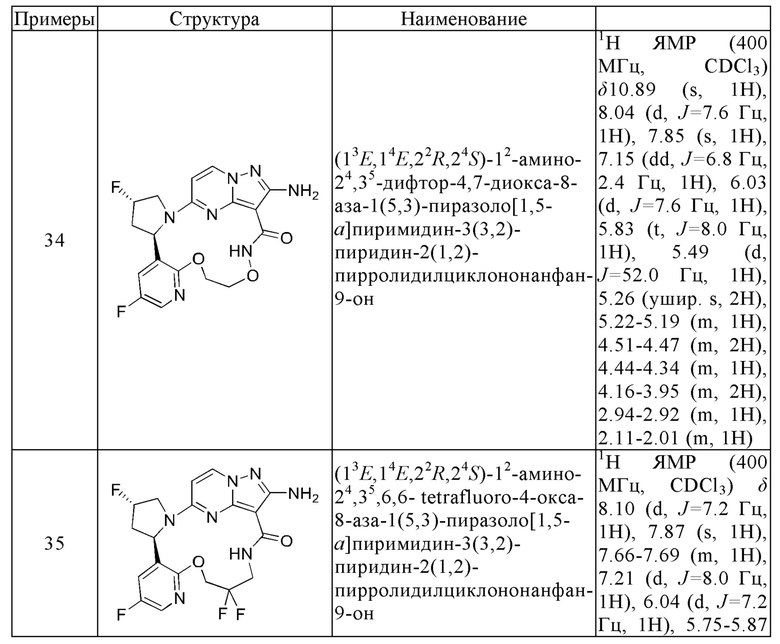
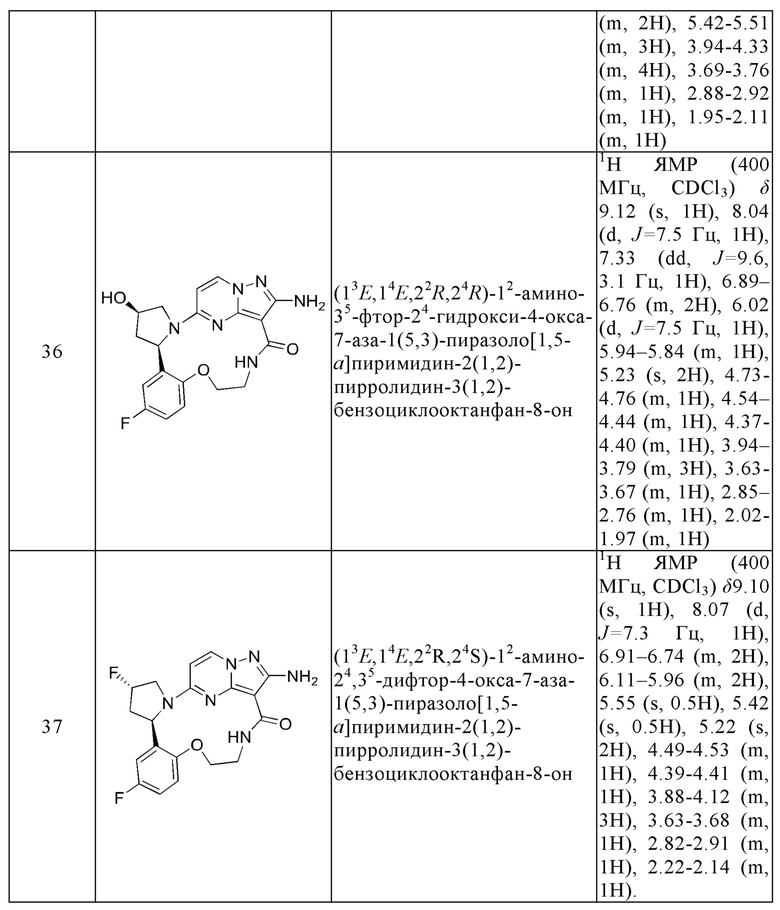
Анализы биологической активности
Анализ ингибиторной активности (IC50) в отношении TrkA-киназы
1. Анализ ингибиторной активности (IC50) в отношении TrkAWT
Платформу для тестирования активности TrkAWT-киназы разрабатывали на основе анализа гомогенной флуоресценции с временным разрешением (ГФВР), и активность соединений тестировали с использованием этой платформы. Готовили восемь пятикратных серийных разведений соединений с 100% DMSO с исходной концентрацией 200 мкМ (всего 9 концентраций). 4 мкл разбавленного образца каждой концентрации добавляли в 96 мкл реакционного буфера (50 мМ HEPES (4-гидроксиэтилпиперазинэтансульфоновая кислота), рН 7,4, 5 мМ MgCl2, 0,1 мМ NaVO3, 0,001% Tween-20 (полисорбат 20), 0,01% BSA (бычий сывороточный альбумин) и 1 мМ DTT (дитиотреитол)) и гомогенно перемешивали для применения в качестве 4*соединения. Реакционный буфер использовали для приготовления 2*TrkA-киназы (ее конечная концентрация составила 1 нМ) и 4*субстрата (АТР+TK пептид) (TK пептид, HTRF® KinEASE™-TK, закупали у Cisbio, и конечная концентрация TK пептида составила 1 мкМ, и конечная концентрация АТР (аденозинтрифосфат) составила 40 мкМ). 2,5 мкл 4*соединения добавляли в 384-луночный планшет (OptiPlate-384, закупленный у PerkinElmer), и затем добавляли 5 мкл 2*TrkA-киназы, и гомогенно перемешивали посредством центрифугирования. Затем добавляли 2,5 мкл 4*субстратной смеси для инициирования реакции (общий объем реакционной смеси составил 10 мкл). 384-луночный планшет помещали в инкубатор для проведения взаимодействия в течение 60 минут при 23°С. Затем реакцию останавливали посредством добавления 5 мкл меченого криптатом Eu3+ анти-фосфотирозинового антитела (HTRF® KinEASE™-TK, закупленное у Cisbio) и 5 мкл стрептавидин-XL-665 (HTRF® KinEASE™-TK, закупленного у Cisbio). После инкубирования в течение 1 часа в инкубаторе величину флуоресценции считывали на планшетном анализаторе Envision (закупленном у PerkinElmer). Длина волны возбуждения составляла 320 нм, и длины волн излучения для детектирования составляли 665 нм и 620 нм. Ферментативную активность представляли в виде соотношения двух считанных данных при двух длинах волн излучения. Ферментативную активность каждого соединения тестировали при 9 концентрациях, и значения IC50 соединений получали посредством подсчета данных при использовании программного обеспечения GraFit6.0 (Erithacus Software).
