Область изобретения
В настоящем изобретении описаны новые 5,7-дизамещенные производные [1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она, а также способы их получения, фармацевтические композиции, содержащие их, и их применение в терапии.
Предпосылки изобретения
Хемокины играют важную роль в иммунном и воспалительном ответах при различных заболеваниях и расстройствах, включая астму, атеросклероз и аллергические заболевания, а также аутоиммунные патологии, такие как ревматоидный артрит и рассеянный склероз. Эти малые секретируемые молекулы представляют собой растущее надсемейство белков массой 8-14 кДа, характеризующихся консервативным цистеиновым мотивом. В настоящее время надсемейство хемокинов содержит четыре группы, проявляющие структурные характеристические мотивы, семейства С-Х-С, С-С, С-Х3-С и ХС. Семейства С-Х-С и С-С обладают подобием последовательностей и отличаются друг от друга на основании единственной аминокислотной вставки между NH-проксимальной парой цистеиновых остатков. Семейство С-Х3-С отличается от других двух семейств на том основании, что имеет тройную аминокислотную вставку между NH-проксимальной парой цистеиновых остатков. Напротив, у членов семейства ХС отсутствует один из первых двух цистеиновых остатков.
Хемокины С-Х-С включают несколько эффективных хемоаттрактантов и активаторов нейтрофилов, таких как интерлейкин-8 (IL-8) и активирующий нейтрофилы пептид 2 (NAP-2, neutrophil-activating peptide 2).
Хемокины С-С включают эффективные хемоаттрактанты моноцитов, лимфоцитов и нейтрофилов. Примеры включают хемотаксические белки моноцитов человека 1-3 (МСР-1, МСР-2 и МСР-3, monocyte chemotactic proteins 1-3), RANTES (регулируемые при активации, экспрессируемые и секретируемые нормальными Т-клетками, Regulated on Activation, Normal Т Expressed и Secreted), эотаксин и воспалительные белки макрофагов 1α и 1β (МIР-1α и MIP-1β).
Хемокин С-Х3-С (также известный как фракталкин) является эффективным хемоаттрактантом и активатором микроглии в центральной нервной системе (ЦНС), а также моноцитов, Т клеток, NK клеток и тучных клеток.
Исследования показали, что действия хемокинов опосредованы подсемействами рецепторов, связанных с G-белком, среди которых находятся рецепторы, обозначаемые CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для семейства С-С); CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для семейства С-Х-С) и СХ3CR1 для семейства С-Х3-С. Эти рецепторы представляют собой хорошие мишени для разработки лекарственных средств, поскольку агенты, которые модулируют эти рецепторы, могут быть полезны при лечении расстройств и заболеваний, таких как упомянутые выше.
В WO 01/25242 описаны некоторые производные тиазоло[4,5-d]пиримидина, которые полезны в качестве антагонистов рецепторов, связанных с семействами хемокинов С-Х-С и С-С, в частности в качестве антагонистов рецептора CXCR2.
Настоящее изобретение относится к группе соединений, которые родственны соединениям, описанным в WO 01/25242, но имеют структурный тип, конкретно там не описанный. По сравнению с Примерами, описанными в WO 01/58907, соединения по настоящему изобретению обладают неожиданно полезными свойствами в качестве антагонистов рецептора CX3CR1.
Описание изобретения
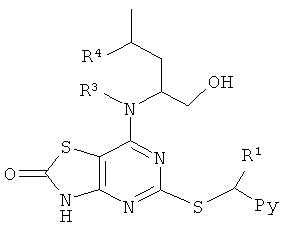
В настоящем изобретении предложены соединения формулы (I)
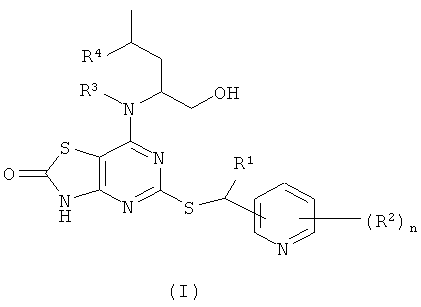
где:
R1 представляет собой СН3 или CF3;
R2 представляет собой галогено, CN или C1-6алкил;
R3 представляет собой Н или СН3;
R4 представляет собой Н или СН3;
n представляет собой 0, 1 или 2;
в виде свободного основания или его фармацевтически приемлемых соли, сольвата или сольвата соли.
В одном воплощении изобретения предложены соединения формулы (I), где n представляет собой 1.
В другом воплощении изобретения предложены соединения формулы (I), где R1 представляет собой СН3.
В еще одном воплощении изобретения предложены соединения формулы (I), где R2 представляет собой галогено или CN.
В еще одном воплощении изобретения предложены соединения формулы (I), где R2 представляет собой F или Cl.
В еще одном воплощении изобретения предложены соединения формулы (I), где R2 представляет собой CN.
В еще одном воплощении изобретения предложены соединения формулы (I), где n представляет собой 1; R1 представляет собой СН3 и R2 представляет собой F, Cl или CN.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 5 и содержит Cl в положении 2.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 2 и содержит CN в положении 4.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 2 и содержит F в положении 5.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 2 и содержит Cl в положении 5.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 2 и содержит F в положении 3.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 4 и содержит F в положении 3.
В еще одном воплощении изобретения предложены соединения формулы (I), где R3 представляет собой Н.
В еще одном воплощении изобретения предложены соединения формулы (I), где R4 представляет собой СН3.
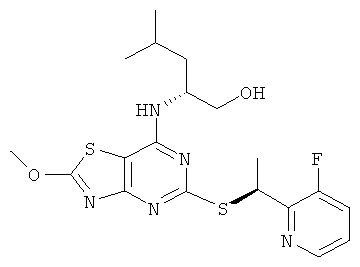
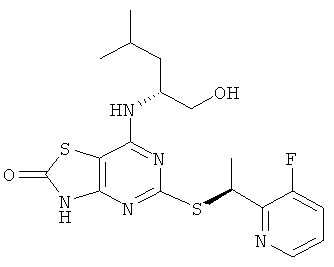
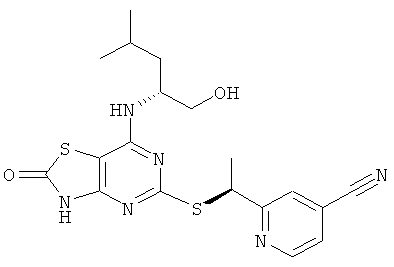
В еще одном воплощении изобретения предложены соединения формулы (I), выбранные из:
5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3H)-она;
5-{[1-(3-фторпиридин-4-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3H)-она;
5-{[(1S)-1-(3-фторпиридин-4-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
5-{[(1S)-1-(3-фторпиридин-4-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
2-{(1S)-1-[(7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}-2-оксо-2,3-дигидро[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}изоникотинонитрила;
5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-бутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3H)-она и
5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио}-7-[[(1R)-1-(гидроксиметил)-бутил](метил)амино][1,3]тиазоло[4,5-d]пиримидин-2(3H)-она;
в виде свободного основания или его фармацевтически приемлемых соли, сольвата или сольвата соли.
Соединения формулы (I) могут существовать в стереоизомерных и/или таутомерных формах. Следует понимать, что все энантиомеры, диастереомеры, рацематы, таутомеры и их смеси входят в объем изобретения.
По сравнению с соединениями, описанными в WO 01/25242, соединения по настоящему изобретению характеризуются наличием разветвленной тиоалкилпиридильной группы в положении 5 тиазолпиримидиновой кольцевой системы. То есть соединения по настоящему изобретению включают группу R1, отличную от водорода.
Согласно изобретению также предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, который включает:
а) взаимодействие соединения формулы (II)
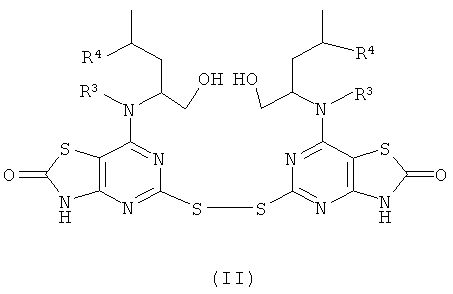
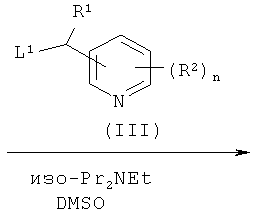
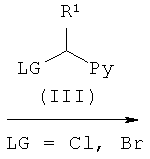
где R3 и R4 являются такими, как определено в формуле (I), с соединением формулы (III)
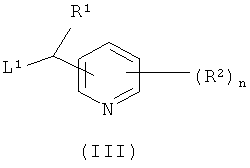
где R1, R2 и n являются такими, как определено в формуле (I), и L1 представляет собой уходящую группу; или
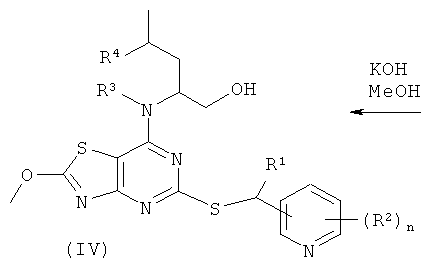
б) гидролиз соединения формулы (IV)
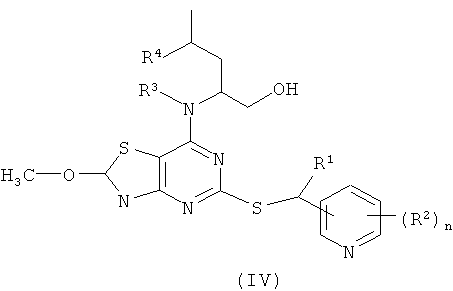
где R1, R2, R3, R4 и n являются такими, как определено в формуле (I);
и, когда это необходимо, превращение полученного соединения формулы (I) или его другой соли в его фармацевтически приемлемую соль; или превращение полученного соединения формулы (I) в другое соединение формулы (I); и, когда это желательно, превращение полученного соединения формулы (I) в его оптический изомер.
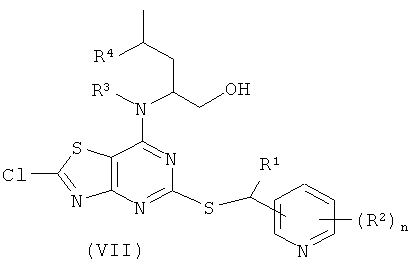
В способе (а) реагенты (II) и (III) подвергают сочетанию в подходящем органическом растворителе, таком как диметилсульфоксид (DMSO), ацетонитрил или 1-метил-2-пирролидинон (NMP). Взаимодействие возможно проводят в присутствии добавленного органического или неорганического основания, такого как триэтиламин, N,N-диизопропилэтиламин (DIPEA) или гидрид натрия. Взаимодействие осуществляют в присутствии умеренного восстанавливающего агента, такого как боргидрид натрия. Взаимодействие проводят при подходящей температуре, обычно между комнатной температурой и температурой кипения растворителя. Реакция обычно продолжается в течение периода времени примерно от одного часа до одной недели, либо до тех пор, пока анализ не покажет, что образование целевого продукта завершено. Подходящие уходящие группы L1 представляют собой галоген, в частности хлор или бром. В одном воплощении L1 представляет собой хлор.
В способе (б) реагент (IV) подвергают катализируемому кислотой гидролизу в подходящем органическом растворителе, таком как 1,4-диоксан, тетрагидрофуран (THF), диметилсульфоксид (DMSO) или 1-метил-2-пирролидинон (NMP). Подходящие кислоты включают неорганические кислоты, такие как соляная кислота или бромистоводородная кислота, либо сильные органические кислоты, такие как трифторуксусная кислота. Взаимодействие проводят при подходящей температуре, обычно между комнатной температурой и температурой кипения растворителя. Реакция обычно протекает в течение периода времени примерно от одного часа до одного дня, либо до тех пор, пока анализ не покажет, что образование целевого продукта завершено.
Специалисту в данной области техники должно быть понятно, что в вышеописанных способах может быть желательно или необходимо защитить группы амино, гидрокси или другие потенциально реакционноспособные группы. Подходящие защитные группы и подробности способов введения и удаления таких групп в общем хорошо известны из уровня техники (см., например, "Protective Groups in Organic Synthesis", 3rd Edition (1999), Greene и Wuts).
Настоящее изобретение включает соединения формулы (I) в форме солей. Подходящие соли включают соли, образованные с органическими или неорганическими кислотами либо с органическими или неорганическими основаниями. Такие соли обычно являются фармацевтически приемлемыми, хотя соли не фармацевтически приемлемых кислот или оснований могут находить применение в получении и очистке рассматриваемого соединения.
Соли соединений формулы (I) смогут быть получены посредством взаимодействия свободного соединения или его соли, энантиомера или рацемата с одним или более эквивалентами соответствующих кислоты или основания. Взаимодействие может быть осуществлено в растворителе или среде, в которых соль не растворима, либо в растворителе, в котором соль растворима, например воде, диоксане, этаноле, тетрагидрофуране или диэтиловом эфире, либо в смеси растворителей, которые могут быть удалены в вакууме или посредством сублимационной сушки. Это взаимодействие также может представлять собой обменный процесс или может быть осуществлено на ионообменной смоле.
Соединения формулы (II) могут в общем быть получены известными способами, которые очевидны для специалиста в данной области. Один такой подходящий путь показан на Схеме 1.
Схема 1
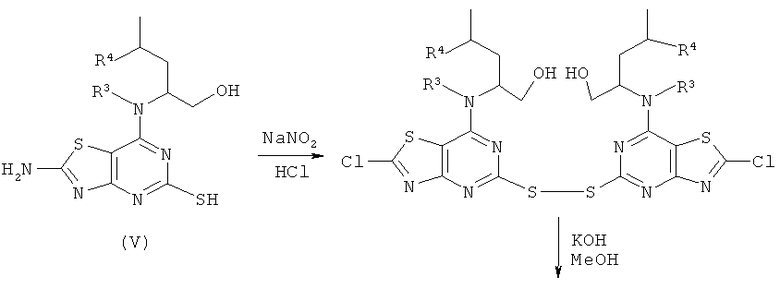
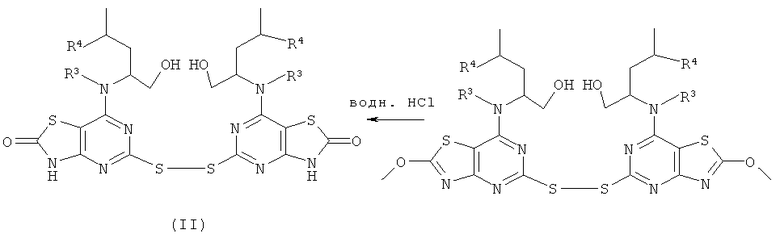
Соединения формулы (III) либо имеются в продаже, либо известны из литературы, либо могут быть получены с использованием известных способов, которые будут очевидны специалисту в данной области.
