ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к новым синтетическим методам получения соединений с высокой хиральной чистотой. В частности, оно относится к способам получения хиральных пиримидинов и их применению в качестве промежуточных продуктов при получении фармацевтически активных соединений.
УРОВЕНЬ ТЕХНИКИ
В данном описании перечисление или обсуждение ранее опубликованных в явном виде документов не следует обязательно понимать как признание того, что данный документ является частью известного уровня техники или общих знаний.
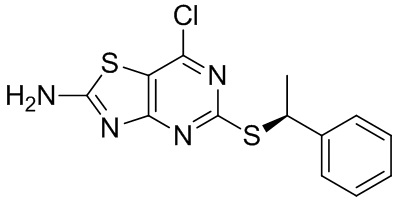
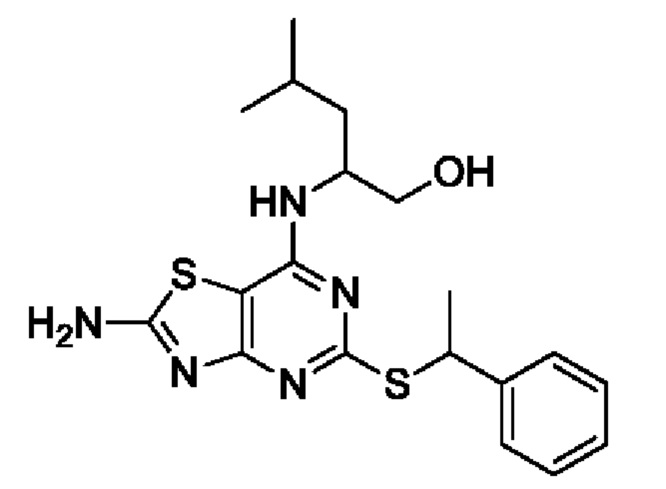
Известно, что 7-амино-5-тио-тиазоло[4,5-d]пиримидины являются антагонистами рецептора фракталкина (Karlström et al. J. Med. Chem., 2013, 56, 3177-3190). Среди данных соединений значительный интерес представляют соединения, содержащие хиральные α-алкильные разветвленные бензилтиоэфирные группы. В частности, соединение (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол, как известно, является сильным антагонистом.
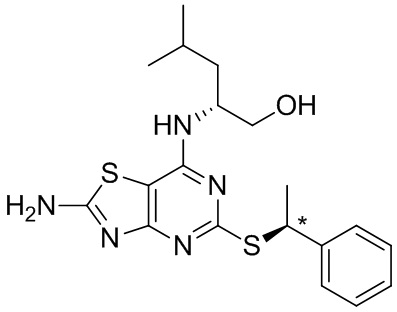
Стереоцентр в верхней части молекулы может быть установлен с помощью желаемого энантиомера лейцинола (который происходит из аминокислоты лейцина и доступен с очень высокой энантиомерной чистотой). Однако предложение масштабируемого синтетического пути, который обеспечивает достаточную хиральную чистоту на втором стереоцентре (отмеченном *), оказалось серьезной проблемой.
В опубликованном лабораторном синтетическом пути (Karlström и др.) стереохимию данного хирального центра устанавливают на последней стадии посредством реакции тиотиазолопиримидинового промежуточного соединения с (R)-1-хлорэтилбензолом, что приводит к соотношению диастереомеров 9:1 в пользу желаемого R,S-диастереомера. Затем очистка хиральной ВЭЖХ приводила к желаемому соединению с хиральной чистотой 99,7%. Однако выполнить хиральную ВЭЖХ в масштабе, необходимом для производства активных фармацевтических ингредиентов, невозможно.
Основной синтетический путь к 6-амино-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-4-олу опубликован в WO 2006/107258.
Однако увеличение масштабов известных методов может привести к материалам с более низкой хиральной чистотой. Хотя более чистые материалы могут быть получены до определенного предела, например, последовательными стадиями перекристаллизации, такие стадии очистки могут значительно снизить выход способа и также крайне нежелательны в процессе химического производства.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Мы неожиданно обнаружили, что (гетеро)арилалкилсульфанилпиримидины, такие как 6-амино-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-4-ол, могут быть получены с хорошими выходами и с превосходной хиральной чистотой без необходимости дополнительных стадий перекристаллизации путем получения соответствующего (гетеро)арилалкилсульфонатного эфира в тщательно выбранных условиях реакции и последующей реакции данного соединения с требуемым анионом тиолата пиримидина.
Данный способ обеспечивает простой и масштабируемый синтетический путь к синтетическим промежуточным соединениям, которые позволяют получать соединения, представляющие фармацевтический интерес, такие как (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол с очень высокой хиральной чистотой.
Новые способы
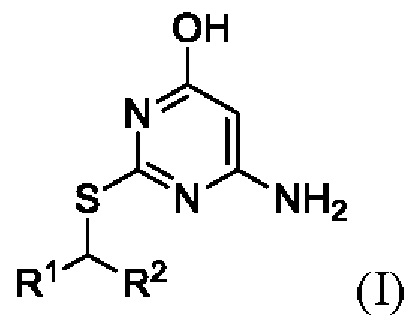
В первом аспекте данного изобретения предложен способ получения соединения формулы I,
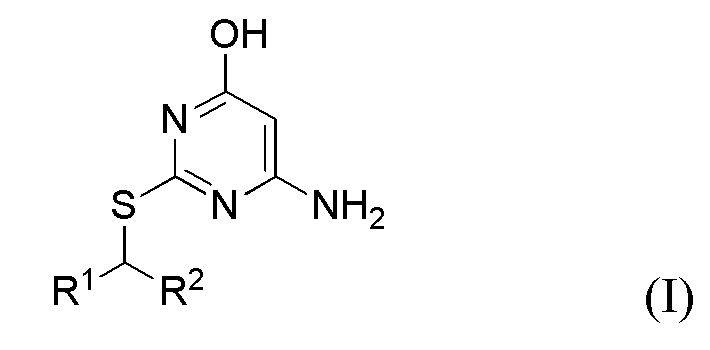
или его соли, где
R1 представляет собой арил или пиридил, необязательно замещенный одной или несколькими группами, выбранными из галогена, -CN, -C(O)NR3R4, -S(O)2R5; C1-6 алкила, C2-6 алкенила, C2-6 алкинила, где последние три группы являются необязательно замещенными одним или несколькими F;
R2 представляет собой C1-6 алкил, необязательно замещенный одним или более F;
каждый R3 и R4 независимо представляет собой H или C1-6 алкил, необязательно замещенный одним или более F;
R5 представляет собой C1-6 алкил, необязательно замещенный одним или более F;
указанный способ включает следующие стадии:
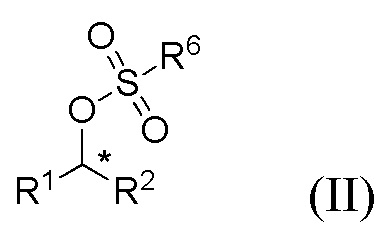
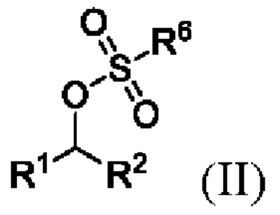
(i) образование соединения формулы II
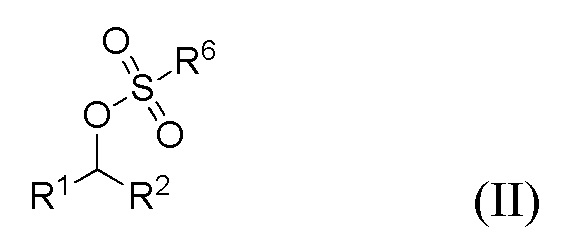
где R1 и R2 представляют собой такие, как указано для соединения формулы I, и R6 представляет собой C1-6 алкил, необязательно замещенный одним или более F или фенилом, необязательно замещенным одной или несколькими группами, выбранными из галогена, метила и -NO2.;
путем приведения в контакт соединения формулы III

где R1 и R2 представляют собой такие, как указано для соединения формулы I или II;
с пригодным сульфирующим агентом в присутствии пригодного основания B1
и пригодного растворителя (S1), в котором соль, образованная между B1 и уходящей группой сульфирующего агента, является нерастворимой,
и затем
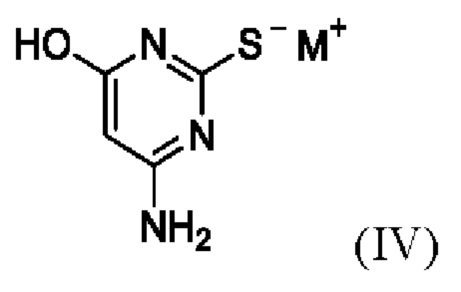
(ii) приведение в контакт соединения формулы II с соединением формулы IV,
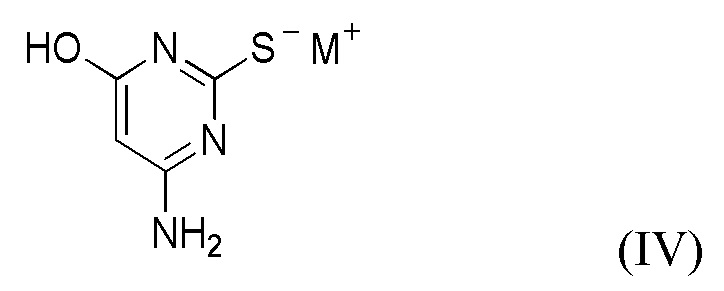
где M+ представляет собой Li+, Na+, K+ или Cs+;
где соединение формулы III представлено в виде одного энантиомера,
указанный способ может быть упомянут в данном документе как «способ по данному изобретению».
Если не указано иное, то все технические и научные термины, используемые в данном документе, имеют значение, общепринятое специалистами в области техники, к которой относится данное изобретение.
Соли соединений, полученных способами, описанными в данном документе (т. е. соединения формулы I, Ia и другие, как определено в данном документе), включают соли присоединения кислоты и основания. Указанные соли могут быть получены традиционными способами, например, путем приведения в контакт формы свободного основания соединения по данному изобретению с одним или более эквивалентами соответствующей кислоты, необязательно в растворителе или в среде, в которой данная соль нерастворима, с последующим удалением указанного растворителя или указанной среды стандартными способами (например, под вакуумом, путем лиофилизации или фильтрования). Соли также могут быть получены с использованием методик, известных специалистам в данной области техники, например, путем замены противоиона в соединении по данному изобретению в форме соли на другой противоион, например, с использованием подходящей ионообменной смолы.
Конкретные соли присоединения кислоты, которые можно упомянуть, включают такие, которые образованы приведением в контакт с соответствующими кислотами, таким образом, протонируя соединение по данному изобретению с образованием солей, таких как карбоксилатные соли (например, формиатные, ацетатные, трифторацетатные, пропионатные, изобутиратные, гептаноатные, деканоатные, капратные, каприлатные, стеаратные, акрилатные, капроатные, пропиолатные, аскорбатные, цитратные, глюкуронатные, глутаматные, гликолятные, α-гидроксибутиратные, лактатные, тартратные, фенилацетатные, манделатные, фенилпропионатные, фенилбутиратные, бензоатные, хлорбензоатные, метилбензоатные, гидроксибензоатные, метоксибензоатные, динитробензоатные, о-ацетоксибензоатные, салицилатные, никотинатные, изоникотинатные, циннаматные, оксалатные, малонатные, сукцинатные, субератные, себакатные, фумаратные, малатные, малеатные, гидроксималеатные, гиппуратные, фталатные или терефталатные соли), гидрогалогенидные соли (например, гидрохлоридные, гидробромидные или гидройодидные соли), сульфонатные соли (например, бензолсульфонатные, метил-, бром- или хлор-бензолсульфонатные, ксилолсульфонатные, метансульфонатные, этансульфонатные, пропансульфонатные, гидроксиэтансульфонатные, 1- или 2-нафталинсульфонатные или 1,5-нафталиндисульфонатные соли) или сульфатные, пиросульфатные, бисульфатные, сульфитные, бисульфитные, фосфатные, моногидрофосфатные, дигидрофосфатные, метафосфатные, пирофосфатные или нитратные соли, и т. д.
Конкретные соли присоединения основания, которые можно упомянуть, включают соли, образованные приведением в контакт с соответствующими основаниями, таким образом, снимая протон из соединений по данному изобретению с образованием солей с щелочными металлами (таких как соли Na и K), щелочноземельными металлами (таких как соли Mg и Ca), органическими основаниями (такими как этаноламин, диэтаноламин, триэтаноламин, трометамин и лизин) и неорганическими основаниями (такими как аммиак и гидроксид алюминия). Более конкретно, соли присоединения основания, которые могут быть упомянуты, включают соли Mg, Ca и, наиболее конкретно, соли K и Na.
Более конкретные соли, которые можно упомянуть, включают гидрохлорид, гидробромид, соли натрия, калия и лития.
Если не указано иное, C1-z алкильные группы (где z представляет собой верхний предел диапазона), определенные в данном документе, могут быть неразветвленными или, если присутствует достаточное количество (т. е. не менее двух или трех, по обстоятельствам) атомов углерода, могут быть разветвленными и/или циклическими (образуя C3-z циклоалкильную группу). Если присутствует достаточное количество (т. е. не менее четырех) атомов углерода, такие группы также могут быть частично циклическими (образуя C4-z частично циклоалкильную группу). Например, можно указать циклоалкильные группы, включающие циклопропил, циклопентил и циклогексил. Аналогично, частично циклические алкильные группы (которые также могут быть упомянуты как «частично циклоалкильные» группы), которые могут быть упомянуты, включают циклопропилметил. Если присутствует достаточное количество атомов углерода, то указанные группы также могут быть полициклическими (например, бициклическими или трициклическими) и/или спироциклическими. Во избежание неопределенности, конкретные алкильные группы, которые могут быть упомянуты, включают алкильные группы с прямой цепью (т. е. неразветвленные и/или ациклические).