2. Анализ ингибиторной активности (IC50) в отношении TrkAG667C
TrkAG667C (киназный домен) киназу экспрессировали в клетках Sf9 при использовании вектора pIEX-Вас-4 и очищали при использовании аффинной хроматографии на хроматографе AKTA Purifier (компания GE). Платформу тестирования активности TrkAG667C-киназы разрабатывали на основе анализа гомогенной флуоресценции с временным разрешением (ГФВР), и активность соединений тестировали с использованием этой платформы. Готовили пятикратные серийные разведения соединений с 100% DMSO с исходной концентрацией 200 мкМ (всего 8 концентраций). 4 мкл разбавленного образца каждой концентрации добавляли в 96 мкл реакционного буфера (50 мМ HEPES, рН 7,4, 5 мМ MgCl2, 0,1 мМ NaVO3, 0,001% Tween-20, 0,01% BSA и 1 мМ DTT) и гомогенно перемешивали для применения в качестве 4*соединения. Реакционный буфер использовали для приготовления 2*TrkAG667C-киназы (ее конечная концентрация составила 0,5 нМ) и 4*субстрата (АТР+TK пептид) (TK пептид, HTRF® KinEASE™ -TK, закупали у Cisbio, и его конечная концентрация составила 1 мкМ, и конечная концентрация АТР составила 15 мкМ) для использования. 2,5 мкл 4*соединения затем добавляли в 384-луночный планшет (OptiPlate-384, закупленный у PerkinElmer), и добавляли 5 мкл 2*TrkAG667C-киназы, и гомогенно перемешивали посредством центрифугирования. Затем добавляли 2,5 мкл 4*субстратной смеси для инициирования реакции (общий объем реакционной смеси составил 10 мкл). 384-луночный планшет помещали в инкубатор для проведения взаимодействия в течение 60 минут при 23°С. Затем реакцию останавливали посредством добавления 5 мкл меченого криптатом Eu3+ анти-фосфотирозинового антитела (HTRF® KinEASE™-TK, закупленное у Cisbio) и 5 мкл стрептавидин-XL-665 (HTRF® KinEASE™-TK, закупленного у Cisbio). После инкубирования в течение 60 минут в инкубаторе величину флуоресценции считывали на планшетном анализаторе Envision (закупленном у PerkinElmer). Длина волны возбуждения составляла 320 нм, и длины волн излучения для детектирования составляли 665 нм и 620 нм. Ферментативную активность представляли в виде соотношения двух считанных данных при двух длинах волн излучения. Ферментативную активность каждого соединения тестировали при 8 концентрациях, и значения IC50 соединений получали посредством подсчета данных при использовании программного обеспечения GraFit6.0 (Erithacus Software).
3. Анализ ингибиторной активности (IC50) в отношении TrkAG595R-киназы
TrkAG595R (киназный домен) киназу экспрессировали в клетках Sf9 с использованием вектора pIEX-Вас-4 и очищали при использовании аффинной хроматографии на хроматографе AKTA Purifier (компания GE). Платформу тестирования активности TrkAG595R-киназы разрабатывали на основе анализа гомогенной флуоресценции с временным разрешением (ГФВР), и активность соединений тестировали с использованием этой платформы. Готовили пятикратные серийные разведения соединений с 100% DMSO с исходной концентрацией 200 мкМ (всего 8 концентраций). 4 мкл разбавленного образца каждой концентрации добавляли в 96 мкл реакционного буфера (50 мМ HEPES, рН 7,4, 5 мМ MgCl2, 0,1 мМ NaVO3, 0,001% Tween-20, 0,01% BSA, 1 мМ DTT и 50 нМ SEB) и гомогенно перемешивали для использования в качестве 4*соединения. Реакционный буфер использовали для приготовления 2*TrkAG595R-киназы (конечная концентрация составила 0,2 нМ) и 4*субстрата (АТР+TK пептид) (TK пептид, HTRF® KinEASE™-TK, закупали у Cisbio, и его конечная концентрация составила 1 мкМ, и конечная концентрация АТР составила 5 мкМ) для использования. 2,5 мкл 4*соединения добавляли в 384-луночный планшет (OptiPlate-384, закупленный у PerkinElmer), и затем добавляли 5 мкл 2*TrkAG595R-киназы, и гомогенно перемешивали посредством центрифугирования. Затем добавляли 2,5 мкл 4*субстратной смеси для инициирования реакции (общий объем реакционной смеси составил 10 мкл). 384-луночный планшет помещали в инкубатор для проведения взаимодействия в течение 120 минут при 23°С. Затем реакцию останавливали посредством добавления 5 мкл меченого криптатом Eu3+ анти-фосфотирозинового антитела (HTRF® KinEASE™-TK, закупленное у Cisbio) и 5 мкл стрептавидин-XL-665 (HTRF® KinEASE™-TK, закупленного у Cisbio). После инкубирования в течение 60 минут в инкубаторе величину флуоресценции считывали на планшетном анализаторе Envision (закупленном у PerkinElmer). Длина волны возбуждения составляла 320 нм, и длины волн излучения для детектирования составляли 665 нм и 620 нм. Ферментативную активность представляли в виде соотношения двух считанных данных при двух длинах волн излучения. Ферментативную активность каждого соединения тестировали при 8 концентрациях, и значения IC50 соединений получали посредством подсчета данных при использовании программного обеспечения GraFit6.0 (Erithacus Software).