Соединения формулы (IV) либо известны из, например, WO 01/25242 или WO 05/33115, либо могут быть получены с использованием известных способов, которые будут очевидны специалисту в данной области. Один такой подходящий путь показан на Схеме 2.
Схема 2
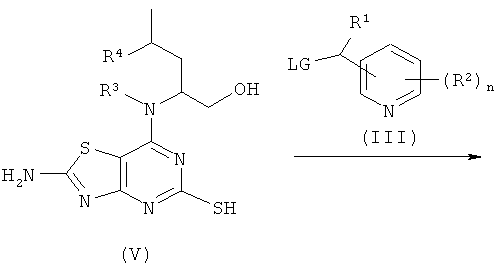
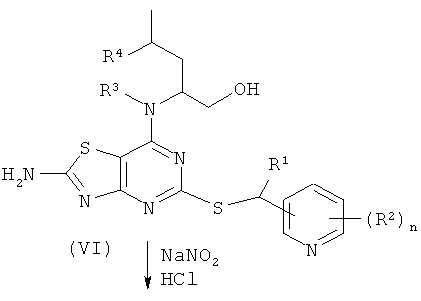
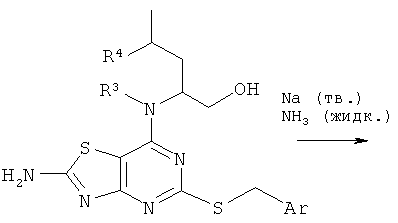
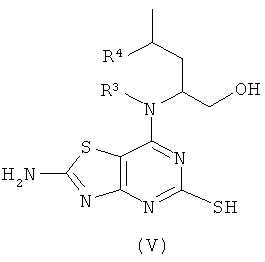
Соединения формулы (V) либо известны из WO 01/58907, WO 01/25242 или WO 02/76990, либо могут быть получены с использованием известных способов, которые будут очевидны специалисту в данной области.
Например, соединения формулы (V) и, соответственно, соединения формулы (VI) могут быть получены так, как показано на Схеме 3.
Схема 3
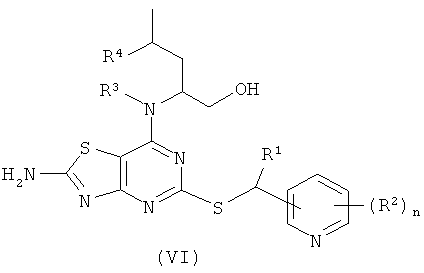
Подходящие конкретные способы получения соединений формулы (II), (III), (IV), (V) и (VI) детализированы в разделе «Примеры» настоящей заявки, и такие способы составляют конкретные воплощения способов по изобретению.
Промежуточные соединения могут быть использованы как таковые или в защищенной форме. Подходящие защитные группы и особенности способов введения и удаления таких групп в общем хорошо известны из уровня техники (см., например, "Protective Groups in Organic Synthesis", 3rd Edition (1999), Greene и Wuts).
Соединения по изобретению и их промежуточные соединения могут быть выделены из их реакционных смесей и при необходимости подвергнуты дополнительной очистке с использованием стандартных методик.
Соединения формулы (I) могут существовать в стереоизомерных формах. Соответственно, все энантиомеры, диастереомеры, рацематы и их смеси включены в объем изобретения. Различные оптические изомеры могут быть выделены посредством разделения стереоизомерной смеси соединений с помощью традиционных методик, например фракционной кристаллизации или HPLC. Альтернативно, разные оптические изомеры могут быть получены непосредственно с использованием оптически активных исходных веществ.
Соединения формулы (I) содержат два стереогенных центра и поэтому могут существовать в четырех дискретных стереоизомерных формах, как показано в формулах (Ia)-(Id)
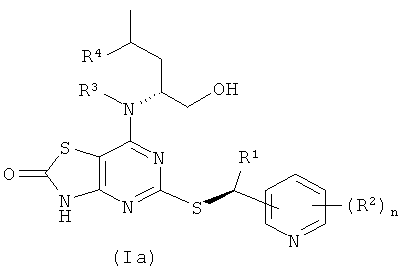


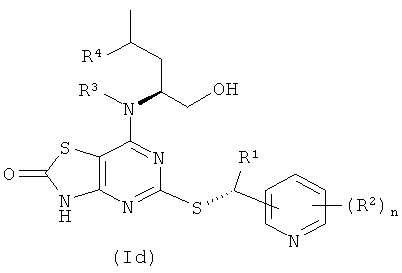
Все такие четыре стереоизомера и любые их смеси включены в объем изобретения. В одном воплощении соединения формулы (I) имеют стереохимию, показанную в формуле (Ia). В другом воплощении соединения формулы (I) имеют стереохимию, показанную в формуле (Ib).
Промежуточные соединения также могут существовать в стереоизомерных формах и могут быть использованы в виде очищенных энантиомеров, диастереомеров, рацематов или смесей.
В данном описании термин "С1-6алкил" включает как прямую, так и разветвленную цепь, а также циклические алкильные группы. С1-6алкил имеет от 1 до 6 атомов углерода и может представлять собой, без ограничения ими, метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, н-пентил, изо-пентил, трет-пентил, нео-пентил, н-гексил, изо-гексил или циклогексил.
В данном описании термин "галогено" или "галоген" относится к фтору, хлору, брому и иоду.
Соединения формулы (I) и их фармацевтически приемлемые соли полезны, поскольку они обладают фармакологической активностью в качестве антагонистов CX3CR1 рецептора. В частности, по сравнению с соединениями, специфически раскрытыми в примерах в WO 01/25242, соединения формулы (I) по настоящему изобретению обладают значительно более выраженной способностью в плане ингибирования CX3CR1 рецептора и/или пониженной способностью к ингибированию CXCR2 рецептора. Предпочтительные соединения по настоящему изобретению обладают усиленной способностью к ингибированию CX3CR1 и пониженной способностью к ингибированию CXCR2.
В одном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в качестве лекарственного средства.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики заболеваний или состояний, при которых благоприятным является антагонизм в отношении CX3CR1 рецептора.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики нейродегенеративных расстройств, демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий, ревматоидного артрита, легочных заболеваний, таких как COPD (хроническое обструктивное легочное заболевание), астмы или боли.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики рассеянного склероза (MS).
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики атеросклероза путем предупреждения и/или уменьшения формирования новых атеросклеротических поражений или бляшек и/или путем предупреждения или замедления развития уже существующих поражений и бляшек.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики атеросклероза путем изменения состава бляшек с целью уменьшения риска разрыва бляшек и атеротромботических явлений.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики инсульта или транзиторного повреждения головного мозга (TBI).
Согласно изобретению также предложен способ лечения или уменьшения риска заболеваний или состояний, при которых благоприятным является антагонизм в отношении CX3CR1 рецептора, который включает введение субъекту, страдающему указанным заболеванием или состоянием, или подверженному ему, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Предложен также способ лечения или уменьшения риска нейродегенеративных расстройств, демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий, ревматоидного артрита, легочных заболеваний, таких как COPD, астмы или боли у субъекта, страдающего указанным заболеванием или состоянием или подверженного ему, включающий введение этому субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Предложен также способ лечения или уменьшения риска рассеянного склероза (MS) у субъекта, страдающего указанным заболеванием или состоянием или подверженного ему, включающий введение этому субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Предложен также способ лечения или уменьшения риска атеросклероза путем предупреждения и/или уменьшения формирования новых атеросклеротических поражений или бляшек и/или путем предупреждения или замедления развития уже существующих поражений и бляшек у субъекта, страдающего указанным заболеванием или состоянием или подверженного ему, включающий введение этому субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Предложен также способ лечения или уменьшения риска атеросклероза путем изменения состава бляшек с целью уменьшения риска разрыва бляшек и атеротромботических явлений у субъекта, страдающего указанным заболеванием или состоянием или подверженного ему, включающий введение этому субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В другом аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемыми адъювантом, разбавителем или носителем, для применения в лечении или профилактике заболеваний или состояний, при которых благоприятным является антагонизм в отношении CX3CR1 рецептора.
В другом аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемыми адъювантом, разбавителем или носителем, для применения в лечении или профилактике нейродегенеративных расстройств, демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий, ревматоидного артрита, COPD, астмы или боли.
В другом аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемыми адъювантом, разбавителем или носителем, для применения в лечении или профилактике рассеянного склероза.
В другом аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемыми адъювантом, разбавителем или носителем, для применения в лечении или профилактике атеросклероза путем предупреждения и уменьшения формирования новых атеросклеротических поражений и/или бляшек и/или путем предупреждения или замедления развития уже существующих поражений и бляшек.
Соединения могут быть использованы в виде монотерапии или в комбинациях, в качестве профилактического или терапевтического лечения воспалительных состояний и заболеваний центральной нервной системы, таких как инсульт или транзиторное повреждение головного мозга (TBI) (Soriano et al. J. Neuroimmunology 2002, 125, 59-65).
В другом аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемыми адъювантом, разбавителем или носителем, для применения в лечении или профилактике атеросклероза путем изменения состава бляшек с целью уменьшения риска разрыва бляшек и атеротромботических явлений.
Соединения формулы (I) и их фармацевтически приемлемые соли показаны для применения в лечении или профилактике заболеваний или состояний, при которых желательна модуляция активности CX3CRI рецептора. В частности, соединения показаны для применения в лечении нейродегенеративных расстройств или демиелинизирующего заболевания у млекопитающих, включая человека. Более конкретно, эти соединения показаны для применения в лечении рассеянного склероза. Эти соединения также показаны для применения в лечении боли, ревматоидного артрита, остеоартрита, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий и легочной артериальной гипертензии.
Состояниями, которые могут быть конкретно упомянуты, являются: нейродегенеративные заболевания и деменции, например болезнь Альцгеймера, амиотрофический боковой склероз и другие заболевания моторных нейронов, болезнь Крейцфельда-Якоба и другие прионные заболевания, ВИЧ (вирус иммунодефицита человека)-ассоциированная энцефалопатия, болезнь Гентингтона, фронтотемпоральная деменция, деменция с тельцами Леви и васкулярная деменция; полиневропатии, например синдром Гийена-Барре, хроническая воспалительная демиелинизирующая полирадикулоневропатия, мультифокальная моторная нейропатия и плексопатии; демиелинизация ЦНС, например острый диссеминированный/геморрагический энцефаломиелит и подострый склерозирующий панэнцефалит; нейромышечные расстройства, например тяжелая псевдопаралитическая миастения и синдром Ламберта-Итона; спинномозговые расстройства, например тропический спастический парапарез и синдром мышечной скованности; паранеопластические синдромы, например мозжечковая дегенерация и энцефаломиелит; травматическое повреждение головного мозга; мигрень; рак; отторжение аллотрансплантата; системный склероз; вирусные инфекции; передающиеся через паразитов заболевания, например малярия; заболевание периодонта; инфаркт миокарда; инсульт; коронарная болезнь сердца; ишемическая болезнь сердца; рестеноз; ревматоидный артрит; легочные заболевания, такие как COPD; астма и боль.
Соединения по изобретению также показаны для применения в лечении атеросклероза путем предупреждения и/или уменьшения формирования новых атеросклеротических поражений или бляшек и/или путем предупреждения или замедления развития уже существующих поражений и бляшек.
Соединения по изобретению также показаны для применения в лечении атеросклероза путем изменения состава бляшек с целью уменьшения риска разрыва бляшек и атеротромботических явлений.
Соединения по изобретению также показаны для применения в лечении воспалительного заболевания кишечника (IBD), например болезни Крона и неспецифического язвенного колита, путем индуцирования ремиссии и/или поддержания ремиссии IBD.
Ожидается, что профилактика является особенно целесообразной для лечения индивидуумов, у которых имел место предшествующий эпизод, или которые иным образом считаются имеющими повышенный риск рассматриваемого заболевания или состояния. Индивидуумы с риском развития конкретного заболевания или состояния обычно включают индивидуумов с семейной историей этого заболевания или состояния и индивидуумов, которые с помощью генетического тестирования или скрининга были идентифицированы как являющиеся особенно чувствительными к развитию этого заболевания или состояния.
Вводимые дозировки для вышеуказанных терапевтических показаний, конечно, будут варьироваться в зависимости от используемого соединения, пути введения и желаемого лечения. Однако в общем удовлетворительные результаты получают, когда соединения вводят в дозировке твердой формы между 1 мг и 2000 мг в сутки.
Соединения формулы (I) и их фармацевтически приемлемые производные могут быть использованы сами по себе или в форме соответствующих фармацевтических композиций, в которых соединение или производное находится в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем. Введение может быть осуществлено, без ограничения ими, парентеральным (включая пероральный, сублингвальный или ректальный), интраназальным, внутривенным, местным или другими парентеральными путями. Традиционные процедуры для выбора и изготовления подходящих фармацевтических композиций описаны, например, в "Pharmaceuticals - The Science of Dosage Form Designs", M.E.Aulton, Churchill Livingstone, 1988. Фармацевтическая композиция предпочтительно содержит менее 80% и более предпочтительно менее 50% соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ изготовления такой фармацевтической композиции, который включает смешивание ингредиентов.
Кроме того, изобретение относится к комбинационной терапии, где соединение формулы (I) или его фармацевтически приемлемую соль либо фармацевтическую композицию или препарат, содержащие соединение формулы (I), вводят одновременно или последовательно с терапией и/или агентом для лечения любых кардио- или цереброваскулярных атеросклеротических расстройств и заболевания периферических артерий.
В частности, соединение формулы (I) или его фармацевтически приемлемая соль могут быть введены вместе с соединением из одной или более следующих групп:
1) противовоспалительные агенты, например
а) NSAIDs (нестероидные противовоспалительные средства, например ацетилсалициловая кислота, ибупрофен, напроксен, флурбипрофен, диклофенак, индометацин);
б) ингибиторы синтеза лейкотриенов (5-LO (5-липоксигеназа) ингибиторы, например AZD4407, зилеутон (Zileuton), ликофелон, CJ13610, CJ13454; FLAP (белок, активирующий 5-липоксигеназу) ингибиторы, например BAY-Y-1015, DG-031, МК591, МК886, А81834; ингибиторы LTA4 (лейкотриен А4) гидролазы, например SC56938, SC57461A);
в) антагонисты рецепторов лейкотриенов (например, СР195543, амелубант, LY293111, акколат, МК571);
2) антигипертензивные агенты, например
а) бета-блокаторы (например, метопролол, атенолол, соталол);
б) ингибиторы ангиотензинпревращающего фермента (например, каптоприл, рамиприл, хинаприл, эналаприл);
в) блокаторы кальциевых каналов (например, верапамил, дилтиазем, фелодипин, амлодипин);
г) антагонисты рецепторов ангиотензина II (например, ирбесартан, кандесартан, телемисартан, лосартан);
3) антикоагулянты, например
а) ингибиторы тромбина (например, ксимелагатран), гепарины, ингибиторы фактора Ха;
б) ингибиторы агрегации тромбоцитов (например, клопидогрель, тиклопидин, празугель, AZ4160);
4) модуляторы липидного метаболизма, например
активаторы сенсибилизаторов инсулина, такие как агонисты PPAR (ядерные рецепторы, активируемые пролифератором пероксисом) (например, пиоглитазон, розиглитазон, галида (Galida), мураглитазаар, гефемрозил, фенофибрат);
а) ингибиторы HMG-CoA редуктазы (гидроксиметилглутарил-коэнзим-А редуктазы), статины (например, симвастатин, правастатин, аторвастатин, розувастатин, флувастатин, питавастатин);
б) ингибиторы всасывания холестерина (например, эзетимид);
в) ингибиторы IBAT (транспорта желчных кислот в подвздошной кишке, например AZD-7806);
г) агонисты LXR (печеночных Х-рецепторов, например GW-683965A, Т-0901317);
д) модуляторы FXR рецепторов (фарнезиодных Х-рецепторов);
е) ингибиторы фосфолипазы;
5) агенты против стенокардии, например нитраты и нитриты;
6) модуляторы окислительного стресса, например антиоксиданты (пробукол), ингибиторы миелопероксидазы.