Если не указано иное, C2-z алкенильные группы (где z представляет собой верхний предел диапазона), определенные в данном документе, могут быть неразветвленными или, если присутствует достаточное количество (т. е. не менее трех) атомов углерода, могут быть разветвленными и/или циклическими (образуя C4-z циклоалкенильную группу). Если присутствует достаточное количество (т. е. не менее пяти) атомов углерода, такие группы также могут быть частично циклическими. Например, частично циклические алкенильные группы (которые также могут быть упомянуты как «частично циклоалкенильные» группы), которые могут быть упомянуты, включают циклопентенилметил и циклогексенилметил. Если присутствует достаточное количество атомов углерода, то указанные группы также могут быть полициклическими (например, бициклическими или трициклическими) или спироциклическими. Во избежание неопределенности, конкретные алкенильные группы, которые могут быть упомянуты, включают алкенильные группы с прямой цепью (т. е. неразветвленные и/или ациклические).
Если не указано иное, C2-z алкинильные группы (где z представляет собой верхний предел диапазона), определенные в данном документе, могут быть неразветвленными или, если присутствует достаточное количество (т. е. не менее четырех) атомов углерода, могут быть разветвленными. Во избежание неопределенности, конкретные алкинильные группы, которые могут быть упомянуты, включают алкинильные группы с прямой цепью (т. е. неразветвленные и/или ациклические).
Во избежание неопределенности, если не указано иное, группы, упоминаемые в данном документе как «алкил», «алкенил» и/или «алкинил», будут рассматриваться как относящиеся к наивысшей степени ненасыщенности в связи, присутствующей в таких группах. Например, такая группа, имеющая двойную связь углерод-углерод и в той же группе тройную связь углерод-углерод, будет называться «алкинил». Альтернативно, может быть конкретно указано, что такие группы будут содержать только указанную степень ненасыщенности (т. е. в одной или нескольких связях в них, в зависимости от обстоятельств; например, в одной связи в них).
Специалист в данной области техники поймет, что заместители, присутствующие в соединении формулы I, полученном способом по данному изобретению, будут зависеть от заместителей, присутствующих в соединениях формулы II, которые приводят в контакт, и сможет соответственно выбрать пригодные исходные материалы.
Соединения формул I, II и III содержат асимметричный атом углерода (*) и поэтому существуют в двух энантиомерных формах.
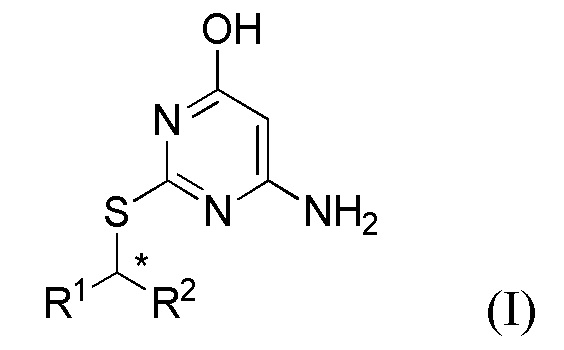
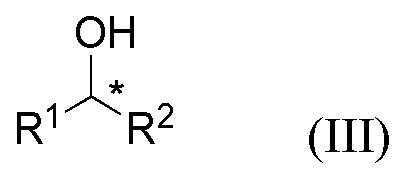
Стереохимию данного хирального центра можно определить в соответствии с правилом об использовании клиновидной связи или штрихованой связи для обозначения группы, обращенной наружу или внутрь плоскости бумаги. Если такая связь используется, ее можно понять как указание на конкретный единственный энантиомер соединения формулы I.
Как описано в данном документе, способ по данному изобретению приводит к соединениям формулы I с высокой хиральной чистотой (т. е. одиночным энантиомерам). Соответственно, если стереохимия хирального центра не определена (т. е. все группы, присоединенные к хиральному центру, соединены с ним плоскими связями), если не указано иное, это можно понимать как указание на то, что любой энантиомер может присутствовать в чистой форме.
Если не указано иное, ссылки на отдельные энантиомеры в данном документе (и аналогично энантиомерам, присутствующим в чистой форме и т. п.) могут быть поняты как указывающие на энантиомерную чистоту по меньшей мере около 90% (т. е. энантиомерный избыток (ее) по меньшей мере около 80%), например, по меньшей мере около 95%, например, по меньшей мере около 97% (например, по меньшей мере около 99% (около 98% ее)). Более конкретно, один энантиомер может иметь энантиомерную чистоту по меньшей мере около 99,5%, например, по меньшей мере около 99,7% (99,4% ее).
Предыдущие два абзаца аналогичным образом применимы к эпимерной чистоте в соответствующем хиральном центре в соединениях, содержащих более одного хирального центра.
Как используется в данном документе, по отношению к определенному значению (например, количеству) термин «около» (или аналогичные термины, такие как «приблизительно») будет пониматься как указывающий на то, что такие значения могут варьироваться до 10% (в частности, до 5%, например до 1%) от определенного значения. Предполагается, что в каждом случае такие термины могут быть заменены обозначением «± 10%» или тому подобным (или путем указания конкретного значения отклонения, рассчитанного на основе соответствующего значения). Также предполагается, что в каждом случае такие термины могут быть удалены.
В конкретных вариантах реализации изобретения указанное соединение формулы II не выделяют из реакционной смеси со стадии (i) до его использования на стадии (ii).
Специалист в данной области техники поймет, что ссылки на соединения, которые не выделяют из реакционной смеси (например, соединение формулы II, не выделяемое из реакционной смеси со стадии (i) до его использования на стадии (ii)), указывают на то, что не предпринимали никаких попыток получения соответствующего соединения в чистом виде перед его использованием в реакции следующей стадии. Однако при необходимости могут быть выполнены определенные стадии очистки и/или другие манипуляции, такие как отмывка водой, упаривание или разбавление реакционной смеси и/или фильтрация реакционной смеси для удаления нерастворимых побочных продуктов. В частности, данную фразу можно понимать как указание на то, что соответствующее соединение (например, соединение формулы II) вводят в реакцию следующей стадии в форме раствора в реакционном растворителе предыдущей стадии синтеза.
Стадия (ii) способа по данному изобретению происходит с инверсией стереохимии на хиральном центре. Специалист в данной области техники поймет, что это означает, что относительная стереохимия хирального центра в продукте (т. е. соединении формулы I) является обратной по сравнению с исходным веществом (т. е. соединением формулы II (или III)). Поскольку приоритет заместителей в соответствии с правилом Кана-Ингольда-Прелога для определения абсолютной стереохимии не меняется во время стадии (ii), абсолютная стереохимия соединения формулы I также будет противоположна таковой у соединения формулы II (или III).
Не ограничиваясь теорией, считается, что стадия (ii) происходит посредством реакции бимолекулярного нуклеофильного замещения (SN2). Как поймет специалист в данной области техники, данная реакция является стереоспецифической, и ее механизм требует, чтобы инверсия стереохимии хирального центра происходила, когда уходящая группа (на стадии (ii) -OS(O)2R6) заменяется на нуклеофил (на стадии (ii) анионные фрагменты в соединении формулы IV).
В конкретных вариантах реализации изобретения стадию (i) способа по данному изобретению выполняют в растворителе (S1), в котором соль, образованная между основанием, присутствующим в реакционной смеси (B1), и уходящей группой сульфирующего агента, является нерастворимой.
Например, если стадию (i) выполняют с использованием мезилхлорида (метансульфонилхлорида) в качестве сульфирующего (или, в частности, мезилирующего) агента и триэтиламина в качестве основания (B1), соль гидрохлорида триэтиламина будет образовываться в качестве побочного продукта в реакционной смеси, и растворитель, в котором данная соль является нерастворимой, выбран как S1.
Как используется в данном документе, термин «нерастворимый» можно понимать как означающий, что указанное химическое вещество (например, соль, ион или нейтральное соединение) по существу не растворяется в соответствующем растворителе при комнатной температуре. Например, менее, чем около 10% от общего количества вещества находится в растворе, например, менее, чем около 5% (например, менее, чем около 2%).
Как используется в данном документе, термин «уходящая группа» можно понимать как означающий группу, присутствующую в соответствующем сульфирующем агенте до прохождения реакции, которая замещается связью с атомом кислорода соединения формулы II.
Не ограничиваясь теорией, полагают, что выбор в качестве S1 растворителя, в котором соль, образованная между B1 и уходящей группой сульфирующего агента, является нерастворимой, может способствовать высокой энантиомерной чистоте, наблюдаемой в соединении формулы I, поскольку количество свободного аниона уходящей группы сульфирующего агента (например, хлорида) в растворе уменьшается. Если в реакционной смеси присутствует анион, такой как хлорид, он может принимать участие в нежелательных процессах нуклеофильного замещения, приводящих к ухудшению стереохимической чистоты из-за множественных инверсий стереохимии.
В конкретных вариантах реализации способа по данному изобретению стадия (ii) включает приведение раствора соединения формулы II в растворителе S1 (т. е. реакционном растворителе, в котором образуется соединение формулы II) в контакт с раствором соединения формулы IV в пригодном растворителе S2. В более конкретных вариантах реализации изобретения S2 представляет собой растворитель, в котором образуется соединение формулы IV.
Как используется в данном документе, ссылки на приведение в контакт растворов можно понимать как указывающие, что растворы смешивают вместе в одном реакционном сосуде. Такое смешивание может включать добавление одного из растворов к другому, и указанное добавление может происходить в течение короткого периода времени (например, один раствор можно вливать в другой), или добавление можно контролировать для прохождения в течение более длительного периода времени (например, один раствор можно добавлять по каплям к другому).
Ссылки на добавление по каплям можно понимать как означающие постепенное контролируемое добавление раствора или жидкого реагента в течение продолжительного периода времени. Специалист в данной области техники сможет определить подходящую скорость добавления для данной экспериментальной установки и набора условий реакции.
В конкретных вариантах реализации изобретения указанный способ дополнительно включает следующую стадию:
(iii) получение соединения формулы IV приведением в контакт соединения формулы V
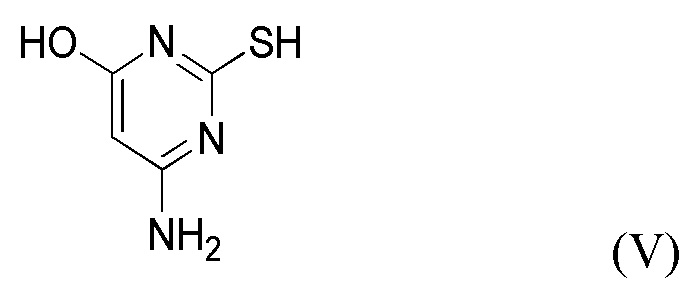
с пригодным основанием B2 в присутствии пригодного растворителя S2.
В таких случаях соединение формулы IV можно не выделять из реакционной смеси стадии (iii) до использования на стадии (ii). В таких вариантах реализации изобретения раствор соединения формулы IV получают на стадии (iii), необязательно, после одной или нескольких стадий очистки.
В более конкретных вариантах реализации изобретения раствор соединения формулы II в S1 добавляют к раствору соединения формулы IV в S2. Более конкретно, раствор соединения формулы II добавляют по каплям.
В конкретных вариантах реализации изобретения стадии (i) и (iii) выполняют одновременно (т. е. реакции проводят параллельно).
В конкретных вариантах реализации изобретения стадию (ii) выполняют при температуре около комнатной. Как используется в данном документе, термин «комнатная температура» может означать температуру окружающей среды в помещении, которая как правило составляет от около 15°C до около 25°C, например, от около 20°C до около 25°C (например, около 25°C).
Как описано выше в данном документе, S1 может представлять собой растворитель, в котором соль, образованная между B1 и уходящей группой сульфирующего агента, является нерастворимой.
Пригодные в качестве S1 растворители включают простые эфиры, такие как диэтиловый эфир, метил трет-бутиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран, 1,4-диоксан и 2-метилтетрагидрофуран. В конкретных вариантах реализации изобретения S1 представляет собой диэтиловый эфир, метил трет-бутиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран. В предпочтительных вариантах реализации изобретения указанный растворитель представляет собой метил трет-бутиловый эфир.
В конкретных вариантах реализации изобретения B1 представляет собой основание органический амин. Примеры пригодных оснований включают триэтиламин, N,N-диизопропилэтиламин, пиридин, пиперидин, N-метилпиперидин и пирролидин. Более конкретные основания включают триэтиламин и N,N-диизопропилэтиламин (например, триэтиламин).
В конкретных вариантах реализации изобретения стадию (i) выполняют при температуре от около -10°C до около 20°C, например, от около -5°C до около 10°C (например, около 0°C). Стадию (i) можно выполнять с охлаждением ледяной баней. В таких случаях понятно, что реакционную смесь охлаждают до около 0 ± около 5°С.
В конкретных вариантах реализации изобретения S2 представляет собой полярный апротонный растворитель. Примеры пригодных растворителей включают N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и N-метил-2-пирролидон. В более конкретных вариантах реализации изобретения указанный растворитель представляет собой N,N-диметилформамид.
B2 представляет собой соль металла Группы I (т. е. щелочного металла). В конкретных вариантах реализации изобретения B2 выбран из группы, включающей гидроксид лития, карбонат лития, гидроксид натрия, карбонат натрия, гидроксид калия, карбонат калия, гидроксид цезия и карбонат цезия. В более конкретных вариантах реализации изобретения B2 выбран из гидроксида лития, гидроксида натрия, гидроксида калия и гидроксида цезия. В еще более конкретных вариантах реализации изобретения B2 представляет собой гидроксид натрия.