В анализах ингибиторной активности в отношении киназ по настоящему изобретению, знак "*" означает умножение и указывает на кратные количества. Для иллюстрации, "2*TrkAG595R-киназа (ее конечная концентрация составляет 0,2 нМ)" означает TrkAG595R-киназу в концентрации 0,4 нМ.
Типичное значение серийного разведения: например "5-кратное серийное разведение" означает, что 4 объема раствора-разбавителя добавляли в 1 объем исходного раствора 1 для получения исходного раствора 2; и затем брали 1 объем исходного раствора 2 и добавляли туда 4 объема раствора-разбавителя для получения исходного раствора 3. Аналогичным образом получали растворы, имеющие другие концентрации.
Термин "TrkAWT" означает тропомиозин-подобную киназу А дикого типа.
Термин "TrkAG667C" означает TrkAWT, где глицин в положении 667 мутирован до цистеина.
Термин "TrkAG595R" означает TrkAWT, где глицин в положении 595 мутирован до аргинина.
Термин "HEPES" означает 4-гидроксиэтилпиперазинэтансульфоновую кислоту.
Термин "MgCl2" означает хлорид магния.
Термин "NaVO3" означает ванадат натрия.
Термин "0,001% Tween-20" означает, что объемное соотношение Tween 20 к реакционному буферу составляет 0,001%.
Термин "0,01% BSA" означает массо-объемное соотношение бычьего сывороточного альбумина к реакционному буферу, такое как 0,01 г BSA в 100 мл буфера.
Термин "DTT" означает дитиотреитол.
Термин "SEB" означает добавку в реакционный буфер для ферментов.
"мМ" означает миллимоли на литр.
Соединения, полученные в вышеприведенных примерах, анализировали согласно биологическим способам, описанным в настоящем изобретении, и результаты показаны в таблице 1:
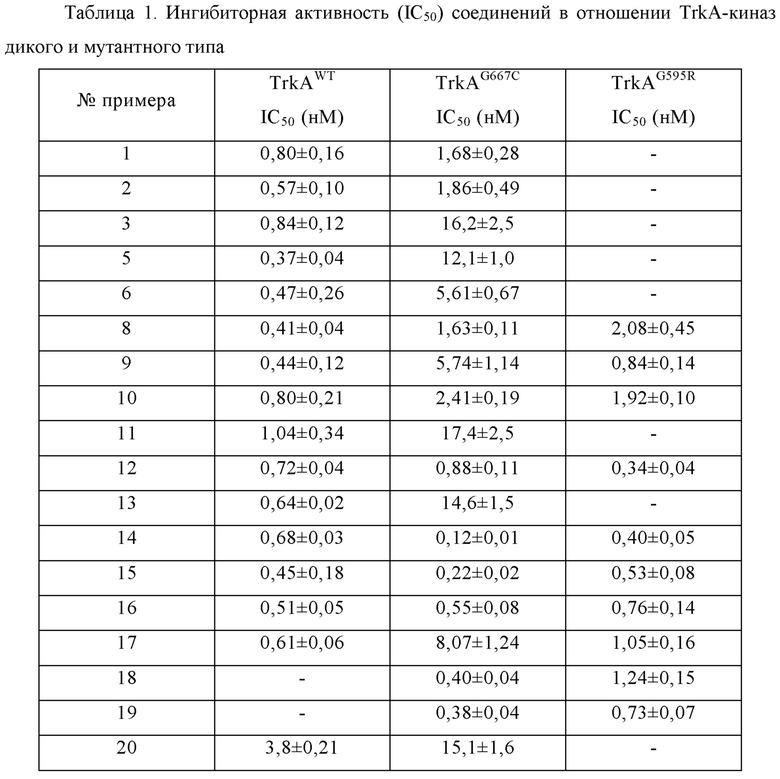
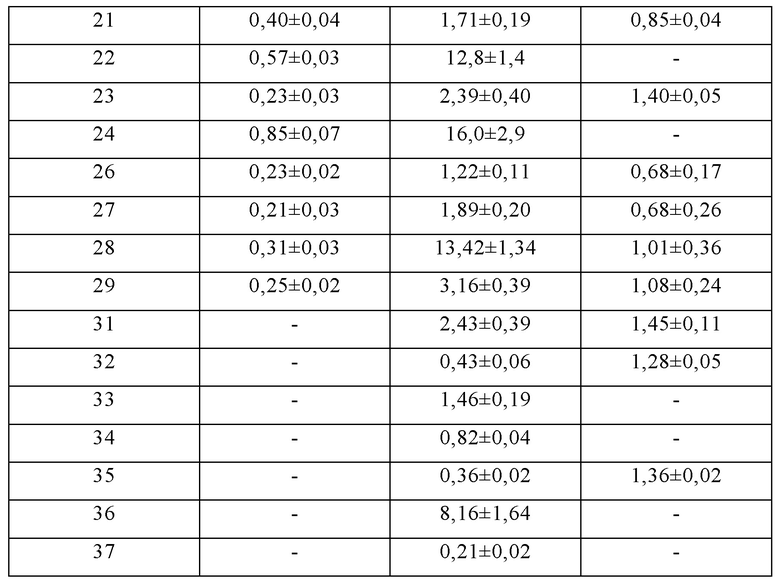
где "-" означает "не определена".