Изобретение проиллюстрировано следующими неограничивающими примерами.
Общие методы
Все использованные растворители были аналитической степени чистоты, и для реакций обычно использовали коммерчески доступные безводные растворители. Реакции обычно проводили в инертной атмосфере азота или аргона.
1H и 13С ЯМР-спектры записывали при 400 МГц для протона и 100 МГц для углерода-13 на ЯМР-спектрометре Varian Unity+ 400, оснащенном 5 мм ВВО датчиком с Z-градиентами, либо на ЯМР-спектрометре Bruker Avance 400, оснащенном 60 мкл двойным инверсным проточным датчиком с Z-градиентами, либо на ЯМР-спектрометре Bruker DPX400, оснащенном 4-ядерным датчиком с Z-градиентами. 600 МГц 1H ЯМР-спектры снимали на ЯМР-спектрометре Bruker av600, оснащенном 5 мм BBI датчиком с Z-градиентами. 300 МГц 1H ЯМР-спектры снимали на Varian Gemini 300 ЯМР, снабженном 5 мм BBI датчиком. 500 МГц 1H ЯМР-спектры снимали на Varian Inova 500 Spectrometer, оперируя при магнитном поле 11,74 Т, снабженном 5 мм зондом с ядерным градиентом. Если особо не указано в примерах, спектры снимали при 400 МГц для протона и 100 МГц для углерода-13. Использовали следующие референсные сигналы: средняя линия DMSO-d6 δ 2.50 (1Н), δ 39.51 (13С); средняя линия CD3OD δ 3.31 (1Н) или δ 49.15 (13С); ацетон-d6 2.04 (1Н), 206.5 (13С) и CDCl3 δ 7.26 (1H), средняя линия CDCl3 δ 77.16 (13С) (если не указано иное).
Энантиомерный избыток (ее) определяли посредством GC (газовая хроматография) на колонке Cyclodex В (изотермическая элюция 100°С) или на колонке Cyclosil В (температурный градиент 110-130°С). Диастереомерный избыток (de) определяли посредством HPLC (высокоэффективная жидкостная хроматография).
Масс-спектры снимали на аппарате Waters LCMS, состоящем из Alliance 2795 (LC) и одноквадрупольного масс-спектрометра ZQ. Масс-спектрометр был оснащен источником ионизации электрораспылением (ESI), управляемым в режиме положительных или отрицательных ионов. Капиллярное напряжение составляло 3 кВ, и масс-спектрометр сканировал в диапазоне m/z 100-700 с временем сканирования 0,3 или 0,8 сек. Разделения проводили либо на Waters X-Terra MS, колонка С8 (3,5 мкм, 50 или 100 мм × 2,1 мм в.д.), либо на АСЕ 3 AQ (100 мм × 2,1 мм в.д.), полученными от ScantecLab. Температуру колонки устанавливали равной 40°С. Применяли линейный градиент, используя нейтральную или кислотную подвижную фазовую систему, прогоняя от 0% до 100% органической фазы за 4-5 минут, скорость протока 0,3 мл/мин. Нейтральная подвижная фазовая система: ацетонитрил/[10 мМ NH4OAc (водн.) / MeCN (95:5)], или [10 мМ NH4OAc (водн.) / MeCN (1/9)] / [10 мМ NH4OAc (водн.) / MeCN (9/1)]. Кислотная подвижная фазовая система: [133 мМ НСООН (водн.) / MeCN (5/95)] / [8 мМ НСООН (водн.) / MeCN (98/2)]. Альтернативно масс-спекты снимали на масс-спектрометре Micromass LCT, оснащенном источником ионизации электрораспылением (ESI), управляемым в режиме положительных ионов.
Идентификацию соединений осуществляли на GC-MS (сочетание газовой хроматографии и масс-спектроскопии, GC 6890, 5973N MSD), поставляемом Agilent Technologies. Используемая колонка представляла собой VF-5 MS, в.д. 0,25 мм × 30 м, 0,25 мкм (Varian Inc.). Линейный температурный градиент прилагали, начиная с 40°С (выдерживали 1 минуту) и заканчивая при 300°С (выдерживали 1 минуту), 25°С/минуту. MS был оснащен EI (электронная ионизация) источником ионов. MS сканировали между m/z 50-500, а скорость сканирования составляла 3,25 сканов/с. Электронное напряжение устанавливали на 70 эВ.
HPLC анализы выполняли в системе Agilent HP1000, состоявшей из микровакуумного дегазатора G1379A, бинарного насоса G1312A, автосемлера G1367A Wellplate, С1316А термостатируемого колоночного отделения и G1315B диодно-матричного детектора. Колонка: X-Terra MS, Waters, 4,6×50 мм, 3,5 мкм. Температуру колонки устанавливали равной 40°С, а скорость потока - 1,5 мл/минуту. Диодно-матричный детектор сканировал в диапазоне 210-300 нм, шаг и ширину пика устанавливали равными 2 нм и 0,05 минуты, соответственно. Прилагали линейный градиент, прогоняли от 0% до 100% ацетонитрила за 4 минуты. Подвижная фаза: ацетонитрил/10 мМ ацетат аммония в 5% ацетонитрила в MilliQ Water.
Типичная процедура обработки после завершения реакции состояла из экстракции продукта растворителем, таким как этилацетат, промывания водой с последующей сушкой органической фазы над MgSO4 или Na2SO4 и концентрирования раствора в вакууме.
Тонкослойную хроматографияю (TLC) выполняли на пластинах Merck TLC (силикагель 60 F254), а для визуализации пятен использовали ультрафиолет. Флэш-хроматографию проводили на Combi Flash® Companion™, используя флэш-колонки с нормальной фазой RediSep™, или на Merck Silica gel 60 (0,040-0,063 мм). Типичные растворители, использованные для флэш-хроматографии, представляли собой смеси хлороформ/метанол, толуол/этилацетат и этилацетат/гексаны.
Препаративную хроматографию проводили на установке Gilson ауто-препаративной HPLC с диодно-матричным детектором, используя колонку XTerra MS (C8, 19×300 мм, 7 мкм) и градиент ацетонитрил/0,1 М ацетат аммония в 5% ацетонитрила, в MilliQ Water, прогоняли от 20% до 60% ацетонитрила за 13 минут при скорости потока 20 мл/минуту, если в примерах не указано иное. Альтернативно очистку осуществляли на полупрепаративной установке Shimadzu LC-8A HPLC с Shimadzu SPD-10A UV-vis.-детектором, снабженной колонкой Waters Symmetry® (C18,5 мкм, 100 мм × 19 мм). Градиент ацетонитрил/0,1% трифторуксусная кислота в MilliQ Water, прогоняли от 35% до 60% ацетонитрила за 20 минут. Скорость потока: 10 мл/минуту. Альтернативно препаративную HPLC проводили на установке Agilent 1100 с УФ-детекцией. Колонка: Kromasil-C18, 20×250 мм, 10 мкм. Изократическая элюция подвижной фазой ацетонитрил/MilliQ Water/муравьиная кислота (46/54/0,1). Скорость потока: 19 мл/минуту.
Перекристаллизацию обычно проводили в растворителях или смесях растворителей, таких как диэтиловый эфир, этилацетат/гептаны и метанол/вода.
Использовали следующие аббревиатуры: DCM = дихлорметан; de = диастереомерный избыток; DIPCI = β-хлордиизопинокамфенилборан (DIP-Chloride™); DIPEA = N,N-диизопропилэтиламин; DMF = N,N-диметилформамид; DMSO = диметилсульфоксид; ее = энантиомерный избыток; NCS = N-хлорсукцинимид; NMP = 1-метил-2-пирролидинон; THF = тетрагидрофуран; водн. = водный; конц. = концентрированный.
Использованные исходные вещества были либо доступны из коммерческих источников, либо их получали согласно описанным в литературе процедурам, и они имели экспериментальные данные в соответствии с сообщавшимися. Нижеследующие соединения представляют собой примеры исходные веществ, которые были получены:
(2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-фиримидин-7-ил)амино]-4-метилпентан-1-ол: WO 02/076990 (Примеры 1-4);
5-(бензилтио)-7-хлор[1,3]тиазоло[4,5-d]пиримидин-2-амин: WO 00/09511 (Примеры 6 и 7);
5-фторпиридин-2-карбонитрил: WO 2005/066155 (Пример 2);
1-(3-фторпиридин-4-ил)этанол: Marsais, F. et al. Tetrahedron 1983, 39, 2009-2021 (Пример 3);
2-ацетил-изоникотинонитрил: Citterio et al. J. Chem. Res. Synopses 1982, 10, 272-273 (Пример 5);
1-(6-хлорпиридин-3-ил)этанон: Lee, С. et al. J. Med. Chem. 2001, 44, 2133 (Примеры 6 и 7).
В общих способах, которые следуют далее, R3 и R4 являются такими, как определено в формуле (I); Ру представляет собой возможно замещенный пиридил и LG представляет собой уходящую группу.
Общий способ А
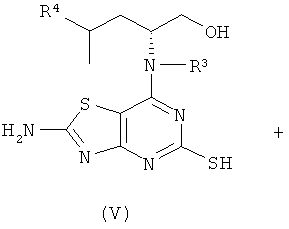
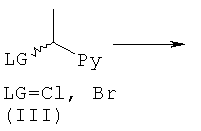
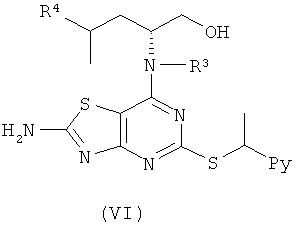
Боргидрид натрия (0,1 эквив.), DIPEA (1,5 эквив.) и (III) (1,2 эквив.) добавляли к (V) (1,0 эквив.) в DMSO в атмосфере азота. Полученную реакционную смесь перемешивали при 40°С до завершения реакции (контролировали посредством LC-MS, HPLC или TLC). Смесь выливали в ледяную воду и продукт экстрагировали DCM или EtOAc. Объединенные органические фазы сушили и концентрировали в вакууме. Сырой продукт, если необходимо, очищали с помощью препаративной HPLC или посредством колоночной флэш-хроматографии.
Общий способ В
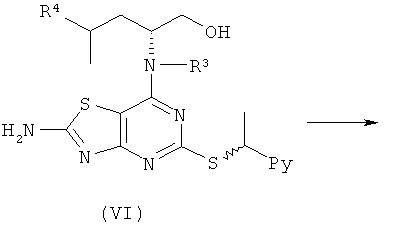
Конц. HCl (2,5 мл/ммоль (VI)) добавляли к (VI) (1,0 эквив.) в CH3CN. Реакционную смесь охлаждали на ледяной бане и по каплям добавляли нитрит натрия (2,0 эквив.), растворенный в минимальном количестве воды. Реакционную смесь перемешивали при 0°С до завершения реакции (контролировали посредством LC-MS, HPLC или TLC) и затем выливали в ледяную воду, нейтрализовали бикарбонатом натрия и экстрагировали DCM или EtOAc. Объединенные органические фазы сушили и концентрировали в вакууме с получением продукта.
Общий способ С
Гидроксид калия (2,0 эквив.), растворенный в метаноле, по каплям добавляли к охлажденному (0°С) раствору (VII) (1,0 эквив.) в метаноле. Полученную смесь перемешивали при 0°С до завершения реакции (контролировали посредством LC-MS, HPLC или TLC). Растворитель выпаривали и продукт использовали на следующей реакционной стадии без дополнительной очистки.
Общий способ D
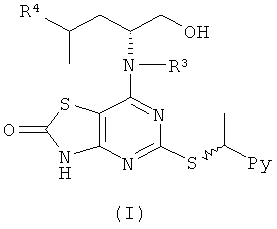
Раствор концентрированной HCl (1,0 эквив.) добавляли к охлажденному (0°С) раствору (IV) (1,0 эквив.) в 1,4-диоксане. Полученную смесь перемешивали при 40°С до завершения реакции (контролировали посредством LC-MS, HPLC или TLC). Реакционную смесь нейтрализовали насыщенным NaHCO3 (водн.) и диоксан выпаривали. Остаток растворяли в DCM или EtOAc, промывали рассолом, сушили и концентрировали в вакууме. Сырой продукт, если необходимо, очищали с помощью препаративной HPLC или посредством колоночной флэш-хроматографии.
Общий способ Е1
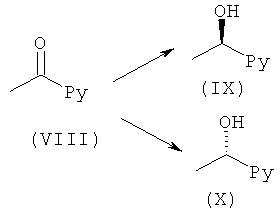
(VIII) (1,0 эквив.) в THF добавляли при 0°С к (+)-DIPCI (с получением (IX)) или (-)-DIPCI (с получением (X)) (1,5 эквив.) в THF в атмосфере аргона. Реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение ночи. Растворитель выпаривали с последующим добавлением Et2O и диэтаноламина (2,2 эквив.). Смесь перемешивали до завершения реакции (контролировали посредством LC-MS, HPLC или TLC). Образовавшийся осадок отфильтровывали, промывали Et2O и фильтрат концентрировали в вакууме. Сырой продукт, если необходимо, очищали с помощью препаративной HPLC или посредством колоночной флэш-хроматографии.
Общий способ Е2
(R)-(+)-2-метил-CBS-оксазаборолидин (1 М в толуоле, 0,1-1 эквив.) растворяли в THF и охлаждали до 0°С. По каплям добавляли комплекс боран-метилсульфид (2 М в THF, 1 эквив.) и реакционную смесь перемешивали в течение 1 ч. Реакционную смесь охлаждали до -10°С и по каплям добавляли (VIII) (1 эквив.), растворенный в THF, на протяжении 0,5 ч. Полученную смесь перемешивали в течение 1 ч или до завершения реакции и температуру медленно повышали до 10°С. Для того, чтобы погасить реакцию, добавляли 1 М водн. HCl. Добавляли насыщенный водн. NaHCO3 до тех пор, пока pH не достигал приблизительно 8. Продукт экстрагировали DCM. Объединенные органический экстракты сушили над Na2SO4 и концентрировали в вакууме с получением (X). Продукт возможно очищали посредством колоночной хроматографии.