В конкретных вариантах реализации изобретения стадию (iii) выполняют при температуре от около 40°C до около 80°C, например, от около 50°C до около 75°C, например, от около 55°C до около 70°C (например, около 65°С).
В конкретных вариантах реализации изобретения R1 представляет собой арил (например, фенил) или пиридил, необязательно замещенный одной или несколькими группами, выбранными из галогена (например, брома, хлора или фтора (например, хлора или фтора), -CN, -SO2Me или -CONH2. В более конкретных вариантах реализации изобретения R1 представляет собой фенил (т. е. незамещенный).
В конкретных вариантах реализации изобретения R2 представляет собой C1-6 алкил (т. е. незамещенный). В более конкретных вариантах реализации изобретения R2 представляет собой C1-3 алкил, необязательно замещенный одним или более F (например, незамещенный). В еще более конкретных вариантах реализации изобретения R2 представляет собой трифторметил, дифторметил, фторметил или, в частности, метил.
В конкретных вариантах реализации изобретения каждый из R3 и R4 независимо представляет собой H или C1-6 алкил (т. е. незамещенный). В более конкретных вариантах реализации изобретения каждый из R3 и R4 независимо представляет собой C1-3 алкил, необязательно замещенный одним или более F (например, незамещенный). В еще более конкретных вариантах реализации изобретения каждый из R3 и R4 независимо представляет собой H, трифторметил, дифторметил, фторметил или метил. В В дополнительных конкретных вариантах реализации R3 и R4 оба представляют собой Н.
В конкретных вариантах реализации изобретения R5 представляет собой C1-6 алкил (т. е. незамещенный). В более конкретных вариантах реализации изобретения R5 представляет собой C1-3 алкил, необязательно замещенный одним или более F (например, незамещенный). В еще более конкретных вариантах реализации изобретения R5 представляет собой трифторметил, дифторметил, фторметил или, в частности, метил.
В конкретных вариантах реализации изобретения R6 представляет собой C1-4 алкил, необязательно замещенный одним или более F или фенилом, необязательно замещенным одной или несколькими группами, выбранными из F, Cl, Br, метила и -NO2. В более конкретных вариантах реализации изобретения R6 представляет собой метил, трифторметил, 2,2,2-трифторэтил, нонафторбутил (CF3CF2CF2CF2-), п-толил, п-нитрофенил или п-бромфенил. В более конкретных вариантах реализации изобретения R6 представляет собой метил.
Как используется в данном документе, термин «сульфирующие агенты» можно понимать как реагенты, которые образуют сложные эфиры сульфоновой кислоты (RSO3R') при реакции с соединением формулы III. Такие сложные эфиры сульфоновой кислоты образуются за счет замещения уходящей группы (например, хлорида), присоединенной к атому серы сульфирующего агента, атомом кислорода соединения формулы III.
Пригодные сульфирующие агенты включают ангидриды сульфоновых кислот и, в частности, сульфонилгалогениды (например, сульфонилхлориды). Конкретные сульфонирующие агенты, которые можно упомянуть, включают метансульфоновый ангидрид (мезилангидрид), метансульфонилхлорид (мезилхлорид), трифторметансульфонилхлорид (трифлилхлорид), трифторметилсульфоновый ангидрид (трифлилангидрид), 2,2,2-трифторэтансульфонилхлорид (трезилхлорид), перфтор-1-бутансульфонилфторид, перфтор-1-бутансульфонилхлорид, п-толуолсульфонилхлорид (тозилхлорид), п-толуолсульфоновый ангидрид (тозилангидрид), п-нитробензолсульфонилхлорид (нозилхлорид) и 4-бромбензолсульфонилхлорид (брозилхлорид). Более конкретные сульфонирующие агенты, которые можно упомянуть, включают метансульфоновый ангидрид (мезилангидрид), метансульфонилхлорид (мезилхлорид), п-толуолсульфонилхлорид (тозилхлорид) и п-толуолсульфоновый ангидрид (тозилангидрид). В конкретных вариантах реализации изобретения указанный сульфирующий агент представляет собой метансульфонилхлорид (мезилхлорид).
Другие стадии способа
В некоторых вариантах реализации изобретения способ по данному изобретению включает дополнительные стадии способа, которые можно выполнять между, во время или после стадий (i)-(iii), в зависимости от ситуации.
В конкретных вариантах реализации изобретения указанный способ дополнительно включает следующую стадию:
(ib) удаление (например, фильтрацией) соли, образованной между B1 и уходящей группой сульфирующего агента, из раствора соединения формулы II в S1, полученного на стадии (i).
Специалист в данной области техники поймет, что стадию (ib) выполняют после завершения реакции стадии (i) и перед началом стадии (ii).
Не желая ограничиваться теорией, считается, что стадия (ib) дает преимущество предотвращения участия аниона, образованного из уходящей группы сульфирующего агента, (например, хлорида) в конкурирующих процессах нуклеофильного замещения во время стадии (ii) (с, например, образованием соответствующего бензилхлорида соединения формулы II), которое может привести к рацемизации соединения формулы I (посредством повторяющихся обращений стереохимии).
В более конкретных вариантах реализации изобретения указанный способ дополнительно включает следующую стадию:
(ic) уменьшение объема раствора соединения формулы II в S1 перед стадией (ii).
Специалист в данной области техники поймет, что ссылки на уменьшение объема раствора относятся к упариванию раствора путем удаления части растворителя. Это может быть достигнуто с помощью стандартных лабораторных методов, таких как перегонка или, в частности, упаривание на роторном испарителе.
Уменьшение объема раствора таким образом имеет то преимущество, что соединение формулы IV остается в растворе во время стадии (ii). Если слишком высокая доля S1 присутствует в реакционной смеси на стадии (ii), соединение формулы IV может начать выпадать в осадок и, таким образом, стать неспособным участвовать в реакции.
Степень, до которой необходимо уменьшить объем раствора соединения формулы II в S1, может легко определить специалист в данной области техники, и она будет зависеть от концентрации, при которой выполняли стадию (i). Например, объем раствора может быть уменьшен на около 30%, например, на около 50%, например, на около 60-70% (т. е. объем раствора может быть уменьшен до около одной трети от его исходного объема).
После завершения стадии (ii) неочищенный продукт получают стандартными лабораторными методами, такими как удаление растворителей (например, смеси S1 и S2) упариванием на роторном испарителе (или перегонкой, если необходимо), отмывка водой и/или осаждение/фильтрация побочных продуктов, таких как непрореагировавшие соединения меркаптопиримидина. Когда присутствуют высококипящие растворители, такие как N,N-диметилформамид, удаление реакционных растворителей может включать несколько манипуляций, таких как: упаривание реакционной смеси на роторном испарителе, отмывка водой и, если необходимо, перегонка при пониженном давлении.
В конкретных вариантах реализации изобретения указанный способ включает одну или более следующих стадий:
(iib) упаривание реакционной смеси роторным испарителем;
(iic) растворение остатка, полученного после стадии (iib), в пригодном растворителе (S3), чтобы вызвать осаждение любых непрореагировавших частиц меркаптопиримидина (соединения формулы IV/V) и фильтрование суспензии для удаления осадка;
(iid) растворение осадка, полученного на стадии (iic), в смеси воды или водного раствора (например, раствора основания (например, насыщенного раствора Na2CO3) и S3, последующее разделение водного и органического слоев и объединение органического слоя с фильтратом со стадии (iiic);
(iie) упаривание объединенных органических слоев роторным испарителем; и
(iif) при необходимости, удаление остаточного растворителя перегонкой при пониженном давлении с целью получения неочищенного продукта.
В некоторых вариантах реализации изобретения S3 представляет собой этилацетат.
В конкретных вариантах реализации изобретения способ по данному изобретению дополнительно включает следующую стадию:
(iiib) обработка реакционной смеси стадии (iii) пригодным восстанавливающим агентом для восстановления любого дисульфида (VI), образующегося в реакционной смеси.
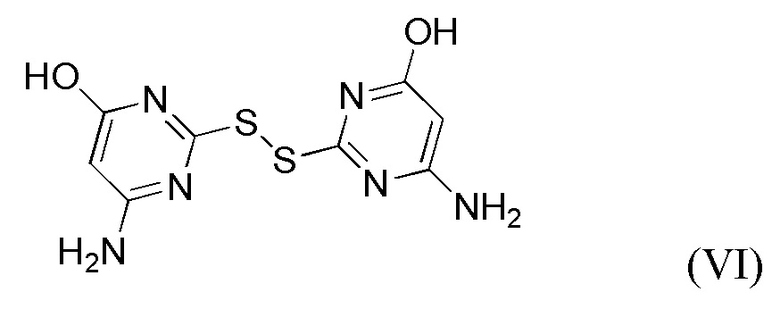
Пригодные восстанавливающие агенты для применения на стадии (iiib) включают NaBH4 , NaB(OAc)3H, NaB(CN)H3 и LiAlH4. В предпочтительных вариантах реализации изобретения указанный восстанавливающий агент представляет собой NaBH4.
Включение стадии (iiib) в способ по данному изобретению преобразует дисульфид обратно в тиол/тиолат, который затем может реагировать на стадии (ii).
В конкретных вариантах реализации изобретения стадию (iiib) выполняют при комнатной температуре. В таких вариантах реализации изобретения реакционной смеси со стадии (iii) дают остыть до комнатной температуры перед добавлением восстанавливающего агента в реакцию.
В конкретных вариантах реализации изобретения способ по данному изобретению включает следующую стадию:
(iv) обработка неочищенного материала, полученного на стадии (ii) (например, после того, как была выполнена одна или несколько стадий (iib)-(iif)) пригодным растворителем S4, чтобы вызвать осаждение соединения формулы I.
Специалисту в данной области техники будет понятно, что стадия (iv) также может быть описана как очистка неочищенного продукта растиранием в пригодном растворителе (S4).
Специалист в данной области техники поймет, что ссылки на неочищенный материал относятся к материалу, полученному после того, как с реакционной смесью были выполнены стандартные манипуляции в области органической химии (например, описанные в стадиях (iib)-(iif)) для, например, удаления растворителей и/или водорастворимых побочных продуктов, чтобы получить материал, пригодный для дальнейшей очистки. В частности, под неочищенным материалом можно понимать материал, полученный после удаления реакционных растворителей и любых побочных продуктов, которые можно удалить путем осаждения/фильтрации и/или отмывки водой.
В конкретных вариантах реализации изобретения S4 представляет собой ацетонитрил.
Включение стадии (iv) в способ по данному изобретению может дополнительно повысить энантиомерную чистоту продукта, так как осадок, оказывается обогащенным мажорным энантиомером по сравнению с неочищенным продуктом.
В конкретном варианте реализации изобретения, который можно упомянуть, указанный способ включает следующую комбинацию стадий, которые определены в данном документе:
I. стадии (i) (сульфирование) и (iii) (образование тиолат-аниона), которые выполняются одновременно; затем
II. стадия (ib) (фильтрация для удаления нерастворимой соли, образовавшейся на стадии (i)) и, необязательно, стадия (ic) (уменьшение объема раствора соединения формулы II в S1); и
III. необязательно, стадия (iiib) (восстановление дисульфида);
IV. стадия (ii) (нуклеофильное замещение для получения соединения формулы I), включающая добавление по каплям раствора, полученного после стадии (ib) (или, если она включена, после стадии (ic)), к раствору, полученному после стадии (iii) (или, если она включен, после стадии (iiib));
V. стадия (iv) (осаждение соединения формулы I).
Конкретные энантиомеры
Как описано выше, в способе по данному изобретению образуются отдельные энантиомеры соединения формулы I в зависимости от энантиомерной чистоты исходного материала (соединения формулы II и, в конечном итоге, соединения формулы III, из которого оно получено).
Соответственно, указанный способ предназначен для получения соединения формулы Ia или Ib или его соли,
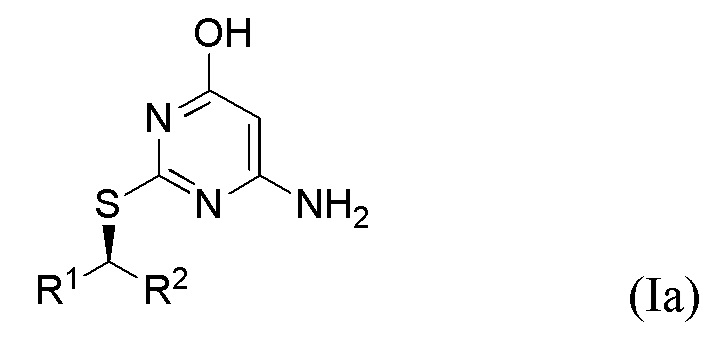
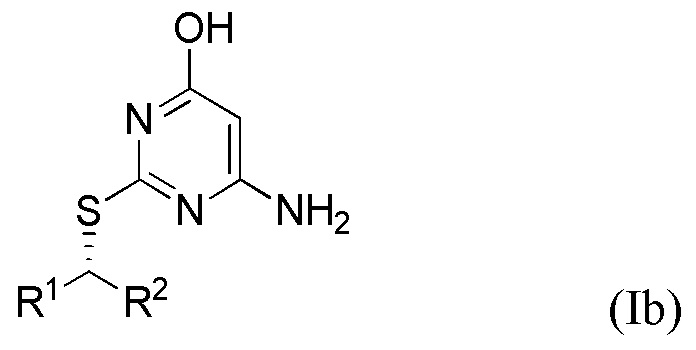
где R1 и R2 являются такими, как определено выше в данном документе.