Фармакокинетический анализ
Самцов крыс линии SD получали от Beijing Vital River Laboratory Animal Technology Co., Ltd.. Крыс распределяли по три крысы на группу и перорально вводили суспензию образца для тестирования (5 мг/кг, суспензия представляет собой раствор смеси 10% EtOH, 40% PEG (полиэтиленгликоль) 400 и 50% H2O) посредством однократного внутрижелудочного введения, соответственно. Перед экспериментом животные были лишены пищи в течение ночи, и время голодания составляло от 10 часов до введения до 4 часов после введения. После введения брали образцы крови через 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа, соответственно. После наркотизации животных изофлураном с использованием аппарата для наркоза мелких животных отбирали 0,3 мл цельной крови из нижнего венозного сплетения и помещали в пробирку с антикоагулянтом - гепарином. Образец центрифугировали при 4000 об/мин в течение 5 минут при 4°С, и плазму переносили в центрифужную пробирку и хранили при -80°С до начала анализа. Образец в плазме экстрагировали посредством способа осаждения белков, и экстрагированную жидкость анализировали посредством ЖХ/МС/МС. Результаты показаны в таблице 2.

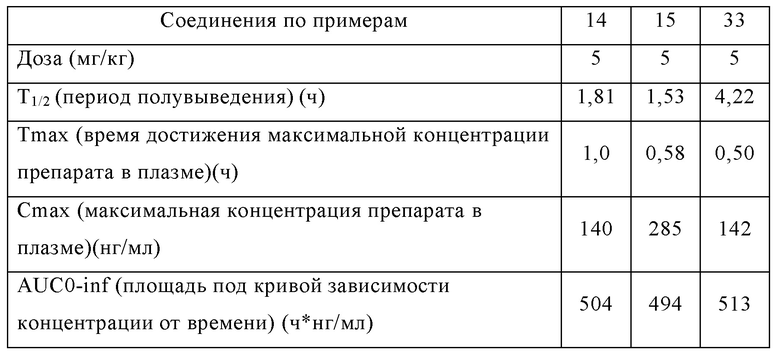
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ АМИНОПИРАЗОЛОПИРИМИДИНА, ИСПОЛЬЗУЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ТИРОЗИНКИНАЗНОГО РЕЦЕПТОРА НЕЙРОТРОФИЧЕСКОГО ФАКТОРА | 2017 |
|
RU2764523C2 |
| УСИЛИТЕЛЬ ПРОТИВООПУХОЛЕВОГО ВОЗДЕЙСТВИЯ, СОДЕРЖАЩИЙ СОЕДИНЕНИЕ ПИРРОЛОПИРИМИДИНА | 2016 |
|
RU2710380C1 |
| Макроциклический ингибитор тирозинкиназы и его применение | 2019 |
|
RU2798231C2 |
| ПРОИЗВОДНЫЕ АНИЛИНПИРИМИДИНА И ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2734849C2 |
| Селективный ингибитор киназы JAK1 | 2020 |
|
RU2818002C2 |
| ЗАМЕЩЕННЫЙ 2-АМИНОПИРИДИН В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2671212C2 |
| ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ | 2015 |
|
RU2701206C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОГО ЭФФЕКТА ИНГИБИТОРА LSD1 НА ОСНОВЕ ЭКСПРЕССИИ INSM1 | 2018 |
|
RU2789449C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2726622C2 |
Настоящее изобретение относится к макроциклу формулы (I) или к его фармацевтически приемлемой соли, к содержащей их фармацевтической композиции, и к их применению при ингибировании активности тропомиозин-рецепторной киназы А (TrkА-киназы) и в лечении заболеваний, опосредованных TrkА-киназой. Технический результат – разработка нового эффективного ингибитора активности TrkА-киназы.