Общий способ F1
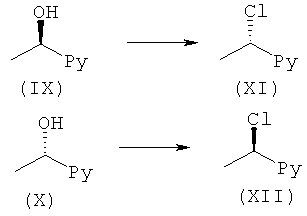
Трифенилфосфин (1,3 эквив.) в THF добавляли при 0°С к NCS (1,3 эквив.) в THF в атмосфере аргона. Полученную смесь перемешивали при температуре окружающей среды в течение 30 минут. При 0°С добавляли (IX) или (X) (1 эквив.) и реакционную смесь перемешивали при температуре окружающей среды до завершения реакции (контролировали посредством LC-MS, HPLC или TLC). Растворитель выпаривали, после чего добавляли гексан и осадок удаляли фильтрованием. Фильтрат концентрировали в вакууме и сырой продукт, если необходимо, очищали с помощью препаративной HPLC или посредством колоночной флэш-хроматографии.
Общий способ F2
Цианурхлорид (0,6 эквив.) растворяли в этилацетате. Добавляли DMF (1,5 эквив.) и смесь перемешивали при комнатной температуре в течение 10 минут. Реакционную смесь охлаждали до 0°С. (IX) или (X) (1 эквив.) растворяли в этилацетате и по каплям добавляли на протяжении 10 минут. Полученную смесь перемешивали при комнатной температуре в течение ночи. Добавляли изопропанол (приблизит. 0,25 мл/ммоль (IX) или (X)). Осадок отфильтровывали и промывали EtOAc. Фильтрат концентрировали с получением (XI) или (XII).
Общий способ G
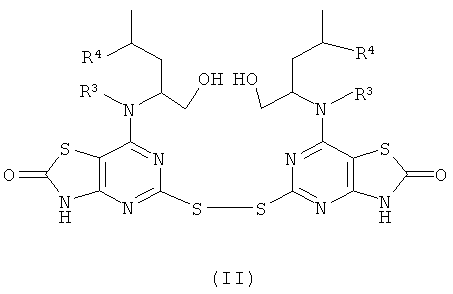
Боргидрид натрия (от 1 до 2 эквив.) добавляли к (II) (1.0 эквив.) в DMSO. После завершения выделения газа добавляли (III) (2-2,5 эквив.). Полученную реакционную смесь перемешивали при 40°С до завершения реакции (контролировали посредством LC-MS, HPLC или TLC). Очистку, если необходимо, осуществляли с помощью препаративной HPLC или посредством колоночной флэш-хроматографии.
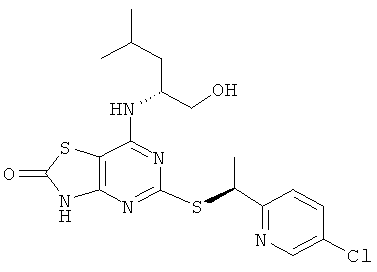
Пример 1
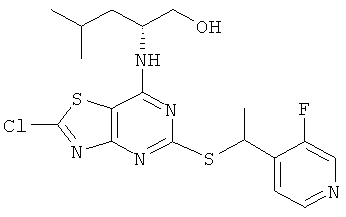
5-{[(1S)-1-(5-Хлорпиридин-2-ил)этил]тио}-7-{[1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
а) 1-(5-Хлорпиридин-2-ил)этанон
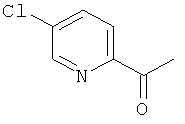
5-Хлорпиридин-2-карбонитрил (10,71 г, 77 ммоль) растворяли в диэтиловом эфире (65 мл) и THF (35 мл) в атмосфере азота. Смесь охлаждали до тех пор, пока внутренняя температура не достигала -63°С. На протяжении 30 минут добавляли метилбромид магния (3 М в THF, 35 мл, 105 ммоль). Реакционную смесь затем оставляли перемешиваться при -60°С в течение 45 минут и затем нагревали до комнатной температуры. Для растворения выпавшего в осадок вещества добавляли 50 мл THF. Через 1 ч при комнатной температуре завершение реакции проверяли посредством HPLC. Добавляли 2 М соляную кислоту (водн., 100 мл) и реакционную смесь перемешивали в течение 4 ч. pH доводили до 7 с помощью бикарбоната натрия. Фазы разделяли и продукт экстрагировали из водной фазы дважды посредством DCM. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме. Продукт очищали посредством колоночной хроматографии (элюент - градиент гептан:этилацетат) с получением 7,9 г (выход 64%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDCl3): δ м.д. 8.62 (m, 1Н); 8.00 (m, 1H); 7.80 (m, 1H); 2.70 (s, 3H).
б) (1S)-1-(5-Хлорпиридин-2-ил)этанол

Указанное в заголовке соединение получали общим способом Е2, начиная с 1-(5-хлорпиридин-2-ил)этанона (780 мг, 5 ммоль). Очистка посредством колоночной флэш-хроматографии давала 695 мг (выход 88%) указанного в заголовке соединения с 92%-ным ее.
1H ЯМР (300 МГц, CDCl3): 8.47 (s, 1H); 7.65 (d, 1H); 7.26 (d, 1H); 4.87 (q, 1H); 3.87 (br s, 1H); 1.47 (d, 3H); MS (ESI) m/z 140 и 142 [M+1]+.
в) 5-Хлор-2-[(1R)-1 -хлорэтил]пиридин
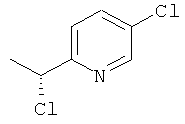
Указанное в заголовке соединение получали общим способом F2, начиная с (1S)-1-(5-хлорпиридин-2-ил)этанола (695 мг, 4,41 ммоль). Сырой продукт использовали на следующей стадии без очистки.
1H ЯМР (400 МГц, CDCl3): δ м.д. 8.46 (d, 1H), 7.64 (dd, 1H), 7.41 (d, 1H), 5.08 (q, 1H), 1.80 (d, 3H); MS (ESI) m/z 176 и 178 [М+1]+.
г) (2R)-2-[(2-Амино-5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
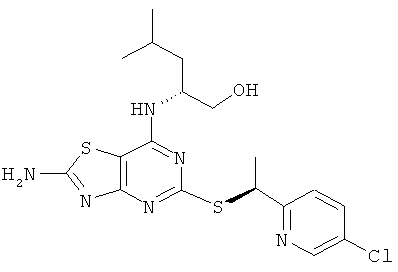
Указанное в заголовке соединение получали общим способом А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (823 мг, 2,75 ммоль) и 5-хлор-2-[(1R)-1-хлорэтил]пиридина (менее 4,4 ммоль). Очистка посредством колоночной флэш-хроматографии (элюент - градиент ОСМ:метанол) давала 350 мг (30%-ный выход) указанного в заголовке соединения.
1H ЯМР (400 МГц, CD3OD): δ м.д. 8.49 (d, 1H), 7.79 (dd, 1H), 7.66 (d, 1H), 5.22 (q, 1H), 4.46 (br s, 1H), 3.40-3.57 (m, 2H), 1.66-1.78 (m, 4H), 1.40-1.61 (m, 2H), 0.93-1.03 (m, 6H); MS (ESI) m/z 439 и 441 [М+1]+.
д) (2R)-2-[(2-Хлор-5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}[1,3]тиазоло-[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
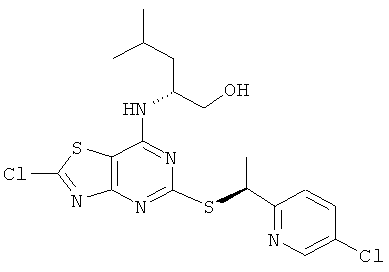
Указанное в заголовке соединение получали общим способом В, начиная с (2R)-2-[(2-амино-5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (340 мг, 0,77 ммоль).
MS (ESI) m/z 458 и 460 [M+1]+.
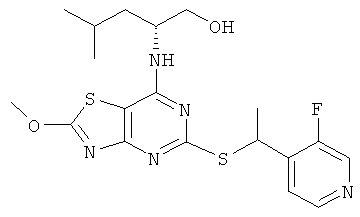
е) (2R)-2-[(5-{[(1S)-1-(5-Хлорпиридин-2-ил)этил]тио}-2-метокси-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
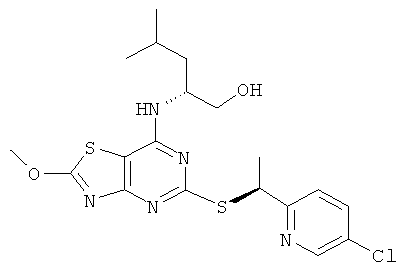
Указанное в заголовке соединение получали из (2R)-2-[(2-хлор-5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола с предыдущей стадии, используя общий способ С, за исключением того, что реакционную смесь нагревали до 50°С в течение 1 ч. После завершения реакции реакционную смесь разбавляли водой и продукт экстрагировали DCM (четыре раза). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения, которое использовали на следующей стадии без очистки.
MS (ESI) m/z 453 и 455 [М+1]+.
ж) 5-{[(1S)-1-(5-Хлорпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
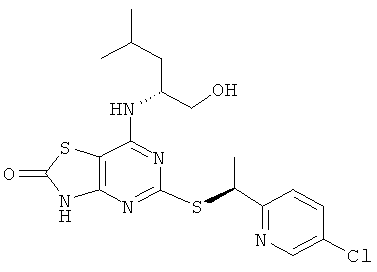
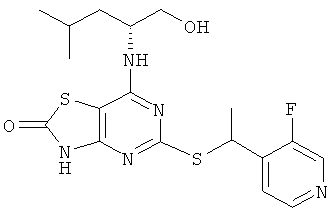
Указанное в заголовке соединение получали из (2R)-2-[(5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}-2-метокси[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола с предыдущей стадии, используя общий способ D, за исключением того, что реакционную смесь перемешивали при 50°С в течение 2,5 ч и затем при комнатной температуре в течение ночи. После завершения реакции реакционную смесь разбавляли рассолом и экстрагировали DCM (три раза). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме. Продукт очищали флэш-хроматографией (элюент - градиент DСМ:метанол) с получением 160 мг. Дополнительная очистка посредством препаративной HPLC (колонка: Chiralcel OJ, элюент: этанол/гептан 30/70, поток: 12 мл/минуту) давала 82 мг указанного в заголовке соединения.
1H ЯМР (400 МГц, CD3OD): δ м.д. 8.24 (d, 1Н), 7.56 (dd, 1H), 7.38 (d, 1H), 4.90 (q, 1H), 4.19 (br s, 1H), 3.16-3.30 (m, 2H), 1.39-1.51 (m, 4H), 1.15-1.34 (m, 2H), 0.68-0.76 (m, 6H); 1H ЯМР (DMSO-d6) δ м.д. 12.36 (br s, 1H), 8.57 (d, 1H), 7.86 (dd, 1H); 7.57 (d, 1H); 7.23 (d, 1H); 5.03 (q, 1H); 4.69 (t, 1H); 4.29 (br s, 1H); 3.40-3.25 (m, 2H), 1.66 (d, 3H), 1.63-1.52 (m, 1H); 1.48-1.32 (m, 2H), 0.88 (d, 3H), 0.85 (d, 3H); MS (ESI) m/z 440 и 442 [M+1]+, 438 и 440 [М-1]+.
Пример 2
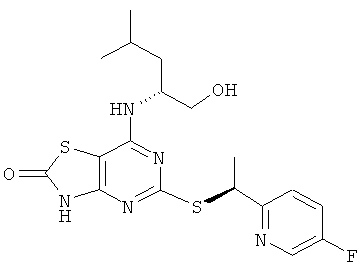
5-{[(1S)-1-(5-Фторпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}][1,31]тиазоло[4,5-d]пиримидин-2(3Н)-он
а) 1-(5-Фторпиридин-2-ил)этанон
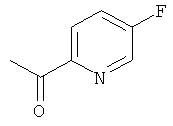
5-Фторпиридин-2-карбонитрил (29 г, 240 ммоль) растворяли в THF (150 мл) в атмосфере азота. Реакционную смесь охлаждали до внутренней температуры -64°С. На протяжении 40 минут добавляли метилбромид магния (3 М в THF, 105 мл, 315 ммоль). Реакционную смесь перемешивали при -65°С в течение 1,5 ч, затем ее нагревали до комнатной температуры. Добавляли THF (50 мл) и смесь перемешивали еще в течение 3 ч. Добавляли 2 М соляную кислоту (водн., 100 мл) до тех пор, пока смесь не становилась слабо-кислой, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем для нейтрализации реакционной смеси добавляли бикарбонат натрия. Фазы разделяли и водную фазу экстрагировали DCM. Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали посредством колоночной флэш-хроматографии с получением 18 г (55%-ный выход) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDCl3): 8.50 (m, 1Н); 8.10 (m, 1H); 7.52 (m, 1H); 2.70 (s, 3Н).
б) (1S)-1-(5-Фторпиридин-2-ил)этанол
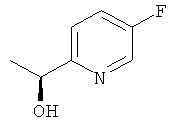
Указанное в заголовке соединение получали общим способом Е2, начиная с 1-(5-фторпиридин-2-ил)этанона (3,18 г, 22,9 ммоль). Очистка посредством колоночной флэш-хроматографии давала 2,73 г (84%-ный выход) указанного в заголовке соединения с 84%-ным ее.
1H ЯМР (300 МГц, CDCl3): 8.38 (m, 1H); 7.5-7.2 (m, 2H); 4.89 (q, 1H); 3.9 (br s, 1H); 1.49 (d, 3H).
в) 2-[(1R)-1-Хлорэтил]-5-фторпиридин
Указанное в заголовке соединение с 80%-ным ее получали общим способом F2, начиная с (1S)-1-(5-фторпиридин-2-ил)этанола (720 мг, 5,1 ммоль). Сырой продукт использовали на следующей стадии без очистки.
1H ЯМР (300 МГц, CDCl3): 8.44-8.40 (m, 1H); 7.6-7.4 (m, 2H); 5.16 (q, 1H), 1.86 (d, 3H).
г) (2R)-2-[(2-Амино-5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали общим способом А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (940 мг, 3,1 ммоль) и 2-[(1R)-1-хлорэтил]-5-фторпиридина (0,81 г, 5,1 ммоль). Продукт очищали колоночной флэш-хроматографией с получением 0,75 г (56%-ный выход) указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-d6) δ м.д. 8.51 (d, 1Н), 7.98 (s, 2H), 7.65 (dt, 1H); 7.58 (dd, 1H), 6.88 (d, 1H); 5.12 (q, 1H); 4.66 (t, 1H); 4.27 (br s, 1H); 3.41-3.27 (m, 2H), 1.66 (d, 3H), 1.65-1.55 (m, 1H); 1.48-1.35 (m, 2H), 0.88 (d, 3H), 0.85 (d, 3H); MS (ESI) m/z 423 [M+1]+.