Поскольку инверсия стереохимии происходит во время стадии (ii), специалист в данной области техники поймет, что соединение формулы Ia получают в результате реакции соединения формулы IIa, и что соединение формулы Ib получают в результате реакции соединения формулы IIb,
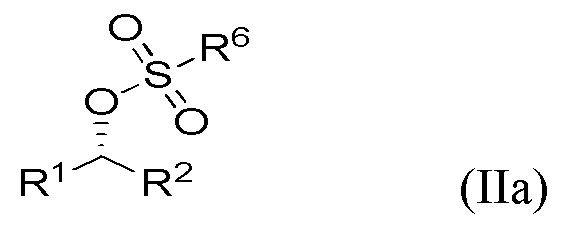
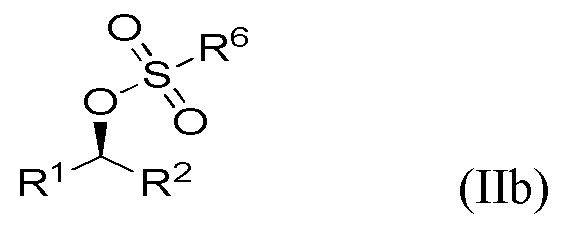
где R1 и R2 являются такими, как определено выше в данном документе.
Соединения IIa и IIb получают на стадии (i) указанного способа из соответствующих соединений формул IIIa и IIIb.


где R1 и R2 являются такими, как определено выше в данном документе.
В конкретном варианте реализации изобретения указанный способ предназначен для получения соединения формулы Ia.
В конкретных вариантах реализации изобретения соединение формулы I (т. е. соединение формулы Ia или Ib (например, Ia)) имеет хиральную чистоту более 96%, предпочтительно, более 99%, более предпочтительно, более 99,5%, например 99,7%.
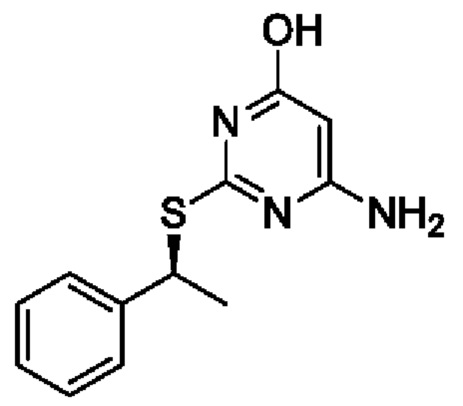
В более конкретных вариантах реализации изобретения, которые можно упомянуть, указанный способ предназначен для получения 6-амино-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-4-ола или его соли.
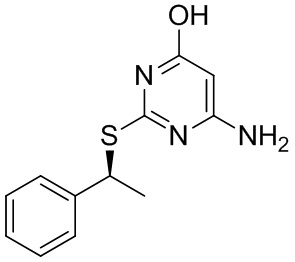
Согласно способу по данному изобретению 6-амино-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-4-ол или его соль могут быть получены с хиральной чистотой более 96%, предпочтительно, более 99%, более предпочтительно, более 99,5%, например 99,7%.
Дальнейшие способы
Соединения, полученные способом по данному изобретению (т. е. соединения формулы I, Ia или Ib (в частности, Ia)), могут быть полезны в качестве синтетических промежуточных продуктов при получении фармацевтически активных молекул.
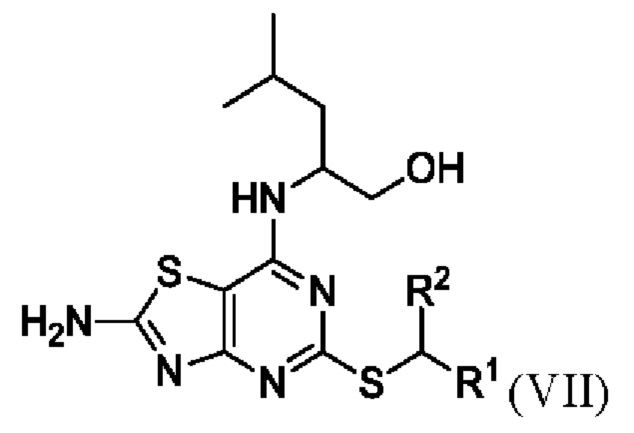
Соответственно, во втором аспекте данного изобретения предложен способ получения соединения формулы VII или его соли,
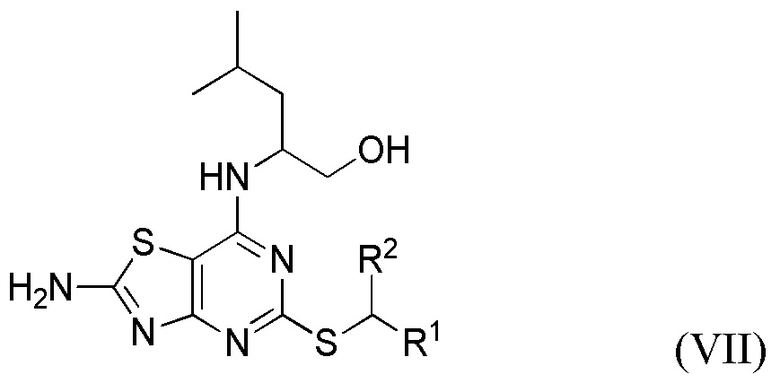
где R1 и R2 являются такими, как определено выше в данном документе,
где указанный способ включает способ по данному изобретению, как определено выше в данном документе (т. е. способ, включающий стадии (i)-(iii) (необязательно, включающий одну или несколько дополнительных стадий способа, описанных в данном документе) и, необязательно, стадию (iv), как определено выше в данном документе).
В конкретных вариантах реализации второго аспекта данного изобретения указанный способ дополнительно включает следующие стадии:
(v) Получение соединения формулы VIII или его соли,
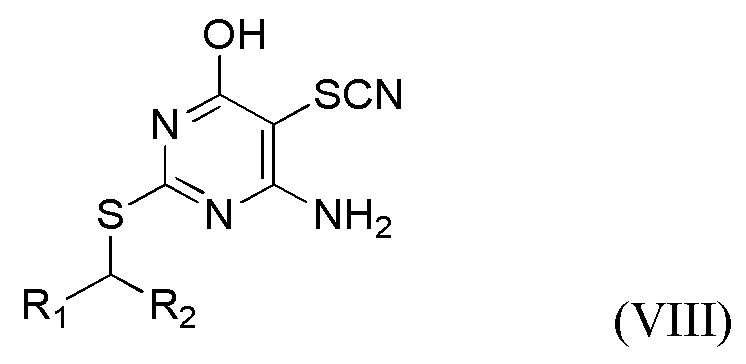
где R1 и R2 являются такими, как определено выше в данном документе, путем приведения в контакт соединения формулы I, определенного выше в данном документе, с пригодной тиоцианатной солью (например, LiSCN, NaSCN или, в частности, KSCN) в присутствии брома, подходящего основания (например, 2,6-лутидина или, в частности, пиридина) и пригодного растворителя (например, полярного апротонного растворителя, такого как упомянутые выше в данном документе, в частности, N,N-диметилформамида).
(vi) Получение соединения формулы IX или его соли,
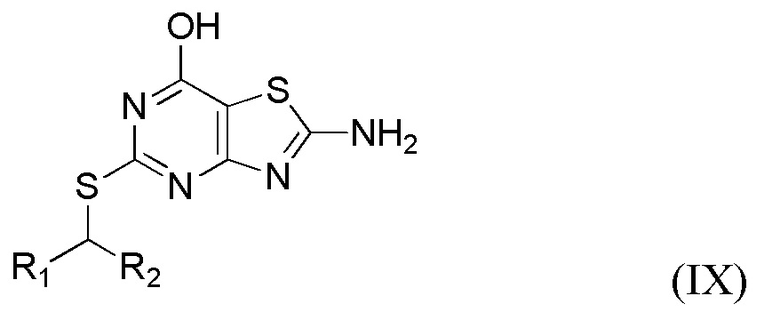
где R1 и R2 являются такими, как определено выше в данном документе, путем приведения в контакт соединения формулы VIII в присутствии пригодного основания (например, LiOH, KOH, CsOH или, в частности, NaOH) и пригодного растворителя (например, полярного апротонного растворителя, такого как упомянутые выше в данном документе, в частности, N,N-диметилформамида) при температуре от около 80°C до около 150°C (например, от около 100°C до около 140°C, например, от около 110°C до около 130°C (например, около 120°C)).
(vii) Получение соединения формулы X или его соли,
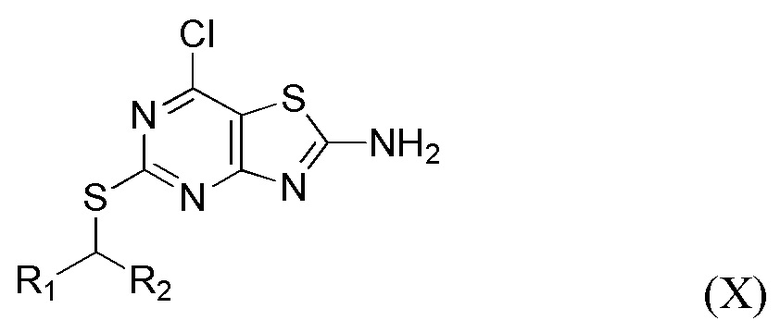
где R1 и R2 являются такими, как определено выше в данном документе, путем приведения в контакт соединения формулы IX в присутствии пригодного хлорирующего агента (например, SOCl2 или, в частности, POCl3), N,N-диметилформамида и пригодного растворителя (например, тетрагидрофурана, 2-метилтетрагидрофурана или, в частности, 1,4-диоксана) при температуре от около 40°C до около 80°C (например, от около 50°C до около 75°C, например, от около 60°C до около 70°C (например, около 65°C)).
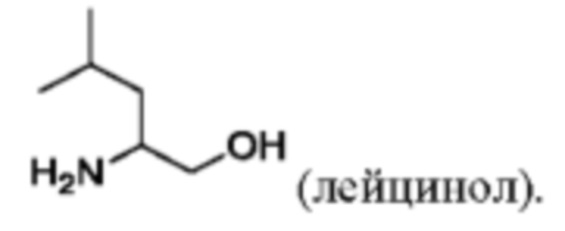
(viii) Получение соединения VII, как определено выше в данном документе, путем приведения в контакт соединения формулы X с лейцинолом
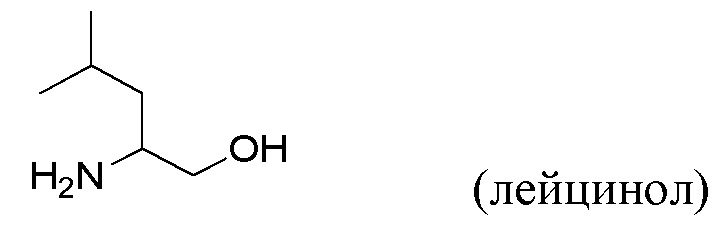
в присутствии пригодного основания (например, триэтиламина или, в частности, N,N-диизопропилэтиламина) и пригодного растворителя (например, полярного апротонного растворителя, такого как указанные выше в данном документе, в частности N-метил-2-пирролидона) при температуре от около 80°С до около 150°С (например, от около 100°С до около 140°С, например, от около 110°С до около 130°С (например, около 120°С)).
В дополнительных вариантах реализации изобретения, которые можно упомянуть, указанный способ дополнительно включает образование соли HCl (например, моно HCl соли) соединения формулы VII путем обработки соединения формулы VII HCl в присутствии пригодного растворителя (например, метилэтилкетона).
В конкретных вариантах реализации изобретения соединение формулы VII представляет собой единственный стереоизомер.
Ссылки в данном документе на соединения, получаемые в виде отдельных стереоизомеров, можно понимать как указание на то, что соединение получают в виде единственного диастереомера (например, в случае соединения VII, (R,S)-диастереомера) и единственного энантиомера данного диастереомера.
Если не указано иное, ссылки на отдельные стереоизомеры в данном документе (и аналогично стереоизомерам, присутствующим в чистой форме и т. п.) можно понимать как указывающие на хиральную чистоту по меньшей мере около 90%, например, по меньшей мере около 95%, например по меньшей мере около 97%. Более конкретно, один стереоизомер может иметь хиральную чистоту по меньшей мере около 99,5%, например, по меньшей мере около 99,7%.
Как используется в данном документе, термин "хиральная чистота" может означать количество (% мас./мас.) одного стереоизомера (т. е. диастереомера и/или энантиомера) в соответствующей партии соединения.
В конкретных вариантах реализации изобретения соединение формулы VII представляет собой 2-[(2-амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол или его соль.
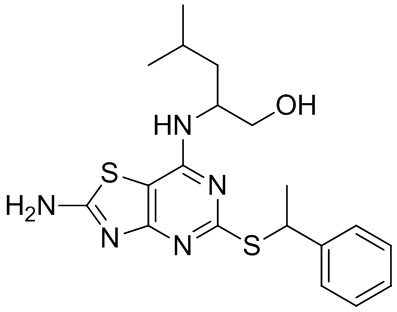
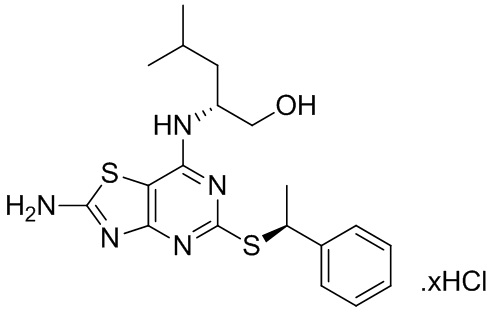
В более конкретных вариантах реализации изобретения соединение формулы VII представляет собой (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол или его соль.