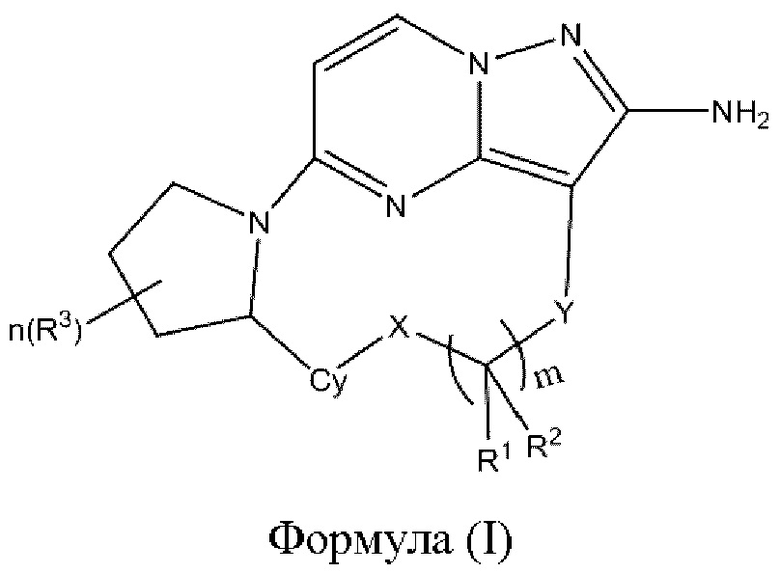 , где X выбран из группы, состоящей из связи, -О-, -S- и -NR4-;
, где X выбран из группы, состоящей из связи, -О-, -S- и -NR4-;
Y выбран из группы, состоящей из 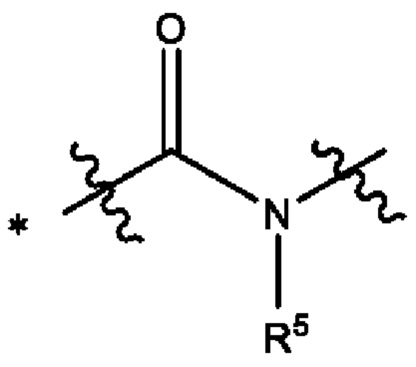 и
и 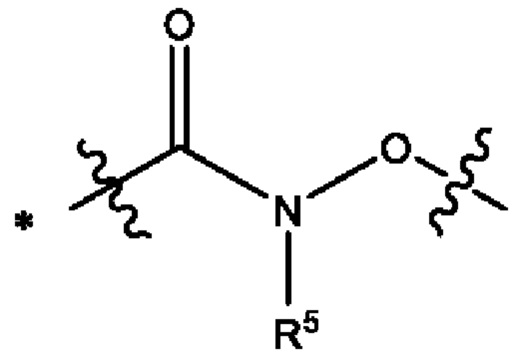 , где "*" обозначает конец группы Y, присоединенный к аминопиразолопиримидиновому кольцу, а R1- R5, m, n, Cy имеют значения, указанные в формуле изобретения. 4 н. и 22 з.п. ф-лы, 2 табл., 37 пр.
, где "*" обозначает конец группы Y, присоединенный к аминопиразолопиримидиновому кольцу, а R1- R5, m, n, Cy имеют значения, указанные в формуле изобретения. 4 н. и 22 з.п. ф-лы, 2 табл., 37 пр.
1. Соединение формулы (I)
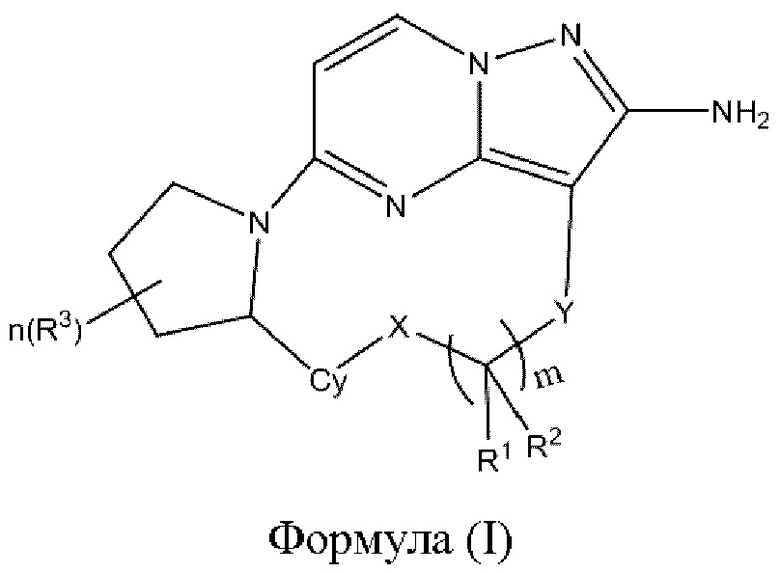
или его фармацевтически приемлемая соль, где
X выбран из группы, состоящей из связи, -О-, -S- и -NR4-;
Y выбран из группы, состоящей из 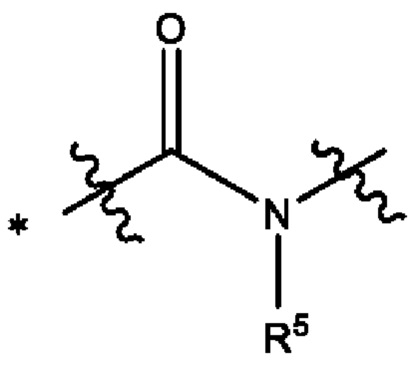 и
и  , где "*" обозначает конец группы Y, присоединенный к аминопиразолопиримидиновому кольцу;
, где "*" обозначает конец группы Y, присоединенный к аминопиразолопиримидиновому кольцу;
R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-С6алкила, C1-С6алкокси, галогена, гидрокси и амино, где C1-С6алкил и C1-С6алкокси возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из галогена, гидрокси и амино; или
R1 и R2 взяты вместе с образованием  или
или  ;
;
R3 выбран из группы, состоящей из C1-С6алкила, галогена, гидрокси и амино, где C1-С6алкил возможно замещен одним или более чем одним заместителем, независимо выбранным из группы, состоящей из галогена, гидрокси и амино;
R4 и R5 независимо выбраны из группы, состоящей из водорода и C1-С6алкила;
m выбран из 0, 1, 2, 3, 4, 5 или 6;
n выбран из 0, 1, 2, 3 или 4;
Су выбран из группы, состоящей из 6-10-членного ароматического кольца, 5-10-членного ароматического гетероцикла и 3-10-членного алифатического гетероцикла, где 6-10-членное ароматическое кольцо, 5-10-членный ароматический гетероцикл и 3-10-членный алифатический гетероцикл возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из C1-С6алкила, C1-С6алкокси, 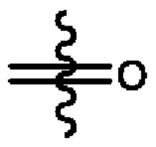 , галогена, гидрокси и амино,
, галогена, гидрокси и амино,
и где указанный "алифатический гетероциклил" или "ароматический гетероциклил" содержит от 1 до 3 гетероатомов, независимо выбранных из серы, кислорода и/или азота.