д) (2R)-2-[(2-Хлор-5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали, используя общий способ В, начиная с (2R)-2-[(2-амино-5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (750 мг, 1,77 ммоль).
MS (ESI) m/z 442 и 444 [М+1]+.
е) (2R)-2-[(5-{[(1S)-1-(5-Фторпиридин-2-ил)этил]тио}-2-метокси-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали из (2R)-2-[(2-хлор-5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола с предыдущей стадии, используя общий способ С, за исключением того, что реакционную смесь нагревали до 50°С в течение 1,5 ч. После завершения реакции реакционную смесь разбавляли водой и рассолом и продукт экстрагировали хлороформом (три раза). Объединенные органические экстракты сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения, которое использовали без очистки.
MS(ESI) m/z 438 [M+1]+.
ж) 5-{[(1S)-1-(5-Фторпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
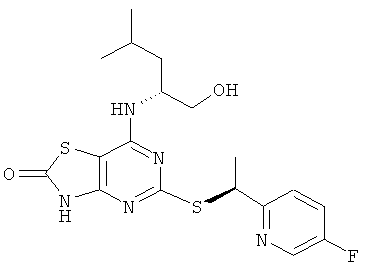
Указанное в заголовке соединение получали из (2R)-2-[(5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}-2-метокси[1,3]тиазоло[4,5-d]пиримидин-7-ил)-амино]-4-метилпентан-1-ола с предыдущей стадии, используя общий способ D, с тем исключением, что реакционную смесь перемешивали при 50°С в течение 3 ч. После завершения реакции реакционную смесь разбавляли рассолом и экстрагировали DCM (три раза). Объединенные органические экстракты сушили над сульфатом магния и концентрировали в вакууме. Продукт очищали посредством флэш-хроматографии (элюент - градиент DCM:метанол). Дополнительная очистка посредством препаративной HPLC (колонка Chiralcel OJ, элюент: этанол, поток: 8 мл/минуту) давала 113 мг указанного в заголовке соединения.
1H ЯМР (CD3OD): δ м.д. 8.19 (d, 1H), 7.46 (dd, 1H), 7.36 (dt, 1H), 4.97 (q, 1H), 4.26 (br s, 1H), 3.23-3.34 (m, 2H), 1.44-1.55 (m, 4H), 1.19-1.37 (m, 2H), 0.75 (dd, 6H); 1H ЯМР (DMSO-d6) δ м.д. 12.36 (br s, 1H), 8.52 (d, 1H), 7.66 (dt, 1H); 7.60 (dd, 1H), 7.23 (d, 1H); 5.07 (q, 1H); 4.69 (t, 1H); 4.30 (br s, 1H); 3.40-3.26 (m, 2H), 1.67 (d, 3H), 1.64-1.53 (m, 1H); 1.48-1.33 (m, 2H), 0.88 (d, 3H), 0.85 (d, 3H); MS (ESI) m/z 424 [M+1]+. OK
Пример 3
5-{[1-(3-Фторпиридин-4-ил)этил]тио}-7-{[1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
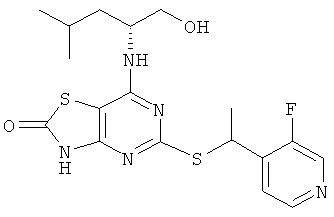
а) 4-(1-Хлорэтил)-3-фторпиридин
1-(3-Фторпиридин-4-ил)этанол (0,8 г, 5,7 ммоль) обрабатывали тионилхлоридом (5 мл) и полученную смесь нагревали до 80°С в течение 2 ч. Добавляли воду (10 мл) и насыщ. бикарбонат натрия (водн., 10 мл). Продукт экстрагировали DCM (три раза). Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (элюент - градиент гептан:этилацетат) с получением 0,36 г (39%-ный выход) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDCl3) 8.45 (m, 2H), 7.50 (m, 1H), 5.34 (q, 1H), 1.83 (d, 3H).
б) (2R)-2-[(2-Амино-5-{[1-(3-фторпиридин-4-ил)этил]тио}[1,3]тиазоло-[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
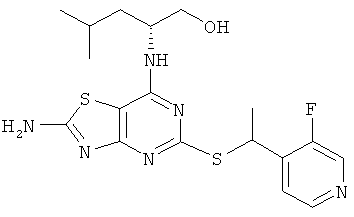
Указанное в заголовке соединение (370 мг, 47%-ный выход) получали, используя общий способ А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (560 мг, 1,87 ммоль).
MS (ESI) m/z 423 [M+1]+.
в) (2R)-2-[(2-Хлор-5-{[1-(3-фторпиридин-4-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали, используя общий способ В, начиная с (2R)-2-[(2-амино-5-{[1-(3-фторпиридин-4-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (370 мг, 0,84 ммоль).
г) (2R)-2-[(5-{[1-(3-Фторпиридин-4-ил)этил]тио}-2-метокси[1,3]тиазоло-[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали из (2R)-2-[(2-хлор-5-{[1-(3-фторпиридин-4-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола с предыдущей стадии, используя общий способ С, с тем исключением, что реакционную смесь нагревали до 50°С в течение 1,5 ч. После завершения реакции реакционную смесь разбавляли водой и рассолом (1:1) и продукт экстрагировали DCM (дважды). Затем pH водной фазы доводили до 7 с помощью хлорида аммония и продукт экстрагировали DCM (дважды). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения.
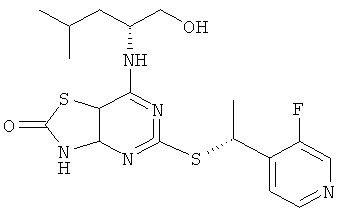
е) 5-{[1-(3-Фторпиридин-4-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
Указанное в заголовке соединение получали, начиная с (2R)-2-[(5-{[1-(3-фторпиридин-4-ил)этил]тио}-2-метокси[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола с предыдущей стадии, используя общий способ D, с тем исключением, что реакционную смесь перемешивали при 50°С в течение 2 ч. После завершения реакции реакционную смесь разбавляли насыщ. бикарбонатом натрия (водн.) и водой (1:1) и экстрагировали DCM (три раза). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме. Продукт очищали посредством колоночной флэш-хроматографии (элюент - градиент гептан:этилацетат) с получением указанного в заголовке соединения в виде смеси диастереомеров (194 мг).
MS (ESI) m/z 424 [M+1]+.
Пример 4
5-{(1S)-1-(3-Фторпиридин-2-ил)этил]тио}-7-{(1R)-1-(гидроксиметил)-3-метилбутил]амино[1,3]тиазоло[4,5-d]пиримидин-2(3H)-он

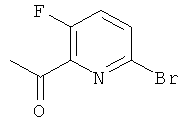
а) 1-(6-Бром-3-фторпиридин-2-ил)этанон
2-Бром-5-фторпиридин (11 г, 62,5 ммоль) растворяли в диэтиловом эфире при комнатной температуре в атмосфере азота. Реакционную смесь охлаждали до тех пор, пока внутренняя температура не достигала -66°С. По каплям на протяжении 0,5 ч добавляли бутиллитий (2,5 М в гексанах, 26 мл, 65 ммоль). Полученную реакционную смесь оставляли при -65°С в течение 1 ч. На протяжении 10 минут добавляли N,N-диметилацетамид (6,5 мл, 70 ммоль) и реакционную смесь перемешивали при -65°С в течение 2 ч. Добавляли 1 М соляную кислоту (водн., 50 мл) и смесь нагревали до комнатной температуры. pH доводили до 7 добавлением соляной кислоты. Водную фазу три раза экстрагировали диэтиловым эфиром. Объединенные органические фазы промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Очистка посредством колоночной флэш-хроматографии (элюент - градиент гептан:диэтиловый эфир) давала 4,6 г (34%-ный выход) указанного в заголовке соединения.
1H ЯМР (300 МГц, DMSO-d6): 8.0-7.8 (m, 2H); 2.57 (s, 3Н); MS (ESI) m/z 218 и 220 [М+1]+.
б) (1S)-1-(6-Бром-3-фторпиридин-2-ил)этанол

Указанное в заголовке соединение получали, используя общий способ Е2, начиная с 1-(6-бром-3-фторпиридин-2-ил)этанона (1,76 г, 8,19 ммоль). Продукт очищали посредством колоночной флэш-хроматографии (элюент - градиент гептан:этилацетат) с получением 1,31 г (73%-ный выход) указанного в заголовке соединения с 80%-ным ее.
1H ЯМР (300 МГц, CDCl3) 7.38 (m, 1Н); 7.26 (m, 1H); 5.06 (q, 1H); 3.38 (br s, 1H); 1.47 (d, 3H); MS (ESI) m/z 220 и 222 [M+1]+, m/z 202 [M-H2O]+.
в) (1S)-1-(3-Фторпиридин-2-ил)этанол
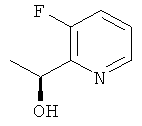
(1S)-1-(6-Бром-3-фторпиридин-2-ил)этанол (1,3 г, 5,9 ммоль), триэтиламин (1,6 мл, 11,5 ммоль) и палладий на углероде (0,64 г, 0,34 ммоль) смешивали в DCM (25 мл). Колбу вакуумировали/заполняли газообразным водородом в течение 4 циклов и затем оставляли под давлением водорода 2,5 атм (253,25 кПа) при комнатной температуре в течение 24 ч. Смесь фильтровали и твердое вещество промывали DCM. Фильтрат промывали водой и рассолом и сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (элюент - градиент DСМ:метанол) с получением 0,54 г (65%-ный выход) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDCl3): 8.38 (m, 1Н); 7.39 (m, 1H); 7.26 (m, 1H); 5.11 (q, 1H); 4.16 (br s, 1H); 1.49 (d, 3H).
г) 2-((R)-1-Хлорэтил)-3-фторпиридин
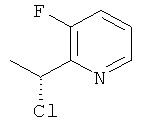
Указанное в заголовке соединение (0,24 г) получали, используя общий способ F2, начиная с (1S)-1-(3-фторпиридин-2-ил)этанола (254 мг, 1,8 ммоль).
1H ЯМР (300 МГц, CDCl3): 8.46 (m, 1H); 7.47 (m, 1H); 7.34 (m, 1H); 5.48 (q, 1H), 1.94 (d, 3H); MS (ESI) m/z 160 и 162 [M+1]+.
д) (2R)-2-[(2-Амино-5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
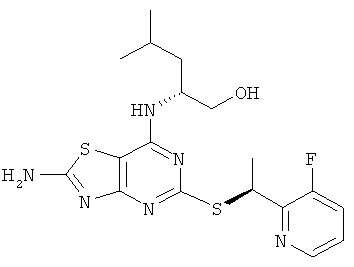
Указанное в заголовке соединение получали, используя общий способ А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]фиримидин-7-ил)-амино]-4-метилпентан-1-ола (348 мг, 1,16 ммоль) и 2-((R)-1-хлорэтил)-3-фторпиридина (240 мг, 1,5 ммоль). В результате очистки посредством колоночной флэш-хроматографии (элюент - градиент DСМ:метанол) получали 190 мг (47%-ный выход) указанного в заголовке соединения с диастереомерным избытком 60%.
1H ЯМР (400 МГц, DMSO-d6) δ м.д. 8.40 (dt, 1Н), 7.98 (s, 2Н), 7.70 (m, 1H), 7.40 (m, 1H); 6.92 (d, 1H); 5.45 (q, 1H); 4.65 (t, 1H); 4.27 (br s, 1H); 3.45-3.30 (m, 2Н), 1.69 (d, 3Н), 1.66-1.58 (m, 1H), 1.50-1.35 (m, 2Н), 0.88 (d, 3Н), 0.85 (d, 3Н); MS (ESI) m/z 423 [M+1]+. MS (ESI) m/z 423 [M+]+.
e) (2R)-2-[(2-Хлор-5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
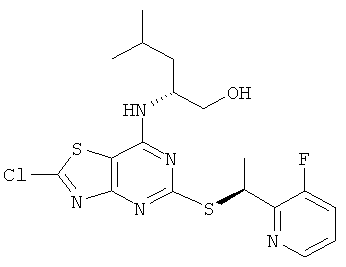
Указанное в заголовке соединение получали, используя общий способ В, начиная с (2R)-2-[(2-амино-5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (135 мг, 0,32 ммоль).
MS (ESI) m/z 442 и 444 [М+1]+.
ж) (2R)-2-[(5-{[(1S)-1-(3-Фторпиридин-2-ил)этил]тио}-2-метокси-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали из (2R)-2-[(2-хлор-5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]фиримидин-7-ил)амино]-4-метилпентан-1-ола с предыдущей стадии, используя общий способ С, за исключением того, что реакционную смесь нагревали до 50°С в течение 1,5 ч. После завершения реакции реакционную смесь разбавляли водой и рассолом (2:1) и продукт экстрагировали хлороформом (три раза). Объединенные органические экстракты сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения.
MS (ESI) m/z 438 [M+1]+.
з) 5-{[(1S)-1-(3-Фторпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
Указанное в заголовке соединение получали из (2R)-2-[(5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}-2-метокси[1,3]тиазоло[4,5-d]пиримидин-7-ил)-амино]-4-метилпентан-1-ола, используя общий способ D, с тем исключением, что реакционную смесь нагревали до 50°С в течение 1,5 ч. После завершения реакции реакционную смесь разбавляли рассолом и экстрагировали DCM (три раза). Объединенные органические экстракты сушили над сульфатом магния и концентрировали в вакууме. Продукт очищали колоночной флэш-хроматографией (элюент - градиент DСМ:метанол), а затем препаративной HPLC с получением 20 мг указанного в заголовке соединения.
1H ЯМР (DMSO-d6) δ м.д. 12.37 (br s, 1Н), 8.41 (dt, 1H), 7.72 (m, 1H); 7.42 (m, 1H); 7.27 (br s, 1H); 5.43 (q, 1H); 4.67 (t, 1H); 4.30 (br s, 1H); 3.44-3.30 (m, 2H), 1.70 (d, 3H), 1.65-1.55 (m, 1H); 1.52-1.32 (m, 2H), 0.89 (d, 3Н), 0.86 (d, 3H); MS (ESI) m/z 424 [M+1]+.
Пример 5
2-{(1S)-1-[(7-[{(1R)-1-(Гидроксиметил)-3-метилбутил1амино}-2-оксо-2,3-дигидро[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}изоникотинонитрил
a) 2-((S)-1-Гидроксиэтил)-изоникотинонитрил
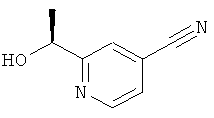
Указанное в заголовке соединение (1,13 г, 7,63 ммоль) получали в соответствии с общим способом Е1, начиная с 2-ацетил-изоникотинонитрила (1,42 г, 9,72 ммоль) и (-)-DIPCI (4,67 г, 14,57 ммоль).