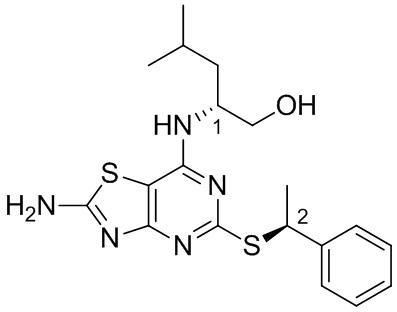
Специалист в данной области техники поймет, что стереохимию хирального центра, обозначенного «1», можно контролировать, выбирая любой энантиомер лейцинола (для обеспечения желаемой (R)-стереохимии на данном центре применяют d-лейцинол). Как описано выше, стереохимия на хиральном центре, обозначенном «2», зависит от стереохимии хирального бензилового спирта, применяемого в качестве исходного материала на стадии (i) (соединение формулы III). В способе по данному изобретению предложен масштабируемый синтетический путь для достижения более высокой хиральной чистоты на данном стереоцентре.
В конкретных вариантах реализации второго аспекта данного изобретения (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол имеет хиральную чистоту более 99,2%, предпочтительно, более 99,5%. (например 99,7%).
Соединения с улучшенной хиральной чистотой
В способах, описанных в данном документе, предложен масштабируемый синтетический путь к соединениям с более высокой хиральной чистотой, чем те, которые можно получить способами, описанными ранее.
Соответственно, в третьем аспекте данного изобретения предложено соединение 6-амино-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-4-ол или его соль,
имеющее хиральную чистоту более 99,1%, предпочтительно, более 99,5%.
В четвертом аспекте данного изобретения предложено соединение (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол или его соль,
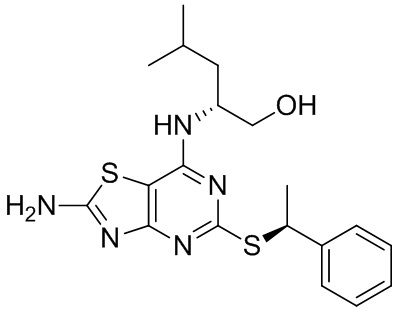
имеющее хиральную чистоту более 99,8%.
Фармацевтические составы
Соединения формулы VII, в частности, (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол или его соль, могут быть полезными в качестве фармацевтических препаратов. По существу, данные соединения могут быть составлены в фармацевтически приемлемый состав стандартными методиками.
В пятом аспекте данного изобретения предложен способ получения фармацевтического состава, содержащего (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол или его соль, (или другое пригодное соединение формулы VII или его соль) характеризующийся тем, что включает стадии способа (в частности, стадии (i)-(iv), включая дополнительные стадии, такие как стадия (ib) и (iiib) и другие). Специалист в данной области техники будет знать, что такие фармацевтические составы будут содержать/состоять из смеси активного ингредиента и, необязательно, одного или более фармацевтически приемлемых вспомогательных веществ, адъювантов, разбавителей и/или носителей).
В шестом аспекте данного изобретения предложен способ получения фармацевтического состава, содержащего (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол или его соль, (или другое пригодное соединение формулы VII или его соль) включающий приведение в контакт (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола или его соли, (или другого пригодного соединения формулы VII или его соли) (которое может быть образовано способом, описанным выше в данном документе) с (а) одним или несколькими фармацевтически приемлемыми вспомогательными веществами (например, адъювантом, разбавителем и/или носителем).
Способы, описанные в данном документе, имеют то преимущество, что они более эффективны и более подходят для применения в крупномасштабном синтезе, чем способы предшествующего уровня техники. В частности, преимущества данных способов заключаются в обеспечении масштабируемого способа производства фармацевтически активных соединений с превосходной хиральной чистотой.
Полученные соединения обладают более высокой хиральной чистотой, чем те, которые доступны другими масштабируемыми синтетическими путями.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
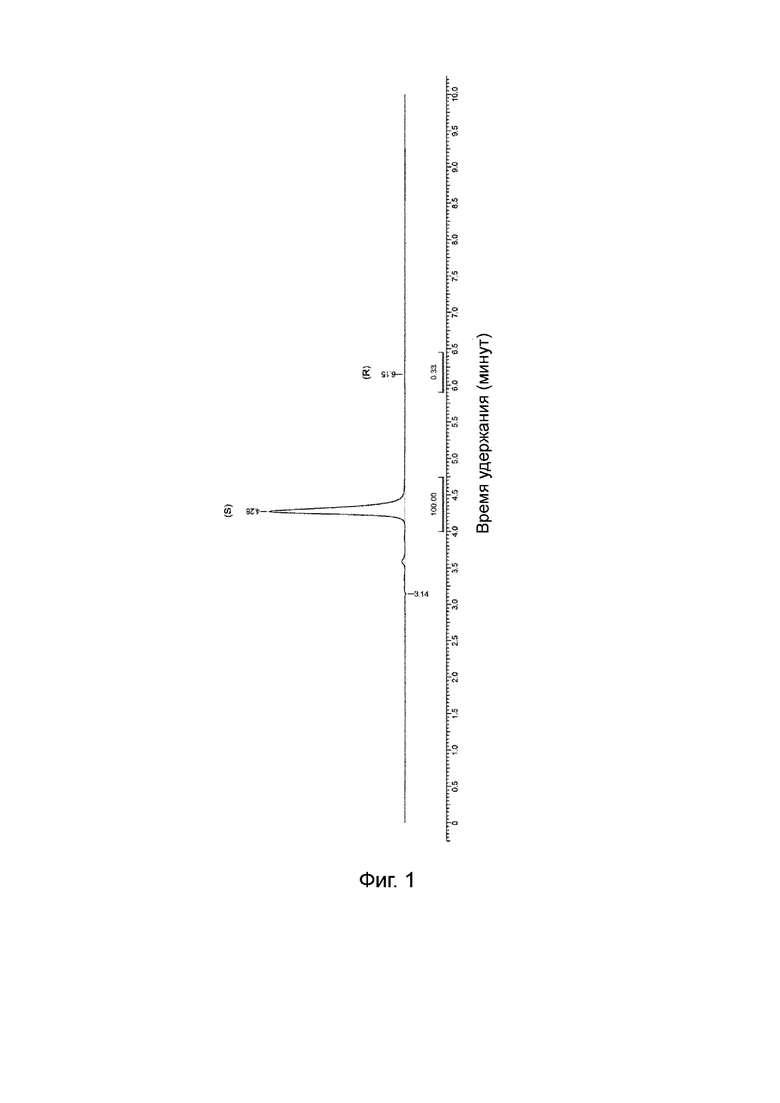
На Фиг. 1 показана хроматограмма хиральной ВЭЖХ для продукта, полученного способом, описанным в Примере 1.
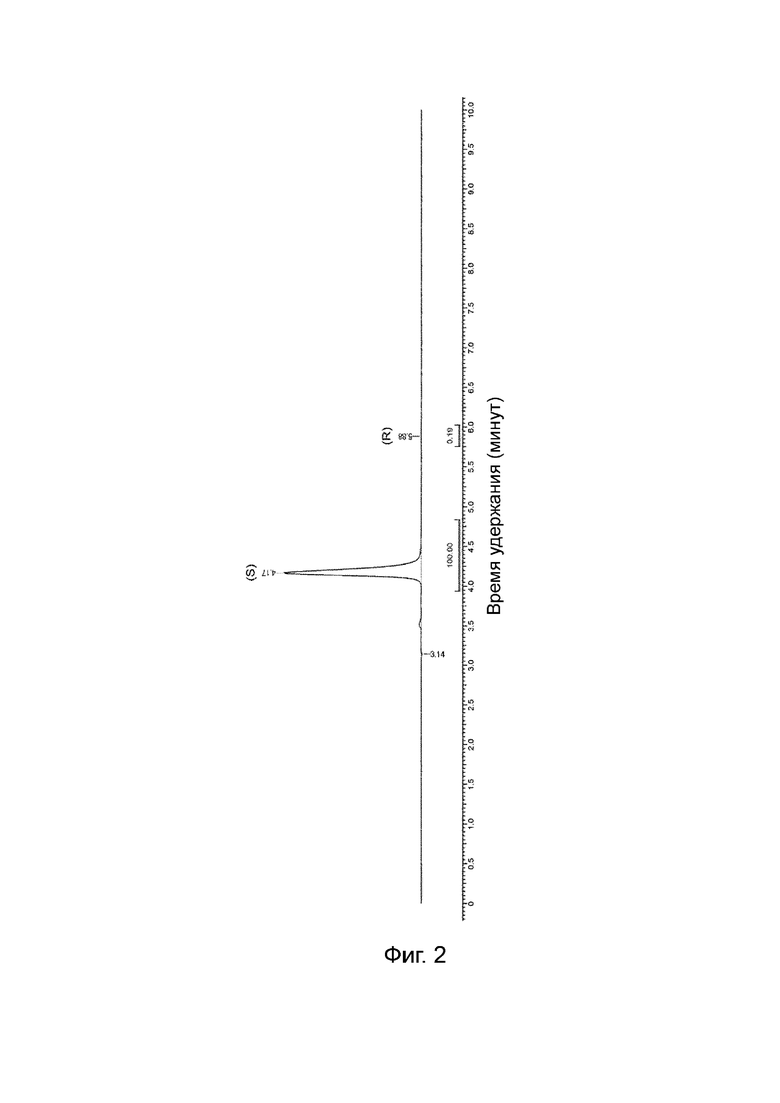
На Фиг. 2 показана хроматограмма хиральной ВЭЖХ для продукта, полученного способом, описанным в Примере 2.
Примеры
Сокращения
ДИПЭА N,N-диизопропилэтиламин
ДМФА N,N-диметилформамид
ВЭЖХ высокоэффективная жидкостная хроматография
МЭК метилэтилкетон
МТБЭ метил трет-бутиловый эфир
НМП N-метил-2-пирролидон
ЯМР ядерный магнитный резонанс
МС масс-спектрометрия
Пример 1 - Получение 6-амино-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-4-ола (1)
Образование тиолат-аниона:
В круглую колбу объемом 250 мл загружали хлопья NaOH (600 мг, 15,00 ммоль),
6-амино-2-тиоксо-1H-пиримидин-4-он (Ark Pharm, 95+%, 2,37 г, 15,75 ммоль) и ДМФА (30 мл). Колба была снабжена холодильником. Смесь перемешивали при 65°C (начало в 13:25). Через 1 ч 30 минут смеси давали остыть до комнатной температуры и добавляли NaBH4 (174 мг, 4,50 ммоль, 30%) (15:10) для уменьшения количества образовавшегося дисульфида. Смесь перемешивали при комнатной температуре в течение 1 часа 50 минут.
Образование мезилата:
Параллельно с реакцией, описанной выше, следующим образом получали (R)-1-фенилэтилмезилат в МТБЭ:
К перемешиваемому раствору (R)-1-фенилэтанола (Ark Pharm, 98%, 1,87 г, 15,00 ммоль) и триэтиламина (Aldrich, 99,5%, 1,60 г, 15,75 ммоль) в МТБЭ (15 мл) добавляли по каплям раствор мезилхлорида (Lancaster, 98%, 1,84 г, 15,75 ммоль) в МТБЭ (10 мл) в течение 14 минут при температуре ледяной бани. Смесь перемешивали на ледяной бане в течение 20 минут. Образовавшуюся соль Et3NHCl отфильтровывали и осадок на фильтре промывали около 10 мл МТБЭ. Раствор МТБЭ упаривали до около 10 мл.
Стадия алкилирования:
Раствор МТБЭ, содержащий мезилат, добавляли по каплям к раствору ДМФА, содержащему анион 6-амино-2-тиоксо-1H-пиримидин-4-она, в течение 10 минут при температуре ледяной бани (17:10). Смеси давали медленно нагреться до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Отмывали через 17,5 часов: Смесь упаривали до небольшого объема. Добавляли EtOAc (около 50 мл), что привело к сильному осаждению 6-амино-2-тиоксо-1H-пиримидин-4-она. Смесь обрабатывали ультразвуком, затем твердое вещество отфильтровывали и осадок на фильтре промывали EtOAc (около 10 мл). Твердое вещество суспендировали в насыщенном растворе NaHCO3 (15 мл) и добавляли EtOAc (20 мл). Полученную суспензию обрабатывали ультразвуком и фильтровали в делительную воронку. Фазы разделяли и водную фазу отбрасывали. Органическую фазу, содержащую продукт, объединяли с фильтратом EtOAc. Объединенные органические фазы промывали насыщенным раствором NaHCO3 (10 мл), сушили над MgSO4, фильтровали и упаривали. Последний остаток ДМФА отгоняли (78°C/18 мбар) с получением 4,38 г неочищенного продукта в виде светло-желтого вязкого масла. К данному маслу добавляли CH3CN (11 мл) и полученный раствор обрабатывали ультразвуком, и через несколько минут наблюдалось сильное осаждение. Смесь нагревали при 75°C в течение 45 минут. Смеси давали медленно остыть до комнатной температуры, твердое вещество собирали фильтрованием и промывали ледяным CH3CN (около 5 мл). Полученный белый твердый материал сушили в вакуумной печи при 40°C в течение 2,5 дней. Выход: 1,861 г. 1H ЯМР (600 МГц, CD3OD) δ м.д. 7,43-7,47 (м, 2H) 7,31-7,35 (м, 2H) 7,23-7,28 (м, 1H), 5,12 (к, J=7,1 Гц, 1H) 5,06 (с, 1H) 1,75 (д, J=7,0 Гц, 3H). МС (ESI+) m/z 248 [M+H]+.