2. Соединение по п. 1, где X выбран из группы, состоящей из связи и -О-.
3. Соединение по п. 1 или 2, где R5 выбран из группы, состоящей из водорода и С1-С3алкила.
4. Соединение по п. 3, где R5 выбран из группы, состоящей из водорода и метила.
5. Соединение по любому из пп. 1-4, где R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-С3алкила, C1-С3алкокси, фтора, хлора, брома, иода, гидрокси и амино, где C1-С3алкил и C1-С3алкокси возможно замещены одним или более чем одним заместителем, независимо выбранным из группы, состоящей из фтора, хлора, брома, иода, гидрокси и амино.
6. Соединение по п. 5, где R1 и R2 независимо выбраны из группы, состоящей из водорода, фтора и C1-С3алкила.
7. Соединение по п. 6, где R1 и R2 независимо выбраны из группы, состоящей из водорода, фтора и метила.
8. Соединение по любому из пп. 1-7, где m выбран из 1, 2, 3, 4 или 5.
9. Соединение по п. 8, где m выбран из 2, 3 или 4.
10. Соединение по любому из пп. 1-9, где R3 выбран из группы, состоящей из C1-С3алкила, фтора, хлора, брома, иода, гидрокси и амино, где C1-С3алкил возможно замещен одним или более чем одним заместителем, независимо выбранным из группы, состоящей из фтора, хлора, брома, иода, гидрокси и амино.
11. Соединение по п. 10, где R3 выбран из группы, состоящей из фтора, хлора, брома, иода и гидрокси.
12. Соединение по п. 11, где R3 выбран из группы, состоящей из фтора и гидрокси.
13. Соединение по любому из пп. 1-12, где n выбран из 0, 1, 2 или 3.
14. Соединение по п. 13, где n выбран из 0 или 1.
15. Соединение по любому из пп. 1-14, где Су выбран из группы, состоящей из бензольного кольца, нафталинового кольца, пиррола, фурана, тиофена, имидазола, оксазола, пиразола, пиридина, пиримидина, пиразина, хинолина, изохинолина, бензофурана, бензотиофена, индола, изоиндола, оксирана, тетрагидрофурана, дигидрофурана, пирролидина, дигидропирролидина, 2H-пиридина, пиперидина, пиперазина, пиразолидина, тетрагидропирана, морфолина, тиоморфолина и тетрагидротиофена, каждый из которых возможно замещен одним или более чем одним заместителем, независимо выбранным из группы, состоящей из C1-С3алкила, C1-С3алкокси, 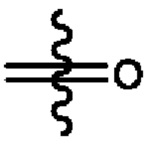 , фтора, хлора, брома, иода, гидрокси и амино.
, фтора, хлора, брома, иода, гидрокси и амино.