1H ЯМР (500 МГц, CDCl3) δ 8.72 (d, 1H), 7.62 (s, 1H), 7.44 (dd, 1H), 4.96 (q, 1H), 1.54 (d, 3H).
б) 2-((R)-1-Хлорэтил)-изоникотинонитрил
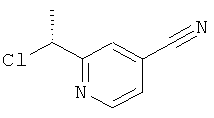
Указанное в заголовке соединение (32,2 мг, 0,19 ммоль) получали в соответствии с общим способом F1, начиная с 2-((S)-1-гидрокси-этил)-изоникотинонитрила (400 мг, 2,7 ммоль).
1H ЯМР (500 МГц, CDCl3) δ 8.74 (d, 1H), 7.76 (s, 1H), 7.46 (dd, 1H), 5.16 (q, 1H), 1.88 (d, 3H).
в) (2R)-2-{2-Хлор-5-[2-хлор-7-((1R)-1-гидроксиметил-3-метил-бутиламино)-тиазоло[4,5-d]пиримидин-5-илдисульфанил]-тиазоло[4,5-d]пиримидин-7-иламино}-4-метил-пентан-1-ол
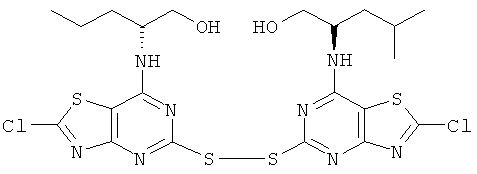
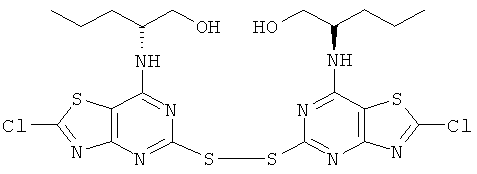
Нитрит натрия (5,19 г, 75 ммоль) в воде (25 мл) при 0°С по каплям добавляли к (2R)-2-[[2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил]-амино]-4-метилпентан-1-олу (7,50 г, 25 ммоль) в конц. соляной кислоте (150 мл) и ацетонитриле (150 мл). Реакционную смесь перемешивали в течение 18 ч при 0-5°С и затем выливали на лед (500 мл) и экстрагировали этилацетатом. Все оставшееся твердое вещество отфильтровывали. Объединенные органические фазы промывали последовательно рассолом и насыщенным водным раствором бикарбона натрия. Органическую фазу сушили и выпаривали и к ней добавляли предварительно отфильтрованное твердое вещество. Совокупное твердое вещество диспергировали в этилацетате, из которого после фильтрования получали указанное в заголовке соединение (6,3 г, 80%-ный выход).
1H ЯМР (DMSO-d6) δ 8.25 (d, 2H), 4.19 (m, 2H), 3.35 (m, 4H), 1.40 (m, 4H), 1.21 (m, 2H), 0.68 (d, 6H), 0.51 (d, 6H); MS (ESI) m/z 635 [М+1]+.
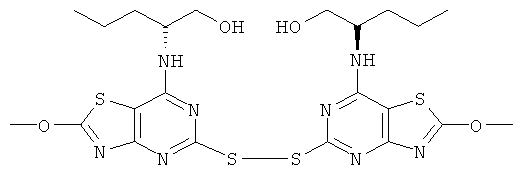
г) (2R)-2-{5-[7-((1R)-1-Гидроксиметил-3-метил-бутиламино)-2-метокси-тиазоло[4,5-d]пиримидин-5-илдисульфанил]-2-метокси-тиазоло[4,5-d]пиримидин-7-иламино}-4-метил-пентан-1-ол
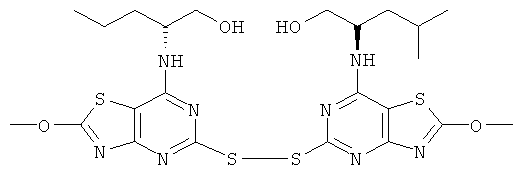
Гидроксид калия (0,53 г, 9,4 ммоль) в метаноле (5 мл) добавляли при 0°С к раствору (2R)-2-{2-хлор-5-[2-хлор-7-((1R)-1-гидроксиметил-3-метил-бутил-амино)-тиазоло[4,5-d]пиримидин-5-илдисульфанил]-тиазоло[4,5-d]пиримидин-7-иламино}-4-метил-пентан-1-ола (3,0 г, 4,7 ммоль) в метаноле (200 мл). Реакцию поддерживали при 0-5°С в течение 18 ч. Растворитель выпаривали и остаток переносили в смесь метанол/этилацетат (1:1). Этот раствор быстро хроматографировали (элюент этилацетат) с получением указанного в заголовке соединения (2,0 г, 68%-ный выход).
MS (ESI) m/z 627 [M+1]+.
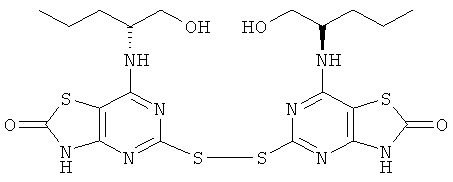
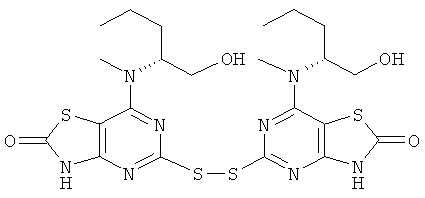
д) 5-[7-{[(1R)-1-(Гидроксиметил)-3-метилбутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он-5-илдисульфанил]-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
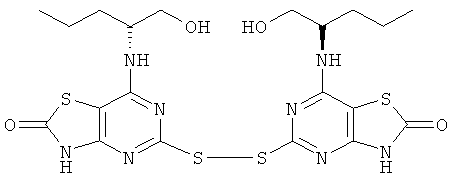
Смесь конц. соляной кислоты (20 мл) и воды (20 мл) добавляли к раствору (2R)-2-{5-[7-((1R)-1-гидроксиметил-3-метил-бутиламино)-2-метокси-тиазоло[4,5-d]пиримидин-5-илдисульфанил]-2-метокси-тиазоло[4,5-d]пиримидин-7-иламино}-4-метил-пентан-1-ола (1,5 г, 2,4 ммоль) в 1,4-диоксане (20 мл). Этот раствор затем перемешивали при 45°С в течение 18 ч. Растворитель выпаривали и остаток переносили в этилацетат. Весь нерастворенный остаток собирали фильтрованием. Фильтрат подвергали колоночной флэш-хроматографии (элюент - смесь этилацетат:метанол, 95:5). Твердый остаток и продукт, собранный после хроматографии, объединяли с получением указанного в заголовке соединения (600 мг, 42%-ный выход).
1H ЯМР (DMSO-d6) δ 12.45 (s, 2Н), 7.33 (d, 2H), 4.62 (t, 2H), 4.17 (br s, 2H), 1.48-1.31 (m, 4H), 1.25-1.14 (m, 2H), 0.72 (d, 6H), 0.56 (d, 6H); MS (ESI) m/z 599 [М+1]+.
e) 2-{(1S)-1-[(7-{[(1R)-1-(Гидроксиметил)-3-метилбутил]амино}-2-оксо-2,3-дигидро[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}изоникотинонитрил
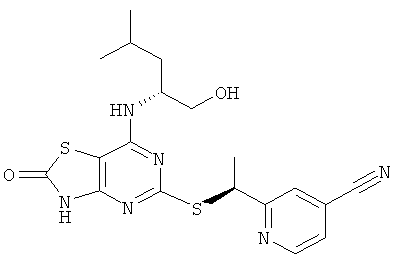
Указанное в заголовке соединение получали в соответствии с общим способом G с добавлением DIPEA (2 эквив.), начиная с соединения 5,5'-дитиобис[7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он] (64 мг, 0,096 ммоль) и 2-((R)-1-хлор-этил)-изоникотинонитрила (32 мг, 0,192 ммоль). Указанное в заголовке соединение (39 мг) получали в виде диастереомерной смеси. Очистка посредством препаративной HPLC (колонка: Kromasil-C18) давала 15 мг (36%-ный выход) указанного в заголовке соединения с 98%-ным de.
1H ЯМР (500 МГц, CD3OD) δ 8.71 (d, 1H), 7.92 (s, 1H), 7.56 (d, 1H), 5.17 (q, 1H), 4.4 (s, 1H), 3.40-3.52 (m, 2H), 1.72 (d 3Н), 1.60-1.71 (m 1H), 1.38-1.54 (m, 2H), 0.90-0.98 (m 6H); MS (ESI+ m/z 431 [M+H]+.
Пример 6
5-{[(1S)-1-(6-Хлорпиридин-3-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-бутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3H)-он
а) (2R)-2-{[2-Амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}пентан-1-ол
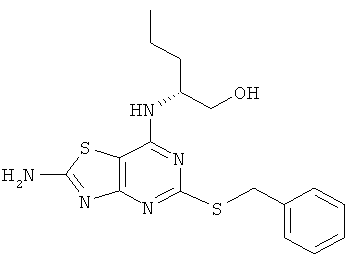
5-(Бензилтио)-7-хлор[1,3]тиазоло[4,5-d]пиримидин-2-амин (6,0 г, 19,4 ммоль) растворяли в NMP (30 мл). Добавляли DIPEA (8,4 мл, 48,5 ммоль) и 2-амино-(2R)-1-пентанол (3,5 г, 33,9 ммоль) и смесь нагревали до 110°С в течение 4 дней. После охлаждения до комнатной температуры смесь выливали в воду (200 мл). Выпавший в осадок продукт собирали фильтрованием, промывали водой и использовали на следующей стадии без дальнейшей очистки (7,0 г, 97%-ный выход).
MS (ESI+) m/z 376 [M+H]+.
б) (2R)-2-[(2-Амино-5-меркапппо[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол
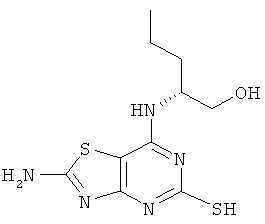
Круглодонную колбу снабжали холодильником сухой лед-этанол и погружали в охлаждающую баню сухой лед-этанол. В колбе конденсировали аммиак (250 мл), а затем добавляли (2R)-2-{[2-амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}пентан-1-ол (6,8 г, 18,1 ммоль). Полученной смеси давали нагреться до -33°С и небольшими кусочками добавляли металлический натрий до тех пор, пока не появлялось синее окрашивание, которое сохранялось в течение 30 секунд. Затем реакцию гасили добавлением ложки твердого хлорида аммония. Аммиак выпаривали и к остатку добавляли воду (250 мл). Полученную смесь нейтрализовали 1 М соляной кислотой (водн.). Выпавший в осадок продукт собирали фильтрованием, промывали водой и сушили в вакууме с получением 4,15 г (80%-ный выход) указанного в заголовке соединения.
MS (ESI+) m/z 286 [M+H]+.
в) (2R)-2-{2-Хлор-5-[2-хлор-7-((1R)-1-гидроксиметилбутиламино)-тиазоло[4,5-d]пиримидин-5-илдисульфанил]-тиазоло[4,5-d]пиримидин-7-иламино}-пентан-1-ол
(2R)-2-[(2-Амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-пентан-1-ол (4,0 г, 14 ммоль) растворяли в ацетонитриле (100 мл) и концентрированной соляной кислоте (150 мл). Нитрит натрия (1,93 г, 28 ммоль) растворяли в воде (10 мл) и добавляли при 0°С. Реакционную смесь оставляли при 0°С на 2 дня до завершения реакции согласно LCMS. Реакционную смесь выливали на лед и выпавший в осадок продукт собирали фильтрованием. Твердое вещество сушили в вакууме с получением 3,3 г (78%-ный выход) указанного в заголовке соединения.
1H ЯМР (DMSO-d6) δ 8.27 (d, 1H), 4.32-3.81 (m, 2H), 3.50-3.23 (m, 2H), 1.37-1.19 (m, 2H), 1.10-0.93 (m, 1H), 0.94-0.78 (m, 1H), 0.49 (t, 3H); MS (ESI) m/z 607 [М+1]+.
г) (2R)-2-{5-[7-((1R)-1-Гидроксиметилбутиламино)-2-метокси-тиазоло[4,5-d]пиримидин-5-илдисульфанил]-2-метокси-тиазоло[4,5-d]пиримидин-7-иламино}-пентан-1-ол
Гидроксид калия (495 мг, 8,8 ммоль) добавляли к (2R)-2-{2-хлор-5-[2-хлор-7-((1R)-1-гидроксиметилбутиламино)-тиазоло[4,5-d]пиримидин-5-илдисульфанил]-тиазоло[4,5-d]пиримидин-7-иламино}-пентан-1-олу (2,68 г, 4,41 ммоль) в метаноле (200 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение ночи и затем метанол выпаривали. Остаток выливали в воду и полученный в результате осадок собирали фильтрованием. Сырой влажный продукт использовали на следующей стадии без дополнительной очистки.
MS (ESI) m/z 599 [М+1]+.
д) 5-[7-{[(1R)-1-(Гидроксиметил)]амино}-[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он-5-илдисульфанил]-7-{[(1R)-1-(гидроксиметилбутил]амино}[1,3]-тиазоло[4,5-d]пиримидин-2(3Н)-он
Сырой (2R)-2-{5-[7-((1R)-1-гидроксиметилбутиламино)-2-метокси-тиазоло[4,5-d]пиримидин-5-илдисульфанил]-2-метокси-тиазоло[4,5-d]пиримидин-7-иламино}-пентан-1-ол (4,41 ммоль) с предыдущей стадии растворяли в 1,4-диоксане (100 мл). Добавляли конц. соляную кислоту (2 мл) и воду (2 мл) и полученную смесь перемешивали при 45°С в течение ночи. Растворитель выпаривали в вакууме и продукт осаждали добавлением воды. Осадок собирали фильтрованием и промывали водой. Сырой продукт очищали колоночной флэш-хроматографией (элюент - градиент DCM:этилацетат) с получением 1,5 г (59%-ный выход за две стадии) указанного в заголовке соединения.
1H ЯМР (DMSO-d6) δ 12.46 (s, 1H), 7.33 (d, 1H), 4.61 (t, 1H), 4.10 (br s, 1H), 3.35 (t, 2H), 1.37-1.20 (m, 2H), 1.13-1.10 (m, 1H), 0.96-0.82 (m, 1H), 0.59 (t, 3H); MS (ESI) m/z 571 [M+1]+.
e) (1S)-1-(6-Хлорпиридин-3-ил)этанол
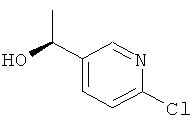
Указанное в заголовке соединение получали в соответствии с общим способом Е1, используя 1-(6-хлорпиридин-3-ил)этанон (0,80 г, 5,14 ммоль), с получением 0,71 г (88%-ный выход) указанного в заголовке соединения.