Степень чистоты по ВЭЖХ: около 94%
Хиральная чистота по ВЭЖХ: 99,7% (S), т. е. 99,4% ее. Хроматограмма хиральной ВЭЖХ проиллюстрирована на Фиг. 1.
Измерено на колонке Astec CHIROBIOTIC T (250x4,6 мм, 5 мкм), Элюент: 100% MeOH, система Agilent 1100.
1H ЯМР: выглядит чистым, за исключением некоторого остаточного растворителя (MeCN: 0,03% (мас./мас.), ДМФА: 0,16% (мас./мас.)).
Пример 2 - Получение 6-амино-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-4-ола (1) (масштаб 50 ммоль)
Образование тиолат-аниона:
В круглодонную колбу на 250 мл загружали хлопья NaOH (2,00 г, 50,00 ммоль),
6-амино-2-тиоксо-1H-пиримидин-4-он (Ark Pharm, 95+%, 7,91 г, 52,50 ммоль) и ДМФА (100 мл). Колба была снабжена холодильником. Смесь перемешивали при 65°C (начало в 11:00). Через 2 ч 30 минут смеси давали остыть до комнатной температуры и добавляли NaBH4 (567 мг, 15,00 ммоль, 30%) (14:10) для уменьшения количества образовавшегося дисульфида. Смесь перемешивали при комнатной температуре в течение 40 минут.
Образование мезилата:
Параллельно с реакцией, описанной выше, следующим образом получали (R)-1-фенилэтилмезилат в МТБЭ:
К перемешиваемому раствору (R)-1-фенилэтанола (Ark Pharm, 98%, 6,23 г, 50,00 ммоль) и триэтиламина (Aldrich, 99,5%, 5,34 г, 52,50 ммоль) в МТБЭ (50 мл) добавляли по каплям раствор мезилхлорида (Lancaster, 98%, 6,14 г, 52,50 ммоль) в МТБЭ (30 мл) в течение 10 минут при температуре ледяной бани. Смесь перемешивали на ледяной бане в течение 20 минут. Образовавшуюся соль Et3NHCl отфильтровывали и осадок на фильтре промывали около 20 мл МТБЭ. Раствор МТБЭ упаривали до около 25 мл.
Стадия алкилирования:
Раствор МТБЭ, содержащий мезилат, добавляли по каплям к раствору ДМФА, содержащему анион 6-амино-2-тиоксо-1H-пиримидин-4-она, в течение 20 минут при температуре ледяной бани (15:05). Смеси давали медленно нагреться до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Отмывали через 19 часов: Смесь упаривали до небольшого объема, так что почти весь ДМФА был удален. Добавляли EtOAc (120 мл), что привело к сильному осаждению 6-амино-2-тиоксо-1H-пиримидин-4-она. Смесь обрабатывали ультразвуком, затем твердое вещество отфильтровывали и осадок на фильтре промывали EtOAc (около 30 мл). Твердое вещество суспендировали в насыщенном растворе Na2CO3 (50 мл) и добавляли EtOAc (70 мл). Полученную суспензию перемешивали до растворения всего материала и затем его переносили в делительную воронку. Фазы разделяли и водную фазу отбрасывали. Органическую фазу, содержащую продукт, объединяли с фильтратом EtOAc. Объединенные органические фазы промывали насыщенным раствором NaHCO3 (25 мл), водой (15 мл), солевым раствором (15 мл), сушили над MgSO4, фильтровали и упаривали. Последний остаток ДМФА отгоняли (78°C/18 мбар) с получением 13,32 г неочищенного продукта в виде светло-желтого полутвердого вещества. К неочищенному материалу добавляли CH3CN (35 мл) и полученную суспензию обрабатывали ультразвуком. Смесь нагревали при 75°C в течение 30 минут. Смеси давали медленно остыть до комнатной температуры, твердое вещество собирали фильтрованием и промывали ледяным CH3CN (около 10 мл). Полученный белый твердый материал сушили в вакуумной печи при 40°C в течение 2 дней. Выход: 6,168 г (50%) продукта в виде белого твердого вещества. 1H ЯМР (600 МГц, CD3OD) δ м.д. 7,43-7,47 (м, 2H) 7,31-7,35 (м, 2H) 7,23-7,28 (м, 1H), 5,13 (к, J=7,0 Гц, 1H) 5,06 (с, 1H) 1,75 (д, J=7,0 Гц, 3H). МС (ESI+) m/z 248 [M+H]+.
Степень чистоты по ВЭЖХ: 96,5%
Хиральная чистота по ВЭЖХ: 99,8% (S), т. е. 99,6% ее. (Пик, соответствующий R-изомеру, трудно обнаружить. Интеграл является неопределенным из-за шума базовой линии). Хроматограмма хиральной ВЭЖХ проиллюстрирована на Фиг. 2.
Измерено на колонке Astec CHIROBIOTIC T (250x4,6 мм, 5 мкм), Элюент: 100% MeOH, система Agilent 1100.
1H ЯМР: выглядит чистым, за исключением некоторого остаточного растворителя (MeCN: 0,41% (мас./мас.), ДМФА: 0,23% (мас./мас.)).
Пример 3 - Получение (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола.HCl
3.1 Получение 4-амино-6-гидрокси-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-5-ил тиоцианата (2)
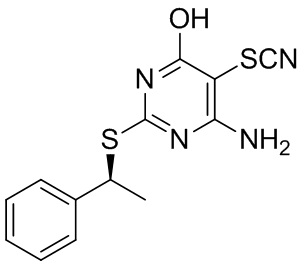
Соединение 1 (1,63 г, 6,59 ммоль) и KSCN (1,92 г, 19,8 ммоль) растворяли в ДМФА (20 мл) в колбе на 100 мл. Смесь нагревали до 65°C до растворения всего исходного материала. Добавляли пиридин (886 мг, 12,35 ммоль) в ДМФА (2 мл). Реакционную смесь охлаждали до 4°C и по каплям добавляли бром (1,219 г, 7,62 ммоль) в ДМФА (3 мл) (15:12) в течение 10 минут, что приводило к некоторому осаждению. Смесь перемешивали при 4-10°C в течение 1 часа 15 минут, когда ВЭЖХ показала полную конверсию в продукт. Реакционной смеси давали нагреться до комнатной температуры и добавляли 5 мл 15% Na2S2O5 (метабисульфит натрия) вместе с водой (2 мл). Смесь перемешивали при 60°C в течение 10 минут. Затем при комнатной температуре дополнительно добавляли воду (25 мл). Перемешивали при комнатной температуре в течение ночи. Твердый продукт собирали фильтрацией, промывали холодной водой и сушили 4,5 часов под вакуумом при 60°C. Выход: 1,806 г (90%) продукта со степенью чистоты 98% в виде светло-желтого твердого вещества. 1H ЯМР (600 МГц, CD3OD) δ м.д. 7,44-7,48 (м, 2H) 7,32-7,36 (м, 2H) 7,25-7,29 (м, 1H) 5,14 (к, J=7,1 Гц, 1H) 1,77 (д, J=7,3 Гц, 3H). МС (ESI+) m/z 305 [M+H]+.
1H ЯМР: выглядит очень чистым. Единственное видимое загрязнение представляет собой ДМФА (около 0,36% мас./мас.)
3.2 Получение 2-амино-5-{[(1S)-1-фенилэтил]сульфанил}[1,3]тиазоло[4,5-d]пиримидин-7-ола (3)
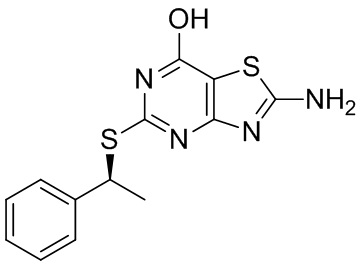
Соединение 2 (1,80 г, 5,91 ммоль) суспендировали в ДМФА (16 мл) и по каплям добавляли 2 М NaOH (6 мл, 12 ммоль) при комнатной температуре. После добавления NaOH смесь стала красно-коричневой, и суспензия растворилась. Нагревали при 120°C (13:20) (ВЭЖХ через 3 часа: 100% конверсия в продукт).
Отмывали: смеси давали остыть до комнатной температуры и добавляли воду (20 мл) => небольшое осаждение. pH реакционной смеси доводили до около pH 4-5 путем добавления конц. HCl. Твердое вещество собирали фильтрованием, промывали двумя порциями воды (2×8 мл) и сушили под вакуумом при 60°C с получением 1,687 г (94%) продукта в виде сероватого твердого вещества. 1H ЯМР (600 МГц, CD3OD) δ м.д. 7,45-7,49 (м, 2H) 7,31-7,35 (м, 2H) 7,24-7,28 (м, 1H) 5,21 (к, J=7,1 Гц, 1H) 1,79 (д, J=7,0 Гц, 3H). МС (ESI+) m/z 305 [M+H]+.
ВЭЖХ: около 94% степень чистоты
1H ЯМР: выглядит чистым, но присутствует некоторое количество ДМФА. Интегралы указывают 0,55% (мас./мас.).
3.3 Получение 7-хлор-5-{[(1S)-1-фенилэтил]сульфанил}[1,3]тиазоло[4,5-d]пиримидин-2-амина (4)
К суспензии соединения 3 (1,82 г, 5,98 ммоль) в диоксане (15 мл) добавляли ДМФА (962 мг, 13,2 ммоль) в диоксане (2,5 мл) и POCl3 (3,03 г, 19,7 ммоль) в диоксане (2,5 мл) при комнатной температуре (начало: 14:00). Через 30 минут суспензия растворилась с образованием светло-коричневого гомогенного раствора. ВЭЖХ через 0,5 часа при комнатной температуре показала имин, образованный ДМФА и исходным материалом (68%), и имин, образованный продуктом и ДМФА (32%). Затем реакционную смесь нагревали при 65°C (начало: 13:40). ВЭЖХ через 3 часа при 65°C показала полную конверсию в имин, образованный ДМФА и продуктом. Реакционной смеси давали остыть до комнатной температуры и добавляли воду (5 мл). Температуру повышали до 65°C (17:40). После перемешивания в течение 10 минут при данной температуре весь продукт-имин гидролизовался. Смесь охлаждали до комнатной температуры. Добавляли воду (10 мл) и смесь упаривали до около половины объема. pH доводили до около 6-7 с использованием насыщенного раствора NaHCO3 и твердого NaHCO3. Полученную фазу экстрагировали EtOAc (3×25 мл) и объединенные органические фазы промывали насыщенным раствором Na2CO3 (2×6 мл), водой (6 мл) и солевым раствором (6 мл), сушили над Na2SO4 и упаривали с получением 1,746 г (98%) продукта со степенью чистоты >97% в виде светло-оранжевого твердого вещества. 1H ЯМР (600 МГц, CD3OD) δ м.д. 7,44-7,49 (м, 2H) 7,28-7,33 (м, 2H) 7,20-7,25 (м, 1H) 5,06 (к, J=7,1 Гц, 1H) 1,75 (д, J=7,3 Гц, 3H). МС (ESI+) m/z 323 [M+H]+.
1H ЯМР: выглядит очень чистым, но еще присутствует растворитель. ДМФА: 2,7% (мас./мас.), EtOAc: 1,8% (мас./мас.), диоксан: 1,8% (мас./мас.).
Выход с поправкой на содержание растворителя: около 5,065 г (92%).
3.4 Получение (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола.xHCl (5)
Соединение 4 (1,852 г, 5,74 ммоль), ДИПЭА (1,112 г, 8,61 ммоль) и D-лейцинол (1,008 г, 8,61 ммоль) растворяли в НМП (12 мл) и смесь перемешивали при 120°C в герметичной пробирке из пирекса (начало: 17:40).
ВЭЖХ через 15,5 часов: конверсия около 98%
ВЭЖХ через 19,5 часов: конверсия >99%
Отмывали: в смесь наливали ледяную воду. Первоначально образовывалось твердое вещество, но в конце добавления твердое вещество преобразовалось в темно-коричневое масло. Добавляли EtOAc (50 мл) и фазы разделяли. Водную фазу экстрагировали EtOAc (2×25 мл) и объединенные органические фазы промывали водой (8 мл), насыщенным раствором NaHCO3 (3×8 мл), водой (8 мл) и солевым раствором (8 мл), сушили над MgSO4, фильтровали и упаривали. Сушили под вакуумом с получением 2,697 г неочищенного материала в виде коричневого масла. Степень чистоты по ВЭЖХ: около 92% Масло растворяли в МЭК (около 18 мл) и добавляли конц. HCl (12,5 M, 574 мкл, 7,18 ммоль). Самопроизвольного осаждения соли HCl не наблюдалось. Смесь осторожно перемешивали при комнатной температуре и через около 20 минут выпал осадок. Смесь осторожно перемешивали в течение 2,5 часов и твердое вещество выделяли фильтрованием на фильтре из пористого стекла P3. Твердое вещество промывали тремя порциями МЭК и затем сушили под вакуумом при 60°C в течение 2,5 дней. Выход (партия 1): 1,224 г (48,5%) продукта в виде гидрохлоридной соли.
Чистота по ВЭЖХ: 99,0% (основный способ);
97,4% (кислотный способ).
Значительное количество твердых частиц прошло через фильтр в фильтрат. Твердые частицы выделяли центрифугированием, а супернатант удаляли пипеткой. Твердое вещество промывали двумя порциями (около 2x5 мл) МЭК. После удаления последнего супернатанта продукт сушили под вакуумом при 60°C в течение 2,5 дней. Выход (партия 2): 324 мг (12,8%) продукта в виде гидрохлоридной соли.
Чистота по ВЭЖХ: 99,0% (основный способ);
97,5% (кислотный способ).
Общий выход: 1,548 г (61,3%).
Обе партии содержат около 0,07% ДМФА (мас./мас.). ДМФА уже присутствовал в исходном материале.