16. Соединение по п. 15, где Су выбран из группы, состоящей из бензольного кольца, пиридина и 1,2-2H-пиридина, каждый из которых возможно замещен одним или более чем одним заместителем, независимо выбранным из группы, состоящей из фтора и 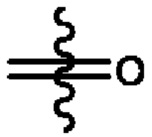
17. Соединение по п. 16, где Су выбран из группы, состоящей из 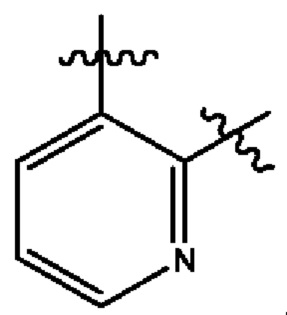 ,
,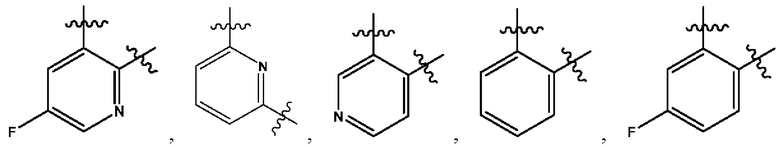 и
и
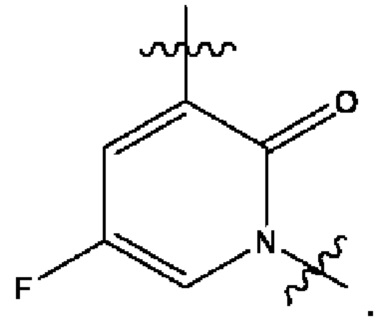
18. Соединение по п. 1, где структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из 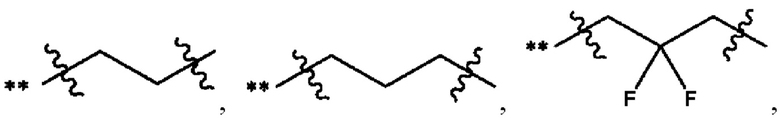
 и
и  , где "*" обозначает конец структурной единицы
, где "*" обозначает конец структурной единицы 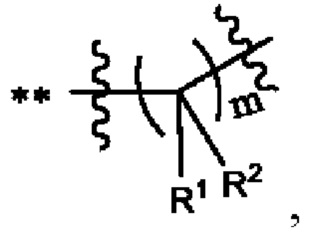 присоединенный к X.
присоединенный к X.
19. Соединение по любому из пп. 1-18, где соединение формулы (I) представляет собой соединение формулы (II)
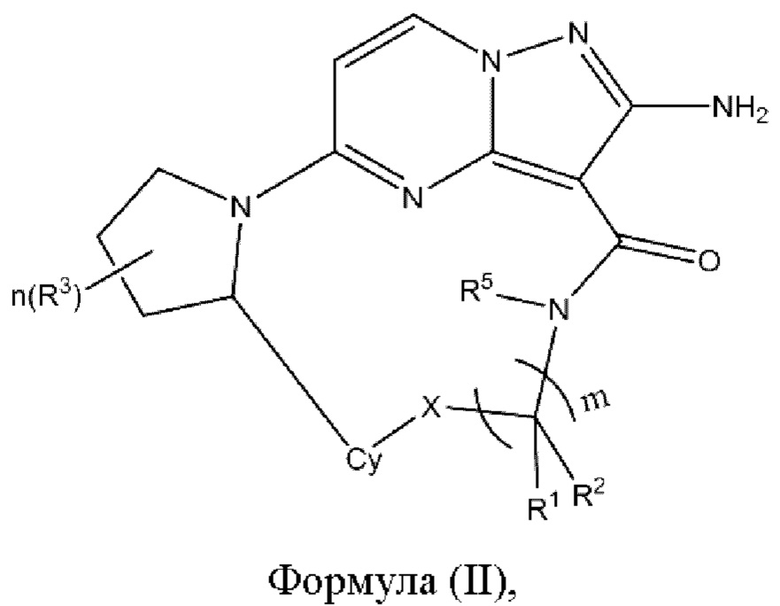
где X, R1, R2, R3, R5, Су, m и n являются такими, как определено в любом из пп. 1-18.
20. Соединение по любому из пп. 1-18, где соединение формулы (I) или его фармацевтически приемлемая соль выбраны из группы, состоящей из 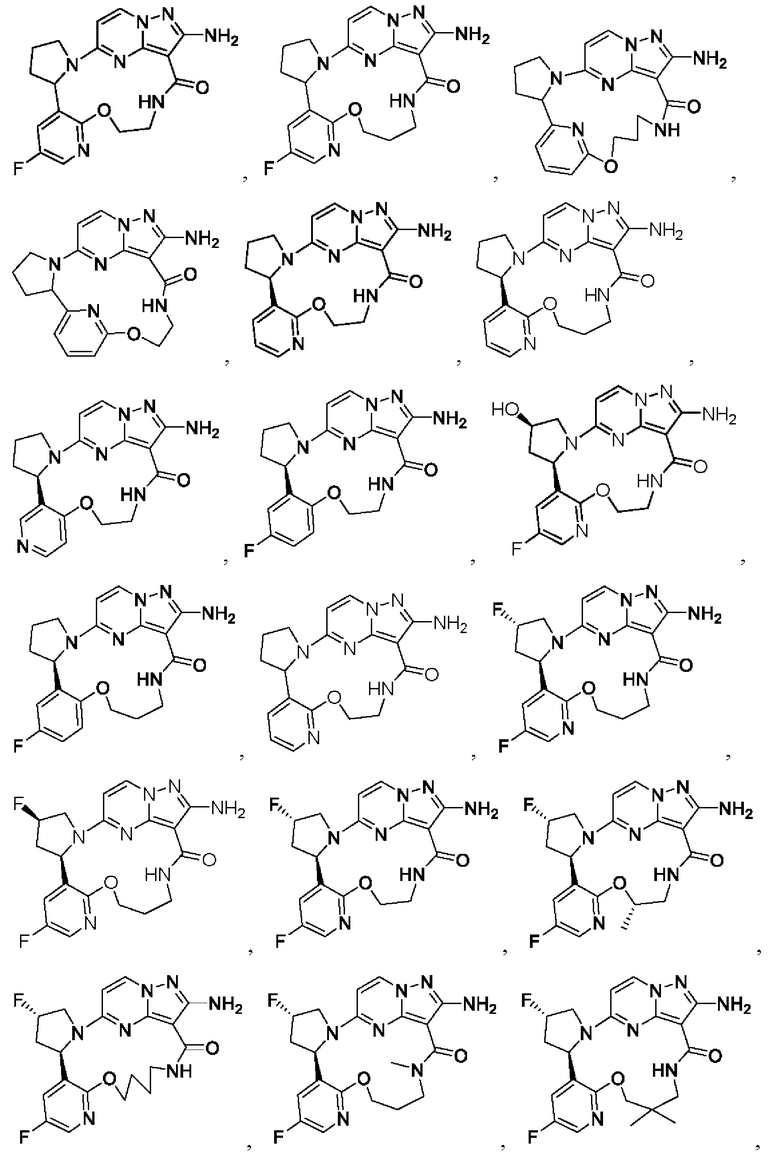
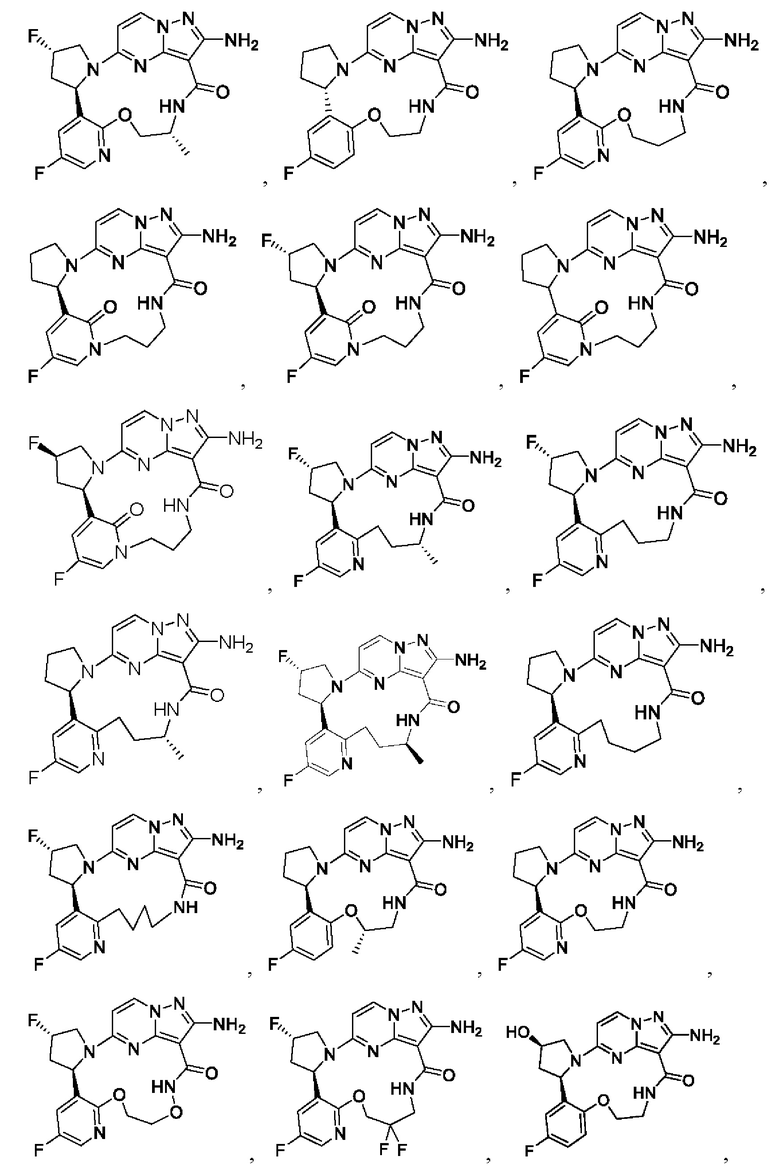

и их фармацевтически приемлемой соли.