1H ЯМР (CDCl3) δ м.д. 8.40-8.28 (m, 1H), 7.75-7.63 (m, 1H), 7.35-7.24 (m, 1H), 5.04-4.79 (m, 1H), 1.63-1.45 (m, 3H); MS (ESI) m/z 158 и 160 [M+1]+.
ж) 2-Хлор-5-[(1R)-1-хлорэтил]пиридин
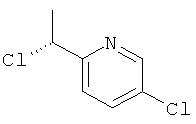
Указанное в заголовке соединение получали в соответствии с общим способом F1, используя (1S)-1-(6-хлорпиридин-3-ил)этанол (0,20 г, 1,27 ммоль), с получением 0,16 г (72%-ный выход) указанного в заголовке соединения.
1H ЯМР (CDCl3) δ м.д. 8.45-8.35 (m, 1H), 7.79-7.70 (m, 1H), 7.39-7.29 (m, 1H), 5.07 (q, 1H), 1.85-1.78 (m, 3H); MS (ESI) m/z 176 и 178 [М+1]+.
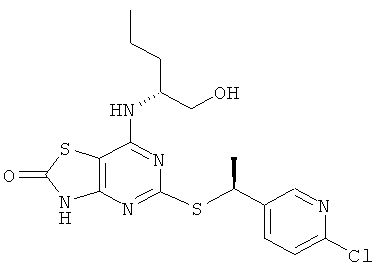
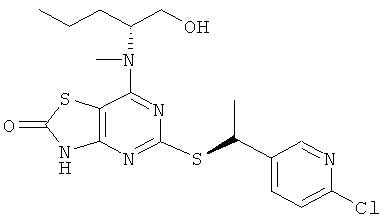
з) 5-{[(1S)-1-(6-Хлорпиридин-3-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-бутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
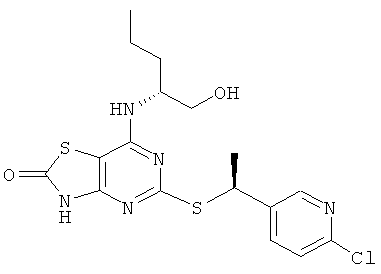
Указанное в заголовке соединение получали в соответствии с общим способ G, используя 5-[7-{[(1R)-1-(гидроксиметил)]амино}-[1,3]тиазоло[4,5-d]пиримидин-2(3H)-он-5-илдисульфанил]-7-{[(1R)-1-(гидроксиметилбутил]-амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он (0,10 г, 0,175 ммоль), 2-хлор-5-[(1R)-1-хлорэтил]пиридин (0,069 г, 0,39 ммоль) и боргидрид натрия (0,040 г, 1,05 ммоль), с получением 0,055 г (37%-ный выход) указанного в заголовке соединения.
1H ЯМР (CDCl3) δ м.д. 8.52-8.38 (m, 1H), 7.87-7.72 (m, 1H), 7.30-7.26 (m, 1H), 4.91-4.81 (m, 1H), 4.74-4.65 (m, 1H), 4.29-4.17 (m, 1H), 3.68-3.52 (m, 2H), 1.69-1.64 (m, 3H), 1.56-1.46 (m, 2H), 1.46-1.32 (m, 2H), 0.98-0.90 (m, 3Н);
MS (ESI) m/z 426 и 428 [M+1]+.
Пример 7
5-{[(1S)-1-(6-Хлорпиридин-3-ил)этил]тио}-7-[[(1R)-1-(гидроксиметил)-бутил](метил)амино][1,3]тиазоло[4,5-d]-пиримидин-2(3H)-он

a) N-(Этоксикарбонил)-D-норвалин
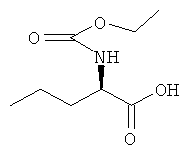
D-Норвалин (10,0 г, 85,3 ммоль) растворяли в водном гидроксиде натрия (4 M, 25 мл). На протяжении 15 минут при 0°С добавляли этилхлорформиат (10,6 мл, 111 ммоль) и водный гидроксид натрия (4 M, 25 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали при этой температуре в течение 4 ч. Реакционную смесь промывали диэтиловым эфиром три раза и затем подкисляли водной соляной кислотой (2 M). Продукт экстрагировали диэтиловым эфиром три раза. Объединенные органические фазы сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения с количественным выходом.
1H ЯМР (CDСl3) δ м.д. 6.43 (br s, 1Н), 5.22 (d, 1Н), 4.37 (q, 1H), 4.13 (q, 2H), 1.84 (m, 1Н), 1.68 (секстет, 1Н), 1.42 (секстет, 1Н), 1.25 (t, 3H), 0.95 (t, 3H); MS (Cl) 144 (100%), 190 [М+1]+.
б) (2R)-2-(Метиламино)пентан-1-ол
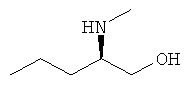
Алюмогидрид лития (6,5 г, 171 ммоль) суспендировали в THF при 0°С в атмосфере азота. N-(этоксикарбонил)-D-норвалин растворяли в THF и по каплям добавляли при 0°С. Реакционную смесь кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры добавляли насыщенный водный сульфат натрия с образованием суспензии. Полученную смесь фильтровали через целит. Твердое вещество промывали DCM до тех пор, пока не экстрагировали весь продукт. Объединенные фильтраты сушили над сульфатом натрия и концентрировали в вакууме. В результате перегонки "из колбы в колбу" (bulb-to-bulb distiltion) при 0,1 мбар (10 Па) со сбором фракции между 75-85°С получали 7,1 г (71%-ный выход) указанного в заголовке соединения.
1H ЯМР (CDCl3) 3.63 (dd, 1Н); 3.30 (dd, 1Н); 2.51 (m, 1Н); 2.41 (s, 3H); 2.09 (br s, 2H); 1.50-1.28 (m, 4H); 0.93 (t, 3H); MS (Cl) 86 (100%), 118 [M+1]+.
в) (2R)-2-{[2-Амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил](метил)амино}пентан-1-ол
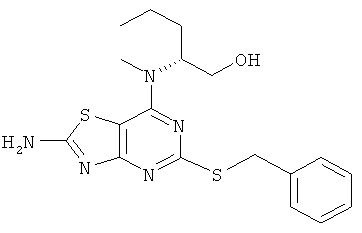
5-(Бензилтио)-7-хлор[1,3]тиазоло[4,5d]-пиримидин-2-амин (6,0 г, 19,4 ммоль) растворяли в NMP (25 мл). Добавляли DIPEA (6,8 мл, 38,8 ммоль) и (2R)-2-(метиламино)пентан-1-ол (3,4 г, 29,1 ммоль) и смесь нагревали до 120°С в течение 3 дней. Добавляли дополнительное количество (2R)-2-(метиламино)пентан-1-ола (350 мг, 2,99 ммоль) и DIPEA (1 мл, 5,74 ммоль) и реакционную смесь нагревали в течение 6 ч при 120°С. После охлаждения до комнатной температуры смесь выливали на лед. Выпавший в осадок продукт собирали фильтрованием и очищали колоночной флэш-хроматографией (элюент - градиент DCM:этилацетат) с получением указанного в заголовке соединения (5,74 г, 76%-ный выход).
1H ЯМР (DMSO-d6) 7.98 (br s, 2H), 7.41 (m, 2H), 7.29 (m, 2H), 7.22 (m, 1H), 4.73 (t, 1H), 4.54 (br s, 1H), 4.33 (m, 2H), 3.55-3.40 (m, 2H), 3.01 (s, 3Н), 1.52-1.44 (m, 2H), 1.25-1.10 (m, 2H), 0.84 (t, 3Н); MS (ESI) m/z 390 [M+1]+.
г) (2R)-2-[(2-Амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)(метил)амино]пентан-1-ол
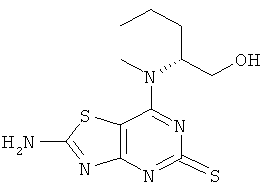
Круглодонную колбу снабжали холодильником сухой лед-этанол и погружали в охлаждающую баню сухой лед-этанол. В колбе конденсировали аммиак (200 мл), а затем добавляли (2R)-2-{[2-амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил](метил)амино}пентан-1-ол (5,43 г, 13,9 ммоль). Полученную смесь оставляли нагреваться до -33°С и небольшими кусочками добавляли металлический натрий до тех пор, пока не появлялось синее окрашивание, которое сохранялось в течение 30 секунд. Затем реакцию гасили добавлением ложки твердого хлорида аммония. Аммиак выпаривали и к остатку добавляли воду (250 мл). Полученную смесь нейтрализовали 1 М соляной кислотой (водн.). Выпавший в осадок продукт собирали фильтрованием, промывали водой и ацетонитрилом и сушили в вакууме с получением 3,38 г (81%-ный выход) указанного в заголовке соединения.
1H ЯМР (DMSO-d6) 12.81 (br s, 1H); 8.45 (br s, 2H), 4.84 (br s, 1H), 3.55-3.40 (m, 2H), 3.02 (s, 3Н), 1.48 (m, 2H), 1.21 (m, 2H), 0.87 (t, 3Н); MS (ESI) m/z 300 [М+1]+.
д) (2R,2'R)-2,2'-{Дитиобис[(2-хлор[1,3]тиазоло[4,5-d]пиримидин-5,7-диил)(метилимино)]}дипентан-1-ол
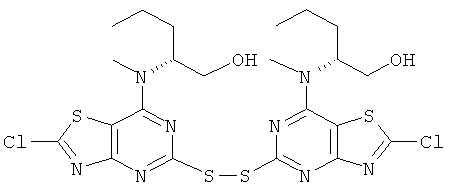
(2R)-2-[(2-Амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)(метил)-амино]пентан-1-ол (1,0 г, 3,34 ммоль) растворяли в ацетонитриле (25 мл) и концентрированной соляной кислоте (40 мл). Нитрит натрия (461 мг, 6,67 ммоль) растворяли в воде (2 мл) и добавляли при 0°С. Реакционную смесь выдерживали при 0°С в течение трех дней. Реакционную смесь выливали на лед и выпавший в осадок продукт собирали фильтрованием и промывали водой. Сушка в вакууме давала указанное в заголовке соединение (800 мг, 75%-ный выход).
MS (ESI) m/z 635 и 637 [M+1]+.
е) (2R,2'R)-2,2'-{Дитиобис[(2-метокси[1,3]тиазоло[4,5-d]пиримидин-5,7-диил)(метилимино)]}дипентан-1-ол
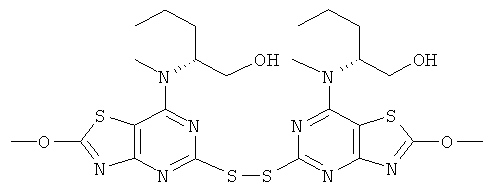
Гидроксид калия (210 мг, 3,75 ммоль), растворенный в метаноле (20 мл), добавляли к (2R,2'R)-2,2'-{дитиобис[(2-хлор[1,3]тиазоло[4,5-d]пиримидин-5,7-диил)(метилимино)]}дипентан-1-олу (795 мг, 1,25 ммоль) в метаноле (40 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение ночи и затем метанол выпаривали. Остаток выливали на лед и получившийся осадок собирали фильтрованием. Фильтрат экстрагировали этилацетатом. Органическую фазу сушили над сульфатом натрия и концентрировали в вакууме, а остаток объединяли с ранее собранным твердым веществом с получением указанного в заголовке соединения, которое использовали на следующей стадии без какой-либо дополнительной очистки.
MS (ESI) m/z 627 [M+1]+.
ж) 5-[7-{[(1R)-1-(Гидроксиметил)](метил)амино}-[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он-5-илдисульфанил]-7-{[(1R)-1-(гидроксиметилбутил]-амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
Сырой (2R,2'R)-2,2'-{дитиобис[(2-метокси[1,3]тиазоло[4,5-d]пиримидин-5,7-диил)(метилимино)]}дипентан-1-ол (1,25 ммоль) с предыдущей стадии растворяли в 1,4-диоксане (25 мл). Добавляли конц. соляную кислоту (0,5 мл) и воду (0,5 мл) и полученную смесь перемешивали при 45°С в течение ночи. Диоксан выпаривали в вакууме и остаток выливали на лед с осаждением продукта, который собирали фильтрованием. Сушка в вакууме давала 590 мг (78%-ный выход за две стадии) указанного в заголовке соединения.
MS (ESI) m/z 599 [M+1]+.
ж) 5-{[(1S)-1-(6-Хлорпиридин-3-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-бутил](метил)амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
Указанное в заголовке соединение получали в соответствии с общим способом G, используя 5-[7-{[(1R)-1-(гидроксиметил)](метил)амино}-[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он-5-илдисульфанил]-7-{[(1R)-1-(гидрокси-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он (0,10 г, 0,167 ммоль), 2-хлор-5-[(1R)-1-хлорэтил]пиридин (Пример 6(е), 0,065 г, 0,37 ммоль) и боргидрид натрия (0,038 г, 1,00 ммоль), получая 0,060 г (41%-ный выход) указанного в заголовке соединения.
1H ЯМР (CDCl3) δ м.д. 8.56-8.38 (m, 1H), 7.87-7.73 (m, 1H), 7.28-7.26 (m, 1H), 4.86 (q, 1H), 4.75-4.62 (m, 1H), 3.76-3.55 (m, 3Н), 3.03 (s, 3H), 1.70-1.63 (m, 3Н), 1.53-1.45 (m, 2H), 1.26-1.21 (m, 2H), 0.95-0.88 (m, 3H);
MS (ESI) m/z 440 и 442 [М+1]+.
Пример 8
Пример 8а
5-{[(1R)1-(3-Фторпиридин-4-ил)этил]тио}-7-{([1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3H)-он и
Пример 8б
5-{[(1S)-1-(3-Фторпиридин-4-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-он
 и
и 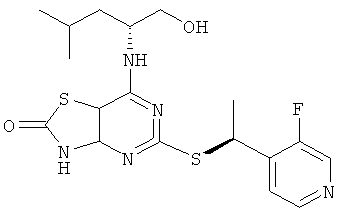
Диастереомерную смесь 5-{[1-(3-фторпиридин-4-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3H)-она (179 мг) из Примера 3 разделяли посредством препаративной HPLC с получением 25 мг первого элюирующегося изомера:
1H ЯМР (DMSO-d6) δ м.д. 12.31 (br s, 1H), 8.51 (m, 1H), 8.38 (d, 1H); 7.62 (m, 1H); 6.97 (br s, 1H); 5.16 (q, 1H); 4.66 (t, 1H); 4.12 (m, 1H); 3.44-3.30 (m, 2H, скрыт сигналом воды), 1.66 (d, 3Н), 1.61-1.27 (m, 3Н), 0.84 (d, 3Н), 0.74 (d, 3Н);
MS (ESI) m/z 424 [M+1]+;
и 45 мг элюирующегося последним изомера:
1H ЯМР (DMSO-d6) δ м.д. 12.35 (br s, 1H), 8.52 (d, 1H), 8.38 (d, 1H); 7.62 (dd, 1H); 7.12 (br s, 1H); 5.15 (q, 1H); 4.62 (t, 1H); 4.21 (m, 1H); 3.35-3.15 (m, 2H, частично скрыт сигналом воды), 1.65 (d, 3Н), 1.63-1.29 (m, 3Н), 0.88 (d, 3Н), 0.85 (d, 3Н); MS (ESI) m/z 424 [М+1]+.