Дальнейшая очистка объединенных партий
Две партии соединения 5 объединяли (1,338 г, 3,041 ммоль) в круглодонной колбе на 50 мл и добавляли воду (6 мл) с последующим добавлением 2 М NaOH (1,6 мл, 3,2 ммоль). Смесь перемешивали и добавляли EtOAc (40 мл). При перемешивании дополнительно добавляли 0,5 мл (1 ммоль) 2 M NaOH. Через 15 минут все твердые вещества растворились и фазы разделились. pH водной фазы измеряли с помощью индикатора pH => pH=7. К водной фазе добавляли еще 2 М NaOH (0,4 мл, 0,8 ммоль), что приводило к pH 10. Водную фазу экстрагировали EtOAc (25 мл) и фазы разделяли. Объединенные органические фазы сушили над Na2SO4, фильтровали и упаривали с получением свободного основания в виде кристаллического твердого бежевого вещества. Свободное основание растворяли в МЭК (15 мл) и при перемешивании добавляли HCl (37%, 12,5 М, 255 мкл, 3,19 ммоль). Сразу образовался белый осадок. Смесь осторожно перемешивали в течение 2 часов и твердое вещество собирали фильтрованием на фильтре из пористого стекла P4. Твердые вещества промывали МЭК (5 мл) и сушили под вакуумом при 60°C в течение 3 часов. Выход: 1,187 г (89% в пересчете на неочищенный материал) продукта со степенью чистоты 99% в виде белого твердого вещества. 1H ЯМР (600 МГц, CD3OD) δ м.д. 7,49 (д, J=7,3 Гц, 2H) 7,37 (т, J=7,6 Гц, 2H) 7,27-7,32 (м, 1H) 5,23 (к, J=7,0 Гц, 1H) 4,60-4,70 (м, 1H) 3,55 (д, J=5,5 Гц, 2H) 1,83 (д, J=7,3 Гц, 3H) 1,67-1,76 (м, 1H) 1,58 -1,65 (м, 1H) 1,48-1,54 (м, 1H) 1,00 (д, J=6,7 Гц, 3H) 0,98 (д, J=6,7 Гц, 3H). МС (ESI+) m/z 404 [M+H]+.
Соотношение диастереомеров конечного продукта отражает соотношение энантиомеров исходного материала (соединение 1 ), которое составляло 99,7% (S).
1H ЯМР: спектр выглядит очень чистым. Однако были обнаружены следовые количества ДМФА.
Сравнительный Пример 4 - Двухстадийная методика синтеза в масштабе производственного процесса 6-амино-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-4-ола (1)
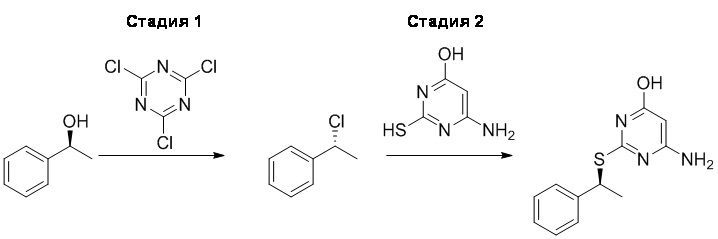
Стадия 1
В эмалированный реактор объемом 1380 л загружали при Ti=20°C этилацетат и (S)-фенилэтанол в атмосфере азота. Прозрачно-желтый раствор выдерживали при Ti=20°C в течение 30 минут, затем выгружали в бочки с полимерным покрытием на 200 л. В эмалированный реактор объемом 1380 л загружали хлорангидрид циануровой кислоты, этилацетат (1,2) и N,N-диметилформамид в течение 60 минут. Раствор нагревали от Ti=6,3°C до Ti=25°C в течение 25 минут, затем охлаждали до Ti=-5°C в течение 180 минут. При использовании 1-дюймового мембранного насоса предварительно приготовленный раствор (S)-фенилэтанола в этилацетате загружали в течение 12 часов при максимальной температуре Ti=-0,4°C, затем выдерживали при Ti=-5°C в течение 3 часов. Образец КПП показал содержание (S)-фенилэтанола = 0,35% (цель <1%). Загружали пропан-2-ол в течение 120 минут при Ti=-5°C и выдерживали в течение 30 минут, загружали целит и затем фильтровали на нутч-фильтре из нержавеющей стали, снабженном тканью 50 мкм, возникли значительные проблемы с фильтрацией из-за засорения ткани, материал загружали порциями в фильтр под давлением на 50 л с тканью 50 мкм со слоем целита. Твердое вещество промывали этилацетатом при пропорциональной фильтрации. Маточные растворы повторно загружали в сосуд емкостью 1380 л и устанавливали для перегонки при максимальной Ti=40,9°C и вакууме 200 мбар с удалением 750 кг дистиллята. Сосуд охлаждали до Ti<25°C и фильтровали порциями на фильтре под давлением на 50 л с тканью 50 мкм со слоем целита, порциями промывая этилацетатом. Маточные растворы повторно загружали в сосуд емкостью 330 л и устанавливали для перегонки при максимальной Ti=35,1°C и вакууме 24 мбар с удалением 500 кг дистиллята. Было выделено 275 кг в виде бледно-желтой жидкости.
Результат:
275 кг без поправки:
Степень чистоты по ГХ: 73,87% (% площади)
Хиральная чистота по ГХ: 94,9% (% площади, R-изомер).
Стадия 2
В сухой эмалированный реактор емкостью 2800 л загружали при Ti=20°C N,N-диметилформамид, гранулы гидроксида натрия и 4-амино-6-гидрокси-2-меркапропиримидин в течение 60 минут, поддерживая температуру ниже Ti=25°C. Нагревали до Ti=62,5°C в течение 60 минут и выдерживали 30 минут. Загружали (1R)-фенилэтилхлорид в течение 75 минут, поддерживая Ti=60-65°C. Реакцию выдерживали при Ti=60-70°C в течение 6 часов. Образец КПП показал содержание 4-амино-6-гидрокси-2-меркапропиримидина = <0,1% (цель <1%). Реакционную смесь охлаждали до Ti<25°C и добавляли воду, pH доводили до 12,01 32% раствором гидроксида натрия в течение 90 минут. Загружали толуол и выдерживали в течение 30 минут с последующей фильтрацией для удаления межфазных твердых частиц с использованием нутч-фильтра из нержавеющей стали, снабженного тканью 50 мкм со слоем целита, в течение 3 часов. После повторной загрузки фазу фильтрата разделяли для удаления нижней водной фазы (1500 л), органическую фазу оставляли как отходы. Водную фазу загружали в эмалированный реактор на 1380 л и устанавливали для перегонки при максимальной Ti=61,3°C и вакууме 180 мбар с удалением 1800 л дистиллята. Загружали воду и затем нагревали до Ti=65,3°C в течение 90 минут. pH доводили 6,2% раствором соляной кислоты до pH 5,5 в течение 180 минут. Охлаждали до Ti<20°C в течение 180 минут, затем фильтровали на нутч-фильтре из нержавеющей стали, снабженном тканью 50 мкм, в течение 10 часов. Сушили вытягиванием и промывали вытеснением с выделением 588 кг влажного сероватого твердого вещества.
Результат:
588 кг без поправки (потери при сушке 30% мас./мас. = 176,4 кг, выход (попр): 48,1%)
Степень чистоты по ВЭЖХ: 81,26% (% площади)
Хиральная чистота по ВЭЖХ: 95,03% (% площади S-изомер)
Первая перекристаллизация
В сухой эмалированный реактор на 2800 л загружали при Ti=20°C 4-амино-6-гидрокси-2-[(1S)-1-(фенилэтил)тио]пиримидин и метанол, нагревали до Ti=55°C в течение 120 минут
и выдерживали в течение 1 часа. Методом с проточным фильтром GAFF раствор переносили при Ti=55°C в эмалированный сосуд на 4500 л, после чего промывали линии метанолом. Нагревали до Ti=65°C (кипячение с обратным холодильником) и выдерживали в течение 1 часа, загружали воду в течение 3 часов, поддерживая Ti>60°C. Перемешивание уменьшали и медленно охлаждали до Ti=22,5°C в течение 6 часов. Фильтровали на нутч-фильтре из нержавеющей стали, снабженном тканью 50 мкм, в течение 12 часов, сушили вытягиванием и промывали вытеснением, сушили вытягиванием с выделением 302 кг сероватого твердого вещества.
Результат:
302 кг без поправки
Степень чистоты по ВЭЖХ: 95,11% (% площади)
Хиральная чистота по ВЭЖХ: 97,09% (% площади R-изомер) (% площади S-изомер)
Вторая перекристаллизация
В сухой эмалированный реактор на 4500 л загружали при Ti=20°C 4-амино-6-гидрокси-2-[(1S)-1-(фенилэтил)тио]пиримидин и метанол, нагревали до Ti=65°C (кипячение с обратным холодильником) в течение 50 минут и выдерживали 30 минут, загружали воду в течение 3 часов, поддерживая Ti>60°C. Перемешивание уменьшали и медленно охлаждали до Ti=22,5°C в течение 6 часов. Фильтровали на нутч-фильтре из нержавеющей стали, снабженном тканью 50 мкм, в течение 12 часов, сушили вытягиванием и промывали вытеснением, сушили вытягиванием с выделением 142 кг сероватого твердого вещества. Сушили в сушилке Cone Vac при максимальной температуре Ti=60°C и давлении 20 мбар.
Результат:
106 кг без поправки (потери при сушке 99,1% мас./мас. = 105,1 кг, выход (попр): 61,1%)
Степень чистоты по ВЭЖХ: 94,07% (% площади)
Хиральная чистота по ВЭЖХ: 99,04% (% площади S-изомер)
Резюме
Для достижения хиральной чистоты >99% потребовались две стадии синтеза и две перекристаллизации.
Общий выход способа составил 29,3% (включая стадии перекристаллизации).
| название | год | авторы | номер документа |
|---|---|---|---|
| ФОСФАТНЫЕ И ФОСФОНАТНЫЕ ПРОИЗВОДНЫЕ 7-АМИНО-5-ТИОТИАЗОЛО[4,5-d]ПИРИМИДИНОВ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ, СВЯЗАННЫХ С ПОВЫШЕННЫМИ УРОВНЯМИ CX3CR1 И/ИЛИ CX3CL1 | 2019 |
|
RU2801664C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРИМИДИНИЛЦИКЛОПЕНТАНОВЫХ СОЕДИНЕНИЙ | 2014 |
|
RU2826179C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРИМИДИНИЛЦИКЛОПЕНТАНОВЫХ СОЕДИНЕНИЙ | 2014 |
|
RU2702355C1 |
| ПРОИЗВОДНЫЕ 5-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-5-АЗАСПИРО[2.5]ОКТАН-8-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2761626C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ (ЦИКЛОПЕНТИЛ[d]ПИРИМИДИН-4-ИЛ)ПИПЕРАЗИНОВЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2732404C2 |
| ИНГИБИТОРЫ ДЕМЕТИЛАЗЫ LSD1 НА ОСНОВЕ АРИЛЦИКЛОПРОПИЛАМИНА И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2011 |
|
RU2611437C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ (ЦИКЛОПЕНТИЛ[D]ПИРИМИДИН-4-ИЛ)ПИПЕРАЗИНОВЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2712224C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-1Н-ИМИДАЗО[1,2-а]ПИРИМИДИН-5-ОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ФАРМАЦИИ | 2010 |
|
RU2554868C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛИРОВАННЫХ ЦИКЛОПЕНТИЛПИРИМИДИНОВЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2643811C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСАСПИРОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2018 |
|
RU2777983C2 |
Изобретение относится к способу получения соединения Формулы I или его соли, где R1 представляет собой арил или пиридил, необязательно замещенный одной или несколькими группами, выбранными из галогена, -CN, -C(O)NR3R4, -S(O)2R5; C1-6 алкила, С2-6 алкенила, С2-6 алкинила, где последние три группы являются необязательно замещенными одним или несколькими F; R2 представляет собой C1-6 алкил, необязательно замещенный одним или более F; каждый R3 и R4 независимо представляет собой Н или C1-6 алкил, необязательно замещенный одним или более F; R5 представляет собой C1-6 алкил, необязательно замещенный одним или более F; причем указанный способ включает следующие стадии: (i) образование соединения Формулы II, где R1 и R2 представляют собой такие, как указано для соединения Формулы I, и R6 представляет собой C1-6 алкил, необязательно замещенный одним или более F или фенилом, необязательно замещенным одной или несколькими группами, выбранными из галогена, метила и -NO2., путем приведения в контакт соединения Формулы III с подходящим сульфирующим агентом в присутствии подходящего основания В1 и подходящего растворителя S1, причем S1 является растворителем, в котором соль, образованная между В1 и уходящей группой сульфирующего агента, является нерастворимой, и затем (ii) приведение в контакт соединения Формулы II с соединением Формулы IV, где М+ представляет собой Li+, Na+, K+ или Cs+, и где соединение Формулы III представлено в виде одного энантиомера. Изобретение также относится к способу получения соединения формулы (VII), включающему способ получения соединения формулы (I). Технический результат – разработан способ получения соединения формулы (I) с высокими выходом и степенью хиральной чистоты. 3 н. и 24 з.п. ф-лы, 2 ил., 4 пр.