21. Фармацевтическая композиция для ингибирования активности TrkA-киназы (тропомиозин-рецепторной киназы А), содержащая терапевтически эффективное количество соединения формулы (I) по любому из пп. 1-20 или его фармацевтически приемлемой соли.
22. Способ лечения заболевания, опосредованного TrkA-киназой, у млекопитающего, включающий введение млекопитающему, нуждающемуся в этом, предпочтительно человеку, терапевтически эффективного количества соединения формулы (I) по любому из пп. 1-20 или его фармацевтически приемлемой соли, или фармацевтической композиции по п. 21.
23. Применение соединения формулы (I) по любому из пп. 1-20, или его фармацевтически приемлемой соли, или фармацевтической композиции по п. 21 в изготовлении лекарственного средства для профилактики или лечения заболевания, опосредованного TrkA-киназой.
24. Соединение формулы (I) по любому из пп. 1-20, или его фармацевтически приемлемая соль, или фармацевтическая композиция по п. 21 для использования в профилактике или лечении заболевания, опосредованного TrkA-киназой.
25. Способ по п. 22, где заболевание представляет собой опухоль.
26. Применение по п. 23, где заболевание представляет собой опухоль.
| WO 2011146336 A1, 24.11.2011 | |||
| WO 2017004342 A1, 05.01.2017 | |||
| WO 2010048314 A1, 29.04.2010 | |||
| Приспособление для указания отклонений судна от фарватера реки | 1931 |
|
SU25881A1 |
| Реактивная турбина внутреннего горения | 1930 |
|
SU26155A1 |