Фармакологические скрининги
Материалы
Рекомбинантный фракталкин человека (hCX3CL1) и рекомбинантный интерлейкин-8 человека (IL-8 или hCXCL8) получали от PeproTech Inc., UK. Рекомбинантный [125I]-фракталкин (человеческий) и [125I] hIL-8 с удельной активностью 2200 Ки/ммоль получали от NEN® Life Science Products, Inc., UK. Fluo4-AM получали от Molecular Probes, US. Все другие химические реактивы имели аналитическую чистоту.
Клетки
Полную кДНК СХ3CR1 человека (GenBank каталожный номер U20350) экстрагировали из мРНК головного мозга человека (Superscript, Life Technologies) и лигировали в вектор pCR-Blunt II ТОРО (InVitrogen). Вставку, соответствующую hCX3CR1, выделяли и затем субклонировали в pcDNA3.1zeo. Плазмидную ДНК получали с помощью набора Plasmid Midi Kit (Qiagen). Используя Superfect Transfection Reagent (Qiagen) согласно инструкциям изготовителя, экспрессионную плазмиду для hCX3CR1 затем вводили в суспензию линии клеток почек человека (HEKS) 293, содержащих вектор для стабильной экспрессии химерного G-белка Gαqi5. Стабильный клон получали посредством селекции с помощью зеоцина (500 мкг/мл) и гигромицина (100 мкг/мл). Для последующего использования клетки поддерживали в модифицированной по Дульбекко среде Игла/питательной смеси Хэма F12 (DMEM/F12), содержащей пиридоксин и дополненной 10% (об./об.) фетальной телячьей сыворотки, 2 мМ L-глутамина, 100 ед./мл пенициллина и 100 мг/мл стрептомицина, 250 мкг/мл зеоцина и 100 мкг/мл гигромицина.
Клетки, экспресирующие человеческий CXCR2, полученные от AstraZeneca Charnwood, культивируют в ЕМЕМ, содержащей глутамакс и дополненной 10% FBS (фетальная телячья сыворотка, от РАА, Austria), 1% заменимых аминокислот (NEAA), 100 ед./мл пенициллина и 100 мкг/мл стрептомицина (PEST) и 500 мкг/мл генетицина/G418.
Получение мембран
Клетки выращивают при 37°С и 5% CO2 и собирают при слиянии 60-80% в буфере, содержащем 10 мМ Трис-HCl, pH 7,4, 5 мМ EDTA, 0,1 мг/мл бацитрацина. Клетки центрифугируют при 300×g в течение 10 минут и осадок ресуспендируют в буфере для сбора (10 мМ Трис-HCl, pH 7,4, 5 мМ этилендиаминтетрауксусной кислоты (EDTA) и 0,1 мг/мл бацитрацина), объединяют и гомогенизируют с помощью гомогенизатора Dounce. Гомогенат центрифугируют при 48000×g в течение 10 минут и ресуспендируют в буфере для сбора, используя Ultra-Turrax T8. Аликвоты мембран хранят при -80°С. Концентрацию белка определяли в микротитрационных планшетах, как описано в Harrington (1990, Anal. Biochem. 186, 285-287).
In vitro анализ связывания с рецептором
Исследования конкурентного связывания [125I]-фракталкина выполняли в планшетах с 96 глубокими лунками на 2 мл (Beckman, Germany) в общем объеме 1000 мкл/лунку. Каждая лунка содержала 10 пМ [125I]-фракталкина и мембраны в количестве, эквивалентном концентрации рецепторов 1 пМ, в буфере для анализа (50 мМ Hepes-KOH, pH 7,4, 10 мМ MgCl2, 1 мМ EDTA, 0,1% (масс./об.) желатина). Десять концентраций (2 точки/log единицу) тестируемого соединения предварительно растворяли в DMSO и добавляли для достижения конечной концентрации 1% (об./об.) DMSO. Анализ инициировали добавлением мембран и инкубировали при 25°С в течение 24 ч. Реакции останавливали быстрым фильтрованием через фильтры со стеклянным волокном Whatman GF/B, предварительно обработанные 0,3%-ным полиэтилимином и затем промытые охлажденным на льду буфером (10 мМ Hepes-KOH, pH 7,4, 500 мМ NaCl), используя харвестер для связывания с рецепторами Brandel. Добавляли сцинтилляционную смесь и определяли радиоактивность в жидкостном сцинтилляционном счетчике Packard 2500TR (Perkin Elmer, USA).
Исследования конкурентного связывания [125I]-hIL-8 выполняют однократно в 96-луночных белых изопланшетах с прозрачным дном с конечным объемом 200 мкл, причем каждая лунка содержит 150 пМ [125I]-hlL-8 (удельная активность 2200 Ки/ммоль), препарат мембраны-SPA (SPA - анализ сцинтилляционной близости), эквивалентный 20 пМ рецепторов, и 1,5 мг SPA-шариков в буфере для анализа [50 мМ HEPES-KOH pH 7,4, 10 мМ MgCl2, 1 мМ EDTA, 0,5% (масс./об.) желатина]. Тестируемые соединения обрабатывали, как ранее. Неспецифическое связывание определяют в присутствии 500 нМ немеченого hIL-8. В качестве эталонного соединения в каждом случае используют агонист hIL-8 (кривая концентрация-ответ от 3 пМ до 30 нМ). Пептидная кривая не содержит DMSO. Реакцию связывания инициируют добавлением 140 мкл препарата мембраны-SPA и образцы инкубируют в темноте при КГ в течение 4 ч. Планшеты для анализа обсчитывают в жидкостном сцинтилляционном счетчике (Wallac MicroBeta® TriLux 1450 от PerkinElmer, USA).
[35S]GTPγS связывание
Исследования [35S]GTPγS связывания проводили в микротитрационных планшетах с прозрачным дном в двух повторностях с 10 концентрациями ингибитора (2 конц./log единицы), разведенного в DMSO (конечная концентрация 1%) и при комнатной температуре. Мембраны, экспрессирующие hCX3CR1 рецептор (конечная концентрация 20 мкг белка/лунку), добавляли вместе со SPA-шариками (конечная концентрация 1 мг/лунку). Все суспендировали в буфере для связывания GTPγS (50 мМ Tris-HCl, 100 мМ NaCl, 0,1% желатина, 15 мкг сапонина/мл и 3 мкл GDP (гуанозин-5'-дифосфат), pH 7,4 при к.т.). Мембраны, SPA-шарики и лекарственные средства подвергали предварительной инкубации в течение 30 минут перед добавлением 310 пМ фракталкина для максимальной стимуляции. Базовую активность определяли как активность, обнаруженную без стимуляции фракталкином (буфер для связывания GTPγS). Спустя еще 30 минут реакцию инициировали добавлением [35S]GTPγS до конечной концентрации 0,1 нМ и конечного объема анализа 0,2 мл. Эксперимент заканчивали через 30 минут центрифугированием при 2000 об/минуту в течение 2×5 минут (в разных направлениях) и радиоактивность определяли в жидкостном сцинтилляционном счетчике (Wallac MicroBeta® TriLux 1450).
Результаты
Типичные величины СХ3CR1 Ki для соединений по настоящему изобретению находятся в диапазоне от приблизительно 0,1 до приблизительно 1000 нМ. Другие величины для CX3CR1 Ki находятся в диапазоне от приблизительно 0,1 нМ до приблизительно 500 нМ. Другие величины для CX3CR1 Ki находятся в диапазоне от приблизительно 0,1 нМ до приблизительно 25 нМ. Результаты анализа in vitro hCX3CR1 связывания для ряда соединений показаны в Таблице 1.
Соединения по настоящему изобретению, где R1 представляет собой Me (содержащие разветвленную тиоалкилпиридильную группу в положении 5), являются как более эффективными антагонистами СХ3CR1 рецептора, так и/или менее эффективными антагонистами CXCR2 рецептора, чем соответствующие эталонные соединения, где R1 представляет собой Н. Такая улучшенная селективность в отношении антагонизма CX3CR1 рецептора, как ожидается, будет приносить значительную терапевтическую пользу.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ 5,7-ДИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ [1,3]ТИАЗОЛО[4,5-d]ПИРИМИДИН-2-(3Н)-АМИНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2007 |
|
RU2437889C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 5,7-ДИЗАМЕЩЕННОГО [1,3] ТИАЗОЛО[4,5-D]ПИРИМИДИН-2(3H)-ОНА | 2006 |
|
RU2411245C9 |
| НОВЫЕ ПРОИЗВОДНЫЕ 5-ЗАМЕЩЕННОГО 7-АМИНО-[1,3]ТИАЗОЛО[4,5-d] ПИРИМИДИНА | 2006 |
|
RU2419623C2 |
| ФОСФАТНЫЕ И ФОСФОНАТНЫЕ ПРОИЗВОДНЫЕ 7-АМИНО-5-ТИОТИАЗОЛО[4,5-d]ПИРИМИДИНОВ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ, СВЯЗАННЫХ С ПОВЫШЕННЫМИ УРОВНЯМИ CX3CR1 И/ИЛИ CX3CL1 | 2019 |
|
RU2801664C2 |
| АЗАИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ПОВРЕЖДЕНИЯХ СУХОЖИЛИЙ И/ИЛИ СВЯЗОК | 2017 |
|
RU2758256C2 |
| Новые модулирующие регуляцию дыхания соединения и способы их получения и применения | 2016 |
|
RU2734506C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ЦИКЛОАЛКАНОВОЕ ПРОИЗВОДНОЕ | 2013 |
|
RU2635354C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| НОВЫЙ ИНГИБИТОР ЦИКЛИНЗАВИСИМОЙ КИНАЗЫ CDK9 | 2018 |
|
RU2738654C1 |
Изобретение относится к соединениям общей формулы (I), где R1 представляет собой СН3; R2 представляет собой галогено или CN; R3 представляет собой H или CH3; R4 представляет собой H или CH3; n представляет собой 0, 1 или 2; и к их фармацевтически приемлемым солям. Также изобретение относится к фармацевтической композиции и к применению соединений формулы (I) в изготовлении лекарственного средства, обладающего антагонистической активностью в отношении CX3CR1 рецептора. Технический результат - соединения формулы (I) в качестве антагонистов CX3CR1 рецептора. 7 н. и 13 з.п. ф-лы, 1 табл.
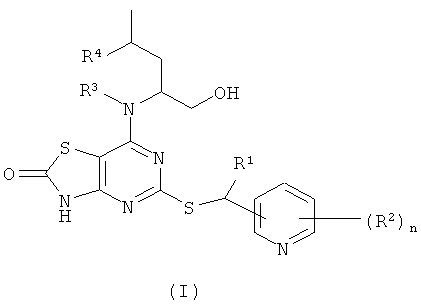
1. Соединение формулы (I)
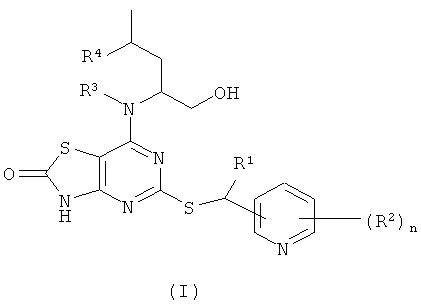
где R1 представляет собой CH3;
R2 представляет собой галогено или CN;
R3 представляет собой H или CH3;
R4 представляет собой H или CH3;
n представляет собой 0, 1 или 2;
в виде свободного основания или его фармацевтически приемлемой соли.
2. Соединение по п.1, где n представляет собой 1.
3. Соединение по п.1, где R2 представляет собой F или Cl.
4. Соединение по п.1, где R2 представляет собой CN.
5. Соединение по п.1, где n представляет собой 1; и R2 представляет собой F, Cl или CN.
6. Соединение по п.1, где пиридин присоединен по своему положению 5 и содержит Cl в положении 2.
7. Соединение по п.1, где пиридин присоединен по своему положению 2 и содержит CN в положении 4.
8. Соединение по п.1, где пиридин присоединен по своему положению 2 и содержит F в положении 5.
9. Соединение по п.1, где пиридин присоединен по своему положению 2 и содержит Cl в положении 5.
10. Соединение по п.1, где пиридин присоединен по своему положению 2 и содержит F в положении 3.
11. Соединение по п.1, где пиридин присоединен по своему положению 4 и содержит F в положении 3.
12. Соединение по п.1, где R3 представляет собой H.
13. Соединение по п.1, где R4 представляет собой CH3.
14. Соединение, выбранное из:
5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
5-{[1-(3-фторпиридин-4-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
5-{[(1S)-1-(3-фторпиридин-4-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
5-{[(1R)-1-(3-фторпиридин-4-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
2-{(1S)-1-[(7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}-2-оксо-2,3-дигидро[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}изоникотинонитрила;
5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио}-7-{[(1R)-1-(гидроксиметил)-бутил]амино}[1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она и
5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио}-7-[[(1R)-1-(гидроксиметил)-бутил](метил)амино][1,3]тиазоло[4,5-d]пиримидин-2(3Н)-она;
в виде свободного основания или фармацевтически приемлемой соли.
15. Соединение по любому из пп.1-14 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства для лечения или профилактики заболеваний или состояний, при которых благоприятным является антагонизм в отношении CX3CR1 рецептора.
16. Фармацевтическая композиция, обладающая антагонистической активностью в отношении CX3CR1 рецептора, содержащая терапевтически эффективное количество соединения по любому из пп.1-14 или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым разбавителем или носителем.
17. Применение соединения формулы (I), как оно определено в любом из пп.1-14, или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий, ревматоидного артрита или астмы.
18. Применение соединения формулы (I), как оно определено в любом из пп.1-14, или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики рассеянного склероза.
19. Применение соединения формулы (I), как оно определено в любом из пп.1-14, или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики атеросклероза путем изменения состава бляшек с целью уменьшения риска разрыва бляшек и атеротромботических явлений.
20. Применение соединения формулы (I), как оно определено в любом из пп.1-14, или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики атеросклероза путем предупреждения и/или уменьшения формирования новых атеросклеротических поражений или бляшек и/или путем предупреждения или замедления развития уже существующих поражений и бляшек.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2002125451 A, 10.01.2004 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |