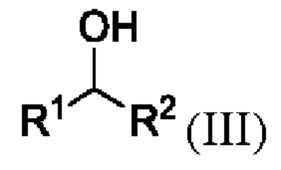
1. Способ получения соединения Формулы I
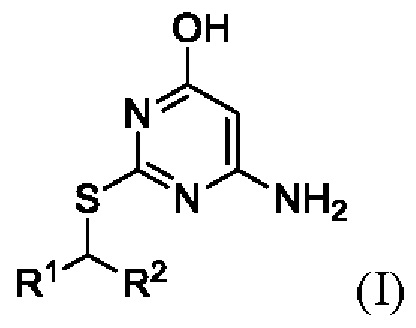
или его соли, где
R1 представляет собой арил или пиридил, необязательно замещенный одной или несколькими группами, выбранными из галогена, -CN, -C(O)NR3R4, -S(O)2R5; C1-6 алкила, С2-6 алкенила, С2-6 алкинила, где последние три группы являются необязательно замещенными одним или несколькими F;
R2 представляет собой C1-6 алкил, необязательно замещенный одним или более F;
каждый R3 и R4 независимо представляет собой Н или C1-6 алкил, необязательно замещенный одним или более F;
R5 представляет собой C1-6 алкил, необязательно замещенный одним или более F;
причем указанный способ включает следующие стадии:
(i) образование соединения Формулы II
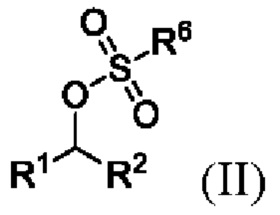 ,
,
где R1 и R2 представляют собой такие, как указано для соединения Формулы I, и R6 представляет собой C1-6 алкил, необязательно замещенный одним или более F или фенилом, необязательно замещенным одной или несколькими группами, выбранными из галогена, метила и -NO2.;
путем приведения в контакт соединения Формулы III
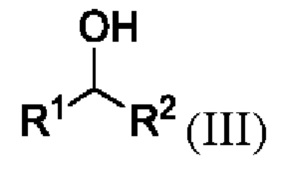 ,
,
где R1 и R2 представляют собой такие, как указано для соединения Формулы I или II;
с подходящим сульфирующим агентом в присутствии подходящего основания В1, такого как, но не ограничиваясь этим, триэтиламин и подходящего растворителя S1, такого как, но не ограничиваясь этим, метил трет-бутиловый эфир, причем S1 является растворителем, в котором соль, образованная между В1 и уходящей группой сульфирующего агента, является нерастворимой
и затем
(ii) приведение в контакт соединения Формулы II с соединением Формулы IV
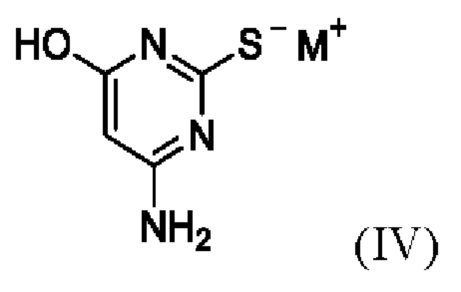 ,
,
где М+ представляет собой Li+, Na+, K+ или Cs+,
где соединение Формулы III представлено в виде одного энантиомера.
2. Способ по п. 1, отличающийся тем, что соединение Формулы II не выделяют из реакционной смеси со стадии (i) до его использования на стадии (ii).
3. Способ по любому из пп. 1-2, отличающийся тем, что стадия (ii) включает приведение раствора соединения Формулы II в подходящем растворителе S1 в контакт с раствором соединения Формулы IV в подходящем растворителе S2.
4. Способ по любому из пп. 1-3, дополнительно включающий следующую стадию:
(iii) получение соединения Формулы IV приведением в контакт соединения Формулы V
с подходящим основанием В2 в присутствии подходящего растворителя S2.
5. Способ по п. 3, отличающийся тем, что раствор соединения Формулы IV получают на стадии (iii), необязательно, после одной или нескольких стадий очистки.
6. Способ по любому из предшествующих пунктов, отличающийся тем, что S1 выбран из группы, включающей диэтиловый эфир, метил трет-бутиловый эфир, 1,2-диметоксиэтан, 1,4-диоксан, тетрагидрофуран и 2-метилтетрагидрофуран.
7. Способ по любому из пп. 1-6, отличающийся тем, что В1 представляет собой основание органического амина.
8. Способ по любому из пп. 3-7, отличающийся тем, что S2 представляет собой полярный апротонный растворитель.
9. Способ по п. 8, отличающийся тем, что S2 представляет собой N,N-диметилформамид.
10. Способ по любому из пп. 1-9, отличающийся тем, что В2 выбран из группы, включающей гидроксид лития, карбонат лития, гидроксид натрия, карбонат натрия, гидроксид калия, карбонат калия, гидроксид цезия и карбонат цезия.
11. Способ по п. 10, отличающийся тем, что В2 представляет собой гидроксид натрия.
12. Способ по любому из пп. 1-11, отличающийся тем, что R1 представляет собой арил или пиридил, необязательно замещенный одной или несколькими группами, выбранными из галогена, -CN, -SO2Me или -CONH2.
13. Способ по п. 12, отличающийся тем, что R1 представляет собой фенил.
14. Способ по любому из пп. 1-13, отличающийся тем, что R2 представляет собой С1-3 алкил.
15. Способ по любому из пп. 1-14, отличающийся тем, что R2 представляет собой метил.
16. Способ по любому из пп. 1-15, дополнительно включающий следующую стадию:
(ib) удаление соли, образованной между В1 и уходящей группой сульфирующего агента, из раствора соединения Формулы II в S1, полученного на стадии (i).
17. Способ по любому из пп. 1-16, дополнительно включающий следующую стадию:
(iv) обработка неочищенного материала, полученного на стадии (ii), подходящим растворителем S4, чтобы вызвать осаждение соединения Формулы I.
18. Способ по п. 17, отличающийся тем, что S4 представляет собой ацетонитрил.
19. Способ по любому из пп. 1-18, отличающийся тем, что указанный сульфирующий агент представляет собой мезилхлорид или тозилхлорид.
20. Способ по любому из предшествующих пунктов, отличающийся тем, что представляет собой способ получения соединения Формулы Ia или его соли,
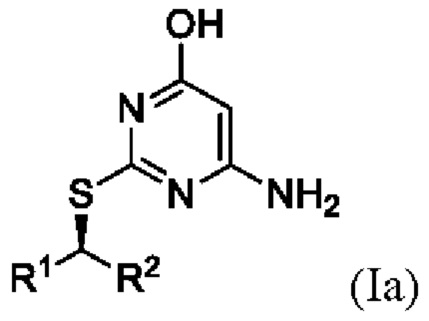
где R1 представляет собой арил или пиридил, необязательно замещенный одной или несколькими группами, выбранными из галогена, -CN, -C(O)NR3R4, -S(O)2R5; C1-6 алкила, С2-6 алкенила, С2-6 алкинила, где последние три группы являются необязательно замещенными одним или несколькими F и R2 представляет собой C1-6 алкил, необязательно замещенный одним или более F.
21. Способ по п. 20, отличающийся тем, что соединение Формулы Ia представляет собой 6-амино-2-{[(1S)-1-фенилэтил]сульфанил}пиримидин-4-ол или его соль.
22. Способ по п. 20 или 21, отличающийся тем, что соединение Формулы I или Ia имеет хиральную чистоту более 96%, предпочтительно, более 99%.
23. Способ получения соединения Формулы VII или его соли
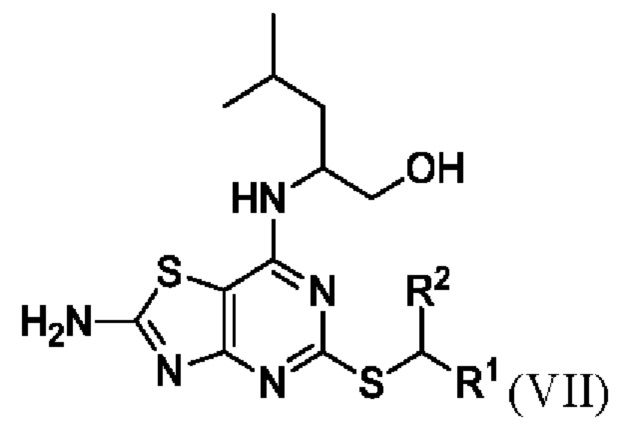 ,
,
где R1 представляет собой арил или пиридил, необязательно замещенный одной или несколькими группами, выбранными из галогена, -CN, -C(O)NR3R4, -S(O)2R5; C1-6 алкила, С2-6 алкенила, С2-6 алкинила, где последние три группы являются необязательно замещенными одним или несколькими F, и R2 представляет собой C1-6 алкил, необязательно замещенный одним или более F,
причем способ включает следующие стадии:
(i) образование соединения Формулы VIII
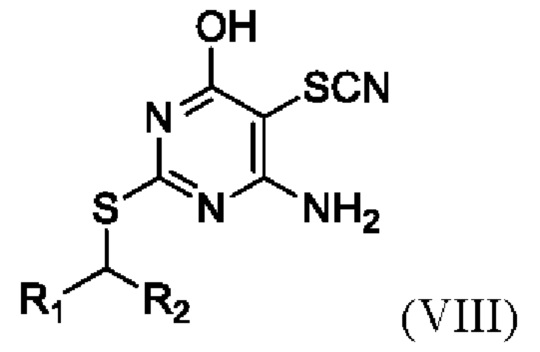 ,
,
где R1 и R2 представляют собой такие, как указано для соединения Формулы VII;
путем приведения в контакт соединения формулы I, как определено в любом из пп. 1-22, с подходящей тиоцианатной солью;
(ii) образование соединения Формулы IX
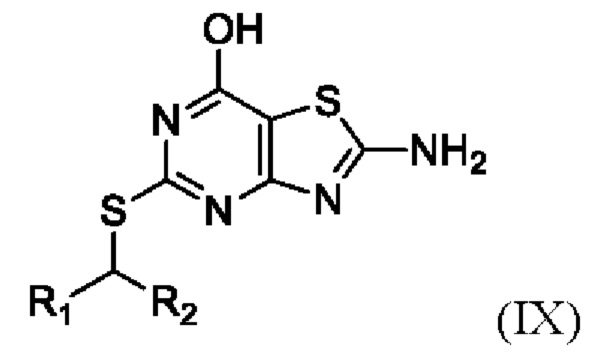 ,
,
где R1 и R2 представляют собой такие, как указано для соединения Формулы VII из соединения Формулы VIII;
(iii) приведение в контакт формулы IX в присутствии подходящего хлорирующего агента для получения соединения Формулы X
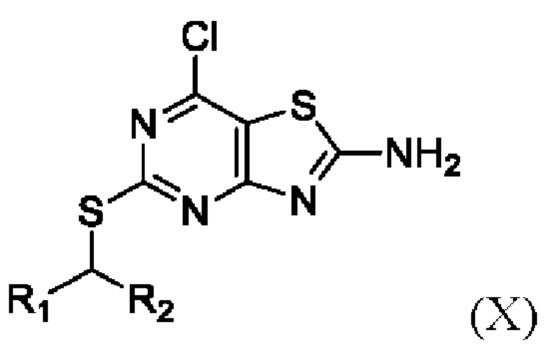 ,
,
где R1 и R2 представляют собой такие, как указано для соединения Формулы VII,
затем
(vi) приведение в контакт формулы X с лейцинолом
24. Способ по п. 23, отличающийся тем, что предназначен для получения 2-[(2-амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола или его соли.
25. Способ по п. 24, отличающийся тем, что представляет собой способ получения (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола или его соли.
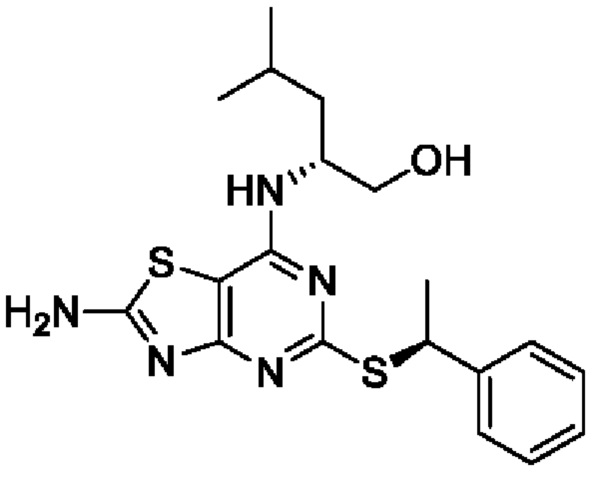
26. Способ по п. 25, отличающийся тем, что указанный (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол имеет хиральную чистоту более 99,2%.
27. Способ получения фармацевтического состава, обладающего активностью антагонистов рецептора фракталкина, содержащего эффективное количество соединения Формулы I или его соль, который включает использование способа по пп. 1-19.
| WO 2006107258 A1, 12.10.2006 | |||
| SOFIA KARLSTROM ET AL, Journal of Medicinal Chemistry, vol | |||
| Приспособление для разматывания лент с семенами при укладке их в почву | 1922 |
|
SU56A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| ВИСЯЧИЙ ЗАМОК С КОНТРОЛЬНОЙ ПЕЧАТЬЮ | 1925 |
|
SU3177A1 |
| WO 2000009511 A1, 24.02.2000 | |||
| ПРОИЗВОДНЫЕ ПИРИМИДИНСУЛЬФОНАМИДА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ХЕМОКИНОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ПРИМЕНЕНИЕ | 2003 |
|
RU2342366C2 |
| НОВЫЕ 5,7-ДИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ [1,3]ТИАЗОЛО[4,5-d]ПИРИМИДИН-2(3Н)-ОНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2007 |
|
RU2441012C2 |
| СПОСОБ ПОЛУЧЕНИЯ 6-ЗАМЕЩЕННЫХ 2-(АЛКИЛСУЛЬФАНИЛ)-4(3Н)-ПИРИМИДИНОНОВ | 2003 |
|
RU2238269C1 |
